
組換えタンパク質を工業規模で製造する方法
工業規模の目的タンパク質の製造方法は、目的タンパク質をコードするDNAの、事前選択された部位における細菌細胞のゲノムの相同組換えによる組み込みに基づく。工業規模の組換えタンパク質の製造は、流加培養方式、半連続方式またはケモスタットで行われる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、細菌宿主細胞における工業規模における組換えタンパク質の製造に関する。
【背景技術】
【0002】
現在、細菌宿主、特に大腸菌(Escherichia coli)における組換えタンパク質の製造には、主としてプラスミドベースの発現系が用いられている。これらの系は遺伝子量が多く、樹立されており、利用可能なクローニングプロトコルは扱いやすいため、広く受け入れられるようになった。
【0003】
プラスミドベースの発現は、1細胞当たり最大数百までのプラスミドコピー数を特徴とする(Baneyx、1999年)。発現プラスミドは、通例、プロモーターの制御下に有る目的遺伝子、複製起点(ori)およびプラスミド含有クローンの選択のためのマーカー遺伝子を有する。加えて、しばしば、前記プラスミド(すなわちベクター)上にコード配列または非コード配列または非機能性バックボーン配列が存在する。プラスミドの存在および対応するその複製機構により、宿主細胞の代謝は改変され(Diaz-RizziおよびHernandez、2000年)、その細胞に高い代謝負荷が課され、それによって組換えタンパク質製造のためのそれらの資源が限定を受ける。加えて、遺伝子量の多さと強力なプロモーターの組み合わせの適用は、組換えタンパク質の製造速度の増大を引き起こすが、宿主細胞にとってこの速度は通例高すぎるため、急速で不可逆的な細胞代謝の崩壊をもたらす恐れがある。その結果として、宿主細胞の潜在能力は、プラスミドベースの系では十分に活用できず、その結果として組換えタンパク質の収量は低くなり、品質は低下する。このように、プラスミドベースの発現系の主な欠点の1つは、プラスミドの複製および維持に必要な栄養素およびエネルギーの必要量の増加にあるとすることができる。
【0004】
プラスミドベースの系における他の典型的な現象には、培養過程におけるプラスミドコピー数の変化がある。組換えタンパク質製造には、高い発現率で、チャージされていないtRNAのプールの増加をもたらす飢餓および細胞ストレスが付随する。このことは、プラスミドコピー数(PCN)の制御機構との抵触をもたらす。その結果として、PCNは急速に増加し、培養プロセスの崩壊(いわゆる“暴走(run-away)効果”)を引き起こす。組換え遺伝子発現の誘導後のColE1型プラスミドの暴走現象は、遺伝子量の顕著な増加をもたらしうる(Grabherrら、2002年)。
【0005】
分離不安定性(すなわち無プラスミド宿主細胞の生成)および構造不安定性(すなわちプラスミドの配列における変異)は、プラスミドベースの系においてしばしば見られるさらなる問題である。細胞分裂の間に、細胞はプラスミドを失う場合があり、その結果として目的遺伝子も失う場合がある。このようなプラスミド喪失は、いくつかの外部要因に依存し、細胞分裂回数(世代数)と共に増加する。このことは、プラスミドベースの発酵が、世代数または細胞倍加数に関して限定を受けていることを意味する(Summers、1991年)。
【0006】
すべてを考慮に入れれば、プラスミドベースの発現系のこれらの特性により、組換えタンパク質の収量は限定され、プロセス操作の制御性およびプロセスの経済性は低い。それにもかかわらず、より効率的な代替法を欠くため、プラスミドベースの細菌発現系は、工業規模の異種組換えタンパク質の製造および単離のための最新技術となった。
【0007】
従って、プラスミドベースの発現に代わるものとして、ゲノムベースの発現系が探求されてきた。大腸菌(E. coli)内で染色体により発現される異種タンパク質の、公知で広く利用されている例には、T7ファージのRNAポリメラーゼがあり、これはプラスミドベースの目的遺伝子のプラスミドベースの転写の目的に役立つ。米国特許第4,952,496号に初めて記載されたこの系は部位非特異的組み込みに基づいており、得られた細菌株(例えば大腸菌株BL21(DE3)またはHMS174(DE3))は溶原菌となっている。望ましくないファージ放出に続いて起こりうる細胞溶解を防ぐために、ファージ溶原菌を生じることなくT7ポリメラーゼ遺伝子が染色体に組み込まれた(すなわちT7ポリメラーゼ遺伝子が相同組換えにより挿入された)(WO2003/050240)。同様に、組換えタンパク質のプラスミドベースの発現の目的で、最近、コリネバクテリウム・アセトアシドフィラム(Corynebacterium acetoacidophilum)のゲノムへのT7 RNAポリメラーゼ遺伝子の組み込みが記載された(米国特許第2006/0003404号)。
【0008】
タンパク質発現研究、配列挿入(例えば制限部位、部位特異的組換え酵素標的部位、タンパク質タグ、機能性遺伝子、プロモーターの)、欠失および置換のために、λファージのRed組換え酵素機能(Murphy、1998年)またはRacプロファージのRecE/RecT組換え酵素機能(Zhangら、1998年)により組換えが仲介される、核酸配列のゲノム組み込みのための他の方法が提案されている(Muyrersら、2000年)。
【0009】
WO2001/18222は、エタノール生産特性を宿主に付与するために、大腸菌の染色体プロモーターの下流に、ザイモモナス・モビリス(Zymomonas mobilis)由来のエタノール経路遺伝子を挿入するための、サッカロミセス・セレビジエ(Saccharomyces cerevisiae)FLP(フリッパーゼ)/FRT(フリッパーゼ認識標的)組換え系(Posfaiら、1994年)に基づく染色体組み込み方法の利用を記載している。環状ベクターの染色体組み込み後に、正確に配列(マーカーおよびレプリコン)を除去するためにFLP系が用いられた(Martinez-Moralesら、1999年)。
【0010】
染色体遺伝子置換(Murphy、1998年)または遺伝子破壊(DatsenkoおよびWanner、2000年)のための抗生物質耐性遺伝子の挿入のために、FLPベースのマーカー切除戦略と組み合わせたλ Red法を用いる、短い相同性配列を有する直線状DNAフラグメントを挿入する、DatsenkoおよびWanner(2000年)により開発された方法が使用された。同様に、この方法による、前記タンパク質の前(すなわち上流)でのマーカー、プロモーターおよびHisタグの染色体挿入によるタグ付き相同大腸菌タンパク質の過剰発現が提案されている(Jain、2005年)。
【0011】
抗生物質耐性マーカーなしでプラスミドを増殖させるための宿主/ベクター選択系を確立するために、大腸菌の染色体にリプレッサー分子を組み込むゲノムベースの発現も提案された(WO2006/029985)。その選択系に関連して、プラスミド由来RNAI/IIアンチセンス反応を明らかにするために、モデル系としてレポータータンパク質(緑色蛍光タンパク質)のゲノム挿入が用いられた(Pfaffenzellerら、2006年)。同様に、Zhouら(2004年)はレポーター分子として緑色蛍光タンパク質を含む環状ベクターの組み込みを記載した。
【0012】
WO1996/40722においては、細菌染色体への選択マーカー(すなわち大腸菌のattB部位に)を含む環状ベクター(いわゆる“環状染色体導入DNA”(CTD))の組み込みを使用する方法が記載されている。その方法において、選択マーカーに隣接する二倍のDNA配列を用いることにより、染色体の遺伝子量の増幅が達成された。それによって、得られた染色体の遺伝子量は1細胞当たりおおよそ15〜40コピーであったが、これは一般的に用いられるプラスミドベクターにより達成されるものと同様であった。細菌ゲノムに組み込まれた染色体導入DNAを含むクローンの培養により、プラスミドベースの系で得られるものと同様な組換えタンパク質のレベルが得られた(Olsonら、1998年)。この方法は、CTDのin vitro連結反応を必要とし、組み込みに関してはattB部位に限定されている。
【発明の概要】
【0013】
本発明の目的は、組換えタンパク質を製造するための、新規な工業規模の発現方法を提供することであった。
【0014】
本発明は、
a)細菌発現宿主細胞の集団を培養し、
b)目的タンパク質を採取し、
c)それを分離及び精製する
工程を含む、目的組換えタンパク質を工業規模で製造する方法であって、
工程a)において、培養はフィード培地の添加を用いる方式で行われ、前記方式は流加培養方式、半連続方式または連続方式から選択され、かつ、前記発現宿主細胞は、前記タンパク質の発現を可能にするプロモーターの制御下にある、目的タンパク質をコードするDNA配列を有するDNA構築物を、ゲノム中に組み込まれて含む前記方法に関する。
【0015】
次に、用語“宿主細胞”は、目的遺伝子を含むDNA構築物で形質転換する前の出発細胞として用いられる細菌細胞を意味する。形質転換により目的遺伝子をそのゲノムに組み込んで含む直線状DNA構築物を有する細胞を“発現宿主細胞”と呼ぶ。
【0016】
発現カセットは、その重要な要素として、目的遺伝子、プロモーターおよびリボソーム結合部位(RBS)を含む。さらに、発現カセットは、“ヘルパータンパク質(helper protein)”、例えばマーカーまたは発現宿主細胞の増殖を助けるタンパク質、例えば糖またはタンパク質の代謝に関連する遺伝子および/または目的遺伝子の発現を改善するタンパク質(例えばシャペロン分子(GroElまたはGroEsなど)をコードする配列または転写および翻訳に関与する遺伝子をコードする配列を含むことができる。
【0017】
発明の詳細な説明
目的タンパク質
目的タンパク質に関しては、制限はない。原則として、工業規模で製造される任意のタンパク質、例えば工業用タンパク質または治療用タンパク質であることができる。本発明の方法により製造できるタンパク質の例は、限定するものではないが、酵素、制御タンパク質、受容体、ペプチド、例えばペプチドホルモン、サイトカイン、膜または輸送タンパク質である。目的タンパク質はまた、予防接種に使用する抗原、ワクチン、抗原結合タンパク質、免疫刺激タンパク質、アレルゲン、完全長抗体または抗体フラグメントもしくは誘導体であることもできる。抗体誘導体は、一本鎖抗体、(scFV)、Fabフラグメント、FVフラグメント、単一ドメイン抗体(VHまたはVLフラグメント)、ドメイン抗体、例えばラクダ科動物単一ドメイン抗体(VHH、ナノボディ)または他の抗体フォーマット、例えばAndersenおよびReilly(2004年)またはHolligerおよびHudson(2005年)に記載されているものの群から選択することができる。目的タンパク質をコードするDNA分子を“目的遺伝子”とも呼ぶ。
【0018】
プロモーター
本発明の意味において、“プロモーター”は、RNAポリメラーゼの結合および転写開始を可能にする発現調節因子である。本発明の一実施形態において、目的遺伝子は“強力な”プロモーターの制御下に有る。強力なプロモーターは、一方ではRNAポリメラーゼ、通例天然に存在する対応するRNAポリメラーゼに対するプロモーター配列の高い結合親和性を特徴とし、他方ではそのRNAポリメラーゼによるmRNAの高い産生速度を特徴とする。
【0019】
好ましくは、目的遺伝子は誘導性プロモーターの制御下に有る。誘導性プロモーターは、外部要因、例えば誘導原(“誘導物質”とも呼ばれる)分子の存在もしくはリプレッサー分子の非存在または物理的要因、例えば温度、浸透圧もしくはpH値の上昇もしくは低下により制御できるプロモーターである。種々のプロモーターおよびそれぞれの誘導原理は、Makrides(1996年)により概説されている。
【0020】
本発明において、プラスミドベースの発現のために開発されてきたプロモーターを使用することができるが、強力なプロモーターの中で、バクテリオファージT7のT7プロモーターが最も広く使用されてきた。
【0021】
好ましい側面において、プロモーターはT7プロモーターである。T7プロモーターおよび、lacプロモーターの制御下に有るT7ポリメラーゼ遺伝子の組み合わせを含むT7ベース発現系は、細菌細胞および真核細胞のどちらにおいても、組換えタンパク質の大規模発現に広く使用されている。この系はT7 RNAポリメラーゼを使用し、その転写速度は大腸菌のRNAポリメラーゼよりも数倍高い。T7プロモーターからの発現は、T7プロモーターに厳密に特異的なT7 RNAポリメラーゼの制御下に有る(Chamberlinら、1970年)。すなわち、T7プロモーターは、バクテリオファージT7のポリメラーゼによってのみ利用されうる。IPTGを培地に添加するとき、lacプロモーターからの転写によりT7 RNAポリメラーゼが発現される。
【0022】
本発明の意味において、用語“T7プロモーター"は、バクテリオファージT7のゲノムに存在するプロモーターばかりでなく、T7 RNAポリメラーゼによる転写を仲介する能力を有するこのようなプロモーターのコンセンサス配列およびバリアントを含む。バクテリオファージT7は、17の異なるプロモーター配列を含有し、そのすべてが高度に保存されたヌクレオチド配列を含む(OakleyおよびColeman、1977年;PanayotatosおよびWells、1979年)。
【0023】
本発明の特定の実施形態によれば、目的遺伝子は、tacもしくはtrcプロモーター、lacもしくはlacUV5プロモーター(すべてラクトースまたはそのアナログであるIPTG(イソプロピルチオール-β-D-ガラクトシドにより誘導される))、強力に調節できるaraBADプロモーター(PBAD;Guzmanら、1995年、アラビノースにより調節される)、trpプロモーター(β-インドールアクリル酸添加またはトリプトファン飢餓により誘導され、トリプトファン添加により抑制される)、λプロモーターpL(λ)(温度上昇による誘導)、phoAプロモーター(リン酸飢餓により誘導される)または組換えタンパク質発現に適した他のプロモーター(Makrides、1996年)の制御下に有ることができ、これらはすべて大腸菌のRNAポリメラーゼを利用する。
【0024】
誘導性プロモーターには、“リーキー”発現行動を示すものがある。このようなプロモーター(いわゆる“リーキープロモーター(leaky promoter)”)は、原則として誘導性であるが、それにもかかわらず外部からの誘導なしに、基礎発現も示す。非誘導条件下でリーキー発現を示す誘導性プロモーターは、同様に構成的プロモーターとして作用することができる(すなわちこれらは定常的・連続的に活性であり、特定の培養条件の結果として活性化または促進されることもできる)。リーキープロモーターは、連続運転培養プロセスに特に有用な可能性がある。例には、T7プロモーターおよびtrpプロモーターがある。
【0025】
本発明の方法において、プロモーターは構成的であることができる。すなわち一方では誘導を、他方ではおそらく抑制を必要とせずに発現を調節するプロモーターであることができる。従って、継続的で定常的な発現が特定のレベルで行われる。誘導物質を必要としないという利点は別にして、構成的プロモーターは、方法が連続的な(下記のように)実施形態に特に有用である。例として、強力な構成的HCDプロモーター(Pooら、2002;Jeongら、2004年)を構成発現に適用することができる。
【0026】
細菌細胞のゲノムに組み込まれる直線状カセットは、目的遺伝子のすぐ上流にシャイン・ダルガーノ(SD)配列(リボソーム結合部位(RBS)とも呼ばれる)も含む。
【0027】
発現カートリッジ
次に、細菌ゲノムに組み込まれる直線状または環状DNA構築物は、“発現カートリッジ”または“カートリッジ”とも呼ばれる。組み込みの結果として、発現宿主細胞は組み込み“発現カセット”を有する。好ましくは、カートリッジは、本質的に、プロモーター、目的遺伝子、リボソーム結合部位およびゲノム領域に相同性を示し、相同組換えを可能にする、末端で隣接する2つの領域(図10に示す挿入カートリッジ参照)を含む直線状DNA構築物である。加えて、以下に詳しく述べるように、他の配列を含むことができる;例えば、抗生物質選択マーカー、原栄養性選択マーカーまたは蛍光マーカーをコードする配列、代謝遺伝子、タンパク質発現(例えばT7 RNAポリメラーゼ遺伝子など)を改善する遺伝子または、組み込み後に特定の配列(例えば抗生物質耐性遺伝子)を除去することを可能にする2つのフリッパーゼ認識標的部位(FRT)をコードするマーカー。
【0028】
カートリッジは、当該技術分野で公知の方法により合成され、増幅される。直線状カートリッジの場合は、通例ポリメラーゼ連鎖反応(PCR)(WilsonおよびWalker、2005年)による。直線状カートリッジは、通例構築するのがより容易であるため、本発明の方法に用いる発現宿主細胞を得るには直線状カートリッジが好ましい。さらに、直線状発現カートリッジの使用には、カートリッジに隣接する相同領域のそれぞれの設計によりゲノム組み込み部位を自由に選択できるという利点がある。それによって、直線状発現カートリッジの組み込みにより、ゲノム領域に関するより大きな可変性が可能となる。
【0029】
宿主細胞
細菌宿主細胞に関しては、原則として制限はない。目的遺伝子の挿入のための遺伝子操作、好都合には部位特異的組み込みが可能であり、工業規模で培養することができる限り、細菌宿主細胞は真正細菌(グラム陽性またはグラム陰性)であることもできるし、古細菌であることもできる。好ましくは、宿主細胞は、高い細胞密度まで培養することが可能な性質を有する。
【0030】
工業用組換えタンパク質の製造に適することが示された細菌宿主細胞の例には、大腸菌(Lee、1996年;HannigおよびMakrides、1998年)、バチルス・ズブチリス(Bacillus subtilis)、シュードモナス・フルオレッセンス(Pseudomonas fluorescens)(Squiresら、2004年;Retallackら、2006年)ばかりでなく、種々のコリネバクテリウム(Corynebacterium)(米国特許第2006/0003404 A1号)およびラクトコッカス・ラクティス(Lactococcus lactis)株(Mierauら、2005年)がある。好ましくは、宿主細胞は大腸菌細胞である。
【0031】
宿主細胞に対する他の必要条件は、目的遺伝子を調節するプロモーターに結合できるRNAポリメラーゼをそれが含むことである。RNAポリメラーゼは、宿主細胞に対して内在性であることもできるし、外来性であることもできる。
【0032】
好ましくは、外来性の強力なRNAポリメラーゼを有する宿主細胞が用いられる。最も好ましくは、外来性RNAポリメラーゼ(例えば、T7プロモーターを用いる場合は、いわゆる“T7株”におけるT7様RNAポリメラーゼなど)をそのゲノムに組み込んで含むように操作された宿主株、例えば大腸菌株が用いられる。広く使用され、市販されているT7株の例には、例えばBL21(DE3)、HMS174(DE3)およびそれらの派生株または近縁株がある(Novagen社、pET Systemmanual、第11版)。これらの株は、lacUV5プロモーターの制御下にあるT7 RNAポリメラーゼ遺伝子を含むDE3溶原菌である。IPTGでの誘導により、T7 RNAポリメラーゼの産生が可能となり、次いでそれがT7プロモーターの制御下に有る目的遺伝子の発現を指示する。
【0033】
好ましくは、例えばWO2003/050240に記載されているように、ファージ溶原菌を生じることなしにゲノムに組み込まれたT7 RNAポリメラーゼを含む宿主細胞が用いられる(ファージ溶原菌を生じない細胞は“非溶原性”である)。
【0034】
"T7様RNAポリメラーゼ"は、限定するものではないが、他のT7様ファージ由来のRNAポリメラーゼ、例えば米国特許第4,952,496号に開示されているT3 RNAポリメラーゼを含む。T7様RNAポリメラーゼの必要条件は、高度に特異的な同種プロモーターが存在しなければならないことおよび前記RNAポリメラーゼの存在下で目的遺伝子が急速に転写されることである。
【0035】
あるいは、T7 RNAポリメラーゼを含む前記の株を使用するために、例えば、米国特許第4,952,496号、米国特許第2006/0003404号またはWO2003/050240に記載されているようにして、このような株をあらたに作成することができる。
【0036】
ファージDE3によりゲノムベースのT7 RNAポリメラーゼを受け取った大腸菌宿主細胞株BL21(DE3)またはHMS174(DE3)は溶原性である。溶原株は、望ましくないファージ放出および細胞溶解を引き起こす溶菌特性を潜在的に示す不都合を有する。従って、本発明に関しては、宿主細胞に含まれるT7 RNAポリメラーゼが、宿主細胞ゲノムにおける残りのファージ配列の挿入をさけるか、好ましくは排除する方法により組み込まれていることが好ましい。本発明の好ましい実施形態において、例えば、DatsenkoおよびWanner(2000年)により記載された方法に従って、部位特異的組み込みにより細胞のゲノムにT7ポリメラーゼを組み込むことにより宿主細胞が得られた。
【0037】
T7系を用いる場合、T7 RNAポリメラーゼと組み合わせて、そのプロモーター(好ましくはlacUV5プロモーター)をそのゲノムに組み込んで有する宿主細胞を用いることができる。
【0038】
あるいは、T7 RNAポリメラーゼは、発現カートリッジの要素、すなわち発現カセットの一部として細胞のゲノムに組み込まれた要素として提供できる。
【0039】
宿主細胞に対する他の必要条件は、相同組換えを行うその能力である(これは、ゲノムへの発現カートリッジの組み込みおよび場合によりP1形質導入に関連する)。
【0040】
従って、宿主細胞は組換えタンパク質RecAの機能を有することが好ましい。しかしながら、RecAは培養中に望ましくない組換え事象を引き起こす恐れがあるので、宿主細胞は、そのゲノムrecA部位にゲノム変異(機能不全にする)を有し、その代わりに、ヘルパープラスミドに存在するrecA配列により提供されるRecA機能を有することが好ましいが、これはヘルパープラスミドの温度感受性レプリコン(DatsenkoおよびWanner、2000年)を用いて組換え後に除去(治癒)することができる。
【0041】
組換えを考慮すれば、宿主細胞は、RecAに加えて、組換えタンパク質(例えばExo、BetaおよびGam)をコードするDNA配列をそのゲノムに含むことが好ましい。この場合は、すでにその特徴を有する宿主細胞を選択することもできるし、遺伝子工学によりこれらの配列を挿入して、あらたに宿主細胞を作成することもできる。
【0042】
部位特異的遺伝子挿入を考慮すれば、宿主細胞に対する他の必要条件は、その配列が知られ、破壊または別なふうに操作して、細胞に不利益を与えることなく異種配列を挿入することができる少なくとも1つのゲノム領域(コードまたは任意の非コード・機能性または非機能性領域あるいは未知の機能を有する領域のどちらか)を宿主細胞が含むことである。
【0043】
特定の実施形態において、宿主細胞は、選択を考慮してそのゲノムにマーカー遺伝子を含む。
【0044】
組み込み遺伝子座
組み込み遺伝子座に関しては、本発明に用いられる発現系は、幅広いバリエーションが可能である。原則として、既知の配列を有する任意の遺伝子座を選択することができるが、ただし、その配列の機能が重要でないか、あるいは重要である場合、補完することができる(例えば栄養要求性の場合など)ことを条件とする。
【0045】
組み込み遺伝子座を選択する場合には、いわゆる“適応進化”(Herringら、2006年)によって引き起こされるDNAの変異頻度が大腸菌のゲノムの全域で変化することおよび、染色体にコードされた組換え遺伝子発現により引き起こされる代謝負荷が組み込み部位での変異頻度の増加を引き起こす恐れがあることを考慮する必要がある。安定で頑健な発現宿主細胞を得るためには、組み込み部位として、より低い変異頻度をもたらす高度に保存されたゲノム領域を選択することが好ましい。このような高度に保存された大腸菌ゲノム領域には、例えば、リボソームの成分をコードする遺伝子またはペプチドグリカンの生合成に関与する遺伝子があり(Mauら、2006年)、発現カートリッジの組み込みのためにこれらの領域が選択されることが好ましい。その際、正確な組み込み遺伝子座は、機能性遺伝子が破壊も障害も受けず、どちらかと言えば組み込み部位が非機能性領域に位置するように選択される。
【0046】
カートリッジの組み込みのために選択できる既知の配列を有するゲノム領域は、非必須遺伝子またはその一部のコード領域;重要でない非コード機能性領域(すなわちプロモーター、トランスポゾンなど)、その欠失が特定の目的タンパク質の製造を考慮して有利な可能性のある遺伝子、例えば特定のプロテアーゼ、OmpTのような外膜タンパク質、産物の潜在的汚染物質、代謝のタンパク質、(例えば所定の宿主株および/または発酵プロセスに望ましくないかまたは重要でない糖分子の代謝に関連する)またはストレスシグナル伝達経路、例えばアミノ酸飢餓の間tRNAおよびrRNAの合成を抑制する、原核生物の転写制御機構である緊縮応答において生じる経路をコードする遺伝子から選択できる(Cashel、1969年)。
【0047】
あるいは、組み込み部位は、組み込み後にマーカー表現型の消失についての選択を可能にするマーカー遺伝子であることができる。その欠失が蛍光の消失をもたらし、発現カセットを含むクローンの視覚的検出を可能にする、緑色蛍光タンパク質などの蛍光マーカーが使用されることが好ましい(図10)。
【0048】
あるいは、組み込みのために選択するのに有用な部位は、欠失したとき栄養要求性を示す機能である。この場合は、組み込み部位は、生合成または代謝経路に関与する酵素であって、その欠失が栄養要求株をもたらす酵素であることができる。陽性クローン、すなわち発現カセットを含むクローンは、前記酵素の基質または前駆体分子に対する栄養要求性に関して選択することができる(図10)。
【0049】
あるいは、組み込み部位は、発現カセットに存在する対応原栄養性マーカー(すなわち欠損配列を補完または置換する配列)により置換/補完し、それにより原栄養性選択を可能にする栄養要求性マーカー(非機能性遺伝子、すなわち欠損遺伝子)であることができる。
【0050】
一側面において、領域は非必須遺伝子である。一側面によれば、領域は、それ自体細胞に非必須な遺伝子であることができる。
【0051】
非必須細菌遺伝子は文献で知られ、例えばGerdesら(2002年および2003年)、PEC(大腸菌ゲノム情報(Profiling the E. coli Chromosome))データベース(http://www.shigen.nig.ac.jp/ecoli/pec/genes.jsp)またはいわゆる“Keio collection”(Babaら、2006年)から知られる。
【0052】
非必須遺伝子の1例にRecAがある。この部位に発現カセットを組み込むことにより、宿主細胞の必要条件との関連で前述したゲノム変異が得られる。
【0053】
適切な組み込み部位、例えば利用しやすいおよび/またはより高い発現率を得ることが予想される部位は予備スクリーニングで決定することができる。組み込みカートリッジが、例えば実施例5で用いられた、緑色蛍光タンパク質をコードするカートリッジのような、可変要素として特定の組み込み部位を考慮して事前選択された種々の組換え配列を特徴とし、不変要素として、誘導性プロモーターの制御下に有る、容易に検出できるタンパク質をコードするDNA配列であるサロゲイト“目的遺伝子”を含む組み込みおよび選択のための基本配列を特徴とする、Keio collection(Babaら、2006年)による一連の単一変異欠失を作成することにより、このようなスクリーニングを行うことができる。このようにして作成した単一ノックアウト変異体の発現レベルは、蛍光測定により容易に定量できる。この手順の結果に基づき、所望の標的タンパク質の発現レベルのカスタマイズは、組み込み部位および/または組み込みカートリッジ数を変化させることにより達成できる。
【0054】
宿主細胞が、出発細胞の特徴として、あるいは遺伝子工学により得られたものとして、組換えタンパク質(例えばExo、BetaおよびGam)をそのゲノム中にコードするDNA配列を含む実施形態において、これらの組換えタンパク質配列が位置するゲノム部位で組み込みを起こすことができる。発現カートリッジの組み込みにより、組換えタンパク質をコードする配列は破壊または除去され、その結果として、プラスミドがコードするヘルパータンパク質の場合は、別の段階で除去する必要がない(図9)。
【0055】
組み込み方法
前述のように、組み込み遺伝子座に関しては、既知の配列を有する任意の遺伝子座を選択することができる。ただし、その配列の機能が重要でないか、あるいは重要である場合、補完することができることを条件とする。好ましい実施形態において、直線状カートリッジの組み込みは、attB部位またはattTn7部位などの結合部位で行われ、これらはよく立証された組み込み部位である。しかしながら、前述のように、この系の順応性により、任意の事前選択された遺伝子座で目的遺伝子を挿入することができる。
【0056】
細菌ゲノムへの目的遺伝子の組み込みは、慣用法により、例えば、attTn7部位に関して記載されているように(Rogersら、1986年;WaddellおよびCraig、1988年;Craig、1989年)、染色体の特定部位に相同な隣接する配列を含む直線状カートリッジを用いることにより達成できる。カートリッジは、大腸菌株、例えばプラスミドpKD46を含む大腸菌MG1655(DatsenkoおよびWanner;2000年)の細胞に形質転換される。このプラスミドは、in vivoで組換えを促進するγファージ由来Red機能(γβ exo)を有する。
【0057】
あるいは、最初に中間ドナー宿主細胞のゲノムに発現カセットを組み込むことができ、それからファージP1での形質導入(SternbergおよびHoess、1983;およびLennox、1955年)により宿主細胞にそれを移すことができるが、ドナー細胞には、例えば、欠損γプロファージを有する大腸菌K12株MG1655またはDY378(Yuら、2000年)がある。手短かに言えば、P1形質導入は、1つの"ドナー"株からもう1つの"レシピエント"株に選択遺伝子マーカーを移すために用いられる方法である。細菌培養におけるファージの複製および溶菌中に、低い割合のファージ粒子が、発現カセットを含むゲノムセグメントを含む。ひとたびドナー細胞からファージ集団が作成されれば、レシピエント細胞(本発明の場合は宿主細胞)を感染させるためにこのファージが用いられる。P1ゲノムをパッケージングしたファージにより細菌の大部分は溶菌するが、ファージの一部はドナー宿主由来のゲノムセグメントを注入する。次いで、相同組換えにより、既存の相同のセグメントと入ってくるゲノムセグメントとを置換することができる。ドナー細菌のゲノムセグメントを選択するための培地に感染したレシピエント細菌をプレーティングする(抗生物質耐性、原栄養性、蛍光標識など)。ファージの感染性を調節しなければならないことは重要である。そうでなければ、近くの細胞から放出されたファージが、形質導入粒子に感染した細菌に感染し、溶菌してしまうからである。ファージP1は感染性にカルシウムを必要とするので、カルシウムの存在および非存在において増殖させることによりファージの感染性を調節することができる。カルシウムキレート化剤であるクエン酸塩が通例用いられる。なぜならば、クエン酸塩は遊離カルシウムの濃度を(クエン酸カルシウムの形成により)P1感染を抑制するのに十分低い濃度に低下させるが、細胞をカルシウム飢餓にさせるまでは低下させないからである。
【0058】
組み込み手順の簡単化のために、図9に示すように、DatsenkoおよびWanner(2000年)に従ってプラスミドpKD20またはpKD46を用いて組換えタンパク質を得る代わりに、組換え機能をコードする遺伝子exo、betaおよびgamを宿主細胞の染色体に事前に存在させることができる。組換えタンパク質の発現は誘導性プロモーター、例えばfrt、lac、tac、T7またはaraPプロモーターにより調節できる。対応する誘導物質の適用により、こうして得られた改変株を、組換えタンパク質の部位での直線状発現カートリッジの組み込みに使用することができる。それによって、組換えタンパク質が最初に発現され、次いで前記組換えタンパク質をコードするゲノム領域に発現カートリッジを組み込むのに利用される。それによって、組換えの結果として、かつ本実施形態の特別な利点として、組換えタンパク質をコードする遺伝子が除去(切除)または破壊される(図9)。さらにまた、本実施形態は、プラスミドがこれらの組換え機能をコードする必要性がないという利点を有する。
【0059】
本発明に有用な他の組み込み方法の例には、限定するものではないが、例えばRed/ET組換えに基づく例がある(Muyrersら、2002年;Wenzelら、2005年;Vetcherら、2005年)。
【0060】
選択マーカー
陽性クローン、すなわち発現カセットを含むクローンは、マーカー遺伝子によって選択できる。
【0061】
いくつかの実施形態において、マーカー遺伝子、例えば抗生物質耐性遺伝子または蛍光タンパク質、例えばGFPをコードする遺伝子を事前にそのゲノムに組み込んで含む宿主細胞が用いられる(図10に示すように)。この場合は、選択マーカーを含まない発現カートリッジが、染色体のマーカー遺伝子の遺伝子座に組み込まれ、それぞれの表現型の喪失/消失によって陽性クローンが選択される。例えば、陽性クローンは、抗生物質の感受性または蛍光の消失によって選択され、培養プレート上で直接に可視化できる。マーカーは発現カセットにより中断されるかまたは完全に置換され、組み込み後に機能性マーカー配列は存在せず、抗生物質耐性遺伝子の場合のように望ましくない場合でも除去する必要はないという利点をこれらの実施形態は有する。
【0062】
あるいは、マーカー遺伝子は発現カートリッジの一部である。選択に用いられるマーカーが抗生物質耐性(例えばカナマイシンまたはクロラムフェニコール耐性)を付与する遺伝子である場合、図7に示すように、陽性クローンは抗生物質耐性(すなわちそれぞれの抗生物質の存在下での増殖)により選択される。好ましくは、抗生物質によるクローンの選択には、カナマイシンが用いられる。
【0063】
マーカー遺伝子(宿主細胞のゲノムに存在するものであるか、または発現カートリッジを用いて導入されたものであるかにかかわらず)は、図7に概略を示すように、例えば、酵母2μプラスミドの部位特異的組換え系であるFLP組換え酵素およびその組換え標的部位FRT(DatsenkoおよびWanner、2000年)に基づくFLP組換え酵素機能を用いることによるカセットの組み込みにより除去できる。
【0064】
特定の実施形態において、発現細胞は、選択マーカー遺伝子、例えばクロラムフェニコールまたはカナマイシンなどの抗生物質耐性遺伝子、蛍光マーカーまたは糖もしくはアミノ酸の代謝経路に関与する遺伝子の欠損を有するように操作されている。この場合、目的遺伝子を有するカートリッジは、マーカー遺伝子の欠失部分を有し、マーカー遺伝子の組み込みによりその機能性を回復する。一例として、カートリッジはその末端の1つにマーカー遺伝子の欠失部分を含み、ゲノムに組み込まれた欠損マーカー遺伝子に隣接して直接に組み込まれ、それにより2つのフラグメントの融合によりマーカー遺伝子が完全となり、その機能的発現が可能となる。抗生物質耐性遺伝子の場合は、発現カセットを含む細胞は特定の抗生物質に対して耐性であり、蛍光マーカーの場合は、蛍光により細胞を可視化することができ、代謝経路遺伝子の場合は、細胞はそれぞれの成分を代謝する能力を得る(図11に概略を示す)。この実施形態の利点は、先行技術と比較して、カートリッジのマーカー遺伝子のほんの短い部分しか合成を必要とせず、より短いかまたは小さい挿入カートリッジを可能とすることである。
【0065】
特定の実施形態において、陽性クローン(すなわち発現カセットを有するクローン)の選択は、宿主細胞の栄養要求性の補正(すなわち補完)により行われる。図8に概略を示すこの実施形態において、簡単な方法で陽性形質転換細胞コロニーの選択を可能にするように選択された変異を有する宿主細胞、例えばその増殖に必須である化合物を合成できないようにする欠失または変異(このような変異を“栄養要求性マーカー”と呼ぶ)を有する株が用いられる。例えば、プロリン合成経路の遺伝子が不活性化されている細菌変異体はプロリン栄養要求株である。このような株はプロリンを合成することができず、従って、プロリンの非存在下で増殖できるプロリン原栄養株とは対照的に、環境からプロリンを吸収することができる場合にのみ増殖できる。
【0066】
栄養要求性マーカーを有する任意の宿主細胞を用いることができる。好ましくは、栄養要求性マーカーとして、アミノ酸合成に必要な遺伝子における変異、例えばプロリン、ロイシンもしくはトレオニンの合成またはチアミンなどの補因子に関連する遺伝子における変異が用いられる。本発明によれば、目的遺伝子を組み込むことに加えて、発現カートリッジの構成要素として欠失/欠損遺伝子をゲノムに組み込むことにより、宿主細胞の栄養要求性が補正される。このようにして得られた原栄養性細胞は、元の宿主細胞が栄養要求する化合物を含まず、従って陽性クローンのみを増殖させることが可能ないわゆる“最小培地”(原栄養性選択)で増殖させることにより容易に選択できる。
【0067】
原栄養性選択は組み込み遺伝子座とは無関係である。原栄養性選択の組み込み遺伝子座はゲノムにおける任意の遺伝子であることもでき、栄養要求性マーカーを含む遺伝子座であることもできる。原栄養性選択の特別な利点は、組み込みに成功した後に、宿主に対して異物である抗生物質耐性マーカーも任意の他のマーカーもゲノムに残らないことである。その結果として、前記マーカー遺伝子を除去する必要がなく、迅速で簡単なクローニングおよび選択手順が得られる。他の利点は、遺伝子機能を回復させることは細胞にとって有益であり、その系に高い安定性を与えるという点である。
【0068】
あるいは、同様に図8に概略で示すように、発現カートリッジと共にゲノムに挿入されたマーカー遺伝子は、特定の選択方法を可能にする代謝遺伝子であることができる。このような代謝遺伝子は、特定の(まれな)糖または他の炭素源で細胞を増殖させることができ、唯一の炭素源として前記糖で細胞を増殖させることにより陽性クローンの選択を行うことができる。
【0069】
前述のように、細菌の長期培養の間に、適応進化(Herringら、2006年)は、染色体にコードされた組換えタンパク質の発現中に組み込み部位での変異頻度の増加を引き起こす恐れがある。変異株の欠失した機能を補う発現カートリッジと組み合わせて栄養要求性ノックアウト変異株を使用すること(それによって栄養要求性変異株から原栄養株を作成する)は、適応進化が生じた細胞が抑制される競争的利点を細胞が獲得することによる利点を、回復された遺伝子が細胞に与えるというさらなる利点を有する。それによって、変異クローンに負の選択という手段が与えられる。
【0070】
いくつかの実施形態において(例えばFACS分析または免疫学的分析(ELISA)による、単一細胞または単一コロニーベースでの目的タンパク質の検出が可能な場合)、マーカー遺伝子は必要ではない。なぜなら、目的タンパク質の直接検出により陽性クローンが検出できるからである。
【0071】
他の特徴
発現宿主細胞を得るための組み込み方法は、ゲノムの1つの部位に目的遺伝子を1つ組み込むことに限定されない。組み込み部位および発現カセットの両方に関して組み込み方法を変えることが可能である。一例として、2以上の目的遺伝子を挿入することができる。すなわち、同一または異なるプロモーターの制御下に有る2以上の同一または異なる配列を、ゲノムの1以上の種々の遺伝子座に組み込むことができる。一例として、ヘテロ二量体複合体を形成する2種の異なるタンパク質の発現が可能である。ヘテロ二量体タンパク質は、2つの個別に発現したタンパク質サブユニット、例えばモノクローナル抗体または抗体フラグメントのH鎖およびL鎖からなる(AndersenおよびReilly、2004年;HolligerおよびHudson、2005年)。他のヘテロ二量体タンパク質の例には、例えばCapZ(Remmertら、2000年)、Rasファルネシルトランスフェラーゼ(TsaoおよびWaugh、1997年)、血小板活性化因子アセチルヒドロラーゼIb(Sheffieldら、2001年)またはヒトDNAヘリカーゼII(Ochemら、1997年)がある。モノマーをコードするこれらの2つの配列は、1つの組み込み遺伝子座に挿入された1つの発現カートリッジに存在することができる。あるいは、これらの2つの配列は、2つの異なる組み込み遺伝子座に互いに独立して挿入された2つの異なる発現カートリッジに存在することもできる。いずれにせよ、プロモーターおよび誘導方法は、同一であることもできるし、異なることもできる。
【0072】
本発明は、無プラスミドでの目的タンパク質の製造が可能であるが、目的遺伝子以外に発現配列、例えば前述のヘルパータンパク質および/または組換えタンパク質を含むプラスミドが発現宿主細胞に存在することを排除するものではない。当然、このような実施形態において、プラスミドの存在により本発明の利点が覆されないように注意が必要である。すなわち、プラスミドは低コピー数で存在することが必要であり、細胞に代謝負荷を与えてはならない。
【0073】
組換えタンパク質のゲノムベースの発現は、以下の主な利点を提供する:
【0074】
発現宿主の構築方法に関しては、利点は、(i)直線状挿入カートリッジの簡単な合成および増幅方法、(ii)組み込み遺伝子座に関する高い柔軟性(すなわち制限なし)、(iii)選択マーカーおよび選択原理に関する高い柔軟性、(iv)後の選択マーカーの除去の選択肢、(v)不連続で定義された数の挿入された発現カートリッジ(通例1または2)である。
【0075】
本発明の方法に有用な発現系は、本質的にまたは完全にファージ機能のないように設計されたものであることができる。
【0076】
ゲノムへの1以上の組換え遺伝子の組み込みにより、1細胞当たり不連続であらかじめ定義された数の目的遺伝子がもたらされる。遺伝子1コピーを挿入する本発明の実施形態において、最大数百までのコピー数を伴うプラスミドベースの発現と比較して、この数は通例1である(細胞分裂中に一過性に生じる、細胞が2以上のゲノムを含む場合を除く)。本発明の方法に用いられる発現系において、プラスミドの複製による宿主の代謝を軽減することにより、組換えタンパク質製造に細胞の合成能力の増加した部分が用いられる。遺伝子量を減らすことにより、宿主の代謝に有害な効果を与えることなく、T7系などの強力なプロモーターを適用することができる。
【0077】
発現系が誘導レベルに関して制限を有さないことは特別な利点である。このことは、プラスミドベースの系においてしばしば生じる、誘導によりPCNが大幅に増加する“過剰誘導”をこの系が起し得ないことを意味する(Teichら、1998;WrobelおよびWegrzyn、1998年)。遺伝子量の多さおよび/または増加と組み合わせた強力な発現系は、代謝過負荷を引き起こし、細胞生存率の顕著な低下をもたらし(Bentlyら、1990;Glick、1995;Cserjan-Puschmannら、1999年)、産物生成期間の顕著な短縮および全収量の低下を引き起こす。
【0078】
上記のように、プラスミドベースの発現系は、細胞が、細胞分裂の間にプラスミド、それとともに目的遺伝子を失う恐れがあるという欠点を有する。このようなプラスミドの喪失はいくつかの外部要因に依存し、細胞分裂回数(世代数)と共に増加する。このことは、プラスミドベースの発酵は、世代数(従来の発酵においては、この数はおおよそ20〜50である)に関して制限されていることを意味する。対照的に、本発明の方法に用いられるゲノムベースの発現系は、事実上無限の世代数、従って調節条件下で理論上は無限の培養時間にわたって安定であらかじめ定義された遺伝子量を確実にする(目的タンパク質を産生しない細胞が生じるという不都合はなく、ただ任意の遺伝子に生じる自然突然変異が生じうるという制限があるだけである)。
【0079】
化学的誘導性プロモーターの場合は、例えばStriednerら(2003年)に従って連続方式で添加されるとき、誘導分子の量が、全培養にわたって一定であるか、あるいは培養時間にわたってあらかじめ定義された値に変化させるかにより、1細胞当たりの遺伝子量に直接に比例しているという利点を本発明は提供する。それによって、組換え発現速度の調節を達成でき、このことが遺伝子発現速度を調節するために、大変関心の持たれるところである。
【0080】
ゲノムベースの発現系は、タンパク質発現の正確な調節が可能なので、よく調節された発現に依存または頼る発現標的経路と組み合わせると特に有利である。好ましい実施形態において、本発明の方法は、細菌細胞質から培地への目的タンパク質の分泌(排泄)を含む。この実施形態の利点は、先行技術の分泌系と比較してより高い力価の分泌タンパク質をもたらす、最適化され、維持されたタンパク質分泌速度である。細菌細胞壁を通しての受動拡散または細胞膜周辺腔への能動分泌と、それに続く拡散または物理化学的放出(例えば米国特許第4,680,262号および米国特許第4,963,495号に記載されているようなStIIシグナル配列を用いる)、あるいは細胞質から培地への目的タンパク質の直接移動(例えばChoiおよびLee(2004年)に記載されている方法)により分泌を生じることができる。
【0081】
前述のように、本発明は、簡略プロセス、改善されたプロセス予測性および発酵間の高い再現性を設計することを可能にする。前述の発現系を用いる本発明のプロセスは、流加培養方式、半連続方式または連続方式(すなわちケモスタット)で行われ、それによって、ゲノムにコードされた発現系の利点が最適に活用される。宿主細胞の必要条件により定義され、選択されるプロモーターによりあらかじめ定義される場合を除き、増殖速度、温度および培地成分などのプロセスパラメータに関して制限はない。
【0082】
他の利点は、誘導分子の選択に関する。大腸菌内での組換え遺伝子の高レベルの発現のための利用可能な系の大部分は、lacベースのプロモーター・オペレーター系(Makrides、1996年)である。天然の誘導物質1,6-アロラクトースの前駆体であり、容易に大量に入手できる安価な天然炭水化物であるラクトースは、IPTGに代わるものとして考えられてきた。しかしながら、かっては、グルコース培地による誘導物質排除(inducer exclusion)および細胞内での分解の問題により、ラクトースは誘導には不適当であると考えられた。本発明に用いられる発現系は、誘導物質としてのラクトースの連続供給を用いる炭素制限培養(Striednerら、2003年)を可能にし、それと共にラクトース排除を防ぎ、ラクトースを用いる従来のパルス誘導中に観察される誘導レベルの低下(Hoffmanら、1995年)を排除し、誘導物質としてラクトースを用いる場合でも厳密な発現速度調節を可能にする。
【0083】
重要なことに、本発明に用いる発現系は、培地容量あたりのタンパク質濃度(すなわち力価)および得られたバイオマス中のタンパク質含量の両方に関して高収量の組換えタンパク質を提供する利点を有する。この特徴により、本発明に用いられる発現系が先行技術の発現系より優れたものとなる。
【0084】
さらにまた、本発明は、発現宿主細胞の選択および/または発現カートリッジの最適設計を予備スクリーニング検査で容易に達成できるという利点を提供する。一例として、このような予備スクリーニングにおいて、目的タンパク質の発現特性(発現レベルまたは生物活性のような定性的特徴)に影響を有する少なくとも1つの因子、すなわち調節因子(例えばプロモーターおよび/またはポリメラーゼ結合部位)および/または目的遺伝子の配列(すなわち異なるコドン使用バリアント)および/または組換えのターゲッティング配列および/または分泌リーダーなどのカートリッジ上の任意の他の因子の異なる一連の直線状発現カートリッジが構築される。このカートリッジバリアントは、事前選択された宿主細胞のゲノムに組み込まれ、得られた発現宿主バリアントは、調節条件下でのタンパク質発現の誘導を含めて培養される。タンパク質発現を比較し、工業生産プロセスを考慮して最も好ましい結果を示す宿主細胞バリアントが選択される。この予備スクリーニングアプローチの変形において、最適の発現カートリッジを決定するかわりに、1群の異なる宿主細胞に異なるカートリッジを組み込むことにより最適の細菌株を同定することができる。この組み込み戦略は、不連続な遺伝子コピー数(例えば1つのみ)をゲノムに組み込むことを可能にする利点を有するので、プラスミドの複製またはプラスミドコピー数の変化により抵触されることなく種々のパラメータの予備スクリーニングを行うことができる。
【0085】
工業規模の製造
本発明において、用語“培養すること”(または“培養”、“発酵”ともいう)は、当業界に公知の方法による制御されたバイオリアクターにおける細菌発現細胞の増殖に関する。
【0086】
組換えタンパク質の生産は、一般的には、より大容量で培養を行うことにより達成される。本発明において、用語"工業(manufacturing)"および"工業規模(manufacturing scale)"は、最小容量5Lの培養液での発酵を定義する。通例、“工業規模”プロセスは、目的組換えタンパク質を含む大容量の調製物を処理し、例えば治療用タンパク質の場合は、臨床試験および市場供給に対する要求を満たす量の目的タンパク質を産生することができることにより定義される。振盪フラスコ培養などの簡単な実験室規模の方法とは対照的に、生産規模の方法は、大容量に加えて、撹拌、通気、栄養素フィード、モニタリングおよびプロセスパラメータ制御(pH、温度、溶存酸素圧、背圧などのための装置を備えるバイオリアクター(発酵槽)の技術システムの使用を特徴とする。実験室規模の方法における発現系の行動からは、バイオリアクターの複雑な環境におけるその系の行動を予測できない。
【0087】
驚くべきことに、本発明の実験において、発現系に基づくゲノム組み込みは、栄養素フィードに基づく培養方式、特に流加プロセスまたは連続プロセスまたは半連続プロセス(例えばケモスタット)と組み合わせた工業規模の方法(容量および技術システムの両方に関して)に特に有利であることが見出された。
【0088】
流加培養
特定の実施形態において、本発明の方法は流加培養プロセスである。
【0089】
バッチプロセスが、発酵中にさらなる栄養素のさらなる供給をせずに、細胞培養に必要なすべての栄養素が初期培地に含まれる培養方式であるのに対し、流加プロセスにおいては、バッチ段階(batch phase)後、フィードにより培養物に1以上の栄養素が供給されるフィード段階(feeding phase)が行われる。栄養素フィードの目的は、バイオマスの量を増加させて(いわゆる“高細胞密度培養プロセス”すなわち“HCDC”)、組換えタンパク質の量もまた増加させることである。大部分の培養プロセスにおいて、フィード方式は重要かつ不可欠であるが、本発明は特定のフィード方式には限定されない。
【0090】
栄養素フィードは、当該技術分野で公知の方法に従って、連続方式または不連続方式で行うことができる。フィード方式はあらかじめ定義された方式であることができ(すなわち実際のプロセスパラメータとは独立してフィードが添加される)、例えば線形定係数方式、線形増加方式、段階的増加方式または数学関数に従った方式、例えば指数関数的フィードであることができる。
【0091】
好ましい実施形態において、本発明の方法は、フィード方式があらかじめ定義された指数関数に従った方式の流加プロセスである。指数関数的フィード方式を適用することにより、細胞集団の比増殖速度μを一定レベルにあらかじめ定義することができ、最大組換えタンパク質発現に関して最適化することができる。フィード速度の制御は、所望の比増殖速度μに基づく。下記のような定義された培地が用いられる場合、与えられる基質単位に基づいて形成されるバイオマスアリコートの推定により増殖を正確に予想し、あらかじめ定義できる。
【0092】
他の好ましい実施形態において、培養の最終段階において、線形定係数フィード方式後に指数関数的フィード方式を続けることができる。
【0093】
流加プロセスの他の実施形態において、線形定係数フィードが適用される。線形定係数フィードは、特定の培養段階を通じて一定の(すなわち不変の)フィード速度(時間単位当たりのフィード培地容量)を特徴とする。
【0094】
流加プロセスの他の実施形態において、線形増加フィードが適用される。線形増加フィードは、線形関数に従ったフィード培地のフィード速度を特徴とする。線形増加関数に従ったフィードは、定義された時間増加量当たりの定義されたフィード速度増加量を特徴とする。
【0095】
本発明の流加プロセスの他の実施形態において、フィードに、フィードバック制御アルゴリズムが適用される(あらかじめ定義されたフィード方式とは対照的に)。フィードバック制御された流加プロセスにおいて、フィード速度は、特定の培養パラメータの実際のレベルに依存する。フィードバック制御されたフィードに適した培養パラメータは、例えばバイオマス(およびそれから誘導される化学的または物理的パラメータ)、溶存酸素、呼吸係数、pHまたは温度である(例えばJahicら(2003年)に記載されているもの)。フィードバック制御されたフィード方式の他の例は、バイオリアクター内の実際のグルコース濃度に基づく(Klemanら、1991年)。
【0096】
連続および半連続培養方式
他の実施形態において、本発明によるゲノムベースの発現カセットを含む細菌細胞は連続方式(ケモスタット)で培養される。連続発酵プロセスは、バイオリアクターへの定義された一定の連続速度の新鮮な培地のフィードを特徴とし、それによって、同じ定義された一定の連続除去速度でバイオリアクターから培養液が同時に除去される。培地、フィード速度および除去速度を同じ一定のレベルに保つことにより、バイオリアクターにおける培養パラメータおよび条件は一定のままである(いわゆる“定常状態”)。比増殖速度μはあらかじめ定義されたものであることができ、もっぱらバイオリアクターにおけるフィード速度および培地容量の結果である。1以上のゲノムベースの発現カセットを有する細胞は遺伝的に大変安定なため(構造不安定なおよび分離不安定なプラスミドベースの発現系またはゲノムに挿入されたカセットがゲノム増幅に依存する発現系とは対照的に)、本発明による細胞の世代数(細胞倍加数)は理論上は制限がなく、同様に、その結果として培養時間にも制限がない。連続方式で遺伝的に安定なゲノムベースの発現系を培養する利点は、遺伝的に不安定な先行技術システムと比較して、より高い、期間あたりの組換えタンパク質の総量を得ることができることである。加えて、理論上無期限の培養により、本発明による細胞の連続培養は、流加培養プロセスと比較してさえ、より高い、期間当たりの総タンパク質量をもたらすことができる。実施例7において、ゲノムベースの発現構築物の高い安定性および生産性を示す。
【0097】
他の好ましい実施形態には、細胞の半連続培養が言及される。本発明において、半連続培養プロセスは、その第1段階で流加プロセスとしてはたらくプロセスである(すなわちバッチ段階とそれに続くフィード段階)。特定の容量またはバイオマスが得られた後(すなわち通例発酵槽容量の上限が得られたとき)、目的組換えタンパク質を含む細胞培養液のかなりの部分がバイオリアクターから取り除かれる。続いて、フィードを再開し、バイオマスまたは培養液の容量が特定の値に達するまで加える。この方法(培養液の排出およびフィードによる再充填)を、少なくとも1回、理論上は無限回繰り返すことができる。
【0098】
培地
この発酵プロセスに用いられる培地の種類に関しては制限はない。培地は、半合成培地、すなわち複合培地化合物(例えば酵母抽出物、大豆ペプトン、カザミノ酸)を含む培地であることもでき、複合化合物を何も含まない化学的に定義された培地であることもできる。
【0099】
好ましくは、“定義された培地”が用いられる。"定義された"培地(“最小”または"合成"培地とも呼ばれる)は、もっぱら化学的に定義された物質、すなわちグルコースまたはグリセロールなどの炭素源、塩、ビタミンならびに、可能性のある株の栄養要求性を考慮した特定のアミノ酸または他の物質、例えばチアミンで構成される。最も好ましくは、炭素源としてグルコースが用いられる。通例、フィード培地の炭素源は、比増殖速度を制御する増殖制限物質として役立つ。
【0100】
本発明の方法において、細菌の増殖は高度であるが、全生産プロセスの間に生理学的に許容される組換え遺伝子の発現速度を維持できるので、かなり高い収量が得られる。
【0101】
誘導方法
前述のように、本発明の最も好ましい実施形態において、目的タンパク質は、“誘導性”または“制御性”プロモーターの制御下にある。
【0102】
タンパク質発現を誘導する方法については制限はない。一例として、1回もしくは複数回ボーラスとして、または連続フィードにより誘導原を添加でき、後者は別名“誘導原フィード(フィーディング)”である。誘導を行う時点に関しては制限はない。培養の開始時点、連続栄養素フィードの開始時点またはフィード開始後(を過ぎて)に誘導原を添加できる。
【0103】
誘導原を培地に含有させるか、それを別にフィードするかのいずれかにより、誘導原フィードを行うことができる。
【0104】
誘導原フィードの利点は、誘導原量を調節できること、すなわち、産生系における一定数の目的遺伝子に対して定義されたまたは一定量の誘導原量を維持できることである。例えば、誘導原フィードは、誘導原とバイオマスの比率が一定になるように、バイオマスに比例した誘導原量を可能にする。誘導原量の基礎となりうるバイオマス単位は、例えば細胞乾燥重量(CDW)、湿細胞重量(WCW)、吸光度、全細胞数(TCN;容量当たりの細胞数)またはコロニー形成単位(容量当たりのCFU)であることができ、あるいはバイオマスに比例したオンラインでモニターされる信号(例えば蛍光、濁度、誘電容量など)であることができる。本質的に、本発明の方法は、信号がオフラインで測定されたかオンラインで測定されたかにかかわらず、バイオマスに比例した任意のパラメータまたは信号当たりの誘導原の正確な量が可能である。目的遺伝子数が定義され、バイオマス単位当たり一定であるため(1細胞当たり1以上の遺伝子)、この誘導方法の結果は、目的遺伝子当たり一定量の誘導原である。さらなる利点として、バイオマス量に対する誘導原の正確で最適な量は、実施例8および図16および図17において明らかにするように、実験的に決定し、最適化することができる。
【0105】
実際のバイオマスレベルを分析方法により測定することは、必ずしも必要ではない。例えば、以前の培養(バイオマスの履歴データ)に基づく誘導原量を添加することで十分な場合がある。他の実施形態において、理論的に算出または予測されたバイオマス1単位当たりの誘導原量を添加することが好ましい場合がある。例えば、フィードベースの培養(流加培養または連続培養など)に関しては、フィード培地における増殖制限物質(通例は炭素源)1単位が特定量のバイオマスをもたらすことは公知である。例として、増殖制限基質としてのグルコース1gは、おおよそ細胞乾燥重量0.33g(基質収率係数YX/S=0.33としても表される)をもたらす。その結果として、目的遺伝子に対する定義された誘導原量は、単位増殖制限物質当たりの定義された量の誘導原によっても達成できる。なぜなら、特定単位の増殖制限物質は定義された単位のバイオマスをもたらし、定義された単位のバイオマスは本発明の方法による目的タンパク質の定義された分子数を含むからである。
【0106】
IPTGの濃度は、CDW 1g当たり0.1〜30μgの範囲であることができ、好ましくは、IPTGの濃度はCDW 1g当たり0.5〜20μgの範囲である。
【0107】
実施例8において、フィード培地における最大誘導原量は、CDW 1g当たりIPTG 20μmolであり、基質収率係数YX/S=0.33と仮定すれば、これはグルコース1g当たりIPTG 6.6μmolと等価である。その結果として、フィード培地のグルコース濃度が128g/Lなので、同じ培地のIPTG濃度は844μMであった。
【0108】
本質的な利点として、制限された量の誘導原のフィードは、代謝負荷を防ぎ、ストレスを低減し、タンパク質合成の能力を最大限にする利益となる。
【0109】
誘導原とバイオマス(または遺伝子または単位増殖制限基質)の比は必ずしも一定である必要はない。線形増加、線形減少、指数関数もしくは他の数学関数に従って増加または減少などであることもできる。本発明による本質的な特徴は、目的遺伝子当たりの誘導原量の値が定義されているということである。
【0110】
特定の実施形態において、本発明の方法は流加プロセスであり、誘導原は培養の開始からバッチ培地に存在している。
【0111】
発現の誘導方法は構成的であることもできる。これは、化学的にまたは他の刺激により誘導が引き起こされるのではなく、培養の開始から誘導は不変であることを意味する。構成的誘導は連続培養にとって好ましい誘導方法であり、流加培養にとっても有用である。
【図面の簡単な説明】
【0112】
【図1】不溶性自己プロテアーゼ融合タンパク質の製造のための、ゲノムにコードされた発現系HMS174(DE3)<Cam:T7:6xHis-NproEddieGFPmut3.1>(ES4)を用いる流加培養を示す図である。
【図2】不溶性自己プロテアーゼ融合タンパク質の製造のための、プラスミドにコードされた発現系HMS174(DE3)(pET30a6xHis-NproEddie-GFPmut3.1)(ES1)を用いる流加培養を示す図である。
【図3】ゲノムにコードされた発現系BL21(DE3)<Cam:T7:6xHis-NproEddieGFPmut3.1>(ES5)を用いる流加培養を示す図である。
【図4】不溶性自己プロテアーゼ融合タンパク質の製造のための、プラスミドにコードされた発現系BL21(DE3)(pET30a6xHis-Npro-GFPmut3.1)(ES2)を用いる流加培養を示す図である。
【図5】可溶性緑色蛍光タンパク質の製造のための、ゲノムにコードされた発現系HMS174(DE3TN7::<Kan:T7GFPmut3.1>(ES6)を用いる流加培養を示す図である。
【図6】可溶性緑色蛍光タンパク質の製造のための、プラスミドにコードされた発現系HMS174(DE3)(pET30a GFPmut3.1)(ES3)を用いる流加培養を示す図である。
【図7】抗生物質耐性マーカーを挿入し、場合により続いてマーカーを除去(切除)することによる選択を示す図である。
【図8】マーカーとして非必須代謝遺伝子または原栄養性補完遺伝子を提供することによる選択を示す図である。
【図9】組換えタンパク質のゲノム部位に発現カートリッジを挿入し、それによってそれらを除去することを示す図である。
【図10】マーカー遺伝子の部位に発現カートリッジを挿入し、マーカー表現型の消失に関して選択することを示す図である。
【図11】非機能性マーカーの欠失部分を完全にして機能性選択マーカーを作成することによる選択を示す図である。
【図12】可溶性hSODの製造のための、プラスミドにコードされた発現系HMS174(DE3)(ES7)を用いる流加培養を示す図である。
【図13】可溶性hSODの製造のための、ゲノムにコードされた発現系HMS174(DE3)(ES8)を用いる流加培養を示す図である。
【図14】可溶性hSODの製造のための、ゲノムにコードされた発現系BL21(DE3)(ES9)を用いる流加培養を示す図である。
【図15】連続方式(ケモスタット)での大腸菌HMS174(DE3)を用いる可溶性GFPmut3.1の発現(ES6)を示す図である。
【図16】可溶性GFPmut3.1の製造のための、制御された誘導原量と組み合わせた、ゲノムにコードされた発現系BL21(DE3)を用いる流加培養を示す図である。
【図17】誘導原の濃度と相関する、ゲノムにコードされた発現系BL21(DE3)を用いる流加培養から得られる可溶性GFPmut3.1の正規化平均産物生成速度(qP)を示す図である。
【実施例1】
【0113】
細菌株の作成および組換えタンパク質の説明
Novagen社により提供された大腸菌recA-K12株HMS174(DE3)またはB株BL21(DE3)のいずれかを用いて実験を行う。記号表示(DE3)は、宿主がlacUV5プロモーターの制御下にあるT7 RNAポリメラーゼ遺伝子の染色体コピーを含むλ DE3プロファージの溶原菌であり、これらの株をT7またはT7lacプロモーターを用いるタンパク質発現に適したものにしたものであることを示している(StudierおよびMoffat、1986年;Studierら、1990年)。細菌染色体への直線状DNAカートリッジの組み込みは、DatsenkoおよびWanner(2000年)に記載されているように、RedヘルパープラスミドpKD46を含む大腸菌MG1655で行われる。直線状発現カートリッジは、大腸菌ゲノムのglmS遺伝子とpstS遺伝子の間にあるattTn7部位に組み込まれる。ゲノム上のカセットのPCRによる確認は、ゲノムにアニーリングする外部プライマーと内部プライマーを組み合わせて行う。P1形質導入(SternbergおよびHoess、1983年;およびLennox、1955年)により、組換え染色体部分が発現宿主HMS174(DE3)およびBL21(DE3)に導入される。レシピエントとしての機能を果たすために、RecAタンパク質をもたらす温度感受性pTSA29-recAヘルパープラスミドを含むrecA欠損株MS174(DE3)が用いられる(Phillips、1998年)。
【0114】
先行技術である標的タンパク質のプラスミドベースの発現を用いる参照実験は、Novagen社製pET30aおよびpET11aプラスミド(pET Systemmanual、第11版)を用いて行う。
【0115】
本発明の発現系の潜在能力を示すために、可溶性タンパク質(緑色蛍光タンパク質GFP;Shimomuraら、1962年))および封入体形成タンパク質(Npro)を実験に用いる。GFPmut3.1は、置換S65GおよびS72Aを有し、488nmで励起され、フォールディングが改善された、FACS用に最適化したバリアントである(Cormackら、1996年)。封入体形成タンパク質6xHis-NproEddie-GFPmut3.1は、上記のGFPmut3.1と、豚コレラウイルス由来の天然のNpro自己プロテアーゼの改変されたバリアントであるNpro Eddie(WO2006/113959に記載されている)との融合体である(Thielら、1991年)。Nproは168アミノ酸(見かけの分子量23000)からなる非構造タンパク質であり、新生ポリタンパク質から翻訳と同時にそれ自身を切断し、それによってカプシドタンパク質Cの正しいN末端が生成される(Wiskerchenら、1991;Starkら、1993年)。
【0116】
表1:作成した発現系のリスト
【表1】
【実施例2】
【0117】
培養方式およびプロセス分析
標準的な制御装置を備えた、7L(正味容量5L、バッチ容量2.5L)または20L(正味容量12L、バッチ容量8L)のコンピュータ制御のバイオリアクター(MBR社;チューリッヒ・ヴェッツィコーン)のいずれかで細胞を増殖させる。25%アンモニア溶液(ACROS Organics社)の添加によりpHを設定値7.0±0.05に維持し、温度を37℃±0.5℃に調節する。酸素制限を避けるために、攪拌機速度および通気速度を制御することにより、溶存酸素濃度を飽和状態の30%以上に安定化させる。吹出空気におけるO2およびCO2含有量を、Hartmann andBraun AdvancedOptimaガス分析装置により測定する。Biomass monitorモデル214M(Aber Instruments社、英国アベリストウィス)セットで誘電容量および伝導率を測定する。産業環境におけるオンライン測定のために特別に設計された多波長蛍光分光光度計であるBioView(登録商標)(DELTA Light&Optics社、デンマーク・リンビー)を用いて蛍光測定を行う。消泡剤懸濁液(Glanapon 2000、Bussetti社、ウイーン)を1ml/フィード培地1lの濃度で添加することにより発泡を抑える。接種のために、強冷凍した(-80℃)ワーキングセルバンクバイアルを解凍し、1ml(吸光度OD600=1)を無菌でバイオリアクターに導入する。培養物が、バッチ培地2.5L中細菌乾燥物質12.5g(またはバッチ培地4L中細菌乾燥物質30g)に増殖して定常期に入ったときフィードを開始する。4倍加時間の間一定の増殖速度0.1h-1を得るために指数関数的基質フィードによる流加法を用いる。基質タンクにおける重量減少の重ねフィードバック制御を用い、対数増殖アルゴリズム、x=xo.eμt、に従ってポンプ速度を増加させることにより基質フィードを制御する(Cserjan-Puschmannら、1999年)。さらなる細菌乾燥物質202g(または450g)を得るために必要な成分をフィード培地は提供した。
【0118】
誘導
バイオリアクターに直接に1回パルスする従来の方式で誘導を行う。完全誘導系を得るために、プロセスの終わりにIPTGの濃度が1μmolになるようにIPTGの供給量を算出する。
【0119】
培地組成
本研究に用いる最小培地は、1リットル当たりKH2PO4 3gおよびK2HPO4*3H2O 6gを含む。これらの濃度により必要な緩衝能が得られ、PおよびK源としても役立つ。産生されるグラム細菌乾燥物質に関連して他の成分が添加される:クエン酸ナトリウム(三ナトリウム塩*2H2O;ACROS organics社)0.25g、MgSO4*7H2O 0.10g、CaCl2*2H2O 0.02g、微量元素溶液50μlおよびグルコース*H2O 3g。発現系ES6、ES7およびES8を用いるすべての実験において、細菌乾燥物質1g当たりCuCl2*2H20 4mgおよびZnSO4*7H20 3.2mgを培地に添加する。集団の初期増殖を促進するために、複合成分である酵母抽出物0.15gを最小培地に加えてバッチ培地を得る。フィード段階に関して、フィード段階において、産生される生物学的乾燥物質量202g(または450g)に従って、1リットル当たりP塩をさらに加え、最小培地2.5Lを調製する。微量元素溶液:5N HCl溶液(g/L):FeSO4*7H2O 40.0、MnSO4*H2O 10.0、AlCl3*6H2O 10.0、CoCl2(Fluka社) 4.0、ZnSO4*7H2O 2.0、Na2MoO2*2H2O 2.0、CuCl2*2H2O 1.0、H3BO3 0.50。
【0120】
オフライン分析
600nmで吸光度(OD)を測定する。細胞浮遊液10mlを遠心分離し、蒸留水に再懸濁した後遠心分離し、再懸濁してあらかじめ計量したビーカーに移し、次いでこれを105℃で24時間乾燥して再計量して細菌乾燥物質を測定する。
【0121】
バイオマスの総量(全細菌乾燥物質BDM;細胞乾燥重量CDWとも呼ぶ)を算出することにより、細菌増殖の進行を測定する。
【0122】
フローサイトメトリーを用いて、全細胞数(TCN)および死細胞(DC)の百分率を測定する。488nmで15mWの出力で標準的なフィルター設定を用いる空冷レーザーを備えたFACSCaliburフローサイトメーター(4カラーシステム;Becton Dickinson社)を用いてすべての測定を行う。絶対細胞数を逆算できるように、既知量の蛍光計数ビーズ(Becton Dickinson社、米国)を試料に加える。
【0123】
ELISAおよびAgilent Bioanalyserを用いる電気泳動によるタンパク質定量の組み合わせにより、組換えタンパク質Npro-Eddie-GFPmut3.1の含有量を測定する。可溶性組換え産物は、Reischerら(2004年)に従ってGFP-ELISAにより定量し、そして、封入体における組換え産物は、Protein 200 LabChip(登録商標)キットを用い、Agilent Bioanalyser 2100で定量する。
【0124】
組換えGFPの含有量は、Reischerら(2004年)に従ってGFP-ELISAにより定量する。hSODの含有量は、Bayerら(1990年)に従ってSOD ELISAにより定量する。
【0125】
プラスミドを含有する細胞ばかりでなく、組み込みカセットを含む細胞もまた、100mg/mlカナマイシンおよび100mg/mlアンピシリンをそれぞれ含むLB寒天平板上で培養し、24時間後にコロニー形成単位(CFU)を計数することにより測定する。
【実施例3】
【0126】
流加培養方式における大腸菌HMS174(DE3)を用いる不溶性封入体としての6xHis-NproEddie-GFPmut3.1の発現
不溶性標的タンパク質の製造(すなわち封入体として)に関する本発明の発現系(ES4)の行動を特徴づけ、プラスミドベースの発現系(ES1)と比較して経済性を評価するために、表1に記載した発現系ES1およびES4を用いる。本発明の発現系を用いる実験により、高いが生理学的に許容される遺伝子発現速度が示される。20時間を超えて産物生成を維持できる。完全誘導系を得るために、フィード開始後1回の倍加での1回のパルスによる誘導を行う(図1)。
【0127】
これらの結果と対照的に、プラスミドベースの発現系により引き起こされる遺伝子発現速度により、細胞の代謝能は酷使され、フィード開始後3回の倍加での1回のIPTGパルスにより行う誘導の4時間後に細胞系は崩壊する(図2)。本発明による発現系ES4は、わずかに高い容量産物収量を示した(表2)。これらの実験からわかるように、本発明の発現系は、産物生成の全期間を通じてプロセス制御および定義された条件を維持できるという利点を提供する。
【0128】
表2:実施例3からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質6xHis-NproEddie-GFPmut3.1およびプラスミドにコードされたタンパク質6xHis-NproEddie-GFPmut3.1の製造の比較)
【表2】
【実施例4】
【0129】
流加培養方式における、大腸菌BL21(DE3)を用いる、不溶性封入体としての6xHis-NproEddie-GFPmut3.1の発現
本発明の発現系の一般適用性を証明するために、発現宿主として大腸菌株BL21(DE3)を用いる。前述の手順に従って、5L規模で表1のES5を培養する。この系の結果(図3;フィード後1回の倍加での1回のパルスにより誘導が行われる)は、K12株HMS174を用いた結果よりもよく、プラスミドベースのES2を用いて得られた結果(図4)と比較してもよい;フィード開始後3回の倍加での1回のパルスにより誘導が行われる)。本発明の系は、プラスミドベースの系よりも有意に高い産物収量およびプロセス安定性を示す(表3)。株BL21のプラスミドベースの系は、株HMS174について記載したものと同じ特性を示し、高すぎる産物生成速度により、系は崩壊し、プロセス制御は失われる。
【0130】
表3:実施例4からの結果の概要
(宿主株BL21(DE3)を用いる、ゲノムにコードされたタンパク質6xHis-NproEddie-GFPmut3.1およびプラスミドにコードされたタンパク質6xHis-NproEddie-GFPmut3.1の製造の比較)
【表3】
【実施例5】
【0131】
流加培養方式における大腸菌HMS174(DE3)を用いる可溶性タンパク質としてのGFPmut3.1の発現
可溶性標的タンパク質の製造のための本発明の発現系の行動を特徴づけるために、表1に記載した発現系ES3およびES6を用いる(表1の他の発現系は、不溶性タンパク質凝集体、すなわち封入体を形成する)。異なる細胞密度により引き起こされる影響を排除するために、フィード開始後1回の倍加での誘導による同じ培養を行うが、他の実験の結果から、プラスミドベースの系は引き起こされ発現速度が高すぎるので、この系では細胞活性を維持できないことが明らかとなった。要求される比較性に対処するために、表4には“プラスミドにコードされた(最適化後)”と呼ぶ第3列目が存在するが、これは、“プラスミドにコードされた発現系”ES2の結果が、フィード開始後1回の倍加での誘導をまねた実験設定に外挿されていることを意味する。図5および図6(フィード開始後3回の倍加での1回のパルスにより誘導が行われる)ならびに表4から導くことができるように、本発明の発現系はより効率的であることが明らかとなった。このことは、特に、組換えタンパク質のより高い比収量および容量収量ならびにバイオマスへの栄養素のより優れた転化率により明らかにされる(すなわちより高いBDM収量または理論値からの下限逸脱率)。
【0132】
表4:実施例5からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質HMS174(DE3)およびプラスミドにコードされたタンパク質HMS174(DE3)の製造の比較)
【表4】
【実施例6】
【0133】
流加培養方式における大腸菌HMS174(DE3)およびBL21(DE3)を用いる可溶性hSODの発現
大腸菌HMS174(DE3)(ES7、ES8)を用いるプラスミドベースおよびゲノムベースの発現ならびに大腸菌BL21(DE3)(ES9)を用いるゲノムベースの発現により組換えヒトスーパーオキシドジスムターゼ(hSOD)を製造する。ヒトSODを3番目のモデルタンパク質として選択する。なぜなら、この酵素は細菌サイトゾル内で高度に相互作用的であることが記載されており、それによって、ゲノムベースの発現を用いる製造方法の頑健性および性能を研究するための適切なタンパク質候補を代表する。ES7を用いる実験(図12)において、製造される生物学的乾燥物質の量は、BDM 225gにあらかじめ定義され、誘導はフィード開始後2.5回の倍加で行われる。発現系ES8(図13)およびES9(図14)を用いる培養において、計画されるBDMはBDM 360gにあらかじめ定義され、誘導はフィード開始後1回の倍加で行われる。この実験は、より低い細胞密度とより低い容量で行うので、ES7とES8の適切な比較を可能にするために、ES7を用いる実験において得られる全収量および容量収量の全収量および容量収量の計算値を用いる。表5における結果は、ES8ベースのゲノムで得られる収量は約3倍高く、製造中での行動はより頑健で安定であることを示している(BDMの計算値とBDM実際の値との比較的小さな差異により示される)。計算過程とはわずかなBDM偏差値があるが、全プロセスを通じて増殖を維持でき、プラスミドにコードされたSODの発現と対照的に、ゲノムにコードされたhSODの発現は細胞の代謝能を超えないことを示している。ゲノムベースのES9を用いる実験により得られる結果(表5第4列目)は、おおよそ20%のさらなる収量増加を示しており、このことは、BL21(DE3)株が、K12株よりもhSODの製造により適していることを意味する。
【0134】
表5:実施例5からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質hSODおよびプラスミドにコードされたタンパク質hSODの製造と、株BL21(DE3)を用いる、ゲノムにコードされたhSODの製造の比較。*を付けた値は、比較性を確かにするための計算値である(プラスミドにコードされた系は低めの細胞密度で運転されるという事実による。)
【表5】
【実施例7】
【0135】
連続(ケモスタット)方式での大腸菌HMS174(DE3)を用いる可溶性GFPmut3.1の発現
ゲノムベース(すなわち無プラスミド)の発現系の遺伝的安定性を確認するために、発現系ES6を用いる連続培養実験(ケモスタット)を行う。希釈速度0.1h-1および1リットル当たりBDM 10gの細胞密度を設定し、実施例2記載の定義された培地を本培養に用いる。この培養の結果を図15に示す。実験の最初の部分で、非誘導状態で細胞を増殖させる。本培養の非誘導状態を、継代培養の行動になんら検出可能な変化なしに42回の倍加を超えて維持する。産生細胞の割合を75〜95%のおよそ一定に保つ((正方形図15)。続いて、フィード300時間近くで、IPTGを用いて培養物を完全に誘導し、誘導後約5時間で、オンラインでの蛍光はこの実験設定で得られる最高レベルに近づく。Reischerら(2004年)によれば、特異的GFP細胞含有量は、BDM 1g当たりGFPおおよそ140〜170mgと計算される。13.5回の倍加を超えて、この誘導状態で細胞を維持する。この期間中のオンラインでの蛍光の推移は、6000〜7000単位のrfu範囲を示す。ゲノムベースの発現宿主の系の安定性は、誘導状態で長期の世代数までも大変高いことをこれらの結果は示している。プラスミドベースの系と対照的に、ゲノム組み込みに基づく系を用いて大変高い発現速度を維持でき、このことは、連続製造工程の開発のための明確な利益を意味する。
【実施例8】
【0136】
あらかじめ定義された誘導物質濃度による改善された誘導制御
組換え遺伝子発現の厳密な誘導制御は、バイオプロセス制御における大変重要な側面である。大変高く、さまざまな遺伝子量(プラスミドコピー数)を有するプラスミドベースのpET/T7系とは異なり、本発明の方法に用いられるゲノムベース(無プラスミド)のT7系は、厳密に、標的遺伝子の定義された遺伝子コピー数(1以上)を特徴とする。この事実により、ゲノムベースの系の発現速度は、より効率的に制御できる。この利益を明らかにするために、異なる誘導レベル(すなわち誘導物質濃度)を適用する、ES10を用いる一連の流加培養を行う。バイオリアクターへのIPTGの単回ボーラスにより誘導を行い、細胞乾燥重量(CDW)に対する誘導物質の最初の比率を設定し、次いでIPTGを連続的にフィードして、定義された濃度を一定のレベルに保つ。CDWに対するIPTGの比率は0.5、0.75、1.0、2.0μmol gCDW-1から、lacリプレッサー分子の完全滴定をもたらす20μmol gCDW-1のレベルまで(すなわち最大級の誘導レベル)である。誘導中の産物の蓄積をモニターし、比産物生成速度(qPF)を:
【数1】
に従って算出するために、GFPの比含量と強く相関する、オフラインで測定される比細胞蛍光(spec.F)(Reischerら、2004年)を用いる。
【0137】
適用した範囲のCDW対IPTG比に応じて、プロセス終了時点で2000〜10000rfu g-1CDWの細胞蛍光レベルがもたらされ、このことは、転写の同調戦略の有効性を印象的に証明している(図16)。完全誘導系のqPF(20μmol IPTG/gCDW)を最高レベルの100%と定め、限定的誘導を用いる実験で得られる平均qPF値は、誘導レベル(誘導物質濃度)と強く相関し、対数トレンドに従う(図17は正規化pPを示す)。低レベルの誘導は、IPTG/CDW比の変化により影響を受けやすく、リアルタイムで推定されるCDWに基づく厳密に制御された誘導物質フィード法を必要とすることをこれらの結果は意味する。さらに、系の完全誘導に必要なIPTG/CDW比は、図17におけるトレンドの外挿により推定でき、3〜5μmol g-1の範囲であり、これは実験に用いられる20μmol g-1よりも実質的に低い。それぞれの適用するIPTG/CDW比は、以下のような一定のバンド幅を有する対応するqPFレベルをもたらす:
【0138】
【表6】
【0139】
結論として、本発明によるゲノムベースの発現に基づく方法は、産物収量および産物生成速度のより優れた制御性を可能にすることをこの実施例は示している。
【0140】
参考文献
【技術分野】
【0001】
本発明は、細菌宿主細胞における工業規模における組換えタンパク質の製造に関する。
【背景技術】
【0002】
現在、細菌宿主、特に大腸菌(Escherichia coli)における組換えタンパク質の製造には、主としてプラスミドベースの発現系が用いられている。これらの系は遺伝子量が多く、樹立されており、利用可能なクローニングプロトコルは扱いやすいため、広く受け入れられるようになった。
【0003】
プラスミドベースの発現は、1細胞当たり最大数百までのプラスミドコピー数を特徴とする(Baneyx、1999年)。発現プラスミドは、通例、プロモーターの制御下に有る目的遺伝子、複製起点(ori)およびプラスミド含有クローンの選択のためのマーカー遺伝子を有する。加えて、しばしば、前記プラスミド(すなわちベクター)上にコード配列または非コード配列または非機能性バックボーン配列が存在する。プラスミドの存在および対応するその複製機構により、宿主細胞の代謝は改変され(Diaz-RizziおよびHernandez、2000年)、その細胞に高い代謝負荷が課され、それによって組換えタンパク質製造のためのそれらの資源が限定を受ける。加えて、遺伝子量の多さと強力なプロモーターの組み合わせの適用は、組換えタンパク質の製造速度の増大を引き起こすが、宿主細胞にとってこの速度は通例高すぎるため、急速で不可逆的な細胞代謝の崩壊をもたらす恐れがある。その結果として、宿主細胞の潜在能力は、プラスミドベースの系では十分に活用できず、その結果として組換えタンパク質の収量は低くなり、品質は低下する。このように、プラスミドベースの発現系の主な欠点の1つは、プラスミドの複製および維持に必要な栄養素およびエネルギーの必要量の増加にあるとすることができる。
【0004】
プラスミドベースの系における他の典型的な現象には、培養過程におけるプラスミドコピー数の変化がある。組換えタンパク質製造には、高い発現率で、チャージされていないtRNAのプールの増加をもたらす飢餓および細胞ストレスが付随する。このことは、プラスミドコピー数(PCN)の制御機構との抵触をもたらす。その結果として、PCNは急速に増加し、培養プロセスの崩壊(いわゆる“暴走(run-away)効果”)を引き起こす。組換え遺伝子発現の誘導後のColE1型プラスミドの暴走現象は、遺伝子量の顕著な増加をもたらしうる(Grabherrら、2002年)。
【0005】
分離不安定性(すなわち無プラスミド宿主細胞の生成)および構造不安定性(すなわちプラスミドの配列における変異)は、プラスミドベースの系においてしばしば見られるさらなる問題である。細胞分裂の間に、細胞はプラスミドを失う場合があり、その結果として目的遺伝子も失う場合がある。このようなプラスミド喪失は、いくつかの外部要因に依存し、細胞分裂回数(世代数)と共に増加する。このことは、プラスミドベースの発酵が、世代数または細胞倍加数に関して限定を受けていることを意味する(Summers、1991年)。
【0006】
すべてを考慮に入れれば、プラスミドベースの発現系のこれらの特性により、組換えタンパク質の収量は限定され、プロセス操作の制御性およびプロセスの経済性は低い。それにもかかわらず、より効率的な代替法を欠くため、プラスミドベースの細菌発現系は、工業規模の異種組換えタンパク質の製造および単離のための最新技術となった。
【0007】
従って、プラスミドベースの発現に代わるものとして、ゲノムベースの発現系が探求されてきた。大腸菌(E. coli)内で染色体により発現される異種タンパク質の、公知で広く利用されている例には、T7ファージのRNAポリメラーゼがあり、これはプラスミドベースの目的遺伝子のプラスミドベースの転写の目的に役立つ。米国特許第4,952,496号に初めて記載されたこの系は部位非特異的組み込みに基づいており、得られた細菌株(例えば大腸菌株BL21(DE3)またはHMS174(DE3))は溶原菌となっている。望ましくないファージ放出に続いて起こりうる細胞溶解を防ぐために、ファージ溶原菌を生じることなくT7ポリメラーゼ遺伝子が染色体に組み込まれた(すなわちT7ポリメラーゼ遺伝子が相同組換えにより挿入された)(WO2003/050240)。同様に、組換えタンパク質のプラスミドベースの発現の目的で、最近、コリネバクテリウム・アセトアシドフィラム(Corynebacterium acetoacidophilum)のゲノムへのT7 RNAポリメラーゼ遺伝子の組み込みが記載された(米国特許第2006/0003404号)。
【0008】
タンパク質発現研究、配列挿入(例えば制限部位、部位特異的組換え酵素標的部位、タンパク質タグ、機能性遺伝子、プロモーターの)、欠失および置換のために、λファージのRed組換え酵素機能(Murphy、1998年)またはRacプロファージのRecE/RecT組換え酵素機能(Zhangら、1998年)により組換えが仲介される、核酸配列のゲノム組み込みのための他の方法が提案されている(Muyrersら、2000年)。
【0009】
WO2001/18222は、エタノール生産特性を宿主に付与するために、大腸菌の染色体プロモーターの下流に、ザイモモナス・モビリス(Zymomonas mobilis)由来のエタノール経路遺伝子を挿入するための、サッカロミセス・セレビジエ(Saccharomyces cerevisiae)FLP(フリッパーゼ)/FRT(フリッパーゼ認識標的)組換え系(Posfaiら、1994年)に基づく染色体組み込み方法の利用を記載している。環状ベクターの染色体組み込み後に、正確に配列(マーカーおよびレプリコン)を除去するためにFLP系が用いられた(Martinez-Moralesら、1999年)。
【0010】
染色体遺伝子置換(Murphy、1998年)または遺伝子破壊(DatsenkoおよびWanner、2000年)のための抗生物質耐性遺伝子の挿入のために、FLPベースのマーカー切除戦略と組み合わせたλ Red法を用いる、短い相同性配列を有する直線状DNAフラグメントを挿入する、DatsenkoおよびWanner(2000年)により開発された方法が使用された。同様に、この方法による、前記タンパク質の前(すなわち上流)でのマーカー、プロモーターおよびHisタグの染色体挿入によるタグ付き相同大腸菌タンパク質の過剰発現が提案されている(Jain、2005年)。
【0011】
抗生物質耐性マーカーなしでプラスミドを増殖させるための宿主/ベクター選択系を確立するために、大腸菌の染色体にリプレッサー分子を組み込むゲノムベースの発現も提案された(WO2006/029985)。その選択系に関連して、プラスミド由来RNAI/IIアンチセンス反応を明らかにするために、モデル系としてレポータータンパク質(緑色蛍光タンパク質)のゲノム挿入が用いられた(Pfaffenzellerら、2006年)。同様に、Zhouら(2004年)はレポーター分子として緑色蛍光タンパク質を含む環状ベクターの組み込みを記載した。
【0012】
WO1996/40722においては、細菌染色体への選択マーカー(すなわち大腸菌のattB部位に)を含む環状ベクター(いわゆる“環状染色体導入DNA”(CTD))の組み込みを使用する方法が記載されている。その方法において、選択マーカーに隣接する二倍のDNA配列を用いることにより、染色体の遺伝子量の増幅が達成された。それによって、得られた染色体の遺伝子量は1細胞当たりおおよそ15〜40コピーであったが、これは一般的に用いられるプラスミドベクターにより達成されるものと同様であった。細菌ゲノムに組み込まれた染色体導入DNAを含むクローンの培養により、プラスミドベースの系で得られるものと同様な組換えタンパク質のレベルが得られた(Olsonら、1998年)。この方法は、CTDのin vitro連結反応を必要とし、組み込みに関してはattB部位に限定されている。
【発明の概要】
【0013】
本発明の目的は、組換えタンパク質を製造するための、新規な工業規模の発現方法を提供することであった。
【0014】
本発明は、
a)細菌発現宿主細胞の集団を培養し、
b)目的タンパク質を採取し、
c)それを分離及び精製する
工程を含む、目的組換えタンパク質を工業規模で製造する方法であって、
工程a)において、培養はフィード培地の添加を用いる方式で行われ、前記方式は流加培養方式、半連続方式または連続方式から選択され、かつ、前記発現宿主細胞は、前記タンパク質の発現を可能にするプロモーターの制御下にある、目的タンパク質をコードするDNA配列を有するDNA構築物を、ゲノム中に組み込まれて含む前記方法に関する。
【0015】
次に、用語“宿主細胞”は、目的遺伝子を含むDNA構築物で形質転換する前の出発細胞として用いられる細菌細胞を意味する。形質転換により目的遺伝子をそのゲノムに組み込んで含む直線状DNA構築物を有する細胞を“発現宿主細胞”と呼ぶ。
【0016】
発現カセットは、その重要な要素として、目的遺伝子、プロモーターおよびリボソーム結合部位(RBS)を含む。さらに、発現カセットは、“ヘルパータンパク質(helper protein)”、例えばマーカーまたは発現宿主細胞の増殖を助けるタンパク質、例えば糖またはタンパク質の代謝に関連する遺伝子および/または目的遺伝子の発現を改善するタンパク質(例えばシャペロン分子(GroElまたはGroEsなど)をコードする配列または転写および翻訳に関与する遺伝子をコードする配列を含むことができる。
【0017】
発明の詳細な説明
目的タンパク質
目的タンパク質に関しては、制限はない。原則として、工業規模で製造される任意のタンパク質、例えば工業用タンパク質または治療用タンパク質であることができる。本発明の方法により製造できるタンパク質の例は、限定するものではないが、酵素、制御タンパク質、受容体、ペプチド、例えばペプチドホルモン、サイトカイン、膜または輸送タンパク質である。目的タンパク質はまた、予防接種に使用する抗原、ワクチン、抗原結合タンパク質、免疫刺激タンパク質、アレルゲン、完全長抗体または抗体フラグメントもしくは誘導体であることもできる。抗体誘導体は、一本鎖抗体、(scFV)、Fabフラグメント、FVフラグメント、単一ドメイン抗体(VHまたはVLフラグメント)、ドメイン抗体、例えばラクダ科動物単一ドメイン抗体(VHH、ナノボディ)または他の抗体フォーマット、例えばAndersenおよびReilly(2004年)またはHolligerおよびHudson(2005年)に記載されているものの群から選択することができる。目的タンパク質をコードするDNA分子を“目的遺伝子”とも呼ぶ。
【0018】
プロモーター
本発明の意味において、“プロモーター”は、RNAポリメラーゼの結合および転写開始を可能にする発現調節因子である。本発明の一実施形態において、目的遺伝子は“強力な”プロモーターの制御下に有る。強力なプロモーターは、一方ではRNAポリメラーゼ、通例天然に存在する対応するRNAポリメラーゼに対するプロモーター配列の高い結合親和性を特徴とし、他方ではそのRNAポリメラーゼによるmRNAの高い産生速度を特徴とする。
【0019】
好ましくは、目的遺伝子は誘導性プロモーターの制御下に有る。誘導性プロモーターは、外部要因、例えば誘導原(“誘導物質”とも呼ばれる)分子の存在もしくはリプレッサー分子の非存在または物理的要因、例えば温度、浸透圧もしくはpH値の上昇もしくは低下により制御できるプロモーターである。種々のプロモーターおよびそれぞれの誘導原理は、Makrides(1996年)により概説されている。
【0020】
本発明において、プラスミドベースの発現のために開発されてきたプロモーターを使用することができるが、強力なプロモーターの中で、バクテリオファージT7のT7プロモーターが最も広く使用されてきた。
【0021】
好ましい側面において、プロモーターはT7プロモーターである。T7プロモーターおよび、lacプロモーターの制御下に有るT7ポリメラーゼ遺伝子の組み合わせを含むT7ベース発現系は、細菌細胞および真核細胞のどちらにおいても、組換えタンパク質の大規模発現に広く使用されている。この系はT7 RNAポリメラーゼを使用し、その転写速度は大腸菌のRNAポリメラーゼよりも数倍高い。T7プロモーターからの発現は、T7プロモーターに厳密に特異的なT7 RNAポリメラーゼの制御下に有る(Chamberlinら、1970年)。すなわち、T7プロモーターは、バクテリオファージT7のポリメラーゼによってのみ利用されうる。IPTGを培地に添加するとき、lacプロモーターからの転写によりT7 RNAポリメラーゼが発現される。
【0022】
本発明の意味において、用語“T7プロモーター"は、バクテリオファージT7のゲノムに存在するプロモーターばかりでなく、T7 RNAポリメラーゼによる転写を仲介する能力を有するこのようなプロモーターのコンセンサス配列およびバリアントを含む。バクテリオファージT7は、17の異なるプロモーター配列を含有し、そのすべてが高度に保存されたヌクレオチド配列を含む(OakleyおよびColeman、1977年;PanayotatosおよびWells、1979年)。
【0023】
本発明の特定の実施形態によれば、目的遺伝子は、tacもしくはtrcプロモーター、lacもしくはlacUV5プロモーター(すべてラクトースまたはそのアナログであるIPTG(イソプロピルチオール-β-D-ガラクトシドにより誘導される))、強力に調節できるaraBADプロモーター(PBAD;Guzmanら、1995年、アラビノースにより調節される)、trpプロモーター(β-インドールアクリル酸添加またはトリプトファン飢餓により誘導され、トリプトファン添加により抑制される)、λプロモーターpL(λ)(温度上昇による誘導)、phoAプロモーター(リン酸飢餓により誘導される)または組換えタンパク質発現に適した他のプロモーター(Makrides、1996年)の制御下に有ることができ、これらはすべて大腸菌のRNAポリメラーゼを利用する。
【0024】
誘導性プロモーターには、“リーキー”発現行動を示すものがある。このようなプロモーター(いわゆる“リーキープロモーター(leaky promoter)”)は、原則として誘導性であるが、それにもかかわらず外部からの誘導なしに、基礎発現も示す。非誘導条件下でリーキー発現を示す誘導性プロモーターは、同様に構成的プロモーターとして作用することができる(すなわちこれらは定常的・連続的に活性であり、特定の培養条件の結果として活性化または促進されることもできる)。リーキープロモーターは、連続運転培養プロセスに特に有用な可能性がある。例には、T7プロモーターおよびtrpプロモーターがある。
【0025】
本発明の方法において、プロモーターは構成的であることができる。すなわち一方では誘導を、他方ではおそらく抑制を必要とせずに発現を調節するプロモーターであることができる。従って、継続的で定常的な発現が特定のレベルで行われる。誘導物質を必要としないという利点は別にして、構成的プロモーターは、方法が連続的な(下記のように)実施形態に特に有用である。例として、強力な構成的HCDプロモーター(Pooら、2002;Jeongら、2004年)を構成発現に適用することができる。
【0026】
細菌細胞のゲノムに組み込まれる直線状カセットは、目的遺伝子のすぐ上流にシャイン・ダルガーノ(SD)配列(リボソーム結合部位(RBS)とも呼ばれる)も含む。
【0027】
発現カートリッジ
次に、細菌ゲノムに組み込まれる直線状または環状DNA構築物は、“発現カートリッジ”または“カートリッジ”とも呼ばれる。組み込みの結果として、発現宿主細胞は組み込み“発現カセット”を有する。好ましくは、カートリッジは、本質的に、プロモーター、目的遺伝子、リボソーム結合部位およびゲノム領域に相同性を示し、相同組換えを可能にする、末端で隣接する2つの領域(図10に示す挿入カートリッジ参照)を含む直線状DNA構築物である。加えて、以下に詳しく述べるように、他の配列を含むことができる;例えば、抗生物質選択マーカー、原栄養性選択マーカーまたは蛍光マーカーをコードする配列、代謝遺伝子、タンパク質発現(例えばT7 RNAポリメラーゼ遺伝子など)を改善する遺伝子または、組み込み後に特定の配列(例えば抗生物質耐性遺伝子)を除去することを可能にする2つのフリッパーゼ認識標的部位(FRT)をコードするマーカー。
【0028】
カートリッジは、当該技術分野で公知の方法により合成され、増幅される。直線状カートリッジの場合は、通例ポリメラーゼ連鎖反応(PCR)(WilsonおよびWalker、2005年)による。直線状カートリッジは、通例構築するのがより容易であるため、本発明の方法に用いる発現宿主細胞を得るには直線状カートリッジが好ましい。さらに、直線状発現カートリッジの使用には、カートリッジに隣接する相同領域のそれぞれの設計によりゲノム組み込み部位を自由に選択できるという利点がある。それによって、直線状発現カートリッジの組み込みにより、ゲノム領域に関するより大きな可変性が可能となる。
【0029】
宿主細胞
細菌宿主細胞に関しては、原則として制限はない。目的遺伝子の挿入のための遺伝子操作、好都合には部位特異的組み込みが可能であり、工業規模で培養することができる限り、細菌宿主細胞は真正細菌(グラム陽性またはグラム陰性)であることもできるし、古細菌であることもできる。好ましくは、宿主細胞は、高い細胞密度まで培養することが可能な性質を有する。
【0030】
工業用組換えタンパク質の製造に適することが示された細菌宿主細胞の例には、大腸菌(Lee、1996年;HannigおよびMakrides、1998年)、バチルス・ズブチリス(Bacillus subtilis)、シュードモナス・フルオレッセンス(Pseudomonas fluorescens)(Squiresら、2004年;Retallackら、2006年)ばかりでなく、種々のコリネバクテリウム(Corynebacterium)(米国特許第2006/0003404 A1号)およびラクトコッカス・ラクティス(Lactococcus lactis)株(Mierauら、2005年)がある。好ましくは、宿主細胞は大腸菌細胞である。
【0031】
宿主細胞に対する他の必要条件は、目的遺伝子を調節するプロモーターに結合できるRNAポリメラーゼをそれが含むことである。RNAポリメラーゼは、宿主細胞に対して内在性であることもできるし、外来性であることもできる。
【0032】
好ましくは、外来性の強力なRNAポリメラーゼを有する宿主細胞が用いられる。最も好ましくは、外来性RNAポリメラーゼ(例えば、T7プロモーターを用いる場合は、いわゆる“T7株”におけるT7様RNAポリメラーゼなど)をそのゲノムに組み込んで含むように操作された宿主株、例えば大腸菌株が用いられる。広く使用され、市販されているT7株の例には、例えばBL21(DE3)、HMS174(DE3)およびそれらの派生株または近縁株がある(Novagen社、pET Systemmanual、第11版)。これらの株は、lacUV5プロモーターの制御下にあるT7 RNAポリメラーゼ遺伝子を含むDE3溶原菌である。IPTGでの誘導により、T7 RNAポリメラーゼの産生が可能となり、次いでそれがT7プロモーターの制御下に有る目的遺伝子の発現を指示する。
【0033】
好ましくは、例えばWO2003/050240に記載されているように、ファージ溶原菌を生じることなしにゲノムに組み込まれたT7 RNAポリメラーゼを含む宿主細胞が用いられる(ファージ溶原菌を生じない細胞は“非溶原性”である)。
【0034】
"T7様RNAポリメラーゼ"は、限定するものではないが、他のT7様ファージ由来のRNAポリメラーゼ、例えば米国特許第4,952,496号に開示されているT3 RNAポリメラーゼを含む。T7様RNAポリメラーゼの必要条件は、高度に特異的な同種プロモーターが存在しなければならないことおよび前記RNAポリメラーゼの存在下で目的遺伝子が急速に転写されることである。
【0035】
あるいは、T7 RNAポリメラーゼを含む前記の株を使用するために、例えば、米国特許第4,952,496号、米国特許第2006/0003404号またはWO2003/050240に記載されているようにして、このような株をあらたに作成することができる。
【0036】
ファージDE3によりゲノムベースのT7 RNAポリメラーゼを受け取った大腸菌宿主細胞株BL21(DE3)またはHMS174(DE3)は溶原性である。溶原株は、望ましくないファージ放出および細胞溶解を引き起こす溶菌特性を潜在的に示す不都合を有する。従って、本発明に関しては、宿主細胞に含まれるT7 RNAポリメラーゼが、宿主細胞ゲノムにおける残りのファージ配列の挿入をさけるか、好ましくは排除する方法により組み込まれていることが好ましい。本発明の好ましい実施形態において、例えば、DatsenkoおよびWanner(2000年)により記載された方法に従って、部位特異的組み込みにより細胞のゲノムにT7ポリメラーゼを組み込むことにより宿主細胞が得られた。
【0037】
T7系を用いる場合、T7 RNAポリメラーゼと組み合わせて、そのプロモーター(好ましくはlacUV5プロモーター)をそのゲノムに組み込んで有する宿主細胞を用いることができる。
【0038】
あるいは、T7 RNAポリメラーゼは、発現カートリッジの要素、すなわち発現カセットの一部として細胞のゲノムに組み込まれた要素として提供できる。
【0039】
宿主細胞に対する他の必要条件は、相同組換えを行うその能力である(これは、ゲノムへの発現カートリッジの組み込みおよび場合によりP1形質導入に関連する)。
【0040】
従って、宿主細胞は組換えタンパク質RecAの機能を有することが好ましい。しかしながら、RecAは培養中に望ましくない組換え事象を引き起こす恐れがあるので、宿主細胞は、そのゲノムrecA部位にゲノム変異(機能不全にする)を有し、その代わりに、ヘルパープラスミドに存在するrecA配列により提供されるRecA機能を有することが好ましいが、これはヘルパープラスミドの温度感受性レプリコン(DatsenkoおよびWanner、2000年)を用いて組換え後に除去(治癒)することができる。
【0041】
組換えを考慮すれば、宿主細胞は、RecAに加えて、組換えタンパク質(例えばExo、BetaおよびGam)をコードするDNA配列をそのゲノムに含むことが好ましい。この場合は、すでにその特徴を有する宿主細胞を選択することもできるし、遺伝子工学によりこれらの配列を挿入して、あらたに宿主細胞を作成することもできる。
【0042】
部位特異的遺伝子挿入を考慮すれば、宿主細胞に対する他の必要条件は、その配列が知られ、破壊または別なふうに操作して、細胞に不利益を与えることなく異種配列を挿入することができる少なくとも1つのゲノム領域(コードまたは任意の非コード・機能性または非機能性領域あるいは未知の機能を有する領域のどちらか)を宿主細胞が含むことである。
【0043】
特定の実施形態において、宿主細胞は、選択を考慮してそのゲノムにマーカー遺伝子を含む。
【0044】
組み込み遺伝子座
組み込み遺伝子座に関しては、本発明に用いられる発現系は、幅広いバリエーションが可能である。原則として、既知の配列を有する任意の遺伝子座を選択することができるが、ただし、その配列の機能が重要でないか、あるいは重要である場合、補完することができる(例えば栄養要求性の場合など)ことを条件とする。
【0045】
組み込み遺伝子座を選択する場合には、いわゆる“適応進化”(Herringら、2006年)によって引き起こされるDNAの変異頻度が大腸菌のゲノムの全域で変化することおよび、染色体にコードされた組換え遺伝子発現により引き起こされる代謝負荷が組み込み部位での変異頻度の増加を引き起こす恐れがあることを考慮する必要がある。安定で頑健な発現宿主細胞を得るためには、組み込み部位として、より低い変異頻度をもたらす高度に保存されたゲノム領域を選択することが好ましい。このような高度に保存された大腸菌ゲノム領域には、例えば、リボソームの成分をコードする遺伝子またはペプチドグリカンの生合成に関与する遺伝子があり(Mauら、2006年)、発現カートリッジの組み込みのためにこれらの領域が選択されることが好ましい。その際、正確な組み込み遺伝子座は、機能性遺伝子が破壊も障害も受けず、どちらかと言えば組み込み部位が非機能性領域に位置するように選択される。
【0046】
カートリッジの組み込みのために選択できる既知の配列を有するゲノム領域は、非必須遺伝子またはその一部のコード領域;重要でない非コード機能性領域(すなわちプロモーター、トランスポゾンなど)、その欠失が特定の目的タンパク質の製造を考慮して有利な可能性のある遺伝子、例えば特定のプロテアーゼ、OmpTのような外膜タンパク質、産物の潜在的汚染物質、代謝のタンパク質、(例えば所定の宿主株および/または発酵プロセスに望ましくないかまたは重要でない糖分子の代謝に関連する)またはストレスシグナル伝達経路、例えばアミノ酸飢餓の間tRNAおよびrRNAの合成を抑制する、原核生物の転写制御機構である緊縮応答において生じる経路をコードする遺伝子から選択できる(Cashel、1969年)。
【0047】
あるいは、組み込み部位は、組み込み後にマーカー表現型の消失についての選択を可能にするマーカー遺伝子であることができる。その欠失が蛍光の消失をもたらし、発現カセットを含むクローンの視覚的検出を可能にする、緑色蛍光タンパク質などの蛍光マーカーが使用されることが好ましい(図10)。
【0048】
あるいは、組み込みのために選択するのに有用な部位は、欠失したとき栄養要求性を示す機能である。この場合は、組み込み部位は、生合成または代謝経路に関与する酵素であって、その欠失が栄養要求株をもたらす酵素であることができる。陽性クローン、すなわち発現カセットを含むクローンは、前記酵素の基質または前駆体分子に対する栄養要求性に関して選択することができる(図10)。
【0049】
あるいは、組み込み部位は、発現カセットに存在する対応原栄養性マーカー(すなわち欠損配列を補完または置換する配列)により置換/補完し、それにより原栄養性選択を可能にする栄養要求性マーカー(非機能性遺伝子、すなわち欠損遺伝子)であることができる。
【0050】
一側面において、領域は非必須遺伝子である。一側面によれば、領域は、それ自体細胞に非必須な遺伝子であることができる。
【0051】
非必須細菌遺伝子は文献で知られ、例えばGerdesら(2002年および2003年)、PEC(大腸菌ゲノム情報(Profiling the E. coli Chromosome))データベース(http://www.shigen.nig.ac.jp/ecoli/pec/genes.jsp)またはいわゆる“Keio collection”(Babaら、2006年)から知られる。
【0052】
非必須遺伝子の1例にRecAがある。この部位に発現カセットを組み込むことにより、宿主細胞の必要条件との関連で前述したゲノム変異が得られる。
【0053】
適切な組み込み部位、例えば利用しやすいおよび/またはより高い発現率を得ることが予想される部位は予備スクリーニングで決定することができる。組み込みカートリッジが、例えば実施例5で用いられた、緑色蛍光タンパク質をコードするカートリッジのような、可変要素として特定の組み込み部位を考慮して事前選択された種々の組換え配列を特徴とし、不変要素として、誘導性プロモーターの制御下に有る、容易に検出できるタンパク質をコードするDNA配列であるサロゲイト“目的遺伝子”を含む組み込みおよび選択のための基本配列を特徴とする、Keio collection(Babaら、2006年)による一連の単一変異欠失を作成することにより、このようなスクリーニングを行うことができる。このようにして作成した単一ノックアウト変異体の発現レベルは、蛍光測定により容易に定量できる。この手順の結果に基づき、所望の標的タンパク質の発現レベルのカスタマイズは、組み込み部位および/または組み込みカートリッジ数を変化させることにより達成できる。
【0054】
宿主細胞が、出発細胞の特徴として、あるいは遺伝子工学により得られたものとして、組換えタンパク質(例えばExo、BetaおよびGam)をそのゲノム中にコードするDNA配列を含む実施形態において、これらの組換えタンパク質配列が位置するゲノム部位で組み込みを起こすことができる。発現カートリッジの組み込みにより、組換えタンパク質をコードする配列は破壊または除去され、その結果として、プラスミドがコードするヘルパータンパク質の場合は、別の段階で除去する必要がない(図9)。
【0055】
組み込み方法
前述のように、組み込み遺伝子座に関しては、既知の配列を有する任意の遺伝子座を選択することができる。ただし、その配列の機能が重要でないか、あるいは重要である場合、補完することができることを条件とする。好ましい実施形態において、直線状カートリッジの組み込みは、attB部位またはattTn7部位などの結合部位で行われ、これらはよく立証された組み込み部位である。しかしながら、前述のように、この系の順応性により、任意の事前選択された遺伝子座で目的遺伝子を挿入することができる。
【0056】
細菌ゲノムへの目的遺伝子の組み込みは、慣用法により、例えば、attTn7部位に関して記載されているように(Rogersら、1986年;WaddellおよびCraig、1988年;Craig、1989年)、染色体の特定部位に相同な隣接する配列を含む直線状カートリッジを用いることにより達成できる。カートリッジは、大腸菌株、例えばプラスミドpKD46を含む大腸菌MG1655(DatsenkoおよびWanner;2000年)の細胞に形質転換される。このプラスミドは、in vivoで組換えを促進するγファージ由来Red機能(γβ exo)を有する。
【0057】
あるいは、最初に中間ドナー宿主細胞のゲノムに発現カセットを組み込むことができ、それからファージP1での形質導入(SternbergおよびHoess、1983;およびLennox、1955年)により宿主細胞にそれを移すことができるが、ドナー細胞には、例えば、欠損γプロファージを有する大腸菌K12株MG1655またはDY378(Yuら、2000年)がある。手短かに言えば、P1形質導入は、1つの"ドナー"株からもう1つの"レシピエント"株に選択遺伝子マーカーを移すために用いられる方法である。細菌培養におけるファージの複製および溶菌中に、低い割合のファージ粒子が、発現カセットを含むゲノムセグメントを含む。ひとたびドナー細胞からファージ集団が作成されれば、レシピエント細胞(本発明の場合は宿主細胞)を感染させるためにこのファージが用いられる。P1ゲノムをパッケージングしたファージにより細菌の大部分は溶菌するが、ファージの一部はドナー宿主由来のゲノムセグメントを注入する。次いで、相同組換えにより、既存の相同のセグメントと入ってくるゲノムセグメントとを置換することができる。ドナー細菌のゲノムセグメントを選択するための培地に感染したレシピエント細菌をプレーティングする(抗生物質耐性、原栄養性、蛍光標識など)。ファージの感染性を調節しなければならないことは重要である。そうでなければ、近くの細胞から放出されたファージが、形質導入粒子に感染した細菌に感染し、溶菌してしまうからである。ファージP1は感染性にカルシウムを必要とするので、カルシウムの存在および非存在において増殖させることによりファージの感染性を調節することができる。カルシウムキレート化剤であるクエン酸塩が通例用いられる。なぜならば、クエン酸塩は遊離カルシウムの濃度を(クエン酸カルシウムの形成により)P1感染を抑制するのに十分低い濃度に低下させるが、細胞をカルシウム飢餓にさせるまでは低下させないからである。
【0058】
組み込み手順の簡単化のために、図9に示すように、DatsenkoおよびWanner(2000年)に従ってプラスミドpKD20またはpKD46を用いて組換えタンパク質を得る代わりに、組換え機能をコードする遺伝子exo、betaおよびgamを宿主細胞の染色体に事前に存在させることができる。組換えタンパク質の発現は誘導性プロモーター、例えばfrt、lac、tac、T7またはaraPプロモーターにより調節できる。対応する誘導物質の適用により、こうして得られた改変株を、組換えタンパク質の部位での直線状発現カートリッジの組み込みに使用することができる。それによって、組換えタンパク質が最初に発現され、次いで前記組換えタンパク質をコードするゲノム領域に発現カートリッジを組み込むのに利用される。それによって、組換えの結果として、かつ本実施形態の特別な利点として、組換えタンパク質をコードする遺伝子が除去(切除)または破壊される(図9)。さらにまた、本実施形態は、プラスミドがこれらの組換え機能をコードする必要性がないという利点を有する。
【0059】
本発明に有用な他の組み込み方法の例には、限定するものではないが、例えばRed/ET組換えに基づく例がある(Muyrersら、2002年;Wenzelら、2005年;Vetcherら、2005年)。
【0060】
選択マーカー
陽性クローン、すなわち発現カセットを含むクローンは、マーカー遺伝子によって選択できる。
【0061】
いくつかの実施形態において、マーカー遺伝子、例えば抗生物質耐性遺伝子または蛍光タンパク質、例えばGFPをコードする遺伝子を事前にそのゲノムに組み込んで含む宿主細胞が用いられる(図10に示すように)。この場合は、選択マーカーを含まない発現カートリッジが、染色体のマーカー遺伝子の遺伝子座に組み込まれ、それぞれの表現型の喪失/消失によって陽性クローンが選択される。例えば、陽性クローンは、抗生物質の感受性または蛍光の消失によって選択され、培養プレート上で直接に可視化できる。マーカーは発現カセットにより中断されるかまたは完全に置換され、組み込み後に機能性マーカー配列は存在せず、抗生物質耐性遺伝子の場合のように望ましくない場合でも除去する必要はないという利点をこれらの実施形態は有する。
【0062】
あるいは、マーカー遺伝子は発現カートリッジの一部である。選択に用いられるマーカーが抗生物質耐性(例えばカナマイシンまたはクロラムフェニコール耐性)を付与する遺伝子である場合、図7に示すように、陽性クローンは抗生物質耐性(すなわちそれぞれの抗生物質の存在下での増殖)により選択される。好ましくは、抗生物質によるクローンの選択には、カナマイシンが用いられる。
【0063】
マーカー遺伝子(宿主細胞のゲノムに存在するものであるか、または発現カートリッジを用いて導入されたものであるかにかかわらず)は、図7に概略を示すように、例えば、酵母2μプラスミドの部位特異的組換え系であるFLP組換え酵素およびその組換え標的部位FRT(DatsenkoおよびWanner、2000年)に基づくFLP組換え酵素機能を用いることによるカセットの組み込みにより除去できる。
【0064】
特定の実施形態において、発現細胞は、選択マーカー遺伝子、例えばクロラムフェニコールまたはカナマイシンなどの抗生物質耐性遺伝子、蛍光マーカーまたは糖もしくはアミノ酸の代謝経路に関与する遺伝子の欠損を有するように操作されている。この場合、目的遺伝子を有するカートリッジは、マーカー遺伝子の欠失部分を有し、マーカー遺伝子の組み込みによりその機能性を回復する。一例として、カートリッジはその末端の1つにマーカー遺伝子の欠失部分を含み、ゲノムに組み込まれた欠損マーカー遺伝子に隣接して直接に組み込まれ、それにより2つのフラグメントの融合によりマーカー遺伝子が完全となり、その機能的発現が可能となる。抗生物質耐性遺伝子の場合は、発現カセットを含む細胞は特定の抗生物質に対して耐性であり、蛍光マーカーの場合は、蛍光により細胞を可視化することができ、代謝経路遺伝子の場合は、細胞はそれぞれの成分を代謝する能力を得る(図11に概略を示す)。この実施形態の利点は、先行技術と比較して、カートリッジのマーカー遺伝子のほんの短い部分しか合成を必要とせず、より短いかまたは小さい挿入カートリッジを可能とすることである。
【0065】
特定の実施形態において、陽性クローン(すなわち発現カセットを有するクローン)の選択は、宿主細胞の栄養要求性の補正(すなわち補完)により行われる。図8に概略を示すこの実施形態において、簡単な方法で陽性形質転換細胞コロニーの選択を可能にするように選択された変異を有する宿主細胞、例えばその増殖に必須である化合物を合成できないようにする欠失または変異(このような変異を“栄養要求性マーカー”と呼ぶ)を有する株が用いられる。例えば、プロリン合成経路の遺伝子が不活性化されている細菌変異体はプロリン栄養要求株である。このような株はプロリンを合成することができず、従って、プロリンの非存在下で増殖できるプロリン原栄養株とは対照的に、環境からプロリンを吸収することができる場合にのみ増殖できる。
【0066】
栄養要求性マーカーを有する任意の宿主細胞を用いることができる。好ましくは、栄養要求性マーカーとして、アミノ酸合成に必要な遺伝子における変異、例えばプロリン、ロイシンもしくはトレオニンの合成またはチアミンなどの補因子に関連する遺伝子における変異が用いられる。本発明によれば、目的遺伝子を組み込むことに加えて、発現カートリッジの構成要素として欠失/欠損遺伝子をゲノムに組み込むことにより、宿主細胞の栄養要求性が補正される。このようにして得られた原栄養性細胞は、元の宿主細胞が栄養要求する化合物を含まず、従って陽性クローンのみを増殖させることが可能ないわゆる“最小培地”(原栄養性選択)で増殖させることにより容易に選択できる。
【0067】
原栄養性選択は組み込み遺伝子座とは無関係である。原栄養性選択の組み込み遺伝子座はゲノムにおける任意の遺伝子であることもでき、栄養要求性マーカーを含む遺伝子座であることもできる。原栄養性選択の特別な利点は、組み込みに成功した後に、宿主に対して異物である抗生物質耐性マーカーも任意の他のマーカーもゲノムに残らないことである。その結果として、前記マーカー遺伝子を除去する必要がなく、迅速で簡単なクローニングおよび選択手順が得られる。他の利点は、遺伝子機能を回復させることは細胞にとって有益であり、その系に高い安定性を与えるという点である。
【0068】
あるいは、同様に図8に概略で示すように、発現カートリッジと共にゲノムに挿入されたマーカー遺伝子は、特定の選択方法を可能にする代謝遺伝子であることができる。このような代謝遺伝子は、特定の(まれな)糖または他の炭素源で細胞を増殖させることができ、唯一の炭素源として前記糖で細胞を増殖させることにより陽性クローンの選択を行うことができる。
【0069】
前述のように、細菌の長期培養の間に、適応進化(Herringら、2006年)は、染色体にコードされた組換えタンパク質の発現中に組み込み部位での変異頻度の増加を引き起こす恐れがある。変異株の欠失した機能を補う発現カートリッジと組み合わせて栄養要求性ノックアウト変異株を使用すること(それによって栄養要求性変異株から原栄養株を作成する)は、適応進化が生じた細胞が抑制される競争的利点を細胞が獲得することによる利点を、回復された遺伝子が細胞に与えるというさらなる利点を有する。それによって、変異クローンに負の選択という手段が与えられる。
【0070】
いくつかの実施形態において(例えばFACS分析または免疫学的分析(ELISA)による、単一細胞または単一コロニーベースでの目的タンパク質の検出が可能な場合)、マーカー遺伝子は必要ではない。なぜなら、目的タンパク質の直接検出により陽性クローンが検出できるからである。
【0071】
他の特徴
発現宿主細胞を得るための組み込み方法は、ゲノムの1つの部位に目的遺伝子を1つ組み込むことに限定されない。組み込み部位および発現カセットの両方に関して組み込み方法を変えることが可能である。一例として、2以上の目的遺伝子を挿入することができる。すなわち、同一または異なるプロモーターの制御下に有る2以上の同一または異なる配列を、ゲノムの1以上の種々の遺伝子座に組み込むことができる。一例として、ヘテロ二量体複合体を形成する2種の異なるタンパク質の発現が可能である。ヘテロ二量体タンパク質は、2つの個別に発現したタンパク質サブユニット、例えばモノクローナル抗体または抗体フラグメントのH鎖およびL鎖からなる(AndersenおよびReilly、2004年;HolligerおよびHudson、2005年)。他のヘテロ二量体タンパク質の例には、例えばCapZ(Remmertら、2000年)、Rasファルネシルトランスフェラーゼ(TsaoおよびWaugh、1997年)、血小板活性化因子アセチルヒドロラーゼIb(Sheffieldら、2001年)またはヒトDNAヘリカーゼII(Ochemら、1997年)がある。モノマーをコードするこれらの2つの配列は、1つの組み込み遺伝子座に挿入された1つの発現カートリッジに存在することができる。あるいは、これらの2つの配列は、2つの異なる組み込み遺伝子座に互いに独立して挿入された2つの異なる発現カートリッジに存在することもできる。いずれにせよ、プロモーターおよび誘導方法は、同一であることもできるし、異なることもできる。
【0072】
本発明は、無プラスミドでの目的タンパク質の製造が可能であるが、目的遺伝子以外に発現配列、例えば前述のヘルパータンパク質および/または組換えタンパク質を含むプラスミドが発現宿主細胞に存在することを排除するものではない。当然、このような実施形態において、プラスミドの存在により本発明の利点が覆されないように注意が必要である。すなわち、プラスミドは低コピー数で存在することが必要であり、細胞に代謝負荷を与えてはならない。
【0073】
組換えタンパク質のゲノムベースの発現は、以下の主な利点を提供する:
【0074】
発現宿主の構築方法に関しては、利点は、(i)直線状挿入カートリッジの簡単な合成および増幅方法、(ii)組み込み遺伝子座に関する高い柔軟性(すなわち制限なし)、(iii)選択マーカーおよび選択原理に関する高い柔軟性、(iv)後の選択マーカーの除去の選択肢、(v)不連続で定義された数の挿入された発現カートリッジ(通例1または2)である。
【0075】
本発明の方法に有用な発現系は、本質的にまたは完全にファージ機能のないように設計されたものであることができる。
【0076】
ゲノムへの1以上の組換え遺伝子の組み込みにより、1細胞当たり不連続であらかじめ定義された数の目的遺伝子がもたらされる。遺伝子1コピーを挿入する本発明の実施形態において、最大数百までのコピー数を伴うプラスミドベースの発現と比較して、この数は通例1である(細胞分裂中に一過性に生じる、細胞が2以上のゲノムを含む場合を除く)。本発明の方法に用いられる発現系において、プラスミドの複製による宿主の代謝を軽減することにより、組換えタンパク質製造に細胞の合成能力の増加した部分が用いられる。遺伝子量を減らすことにより、宿主の代謝に有害な効果を与えることなく、T7系などの強力なプロモーターを適用することができる。
【0077】
発現系が誘導レベルに関して制限を有さないことは特別な利点である。このことは、プラスミドベースの系においてしばしば生じる、誘導によりPCNが大幅に増加する“過剰誘導”をこの系が起し得ないことを意味する(Teichら、1998;WrobelおよびWegrzyn、1998年)。遺伝子量の多さおよび/または増加と組み合わせた強力な発現系は、代謝過負荷を引き起こし、細胞生存率の顕著な低下をもたらし(Bentlyら、1990;Glick、1995;Cserjan-Puschmannら、1999年)、産物生成期間の顕著な短縮および全収量の低下を引き起こす。
【0078】
上記のように、プラスミドベースの発現系は、細胞が、細胞分裂の間にプラスミド、それとともに目的遺伝子を失う恐れがあるという欠点を有する。このようなプラスミドの喪失はいくつかの外部要因に依存し、細胞分裂回数(世代数)と共に増加する。このことは、プラスミドベースの発酵は、世代数(従来の発酵においては、この数はおおよそ20〜50である)に関して制限されていることを意味する。対照的に、本発明の方法に用いられるゲノムベースの発現系は、事実上無限の世代数、従って調節条件下で理論上は無限の培養時間にわたって安定であらかじめ定義された遺伝子量を確実にする(目的タンパク質を産生しない細胞が生じるという不都合はなく、ただ任意の遺伝子に生じる自然突然変異が生じうるという制限があるだけである)。
【0079】
化学的誘導性プロモーターの場合は、例えばStriednerら(2003年)に従って連続方式で添加されるとき、誘導分子の量が、全培養にわたって一定であるか、あるいは培養時間にわたってあらかじめ定義された値に変化させるかにより、1細胞当たりの遺伝子量に直接に比例しているという利点を本発明は提供する。それによって、組換え発現速度の調節を達成でき、このことが遺伝子発現速度を調節するために、大変関心の持たれるところである。
【0080】
ゲノムベースの発現系は、タンパク質発現の正確な調節が可能なので、よく調節された発現に依存または頼る発現標的経路と組み合わせると特に有利である。好ましい実施形態において、本発明の方法は、細菌細胞質から培地への目的タンパク質の分泌(排泄)を含む。この実施形態の利点は、先行技術の分泌系と比較してより高い力価の分泌タンパク質をもたらす、最適化され、維持されたタンパク質分泌速度である。細菌細胞壁を通しての受動拡散または細胞膜周辺腔への能動分泌と、それに続く拡散または物理化学的放出(例えば米国特許第4,680,262号および米国特許第4,963,495号に記載されているようなStIIシグナル配列を用いる)、あるいは細胞質から培地への目的タンパク質の直接移動(例えばChoiおよびLee(2004年)に記載されている方法)により分泌を生じることができる。
【0081】
前述のように、本発明は、簡略プロセス、改善されたプロセス予測性および発酵間の高い再現性を設計することを可能にする。前述の発現系を用いる本発明のプロセスは、流加培養方式、半連続方式または連続方式(すなわちケモスタット)で行われ、それによって、ゲノムにコードされた発現系の利点が最適に活用される。宿主細胞の必要条件により定義され、選択されるプロモーターによりあらかじめ定義される場合を除き、増殖速度、温度および培地成分などのプロセスパラメータに関して制限はない。
【0082】
他の利点は、誘導分子の選択に関する。大腸菌内での組換え遺伝子の高レベルの発現のための利用可能な系の大部分は、lacベースのプロモーター・オペレーター系(Makrides、1996年)である。天然の誘導物質1,6-アロラクトースの前駆体であり、容易に大量に入手できる安価な天然炭水化物であるラクトースは、IPTGに代わるものとして考えられてきた。しかしながら、かっては、グルコース培地による誘導物質排除(inducer exclusion)および細胞内での分解の問題により、ラクトースは誘導には不適当であると考えられた。本発明に用いられる発現系は、誘導物質としてのラクトースの連続供給を用いる炭素制限培養(Striednerら、2003年)を可能にし、それと共にラクトース排除を防ぎ、ラクトースを用いる従来のパルス誘導中に観察される誘導レベルの低下(Hoffmanら、1995年)を排除し、誘導物質としてラクトースを用いる場合でも厳密な発現速度調節を可能にする。
【0083】
重要なことに、本発明に用いる発現系は、培地容量あたりのタンパク質濃度(すなわち力価)および得られたバイオマス中のタンパク質含量の両方に関して高収量の組換えタンパク質を提供する利点を有する。この特徴により、本発明に用いられる発現系が先行技術の発現系より優れたものとなる。
【0084】
さらにまた、本発明は、発現宿主細胞の選択および/または発現カートリッジの最適設計を予備スクリーニング検査で容易に達成できるという利点を提供する。一例として、このような予備スクリーニングにおいて、目的タンパク質の発現特性(発現レベルまたは生物活性のような定性的特徴)に影響を有する少なくとも1つの因子、すなわち調節因子(例えばプロモーターおよび/またはポリメラーゼ結合部位)および/または目的遺伝子の配列(すなわち異なるコドン使用バリアント)および/または組換えのターゲッティング配列および/または分泌リーダーなどのカートリッジ上の任意の他の因子の異なる一連の直線状発現カートリッジが構築される。このカートリッジバリアントは、事前選択された宿主細胞のゲノムに組み込まれ、得られた発現宿主バリアントは、調節条件下でのタンパク質発現の誘導を含めて培養される。タンパク質発現を比較し、工業生産プロセスを考慮して最も好ましい結果を示す宿主細胞バリアントが選択される。この予備スクリーニングアプローチの変形において、最適の発現カートリッジを決定するかわりに、1群の異なる宿主細胞に異なるカートリッジを組み込むことにより最適の細菌株を同定することができる。この組み込み戦略は、不連続な遺伝子コピー数(例えば1つのみ)をゲノムに組み込むことを可能にする利点を有するので、プラスミドの複製またはプラスミドコピー数の変化により抵触されることなく種々のパラメータの予備スクリーニングを行うことができる。
【0085】
工業規模の製造
本発明において、用語“培養すること”(または“培養”、“発酵”ともいう)は、当業界に公知の方法による制御されたバイオリアクターにおける細菌発現細胞の増殖に関する。
【0086】
組換えタンパク質の生産は、一般的には、より大容量で培養を行うことにより達成される。本発明において、用語"工業(manufacturing)"および"工業規模(manufacturing scale)"は、最小容量5Lの培養液での発酵を定義する。通例、“工業規模”プロセスは、目的組換えタンパク質を含む大容量の調製物を処理し、例えば治療用タンパク質の場合は、臨床試験および市場供給に対する要求を満たす量の目的タンパク質を産生することができることにより定義される。振盪フラスコ培養などの簡単な実験室規模の方法とは対照的に、生産規模の方法は、大容量に加えて、撹拌、通気、栄養素フィード、モニタリングおよびプロセスパラメータ制御(pH、温度、溶存酸素圧、背圧などのための装置を備えるバイオリアクター(発酵槽)の技術システムの使用を特徴とする。実験室規模の方法における発現系の行動からは、バイオリアクターの複雑な環境におけるその系の行動を予測できない。
【0087】
驚くべきことに、本発明の実験において、発現系に基づくゲノム組み込みは、栄養素フィードに基づく培養方式、特に流加プロセスまたは連続プロセスまたは半連続プロセス(例えばケモスタット)と組み合わせた工業規模の方法(容量および技術システムの両方に関して)に特に有利であることが見出された。
【0088】
流加培養
特定の実施形態において、本発明の方法は流加培養プロセスである。
【0089】
バッチプロセスが、発酵中にさらなる栄養素のさらなる供給をせずに、細胞培養に必要なすべての栄養素が初期培地に含まれる培養方式であるのに対し、流加プロセスにおいては、バッチ段階(batch phase)後、フィードにより培養物に1以上の栄養素が供給されるフィード段階(feeding phase)が行われる。栄養素フィードの目的は、バイオマスの量を増加させて(いわゆる“高細胞密度培養プロセス”すなわち“HCDC”)、組換えタンパク質の量もまた増加させることである。大部分の培養プロセスにおいて、フィード方式は重要かつ不可欠であるが、本発明は特定のフィード方式には限定されない。
【0090】
栄養素フィードは、当該技術分野で公知の方法に従って、連続方式または不連続方式で行うことができる。フィード方式はあらかじめ定義された方式であることができ(すなわち実際のプロセスパラメータとは独立してフィードが添加される)、例えば線形定係数方式、線形増加方式、段階的増加方式または数学関数に従った方式、例えば指数関数的フィードであることができる。
【0091】
好ましい実施形態において、本発明の方法は、フィード方式があらかじめ定義された指数関数に従った方式の流加プロセスである。指数関数的フィード方式を適用することにより、細胞集団の比増殖速度μを一定レベルにあらかじめ定義することができ、最大組換えタンパク質発現に関して最適化することができる。フィード速度の制御は、所望の比増殖速度μに基づく。下記のような定義された培地が用いられる場合、与えられる基質単位に基づいて形成されるバイオマスアリコートの推定により増殖を正確に予想し、あらかじめ定義できる。
【0092】
他の好ましい実施形態において、培養の最終段階において、線形定係数フィード方式後に指数関数的フィード方式を続けることができる。
【0093】
流加プロセスの他の実施形態において、線形定係数フィードが適用される。線形定係数フィードは、特定の培養段階を通じて一定の(すなわち不変の)フィード速度(時間単位当たりのフィード培地容量)を特徴とする。
【0094】
流加プロセスの他の実施形態において、線形増加フィードが適用される。線形増加フィードは、線形関数に従ったフィード培地のフィード速度を特徴とする。線形増加関数に従ったフィードは、定義された時間増加量当たりの定義されたフィード速度増加量を特徴とする。
【0095】
本発明の流加プロセスの他の実施形態において、フィードに、フィードバック制御アルゴリズムが適用される(あらかじめ定義されたフィード方式とは対照的に)。フィードバック制御された流加プロセスにおいて、フィード速度は、特定の培養パラメータの実際のレベルに依存する。フィードバック制御されたフィードに適した培養パラメータは、例えばバイオマス(およびそれから誘導される化学的または物理的パラメータ)、溶存酸素、呼吸係数、pHまたは温度である(例えばJahicら(2003年)に記載されているもの)。フィードバック制御されたフィード方式の他の例は、バイオリアクター内の実際のグルコース濃度に基づく(Klemanら、1991年)。
【0096】
連続および半連続培養方式
他の実施形態において、本発明によるゲノムベースの発現カセットを含む細菌細胞は連続方式(ケモスタット)で培養される。連続発酵プロセスは、バイオリアクターへの定義された一定の連続速度の新鮮な培地のフィードを特徴とし、それによって、同じ定義された一定の連続除去速度でバイオリアクターから培養液が同時に除去される。培地、フィード速度および除去速度を同じ一定のレベルに保つことにより、バイオリアクターにおける培養パラメータおよび条件は一定のままである(いわゆる“定常状態”)。比増殖速度μはあらかじめ定義されたものであることができ、もっぱらバイオリアクターにおけるフィード速度および培地容量の結果である。1以上のゲノムベースの発現カセットを有する細胞は遺伝的に大変安定なため(構造不安定なおよび分離不安定なプラスミドベースの発現系またはゲノムに挿入されたカセットがゲノム増幅に依存する発現系とは対照的に)、本発明による細胞の世代数(細胞倍加数)は理論上は制限がなく、同様に、その結果として培養時間にも制限がない。連続方式で遺伝的に安定なゲノムベースの発現系を培養する利点は、遺伝的に不安定な先行技術システムと比較して、より高い、期間あたりの組換えタンパク質の総量を得ることができることである。加えて、理論上無期限の培養により、本発明による細胞の連続培養は、流加培養プロセスと比較してさえ、より高い、期間当たりの総タンパク質量をもたらすことができる。実施例7において、ゲノムベースの発現構築物の高い安定性および生産性を示す。
【0097】
他の好ましい実施形態には、細胞の半連続培養が言及される。本発明において、半連続培養プロセスは、その第1段階で流加プロセスとしてはたらくプロセスである(すなわちバッチ段階とそれに続くフィード段階)。特定の容量またはバイオマスが得られた後(すなわち通例発酵槽容量の上限が得られたとき)、目的組換えタンパク質を含む細胞培養液のかなりの部分がバイオリアクターから取り除かれる。続いて、フィードを再開し、バイオマスまたは培養液の容量が特定の値に達するまで加える。この方法(培養液の排出およびフィードによる再充填)を、少なくとも1回、理論上は無限回繰り返すことができる。
【0098】
培地
この発酵プロセスに用いられる培地の種類に関しては制限はない。培地は、半合成培地、すなわち複合培地化合物(例えば酵母抽出物、大豆ペプトン、カザミノ酸)を含む培地であることもでき、複合化合物を何も含まない化学的に定義された培地であることもできる。
【0099】
好ましくは、“定義された培地”が用いられる。"定義された"培地(“最小”または"合成"培地とも呼ばれる)は、もっぱら化学的に定義された物質、すなわちグルコースまたはグリセロールなどの炭素源、塩、ビタミンならびに、可能性のある株の栄養要求性を考慮した特定のアミノ酸または他の物質、例えばチアミンで構成される。最も好ましくは、炭素源としてグルコースが用いられる。通例、フィード培地の炭素源は、比増殖速度を制御する増殖制限物質として役立つ。
【0100】
本発明の方法において、細菌の増殖は高度であるが、全生産プロセスの間に生理学的に許容される組換え遺伝子の発現速度を維持できるので、かなり高い収量が得られる。
【0101】
誘導方法
前述のように、本発明の最も好ましい実施形態において、目的タンパク質は、“誘導性”または“制御性”プロモーターの制御下にある。
【0102】
タンパク質発現を誘導する方法については制限はない。一例として、1回もしくは複数回ボーラスとして、または連続フィードにより誘導原を添加でき、後者は別名“誘導原フィード(フィーディング)”である。誘導を行う時点に関しては制限はない。培養の開始時点、連続栄養素フィードの開始時点またはフィード開始後(を過ぎて)に誘導原を添加できる。
【0103】
誘導原を培地に含有させるか、それを別にフィードするかのいずれかにより、誘導原フィードを行うことができる。
【0104】
誘導原フィードの利点は、誘導原量を調節できること、すなわち、産生系における一定数の目的遺伝子に対して定義されたまたは一定量の誘導原量を維持できることである。例えば、誘導原フィードは、誘導原とバイオマスの比率が一定になるように、バイオマスに比例した誘導原量を可能にする。誘導原量の基礎となりうるバイオマス単位は、例えば細胞乾燥重量(CDW)、湿細胞重量(WCW)、吸光度、全細胞数(TCN;容量当たりの細胞数)またはコロニー形成単位(容量当たりのCFU)であることができ、あるいはバイオマスに比例したオンラインでモニターされる信号(例えば蛍光、濁度、誘電容量など)であることができる。本質的に、本発明の方法は、信号がオフラインで測定されたかオンラインで測定されたかにかかわらず、バイオマスに比例した任意のパラメータまたは信号当たりの誘導原の正確な量が可能である。目的遺伝子数が定義され、バイオマス単位当たり一定であるため(1細胞当たり1以上の遺伝子)、この誘導方法の結果は、目的遺伝子当たり一定量の誘導原である。さらなる利点として、バイオマス量に対する誘導原の正確で最適な量は、実施例8および図16および図17において明らかにするように、実験的に決定し、最適化することができる。
【0105】
実際のバイオマスレベルを分析方法により測定することは、必ずしも必要ではない。例えば、以前の培養(バイオマスの履歴データ)に基づく誘導原量を添加することで十分な場合がある。他の実施形態において、理論的に算出または予測されたバイオマス1単位当たりの誘導原量を添加することが好ましい場合がある。例えば、フィードベースの培養(流加培養または連続培養など)に関しては、フィード培地における増殖制限物質(通例は炭素源)1単位が特定量のバイオマスをもたらすことは公知である。例として、増殖制限基質としてのグルコース1gは、おおよそ細胞乾燥重量0.33g(基質収率係数YX/S=0.33としても表される)をもたらす。その結果として、目的遺伝子に対する定義された誘導原量は、単位増殖制限物質当たりの定義された量の誘導原によっても達成できる。なぜなら、特定単位の増殖制限物質は定義された単位のバイオマスをもたらし、定義された単位のバイオマスは本発明の方法による目的タンパク質の定義された分子数を含むからである。
【0106】
IPTGの濃度は、CDW 1g当たり0.1〜30μgの範囲であることができ、好ましくは、IPTGの濃度はCDW 1g当たり0.5〜20μgの範囲である。
【0107】
実施例8において、フィード培地における最大誘導原量は、CDW 1g当たりIPTG 20μmolであり、基質収率係数YX/S=0.33と仮定すれば、これはグルコース1g当たりIPTG 6.6μmolと等価である。その結果として、フィード培地のグルコース濃度が128g/Lなので、同じ培地のIPTG濃度は844μMであった。
【0108】
本質的な利点として、制限された量の誘導原のフィードは、代謝負荷を防ぎ、ストレスを低減し、タンパク質合成の能力を最大限にする利益となる。
【0109】
誘導原とバイオマス(または遺伝子または単位増殖制限基質)の比は必ずしも一定である必要はない。線形増加、線形減少、指数関数もしくは他の数学関数に従って増加または減少などであることもできる。本発明による本質的な特徴は、目的遺伝子当たりの誘導原量の値が定義されているということである。
【0110】
特定の実施形態において、本発明の方法は流加プロセスであり、誘導原は培養の開始からバッチ培地に存在している。
【0111】
発現の誘導方法は構成的であることもできる。これは、化学的にまたは他の刺激により誘導が引き起こされるのではなく、培養の開始から誘導は不変であることを意味する。構成的誘導は連続培養にとって好ましい誘導方法であり、流加培養にとっても有用である。
【図面の簡単な説明】
【0112】
【図1】不溶性自己プロテアーゼ融合タンパク質の製造のための、ゲノムにコードされた発現系HMS174(DE3)<Cam:T7:6xHis-NproEddieGFPmut3.1>(ES4)を用いる流加培養を示す図である。
【図2】不溶性自己プロテアーゼ融合タンパク質の製造のための、プラスミドにコードされた発現系HMS174(DE3)(pET30a6xHis-NproEddie-GFPmut3.1)(ES1)を用いる流加培養を示す図である。
【図3】ゲノムにコードされた発現系BL21(DE3)<Cam:T7:6xHis-NproEddieGFPmut3.1>(ES5)を用いる流加培養を示す図である。
【図4】不溶性自己プロテアーゼ融合タンパク質の製造のための、プラスミドにコードされた発現系BL21(DE3)(pET30a6xHis-Npro-GFPmut3.1)(ES2)を用いる流加培養を示す図である。
【図5】可溶性緑色蛍光タンパク質の製造のための、ゲノムにコードされた発現系HMS174(DE3TN7::<Kan:T7GFPmut3.1>(ES6)を用いる流加培養を示す図である。
【図6】可溶性緑色蛍光タンパク質の製造のための、プラスミドにコードされた発現系HMS174(DE3)(pET30a GFPmut3.1)(ES3)を用いる流加培養を示す図である。
【図7】抗生物質耐性マーカーを挿入し、場合により続いてマーカーを除去(切除)することによる選択を示す図である。
【図8】マーカーとして非必須代謝遺伝子または原栄養性補完遺伝子を提供することによる選択を示す図である。
【図9】組換えタンパク質のゲノム部位に発現カートリッジを挿入し、それによってそれらを除去することを示す図である。
【図10】マーカー遺伝子の部位に発現カートリッジを挿入し、マーカー表現型の消失に関して選択することを示す図である。
【図11】非機能性マーカーの欠失部分を完全にして機能性選択マーカーを作成することによる選択を示す図である。
【図12】可溶性hSODの製造のための、プラスミドにコードされた発現系HMS174(DE3)(ES7)を用いる流加培養を示す図である。
【図13】可溶性hSODの製造のための、ゲノムにコードされた発現系HMS174(DE3)(ES8)を用いる流加培養を示す図である。
【図14】可溶性hSODの製造のための、ゲノムにコードされた発現系BL21(DE3)(ES9)を用いる流加培養を示す図である。
【図15】連続方式(ケモスタット)での大腸菌HMS174(DE3)を用いる可溶性GFPmut3.1の発現(ES6)を示す図である。
【図16】可溶性GFPmut3.1の製造のための、制御された誘導原量と組み合わせた、ゲノムにコードされた発現系BL21(DE3)を用いる流加培養を示す図である。
【図17】誘導原の濃度と相関する、ゲノムにコードされた発現系BL21(DE3)を用いる流加培養から得られる可溶性GFPmut3.1の正規化平均産物生成速度(qP)を示す図である。
【実施例1】
【0113】
細菌株の作成および組換えタンパク質の説明
Novagen社により提供された大腸菌recA-K12株HMS174(DE3)またはB株BL21(DE3)のいずれかを用いて実験を行う。記号表示(DE3)は、宿主がlacUV5プロモーターの制御下にあるT7 RNAポリメラーゼ遺伝子の染色体コピーを含むλ DE3プロファージの溶原菌であり、これらの株をT7またはT7lacプロモーターを用いるタンパク質発現に適したものにしたものであることを示している(StudierおよびMoffat、1986年;Studierら、1990年)。細菌染色体への直線状DNAカートリッジの組み込みは、DatsenkoおよびWanner(2000年)に記載されているように、RedヘルパープラスミドpKD46を含む大腸菌MG1655で行われる。直線状発現カートリッジは、大腸菌ゲノムのglmS遺伝子とpstS遺伝子の間にあるattTn7部位に組み込まれる。ゲノム上のカセットのPCRによる確認は、ゲノムにアニーリングする外部プライマーと内部プライマーを組み合わせて行う。P1形質導入(SternbergおよびHoess、1983年;およびLennox、1955年)により、組換え染色体部分が発現宿主HMS174(DE3)およびBL21(DE3)に導入される。レシピエントとしての機能を果たすために、RecAタンパク質をもたらす温度感受性pTSA29-recAヘルパープラスミドを含むrecA欠損株MS174(DE3)が用いられる(Phillips、1998年)。
【0114】
先行技術である標的タンパク質のプラスミドベースの発現を用いる参照実験は、Novagen社製pET30aおよびpET11aプラスミド(pET Systemmanual、第11版)を用いて行う。
【0115】
本発明の発現系の潜在能力を示すために、可溶性タンパク質(緑色蛍光タンパク質GFP;Shimomuraら、1962年))および封入体形成タンパク質(Npro)を実験に用いる。GFPmut3.1は、置換S65GおよびS72Aを有し、488nmで励起され、フォールディングが改善された、FACS用に最適化したバリアントである(Cormackら、1996年)。封入体形成タンパク質6xHis-NproEddie-GFPmut3.1は、上記のGFPmut3.1と、豚コレラウイルス由来の天然のNpro自己プロテアーゼの改変されたバリアントであるNpro Eddie(WO2006/113959に記載されている)との融合体である(Thielら、1991年)。Nproは168アミノ酸(見かけの分子量23000)からなる非構造タンパク質であり、新生ポリタンパク質から翻訳と同時にそれ自身を切断し、それによってカプシドタンパク質Cの正しいN末端が生成される(Wiskerchenら、1991;Starkら、1993年)。
【0116】
表1:作成した発現系のリスト
【表1】
【実施例2】
【0117】
培養方式およびプロセス分析
標準的な制御装置を備えた、7L(正味容量5L、バッチ容量2.5L)または20L(正味容量12L、バッチ容量8L)のコンピュータ制御のバイオリアクター(MBR社;チューリッヒ・ヴェッツィコーン)のいずれかで細胞を増殖させる。25%アンモニア溶液(ACROS Organics社)の添加によりpHを設定値7.0±0.05に維持し、温度を37℃±0.5℃に調節する。酸素制限を避けるために、攪拌機速度および通気速度を制御することにより、溶存酸素濃度を飽和状態の30%以上に安定化させる。吹出空気におけるO2およびCO2含有量を、Hartmann andBraun AdvancedOptimaガス分析装置により測定する。Biomass monitorモデル214M(Aber Instruments社、英国アベリストウィス)セットで誘電容量および伝導率を測定する。産業環境におけるオンライン測定のために特別に設計された多波長蛍光分光光度計であるBioView(登録商標)(DELTA Light&Optics社、デンマーク・リンビー)を用いて蛍光測定を行う。消泡剤懸濁液(Glanapon 2000、Bussetti社、ウイーン)を1ml/フィード培地1lの濃度で添加することにより発泡を抑える。接種のために、強冷凍した(-80℃)ワーキングセルバンクバイアルを解凍し、1ml(吸光度OD600=1)を無菌でバイオリアクターに導入する。培養物が、バッチ培地2.5L中細菌乾燥物質12.5g(またはバッチ培地4L中細菌乾燥物質30g)に増殖して定常期に入ったときフィードを開始する。4倍加時間の間一定の増殖速度0.1h-1を得るために指数関数的基質フィードによる流加法を用いる。基質タンクにおける重量減少の重ねフィードバック制御を用い、対数増殖アルゴリズム、x=xo.eμt、に従ってポンプ速度を増加させることにより基質フィードを制御する(Cserjan-Puschmannら、1999年)。さらなる細菌乾燥物質202g(または450g)を得るために必要な成分をフィード培地は提供した。
【0118】
誘導
バイオリアクターに直接に1回パルスする従来の方式で誘導を行う。完全誘導系を得るために、プロセスの終わりにIPTGの濃度が1μmolになるようにIPTGの供給量を算出する。
【0119】
培地組成
本研究に用いる最小培地は、1リットル当たりKH2PO4 3gおよびK2HPO4*3H2O 6gを含む。これらの濃度により必要な緩衝能が得られ、PおよびK源としても役立つ。産生されるグラム細菌乾燥物質に関連して他の成分が添加される:クエン酸ナトリウム(三ナトリウム塩*2H2O;ACROS organics社)0.25g、MgSO4*7H2O 0.10g、CaCl2*2H2O 0.02g、微量元素溶液50μlおよびグルコース*H2O 3g。発現系ES6、ES7およびES8を用いるすべての実験において、細菌乾燥物質1g当たりCuCl2*2H20 4mgおよびZnSO4*7H20 3.2mgを培地に添加する。集団の初期増殖を促進するために、複合成分である酵母抽出物0.15gを最小培地に加えてバッチ培地を得る。フィード段階に関して、フィード段階において、産生される生物学的乾燥物質量202g(または450g)に従って、1リットル当たりP塩をさらに加え、最小培地2.5Lを調製する。微量元素溶液:5N HCl溶液(g/L):FeSO4*7H2O 40.0、MnSO4*H2O 10.0、AlCl3*6H2O 10.0、CoCl2(Fluka社) 4.0、ZnSO4*7H2O 2.0、Na2MoO2*2H2O 2.0、CuCl2*2H2O 1.0、H3BO3 0.50。
【0120】
オフライン分析
600nmで吸光度(OD)を測定する。細胞浮遊液10mlを遠心分離し、蒸留水に再懸濁した後遠心分離し、再懸濁してあらかじめ計量したビーカーに移し、次いでこれを105℃で24時間乾燥して再計量して細菌乾燥物質を測定する。
【0121】
バイオマスの総量(全細菌乾燥物質BDM;細胞乾燥重量CDWとも呼ぶ)を算出することにより、細菌増殖の進行を測定する。
【0122】
フローサイトメトリーを用いて、全細胞数(TCN)および死細胞(DC)の百分率を測定する。488nmで15mWの出力で標準的なフィルター設定を用いる空冷レーザーを備えたFACSCaliburフローサイトメーター(4カラーシステム;Becton Dickinson社)を用いてすべての測定を行う。絶対細胞数を逆算できるように、既知量の蛍光計数ビーズ(Becton Dickinson社、米国)を試料に加える。
【0123】
ELISAおよびAgilent Bioanalyserを用いる電気泳動によるタンパク質定量の組み合わせにより、組換えタンパク質Npro-Eddie-GFPmut3.1の含有量を測定する。可溶性組換え産物は、Reischerら(2004年)に従ってGFP-ELISAにより定量し、そして、封入体における組換え産物は、Protein 200 LabChip(登録商標)キットを用い、Agilent Bioanalyser 2100で定量する。
【0124】
組換えGFPの含有量は、Reischerら(2004年)に従ってGFP-ELISAにより定量する。hSODの含有量は、Bayerら(1990年)に従ってSOD ELISAにより定量する。
【0125】
プラスミドを含有する細胞ばかりでなく、組み込みカセットを含む細胞もまた、100mg/mlカナマイシンおよび100mg/mlアンピシリンをそれぞれ含むLB寒天平板上で培養し、24時間後にコロニー形成単位(CFU)を計数することにより測定する。
【実施例3】
【0126】
流加培養方式における大腸菌HMS174(DE3)を用いる不溶性封入体としての6xHis-NproEddie-GFPmut3.1の発現
不溶性標的タンパク質の製造(すなわち封入体として)に関する本発明の発現系(ES4)の行動を特徴づけ、プラスミドベースの発現系(ES1)と比較して経済性を評価するために、表1に記載した発現系ES1およびES4を用いる。本発明の発現系を用いる実験により、高いが生理学的に許容される遺伝子発現速度が示される。20時間を超えて産物生成を維持できる。完全誘導系を得るために、フィード開始後1回の倍加での1回のパルスによる誘導を行う(図1)。
【0127】
これらの結果と対照的に、プラスミドベースの発現系により引き起こされる遺伝子発現速度により、細胞の代謝能は酷使され、フィード開始後3回の倍加での1回のIPTGパルスにより行う誘導の4時間後に細胞系は崩壊する(図2)。本発明による発現系ES4は、わずかに高い容量産物収量を示した(表2)。これらの実験からわかるように、本発明の発現系は、産物生成の全期間を通じてプロセス制御および定義された条件を維持できるという利点を提供する。
【0128】
表2:実施例3からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質6xHis-NproEddie-GFPmut3.1およびプラスミドにコードされたタンパク質6xHis-NproEddie-GFPmut3.1の製造の比較)
【表2】
【実施例4】
【0129】
流加培養方式における、大腸菌BL21(DE3)を用いる、不溶性封入体としての6xHis-NproEddie-GFPmut3.1の発現
本発明の発現系の一般適用性を証明するために、発現宿主として大腸菌株BL21(DE3)を用いる。前述の手順に従って、5L規模で表1のES5を培養する。この系の結果(図3;フィード後1回の倍加での1回のパルスにより誘導が行われる)は、K12株HMS174を用いた結果よりもよく、プラスミドベースのES2を用いて得られた結果(図4)と比較してもよい;フィード開始後3回の倍加での1回のパルスにより誘導が行われる)。本発明の系は、プラスミドベースの系よりも有意に高い産物収量およびプロセス安定性を示す(表3)。株BL21のプラスミドベースの系は、株HMS174について記載したものと同じ特性を示し、高すぎる産物生成速度により、系は崩壊し、プロセス制御は失われる。
【0130】
表3:実施例4からの結果の概要
(宿主株BL21(DE3)を用いる、ゲノムにコードされたタンパク質6xHis-NproEddie-GFPmut3.1およびプラスミドにコードされたタンパク質6xHis-NproEddie-GFPmut3.1の製造の比較)
【表3】
【実施例5】
【0131】
流加培養方式における大腸菌HMS174(DE3)を用いる可溶性タンパク質としてのGFPmut3.1の発現
可溶性標的タンパク質の製造のための本発明の発現系の行動を特徴づけるために、表1に記載した発現系ES3およびES6を用いる(表1の他の発現系は、不溶性タンパク質凝集体、すなわち封入体を形成する)。異なる細胞密度により引き起こされる影響を排除するために、フィード開始後1回の倍加での誘導による同じ培養を行うが、他の実験の結果から、プラスミドベースの系は引き起こされ発現速度が高すぎるので、この系では細胞活性を維持できないことが明らかとなった。要求される比較性に対処するために、表4には“プラスミドにコードされた(最適化後)”と呼ぶ第3列目が存在するが、これは、“プラスミドにコードされた発現系”ES2の結果が、フィード開始後1回の倍加での誘導をまねた実験設定に外挿されていることを意味する。図5および図6(フィード開始後3回の倍加での1回のパルスにより誘導が行われる)ならびに表4から導くことができるように、本発明の発現系はより効率的であることが明らかとなった。このことは、特に、組換えタンパク質のより高い比収量および容量収量ならびにバイオマスへの栄養素のより優れた転化率により明らかにされる(すなわちより高いBDM収量または理論値からの下限逸脱率)。
【0132】
表4:実施例5からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質HMS174(DE3)およびプラスミドにコードされたタンパク質HMS174(DE3)の製造の比較)
【表4】
【実施例6】
【0133】
流加培養方式における大腸菌HMS174(DE3)およびBL21(DE3)を用いる可溶性hSODの発現
大腸菌HMS174(DE3)(ES7、ES8)を用いるプラスミドベースおよびゲノムベースの発現ならびに大腸菌BL21(DE3)(ES9)を用いるゲノムベースの発現により組換えヒトスーパーオキシドジスムターゼ(hSOD)を製造する。ヒトSODを3番目のモデルタンパク質として選択する。なぜなら、この酵素は細菌サイトゾル内で高度に相互作用的であることが記載されており、それによって、ゲノムベースの発現を用いる製造方法の頑健性および性能を研究するための適切なタンパク質候補を代表する。ES7を用いる実験(図12)において、製造される生物学的乾燥物質の量は、BDM 225gにあらかじめ定義され、誘導はフィード開始後2.5回の倍加で行われる。発現系ES8(図13)およびES9(図14)を用いる培養において、計画されるBDMはBDM 360gにあらかじめ定義され、誘導はフィード開始後1回の倍加で行われる。この実験は、より低い細胞密度とより低い容量で行うので、ES7とES8の適切な比較を可能にするために、ES7を用いる実験において得られる全収量および容量収量の全収量および容量収量の計算値を用いる。表5における結果は、ES8ベースのゲノムで得られる収量は約3倍高く、製造中での行動はより頑健で安定であることを示している(BDMの計算値とBDM実際の値との比較的小さな差異により示される)。計算過程とはわずかなBDM偏差値があるが、全プロセスを通じて増殖を維持でき、プラスミドにコードされたSODの発現と対照的に、ゲノムにコードされたhSODの発現は細胞の代謝能を超えないことを示している。ゲノムベースのES9を用いる実験により得られる結果(表5第4列目)は、おおよそ20%のさらなる収量増加を示しており、このことは、BL21(DE3)株が、K12株よりもhSODの製造により適していることを意味する。
【0134】
表5:実施例5からの結果の概要
(宿主株HMS174(DE3)を用いる、ゲノムにコードされたタンパク質hSODおよびプラスミドにコードされたタンパク質hSODの製造と、株BL21(DE3)を用いる、ゲノムにコードされたhSODの製造の比較。*を付けた値は、比較性を確かにするための計算値である(プラスミドにコードされた系は低めの細胞密度で運転されるという事実による。)
【表5】
【実施例7】
【0135】
連続(ケモスタット)方式での大腸菌HMS174(DE3)を用いる可溶性GFPmut3.1の発現
ゲノムベース(すなわち無プラスミド)の発現系の遺伝的安定性を確認するために、発現系ES6を用いる連続培養実験(ケモスタット)を行う。希釈速度0.1h-1および1リットル当たりBDM 10gの細胞密度を設定し、実施例2記載の定義された培地を本培養に用いる。この培養の結果を図15に示す。実験の最初の部分で、非誘導状態で細胞を増殖させる。本培養の非誘導状態を、継代培養の行動になんら検出可能な変化なしに42回の倍加を超えて維持する。産生細胞の割合を75〜95%のおよそ一定に保つ((正方形図15)。続いて、フィード300時間近くで、IPTGを用いて培養物を完全に誘導し、誘導後約5時間で、オンラインでの蛍光はこの実験設定で得られる最高レベルに近づく。Reischerら(2004年)によれば、特異的GFP細胞含有量は、BDM 1g当たりGFPおおよそ140〜170mgと計算される。13.5回の倍加を超えて、この誘導状態で細胞を維持する。この期間中のオンラインでの蛍光の推移は、6000〜7000単位のrfu範囲を示す。ゲノムベースの発現宿主の系の安定性は、誘導状態で長期の世代数までも大変高いことをこれらの結果は示している。プラスミドベースの系と対照的に、ゲノム組み込みに基づく系を用いて大変高い発現速度を維持でき、このことは、連続製造工程の開発のための明確な利益を意味する。
【実施例8】
【0136】
あらかじめ定義された誘導物質濃度による改善された誘導制御
組換え遺伝子発現の厳密な誘導制御は、バイオプロセス制御における大変重要な側面である。大変高く、さまざまな遺伝子量(プラスミドコピー数)を有するプラスミドベースのpET/T7系とは異なり、本発明の方法に用いられるゲノムベース(無プラスミド)のT7系は、厳密に、標的遺伝子の定義された遺伝子コピー数(1以上)を特徴とする。この事実により、ゲノムベースの系の発現速度は、より効率的に制御できる。この利益を明らかにするために、異なる誘導レベル(すなわち誘導物質濃度)を適用する、ES10を用いる一連の流加培養を行う。バイオリアクターへのIPTGの単回ボーラスにより誘導を行い、細胞乾燥重量(CDW)に対する誘導物質の最初の比率を設定し、次いでIPTGを連続的にフィードして、定義された濃度を一定のレベルに保つ。CDWに対するIPTGの比率は0.5、0.75、1.0、2.0μmol gCDW-1から、lacリプレッサー分子の完全滴定をもたらす20μmol gCDW-1のレベルまで(すなわち最大級の誘導レベル)である。誘導中の産物の蓄積をモニターし、比産物生成速度(qPF)を:
【数1】
に従って算出するために、GFPの比含量と強く相関する、オフラインで測定される比細胞蛍光(spec.F)(Reischerら、2004年)を用いる。
【0137】
適用した範囲のCDW対IPTG比に応じて、プロセス終了時点で2000〜10000rfu g-1CDWの細胞蛍光レベルがもたらされ、このことは、転写の同調戦略の有効性を印象的に証明している(図16)。完全誘導系のqPF(20μmol IPTG/gCDW)を最高レベルの100%と定め、限定的誘導を用いる実験で得られる平均qPF値は、誘導レベル(誘導物質濃度)と強く相関し、対数トレンドに従う(図17は正規化pPを示す)。低レベルの誘導は、IPTG/CDW比の変化により影響を受けやすく、リアルタイムで推定されるCDWに基づく厳密に制御された誘導物質フィード法を必要とすることをこれらの結果は意味する。さらに、系の完全誘導に必要なIPTG/CDW比は、図17におけるトレンドの外挿により推定でき、3〜5μmol g-1の範囲であり、これは実験に用いられる20μmol g-1よりも実質的に低い。それぞれの適用するIPTG/CDW比は、以下のような一定のバンド幅を有する対応するqPFレベルをもたらす:
【0138】
【表6】
【0139】
結論として、本発明によるゲノムベースの発現に基づく方法は、産物収量および産物生成速度のより優れた制御性を可能にすることをこの実施例は示している。
【0140】
参考文献
【特許請求の範囲】
【請求項1】
a)細菌発現宿主細胞の集団を培養し、
b)目的タンパク質を採取し、
c)それを分離及び精製する
工程を含む、目的組換えタンパク質を工業規模で製造する方法であって、
工程a)において、培養はフィード培地を添加する方式で行われ、前記方式は流加培養方式、半連続方式または連続方式から選択され、かつ、前記発現宿主細胞は、前記タンパク質の発現を可能にするプロモーターの制御下にある、目的タンパク質をコードするDNA配列を有するDNA構築物を、ゲノム中に組み込まれて含む前記方法。
【請求項2】
前記プロモーターが誘導性プロモーターである、請求項1記載の方法。
【請求項3】
前記細菌発現宿主細胞が大腸菌細胞である、請求項1記載の方法。
【請求項4】
前記大腸菌発現宿主細胞がそのゲノムにT7 RNAポリメラーゼ遺伝子を含み、前記プロモーターがT7プロモーターである、請求項2または3記載の方法。
【請求項5】
前記大腸菌発現宿主細胞が、既に生来的にゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞の中に、前記DNA構築物を組み込むことにより得られたものである、請求項4記載の方法。
【請求項6】
前記大腸菌発現宿主細胞が、ゲノム中にT7 RNAポリメラーゼ遺伝子を含まない大腸菌宿主細胞の中に、さらなる因子としてT7 RNAポリメラーゼ遺伝子を含むDNA構築物を組み込むことにより得られたものである、請求項4記載の方法。
【請求項7】
既に生来的にゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞が、BL21(DE3)株、HMS174(DE3)株またはそれらの派生株から選択される、請求項5記載の方法。
【請求項8】
ゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞が非溶原性である、請求項5記載の方法。
【請求項9】
前記誘導性プロモーターが、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーター、trpプロモーター、λプロモーターpL、phoAプロモーターまたはPBADプロモーターから選択される、請求項2記載の方法。
【請求項10】
前記プロモーターが構成的プロモーターである、請求項1記載の方法。
【請求項11】
前記プロモーターがリーキープロモーターである、請求項2記載の方法。
【請求項12】
前記発現宿主細胞が、結合部位に組み込まれた前記DNA構築物を含む、請求項1記載の方法。
【請求項13】
前記結合部位がattTn7部位である、請求項12記載の方法。
【請求項14】
前記発現宿主細胞が、細菌宿主細胞のゲノムに含まれるDNAマーカー配列部位に組み込まれた前記DNA構築物を含む、請求項1記載の方法。
【請求項15】
前記マーカーDNA配列が、抗生物質耐性を与えるタンパク質をコードする、請求項14に記載の方法。
【請求項16】
前記マーカーのマーカーDNA配列が蛍光タンパク質をコードする、請求項14記載の方法。
【請求項17】
前記DNA構築物が、さらなる因子として、マーカータンパク質をコードするマーカー配列を含む、請求項1記載の方法。
【請求項18】
前記マーカータンパク質が細菌宿主細胞の栄養要求変異を補完するタンパク質であり、それによって発現宿主細胞に原栄養性を与える、請求項17記載の方法。
【請求項19】
前記マーカーDNA配列が、抗生物質耐性を与えるタンパク質をコードする、請求項17記載の方法。
【請求項20】
前記マーカーDNA配列が蛍光タンパク質をコードする、請求項17記載の方法。
【請求項21】
あらかじめ定義されたフィード方式による流加培養方式で培養が行われる、請求項1記載の方法。
【請求項22】
あらかじめ定義されたフィード方式が指数関数に従って行われる、請求項21記載の方法。
【請求項23】
フィードバック制御されたフィード方式による流加培養方式で培養が行われる、請求項1記載の方法。
【請求項24】
誘導原の存在により前記目的タンパク質の発現が誘導される、請求項2記載の方法。
【請求項25】
誘導原が連続的に添加される、請求項24記載の方法。
【請求項26】
プロモーターが、T7プロモーター、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーターから選択され、誘導原がIPTGである、請求項24または25記載の方法。
【請求項27】
プロモーターが、T7プロモーター、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーターから選択され、誘導原がラクトースである、請求項24または25記載の方法。
【請求項28】
誘導原とバイオマスの比率が一定になるように、バイオマスに比例した濃度で誘導原が添加される、請求項25記載の方法。
【請求項29】
IPTGの培地中濃度が細胞乾燥重量1g当たり0.1〜30μgの範囲内である、請求項26および28記載の方法。
【請求項30】
前記範囲がCDW 1g当たり0.5〜20μgである、請求項29記載の方法。
【請求項31】
培養の開始時から誘導原がバッチ段階に存在する、請求項24記載の方法。
【請求項32】
細胞質から培地に目的タンパク質が分泌される、請求項23記載の方法。
【請求項33】
目的組換えタンパク質がヘテロ二量体であり、2つのモノマーをコードするDNA分子が同一または異なるDNA構築物中に存在する、請求項1記載の方法。
【請求項1】
a)細菌発現宿主細胞の集団を培養し、
b)目的タンパク質を採取し、
c)それを分離及び精製する
工程を含む、目的組換えタンパク質を工業規模で製造する方法であって、
工程a)において、培養はフィード培地を添加する方式で行われ、前記方式は流加培養方式、半連続方式または連続方式から選択され、かつ、前記発現宿主細胞は、前記タンパク質の発現を可能にするプロモーターの制御下にある、目的タンパク質をコードするDNA配列を有するDNA構築物を、ゲノム中に組み込まれて含む前記方法。
【請求項2】
前記プロモーターが誘導性プロモーターである、請求項1記載の方法。
【請求項3】
前記細菌発現宿主細胞が大腸菌細胞である、請求項1記載の方法。
【請求項4】
前記大腸菌発現宿主細胞がそのゲノムにT7 RNAポリメラーゼ遺伝子を含み、前記プロモーターがT7プロモーターである、請求項2または3記載の方法。
【請求項5】
前記大腸菌発現宿主細胞が、既に生来的にゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞の中に、前記DNA構築物を組み込むことにより得られたものである、請求項4記載の方法。
【請求項6】
前記大腸菌発現宿主細胞が、ゲノム中にT7 RNAポリメラーゼ遺伝子を含まない大腸菌宿主細胞の中に、さらなる因子としてT7 RNAポリメラーゼ遺伝子を含むDNA構築物を組み込むことにより得られたものである、請求項4記載の方法。
【請求項7】
既に生来的にゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞が、BL21(DE3)株、HMS174(DE3)株またはそれらの派生株から選択される、請求項5記載の方法。
【請求項8】
ゲノム中にT7 RNAポリメラーゼ遺伝子を含む大腸菌宿主細胞が非溶原性である、請求項5記載の方法。
【請求項9】
前記誘導性プロモーターが、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーター、trpプロモーター、λプロモーターpL、phoAプロモーターまたはPBADプロモーターから選択される、請求項2記載の方法。
【請求項10】
前記プロモーターが構成的プロモーターである、請求項1記載の方法。
【請求項11】
前記プロモーターがリーキープロモーターである、請求項2記載の方法。
【請求項12】
前記発現宿主細胞が、結合部位に組み込まれた前記DNA構築物を含む、請求項1記載の方法。
【請求項13】
前記結合部位がattTn7部位である、請求項12記載の方法。
【請求項14】
前記発現宿主細胞が、細菌宿主細胞のゲノムに含まれるDNAマーカー配列部位に組み込まれた前記DNA構築物を含む、請求項1記載の方法。
【請求項15】
前記マーカーDNA配列が、抗生物質耐性を与えるタンパク質をコードする、請求項14に記載の方法。
【請求項16】
前記マーカーのマーカーDNA配列が蛍光タンパク質をコードする、請求項14記載の方法。
【請求項17】
前記DNA構築物が、さらなる因子として、マーカータンパク質をコードするマーカー配列を含む、請求項1記載の方法。
【請求項18】
前記マーカータンパク質が細菌宿主細胞の栄養要求変異を補完するタンパク質であり、それによって発現宿主細胞に原栄養性を与える、請求項17記載の方法。
【請求項19】
前記マーカーDNA配列が、抗生物質耐性を与えるタンパク質をコードする、請求項17記載の方法。
【請求項20】
前記マーカーDNA配列が蛍光タンパク質をコードする、請求項17記載の方法。
【請求項21】
あらかじめ定義されたフィード方式による流加培養方式で培養が行われる、請求項1記載の方法。
【請求項22】
あらかじめ定義されたフィード方式が指数関数に従って行われる、請求項21記載の方法。
【請求項23】
フィードバック制御されたフィード方式による流加培養方式で培養が行われる、請求項1記載の方法。
【請求項24】
誘導原の存在により前記目的タンパク質の発現が誘導される、請求項2記載の方法。
【請求項25】
誘導原が連続的に添加される、請求項24記載の方法。
【請求項26】
プロモーターが、T7プロモーター、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーターから選択され、誘導原がIPTGである、請求項24または25記載の方法。
【請求項27】
プロモーターが、T7プロモーター、tacプロモーター、trcプロモーター、lacプロモーター、lacUV5プロモーターから選択され、誘導原がラクトースである、請求項24または25記載の方法。
【請求項28】
誘導原とバイオマスの比率が一定になるように、バイオマスに比例した濃度で誘導原が添加される、請求項25記載の方法。
【請求項29】
IPTGの培地中濃度が細胞乾燥重量1g当たり0.1〜30μgの範囲内である、請求項26および28記載の方法。
【請求項30】
前記範囲がCDW 1g当たり0.5〜20μgである、請求項29記載の方法。
【請求項31】
培養の開始時から誘導原がバッチ段階に存在する、請求項24記載の方法。
【請求項32】
細胞質から培地に目的タンパク質が分泌される、請求項23記載の方法。
【請求項33】
目的組換えタンパク質がヘテロ二量体であり、2つのモノマーをコードするDNA分子が同一または異なるDNA構築物中に存在する、請求項1記載の方法。
【図1】

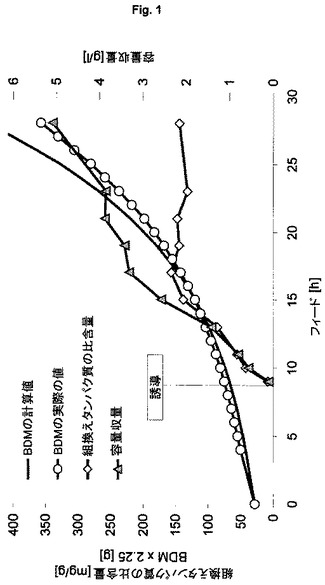
【図2】
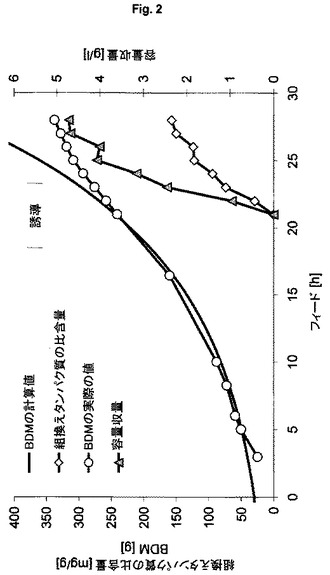
【図3】
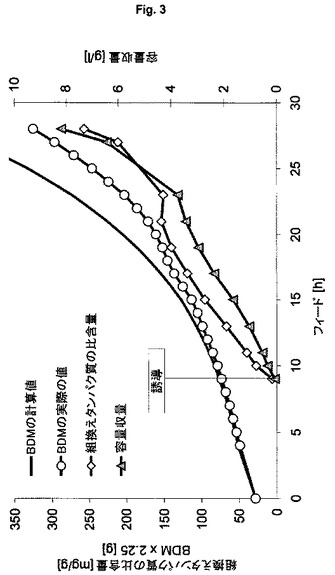
【図4】
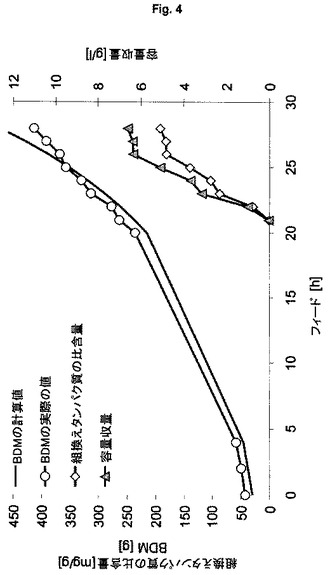
【図5】
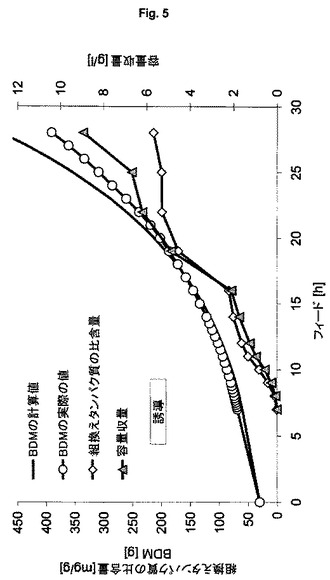
【図6】
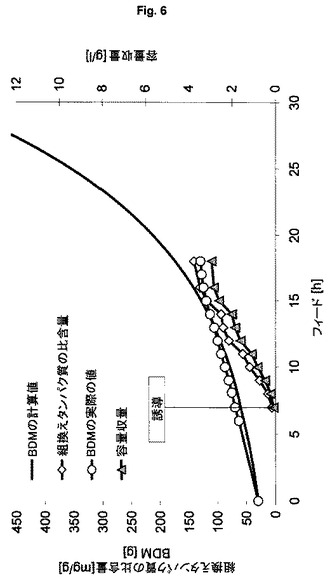
【図7】
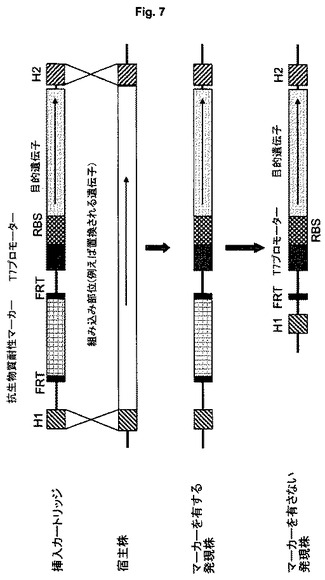
【図8】
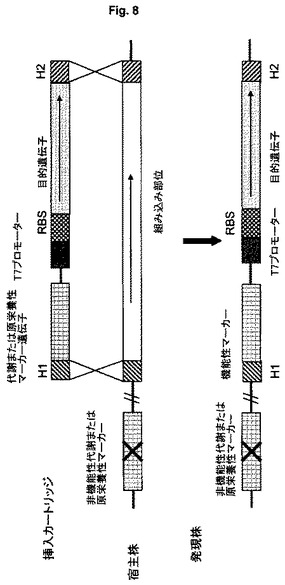
【図9】
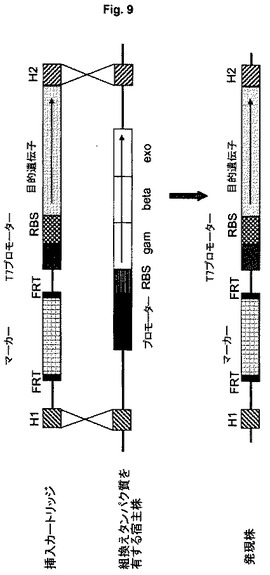
【図10】
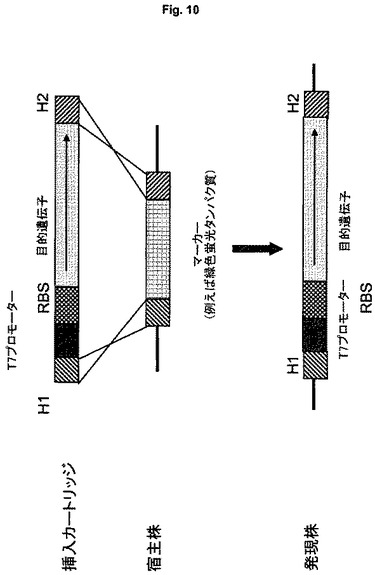
【図11】
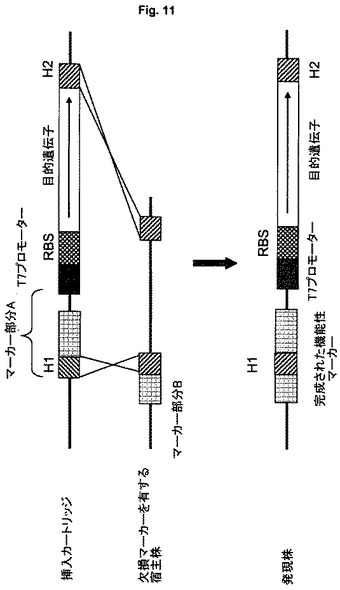
【図12】
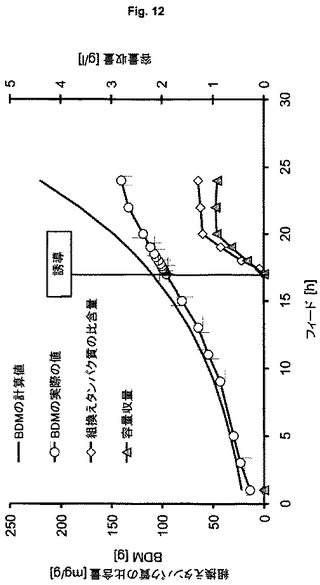
【図13】
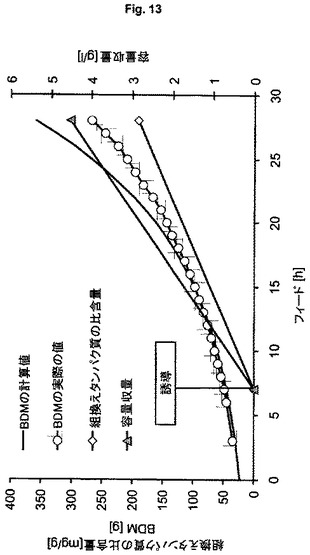
【図14】
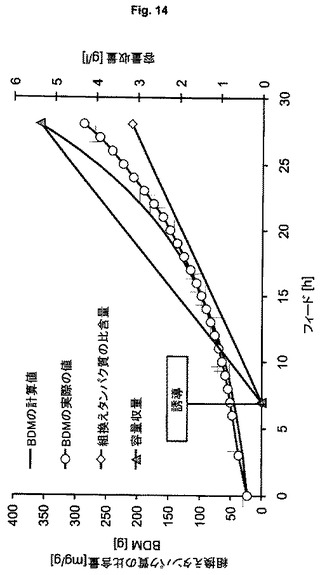
【図15】
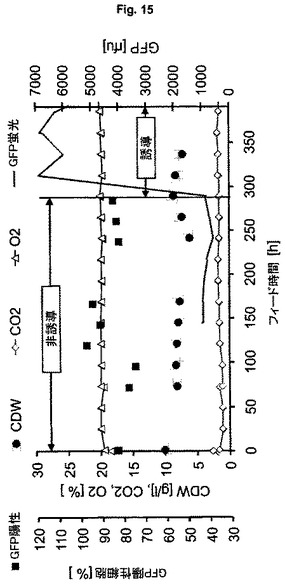
【図16】
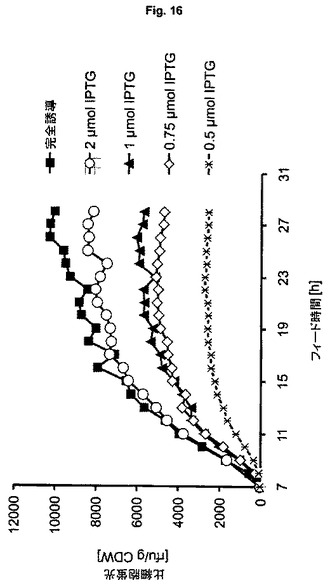
【図17】
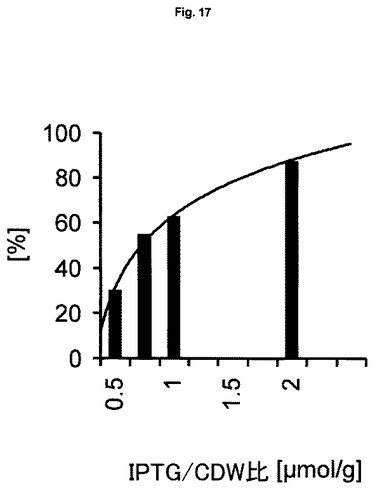
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公表番号】特表2010−527239(P2010−527239A)
【公表日】平成22年8月12日(2010.8.12)
【国際特許分類】
【出願番号】特願2010−507934(P2010−507934)
【出願日】平成20年5月16日(2008.5.16)
【国際出願番号】PCT/EP2008/056062
【国際公開番号】WO2008/142028
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(504135837)ベーリンガー インゲルハイム エルツェーファウ ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (10)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
【公表日】平成22年8月12日(2010.8.12)
【国際特許分類】
【出願日】平成20年5月16日(2008.5.16)
【国際出願番号】PCT/EP2008/056062
【国際公開番号】WO2008/142028
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(504135837)ベーリンガー インゲルハイム エルツェーファウ ゲゼルシャフト ミット ベシュレンクテル ハフツング ウント コンパニー コマンディトゲゼルシャフト (10)
【出願人】(504389991)ノバルティス アーゲー (806)
【Fターム(参考)】
[ Back to top ]