
組換えヒトアルブミン−ヒト顆粒球コロニー刺激因子融合タンパク質の安定な製剤
白血球減少症および好中球減少症などの正常より低い白血球数を特徴とする病気または症状の治療、予防、または改善用の組成物および方法を本明細書に記載する。組成物および方法は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を含む。組換え融合タンパク質を含む医薬製剤、およびこのような製剤を製造する方法も記載する。

【発明の詳細な説明】
【技術分野】
【0001】
本願は、2009年1月16日に出願された米国特許仮出願第61/145,440号および2009年1月16日に出願された米国特許仮出願第61/145,436号の恩典を主張する。米国特許仮出願第61/145,440号および米国特許仮出願第61/145,436号の内容は、その全体において参照により本明細書に組み入れられる。
【背景技術】
【0002】
背景
白血球減少症は、循環する白血球(WBC)の減少であり、多くの場合、WBC数4000/mL未満と規定される。白血球減少症に関与する主な細胞は、好中球である。しかしながら、リンパ球、単球、好酸球、または好塩基球の数の減少も総白血球数の低下に寄与する場合がある(メルクマニュアル17版(非特許文献1))。
【0003】
好中球減少症は、しばしば細菌感染や真菌感染に対する感受性の増加につながる、血中好中球数の減少を特徴とする。好中球減少症は、好中球数および相対感染危険度により次のように分類される。軽度(1000から1500/mL)、中程度(グレード3、500から1000/mL)、または重度(グレード4、<500/mL)。急性かつ重度の好中球減少症は、患者が急速に死に至る感染症に罹患しやすくなるので、生命にかかわる疾患である(メルクマニュアル17版(非特許文献1))。
【0004】
好中球減少症は、骨髄中の好中球産生の低下または好中球破壊が加速することにより引き起こされ得る。急性好中球減少症は、好中球の消費が速く、産生が著しく低下すると数日にわたって起きる場合がある。慢性好中球減少症は、何か月もの間持続する場合があり、しばしば脾臓における好中球産生の減少または好中球の捕捉によって引き起こされる。好中球減少症は、骨髄細胞に対して外因性の因子に副次的に生じるかどうか、あるいは内因性の欠陥が骨髄性前駆細胞中に存在するかどうかによって分類される場合がある(メルクマニュアル17版(非特許文献1))。
【0005】
好中球減少症およびその感染性合併症は、細胞毒性化学療法ならびに、放射線治療、生物療法、および骨髄移植などの他の癌治療の最も一般的で重篤な副作用の一つである。細胞毒性化学療法(成長の速い細胞を探し出し破壊することにより作用する)は、好中球前駆体の増殖速度が速くかつ血中好中球のターンオーバーが速いので、好中球減少症をもたらす(メルクマニュアル17版(非特許文献1))。化学療法を受けている患者における好中球減少症の最も一般的な症状としては、発熱、口腔潰瘍、および耳部感染が挙げられる。重度の好中球減少症の患者は、しばしば、敗血症、皮膚の蜂巣炎、肝膿瘍、フルンケル症、肺炎、口内炎、歯肉炎、直腸周囲炎、大腸炎、副鼻腔炎、および中耳炎などの化膿性感染症を患う。身体がより多くの好中球を産生できるまで、化学療法を遅延させなければならない場合があり、そしてより低用量で投与されなければならない場合があり、治療の効果が低下してしまう。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】メルクマニュアル17版
【発明の概要】
【0007】
概要
本発明は、白血球減少症および好中球減少症などの正常より低い白血球数を特徴とする病気または症状の治療、予防、または改善に有用な組成物および方法に関する。いくつかの態様では、組成物および方法は、図9に示す組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子またはその変種もしくは断片を含む。いくつかの態様では、組成物および方法は、好中球減少症および/または白血球減少症、例えば、薬剤(癌の治療のために投与される化学療法剤など)の投与が原因となる好中球減少症を治療、予防、または改善するために使用され、これは、本発明の組成物を使用して治療され得る。
【0008】
いくつかの態様では、組成物は医薬製剤であり、図9に示す組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子またはその断片もしくは変種を含む。いくつかの態様では、医薬製剤は、少なくとも一種類の薬学的に許容される担体を含み、pHが約5から約8.0、約5から約7.5、約5から約7.2、約5から約7.0、約5から約6.8、約5から約6.6、約5から約6.4、約5から約6.2、約5から約6、約6から約7.5、約6.0から約7.2、約6から約7である。他の態様では、pHは、約4、約4.2、約4.4、約4.5、約4.6、約4.8、約5、約5.2、約5.4、約5.5、約5.6、約5.8、約6.0、約6.2、約6.4、約6.5、約6.6、約6.8、約7.0、約7.2、約7.4、約7.5、約7.6、約7.8、または約8.0である。
【0009】
いくつかの態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、約2.5mg/mlから約240mg/ml、約30mg/mlから約120mg/ml、約60mg/mlから約120mg/ml、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlの濃度で含む。
【0010】
いくつかの態様では、薬学的組成物は、少なくとも一種類の薬学的に許容される塩を含む。いくつかの態様では、塩は、組成物中に約5mMから約50mM、約10mMから約40mM、約15mMから約30mM、約20mMから約25mMの濃度で存在する。いくつかの態様では、塩は、組成物中に約5mM、約10mM、約15mM、約20mM、約25mM、約30mM、約35mM、約40mM、約45mM、および約50mMの濃度で存在する。
【0011】
いくつかの態様では、薬学的組成物は、少なくとも一種類の薬学的に許容される緩衝剤を含む。いくつかの態様では、緩衝剤は、組成物中に約5mMから約50mM、約mM10から約50mM、約15mMから約50mM、約5mMから約10mM、約10mMから約20mM、約20mMから約30mM、約15mMから約25mM、または約20mMの濃度で存在する。いくつかの態様では、緩衝剤は、組成物の中に約5mM、約10mM、約15mM、約20mM、約25mM、約30mM、約35mM、約40mM、約45mM、約50mM、約55mM、または約60mMの濃度で存在する。いくつかの態様では、緩衝剤は、リン酸塩、クエン酸塩、またはそれらの組み合わせである。いくつかの態様では、緩衝剤としては、リン酸ナトリウム、リン酸一ナトリウム、リン酸二ナトリウム、またはそれらの組み合わせが挙げられる。
【0012】
いくつかの態様では、薬学的組成物は、凍結乾燥安定剤を含む。いくつかの態様では、凍結乾燥安定剤は、トレハロース二水和物である。いくつかの態様では、トレハロース二水和物の濃度は、約20mMから約100mM、約40mMから約80mM、約50mMから約70mM、または約60mMである。いくつかの態様では、安定剤の濃度は、約20mM、約30mM、約40mM、約50mM、約60mM、約70mM、約80mM、約90mM、約100mM、約110mM、または約120mMである。
【0013】
いくつかの態様では、薬学的組成物は、増量剤を含む。いくつかの態様では、増量剤は、多価アルコールである。いくつかの態様では、多価アルコールは、マンニトールである。さらなる態様では、薬学的組成物は、薬学的に許容される担体を含む。いくつかの態様では、担体は、ポリソルベートである。
【0014】
本明細書に記載の薬学的組成物は、様々な形態での投与のために製剤化されてもよい。例えば、いくつかの態様では、薬学的組成物は、経口、経肺、静脈内、筋肉内、皮下、直腸、眼(ophthalmic)、結腸、腸管外、大槽内、膣内、腹腔内、眼(ocular)、耳、局所(local)、口腔、経鼻、または局所(topical)投与用に調製される。組成物はまた、特定の剤形用に製剤化されてもよい。例えば、いくつかの態様では、薬学的組成物は、液剤、ゲル、エアロゾル、軟膏、クリーム、凍結乾燥製剤、粉末、ケーキ、錠剤、またはカプセルとして製剤化されてもよい。他の態様では、薬学的組成物は、制御放出製剤、速溶性製剤、遅延放出性製剤、徐放性製剤、拍動放出性製剤、混合即時放出性製剤として製剤化される。いくつかの態様では、薬学的組成物は、液剤として提供される。他の態様では、薬学的組成物は、凍結乾燥粉末として提供される。さらに別の態様では、薬学的組成物は、凍結乾燥ケーキとして提供される。
【0015】
本明細書に記載の薬学的組成物は、様々な様式で保存されてもよい。いくつかの態様では、薬学的組成物は、バイアルに保存され、他の態様では、薬学的組成物は、シリンジに保存される。
【0016】
いくつかの態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、少なくとも一種類の緩衝剤および/またはpH調整剤、ならびに任意で少なくとも一種類のさらなる薬学的に許容される担体を含み、25℃で24時間インキュベーションした後の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の溶液中のモノマー純度の減少は10%未満である。いくつかの態様では、緩衝剤は、pH調整剤と同じである。いくつかの態様では、25℃で24時間インキュベーションした後の溶液中のモノマー純度の減少は、約1%未満、約5%未満、約15%未満、約20%未満、または約25%未満である。
【0017】
他の態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、リン酸ナトリウム約20mM、マンニトール約180mM、トレハロース二水和物約60mM、ポリソルベート80約0.01%(w/v)を含み、pHが約6.0であり、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約2.5mg/mlから約120mg/ml、または約30mg/mlから約60mg/mlである。いくつかの態様では、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlである。
【0018】
さらに別の態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子およびPMTT20/6.0を含み、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約2.5mg/mlから約120mg/ml、または約30mg/mlから約60mg/mlである。いくつかの態様では、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlである。
【0019】
上記の概要および以下の図面の簡単な説明および詳細な説明はいずれも例示および説明のためのものであり、請求項に記載の本発明をさらに説明することを意図するものである。その他の目的、利点、および新規な特徴は、以下の本発明の詳細な説明から当業者には容易に分かると考えられる。
【図面の簡単な説明】
【0020】
【図1】rHSA-G-CSF濃度を上げるとモノマー純度が減少することを示すグラフである。2.5mg/mlから240mg/mlまでの濃度範囲のrHSA-G-CSF試料のモノマー純度は、PMTT10/7.2中で25℃で24時間インキュベーションした後に、サイズ排除高速液体クロマトグラフィー(「SE-HPLC」)により測定した。
【図2】pHを上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が15mg/mlまたは60mg/mlのrHSA-G-CSFの凝集は、pHが6.0、6.8、7.2、または8.0のPMTT10中で25℃にて7日間インキュベーションした後に、SE-HPLCにより測定した。
【図3】温度を上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が15mg/mlまたは60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2中で4℃、25℃、または40℃で24時間インキュベーションした後にSE-HPLCにより測定した。
【図4】pHを上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が48mg/mlのrHSA-G-CSFの凝集は、pHが5.8、6.3、6.4、もしくは7.0のPMTT10中でまたはpHが6.2のCMTT10中で25℃にて最大で3日間までインキュベーションした後に、SE-HPLCにより測定した。一番上の線は、10mM PMTT pH7.0であり、上からの2番目の線は、10mM PMTT pH6.4であり、上からの3番目の線は、10mm PMTT pH6.3であり、一番下の線の暗い方は、10mM PMTT pH5.8であり、一番下の線の明るい方は、10mM CMTT pH6.2である。
【図5】塩濃度を上げるとrHSA-G-CSFの凝集が低下することを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2中で1日間25℃/60%RHおよび5mM、10mM、20mM、または50mMの塩化ナトリウム濃度でインキュベーションした後にSE-HPLCにより測定した。
【図6】リン酸塩濃度を上げるとrHSA-G-CSFの凝集が低下することを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT/7.2中で1日間25℃/60%相対湿度(「RH」)および15mM、20mM、25mM、30mM、40mM、または50mMのリン酸塩濃度でインキュベーションした後にSE-HPLCにより測定した。
【図7】PMTT20/6.0調製用緩衝剤中で最大で120mg/mlまでの濃度でrHSA-G-CSFの純度が維持されることを示すグラフである。2.5mg/mlから120mg/mlまでの濃度範囲のrHSA-G-CSF試料のモノマー純度は、25℃で24時間PMTT10/7.2(従来の緩衝剤)またはPMTT20/6.0(新規な緩衝剤)中でインキュベーションした後に、SE-HPLCにより測定した。
【図8】pHおよびリン酸塩濃度がどのようにrHSA-G-CSFの凝集をもたらすかを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2(従来の緩衝剤)またはPMTT20/6.0(新規な緩衝剤)中で4℃または25℃/60%RHにて最大14日間インキュベーションした後に、SE-HPLCにより測定した。
【図9】図9Aは、Neugranin(商標)(「NEUG」)と命名されたrHSA-G-CSF融合ポリペプチドの核酸およびアミノ酸配列を示し、図9Bは、ヒトG-CSFのアミノ酸配列を示し、図9Cは、ヒト血清アルブミンのアミノ酸配列を示す。
【図10】48mg/mlの濃度および25℃で最大で3日間まで、異なるpHでインキュベーションしたrHSA-G-CSFの、SE-HPLCで測定した凝集を示す表である。
【図11】SE-HPLCにより測定した、異なるpH、タンパク質濃度、および温度でのrHSA-G-CSFの凝集を示す表である。表の上半分(最初の3つの項目)は、15mg/ml rHSA-G-CSFである。表の下半分(最後の3つの項目)は、60mg/ml rHSA-G-CSFである。
【図12】異なるpH、温度、および濃度でインキュベーションした緩衝剤後のrHSA-G-CSFの活性を示す表である。
【図13】NEUGの製造の概略例を示すフローチャートである。
【図14】図14Aおよび図14Bは、NEUG-1(「1P」)およびNEUG-2(「2P」)を比較する、SEC-HPLCおよびRP-HPLCそれぞれの結果を示す。
【図15】図15Aおよび図15Bは、NEUG-1(「1P」)およびNEUG-2(「2P」)それぞれの、IEC-HPLCによる電荷不均一性の比較、ならびにペプチドマッピングによる同一性の比較を示す。
【図16】クーマシーブルーで染色した、SDS-PAGEゲル上でのNEUG-1およびNEUG-2の純度の比較を示す。
【図17】図17Aおよび図17Bは、ヒトにおける臨床での使用のために調製した、NEUG-1の3種類の異なるロットおよびNEUG-2の5種類の異なるロットの試験結果をまとめた表を示す。
【図18】ロット2378-R NEUG-1標準試料のクーマシー染色SDS-PAGE分析(還元)を示す。
【図19】図19Aおよび図19Bは、ロット2378-R NEUG-1標準試料のSE-HPLCおよびRP-HPLCのクロマトグラムを示す。
【図20】NEUG-2の最終製剤の典型的な開発ロットに対して行った分析の結果をまとめたものである。
【図21】過酸化水素とTBOまたはTBPとで処理したNEUGの逆相(「RP」)、イオン交換(「IE」)、およびサイズ排除(「SEC」)クロマトグラフィーのクロマトグラムを示し、試験はNEUGの酸化をモニタリングするために行った。NEUG対照=ミディアムグレー、過酸化水素で処理したNEUG=ライトグレー、TBPで処理したNEUG=黒。
【図22】過酸化水素とTBOまたはTPBとで処理したNEUGの結果を示す表である。試験はNEUGの酸化をモニタリングするために行った。
【図23】治療から14日目までの、化学療法前(サイクル0)にNEUG-1を投与される被験者の好中球絶対数(ANC)のメジアンを示すグラフである。4日目では、最も高い線から最も低い線の順に、300μg/kg NEUG、450μg/kg NEUG、150μg/kg、および50μg/kg NEUGである。
【図24】図24Aおよび図24Bは、化学療法サイクル前と24時間後にNEUG-1を投与された被験者のANCおよび白血球(「WBC」)数を示すグラフである。NEUGの用量は、50μg/kg、150μg/kg、300μg/kg、または450μg/kgであった。グレード3およびグレード4好中球減少症の好中球カットオフは、図24Aにおいて点線で示し、正常な好中球範囲も図24Aおよび図24Bの両方に示す。
【図25】サイクル1中の化学療法の約24時間後にNEUG300μg/kg、NEUG450μg/kg、またはペグフィルグラスチム(Neulasta(登録商標))6mgを投与された被験者の好中球絶対数(ANC)を示すグラフである。
【図26】第I相試験用化学療法サイクルを示す。
【図27】ヒト被験者における第I相試験でのNEUGの薬物動態を示すグラフである。記載の用量(450μg/kg、300μg/kg、または150μg/kg)で皮下投与したNEUGの血清濃度は、化学療法を受けていない乳癌被験者において測定した。正方形:450μg/kgサイクル0;三角形:300μg/kgサイクル0;丸:150μg/kgサイクル0。
【図28】第I相における被験者の好中球絶対数(「ANC」)を示すグラフである。被験者は、試験化学療法に続いて、サイクル1でNEUG300μg/kg(n=19)、NEUG450μg/kg(n=20)、またはペグフィルグラスチム(Neulasta(登録商標))6mg(n=9)を投与された。
【図29】化学療法(第I相試験)のサイクル1におけるNEUGの薬物動態/薬力(「PK/PD」)を示すグラフである。患者は、サイクル1において、ドキソルビシン/ドセタキセル投与の1日後にNEUGを450μg/kg投与された。ANCを白抜きのひし形で示し、NEUG濃度を黒の正方形で示す。グレード3およびグレード4好中球減少症のカットオフを点線で示す。NEUGの定量下限(「LLOQ」)は、6ng/mlにて点線で示す。
【図30】図30Aおよび図30Bは、0日目から15日目の間に得られたANC値に基づく、第1相試験B部において治療した各被験者の曲線下面積(AUC)を示す。図30Aはグラフであり、図23Aからのデータを表(図30B)にまとめている。
【発明を実施するための形態】
【0021】
発明の詳細な説明
本明細書には、白血球数の低下を特徴とする症状および病気を治療、予防、および改善する組成物および方法が開示される。本明細書に記載の組成物および方法は、ヒト血清アルブミンタンパク質(「HSA」)およびヒト顆粒球コロニー刺激因子(「G-CSF」)から形成された融合ポリペプチドを含む。本発明の一態様では、融合ポリペプチドはアミノ酸759個の長さがあり、当該融合物のアミノ酸1〜585は、成熟型HSAからのアミノ酸に対応し、当該融合物のアミノ酸586〜759は、成熟型ヒトG-CSFのアミノ酸に対応する。融合タンパク質のアミノ酸配列を図9Aから図9Cに示す。
【0022】
本明細書に記載の組成物および方法はまた、組換えHSA-G-CSFポリペプチドを含む治療製剤および薬学的組成物を含む。いくつかの態様では、これら製剤および組成物は、当該ポリペプチドをより低用量で投与するために構成されるが、他の態様では、当該製剤は当該ポリペプチドをより高用量で投与するために構成される。
【0023】
本発明はまた、G-CSFの変種または断片を含む融合タンパク質とアルブミンまたはアルブミンの断片もしくは変種とを含む融合タンパク質を包含する。本発明はまた、本発明の治療用アルブミン融合タンパク質をコードするポリヌクレオチド、治療用アルブミン融合タンパク質、組成物、薬学的組成物、製剤、およびキットを包含する。治療用アルブミン融合タンパク質をコードするポリヌクレオチドで形質転換された宿主細胞も本発明に包含され、当該ポリヌクレオチドおよび/または宿主細胞を使用して本発明のアルブミン融合タンパク質を作製する方法もまた包含される。
【0024】
一態様では、本発明によるアルブミン融合タンパク質は、保存期間が長い。
【0025】
第二の態様では、本発明によるアルブミン融合タンパク質は、対応する未融合のG-CSF分子よりも安定している。
【0026】
本発明は、本発明の核酸分子を含むように修飾された(好ましくは本発明のアルブミン融合タンパク質を発現するように修飾された)、遺伝子組換え生物をさらに含む。
【0027】
本発明は概して、アルブミン融合タンパク質をコードするポリヌクレオチド;アルブミン融合タンパク質;ならびにアルブミン融合タンパク質またはアルブミン融合タンパク質をコードするポリヌクレオチドを使用して病気や疾患を治療、予防、または改善する方法に関する。本明細書では、「アルブミン融合タンパク質」とは、少なくとも1個のアルブミン(またはその断片もしくは変種)分子の、少なくとも1個のG-CSF(またはその断片もしくはその変種)分子に対する融合により形成されたタンパク質を指す。本発明のアルブミン融合タンパク質は、遺伝子融合により互いに会合した、少なくともG-CSFの断片または変種と少なくともヒト血清アルブミンの断片または変種とを含む(すなわち、アルブミン融合タンパク質は、G-CSFの全てまたは一部をコードするポリヌクレオチドがインフレームでアルブミンの全てまたは一部をコードするポリヌクレオチドと結合した核酸の翻訳により生じる)。一旦アルブミン融合タンパク質の一部となったG-CSFおよびアルブミンタンパク質は、それぞれ、アルブミン融合タンパク質の「部分(portion)」、「領域」、または「部分(moiety)」と呼ばれる場合がある(例えば、「G-CSFタンパク質部分」または「アルブミンタンパク質部分」)。非常に好ましい態様では、本発明のアルブミン融合タンパク質は、G-CSFの分子またはその断片もしくは変種(成熟型G-CSFタンパク質が挙げられるが、これに限定されるわけではない)を少なくとも1個とアルブミンの分子またはその断片もしくは変種(成熟型アルブミンが挙げられるが、これに限定されるわけではない)を少なくとも1個とを含む。
【0028】
さらに好ましい態様では、本発明のアルブミン融合タンパク質は、宿主細胞によりプロセシングされ、周囲の培地へ分泌される。発現に使用される宿主の分泌経路で発生する新生アルブミン融合タンパク質のプロセシングとしては、シグナルペプチド切断;ジスルフィド結合の形成;適切な折り畳み;糖質の付加およびプロセシング(例えば、N結合およびO結合によるグリコシル化);特異的タンパク質切断;および多量体タンパク質への組み立てが挙げられるが、これらに限定されるわけではない。本発明のアルブミン融合タンパク質は、好ましくはプロセシングされた形態である。最も好ましい態様では、「アルブミン融合タンパク質のプロセシングされた形態」とは、N末端シグナルペプチド切断を経たアルブミン融合タンパク質産物を意味し、本明細書ではまた「成熟アルブミン融合タンパク質」とも呼ばれる。
【0029】
一態様では、本発明は、G-CSFおよび血清アルブミンタンパク質を含むかあるいはそれらからなるアルブミン融合タンパク質をコードするポリヌクレオチドを提供する。さらなる態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質を含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。別の態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質の生物学的活性がありかつ/または治療上活性がある断片を含むかあるいはそれからなるアルブミン融合タンパク質を提供する。別の態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質の生物学的活性がありかつ/または治療上活性がある変種を含むかあるいはそれからなるアルブミン融合タンパク質を提供する。好ましい態様では、アルブミン融合タンパク質の血清アルブミンタンパク質成分は、血清アルブミンの成熟部分である。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0030】
さらなる態様では、本発明は、G-CSFタンパク質と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある断片とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。さらなる態様では、本発明は、G-CSFタンパク質と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある変種とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、G-CSFタンパク質の成熟部分である。さらに好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、G-CSFタンパク質の細胞外可溶性ドメインである。他の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、活性型G-CSFタンパク質である。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0031】
さらなる態様では、本発明は、G-CSFタンパク質の生物学的活性がありかつ/または治療上活性がある断片または変種と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある断片または変種とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。好ましい態様では、本発明は、G-CSFタンパク質の成熟部分および血清アルブミンの成熟部分を含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0032】
I.定義
本明細書において、下記および明細書全体にわたって示すようにいくつかの定義を用いて本発明を記載する。
【0033】
本明細書では、「ポリヌクレオチド」は、少なくとも1個の顆粒球コロニー刺激因子(G-CSF)分子(またはその断片もしくは変種)にインフレームで結合した少なくとも1個のアルブミン分子(またはその断片もしくは変種)を含むかあるいはそれらからなる融合タンパク質をコードするヌクレオチド配列を有する核酸分子を指す。
【0034】
本明細書では、「アルブミン融合コンストラクト」は、少なくとも1個のG-CSF(またはその断片もしくは変種)分子をコードする少なくとも1個のポリヌクレオチドにインフレームで結合した少なくとも1個のアルブミン分子(またはその断片もしくは変種)をコードするポリヌクレオチドを含むかあるいはそれらからなり、さらに例えば一種類以上の次の要素を含む、核酸分子を指す:(1)機能性の自己増殖ベクター(シャトルベクター、発現ベクター、組込み型ベクター、および/または複製系が挙げられるが、これらに限定されるわけではない)、(2)転写開始領域(例えば、調節的または誘導的プロモーター、構成的プロモーターなどのプロモーター領域)、(3)転写の終了領域、(4)リーダー配列、および(5)選択マーカー。一旦アルブミン融合コンストラクトの一部となったG-CSFおよびアルブミンタンパク質をコードするポリヌクレオチドは、それぞれ、アルブミン融合コンストラクトの「部分(portion)」、「領域」、または「部分(moiety)」と呼ばれる場合がある。
【0035】
「治療活性」を示すG-CSFポリペプチドまたは「治療上活性のある」G-CSFタンパク質とは、G-CSFタンパク質に伴う一つ以上の公知の生物学的および/または治療的な活性を有するG-CSFポリペプチドを意味する。非限定的な例として、「G-CSF治療タンパク質」は、病気、症状、もしくは疾患を治療、予防、または改善するのに有用なG-CSFタンパク質である。非限定的な例として、「G-CSF治療タンパク質」は、特定の細胞型(正常(例えばリンパ球)または異常(例えば癌細胞))に特異的に結合するものであってもよく、したがって、化合物(薬剤または細胞毒性薬)が当該細胞型を特異的に標的にするように使用されてもよい。
【0036】
本明細書では、「被験者」なる語は、動物、好ましくは哺乳類、より好ましくはヒトを指す。「被験者」および「患者」なる語は、区別なく使用されてもよい。
【0037】
「薬学的に許容される担体」なる語は、個体に投与されても長期的または永続的な有害作用が実質的に無い任意の担体を指す。薬学的に許容される担体としては、希釈剤、充填剤、塩、分散媒、コーティング剤、乳化剤、湿潤剤、甘味もしくは香味剤、等張化剤、吸収遅延剤、防腐剤、抗菌抗真菌剤、緩衝剤、pH調整剤、酸化防止剤、安定剤、溶解剤、増量剤、凍結保護物質、凝集阻害剤、または任意の種類の製剤助剤が挙げられる。適切な担体は、Remington's Pharmaceutical Sciences (Remington's Pharmaceutical Sciences, 2000, 20th Ed., Lippincott, Williams & Wilkins)に記載されており、これは参照により本明細書に組み入れられる。そのような担体または希釈剤の好ましい例としては、水、塩化ナトリウム、マンニトール、トレハロース無水物、ポリソルベート(ポリソルベート80など)、pHを調整するための様々な薬学的に許容される緩衝剤(例えば、リン酸塩緩衝剤、クエン酸塩緩衝剤、酢酸塩緩衝剤、およびホウ酸塩緩衝剤)が挙げられるが、これらに限定されるわけではない。
【0038】
「薬学的に許容される塩」なる語は、塩酸、リン酸、酢酸、シュウ酸、酒石酸などに由来するものなどのアニオンと形成されたものや、ナトリウム、カリウム、アンモニウム、カルシウム、水酸化鉄、イソプロピルアミン、トリエチルアミン、2-エチルアミノエタノール、ヒスチジン、プロカインなどに由来するものなどのカチオンと形成されたものを含む。
【0039】
「凍結乾燥安定剤」なる語は、凍結乾燥された物質の化学的および/または物理的な不安定さを保護するか減少させる分子を指す。凍結乾燥安定剤の好ましい例としては、スクロース、トレハロース、グルタミン酸一ナトリウム、ヒスチジン、ベタイン、硫酸マグネシウム、グリセリン、エリトリトール、グリセロール、アラビトール、キシリトール、ソルビトール、マンニトールプロピレングリコール、ポリエチレングリコール、プルロニック、およびこれらの組み合わせが挙げられるが、これらに限定されるわけではない。好ましい凍結乾燥安定剤は、トレハロース二水和物やスクロースなどの非還元糖である。
【0040】
「増量剤」なる語は、凍結乾燥混合物に大きさを加え、凍結乾燥ケーキの物理的構造に寄与する化合物を指す。増量剤の例としては、ソルビトール、グリシン、マンニトール、およびポリエチレングリコールが挙げられる。
【0041】
「PMTT20/6.0」なる語は、リン酸一ナトリウム(2.42mg/mL、17.4mM)、リン酸二ナトリウム(0.35mg/mL、2.5mM)、マンニトール(32.79mg/mL、180mM)、トレハロース無水物(22.70mg/mL、60mM)、ポリソルベート80(0.1mg/mL、0.01%)を含む組成物を指し、組成物の最終pHが6.0である。これはまた「新規な緩衝剤」、「新規なPMTT」、または「新規な調製用緩衝剤」と呼ばれ、20mMリン酸塩、180mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80を含み、pHが6.0であるとも記載される。
【0042】
「PMTT10/7.2」なる語は、10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80を含む組成物を指し、組成物の最終pHが7.2である。これはまた、「従来の緩衝剤」、「従来のPMTT」、または「従来の調製用緩衝剤」とも呼ばれる。
【0043】
「PMTT」なる語は、リン酸塩、マンニトール、トレハロース無水物、およびポリソルベートを含む組成物を指す。
【0044】
「CMTT10」なる語は、10mMクエン酸ナトリウム、190mMマンニトール、60mMトレハロース無水物、0.01%(W/V)ポリソルベート80を含む組成物(pHが6.2の緩衝剤)を指す。
【0045】
II.顆粒球コロニー刺激因子
顆粒球コロニー刺激因子(G-CSF)は、好中球の産生を促す造血成長因子である。G-CSFの投与は、好中球の増加を迅速に誘導する。G-CSFの他の重要なインビボ活性は、末梢血中への造血前駆細胞の動員である(Duhrsen et al, 1988; Molineux et al, 1999; Roberts et al, 1994)。この効果は、好中球系統だけを含むものではなく、他の単一系統および多系統前駆細胞ならびに造血多能性幹細胞にまで及ぶ(Molineux et al, 1999)。G-CSFはまた、好中球をプライミングすることによって感染に対する防衛機構の一部である細胞事象を高め、これによって、オプソニン化黄色ブドウ球菌に対する食作用および抗菌作用の両方が高まる。G-CSFはまた、好中球および単球の走化性および好中球の接着性を誘導する(Yuo et al, 1989; Wang et al, 1988)。
【0046】
組換えG-CSF産物は現在、好中球の増殖および分化を促すために多数の臨床上の適応症に関して承認されている。臨床試験では、フィルグラスチム(組換えメチオニルヒトG-CSF;ニューポジェン(登録商標)、アムジェン社、サウサンドオークス、カリフォルニア州)は、末梢好中球の数を増やし、このため骨髄抑制性の化学療法後の好中球減少症の期間を短縮させた。フィルグラスチムは、毎日の皮下(SC)注射によって投与される。ポリエチレングリコール共役rG-CSF(Neulasta(登録商標))であるペグフィルグラスチムは、毎日のrG-CSF投与に代わるサイクル毎に一回投与される代替物として、骨髄抑制性の抗癌剤が投与される患者における発熱性好中球減少症の発生率を減少させるのに安全かつ効果的であることが証明されている(Holmes, O'Shaughnessy et al, 2002; Green et al, 2003;Neulasta(登録商標)SmPC 2007)。
【0047】
III.ヒト血清アルブミン
ヒト血清アルブミン(HSA)は、ヒト循環系で最も一般的な天然の血液タンパク質であり、1リットル当たり約40グラムのアルブミンが測定され、20日以上の間循環系に存続する。HSAは、酵素的または免疫的な機能を有さず、生体内に広く分布し、血液中で治療物質の担体となることが知られている。アルブミンは、生理的濃度で最小限の活性を有する担体タンパク質である。HSAおよび組換えヒトアルブミン(rHSA)はいずれも、ヒトにおいて同様の長期の血中半減期を有する。ヒトアルブミンに遺伝子的に融合した治療用タンパク質は、アルブミンの血中半減期特性を獲得し得ることが研究で示されている(Syed et al, 1997)。例えば、ウサギでの研究では、アルブミンに融合したCD4の半減期が、未融合のCD4よりも140倍長いことが示されている(Yeh et al, 1992)。
【0048】
成熟型では585個のアミノ酸からなるタンパク質であるヒト血清アルブミン(米国特許第7,592,010号の図1に示される)は、血清の浸透圧の大部分を担っており、また内因性および外因性リガンドの担体として機能する。現在、臨床で使用されるHSAは、ヒトの血液から抽出されて製造される。微生物中の組換えHSA(rHSA)の産生は、EP 330 451およびEP 361 991に開示されている。
【0049】
IV.G-CSFを用いた治療
顆粒球コロニー刺激因子(G-CSF)を用いた一次予防は、年齢、病歴、病気の特徴、および化学療法レジメンの骨髄毒性に基づくリスクが高い患者における発熱性好中球減少症の防止に推奨される。米国臨床腫瘍学会(ASCO)および欧州癌研究治療機関(EORTC)は、発熱性好中球減少症のリスクが約20%ある場合、G-CSFの利用を推奨している。発熱性好中球減少症のリスクが10%から20%である場合、米国総合癌センターネットワーク(NCCN)は、G-CSFによる予防を任意で適用することを推奨し、発熱性好中球減少症のリスクが少なくとも20%である場合、G-CSFによる予防を確実に適用することを推奨している(Smith et al, 2006, Vogel et al 2005, Timmer-Bonte et al 2006, NCCN Guidelines)。
【0050】
コロニー刺激因子(CSF)を用いた予防が、特定の化学療法レジメンの毒性を緩和するために推奨される。しかしながら、当該治療のコストが増加することは、米国においてそして特にEUの一部において重要な検討事項であり、予防的なG-CSF治療の利用が不十分になる可能性があり、また高用量化学療法レジメンに対する患者の適格性が制限される可能性がある(Timmer-Bonte et al, 2006; Adams et al, 2006, NCCN Guidelines)。
【0051】
V.ポリペプチドならびにポリヌクレオチドの断片および変種
A.断片
本発明はさらに、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片に関する。本発明はまた、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片をコードするポリヌクレオチドに関する。タンパク質のN末端から1個以上のアミノ酸が欠失して、G-CSFタンパク質、アルブミンタンパク質、および/または本発明のアルブミン融合タンパク質の1つ以上の生物学的機能が修飾または喪失しても、他の治療的活性および/または機能的活性(例えば、生物学的活性、多量体化能、リガンド結合能)はなお保持され得る。例えば、完全型または成熟型のポリペプチドを認識する抗体を誘導しかつ/またはそれに結合するN末端欠失を有するポリペプチドの能力は、N末端から除去される完全型ポリペプチドの残基が過半数未満であれば、一般に保持される。完全型のポリペプチドのN末端が欠失した特定のポリペプチドが、そのような免疫学的活性を保持するかどうかは、本明細書に記載されるかまたは当技術分野で公知である常法によって容易に決定することができる。N末端アミノ酸残基が多数欠失した変異タンパク質が、一定の生物学的または免疫原的活性を保持している可能性がある。事実、わずか6個のアミノ酸残基からなるペプチドがしばしば免疫反応を誘発する場合がある。
【0052】
したがって、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質の断片は、完全長のタンパク質、ならびに参照ポリペプチド(reference polypeptide)のアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチド(つまり、G-CSFタンパク質、あるいはポリヌクレオチドまたはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質のG-CSFタンパク質部分)を含む。特に、N末端欠失は、一般式m〜qで記すことができ、式中、qは、参照ポリペプチド(例えば、G-CSFタンパク質、または本発明のアルブミン融合タンパク質のG-CSFタンパク質部分)中のアミノ酸残基の総数を表す整数であり、mは、2〜qマイナス6までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0053】
さらに、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応する血清アルブミンポリペプチドの断片は、完全長のタンパク質、ならびに参照ポリペプチドのアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチド(つまり、血清アルブミン、またはアルブミン融合タンパク質の血清アルブミン部分)を含む。好ましい態様では、N末端欠失は、一般式m〜585により記すことができ、式中、585は、成熟ヒト血清アルブミン中のアミノ酸残基の総数を表す整数であり、mは、2から579までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。さらなる態様では、N末端の欠失は、一般式m〜609により記すことができ、式中609は、完全長のヒト血清アルブミン中のアミノ酸残基の総数を表す整数であり、mは、2から603までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0054】
さらに、本発明のアルブミン融合タンパク質の断片は、完全長のアルブミン融合タンパク質、ならびにアルブミン融合タンパク質のアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチドを含む。特に、N末端欠失は、一般式m〜qにより記すことができ、式中、qは、アルブミン融合タンパク質中のアミノ酸残基の総数を表す整数であり、mは、2〜qマイナス6までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0055】
また、上記のように、参照ポリペプチド(例えば、G-CSFタンパク質;血清アルブミンタンパク質;または本発明のアルブミン融合タンパク質)のN末端またはC末端から1個以上のアミノ酸が欠失して、当該タンパク質の1つ以上の生物学的機能が修飾または喪失しても、他の機能的活性(例えば、生物学的活性、多量体化能、リガンド結合能)および/または治療的活性はなお保持され得る。例えば、完全型または成熟型のポリペプチドを認識する抗体を誘導しかつ/またはそれに結合するC末端欠失を有するポリペプチドの能力は、C末端から除去される完全型または成熟型のポリペプチドの残基が過半数未満であれば、一般に保持される。参照ポリペプチドのN末端および/またはC末端が欠失した特定のポリペプチドが治療的活性を保持するかどうかは、本明細書に記載および/またはそうでなければ当技術分野で公知である常法によって容易に決定することができる。
【0056】
本発明はさらに、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質のアミノ酸配列のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6〜qマイナス1までの範囲の任意の整数であり、qは、参照ポリペプチド(例えば、G-CSFタンパク質、あるいはポリヌクレオチドまたはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質のG-CSFタンパク質部分)中のアミノ酸残基の総数を表す整数である。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0057】
さらに、本発明は、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質のアミノ酸配列のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6から584までの範囲の任意の整数であり、584は成熟型ヒト血清アルブミン中のアミノ酸残基の総数を表す整数から1を引いたものである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6から608までの範囲の任意の整数であり、608は血清アルブミン中のアミノ酸残基の総数を表す整数から1を引いたものである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0058】
さらに、本発明は、本発明のアルブミン融合タンパク質のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6〜qマイナス1までの範囲の任意の整数であり、qは本発明のアルブミン融合タンパク質中のアミノ酸残基の総数を表す整数である。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0059】
さらに、上記NまたはC末端欠失のうちの任意のものを、NおよびC末端欠失参照ポリペプチドを製造するために組み合わせてもよい。本発明はまた、参照ポリペプチド(例えば、G-CSFタンパク質、または本発明のアルブミン融合タンパク質のG-CSFタンパク質部分、または血清アルブミン、または本発明のアルブミン融合タンパク質のアルブミンタンパク質部分、またはアルブミン融合タンパク質、または本発明のポリヌクレオチドもしくはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質)の残基mからn(nおよびmは、上に記載の整数である)を有すると一般に記載される場合がある、アミノ末端およびカルボキシル末端の両方から1個以上のアミノ酸が欠失したポリペプチドを提供する。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0060】
本願はまた、本明細書に記載の参照G-CSFポリペプチドまたは参照アルブミンポリペプチドあるいはそれらの断片と少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるポリペプチドを含むタンパク質に関する。好ましい態様では、本願は、上記のNおよびC末端欠失のアミノ酸配列を有する参照ポリペプチドと少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるポリペプチドを含むタンパク質に関する。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0061】
好ましい本発明のポリペプチド断片は、G-CSFタンパク質または血清アルブミンタンパク質のポリペプチド配列の治療的活性および/または機能的活性(例えば、生物学的活性)を示すアミノ酸配列を含むかあるいはそれからなる断片であり、当該タンパク質のアミノ酸配列が断片である。
【0062】
他の好ましいポリペプチド断片は、生物学的に活性のある断片である。生物学的に活性のある断片は、本発明のポリペプチドの活性に類似しているが必ずしも同一ではない活性を示すものである。当該断片の生物学的活性には、所望の活性の向上、または望ましくない活性の減少が含まれてもよい。
【0063】
B.変種
「変種」は、参照核酸またはポリペプチドとは異なるがそれらの本質的な特性を保持するポリヌクレオチドまたは核酸を指す。一般的に、変種は、全体として密接に類似し、多くの領域で参照核酸またはポリペプチドと同一である。
【0064】
本明細書では、「変種」は、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分、本発明のアルブミン融合タンパク質のアルブミン部分、または本発明のアルブミン融合タンパク質を指し、これらは、G-CSFタンパク質、アルブミンタンパク質、および/またはアルブミン融合タンパク質それぞれと配列が異なるが、本明細書の他の部分で記載するかまたはそうでなければ当技術分野で公知であるその少なくとも1つの機能的および/または治療的性質を保持する。一般に、変種は、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質、アルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質、および/またはアルブミン融合タンパク質のアミノ酸配列に全体として非常に類似し、多くの領域で同一である。当該変種をコードする核酸も、本発明に包含される。
【0065】
本発明はまた、例えば、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質、および/またはアルブミン融合タンパク質のアミノ酸配列に対して少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、または約100%同一であるアミノ酸配列を含むかあるいはそれからなるタンパク質に関する。これらのポリペプチドの断片も提供される。本発明により包含されるさらなるポリペプチドは、ストリンジェントなハイブリダイゼーション条件下(例えば、6xの塩化ナトリウム/クエン酸ナトリウム(SSC)中で約45℃にて、フィルターに結合したDNAにハイブリダイゼーションさせ、次いで0.2xのSSC、0.1%SDS中で約50から65℃にて1回以上洗浄する)、非常にストリンジェントな条件下(例えば、6xの塩化ナトリウム/クエン酸ナトリウム(SSC)中で約45℃にて、フィルターに結合したDNAにハイブリダイゼーションさせ、次いで0.1xのSSC、0.2%SDS中で約68℃にて1回以上洗浄する)、または当業者に公知の他のストリンジェントなハイブリダイゼーション条件下(例えば、Ausubel, F. M. et al., eds., 1989 Current protocol in Molecular Biology, Green publishing associates, Inc.およびJohn Wiley & Sons Inc., New York, 6.3.1-6.3.6および2.10.3頁参照)で本発明のアルブミン融合タンパク質をコードする核酸分子の補体にハイブリダイズするポリヌクレオチドによりコードされるポリペプチドである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0066】
問い合わせアミノ酸配列に少なくとも、例えば、95%「同一の」アミノ酸配列を有するポリペプチドにより、対象ポリペプチド配列が、問い合わせアミノ酸配列のアミノ酸100個当たりアミノ酸改変を最大で5個まで含み得るということを除いて、対象ポリペプチドのアミノ酸配列が問い合わせ配列と同一であることが意図される。言いかえれば、問い合わせアミノ酸配列に対して少なくとも95%同一のアミノ酸配列を有するポリペプチドを得るため、対象配列のアミノ酸残基の最大5%までが、他のアミノ酸で、挿入、欠失、置換されてもよい。参照配列の当該改変は、参照アミノ酸配列のアミノもしくはカルボキシ端末位置、またはこれらの端末位置の間の任意の場所において、参照配列における残基が個々にまたは参照配列内の1つ以上の隣接する群で点在して起こってもよい。
【0067】
実際問題としては、任意の特定のポリペプチドが、本発明のアルブミン融合タンパク質のアミノ酸配列またはその断片(例えば、アルブミン融合タンパク質のG-CSFタンパク質部分またはアルブミン融合タンパク質のアルブミン部分)に対して少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるかどうかは、公知のコンピュータプログラムを用いて従来通りに決定することができる。グローバル配列アラインメントとも呼ばれる、問い合わせ配列(本発明の配列)と対象配列との間の最大の全体の整合性を決定する好ましい方法は、Brutlagらのアルゴリズムに基づいたFASTDBコンピュータプログラムを用いて決定することができる(Comp. App. Biosci. 6:237-245 (1990))。配列アラインメントにおいて、問い合わせ配列および対象配列は両方がヌクレオチド配列であるかまたは両方がアミノ酸配列のいずれかである。グローバル配列アラインメントの結果では、同一性はパーセントで表される。FASTDBアミノ酸アライメントにおいて使用される好ましいパラメータは次のとおりである。Matrix=PAM 0, k-tuple=2, Mismatch Penalty=1, Joining Penalty=20, Randomization Group Length=0, Cutoff Score=1, Window Size=sequence length, Gap Penalty=5, Gap Size Penalty=0.05, Window Size=500または対象アミノ酸配列の長さのどちらか短い方。
【0068】
対象配列が、内部欠失のせいではなくNまたはC末端欠失により問い合わせ配列より短い場合、結果をマニュアル補正しないといけない。これは、全体的な同一性をパーセントで計算する際に、FASTDBプログラムは対象配列のNおよびC末端切断を考慮しないからである。NおよびC末端で切断された対象配列について、問い合わせ配列に対して、同一性のパーセントは、対象配列のNおよびC末端側であり、対応する対象残基と一致/アライメントしない、問い合わせ配列の残基の数を、問い合わせ配列の塩基全体のパーセントとして計算することにより補正される。残基が一致/アライメントするかどうかはFASTDB配列アラインメントの結果により決定される。次にこのパーセントは、特定のパラメータを用いた上記のFASTDBプログラムにより計算された同一性のパーセントから減じられ、最終的なパーセント同一性スコアに達する。この最終的なパーセント同一性スコアは、本発明の目的に使用されるものである。問い合わせ配列と一致/アライメントしない対象配列のNおよびC末端側に対する残基のみ、パーセント同一性スコアをマニュアル補正する目的のために考慮される。すなわち、対象配列の最も遠いNおよびC末端残基の外側の問い合わせ残基位置のみである。
【0069】
例えば、90個のアミノ酸残基を有する対象配列は、同一性のパーセントを決定するために100個の残基を有する問い合わせ配列とアライメントされる。対象配列のN末端にて欠失が起こり、したがって、FASTDBアライメントは、N末端で最初の10残基の一致/アライメントを示さない。10個の不対残基は、配列の10%に相当し(一致しないNおよびC末端における残基数/問い合わせ配列中の残基の総数)、したがって、FASTDBプログラムにより計算されたパーセント同一性スコアから10%減じられる。残りの90残基が完全に一致する場合は、最終的なパーセント同一性は90%になる。別の例では、90残基の対象配列は、100残基の問い合わせ配列と比較される。このとき、欠失は内部欠失であり、したがって、対象配列のNまたはC末端には、問い合わせ配列と一致/アライメントしない残基はない。この場合、FASTDBにより計算されたパーセント同一性は、マニュアル補正されない。再び、FASTDBアライメントで示されるように、問い合わせ配列と一致/アライメントしない対象配列のNおよびC末端の外側の残基位置のみ、マニュアル補正される。本発明の目的のためには、他のマニュアル補正は行われない。
【0070】
変種は、変種と同じ長さである正常HAまたはG-CSFタンパク質の長さと少なくとも約75%(他の態様では、少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%)の配列同一性を通常有する。ヌクレオチドまたはアミノ酸配列レベルでの相同性または同一性は、配列類似性検索のために作られた、blastp、blastn、blastx、tblastn、およびtblastxプログラムにより利用されるアルゴリズムを用いて、BLAST(Basic Local Alignment Search Tool)分析により決定される(Karlin et al, Proc. Natl. Acad. Sci. USA 87: 2264-2268 (1990)およびAltschul, J. MoI. Evol. 36: 290-300 (1993)、全体として参照により組み入れられる)。
【0071】
BLASTプログラムにより使用される手法は、まず問い合わせ配列とデータベース配列との間の類似のセグメントを検討し、次に、同定された一致すべての統計的有意性を評価し、最後に、所定の重要性の閾値を満たす一致のみをまとめる。配列データベースの類似性検索に関する基本的な事柄の考察については、Altschulら(Nature Genetics 6: 119-129 (1994))参照。本書は全体として参照により組み入れられる。histogram、descriptions、alignments、expect(すなわち、データベース配列に対する一致を報告するための統計的有意性の閾値)、cutoff、matrix、およびfilterに対する探索パラメータは初期値である。blastp、blastx、tblastn、およびtblastxにより用いられるデフォルトスコアリングマトリクスは、BLOSUM62マトリクスである(Henikoff et al., Proc. Natl. Acad. Sci. USA 89: 10915-10919 (1992)、この文献は、全体として参照により組み入れられる)。blastnについては、スコアリングマトリクスは、N(すなわち、ミスマッチ残基のペナルティスコア)に対するM(すなわち、一対の一致する残基のリワードスコア)の比であり、ここで、MおよびNの初期値は、それぞれ5および-4である。4つのblastnパラメータは、以下のように調整されてもよい。Q-10(ギャップ生成ペナルティー);R=1O(ギャップ伸張ペナルティー);wink=1(問い合わせに沿ってwinkth位置でワードヒットを生じる);およびgapw=16(これは分離したアラインメントが生成されるウィンドウ幅を設定する)。等価のBlastpパラメータ設定は、Q=9;R=2;wink=1;およびgapw=32である。GCGパッケージバージョン10.0で利用可能である、配列間のBestfit比較は、DNAパラメータGAP=50(ギャップ形成ペナルティー)およびLEN=3(ギャップ延長ペナルティー)を用い、タンパク質の比較における等価な設定は、GAP=8およびLEN=2である。
【0072】
本発明のポリヌクレオチド変種は、コード領域、非コード領域、または両方において改変を含んでもよい。特に好ましいものは、サイレント置換、付加、または欠失を生ずるが、コードされたポリペプチドの特性や活性を改変しない改変を含むポリヌクレオチド変種である。遺伝コードの縮重によるサイレント置換により産生したヌクレオチド変種が好ましい。さらに、50個未満、40個未満、30個未満、20個未満、10個未満、または5〜50個、5〜25個、5〜10個、1〜5個、または1〜2個のアミノ酸が任意の組み合わせで置換、欠失、あるいは付加したポリペプチド変種も好ましい。ポリヌクレオチド変種は、例えば、特定の宿主に対してコドン発現を最適化するため(酵母または大腸菌などの細菌宿主に好まれるコドンにヒトmRNA中のコドンを変化させるため)などの種々の理由で産生され得る。
【0073】
好ましい態様では、アルブミン融合タンパク質のアルブミン部分をコードする本発明のポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。さらなる好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードする本発明のポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。またさらに好ましい態様では、本発明のアルブミン融合タンパク質をコードするポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。
【0074】
別の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではG-CSFタンパク質をコードする野生型ポリヌクレオチドにはハイブリダイズしない。さらなる態様では、アルブミン融合タンパク質のアルブミン部分をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではアルブミンタンパク質をコードする野生型ポリヌクレオチドにはハイブリダイズしない。他の態様では、アルブミン融合タンパク質をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではG-CSFタンパク質部分またはアルブミンタンパク質部分をコードする野生型ポリヌクレオチドにはハイブリダイズしない。
【0075】
さらなる態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードするポリヌクレオチドは、そのG-CSFタンパク質の天然に存在する配列を含まないかあるいはそれから成り立っていない。さらなる態様では、アルブミン融合タンパク質のアルブミンタンパク質部分をコードするポリヌクレオチドは、アルブミンタンパク質の天然に存在する配列を含まないかあるいはそれから成り立っていない。別の態様では、アルブミン融合タンパク質をコードするポリヌクレオチドは、G-CSFタンパク質部分またはアルブミンタンパク質部分の天然に存在する配列を含まないかあるいはそれらから成り立っていない。
【0076】
本発明のポリペプチドの特徴を向上または改変させるために、タンパク質工学および組換えDNA技術の公知の方法を使用して、変種を発生させてもよい。例えば、1個以上のアミノ酸を、生体機能の本質的な喪失を伴わないで本発明のポリペプチドのN末端またはC末端から欠失させることができる。
【0077】
好ましい態様では、本発明の変種は同類置換を有してもよい。「同類置換」とは、脂肪族または疎水性アミノ酸Ala、VaI、Leu、およびIleの置換;水酸基残基SerおよびTarの置換;酸性残基AspおよびGluの置換;アミド残基AsnおよびGlnの置換、塩基性残基Lys、Arg、およびHisの置換;芳香族残基Phe、Tyr、およびTrpの置換、ならびに低分子アミノ酸Ala、Ser、Thr、Met、およびGlyの置換などのグループ内での交換を指す。
【0078】
表現型的にサイレントなアミノ酸置換を行う方法に関する指針は、例えば、Bowie et al., "Deciphering the Message in Protein Sequences: Tolerance to Amino Acid Substitutions," Science 247:1306-1310 (1990)に提供されており、ここで著者らは、変化に対するアミノ酸配列の耐性を研究するには2つの主な戦略があることを示している。
【0079】
第一の戦略は、進化の過程での自然淘汰によるアミノ酸置換に対する耐性を利用する。異なる種におけるアミノ酸配列の比較により、保存されたアミノ酸を同定することができる。保存されたアミノ酸は、タンパク質機能にとって重要であると考えられる。対照的に、自然淘汰によって置換が許容されてきたアミノ酸の位置は、当該位置がタンパク質機能にとって重要でないことを示す。したがって、タンパク質の生物学的活性を維持しつつ、アミノ酸の置換が許容される位置が修飾され得る。
【0080】
第二の戦略は、タンパク質機能に重要な領域を同定するために、クローン化遺伝子の特定位置にアミノ酸の変更を導入する遺伝子工学を使用する。例えば、部位特異的変異誘発またはアラニン走査突然変異誘発(分子中の残基毎に単一アラニン変異を導入する)を使用することができる。Cunningham and Wells, Science 244:1081-1085 (1989)参照。次いで、得られた変異体分子の生物学的活性を試験することができる。
【0081】
著者らが述べるように、これらの二つの戦略は、タンパク質がアミノ酸置換に対して驚くほど耐性があることを示した。著者らは、さらに、どのアミノ酸の変更が、タンパク質中の特定のアミノ酸位置で許容される可能性があるかを示す。例えば、最も深く埋まった(タンパク質の三次構造内で)アミノ酸残基は、非極性の側鎖を必要とする一方で、表面の側鎖の特徴は一般にほとんど保存されない。さらに、許容される同類アミノ酸置換は、脂肪族または疎水性アミノ酸Ala、Val、Leu、およびBeの置換;水酸基残基SerおよびThrの置換;酸性残基AspおよびGluの置換;アミド残基AsnおよびGlnの置換、塩基性残基Lys、Arg、およびHisの置換;芳香族残基Phe、Tyr、およびTrpの置換、ならびに低分子アミノ酸Ala、Ser、Thr、Met、およびGlyの置換を伴う。同類アミノ酸置換の他に、本発明の変種としては、(i)一種類以上の非保存アミノ酸残基の置換を含むポリペプチドであって、置換アミノ酸残基が遺伝コードによりコードされたものであってもなくてもよいポリペプチド、あるいは(ii)置換基を有する一種類以上のアミノ酸残基の置換を含むポリペプチド、あるいは(iii)ポリペプチドの安定性および/または溶解性を高める化合物(例えば、ポリエチレングリコール)などの他の化合物と融合したかまたは化学的に共役したポリペプチド、あるいは(iv)例えば、IgG Fc融合領域ペプチドなどのさらなるアミノ酸を含むポリペプチドが挙げられる。このような変種ポリペプチドは、本明細書の教示から当業者の範疇にあると見なされる。
【0082】
例えば、電荷を有するアミノ酸の、電荷を有するか中性の他のアミノ酸とのアミノ酸置換を含むポリペプチド変種は、改善した特徴(例えば、凝集しにくい)を有するタンパク質を産生する場合がある。医薬製剤の凝集は、凝集体の免疫原的活性によって、活性の低下およびクリアランスの増加の両方を起こす。Pinckard et al, Clin. Exp. Immunol. 2:331-340 (1967); Robbins et al, Diabetes 36: 838-845 (1987); Cleland et al., Crit. Rev. Therapeutic Drug Carrier Systems 10:307-377 (1993)参照。
【0083】
特定の態様では、本発明のポリペプチドは、アルブミン融合タンパク質のアミノ酸配列、G-CSFタンパク質のアミノ酸配列、および/またはヒト血清アルブミンの断片または変種を含むかあるいはそれからなり、ここで、当該断片または変種は、参照アミノ酸配列と比較すると、1〜5、5〜10、5〜25、5〜50、10〜50、または50〜150のアミノ酸残基の付加、置換、および/または欠失を有する。好ましい態様では、アミノ酸置換は同類的である。当該ポリペプチドをコードする核酸も本発明に包含される。
【0084】
本発明のポリペプチドは、ペプチド結合または修飾ペプチド結合(すなわちペプチド同配体)により互いに連結されたアミノ酸からなることができ、20個の遺伝子コード化アミノ酸とは別のアミノ酸を含んでもよい。ポリペプチドは、翻訳後プロセシングなどの天然のプロセス、または当技術分野で周知の化学的修飾技術により修飾されてもよい。このような修飾は、基礎的な教科書やさらに詳しい学術論文、また多数の研究文献に十分に記載されている。修飾は、ペプチド主鎖、アミノ酸側鎖、およびアミノもしくはカルボキシル末端を含むポリペプチド中の任意の場所で起こり得る。なお、同じタイプの修飾が、所与のポリペプチド中のいくつかの部位において同一または異なった程度で存在する場合がある。また、所与のポリペプチドは、様々なタイプの修飾を含む場合がある。ポリペプチドは、例えばユビキチン化の結果、分岐してもよく、ポリペプチドは、分岐の有無にかかわらず環状であってもよい。環状、分岐、および環状分岐ポリペプチドは、翻訳後の天然のプロセスに起因してもよいし、人工的な方法により作製されてもよい。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチドまたはヌクレオチド誘導体の共有結合、脂質または脂質の誘導体の共有結合、ホスファチジルイノシトール(phosphotidylinositol)の共有結合、架橋、環化、ジスルフィド結合形成、脱メチル化、共有結合による架橋の形成、システインの形成、ピログルタミン酸の形成、ホルミル化、ガンマ-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリスチル化、酸化、ペグ化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、トランスファーRNAが仲介するタンパク質へのアミノ酸の付加(例えば、アルギニル化)、およびユビキチン化が挙げられる。(例えば、PROTEINS--STRUCTURE AND MOLECULAR PROPERTIES, 2nd Ed., T. E. Creighton, W. H. Freeman and Company, New York (1993); POST-TRANSLATIONAL COVALENT MODIFICATION OF PROTEINS, B. C. Johnson, Ed., Academic Press, New York, pgs. 1-12 (1983); Seifter et al, Meth. Enzymol. 182:626-646 (1990); Rattan et al., Ann. N.Y. Acad. Sci. 663:4862 (1992)参照)。
【0085】
C.機能的活性
「機能的活性を有するポリペプチド」は、完全長、前駆タンパク質、および/または成熟型G-CSFタンパク質に関連する1つ以上の公知の機能的活性を示すことができるポリペプチドを指す。このような機能的活性としては、生物学的活性、抗原性[抗ポリペプチド抗体に結合する(または結合をめぐってポリペプチドと競合する)能力])、免疫原性(本発明の特定のポリペプチドに結合する抗体を生じる能力)、本発明のポリペプチドで多量体を形成する能力、およびポリペプチドの受容体またはリガンドに結合する能力が挙げられるが、これらに限定されるわけではない。
【0086】
「生物学的活性を有するポリペプチド」は、用量依存性に関わらず、特定のバイオアッセイ中で測定される、本発明のG-CSFタンパク質(成熟型を含む)の活性に類似(必ずしも同一ではない)した活性を示すポリペプチドを指す。
【0087】
好ましい態様では、本発明のアルブミン融合タンパク質は、アルブミンに融合していない場合のG-CSFタンパク質部分(またはその断片もしくは変種)に関連する少なくとも1つの生物学的および/または治療的活性を有する。
【0088】
本発明のアルブミン融合タンパク質は、当技術分野で公知のアッセイや本明細書に記載のアッセイを使用するかまたは常法によって変更して、機能的活性(例えば、生物学的活性)を分析することができる。さらに、当業者であれば、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質の断片を常法によって測定し得る。さらに、当業者であれば、当技術分野で公知のおよび/または下記の実施例部分に記載のアッセイを使用して、アルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質の断片の活性を常法によって測定し得る。
【0089】
例えば、一態様では、アルブミン融合タンパク質の、抗G-CSFポリペプチド抗体および/または抗アルブミン抗体へ結合する能力、あるいはそれらへの結合をめぐってG-CSFタンパク質と競合する能力を測定する場合、当技術分野において公知の下記の様々な免疫測定法を使用することができるが、それらに限定されるわけではない:放射免疫測定、ELISA(酵素結合免疫吸着測定)、「サンドイッチ」免疫測定、免疫放射線測定、ゲル拡散沈降反応、免疫拡散測定、インサイチュー免疫測定(例えば、コロイド金、酵素、または放射性同位体標識を使用)、ウエスタンブロット、沈降反応、凝集アッセイ(例えば、ゲル凝集測定、赤血球凝集反応測定)、補体結合測定、免疫蛍光測定、プロテインA測定、および免疫電気泳動法測定などの技術を使用した拮抗的および非拮抗的アッセイ系が挙げられる。一態様では、抗体結合は、一次抗体上の標識を検出することにより検出される。別の態様では、一次抗体は、一次抗体への二次抗体または試薬の結合を検出することにより検出される。さらなる態様では、二次抗体が標識される。免疫測定における結合を検出するための多くの手段が当技術分野において公知であり、当該手段は本発明の範囲内である。
【0090】
好ましい態様では、G-CSFタンパク質の結合相手(例えば、受容体またはリガンド)が同定される場合、アルブミン融合タンパク質のG-CSFタンパク質部分として当該G-CSFタンパク質を含むアルブミン融合タンパク質による当該結合相手への結合は、例えば、還元および非還元ゲルクロマトグラフィー、タンパク質アフィニティークロマトグラフィー、ならびにアフィニティーブロッティングなどの当技術分野において周知の手段によりアッセイすることができる。一般的にはPhizicky et al., Microbiol. Rev. 59:94-123 (1995)参照。他の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFポリペプチドの受容体に結合する、アルブミン融合タンパク質の生理学的相関物の能力は、当技術分野において公知の技術を使用して常法によって測定することができる。
【0091】
別の態様では、アルブミン融合タンパク質が多量体を形成する能力が評価される場合、多量体の他の成分との会合は、例えば、還元および非還元ゲルクロマトグラフィー、タンパク質アフィニティークロマトグラフィー、ならびにアフィニティーブロッティングなどの当技術分野において周知の手段によりアッセイすることができる。一般的には上述のPhizickyら参照。
【0092】
結合および交差反応性を分析するためならびにHSA-G-CSF融合物の同一性を確認するために使用することができる免疫測定法としては、下記のものが挙げられるが、これらに限定されるわけではない:ウエスタンブロット、放射免疫測定、ELISA(酵素結合免疫吸着測定)、「サンドイッチ」免疫測定、免疫沈降測定、沈降素反応、ゲル拡散沈降素反応、免疫拡散測定、凝集測定、補体結合測定、免疫放射線測定、蛍光免疫測定、およびプロテインA免疫測定などの技術を使用した拮抗的および非拮抗的アッセイ系が挙げられる。このようなアッセイは、当該技術分野において常法であり周知である(Ausubel et al, eds, 1994, Current Protocols in Molecular Biology, Vol. 1, John Wiley & Sons, Inc., New York参照。当該文献はその全体において参照により本明細書に組み入れられる)。
【0093】
アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質に結合する抗体も、所与のタンパク質または抗原、好ましくは抗体が特異的に結合する抗原、に対する結合親和性の点から説明または規定されてもよい。好ましい結合親和性としては、5x10-2M、10-2M、5x10-3M、10-3M、5x10-4M、10-4M未満の解離定数またはKdが挙げられる。より好ましい結合親和性としては、5x10-5M、10-5M、5x10-6M、10-6M、5x10-7M、10-7M、5x10-8M、もしくは10-8M未満の解離定数またはKdが挙げられる。さらに好ましい結合親和性としては、5x10-9M、10-9M、5x10-10M、10-10M、5x10-11M、10-11M、5x10-12M、10-12M、5x10-13M、10-13M、5x10-14M、10-14M、5x10-15M、もしくは10-15M未満の解離定数またはKdが挙げられる。さらに、本明細書に記載されているかそうでなければ当技術分野で公知のアッセイが、アルブミン融合タンパク質ならびにその断片、変種、および誘導体の、アルブミン融合タンパク質のG-CSFタンパク質部分および/またはアルブミン部分のいずれかに関連する生物学的活性および/またはG-CSF活性を誘発(インビトロやインビボのいずれかで)する能力を測定するために慣例的に適用され得る。他の方法も当業者には公知であり、本発明の範囲内である。
【0094】
VI.HSA-G-CSF融合タンパク質
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子(rHSA-G-CSF)はG-CSFアナログである。rHSA-G-CSFの例は、米国特許第5,665,863号および米国特許第7,041,478号に記載されており、これらは参照により本明細書に組み入れられる。
【0095】
rHSA-G-CSFの別の例は、Teva Biopharmaceuticals USA LTDにより開発されているNeugranin(商標)である。Neugranin(商標)(「NEUG」)は、一本鎖に結合した約85kDaの分子量を有する759個のアミノ酸の融合ポリペプチドである。残基1〜585は、成熟型HSAに対応し、残基586〜759は、成熟型ヒトG-CSFに対応する。Neugranin(商標)融合ポリペプチドを図9に示す。
【0096】
VII.融合タンパク質の産生
rHSA-G-CSFの合成方法の例は、米国特許出願番号第11/929,828号に記載されており、これはその全体において参照により本明細書に組み入れられる。いくつかの態様では、NEUGは、NEUG融合タンパク質を発現するように遺伝子操作された酵母宿主系(サッカロミセス・セレビジエ(Saccharomyces cerevisiae))を用いて産生される。NEUGは、酵母培養物の発酵培地から集菌され、当技術分野で周知の方法を用いて精製される(例えば、一連のクロマトグラフィーおよびろ過工程、例えば、アフィニティークロマトグラフィーおよびイオン交換クロマトグラフィーなど)。
【0097】
非限定的な一例では、NEUG融合コンストラクトは以下のように開発された。完全長アルブミンcDNAは、英国オックスフォード大学にてF. E. Baralle博士の実験室でヒトcDNAライブラリーから単離された。このクローンは、プラスミドpAT153ALBとして、英国ノッティンガムのDelta Biotechnology Limitedへ送付された。さらに、6-アミノ酸HSAプロペプチド(RGVFRR)は、酵母(RSLDKR)でのより効率的なプロセシングを促進するように修飾された。
【0098】
Neugranin産生プラスミドは、野生型サッカロミセス・セレビジエで発見された2μプラスミドに基づいている。pSAC35に基づいた発現ベクター(特許EP 286 424 B、US 5,637,504)は、産生宿主のロイシン欠乏を補完する選択マーカーとしてサッカロミセス・セレビジエからのLEU2遺伝子を含む。この産生プラスミドはまた、強力な酵母プロモーターであるPRB1ならびに大腸菌におけるクローニングおよび増殖を可能にするプラスミドpUC9からの配列を含む。さらに、このプラスミドは、酵母に形質転換されると、大腸菌における増殖に必要なpUC9由来配列を排除する特有の性質を示す。これは、酵母においては、隣接するFLP認識標的とプラスミドからの酵母FLPリコンビナーゼの発現により達成される。したがって、NEUGの産生に使用される生体中には細菌DNAは存在しない。これは、マスターセルバンクを作成した後、酵母からの2μmプラスミドのレスキュー実験および配列決定により確認される。
【0099】
上に記載のように、CID1643(pSAC35:HSA.GCSF(T31-P204))と命名されたNEUG産生プラスミドは、pSAC35に基づいた発現ベクターに由来した。ヒトG-CSFのT31-P204に対応する領域は、HSAオープンリーディングフレームの3'末端へのシームレスな融合を可能にするために適切な5'および3'制限部位を加えながら、PCRにより増幅した。
【0100】
メリーランド州ロックビルのHuman Genome Sciences, Inc.(HGS)では、cGMPマスターセルバンク(MCB)を作成するためにNEUGシードバイアルを使用した。NEUG MCBの試験および特性評価は、ICHガイドラインQ5D(Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biologicals Products)に従ってCharles River Laboratories(マルバーン、ペンシルベニア州、米国)およびLark Technologies(ヒューストン、テキサス州、米国)にて行われた。
【0101】
次いで、このMCBに由来したcGMPのワーキングセルバンク(WCB)が、Charles River Laboratories(マルバーン、ペンシルベニア州、米国)にて作成され試験された。
【0102】
NEUG細胞株バンクの製造で使用した培地成分はすべて、合成されたか、生合成されたか、または植物由来のものであった。細胞株または細胞バンク作成の際には、動物またはヒト起源の成分は使用されなかった。
【0103】
セルバンクは、めねじ付の蓋を有する予め殺菌した1.8mL Nuncポリプロピレンチューブにおいて低温保存培地中で<-135℃にて保存される。
【0104】
医薬用途向けのrHSA-G-CSF融合タンパク質の単離、精製、調製方法の非限定的な例を、実験例において下に記載する。
【0105】
VIII.治療用タンパク質の調製
未変性の状態または組換え技術によって作られた治療用タンパク質、例えば、インターフェロンおよび成長ホルモンは、特に水溶液中に調製された場合、保存期間の短い典型的な不安定な分子である。投与用に調製された場合の当該分子の不安定さのため、分子の多くが保存の際には常に凍結乾燥または冷蔵されなければならず、このため分子の輸送および/または運搬が困難である。院内環境外で医薬製剤を保存および投与しなければならない場合、保存に関する問題は特に深刻である。治療用タンパク質を含むアルブミン融合タンパク質は、アルブミン融合していない同様の治療用タンパク質の保存期間と比べて長い保存期間を示している。保存期間とは、典型的には、溶液または他の保存剤形の治療用タンパク質の治療的活性が、治療的活性の過度な喪失を受けずに安定している期間を指す。
【0106】
しかしながら、凝集および化学的な不安定さが、治療用タンパク質を含むアルブミン融合タンパク質製剤の開発における大きな障害である。医薬製剤の凝集は活性を低下させ、凝集体の免疫原的活性のために薬物クリアランスを増加させるので、医薬製剤の凝集は非常に有害となり得る(Pinckard et al. 1967; Robbins et al. 1987; Cleland et aL, 1993)。
【0107】
本明細書に記載の方法および組成物の様々な態様を記載した非限定的ないくつかの実施例を下記に示す。
【実施例】
【0108】
IX.実験例
次の実施例は本発明を例証するものである。しかしながら、本発明は、当該実施例に記載の特定の条件または詳細に限定されるわけではないことを理解されたい。限定されるわけではないが米国特許を含む本明細書で参照する公開されている文書はいずれも参照により本明細書に組み入れられる。
【0109】
実施例1、2、および6では、三種類の非限定的なNeugranin(商標)(「NEUG」)薬学的組成物を示す。各製剤は、図9に示すNEUG融合ポリペプチドを含む。セクションAに記載の方法および得られた組成物は、広く「NEUG-0」と呼ぶ。セクションBに記載の方法および得られた組成物は、広く「NEUG-1」と呼び、セクションEに記載の方法および得られた組成物は、広く「NEUG-2」と呼ぶ。
【0110】
NEUGは、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合ポリペプチドである。NEUG融合タンパク質のアミノ酸配列を図9に示す。
【0111】
実施例1:治療用NEUG-0の調製
NEUG-0は毒物学実験用に製造した。製剤は、10mMクエン酸塩、75mM塩化ナトリウム、100mMスクロース、0.01%ポリソルベート80中にNEUGを4.0mg/ml含み、pH6.5まで緩衝化された。
【0112】
安全性/毒性
NEUGの非臨床的安全性は、サルで評価した。100、500、および1000μg/kgのNEUGへの反復暴露(週1回4週間)では良好な耐容性が示され、死亡、罹患、または体重もしくは摂食量の変化は無かった。有害な毒性と考えられる肉眼的または微視的な変化は無かった。治療に関連した所見は、G-CSF投与で予想される薬理作用に限定され(例えば、好中球増加、脾臓重量のわずかな増加、骨髄増生)、毒性とは考えられなかった。サルにおけるNOAELは、>1mg/kg/週であり、これは、ヒトにおける0.45mg/kgの投与量に伴って観察されるものより約12倍高かった。処置した動物はまた、ANCおよびWBCの増加を示した(データ示さず)。
【0113】
実施例2:治療用NEUG-1の調製
NEUG-1は、ヒトでの臨床研究用に製造された。製剤は、PMMT10/7.2(10mMリン酸ナトリウム、200mMマンニトール、60mmトレハロース無水物、0.01%(W/V)ポリソルベート80、pH7.2)中にNEUGを15.0mg/mlを含んでいた。
【0114】
NEUG-1の組成を表1に示す。
【0115】
(表1)NEUG-1製剤の組成
a各バイアルは、1.0mL送達可能容量を確保するため0.16mL過剰に含む。
【0116】
NEUG-1用の原薬(「BDS」)の製造方法の一例として、図13の非限定的な工程を示す。次に、BDSは、約-80℃で保存される(公称値、保存温度の許容範囲は約-65℃である)。
【0117】
輸送や臨床現場での保存のための製剤のロバスト性を向上させるためや、期待される長期保存性を有する安定した製品を提供するため、NEUG-1の凍結乾燥形態が開発されている。凍結乾燥の方法は、当技術分野において周知であり、洗練された凍結乾燥ケーキを形成する方法の一例を、下記工程に記載のように行った。
【0118】
工程1:原薬の解凍および希釈
NEUG原薬の容器を15〜30℃で解凍する。原薬をプールし(必要であれば)、混ぜ、280nmでの吸光度によりタンパク質濃度を決定する。原薬の濃度を使用して、必要な希釈用緩衝剤の量を計算する。密度およびpHも測定する。調製用緩衝剤は、所望の最終濃度まで原薬を希釈するために使用される。
【0119】
工程2:無菌ろ過
希釈溶液は、完全性試験を行った滅菌フィルターを介して、調製領域から無菌の受容容器中へ移される。ろ過トレインは、直列に繋いだ0.2μmPVDFフィルターからなる。フィルターはすべて未使用であり、完全性試験がなされ、製造後には廃棄される。
【0120】
工程3:充填および部分的な付栓
Neugraninを、発熱性物質を除去した3mL USP Type Iガラスバイアルへ充填し、部分的に付栓し、底のない凍結乾燥トレーに載せた。充填した容量を確認するために、充填操作全体にわたって重量をチェックする。
【0121】
工程4:凍結乾燥
凍結乾燥サイクルは、室圧および棚温度により制御する。製品温度は、凍結乾燥室全体に配置した製品用熱電対によりモニタリングする。部分的に栓をしたバイアルを、予め冷却した棚に載せ、制御した速度で凍結する。アニーリング工程を行い、マンニトール(増量剤)の結晶化を増進させる。次に、一時乾燥を行い、熱電対をモニタリングして確認する。二次乾燥を行い、所望の含水率を達成する。二次乾燥の終わりに、0.2μmの無菌ろ過をした窒素を通気して凍結乾燥室の真空を開放する。
【0122】
工程5:密封およびキャップ付け
部分真空下(約11〜12psia)でバイアルに栓を置く。製品トレーからバイアルを取り、バイアルにキャップを付ける。
【0123】
最初の凍結乾燥臨床材料(NEUG-1)は、きわめて安定していることを示し、現在のところ2年間の保存期間を有し(2〜8℃で保存した場合)、これは、継続している安定性研究のデータが検討されるにつれさらに延びると考えられる。
【0124】
実施例3:NEUG-1参照標準物(ロット2378-R)の一般的特性
NEUGの生理化学的および生物学的特徴を調べるために様々な方法が開発されている。参照標準物ロット2378-Rと呼ぶNEUGの1ロットは、NEUGを21.3mg/mL含み、pH7.2で、10mMリン酸ナトリウム、20OmMマンニトール、60mMトレハロース無水物、および0.01%(w/v)ポリソルベート80(PMTT10/7.2)中で調製された。物理化学的特性評価を参照標準物ロットに対して行い、試験性状、分析法、および結果の概要を下記表2に示す。
【0125】
(表2)NEUG-1ロット2378-Rの特性評価
【0126】
クーマシーブルーで染色したNEUG-1ロット2378-RのSDS-PAGE分析は、単一バンドという結果になった(図19)。NEUG-1ロット2378-Rの代表的なSEC-HPLCおよびRP-HPLCクロマトグラムも、それぞれ図19Aおよび図19Bに示す。
【0127】
実施例4:NEUG-0製剤およびNEUG-1製剤の試験
1.rHSA-G-CSFモノマー純度に対するタンパク質濃度の影響
2.5mg/mlから240mg/mlの濃度範囲の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子(rHSA-G-CSF)をPMTT10/7.2(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH7.2)中にて25℃で24時間インキュベーションした。インキュベーションに続き、SE-HPLCによりモノマー純度を測定した。
【0128】
結果(図1)は、rHSA-G-CSFの濃度が増加するにつれて凝集が増加することを示す。
【0129】
2.rHSA-G-CSF凝集に対するpHの影響
15mg/mlおよび60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、25℃で7日間、PMTT10(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)中、pH6.0、6.8、7.2、および8.0でインキュベーションした。インキュベーションに続き、SE-HPLCによりrHA-G-CSFモノマー純度を測定した。
【0130】
結果(図2)は、pHが増加するにつれて凝集が増加し、タンパク質濃度の増加により凝集が加速したことを示す。
【0131】
3.rHSA-G-CSFの凝集に対する温度の影響
60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、PMTT 10/7.2緩衝剤中で、4℃、25℃、または40℃で24時間インキュベーションした。インキュベーションに続き、SE-HPLCによりrHSA-G-CSFモノマー純度を測定した。
【0132】
結果(図3)は、温度が上昇すると凝集が加速したことを示す。
【0133】
4.rHSA-G-CSFの凝集に対するpHの影響
48mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、25℃で最大3日間まで、PMTT10(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)中、pH5.8、6.3、6.4、または7.0、あるいはCMTT10(10mMクエン酸ナトリウム、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)緩衝剤中でpH6.2でインキュベーションした。組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子のモノマー純度を、インキュベーション後0、1、2、および3日目にSE-HPLCにより測定した。
【0134】
結果(図4および図10)は、凝集のほとんどがインキュベーションの1日目に起こり、凝集速度がpHの増加により増加したことを示す。
【0135】
5.rHSA-G-CSFの凝集に対するpHの影響
15mg/mlまたは60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、pHが6.0、6.8、7.2、または8.0のPMTT10中で4℃、25℃、または40℃で最大10日間までインキュベーションした。モノマー純度は、インキュベーション後に0、1、7、および10日目にSE-HPLCで測定した。
【0136】
結果(図11)は、pHが増加するにつれて凝集が増加し、温度およびタンパク質濃度の増加により凝集が加速したことを示す。
【0137】
6.rHSA-G-CSF生物活性に対するpHの影響
15mg/mlまたは60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、pHが6.0、6.8、7.2、または8.0のPMTT10中で4℃、25℃、または40℃で最大10日間までインキュベーションした。生物活性を、インキュベーション後、0日目と、7日目または10日目のいずれかとでバイオアッセイにより測定した。
【0138】
手短に説明すると、NFS-60細胞は、G-CSFおよびNEUGの両方に応答して増殖する。このアッセイでは、96ウェルマイクロプレートに細胞10000個/ウェルで蒔き、NEUGタンパク質で20時間37℃で処理した。20時間インキュベーションした後、NFS-60細胞を、0.5μCi/ウェルの[3H]-チミジンで4時間パルス化し、[3H]-チミジン取込みにより測定されるDNAの合成レベルを測定した。統計分析については、NEUG濃度の関数としての[3H]-チミジン取込みの測定を、4パラメータ・ロジスティックモデルを用いてモデル化し、EC50値(pg/mL)を決定した。試験タンパク質の結果は、参照標準物のEC50値を同一のアッセイ内で分析される試験サンプルのEC50と比較することにより得られた相対効力(RP%)値[RP%=(EC50参照物/EC50試料)*100]として報告される。
【0139】
結果(図12)は、低pHでインキュベーションされるとrHSA-G-CSFの活性がよりよく維持されたことを示す。
【0140】
7.rHSA-G-CSFの凝集に対する塩濃度の影響
rHA-G-CSFの60mg/ml溶液のイオン強度を、溶液中の塩化ナトリウムの量を変更することにより制御した。調製用緩衝剤はPMTT10/7.2であった。試料を、25℃/60%RHにて1日間、5mM、10mM、20mM、または50mMの食塩濃度でインキュベーションし、続いてSE-HPLCにより試験した。
【0141】
結果は、塩濃度を増加させると、rHSA-G-CSFの凝集が低下したことを示す(図5)。
【0142】
8.rHSA-G-CSFの凝集に対するリン酸塩濃度の影響
rHSA-G-CSFの60mg/ml溶液のイオン強度を、溶液中のリン酸塩の量を変更することにより制御した。調製用緩衝剤はpH7.2のPMTTであった。試料を、25℃/60%RHにて1日間、15mM、20mM、25mM、30mM、40mM、または50mMのリン酸塩濃度でインキュベーションし、続いてSE-HPLCにより試験した。
【0143】
結果は、リン酸塩濃度を増加させると、rHSA-G-CSFの凝集が低下したことを示す(図6)。
【0144】
実施例5:治療用NEUG-2の調製
より高濃度での使用および投与の臨床研究のためにNEUG-2を製造した。製剤は、PMMT20/6.0(20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース、0.01%ポリソルベート80)中にNEUGを50.0mg/mlで含んでいた。
【0145】
本質的には、NEUG-1に関して示したのと同じ発酵および精製工程を行った(例えば図13参照)。
【0146】
NEUG-2は、液体形態および凍結乾燥形態の両方で調製した。輸送や臨床現場での保存のための製剤のロバスト性を向上させるため、および期待される長期保存性を有する安定した製品を提供するために凍結乾燥形態が調製された。NEUG-2は、NEUG-1に関して記載したのと同じ凍結乾燥サイクルを用いて凍結乾燥した。NEUG-2製剤も良好に形成され、薬学的に洗練されたケーキを生じた。
【0147】
実施例6:NEUG-0、NEUG-1、およびNEUG-2製剤の比較
表3は、NEUG-0、NEUG-1の標準ロット(参照標準物2378-R)、およびNEUG-2の3種類の異なるロットの生理化学的な比較を示す。表中、「CP」は透明な淡黄色を指す。「ND」は測定されなかったことを指す。
【0148】
(表3)NEUG-0、NEUG-1、およびNEUG-2の生理化学的な比較
【0149】
NEUG-0〜NEUG-2を製造する方法が開発されるにつれ、方法に関連した不純物(例えば、酵母宿主細胞タンパク質)および産物に関連した変種(SEC、RP、およびIEC-HPLC)は、一貫して減少している。
【0150】
表4および表5は、NEUG-0、NEUG-1、およびNEUG-2の調製履歴および調製比較の概要を示す。
【0151】
(表4)調製履歴
【0152】
(表5)cGMP製剤の比較
【0153】
実施例7:NEUG-1およびNEUG-2の試験
1.rHSA-G-CSFのモノマー純度に対するpHおよびリン酸塩濃度の影響
図7に示す結果は、PMTT10/7.2およびPMTT20/6.0(20mMリン酸塩、180mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH6.0)それぞれにおけるNEUGのSE-HPLCにより測定されたモノマー純度を示す。試料は、2.5mg/mlから120mg/mlの範囲で異なるタンパク質濃度を有し、純度分析前に25℃で24時間インキュベーションした。
【0154】
結果は、SE-HPLCにより測定されたようにPMTT20/6.0調製用緩衝剤中で最大120mg/mlまでの濃度のrHA-G-CSFでは純度の有意な減少は観察されなかったが、PMTT10/7.2中では10%の凝集の増加があったことを示す。
【0155】
2.rHA-G-CSFの凝集に対するpHおよびリン酸塩濃度の影響
図8に示す結果は、PMTT20/6.0またはPMTT10/7.2中で4℃または25℃/60%RHで最大14日間インキュベーションしたrHA-G-CSF60mg/mlのSE-HPLCにより測定された凝集を示す。
【0156】
結果は、PMTT20/6.0調製用緩衝剤中でインキュベーションした試料では25℃で14日間インキュベーションした後には有意な凝集の増加は観察されなかったが、PMTT10/7.2調製用緩衝剤中で同様の条件下でインキュベーションした試料は、6%の凝集の増加を示したことを示した。
【0157】
3.NEUG-1対NEUG-2のHPLC比較
NEUG-1およびNEUG-2のSEC-HPLC分析およびRP-HPLC分析を図14Aおよび図14Bに示す。
【0158】
4.NEUG-1およびNEUG-2の電荷の比較、ペプチドマッピング、および純度
NEUG-1(図中「1P」として示す)およびNEUG-2(図中「2P」として示す)の電荷不均一性をIEC-HPLCにより比較した。結果を図15Aに示す。二種類のペプチド(NEUG-1「1P」およびNEUG-2「2P」)の同一性も、図15Bに示すようにペプチドマッピングにより比較した。
【0159】
NEUG-1およびNEUG-2の純度も、図16に示すようにSDS-PAGEおよびクーマシーブルー染色により比較した。各レーンの試料を下記表6に示す。
【0160】
(表6)図16のSDS-PAGEにロードした試料
【0161】
5.凍結融解
凍結融解の影響を、濃度が15、60、120mg/mlのNEUG-2(PMTT20/6.0中で)緩衝剤を用いて評価した。試料を0回から10回まで凍結および融解し、その後、目視およびSEC、RP、IECにより分析した。
【0162】
結果は、従来の(PMTT10/7.2)および新規な(PMTT20/6.0)調製用緩衝剤に関しては、Neugraninが、凍結融解誘導性の分解に感受性がないようであることを示す(データ示さず)。
【0163】
6.短期安定性
従来の緩衝剤(PMTT10/7.2)および新規な緩衝剤(PMTT20/6.0)中のNEUG試料(60mg/ml)を、4℃および25℃/60%RHで1か月間維持し、次いで、目視、SEC、RP、IEC、およびバイオアッセイにより試験した。モノマー純度はSE-HPLCにより評価した。
【0164】
14日目に、従来の緩衝剤中のNEUGは、6%の凝集体の増加を示したが、新規な緩衝剤中のNEUGは変化を示さなかった。目視、RP、IEC、およびバイオアッセイによると1か月間有意な変化はなかった。
【0165】
7.振とう
振とうにより誘導された凝集の影響を、従来の調製用緩衝剤(PMTT10/7.2)、新規な調製用緩衝剤(MPTT20/6.0)中のNEUGを用いて、3つの条件(15、60、および120mg/ml)、pH6.0で評価した。試料をそれぞれ、120rpmで0分から30分まで25℃で水平に振とうした。目視およびSE-HPLCを行った。
【0166】
SE-HPLC、RP-HPLC、IE-HPLCの結果は、モノマーの有意な喪失を示さなかった(データ示さず)。さらに、目視からの結果は、振とうの結果としての外観の変化を示さない。これは、従来および新規な調製用緩衝剤の両方に関して、Neugraninが、振とうにより誘導された凝集に対し感受性ではない可能性があることを示唆している。
【0167】
8.酸化
イオン交換高速液体クロマトグラフィー(「IEC」)および逆相高速液体クロマトグラフィー(「RPLC」)を、Neugraninの酸化をモニタリングするために使用することができる。過酸化水素試験のための実験条件は以下のとおりである。8mg/mLのNeugraninを0.03%過酸化水素およびTBOに暴露した。試料を、25℃で0、1、3、5、8、20、および24時間インキュベーションした後、サイズ排除クロマトグラフィー(「SEC」)、逆相(「RP」)、およびIECにより分析した。過酸化水素およびTPB誘導酸化の評価のために、RPLC、IECを用いた。クロマトグラフおよび表形式のデータを図21および図22に示す。
【0168】
HSAは、過酸化水素の存在下で酸化されることが示されている。同定された酸化部位は、Cys-34、Met-123、Met-298、Met-446、およびMet-548を含む。
【0169】
クロマトグラム(図21)は、1時間RTで過酸化水素(薄いグレー)およびTPB(黒)への暴露後、NEUG対照(灰色)のオーバーレイを示す。RPおよびIECによれば、H2O2で処理された試料ではメインピークの3分後に溶出してピークの有意な増加が示され、RPによれば、TPBで処理された試料は、メインピークの3分後に溶出するピークを示し、IECによれば、ピークはわずかに増加し、メインピークより2、3分後に溶出した。
【0170】
要約すると、結果は、RPおよびIECが異なる酸化形態を検出することができ、H2O2およびTBPのいずれも、メチオニンおよびシステイン残基であるNEUGの酸化を誘導し得たことを示唆する。NEUGの酸化は負荷のかかる条件下で起こるが、それが保存の際のNEUGの主要な分解経路を表しているということ示すものはなく、それらの活性に対する有意な変化はない。
【0171】
実施例8:薬学的利用のためのNEUGの特性評価の例
NEUG-1の工程を用いて三種類のcGMP薬剤成分ロットを製造し、NEUG-2の工程を用いて五種類のロットを製造した。一般に、NEUG-2製剤からは、NEUG-0およびNEUG-1に比べて同等かまたはより優れた特性を有するタンパク質が得られた。すべてのcGMPロットの試験結果を図17A-B中の表に示す。NEUG-1のロットは、060615001、060628001、および060707001である。NEUG-2のロットは、071008005、071025001、071026004、071106001、および07116001である。ロットの試験は以下のように行った。
【0172】
1.目視による外観
Neugranin(商標)(「NEUG」)の外観は、白黒の背景で前方に向けて照射する蛍光灯を用いて、目視により評価した。
【0173】
2.pH
pHメーターは、pHが4.0、7.0、および10.0の緩衝剤を用いて標準化した。標準化後、NEUG試料のpHを測定し記録した。
【0174】
3.凝固点によるオスモル濃度
オスモル濃度は凝固点降下により決定した。測定前に290mOsm/kg較正基準を用いて標準化を行った。
【0175】
4.吸光度(280nm)の測定による濃度
NEUGの濃度は、280nmで吸光度を測定することにより測定した。タンパク質濃度は、実験的に測定した0.54mg-1*mL*AUの吸光係数を用いて計算した。
【0176】
5.ELISAによる同一性
同一性アッセイは、当技術分野で周知の方法を用いて、標準的なサンドイッチELISAを使用した。アッセイは、NEUG分子のG-CSF部分およびアルブミン部分の両方に対する抗体の特異性に基づくものであった。一緒になってこの抗体対は、NEUGと、G-CSF、HSA、または他のアルブミン結合タンパク質とを識別することができる。
【0177】
6.クーマシー染色SDS-PAGEによる純度
変性条件下のNEUGの純度は、クーマシー染色SDS-PAGEを用いて評価した。試料は、分析前に、還元剤を用いてまたは用いないで試料緩衝剤で希釈した。分子量マーカーおよびNEUG参照標準物も各ゲルに添加した。電気泳動後、ゲルをクーマシーで染色し、濃度計を用いて純度を測定した。この方法は、二量体などの産物に関連した不純物を検出するために主に使用される。
【0178】
7.銀染色SDS-PAGEによる純度
変性条件下のNEUGの純度は、銀染色過負荷SDS-PAGEを用いて評価した。試料は、分析前に、還元剤を用いてまたは用いないで試料緩衝剤で希釈した。試料は、最大10μg/レーンまで添加した。分子量マーカーおよびNEUG参照標準物も各ゲルに添加した。電気泳動後、ゲルを銀染色した。NEUG試料と参照標準物との間の同一性を測定するために、泳動距離とバンドパターンとの間の比較を行う。低レベルの産物関連不純物および方法関連不純物を検出するためにこの方法を使用した。
【0179】
8.サイズ排除HPLCによる純度
未変性(native)の条件下のNEUGの純度は、サイズ排除HPLCを用いて評価した。試料は、リン酸塩および硫酸塩からなる無勾配移動相を用いて、TosoHaas G3000SWXLカラム(7.8mm x 30cm)を使用して分析した。NEUGの純度は、280nmでの総ピーク面積に対するメインピーク下面積の比を計算することにより決定した。このアッセイでは、二量体および凝集体を含む産物関連サイジング不純物が検出される。
【0180】
9.逆相HPLCによる純度
逆相HPLCは、NEUGの純度を測定するために使用した。試料は、アセトニトリル濃度が上昇する溶離勾配で、Agilent Zorbax逆相カラム(StableBond C8、300A、5μm、250x4.6mm)を用いて分析した。純度は、215nmの検出波長での総関連ピーク面積に対するメインピーク面積の割合を計算することにより決定した。
【0181】
RP-HPLCアッセイにおけるメインピークの前のピークは、G-CSF部分のThr-133にてジヘキソース修飾を含むNeugranin変種と一致する大きさを有する。この修飾は、薬効には影響せず、現在まで製造された開発バッチ、毒性バッチ、およびcGMPバッチすべてにおいて一定のレベルで存在した。Thr-133は、G-CSFの修飾の公知の部位である。
【0182】
10.バイオアッセイ(NFS-60細胞増殖)による薬効
NEUGの薬効アッセイは、NSF-60細胞増殖の誘発に基づく、細胞に基づいた増殖アッセイである。NFS-60細胞は、G-CSFおよびNEUGの両方に応答して増殖する。このアッセイでは、96ウェルマイクロプレートに細胞10,000個/ウェルで蒔き、NEUGタンパク質で37℃で20時間処理した。20時間インキュベーションした後、NFS-60細胞を、0.5μCi/ウェルの[3H]-チミジンで4時間パルス化し、[3H]-チミジン取込みにより測定されるDNAの合成レベルを測定した。統計分析については、NEUG濃度の関数としての[3H]-チミジン取込みの測定を、4パラメータ・ロジスティックモデルを用いてモデル化し、EC50値(pg/mL)を決定する。試験タンパク質の結果は、標準試料のEC50値を同一のアッセイ内で分析される試験サンプルのEC50と比較することにより得られた相対効力(RP%)値[RP%=(EC50参照物/EC50試料)*100]として報告した。
【0183】
11.閾値法による残存DNA
試料に残存するDNAの測定方法は、市販のキット(Molecular Devices Threshold 総DNA測定キット)に基づく。試料をまず希釈し、タンパク質を消化し、DNAを抽出した。次に、DNAの濃度をThresholdキットを用いて定量した。このキットは、DNAを免疫標識し、固定し、子ウシ胸腺DNA標準曲線に対するDNAの濃度を電気化学的に測定する。結果は、許容範囲内のDNA標準物の添加回収率により内部的に適格とされた。
【0184】
12.比濁時間分析法によるエンドトキシン
NEUG溶液中のエンドトキシン濃度は、標準的なエンドトキシン自動比濁時間分析システムを用いて測定した。比濁時間分析システムは、ゲルクロット法を改良したものであり、ゲルクロット形成に先行する濁度増加を測定する。リムルスアメーバ様細胞溶解物(LAL)を標準物および被験物質に加え、37℃でインキュベーションした。インキュベーション時に、反応混合物の濁度を分光測光法を用いてモニタリングした。濁度はエンドトキシン濃度に比例した。被験物質中の濁り形成速度を、公知の標準濃度の標準曲線における濁り形成と比較する。
【0185】
13.膜ろ過によるバイオバーデン
膜ろ過法は、試料中の生存生物の数を推定するアメリカ薬局方(USP<61>)微生物限度試験に類似している。膜ろ過方法では、生物は、試験液からメンブレンフィルター上に捕えられ、次に、適切な増殖培地に蒔かれる。試験前に、試料体積が10mLの被験物質を、無菌リン酸緩衝生理食塩水(PBS)90mLに希釈する。希釈した被験物質100mLをすべてろ過する。ろ過工程後、フィルター膜を、一般細菌数(HPC)寒天に移し、30℃から35℃で2日から3日間インキュベーションする。HPC寒天は、USP<61>に記載の大豆カゼイン消化物寒天(SCD、これはTSAとも呼ばれる))とは異なる。TSAと同様に、HPC寒天は、広範な好気性従属栄養細菌を回収するように設計されている。しかしながら、HPC寒天もまた、負荷のかかった生物を最大限回収するように設計されている。
【0186】
14.タングステン調査
特定の注射器の銘柄は、賦形剤としてタングステンを含むことが知られている。液体のNEUG試料を、様々な形態のタングステン(タングステン酸ナトリウム無水物、ポリタングステン酸ナトリウム、タングステン酸アンモニウム、および酸化タングステンWO3およびWO2)の存在下、様々な濃度(500ppb、1000ppb、および2500ppb)で2〜8℃および25℃で最大3か月まで試験した。試料を、SE-HPLC、RP-HPLC、およびバイオアッセイにより分析した。3か月の安定性試験後、NEUG試料に対するタングステンの有意な影響はなかった。
【0187】
15.特性評価アッセイ
下記分析も、特性評価情報のために使用した。
【0188】
N末端配列
NeugraninのN末端のアミノ酸配列は、PE Applied Biosystems Procise 494 cLCプロテインシークエンサーを用いて同定した。当該方法は、エドマン分解化学作用を組み入れ、気相ブロットサイクル、オンラインPTHアミノ酸HPLC分析器、およびSequenceProデータ分析ソフトウェアを利用する。
【0189】
IE-HPLCによる電荷不均一性
IEC-HPLCを使用して、NEUGにおける電荷不均一性の相対存在量を測定した。試料は、Dionex Pro-Pac Wax 10イオン交換カラム(250x4.6mm)を用いて分析し、移動相はBis-TrisおよびNaClからなった。この方法は、試料非依存性の変動を示し、展開のこの時点では、定量分析というよりむしろ定性分析として使用される。
【0190】
ペプチドマッピング
ペプチドマッピングは、NEUGの構造の完全性(integrity)を測定するために使用される。ペプチドマッピングは、消化のためにLys-Cを使用し、ペプチド分離のために勾配溶出する逆相HPLCを用いて行った。タンパク質試料をまず還元し、変性し、次いでアルキル化した。緩衝剤交換を行い、続いて1時間、その後一晩Lys-C消化を行った。消化に起因するLys-Cペプチド混合物を逆相HPLCにより分離し、Agilent 1100 HPLC系と共にYMC Pack ODS-A C18カラム(4.6mm x 250mm)を用いてペプチドプロファイルを得た。HPLC溶出は、ダイオードアレーUV検出器およびエレクトロスプレーイオン化Finigan LCQ DUO-Ion Trap質量分析装置を用いてモニタリングした。質量分析データは、Xcalibur V2.3ソフトウェアを用いて処理した。この方法の質量精度は、20ppm未満であった。HPLCピークはLC-MSにより同定した。
【0191】
エレクトロスプレーイオン化質量分析
質量分析は、Pico Viewナノエレクトロスプレー源を装備したApplied Biosystems QStar ESI-Quadrapole Time of Flight(QTOF)質量分析計を用いて行った。試料は、HamilitonシリンジおよびHarvardシリンジポンプによりQStarへ導入した。エレクトロスプレイーイオン源は、カーテンガス値25で、2500Vで操作した。器具は、ミオグロビンで較正した。質量電荷比は、割り当てられた分析時間全体にわたって、一秒ごとに質量分析器内の質量範囲全体(500〜3000m/z)を走査することにより記録した。得られた総イオン流は、Analyst v1.4ソフトウェアを用いて分析した。この器具の質量精度は、10ppm未満である。
【0192】
残存S.セレビジエ(酵母)宿主細胞タンパク質
原薬中に存在する酵母宿主細胞タンパク質(yHCP)の量を、NEUGの発現に使用したのと同じ酵母宿主のヌル(null)株に由来した宿主細胞タンパク質での免疫付与により産生されたカスタム抗体に基づくELISAを用いて測定した。
【0193】
エルマンアッセイによる遊離チオール
エルマン試薬である5,5'-ジチオ-ビス-(2-ニトロ安息香酸)(DTNB)を用いて、NEUG試料中の遊離チオール含量を測定した。DTNBは、遊離スルフヒドリル基と反応し、ジスルフィドおよび2-ニトロ-5-チオ安息香酸(TNB)の混合物が得られる。TNBは412nmに強い吸収があり、N-アセチルシステイン(NAC)標準曲線と比較することにより容易に反応を定量することができる。この方法の精度を高めるため、部分的に変性条件下で反応を行った。これにより、未変性条件下では完全には暴露されない場合のある遊離スルフヒドリル基にDTNBがアクセスできる。
【0194】
16.安定性
原薬(「BDS」)は、-80℃で保存され輸送される(公称値、許容温度は≦-65℃である)。BDSの安定性は、-80℃および加速貯蔵条件で製品を保存することにより試験した。参照標準物(NEUG-1、ロット2378-R)をまず安定した環境に配置して、GMP BDSに先導的な安定性(leading stability)を与えかつ参照標準物自体の安定性についての理解を得た。各工程(NEUG-1、NEUG-2)には典型的に、少なくとも1つの開発ロットおよび1つのcGMPロットを安定した環境に配置し、各試験は、ICHガイドラインに従った試験に特化したプロトコルに詳述されている。アッセイの適性および可能性の高い分解経路を規定し、かつ有効期限の予測に役立てるため標準加速条件を使用した。安定性は、外観、pH、タンパク質濃度、オスモル濃度、SEC-HPLC、RP-HPLC、SDS-PAGE、およびバイオアッセイによりモニタリングした。
【0195】
NEUG1参照標準ロット2378の代表的な安定性結果の概略を下記表7から表9にまとめる。
【0196】
(表7)NEUG-1ロット2378-Rの安定性データ(-80℃)
CPF=透明、淡黄色、外来の粉粒体が本質的に含まれていない
【0197】
(表8)NEUG-1ロット2378-Rの安定性データ(-20℃)
【0198】
(表9)NEUG-1ロット2378-Rの安定性データ(2〜8℃)
【0199】
どのパラメータの有意な変化も、推奨される保存条件で36か月まで、-20℃で18か月まで、または2〜8℃で12か月まで見られなかった。
【0200】
NEUG-2の1か月データを示す。推奨される保存条件下、-2O℃、または2〜8℃では有意な変化は認められない。表10。
【0201】
(表10)NEUG-2の安定性データ
【0202】
2〜8℃での液状のNEUG-2の2つのロットの安定性データを、下記表10.1および表10.2に示す。
【0203】
(表10.1)ポリプロピレンチューブ中の液体NEUG-2(ロット3029)1.2mlの安定性
【0204】
(表10.2)テフロンバイアル中の液体NEUG-2(cGMPロット071026004)1.0mlの安定性
【0205】
上に記載のように、NEUG-1工程からNEUG-2工程への変更を製剤に対して行った。当該変更は、より高いNEUG濃度で製品品質を確保するために行い、新たな賦形剤は含まれなかった。主な変更点は、イオン強度の増加(10mMから20mM)およびpHの低下(7.2から6.0)であった。BDS製剤は、NEUG-1工程からNEUG-2工程になり、濃度依存性の凝集が低下するように改善した。したがって、NEUG-2製剤は、既にロバスト性を有するNEUG-1と比較して向上した安定性を示すことが予想される。
【0206】
さらに、凍結乾燥形態または液状形態のいずれであっても、2〜8℃での推奨される保存条件で3か月間の安定性試験後にNEUG-2に対するタングステンの有意な影響はなかった(SE-HPLC、RP-HPLC、および効力による分析;データ示さず)。
【0207】
実施例9:例示的製剤(NEUG-2)の説明および組成
1.剤形の説明
一態様では、Neugranin(商標)(「NEUG」)は、コーティングされたゴム栓およびフリップオフシール(flip-off seal)で密閉した使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給される。NEUGは2〜8℃で保存することができる。注射用滅菌水(WFI)1.0mLで再構成する際、各バイアルは、20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース二水和物、0.01%(w/v)ポリソルベート80、pH6.0中に、NEUGを50mg/mL(バイアル毎に50mgが送達可能)含む。再構成後、バイアルは室温で保持することができ、8時間以内に使用するのがよい。
【0208】
2.定量的組成
1つのNEUG製剤(NEUG-2)の例示的な定量的組成を下記表11に示す。なお、液状形態のNEUG-2は、凍結乾燥形態と同じ定量的組成を有している。液状形態のものは、充填済みシリンジ中に提供することができる(下記セクション9参照)。
【0209】
(表11)NEUG-2製剤の定量的組成
a各バイアルは、1.0ml送達可能容量を確保するため0.11ml過剰に充填される。
【0210】
3.再構成用緩衝剤
凍結乾燥形態のNEUGの再構成に注射用滅菌水(WFI)を使用することができる。
【0211】
4.賦形剤
NEUG製剤の例示的組成は次のとおりである。2.77mg/mLリン酸ナトリウム、33.79mg/mLマンニトール、22.7mg/mLトレハロース二水和物、および0.1mg/mLポリソルベート80で、pHが6.0(上記表5参照)。リン酸ナトリウムは緩衝剤であり、マンニトールは、良好なケーキ構造を提供しかつ張性を調整するための増量剤として使用され、トレハロースは、抗凍結剤として使用され、ポリソルベート80は、凝集および吸着の可能性を低下させるために使用される。50mg/mLのNEUG-2における有効成分(「API」)の濃度では、タンパク質自体がロバスト性を有する緩衝剤として作用する。製剤に使用される公定賦形剤は、複数の公定書(MC)に準拠している。例外としては、現在利用可能な公定書がないトレハロース二水和物である。しかしながら、トレハロース二水和物は、公定書ではないが厳格な一連の仕様書に準拠している。トレハロースは、米国FDAでGRASとしてリストに記載されており、多数の市販されている非経口薬剤(例えば、ベバシズマブ(アバスチン)およびトラスツズマブ(ハーセプチン))の製剤に使用されている。
【0212】
5.製剤開発
毒性研究に使用された最初の製剤は、凍結させた液体として-20℃での保存を支援するように設計された(NEUG-0)。輸送および臨床現場での保存に対する製剤のロバスト性を向上させ、および期待される長い保存期間を有する製品を提供するために、全臨床材料に対して凍結乾燥形態が現在まで使用されている。最初の臨床材料(NEUG-1)は、きわめて安定していることを示し、現在は2年間の保存期間を有し、これはさらに延びると考えられる。後の製剤開発では、より大きなイオン強度およびより低いpHが、より高い濃度(>25mg/mL)でAPIを安定させることを示した。このために、1つの製剤(NEUG-2)は、より低いpH(6.0対7.2)およびより高いリン酸塩濃度(20対10mM)を有する。強制分解試験は、この製剤が、激しい振とう、凍結融解の繰り返し、および濃度誘導凝集から液体状態の薬剤成分を保護することを示した。NEUG-1に関して記載したのと同一の凍結乾燥サイクルをNEUG-2製剤に対して使用すると、いずれも良好に形成されたケーキを生成した。
【0213】
6.製造工程開発
いくつかの態様では、NEUGの最終製剤は、調製用緩衝剤で原薬を所望の濃度(50mg/mL)まで希釈し、充填し、製品を凍結乾燥することにより得られる。
【0214】
毒性と臨床材料とを比較し易くするために、両方の工程(NEUG-1およびNEUG-2)からの多くの最終製剤(「FDP」)を物理化学的に特性評価した。
【0215】
物理化学的な比較試験の結果が下記表12に含まれており、NEUG-2製品の品質は、NEUG-1製品とくらべて、それ以上ではなくても少なくとも同程度であることを示している。凍結乾燥工程は、製品に対する悪影響はない。
【0216】
(表12)NEUG-1およびNEUG-2の比較
【0217】
最初の臨床試験に使用したNEUG(NEUG-1)は、現在調製されているNEUG(NEUG-2)に物理化学的に同等である。製造工程および剤形に対して行った変更は、臨床有効性または安全性に影響を及ぼさなかった(データ示さず)。
【0218】
7.バッチ分析
NEUG-2の最終製剤の代表的な開発ロットに対して行われた分析の結果を図20にまとめる。表中、「ND」は、開発ロットに対して試験が「行われなかった」ことを意味する。
【0219】
8.最終製剤の安定性
NEUG-2の最終製剤のロットに対して安定性試験を開始した。1か月の時点では、どの試料にも有意な変化は観察されなかった。(データ示さず)。安定性条件は以下のとおりであった:PMTT10/6.0中にNEUGが50.0mg/mlのNEUG-2開発ロット;3mlガラスバイアル中に1.2mlsの溶液;2〜8℃、25℃/60%RH、または40℃/75%RHで保存。
【0220】
9.充填済みシリンジ中のNEUG-2溶液の試験
市販用の製品とするために、充填済みシリンジ中の溶液としてNEUG-2(PMTT、pH6.0)を提供することの実現可能性を評価するため、次の試験を行った。
【0221】
いくつかのシリンジ(例えば、Becton Dickinson製のもの)は、製品および/または緩衝剤と相互作用し得る二種類の主要な賦形剤であるタングステンおよびシリコーンを含むことが知られている。シリンジの賦形剤および製品の相互作用を評価するため、シリコーン油移動試験ならびにシリコーン添加およびタングステン添加試験、そして充填済みシリンジの長期安定性試験を含むいくつかの試験を行った。液体状態のCG10639(NEUG-2)の安定性の評価に使用した方法には、目視、SE-HPLC、RP-HPLC、IE-HPLC、バイオアッセイなどが含まれる。
【0222】
結果をまとめると、三種類の異なる用量(500、1000、および2500ppb)で五種類の形態のタングステン(タングステン酸ナトリウム無水物、ポリタングステン酸ナトリウム、タングステン酸アンモニウム、WO3、およびWO2)を添加し、3か月にわたって2〜8℃または25℃のいずれかで保存した試料には、有意な変化は見つからなかった。SE-HPLC、RP-HPLC、およびバイオアッセイにより試料を分析した(データ示さず)。
【0223】
シリコーン適合性試験では、異なる二種類の用量のシリコーン(0.4mgまたは0.8mg)でコーティングされたシリンジ中で2〜8℃で4週間試料を保存した場合、あるいは異なる二種類の用量のシリコーンを添加し2〜8℃で3か月試料を保存した場合、製品品質に対するシリコーンの影響は見られなかった(データ示さず)。調製用緩衝剤および製剤溶液の両方におけるシリコーン油分散は適合性があり、本質的に均等なままであった(データ示さず)。2か月保管後、充填済みシリンジに対する滑り力(gliding force)の変化はなかった(データ示さず)。さらに、CG10639は、撹拌および針によるせん断に対して感受性がないようであった(データ示さず)。
【0224】
少なくとも最大9か月までの長期安定性試験は、液状のNEUG-2は、2〜8℃で充填済みシリンジ中で安定であり、少なくとも最大18か月まで4℃で安定であり、製品と接触する物質と適合性があるということを示す。充填済みシリンジ中の液状のNEUG-2の長期安定性のデータを下記表13に示す。
【0225】
(表13)2〜8℃充填済みシリンジ中の液状NEUG-2の安定性
CPF=透明、淡黄色、外来の粉粒体が本質的に含まれていない
【0226】
これらの試験に基づくと、タングステンまたはシリコーンとPMTT製剤(20mMリン酸塩、180mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH6.0)を用いたNEUG製品との間に有意な相互作用はない。
【0227】
ヒト患者におけるNEUGの臨床評価
次の実施例は、「第I相」および「第II相」と題した2つのメインセクションにおいて記載する。各相は、2つの部分、即ちA部およびB部を含む。第I相および第II相の実施例は、下記表14にまとめる。なお、NEUG-1は第I相で試験し、NEUG-2は第II相で試験した。
【0228】
(表14)ヒト臨床NEUG試験の概要
【0229】
各相は、1)目的、2)患者の特徴、3)試験薬剤、4)試験の特徴、および5)A部およびB部の結果という5つのセクションに分かれる。
【0230】
実施例10:第I相
1.目的
第I相A/B、第II相A/B試験は、骨髄抑制性化学療法剤(ドキソルビシン/ドセタキセル)が投与された被験者における、皮下投与したNeugranin(商標)(「NEUG」)(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子)の安全性、耐容性、免疫原性、薬物動態、および薬力を評価するために行った。
【0231】
第I相では、第一の試験目的は、治療により発現した有害事象の頻度、重度、および継続時間を測定し、これらをNEUG投与の時間および用量と相関させることにより、ペグフィルグラスチムと比較して、一連の可能性のある治療用量にわたって皮下投与されたNEUGの安全性プロファイルを評価することであった。
【0232】
第二の試験目的は、NEUGの薬物動態および免疫原性を評価すること、ならびにドキソルビシン/ドセタキセルが投与される患者における、好中球減少症の罹患率、重症度、および継続期間に対するNEUG投与の効果をペグフィルグラスチムと比較することである。
【0233】
第I相は、上記表2に記載のように、A部およびB部という二部構成で行った。
【0234】
2.患者の特徴
第I相では、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
【0235】
採用:
1.ドキソルビシンおよびドセタキセルを投与される予定の組織学的に確認された乳癌患者。
2.18歳以上。
3.十分な血液学的機能。
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<2.0x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.ECOG一般状態0または1
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格。
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
【0236】
不採用:
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む);試験開始に先立つ4週間以内の任意の化学療法/免疫療法;この試験におけるドキソルビシンの2度の全量サイクルを除く累積的なアントラサイクリン投与。
2.試験化学療法の6週間以内の事前のニトロソウレア(BCNU、CCNU)またはマイトマイシンCのいずれかの使用。
3.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状。
4.試験化学療法の2週間以内の事前の手術または放射線治療。
5.骨盤に対するまたは骨髄支持面積の20%を超える面積に対する、または骨髄障害に対する事前の広範囲照射。
6.造血幹細胞移植による事前の高用量化学療法。
7.試験化学療法の4週間以内の骨髄性(G-CSFまたはGM-CSF)成長因子の事前使用。
8.試験化学療法の4週間以内のエリスロポエチンの事前使用。
9.骨髄性の悪性腫瘍または脊髄形成異常の病歴。
10.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
11.既知の鎌状赤血球症。
12.成人型呼吸窮迫症候群(ARDS)の診断。
13.静脈内または経口抗生物質を必要とする現行の感染症。
14.酵母由来製品に対するアレルギーの既知の履歴。
15.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分(第2相のみ)に対する既知の過敏症。
16.妊娠女性または授乳中の女性(試験期間中は、女性は全員、確実性が90%を超える避妊法を実施するか、不妊または閉経後でなければならない)。
17.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)。
18.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性。
【0237】
次の理由で被験者をさらなる治療から外した:
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
【0238】
NEUGによる治療を停止した場合、被験者は試験にとどまり、予定した安全性およびPK評価のために任意の治験薬の最後の投与後少なくとも30日間経過観察した。
【0239】
3.試験薬剤
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-G-CSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
【0240】
NEUGは、使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給され、2〜8℃で保存される。注射用滅菌水1.1mLで再構成する際、各バイアルは、pH7.2の10mMリン酸ナトリウム、200mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80中に、NEUGを15mg/ml(バイアル毎に15mgが送達可能)含む。
【0241】
第I相で使用したNEUG製剤の組成を表1に示す。
【0242】
市販のNeulasta(登録商標)(ペグフィルグラスチム)は、皮下注射用0.6ml充填済みシリンジで供給された。各シリンジは、注射用水中に酢酸塩(0.35mg)、ソルビトール(30.0mg)、ポリソルベート20(0.02mg)、ナトリウム(0.102mg)を含む無菌、透明、無色、防腐剤無添加の溶液(pH4.0)にペグフィルグラスチムを6mg(タンパク質重量に基づく)含む。USP。
【0243】
NEUG(50、150、300、もしくは450μg)またはNeulasta(登録商標)(ペグフィルグラスチム)(6mg)を皮下投与した。
【0244】
4.試験の特徴
a.試験スケジュールおよび継続期間
この試験は、ドキソルビシン/ドセタキセルを投与される予定である乳癌患者62人において行われた、ヒト初回投与、多施設、非盲検、非対照、逐次用量漸進試験であり、続いて対照無作為化試験を行った。この試験は、二部構成であった。A部は、13人の被験者、4つの用量コホート(50、150、300、または450μg/kg)における逐次用量漸進試験であり、50、150、および450μg/kgのコホートのそれぞれには3人の被験者で、300μg/kgのコホートには4人の被験者で、無作為化の実験B部を行う前に安全性を評価した。
【0245】
A部では、被験者は、細胞毒性を有する化学療法剤の非存在下で、安全性および好中球絶対数(「ANC」)に対する効果の初期評価のために、化学療法の開始(サイクル0)の少なくとも2週間前にNEUGの最初の投与を受けた。最低2週間の経過観察後、被験者は、サイクル0のNEUGに関連すると考えられる用量制限的な有害事象がなく、被験者がすべての適格基準を満たす場合、サイクル1およびサイクル2において化学療法後に同じ用量でNEUGの投与を受けた。
【0246】
A部では、用量制限毒性(DLT)は、グレード2骨痛を除いて、試験薬剤と、場合によっては関連する、恐らく関連する、または明確に関連すると考えられるグレード2以上の臨床的に有意な有害事象として規定された。各A部コホート内では、実験に参加した各被験者に対する最初の試験薬剤の投与は、急性有害事象をモニタリングするために、最低24時間隔てた。
【0247】
次の用量レベルまで漸進させる決定は、所与のコホート中の被験者全員についてNEUGの最初の投与後少なくとも7日間の安全データの検討に基づいて行った。被験者3人がだれもDLTを経験しない場合は、次の用量レベルにおいて被験者3人を加えて用量漸進を継続した。所与のコホートにおいて被験者3人の1人がDLTの兆候を示した場合、別の被験者3人をその用量レベルで補充し、1コホート当たり全部で6人の被験者になる。被験者6人の1人のみにDLTが起こった場合、用量漸進を続けた。被験者6人の2人にDLTが起きた場合、用量漸進を停止し、それ以上NEUG治療薬は投与しなかった。
【0248】
残りの被験者は、予定された安全性、薬物動態、および薬力学的評価を完了した。
【0249】
最初のA部コホートにおいて安全性が実証された後、B部を行った。B部では、被験者は3つの無作為化並行治療群の1つに分けられ、NEUG300μg/kg(n=20)、NEUG450μg/kg(n=21)、または承認された用量であるペグフィルグラスチム6mg(n=10)を試験化学療法の約24時間後に投与した。
【0250】
下記表15および表16は、第I相A部およびB部における被験者の割付をまとめたものである。図26は、第I相A部およびB部の化学療法サイクルを示す。
【0251】
(表15)第I相における被験者の割付
【0252】
(表16)第I相における被験者の割付
【0253】
b.第I相A部およびB部中の併用療法
化学療法
この実験の化学療法レジメンは、最大2回までの21日サイクルを、治療の1日目に静脈注射により逐次投与するドキソルビシン50mg/m2およびドセタキセル75mg/m2からなった。
【0254】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1.5x109/Lであり、血小板が>100x109/Lでなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることもあり得た。
【0255】
ドキソルビシンおよびドセタキセルの組み合わせは、乳癌患者において有意な臨床上の活性を有すると報告されている。しかしながら、この組み合わせは、骨髄抑制性であり、他の標準的なレジメンよりも高い割合でグレード3またはグレード4の好中球減少症が起きる。
【0256】
CSFを添加しても、ドキソルビシンおよびドセタキセルの組み合わせは、患者の79%にグレード4好中球減少症誘発し、9〜18%の割合で発熱性好中球減少症を誘発する。このドキソルビシン/ドセタキセルレジメンは、好中球減少症およびその合併症を予防するための新規な薬剤の研究に使用されている。したがって、ドキソルビシンおよびドセタキセルの組み合わせは、NEUGのような新規の薬剤の可能性を研究するのに適切な化学療法レジメンである。
【0257】
ドキソルビシン
薬理学的データ
塩酸ドキソルビシンは、DNAおよびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウス(Streptomyces peucetius var caesius)から得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
【0258】
医薬データ
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
【0259】
副作用および毒性
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。著しい脱毛症および中程度の悪心/嘔吐は、予想される毒性である。偶発的な溢出の部位での局所的な皮膚および組織損傷を生ずる溢出反応、口内炎、皮膚(特に爪床)の色素過剰症、ならびに過去に照射した部位での「メモリ」現象が報告されている。
【0260】
ドセタキセル
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
【0261】
医薬データ
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
【0262】
副作用および毒性
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
【0263】
激しい過敏反応を経験する患者には再投薬すべきではない。下に概説するように、ドセタキセルが投与される患者には、コルチコステロイドを前投与するのがよい。
【0264】
軽度から中程度の肝機能障害では、代謝が27%遅延し、全身暴露(AUC)が38%増加する。ドセタキセルは、SGOTおよび/またはSGPTが正常範囲の>1.5倍、およびアルカリフォスファターゼが正常範囲の>2.5倍の患者には投与すべきではない。体液鬱滞が、コルチコステロイドを前投与したにもかかわらず、第III相試験における患者の17%(中程度)および6%(重度鬱滞)で起こった。重度の知覚神経症(感覚異常、錯感覚、疼痛)が観察されている。
【0265】
予想される副作用としては、最下点が約9日間あり15〜21日目までには回復する骨髄抑制(主に白血球減少症)が挙げられる。脱毛症、爪および皮膚の変化、口内炎、筋肉痛/関節痛、悪心/嘔吐、ならびに低血圧が報告されている。
【0266】
化学療法剤の用量、投与、および用量の変更
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
【0267】
ドキソルビシンは、管外溢出による傷害を回避するため、点滴用静脈ラインまたは中心静脈カテーテルのサイドアームを介してIVボーラスにより50mg/m2の用量で投与した。
【0268】
75mg/m2のドセタキセルは、0.9%食塩水または5%デキストロース溶液250mLで希釈し、ポリエチレンライニングを有する輸液セットを介して約1時間にわたって静脈内投与した。バイタルサインは、ドセタキセル点滴の直前および直後に得た。
【0269】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1500/mm3であり、血小板が>100,000/mm3でなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることもあり得た。グレード3〜4の非血液毒性、グレード3〜4の感染エピソード、またはグレード4の血小板減少症に対しては化学療法剤の用量を25%減少させた。
【0270】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者は試験治療から除外されたが、経過観察は完了した。
【0271】
化学療法剤の前投薬
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
【0272】
制吐薬または他の前投薬(例えば、H2拮抗薬)の使用および選択は、治療を行う医師の裁量に任せた。
【0273】
禁止薬剤
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けないのがよい。
1.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
2.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
3.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療された場合、NEUGの投与に適格ではなく、試験を中断した。
【0274】
許可薬剤
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症は、CRFの適切なページに記録した。
【0275】
抗生物質
被験者は全員、感染の可能性を減らすため、化学療法の各コースに続いて予防用経口抗生物質(例えば、シプロフロキサシン)を投与された。発熱性好中球減少症または持続性の重度の好中球減少症(ANC<0.5x109/L、≧5日)が起こった場合、被験者は、治療不成功と考えられ、試験から除外され、経過観察を完了し、研究者の裁量による成長因子支持を含む標準的な支持療法すべて受けた。
【0276】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者も試験治療から除外され、そして経過観察を完了した。
【0277】
c.安全性評価
NEUGの安全性は、有害事象(「AE」)の種類、頻度、および重度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準に基づいて等級分けした(NCI-CTCAEバージョン3.0、2003年12月12日)。有害事象(重度有害事象「SAE」を含む)は、試験薬剤の投与開始から任意の試験薬剤の最後の投与後30日まで記録した。実験的評価は、評価スケジュール(Schedule of Assessments)で概説されているように得た。グレード4好中球減少症毒性の場合には、ANCが>500になるまで毎日実験的評価を得た。被験者の次の治療サイクルが遅延した場合(そして最後の処置サイクルの後)、ANCが>1500になるまで、鑑別を伴う全血球計算(CBC)を毎週少なくとも2回行った。
【0278】
5.第I相A部およびB部の結果
a.概論
統計的方法:
安全性、薬物動態(PK)、薬力(PD)、および免疫原性パラメータに関連するデータは、記述統計法を用いて分析した。
【0279】
有害事象の頻度および重度、ならびに実験室毒性の等級分けに関して、数および割合を示す。
【0280】
効能分析には、グレード4およびグレード3〜4の好中球減少症の発生率および持続期間、最下点ANC、最下点ANCまでの時間、回復するまでの時間(ANC>0.5x109/LおよびANC>1.0x109/Lまで)、ならびに発熱性好中球減少症の発生率が含まれた。
【0281】
この試験には、サンプルサイズを選ぶためには厳密な統計的検出力要件は使用しなかった。5%有意水準でペグフィルグラスチムと比較してNEUGが劣っていないことを示す80%検出力を用いた試験では、一治療群当たり被験者約37人を必要とすると計算された。これは、主に安全性に関して行った第l/2a相試験であったので、効果が検出されるために必要なサンプルサイズは適切なものよりも大きいと決定された。このように、効能傾向を評価した。
【0282】
内訳/個体群統計:
合計13人の被験者が、A部(実験において用量が逐次的に漸進する部分)に参加した。合計51人の被験者がB部に参加し、無作為にNEUG300μg/kg(n=20)、NEUG450μg/kg(N=21)、またはペグフィルグラスチム6mg(n=10)に分けられた。
【0283】
b.試験結果
最初の用量設定試験では、化学療法剤の非存在下で、NEUGは、良好な耐容性を示し、予想されたようにANCが上昇し、これは2日目と4日目の間にピークに達し、14日目までに正常に戻った(図23)。
【0284】
A部では、50μg/kgNEUG投与群中の被験者3人全員および450μg/kgNeugranin投与群中の被験者1人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。B部では、300μg/kgNEUG投与群中の被験者1人および450μg/kgNEUG投与群中の被験者2人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。ペグフィルグラスチム群中の被験者1人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。
【0285】
c.免疫原性
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目での投与前および治療訪問の終わり(最後の投与の少なくとも15日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得た。
【0286】
A部およびB部の両方の治療の終わりをもって被験者全員に対する試験を完了した。試料はすべて、NEUGに対する抗体には陰性であった。
【0287】
d.有害事象
A部では、用量制限毒性(DLT)は、グレード2髄骨痛を除いて、試験薬剤と、場合によっては関連する、恐らく関連する、または明確に関連すると考えられるグレード2以上の臨床的に有意な有害事象として規定された。
【0288】
A部のコホートのいずれもサイクル0ではDLTに達しなかった。NEUG投与に関連して、骨痛と既存の高血圧の悪化(後者はNEUG投与の7日後に起きた)という2つの有害事象のみ報告された。いずれの事象も後遺症なく解消した。
【0289】
NEUG治療被験者41人のうち31人は少なくとも1つの有害事象を経験した。NEUGおよびペグフィルグラスチム治療被験者中のAEの発生率は同程度であった(それぞれ75.6%および70%)。
【0290】
B部で一般に報告される有害事象(被験者全員のうちの5%以上のAE)をまとめたものを表17に示す。
【0291】
(表17)第1相B部集団における治療関連有害事象のまとめ
1関連=場合によっては関連している、恐らく関連している、または明確に関連していると考えられる
2非関連=恐らく関連していないか、関連していないと考えられる
【0292】
NEUGに関連すると考えられる最も多く報告される有害事象は、すべてのG-CSF製品に伴う典型的な有害反応である骨痛であり、これは上記表に記載の4人および450μg/kg投与されるA部被験者1人の5人の患者において報告された。すべての事例で、骨痛は、強度がNCI-CTCAEグレード1〜2であり、持続期間としては一過性で、後遺症なく解消した。アルカリフォスファターゼおよび尿酸におけるグレード1上昇は、サイクル0におけるNEUGの投与後に起こり;これらの事象は、研究者によって臨床的に有意でないと考えられ、処置せずに解消した。これらは、G-CSF(例えば、Neulasta(登録商標))を投与される患者における予想される作用である。
【0293】
化学療法サイクル中の他の一般に報告される有害事象(悪心、嘔吐、脱毛症、口内炎)は、ドキソルビシン/ドセタキセルレジメンが投与される患者において予想される有害事象と一致していた。
【0294】
大多数の報告されたAEは、NCI CTCグレード1またはグレード2の重篤度であった。4つのAEは、重篤な有害事象として報告された。被験者2人(1人は150μg/kg投与されもう1人は450μg/kg投与された)は嘔吐のために入院し、これらの被験者の1人は、次の化学療法サイクルで二度目のSAEを経験した;嘔吐の程度は軽かったが、入院の原因となったかまたは入院を長引かせた。450μg/kg投与された3人目の被験者は、発熱性好中球減少症のため入院した。当該事象は、NEUGと無関係であると考えられた。
【0295】
e.薬物動態
NEUGが投与される被験者は全員、試験期間中に渡って血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン4.1以上)を用いてPK分析に供した。
【0296】
以下のPKパラメータを得た:曲線下面積(AUC0-∞)、クリアランス(CL/F)、分布容積(Vz/F)、最大濃度(Cmax)、吸収半減期(t1/2、abs)、排出半減期(t1/2、elim)、および平均滞留時間(MRT)。薬物動態データは、プロトコル中で用いた用量範囲にわたって直線性について評価した。
【0297】
サイクル0(化学療法前)からの薬物動態パラメータは表18にまとめてあり、サイクル0のPKプロファイルは図27に示してある。
【0298】
(表18)ヒト被験者におけるNeugranin薬物動態(第1相サイクル0)
【0299】
最大血清NEUG濃度および時間濃度曲線下面積により測定される薬物暴露は、用量依存的に増加した。最初の50μg/kg用量コホート中の被験者の血清濃度は、一貫して量子化の下限(6.3ng/mL)未満であった。Tmaxは、150から450μg/kgまでのすべての用量で6〜24時間の範囲であった。Cmaxは、150μg/kgの用量で72.7±59.7(平均±SD)ng/mLから450μg/kgの用量で294±351ng/mLまでであった。対応して、AUC0-∞は、150mcg/kgの用量で1758±1675ng/mL*hrから450μg/kgの用量で10131±9563ng/mL*hrであった。サイクル1の範囲は類似していた。NEUGの平均排出半減期は、14〜30時間の範囲であった。
【0300】
「試験の特徴」(上のセクション4)で記載したように、A部の被験者は、細胞毒性を有する化学療法の非存在下で、安全性および好中球絶対数(「ANC」)に対する影響の初期評価のために、化学療法の開始(サイクル0)の少なくとも2週間前にNEUGの最初の投与を受けた。最低2週間の経過観察後、サイクル0においてNEUGに関連すると考えられる用量制限的な有害事象がなく、被験者がすべての適格基準をみたす場合は、被験者は、サイクル1およびサイクル2における化学療法後に、同じ用量のNEUGの投与を受けた。NEUGは、化学療法剤投与の24時間後に投与された。図24Aおよび図24Bは、サイクル1およびサイクル2中でNEUGが投与された被験者のANC数およびWBC数を示す。
【0301】
f.B部の用量の薬力学および設定
試験の第I相A部からのデータの分析によって次の所見が得られた:
1.NEUGは、サイクル0(化学療法前)においてWBCおよびANCの用量依存的な上昇を誘導する(図24Aおよび図24Bにおけるサイクル0データ参照)。
2.サイクル0におけるANCの増加は、等モル量のペグフィルグラスチムの病歴データと同程度であった
3.予想されたように、化学療法後にWBCおよびANCは低下する
4.最下点ANCからの回復は、用量依存依存的であると考えられる
5.ANCおよびWBCは、15日目までに正常値まで回復した
【0302】
A部でのすべての用量レベルでの安全性のこれらの所見および実証に基づいて、B部の評価のための選択した用量は300および450μg/kgであった。上に記載のように、被験者は、NEUG300μg/kg、NEUG450μg/kg、またはペグフィルグラスチムを認可された固定投与量である6mgに無作為に分けた。被験者は、ドキソルビシン/ドセタキセルの投与1日後にNEUGまたはペグフィルグラスチムを投与された(2サイクルにわたる投与、21日隔てて)。B部のデータは、集団のサイクル1ANCプロファイルを含む。結果を図28および下記表18にまとめる。
【0303】
グレード3およびグレード4好中球減少症の発生率およびサイクル1中のANCプロファイルは、表19に示すように、治療した被験者51人中の48人で測定された。なお、予防的なG-CSF治療が無いと、ドキソルビシン/ドセタキセルで治療した患者の70〜80%が平均で5日間の持続期間を有するグレード4好中球減少症になる。
【0304】
(表19)化学療法のサイクル1後の第I相B部におけるグレード4好中球減少症の発生率および持続期間
【0305】
治療群の平均ANC曲線を図28に示す。
【0306】
NEUGは、グレード3、グレード4、および発熱性好中球減少症の治療に効果がある。この化学療法レジメンに対してG-CSF治療がないと、発熱性好中球減少症の発生率は約40%になる。ANCの用量相関性の上昇およびドキソルビシン/ドセタキセルで予想されるよりも低い好中球減少症の割合がNEUGの投与後に観察された。NEUGに起因する予想外または重篤な有害事象はなかった。
【0307】
グレード3およびグレード4好中球減少症の発生率は、ペグフィルグラスチム(Neulasta(登録商標))が投与された患者よりも300μg/kgのNEUGが投与された患者でより高く、正常なANCまで回復する速度も、ペグフィルグラスチムが投与される被験者よりもNEUGが300μg/kg投与される患者でより遅いようであった。NEUGが450μg/kg投与される患者のANCプロファイルは、ペグフィルグラスチムが投与される患者と類似していたが、好中球減少症からの回復の際のANCは、ペグフィルグラスチムが投与される患者よりもNEUGが投与される患者において一般により低かった。要約すると、これらの用量のNEUGは、ペグフィルグラスチムと同様の効果をもたらすようである。
【0308】
f.PK/PDプロファイル、第I相、B部
乳癌治療のサイクル1におけるドキソルビシン/ドセタキセル投与の1日後にNEUGが450μg/kg投与された患者からのPK/PDプロファイルを図29に示す。NEUGのCmaxは、投与の1日以内に達成され、10日目までに検出できないレベルにまで徐々に低下した。NEUGの投与後に、ANCは4日目までにピークにまで上昇し、ANCは、ドキソルビシン/ドセタキセルおよびG-CSF治療を受ける患者において予想される通りに、8日目に最下点まで低下し、10日目に正常値にまで戻る。12日目までには、ANC値は正常範囲にあり、NEUGは検出できない。なお、予防的なG-CSF治療を受けない患者では、最下点ANCの期間およびANCが回復するまでにかかる時間はずっと長い(例えば、5〜7日)。450μg/kg投与後、NEUG排出半減期のメジアンは約30時間であり、これに対し、ペグフィルグラスチムの標準用量では15〜80時間と報告されている。
【0309】
g.NEUGとペグフィルグラスチムとの間のさらなる差異
好中球減少症からの回復を促進させる効果における試験用量でのNEUGとペグフィルグラスチムとの間のより詳細な差異は、治療のサイクル1における個々のANCプロファイルを比較すれば明らかである。すべての群のピークANCは非常に類似しており、NEUGが300μg/kg投与された被験者の最下点ANCは、NEUGが450μg/kg投与された被験者より低く、ペグフィルグラスチムが投与された被験者のANC最下点は平均して最も高かった。最下点ANCからベースラインまでの回復がすべての治療群で14日目までに起きたが、NEUGが450μg/kg投与された被験者よりもNEUGが300μg/kg投与された被験者では遅く、ペグフィルグラスチムが投与された被験者で最も速かった。
【0310】
類似の化学療法前の投与を伴うペグフィルグラスチム実験の入手可能な公表済みデータを、予定したサイクル0(化学療法前)までの第I相を完了した患者からNEUGのPK/PDデータと比較した。この比較の結果は以下のとおりであった:
1.NEUG用量が150μg/kgのEmax(最大観測ANC)は、サイクル0におけるペグフィルグラスチム用量30μg/kg(確認された有効なペグフィルグラスチム用量である100μg/kgに対して効能で劣ることが後に示された用量)と一致する。
2.Neugranin用量が300および450μg/kgのEmaxは、ペグフィルグラスチム用量が100μg/kgのサイクル0レベルとより一致していた。
3.NEUG300μg/kgおよび450μg/kgのCmaxのメジアンおよびEmaxのメジアンはほぼ同じであり、したがって、Cmaxによって引き続きEmaxが予測される。
4.ANC増加は、等モル量のペグフィルグラスチムの出版済みデータと同程度であった。
【0311】
上に記載したように、動物およびヒトにおけるPK/PD評価は、等モル基準で投与したNEUGおよびペグフィルグラスチム用量等価性の推定値と一致していた。マウスでは、同等のAUCANCが、ペグフィルグラスチムより7.7倍高い用量で達成された。アルブミンがNEUGの分子量に著しく寄与し、Neulasta(登録商標)(ペグフィルグラスチム)が、rhG-CSFの重量(ペグフィルグラスチム中のポリエチレングリコールの寄与を含まない)に基づいて投与されるので、4.5倍の用量のNEUG(重量基準)は、等量のNeulasta(登録商標)(ペグフィルグラスチム)と同程度に効果的であると予測される。動物における効能データは、ペグフィルグラスチムの4.5〜7.7倍と一致した(ペグフィルグラスチム1mg=NEUG4.5〜7.7)。非臨床上の安全性および効果のデータは、この用量推定値と一致しており、入手可能な臨床データと検討すると、臨床評価のための用量を選ぶ際の根拠になる。
【0312】
h.第I相の結果
第I相薬物動態評価の結果は次のとおりである。
NEUGは、サイクル0およびサイクル1で、150μg/kg、300μg/kg、および450μg/kgの用量にてNEUGで処理されたすべての被験者からの血清試料において検出された。
サイクル1では、NEUGは、150mg/kg、300μg/kg、および450μg/kg用量群において、ほとんどの被験者(45/50でサンプリングされた)で最大144時間まで検出された。実質的に、サイクル間での薬剤蓄積は観察されなかった。
薬物暴露は、各用量群でサイクル1およびサイクル0(前化学療法)においてより高かった。サイクル1におけるNEUGへの暴露の増加は、受容体が仲介するG-CSFクリアランスにおいて役割を果たす好中球の数の減少による可能性がある。
サイクル1におけるNEUGの排出半減期のメジアンは、300μg/kg用量群で約36時間であり、450μg/kg用量群で30時間であった。排出半減期は、フィルグラスチムで3〜4時間であり、ペグフィルグラスチムで、用量に応じて42〜67.5時間であると報告されている。
用量間の統計的有意差は、最大血清濃度(tmax)までの時間および吸収半減期(t1/2、abs)に見られた。これらのパラメータはいずれも、NEUG用量の増加と共に増加した。他の用量正規化PKパラメータは、用量間の統計的有意差を示さなかった。
【0313】
実施例11:第II相
試験の第II相は、ドキソルビシン/ドセタキセルを最大4回まで投与した乳癌を有する被験者334人で行われた対照無作為化試験であった。試験は、臨床現場約50カ所で行われ、皮下投与したNEUGとペグフィルグラスチムとの安全性および効果を評価するための二元配置無作為化パイロットフェーズ、その後の、被験者がパイロットフェーズに基づいて選ばれるペグフィルグラスチムおよび2つの良好な耐性用量のNEUGに無作為に分けられる(1:1:1)メインフェーズからなった。主要評価項目である化学療法サイクル1中の重度(グレード4)の好中球減少(DSN)の持続期間に関してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、メインフェーズのサンプルサイズを検出した。試験の設計を下に概略的に示す。
【0314】
1.目的
第II相の第一の目的は、ペグフィルグラスチムに対して同等の効果を示すNEUG用量を選択すること、およびNEUGでの治療後の化学療法のサイクル1において重度の好中球減少(DSN)の持続期間を評価することであった。第二の目的は、サイクル2〜4におけるDSNを評価すること、好中球絶対数が回復するまでの時間およびサイクル1〜4における発熱性好中球減少症の割合を評価すること;ならびにNEUGの安全性、耐容性、薬物動態(サイクル1において)、および免疫原性を評価することであった。
【0315】
2.患者の特徴
第II相に関連して、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
【0316】
採用:
1.ドキソルビシンを60mg/m2およびドセタキセルを75mg/m2投与される予定の組織学的に確認された乳癌患者。
2.18歳以上
3.十分な血液学的機能:
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<1.5x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.米国東海岸癌臨床試験グループ(「ECOG」)一般状態0〜2
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
【0317】
不採用:
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む)
2.この試験におけるドキソルビシンの4度の全量サイクルを除く累積的なアントラサイクリン投与
3.試験化学療法剤の30日以内の事前の化学療法/免疫療法(ニトロソウレア(BCNU、CCNU)またはマイトマイシンCの試験化学療法剤の6週間以内)
4.トラスツズマブ(ハーセプチン)併用療法
5.過去30日での治験薬の投与
6.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状
7.試験化学療法の2週間以内の事前手術
8.試験化学療法の4週間以内の事前手術(骨転移のためのスポット照射を除く)
9.造血幹細胞移植による事前の高用量化学療法
10.試験化学療法の4週間以内のG-CSF、GM-CSF、またはエリスロポエチンの事前使用
11.試験化学療法の72時間以内の抗生物質の全身投与
12.骨髄性の悪性腫瘍または脊髄形成異常の病歴
13.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
14.既知の鎌状赤血球症
15.成人型呼吸窮迫症候群(ARDS)の診断
16.酵母由来製品に対するアレルギーの既知の履歴
17.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分に対する既知の過敏症
18.妊娠女性または授乳中の女性。(子宮が無傷である女性は全員スクリーニングされて血清妊娠テストが陰性でなければならない。不妊でないかまたは非閉経前の女性は全員、試験期間中および試験薬剤の最後の投与後の30日間医学的に認められた避妊法を実施しなければならない。)
19.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性
20.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)
【0318】
次の理由で被験者をさらなる治療から外した:
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
【0319】
試験薬剤による治療を停止した場合、試験にとどまった被験者は、予定した安全性およびPK評価のために試験薬剤の最後の投与後少なくとも30日間経過観察した。
【0320】
3.研究薬剤
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-GCSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
【0321】
NEUGは、使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給され、2〜8℃で保存される。注射用滅菌水1.0mlで再構成する際、各バイアルは、20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80、pH6.0中に、NEUGを50mg/ml(バイアル毎に50mgが送達可能)含む。
【0322】
第II相で使用した製剤の組成を表11に示す。第I相および第II相で使用したNEUG製剤間の差異を下記表20に示す。
【0323】
(表20)cGMP製剤の比較
【0324】
第I相で使用された製剤はきわめて安定しており、少なくとも2年間の保存期間を有していた。試験により、より大きなイオン強度およびより低いpHが、より高い濃度(>25mg/mL)でAPIを安定させることが示された(データ示さず)。このために、第II相の製剤は、より低いpH(6.0対7.2)およびより高いリン酸塩濃度(20対10 mM)を有する。強制分解試験は、この製剤が、激しい振とう、凍結融解の繰り返し、および濃度により誘導される凝集から液体状態の薬剤成分を保護することを示した。第II相の製剤の凍結乾燥はまた、良好に形成されたケーキを生じる。
【0325】
市販のNeulasta(登録商標)(ペグフィルグラスチム)は、皮下注射用0.6ml充填済みシリンジで供給される。各シリンジは、注射用水中に酢酸塩(0.35mg)、ソルビトール(30.0mg)、ポリソルベート20(0.02mg)、ナトリウム(0.102mg)を含む無菌、透明、無色、防腐剤無添加の溶液(pH4.0)にペグフィルグラスチムを6mg(タンパク質重量に基づく)含む。USP。
【0326】
NEUG(30、40、50、もしくは60mg)またはNeulasta(登録商標)(ペグフィルグラスチム)(6mg)を皮下投与した。
【0327】
用量の論理的根拠
第I相からのデータは、300および450μg/kgのNEUG用量が安全であり良好な耐容性があることを示した。さらに、ペグフィルグラスチムの承認された固定用量と比較して、NEUGのいずれの用量も、細胞毒性を有する化学療法を受ける乳癌患者におけるANCプロファイルに対して同様の効果をもたらした。ANCプロファイルのAUCは、効果の一点計測値(single-point measure of effect)としての役割を果たす。AUCANCについてはこれらの治療群間で統計的有意差はなかったが、450μg/kg群のAUCは、300μg/kg群で観察されたものよりもわずかに高く、ペグフィルグラスチム群で観察されたものとほとんど同じである(図30)。入手可能なデータに基づくと、NEUGは、300μg/kgではペグフィルグラスチムより効果が低く、450μg/kgでペグフィルグラスチムと同様の効果を与えるためのおおよその最小必要用量であると推定された。
【0328】
固定用量の目的は、患者の体重にかかわらず効能および安全性をもたらすのに十分な用量を患者に提供する用量を確認することである。第I相の結果に基づくと、NEUGは450μg/kgでペグフィルグラスチムと類似の効果を与える最小必要用量であり得ると推定され、>300μg/kgが第II相でさらに評価するための最小量として設定された。NEUGの固定用量を選択するため、第II相の患者集団(乳癌)をモデリングした。40〜100kgの体重範囲を用いて、最も重い患者には30mgの固定用量で最小量(300μg/kgまたは0.3mg/kg)が与えられる一方で、約75%の患者には、40mgの固定用量で、少なくとも標的用量である450mg/kgが投与された。したがって、第II相での評価に選択した用量は、30mg、40mgおよび50mgであった。これらの用量は、平均70kgの患者にそれぞれ0.42、0.57、および0.71mg/kgの用量を提供する。
【0329】
この実験で評価された固定用量に基づいたキログラム当たりの等価用量を表21に示す。
【0330】
(表21)予測される被験者体重範囲のキログラム当たりの等価用量
【0331】
NEUGの非臨床安全性は、これらの用量での安全性の予想に対するさらなるサポートを提供する。これらの固定用量(AUCおよびCmax)での患者の暴露は、サルで良好な耐容性がある用量での暴露より低いと予想される。例えば、良好な耐性用量である1mg/kgでのサルにおけるCmaxおよびAUCは、0.45mg/kgでの患者における暴露より12倍高く、これは、患者におけるより高い用量評価およびサルにおける反復用量毒性試験でさらなる安全域が存在することを示唆しており、10mg/kg以下の用量で良好な耐容性があった。0.3mg/kgもの高いペグフィルグラスチム用量が患者において安全であることが示されている。
【0332】
4.試験の特徴
a.試験スケジュールおよび継続時間
この試験は、ドキソルビシン/ドセタキセルが最大4回まで投与される予定の乳癌患者被験者約330人で行われた対照無作為化試験であった。約45カ所の臨床現場で行ったこの試験は、パイロットフェーズおよびメインフェーズの2相からなった。
【0333】
パイロットフェーズ(A部)は、二元配置無作為化試験からなり、次の用量を順次行ってNEUG対ペグフィルグラスチムの安全性および効果を評価した:NEUG30mg(N=10)対ペグフィルグラスチム(N=5);NEUG40mg(N=20)対ペグフィルグラスチム(N=10)、およびNEUG50mg(N=20)対ペグフィルグラスチム(N=10)。さらなる試験では、NEUG60mg(N=20)対ペグフィルグラスチム(N=10)も検査し得る。A部パイロットフェーズでは、被験者は、30mgコホート中に全部で被験者10人および他の各コホートに被験者20人で、NEUGとペグフィルグラスチムとに2:1の割合で無作為に分けた。NEUGまたはペグフィルグラスチムは、各サイクルの化学療法治療の24時間後に被験者に投与した。被験者は、体重(<50kg、≧50kg-<80kg、または≧80kg)、事前の化学療法暴露、および地球上の位置に基づいて治療群中でバランスをとるために層別無作為化を用いて、治療群に割り振られた。
【0334】
パイロットフェーズに続いて、被験者255人を、ペグフィルグラスチム、およびパイロットフェーズにおいてよりもペグフィルグラスチムにより類似する効果を有する2つの良好な耐性用量のNEUGに無作為に分類した(1:1:1)(3治療群、平衡、並行無作為化相)。NEUGまたはペグフィルグラスチムは、各サイクルの化学療法治療の24時間後に投与した。被験者は、体重(<50kg、≧50kg-<80kg、または≧80kg)に基づいて治療群中でバランスをとるために層別無作為化を用いて、治療群に割り振られた。
【0335】
パイロットフェーズ中、有害事象は継続的に評価した。継続的なデータの評価によって安全性の懸念が示唆されなければ、30から50mgまで用量を増加させた。Neugranin40mgのサイクル1ANCプロファイルが、ペグフィルグラスチム患者に見られるプロファイルより劣っていると考えられ、Neugraninが50mgで安全である場合、さらなる治療群を、コホート中の患者総勢30人で、Neugranin60mgとペグフィルグラスチムとを2:1の割合で無作為に分けてもよい。
【0336】
NEUGの各用量レベルを、安全性および効能についてペグフィルグラスチムと比較する。表22は、第II相のA部およびB部の患者の割付をまとめたものである。
【0337】
(表22)第II相A部およびB部における被験者の割付
【0338】
安全性評価:
NEUGの安全性は、AEの種類、頻度、および重症度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準(NCI-CTCAEバージョン3.0、2003年12月12日)に基づいて等級分けした。
【0339】
有害事象は、試験薬剤の投与開始から任意の試験薬剤の最後の投与後30日まで記録した。重篤な有害事象(SAE)は、同意時から任意の試験薬剤の最後の投与後30日まで記録した。実験室評価は、評価スケジュールで概説されているように入手した。
【0340】
c.併用療法
化学療法
この実験の化学療法レジメンは、最大4回の21日サイクルを、治療の1日目に静脈注射により逐次的に投与するドキソルビシン60mg/m2およびドセタキセル75mg/m2からなった。
【0341】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1000/mm3であり、血小板が>100,000/mm3でなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることができた。グレード3〜4の非血液毒性、グレード3〜4の感染エピソード、またはグレード4の血小板減少症に対して、化学療法剤の用量を25%減少させることが可能であった。予防用抗生物質または他の造血成長因子の使用は試験に参加している間は禁止された。
【0342】
ドキソルビシンおよびドセタキセルの組み合わせは、乳癌患者において有意な臨床上の活性を有すると報告されている。しかしながら、この組み合わせは、骨髄抑制性であり、他の標準的なレジメンよりも高い割合でグレード3またはグレード4の好中球減少症が起きる。
【0343】
CSFを添加しても、ドキソルビシンおよびドセタキセルの組み合わせは、患者の79%にグレード4好中球減少症を誘発し、9〜18%の割合で発熱性好中球減少症を誘発する。このドキソルビシン/ドセタキセルレジメンは、好中球減少症およびその合併症を予防するための新規な薬剤の研究に使用されている。したがって、ドキソルビシンおよびドセタキセルの組み合わせは、NEUGのような新規の薬剤の可能性を研究するのに適切な化学療法レジメンである。
【0344】
ドキソルビシン
薬理学的データ
塩酸ドキソルビシンは、DNA塩基対に直接結合し(インターカレートし)、DNA合成およびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウスから得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
【0345】
医薬データ
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
【0346】
副作用および毒性
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。
【0347】
著しい脱毛症および中程度の悪心/嘔吐は、予想される毒性である。偶発的な溢出の部位での局所的な皮膚および組織損傷を生ずる溢出反応、口内炎、皮膚(特に爪床)の色素過剰症、ならびに過去に照射した部位での「メモリ」現象が報告されている。
【0348】
ドセタキセル
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
【0349】
医薬データ
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
【0350】
副作用および毒性
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
【0351】
激しい過敏反応を経験する患者には再投薬すべきではない。下に概説するように、ドセタキセルが投与される患者には全員、コルチコステロイドを前投与するのがよい。
【0352】
軽度から中程度の肝機能障害では、代謝が27%遅延し、全身暴露(AUC)が38%増加する。ドセタキセルは、SGOTおよび/またはSGPTが正常範囲の>1.5倍、およびアルカリフォスファターゼが正常範囲の>2.5倍の患者には投与すべきではない。体液鬱滞が、コルチコステロイドを前投与したにもかかわらず、第III相試験における患者の17%(中程度)および6%(重度鬱滞)で起こった。重度の知覚神経症(感覚異常、錯感覚、疼痛)が観察されている。
【0353】
予想される副作用としては、最下点が約9日間あり15〜21日目までには回復する骨髄抑制(主に白血球減少症)が挙げられる。脱毛症、爪および皮膚の変化、口内炎、筋肉痛/関節痛、悪心/嘔吐、ならびに低血圧が報告されている。
【0354】
化学療法剤の用量、投与、および用量の変更
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
【0355】
ドキソルビシンは、管外溢出による傷害を回避するため、点滴用静脈ラインまたは中心静脈カテーテルのサイドアームを介してIVボーラスにより60mg/m2の用量で投与した。
【0356】
75mg/m2のドセタキセルは、0.9%食塩水または5%デキストロース溶液250mlで希釈し、ポリエチレンライニングを有する輸液セットを介して約1時間にわたって静脈内投与した。バイタルサインは、ドセタキセル点滴の直前および直後に得た。
【0357】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者は試験治療から除外され、経過観察を完了することになっていた。
【0358】
化学療法剤の前投薬
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
【0359】
制吐薬または他の前投薬(例えば、H2拮抗薬)の使用および選択は、治療を行う医師の裁量に任せた。
【0360】
禁止薬剤
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けてはならない。
1.サイクル1化学療法の72時間以内の抗生物質の全身投与。
2.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
3.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
4.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療される場合、NEUGの投与に適格ではなく、試験を中断する。
【0361】
許可薬剤
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症、を記録した。
【0362】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験した被験者は試験治療から除外され、経過観察を完了した。
【0363】
d.薬物動態
NEUGが投与される被験者は全員、サイクル1中に血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン5.0以上)を用いてPK分析に供した。次のPKパラメータを測定した:曲線下面積(AUC0-∞)、クリアランス(CL/F)、分布容積(Vz/F)、最大濃度(Cmax)、吸収半減期(t1/2、abs)、排出半減期(t1/2、elim)、および平均滞留時間(MRT)。
【0364】
e.免疫原性
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目における投与前および治療訪問の終わり(最後の投与の約30日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得、この試料が陽性であった場合、12か月目に試料を得た。プロトコルは、被験者全員からの6か月および12か月免疫原性試料を必要とするようにその後修正された。
【0365】
5.結果
a.概論
統計的方法
サイクル1における重度の好中球減少(DSN)の平均持続期間の主要評価項目に関連してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、この実験のメインフェーズ(B部)における1治療群当たり被験者約85人のサンプルサイズは、非劣性マージンが1日で、複数回の試験(Hochberg法による)のために調整した片側有意水準が全体で0.025で、91%の検出を提供するように選択した。サンプルサイズは、2つの独立群に対する正規近似、サイクル1 DSNの治療内標準偏差として推定1.6日、およびサイクル1 DSNの主要評価項目に関して評価できない最高率20%に基づいて計算した。
【0366】
3治療群無作為化フェーズ(A部)中の被験者に基づいて、2つの選択されたNEUG用量(40mgと50mgのいずれか)およびペグフィルグラスチムの効能を比較した。
【0367】
第二の効能分析は、化学療法サイクル2から4のそれぞれにおけるDSN、サイクル1から4のそれぞれにおける最下点ANCの低さ、サイクルによるそしてすべてのサイクルにわたるFN(その時の口腔相当温度>38.2℃でANC<0.5x109/Lと規定)の割合、および全サイクルにおいてANCが>1.5x109/Lに回復するまでの時間を含む。
【0368】
第二の効能分析に関連するデータは、適切な統計的方法を用いて分析した。安全性、PKおよび、免疫原性パラメータは記述統計法を用いて分析した。
【0369】
有害事象の頻度および重度、ならびに実験室毒性の等級分けに関して、数および割合を示す。
【0370】
効能の測定
全血球計算値(「CBC」)は、1日目、3日目、そして5日目以降、最下点後にANCが>2.0x109Lになるまで、その後、週2回、および治療の終わりに得た。
【0371】
b.第II相のA部の効能
試験のパイロットフェーズに参加した被験者78人のうち、被験者13人が試験を完了せず、3人(27.3%)がNEUG30mgで治療され、3人(14.3%)がNEUG40mgで治療され、3人(15.0%)がNEUG50mgで治療され、4人(15.4%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および研究者の判断によるもの(被験者3人)であった。NEUGを30mg投与した被験者の1人は、有害事象(糖尿病性足病変)によるとりやめであった。
【0372】
重度の好中球減少症の発生率および重度の好中球減少(DSN)の平均継続期間は、各化学療法サイクルの治療群にわたって類似していたが、ANC回復までの時間および発熱性好中球減少症の発生率は、NEUG30mgが、NEUG40mg、NEUG50mg、またはペグフィルグラスチムほど効果的ではないことを示唆した。
【0373】
サイクル1中に、発熱性好中球減少症を経験する被験者の割合は、NEUG30mg、40mg、50mg、およびペグフィルグラスチム群でそれぞれ20.0%、9.5%、10.0%、および8.0%であった。発熱性好中球減少症は、サイクル2〜4においてさらに被験者3人のみに観察された(NEUG30mg、NEUG40mg、およびペグフィルグラスチム群で1人ずつ)。図5は、NEUG30またはペグフィルグラスチムのいずれかを投与された患者のサブセットで、後にグレード4好中球減少症を発症した者のANCプロファイルを示す。
【0374】
サイクル1では、平均DSNは、NEUG30mg(0.9日)、NEUG50mg(1.1日)、およびペグフィルグラスチム(0.9日)に関しては類似していた。平均DSNは、NEUG40mgでは他の3つの治療よりもわずかに長かった(1.6日)が、治療間の違いはすべて1日未満で、メインフェーズにおいて同等な治療を検討する基準である。DSN中間値は、4つの治療群すべてで0日または1日であった。
【0375】
グレード3またはグレード4好中球減少症の発生率および持続期間に関する要約データは、類似のパターンをたどる、つまり、NEUG30mg、NEUG50mg、およびペグフィルグラスチム群は、類似の結果になった。その一方で、グレード3またはグレード4好中球減少症の発生率および持続期間は、NEUG40mg群で他の治療群よりわずかに高かった。パイロットフェーズ(A部)の被験者数はきわめて少なく、観察された差異は統計的に有意ではなかった。NEUG40mgおよびNEUG50mgは、B部(試験の3治療群無作為化フェーズ)におけるさらなる評価のために選択された。
【0376】
c.第II相のB部の効能
試験のメインフェーズに参加した被験者256人のうち、被験者18人が試験を完了せず、10人(11.6%)がNEUG40mgで治療され、5人(6.0%)がNEUG50mgで治療され、3人(3.5%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および2人の死者を含むAE(被験者4人)であった。研究者は、AEはすべて、試験薬剤または化学療法と関連しないと見なした。メインフェーズでは、NEUG40mgが投与された被験者の1人(1.2%)が、試験薬剤で治療される前に除かれた。
【0377】
サイクル1における重度の好中球減少症の発生率および持続期間を表23にまとめている。
【0378】
(表23)第II相B部:サイクル1における重度の好中球減少症の発生率および持続期間
【0379】
重度の好中球減少症の発生率は、ペグフィルグラスチム群で58.1%からNEUG50mg群で65.5%の範囲であった。治療効果は、統計的に有意ではなかった(p=0.559)。治療群は、サイクル1 DSNに関して、NEUG40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ平均値が1.0、1.3および1.2日であり同程度であった。NEUGとペグフィルグラスチムとの差異の95%および97.5%両側信頼区間は、両方のNEUG用量で正確に1日未満だった。この分析は、ペグフィルグラスチムに対してNEUGが劣っていないことを実証した。治療サイクルにわたって、重度の好中球減少症およびグレード3またはグレード4好中球減少症の発生率は、サイクル1においてよりもサイクル2〜4において低かった。グレード3またはグレード4好中球減少症の平均DSNおよび平均持続期間は、サイクル1においてよりもサイクル2〜4において小さかった。治療サイクル内では、治療は類似しており、処理効果は、任意の化学療法サイクルにおけるこれらパラメータのどれについても有意な差異はなかった。
【0380】
体重で四分した患者でDSNを比較して、NEUGの固定用量がすべての体重の患者に十分なサポートを提供するかどうかを決定した。当該結果は、体重サブグループの平均DSNにおける有意差は無く(表24)、すべての体重群で十分にサポートされたことを示す。
【0381】
(表24)体重別の、重度の好中球減少症のサイクル1での持続期間(日数)
【0382】
すべてのサイクルにおける発熱性好中球減少症を表25にまとめる。サイクル1中、発熱性好中球減少症を経験する被験者の割合は、NEUG40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ、被験者2人(3.5%)、被験者5人(6.0%)、および被験者2人(2.3%)であった。発熱性好中球減少症は、サイクル2〜4中でさらに被験者3人のみ観察された(NEUG40mg群で被験者2人およびペグフィルグラスチム群で被験者1人)。治療効果は、どの化学療法サイクルでも統計的に有意ではなかった。
【0383】
(表25)サイクル1〜4における好中球減少症の発生率
【0384】
サイクル2〜4において重度の好中球減少症の持続期間中の治療間で有意差はなかった(表26)。
【0385】
(表26)サイクル2〜4における重度の好中球減少症の平均持続期間
【0386】
ANC回復(>1.5x109/L)までの平均時間は、Neugranin40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ、2.0、2.1、および2.6日であった(表27)。ANC最下点の低さまたは最下点までの時間に関して治療群間で有意差はなかった。
【0387】
(表27)ANC最下点、ANC最下点までの時間、および回復までの時間
【0388】
d.第II相B部の薬物動態
血清Neugranin濃度は、6.312ng/mLの定量範囲の下限値(LLQ)で、有効なサンドイッチELISAを用いて測定した。薬物動態パラメータは、吸収半減期以外はノンコンパートメントモデル法を用いて計算し、吸収半減期は、一次吸収、一次排出1コンパートメントモデルを用いて測定した。モデリングは、WinNonlin Professional(バージョン5.0.1)を用いて行った。血清NEUG濃度は、第II相においてNEUGで治療されたすべての被験者における化学療法サイクル1で測定した。第II相A部では、NEUGの排出半減期の中間値は、30mg投与群で33時間であり、40mg投与群で46時間であり、50mg投与群で18時間であった(表28)。B部では、NEUGの排出半減期の中間値は、40mg投与群で40時間であり、50mg投与群で39時間であった(表29)。A部中は、PKサンプリングは、B部と比較して(投与前、3日目、5〜8日目)、より頻繁に行われた(投与前、3時間、6時間、12時間、24時間、3日目、5〜9日目、11日目)。
【0389】
(表28)第II相A部の治療による排出半減期の中間値
【0390】
(表29)第II相B部の治療による排出半減期の中間値
【0391】
血清ペグフィルグラスチム濃度は、第II相において、ペグフィルグラスチムで治療された被験者全員における化学療法サイクル1で、有効なサンドイッチELISAを用いて測定した。A部では、ペグフィルグラスチムの排出半減期の中間値は、約40時間であった。B部では、ペグフィルグラスチムの排出半減期の中間値は、約50時間であった。排出半減期は、フィルグラスチムで3〜4時間、ペグフィルグラスチムで42〜67.5時間(用量に応じて)であると報告される。
【0392】
e.免疫原性
試験参加者間で、Neugraninで治療された被験者において、確認された抗G-CSF/新エピトープ抗体反応1つおよびペグフィルグラスチム治療群において抗G-CSF反応が1つ、すなわちそれぞれ0.5%および0.9%があった(表30)。どちらの場合も、被験者は、前投与試料中で非特異的結合の上昇があった。
【0393】
(表30)NEUGおよびペグフィルグラスチムに対するG-CSFに特異的な治療により発現した免疫反応の概要
【0394】
NEUG治療後、患者において、非常に低レベルの確認された陽性抗体が見られ、反復投与後の応答の程度に明確な増加はなかった(データ示さず)。ペグフィルグラスチムで治療された患者では、異常に高い非特異的バックグラウンド結合が観察されたが、サイクル2治療後は確認された一過性抗体反応のみが見られた(データ示さず)。抗体反応は中和的ではなかった。
【0395】
抗HSA抗体はこの集団において低レベルで天然に存在し、6.9%の被験者が投与前評価でHSA抗体に陽性であった。治療により発現した抗HSA抗体は、NEUG治療被験者4人(1.8%)で観察された(表31)。反応はすべて一過性で弱かった。最初の治療サイクル後に3つの反応が現われ、サイクル2、3、および4の後は検出できなかった。3度目の治療後に反応が1つ起きたが、4度目の治療後の30日の経過観察では検出できなかった(データ示さず)。
【0396】
(表31)NEUGに対するHSAに特異的な治療により発現した免疫反応の概要
【0397】
f.第II相B部における治療により発現した有害事象
第II相B部では、各治療群中の被験者の>90%が、治療により発現した有害事象(TEAE)を少なくとも1つ経験し、試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、ペグフィルグラスチム群で23.1%からNeugranin50mg群で35.0%の範囲であった。少なくとも1つのSAEを有する被験者のパーセントは、NEUG30mg群で最も高かった(30%)が、他の3つの治療群では約15%であった。SAEはどれも試験薬剤と関連していなかった。患者1人(NEUG30mg)は、糖尿病性足病変により試験から除かれ、これは試験薬剤とは関連しないと考えられた。B部では、被験者8人(NEUG40mg2人、NEUG50mg3人、ペグフィルグラスチム3人)以外はみな少なくとも1つのTEAEを有した。試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、NEUG50mg群で20.2%、NEUG 40mg群で22.4%、ペグフィルグラスチムが投与される被験者で22.1%であった。被験者2人(NEUG 40mg 1人、ペグフィルグラスチム1人)が試験中に死亡し、各治療群の被験者6〜8人がSAEを少なくとも1つ経験した。死亡やSAEは試験薬剤と関連しないと考えられた。
【0398】
NEUG30mg用量についてサンプルサイズを考慮すると、TEAEの総数は、A部およびB部の治療群にわたって類似していた。A部およびB部の両方において、CTCグレード3以上のTEAEのパーセントは、試験薬剤に関連するTEAEのパーセントと同様に、NEUGおよびペグフィルグラスチムの場合で類似していた。
【0399】
g.用量反応
第II相の結果は、NEUGの40および50mg固定用量がいずれも、骨髄毒性の化学療法で治療される乳癌被験者においてペグフィルグラスチム6mgと同等の安全性および効能をもたらすことを実証した。40mg治療群の平均DSNは、50mg群の平均DSNよりわずかに低かったが、これらの差異は統計的に有意ではなかった。用量応答は、体重により調整した用量を考慮した場合と固定用量コホートに対してとの両方でAUCANCについて観察した(サイクル1における0〜15日目)(図24)。30mgコホートのAUCANCは、ペグフィルグラスチムのそれよりわずかに低く、これは、30mg固定用量がこの研究では効果が劣るが、40mgおよび50mgコホートのAUCANCは用量相関性があり、ペグフィルグラスチム治療被験者のAUCANCよりも高い(有意ではないが)ことを示している。上の分析から、NEUGが体重調節基準(mg/kg)で投与されると、用量反応が明らかである。しかしながら、第II相B部のサイクル1におけるDSNの比較によって、治療群(40および50mgNEUGおよびペグフィルグラスチム)において、そして体重調節用量(mg/kg)で、DSNが有意に異ならなかったので、体重で四分した患者はすべて十分にサポートされることが示唆された。さらに、関連する有害事象(特に骨痛;データ示さず)の発生率および重症度が、体重1キログラム当たりに投与される用量と相関せず、またペグフィルグラスチムでの有害事象と差異はなかったので、固定用量が、より低体重の患者において安全性プロファイルに変更をもたらす可能性があるという証拠はなかった。
【0400】
実施例12:さらなるNEUG製剤の例
さらなるNEUG製剤の開発が行われ、pH(4〜7)、緩衝剤の種類(L-ヒスチジン、クエン酸塩、酢酸塩)、等張化剤(ソルビトール対NaCl)、およびタンパク質濃度(最大で70〜90mg/mlまで濃縮)の影響を分析し測定した。試料は、SEC(モノマー%)、RH-HPLC(純度%)、エルマンアッセイ(-SH量)、A280(タンパク質)、およびIE-HPLC(電荷アイソフォーム)によって分析/モニタリングした。試験した製剤を表32に示す。
【0401】
試験後、製剤A40S、A45S、C55N、およびC55Sが(検討した側面においては)最も安定しているようであった。
【0402】
(表32)さらなるNEUG製剤
【0403】
本発明の趣旨または範囲から逸脱することなく、本発明の方法および組成物において多くの改変および変更をなし得ることが当業者には明らかである。したがって、本発明は、添付の特許請求の範囲およびその同等物の範囲内であれば、本発明の改変および変更を包含することが意図される。
【技術分野】
【0001】
本願は、2009年1月16日に出願された米国特許仮出願第61/145,440号および2009年1月16日に出願された米国特許仮出願第61/145,436号の恩典を主張する。米国特許仮出願第61/145,440号および米国特許仮出願第61/145,436号の内容は、その全体において参照により本明細書に組み入れられる。
【背景技術】
【0002】
背景
白血球減少症は、循環する白血球(WBC)の減少であり、多くの場合、WBC数4000/mL未満と規定される。白血球減少症に関与する主な細胞は、好中球である。しかしながら、リンパ球、単球、好酸球、または好塩基球の数の減少も総白血球数の低下に寄与する場合がある(メルクマニュアル17版(非特許文献1))。
【0003】
好中球減少症は、しばしば細菌感染や真菌感染に対する感受性の増加につながる、血中好中球数の減少を特徴とする。好中球減少症は、好中球数および相対感染危険度により次のように分類される。軽度(1000から1500/mL)、中程度(グレード3、500から1000/mL)、または重度(グレード4、<500/mL)。急性かつ重度の好中球減少症は、患者が急速に死に至る感染症に罹患しやすくなるので、生命にかかわる疾患である(メルクマニュアル17版(非特許文献1))。
【0004】
好中球減少症は、骨髄中の好中球産生の低下または好中球破壊が加速することにより引き起こされ得る。急性好中球減少症は、好中球の消費が速く、産生が著しく低下すると数日にわたって起きる場合がある。慢性好中球減少症は、何か月もの間持続する場合があり、しばしば脾臓における好中球産生の減少または好中球の捕捉によって引き起こされる。好中球減少症は、骨髄細胞に対して外因性の因子に副次的に生じるかどうか、あるいは内因性の欠陥が骨髄性前駆細胞中に存在するかどうかによって分類される場合がある(メルクマニュアル17版(非特許文献1))。
【0005】
好中球減少症およびその感染性合併症は、細胞毒性化学療法ならびに、放射線治療、生物療法、および骨髄移植などの他の癌治療の最も一般的で重篤な副作用の一つである。細胞毒性化学療法(成長の速い細胞を探し出し破壊することにより作用する)は、好中球前駆体の増殖速度が速くかつ血中好中球のターンオーバーが速いので、好中球減少症をもたらす(メルクマニュアル17版(非特許文献1))。化学療法を受けている患者における好中球減少症の最も一般的な症状としては、発熱、口腔潰瘍、および耳部感染が挙げられる。重度の好中球減少症の患者は、しばしば、敗血症、皮膚の蜂巣炎、肝膿瘍、フルンケル症、肺炎、口内炎、歯肉炎、直腸周囲炎、大腸炎、副鼻腔炎、および中耳炎などの化膿性感染症を患う。身体がより多くの好中球を産生できるまで、化学療法を遅延させなければならない場合があり、そしてより低用量で投与されなければならない場合があり、治療の効果が低下してしまう。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】メルクマニュアル17版
【発明の概要】
【0007】
概要
本発明は、白血球減少症および好中球減少症などの正常より低い白血球数を特徴とする病気または症状の治療、予防、または改善に有用な組成物および方法に関する。いくつかの態様では、組成物および方法は、図9に示す組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子またはその変種もしくは断片を含む。いくつかの態様では、組成物および方法は、好中球減少症および/または白血球減少症、例えば、薬剤(癌の治療のために投与される化学療法剤など)の投与が原因となる好中球減少症を治療、予防、または改善するために使用され、これは、本発明の組成物を使用して治療され得る。
【0008】
いくつかの態様では、組成物は医薬製剤であり、図9に示す組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子またはその断片もしくは変種を含む。いくつかの態様では、医薬製剤は、少なくとも一種類の薬学的に許容される担体を含み、pHが約5から約8.0、約5から約7.5、約5から約7.2、約5から約7.0、約5から約6.8、約5から約6.6、約5から約6.4、約5から約6.2、約5から約6、約6から約7.5、約6.0から約7.2、約6から約7である。他の態様では、pHは、約4、約4.2、約4.4、約4.5、約4.6、約4.8、約5、約5.2、約5.4、約5.5、約5.6、約5.8、約6.0、約6.2、約6.4、約6.5、約6.6、約6.8、約7.0、約7.2、約7.4、約7.5、約7.6、約7.8、または約8.0である。
【0009】
いくつかの態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、約2.5mg/mlから約240mg/ml、約30mg/mlから約120mg/ml、約60mg/mlから約120mg/ml、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlの濃度で含む。
【0010】
いくつかの態様では、薬学的組成物は、少なくとも一種類の薬学的に許容される塩を含む。いくつかの態様では、塩は、組成物中に約5mMから約50mM、約10mMから約40mM、約15mMから約30mM、約20mMから約25mMの濃度で存在する。いくつかの態様では、塩は、組成物中に約5mM、約10mM、約15mM、約20mM、約25mM、約30mM、約35mM、約40mM、約45mM、および約50mMの濃度で存在する。
【0011】
いくつかの態様では、薬学的組成物は、少なくとも一種類の薬学的に許容される緩衝剤を含む。いくつかの態様では、緩衝剤は、組成物中に約5mMから約50mM、約mM10から約50mM、約15mMから約50mM、約5mMから約10mM、約10mMから約20mM、約20mMから約30mM、約15mMから約25mM、または約20mMの濃度で存在する。いくつかの態様では、緩衝剤は、組成物の中に約5mM、約10mM、約15mM、約20mM、約25mM、約30mM、約35mM、約40mM、約45mM、約50mM、約55mM、または約60mMの濃度で存在する。いくつかの態様では、緩衝剤は、リン酸塩、クエン酸塩、またはそれらの組み合わせである。いくつかの態様では、緩衝剤としては、リン酸ナトリウム、リン酸一ナトリウム、リン酸二ナトリウム、またはそれらの組み合わせが挙げられる。
【0012】
いくつかの態様では、薬学的組成物は、凍結乾燥安定剤を含む。いくつかの態様では、凍結乾燥安定剤は、トレハロース二水和物である。いくつかの態様では、トレハロース二水和物の濃度は、約20mMから約100mM、約40mMから約80mM、約50mMから約70mM、または約60mMである。いくつかの態様では、安定剤の濃度は、約20mM、約30mM、約40mM、約50mM、約60mM、約70mM、約80mM、約90mM、約100mM、約110mM、または約120mMである。
【0013】
いくつかの態様では、薬学的組成物は、増量剤を含む。いくつかの態様では、増量剤は、多価アルコールである。いくつかの態様では、多価アルコールは、マンニトールである。さらなる態様では、薬学的組成物は、薬学的に許容される担体を含む。いくつかの態様では、担体は、ポリソルベートである。
【0014】
本明細書に記載の薬学的組成物は、様々な形態での投与のために製剤化されてもよい。例えば、いくつかの態様では、薬学的組成物は、経口、経肺、静脈内、筋肉内、皮下、直腸、眼(ophthalmic)、結腸、腸管外、大槽内、膣内、腹腔内、眼(ocular)、耳、局所(local)、口腔、経鼻、または局所(topical)投与用に調製される。組成物はまた、特定の剤形用に製剤化されてもよい。例えば、いくつかの態様では、薬学的組成物は、液剤、ゲル、エアロゾル、軟膏、クリーム、凍結乾燥製剤、粉末、ケーキ、錠剤、またはカプセルとして製剤化されてもよい。他の態様では、薬学的組成物は、制御放出製剤、速溶性製剤、遅延放出性製剤、徐放性製剤、拍動放出性製剤、混合即時放出性製剤として製剤化される。いくつかの態様では、薬学的組成物は、液剤として提供される。他の態様では、薬学的組成物は、凍結乾燥粉末として提供される。さらに別の態様では、薬学的組成物は、凍結乾燥ケーキとして提供される。
【0015】
本明細書に記載の薬学的組成物は、様々な様式で保存されてもよい。いくつかの態様では、薬学的組成物は、バイアルに保存され、他の態様では、薬学的組成物は、シリンジに保存される。
【0016】
いくつかの態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、少なくとも一種類の緩衝剤および/またはpH調整剤、ならびに任意で少なくとも一種類のさらなる薬学的に許容される担体を含み、25℃で24時間インキュベーションした後の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の溶液中のモノマー純度の減少は10%未満である。いくつかの態様では、緩衝剤は、pH調整剤と同じである。いくつかの態様では、25℃で24時間インキュベーションした後の溶液中のモノマー純度の減少は、約1%未満、約5%未満、約15%未満、約20%未満、または約25%未満である。
【0017】
他の態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、リン酸ナトリウム約20mM、マンニトール約180mM、トレハロース二水和物約60mM、ポリソルベート80約0.01%(w/v)を含み、pHが約6.0であり、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約2.5mg/mlから約120mg/ml、または約30mg/mlから約60mg/mlである。いくつかの態様では、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlである。
【0018】
さらに別の態様では、薬学的組成物は、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子およびPMTT20/6.0を含み、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約2.5mg/mlから約120mg/ml、または約30mg/mlから約60mg/mlである。いくつかの態様では、組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度は、約5mg/ml、約10mg/ml、約15mg/ml、約20mg/ml、約25mg/ml、約30mg/ml、約35mg/ml、約40mg/ml、約45mg/ml、約50mg/ml、約55mg/ml、約60mg/ml、約70mg/ml、約80mg/ml、約90mg/ml、約100mg/ml、約120mg/ml、約150mg/ml、約100mg/ml、約150mg/ml、約200mg/ml、約240mg/ml、または約250mg/mlである。
【0019】
上記の概要および以下の図面の簡単な説明および詳細な説明はいずれも例示および説明のためのものであり、請求項に記載の本発明をさらに説明することを意図するものである。その他の目的、利点、および新規な特徴は、以下の本発明の詳細な説明から当業者には容易に分かると考えられる。
【図面の簡単な説明】
【0020】
【図1】rHSA-G-CSF濃度を上げるとモノマー純度が減少することを示すグラフである。2.5mg/mlから240mg/mlまでの濃度範囲のrHSA-G-CSF試料のモノマー純度は、PMTT10/7.2中で25℃で24時間インキュベーションした後に、サイズ排除高速液体クロマトグラフィー(「SE-HPLC」)により測定した。
【図2】pHを上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が15mg/mlまたは60mg/mlのrHSA-G-CSFの凝集は、pHが6.0、6.8、7.2、または8.0のPMTT10中で25℃にて7日間インキュベーションした後に、SE-HPLCにより測定した。
【図3】温度を上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が15mg/mlまたは60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2中で4℃、25℃、または40℃で24時間インキュベーションした後にSE-HPLCにより測定した。
【図4】pHを上げるとrHSA-G-CSFの凝集が増加することを示すグラフである。濃度が48mg/mlのrHSA-G-CSFの凝集は、pHが5.8、6.3、6.4、もしくは7.0のPMTT10中でまたはpHが6.2のCMTT10中で25℃にて最大で3日間までインキュベーションした後に、SE-HPLCにより測定した。一番上の線は、10mM PMTT pH7.0であり、上からの2番目の線は、10mM PMTT pH6.4であり、上からの3番目の線は、10mm PMTT pH6.3であり、一番下の線の暗い方は、10mM PMTT pH5.8であり、一番下の線の明るい方は、10mM CMTT pH6.2である。
【図5】塩濃度を上げるとrHSA-G-CSFの凝集が低下することを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2中で1日間25℃/60%RHおよび5mM、10mM、20mM、または50mMの塩化ナトリウム濃度でインキュベーションした後にSE-HPLCにより測定した。
【図6】リン酸塩濃度を上げるとrHSA-G-CSFの凝集が低下することを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT/7.2中で1日間25℃/60%相対湿度(「RH」)および15mM、20mM、25mM、30mM、40mM、または50mMのリン酸塩濃度でインキュベーションした後にSE-HPLCにより測定した。
【図7】PMTT20/6.0調製用緩衝剤中で最大で120mg/mlまでの濃度でrHSA-G-CSFの純度が維持されることを示すグラフである。2.5mg/mlから120mg/mlまでの濃度範囲のrHSA-G-CSF試料のモノマー純度は、25℃で24時間PMTT10/7.2(従来の緩衝剤)またはPMTT20/6.0(新規な緩衝剤)中でインキュベーションした後に、SE-HPLCにより測定した。
【図8】pHおよびリン酸塩濃度がどのようにrHSA-G-CSFの凝集をもたらすかを示すグラフである。濃度が60mg/mlのrHSA-G-CSFの凝集は、PMTT10/7.2(従来の緩衝剤)またはPMTT20/6.0(新規な緩衝剤)中で4℃または25℃/60%RHにて最大14日間インキュベーションした後に、SE-HPLCにより測定した。
【図9】図9Aは、Neugranin(商標)(「NEUG」)と命名されたrHSA-G-CSF融合ポリペプチドの核酸およびアミノ酸配列を示し、図9Bは、ヒトG-CSFのアミノ酸配列を示し、図9Cは、ヒト血清アルブミンのアミノ酸配列を示す。
【図10】48mg/mlの濃度および25℃で最大で3日間まで、異なるpHでインキュベーションしたrHSA-G-CSFの、SE-HPLCで測定した凝集を示す表である。
【図11】SE-HPLCにより測定した、異なるpH、タンパク質濃度、および温度でのrHSA-G-CSFの凝集を示す表である。表の上半分(最初の3つの項目)は、15mg/ml rHSA-G-CSFである。表の下半分(最後の3つの項目)は、60mg/ml rHSA-G-CSFである。
【図12】異なるpH、温度、および濃度でインキュベーションした緩衝剤後のrHSA-G-CSFの活性を示す表である。
【図13】NEUGの製造の概略例を示すフローチャートである。
【図14】図14Aおよび図14Bは、NEUG-1(「1P」)およびNEUG-2(「2P」)を比較する、SEC-HPLCおよびRP-HPLCそれぞれの結果を示す。
【図15】図15Aおよび図15Bは、NEUG-1(「1P」)およびNEUG-2(「2P」)それぞれの、IEC-HPLCによる電荷不均一性の比較、ならびにペプチドマッピングによる同一性の比較を示す。
【図16】クーマシーブルーで染色した、SDS-PAGEゲル上でのNEUG-1およびNEUG-2の純度の比較を示す。
【図17】図17Aおよび図17Bは、ヒトにおける臨床での使用のために調製した、NEUG-1の3種類の異なるロットおよびNEUG-2の5種類の異なるロットの試験結果をまとめた表を示す。
【図18】ロット2378-R NEUG-1標準試料のクーマシー染色SDS-PAGE分析(還元)を示す。
【図19】図19Aおよび図19Bは、ロット2378-R NEUG-1標準試料のSE-HPLCおよびRP-HPLCのクロマトグラムを示す。
【図20】NEUG-2の最終製剤の典型的な開発ロットに対して行った分析の結果をまとめたものである。
【図21】過酸化水素とTBOまたはTBPとで処理したNEUGの逆相(「RP」)、イオン交換(「IE」)、およびサイズ排除(「SEC」)クロマトグラフィーのクロマトグラムを示し、試験はNEUGの酸化をモニタリングするために行った。NEUG対照=ミディアムグレー、過酸化水素で処理したNEUG=ライトグレー、TBPで処理したNEUG=黒。
【図22】過酸化水素とTBOまたはTPBとで処理したNEUGの結果を示す表である。試験はNEUGの酸化をモニタリングするために行った。
【図23】治療から14日目までの、化学療法前(サイクル0)にNEUG-1を投与される被験者の好中球絶対数(ANC)のメジアンを示すグラフである。4日目では、最も高い線から最も低い線の順に、300μg/kg NEUG、450μg/kg NEUG、150μg/kg、および50μg/kg NEUGである。
【図24】図24Aおよび図24Bは、化学療法サイクル前と24時間後にNEUG-1を投与された被験者のANCおよび白血球(「WBC」)数を示すグラフである。NEUGの用量は、50μg/kg、150μg/kg、300μg/kg、または450μg/kgであった。グレード3およびグレード4好中球減少症の好中球カットオフは、図24Aにおいて点線で示し、正常な好中球範囲も図24Aおよび図24Bの両方に示す。
【図25】サイクル1中の化学療法の約24時間後にNEUG300μg/kg、NEUG450μg/kg、またはペグフィルグラスチム(Neulasta(登録商標))6mgを投与された被験者の好中球絶対数(ANC)を示すグラフである。
【図26】第I相試験用化学療法サイクルを示す。
【図27】ヒト被験者における第I相試験でのNEUGの薬物動態を示すグラフである。記載の用量(450μg/kg、300μg/kg、または150μg/kg)で皮下投与したNEUGの血清濃度は、化学療法を受けていない乳癌被験者において測定した。正方形:450μg/kgサイクル0;三角形:300μg/kgサイクル0;丸:150μg/kgサイクル0。
【図28】第I相における被験者の好中球絶対数(「ANC」)を示すグラフである。被験者は、試験化学療法に続いて、サイクル1でNEUG300μg/kg(n=19)、NEUG450μg/kg(n=20)、またはペグフィルグラスチム(Neulasta(登録商標))6mg(n=9)を投与された。
【図29】化学療法(第I相試験)のサイクル1におけるNEUGの薬物動態/薬力(「PK/PD」)を示すグラフである。患者は、サイクル1において、ドキソルビシン/ドセタキセル投与の1日後にNEUGを450μg/kg投与された。ANCを白抜きのひし形で示し、NEUG濃度を黒の正方形で示す。グレード3およびグレード4好中球減少症のカットオフを点線で示す。NEUGの定量下限(「LLOQ」)は、6ng/mlにて点線で示す。
【図30】図30Aおよび図30Bは、0日目から15日目の間に得られたANC値に基づく、第1相試験B部において治療した各被験者の曲線下面積(AUC)を示す。図30Aはグラフであり、図23Aからのデータを表(図30B)にまとめている。
【発明を実施するための形態】
【0021】
発明の詳細な説明
本明細書には、白血球数の低下を特徴とする症状および病気を治療、予防、および改善する組成物および方法が開示される。本明細書に記載の組成物および方法は、ヒト血清アルブミンタンパク質(「HSA」)およびヒト顆粒球コロニー刺激因子(「G-CSF」)から形成された融合ポリペプチドを含む。本発明の一態様では、融合ポリペプチドはアミノ酸759個の長さがあり、当該融合物のアミノ酸1〜585は、成熟型HSAからのアミノ酸に対応し、当該融合物のアミノ酸586〜759は、成熟型ヒトG-CSFのアミノ酸に対応する。融合タンパク質のアミノ酸配列を図9Aから図9Cに示す。
【0022】
本明細書に記載の組成物および方法はまた、組換えHSA-G-CSFポリペプチドを含む治療製剤および薬学的組成物を含む。いくつかの態様では、これら製剤および組成物は、当該ポリペプチドをより低用量で投与するために構成されるが、他の態様では、当該製剤は当該ポリペプチドをより高用量で投与するために構成される。
【0023】
本発明はまた、G-CSFの変種または断片を含む融合タンパク質とアルブミンまたはアルブミンの断片もしくは変種とを含む融合タンパク質を包含する。本発明はまた、本発明の治療用アルブミン融合タンパク質をコードするポリヌクレオチド、治療用アルブミン融合タンパク質、組成物、薬学的組成物、製剤、およびキットを包含する。治療用アルブミン融合タンパク質をコードするポリヌクレオチドで形質転換された宿主細胞も本発明に包含され、当該ポリヌクレオチドおよび/または宿主細胞を使用して本発明のアルブミン融合タンパク質を作製する方法もまた包含される。
【0024】
一態様では、本発明によるアルブミン融合タンパク質は、保存期間が長い。
【0025】
第二の態様では、本発明によるアルブミン融合タンパク質は、対応する未融合のG-CSF分子よりも安定している。
【0026】
本発明は、本発明の核酸分子を含むように修飾された(好ましくは本発明のアルブミン融合タンパク質を発現するように修飾された)、遺伝子組換え生物をさらに含む。
【0027】
本発明は概して、アルブミン融合タンパク質をコードするポリヌクレオチド;アルブミン融合タンパク質;ならびにアルブミン融合タンパク質またはアルブミン融合タンパク質をコードするポリヌクレオチドを使用して病気や疾患を治療、予防、または改善する方法に関する。本明細書では、「アルブミン融合タンパク質」とは、少なくとも1個のアルブミン(またはその断片もしくは変種)分子の、少なくとも1個のG-CSF(またはその断片もしくはその変種)分子に対する融合により形成されたタンパク質を指す。本発明のアルブミン融合タンパク質は、遺伝子融合により互いに会合した、少なくともG-CSFの断片または変種と少なくともヒト血清アルブミンの断片または変種とを含む(すなわち、アルブミン融合タンパク質は、G-CSFの全てまたは一部をコードするポリヌクレオチドがインフレームでアルブミンの全てまたは一部をコードするポリヌクレオチドと結合した核酸の翻訳により生じる)。一旦アルブミン融合タンパク質の一部となったG-CSFおよびアルブミンタンパク質は、それぞれ、アルブミン融合タンパク質の「部分(portion)」、「領域」、または「部分(moiety)」と呼ばれる場合がある(例えば、「G-CSFタンパク質部分」または「アルブミンタンパク質部分」)。非常に好ましい態様では、本発明のアルブミン融合タンパク質は、G-CSFの分子またはその断片もしくは変種(成熟型G-CSFタンパク質が挙げられるが、これに限定されるわけではない)を少なくとも1個とアルブミンの分子またはその断片もしくは変種(成熟型アルブミンが挙げられるが、これに限定されるわけではない)を少なくとも1個とを含む。
【0028】
さらに好ましい態様では、本発明のアルブミン融合タンパク質は、宿主細胞によりプロセシングされ、周囲の培地へ分泌される。発現に使用される宿主の分泌経路で発生する新生アルブミン融合タンパク質のプロセシングとしては、シグナルペプチド切断;ジスルフィド結合の形成;適切な折り畳み;糖質の付加およびプロセシング(例えば、N結合およびO結合によるグリコシル化);特異的タンパク質切断;および多量体タンパク質への組み立てが挙げられるが、これらに限定されるわけではない。本発明のアルブミン融合タンパク質は、好ましくはプロセシングされた形態である。最も好ましい態様では、「アルブミン融合タンパク質のプロセシングされた形態」とは、N末端シグナルペプチド切断を経たアルブミン融合タンパク質産物を意味し、本明細書ではまた「成熟アルブミン融合タンパク質」とも呼ばれる。
【0029】
一態様では、本発明は、G-CSFおよび血清アルブミンタンパク質を含むかあるいはそれらからなるアルブミン融合タンパク質をコードするポリヌクレオチドを提供する。さらなる態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質を含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。別の態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質の生物学的活性がありかつ/または治療上活性がある断片を含むかあるいはそれからなるアルブミン融合タンパク質を提供する。別の態様では、本発明は、G-CSFタンパク質および血清アルブミンタンパク質の生物学的活性がありかつ/または治療上活性がある変種を含むかあるいはそれからなるアルブミン融合タンパク質を提供する。好ましい態様では、アルブミン融合タンパク質の血清アルブミンタンパク質成分は、血清アルブミンの成熟部分である。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0030】
さらなる態様では、本発明は、G-CSFタンパク質と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある断片とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。さらなる態様では、本発明は、G-CSFタンパク質と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある変種とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、G-CSFタンパク質の成熟部分である。さらに好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、G-CSFタンパク質の細胞外可溶性ドメインである。他の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分は、活性型G-CSFタンパク質である。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0031】
さらなる態様では、本発明は、G-CSFタンパク質の生物学的活性がありかつ/または治療上活性がある断片または変種と、血清アルブミンの生物学的活性がありかつ/または治療上活性がある断片または変種とを含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。好ましい態様では、本発明は、G-CSFタンパク質の成熟部分および血清アルブミンの成熟部分を含むかあるいはそれらからなるアルブミン融合タンパク質を提供する。本発明は、これらアルブミン融合タンパク質をコードするポリヌクレオチドをさらに包含する。
【0032】
I.定義
本明細書において、下記および明細書全体にわたって示すようにいくつかの定義を用いて本発明を記載する。
【0033】
本明細書では、「ポリヌクレオチド」は、少なくとも1個の顆粒球コロニー刺激因子(G-CSF)分子(またはその断片もしくは変種)にインフレームで結合した少なくとも1個のアルブミン分子(またはその断片もしくは変種)を含むかあるいはそれらからなる融合タンパク質をコードするヌクレオチド配列を有する核酸分子を指す。
【0034】
本明細書では、「アルブミン融合コンストラクト」は、少なくとも1個のG-CSF(またはその断片もしくは変種)分子をコードする少なくとも1個のポリヌクレオチドにインフレームで結合した少なくとも1個のアルブミン分子(またはその断片もしくは変種)をコードするポリヌクレオチドを含むかあるいはそれらからなり、さらに例えば一種類以上の次の要素を含む、核酸分子を指す:(1)機能性の自己増殖ベクター(シャトルベクター、発現ベクター、組込み型ベクター、および/または複製系が挙げられるが、これらに限定されるわけではない)、(2)転写開始領域(例えば、調節的または誘導的プロモーター、構成的プロモーターなどのプロモーター領域)、(3)転写の終了領域、(4)リーダー配列、および(5)選択マーカー。一旦アルブミン融合コンストラクトの一部となったG-CSFおよびアルブミンタンパク質をコードするポリヌクレオチドは、それぞれ、アルブミン融合コンストラクトの「部分(portion)」、「領域」、または「部分(moiety)」と呼ばれる場合がある。
【0035】
「治療活性」を示すG-CSFポリペプチドまたは「治療上活性のある」G-CSFタンパク質とは、G-CSFタンパク質に伴う一つ以上の公知の生物学的および/または治療的な活性を有するG-CSFポリペプチドを意味する。非限定的な例として、「G-CSF治療タンパク質」は、病気、症状、もしくは疾患を治療、予防、または改善するのに有用なG-CSFタンパク質である。非限定的な例として、「G-CSF治療タンパク質」は、特定の細胞型(正常(例えばリンパ球)または異常(例えば癌細胞))に特異的に結合するものであってもよく、したがって、化合物(薬剤または細胞毒性薬)が当該細胞型を特異的に標的にするように使用されてもよい。
【0036】
本明細書では、「被験者」なる語は、動物、好ましくは哺乳類、より好ましくはヒトを指す。「被験者」および「患者」なる語は、区別なく使用されてもよい。
【0037】
「薬学的に許容される担体」なる語は、個体に投与されても長期的または永続的な有害作用が実質的に無い任意の担体を指す。薬学的に許容される担体としては、希釈剤、充填剤、塩、分散媒、コーティング剤、乳化剤、湿潤剤、甘味もしくは香味剤、等張化剤、吸収遅延剤、防腐剤、抗菌抗真菌剤、緩衝剤、pH調整剤、酸化防止剤、安定剤、溶解剤、増量剤、凍結保護物質、凝集阻害剤、または任意の種類の製剤助剤が挙げられる。適切な担体は、Remington's Pharmaceutical Sciences (Remington's Pharmaceutical Sciences, 2000, 20th Ed., Lippincott, Williams & Wilkins)に記載されており、これは参照により本明細書に組み入れられる。そのような担体または希釈剤の好ましい例としては、水、塩化ナトリウム、マンニトール、トレハロース無水物、ポリソルベート(ポリソルベート80など)、pHを調整するための様々な薬学的に許容される緩衝剤(例えば、リン酸塩緩衝剤、クエン酸塩緩衝剤、酢酸塩緩衝剤、およびホウ酸塩緩衝剤)が挙げられるが、これらに限定されるわけではない。
【0038】
「薬学的に許容される塩」なる語は、塩酸、リン酸、酢酸、シュウ酸、酒石酸などに由来するものなどのアニオンと形成されたものや、ナトリウム、カリウム、アンモニウム、カルシウム、水酸化鉄、イソプロピルアミン、トリエチルアミン、2-エチルアミノエタノール、ヒスチジン、プロカインなどに由来するものなどのカチオンと形成されたものを含む。
【0039】
「凍結乾燥安定剤」なる語は、凍結乾燥された物質の化学的および/または物理的な不安定さを保護するか減少させる分子を指す。凍結乾燥安定剤の好ましい例としては、スクロース、トレハロース、グルタミン酸一ナトリウム、ヒスチジン、ベタイン、硫酸マグネシウム、グリセリン、エリトリトール、グリセロール、アラビトール、キシリトール、ソルビトール、マンニトールプロピレングリコール、ポリエチレングリコール、プルロニック、およびこれらの組み合わせが挙げられるが、これらに限定されるわけではない。好ましい凍結乾燥安定剤は、トレハロース二水和物やスクロースなどの非還元糖である。
【0040】
「増量剤」なる語は、凍結乾燥混合物に大きさを加え、凍結乾燥ケーキの物理的構造に寄与する化合物を指す。増量剤の例としては、ソルビトール、グリシン、マンニトール、およびポリエチレングリコールが挙げられる。
【0041】
「PMTT20/6.0」なる語は、リン酸一ナトリウム(2.42mg/mL、17.4mM)、リン酸二ナトリウム(0.35mg/mL、2.5mM)、マンニトール(32.79mg/mL、180mM)、トレハロース無水物(22.70mg/mL、60mM)、ポリソルベート80(0.1mg/mL、0.01%)を含む組成物を指し、組成物の最終pHが6.0である。これはまた「新規な緩衝剤」、「新規なPMTT」、または「新規な調製用緩衝剤」と呼ばれ、20mMリン酸塩、180mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80を含み、pHが6.0であるとも記載される。
【0042】
「PMTT10/7.2」なる語は、10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80を含む組成物を指し、組成物の最終pHが7.2である。これはまた、「従来の緩衝剤」、「従来のPMTT」、または「従来の調製用緩衝剤」とも呼ばれる。
【0043】
「PMTT」なる語は、リン酸塩、マンニトール、トレハロース無水物、およびポリソルベートを含む組成物を指す。
【0044】
「CMTT10」なる語は、10mMクエン酸ナトリウム、190mMマンニトール、60mMトレハロース無水物、0.01%(W/V)ポリソルベート80を含む組成物(pHが6.2の緩衝剤)を指す。
【0045】
II.顆粒球コロニー刺激因子
顆粒球コロニー刺激因子(G-CSF)は、好中球の産生を促す造血成長因子である。G-CSFの投与は、好中球の増加を迅速に誘導する。G-CSFの他の重要なインビボ活性は、末梢血中への造血前駆細胞の動員である(Duhrsen et al, 1988; Molineux et al, 1999; Roberts et al, 1994)。この効果は、好中球系統だけを含むものではなく、他の単一系統および多系統前駆細胞ならびに造血多能性幹細胞にまで及ぶ(Molineux et al, 1999)。G-CSFはまた、好中球をプライミングすることによって感染に対する防衛機構の一部である細胞事象を高め、これによって、オプソニン化黄色ブドウ球菌に対する食作用および抗菌作用の両方が高まる。G-CSFはまた、好中球および単球の走化性および好中球の接着性を誘導する(Yuo et al, 1989; Wang et al, 1988)。
【0046】
組換えG-CSF産物は現在、好中球の増殖および分化を促すために多数の臨床上の適応症に関して承認されている。臨床試験では、フィルグラスチム(組換えメチオニルヒトG-CSF;ニューポジェン(登録商標)、アムジェン社、サウサンドオークス、カリフォルニア州)は、末梢好中球の数を増やし、このため骨髄抑制性の化学療法後の好中球減少症の期間を短縮させた。フィルグラスチムは、毎日の皮下(SC)注射によって投与される。ポリエチレングリコール共役rG-CSF(Neulasta(登録商標))であるペグフィルグラスチムは、毎日のrG-CSF投与に代わるサイクル毎に一回投与される代替物として、骨髄抑制性の抗癌剤が投与される患者における発熱性好中球減少症の発生率を減少させるのに安全かつ効果的であることが証明されている(Holmes, O'Shaughnessy et al, 2002; Green et al, 2003;Neulasta(登録商標)SmPC 2007)。
【0047】
III.ヒト血清アルブミン
ヒト血清アルブミン(HSA)は、ヒト循環系で最も一般的な天然の血液タンパク質であり、1リットル当たり約40グラムのアルブミンが測定され、20日以上の間循環系に存続する。HSAは、酵素的または免疫的な機能を有さず、生体内に広く分布し、血液中で治療物質の担体となることが知られている。アルブミンは、生理的濃度で最小限の活性を有する担体タンパク質である。HSAおよび組換えヒトアルブミン(rHSA)はいずれも、ヒトにおいて同様の長期の血中半減期を有する。ヒトアルブミンに遺伝子的に融合した治療用タンパク質は、アルブミンの血中半減期特性を獲得し得ることが研究で示されている(Syed et al, 1997)。例えば、ウサギでの研究では、アルブミンに融合したCD4の半減期が、未融合のCD4よりも140倍長いことが示されている(Yeh et al, 1992)。
【0048】
成熟型では585個のアミノ酸からなるタンパク質であるヒト血清アルブミン(米国特許第7,592,010号の図1に示される)は、血清の浸透圧の大部分を担っており、また内因性および外因性リガンドの担体として機能する。現在、臨床で使用されるHSAは、ヒトの血液から抽出されて製造される。微生物中の組換えHSA(rHSA)の産生は、EP 330 451およびEP 361 991に開示されている。
【0049】
IV.G-CSFを用いた治療
顆粒球コロニー刺激因子(G-CSF)を用いた一次予防は、年齢、病歴、病気の特徴、および化学療法レジメンの骨髄毒性に基づくリスクが高い患者における発熱性好中球減少症の防止に推奨される。米国臨床腫瘍学会(ASCO)および欧州癌研究治療機関(EORTC)は、発熱性好中球減少症のリスクが約20%ある場合、G-CSFの利用を推奨している。発熱性好中球減少症のリスクが10%から20%である場合、米国総合癌センターネットワーク(NCCN)は、G-CSFによる予防を任意で適用することを推奨し、発熱性好中球減少症のリスクが少なくとも20%である場合、G-CSFによる予防を確実に適用することを推奨している(Smith et al, 2006, Vogel et al 2005, Timmer-Bonte et al 2006, NCCN Guidelines)。
【0050】
コロニー刺激因子(CSF)を用いた予防が、特定の化学療法レジメンの毒性を緩和するために推奨される。しかしながら、当該治療のコストが増加することは、米国においてそして特にEUの一部において重要な検討事項であり、予防的なG-CSF治療の利用が不十分になる可能性があり、また高用量化学療法レジメンに対する患者の適格性が制限される可能性がある(Timmer-Bonte et al, 2006; Adams et al, 2006, NCCN Guidelines)。
【0051】
V.ポリペプチドならびにポリヌクレオチドの断片および変種
A.断片
本発明はさらに、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片に関する。本発明はまた、G-CSFタンパク質の、アルブミンタンパク質の、および/または本発明のアルブミン融合タンパク質の断片をコードするポリヌクレオチドに関する。タンパク質のN末端から1個以上のアミノ酸が欠失して、G-CSFタンパク質、アルブミンタンパク質、および/または本発明のアルブミン融合タンパク質の1つ以上の生物学的機能が修飾または喪失しても、他の治療的活性および/または機能的活性(例えば、生物学的活性、多量体化能、リガンド結合能)はなお保持され得る。例えば、完全型または成熟型のポリペプチドを認識する抗体を誘導しかつ/またはそれに結合するN末端欠失を有するポリペプチドの能力は、N末端から除去される完全型ポリペプチドの残基が過半数未満であれば、一般に保持される。完全型のポリペプチドのN末端が欠失した特定のポリペプチドが、そのような免疫学的活性を保持するかどうかは、本明細書に記載されるかまたは当技術分野で公知である常法によって容易に決定することができる。N末端アミノ酸残基が多数欠失した変異タンパク質が、一定の生物学的または免疫原的活性を保持している可能性がある。事実、わずか6個のアミノ酸残基からなるペプチドがしばしば免疫反応を誘発する場合がある。
【0052】
したがって、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質の断片は、完全長のタンパク質、ならびに参照ポリペプチド(reference polypeptide)のアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチド(つまり、G-CSFタンパク質、あるいはポリヌクレオチドまたはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質のG-CSFタンパク質部分)を含む。特に、N末端欠失は、一般式m〜qで記すことができ、式中、qは、参照ポリペプチド(例えば、G-CSFタンパク質、または本発明のアルブミン融合タンパク質のG-CSFタンパク質部分)中のアミノ酸残基の総数を表す整数であり、mは、2〜qマイナス6までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0053】
さらに、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応する血清アルブミンポリペプチドの断片は、完全長のタンパク質、ならびに参照ポリペプチドのアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチド(つまり、血清アルブミン、またはアルブミン融合タンパク質の血清アルブミン部分)を含む。好ましい態様では、N末端欠失は、一般式m〜585により記すことができ、式中、585は、成熟ヒト血清アルブミン中のアミノ酸残基の総数を表す整数であり、mは、2から579までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。さらなる態様では、N末端の欠失は、一般式m〜609により記すことができ、式中609は、完全長のヒト血清アルブミン中のアミノ酸残基の総数を表す整数であり、mは、2から603までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0054】
さらに、本発明のアルブミン融合タンパク質の断片は、完全長のアルブミン融合タンパク質、ならびにアルブミン融合タンパク質のアミノ酸配列のアミノ末端から1個以上の残基が欠失したポリペプチドを含む。特に、N末端欠失は、一般式m〜qにより記すことができ、式中、qは、アルブミン融合タンパク質中のアミノ酸残基の総数を表す整数であり、mは、2〜qマイナス6までの範囲の任意の整数として規定される。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0055】
また、上記のように、参照ポリペプチド(例えば、G-CSFタンパク質;血清アルブミンタンパク質;または本発明のアルブミン融合タンパク質)のN末端またはC末端から1個以上のアミノ酸が欠失して、当該タンパク質の1つ以上の生物学的機能が修飾または喪失しても、他の機能的活性(例えば、生物学的活性、多量体化能、リガンド結合能)および/または治療的活性はなお保持され得る。例えば、完全型または成熟型のポリペプチドを認識する抗体を誘導しかつ/またはそれに結合するC末端欠失を有するポリペプチドの能力は、C末端から除去される完全型または成熟型のポリペプチドの残基が過半数未満であれば、一般に保持される。参照ポリペプチドのN末端および/またはC末端が欠失した特定のポリペプチドが治療的活性を保持するかどうかは、本明細書に記載および/またはそうでなければ当技術分野で公知である常法によって容易に決定することができる。
【0056】
本発明はさらに、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質のアミノ酸配列のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6〜qマイナス1までの範囲の任意の整数であり、qは、参照ポリペプチド(例えば、G-CSFタンパク質、あるいはポリヌクレオチドまたはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質のG-CSFタンパク質部分)中のアミノ酸残基の総数を表す整数である。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0057】
さらに、本発明は、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質のアミノ酸配列のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6から584までの範囲の任意の整数であり、584は成熟型ヒト血清アルブミン中のアミノ酸残基の総数を表す整数から1を引いたものである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6から608までの範囲の任意の整数であり、608は血清アルブミン中のアミノ酸残基の総数を表す整数から1を引いたものである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0058】
さらに、本発明は、本発明のアルブミン融合タンパク質のカルボキシ末端から1個以上の残基が欠失したポリペプチドを提供する。特に、C末端欠失は、一般式1〜nで記すことができ、式中、nは、6〜qマイナス1までの範囲の任意の整数であり、qは本発明のアルブミン融合タンパク質中のアミノ酸残基の総数を表す整数である。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0059】
さらに、上記NまたはC末端欠失のうちの任意のものを、NおよびC末端欠失参照ポリペプチドを製造するために組み合わせてもよい。本発明はまた、参照ポリペプチド(例えば、G-CSFタンパク質、または本発明のアルブミン融合タンパク質のG-CSFタンパク質部分、または血清アルブミン、または本発明のアルブミン融合タンパク質のアルブミンタンパク質部分、またはアルブミン融合タンパク質、または本発明のポリヌクレオチドもしくはアルブミン融合コンストラクトによりコードされるアルブミン融合タンパク質)の残基mからn(nおよびmは、上に記載の整数である)を有すると一般に記載される場合がある、アミノ末端およびカルボキシル末端の両方から1個以上のアミノ酸が欠失したポリペプチドを提供する。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0060】
本願はまた、本明細書に記載の参照G-CSFポリペプチドまたは参照アルブミンポリペプチドあるいはそれらの断片と少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるポリペプチドを含むタンパク質に関する。好ましい態様では、本願は、上記のNおよびC末端欠失のアミノ酸配列を有する参照ポリペプチドと少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるポリペプチドを含むタンパク質に関する。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0061】
好ましい本発明のポリペプチド断片は、G-CSFタンパク質または血清アルブミンタンパク質のポリペプチド配列の治療的活性および/または機能的活性(例えば、生物学的活性)を示すアミノ酸配列を含むかあるいはそれからなる断片であり、当該タンパク質のアミノ酸配列が断片である。
【0062】
他の好ましいポリペプチド断片は、生物学的に活性のある断片である。生物学的に活性のある断片は、本発明のポリペプチドの活性に類似しているが必ずしも同一ではない活性を示すものである。当該断片の生物学的活性には、所望の活性の向上、または望ましくない活性の減少が含まれてもよい。
【0063】
B.変種
「変種」は、参照核酸またはポリペプチドとは異なるがそれらの本質的な特性を保持するポリヌクレオチドまたは核酸を指す。一般的に、変種は、全体として密接に類似し、多くの領域で参照核酸またはポリペプチドと同一である。
【0064】
本明細書では、「変種」は、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分、本発明のアルブミン融合タンパク質のアルブミン部分、または本発明のアルブミン融合タンパク質を指し、これらは、G-CSFタンパク質、アルブミンタンパク質、および/またはアルブミン融合タンパク質それぞれと配列が異なるが、本明細書の他の部分で記載するかまたはそうでなければ当技術分野で公知であるその少なくとも1つの機能的および/または治療的性質を保持する。一般に、変種は、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質、アルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質、および/またはアルブミン融合タンパク質のアミノ酸配列に全体として非常に類似し、多くの領域で同一である。当該変種をコードする核酸も、本発明に包含される。
【0065】
本発明はまた、例えば、本発明のアルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質、本発明のアルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質、および/またはアルブミン融合タンパク質のアミノ酸配列に対して少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、または約100%同一であるアミノ酸配列を含むかあるいはそれからなるタンパク質に関する。これらのポリペプチドの断片も提供される。本発明により包含されるさらなるポリペプチドは、ストリンジェントなハイブリダイゼーション条件下(例えば、6xの塩化ナトリウム/クエン酸ナトリウム(SSC)中で約45℃にて、フィルターに結合したDNAにハイブリダイゼーションさせ、次いで0.2xのSSC、0.1%SDS中で約50から65℃にて1回以上洗浄する)、非常にストリンジェントな条件下(例えば、6xの塩化ナトリウム/クエン酸ナトリウム(SSC)中で約45℃にて、フィルターに結合したDNAにハイブリダイゼーションさせ、次いで0.1xのSSC、0.2%SDS中で約68℃にて1回以上洗浄する)、または当業者に公知の他のストリンジェントなハイブリダイゼーション条件下(例えば、Ausubel, F. M. et al., eds., 1989 Current protocol in Molecular Biology, Green publishing associates, Inc.およびJohn Wiley & Sons Inc., New York, 6.3.1-6.3.6および2.10.3頁参照)で本発明のアルブミン融合タンパク質をコードする核酸分子の補体にハイブリダイズするポリヌクレオチドによりコードされるポリペプチドである。当該ポリペプチドをコードするポリヌクレオチドも本発明に包含される。
【0066】
問い合わせアミノ酸配列に少なくとも、例えば、95%「同一の」アミノ酸配列を有するポリペプチドにより、対象ポリペプチド配列が、問い合わせアミノ酸配列のアミノ酸100個当たりアミノ酸改変を最大で5個まで含み得るということを除いて、対象ポリペプチドのアミノ酸配列が問い合わせ配列と同一であることが意図される。言いかえれば、問い合わせアミノ酸配列に対して少なくとも95%同一のアミノ酸配列を有するポリペプチドを得るため、対象配列のアミノ酸残基の最大5%までが、他のアミノ酸で、挿入、欠失、置換されてもよい。参照配列の当該改変は、参照アミノ酸配列のアミノもしくはカルボキシ端末位置、またはこれらの端末位置の間の任意の場所において、参照配列における残基が個々にまたは参照配列内の1つ以上の隣接する群で点在して起こってもよい。
【0067】
実際問題としては、任意の特定のポリペプチドが、本発明のアルブミン融合タンパク質のアミノ酸配列またはその断片(例えば、アルブミン融合タンパク質のG-CSFタンパク質部分またはアルブミン融合タンパク質のアルブミン部分)に対して少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%同一であるかどうかは、公知のコンピュータプログラムを用いて従来通りに決定することができる。グローバル配列アラインメントとも呼ばれる、問い合わせ配列(本発明の配列)と対象配列との間の最大の全体の整合性を決定する好ましい方法は、Brutlagらのアルゴリズムに基づいたFASTDBコンピュータプログラムを用いて決定することができる(Comp. App. Biosci. 6:237-245 (1990))。配列アラインメントにおいて、問い合わせ配列および対象配列は両方がヌクレオチド配列であるかまたは両方がアミノ酸配列のいずれかである。グローバル配列アラインメントの結果では、同一性はパーセントで表される。FASTDBアミノ酸アライメントにおいて使用される好ましいパラメータは次のとおりである。Matrix=PAM 0, k-tuple=2, Mismatch Penalty=1, Joining Penalty=20, Randomization Group Length=0, Cutoff Score=1, Window Size=sequence length, Gap Penalty=5, Gap Size Penalty=0.05, Window Size=500または対象アミノ酸配列の長さのどちらか短い方。
【0068】
対象配列が、内部欠失のせいではなくNまたはC末端欠失により問い合わせ配列より短い場合、結果をマニュアル補正しないといけない。これは、全体的な同一性をパーセントで計算する際に、FASTDBプログラムは対象配列のNおよびC末端切断を考慮しないからである。NおよびC末端で切断された対象配列について、問い合わせ配列に対して、同一性のパーセントは、対象配列のNおよびC末端側であり、対応する対象残基と一致/アライメントしない、問い合わせ配列の残基の数を、問い合わせ配列の塩基全体のパーセントとして計算することにより補正される。残基が一致/アライメントするかどうかはFASTDB配列アラインメントの結果により決定される。次にこのパーセントは、特定のパラメータを用いた上記のFASTDBプログラムにより計算された同一性のパーセントから減じられ、最終的なパーセント同一性スコアに達する。この最終的なパーセント同一性スコアは、本発明の目的に使用されるものである。問い合わせ配列と一致/アライメントしない対象配列のNおよびC末端側に対する残基のみ、パーセント同一性スコアをマニュアル補正する目的のために考慮される。すなわち、対象配列の最も遠いNおよびC末端残基の外側の問い合わせ残基位置のみである。
【0069】
例えば、90個のアミノ酸残基を有する対象配列は、同一性のパーセントを決定するために100個の残基を有する問い合わせ配列とアライメントされる。対象配列のN末端にて欠失が起こり、したがって、FASTDBアライメントは、N末端で最初の10残基の一致/アライメントを示さない。10個の不対残基は、配列の10%に相当し(一致しないNおよびC末端における残基数/問い合わせ配列中の残基の総数)、したがって、FASTDBプログラムにより計算されたパーセント同一性スコアから10%減じられる。残りの90残基が完全に一致する場合は、最終的なパーセント同一性は90%になる。別の例では、90残基の対象配列は、100残基の問い合わせ配列と比較される。このとき、欠失は内部欠失であり、したがって、対象配列のNまたはC末端には、問い合わせ配列と一致/アライメントしない残基はない。この場合、FASTDBにより計算されたパーセント同一性は、マニュアル補正されない。再び、FASTDBアライメントで示されるように、問い合わせ配列と一致/アライメントしない対象配列のNおよびC末端の外側の残基位置のみ、マニュアル補正される。本発明の目的のためには、他のマニュアル補正は行われない。
【0070】
変種は、変種と同じ長さである正常HAまたはG-CSFタンパク質の長さと少なくとも約75%(他の態様では、少なくとも約80%、約85%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、または約99%)の配列同一性を通常有する。ヌクレオチドまたはアミノ酸配列レベルでの相同性または同一性は、配列類似性検索のために作られた、blastp、blastn、blastx、tblastn、およびtblastxプログラムにより利用されるアルゴリズムを用いて、BLAST(Basic Local Alignment Search Tool)分析により決定される(Karlin et al, Proc. Natl. Acad. Sci. USA 87: 2264-2268 (1990)およびAltschul, J. MoI. Evol. 36: 290-300 (1993)、全体として参照により組み入れられる)。
【0071】
BLASTプログラムにより使用される手法は、まず問い合わせ配列とデータベース配列との間の類似のセグメントを検討し、次に、同定された一致すべての統計的有意性を評価し、最後に、所定の重要性の閾値を満たす一致のみをまとめる。配列データベースの類似性検索に関する基本的な事柄の考察については、Altschulら(Nature Genetics 6: 119-129 (1994))参照。本書は全体として参照により組み入れられる。histogram、descriptions、alignments、expect(すなわち、データベース配列に対する一致を報告するための統計的有意性の閾値)、cutoff、matrix、およびfilterに対する探索パラメータは初期値である。blastp、blastx、tblastn、およびtblastxにより用いられるデフォルトスコアリングマトリクスは、BLOSUM62マトリクスである(Henikoff et al., Proc. Natl. Acad. Sci. USA 89: 10915-10919 (1992)、この文献は、全体として参照により組み入れられる)。blastnについては、スコアリングマトリクスは、N(すなわち、ミスマッチ残基のペナルティスコア)に対するM(すなわち、一対の一致する残基のリワードスコア)の比であり、ここで、MおよびNの初期値は、それぞれ5および-4である。4つのblastnパラメータは、以下のように調整されてもよい。Q-10(ギャップ生成ペナルティー);R=1O(ギャップ伸張ペナルティー);wink=1(問い合わせに沿ってwinkth位置でワードヒットを生じる);およびgapw=16(これは分離したアラインメントが生成されるウィンドウ幅を設定する)。等価のBlastpパラメータ設定は、Q=9;R=2;wink=1;およびgapw=32である。GCGパッケージバージョン10.0で利用可能である、配列間のBestfit比較は、DNAパラメータGAP=50(ギャップ形成ペナルティー)およびLEN=3(ギャップ延長ペナルティー)を用い、タンパク質の比較における等価な設定は、GAP=8およびLEN=2である。
【0072】
本発明のポリヌクレオチド変種は、コード領域、非コード領域、または両方において改変を含んでもよい。特に好ましいものは、サイレント置換、付加、または欠失を生ずるが、コードされたポリペプチドの特性や活性を改変しない改変を含むポリヌクレオチド変種である。遺伝コードの縮重によるサイレント置換により産生したヌクレオチド変種が好ましい。さらに、50個未満、40個未満、30個未満、20個未満、10個未満、または5〜50個、5〜25個、5〜10個、1〜5個、または1〜2個のアミノ酸が任意の組み合わせで置換、欠失、あるいは付加したポリペプチド変種も好ましい。ポリヌクレオチド変種は、例えば、特定の宿主に対してコドン発現を最適化するため(酵母または大腸菌などの細菌宿主に好まれるコドンにヒトmRNA中のコドンを変化させるため)などの種々の理由で産生され得る。
【0073】
好ましい態様では、アルブミン融合タンパク質のアルブミン部分をコードする本発明のポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。さらなる好ましい態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードする本発明のポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。またさらに好ましい態様では、本発明のアルブミン融合タンパク質をコードするポリヌクレオチドは、酵母または哺乳類細胞における発現のために最適化される。
【0074】
別の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではG-CSFタンパク質をコードする野生型ポリヌクレオチドにはハイブリダイズしない。さらなる態様では、アルブミン融合タンパク質のアルブミン部分をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではアルブミンタンパク質をコードする野生型ポリヌクレオチドにはハイブリダイズしない。他の態様では、アルブミン融合タンパク質をコードするコドン最適化ポリヌクレオチドは、本明細書に記載のようなストリンジェントなハイブリダイゼーション条件下ではG-CSFタンパク質部分またはアルブミンタンパク質部分をコードする野生型ポリヌクレオチドにはハイブリダイズしない。
【0075】
さらなる態様では、アルブミン融合タンパク質のG-CSFタンパク質部分をコードするポリヌクレオチドは、そのG-CSFタンパク質の天然に存在する配列を含まないかあるいはそれから成り立っていない。さらなる態様では、アルブミン融合タンパク質のアルブミンタンパク質部分をコードするポリヌクレオチドは、アルブミンタンパク質の天然に存在する配列を含まないかあるいはそれから成り立っていない。別の態様では、アルブミン融合タンパク質をコードするポリヌクレオチドは、G-CSFタンパク質部分またはアルブミンタンパク質部分の天然に存在する配列を含まないかあるいはそれらから成り立っていない。
【0076】
本発明のポリペプチドの特徴を向上または改変させるために、タンパク質工学および組換えDNA技術の公知の方法を使用して、変種を発生させてもよい。例えば、1個以上のアミノ酸を、生体機能の本質的な喪失を伴わないで本発明のポリペプチドのN末端またはC末端から欠失させることができる。
【0077】
好ましい態様では、本発明の変種は同類置換を有してもよい。「同類置換」とは、脂肪族または疎水性アミノ酸Ala、VaI、Leu、およびIleの置換;水酸基残基SerおよびTarの置換;酸性残基AspおよびGluの置換;アミド残基AsnおよびGlnの置換、塩基性残基Lys、Arg、およびHisの置換;芳香族残基Phe、Tyr、およびTrpの置換、ならびに低分子アミノ酸Ala、Ser、Thr、Met、およびGlyの置換などのグループ内での交換を指す。
【0078】
表現型的にサイレントなアミノ酸置換を行う方法に関する指針は、例えば、Bowie et al., "Deciphering the Message in Protein Sequences: Tolerance to Amino Acid Substitutions," Science 247:1306-1310 (1990)に提供されており、ここで著者らは、変化に対するアミノ酸配列の耐性を研究するには2つの主な戦略があることを示している。
【0079】
第一の戦略は、進化の過程での自然淘汰によるアミノ酸置換に対する耐性を利用する。異なる種におけるアミノ酸配列の比較により、保存されたアミノ酸を同定することができる。保存されたアミノ酸は、タンパク質機能にとって重要であると考えられる。対照的に、自然淘汰によって置換が許容されてきたアミノ酸の位置は、当該位置がタンパク質機能にとって重要でないことを示す。したがって、タンパク質の生物学的活性を維持しつつ、アミノ酸の置換が許容される位置が修飾され得る。
【0080】
第二の戦略は、タンパク質機能に重要な領域を同定するために、クローン化遺伝子の特定位置にアミノ酸の変更を導入する遺伝子工学を使用する。例えば、部位特異的変異誘発またはアラニン走査突然変異誘発(分子中の残基毎に単一アラニン変異を導入する)を使用することができる。Cunningham and Wells, Science 244:1081-1085 (1989)参照。次いで、得られた変異体分子の生物学的活性を試験することができる。
【0081】
著者らが述べるように、これらの二つの戦略は、タンパク質がアミノ酸置換に対して驚くほど耐性があることを示した。著者らは、さらに、どのアミノ酸の変更が、タンパク質中の特定のアミノ酸位置で許容される可能性があるかを示す。例えば、最も深く埋まった(タンパク質の三次構造内で)アミノ酸残基は、非極性の側鎖を必要とする一方で、表面の側鎖の特徴は一般にほとんど保存されない。さらに、許容される同類アミノ酸置換は、脂肪族または疎水性アミノ酸Ala、Val、Leu、およびBeの置換;水酸基残基SerおよびThrの置換;酸性残基AspおよびGluの置換;アミド残基AsnおよびGlnの置換、塩基性残基Lys、Arg、およびHisの置換;芳香族残基Phe、Tyr、およびTrpの置換、ならびに低分子アミノ酸Ala、Ser、Thr、Met、およびGlyの置換を伴う。同類アミノ酸置換の他に、本発明の変種としては、(i)一種類以上の非保存アミノ酸残基の置換を含むポリペプチドであって、置換アミノ酸残基が遺伝コードによりコードされたものであってもなくてもよいポリペプチド、あるいは(ii)置換基を有する一種類以上のアミノ酸残基の置換を含むポリペプチド、あるいは(iii)ポリペプチドの安定性および/または溶解性を高める化合物(例えば、ポリエチレングリコール)などの他の化合物と融合したかまたは化学的に共役したポリペプチド、あるいは(iv)例えば、IgG Fc融合領域ペプチドなどのさらなるアミノ酸を含むポリペプチドが挙げられる。このような変種ポリペプチドは、本明細書の教示から当業者の範疇にあると見なされる。
【0082】
例えば、電荷を有するアミノ酸の、電荷を有するか中性の他のアミノ酸とのアミノ酸置換を含むポリペプチド変種は、改善した特徴(例えば、凝集しにくい)を有するタンパク質を産生する場合がある。医薬製剤の凝集は、凝集体の免疫原的活性によって、活性の低下およびクリアランスの増加の両方を起こす。Pinckard et al, Clin. Exp. Immunol. 2:331-340 (1967); Robbins et al, Diabetes 36: 838-845 (1987); Cleland et al., Crit. Rev. Therapeutic Drug Carrier Systems 10:307-377 (1993)参照。
【0083】
特定の態様では、本発明のポリペプチドは、アルブミン融合タンパク質のアミノ酸配列、G-CSFタンパク質のアミノ酸配列、および/またはヒト血清アルブミンの断片または変種を含むかあるいはそれからなり、ここで、当該断片または変種は、参照アミノ酸配列と比較すると、1〜5、5〜10、5〜25、5〜50、10〜50、または50〜150のアミノ酸残基の付加、置換、および/または欠失を有する。好ましい態様では、アミノ酸置換は同類的である。当該ポリペプチドをコードする核酸も本発明に包含される。
【0084】
本発明のポリペプチドは、ペプチド結合または修飾ペプチド結合(すなわちペプチド同配体)により互いに連結されたアミノ酸からなることができ、20個の遺伝子コード化アミノ酸とは別のアミノ酸を含んでもよい。ポリペプチドは、翻訳後プロセシングなどの天然のプロセス、または当技術分野で周知の化学的修飾技術により修飾されてもよい。このような修飾は、基礎的な教科書やさらに詳しい学術論文、また多数の研究文献に十分に記載されている。修飾は、ペプチド主鎖、アミノ酸側鎖、およびアミノもしくはカルボキシル末端を含むポリペプチド中の任意の場所で起こり得る。なお、同じタイプの修飾が、所与のポリペプチド中のいくつかの部位において同一または異なった程度で存在する場合がある。また、所与のポリペプチドは、様々なタイプの修飾を含む場合がある。ポリペプチドは、例えばユビキチン化の結果、分岐してもよく、ポリペプチドは、分岐の有無にかかわらず環状であってもよい。環状、分岐、および環状分岐ポリペプチドは、翻訳後の天然のプロセスに起因してもよいし、人工的な方法により作製されてもよい。修飾としては、アセチル化、アシル化、ADP-リボシル化、アミド化、フラビンの共有結合、ヘム部分の共有結合、ヌクレオチドまたはヌクレオチド誘導体の共有結合、脂質または脂質の誘導体の共有結合、ホスファチジルイノシトール(phosphotidylinositol)の共有結合、架橋、環化、ジスルフィド結合形成、脱メチル化、共有結合による架橋の形成、システインの形成、ピログルタミン酸の形成、ホルミル化、ガンマ-カルボキシル化、グリコシル化、GPIアンカー形成、ヒドロキシル化、ヨウ素化、メチル化、ミリスチル化、酸化、ペグ化、タンパク質分解処理、リン酸化、プレニル化、ラセミ化、セレノイル化、硫酸化、トランスファーRNAが仲介するタンパク質へのアミノ酸の付加(例えば、アルギニル化)、およびユビキチン化が挙げられる。(例えば、PROTEINS--STRUCTURE AND MOLECULAR PROPERTIES, 2nd Ed., T. E. Creighton, W. H. Freeman and Company, New York (1993); POST-TRANSLATIONAL COVALENT MODIFICATION OF PROTEINS, B. C. Johnson, Ed., Academic Press, New York, pgs. 1-12 (1983); Seifter et al, Meth. Enzymol. 182:626-646 (1990); Rattan et al., Ann. N.Y. Acad. Sci. 663:4862 (1992)参照)。
【0085】
C.機能的活性
「機能的活性を有するポリペプチド」は、完全長、前駆タンパク質、および/または成熟型G-CSFタンパク質に関連する1つ以上の公知の機能的活性を示すことができるポリペプチドを指す。このような機能的活性としては、生物学的活性、抗原性[抗ポリペプチド抗体に結合する(または結合をめぐってポリペプチドと競合する)能力])、免疫原性(本発明の特定のポリペプチドに結合する抗体を生じる能力)、本発明のポリペプチドで多量体を形成する能力、およびポリペプチドの受容体またはリガンドに結合する能力が挙げられるが、これらに限定されるわけではない。
【0086】
「生物学的活性を有するポリペプチド」は、用量依存性に関わらず、特定のバイオアッセイ中で測定される、本発明のG-CSFタンパク質(成熟型を含む)の活性に類似(必ずしも同一ではない)した活性を示すポリペプチドを指す。
【0087】
好ましい態様では、本発明のアルブミン融合タンパク質は、アルブミンに融合していない場合のG-CSFタンパク質部分(またはその断片もしくは変種)に関連する少なくとも1つの生物学的および/または治療的活性を有する。
【0088】
本発明のアルブミン融合タンパク質は、当技術分野で公知のアッセイや本明細書に記載のアッセイを使用するかまたは常法によって変更して、機能的活性(例えば、生物学的活性)を分析することができる。さらに、当業者であれば、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質の断片を常法によって測定し得る。さらに、当業者であれば、当技術分野で公知のおよび/または下記の実施例部分に記載のアッセイを使用して、アルブミン融合タンパク質のアルブミンタンパク質部分に対応するアルブミンタンパク質の断片の活性を常法によって測定し得る。
【0089】
例えば、一態様では、アルブミン融合タンパク質の、抗G-CSFポリペプチド抗体および/または抗アルブミン抗体へ結合する能力、あるいはそれらへの結合をめぐってG-CSFタンパク質と競合する能力を測定する場合、当技術分野において公知の下記の様々な免疫測定法を使用することができるが、それらに限定されるわけではない:放射免疫測定、ELISA(酵素結合免疫吸着測定)、「サンドイッチ」免疫測定、免疫放射線測定、ゲル拡散沈降反応、免疫拡散測定、インサイチュー免疫測定(例えば、コロイド金、酵素、または放射性同位体標識を使用)、ウエスタンブロット、沈降反応、凝集アッセイ(例えば、ゲル凝集測定、赤血球凝集反応測定)、補体結合測定、免疫蛍光測定、プロテインA測定、および免疫電気泳動法測定などの技術を使用した拮抗的および非拮抗的アッセイ系が挙げられる。一態様では、抗体結合は、一次抗体上の標識を検出することにより検出される。別の態様では、一次抗体は、一次抗体への二次抗体または試薬の結合を検出することにより検出される。さらなる態様では、二次抗体が標識される。免疫測定における結合を検出するための多くの手段が当技術分野において公知であり、当該手段は本発明の範囲内である。
【0090】
好ましい態様では、G-CSFタンパク質の結合相手(例えば、受容体またはリガンド)が同定される場合、アルブミン融合タンパク質のG-CSFタンパク質部分として当該G-CSFタンパク質を含むアルブミン融合タンパク質による当該結合相手への結合は、例えば、還元および非還元ゲルクロマトグラフィー、タンパク質アフィニティークロマトグラフィー、ならびにアフィニティーブロッティングなどの当技術分野において周知の手段によりアッセイすることができる。一般的にはPhizicky et al., Microbiol. Rev. 59:94-123 (1995)参照。他の態様では、アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFポリペプチドの受容体に結合する、アルブミン融合タンパク質の生理学的相関物の能力は、当技術分野において公知の技術を使用して常法によって測定することができる。
【0091】
別の態様では、アルブミン融合タンパク質が多量体を形成する能力が評価される場合、多量体の他の成分との会合は、例えば、還元および非還元ゲルクロマトグラフィー、タンパク質アフィニティークロマトグラフィー、ならびにアフィニティーブロッティングなどの当技術分野において周知の手段によりアッセイすることができる。一般的には上述のPhizickyら参照。
【0092】
結合および交差反応性を分析するためならびにHSA-G-CSF融合物の同一性を確認するために使用することができる免疫測定法としては、下記のものが挙げられるが、これらに限定されるわけではない:ウエスタンブロット、放射免疫測定、ELISA(酵素結合免疫吸着測定)、「サンドイッチ」免疫測定、免疫沈降測定、沈降素反応、ゲル拡散沈降素反応、免疫拡散測定、凝集測定、補体結合測定、免疫放射線測定、蛍光免疫測定、およびプロテインA免疫測定などの技術を使用した拮抗的および非拮抗的アッセイ系が挙げられる。このようなアッセイは、当該技術分野において常法であり周知である(Ausubel et al, eds, 1994, Current Protocols in Molecular Biology, Vol. 1, John Wiley & Sons, Inc., New York参照。当該文献はその全体において参照により本明細書に組み入れられる)。
【0093】
アルブミン融合タンパク質のG-CSFタンパク質部分に対応するG-CSFタンパク質に結合する抗体も、所与のタンパク質または抗原、好ましくは抗体が特異的に結合する抗原、に対する結合親和性の点から説明または規定されてもよい。好ましい結合親和性としては、5x10-2M、10-2M、5x10-3M、10-3M、5x10-4M、10-4M未満の解離定数またはKdが挙げられる。より好ましい結合親和性としては、5x10-5M、10-5M、5x10-6M、10-6M、5x10-7M、10-7M、5x10-8M、もしくは10-8M未満の解離定数またはKdが挙げられる。さらに好ましい結合親和性としては、5x10-9M、10-9M、5x10-10M、10-10M、5x10-11M、10-11M、5x10-12M、10-12M、5x10-13M、10-13M、5x10-14M、10-14M、5x10-15M、もしくは10-15M未満の解離定数またはKdが挙げられる。さらに、本明細書に記載されているかそうでなければ当技術分野で公知のアッセイが、アルブミン融合タンパク質ならびにその断片、変種、および誘導体の、アルブミン融合タンパク質のG-CSFタンパク質部分および/またはアルブミン部分のいずれかに関連する生物学的活性および/またはG-CSF活性を誘発(インビトロやインビボのいずれかで)する能力を測定するために慣例的に適用され得る。他の方法も当業者には公知であり、本発明の範囲内である。
【0094】
VI.HSA-G-CSF融合タンパク質
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子(rHSA-G-CSF)はG-CSFアナログである。rHSA-G-CSFの例は、米国特許第5,665,863号および米国特許第7,041,478号に記載されており、これらは参照により本明細書に組み入れられる。
【0095】
rHSA-G-CSFの別の例は、Teva Biopharmaceuticals USA LTDにより開発されているNeugranin(商標)である。Neugranin(商標)(「NEUG」)は、一本鎖に結合した約85kDaの分子量を有する759個のアミノ酸の融合ポリペプチドである。残基1〜585は、成熟型HSAに対応し、残基586〜759は、成熟型ヒトG-CSFに対応する。Neugranin(商標)融合ポリペプチドを図9に示す。
【0096】
VII.融合タンパク質の産生
rHSA-G-CSFの合成方法の例は、米国特許出願番号第11/929,828号に記載されており、これはその全体において参照により本明細書に組み入れられる。いくつかの態様では、NEUGは、NEUG融合タンパク質を発現するように遺伝子操作された酵母宿主系(サッカロミセス・セレビジエ(Saccharomyces cerevisiae))を用いて産生される。NEUGは、酵母培養物の発酵培地から集菌され、当技術分野で周知の方法を用いて精製される(例えば、一連のクロマトグラフィーおよびろ過工程、例えば、アフィニティークロマトグラフィーおよびイオン交換クロマトグラフィーなど)。
【0097】
非限定的な一例では、NEUG融合コンストラクトは以下のように開発された。完全長アルブミンcDNAは、英国オックスフォード大学にてF. E. Baralle博士の実験室でヒトcDNAライブラリーから単離された。このクローンは、プラスミドpAT153ALBとして、英国ノッティンガムのDelta Biotechnology Limitedへ送付された。さらに、6-アミノ酸HSAプロペプチド(RGVFRR)は、酵母(RSLDKR)でのより効率的なプロセシングを促進するように修飾された。
【0098】
Neugranin産生プラスミドは、野生型サッカロミセス・セレビジエで発見された2μプラスミドに基づいている。pSAC35に基づいた発現ベクター(特許EP 286 424 B、US 5,637,504)は、産生宿主のロイシン欠乏を補完する選択マーカーとしてサッカロミセス・セレビジエからのLEU2遺伝子を含む。この産生プラスミドはまた、強力な酵母プロモーターであるPRB1ならびに大腸菌におけるクローニングおよび増殖を可能にするプラスミドpUC9からの配列を含む。さらに、このプラスミドは、酵母に形質転換されると、大腸菌における増殖に必要なpUC9由来配列を排除する特有の性質を示す。これは、酵母においては、隣接するFLP認識標的とプラスミドからの酵母FLPリコンビナーゼの発現により達成される。したがって、NEUGの産生に使用される生体中には細菌DNAは存在しない。これは、マスターセルバンクを作成した後、酵母からの2μmプラスミドのレスキュー実験および配列決定により確認される。
【0099】
上に記載のように、CID1643(pSAC35:HSA.GCSF(T31-P204))と命名されたNEUG産生プラスミドは、pSAC35に基づいた発現ベクターに由来した。ヒトG-CSFのT31-P204に対応する領域は、HSAオープンリーディングフレームの3'末端へのシームレスな融合を可能にするために適切な5'および3'制限部位を加えながら、PCRにより増幅した。
【0100】
メリーランド州ロックビルのHuman Genome Sciences, Inc.(HGS)では、cGMPマスターセルバンク(MCB)を作成するためにNEUGシードバイアルを使用した。NEUG MCBの試験および特性評価は、ICHガイドラインQ5D(Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/Biologicals Products)に従ってCharles River Laboratories(マルバーン、ペンシルベニア州、米国)およびLark Technologies(ヒューストン、テキサス州、米国)にて行われた。
【0101】
次いで、このMCBに由来したcGMPのワーキングセルバンク(WCB)が、Charles River Laboratories(マルバーン、ペンシルベニア州、米国)にて作成され試験された。
【0102】
NEUG細胞株バンクの製造で使用した培地成分はすべて、合成されたか、生合成されたか、または植物由来のものであった。細胞株または細胞バンク作成の際には、動物またはヒト起源の成分は使用されなかった。
【0103】
セルバンクは、めねじ付の蓋を有する予め殺菌した1.8mL Nuncポリプロピレンチューブにおいて低温保存培地中で<-135℃にて保存される。
【0104】
医薬用途向けのrHSA-G-CSF融合タンパク質の単離、精製、調製方法の非限定的な例を、実験例において下に記載する。
【0105】
VIII.治療用タンパク質の調製
未変性の状態または組換え技術によって作られた治療用タンパク質、例えば、インターフェロンおよび成長ホルモンは、特に水溶液中に調製された場合、保存期間の短い典型的な不安定な分子である。投与用に調製された場合の当該分子の不安定さのため、分子の多くが保存の際には常に凍結乾燥または冷蔵されなければならず、このため分子の輸送および/または運搬が困難である。院内環境外で医薬製剤を保存および投与しなければならない場合、保存に関する問題は特に深刻である。治療用タンパク質を含むアルブミン融合タンパク質は、アルブミン融合していない同様の治療用タンパク質の保存期間と比べて長い保存期間を示している。保存期間とは、典型的には、溶液または他の保存剤形の治療用タンパク質の治療的活性が、治療的活性の過度な喪失を受けずに安定している期間を指す。
【0106】
しかしながら、凝集および化学的な不安定さが、治療用タンパク質を含むアルブミン融合タンパク質製剤の開発における大きな障害である。医薬製剤の凝集は活性を低下させ、凝集体の免疫原的活性のために薬物クリアランスを増加させるので、医薬製剤の凝集は非常に有害となり得る(Pinckard et al. 1967; Robbins et al. 1987; Cleland et aL, 1993)。
【0107】
本明細書に記載の方法および組成物の様々な態様を記載した非限定的ないくつかの実施例を下記に示す。
【実施例】
【0108】
IX.実験例
次の実施例は本発明を例証するものである。しかしながら、本発明は、当該実施例に記載の特定の条件または詳細に限定されるわけではないことを理解されたい。限定されるわけではないが米国特許を含む本明細書で参照する公開されている文書はいずれも参照により本明細書に組み入れられる。
【0109】
実施例1、2、および6では、三種類の非限定的なNeugranin(商標)(「NEUG」)薬学的組成物を示す。各製剤は、図9に示すNEUG融合ポリペプチドを含む。セクションAに記載の方法および得られた組成物は、広く「NEUG-0」と呼ぶ。セクションBに記載の方法および得られた組成物は、広く「NEUG-1」と呼び、セクションEに記載の方法および得られた組成物は、広く「NEUG-2」と呼ぶ。
【0110】
NEUGは、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合ポリペプチドである。NEUG融合タンパク質のアミノ酸配列を図9に示す。
【0111】
実施例1:治療用NEUG-0の調製
NEUG-0は毒物学実験用に製造した。製剤は、10mMクエン酸塩、75mM塩化ナトリウム、100mMスクロース、0.01%ポリソルベート80中にNEUGを4.0mg/ml含み、pH6.5まで緩衝化された。
【0112】
安全性/毒性
NEUGの非臨床的安全性は、サルで評価した。100、500、および1000μg/kgのNEUGへの反復暴露(週1回4週間)では良好な耐容性が示され、死亡、罹患、または体重もしくは摂食量の変化は無かった。有害な毒性と考えられる肉眼的または微視的な変化は無かった。治療に関連した所見は、G-CSF投与で予想される薬理作用に限定され(例えば、好中球増加、脾臓重量のわずかな増加、骨髄増生)、毒性とは考えられなかった。サルにおけるNOAELは、>1mg/kg/週であり、これは、ヒトにおける0.45mg/kgの投与量に伴って観察されるものより約12倍高かった。処置した動物はまた、ANCおよびWBCの増加を示した(データ示さず)。
【0113】
実施例2:治療用NEUG-1の調製
NEUG-1は、ヒトでの臨床研究用に製造された。製剤は、PMMT10/7.2(10mMリン酸ナトリウム、200mMマンニトール、60mmトレハロース無水物、0.01%(W/V)ポリソルベート80、pH7.2)中にNEUGを15.0mg/mlを含んでいた。
【0114】
NEUG-1の組成を表1に示す。
【0115】
(表1)NEUG-1製剤の組成
a各バイアルは、1.0mL送達可能容量を確保するため0.16mL過剰に含む。
【0116】
NEUG-1用の原薬(「BDS」)の製造方法の一例として、図13の非限定的な工程を示す。次に、BDSは、約-80℃で保存される(公称値、保存温度の許容範囲は約-65℃である)。
【0117】
輸送や臨床現場での保存のための製剤のロバスト性を向上させるためや、期待される長期保存性を有する安定した製品を提供するため、NEUG-1の凍結乾燥形態が開発されている。凍結乾燥の方法は、当技術分野において周知であり、洗練された凍結乾燥ケーキを形成する方法の一例を、下記工程に記載のように行った。
【0118】
工程1:原薬の解凍および希釈
NEUG原薬の容器を15〜30℃で解凍する。原薬をプールし(必要であれば)、混ぜ、280nmでの吸光度によりタンパク質濃度を決定する。原薬の濃度を使用して、必要な希釈用緩衝剤の量を計算する。密度およびpHも測定する。調製用緩衝剤は、所望の最終濃度まで原薬を希釈するために使用される。
【0119】
工程2:無菌ろ過
希釈溶液は、完全性試験を行った滅菌フィルターを介して、調製領域から無菌の受容容器中へ移される。ろ過トレインは、直列に繋いだ0.2μmPVDFフィルターからなる。フィルターはすべて未使用であり、完全性試験がなされ、製造後には廃棄される。
【0120】
工程3:充填および部分的な付栓
Neugraninを、発熱性物質を除去した3mL USP Type Iガラスバイアルへ充填し、部分的に付栓し、底のない凍結乾燥トレーに載せた。充填した容量を確認するために、充填操作全体にわたって重量をチェックする。
【0121】
工程4:凍結乾燥
凍結乾燥サイクルは、室圧および棚温度により制御する。製品温度は、凍結乾燥室全体に配置した製品用熱電対によりモニタリングする。部分的に栓をしたバイアルを、予め冷却した棚に載せ、制御した速度で凍結する。アニーリング工程を行い、マンニトール(増量剤)の結晶化を増進させる。次に、一時乾燥を行い、熱電対をモニタリングして確認する。二次乾燥を行い、所望の含水率を達成する。二次乾燥の終わりに、0.2μmの無菌ろ過をした窒素を通気して凍結乾燥室の真空を開放する。
【0122】
工程5:密封およびキャップ付け
部分真空下(約11〜12psia)でバイアルに栓を置く。製品トレーからバイアルを取り、バイアルにキャップを付ける。
【0123】
最初の凍結乾燥臨床材料(NEUG-1)は、きわめて安定していることを示し、現在のところ2年間の保存期間を有し(2〜8℃で保存した場合)、これは、継続している安定性研究のデータが検討されるにつれさらに延びると考えられる。
【0124】
実施例3:NEUG-1参照標準物(ロット2378-R)の一般的特性
NEUGの生理化学的および生物学的特徴を調べるために様々な方法が開発されている。参照標準物ロット2378-Rと呼ぶNEUGの1ロットは、NEUGを21.3mg/mL含み、pH7.2で、10mMリン酸ナトリウム、20OmMマンニトール、60mMトレハロース無水物、および0.01%(w/v)ポリソルベート80(PMTT10/7.2)中で調製された。物理化学的特性評価を参照標準物ロットに対して行い、試験性状、分析法、および結果の概要を下記表2に示す。
【0125】
(表2)NEUG-1ロット2378-Rの特性評価
【0126】
クーマシーブルーで染色したNEUG-1ロット2378-RのSDS-PAGE分析は、単一バンドという結果になった(図19)。NEUG-1ロット2378-Rの代表的なSEC-HPLCおよびRP-HPLCクロマトグラムも、それぞれ図19Aおよび図19Bに示す。
【0127】
実施例4:NEUG-0製剤およびNEUG-1製剤の試験
1.rHSA-G-CSFモノマー純度に対するタンパク質濃度の影響
2.5mg/mlから240mg/mlの濃度範囲の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子(rHSA-G-CSF)をPMTT10/7.2(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH7.2)中にて25℃で24時間インキュベーションした。インキュベーションに続き、SE-HPLCによりモノマー純度を測定した。
【0128】
結果(図1)は、rHSA-G-CSFの濃度が増加するにつれて凝集が増加することを示す。
【0129】
2.rHSA-G-CSF凝集に対するpHの影響
15mg/mlおよび60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、25℃で7日間、PMTT10(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)中、pH6.0、6.8、7.2、および8.0でインキュベーションした。インキュベーションに続き、SE-HPLCによりrHA-G-CSFモノマー純度を測定した。
【0130】
結果(図2)は、pHが増加するにつれて凝集が増加し、タンパク質濃度の増加により凝集が加速したことを示す。
【0131】
3.rHSA-G-CSFの凝集に対する温度の影響
60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、PMTT 10/7.2緩衝剤中で、4℃、25℃、または40℃で24時間インキュベーションした。インキュベーションに続き、SE-HPLCによりrHSA-G-CSFモノマー純度を測定した。
【0132】
結果(図3)は、温度が上昇すると凝集が加速したことを示す。
【0133】
4.rHSA-G-CSFの凝集に対するpHの影響
48mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、25℃で最大3日間まで、PMTT10(10mMリン酸塩、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)中、pH5.8、6.3、6.4、または7.0、あるいはCMTT10(10mMクエン酸ナトリウム、190mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80)緩衝剤中でpH6.2でインキュベーションした。組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子のモノマー純度を、インキュベーション後0、1、2、および3日目にSE-HPLCにより測定した。
【0134】
結果(図4および図10)は、凝集のほとんどがインキュベーションの1日目に起こり、凝集速度がpHの増加により増加したことを示す。
【0135】
5.rHSA-G-CSFの凝集に対するpHの影響
15mg/mlまたは60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、pHが6.0、6.8、7.2、または8.0のPMTT10中で4℃、25℃、または40℃で最大10日間までインキュベーションした。モノマー純度は、インキュベーション後に0、1、7、および10日目にSE-HPLCで測定した。
【0136】
結果(図11)は、pHが増加するにつれて凝集が増加し、温度およびタンパク質濃度の増加により凝集が加速したことを示す。
【0137】
6.rHSA-G-CSF生物活性に対するpHの影響
15mg/mlまたは60mg/mlの濃度の組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子を、pHが6.0、6.8、7.2、または8.0のPMTT10中で4℃、25℃、または40℃で最大10日間までインキュベーションした。生物活性を、インキュベーション後、0日目と、7日目または10日目のいずれかとでバイオアッセイにより測定した。
【0138】
手短に説明すると、NFS-60細胞は、G-CSFおよびNEUGの両方に応答して増殖する。このアッセイでは、96ウェルマイクロプレートに細胞10000個/ウェルで蒔き、NEUGタンパク質で20時間37℃で処理した。20時間インキュベーションした後、NFS-60細胞を、0.5μCi/ウェルの[3H]-チミジンで4時間パルス化し、[3H]-チミジン取込みにより測定されるDNAの合成レベルを測定した。統計分析については、NEUG濃度の関数としての[3H]-チミジン取込みの測定を、4パラメータ・ロジスティックモデルを用いてモデル化し、EC50値(pg/mL)を決定した。試験タンパク質の結果は、参照標準物のEC50値を同一のアッセイ内で分析される試験サンプルのEC50と比較することにより得られた相対効力(RP%)値[RP%=(EC50参照物/EC50試料)*100]として報告される。
【0139】
結果(図12)は、低pHでインキュベーションされるとrHSA-G-CSFの活性がよりよく維持されたことを示す。
【0140】
7.rHSA-G-CSFの凝集に対する塩濃度の影響
rHA-G-CSFの60mg/ml溶液のイオン強度を、溶液中の塩化ナトリウムの量を変更することにより制御した。調製用緩衝剤はPMTT10/7.2であった。試料を、25℃/60%RHにて1日間、5mM、10mM、20mM、または50mMの食塩濃度でインキュベーションし、続いてSE-HPLCにより試験した。
【0141】
結果は、塩濃度を増加させると、rHSA-G-CSFの凝集が低下したことを示す(図5)。
【0142】
8.rHSA-G-CSFの凝集に対するリン酸塩濃度の影響
rHSA-G-CSFの60mg/ml溶液のイオン強度を、溶液中のリン酸塩の量を変更することにより制御した。調製用緩衝剤はpH7.2のPMTTであった。試料を、25℃/60%RHにて1日間、15mM、20mM、25mM、30mM、40mM、または50mMのリン酸塩濃度でインキュベーションし、続いてSE-HPLCにより試験した。
【0143】
結果は、リン酸塩濃度を増加させると、rHSA-G-CSFの凝集が低下したことを示す(図6)。
【0144】
実施例5:治療用NEUG-2の調製
より高濃度での使用および投与の臨床研究のためにNEUG-2を製造した。製剤は、PMMT20/6.0(20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース、0.01%ポリソルベート80)中にNEUGを50.0mg/mlで含んでいた。
【0145】
本質的には、NEUG-1に関して示したのと同じ発酵および精製工程を行った(例えば図13参照)。
【0146】
NEUG-2は、液体形態および凍結乾燥形態の両方で調製した。輸送や臨床現場での保存のための製剤のロバスト性を向上させるため、および期待される長期保存性を有する安定した製品を提供するために凍結乾燥形態が調製された。NEUG-2は、NEUG-1に関して記載したのと同じ凍結乾燥サイクルを用いて凍結乾燥した。NEUG-2製剤も良好に形成され、薬学的に洗練されたケーキを生じた。
【0147】
実施例6:NEUG-0、NEUG-1、およびNEUG-2製剤の比較
表3は、NEUG-0、NEUG-1の標準ロット(参照標準物2378-R)、およびNEUG-2の3種類の異なるロットの生理化学的な比較を示す。表中、「CP」は透明な淡黄色を指す。「ND」は測定されなかったことを指す。
【0148】
(表3)NEUG-0、NEUG-1、およびNEUG-2の生理化学的な比較
【0149】
NEUG-0〜NEUG-2を製造する方法が開発されるにつれ、方法に関連した不純物(例えば、酵母宿主細胞タンパク質)および産物に関連した変種(SEC、RP、およびIEC-HPLC)は、一貫して減少している。
【0150】
表4および表5は、NEUG-0、NEUG-1、およびNEUG-2の調製履歴および調製比較の概要を示す。
【0151】
(表4)調製履歴
【0152】
(表5)cGMP製剤の比較
【0153】
実施例7:NEUG-1およびNEUG-2の試験
1.rHSA-G-CSFのモノマー純度に対するpHおよびリン酸塩濃度の影響
図7に示す結果は、PMTT10/7.2およびPMTT20/6.0(20mMリン酸塩、180mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH6.0)それぞれにおけるNEUGのSE-HPLCにより測定されたモノマー純度を示す。試料は、2.5mg/mlから120mg/mlの範囲で異なるタンパク質濃度を有し、純度分析前に25℃で24時間インキュベーションした。
【0154】
結果は、SE-HPLCにより測定されたようにPMTT20/6.0調製用緩衝剤中で最大120mg/mlまでの濃度のrHA-G-CSFでは純度の有意な減少は観察されなかったが、PMTT10/7.2中では10%の凝集の増加があったことを示す。
【0155】
2.rHA-G-CSFの凝集に対するpHおよびリン酸塩濃度の影響
図8に示す結果は、PMTT20/6.0またはPMTT10/7.2中で4℃または25℃/60%RHで最大14日間インキュベーションしたrHA-G-CSF60mg/mlのSE-HPLCにより測定された凝集を示す。
【0156】
結果は、PMTT20/6.0調製用緩衝剤中でインキュベーションした試料では25℃で14日間インキュベーションした後には有意な凝集の増加は観察されなかったが、PMTT10/7.2調製用緩衝剤中で同様の条件下でインキュベーションした試料は、6%の凝集の増加を示したことを示した。
【0157】
3.NEUG-1対NEUG-2のHPLC比較
NEUG-1およびNEUG-2のSEC-HPLC分析およびRP-HPLC分析を図14Aおよび図14Bに示す。
【0158】
4.NEUG-1およびNEUG-2の電荷の比較、ペプチドマッピング、および純度
NEUG-1(図中「1P」として示す)およびNEUG-2(図中「2P」として示す)の電荷不均一性をIEC-HPLCにより比較した。結果を図15Aに示す。二種類のペプチド(NEUG-1「1P」およびNEUG-2「2P」)の同一性も、図15Bに示すようにペプチドマッピングにより比較した。
【0159】
NEUG-1およびNEUG-2の純度も、図16に示すようにSDS-PAGEおよびクーマシーブルー染色により比較した。各レーンの試料を下記表6に示す。
【0160】
(表6)図16のSDS-PAGEにロードした試料
【0161】
5.凍結融解
凍結融解の影響を、濃度が15、60、120mg/mlのNEUG-2(PMTT20/6.0中で)緩衝剤を用いて評価した。試料を0回から10回まで凍結および融解し、その後、目視およびSEC、RP、IECにより分析した。
【0162】
結果は、従来の(PMTT10/7.2)および新規な(PMTT20/6.0)調製用緩衝剤に関しては、Neugraninが、凍結融解誘導性の分解に感受性がないようであることを示す(データ示さず)。
【0163】
6.短期安定性
従来の緩衝剤(PMTT10/7.2)および新規な緩衝剤(PMTT20/6.0)中のNEUG試料(60mg/ml)を、4℃および25℃/60%RHで1か月間維持し、次いで、目視、SEC、RP、IEC、およびバイオアッセイにより試験した。モノマー純度はSE-HPLCにより評価した。
【0164】
14日目に、従来の緩衝剤中のNEUGは、6%の凝集体の増加を示したが、新規な緩衝剤中のNEUGは変化を示さなかった。目視、RP、IEC、およびバイオアッセイによると1か月間有意な変化はなかった。
【0165】
7.振とう
振とうにより誘導された凝集の影響を、従来の調製用緩衝剤(PMTT10/7.2)、新規な調製用緩衝剤(MPTT20/6.0)中のNEUGを用いて、3つの条件(15、60、および120mg/ml)、pH6.0で評価した。試料をそれぞれ、120rpmで0分から30分まで25℃で水平に振とうした。目視およびSE-HPLCを行った。
【0166】
SE-HPLC、RP-HPLC、IE-HPLCの結果は、モノマーの有意な喪失を示さなかった(データ示さず)。さらに、目視からの結果は、振とうの結果としての外観の変化を示さない。これは、従来および新規な調製用緩衝剤の両方に関して、Neugraninが、振とうにより誘導された凝集に対し感受性ではない可能性があることを示唆している。
【0167】
8.酸化
イオン交換高速液体クロマトグラフィー(「IEC」)および逆相高速液体クロマトグラフィー(「RPLC」)を、Neugraninの酸化をモニタリングするために使用することができる。過酸化水素試験のための実験条件は以下のとおりである。8mg/mLのNeugraninを0.03%過酸化水素およびTBOに暴露した。試料を、25℃で0、1、3、5、8、20、および24時間インキュベーションした後、サイズ排除クロマトグラフィー(「SEC」)、逆相(「RP」)、およびIECにより分析した。過酸化水素およびTPB誘導酸化の評価のために、RPLC、IECを用いた。クロマトグラフおよび表形式のデータを図21および図22に示す。
【0168】
HSAは、過酸化水素の存在下で酸化されることが示されている。同定された酸化部位は、Cys-34、Met-123、Met-298、Met-446、およびMet-548を含む。
【0169】
クロマトグラム(図21)は、1時間RTで過酸化水素(薄いグレー)およびTPB(黒)への暴露後、NEUG対照(灰色)のオーバーレイを示す。RPおよびIECによれば、H2O2で処理された試料ではメインピークの3分後に溶出してピークの有意な増加が示され、RPによれば、TPBで処理された試料は、メインピークの3分後に溶出するピークを示し、IECによれば、ピークはわずかに増加し、メインピークより2、3分後に溶出した。
【0170】
要約すると、結果は、RPおよびIECが異なる酸化形態を検出することができ、H2O2およびTBPのいずれも、メチオニンおよびシステイン残基であるNEUGの酸化を誘導し得たことを示唆する。NEUGの酸化は負荷のかかる条件下で起こるが、それが保存の際のNEUGの主要な分解経路を表しているということ示すものはなく、それらの活性に対する有意な変化はない。
【0171】
実施例8:薬学的利用のためのNEUGの特性評価の例
NEUG-1の工程を用いて三種類のcGMP薬剤成分ロットを製造し、NEUG-2の工程を用いて五種類のロットを製造した。一般に、NEUG-2製剤からは、NEUG-0およびNEUG-1に比べて同等かまたはより優れた特性を有するタンパク質が得られた。すべてのcGMPロットの試験結果を図17A-B中の表に示す。NEUG-1のロットは、060615001、060628001、および060707001である。NEUG-2のロットは、071008005、071025001、071026004、071106001、および07116001である。ロットの試験は以下のように行った。
【0172】
1.目視による外観
Neugranin(商標)(「NEUG」)の外観は、白黒の背景で前方に向けて照射する蛍光灯を用いて、目視により評価した。
【0173】
2.pH
pHメーターは、pHが4.0、7.0、および10.0の緩衝剤を用いて標準化した。標準化後、NEUG試料のpHを測定し記録した。
【0174】
3.凝固点によるオスモル濃度
オスモル濃度は凝固点降下により決定した。測定前に290mOsm/kg較正基準を用いて標準化を行った。
【0175】
4.吸光度(280nm)の測定による濃度
NEUGの濃度は、280nmで吸光度を測定することにより測定した。タンパク質濃度は、実験的に測定した0.54mg-1*mL*AUの吸光係数を用いて計算した。
【0176】
5.ELISAによる同一性
同一性アッセイは、当技術分野で周知の方法を用いて、標準的なサンドイッチELISAを使用した。アッセイは、NEUG分子のG-CSF部分およびアルブミン部分の両方に対する抗体の特異性に基づくものであった。一緒になってこの抗体対は、NEUGと、G-CSF、HSA、または他のアルブミン結合タンパク質とを識別することができる。
【0177】
6.クーマシー染色SDS-PAGEによる純度
変性条件下のNEUGの純度は、クーマシー染色SDS-PAGEを用いて評価した。試料は、分析前に、還元剤を用いてまたは用いないで試料緩衝剤で希釈した。分子量マーカーおよびNEUG参照標準物も各ゲルに添加した。電気泳動後、ゲルをクーマシーで染色し、濃度計を用いて純度を測定した。この方法は、二量体などの産物に関連した不純物を検出するために主に使用される。
【0178】
7.銀染色SDS-PAGEによる純度
変性条件下のNEUGの純度は、銀染色過負荷SDS-PAGEを用いて評価した。試料は、分析前に、還元剤を用いてまたは用いないで試料緩衝剤で希釈した。試料は、最大10μg/レーンまで添加した。分子量マーカーおよびNEUG参照標準物も各ゲルに添加した。電気泳動後、ゲルを銀染色した。NEUG試料と参照標準物との間の同一性を測定するために、泳動距離とバンドパターンとの間の比較を行う。低レベルの産物関連不純物および方法関連不純物を検出するためにこの方法を使用した。
【0179】
8.サイズ排除HPLCによる純度
未変性(native)の条件下のNEUGの純度は、サイズ排除HPLCを用いて評価した。試料は、リン酸塩および硫酸塩からなる無勾配移動相を用いて、TosoHaas G3000SWXLカラム(7.8mm x 30cm)を使用して分析した。NEUGの純度は、280nmでの総ピーク面積に対するメインピーク下面積の比を計算することにより決定した。このアッセイでは、二量体および凝集体を含む産物関連サイジング不純物が検出される。
【0180】
9.逆相HPLCによる純度
逆相HPLCは、NEUGの純度を測定するために使用した。試料は、アセトニトリル濃度が上昇する溶離勾配で、Agilent Zorbax逆相カラム(StableBond C8、300A、5μm、250x4.6mm)を用いて分析した。純度は、215nmの検出波長での総関連ピーク面積に対するメインピーク面積の割合を計算することにより決定した。
【0181】
RP-HPLCアッセイにおけるメインピークの前のピークは、G-CSF部分のThr-133にてジヘキソース修飾を含むNeugranin変種と一致する大きさを有する。この修飾は、薬効には影響せず、現在まで製造された開発バッチ、毒性バッチ、およびcGMPバッチすべてにおいて一定のレベルで存在した。Thr-133は、G-CSFの修飾の公知の部位である。
【0182】
10.バイオアッセイ(NFS-60細胞増殖)による薬効
NEUGの薬効アッセイは、NSF-60細胞増殖の誘発に基づく、細胞に基づいた増殖アッセイである。NFS-60細胞は、G-CSFおよびNEUGの両方に応答して増殖する。このアッセイでは、96ウェルマイクロプレートに細胞10,000個/ウェルで蒔き、NEUGタンパク質で37℃で20時間処理した。20時間インキュベーションした後、NFS-60細胞を、0.5μCi/ウェルの[3H]-チミジンで4時間パルス化し、[3H]-チミジン取込みにより測定されるDNAの合成レベルを測定した。統計分析については、NEUG濃度の関数としての[3H]-チミジン取込みの測定を、4パラメータ・ロジスティックモデルを用いてモデル化し、EC50値(pg/mL)を決定する。試験タンパク質の結果は、標準試料のEC50値を同一のアッセイ内で分析される試験サンプルのEC50と比較することにより得られた相対効力(RP%)値[RP%=(EC50参照物/EC50試料)*100]として報告した。
【0183】
11.閾値法による残存DNA
試料に残存するDNAの測定方法は、市販のキット(Molecular Devices Threshold 総DNA測定キット)に基づく。試料をまず希釈し、タンパク質を消化し、DNAを抽出した。次に、DNAの濃度をThresholdキットを用いて定量した。このキットは、DNAを免疫標識し、固定し、子ウシ胸腺DNA標準曲線に対するDNAの濃度を電気化学的に測定する。結果は、許容範囲内のDNA標準物の添加回収率により内部的に適格とされた。
【0184】
12.比濁時間分析法によるエンドトキシン
NEUG溶液中のエンドトキシン濃度は、標準的なエンドトキシン自動比濁時間分析システムを用いて測定した。比濁時間分析システムは、ゲルクロット法を改良したものであり、ゲルクロット形成に先行する濁度増加を測定する。リムルスアメーバ様細胞溶解物(LAL)を標準物および被験物質に加え、37℃でインキュベーションした。インキュベーション時に、反応混合物の濁度を分光測光法を用いてモニタリングした。濁度はエンドトキシン濃度に比例した。被験物質中の濁り形成速度を、公知の標準濃度の標準曲線における濁り形成と比較する。
【0185】
13.膜ろ過によるバイオバーデン
膜ろ過法は、試料中の生存生物の数を推定するアメリカ薬局方(USP<61>)微生物限度試験に類似している。膜ろ過方法では、生物は、試験液からメンブレンフィルター上に捕えられ、次に、適切な増殖培地に蒔かれる。試験前に、試料体積が10mLの被験物質を、無菌リン酸緩衝生理食塩水(PBS)90mLに希釈する。希釈した被験物質100mLをすべてろ過する。ろ過工程後、フィルター膜を、一般細菌数(HPC)寒天に移し、30℃から35℃で2日から3日間インキュベーションする。HPC寒天は、USP<61>に記載の大豆カゼイン消化物寒天(SCD、これはTSAとも呼ばれる))とは異なる。TSAと同様に、HPC寒天は、広範な好気性従属栄養細菌を回収するように設計されている。しかしながら、HPC寒天もまた、負荷のかかった生物を最大限回収するように設計されている。
【0186】
14.タングステン調査
特定の注射器の銘柄は、賦形剤としてタングステンを含むことが知られている。液体のNEUG試料を、様々な形態のタングステン(タングステン酸ナトリウム無水物、ポリタングステン酸ナトリウム、タングステン酸アンモニウム、および酸化タングステンWO3およびWO2)の存在下、様々な濃度(500ppb、1000ppb、および2500ppb)で2〜8℃および25℃で最大3か月まで試験した。試料を、SE-HPLC、RP-HPLC、およびバイオアッセイにより分析した。3か月の安定性試験後、NEUG試料に対するタングステンの有意な影響はなかった。
【0187】
15.特性評価アッセイ
下記分析も、特性評価情報のために使用した。
【0188】
N末端配列
NeugraninのN末端のアミノ酸配列は、PE Applied Biosystems Procise 494 cLCプロテインシークエンサーを用いて同定した。当該方法は、エドマン分解化学作用を組み入れ、気相ブロットサイクル、オンラインPTHアミノ酸HPLC分析器、およびSequenceProデータ分析ソフトウェアを利用する。
【0189】
IE-HPLCによる電荷不均一性
IEC-HPLCを使用して、NEUGにおける電荷不均一性の相対存在量を測定した。試料は、Dionex Pro-Pac Wax 10イオン交換カラム(250x4.6mm)を用いて分析し、移動相はBis-TrisおよびNaClからなった。この方法は、試料非依存性の変動を示し、展開のこの時点では、定量分析というよりむしろ定性分析として使用される。
【0190】
ペプチドマッピング
ペプチドマッピングは、NEUGの構造の完全性(integrity)を測定するために使用される。ペプチドマッピングは、消化のためにLys-Cを使用し、ペプチド分離のために勾配溶出する逆相HPLCを用いて行った。タンパク質試料をまず還元し、変性し、次いでアルキル化した。緩衝剤交換を行い、続いて1時間、その後一晩Lys-C消化を行った。消化に起因するLys-Cペプチド混合物を逆相HPLCにより分離し、Agilent 1100 HPLC系と共にYMC Pack ODS-A C18カラム(4.6mm x 250mm)を用いてペプチドプロファイルを得た。HPLC溶出は、ダイオードアレーUV検出器およびエレクトロスプレーイオン化Finigan LCQ DUO-Ion Trap質量分析装置を用いてモニタリングした。質量分析データは、Xcalibur V2.3ソフトウェアを用いて処理した。この方法の質量精度は、20ppm未満であった。HPLCピークはLC-MSにより同定した。
【0191】
エレクトロスプレーイオン化質量分析
質量分析は、Pico Viewナノエレクトロスプレー源を装備したApplied Biosystems QStar ESI-Quadrapole Time of Flight(QTOF)質量分析計を用いて行った。試料は、HamilitonシリンジおよびHarvardシリンジポンプによりQStarへ導入した。エレクトロスプレイーイオン源は、カーテンガス値25で、2500Vで操作した。器具は、ミオグロビンで較正した。質量電荷比は、割り当てられた分析時間全体にわたって、一秒ごとに質量分析器内の質量範囲全体(500〜3000m/z)を走査することにより記録した。得られた総イオン流は、Analyst v1.4ソフトウェアを用いて分析した。この器具の質量精度は、10ppm未満である。
【0192】
残存S.セレビジエ(酵母)宿主細胞タンパク質
原薬中に存在する酵母宿主細胞タンパク質(yHCP)の量を、NEUGの発現に使用したのと同じ酵母宿主のヌル(null)株に由来した宿主細胞タンパク質での免疫付与により産生されたカスタム抗体に基づくELISAを用いて測定した。
【0193】
エルマンアッセイによる遊離チオール
エルマン試薬である5,5'-ジチオ-ビス-(2-ニトロ安息香酸)(DTNB)を用いて、NEUG試料中の遊離チオール含量を測定した。DTNBは、遊離スルフヒドリル基と反応し、ジスルフィドおよび2-ニトロ-5-チオ安息香酸(TNB)の混合物が得られる。TNBは412nmに強い吸収があり、N-アセチルシステイン(NAC)標準曲線と比較することにより容易に反応を定量することができる。この方法の精度を高めるため、部分的に変性条件下で反応を行った。これにより、未変性条件下では完全には暴露されない場合のある遊離スルフヒドリル基にDTNBがアクセスできる。
【0194】
16.安定性
原薬(「BDS」)は、-80℃で保存され輸送される(公称値、許容温度は≦-65℃である)。BDSの安定性は、-80℃および加速貯蔵条件で製品を保存することにより試験した。参照標準物(NEUG-1、ロット2378-R)をまず安定した環境に配置して、GMP BDSに先導的な安定性(leading stability)を与えかつ参照標準物自体の安定性についての理解を得た。各工程(NEUG-1、NEUG-2)には典型的に、少なくとも1つの開発ロットおよび1つのcGMPロットを安定した環境に配置し、各試験は、ICHガイドラインに従った試験に特化したプロトコルに詳述されている。アッセイの適性および可能性の高い分解経路を規定し、かつ有効期限の予測に役立てるため標準加速条件を使用した。安定性は、外観、pH、タンパク質濃度、オスモル濃度、SEC-HPLC、RP-HPLC、SDS-PAGE、およびバイオアッセイによりモニタリングした。
【0195】
NEUG1参照標準ロット2378の代表的な安定性結果の概略を下記表7から表9にまとめる。
【0196】
(表7)NEUG-1ロット2378-Rの安定性データ(-80℃)
CPF=透明、淡黄色、外来の粉粒体が本質的に含まれていない
【0197】
(表8)NEUG-1ロット2378-Rの安定性データ(-20℃)
【0198】
(表9)NEUG-1ロット2378-Rの安定性データ(2〜8℃)
【0199】
どのパラメータの有意な変化も、推奨される保存条件で36か月まで、-20℃で18か月まで、または2〜8℃で12か月まで見られなかった。
【0200】
NEUG-2の1か月データを示す。推奨される保存条件下、-2O℃、または2〜8℃では有意な変化は認められない。表10。
【0201】
(表10)NEUG-2の安定性データ
【0202】
2〜8℃での液状のNEUG-2の2つのロットの安定性データを、下記表10.1および表10.2に示す。
【0203】
(表10.1)ポリプロピレンチューブ中の液体NEUG-2(ロット3029)1.2mlの安定性
【0204】
(表10.2)テフロンバイアル中の液体NEUG-2(cGMPロット071026004)1.0mlの安定性
【0205】
上に記載のように、NEUG-1工程からNEUG-2工程への変更を製剤に対して行った。当該変更は、より高いNEUG濃度で製品品質を確保するために行い、新たな賦形剤は含まれなかった。主な変更点は、イオン強度の増加(10mMから20mM)およびpHの低下(7.2から6.0)であった。BDS製剤は、NEUG-1工程からNEUG-2工程になり、濃度依存性の凝集が低下するように改善した。したがって、NEUG-2製剤は、既にロバスト性を有するNEUG-1と比較して向上した安定性を示すことが予想される。
【0206】
さらに、凍結乾燥形態または液状形態のいずれであっても、2〜8℃での推奨される保存条件で3か月間の安定性試験後にNEUG-2に対するタングステンの有意な影響はなかった(SE-HPLC、RP-HPLC、および効力による分析;データ示さず)。
【0207】
実施例9:例示的製剤(NEUG-2)の説明および組成
1.剤形の説明
一態様では、Neugranin(商標)(「NEUG」)は、コーティングされたゴム栓およびフリップオフシール(flip-off seal)で密閉した使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給される。NEUGは2〜8℃で保存することができる。注射用滅菌水(WFI)1.0mLで再構成する際、各バイアルは、20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース二水和物、0.01%(w/v)ポリソルベート80、pH6.0中に、NEUGを50mg/mL(バイアル毎に50mgが送達可能)含む。再構成後、バイアルは室温で保持することができ、8時間以内に使用するのがよい。
【0208】
2.定量的組成
1つのNEUG製剤(NEUG-2)の例示的な定量的組成を下記表11に示す。なお、液状形態のNEUG-2は、凍結乾燥形態と同じ定量的組成を有している。液状形態のものは、充填済みシリンジ中に提供することができる(下記セクション9参照)。
【0209】
(表11)NEUG-2製剤の定量的組成
a各バイアルは、1.0ml送達可能容量を確保するため0.11ml過剰に充填される。
【0210】
3.再構成用緩衝剤
凍結乾燥形態のNEUGの再構成に注射用滅菌水(WFI)を使用することができる。
【0211】
4.賦形剤
NEUG製剤の例示的組成は次のとおりである。2.77mg/mLリン酸ナトリウム、33.79mg/mLマンニトール、22.7mg/mLトレハロース二水和物、および0.1mg/mLポリソルベート80で、pHが6.0(上記表5参照)。リン酸ナトリウムは緩衝剤であり、マンニトールは、良好なケーキ構造を提供しかつ張性を調整するための増量剤として使用され、トレハロースは、抗凍結剤として使用され、ポリソルベート80は、凝集および吸着の可能性を低下させるために使用される。50mg/mLのNEUG-2における有効成分(「API」)の濃度では、タンパク質自体がロバスト性を有する緩衝剤として作用する。製剤に使用される公定賦形剤は、複数の公定書(MC)に準拠している。例外としては、現在利用可能な公定書がないトレハロース二水和物である。しかしながら、トレハロース二水和物は、公定書ではないが厳格な一連の仕様書に準拠している。トレハロースは、米国FDAでGRASとしてリストに記載されており、多数の市販されている非経口薬剤(例えば、ベバシズマブ(アバスチン)およびトラスツズマブ(ハーセプチン))の製剤に使用されている。
【0212】
5.製剤開発
毒性研究に使用された最初の製剤は、凍結させた液体として-20℃での保存を支援するように設計された(NEUG-0)。輸送および臨床現場での保存に対する製剤のロバスト性を向上させ、および期待される長い保存期間を有する製品を提供するために、全臨床材料に対して凍結乾燥形態が現在まで使用されている。最初の臨床材料(NEUG-1)は、きわめて安定していることを示し、現在は2年間の保存期間を有し、これはさらに延びると考えられる。後の製剤開発では、より大きなイオン強度およびより低いpHが、より高い濃度(>25mg/mL)でAPIを安定させることを示した。このために、1つの製剤(NEUG-2)は、より低いpH(6.0対7.2)およびより高いリン酸塩濃度(20対10mM)を有する。強制分解試験は、この製剤が、激しい振とう、凍結融解の繰り返し、および濃度誘導凝集から液体状態の薬剤成分を保護することを示した。NEUG-1に関して記載したのと同一の凍結乾燥サイクルをNEUG-2製剤に対して使用すると、いずれも良好に形成されたケーキを生成した。
【0213】
6.製造工程開発
いくつかの態様では、NEUGの最終製剤は、調製用緩衝剤で原薬を所望の濃度(50mg/mL)まで希釈し、充填し、製品を凍結乾燥することにより得られる。
【0214】
毒性と臨床材料とを比較し易くするために、両方の工程(NEUG-1およびNEUG-2)からの多くの最終製剤(「FDP」)を物理化学的に特性評価した。
【0215】
物理化学的な比較試験の結果が下記表12に含まれており、NEUG-2製品の品質は、NEUG-1製品とくらべて、それ以上ではなくても少なくとも同程度であることを示している。凍結乾燥工程は、製品に対する悪影響はない。
【0216】
(表12)NEUG-1およびNEUG-2の比較
【0217】
最初の臨床試験に使用したNEUG(NEUG-1)は、現在調製されているNEUG(NEUG-2)に物理化学的に同等である。製造工程および剤形に対して行った変更は、臨床有効性または安全性に影響を及ぼさなかった(データ示さず)。
【0218】
7.バッチ分析
NEUG-2の最終製剤の代表的な開発ロットに対して行われた分析の結果を図20にまとめる。表中、「ND」は、開発ロットに対して試験が「行われなかった」ことを意味する。
【0219】
8.最終製剤の安定性
NEUG-2の最終製剤のロットに対して安定性試験を開始した。1か月の時点では、どの試料にも有意な変化は観察されなかった。(データ示さず)。安定性条件は以下のとおりであった:PMTT10/6.0中にNEUGが50.0mg/mlのNEUG-2開発ロット;3mlガラスバイアル中に1.2mlsの溶液;2〜8℃、25℃/60%RH、または40℃/75%RHで保存。
【0220】
9.充填済みシリンジ中のNEUG-2溶液の試験
市販用の製品とするために、充填済みシリンジ中の溶液としてNEUG-2(PMTT、pH6.0)を提供することの実現可能性を評価するため、次の試験を行った。
【0221】
いくつかのシリンジ(例えば、Becton Dickinson製のもの)は、製品および/または緩衝剤と相互作用し得る二種類の主要な賦形剤であるタングステンおよびシリコーンを含むことが知られている。シリンジの賦形剤および製品の相互作用を評価するため、シリコーン油移動試験ならびにシリコーン添加およびタングステン添加試験、そして充填済みシリンジの長期安定性試験を含むいくつかの試験を行った。液体状態のCG10639(NEUG-2)の安定性の評価に使用した方法には、目視、SE-HPLC、RP-HPLC、IE-HPLC、バイオアッセイなどが含まれる。
【0222】
結果をまとめると、三種類の異なる用量(500、1000、および2500ppb)で五種類の形態のタングステン(タングステン酸ナトリウム無水物、ポリタングステン酸ナトリウム、タングステン酸アンモニウム、WO3、およびWO2)を添加し、3か月にわたって2〜8℃または25℃のいずれかで保存した試料には、有意な変化は見つからなかった。SE-HPLC、RP-HPLC、およびバイオアッセイにより試料を分析した(データ示さず)。
【0223】
シリコーン適合性試験では、異なる二種類の用量のシリコーン(0.4mgまたは0.8mg)でコーティングされたシリンジ中で2〜8℃で4週間試料を保存した場合、あるいは異なる二種類の用量のシリコーンを添加し2〜8℃で3か月試料を保存した場合、製品品質に対するシリコーンの影響は見られなかった(データ示さず)。調製用緩衝剤および製剤溶液の両方におけるシリコーン油分散は適合性があり、本質的に均等なままであった(データ示さず)。2か月保管後、充填済みシリンジに対する滑り力(gliding force)の変化はなかった(データ示さず)。さらに、CG10639は、撹拌および針によるせん断に対して感受性がないようであった(データ示さず)。
【0224】
少なくとも最大9か月までの長期安定性試験は、液状のNEUG-2は、2〜8℃で充填済みシリンジ中で安定であり、少なくとも最大18か月まで4℃で安定であり、製品と接触する物質と適合性があるということを示す。充填済みシリンジ中の液状のNEUG-2の長期安定性のデータを下記表13に示す。
【0225】
(表13)2〜8℃充填済みシリンジ中の液状NEUG-2の安定性
CPF=透明、淡黄色、外来の粉粒体が本質的に含まれていない
【0226】
これらの試験に基づくと、タングステンまたはシリコーンとPMTT製剤(20mMリン酸塩、180mMマンニトール、60mMトレハロース二水和物、0.01%(W/V)ポリソルベート80、pH6.0)を用いたNEUG製品との間に有意な相互作用はない。
【0227】
ヒト患者におけるNEUGの臨床評価
次の実施例は、「第I相」および「第II相」と題した2つのメインセクションにおいて記載する。各相は、2つの部分、即ちA部およびB部を含む。第I相および第II相の実施例は、下記表14にまとめる。なお、NEUG-1は第I相で試験し、NEUG-2は第II相で試験した。
【0228】
(表14)ヒト臨床NEUG試験の概要
【0229】
各相は、1)目的、2)患者の特徴、3)試験薬剤、4)試験の特徴、および5)A部およびB部の結果という5つのセクションに分かれる。
【0230】
実施例10:第I相
1.目的
第I相A/B、第II相A/B試験は、骨髄抑制性化学療法剤(ドキソルビシン/ドセタキセル)が投与された被験者における、皮下投与したNeugranin(商標)(「NEUG」)(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子)の安全性、耐容性、免疫原性、薬物動態、および薬力を評価するために行った。
【0231】
第I相では、第一の試験目的は、治療により発現した有害事象の頻度、重度、および継続時間を測定し、これらをNEUG投与の時間および用量と相関させることにより、ペグフィルグラスチムと比較して、一連の可能性のある治療用量にわたって皮下投与されたNEUGの安全性プロファイルを評価することであった。
【0232】
第二の試験目的は、NEUGの薬物動態および免疫原性を評価すること、ならびにドキソルビシン/ドセタキセルが投与される患者における、好中球減少症の罹患率、重症度、および継続期間に対するNEUG投与の効果をペグフィルグラスチムと比較することである。
【0233】
第I相は、上記表2に記載のように、A部およびB部という二部構成で行った。
【0234】
2.患者の特徴
第I相では、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
【0235】
採用:
1.ドキソルビシンおよびドセタキセルを投与される予定の組織学的に確認された乳癌患者。
2.18歳以上。
3.十分な血液学的機能。
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<2.0x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.ECOG一般状態0または1
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格。
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
【0236】
不採用:
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む);試験開始に先立つ4週間以内の任意の化学療法/免疫療法;この試験におけるドキソルビシンの2度の全量サイクルを除く累積的なアントラサイクリン投与。
2.試験化学療法の6週間以内の事前のニトロソウレア(BCNU、CCNU)またはマイトマイシンCのいずれかの使用。
3.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状。
4.試験化学療法の2週間以内の事前の手術または放射線治療。
5.骨盤に対するまたは骨髄支持面積の20%を超える面積に対する、または骨髄障害に対する事前の広範囲照射。
6.造血幹細胞移植による事前の高用量化学療法。
7.試験化学療法の4週間以内の骨髄性(G-CSFまたはGM-CSF)成長因子の事前使用。
8.試験化学療法の4週間以内のエリスロポエチンの事前使用。
9.骨髄性の悪性腫瘍または脊髄形成異常の病歴。
10.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
11.既知の鎌状赤血球症。
12.成人型呼吸窮迫症候群(ARDS)の診断。
13.静脈内または経口抗生物質を必要とする現行の感染症。
14.酵母由来製品に対するアレルギーの既知の履歴。
15.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分(第2相のみ)に対する既知の過敏症。
16.妊娠女性または授乳中の女性(試験期間中は、女性は全員、確実性が90%を超える避妊法を実施するか、不妊または閉経後でなければならない)。
17.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)。
18.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性。
【0237】
次の理由で被験者をさらなる治療から外した:
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
【0238】
NEUGによる治療を停止した場合、被験者は試験にとどまり、予定した安全性およびPK評価のために任意の治験薬の最後の投与後少なくとも30日間経過観察した。
【0239】
3.試験薬剤
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-G-CSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
【0240】
NEUGは、使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給され、2〜8℃で保存される。注射用滅菌水1.1mLで再構成する際、各バイアルは、pH7.2の10mMリン酸ナトリウム、200mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80中に、NEUGを15mg/ml(バイアル毎に15mgが送達可能)含む。
【0241】
第I相で使用したNEUG製剤の組成を表1に示す。
【0242】
市販のNeulasta(登録商標)(ペグフィルグラスチム)は、皮下注射用0.6ml充填済みシリンジで供給された。各シリンジは、注射用水中に酢酸塩(0.35mg)、ソルビトール(30.0mg)、ポリソルベート20(0.02mg)、ナトリウム(0.102mg)を含む無菌、透明、無色、防腐剤無添加の溶液(pH4.0)にペグフィルグラスチムを6mg(タンパク質重量に基づく)含む。USP。
【0243】
NEUG(50、150、300、もしくは450μg)またはNeulasta(登録商標)(ペグフィルグラスチム)(6mg)を皮下投与した。
【0244】
4.試験の特徴
a.試験スケジュールおよび継続期間
この試験は、ドキソルビシン/ドセタキセルを投与される予定である乳癌患者62人において行われた、ヒト初回投与、多施設、非盲検、非対照、逐次用量漸進試験であり、続いて対照無作為化試験を行った。この試験は、二部構成であった。A部は、13人の被験者、4つの用量コホート(50、150、300、または450μg/kg)における逐次用量漸進試験であり、50、150、および450μg/kgのコホートのそれぞれには3人の被験者で、300μg/kgのコホートには4人の被験者で、無作為化の実験B部を行う前に安全性を評価した。
【0245】
A部では、被験者は、細胞毒性を有する化学療法剤の非存在下で、安全性および好中球絶対数(「ANC」)に対する効果の初期評価のために、化学療法の開始(サイクル0)の少なくとも2週間前にNEUGの最初の投与を受けた。最低2週間の経過観察後、被験者は、サイクル0のNEUGに関連すると考えられる用量制限的な有害事象がなく、被験者がすべての適格基準を満たす場合、サイクル1およびサイクル2において化学療法後に同じ用量でNEUGの投与を受けた。
【0246】
A部では、用量制限毒性(DLT)は、グレード2骨痛を除いて、試験薬剤と、場合によっては関連する、恐らく関連する、または明確に関連すると考えられるグレード2以上の臨床的に有意な有害事象として規定された。各A部コホート内では、実験に参加した各被験者に対する最初の試験薬剤の投与は、急性有害事象をモニタリングするために、最低24時間隔てた。
【0247】
次の用量レベルまで漸進させる決定は、所与のコホート中の被験者全員についてNEUGの最初の投与後少なくとも7日間の安全データの検討に基づいて行った。被験者3人がだれもDLTを経験しない場合は、次の用量レベルにおいて被験者3人を加えて用量漸進を継続した。所与のコホートにおいて被験者3人の1人がDLTの兆候を示した場合、別の被験者3人をその用量レベルで補充し、1コホート当たり全部で6人の被験者になる。被験者6人の1人のみにDLTが起こった場合、用量漸進を続けた。被験者6人の2人にDLTが起きた場合、用量漸進を停止し、それ以上NEUG治療薬は投与しなかった。
【0248】
残りの被験者は、予定された安全性、薬物動態、および薬力学的評価を完了した。
【0249】
最初のA部コホートにおいて安全性が実証された後、B部を行った。B部では、被験者は3つの無作為化並行治療群の1つに分けられ、NEUG300μg/kg(n=20)、NEUG450μg/kg(n=21)、または承認された用量であるペグフィルグラスチム6mg(n=10)を試験化学療法の約24時間後に投与した。
【0250】
下記表15および表16は、第I相A部およびB部における被験者の割付をまとめたものである。図26は、第I相A部およびB部の化学療法サイクルを示す。
【0251】
(表15)第I相における被験者の割付
【0252】
(表16)第I相における被験者の割付
【0253】
b.第I相A部およびB部中の併用療法
化学療法
この実験の化学療法レジメンは、最大2回までの21日サイクルを、治療の1日目に静脈注射により逐次投与するドキソルビシン50mg/m2およびドセタキセル75mg/m2からなった。
【0254】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1.5x109/Lであり、血小板が>100x109/Lでなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることもあり得た。
【0255】
ドキソルビシンおよびドセタキセルの組み合わせは、乳癌患者において有意な臨床上の活性を有すると報告されている。しかしながら、この組み合わせは、骨髄抑制性であり、他の標準的なレジメンよりも高い割合でグレード3またはグレード4の好中球減少症が起きる。
【0256】
CSFを添加しても、ドキソルビシンおよびドセタキセルの組み合わせは、患者の79%にグレード4好中球減少症誘発し、9〜18%の割合で発熱性好中球減少症を誘発する。このドキソルビシン/ドセタキセルレジメンは、好中球減少症およびその合併症を予防するための新規な薬剤の研究に使用されている。したがって、ドキソルビシンおよびドセタキセルの組み合わせは、NEUGのような新規の薬剤の可能性を研究するのに適切な化学療法レジメンである。
【0257】
ドキソルビシン
薬理学的データ
塩酸ドキソルビシンは、DNAおよびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウス(Streptomyces peucetius var caesius)から得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
【0258】
医薬データ
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
【0259】
副作用および毒性
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。著しい脱毛症および中程度の悪心/嘔吐は、予想される毒性である。偶発的な溢出の部位での局所的な皮膚および組織損傷を生ずる溢出反応、口内炎、皮膚(特に爪床)の色素過剰症、ならびに過去に照射した部位での「メモリ」現象が報告されている。
【0260】
ドセタキセル
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
【0261】
医薬データ
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
【0262】
副作用および毒性
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
【0263】
激しい過敏反応を経験する患者には再投薬すべきではない。下に概説するように、ドセタキセルが投与される患者には、コルチコステロイドを前投与するのがよい。
【0264】
軽度から中程度の肝機能障害では、代謝が27%遅延し、全身暴露(AUC)が38%増加する。ドセタキセルは、SGOTおよび/またはSGPTが正常範囲の>1.5倍、およびアルカリフォスファターゼが正常範囲の>2.5倍の患者には投与すべきではない。体液鬱滞が、コルチコステロイドを前投与したにもかかわらず、第III相試験における患者の17%(中程度)および6%(重度鬱滞)で起こった。重度の知覚神経症(感覚異常、錯感覚、疼痛)が観察されている。
【0265】
予想される副作用としては、最下点が約9日間あり15〜21日目までには回復する骨髄抑制(主に白血球減少症)が挙げられる。脱毛症、爪および皮膚の変化、口内炎、筋肉痛/関節痛、悪心/嘔吐、ならびに低血圧が報告されている。
【0266】
化学療法剤の用量、投与、および用量の変更
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
【0267】
ドキソルビシンは、管外溢出による傷害を回避するため、点滴用静脈ラインまたは中心静脈カテーテルのサイドアームを介してIVボーラスにより50mg/m2の用量で投与した。
【0268】
75mg/m2のドセタキセルは、0.9%食塩水または5%デキストロース溶液250mLで希釈し、ポリエチレンライニングを有する輸液セットを介して約1時間にわたって静脈内投与した。バイタルサインは、ドセタキセル点滴の直前および直後に得た。
【0269】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1500/mm3であり、血小板が>100,000/mm3でなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることもあり得た。グレード3〜4の非血液毒性、グレード3〜4の感染エピソード、またはグレード4の血小板減少症に対しては化学療法剤の用量を25%減少させた。
【0270】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者は試験治療から除外されたが、経過観察は完了した。
【0271】
化学療法剤の前投薬
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
【0272】
制吐薬または他の前投薬(例えば、H2拮抗薬)の使用および選択は、治療を行う医師の裁量に任せた。
【0273】
禁止薬剤
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けないのがよい。
1.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
2.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
3.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療された場合、NEUGの投与に適格ではなく、試験を中断した。
【0274】
許可薬剤
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症は、CRFの適切なページに記録した。
【0275】
抗生物質
被験者は全員、感染の可能性を減らすため、化学療法の各コースに続いて予防用経口抗生物質(例えば、シプロフロキサシン)を投与された。発熱性好中球減少症または持続性の重度の好中球減少症(ANC<0.5x109/L、≧5日)が起こった場合、被験者は、治療不成功と考えられ、試験から除外され、経過観察を完了し、研究者の裁量による成長因子支持を含む標準的な支持療法すべて受けた。
【0276】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者も試験治療から除外され、そして経過観察を完了した。
【0277】
c.安全性評価
NEUGの安全性は、有害事象(「AE」)の種類、頻度、および重度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準に基づいて等級分けした(NCI-CTCAEバージョン3.0、2003年12月12日)。有害事象(重度有害事象「SAE」を含む)は、試験薬剤の投与開始から任意の試験薬剤の最後の投与後30日まで記録した。実験的評価は、評価スケジュール(Schedule of Assessments)で概説されているように得た。グレード4好中球減少症毒性の場合には、ANCが>500になるまで毎日実験的評価を得た。被験者の次の治療サイクルが遅延した場合(そして最後の処置サイクルの後)、ANCが>1500になるまで、鑑別を伴う全血球計算(CBC)を毎週少なくとも2回行った。
【0278】
5.第I相A部およびB部の結果
a.概論
統計的方法:
安全性、薬物動態(PK)、薬力(PD)、および免疫原性パラメータに関連するデータは、記述統計法を用いて分析した。
【0279】
有害事象の頻度および重度、ならびに実験室毒性の等級分けに関して、数および割合を示す。
【0280】
効能分析には、グレード4およびグレード3〜4の好中球減少症の発生率および持続期間、最下点ANC、最下点ANCまでの時間、回復するまでの時間(ANC>0.5x109/LおよびANC>1.0x109/Lまで)、ならびに発熱性好中球減少症の発生率が含まれた。
【0281】
この試験には、サンプルサイズを選ぶためには厳密な統計的検出力要件は使用しなかった。5%有意水準でペグフィルグラスチムと比較してNEUGが劣っていないことを示す80%検出力を用いた試験では、一治療群当たり被験者約37人を必要とすると計算された。これは、主に安全性に関して行った第l/2a相試験であったので、効果が検出されるために必要なサンプルサイズは適切なものよりも大きいと決定された。このように、効能傾向を評価した。
【0282】
内訳/個体群統計:
合計13人の被験者が、A部(実験において用量が逐次的に漸進する部分)に参加した。合計51人の被験者がB部に参加し、無作為にNEUG300μg/kg(n=20)、NEUG450μg/kg(N=21)、またはペグフィルグラスチム6mg(n=10)に分けられた。
【0283】
b.試験結果
最初の用量設定試験では、化学療法剤の非存在下で、NEUGは、良好な耐容性を示し、予想されたようにANCが上昇し、これは2日目と4日目の間にピークに達し、14日目までに正常に戻った(図23)。
【0284】
A部では、50μg/kgNEUG投与群中の被験者3人全員および450μg/kgNeugranin投与群中の被験者1人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。B部では、300μg/kgNEUG投与群中の被験者1人および450μg/kgNEUG投与群中の被験者2人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。ペグフィルグラスチム群中の被験者1人が、5日を超えて継続する発熱性好中球減少症または重度の好中球減少症になった。
【0285】
c.免疫原性
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目での投与前および治療訪問の終わり(最後の投与の少なくとも15日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得た。
【0286】
A部およびB部の両方の治療の終わりをもって被験者全員に対する試験を完了した。試料はすべて、NEUGに対する抗体には陰性であった。
【0287】
d.有害事象
A部では、用量制限毒性(DLT)は、グレード2髄骨痛を除いて、試験薬剤と、場合によっては関連する、恐らく関連する、または明確に関連すると考えられるグレード2以上の臨床的に有意な有害事象として規定された。
【0288】
A部のコホートのいずれもサイクル0ではDLTに達しなかった。NEUG投与に関連して、骨痛と既存の高血圧の悪化(後者はNEUG投与の7日後に起きた)という2つの有害事象のみ報告された。いずれの事象も後遺症なく解消した。
【0289】
NEUG治療被験者41人のうち31人は少なくとも1つの有害事象を経験した。NEUGおよびペグフィルグラスチム治療被験者中のAEの発生率は同程度であった(それぞれ75.6%および70%)。
【0290】
B部で一般に報告される有害事象(被験者全員のうちの5%以上のAE)をまとめたものを表17に示す。
【0291】
(表17)第1相B部集団における治療関連有害事象のまとめ
1関連=場合によっては関連している、恐らく関連している、または明確に関連していると考えられる
2非関連=恐らく関連していないか、関連していないと考えられる
【0292】
NEUGに関連すると考えられる最も多く報告される有害事象は、すべてのG-CSF製品に伴う典型的な有害反応である骨痛であり、これは上記表に記載の4人および450μg/kg投与されるA部被験者1人の5人の患者において報告された。すべての事例で、骨痛は、強度がNCI-CTCAEグレード1〜2であり、持続期間としては一過性で、後遺症なく解消した。アルカリフォスファターゼおよび尿酸におけるグレード1上昇は、サイクル0におけるNEUGの投与後に起こり;これらの事象は、研究者によって臨床的に有意でないと考えられ、処置せずに解消した。これらは、G-CSF(例えば、Neulasta(登録商標))を投与される患者における予想される作用である。
【0293】
化学療法サイクル中の他の一般に報告される有害事象(悪心、嘔吐、脱毛症、口内炎)は、ドキソルビシン/ドセタキセルレジメンが投与される患者において予想される有害事象と一致していた。
【0294】
大多数の報告されたAEは、NCI CTCグレード1またはグレード2の重篤度であった。4つのAEは、重篤な有害事象として報告された。被験者2人(1人は150μg/kg投与されもう1人は450μg/kg投与された)は嘔吐のために入院し、これらの被験者の1人は、次の化学療法サイクルで二度目のSAEを経験した;嘔吐の程度は軽かったが、入院の原因となったかまたは入院を長引かせた。450μg/kg投与された3人目の被験者は、発熱性好中球減少症のため入院した。当該事象は、NEUGと無関係であると考えられた。
【0295】
e.薬物動態
NEUGが投与される被験者は全員、試験期間中に渡って血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン4.1以上)を用いてPK分析に供した。
【0296】
以下のPKパラメータを得た:曲線下面積(AUC0-∞)、クリアランス(CL/F)、分布容積(Vz/F)、最大濃度(Cmax)、吸収半減期(t1/2、abs)、排出半減期(t1/2、elim)、および平均滞留時間(MRT)。薬物動態データは、プロトコル中で用いた用量範囲にわたって直線性について評価した。
【0297】
サイクル0(化学療法前)からの薬物動態パラメータは表18にまとめてあり、サイクル0のPKプロファイルは図27に示してある。
【0298】
(表18)ヒト被験者におけるNeugranin薬物動態(第1相サイクル0)
【0299】
最大血清NEUG濃度および時間濃度曲線下面積により測定される薬物暴露は、用量依存的に増加した。最初の50μg/kg用量コホート中の被験者の血清濃度は、一貫して量子化の下限(6.3ng/mL)未満であった。Tmaxは、150から450μg/kgまでのすべての用量で6〜24時間の範囲であった。Cmaxは、150μg/kgの用量で72.7±59.7(平均±SD)ng/mLから450μg/kgの用量で294±351ng/mLまでであった。対応して、AUC0-∞は、150mcg/kgの用量で1758±1675ng/mL*hrから450μg/kgの用量で10131±9563ng/mL*hrであった。サイクル1の範囲は類似していた。NEUGの平均排出半減期は、14〜30時間の範囲であった。
【0300】
「試験の特徴」(上のセクション4)で記載したように、A部の被験者は、細胞毒性を有する化学療法の非存在下で、安全性および好中球絶対数(「ANC」)に対する影響の初期評価のために、化学療法の開始(サイクル0)の少なくとも2週間前にNEUGの最初の投与を受けた。最低2週間の経過観察後、サイクル0においてNEUGに関連すると考えられる用量制限的な有害事象がなく、被験者がすべての適格基準をみたす場合は、被験者は、サイクル1およびサイクル2における化学療法後に、同じ用量のNEUGの投与を受けた。NEUGは、化学療法剤投与の24時間後に投与された。図24Aおよび図24Bは、サイクル1およびサイクル2中でNEUGが投与された被験者のANC数およびWBC数を示す。
【0301】
f.B部の用量の薬力学および設定
試験の第I相A部からのデータの分析によって次の所見が得られた:
1.NEUGは、サイクル0(化学療法前)においてWBCおよびANCの用量依存的な上昇を誘導する(図24Aおよび図24Bにおけるサイクル0データ参照)。
2.サイクル0におけるANCの増加は、等モル量のペグフィルグラスチムの病歴データと同程度であった
3.予想されたように、化学療法後にWBCおよびANCは低下する
4.最下点ANCからの回復は、用量依存依存的であると考えられる
5.ANCおよびWBCは、15日目までに正常値まで回復した
【0302】
A部でのすべての用量レベルでの安全性のこれらの所見および実証に基づいて、B部の評価のための選択した用量は300および450μg/kgであった。上に記載のように、被験者は、NEUG300μg/kg、NEUG450μg/kg、またはペグフィルグラスチムを認可された固定投与量である6mgに無作為に分けた。被験者は、ドキソルビシン/ドセタキセルの投与1日後にNEUGまたはペグフィルグラスチムを投与された(2サイクルにわたる投与、21日隔てて)。B部のデータは、集団のサイクル1ANCプロファイルを含む。結果を図28および下記表18にまとめる。
【0303】
グレード3およびグレード4好中球減少症の発生率およびサイクル1中のANCプロファイルは、表19に示すように、治療した被験者51人中の48人で測定された。なお、予防的なG-CSF治療が無いと、ドキソルビシン/ドセタキセルで治療した患者の70〜80%が平均で5日間の持続期間を有するグレード4好中球減少症になる。
【0304】
(表19)化学療法のサイクル1後の第I相B部におけるグレード4好中球減少症の発生率および持続期間
【0305】
治療群の平均ANC曲線を図28に示す。
【0306】
NEUGは、グレード3、グレード4、および発熱性好中球減少症の治療に効果がある。この化学療法レジメンに対してG-CSF治療がないと、発熱性好中球減少症の発生率は約40%になる。ANCの用量相関性の上昇およびドキソルビシン/ドセタキセルで予想されるよりも低い好中球減少症の割合がNEUGの投与後に観察された。NEUGに起因する予想外または重篤な有害事象はなかった。
【0307】
グレード3およびグレード4好中球減少症の発生率は、ペグフィルグラスチム(Neulasta(登録商標))が投与された患者よりも300μg/kgのNEUGが投与された患者でより高く、正常なANCまで回復する速度も、ペグフィルグラスチムが投与される被験者よりもNEUGが300μg/kg投与される患者でより遅いようであった。NEUGが450μg/kg投与される患者のANCプロファイルは、ペグフィルグラスチムが投与される患者と類似していたが、好中球減少症からの回復の際のANCは、ペグフィルグラスチムが投与される患者よりもNEUGが投与される患者において一般により低かった。要約すると、これらの用量のNEUGは、ペグフィルグラスチムと同様の効果をもたらすようである。
【0308】
f.PK/PDプロファイル、第I相、B部
乳癌治療のサイクル1におけるドキソルビシン/ドセタキセル投与の1日後にNEUGが450μg/kg投与された患者からのPK/PDプロファイルを図29に示す。NEUGのCmaxは、投与の1日以内に達成され、10日目までに検出できないレベルにまで徐々に低下した。NEUGの投与後に、ANCは4日目までにピークにまで上昇し、ANCは、ドキソルビシン/ドセタキセルおよびG-CSF治療を受ける患者において予想される通りに、8日目に最下点まで低下し、10日目に正常値にまで戻る。12日目までには、ANC値は正常範囲にあり、NEUGは検出できない。なお、予防的なG-CSF治療を受けない患者では、最下点ANCの期間およびANCが回復するまでにかかる時間はずっと長い(例えば、5〜7日)。450μg/kg投与後、NEUG排出半減期のメジアンは約30時間であり、これに対し、ペグフィルグラスチムの標準用量では15〜80時間と報告されている。
【0309】
g.NEUGとペグフィルグラスチムとの間のさらなる差異
好中球減少症からの回復を促進させる効果における試験用量でのNEUGとペグフィルグラスチムとの間のより詳細な差異は、治療のサイクル1における個々のANCプロファイルを比較すれば明らかである。すべての群のピークANCは非常に類似しており、NEUGが300μg/kg投与された被験者の最下点ANCは、NEUGが450μg/kg投与された被験者より低く、ペグフィルグラスチムが投与された被験者のANC最下点は平均して最も高かった。最下点ANCからベースラインまでの回復がすべての治療群で14日目までに起きたが、NEUGが450μg/kg投与された被験者よりもNEUGが300μg/kg投与された被験者では遅く、ペグフィルグラスチムが投与された被験者で最も速かった。
【0310】
類似の化学療法前の投与を伴うペグフィルグラスチム実験の入手可能な公表済みデータを、予定したサイクル0(化学療法前)までの第I相を完了した患者からNEUGのPK/PDデータと比較した。この比較の結果は以下のとおりであった:
1.NEUG用量が150μg/kgのEmax(最大観測ANC)は、サイクル0におけるペグフィルグラスチム用量30μg/kg(確認された有効なペグフィルグラスチム用量である100μg/kgに対して効能で劣ることが後に示された用量)と一致する。
2.Neugranin用量が300および450μg/kgのEmaxは、ペグフィルグラスチム用量が100μg/kgのサイクル0レベルとより一致していた。
3.NEUG300μg/kgおよび450μg/kgのCmaxのメジアンおよびEmaxのメジアンはほぼ同じであり、したがって、Cmaxによって引き続きEmaxが予測される。
4.ANC増加は、等モル量のペグフィルグラスチムの出版済みデータと同程度であった。
【0311】
上に記載したように、動物およびヒトにおけるPK/PD評価は、等モル基準で投与したNEUGおよびペグフィルグラスチム用量等価性の推定値と一致していた。マウスでは、同等のAUCANCが、ペグフィルグラスチムより7.7倍高い用量で達成された。アルブミンがNEUGの分子量に著しく寄与し、Neulasta(登録商標)(ペグフィルグラスチム)が、rhG-CSFの重量(ペグフィルグラスチム中のポリエチレングリコールの寄与を含まない)に基づいて投与されるので、4.5倍の用量のNEUG(重量基準)は、等量のNeulasta(登録商標)(ペグフィルグラスチム)と同程度に効果的であると予測される。動物における効能データは、ペグフィルグラスチムの4.5〜7.7倍と一致した(ペグフィルグラスチム1mg=NEUG4.5〜7.7)。非臨床上の安全性および効果のデータは、この用量推定値と一致しており、入手可能な臨床データと検討すると、臨床評価のための用量を選ぶ際の根拠になる。
【0312】
h.第I相の結果
第I相薬物動態評価の結果は次のとおりである。
NEUGは、サイクル0およびサイクル1で、150μg/kg、300μg/kg、および450μg/kgの用量にてNEUGで処理されたすべての被験者からの血清試料において検出された。
サイクル1では、NEUGは、150mg/kg、300μg/kg、および450μg/kg用量群において、ほとんどの被験者(45/50でサンプリングされた)で最大144時間まで検出された。実質的に、サイクル間での薬剤蓄積は観察されなかった。
薬物暴露は、各用量群でサイクル1およびサイクル0(前化学療法)においてより高かった。サイクル1におけるNEUGへの暴露の増加は、受容体が仲介するG-CSFクリアランスにおいて役割を果たす好中球の数の減少による可能性がある。
サイクル1におけるNEUGの排出半減期のメジアンは、300μg/kg用量群で約36時間であり、450μg/kg用量群で30時間であった。排出半減期は、フィルグラスチムで3〜4時間であり、ペグフィルグラスチムで、用量に応じて42〜67.5時間であると報告されている。
用量間の統計的有意差は、最大血清濃度(tmax)までの時間および吸収半減期(t1/2、abs)に見られた。これらのパラメータはいずれも、NEUG用量の増加と共に増加した。他の用量正規化PKパラメータは、用量間の統計的有意差を示さなかった。
【0313】
実施例11:第II相
試験の第II相は、ドキソルビシン/ドセタキセルを最大4回まで投与した乳癌を有する被験者334人で行われた対照無作為化試験であった。試験は、臨床現場約50カ所で行われ、皮下投与したNEUGとペグフィルグラスチムとの安全性および効果を評価するための二元配置無作為化パイロットフェーズ、その後の、被験者がパイロットフェーズに基づいて選ばれるペグフィルグラスチムおよび2つの良好な耐性用量のNEUGに無作為に分けられる(1:1:1)メインフェーズからなった。主要評価項目である化学療法サイクル1中の重度(グレード4)の好中球減少(DSN)の持続期間に関してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、メインフェーズのサンプルサイズを検出した。試験の設計を下に概略的に示す。
【0314】
1.目的
第II相の第一の目的は、ペグフィルグラスチムに対して同等の効果を示すNEUG用量を選択すること、およびNEUGでの治療後の化学療法のサイクル1において重度の好中球減少(DSN)の持続期間を評価することであった。第二の目的は、サイクル2〜4におけるDSNを評価すること、好中球絶対数が回復するまでの時間およびサイクル1〜4における発熱性好中球減少症の割合を評価すること;ならびにNEUGの安全性、耐容性、薬物動態(サイクル1において)、および免疫原性を評価することであった。
【0315】
2.患者の特徴
第II相に関連して、患者は、下記の特徴またはパラメータに基づいてスクリーニングされた。
【0316】
採用:
1.ドキソルビシンを60mg/m2およびドセタキセルを75mg/m2投与される予定の組織学的に確認された乳癌患者。
2.18歳以上
3.十分な血液学的機能:
4.ANC>1500/mm3
5.血小板>100,000/mm3
6.十分な肝機能および腎機能:
7.血清クレアチニン<1.5x正常上限値
8.検査室(local laboratory)での正常範囲内(WNL)の総ビリルビン
9.血清トランスアミナーゼ(SGOT/SGPT)<1.5x正常上限値
10.アルカリフォスファターゼ<2.5x正常上限値
11.米国東海岸癌臨床試験グループ(「ECOG」)一般状態0〜2
12.正常範囲内の左心室駆出分画(LVEF)に基づいたドキソルビシン投与を受ける資格
13.試験条件を理解する能力を有すること、書面によるインフォームドコンセント(研究に関連する健康情報の使用および開示に関する同意書を含む)を提供すること、ならびに試験プロトコル手順を順守すること。
【0317】
不採用:
1.過去に1を超える化学療法レジメン(過去12か月以内に行われたのであればアジュバント療法も含む)
2.この試験におけるドキソルビシンの4度の全量サイクルを除く累積的なアントラサイクリン投与
3.試験化学療法剤の30日以内の事前の化学療法/免疫療法(ニトロソウレア(BCNU、CCNU)またはマイトマイシンCの試験化学療法剤の6週間以内)
4.トラスツズマブ(ハーセプチン)併用療法
5.過去30日での治験薬の投与
6.アントラサイクリンに基づいた化学療法レジメンの使用を除く、研究者の見解における心臓の病歴、兆候、または症状
7.試験化学療法の2週間以内の事前手術
8.試験化学療法の4週間以内の事前手術(骨転移のためのスポット照射を除く)
9.造血幹細胞移植による事前の高用量化学療法
10.試験化学療法の4週間以内のG-CSF、GM-CSF、またはエリスロポエチンの事前使用
11.試験化学療法の72時間以内の抗生物質の全身投与
12.骨髄性の悪性腫瘍または脊髄形成異常の病歴
13.既知の脳転移(十分に治療(手術または放射線療法)され、最低3週間の観察で進行が見られず、抗痙攣薬およびステロイドを使用しないで神経学的に安定している場合を除く)。
14.既知の鎌状赤血球症
15.成人型呼吸窮迫症候群(ARDS)の診断
16.酵母由来製品に対するアレルギーの既知の履歴
17.大腸菌由来タンパク質、ペグフィルグラスチム、フィルグラスチム、またはペグフィルグラスチムの任意の他の成分に対する既知の過敏症
18.妊娠女性または授乳中の女性。(子宮が無傷である女性は全員スクリーニングされて血清妊娠テストが陰性でなければならない。不妊でないかまたは非閉経前の女性は全員、試験期間中および試験薬剤の最後の投与後の30日間医学的に認められた避妊法を実施しなければならない。)
19.試験期間中および試験薬剤の最後の投与後30日間効果的な避妊を使用することに合意しない男性
20.HIV陽性または活動性肝炎が既知(未知の状態の患者は試験されない)
【0318】
次の理由で被験者をさらなる治療から外した:
1.疾患進行
2.最善の治療にもかかわらず容認できない毒性
3.研究者の裁量による併発性の病気
4.ドキソルビシンレジメン-寿命中で許容される最大蓄積量に到達(適格基準参照)
5.同意の撤回
6.経過観察への非遵守/喪失
7.妊娠
【0319】
試験薬剤による治療を停止した場合、試験にとどまった被験者は、予定した安全性およびPK評価のために試験薬剤の最後の投与後少なくとも30日間経過観察した。
【0320】
3.研究薬剤
NEUG(組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、rHSA-GCSF)は、成熟型HSAに対応する残基1〜585および成熟型ヒトG-CSFに対応する残基586〜759を含む一本鎖に繋がった約85kDaの分子量を有する融合タンパク質である。NEUGの治療有効部分は、組換えヒトDNA由来G-CSFである。
【0321】
NEUGは、使い捨てのタイプ1ガラスバイアル中で無菌の凍結乾燥製剤として供給され、2〜8℃で保存される。注射用滅菌水1.0mlで再構成する際、各バイアルは、20mMリン酸ナトリウム、180mMマンニトール、60mMトレハロース無水物、0.01%(w/v)ポリソルベート80、pH6.0中に、NEUGを50mg/ml(バイアル毎に50mgが送達可能)含む。
【0322】
第II相で使用した製剤の組成を表11に示す。第I相および第II相で使用したNEUG製剤間の差異を下記表20に示す。
【0323】
(表20)cGMP製剤の比較
【0324】
第I相で使用された製剤はきわめて安定しており、少なくとも2年間の保存期間を有していた。試験により、より大きなイオン強度およびより低いpHが、より高い濃度(>25mg/mL)でAPIを安定させることが示された(データ示さず)。このために、第II相の製剤は、より低いpH(6.0対7.2)およびより高いリン酸塩濃度(20対10 mM)を有する。強制分解試験は、この製剤が、激しい振とう、凍結融解の繰り返し、および濃度により誘導される凝集から液体状態の薬剤成分を保護することを示した。第II相の製剤の凍結乾燥はまた、良好に形成されたケーキを生じる。
【0325】
市販のNeulasta(登録商標)(ペグフィルグラスチム)は、皮下注射用0.6ml充填済みシリンジで供給される。各シリンジは、注射用水中に酢酸塩(0.35mg)、ソルビトール(30.0mg)、ポリソルベート20(0.02mg)、ナトリウム(0.102mg)を含む無菌、透明、無色、防腐剤無添加の溶液(pH4.0)にペグフィルグラスチムを6mg(タンパク質重量に基づく)含む。USP。
【0326】
NEUG(30、40、50、もしくは60mg)またはNeulasta(登録商標)(ペグフィルグラスチム)(6mg)を皮下投与した。
【0327】
用量の論理的根拠
第I相からのデータは、300および450μg/kgのNEUG用量が安全であり良好な耐容性があることを示した。さらに、ペグフィルグラスチムの承認された固定用量と比較して、NEUGのいずれの用量も、細胞毒性を有する化学療法を受ける乳癌患者におけるANCプロファイルに対して同様の効果をもたらした。ANCプロファイルのAUCは、効果の一点計測値(single-point measure of effect)としての役割を果たす。AUCANCについてはこれらの治療群間で統計的有意差はなかったが、450μg/kg群のAUCは、300μg/kg群で観察されたものよりもわずかに高く、ペグフィルグラスチム群で観察されたものとほとんど同じである(図30)。入手可能なデータに基づくと、NEUGは、300μg/kgではペグフィルグラスチムより効果が低く、450μg/kgでペグフィルグラスチムと同様の効果を与えるためのおおよその最小必要用量であると推定された。
【0328】
固定用量の目的は、患者の体重にかかわらず効能および安全性をもたらすのに十分な用量を患者に提供する用量を確認することである。第I相の結果に基づくと、NEUGは450μg/kgでペグフィルグラスチムと類似の効果を与える最小必要用量であり得ると推定され、>300μg/kgが第II相でさらに評価するための最小量として設定された。NEUGの固定用量を選択するため、第II相の患者集団(乳癌)をモデリングした。40〜100kgの体重範囲を用いて、最も重い患者には30mgの固定用量で最小量(300μg/kgまたは0.3mg/kg)が与えられる一方で、約75%の患者には、40mgの固定用量で、少なくとも標的用量である450mg/kgが投与された。したがって、第II相での評価に選択した用量は、30mg、40mgおよび50mgであった。これらの用量は、平均70kgの患者にそれぞれ0.42、0.57、および0.71mg/kgの用量を提供する。
【0329】
この実験で評価された固定用量に基づいたキログラム当たりの等価用量を表21に示す。
【0330】
(表21)予測される被験者体重範囲のキログラム当たりの等価用量
【0331】
NEUGの非臨床安全性は、これらの用量での安全性の予想に対するさらなるサポートを提供する。これらの固定用量(AUCおよびCmax)での患者の暴露は、サルで良好な耐容性がある用量での暴露より低いと予想される。例えば、良好な耐性用量である1mg/kgでのサルにおけるCmaxおよびAUCは、0.45mg/kgでの患者における暴露より12倍高く、これは、患者におけるより高い用量評価およびサルにおける反復用量毒性試験でさらなる安全域が存在することを示唆しており、10mg/kg以下の用量で良好な耐容性があった。0.3mg/kgもの高いペグフィルグラスチム用量が患者において安全であることが示されている。
【0332】
4.試験の特徴
a.試験スケジュールおよび継続時間
この試験は、ドキソルビシン/ドセタキセルが最大4回まで投与される予定の乳癌患者被験者約330人で行われた対照無作為化試験であった。約45カ所の臨床現場で行ったこの試験は、パイロットフェーズおよびメインフェーズの2相からなった。
【0333】
パイロットフェーズ(A部)は、二元配置無作為化試験からなり、次の用量を順次行ってNEUG対ペグフィルグラスチムの安全性および効果を評価した:NEUG30mg(N=10)対ペグフィルグラスチム(N=5);NEUG40mg(N=20)対ペグフィルグラスチム(N=10)、およびNEUG50mg(N=20)対ペグフィルグラスチム(N=10)。さらなる試験では、NEUG60mg(N=20)対ペグフィルグラスチム(N=10)も検査し得る。A部パイロットフェーズでは、被験者は、30mgコホート中に全部で被験者10人および他の各コホートに被験者20人で、NEUGとペグフィルグラスチムとに2:1の割合で無作為に分けた。NEUGまたはペグフィルグラスチムは、各サイクルの化学療法治療の24時間後に被験者に投与した。被験者は、体重(<50kg、≧50kg-<80kg、または≧80kg)、事前の化学療法暴露、および地球上の位置に基づいて治療群中でバランスをとるために層別無作為化を用いて、治療群に割り振られた。
【0334】
パイロットフェーズに続いて、被験者255人を、ペグフィルグラスチム、およびパイロットフェーズにおいてよりもペグフィルグラスチムにより類似する効果を有する2つの良好な耐性用量のNEUGに無作為に分類した(1:1:1)(3治療群、平衡、並行無作為化相)。NEUGまたはペグフィルグラスチムは、各サイクルの化学療法治療の24時間後に投与した。被験者は、体重(<50kg、≧50kg-<80kg、または≧80kg)に基づいて治療群中でバランスをとるために層別無作為化を用いて、治療群に割り振られた。
【0335】
パイロットフェーズ中、有害事象は継続的に評価した。継続的なデータの評価によって安全性の懸念が示唆されなければ、30から50mgまで用量を増加させた。Neugranin40mgのサイクル1ANCプロファイルが、ペグフィルグラスチム患者に見られるプロファイルより劣っていると考えられ、Neugraninが50mgで安全である場合、さらなる治療群を、コホート中の患者総勢30人で、Neugranin60mgとペグフィルグラスチムとを2:1の割合で無作為に分けてもよい。
【0336】
NEUGの各用量レベルを、安全性および効能についてペグフィルグラスチムと比較する。表22は、第II相のA部およびB部の患者の割付をまとめたものである。
【0337】
(表22)第II相A部およびB部における被験者の割付
【0338】
安全性評価:
NEUGの安全性は、AEの種類、頻度、および重症度の評価、臨床検査(血液学および臨床化学)の変化、免疫原性、身体検査、ならびに継時的なバイタルサインのモニタリングにより評価した。AEおよび実験室毒性はすべて、国立癌研究所有害事象共通用語基準(NCI-CTCAEバージョン3.0、2003年12月12日)に基づいて等級分けした。
【0339】
有害事象は、試験薬剤の投与開始から任意の試験薬剤の最後の投与後30日まで記録した。重篤な有害事象(SAE)は、同意時から任意の試験薬剤の最後の投与後30日まで記録した。実験室評価は、評価スケジュールで概説されているように入手した。
【0340】
c.併用療法
化学療法
この実験の化学療法レジメンは、最大4回の21日サイクルを、治療の1日目に静脈注射により逐次的に投与するドキソルビシン60mg/m2およびドセタキセル75mg/m2からなった。
【0341】
療法の各サイクルを受ける前に、被験者は、好中球絶対数(ANC)が>1000/mm3であり、血小板が>100,000/mm3でなければならなかった。血液学的な回復のため、最大2週間まで治療を遅らせることができた。グレード3〜4の非血液毒性、グレード3〜4の感染エピソード、またはグレード4の血小板減少症に対して、化学療法剤の用量を25%減少させることが可能であった。予防用抗生物質または他の造血成長因子の使用は試験に参加している間は禁止された。
【0342】
ドキソルビシンおよびドセタキセルの組み合わせは、乳癌患者において有意な臨床上の活性を有すると報告されている。しかしながら、この組み合わせは、骨髄抑制性であり、他の標準的なレジメンよりも高い割合でグレード3またはグレード4の好中球減少症が起きる。
【0343】
CSFを添加しても、ドキソルビシンおよびドセタキセルの組み合わせは、患者の79%にグレード4好中球減少症を誘発し、9〜18%の割合で発熱性好中球減少症を誘発する。このドキソルビシン/ドセタキセルレジメンは、好中球減少症およびその合併症を予防するための新規な薬剤の研究に使用されている。したがって、ドキソルビシンおよびドセタキセルの組み合わせは、NEUGのような新規の薬剤の可能性を研究するのに適切な化学療法レジメンである。
【0344】
ドキソルビシン
薬理学的データ
塩酸ドキソルビシンは、DNA塩基対に直接結合し(インターカレートし)、DNA合成およびDNA依存RNA合成ならびにタンパク質合成を阻害するストレプトミセス・ペウセティウス var カエシウスから得られるアントラサイクリン系抗生物質である。ドキソルビシンは、細胞周期のどの期においても活性があるが、S期において最も細胞毒性がある。当該薬物の排出は、主に肝臓によるものであり、腎クリアランスは小さい。
【0345】
医薬データ
前記薬物は、10、20、50、100、または200mgバイアル中で市販されている。凍結乾燥製剤は、注射用滅菌水、5%デキストロース溶液、または0.9%注射用生理食塩水で再構成されてもよい。
【0346】
副作用および毒性
約10〜14日間に最下点を有する骨髄抑制(主に白血球減少症)、ならびにまれな急性心嚢炎心筋炎症候群および累積的な容量に伴う後発性の心筋症を含む心毒性が、ドキソルビシンの用量制限毒性である。
【0347】
著しい脱毛症および中程度の悪心/嘔吐は、予想される毒性である。偶発的な溢出の部位での局所的な皮膚および組織損傷を生ずる溢出反応、口内炎、皮膚(特に爪床)の色素過剰症、ならびに過去に照射した部位での「メモリ」現象が報告されている。
【0348】
ドセタキセル
薬理学的データ
ドセタキセルは、遊離チューブリンに結合する半合成タキソイドであり、安定した微小管の構築を促進し、有糸分裂および細胞複製(M期に特異的な細胞周期)を妨害する。ドセタキセルは、タンパク質に広く結合し、肝臓において多くは代謝され、7日以内に用量の約75%が糞便中に排泄される。
【0349】
医薬データ
ドセタキセル(Taxotere(商標)、Sanofi Aventis)は、80mg/2mLまたは20mg/0.5mlの単回投与バイアル中に提供され、希釈剤(注射用水中13%エタノール)バイアルが付帯される。Taxotereは1ml毎にドセタキセル(無水)を40mgおよびポリソルベート80を1080mg含む。
【0350】
副作用および毒性
ドセタキセルは、ドセタキセルまたはエトポシドおよびビタミンEなどのポリソルベート80と調製された他の薬剤に対して激しい過敏反応の既往歴のある患者には投薬すべきではない。
【0351】
激しい過敏反応を経験する患者には再投薬すべきではない。下に概説するように、ドセタキセルが投与される患者には全員、コルチコステロイドを前投与するのがよい。
【0352】
軽度から中程度の肝機能障害では、代謝が27%遅延し、全身暴露(AUC)が38%増加する。ドセタキセルは、SGOTおよび/またはSGPTが正常範囲の>1.5倍、およびアルカリフォスファターゼが正常範囲の>2.5倍の患者には投与すべきではない。体液鬱滞が、コルチコステロイドを前投与したにもかかわらず、第III相試験における患者の17%(中程度)および6%(重度鬱滞)で起こった。重度の知覚神経症(感覚異常、錯感覚、疼痛)が観察されている。
【0353】
予想される副作用としては、最下点が約9日間あり15〜21日目までには回復する骨髄抑制(主に白血球減少症)が挙げられる。脱毛症、爪および皮膚の変化、口内炎、筋肉痛/関節痛、悪心/嘔吐、ならびに低血圧が報告されている。
【0354】
化学療法剤の用量、投与、および用量の変更
各治療サイクルの1日目に、化学療法剤(ドキソルビシンに続いてドセタキセル)を投与した。
【0355】
ドキソルビシンは、管外溢出による傷害を回避するため、点滴用静脈ラインまたは中心静脈カテーテルのサイドアームを介してIVボーラスにより60mg/m2の用量で投与した。
【0356】
75mg/m2のドセタキセルは、0.9%食塩水または5%デキストロース溶液250mlで希釈し、ポリエチレンライニングを有する輸液セットを介して約1時間にわたって静脈内投与した。バイタルサインは、ドセタキセル点滴の直前および直後に得た。
【0357】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験する被験者は試験治療から除外され、経過観察を完了することになっていた。
【0358】
化学療法剤の前投薬
経口(必要であればIV)コルチコステロイド(例えば、デキサメタゾン8mgBID)を、体液鬱滞および過敏反応の発生および重篤さを低下させるため、ドセタキセル投与の1日前に開始して3日間投与した。
【0359】
制吐薬または他の前投薬(例えば、H2拮抗薬)の使用および選択は、治療を行う医師の裁量に任せた。
【0360】
禁止薬剤
被験者は、この試験中および下に規定するさらなる期間中は、次の薬剤および/または処置のいずれも受けてはならない。
1.サイクル1化学療法の72時間以内の抗生物質の全身投与。
2.試験薬剤の開始の30日以内および実験期間中における他の治験薬。
3.後続の化学療法サイクルは、NEUGの投与後14日まで開始すべきではない。
4.持続性または発熱性の好中球減少症が起こらない限り、実験期間中におけるサイトカイン、他の造血成長因子、および予防用抗生物質。被験者が、スクリーニング期間から0日目の間の任意の時点でG-CSFで治療される場合、NEUGの投与に適格ではなく、試験を中断する。
【0361】
許可薬剤
被験者にベースライン薬剤を継続させた。可能であれば、試験全体にわたって各薬剤の一日量を維持した。何らかの理由で研究者によって必要であると認められた場合、さらなる薬剤、または用量、薬剤、投与経路の変更を必要とした被験者、および薬剤が投与された適応症、を記録した。
【0362】
化学療法のさらなるサイクルの妨げになる重度の過敏反応または非血液毒性を経験した被験者は試験治療から除外され、経過観察を完了した。
【0363】
d.薬物動態
NEUGが投与される被験者は全員、サイクル1中に血清NEUG濃度をサンプリングされた。薬剤は、NEUG特異的なサンドイッチ酵素結合免疫吸着測定(ELISA)を用いて検出した。血清薬剤濃度時間データは、ノンコンパートメントまたはモデルベース分析を用い、WinNonlin Enterprise Edition(バージョン5.0以上)を用いてPK分析に供した。次のPKパラメータを測定した:曲線下面積(AUC0-∞)、クリアランス(CL/F)、分布容積(Vz/F)、最大濃度(Cmax)、吸収半減期(t1/2、abs)、排出半減期(t1/2、elim)、および平均滞留時間(MRT)。
【0364】
e.免疫原性
NEUGが投与される被験者において、NEUGに対する抗体の血清試料は、毎NEUGサイクルの1日目における投与前および治療訪問の終わり(最後の投与の約30日後)に得た。被験者が試験中の任意の時点で陽性の抗NEUG抗体反応を生じた場合、最後のNEUG投与の約6か月後に反復試料を得、この試料が陽性であった場合、12か月目に試料を得た。プロトコルは、被験者全員からの6か月および12か月免疫原性試料を必要とするようにその後修正された。
【0365】
5.結果
a.概論
統計的方法
サイクル1における重度の好中球減少(DSN)の平均持続期間の主要評価項目に関連してペグフィルグラスチムに対してNEUGが劣っていないことを実証するため、この実験のメインフェーズ(B部)における1治療群当たり被験者約85人のサンプルサイズは、非劣性マージンが1日で、複数回の試験(Hochberg法による)のために調整した片側有意水準が全体で0.025で、91%の検出を提供するように選択した。サンプルサイズは、2つの独立群に対する正規近似、サイクル1 DSNの治療内標準偏差として推定1.6日、およびサイクル1 DSNの主要評価項目に関して評価できない最高率20%に基づいて計算した。
【0366】
3治療群無作為化フェーズ(A部)中の被験者に基づいて、2つの選択されたNEUG用量(40mgと50mgのいずれか)およびペグフィルグラスチムの効能を比較した。
【0367】
第二の効能分析は、化学療法サイクル2から4のそれぞれにおけるDSN、サイクル1から4のそれぞれにおける最下点ANCの低さ、サイクルによるそしてすべてのサイクルにわたるFN(その時の口腔相当温度>38.2℃でANC<0.5x109/Lと規定)の割合、および全サイクルにおいてANCが>1.5x109/Lに回復するまでの時間を含む。
【0368】
第二の効能分析に関連するデータは、適切な統計的方法を用いて分析した。安全性、PKおよび、免疫原性パラメータは記述統計法を用いて分析した。
【0369】
有害事象の頻度および重度、ならびに実験室毒性の等級分けに関して、数および割合を示す。
【0370】
効能の測定
全血球計算値(「CBC」)は、1日目、3日目、そして5日目以降、最下点後にANCが>2.0x109Lになるまで、その後、週2回、および治療の終わりに得た。
【0371】
b.第II相のA部の効能
試験のパイロットフェーズに参加した被験者78人のうち、被験者13人が試験を完了せず、3人(27.3%)がNEUG30mgで治療され、3人(14.3%)がNEUG40mgで治療され、3人(15.0%)がNEUG50mgで治療され、4人(15.4%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および研究者の判断によるもの(被験者3人)であった。NEUGを30mg投与した被験者の1人は、有害事象(糖尿病性足病変)によるとりやめであった。
【0372】
重度の好中球減少症の発生率および重度の好中球減少(DSN)の平均継続期間は、各化学療法サイクルの治療群にわたって類似していたが、ANC回復までの時間および発熱性好中球減少症の発生率は、NEUG30mgが、NEUG40mg、NEUG50mg、またはペグフィルグラスチムほど効果的ではないことを示唆した。
【0373】
サイクル1中に、発熱性好中球減少症を経験する被験者の割合は、NEUG30mg、40mg、50mg、およびペグフィルグラスチム群でそれぞれ20.0%、9.5%、10.0%、および8.0%であった。発熱性好中球減少症は、サイクル2〜4においてさらに被験者3人のみに観察された(NEUG30mg、NEUG40mg、およびペグフィルグラスチム群で1人ずつ)。図5は、NEUG30またはペグフィルグラスチムのいずれかを投与された患者のサブセットで、後にグレード4好中球減少症を発症した者のANCプロファイルを示す。
【0374】
サイクル1では、平均DSNは、NEUG30mg(0.9日)、NEUG50mg(1.1日)、およびペグフィルグラスチム(0.9日)に関しては類似していた。平均DSNは、NEUG40mgでは他の3つの治療よりもわずかに長かった(1.6日)が、治療間の違いはすべて1日未満で、メインフェーズにおいて同等な治療を検討する基準である。DSN中間値は、4つの治療群すべてで0日または1日であった。
【0375】
グレード3またはグレード4好中球減少症の発生率および持続期間に関する要約データは、類似のパターンをたどる、つまり、NEUG30mg、NEUG50mg、およびペグフィルグラスチム群は、類似の結果になった。その一方で、グレード3またはグレード4好中球減少症の発生率および持続期間は、NEUG40mg群で他の治療群よりわずかに高かった。パイロットフェーズ(A部)の被験者数はきわめて少なく、観察された差異は統計的に有意ではなかった。NEUG40mgおよびNEUG50mgは、B部(試験の3治療群無作為化フェーズ)におけるさらなる評価のために選択された。
【0376】
c.第II相のB部の効能
試験のメインフェーズに参加した被験者256人のうち、被験者18人が試験を完了せず、10人(11.6%)がNEUG40mgで治療され、5人(6.0%)がNEUG50mgで治療され、3人(3.5%)がペグフィルグラスチムで治療された。早期中断の最も多い理由は、同意の撤回(被験者7人)および2人の死者を含むAE(被験者4人)であった。研究者は、AEはすべて、試験薬剤または化学療法と関連しないと見なした。メインフェーズでは、NEUG40mgが投与された被験者の1人(1.2%)が、試験薬剤で治療される前に除かれた。
【0377】
サイクル1における重度の好中球減少症の発生率および持続期間を表23にまとめている。
【0378】
(表23)第II相B部:サイクル1における重度の好中球減少症の発生率および持続期間
【0379】
重度の好中球減少症の発生率は、ペグフィルグラスチム群で58.1%からNEUG50mg群で65.5%の範囲であった。治療効果は、統計的に有意ではなかった(p=0.559)。治療群は、サイクル1 DSNに関して、NEUG40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ平均値が1.0、1.3および1.2日であり同程度であった。NEUGとペグフィルグラスチムとの差異の95%および97.5%両側信頼区間は、両方のNEUG用量で正確に1日未満だった。この分析は、ペグフィルグラスチムに対してNEUGが劣っていないことを実証した。治療サイクルにわたって、重度の好中球減少症およびグレード3またはグレード4好中球減少症の発生率は、サイクル1においてよりもサイクル2〜4において低かった。グレード3またはグレード4好中球減少症の平均DSNおよび平均持続期間は、サイクル1においてよりもサイクル2〜4において小さかった。治療サイクル内では、治療は類似しており、処理効果は、任意の化学療法サイクルにおけるこれらパラメータのどれについても有意な差異はなかった。
【0380】
体重で四分した患者でDSNを比較して、NEUGの固定用量がすべての体重の患者に十分なサポートを提供するかどうかを決定した。当該結果は、体重サブグループの平均DSNにおける有意差は無く(表24)、すべての体重群で十分にサポートされたことを示す。
【0381】
(表24)体重別の、重度の好中球減少症のサイクル1での持続期間(日数)
【0382】
すべてのサイクルにおける発熱性好中球減少症を表25にまとめる。サイクル1中、発熱性好中球減少症を経験する被験者の割合は、NEUG40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ、被験者2人(3.5%)、被験者5人(6.0%)、および被験者2人(2.3%)であった。発熱性好中球減少症は、サイクル2〜4中でさらに被験者3人のみ観察された(NEUG40mg群で被験者2人およびペグフィルグラスチム群で被験者1人)。治療効果は、どの化学療法サイクルでも統計的に有意ではなかった。
【0383】
(表25)サイクル1〜4における好中球減少症の発生率
【0384】
サイクル2〜4において重度の好中球減少症の持続期間中の治療間で有意差はなかった(表26)。
【0385】
(表26)サイクル2〜4における重度の好中球減少症の平均持続期間
【0386】
ANC回復(>1.5x109/L)までの平均時間は、Neugranin40mg、NEUG50mg、およびペグフィルグラスチム群でそれぞれ、2.0、2.1、および2.6日であった(表27)。ANC最下点の低さまたは最下点までの時間に関して治療群間で有意差はなかった。
【0387】
(表27)ANC最下点、ANC最下点までの時間、および回復までの時間
【0388】
d.第II相B部の薬物動態
血清Neugranin濃度は、6.312ng/mLの定量範囲の下限値(LLQ)で、有効なサンドイッチELISAを用いて測定した。薬物動態パラメータは、吸収半減期以外はノンコンパートメントモデル法を用いて計算し、吸収半減期は、一次吸収、一次排出1コンパートメントモデルを用いて測定した。モデリングは、WinNonlin Professional(バージョン5.0.1)を用いて行った。血清NEUG濃度は、第II相においてNEUGで治療されたすべての被験者における化学療法サイクル1で測定した。第II相A部では、NEUGの排出半減期の中間値は、30mg投与群で33時間であり、40mg投与群で46時間であり、50mg投与群で18時間であった(表28)。B部では、NEUGの排出半減期の中間値は、40mg投与群で40時間であり、50mg投与群で39時間であった(表29)。A部中は、PKサンプリングは、B部と比較して(投与前、3日目、5〜8日目)、より頻繁に行われた(投与前、3時間、6時間、12時間、24時間、3日目、5〜9日目、11日目)。
【0389】
(表28)第II相A部の治療による排出半減期の中間値
【0390】
(表29)第II相B部の治療による排出半減期の中間値
【0391】
血清ペグフィルグラスチム濃度は、第II相において、ペグフィルグラスチムで治療された被験者全員における化学療法サイクル1で、有効なサンドイッチELISAを用いて測定した。A部では、ペグフィルグラスチムの排出半減期の中間値は、約40時間であった。B部では、ペグフィルグラスチムの排出半減期の中間値は、約50時間であった。排出半減期は、フィルグラスチムで3〜4時間、ペグフィルグラスチムで42〜67.5時間(用量に応じて)であると報告される。
【0392】
e.免疫原性
試験参加者間で、Neugraninで治療された被験者において、確認された抗G-CSF/新エピトープ抗体反応1つおよびペグフィルグラスチム治療群において抗G-CSF反応が1つ、すなわちそれぞれ0.5%および0.9%があった(表30)。どちらの場合も、被験者は、前投与試料中で非特異的結合の上昇があった。
【0393】
(表30)NEUGおよびペグフィルグラスチムに対するG-CSFに特異的な治療により発現した免疫反応の概要
【0394】
NEUG治療後、患者において、非常に低レベルの確認された陽性抗体が見られ、反復投与後の応答の程度に明確な増加はなかった(データ示さず)。ペグフィルグラスチムで治療された患者では、異常に高い非特異的バックグラウンド結合が観察されたが、サイクル2治療後は確認された一過性抗体反応のみが見られた(データ示さず)。抗体反応は中和的ではなかった。
【0395】
抗HSA抗体はこの集団において低レベルで天然に存在し、6.9%の被験者が投与前評価でHSA抗体に陽性であった。治療により発現した抗HSA抗体は、NEUG治療被験者4人(1.8%)で観察された(表31)。反応はすべて一過性で弱かった。最初の治療サイクル後に3つの反応が現われ、サイクル2、3、および4の後は検出できなかった。3度目の治療後に反応が1つ起きたが、4度目の治療後の30日の経過観察では検出できなかった(データ示さず)。
【0396】
(表31)NEUGに対するHSAに特異的な治療により発現した免疫反応の概要
【0397】
f.第II相B部における治療により発現した有害事象
第II相B部では、各治療群中の被験者の>90%が、治療により発現した有害事象(TEAE)を少なくとも1つ経験し、試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、ペグフィルグラスチム群で23.1%からNeugranin50mg群で35.0%の範囲であった。少なくとも1つのSAEを有する被験者のパーセントは、NEUG30mg群で最も高かった(30%)が、他の3つの治療群では約15%であった。SAEはどれも試験薬剤と関連していなかった。患者1人(NEUG30mg)は、糖尿病性足病変により試験から除かれ、これは試験薬剤とは関連しないと考えられた。B部では、被験者8人(NEUG40mg2人、NEUG50mg3人、ペグフィルグラスチム3人)以外はみな少なくとも1つのTEAEを有した。試験薬剤と関連する少なくとも1つのTEAEを有する被験者のパーセントは、NEUG50mg群で20.2%、NEUG 40mg群で22.4%、ペグフィルグラスチムが投与される被験者で22.1%であった。被験者2人(NEUG 40mg 1人、ペグフィルグラスチム1人)が試験中に死亡し、各治療群の被験者6〜8人がSAEを少なくとも1つ経験した。死亡やSAEは試験薬剤と関連しないと考えられた。
【0398】
NEUG30mg用量についてサンプルサイズを考慮すると、TEAEの総数は、A部およびB部の治療群にわたって類似していた。A部およびB部の両方において、CTCグレード3以上のTEAEのパーセントは、試験薬剤に関連するTEAEのパーセントと同様に、NEUGおよびペグフィルグラスチムの場合で類似していた。
【0399】
g.用量反応
第II相の結果は、NEUGの40および50mg固定用量がいずれも、骨髄毒性の化学療法で治療される乳癌被験者においてペグフィルグラスチム6mgと同等の安全性および効能をもたらすことを実証した。40mg治療群の平均DSNは、50mg群の平均DSNよりわずかに低かったが、これらの差異は統計的に有意ではなかった。用量応答は、体重により調整した用量を考慮した場合と固定用量コホートに対してとの両方でAUCANCについて観察した(サイクル1における0〜15日目)(図24)。30mgコホートのAUCANCは、ペグフィルグラスチムのそれよりわずかに低く、これは、30mg固定用量がこの研究では効果が劣るが、40mgおよび50mgコホートのAUCANCは用量相関性があり、ペグフィルグラスチム治療被験者のAUCANCよりも高い(有意ではないが)ことを示している。上の分析から、NEUGが体重調節基準(mg/kg)で投与されると、用量反応が明らかである。しかしながら、第II相B部のサイクル1におけるDSNの比較によって、治療群(40および50mgNEUGおよびペグフィルグラスチム)において、そして体重調節用量(mg/kg)で、DSNが有意に異ならなかったので、体重で四分した患者はすべて十分にサポートされることが示唆された。さらに、関連する有害事象(特に骨痛;データ示さず)の発生率および重症度が、体重1キログラム当たりに投与される用量と相関せず、またペグフィルグラスチムでの有害事象と差異はなかったので、固定用量が、より低体重の患者において安全性プロファイルに変更をもたらす可能性があるという証拠はなかった。
【0400】
実施例12:さらなるNEUG製剤の例
さらなるNEUG製剤の開発が行われ、pH(4〜7)、緩衝剤の種類(L-ヒスチジン、クエン酸塩、酢酸塩)、等張化剤(ソルビトール対NaCl)、およびタンパク質濃度(最大で70〜90mg/mlまで濃縮)の影響を分析し測定した。試料は、SEC(モノマー%)、RH-HPLC(純度%)、エルマンアッセイ(-SH量)、A280(タンパク質)、およびIE-HPLC(電荷アイソフォーム)によって分析/モニタリングした。試験した製剤を表32に示す。
【0401】
試験後、製剤A40S、A45S、C55N、およびC55Sが(検討した側面においては)最も安定しているようであった。
【0402】
(表32)さらなるNEUG製剤
【0403】
本発明の趣旨または範囲から逸脱することなく、本発明の方法および組成物において多くの改変および変更をなし得ることが当業者には明らかである。したがって、本発明は、添付の特許請求の範囲およびその同等物の範囲内であれば、本発明の改変および変更を包含することが意図される。
【特許請求の範囲】
【請求項1】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子および少なくとも一種類の薬学的に許容される担体を含む薬学的組成物であって、pHが約4から約6.8である、薬学的組成物。
【請求項2】
pHが約6.0である、請求項1記載の薬学的組成物。
【請求項3】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約2.5mg/mlから約240mg/mlである、請求項1記載の薬学的組成物。
【請求項4】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約30mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【請求項5】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約60mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【請求項6】
少なくとも一種類の薬学的に許容される塩を含む、請求項1〜5のいずれか一項記載の薬学的組成物。
【請求項7】
塩の濃度が約5mMから約50mMである、請求項6記載の薬学的組成物。
【請求項8】
薬学的に許容される緩衝剤を含む、請求項1〜7のいずれか一項記載の薬学的組成物。
【請求項9】
緩衝剤の濃度が約15mMから約50mMである、請求項8記載の薬学的組成物。
【請求項10】
緩衝剤の濃度が約20mMである、請求項8記載の薬学的組成物。
【請求項11】
前記緩衝剤がリン酸塩またはクエン酸塩である、請求項8〜10のいずれか一項記載の薬学的組成物。
【請求項12】
前記緩衝剤がリン酸ナトリウムである、請求項8〜11のいずれか一項記載の薬学的組成物。
【請求項13】
前記緩衝剤がリン酸一ナトリウムまたはリン酸二ナトリウムを含む、請求項12記載の薬学的組成物。
【請求項14】
凍結乾燥安定剤を含む、請求項1〜13のいずれか一項記載の薬学的組成物。
【請求項15】
前記凍結乾燥安定剤がトレハロース二水和物を含む、請求項14記載の薬学的組成物。
【請求項16】
トレハロース二水和物の濃度が約6OmMである、請求項15記載の薬学的組成物。
【請求項17】
増量剤を含む、請求項1〜16のいずれか一項記載の薬学的組成物。
【請求項18】
前記増量剤が多価アルコールを含む、請求項17記載の薬学的組成物。
【請求項19】
前記多価アルコールがマンニトールを含む、請求項18記載の薬学的組成物。
【請求項20】
凍結乾燥粉末の形態である、請求項1〜19のいずれか一項に記載の薬学的組成物。
【請求項21】
前記粉末がバイアルに保存される、請求項20記載の薬学的組成物。
【請求項22】
液体の形態である、請求項1〜19のいずれか一項記載の薬学的組成物。
【請求項23】
前記液体がシリンジに保存される、請求項22記載の薬学的組成物。
【請求項24】
(a)組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、および
(b)少なくとも一種類の緩衝剤
を含む薬学的組成物であって、
25℃で24時間インキュベーションした後の前記組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の溶液中のモノマー純度の減少が10%未満である、
請求項1〜23のいずれか一項に記載の薬学的組成物。
【請求項25】
(a)組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、
(b)20mMリン酸ナトリウム、
(c)180mmマンニトール、
(d)60mMトレハロース二水和物、
(e)0.01%(W/V)ポリソルベート80
を含む薬学的組成物であって、
前記組成物のpHが約6.0であり、前記組換え融合タンパク質の濃度が2.5mg/mlから120mg/mlである、
請求項1記載の薬学的組成物。
【請求項26】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子およびPMTT20/6.0を含む薬学的組成物であって、前記組換え融合タンパク質の濃度が約2.5mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【請求項1】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子および少なくとも一種類の薬学的に許容される担体を含む薬学的組成物であって、pHが約4から約6.8である、薬学的組成物。
【請求項2】
pHが約6.0である、請求項1記載の薬学的組成物。
【請求項3】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約2.5mg/mlから約240mg/mlである、請求項1記載の薬学的組成物。
【請求項4】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約30mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【請求項5】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の濃度が、約60mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【請求項6】
少なくとも一種類の薬学的に許容される塩を含む、請求項1〜5のいずれか一項記載の薬学的組成物。
【請求項7】
塩の濃度が約5mMから約50mMである、請求項6記載の薬学的組成物。
【請求項8】
薬学的に許容される緩衝剤を含む、請求項1〜7のいずれか一項記載の薬学的組成物。
【請求項9】
緩衝剤の濃度が約15mMから約50mMである、請求項8記載の薬学的組成物。
【請求項10】
緩衝剤の濃度が約20mMである、請求項8記載の薬学的組成物。
【請求項11】
前記緩衝剤がリン酸塩またはクエン酸塩である、請求項8〜10のいずれか一項記載の薬学的組成物。
【請求項12】
前記緩衝剤がリン酸ナトリウムである、請求項8〜11のいずれか一項記載の薬学的組成物。
【請求項13】
前記緩衝剤がリン酸一ナトリウムまたはリン酸二ナトリウムを含む、請求項12記載の薬学的組成物。
【請求項14】
凍結乾燥安定剤を含む、請求項1〜13のいずれか一項記載の薬学的組成物。
【請求項15】
前記凍結乾燥安定剤がトレハロース二水和物を含む、請求項14記載の薬学的組成物。
【請求項16】
トレハロース二水和物の濃度が約6OmMである、請求項15記載の薬学的組成物。
【請求項17】
増量剤を含む、請求項1〜16のいずれか一項記載の薬学的組成物。
【請求項18】
前記増量剤が多価アルコールを含む、請求項17記載の薬学的組成物。
【請求項19】
前記多価アルコールがマンニトールを含む、請求項18記載の薬学的組成物。
【請求項20】
凍結乾燥粉末の形態である、請求項1〜19のいずれか一項に記載の薬学的組成物。
【請求項21】
前記粉末がバイアルに保存される、請求項20記載の薬学的組成物。
【請求項22】
液体の形態である、請求項1〜19のいずれか一項記載の薬学的組成物。
【請求項23】
前記液体がシリンジに保存される、請求項22記載の薬学的組成物。
【請求項24】
(a)組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、および
(b)少なくとも一種類の緩衝剤
を含む薬学的組成物であって、
25℃で24時間インキュベーションした後の前記組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子の溶液中のモノマー純度の減少が10%未満である、
請求項1〜23のいずれか一項に記載の薬学的組成物。
【請求項25】
(a)組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子、
(b)20mMリン酸ナトリウム、
(c)180mmマンニトール、
(d)60mMトレハロース二水和物、
(e)0.01%(W/V)ポリソルベート80
を含む薬学的組成物であって、
前記組成物のpHが約6.0であり、前記組換え融合タンパク質の濃度が2.5mg/mlから120mg/mlである、
請求項1記載の薬学的組成物。
【請求項26】
組換えヒトアルブミン-ヒト顆粒球コロニー刺激因子およびPMTT20/6.0を含む薬学的組成物であって、前記組換え融合タンパク質の濃度が約2.5mg/mlから約120mg/mlである、請求項1記載の薬学的組成物。
【図1】

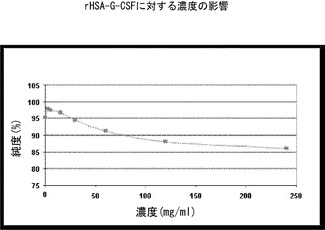
【図2】

【図3】
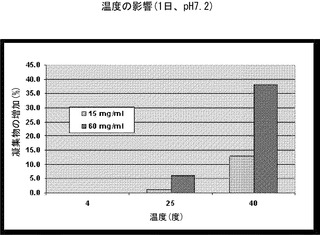
【図4】
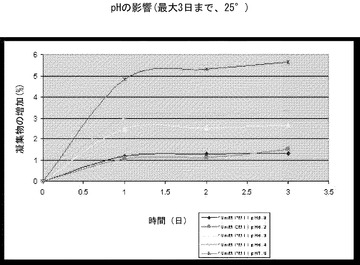
【図5】

【図6】

【図7】

【図8】
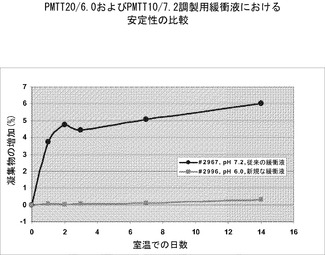
【図9A−1】

【図9A−2】

【図9B】

【図9C】

【図10】

【図11】
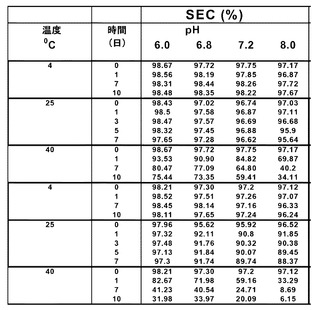
【図12】
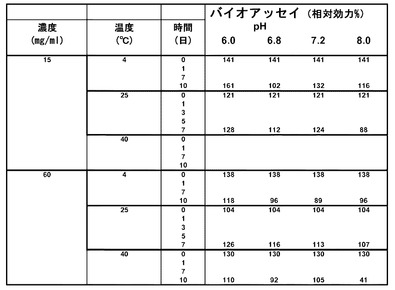
【図13】

【図14A】
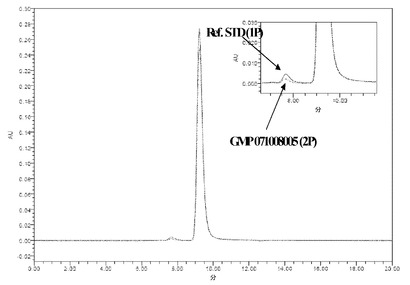
【図14B】

【図15A】
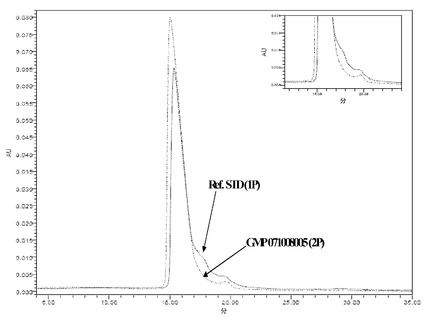
【図15B】
【図16】

【図17A−1】
【図17A−2】

【図17B−1】

【図17B−2】
【図18】

【図19】
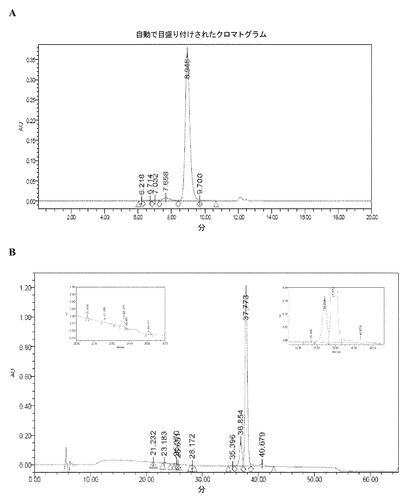
【図20】
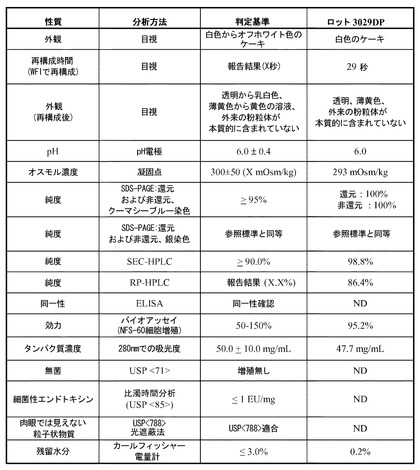
【図21】

【図22】

【図23】
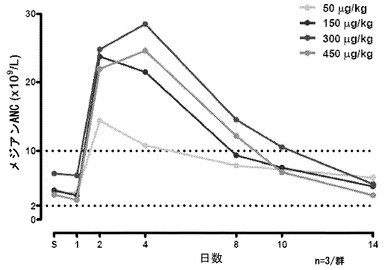
【図24A】

【図24B】

【図25】
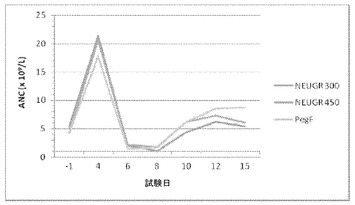
【図26】

【図27】
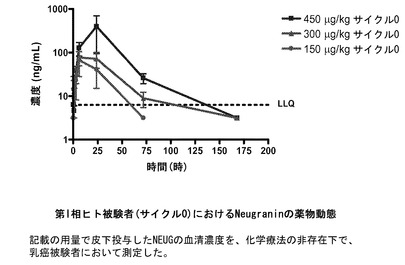
【図28】

【図29】

【図30】
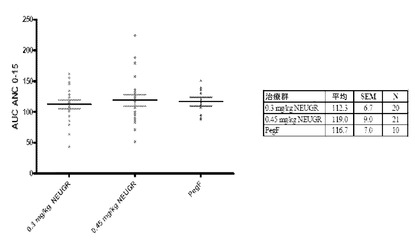
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9A−1】
【図9A−2】
【図9B】
【図9C】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15A】
【図15B】
【図16】
【図17A−1】
【図17A−2】
【図17B−1】
【図17B−2】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24A】
【図24B】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【公表番号】特表2012−515221(P2012−515221A)
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願番号】特願2011−546396(P2011−546396)
【出願日】平成22年1月15日(2010.1.15)
【国際出願番号】PCT/US2010/021235
【国際公開番号】WO2010/083434
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(511173228)テバ ファーマシューティカル インダストリーズ リミテッド (2)
【Fターム(参考)】
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願日】平成22年1月15日(2010.1.15)
【国際出願番号】PCT/US2010/021235
【国際公開番号】WO2010/083434
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(511173228)テバ ファーマシューティカル インダストリーズ リミテッド (2)
【Fターム(参考)】
[ Back to top ]