
組換え微生物、Tat分泌系阻害剤のスクリーニング方法及びTat分泌系阻害剤のスクリーニングキット

【課題】種々の供試物質のなかからTat分泌系を標的とする、すなわち、Tat分泌系を阻害する活性を有する物質をスクリーニングする。
【解決手段】Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成する。
【解決手段】Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、例えば緑膿菌におけるTat分泌系を標的とする阻害剤をスクリーニングする際に使用することができる組換え微生物、当該組換え微生物を使用したTat分泌系阻害剤のスクリーニング方法及び当該組換え微生物を含むTat分泌系阻害剤のスクリーニングキットに関する。
【背景技術】
【0002】
微生物のなかには、抗生物質等の菌体外への排出を担う薬剤排出ポンプを備えているものがあり、これにより元来自然耐性並びに獲得多剤耐性を有するものがある。例えば、緑膿菌は構造的に関連性のない各種薬剤に対し、元来自然耐性ならびに獲得多剤耐性能を有しているために臨床上大きな問題となっている。すなわち、日和見感染症の主要起因菌である緑膿菌における多剤耐性の獲得は、院内感染の原因となり、社会問題として認識されている。
【0003】
緑膿菌には、MexA,B-OprMという3つのコンポーネントからなるエネルギー依存性の薬剤排出ポンプが備わっている。この薬剤排出ポンプは、抗生物質などの抗菌性薬剤を菌体外に排出する役割を担っており薬剤耐性に深く関与している。この薬剤排出ポンプは、ポンプ本体である内膜サブユニットMexB、外膜に存在して薬剤排出チャネルを形成するOprM、及び修飾リピドを介して内膜にアンカリングし、MexBとOprMの相互作用を補強していると考えられているMexAから構成される。これらの各サブユニットは、一般的な分泌システム(general secretion pathway)であるSec系を介して、各々の局在部位に輸送され排出ポンプ複合体を形成することが知られている。
【0004】
一方、新たな分泌システムであるTat(twin arginine translocation)系が、植物のチラコイド膜を介する分泌系で見出され、このTat分泌系システムが広く細菌界に存在することが最近明らかとなってきた(非特許文献1)。緑膿菌もこのTat分泌系を有していることが明らかとなり、本菌の病原性因子であるフォスフォリパーゼや鉄キレーターの産生に関与する因子などが、このTat系を介して分泌されることが最近明らかとなってきた(非特許文献2及び3)。また、Tat分泌系が欠損した緑膿菌変異株においては、その病原性が低下することが明らかとなった(非特許文献4)。
【0005】
このことから、例えば緑膿菌におけるTat分泌系を標的とする物質は、緑膿菌における病原因子の分泌を抑制し、当該病原因子に起因する種々の疾患を予防及び治療できる薬剤として期待される。
【0006】
広く細菌界に分布するTat分泌系は、動物細胞には存在しないことが知られており(非特許文献5)、抗菌剤が具備すべき最重要事項である選択毒性の観点から理想的な標的としての特性を有している。さらに、緑膿菌をはじめ詳細な研究がなされている大腸菌等においてTat分泌系を構成する遺伝子破壊株が作製されていることから、このTat分泌系は細菌の生存に必須ではないことは明らかである。ペニシリン以来多くの抗生物質が発見され臨床的に使用されてきたが、病原細菌はそのすべての薬剤に対し耐性能を獲得し、社会的に大きな問題となっている。これは、既存の抗生物質がいずれも細菌の生存にとって必須の代謝プロセスを阻害するからであり、変異によって耐性能を獲得した耐性菌が抗生物質による選択圧のため選択され生き残ったからである。一方、Tat系を標的とした場合、Tat系は細菌の生存に必須でないため、薬剤に対する耐性菌が選択される可能性は既存の抗生物質に比べ非常に低いことが期待されることから、臨床現場において危惧される耐性菌問題を回避できる可能性がある。しかしながら、Tat分泌系を標的とする物質のスクリーニング系は知られておらず、このようなスクリーニング系、特にハイスループットなスクリーニング系が求められていた。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Prokaryotic utilization of the Twin-arginine translocation pathway: a genomic survey. Dilks K, Rose R W, Hartmann E, Pohlschroder M. J. Bacteriol. 2003, 185, 1478-1483.
【非特許文献2】Role of the Pseudomonas aeruginosa PlcH Tat signal peptide in protein secretion, transcription, and cross-species Tat secretion system compatibility. Snyder A, Vasil A I, Zajdowicz, S L, Wilson Z R, Vasil M L. J. Bacteriol. 2006, 188, 1762-1774.
【非特許文献3】Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: the Tat system is required for PvdN but not for FpvA transport. Voulhoux R, Filloux A, Schalk I J. J. Bacteriol. 2006, 188, 3317-3323.
【非特許文献4】Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Ochsner U A, Snyder A, Vasil A I, Vasil M L. Proc. Natl. Acad. Sci. USA. 2002, 99, 8312-8317.
【非特許文献5】Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Yen M-R, Tseng, Y-H, Nguyen E, Wu L-Fe, Saier M H Jr. Arch. Microbiol. 2002, 177, 441-450
【発明の概要】
【発明が解決しようとする課題】
【0008】
そこで、本発明は、種々の供試物質のなかからTat分泌系を標的とする、すなわち、Tat分泌系を阻害する活性を有する物質をスクリーニングする際に使用する組換え微生物、当該組換え微生物を使用したスクリーニング方法、及び当該組換え微生物を含むスクリーニングキットを提供することを目的とする。
【課題を解決するための手段】
【0009】
Tat分泌系を阻害する(標的とする)物質のハイスループットスクリーニング系を確立するために必要な要件としては、Tat分泌系依存性の感度の高い生物学的評価系を確立することが挙げられる。Tat分泌系については、フォールドしたタンパク質を分泌、輸送することが知られており、Tat分泌系の基質であるシグナル配列を融合した細菌のアルカリフォスファターゼを発現させても、その機能は保持されないことが示されている(Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. Richter S and Bruse T. J. Biol. Chem. 2005, 280, 42723-42730)。したがって、この文献に記載されるように、Tat分泌系の基質であるシグナル配列を融合した細菌のアルカリフォスファターゼは、Tat分泌系の機能評価をするためのレポーターとして使用することはできない。以上のように、Tat分泌系の機能を、Tat分泌系の基質であるシグナル配列を利用したレポーター系で評価することは非常に困難であると認識されていた。
【0010】
しかしながら、上述した実情に鑑み、本発明者らが鋭意検討した結果、薬剤排出ポンプを構成するサブユニットにおけるシグナル配列を、Tat分泌系の基質となるタンパク質のシグナル配列で置換したキメラタンパク質をコードする融合遺伝子を発現し、当該サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成する組換え微生物を使用することで、上述したハイスループットなスクリーニング系を構築できることを見いだし、本発明を完成するに至った。すなわち、本発明は以下を包含する。
【0011】
(1)Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成することを特徴とする組換え微生物。
(2)上記融合遺伝子を、上記サブユニットをコードする内在遺伝子を欠損した上記微生物に導入したものであることを特徴とする(1)記載の組換え微生物。
(3)上記微生物は内在性のTat分泌系を有する細菌であることを特徴とする(1)記載の組換え微生物。
(4)上記微生物はグラム陰性菌であることを特徴とする(1)記載の組換え微生物。
(5)上記微生物は緑膿菌(Pseudomonas aeruginosa)であることを特徴とする(1)記載の組換え微生物。
(6)上記薬剤排出ポンプは、サブユニット:MexA、MexB及びOprMから構成されることを特徴とする(1)記載の組換え微生物。
(7)上記融合遺伝子は、上記サブユニットとしてMexAにおける成熟領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(8)上記融合遺伝子は、上記サブユニットとしてOprMにおける成熟領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(9)上記融合遺伝子は、上記シグナル配列として病原性因子フォスフォリパーゼC(plcH)のシグナル配列を含む領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(10)互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現することを特徴とする(1)記載の組換え微生物。
(11)上記(1)乃至(10)いずれか一記載の組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる工程と、上記組換え微生物の上記薬剤に対する感受性を評価する工程と、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断する、Tat分泌系阻害剤のスクリーニング方法。
(12)上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする(11)記載のスクリーニング方法。
(13)上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする(11)記載のスクリーニング方法。
(14)上記(1)乃至(10)いずれか一記載の組換え微生物と、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤とを含む、Tat分泌系阻害剤のスクリーニングキット。
(15)上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする(14)記載のTat分泌系阻害剤のスクリーニングキット。
(16)上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする(14)記載のスクリーニングキット。
【発明の効果】
【0012】
本発明によれば、微生物におけるTat分泌系を標的とする、すなわち、Tat分泌系を阻害する活性を有する物質の存在下で、薬剤感受性が大となる組換え微生物を提供することができる。本発明に係る組換え微生物を使用することによって、薬剤感受性を指標としたTat分泌系阻害物質を効率良くスクリーニングすることができる。
【図面の簡単な説明】
【0013】
【図1】plcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子の塩基配列を示す特性図であり、キメラ遺伝子のオープンリーディングフレームを大文字で示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、5'末端側の下線付き小文字はBamHI部位を示し、3'末端側の下線付き小文字はKpnI部位を示している。
【図2】TNP070及びTNP080におけるpyoverdine産生能を示す特性図である。
【図3】plcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子がtatC遺伝子の下流に配置したプラスミドの塩基配列を示す特性図であり、枠で囲った領域は由来不明の塩基配列を示し、5'末端側の大文字はtatC遺伝子のオープンリーディングフレームを示し、3'末端側の大文字はキメラ遺伝子のオープンリーディングフレームを示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、下線付き小文字(4箇所)は5'末端側から順にHindIII部位、BamHI部位、BamHI部位及びKpnI部位を示している。
【図4】形質転換体TNP080/pTatPlcMexAにおけるpyoverdine産生能を示す特性図である。
【図5】抗MexA抗体を用いたウエスタンブロット解析の結果を示す電気泳動写真である。
【図6】plcHシグナル配列遺伝子とoprM遺伝子の成熟領域とを融合したキメラ遺伝子の塩基配列を示す特性図であり、キメラ遺伝子のオープンリーディングフレームを大文字で示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、5'末端側の下線付き小文字はXbaI部位を示し、3'末端側の下線付き小文字はEcoRI部位を示している。
【図7】野生株(PAO4290)、TNP072、TNP082及びTNP082/pTatC-PlcH-OprMにおけるpyoverdine産生能を示す特性図である。
【図8】抗OprM抗体を用いたウエスタンブロット解析の結果を示す電気泳動写真である。
【発明を実施するための形態】
【0014】
以下、本発明に係る組換え微生物、当該組換え微生物を使用したスクリーニング方法及びスクリーニングキットを詳細に説明する。本発明に係る組換え微生物は、Tat分泌系による分泌能に依存して、詳細を後述する融合遺伝子産物が関与する薬剤排出ポンプの機能が変化するといった特徴を有する。従って、本発明に係る組換え微生物は、Tat分泌系を標的とする物質の存在下で、当該薬剤排出ポンプから菌体外に排出される薬剤に対する感受性が変化することとなる。
【0015】
融合遺伝子及び組換え微生物
本発明において融合遺伝子は、薬剤排出ポンプを構成するサブユニットの成熟領域と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域とを融合したキメラタンパク質をコードしている。融合遺伝子の産物であるキメラタンパク質は、Tat分泌系の基質となりTat分泌系を介して分泌されることとなる。したがって、融合タンパク質に含まれる薬剤排出ポンプのサブユニットは、Tat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成することになる。
【0016】
本発明に係る組換え微生物とは、この融合遺伝子を発現することで、当該融合遺伝子に含まれるサブユニットがTat分泌系を介して分泌され薬剤排出ポンプの複合体が形成されるものである。組換え微生物は、宿主微生物における染色体上に内在する、上記サブユニットをコードする遺伝子を欠損させるとともに、上記融合遺伝子を発現可能に有するベクターを当該宿主微生物に導入することで作製することができる。すなわち、本発明に係る組換え微生物は、上記融合遺伝子を発現可能なベクターを使用した形質転換微生物を包含する。ここで、ベクターとしては、宿主微生物内において複製可能なベクターであれば何ら制限することなく使用することができる。
【0017】
また、組換え微生物は、染色体上に内在する、上記サブユニットをコードする遺伝子におけるシグナル配列をコードする領域を、Tat分泌系を介して分泌されるタンパク質のシグナル配列をコードする領域で置換することで作製することもできる。例えば、Tat分泌系を介して分泌されるタンパク質のシグナル配列をコードする核酸断片を相同組み換えによって、上記サブユニットをコードする遺伝子におけるシグナル配列をコードする領域と置換することで、上記融合遺伝子を発現する組換え微生物を作製してもよい。すなわち、本発明に係る組換え微生物は、上述した相同組み換え技術によって作製されたものも包含する。
【0018】
なお、本発明に係る組換え微生物は、所定の薬剤排出ポンプを構成する複数のサブユニットのうち、少なくとも1つのサブユニットがTat分泌系の基質となるように上記融合遺伝子を発現すればよいが、全てのサブユニットがTat分泌系の基質となるように全てのサブユニットについて上記融合遺伝子を発現するものであってもよい。
【0019】
先ず、宿主微生物について説明する。本発明に係る組換え微生物の宿主微生物としては、薬剤排出ポンプ及びTat分泌系を備え、当該薬剤排出ポンプにより所定の薬剤に対する耐性(低い感受性と同義)を示す微生物である。このような微生物としては、特に限定されないが、緑膿菌(Pseudomonas aeruginosa)、大腸菌(Escherichia coli)、黄色ブドウ球菌(Staphylococcus aureus)、結核菌(Mycobacterium tuberculosis)、ペスト菌(Yersinia pestis)、インフルエンザ菌(Haemophilus influenzae)、コレラ菌(Vibrio cholerae)、ピロリ菌(Helicobacter pylori)、リステリア菌(Listeria monocytogenes)、キャンピロバクター(Campylobacter jejuni)、サルモネラ菌(Salmonella enterica)、髄膜炎菌(Neisseria meningitidis)、らい菌(Mycobacterium leprae)、枯草菌(Bacillus subtilis)、ブルセラ菌(Brucella属)、リケッチア(Rickettisia prowazekii)、炭疽菌(Bacillus anthracis)等を挙げることができる。特に本発明においては、日和見感染症等の感染症の原因となる緑膿菌を宿主微生物として使用することが好ましい。なお、宿主微生物としては、上記で列挙した細菌に限定されず、例えばDilks, K. et al. (2003) Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J. Bacteriol. 185, 1478-1483.に開示されている微生物や、ゲノムの解読の進展により新たにTat分泌系を有する微生物として同定される全ての微生物を包含する。また、宿主微生物としては、Tat分泌系を本来的に有しない細菌に対して、Tat分泌系を備えるように、Tat分泌系に関与する遺伝子群を外来遺伝子として導入した微生物を使用することもできる。
【0020】
特に、宿主微生物としては、グラム陰性菌とすることが好ましい。なお、上記で列挙した細菌のうち、グラム陰性菌は、緑膿菌(Pseudomonas aeruginosa)、大腸菌(Escherichia coli)、ペスト菌(Yersinia pestis)、インフルエンザ菌(Haemophilus influenzae)、コレラ菌(Vibrio cholerae)、ピロリ菌(Helicobacter pylori)、キャンピロバクター(Campylobacter jejuni)、サルモネラ菌(Salmonella enterica)、髄膜炎菌(Neisseria meningitidis)、ブルセラ菌(Brucella属)、リケッチア(Rickettisia prowazekii)である。グラム陰性菌は、外膜を有するためマルチコンポーネント型の薬剤排出ポンプを有しており、Tat分泌系を介して分泌、輸送しうるサブユニットを適宜選択できる。ここで、薬剤排出ポンプとは、菌体内に存在する抗生物質などの抗菌性薬剤を菌体外に排出する機能を有するポンプであり、グラム陰性菌の場合、通常、微生物の内膜、ペリプラズム及び外膜を貫通するように局在している。薬剤排出ポンプは、通常、複数のサブユニットから構成される複合体である。なお、薬剤排出ポンプは、通常、排出対象の薬剤について特異性がある。
【0021】
上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしては、上述した薬剤排出ポンプのサブユニットを何ら限定されず使用することができる。すなわち、宿主微生物に本来的に備わっている薬剤排出ポンプを構成するサブユニットであれば、如何なるサブユニットを使用しても良い。
【0022】
一例として、緑膿菌の薬剤排出ポンプとしては、主要なものとして、「MexA-MexB-OprM」、「MexC-MexD-OprJ」、「MexE-MexF-OprN」及び「MexX-MexY-OprM」の4種類が知られている。したがって、宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしては、MexA、MexB、MexC、MexD、MexE、MexF、MexX、MexY、OprM、OprJ及びOprNのいずれを使用しても良い。これら薬剤排出ポンプを構成するサブユニットは、従来公知のデータベース(例えば、Pseudomonas Genome Database v2)を検索することでそのアミノ酸配列を取得することができる。また、サブユニットの成熟領域は、例えば、シグナル配列を予測するソフトウェア「SignalP 3.0 Server」を使用してシグナル配列を予測し、当該シグナル配列を除いた領域として特定することができる。
【0023】
宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはMexAを使用することが好ましい。MexA、MexB及びOprMからなる薬剤排出ポンプにおいて、MexAサブユニットは、基質薬剤を認識するポンプ本体であるMexBと、基質薬剤を菌体外へ放出する膜チャンネル機能を有するOprMとを連結する内膜タンパク質である。このように、MexAサブユニットは膜貫通ドメイン等の高次構造を有しないため、折りたたまれたタンパク質を分泌、輸送するTat分泌系を介して確実に分泌することができる。
【0024】
また、宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用することが好ましい。膜チャンネルとして機能するOprMサブユニットは、MexA、MexB及びOprMからなる薬剤排出ポンプのみならず、MexX、MexY及びOprMからなる薬剤排出ポンプにおけるサブユニットとしても機能している。よって、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用した場合には、詳細を後述するスクリーニング方法及びキットにおいて使用可能な薬剤の選択肢を広げることができる。また、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用した場合には、薬剤に対する感受性が大きく変動することとなり、より高精度にスクリーニングすることができる。特に、上掲の論文(J. Biol. Chem. 2005, 280, 42723-42730)によれば、膜貫通ドメインを有するOprMをTat分泌系を介して分泌、輸送することについて困難性があると考えれる。しかしながら、後述の実施例に示すように、OprMをキメラタンパク質としてTat分泌系の基質として発現させても、OprMを含む薬剤排出ポンプを形成できたことは驚くべきである。
【0025】
さらにまた、本発明に係る組換え微生物は、互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現するものであってもよい。すなわち、宿主となる微生物が複数の異なる薬剤排出ポンプを備える場合、これら複数の薬剤排出ポンプのうち、2以上の薬剤排出ポンプのサブユニットが、Tat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成するものであってもよい。特に、Tat分泌系を介して形成される複数の薬剤排出ポンプは、それぞれ異なる種類の薬剤を排出するものを選択することが好ましい。
【0026】
一方、Tat分泌系を介して分泌されるタンパク質のシグナル配列とは、Tat分泌系を介して分泌されるタンパク質におけるN末端側に位置し、シグナルペプチターゼにより成熟タンパク質から切断される領域を意味する。上述した融合遺伝子においては、上述したシグナル配列を含んでいれば良く、Tat分泌系を介して分泌されるタンパク質におけるシグナル配列とこれに続く成熟タンパク質のN末端側の一部とを含んでいても良い。すなわち、上述した融合遺伝子においては、Tat分泌系の基質となりうる領域と薬剤排出ポンプのサブユニットとを融合した融合タンパク質をコードしていればよい。
【0027】
一例として、緑膿菌におけるTat分泌系を介して分泌されるタンパク質としては、例えば表1に示す遺伝子によりコードされるタンパク質を挙げることができる。
【0028】
【表1】

【0029】
なお、表1において、『Locus Tag』とは、Pseudomonas Genome Database v2に登録されているIDであり、PA numberとも称されている。また、表1において、Gene nameの欄には、当該Locus Tagで特定される遺伝子の機能が明らかになっている場合には当該機能に基づく名称が記載されており、機能未知の遺伝子については空欄となっている。また、表1中、1〜26の遺伝子は、Genome Research (2006) 15, 321-329においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。表1中、27〜34の遺伝子は、Proc. Nat. Acad. Sci. (2002) 99, 8312-8317においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。表1中、35の遺伝子は、The EMBO Journal (2001) 20, 6735-3741においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。
【0030】
一旦、Tat分泌系を介して分泌されるタンパク質をコードする遺伝子が同定されれば、当該タンパク質におけるシグナル配列は、当業者により容易に特定することができる。例えば、シグナル配列を予測するソフトウェア「SignalP 3.0 Server」を使用することによりシグナル配列をコードする領域を特定することができる。
【0031】
また、Tat分泌系を介して分泌されるタンパク質をコードする遺伝子が同定されれば、上述したようにソフトウェアを利用してシグナル配列を特定することなく、当該タンパク質のN末端から数えて30〜50残基のアミノ酸配列を、シグナル配列を含む領域として特定しても良い。通常、シグナル配列は、タンパク質のN末端から数えて30〜50残基のアミノ酸配列に含まれると考えられるからである。
【0032】
すなわち、上述した融合遺伝子には、ソフトウェアを利用して特定したシグナル配列を含む領域をコードする遺伝子、又はN末端から数えて30〜50残基のアミノ酸配列をコードする遺伝子のいずれを含んでいても良い。一例として、Tat分泌系を介して分泌されるplcH(PA0844)におけるシグナル配列を含む領域を配列番号1に示す。Tat分泌系を介して分泌されるplcN(PA3319)におけるシグナル配列を含む領域を配列番号2に示す。Tat分泌系を介して分泌されるpvdN(PA2394)におけるシグナル配列を含む領域を配列番号3に示す。Tat分泌系を介して分泌されるglpQ(PA0347)におけるシグナル配列を含む領域を配列番号4に示す。
【0033】
Tat分泌系を標的とする物質のスクリーニング方法及びキット
本発明に係る組換え微生物においては、上述した融合遺伝子を発現することで、薬剤排出ポンプを構成する所定のサブユニットがTat分泌系を介して分泌され、当該薬剤排出ポンプ複合体を形成することとなる。したがって、本発明に係る組換え微生物は、Tat分泌系を阻害する物質の存在下において当該薬剤排出ポンプの構築が阻害されることとなり、当該薬剤排出ポンプにより菌体外へ排出される薬剤への感受性が高くなるといった特徴を有する。
【0034】
すなわち、本発明に係るTat分泌系阻害剤のスクリーニング方法では、先ず、上述した組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる。次に、上記組換え微生物の上記薬剤に対する感受性を評価する。そして、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断することができる。
【0035】
なお、供試物質の存在下における上記感受性が大である場合には、上記供試物質は、上記薬剤排出ポンプに対する阻害作用を有している可能性もある。しかしながら、当該供試物質を上記薬剤の存在下で野生型微生物に作用させ、当該薬剤に対する感受性を評価することで、当該供試物質がTat分泌系及び薬剤排出ポンプのいずれに対して阻害作用を有するのかを判断することができる。すなわち、当該供試物質を上記薬剤の存在下で野生型微生物に作用させたときに、薬剤に対する感受性が高くなるならば当該供試物質は薬剤排出ポンプに対して阻害作用を有することがわかり、薬剤に対する感受性が変動しないならば当該供試物質はTat分泌系に対して特異的に阻害作用を有していることがわかる。
【0036】
ここで、供試物質としては、何ら限定されず、如何なる物質であってもよい。供試物質としては、単独の物質であってもよいし、複数の構成成分からなる混合物であってもよい。供試物質としては、例えば微生物若しくは培養液からの抽出物のように未同定の物質を含むような構成であってもよいし、既知の組成物を所定の組成比で含むような構成であってもよい。また、候補物質としては、タンパク質、核酸、脂質、多糖類、有機化合物及び無機化合物のいずれでもよい。
【0037】
また、上記薬剤としては、例えば、上記融合遺伝子として緑膿菌における薬剤排出ポンプMexA-MexB-OprMのサブユニットを含む場合には、薬剤排出ポンプMexA-MexB-OprMにより菌体外に排出され、他の薬剤排出ポンプ(例えば、「MexC-MexD-OprJ」、「MexE-MexF-OprN」及び「MexX-MexY-OprM」)により菌体外に排出されない薬剤を使用する。換言すれば、薬剤として、薬剤排出ポンプMexA-MexB-OprMのみならず他の薬剤排出ポンプによっても菌体外へ排出される薬剤を使用した場合、当該薬剤に対する感受性は、薬剤排出ポンプMexA-MexB-OprM以外の他の薬剤排出ポンプによる影響により変動しないおそれがある。
【0038】
さらに、上記薬剤としては、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤を使用することができる。例えば、上記融合遺伝子として緑膿菌における薬剤排出ポンプMexA-MexB-OprMのサブユニットを含む場合には、上記変異体としては、例えばMexA欠損変異株を使用することができる。所定の薬剤を野生型緑膿菌及びMexA欠損変異株にそれぞれ作用させ、MexA欠損変異株の薬剤感受性が野生型緑膿菌の薬剤感受性と比較して有意に高まっている場合、当該薬剤は、薬剤排出ポンプMexA-MexB-OprMを介して菌体外に排出される薬剤と考えられる。したがって、当該薬剤を使用することによって、上述したような供試物質の性質について判断を行うことができる。
【0039】
ここで、有意に低下するとは、薬剤排出ポンプを欠損した変異体における薬剤耐性の低下度合いについて統計学的に有意差があることを意味する。特に、薬剤としては、薬剤排出ポンプを欠損した変異体における最小発育阻止濃度が、野生型と比較して1/2以上低下するものが好ましく、1/4以上低下するものがより好ましく、1/8以上低下するものが最も好ましい。薬剤排出ポンプを欠損した変異体における最小発育阻止濃度がこの範囲で低下する薬剤を使用すると、感受性の変動をより高精度に検出することができる。
【0040】
また、緑膿菌における薬剤排出ポンプのなかで、最も基質特異性の広いものは薬剤排出ポンプMexA-MexB-OprMである。したがって、他の薬剤排出ポンプMexC-MexD-OprJ、MexE-MexF-OprN及びMexX-MexY-OprMのサブユニットを上述した融合遺伝子により発現させるよりも、薬剤排出ポンプMexA-MexB-OprMのサブユニットを上述した融合遺伝子として発現させることが、使用可能な薬剤の選択肢を広くする意味で好ましい。
【0041】
上述した融合遺伝子に含まれるサブユニットをMexAとした場合、使用可能な薬剤としては例えば、アズトレオナム、クロラムフェニコール、キノロン剤(ノルフロキサシン)、ノボビオシン、セフォペラゾン、セフスロジンを挙げることができる。これらの薬剤は、MexA変異により最小発育阻止濃度が1/4〜1/8に低下することが知られている。したがって、これら薬剤を使用することで、感受性の変動をより高精度に検出することができる。
【0042】
さらに、上述した融合遺伝子に含まれるサブユニットをOprMとした場合、薬剤排出ポンプのうちMexA-MexB-OprM及びMexX-MexY-OprMがTat分泌系に依存することとなる。よって、この場合、使用可能な薬剤としては、オフロキサシン、ノルフロキサシン、シプロフロキサシン、クロラムフェニコール、ゲンタミシン、ノボビオシン、セフタジジム、セファペラゾン、セフスロジン及びアズトレオナムを挙げることができる。
【0043】
なお、上記で列挙した具体的な薬剤以外の薬剤であっても、例えば、新しい第四世代のセファロスポリン等、使用可能な薬剤を挙げることができる。例えば、使用可能な薬剤としては、参考文献(The role of mex-gene products in antibiotic extrusion in Pseudomonas aeruginosa. Yoneyama H, Ocaktan A, Tsuda M, Nakae T. Biochem Biophys Res Commun. 1997, 233, 611-618)に開示されている薬剤を使用することができる。
【0044】
上記薬剤に対する感受性は、従来公知の手法を適用して測定することができる。例えば、使用する薬剤を段階希釈した複数の培地を用意し、供試物質の存在下及び非存在下における組換え微生物の最小発育阻止濃度を決定するといった方法を適用することができる。また、より簡便には、組換え微生物の生育を阻止する一歩手前の濃度の薬剤を含んだ寒天培地に組換え微生物を混合し、その後、プラスチックシャーレにまき、寒天が固化した平板上に供試物質を一定量載せて培養するといった方法を適用することができる。もし供試物質がTat分泌系を阻害するならば薬剤排出ポンプが機能せず、組換え微生物が薬剤に感受性となり、供試物質の周りには発育阻止ゾーンが観察されることとなる。以上のように、供試物質の周りに形成される発育阻止ゾーンの有無を指標として、Tat分泌系に対して阻害作用のあるリード化合物を供試物質のなかから簡便に判別することができる。なお、上述した融合遺伝子に含まれるサブユニットとしてMexAを使用した場合、アズトレオナムの濃度を1.0〜1.5μg/ml程度とする。
【0045】
また、この簡便な系は、96穴のプラスチック容器を用いた液体培地を用いる系として適用することもできる。すなわち、この場合、組換え微生物を薬剤と混合し評価系を作製する操作が簡便であり、且つ、培養後の評価はマイクロタイター分光光度計を用いて半定量化して判定することができる。
【0046】
以上のように、本発明に係る組換え微生物を使用することによって、新規なTat分泌系阻害剤をハイスループットにスクリーニングすることができる。すなわち、本発明に係るTat分泌系阻害剤のスクリーニングキットは、本発明に係る組換え微生物と上記薬剤とを含むものである。なお、スクリーニングキットにおいて、上記薬剤は、段階的に希釈された濃度で複数の培地として提供されても良い。
【0047】
また、本発明に係る組換え微生物において、2以上の薬剤排出ポンプのサブユニットがTat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成するものである場合には、供試物質とともに各薬剤排出ポンプに対応する複数の薬剤についてそれぞれ感受性を独立して評価する。例えば、第1の薬剤排出ポンプ及び第2の薬剤排出ポンプがTat分泌系を介して形成されるよう2種類の融合遺伝子を導入した組換え微生物を使用する場合、第1の薬剤排出ポンプにより排出される薬剤Aに対する感受性を供試物質の存在/非存在下で評価し、第2の薬剤排出ポンプにより排出される薬剤Bに対する感受性を供試物質の存在/非存在下で評価する。このとき、薬剤Aに対する感受性が供試物質の存在下で高くなり、薬剤Bに対する感受性が供試物質の存在/非存在下で変動していないならば、当該供試物質は、Tat分泌系に対して阻害的に作用しているとは判断できず、第1の薬剤排出ポンプに阻害作用を有すると判断する。一方、供試物質の存在下で薬剤Aに対する感受性及び薬剤Bに対する感受性ともに高くなっている場合には、当該供試物質が、第1の薬剤排出ポンプ、第2の薬剤排出ポンプ及びTat分泌系の全てに阻害作用を示している可能性もあるが、Tat分泌系を特異的に阻害している蓋然性が高いと判断できる。
【実施例】
【0048】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【0049】
〔実施例1〕
本実施例では、薬剤排出ポンプ:MexA-MexB-OprMを構成するサブユニットのうちMexAのシグナル配列を、Tat分泌系を介して外界へ分泌される病原性因子フォスフォリパーゼC(PlcH)のシグナル配列を含む領域と交換した融合遺伝子を作製した。また、本実施例では、染色体上のmexA遺伝子を欠失した宿主細胞に上記融合遺伝子を導入して形質転換微生物を作製した。さらに、本実施例では、染色体上のmexA遺伝子とともに、Tat分泌系を構成するサブユニットであるtatC遺伝子を更に欠失した宿主細胞に上記融合遺伝子を導入して形質転換微生物を作製した。薬剤排出ポンプ(MexA-MexB-OprM)を介して排出される薬剤に対する感受性を、得られた2種類の形質転換微生物ついて比較した。
【0050】
(1) Tat系シグナル配列を有するキメラmexA遺伝子(plcH-mexA)の構築
(1-1) オーバーラップPCR法によるplcH-mexAキメラ遺伝子断片の増幅
(1-1-1) plcHのシグナル配列領域の増幅
plcHのシグナル配列をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-GAGGATCCTCGCTGTATTCCCTGCC-3’:配列番号5)およびリバースプライマー(5’-CGGCGCCTCGCTTTTTCCCTCGACGGCCAGGGCGCG -3’:配列番号6)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taq(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、40秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約230塩基対の増幅産物が確認できた。
【0051】
(1-1-2) mexA遺伝子の成熟領域の増幅
mexA遺伝子の成熟領域をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-CGCGCCCTGGCCGTCGAGGGAAAAAGCGAGGCGCCG-3’:配列番号7)およびリバースプライマー(5’-AGGGTACCCTTGATCAGCCCTTGC-3’:配列番号8)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロース電気泳動にて確認したところ約1,100塩基対の増幅産物が確認できた。
【0052】
(1-1-3) plcH-mexAキメラ遺伝子の増幅
plcHシグナル配列の増幅断片1μl、mexA成熟領域の増幅断片1μlおよび滅菌蒸留水8μlを混合し、この混合液を滅菌蒸留水で100倍希釈した溶液を鋳型として、2段階目のPCR反応を行った。プライマーは、フォワードプライマー(5’- GAGGATCCTCGCTGTATTCCCTGCC -3’:配列番号5)およびリバースプライマー(5’- AGGGTACCCTTGATCAGCCCTTGC -3’:配列番号8)を用いた。PCR増幅反応条件は、項目(1-1-2)と同じである。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。反応後、増幅断片をアガロース電気泳動にて確認したところ約1,300塩基対の増幅産物が確認できた。
【0053】
(1-2) pUC18ベクターへのキメラ遺伝子(plcH-mexA)のクローニング
(1-2-1) キメラ遺伝子(plcH-mexA)の連結反応と形質転換
前項で得たキメラ遺伝子(plcH-mexA)を順次KpnI(TOYOBO)、BamHI(TOYOBO)処理し(前項1-1-3で用いたプライマー末端には、各々KpnIあるいはBamHI部位が付加してある)、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpUC18(TOYOBO)と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を100μg/mlのアンピシリンを含むL寒天平板培地(isopropyl β-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0054】
(1-2-2) 形質転換体からのプラスミド分離と組換えプラスミドの解析
生育した白色コロニーを4 mlの100μg/mlのアンピシリンを含むL液体培地に植菌し、37℃にて一夜培養後、1.5 mlの菌液をミクロチューブに移し、遠心(12,000 rpm、23℃、5分間)して細胞を回収した。この細胞を氷冷した100μlの溶液1(25 mM Tris-HCl、10 mM EDTA、50 mM Glucose、pH 8.0)に懸濁後、200μlの溶液2(0.2 N NaOH、1% SDS)を加えて穏やかに混和した(室温、5分間)。その後、150μlの溶液3(3 M potassium-acetate、acetic acid)添加後、氷上にて5分間静置した。この溶液を遠心後(12,000 rpm、23℃、5分間)、上清を新しいミクロチューブに回収し、400μlのフェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液を加えて、室温にて約5分間混合した。溶液を遠心分離(12,000 rpm、23℃、5分間)後、回収した上清に900μlのエタノールを添加混合後室温にて5分放置してから遠心分離(12,000 rpm、23℃、5分間)した。得られた沈殿を500μlの70%エタノールで洗浄した後乾燥させ、RNaseA (20μg/ml)を含むTE (10 mM Tris-HCl、1 mM EDTA、pH 8.0) 30μlに溶解した。得られたプラスミドをEcoRIおよびEcoRI + BamHIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。このようにして得られたplcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子の配列を図1及び配列番号9に示す。なお、キメラ遺伝子がコードするキメラタンパク質のアミノ酸配列を配列番号10に示す。
【0055】
(1-3) シャトルベクターpMMB67HEへのキメラ遺伝子(plcH-mexA)のクローニング
(1-3-1) カナマイシン耐性マーカーをもつpTH18ks1ベクターへのplcH-mexAキメラ遺伝子のクローニング
緑膿菌へのキメラ遺伝子の形質転換に使用するシャトルベクターは、アンピシリン耐性マーカーをもっている。このシャトルベクターへのクローニングをする際、前項(1-2-2)で作製したプラスミドより断片を切り出してpMMB67HEにクローニングする場合、pUC18が同じアンピシリン耐性マーカーをもっているので組換え体を選別する際、pUC18誘導体をもつ形質転換体のバックグランドが高くなる。そこで、いったんカナマイシン耐性マーカーをもつpTH18ks1ベクターへplcH-mexAキメラ遺伝子を組換えることにした。塩基配列を確認済みのplcH-mexAキメラ遺伝子をもつ組換えプラスミドをKpnIおよびBamHIで消化後、アガロースゲル電気泳動に供し、キメラ遺伝子断片(約1.3 kb)をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。このDNA断片を、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpTH18ks1と混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。pTH18ks1にplcH-mexAキメラ遺伝子断片がクローニングされたことは、形質転換体より抽出したプラスミドをEcoRIおよびPstI消化後、アガロースゲル電気泳動を行い断片の挿入を確認した。
【0056】
(1-3-2) pMMB67HEへのキメラ遺伝子(plcH-mexA)のクローニング
pMMB67HEへ挿入するキメラ遺伝子断片の調整は、項目(1-3-1)で作製した組換えプラスミドを鋳型とし、フォワードプライマー(5’- GAGGATCCTCGCTGTATTCCCTGCC -3’:配列番号5)およびリバースプライマー(5’- AGGGTACCCTTGATCAGCCCTTGC -3’:配列番号8)を用いて、項目(1-1-2)の反応条件でPCR反応を行って得た。この増幅断片をKpnIおよびBamHI消化し、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、キメラ遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpPlcH-MexAとよぶことにする。
【0057】
(2) キメラmexA遺伝子(plcH-mexA)を導入した形質転換体の作製
(2-1) mexAを欠損した緑膿菌変異株(P. aeruginosa TNP070)のコンピタント細胞の調整
TNP070株(The role of mex-gene products in antibiotic extrusion in Pseudomonas aeruginosa. Yoneyama H, Ocaktan A, Tsuda M, Nakae T. Biochem Biophys Res Commun. 1997, 233, 611-618参照)を4 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で一夜振とう培養した。増殖した菌液800μlを40 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で約7時間培養して得た対数増殖期の細胞を、プラスチックチューブに移し、7,000 rpm、4℃、10分間遠心し(ローター、AG-508;遠心機、KUBOTA 6930)集菌した。この菌体を氷冷した40 mlの0.3 Mシュークロース溶液で洗浄した後、遠心(7,000 rpm、4℃、5分間)して集菌した。この遠心洗浄操作を3回繰り返して得た菌体を、少量(200〜400μl)の氷冷した0.3 Mシュークロース溶液に懸濁しコンピタント細胞としてエレクトロポレーションに供した。
【0058】
(2-2) 前項で調整したコンピタントTNP070細胞40μlに1μlのキメラ遺伝子(plcH-mexA)を挿入したpMMB67HEを加え、緩やかに混合した菌液をエレクトロポレーション用キュベット 2 mmギャップ(Molecular BioProducts)に注入し、エレクトロポレーション装置に装填した(GenePulser II、Bio-Rad)。エレクトロポレーションを1.6 kV、25μF、400Ωの条件下で行った後、菌液を100μg/mlのスルベニシリン(武田薬品工業)を含むL平板培地に塗布し、37℃で2夜培養した。生育したコロニーより項目(1-2-2)の方法に従いプラスミドを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位の両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCR反応を行い、組換えプラスミドの存在を確認した。
【0059】
(3) キメラ遺伝子(plcH-mexA)を保有する染色体上mexA遺伝子が欠損した形質転換体の薬剤感受性の評価
(3-1) 段階希釈した指標薬剤を含む寒天平板の作製
指標薬剤であるアズトレオナム(Sigma)およびクロラムフェニコール(和光純薬)の原液を、おのおの滅菌蒸留水および10%エタノールで溶解して調整した。この各薬剤の原液を滅菌蒸留水で希釈し、アズトレオナムを16〜0.0625μg/ml、クロラムフェニコールを64〜1μg/mlの範囲の2倍希釈系列を含むミューラー ヒントン寒天培地(Difco)を作製した。
【0060】
(3-2) 最小発育阻止濃度評価用菌株の培養
最小発育阻止濃度を評価する菌株を表2に示した。各菌株をアクティブプレートより一白金耳、0.4% (w/v)硝酸カリウムを含む2 mlのミューラー ヒントン液体培地(Difco)に接種し、37℃にて一夜静置培養した。なお、プラスミド保有株を培養する際の液体培地にはスルべニシリン(50μg/ml)を含む0.4% (w/v)硝酸カリウム添加ミューラー ヒントン液体培地を用いた。一夜培養後の菌液を、同液体培地で菌体密度が約106 cell/mlになるように希釈して薬剤感受性評価に供した。
【0061】
(3-3) 最小発育阻止濃度の評価
前項(3-2)で調整した菌株をガラス製小試験管に0.5〜1.0 ml分注し、最小発育阻止濃度測定用装置ミクロプランター(佐久間製作所)に装着し、薬剤を含む寒天平板に接種した。この平板培地を37℃、約20時間培養後に各菌株の生育を評価した。
【0062】
(4) PlcH-MexAキメラタンパク質がTat分泌系依存性であることの検証
(4-1) tatC遺伝子内部配列をもつ組換えプラスミドの作製
(4-1-1) tatC遺伝子の内部配列断片の増幅
tatC遺伝子の内部配列をコードする遺伝子断片(446 bp)を増幅するためにフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)およびリバースプライマー(5’-ACGGATCCGTCAGGACGAAGTCCAGGTAG-3’:配列番号14)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taq(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約450塩基対の増幅産物が確認できた。
【0063】
(4-1-2) pK18mobベクターへのtatC遺伝子内部配列のクローニング
前項で得たtatC遺伝子の内部配列断片をBamHI(TOYOBO)で処理し(前項4-1-1で用いたプライマー末端には、BamHI部位が付加してある)、予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpK18mobと混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を12.5μg/mlのカナマイシンを含むL寒天平板培地(isopropylβ-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0064】
(4-1-3) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
白色コロニーを4 mlの12.5μg/mlのカナマイシンを含むL液体培地に植菌し、37℃にて一夜培養後、項目(1-2-2)の方法に従いプラスミドを分離した。得られたプラスミドをBamHI、EcoRIあるいはHindIIIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。
【0065】
(4-2) tatC遺伝子破壊株の作製
(4-2-1) tatC内部配列をもつプラスミドのE. coli S17-1への形質転換
E. coli S17-1コンピタント細胞は塩化カルシウム法にしたがって作製した。一夜L液体培地で培養した200μlのE. coli S17-1を4 mlの新鮮なL液体培地に接種後、37℃にて約3時間培養した。対数増殖期の細胞を遠心集菌後(12,000 rpm、4℃、5分間)、1 mlの氷冷した0.1 M CaCl2溶液で2回遠心洗浄(12,000 rpm、4℃、5分間)をし、菌体を200μlの同溶液に懸濁した。この菌懸濁液と2μlの項目(4-1-3)で分離したプラスミドDNAを、キャップ付きプラスチック製試験管に移し混合した後氷中で30分間静置した。その後、試験管を42℃の水槽で90秒間インキュベーションして再度氷中で2分間静置した。この菌懸濁液に室温のSOC培地1 mlを加え37℃で約1時間振とう培養をした後、12.5μg/mlのカナマイシンを含むL寒天培地に塗布して37℃にて一夜培養した。生育したコロニーから項目(1-2-2)の方法に従いプラスミドを分離し、得られたプラスミドをBamHIあるいはBglIIで消化後、アガロースゲル電気泳動により組換えプラスミドの存在を確認した。
【0066】
(4-2-2) 組換えプラスミドのP. aeruginosa TNP070への接合伝達とtatC破壊株の選択
組換えプラスミドをもつE. coli S17-1とP. aeruginosa TNP070を、それぞれ12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地で一夜37℃にて培養した。生育したそれぞれの菌液80μlを12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地に添加し、37℃で約8時間振とう培養をした後、700μlの大腸菌と700μlの緑膿菌を同じミクロチューブに添加混合後、遠心集菌(12,000 rpm、室温、5分間)した。抗生物質を除去するために、1 mlのL液体培地で菌体を2回遠心洗浄(12,000 rpm、室温、5分間)し100μlのL液体培地に再懸濁した。この菌液を12.5μg/mlのカナマイシンと50μg/mlのテトラサイクリンを含むL寒天平板に塗布して37℃にて3日培養し、tatC遺伝子の相同配列領域で組換えが起こった破壊株を得た。
【0067】
(4-2-3) tatC遺伝子破壊株の染色体DNAの分離
前項で得た破壊株のtatC遺伝子領域のサザンブロット解析を実施するために、破壊株の染色体を以下の方法にしたがって分離した。破壊株を4 mlの12.5μg/mlのカナマイシン添加L液体培地で一夜37℃にて培養後集菌(12,000 rpm、室温、5分間)し、1 mlのSTE緩衝液(0.3 M sucrose、25 mM Tris-HCl、25 mM EDTA、pH 8.0)で1回遠心洗浄した後、5 mlの同緩衝液に懸濁した。この菌懸濁液に556μlの10% SDS (sodium dodecylsulfate、和光純薬)と28μlの20 mg/ml proteinase K(ナカライテスク株式会社)溶液を加え、穏やかに混合後、37℃にて一夜インキュベーションした。溶液を2 mlのフェノール・クロロフォルム・イソアミルアルコール (25:24:1)混合溶液で抽出操作を室温にて5分間行い、遠心(3,000 rpm、室温、15分間)後、上清を新しいプラスチック試験管に回収した。この溶液に1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加混合し核酸を沈殿させた。上清を遠心(3,000 rpm、室温、5分間)除去後、核酸を1 mlの70%エタノールで1回遠心洗浄し、溶液をデカント操作で除去した。沈殿した核酸を風乾後、4 mlの20μg/mlのRNaseA(ナカライテスク株式会社)を含むTE溶液(10 mM Tris-HCl、1 mM EDTA、pH 8.0)に溶解し、37℃にて約1時間インキュベーションをした。その後、上記同様フェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液で抽出操作を1回行い、1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加混合してDNAを沈殿させ、70%エタノールで2回遠心洗浄後、DNAを1 mlのTE溶液に溶解した。
【0068】
(4-2-4) tatC遺伝子破壊株染色体DNAのサザンブロット解析
前項で分離した染色体約1μgをBamHI、PstI、SacIIおよびSalIで37℃、15時間切断した後、1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加してDNAを沈殿、70%エタノールで1回遠心洗浄後、風乾したDNAを8μlのTE溶液に溶解した。この制限酵素消化後の各DNAサンプルを1.6μlのローディング色素液(0.25% bromophenol blue、40% sucrose)と混合し、全量をTBE緩衝液(45 mM Tris-HCl、45 mM Boric acid、10 mM EDTA、pH 8.0)で作製したアガロースゲル(0.6% w/v)に供し電気泳動を行った。泳動緩衝液にはTBE緩衝液を用いた。アガロース電気泳動後、ゲルをアルカリ変性(1.5 M NaCl、0.5 M NaOH)、蒸留水による洗浄、中和(1 M Tris-HCl、1.5 M NaCl、pH 7.4)処理した後、分画した断片をナイロンメンブランHiBond+ (Amersham)にキャピラリーブロッティング法を用いて一夜転写した後、2xSSC(0.3 M NaCl、30 mM sodium citrate、pH 7.0)にてメンブランを洗浄した。以降のシグナルの検出操作は、DIG High Prime DNA Labeling and Detection Starter Kit I (Roche社)の実験操作法にしたがった。検出に用いたプローブはtatC遺伝子全体をカバーするフォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)とリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用いて増幅したDNA断片(約910 bp)を使用した。その結果、組換えプラスミドがもつ内部配列の相同領域と染色体の同領域間での組換えが起こり、pK18mobベクターのtatC遺伝子内への挿入による破壊が確認できた。
【0069】
(4-2-5) シデロフォア(pyoverdine)産生能の評価
tatC遺伝子破壊株は緑膿菌のシデロフォアであるpyoverdine産生能が消失することが報告されている。そこで、項目(4-2-2)で作製したtatC遺伝子破壊株を12.5μg/mlのカナマイシンを含む5 mlのCAA液体培地(5 mMリン酸カリウム、1 mM 硫酸マグネシウム、5 g/lカザミノ酸、pH 7.0)で37℃にて一夜培養し、遠心(12,000 rpm、室温、5分間)により培養上清を回収した。この上清の吸収を分光光度計(V-630BIO、日本分光)を用いて測定した。その結果、親株TNP070で認められる典型的なpyoverdineの吸収が、tatC遺伝子破壊株では認められず、tatC遺伝子が破壊されていることが確認できた(図2)。このtatC遺伝子破壊株をP. aeruginosa TNP080とよぶことにする。なお、図2において(a)は親株TNP070を使用した際の結果を示し、(b)はTNP080を使用した際の結果を示している。
【0070】
(4-3) mexA/tatC二重変異株でのPlcH-MexA融合タンパク質の機能評価
(4-3-1) P. aeruginosa TNP080へのpPlcH-MexA発現ベクターの形質転換
P. aeruginosa TNP080 コンピタント細胞は、項目(2-1)の方法に準じて調製した。なお、L液体培地には12.5μg/mlのカナマイシンを添加して培養した。このmexA/tatC二重変異株のコンピタント細胞40μlに、項目(1-3-2)で調製したpPlcH-MexA発現ベクターを1μl添加し、エレクトロポレーション法にて形質転換した。条件は、項目(2-2)と同じである。
【0071】
(4-3-2) pPlcH-MexA発現ベクターを有するmexA/tatC二重変異株の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0072】
(4-4) tatC遺伝子のクローニングとtatC遺伝子破壊株への導入および機能評価
(4-4-1) tatC遺伝子の増幅
野生型のtatC遺伝子を増幅するために、フォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)およびリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTAR(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約910 bpの増幅産物が確認できた。
【0073】
(4-4-2) pUC18ベクターへのtatC遺伝子のクローニング
前項(4-4-1)で得たtatC遺伝子断片をBamHI(TOYOBO)で処理し(前項4-4-1で用いたプライマー末端には、BamHI部位が付加してある)、予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpUC18と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を100μg/mlのアンピシリンを含むL寒天平板培地(isopropylβ-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0074】
(4-4-3) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
白色コロニーを4 mlの100μg/mlのアンピシリンを含むL液体培地に植菌し、37℃にて一夜培養後、項目(1-2-2)の方法に従いプラスミドを分離した。得られたプラスミドをBamHIおよびEcoRIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析し、野生型のtatC遺伝子と同じ塩基配列であることを確認した。
【0075】
(4-4-4) tatC遺伝子とplcH-mexAキメラ遺伝子をもつシャトルベクターの作製
項目(1-3-1)で述べたようにシャトルベクターの作製効率をあげるために、クローン化したtatC遺伝子をシャトルベクターに載せる前に、一旦、plcH-mexAキメラ遺伝子をもつpTH18ks1(カナマイシン耐性マーカーをもつ)ベクターにtatC遺伝子をクローン化することにした。項目(4-4-3)で作製したプラスミドをBamHIで消化後、アガロースゲル電気泳動を行い約910 bpのtatC遺伝子断片をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。このDNA断片を、項目(1-3-1)で作製したplcH-mexAキメラ遺伝子保有pTH18ks1を予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したDNAと混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。この実験デザインではplcH-mexAキメラ遺伝子の直前にtatC遺伝子が順向きあるいは逆向きに挿入される可能性がある。そこで、順向きに挿入された場合のみ連結した2遺伝子(tatC-plcH-mexA)が増幅されるフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)とリバースプライマー(5’-AGGGTACCCTTGATCAGCCCTTGC-3’:配列番号8)を用いたコロニーPCR法にて個々の形質転換体を直接鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、2分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロース電気泳動にかけ、約2.2 kbの増幅産物が認められるクローンを選抜した。
【0076】
このようにしてtatC遺伝子が順向きにplcH-mexAキメラ遺伝子の直前に挿入されたクローンを選び、(tatC-plcH-mexA)遺伝子の全塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。その結果得られた(tatC-plcH-mexA)遺伝子の配列を図3及び配列番号17に示す。なお、tatC遺伝子によりコードされるTatCタンパク質のアミノ酸配列を配列番号18に示す。
【0077】
予期しなかったことだが、PCR増幅断片をpTH18ks1ベクターにクローニングする際、由来不明の130 bpからなるDNA断片がtatC遺伝子とベクターのマルチクローニング部位であるHindIII部位の間に挿入されていることが判明した。この組換えプラスミドをHindIIIおよびKpnI消化し、約2.2 kbの(tatC-plcH-mexA)遺伝子断片を、予めKpnI、HindIIIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、(tatC-plcH-mexA)遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpTatC-PlcH-MexAをよぶことにする。
【0078】
(4-4-5) P. aeruginosa TNP080へのpTatC-PlcH-MexA発現ベクターの形質転換
項目(4-3-1)と同様の操作にて、pTatC-PlcH-MexA発現ベクターを染色体上のtatCとmexAが欠失した二重変異株TNP080に形質転換し、スルベニシリン(武田薬品工業)50μg/mlを添加したL寒天平板にて形質転換体を取得した。得られた形質転換体よりプラスミドを項目(1-2-2)の方法に従いを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位の両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCRを行い、組換えプラスミドの存在を確認した。
【0079】
(4-4-6) クローン化したtatC遺伝子産物の機能評価
クローン化したtatC遺伝子の導入によりTat分泌系の機能が回復することが期待できる。この点を評価するために項目(4-2-5)の方法にしたがい、前項目(4-4-5)で構築した形質転換体をCAA培地で生育させ、その培養上清中へのpyoverdineの分泌生産能の評価を行った。その結果、クローン化したtatC遺伝子をもつ形質転換体TNP080/pTatC-PlcH-MexAは、親株TNP070と同様なpyoverdineの吸収が認められ、Tat分泌系が機能していることが明らかとなった(図4)。
【0080】
(4-4-7) pTatC-PlcH-MexA発現ベクターを有するmexA/tatC二重変異株の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0081】
(4-4-8)ペリプラズム画分の分離
緑膿菌の各菌株を20mlのL液体培地(プラスミド保有株の場合は50μg/mlのスルベニシリンを含む)に接種し、37℃にて一夜培養した。この菌液を200mlの37℃に保温しておいた同培地に接種し、対数期後期(OD660nm、約1.0)まで振とう培養した後、プラスチック遠心管に移し室温にて遠心集菌をした(7500rpm、10分、室温)。この菌体を湿重量あたり5倍量の溶液(0.2M MgCl2, 50mM Tris-HCl, pH7.3)に懸濁しガラス試験管に移した後、37℃の水槽で10分間静置後、氷水中に15分間静置した。さらに、この37℃、10分間保温した後、氷水中、15分間インキュベーションする操作を繰り返し、細胞を12,000rpm、15分間、10℃にて遠心した。集菌後の上清0.5mlを超遠心用のプラスチックチューブ(Beckman, Cat. No. 343776)に移し、55,000rpm、10℃、1時間遠心(ローター、Beckman TLA-120.1;遠心機、Beckman Optima TLX)した後、上清を回収してペリプラズム画分とした。
【0082】
(4-4-9)ウエスタンブロット解析
項目(4-4-8)で調整したペリプラズム画分をSDS-ポリアクリルアミドゲル(12%)に供し電気泳動を行った後、Immobilon-Pメンブラン(Millipore社)に分離したタンパク質を転写し、一次抗体として抗MexAウサギ抗体、二次抗体としてアルカリフォスファターゼ結合抗ウサギーヒツジ抗体を用いて免疫染色を行った。
【0083】
<結果>
本実施例で行った薬剤感受性の評価結果を表2に示す。
【0084】
【表2】
【0085】
(1) PlcH-MexAキメラタンパク質はMexA欠損緑膿菌で機能する
pPlcH-MexA発現ベクターを有するP. aeruginosa TNP070形質転換体のアズトレオナムおよびクロラムフェニコールに対する感受性を評価した結果を表2に示した。pPlcH-MexAをもつ形質転換体のアズトレオナムおよびクロラムフェニコールに対する最小発育阻止濃度(MIC)は、それぞれ2.0μg/mlおよび32μg/mlであった。この感受性は野生株TNP070に比べ若干低い値であるが、クローン化した野生型のmexA遺伝子を導入した形質転換体(TNP070/pMexA)と同じ薬剤感受性を示したことから、キメラタンパク質PlcH-MexAは機能を保持していることが明らかとなった。このようにTNP070株の薬剤排出ポンプの機能が回復したことから、PlcH-MexAはTat分泌系を介してペリプラズムに分泌されるものと思われる。実際、形質転換体(TNP070/pPlcH-MexA)が発現したキメラタンパク質(PlcH-MexA)が成熟型MexAとしてペリプラズム画分に輸送されているか否かを、抗MexA抗体で検出したところ、シグナル配列が切断されたと思われる成熟型のMexAが形質転換体(TNP070/pPlcH-MexA)で認められた(図5、レーン8)。野生型細胞(PAO4290)は全菌体中に成熟型MexAのシグナルが認められるが(図5、レーン1)、ペリプラズムには存在しなかった(図5、レーン6)。また、mexA欠損変異株TNP070は全菌体中およびペリプラズム中いずれもMexAは発現しておらず(図5、レーン2および7)、このペリプラズム分画法によってMexAの局在が正確に評価されていることが確認できた。
【0086】
(2) PlcH-MexAキメラタンパク質はTat分泌系依存的に機能する
PlcH-MexAキメラタンパク質が機能を有していることから、本キメラタンパク質は翻訳後にTat系を介して本来の局在場所であるペリプラズムに分泌され、MexBおよびOprMと複合体を形成して薬剤を排出しているものと考えられる。この仮説を検証するために、MexA欠損変異株(TNP070)のTat分泌系を構成するtatC遺伝子を破壊した二重変異株TNP080(MexA-, TatC-)を作製した。この変異株にpPlcH-MexA発現ベクターを導入した形質転換株のアズトレオナムおよびクロラムフェニコールに対する感受性を調べた結果、それぞれの薬剤に対する形質転換体の感受性は、0.25μg/mlおよび4.0μg/mlであった(表2)。これは、TNP070とほぼ同じ感受性であり、薬剤排出ポンプが機能していないことを示唆している。すなわち、Tat分泌系が機能を失うとPlcH-MexAキメラタンパク質は正常に分泌されないことを意味しており、このキメラタンパク質はTat系依存的に分泌されていることが強く示唆された。このことをさらに検討するために、tatC破壊変異株より誘導した形質転換体(TNP080 /pPlcH-MexA)のペリプラズム画分を抗MexA抗体で検出した結果、全菌体中にはIPTG誘導による過剰なキメラタンパク質の発現が認められたが(図5、レーン4)、ペリプラズムには抗MexA抗体で染色されるタンパク質は認められず(図5、レーン9)、キメラPlcH-MexAはTat系に依存してペリプラズムに輸送されることが明らかとなった。おもしろいことに全菌体中のシグナルは成熟型より大きなMexAが認められたが、これはシグナルペプチダーゼで切断されていない状態のMexAと考えられた。
【0087】
なお、図5に示したウエスタンブロット解析の結果は、全菌体タンパク質(4μg)又はペリプラズム画分(1μg)をSDS-PAGE (12%ゲル)にて泳動後、抗MexA抗体にて検出した結果である。図5中、白三角は成熟型MexA、黒三角はシグナルペプチダーゼで切断されていないMexAを示す。
【0088】
この結果をさらに確認するために、野生型のtatC遺伝子をクローニングし、二重変異株TNP080にpPlcH-MexAとともに導入した(TNP080/pTatC-PlcH-MexA)。この形質転換体のアズトレオナムとクロラムフェニコールに対する感受性(それぞれ2.0μg/ml、32μg/ml)は、ほぼ野生株と同等レベルであったことから、クローン化したTatCが供給されることによってTat分泌系が機能回復していることが明らかとなった。このことから、PlcH-MexAキメラタンパク質が機能を回復したTat分泌系を介してペリプラズムに輸送されたことが強く示唆された。実際、この形質転換体(TNP080/pTatC-PlcH-MexA)のペリプラズム画分を抗MexA抗体で染色すると、発現量は少ないものの成熟型のMexAが輸送されていることが確認できた(図5、レーン10)。
【0089】
以上の結果より、今回作製したPlcH-MexAキメラタンパク質は予想したとおりTat分泌系を介してペリプラズムに輸送され、MexBおよびOprMと複合体を形成しMexA,B-OprM排出ポンプとして機能することが明らかとなった。この基礎的な知見を応用した新しい評価系を用いて天然化合物ライブラリーや低分子有機化合物ライブラリーといった供試物質を簡便に評価するハイスループットスクリーニング系の構築が可能である。本評価系においてTat分泌系を阻害する物質が指示薬剤と共存すれば、PlcH-MexA融合タンパク質は翻訳後にペリプラズムに分泌されないことから、pPlcH-MexA発現ベクターを有する指示菌は抗生物質に高感受性を示すことになる。
【0090】
Tat分泌系を標的とする抗菌剤は現在見出されておらず、この新しい評価系により効率的に新規標的であるTat系を阻害するリード化合物の探索が可能となる。また、本スクリーニング系で陽性を示す化合物は、Tat分泌系及び/又はMexA-MexB-OprM排出ポンプに対する阻害剤である可能性がある。換言すれば、本スクリーニング系によれば、Tat分泌系と薬剤排出ポンプというこれまでにない新しい二つの標的を同時にターゲットとした非常に効率のよいスクリーニング系である。
【0091】
〔実施例2〕
本実施例では、薬剤排出ポンプ:MexA-MexB-OprMを構成するサブユニットのうちOprMのシグナル配列を、Tat分泌系を介して外界へ分泌される病原性因子フォスフォリパーゼC(PlcH)のシグナル配列を含む領域と交換した融合遺伝子を作製した。そして、実施例1と同様に、染色体上のoprM遺伝子を欠失した宿主細胞、および染色体上のoprM遺伝子とともにtatC遺伝子を欠失した二重変異宿主細胞に本実施例で作製した融合遺伝子を導入し、薬剤排出ポンプ(MexA-MexB-OprM)を介して排出される薬剤に対する感受性を、得られた2種類の形質転換微生物について評価した。
【0092】
(1) Tat系シグナル配列を有するキメラoprM遺伝子(plcH-oprM)の構築
(1-1) オーバーラップPCR法によるplcH-oprMキメラ遺伝子断片の増幅
(1-1-1) plcHのシグナル配列領域の増幅
plcHのシグナル配列をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-AGTCTAGACTCGCTGTATTCCCTGCC-3’:配列番号19)およびリバースプライマー(5’-GTAGTCGGGGATCAGCGAGGTTCCGGTGCGGATGTC-3’:配列番号20)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTAR(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、40秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロースゲル電気泳動にて確認したところ約230塩基対の増幅産物が確認できた。
【0093】
(1-1-2) oprM遺伝子の成熟領域の増幅
oprM遺伝子の成熟領域をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-GACATCCGCACCGGAACCTCGCTGATCCCCGACTAC-3’:配列番号21)およびリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTARを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;65℃、5秒間;72℃、2分間の反応を30サイクル行った後、72℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にて確認したところ約1,500塩基対の増幅産物が確認できた。
【0094】
(1-1-3) plcH-oprMキメラ遺伝子の増幅
plcHシグナル配列の増幅断片1μl、oprM成熟領域の増幅断片1μlおよび滅菌蒸留水8μlを混合し、この混合液を滅菌蒸留水で100倍希釈した溶液を鋳型として、2段階目のPCR反応を行った。プライマーは、フォワードプライマー(5’- AGTCTAGACTCGCTGTATTCCCTGCC -3’:配列番号19)およびリバースプライマー(5’- CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いた。PCR増幅反応条件は、項目(1-1-2)と同じである。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。
【0095】
(1-2) pHSG298ベクターへのキメラ遺伝子(plcH-oprM)のクローニング
(1-2-1) キメラ遺伝子(plcH- oprM)の連結反応と形質転換
前項で得たキメラ遺伝子(plcH-oprM)を順次XbaI(TOYOBO)、EcoRI(TOYOBO)処理し(前項1-1-3で用いたプライマー末端には、各々XbaIあるいはEcoRI部位が付加してある)、予めXbaI、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpHSG298(TOYOBO)と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200 Ωである。回収した細胞を12.5μg/mlのカナマイシンを含むL寒天平板培地(isopropyl β-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0096】
(1-2-2) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
生育した白色コロニーを4mlの12.5μg/mlのカナマイシンを含むL液体培地に植菌し、37℃にて一夜培養後、1.5 mlの菌液をミクロチューブに移し、遠心(12,000 rpm、23℃、5分間)して細胞を回収した。この細胞を氷冷した100μlの溶液1(25 mM Tris-Hcl、10 mM EDTA、50 mM Glucose、pH 8.0)に懸濁後、200μlの溶液2(0.2 N NaOH、1% SDS)を加えて穏やかに混和した(室温、5分間)。その後、150μlの溶液3(3 M potassium-acetate、acetic acid)添加後、氷上にて5分間静置した。この溶液を遠心後(12,000 rpm、23℃、5分間)、上清を新しいミクロチューブに回収し、400μlのフェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液を加えて、室温にて約5分間混合した。溶液を遠心分離(12,000 rpm、23℃、5分間)後、回収した上清に900μlのエタノールを添加混合後室温にて5分放置してから遠心分離(12,000 rpm、23℃、5分間)した。得られた沈殿を500μlの70%エタノールで洗浄した後乾燥させ、RNase (20μg/ml)を含むTE (10 mM Tris-HCl、1 mM EDTA、pH 8.0) 30μlに溶解した。得られたプラスミドをEcoRIおよびEcoRI + XbaIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。このようにして得られたplcHシグナル配列遺伝子とoprM遺伝子の成熟領域とを融合したキメラ遺伝子の配列を図6及び配列番号23に示す。なお、このキメラ遺伝子がコードするキメラタンパク質のアミノ酸配列を配列番号24に示す。
【0097】
(1-3) シャトルベクターpMMB67HEへのキメラ遺伝子(plcH-oprM)のクローニング
pMMB67HEへ挿入するキメラ遺伝子断片の調整は、項目(1-2-2)で作製した組換えプラスミドをXbaIおよびEcoRIで消化後、アガロースゲル電気泳動によりキメラ遺伝子(plcH-oprM)を含むDNA断片(約1.7 kb)を分離した。この断片をQIAEX II Gel Extraction Kit (QIAGEN)を用いて分離精製し、予めXbaI、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を16℃にて一夜行った。その後、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、キメラ遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpPlcH-OprMと呼ぶことにする。
【0098】
(2) キメラoprM遺伝子(plcH-oprM)を導入した形質転換体の作製
(2-1) oprMを欠損した緑膿菌変異株(P. aeruginosa TNP072)のコンピタント細胞の調整
TNP072株を4 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で一夜振とう培養した。増殖した菌液800μlを40mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で約7時間培養して得た対数増殖期の細胞を、プラスチックチューブに移し、7,500 rpm、4℃、10分間遠心し(ローター、AG-508;遠心機、KUBOTA 6930)集菌した。この菌体を氷冷した40mlの0.3 Mシュークロース溶液で洗浄した後、遠心(7,500 rpm、4℃、5分間)して集菌した。この遠心洗浄操作を3回繰り返して得た菌体を、少量(200〜400μl)の氷冷した0.3 Mシュークロース溶液に懸濁しコンピタント細胞としてエレクトロポレーションに供した。
【0099】
(2-2) 前項(2-1)で調整したコンピタントTNP072細胞40μlに1μlのキメラ遺伝子(plcH-oprM)を挿入したpMMB67HEを加え、緩やかに混合した菌液をエレクトロポレーション用キュベット 2 mmギャップ(Molecular BioProducts)に注入し、エレクトロポレーション装置に装填した(GenePulser II、Bio-Rad)。エレクトロポレーションを2.5 kV、25μF、200Ωの条件下で行った後、菌液を100μg/mlのスルベニシリン(武田薬品工業)を含むL平板培地に塗布し、37℃で2夜培養した。生育したコロニーより項目(1-2-2)の方法に従いプラスミドを抽出し、分離したプラスミドを鋳型プライマーとしてフォワードプライマー(5’-AGTCTAGACTCGCTGTATTCCCTGCC-3’:配列番号19)およびリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いてPCR反応を行い、組換えプラスミドの存在を確認した。
【0100】
(3) キメラ遺伝子(plcH-oprM)を保有する染色体上oprM遺伝子が欠損した形質転換体の薬剤感受性の評価
(3-1) 段階希釈した指標薬剤を含む寒天平板の作製
指標薬剤であるアズトレオナム(Sigma)およびクロラムフェニコール(和光純薬)の原液を、おのおの滅菌蒸留水および10%エタノールで溶解して調整した。この各薬剤の原液を滅菌蒸留水で希釈し、アズトレオナムを16〜0.0625μg/ml、クロラムフェニコールを64〜1μg/mlの濃度範囲の2倍希釈系列を含むミューラー ヒントン寒天培地(Difco)を作製した。
【0101】
(3-2) 最小発育阻止濃度評価用菌株の培養
最小発育阻止濃度を評価する際には、各菌株をアクティブプレートより一白金耳、0.4% (w/v)硝酸カリウムを含む2mlのミューラー ヒントン液体培地(Difco)に接種し、37℃にて一夜静置培養した。なお、プラスミド保有株を培養する際の液体培地にはスルべニシリン(50μg/ml)を含む0.4%(w/v)硝酸カリウム添加ミューラー ヒントン液体培地を用いた。一夜培養後の菌液を、同液体培地で菌体密度が約106 cells/mlになるように希釈して薬剤感受性評価に供した。
【0102】
(4) PlcH-OprMキメラタンパク質の機能発現がTat分泌系依存性であることの検証
(4-1) oprM遺伝子破壊株(TNP072)のtatC遺伝子を破壊した二重変異株(TNP082)の作製
mexA破壊株(TNP070)のtatC遺伝子を破壊したときに使用した自殺ベクターを用い、以下の方法にてoprM破壊株(TNP072)のtatC遺伝子を破壊した。
【0103】
(4-1-1) tatC遺伝子破壊用自殺ベクターのP. aeruginosa TNP072への接合伝達とtatC破壊株の選択
tatC遺伝子破壊用自殺ベクターをもつE. coli S17-1とP. aeruginosa TNP072を、それぞれ12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4mlのL液体培地で一夜37℃にて培養した。生育したそれぞれの菌液40μlを12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地に添加し、37℃で約8時間振とう培養をした後、700μlの大腸菌と700μlの緑膿菌を同じミクロチューブに添加混合後、遠心集菌(12,000 rpm、室温、5分間)した。抗生物質を除去するために、1 mlのL液体培地で菌体を2回遠心洗浄(12,000 rpm、室温、5分間)し100μlのL液体培地に再懸濁した。この菌液を12.5μg/mlのカナマイシンと50μg/mlのテトラサイクリンを含むL寒天平板に塗布して37℃にて2日培養し、tatC遺伝子の相同配列領域で組換えが起こった破壊株を得た。
【0104】
(4-1-2) tatC遺伝子破壊株ゲノムのtatC遺伝子領域の構造解析
前項目(4-1-1)においてtatC遺伝子の内部配列をもつ自殺ベクターの一点交叉による挿入によりtatC遺伝子を破壊した。そこで、染色体上のtatC遺伝子が破壊されていることを検証するためにtatC遺伝子に対応したフォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)とリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用いPCR反応にて増幅した。PCR反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、5分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にて確認したところtatC遺伝子内に自殺ベクターが挿入した結果生じる約5.0キロ塩基対の増幅産物が確認できた。
【0105】
(4-1-3) シデロフォア(pyoverdine)産生能の評価
前項目(4-1-1)で取得したoprM、tatC二重変異株のTat分泌系が機能消失したことを検証するために、mexA、tatC二重欠損変異株の項で記載したように緑膿菌のシデロフォアであるpyoverdine産生能を調べることによって評価した。前項で取得したtatC遺伝子破壊株を12.5μg/mlのカナマイシンを含む5 mlのCAA液体培地(5 mMリン酸カリウム、1 mM 硫酸マグネシウム、5 g/lカザミノ酸、pH 7.0)で37℃にて1日〜2日間培養し十分に菌体を生育させ後、遠心(12,000 rpm、室温、5分間)により培養上清を回収した。この上清の吸収を分光光度計(V-630BIO、日本分光)を用いて測定した。その結果、野生株および親株TNP072で認められる典型的なpyoverdineの吸収が、tatC遺伝子破壊株では認められず、tatC遺伝子が破壊されていることが確認できた(図7)。このtatC遺伝子が破壊された二重変異株をP. aeruginosa TNP082とよぶことにする。なお、図7において(a)は野生株、(b)は親株TNP072を使用した際の結果を示し、(c)はTNP082を使用した際の結果を示している。
【0106】
(4-2) oprM/tatC二重変異株でのPlcH-OprM融合タンパク質の機能評価
PlcH-OprM融合タンパク質がTat分泌系に依存してトランスロケートされるか否かを評価するために、pPlcH-OprM発現ベクターを前項で作製したtatC遺伝子破壊株TNP082(oprM/tatC二重変異株)に形質転換しアズトレオナムおよびクロラムフェニコールに対する感受性を項目(3)に記載した寒天希釈法によって調べた。
【0107】
(4-3) tatC遺伝子とplcH-oprMキメラ遺伝子をもつシャトルベクターの作製
シャトルベクターの作製効率をあげるために、クローン化したtatC遺伝子をシャトルベクターに載せる前に、一旦、plcH-oprMキメラ遺伝子をもつpHSG298(カナマイシン耐性マーカーをもつ)ベクターにtatC遺伝子をクローン化することにした。実施例1の項目(4-4-3)で作製したプラスミドtatC / pUC18をSphIとSmaIで消化後、アガロースゲル電気泳動を行い約910 bpのtatC遺伝子断片をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。これと並行して、項目(1-3)で作製したplcH-oprMキメラ遺伝子保有pHSG298を予めXbaI処理し、T4DNAポリメラーゼによる平滑末端化を行った。その後SphI処理およびバクテリアルアルカリフォスファターゼ(TOYOBO)処理を施した。このように調製したベクターと精製したtatC遺伝子断片とを混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。この実験デザインではplcH-oprMキメラ遺伝子の直前にtatC遺伝子が順向きに挿入されることになる。そこで、順向きに連結した2遺伝子(tatC-plcH-oprM)が増幅されるフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)とリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いた直接コロニーPCR法を用いて目的とする組換え体を選抜した。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、3分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にかけ、約2.5 kbの増幅産物が認められるクローンを選抜した。
【0108】
このようにしてtatC遺伝子が順向きにplcH-oprMキメラ遺伝子の直前に挿入されたクローンを選び、組換えプラスミドをHindIIIおよびEcoRI消化し、約2.6 kbの遺伝子断片(tatC-plcH-oprM)をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。そして予めHindIII、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、(tatC-plcH-oprM)遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpTatC-PlcH-OprMをよぶことにする。
【0109】
(4-4) P. aeruginosa TNP082へのpTatC-PlcH-OprM発現ベクターの形質転換
項目(2-2)と同様の操作にて、pTatC-PlcH-OprM発現ベクターを染色体上のtatCとoprMが欠失した二重変異株TNP082に形質転換し、スルベニシリン(武田薬品工業)50μg/mlを添加したL寒天平板にて形質転換体を取得した。得られた形質転換体よりプラスミドを項目(1-2-2)の方法に従いを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCRを行い、組換えプラスミドの存在を確認した。
【0110】
(4-5) クローン化したtatC遺伝子産物の機能評価
クローン化したtatC遺伝子の導入によりTat分泌系の機能が回復することが期待できる。この点を評価するために項目(4-1-3)の方法にしたがい、前項目(4-4)で構築した形質転換体をCAA培地で生育させ、その培養上清中へのpyoverdineの分泌生産能の評価を行った。その結果、クローン化したtatC遺伝子をもつ形質転換体TNP082/pTatC-PlcH-OprMは、野生株および親株TNP072と同様なpyoverdineの吸収が認められ、Tat分泌系が機能していることが明らかとなった(図7(d))。
【0111】
(4-6) pTatC-PlcH-OprM発現ベクターを有するoprM/tatC二重変異株(TNP082/pTatC-PlcH-OprM)の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0112】
(4-7)外膜画分の分離
緑膿菌の各菌株を4 mlのL液体培地(プラスミド保有株の場合は50μg/mlのスルベニシリンを含む)に接種し、37℃にて一夜培養した。この菌液を100 mlの37℃に保温しておいた同培地に接種し、対数期後期(OD660nm、約1.0)まで振とう培養した後、プラスチック遠心管に移し遠心集菌をした(7500 rpm、10分、室温)。この菌体を100 mlの溶液(200 mM Na-Phosphate Buffer, pH 7.6)で2回洗浄した後、1.5 mlの同溶液に懸濁し、超音波処理(Bioruputor, コスモバイオ) を氷冷下5分間行った。この処理液を12,000 rpm、1分、室温で遠心集菌し、集菌後の上清0.5 mlを超遠心用のプラスチックチューブ(Beckman, Cat. No. 343776)に移し、45,000 rpm、30分、10℃で遠心(ローター、Beckman TLA-120.1; 遠心機、Beckman Optima TLX)した。遠心後、ペレットを溶液 (20 mM Na-Phosphate Buffer, pH 7.6) 200μlで懸濁し、その後同緩衝液で2回洗浄した。この膜画分を1% Na-Salcosinate(和光純薬)を加えた同溶液1 mlに懸濁して30℃、30分静置した後、45,000 rpm、30分、10℃で遠心(ローター、Beckman TLA-120.1;遠心機、Beckman Optima TLX)した。得られたペレットは溶液 (200 mM Na-Phosphate Buffer, pH 7.6) で2回洗浄し、最終的に100μlの水に懸濁して外膜画分とした。
【0113】
(4-8)ウエスタンブロット解析
項目(4-7)で調整した外膜画分をSDS-ポリアクリルアミドゲル(12%)に供し電気泳動を行った後、Immobilon-Pメンブラン(Millipore社)に分離したタンパク質を転写し、一次抗体として抗OprMウサギ抗体、二次抗体としてアルカリフォスファターゼ結合抗ウサギーヒツジ抗体を用いて免疫染色を行った(図8)。なお、野生型のoprM遺伝子をもつプラスミド(pOprM)保有株はタンパク量として5μgの各画分を、キメラoprM遺伝子および野生型tatC遺伝子を有する各プラスミド(pPlcH-OprMおよびpTatC-PlcH-OprM)保有株はタンパク量として7.5μgの各画分を電気泳動した。
【0114】
<結果>
本実施例で行った薬剤感受性の評価結果を表3に示す。
【0115】
【表3】
【0116】
(1) PlcH-OprMキメラタンパク質はOprM欠損緑膿菌で機能する
pPlcH-OprM発現ベクターを有するP. aeruginosa TNP072形質転換体のアズトレオナムおよびクロラムフェニコールに対する感受性を評価した結果を表3に示した。oprM欠損変異株(TNP072)はMexAB-OprM排出ポンプが機能しないため薬剤を排出することができない。従って、野生株PAO4290に比べTNP072はアズトレオナムおよびクロラムフェニコールに対して薬剤感受性が著しく上昇する(MICが低下する)。事実、oprM欠損変異株(TNP072)の各薬剤に対するMIC値は0.125μg/mlおよび1.0μg/mlである。このoprM欠損変異株にキメラoprM遺伝子をクローン化したプラスミド(pPlcH-OprM)を形質転換した株のアズトレオナムおよびクロラムフェニコールに対するMICはそれぞれ4.0μg/mlおよび16μg/mlであった。この値は野生株およびクローン化した野生型のoprM遺伝子をもつoprM欠損変異株(TNP072/pOprM)のMIC値とほぼ同等であることから、キメラOprMは機能的な発現をしていることが明らかとなった。実際、キメラPlcH-OprMを発現する全菌体タンパク質をウエスタンブロット解析すると、シグナル配列が未切断のキメラタンパク質に加え成熟型OprMに相当するシグナルが観察され(図8,レーン2)。また外膜画分ではシグナルは弱いものの成熟型OprMの発現が確認できた(図8,レーン3)。このことから、キメラOprMは内膜をトランスロケートし外膜に達してポンプ複合体を形成していることが明らかとなった。
【0117】
(2) PlcH-OprMキメラタンパク質はTat分泌系依存的に機能する
PlcH-OprMキメラタンパク質が機能を有していることから、本キメラタンパク質は翻訳後にTat系を介して本来の局在場所である外膜に分泌され、MexAおよびMexBと共にポンプ複合体を形成して薬剤を排出しているものと考えられる。この仮説を遺伝学的に検証するために、OprM欠損変異株(TNP072)のTat分泌系を構成するtatC遺伝子を破壊した二重変異株TNP082(OprM-, TatC-)を作製した。この変異株にpPlcH-OprM発現ベクターを導入した形質転換株のアズトレオナムおよびクロラムフェニコールに対する感受性を調べた結果、それぞれの薬剤に対する形質転換体の感受性は、0.125μg/mlおよび1.0μg/mlであった(表3)。これは、OprM欠損変異株TNP072と同じ感受性であり、薬剤排出ポンプが機能していないことを示唆している。すなわち、Tat分泌系が機能を失うことによりPlcH-OprMキメラタンパク質は正常に分泌されないことを意味しており、このキメラタンパク質はTat系依存的に分泌されていることが強く示唆された。一方、この二重変異株に野生型のoprM遺伝子をもつpOprMを導入した形質転換体の両薬剤に対する感受性は、2.0μg/ml(アズトレオナム)および16μg/ml(クロラムフェニコール)と野生株と同等のMIC値を示したことから、先のキメラOprMが機能しない原因はTat分泌系を介してキメラOprMがトランスロケートできないことによるものであることが明らかとなった。
【0118】
この結果をさらに検証するために、野生型のtatC遺伝子をクローニングし、キメラOprM遺伝子(plcH-oprM)とともに二重変異株TNP082に導入した形質転換体(TNP082/ pTatC-PlcH-OprM)を得た。この形質転換体のアズトレオナムとクロラムフェニコールに対する感受性は、それぞれ4.0μg/mlおよび32μg/mlであり、野生株と同等のMIC値を示した。このことから、クローン化したTatCが供給されることによってTat分泌系が機能回復し、PlcH-OprMキメラタンパク質がこの機能回復したTat分泌系を介してトランスロケートし外膜に輸送されたことが確認できた。事実、クローン化した野生型のtatC遺伝子を染色体上のtatC遺伝子破壊株(TNP082)に導入すると、成熟型のOprMが全菌体および外膜画分に検出され(図8,レーン6,7)、本形質転換体(TNP082/pTatC-PlcH-OprM)においてキメラOprMがTat系依存的に内膜をトランスロケートする結果と一致していた。
【0119】
以上の結果より、フォスフォリパーゼCのシグナル配列をもつキメラOprM(PlcH-OprM)を染色体上のoprM遺伝子が欠損した宿主で発現させる本評価システムは、〔実施例1〕で詳述したキメラMexAを利用したシステムに比べ、多くの安価な抗生物質を指標薬剤として用いることができるため、天然化合物ライブラリーおよび低分子有機化合物ライブラリーを効率よくスクリーニングするためのより優れた評価系である。また、本スクリーニング系で陽性を示す化合物は、Tat分泌系阻害剤以外に、MexA-MexB-OprM排出ポンプ自体の阻害剤の可能性もあるので、この新規スクリーニング系はTat分泌系と薬剤排出ポンプというこれまでにない新しい二つの標的を同時にターゲットとした非常に効率のよいスクリーニング系である。
【技術分野】
【0001】
本発明は、例えば緑膿菌におけるTat分泌系を標的とする阻害剤をスクリーニングする際に使用することができる組換え微生物、当該組換え微生物を使用したTat分泌系阻害剤のスクリーニング方法及び当該組換え微生物を含むTat分泌系阻害剤のスクリーニングキットに関する。
【背景技術】
【0002】
微生物のなかには、抗生物質等の菌体外への排出を担う薬剤排出ポンプを備えているものがあり、これにより元来自然耐性並びに獲得多剤耐性を有するものがある。例えば、緑膿菌は構造的に関連性のない各種薬剤に対し、元来自然耐性ならびに獲得多剤耐性能を有しているために臨床上大きな問題となっている。すなわち、日和見感染症の主要起因菌である緑膿菌における多剤耐性の獲得は、院内感染の原因となり、社会問題として認識されている。
【0003】
緑膿菌には、MexA,B-OprMという3つのコンポーネントからなるエネルギー依存性の薬剤排出ポンプが備わっている。この薬剤排出ポンプは、抗生物質などの抗菌性薬剤を菌体外に排出する役割を担っており薬剤耐性に深く関与している。この薬剤排出ポンプは、ポンプ本体である内膜サブユニットMexB、外膜に存在して薬剤排出チャネルを形成するOprM、及び修飾リピドを介して内膜にアンカリングし、MexBとOprMの相互作用を補強していると考えられているMexAから構成される。これらの各サブユニットは、一般的な分泌システム(general secretion pathway)であるSec系を介して、各々の局在部位に輸送され排出ポンプ複合体を形成することが知られている。
【0004】
一方、新たな分泌システムであるTat(twin arginine translocation)系が、植物のチラコイド膜を介する分泌系で見出され、このTat分泌系システムが広く細菌界に存在することが最近明らかとなってきた(非特許文献1)。緑膿菌もこのTat分泌系を有していることが明らかとなり、本菌の病原性因子であるフォスフォリパーゼや鉄キレーターの産生に関与する因子などが、このTat系を介して分泌されることが最近明らかとなってきた(非特許文献2及び3)。また、Tat分泌系が欠損した緑膿菌変異株においては、その病原性が低下することが明らかとなった(非特許文献4)。
【0005】
このことから、例えば緑膿菌におけるTat分泌系を標的とする物質は、緑膿菌における病原因子の分泌を抑制し、当該病原因子に起因する種々の疾患を予防及び治療できる薬剤として期待される。
【0006】
広く細菌界に分布するTat分泌系は、動物細胞には存在しないことが知られており(非特許文献5)、抗菌剤が具備すべき最重要事項である選択毒性の観点から理想的な標的としての特性を有している。さらに、緑膿菌をはじめ詳細な研究がなされている大腸菌等においてTat分泌系を構成する遺伝子破壊株が作製されていることから、このTat分泌系は細菌の生存に必須ではないことは明らかである。ペニシリン以来多くの抗生物質が発見され臨床的に使用されてきたが、病原細菌はそのすべての薬剤に対し耐性能を獲得し、社会的に大きな問題となっている。これは、既存の抗生物質がいずれも細菌の生存にとって必須の代謝プロセスを阻害するからであり、変異によって耐性能を獲得した耐性菌が抗生物質による選択圧のため選択され生き残ったからである。一方、Tat系を標的とした場合、Tat系は細菌の生存に必須でないため、薬剤に対する耐性菌が選択される可能性は既存の抗生物質に比べ非常に低いことが期待されることから、臨床現場において危惧される耐性菌問題を回避できる可能性がある。しかしながら、Tat分泌系を標的とする物質のスクリーニング系は知られておらず、このようなスクリーニング系、特にハイスループットなスクリーニング系が求められていた。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Prokaryotic utilization of the Twin-arginine translocation pathway: a genomic survey. Dilks K, Rose R W, Hartmann E, Pohlschroder M. J. Bacteriol. 2003, 185, 1478-1483.
【非特許文献2】Role of the Pseudomonas aeruginosa PlcH Tat signal peptide in protein secretion, transcription, and cross-species Tat secretion system compatibility. Snyder A, Vasil A I, Zajdowicz, S L, Wilson Z R, Vasil M L. J. Bacteriol. 2006, 188, 1762-1774.
【非特許文献3】Pyoverdine-mediated iron uptake in Pseudomonas aeruginosa: the Tat system is required for PvdN but not for FpvA transport. Voulhoux R, Filloux A, Schalk I J. J. Bacteriol. 2006, 188, 3317-3323.
【非特許文献4】Effects of the twin-arginine translocase on secretion of virulence factors, stress response, and pathogenesis. Ochsner U A, Snyder A, Vasil A I, Vasil M L. Proc. Natl. Acad. Sci. USA. 2002, 99, 8312-8317.
【非特許文献5】Sequence and phylogenetic analyses of the twin-arginine targeting (Tat) protein export system. Yen M-R, Tseng, Y-H, Nguyen E, Wu L-Fe, Saier M H Jr. Arch. Microbiol. 2002, 177, 441-450
【発明の概要】
【発明が解決しようとする課題】
【0008】
そこで、本発明は、種々の供試物質のなかからTat分泌系を標的とする、すなわち、Tat分泌系を阻害する活性を有する物質をスクリーニングする際に使用する組換え微生物、当該組換え微生物を使用したスクリーニング方法、及び当該組換え微生物を含むスクリーニングキットを提供することを目的とする。
【課題を解決するための手段】
【0009】
Tat分泌系を阻害する(標的とする)物質のハイスループットスクリーニング系を確立するために必要な要件としては、Tat分泌系依存性の感度の高い生物学的評価系を確立することが挙げられる。Tat分泌系については、フォールドしたタンパク質を分泌、輸送することが知られており、Tat分泌系の基質であるシグナル配列を融合した細菌のアルカリフォスファターゼを発現させても、その機能は保持されないことが示されている(Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. Richter S and Bruse T. J. Biol. Chem. 2005, 280, 42723-42730)。したがって、この文献に記載されるように、Tat分泌系の基質であるシグナル配列を融合した細菌のアルカリフォスファターゼは、Tat分泌系の機能評価をするためのレポーターとして使用することはできない。以上のように、Tat分泌系の機能を、Tat分泌系の基質であるシグナル配列を利用したレポーター系で評価することは非常に困難であると認識されていた。
【0010】
しかしながら、上述した実情に鑑み、本発明者らが鋭意検討した結果、薬剤排出ポンプを構成するサブユニットにおけるシグナル配列を、Tat分泌系の基質となるタンパク質のシグナル配列で置換したキメラタンパク質をコードする融合遺伝子を発現し、当該サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成する組換え微生物を使用することで、上述したハイスループットなスクリーニング系を構築できることを見いだし、本発明を完成するに至った。すなわち、本発明は以下を包含する。
【0011】
(1)Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成することを特徴とする組換え微生物。
(2)上記融合遺伝子を、上記サブユニットをコードする内在遺伝子を欠損した上記微生物に導入したものであることを特徴とする(1)記載の組換え微生物。
(3)上記微生物は内在性のTat分泌系を有する細菌であることを特徴とする(1)記載の組換え微生物。
(4)上記微生物はグラム陰性菌であることを特徴とする(1)記載の組換え微生物。
(5)上記微生物は緑膿菌(Pseudomonas aeruginosa)であることを特徴とする(1)記載の組換え微生物。
(6)上記薬剤排出ポンプは、サブユニット:MexA、MexB及びOprMから構成されることを特徴とする(1)記載の組換え微生物。
(7)上記融合遺伝子は、上記サブユニットとしてMexAにおける成熟領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(8)上記融合遺伝子は、上記サブユニットとしてOprMにおける成熟領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(9)上記融合遺伝子は、上記シグナル配列として病原性因子フォスフォリパーゼC(plcH)のシグナル配列を含む領域をコードする遺伝子を含むことを特徴とする(1)記載の組換え微生物。
(10)互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現することを特徴とする(1)記載の組換え微生物。
(11)上記(1)乃至(10)いずれか一記載の組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる工程と、上記組換え微生物の上記薬剤に対する感受性を評価する工程と、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断する、Tat分泌系阻害剤のスクリーニング方法。
(12)上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする(11)記載のスクリーニング方法。
(13)上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする(11)記載のスクリーニング方法。
(14)上記(1)乃至(10)いずれか一記載の組換え微生物と、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤とを含む、Tat分泌系阻害剤のスクリーニングキット。
(15)上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする(14)記載のTat分泌系阻害剤のスクリーニングキット。
(16)上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする(14)記載のスクリーニングキット。
【発明の効果】
【0012】
本発明によれば、微生物におけるTat分泌系を標的とする、すなわち、Tat分泌系を阻害する活性を有する物質の存在下で、薬剤感受性が大となる組換え微生物を提供することができる。本発明に係る組換え微生物を使用することによって、薬剤感受性を指標としたTat分泌系阻害物質を効率良くスクリーニングすることができる。
【図面の簡単な説明】
【0013】
【図1】plcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子の塩基配列を示す特性図であり、キメラ遺伝子のオープンリーディングフレームを大文字で示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、5'末端側の下線付き小文字はBamHI部位を示し、3'末端側の下線付き小文字はKpnI部位を示している。
【図2】TNP070及びTNP080におけるpyoverdine産生能を示す特性図である。
【図3】plcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子がtatC遺伝子の下流に配置したプラスミドの塩基配列を示す特性図であり、枠で囲った領域は由来不明の塩基配列を示し、5'末端側の大文字はtatC遺伝子のオープンリーディングフレームを示し、3'末端側の大文字はキメラ遺伝子のオープンリーディングフレームを示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、下線付き小文字(4箇所)は5'末端側から順にHindIII部位、BamHI部位、BamHI部位及びKpnI部位を示している。
【図4】形質転換体TNP080/pTatPlcMexAにおけるpyoverdine産生能を示す特性図である。
【図5】抗MexA抗体を用いたウエスタンブロット解析の結果を示す電気泳動写真である。
【図6】plcHシグナル配列遺伝子とoprM遺伝子の成熟領域とを融合したキメラ遺伝子の塩基配列を示す特性図であり、キメラ遺伝子のオープンリーディングフレームを大文字で示し、シグナル配列遺伝子の部分を下線付き斜体文字で示し、5'末端側の下線付き小文字はXbaI部位を示し、3'末端側の下線付き小文字はEcoRI部位を示している。
【図7】野生株(PAO4290)、TNP072、TNP082及びTNP082/pTatC-PlcH-OprMにおけるpyoverdine産生能を示す特性図である。
【図8】抗OprM抗体を用いたウエスタンブロット解析の結果を示す電気泳動写真である。
【発明を実施するための形態】
【0014】
以下、本発明に係る組換え微生物、当該組換え微生物を使用したスクリーニング方法及びスクリーニングキットを詳細に説明する。本発明に係る組換え微生物は、Tat分泌系による分泌能に依存して、詳細を後述する融合遺伝子産物が関与する薬剤排出ポンプの機能が変化するといった特徴を有する。従って、本発明に係る組換え微生物は、Tat分泌系を標的とする物質の存在下で、当該薬剤排出ポンプから菌体外に排出される薬剤に対する感受性が変化することとなる。
【0015】
融合遺伝子及び組換え微生物
本発明において融合遺伝子は、薬剤排出ポンプを構成するサブユニットの成熟領域と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域とを融合したキメラタンパク質をコードしている。融合遺伝子の産物であるキメラタンパク質は、Tat分泌系の基質となりTat分泌系を介して分泌されることとなる。したがって、融合タンパク質に含まれる薬剤排出ポンプのサブユニットは、Tat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成することになる。
【0016】
本発明に係る組換え微生物とは、この融合遺伝子を発現することで、当該融合遺伝子に含まれるサブユニットがTat分泌系を介して分泌され薬剤排出ポンプの複合体が形成されるものである。組換え微生物は、宿主微生物における染色体上に内在する、上記サブユニットをコードする遺伝子を欠損させるとともに、上記融合遺伝子を発現可能に有するベクターを当該宿主微生物に導入することで作製することができる。すなわち、本発明に係る組換え微生物は、上記融合遺伝子を発現可能なベクターを使用した形質転換微生物を包含する。ここで、ベクターとしては、宿主微生物内において複製可能なベクターであれば何ら制限することなく使用することができる。
【0017】
また、組換え微生物は、染色体上に内在する、上記サブユニットをコードする遺伝子におけるシグナル配列をコードする領域を、Tat分泌系を介して分泌されるタンパク質のシグナル配列をコードする領域で置換することで作製することもできる。例えば、Tat分泌系を介して分泌されるタンパク質のシグナル配列をコードする核酸断片を相同組み換えによって、上記サブユニットをコードする遺伝子におけるシグナル配列をコードする領域と置換することで、上記融合遺伝子を発現する組換え微生物を作製してもよい。すなわち、本発明に係る組換え微生物は、上述した相同組み換え技術によって作製されたものも包含する。
【0018】
なお、本発明に係る組換え微生物は、所定の薬剤排出ポンプを構成する複数のサブユニットのうち、少なくとも1つのサブユニットがTat分泌系の基質となるように上記融合遺伝子を発現すればよいが、全てのサブユニットがTat分泌系の基質となるように全てのサブユニットについて上記融合遺伝子を発現するものであってもよい。
【0019】
先ず、宿主微生物について説明する。本発明に係る組換え微生物の宿主微生物としては、薬剤排出ポンプ及びTat分泌系を備え、当該薬剤排出ポンプにより所定の薬剤に対する耐性(低い感受性と同義)を示す微生物である。このような微生物としては、特に限定されないが、緑膿菌(Pseudomonas aeruginosa)、大腸菌(Escherichia coli)、黄色ブドウ球菌(Staphylococcus aureus)、結核菌(Mycobacterium tuberculosis)、ペスト菌(Yersinia pestis)、インフルエンザ菌(Haemophilus influenzae)、コレラ菌(Vibrio cholerae)、ピロリ菌(Helicobacter pylori)、リステリア菌(Listeria monocytogenes)、キャンピロバクター(Campylobacter jejuni)、サルモネラ菌(Salmonella enterica)、髄膜炎菌(Neisseria meningitidis)、らい菌(Mycobacterium leprae)、枯草菌(Bacillus subtilis)、ブルセラ菌(Brucella属)、リケッチア(Rickettisia prowazekii)、炭疽菌(Bacillus anthracis)等を挙げることができる。特に本発明においては、日和見感染症等の感染症の原因となる緑膿菌を宿主微生物として使用することが好ましい。なお、宿主微生物としては、上記で列挙した細菌に限定されず、例えばDilks, K. et al. (2003) Prokaryotic utilization of the twin-arginine translocation pathway: a genomic survey. J. Bacteriol. 185, 1478-1483.に開示されている微生物や、ゲノムの解読の進展により新たにTat分泌系を有する微生物として同定される全ての微生物を包含する。また、宿主微生物としては、Tat分泌系を本来的に有しない細菌に対して、Tat分泌系を備えるように、Tat分泌系に関与する遺伝子群を外来遺伝子として導入した微生物を使用することもできる。
【0020】
特に、宿主微生物としては、グラム陰性菌とすることが好ましい。なお、上記で列挙した細菌のうち、グラム陰性菌は、緑膿菌(Pseudomonas aeruginosa)、大腸菌(Escherichia coli)、ペスト菌(Yersinia pestis)、インフルエンザ菌(Haemophilus influenzae)、コレラ菌(Vibrio cholerae)、ピロリ菌(Helicobacter pylori)、キャンピロバクター(Campylobacter jejuni)、サルモネラ菌(Salmonella enterica)、髄膜炎菌(Neisseria meningitidis)、ブルセラ菌(Brucella属)、リケッチア(Rickettisia prowazekii)である。グラム陰性菌は、外膜を有するためマルチコンポーネント型の薬剤排出ポンプを有しており、Tat分泌系を介して分泌、輸送しうるサブユニットを適宜選択できる。ここで、薬剤排出ポンプとは、菌体内に存在する抗生物質などの抗菌性薬剤を菌体外に排出する機能を有するポンプであり、グラム陰性菌の場合、通常、微生物の内膜、ペリプラズム及び外膜を貫通するように局在している。薬剤排出ポンプは、通常、複数のサブユニットから構成される複合体である。なお、薬剤排出ポンプは、通常、排出対象の薬剤について特異性がある。
【0021】
上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしては、上述した薬剤排出ポンプのサブユニットを何ら限定されず使用することができる。すなわち、宿主微生物に本来的に備わっている薬剤排出ポンプを構成するサブユニットであれば、如何なるサブユニットを使用しても良い。
【0022】
一例として、緑膿菌の薬剤排出ポンプとしては、主要なものとして、「MexA-MexB-OprM」、「MexC-MexD-OprJ」、「MexE-MexF-OprN」及び「MexX-MexY-OprM」の4種類が知られている。したがって、宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしては、MexA、MexB、MexC、MexD、MexE、MexF、MexX、MexY、OprM、OprJ及びOprNのいずれを使用しても良い。これら薬剤排出ポンプを構成するサブユニットは、従来公知のデータベース(例えば、Pseudomonas Genome Database v2)を検索することでそのアミノ酸配列を取得することができる。また、サブユニットの成熟領域は、例えば、シグナル配列を予測するソフトウェア「SignalP 3.0 Server」を使用してシグナル配列を予測し、当該シグナル配列を除いた領域として特定することができる。
【0023】
宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはMexAを使用することが好ましい。MexA、MexB及びOprMからなる薬剤排出ポンプにおいて、MexAサブユニットは、基質薬剤を認識するポンプ本体であるMexBと、基質薬剤を菌体外へ放出する膜チャンネル機能を有するOprMとを連結する内膜タンパク質である。このように、MexAサブユニットは膜貫通ドメイン等の高次構造を有しないため、折りたたまれたタンパク質を分泌、輸送するTat分泌系を介して確実に分泌することができる。
【0024】
また、宿主微生物として緑膿菌を使用する場合、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用することが好ましい。膜チャンネルとして機能するOprMサブユニットは、MexA、MexB及びOprMからなる薬剤排出ポンプのみならず、MexX、MexY及びOprMからなる薬剤排出ポンプにおけるサブユニットとしても機能している。よって、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用した場合には、詳細を後述するスクリーニング方法及びキットにおいて使用可能な薬剤の選択肢を広げることができる。また、上述した融合タンパク質に含まれる薬剤排出ポンプのサブユニットとしてはOprMを使用した場合には、薬剤に対する感受性が大きく変動することとなり、より高精度にスクリーニングすることができる。特に、上掲の論文(J. Biol. Chem. 2005, 280, 42723-42730)によれば、膜貫通ドメインを有するOprMをTat分泌系を介して分泌、輸送することについて困難性があると考えれる。しかしながら、後述の実施例に示すように、OprMをキメラタンパク質としてTat分泌系の基質として発現させても、OprMを含む薬剤排出ポンプを形成できたことは驚くべきである。
【0025】
さらにまた、本発明に係る組換え微生物は、互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現するものであってもよい。すなわち、宿主となる微生物が複数の異なる薬剤排出ポンプを備える場合、これら複数の薬剤排出ポンプのうち、2以上の薬剤排出ポンプのサブユニットが、Tat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成するものであってもよい。特に、Tat分泌系を介して形成される複数の薬剤排出ポンプは、それぞれ異なる種類の薬剤を排出するものを選択することが好ましい。
【0026】
一方、Tat分泌系を介して分泌されるタンパク質のシグナル配列とは、Tat分泌系を介して分泌されるタンパク質におけるN末端側に位置し、シグナルペプチターゼにより成熟タンパク質から切断される領域を意味する。上述した融合遺伝子においては、上述したシグナル配列を含んでいれば良く、Tat分泌系を介して分泌されるタンパク質におけるシグナル配列とこれに続く成熟タンパク質のN末端側の一部とを含んでいても良い。すなわち、上述した融合遺伝子においては、Tat分泌系の基質となりうる領域と薬剤排出ポンプのサブユニットとを融合した融合タンパク質をコードしていればよい。
【0027】
一例として、緑膿菌におけるTat分泌系を介して分泌されるタンパク質としては、例えば表1に示す遺伝子によりコードされるタンパク質を挙げることができる。
【0028】
【表1】
【0029】
なお、表1において、『Locus Tag』とは、Pseudomonas Genome Database v2に登録されているIDであり、PA numberとも称されている。また、表1において、Gene nameの欄には、当該Locus Tagで特定される遺伝子の機能が明らかになっている場合には当該機能に基づく名称が記載されており、機能未知の遺伝子については空欄となっている。また、表1中、1〜26の遺伝子は、Genome Research (2006) 15, 321-329においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。表1中、27〜34の遺伝子は、Proc. Nat. Acad. Sci. (2002) 99, 8312-8317においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。表1中、35の遺伝子は、The EMBO Journal (2001) 20, 6735-3741においてTat分泌系を介して分泌されるタンパク質をコードする遺伝子であることが示されている。
【0030】
一旦、Tat分泌系を介して分泌されるタンパク質をコードする遺伝子が同定されれば、当該タンパク質におけるシグナル配列は、当業者により容易に特定することができる。例えば、シグナル配列を予測するソフトウェア「SignalP 3.0 Server」を使用することによりシグナル配列をコードする領域を特定することができる。
【0031】
また、Tat分泌系を介して分泌されるタンパク質をコードする遺伝子が同定されれば、上述したようにソフトウェアを利用してシグナル配列を特定することなく、当該タンパク質のN末端から数えて30〜50残基のアミノ酸配列を、シグナル配列を含む領域として特定しても良い。通常、シグナル配列は、タンパク質のN末端から数えて30〜50残基のアミノ酸配列に含まれると考えられるからである。
【0032】
すなわち、上述した融合遺伝子には、ソフトウェアを利用して特定したシグナル配列を含む領域をコードする遺伝子、又はN末端から数えて30〜50残基のアミノ酸配列をコードする遺伝子のいずれを含んでいても良い。一例として、Tat分泌系を介して分泌されるplcH(PA0844)におけるシグナル配列を含む領域を配列番号1に示す。Tat分泌系を介して分泌されるplcN(PA3319)におけるシグナル配列を含む領域を配列番号2に示す。Tat分泌系を介して分泌されるpvdN(PA2394)におけるシグナル配列を含む領域を配列番号3に示す。Tat分泌系を介して分泌されるglpQ(PA0347)におけるシグナル配列を含む領域を配列番号4に示す。
【0033】
Tat分泌系を標的とする物質のスクリーニング方法及びキット
本発明に係る組換え微生物においては、上述した融合遺伝子を発現することで、薬剤排出ポンプを構成する所定のサブユニットがTat分泌系を介して分泌され、当該薬剤排出ポンプ複合体を形成することとなる。したがって、本発明に係る組換え微生物は、Tat分泌系を阻害する物質の存在下において当該薬剤排出ポンプの構築が阻害されることとなり、当該薬剤排出ポンプにより菌体外へ排出される薬剤への感受性が高くなるといった特徴を有する。
【0034】
すなわち、本発明に係るTat分泌系阻害剤のスクリーニング方法では、先ず、上述した組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる。次に、上記組換え微生物の上記薬剤に対する感受性を評価する。そして、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断することができる。
【0035】
なお、供試物質の存在下における上記感受性が大である場合には、上記供試物質は、上記薬剤排出ポンプに対する阻害作用を有している可能性もある。しかしながら、当該供試物質を上記薬剤の存在下で野生型微生物に作用させ、当該薬剤に対する感受性を評価することで、当該供試物質がTat分泌系及び薬剤排出ポンプのいずれに対して阻害作用を有するのかを判断することができる。すなわち、当該供試物質を上記薬剤の存在下で野生型微生物に作用させたときに、薬剤に対する感受性が高くなるならば当該供試物質は薬剤排出ポンプに対して阻害作用を有することがわかり、薬剤に対する感受性が変動しないならば当該供試物質はTat分泌系に対して特異的に阻害作用を有していることがわかる。
【0036】
ここで、供試物質としては、何ら限定されず、如何なる物質であってもよい。供試物質としては、単独の物質であってもよいし、複数の構成成分からなる混合物であってもよい。供試物質としては、例えば微生物若しくは培養液からの抽出物のように未同定の物質を含むような構成であってもよいし、既知の組成物を所定の組成比で含むような構成であってもよい。また、候補物質としては、タンパク質、核酸、脂質、多糖類、有機化合物及び無機化合物のいずれでもよい。
【0037】
また、上記薬剤としては、例えば、上記融合遺伝子として緑膿菌における薬剤排出ポンプMexA-MexB-OprMのサブユニットを含む場合には、薬剤排出ポンプMexA-MexB-OprMにより菌体外に排出され、他の薬剤排出ポンプ(例えば、「MexC-MexD-OprJ」、「MexE-MexF-OprN」及び「MexX-MexY-OprM」)により菌体外に排出されない薬剤を使用する。換言すれば、薬剤として、薬剤排出ポンプMexA-MexB-OprMのみならず他の薬剤排出ポンプによっても菌体外へ排出される薬剤を使用した場合、当該薬剤に対する感受性は、薬剤排出ポンプMexA-MexB-OprM以外の他の薬剤排出ポンプによる影響により変動しないおそれがある。
【0038】
さらに、上記薬剤としては、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤を使用することができる。例えば、上記融合遺伝子として緑膿菌における薬剤排出ポンプMexA-MexB-OprMのサブユニットを含む場合には、上記変異体としては、例えばMexA欠損変異株を使用することができる。所定の薬剤を野生型緑膿菌及びMexA欠損変異株にそれぞれ作用させ、MexA欠損変異株の薬剤感受性が野生型緑膿菌の薬剤感受性と比較して有意に高まっている場合、当該薬剤は、薬剤排出ポンプMexA-MexB-OprMを介して菌体外に排出される薬剤と考えられる。したがって、当該薬剤を使用することによって、上述したような供試物質の性質について判断を行うことができる。
【0039】
ここで、有意に低下するとは、薬剤排出ポンプを欠損した変異体における薬剤耐性の低下度合いについて統計学的に有意差があることを意味する。特に、薬剤としては、薬剤排出ポンプを欠損した変異体における最小発育阻止濃度が、野生型と比較して1/2以上低下するものが好ましく、1/4以上低下するものがより好ましく、1/8以上低下するものが最も好ましい。薬剤排出ポンプを欠損した変異体における最小発育阻止濃度がこの範囲で低下する薬剤を使用すると、感受性の変動をより高精度に検出することができる。
【0040】
また、緑膿菌における薬剤排出ポンプのなかで、最も基質特異性の広いものは薬剤排出ポンプMexA-MexB-OprMである。したがって、他の薬剤排出ポンプMexC-MexD-OprJ、MexE-MexF-OprN及びMexX-MexY-OprMのサブユニットを上述した融合遺伝子により発現させるよりも、薬剤排出ポンプMexA-MexB-OprMのサブユニットを上述した融合遺伝子として発現させることが、使用可能な薬剤の選択肢を広くする意味で好ましい。
【0041】
上述した融合遺伝子に含まれるサブユニットをMexAとした場合、使用可能な薬剤としては例えば、アズトレオナム、クロラムフェニコール、キノロン剤(ノルフロキサシン)、ノボビオシン、セフォペラゾン、セフスロジンを挙げることができる。これらの薬剤は、MexA変異により最小発育阻止濃度が1/4〜1/8に低下することが知られている。したがって、これら薬剤を使用することで、感受性の変動をより高精度に検出することができる。
【0042】
さらに、上述した融合遺伝子に含まれるサブユニットをOprMとした場合、薬剤排出ポンプのうちMexA-MexB-OprM及びMexX-MexY-OprMがTat分泌系に依存することとなる。よって、この場合、使用可能な薬剤としては、オフロキサシン、ノルフロキサシン、シプロフロキサシン、クロラムフェニコール、ゲンタミシン、ノボビオシン、セフタジジム、セファペラゾン、セフスロジン及びアズトレオナムを挙げることができる。
【0043】
なお、上記で列挙した具体的な薬剤以外の薬剤であっても、例えば、新しい第四世代のセファロスポリン等、使用可能な薬剤を挙げることができる。例えば、使用可能な薬剤としては、参考文献(The role of mex-gene products in antibiotic extrusion in Pseudomonas aeruginosa. Yoneyama H, Ocaktan A, Tsuda M, Nakae T. Biochem Biophys Res Commun. 1997, 233, 611-618)に開示されている薬剤を使用することができる。
【0044】
上記薬剤に対する感受性は、従来公知の手法を適用して測定することができる。例えば、使用する薬剤を段階希釈した複数の培地を用意し、供試物質の存在下及び非存在下における組換え微生物の最小発育阻止濃度を決定するといった方法を適用することができる。また、より簡便には、組換え微生物の生育を阻止する一歩手前の濃度の薬剤を含んだ寒天培地に組換え微生物を混合し、その後、プラスチックシャーレにまき、寒天が固化した平板上に供試物質を一定量載せて培養するといった方法を適用することができる。もし供試物質がTat分泌系を阻害するならば薬剤排出ポンプが機能せず、組換え微生物が薬剤に感受性となり、供試物質の周りには発育阻止ゾーンが観察されることとなる。以上のように、供試物質の周りに形成される発育阻止ゾーンの有無を指標として、Tat分泌系に対して阻害作用のあるリード化合物を供試物質のなかから簡便に判別することができる。なお、上述した融合遺伝子に含まれるサブユニットとしてMexAを使用した場合、アズトレオナムの濃度を1.0〜1.5μg/ml程度とする。
【0045】
また、この簡便な系は、96穴のプラスチック容器を用いた液体培地を用いる系として適用することもできる。すなわち、この場合、組換え微生物を薬剤と混合し評価系を作製する操作が簡便であり、且つ、培養後の評価はマイクロタイター分光光度計を用いて半定量化して判定することができる。
【0046】
以上のように、本発明に係る組換え微生物を使用することによって、新規なTat分泌系阻害剤をハイスループットにスクリーニングすることができる。すなわち、本発明に係るTat分泌系阻害剤のスクリーニングキットは、本発明に係る組換え微生物と上記薬剤とを含むものである。なお、スクリーニングキットにおいて、上記薬剤は、段階的に希釈された濃度で複数の培地として提供されても良い。
【0047】
また、本発明に係る組換え微生物において、2以上の薬剤排出ポンプのサブユニットがTat分泌系を介して分泌され、各々の局在部位に輸送され薬剤排出ポンプを形成するものである場合には、供試物質とともに各薬剤排出ポンプに対応する複数の薬剤についてそれぞれ感受性を独立して評価する。例えば、第1の薬剤排出ポンプ及び第2の薬剤排出ポンプがTat分泌系を介して形成されるよう2種類の融合遺伝子を導入した組換え微生物を使用する場合、第1の薬剤排出ポンプにより排出される薬剤Aに対する感受性を供試物質の存在/非存在下で評価し、第2の薬剤排出ポンプにより排出される薬剤Bに対する感受性を供試物質の存在/非存在下で評価する。このとき、薬剤Aに対する感受性が供試物質の存在下で高くなり、薬剤Bに対する感受性が供試物質の存在/非存在下で変動していないならば、当該供試物質は、Tat分泌系に対して阻害的に作用しているとは判断できず、第1の薬剤排出ポンプに阻害作用を有すると判断する。一方、供試物質の存在下で薬剤Aに対する感受性及び薬剤Bに対する感受性ともに高くなっている場合には、当該供試物質が、第1の薬剤排出ポンプ、第2の薬剤排出ポンプ及びTat分泌系の全てに阻害作用を示している可能性もあるが、Tat分泌系を特異的に阻害している蓋然性が高いと判断できる。
【実施例】
【0048】
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【0049】
〔実施例1〕
本実施例では、薬剤排出ポンプ:MexA-MexB-OprMを構成するサブユニットのうちMexAのシグナル配列を、Tat分泌系を介して外界へ分泌される病原性因子フォスフォリパーゼC(PlcH)のシグナル配列を含む領域と交換した融合遺伝子を作製した。また、本実施例では、染色体上のmexA遺伝子を欠失した宿主細胞に上記融合遺伝子を導入して形質転換微生物を作製した。さらに、本実施例では、染色体上のmexA遺伝子とともに、Tat分泌系を構成するサブユニットであるtatC遺伝子を更に欠失した宿主細胞に上記融合遺伝子を導入して形質転換微生物を作製した。薬剤排出ポンプ(MexA-MexB-OprM)を介して排出される薬剤に対する感受性を、得られた2種類の形質転換微生物ついて比較した。
【0050】
(1) Tat系シグナル配列を有するキメラmexA遺伝子(plcH-mexA)の構築
(1-1) オーバーラップPCR法によるplcH-mexAキメラ遺伝子断片の増幅
(1-1-1) plcHのシグナル配列領域の増幅
plcHのシグナル配列をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-GAGGATCCTCGCTGTATTCCCTGCC-3’:配列番号5)およびリバースプライマー(5’-CGGCGCCTCGCTTTTTCCCTCGACGGCCAGGGCGCG -3’:配列番号6)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taq(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、40秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約230塩基対の増幅産物が確認できた。
【0051】
(1-1-2) mexA遺伝子の成熟領域の増幅
mexA遺伝子の成熟領域をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-CGCGCCCTGGCCGTCGAGGGAAAAAGCGAGGCGCCG-3’:配列番号7)およびリバースプライマー(5’-AGGGTACCCTTGATCAGCCCTTGC-3’:配列番号8)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロース電気泳動にて確認したところ約1,100塩基対の増幅産物が確認できた。
【0052】
(1-1-3) plcH-mexAキメラ遺伝子の増幅
plcHシグナル配列の増幅断片1μl、mexA成熟領域の増幅断片1μlおよび滅菌蒸留水8μlを混合し、この混合液を滅菌蒸留水で100倍希釈した溶液を鋳型として、2段階目のPCR反応を行った。プライマーは、フォワードプライマー(5’- GAGGATCCTCGCTGTATTCCCTGCC -3’:配列番号5)およびリバースプライマー(5’- AGGGTACCCTTGATCAGCCCTTGC -3’:配列番号8)を用いた。PCR増幅反応条件は、項目(1-1-2)と同じである。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。反応後、増幅断片をアガロース電気泳動にて確認したところ約1,300塩基対の増幅産物が確認できた。
【0053】
(1-2) pUC18ベクターへのキメラ遺伝子(plcH-mexA)のクローニング
(1-2-1) キメラ遺伝子(plcH-mexA)の連結反応と形質転換
前項で得たキメラ遺伝子(plcH-mexA)を順次KpnI(TOYOBO)、BamHI(TOYOBO)処理し(前項1-1-3で用いたプライマー末端には、各々KpnIあるいはBamHI部位が付加してある)、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpUC18(TOYOBO)と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を100μg/mlのアンピシリンを含むL寒天平板培地(isopropyl β-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0054】
(1-2-2) 形質転換体からのプラスミド分離と組換えプラスミドの解析
生育した白色コロニーを4 mlの100μg/mlのアンピシリンを含むL液体培地に植菌し、37℃にて一夜培養後、1.5 mlの菌液をミクロチューブに移し、遠心(12,000 rpm、23℃、5分間)して細胞を回収した。この細胞を氷冷した100μlの溶液1(25 mM Tris-HCl、10 mM EDTA、50 mM Glucose、pH 8.0)に懸濁後、200μlの溶液2(0.2 N NaOH、1% SDS)を加えて穏やかに混和した(室温、5分間)。その後、150μlの溶液3(3 M potassium-acetate、acetic acid)添加後、氷上にて5分間静置した。この溶液を遠心後(12,000 rpm、23℃、5分間)、上清を新しいミクロチューブに回収し、400μlのフェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液を加えて、室温にて約5分間混合した。溶液を遠心分離(12,000 rpm、23℃、5分間)後、回収した上清に900μlのエタノールを添加混合後室温にて5分放置してから遠心分離(12,000 rpm、23℃、5分間)した。得られた沈殿を500μlの70%エタノールで洗浄した後乾燥させ、RNaseA (20μg/ml)を含むTE (10 mM Tris-HCl、1 mM EDTA、pH 8.0) 30μlに溶解した。得られたプラスミドをEcoRIおよびEcoRI + BamHIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。このようにして得られたplcHシグナル配列遺伝子とmexA遺伝子の成熟領域とを融合したキメラ遺伝子の配列を図1及び配列番号9に示す。なお、キメラ遺伝子がコードするキメラタンパク質のアミノ酸配列を配列番号10に示す。
【0055】
(1-3) シャトルベクターpMMB67HEへのキメラ遺伝子(plcH-mexA)のクローニング
(1-3-1) カナマイシン耐性マーカーをもつpTH18ks1ベクターへのplcH-mexAキメラ遺伝子のクローニング
緑膿菌へのキメラ遺伝子の形質転換に使用するシャトルベクターは、アンピシリン耐性マーカーをもっている。このシャトルベクターへのクローニングをする際、前項(1-2-2)で作製したプラスミドより断片を切り出してpMMB67HEにクローニングする場合、pUC18が同じアンピシリン耐性マーカーをもっているので組換え体を選別する際、pUC18誘導体をもつ形質転換体のバックグランドが高くなる。そこで、いったんカナマイシン耐性マーカーをもつpTH18ks1ベクターへplcH-mexAキメラ遺伝子を組換えることにした。塩基配列を確認済みのplcH-mexAキメラ遺伝子をもつ組換えプラスミドをKpnIおよびBamHIで消化後、アガロースゲル電気泳動に供し、キメラ遺伝子断片(約1.3 kb)をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。このDNA断片を、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpTH18ks1と混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。pTH18ks1にplcH-mexAキメラ遺伝子断片がクローニングされたことは、形質転換体より抽出したプラスミドをEcoRIおよびPstI消化後、アガロースゲル電気泳動を行い断片の挿入を確認した。
【0056】
(1-3-2) pMMB67HEへのキメラ遺伝子(plcH-mexA)のクローニング
pMMB67HEへ挿入するキメラ遺伝子断片の調整は、項目(1-3-1)で作製した組換えプラスミドを鋳型とし、フォワードプライマー(5’- GAGGATCCTCGCTGTATTCCCTGCC -3’:配列番号5)およびリバースプライマー(5’- AGGGTACCCTTGATCAGCCCTTGC -3’:配列番号8)を用いて、項目(1-1-2)の反応条件でPCR反応を行って得た。この増幅断片をKpnIおよびBamHI消化し、予めKpnI、BamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、キメラ遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpPlcH-MexAとよぶことにする。
【0057】
(2) キメラmexA遺伝子(plcH-mexA)を導入した形質転換体の作製
(2-1) mexAを欠損した緑膿菌変異株(P. aeruginosa TNP070)のコンピタント細胞の調整
TNP070株(The role of mex-gene products in antibiotic extrusion in Pseudomonas aeruginosa. Yoneyama H, Ocaktan A, Tsuda M, Nakae T. Biochem Biophys Res Commun. 1997, 233, 611-618参照)を4 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で一夜振とう培養した。増殖した菌液800μlを40 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で約7時間培養して得た対数増殖期の細胞を、プラスチックチューブに移し、7,000 rpm、4℃、10分間遠心し(ローター、AG-508;遠心機、KUBOTA 6930)集菌した。この菌体を氷冷した40 mlの0.3 Mシュークロース溶液で洗浄した後、遠心(7,000 rpm、4℃、5分間)して集菌した。この遠心洗浄操作を3回繰り返して得た菌体を、少量(200〜400μl)の氷冷した0.3 Mシュークロース溶液に懸濁しコンピタント細胞としてエレクトロポレーションに供した。
【0058】
(2-2) 前項で調整したコンピタントTNP070細胞40μlに1μlのキメラ遺伝子(plcH-mexA)を挿入したpMMB67HEを加え、緩やかに混合した菌液をエレクトロポレーション用キュベット 2 mmギャップ(Molecular BioProducts)に注入し、エレクトロポレーション装置に装填した(GenePulser II、Bio-Rad)。エレクトロポレーションを1.6 kV、25μF、400Ωの条件下で行った後、菌液を100μg/mlのスルベニシリン(武田薬品工業)を含むL平板培地に塗布し、37℃で2夜培養した。生育したコロニーより項目(1-2-2)の方法に従いプラスミドを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位の両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCR反応を行い、組換えプラスミドの存在を確認した。
【0059】
(3) キメラ遺伝子(plcH-mexA)を保有する染色体上mexA遺伝子が欠損した形質転換体の薬剤感受性の評価
(3-1) 段階希釈した指標薬剤を含む寒天平板の作製
指標薬剤であるアズトレオナム(Sigma)およびクロラムフェニコール(和光純薬)の原液を、おのおの滅菌蒸留水および10%エタノールで溶解して調整した。この各薬剤の原液を滅菌蒸留水で希釈し、アズトレオナムを16〜0.0625μg/ml、クロラムフェニコールを64〜1μg/mlの範囲の2倍希釈系列を含むミューラー ヒントン寒天培地(Difco)を作製した。
【0060】
(3-2) 最小発育阻止濃度評価用菌株の培養
最小発育阻止濃度を評価する菌株を表2に示した。各菌株をアクティブプレートより一白金耳、0.4% (w/v)硝酸カリウムを含む2 mlのミューラー ヒントン液体培地(Difco)に接種し、37℃にて一夜静置培養した。なお、プラスミド保有株を培養する際の液体培地にはスルべニシリン(50μg/ml)を含む0.4% (w/v)硝酸カリウム添加ミューラー ヒントン液体培地を用いた。一夜培養後の菌液を、同液体培地で菌体密度が約106 cell/mlになるように希釈して薬剤感受性評価に供した。
【0061】
(3-3) 最小発育阻止濃度の評価
前項(3-2)で調整した菌株をガラス製小試験管に0.5〜1.0 ml分注し、最小発育阻止濃度測定用装置ミクロプランター(佐久間製作所)に装着し、薬剤を含む寒天平板に接種した。この平板培地を37℃、約20時間培養後に各菌株の生育を評価した。
【0062】
(4) PlcH-MexAキメラタンパク質がTat分泌系依存性であることの検証
(4-1) tatC遺伝子内部配列をもつ組換えプラスミドの作製
(4-1-1) tatC遺伝子の内部配列断片の増幅
tatC遺伝子の内部配列をコードする遺伝子断片(446 bp)を増幅するためにフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)およびリバースプライマー(5’-ACGGATCCGTCAGGACGAAGTCCAGGTAG-3’:配列番号14)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taq(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約450塩基対の増幅産物が確認できた。
【0063】
(4-1-2) pK18mobベクターへのtatC遺伝子内部配列のクローニング
前項で得たtatC遺伝子の内部配列断片をBamHI(TOYOBO)で処理し(前項4-1-1で用いたプライマー末端には、BamHI部位が付加してある)、予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpK18mobと混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を12.5μg/mlのカナマイシンを含むL寒天平板培地(isopropylβ-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0064】
(4-1-3) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
白色コロニーを4 mlの12.5μg/mlのカナマイシンを含むL液体培地に植菌し、37℃にて一夜培養後、項目(1-2-2)の方法に従いプラスミドを分離した。得られたプラスミドをBamHI、EcoRIあるいはHindIIIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。
【0065】
(4-2) tatC遺伝子破壊株の作製
(4-2-1) tatC内部配列をもつプラスミドのE. coli S17-1への形質転換
E. coli S17-1コンピタント細胞は塩化カルシウム法にしたがって作製した。一夜L液体培地で培養した200μlのE. coli S17-1を4 mlの新鮮なL液体培地に接種後、37℃にて約3時間培養した。対数増殖期の細胞を遠心集菌後(12,000 rpm、4℃、5分間)、1 mlの氷冷した0.1 M CaCl2溶液で2回遠心洗浄(12,000 rpm、4℃、5分間)をし、菌体を200μlの同溶液に懸濁した。この菌懸濁液と2μlの項目(4-1-3)で分離したプラスミドDNAを、キャップ付きプラスチック製試験管に移し混合した後氷中で30分間静置した。その後、試験管を42℃の水槽で90秒間インキュベーションして再度氷中で2分間静置した。この菌懸濁液に室温のSOC培地1 mlを加え37℃で約1時間振とう培養をした後、12.5μg/mlのカナマイシンを含むL寒天培地に塗布して37℃にて一夜培養した。生育したコロニーから項目(1-2-2)の方法に従いプラスミドを分離し、得られたプラスミドをBamHIあるいはBglIIで消化後、アガロースゲル電気泳動により組換えプラスミドの存在を確認した。
【0066】
(4-2-2) 組換えプラスミドのP. aeruginosa TNP070への接合伝達とtatC破壊株の選択
組換えプラスミドをもつE. coli S17-1とP. aeruginosa TNP070を、それぞれ12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地で一夜37℃にて培養した。生育したそれぞれの菌液80μlを12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地に添加し、37℃で約8時間振とう培養をした後、700μlの大腸菌と700μlの緑膿菌を同じミクロチューブに添加混合後、遠心集菌(12,000 rpm、室温、5分間)した。抗生物質を除去するために、1 mlのL液体培地で菌体を2回遠心洗浄(12,000 rpm、室温、5分間)し100μlのL液体培地に再懸濁した。この菌液を12.5μg/mlのカナマイシンと50μg/mlのテトラサイクリンを含むL寒天平板に塗布して37℃にて3日培養し、tatC遺伝子の相同配列領域で組換えが起こった破壊株を得た。
【0067】
(4-2-3) tatC遺伝子破壊株の染色体DNAの分離
前項で得た破壊株のtatC遺伝子領域のサザンブロット解析を実施するために、破壊株の染色体を以下の方法にしたがって分離した。破壊株を4 mlの12.5μg/mlのカナマイシン添加L液体培地で一夜37℃にて培養後集菌(12,000 rpm、室温、5分間)し、1 mlのSTE緩衝液(0.3 M sucrose、25 mM Tris-HCl、25 mM EDTA、pH 8.0)で1回遠心洗浄した後、5 mlの同緩衝液に懸濁した。この菌懸濁液に556μlの10% SDS (sodium dodecylsulfate、和光純薬)と28μlの20 mg/ml proteinase K(ナカライテスク株式会社)溶液を加え、穏やかに混合後、37℃にて一夜インキュベーションした。溶液を2 mlのフェノール・クロロフォルム・イソアミルアルコール (25:24:1)混合溶液で抽出操作を室温にて5分間行い、遠心(3,000 rpm、室温、15分間)後、上清を新しいプラスチック試験管に回収した。この溶液に1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加混合し核酸を沈殿させた。上清を遠心(3,000 rpm、室温、5分間)除去後、核酸を1 mlの70%エタノールで1回遠心洗浄し、溶液をデカント操作で除去した。沈殿した核酸を風乾後、4 mlの20μg/mlのRNaseA(ナカライテスク株式会社)を含むTE溶液(10 mM Tris-HCl、1 mM EDTA、pH 8.0)に溶解し、37℃にて約1時間インキュベーションをした。その後、上記同様フェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液で抽出操作を1回行い、1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加混合してDNAを沈殿させ、70%エタノールで2回遠心洗浄後、DNAを1 mlのTE溶液に溶解した。
【0068】
(4-2-4) tatC遺伝子破壊株染色体DNAのサザンブロット解析
前項で分離した染色体約1μgをBamHI、PstI、SacIIおよびSalIで37℃、15時間切断した後、1/10容の3 M酢酸ナトリウム溶液(pH 5.2)と2.5倍容量のエタノールを添加してDNAを沈殿、70%エタノールで1回遠心洗浄後、風乾したDNAを8μlのTE溶液に溶解した。この制限酵素消化後の各DNAサンプルを1.6μlのローディング色素液(0.25% bromophenol blue、40% sucrose)と混合し、全量をTBE緩衝液(45 mM Tris-HCl、45 mM Boric acid、10 mM EDTA、pH 8.0)で作製したアガロースゲル(0.6% w/v)に供し電気泳動を行った。泳動緩衝液にはTBE緩衝液を用いた。アガロース電気泳動後、ゲルをアルカリ変性(1.5 M NaCl、0.5 M NaOH)、蒸留水による洗浄、中和(1 M Tris-HCl、1.5 M NaCl、pH 7.4)処理した後、分画した断片をナイロンメンブランHiBond+ (Amersham)にキャピラリーブロッティング法を用いて一夜転写した後、2xSSC(0.3 M NaCl、30 mM sodium citrate、pH 7.0)にてメンブランを洗浄した。以降のシグナルの検出操作は、DIG High Prime DNA Labeling and Detection Starter Kit I (Roche社)の実験操作法にしたがった。検出に用いたプローブはtatC遺伝子全体をカバーするフォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)とリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用いて増幅したDNA断片(約910 bp)を使用した。その結果、組換えプラスミドがもつ内部配列の相同領域と染色体の同領域間での組換えが起こり、pK18mobベクターのtatC遺伝子内への挿入による破壊が確認できた。
【0069】
(4-2-5) シデロフォア(pyoverdine)産生能の評価
tatC遺伝子破壊株は緑膿菌のシデロフォアであるpyoverdine産生能が消失することが報告されている。そこで、項目(4-2-2)で作製したtatC遺伝子破壊株を12.5μg/mlのカナマイシンを含む5 mlのCAA液体培地(5 mMリン酸カリウム、1 mM 硫酸マグネシウム、5 g/lカザミノ酸、pH 7.0)で37℃にて一夜培養し、遠心(12,000 rpm、室温、5分間)により培養上清を回収した。この上清の吸収を分光光度計(V-630BIO、日本分光)を用いて測定した。その結果、親株TNP070で認められる典型的なpyoverdineの吸収が、tatC遺伝子破壊株では認められず、tatC遺伝子が破壊されていることが確認できた(図2)。このtatC遺伝子破壊株をP. aeruginosa TNP080とよぶことにする。なお、図2において(a)は親株TNP070を使用した際の結果を示し、(b)はTNP080を使用した際の結果を示している。
【0070】
(4-3) mexA/tatC二重変異株でのPlcH-MexA融合タンパク質の機能評価
(4-3-1) P. aeruginosa TNP080へのpPlcH-MexA発現ベクターの形質転換
P. aeruginosa TNP080 コンピタント細胞は、項目(2-1)の方法に準じて調製した。なお、L液体培地には12.5μg/mlのカナマイシンを添加して培養した。このmexA/tatC二重変異株のコンピタント細胞40μlに、項目(1-3-2)で調製したpPlcH-MexA発現ベクターを1μl添加し、エレクトロポレーション法にて形質転換した。条件は、項目(2-2)と同じである。
【0071】
(4-3-2) pPlcH-MexA発現ベクターを有するmexA/tatC二重変異株の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0072】
(4-4) tatC遺伝子のクローニングとtatC遺伝子破壊株への導入および機能評価
(4-4-1) tatC遺伝子の増幅
野生型のtatC遺伝子を増幅するために、フォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)およびリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTAR(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、1分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロース電気泳動にて確認したところ約910 bpの増幅産物が確認できた。
【0073】
(4-4-2) pUC18ベクターへのtatC遺伝子のクローニング
前項(4-4-1)で得たtatC遺伝子断片をBamHI(TOYOBO)で処理し(前項4-4-1で用いたプライマー末端には、BamHI部位が付加してある)、予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpUC18と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200Ωである。回収した細胞を100μg/mlのアンピシリンを含むL寒天平板培地(isopropylβ-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0074】
(4-4-3) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
白色コロニーを4 mlの100μg/mlのアンピシリンを含むL液体培地に植菌し、37℃にて一夜培養後、項目(1-2-2)の方法に従いプラスミドを分離した。得られたプラスミドをBamHIおよびEcoRIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析し、野生型のtatC遺伝子と同じ塩基配列であることを確認した。
【0075】
(4-4-4) tatC遺伝子とplcH-mexAキメラ遺伝子をもつシャトルベクターの作製
項目(1-3-1)で述べたようにシャトルベクターの作製効率をあげるために、クローン化したtatC遺伝子をシャトルベクターに載せる前に、一旦、plcH-mexAキメラ遺伝子をもつpTH18ks1(カナマイシン耐性マーカーをもつ)ベクターにtatC遺伝子をクローン化することにした。項目(4-4-3)で作製したプラスミドをBamHIで消化後、アガロースゲル電気泳動を行い約910 bpのtatC遺伝子断片をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。このDNA断片を、項目(1-3-1)で作製したplcH-mexAキメラ遺伝子保有pTH18ks1を予めBamHIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したDNAと混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。この実験デザインではplcH-mexAキメラ遺伝子の直前にtatC遺伝子が順向きあるいは逆向きに挿入される可能性がある。そこで、順向きに挿入された場合のみ連結した2遺伝子(tatC-plcH-mexA)が増幅されるフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)とリバースプライマー(5’-AGGGTACCCTTGATCAGCCCTTGC-3’:配列番号8)を用いたコロニーPCR法にて個々の形質転換体を直接鋳型としてPCRを行った。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、2分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロース電気泳動にかけ、約2.2 kbの増幅産物が認められるクローンを選抜した。
【0076】
このようにしてtatC遺伝子が順向きにplcH-mexAキメラ遺伝子の直前に挿入されたクローンを選び、(tatC-plcH-mexA)遺伝子の全塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。その結果得られた(tatC-plcH-mexA)遺伝子の配列を図3及び配列番号17に示す。なお、tatC遺伝子によりコードされるTatCタンパク質のアミノ酸配列を配列番号18に示す。
【0077】
予期しなかったことだが、PCR増幅断片をpTH18ks1ベクターにクローニングする際、由来不明の130 bpからなるDNA断片がtatC遺伝子とベクターのマルチクローニング部位であるHindIII部位の間に挿入されていることが判明した。この組換えプラスミドをHindIIIおよびKpnI消化し、約2.2 kbの(tatC-plcH-mexA)遺伝子断片を、予めKpnI、HindIIIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E.coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、(tatC-plcH-mexA)遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpTatC-PlcH-MexAをよぶことにする。
【0078】
(4-4-5) P. aeruginosa TNP080へのpTatC-PlcH-MexA発現ベクターの形質転換
項目(4-3-1)と同様の操作にて、pTatC-PlcH-MexA発現ベクターを染色体上のtatCとmexAが欠失した二重変異株TNP080に形質転換し、スルベニシリン(武田薬品工業)50μg/mlを添加したL寒天平板にて形質転換体を取得した。得られた形質転換体よりプラスミドを項目(1-2-2)の方法に従いを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位の両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCRを行い、組換えプラスミドの存在を確認した。
【0079】
(4-4-6) クローン化したtatC遺伝子産物の機能評価
クローン化したtatC遺伝子の導入によりTat分泌系の機能が回復することが期待できる。この点を評価するために項目(4-2-5)の方法にしたがい、前項目(4-4-5)で構築した形質転換体をCAA培地で生育させ、その培養上清中へのpyoverdineの分泌生産能の評価を行った。その結果、クローン化したtatC遺伝子をもつ形質転換体TNP080/pTatC-PlcH-MexAは、親株TNP070と同様なpyoverdineの吸収が認められ、Tat分泌系が機能していることが明らかとなった(図4)。
【0080】
(4-4-7) pTatC-PlcH-MexA発現ベクターを有するmexA/tatC二重変異株の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0081】
(4-4-8)ペリプラズム画分の分離
緑膿菌の各菌株を20mlのL液体培地(プラスミド保有株の場合は50μg/mlのスルベニシリンを含む)に接種し、37℃にて一夜培養した。この菌液を200mlの37℃に保温しておいた同培地に接種し、対数期後期(OD660nm、約1.0)まで振とう培養した後、プラスチック遠心管に移し室温にて遠心集菌をした(7500rpm、10分、室温)。この菌体を湿重量あたり5倍量の溶液(0.2M MgCl2, 50mM Tris-HCl, pH7.3)に懸濁しガラス試験管に移した後、37℃の水槽で10分間静置後、氷水中に15分間静置した。さらに、この37℃、10分間保温した後、氷水中、15分間インキュベーションする操作を繰り返し、細胞を12,000rpm、15分間、10℃にて遠心した。集菌後の上清0.5mlを超遠心用のプラスチックチューブ(Beckman, Cat. No. 343776)に移し、55,000rpm、10℃、1時間遠心(ローター、Beckman TLA-120.1;遠心機、Beckman Optima TLX)した後、上清を回収してペリプラズム画分とした。
【0082】
(4-4-9)ウエスタンブロット解析
項目(4-4-8)で調整したペリプラズム画分をSDS-ポリアクリルアミドゲル(12%)に供し電気泳動を行った後、Immobilon-Pメンブラン(Millipore社)に分離したタンパク質を転写し、一次抗体として抗MexAウサギ抗体、二次抗体としてアルカリフォスファターゼ結合抗ウサギーヒツジ抗体を用いて免疫染色を行った。
【0083】
<結果>
本実施例で行った薬剤感受性の評価結果を表2に示す。
【0084】
【表2】
【0085】
(1) PlcH-MexAキメラタンパク質はMexA欠損緑膿菌で機能する
pPlcH-MexA発現ベクターを有するP. aeruginosa TNP070形質転換体のアズトレオナムおよびクロラムフェニコールに対する感受性を評価した結果を表2に示した。pPlcH-MexAをもつ形質転換体のアズトレオナムおよびクロラムフェニコールに対する最小発育阻止濃度(MIC)は、それぞれ2.0μg/mlおよび32μg/mlであった。この感受性は野生株TNP070に比べ若干低い値であるが、クローン化した野生型のmexA遺伝子を導入した形質転換体(TNP070/pMexA)と同じ薬剤感受性を示したことから、キメラタンパク質PlcH-MexAは機能を保持していることが明らかとなった。このようにTNP070株の薬剤排出ポンプの機能が回復したことから、PlcH-MexAはTat分泌系を介してペリプラズムに分泌されるものと思われる。実際、形質転換体(TNP070/pPlcH-MexA)が発現したキメラタンパク質(PlcH-MexA)が成熟型MexAとしてペリプラズム画分に輸送されているか否かを、抗MexA抗体で検出したところ、シグナル配列が切断されたと思われる成熟型のMexAが形質転換体(TNP070/pPlcH-MexA)で認められた(図5、レーン8)。野生型細胞(PAO4290)は全菌体中に成熟型MexAのシグナルが認められるが(図5、レーン1)、ペリプラズムには存在しなかった(図5、レーン6)。また、mexA欠損変異株TNP070は全菌体中およびペリプラズム中いずれもMexAは発現しておらず(図5、レーン2および7)、このペリプラズム分画法によってMexAの局在が正確に評価されていることが確認できた。
【0086】
(2) PlcH-MexAキメラタンパク質はTat分泌系依存的に機能する
PlcH-MexAキメラタンパク質が機能を有していることから、本キメラタンパク質は翻訳後にTat系を介して本来の局在場所であるペリプラズムに分泌され、MexBおよびOprMと複合体を形成して薬剤を排出しているものと考えられる。この仮説を検証するために、MexA欠損変異株(TNP070)のTat分泌系を構成するtatC遺伝子を破壊した二重変異株TNP080(MexA-, TatC-)を作製した。この変異株にpPlcH-MexA発現ベクターを導入した形質転換株のアズトレオナムおよびクロラムフェニコールに対する感受性を調べた結果、それぞれの薬剤に対する形質転換体の感受性は、0.25μg/mlおよび4.0μg/mlであった(表2)。これは、TNP070とほぼ同じ感受性であり、薬剤排出ポンプが機能していないことを示唆している。すなわち、Tat分泌系が機能を失うとPlcH-MexAキメラタンパク質は正常に分泌されないことを意味しており、このキメラタンパク質はTat系依存的に分泌されていることが強く示唆された。このことをさらに検討するために、tatC破壊変異株より誘導した形質転換体(TNP080 /pPlcH-MexA)のペリプラズム画分を抗MexA抗体で検出した結果、全菌体中にはIPTG誘導による過剰なキメラタンパク質の発現が認められたが(図5、レーン4)、ペリプラズムには抗MexA抗体で染色されるタンパク質は認められず(図5、レーン9)、キメラPlcH-MexAはTat系に依存してペリプラズムに輸送されることが明らかとなった。おもしろいことに全菌体中のシグナルは成熟型より大きなMexAが認められたが、これはシグナルペプチダーゼで切断されていない状態のMexAと考えられた。
【0087】
なお、図5に示したウエスタンブロット解析の結果は、全菌体タンパク質(4μg)又はペリプラズム画分(1μg)をSDS-PAGE (12%ゲル)にて泳動後、抗MexA抗体にて検出した結果である。図5中、白三角は成熟型MexA、黒三角はシグナルペプチダーゼで切断されていないMexAを示す。
【0088】
この結果をさらに確認するために、野生型のtatC遺伝子をクローニングし、二重変異株TNP080にpPlcH-MexAとともに導入した(TNP080/pTatC-PlcH-MexA)。この形質転換体のアズトレオナムとクロラムフェニコールに対する感受性(それぞれ2.0μg/ml、32μg/ml)は、ほぼ野生株と同等レベルであったことから、クローン化したTatCが供給されることによってTat分泌系が機能回復していることが明らかとなった。このことから、PlcH-MexAキメラタンパク質が機能を回復したTat分泌系を介してペリプラズムに輸送されたことが強く示唆された。実際、この形質転換体(TNP080/pTatC-PlcH-MexA)のペリプラズム画分を抗MexA抗体で染色すると、発現量は少ないものの成熟型のMexAが輸送されていることが確認できた(図5、レーン10)。
【0089】
以上の結果より、今回作製したPlcH-MexAキメラタンパク質は予想したとおりTat分泌系を介してペリプラズムに輸送され、MexBおよびOprMと複合体を形成しMexA,B-OprM排出ポンプとして機能することが明らかとなった。この基礎的な知見を応用した新しい評価系を用いて天然化合物ライブラリーや低分子有機化合物ライブラリーといった供試物質を簡便に評価するハイスループットスクリーニング系の構築が可能である。本評価系においてTat分泌系を阻害する物質が指示薬剤と共存すれば、PlcH-MexA融合タンパク質は翻訳後にペリプラズムに分泌されないことから、pPlcH-MexA発現ベクターを有する指示菌は抗生物質に高感受性を示すことになる。
【0090】
Tat分泌系を標的とする抗菌剤は現在見出されておらず、この新しい評価系により効率的に新規標的であるTat系を阻害するリード化合物の探索が可能となる。また、本スクリーニング系で陽性を示す化合物は、Tat分泌系及び/又はMexA-MexB-OprM排出ポンプに対する阻害剤である可能性がある。換言すれば、本スクリーニング系によれば、Tat分泌系と薬剤排出ポンプというこれまでにない新しい二つの標的を同時にターゲットとした非常に効率のよいスクリーニング系である。
【0091】
〔実施例2〕
本実施例では、薬剤排出ポンプ:MexA-MexB-OprMを構成するサブユニットのうちOprMのシグナル配列を、Tat分泌系を介して外界へ分泌される病原性因子フォスフォリパーゼC(PlcH)のシグナル配列を含む領域と交換した融合遺伝子を作製した。そして、実施例1と同様に、染色体上のoprM遺伝子を欠失した宿主細胞、および染色体上のoprM遺伝子とともにtatC遺伝子を欠失した二重変異宿主細胞に本実施例で作製した融合遺伝子を導入し、薬剤排出ポンプ(MexA-MexB-OprM)を介して排出される薬剤に対する感受性を、得られた2種類の形質転換微生物について評価した。
【0092】
(1) Tat系シグナル配列を有するキメラoprM遺伝子(plcH-oprM)の構築
(1-1) オーバーラップPCR法によるplcH-oprMキメラ遺伝子断片の増幅
(1-1-1) plcHのシグナル配列領域の増幅
plcHのシグナル配列をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-AGTCTAGACTCGCTGTATTCCCTGCC-3’:配列番号19)およびリバースプライマー(5’-GTAGTCGGGGATCAGCGAGGTTCCGGTGCGGATGTC-3’:配列番号20)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTAR(タカラバイオ株式会社)を用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、40秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradient(タカラバイオ株式会社)を用いた。増幅断片をアガロースゲル電気泳動にて確認したところ約230塩基対の増幅産物が確認できた。
【0093】
(1-1-2) oprM遺伝子の成熟領域の増幅
oprM遺伝子の成熟領域をコードする遺伝子断片を増幅するために、フォワードプライマー(5’-GACATCCGCACCGGAACCTCGCTGATCCCCGACTAC-3’:配列番号21)およびリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用い、緑膿菌より分離した染色体を鋳型としてPCRを行った。PCR増幅反応は、PrimeSTARを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;65℃、5秒間;72℃、2分間の反応を30サイクル行った後、72℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にて確認したところ約1,500塩基対の増幅産物が確認できた。
【0094】
(1-1-3) plcH-oprMキメラ遺伝子の増幅
plcHシグナル配列の増幅断片1μl、oprM成熟領域の増幅断片1μlおよび滅菌蒸留水8μlを混合し、この混合液を滅菌蒸留水で100倍希釈した溶液を鋳型として、2段階目のPCR反応を行った。プライマーは、フォワードプライマー(5’- AGTCTAGACTCGCTGTATTCCCTGCC -3’:配列番号19)およびリバースプライマー(5’- CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いた。PCR増幅反応条件は、項目(1-1-2)と同じである。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。
【0095】
(1-2) pHSG298ベクターへのキメラ遺伝子(plcH-oprM)のクローニング
(1-2-1) キメラ遺伝子(plcH- oprM)の連結反応と形質転換
前項で得たキメラ遺伝子(plcH-oprM)を順次XbaI(TOYOBO)、EcoRI(TOYOBO)処理し(前項1-1-3で用いたプライマー末端には、各々XbaIあるいはEcoRI部位が付加してある)、予めXbaI、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理したpHSG298(TOYOBO)と混合し、DNAライゲーションキットMighty Mix(タカラバイオ株式会社)を用いて連結反応を16℃にて一夜行った。連結反応後のDNA溶液を、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。エレクトロポレーション装置は、GenePulser II(Bio-Rad)を用い、設定条件は2.5 kV、25μF、200 Ωである。回収した細胞を12.5μg/mlのカナマイシンを含むL寒天平板培地(isopropyl β-thiogalactopyranoside 80μg/ml、5-bromo-4-chloro-3-indolyl-β-galactoside 80μg/ml含有)に塗布し、37℃にて一夜培養を行った。
【0096】
(1-2-2) 形質転換体からのプラスミドの分離と組換えプラスミドの解析
生育した白色コロニーを4mlの12.5μg/mlのカナマイシンを含むL液体培地に植菌し、37℃にて一夜培養後、1.5 mlの菌液をミクロチューブに移し、遠心(12,000 rpm、23℃、5分間)して細胞を回収した。この細胞を氷冷した100μlの溶液1(25 mM Tris-Hcl、10 mM EDTA、50 mM Glucose、pH 8.0)に懸濁後、200μlの溶液2(0.2 N NaOH、1% SDS)を加えて穏やかに混和した(室温、5分間)。その後、150μlの溶液3(3 M potassium-acetate、acetic acid)添加後、氷上にて5分間静置した。この溶液を遠心後(12,000 rpm、23℃、5分間)、上清を新しいミクロチューブに回収し、400μlのフェノール・クロロフォルム・イソアミルアルコール(25:24:1)混合溶液を加えて、室温にて約5分間混合した。溶液を遠心分離(12,000 rpm、23℃、5分間)後、回収した上清に900μlのエタノールを添加混合後室温にて5分放置してから遠心分離(12,000 rpm、23℃、5分間)した。得られた沈殿を500μlの70%エタノールで洗浄した後乾燥させ、RNase (20μg/ml)を含むTE (10 mM Tris-HCl、1 mM EDTA、pH 8.0) 30μlに溶解した。得られたプラスミドをEcoRIおよびEcoRI + XbaIで消化後、アガロースゲル電気泳動により挿入断片の存在を確認した。このプラスミドの挿入断片の塩基配列をBigDye Terminator v1.1 (AB Applied Biosystems)のプロトコールに従い、ABI PRISM 377 DNA sequencing system (AB Applied Biosystems)にて解析した。このようにして得られたplcHシグナル配列遺伝子とoprM遺伝子の成熟領域とを融合したキメラ遺伝子の配列を図6及び配列番号23に示す。なお、このキメラ遺伝子がコードするキメラタンパク質のアミノ酸配列を配列番号24に示す。
【0097】
(1-3) シャトルベクターpMMB67HEへのキメラ遺伝子(plcH-oprM)のクローニング
pMMB67HEへ挿入するキメラ遺伝子断片の調整は、項目(1-2-2)で作製した組換えプラスミドをXbaIおよびEcoRIで消化後、アガロースゲル電気泳動によりキメラ遺伝子(plcH-oprM)を含むDNA断片(約1.7 kb)を分離した。この断片をQIAEX II Gel Extraction Kit (QIAGEN)を用いて分離精製し、予めXbaI、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を16℃にて一夜行った。その後、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、キメラ遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpPlcH-OprMと呼ぶことにする。
【0098】
(2) キメラoprM遺伝子(plcH-oprM)を導入した形質転換体の作製
(2-1) oprMを欠損した緑膿菌変異株(P. aeruginosa TNP072)のコンピタント細胞の調整
TNP072株を4 mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で一夜振とう培養した。増殖した菌液800μlを40mlの100μg/mlのテトラサイクリンを含むL液体培地に接種し、37℃で約7時間培養して得た対数増殖期の細胞を、プラスチックチューブに移し、7,500 rpm、4℃、10分間遠心し(ローター、AG-508;遠心機、KUBOTA 6930)集菌した。この菌体を氷冷した40mlの0.3 Mシュークロース溶液で洗浄した後、遠心(7,500 rpm、4℃、5分間)して集菌した。この遠心洗浄操作を3回繰り返して得た菌体を、少量(200〜400μl)の氷冷した0.3 Mシュークロース溶液に懸濁しコンピタント細胞としてエレクトロポレーションに供した。
【0099】
(2-2) 前項(2-1)で調整したコンピタントTNP072細胞40μlに1μlのキメラ遺伝子(plcH-oprM)を挿入したpMMB67HEを加え、緩やかに混合した菌液をエレクトロポレーション用キュベット 2 mmギャップ(Molecular BioProducts)に注入し、エレクトロポレーション装置に装填した(GenePulser II、Bio-Rad)。エレクトロポレーションを2.5 kV、25μF、200Ωの条件下で行った後、菌液を100μg/mlのスルベニシリン(武田薬品工業)を含むL平板培地に塗布し、37℃で2夜培養した。生育したコロニーより項目(1-2-2)の方法に従いプラスミドを抽出し、分離したプラスミドを鋳型プライマーとしてフォワードプライマー(5’-AGTCTAGACTCGCTGTATTCCCTGCC-3’:配列番号19)およびリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いてPCR反応を行い、組換えプラスミドの存在を確認した。
【0100】
(3) キメラ遺伝子(plcH-oprM)を保有する染色体上oprM遺伝子が欠損した形質転換体の薬剤感受性の評価
(3-1) 段階希釈した指標薬剤を含む寒天平板の作製
指標薬剤であるアズトレオナム(Sigma)およびクロラムフェニコール(和光純薬)の原液を、おのおの滅菌蒸留水および10%エタノールで溶解して調整した。この各薬剤の原液を滅菌蒸留水で希釈し、アズトレオナムを16〜0.0625μg/ml、クロラムフェニコールを64〜1μg/mlの濃度範囲の2倍希釈系列を含むミューラー ヒントン寒天培地(Difco)を作製した。
【0101】
(3-2) 最小発育阻止濃度評価用菌株の培養
最小発育阻止濃度を評価する際には、各菌株をアクティブプレートより一白金耳、0.4% (w/v)硝酸カリウムを含む2mlのミューラー ヒントン液体培地(Difco)に接種し、37℃にて一夜静置培養した。なお、プラスミド保有株を培養する際の液体培地にはスルべニシリン(50μg/ml)を含む0.4%(w/v)硝酸カリウム添加ミューラー ヒントン液体培地を用いた。一夜培養後の菌液を、同液体培地で菌体密度が約106 cells/mlになるように希釈して薬剤感受性評価に供した。
【0102】
(4) PlcH-OprMキメラタンパク質の機能発現がTat分泌系依存性であることの検証
(4-1) oprM遺伝子破壊株(TNP072)のtatC遺伝子を破壊した二重変異株(TNP082)の作製
mexA破壊株(TNP070)のtatC遺伝子を破壊したときに使用した自殺ベクターを用い、以下の方法にてoprM破壊株(TNP072)のtatC遺伝子を破壊した。
【0103】
(4-1-1) tatC遺伝子破壊用自殺ベクターのP. aeruginosa TNP072への接合伝達とtatC破壊株の選択
tatC遺伝子破壊用自殺ベクターをもつE. coli S17-1とP. aeruginosa TNP072を、それぞれ12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4mlのL液体培地で一夜37℃にて培養した。生育したそれぞれの菌液40μlを12.5μg/mlのカナマイシンあるいは12.5μg/mlのテトラサイクリンを含む4 mlのL液体培地に添加し、37℃で約8時間振とう培養をした後、700μlの大腸菌と700μlの緑膿菌を同じミクロチューブに添加混合後、遠心集菌(12,000 rpm、室温、5分間)した。抗生物質を除去するために、1 mlのL液体培地で菌体を2回遠心洗浄(12,000 rpm、室温、5分間)し100μlのL液体培地に再懸濁した。この菌液を12.5μg/mlのカナマイシンと50μg/mlのテトラサイクリンを含むL寒天平板に塗布して37℃にて2日培養し、tatC遺伝子の相同配列領域で組換えが起こった破壊株を得た。
【0104】
(4-1-2) tatC遺伝子破壊株ゲノムのtatC遺伝子領域の構造解析
前項目(4-1-1)においてtatC遺伝子の内部配列をもつ自殺ベクターの一点交叉による挿入によりtatC遺伝子を破壊した。そこで、染色体上のtatC遺伝子が破壊されていることを検証するためにtatC遺伝子に対応したフォワードプライマー(5’-TCGGATCCGAGACGCCGCGTAATCCATGAG-3’:配列番号15)とリバースプライマー(5’-GTGGATCCACTTCGTGCAGGTGCTTGAGG-3’:配列番号16)を用いPCR反応にて増幅した。PCR反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、5分30秒間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にて確認したところtatC遺伝子内に自殺ベクターが挿入した結果生じる約5.0キロ塩基対の増幅産物が確認できた。
【0105】
(4-1-3) シデロフォア(pyoverdine)産生能の評価
前項目(4-1-1)で取得したoprM、tatC二重変異株のTat分泌系が機能消失したことを検証するために、mexA、tatC二重欠損変異株の項で記載したように緑膿菌のシデロフォアであるpyoverdine産生能を調べることによって評価した。前項で取得したtatC遺伝子破壊株を12.5μg/mlのカナマイシンを含む5 mlのCAA液体培地(5 mMリン酸カリウム、1 mM 硫酸マグネシウム、5 g/lカザミノ酸、pH 7.0)で37℃にて1日〜2日間培養し十分に菌体を生育させ後、遠心(12,000 rpm、室温、5分間)により培養上清を回収した。この上清の吸収を分光光度計(V-630BIO、日本分光)を用いて測定した。その結果、野生株および親株TNP072で認められる典型的なpyoverdineの吸収が、tatC遺伝子破壊株では認められず、tatC遺伝子が破壊されていることが確認できた(図7)。このtatC遺伝子が破壊された二重変異株をP. aeruginosa TNP082とよぶことにする。なお、図7において(a)は野生株、(b)は親株TNP072を使用した際の結果を示し、(c)はTNP082を使用した際の結果を示している。
【0106】
(4-2) oprM/tatC二重変異株でのPlcH-OprM融合タンパク質の機能評価
PlcH-OprM融合タンパク質がTat分泌系に依存してトランスロケートされるか否かを評価するために、pPlcH-OprM発現ベクターを前項で作製したtatC遺伝子破壊株TNP082(oprM/tatC二重変異株)に形質転換しアズトレオナムおよびクロラムフェニコールに対する感受性を項目(3)に記載した寒天希釈法によって調べた。
【0107】
(4-3) tatC遺伝子とplcH-oprMキメラ遺伝子をもつシャトルベクターの作製
シャトルベクターの作製効率をあげるために、クローン化したtatC遺伝子をシャトルベクターに載せる前に、一旦、plcH-oprMキメラ遺伝子をもつpHSG298(カナマイシン耐性マーカーをもつ)ベクターにtatC遺伝子をクローン化することにした。実施例1の項目(4-4-3)で作製したプラスミドtatC / pUC18をSphIとSmaIで消化後、アガロースゲル電気泳動を行い約910 bpのtatC遺伝子断片をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。これと並行して、項目(1-3)で作製したplcH-oprMキメラ遺伝子保有pHSG298を予めXbaI処理し、T4DNAポリメラーゼによる平滑末端化を行った。その後SphI処理およびバクテリアルアルカリフォスファターゼ(TOYOBO)処理を施した。このように調製したベクターと精製したtatC遺伝子断片とを混合し、連結反応をした後、前項(1-2)と同様にして形質転換体を得た。この実験デザインではplcH-oprMキメラ遺伝子の直前にtatC遺伝子が順向きに挿入されることになる。そこで、順向きに連結した2遺伝子(tatC-plcH-oprM)が増幅されるフォワードプライマー(5’-GTGGATCCACCTGACCGAACTGCGTACG-3’:配列番号13)とリバースプライマー(5’-CGGAATTCCGCGAGGATAAAAAAACG-3’:配列番号22)を用いた直接コロニーPCR法を用いて目的とする組換え体を選抜した。PCR増幅反応は、TaKaRa Ex Taqを用い、94℃、2分間鋳型DNAを変性し、98℃、10秒間;68℃、3分間の反応を30サイクル行った後、68℃、5分間の反応で断片を完全伸長させた後、4℃で増幅産物を保持した。PCR装置は、PCR Thermal Cycler Dice Gradientを用いた。増幅断片をアガロースゲル電気泳動にかけ、約2.5 kbの増幅産物が認められるクローンを選抜した。
【0108】
このようにしてtatC遺伝子が順向きにplcH-oprMキメラ遺伝子の直前に挿入されたクローンを選び、組換えプラスミドをHindIIIおよびEcoRI消化し、約2.6 kbの遺伝子断片(tatC-plcH-oprM)をQIAEX II Gel Extraction Kit(QIAGEN)を用いて精製した。そして予めHindIII、EcoRIおよびバクテリアルアルカリフォスファターゼ(TOYOBO)処理をしたpMMB67HEと混合し、連結反応を行った後、E. coli JM109のコンピタント細胞にエレクトロポレーション法にて形質転換した。条件は、項目(1-2-1)と同じである。生育したコロニーよりプラスミドDNAを調整し、上記プライマーを用いたPCR反応を行うことによって、(tatC-plcH-oprM)遺伝子がpMMB67HEのマルチクローニング部位に挿入されている組換えプラスミドを取得した。この組換えプラスミドをpTatC-PlcH-OprMをよぶことにする。
【0109】
(4-4) P. aeruginosa TNP082へのpTatC-PlcH-OprM発現ベクターの形質転換
項目(2-2)と同様の操作にて、pTatC-PlcH-OprM発現ベクターを染色体上のtatCとoprMが欠失した二重変異株TNP082に形質転換し、スルベニシリン(武田薬品工業)50μg/mlを添加したL寒天平板にて形質転換体を取得した。得られた形質転換体よりプラスミドを項目(1-2-2)の方法に従いを抽出し、分離したプラスミドを鋳型、プライマーとしてpMMB67HEのマルチクローニング部位両端の外側に設定したフォワードプライマー(5’-GAGCGGATAACAATTTCACACAGG-3’:配列番号11)とリバースプライマー(5’-CTCTCATCCGCCAAAACAGC-3’:配列番号12)を用いてPCRを行い、組換えプラスミドの存在を確認した。
【0110】
(4-5) クローン化したtatC遺伝子産物の機能評価
クローン化したtatC遺伝子の導入によりTat分泌系の機能が回復することが期待できる。この点を評価するために項目(4-1-3)の方法にしたがい、前項目(4-4)で構築した形質転換体をCAA培地で生育させ、その培養上清中へのpyoverdineの分泌生産能の評価を行った。その結果、クローン化したtatC遺伝子をもつ形質転換体TNP082/pTatC-PlcH-OprMは、野生株および親株TNP072と同様なpyoverdineの吸収が認められ、Tat分泌系が機能していることが明らかとなった(図7(d))。
【0111】
(4-6) pTatC-PlcH-OprM発現ベクターを有するoprM/tatC二重変異株(TNP082/pTatC-PlcH-OprM)の薬剤感受性の評価
項目(3)に記した寒天希釈法を用いてアズトレオナムとクロラムフェニコールに対する最小発育阻止濃度を測定した。
【0112】
(4-7)外膜画分の分離
緑膿菌の各菌株を4 mlのL液体培地(プラスミド保有株の場合は50μg/mlのスルベニシリンを含む)に接種し、37℃にて一夜培養した。この菌液を100 mlの37℃に保温しておいた同培地に接種し、対数期後期(OD660nm、約1.0)まで振とう培養した後、プラスチック遠心管に移し遠心集菌をした(7500 rpm、10分、室温)。この菌体を100 mlの溶液(200 mM Na-Phosphate Buffer, pH 7.6)で2回洗浄した後、1.5 mlの同溶液に懸濁し、超音波処理(Bioruputor, コスモバイオ) を氷冷下5分間行った。この処理液を12,000 rpm、1分、室温で遠心集菌し、集菌後の上清0.5 mlを超遠心用のプラスチックチューブ(Beckman, Cat. No. 343776)に移し、45,000 rpm、30分、10℃で遠心(ローター、Beckman TLA-120.1; 遠心機、Beckman Optima TLX)した。遠心後、ペレットを溶液 (20 mM Na-Phosphate Buffer, pH 7.6) 200μlで懸濁し、その後同緩衝液で2回洗浄した。この膜画分を1% Na-Salcosinate(和光純薬)を加えた同溶液1 mlに懸濁して30℃、30分静置した後、45,000 rpm、30分、10℃で遠心(ローター、Beckman TLA-120.1;遠心機、Beckman Optima TLX)した。得られたペレットは溶液 (200 mM Na-Phosphate Buffer, pH 7.6) で2回洗浄し、最終的に100μlの水に懸濁して外膜画分とした。
【0113】
(4-8)ウエスタンブロット解析
項目(4-7)で調整した外膜画分をSDS-ポリアクリルアミドゲル(12%)に供し電気泳動を行った後、Immobilon-Pメンブラン(Millipore社)に分離したタンパク質を転写し、一次抗体として抗OprMウサギ抗体、二次抗体としてアルカリフォスファターゼ結合抗ウサギーヒツジ抗体を用いて免疫染色を行った(図8)。なお、野生型のoprM遺伝子をもつプラスミド(pOprM)保有株はタンパク量として5μgの各画分を、キメラoprM遺伝子および野生型tatC遺伝子を有する各プラスミド(pPlcH-OprMおよびpTatC-PlcH-OprM)保有株はタンパク量として7.5μgの各画分を電気泳動した。
【0114】
<結果>
本実施例で行った薬剤感受性の評価結果を表3に示す。
【0115】
【表3】
【0116】
(1) PlcH-OprMキメラタンパク質はOprM欠損緑膿菌で機能する
pPlcH-OprM発現ベクターを有するP. aeruginosa TNP072形質転換体のアズトレオナムおよびクロラムフェニコールに対する感受性を評価した結果を表3に示した。oprM欠損変異株(TNP072)はMexAB-OprM排出ポンプが機能しないため薬剤を排出することができない。従って、野生株PAO4290に比べTNP072はアズトレオナムおよびクロラムフェニコールに対して薬剤感受性が著しく上昇する(MICが低下する)。事実、oprM欠損変異株(TNP072)の各薬剤に対するMIC値は0.125μg/mlおよび1.0μg/mlである。このoprM欠損変異株にキメラoprM遺伝子をクローン化したプラスミド(pPlcH-OprM)を形質転換した株のアズトレオナムおよびクロラムフェニコールに対するMICはそれぞれ4.0μg/mlおよび16μg/mlであった。この値は野生株およびクローン化した野生型のoprM遺伝子をもつoprM欠損変異株(TNP072/pOprM)のMIC値とほぼ同等であることから、キメラOprMは機能的な発現をしていることが明らかとなった。実際、キメラPlcH-OprMを発現する全菌体タンパク質をウエスタンブロット解析すると、シグナル配列が未切断のキメラタンパク質に加え成熟型OprMに相当するシグナルが観察され(図8,レーン2)。また外膜画分ではシグナルは弱いものの成熟型OprMの発現が確認できた(図8,レーン3)。このことから、キメラOprMは内膜をトランスロケートし外膜に達してポンプ複合体を形成していることが明らかとなった。
【0117】
(2) PlcH-OprMキメラタンパク質はTat分泌系依存的に機能する
PlcH-OprMキメラタンパク質が機能を有していることから、本キメラタンパク質は翻訳後にTat系を介して本来の局在場所である外膜に分泌され、MexAおよびMexBと共にポンプ複合体を形成して薬剤を排出しているものと考えられる。この仮説を遺伝学的に検証するために、OprM欠損変異株(TNP072)のTat分泌系を構成するtatC遺伝子を破壊した二重変異株TNP082(OprM-, TatC-)を作製した。この変異株にpPlcH-OprM発現ベクターを導入した形質転換株のアズトレオナムおよびクロラムフェニコールに対する感受性を調べた結果、それぞれの薬剤に対する形質転換体の感受性は、0.125μg/mlおよび1.0μg/mlであった(表3)。これは、OprM欠損変異株TNP072と同じ感受性であり、薬剤排出ポンプが機能していないことを示唆している。すなわち、Tat分泌系が機能を失うことによりPlcH-OprMキメラタンパク質は正常に分泌されないことを意味しており、このキメラタンパク質はTat系依存的に分泌されていることが強く示唆された。一方、この二重変異株に野生型のoprM遺伝子をもつpOprMを導入した形質転換体の両薬剤に対する感受性は、2.0μg/ml(アズトレオナム)および16μg/ml(クロラムフェニコール)と野生株と同等のMIC値を示したことから、先のキメラOprMが機能しない原因はTat分泌系を介してキメラOprMがトランスロケートできないことによるものであることが明らかとなった。
【0118】
この結果をさらに検証するために、野生型のtatC遺伝子をクローニングし、キメラOprM遺伝子(plcH-oprM)とともに二重変異株TNP082に導入した形質転換体(TNP082/ pTatC-PlcH-OprM)を得た。この形質転換体のアズトレオナムとクロラムフェニコールに対する感受性は、それぞれ4.0μg/mlおよび32μg/mlであり、野生株と同等のMIC値を示した。このことから、クローン化したTatCが供給されることによってTat分泌系が機能回復し、PlcH-OprMキメラタンパク質がこの機能回復したTat分泌系を介してトランスロケートし外膜に輸送されたことが確認できた。事実、クローン化した野生型のtatC遺伝子を染色体上のtatC遺伝子破壊株(TNP082)に導入すると、成熟型のOprMが全菌体および外膜画分に検出され(図8,レーン6,7)、本形質転換体(TNP082/pTatC-PlcH-OprM)においてキメラOprMがTat系依存的に内膜をトランスロケートする結果と一致していた。
【0119】
以上の結果より、フォスフォリパーゼCのシグナル配列をもつキメラOprM(PlcH-OprM)を染色体上のoprM遺伝子が欠損した宿主で発現させる本評価システムは、〔実施例1〕で詳述したキメラMexAを利用したシステムに比べ、多くの安価な抗生物質を指標薬剤として用いることができるため、天然化合物ライブラリーおよび低分子有機化合物ライブラリーを効率よくスクリーニングするためのより優れた評価系である。また、本スクリーニング系で陽性を示す化合物は、Tat分泌系阻害剤以外に、MexA-MexB-OprM排出ポンプ自体の阻害剤の可能性もあるので、この新規スクリーニング系はTat分泌系と薬剤排出ポンプというこれまでにない新しい二つの標的を同時にターゲットとした非常に効率のよいスクリーニング系である。
【特許請求の範囲】
【請求項1】
Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成することを特徴とする組換え微生物。
【請求項2】
上記融合遺伝子を、上記サブユニットをコードする内在遺伝子を欠損した上記微生物に導入したものであることを特徴とする請求項1記載の組換え微生物。
【請求項3】
上記微生物は内在性のTat分泌系を有する細菌であることを特徴とする請求項1記載の組換え微生物。
【請求項4】
上記微生物はグラム陰性菌であることを特徴とする請求項1記載の組換え微生物。
【請求項5】
上記微生物は緑膿菌(Pseudomonas aeruginosa)であることを特徴とする請求項1記載の組換え微生物。
【請求項6】
上記薬剤排出ポンプは、サブユニット:MexA、MexB及びOprMから構成されることを特徴とする請求項1記載の組換え微生物。
【請求項7】
上記融合遺伝子は、上記サブユニットとしてMexAにおける成熟領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項8】
上記融合遺伝子は、上記サブユニットとしてOprMにおける成熟領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項9】
上記融合遺伝子は、上記シグナル配列として病原性因子フォスフォリパーゼC(plcH)のシグナル配列を含む領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項10】
互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現することを特徴とする請求項1記載の組換え微生物。
【請求項11】
請求項1乃至10いずれか一項記載の組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる工程と、上記組換え微生物の上記薬剤に対する感受性を評価する工程と、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断する、Tat分泌系阻害剤のスクリーニング方法。
【請求項12】
上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする請求項11記載のスクリーニング方法。
【請求項13】
上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする請求項11記載のスクリーニング方法。
【請求項14】
請求項1乃至10いずれか一項記載の組換え微生物と、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤とを含む、Tat分泌系阻害剤のスクリーニングキット。
【請求項15】
上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする請求項14記載のTat分泌系阻害剤のスクリーニングキット。
【請求項16】
上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする請求項14記載のスクリーニングキット。
【請求項1】
Tat(Twin Arginine Translocation)分泌系を有する微生物における、薬剤排出ポンプを構成するサブユニットにおける成熟領域をコードする遺伝子と、Tat分泌系を介して分泌されるタンパク質のシグナル配列を含む領域をコードする遺伝子とを融合させた融合遺伝子を発現し、上記サブユニットがTat分泌系を介して輸送され上記薬剤排出ポンプを形成することを特徴とする組換え微生物。
【請求項2】
上記融合遺伝子を、上記サブユニットをコードする内在遺伝子を欠損した上記微生物に導入したものであることを特徴とする請求項1記載の組換え微生物。
【請求項3】
上記微生物は内在性のTat分泌系を有する細菌であることを特徴とする請求項1記載の組換え微生物。
【請求項4】
上記微生物はグラム陰性菌であることを特徴とする請求項1記載の組換え微生物。
【請求項5】
上記微生物は緑膿菌(Pseudomonas aeruginosa)であることを特徴とする請求項1記載の組換え微生物。
【請求項6】
上記薬剤排出ポンプは、サブユニット:MexA、MexB及びOprMから構成されることを特徴とする請求項1記載の組換え微生物。
【請求項7】
上記融合遺伝子は、上記サブユニットとしてMexAにおける成熟領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項8】
上記融合遺伝子は、上記サブユニットとしてOprMにおける成熟領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項9】
上記融合遺伝子は、上記シグナル配列として病原性因子フォスフォリパーゼC(plcH)のシグナル配列を含む領域をコードする遺伝子を含むことを特徴とする請求項1記載の組換え微生物。
【請求項10】
互いに異なる複数の薬剤排出ポンプについて、複数の上記融合遺伝子を発現することを特徴とする請求項1記載の組換え微生物。
【請求項11】
請求項1乃至10いずれか一項記載の組換え微生物に対して、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤と供試物資とを作用させる工程と、上記組換え微生物の上記薬剤に対する感受性を評価する工程と、上記供試物質の非存在下における上記感受性と比較して、上記供試物質の存在下における上記感受性が大である場合には、上記供試物質はTat分泌系の阻害作用を有するものであると判断する、Tat分泌系阻害剤のスクリーニング方法。
【請求項12】
上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする請求項11記載のスクリーニング方法。
【請求項13】
上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする請求項11記載のスクリーニング方法。
【請求項14】
請求項1乃至10いずれか一項記載の組換え微生物と、上記融合遺伝子の産物をサブユニットとして含む薬剤排出ポンプにより微生物外部へ排出される薬剤とを含む、Tat分泌系阻害剤のスクリーニングキット。
【請求項15】
上記薬剤は、上記薬剤排出ポンプを欠損した変異体における薬剤耐性が当該薬剤排出ポンプを有する野生型微生物における薬剤耐性と比較して有意に低下する薬剤であることを特徴とする請求項14記載のTat分泌系阻害剤のスクリーニングキット。
【請求項16】
上記薬剤は、アズトレオナム、クロラムフェニコール、ノルフロキサシン、ノボビオシン、セフォペラゾン、セフスロジン、オフロキサシン、シプロフロキサシン、ゲンタミシン及びセフタジジムからなる群から選ばれる少なくとも1種の抗生物質であることを特徴とする請求項14記載のスクリーニングキット。
【図1】


【図2】
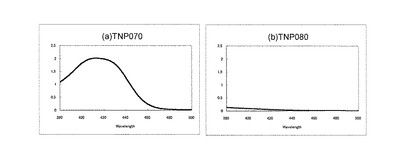
【図3】

【図4】
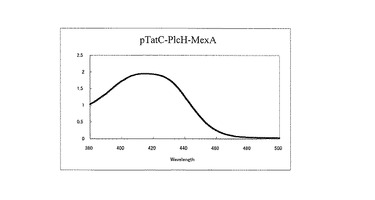
【図5】
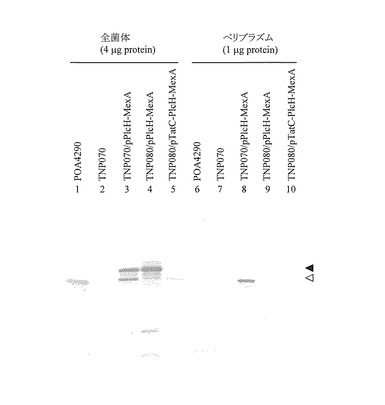
【図6】

【図7】
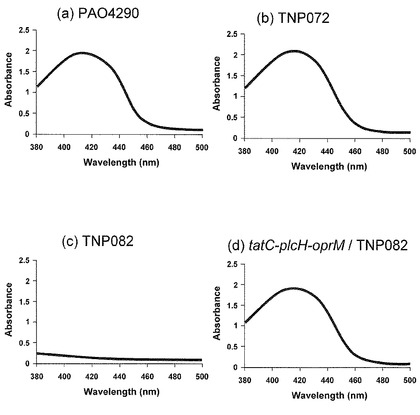
【図8】
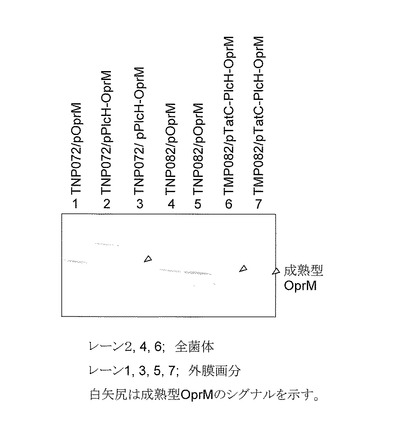
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2011−78408(P2011−78408A)
【公開日】平成23年4月21日(2011.4.21)
【国際特許分類】
【出願番号】特願2010−202114(P2010−202114)
【出願日】平成22年9月9日(2010.9.9)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
【公開日】平成23年4月21日(2011.4.21)
【国際特許分類】
【出願日】平成22年9月9日(2010.9.9)
【出願人】(504157024)国立大学法人東北大学 (2,297)
【Fターム(参考)】
[ Back to top ]