
結晶化度を改善する方法
本発明は、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、固体物質を、その固体物質が不溶性であるか又は難溶性である溶媒(非溶媒)と接触させる工程と、固体物質に、非溶媒と接触したときに超音波を適用する工程とを含む方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、非晶質固体物質の、結晶化度のレベルを増加させて、表面特性を改質する方法に関する。本発明は、活性成分化合物のような化学品の製造、吸入製剤のような医薬製剤に使用される賦形剤の製造、及び、液体系の懸濁液のような農芸化学的薬剤の製造において用途を有する。
【0002】
本発明は、また、例えば乾燥粉末吸入器(DPI)装置を使用して、肺に投与される乾燥粉末製剤を形成する、活性薬剤粒子の製造に関する。特に、本発明は、その機能が従来のDPI、加圧式定量噴霧吸入器(pMDI)及び鼻懸濁粉末剤、特にはDPI及びpMDI粉末剤、とりわけDPI粉末剤よりも有意に卓越している、粒子の特性及び好ましい加工を提供する。
【背景技術】
【0003】
薬剤を気道に投与するために広く使用されている2つの系は、通常ラクトンのような製薬学的に不活性な物質の粗賦形剤粒子と混合される乾燥粉末剤として微粉化薬剤粒子を含む乾燥粉末剤吸入器(DPI)及び噴射ガス中の微粉化薬剤粒子の懸濁剤を含むことができる加圧式定量噴霧吸入器(pMDI)である。本発明は、これらの送達方法の両方に関連する。
【0004】
鼻送達は、薬剤粒子を中枢神経系(CNS−鼻から脳)に投与することができる手段であり、粉末剤又は液体懸濁剤のいずれかによる全身的又は局所的な鼻製剤である。多様な呼吸作動式装置は、肺への付着を生じることなく、副鼻腔及び嗅部を含む鼻腔の標的領域に鼻腔内薬剤を送達する。本発明は、この送達方法に関連する。
【0005】
活性及び他の組成成分の結晶及び沈殿粒径の制御は、製薬産業や農芸化学産業のような、目的の活性成分の最終製品形態が微粉末の形態である産業において必要である。活性成分が生体系において作用する方法は、多くの要因、とりわけ粒径及び結晶形態によって決まる。小さな粒子は、粉砕のような方法により作製することができるが、そのような方法は、粉砕粒子の材質に有害な影響を与える場合がある。更に、粒子の有意な割合が、所定の最終用途に不適切な形状で製造される場合がある。粒子が粉砕されるとき、これらは形態変化を受け、望ましくない表面多形態変換をもたら素場合があり、このことは、吸入のために設計される医薬製剤のような最終目的用途に不適切である非晶構造の形成を生じる場合がある。加えて、粉砕は、例えば活性成分が低融点固体である場合、粒状粉砕を不適切にしうる相当量の熱を発生する。加えて、エアロゾルに使用される目的の粒子の物理的機能は、粉砕の結果として高度に荷電されると損なわれる。
【0006】
薬剤粒子の製造技術には、薬剤の溶液から小さな滴のエアロゾルを生成し、続いて小さな滴を噴霧乾燥して、粒子を固化することが含まれうる。噴霧乾燥は、粒子形成及び乾燥に関わる最も広く使用されている工業的方法の1つである。これは、例えば溶液、乳液又はポンプ移動可能な懸濁液のような液体供給原料から、粉末、顆粒又は凝集形態のいずれかで乾燥固体を連続的に製造するために特に適している。したがって、乾燥噴霧は、最終製品が、粒径分布、残留含水量、嵩密度、粒子形状などのパラメーターに関する品質標準に適合する場合に理想的な方法である。従来の連続噴霧乾燥技術の欠点は、固化が典型的には急速であり、加えて加工が乾燥粒子の高度な凝集をもたらすので、乾燥される粒子が結晶質粒状形態ではなく、おそらく100%の高さで非晶質形態になる傾向があることである。エアロゾルの小さな滴の凍結乾燥も粒子を得るために当該技術において使用されるが、ここでも、生じる典型的な急速固化は、一般に非晶質粒子の生成をもたらす。
【0007】
特許文献1は、適切な溶媒中で所望の物質の溶液を形成する工程、その溶液からエアロゾルを生成する工程、その物質からエアロゾルの小さな滴を非溶媒で収集する工程、及び、非溶媒に分散した液滴に超音波を適用して、物質の結晶化を実施する工程を含む、SAXと呼ばれる方法による粒子の製造を記載する。この技術の欠点は、エアロゾルから蒸発する溶媒の程度に対して臨界制御が必要なことである。
【0008】
吸入は、全身作用薬、ならびに喘息、慢性閉塞性肺疾患及び炎症のような、肺自体に局所的に作用するように設計された薬剤の送達にとって、非常に魅力的で迅速でさらに患者に優しい経路である。予測可能及び再現可能な方法で肺に薬剤を送達する技術を開発することが特に望ましく、有利である。薬剤吸入の利益には、素早い開始速度;非侵襲的全身経路に対する患者許容度及び服薬順守の改善;副作用の低減;製品寿命の延長;送達の一貫性の改善;より高い用量、大きな効能及び標的化の正確さを含む新しい治療形態の利用が含まれる。
【0009】
乾燥粉末吸引(DPI)は、肺疾患の治療において重要な役割を果たす。第一には、これらは定量吸入器(MDI)の使用において遭遇した問題を克服するため、後には、これらが噴射剤無含有であり、したがって環境により優しいために開発された。MDIを使用すると、患者は、エアロゾルが肺に達することができるように、吸入と吸入器の作動を同調させなければならない。乾燥粉末剤吸入器(DPI)は、呼吸作動式であり、そのため理論的には、問題なく肺に達することができる/するはずである。しかし、問題は、取扱い、用量の内容均一性及び用量の制御に関する技術的な制限によって生じる。また、吸気流速は、患者によって異なり、DPIの機械的原理に応じて決まる。エアロゾルの肺への付着率が吸気流速に依存する可能性があるので、高い流れ抵抗により吸気流速を顕著に低減するDPIは、あまり適していない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO2004/073827
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかし、成功した乾燥粉末剤及びDPI製品のための粉末技術は、依然として大きな技術的問題を残している。製剤は、製造者及び粉末剤の定量を助けるばかりでなく、予想可能な再懸濁及び流動化を提供し、分配装置内の粉末剤の過剰な保持を回避するためにも、適切な流動特性を有さなければならない。懸濁粉末剤における薬剤粒子又は製薬学的に活性な物質の粒子(本明細書において、API粒子とも呼ぶ)は、肺の中の適切な標的に輸送されうるように、適切にエアロゾル化されなければならない。典型的には、肺への付着のためには、活性粒子は10μm未満、しばしば0.1〜7μm又は0.1〜5μm未満の直径を有する。
【0012】
この種類の系において、薬剤−薬剤及び薬剤−担体粒子、並びに粒子−壁の相互作用は、肺深部への薬剤送達の成功のために極めて重要である。粒子間の相互作用は、ファンデルワールス、毛細管作用及びクーロン力のような接着力によって決定される。これらの力の強さは、粒子の大きさ、形状及び形態によって影響を受ける。粗面を有する球状又は球形粒子は、小さい接触域及び粒子間の分離距離の増加によって、肺薬剤送達にとって最適であると考慮される。大きな分離距離は、結合力を減少し、粉末分散を改善する。最適薬剤粒子のための粒子工学は、DPI装置工学と共に、肺を介する効率的な薬剤送達にために必須である。WO2006/056812は、乾燥粉末剤吸入器(DPI)装置を使用して肺に投与される乾燥粉末製剤を形成する粒子加工の改善に関する発明を報告し、それによると、活性物質の粒子及び担体物質の粒子の加工が添加剤物質の存在下で実施され、優れた粉末特性を示す粉末組成物を提供する。
【0013】
乾燥粉末剤が従来の方法により製造される場合、活性粒子は大きさが異なり、多くの場合にこの差異は顕著である。このことは、十分に高い割合の粒子が適切な部位への投与に適した大きさになるのを確実にすることを困難にする。したがって、活性粒子の粒度分布が可能な限り狭い乾燥粉末製剤を有することが望ましい。例えば、好ましくは粒子分布はガウスであり、好ましくは粒子分布は単峰性である。更に、例えば、活性粒子の空気力学的及び体積的な粒度分布の幾何学的標準偏差は、好ましくは2以下、より好ましくは1.8以下、1.6以下、1.5以下、1.4以下又はさらには1.2以下である。このことは、用量効率及び再現性を改善する。
【0014】
平均中央空気力学的直径(MMAD)とは、50%の粒子が、吸入薬粒子のインビトロ機能を決定するのに適しているインパクターに入るのよりも小さい粒子直径であって、形状と密度との両方を考慮する。(例えば)5μmのMMADを有するサンプルは、粒子の全質量(すなわち、総数ではない)の50パーセントが5μmを超える直径を有し、50パーセントが5μm未満の直径を有する。
【0015】
MMADが10μm以下であるより小さい微粒子は、表面積と体積との比が増加するにつれてますます熱力学的に不安定になる傾向があり、そのことは、粒径が減少すると表面自由エネルギーの増加をもたらし、その結果、粒子が凝集する傾向及び凝集塊の強度を増加する。吸入器では、微粒子の凝集及びそのような粒子の吸入器の壁への接着が問題であり、微粒子が大型で安定した凝集塊で吸入器から遊離されるか又は吸入器から離れることができず、吸入器の内部に接着したままになっているか又はさらには吸入器を目詰まり若しくは閉塞させる結果をもたらす。
【0016】
吸入器のそれぞれの作動の間、また異なる吸入器及び異なる粒子バッチの間に粒子の安定した凝集塊が形成される程度が不確実であるので、不十分な再現性がもたらされる。更に、凝集塊の形成は、活性粒子のMMADが著しく増加する可能性があり、活性粒子の凝集塊が肺の必要な領域に到達しないことを意味する。肺深部又は全身送達に必要なこれらのμmからサブμmの粒径は、呼吸に適した活性粒子が高い凝集性の傾向を有するという問題をもたらし、このことは、これらが一般に不十分な流動性及び不十分なエアロゾル化を示すことを意味する。
【0017】
そのような呼吸に適した活性粒子の高い凝集性を克服するために、過去に製剤には流動性及び薬剤エアロゾル化の両方を助けるために不活性賦形剤の大型の担体粒子を含ませた。微細活性粒子が、互いに固着するのではなく、吸入器装置の中にある間に大型の担体粒子に付着する傾向があるので、これらの大型の担体粒子は、粉末製剤に有益な効果を有する。活性粒子は、担体粒子表面から遊離し、分配装置の作動の際に分散して、気道に吸入されうる微細懸濁剤を生じる。
【0018】
比較的大型の担体粒子の添加は、粉末特性を改善する傾向があるが、通常は、製剤の総重量の95%以上が担体であるという程度まで薬剤を希釈する効果も有する。大部分の微細又は超微細活性成分が担体粒子の表面に付着する必要があり、そうでなければ、活性粒子の凝集性が依然として粉末において優位を占め、不十分な流動性をもたらすので、粉末特性に所望の効果を有するために比較的多量の担体が必要となる。微細粒子が接着するために利用可能な担体粒子の表面積は、担体粒子の直径が増加すると減少する。しかし、流動性は、直径が減少すると悪化する傾向がある。したがって、満足できる担体粒子を得るために適切な平衡を見出す必要がある。更なる考慮は、少なすぎる担体粒子が含まれると分離を生じる可能性があることであり、これは極めて望ましくない。
【0019】
製剤の際に経験する更なる問題は、薬剤及び賦形剤粒子の表面特性の差異である。それぞれの活性剤粉末は、独自の固有粘着性又は表面エネルギーを有し、化合物毎に著しく変動する。更に、表面エネルギーの性質は、与えられた化合物によって加工の方法に応じて変わる可能性がある。例えば、噴射粉砕は、用いる衝突の攻撃的な性質のために、表面特性に有意な差異を生じることで悪名高い。そのような差異は、表面エネルギーの増加、並びに凝集性及び接着性の増加をもたらす可能性がある。高度に規則的な結晶質粉末でも、近距離のリフシッツ−ファンデルワールス力は、凝集性及び接着性の高い粉末をもたらす可能性がある。
【0020】
担体賦形剤を使用しない場合、微粉化薬剤粒子は、リフシッツ−ファンデルワールス力のみにより緩く凝集される。粒子凝集塊は空気流により解凝集されなければならないので、毛細管力を形成しないことがそのような製剤の機能にとって重要である。毛細管力は、通常、例えばリフシッツ−ファンデルワールス力の数倍大きく、そのような凝集塊が単一の粒子に分かれる能力は、凝集塊を一緒に保持する自己付着力が増加するにつれて減少する。そのような緩い凝集は、球形化の方法を使用して達成することができる。
【0021】
空気流の中に設置されると、担体粒子に接着した粒子に対して作用する力は、揚力(担体粒子から小さな粒子を持ち上げる揚力;これは微粉化粉末では無視することができる)、抵抗力(接着力及び摩擦力を相殺する)、接着力及び摩擦力(接触する2つの表面の接線変位を防ぐ力)と記載することができる。これらの最後の2つは、担体表面からの薬剤粒子の脱離を妨ぐことができる。乾燥粉末剤吸入における相互作用混合物の成功又は失敗は、主に、薬剤粒子を担体表面に固定する接着力の大きさによって決まる。
【0022】
明らかに、薬剤−担体単位が抵抗力により単一構成成分に分けることができなければ、薬剤−担体単位の全体が嚥下されるので、非常に高い接着力は不要である。平衡した接着力は、吸入されたときには、薬剤−担体単位の微粉化薬剤粒子への分離を促進し、そして、嚥下されたときには、粗担体粒子への分離を促進する。一方、薬剤と担体粒子との間の接着力が小さすぎると、粒子分離をもたらす場合があり、したがって、用量の含有均一性に大きな差異をもたらす場合がある。また、薬剤粒子は、強く接着する傾向がある吸入器装置壁との滑り接触の際に担体粒子から容易に除去される。したがって、より多くの薬剤が吸入器装置の中に失われる。
【0023】
従来技術は、吸入用の相互作用粉末混合物における接着力は、幾つかの方法により操作できると教示している。第1には、担体粒子を、粒子の大きさの中央値、形状及び表面粗さに従って選択することができ、このことは、規定された混合手順において接着力に大きな差をもたらし、したがって、異なるエアロゾル化特性をもたらす。
【0024】
中央粒径値の減少は、薬剤と担体粒子との間の接着力を増加する。大きな接着力は、不規則形状又は伸長担体粒子においても見出される。この効果は、混合の際の摩擦の増加によって説明することができる。表面粗さは、粗さの程度に応じて接着力を増加又は減少させる。接着力の増加は、真接触領域の増加に起因して極めて平滑な担体粒子表面において見出され、又は非常に粗い担体粒子表面に見出され、それは、凸凹の間の広い間隔が微粉化薬剤粒子の機械的捕捉を可能にするからである。
【0025】
典型的なDPI製剤において、粉末はプレブレンドされ、それは微細及び粗担体粒子の間に自己接着をもたらす。微細担体粒子は、主に、粗担体粒子表面の溝及び割れ目による機械的捕捉によって自己接着する。したがって、微細粒子の量は物理的に除去され、担体粉末の流動特性は改善される。磨食(谷を埋めることを意味する地質学用語)は、起伏の少ない担体粒子表面をもたらし、それによって微粉化薬剤粒子は、担体粒子表面に機械的に捕捉又は埋め込まれることが少なくなる。磨食は、また、担体粒子表面の微小粗さを増加し、したがって、真接触領域の低減に起因して薬剤と担体粒子との間の接着力を低減する。しかし、接着力、したがって乾燥粉末吸入機能に関して、磨食は必ずしも有利ではないことが見出されている。粗担体粒子の最小表面粗さが、磨食の意味において微細担体粒子の埋め込みを可能にするために必要である。粗担体粒子表面が比較的平滑である場合、微細担体粒子は、担体粒子表面の見掛巨大粗さが増加するように自己接着し、そのことは、薬剤粒子が機械的に捕捉される部位をより多く提供する。この場合、薬剤粒子を、再懸濁の際に微細担体粒子を有する凝集塊としてのみ担体粒子表面から除去することができ、肺への薬剤の付着は、この凝集塊の大きさに応じて決まる。
【0026】
担体物質の選択は、薬剤と担体粒子との間の接着力の強さに明らかに影響を与える。しかし、適用箇所、すなわち、肺への吸入は、この選択肢を著しく制限する。現在まで、ラクトース一水和物及びグルコースが市販の乾燥粉末剤の吸入において担体物質として使用されている。グルコースは、55%を超える相対湿度の保存空気の環境に保存されると、急速に水分を吸収する。これは、薬剤と担体粒子との間に強力な毛細管力をもたらす。ラクトース一水和物は、湿度レベルを増加する薬剤−担体単位の弱点を低減したと主張されてきた。しかし、異なる湿度条件下で保存した後の微粉化薬剤とラクトース一水和物担体粒子と間の接着力測定は、この意見に対して疑いを投げかけている。
【0027】
相互作用粉末混合物の使用は、非常に低用量の吸入用薬剤(例えば、サルメテロールキキシナホ酸塩:50マイクログラム)の取扱いを容易にし、これらを1回の投与単位、例えばホイルブリスター(例えば、Advair Discus吸入器装置)又はカプセルで提供することができる。また、均質性の増加及びそのような混合物の分離の低減は、内容物にとって有利である。
【0028】
DPIの微細粒子を製造する2つ慣用の技術は、機械的微粉化及び噴霧乾燥である。高エネルギー粉砕操作は、高度に荷電された粒子を生成し、したがって非常に凝集性がある。凝集性を減少させるために、界面活性剤を例えば湿式粉砕に使用する。粉砕方法は、粉末安定性に影響を与える表面及び結晶学的損傷も導入する。
【0029】
製造された粒子は、多くの場合、強力な凝集体を形成することができる不規則な破片を含有する。加えて、多工程の加工は、粉末製造の際に材料の有意な損失を引き起こす場合があり、バッチ毎に生成物の特性に差異を引き起こす場合がある。粉砕と異なり、噴霧乾燥技術は、所望の大きさの医薬粒子を直接製造することができる1工程連続方法である。この方法には界面活性剤又は他の可溶化剤の必要がない。しかし、この方法に必要な高い流速、及び制御可能なパラメーターが限定されていることに起因して、それぞれの粒子の熱履歴及び乾燥速度は、制御することが困難である。それにより、製造された粒子は、通常は非晶質であり、したがって、粉末の保存の間に粒子の構造変化及び焼結を引き起こしうる温度及び湿度の変化に対して、影響されやすい。
【課題を解決するための手段】
【0030】
本発明の第1の態様によると、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、固体物質を、その固体物質が不溶性であるか又は難溶性である溶媒と接触させる工程と;その固体物質に、上記溶媒と接触したときに超音波を適用する工程とを含む方法が提供される。
【0031】
本発明の第2態様において、本明細書に記載されている方法により得られる、少なくとも1つの物質を含む粒子が提供される。また、本明細書に記載されている方法により得られる、少なくとも1つの粒状物質を含む粒子を有する製剤が提供される。
【0032】
そのような粒子及びそれらを含有する製剤は、特に、吸入可能な医薬製剤に有用である。そのような粒子及びそのような粒子を含む製剤は、従来どおりに調製された粒子と比較して驚くべきインビトロ性能を示す。この有意な性能増加は、細粒分(Fine Particle Fraction,FPF、送達用量に対する%、インピンジャー及び咽喉の全ての段階の合計として定義)の比例増加により定量化される。これらの粒子は、DPIの薬剤配合にとって優れた性能特性を有する。これらの粒子は、また、溶解速度及び肺に送達されたFPFに関して、従来の粒子と比較して驚くべきインビボ性能を示す。
【0033】
本明細書の以降では、固体物質が不溶性又は難溶性である溶媒を非溶媒と呼ぶ。本明細書で使用されるとき、非溶媒とは、固体物質が25℃で1mlあたり0.1mg未満、好ましくは25℃で1mlあたり0.05mg未満、好ましくは25℃で1mlあたり0.01mg未満の量で可溶性であるものである。
【0034】
逆に、本発明で使用されるとき、溶媒とは、固体物質が25℃で1mlあたり0.1mgより多く、好ましくは25℃で1mlあたり0.5mgより多く、好ましくは25℃で1mlあたり1mgより多く、好ましくは25℃で1mlあたり5mgより多く、好ましくは25℃で1mlあたり10mgより多くの量で可溶性であるものである。
【0035】
好ましくは、本発明において利用される固形物質は、粒状固形物質である。粒子は、好ましくは、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有する。
【0036】
空気力学的直径は、試験物質が粒子として空気力学的に挙動する、単位密度あたりの球体の直径である。異なる大きさ、形状及び密度の粒子を比較するため、並びにそのような粒子が気道のどこに付着しうるかを予測するために使用される。この用語は、それ自体気道内の付着と関連することがない実際の直径を表す、等体積球相当径、光学的直径、測定直径又は幾何学的直径に対比して使用される。
【0037】
多数の方法が、呼吸粒子の分布及び(程度は低いが)吸入粒子の分布を決定するために利用可能であり、粒径を示すために、平均中央空気力学的直径(MMAD)及び幾何学的標準偏差(GSD)を計算することができる。MMADは、粒子サンプルの統計由来の数字であり、例えば、5μmのMMADは、総サンプル質量の50%が、5μm未満の空気力学的直径を有する粒子として存在し、総サンプル質量の50%が、5μmより大きい空気力学的直径を有する粒子として存在することを意味する。
【0038】
Anderson Cascade lmpactor又はNext Generation Impactor、好ましくはNext Generation Impactorのようなカスケードインパクターを使用して、エアロゾル(又は砂塵雲)の粒度分布を得ることができる。粒子が例えばガラス又はガラス繊維に付着した幾つかの段階から構成される空気サンプルを、装置から取り出す。粒子は、その大きさに応じて、特定の段階に対してインパクトする。サンプル採取前後の各段階を計量することによって、各段階の噴射速度から分離粒径を計算することができ、MMADをこれらの計算から導くことができる。この方法の限界、すなわち粒子の跳ね返り、流速の過負荷及び変動などにもかかわらず、これは、エアロゾルの空中粒度分布及びそのMMADを測定する十分に確立された技術である。
【0039】
粒径は、レーザー回折技術により測定することができる。レーザーからの光線は、空気のような透明ガスの中に懸濁されている、粒子雲の中に照射される。粒子は光線を散乱し、小さな粒子は大きな粒子に比べて光線を大きな角度で散乱する。散乱光線を、異なる角度で設置された一連の光検出器により測定することができる。これはサンプルの回折パターンとして知られている。回折パターンを、十分に立証された光散乱理論を使用して粒子の大きさを測定するために使用することができる。粒子は球状であると想定されるが、実際にはほんの少しの粒子しか球状ではない。粒子の直径は、粒子の測定体積から計算されるが、等体積球相当径の球体であると想定される。
【0040】
好ましくは、本発明において利用される固体物質は、機械的微粉化、粉砕、噴射粉砕、摩砕、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法によって得られる。最も好ましくは、本発明において利用される固体物質は、噴霧乾燥の方法により得られる。従来の噴霧乾燥技術を使用することができる。好ましくは、WO2004/073827に開示されているようなSAX方法は、使用されない。
【0041】
好ましくは、上記の方法の1つを適用する前に、固体物質は実質的に非晶質であり、例えば、50%未満の結晶度、より好ましくは40%未満の結晶度、より好ましくは25%未満の結晶度、より好ましくは10%未満の結晶度、より好ましくは5%未満の結晶度、例えば1%未満の結晶度である。
【0042】
本発明において利用される固体物質が機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合により得られる場合、これらの方法の1つの前に、固体物質は、50%より高い結晶度、例えば60%より高い結晶度、例えば75%より高い結晶度、例えば90%より高い結晶度、例えば95%より高い結晶度、例えば99%より高い結晶度のように、実質的に結晶質であることができる。4つの方法の1つ又はこれらの混合の後、固体物質は、粒子の核が実質的に結晶質であり、粒子の外層が実質的に非晶質であることができる。
【0043】
多数の技術を使用して結晶質の内容を決定することができる。例えば、PXRD(粉末X線回折)は、固体物質のX線回折パターンを見る技術である。結晶質粒子は、個別の多形体に特有の「指紋」パターンを有する。逆に、非晶質化合物は、ほとんど又は全く特徴的なパターンを示さず、単に幅広の円丘又は雑音として表れる。示差走査熱量測定(DSC)も、所定のサンプルの結晶化度のレベルと等価でありうる、明確な融点及び融解熱測定値を明らかにする。非晶質物質は、DSCプロフィールと一致しない挙動を示す。結晶質物質のDSCは、結晶質を示す鮮明な吸熱を描く。動的蒸気吸着(DVS)は、結晶質及び非晶質物質の等温線及び吸湿挙動を測定する、急速で連続した方法を提供する。DSCと共にこれを使用して、生成物の安定性を測定することができる。最後に、ラマン分析は、結晶質物質を表示することができ、実際、異なる多形体を区別することができる。非晶質物質は、同じ特徴的なパターンを有さず、そのため結晶相と区別される。本出願の目的において、示差走査熱量測定(DSC)は結晶化度の測定に好ましい方法である。DSC実験は、TA InstrumentsのDSC Q2000 V24.2 build 107を含む多数の市販の装置により実施することができ、前者は、本発明のDCSを測定する好ましい器具である。典型的には、正確な量の物質をDSC器具のサンプルパンに導入し、およそ275℃になるまで、100℃/分までの加熱傾斜に付す。融解熱の測定値としての融点吸熱及び熱流の積分は、結晶化度の定性的及び定量的なそれぞれの測定値である。特に、所定の固体物質では、DSCは、2つのサンプルの直接的な比較を提供し、一方が他方よりも結晶度が多いか又は少ないかを明確に示す。
【0044】
追加的に又は代替的に、本発明の方法を適用する前に、固体物質は、準安定結晶質物質を含むことができる。
【0045】
任意の特定の物質では、当業者は、固体物質が不溶性又は難溶性であるかを容易に決定することができる。例えば、高速液体クロマトグラフィー(HPLC)又は気液クロマトグラフィー(GLC)は、既知の濃度の溶液に対して基準を作製し、澄んだサンプルを分析することにより、飽和したときの液体サンプル中の可溶化物質のレベルを決定することを可能にする。前者の方法は、医薬生成物のためにより典型的に使用され、一方、後者は、分析される物質が300℃までの温度で蒸発するのに十分に揮発性である場合に使用され、大部分の医薬生成物が除外される。好ましくは、水を水難溶性物質に非溶媒として使用する。水溶性物質には、好ましくは非溶媒炭化水素、例えばヘプタンが使用される。水溶性物質の更なる非溶媒には、適切であれば、エーテル(メチルtert−ブチルエーテル)、アルコール(エタノール)及びケトン(ブタノン)が含まれうる。
【0046】
超音波は、好ましくは、非晶質物質の少なくとも一部分が結晶質物質に変換される又は準安定物質がより安定した物質に変換されるのに必要な、適切な時間及び温度で適用される。例えば、方法は、好ましくは0.1ミリ秒より長く、より好ましくは1ミリ秒より長く、より好ましくは1分より長く、例えば1秒〜24時間、より好ましくは1分〜6時間、より好ましくは5分〜1時間の間実施される。
【0047】
好ましくは、本発明において使用される固体物質は乾燥している。このことは、好ましくは、非溶媒、水及び有機溶媒を含む溶媒を実質的に含有しないことを意味する。このことは、固体物質は実質的に水又は溶媒を含有しないことを意味する。溶媒を実質的に含有しないとは、固体物質が、5重量%未満の溶媒、より好ましくは4重量%未満、より好ましくは3重量%未満、より好ましくは2重量%未満、より好ましくは1重量%未満、より好ましくは0.5重量%未満、より好ましくは0.1重量%の溶媒を含有することを意味する。
【0048】
水和の水を含有する固体物質、及び分子溶媒和物は、結晶の単位格子を組み込むために必要な必須量の水又は溶媒のみを含有するので、実質的に溶媒無含有であるということができる。それ以外は、これらは実質的に水又は溶媒を含有しない。
【0049】
本発明の方法は、活性製薬学的成分、活性農芸化学的成分、製薬学的賦形剤、農芸化学的賦形剤及びこれらの2つ以上の適切な混合物からなる群より選択される物質を含む、噴霧乾燥粒子の加工に特に有用性を見出す。「適切な」とは、活性製薬学的成分を、他の活性製薬学的成分及び/又は製薬学的賦形剤と組み合わせうることを意味するが、通常は、製薬学的活性成分を例えば農芸化学的賦形剤と組み合わせることはない。
【0050】
本発明の好ましい実施態様において、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、
(i)溶媒中で、少なくとも1つの固体物質の溶液を形成する工程;
(ii)溶液を、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法に付し、溶解させた固体物質が実質的に乾燥固体物質に変換される工程;
(iii)任意に、固体物質を工程(ii)の方法の液体及び/又は気体成分から単離する工程;
(iv)工程(ii)又は工程(iii)の乾燥固体物質を非溶媒で処理する工程;
(v)工程(iv)の固体物質に、上記非溶媒と接触したときに超音波を適用する工程;及び
(vi)任意に、工程(v)で得られた固体物質を、分離及び/又は乾燥する工程
を含む方法が提供される。
【0051】
そのような方法において、工程(ii)は、好ましくは、固体物質の溶液の噴霧乾燥を含む。従来の噴霧乾燥を使用することができる。噴霧乾燥方法において、生成された固体物質は、通常、実質的に非晶質である。
【0052】
好ましくは、工程(ii)を適用する後に、工程(iii)又は(iv)に入る固体物質は実質的に非晶質であり、例えば、50%未満の結晶度、より好ましくは40%未満の結晶度、より好ましくは25%未満の結晶度、より好ましくは10%未満の結晶度、より好ましくは5%未満の結晶度、例えば1%未満の結晶度である。
【0053】
工程(iv)において、処理するという用語は、乾燥固体物質を非溶媒に暴露することを意味する。これを、工程(ii)により生成された物質を収集するのに使用されるものと同じ又は別の容器において実施することができる。好ましくは、非溶媒の量は、固体物質の量よりも多い。例えば、工程(iv)における固体物質と非溶媒の重量比は、好ましくは1:100、より好ましくは1:10、例えば、1:2、1:3、1:4、1:5などの範囲である。
【0054】
好ましくは、工程(ii)及び/又は工程(iii)により生成された固体物質は、実質的に乾燥している。これは、工程(iv)の方法に入る固体物質の好ましくは全て(100%)が、好ましくは、水及び有機溶媒を含む溶媒を実質的に含有しないことを意味する(ここで用語「溶媒を実質的に含有しない」は、上記に定義されている)。
【0055】
任意の所定の固体物質において、当業者は、負担となることなく適切な溶媒を決定することができる。特定の固体物質に適した溶媒の幾つかの例は以下の通りである。メタノール、エタノール、ジクロロメタン、酢酸エチル、アセトン、2−プロパノールのような揮発性有機溶媒及び水のような非有機溶媒は、製薬学的に活性な成分の典型的な溶媒である。
【0056】
好ましい賦形剤には、例えば、ラクトース及びステアリン酸が含まれうる。ラクトースは、水又はエタノール/水混合物に溶解しうる。ステアリン酸は、酢酸エチル又はエタノールに溶解しうる。
【0057】
非溶媒(例えば、工程(iv)の方法のもの)は、好ましくは、無水結晶が望ましい場合には実質的に遊離の水(すなわち、予め固体物質と結合して水和物などを形成していない水が)を含有せず、また、固体物質が実質的に可溶性である任意の溶媒も含有しない。非溶媒は、固体物質が実質的に不溶性である場合、水であることができる。
【0058】
網羅的なリストではないが、溶媒及び非溶媒の組み合わせの幾つかの例を表1に示す。
【0059】
【表1】

【0060】
本発明の粒子を調製するのに適した他の非溶媒には、1,1−ジフルオロエタン、1,1,1−トリフルオロエタン、1,1,1,2−テトラフルオロエタン、ペンタフルオロエタン、1,1,1,3,3−ペンタフルオロプロパン、1,1,1,3,3,3−ヘキサフルオロプロパン、1,1,1,2,3,3,3−ヘプタフルオロプロパン、1,1,1,3,3−ペンタフルオロブタン及び1,1,1,2,3,4,4,5,5,5−デカフルオロペンタンからなる群より選択されるヒドロフルオロアルカンが含まれる。そのような非溶媒の使用は、PMDIに使用される直接配合を促進する。別の実施態様において、ペルフルオロデカリンのような揮発性の低いフッ素化化合物を非溶媒として使用することができる。
【0061】
工程(i)の方法で形成された溶液中の固体物質(好ましくは、製薬学的に許容される物質、製薬学的に許容される賦形剤又はこれらの混合物)の濃度は、好ましくは10mg/ml〜800mg/ml、より好ましくは50mg/ml〜600mg/mlの範囲、より好ましくは100mg/ml〜400mg/mlである。
【0062】
本発明の方法の間、非溶媒の温度は、好ましくは−10℃〜+120℃の間にあり、非溶媒が液体形態のままであることが前提である。好ましくは、非溶媒の温度は、好ましくは0〜80℃、より好ましくは20℃〜60℃の間にある。
【0063】
好ましくは、上記の方法は、順次に行われ、工程(iv)及び(v)は、工程(ii)の直後(又は行われる場合は、任意の工程(iii)の直後)に実施される。「直後」とは、工程(ii)(又は行われる場合は、工程(iii))の噴霧乾燥粒子が工程(ii)を受けてから1時間以内、好ましくは工程(ii)を受けてから30分以内、好ましくは5分以内、好ましくは1分以内に工程(iv)及び(v)において加工されることを意味する。好ましくは「直」とは、任意の中間工程がないことを意味する。好ましくは、上記の方法は、連続方法である。例えば、方法は、未加工物質を連続的に供給することができ、加工物質を連続的に又は漸増的に取り出すことができる。あるいは、方法は、バッチ型方法であることができ、ここで方法は、未加工物質をバッチ様に供給し、加工物質をバッチで取り出すことができる。
【0064】
あるいは、工程(ii)及び工程(iii)を、工程(iv)の6か月前、より好ましくは3か月前、より好ましくは1か月前、より好ましくは1週間前、より好ましくは1日前のように、工程(iv)の前に実施することができる。
【0065】
工程(v)で得られた固体物質を、本明細書のこれ以降、「活性粒子」と呼ぶ場合がある。
【0066】
本発明の更なる実施態様において、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、
(a)少なくとも1つの固体物質を、機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合に付す工程;
(b)工程(a)で得られた固体物質を非溶媒で処理する工程;
(c)工程(b)で得られた固体物質に、非溶媒と接触したときに超音波を適用する工程;及び
(d)任意に、工程(c)で得られた固体物質を、分離及び/又は乾燥する工程
を含む方法が提供される。
【0067】
工程(a)を適用した後、工程(b)に用いられる物質は、例えば50%より高い結晶度、例えば60%より高い結晶度、例えば75%より高い結晶度、例えば90%より高い結晶度、例えば95%より高い結晶度、例えば99%より高い結晶度、又は、例えば50%未満の結晶度、例えば40%未満の結晶度、例えば25%未満の結晶度、例えば10%未満の結晶度、例えば5%未満の結晶度、例えば1%未満の結晶度である。
【0068】
工程(b)において、処理するという用語は、乾燥固体物質を非溶媒に暴露することを意味する。これを、工程(a)により生成された物質を収集するのに使用されるものと同じ又は別の容器において実施することができる。好ましくは、非溶媒の量は、固体物質の量よりも多い。例えば、工程(b)における固体物質と非溶媒の重量比は、好ましくは1:100、より好ましくは1:10、例えば、1:2、1:3、1:4、1:5などの範囲である。
【0069】
工程(b)は、工程(a)の直後に実施することができ、ここで「直後」とは、上記に定義されたとおりである。あるいは、工程(a)を、工程(b)の6か月前、好ましくは3か月前、より好ましくは1か月前、より好ましくは1週間前のように、工程(b)の前に実施することができる。
【0070】
吸入により肺深部又は血流に到達する製剤において、製剤の活性剤は、非常に微細な粒子の形態、例えば10μm未満の平均中央空気力学的直径(MMAD)を有さなければならない。10μmを超えるMMADを有する粒子は、咽喉の壁に衝突する可能性があり、一般に肺に到達しないことが十分に確立されている。5〜2μmの範囲のMMADを有する粒子は、一般に呼吸細気管支に付着し、一方、3〜0.05μmの範囲のMMADを有する粒子は、肺胞に付着し、血流に吸収される可能性がある。
【0071】
理想的には、乾燥粉末製剤の活性粒子は、10μm以下、好ましくは5μm以下、より好ましくは3μm以下、より好ましくは2.5μm以下、より好ましくは2.0μm以下、より好ましくは1.5μm以下又は好ましくは1.0μm以下のMMADを有するべきである。
【0072】
重要性が高いのは、乾燥粉末剤吸入における組成である。乾燥粉末剤吸入器(DPI)において、活性粒子(1〜5μm)とラクトースのような粗担体粒子(50〜500μm)の混合物を使用して、効果的な薬剤粒子投入を得ることができる。
【0073】
噴霧乾燥粒子は、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有する。
【0074】
方法の最終生成物である活性粒子も、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有することができる。
【0075】
本発明の方法に使用される超音波の周波数は、好ましくは、16kHz〜1MHz、好ましくは10〜500kHz、より好ましくは10〜100kHzの範囲、例えば10、20、40、60、80若しくは100kHz又はその間の任意の周波数である。
【0076】
本発明の方法によって生成される固体物質の結晶化度を増加させることに加えて、超音波の適用は、凝集粒状物質の量を低減するために使用することもできる。この凝集低減は、好ましくは、上記に言及した工程(v)又は(c)と同時に実施される。
【0077】
超音波照射に付される、非溶媒と接触する固体物質の非晶質、部分的に非晶質又は準安定結晶質形態の種類に応じて、粒子は、より小型の形態及び/又はより安定した形態に変換されうる。例えば、活性成分はより安定した結晶質形態に変換されうるか、又は超音波照射の前に粒子が不安定な非晶質形態で存在する物質であると、より安定した非晶質形態に変換されうる。非溶媒と接触したときに粒子がどのような形態であろうとも、本明細書において概説した超音波照射が適用されたとき、粒子特性は変化し、より安定した粒子の形成をもたらし、これらをより効率的な方法で医薬又は農薬用途のような他の用途に使用することができる。好ましくは、この方法で得られた粒子は、高度に結晶質であり安定している。
【0078】
超音波照射工程が適用されると、粒状スラリーからの結晶の単離を、濾過、遠心分離、噴霧乾燥、超臨界二酸化炭素抽出、単なる蒸発又はこのような技術の2つ以上の混合のような、任意の従来の方法により実施することができる。典型的には、結晶は従来の蒸発方法を使用して単離される。
【0079】
本発明の方法において噴霧乾燥条件及び超音波処理方式を操作することによって、発明者たちは、今、所定の特性を有する結晶又は非晶質体を提供することを可能とした。噴霧乾燥物質を非溶媒中で超音波により所定の時間及び温度で処理することによって、特定の特性を再現性をもって得ることができる。これらの特性には、粒子形態、表面自由エネルギー粒径分布、所望の多形体、並びに単離粒子の流動性に関して低減された静電気及び凝集性/接着性が含まれうる。
【0080】
固体物質、好ましくは本発明の方法に付される粒状固体物質は、好ましくは、結晶を形成することができるか又は粒子のより安定した形態をもたらす形態の変化を受けることができる、活性成分又は薬剤若しくはプロドラッグ若しくは目的の農薬のような所望のその前駆体である。典型的には、そのような改質粒子は、従来の薬剤送達ビヒクル又は少なくとも1個の所定の改質粒子のために特に設計されうる薬剤送達ビヒクルのような所望の状況における使用のために、より作製し易くなる物理的特性を有する。本明細書において示唆されているように、従来の噴霧乾燥により調製された初期溶液(又は本明細書に参照された他の方法技術のいずれかの初期溶液又は固体物質)には、2個以上の目的の粒子の混合物のような、1個以上の目的の粒子が含まれうる。そのような状況において、超音波処理後の所望の最終用途に応じて、2つ以上の目的の活性成分、又は、少なくとも1つのプロドラッグと少なくとも1つの薬剤若しくは2つ以上の薬剤若しくは2つ以上の農薬との組み合わせが、溶質として又は初期固体物質として初期溶液に存在することができる。本発明の方法条件下で結晶化することができる適切な粒子は、コルチコステロイド、β2アゴニスト、抗コリン作用剤、ロイコトリエンアンタゴニスト、吸入タンパク質又はペプチド、フランカルボン酸モメタゾン;ジプロピオン酸ベクロメタゾン;ブデソニド;フルチカゾン;デキサメタゾン;フルニソリド;トリアムシノロン;サルブタモール;アルブテロール;テルブタリン;サルメテロール;ビトルテロール;イプラトロピウム臭化物;オキシトロピウム臭化物;クロモグリク酸ナトリウム;ネドクロミルナトリウム;ザフィルルカスト;プランルカスト;ホルモテロール;エホルモテロール;バンブテロール;フェノテロール;クレンブテロール;プロカテロール;ブロキサテロール;(22R)−6a,9a−ジフルオロ−llb,21−ジヒドロキシ−16a,17a−プロピルメチレンジオキシ−4−プレグネン−3,20−ジオン;TA−2005;チプレダン;インスリン;インターフェロン;カルシトニン;副甲状腺ホルモン;及び顆粒球コロニー刺激因子のような、本明細書の方法により結晶質粒子に形成されうる活性成分又は薬剤を含む。
【0081】
2つ以上の固体物質が使用される場合、共結晶が形成されうる。共結晶は、2つ以上の同一ではない中性分子成分の結晶複合体として定義することができ、例えば2つ以上の同一ではない中性分子成分は、活性主成分又はその所望の前駆体、及び、非共有結合、好ましくは主に水素結合を通じて、結晶格子中で互いに結合しているゲストである。ゲストは、別の活性主成分若しくはその所望の前駆体、又は共結晶形成剤であることができる。
【0082】
医薬共結晶の形成には、別の製薬学的に許容される分子と、所定の活性医薬とを、結晶格子中に組み込むことを伴う。得られた多成分結晶相は、親活性医薬の固有の活性を維持し、同時に特有の生理化学的プロフィールを有する。
【0083】
本明細書で使用されるとき、用語「共結晶形成剤」は、活性主成分又はその所望の前駆体として同じ結晶構造に存在する1つ以上の追加の分子を意味し、1つ以上の追加の分子は、共結晶中の分子間相互結合作用によって、活性主成分又はその所望の前駆体と超分子シントンを形成することができる。
【0084】
一つの実施態様において、共結晶形成剤は、以下の群から選択される少なくとも1つのシントン形成部分を有する少なくとも1つの分子を含む:エーテル、チオエーテル、アルコール、カルボニル、チオール、アルデヒド、ケトン、チオケトン、硝酸エステル、リン酸エステル、チオリン酸エステル、エスエル、チオエステル、硫酸エステル、カルボン酸、ホスホン酸、ホスフィン酸、スルホン酸、スルホンアミド、アミド、第一級アミン、第二級アミン、アンモニア、第三級アミン、イミン、チオシアネート、シアンアミド、オキシム、ニトリル、ジアゾ、有機ハロゲン化物、S含有複素環(例えば、チオフェン)、N含有複素環(例えば、ピロール、イミダゾール又はピリジン)、O含有複素環(例えば、フラン、エポキシド又はペルオキシド)及びヒドロキサム酸部分。
【0085】
更なる実施態様において、ゲストは、例えば活性主成分又はその所望の前駆体と共結晶を形成するために存在することができる。1つ以上のゲストが共結晶に含まれうることが考慮される。したがって、ゲストはそれ自体の活性を有する必要がないが、活性剤の所望の活性を過度に減じない幾つかの活性を有することができる。非活性ゲストは、有益な薬理学的活性を示したことがない、明らかに生物学的に非毒性であるか又は薬理学的に良性である、化合物であることができる。幾つかの場合において、ゲストは、活性剤と同一の活性又は相補的な活性を有することができる。ゲストは、別の活性主成分又はその所望の前駆体であることができる。例えば、幾つかのゲストは、活性主成分又はその所望の前駆体の治療効果を促進することができる。医薬製剤において、ゲストは、活性主成分又はその所望の前駆体又はそれらの塩と共結晶を形成する任意の製薬学的に許容される分子であることができる。
【0086】
ゲスト又は共結晶形成剤は、酸であることができ、両方とも中性態様であるが、シュウ酸又は適切なカルボン酸のような場合では、カフェインとの共結晶として調製されるとき、非共有相互作用(主に水素結合)により挙動することができ、例えばアミンとの反応又はプロトン交換のようなイオン塩を形成する場合では、プロトン供与体として挙動することができる。同様に、安息香酸及びコハク酸は、フルオキセチン塩酸塩と共結晶を形成する場合では、中性態様で挙動し(ホルマールプロトン交換なし)又は安息香酸ナトリウム若しくはコハク酸ナトリウムのようなイオン塩を形成するプロトン交換態様で挙動する。これらの化合物は、それ自体イオン性のゲストであることができる。中性ゲストは、好ましくは非イオン性のゲストである。イオン性ゲストは、イオン結合を有する化合物又は錯体である。ゲストは、塩化物(又は他のアニオン)と水素結合を形成する酸であることができる。イオン性ゲストは、イオン相互作用及び引力により例示されるようなイオン性を有する化合物又は錯体である。ゲストは、医薬成分と水素結合を形成する酸であることができる。例えば、酸である適切なゲストには、アスコルビン酸、グルコヘプタン酸、セバシン酸、アルギン酸、シクラミン酸、エタン−1,2−ジスルホン酸、2−ヒドロキシエタンスルホン酸、2−オキソ−5−グルタル酸、ナフタレン−1,5−ジスルホン酸、ニコチン酸、ピログルタミン酸及び4−アセトアミド安息香酸が含まれる(がこれらに限定されない)。本出願に提示されている溶質及び活性主成分には、その塩及び/又は溶媒和物が含まれる。共結晶は、WO2005/089375に記載されている。
【0087】
本発明の共結晶の例は、シルデナフィル又はその製薬学的に許容される塩、及びアセチルサリチル酸(アスピリン)である。
【0088】
本発明により作製されうる他の粒子は、鎮痛薬、例えばコデイン、ジヒドロモルヒネ、エルゴタミン、フェンタニル又はモルヒネ;アンギナール調合剤、例えばジルチアゼム;抗アレルギー薬、例えばクロモグリク酸、ケトチフェン又はネドクロミル;抗感染症薬、例えばセファロスポリン、ペニシリン、ストレプトマイシン、スルホンアミド、テトラサイクリン又はペンタミジン;抗ヒスタミン薬、例えばメタピリレン;抗炎症薬、例えばベクロメタゾン、フルニソリド、ブデソニド、チプレダン、トリアムシノロン、アセトニド又はフルチカゾン;鎮咳薬、例えばノスカピン;気管支拡張薬、例えばエフェドリン、アドレナリン、フェノテロール、ホルモテロール、イソプレナリン、メタプロテレノール、フェニレフリン、フェニルプロパノールアミン、ピルブテロール、レプロテロール、リミテロール、サルブタモール、 サルメテロール、テルブタリン、イソエタリン、ツロブテロール、オルシプレナリン又は(−)−4−アミノ−3,5−ジクロロ−a[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]メチル]ベンゼンメタノール;利尿薬、例えばアミロライド;抗コリン作用薬、例えばイプラトロピウム、アトロピン又はオキシトロピウム;ホルモン、例えば コルチゾン、ヒドロコルチゾン又はプレドニゾロン; キサンチン、例えばアミノフィリン、コリンテオフィリン、リシンテオフィリン酸塩又はテオフィリン;並びに治療タンパク質及びペプチド、例えばインスリン又はグルカゴンのような吸入により有用に送達されうる任意の薬剤又は活性成分を含む。活性成分又は薬剤を含む適切な医薬を、塩の形態で(例えば、アルカリ金属若しくはアミン塩として又は酸付加塩として)、又はエステル(例えば、低級アルカリエステル)として、又は溶媒和物(例えば水和物)として使用して、医薬の活性及び/又は安定性を最適化できることが、当業者には理解される。
【0089】
本発明の方法により得られる粒子の調製に特に適した医薬には、吸入治療による喘息のような呼吸障害の治療において使用される抗アレルギー薬、気管支拡張薬及び抗炎症性ステロイド、例えば、クロモグリク酸(例えば、そのナトリウム塩)、サルブタモール(例えば、その遊離塩基若しくはその硫酸塩)、サルメテロール(例えば、そのキシナホ酸塩)、テルブタリン(例えば、その硫酸塩)、レプロテロール(例えば、その塩酸塩)、ジプロピオン酸ベクロメタゾン(例えば、その一水和物)、フルチカゾンプロピオン酸エステル、(−)−4−アミノ−3,5−ジクロロ−α−[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]−メチル]ベンゼンメタノール、グリコピロニウム臭化物、ダロトロピウム、アクリジニウム、チオトロピウム(例えば、その臭化物塩)、テオフィリン、アロフィリン、ザフィルルカスト、モンテルカスト、カルモテロール(例えば、その塩酸塩)、ホルモテロール(例えば、そのフマル酸塩)又はインダカテロール、並びにその生理学的に許容される塩及び溶媒和物が含まれる。
【0090】
好ましい実施態様において、固体物質は、フルチカゾン化合物(例えば、そのプロピオン酸エステル又はフランカルボン酸エステル)とサルメテロール(例えば、そのフマル酸塩)の混合物を含む。
【0091】
ここでも、本発明の方法により作製される粒子は、本明細書において示唆されている2つ以上の活性成分の組み合わせを含有できることが、当業者には理解される。活性成分は、本明細書前記に記述された活性成分の適切な組み合わせから選択することができる。したがって、適切な気管支拡張性剤の組み合わせには、エフェドリンとテオフェリン、フェノテロールとイプラトロピウム及びイソエタリンとフェニレフリンが含まれる。
【0092】
本発明の方法により作製される活性成分の粒子の更なる適切な組み合わせには、ブデソニド、ジプロピオン酸ベクロメタゾン及びフルチカゾンプロピオン酸エステルのようなコルチコステロイドと、サルブタモール、テルブタリン、サルメテロール及びホルモテロールのようなβ2アゴニスト、並びにそれらの生理学的に許容される誘導体、特に硫酸塩を含む塩との組み合わせが含まれる。
【0093】
本発明の方法により作製される活性成分の粒子の特に適切な組み合わせには、ホルモテロールとフルチカゾン;ベクロメタゾンとホルモテロール;ホルモテロールとメタゾン;インダカテロールとメタゾン;イプラトロピウム臭化物とアルブテロール;サルブタモールとアルブテロール;チオトロピウム臭化物とホルモテロール;グリコピロニウム臭化物とインダカテロール;ホルモテロールとシクレソニド;ベクロメタゾン/サルメテロールのような組み合わせが含まれる。
【0094】
別の実施態様において、3つの成分を組み合わせることができ、コルチコステロイド、気管支拡張薬(例えば、ベータアゴニスト)及び抗コリン作用薬の組み合わせが含まれる。一例は、フルチカゾン/サルメテロール/チオトロピウム臭化物である。
【0095】
本発明の方法により得られる粒子の他の例は、クロモグリク酸ナトリウム若しくはネドクロミルでありうるクロモン又は炭水化物、例えばヘパリンを含むことができる。
【0096】
本発明の方法により作製される粒子は、吸入に適した活性成分を含むことができ、全身用途の薬理学的活性剤であることができる。例えば、そのような活性粒子は、デオキシリボヌクレアーゼ(DNase)、ロイコチン又はインスリン(プロインスリンを含む)、シクロスポリン、インターロイキン、サイトカイン、抗サイトカイン及びサイトカインレセプター、ワクチン、成長ホルモン、ロイプロリド及び関連する類似体、インターフェロン、デスモプレシン、免疫グロブリン、エリトロポエチン及びカルシトニンのような、ペプチド又はポリペプチド又はタンパク質を含むことができる。
【0097】
あるいは、本発明の方法により作製される活性成分は、経口投与に適している場合がある。経口投与用薬剤は、上記に記述した全身用薬剤の1つでありうる。活性成分は、消化管において低い可溶性を示す物質、例えば、三ケイ酸マグネシウム、炭酸カルシウム及び次硝酸ビスマスであることができる。有機化合物は、例えば、コンビナトリアル化学、ロシグリタゾン及び他の関連するグリタゾン薬、ヒドロクロロチアジド、グリセオフルビン、ラミブジン及び他のヌクレアーゼ逆転写インヒビター、シンバスタチン及び他のスタチン薬、ベザフィブレート及び他のフィブレート薬、ロラタジン、並びに他の任意の生理学的に許容される塩及びそれらの誘導体の全ての生成物を含むことができる。
【0098】
本発明の方法による加工に適した製薬学的賦形剤には、例えば、炭水化物、特にフルクトース、グルコース及びガラクトースのような単糖類;スクロース、ラクトース及びトレハロースのような非還元二糖類;ラフィノース及びメレジトースのような非還元オリゴ糖類;マルトデキストリン、デキストリン及びシクロデキストリンのような非還元デンプン由来多糖類生成物;並びにマンニトール及びキシリトールのような非還元アルジトールが含まれる。更に適した賦形剤には、例えばトウモロコシデンプン、コムギデンプン、コメデンプン、ジャガイモデンプン、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ナトリウムカルボキシメチルセルロース及び/又はポリビニルピロリドン(PVP)のようなセルロース調製物が含まれる。上記の賦形剤のいずれかの2つ以上の混合物も考慮される。
【0099】
医療における使用では、本発明の化合物の塩は、非毒性の「製薬学的に許容される塩」と呼ばれる。FDAにより認証された製薬学的に許容される塩形態(International J. Pharm. 1986, 33,201 217; J. Pharm. Sci, 1977, Jan, 66 (1), p1)には、製薬学的に許容される酸性/アニオン性又は塩基性/カチオン性の塩が含まれる。
【0100】
本発明の酸性又は塩基性化合物の製薬学的に許容される塩は、当然のことながら、遊離塩基又は酸を少なくとも1つの理論量の所望の塩形成酸又は塩基と反応させるような、従来の手順により作製することができる。
【0101】
本発明の酸性化合物の製薬学的に許容される塩には、ナトリム、カリウム、カルシウム、マグネシウム、亜鉛及びアンモニウムのような無機カチオンとの塩、並びに有機塩基との塩が含まれる。適切な有機塩基には、NメチルDグルカミン、アルギニン、ベンザチン、ジオラミン、オーラミン、プロカイン及びトロメタミンが含まれる。
【0102】
本発明の塩基性化合物の製薬学的に許容される塩には、有機又は無機酸から誘導される塩が含まれる。適切なアニオンには、酢酸塩、アジピン酸塩、ベシル酸塩、臭化物、カンシラート、塩化物、クエン酸塩、エジシル酸塩、エストレート、フマル酸塩、グルセプト酸、グルコン酸塩、グルクロン酸塩、馬尿酸塩、ヒクレート、臭化水素酸塩、塩酸塩、ヨウ化物、イセチオン酸塩、乳酸塩、ラクトビオン酸塩、マレイン酸塩、メシレート、臭化メチル、硫酸メチル、ナプシレート、硝酸塩、オレイン酸塩、パモ酸塩、リン酸塩、ポリガラクツロ酸塩、ステアリン酸塩、コハク酸塩、硫酸塩、スルホサリチレート、タンニン酸塩、テレフタル酸塩、トシレート及びトリトヨージドが含まれる。
【0103】
本発明の方法により調製される活性成分の粒子が農芸化学的に活性である場合、活性成分は、例えば、植物成長調整剤、除草剤、及び/又は病虫害防除剤、例えば殺虫剤、殺真菌剤、ダニ駆除剤、殺線虫剤、殺ダニ剤、殺鼠剤、殺菌剤、軟体動物駆除剤又は鳥忌避剤であることができる。
【0104】
本発明の方法により作製される有機水不溶性農芸化学的活性成分の例には、例えば、メトミル、カルバリル、カルボフラン又はアルディカーブのようなカルバメート;EPN、イソフェンホス、イソキサチオン、クロルピリホス又はクロルメホスのような有機チオリン酸塩;テルブホス、モノクロトホス又はテトラクロルビンホスのような有機リン酸塩;メトキシクロルのような過塩素過有機化合物;フェンバレレートのような合成ピレスロイドからなる群より選択される殺虫剤;例えば、メトリブジン、ヘキサキシノン又はアトラジンのようなトリアジン;2−クロロ−N−[(4−メトキシ−6−メチル−1,3,5−トリアジン−2−イル)アミノカルボニル]−ベンゼンスルホンアミドのようなスルホニル尿素;レナシル、ブロマシル又はテルバシルのようなウラシル(ピリミジン);リニュロン、ジウロン、シズロン又はネブロンのような尿素;アラクロール又はメトラクロールのようなアセトアニリド;ベンチオカルブ(SATURN)、トリアレートのようなチオカルバメート;オキサジアゾンのようなオキサジアゾール−オン;2,4−Dのようなフェノキシ酢酸;フルアジホップ−ブチル、アシフルオルフェン、ビフェノックス又はオキシフルオルフェンのようなジフェニルエーテル;トリフルラリンのようなジニトロアニリン;グリフォセート塩及びエステルのようなグリシンホスホネート;ブロモキシニル又はイオキシニルのようなジハロベンゾニトリルからなる群より選択されるオキサミル除草剤のような殺線虫剤カルバメート;例えば、シモキサニル(クルゼート)のようなニトリロオキシム;ベノミル、カルベンダジム又はチオフェネート−mエチルのようなイミダゾール;トリアジメホンのようなトリアゾール;カプタンのようなスルフェンアミド;マネブ、マンコゼブ又はチラムのようなジチオカルバメート;クロロネブのような塩素過芳香族化合物;イプロジオンのようなジクロロアニリンからなる群より選択される殺真菌剤;ピリミカーブのようなカルバメートからなる群より選択されるアブラムシ駆除剤;,例えば、プロパルギットのような亜硫酸プロピニル;アミトラズのようなトリアザペンタジエン;クロロベンジレート又はテトラジホンのような塩素過芳香族化合物;及びビナプアクリルのようなジニトロフェノールからなる群より選択される殺ダニ剤が挙げられる。
【0105】
有機水不溶性農芸化学的活性成分は、幾つかの成分の混合物として、本発明により製造される粒子に含まれうる。特に好ましい有機水不溶性農薬活性成分は、アトラジン、シモキサニル、クロロタロニル、シプロコナゾール及びテブコナゾールである。
【0106】
非溶媒及び溶媒は、特定の活性成分又は活性前駆体に適しているように選択されるべきであることが理解される。ブデソニド、ジプロピオン酸ベクロメタゾン及びフルチカゾンプロピオン酸エステルのようなコルチコステロイドを、ジクロロメタン又はメタノールに溶解し、ヘプタンのような非溶媒中で超音波処理することができる。サルメテロールキシナホ酸塩及びサルメテロールフマル酸塩のようなβ2−アゴニストをメタノールに溶解し、アセトン、酢酸エチル又はヘプタンのような非溶媒中で超音波処理することができる。
【0107】
サイクロン分離のような従来の分離工程に従い、乾燥粒子を非溶媒と接触させ、次に超音波照射に付して、結晶を形成するか又は本明細書前記に記載した所望のMMADの非晶質構造を焼還及び/若しくは安定化する。粒子を、使用される場合は超音波プローブの又はこの構造が用いられる場合は巻き付け超音波エネルギー変換器アセンブリーのような超音波エネルギー変換器の稼働近辺に付される。そのような装置の適切な例は、WO00/35579において確認されている。超音波エネルギーを、連続的方法又はパルス適用のように非連続的方法により適用することができる。任意の適切な超音波照射供給源を使用することができる。連続超音波フローセルのような超音波プローブを、例えば混合容器の中に挿入することができるか、超音波発信器を混合容器の中に含めることができるか、或いは混合容器を超音波浴の中に収容することができるか又は超音波変換器を混合容器の外壁に固定することができる。超音波の振幅及び周波数は、核生成及び結晶成長に影響を与える。超音波の周波数は、例えば、16kHz〜1MHz、好ましくは10〜500kHz、より好ましくは10〜100MHz、例えば、10、20、40、60、80若しくは100kHz又は30kHz〜50kHzのようなその間の任意の周波数であることができる。
【0108】
超音波照射は、所定の用途において、所望の大きさの結晶の生成に適した振幅又は出力密度で用いられる。発射面が例えば80cm2の実験室プローブ系では、選択される振幅は、約1〜30μm 、典型的には3〜20μm 、好ましくは5〜10μm、例えば6μmであることができる。8cm2のプローブ面及び5〜80Wの所要電力を有するプローブは、2〜15μmの振幅を使用して約0.6〜12.5W/cm2の出力密度をもたらす。好ましくは、フローセル、例えば6リットルのフローセルに結合している変換器を含むWO03/101577に記載されているような大型の系では、用いられる変換器の出力密度は、10〜100W/L、好ましくは30〜80W/L、より好ましくは50〜75W/L、例えば60W/L〜70W/Lであることができる。本発明は、工業規模の生産に特に適している。
【0109】
超音波フローセルにおける混合成分の滞留時間は、好ましくは0.1ミリ秒より長く、より好ましくは1ミリ秒より長く、より好ましくは1分より長く、例えば1秒〜24時間、より好ましくは1分〜6時間、より好ましくは5分〜1時間であることができる。
【0110】
生成された結晶を、当該技術の従来の方法を使用して結晶を引き出すことによりバッチチャンバーから又は水性懸濁剤として収集又は採取することができる。
【0111】
本発明により生成される粒子は、実質的に結晶質であり、低減された吸湿の傾向を示し、このこと物理的及び化学的安定性を増加することに寄与する。「実質的に結晶質」とは、粒子の総重量に対して結晶粒子重量%として表される粒子の結晶化度が、90%より高く、好ましくは93%より高く、さらにより好ましくは95%より高いことを意味する。前記粒子は、特に粒子が吸入用の乾燥粉末剤として配合される場合、均質の製剤を容易に得ることを可能にする優れた分散特性も示す。粒子の結晶化度は、示差走査熱量測定(DSC)、X線粉末回折又はマイクロ熱量測定法のような当業者に既知の他の技術、好ましくはDSCを使用して決定することができる。
【0112】
一つの実施態様において、固体物質は、コルチコステロイドであり、好ましくは、呼吸疾患の予防及び/又は治療のために吸入用にとって有用でありうる、ヨーロッパ薬局方、第4版、2002年に示されている溶解度の定義に従って水において不溶性又は難溶性である任意のコルチコステロイドである。好ましくは、コルチコステロイドは、50μgを超える、好ましくは80μg以上、より好ましくは100μg以上の単回治療用量を有する。
【0113】
好ましくは、コルチコステロイドは、50μgを超える、好ましくは80μg以上、より好ましくは100μg以上の単回治療用量を有する。
【0114】
好ましくは、コルチコステロイドは、ジプロピオン酸ベクロメタゾン(BDP)、ブデソニド、シクレソニド、モメタゾン及びフロン酸エステルのようなそのエステル、並びにフルチカゾン及びプロピオン酸エステルとフロン酸エステルのようなそのエステルからなる群より選択される。本発明の好ましい実施態様において、コルチコステロイドは、ブデソニド又はフルチカゾン及びその塩又はエステルである。
【0115】
好ましくは、本発明の活性粒子は10μm未満の体積直径を有し、より好ましくは、所定の組成物において少なくとも90重量%の活性成分粒子は、上記に記載されたレーザー回折により、好ましくはMalvern又は同等の装置を使用して、体積直径として知られている特徴的な等体積球相当径を測定して決定された10μm以下の直径を有する。考慮されるパラメーターは、粒子の大きさ独立密度を推定する質量直径に対応する、それぞれd(10)、d(50)及びd(90)と表される10%、50%及び90%の粒子の体積直径(VD)(μm)である。
【0116】
好ましくは、10重量%以下の前記粒子が、0.8μm未満の体積直径d(10)を有し、好ましくは、50重量%以下の前記粒子が、2.0μm未満の体積直径d(50)を有し、好ましくは、90重量%以下の前記粒子が、10μm以下の体積直径d(90)を有する。好ましくは、100重量%の前記粒子が、10μm以下の体積直径を有する。
【0117】
本発明の粒子の活性成分は、実質的に純粋な形態である。「実質的に純粋な形態」とは、少なくとも95%w/w、好ましくは少なくとも98%又は少なくとも99%w/wの純度を意味する。化学的な純度は、高速液体クロマトグラフィー(HPLC)のような当業者に既知の方法に従って決定することができる。
【0118】
別の態様において、本発明は、本発明の粒子を含む、吸入による投与用の製剤を提供する。粒子を、1つ以上の製薬学的に許容される賦形剤、添加剤、希釈剤又は担体と一緒に、前記製剤に配合することができる。例えば、製剤は、加圧式定量噴霧吸入器(pMDI)により投与される、エアロゾル担体としての噴射剤中の懸濁形態で提供される。
【0119】
懸濁製剤は、界面活性剤及び湿潤剤のような追加の賦形剤を含むことができる。
【0120】
好ましい実施態様において、製剤は、乾燥吸入粉末剤の形態で、より好ましくは相互作用規則混合物の形態で、すなわち、本発明の粒子を、粗粒子からなる薬理学的に不活性で生理学的に許容される賦形剤により希釈することによって、提供される。
【0121】
有利には、吸入用の前記粉末製剤は、本発明の粒子と、生理学的に許容される賦形剤の粗粒子(本明細書以降、「担体粒子」と呼ぶ)、例えば50μmより大きい中央粒子径(MMD)、好ましくは50μm〜500μm、より好ましくは150〜400μm、さらにより好ましくは210〜355μmの中央粒子径(MMD)を有する粒子とを含むことができる。別の実施態様において、粗粒子は、90〜150μmを含むMMDを有する。MMDは、頻度分布を半分に割った粒子直径であり、エアロゾル質量の50パーセントが、大きな直径の粒子を有し、エアロゾル質量の50パーセントが、小さな直径の粒子を有する。
【0122】
好ましくは、少なくとも50重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、少なくとも80重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、少なくとも90重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、100重量%の担体粒子が、500μm未満の直径を有する。
【0123】
生理学的に許容される賦形剤を、動物若しくは植物供給源又はこれらの組み合わせの任意の非晶質又は結晶質の生理学的に許容される薬理学的に不活性な物質から構成することができる。好ましい物質は結晶糖であり、例えば、グルコース若しくはアラビノースのような単糖類、又はマルトース、サッカロース、デキストロース若しくはラクトースのような二糖類である。マンニトール、ソルビトール、マルチトール、ラクチトールのようなポリアルコールを使用することもできる。最も好ましい物質は、α−ラクトース一水和物である。
【0124】
市販のラクトースの例は、Capsulac(商標)及びPharmatose(商標)である。市販のマンニトールの例は、Pearlitol(商標)である。
【0125】
製剤を、呼吸作動式鼻吸入器により投与される懸濁剤又は粉末剤の形態で提供することもできる。
【0126】
前記粉末製剤を、当該技術に既知の任意の種類のDPIを用いて吸入により投与することができる。
【0127】
DPIは、2つの基本的な種類に分けることができる:i)活性化合物の予め細分化された単回用量を投与するための単回用量吸入器;ii)予め細分化された単回用量を有するか又は多回用量に十分な活性成分の量を予め装填した多用量乾燥粉末剤吸入器(MDPI)。必要とされる吸気流速(l/分)に基づき、設計及び機械的特徴に厳密に依存して、DPIは、i)低抵抗装置(>90l/分);ii)中抵抗装置(約60l/分);iii)高抵抗装置(約30l/分)に分けられる。
【0128】
活性成分の薬理学的活性に関して、本発明の粒子を、軽度、中程度若しくは重篤な急性若しくは慢性の症状の予防及び/若しくは治療のため又は喘息及び慢性閉塞性肺疾患(COPD)のような呼吸疾患の予防的治療のために指示することができる。慢性閉塞性細気管支炎及び慢性気管支炎のような炎症及び粘液の存在よりもたらされる末梢気道の閉塞によって特徴付けられる他の呼吸障害も、これらの使用により利益を受ける。
【0129】
吸入による投与では、本発明の方法により生成される粒状活性成分は、好ましくは担体粒子と配合される。前記活性成分は、製剤の0.1〜90重量%、好ましくは製剤の0.25〜50重量%、より好ましくは1〜25重量%で存在する。好ましくは、担体粒子は、製剤の10〜99.9重量%、より好ましくは製剤の50〜99.75重量%、より好ましくは75〜99重量%の量で存在することができる。
【0130】
特に好ましい実施態様において、本発明により生成された粒子の活性成分は、フルカチゾンプロピオン酸エステル、ブデソニド、ホルモテロール、サルメテロール、ベクロメタゾン又はベタメタゾン、並びにこれらの混合物及び共結晶を含む(好ましくは、前記物質から実質的になる)。このリストは、前記化合物の塩、水和物及び溶媒和物も包含する。
【0131】
走査電子顕微鏡法(SEM)により、前記活性粒子が、出発物質のSEM画像と比較したとき、有意に特徴的であることを明確に観察することができる。本発明の粒子が、より均一で規則的な球状を示し、少量の微細粒子も存在する出発物質と同じように破損して不規則であるようには見えない。理論に束縛されることなく、表面形態の前記の差は、本発明の粒子の低い凝集傾向、したがって優れた分散特性の説明に寄与すると考えられる。
【0132】
本発明により生成される活性成分の粒子は、好ましくは実質的に球状である。このことは、粗面を有する粒子を排除するものではない。好ましくは、本発明により生成される粒子は、最大直径と最小直径の平均比が、1.3〜1:1、より好ましくは1.25〜1:1、より好ましくは1.2〜1.01:1、より好ましくは1.15〜1.02:1、より好ましくは1.1〜1.03:1、より好ましくは1.075〜1.05:1である。したがって、本発明の粒子は、実質的に球状であることが分かる。
【0133】
粒径と形状分析を組み合わせたSympatec QICPIC画像分析センサーのような、多数の粒径及び形状分析器具が利用可能である。この1ナノ秒未満の極めて短い暴露時間を用いる技術は、100m/秒までの速度の最速粒子の鮮明な画像も提供する分散単位の使用を可能にする。このことは、凝集した微細及び凝集性粉末の正確な分散を保証する。1μm〜20μmの粒径を測定することができる。一次測定データは、30000の一次分類に記憶され、個別の限定フォーマットにより評価することができる。予め定義された粒度分類のセットを、存在する測定仕様に容易に適合させることができる。高速データ圧縮モジュールは、1秒あたり500枚までの画像の取得を支援する。1μm以下の粒子は、前記に記載されたMalvern又はSympactec回折、好ましくはMalvernレーザー回折のようなレーザー回折技術により測定することができる。
【0134】
接着API薬剤粒子をエアロゾル化するのに必要な力が、接触面の表面エネルギーの合成に比例し、投影接触面に反比例することは、良く知られている。したがって、DPIのエアロゾル化効率を改善する大部分の一般的な手法は、接触面の表面自由エネルギーを低減すること又は粒子形状を変えて接触面を制限することである。表面積は、粒径及び形状のみにより決定されるものではなく、表面形態も表面積に寄与し、波形(すなわち、粗い)粒子は、同じ体積を占める平滑粒子よりも大きな表面積を有する。
【0135】
本発明の方法により調製される薬剤粒子は、特定の表面形態により定義することができる。粒子間力を調節して、肺への付着を向上させることができる。理想的には、接触面、したがって力は、安定した製剤をもたらし、それでも吸入時には容易な分離を可能にするために、薬剤と担体との間に十分な接着をもたらすようなレベルに調整されるべきである。微細粒子画分に対する表面波形の影響を、明確にすることができる。
【0136】
平滑表面ラクトン担体粒子は、微細粒子画分及び微粉化薬剤の分散性を増加することが示されており、一方、他の研究は、波形担体粒子が微細粒子画分を増加することを示した。これらの明らかに矛盾する結果は、表面力平衡は、表面構造だけではなく幾つかの変数によって決まるという仮定により説明することができる。
【0137】
例として記載される粒子では、表面積及び形態測定値は、表面積が粒子相互作用と高度に相関することを明らかにしている。粉末表面積の決定は、所定の圧力で粉末表面に吸着されたガスの量を測定することを含む。過去数十年間にわたって、表面を研究する新たな技術が出現してきた。
【0138】
本発明の粒子の表面積は、吸着性ガスとして窒素を使用するAccelerated Surface Area and Porosimetry Analyser(model ASAP 2000, Micromeritics, Norcross, GA)により決定した。粉末物質(0.3〜0.7g)を窒素下、45℃でおよそ24時間脱ガスして、予め吸着したガス及び蒸気をサンプルの表面から除去した。表面積は、0.07〜0.22の相対圧力(P/Po)で吸着データを使用する、マルチポイントBrunauer, Emmett and Teller(BET)法により決定した。
【0139】
好ましくは、本発明の粒子は、6〜22m2/g、好ましくは9〜18m2/g、より好ましくは10〜13m2/gの範囲、より好ましくは12m2/gの表面積を有する。
【0140】
逆ガスクロマトグラフィー(Inverse Gas Chromatography, IGC)は、固体物質の表面及び嵩特性を特徴決定するガス相技術である。ICGの原理は非常に簡単であり、従来のガスクロマトグラフィー(Gas Chromatography, GC)実験の逆である。円柱カラムを、目的の固体物質、典型的には粉末、繊維又は膜で均一に充填し、保持時間及び溶出ピーク形状を、一連の十分に特徴決定された非極性及び極性ガスによって研究する。パルス又は一定濃度のガスを、固定した担体ガス流速でカラムの注入し、パルス又は濃度が先からカラムの底に溶出するのに掛かる時間を検出器により測定する。異なるガス相プローブ分子による一連のIGC測定は、広範囲の接触を可能にする。IGCを、表面エネルギーを測定するため、同様に、加工により引き起こされる表面特性の小さな変化を研究するために使用する。
【0141】
IGCを、本発明の粒子の表面エネルギーを測定するために使用した。IGCは、2セットの条件により実施することができる。有限希釈では、吸着等温線をピークプロフィールから誘導することができ、吸着エネルギー分布を計算するために使用することができる。第2に、無限希釈では、器具の検出限界に近い溶質の量を注入し、この場合、溶質−溶質相互作用は小さく、溶質−吸着剤相互作用のみが測定保持時間に影響を与える。これは、プローブ(ガス)分子と固定粉末との間の分散的と特異的の両方の相互作用を生じることができる。
本発明の粒子は、図21において非常に明瞭に示されるように、表面エネルギーの等エネルギー分布を有することによって特徴付けられる。この表面エネルギーは、本発明の好ましい方法により調製される粒子の有限及び無限希釈の両方において、極めて類似し、ほぼ同一であり、一方、典型的な微粉化粒子は、有限及び無限希釈において顕著な差を示す。
【0142】
原子間力顕微鏡法(Atomic Force Microscopy, AFM)を使用して凝集−接着平衡を測定することができ、サンプルの表面にプローブチップを接触して設置する(接触モード原子間力顕微鏡法)又はサンプルの表面に非常に近接して設置する(非接触及びタッピングモード原子間力顕微鏡法)ことによって、高さを測定するように機能する。プローブチップは、チップから受ける力に比例して湾曲する弾性カンチレバーに結合している。原子間力顕微鏡はサンプルをラスター走査して、データポイントのマトリックスを生成し、それから定量的な高さ及び粗さ測定値を抽出することができる。タッピングモード原子間力顕微鏡法は、薬剤を含む多様な有機化合物の結晶及び担体粒子ラクトースの接着特性を効果的に画像化する。コロイドプローブ顕微鏡法(Colloid Probe Microscopy, CPM)は、微晶質粒子の間の粒子−粒子接着を測定するために、日常的に使用される。
【0143】
サンプルの波形及び表面形態の程度を、AFMを使用して定量化した。本発明の粒子の表面様相及び粗さ測定値を、Nanoscope IIIコントローラー、Multimode AFM及びJ型スキャナー(全て、Dl, Santa Babara, CA, USA)を使用して調査した。全てのAFM表面様相画像を、画像化が1Hzの走査速度のTESP Olympusチップ(Di, Cambridge, UK)を使用して実施される、タッピングモード操作により記録した。表面粗さ測定値を、1μm×1μm領域で分析した。結晶表面の表面特性の差を定性化するために、表面凹凸の高さの差の実効値表面粗さ測定(Rq)及び平均表面粗さ(Ra)を計算した。
【0144】
力測定の前に、サンプルの各バッチの粒子を、エポキシ樹脂接着剤(Araldite, Cambridge, UK)を使用して、予め定義されたばね定数を有する標準V字形チップレスカンチレバー(DNP-020, Dl, CA, USA)に結合した。3つのチップを各サンプルのために調製し、接着剤の薄層が乾燥する前に、全てのプローブを、光学顕微鏡(倍率50×)により検査して、結合粒子の一体性を確実にした。
【0145】
基材を、周囲条件が25℃(±1.5℃)の一定温度及び35%RH(±3%)の相対湿度で維持して特注の環境チャンバーに収納されているAFMスキャナー台に設置した。相互作用力は、フックの法則を適用し(F=−kz)、基材の変位の関数としてAFMカンチレバーのたわみを記録することによって測定した。個別の力曲線(n=1024)を、4Hzの走査速度及び40nNの圧縮荷重により10μm×10μmにおいて実施した。パラメーターを一定に保持した。
【0146】
本発明の粒子は、図22に示されるように、実質的に波形表面を有することによって特徴付けられる。好ましくは、本発明の粒子は、ナノメートル規模の表面波形を有する。好ましくは、Rq値は、10〜100nm、より好ましくは20〜90nmである。好ましくは、Ra値は、10〜100nm、より好ましくは20〜90nmである。
【0147】
AFMコロイドプローブ技術の凝集−接着平衡(CAB)手法は、凝集性及び接着性の直接的な定量化−製剤内のAPIの「力平衡]−を可能にする開発である。ミリグラム以下の材料を使用して潜在的な製剤成分の凝集/接着力相互作用を決定する、市販のスクリーニングツールである。
【0148】
CAB手法は、コロイドAFMプローブとして設置されているAPI粒子と、明確に画定されたAPIの結晶質表面と担体基材との間の相互作用力を測定する。CABプロットは、定量化された、賦形剤物質へのAPIの接着傾向に対する凝集性の特徴的測定を可能にする多数のプローブの相互作用から生成される。CAB手法の開発は、器具の検証についての問題及び相互作用表面の真接触領域を決定する必要性を含む、従来のAFMコロイドプローブ方法論に関連する多数の制限を克服した。CAB値の1は、粒子−担体接着の力が粒子−粒子凝集の力と等しいことを示す。CAB比<1は、他の変数が等しいと仮定して、薬剤がそれ自体と凝集性であるよりも担体に対して接着性があることを示し、それによって、ブレンドすると安定した規則的な混合物を形成するとを予測することができる。しかし、CAB比>1は、薬剤が担体に対して接着性があるよりも、それ自体に凝集性があることを示し、ブレンドすると薬剤の凝集塊を含有する均一性の少ない混合物を生成しうることを示唆する。>1のような高いCAB比を有する薬剤−担体の組み合わせは、大きさ、形状、粗さ及び流動性に関して担体の間に潜在的な差があるにもかかわらず、エアロゾル化によってより多くの微細粒子画分(FPF)をもたらす。<1のような低いCAB比を有する薬剤−担体の組み合わせは、担体に対する活性粒子の大きな凝集をもたらし、したがって、活性粒子は、担体に結合したままの可能性が多くなる。このことは、吸入組成物において、担体粒子に結合したままの活性粒子が肺に到達せずに、咽喉又は舌に付着しうることを意味する。好ましくは、本発明により生成される粒子は、微粉化及び粉砕を含む他の方法により調製される粒子よりもはるかに低い凝集性を有する。凝集性の測定に関連し、全ての基材が凝集性及び接着性に関して異なっているので、CAB比は、無次元値であり、比較にとってより有用な測定値である。好ましくは、本発明により生成される粒子は、担体の0.8〜1.3、より好ましくは0.9〜1.2、より好ましくは1.0〜1.1のCAB比を有する。これは、薬剤のそれ自体の凝集及び薬剤と担体の接着の注意深い平衡である。
【0149】
本発明のエアロゾル製剤の化学的及び物理的安定性、並びに製薬学的許容性は、当業者に周知の技術によって決定することができる。したがって、例えば、成分の化学的安定性は、例えば生成物の長期保存後に、HPLCアッセイにより決定することができる。物理的安定性データは、例えば、漏れ試験、バルブ送達アッセイ(1作動あたりの平均ショット重量)、用量再現性アッセイ(1作動あたりの活性成分)及び噴霧分布分析のような、他の従来の分析技術によって得ることができる。
【0150】
本発明のエアロゾル製剤の粒径分布は、例えばカスケード衝撃又は「Twin Impinger」分析方法によるプレセパレーターを用いる、例えばNext Generation lmpactor(NGI)を使用する従来の技術によって測定することができる。本明細書で使用されるとき、「Twin Impinger」アッセイの参照は、英国薬局方1988年A204〜207頁、付録XVII Cにおいて定義されている“Determination of the deposition of the emitted dose in pressurised inhalations using apparatus A”を意味する。そのような方法は、HPLC移動相でプレセパレーターを充填することを含み、NGIカップのカップは、ヘキサン中の1%v/vシリコーンで被覆されて、粒子の跳ね返りを排除している。典型的には同じ製剤の4つの個別のカプセルを、所定の条件下でNGIに投入する。エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流す。次にNGIの各部品に付着した薬剤の量を、HPLCにより決定することができる。FPD決定は、NGIの段階3〜8で収集した薬剤量を表す。FPF発射用量も決定される。微細粒子画分の率(FPF%)又は呼吸画分により決定されるエアロゾル化効率も、評価される。
【0151】
そのような技術は、エアロゾル製剤の「呼吸画分」の計算も可能にする。本明細書で使用されるとき、「呼吸画分」への参照は、上記に記載された方法を使用して、1回の作動あたり送達される活性成分の総量の率として表される、1回の作動あたりのNGIの下部チャンバーに収集された活性成分の量を意味する。本発明の製剤は、医薬の発射用量の10〜30重量%以上、好ましくは14〜26重量%、例えば約15.9重量%及び約25.9重量%の呼吸画分を有することが見出されており、実施例2及び3により例示されている(図13及び19に示されている)。実施例1(ブデソニド)、実施例2(フルチカゾンプロピオン酸エステル)及び実施例8(フェノテロール臭化水素酸塩)では、従来技術と比較して、それぞれ、本発明の粒子に53%、50〜60%及び30〜50%のFPFの増加があった。
【0152】
本発明の粒子を含む吸入製剤に使用される噴射剤は、噴射剤として有効にするのに十分な蒸気圧を有する任意のフルオロカーボン又は水素含有クロロフルオロカーボン又はこれらの混合物を含む。好ましくは、噴射剤は、医薬の非溶媒である。適切な噴射剤には、従来の水素含有クロロフルオロカーボン、非クロロフルオロカーボン、水素含有フルオロカーボン及びペルフルオロカーボンなどが含まれる。特に、噴射剤HFA134a及びHFA227又はこれらの混合物を有利に使用することができる。
【0153】
本発明の製剤を、医薬エアロゾル製剤の送達に適したキャニスターに充填することができる。キャニスターは、一般に、プラスチック又はプラスチック被覆ガラス瓶又は好ましくは金属缶、例えば任意に陽極酸化処理、ラッカー被覆及び/若しくはプラスチック被覆されうるアルミニウム缶のような、使用される噴射剤の蒸気圧に耐えることができる容器を含み、容器は、計量バルブで密閉されている。計量バルブは、作動毎に計量された量の製剤を送達するように設計され、バルブを通した噴射剤の漏れを防止するためにガスケットを組み込む。ガスケットは、例えば、低密度ポリエチレン、クロロブチル、白黒ブタジエン−アクリロニトリルゴム、ブチルゴム及びネオプレンのような、任意の適切なエラストマー材料を含むことができる。
【0154】
適切なバルブは、エアロゾル産業において周知の製造者、例えば、Valois, France(例えば、DF10、DF30、DF60)、Bespak pic, UK(例えば、BK300、BK356)及び3M-Neotechnic Ltd, UK(例えば、SpraymiserW)により市販されている。
【0155】
医薬エアロゾル製造の当業者に周知の従来のバルク製造法及び機械を、充填キャニスターの商業的生産のための大規模バッチの調製に用いることができる。
【0156】
典型的には、医薬用途のために調製されるバッチでは、それぞれの充填キャニスターの重量を検査し、バッチ番号をコード化し、放出試験の前に保存のためにトレーに積み重ねる。
【0157】
各充填キャニスターを、使用前に適切なチャネリング装置に都合良く取り付けて、患者の肺又は鼻腔への医薬の投与用の定量吸入器を形成する。適切なチャネリング装置は、例えば医薬がそれを通って充填キャニスターから計量バルブを介して患者の鼻又は口に送達されうる、バルブ作動器及び円柱又は円錐様経路、例えばマウスピース作動器を含む。定量吸入器は、1回の作動又は「吸い」あたりに医薬の固定単位投与量を送達するように設計され、例えば1吸いあたり10〜5000マイクログラムの範囲の医薬である。医薬の投与を、軽度、中程度又重篤な急性又は慢性症状の治療又は予防的治療のために指示することができる。投与される正確な用量は、患者の年齢及び状態、使用される特定の粒状医薬、投与頻度、最終的には担当医の裁量に応じて決まることが理解される。医薬の組み合わせが用いられる場合、組み合わせの各成分の用量は、一般に、各成分が単独で使用される場合に用いられるものである。典型的には、投与は、1日あたり1回以上、例えば1〜8回であることができ、例として、1回あたり1、2、3又は4回の吸いである。
【0158】
適切な1日の用量は、例えば、疾患の重篤度に応じて、50〜200μmの範囲のサルメテロール、100〜1000μmの範囲のサルブタモール、50〜2000μmの範囲のフルチカゾンプロピオン酸エステル、又は、100〜2000μmの範囲のジプロピオン酸ベクロメタゾンである。
【0159】
したがって、例えば、各バルブ作動は、25μgのサルメテロール、100μgのサルブタモール、25、50、125又は250μgのフルチカゾンプロピオン酸エステル又は50、100、200又は250μgのジプロピオン酸ベクロメタゾンを送達することができる。典型的には、定量吸入器に使用される各充填キャニスターは、医薬の100、160又は240の計量用量又は吸いを含有する。
【0160】
本明細書に記載される充填キャニスター及び定量吸入器は、本発明の更なる態様を含む。
【0161】
ここで本発明は、添付の実施例及び図面を参照して記載される。実施例及び図面は本発明の範囲をいかなる方法によっても制限すると解釈されるべきではないことが理解されるべきである。
【図面の簡単な説明】
【0162】
本発明の方法は、添付の図面に示されているように、従来の装置を使用して実施することができる。
【図1】従来の噴霧乾燥ブデソニド(超音波処理あり)を示す。
【図2】本発明の超音波処理を有する噴霧乾燥ブデソニドを示す。
【図3】従来の噴霧乾燥ブデソニドのDSCを示す。
【図4】本発明の乾燥粉末超音波処理を有する噴霧乾燥ブデソニドのDSCを示す。
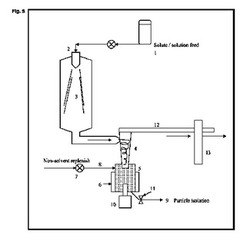
【図5】乾燥固体収集チャンバーが、セルに挿入されている超音波プローブを有する超音波セルに代えられている、従来の噴霧乾燥装置の図表示を示す。
【図6】図5と同様の構造において結合した変換装置を示す。
【図7】図5及び6と同様の構造において複数の結合した変換装置を示す。この場合、複数の変換器は円筒ダクトの周りを囲むように取り付けられている。
【図8】複数の変換器装置が再循環ループを構成している、図7と同様の複数の変換器装置を示す。
【図9】(本発明の)エアロゾル化により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図10】(本発明の)エアロゾル化により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図11】超音波を用いる分散及び沈殿により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図12】微粉化/粉砕されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図13】多様なサンプルのエアロゾル効率を示す。
【図14】微細粒子画分を表す棒グラフを示す。
【図15】GSKのFlixotideと比較したエアロゾル効率を示す。
【図16】超音波処理後のフェノテロール臭化水素酸塩の粒子を示す。
【図17】微粉化フェノテロール臭化水素酸塩を示す。
【図18】粒径分布データGR005/180/A4及びGR005/179/Cは、本発明により調製される粒子である。
【図19】吸入装置を使用する比較微細粒子画分(FPF)データを示す。
【図20】吸入専用検査用具を使用する比較微細粒子画分(FPF)データを示す。
【図21】有限及び無限希釈でのIGCによる表面エネルギー測定を示す。
【図22】ブデソニド微粉化粒子と本発明の粒子のAFMトポロジープロフィールを示す。
【図23】1及び3か月の保存後の多様なFPサンプルのエアロゾル効率を示す。
【図24】1及び3か月の保存後の多様なFPサンプルのエアロゾル効率を比較した棒グラフを示す。
【図25】Next Generation Impactorの多様な段階の粒子画分分布グラフを示す。
【図26】1か月の保存後のフルチカゾンプロピオン酸エステルの多様なサンプルの微細粒子画分を示す。
【図27】微細粒子画分を表す棒グラフを示す。
【図28】サルメテロールキシナホ酸塩粒子(10重量%)と組み合わせた、本発明により調製されたフルチカゾンプロピオン酸エステル粒子(90重量%)を示す。
【図29】フルチカゾンプロピオン酸エステルとラクトンの凝集−接着平衡を示す。
【図30】微粉化SXとブレンドしたときの、本発明のFP粒子の優れた均質性を示す。
【図31】Advarと比較した、微粉化SXとブレンドしたときの本発明のFP操作のFPFLDを示す。
【図32】サンプル3のFPの表面粗さのAFM輪郭プロットを示す。
【図33】サンプル4のFPの表面粗さのAFM輪郭プロットを示す。
【発明を実施するための形態】
【0163】
図3及び4を参照すると、2つのバッチのDSC追跡の比較では、噴霧乾燥粒子への超音波の適用が、粒子の物理的特性を変更することを明確に示している。120℃での発熱(正のピーク)は、DSC装置における非晶質から結晶質への変換を示す。一般的に、加工された物質の結晶特性には明確な改善がある。
【0164】
図5を見ると、超音波装置を用いる噴霧乾燥は、液体供給チャンバー1、噴霧乾燥噴霧器及び加熱ガス入口2、蒸発チャンバー3、サイクロン分離器4、連続超音波処理チャンバー5(熱ジャケット6で囲まれている)を含む。従来の処理噴霧乾燥粉末は、超音波フローセルチャンバー5に直接入れられる。同時に、非溶媒7の連続供給が、粒子スラリー9の流速と平衡した適切な速度により、ポンプ8を介して、濾過又は乾燥による後加工にポンプで送られる。超音波プローブ10は、超音波エネルギーを混合物に照射し、混合物は、出口11を通って流れる。溶媒蒸気、超微細粒子及びガス12は、フィルター13を介して排出される。超音波放射は、所望の粒径及び結晶化度が達成されるまで、必要な限り続けられる。当然のことながら、噴霧乾燥器の供給流は、粒子スラリーが取り出せる速度と平衡している。流速は、超音波フローセルチャンバー5における滞留時間が、例えば10秒〜1時間になるように制御される。顕微鏡規模で生じる局所キャビテーションは、前述の固体状態効果を誘導する流体温度及び圧力において変化を促進する。超音波の出力及びチャンバー5の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー5内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0165】
図6を見ると、超音波装置による噴霧乾燥は、チャンバー21が外面に位置する単一の結合超音波変換器22を有する以外は、図5と同様の構造である。変換器22は、チャンバー21の全容量に分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で受声波作用を与え、超音波の出力及びチャンバー21の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー21内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0166】
図7を見ると、超音波装置による噴霧乾燥は、チャンバー31が外面に位置する単一の結合超音波変換器32を有する以外は、図5及び6と同様の構造である。巻き付け変換器32は、チャンバー31の全容量に分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で受声波作用を与え、超音波の出力及びチャンバー31の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー31内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0167】
図8を見ると、これは、チャンバー31が、熱調節ジャケット43及びポンプ42を介した任意の撹拌羽根車44を備えた一次粒子収集容器41に結合して、連続閉ループ加工系を作り出す以外は、図7と同様の構造の、超音波装置を有する噴霧乾燥装置を示す。超音波は、分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で適用され、超音波の出力及び再循環加工ループ31、41、42の滞留時間を調整することによって、粒径及び形態を制御することができる。
【0168】
当業者は、熱ジャケットが非溶媒の温度を設計に応じて所望の温度に維持するのを助けることを理解する。
【0169】
用語「含む」は、「含まれる」ならびに「からなる」を意味し、例えば、Xを「含む」組成物は、Xのみからなることができるか又は何か別のものを含むことができ、例えばX+Yである。
【0170】
特に定義されない限り、語「実質的に」は、「完全に」を除外せず、例えばYを「実質的に含有しない」組成物は、Yを完全に含有しない場合がある。必要であれば、語「実質的に」を本発明の定義から省くことができる。
【0171】
「任意」又は「任意に」は、後に続く記載の事象又は状況が起こってもよいが起こる必要もなく、そしてその記載が、前記事象又は状況が起こる場合と起こらない場合とを含むことを意味する。
【実施例】
【0172】
実施例1
ブデソニド(5g)を100mLのジクロロメタンに溶解した。超音波チャンバーで収集したブデソニド粉末のサンプルは、Buchi-290実験室規模噴霧乾燥器(Buchi, Switzerland)を使用して生成した。溶液を、およそ10Lpm(リットル/分)で流れる窒素を7barで使用して噴霧した。吸引器を100%に設定し、溶液の流速を10Lpmに設定した。ガス温度を120℃に設定した。ブデソニド粒子を、高速サイクロン分離器の末端に連結されている超音波チャンバーで収集した。超音波を噴霧乾燥粒子に適用するために、超音波収集チャンバーを、25℃に温度調節したヘプタンで充填し、20kHzで共振する超音波プローブを取り付けた。20Wの出力の超音波を30分〜1時間適用した。得られた粒子スラリーを噴霧乾燥し、粒子を、光学顕微鏡及びDSC(示差走査熱量測定)により特徴決定した。粒子の大きさは、典型的には、1〜7μmの範囲であった。
【0173】
2つの代表的な試料のD(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、それぞれ、1.21、3.03、4.63μm及び1.05、2.99、3.76μmであった。
【0174】
示差走査熱量測定
DSC実験は、DSC Q2000 V24.2ビルド107(TA Instruments, UK)により実施した。およそ3mgの物質をDSCのサンプルパンで計量し、275℃まで熱付加する100℃/分の加熱傾斜に付した。DSC測定を、以下の工程に従って実施した。
→稼働9(本発明の超音波により処理していない噴霧乾燥物質)
→機器 DSC Q2000 V24.2ビルド107
→モジュール DSC標準セルRC
→サンプル 噴霧乾燥したpxO2−262
→大きさ 2.140mg
→方法 100℃/分の高速加熱実験 計量サンプルを100℃/分の速度で275℃まで加熱する
→稼働10(本発明の方法に従って超音波で処理した物質)
→機器 DSC Q2000 V24.2ビルド107
→モジュール DSC標準セルRC
→サンプル 超音波後のpx02−262
→大きさ 3.590mg
→方法 100℃/分の高速加熱実験 計量サンプルを100℃/分の速度で275℃まで加熱する
【0175】
実施例2
実施例2は、本発明により生成された粒子の利点を示す。
【0176】
多様な加工により生成された操作フルチカゾンプロピオン酸エステル(FP)のエアロゾル化効率を、2成分乾燥粉末剤吸入器(DPI)製剤において評価した。バッチ研究は以下を含む:
サンプル2 図9のSEMに示されているように、本発明により例示されるエアロゾル化方法により調製。フルチカゾンプロピオン酸エステル(4g)を100mLのアセトンに溶解した。超音波チャンバーで収集したフルチカゾンプロピオン酸エステル粉末のサンプルは、Buechi-290実験室規模噴霧乾燥機(Buechi, Switzerland)を使用して生成した。溶液を、窒素をおよそ10Lpm(リットル/分)で流れる窒素を7barで使用して噴霧した。吸引器を100%の流速に設定し、溶液の流速を10Lpmに設定した。ガス温度を120℃に設定した。フルチカゾンプロピオン酸エステルを、高速サイクロン分離器の末端に連結されている超音波チャンバーで収集した。超音波を噴霧乾燥粒子に適用するために、超音波収集チャンバーを、25℃に温度調節したヘプタンで充填し、20kHzで共振する複数の結合変換器(図7と同じ)を取り付けた。20Wの出力の超音波を30分〜1時間適用した。得られた粒子スラリーを噴霧乾燥し、粒子を、光学顕微鏡及びDSC(示差走査熱量測定)により特徴決定した。粒子の大きさは、典型的には、1〜6μmの範囲であった。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、1.35、3.25、5.63μmであった。
サンプル3 図10のSEMに示されているように、本発明により例示されるエアロゾル化方法により調製。サンプル3は、3gのFPをサンプル3のために使用した以外は、サンプル2と同じ方法により調製した。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、0.99、2.55、4.97μmであった。
サンプル4 図11のSEMに示されているように、代替的な沈殿手法により調製。サンプル4は、WO2008/114052A1に記載されたとおりに調製した。この方法は、初期溶液噴霧を使用しない。代わりに、この従来技術は、アセトン中のフルチカゾンプロピオン酸エステルの溶液を超音波音場の存在下でヘプタン抗溶媒に加えることにより生じる、分散性抗溶媒結晶化を伴う。このことは、本発明の方法を使用して形成される粒子よりも有意に平滑な粒子をもたらす。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、1.14、2.67、5.11μmであった。
【0177】
このサンプルを、微粉化FPを含有する追加の2成分DPI製剤及び Flixotide Discus吸入器から抽出した製剤と比較した。
【0178】
操作FPのサンプル2、3及び4のエアロゾル化効率を、0.4%w/wのFPを含有する2成分製剤を使用して評価した。
【0179】
各2成分製剤は、0.016gのFP及び3.984gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いてブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。
【0180】
含有均一性試験の後、12.5±1mgの各ブレンドをサイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC, Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0181】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0182】
各実験において、同じ製剤の4個の個別のカプセルを、Rotahaler(GSK, Ware, UK)DPI装置により60Lpmで4秒間NGIに投入した。加えて、Flixotide Diskus(GSK, Ware, UK)のブリスターを空にして、サイズ3のHPMSカプセルを装填し、Rotahalerにより60Lpmで1〜4秒間NGIに投入した。エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。
【0183】
NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドについて3回繰り返し、続いて、平均中央空気力学的直径(MMAD)、幾何学的標準偏差(GSD)、微細粒子用量(FPD)及び発射用量の微細粒子画分(PFPED)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0184】
エアロゾル化効率を図13に示す。微細粒子画分の率を図14に示す。
【0185】
サンプル2及び3の微細粒子画分の率(FPF%)により決定されたエアロゾル化効率は、微粉化FPよりも有意に大きかった。このデータを図15に示し、Flixotideから形成したものと比較したエアロゾル効率を明確に示している。これらの物質を含めたことによる性能の増加は、この研究に使用した吸入器にとって顕著であった。サンプル4は、微粉化FPよりも有意に低いFPF%を有し、これは粒子の表面形態と関連していた。これらのデータは、サンプル2と3のエアロゾル化効率にほとんど差がないことを示唆した。
【0186】
比較インビトロデータは、本発明により生成された粒子の性能が、従来の粉砕粒子及び非常に異なる表面と幾何学的特性を有する特別に調製された(超音波沈殿)粒子の両方の性能に対して圧倒的に、そして驚くほど優れている。全ての場合において、粒子は同じ粒径範囲を有する。フルチカゾンプロピオン酸エステルの特定の実施例において、最適粒子のFPFは、微粉化/粉砕物質よりも54%大きく、沈殿物質よりも200%大きかった。したがって、これらの最適粒子の有意な差及び改善された機能は、大きさではなく(重要な設計基準であるが)、これらの非常に改善された粒子を記載する他の特性の範囲が寄与している可能性がある。
【0187】
図9及び10(本発明)に示されているSEM画像は、図11及び12(本発明ではない)にそれぞれ示されている沈殿及び粉砕材料と比較して、形状及び表面粗さについて明確な対照を明確に示す。粗さ及び円形/球状3D形状は、性能に対して顕著な影響を与える。
【0188】
全ての製剤を、25℃/75%RHで1〜3か月間保存した。微粉化フルチカゾンプロピオン酸エステルを含有する製剤のFPFは、t=0でのFPFと比較したとき、1か月保存した後にほぼ50%、3か月保存した後にほぼ70%減少した。しかし、本発明により調製された粒子を含有する製剤のFPFは、ストレス条件下での保存後に影響を受けなかった。サンプル3のFPFは、t=0でのFPFと比較したとき、1か月保存した後に6%、3か月保存した後に3%減少した。サンプル2のFPFは、t=0でのFPFと比較したとき、1か月保存した後に7%、3か月保存した後に28%減少した。代替的沈殿手法により調製したサンプル4を含有する製剤の微細粒子画分に、t=0でのFPFと比較したとき、1か月後に29%、3か月保存した後に24%の、小さいが統計的に有意な減少があった。このことは、粒子の平坦な形態に関連する場合があり、したがって、粒子は、毛細管力によってラクトースに対してより大きな接着を生じる可能性がある。結果を図23及び24に示す。サンプル2及び3のFPFは、記載された条件下で1〜3か月間保存したとき微粉化サンプル又はサンプル4よりも高いFPFを維持したことが明らかである。サンプル2及び3のFPFは、記載された条件下で1〜3か月間保存したとき微粉化サンプルより小さい率で減少した。このことは、サンプル2及び3が、1〜3か月間保存した後、粉砕サンプルよりも安定していたことを示す。サンプル4は、1〜3か月間保存した後、サンプル2及び3よりもかなり低いFPFを有する。
【0189】
実施例3
実施例3は、pMDIの製剤のために本発明により生成された粒子の利点を示す。図25は、NGIの多様な段階における粒子画分分布グラフであり、サンプル1及びサンプル5のFPFに関して優れた性能を明確に示している。両方のサンプルは、実施例2のサンプル2と同様の方法により生成した。サインプル1は、メタノール中20%ジクロロメタン中のフルチカゾンプロピオン酸エステルの3%溶液(100ml中3g)の噴霧により調製し、サンプル5は、アセトン中3%の溶液(100ml中3g)の噴霧により調製した。図25に示されているように、両方のサンプルの段階5(カットオフ1.36μm)のNGIのFPFは、段階4(カットオフ2.30μm)の同様のFPFと共に、粉砕物質と比較して、100%を超える増加を示した。FPF値は、段階5のサンプル5及び1では、粉砕物質の6.20%と比較して、それぞれ11.80%及び13.80%であった。したがって、サンプル1及び5は、段階4では粉砕生成物と同様のFPFを有し、段階5では粉砕生成物よりも大きなFPFを有することが明らかである。
【0190】
実施例4
実施例2に記載されたように生成されたフルチカゾンプロピオン酸エステル(FP)のサンプル2、3及び4のエアロゾル化効率を、Rotahaler(登録商標)単位用量DPI装置(GSK, Ware, UK)及びCyclohaler(登録商標)単位用量DPI装置(TEVA Pharmaceuticals, Netherlands)を使用して、微粉化サルメテロールキシナホ酸塩(SX)を含有する複合乾燥粉末剤吸入器(DPI)製剤で評価した。
【0191】
エアロゾル化効率を、微粉化SXも含有する複合DPI製剤で評価した。それぞれの複合製剤は、0.16000gのFP(サンプル2、3又は4)、0.02320gのSX及び3.8168gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いて各ブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。それぞれの複合製剤のブレンド強度は、500μgのFP及び50μgのサルメテロール基剤に等しかった。これはAdvair 500/50の用量強度と適合する。Advair 500/50のサンプルを微粉化サンプルとして使用した。FPサンプル2、3、4又は微粉化FPを含有する4つの異なる製剤のエアロゾル化効率を測定した。
【0192】
図26に示されているように、t=0では、Cyclohaler単位用量DPI装置を使用するサンプル2、3及び4のFPFは、微粉化サンプルよりも大きい。Rolahaler単位用量DPI装置を使用する場合、サンプル2及び3のFPFは、サンプル4及び微粉化サンプルよりも大きい。このことは、本発明のフルチカゾンプロピオン酸エステル粒子、サンプル2及び3が、全体的に、2つの装置において高いFPFを有することを示す。サンプル4は、Cyclohaler単位において良好に機能したが、Rolahaler単位は機能が不十分である。サンプル2及び3は両方のユニットにおいて高いFPFを有する。
【0193】
4つのサンプルを25℃/75%RHで1か月保存した。微粉化FP及びサンプル2、3及び4のFPF%は、ストレス保存条件下で有意に影響を受けなかった。これらのデータは、本発明により調製される粒子が、DPI製剤に対して安定性をもたらすはずであることを示す。
【0194】
含有均一性試験の後、12.5±0.5mgの各ブレンドをサイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC, Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0195】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0196】
各実験において、同じ製剤の2個の個別のカプセルを、Rotahaler(GSK, Ware, UK)により60Lpmで4秒間、Cyclohaler(TEVA Pharmaceuticals, Netherlands)により90Lpmで2.8秒間NGIに投入して、両方の装置が4kPa圧力低下を生じるように操作されることを確実にした。
【0197】
加えて、市販のAdvair 500/50 Diskus(GSK, USA)のブリスターを空にして、1.25mgの製剤をサイズ3のHPMCカプセルに移し、Rotahalerにより60Lpmで4秒間、Cyclohalerにより90Lpmで2.8秒間NGIに投入した。
【0198】
エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドにおいて3回繰り返し、続いて、微細粒子用量(FPD)及び装填用量の微細粒子画分(FPFLD)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0199】
図26は、この実施例に従って調製したFPのAdvair 500/50同等ブレンド及び機械的に微粉化したSX粒子の、特にFPについてのFRFを示す。図27は、粒子のFPF性能をグラフで示し、ここでFPは本発明により調製した。サンプル2及び3(cyclo2とcyclo3及びrota2とrota3)は、FPに関して優れた性能を明確に示し、したがって、FP粒子とFP粒子の間の凝集及びラクトースとFPの接着を制御できることを明確に示している。とりわけ、この研究に使用した微粉化成分は、市販の装置からのものであり、微粉化物質は、数か月でなければ数週間の条件付けを受けている。逆に、本発明により作製される粒子は、新たに調製される場合でも高度に安定しており、繰り返しになるが、このことは、本発明により調製される粒子はDPI製剤に対して安定性をもたらすはずであることがここでも記述される。
【0200】
図28は、本発明により作製されるFPサンプル3の粒子を示す。次にこれらをサルメテロールフマル酸塩(10重量%)及びフルチカゾンプロピオン酸エステル(90重量%)からなる組み合わせとブレンドし、ここでサルメテロールフマル酸塩粒は微粉化されており、フルチカゾンプロピオン酸エステルは、本発明により調製されている。
【0201】
実施例5
微粉化フルチカゾンプロピオン酸エステル(FP)、並びに、本発明の方法により(サンプル2及び3)及び実施例1に記されるようにWO2008/114052A1に例示されている異なる方法により(サンプル4)操作されたFPのバッチの、凝集−接着平衡(CAB)を、FP及びラクトース一水和物の結晶質基材に対して決定した。結晶質基材に対する異なるバッチのCAB力平衡は、以下のようにして決定した:
プローブ調製:
FPの全てのバッチの粒子(n=3)を、エポキシ樹脂接着剤(Araldite, Cambridge, UK)を使用して、標準V字形チップレスカンチレバー(DNP-020, Dl, CA, USA)に結合した。
平滑ラクトース及び薬剤結晶の生成:
ラクトースの平滑結晶は、カバースリップで挟んだ加熱飽和液滴を冷却することにより生成した。平滑薬剤結晶は、活性物質をアセトンに溶解し、使用した抗溶媒が水である、静止液滴抗溶媒結晶化を使用して生成した。
AFM力測定:
個別の力曲線(n=1024)を、4Hzの走査速度及び40nNの圧縮荷重により10μm×10μm領域において実施した。環境条件は、20℃(±1.5℃)の一定温度及び45±3%の相対湿度を維持した。
【0202】
微粉化FPのCAB分析は、同等の接触幾何学では、微粉化FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.36倍大きいことを示唆した。
【0203】
サンプル2FPのCAB分析は、同等の接触幾何学では、サンプルFPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.17倍大きいことを示唆した。
【0204】
サンプル3FPのCAB分析は、同等の接触幾何学では、サンプル3FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.16倍大きいことを示唆した。
【0205】
サンプル4FPのCAB分析は、同等の接触幾何学では、サンプル4FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用とほぼ同等であることを示唆した。
【0206】
図29に示されているように、微粉化FPは、調製されたFPのサンプル2、3及び4よりもラクトースに対して有意に接着性があった。サンプル4は、ラクトース及びSXに対して最小の接着性があり、したがって、この物質の表面エネルギーは、他のバッチと有意に異なることを示唆している。しかし、接着値は400〜800nNの範囲であり、このことは、異なる基材と接触したときのこの物質の大きな接触半径を反映している。この物質の大きな接触半径は、乾燥粉末剤吸入器(DPI)製剤のサンプル4の配合の際に限定されたエアロゾル化をもたらす。
【0207】
対照的に、サンプル3のFP−ラクトース交互作用に関する接着値は、35〜89nNの範囲であり、サンプル2の接着値は、128〜169nNの範囲であり、このことは、異なる基材と接触したときの本発明の粒子の小さな接触半径を反映している。この物質の小さな接触半径は、接着値が169〜249nMの範囲である微粉化FPと比較して、担体に基づいたDPI製剤のサンプル2及び3の配合の際に大きなFPエアロゾル化をもたらす。
【0208】
CAB分析は、サンプル3が他の物質よりも小さい接触半径を有しうること、一方、サンプル4が大きな接触半径を有しうることを確認し、このことは、これらの粒子の表面形状に関連している。
【0209】
これらのデータは、本発明により例示された粒子操作戦略が、表面エネルギー及び粒子接触形状の両方の制御をもたらすことができることを示し、これらは両方ともDPIS製剤の薬剤粒子に寄与する重要な品質である。
【0210】
図29は、サンプル4の接着及び凝集の両方の比較的高い力に注目して、粒子の異なる接触形状に関するFP−ラクトース相互作用を示す。
【0211】
実施例6
実施例2に記載されたように生成されたフルチカゾンプロピオン酸エステル(FP)のサンプル3のエアロゾル化効率を、Rotahaler(登録商標)単位用量DPI装置(GSK, Ware, UK)及びCyclohaler(登録商標)単位用量DPI装置(TEVA Pharmaceuticals, Netherlands)を使用して、微粉化サルメテロールキシナホ酸塩(SX)を含有する複合乾燥粉末剤吸入器(DPI)製剤で評価した。
【0212】
エアロゾル化効率を、微粉化SXも含有する複合DPI製剤で評価した。各複合製剤は、0.16000gのFP、0.01160gのSX及び3.8284gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いてブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。複合製剤のブレンド強度は、500μgのFP及び25μgのサルメテロール基剤に等しく、したがって、これは500/25製剤であった。
【0213】
500/25製剤を有する、微粉化FP及び微粉化SXを含有する製剤の含有均一性の評価は、不十分な均質性を示唆し、したがて、この製剤は、インビトロ衝突研究により特徴決定しなかった。対照的に、FPサンプル3及び微粉化SXを含有する製剤は、図20に示されているように、非常に良好な均質性を示した。これは、FPのサンプル3の相対標準偏差%が、3.43であり、SXでは、4.55であったことを示す。これを微粉化FP−SX製剤と比較し、FPの相対標準偏差%は8.76であり、SXでは15.95であった。
【0214】
含有均一性の試験の後、12.5±0.5μgの、FPサンプル3ブレンドを含有するブレンド又はAdvairを、サイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC、Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0215】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0216】
各実験において、同じ製剤の2個の個別のカプセルを、Cyclohaler(TEVA Pharmaceuticals, Netherlands)により90Lpmで2.8秒間NGIに投入して、両方の装置が4kPa圧力低下を生じるように操作されることを確実にした。
【0217】
エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドにおいて3回繰り返し、続いて、微細粒子用量(FPD)及び装填用量の微細粒子画分(FPFLD)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0218】
性能データは、図30に示されているように、それぞれ79μg及び11μgのFP及びSXの500/25製剤におけるサンプル3のFPDを示唆した。図31に示されているように、これは、微細粒子画分が操作FPの15.8%と比較してSXでは44%であると解釈された
【0219】
図21は、500μgのサンプル3FP及び50μgのSX(サンプル3 300/50)、500μgのサンプル3FP及び25μgのSX(サンプル3 500/25)及び500μgのFPと50μgのSXを含有するAdvair(Advair 500/50)を含有する製剤において、上記に記載したようなFPFLD測定を示す。この図は、サンプル3 500/50及びサンプル3 500/25では、SXはAdvair 500/50よりもかなり高いFPFLDを有したことを示す。サンプル3 500/25のSXのFPFLDは、サンプル3 500/50よりも高かった。
【0220】
このことは、操作FPがブレンドの含有均一性に対して顕著な効果を有し、FPFに関してSCの増加を促進することを意味し、また、操作FPを使用した所定の製剤では、有意に少ないSXをブレンドに使用して、FP及びSXの両方において匹敵するFPFを達成できることを意味している。これらのデータは、本発明により調製した操作FPを使用して、Advair製品に現在用いられている名目強度の半分を含有する複合DPI製品を配合することが可能であることを示唆している。
【0221】
実施例7
微粉化フルチカゾンプロピオン酸エステル(FP)、本発明により調製されるFPのサンプル3及び実施例2に記載されたFPのサンプル4の表面粗さ及び表面積は、原子間力顕微鏡法(AFM)及びBET表面積分析をそれぞれ使用して決定した。画像化領域の粗さは、画像化表面の高さにおける差の平均(Ra)及び二乗平均平方根(Rq)を使用して定量化した。更に、サンプルの表面積は、5ポイントBET窒素吸着分析により決定した。
【0222】
FPサンプルの表面様相は、Multimode AFM、J型スキャナー、Nanoscope IIIaコントローラー(全て、Dl, Cambridge, UK)及びシリコンチップ(型番OMCL- AC240TS, Olympus, Japan)を使用して、各物質の粒子の表面から3つの無作為に選択した1μm×1μm正方形領域を、512×512ピクセルの解像度及び1Hzの走査測度で画像化する、TappingMode(商標)原子間力顕微鏡法(AFM)により調査した。画像化領域の粗さは、以下の方程式を使用するAFMソフトウエアにより計算して、画像化表面の高さにおける差の平均(Ra)及び二乗平均平方根(Rq)を使用して定量化した:
【0223】
【数1】
【0224】
ここで、npは、画像におけるポイントの数であり、yiは、中央線からのポイントiの距離である。
【0225】
FPサンプルの比表面積を、Gemini 2360表面積分析器(Micromeritics Instrument Corporation, Norcross, USA)を使用して測定した。5ポイントBET窒素吸着分析を、FlowPrep 060脱ガス器((Micromeritics Instrument Corporation, Norcross, USA)によりサンプルを24時間脱ガスした後で実施した。
【0226】
結果を下記の表2にまとめる。
【0227】
【表2】
【0228】
サンプルの表面粗さ分析は、サンプル3が最大の表面粗さを有し、一方、サンプル4(本発明によって調製されたものではない)が、最も平滑であったことを示唆した。サンプル3は、他のサンプルよりも大きな表面積を有し、これは物質の粗さに関連しうる。サンプル3のRa及びRq値、並びに表面積は、微粉化サンプル及びサンプル4よりも大きい。
【0229】
微粉化サンプルのRa及びRq値の平均偏差は、サンプル3及び4よりもかなり大きい。このことは、サンプル3及び4よりも微粉化サンプルの方が表面粗さに大きな差があることを示す可能性がある。
【0230】
図32及び33は、サンプル3及び4それぞれの表面粗さのAFM輪郭プロットを示す。これらは、サンプル4がサンプル3よりもかなり平滑であることを示す。輪郭プロットにおける差は、サンプル3がサンプル4と異なる表面粗さを有することを示し、このことは、Ra及びRq値に反映されている。
【0231】
実施例8
次にメタノール(200mL)中のフェノテロール臭化水素酸塩(10g)の溶液を調製し、二流体ノズル及び0.7mmオリフィスを使用し、支援窒素流速が35〜40m3/時間(吸引器100%)のBuchi-B290を用いて、流速9mU/分(30%ポンプ)及びノズルクリーン設定2で噴霧乾燥した。入口温度は78℃であり、出口温度は38℃である。ジイソプロピルエーテル(300mL)を、底がB-290サイクロンに連結しており、5℃に温度調節された最大容量500mLの撹拌超音波容器に投入した。噴霧乾燥生成物を、40Wの連続出力で2時間稼働している超音波容器の中に収集し、続いて非晶質フェノテロール臭化水素酸塩の第1粒子を加えた。上記のBuchi-B290を用いて、入口温度110℃及び出口温度50℃で懸濁液を噴霧乾燥することによって、粒子を回収した。この実施例のデータ及び粒子のSEM画像を、図17〜20に示す。
【0232】
図17は、市販の微粉化フェノテロール臭化水素酸のSEM画像を示す。図18は、本発明により加工された粒子のサンプル、すなわちGR005/180/A4及びGR005/179/Cの粒径分布データを示す。図19は、検査用具として市販のHandiHaler吸入装置を使用した、比較の微細粒子画分(FPF)を示す。図19及び20のFPF〔%〕の横列を比較するとFPFの増加は30〜117%に異なっていた。
【0233】
図21は、微粉化ブデソニドと本発明のブデソニド粒子の表面エネルギーに対する分散被覆率を示す。IGCを、本発明の粒子の表面エネルギーを測定するために使用した。IGCは、2セットの条件により実施することができる。有限希釈では、吸着等温線をピークプロフィールから誘導することができ、吸着エネルギー分布を計算するために使用することができる。第2に、無限希釈では、器具の検出限界に近い溶質の量を注入し、この場合、溶質−溶質相互作用は小さく、溶質−吸着剤相互作用のみが測定保持時間に影響を与える。図21の左側に示されているように、特に高いエネルギー部位においてのみ限定された吸着率(被覆率)が分析される。溶質の量が有限希釈まで増加するので、最終的に100%の被覆率が達成され、表面エネルギーが異なっているにもかかわらず、粒子の全ての部位において吸着率が上昇する。本発明の粒子は、図21に明確に示されているように、表面エネルギーの等エネルギー分布を有することによって特徴付けられる。表面エネルギーは、本発明の好ましい方法により調製される粒子の有限及び無限希釈の両方において極めて類似し、ほぼ同一であり、一方、典型的な微粉化粒子は、有限及び無限希釈において顕著な差を示す。
【技術分野】
【0001】
本発明は、非晶質固体物質の、結晶化度のレベルを増加させて、表面特性を改質する方法に関する。本発明は、活性成分化合物のような化学品の製造、吸入製剤のような医薬製剤に使用される賦形剤の製造、及び、液体系の懸濁液のような農芸化学的薬剤の製造において用途を有する。
【0002】
本発明は、また、例えば乾燥粉末吸入器(DPI)装置を使用して、肺に投与される乾燥粉末製剤を形成する、活性薬剤粒子の製造に関する。特に、本発明は、その機能が従来のDPI、加圧式定量噴霧吸入器(pMDI)及び鼻懸濁粉末剤、特にはDPI及びpMDI粉末剤、とりわけDPI粉末剤よりも有意に卓越している、粒子の特性及び好ましい加工を提供する。
【背景技術】
【0003】
薬剤を気道に投与するために広く使用されている2つの系は、通常ラクトンのような製薬学的に不活性な物質の粗賦形剤粒子と混合される乾燥粉末剤として微粉化薬剤粒子を含む乾燥粉末剤吸入器(DPI)及び噴射ガス中の微粉化薬剤粒子の懸濁剤を含むことができる加圧式定量噴霧吸入器(pMDI)である。本発明は、これらの送達方法の両方に関連する。
【0004】
鼻送達は、薬剤粒子を中枢神経系(CNS−鼻から脳)に投与することができる手段であり、粉末剤又は液体懸濁剤のいずれかによる全身的又は局所的な鼻製剤である。多様な呼吸作動式装置は、肺への付着を生じることなく、副鼻腔及び嗅部を含む鼻腔の標的領域に鼻腔内薬剤を送達する。本発明は、この送達方法に関連する。
【0005】
活性及び他の組成成分の結晶及び沈殿粒径の制御は、製薬産業や農芸化学産業のような、目的の活性成分の最終製品形態が微粉末の形態である産業において必要である。活性成分が生体系において作用する方法は、多くの要因、とりわけ粒径及び結晶形態によって決まる。小さな粒子は、粉砕のような方法により作製することができるが、そのような方法は、粉砕粒子の材質に有害な影響を与える場合がある。更に、粒子の有意な割合が、所定の最終用途に不適切な形状で製造される場合がある。粒子が粉砕されるとき、これらは形態変化を受け、望ましくない表面多形態変換をもたら素場合があり、このことは、吸入のために設計される医薬製剤のような最終目的用途に不適切である非晶構造の形成を生じる場合がある。加えて、粉砕は、例えば活性成分が低融点固体である場合、粒状粉砕を不適切にしうる相当量の熱を発生する。加えて、エアロゾルに使用される目的の粒子の物理的機能は、粉砕の結果として高度に荷電されると損なわれる。
【0006】
薬剤粒子の製造技術には、薬剤の溶液から小さな滴のエアロゾルを生成し、続いて小さな滴を噴霧乾燥して、粒子を固化することが含まれうる。噴霧乾燥は、粒子形成及び乾燥に関わる最も広く使用されている工業的方法の1つである。これは、例えば溶液、乳液又はポンプ移動可能な懸濁液のような液体供給原料から、粉末、顆粒又は凝集形態のいずれかで乾燥固体を連続的に製造するために特に適している。したがって、乾燥噴霧は、最終製品が、粒径分布、残留含水量、嵩密度、粒子形状などのパラメーターに関する品質標準に適合する場合に理想的な方法である。従来の連続噴霧乾燥技術の欠点は、固化が典型的には急速であり、加えて加工が乾燥粒子の高度な凝集をもたらすので、乾燥される粒子が結晶質粒状形態ではなく、おそらく100%の高さで非晶質形態になる傾向があることである。エアロゾルの小さな滴の凍結乾燥も粒子を得るために当該技術において使用されるが、ここでも、生じる典型的な急速固化は、一般に非晶質粒子の生成をもたらす。
【0007】
特許文献1は、適切な溶媒中で所望の物質の溶液を形成する工程、その溶液からエアロゾルを生成する工程、その物質からエアロゾルの小さな滴を非溶媒で収集する工程、及び、非溶媒に分散した液滴に超音波を適用して、物質の結晶化を実施する工程を含む、SAXと呼ばれる方法による粒子の製造を記載する。この技術の欠点は、エアロゾルから蒸発する溶媒の程度に対して臨界制御が必要なことである。
【0008】
吸入は、全身作用薬、ならびに喘息、慢性閉塞性肺疾患及び炎症のような、肺自体に局所的に作用するように設計された薬剤の送達にとって、非常に魅力的で迅速でさらに患者に優しい経路である。予測可能及び再現可能な方法で肺に薬剤を送達する技術を開発することが特に望ましく、有利である。薬剤吸入の利益には、素早い開始速度;非侵襲的全身経路に対する患者許容度及び服薬順守の改善;副作用の低減;製品寿命の延長;送達の一貫性の改善;より高い用量、大きな効能及び標的化の正確さを含む新しい治療形態の利用が含まれる。
【0009】
乾燥粉末吸引(DPI)は、肺疾患の治療において重要な役割を果たす。第一には、これらは定量吸入器(MDI)の使用において遭遇した問題を克服するため、後には、これらが噴射剤無含有であり、したがって環境により優しいために開発された。MDIを使用すると、患者は、エアロゾルが肺に達することができるように、吸入と吸入器の作動を同調させなければならない。乾燥粉末剤吸入器(DPI)は、呼吸作動式であり、そのため理論的には、問題なく肺に達することができる/するはずである。しかし、問題は、取扱い、用量の内容均一性及び用量の制御に関する技術的な制限によって生じる。また、吸気流速は、患者によって異なり、DPIの機械的原理に応じて決まる。エアロゾルの肺への付着率が吸気流速に依存する可能性があるので、高い流れ抵抗により吸気流速を顕著に低減するDPIは、あまり適していない。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】WO2004/073827
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかし、成功した乾燥粉末剤及びDPI製品のための粉末技術は、依然として大きな技術的問題を残している。製剤は、製造者及び粉末剤の定量を助けるばかりでなく、予想可能な再懸濁及び流動化を提供し、分配装置内の粉末剤の過剰な保持を回避するためにも、適切な流動特性を有さなければならない。懸濁粉末剤における薬剤粒子又は製薬学的に活性な物質の粒子(本明細書において、API粒子とも呼ぶ)は、肺の中の適切な標的に輸送されうるように、適切にエアロゾル化されなければならない。典型的には、肺への付着のためには、活性粒子は10μm未満、しばしば0.1〜7μm又は0.1〜5μm未満の直径を有する。
【0012】
この種類の系において、薬剤−薬剤及び薬剤−担体粒子、並びに粒子−壁の相互作用は、肺深部への薬剤送達の成功のために極めて重要である。粒子間の相互作用は、ファンデルワールス、毛細管作用及びクーロン力のような接着力によって決定される。これらの力の強さは、粒子の大きさ、形状及び形態によって影響を受ける。粗面を有する球状又は球形粒子は、小さい接触域及び粒子間の分離距離の増加によって、肺薬剤送達にとって最適であると考慮される。大きな分離距離は、結合力を減少し、粉末分散を改善する。最適薬剤粒子のための粒子工学は、DPI装置工学と共に、肺を介する効率的な薬剤送達にために必須である。WO2006/056812は、乾燥粉末剤吸入器(DPI)装置を使用して肺に投与される乾燥粉末製剤を形成する粒子加工の改善に関する発明を報告し、それによると、活性物質の粒子及び担体物質の粒子の加工が添加剤物質の存在下で実施され、優れた粉末特性を示す粉末組成物を提供する。
【0013】
乾燥粉末剤が従来の方法により製造される場合、活性粒子は大きさが異なり、多くの場合にこの差異は顕著である。このことは、十分に高い割合の粒子が適切な部位への投与に適した大きさになるのを確実にすることを困難にする。したがって、活性粒子の粒度分布が可能な限り狭い乾燥粉末製剤を有することが望ましい。例えば、好ましくは粒子分布はガウスであり、好ましくは粒子分布は単峰性である。更に、例えば、活性粒子の空気力学的及び体積的な粒度分布の幾何学的標準偏差は、好ましくは2以下、より好ましくは1.8以下、1.6以下、1.5以下、1.4以下又はさらには1.2以下である。このことは、用量効率及び再現性を改善する。
【0014】
平均中央空気力学的直径(MMAD)とは、50%の粒子が、吸入薬粒子のインビトロ機能を決定するのに適しているインパクターに入るのよりも小さい粒子直径であって、形状と密度との両方を考慮する。(例えば)5μmのMMADを有するサンプルは、粒子の全質量(すなわち、総数ではない)の50パーセントが5μmを超える直径を有し、50パーセントが5μm未満の直径を有する。
【0015】
MMADが10μm以下であるより小さい微粒子は、表面積と体積との比が増加するにつれてますます熱力学的に不安定になる傾向があり、そのことは、粒径が減少すると表面自由エネルギーの増加をもたらし、その結果、粒子が凝集する傾向及び凝集塊の強度を増加する。吸入器では、微粒子の凝集及びそのような粒子の吸入器の壁への接着が問題であり、微粒子が大型で安定した凝集塊で吸入器から遊離されるか又は吸入器から離れることができず、吸入器の内部に接着したままになっているか又はさらには吸入器を目詰まり若しくは閉塞させる結果をもたらす。
【0016】
吸入器のそれぞれの作動の間、また異なる吸入器及び異なる粒子バッチの間に粒子の安定した凝集塊が形成される程度が不確実であるので、不十分な再現性がもたらされる。更に、凝集塊の形成は、活性粒子のMMADが著しく増加する可能性があり、活性粒子の凝集塊が肺の必要な領域に到達しないことを意味する。肺深部又は全身送達に必要なこれらのμmからサブμmの粒径は、呼吸に適した活性粒子が高い凝集性の傾向を有するという問題をもたらし、このことは、これらが一般に不十分な流動性及び不十分なエアロゾル化を示すことを意味する。
【0017】
そのような呼吸に適した活性粒子の高い凝集性を克服するために、過去に製剤には流動性及び薬剤エアロゾル化の両方を助けるために不活性賦形剤の大型の担体粒子を含ませた。微細活性粒子が、互いに固着するのではなく、吸入器装置の中にある間に大型の担体粒子に付着する傾向があるので、これらの大型の担体粒子は、粉末製剤に有益な効果を有する。活性粒子は、担体粒子表面から遊離し、分配装置の作動の際に分散して、気道に吸入されうる微細懸濁剤を生じる。
【0018】
比較的大型の担体粒子の添加は、粉末特性を改善する傾向があるが、通常は、製剤の総重量の95%以上が担体であるという程度まで薬剤を希釈する効果も有する。大部分の微細又は超微細活性成分が担体粒子の表面に付着する必要があり、そうでなければ、活性粒子の凝集性が依然として粉末において優位を占め、不十分な流動性をもたらすので、粉末特性に所望の効果を有するために比較的多量の担体が必要となる。微細粒子が接着するために利用可能な担体粒子の表面積は、担体粒子の直径が増加すると減少する。しかし、流動性は、直径が減少すると悪化する傾向がある。したがって、満足できる担体粒子を得るために適切な平衡を見出す必要がある。更なる考慮は、少なすぎる担体粒子が含まれると分離を生じる可能性があることであり、これは極めて望ましくない。
【0019】
製剤の際に経験する更なる問題は、薬剤及び賦形剤粒子の表面特性の差異である。それぞれの活性剤粉末は、独自の固有粘着性又は表面エネルギーを有し、化合物毎に著しく変動する。更に、表面エネルギーの性質は、与えられた化合物によって加工の方法に応じて変わる可能性がある。例えば、噴射粉砕は、用いる衝突の攻撃的な性質のために、表面特性に有意な差異を生じることで悪名高い。そのような差異は、表面エネルギーの増加、並びに凝集性及び接着性の増加をもたらす可能性がある。高度に規則的な結晶質粉末でも、近距離のリフシッツ−ファンデルワールス力は、凝集性及び接着性の高い粉末をもたらす可能性がある。
【0020】
担体賦形剤を使用しない場合、微粉化薬剤粒子は、リフシッツ−ファンデルワールス力のみにより緩く凝集される。粒子凝集塊は空気流により解凝集されなければならないので、毛細管力を形成しないことがそのような製剤の機能にとって重要である。毛細管力は、通常、例えばリフシッツ−ファンデルワールス力の数倍大きく、そのような凝集塊が単一の粒子に分かれる能力は、凝集塊を一緒に保持する自己付着力が増加するにつれて減少する。そのような緩い凝集は、球形化の方法を使用して達成することができる。
【0021】
空気流の中に設置されると、担体粒子に接着した粒子に対して作用する力は、揚力(担体粒子から小さな粒子を持ち上げる揚力;これは微粉化粉末では無視することができる)、抵抗力(接着力及び摩擦力を相殺する)、接着力及び摩擦力(接触する2つの表面の接線変位を防ぐ力)と記載することができる。これらの最後の2つは、担体表面からの薬剤粒子の脱離を妨ぐことができる。乾燥粉末剤吸入における相互作用混合物の成功又は失敗は、主に、薬剤粒子を担体表面に固定する接着力の大きさによって決まる。
【0022】
明らかに、薬剤−担体単位が抵抗力により単一構成成分に分けることができなければ、薬剤−担体単位の全体が嚥下されるので、非常に高い接着力は不要である。平衡した接着力は、吸入されたときには、薬剤−担体単位の微粉化薬剤粒子への分離を促進し、そして、嚥下されたときには、粗担体粒子への分離を促進する。一方、薬剤と担体粒子との間の接着力が小さすぎると、粒子分離をもたらす場合があり、したがって、用量の含有均一性に大きな差異をもたらす場合がある。また、薬剤粒子は、強く接着する傾向がある吸入器装置壁との滑り接触の際に担体粒子から容易に除去される。したがって、より多くの薬剤が吸入器装置の中に失われる。
【0023】
従来技術は、吸入用の相互作用粉末混合物における接着力は、幾つかの方法により操作できると教示している。第1には、担体粒子を、粒子の大きさの中央値、形状及び表面粗さに従って選択することができ、このことは、規定された混合手順において接着力に大きな差をもたらし、したがって、異なるエアロゾル化特性をもたらす。
【0024】
中央粒径値の減少は、薬剤と担体粒子との間の接着力を増加する。大きな接着力は、不規則形状又は伸長担体粒子においても見出される。この効果は、混合の際の摩擦の増加によって説明することができる。表面粗さは、粗さの程度に応じて接着力を増加又は減少させる。接着力の増加は、真接触領域の増加に起因して極めて平滑な担体粒子表面において見出され、又は非常に粗い担体粒子表面に見出され、それは、凸凹の間の広い間隔が微粉化薬剤粒子の機械的捕捉を可能にするからである。
【0025】
典型的なDPI製剤において、粉末はプレブレンドされ、それは微細及び粗担体粒子の間に自己接着をもたらす。微細担体粒子は、主に、粗担体粒子表面の溝及び割れ目による機械的捕捉によって自己接着する。したがって、微細粒子の量は物理的に除去され、担体粉末の流動特性は改善される。磨食(谷を埋めることを意味する地質学用語)は、起伏の少ない担体粒子表面をもたらし、それによって微粉化薬剤粒子は、担体粒子表面に機械的に捕捉又は埋め込まれることが少なくなる。磨食は、また、担体粒子表面の微小粗さを増加し、したがって、真接触領域の低減に起因して薬剤と担体粒子との間の接着力を低減する。しかし、接着力、したがって乾燥粉末吸入機能に関して、磨食は必ずしも有利ではないことが見出されている。粗担体粒子の最小表面粗さが、磨食の意味において微細担体粒子の埋め込みを可能にするために必要である。粗担体粒子表面が比較的平滑である場合、微細担体粒子は、担体粒子表面の見掛巨大粗さが増加するように自己接着し、そのことは、薬剤粒子が機械的に捕捉される部位をより多く提供する。この場合、薬剤粒子を、再懸濁の際に微細担体粒子を有する凝集塊としてのみ担体粒子表面から除去することができ、肺への薬剤の付着は、この凝集塊の大きさに応じて決まる。
【0026】
担体物質の選択は、薬剤と担体粒子との間の接着力の強さに明らかに影響を与える。しかし、適用箇所、すなわち、肺への吸入は、この選択肢を著しく制限する。現在まで、ラクトース一水和物及びグルコースが市販の乾燥粉末剤の吸入において担体物質として使用されている。グルコースは、55%を超える相対湿度の保存空気の環境に保存されると、急速に水分を吸収する。これは、薬剤と担体粒子との間に強力な毛細管力をもたらす。ラクトース一水和物は、湿度レベルを増加する薬剤−担体単位の弱点を低減したと主張されてきた。しかし、異なる湿度条件下で保存した後の微粉化薬剤とラクトース一水和物担体粒子と間の接着力測定は、この意見に対して疑いを投げかけている。
【0027】
相互作用粉末混合物の使用は、非常に低用量の吸入用薬剤(例えば、サルメテロールキキシナホ酸塩:50マイクログラム)の取扱いを容易にし、これらを1回の投与単位、例えばホイルブリスター(例えば、Advair Discus吸入器装置)又はカプセルで提供することができる。また、均質性の増加及びそのような混合物の分離の低減は、内容物にとって有利である。
【0028】
DPIの微細粒子を製造する2つ慣用の技術は、機械的微粉化及び噴霧乾燥である。高エネルギー粉砕操作は、高度に荷電された粒子を生成し、したがって非常に凝集性がある。凝集性を減少させるために、界面活性剤を例えば湿式粉砕に使用する。粉砕方法は、粉末安定性に影響を与える表面及び結晶学的損傷も導入する。
【0029】
製造された粒子は、多くの場合、強力な凝集体を形成することができる不規則な破片を含有する。加えて、多工程の加工は、粉末製造の際に材料の有意な損失を引き起こす場合があり、バッチ毎に生成物の特性に差異を引き起こす場合がある。粉砕と異なり、噴霧乾燥技術は、所望の大きさの医薬粒子を直接製造することができる1工程連続方法である。この方法には界面活性剤又は他の可溶化剤の必要がない。しかし、この方法に必要な高い流速、及び制御可能なパラメーターが限定されていることに起因して、それぞれの粒子の熱履歴及び乾燥速度は、制御することが困難である。それにより、製造された粒子は、通常は非晶質であり、したがって、粉末の保存の間に粒子の構造変化及び焼結を引き起こしうる温度及び湿度の変化に対して、影響されやすい。
【課題を解決するための手段】
【0030】
本発明の第1の態様によると、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、固体物質を、その固体物質が不溶性であるか又は難溶性である溶媒と接触させる工程と;その固体物質に、上記溶媒と接触したときに超音波を適用する工程とを含む方法が提供される。
【0031】
本発明の第2態様において、本明細書に記載されている方法により得られる、少なくとも1つの物質を含む粒子が提供される。また、本明細書に記載されている方法により得られる、少なくとも1つの粒状物質を含む粒子を有する製剤が提供される。
【0032】
そのような粒子及びそれらを含有する製剤は、特に、吸入可能な医薬製剤に有用である。そのような粒子及びそのような粒子を含む製剤は、従来どおりに調製された粒子と比較して驚くべきインビトロ性能を示す。この有意な性能増加は、細粒分(Fine Particle Fraction,FPF、送達用量に対する%、インピンジャー及び咽喉の全ての段階の合計として定義)の比例増加により定量化される。これらの粒子は、DPIの薬剤配合にとって優れた性能特性を有する。これらの粒子は、また、溶解速度及び肺に送達されたFPFに関して、従来の粒子と比較して驚くべきインビボ性能を示す。
【0033】
本明細書の以降では、固体物質が不溶性又は難溶性である溶媒を非溶媒と呼ぶ。本明細書で使用されるとき、非溶媒とは、固体物質が25℃で1mlあたり0.1mg未満、好ましくは25℃で1mlあたり0.05mg未満、好ましくは25℃で1mlあたり0.01mg未満の量で可溶性であるものである。
【0034】
逆に、本発明で使用されるとき、溶媒とは、固体物質が25℃で1mlあたり0.1mgより多く、好ましくは25℃で1mlあたり0.5mgより多く、好ましくは25℃で1mlあたり1mgより多く、好ましくは25℃で1mlあたり5mgより多く、好ましくは25℃で1mlあたり10mgより多くの量で可溶性であるものである。
【0035】
好ましくは、本発明において利用される固形物質は、粒状固形物質である。粒子は、好ましくは、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有する。
【0036】
空気力学的直径は、試験物質が粒子として空気力学的に挙動する、単位密度あたりの球体の直径である。異なる大きさ、形状及び密度の粒子を比較するため、並びにそのような粒子が気道のどこに付着しうるかを予測するために使用される。この用語は、それ自体気道内の付着と関連することがない実際の直径を表す、等体積球相当径、光学的直径、測定直径又は幾何学的直径に対比して使用される。
【0037】
多数の方法が、呼吸粒子の分布及び(程度は低いが)吸入粒子の分布を決定するために利用可能であり、粒径を示すために、平均中央空気力学的直径(MMAD)及び幾何学的標準偏差(GSD)を計算することができる。MMADは、粒子サンプルの統計由来の数字であり、例えば、5μmのMMADは、総サンプル質量の50%が、5μm未満の空気力学的直径を有する粒子として存在し、総サンプル質量の50%が、5μmより大きい空気力学的直径を有する粒子として存在することを意味する。
【0038】
Anderson Cascade lmpactor又はNext Generation Impactor、好ましくはNext Generation Impactorのようなカスケードインパクターを使用して、エアロゾル(又は砂塵雲)の粒度分布を得ることができる。粒子が例えばガラス又はガラス繊維に付着した幾つかの段階から構成される空気サンプルを、装置から取り出す。粒子は、その大きさに応じて、特定の段階に対してインパクトする。サンプル採取前後の各段階を計量することによって、各段階の噴射速度から分離粒径を計算することができ、MMADをこれらの計算から導くことができる。この方法の限界、すなわち粒子の跳ね返り、流速の過負荷及び変動などにもかかわらず、これは、エアロゾルの空中粒度分布及びそのMMADを測定する十分に確立された技術である。
【0039】
粒径は、レーザー回折技術により測定することができる。レーザーからの光線は、空気のような透明ガスの中に懸濁されている、粒子雲の中に照射される。粒子は光線を散乱し、小さな粒子は大きな粒子に比べて光線を大きな角度で散乱する。散乱光線を、異なる角度で設置された一連の光検出器により測定することができる。これはサンプルの回折パターンとして知られている。回折パターンを、十分に立証された光散乱理論を使用して粒子の大きさを測定するために使用することができる。粒子は球状であると想定されるが、実際にはほんの少しの粒子しか球状ではない。粒子の直径は、粒子の測定体積から計算されるが、等体積球相当径の球体であると想定される。
【0040】
好ましくは、本発明において利用される固体物質は、機械的微粉化、粉砕、噴射粉砕、摩砕、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法によって得られる。最も好ましくは、本発明において利用される固体物質は、噴霧乾燥の方法により得られる。従来の噴霧乾燥技術を使用することができる。好ましくは、WO2004/073827に開示されているようなSAX方法は、使用されない。
【0041】
好ましくは、上記の方法の1つを適用する前に、固体物質は実質的に非晶質であり、例えば、50%未満の結晶度、より好ましくは40%未満の結晶度、より好ましくは25%未満の結晶度、より好ましくは10%未満の結晶度、より好ましくは5%未満の結晶度、例えば1%未満の結晶度である。
【0042】
本発明において利用される固体物質が機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合により得られる場合、これらの方法の1つの前に、固体物質は、50%より高い結晶度、例えば60%より高い結晶度、例えば75%より高い結晶度、例えば90%より高い結晶度、例えば95%より高い結晶度、例えば99%より高い結晶度のように、実質的に結晶質であることができる。4つの方法の1つ又はこれらの混合の後、固体物質は、粒子の核が実質的に結晶質であり、粒子の外層が実質的に非晶質であることができる。
【0043】
多数の技術を使用して結晶質の内容を決定することができる。例えば、PXRD(粉末X線回折)は、固体物質のX線回折パターンを見る技術である。結晶質粒子は、個別の多形体に特有の「指紋」パターンを有する。逆に、非晶質化合物は、ほとんど又は全く特徴的なパターンを示さず、単に幅広の円丘又は雑音として表れる。示差走査熱量測定(DSC)も、所定のサンプルの結晶化度のレベルと等価でありうる、明確な融点及び融解熱測定値を明らかにする。非晶質物質は、DSCプロフィールと一致しない挙動を示す。結晶質物質のDSCは、結晶質を示す鮮明な吸熱を描く。動的蒸気吸着(DVS)は、結晶質及び非晶質物質の等温線及び吸湿挙動を測定する、急速で連続した方法を提供する。DSCと共にこれを使用して、生成物の安定性を測定することができる。最後に、ラマン分析は、結晶質物質を表示することができ、実際、異なる多形体を区別することができる。非晶質物質は、同じ特徴的なパターンを有さず、そのため結晶相と区別される。本出願の目的において、示差走査熱量測定(DSC)は結晶化度の測定に好ましい方法である。DSC実験は、TA InstrumentsのDSC Q2000 V24.2 build 107を含む多数の市販の装置により実施することができ、前者は、本発明のDCSを測定する好ましい器具である。典型的には、正確な量の物質をDSC器具のサンプルパンに導入し、およそ275℃になるまで、100℃/分までの加熱傾斜に付す。融解熱の測定値としての融点吸熱及び熱流の積分は、結晶化度の定性的及び定量的なそれぞれの測定値である。特に、所定の固体物質では、DSCは、2つのサンプルの直接的な比較を提供し、一方が他方よりも結晶度が多いか又は少ないかを明確に示す。
【0044】
追加的に又は代替的に、本発明の方法を適用する前に、固体物質は、準安定結晶質物質を含むことができる。
【0045】
任意の特定の物質では、当業者は、固体物質が不溶性又は難溶性であるかを容易に決定することができる。例えば、高速液体クロマトグラフィー(HPLC)又は気液クロマトグラフィー(GLC)は、既知の濃度の溶液に対して基準を作製し、澄んだサンプルを分析することにより、飽和したときの液体サンプル中の可溶化物質のレベルを決定することを可能にする。前者の方法は、医薬生成物のためにより典型的に使用され、一方、後者は、分析される物質が300℃までの温度で蒸発するのに十分に揮発性である場合に使用され、大部分の医薬生成物が除外される。好ましくは、水を水難溶性物質に非溶媒として使用する。水溶性物質には、好ましくは非溶媒炭化水素、例えばヘプタンが使用される。水溶性物質の更なる非溶媒には、適切であれば、エーテル(メチルtert−ブチルエーテル)、アルコール(エタノール)及びケトン(ブタノン)が含まれうる。
【0046】
超音波は、好ましくは、非晶質物質の少なくとも一部分が結晶質物質に変換される又は準安定物質がより安定した物質に変換されるのに必要な、適切な時間及び温度で適用される。例えば、方法は、好ましくは0.1ミリ秒より長く、より好ましくは1ミリ秒より長く、より好ましくは1分より長く、例えば1秒〜24時間、より好ましくは1分〜6時間、より好ましくは5分〜1時間の間実施される。
【0047】
好ましくは、本発明において使用される固体物質は乾燥している。このことは、好ましくは、非溶媒、水及び有機溶媒を含む溶媒を実質的に含有しないことを意味する。このことは、固体物質は実質的に水又は溶媒を含有しないことを意味する。溶媒を実質的に含有しないとは、固体物質が、5重量%未満の溶媒、より好ましくは4重量%未満、より好ましくは3重量%未満、より好ましくは2重量%未満、より好ましくは1重量%未満、より好ましくは0.5重量%未満、より好ましくは0.1重量%の溶媒を含有することを意味する。
【0048】
水和の水を含有する固体物質、及び分子溶媒和物は、結晶の単位格子を組み込むために必要な必須量の水又は溶媒のみを含有するので、実質的に溶媒無含有であるということができる。それ以外は、これらは実質的に水又は溶媒を含有しない。
【0049】
本発明の方法は、活性製薬学的成分、活性農芸化学的成分、製薬学的賦形剤、農芸化学的賦形剤及びこれらの2つ以上の適切な混合物からなる群より選択される物質を含む、噴霧乾燥粒子の加工に特に有用性を見出す。「適切な」とは、活性製薬学的成分を、他の活性製薬学的成分及び/又は製薬学的賦形剤と組み合わせうることを意味するが、通常は、製薬学的活性成分を例えば農芸化学的賦形剤と組み合わせることはない。
【0050】
本発明の好ましい実施態様において、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、
(i)溶媒中で、少なくとも1つの固体物質の溶液を形成する工程;
(ii)溶液を、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法に付し、溶解させた固体物質が実質的に乾燥固体物質に変換される工程;
(iii)任意に、固体物質を工程(ii)の方法の液体及び/又は気体成分から単離する工程;
(iv)工程(ii)又は工程(iii)の乾燥固体物質を非溶媒で処理する工程;
(v)工程(iv)の固体物質に、上記非溶媒と接触したときに超音波を適用する工程;及び
(vi)任意に、工程(v)で得られた固体物質を、分離及び/又は乾燥する工程
を含む方法が提供される。
【0051】
そのような方法において、工程(ii)は、好ましくは、固体物質の溶液の噴霧乾燥を含む。従来の噴霧乾燥を使用することができる。噴霧乾燥方法において、生成された固体物質は、通常、実質的に非晶質である。
【0052】
好ましくは、工程(ii)を適用する後に、工程(iii)又は(iv)に入る固体物質は実質的に非晶質であり、例えば、50%未満の結晶度、より好ましくは40%未満の結晶度、より好ましくは25%未満の結晶度、より好ましくは10%未満の結晶度、より好ましくは5%未満の結晶度、例えば1%未満の結晶度である。
【0053】
工程(iv)において、処理するという用語は、乾燥固体物質を非溶媒に暴露することを意味する。これを、工程(ii)により生成された物質を収集するのに使用されるものと同じ又は別の容器において実施することができる。好ましくは、非溶媒の量は、固体物質の量よりも多い。例えば、工程(iv)における固体物質と非溶媒の重量比は、好ましくは1:100、より好ましくは1:10、例えば、1:2、1:3、1:4、1:5などの範囲である。
【0054】
好ましくは、工程(ii)及び/又は工程(iii)により生成された固体物質は、実質的に乾燥している。これは、工程(iv)の方法に入る固体物質の好ましくは全て(100%)が、好ましくは、水及び有機溶媒を含む溶媒を実質的に含有しないことを意味する(ここで用語「溶媒を実質的に含有しない」は、上記に定義されている)。
【0055】
任意の所定の固体物質において、当業者は、負担となることなく適切な溶媒を決定することができる。特定の固体物質に適した溶媒の幾つかの例は以下の通りである。メタノール、エタノール、ジクロロメタン、酢酸エチル、アセトン、2−プロパノールのような揮発性有機溶媒及び水のような非有機溶媒は、製薬学的に活性な成分の典型的な溶媒である。
【0056】
好ましい賦形剤には、例えば、ラクトース及びステアリン酸が含まれうる。ラクトースは、水又はエタノール/水混合物に溶解しうる。ステアリン酸は、酢酸エチル又はエタノールに溶解しうる。
【0057】
非溶媒(例えば、工程(iv)の方法のもの)は、好ましくは、無水結晶が望ましい場合には実質的に遊離の水(すなわち、予め固体物質と結合して水和物などを形成していない水が)を含有せず、また、固体物質が実質的に可溶性である任意の溶媒も含有しない。非溶媒は、固体物質が実質的に不溶性である場合、水であることができる。
【0058】
網羅的なリストではないが、溶媒及び非溶媒の組み合わせの幾つかの例を表1に示す。
【0059】
【表1】
【0060】
本発明の粒子を調製するのに適した他の非溶媒には、1,1−ジフルオロエタン、1,1,1−トリフルオロエタン、1,1,1,2−テトラフルオロエタン、ペンタフルオロエタン、1,1,1,3,3−ペンタフルオロプロパン、1,1,1,3,3,3−ヘキサフルオロプロパン、1,1,1,2,3,3,3−ヘプタフルオロプロパン、1,1,1,3,3−ペンタフルオロブタン及び1,1,1,2,3,4,4,5,5,5−デカフルオロペンタンからなる群より選択されるヒドロフルオロアルカンが含まれる。そのような非溶媒の使用は、PMDIに使用される直接配合を促進する。別の実施態様において、ペルフルオロデカリンのような揮発性の低いフッ素化化合物を非溶媒として使用することができる。
【0061】
工程(i)の方法で形成された溶液中の固体物質(好ましくは、製薬学的に許容される物質、製薬学的に許容される賦形剤又はこれらの混合物)の濃度は、好ましくは10mg/ml〜800mg/ml、より好ましくは50mg/ml〜600mg/mlの範囲、より好ましくは100mg/ml〜400mg/mlである。
【0062】
本発明の方法の間、非溶媒の温度は、好ましくは−10℃〜+120℃の間にあり、非溶媒が液体形態のままであることが前提である。好ましくは、非溶媒の温度は、好ましくは0〜80℃、より好ましくは20℃〜60℃の間にある。
【0063】
好ましくは、上記の方法は、順次に行われ、工程(iv)及び(v)は、工程(ii)の直後(又は行われる場合は、任意の工程(iii)の直後)に実施される。「直後」とは、工程(ii)(又は行われる場合は、工程(iii))の噴霧乾燥粒子が工程(ii)を受けてから1時間以内、好ましくは工程(ii)を受けてから30分以内、好ましくは5分以内、好ましくは1分以内に工程(iv)及び(v)において加工されることを意味する。好ましくは「直」とは、任意の中間工程がないことを意味する。好ましくは、上記の方法は、連続方法である。例えば、方法は、未加工物質を連続的に供給することができ、加工物質を連続的に又は漸増的に取り出すことができる。あるいは、方法は、バッチ型方法であることができ、ここで方法は、未加工物質をバッチ様に供給し、加工物質をバッチで取り出すことができる。
【0064】
あるいは、工程(ii)及び工程(iii)を、工程(iv)の6か月前、より好ましくは3か月前、より好ましくは1か月前、より好ましくは1週間前、より好ましくは1日前のように、工程(iv)の前に実施することができる。
【0065】
工程(v)で得られた固体物質を、本明細書のこれ以降、「活性粒子」と呼ぶ場合がある。
【0066】
本発明の更なる実施態様において、100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、
(a)少なくとも1つの固体物質を、機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合に付す工程;
(b)工程(a)で得られた固体物質を非溶媒で処理する工程;
(c)工程(b)で得られた固体物質に、非溶媒と接触したときに超音波を適用する工程;及び
(d)任意に、工程(c)で得られた固体物質を、分離及び/又は乾燥する工程
を含む方法が提供される。
【0067】
工程(a)を適用した後、工程(b)に用いられる物質は、例えば50%より高い結晶度、例えば60%より高い結晶度、例えば75%より高い結晶度、例えば90%より高い結晶度、例えば95%より高い結晶度、例えば99%より高い結晶度、又は、例えば50%未満の結晶度、例えば40%未満の結晶度、例えば25%未満の結晶度、例えば10%未満の結晶度、例えば5%未満の結晶度、例えば1%未満の結晶度である。
【0068】
工程(b)において、処理するという用語は、乾燥固体物質を非溶媒に暴露することを意味する。これを、工程(a)により生成された物質を収集するのに使用されるものと同じ又は別の容器において実施することができる。好ましくは、非溶媒の量は、固体物質の量よりも多い。例えば、工程(b)における固体物質と非溶媒の重量比は、好ましくは1:100、より好ましくは1:10、例えば、1:2、1:3、1:4、1:5などの範囲である。
【0069】
工程(b)は、工程(a)の直後に実施することができ、ここで「直後」とは、上記に定義されたとおりである。あるいは、工程(a)を、工程(b)の6か月前、好ましくは3か月前、より好ましくは1か月前、より好ましくは1週間前のように、工程(b)の前に実施することができる。
【0070】
吸入により肺深部又は血流に到達する製剤において、製剤の活性剤は、非常に微細な粒子の形態、例えば10μm未満の平均中央空気力学的直径(MMAD)を有さなければならない。10μmを超えるMMADを有する粒子は、咽喉の壁に衝突する可能性があり、一般に肺に到達しないことが十分に確立されている。5〜2μmの範囲のMMADを有する粒子は、一般に呼吸細気管支に付着し、一方、3〜0.05μmの範囲のMMADを有する粒子は、肺胞に付着し、血流に吸収される可能性がある。
【0071】
理想的には、乾燥粉末製剤の活性粒子は、10μm以下、好ましくは5μm以下、より好ましくは3μm以下、より好ましくは2.5μm以下、より好ましくは2.0μm以下、より好ましくは1.5μm以下又は好ましくは1.0μm以下のMMADを有するべきである。
【0072】
重要性が高いのは、乾燥粉末剤吸入における組成である。乾燥粉末剤吸入器(DPI)において、活性粒子(1〜5μm)とラクトースのような粗担体粒子(50〜500μm)の混合物を使用して、効果的な薬剤粒子投入を得ることができる。
【0073】
噴霧乾燥粒子は、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有する。
【0074】
方法の最終生成物である活性粒子も、約10μmまで、好ましくは約100nm〜約10μm、好ましくは約100nm〜約5μm、最も好ましくは約100nm〜約2μm、例えば約110nm、約250nm、約400nm、約700nm又は約1μmなどのMMADを有することができる。
【0075】
本発明の方法に使用される超音波の周波数は、好ましくは、16kHz〜1MHz、好ましくは10〜500kHz、より好ましくは10〜100kHzの範囲、例えば10、20、40、60、80若しくは100kHz又はその間の任意の周波数である。
【0076】
本発明の方法によって生成される固体物質の結晶化度を増加させることに加えて、超音波の適用は、凝集粒状物質の量を低減するために使用することもできる。この凝集低減は、好ましくは、上記に言及した工程(v)又は(c)と同時に実施される。
【0077】
超音波照射に付される、非溶媒と接触する固体物質の非晶質、部分的に非晶質又は準安定結晶質形態の種類に応じて、粒子は、より小型の形態及び/又はより安定した形態に変換されうる。例えば、活性成分はより安定した結晶質形態に変換されうるか、又は超音波照射の前に粒子が不安定な非晶質形態で存在する物質であると、より安定した非晶質形態に変換されうる。非溶媒と接触したときに粒子がどのような形態であろうとも、本明細書において概説した超音波照射が適用されたとき、粒子特性は変化し、より安定した粒子の形成をもたらし、これらをより効率的な方法で医薬又は農薬用途のような他の用途に使用することができる。好ましくは、この方法で得られた粒子は、高度に結晶質であり安定している。
【0078】
超音波照射工程が適用されると、粒状スラリーからの結晶の単離を、濾過、遠心分離、噴霧乾燥、超臨界二酸化炭素抽出、単なる蒸発又はこのような技術の2つ以上の混合のような、任意の従来の方法により実施することができる。典型的には、結晶は従来の蒸発方法を使用して単離される。
【0079】
本発明の方法において噴霧乾燥条件及び超音波処理方式を操作することによって、発明者たちは、今、所定の特性を有する結晶又は非晶質体を提供することを可能とした。噴霧乾燥物質を非溶媒中で超音波により所定の時間及び温度で処理することによって、特定の特性を再現性をもって得ることができる。これらの特性には、粒子形態、表面自由エネルギー粒径分布、所望の多形体、並びに単離粒子の流動性に関して低減された静電気及び凝集性/接着性が含まれうる。
【0080】
固体物質、好ましくは本発明の方法に付される粒状固体物質は、好ましくは、結晶を形成することができるか又は粒子のより安定した形態をもたらす形態の変化を受けることができる、活性成分又は薬剤若しくはプロドラッグ若しくは目的の農薬のような所望のその前駆体である。典型的には、そのような改質粒子は、従来の薬剤送達ビヒクル又は少なくとも1個の所定の改質粒子のために特に設計されうる薬剤送達ビヒクルのような所望の状況における使用のために、より作製し易くなる物理的特性を有する。本明細書において示唆されているように、従来の噴霧乾燥により調製された初期溶液(又は本明細書に参照された他の方法技術のいずれかの初期溶液又は固体物質)には、2個以上の目的の粒子の混合物のような、1個以上の目的の粒子が含まれうる。そのような状況において、超音波処理後の所望の最終用途に応じて、2つ以上の目的の活性成分、又は、少なくとも1つのプロドラッグと少なくとも1つの薬剤若しくは2つ以上の薬剤若しくは2つ以上の農薬との組み合わせが、溶質として又は初期固体物質として初期溶液に存在することができる。本発明の方法条件下で結晶化することができる適切な粒子は、コルチコステロイド、β2アゴニスト、抗コリン作用剤、ロイコトリエンアンタゴニスト、吸入タンパク質又はペプチド、フランカルボン酸モメタゾン;ジプロピオン酸ベクロメタゾン;ブデソニド;フルチカゾン;デキサメタゾン;フルニソリド;トリアムシノロン;サルブタモール;アルブテロール;テルブタリン;サルメテロール;ビトルテロール;イプラトロピウム臭化物;オキシトロピウム臭化物;クロモグリク酸ナトリウム;ネドクロミルナトリウム;ザフィルルカスト;プランルカスト;ホルモテロール;エホルモテロール;バンブテロール;フェノテロール;クレンブテロール;プロカテロール;ブロキサテロール;(22R)−6a,9a−ジフルオロ−llb,21−ジヒドロキシ−16a,17a−プロピルメチレンジオキシ−4−プレグネン−3,20−ジオン;TA−2005;チプレダン;インスリン;インターフェロン;カルシトニン;副甲状腺ホルモン;及び顆粒球コロニー刺激因子のような、本明細書の方法により結晶質粒子に形成されうる活性成分又は薬剤を含む。
【0081】
2つ以上の固体物質が使用される場合、共結晶が形成されうる。共結晶は、2つ以上の同一ではない中性分子成分の結晶複合体として定義することができ、例えば2つ以上の同一ではない中性分子成分は、活性主成分又はその所望の前駆体、及び、非共有結合、好ましくは主に水素結合を通じて、結晶格子中で互いに結合しているゲストである。ゲストは、別の活性主成分若しくはその所望の前駆体、又は共結晶形成剤であることができる。
【0082】
医薬共結晶の形成には、別の製薬学的に許容される分子と、所定の活性医薬とを、結晶格子中に組み込むことを伴う。得られた多成分結晶相は、親活性医薬の固有の活性を維持し、同時に特有の生理化学的プロフィールを有する。
【0083】
本明細書で使用されるとき、用語「共結晶形成剤」は、活性主成分又はその所望の前駆体として同じ結晶構造に存在する1つ以上の追加の分子を意味し、1つ以上の追加の分子は、共結晶中の分子間相互結合作用によって、活性主成分又はその所望の前駆体と超分子シントンを形成することができる。
【0084】
一つの実施態様において、共結晶形成剤は、以下の群から選択される少なくとも1つのシントン形成部分を有する少なくとも1つの分子を含む:エーテル、チオエーテル、アルコール、カルボニル、チオール、アルデヒド、ケトン、チオケトン、硝酸エステル、リン酸エステル、チオリン酸エステル、エスエル、チオエステル、硫酸エステル、カルボン酸、ホスホン酸、ホスフィン酸、スルホン酸、スルホンアミド、アミド、第一級アミン、第二級アミン、アンモニア、第三級アミン、イミン、チオシアネート、シアンアミド、オキシム、ニトリル、ジアゾ、有機ハロゲン化物、S含有複素環(例えば、チオフェン)、N含有複素環(例えば、ピロール、イミダゾール又はピリジン)、O含有複素環(例えば、フラン、エポキシド又はペルオキシド)及びヒドロキサム酸部分。
【0085】
更なる実施態様において、ゲストは、例えば活性主成分又はその所望の前駆体と共結晶を形成するために存在することができる。1つ以上のゲストが共結晶に含まれうることが考慮される。したがって、ゲストはそれ自体の活性を有する必要がないが、活性剤の所望の活性を過度に減じない幾つかの活性を有することができる。非活性ゲストは、有益な薬理学的活性を示したことがない、明らかに生物学的に非毒性であるか又は薬理学的に良性である、化合物であることができる。幾つかの場合において、ゲストは、活性剤と同一の活性又は相補的な活性を有することができる。ゲストは、別の活性主成分又はその所望の前駆体であることができる。例えば、幾つかのゲストは、活性主成分又はその所望の前駆体の治療効果を促進することができる。医薬製剤において、ゲストは、活性主成分又はその所望の前駆体又はそれらの塩と共結晶を形成する任意の製薬学的に許容される分子であることができる。
【0086】
ゲスト又は共結晶形成剤は、酸であることができ、両方とも中性態様であるが、シュウ酸又は適切なカルボン酸のような場合では、カフェインとの共結晶として調製されるとき、非共有相互作用(主に水素結合)により挙動することができ、例えばアミンとの反応又はプロトン交換のようなイオン塩を形成する場合では、プロトン供与体として挙動することができる。同様に、安息香酸及びコハク酸は、フルオキセチン塩酸塩と共結晶を形成する場合では、中性態様で挙動し(ホルマールプロトン交換なし)又は安息香酸ナトリウム若しくはコハク酸ナトリウムのようなイオン塩を形成するプロトン交換態様で挙動する。これらの化合物は、それ自体イオン性のゲストであることができる。中性ゲストは、好ましくは非イオン性のゲストである。イオン性ゲストは、イオン結合を有する化合物又は錯体である。ゲストは、塩化物(又は他のアニオン)と水素結合を形成する酸であることができる。イオン性ゲストは、イオン相互作用及び引力により例示されるようなイオン性を有する化合物又は錯体である。ゲストは、医薬成分と水素結合を形成する酸であることができる。例えば、酸である適切なゲストには、アスコルビン酸、グルコヘプタン酸、セバシン酸、アルギン酸、シクラミン酸、エタン−1,2−ジスルホン酸、2−ヒドロキシエタンスルホン酸、2−オキソ−5−グルタル酸、ナフタレン−1,5−ジスルホン酸、ニコチン酸、ピログルタミン酸及び4−アセトアミド安息香酸が含まれる(がこれらに限定されない)。本出願に提示されている溶質及び活性主成分には、その塩及び/又は溶媒和物が含まれる。共結晶は、WO2005/089375に記載されている。
【0087】
本発明の共結晶の例は、シルデナフィル又はその製薬学的に許容される塩、及びアセチルサリチル酸(アスピリン)である。
【0088】
本発明により作製されうる他の粒子は、鎮痛薬、例えばコデイン、ジヒドロモルヒネ、エルゴタミン、フェンタニル又はモルヒネ;アンギナール調合剤、例えばジルチアゼム;抗アレルギー薬、例えばクロモグリク酸、ケトチフェン又はネドクロミル;抗感染症薬、例えばセファロスポリン、ペニシリン、ストレプトマイシン、スルホンアミド、テトラサイクリン又はペンタミジン;抗ヒスタミン薬、例えばメタピリレン;抗炎症薬、例えばベクロメタゾン、フルニソリド、ブデソニド、チプレダン、トリアムシノロン、アセトニド又はフルチカゾン;鎮咳薬、例えばノスカピン;気管支拡張薬、例えばエフェドリン、アドレナリン、フェノテロール、ホルモテロール、イソプレナリン、メタプロテレノール、フェニレフリン、フェニルプロパノールアミン、ピルブテロール、レプロテロール、リミテロール、サルブタモール、 サルメテロール、テルブタリン、イソエタリン、ツロブテロール、オルシプレナリン又は(−)−4−アミノ−3,5−ジクロロ−a[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]メチル]ベンゼンメタノール;利尿薬、例えばアミロライド;抗コリン作用薬、例えばイプラトロピウム、アトロピン又はオキシトロピウム;ホルモン、例えば コルチゾン、ヒドロコルチゾン又はプレドニゾロン; キサンチン、例えばアミノフィリン、コリンテオフィリン、リシンテオフィリン酸塩又はテオフィリン;並びに治療タンパク質及びペプチド、例えばインスリン又はグルカゴンのような吸入により有用に送達されうる任意の薬剤又は活性成分を含む。活性成分又は薬剤を含む適切な医薬を、塩の形態で(例えば、アルカリ金属若しくはアミン塩として又は酸付加塩として)、又はエステル(例えば、低級アルカリエステル)として、又は溶媒和物(例えば水和物)として使用して、医薬の活性及び/又は安定性を最適化できることが、当業者には理解される。
【0089】
本発明の方法により得られる粒子の調製に特に適した医薬には、吸入治療による喘息のような呼吸障害の治療において使用される抗アレルギー薬、気管支拡張薬及び抗炎症性ステロイド、例えば、クロモグリク酸(例えば、そのナトリウム塩)、サルブタモール(例えば、その遊離塩基若しくはその硫酸塩)、サルメテロール(例えば、そのキシナホ酸塩)、テルブタリン(例えば、その硫酸塩)、レプロテロール(例えば、その塩酸塩)、ジプロピオン酸ベクロメタゾン(例えば、その一水和物)、フルチカゾンプロピオン酸エステル、(−)−4−アミノ−3,5−ジクロロ−α−[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]−メチル]ベンゼンメタノール、グリコピロニウム臭化物、ダロトロピウム、アクリジニウム、チオトロピウム(例えば、その臭化物塩)、テオフィリン、アロフィリン、ザフィルルカスト、モンテルカスト、カルモテロール(例えば、その塩酸塩)、ホルモテロール(例えば、そのフマル酸塩)又はインダカテロール、並びにその生理学的に許容される塩及び溶媒和物が含まれる。
【0090】
好ましい実施態様において、固体物質は、フルチカゾン化合物(例えば、そのプロピオン酸エステル又はフランカルボン酸エステル)とサルメテロール(例えば、そのフマル酸塩)の混合物を含む。
【0091】
ここでも、本発明の方法により作製される粒子は、本明細書において示唆されている2つ以上の活性成分の組み合わせを含有できることが、当業者には理解される。活性成分は、本明細書前記に記述された活性成分の適切な組み合わせから選択することができる。したがって、適切な気管支拡張性剤の組み合わせには、エフェドリンとテオフェリン、フェノテロールとイプラトロピウム及びイソエタリンとフェニレフリンが含まれる。
【0092】
本発明の方法により作製される活性成分の粒子の更なる適切な組み合わせには、ブデソニド、ジプロピオン酸ベクロメタゾン及びフルチカゾンプロピオン酸エステルのようなコルチコステロイドと、サルブタモール、テルブタリン、サルメテロール及びホルモテロールのようなβ2アゴニスト、並びにそれらの生理学的に許容される誘導体、特に硫酸塩を含む塩との組み合わせが含まれる。
【0093】
本発明の方法により作製される活性成分の粒子の特に適切な組み合わせには、ホルモテロールとフルチカゾン;ベクロメタゾンとホルモテロール;ホルモテロールとメタゾン;インダカテロールとメタゾン;イプラトロピウム臭化物とアルブテロール;サルブタモールとアルブテロール;チオトロピウム臭化物とホルモテロール;グリコピロニウム臭化物とインダカテロール;ホルモテロールとシクレソニド;ベクロメタゾン/サルメテロールのような組み合わせが含まれる。
【0094】
別の実施態様において、3つの成分を組み合わせることができ、コルチコステロイド、気管支拡張薬(例えば、ベータアゴニスト)及び抗コリン作用薬の組み合わせが含まれる。一例は、フルチカゾン/サルメテロール/チオトロピウム臭化物である。
【0095】
本発明の方法により得られる粒子の他の例は、クロモグリク酸ナトリウム若しくはネドクロミルでありうるクロモン又は炭水化物、例えばヘパリンを含むことができる。
【0096】
本発明の方法により作製される粒子は、吸入に適した活性成分を含むことができ、全身用途の薬理学的活性剤であることができる。例えば、そのような活性粒子は、デオキシリボヌクレアーゼ(DNase)、ロイコチン又はインスリン(プロインスリンを含む)、シクロスポリン、インターロイキン、サイトカイン、抗サイトカイン及びサイトカインレセプター、ワクチン、成長ホルモン、ロイプロリド及び関連する類似体、インターフェロン、デスモプレシン、免疫グロブリン、エリトロポエチン及びカルシトニンのような、ペプチド又はポリペプチド又はタンパク質を含むことができる。
【0097】
あるいは、本発明の方法により作製される活性成分は、経口投与に適している場合がある。経口投与用薬剤は、上記に記述した全身用薬剤の1つでありうる。活性成分は、消化管において低い可溶性を示す物質、例えば、三ケイ酸マグネシウム、炭酸カルシウム及び次硝酸ビスマスであることができる。有機化合物は、例えば、コンビナトリアル化学、ロシグリタゾン及び他の関連するグリタゾン薬、ヒドロクロロチアジド、グリセオフルビン、ラミブジン及び他のヌクレアーゼ逆転写インヒビター、シンバスタチン及び他のスタチン薬、ベザフィブレート及び他のフィブレート薬、ロラタジン、並びに他の任意の生理学的に許容される塩及びそれらの誘導体の全ての生成物を含むことができる。
【0098】
本発明の方法による加工に適した製薬学的賦形剤には、例えば、炭水化物、特にフルクトース、グルコース及びガラクトースのような単糖類;スクロース、ラクトース及びトレハロースのような非還元二糖類;ラフィノース及びメレジトースのような非還元オリゴ糖類;マルトデキストリン、デキストリン及びシクロデキストリンのような非還元デンプン由来多糖類生成物;並びにマンニトール及びキシリトールのような非還元アルジトールが含まれる。更に適した賦形剤には、例えばトウモロコシデンプン、コムギデンプン、コメデンプン、ジャガイモデンプン、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチルセルロース、ナトリウムカルボキシメチルセルロース及び/又はポリビニルピロリドン(PVP)のようなセルロース調製物が含まれる。上記の賦形剤のいずれかの2つ以上の混合物も考慮される。
【0099】
医療における使用では、本発明の化合物の塩は、非毒性の「製薬学的に許容される塩」と呼ばれる。FDAにより認証された製薬学的に許容される塩形態(International J. Pharm. 1986, 33,201 217; J. Pharm. Sci, 1977, Jan, 66 (1), p1)には、製薬学的に許容される酸性/アニオン性又は塩基性/カチオン性の塩が含まれる。
【0100】
本発明の酸性又は塩基性化合物の製薬学的に許容される塩は、当然のことながら、遊離塩基又は酸を少なくとも1つの理論量の所望の塩形成酸又は塩基と反応させるような、従来の手順により作製することができる。
【0101】
本発明の酸性化合物の製薬学的に許容される塩には、ナトリム、カリウム、カルシウム、マグネシウム、亜鉛及びアンモニウムのような無機カチオンとの塩、並びに有機塩基との塩が含まれる。適切な有機塩基には、NメチルDグルカミン、アルギニン、ベンザチン、ジオラミン、オーラミン、プロカイン及びトロメタミンが含まれる。
【0102】
本発明の塩基性化合物の製薬学的に許容される塩には、有機又は無機酸から誘導される塩が含まれる。適切なアニオンには、酢酸塩、アジピン酸塩、ベシル酸塩、臭化物、カンシラート、塩化物、クエン酸塩、エジシル酸塩、エストレート、フマル酸塩、グルセプト酸、グルコン酸塩、グルクロン酸塩、馬尿酸塩、ヒクレート、臭化水素酸塩、塩酸塩、ヨウ化物、イセチオン酸塩、乳酸塩、ラクトビオン酸塩、マレイン酸塩、メシレート、臭化メチル、硫酸メチル、ナプシレート、硝酸塩、オレイン酸塩、パモ酸塩、リン酸塩、ポリガラクツロ酸塩、ステアリン酸塩、コハク酸塩、硫酸塩、スルホサリチレート、タンニン酸塩、テレフタル酸塩、トシレート及びトリトヨージドが含まれる。
【0103】
本発明の方法により調製される活性成分の粒子が農芸化学的に活性である場合、活性成分は、例えば、植物成長調整剤、除草剤、及び/又は病虫害防除剤、例えば殺虫剤、殺真菌剤、ダニ駆除剤、殺線虫剤、殺ダニ剤、殺鼠剤、殺菌剤、軟体動物駆除剤又は鳥忌避剤であることができる。
【0104】
本発明の方法により作製される有機水不溶性農芸化学的活性成分の例には、例えば、メトミル、カルバリル、カルボフラン又はアルディカーブのようなカルバメート;EPN、イソフェンホス、イソキサチオン、クロルピリホス又はクロルメホスのような有機チオリン酸塩;テルブホス、モノクロトホス又はテトラクロルビンホスのような有機リン酸塩;メトキシクロルのような過塩素過有機化合物;フェンバレレートのような合成ピレスロイドからなる群より選択される殺虫剤;例えば、メトリブジン、ヘキサキシノン又はアトラジンのようなトリアジン;2−クロロ−N−[(4−メトキシ−6−メチル−1,3,5−トリアジン−2−イル)アミノカルボニル]−ベンゼンスルホンアミドのようなスルホニル尿素;レナシル、ブロマシル又はテルバシルのようなウラシル(ピリミジン);リニュロン、ジウロン、シズロン又はネブロンのような尿素;アラクロール又はメトラクロールのようなアセトアニリド;ベンチオカルブ(SATURN)、トリアレートのようなチオカルバメート;オキサジアゾンのようなオキサジアゾール−オン;2,4−Dのようなフェノキシ酢酸;フルアジホップ−ブチル、アシフルオルフェン、ビフェノックス又はオキシフルオルフェンのようなジフェニルエーテル;トリフルラリンのようなジニトロアニリン;グリフォセート塩及びエステルのようなグリシンホスホネート;ブロモキシニル又はイオキシニルのようなジハロベンゾニトリルからなる群より選択されるオキサミル除草剤のような殺線虫剤カルバメート;例えば、シモキサニル(クルゼート)のようなニトリロオキシム;ベノミル、カルベンダジム又はチオフェネート−mエチルのようなイミダゾール;トリアジメホンのようなトリアゾール;カプタンのようなスルフェンアミド;マネブ、マンコゼブ又はチラムのようなジチオカルバメート;クロロネブのような塩素過芳香族化合物;イプロジオンのようなジクロロアニリンからなる群より選択される殺真菌剤;ピリミカーブのようなカルバメートからなる群より選択されるアブラムシ駆除剤;,例えば、プロパルギットのような亜硫酸プロピニル;アミトラズのようなトリアザペンタジエン;クロロベンジレート又はテトラジホンのような塩素過芳香族化合物;及びビナプアクリルのようなジニトロフェノールからなる群より選択される殺ダニ剤が挙げられる。
【0105】
有機水不溶性農芸化学的活性成分は、幾つかの成分の混合物として、本発明により製造される粒子に含まれうる。特に好ましい有機水不溶性農薬活性成分は、アトラジン、シモキサニル、クロロタロニル、シプロコナゾール及びテブコナゾールである。
【0106】
非溶媒及び溶媒は、特定の活性成分又は活性前駆体に適しているように選択されるべきであることが理解される。ブデソニド、ジプロピオン酸ベクロメタゾン及びフルチカゾンプロピオン酸エステルのようなコルチコステロイドを、ジクロロメタン又はメタノールに溶解し、ヘプタンのような非溶媒中で超音波処理することができる。サルメテロールキシナホ酸塩及びサルメテロールフマル酸塩のようなβ2−アゴニストをメタノールに溶解し、アセトン、酢酸エチル又はヘプタンのような非溶媒中で超音波処理することができる。
【0107】
サイクロン分離のような従来の分離工程に従い、乾燥粒子を非溶媒と接触させ、次に超音波照射に付して、結晶を形成するか又は本明細書前記に記載した所望のMMADの非晶質構造を焼還及び/若しくは安定化する。粒子を、使用される場合は超音波プローブの又はこの構造が用いられる場合は巻き付け超音波エネルギー変換器アセンブリーのような超音波エネルギー変換器の稼働近辺に付される。そのような装置の適切な例は、WO00/35579において確認されている。超音波エネルギーを、連続的方法又はパルス適用のように非連続的方法により適用することができる。任意の適切な超音波照射供給源を使用することができる。連続超音波フローセルのような超音波プローブを、例えば混合容器の中に挿入することができるか、超音波発信器を混合容器の中に含めることができるか、或いは混合容器を超音波浴の中に収容することができるか又は超音波変換器を混合容器の外壁に固定することができる。超音波の振幅及び周波数は、核生成及び結晶成長に影響を与える。超音波の周波数は、例えば、16kHz〜1MHz、好ましくは10〜500kHz、より好ましくは10〜100MHz、例えば、10、20、40、60、80若しくは100kHz又は30kHz〜50kHzのようなその間の任意の周波数であることができる。
【0108】
超音波照射は、所定の用途において、所望の大きさの結晶の生成に適した振幅又は出力密度で用いられる。発射面が例えば80cm2の実験室プローブ系では、選択される振幅は、約1〜30μm 、典型的には3〜20μm 、好ましくは5〜10μm、例えば6μmであることができる。8cm2のプローブ面及び5〜80Wの所要電力を有するプローブは、2〜15μmの振幅を使用して約0.6〜12.5W/cm2の出力密度をもたらす。好ましくは、フローセル、例えば6リットルのフローセルに結合している変換器を含むWO03/101577に記載されているような大型の系では、用いられる変換器の出力密度は、10〜100W/L、好ましくは30〜80W/L、より好ましくは50〜75W/L、例えば60W/L〜70W/Lであることができる。本発明は、工業規模の生産に特に適している。
【0109】
超音波フローセルにおける混合成分の滞留時間は、好ましくは0.1ミリ秒より長く、より好ましくは1ミリ秒より長く、より好ましくは1分より長く、例えば1秒〜24時間、より好ましくは1分〜6時間、より好ましくは5分〜1時間であることができる。
【0110】
生成された結晶を、当該技術の従来の方法を使用して結晶を引き出すことによりバッチチャンバーから又は水性懸濁剤として収集又は採取することができる。
【0111】
本発明により生成される粒子は、実質的に結晶質であり、低減された吸湿の傾向を示し、このこと物理的及び化学的安定性を増加することに寄与する。「実質的に結晶質」とは、粒子の総重量に対して結晶粒子重量%として表される粒子の結晶化度が、90%より高く、好ましくは93%より高く、さらにより好ましくは95%より高いことを意味する。前記粒子は、特に粒子が吸入用の乾燥粉末剤として配合される場合、均質の製剤を容易に得ることを可能にする優れた分散特性も示す。粒子の結晶化度は、示差走査熱量測定(DSC)、X線粉末回折又はマイクロ熱量測定法のような当業者に既知の他の技術、好ましくはDSCを使用して決定することができる。
【0112】
一つの実施態様において、固体物質は、コルチコステロイドであり、好ましくは、呼吸疾患の予防及び/又は治療のために吸入用にとって有用でありうる、ヨーロッパ薬局方、第4版、2002年に示されている溶解度の定義に従って水において不溶性又は難溶性である任意のコルチコステロイドである。好ましくは、コルチコステロイドは、50μgを超える、好ましくは80μg以上、より好ましくは100μg以上の単回治療用量を有する。
【0113】
好ましくは、コルチコステロイドは、50μgを超える、好ましくは80μg以上、より好ましくは100μg以上の単回治療用量を有する。
【0114】
好ましくは、コルチコステロイドは、ジプロピオン酸ベクロメタゾン(BDP)、ブデソニド、シクレソニド、モメタゾン及びフロン酸エステルのようなそのエステル、並びにフルチカゾン及びプロピオン酸エステルとフロン酸エステルのようなそのエステルからなる群より選択される。本発明の好ましい実施態様において、コルチコステロイドは、ブデソニド又はフルチカゾン及びその塩又はエステルである。
【0115】
好ましくは、本発明の活性粒子は10μm未満の体積直径を有し、より好ましくは、所定の組成物において少なくとも90重量%の活性成分粒子は、上記に記載されたレーザー回折により、好ましくはMalvern又は同等の装置を使用して、体積直径として知られている特徴的な等体積球相当径を測定して決定された10μm以下の直径を有する。考慮されるパラメーターは、粒子の大きさ独立密度を推定する質量直径に対応する、それぞれd(10)、d(50)及びd(90)と表される10%、50%及び90%の粒子の体積直径(VD)(μm)である。
【0116】
好ましくは、10重量%以下の前記粒子が、0.8μm未満の体積直径d(10)を有し、好ましくは、50重量%以下の前記粒子が、2.0μm未満の体積直径d(50)を有し、好ましくは、90重量%以下の前記粒子が、10μm以下の体積直径d(90)を有する。好ましくは、100重量%の前記粒子が、10μm以下の体積直径を有する。
【0117】
本発明の粒子の活性成分は、実質的に純粋な形態である。「実質的に純粋な形態」とは、少なくとも95%w/w、好ましくは少なくとも98%又は少なくとも99%w/wの純度を意味する。化学的な純度は、高速液体クロマトグラフィー(HPLC)のような当業者に既知の方法に従って決定することができる。
【0118】
別の態様において、本発明は、本発明の粒子を含む、吸入による投与用の製剤を提供する。粒子を、1つ以上の製薬学的に許容される賦形剤、添加剤、希釈剤又は担体と一緒に、前記製剤に配合することができる。例えば、製剤は、加圧式定量噴霧吸入器(pMDI)により投与される、エアロゾル担体としての噴射剤中の懸濁形態で提供される。
【0119】
懸濁製剤は、界面活性剤及び湿潤剤のような追加の賦形剤を含むことができる。
【0120】
好ましい実施態様において、製剤は、乾燥吸入粉末剤の形態で、より好ましくは相互作用規則混合物の形態で、すなわち、本発明の粒子を、粗粒子からなる薬理学的に不活性で生理学的に許容される賦形剤により希釈することによって、提供される。
【0121】
有利には、吸入用の前記粉末製剤は、本発明の粒子と、生理学的に許容される賦形剤の粗粒子(本明細書以降、「担体粒子」と呼ぶ)、例えば50μmより大きい中央粒子径(MMD)、好ましくは50μm〜500μm、より好ましくは150〜400μm、さらにより好ましくは210〜355μmの中央粒子径(MMD)を有する粒子とを含むことができる。別の実施態様において、粗粒子は、90〜150μmを含むMMDを有する。MMDは、頻度分布を半分に割った粒子直径であり、エアロゾル質量の50パーセントが、大きな直径の粒子を有し、エアロゾル質量の50パーセントが、小さな直径の粒子を有する。
【0122】
好ましくは、少なくとも50重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、少なくとも80重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、少なくとも90重量%の担体粒子が、500μm未満の直径を有し、より好ましくは、100重量%の担体粒子が、500μm未満の直径を有する。
【0123】
生理学的に許容される賦形剤を、動物若しくは植物供給源又はこれらの組み合わせの任意の非晶質又は結晶質の生理学的に許容される薬理学的に不活性な物質から構成することができる。好ましい物質は結晶糖であり、例えば、グルコース若しくはアラビノースのような単糖類、又はマルトース、サッカロース、デキストロース若しくはラクトースのような二糖類である。マンニトール、ソルビトール、マルチトール、ラクチトールのようなポリアルコールを使用することもできる。最も好ましい物質は、α−ラクトース一水和物である。
【0124】
市販のラクトースの例は、Capsulac(商標)及びPharmatose(商標)である。市販のマンニトールの例は、Pearlitol(商標)である。
【0125】
製剤を、呼吸作動式鼻吸入器により投与される懸濁剤又は粉末剤の形態で提供することもできる。
【0126】
前記粉末製剤を、当該技術に既知の任意の種類のDPIを用いて吸入により投与することができる。
【0127】
DPIは、2つの基本的な種類に分けることができる:i)活性化合物の予め細分化された単回用量を投与するための単回用量吸入器;ii)予め細分化された単回用量を有するか又は多回用量に十分な活性成分の量を予め装填した多用量乾燥粉末剤吸入器(MDPI)。必要とされる吸気流速(l/分)に基づき、設計及び機械的特徴に厳密に依存して、DPIは、i)低抵抗装置(>90l/分);ii)中抵抗装置(約60l/分);iii)高抵抗装置(約30l/分)に分けられる。
【0128】
活性成分の薬理学的活性に関して、本発明の粒子を、軽度、中程度若しくは重篤な急性若しくは慢性の症状の予防及び/若しくは治療のため又は喘息及び慢性閉塞性肺疾患(COPD)のような呼吸疾患の予防的治療のために指示することができる。慢性閉塞性細気管支炎及び慢性気管支炎のような炎症及び粘液の存在よりもたらされる末梢気道の閉塞によって特徴付けられる他の呼吸障害も、これらの使用により利益を受ける。
【0129】
吸入による投与では、本発明の方法により生成される粒状活性成分は、好ましくは担体粒子と配合される。前記活性成分は、製剤の0.1〜90重量%、好ましくは製剤の0.25〜50重量%、より好ましくは1〜25重量%で存在する。好ましくは、担体粒子は、製剤の10〜99.9重量%、より好ましくは製剤の50〜99.75重量%、より好ましくは75〜99重量%の量で存在することができる。
【0130】
特に好ましい実施態様において、本発明により生成された粒子の活性成分は、フルカチゾンプロピオン酸エステル、ブデソニド、ホルモテロール、サルメテロール、ベクロメタゾン又はベタメタゾン、並びにこれらの混合物及び共結晶を含む(好ましくは、前記物質から実質的になる)。このリストは、前記化合物の塩、水和物及び溶媒和物も包含する。
【0131】
走査電子顕微鏡法(SEM)により、前記活性粒子が、出発物質のSEM画像と比較したとき、有意に特徴的であることを明確に観察することができる。本発明の粒子が、より均一で規則的な球状を示し、少量の微細粒子も存在する出発物質と同じように破損して不規則であるようには見えない。理論に束縛されることなく、表面形態の前記の差は、本発明の粒子の低い凝集傾向、したがって優れた分散特性の説明に寄与すると考えられる。
【0132】
本発明により生成される活性成分の粒子は、好ましくは実質的に球状である。このことは、粗面を有する粒子を排除するものではない。好ましくは、本発明により生成される粒子は、最大直径と最小直径の平均比が、1.3〜1:1、より好ましくは1.25〜1:1、より好ましくは1.2〜1.01:1、より好ましくは1.15〜1.02:1、より好ましくは1.1〜1.03:1、より好ましくは1.075〜1.05:1である。したがって、本発明の粒子は、実質的に球状であることが分かる。
【0133】
粒径と形状分析を組み合わせたSympatec QICPIC画像分析センサーのような、多数の粒径及び形状分析器具が利用可能である。この1ナノ秒未満の極めて短い暴露時間を用いる技術は、100m/秒までの速度の最速粒子の鮮明な画像も提供する分散単位の使用を可能にする。このことは、凝集した微細及び凝集性粉末の正確な分散を保証する。1μm〜20μmの粒径を測定することができる。一次測定データは、30000の一次分類に記憶され、個別の限定フォーマットにより評価することができる。予め定義された粒度分類のセットを、存在する測定仕様に容易に適合させることができる。高速データ圧縮モジュールは、1秒あたり500枚までの画像の取得を支援する。1μm以下の粒子は、前記に記載されたMalvern又はSympactec回折、好ましくはMalvernレーザー回折のようなレーザー回折技術により測定することができる。
【0134】
接着API薬剤粒子をエアロゾル化するのに必要な力が、接触面の表面エネルギーの合成に比例し、投影接触面に反比例することは、良く知られている。したがって、DPIのエアロゾル化効率を改善する大部分の一般的な手法は、接触面の表面自由エネルギーを低減すること又は粒子形状を変えて接触面を制限することである。表面積は、粒径及び形状のみにより決定されるものではなく、表面形態も表面積に寄与し、波形(すなわち、粗い)粒子は、同じ体積を占める平滑粒子よりも大きな表面積を有する。
【0135】
本発明の方法により調製される薬剤粒子は、特定の表面形態により定義することができる。粒子間力を調節して、肺への付着を向上させることができる。理想的には、接触面、したがって力は、安定した製剤をもたらし、それでも吸入時には容易な分離を可能にするために、薬剤と担体との間に十分な接着をもたらすようなレベルに調整されるべきである。微細粒子画分に対する表面波形の影響を、明確にすることができる。
【0136】
平滑表面ラクトン担体粒子は、微細粒子画分及び微粉化薬剤の分散性を増加することが示されており、一方、他の研究は、波形担体粒子が微細粒子画分を増加することを示した。これらの明らかに矛盾する結果は、表面力平衡は、表面構造だけではなく幾つかの変数によって決まるという仮定により説明することができる。
【0137】
例として記載される粒子では、表面積及び形態測定値は、表面積が粒子相互作用と高度に相関することを明らかにしている。粉末表面積の決定は、所定の圧力で粉末表面に吸着されたガスの量を測定することを含む。過去数十年間にわたって、表面を研究する新たな技術が出現してきた。
【0138】
本発明の粒子の表面積は、吸着性ガスとして窒素を使用するAccelerated Surface Area and Porosimetry Analyser(model ASAP 2000, Micromeritics, Norcross, GA)により決定した。粉末物質(0.3〜0.7g)を窒素下、45℃でおよそ24時間脱ガスして、予め吸着したガス及び蒸気をサンプルの表面から除去した。表面積は、0.07〜0.22の相対圧力(P/Po)で吸着データを使用する、マルチポイントBrunauer, Emmett and Teller(BET)法により決定した。
【0139】
好ましくは、本発明の粒子は、6〜22m2/g、好ましくは9〜18m2/g、より好ましくは10〜13m2/gの範囲、より好ましくは12m2/gの表面積を有する。
【0140】
逆ガスクロマトグラフィー(Inverse Gas Chromatography, IGC)は、固体物質の表面及び嵩特性を特徴決定するガス相技術である。ICGの原理は非常に簡単であり、従来のガスクロマトグラフィー(Gas Chromatography, GC)実験の逆である。円柱カラムを、目的の固体物質、典型的には粉末、繊維又は膜で均一に充填し、保持時間及び溶出ピーク形状を、一連の十分に特徴決定された非極性及び極性ガスによって研究する。パルス又は一定濃度のガスを、固定した担体ガス流速でカラムの注入し、パルス又は濃度が先からカラムの底に溶出するのに掛かる時間を検出器により測定する。異なるガス相プローブ分子による一連のIGC測定は、広範囲の接触を可能にする。IGCを、表面エネルギーを測定するため、同様に、加工により引き起こされる表面特性の小さな変化を研究するために使用する。
【0141】
IGCを、本発明の粒子の表面エネルギーを測定するために使用した。IGCは、2セットの条件により実施することができる。有限希釈では、吸着等温線をピークプロフィールから誘導することができ、吸着エネルギー分布を計算するために使用することができる。第2に、無限希釈では、器具の検出限界に近い溶質の量を注入し、この場合、溶質−溶質相互作用は小さく、溶質−吸着剤相互作用のみが測定保持時間に影響を与える。これは、プローブ(ガス)分子と固定粉末との間の分散的と特異的の両方の相互作用を生じることができる。
本発明の粒子は、図21において非常に明瞭に示されるように、表面エネルギーの等エネルギー分布を有することによって特徴付けられる。この表面エネルギーは、本発明の好ましい方法により調製される粒子の有限及び無限希釈の両方において、極めて類似し、ほぼ同一であり、一方、典型的な微粉化粒子は、有限及び無限希釈において顕著な差を示す。
【0142】
原子間力顕微鏡法(Atomic Force Microscopy, AFM)を使用して凝集−接着平衡を測定することができ、サンプルの表面にプローブチップを接触して設置する(接触モード原子間力顕微鏡法)又はサンプルの表面に非常に近接して設置する(非接触及びタッピングモード原子間力顕微鏡法)ことによって、高さを測定するように機能する。プローブチップは、チップから受ける力に比例して湾曲する弾性カンチレバーに結合している。原子間力顕微鏡はサンプルをラスター走査して、データポイントのマトリックスを生成し、それから定量的な高さ及び粗さ測定値を抽出することができる。タッピングモード原子間力顕微鏡法は、薬剤を含む多様な有機化合物の結晶及び担体粒子ラクトースの接着特性を効果的に画像化する。コロイドプローブ顕微鏡法(Colloid Probe Microscopy, CPM)は、微晶質粒子の間の粒子−粒子接着を測定するために、日常的に使用される。
【0143】
サンプルの波形及び表面形態の程度を、AFMを使用して定量化した。本発明の粒子の表面様相及び粗さ測定値を、Nanoscope IIIコントローラー、Multimode AFM及びJ型スキャナー(全て、Dl, Santa Babara, CA, USA)を使用して調査した。全てのAFM表面様相画像を、画像化が1Hzの走査速度のTESP Olympusチップ(Di, Cambridge, UK)を使用して実施される、タッピングモード操作により記録した。表面粗さ測定値を、1μm×1μm領域で分析した。結晶表面の表面特性の差を定性化するために、表面凹凸の高さの差の実効値表面粗さ測定(Rq)及び平均表面粗さ(Ra)を計算した。
【0144】
力測定の前に、サンプルの各バッチの粒子を、エポキシ樹脂接着剤(Araldite, Cambridge, UK)を使用して、予め定義されたばね定数を有する標準V字形チップレスカンチレバー(DNP-020, Dl, CA, USA)に結合した。3つのチップを各サンプルのために調製し、接着剤の薄層が乾燥する前に、全てのプローブを、光学顕微鏡(倍率50×)により検査して、結合粒子の一体性を確実にした。
【0145】
基材を、周囲条件が25℃(±1.5℃)の一定温度及び35%RH(±3%)の相対湿度で維持して特注の環境チャンバーに収納されているAFMスキャナー台に設置した。相互作用力は、フックの法則を適用し(F=−kz)、基材の変位の関数としてAFMカンチレバーのたわみを記録することによって測定した。個別の力曲線(n=1024)を、4Hzの走査速度及び40nNの圧縮荷重により10μm×10μmにおいて実施した。パラメーターを一定に保持した。
【0146】
本発明の粒子は、図22に示されるように、実質的に波形表面を有することによって特徴付けられる。好ましくは、本発明の粒子は、ナノメートル規模の表面波形を有する。好ましくは、Rq値は、10〜100nm、より好ましくは20〜90nmである。好ましくは、Ra値は、10〜100nm、より好ましくは20〜90nmである。
【0147】
AFMコロイドプローブ技術の凝集−接着平衡(CAB)手法は、凝集性及び接着性の直接的な定量化−製剤内のAPIの「力平衡]−を可能にする開発である。ミリグラム以下の材料を使用して潜在的な製剤成分の凝集/接着力相互作用を決定する、市販のスクリーニングツールである。
【0148】
CAB手法は、コロイドAFMプローブとして設置されているAPI粒子と、明確に画定されたAPIの結晶質表面と担体基材との間の相互作用力を測定する。CABプロットは、定量化された、賦形剤物質へのAPIの接着傾向に対する凝集性の特徴的測定を可能にする多数のプローブの相互作用から生成される。CAB手法の開発は、器具の検証についての問題及び相互作用表面の真接触領域を決定する必要性を含む、従来のAFMコロイドプローブ方法論に関連する多数の制限を克服した。CAB値の1は、粒子−担体接着の力が粒子−粒子凝集の力と等しいことを示す。CAB比<1は、他の変数が等しいと仮定して、薬剤がそれ自体と凝集性であるよりも担体に対して接着性があることを示し、それによって、ブレンドすると安定した規則的な混合物を形成するとを予測することができる。しかし、CAB比>1は、薬剤が担体に対して接着性があるよりも、それ自体に凝集性があることを示し、ブレンドすると薬剤の凝集塊を含有する均一性の少ない混合物を生成しうることを示唆する。>1のような高いCAB比を有する薬剤−担体の組み合わせは、大きさ、形状、粗さ及び流動性に関して担体の間に潜在的な差があるにもかかわらず、エアロゾル化によってより多くの微細粒子画分(FPF)をもたらす。<1のような低いCAB比を有する薬剤−担体の組み合わせは、担体に対する活性粒子の大きな凝集をもたらし、したがって、活性粒子は、担体に結合したままの可能性が多くなる。このことは、吸入組成物において、担体粒子に結合したままの活性粒子が肺に到達せずに、咽喉又は舌に付着しうることを意味する。好ましくは、本発明により生成される粒子は、微粉化及び粉砕を含む他の方法により調製される粒子よりもはるかに低い凝集性を有する。凝集性の測定に関連し、全ての基材が凝集性及び接着性に関して異なっているので、CAB比は、無次元値であり、比較にとってより有用な測定値である。好ましくは、本発明により生成される粒子は、担体の0.8〜1.3、より好ましくは0.9〜1.2、より好ましくは1.0〜1.1のCAB比を有する。これは、薬剤のそれ自体の凝集及び薬剤と担体の接着の注意深い平衡である。
【0149】
本発明のエアロゾル製剤の化学的及び物理的安定性、並びに製薬学的許容性は、当業者に周知の技術によって決定することができる。したがって、例えば、成分の化学的安定性は、例えば生成物の長期保存後に、HPLCアッセイにより決定することができる。物理的安定性データは、例えば、漏れ試験、バルブ送達アッセイ(1作動あたりの平均ショット重量)、用量再現性アッセイ(1作動あたりの活性成分)及び噴霧分布分析のような、他の従来の分析技術によって得ることができる。
【0150】
本発明のエアロゾル製剤の粒径分布は、例えばカスケード衝撃又は「Twin Impinger」分析方法によるプレセパレーターを用いる、例えばNext Generation lmpactor(NGI)を使用する従来の技術によって測定することができる。本明細書で使用されるとき、「Twin Impinger」アッセイの参照は、英国薬局方1988年A204〜207頁、付録XVII Cにおいて定義されている“Determination of the deposition of the emitted dose in pressurised inhalations using apparatus A”を意味する。そのような方法は、HPLC移動相でプレセパレーターを充填することを含み、NGIカップのカップは、ヘキサン中の1%v/vシリコーンで被覆されて、粒子の跳ね返りを排除している。典型的には同じ製剤の4つの個別のカプセルを、所定の条件下でNGIに投入する。エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流す。次にNGIの各部品に付着した薬剤の量を、HPLCにより決定することができる。FPD決定は、NGIの段階3〜8で収集した薬剤量を表す。FPF発射用量も決定される。微細粒子画分の率(FPF%)又は呼吸画分により決定されるエアロゾル化効率も、評価される。
【0151】
そのような技術は、エアロゾル製剤の「呼吸画分」の計算も可能にする。本明細書で使用されるとき、「呼吸画分」への参照は、上記に記載された方法を使用して、1回の作動あたり送達される活性成分の総量の率として表される、1回の作動あたりのNGIの下部チャンバーに収集された活性成分の量を意味する。本発明の製剤は、医薬の発射用量の10〜30重量%以上、好ましくは14〜26重量%、例えば約15.9重量%及び約25.9重量%の呼吸画分を有することが見出されており、実施例2及び3により例示されている(図13及び19に示されている)。実施例1(ブデソニド)、実施例2(フルチカゾンプロピオン酸エステル)及び実施例8(フェノテロール臭化水素酸塩)では、従来技術と比較して、それぞれ、本発明の粒子に53%、50〜60%及び30〜50%のFPFの増加があった。
【0152】
本発明の粒子を含む吸入製剤に使用される噴射剤は、噴射剤として有効にするのに十分な蒸気圧を有する任意のフルオロカーボン又は水素含有クロロフルオロカーボン又はこれらの混合物を含む。好ましくは、噴射剤は、医薬の非溶媒である。適切な噴射剤には、従来の水素含有クロロフルオロカーボン、非クロロフルオロカーボン、水素含有フルオロカーボン及びペルフルオロカーボンなどが含まれる。特に、噴射剤HFA134a及びHFA227又はこれらの混合物を有利に使用することができる。
【0153】
本発明の製剤を、医薬エアロゾル製剤の送達に適したキャニスターに充填することができる。キャニスターは、一般に、プラスチック又はプラスチック被覆ガラス瓶又は好ましくは金属缶、例えば任意に陽極酸化処理、ラッカー被覆及び/若しくはプラスチック被覆されうるアルミニウム缶のような、使用される噴射剤の蒸気圧に耐えることができる容器を含み、容器は、計量バルブで密閉されている。計量バルブは、作動毎に計量された量の製剤を送達するように設計され、バルブを通した噴射剤の漏れを防止するためにガスケットを組み込む。ガスケットは、例えば、低密度ポリエチレン、クロロブチル、白黒ブタジエン−アクリロニトリルゴム、ブチルゴム及びネオプレンのような、任意の適切なエラストマー材料を含むことができる。
【0154】
適切なバルブは、エアロゾル産業において周知の製造者、例えば、Valois, France(例えば、DF10、DF30、DF60)、Bespak pic, UK(例えば、BK300、BK356)及び3M-Neotechnic Ltd, UK(例えば、SpraymiserW)により市販されている。
【0155】
医薬エアロゾル製造の当業者に周知の従来のバルク製造法及び機械を、充填キャニスターの商業的生産のための大規模バッチの調製に用いることができる。
【0156】
典型的には、医薬用途のために調製されるバッチでは、それぞれの充填キャニスターの重量を検査し、バッチ番号をコード化し、放出試験の前に保存のためにトレーに積み重ねる。
【0157】
各充填キャニスターを、使用前に適切なチャネリング装置に都合良く取り付けて、患者の肺又は鼻腔への医薬の投与用の定量吸入器を形成する。適切なチャネリング装置は、例えば医薬がそれを通って充填キャニスターから計量バルブを介して患者の鼻又は口に送達されうる、バルブ作動器及び円柱又は円錐様経路、例えばマウスピース作動器を含む。定量吸入器は、1回の作動又は「吸い」あたりに医薬の固定単位投与量を送達するように設計され、例えば1吸いあたり10〜5000マイクログラムの範囲の医薬である。医薬の投与を、軽度、中程度又重篤な急性又は慢性症状の治療又は予防的治療のために指示することができる。投与される正確な用量は、患者の年齢及び状態、使用される特定の粒状医薬、投与頻度、最終的には担当医の裁量に応じて決まることが理解される。医薬の組み合わせが用いられる場合、組み合わせの各成分の用量は、一般に、各成分が単独で使用される場合に用いられるものである。典型的には、投与は、1日あたり1回以上、例えば1〜8回であることができ、例として、1回あたり1、2、3又は4回の吸いである。
【0158】
適切な1日の用量は、例えば、疾患の重篤度に応じて、50〜200μmの範囲のサルメテロール、100〜1000μmの範囲のサルブタモール、50〜2000μmの範囲のフルチカゾンプロピオン酸エステル、又は、100〜2000μmの範囲のジプロピオン酸ベクロメタゾンである。
【0159】
したがって、例えば、各バルブ作動は、25μgのサルメテロール、100μgのサルブタモール、25、50、125又は250μgのフルチカゾンプロピオン酸エステル又は50、100、200又は250μgのジプロピオン酸ベクロメタゾンを送達することができる。典型的には、定量吸入器に使用される各充填キャニスターは、医薬の100、160又は240の計量用量又は吸いを含有する。
【0160】
本明細書に記載される充填キャニスター及び定量吸入器は、本発明の更なる態様を含む。
【0161】
ここで本発明は、添付の実施例及び図面を参照して記載される。実施例及び図面は本発明の範囲をいかなる方法によっても制限すると解釈されるべきではないことが理解されるべきである。
【図面の簡単な説明】
【0162】
本発明の方法は、添付の図面に示されているように、従来の装置を使用して実施することができる。
【図1】従来の噴霧乾燥ブデソニド(超音波処理あり)を示す。
【図2】本発明の超音波処理を有する噴霧乾燥ブデソニドを示す。
【図3】従来の噴霧乾燥ブデソニドのDSCを示す。
【図4】本発明の乾燥粉末超音波処理を有する噴霧乾燥ブデソニドのDSCを示す。
【図5】乾燥固体収集チャンバーが、セルに挿入されている超音波プローブを有する超音波セルに代えられている、従来の噴霧乾燥装置の図表示を示す。
【図6】図5と同様の構造において結合した変換装置を示す。
【図7】図5及び6と同様の構造において複数の結合した変換装置を示す。この場合、複数の変換器は円筒ダクトの周りを囲むように取り付けられている。
【図8】複数の変換器装置が再循環ループを構成している、図7と同様の複数の変換器装置を示す。
【図9】(本発明の)エアロゾル化により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図10】(本発明の)エアロゾル化により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図11】超音波を用いる分散及び沈殿により調製されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図12】微粉化/粉砕されたフルチカゾンプロピオン酸エステルのサンプルを示す。
【図13】多様なサンプルのエアロゾル効率を示す。
【図14】微細粒子画分を表す棒グラフを示す。
【図15】GSKのFlixotideと比較したエアロゾル効率を示す。
【図16】超音波処理後のフェノテロール臭化水素酸塩の粒子を示す。
【図17】微粉化フェノテロール臭化水素酸塩を示す。
【図18】粒径分布データGR005/180/A4及びGR005/179/Cは、本発明により調製される粒子である。
【図19】吸入装置を使用する比較微細粒子画分(FPF)データを示す。
【図20】吸入専用検査用具を使用する比較微細粒子画分(FPF)データを示す。
【図21】有限及び無限希釈でのIGCによる表面エネルギー測定を示す。
【図22】ブデソニド微粉化粒子と本発明の粒子のAFMトポロジープロフィールを示す。
【図23】1及び3か月の保存後の多様なFPサンプルのエアロゾル効率を示す。
【図24】1及び3か月の保存後の多様なFPサンプルのエアロゾル効率を比較した棒グラフを示す。
【図25】Next Generation Impactorの多様な段階の粒子画分分布グラフを示す。
【図26】1か月の保存後のフルチカゾンプロピオン酸エステルの多様なサンプルの微細粒子画分を示す。
【図27】微細粒子画分を表す棒グラフを示す。
【図28】サルメテロールキシナホ酸塩粒子(10重量%)と組み合わせた、本発明により調製されたフルチカゾンプロピオン酸エステル粒子(90重量%)を示す。
【図29】フルチカゾンプロピオン酸エステルとラクトンの凝集−接着平衡を示す。
【図30】微粉化SXとブレンドしたときの、本発明のFP粒子の優れた均質性を示す。
【図31】Advarと比較した、微粉化SXとブレンドしたときの本発明のFP操作のFPFLDを示す。
【図32】サンプル3のFPの表面粗さのAFM輪郭プロットを示す。
【図33】サンプル4のFPの表面粗さのAFM輪郭プロットを示す。
【発明を実施するための形態】
【0163】
図3及び4を参照すると、2つのバッチのDSC追跡の比較では、噴霧乾燥粒子への超音波の適用が、粒子の物理的特性を変更することを明確に示している。120℃での発熱(正のピーク)は、DSC装置における非晶質から結晶質への変換を示す。一般的に、加工された物質の結晶特性には明確な改善がある。
【0164】
図5を見ると、超音波装置を用いる噴霧乾燥は、液体供給チャンバー1、噴霧乾燥噴霧器及び加熱ガス入口2、蒸発チャンバー3、サイクロン分離器4、連続超音波処理チャンバー5(熱ジャケット6で囲まれている)を含む。従来の処理噴霧乾燥粉末は、超音波フローセルチャンバー5に直接入れられる。同時に、非溶媒7の連続供給が、粒子スラリー9の流速と平衡した適切な速度により、ポンプ8を介して、濾過又は乾燥による後加工にポンプで送られる。超音波プローブ10は、超音波エネルギーを混合物に照射し、混合物は、出口11を通って流れる。溶媒蒸気、超微細粒子及びガス12は、フィルター13を介して排出される。超音波放射は、所望の粒径及び結晶化度が達成されるまで、必要な限り続けられる。当然のことながら、噴霧乾燥器の供給流は、粒子スラリーが取り出せる速度と平衡している。流速は、超音波フローセルチャンバー5における滞留時間が、例えば10秒〜1時間になるように制御される。顕微鏡規模で生じる局所キャビテーションは、前述の固体状態効果を誘導する流体温度及び圧力において変化を促進する。超音波の出力及びチャンバー5の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー5内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0165】
図6を見ると、超音波装置による噴霧乾燥は、チャンバー21が外面に位置する単一の結合超音波変換器22を有する以外は、図5と同様の構造である。変換器22は、チャンバー21の全容量に分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で受声波作用を与え、超音波の出力及びチャンバー21の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー21内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0166】
図7を見ると、超音波装置による噴霧乾燥は、チャンバー31が外面に位置する単一の結合超音波変換器32を有する以外は、図5及び6と同様の構造である。巻き付け変換器32は、チャンバー31の全容量に分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で受声波作用を与え、超音波の出力及びチャンバー31の滞留時間を調整することによって、粒径及び形態を制御することができる。超音波は、チャンバー31内の任意の結晶付着が表面から除去される傾向があるという、更なる利点を有する。
【0167】
図8を見ると、これは、チャンバー31が、熱調節ジャケット43及びポンプ42を介した任意の撹拌羽根車44を備えた一次粒子収集容器41に結合して、連続閉ループ加工系を作り出す以外は、図7と同様の構造の、超音波装置を有する噴霧乾燥装置を示す。超音波は、分散、解凝集及び非晶質から結晶質への又は準安定から安定した結晶質への変換を引き起こすのに十分な強度で適用され、超音波の出力及び再循環加工ループ31、41、42の滞留時間を調整することによって、粒径及び形態を制御することができる。
【0168】
当業者は、熱ジャケットが非溶媒の温度を設計に応じて所望の温度に維持するのを助けることを理解する。
【0169】
用語「含む」は、「含まれる」ならびに「からなる」を意味し、例えば、Xを「含む」組成物は、Xのみからなることができるか又は何か別のものを含むことができ、例えばX+Yである。
【0170】
特に定義されない限り、語「実質的に」は、「完全に」を除外せず、例えばYを「実質的に含有しない」組成物は、Yを完全に含有しない場合がある。必要であれば、語「実質的に」を本発明の定義から省くことができる。
【0171】
「任意」又は「任意に」は、後に続く記載の事象又は状況が起こってもよいが起こる必要もなく、そしてその記載が、前記事象又は状況が起こる場合と起こらない場合とを含むことを意味する。
【実施例】
【0172】
実施例1
ブデソニド(5g)を100mLのジクロロメタンに溶解した。超音波チャンバーで収集したブデソニド粉末のサンプルは、Buchi-290実験室規模噴霧乾燥器(Buchi, Switzerland)を使用して生成した。溶液を、およそ10Lpm(リットル/分)で流れる窒素を7barで使用して噴霧した。吸引器を100%に設定し、溶液の流速を10Lpmに設定した。ガス温度を120℃に設定した。ブデソニド粒子を、高速サイクロン分離器の末端に連結されている超音波チャンバーで収集した。超音波を噴霧乾燥粒子に適用するために、超音波収集チャンバーを、25℃に温度調節したヘプタンで充填し、20kHzで共振する超音波プローブを取り付けた。20Wの出力の超音波を30分〜1時間適用した。得られた粒子スラリーを噴霧乾燥し、粒子を、光学顕微鏡及びDSC(示差走査熱量測定)により特徴決定した。粒子の大きさは、典型的には、1〜7μmの範囲であった。
【0173】
2つの代表的な試料のD(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、それぞれ、1.21、3.03、4.63μm及び1.05、2.99、3.76μmであった。
【0174】
示差走査熱量測定
DSC実験は、DSC Q2000 V24.2ビルド107(TA Instruments, UK)により実施した。およそ3mgの物質をDSCのサンプルパンで計量し、275℃まで熱付加する100℃/分の加熱傾斜に付した。DSC測定を、以下の工程に従って実施した。
→稼働9(本発明の超音波により処理していない噴霧乾燥物質)
→機器 DSC Q2000 V24.2ビルド107
→モジュール DSC標準セルRC
→サンプル 噴霧乾燥したpxO2−262
→大きさ 2.140mg
→方法 100℃/分の高速加熱実験 計量サンプルを100℃/分の速度で275℃まで加熱する
→稼働10(本発明の方法に従って超音波で処理した物質)
→機器 DSC Q2000 V24.2ビルド107
→モジュール DSC標準セルRC
→サンプル 超音波後のpx02−262
→大きさ 3.590mg
→方法 100℃/分の高速加熱実験 計量サンプルを100℃/分の速度で275℃まで加熱する
【0175】
実施例2
実施例2は、本発明により生成された粒子の利点を示す。
【0176】
多様な加工により生成された操作フルチカゾンプロピオン酸エステル(FP)のエアロゾル化効率を、2成分乾燥粉末剤吸入器(DPI)製剤において評価した。バッチ研究は以下を含む:
サンプル2 図9のSEMに示されているように、本発明により例示されるエアロゾル化方法により調製。フルチカゾンプロピオン酸エステル(4g)を100mLのアセトンに溶解した。超音波チャンバーで収集したフルチカゾンプロピオン酸エステル粉末のサンプルは、Buechi-290実験室規模噴霧乾燥機(Buechi, Switzerland)を使用して生成した。溶液を、窒素をおよそ10Lpm(リットル/分)で流れる窒素を7barで使用して噴霧した。吸引器を100%の流速に設定し、溶液の流速を10Lpmに設定した。ガス温度を120℃に設定した。フルチカゾンプロピオン酸エステルを、高速サイクロン分離器の末端に連結されている超音波チャンバーで収集した。超音波を噴霧乾燥粒子に適用するために、超音波収集チャンバーを、25℃に温度調節したヘプタンで充填し、20kHzで共振する複数の結合変換器(図7と同じ)を取り付けた。20Wの出力の超音波を30分〜1時間適用した。得られた粒子スラリーを噴霧乾燥し、粒子を、光学顕微鏡及びDSC(示差走査熱量測定)により特徴決定した。粒子の大きさは、典型的には、1〜6μmの範囲であった。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、1.35、3.25、5.63μmであった。
サンプル3 図10のSEMに示されているように、本発明により例示されるエアロゾル化方法により調製。サンプル3は、3gのFPをサンプル3のために使用した以外は、サンプル2と同じ方法により調製した。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、0.99、2.55、4.97μmであった。
サンプル4 図11のSEMに示されているように、代替的な沈殿手法により調製。サンプル4は、WO2008/114052A1に記載されたとおりに調製した。この方法は、初期溶液噴霧を使用しない。代わりに、この従来技術は、アセトン中のフルチカゾンプロピオン酸エステルの溶液を超音波音場の存在下でヘプタン抗溶媒に加えることにより生じる、分散性抗溶媒結晶化を伴う。このことは、本発明の方法を使用して形成される粒子よりも有意に平滑な粒子をもたらす。D(10)、D(50)、D(90)は、Sympatec HELOSレーザー回折により決定して、1.14、2.67、5.11μmであった。
【0177】
このサンプルを、微粉化FPを含有する追加の2成分DPI製剤及び Flixotide Discus吸入器から抽出した製剤と比較した。
【0178】
操作FPのサンプル2、3及び4のエアロゾル化効率を、0.4%w/wのFPを含有する2成分製剤を使用して評価した。
【0179】
各2成分製剤は、0.016gのFP及び3.984gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いてブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。
【0180】
含有均一性試験の後、12.5±1mgの各ブレンドをサイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC, Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0181】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0182】
各実験において、同じ製剤の4個の個別のカプセルを、Rotahaler(GSK, Ware, UK)DPI装置により60Lpmで4秒間NGIに投入した。加えて、Flixotide Diskus(GSK, Ware, UK)のブリスターを空にして、サイズ3のHPMSカプセルを装填し、Rotahalerにより60Lpmで1〜4秒間NGIに投入した。エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。
【0183】
NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドについて3回繰り返し、続いて、平均中央空気力学的直径(MMAD)、幾何学的標準偏差(GSD)、微細粒子用量(FPD)及び発射用量の微細粒子画分(PFPED)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0184】
エアロゾル化効率を図13に示す。微細粒子画分の率を図14に示す。
【0185】
サンプル2及び3の微細粒子画分の率(FPF%)により決定されたエアロゾル化効率は、微粉化FPよりも有意に大きかった。このデータを図15に示し、Flixotideから形成したものと比較したエアロゾル効率を明確に示している。これらの物質を含めたことによる性能の増加は、この研究に使用した吸入器にとって顕著であった。サンプル4は、微粉化FPよりも有意に低いFPF%を有し、これは粒子の表面形態と関連していた。これらのデータは、サンプル2と3のエアロゾル化効率にほとんど差がないことを示唆した。
【0186】
比較インビトロデータは、本発明により生成された粒子の性能が、従来の粉砕粒子及び非常に異なる表面と幾何学的特性を有する特別に調製された(超音波沈殿)粒子の両方の性能に対して圧倒的に、そして驚くほど優れている。全ての場合において、粒子は同じ粒径範囲を有する。フルチカゾンプロピオン酸エステルの特定の実施例において、最適粒子のFPFは、微粉化/粉砕物質よりも54%大きく、沈殿物質よりも200%大きかった。したがって、これらの最適粒子の有意な差及び改善された機能は、大きさではなく(重要な設計基準であるが)、これらの非常に改善された粒子を記載する他の特性の範囲が寄与している可能性がある。
【0187】
図9及び10(本発明)に示されているSEM画像は、図11及び12(本発明ではない)にそれぞれ示されている沈殿及び粉砕材料と比較して、形状及び表面粗さについて明確な対照を明確に示す。粗さ及び円形/球状3D形状は、性能に対して顕著な影響を与える。
【0188】
全ての製剤を、25℃/75%RHで1〜3か月間保存した。微粉化フルチカゾンプロピオン酸エステルを含有する製剤のFPFは、t=0でのFPFと比較したとき、1か月保存した後にほぼ50%、3か月保存した後にほぼ70%減少した。しかし、本発明により調製された粒子を含有する製剤のFPFは、ストレス条件下での保存後に影響を受けなかった。サンプル3のFPFは、t=0でのFPFと比較したとき、1か月保存した後に6%、3か月保存した後に3%減少した。サンプル2のFPFは、t=0でのFPFと比較したとき、1か月保存した後に7%、3か月保存した後に28%減少した。代替的沈殿手法により調製したサンプル4を含有する製剤の微細粒子画分に、t=0でのFPFと比較したとき、1か月後に29%、3か月保存した後に24%の、小さいが統計的に有意な減少があった。このことは、粒子の平坦な形態に関連する場合があり、したがって、粒子は、毛細管力によってラクトースに対してより大きな接着を生じる可能性がある。結果を図23及び24に示す。サンプル2及び3のFPFは、記載された条件下で1〜3か月間保存したとき微粉化サンプル又はサンプル4よりも高いFPFを維持したことが明らかである。サンプル2及び3のFPFは、記載された条件下で1〜3か月間保存したとき微粉化サンプルより小さい率で減少した。このことは、サンプル2及び3が、1〜3か月間保存した後、粉砕サンプルよりも安定していたことを示す。サンプル4は、1〜3か月間保存した後、サンプル2及び3よりもかなり低いFPFを有する。
【0189】
実施例3
実施例3は、pMDIの製剤のために本発明により生成された粒子の利点を示す。図25は、NGIの多様な段階における粒子画分分布グラフであり、サンプル1及びサンプル5のFPFに関して優れた性能を明確に示している。両方のサンプルは、実施例2のサンプル2と同様の方法により生成した。サインプル1は、メタノール中20%ジクロロメタン中のフルチカゾンプロピオン酸エステルの3%溶液(100ml中3g)の噴霧により調製し、サンプル5は、アセトン中3%の溶液(100ml中3g)の噴霧により調製した。図25に示されているように、両方のサンプルの段階5(カットオフ1.36μm)のNGIのFPFは、段階4(カットオフ2.30μm)の同様のFPFと共に、粉砕物質と比較して、100%を超える増加を示した。FPF値は、段階5のサンプル5及び1では、粉砕物質の6.20%と比較して、それぞれ11.80%及び13.80%であった。したがって、サンプル1及び5は、段階4では粉砕生成物と同様のFPFを有し、段階5では粉砕生成物よりも大きなFPFを有することが明らかである。
【0190】
実施例4
実施例2に記載されたように生成されたフルチカゾンプロピオン酸エステル(FP)のサンプル2、3及び4のエアロゾル化効率を、Rotahaler(登録商標)単位用量DPI装置(GSK, Ware, UK)及びCyclohaler(登録商標)単位用量DPI装置(TEVA Pharmaceuticals, Netherlands)を使用して、微粉化サルメテロールキシナホ酸塩(SX)を含有する複合乾燥粉末剤吸入器(DPI)製剤で評価した。
【0191】
エアロゾル化効率を、微粉化SXも含有する複合DPI製剤で評価した。それぞれの複合製剤は、0.16000gのFP(サンプル2、3又は4)、0.02320gのSX及び3.8168gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いて各ブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。それぞれの複合製剤のブレンド強度は、500μgのFP及び50μgのサルメテロール基剤に等しかった。これはAdvair 500/50の用量強度と適合する。Advair 500/50のサンプルを微粉化サンプルとして使用した。FPサンプル2、3、4又は微粉化FPを含有する4つの異なる製剤のエアロゾル化効率を測定した。
【0192】
図26に示されているように、t=0では、Cyclohaler単位用量DPI装置を使用するサンプル2、3及び4のFPFは、微粉化サンプルよりも大きい。Rolahaler単位用量DPI装置を使用する場合、サンプル2及び3のFPFは、サンプル4及び微粉化サンプルよりも大きい。このことは、本発明のフルチカゾンプロピオン酸エステル粒子、サンプル2及び3が、全体的に、2つの装置において高いFPFを有することを示す。サンプル4は、Cyclohaler単位において良好に機能したが、Rolahaler単位は機能が不十分である。サンプル2及び3は両方のユニットにおいて高いFPFを有する。
【0193】
4つのサンプルを25℃/75%RHで1か月保存した。微粉化FP及びサンプル2、3及び4のFPF%は、ストレス保存条件下で有意に影響を受けなかった。これらのデータは、本発明により調製される粒子が、DPI製剤に対して安定性をもたらすはずであることを示す。
【0194】
含有均一性試験の後、12.5±0.5mgの各ブレンドをサイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC, Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0195】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0196】
各実験において、同じ製剤の2個の個別のカプセルを、Rotahaler(GSK, Ware, UK)により60Lpmで4秒間、Cyclohaler(TEVA Pharmaceuticals, Netherlands)により90Lpmで2.8秒間NGIに投入して、両方の装置が4kPa圧力低下を生じるように操作されることを確実にした。
【0197】
加えて、市販のAdvair 500/50 Diskus(GSK, USA)のブリスターを空にして、1.25mgの製剤をサイズ3のHPMCカプセルに移し、Rotahalerにより60Lpmで4秒間、Cyclohalerにより90Lpmで2.8秒間NGIに投入した。
【0198】
エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドにおいて3回繰り返し、続いて、微細粒子用量(FPD)及び装填用量の微細粒子画分(FPFLD)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0199】
図26は、この実施例に従って調製したFPのAdvair 500/50同等ブレンド及び機械的に微粉化したSX粒子の、特にFPについてのFRFを示す。図27は、粒子のFPF性能をグラフで示し、ここでFPは本発明により調製した。サンプル2及び3(cyclo2とcyclo3及びrota2とrota3)は、FPに関して優れた性能を明確に示し、したがって、FP粒子とFP粒子の間の凝集及びラクトースとFPの接着を制御できることを明確に示している。とりわけ、この研究に使用した微粉化成分は、市販の装置からのものであり、微粉化物質は、数か月でなければ数週間の条件付けを受けている。逆に、本発明により作製される粒子は、新たに調製される場合でも高度に安定しており、繰り返しになるが、このことは、本発明により調製される粒子はDPI製剤に対して安定性をもたらすはずであることがここでも記述される。
【0200】
図28は、本発明により作製されるFPサンプル3の粒子を示す。次にこれらをサルメテロールフマル酸塩(10重量%)及びフルチカゾンプロピオン酸エステル(90重量%)からなる組み合わせとブレンドし、ここでサルメテロールフマル酸塩粒は微粉化されており、フルチカゾンプロピオン酸エステルは、本発明により調製されている。
【0201】
実施例5
微粉化フルチカゾンプロピオン酸エステル(FP)、並びに、本発明の方法により(サンプル2及び3)及び実施例1に記されるようにWO2008/114052A1に例示されている異なる方法により(サンプル4)操作されたFPのバッチの、凝集−接着平衡(CAB)を、FP及びラクトース一水和物の結晶質基材に対して決定した。結晶質基材に対する異なるバッチのCAB力平衡は、以下のようにして決定した:
プローブ調製:
FPの全てのバッチの粒子(n=3)を、エポキシ樹脂接着剤(Araldite, Cambridge, UK)を使用して、標準V字形チップレスカンチレバー(DNP-020, Dl, CA, USA)に結合した。
平滑ラクトース及び薬剤結晶の生成:
ラクトースの平滑結晶は、カバースリップで挟んだ加熱飽和液滴を冷却することにより生成した。平滑薬剤結晶は、活性物質をアセトンに溶解し、使用した抗溶媒が水である、静止液滴抗溶媒結晶化を使用して生成した。
AFM力測定:
個別の力曲線(n=1024)を、4Hzの走査速度及び40nNの圧縮荷重により10μm×10μm領域において実施した。環境条件は、20℃(±1.5℃)の一定温度及び45±3%の相対湿度を維持した。
【0202】
微粉化FPのCAB分析は、同等の接触幾何学では、微粉化FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.36倍大きいことを示唆した。
【0203】
サンプル2FPのCAB分析は、同等の接触幾何学では、サンプルFPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.17倍大きいことを示唆した。
【0204】
サンプル3FPのCAB分析は、同等の接触幾何学では、サンプル3FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用よりも1.16倍大きいことを示唆した。
【0205】
サンプル4FPのCAB分析は、同等の接触幾何学では、サンプル4FPの接着性FP−ラクトース相互作用は、凝集性FP−FP相互作用とほぼ同等であることを示唆した。
【0206】
図29に示されているように、微粉化FPは、調製されたFPのサンプル2、3及び4よりもラクトースに対して有意に接着性があった。サンプル4は、ラクトース及びSXに対して最小の接着性があり、したがって、この物質の表面エネルギーは、他のバッチと有意に異なることを示唆している。しかし、接着値は400〜800nNの範囲であり、このことは、異なる基材と接触したときのこの物質の大きな接触半径を反映している。この物質の大きな接触半径は、乾燥粉末剤吸入器(DPI)製剤のサンプル4の配合の際に限定されたエアロゾル化をもたらす。
【0207】
対照的に、サンプル3のFP−ラクトース交互作用に関する接着値は、35〜89nNの範囲であり、サンプル2の接着値は、128〜169nNの範囲であり、このことは、異なる基材と接触したときの本発明の粒子の小さな接触半径を反映している。この物質の小さな接触半径は、接着値が169〜249nMの範囲である微粉化FPと比較して、担体に基づいたDPI製剤のサンプル2及び3の配合の際に大きなFPエアロゾル化をもたらす。
【0208】
CAB分析は、サンプル3が他の物質よりも小さい接触半径を有しうること、一方、サンプル4が大きな接触半径を有しうることを確認し、このことは、これらの粒子の表面形状に関連している。
【0209】
これらのデータは、本発明により例示された粒子操作戦略が、表面エネルギー及び粒子接触形状の両方の制御をもたらすことができることを示し、これらは両方ともDPIS製剤の薬剤粒子に寄与する重要な品質である。
【0210】
図29は、サンプル4の接着及び凝集の両方の比較的高い力に注目して、粒子の異なる接触形状に関するFP−ラクトース相互作用を示す。
【0211】
実施例6
実施例2に記載されたように生成されたフルチカゾンプロピオン酸エステル(FP)のサンプル3のエアロゾル化効率を、Rotahaler(登録商標)単位用量DPI装置(GSK, Ware, UK)及びCyclohaler(登録商標)単位用量DPI装置(TEVA Pharmaceuticals, Netherlands)を使用して、微粉化サルメテロールキシナホ酸塩(SX)を含有する複合乾燥粉末剤吸入器(DPI)製剤で評価した。
【0212】
エアロゾル化効率を、微粉化SXも含有する複合DPI製剤で評価した。各複合製剤は、0.16000gのFP、0.01160gのSX及び3.8284gのラクトース(ML001, DMV-Fonterra, Vehgel, Netherlands)を含有し、幾何学的混合により調製した。この後、続いてブレンドを、Turbula T2F(Willy A Bachofen AG, Basel, Switzerland)を46rpmで45分間使用して調製した。複合製剤のブレンド強度は、500μgのFP及び25μgのサルメテロール基剤に等しく、したがって、これは500/25製剤であった。
【0213】
500/25製剤を有する、微粉化FP及び微粉化SXを含有する製剤の含有均一性の評価は、不十分な均質性を示唆し、したがて、この製剤は、インビトロ衝突研究により特徴決定しなかった。対照的に、FPサンプル3及び微粉化SXを含有する製剤は、図20に示されているように、非常に良好な均質性を示した。これは、FPのサンプル3の相対標準偏差%が、3.43であり、SXでは、4.55であったことを示す。これを微粉化FP−SX製剤と比較し、FPの相対標準偏差%は8.76であり、SXでは15.95であった。
【0214】
含有均一性の試験の後、12.5±0.5μgの、FPサンプル3ブレンドを含有するブレンド又はAdvairを、サイズ3のヒドロキシプロピルメチルセルロースカプセル(HPMC、Shionogi Qualicaps SA, Basingstoke, UK)に装填した。カプセルを、インビトロ性能試験の前に、44%のRHで24時間保存した。
【0215】
試験を、真空ポンプ(GE Motors)に連結している、プレセパレーターを有するNext Generation lmpactor(NGI)を使用して実施した。試験の前に、プレセパレーターを15mlの移動相で充填し、NGIカップのカップを、ヘキサン中の1%v/vシリコーン油で被覆して、粒子の跳ね返りを排除した。
【0216】
各実験において、同じ製剤の2個の個別のカプセルを、Cyclohaler(TEVA Pharmaceuticals, Netherlands)により90Lpmで2.8秒間NGIに投入して、両方の装置が4kPa圧力低下を生じるように操作されることを確実にした。
【0217】
エアロゾル化の後、NGI装置を解体し、吸入器、カプセル及びNGIの各部品を既知の量のHPLC移動相で洗い流した。NGIの各部品に付着した薬剤の量を、HPLCにより決定した。このプロトコールを各ブレンドにおいて3回繰り返し、続いて、微細粒子用量(FPD)及び装填用量の微細粒子画分(FPFLD)を決定した。FPDは、NGIの段階3〜8で収集した薬剤量を表した。
【0218】
性能データは、図30に示されているように、それぞれ79μg及び11μgのFP及びSXの500/25製剤におけるサンプル3のFPDを示唆した。図31に示されているように、これは、微細粒子画分が操作FPの15.8%と比較してSXでは44%であると解釈された
【0219】
図21は、500μgのサンプル3FP及び50μgのSX(サンプル3 300/50)、500μgのサンプル3FP及び25μgのSX(サンプル3 500/25)及び500μgのFPと50μgのSXを含有するAdvair(Advair 500/50)を含有する製剤において、上記に記載したようなFPFLD測定を示す。この図は、サンプル3 500/50及びサンプル3 500/25では、SXはAdvair 500/50よりもかなり高いFPFLDを有したことを示す。サンプル3 500/25のSXのFPFLDは、サンプル3 500/50よりも高かった。
【0220】
このことは、操作FPがブレンドの含有均一性に対して顕著な効果を有し、FPFに関してSCの増加を促進することを意味し、また、操作FPを使用した所定の製剤では、有意に少ないSXをブレンドに使用して、FP及びSXの両方において匹敵するFPFを達成できることを意味している。これらのデータは、本発明により調製した操作FPを使用して、Advair製品に現在用いられている名目強度の半分を含有する複合DPI製品を配合することが可能であることを示唆している。
【0221】
実施例7
微粉化フルチカゾンプロピオン酸エステル(FP)、本発明により調製されるFPのサンプル3及び実施例2に記載されたFPのサンプル4の表面粗さ及び表面積は、原子間力顕微鏡法(AFM)及びBET表面積分析をそれぞれ使用して決定した。画像化領域の粗さは、画像化表面の高さにおける差の平均(Ra)及び二乗平均平方根(Rq)を使用して定量化した。更に、サンプルの表面積は、5ポイントBET窒素吸着分析により決定した。
【0222】
FPサンプルの表面様相は、Multimode AFM、J型スキャナー、Nanoscope IIIaコントローラー(全て、Dl, Cambridge, UK)及びシリコンチップ(型番OMCL- AC240TS, Olympus, Japan)を使用して、各物質の粒子の表面から3つの無作為に選択した1μm×1μm正方形領域を、512×512ピクセルの解像度及び1Hzの走査測度で画像化する、TappingMode(商標)原子間力顕微鏡法(AFM)により調査した。画像化領域の粗さは、以下の方程式を使用するAFMソフトウエアにより計算して、画像化表面の高さにおける差の平均(Ra)及び二乗平均平方根(Rq)を使用して定量化した:
【0223】
【数1】
【0224】
ここで、npは、画像におけるポイントの数であり、yiは、中央線からのポイントiの距離である。
【0225】
FPサンプルの比表面積を、Gemini 2360表面積分析器(Micromeritics Instrument Corporation, Norcross, USA)を使用して測定した。5ポイントBET窒素吸着分析を、FlowPrep 060脱ガス器((Micromeritics Instrument Corporation, Norcross, USA)によりサンプルを24時間脱ガスした後で実施した。
【0226】
結果を下記の表2にまとめる。
【0227】
【表2】
【0228】
サンプルの表面粗さ分析は、サンプル3が最大の表面粗さを有し、一方、サンプル4(本発明によって調製されたものではない)が、最も平滑であったことを示唆した。サンプル3は、他のサンプルよりも大きな表面積を有し、これは物質の粗さに関連しうる。サンプル3のRa及びRq値、並びに表面積は、微粉化サンプル及びサンプル4よりも大きい。
【0229】
微粉化サンプルのRa及びRq値の平均偏差は、サンプル3及び4よりもかなり大きい。このことは、サンプル3及び4よりも微粉化サンプルの方が表面粗さに大きな差があることを示す可能性がある。
【0230】
図32及び33は、サンプル3及び4それぞれの表面粗さのAFM輪郭プロットを示す。これらは、サンプル4がサンプル3よりもかなり平滑であることを示す。輪郭プロットにおける差は、サンプル3がサンプル4と異なる表面粗さを有することを示し、このことは、Ra及びRq値に反映されている。
【0231】
実施例8
次にメタノール(200mL)中のフェノテロール臭化水素酸塩(10g)の溶液を調製し、二流体ノズル及び0.7mmオリフィスを使用し、支援窒素流速が35〜40m3/時間(吸引器100%)のBuchi-B290を用いて、流速9mU/分(30%ポンプ)及びノズルクリーン設定2で噴霧乾燥した。入口温度は78℃であり、出口温度は38℃である。ジイソプロピルエーテル(300mL)を、底がB-290サイクロンに連結しており、5℃に温度調節された最大容量500mLの撹拌超音波容器に投入した。噴霧乾燥生成物を、40Wの連続出力で2時間稼働している超音波容器の中に収集し、続いて非晶質フェノテロール臭化水素酸塩の第1粒子を加えた。上記のBuchi-B290を用いて、入口温度110℃及び出口温度50℃で懸濁液を噴霧乾燥することによって、粒子を回収した。この実施例のデータ及び粒子のSEM画像を、図17〜20に示す。
【0232】
図17は、市販の微粉化フェノテロール臭化水素酸のSEM画像を示す。図18は、本発明により加工された粒子のサンプル、すなわちGR005/180/A4及びGR005/179/Cの粒径分布データを示す。図19は、検査用具として市販のHandiHaler吸入装置を使用した、比較の微細粒子画分(FPF)を示す。図19及び20のFPF〔%〕の横列を比較するとFPFの増加は30〜117%に異なっていた。
【0233】
図21は、微粉化ブデソニドと本発明のブデソニド粒子の表面エネルギーに対する分散被覆率を示す。IGCを、本発明の粒子の表面エネルギーを測定するために使用した。IGCは、2セットの条件により実施することができる。有限希釈では、吸着等温線をピークプロフィールから誘導することができ、吸着エネルギー分布を計算するために使用することができる。第2に、無限希釈では、器具の検出限界に近い溶質の量を注入し、この場合、溶質−溶質相互作用は小さく、溶質−吸着剤相互作用のみが測定保持時間に影響を与える。図21の左側に示されているように、特に高いエネルギー部位においてのみ限定された吸着率(被覆率)が分析される。溶質の量が有限希釈まで増加するので、最終的に100%の被覆率が達成され、表面エネルギーが異なっているにもかかわらず、粒子の全ての部位において吸着率が上昇する。本発明の粒子は、図21に明確に示されているように、表面エネルギーの等エネルギー分布を有することによって特徴付けられる。表面エネルギーは、本発明の好ましい方法により調製される粒子の有限及び無限希釈の両方において極めて類似し、ほぼ同一であり、一方、典型的な微粉化粒子は、有限及び無限希釈において顕著な差を示す。
【特許請求の範囲】
【請求項1】
100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、前記固体物質を、固体物質が不溶性であるか又は難溶性である溶媒(非溶媒)と接触させる工程と、前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程とを含む方法。
【請求項2】
前記固体物質が、約10μmまで、好ましくは5μmまでの平均中央空気力学的直径を有する粒状固体物質である、請求項1に記載の方法。
【請求項3】
前記固体物質が、機械的微粉化、粉砕、噴射粉砕、摩砕、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法により、好ましくは噴霧乾燥により得られる、請求項1又は請求項2に記載の方法。
【請求項4】
上記の方法を適用する前の、前記固体物質の結晶度が50%未満である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記方法が0.1ミリ秒より長い時間実施される、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
本発明に使用される前記固体物質が、5重量%未満の溶媒を含有する、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記固体物質が、活性製薬学的成分、活性農芸化学的成分、製薬学的賦形剤、農芸化学的賦形剤及びこれらの2つ以上の適切な混合物からなる群より選択される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
(i)溶媒中で、少なくとも1つの固体物質の溶液を形成する工程;
(ii)前記溶液を、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法に付し、溶解させた前記固体物質が実質的に乾燥固体物質に変換される工程;
(iii)任意に、前記固体物質を工程(ii)の方法の液体及び/又は気体成分から単離する工程;
(iv)工程(ii)又は工程(iii)の前記乾燥固体物質を、前記乾燥固体物質の非溶媒で処理する工程;
(v)工程(iv)の前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程;及び
(vi)任意に、工程(v)で得られた前記固体物質を、分離及び/又は乾燥する工程
を含む、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
方法が順次であり、工程(iv)及び(v)が工程(ii)の直後に実施される、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
(a)少なくとも1つの固体物質を、機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合に付す工程;
(b)工程(a)で得られた前記固体物質を、前記固体物質の非溶媒で処理する工程;
(c)工程(b)で得られた前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程;及び
(d)任意に、工程(c)で得られた前記固体物質を、分離及び/又は乾燥する工程
を含む、請求項1〜7のいずれか1項に記載の方法。
【請求項11】
本発明の方法に使用される超音波の周波数が、好ましくは10〜100kHzの範囲である、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
前記固体物質が、抗アレルギー薬、気管支拡張薬、抗炎症性ステロイド及びこれらの混合物からなる群より選択される製薬学的に活性な成分である、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
前記非溶媒が、1つの以上の添加剤を0.01%w/w〜10%w/wの量で含む、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
前記固体物質が、吸入製剤における使用に適した製薬学的に活性な成分、好ましくはブデソニド、サルブタモール、ホルモテロール又はこれらの2つ以上の混合物である、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
前記固体物質が、少なくとも1つの製薬学的に活性な成分又はその前駆体、及び、担体基材を含む、請求項1〜14のいずれか1項に記載の方法。
【請求項16】
周囲及び長手軸方向の両方に延びている列のかたちで、容器の壁に取り付けられている複数の超音波変換器を使用して、超音波が前記容器内の前記過飽和溶液に提供される方法であって、前記変換器が3W/cm2以下で照射するように各変換器が単一の発生器に連結されており、前記変換器が互いに十分に近接しており、かつ、前記変換器の数が、前記容器内の電力損失が25〜150W/リットルであるように十分に多い、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
製薬学的な又は農芸化学的な化合物を含み、請求項1〜16のいずれか1項に記載の方法により得られる粒子。
【請求項18】
前記粒子が、10μm未満、好ましくは0.05〜5μm、より好ましくは0.05〜2μmの平均中央空気力学的直径(MMAD)を有する、請求項17に記載の粒子。
【請求項19】
前記粒子が、ナノメートル規模の表面波形を有し、好ましくはRqの値が10〜100nmである、請求項17又は18に記載の粒子。
【請求項20】
最大直径と最小直径との平均比が1.3〜1:1である、請求項17〜19のいずれか1項に記載の粒子。
【請求項21】
前記粒子の表面積が6〜22m2/gである、請求項17〜20のいずれか1項に記載の粒子。
【請求項22】
請求項17〜21のいずれか1項に記載の粒子を含む、医薬組成物又は農芸化学的組成物。
【請求項23】
好ましくはラクトース、グルコース及びこれらの水和物から選択される、担体粒子を更に含む、請求項21に記載の医薬組成物。
【請求項24】
前記粒子と前記担体粒子との凝集−接着平衡比が0.8〜1.3である、請求項23に記載の医薬組成物。
【請求項25】
前記製薬学的に活性な成分が、カルモテロール、並びに、その生理学的に許容される塩及び溶媒和物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項26】
好ましくは、コルチコステロイドが、ブデソニド、ジプロピオン酸ベクロメタゾン、シクレソニド又はモメタゾンである時に、前記製薬学的に活性な成分が、カルモテロール並びにその生理学的に許容される塩及び溶媒和物と、前記コルチコステロイドとの組み合わせである、請求項17〜21及び25のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項27】
前記製薬学的に活性な成分が、グリコピロニウム臭化物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項28】
前記製薬学的に活性な成分が、フェノテロールである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項29】
好ましくは、コルチコステロイドが、ジプロピオン酸ベクロメタゾン、シクレソニド又はモメタゾンである時に、前記製薬学的に活性な成分が、ホルモテロールと前記コルチコステロイドとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項30】
前記製薬学的に活性な成分が、インダカテロールとモメタゾンとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項31】
前記製薬学的に活性な成分が、グリコピロニウム臭化物とインダカテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項32】
前記製薬学的に活性な成分が、チオトロピウム臭化物とホルモテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項33】
前記製薬学的に活性な成分が、ベクロメタゾンとサルメテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項34】
前記製薬学的に活性な成分が、イプラトロピウム臭化物とアルブテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項35】
前記製薬学的に活性な成分が、イプラトロピウム、好ましくはイプラトロピウム臭化物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項36】
前記製薬学的に活性な成分が、ダロトロピウム又はアクリジニウムである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項37】
前記製薬学的に活性な成分が、フェノテロールとイプラトロピウムとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項38】
前記製薬学的に活性な成分が、クロモグリク酸塩(例えば、そのナトリウム塩)、サルブタモール(例えば、その遊離塩基若しくはその硫酸塩)、サルメテロール(例えば、そのキシナホ酸塩)、チオトロピウム(例えば、その臭化物塩)、テオフィリン、テルブタリン(例えば、その硫酸塩)、レプロテロール(例えば、その塩酸塩)、ジプロピオン酸ベクロメタゾン(例えば、その一水和物)、(−)−4−アミノ−3,5−ジクロロ−α−[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]−メチル]ベンゼンメタノール、並びにその生理学的に許容される塩及び溶媒和物、又はこれらの混合物からなる群から選択される、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22又は24のいずれか1項に記載の医薬組成物。
【請求項39】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する乾燥粉末吸入器。
【請求項40】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する加圧式定量噴霧吸入器(pMDI)。
【請求項41】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する呼吸作動式鼻吸入器。
【請求項1】
100%未満の結晶度である少なくとも1つの固体物質の結晶化度を増加させる方法であって、前記固体物質を、固体物質が不溶性であるか又は難溶性である溶媒(非溶媒)と接触させる工程と、前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程とを含む方法。
【請求項2】
前記固体物質が、約10μmまで、好ましくは5μmまでの平均中央空気力学的直径を有する粒状固体物質である、請求項1に記載の方法。
【請求項3】
前記固体物質が、機械的微粉化、粉砕、噴射粉砕、摩砕、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法により、好ましくは噴霧乾燥により得られる、請求項1又は請求項2に記載の方法。
【請求項4】
上記の方法を適用する前の、前記固体物質の結晶度が50%未満である、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
前記方法が0.1ミリ秒より長い時間実施される、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
本発明に使用される前記固体物質が、5重量%未満の溶媒を含有する、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記固体物質が、活性製薬学的成分、活性農芸化学的成分、製薬学的賦形剤、農芸化学的賦形剤及びこれらの2つ以上の適切な混合物からなる群より選択される、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
(i)溶媒中で、少なくとも1つの固体物質の溶液を形成する工程;
(ii)前記溶液を、急速沈殿、冷凍乾燥、凍結乾燥、超臨界溶液の急速膨張、噴霧乾燥又はこれらの混合からなる群より選択される方法に付し、溶解させた前記固体物質が実質的に乾燥固体物質に変換される工程;
(iii)任意に、前記固体物質を工程(ii)の方法の液体及び/又は気体成分から単離する工程;
(iv)工程(ii)又は工程(iii)の前記乾燥固体物質を、前記乾燥固体物質の非溶媒で処理する工程;
(v)工程(iv)の前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程;及び
(vi)任意に、工程(v)で得られた前記固体物質を、分離及び/又は乾燥する工程
を含む、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
方法が順次であり、工程(iv)及び(v)が工程(ii)の直後に実施される、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
(a)少なくとも1つの固体物質を、機械的微粉化、粉砕、噴射粉砕、摩砕又はこれらの混合に付す工程;
(b)工程(a)で得られた前記固体物質を、前記固体物質の非溶媒で処理する工程;
(c)工程(b)で得られた前記固体物質に、前記非溶媒と接触したときに超音波を適用する工程;及び
(d)任意に、工程(c)で得られた前記固体物質を、分離及び/又は乾燥する工程
を含む、請求項1〜7のいずれか1項に記載の方法。
【請求項11】
本発明の方法に使用される超音波の周波数が、好ましくは10〜100kHzの範囲である、請求項1〜10のいずれか1項に記載の方法。
【請求項12】
前記固体物質が、抗アレルギー薬、気管支拡張薬、抗炎症性ステロイド及びこれらの混合物からなる群より選択される製薬学的に活性な成分である、請求項1〜11のいずれか1項に記載の方法。
【請求項13】
前記非溶媒が、1つの以上の添加剤を0.01%w/w〜10%w/wの量で含む、請求項1〜12のいずれか1項に記載の方法。
【請求項14】
前記固体物質が、吸入製剤における使用に適した製薬学的に活性な成分、好ましくはブデソニド、サルブタモール、ホルモテロール又はこれらの2つ以上の混合物である、請求項1〜13のいずれか1項に記載の方法。
【請求項15】
前記固体物質が、少なくとも1つの製薬学的に活性な成分又はその前駆体、及び、担体基材を含む、請求項1〜14のいずれか1項に記載の方法。
【請求項16】
周囲及び長手軸方向の両方に延びている列のかたちで、容器の壁に取り付けられている複数の超音波変換器を使用して、超音波が前記容器内の前記過飽和溶液に提供される方法であって、前記変換器が3W/cm2以下で照射するように各変換器が単一の発生器に連結されており、前記変換器が互いに十分に近接しており、かつ、前記変換器の数が、前記容器内の電力損失が25〜150W/リットルであるように十分に多い、請求項1〜15のいずれか1項に記載の方法。
【請求項17】
製薬学的な又は農芸化学的な化合物を含み、請求項1〜16のいずれか1項に記載の方法により得られる粒子。
【請求項18】
前記粒子が、10μm未満、好ましくは0.05〜5μm、より好ましくは0.05〜2μmの平均中央空気力学的直径(MMAD)を有する、請求項17に記載の粒子。
【請求項19】
前記粒子が、ナノメートル規模の表面波形を有し、好ましくはRqの値が10〜100nmである、請求項17又は18に記載の粒子。
【請求項20】
最大直径と最小直径との平均比が1.3〜1:1である、請求項17〜19のいずれか1項に記載の粒子。
【請求項21】
前記粒子の表面積が6〜22m2/gである、請求項17〜20のいずれか1項に記載の粒子。
【請求項22】
請求項17〜21のいずれか1項に記載の粒子を含む、医薬組成物又は農芸化学的組成物。
【請求項23】
好ましくはラクトース、グルコース及びこれらの水和物から選択される、担体粒子を更に含む、請求項21に記載の医薬組成物。
【請求項24】
前記粒子と前記担体粒子との凝集−接着平衡比が0.8〜1.3である、請求項23に記載の医薬組成物。
【請求項25】
前記製薬学的に活性な成分が、カルモテロール、並びに、その生理学的に許容される塩及び溶媒和物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項26】
好ましくは、コルチコステロイドが、ブデソニド、ジプロピオン酸ベクロメタゾン、シクレソニド又はモメタゾンである時に、前記製薬学的に活性な成分が、カルモテロール並びにその生理学的に許容される塩及び溶媒和物と、前記コルチコステロイドとの組み合わせである、請求項17〜21及び25のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項27】
前記製薬学的に活性な成分が、グリコピロニウム臭化物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項28】
前記製薬学的に活性な成分が、フェノテロールである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項29】
好ましくは、コルチコステロイドが、ジプロピオン酸ベクロメタゾン、シクレソニド又はモメタゾンである時に、前記製薬学的に活性な成分が、ホルモテロールと前記コルチコステロイドとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項30】
前記製薬学的に活性な成分が、インダカテロールとモメタゾンとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項31】
前記製薬学的に活性な成分が、グリコピロニウム臭化物とインダカテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項32】
前記製薬学的に活性な成分が、チオトロピウム臭化物とホルモテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項33】
前記製薬学的に活性な成分が、ベクロメタゾンとサルメテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項34】
前記製薬学的に活性な成分が、イプラトロピウム臭化物とアルブテロールとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項35】
前記製薬学的に活性な成分が、イプラトロピウム、好ましくはイプラトロピウム臭化物である、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項36】
前記製薬学的に活性な成分が、ダロトロピウム又はアクリジニウムである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項37】
前記製薬学的に活性な成分が、フェノテロールとイプラトロピウムとの組み合わせである、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22〜24のいずれか1項に記載の医薬組成物。
【請求項38】
前記製薬学的に活性な成分が、クロモグリク酸塩(例えば、そのナトリウム塩)、サルブタモール(例えば、その遊離塩基若しくはその硫酸塩)、サルメテロール(例えば、そのキシナホ酸塩)、チオトロピウム(例えば、その臭化物塩)、テオフィリン、テルブタリン(例えば、その硫酸塩)、レプロテロール(例えば、その塩酸塩)、ジプロピオン酸ベクロメタゾン(例えば、その一水和物)、(−)−4−アミノ−3,5−ジクロロ−α−[[[6−[2−(2−ピリジニル)エトキシ]ヘキシル]アミノ]−メチル]ベンゼンメタノール、並びにその生理学的に許容される塩及び溶媒和物、又はこれらの混合物からなる群から選択される、請求項17〜21のいずれか1項に記載の粒子、又は、請求項22又は24のいずれか1項に記載の医薬組成物。
【請求項39】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する乾燥粉末吸入器。
【請求項40】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する加圧式定量噴霧吸入器(pMDI)。
【請求項41】
請求項22〜38のいずれか1項に記載の医薬組成物を含有する呼吸作動式鼻吸入器。
【図1】

【図2】

【図3】
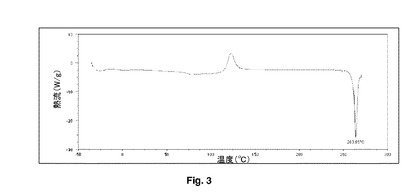
【図4】
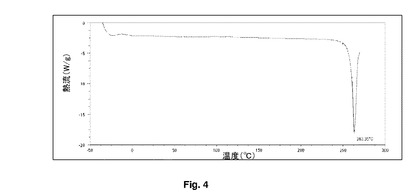
【図5】
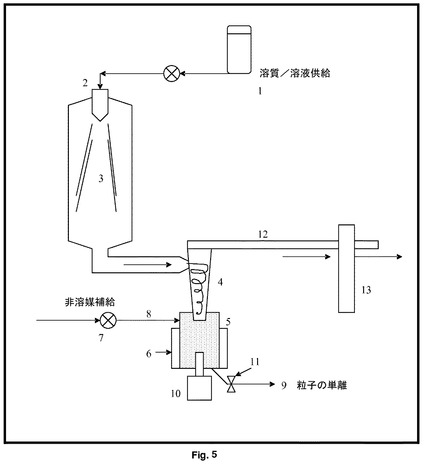
【図6】
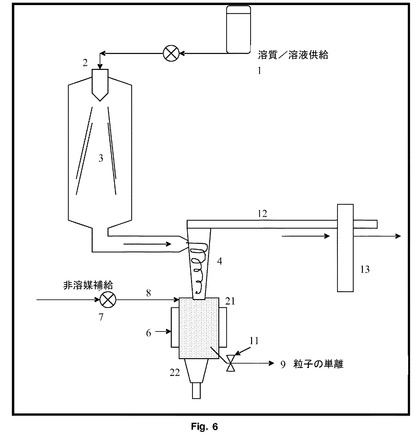
【図7】
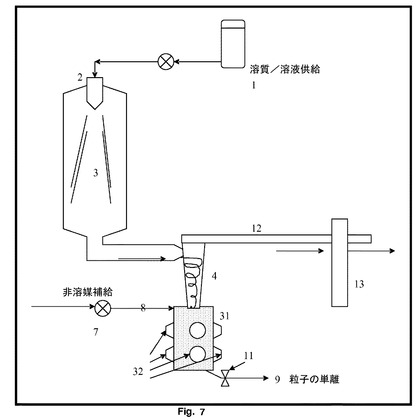
【図8】
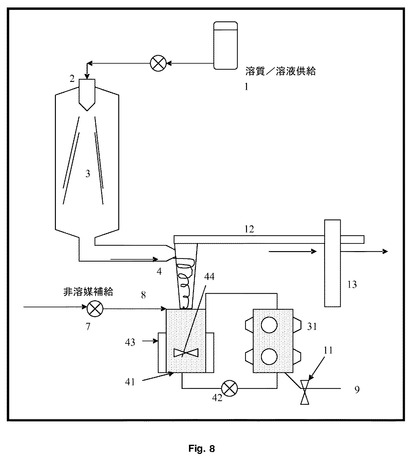
【図9】

【図10】

【図11】

【図12】

【図13】
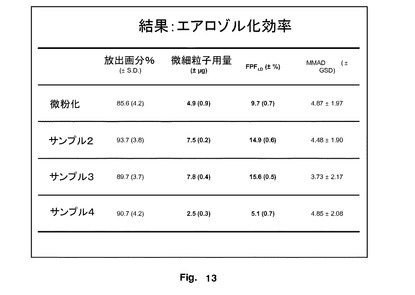
【図14】
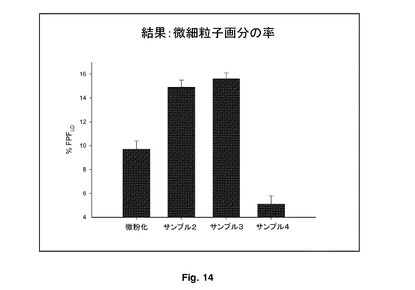
【図15】
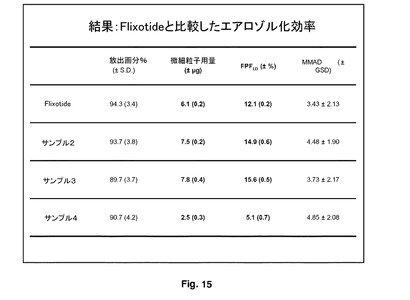
【図16】

【図17】

【図18】
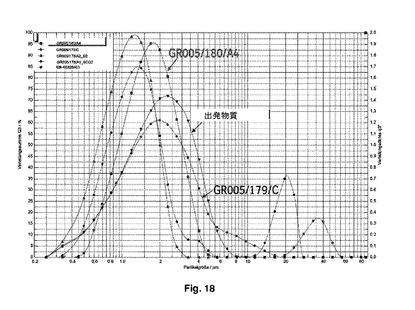
【図19】
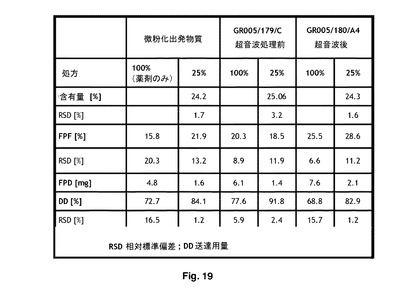
【図20】
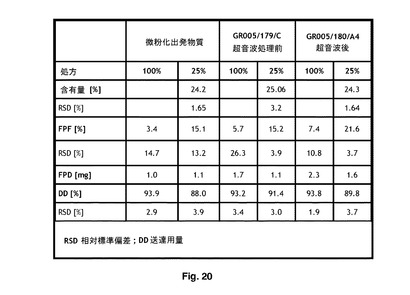
【図21】
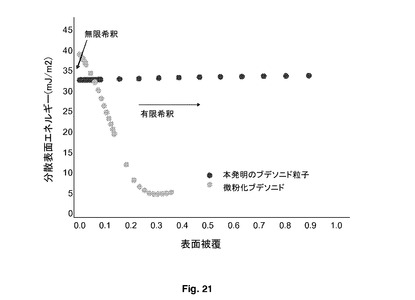
【図22】
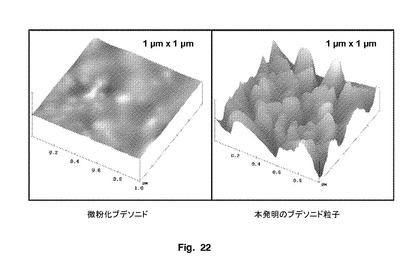
【図23】

【図24】
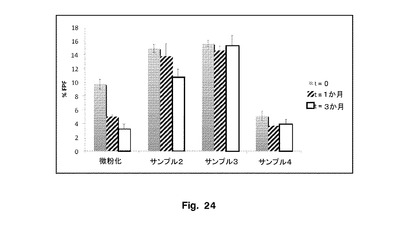
【図25】
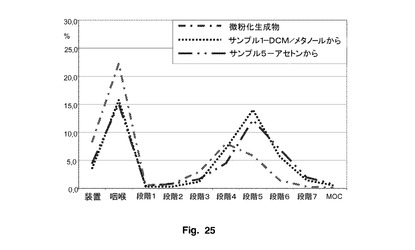
【図26】
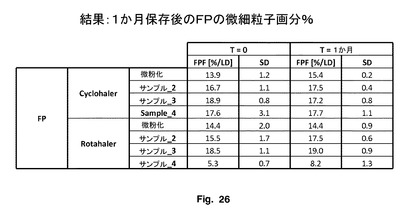
【図27】
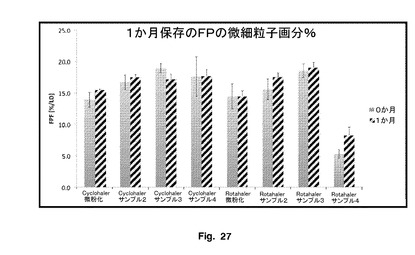
【図28】

【図29】
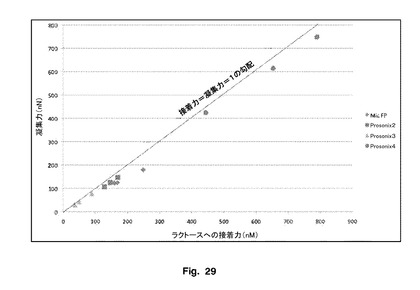
【図30】
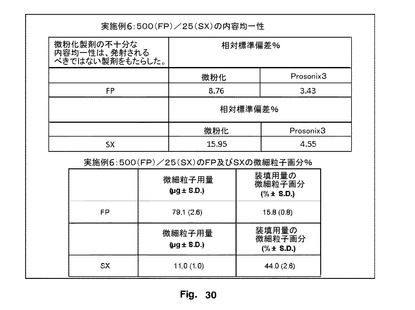
【図31】
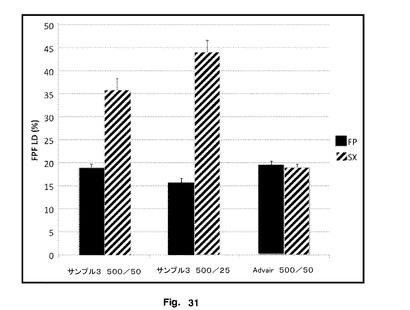
【図32】

【図33】
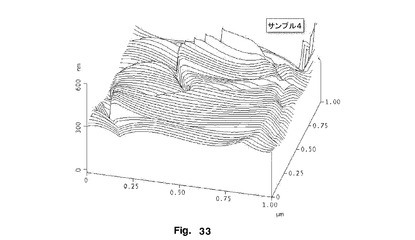
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【公表番号】特表2011−528343(P2011−528343A)
【公表日】平成23年11月17日(2011.11.17)
【国際特許分類】
【出願番号】特願2011−518014(P2011−518014)
【出願日】平成21年7月20日(2009.7.20)
【国際出願番号】PCT/GB2009/050885
【国際公開番号】WO2010/007447
【国際公開日】平成22年1月21日(2010.1.21)
【出願人】(511015548)プロソニックス リミテッド (2)
【氏名又は名称原語表記】PROSONIX LIMITED
【住所又は居所原語表記】Magdalen Centre,Oxford Science Park,Robert Robinson Avenue,Oxford,Oxfordshire OX4 4GA(GB).
【Fターム(参考)】
【公表日】平成23年11月17日(2011.11.17)
【国際特許分類】
【出願日】平成21年7月20日(2009.7.20)
【国際出願番号】PCT/GB2009/050885
【国際公開番号】WO2010/007447
【国際公開日】平成22年1月21日(2010.1.21)
【出願人】(511015548)プロソニックス リミテッド (2)
【氏名又は名称原語表記】PROSONIX LIMITED
【住所又は居所原語表記】Magdalen Centre,Oxford Science Park,Robert Robinson Avenue,Oxford,Oxfordshire OX4 4GA(GB).
【Fターム(参考)】
[ Back to top ]