
結晶性の〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸(アトルバスタチン)の製法
【課題】 高脂血症および高コレステロール血症を治療する薬剤であるアトルバスタチンの安定な結晶性形態の製法を提供する。
【解決手段】 結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸の塩基性塩の水溶液をカルシウム塩で処理する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【解決手段】 結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸の塩基性塩の水溶液をカルシウム塩で処理する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬として有用である新規な結晶性形態の化学名〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩によって知られているアトルバスタチンを製造および単離する方法に関するものである。本発明の新規な結晶性化合物は、酵素3−ヒドロキシ−3−メチルグルタリル−コエンザイムAレダクターゼ(HMG-CoAレダクターゼ)の阻害剤として有用であり、そして結果として、有用な血中脂質性低下剤および血中コレステロール低下剤である。
【背景技術】
【0002】
参照により本明細書に加入した特許文献1は、トランス(±)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔(2−テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミドを包含するあるトランス−6−〔2−(3−または4−カルボキサミド−置換されたピロール−1−イル)アルキル〕−4−ヒドロキシ−ピラン−2−オンを開示している。
【0003】
参照により本明細書に加入した特許文献2は、トランス−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔(2−テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミドのR形態の開環した酸を有するエナンチオマーすなわち〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸を開示している。
【0004】
参照により本明細書に加入した特許文献3、特許文献4、特許文献5、特許文献6、特許文献7、特許文献8、特許文献9、特許文献10、特許文献11、特許文献12、特許文献13および特許文献14は、アトルバスタチンを製造する種々な方法および重要な中間体を開示している。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第4681893号明細書
【特許文献2】米国特許第5273995号明細書
【特許文献3】米国特許第5003080号明細書
【特許文献4】米国特許第5097045号明細書
【特許文献5】米国特許第5103024号明細書
【特許文献6】米国特許第5124482号明細書
【特許文献7】米国特許第5149837号明細書
【特許文献8】米国特許第5155251号明細書
【特許文献9】米国特許第5216174号明細書
【特許文献10】米国特許第5245047号明細書
【特許文献11】米国特許第5248793号明細書
【特許文献12】米国特許第5280126号明細書
【特許文献13】米国特許第5397792号明細書
【特許文献14】米国特許第5342952号明細書
【発明の概要】
【発明が解決しようとする課題】
【0006】
アトルバスタチンは、そのカルシウム塩、すなわち、〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸カルシウム塩(2:1)として製造される。カルシウム塩は、アトルバスタチンを例えば経口的投与のための錠剤、カプセル、ロゼンジ、粉末などに有利に処方することを可能にするので、望ましい。さらに、厳密な製剤上の必要条件および規格を満足する処方を可能にするために、純粋な且つ結晶性の形態でアトルバスタチンを製造することが必要である。
【0007】
さらに、アトルバスタチンを製造する方法は、大規模な生産に耐えられる方法であることが必要である。さらに、生成物は、容易に濾過することができそして容易に乾燥することができる形態にあることが望ましい。最後に、生成物は、特別な貯蔵条件を必要とすることなしに長期間安定であることが経済的に望ましい。
【0008】
上記米国特許の方法は、大規模な生産に対して適当でない濾過および乾燥特性を有しそして熱、光、酸素および湿気から保護しなければならない無定形のアトルバスタチンを開示している。
【課題を解決するための手段】
【0009】
驚くべきことにはそして意外にも、アトルバスタチンは結晶性の形態で製造することができるということが見出された。すなわち、本発明は、形態I、形態IIおよび形態IVと称される新規な結晶性形態のアトルバスタチンを提供する。
【発明の効果】
【0010】
形態Iのアトルバスタチンは、従来の無定形の生成物よりも小さい粒子およびより一様な大きさの分布からなり、そしてより有利な濾過および乾燥特性を示す。さらに、形態Iのアトルバスタチンは、無定形の生成物よりも純粋でありそしてより安定である。
【図面の簡単な説明】
【0011】
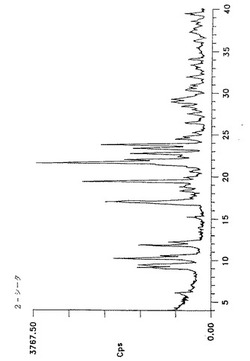
【図1】2分粉砕した形態Iのアトルバスタチンのジフラクトグラム(Y軸=0〜3767.50カウント/秒(cps)の最大強度)を示す。
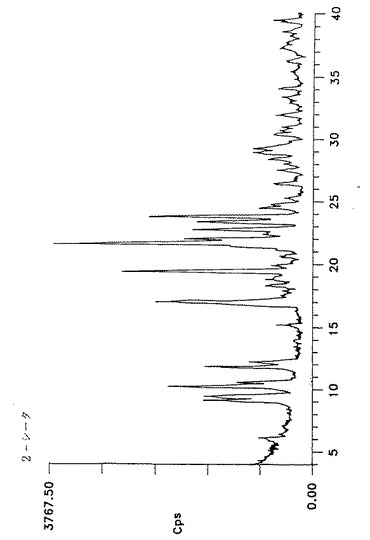
【図2】2分粉砕した形態IIのアトルバスタチンのジフラクトグラム(Y軸=0〜1500cpsの最大強度)を示す。
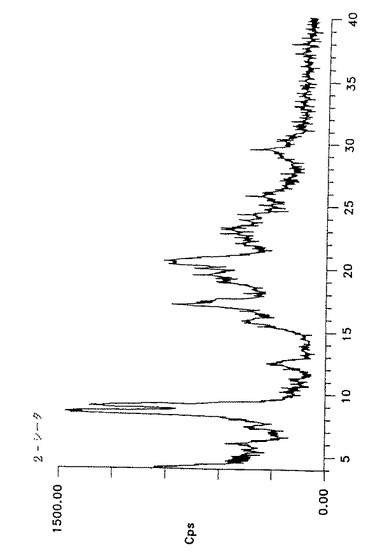
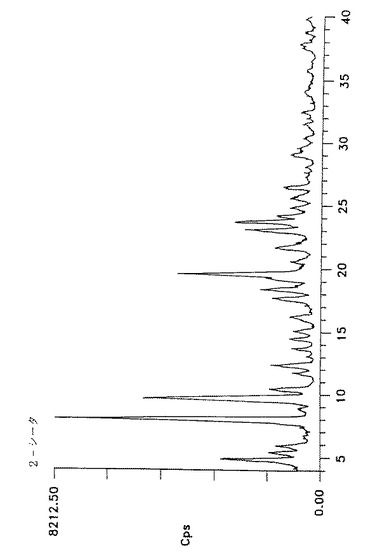
【図3】形態IVのアトルバスタチンのジフラクトグラム(Y軸=0〜8212.5cpsの最大強度)を示す。
【図4】形態Iのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【図5】形態IIのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【図6】形態IVのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【発明を実施するための形態】
【0012】
本発明は、2分の粉砕後に測定したおよびCuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔、および>20%の相対強度の相対強度によって表した次のX線粉末回折パターンを特徴とする結晶性の形態Iのアトルバスタチンおよびその水和物に関するものである。
【0013】
【表1】

【0014】
さらに、本発明は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする結晶性の形態Iのアトルバスタチンおよびその水和物に関するものである。
【0015】
【表2】
【0016】
本発明の第1の見地の好ましい実施化においては、結晶性の形態Iのアトルバスタチンは、三水和物である。
第2の見地においては、本発明は、2分の粉砕後に測定したそしてCuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔、および>20%の相対強度を有する強度によって表した次のX線粉末回折パターンを特徴とする結晶性の形態IIのアトルバスタチンおよびその水和物に関するものである。
【0017】
【表3】
【0018】
さらに、本発明の第2の見地は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする結晶性の形態IIのアトルバスタチンおよびその水和物に関するものである。
【0019】
【表4】
【0020】
第3の見地においては、本発明は、CuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔および>15%の相対強度の相対強度によって表示された次のX線粉末回折パターンを特徴とする結晶性の形態IVのアトルバスタチンおよびその水和物に関するものである。
【0021】
【表5】
【0022】
さらに、本発明の第4の見地は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする形態IVのアトルバスタチンおよびその水和物に関するものである。
【0023】
【表6】
【0024】
HMG-CoAの阻害剤として、新規な結晶性形態のアトルバスタチンは、有用な血中脂質低下剤および血中コレストロール低下剤である。
さらに、本発明の別の実施態様は、上述した治療方法に、単位投与形態の結晶性の形態I、形態IIまたは形態IVのアトルバスタチンの有効量を投与するための医薬組成物である。最後に、本発明は、形態I、形態IIまたは形態IVのアトルバスタチンを製造する方法に関するものである。
【0025】
本発明を、さらに添付図面図1〜6に関する以下の実施例によって説明する。
結晶性の形態I、形態IIまたは形態IVのアトルバスタチンは、これらの化合物のX線粉末回折パターンおよび(または)これらの化合物の固体状態の核磁気共鳴スペクトル(NMR)によって特徴づけることができる。
【0026】
X線粉末回折
形態I、IIおよびIVのアトルバスタチン
形態I、形態IIまたは形態IVのアトルバスタチンは、それらのX線粉末回折パターンにより特徴づけられる。したがって、形態I、形態IIおよび形態IVのアトルバスタチンのX線回折パターンを、CuKα放射線を使用してSiemens D-500回折計上で測定した。
装 置
IBM−コンパーチブルインターフェイスを具備したSiemens D-500回折計−Kristalloflex、ソアトウエアー=DIFFRAC AT (SOCABIM 1986,1992)。
CuKα放射線(20mA、40kv、 λ=1.5406Å)。Kevex Psi Peltier冷却シリコーン〔Si(Li)〕検出器によって電子的にフィルター処理した(スリット:1°でのIおよびII)(1°でのIII、および0.15°でのIV)。
方 法
シリコン標準は、毎日X線管アラインメントを検査するために実験する。
連続θ/2θカップル走査:2θにおいて4.00°〜40.00°。6°/分の走査速度:0.4秒/0.04°ステップ。
試料は、バイアルから取出しそしてアルミニウムホルダー中のゼロ−バックグランド石英上に圧縮する。試料の幅13〜15mm。
試料は、室温で貯蔵しそして実験する。
粉砕/ふるい分け
粉砕は、本明細書に記載したジフラクトグラムに対する強度変化を最小にするために使用する。しかしながら、粉砕がジフラクトグラムを有意に変化するかまたは試料の無定形含量を有意に増加する場合は、粉砕しない試料のジフラクトグラムを使用した。粉砕は、小さなめのう乳ばちおよび乳棒において実施した。乳ばちは粉砕中保持しそして軽い圧力を乳棒に適用した。
粉砕した形態IIのアトルバスタチンは、X線回折による分析の前に、230メッシュのスクリーンを通してふるい分けした。
表1は、結晶性形態Iのアトルバスタチンに対する、>20%の相対強度を有する未粉砕試料におけるすべての線の2θ、d−面間隔および相対強度を示す。表1はまた、2分の粉砕後に測定したジフラクトグラムにおける同じ線の相対強度を示す。2分間粉砕した試料の強度は、選択方位のない回折パターンの代表的なものである。またコンピューターにより得られた端数のある数字がこの表に示されていることは注目されなければならない。
【0027】
【表7】
【0028】
表2は、結晶性形態IIのアトルバスタチンに対する、>20%の相対強度を有する粉砕/ふるい分けした試料におけるすべての線の2θ、d−面間隔および相対強度を示す。またコンピューターにより得られた端数のある数字がこの表に示されていることは注目されなければならない。
【0029】
【表8】
【0030】
表3は、結晶性形態IVのアトルバスタチンに対する、>15%の相対強度を有する未粉砕試料におけるすべての線の2θ、d−面間隔および相対強度を示す。また、コンピューターにより得られた端数のある数字がこの表に示されていることは、注目されなければならない。
【0031】
【表9】
【0032】
固体状態の核磁気共鳴(NMR)
方 法
すべての固体状態の13CNMR測定は、Bruker AX-250、 250MHz NMR分光計を使用して行った。高分解能スペクトルは、高電力プロトンデカップリングおよびクロス−ポーラリゼーション(CP)(約5KHzでのマジック−アングル回転(MAS)を使用)を使用して得た。マジック−アングルは、FryeおよびMaciel(Frye J.S.およびMaciel G.E., J. Mag. Res.,1982;48:125)によって記載されているようにサイドバンドを検出することによりKBrのBrシグナルを使用して調節した。カニスター−デザインローターに充填した試料約300〜450mgを、それぞれの実験に対して使用した。化学シフトは、外部テトラキス(トリメチルシリル)シラン(3.50ppmでメチルシグナル)(Muntean J.V.およびStock L.M., J. Mag.Res., 1988;76:54)を参照した。
【0033】
表4は、結晶性形態Iのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【化1】
【0034】
【表10】
【0035】
表5は、結晶性形態IIのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【0036】
【表11】
【0037】
表6は、結晶性形態IVのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【0038】
【表12】
【0039】
本発明の結晶性形態I、形態IIおよび形態IVのアトルバスタチンは、無水の形態ならびに水和形態で存在することができる。一般に、水和形態は、非水和形態と均等でありそして本発明の範囲内に包含される。結晶性形態Iのアトルバスタチンは、水約1〜8モルを含有する。好ましくは、形態Iのアトルバスタチンは、水3モルを含有する。
【0040】
本発明は、結晶性形態Iのアトルバスタチンを与える条件下で溶剤中の溶液からアトルバスタチンを結晶化させることからなる結晶性形態Iのアトルバスタチンの製法を提供する。
結晶性形態Iのアトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される多数の方法を与えることができる。
【0041】
すなわち、例えば、結晶性形態Iのアトルバスタチンは、調節された条件下における結晶化によって製造することができる。特に、それは、例えば酢酸カルシウムなどのようなカルシウム塩の添加によって、相当する塩基性塩、例えばアルカリ金属塩、例えばリチウム、カリウム、ナトリウム塩など;アンモニアまたはアミン塩;好ましくはナトリウム塩の水溶液から、または、無定形のアトルバスタチンを水に懸濁することによって製造することができる。一般に、ヒドロキシル性補助溶剤、例えば低級アルカノール、例えばメタノールなどの使用が好ましい。
【0042】
所望の結晶性形態Iのアトルバスタチンを製造するための出発物質が相当するナトリウム塩の溶液である場合は、一つの好ましい製法は、約5v/v%以上のメタノール、好ましくは約5〜33v/v%のメタノール、特に好ましくは約10〜15v/v%のメタノールを含有する水中のナトリウム塩の溶液を、約70℃までの高い温度、例えば約45〜60℃、特に好ましくは約47〜52℃で、酢酸カルシウムの水溶液で処理することからなる。一般に、アトルバスタチンのナトリウム塩2モルに対して酢酸カルシウム1モルを使用することが好ましい。これらの条件下において、カルシウム塩形成ならびに結晶化は、好ましくは高い温度、例えば上述した温度範囲内で実施しなければならない。出発溶液に、例えば約7w/w%のような少量のメチル第3ブチルエーテル(MTBE)を含有させることが有利であるということが見出された。しばしば、結晶性形態Iのアトルバスタチンを一貫して製造するために、結晶性形態Iのアトルバスタチンの“種子(seeds)”を結晶化溶液に加えるのが望ましいということが見出されている。
【0043】
出発物質が無定形のアトルバスタチンまたは無定形および結晶性形態Iのアトルバスタチンの組み合わせである場合は、所望の結晶性形態Iのアトルバスタチンは、必要な形態への変換が完了するまで、約40v/v%まで、例えば約0〜20v/v%、特に好ましくは約5〜15v/v%の補助溶剤、例えばメタノール、エタノール、2−プロパノール、アセトンなどを含有する水中に固体を懸濁し次いで濾過することによって得ることができる。しばしば、結晶性形態Iのアトルバスタチンへの完全な変換を確保するために、結晶性形態Iのアトルバスタチンの"種子"を懸濁液に添加することが望ましいということが見出されている。このようにする代わりに、主として無定形のアトルバスタチンからなる水湿潤ケーキを、有意な量の結晶性形態Iのアトルバスタチンが存在するまで、高い温度、例えば約75℃まで、特に好ましくは約65〜70℃の温度で加熱し、それによって無定形/懸濁液形態Iの混合物を上述したようにスラリー化することができる。
【0044】
結晶性形態Iのアトルバスタチンは、無定形のアトルバスタチンよりも著しく容易に単離し、そして冷却後結晶化媒質から濾過し、洗浄しそして乾燥することができる。例えば、結晶性形態Iのアトルバスタチンの50mlのスラリーの濾過は、10秒以内に完了した。無定形のアトルバスタチンの同様な量の試料は、濾過するのに1時間以上を必要とした。
【0045】
本発明は、また結晶性形態IIのアトルバスタチンを与える条件下でアトルバスタチンを溶剤に懸濁することからなる結晶性懸濁IIのアトルバスタチンの製法を提供する。
形態IIの結晶性アトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される方法を与えることができる。
【0046】
すなわち、例えば、出発物質が無定形、無定形および形態Iの組み合わせまたは結晶性形態Iのアトルバスタチンである場合は、所望の形態IIの結晶性アトルバスタチンは、必要な形態への変換が完了するまで、固体を、約40〜50%の水を含有するメタノールに懸濁し次いで濾過することによって得ることができる。
【0047】
本発明は、また結晶性形態IVのアトルバスタチンを与える条件下で溶剤中のアトルバスタチンの溶液からアトルバスタチンを結晶化することからなる結晶性形態IVのアトルバスタチンの製法を提供する。
形態IVの結晶性アトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される方法を与えることができる。
【0048】
すなわち、例えば出発物質が形態Iの結晶性アトルバスタチンである場合は、所望の形態IVの結晶性アトルバスタチンは、固体をメタノールに溶解し、それによって結晶性形態IVを沈澱させることによって得ることができる。
【0049】
本発明の化合物は、広範囲の種々な経口的および非経口的投与形態で製剤化そして投与することができる。すなわち、本発明の化合物は、注射によって、すなわち、静脈内に、筋肉内に、皮内に、皮下に、十二指腸内にまたは腹腔内に投与することができる。また、本発明の化合物は、吸入によって、例えば鼻内に投与することができる。さらに、本発明の化合物は、経皮的に投与することができる。当該技術に精通する者に明らかであるように、以下の投与形態は、活性成分として、化合物または本発明の化合物の相当する医薬的に許容し得る塩を含有することができる。
【0050】
本発明の化合物から医薬組成物を製造するに際しては、医薬的に許容し得る担体は、固体または液体であることができる。固体形態の製剤としては、粉末、錠剤、ピル、カプセル、カシエー、坐剤および分散性顆粒が含まれる。固体の担体は、希釈剤、風味剤、可溶化剤、滑沢剤、懸濁剤、結合剤、防腐剤、錠剤崩壊剤または封入物質として作用することもできる1種以上の物質であることができる。
【0051】
粉末においては、担体は、微細な活性成分と混合される微細な固体である。
錠剤においては、活性成分を適当な割合で必要な結合性を有する担体と混合し、そして所望の形状および大きさに圧縮する。
【0052】
粉末および錠剤は、好ましくは約2%または10%〜約70%の活性化合物を含有する。適当な担体は、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、ペクチン、デキストリン、澱粉、ゼラチン、トラガカントゴム、メチルセルロース、ナトリウムカルボキシメチルセルロース、低融点ワックス、ココアバターなどである。“製剤”なる用語は、他の担体を有しているかまたは有していない活性成分が担体によって囲まれそして活性成分が該担体と一緒になっているカプセルを与える、担体としての封入物質と活性化合物との処方を包含するように企図する。同様に、カシエーおよびロゼンジも包含される。錠剤、粉末、カプセル、ピル、カシエーおよびロゼンジは、経口投与に適した固体の投与形態として使用することができる。
【0053】
坐剤の製造に際しては、低融点のワックス、例えば脂肪酸グリセリドの混合物またはココアバターをはじめに融解し、そして活性成分を例えば撹拌によってその中に一様に分散する。それから、融解した均質な混合物を都合のよい大きさの型に注入し、冷却しそしてそれによって固化させる。
【0054】
液状の製剤は、溶液、懸濁液、保持浣腸およびエマルジョン、例えば水またはプロピレングリコール水溶液を包含する。非経口的注射に際しては、液状製剤は、ポリエチレングリコール水溶液中の溶液として処方することができる。
【0055】
経口的使用に適した水溶液は、活性成分を水に溶解しそして必要に応じて適当な着色剤、風味剤、安定剤および環化剤を加えることによって製造することができる。
【0056】
経口的使用に適した水性懸濁液は、微細な活性成分を、粘稠物質、例えば天然または合成ゴム、樹脂、メチルセルロース、ナトリウムカルボキシメチルセルロースおよび他の公知の懸濁剤と一緒に、水に分散することによって製造することができる。
【0057】
また、使用直前に経口的投与用の液状の製剤に変換すべく企図された固体の形態の製剤も包含される。このような液状の形態としては溶液、懸濁液およびエマルジョンが含まれる。これらの製剤は、活性成分に加えて、着色料、風味料、安定剤、緩衝剤、人工および天然甘味剤、分散剤、環化剤、可溶化剤などを含有することができる。
【0058】
医薬製剤は、好ましくは単位使用形態にある。このような形態においては、製剤は、活性成分の適当な量を含有する単位投与量に小分けされる。単位使用形態は、包装された錠剤、カプセル、およびバイアルまたはアンプル中の粉末のような、分離した量の製剤を含有する包装された製剤であることができる。また単位使用形態は、カプセル、錠剤、カシエーまたはロゼンジそれ自体であることができ、またはそれは、これらの何れかが適当な数で包装された形態であることができる。
【0059】
単位投与製剤中の活性成分の量は、特定の適用および活性成分の力価によって、0.5〜100mg、好ましくは2.5〜80mgに変化または調節することができる。必要に応じて、組成物はまた、他の相容性の治療剤を含有することもできる。
【0060】
血中脂質低下剤および(または)血中コレステロール低下剤としての治療的使用において、本発明の医薬的方法に利用される結晶性形態I、形態IIおよび形態IVのアトルバスタチンは、1日につき約2.5mg〜約80mgの初期使用量で投与される。約2.5mg〜約20mgの1日当たりの投与量範囲が好ましい。しかしながら、使用量は、患者の必要条件、処置される疾患の程度、および使用される化合物によって変えることができる。特定の情況に対する適当な使用量は、当該技術の熟練度の範囲内で決定される。一般に、治療は、化合物の最適の投与量より低い使用量で開始される。その後、使用量は、情況下における最適の作用に達するまで少量ずつ増加される。便宜上、必要に応じて、1日当たりの全使用量を分割しそして1日のうち数回投与することができる。
【0061】
以下の非限定的実施例は、本発明の化合物を製造する好ましい方法を説明する。
【実施例】
【0062】
実施例 1
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態Iのアトルバスタチン)
方法A
(2R−トランス)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔2−(テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミド(アトルバスタチンラクトン)(米国特許第5,273,995号)(75kg)、メチル第3ブチルエーテル(MTBE)(308kg)、メタノール(190L)の混合物を、48〜58℃で水酸化ナトリウムの水溶液(950L中 5.72kg)と40〜60分反応させて開環したナトリウム塩を形成させる。25〜35℃に冷却した後、有機層を捨てそして水性層を再びMTBE(230kg)で抽出する。有機層を捨て、そしてナトリウム塩のMTBE飽和水溶液を、47〜52℃に加熱する。この溶液に、水(410L)に溶解した酢酸カルシウム半水和物(11.94kg)の溶液を、少なくとも30分にわたって加える。
酢酸カルシウム溶液の添加後直ぐに、結晶性形態Iのアトルバスタチンのスラリー(水11Lおよびメタノール5L中の1.1kg)を、種子として混合物に加える。それから、混合物を少なくとも10分51〜57℃に加熱し、そしてそれから15〜40℃に冷却する。混合物を濾過し、水(300L)およびメタノール(150L)の溶液で洗浄し、次いで水(450L)で洗浄する。固体を、真空下60〜70℃で3〜4日間乾燥して結晶性形態Iのアトルバスタチン(72.2kg)を得た。
方法B
無定形のアトルバスタチン(9g)および結晶性形態Iのアトルバスタチン(1g)を、水(170ml)およびメタノール(30ml)の混合物中において約40℃で全体で17時間撹拌する。混合物を濾過し、水ですすぎ、そして減圧下70℃で乾燥して結晶性形態Iのアトルバスタチン(9.7g)を得た。
【0063】
実施例 2
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態IIのアトルバスタチン)
無定形および結晶性形態Iのアトルバスタチンの混合物(100g)を、メタノール(1200ml)および水(800ml)の混合物に懸濁し、そして3日間撹拌する。この物質を濾過し、減圧下70℃で乾燥して結晶性形態IIのアトルバスタチンを得た。
【0064】
実施例 3
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態IVのアトルバスタチン)
(2R−トランス)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔2−(テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミド(アトルバスタチンラクトン)(米国特許第5,273,995号)(12kg)、MTBE(50kg)、メタノール(30L)の混合物を、50〜55℃で水酸化ナトリウムの水溶液(150L中の1.83kg)と30〜45分反応させて開環したナトリウム塩を形成させる。20〜25℃に冷却した後、有機層を捨てそして水性層を再びMTBE(37kg)で抽出する。有機層を捨て、ナトリウム塩の水溶液を70〜80℃に加熱し、そして残留MTBEを蒸溜によって除去する。それから、溶液を60〜70℃に冷却する。この溶液に、水/メタノール(水72L+メタノール16L)に溶解した酢酸カルシウム半水和物(1.91kg)の溶液を加える。酢酸カルシウム溶液の添加後直ぐに、結晶性形態Iのアトルバスタチン(180g)を、種子として混合物に加える。混合物を、65〜75℃に少なくとも5分加熱し、そしてそれから50〜55℃に冷却する。混合物を濾過し、メタノール(約200L)中で55〜65℃でスラリー化し、そしてそれから25〜30℃に冷却して濾過する。固体を、真空下66〜70℃で乾燥して、形態IVの結晶性アトルバスタチン(約3kgを単離)を得た。
【産業上の利用可能性】
【0065】
本発明に係るアトルバスタチンは、高脂血症および高コレステロール血症を治療する薬剤として有用である。
【技術分野】
【0001】
本発明は、医薬として有用である新規な結晶性形態の化学名〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩によって知られているアトルバスタチンを製造および単離する方法に関するものである。本発明の新規な結晶性化合物は、酵素3−ヒドロキシ−3−メチルグルタリル−コエンザイムAレダクターゼ(HMG-CoAレダクターゼ)の阻害剤として有用であり、そして結果として、有用な血中脂質性低下剤および血中コレステロール低下剤である。
【背景技術】
【0002】
参照により本明細書に加入した特許文献1は、トランス(±)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔(2−テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミドを包含するあるトランス−6−〔2−(3−または4−カルボキサミド−置換されたピロール−1−イル)アルキル〕−4−ヒドロキシ−ピラン−2−オンを開示している。
【0003】
参照により本明細書に加入した特許文献2は、トランス−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔(2−テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミドのR形態の開環した酸を有するエナンチオマーすなわち〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸を開示している。
【0004】
参照により本明細書に加入した特許文献3、特許文献4、特許文献5、特許文献6、特許文献7、特許文献8、特許文献9、特許文献10、特許文献11、特許文献12、特許文献13および特許文献14は、アトルバスタチンを製造する種々な方法および重要な中間体を開示している。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第4681893号明細書
【特許文献2】米国特許第5273995号明細書
【特許文献3】米国特許第5003080号明細書
【特許文献4】米国特許第5097045号明細書
【特許文献5】米国特許第5103024号明細書
【特許文献6】米国特許第5124482号明細書
【特許文献7】米国特許第5149837号明細書
【特許文献8】米国特許第5155251号明細書
【特許文献9】米国特許第5216174号明細書
【特許文献10】米国特許第5245047号明細書
【特許文献11】米国特許第5248793号明細書
【特許文献12】米国特許第5280126号明細書
【特許文献13】米国特許第5397792号明細書
【特許文献14】米国特許第5342952号明細書
【発明の概要】
【発明が解決しようとする課題】
【0006】
アトルバスタチンは、そのカルシウム塩、すなわち、〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸カルシウム塩(2:1)として製造される。カルシウム塩は、アトルバスタチンを例えば経口的投与のための錠剤、カプセル、ロゼンジ、粉末などに有利に処方することを可能にするので、望ましい。さらに、厳密な製剤上の必要条件および規格を満足する処方を可能にするために、純粋な且つ結晶性の形態でアトルバスタチンを製造することが必要である。
【0007】
さらに、アトルバスタチンを製造する方法は、大規模な生産に耐えられる方法であることが必要である。さらに、生成物は、容易に濾過することができそして容易に乾燥することができる形態にあることが望ましい。最後に、生成物は、特別な貯蔵条件を必要とすることなしに長期間安定であることが経済的に望ましい。
【0008】
上記米国特許の方法は、大規模な生産に対して適当でない濾過および乾燥特性を有しそして熱、光、酸素および湿気から保護しなければならない無定形のアトルバスタチンを開示している。
【課題を解決するための手段】
【0009】
驚くべきことにはそして意外にも、アトルバスタチンは結晶性の形態で製造することができるということが見出された。すなわち、本発明は、形態I、形態IIおよび形態IVと称される新規な結晶性形態のアトルバスタチンを提供する。
【発明の効果】
【0010】
形態Iのアトルバスタチンは、従来の無定形の生成物よりも小さい粒子およびより一様な大きさの分布からなり、そしてより有利な濾過および乾燥特性を示す。さらに、形態Iのアトルバスタチンは、無定形の生成物よりも純粋でありそしてより安定である。
【図面の簡単な説明】
【0011】
【図1】2分粉砕した形態Iのアトルバスタチンのジフラクトグラム(Y軸=0〜3767.50カウント/秒(cps)の最大強度)を示す。
【図2】2分粉砕した形態IIのアトルバスタチンのジフラクトグラム(Y軸=0〜1500cpsの最大強度)を示す。
【図3】形態IVのアトルバスタチンのジフラクトグラム(Y軸=0〜8212.5cpsの最大強度)を示す。
【図4】形態Iのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【図5】形態IIのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【図6】形態IVのアトルバスタチンの星印によって確認された回転サイドバンドを有する固体状態の13C核磁気共鳴スペクトルを示す。
【発明を実施するための形態】
【0012】
本発明は、2分の粉砕後に測定したおよびCuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔、および>20%の相対強度の相対強度によって表した次のX線粉末回折パターンを特徴とする結晶性の形態Iのアトルバスタチンおよびその水和物に関するものである。
【0013】
【表1】
【0014】
さらに、本発明は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする結晶性の形態Iのアトルバスタチンおよびその水和物に関するものである。
【0015】
【表2】
【0016】
本発明の第1の見地の好ましい実施化においては、結晶性の形態Iのアトルバスタチンは、三水和物である。
第2の見地においては、本発明は、2分の粉砕後に測定したそしてCuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔、および>20%の相対強度を有する強度によって表した次のX線粉末回折パターンを特徴とする結晶性の形態IIのアトルバスタチンおよびその水和物に関するものである。
【0017】
【表3】
【0018】
さらに、本発明の第2の見地は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする結晶性の形態IIのアトルバスタチンおよびその水和物に関するものである。
【0019】
【表4】
【0020】
第3の見地においては、本発明は、CuKα放射線を使用してSiemens D-500回折計上で測定した、2θ、d−面間隔および>15%の相対強度の相対強度によって表示された次のX線粉末回折パターンを特徴とする結晶性の形態IVのアトルバスタチンおよびその水和物に関するものである。
【0021】
【表5】
【0022】
さらに、本発明の第4の見地は、Bruker AX-250分光計上で測定した化学シフトが100万部当たりの部数で表された次の固体状態の13C核磁気共鳴スペクトルを特徴とする形態IVのアトルバスタチンおよびその水和物に関するものである。
【0023】
【表6】
【0024】
HMG-CoAの阻害剤として、新規な結晶性形態のアトルバスタチンは、有用な血中脂質低下剤および血中コレストロール低下剤である。
さらに、本発明の別の実施態様は、上述した治療方法に、単位投与形態の結晶性の形態I、形態IIまたは形態IVのアトルバスタチンの有効量を投与するための医薬組成物である。最後に、本発明は、形態I、形態IIまたは形態IVのアトルバスタチンを製造する方法に関するものである。
【0025】
本発明を、さらに添付図面図1〜6に関する以下の実施例によって説明する。
結晶性の形態I、形態IIまたは形態IVのアトルバスタチンは、これらの化合物のX線粉末回折パターンおよび(または)これらの化合物の固体状態の核磁気共鳴スペクトル(NMR)によって特徴づけることができる。
【0026】
X線粉末回折
形態I、IIおよびIVのアトルバスタチン
形態I、形態IIまたは形態IVのアトルバスタチンは、それらのX線粉末回折パターンにより特徴づけられる。したがって、形態I、形態IIおよび形態IVのアトルバスタチンのX線回折パターンを、CuKα放射線を使用してSiemens D-500回折計上で測定した。
装 置
IBM−コンパーチブルインターフェイスを具備したSiemens D-500回折計−Kristalloflex、ソアトウエアー=DIFFRAC AT (SOCABIM 1986,1992)。
CuKα放射線(20mA、40kv、 λ=1.5406Å)。Kevex Psi Peltier冷却シリコーン〔Si(Li)〕検出器によって電子的にフィルター処理した(スリット:1°でのIおよびII)(1°でのIII、および0.15°でのIV)。
方 法
シリコン標準は、毎日X線管アラインメントを検査するために実験する。
連続θ/2θカップル走査:2θにおいて4.00°〜40.00°。6°/分の走査速度:0.4秒/0.04°ステップ。
試料は、バイアルから取出しそしてアルミニウムホルダー中のゼロ−バックグランド石英上に圧縮する。試料の幅13〜15mm。
試料は、室温で貯蔵しそして実験する。
粉砕/ふるい分け
粉砕は、本明細書に記載したジフラクトグラムに対する強度変化を最小にするために使用する。しかしながら、粉砕がジフラクトグラムを有意に変化するかまたは試料の無定形含量を有意に増加する場合は、粉砕しない試料のジフラクトグラムを使用した。粉砕は、小さなめのう乳ばちおよび乳棒において実施した。乳ばちは粉砕中保持しそして軽い圧力を乳棒に適用した。
粉砕した形態IIのアトルバスタチンは、X線回折による分析の前に、230メッシュのスクリーンを通してふるい分けした。
表1は、結晶性形態Iのアトルバスタチンに対する、>20%の相対強度を有する未粉砕試料におけるすべての線の2θ、d−面間隔および相対強度を示す。表1はまた、2分の粉砕後に測定したジフラクトグラムにおける同じ線の相対強度を示す。2分間粉砕した試料の強度は、選択方位のない回折パターンの代表的なものである。またコンピューターにより得られた端数のある数字がこの表に示されていることは注目されなければならない。
【0027】
【表7】
【0028】
表2は、結晶性形態IIのアトルバスタチンに対する、>20%の相対強度を有する粉砕/ふるい分けした試料におけるすべての線の2θ、d−面間隔および相対強度を示す。またコンピューターにより得られた端数のある数字がこの表に示されていることは注目されなければならない。
【0029】
【表8】
【0030】
表3は、結晶性形態IVのアトルバスタチンに対する、>15%の相対強度を有する未粉砕試料におけるすべての線の2θ、d−面間隔および相対強度を示す。また、コンピューターにより得られた端数のある数字がこの表に示されていることは、注目されなければならない。
【0031】
【表9】
【0032】
固体状態の核磁気共鳴(NMR)
方 法
すべての固体状態の13CNMR測定は、Bruker AX-250、 250MHz NMR分光計を使用して行った。高分解能スペクトルは、高電力プロトンデカップリングおよびクロス−ポーラリゼーション(CP)(約5KHzでのマジック−アングル回転(MAS)を使用)を使用して得た。マジック−アングルは、FryeおよびMaciel(Frye J.S.およびMaciel G.E., J. Mag. Res.,1982;48:125)によって記載されているようにサイドバンドを検出することによりKBrのBrシグナルを使用して調節した。カニスター−デザインローターに充填した試料約300〜450mgを、それぞれの実験に対して使用した。化学シフトは、外部テトラキス(トリメチルシリル)シラン(3.50ppmでメチルシグナル)(Muntean J.V.およびStock L.M., J. Mag.Res., 1988;76:54)を参照した。
【0033】
表4は、結晶性形態Iのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【化1】
【0034】
【表10】
【0035】
表5は、結晶性形態IIのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【0036】
【表11】
【0037】
表6は、結晶性形態IVのアトルバスタチンに対する固体状態のNMRスペクトルを示す。
【0038】
【表12】
【0039】
本発明の結晶性形態I、形態IIおよび形態IVのアトルバスタチンは、無水の形態ならびに水和形態で存在することができる。一般に、水和形態は、非水和形態と均等でありそして本発明の範囲内に包含される。結晶性形態Iのアトルバスタチンは、水約1〜8モルを含有する。好ましくは、形態Iのアトルバスタチンは、水3モルを含有する。
【0040】
本発明は、結晶性形態Iのアトルバスタチンを与える条件下で溶剤中の溶液からアトルバスタチンを結晶化させることからなる結晶性形態Iのアトルバスタチンの製法を提供する。
結晶性形態Iのアトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される多数の方法を与えることができる。
【0041】
すなわち、例えば、結晶性形態Iのアトルバスタチンは、調節された条件下における結晶化によって製造することができる。特に、それは、例えば酢酸カルシウムなどのようなカルシウム塩の添加によって、相当する塩基性塩、例えばアルカリ金属塩、例えばリチウム、カリウム、ナトリウム塩など;アンモニアまたはアミン塩;好ましくはナトリウム塩の水溶液から、または、無定形のアトルバスタチンを水に懸濁することによって製造することができる。一般に、ヒドロキシル性補助溶剤、例えば低級アルカノール、例えばメタノールなどの使用が好ましい。
【0042】
所望の結晶性形態Iのアトルバスタチンを製造するための出発物質が相当するナトリウム塩の溶液である場合は、一つの好ましい製法は、約5v/v%以上のメタノール、好ましくは約5〜33v/v%のメタノール、特に好ましくは約10〜15v/v%のメタノールを含有する水中のナトリウム塩の溶液を、約70℃までの高い温度、例えば約45〜60℃、特に好ましくは約47〜52℃で、酢酸カルシウムの水溶液で処理することからなる。一般に、アトルバスタチンのナトリウム塩2モルに対して酢酸カルシウム1モルを使用することが好ましい。これらの条件下において、カルシウム塩形成ならびに結晶化は、好ましくは高い温度、例えば上述した温度範囲内で実施しなければならない。出発溶液に、例えば約7w/w%のような少量のメチル第3ブチルエーテル(MTBE)を含有させることが有利であるということが見出された。しばしば、結晶性形態Iのアトルバスタチンを一貫して製造するために、結晶性形態Iのアトルバスタチンの“種子(seeds)”を結晶化溶液に加えるのが望ましいということが見出されている。
【0043】
出発物質が無定形のアトルバスタチンまたは無定形および結晶性形態Iのアトルバスタチンの組み合わせである場合は、所望の結晶性形態Iのアトルバスタチンは、必要な形態への変換が完了するまで、約40v/v%まで、例えば約0〜20v/v%、特に好ましくは約5〜15v/v%の補助溶剤、例えばメタノール、エタノール、2−プロパノール、アセトンなどを含有する水中に固体を懸濁し次いで濾過することによって得ることができる。しばしば、結晶性形態Iのアトルバスタチンへの完全な変換を確保するために、結晶性形態Iのアトルバスタチンの"種子"を懸濁液に添加することが望ましいということが見出されている。このようにする代わりに、主として無定形のアトルバスタチンからなる水湿潤ケーキを、有意な量の結晶性形態Iのアトルバスタチンが存在するまで、高い温度、例えば約75℃まで、特に好ましくは約65〜70℃の温度で加熱し、それによって無定形/懸濁液形態Iの混合物を上述したようにスラリー化することができる。
【0044】
結晶性形態Iのアトルバスタチンは、無定形のアトルバスタチンよりも著しく容易に単離し、そして冷却後結晶化媒質から濾過し、洗浄しそして乾燥することができる。例えば、結晶性形態Iのアトルバスタチンの50mlのスラリーの濾過は、10秒以内に完了した。無定形のアトルバスタチンの同様な量の試料は、濾過するのに1時間以上を必要とした。
【0045】
本発明は、また結晶性形態IIのアトルバスタチンを与える条件下でアトルバスタチンを溶剤に懸濁することからなる結晶性懸濁IIのアトルバスタチンの製法を提供する。
形態IIの結晶性アトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される方法を与えることができる。
【0046】
すなわち、例えば、出発物質が無定形、無定形および形態Iの組み合わせまたは結晶性形態Iのアトルバスタチンである場合は、所望の形態IIの結晶性アトルバスタチンは、必要な形態への変換が完了するまで、固体を、約40〜50%の水を含有するメタノールに懸濁し次いで濾過することによって得ることができる。
【0047】
本発明は、また結晶性形態IVのアトルバスタチンを与える条件下で溶剤中のアトルバスタチンの溶液からアトルバスタチンを結晶化することからなる結晶性形態IVのアトルバスタチンの製法を提供する。
形態IVの結晶性アトルバスタチンが形成される正確な条件は、経験的に決定することができそして実施に適当であることが見出される方法を与えることができる。
【0048】
すなわち、例えば出発物質が形態Iの結晶性アトルバスタチンである場合は、所望の形態IVの結晶性アトルバスタチンは、固体をメタノールに溶解し、それによって結晶性形態IVを沈澱させることによって得ることができる。
【0049】
本発明の化合物は、広範囲の種々な経口的および非経口的投与形態で製剤化そして投与することができる。すなわち、本発明の化合物は、注射によって、すなわち、静脈内に、筋肉内に、皮内に、皮下に、十二指腸内にまたは腹腔内に投与することができる。また、本発明の化合物は、吸入によって、例えば鼻内に投与することができる。さらに、本発明の化合物は、経皮的に投与することができる。当該技術に精通する者に明らかであるように、以下の投与形態は、活性成分として、化合物または本発明の化合物の相当する医薬的に許容し得る塩を含有することができる。
【0050】
本発明の化合物から医薬組成物を製造するに際しては、医薬的に許容し得る担体は、固体または液体であることができる。固体形態の製剤としては、粉末、錠剤、ピル、カプセル、カシエー、坐剤および分散性顆粒が含まれる。固体の担体は、希釈剤、風味剤、可溶化剤、滑沢剤、懸濁剤、結合剤、防腐剤、錠剤崩壊剤または封入物質として作用することもできる1種以上の物質であることができる。
【0051】
粉末においては、担体は、微細な活性成分と混合される微細な固体である。
錠剤においては、活性成分を適当な割合で必要な結合性を有する担体と混合し、そして所望の形状および大きさに圧縮する。
【0052】
粉末および錠剤は、好ましくは約2%または10%〜約70%の活性化合物を含有する。適当な担体は、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、ペクチン、デキストリン、澱粉、ゼラチン、トラガカントゴム、メチルセルロース、ナトリウムカルボキシメチルセルロース、低融点ワックス、ココアバターなどである。“製剤”なる用語は、他の担体を有しているかまたは有していない活性成分が担体によって囲まれそして活性成分が該担体と一緒になっているカプセルを与える、担体としての封入物質と活性化合物との処方を包含するように企図する。同様に、カシエーおよびロゼンジも包含される。錠剤、粉末、カプセル、ピル、カシエーおよびロゼンジは、経口投与に適した固体の投与形態として使用することができる。
【0053】
坐剤の製造に際しては、低融点のワックス、例えば脂肪酸グリセリドの混合物またはココアバターをはじめに融解し、そして活性成分を例えば撹拌によってその中に一様に分散する。それから、融解した均質な混合物を都合のよい大きさの型に注入し、冷却しそしてそれによって固化させる。
【0054】
液状の製剤は、溶液、懸濁液、保持浣腸およびエマルジョン、例えば水またはプロピレングリコール水溶液を包含する。非経口的注射に際しては、液状製剤は、ポリエチレングリコール水溶液中の溶液として処方することができる。
【0055】
経口的使用に適した水溶液は、活性成分を水に溶解しそして必要に応じて適当な着色剤、風味剤、安定剤および環化剤を加えることによって製造することができる。
【0056】
経口的使用に適した水性懸濁液は、微細な活性成分を、粘稠物質、例えば天然または合成ゴム、樹脂、メチルセルロース、ナトリウムカルボキシメチルセルロースおよび他の公知の懸濁剤と一緒に、水に分散することによって製造することができる。
【0057】
また、使用直前に経口的投与用の液状の製剤に変換すべく企図された固体の形態の製剤も包含される。このような液状の形態としては溶液、懸濁液およびエマルジョンが含まれる。これらの製剤は、活性成分に加えて、着色料、風味料、安定剤、緩衝剤、人工および天然甘味剤、分散剤、環化剤、可溶化剤などを含有することができる。
【0058】
医薬製剤は、好ましくは単位使用形態にある。このような形態においては、製剤は、活性成分の適当な量を含有する単位投与量に小分けされる。単位使用形態は、包装された錠剤、カプセル、およびバイアルまたはアンプル中の粉末のような、分離した量の製剤を含有する包装された製剤であることができる。また単位使用形態は、カプセル、錠剤、カシエーまたはロゼンジそれ自体であることができ、またはそれは、これらの何れかが適当な数で包装された形態であることができる。
【0059】
単位投与製剤中の活性成分の量は、特定の適用および活性成分の力価によって、0.5〜100mg、好ましくは2.5〜80mgに変化または調節することができる。必要に応じて、組成物はまた、他の相容性の治療剤を含有することもできる。
【0060】
血中脂質低下剤および(または)血中コレステロール低下剤としての治療的使用において、本発明の医薬的方法に利用される結晶性形態I、形態IIおよび形態IVのアトルバスタチンは、1日につき約2.5mg〜約80mgの初期使用量で投与される。約2.5mg〜約20mgの1日当たりの投与量範囲が好ましい。しかしながら、使用量は、患者の必要条件、処置される疾患の程度、および使用される化合物によって変えることができる。特定の情況に対する適当な使用量は、当該技術の熟練度の範囲内で決定される。一般に、治療は、化合物の最適の投与量より低い使用量で開始される。その後、使用量は、情況下における最適の作用に達するまで少量ずつ増加される。便宜上、必要に応じて、1日当たりの全使用量を分割しそして1日のうち数回投与することができる。
【0061】
以下の非限定的実施例は、本発明の化合物を製造する好ましい方法を説明する。
【実施例】
【0062】
実施例 1
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態Iのアトルバスタチン)
方法A
(2R−トランス)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔2−(テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミド(アトルバスタチンラクトン)(米国特許第5,273,995号)(75kg)、メチル第3ブチルエーテル(MTBE)(308kg)、メタノール(190L)の混合物を、48〜58℃で水酸化ナトリウムの水溶液(950L中 5.72kg)と40〜60分反応させて開環したナトリウム塩を形成させる。25〜35℃に冷却した後、有機層を捨てそして水性層を再びMTBE(230kg)で抽出する。有機層を捨て、そしてナトリウム塩のMTBE飽和水溶液を、47〜52℃に加熱する。この溶液に、水(410L)に溶解した酢酸カルシウム半水和物(11.94kg)の溶液を、少なくとも30分にわたって加える。
酢酸カルシウム溶液の添加後直ぐに、結晶性形態Iのアトルバスタチンのスラリー(水11Lおよびメタノール5L中の1.1kg)を、種子として混合物に加える。それから、混合物を少なくとも10分51〜57℃に加熱し、そしてそれから15〜40℃に冷却する。混合物を濾過し、水(300L)およびメタノール(150L)の溶液で洗浄し、次いで水(450L)で洗浄する。固体を、真空下60〜70℃で3〜4日間乾燥して結晶性形態Iのアトルバスタチン(72.2kg)を得た。
方法B
無定形のアトルバスタチン(9g)および結晶性形態Iのアトルバスタチン(1g)を、水(170ml)およびメタノール(30ml)の混合物中において約40℃で全体で17時間撹拌する。混合物を濾過し、水ですすぎ、そして減圧下70℃で乾燥して結晶性形態Iのアトルバスタチン(9.7g)を得た。
【0063】
実施例 2
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態IIのアトルバスタチン)
無定形および結晶性形態Iのアトルバスタチンの混合物(100g)を、メタノール(1200ml)および水(800ml)の混合物に懸濁し、そして3日間撹拌する。この物質を濾過し、減圧下70℃で乾燥して結晶性形態IIのアトルバスタチンを得た。
【0064】
実施例 3
〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸ヘミカルシウム塩(形態IVのアトルバスタチン)
(2R−トランス)−5−(4−フルオロフェニル)−2−(1−メチルエチル)−N,4−ジフェニル−1−〔2−(テトラヒドロ−4−ヒドロキシ−6−オキソ−2H−ピラン−2−イル)エチル〕−1H−ピロール−3−カルボキサミド(アトルバスタチンラクトン)(米国特許第5,273,995号)(12kg)、MTBE(50kg)、メタノール(30L)の混合物を、50〜55℃で水酸化ナトリウムの水溶液(150L中の1.83kg)と30〜45分反応させて開環したナトリウム塩を形成させる。20〜25℃に冷却した後、有機層を捨てそして水性層を再びMTBE(37kg)で抽出する。有機層を捨て、ナトリウム塩の水溶液を70〜80℃に加熱し、そして残留MTBEを蒸溜によって除去する。それから、溶液を60〜70℃に冷却する。この溶液に、水/メタノール(水72L+メタノール16L)に溶解した酢酸カルシウム半水和物(1.91kg)の溶液を加える。酢酸カルシウム溶液の添加後直ぐに、結晶性形態Iのアトルバスタチン(180g)を、種子として混合物に加える。混合物を、65〜75℃に少なくとも5分加熱し、そしてそれから50〜55℃に冷却する。混合物を濾過し、メタノール(約200L)中で55〜65℃でスラリー化し、そしてそれから25〜30℃に冷却して濾過する。固体を、真空下66〜70℃で乾燥して、形態IVの結晶性アトルバスタチン(約3kgを単離)を得た。
【産業上の利用可能性】
【0065】
本発明に係るアトルバスタチンは、高脂血症および高コレステロール血症を治療する薬剤として有用である。
【特許請求の範囲】
【請求項1】
結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸の塩基性塩の水溶液をカルシウム塩で処理する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【請求項2】
工程(a)での処理後に、結晶性形態Iのアトルバスタチンの種結晶を加える請求項1記載の方法。
【請求項3】
工程(a)において、水溶液がヒドロキシル性補助溶剤およびメチル第3ブチルエーテルを含有する請求項1または2記載の方法。
【請求項4】
ヒドロキシル性補助溶剤がメタノールである請求項3記載の方法。
【請求項5】
工程(a)において、カルシウム塩が酢酸カルシウムである請求項1〜4のいずれかに記載の方法。
【請求項6】
工程(a)において、塩基性塩がアルカリ金属塩、アンモニアおよびアミン塩からなる群より選択される請求項1〜5のいずれかに記載の方法。
【請求項7】
塩基性塩がナトリウム塩である請求項6記載の方法。
【請求項8】
結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)無定形のアトルバスタチンおよび結晶性形態Iのアトルバスタチンの混合物を、補助溶剤を含有する水に懸濁して、攪拌する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【請求項9】
工程(a)において、補助溶剤がメタノール、エタノール、2−プロパノールおよびアセトンからなる群より選択される請求項8記載の方法。
【請求項10】
補助溶剤がメタノールである請求項9記載の方法。
【請求項11】
結晶性形態IIのアトルバスタチンまたはその水和物の製法であって、
(a)無定形のアトルバスタチンおよび結晶性形態Iのアトルバスタチンの混合物を、水を含有するメタノールに懸濁して、攪拌する工程、および
(b)結晶性形態IIのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態IIのアトルバスタチンまたはその水和物の製法。
【請求項12】
結晶性形態IVのアトルバスタチンまたはその水和物の製法であって、
(a)結晶性形態Iのアトルバスタチンをメタノールに懸濁する工程、および
(b)冷却して結晶性形態IVのアトルバスタチンまたはその水和物を単離する工程
を含む結晶性形態IVのアトルバスタチンまたはその水和物の製法。
【請求項13】
請求項1〜10のいずれかに記載の方法によって製造される結晶性形態Iのアトルバスタチン水和物。
【請求項14】
請求項1〜10のいずれかに記載の方法によって製造される結晶性形態Iのアトルバスタチン3水和物。
【請求項15】
請求項11に記載の方法によって製造される結晶性形態IIのアトルバスタチン水和物。
【請求項16】
請求項12に記載の方法によって製造される結晶性形態IVのアトルバスタチン水和物。
【請求項17】
活性成分としてアトルバスタチンを含有する血中脂質低下剤または血中コレステロール低下剤であって、
前記アトルバスタチンが結晶性形態Iのアトルバスタチン、その水和物、結晶性形態IIのアトルバスタチン、その水和物、結晶性形態IVのアトルバスタチンおよびその水和物からなる群より選択されることを特徴とする血中脂質低下剤または血中コレステロール低下剤。
【請求項1】
結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)〔R−(R*,R*)〕−2−(4−フルオロフェニル)−β,δ−ジヒドロキシ−5−(1−メチルエチル)−3−フェニル−4−〔(フェニルアミノ)カルボニル〕−1H−ピロール−1−ヘプタン酸の塩基性塩の水溶液をカルシウム塩で処理する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【請求項2】
工程(a)での処理後に、結晶性形態Iのアトルバスタチンの種結晶を加える請求項1記載の方法。
【請求項3】
工程(a)において、水溶液がヒドロキシル性補助溶剤およびメチル第3ブチルエーテルを含有する請求項1または2記載の方法。
【請求項4】
ヒドロキシル性補助溶剤がメタノールである請求項3記載の方法。
【請求項5】
工程(a)において、カルシウム塩が酢酸カルシウムである請求項1〜4のいずれかに記載の方法。
【請求項6】
工程(a)において、塩基性塩がアルカリ金属塩、アンモニアおよびアミン塩からなる群より選択される請求項1〜5のいずれかに記載の方法。
【請求項7】
塩基性塩がナトリウム塩である請求項6記載の方法。
【請求項8】
結晶性形態Iのアトルバスタチンまたはその水和物の製法であって、
(a)無定形のアトルバスタチンおよび結晶性形態Iのアトルバスタチンの混合物を、補助溶剤を含有する水に懸濁して、攪拌する工程、および
(b)結晶性形態Iのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態Iのアトルバスタチンまたはその水和物の製法。
【請求項9】
工程(a)において、補助溶剤がメタノール、エタノール、2−プロパノールおよびアセトンからなる群より選択される請求項8記載の方法。
【請求項10】
補助溶剤がメタノールである請求項9記載の方法。
【請求項11】
結晶性形態IIのアトルバスタチンまたはその水和物の製法であって、
(a)無定形のアトルバスタチンおよび結晶性形態Iのアトルバスタチンの混合物を、水を含有するメタノールに懸濁して、攪拌する工程、および
(b)結晶性形態IIのアトルバスタチンまたはその水和物を単離する工程
を含む、結晶性形態IIのアトルバスタチンまたはその水和物の製法。
【請求項12】
結晶性形態IVのアトルバスタチンまたはその水和物の製法であって、
(a)結晶性形態Iのアトルバスタチンをメタノールに懸濁する工程、および
(b)冷却して結晶性形態IVのアトルバスタチンまたはその水和物を単離する工程
を含む結晶性形態IVのアトルバスタチンまたはその水和物の製法。
【請求項13】
請求項1〜10のいずれかに記載の方法によって製造される結晶性形態Iのアトルバスタチン水和物。
【請求項14】
請求項1〜10のいずれかに記載の方法によって製造される結晶性形態Iのアトルバスタチン3水和物。
【請求項15】
請求項11に記載の方法によって製造される結晶性形態IIのアトルバスタチン水和物。
【請求項16】
請求項12に記載の方法によって製造される結晶性形態IVのアトルバスタチン水和物。
【請求項17】
活性成分としてアトルバスタチンを含有する血中脂質低下剤または血中コレステロール低下剤であって、
前記アトルバスタチンが結晶性形態Iのアトルバスタチン、その水和物、結晶性形態IIのアトルバスタチン、その水和物、結晶性形態IVのアトルバスタチンおよびその水和物からなる群より選択されることを特徴とする血中脂質低下剤または血中コレステロール低下剤。
【図1】

【図2】
【図3】
【図4】
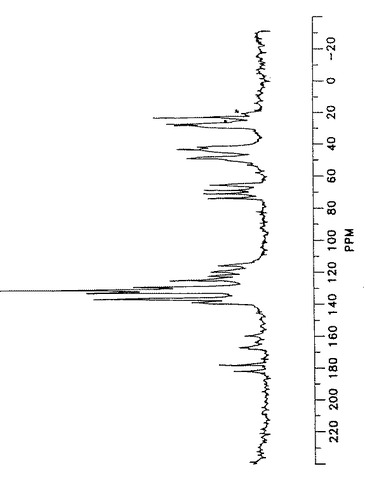
【図5】
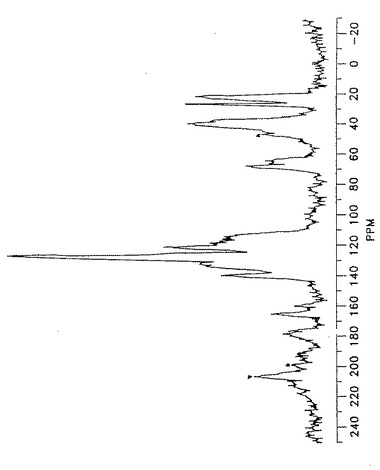
【図6】
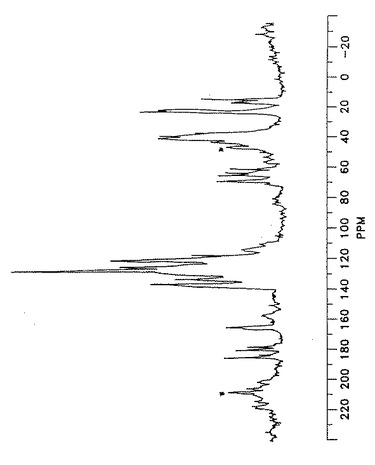
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2011−195592(P2011−195592A)
【公開日】平成23年10月6日(2011.10.6)
【国際特許分類】
【出願番号】特願2011−130841(P2011−130841)
【出願日】平成23年6月13日(2011.6.13)
【分割の表示】特願2002−8746(P2002−8746)の分割
【原出願日】平成8年7月8日(1996.7.8)
【出願人】(503181266)ワーナー−ランバート カンパニー リミテッド ライアビリティー カンパニー (167)
【Fターム(参考)】
【公開日】平成23年10月6日(2011.10.6)
【国際特許分類】
【出願日】平成23年6月13日(2011.6.13)
【分割の表示】特願2002−8746(P2002−8746)の分割
【原出願日】平成8年7月8日(1996.7.8)
【出願人】(503181266)ワーナー−ランバート カンパニー リミテッド ライアビリティー カンパニー (167)
【Fターム(参考)】
[ Back to top ]