
結晶性の且つ純粋なモダフィニルおよびそれを製造する方法
【課題】本発明は、1回の結晶化により高純度で単離できる、モダフィニルを製造する改良法を提供する。
【解決手段】本方法は、過剰酸化によるスルホン生成物およびその他の副産物を含まないモダフィニルを生成する。本発明はさらに、モダフィニルの新たな結晶形II-VIおよびそれらを製造する方法を提供する。この新たな結晶形の各々は特異な粉末X線回折パターンにより識別する。本発明はさらに、新規なモダフィニルII-IVおよびVI型を含有する薬用組成物を提供する。
【解決手段】本方法は、過剰酸化によるスルホン生成物およびその他の副産物を含まないモダフィニルを生成する。本発明はさらに、モダフィニルの新たな結晶形II-VIおよびそれらを製造する方法を提供する。この新たな結晶形の各々は特異な粉末X線回折パターンにより識別する。本発明はさらに、新規なモダフィニルII-IVおよびVI型を含有する薬用組成物を提供する。
【発明の詳細な説明】
【発明の開示】
【0001】
発明の分野
本発明は、不純物を含まないモダフィニルの製造方法、ならびにモダフィニルの新しい結晶形およびそれらを製造するための方法に関するものである。
【0002】
発明の背景
モダフィニルとしても知られる、式1で示される(±)2-[(ジフェニルメチル)スルフィニル]アセトアミドは、人間および動物に覚醒促進効果を表す。
【化1】

【0003】
モダフィニルの向精神活性は、米国特許第4177290号(「’290特許」)に記載のように動物試験で、そして人間の患者での臨床試験で証明された。モダフィニルのラセミ化合物はナルコレプシーの治療用にF.D.A.で承認されている。
【0004】
‘290特許はモダフィニルの製造を記載している。’290特許の実施例1では、2-[(ジフェニルメチル)チオ]酢酸クロリドをアンモニアと反応させ、アミド生成物を単離し、次いでそのスルフィド基を酢酸中の過酸化水素で酸化することによってモダフィニルを製造する。’290特許の実施例1aは、工業的規模のモダフィニル製造のため、異なる合成法を記載している。ベンズヒドロールをチオ尿素と反応させて中間体を生成し、次いでこれを加水分解して2-[(ジフェニルメチル)チオ]酢酸とする。次にこの酸を、クロロ酢酸および水を含有する混合物中、過酸化水素でin situ酸化する。得られたスルホキシドを次いで硫酸ジメチルで処理してカルボン酸基をメチル化する。得られたエステルをアンモニアで誘導体化してモダフィニルを得る。
【0005】
これらの方法の各々は、スルフィド基をスルホキシドに酸化するために過酸化水素を使用している。Drabowicz,J et al. Synthesis, 1990, 37-38は、立体障害されたスルフィドをスルホキシドに酸化するための方法を記載している。その方法は、酸化剤として過酸化水素を、溶媒としてメタノールを、そして触媒として、硫酸および幾つかの分枝脂肪族アルコールのうちの1つの混合物を使用する。この方法は立体障害されたスルフィドの酸化に良く適合している。反応混合物の薄層クロマトグラフィーでは過剰酸化生成物は観察されなかった。モダフィニルを製造するためのこの方法の使用は文献に記載されていない。
【0006】
スルフィドは、過ヨウ素酸ナトリウム、次亜塩素酸t-ブチル、次亜塩素酸カルシウム、亜塩素酸ナトリウム、次亜塩素酸ナトリウム、メタ−クロロ過安息香酸および過硼酸ナトリウムといった他の酸化剤でスルホキシドに酸化することもできる。March J. Advanced Organic Chemistry 1201-02(4th ed. 1992)。
本発明者等は、’290特許の実施例1の方法が、スルフィドからスルホン2への過剰酸化の問題を有することを発見した。
【化2】
【0007】
上に記載した化学構造を比較することにより、いったん生成したスルホンをモダフィニルから分離することは困難な仕事であることが容易に理解できる。故に、1回またはそれ以上の再結晶後にスルホンを含まないモダフィニルを得るため、選択的酸化法を開発することが求められる。
【0008】
実施例1aに記載の方法では、工程(b)および(c)における出発物質の不完全な変換のため、かなりの量の中間体2-[(ジフェニルメチル)スルフィニル]酢酸3およびメチル2-[(ジフェニルメチル)スルフィニル]酢酸4が得られる。Becue,T; Broquaire,M. J.Chromatography 1991, 557, 489-494。これらの化合物もまたモダフィニルから分離することが困難である。
【化3】
【0009】
工業的規模の方法で使用する溶媒の容量のため、そして大量の有機溶媒の廃棄により生ずる環境問題のため、本質上不純物を含まないモダフィニルを産生し且つ検出限界内の不純物を含まないモダフィニルを取得するために最終生成物を1回しか結晶化しなくてもよい工業生産は、同等の純度でモダフィニルを得るために再結晶を何回も必要とする別の方法よりもはるかに有利である。’290特許の実施例1aは工業的方法として記載されているが、生成物を白色結晶粉末として得るために2回の再結晶を使用した。その粉末の組成は報告されていない。
【0010】
スルホン2を本質上含まないモダフィニルを産生し、その結果、1回の結晶化によってこれが高純度で取得できる改良法を得ることが極めて望ましい。加えて、毒性が高いことから、実施例1aにおける試薬の1つである硫酸ジメチルの使用を回避することもまた極めて望ましい。
【0011】
高純度のモダフィニルを効率的に生産するという目的を追求する間に、本発明者等は、モダフィニルが幾つかの別個の固体状態の結晶性多形に結晶化できることを発見した。化合物の結晶形は、固体化した化合物の単位格子中の原子核の位置によって識別する。この相違は、薬学において実用上の重要性を持つ、熱による挙動、蒸気透過性および溶解度といった異なる巨視的性質を産む。化合物の結晶形はX線解析によって最も容易に識別できる。単結晶X線結晶学は、核の位置決定に利用できるデータをもたらし、次いでこれをコンピューターまたは機械的モデルによって視覚化し、このようにして当該化合物の三次元画像を得ることができる。単結晶X線解析は無類の構造情報を提供するものの、これらは高価で、特性データは時に獲得が困難である。粉末X線回折分光法は、薬物の新しい結晶形を特性決定するために、単結晶X線解析よりも頻繁に製薬工業で使用されている。粉末X線回折分光法はその結晶形に特異なフィンガープリントを生成し、それをアモルファス化合物および該化合物の他の全ての結晶形から識別することができる。
【0012】
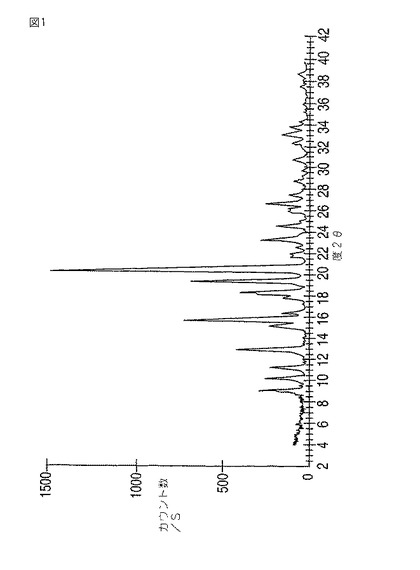
異なる結晶形の化合物を生成する可能性を持つ技術は多岐にわたっている。例として、結晶化、結晶温浸、昇華および熱処理が包含される。’290特許の実施例1における実験室的製造では、モダフィニル、水および過剰の過酸化水素を含有する反応混合物に水を添加することにより、まずモダフィニルを沈殿させる。次にモダフィニルをメタノールから再結晶する。実施例1aの工業的規模の製造では、最初にメタノールと水の1:4混合物から、そして次に1:9のメタノール/水混合物から再度結晶化することにより、モダフィニルを白色粉末として得る。メタノールおよび1:9メタノール/水混合物からの結晶化は、多形I型のモダフィニルを生成する。モダフィニルI型は、9.0、10.2、11.2、12.9、15.2、15.8、16.3、17.7、18.2、19.3、20.5、21.6、21.9、23.2、26.6±0.2度の2θにおける反射を伴う粉末X線回折(「PXRD」)パターン(図1)を特徴とする。
【0013】
米国特許第4927855号は、α-メチルベンジルアミンを用いた2-[(ジフェニルメチル)スルフィニル]酢酸のキラル分割によるモダフィニルの左旋性エナンチオマーの製造を記載している。エナンチオマー的に純粋な酸の回収およびアミド化の後、エタノールからの結晶化によって(-)モダフィニルが白色結晶として得られた。
【0014】
薬学上有用な化合物の新しい結晶形の発見は、薬用生成物の挙動特性を改善する機会を提供する。それは、例えば標的を定めた放出プロフィールまたはその他の望ましい特性を持つ薬物の薬用投与型を設計するために利用できる、製剤科学者の有する物質のレパートリーを拡大する。このレパートリーが、有用化合物の新しい結晶形の発見によって拡大するならば、それは明らかに有利である。過去に当分野で記載された結晶化法に従うことによっては入手できないモダフィニルの5種類の新たな結晶形が、ここに発見された。
【0015】
発明の要約
本発明は、1回の結晶化によって高純度の単離ができる、モダフィニルの製造方法を提供する。この方法は、鉱酸、アルコールまたは相転移触媒および所望により不活性液体有機媒質の混合物中で、2-[(ジフェニルメチル)チオ]アセトアミドをH2O2で酸化することを含む。反応混合物からモダフィニルを沈殿させ、次いで≧99.5%純度で結晶化させる。この酸化法は過剰酸化のスルホン生成物を本質上含まないモダフィニルを生成し、2回の結晶化後にUV検出限界内のスルホンを含まないモダフィニルの取得を可能にする。
【0016】
本発明はさらに、モダフィニルの新たな結晶形II-VIおよびそれらを製造するための方法を提供する。この新しい型の各々は特異な粉末X線回折パターンによって識別する。
本発明はさらに、新規なモダフィニルII-IVおよびVI型を含有する薬用組成物を提供する。
【0017】
好ましい態様の詳細な説明
この開示において、合計0.1%未満の不純物含有量を持つモダフィニルを「高純度の」モダフィニルと称する。純度はλ=225nmのUV吸光度により測定する。フェニル環を含む化合物はこの領域のUVスペクトルにおいて強い吸収を持つ。モダフィニルおよび問題となる不純物2-4の各々は2個のフェニルUV発色団を有する。スルホン2のような不純物を0.01%未満含有するモダフィニルを、その不純物を「本質上含まない」と言い、純度分析の検出限界内の不純物を含まない、またはその不純物を0.0001%未満含有するモダフィニルを、その不純物を「含まない」と言う。
【0018】
本発明は、1回の再結晶後に≧99.5%純度、好ましくは≧99.9%純度でモダフィニルが単離できる、モダフィニルの改良された合成製造を提供する。この改良法では2-[(ジフェニルメチル)チオ]アセトアミドがモダフィニルに酸化される。次いでこのモダフィニルを、酸化に使用した試薬から固体として分離し、その後1回の再結晶によって高純度で単離する。
【0019】
酸化工程では、鉱酸および直線状もしくは分枝もしくは環状アルコールまたは相転移触媒の存在下に、所望により不活性液体有機媒質中で、過酸化水素を2-[(ジフェニルメチル)チオ]アセトアミドと反応させる。酸化条件はDrabowicz,J et al. Synthesis, 1990, 37-38に概説されている。2-[(ジフェニルメチル)チオ]アセトアミドの製造の教示に関し、米国特許第4177290号を引用により本明細書の一部とする。
【0020】
過酸化水素は好ましくは10-50wt%水溶液、より好ましくは約30-33wt%水溶液として供給する。このような溶液は市販品が入手可能である(例えば、1998-99 Aldrich Chemical Co. Cat.Nos.42065-4; 42066-2; 31698-9; 21676-3)。
使用できる鉱酸の例はH2SO4、HClO4およびH3PO4を包含する。
好ましいアルコールは7またはそれ以下の炭素原子を持つ炭化水素から誘導され、ヒドロキシ基を除いては置換されていない。分枝アルコールが最も好ましい。イソプロピルアルコール、tert-ブタノールおよび2-メチル-1-ブタノールが、使用できるアルコールの例である。好適な相転移触媒はトリエチルベンジルアンモニウムクロリド(TEBA)およびポリエチレングリコールを包含する。
【0021】
不活性液体有機媒質は、酸化速度を低下させることができ、それでいて2-[(ジフェニルメチル)チオ]アセトアミドのスルフィド基からスルホキシド基への酸化を妨げたりスルフィド基からスルホンへの過剰酸化を惹起したりしない、酸化反応のための希釈剤である。好ましい不活性液体有機媒質は、メタノール、エタノールおよびエチレングリコールのような非分枝アルコール;水を含有していてよい、アセトンのようなケトン;酢酸エチルおよび炭酸ジメチルのようなエステル;ならびにこれらの混合物である。
【0022】
酸化工程では、2-[(ジフェニルメチル)チオ]アセトアミド(「スルフィド」)を過剰の、好ましくは約1.5ないし約4モル当量の過酸化水素と接触させる。鉱酸は触媒量、好ましくはスルフィドに関して約0.02ないし約0.2モル当量の使用が必要とされるに過ぎない。アルコールまたは相転移触媒は好ましくはスルフィドに関して約2ないし約4当量、より好ましくは約3当量の量を使用する。不活性液体有機媒質を使用する場合、酸化反応は好ましくは不活性液体有機媒質1ミリリットルあたりスルフィド約0.07ないし約0.2グラムのスルフィド濃度で実施する。
【0023】
必要な試薬は所望の任意の順序で添加でき、また反応混合物は2-[(ジフェニルメチル)チオ]アセトアミドからモダフィニルへの酸化を惹起する任意の条件に維持できる。以下の方法により、1回の結晶化によって、充分高純度のモダフィニルが、≧99.5%純度の、より好ましくは99.9%より高い純度のモダフィニルがその後得られる反応混合物から、直接沈殿化により実際に生成することが判明した。2-[(ジフェニルメチル)チオ]アセトアミドを不活性液体有機媒質に懸濁する。次いで鉱酸およびアルコールまたは相転移触媒を室温で加える。次に過酸化水素を加える。反応混合物の温度を約30℃に上げ、数時間攪拌する。反応の進行をHPLCで監視する。酸化が完了した後、反応混合物を室温に冷却し、過剰の過酸化水素を例えばメタ重亜硫酸ナトリウム、チオ硫化ナトリウム、硫化ナトリウムまたは硫酸鉄(II)で分解する。
【0024】
酸化が完了し過剰のH2O2が分解した後、モダフィニルを反応混合物から沈殿させる。水を添加することにより沈殿化を加速できる。次いで濾過またはデカンテーションといった常套的手段により反応混合物からモダフィニルを分離する。次いでこのモダフィニルを好ましくは有機溶媒および水で洗浄する。
モダフィニルを製造するこの改良法は、1回の再結晶で除去できる低含有量の2-[(ジフェニルメチル)スルホニル]アセトアミド2、2-[(ジフェニルメチル)スルフィニル]酢酸3、およびメチル2-[(ジフェニルメチル)スルフィニル]酢酸4を含むモダフィニルを生成する。反応混合物から沈殿するモダフィニルは98-99%純度またはそれ以上でなければならず、典型的には0.1%未満のスルホン2を含有する。モダフィニルはスルホン2を0.01%未満混入して反応混合物から直接沈殿化した。酸化反応混合物の組成をHPLCにより定量的に監視し、この反応が円滑に進行することを確認できる。λ=225nmでのUV検出を用いる逆相HPLC法が利用できる。
【0025】
上記方法に従う酸化によって得られるモダフィニルは様々な溶媒から高純度で再結晶できるが、最良の再結晶溶媒はメタノール、エタノール、炭酸ジメチル、アセトン、およびこれらの混合物であることが分かった。最良の多成分溶媒系は、エタノール/炭酸ジメチル、アセトン/炭酸ジメチル、アセトン/水、アセトン/酢酸エチル、アセトン/炭酸ジメチル/水およびメタノール/炭酸ジメチルである。特に好ましい再結晶溶媒は炭酸ジメチルである。
【0026】
結晶化後に得られるモダフィニルは≧99.5%純度、より好ましくは≧99.9%純度であって、0.02%未満、より好ましくは0.01%未満のスルホン2を含有する。好ましい再結晶溶媒からの結晶化の後、スルホン2を含まない、即ち0.0002%または0.0001%以下の混入であるモダフィニルが得られる。このような微量の不純物は多くの分析技術の検出限界にあるかまたはそれを超えるということが理解できるであろう。
【0027】
第二の態様では、本発明は新規な結晶モダフィニル型II-VIおよびそれらの製造方法を提供する。
或る化合物の新規な結晶形の発見を導く一般的技術は当業者に良く知られている。実際それはよくあることである。このような技術は、結晶化、結晶温浸、昇華、熱処理、およびpH調節を包含する。当業者は、化合物の新たな多形を探索する際に、これら技術のどれ1つをとっても化合物の新しい結晶形を生成できる訳ではないと予想されることが理解できるであろう。この探索は、異なる技術および条件を用いる試行錯誤の実験を含む経験的実験である。これらの理由のため、モダフィニル結晶形II-VIを生成するであろう全ての技術および条件を規定することは不可能である。しかしながら、これら望ましい型のうち1つのモダフィニルを成功裏に且つ選択的に生成した方法を提供することは可能である。
【0028】
モダフィニルの新規な結晶形を、特定の結晶形のフィンガープリントを生成する粉末X線回折分光法によって特性決定した。2θ値の測定は典型的には0.2度以内まで正確である。
X線回折データは、Philips粉末X線回折計Goniometer model 1050/70を使用して、毎分2゜のスキャン速度、λ=1.5418ÅのCuKα放射で取得した。試料を穏やかに粉砕し、零バックグラウンド石英プレート上に撒いて薄層とした。
【0029】
モダフィニルI型
本発明はモダフィニルI型の製造方法を提供する。
モダフィニルI型は、アセトン、アセトニトリル、ベンジルアルコール、ジメチルホルムアミド、メタノール、メチルエチルケトンまたは2-ピロリドンから結晶化することにより製造できる。好ましい再結晶溶媒はメタノールおよびアセトンである。結晶化は、溶液の冷却、反溶媒の添加、またはモダフィニルI型の結晶でその溶液に種入れすることによって加速できる。好ましい溶媒/反溶媒の組み合わせはアセトン/水、DMF/水、アセトニトリル/水、エタノール/水およびメタノール/酢酸エチルである。
【0030】
モダフィニルI型はまた、モダフィニルI型とII型の混合物を、変換が完了するに充分な時間酢酸エチル中に懸濁することによっても製造できる。出発モダフィニルがII型である場合、I型への変換を促進するため酢酸エチルに代えて他の幾つかの有機液体を使用できる。特に、II型モダフィニルはまた、これをメチルtert-ブチルエーテル(「MTBE」)、水または酢酸イソブチルに懸濁することによりI型モダフィニルに変換できる。I型を製造するため、モダフィニル(他の任意の型)を酢酸エチル、酢酸イソブチルまたは水と共に、変換が完了するまで単にスラリー化することによってこの技術を実行することがとりわけ簡便である。
【0031】
V型およびVI型は、穏やかに約80℃またはそれ以上に加熱するとモダフィニルI型へと変換する。V型およびVI型は、約100℃に加熱することにより、著明な分解を伴わずにI型に変換できる。
モダフィニルI型は濾過またはデカンテーションおよびその後の乾燥により、溶媒から簡便に分離できる。I型は、他の結晶形またはアモルファス型に変換することなく、そして著明な化学分解を受けることなく、100℃という高温で乾燥した。
【0032】
モダフィニルII型
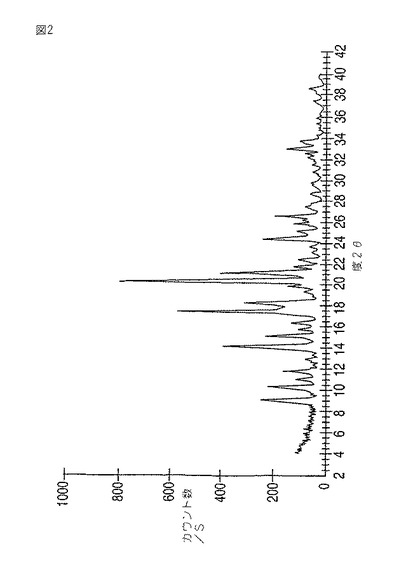
本発明はさらにモダフィニルII型を提供する。モダフィニルII型は、9.1、10.3、11.1、11.9、14.3、15.2、16.4、17.5、18.4、20.5、21.3、24.6、26.6±0.2度の2θにおける反射を伴う粉末X線回折パターン(図2)をもたらす。14.3、17.5、20.5および21.3度の2θにおける強い反射がとりわけ特徴的である。これらの中でも14.3、17.5および21.3度の2θにおける反射が最も特徴的である。
【0033】
以下の技術は結晶形II型のモダフィニルの生成に有効であることが判明した。
モダフィニルIII型はこれを水に懸濁する時モダフィニルII型に変換する。したがって、III型の水への懸濁がモダフィニルII型の取得方法を提供する。
モダフィニルはまた、加熱によって溶媒にモダフィニルを溶解し、冷却して再結晶させることにより、エタノール、イソプロパノール、n-ブタノール、t-ブタノール、メチルイソブチルケトン、エチレングリコール、ジオキソランおよびジオキサンから選択的にII型で結晶化する。モダフィニルII型はさらに、ジクロロエタン中で再スラリー化することによって、そしてメタノールと水の混合物に入れたモダフィニルの溶液を速やかに冷却することによって製造できる。
【0034】
モダフィニルIII型
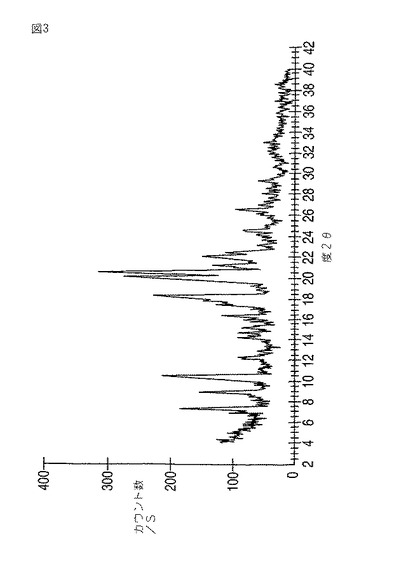
本発明はさらにモダフィニルIII型を提供する。モダフィニルIII型は7.4、9.0、10.5、12.3、14.2、14.7、15.1、16.4、18.3、20.0、20.5、21.1、22.1、24.5±0.2度の2θにおける反射を伴う粉末X線回折パターン(図3)をもたらす。7.4、10.5、18.3、20.0および20.5度の2θにおける強い反射がとりわけ特徴的である。これらの中でも7.4、10.5、18.3および20.0度の2θにおける反射が、それらの強度と、他の型のPXRDパターンでは対応位置に反射が無いことの故に特徴的である。
【0035】
モダフィニルIII型はトルエンからの結晶化によって生成する。III型はまた、炭酸ジメチルおよびエタノールの混合物からも結晶化するが、但しこの混合溶媒系から結晶化した場合、V型との混合物として得られる例もあった。
モダフィニルIV型
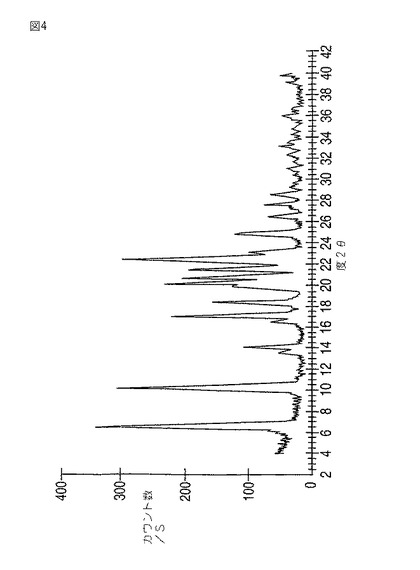
本発明はさらにモダフィニルIV型を提供する。モダフィニルIV型は6.9、10.4、14.1、17.2、18.5、20.3、20.8、21.6、22.7、25.0、26.5、27.6、28.5±0.2度の2θにおける反射を伴う粉末X線回折パターン(図4)をもたらす。6.9、10.4、17.2、20.3および22.7度の2θにおける強い反射がとりわけ特徴的である。
【0036】
モダフィニルはテトラヒドロフランおよびジメチルスルホキシドからIV型結晶形で結晶化する。
【0037】
モダフィニルV型
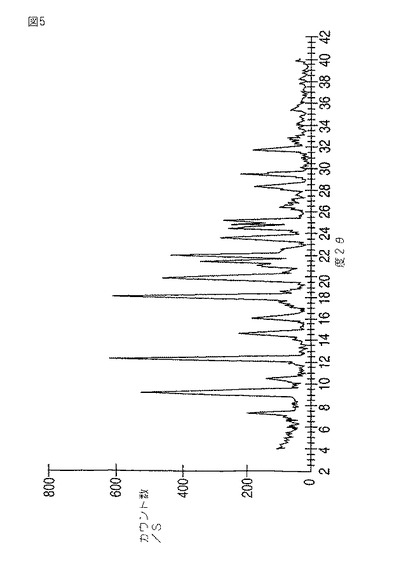
本発明はさらにモダフィニルV型を提供する。V型は7.4、9.3、10.5、12.4、14.7、16.2、18.2、19.9、21.5、22.0、23.6、24.5、25.2、28.4、29.5、31.8±0.2度の2θにおける反射を伴う粉末X線回折パターン(図5)をもたらす。9.3、12.4、18.2、19.9および22.0度の2θにおける強い反射がとりわけ特徴的である。
【0038】
V型は、炭酸ジメチル、ならびに炭酸ジメチルとエタノール、炭酸ジメチルと水および炭酸ジメチルとアセトンの混合物からの結晶化によって製造する。
V型の熱重量分析は、約100℃から出発し150℃までで約12%の質量損失を示した。このLODはモダフィニルと炭酸ジメチルとの半溶媒和物であるV型に一致する。このTGA分析はShimadzu DTG60で、毎分約10℃の速度でほぼ周囲温度から約300℃まで加熱した約10mgの試料について実施した。
【0039】
モダフィニルVI型
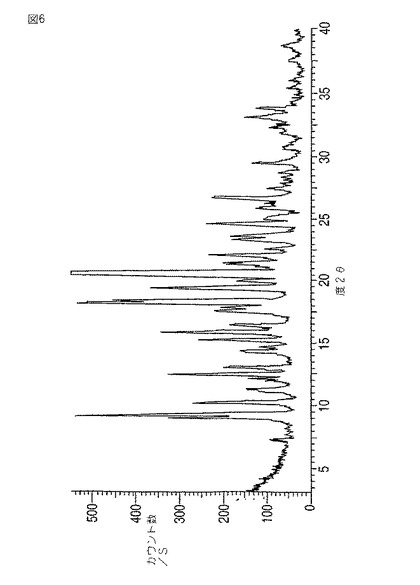
本発明はさらにモダフィニルVI型を提供する。VI型は9.0、9.3、10.2、12.4、14.2、14.5、15.3、17.5、18.1、20.0、20.5、21.5、22.0、23.5、24.5、25.0±0.2度の2θにおける反射を伴う粉末X線回折パターン(図6)をもたらす。9.3、18.1および20.5度の2θにおける反射がそれらの強度の故にとりわけ特徴的である。
【0040】
モダフィニルVI型は、水、エタノールまたは水/エタノール混合物中にモダフィニルV型を変換が完了するに充分な時間懸濁することによって製造できる。好ましくは、モダフィニルVIを水、エタノール、またはエタノール/水混合物中、約28℃でスラリー化し、その後55℃で減圧乾燥する。
アモルファスモダフィニル
モダフィニルはオルト、メタまたはパラキシレンの混合物から結晶化によりアモルファス状態で製造できる。
【0041】
実験室および工業的設定において明瞭なII-VI結晶形のモダフィニルの製造に最も適した技術を記載してきたが、当業者は、これらの結晶形はさらに別の方法で取得できることが理解できるであろう。
【0042】
モダフィニルII-IVおよびVI型を含有する薬用組成物
モダフィニルII-IVおよびVIは、ナルコレプシーに罹患した患者の覚醒を促進するのに有用な種々の薬用組成物および投与型に調合できる。
【0043】
本発明に係る薬用組成物は、所望により互いに混合したモダフィニルII-IVおよびVI型を含有する。本発明に係る薬用組成物はさらに、1またはそれ以上のモダフィニルII-IVおよびVI型と混合した、他のモダフィニル結晶形、アモルファスモダフィニルおよび/または他の活性成分を含有することができる。活性成分に加えて、本発明に係るモダフィニル薬用組成物は1またはそれ以上の賦形剤を含有してよい。賦形剤は様々な目的のために該組成物に添加する。
【0044】
希釈剤は固体の薬用組成物のかさを増やし、該組成物を含有する薬用投与型を、患者および介護者にとって扱い易くする。固体組成物のための希釈剤は、例えば微結晶セルロース(例えばAvicel(登録商標))、ミクロファインセルロース、乳糖、澱粉、α化澱粉、炭酸カルシウム、硫酸カルシウム、糖、デキストラート、デキストリン、デキストロース、二塩基性燐酸カルシウム二水和物、三塩基性燐酸カルシウム、カオリン、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マンニトール、ポリメタクリラート(例えばEudragit(登録商標))、塩化カリウム、粉末セルロース、塩化ナトリウム、ソルビトールおよびタルクを包含する。
【0045】
圧縮して錠剤のような投与型とされる固体薬用組成物は賦形剤を含むことができるが、その機能は活性成分およびその他の賦形剤の、圧縮後の結合を助けることを含む。固体薬用組成物用の結合剤は、アラビアゴム、アルギン酸、carbomer(例えばcarbopol)、カルボキシメチルセルロースナトリウム、デキストリン、エチルセルロース、ゼラチン、グアールガム、水素化植物油、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えばKlucel(登録商標))、ヒドロキシプロピルメチルセルロース(例えばMethocel(登録商標))、液体グルコース、珪酸アルミニウムマグネシウム、マルトデキストリン、メチルセルロース、ポリメタクリラート、ポビドン(例えばKollidon(登録商標)、Plasdone(登録商標))、α化澱粉、アルギン酸ナトリウムおよび澱粉を包含する。
【0046】
患者の胃内での圧縮された固体薬用組成物の溶解速度は、組成物に崩壊剤を添加することにより増大させることができる。崩壊剤は、アルギン酸、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム(例えばAc-Di-Sol(登録商標)、Primellose(登録商標))、コロイド状二酸化珪素、クロスカルメロースナトリウム、クロスポビドン(例えばKollidon(登録商標)、Polyplasdone(登録商標))、グアールガム、珪酸アルミニウムマグネシウム、メチルセルロース、微結晶セルロース、ポラクリリンカリウム、粉末セルロース、α化澱粉、アルギン酸ナトリウム、グリコール酸澱粉ナトリウム(例えばExplotab(登録商標))および澱粉を包含する。
【0047】
非圧縮固体組成物の流動性を改善し投薬の精度を改善するため、潤滑剤を添加できる。潤滑剤として機能し得る賦形剤は、コロイド状二酸化珪素、三珪酸マグネシウム、粉末セルロース、澱粉、タルクおよび三塩基性燐酸カルシウムを包含する。
粉末化した組成物の圧縮によって錠剤のような投与型を製造する場合、この組成物にパンチおよびダイからの圧力をかける。幾つかの賦形剤と活性成分はパンチおよびダイの表面に付着する傾向があり、それが製品に陥凹形成およびその他の表面凹凸を惹起し得る。潤滑剤を組成物に添加して付着を減少させ、ダイからの製品の解放を容易にすることができる。潤滑剤は、ステアリン酸マグネシウム、ステアリン酸カルシウム、モノステアリン酸グリセリル、パルミトステアリン酸グリセリル、水素化ひまし油、水素化植物油、鉱油、ポリエチレングリコール、安息香酸ナトリウム、ラウリル硫酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸、タルクおよびステアリン酸亜鉛を包含する。
【0048】
香料および矯味剤は、患者にとってその剤型をより味の良いものにする。本発明に係る組成物に含有させ得る、薬用生成物のための一般的な香料および矯味剤は、マルトール、バニリン、エチルバニリン、メントール、クエン酸、フマル酸エチルマルトール、および酒石酸を包含する。
組成物はさらに、それらの外観を改善しそして/または製品および単位投薬レベルを患者が同定し易くするために、任意の薬学上許容し得る着色剤を用いて着色できる。
【0049】
賦形剤の選択および使用量は、経験と標準法の考察および当分野における参考となる業績に基づき、製剤科学者によって容易に決定できる。
本発明に係る固体組成物は、粉末、顆粒、凝集物および圧縮組成物を包含する。投与型は、経口、バッカル、直腸、非経口(皮下、筋肉内および静脈内を包含する)、吸入および眼への投与に好適な投薬型を包含する。与えられた任意の症例において最も好適な経路は治療しようとする状態の性格および重篤度に依存するが、本発明に係る最も好ましい経路は経口である。投与型は単位投与型で簡便に提供でき、そして薬学分野で周知の任意の方法によって製造できる。
【0050】
投与型は、錠剤、散剤、カプセル剤、坐剤、サシェー剤、トローチ剤およびlosengeのような固体投与型、ならびに液体シロップ剤、懸濁剤およびエリキシル剤を包含する。本発明に係る特に好ましい剤型は錠剤である。
錠剤、カプセル剤、口内錠およびその他の単位投与型は、好ましくは約50ないし約300mg、より好ましくは約100mgないし約200mgの投与レベルでモダフィニルを含有する。
【0051】
或る好ましい態様を参考にして本発明を記載してきたが、本発明を限定するためではなく例示の目的のために以下の実施例を提供する。
【0052】
実施例
実施例1-8
(高純度モダフィニルの製造)
実施例1:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)をメタノール(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。H2O2の30%溶液(45ml)を加え、温度を30℃に上昇させた。温度を3.5時間30℃に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル40.2gが得られた(収率:75.7%)。
【0053】
実施例2:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)を炭酸ジメチル(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。H2O2の15%溶液(85ml)を加え、温度を30℃に上昇させた。温度を30時間30℃に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル45.1gが得られた(収率:85%)。
【0054】
実施例3:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例1で製造したモダフィニル3gを、5%の水を含有するアセトン32mlに懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を42℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに25℃まで冷却し、濾過した。乾燥後、本質上スルホンを含まない高度に精製されたモダフィニル1.95gが得られた(収率:65%)。
【0055】
実施例4:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例2で製造したモダフィニル1gを、エタノール10.5mlに懸濁した。混合物を窒素雰囲気下で加熱還流した。この懸濁液を25℃まで冷却し、濾過した。乾燥後、高度に精製されたモダフィニル0.83gが得られた(収率:83%)。
【0056】
実施例5:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)を炭酸ジメチル(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。さらに30%H2O249mlを加えた。温度を30℃に上げ、8時間一定に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル45.1gが得られた(収率:85%)。
【0057】
実施例6:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例5で製造したモダフィニル3gを、アセトン100mlおよび炭酸ジメチル20mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を47℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに25℃まで冷却し、濾過した。乾燥後、本質上スルホンを含まない高度に精製されたモダフィニル2.52gが得られた(収率:84%)。
【0058】
実施例7:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例6で得られた未乾燥モダフィニル3.7gを、アセトン123.5mlおよび炭酸ジメチル24.7mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を25℃まで冷却し、濾過した。濾過ケーキを乾燥し再度アセトン94.5mlおよび炭酸ジメチル19mlの混合物に懸濁し、窒素雰囲気下で加熱還流した。得られた溶液を25℃に冷却し濾過した。乾燥後、スルホンを含まない高度に精製されたモダフィニル2.32gが得られた(収率:62.7%)。
【0059】
実施例8:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例5で製造したモダフィニル3gを、アセトン1mlおよび炭酸ジメチル20mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を25℃まで冷却し、濾過した。この湿潤濾過ケーキを再度アセトン100mlおよび炭酸ジメチル.20mlの混合物に懸濁し、窒素雰囲気下で加熱還流した。得られた溶液を25℃に冷却し濾過した。乾燥後、スルホンを含まない高度に精製されたモダフィニル2.1gが得られた(収率:70.5%)。
【0060】
実施例9-13
(モダフィニルI型の製造)
実施例9:モダフィニルIV型を水に懸濁する事による。モダフィニルIV型(0.4g)をpH約5.9の蒸留水(50ml)に懸濁した。この懸濁液を約37℃で約24時間攪拌し、次いで濾過した。濾液をX線粉末回折によって分析し、モダフィニルI型であると決定した。
【0061】
実施例10:モダフィニルVまたはVI型を加熱する事による。モダフィニルVおよびVI型の小アリコートを約100℃の乾燥機で別々に約30分間加熱した。その後モダフィニルVおよびVI型をX線粉末回折により分析し、いずれもI型であると決定した。
【0062】
実施例11:アセトニトリルからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をアセトニトリル(23ml)に懸濁した。混合物を加熱還流した(約80℃)。得られた溶液を約63℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに約25℃に冷却し、濾過した。乾燥後、結晶化したモダフィニル(1.96g)I型が得られた(収率65%)。
【0063】
実施例12:ジメチルホルムアミドからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をジメチルホルムアミド(5.5ml)に懸濁した。混合物を加熱還流した(約60℃)。透明な溶液が得られた。この溶液に水(5ml)を滴下するとモダフィニルが沈殿し始めた。この混合物を約25℃に冷却することにより沈殿化が完了した。生成物を濾過により分離した。乾燥後、結晶化したモダフィニル(2.54g)I型が得られた(収率84.7%)。
【0064】
実施例13:酢酸エチルからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)を酢酸エチル(50ml)に懸濁した。混合物を加熱還流し(約77℃)、約1時間維持した。混合物を約25℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニルI型(1.9g)が得られた(収率63%)。
【0065】
実施例14-15
(モダフィニルII型の製造)
実施例14:イソプロパノールからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をイソプロパノール(34ml)に懸濁した。混合物を加熱還流した(約85℃)。得られた溶液を約58℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニルII型(2.32g)が得られた(収率77.3%)。
【0066】
実施例15:モダフィニルIII型の水中懸濁液より。モダフィニルIII型(0.4g)をpH約5.9の蒸留水(50ml)に懸濁した。この懸濁液を約37℃で約24時間攪拌し、次いで濾過した。濾液を粉末X線回折によって分析し、モダフィニルII型であると決定した。
【0067】
実施例16
(モダフィニルIII型の製造)
実施例16:トルエンからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をトルエン(90ml)に懸濁した。混合物を加熱還流した(約110℃)。得られた溶液を約35℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約17時間約25℃に維持し、約5℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニル(0.6g)III型が得られた(収率19.6%)。
【0068】
実施例17
(モダフィニルIV型の製造)
実施例17:テトラヒドロフランからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をテトラヒドロフラン(90ml)に懸濁した。混合物を加熱還流した(約63℃)。得られた溶液を約53℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、結晶化した(2.4g)モダフィニルIV型が得られた(収率80%)。
【0069】
実施例18
(モダフィニルV型の製造)
実施例18:炭酸ジメチルからの結晶化。モダフィニル(3g)を炭酸ジメチル(105ml)に懸濁した。この混合物を、還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で加熱還流した(約90℃)。約2時間の還流の後、得られた溶液を約79℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、ほぼ結晶化したモダフィニル(3g)V型が得られた(収率約90%)。
【0070】
実施例19
(モダフィニルVI型の製造)
実施例19:V型のエタノール懸濁液より。下降冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3.5g)V型をエタノール(10ml)に懸濁した。混合物を約4.5時間約25℃で攪拌し、次いで濾過した。乾燥後、結晶化したモダフィニル(2.9g)VI型が得られた(収率82%)。
【0071】
実施例20
(アモルファスモダフィニルの製造)
実施例20:キシレンからの結晶化。下降冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(5g)をキシレン(150ml)に懸濁した。混合物を約110℃に加熱し、これを約30分間維持した。得られた溶液を約35℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約17時間約25℃に維持し、次いで約5℃に冷却し、濾過した。乾燥後、アモルファスモダフィニル(1.83g)が得られた(収率36.6%)。
【0072】
このように本発明を或る好ましい態様を参考に記載してきたが、本明細書および実施例の考察から、当業者にはその他の態様もまた自明となろう。実施例を含む本明細書は単に例示であり、本発明の範囲および精神は、以下の請求項により定義するということを意図している。
【図面の簡単な説明】
【0073】
【図1】図1は、モダフィニルI型の粉末X線回折パターンを示す。
【図2】図2は、モダフィニルII型の粉末X線回折パターンを示す。
【図3】図3は、モダフィニルIII型の粉末X線回折パターンを示す。
【図4】図4は、モダフィニルIV型の粉末X線回折パターンを示す。
【図5】図5は、モダフィニルV型の粉末X線回折パターンを示す。
【図6】図6は、モダフィニルVI型の粉末X線回折パターンを示す。
【発明の開示】
【0001】
発明の分野
本発明は、不純物を含まないモダフィニルの製造方法、ならびにモダフィニルの新しい結晶形およびそれらを製造するための方法に関するものである。
【0002】
発明の背景
モダフィニルとしても知られる、式1で示される(±)2-[(ジフェニルメチル)スルフィニル]アセトアミドは、人間および動物に覚醒促進効果を表す。
【化1】
【0003】
モダフィニルの向精神活性は、米国特許第4177290号(「’290特許」)に記載のように動物試験で、そして人間の患者での臨床試験で証明された。モダフィニルのラセミ化合物はナルコレプシーの治療用にF.D.A.で承認されている。
【0004】
‘290特許はモダフィニルの製造を記載している。’290特許の実施例1では、2-[(ジフェニルメチル)チオ]酢酸クロリドをアンモニアと反応させ、アミド生成物を単離し、次いでそのスルフィド基を酢酸中の過酸化水素で酸化することによってモダフィニルを製造する。’290特許の実施例1aは、工業的規模のモダフィニル製造のため、異なる合成法を記載している。ベンズヒドロールをチオ尿素と反応させて中間体を生成し、次いでこれを加水分解して2-[(ジフェニルメチル)チオ]酢酸とする。次にこの酸を、クロロ酢酸および水を含有する混合物中、過酸化水素でin situ酸化する。得られたスルホキシドを次いで硫酸ジメチルで処理してカルボン酸基をメチル化する。得られたエステルをアンモニアで誘導体化してモダフィニルを得る。
【0005】
これらの方法の各々は、スルフィド基をスルホキシドに酸化するために過酸化水素を使用している。Drabowicz,J et al. Synthesis, 1990, 37-38は、立体障害されたスルフィドをスルホキシドに酸化するための方法を記載している。その方法は、酸化剤として過酸化水素を、溶媒としてメタノールを、そして触媒として、硫酸および幾つかの分枝脂肪族アルコールのうちの1つの混合物を使用する。この方法は立体障害されたスルフィドの酸化に良く適合している。反応混合物の薄層クロマトグラフィーでは過剰酸化生成物は観察されなかった。モダフィニルを製造するためのこの方法の使用は文献に記載されていない。
【0006】
スルフィドは、過ヨウ素酸ナトリウム、次亜塩素酸t-ブチル、次亜塩素酸カルシウム、亜塩素酸ナトリウム、次亜塩素酸ナトリウム、メタ−クロロ過安息香酸および過硼酸ナトリウムといった他の酸化剤でスルホキシドに酸化することもできる。March J. Advanced Organic Chemistry 1201-02(4th ed. 1992)。
本発明者等は、’290特許の実施例1の方法が、スルフィドからスルホン2への過剰酸化の問題を有することを発見した。
【化2】
【0007】
上に記載した化学構造を比較することにより、いったん生成したスルホンをモダフィニルから分離することは困難な仕事であることが容易に理解できる。故に、1回またはそれ以上の再結晶後にスルホンを含まないモダフィニルを得るため、選択的酸化法を開発することが求められる。
【0008】
実施例1aに記載の方法では、工程(b)および(c)における出発物質の不完全な変換のため、かなりの量の中間体2-[(ジフェニルメチル)スルフィニル]酢酸3およびメチル2-[(ジフェニルメチル)スルフィニル]酢酸4が得られる。Becue,T; Broquaire,M. J.Chromatography 1991, 557, 489-494。これらの化合物もまたモダフィニルから分離することが困難である。
【化3】
【0009】
工業的規模の方法で使用する溶媒の容量のため、そして大量の有機溶媒の廃棄により生ずる環境問題のため、本質上不純物を含まないモダフィニルを産生し且つ検出限界内の不純物を含まないモダフィニルを取得するために最終生成物を1回しか結晶化しなくてもよい工業生産は、同等の純度でモダフィニルを得るために再結晶を何回も必要とする別の方法よりもはるかに有利である。’290特許の実施例1aは工業的方法として記載されているが、生成物を白色結晶粉末として得るために2回の再結晶を使用した。その粉末の組成は報告されていない。
【0010】
スルホン2を本質上含まないモダフィニルを産生し、その結果、1回の結晶化によってこれが高純度で取得できる改良法を得ることが極めて望ましい。加えて、毒性が高いことから、実施例1aにおける試薬の1つである硫酸ジメチルの使用を回避することもまた極めて望ましい。
【0011】
高純度のモダフィニルを効率的に生産するという目的を追求する間に、本発明者等は、モダフィニルが幾つかの別個の固体状態の結晶性多形に結晶化できることを発見した。化合物の結晶形は、固体化した化合物の単位格子中の原子核の位置によって識別する。この相違は、薬学において実用上の重要性を持つ、熱による挙動、蒸気透過性および溶解度といった異なる巨視的性質を産む。化合物の結晶形はX線解析によって最も容易に識別できる。単結晶X線結晶学は、核の位置決定に利用できるデータをもたらし、次いでこれをコンピューターまたは機械的モデルによって視覚化し、このようにして当該化合物の三次元画像を得ることができる。単結晶X線解析は無類の構造情報を提供するものの、これらは高価で、特性データは時に獲得が困難である。粉末X線回折分光法は、薬物の新しい結晶形を特性決定するために、単結晶X線解析よりも頻繁に製薬工業で使用されている。粉末X線回折分光法はその結晶形に特異なフィンガープリントを生成し、それをアモルファス化合物および該化合物の他の全ての結晶形から識別することができる。
【0012】
異なる結晶形の化合物を生成する可能性を持つ技術は多岐にわたっている。例として、結晶化、結晶温浸、昇華および熱処理が包含される。’290特許の実施例1における実験室的製造では、モダフィニル、水および過剰の過酸化水素を含有する反応混合物に水を添加することにより、まずモダフィニルを沈殿させる。次にモダフィニルをメタノールから再結晶する。実施例1aの工業的規模の製造では、最初にメタノールと水の1:4混合物から、そして次に1:9のメタノール/水混合物から再度結晶化することにより、モダフィニルを白色粉末として得る。メタノールおよび1:9メタノール/水混合物からの結晶化は、多形I型のモダフィニルを生成する。モダフィニルI型は、9.0、10.2、11.2、12.9、15.2、15.8、16.3、17.7、18.2、19.3、20.5、21.6、21.9、23.2、26.6±0.2度の2θにおける反射を伴う粉末X線回折(「PXRD」)パターン(図1)を特徴とする。
【0013】
米国特許第4927855号は、α-メチルベンジルアミンを用いた2-[(ジフェニルメチル)スルフィニル]酢酸のキラル分割によるモダフィニルの左旋性エナンチオマーの製造を記載している。エナンチオマー的に純粋な酸の回収およびアミド化の後、エタノールからの結晶化によって(-)モダフィニルが白色結晶として得られた。
【0014】
薬学上有用な化合物の新しい結晶形の発見は、薬用生成物の挙動特性を改善する機会を提供する。それは、例えば標的を定めた放出プロフィールまたはその他の望ましい特性を持つ薬物の薬用投与型を設計するために利用できる、製剤科学者の有する物質のレパートリーを拡大する。このレパートリーが、有用化合物の新しい結晶形の発見によって拡大するならば、それは明らかに有利である。過去に当分野で記載された結晶化法に従うことによっては入手できないモダフィニルの5種類の新たな結晶形が、ここに発見された。
【0015】
発明の要約
本発明は、1回の結晶化によって高純度の単離ができる、モダフィニルの製造方法を提供する。この方法は、鉱酸、アルコールまたは相転移触媒および所望により不活性液体有機媒質の混合物中で、2-[(ジフェニルメチル)チオ]アセトアミドをH2O2で酸化することを含む。反応混合物からモダフィニルを沈殿させ、次いで≧99.5%純度で結晶化させる。この酸化法は過剰酸化のスルホン生成物を本質上含まないモダフィニルを生成し、2回の結晶化後にUV検出限界内のスルホンを含まないモダフィニルの取得を可能にする。
【0016】
本発明はさらに、モダフィニルの新たな結晶形II-VIおよびそれらを製造するための方法を提供する。この新しい型の各々は特異な粉末X線回折パターンによって識別する。
本発明はさらに、新規なモダフィニルII-IVおよびVI型を含有する薬用組成物を提供する。
【0017】
好ましい態様の詳細な説明
この開示において、合計0.1%未満の不純物含有量を持つモダフィニルを「高純度の」モダフィニルと称する。純度はλ=225nmのUV吸光度により測定する。フェニル環を含む化合物はこの領域のUVスペクトルにおいて強い吸収を持つ。モダフィニルおよび問題となる不純物2-4の各々は2個のフェニルUV発色団を有する。スルホン2のような不純物を0.01%未満含有するモダフィニルを、その不純物を「本質上含まない」と言い、純度分析の検出限界内の不純物を含まない、またはその不純物を0.0001%未満含有するモダフィニルを、その不純物を「含まない」と言う。
【0018】
本発明は、1回の再結晶後に≧99.5%純度、好ましくは≧99.9%純度でモダフィニルが単離できる、モダフィニルの改良された合成製造を提供する。この改良法では2-[(ジフェニルメチル)チオ]アセトアミドがモダフィニルに酸化される。次いでこのモダフィニルを、酸化に使用した試薬から固体として分離し、その後1回の再結晶によって高純度で単離する。
【0019】
酸化工程では、鉱酸および直線状もしくは分枝もしくは環状アルコールまたは相転移触媒の存在下に、所望により不活性液体有機媒質中で、過酸化水素を2-[(ジフェニルメチル)チオ]アセトアミドと反応させる。酸化条件はDrabowicz,J et al. Synthesis, 1990, 37-38に概説されている。2-[(ジフェニルメチル)チオ]アセトアミドの製造の教示に関し、米国特許第4177290号を引用により本明細書の一部とする。
【0020】
過酸化水素は好ましくは10-50wt%水溶液、より好ましくは約30-33wt%水溶液として供給する。このような溶液は市販品が入手可能である(例えば、1998-99 Aldrich Chemical Co. Cat.Nos.42065-4; 42066-2; 31698-9; 21676-3)。
使用できる鉱酸の例はH2SO4、HClO4およびH3PO4を包含する。
好ましいアルコールは7またはそれ以下の炭素原子を持つ炭化水素から誘導され、ヒドロキシ基を除いては置換されていない。分枝アルコールが最も好ましい。イソプロピルアルコール、tert-ブタノールおよび2-メチル-1-ブタノールが、使用できるアルコールの例である。好適な相転移触媒はトリエチルベンジルアンモニウムクロリド(TEBA)およびポリエチレングリコールを包含する。
【0021】
不活性液体有機媒質は、酸化速度を低下させることができ、それでいて2-[(ジフェニルメチル)チオ]アセトアミドのスルフィド基からスルホキシド基への酸化を妨げたりスルフィド基からスルホンへの過剰酸化を惹起したりしない、酸化反応のための希釈剤である。好ましい不活性液体有機媒質は、メタノール、エタノールおよびエチレングリコールのような非分枝アルコール;水を含有していてよい、アセトンのようなケトン;酢酸エチルおよび炭酸ジメチルのようなエステル;ならびにこれらの混合物である。
【0022】
酸化工程では、2-[(ジフェニルメチル)チオ]アセトアミド(「スルフィド」)を過剰の、好ましくは約1.5ないし約4モル当量の過酸化水素と接触させる。鉱酸は触媒量、好ましくはスルフィドに関して約0.02ないし約0.2モル当量の使用が必要とされるに過ぎない。アルコールまたは相転移触媒は好ましくはスルフィドに関して約2ないし約4当量、より好ましくは約3当量の量を使用する。不活性液体有機媒質を使用する場合、酸化反応は好ましくは不活性液体有機媒質1ミリリットルあたりスルフィド約0.07ないし約0.2グラムのスルフィド濃度で実施する。
【0023】
必要な試薬は所望の任意の順序で添加でき、また反応混合物は2-[(ジフェニルメチル)チオ]アセトアミドからモダフィニルへの酸化を惹起する任意の条件に維持できる。以下の方法により、1回の結晶化によって、充分高純度のモダフィニルが、≧99.5%純度の、より好ましくは99.9%より高い純度のモダフィニルがその後得られる反応混合物から、直接沈殿化により実際に生成することが判明した。2-[(ジフェニルメチル)チオ]アセトアミドを不活性液体有機媒質に懸濁する。次いで鉱酸およびアルコールまたは相転移触媒を室温で加える。次に過酸化水素を加える。反応混合物の温度を約30℃に上げ、数時間攪拌する。反応の進行をHPLCで監視する。酸化が完了した後、反応混合物を室温に冷却し、過剰の過酸化水素を例えばメタ重亜硫酸ナトリウム、チオ硫化ナトリウム、硫化ナトリウムまたは硫酸鉄(II)で分解する。
【0024】
酸化が完了し過剰のH2O2が分解した後、モダフィニルを反応混合物から沈殿させる。水を添加することにより沈殿化を加速できる。次いで濾過またはデカンテーションといった常套的手段により反応混合物からモダフィニルを分離する。次いでこのモダフィニルを好ましくは有機溶媒および水で洗浄する。
モダフィニルを製造するこの改良法は、1回の再結晶で除去できる低含有量の2-[(ジフェニルメチル)スルホニル]アセトアミド2、2-[(ジフェニルメチル)スルフィニル]酢酸3、およびメチル2-[(ジフェニルメチル)スルフィニル]酢酸4を含むモダフィニルを生成する。反応混合物から沈殿するモダフィニルは98-99%純度またはそれ以上でなければならず、典型的には0.1%未満のスルホン2を含有する。モダフィニルはスルホン2を0.01%未満混入して反応混合物から直接沈殿化した。酸化反応混合物の組成をHPLCにより定量的に監視し、この反応が円滑に進行することを確認できる。λ=225nmでのUV検出を用いる逆相HPLC法が利用できる。
【0025】
上記方法に従う酸化によって得られるモダフィニルは様々な溶媒から高純度で再結晶できるが、最良の再結晶溶媒はメタノール、エタノール、炭酸ジメチル、アセトン、およびこれらの混合物であることが分かった。最良の多成分溶媒系は、エタノール/炭酸ジメチル、アセトン/炭酸ジメチル、アセトン/水、アセトン/酢酸エチル、アセトン/炭酸ジメチル/水およびメタノール/炭酸ジメチルである。特に好ましい再結晶溶媒は炭酸ジメチルである。
【0026】
結晶化後に得られるモダフィニルは≧99.5%純度、より好ましくは≧99.9%純度であって、0.02%未満、より好ましくは0.01%未満のスルホン2を含有する。好ましい再結晶溶媒からの結晶化の後、スルホン2を含まない、即ち0.0002%または0.0001%以下の混入であるモダフィニルが得られる。このような微量の不純物は多くの分析技術の検出限界にあるかまたはそれを超えるということが理解できるであろう。
【0027】
第二の態様では、本発明は新規な結晶モダフィニル型II-VIおよびそれらの製造方法を提供する。
或る化合物の新規な結晶形の発見を導く一般的技術は当業者に良く知られている。実際それはよくあることである。このような技術は、結晶化、結晶温浸、昇華、熱処理、およびpH調節を包含する。当業者は、化合物の新たな多形を探索する際に、これら技術のどれ1つをとっても化合物の新しい結晶形を生成できる訳ではないと予想されることが理解できるであろう。この探索は、異なる技術および条件を用いる試行錯誤の実験を含む経験的実験である。これらの理由のため、モダフィニル結晶形II-VIを生成するであろう全ての技術および条件を規定することは不可能である。しかしながら、これら望ましい型のうち1つのモダフィニルを成功裏に且つ選択的に生成した方法を提供することは可能である。
【0028】
モダフィニルの新規な結晶形を、特定の結晶形のフィンガープリントを生成する粉末X線回折分光法によって特性決定した。2θ値の測定は典型的には0.2度以内まで正確である。
X線回折データは、Philips粉末X線回折計Goniometer model 1050/70を使用して、毎分2゜のスキャン速度、λ=1.5418ÅのCuKα放射で取得した。試料を穏やかに粉砕し、零バックグラウンド石英プレート上に撒いて薄層とした。
【0029】
モダフィニルI型
本発明はモダフィニルI型の製造方法を提供する。
モダフィニルI型は、アセトン、アセトニトリル、ベンジルアルコール、ジメチルホルムアミド、メタノール、メチルエチルケトンまたは2-ピロリドンから結晶化することにより製造できる。好ましい再結晶溶媒はメタノールおよびアセトンである。結晶化は、溶液の冷却、反溶媒の添加、またはモダフィニルI型の結晶でその溶液に種入れすることによって加速できる。好ましい溶媒/反溶媒の組み合わせはアセトン/水、DMF/水、アセトニトリル/水、エタノール/水およびメタノール/酢酸エチルである。
【0030】
モダフィニルI型はまた、モダフィニルI型とII型の混合物を、変換が完了するに充分な時間酢酸エチル中に懸濁することによっても製造できる。出発モダフィニルがII型である場合、I型への変換を促進するため酢酸エチルに代えて他の幾つかの有機液体を使用できる。特に、II型モダフィニルはまた、これをメチルtert-ブチルエーテル(「MTBE」)、水または酢酸イソブチルに懸濁することによりI型モダフィニルに変換できる。I型を製造するため、モダフィニル(他の任意の型)を酢酸エチル、酢酸イソブチルまたは水と共に、変換が完了するまで単にスラリー化することによってこの技術を実行することがとりわけ簡便である。
【0031】
V型およびVI型は、穏やかに約80℃またはそれ以上に加熱するとモダフィニルI型へと変換する。V型およびVI型は、約100℃に加熱することにより、著明な分解を伴わずにI型に変換できる。
モダフィニルI型は濾過またはデカンテーションおよびその後の乾燥により、溶媒から簡便に分離できる。I型は、他の結晶形またはアモルファス型に変換することなく、そして著明な化学分解を受けることなく、100℃という高温で乾燥した。
【0032】
モダフィニルII型
本発明はさらにモダフィニルII型を提供する。モダフィニルII型は、9.1、10.3、11.1、11.9、14.3、15.2、16.4、17.5、18.4、20.5、21.3、24.6、26.6±0.2度の2θにおける反射を伴う粉末X線回折パターン(図2)をもたらす。14.3、17.5、20.5および21.3度の2θにおける強い反射がとりわけ特徴的である。これらの中でも14.3、17.5および21.3度の2θにおける反射が最も特徴的である。
【0033】
以下の技術は結晶形II型のモダフィニルの生成に有効であることが判明した。
モダフィニルIII型はこれを水に懸濁する時モダフィニルII型に変換する。したがって、III型の水への懸濁がモダフィニルII型の取得方法を提供する。
モダフィニルはまた、加熱によって溶媒にモダフィニルを溶解し、冷却して再結晶させることにより、エタノール、イソプロパノール、n-ブタノール、t-ブタノール、メチルイソブチルケトン、エチレングリコール、ジオキソランおよびジオキサンから選択的にII型で結晶化する。モダフィニルII型はさらに、ジクロロエタン中で再スラリー化することによって、そしてメタノールと水の混合物に入れたモダフィニルの溶液を速やかに冷却することによって製造できる。
【0034】
モダフィニルIII型
本発明はさらにモダフィニルIII型を提供する。モダフィニルIII型は7.4、9.0、10.5、12.3、14.2、14.7、15.1、16.4、18.3、20.0、20.5、21.1、22.1、24.5±0.2度の2θにおける反射を伴う粉末X線回折パターン(図3)をもたらす。7.4、10.5、18.3、20.0および20.5度の2θにおける強い反射がとりわけ特徴的である。これらの中でも7.4、10.5、18.3および20.0度の2θにおける反射が、それらの強度と、他の型のPXRDパターンでは対応位置に反射が無いことの故に特徴的である。
【0035】
モダフィニルIII型はトルエンからの結晶化によって生成する。III型はまた、炭酸ジメチルおよびエタノールの混合物からも結晶化するが、但しこの混合溶媒系から結晶化した場合、V型との混合物として得られる例もあった。
モダフィニルIV型
本発明はさらにモダフィニルIV型を提供する。モダフィニルIV型は6.9、10.4、14.1、17.2、18.5、20.3、20.8、21.6、22.7、25.0、26.5、27.6、28.5±0.2度の2θにおける反射を伴う粉末X線回折パターン(図4)をもたらす。6.9、10.4、17.2、20.3および22.7度の2θにおける強い反射がとりわけ特徴的である。
【0036】
モダフィニルはテトラヒドロフランおよびジメチルスルホキシドからIV型結晶形で結晶化する。
【0037】
モダフィニルV型
本発明はさらにモダフィニルV型を提供する。V型は7.4、9.3、10.5、12.4、14.7、16.2、18.2、19.9、21.5、22.0、23.6、24.5、25.2、28.4、29.5、31.8±0.2度の2θにおける反射を伴う粉末X線回折パターン(図5)をもたらす。9.3、12.4、18.2、19.9および22.0度の2θにおける強い反射がとりわけ特徴的である。
【0038】
V型は、炭酸ジメチル、ならびに炭酸ジメチルとエタノール、炭酸ジメチルと水および炭酸ジメチルとアセトンの混合物からの結晶化によって製造する。
V型の熱重量分析は、約100℃から出発し150℃までで約12%の質量損失を示した。このLODはモダフィニルと炭酸ジメチルとの半溶媒和物であるV型に一致する。このTGA分析はShimadzu DTG60で、毎分約10℃の速度でほぼ周囲温度から約300℃まで加熱した約10mgの試料について実施した。
【0039】
モダフィニルVI型
本発明はさらにモダフィニルVI型を提供する。VI型は9.0、9.3、10.2、12.4、14.2、14.5、15.3、17.5、18.1、20.0、20.5、21.5、22.0、23.5、24.5、25.0±0.2度の2θにおける反射を伴う粉末X線回折パターン(図6)をもたらす。9.3、18.1および20.5度の2θにおける反射がそれらの強度の故にとりわけ特徴的である。
【0040】
モダフィニルVI型は、水、エタノールまたは水/エタノール混合物中にモダフィニルV型を変換が完了するに充分な時間懸濁することによって製造できる。好ましくは、モダフィニルVIを水、エタノール、またはエタノール/水混合物中、約28℃でスラリー化し、その後55℃で減圧乾燥する。
アモルファスモダフィニル
モダフィニルはオルト、メタまたはパラキシレンの混合物から結晶化によりアモルファス状態で製造できる。
【0041】
実験室および工業的設定において明瞭なII-VI結晶形のモダフィニルの製造に最も適した技術を記載してきたが、当業者は、これらの結晶形はさらに別の方法で取得できることが理解できるであろう。
【0042】
モダフィニルII-IVおよびVI型を含有する薬用組成物
モダフィニルII-IVおよびVIは、ナルコレプシーに罹患した患者の覚醒を促進するのに有用な種々の薬用組成物および投与型に調合できる。
【0043】
本発明に係る薬用組成物は、所望により互いに混合したモダフィニルII-IVおよびVI型を含有する。本発明に係る薬用組成物はさらに、1またはそれ以上のモダフィニルII-IVおよびVI型と混合した、他のモダフィニル結晶形、アモルファスモダフィニルおよび/または他の活性成分を含有することができる。活性成分に加えて、本発明に係るモダフィニル薬用組成物は1またはそれ以上の賦形剤を含有してよい。賦形剤は様々な目的のために該組成物に添加する。
【0044】
希釈剤は固体の薬用組成物のかさを増やし、該組成物を含有する薬用投与型を、患者および介護者にとって扱い易くする。固体組成物のための希釈剤は、例えば微結晶セルロース(例えばAvicel(登録商標))、ミクロファインセルロース、乳糖、澱粉、α化澱粉、炭酸カルシウム、硫酸カルシウム、糖、デキストラート、デキストリン、デキストロース、二塩基性燐酸カルシウム二水和物、三塩基性燐酸カルシウム、カオリン、炭酸マグネシウム、酸化マグネシウム、マルトデキストリン、マンニトール、ポリメタクリラート(例えばEudragit(登録商標))、塩化カリウム、粉末セルロース、塩化ナトリウム、ソルビトールおよびタルクを包含する。
【0045】
圧縮して錠剤のような投与型とされる固体薬用組成物は賦形剤を含むことができるが、その機能は活性成分およびその他の賦形剤の、圧縮後の結合を助けることを含む。固体薬用組成物用の結合剤は、アラビアゴム、アルギン酸、carbomer(例えばcarbopol)、カルボキシメチルセルロースナトリウム、デキストリン、エチルセルロース、ゼラチン、グアールガム、水素化植物油、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース(例えばKlucel(登録商標))、ヒドロキシプロピルメチルセルロース(例えばMethocel(登録商標))、液体グルコース、珪酸アルミニウムマグネシウム、マルトデキストリン、メチルセルロース、ポリメタクリラート、ポビドン(例えばKollidon(登録商標)、Plasdone(登録商標))、α化澱粉、アルギン酸ナトリウムおよび澱粉を包含する。
【0046】
患者の胃内での圧縮された固体薬用組成物の溶解速度は、組成物に崩壊剤を添加することにより増大させることができる。崩壊剤は、アルギン酸、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロースナトリウム(例えばAc-Di-Sol(登録商標)、Primellose(登録商標))、コロイド状二酸化珪素、クロスカルメロースナトリウム、クロスポビドン(例えばKollidon(登録商標)、Polyplasdone(登録商標))、グアールガム、珪酸アルミニウムマグネシウム、メチルセルロース、微結晶セルロース、ポラクリリンカリウム、粉末セルロース、α化澱粉、アルギン酸ナトリウム、グリコール酸澱粉ナトリウム(例えばExplotab(登録商標))および澱粉を包含する。
【0047】
非圧縮固体組成物の流動性を改善し投薬の精度を改善するため、潤滑剤を添加できる。潤滑剤として機能し得る賦形剤は、コロイド状二酸化珪素、三珪酸マグネシウム、粉末セルロース、澱粉、タルクおよび三塩基性燐酸カルシウムを包含する。
粉末化した組成物の圧縮によって錠剤のような投与型を製造する場合、この組成物にパンチおよびダイからの圧力をかける。幾つかの賦形剤と活性成分はパンチおよびダイの表面に付着する傾向があり、それが製品に陥凹形成およびその他の表面凹凸を惹起し得る。潤滑剤を組成物に添加して付着を減少させ、ダイからの製品の解放を容易にすることができる。潤滑剤は、ステアリン酸マグネシウム、ステアリン酸カルシウム、モノステアリン酸グリセリル、パルミトステアリン酸グリセリル、水素化ひまし油、水素化植物油、鉱油、ポリエチレングリコール、安息香酸ナトリウム、ラウリル硫酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸、タルクおよびステアリン酸亜鉛を包含する。
【0048】
香料および矯味剤は、患者にとってその剤型をより味の良いものにする。本発明に係る組成物に含有させ得る、薬用生成物のための一般的な香料および矯味剤は、マルトール、バニリン、エチルバニリン、メントール、クエン酸、フマル酸エチルマルトール、および酒石酸を包含する。
組成物はさらに、それらの外観を改善しそして/または製品および単位投薬レベルを患者が同定し易くするために、任意の薬学上許容し得る着色剤を用いて着色できる。
【0049】
賦形剤の選択および使用量は、経験と標準法の考察および当分野における参考となる業績に基づき、製剤科学者によって容易に決定できる。
本発明に係る固体組成物は、粉末、顆粒、凝集物および圧縮組成物を包含する。投与型は、経口、バッカル、直腸、非経口(皮下、筋肉内および静脈内を包含する)、吸入および眼への投与に好適な投薬型を包含する。与えられた任意の症例において最も好適な経路は治療しようとする状態の性格および重篤度に依存するが、本発明に係る最も好ましい経路は経口である。投与型は単位投与型で簡便に提供でき、そして薬学分野で周知の任意の方法によって製造できる。
【0050】
投与型は、錠剤、散剤、カプセル剤、坐剤、サシェー剤、トローチ剤およびlosengeのような固体投与型、ならびに液体シロップ剤、懸濁剤およびエリキシル剤を包含する。本発明に係る特に好ましい剤型は錠剤である。
錠剤、カプセル剤、口内錠およびその他の単位投与型は、好ましくは約50ないし約300mg、より好ましくは約100mgないし約200mgの投与レベルでモダフィニルを含有する。
【0051】
或る好ましい態様を参考にして本発明を記載してきたが、本発明を限定するためではなく例示の目的のために以下の実施例を提供する。
【0052】
実施例
実施例1-8
(高純度モダフィニルの製造)
実施例1:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)をメタノール(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。H2O2の30%溶液(45ml)を加え、温度を30℃に上昇させた。温度を3.5時間30℃に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル40.2gが得られた(収率:75.7%)。
【0053】
実施例2:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)を炭酸ジメチル(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。H2O2の15%溶液(85ml)を加え、温度を30℃に上昇させた。温度を30時間30℃に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル45.1gが得られた(収率:85%)。
【0054】
実施例3:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例1で製造したモダフィニル3gを、5%の水を含有するアセトン32mlに懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を42℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに25℃まで冷却し、濾過した。乾燥後、本質上スルホンを含まない高度に精製されたモダフィニル1.95gが得られた(収率:65%)。
【0055】
実施例4:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例2で製造したモダフィニル1gを、エタノール10.5mlに懸濁した。混合物を窒素雰囲気下で加熱還流した。この懸濁液を25℃まで冷却し、濾過した。乾燥後、高度に精製されたモダフィニル0.83gが得られた(収率:83%)。
【0056】
実施例5:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でジフェニルメチルチオ-2-アセトアミド(50g)を炭酸ジメチル(550ml)に懸濁した。イソプロパノール46.7mlに溶解したH2SO41.2mlを含有する溶液(44ml)を加えた。さらに30%H2O249mlを加えた。温度を30℃に上げ、8時間一定に維持した。反応物を25℃に冷却し、水450mlで希釈した。過剰の未反応H2O2をNa2S2O5で中和し、さらに50mlの水を加えた。濾過によってモダフィニルを分離し、水210mlで再スラリー化した。乾燥後、モダフィニル45.1gが得られた(収率:85%)。
【0057】
実施例6:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例5で製造したモダフィニル3gを、アセトン100mlおよび炭酸ジメチル20mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を47℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに25℃まで冷却し、濾過した。乾燥後、本質上スルホンを含まない高度に精製されたモダフィニル2.52gが得られた(収率:84%)。
【0058】
実施例7:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例6で得られた未乾燥モダフィニル3.7gを、アセトン123.5mlおよび炭酸ジメチル24.7mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を25℃まで冷却し、濾過した。濾過ケーキを乾燥し再度アセトン94.5mlおよび炭酸ジメチル19mlの混合物に懸濁し、窒素雰囲気下で加熱還流した。得られた溶液を25℃に冷却し濾過した。乾燥後、スルホンを含まない高度に精製されたモダフィニル2.32gが得られた(収率:62.7%)。
【0059】
実施例8:還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で、実施例5で製造したモダフィニル3gを、アセトン1mlおよび炭酸ジメチル20mlを含有する混合物に懸濁した。混合物を窒素雰囲気下で加熱還流(〜58℃)した。得られた溶液を25℃まで冷却し、濾過した。この湿潤濾過ケーキを再度アセトン100mlおよび炭酸ジメチル.20mlの混合物に懸濁し、窒素雰囲気下で加熱還流した。得られた溶液を25℃に冷却し濾過した。乾燥後、スルホンを含まない高度に精製されたモダフィニル2.1gが得られた(収率:70.5%)。
【0060】
実施例9-13
(モダフィニルI型の製造)
実施例9:モダフィニルIV型を水に懸濁する事による。モダフィニルIV型(0.4g)をpH約5.9の蒸留水(50ml)に懸濁した。この懸濁液を約37℃で約24時間攪拌し、次いで濾過した。濾液をX線粉末回折によって分析し、モダフィニルI型であると決定した。
【0061】
実施例10:モダフィニルVまたはVI型を加熱する事による。モダフィニルVおよびVI型の小アリコートを約100℃の乾燥機で別々に約30分間加熱した。その後モダフィニルVおよびVI型をX線粉末回折により分析し、いずれもI型であると決定した。
【0062】
実施例11:アセトニトリルからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をアセトニトリル(23ml)に懸濁した。混合物を加熱還流した(約80℃)。得られた溶液を約63℃に冷却するとこの温度で結晶化が始まった。この懸濁液をさらに約25℃に冷却し、濾過した。乾燥後、結晶化したモダフィニル(1.96g)I型が得られた(収率65%)。
【0063】
実施例12:ジメチルホルムアミドからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をジメチルホルムアミド(5.5ml)に懸濁した。混合物を加熱還流した(約60℃)。透明な溶液が得られた。この溶液に水(5ml)を滴下するとモダフィニルが沈殿し始めた。この混合物を約25℃に冷却することにより沈殿化が完了した。生成物を濾過により分離した。乾燥後、結晶化したモダフィニル(2.54g)I型が得られた(収率84.7%)。
【0064】
実施例13:酢酸エチルからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)を酢酸エチル(50ml)に懸濁した。混合物を加熱還流し(約77℃)、約1時間維持した。混合物を約25℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニルI型(1.9g)が得られた(収率63%)。
【0065】
実施例14-15
(モダフィニルII型の製造)
実施例14:イソプロパノールからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をイソプロパノール(34ml)に懸濁した。混合物を加熱還流した(約85℃)。得られた溶液を約58℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニルII型(2.32g)が得られた(収率77.3%)。
【0066】
実施例15:モダフィニルIII型の水中懸濁液より。モダフィニルIII型(0.4g)をpH約5.9の蒸留水(50ml)に懸濁した。この懸濁液を約37℃で約24時間攪拌し、次いで濾過した。濾液を粉末X線回折によって分析し、モダフィニルII型であると決定した。
【0067】
実施例16
(モダフィニルIII型の製造)
実施例16:トルエンからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をトルエン(90ml)に懸濁した。混合物を加熱還流した(約110℃)。得られた溶液を約35℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約17時間約25℃に維持し、約5℃に冷却し、次いで濾過した。乾燥後、結晶化したモダフィニル(0.6g)III型が得られた(収率19.6%)。
【0068】
実施例17
(モダフィニルIV型の製造)
実施例17:テトラヒドロフランからの結晶化。還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3g)をテトラヒドロフラン(90ml)に懸濁した。混合物を加熱還流した(約63℃)。得られた溶液を約53℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、結晶化した(2.4g)モダフィニルIV型が得られた(収率80%)。
【0069】
実施例18
(モダフィニルV型の製造)
実施例18:炭酸ジメチルからの結晶化。モダフィニル(3g)を炭酸ジメチル(105ml)に懸濁した。この混合物を、還流冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中で加熱還流した(約90℃)。約2時間の還流の後、得られた溶液を約79℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約25℃に冷却し、次いで濾過した。乾燥後、ほぼ結晶化したモダフィニル(3g)V型が得られた(収率約90%)。
【0070】
実施例19
(モダフィニルVI型の製造)
実施例19:V型のエタノール懸濁液より。下降冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(3.5g)V型をエタノール(10ml)に懸濁した。混合物を約4.5時間約25℃で攪拌し、次いで濾過した。乾燥後、結晶化したモダフィニル(2.9g)VI型が得られた(収率82%)。
【0071】
実施例20
(アモルファスモダフィニルの製造)
実施例20:キシレンからの結晶化。下降冷却器、温度計および攪拌器を備えた三頸丸底フラスコ中でモダフィニル(5g)をキシレン(150ml)に懸濁した。混合物を約110℃に加熱し、これを約30分間維持した。得られた溶液を約35℃に冷却するとこの時点で結晶化が始まった。この懸濁液を約17時間約25℃に維持し、次いで約5℃に冷却し、濾過した。乾燥後、アモルファスモダフィニル(1.83g)が得られた(収率36.6%)。
【0072】
このように本発明を或る好ましい態様を参考に記載してきたが、本明細書および実施例の考察から、当業者にはその他の態様もまた自明となろう。実施例を含む本明細書は単に例示であり、本発明の範囲および精神は、以下の請求項により定義するということを意図している。
【図面の簡単な説明】
【0073】
【図1】図1は、モダフィニルI型の粉末X線回折パターンを示す。
【図2】図2は、モダフィニルII型の粉末X線回折パターンを示す。
【図3】図3は、モダフィニルIII型の粉末X線回折パターンを示す。
【図4】図4は、モダフィニルIV型の粉末X線回折パターンを示す。
【図5】図5は、モダフィニルV型の粉末X線回折パターンを示す。
【図6】図6は、モダフィニルVI型の粉末X線回折パターンを示す。
【特許請求の範囲】
【請求項1】
a) アルコールまたは相転移触媒を伴う鉱酸の混合物中で2-[(ジフェニルメチル)チオ]アセトアミドをH2O2で酸化し、
b) この混合物からモダフィニルを含有する固体を沈殿させ、そして、
c) この混合物を沈殿した固体から分離する、
工程を含む、モダフィニルを製造する方法。
【請求項2】
沈殿した固体から1回の結晶化によって99.5%より高いまたはこれに等しい純度でモダフィニルを単離することをさらに含む、請求項1に記載の方法。
【請求項3】
沈殿した固体から1回の再結晶によって99.9%より高いまたはこれに等しい純度でモダフィニルを単離する、請求項2に記載の方法。
【請求項4】
モダフィニルを薬学上許容し得る純度で単離する、請求項1に記載の方法。
【請求項5】
モダフィニルの純度を、225nm波長の光を用いる紫外部検出により得られるクロマトグラフィーの相対ピーク面積によって測定する、請求項1に記載の方法。
【請求項6】
沈殿した固体が99%より高いまたはこれに等しい純度のモダフィニルである、請求項1に記載の方法。
【請求項7】
沈殿した固体が99.5%より高いまたはこれに等しい純度のモダフィニルである、請求項6に記載の方法。
【請求項8】
H2O2を10-50重量パーセント水溶液として該混合物に加える、請求項1に記載の方法。
【請求項9】
鉱酸が、硫酸、過塩素酸、および燐酸より成る群から選ばれる、請求項1に記載の方法。
【請求項10】
アルコールが、イソプロパノール、tert-ブタノール、および2-メチル-1-ブタノールより成る群から選ばれる、請求項1に記載の方法。
【請求項11】
該混合物がさらに不活性液体有機媒質を含む、請求項1に記載の方法。
【請求項12】
不活性液体有機媒質が、メタノール、エタノール、エチレングリコール、アセトン、炭酸ジメチル、およびそれらの混合物より成る群から選ばれる、請求項11に記載の方法。
【請求項13】
酸化が、2-[(ジフェニルメチル)チオ]アセトアミド1当量を0.07ないし約0.13グラム/ミリリットルの量で不活性液体有機媒質に懸濁し、鉱酸約0.05ないし約0.2モル当量、アルコール約2ないし4当量、およびH2O2約1.5ないし約4モル当量をこの液体有機媒質に添加することを含む、請求項11に記載の方法。
【請求項14】
酸化が不活性液体有機媒質を加熱することをさらに含む、請求項13に記載の方法。
【請求項15】
請求項2に記載の方法によって製造したモダフィニル。
【請求項16】
0.02%未満の2-[(ジフェニルメチル)スルホニル]アセトアミドを含有するモダフィニル。
【請求項17】
2-[(ジフェニルメチル)スルホニル]アセトアミドを本質上含まない、請求項16に記載のモダフィニル。
【請求項18】
2-[(ジフェニルメチル)スルホニル]アセトアミドを含まない、請求項17に記載のモダフィニル。
【請求項19】
0.02%未満の2-[(ジフェニルメチル)スルフィニル]酢酸を含有するモダフィニル。
【請求項20】
0.02%未満のメチル2-[(ジフェニルメチル)スルフィニル]酢酸塩を含有するモダフィニル。
【請求項21】
a) アセトン、アセトニトリル、ベンジルアルコール、ジメチルホルムアミド、メタノール、メチルエチルケトン、ピロリドンおよびそれらの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) 該液体からモダフィニルを結晶化し、そして、
c) 該液体を分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項22】
該液体がメタノールまたはアセトンである、請求項21に記載の方法。
【請求項23】
a) モダフィニルを、これがモダフィニルI型に変換するに充分な時間酢酸エチルに懸濁し、そして、
b) この酢酸エチルを分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項24】
a) II型結晶形モダフィニルを、モダフィニルII型がモダフィニルI型に変換するに充分な時間、メチルt-ブチルエーテル、水、酢酸イソブチルおよびそれらの混合物より成る群から選ばれる液体に懸濁し、そして、
b) この液体を分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項25】
V型モダフィニルがI型モダフィニルに変換するに充分な時間、V型モダフィニルを約80℃またはこれより高い温度に加熱することによってモダフィニルIを製造する方法。
【請求項26】
V型モダフィニルがI型モダフィニルに変換するに充分な時間、VI型モダフィニルを約80℃またはこれより高い温度に加熱することによってモダフィニルIを製造する方法。
【請求項27】
14.3、17.5、20.5および21.3±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項28】
モダフィニルII型と命名された請求項27に記載の結晶性モダフィニル。
【請求項29】
14.3、17.5、20.5および21.3±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.1、10.3、11.9、15.2、18.4、24.6および26.6±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項27に記載のモダフィニルの結晶形。
【請求項30】
9.1、10.3、11.1、11.9、14.3、15.2、16.4、17.5、18.4、20.5、21.3、24.6、26.6±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項27に記載のモダフィニルの結晶形。
【請求項31】
a) III型モダフィニルが請求項27に記載のモダフィニルに変換するに充分な時間、III型モダフィニルを水に懸濁し、そして、
b) この水を分離して請求項27に記載のモダフィニルを得る、
という工程を含む、請求項27に記載のモダフィニルを製造する方法。
【請求項32】
a) エタノール、イソプロパノール、n-ブタノール、t-ブタノール、メチルイソブチルケトン、エチレングリコール、ジオキソラン、ジオキサンおよびそれらの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項27に記載のモダフィニルを得る、
という工程を含む、請求項27に記載のモダフィニルを製造する方法。
【請求項33】
7.4、10.5、20.0および20.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、モダフィニルの結晶形。
【請求項34】
モダフィニルIII型と命名された請求項33に記載の結晶性モダフィニル。
【請求項35】
7.4、10.5、20.0および20.5±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.0、12.3、22.1および24.5±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項33に記載のモダフィニルの結晶形。
【請求項36】
7.4、9.0、10.5、12.3、14.2、14.7、15.1、16.4、18.3、20.0、20.5、21.1、22.1、24.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項35に記載のモダフィニルの結晶形。
【請求項37】
a) トルエンならびにエタノールおよび炭酸ジメチルの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項33に記載のモダフィニルを得る、
という工程を含む、請求項33に記載のモダフィニルを製造する方法。
【請求項38】
6.9、10.4、17.2、20.3および22.7±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項39】
モダフィニルIV型と命名された請求項38に記載の結晶性モダフィニル。
【請求項40】
6.9、10.4、17.2、20.3および22.7±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、14.1、18.5、20.8、21.6および25.0±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項38に記載のモダフィニルの結晶形。
【請求項41】
6.9、10.4、14.1、17.2、18.5、20.3、20.8、21.6、22.7、25.0、26.5、27.6、28.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項40に記載のモダフィニルの結晶形。
【請求項42】
a) テトラヒドロフランおよびジメチルスルホキシドより成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項38に記載のモダフィニルを得る、
という工程を含む、請求項38に記載のモダフィニルを製造する方法。
【請求項43】
モダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項44】
9.3、12.4、18.2、19.9および22.0±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項43に記載のモダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項45】
モダフィニルV型と命名された、請求項43に記載のモダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項46】
9.3、12.4、18.2、19.9および22.0±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、7.4、24.7、26.2、21.5、23.6、24.5および25.2±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項44に記載のモダフィニルの結晶形。
【請求項47】
7.4、9.3、10.5、12.4、14.7、16.2、18.2、19.9、21.5、22.0、23.6、24.5、25.2、28.4、29.5、31.8±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項46に記載のモダフィニルの結晶形。
【請求項48】
a) 炭酸メチル、エタノールと炭酸ジメチルの混合物、水と炭酸ジメチルの混合物およびアセトンと炭酸ジメチルの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項43に記載のモダフィニルを得る、
という工程を含む、請求項43に記載のモダフィニルを製造する方法。
【請求項49】
9.3、18.2、および20.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項50】
モダフィニルVI型と命名された請求項49に記載の結晶性モダフィニル。
【請求項51】
9.3、18.2、および20.5±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.0、10.2、12.4、15.3、および20.0±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項49に記載のモダフィニルの結晶形。
【請求項52】
9.0、9.3、10.2、12.4、14.2、14.5、15.3、17.5、18.1、20.0、20.5、21.5、22.0、23.5、24.5、25.0±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項51に記載のモダフィニルの結晶形。
【請求項53】
a) 水、エタノールおよびエタノールと水の混合物より成る群から選ばれる液体に、V型モダフィニルを、このV型モダフィニルが請求項49に記載のモダフィニルに変換するに充分な時間懸濁し、そして、
b) この液体を分離して請求項49に記載のモダフィニルを得る、
という工程を含む、請求項49に記載のモダフィニルを製造する方法。
【請求項54】
請求項27に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項55】
請求項54に記載の組成物を含む薬用投与型。
【請求項56】
請求項33に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項57】
請求項56に記載の組成物を含む薬用投与型。
【請求項58】
請求項38に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項59】
請求項58に記載の組成物を含む薬用投与型。
【請求項60】
請求項49に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項61】
請求項60に記載の組成物を含む薬用投与型。
【請求項1】
a) アルコールまたは相転移触媒を伴う鉱酸の混合物中で2-[(ジフェニルメチル)チオ]アセトアミドをH2O2で酸化し、
b) この混合物からモダフィニルを含有する固体を沈殿させ、そして、
c) この混合物を沈殿した固体から分離する、
工程を含む、モダフィニルを製造する方法。
【請求項2】
沈殿した固体から1回の結晶化によって99.5%より高いまたはこれに等しい純度でモダフィニルを単離することをさらに含む、請求項1に記載の方法。
【請求項3】
沈殿した固体から1回の再結晶によって99.9%より高いまたはこれに等しい純度でモダフィニルを単離する、請求項2に記載の方法。
【請求項4】
モダフィニルを薬学上許容し得る純度で単離する、請求項1に記載の方法。
【請求項5】
モダフィニルの純度を、225nm波長の光を用いる紫外部検出により得られるクロマトグラフィーの相対ピーク面積によって測定する、請求項1に記載の方法。
【請求項6】
沈殿した固体が99%より高いまたはこれに等しい純度のモダフィニルである、請求項1に記載の方法。
【請求項7】
沈殿した固体が99.5%より高いまたはこれに等しい純度のモダフィニルである、請求項6に記載の方法。
【請求項8】
H2O2を10-50重量パーセント水溶液として該混合物に加える、請求項1に記載の方法。
【請求項9】
鉱酸が、硫酸、過塩素酸、および燐酸より成る群から選ばれる、請求項1に記載の方法。
【請求項10】
アルコールが、イソプロパノール、tert-ブタノール、および2-メチル-1-ブタノールより成る群から選ばれる、請求項1に記載の方法。
【請求項11】
該混合物がさらに不活性液体有機媒質を含む、請求項1に記載の方法。
【請求項12】
不活性液体有機媒質が、メタノール、エタノール、エチレングリコール、アセトン、炭酸ジメチル、およびそれらの混合物より成る群から選ばれる、請求項11に記載の方法。
【請求項13】
酸化が、2-[(ジフェニルメチル)チオ]アセトアミド1当量を0.07ないし約0.13グラム/ミリリットルの量で不活性液体有機媒質に懸濁し、鉱酸約0.05ないし約0.2モル当量、アルコール約2ないし4当量、およびH2O2約1.5ないし約4モル当量をこの液体有機媒質に添加することを含む、請求項11に記載の方法。
【請求項14】
酸化が不活性液体有機媒質を加熱することをさらに含む、請求項13に記載の方法。
【請求項15】
請求項2に記載の方法によって製造したモダフィニル。
【請求項16】
0.02%未満の2-[(ジフェニルメチル)スルホニル]アセトアミドを含有するモダフィニル。
【請求項17】
2-[(ジフェニルメチル)スルホニル]アセトアミドを本質上含まない、請求項16に記載のモダフィニル。
【請求項18】
2-[(ジフェニルメチル)スルホニル]アセトアミドを含まない、請求項17に記載のモダフィニル。
【請求項19】
0.02%未満の2-[(ジフェニルメチル)スルフィニル]酢酸を含有するモダフィニル。
【請求項20】
0.02%未満のメチル2-[(ジフェニルメチル)スルフィニル]酢酸塩を含有するモダフィニル。
【請求項21】
a) アセトン、アセトニトリル、ベンジルアルコール、ジメチルホルムアミド、メタノール、メチルエチルケトン、ピロリドンおよびそれらの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) 該液体からモダフィニルを結晶化し、そして、
c) 該液体を分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項22】
該液体がメタノールまたはアセトンである、請求項21に記載の方法。
【請求項23】
a) モダフィニルを、これがモダフィニルI型に変換するに充分な時間酢酸エチルに懸濁し、そして、
b) この酢酸エチルを分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項24】
a) II型結晶形モダフィニルを、モダフィニルII型がモダフィニルI型に変換するに充分な時間、メチルt-ブチルエーテル、水、酢酸イソブチルおよびそれらの混合物より成る群から選ばれる液体に懸濁し、そして、
b) この液体を分離してモダフィニルI型を得る、
工程を含む、モダフィニルI型を製造する方法。
【請求項25】
V型モダフィニルがI型モダフィニルに変換するに充分な時間、V型モダフィニルを約80℃またはこれより高い温度に加熱することによってモダフィニルIを製造する方法。
【請求項26】
V型モダフィニルがI型モダフィニルに変換するに充分な時間、VI型モダフィニルを約80℃またはこれより高い温度に加熱することによってモダフィニルIを製造する方法。
【請求項27】
14.3、17.5、20.5および21.3±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項28】
モダフィニルII型と命名された請求項27に記載の結晶性モダフィニル。
【請求項29】
14.3、17.5、20.5および21.3±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.1、10.3、11.9、15.2、18.4、24.6および26.6±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項27に記載のモダフィニルの結晶形。
【請求項30】
9.1、10.3、11.1、11.9、14.3、15.2、16.4、17.5、18.4、20.5、21.3、24.6、26.6±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項27に記載のモダフィニルの結晶形。
【請求項31】
a) III型モダフィニルが請求項27に記載のモダフィニルに変換するに充分な時間、III型モダフィニルを水に懸濁し、そして、
b) この水を分離して請求項27に記載のモダフィニルを得る、
という工程を含む、請求項27に記載のモダフィニルを製造する方法。
【請求項32】
a) エタノール、イソプロパノール、n-ブタノール、t-ブタノール、メチルイソブチルケトン、エチレングリコール、ジオキソラン、ジオキサンおよびそれらの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項27に記載のモダフィニルを得る、
という工程を含む、請求項27に記載のモダフィニルを製造する方法。
【請求項33】
7.4、10.5、20.0および20.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、モダフィニルの結晶形。
【請求項34】
モダフィニルIII型と命名された請求項33に記載の結晶性モダフィニル。
【請求項35】
7.4、10.5、20.0および20.5±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.0、12.3、22.1および24.5±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項33に記載のモダフィニルの結晶形。
【請求項36】
7.4、9.0、10.5、12.3、14.2、14.7、15.1、16.4、18.3、20.0、20.5、21.1、22.1、24.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項35に記載のモダフィニルの結晶形。
【請求項37】
a) トルエンならびにエタノールおよび炭酸ジメチルの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項33に記載のモダフィニルを得る、
という工程を含む、請求項33に記載のモダフィニルを製造する方法。
【請求項38】
6.9、10.4、17.2、20.3および22.7±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項39】
モダフィニルIV型と命名された請求項38に記載の結晶性モダフィニル。
【請求項40】
6.9、10.4、17.2、20.3および22.7±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、14.1、18.5、20.8、21.6および25.0±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項38に記載のモダフィニルの結晶形。
【請求項41】
6.9、10.4、14.1、17.2、18.5、20.3、20.8、21.6、22.7、25.0、26.5、27.6、28.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項40に記載のモダフィニルの結晶形。
【請求項42】
a) テトラヒドロフランおよびジメチルスルホキシドより成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項38に記載のモダフィニルを得る、
という工程を含む、請求項38に記載のモダフィニルを製造する方法。
【請求項43】
モダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項44】
9.3、12.4、18.2、19.9および22.0±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす、請求項43に記載のモダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項45】
モダフィニルV型と命名された、請求項43に記載のモダフィニルと炭酸ジメチルの結晶性半溶媒和物。
【請求項46】
9.3、12.4、18.2、19.9および22.0±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、7.4、24.7、26.2、21.5、23.6、24.5および25.2±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項44に記載のモダフィニルの結晶形。
【請求項47】
7.4、9.3、10.5、12.4、14.7、16.2、18.2、19.9、21.5、22.0、23.6、24.5、25.2、28.4、29.5、31.8±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項46に記載のモダフィニルの結晶形。
【請求項48】
a) 炭酸メチル、エタノールと炭酸ジメチルの混合物、水と炭酸ジメチルの混合物およびアセトンと炭酸ジメチルの混合物より成る群から選ばれる液体にモダフィニルを溶解し、
b) この液体からモダフィニルを結晶化し、そして、
c) この液体を分離して請求項43に記載のモダフィニルを得る、
という工程を含む、請求項43に記載のモダフィニルを製造する方法。
【請求項49】
9.3、18.2、および20.5±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらすモダフィニルの結晶形。
【請求項50】
モダフィニルVI型と命名された請求項49に記載の結晶性モダフィニル。
【請求項51】
9.3、18.2、および20.5±0.2度の2θにおける反射が、強い強度を持つ反射の最初の集合を含み、該結晶形がさらに、9.0、10.2、12.4、15.3、および20.0±0.2度の2θにおけるより低い強度の反射を特徴とする、請求項49に記載のモダフィニルの結晶形。
【請求項52】
9.0、9.3、10.2、12.4、14.2、14.5、15.3、17.5、18.1、20.0、20.5、21.5、22.0、23.5、24.5、25.0±0.2度の2θにおける反射を伴う粉末X線回折パターンをもたらす請求項51に記載のモダフィニルの結晶形。
【請求項53】
a) 水、エタノールおよびエタノールと水の混合物より成る群から選ばれる液体に、V型モダフィニルを、このV型モダフィニルが請求項49に記載のモダフィニルに変換するに充分な時間懸濁し、そして、
b) この液体を分離して請求項49に記載のモダフィニルを得る、
という工程を含む、請求項49に記載のモダフィニルを製造する方法。
【請求項54】
請求項27に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項55】
請求項54に記載の組成物を含む薬用投与型。
【請求項56】
請求項33に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項57】
請求項56に記載の組成物を含む薬用投与型。
【請求項58】
請求項38に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項59】
請求項58に記載の組成物を含む薬用投与型。
【請求項60】
請求項49に記載のモダフィニルおよび薬学上許容し得る賦形剤を含む薬用組成物。
【請求項61】
請求項60に記載の組成物を含む薬用投与型。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−24006(P2009−24006A)
【公開日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願番号】特願2008−179304(P2008−179304)
【出願日】平成20年7月9日(2008.7.9)
【分割の表示】特願2002−516257(P2002−516257)の分割
【原出願日】平成13年7月27日(2001.7.27)
【出願人】(501079705)テバ ファーマシューティカル インダストリーズ リミティド (283)
【Fターム(参考)】
【公開日】平成21年2月5日(2009.2.5)
【国際特許分類】
【出願日】平成20年7月9日(2008.7.9)
【分割の表示】特願2002−516257(P2002−516257)の分割
【原出願日】平成13年7月27日(2001.7.27)
【出願人】(501079705)テバ ファーマシューティカル インダストリーズ リミティド (283)
【Fターム(参考)】
[ Back to top ]