
結晶性ピラゾール誘導体
本発明は、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形及びそのような結晶の製造、相互変換及び単離に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形及びそのような結晶の製造方法、相互変換及び単離に関する。
【背景技術】
【0002】
5−フェニルピラゾール−1−ベンゼンスルホンアミドは、関節炎及び炎症によるその他の症状を治療するために有用な、強力なCOX−2抑制作用を有する新規合成の化合物群である。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドは、以下の構造:
【化1】

を有する5−フェニルピラゾール−1−ベンゼンスルホンアミドである。
【0003】
式Iの化合物は、米国特許第5,466,823号(Tallyら)及び同第5,521,207号(Graneto)に開示されている。これらの特許は参照することにより本明細書に組入れられている。式Iの化合物の製造が開示されているものの、該明細書は、この試薬の結晶形の単離及び性質については全く触れていない。確固たる方法を確保する手段として多形挙動を同定し、濾過性などの化学的製造問題と同様に、錠剤化問題、錠剤破壊、懸濁液中での結晶成長とその結果起こるケーキング、懸濁液からの沈殿を避け、そして解析的再現性を確保する必要性がある(Analysis of Organic Polymorphs, A Review; Threlfall, T.L., Analyst, 120, 2435-2459を参照)。
【発明の開示】
【発明が解決しようとする課題】
【0004】
一つの態様において、本発明は、CuKα1X線(波長=1.5406オングストローム)を用いて得られる2θ角で、14.0、18.9、21.3、21.9及び25.7度(±0.1度)で表されるピークを含む粉末X線回折パターンを有する、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を供する。本発明の他の態様において、i)4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの懸濁液を、好適な溶媒中で約0℃から約60℃の温度で混合し;ii)その懸濁液を約0℃から約60℃の温度で24から72時間撹拌し;そして、iii)形態Iの結晶を集めること;を含む、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態IIをその結晶形態Iへ変換する方法が供される。本発明の他の態様において、該方法における溶媒は、水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン及びテトラヒドロフラン及びそれらの混合物から成るグループから選ばれる。
【0005】
本発明の追加の態様は、(i)該形態IIを、約10℃から約60℃の温度での形態IIの溶解度が2mg/mLより大きな水混和性溶媒中に溶解させ;(ii)水を添加することによって該化合物を沈殿させ;(iii)工程(ii)の懸濁液を約15℃から約45℃の温度で2から72時間撹拌し;そして(iv)形態Iの結晶を集めることを含む、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態IIを結晶形態Iへ変換する方法である。本発明の他の態様において、この方法に対する溶媒はエタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドから成るグループから選ばれる。
【0006】
更なる態様において、本発明は、結晶形態Iを有する式Iの化合物を含む薬剤組成物を供する。本発明の他の態様において、この薬剤組成物は薬学的に許容される担体又は賦形剤を更に含む。本発明の更に他の目的は、本発明の新規組成物を治療的に有効な量で使用することによって、動物における炎症状態を予防又は治療する方法を供することである。本発明の更なる目的は、動物における炎症状態を予防又は治療するための医薬品の製造における、本発明の新規組成物の使用を供することである。
【0007】
本発明の更なる目的は、以下の工程によって、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造することである。
【0008】
工程(a):4−フルオロアセトフェノン1モル当り1.0から1.6モル、又は好ましくは1.2から1.45モル、又はより好ましくは1.25から1.35モルのトリフルオロ酢酸アルキルを、4−フルオロアセトフェノン1モル当り1.0から1.5モル、又は好ましくは1.1から1.35モル、又はより好ましくは1.15から1.25モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンと混合して混合物とする。場合により、1380mlまでの、又は好ましくは900mlまでの、又はより好ましくは490mlまでの適切な溶媒、好ましくは2−プロパノールをこの工程において添加する。場合により、工程(a)の混合物を周囲温度より高く、還流温度までの温度で、又は好ましくは、40℃から70℃の温度で、又はより好ましくは50℃から60℃の温度で、1から24時間、又は好ましくは1から10時間、又はより好ましくは1から4時間、又はより好ましくは該反応が完了するまで加熱する。場合により、そのような加熱の後、該混合物を次いで−5℃から30℃の温度に、又はより好ましくは周囲温度に冷却する。
【0009】
工程(b):工程(a)の混合物を、4−フルオロアセトフェノン1モル当り415から1245ml、又は好ましくは650から870ml、又はより好ましくは725から795mlの水、それに加えて4−フルオロアセトフェノン1モル当り1.1から2.0モル、又は好ましくは1.2から1.7モル、又はより好ましくは1.3から1.5モルの濃塩酸、それに加えて4−フルオロアセトフェノン1モル当り0.8から1.2モル、又は好ましくは0.9から1.1モル、又はより好ましくは0.95から1.05モルの4−スルホンアミドフェニルヒドラジン・塩酸塩、及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が550から1660ml、又は好ましくは600から1000ml、又はより好ましくは650から750mlとなる量の好適な溶媒、好ましくは、C1−C6アルコール、又はより好ましくは2−プロパノールの組合せ物と組合せる、又は好ましくは添加する。場合により、工程(b)の混合物を、周囲温度より高く、還流温度までの温度で、又は好ましくは40℃から70℃の温度で、又は好ましくは50℃から70℃の温度で、1から24時間、又は好ましくは1から10時間、又はより好ましくは1から4時間、又はより好ましくは該反応が完了するまで加熱する。場合により、そのような加熱の後、該混合物を次に周囲温度から71.5℃の温度、又は好ましくは40℃から65℃の温度、又はより好ましくは50℃から60℃の温度で安定化させる。
【0010】
工程(c):次いで該混合物に、4−フルオロアセトフェノンに対してシーディング量、又は好ましくは0.0001質量%から50質量%、又はより好ましくは0.001質量%から5質量%、又はより好ましくは0.01質量%から0.5質量%の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加する。場合により、工程(c)の混合物を40℃から71.5℃より低い温度で、又は好ましくは50℃から71.5℃より低い温度で、又はより好ましくは50℃から60℃の温度で、1から10時間、又は好ましくは3から8時間、又はより好ましくは5から7時間加熱する。場合により、そのような加熱の後、次に該混合物を−5℃から30℃の温度に、又はより好ましくは周囲温度に冷却させる。場合により、この混合物を濾過し、好適な溶媒、好ましくはアルコール、より好ましくは2−プロパノール、又は水又はそれらの混合物で洗浄する。場合により、該溶媒は4−フルオロアセトフェノン1モル当り、300から1500ml、又は好ましくは600から1060ml、又はより好ましくは800から860mlの量であり、そして該水は4−フルオロアセトフェノン1モル当り、100から700ml、又は好ましくは285から545ml、又はより好ましくは385から445mlの量である。
【0011】
工程(d):次いで形態Iの結晶を集める。場合により、この形態Iの結晶を15℃から80℃の温度で、又は好ましくは30℃から65℃の温度で、より好ましくは50℃から60℃の温度で乾燥する。
【0012】
本発明の他の態様において、工程(a)の金属アルコキシドはナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれ、又は好ましくはナトリウムメトキシドである。
【0013】
これらの及びその他の目的は、当業者に容易に明確であろう。
【課題を解決するための手段】
【0014】
米国特許第5,466,823号及び同第5,521,207号に選択的COX−2抑制剤、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミド(式I):
【化2】
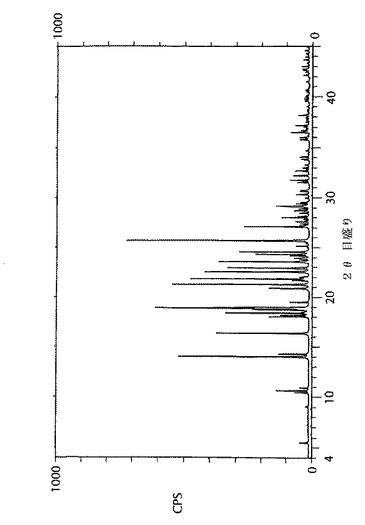
が述べられている。この記述には該化合物の合成が述べられているものの、得られた結晶形に関しては全く触れていない。これらの実施例に述べられているように、これらの特許中に示された手順に従うと、粉末X線回折(PXRD)パターンによって示される形態IIの結晶が得られる。本発明者らはこの結晶形が、もう一つの形態、形態IIに比べて周囲温度及び圧力下で不安定であることを見出した。これらの形態は、それらのPXRD(粉末X線回折)パターンによって区別できる。
【0015】
定義
本出願において言及される「水混和性」とは、あらゆる割合で水に混合又は溶解され得ることを意味する。
【0016】
本出願において言及される「無水結晶性」とは、実質的量の水を含有しない結晶のことである。この水含有量は、例えば、カールフィッシャー滴定を含む従来公知の方法によって求めることができる。好ましくは、無水結晶形は最大で約1質量%、より好ましくは最大で約0.5質量%、そして最も好ましくは最大で約0.1質量%の水を含有する。
【0017】
本出願において言及される、バルク薬物安定性試験における「安定」とは、バルク薬物の少なくとも約90質量%、好ましくは少なくとも約95質量%、より好ましくは少なくとも約99質量%が、指定条件下で指定時間の貯蔵後に変化しないことを意味する。
「DSC」とは、示差走査熱量測定を意味する。
【0018】
単独で又は、「トリフルオロアセテート」などの他の用語と共に使用される用語アルキルは、別途特定されない限り、1から4個の炭素原子、又は好ましくは2個の炭素原子を有する直鎖状又は分枝鎖状のものを包含する。非限定的例としては、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、第三級−ブチル、イソブチル及び第二級−ブチルなどの基が挙げられる。
【0019】
「金属アルコキシド」は、アルコールの金属塩である。一般に、該金属アルコキシドは金属としてナトリウム、リチウム又はカリウムを含有する。本発明において、それらはアルコール溶液の形で使用される。一般に、該アルコールは該アルコキシドのアルコールに対応する。非限定的例としては、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物が挙げられる。
【0020】
シーディングは、混合物、溶液又は懸濁液から多くの結晶の形成を誘起するために、一つ又はそれ以上の結晶を使用する技法である。シーディング量は、混合物、溶液又は懸濁液に添加された際に、化合物の所望の形態を形成させ得るような材料の量である。理論上はこの量は非常に少量であり得るが、実際には、より多くの量が使用される。この量は、合理的に取扱いできそして化合物の所望の形態を形成させ得るために十分である、いかなる量でもよい。非限定的例としては、参照化合物に対して0.0001質量%から50質量%のシーディング化合物がシーディング量として使用できる。
【0021】
温度に関して使用される用語「C」とは、℃又は摂氏を意味する。
「周囲温度」とは、対象物の周りの大気温度である。それは、室内の温度であり、一般に15から25℃である。
【0022】
還流は、長い時間に亘って反応物にエネルギーを与えるために化学において使用される技法である。この技法のために、頂部のみが開いた容器中に液状反応混合物が入れられる。発せられるあらゆる蒸気が冷却されて液体に戻り、この反応容器に落下して戻されるよう、この容器は垂直の凝縮器に連結される。次いで、この容器は反応過程中、激しく加熱される。この技法の利点は、更に溶媒を添加する必要なしに、又は反応容器が沸騰して乾燥する心配なしに長期間放置できることである。加えて、ある溶媒が常に一定の温度で沸騰するため、その反応はその同一温度で進行することになる。溶媒が変われば異なる温度で沸騰するため、溶媒を注意深く選択することによって反応温度が制御できる。本明細書で使用される「還流温度」とは、本還流技法の間、特定の溶媒が沸騰する温度のことである。例えば、海抜ゼロにおいて、2−プロパノールは82℃の沸点を有し、メタノールは65℃の沸点を有する。
【0023】
工程(a)に関して、「反応完了」は、以下のHPLC法を用いて、混合物中に残存する4−フルオロアセトフェノンの量が、4−フルオロアセトフェノンの投入量に対して2質量%以下であることを確認することによって求められる。
カラム: Discover HS F5、5μ、250×4.6mm Supelco Cat #567517-U
移動相(勾配):
【表1】
流速: 1ml/分
注入量: 20μl
検出: 247nm
【0024】
工程(b)に関して、「反応完了」は、以下のHPLC方を用いて、形成される4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの量が90%を越えることを確認することによって求められる。
カラム: Zorbax Eclipse XDBフェニル、3.5μm、150×4.6mm
移動相: メタノール/25mM燐酸(55/45体積%)
pH2.5(NaOHでpH調整)
ラン時間: 30分
カラム温度:35℃
流速: 1.0ml/分
注入量: 15μl
検出: 254nmの紫外線
試料濃度: 0.4mg/ml
【0025】
「治療的に有効な量」とは、症状(例えば、炎症症状)を予防するであろう、又は治療される病気の一つ又はそれ以上の症状がある程度緩和できるような、投与される化合物量のことである。本発明における使用に適する薬剤組成物としては、意図する目的を達成するために十分な量で有効成分を含有する組成物が挙げられる。治療的に有効な量を決定することは、特に本明細書に提供される詳細な開示を参照すれば、十分当業者の能力範囲内にある。
【0026】
本発明の化合物の適切な受容体である動物としては、ヒト又は他の哺乳類又は動物、例えば、牛、羊、豚、馬、山羊及び家禽(例えば、鶏、七面鳥、アヒル及びガチョウ)含む家畜動物及びその他の鳥、及び犬、猫などのペット動物及び外来及び/又は動物園の動物が挙げられるが、これらに限定されるものではない。げっ歯動物及び非げっ歯動物の両者の治療が意図される。
【0027】
実際には、投与される化合物の量は、動物の体重1kg当り約0.001から100mgの範囲であり、そのような総投与量は一度に又は小分けして与えられる。それは、単独で、又は、一つ又はそれ以上の他の薬と組合せて投与されてよい。一般に、それは、一つ又はそれ以上の他の薬学的に許容される賦形剤と共に製剤として投与されることになる。本明細書で用語「賦形剤」は、本発明の化合物(類)以外のあらゆる成分を記述するために使用される。賦形剤の選択は、特定の投与方式、溶解度及び安定性に及ぼす賦形剤の影響及び用量形態の性質などの因子にかなりの程度まで依存するであろう。
【0028】
本発明の化合物の送達に好適な薬剤組成物及びそれらの製造方法は、当業者に容易に明らかであろう。そのような組成物及びそれらの製造方法は、例えば、Remington's Pharmaceutical Sciences, 19th Edition(Mack Publishing Company, 1995)において見出すことができる。
【0029】
動物における消炎剤としての使用に対して、本発明の組成物は経口又は注射によって投与してよい。本発明の組成物を乾燥した、固体の単位剤形にて投与することが望ましい場合には、通常、所望の量の有効化合物を含有するカプセル、丸薬又は錠剤が採用される。これらの剤形は、有効成分を、澱粉、乳糖、タルク、ステアリン酸マグネシウム、植物ゴム質などの細かく粉砕された好適な希釈剤と良く均一に混合することによって製造される。そのようなユニット用量薬剤は、治療されるホスト動物種、炎症の重篤度と種類及び該ホストの体重などの因子に依存して、その総重量及び該消炎剤の含有量を大幅に変更してもよい。錠剤の処方は、Pharmaceutical Dosage Forms: Tablets, Vol.1, H. Lieberman and L. Lachman(Marcel Dekker, New York, 1980)に論じられている。
【0030】
或いは又、本発明の消炎組成物は、非経口的に、例えば、胃内、筋肉内又は皮下注射によって動物に投与されてもよく、その際には有効成分が液体担体賦形剤中に溶解又は分散される。非経口投与に対しては、有効材料が、許容できる賦形剤、好ましくは落花生油、綿実油などの植物油種と好適に混合される。ソルケタール、プロピレングリコール、グリセロールホルマールを用いる有機製剤及び含水非経口製剤などの他の非経口賦形剤も、種々の割合で組合されてしばしば使用される。有効化合物又は化合物類は、投与のために、非経口製剤中に溶解又は懸濁され、そのような製剤は一般に、0.005から5質量%の有効化合物を含有する。
【発明を実施するための最良の形態】
【0031】
本発明の以下の記述において、本発明が実行され得る特定の態様が述べられている。これらの態様は、当業者が本発明を実行し得るように、十分詳細に述べられている。他の態様も利用でき、そして本発明の範囲から逸脱することなく論理的な他の変更がなされ得る。従って、以下の詳細な記述は制限する意味で捉えられるべきではなく、本発明の範囲は、添付の請求範囲及びそのような請求範囲が資格を与える同等物の全範囲によってのみ定義される。
【0032】
形態IIは、米国特許第5,466,823号に述べられた手順から得られる結晶形である。結晶形態Iを得る方法は、以下の非制限的記述及び実施例によって説明される。
【0033】
一つの方法において、結晶形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドは、約0℃から約60℃の温度において、薬物の濃度がその飽和点よりも高い状態で好適な溶媒中に懸濁され混合されてよい。好適な溶媒は、その中でのその薬物の溶解度が約1mg/mLより高く、そして約800mg/mLより低いものである。好適な溶媒の例としては、水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン、及びテトラヒドロフラン、及びこれらの混合物が挙げられるが、これらに限定されるものではない。該懸濁液を約0℃から約60℃の温度で、24から72時間撹拌する。形態I結晶は濾過によって集められる。
【0034】
別の手順において、結晶形態Iは、その中での化合物の溶解度が2mg/mLより高い好適な水混和性溶媒中の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの溶液から、約10℃から約60℃の温度で水を添加することによって沈殿させて製造できる。形態IIから形態Iを製造するための好適な溶媒の例としては、エタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドが挙げられる。水を使用するこの最初の沈殿に続いて、この懸濁液を、約15℃から約45℃の温度で24から72時間撹拌して、形態Iの結晶を濾過によって集める。
【0035】
製造1.形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの製造
以下の手順は、出発物質として4−クロロアセトフェノンの代わりに4−フルオロアセトフェノンを使用するという重大ではない変更以外は、米国特許第5,521,207号の実施例1の手順と本質的に同一である。
【0036】
工程1:4,4,4−トリフルオロ−1−[4−(フルオロ)フェニル]−ブタン−1,3−ジオンの製造
トリフルオロ酢酸エチル(2.35g、1.66mmol)を50mLの丸底フラスコ中に入れてメチル第三級ブチルエーテル(7.5mL)中に溶解した。この撹拌溶液に、25質量%のナトリウムメトキシド(4.0mL、17.7mmol)を添加ロートを介して2分かけて添加した。次に、4−フルオロアセトフェノン(2.1g、15mmol)をメチル第三級ブチルエーテル(2mL)中に溶解させ、反応溶液に5分かけて滴下した。一晩(15.75時間)撹拌した後、3N塩酸(7.0mL)を添加した。この有機層を集め、塩水(7.5mL)で洗浄、MgSO4上で乾燥、濾過、減圧濃縮して、3.2gの淡橙色固体を得た。この固体をイソオクタンから再結晶化させて、2.05gの当ジオンを得た。
【0037】
工程2:4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの製造
4−スルホンアミドフェニルヒドラジン・塩酸塩(982mg、4.4mmol)を4,4,4−トリフルオロ−1−[4−(フルオロ)フェニル]−ブタン−1,3−ジオン(0.936mg、4.0mmol)の撹拌エタノール(50mL)溶液に添加した。この反応溶液を加熱還流しながら20時間撹拌した。室温まで冷却後、この反応混合物を減圧濃縮した。この残留物を酢酸エチル中に採取して水及び塩水で洗浄、MgSO4上で乾燥、濾過、減圧濃縮して、茶色固体を得、それを酢酸エチル及びイソオクタンから再結晶化させて、0.8gの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを得た。
1HNMR(CDCl3):δ:7.92、7.47、7.22、7.09、6.76、4.92;
MS: m/e ES−(M−H):384;
m.p.:167〜171℃。
【0038】
製造2.結晶形態IIを単離しない、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの製造
以下の手順は、結晶形態IIを単離しないで、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造する一般的方法を記述する。
【0039】
パートA
4−フルオロアセトフェノン(1モル当量)を、トリフルオロ酢酸アルキル(1.0から1.6モル当量)、金属アルコキシド(1.0から1.5モル当量)及び、場合により好適な溶媒(4−フルオロアセトフェノン1kg当り10Lまで)の混合物と組合せる。この混合物を還流(反応を完了させるため)状態までの温度に加熱してもよい。このパートAに対する「好適な溶媒」は、直鎖状、分枝鎖状及び環状アルコールを含むC1−C6飽和脂肪族アルコールである。好適な溶媒の非制限的な例としては、メタノール、エタノール、イソプロパノール等のアルコール及びそれらの混合物が挙げられる。
【0040】
パートB
パートAからの混合物を、4−スルホンアミドフェニルヒドラジン・塩酸塩(4−フルオロアセトフェノンに対して0.8から1.2モル当量)、及び水(4−フルオロアセトフェノン1kg当り3から9L)中への塩酸(4−フルオロアセトフェノンに対して1.1から2.0モル当量)溶液の混合物と組合せる。必要ならば、パートB中に存在する溶媒の総量が4−フルオロアセトフェノン1kg当り4から12Lの範囲内になるよう、好適な溶媒を添加してもよい。この反応混合物を還流(反応を完了させるため)状態までの温度に加熱してもよい。反応期間の後、この混合物を71.5℃より低い温度にて、形態I結晶(4−フルオロアセトフェノンに対して0.0001質量%以上)でシーディングする。この固体を濾過によって単離し、そして好適な溶媒を用いて洗浄及び/又は再度スラリー化してもよい。この生成物を80℃までの温度で乾燥してもよい。このパートBに対する「好適な溶媒」は、直鎖状、分枝鎖状及び環状アルコールを含むC1−C6飽和脂肪族アルコール、及び水及びそれらの混合物である。
【0041】
以下の手順は、結晶形態IIを単離することなく、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造する好ましい方法を述べる。2−プロパノール(70ml)及びトリフルオロ酢酸エチル(27.15g、0.191モル)を第一の容器に添加し、続いて配管を2−プロパノール(10ml)で洗浄する。次に、25%のナトリウムメトキシド(37.5g、0.174モル)のメタノール溶液をこの容器に添加し、続いて配管を2−プロパノール(10ml)で洗浄する。次に、4−フルオロアセトフェノン(20g、0.145モル)を添加し、続いて配管を2−プロパノール(10ml)で洗浄する。容器の内容物を55℃に加熱し、その温度に2時間保持し、その後で周囲温度まで冷却する。水(110ml)、濃塩酸(20.0g、0.203モル)及び4−スルホンアミドフェニルヒドラジン・塩酸塩(32.4g、0.145モル)を第二の容器に添加し、続いて配管を水(10ml)で洗浄する。第一容器の内容物を第二容器に添加し、続いて配管を2−プロパノール(60ml)で洗浄する。一体にした内容物を70℃で2時間加熱し、次いで55℃に冷却して、10mg(4−フルオロアセトフェノンに対して0.05質量%)の形態Iの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドでシーディングする。本内容物を55℃で6時間保持した後に、周囲温度まで冷却して濾過する。この生成物を50%の2−プロパノール水溶液(120ml)及び水(60ml)で洗浄し、次いで55℃にて減圧乾燥して、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの所望の形態Iの多形体(44.8g、80%)を得る。
【0042】
本発明を以下の非制限的実施例によって今まで以上に詳細に述べる。
【実施例】
【0043】
〔実施例1〕
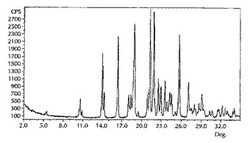
20mLのガラスビン中で、脱イオン水1mLをイソプロパノール1mLと良く混合した。形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミド(258.6mg)をこのビンに添加した。マグネティック撹拌棒を導入して、このビンの蓋をきつく閉めた。この懸濁液を約200rpmで30分間撹拌した。ペースト状の濃い懸濁液が観察された。撹拌を止めてこのビンを3日間放置した。この懸濁液の一部を取出して吸収紙上で乾燥した。この固体の粉末X線回折パターンを図1に示す。いくつかの単離された結晶の、高温顕微鏡下で観察された融点は148から152℃であった。しかしながら、形態Iから形態IIへの140から155℃の間での転移のため、この範囲は再現性よく観察されなかった。
【0044】
〔実施例2〕
380gの形態IIを2mLのエタノール中に懸濁させた。この化合物の一部は溶解したが、いくらかの過剰の固体化合物がこの溶液中に懸濁した。得られた懸濁液を、約20℃から約30℃の温度にて2週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0045】
〔実施例3〕
1.5gの形態IIを2mLのアセトニトリルに添加して懸濁液とした。この懸濁液を、約20℃から約30℃の温度にて2週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0046】
〔実施例4〕
0.5gの形態IIを2mLのメタノール+水混合液(1:1、体積比)に添加して懸濁液とした。この懸濁液を、約20℃から約30℃の温度にて4週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0047】
〔実施例5〕
81.5kgの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを81.5kgのエタノール(6倍量の体積)中に40℃にて完全に溶解し、まだ熱いうちに濾過した。154Lの水(9倍量の体積)をこの溶液に1時間かけて、温度を40℃に保持したまま添加し、続いて20℃まで冷却した。このスラリーを20℃にて24時間撹拌し、濾過して、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを形態Iとして得た。
【0048】
〔実施例6〕
粉末X線回折
Scintag X2最新回折システム(Scintag DMS/NT 1.30a及びマイクロソフトWindows(登録商標) NT 4.0ソフトウエアによって制御される)を使用して粉末X線回折を行った。このシステムは、1.5406オングストロームのCuKα1を放射するための銅X線源(45kV及び40mA)及び固体状のペルチエ冷却検出器を使用する。ビーム絞りは2mm及び4mmの管分岐及び散乱防止スリット、0.5mm及び0.2mm幅の検出器散乱防止及び受信スリットを使用して制御された。各ステップ当り1秒のカウント時間で、0.03度/ステップのステップ走査を用いて2から35度(2θ)からデータが集められた。本実験には、Scintagの丸い頂部に取付けた、直径12mmのキャビティを有するアルミニウム製試料容器が利用された。粉末はこの容器内に充填され、試料表面と試料容器表面の間の共平面性が確保されるよう、ガラススライドによって静かに押付けられた。
【0049】
熟練した結晶学者によって理解されるであろうように、下表内に示される種々のピークの相対強度は、例えば、X線中での結晶の配向効果、又は解析される材料の純度又は試料の結晶化度などの多くの因子があるために変化し得る。これらのピーク位置も、試料の高さにおける変化に対して移動し得るが、実質的にピーク位置は与えられた表中に定義された如くに留まるであろう。
【0050】
熟練した結晶学者は、異なる波長を用いて測定すれば、Braggの式、nλ=2dsinθに従う移動が異なる結果になることも理解するであろう。
代替波長を使用することによって発生するそのような更なるPXRDパターンは、本発明の結晶性材料のPXRDパターンの代替表示であり、そしてそのようなものは、本発明の範囲内にあるものと考えられる。
【0051】
形態Iに対して、角度2θ、間隔d及び相対強度が、Accelrys Materials Studio(登録商標)「バージョン2.2」の「反射粉末回折」モジュールを用いて、単結晶構造から計算された。各ケースにおける適切なシミュレーションパラメータは:
波長=1.540562オングストローム(CuKα);
偏光因子=0.5;
擬Voigtプロファイル(U=0.01、V=-0.001、W=0.002)
であった。
【0052】
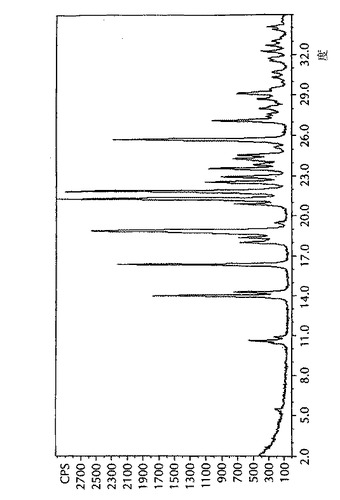
形態Iに対する実験及び計算によるPXRDパターン、及び形態IIに対する実験によるPXRDパターンを、図1、2、及び3に各々示す。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの主要PXRDピークを、表1に示す。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの主要ピークの計算によるPXRDパターンを、表2に示す。形態Iは、2θ角度が14.0、18.9、21.3、21.9及び25.7度±0.1度に独特の回折ピークを示す。
【0053】
【表2】
【0054】
【表3】
【0055】
〔実施例7〕
示差走査熱量測定(DSC)
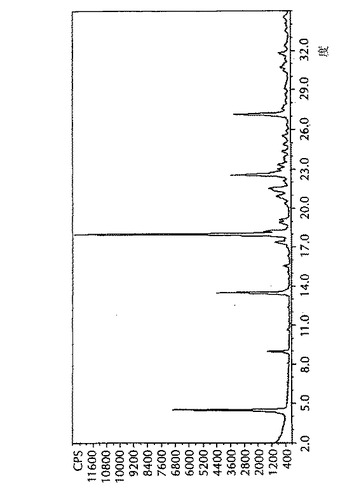
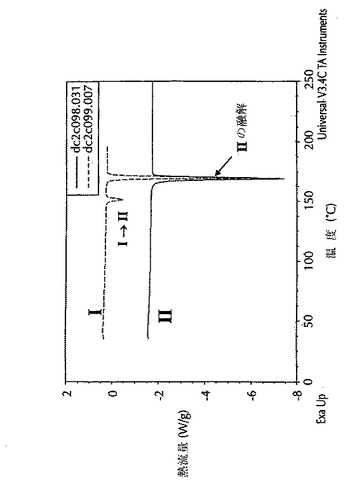
示差走査熱量測定(DSC)データは、DSC熱量測定機(TA Instruments 2920)を用いて得た。粉末(1から5mg)をアルミニウム製皿に充填した。アルミニウム製蓋をその皿の頂部に置いて丸めた。丸めた皿を試料セル中に、参照としての空の皿と共に置いた。特に断らない限り、温度を30℃から250℃へ10℃/分の速度で上昇させた。この熱セルを乾燥窒素で50mL/分でパージした。データ採取にはNT用TA Instruments Thermal Solutions(登録商標)(バージョン1.3L)を使用し、そしてデータ解析にはNT用Universal Analysis(登録商標)(バージョン2.4F)を使用した。図4に見られるように、形態IのDSCは、140℃から155℃で形態Iから形態IIへの吸熱相転移を示す。
【0056】
〔実施例8〕
形態IとIIの間の熱力学的関係
この実施例においては、形態Iと形態IIの間の相転移温度について述べる。形態Iと形態IIは互変性であり、それは一つの多形が転移温度、Tt、より低い温度で安定であるが、もう一方の多形は転移温度、Tt、より高い温度で安定であることを意味する。これら2つの固相は転移温度にて等しい自由エネルギーを有する。熱力学的安定関係の知識は、形態Iを製造するための好適な結晶化条件の選択に対して、及び製剤開発及び商業製造に対して必要である。Ttを求めることは、バルク薬物の製造と加工に対して重要である。
【0057】
材料及び方法
材料
本検討において使用された形態I試料は、HPLCによって試験した結果、純度100%であった。形態IIは、乾燥オーブン中で形態Iを155℃にて3日加熱し、そして室温まで冷却することによって製造された。この形態II試料は、HPLCによって試験した結果、化学的劣化が全くなかった。
【0058】
方法:懸濁結晶化
特定の温度での形態IとIIの間の相対的安定性関係を、2つの多形の混合物を、関心のある温度で平衡状態にした密閉ガラス容器内でトルエン中に懸濁させることによって求めた。一方の多形の過剰固体を最初に添加して、選択された温度でガラス容器中のトルエンを飽和させた。他方の多形を少なくとも30分後に添加した。この懸濁液を少なくとも3日、連続して撹拌した。この懸濁液の一部を抜取って即座に濾過した。この濾過した固体をPXRDを用いて解析し、その温度での平衡固体を同定した。この平衡多形は、その特定温度で熱力学的により安定なものである。この実験を種々の温度で繰返すことにより、該Ttを求めた。
【0059】
結果
懸濁結晶化
懸濁結晶化からの結果は、71.5℃より高い温度では形態I及びIIの混合物が常に形態IIに変換されるが、71.0℃より低い温度では形態Iが平衡固体相であることを示した。従って、形態Iは71.0℃より低い温度で熱力学的により安定であり、形態IIは71.5℃より高い温度でより安定である。該Ttは、71.0と71.5℃の間になければならない。
【0060】
考察
このTtを求めることは、バルク薬物を製造し加工するために重要である。例えば、溶液からの結晶化を含む工程は、しばしばバルク薬物の最終的単離より前に含まれる。もし、望ましい形態に対するTtが結晶化温度より低いところにあれば、その薬物を一貫して純粋相として結晶化させることは困難であろう。別の例は、薬物を含有する顆粒の、湿式造粒後の乾燥である。もし、乾燥温度がTtより高い場合、その薬物の制御できない多形変化が起こり得る。次いでこれらの変化がそれに続く最終製品の加工、品質及び性能に影響を及ぼし得る。懸濁結晶化を用いて、この転移温度、Tt、は約71℃であると求められた。形態Iは約71℃より低い温度でより安定で、約71℃より高い温度で形態IIに対して準安定になるため、温度が71℃より低い限り、形態I結晶は、結晶化と貯蔵の通常の温度範囲を通して、その固体状態安定性を維持する筈である。
【0061】
本発明を詳細にそして種々の特定、且つ、好ましい態様及び技法を参照することによって記述した。しかしながら、本発明の範囲内に留まる多くの変更と修正が成し得ると理解されるべきである。
【図面の簡単な説明】
【0062】
【図1】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの、実験による粉末X線回折(PXRD)パターンである。
【図2】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの、計算による粉末X線回折(PXRD)パターンである。
【図3】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの粉末X線回折(PXRD)パターンである。
【図4】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態I及びIIの示差走査熱量測定(DSC)データである。
【技術分野】
【0001】
本発明は、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形及びそのような結晶の製造方法、相互変換及び単離に関する。
【背景技術】
【0002】
5−フェニルピラゾール−1−ベンゼンスルホンアミドは、関節炎及び炎症によるその他の症状を治療するために有用な、強力なCOX−2抑制作用を有する新規合成の化合物群である。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドは、以下の構造:
【化1】
を有する5−フェニルピラゾール−1−ベンゼンスルホンアミドである。
【0003】
式Iの化合物は、米国特許第5,466,823号(Tallyら)及び同第5,521,207号(Graneto)に開示されている。これらの特許は参照することにより本明細書に組入れられている。式Iの化合物の製造が開示されているものの、該明細書は、この試薬の結晶形の単離及び性質については全く触れていない。確固たる方法を確保する手段として多形挙動を同定し、濾過性などの化学的製造問題と同様に、錠剤化問題、錠剤破壊、懸濁液中での結晶成長とその結果起こるケーキング、懸濁液からの沈殿を避け、そして解析的再現性を確保する必要性がある(Analysis of Organic Polymorphs, A Review; Threlfall, T.L., Analyst, 120, 2435-2459を参照)。
【発明の開示】
【発明が解決しようとする課題】
【0004】
一つの態様において、本発明は、CuKα1X線(波長=1.5406オングストローム)を用いて得られる2θ角で、14.0、18.9、21.3、21.9及び25.7度(±0.1度)で表されるピークを含む粉末X線回折パターンを有する、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を供する。本発明の他の態様において、i)4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの懸濁液を、好適な溶媒中で約0℃から約60℃の温度で混合し;ii)その懸濁液を約0℃から約60℃の温度で24から72時間撹拌し;そして、iii)形態Iの結晶を集めること;を含む、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態IIをその結晶形態Iへ変換する方法が供される。本発明の他の態様において、該方法における溶媒は、水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン及びテトラヒドロフラン及びそれらの混合物から成るグループから選ばれる。
【0005】
本発明の追加の態様は、(i)該形態IIを、約10℃から約60℃の温度での形態IIの溶解度が2mg/mLより大きな水混和性溶媒中に溶解させ;(ii)水を添加することによって該化合物を沈殿させ;(iii)工程(ii)の懸濁液を約15℃から約45℃の温度で2から72時間撹拌し;そして(iv)形態Iの結晶を集めることを含む、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態IIを結晶形態Iへ変換する方法である。本発明の他の態様において、この方法に対する溶媒はエタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドから成るグループから選ばれる。
【0006】
更なる態様において、本発明は、結晶形態Iを有する式Iの化合物を含む薬剤組成物を供する。本発明の他の態様において、この薬剤組成物は薬学的に許容される担体又は賦形剤を更に含む。本発明の更に他の目的は、本発明の新規組成物を治療的に有効な量で使用することによって、動物における炎症状態を予防又は治療する方法を供することである。本発明の更なる目的は、動物における炎症状態を予防又は治療するための医薬品の製造における、本発明の新規組成物の使用を供することである。
【0007】
本発明の更なる目的は、以下の工程によって、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造することである。
【0008】
工程(a):4−フルオロアセトフェノン1モル当り1.0から1.6モル、又は好ましくは1.2から1.45モル、又はより好ましくは1.25から1.35モルのトリフルオロ酢酸アルキルを、4−フルオロアセトフェノン1モル当り1.0から1.5モル、又は好ましくは1.1から1.35モル、又はより好ましくは1.15から1.25モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンと混合して混合物とする。場合により、1380mlまでの、又は好ましくは900mlまでの、又はより好ましくは490mlまでの適切な溶媒、好ましくは2−プロパノールをこの工程において添加する。場合により、工程(a)の混合物を周囲温度より高く、還流温度までの温度で、又は好ましくは、40℃から70℃の温度で、又はより好ましくは50℃から60℃の温度で、1から24時間、又は好ましくは1から10時間、又はより好ましくは1から4時間、又はより好ましくは該反応が完了するまで加熱する。場合により、そのような加熱の後、該混合物を次いで−5℃から30℃の温度に、又はより好ましくは周囲温度に冷却する。
【0009】
工程(b):工程(a)の混合物を、4−フルオロアセトフェノン1モル当り415から1245ml、又は好ましくは650から870ml、又はより好ましくは725から795mlの水、それに加えて4−フルオロアセトフェノン1モル当り1.1から2.0モル、又は好ましくは1.2から1.7モル、又はより好ましくは1.3から1.5モルの濃塩酸、それに加えて4−フルオロアセトフェノン1モル当り0.8から1.2モル、又は好ましくは0.9から1.1モル、又はより好ましくは0.95から1.05モルの4−スルホンアミドフェニルヒドラジン・塩酸塩、及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が550から1660ml、又は好ましくは600から1000ml、又はより好ましくは650から750mlとなる量の好適な溶媒、好ましくは、C1−C6アルコール、又はより好ましくは2−プロパノールの組合せ物と組合せる、又は好ましくは添加する。場合により、工程(b)の混合物を、周囲温度より高く、還流温度までの温度で、又は好ましくは40℃から70℃の温度で、又は好ましくは50℃から70℃の温度で、1から24時間、又は好ましくは1から10時間、又はより好ましくは1から4時間、又はより好ましくは該反応が完了するまで加熱する。場合により、そのような加熱の後、該混合物を次に周囲温度から71.5℃の温度、又は好ましくは40℃から65℃の温度、又はより好ましくは50℃から60℃の温度で安定化させる。
【0010】
工程(c):次いで該混合物に、4−フルオロアセトフェノンに対してシーディング量、又は好ましくは0.0001質量%から50質量%、又はより好ましくは0.001質量%から5質量%、又はより好ましくは0.01質量%から0.5質量%の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加する。場合により、工程(c)の混合物を40℃から71.5℃より低い温度で、又は好ましくは50℃から71.5℃より低い温度で、又はより好ましくは50℃から60℃の温度で、1から10時間、又は好ましくは3から8時間、又はより好ましくは5から7時間加熱する。場合により、そのような加熱の後、次に該混合物を−5℃から30℃の温度に、又はより好ましくは周囲温度に冷却させる。場合により、この混合物を濾過し、好適な溶媒、好ましくはアルコール、より好ましくは2−プロパノール、又は水又はそれらの混合物で洗浄する。場合により、該溶媒は4−フルオロアセトフェノン1モル当り、300から1500ml、又は好ましくは600から1060ml、又はより好ましくは800から860mlの量であり、そして該水は4−フルオロアセトフェノン1モル当り、100から700ml、又は好ましくは285から545ml、又はより好ましくは385から445mlの量である。
【0011】
工程(d):次いで形態Iの結晶を集める。場合により、この形態Iの結晶を15℃から80℃の温度で、又は好ましくは30℃から65℃の温度で、より好ましくは50℃から60℃の温度で乾燥する。
【0012】
本発明の他の態様において、工程(a)の金属アルコキシドはナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれ、又は好ましくはナトリウムメトキシドである。
【0013】
これらの及びその他の目的は、当業者に容易に明確であろう。
【課題を解決するための手段】
【0014】
米国特許第5,466,823号及び同第5,521,207号に選択的COX−2抑制剤、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミド(式I):
【化2】
が述べられている。この記述には該化合物の合成が述べられているものの、得られた結晶形に関しては全く触れていない。これらの実施例に述べられているように、これらの特許中に示された手順に従うと、粉末X線回折(PXRD)パターンによって示される形態IIの結晶が得られる。本発明者らはこの結晶形が、もう一つの形態、形態IIに比べて周囲温度及び圧力下で不安定であることを見出した。これらの形態は、それらのPXRD(粉末X線回折)パターンによって区別できる。
【0015】
定義
本出願において言及される「水混和性」とは、あらゆる割合で水に混合又は溶解され得ることを意味する。
【0016】
本出願において言及される「無水結晶性」とは、実質的量の水を含有しない結晶のことである。この水含有量は、例えば、カールフィッシャー滴定を含む従来公知の方法によって求めることができる。好ましくは、無水結晶形は最大で約1質量%、より好ましくは最大で約0.5質量%、そして最も好ましくは最大で約0.1質量%の水を含有する。
【0017】
本出願において言及される、バルク薬物安定性試験における「安定」とは、バルク薬物の少なくとも約90質量%、好ましくは少なくとも約95質量%、より好ましくは少なくとも約99質量%が、指定条件下で指定時間の貯蔵後に変化しないことを意味する。
「DSC」とは、示差走査熱量測定を意味する。
【0018】
単独で又は、「トリフルオロアセテート」などの他の用語と共に使用される用語アルキルは、別途特定されない限り、1から4個の炭素原子、又は好ましくは2個の炭素原子を有する直鎖状又は分枝鎖状のものを包含する。非限定的例としては、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、第三級−ブチル、イソブチル及び第二級−ブチルなどの基が挙げられる。
【0019】
「金属アルコキシド」は、アルコールの金属塩である。一般に、該金属アルコキシドは金属としてナトリウム、リチウム又はカリウムを含有する。本発明において、それらはアルコール溶液の形で使用される。一般に、該アルコールは該アルコキシドのアルコールに対応する。非限定的例としては、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物が挙げられる。
【0020】
シーディングは、混合物、溶液又は懸濁液から多くの結晶の形成を誘起するために、一つ又はそれ以上の結晶を使用する技法である。シーディング量は、混合物、溶液又は懸濁液に添加された際に、化合物の所望の形態を形成させ得るような材料の量である。理論上はこの量は非常に少量であり得るが、実際には、より多くの量が使用される。この量は、合理的に取扱いできそして化合物の所望の形態を形成させ得るために十分である、いかなる量でもよい。非限定的例としては、参照化合物に対して0.0001質量%から50質量%のシーディング化合物がシーディング量として使用できる。
【0021】
温度に関して使用される用語「C」とは、℃又は摂氏を意味する。
「周囲温度」とは、対象物の周りの大気温度である。それは、室内の温度であり、一般に15から25℃である。
【0022】
還流は、長い時間に亘って反応物にエネルギーを与えるために化学において使用される技法である。この技法のために、頂部のみが開いた容器中に液状反応混合物が入れられる。発せられるあらゆる蒸気が冷却されて液体に戻り、この反応容器に落下して戻されるよう、この容器は垂直の凝縮器に連結される。次いで、この容器は反応過程中、激しく加熱される。この技法の利点は、更に溶媒を添加する必要なしに、又は反応容器が沸騰して乾燥する心配なしに長期間放置できることである。加えて、ある溶媒が常に一定の温度で沸騰するため、その反応はその同一温度で進行することになる。溶媒が変われば異なる温度で沸騰するため、溶媒を注意深く選択することによって反応温度が制御できる。本明細書で使用される「還流温度」とは、本還流技法の間、特定の溶媒が沸騰する温度のことである。例えば、海抜ゼロにおいて、2−プロパノールは82℃の沸点を有し、メタノールは65℃の沸点を有する。
【0023】
工程(a)に関して、「反応完了」は、以下のHPLC法を用いて、混合物中に残存する4−フルオロアセトフェノンの量が、4−フルオロアセトフェノンの投入量に対して2質量%以下であることを確認することによって求められる。
カラム: Discover HS F5、5μ、250×4.6mm Supelco Cat #567517-U
移動相(勾配):
【表1】
流速: 1ml/分
注入量: 20μl
検出: 247nm
【0024】
工程(b)に関して、「反応完了」は、以下のHPLC方を用いて、形成される4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの量が90%を越えることを確認することによって求められる。
カラム: Zorbax Eclipse XDBフェニル、3.5μm、150×4.6mm
移動相: メタノール/25mM燐酸(55/45体積%)
pH2.5(NaOHでpH調整)
ラン時間: 30分
カラム温度:35℃
流速: 1.0ml/分
注入量: 15μl
検出: 254nmの紫外線
試料濃度: 0.4mg/ml
【0025】
「治療的に有効な量」とは、症状(例えば、炎症症状)を予防するであろう、又は治療される病気の一つ又はそれ以上の症状がある程度緩和できるような、投与される化合物量のことである。本発明における使用に適する薬剤組成物としては、意図する目的を達成するために十分な量で有効成分を含有する組成物が挙げられる。治療的に有効な量を決定することは、特に本明細書に提供される詳細な開示を参照すれば、十分当業者の能力範囲内にある。
【0026】
本発明の化合物の適切な受容体である動物としては、ヒト又は他の哺乳類又は動物、例えば、牛、羊、豚、馬、山羊及び家禽(例えば、鶏、七面鳥、アヒル及びガチョウ)含む家畜動物及びその他の鳥、及び犬、猫などのペット動物及び外来及び/又は動物園の動物が挙げられるが、これらに限定されるものではない。げっ歯動物及び非げっ歯動物の両者の治療が意図される。
【0027】
実際には、投与される化合物の量は、動物の体重1kg当り約0.001から100mgの範囲であり、そのような総投与量は一度に又は小分けして与えられる。それは、単独で、又は、一つ又はそれ以上の他の薬と組合せて投与されてよい。一般に、それは、一つ又はそれ以上の他の薬学的に許容される賦形剤と共に製剤として投与されることになる。本明細書で用語「賦形剤」は、本発明の化合物(類)以外のあらゆる成分を記述するために使用される。賦形剤の選択は、特定の投与方式、溶解度及び安定性に及ぼす賦形剤の影響及び用量形態の性質などの因子にかなりの程度まで依存するであろう。
【0028】
本発明の化合物の送達に好適な薬剤組成物及びそれらの製造方法は、当業者に容易に明らかであろう。そのような組成物及びそれらの製造方法は、例えば、Remington's Pharmaceutical Sciences, 19th Edition(Mack Publishing Company, 1995)において見出すことができる。
【0029】
動物における消炎剤としての使用に対して、本発明の組成物は経口又は注射によって投与してよい。本発明の組成物を乾燥した、固体の単位剤形にて投与することが望ましい場合には、通常、所望の量の有効化合物を含有するカプセル、丸薬又は錠剤が採用される。これらの剤形は、有効成分を、澱粉、乳糖、タルク、ステアリン酸マグネシウム、植物ゴム質などの細かく粉砕された好適な希釈剤と良く均一に混合することによって製造される。そのようなユニット用量薬剤は、治療されるホスト動物種、炎症の重篤度と種類及び該ホストの体重などの因子に依存して、その総重量及び該消炎剤の含有量を大幅に変更してもよい。錠剤の処方は、Pharmaceutical Dosage Forms: Tablets, Vol.1, H. Lieberman and L. Lachman(Marcel Dekker, New York, 1980)に論じられている。
【0030】
或いは又、本発明の消炎組成物は、非経口的に、例えば、胃内、筋肉内又は皮下注射によって動物に投与されてもよく、その際には有効成分が液体担体賦形剤中に溶解又は分散される。非経口投与に対しては、有効材料が、許容できる賦形剤、好ましくは落花生油、綿実油などの植物油種と好適に混合される。ソルケタール、プロピレングリコール、グリセロールホルマールを用いる有機製剤及び含水非経口製剤などの他の非経口賦形剤も、種々の割合で組合されてしばしば使用される。有効化合物又は化合物類は、投与のために、非経口製剤中に溶解又は懸濁され、そのような製剤は一般に、0.005から5質量%の有効化合物を含有する。
【発明を実施するための最良の形態】
【0031】
本発明の以下の記述において、本発明が実行され得る特定の態様が述べられている。これらの態様は、当業者が本発明を実行し得るように、十分詳細に述べられている。他の態様も利用でき、そして本発明の範囲から逸脱することなく論理的な他の変更がなされ得る。従って、以下の詳細な記述は制限する意味で捉えられるべきではなく、本発明の範囲は、添付の請求範囲及びそのような請求範囲が資格を与える同等物の全範囲によってのみ定義される。
【0032】
形態IIは、米国特許第5,466,823号に述べられた手順から得られる結晶形である。結晶形態Iを得る方法は、以下の非制限的記述及び実施例によって説明される。
【0033】
一つの方法において、結晶形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドは、約0℃から約60℃の温度において、薬物の濃度がその飽和点よりも高い状態で好適な溶媒中に懸濁され混合されてよい。好適な溶媒は、その中でのその薬物の溶解度が約1mg/mLより高く、そして約800mg/mLより低いものである。好適な溶媒の例としては、水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン、及びテトラヒドロフラン、及びこれらの混合物が挙げられるが、これらに限定されるものではない。該懸濁液を約0℃から約60℃の温度で、24から72時間撹拌する。形態I結晶は濾過によって集められる。
【0034】
別の手順において、結晶形態Iは、その中での化合物の溶解度が2mg/mLより高い好適な水混和性溶媒中の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの溶液から、約10℃から約60℃の温度で水を添加することによって沈殿させて製造できる。形態IIから形態Iを製造するための好適な溶媒の例としては、エタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドが挙げられる。水を使用するこの最初の沈殿に続いて、この懸濁液を、約15℃から約45℃の温度で24から72時間撹拌して、形態Iの結晶を濾過によって集める。
【0035】
製造1.形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの製造
以下の手順は、出発物質として4−クロロアセトフェノンの代わりに4−フルオロアセトフェノンを使用するという重大ではない変更以外は、米国特許第5,521,207号の実施例1の手順と本質的に同一である。
【0036】
工程1:4,4,4−トリフルオロ−1−[4−(フルオロ)フェニル]−ブタン−1,3−ジオンの製造
トリフルオロ酢酸エチル(2.35g、1.66mmol)を50mLの丸底フラスコ中に入れてメチル第三級ブチルエーテル(7.5mL)中に溶解した。この撹拌溶液に、25質量%のナトリウムメトキシド(4.0mL、17.7mmol)を添加ロートを介して2分かけて添加した。次に、4−フルオロアセトフェノン(2.1g、15mmol)をメチル第三級ブチルエーテル(2mL)中に溶解させ、反応溶液に5分かけて滴下した。一晩(15.75時間)撹拌した後、3N塩酸(7.0mL)を添加した。この有機層を集め、塩水(7.5mL)で洗浄、MgSO4上で乾燥、濾過、減圧濃縮して、3.2gの淡橙色固体を得た。この固体をイソオクタンから再結晶化させて、2.05gの当ジオンを得た。
【0037】
工程2:4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの製造
4−スルホンアミドフェニルヒドラジン・塩酸塩(982mg、4.4mmol)を4,4,4−トリフルオロ−1−[4−(フルオロ)フェニル]−ブタン−1,3−ジオン(0.936mg、4.0mmol)の撹拌エタノール(50mL)溶液に添加した。この反応溶液を加熱還流しながら20時間撹拌した。室温まで冷却後、この反応混合物を減圧濃縮した。この残留物を酢酸エチル中に採取して水及び塩水で洗浄、MgSO4上で乾燥、濾過、減圧濃縮して、茶色固体を得、それを酢酸エチル及びイソオクタンから再結晶化させて、0.8gの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを得た。
1HNMR(CDCl3):δ:7.92、7.47、7.22、7.09、6.76、4.92;
MS: m/e ES−(M−H):384;
m.p.:167〜171℃。
【0038】
製造2.結晶形態IIを単離しない、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの製造
以下の手順は、結晶形態IIを単離しないで、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造する一般的方法を記述する。
【0039】
パートA
4−フルオロアセトフェノン(1モル当量)を、トリフルオロ酢酸アルキル(1.0から1.6モル当量)、金属アルコキシド(1.0から1.5モル当量)及び、場合により好適な溶媒(4−フルオロアセトフェノン1kg当り10Lまで)の混合物と組合せる。この混合物を還流(反応を完了させるため)状態までの温度に加熱してもよい。このパートAに対する「好適な溶媒」は、直鎖状、分枝鎖状及び環状アルコールを含むC1−C6飽和脂肪族アルコールである。好適な溶媒の非制限的な例としては、メタノール、エタノール、イソプロパノール等のアルコール及びそれらの混合物が挙げられる。
【0040】
パートB
パートAからの混合物を、4−スルホンアミドフェニルヒドラジン・塩酸塩(4−フルオロアセトフェノンに対して0.8から1.2モル当量)、及び水(4−フルオロアセトフェノン1kg当り3から9L)中への塩酸(4−フルオロアセトフェノンに対して1.1から2.0モル当量)溶液の混合物と組合せる。必要ならば、パートB中に存在する溶媒の総量が4−フルオロアセトフェノン1kg当り4から12Lの範囲内になるよう、好適な溶媒を添加してもよい。この反応混合物を還流(反応を完了させるため)状態までの温度に加熱してもよい。反応期間の後、この混合物を71.5℃より低い温度にて、形態I結晶(4−フルオロアセトフェノンに対して0.0001質量%以上)でシーディングする。この固体を濾過によって単離し、そして好適な溶媒を用いて洗浄及び/又は再度スラリー化してもよい。この生成物を80℃までの温度で乾燥してもよい。このパートBに対する「好適な溶媒」は、直鎖状、分枝鎖状及び環状アルコールを含むC1−C6飽和脂肪族アルコール、及び水及びそれらの混合物である。
【0041】
以下の手順は、結晶形態IIを単離することなく、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの結晶形態Iを製造する好ましい方法を述べる。2−プロパノール(70ml)及びトリフルオロ酢酸エチル(27.15g、0.191モル)を第一の容器に添加し、続いて配管を2−プロパノール(10ml)で洗浄する。次に、25%のナトリウムメトキシド(37.5g、0.174モル)のメタノール溶液をこの容器に添加し、続いて配管を2−プロパノール(10ml)で洗浄する。次に、4−フルオロアセトフェノン(20g、0.145モル)を添加し、続いて配管を2−プロパノール(10ml)で洗浄する。容器の内容物を55℃に加熱し、その温度に2時間保持し、その後で周囲温度まで冷却する。水(110ml)、濃塩酸(20.0g、0.203モル)及び4−スルホンアミドフェニルヒドラジン・塩酸塩(32.4g、0.145モル)を第二の容器に添加し、続いて配管を水(10ml)で洗浄する。第一容器の内容物を第二容器に添加し、続いて配管を2−プロパノール(60ml)で洗浄する。一体にした内容物を70℃で2時間加熱し、次いで55℃に冷却して、10mg(4−フルオロアセトフェノンに対して0.05質量%)の形態Iの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドでシーディングする。本内容物を55℃で6時間保持した後に、周囲温度まで冷却して濾過する。この生成物を50%の2−プロパノール水溶液(120ml)及び水(60ml)で洗浄し、次いで55℃にて減圧乾燥して、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの所望の形態Iの多形体(44.8g、80%)を得る。
【0042】
本発明を以下の非制限的実施例によって今まで以上に詳細に述べる。
【実施例】
【0043】
〔実施例1〕
20mLのガラスビン中で、脱イオン水1mLをイソプロパノール1mLと良く混合した。形態IIの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミド(258.6mg)をこのビンに添加した。マグネティック撹拌棒を導入して、このビンの蓋をきつく閉めた。この懸濁液を約200rpmで30分間撹拌した。ペースト状の濃い懸濁液が観察された。撹拌を止めてこのビンを3日間放置した。この懸濁液の一部を取出して吸収紙上で乾燥した。この固体の粉末X線回折パターンを図1に示す。いくつかの単離された結晶の、高温顕微鏡下で観察された融点は148から152℃であった。しかしながら、形態Iから形態IIへの140から155℃の間での転移のため、この範囲は再現性よく観察されなかった。
【0044】
〔実施例2〕
380gの形態IIを2mLのエタノール中に懸濁させた。この化合物の一部は溶解したが、いくらかの過剰の固体化合物がこの溶液中に懸濁した。得られた懸濁液を、約20℃から約30℃の温度にて2週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0045】
〔実施例3〕
1.5gの形態IIを2mLのアセトニトリルに添加して懸濁液とした。この懸濁液を、約20℃から約30℃の温度にて2週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0046】
〔実施例4〕
0.5gの形態IIを2mLのメタノール+水混合液(1:1、体積比)に添加して懸濁液とした。この懸濁液を、約20℃から約30℃の温度にて4週間、マグネティック撹拌棒で撹拌した。この期間の最後に、この固体を濾過し、PXRDによって形態Iであると同定した。
【0047】
〔実施例5〕
81.5kgの4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを81.5kgのエタノール(6倍量の体積)中に40℃にて完全に溶解し、まだ熱いうちに濾過した。154Lの水(9倍量の体積)をこの溶液に1時間かけて、温度を40℃に保持したまま添加し、続いて20℃まで冷却した。このスラリーを20℃にて24時間撹拌し、濾過して、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドを形態Iとして得た。
【0048】
〔実施例6〕
粉末X線回折
Scintag X2最新回折システム(Scintag DMS/NT 1.30a及びマイクロソフトWindows(登録商標) NT 4.0ソフトウエアによって制御される)を使用して粉末X線回折を行った。このシステムは、1.5406オングストロームのCuKα1を放射するための銅X線源(45kV及び40mA)及び固体状のペルチエ冷却検出器を使用する。ビーム絞りは2mm及び4mmの管分岐及び散乱防止スリット、0.5mm及び0.2mm幅の検出器散乱防止及び受信スリットを使用して制御された。各ステップ当り1秒のカウント時間で、0.03度/ステップのステップ走査を用いて2から35度(2θ)からデータが集められた。本実験には、Scintagの丸い頂部に取付けた、直径12mmのキャビティを有するアルミニウム製試料容器が利用された。粉末はこの容器内に充填され、試料表面と試料容器表面の間の共平面性が確保されるよう、ガラススライドによって静かに押付けられた。
【0049】
熟練した結晶学者によって理解されるであろうように、下表内に示される種々のピークの相対強度は、例えば、X線中での結晶の配向効果、又は解析される材料の純度又は試料の結晶化度などの多くの因子があるために変化し得る。これらのピーク位置も、試料の高さにおける変化に対して移動し得るが、実質的にピーク位置は与えられた表中に定義された如くに留まるであろう。
【0050】
熟練した結晶学者は、異なる波長を用いて測定すれば、Braggの式、nλ=2dsinθに従う移動が異なる結果になることも理解するであろう。
代替波長を使用することによって発生するそのような更なるPXRDパターンは、本発明の結晶性材料のPXRDパターンの代替表示であり、そしてそのようなものは、本発明の範囲内にあるものと考えられる。
【0051】
形態Iに対して、角度2θ、間隔d及び相対強度が、Accelrys Materials Studio(登録商標)「バージョン2.2」の「反射粉末回折」モジュールを用いて、単結晶構造から計算された。各ケースにおける適切なシミュレーションパラメータは:
波長=1.540562オングストローム(CuKα);
偏光因子=0.5;
擬Voigtプロファイル(U=0.01、V=-0.001、W=0.002)
であった。
【0052】
形態Iに対する実験及び計算によるPXRDパターン、及び形態IIに対する実験によるPXRDパターンを、図1、2、及び3に各々示す。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの主要PXRDピークを、表1に示す。4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの主要ピークの計算によるPXRDパターンを、表2に示す。形態Iは、2θ角度が14.0、18.9、21.3、21.9及び25.7度±0.1度に独特の回折ピークを示す。
【0053】
【表2】
【0054】
【表3】
【0055】
〔実施例7〕
示差走査熱量測定(DSC)
示差走査熱量測定(DSC)データは、DSC熱量測定機(TA Instruments 2920)を用いて得た。粉末(1から5mg)をアルミニウム製皿に充填した。アルミニウム製蓋をその皿の頂部に置いて丸めた。丸めた皿を試料セル中に、参照としての空の皿と共に置いた。特に断らない限り、温度を30℃から250℃へ10℃/分の速度で上昇させた。この熱セルを乾燥窒素で50mL/分でパージした。データ採取にはNT用TA Instruments Thermal Solutions(登録商標)(バージョン1.3L)を使用し、そしてデータ解析にはNT用Universal Analysis(登録商標)(バージョン2.4F)を使用した。図4に見られるように、形態IのDSCは、140℃から155℃で形態Iから形態IIへの吸熱相転移を示す。
【0056】
〔実施例8〕
形態IとIIの間の熱力学的関係
この実施例においては、形態Iと形態IIの間の相転移温度について述べる。形態Iと形態IIは互変性であり、それは一つの多形が転移温度、Tt、より低い温度で安定であるが、もう一方の多形は転移温度、Tt、より高い温度で安定であることを意味する。これら2つの固相は転移温度にて等しい自由エネルギーを有する。熱力学的安定関係の知識は、形態Iを製造するための好適な結晶化条件の選択に対して、及び製剤開発及び商業製造に対して必要である。Ttを求めることは、バルク薬物の製造と加工に対して重要である。
【0057】
材料及び方法
材料
本検討において使用された形態I試料は、HPLCによって試験した結果、純度100%であった。形態IIは、乾燥オーブン中で形態Iを155℃にて3日加熱し、そして室温まで冷却することによって製造された。この形態II試料は、HPLCによって試験した結果、化学的劣化が全くなかった。
【0058】
方法:懸濁結晶化
特定の温度での形態IとIIの間の相対的安定性関係を、2つの多形の混合物を、関心のある温度で平衡状態にした密閉ガラス容器内でトルエン中に懸濁させることによって求めた。一方の多形の過剰固体を最初に添加して、選択された温度でガラス容器中のトルエンを飽和させた。他方の多形を少なくとも30分後に添加した。この懸濁液を少なくとも3日、連続して撹拌した。この懸濁液の一部を抜取って即座に濾過した。この濾過した固体をPXRDを用いて解析し、その温度での平衡固体を同定した。この平衡多形は、その特定温度で熱力学的により安定なものである。この実験を種々の温度で繰返すことにより、該Ttを求めた。
【0059】
結果
懸濁結晶化
懸濁結晶化からの結果は、71.5℃より高い温度では形態I及びIIの混合物が常に形態IIに変換されるが、71.0℃より低い温度では形態Iが平衡固体相であることを示した。従って、形態Iは71.0℃より低い温度で熱力学的により安定であり、形態IIは71.5℃より高い温度でより安定である。該Ttは、71.0と71.5℃の間になければならない。
【0060】
考察
このTtを求めることは、バルク薬物を製造し加工するために重要である。例えば、溶液からの結晶化を含む工程は、しばしばバルク薬物の最終的単離より前に含まれる。もし、望ましい形態に対するTtが結晶化温度より低いところにあれば、その薬物を一貫して純粋相として結晶化させることは困難であろう。別の例は、薬物を含有する顆粒の、湿式造粒後の乾燥である。もし、乾燥温度がTtより高い場合、その薬物の制御できない多形変化が起こり得る。次いでこれらの変化がそれに続く最終製品の加工、品質及び性能に影響を及ぼし得る。懸濁結晶化を用いて、この転移温度、Tt、は約71℃であると求められた。形態Iは約71℃より低い温度でより安定で、約71℃より高い温度で形態IIに対して準安定になるため、温度が71℃より低い限り、形態I結晶は、結晶化と貯蔵の通常の温度範囲を通して、その固体状態安定性を維持する筈である。
【0061】
本発明を詳細にそして種々の特定、且つ、好ましい態様及び技法を参照することによって記述した。しかしながら、本発明の範囲内に留まる多くの変更と修正が成し得ると理解されるべきである。
【図面の簡単な説明】
【0062】
【図1】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの、実験による粉末X線回折(PXRD)パターンである。
【図2】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの、計算による粉末X線回折(PXRD)パターンである。
【図3】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの粉末X線回折(PXRD)パターンである。
【図4】4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態I及びIIの示差走査熱量測定(DSC)データである。
【特許請求の範囲】
【請求項1】
CuKα1X線(波長=1.5406オングストローム)を用いて得られる2θ角で14.0、18.9、21.3、21.9及び25.7度(±0.1度)で表されるピークを含む粉末X線回折パターンを有する、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶形。
【請求項2】
4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの結晶形を、請求項1に記載の結晶形へ変換する方法であって、
(a)4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの懸濁液を好適な溶媒中で約0℃〜約60℃の温度で混合し;
(b)その懸濁液を約0℃〜約60℃の温度で24〜72時間撹拌し;そして
(c)形態Iの結晶を集める;
ことを含む、上記の方法。
【請求項3】
溶媒が水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン及びテトラヒドロフラン及びそれらの混合物から成るグループから選ばれる、請求項2に記載の方法。
【請求項4】
4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの結晶形を、請求項1に記載の結晶形へ変換する方法であって、
(a)該形態IIを、約10℃〜約60℃の温度での形態IIの溶解度が2mg/mLより大きな水混和性溶媒中で溶解し;
(b)水を添加することによって該化合物を沈殿させ;
(c)工程(b)の懸濁液を約15℃〜約45℃の温度で2〜72時間撹拌し;そして
(d)形態Iの結晶を集める;
ことを含む、上記の方法。
【請求項5】
溶媒がエタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドから成るグループから選ばれる、請求項4に記載の方法。
【請求項6】
請求項1に記載の結晶形を含む薬剤組成物。
【請求項7】
薬学的に許容できる担体又は賦形剤を更に含む、請求項6に記載の組成物。
【請求項8】
請求項1に記載の結晶形を治療的に有効な量で動物に投与することによって、動物における炎症状態を予防又は治療する方法。
【請求項9】
動物が牛、羊、山羊、馬、豚、鳥、猫、犬及びヒトから成るグループから選ばれる、請求項8に記載の方法。
【請求項10】
動物における炎症状態を予防又は治療するための医薬品の製造における、請求項6に記載の組成物の使用。
【請求項11】
請求項1に記載の結晶形を生成する方法であって、
(a)4−フルオロアセトフェノン1モル当り1.0〜1.6モルのトリフルオロ酢酸アルキル、4−フルオロアセトフェノン1モル当り1.0〜1.5モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンを混合して混合物とし;
(b)工程(a)からの該混合物を、4−フルオロアセトフェノン1モル当り415〜1245mlの水、4−フルオロアセトフェノン1モル当り1.1〜2.0モルの濃塩酸、4−フルオロアセトフェノン1モル当り0.8〜1.2モルの4−スルホンアミドフェニルヒドラジン・塩酸塩及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が550〜1660mlとなる量の好適な溶媒の組合せ物と組合せ;
(c)該混合物に、4−フルオロアセトフェノンに対してシーディング量の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加し; そして
(d)形態Iの結晶を集める;
工程を含む、上記の方法。
【請求項12】
工程(a)において、4−フルオロアセトフェノン1モル当り1380mlまでの好適な溶媒を該混合物に添加することを更に含む、請求項11に記載の方法。
【請求項13】
アルコールが2−プロパノールである、請求項12に記載の方法。
【請求項14】
金属アルコキシドがナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれる、請求項11に記載の方法。
【請求項15】
金属アルコキシドがナトリウムメトキシドである、請求項14に記載の方法。
【請求項16】
工程(a)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項17】
加熱工程に続き、該混合物を−5℃〜30℃に冷却することを含む工程を更に含む、請求項16に記載の方法。
【請求項18】
工程(b)が、該混合物を水、塩酸及び4−スルホンアミドフェニルヒドラジン・塩酸塩の組合せ物に添加することを含む、請求項11に記載の方法。
【請求項19】
工程(b)の該溶媒がアルコールである、請求項11に記載の方法。
【請求項20】
アルコールが2−プロパノールである、請求項19に記載の方法。
【請求項21】
工程(b)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項22】
加熱工程に続き、該混合物を周囲温度から71.5℃の温度で安定化させることを含む工程を更に含む、請求項21に記載の方法。
【請求項23】
シーディング量が0.0001質量%〜50質量%である、請求項11に記載の方法。
【請求項24】
シーディング量が0.001質量%〜5質量%である、請求項23に記載の方法。
【請求項25】
シーディング量が0.01質量%〜0.5質量%である、請求項23に記載の方法。
【請求項26】
工程(c)に続き、該混合物を40℃〜71.5℃より低い温度で、1〜10時間加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項27】
加熱工程に続き、該混合物を−5℃〜30℃に冷却することを含む工程を更に含む、請求項26に記載の方法。
【請求項28】
工程(c)に続き、該混合物を濾過し、そして該形態Iの結晶を好適な溶媒又は水、又はそれらの混合物で洗浄することを含む工程を更に含む、請求項11に記載の方法。
【請求項29】
溶媒がアルコールである、請求項28に記載の方法。
【請求項30】
アルコールが2−プロパノールである、請求項29に記載の方法。
【請求項31】
工程(d)に続き、該形態Iの結晶を15℃〜80℃の温度で乾燥することを含む工程を更に含む、請求項11に記載の方法。
【請求項32】
請求項1に記載の結晶形を生成する方法であって、
(a)4−フルオロアセトフェノン1モル当り1.2〜1.45モルのトリフルオロ酢酸アルキル、4−フルオロアセトフェノン1モル当り1.1〜1.35モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンを混合して混合物とし;
(b)工程(a)からの該混合物を、4−フルオロアセトフェノン1モル当り650〜870mlの水、4−フルオロアセトフェノン1モル当り1.2〜1.7モルの濃塩酸、4−フルオロアセトフェノン1モル当り0.9〜1.1モルの4−スルホンアミドフェニルヒドラジン・塩酸塩及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が600〜1000mlとなる量の好適な溶媒の組合せ物と組合せ;
(c)該混合物に、4−フルオロアセトフェノンに対してシーディング量の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加し; そして
(d)該形態Iの結晶を集める;
工程を含む、上記の方法。
【請求項33】
工程(a)において、4−フルオロアセトフェノン1モル当り900mlまでの好適な溶媒を該混合物に添加することを更に含む、請求項32に記載の方法。
【請求項34】
アルコールが2−プロパノールである、請求項33に記載の方法。
【請求項35】
金属アルコキシドが、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれる、請求項32に記載の方法。
【請求項36】
金属アルコキシドがナトリウムメトキシドである、請求項35に記載の方法。
【請求項37】
工程(a)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項38】
加熱工程に続き、該混合物を周囲温度に冷却することを含む工程を更に含む、請求項37に記載の方法。
【請求項39】
工程(b)が、該混合物を水、塩酸及び4−スルホンアミドフェニルヒドラジン・塩酸塩の組合せ物に添加することを含む、請求項32に記載の方法。
【請求項40】
工程(b)の該溶媒がアルコールである、請求項32に記載の方法。
【請求項41】
アルコールが2−プロパノールである、請求項40に記載の方法。
【請求項42】
工程(b)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項43】
加熱工程に続き、該混合物を40℃〜65℃の温度で安定化させることを含む工程を更に含む、請求項42に記載の方法。
【請求項44】
シーディング量が0.0001質量%〜50質量%である、請求項33に記載の方法。
【請求項45】
シーディング量が0.001質量%〜5質量%である、請求項44に記載の方法。
【請求項46】
シーディング量が0.01質量%〜0.5質量%である、請求項44に記載の方法。
【請求項47】
工程(c)に続き、該混合物を50℃〜71.5℃より低い温度で、3〜8時間加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項48】
加熱工程に続き、該混合物を周囲温度に冷却することを含む工程を更に含む、請求項47に記載の方法。
【請求項49】
工程(c)に続き、該混合物を濾過し、そして該形態Iの結晶を好適な溶媒又は水、又はそれらの混合物で洗浄することを含む工程を更に含む、請求項32に記載の方法。
【請求項50】
溶媒がアルコールである、請求項49に記載の方法。
【請求項51】
アルコールが2−プロパノールである、請求項50に記載の方法。
【請求項52】
工程(d)に続き、該形態Iの結晶を30℃〜65℃の温度で乾燥することを含む工程を更に含む、請求項32に記載の方法。
【請求項1】
CuKα1X線(波長=1.5406オングストローム)を用いて得られる2θ角で14.0、18.9、21.3、21.9及び25.7度(±0.1度)で表されるピークを含む粉末X線回折パターンを有する、4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶形。
【請求項2】
4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの結晶形を、請求項1に記載の結晶形へ変換する方法であって、
(a)4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの懸濁液を好適な溶媒中で約0℃〜約60℃の温度で混合し;
(b)その懸濁液を約0℃〜約60℃の温度で24〜72時間撹拌し;そして
(c)形態Iの結晶を集める;
ことを含む、上記の方法。
【請求項3】
溶媒が水、メタノール、エタノール、イソプロパノール、アセトン、アセトニトリル、塩化メチレン、トルエン及びテトラヒドロフラン及びそれらの混合物から成るグループから選ばれる、請求項2に記載の方法。
【請求項4】
4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態IIの結晶形を、請求項1に記載の結晶形へ変換する方法であって、
(a)該形態IIを、約10℃〜約60℃の温度での形態IIの溶解度が2mg/mLより大きな水混和性溶媒中で溶解し;
(b)水を添加することによって該化合物を沈殿させ;
(c)工程(b)の懸濁液を約15℃〜約45℃の温度で2〜72時間撹拌し;そして
(d)形態Iの結晶を集める;
ことを含む、上記の方法。
【請求項5】
溶媒がエタノール、アセトン、アセトニトリル、テトラヒドロフラン、ジオキサン及びジメチルホルムアミドから成るグループから選ばれる、請求項4に記載の方法。
【請求項6】
請求項1に記載の結晶形を含む薬剤組成物。
【請求項7】
薬学的に許容できる担体又は賦形剤を更に含む、請求項6に記載の組成物。
【請求項8】
請求項1に記載の結晶形を治療的に有効な量で動物に投与することによって、動物における炎症状態を予防又は治療する方法。
【請求項9】
動物が牛、羊、山羊、馬、豚、鳥、猫、犬及びヒトから成るグループから選ばれる、請求項8に記載の方法。
【請求項10】
動物における炎症状態を予防又は治療するための医薬品の製造における、請求項6に記載の組成物の使用。
【請求項11】
請求項1に記載の結晶形を生成する方法であって、
(a)4−フルオロアセトフェノン1モル当り1.0〜1.6モルのトリフルオロ酢酸アルキル、4−フルオロアセトフェノン1モル当り1.0〜1.5モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンを混合して混合物とし;
(b)工程(a)からの該混合物を、4−フルオロアセトフェノン1モル当り415〜1245mlの水、4−フルオロアセトフェノン1モル当り1.1〜2.0モルの濃塩酸、4−フルオロアセトフェノン1モル当り0.8〜1.2モルの4−スルホンアミドフェニルヒドラジン・塩酸塩及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が550〜1660mlとなる量の好適な溶媒の組合せ物と組合せ;
(c)該混合物に、4−フルオロアセトフェノンに対してシーディング量の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加し; そして
(d)形態Iの結晶を集める;
工程を含む、上記の方法。
【請求項12】
工程(a)において、4−フルオロアセトフェノン1モル当り1380mlまでの好適な溶媒を該混合物に添加することを更に含む、請求項11に記載の方法。
【請求項13】
アルコールが2−プロパノールである、請求項12に記載の方法。
【請求項14】
金属アルコキシドがナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれる、請求項11に記載の方法。
【請求項15】
金属アルコキシドがナトリウムメトキシドである、請求項14に記載の方法。
【請求項16】
工程(a)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項17】
加熱工程に続き、該混合物を−5℃〜30℃に冷却することを含む工程を更に含む、請求項16に記載の方法。
【請求項18】
工程(b)が、該混合物を水、塩酸及び4−スルホンアミドフェニルヒドラジン・塩酸塩の組合せ物に添加することを含む、請求項11に記載の方法。
【請求項19】
工程(b)の該溶媒がアルコールである、請求項11に記載の方法。
【請求項20】
アルコールが2−プロパノールである、請求項19に記載の方法。
【請求項21】
工程(b)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項22】
加熱工程に続き、該混合物を周囲温度から71.5℃の温度で安定化させることを含む工程を更に含む、請求項21に記載の方法。
【請求項23】
シーディング量が0.0001質量%〜50質量%である、請求項11に記載の方法。
【請求項24】
シーディング量が0.001質量%〜5質量%である、請求項23に記載の方法。
【請求項25】
シーディング量が0.01質量%〜0.5質量%である、請求項23に記載の方法。
【請求項26】
工程(c)に続き、該混合物を40℃〜71.5℃より低い温度で、1〜10時間加熱することを含む工程を更に含む、請求項11に記載の方法。
【請求項27】
加熱工程に続き、該混合物を−5℃〜30℃に冷却することを含む工程を更に含む、請求項26に記載の方法。
【請求項28】
工程(c)に続き、該混合物を濾過し、そして該形態Iの結晶を好適な溶媒又は水、又はそれらの混合物で洗浄することを含む工程を更に含む、請求項11に記載の方法。
【請求項29】
溶媒がアルコールである、請求項28に記載の方法。
【請求項30】
アルコールが2−プロパノールである、請求項29に記載の方法。
【請求項31】
工程(d)に続き、該形態Iの結晶を15℃〜80℃の温度で乾燥することを含む工程を更に含む、請求項11に記載の方法。
【請求項32】
請求項1に記載の結晶形を生成する方法であって、
(a)4−フルオロアセトフェノン1モル当り1.2〜1.45モルのトリフルオロ酢酸アルキル、4−フルオロアセトフェノン1モル当り1.1〜1.35モルの金属アルコキシド、及び既知量の4−フルオロアセトフェノンを混合して混合物とし;
(b)工程(a)からの該混合物を、4−フルオロアセトフェノン1モル当り650〜870mlの水、4−フルオロアセトフェノン1モル当り1.2〜1.7モルの濃塩酸、4−フルオロアセトフェノン1モル当り0.9〜1.1モルの4−スルホンアミドフェニルヒドラジン・塩酸塩及び4−フルオロアセトフェノン1モル当り、該混合物中の溶媒総量が600〜1000mlとなる量の好適な溶媒の組合せ物と組合せ;
(c)該混合物に、4−フルオロアセトフェノンに対してシーディング量の4−[5−(4−フルオロフェニル)−3−(トリフルオロメチル)−1H−ピラゾール−1−イル]−ベンゼンスルホンアミドの形態Iの結晶を添加し; そして
(d)該形態Iの結晶を集める;
工程を含む、上記の方法。
【請求項33】
工程(a)において、4−フルオロアセトフェノン1モル当り900mlまでの好適な溶媒を該混合物に添加することを更に含む、請求項32に記載の方法。
【請求項34】
アルコールが2−プロパノールである、請求項33に記載の方法。
【請求項35】
金属アルコキシドが、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウムイソプロポキシド、ナトリウム第三級ブトキシド、リチウムメトキシド、リチウムエトキシド、リチウムイソプロポキシド、リチウム第三級ブトキシド、カリウムメトキシド、カリウムエトキシド、カリウムイソプロポキシド、カリウム第三級ブトキシド、及びそれらの混合物から成るグループから選ばれる、請求項32に記載の方法。
【請求項36】
金属アルコキシドがナトリウムメトキシドである、請求項35に記載の方法。
【請求項37】
工程(a)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項38】
加熱工程に続き、該混合物を周囲温度に冷却することを含む工程を更に含む、請求項37に記載の方法。
【請求項39】
工程(b)が、該混合物を水、塩酸及び4−スルホンアミドフェニルヒドラジン・塩酸塩の組合せ物に添加することを含む、請求項32に記載の方法。
【請求項40】
工程(b)の該溶媒がアルコールである、請求項32に記載の方法。
【請求項41】
アルコールが2−プロパノールである、請求項40に記載の方法。
【請求項42】
工程(b)に続き、該混合物を周囲温度より高く、還流温度までの温度で該反応が完了するまで加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項43】
加熱工程に続き、該混合物を40℃〜65℃の温度で安定化させることを含む工程を更に含む、請求項42に記載の方法。
【請求項44】
シーディング量が0.0001質量%〜50質量%である、請求項33に記載の方法。
【請求項45】
シーディング量が0.001質量%〜5質量%である、請求項44に記載の方法。
【請求項46】
シーディング量が0.01質量%〜0.5質量%である、請求項44に記載の方法。
【請求項47】
工程(c)に続き、該混合物を50℃〜71.5℃より低い温度で、3〜8時間加熱することを含む工程を更に含む、請求項32に記載の方法。
【請求項48】
加熱工程に続き、該混合物を周囲温度に冷却することを含む工程を更に含む、請求項47に記載の方法。
【請求項49】
工程(c)に続き、該混合物を濾過し、そして該形態Iの結晶を好適な溶媒又は水、又はそれらの混合物で洗浄することを含む工程を更に含む、請求項32に記載の方法。
【請求項50】
溶媒がアルコールである、請求項49に記載の方法。
【請求項51】
アルコールが2−プロパノールである、請求項50に記載の方法。
【請求項52】
工程(d)に続き、該形態Iの結晶を30℃〜65℃の温度で乾燥することを含む工程を更に含む、請求項32に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公表番号】特表2007−530655(P2007−530655A)
【公表日】平成19年11月1日(2007.11.1)
【国際特許分類】
【出願番号】特願2007−505649(P2007−505649)
【出願日】平成17年3月21日(2005.3.21)
【国際出願番号】PCT/IB2005/000735
【国際公開番号】WO2005/095349
【国際公開日】平成17年10月13日(2005.10.13)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
【公表日】平成19年11月1日(2007.11.1)
【国際特許分類】
【出願日】平成17年3月21日(2005.3.21)
【国際出願番号】PCT/IB2005/000735
【国際公開番号】WO2005/095349
【国際公開日】平成17年10月13日(2005.10.13)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
[ Back to top ]