
結晶性形態のビフェニル化合物
本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を提供する。本発明はまた、そのような塩を含む薬学的組成物、またはそのような塩を使用して調製される薬学的組成物;そのような塩を調製するためのプロセスおよび中間体;およびそのような塩を使用して肺障害を処置する方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、肺障害を処置するための治療因子として有用であると期待されるビフェニル化合物の新規結晶性1,2−エタンジスルホン酸塩に関する。本発明はまた、そのような結晶性化合物を含む薬学的組成物、もしくはそのような結晶性化合物から調製される薬学的組成物、そのような結晶性化合物を調製するためのプロセスおよび中間体、ならびにそのような結晶性化合物を使用して肺障害を処置する方法に関する。
【背景技術】
【0002】
(技術的現状)
同一出願人による2004年2月13日出願の米国特許出願第10/779,157号は、慢性閉塞性肺疾患(COPD)および喘息などの肺障害を処置するための治療因子として有用である新規ビフェニル化合物を開示している。特に、この化合物、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルは、ムスカリンアンタゴニストおよびβ2アドレナリンレセプターアゴニスト活性の両方を有するとして、これらの出願に特異的に開示される。ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの化学構造は、式I:
【0003】
【化2】
によって表される。
【0004】
肺障害を処置するために有用な治療因子は、吸入により、好都合に直接気道へと投与される。そのため、いくつかの種類の薬学的吸入デバイスが、吸入により治療因子を投与するために開発されており、その中には、乾燥粉末吸入器(DPI)、定量吸入器(MDI)、およびネブライザー吸入器が含まれる。そのようなデバイス中での使用のための薬学的組成物および調合物を調製する場合、吸湿性でも潮解性でもなく、比較的高い融点(すなわち、約150℃より高い融点)を有し、それにより結晶性の重大な分壊または喪失なくこの物質を微粉化し得る結晶性形態の治療因子を有することが非常に望ましい。
【発明の開示】
【発明が解決しようとする課題】
【0005】
式Iの化合物の結晶性塩形態は、以前に報告されていない。したがって、受容可能な水準の吸湿性および比較的高い融点を有する式Iの化合物の、安定した、非潮解性の結晶性塩形態に対する必要性が存在する。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を提供する。
【0007】
驚くことに、式Iの化合物のそのような結晶性1,2−エタンジスルホン酸塩は、大気中の湿気に曝露される場合でさえも、潮解性ではないことが見出された。さらに、そのような結晶性塩は、受容可能な水準の吸湿性および非常に高い融点(例えば、約215℃より高い融点)を有する。特定の実施形態において、本発明の結晶性塩は、約230℃より高い融点を有する。
【0008】
いくつかある使用の中で、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、肺障害を処置するために有用であると期待される薬学的組成物を調製するために有用である。したがって、その組成物の別の局面おいて、本発明は、薬学的に受容可能なキャリアおよびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物を提供する。
【0009】
特定の実施形態において、本発明の薬学的組成物は、さらにステロイド抗炎症因子(たとえば、コルチコステロイド(cortecosteroid));もしくはホスホジエステラーゼ−4インヒビター;またはそれらの組み合わせを含む。
【0010】
別の実施形態において、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩を含み、約4〜約6の範囲におけるpHを有する等張生理食塩水溶液を含む薬学的組成物を提供する。
【0011】
さらに別の実施形態において、本発明は:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子
を含む組み合わせを提供する。
【0012】
式Iの化合物は、ムスカリンアンタゴニスト活性およびβ2アドレナリンレセプターアゴニスト活性の両方を有する。したがって、本発明の1,2−エタンジスルホン酸塩は、肺障害(例えば、喘息、および慢性閉塞性肺疾患)を治療するための治療因子として有用であると期待される。
【0013】
さらに、方法の局面のうちの1つにおいて、本発明は、肺障害を処置するための方法を提供し、この方法は、処置を必要としている患者に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を投与する工程を包含する。
【0014】
さらに、方法の別の局面において、本発明は、患者において気管支拡張(bronchodilation)を生じさせる方法を提供し、この方法は、気管支拡張を生じさせる量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を、吸入により患者に投与する工程を包含する。
【0015】
本発明はまた、慢性閉塞性肺疾患または喘息を処置する方法を提供し、この方法は、処置を必要としている患者に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を投与する工程を包含する。
【0016】
本発明はまた、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスに向けられる。したがって、その方法の別の局面において、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を調製するためのプロセス;ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを1,2−エタンジスルホン酸と接触させる工程を包含するプロセスを提供する。
【0017】
その方法のさらに別の局面において、本発明は、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスを提供し、このプロセスは:
(a)式IIの化合物:
【0018】
【化3】
をフッ化物イオンと接触させる工程であって、ここで
R1a、R1b、およびR1cが、C1〜4アルキル、フェニル、−C1〜4アルキル−(フェニル)から独立して選択されるか、またはR1a、R1b、およびR1cのうちの1つが−O−(C1〜4アルキル)である工程;および
(b)工程(a)からの生成物を、1,2−エタンジスルホン酸もしくはその水和物と接触させて式Iの化合物の結晶性1,2−エタンジスルホン酸塩を形成させる工程であって、
工程(a)および(b)が、同じ反応容器中で、工程(a)の生成物を分離せずに実施される工程
を包含する。
【0019】
その方法の別の局面において、本発明は、約230℃より高い融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するプロセスを提供し、このプロセスは、不活性希釈剤に溶解させた式Iの化合物の1,2−エタンジスルホン酸塩を含む溶液に、式Iの化合物の結晶性1,2−エタンジスルホン酸塩の種結晶を加える工程を包含し、ここでこの種結晶は、約230℃より高い融点を有する。
【0020】
このプロセスはまた、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を再結晶化し、約230℃より高い融点を有する結晶性形態を提供するために使用され得る。したがって、本発明は、約230℃より高い融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスをさらに提供し、このプロセスは:
(a)式Iの化合物の結晶性1,2−エタンジスルホン酸塩を、第一の温度にて不活性な希釈剤に溶解させる工程;
(b)工程(a)の生成物を第二の温度に冷却する工程;および
(c)式Iの化合物の1,2−エタンジスルホン酸塩の種結晶を加える工程
を包含し、ここで、種結晶は約230℃より高い融点を有し、第一の温度は、1,2−エタンジスルホン酸塩を溶解させるのに十分な温度であり、そして第二の温度は、種結晶が工程(b)の生成物に加えられる場合に完全に溶解する温度より低い。
【0021】
さらに、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを精製するためのプロセス;ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩を形成させる工程を包含するプロセスに向けられる。本発明はまた、本明細書中に記載されるプロセスによって調製される生成物にも向けられる。
【0022】
本発明はまた、治療における使用または薬としての使用のためのビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物に向けられる。
【0023】
さらに、本発明は、薬の製造;特に肺障害の処置のための薬の製造のためのビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の使用に向けられる。
【0024】
本発明はまた、肺障害の処置のための薬の製造における:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子
の使用に向けられる。
【0025】
本発明はまた、微粉形態のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;ならびに、微粉形態の薬学的に受容可能なキャリアおよびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物に向けられる。
【発明を実施するための最良の形態】
【0026】
(発明の詳細な説明)
本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩またはその溶媒和物を提供する。これらの塩(すなわち、式Iの化合物)中の活性な治療因子は、(R)配置を有する1つのキラル中心を含んでいる。しかしながら、全体として組成物の任意の有用性がそのような異性体の存在によって排除されないならば、他に示さない限り、少量の(S)立体異性体が本発明の組成物中に存在し得ることが、当業者には理解される。
【0027】
式Iの化合物は、市販のAutoNomソフトウェア(MDL,San Leandro,California)を使用して命名された。加えて、1,2−エタンジスルホン酸塩はまた、時にエジシラート(edisylateまたはedisilate)塩として述べられることがある。
【0028】
(定義)
本発明の化合物、組成物、方法、およびプロセスを記載する場合、次の用語は、他に示さない限り、次の意味を有する。
【0029】
用語「融点」は、本明細書中で使用される場合、最大吸熱熱流が、示差走査熱量測定により得られる温度を意味する。
【0030】
用語「微粉形態」は、少なくとも約90%の粒子が約10μmより小さな直径を有する粒子の形態を意味する。
【0031】
用語「溶媒和物」は、溶質の1つ以上の分子によって形成される複合体または凝集物(すなわち、式Iの化合物の1,2−エタンジスルホン酸塩)、および溶媒の1つ以上の分子を意味する。そのような溶媒和物は、典型的に、実質的に固定された溶質と溶媒とのモル比を有する。この用語はまた、水を有する包接化合物を含む包接化合物を含む。代表的な溶媒は、例として、水、メタノール、エタノール、イソプロパノール、酢酸などが挙げられる。溶媒が水の場合、その形成される溶媒は水和物である。
【0032】
用語「治療有効量」は、処置を必要としている患者に投与される場合、処置を達成するのに十分な量を意味する。
【0033】
用語「処置する」または「処置」は、本明細書中で使用される場合:
(a)疾患または医学的状態(medical condition)が生じるのを防ぐ(すなわち、患者の予防的処置)こと;
(b)疾患または医学的状態を改善する(すなわち、患者において、疾患または医学的状態の排除または軽減をもたらす)こと;
(c)疾患または医学的状態を抑制する(すなわち、患者において、疾患または医学的状態の発達を減速させるかまたは阻止する)こと;あるいは
(d)患者において、疾患の症状または医学的状態を緩和すること
を含み、患者(例えば、哺乳動物(特にヒト)において疾患もしくは医学的状態(例えば、COPD)を処置することまたはその処置を意味する。
【0034】
用語「単位投薬形態」は、患者に投薬するのに適した物理的に分離した単位、すなわち、本発明の塩の予め決定された量を含む各単位を表す。この予め決定された量は、単独で、または1つ以上の付加的な単位との組み合わせにおいて所望の治療効果を生むように算定される。例えば、そのような単位投薬形態は、乾燥粉末吸入カプセル、定量吸入器からの一定用量、カプセル、錠剤、丸剤などであり得る。
【0035】
(本発明の1,2−エタンジスルホン酸塩)
本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステル、および1,2−エタンジスルホン酸塩、またはその水和物から調製され得る。
【0036】
本発明の1,2−エタンジスルホン酸塩は、典型的に、式Iの化合物1モル当量当たり約0.90と約1.10モル当量との間の1,2−エタンジスルホン酸を含み;式Iの化合物1モル当量当たり約0.95と約1.05モル当量との間の1,2−エタンジスルホン酸を含む。特定の実施形態において、本発明の1,2−エタンジスルホン酸塩は、式Iの化合物1モル当量当たり約1モル当量の1,2−エタンジスルホン酸を含む。
【0037】
1,2−エタンジスルホン酸のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルに対するモル比率は、当業者が利用し得る種々の方法により、容易に決定され得る。例えば、そのようなモル比率は、1H NMRにより容易に決定され得る。あるいは、元素分析およびHPLC法が、モル比率を決定するために使用され得る。
【0038】
本発明で使用されるビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルは、市販の出発物質および試薬から、以下の実施例に記載される手順を使用するか;または本出願の背景部分に記載される同一出願人による米国特許出願に記載される手順を使用して、容易に調製され得る。
【0039】
1,2−エタンジスルホン酸は、例えば、Alfa Chemicals Ltd.,Berkshire,UKから市販されている。1つの実施形態において、本発明の塩を調製するのに使用される1,2−エタンジスルホン酸は、二水和物である。特定の実施形態において、1,2−エタンジスルホン酸二水和物は、97%以上の純度(HPLCによって決定される場合)を有する。望ましい場合は、本発明に使用される1,2−エタンジスルホン酸二水和物を、使用する前に、例えば酢酸および無水酢酸から再結晶化し得る。
【0040】
本発明の結晶性塩を調製するために、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを、典型的には、約0.75〜約1.3モル当量の1,2−エタンジスルホン酸、またはその水和物と接触させる。一般的に、この反応は、約0℃〜約60℃;約20℃〜約55℃(例えば、約25℃〜約50℃)を含む範囲の温度にて、不活性な希釈剤中で実施される。この反応に適した不活性な希釈剤としては、これらに限定されないが、メタノール、エタノール、イソプロパノール、イソブタノール、酢酸エチル、ジクロロメタンなどが挙げられ、必要に応じて水を含む。特定の実施形態において、エタノール中の1,2−エタンジスルホン酸二水和物の溶液を、イソプロパノールとジクロロメタン(64:1)との混合物中のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの体積の約5倍まで加える。他の特定の実施形態において、1,2−エタンジスルホン酸二水和物の溶液は、希釈剤として水またはエタノールを含み、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステル溶液は、希釈剤としてイソプロパノールまたはエタノールを含む。
【0041】
あるいは、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、式Iの化合物のシリルで保護された誘導体(すなわち、式IIの化合物)を、フッ化物イオンの供給源と接触させ、次いで同じ反応容器中で、その生成物を1,2−エタンジスルホン酸またはその水和物と接触させることにより調製し得る。特定の実施形態において、シリル保護基は、tert−ブチルジメチルシリル基である。他の適切なシリル保護基としては、tert−ブチルジフェニルシリル、ジフェニルメチルシリル、ジ−tert−ブチルメチルシリル(buylmethylsilyl)、tert−ブトキシジフェニルシリルなどが挙げられる。このプロセスで使用されるフッ化物イオンの供給源は、フッ化物イオンまたはフッ化水素を含んでいるか、または包含している任意の試薬であり得る。特定の実施形態において、フッ化物イオンの供給源は、三フッ化水素トリエチルアミンである。フッ化物イオンの他の適切な供給源は、フッ化テトラブチルアンモニウム、18−クラウン−6を有するフッ化カリウム、フッ化水素、ピリジンフッ化水素塩などが挙げられる。
【0042】
一般的に、このプロセスは、約0℃〜約50℃;約20℃〜約35℃(例えば、約25℃〜約30℃)を含む範囲の温度において不活性な希釈剤中で実施される。この反応に適切した不活性な希釈剤としては、これらに限定されないが、ジクロロメタン、メタノール、およびそれらの混合物が挙げられる。特定の実施形態において、ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−tert−ブチルジメチルシラニルオキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロ−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの溶液を、ジクロロメタン中の約2.5〜約3.0モル当量のトリエチルアミン三フッ化水素塩と、周囲温度にて約12〜24時間、またはシリル基の除去が実質的に完了するまで接触させる。得られた溶液を、反応生成物を分離せず、メタノール中の約0.9〜約1.1モル当量の1,2−エタンジスルホン酸二水和物を加え、そしてこの混合物を約25℃〜約35℃にて約2〜約6時間加熱する。この反応の完了の際に、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩は、任意の従来の方法(例えば、沈殿、濃縮、遠心分離など)により、反応混合物から分離される。
【0043】
必要に応じて、本発明の結晶性1,2−エタンジスルホン酸塩は、容量で約15%〜約25%(約20%を含む)の水を含むイソプロパノール中で、塩を攪拌または懸濁(slurring)させることによりさらに精製され得る。特定の実施形態において、1,2−エタンジスルホン酸塩1g当たり約10mLのイソプロパノール/水混合物が使用される。
【0044】
本発明の結晶性1,2−エタンジスルホン酸塩を調製するプロセスは、必要に応じて、特定の結晶性塩を優位に生産するための種結晶の使用を含む。例えば、高温で(すなわち、約230℃より高い温度で)融解する結晶性塩の種結晶を使用することにより、種結晶と本質的に同じ融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製し得る。そのような種結晶は、最初、結晶性塩を形成する場合に使用され得るか、または結晶性もしくは一部が結晶性の塩を再結晶化するために使用され得る。
【0045】
典型的に、種結晶は、攪拌せず、冷却を適用しないゆっくりとした結晶化により、小規模で調製される。種結晶を得るための例として、典型的に、溶解を与えるのに十分な温度で不活性な希釈剤に結晶性塩を溶解させる。一般的に、種結晶を得る最初のプロセスにおいて、少量の、典型的に10g未満(5g未満(例えば1g未満)を含む)の結晶性塩が使用される。特定の実施形態において、希釈剤として約12%〜約20%の水(約13%〜約15%の水を含む)が、約60℃〜約70℃(例えば、約60℃〜約65℃)の範囲の温度にて使用される。この溶液を、室温に冷却させる。約1日〜約3日後、得られた結晶を濾過、または他の慣例的な方法により単離する。あるいは、以前の結晶性物質の調製から、種結晶が得られ得る。
【0046】
種結晶を使用した再結晶化プロセスにおいて、本発明の結晶性1,2−エタンジスルホン酸塩を、種結晶を得るプロセスの場合のように、不活性な希釈剤、典型的には15%の水を含むメタノールに、約60℃〜約65℃の範囲の温度にて溶解させる。この溶液を種結晶が溶解しない温度、例えば、約30℃〜約40℃の範囲における温度に冷却させ、次いで、種結晶を加える。典型的に、この溶液中の結晶性塩の重量に対する種結晶の重量の比は、約1:5と約1:35との間である。この溶液を結晶化が起こる温度、例えば約20度に冷却し、約2時間〜約24時間、攪拌する。得られた結晶を、慣例的な方法により単離する。大量の物質を調製するのに十分な種結晶を得るために、最初の再結晶化によって得られた結晶を、次の再結晶化工程のための種結晶として使用し、この再結晶化プロセスを連続的に実施し得る。再結晶化プロセスの工程が実施される特異的な温度は、希釈剤の性質および希釈剤中の結晶性塩の濃度に従い選択されることが理解される。さらに、冷却する代わりにエバポレーションかまたは抗溶媒を、結晶化を促進するために使用して、この再結晶化プロセスを実施し得る。
【0047】
いくつかある利点の中で、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を形成することが、式Iの化合物を精製するために有用であることが見出された。一般的に、本発明の結晶性1,2−エタンジスルホン酸塩は、高速液体クロマトグラフィーにより決定される場合、95%より高い純度;典型的に98%より高い純度を有する。
【0048】
本発明の結晶性1,2−エタンジスルホン酸塩は、約215℃〜約240℃の範囲で吸熱熱流におけるピークを示す示差走査熱量測定(DSC)の記録によって明示されるように、非常に高い融点を特徴とする。結晶性塩の融点温度は、その結晶性塩が形成されたプロセスに従うことが認められた。攪拌せず、冷却を適用しないゆっくりとした結晶化によって形成される種結晶は、約230℃より高い融点を示す。そのような種結晶を用いた再結晶化を含むプロセスによって形成される結晶性塩は、典型的に、例えば図1に示されるように、約230℃〜約245℃の範囲における融点を示す。約230℃より高い融点を有する種結晶を用いずに形成された結晶性塩は、典型的に、例えば図2に示されるように、約215℃〜約229℃の範囲における融点を示す。したがって、特定の実施形態において、本発明は、実質的に図1または図2に示される記録に従う約200℃より高い温度範囲にDSCの記録を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を提供する。
【0049】
別の実施形態において、本発明の結晶性1,2−エタンジスルホン酸塩は、5.0±0.3および15.0±0.3の2θの値において有意な回析ピークを有する粉末エックス線回析(PXRD)パターンを特徴とする。図3に示されるような融点が高い種結晶からの再結晶化により調製した結晶化塩のPXRDスペクトルにおけるピーク位置と、図4に示されるようなそのような種結晶を使用せずに調製した塩のピーク位置との間に、微妙な違いが認められた。従って、別の実施形態において、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、ピーク位置が実質的に図3または図4に示されるPXRDスペクトルにおけるピーク位置と一致する粉末エックス線回析パターンを特徴とする。
【0050】
別の実施形態において、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、図5に図示されるように、約704cm−1、約748cm−1、約768cm−1、約841cm−1、約900cm−1、約1055cm−1、約1104cm−1、約1166cm−1、約1218cm−1、約1294cm−1、約1408cm−1、約1522cm−1、約1609cm−1、約1655cm−1、および約1701cm−1において有意な吸収帯を示す赤外線(IR)吸収スペクトルを特徴とする。
【0051】
式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、受容可能な適度な水準の吸湿性(すなわち、相対湿度40%〜相対湿度75%の湿度範囲において約2.5%未満重量増)を有する可逆的吸着/脱着プロフィールを有することが示された。
【0052】
本発明の塩のこれらの特性について、以下の実施例においてさらに説明する。
【0053】
(薬学的組成物および調合物)
式Iの化合物の1,2−エタンジスルホン酸塩を、典型的には、薬学的組成物または調合物の形態で患者に投与する。そのような薬学的組成物を、投与の任意の受容可能な経路(これらに限定されないが、吸引、経口、鼻腔、局所(経皮を含む)を含む)、および投与の非経口形態によって患者に投与し得る。しかしながら、本発明の結晶性塩は調合されてしまうと結晶性形態ではなくなり得る(すなわち、この塩を適切なキャリアに溶解し得る)ということが当業者には理解される。
【0054】
したがって、組成物の局面のうちの1つにおいて、本発明は、薬学的に受容可能なキャリアもしくは賦形剤、およびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物に向けられる。必要に応じ、そのような薬学的組成物は、望ましい場合、他の治療剤および/または調合剤を含み得る。
【0055】
本発明の薬学的組成物は、典型的に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む。典型的に、そのような薬学的組成物は、約0.01〜約95重量%;約0.01〜約30重量%;例えば、約0.01〜約10%の活性因子を含む。
【0056】
任意の従来のキャリアもしくは賦形剤を、本発明の薬学的組成物に使用し得る。特定のキャリアもしくは賦形剤の選択、またはキャリアもしくは賦形剤の組み合わせの選択は、特定の患者を治療するために使用されている投与の形態、または医学的状態もしくは病状(disease state)の種類に従う。この点で、特定の形態の投与に適した薬学的組成物の調製は、まさに薬学分野の当業者の範囲内である。さらに、そのような組成物の成分は、例えば、Sigma,P.O.Box 14508、St.Louis, MO 63178から市販されている。さらなる例として、慣例的な調合技術は、Remington:The Science and Pracice of Pharmacy,20th Edition,Lippincott Williams & White,Baltimore,Maryland(2000);およびH.C.Ansel et al.,Pharmaceutical Dosage Forms and Drug Delivery Systems,7th Edition, Lippincott Williams & White,Baltimore,Maryland(1999)に記載されている。
【0057】
薬学的に受容可能なキャリアとなり得る物質の代表的な例としては、これらに限定されないが、次の:(1)糖(例えば、ラクトース、ブドウ糖、およびスクロース);(2)デンプン(例えば、コーンスターチ、およびジャガイモデンプン);(3)セルロース、およびその誘導体(例えば、カルボキシメチルセルロースナトリウム、エチルセルロース、および酢酸セルロース);(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤(例えば、ココアバター、および座剤用ワックス;(9)油(例えば、ピーナツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、コーン油、およびダイズ油;(10)グリコール(例えば、プロピレングリコール);(11)多価アルコール(例えば、グリセリン、ソルビトール、マンニトール、およびポリエチレングリコール);(12)エステル(例えば、オレイン酸エチル、およびラウリン酸エチル);(13)寒天;(14)緩衝剤(例えば、水酸化マグネシウム、および水酸化アルミニウム);(15)アルギン酸;(16)発熱因子非含有水(pyrogen−free water);(17)等張食塩水;(18)リンゲル溶液;(19)エチルアルコール;(20)リン酸緩衝溶液;(21)圧縮噴霧ガス(例えば、クロロフルオロカーボン、およびハイドロフルオロカーボン);そして(22)薬学的組成物に使用される他の無毒性適合性物質が挙げられる。
【0058】
本発明の薬学的組成物は、典型的に、薬学的に受容可能なキャリアおよび1つ以上の任意の成分と本発明の塩とを混合するかまたは組み合わせて調製される。必要な場合、または望ましい場合は、得られた均質に混和した混合物を、慣例的な手順および装置を使用して形作られるか、あるいは錠剤、カプセル剤、丸剤、キャニスター、カートリッジ、ディペンサーなどに詰め込み得る。
【0059】
1つの実施形態において、本発明の薬学的組成物は、吸入投与に適している。吸入投与に適した薬学的組成物は、典型的に、エアゾール、または粉末の形態である。そのような組成物は、一般的に、周知の送達デバイス(例えば、ネブライザー吸入器、定量吸入器(MDI)、乾燥粉末吸入器(DPI)、または類似した送達デバイスを使用して投与される。
【0060】
本発明の特異的な実施形態において、活性因子を含む薬学的組成物は、ネブライザー吸入器を使用して吸入により投与される。そのようなネブライザーデバイスは、典型的に、高速の気流を生成し、活性因子を含む薬学的組成物がミストとしてスプレーされ、患者の気道に運ばれる。したがって、ネブライザー吸入器中での使用のために調合される場合、典型的に、適切なキャリアにこの活性因子を溶解させて溶液を形成する。適切なネブライザーデバイスは、例えば、PARI GmbH(Starnberg,German)により市販されている。他のネブライザーデバイスとしては、Respimat(Boehringer Ingelheim)、ならびに例えば、米国特許第6,123,068号および国際公開第97/12687号で開示されているものが挙げられる。
【0061】
ネブライザー吸入器中での使用のための代表的な薬学的組成物は、約0.05μg/mL〜約10mg/mLの式Iの化合物の1,2−エタンジスルホン酸塩、またはその溶媒和物を含む水溶液を含む。1つの実施形態において、このネブライザー調合水は等張性である。1つの実施形態において、このネブライザー調合水は、約4〜約6の範囲におけるpHを有する。特定の実施形態において、このネブライザー調合水は、pHが約5までのクエン酸緩衝液で緩衝される。別の特定の実施形態において、このネブライザー調合水は約0.1mg/mL〜約1.0mg/mLの遊離塩基当量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを含む。
【0062】
本発明の別の特定の実施形態において、活性因子を含む薬学的組成物は、乾燥粉末吸入器を使用して、吸入により投与される。そのような乾燥粉末吸入器は、典型的に、吸息中に患者の気流中に分散される自由流れ粉末(free−flowing powder)として活性因子を投与する。自由流れ粉末を達成するために、活性因子は、典型的に、適切な賦形剤(例えば、ラクトース、デンプン、マンニトール、ブドウ糖、ポリ乳酸(PLA)、ポリ乳酸・グリコリド共重合体(PLGA)、またはそれらの組み合わせ)と共に調合される。典型的に、この活性因子は、微粉化され、適切なキャリアと組み合わせて呼吸に適した大きさの微粉粒の混合物を形成する。ここで「微粉粒」または「微粉形態」は、少なくとも約90%の粒子が約10μm未満の直径を有することを意味する。
【0063】
乾燥粉末吸入器中での使用のための代表的な薬学的組成物は、約1μmと約100μmとの間の粒径を有するラクトース、および式Iの化合物の1,2−エタンジスルホン酸塩、またはその溶媒和物の微粉粒子を含む。
【0064】
そのような乾燥粉末調合物を、例えば、ラクトースを活性因子と組み合わせ、次いでその成分を乾式混合することにより作製し得る。あるいは、望ましい場合、この活性因子を、賦形剤を用いずに調合し得る。次いで、薬学的組成物を、典型的に、乾燥粉末ディスペンサー、または乾燥粉末送達デバイスと共に使用するための吸入カートリッジ、もしくはカプセル剤に詰める。
【0065】
乾燥粉末吸入器送達デバイスの例としては、Diskhaler(GlaxoSmithKline,Research Triangle Park,NC)(例えば、米国特許第5,035,237号を参照);Diskus(GlaxoSmithKline)(例えば、米国特許第6,378,519号を参照);Turbuhaler(AstraZeneca,Wilmington,DE)(例えば、米国特許第4,524,769号を参照);Rotahaler(GlaxoSmithKline)(例えば、米国特許第4,353,365号を参照)、およびHandihaler(Boehringer Ingelheim)が挙げられる。適切なDPIデバイスのさらなる例には、米国特許第5,415,162号、米国特許第5,239,993号、および米国特許第5,715,810号に記載されており、これらの文献は、本明細書中で引用される。
【0066】
本発明のさらに別の特異的な実施形態において、活性因子を含む薬学的組成物は、定量吸入器を使用して吸入により投与される。そのような定量吸入器は、典型的に、圧縮噴霧ガスを使用して、一定量の活性因子またはその薬学的に受容可能な塩を放出する。したがって、定量吸入器を使用して投与される薬学的組成物は、典型的に、液化された噴霧剤中に、活性因子の溶液または懸濁液を含む。クロロフルオロカーボン(例えば、CCl3F)、およびハイドロフルオロアルカン(HFA)(例えば、1,1,1,2−テトラフルオロエタン(HFA 134a)、および1,1,1,2,3,3,3−へプタフルオロ−n−プロパン(HFA 227))を含む任意の適した液化噴霧剤を使用し得る。オゾン層に影響を及ぼすクロロフルオロカーボンについての懸念から、HFAを含む調合物が、一般的に好ましい。HFA調合物のさらなる任意の成分としては、共溶媒(例えば、エタノールまたはペンタン)および表面活性剤(例えば、ソルビタントリオレエイト(trioleate)、オレイン酸、レシチン、およびグリセリン)が挙げられる。例えば、米国特許第5,225,183号、欧州特許第0717987A2号、および国際公開92/22286号を参照されたい。
【0067】
定量吸入器中における使用のための代表的な薬学的組成物は、約0.01重量%〜約5重量%の式Iの化合物の1,2−エタンジスルホン酸塩またはその溶媒和物;約0重量%〜20重量%のエタノール;そして約0重量%〜約5重量%の表面活性剤を含み、その残りがHFA噴霧剤である。
【0068】
そのような組成物を、典型的に、冷却もしくは加圧ヒドロフルオロアルカンを、活性因子、エタノール(あれば)、および表面活性剤(あれば)を含む適切な容器に加えることにより調製する。懸濁液を調製するために、活性因子を微粉化し、次いで、噴霧剤と組み合わせる。次いで、この調合物をエアゾールキャニスターに詰めて、定量吸入デバイスの一部とする。特異的にHFA噴霧剤と共に使用するために開発された定量吸入器デバイスの例が、米国特許第6,006,745号、および第6,143,277号に提供されている。あるいは、懸濁調合物を、活性因子の微粉粒子上の表面活性剤のコーティングをスプレー乾燥させることにより調製し得る。例えば、国際公開第99/53901号、および国際公開第00/61108号を参照されたい。
【0069】
呼吸に適した粒子を調製するプロセス、ならびに吸入投薬に適した調合物およびデバイスのさらなる例については、米国特許第6,268,533号、第5,983,956号、第5,874,063号、および6,221,398号、ならびに国際公開第99/55319号、および国際公開第00/30614号を参照されたい。
【0070】
別の実施形態において、本発明の薬学的組成物は、経口投与に適している。経口投与に適した薬学的組成物は、カプセル剤、錠剤、丸剤、ドロップ剤、カシェ剤、糖衣剤、粉剤、顆粒剤の形態であるか;あるいは溶液または水性もしくは非水性の液体中の懸濁液として;あるいは水中油もしくは油中水液体乳剤として;あるいはエリキシル剤またはシロップ剤として;などであり得、それぞれ活性成分として所定量の本発明の塩を含む。
【0071】
固形の投薬形態(すなわち、カプセル剤、錠剤、丸剤など)での経口投与に向けられる場合、本発明の薬学的組成物は、典型的に、活性成分として本発明の塩、および1種以上の薬学的に受容可能なキャリア(例えば、クエン酸ナトリウムまたはリン酸二カルシウム)を含む。必要に応じ、または代替的に、そのような固形の投薬形態はまた:(1)充填剤または増量剤(例えば、デンプン、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸);(2)結合剤(例えば、カルボキシメチルセルロース、アルギナート、ゼラチン、ポリビニルピロリドン、スクロース、および/またはアカシア);(3)湿潤剤(例えば、グリセリン);(4)崩壊剤(例えば、寒天、炭酸カルシウム、ジャガイモもしくはタピオカデンプン、アルギン酸、特定のシリケート、および/または炭酸ナトリウム);(5)溶液緩染剤(例えば、パラフィン);(6)吸収促進剤(例えば、第四アンモニウム化合物);(7)加湿剤(例えば、セチルアルコール、および/またはグリセロールモノステアレート);(8)吸収剤(例えば、カロリン、および/またはベントナイトクレー);(9)潤滑剤(例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはそれらの混合物);(10)着色剤;そして(11)緩衝剤を含む。
【0072】
離型剤、加湿剤、コーティング剤、甘味剤、香味剤および芳香剤、保存剤、ならびに酸化防止剤もまた、本発明の薬学的組成物中に存在し得る。薬学的に受容可能な酸化防止剤の例としては:(1)水溶性酸化防止剤(例えば、アスコルビン酸、塩酸システイン、硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウムなど);(2)油溶性酸化防止剤(例えば、アスコルビン酸パルミテート、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、α−トコフェロールなど);そして(3)金属キレート因子(例えば、クエン酸、エチレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸、など)が挙げられる。錠剤、カプセル剤、丸剤などのための被膜因子としては、腸溶コーティング用に使用される被膜因子(例えば、酢酸フタル酸セルロース(CAP)、ポリビニルアセテートフタレート(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、メタクリル酸−メタクリル酸エステルコポリマー、セルソースアセテートトリメリテート(trimellitate)(CAT)、カルボキシメチルエチルセルロース(CMEC),ヒドロキシプロピルメチルセルロースアセテートスクシナート(HPMCAS)など)が挙げられる。
【0073】
望ましい場合、本発明の薬学的組成物は、例として、様々な割合のヒドロキシプロピルメチルセルロース;あるいは他のポリマーマトリックス(例えば、ポリ乳酸(PLA)もしくはポリ乳酸・グリコリド共重合体(PLGA)、リポソームおよび/または小球体を有するようなポリマーマトリクス)を使用して、活性成分のゆっくりとした、もしくは制御された放出を提供するように調合され得る。
【0074】
さらに、本発明の薬学的組成物は、必要に応じて不透明化因子を含み得、胃腸管の特定の部分において、必要に応じ、遅延させる様式で、活性成分のみを、または活性成分を優先的に、放出するように調合され得る。使用し得る包埋組成物(embedding composition)の例としては、高分子物質、およびワックスが挙げられる。この活性成分はまた、適切である場合、1種以上の上記賦形剤を有するマイクロカプセル形態であり得る。
【0075】
経口投与に適した液体投薬形態としては、例として、薬学的に受容可能な乳剤、ミクロエマルジョン、液剤、懸濁剤、シロップ剤、およびエリキシル剤が挙げられる。そのような液体投与形態は、典型的に、活性成分および不活性な希釈剤(例えば、水または他の溶媒など)、可溶化剤、および乳化剤(例えば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセリン、テトラヒドロフリルアルコール、ポリエチレングリコール、およびソルビタン脂肪酸エステル、ならびにこれらの混合物を含む。活性成分に加えて懸濁剤は、懸濁化剤(例えば、エトキシ化したイソステアリールアルコール、ポリオキシエチレンソルビトール、およびソルビタンエステルなど)、微結晶性セルロース、アルミニウムメタヒドロキシド、ベントナイト、寒天、およびトラガカント、ならびにこれらの混合物を含み得る。
【0076】
経口投与に向けられる場合、本発明の薬学的組成物は、好ましくは、単位投薬形態にまとめられる。例えば、そのような単位投薬形態は、カプセル、錠剤、丸剤などであり得る。
【0077】
本発明の塩はまた、公知の経皮送達システムおよび賦形剤を使用して、経皮投与され得る。例えば、本発明の化合物を、浸透促進剤(permeation enhancer)(例えば、プロピレングリコール、モノラウリン酸ポリエチレングリコルム(polyethylene glycolm monolaurate)、アザシクロアルカン−2−オンなど)と混合し、貼付剤もしくは類似の送達システムに組み込み得る。ゲル化剤、乳白剤、および緩衝剤を含むさらなる賦形剤を、望ましい場合、そのような経皮組成物中に使用し得る。
【0078】
本発明の薬学的組成物はまた、他の治療因子を含んで式Iの化合物の1,2−エタンジスルホン酸塩またはその溶媒和物と同時投与し得る。例えば、本発明の薬学的組成物は、抗炎症剤(例えば、ステロイド抗炎症剤(コルチコステロイドなど);および非ステロイド抗炎症剤(NSAID)、ホスホジエステラーゼIV阻害物質、抗感染剤(例えば、抗生物質または抗ウィルス薬)、抗ヒスタミン剤、β2アドレナリンレセプターアゴニスト、ムスカリンレセプターアンタゴニスト(すなわち、抗コリン作用薬)などから選択される1種以上の治療因子をさらに含み得る。他の治療因子は、薬学的に受容可能な塩もしくは溶媒和物の形態で使用され得る。さらに、適切である場合、他の治療因子は、光学的に純粋な立体異性体として使用され得る。
【0079】
望ましい場合、本発明の塩はまた、別の治療因子または本明細書中に記載される因子と組み合せて投与され得る。この実施形態において、成分が物理的に混合しているのではなく、同時にまたは連続的に、別個の組成物として投与される。例えば本発明の塩は、各治療因子に対し別個のコンパートメントを使用する吸入薬送達デバイス(例えば、ブリスターパック)を使用して、ステロイド抗炎症剤(例えば、コルチコステロイド)と共に、吸入により同時にまたは連続的に投与され得る。あるいは、この組み合せは、多数の送達デバイス(すなわち各治療因子に対し1つの送達デバイス)から投与され得る。
【0080】
本発明の化合物と組み合わせて使用し得る代表的なβ2アドレナリンレセプターアゴニストとしては、これらに限定されないが、サルメテロール(salmeterol)、サルブタモール、フォルモテロール、サルメファモール(salmefamol)、フェノテロール、テルブタリン、アルブテロール、イソエタリン、メタプロテレノール、ビトルテロール、ピルブテロール、レバルブテロール(levalbuterol)など、またはそれらの薬学的に受容可能な塩が挙げられる。本発明の化合物と組み合わせて使用し得る他のβ2アドレナリンレセプターアゴニストとしては、これらに限定されないが、3−(4−{[6−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、3−(−3−{[7−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)へプチル]オキシ}プロピル)ベンゼンスルホンアミド、および2002年8月29日公開の国際公開第02/066422号に開示されている関連化合物;3−[3−(4−{[6−([(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル]アミノ)ヘキシル]オキシ}ブチル)フェニル]イミダゾリジン−2,4−ジオン、および2002年9月12日公開の国際公開第02/070490号に開示されている関連化合物;3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、および2002年10月3日公開の国際公開第02/076933号に開示されている関連化合物;4−{(1R)−2−[(6−{2−[(2,6−ジクロロベンジル)オキシ]エトキシ}ヘキシル)アミノ]−1−ヒドロキシエチル}−2−(ヒドロキシメチル)フェノール、および2003年3月27日公開の国際公開第03/024439号に開示されている関連化合物;N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミン、および2003年6月10日発行の米国特許第6,576,793号B1に開示されている関連化合物;N−{2−[4−(3−フェニル−4−メトキシフェニル)アミノフェニル]エチル}−(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2(1H)−キノリノン−5−イル)エチルアミン、および2003年11月25日発行の米国特許第6,653,323号B2に開示されている関連化合物;ならびにそれらの薬学的に受容可能な塩が挙げられる。特定の実施形態において、β2アドレナリンレセプターアゴニストは、N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性モノヒドロクロリド塩である。使用される場合、β2アドレナリンレセプターアゴニストは、治療有効量で薬学的組成物中に存在する。典型的に、このβ2アドレナリンレセプターアゴニストは、1用量あたり約0.05μg〜約500μgを提供するのに十分な量で存在する。
【0081】
本発明の化合物と組み合わせて使用し得る代表的なステロイド抗炎症剤としては、これらに限定されないが、メチルプレドニゾロン、プレドニゾロン、デキサメタゾン、フルチカソンプロピオナート(fluticasone propionate)、6,9−ジフルオロ−17−[(2−フラニルカルボニル)オキシ]−11−ヒドロキシ−16−メチル−3−オキソアンドロスタ−1,4−ジエン−17−カルボチオ酸S−フルオロメチルエステル、6,9−ジフルオロ−11−ヒドロキシ−16−メチル−3−オキソ−17−プロピオニルオキシ−アンドロスタ−1,4−ジエン−17−カルボチオ酸S−(2−オキソテトラヒドロフラン−3S−イル)エステル、ベクロメタゾンエステル(例えば、17−プロピオン酸エステル、または17,21−ジプロピオン酸エステル)、ブデソニド、フルニソリド、モメタソンエステル(mometasone ester)(例えば、フロン酸エステル)、トリアムシノロンアセトニド、ロフレポニド(rofleponide)、シクルソニド(ciclesonide)、ブチキソコルトプロピオナート(butixocort propionate)、RPR−106541、ST−126など、またはそれらの薬学的に受容可能な塩が挙げられる。特定の実施形態において、ステロイド抗炎症剤は、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその薬学的に受容可能な塩もしくは溶媒和物である。使用される場合、ステロイド抗炎症剤は、治療有効量で薬学的組成物中に存在する。典型的に、ステロイド抗炎症剤は、1用量あたり約0.05μg〜約500μgを提供するのに十分な量で存在する。
【0082】
他の適切な組み合わせとしては、例として、他の抗炎症剤、例えば、NSAID(クロモグリク酸ナトリウム;ネドクロミルナトリウム(nedocromil sodium);ホスホジエステラーゼ(PDE)阻害物質(例えば、テオフィリン、PDE4阻害物質、または混合PDE3/PDE4阻害物質);ロイコトリエンアンタゴニスト(例えば、モンテルカスト);ロイコトリエン合成の阻害物質;iNOS阻害物質;プロテアーゼ阻害物質(例えば、トリプターゼ阻害物質、およびエラスターゼ阻害物質);β−2インテグリンアンタゴニスト、およびアデノシンレセプターアゴニストもしくはアンタゴニスト(例えば、アデノシン2aアゴニスト);サイトカインアンタゴニスト(例えば、ケモカインアンタゴニスト(インターロイキン抗体(IL抗体)、特にIL−4療法、IL−13療法、またはそれらの組み合わせなど));あるいはサイトカイン合成の阻害物質が挙げられる。
【0083】
例えば、本発明の化合物と組み合わせて使用し得る代表的なホスホジエステラーゼ−4(PDE4)阻害物質、または混合PDE3/PDE4阻害物質としては、これらに限定されないが、シス4−シアノ−4−(3−シクロペンチルオキシ−4−メトキシフェニル)シクロヘキサン−1−カルボン酸、2−カルボメトキシ−4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オン;シス−[4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オール];シス−4−シアノ−4−[3−(シクロペンチルオキシ)−4−メトキシフェニル]シクロヘキサン−1−カルボン酸など、またはそれらの薬学的に受容可能な塩が挙げられる。他の代表的なPDE4または混合PDE4/PDE3阻害物質としては、AWD−12−281(elbion);NCS−613(INSERM);D−4418(Chiroscience and Schering−Plough);CI−1018、またはPD−168787(Pfizer);国際公開第99/16766号で開示されているベンゾジオキソール化合物(Kyowa Hakko);K−34(Kyowa Hakko);V−11294A(Napp);roflumilast(Byk−Gulden);国際公開第99/47505号で開示されているフタラジノン(pthalazinone)化合物(Byk−Gulden);Pumafentrine(Byk−Gulden、現在はAltana);arofylline(Almirall−Prodesfarma);VM554/UM565(Vernalis);T−440(Tanabe Seiyaku);およびT2585(Tanabe Seiyaku)が挙げられる。
【0084】
本発明の化合物と組み合わせて、そして本発明の化合物に加えて、使用し得る代表的なムスカリンアンタゴニスト(すなわち、抗コリン作用薬)としては、これらに限定されないが、アトロピン、硫酸アトロピン、酸化アトロピン、硝酸メチルアトロピン、臭化水素酸ホマトロピン、臭化水素酸ヒオスシアミン(d,l)、臭化水素酸スコポラミン、臭化イプラトロピウム、臭化オキシトロピウム、臭化チオトロピウム(tiotropium)、メタンテリン、臭化プロパンテリン、臭化メチルアニソトロピン、臭化クリジニウム、copyrrolate(Robinul)、ヨウ化イソプロパミド、臭化メペンゾラート、塩化トリジヘキセチル(Pathilone)、ヘキソシクリウム硫酸メチル(hexocyclium methylsulfate)、塩酸シクロペントレート、トロピカミド、塩酸トリヘキシフェニジル、ピレンゼピン、テレンゼピン(telenzepine)、AF−DX 116、およびメトクトラミン(methoctramine)など、またはそれらの薬学的に受容可能な塩;もしくは、塩として列挙された化合物の代わりに、それらの薬学的に受容可能な塩が挙げられる。
【0085】
本発明の化合物と組み合わせて使用し得る代表的な抗ヒスタミン剤(すなわち、H1−レセプターアンタゴニスト)としては、これらに限定されないが、エタノールアミン(例えば、マレイン酸カルビノキサミン、フマル酸クレマスチン、塩酸ジフェニルヒドラミン、およびジメンヒドリナート;エチレンジアミン(例えば、ピリラミンアムリエート(pyrilamine amleate))、塩酸トリペレナミン(tripelennamine)、クエン酸トリペレンアミン;アルキルアミン(例えば、クロルフェニラミン、およびアクリバスチン(acrivastine);ピペラジン(例えば、塩酸ヒドロキシジン、ヒドロキシジンパモエート、塩酸シクリジン、乳酸シクリジン、塩酸メクリジン、および塩酸セチリジン(cetirizine);ピペリジン(例えば、アステミゾール、塩酸レボカバスチン(levocabastine)、ロラタジン(loratadine)もしくはそのデスカルボエトキシアナログ(descarboethoxy analogue)、テルフェナジン、および塩酸フェキソフェナジン(fexofenadine);塩酸アゼラスチン;など、またはそれらの薬学的に受容可能な塩;もしくは、塩として列挙された化合物の代わりとして、それらの薬学的に受容可能な塩が挙げられる。
【0086】
本発明の化合物と組み合わせて投与される他の治療因子に適した用量は、約0.05mg/日〜約100mg/日の範囲である。
【0087】
次の調合物は、本発明の代表的な薬学的組成物を示している:
(調合物実施例A)
吸入による投与のための乾燥粉末を次のように調製する:
【0088】
【化4】
代表的な手順:本発明の化合物を微粉化し、次いでラクトースと混和する。次いで、この混和した混合物を、ゼラチン吸入カートリッジに詰める。このカートリッジの内容物を、粉末吸入器を使用して投与する。
【0089】
(調合物実施例B)
乾燥粉末吸入デバイス中における使用のための乾燥粉末調合物を次のように調製する:
(代表的な手順)本発明の微粉化した塩とラクトースとの調合容積比(bulk formulation ratio)が1:200である薬学的組成物を調製する。この組成物を1用量当たり約10μgと約100μgとの間の本発明の化合物を送達し得る乾燥粉末吸入デバイスに詰め込む。
【0090】
(調合物実施例C)
定量吸入器での吸入による投与のための乾燥粉末を次のように調製する:
代表的な手順:5重量%の本発明の塩、および0.1重量%のレシチンを含む懸濁液を、0.2gのレシチンを200mLの脱塩水に溶解させて生成した溶液に、10μm未満の平均粒径を有する微粉粒子として10gの本発明の化合物を分散させることにより調製する。この懸濁液を噴霧乾燥(spray dry)させ、得られた物質を1.5μm未満の平均直径を有する粒子に微粉化する。この粒子を加圧した1,1,1,2−テトラフルオロエタンと共にカートリッジに詰める。
【0091】
(調合物実施例D)
定量吸入器中における使用のための薬学的組成物を次のように調製する:
代表的な手順:5%の本発明の塩、0.5%のレシチン、および0.5%のトレハロースを含む懸濁液を、0.5gのトレハロース、および0.5gのレシチンを100mLの脱塩水に溶解させて生成したコロイド溶液に、10μm未満の平均粒径を有する微粉粒子として5gの活性成分を分散させることにより調製する。この懸濁液を噴霧乾燥させ、得られた物質を1.5μm未満の平均直径を有する粒子に微粉化する。この粒子を加圧した1,1,1,2−テトラフルオロエタンと共にキャニスターに詰める。
【0092】
(調合物実施例E)
ネブライザー吸入器中における使用のための薬学的組成物を次のように調製する:
代表的な手順:ネブライザー中における使用のためのエアゾール調合水を、クエン酸を用いて酸性化した1mLの0.9%塩化ナトリム溶液に、0.5mgの本発明の塩を溶解させることにより調製する。この混合物を攪拌し、活性成分が溶解するまで超音波処理する。この溶液のpHを、NaOHのゆっくりとした添加により約5の値に調節する。
【0093】
(調合物実施例F)
経口投与のための硬ゼラチンカプセル剤を、次のように調製する:
【0094】
【化5】
代表的な手順:これらの成分を完全に混和させ、次いで、硬カプセルに詰める(1カプセル当たり460mgの組成物)。
【0095】
(調合物実施例G)
経口投与のための懸濁液を次のように調製する:
【0096】
【化6】
代表的な手順:これらの成分を混合し、10mLの懸濁液につき100mgの活性成分を含む懸濁液を生成する。
【0097】
(調合物実施例H)
注射可能な調合物を次のように調製する:
【0098】
【化7】
代表的な手順:上記成分を混和させ、0.5N HCl、または0.5N NaOHを使用して、pHを4±0.5に調節する。
【0099】
(効用)
式Iの化合物は、β2アドレナリンレセプターアゴニスト、およびムスカリンレセプターアンタゴニスト活性の両方を有するため、本発明の式Iの化合物の1,2−エタンジスルホン酸塩は、β2アドレナリンレセプターもしくはムスカリンレセプターが介在する医学的状態(すなわち、β2アドレナリンレセプターアゴニストもしくはムスカリンレセプターアンタゴニストを用いた処置により改善される医学的状態)を処置するための治療因子として有用であると期待される。そのような医学的状態としては、例として、肺障害、または可逆性気道閉塞と関連する疾患(例えば、慢性閉塞性肺疾患(例えば、慢性的で息苦しい(wheezy)気管支炎、および肺気腫)、喘息、肺線維症、アレルギー性鼻炎、鼻漏など))を含む疾患が挙げられる。処置し得る他の状態としては、早期分娩、鬱病、鬱血性心不全、皮膚病(例えば、炎症性、アレルギー性、乾癬性、および増殖性皮膚疾患)、ペプシンの酸性を低下させることが望ましい状態(例えば、消化性および胃潰瘍)、および筋肉消耗性疾患が挙げられる。
【0100】
したがって、1つの実施形態において本発明は、肺障害を処置するための方法に向けられ、この方法は、処置を必要とする患者に治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を投与する工程を包含する。肺障害を処置するために使用される場合、本発明の塩は、典型的に、1日当たり複数回分の用量、1回で1日分の用量、または1回で1週間分の用量で、吸入により投与される。一般的に、肺障害を処置するための用量は、約10μg/日〜約200μg/日の範囲である。
【0101】
吸入により投与される場合、本発明の化合物は、典型的に気管支拡張を提供する効果を有する。したがって、その方法の別の局面において、本発明は、気管支拡張を必要としている患者に気管支拡張を提供する方法に向けられ、この方法は、気管支拡張を生じる量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を患者に投与する工程を包含する。一般的に、気管支拡張を提供するための用量は、約10μg/日〜約200μg/日の範囲である。
【0102】
1つの実施形態において、本発明は、慢性閉塞性肺疾患または喘息を処置する方法に向けられ、この方法は、処置を必要とする患者に治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を投与する工程を包含する。COPDまたは喘息を処置するために使用される場合、本発明の塩は、典型的に、1日当たり複数回分の用量、または1回で1日分の用量で、吸入により投与される。一般的に、COPDまたは喘息を処置するための用量は、約10μg/日〜約200μg/日の範囲である。本明細書中で使用される場合、COPDは、慢性閉塞性気管支炎および肺気腫を含む(例えば、Barnes,Chronic Obstructive Pulmonary Disease,N Engl J Med 2000:343:269−78を参照)。
【0103】
肺障害を処置するために使用される場合、本発明の塩は、必要に応じ、他の治療因子と組み合せて投与される。したがって、特定の実施形態において、本発明の薬学的組成物および方法は、治療有効量のステロイド抗炎症剤をさらに含む。本発明の1,2−エタンジスルホン酸塩の特性および効用は、当該技術分野で周知の種々のインビトロおよびインビボアッセイを使用して明らかにされ得る。例えば代表的なアッセイは、次の実施例においてさらに詳細に記載されている。
【実施例】
【0104】
次の(調製)および(実施例)は、本発明の具体的な実施例を説明するために提供される。しかしながら、これらの具体的な実施例は、他に示さない限り、決して本発明の範囲を限定することを意図していない。
【0105】
次の略語は、他に示さない限り次の意味を有し、本明細書中で使用され規定されていない任意の他の略語は、それらの標準的な意味を有する:
AC アデニリルシクラーゼ
Ach アセチルコリン
ATCC アメリカン・タイプ・カルチャー・コレクション
BSA ウシ血清アルブミン
cAMP 3’−5’環状アデノシン一リン酸
CHO チャイニーズハムスター卵巣
cM5 クローンチンパンジーのM5レセプター
DCM ジクロロメタン(すなわち、塩化メチレン)
DIPEA N,N−ジイソプロピルエチルアミン
dPBS ダルベッコリン酸緩衝生理食塩水
DMEM ダルベッコ改変イーグル培地
DMSO ジメチルスルホキシド
EDTA エチレンジアミン四酢酸
Emax 最大効果
EtOAc 酢酸エチル
EtOH エタノール
FBS 胎仔ウシ血清
FLIPR 蛍光定量的イメージングプレートリーダー
Gly グリシン
HATU O−(7−アザベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート
HBSS ハンクス緩衝食塩水
HEK ヒト胚腎臓細胞
HEPES 4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸
hM1 クローンヒトM1レセプター
hM2 クローンヒトM2レセプター
hM3 クローンヒトM3レセプター
hM4 クローンヒトM4レセプター
hM5 クローンヒトM5レセプター
HPLC 高速液体クロマトグラフィー
IBMX 3−イソブチル−1−メチルキサンチン
%Eff %効果
PBS リン酸緩衝生理食塩水
PyBOP ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート
rpm 1分当たりの回転数
TFA トリフルオロ酢酸
THF テトラヒドロフラン
Tris トリス(ヒドロキシメチル)アミノメタン
他に断りのない限り、試薬、出発物質、および溶媒は、供給業者(例えば、Aldrich、Fluka、Sigmaなど)から購入され、さらなる精製なしに使用された。
【0106】
下記の実施例において、HPLC分析を、Agilentが提供する3.5ミクロンの粒径を有するZorbax Bonus RP2.1×50mmのカラム(C14カラム)を用いたAgilent(Palo Alto,CA)1100シリーズ機器を使用して実施した。検出は、214nmにおけるUV吸収度による。HPLC10〜70データを、10%〜70%のB0.5mL/分の流速で6分間にわたり入手した。流動相Aは、2%−98%−0.1%のACN−H2O−TFA:そして流動相Bは、90%−10%−0.1%のACN−H2O−TFAであった。上記流動相AおよびBを使用して、HPLC5−35のデータ、およびHPLC10−90のデータを、5分間の勾配で入手した。
【0107】
液体クロマトグラフィー質量分析(LCMS)データを、Applied Biosystems(Foster City,CA)のAPI−150EX型の機器を使用して入手した。LCMS10−90のデータを、10%〜90%の流動相Bを用い、5分の勾配で入手した。
【0108】
小規模な精製を、Applied BiosystemsのAPI 150EX Prep Workstation systemを使用して入手した。流動相は、A:水+0.05容量(v/v)%TFA;そしてB:アセトニトリル+0.05容量(v/v)%TFAであった。アレイ(典型的に約3〜50mgの回収サンプルサイズ)に対し、次の条件が使用された:流速20mL/分;勾配15分、および5ミクロンの粒子を有する20mm×50mmのPrism RPカラム(Thermo Hypersil−Keystone,Bellefonete,PA)。より大規模な精製(典型的に100mgより大きなの粗サンプル)に対しては、次の条件を使用した:流速60mL/分;勾配30分、および10ミクロンの粒子を有する41.4mm×250mmのMicrosorb BDSカラム(Varian,Palo Alto,CA)。
【0109】
タングステンハロゲン光源、および589nmフィルターを用いたJasco Polarimeter(P−1010型)を使用して、20℃にてキラル化合物([α]20Dと示す)についての比旋光度を測定した。テスト化合物のサンプルは、典型的に、1mg/mLの水にて測定された。
【0110】
(調製1)
(4−アミノ−5−クロロ−2−メトキシ安息香酸メチル)
トルエン(9mL)とメタノール(1mL)との混合物中の4−アミノ−5−クロロ−2−メトキシ安息香酸(1.008g、5.0mmol)の溶液に、(トリメチルシリル)ジアゾメタン(ヘキサン中2.0M、3.0mL、6.0mmol)を、0℃にて滴下した。次いで、この反応混合物を室温に温め、16時間、攪拌した。過剰な(トリメチルシリル)ジアゾメタンを、反応混合物の鮮明な黄色が消えるまで、酢酸を加えることによってクエンチした。次いで、この混合物を真空下で濃縮することにより、オフホワイトの固体として表題の化合物を得、これをさらなる精製なしに使用した。
【0111】
(調製2)
(4−アクリロイルアミノ−5−クロロ−2−メトキシ安息香酸メチル)
調製2の粗生成物に、ジクロロメタン(10mL、0.5M)およびトリエチルアミン(2.1mL、15mmol)を加えた。この混合物を0℃に冷却し、塩化アクリロイル(812μL、10mmol)を攪拌しながら滴下した。2時間後、この反応物をメタノール(約2mL)を0℃にて加えることによりクエンチし、得られた混合物を室温にて15分間攪拌し、次いで、真空下で濃縮した。ジクロロメタン(30mL)および水(30mL)をこの残留物に加え、この混合物を十分に混合した。この層を分離し、水層をジクロロメタン(20mL)を用いて抽出した。有機層を合わせて、乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去することにより、褐色の泡沫状固体として表題の化合物を得、これをさらなる精製なしに使用した。
【0112】
(調製3)
(ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル)
ビフェニル−2−イソシアネート(97.5g、521mmol)および4−ヒドロキシ−1−ベンジルピペリジン(105g、549mmol)(共にAldrich、Milwaukee、WIから市販されている)を、12時間、70℃にて一緒に加熱し、その間、ビフェニル−2−イルカルバミン酸1−ベンジルピペリジン−4−イルエステルの形成をLCMSによりモニターした。次いで、この反応混合物を50℃に冷却し、エタノール(1L)を加え、次いで、6M 塩酸(191mL)をゆっくりと加えた。次いで、この反応混合物を周囲温度に冷却し、蟻酸アンモニウム(98.5g、1.56mol)を加え、窒素ガスを20分間溶液に通して活発に泡立てた。次いで、パラジウム(活性炭担持10重量%(乾燥ベース))(20g)を加えた。この反応混合物を、12時間、40℃にて加熱し、次いで、セライトパッドに通して濾過した。次いで、溶媒を減圧下で除去し、1M 塩酸(40mL)を粗残留物に加えた。次いで、水酸化ナトリウム(10N)を加えてpHを12に調節した。水層を酢酸エチル(2×150mL)を用いて抽出し、乾燥させ(硫酸マグネシウム)、次いで、溶媒を減圧下で除去することにより、表題の化合物(155g、100%)を得た。HPLC(10−70)Rt=2.52;MS m/z:[M+H+]C18H20N2O2についての計算値297.15;測定値297.3。
【0113】
(調製4)
(4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ}−5−クロロ−2−メトキシ安息香酸メチル)
調製2からの粗生成物に、調製3の生成物(1.33g、4.5mmol)、およびTHF(22.5mL)とメタノール(2.5mL)との混合物を加えた。この混合物を50℃にて16時間、攪拌しながら加熱し、次いで溶媒を、真空下で除去した。残留物をクロマトグラフィー法(シリカゲル;EtOAc)で分離することにより、オフホワイトの泡沫状固体として、表題の化合物(0.82g;Rf=0.4、3工程にわたる収率29%)を得た。MS m/z566.4(M+H、C30H32ClN3O6についての期待値565.20)。
【0114】
(調製5)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ヒドロキシメチル−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
THF(4.5mL)とメタノール(0.5mL)との混合物中の調製4の生成物(0.82mg、1.45mmol)の溶液に、0℃にてホウ化水素リチウム(32mg、1.45mmol)を加えた。この反応混合物を、室温に温めさせ、41時間、攪拌した。次いで、この反応物を、さらなる泡立ちが観察されなくなるまで、1N塩酸水を0℃にて加えることによりクエンチし、この混合物を10分間、攪拌した。溶媒を真空下で除去し、残留物をアセトニトリル(約2mL)に溶解させた。この溶液を、prep−RP−HPLC(勾配:0.05%のTFAを有する水中に2〜50%のアセトニトリル)により精製した。適切な分画を回収して合わせ、凍結乾燥することにより、トリフルオロ酢酸塩として表題の化合物を得た。この塩を酢酸イソプロピル(10mL)、および1N 水酸化ナトリウム水(10mL)を用いて処理し、有機層を回収し、乾燥させ(Na2SO4)、濾過し、そして溶媒を真空下で除去することにより、白色泡沫状固体として表題の化合物(161mg、収率21%)を得た。MS m/z538.4(M+H、C29H32ClN3O5についての期待値537.20)。
【0115】
(調製6)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイル)エチル]ピペリジン−4−イルエステル)
ジクロロメタン(3mL)中の調製5の生成物(161mg、0.3mmol)の溶液に、ジメチルスルホキシド(213μL、3.0mmol)、およびジイソプロピルエチルアミン(261μL、1.5mmol)を加えた。この混合物を−20℃に冷却し、三酸化硫黄ピリジン錯体(238mg、1.5mmol)をゆっくりと加えた。30分後、この反応混合物を水(約3mL)を加えることによりクエンチした。層を分離して、有機層を乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去することにより、薄黄色の固体として表題の化合物を得た。MS m/z536.3(M+H、C29H30ClN3O5についての期待値535.19)。
【0116】
(調製7)
(8−ベンジルオキシ−5−(2−ブロモアセチル)−1H−キノリン−2−オン)
((a)8−アセトキシ−1H−キノリン−2−オン)
8−ヒドロキシキノリン−N−オキシド(160.0g、1.0mol)(Aldrich、Milwaukee、WIから市販されている)、および無水酢酸(800mL、8.4mol)を、3時間、100℃にて加熱し、次いで、氷中で冷却した。この生成物を、ブフナー漏斗で回収し、無水酢酸(2×100mL)を用いて洗浄し、減圧下で乾燥させることにより、固体として8−アセトキシ−1H−キノリン−2−オン(144g)を得た。
【0117】
((b)5−アセチル−8−ヒドロキシ−1H−キノリン−2−オン)
1,2−ジクロロエタン(280mL)中の塩化アルミニウム(85.7g、640mmol)のスラリーを氷中で冷却し、工程(a)からの生成物(56.8g、280mmol)を加えた。この混合物を室温に温め、次いで、85℃にて加熱した。30分後、塩化アセチル(1.5mL、21mmol)を加え、この混合物をさらに60分間、加熱した。次いで、この反応混合物を冷却し、0℃にてよく攪拌しながら1N 塩酸(3L)に加えた。2時間攪拌後、固体をブフナー漏斗で回収し、水(3×250mL)を用いて洗浄し、減圧下で乾燥させた。いくつかのバッチから分離させた粗生成物(135g)を合わせ、6時間、ジクロロメタン(4L)を用いて粉砕した。得られた固体をブフナー漏斗で回収し、減圧下で乾燥させることにより、表題の化合物(121g)を得た。
【0118】
((c)5−アセチル−8−ベンジルオキシ−1H−キノリン−2−オン)
工程(b)からの生成物(37.7g、186mmol)に、N,N−ジメチルホルムアミド(200mL)、および炭酸カリウム(34.5g、250mmol)を加え、続いて臭化ベンジル(31.8g、186mmol)を加えた。この混合物を2.25時間、室温にて攪拌し、次いで、飽和塩化ナトリウム(3.5L)に0℃にて注ぎ入れ、1時間、攪拌した。この生成物を回収し、1時間、ブフナー漏斗で乾燥させ、得られた固体をジクロロメタン(2L)に溶解させ、この混合物を硫酸ナトリウムで乾燥させた。溶液をセライトパッドに通して濾過し、次いで、ジクロロメタン(5×200mL)を用いて洗浄した。次いで、合わせた濾液を濃縮乾固し、得られた固体を2時間、エーテル(500mL)を用いて粉砕した。この生成物をブフナー漏斗で回収し、エーテル(2×250mL)を用いて洗浄し、減圧下で乾燥させ、粉末として表題の化合物(44g)を得た。
【0119】
((d)8−ベンジルオキシ−5−(2−ブロモアセチル)−1H−キノリン−2−オン)
工程(c)からの生成物(20.0g、68.2mmol)を、ジクロロメタン(200mL)に溶解させ、0℃にて冷却した。三フッ化ホウ素ジエチルエーテラート(10.4mL、82.0mmol)をシリンジによって加え、この混合物を室温に温めることにより、濃厚な懸濁液を得た。この懸濁液を45℃(油浴)にて加熱し、ジクロロメタン(100mL)中の臭素(11.5g、72.0mmol)の溶液を、40分間にわたり加えた。この混合物をさらに15分間、45℃にて保ち、次いで、室温に冷却した。この混合物を減圧下で濃縮し、次いで1時間、10%炭酸ナトリウム水(200mL)を用いて粉砕した。固体をブフナー漏斗で回収し、水(4×100mL)を用いて洗浄し、減圧下で乾燥させた。2処理分の生成物を精製用に合わせた。この粗生成物(52g)を、クロロホルム(500mL)中の50%メタノールを用いて1時間、粉砕した。この生成物をブフナー漏斗で回収し、クロロホルム(2×50mL)およびメタノール(2×50mL)中の50%メタノールを用いて洗浄した。固体を減圧下で乾燥させることにより、粉末として表題の化合物(34.1g)を得た。
【0120】
(調製8)
(8−ベンゾイルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ(butyldimethylsilanyloxy))エチル]−1H−キノリン−2−オン)
((a)8−ベンジルオキシ−5−((R)−2−ブロモ−1−ヒドロキシエチル)−1H−キノリン−2−オン)
(R)−(+)−α,α−ジフェニルプロリノール(30.0g、117mmol)とトリメチルボロキシン(11.1mL、78mmol)とをトルエン(300mL)中で合わせ、30分間、室温にて攪拌した。この混合物を、150℃の油浴中に置き、液体を蒸留により取り除いた。トルエンを20mLのアリコートに加え、4時間、蒸留を続けた。総量で300mLのトルエンを加えた。次いで、この混合物を室温に冷却した。500μLのアリコートをエバポレートして乾固し、計量し(246mg)、この触媒濃度を1.8Mと決定した。
【0121】
8−ベンジルオキシ5−(2−ブロモアセチル)−1H−キノリン−2−オン(90.0g、243mmol)を、窒素下に置き、テトラヒドロフラン(900mL)を加え、続いて上記の触媒(トルエン中1.8M、15mL、27mmol)を加えた。この懸濁液を氷/イソプロパノール浴中で、−10±5℃に冷却した。ボラン(THF中1.0M、294mL、294mmol)を、4時間にわたり加えた。次いでこの反応物を、さらに45分間、−10℃にて攪拌し、次いで、メタノール(250mL)をゆっくりと加えた。この混合物を真空下で濃縮し、残留物を沸騰アセトニトリル(1.3L)に溶解させ、熱いうちに濾過し、次いで、室温に冷却した。結晶を濾過し、アセトニトリルを用いて洗浄し、真空下で乾燥させることにより、表題の化合物を得た(72.5g、196mmol、収率81%、95%ee、HPLCによる純度95%)。
【0122】
((b)8−ベンジルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
工程(b)の生成物(70.2g、189mmol)に、N,N−ジメチルホルムアミド(260mL)を加え、窒素下にてこの混合物を氷浴中で冷却した。2,6−ルチジン(40.3g、376mmol)を、5分間にわたり加え、次いで、tert−ブチルジメチルシリルトリフルオロメタンスルホナート(99.8g、378mmol)を、20℃未満の温度を維持しながらゆっくりと加えた。この混合物を45分間、室温に温めさせた。メタノール(45mL)を混合物に10分にわたって滴下し、この混合物を、酢酸エチル/クロロヘキサン(1:1、500mL)と水/ブライン(1:1、500mL)との間で分割した。有機物(organics)をさらに2度、水/ブライン(1:1、各回500mL)を用いて洗浄した。合わせた有機物を減圧下でエバポレートすることにより、薄黄色の油状体を得た。シクロヘキサン(400mL)を、2度に分けてこの油状体に加え、濃厚な白色のスラリーが生成するまで蒸留を続けた。シクロヘキサン(300mL)をスラリーに加え、得られた白色結晶を濾過し、シクロヘキサン(300mL)を用いて洗浄し、減圧下で乾燥させ、表題の化合物(75.4g、151mmol、収率80%、98.6%ee)を得た。
【0123】
(調製9A)
(8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
DMSO(1.7mL)中の調製8の生成物(1.00g、2.05mmol)とベンジルアミン(493μL、4.51mmol)との攪拌溶液を、105℃にて4時間、加熱した。この反応混合物を冷却させ、次いでEtOAc(10mL)を用いて希釈し、有機層を飽和塩化アンモニア水溶液(5mL)および1N 水酸化ナトリウム(5mL)を用いて洗浄し、乾燥させ(MgSO4)、溶媒を減圧下で除去した。この粗残留物を、カラムクロマトグラフィー(50% EtOAc/ヘキサン)により精製し、表題の化合物(700mg、67%)を得た。MS m/z:[M+H+]C31H38N2O3Siについての計算値515.27;測定値515.5。
【0124】
(調製9B)
(8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
500mLの三ッ口丸底フラスコに、8−ベンジルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン(43g、0.124mol、キラル純度約95%)、1−メチル−2−ピロリジノン(210mL)、およびベンジルアミン(28.3g、0.37mol)を加えた。得られた混合物を窒素を用いて洗い流し、次いで、90℃にて6時間、攪拌した。次いで、この混合物を室温に冷却し、水(300mL)および酢酸エチル(300mL)を加えた。層を分離し、有機層を水(200mL)、水と飽和塩化ナトリウム水溶液との1:1混合物(200mL)、および水(200mL)を用いて洗浄した。次いで、有機層を硫酸マグネシウムで乾燥させ、濾過し、減圧下で濃縮することにより、橙色の油状体として表題の化合物を得た。
【0125】
この橙色の油状体にヘプタン(200mL)および酢酸エチル(200mL)を加え、得られた混合物を65℃に加熱することにより、透明な溶液を得た。この溶液を室温に冷却し、一晩(約16時間)おいた時点で沈殿物を生成した。この沈殿物を濾過により回収し、立体化学的には不純な表題の化合物(8.85g、79.6%ee)を得た。濾液を減圧下で濃縮することにより、表題の化合物(38.6g、99.4%)を得た。この物質を先の物質(19.2g、99.5%ee)のバッチと合わせ、そしてヘプタン(250mL)および酢酸エチル(100mL)を加えた。この混合物を80℃に加熱し(濁りを帯びた〜透明な溶液)、次いで、室温に冷却し、一晩おいた。得られた沈殿物を濾過により回収し、白色固体(36.8g、98.4%ee、化学的純度99.9%)として表題の化合物を得た。濾液を減圧下で濃縮し、残渣をヘプタン(100mL)に溶解させた。得られた固体を回収することにより、黄褐色の固体(24g、キラル純度100%、化学的純度95%)として表題の化合物を得た。
【0126】
(調製10A)
(5−[(R)−2−アミノ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−8−ヒドロキシー1H−キノリン−2−オン)
エタノール(62mL)中の調製9Aの生成物(3.16g、6.15mmol)とパラジウム(活性炭担持10重量%(乾燥ベース))(1.58g)との攪拌溶液を、24時間、水素雰囲気下に置いた。この反応混合物をセライトに通して濾過し、メタノール(15mL)を用いて洗浄し、次いで、溶媒を減圧下で除去することにより、固体として表題の化合物(1.52g、4.55mmol、74%)を得た。
【0127】
(調製10B)
(5−[(R)−2−アミノ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−8−ヒドロキシ−1H−キノリン−2−オン酢酸塩)
8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン(100g、194mmol)および酢酸(17.5mL、291mmol)を、メタノール(1L)に溶解させた。この透明な溶液を窒素を用いてパージし、次いで、炭素担持水酸化パラジウム(20g、20重量%のPd(乾燥ベース)、湿気(約50%の水分))を加えた。室温にて6時間、水素ガスを攪拌溶液に通じて泡立て、その間に、濃厚なスラリーを生じさせた。次いで、この反応混合物を窒素を用いてパージし、メタノール(1L)を加えた。約30分間、得られた混合物を(生成物を溶解させるために)攪拌し、次いで、この混合物をセライトパッドに通して濾過した。濾液を約500mLの容量まで減圧下で濃縮し、得られたスラリーにエタノール(500mL)を加えた。得られた混合物を再度、減圧下で約500mLの容量まで濃縮し、得られた沈殿物を濾過により回収し、乾燥させることにより、黄白色の固体として表題の化合物を得た(65g、収率85%、純度>98%)。
【0128】
(調製11)
(ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−(tert−ブチルジメチルシラニルオキシ)−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロー5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
ジクロロメタン(0.5mL)とメタノール(0.5mL)との混合物中の調製6からの生成物に、調製10Aの生成物(124.1mg、3.1mmol)を加え、得られた混合物を、室温にて1.5時間、攪拌した。トリアセトキシホウ化水素ナトリウム(190.7mg、0.9mmol)を加え、得られた混合物を、室温にて15時間、攪拌した。この反応物を、水(約0.2mL)を加えることによりクエンチし、この混合物を真空下で濃縮することにより、表題の化合物を得、これをさらなる精製なしに使用した。MS m/z854.5(M+H、C46H56ClN5O7Siについての期待値853.36)。
【0129】
(調製12)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル)
ジクロロメタン(1.0mL、0.3M)中の調製11の生成物の懸濁液に、トリエチルアミン三フッ化水素塩(245μL、1.5mmol)を加えた。この混合物を、室温にて45時間、攪拌し、次いで、この混合物を真空下で濃縮した。残留物を、DMF(0.5mL)と、アセトニトリル/水(1:1、0.1%TFAを含む、0.6mL)と、TFA(0.3mL)と、アセトニトリル(約1mL)との混合物に溶解させ、この混合物を、prep−RP−HPLC(勾配:0.05%TFAを含む水中のアセトニトリル2〜50%)によって精製した。適切な分画を回収して合わせ、凍結乾燥させることにより、オフホワイトの固体として、表題の化合物のジトリフルオロ酢酸塩(100mg、収率34%、HPLCによる純度98.7%)を得た。MS m/z740.5(M+H、C40H42ClN5O7についての期待値739.28)。
【0130】
(実施例1)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
エタノール(0.2mL)中の1,2−エタンジスルホン酸二水和物(3.8mg、0.02mmol)の溶液を、イソプロパノールとジクロロメタン(1mL)との64:1v/v混合物中の調製12の生成物(14.3mg、0.02mmol)の溶液にゆっくりと加えた。得られた溶液を、45℃〜50℃にて約30分間、加熱した。次いで、この混合物をゆっくりと室温に冷却し、室温になった時点で溶液は僅かに濁った。この溶液を、一晩、窒素の緩流下で周囲温度においた。得られた沈殿物を濾過により回収し、乾燥させることにより、白色結晶性固体として表題の化合物(13mg、収率72%)を得た。
【0131】
(実施例2)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル(26.8mg、0.0362mmol)の溶液を、エタノール(5.36mL)中に調製し、室温にて、完全な溶解が得られるまで(5分)攪拌した。エタノール(0.2mL)中の1,2エタンジスルホン酸二水和物(8.2mg、0.0362mmol)の溶液を、約1分かけて最初の溶液にゆっくりと加えた。5分間、得られた懸濁液を攪拌し、次いで、窒素下で濾過により分離した。得られた沈殿物を乾燥させることにより、白色固体として表題の化合物(28.5mg、収率85%)を得た。
【0132】
(実施例3)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル(5g、6.75mmol、純度>99%)を、イソプロパノール(100mL)に溶解させ、水(20mL)に溶解させたエタンジスルホン酸二水和物(1.525mg、6.75mmol)を添加した。得られたスラリーを、室温にて1時間攪拌し、次いで約30℃にて一晩攪拌した。表題の化合物(6.0g)を、白色粉末として分離した。この生成物を、30℃にて48時間、イソプロパノール中の20%の水(100mL)中で加熱した。室温に冷却後、得られた沈殿物を濾過により単離し、2時間、風乾させることにより、表題の化合物(5.4g)を得た。
【0133】
(調製13)
(4−アクリロイルアミノ−5−クロロ−2−メトキシ安息香酸メチル)
オーバーヘッドスターラー、温度調節および追加漏斗(addition funnel)を備えた1リットルの3ツ口丸底フラスコに、4−アミノ−5−クロロ−2−メトキシ安息香酸メチル(44.2g、200mmol)、ジクロロメタン(500ml)、およびジイソプロピルエチルアミン(104.5mL、600mmol)を加えた。この得られた混合物を、室温にて成分が溶解するまで攪拌し、次いでこの混合物を0℃に冷却した。次いで、内部の反応混合物温度を10℃未満に維持しながら、塩化アクリロイル(16.25mL、200mmol)を滴下した。添加の合計時間は約30分であった。次いでこの反応混合物を、約2時間かけて0℃〜室温にゆっくりと温めた。次いで、飽和炭酸水素ナトリウム水溶液(200mL)、およびジクロロメタン(200mL)を加え、この混合物を15分間攪拌し、次いで層を分離した。ジクロロメタン層を1M 塩酸(200mL)を用いて洗浄し、次いで、減圧下で元の容量の約3分の1に濃縮し、濃厚なスラリーを得た。このスラリーを濾過し、濾塊をジクロロメタン(100mL)で洗浄し、乾燥させることにより、オフホワイトの固体として表題の化合物(36g、収率67%、HPLCによる純度>98%)を得た。
(調製14)
(4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ}−5−クロロ−2−メトキシ安息香酸メチル)
オーバーヘッドスターラー、温度調節および還流凝縮器を備えた1リットルの3ツ口丸底フラスコに、ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(36.3g、122mmol)、ジクロロメタン(500mL)、およびイソプロパノール(100mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いで、調製10からの生成物(30g、111.5mmol)を加えた。攪拌を、成分が溶解するまで室温で続け、次いでこの混合物を、還流下(50℃〜55℃)で18時間、加熱した。次いで、この反応混合物を室温に冷却し、エタノール(200mL)を加えた。この混合物を減圧下で約150mLの容量に濃縮し、濃厚なスラリーを得た。このスラリーを濾過し、濾塊をエタノール(50mL)で洗浄し、乾燥させることにより、白色固体として表題の化合物(58g、収率92%、HPLCによる純度99.5%)を得た。
【0134】
(調製15)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ヒドロキシメチル−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
2リットルの丸底フラスコに、調製14からの生成物(40g、70.8mmol)およびTHF(400mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いでこのフラスコを、窒素で5分間パージした。次いでこの混合物を0℃(内部温度)に冷却し、THF(106mL、106mmol)中の水素化リチウムアルミニウムの1M溶液を、内部の反応混合物温度を10℃未満に維持しながら、追加漏斗を通じて滴下した。合計添加時間は、約40分であった。次いでこの反応混合物を、0℃にて1時間、攪拌し、次いで内部の反応混合物温度を15℃未満に維持しながら、1M水酸化ナトリウム(200mL)を加えた。次いで、層を分離し、THF層を飽和塩化ナトリウム水溶液(100mL)で洗浄し、硫化ナトリウムで乾燥させ、濾過し、減圧下で濃縮することにより、白色固体として表題の化合物(38g、収率100%、HPLCによる純度94%)を得た。
【0135】
(調製16)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイル)エチル]ピペリジン−4−イルエステル)
1リットルの丸底フラスコに、調製15からの生成物(28g、52mmol)、およびジクロロメタン(500mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いで、活性酸化マンガン(IV)(45g、520mmol)を加えた。この反応混合物を、窒素下で室温にて12時間、攪拌し、次いで、セライトパッドに通して濾過した。次いで、この混合物を減圧下で濃縮し、残留物を真空下で一晩乾燥させることにより、黄色固体として表題の化合物(26g、収率93%、HPLCによる純度約93%)を得た。
【0136】
(調製17)
(ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−(tert−ブチルジメチルシラニルオキシ)−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロ−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
500mLの丸底フラスコに、調製16からの生成物(6g、11.2mmol)、およびジクロロメタン(50mL)を加えた。得られた混合物を成分が溶解するまで室温にて攪拌し、次いで、調製7の生成物(6g、15.0mmol)および乾燥メタノール(50mL)を加えた。この混合物を窒素下で室温にて2時間、攪拌し(透明な黄色から橙色の溶液)、次いで、この混合物を0℃〜5℃に冷却した。固体のナトリウムトリアセトキシボロヒドリド(7.2g、34mmol)を10分間かけて少しずつ加え、次いで、この反応混合物を、約2時間かけて、0℃〜室温にゆっくりと温めた。次いで、この混合物を0℃に冷却し、1M 水酸化ナトリウム水溶液(50mL)およびジクロロメタン(150mL)を加えた。この混合物を十分に攪拌し、次いで、層を分離した。有機層を飽和塩化ナトリウム水溶液(50mL)で洗浄し、濾過し、硫化ナトリウムで乾燥させ、濾過し、減圧下で濃縮することにより、黄色固体として表題の化合物(10.1g、収率100%、HPLCによる純度87%)を得た。
【0137】
(実施例4)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
50mLの丸底フラスコに、調製17からの生成物(2.0g、2.5mmol)およびジクロロメタン(10mL)を加えた。得られた混合物を、成分を溶解するまで室温にて攪拌し、次いで、三フッ化水素トリエチルアミン(1.2mL、7.5mmol)を加え、得られた混合物を、25℃にて20時間、攪拌した。次いで、メタノール(10mL)中の1,2−エタンジスルホン酸二水和物(0.56g、2.5mmol)の溶液を加え、この混合物を30℃にて2時間攪拌した時点で、濃厚な白色スラリーが形成された。このスラリーをゆっくりと濾過し、濾塊をメタノール(10mL)で洗浄し、2時間、風乾し、次いで、真空下で一晩乾燥させることにより、微細な白色粉末として表題の化合物(1.5g、HPLCによる純度>98%)を得た。
【0138】
(実施例5)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の精製)
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(80g)に、イソプロパノール(800mL)中の容量で20%の水の溶液を加えた。得られたスラリーを室温にて一晩おき、次いで濾過することにより、改善した結晶性および純度を有する表題の化合物(74g)を得た。
【0139】
(調製18)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の種結晶)
(工程(a))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(100mg)を、メタノール(20mL)中の13%の水に、約60℃にて溶解させた。得られた透明溶液を、密閉容器中で室温に冷却させた。48時間後、得られた板様結晶を、濾過により分離した。
【0140】
(工程(b))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(1.0g)を、メタノール(100mL)中の15%の水に、60〜65℃にて溶解させた。この透明な攪拌溶液を、30℃に冷却させ、次いで、工程(a)の結晶性生成物(4.2mg)を加えた。この溶液を20℃に冷却し、2時間攪拌した。得られた沈殿物を濾過により分離し、空気中で1時間、乾燥させることにより、表題の化合物(680mg)を得た。
【0141】
(工程(c))
工程(b)の手順を、工程(a)の生成物の代わりに工程(b)の生成物(20mg)を用いて繰り返し、表題の化合物(690mg)を得た。
【0142】
(工程(d))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(10g)を、メタノール(1L)中の15%の水に、60〜65℃にて溶解させた。この透明な攪拌溶液を、30℃に冷却させ、次いで、工程(c)の結晶性生成物(4.2mg)を加えた。この溶液を20℃に冷却し、18時間攪拌した。得られた沈殿物を濾過により分離し、空気中で2時間、乾燥させることにより、表題の化合物(5.5g)を得た。
【0143】
(実施例6)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の種結晶を使用した再結晶化)
12Lの丸底フラスコに、実施例5からの生成物(60g、64.5mmol)、水(0.9L)、およびメタノール(5.1L)を加えた。得られた混合物を、成分が溶解するまで攪拌しながら25℃から61〜65℃に加熱し、さらに20分間、60〜65℃にて攪拌した。この混合物を30℃に冷却させ、次いで、調製18の生成物(2g、2.15mmol)を加えた。この混合物を20℃にゆっくりと冷却し、得られたスラリーを、30℃にてさらに2時間、攪拌した。この生成物をメタノール(500mL)と共に濾過し、2時間空気中で乾燥させ、次いで真空中で25〜30℃にて18時間、乾燥させることにより、表題の化合物(43g、収率72%、純度99.2%)を得た。
【0144】
(実施例7)
(熱分析)
示差走査熱量測定(DSC)を、Thermal Analystコントローラーを備えたTA InstrumentsのQ−10型モジュールを使用して実施した。データを回収し、TA InstrumentsのThermal Solutionsソフトウェアを使用して分析した。約1mgのサンプルを正確に計量して蓋付きのアルミパンに入れた。このサンプルを、周囲温度から約300℃まで5℃/分の一次加熱傾斜(linear heating ramp)を使用して評価した。使用中、DSCセルを、乾燥窒素を用いてパージした。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なDSCの記録を図1に、実施例2のサンプルについての代表的なDSCの記録を図2に示す。
【0145】
熱重量分析(TGA)を、高分解能を備えたTA InstrumentsのQ−50型モジュールを使用して実施した。データを回収し、TA InstrumentsのThermal Solutionsソフトウェアを使用して分析した。約10mgのサンプルをプラチナパンに置き、周囲温度〜300℃の高分解能加熱速度(high resolution−heating rate)でスキャンした。使用中、天秤および加熱炉チャンバーを、窒素フローでパージした。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なTGAの記録を図1に示す。
【0146】
DSCの記録は、本発明の1,2−エタンジスルホン酸塩が、それぞれ約239℃、および約219℃の融点を有して熱安定性に優れており、約200℃未満で熱分解しないことを示している。
【0147】
(実施例8)
(粉末エックス線回析)
粉末エックス線回析パターンを、Si(Li)半導体検出器を備え、1.542Å(45kV、40mA)におけるCuのKα線を使用したThermo ARL X−Ray DiffractometerのX’TRA型(Thermo ARL SA、Switzerland)を用いて得た。この分析を、典型的に、2θ角で2〜30°の範囲にかけて1点当たり0.03°の刻み幅を用い、2°/分のスキャン速度で実施した。入手したままかまたは微細な粉末に磨り潰したサンプルを、機器の分析用のトップローディングカップに合わせて設計された特別注文の小容量インサートに優しく詰め込んだ。機器を週に一度、±0.02°の2θ角内のシリコン金属基準に対応させる。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なPXRDパターンを図3に示し、実施例2のサンプルについてのパターンを図4に示す。
【0148】
(実施例9)
(赤外分析法)
赤外(IR)吸収スペクトルを、Nicolet減衰全反射(ATR)サンプルホルダーを備えたAvatar 360 FT−IR分光計を使用して、4000〜675cm−1の周波数範囲にわたり決定した。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なIR吸収スペクトルは、図5に示されるように、704±1、748±1、768±1、841±1、900±1、1055±1、1104±1、1166±1、1218±1、1294±1、1408±1、1522±1、1609±1、1655±1、および1701±1において有意な吸収帯を有する。
【0149】
(実施例10)
(力学的水分吸着アセスメント)
力学的水分吸着(DMS:dynamic moisture sorption)アセスメント(水分吸着−脱着プロフィールとしても知られる)を、VTI大気マイクロバランス(atmospheric microbalance)、SGA−100システム(VTI Corp.,Hialeah,FL 33016)を使用して、手で砕いた調製18のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについて実施した。約10mgのサンプルサイズを使用し、湿度を分析の開始時に周囲値に設定した。典型的なDMS分析は、3つのスキャンからなる:周囲〜相対湿度(RH)2%、2%RH〜90%RH、90%RH〜5%RH(スキャン速度:5%RH/ステップ)。質量を2分毎に測定し、サンプルの質量が5連続点の間、0.01%以内に安定したときに、RHを次の値(+/−5%RH)に変更した。代表的なDMSの記録を、図6に示す。
【0150】
このDMSの記録は、本発明の1,2−エタンジスルホン酸塩が、中程度(<9%)の吸湿性と共に、可逆的な吸着/脱着プロフィールを有することを示している。この塩は、40%RH〜75%RHの湿度範囲において2.5%未満の増量がある。この可逆的な湿気吸着/脱着プロフィールは、本発明の結晶性塩が、受容可能な吸湿性を有し、潮解性ではないことを示している。
【0151】
(実施例11)
(元素分析、および対イオン比率)
実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの、炭素、水素、窒素、および硫黄の次の元素百分比を、Flash EA 1112 Elemental Analyzer(CE Elantech、Lakewood、NJ)を使用した燃焼分析により決定した:炭素52.95%、水素5.43%、窒素6.83%、および硫黄6.87%。硫黄の測定重量百分比から計算した結晶性サンプル中の1,2−エタンジスルホン酸の重量百分比は、20.4%で理論上の重量百分比20.4%と等しく、対イオン比率1:1が得られた。
【0152】
(調製A)
(細胞培養およびヒトβ1、β2またはβ3アドレナリンレセプターを発現する細胞からの細胞膜の調製)
クローニングしたヒトβ1、β2、またはβ3アドレナリンレセプターをそれぞれ安定的に発現するチャイニーズハムスター卵巣(CHO)細胞株を、500μg/mLのジェネテシンの存在下で10%FBSを用いたHams F−12培地において、ほぼコンフルエンシーまで培養した。細胞単層をPBS中の2mM EDTAで浮かせた。細胞を1,000rpmにおける遠心分離によりペレットにし、この細胞ペレットを、−80℃にて冷凍保存するか、または使用の直前に、細胞膜を調製した。β1およびβ2レセプターを発現する細胞膜の調製に関し、細胞ペレットを溶解緩衝液(10mM HEPES/HCl、10mM EDTA、4℃にてpH7.4)中に再懸濁させ、ぴったりと適合したDounceガラス製ホモジナイザー(30ストローク)を氷上で使用して均質化した。さらなるプロテアーゼ感受性のβ3レセプターを発現する細胞膜に関しては、細胞ペレットを、「Complete Protease Inhibitor Cocktail Tablets with 2mM EDTA」の錠剤(Rocheカタログ番号1697498、Roche Molecular Biochemicals,Indianapolis,IN)を緩衝液50mLにつき1つで補った溶解緩衝液(10mM Tris/HCl、pH7.4)中で均質化した。このホモジェネートを20,000×gにて遠心分離し、得られたペレットを、上記のような再懸濁および遠心分離により、溶解緩衝液を用いて1度、洗浄した。次いでこの最終ペレットを、氷冷の結合アッセイ緩衝液(75mM Tris/HCl pH7.4、12.5mM MgCl2、1mM EDTA)に再懸濁させた。この細胞膜懸濁液のタンパク質濃度を、Lowry et al.,1951,Journal of Biological Chemistry,193,265;およびBradford,Analytical Biochemistry,1976,72,248−54に記載される方法により決定した。全ての細胞膜を、−80℃にて分割して保存するか、または直ちに使用した。
【0153】
(調製B)
(細胞培養およびヒトM1、M2、M3およびM4ムスカリンレセプターを発現する細胞からの細胞膜の調製)
クローニングしたヒトhM1、hM2、hM3、およびhM4ムスカリンレセプターサブタイプをそれぞれ安定的に発現するCHO細胞株を、10%FBS、および250μg/mLのジェネテシンで補ったHams F−12培地において、ほぼコンフルエンシーまで培養した。これらの細胞を、CO25%、37℃のインキュベータ中で増殖させ、dPBS中の2mM EDTAで浮かせた。細胞を650×gにおける5分の遠心分離により回収し、細胞ペレットを、−80℃にて冷凍保存するか、または使用の直前に、細胞膜を調製した。細胞膜の調製に関し、細胞ペレットを溶解緩衝液に再懸濁させ、Polytron PT−2100組織粉砕器(tissue disrupter)(Kinematica AG;20秒×2バースト)を用いて均質化した。粗細胞膜を、40,000×gで4℃にて15分間、遠心分離した。次いでこの細胞膜ペレットを、再懸濁緩衝液を用いて再懸濁させ、Polytron組織粉砕器を用いて再度均質化した。この細胞膜懸濁液のタンパク質濃度を、Lowry et al.,1951,Journal of Biochemistry,193,265に記載される方法により決定した。全ての細胞膜を、分割して−80℃にて保存するか、または直ちに使用した。調製されたhM5レセプター細胞膜のアリコートを、Perkin Elmerから直接購入し、使用まで−80℃にて保存した。
【0154】
(アッセイテスト手順A)
(ヒトβ1、β2およびβ3アドレナリンレセプターについての放射性リガンド結合アッセイ)
結合アッセイを、96ウェルマイクロタイタープレートにおいて、アッセイ緩衝液(75mM Tris/HCl 25℃にてpH7.4、12.5mM MgCl2、1mM EDTA,0.2% BSA)中のヒトβ1、β2、またはβ3アドレナリンレセプターを含む10〜15μgの細胞膜タンパク質を用い、総アッセイ容量100μLで実施した。放射性リガンドのKd値の決定のための飽和結合調査を、β1およびβ2レセプターについては[3H]−ジヒドロアルプレノロール(NET−720、100 Ci/mmol、PerkinElmer Life Sciences Inc.,Boston,MA)を、そして[125I]−(−)−ヨードシアノピンドロール(NEX−189、220 Ci/mmol、PerkinElmer Life Sciences Inc.,Boston,MA)を、0.01nM〜20nMの範囲の10もしくは11の異なる濃度で使用して実施した。テスト化合物のKi値の決定のための置換アッセイを、[3H]−ジヒドロアルプレノロールを1nMにて、そして[125I]−(−)−ヨードシアノピンドロールを0.5nMにて用い、10pM〜10μMの範囲における10もしくは11の異なる濃度のテスト化合物について実施した。非特異的な結合を、10μMのプロプラノロールの存在下で決定した。分析物を37℃にて1時間インキュベートし、次いで、β1およびβ2レセプターについてはGF/B、β3レセプターについてはGF/Cガラス繊維フィルタープレート(Packard BioScience Co.,Meriden,CT)(0.3%のポリエチレンイミンに予浸しておく)を用いた急速濾過により、結合反応を終わらせた。フィルタープレートを濾過緩衝液(75mM Tris/HCl 4℃にてpH7.4、12.5mM MgCl2、1mM EDTA)を用いて3回洗浄し、結合していない放射能を除去した。次いでこのプレートを乾燥させ、50μLのMicroscint−20液体シンチレーション液(Packard BioScience Co.,Meriden,CT)を加え、プレートをPackard Topcount液体シンチレーションカウンター(Packard BioScience Co.,Meriden,CT)で計数した。結合データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、ワンサイト・コンペティション(one−site competition)についての3パラメーターモデルを使用して、非線形回帰分析により分析した。曲線の最小値(curve minimum)を、10μMのプロプラノロールの存在下で決定された非特異的な結合についての値に固定した。テスト化合物についてのKi値を、観測されたIC50値、および放射性リガンドのKd値から、Cheng−Prusoff方程式(Cheng Y,and Prusoff WH.,Biochemical Pharmacology,1973,22,23,3099−108)を使用して計算した。
【0155】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターに対するより高い結合親和力を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトβ2アドレナリンレセプターに対し、10nM未満のKi値を有することが見出された。
【0156】
(アッセイテスト手順B)
(ムスカリンレセプターについての放射性リガンド結合アッセイ)
クローニングしたヒトムスカリンレセプターについての放射性リガンド結合アッセイを、総アッセイ容量100μLで、96ウェルマイクロタイタープレートにて実施した。hM1、hM2、hM3、hM4、またはhM5のいずれかのムスカリンサブタイプを安定的に発現するCHO細胞の細胞膜を、類似したシグナル(cpm)を得るために、次の特異的なターゲットタンパク質濃度(μg/ウェル)までアッセイ緩衝液で希釈した:hM1については10μg、hM2については10〜15μg、hM3については10〜20μg、hM4については10〜20μg、そしてhM5については10〜12μg。これらの細胞膜を、アッセイプレートへの添加前に、Polytron組織粉砕器を使用して(10秒)簡単に均質化した。放射性リガンドのKD値を決定するための飽和結合調査を、0.001nM〜20nMの範囲の濃度でL−[N−メチル−3H]スコポラミンメチルクロリド([3H]−NMS)(TRK666、84.0 Ci/mmol、Amersham Pharmacia Biotech,Buckinghamshire,England)を使用して実施した。テスト化合物のKi値の決定のための置換アッセイを、1nMの[3H]−NMS、および11の異なるテスト化合物濃度を用いて実施した。これらのテスト化合物を、初めに希釈緩衝液に溶解して400μMの濃度にし、次いで、連続的に希釈緩衝液で5×に希釈して、最終濃度を10pM〜100μMの範囲にした。アッセイプレートへの添加順序および容量は次のとおりである:25μLの放射性リガンド、25μLの希釈したテスト化合物、そして50μLの細胞膜。アッセイプレートを、37℃にて60分間、インキュベートした。結合反応を、1%BSA中で予め処理したGF/Bガラス繊維フィルタープレート(PerkinElmer Inc.,Wellesley,MA)を用いた急速濾過により終わらせた。フィルタープレートを、洗浄緩衝液(10mM HEPES)で3回すすぎ、結合していない放射能を除去した。次いで、これらのプレートを風乾させ、50μLのMicroscint−20液体シンチレーション液(PerkinElmer Inc.,Wellesley,MA)を各ウェルに加えた。次いでこれらのプレートを、PerkinElmer Topcount液体シンチレーションカウンター(PerkinElmer Inc.,Wellesley,MA)で計数した。結合データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、ワンサイト・コンペティションモデルを使用して、非線形回帰分析により分析した。テスト化合物についてのKi値を、観測されたIC50値、および放射性リガンドのKD値から、Cheng−Prusoff方程式(Cheng Y;Prusoff WH.(1973)Biochemical Pharmacology,22(23):3099−108)を使用して計算した。Ki値をpKi値に変換し、幾何平均および95%信頼区間を決定した。次いで、これらの要約統計を、データ報告のためにKi値に変換し直した。
【0157】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターに対するより高い結合親和力を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトM2およびM3ムスカリンレセプターに対し、10nM未満のKi値を有することが見出された。
【0158】
(アッセイテスト手順C)
(非相同的にヒトβ1、β2またはβ3アドレナリンレセプターを発現するCHO細胞株における細胞全体cAMPフラッシュアッセイ)
cAMPアッセイを、製造業者の説明に従い、[125I]−cAMPを用いたFlashplate Adenylyl Cyclase Activation Assay System(NEN SMP004,PerkinElmer Life Sciences Inc.,Boston,MA)を使用して、放射線免疫アッセイ形式で実施した。βレセプターアゴニストの効力(EC50)の決定のために、クローニングしたヒトβ1、β2またはβ3アドレナリンレセプターを安定的に発現するCHO−K1細胞株を、10%FBS、およびジェネテシン(250μg/mL)で補ったHams F−12培地において、ほぼコンフルエンシーまで培養した。細胞をPBSですすぎ、2mM EDTAまたはトリプシン−EDTA溶液(0.05%トリプシン/0.53mM EDTA)を含むdPBS(ダルベッコリン酸緩衝生理食塩水、CaCl2およびMgCl2を含まない)で分離した。Coulter細胞カウンターで細胞を計数後、細胞を1,000rpmにおける遠心分離によってペレットにし、予め室温に温めたIBMX(PerkinElmer Kit)を含む刺激緩衝液に再懸濁させ、1.6×106〜2.8×106細胞/mLの濃度にした。1ウェル当たり約60,000〜80,000細胞を、このアッセイで使用した。テスト化合物(DMSO中10mM)をBeckman Biomek−2000中の0.1%BSAを含むPBS中で希釈し、100μM〜1pMの範囲の11の異なる濃度にてテストした。反応物を37℃にて10分間インキュベートし、[125I]−cAMP(NEN SMP004、PerkinElmer Life Sciences,Boston,MA)を含む100μLの冷たい検出緩衝液を加えることにより止めた。生成されたcAMPの量(pmol/ウェル)を、サンプルについて観測された数、および製造業者の使用者マニュアルに記載されるcAMP標準に基づき計算した。データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド方程式(sigmoidal equation)を用いて、非線形回帰分析により分析した。Cheng−Prusoff方程式(Cheng Y,and Prusoff WH.,Biochemical Pharmacology,1973,22,23:3099−108)を使用して、EC50値を計算した。
【0159】
このアッセイにおいて、より低いEC50値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトβ2アドレナリンレセプターに対し、10nM未満のEC50値を有することが見出された。
【0160】
(アッセイテスト手順D)
(ムスカリンレセプターサブタイプについての拮抗作用の機能アッセイ)
(A.アゴニストが媒介する[35S]GTPγS結合の遮断)
テスト化合物の機能的効力を、hM2レセプターを発現するCHO−K1細胞に結合するオキソトレモリンが刺激する[35S]GTPγS結合を遮断する化合物の能力を計測することにより決定した。
使用のときに、冷凍細胞膜を解凍し、次いで、最終ターゲット組織濃度1ウェル当たり5〜10μgのタンパク質を用いてアッセイ緩衝液中で希釈した。この細胞膜を、Polytron PT−2100組織粉砕器を使用して簡単に均質化し、次いで、アッセイプレートに加えた。
【0161】
アゴニストオキソトレモリンによる[35S]GTPγS結合の刺激に対するEC90値(90%の最大応答に対する有効濃度)をそれぞれの実験において決定した。
【0162】
オキソトレモリンが刺激する[35S]GTPγS結合を阻害するテスト化合物の能力を決定するために、次の:[35S]GTPγS(0.4nM)を有する25μLのアッセイ緩衝液、25μLのオキソトレモリン(EC90)およびGDP(3μM)、25μLの希釈したテスト化合物、ならびにhM2レセプターを発現する25μLのCHO細胞の細胞膜を、96ウェルプレートの各ウェルに加えた。次いで、アッセイプレートを37℃にて60分間インキュベートした。これらのアッセイプレートを、PerkinElmer 96ウェルハーベスターを使用し、1%BSAで予め処理したGF/Bフィルターを用いて濾過した。これらのプレートを、3回×3秒間、氷冷の洗浄緩衝液を用いてすすぎ、次いで、風乾または真空乾燥させた。Microscint−20シンチレーション液(50μL)を、各ウェルに加え、各プレートを密封し、放射能をTopcounter(PerkinElmer)で算出した。データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、非線形回帰分析であるワンサイト・コンペティションを使用して分析した。Cheng−Prusoff方程式を、Ki値を計算するために使用し、KDおよび[L]のリガンド濃度として、それぞれアッセイにおけるテスト化合物およびオキソトレモリン濃度についての濃度−応答曲線のIC50値を使用した。
【0163】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、hM2レセプターを発現するCHO−K1細胞におけるオキソトレモリンが刺激する[35S]GTPγS結合の遮断に対して、約10nM未満のKi値を有することが見出された。
【0164】
(B.FLIPRアッセイによるアゴニストが媒介するカルシウム放出の遮断)
Gqタンパク質に結合するムスカリンレセプターサブタイプ(M1、M3、およびM5レセプター)は、アゴニストがレセプターに結合すると、ホスホリパーゼC(PLC)経路を活性化する。その結果、活性化されたPLCはホスファチジル(phosphatyl)イノシトール二リン酸(PIP2)を、ジアシルグリセロール(DAG)とホスファチジル−1,4,5−三リン酸(IP3)とに加水分解し、次いで、細胞内貯蔵(すなわち、小胞体および筋小胞体)からカルシウム放出を生じさせる。FLIPR(Molecular Devices,Sunnyvale,CA)アッセイは、遊離カルシウムが結合するときに蛍光を発するカルシウム感受性色素(Fluo−4AM,Molecular Probes,Eugene,OR)を使用することにより、この細胞内カルシウムの増加を利用する(capitalize)。この蛍光イベントを、ヒトM1およびM3レセプター、ならびにチンパンジーM5レセプターを有するクローンされた細胞の単層から蛍光の変化を検出するFLIPRにより、実時間で計測する。アンタゴニストの効力を、アゴニストが媒介する細胞内カルシウムの増加を阻害するアンタゴニストの能力によって決定し得る。
【0165】
FLIPRカルシウム刺激アッセイに関し、hM1、hM3、およびcM5レセプターを安定的に発現するCHO細胞を、アッセイを実施する前夜に、96ウェルFLIPRプレートに播種した。播種した細胞をFLIPR緩衝液(10mM HEPES、pH7.4、2mM 塩化カルシウム、カルシウムおよびマグネシウムを含まないハンクス緩衝食塩水中(HBSS)の2.5mM プロベネシド)を用いて、Cellwash(MTX Labsystems,Inc.)により2回洗浄し、増殖培地、そしてFLIPR緩衝液50μL/ウェルを残した。次いでこれらの細胞を、二酸化炭素5%で37℃にて40分間、50μL/ウェルの4μM FLUO−4AM(2X溶液が生成された)を用いてインキュベートした。染色インキュベーションに続き、FLIPR緩衝液で2回洗浄し、最終用量50μL/ウェルを残した。
アンタゴニストの効力を決定するために、後でアンタゴニストの効力をEC90濃度におけるオキソトレモリン刺激に対して計測し得るように、まず、オキソトレモリンについての細胞内Ca2+放出の用量依存性刺激を決定した。細胞をまず20分間、化合物希釈緩衝液を用いてインキュベートし、続くアゴニストの添加はFLIPRによって実施された。オキソトレモリンについてのEC90値を、以下のFLIPR測定およびデータ整理の節にて、式ECF=((F/100−F)^1/H)*EC50とともに詳述される方法に従って得た。オキソトレモリンのEC90濃度がアンタゴニスト阻害アッセイプレート中の各ウェルに加えられるように、3×ECFのオキソトレモリン濃度を刺激プレート中に調製した。
【0166】
FLIPRに使用されるパラメーターは:0.4秒の曝露時間、0.5ワットのレーザー強度、488nmの励起波長、550nmの発光波長であった。アゴニストの添加前に、10秒間の蛍光の変化を測定することにより、基準線(baseline)を決定した。アゴニストの刺激に続き、FLIPRは、0.5秒から1秒毎の蛍光の変化を1.5分間、最大蛍光変化を得るために、継続的に計測した。
発光の変化を各ウェルについて、最大蛍光マイナス基準線蛍光として表した。生データを、GraphPad Prism(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド用量反応についてのビルトインモデルを使用して、非線形回帰分析により、薬物濃度の対数に対して分析した。アンタゴニストのK1値を、KDとしてオキソトレモリンEC50値、およびCheng−Prusoff方程式(Cheng&Prusoff,1973)に従うリガンド濃度についてのオキソトレモリンEC90を使用し、Prismにより決定した。
【0167】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、hM1、hM3、およびcM5レセプターを安定的に発現するCHO細胞におけるアゴニストが媒介するカルシウム放出の遮断に対し、約10nM未満のKi値を有することが見出された。
【0168】
(アッセイテスト手順E)
(内因的にヒトβ2アドレナリンレセプターを発現する肺上皮細胞株を用いた細胞全体cAMPフラッシュアッセイ)
内因的濃度のβ2アドレナリンレセプターを発現している細胞株におけるアゴニストの効力および効能(内因性の活性)の決定に関し、ヒト肺上皮細胞株(BEAS−2B)を使用した(ATCC CRL−9609、American Type Culture Collection,Manassas,VA)(January B,et al.,British Journal of Pharmacology,1998,123,4,701−11)。細胞を、完全な無血清培地(エピネフリンおよびレチノイン酸を含むLHC−9 MEDIUM、カタログ番号181−500、Biosource International,Camarillo,CA)において、75〜90%のコンフルエンシーまで培養した。アッセイ前日に、培地をLHC−8(エピネフリンおよびレチノイン酸を含まない、カタログ番号141−500、Biosource International,Camarillo,CA)に変更した。
cAMPアッセイを、製造業者の説明に従い、[125I]−cAMPを用いたFlashplate Adenylyl Cyclase Activation Assay System(NEN SMP004,PerkinElmer Life Sciences Inc.,Boston,MA)を使用して、放射線免疫アッセイ形式で実施した。
【0169】
アッセイの当日、細胞をPBSですすぎ、PBS中の5mM EDTAで擦る(scrape)ことによって浮かせ、計数した。細胞を1,000rpmにて遠心分離によりペレットにし、37℃に予め温めておいた刺激緩衝液に、600,000細胞/mLの最終濃度で再懸濁させた。このアッセイでは、100,000〜120,000細胞/ウェルの最終濃度で、細胞を使用した。テスト化合物を、Beckman Biomek−2000で、アッセイ緩衝液(75mM Tris/HCl 25℃にてpH7.4、12.5mM MgCl2、1mM EDTA、0.2%BSA)で、連続的に希釈した。このアッセイでは、10μM〜10pMの範囲における11の異なる濃度で、テスト化合物をテストした。反応物を37℃にて10分間、インキュベートし、100μLの氷冷の検出緩衝液の添加により止めた。プレートを密閉し、4℃にて一晩中インキュベートし、翌朝、Topcountシンチレーションカウンター(Packard BioScience Co.,Meriden,CT)で計数した。反応物1mL当たりに生成されたcAMP量を、製造業者の使用者マニュアルに記載のとおり、サンプルについて観測された数、およびcAMP標準に基づき算定した。データを、GraphPad Prism Softwareパッケージ(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド用量反応に関する4−パラメーターモデルを使用して、非線形回帰分析により分析した。
【0170】
このアッセイにおいて、より低いEC50値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、β2アドレナリンレセプターに対し、約10nM未満のEC50値を有することが見出された。
【0171】
(アッセイテスト手順F)
(アセチルコリン誘発またはヒスタミン誘発気管支収縮のモルモットモデルにおける気管支保護(bronchoprotection)の持続時間)
これらのインビボアッセイを使用して、ムスカリンレセプターアンタゴニストおよびβ2アドレナリンレセプターアゴニスト活性の両方を示すテスト化合物の気管支保護効果を評価した。アセチルコリン誘発気管支収縮モデルにおけるムスカリンアンタゴニスト活性を分離するために、これらの動物にプロパノロール(βレセプター活性を遮断する化合物)を、アセチルコリンの投与の前に投与した。ヒスタミン誘発気管支収縮モデルにおける気管支保護の持続時間は、β2アドレナリンレセプターアゴニスト活性を反映する。
【0172】
250gと350gとの間の重さの6匹の雄のモルモット(Duncan−Hartley(HsdPoc:DH)Harlan,Madison,WI)群を、それぞれケージカードにより同定した。調査の間中、動物に自由に食料および水を入手させた。
【0173】
テスト化合物を、全身曝露投薬チャンバ(whole−body exposure dosing chamber)(R&S Molds,San Carlos,CA)で10分間にわたり、吸引により投与した。この投薬チャンバを、エアゾールが中央の多岐管から6つの個々のチャンバに同時に送達されるように設計した。モルモットをテスト化合物またはビヒクル(WFI)のエアゾールに曝露させた。これらのエアゾールを、22psiの圧力にて気体の混合物(CO2=5%、O2=21%、およびN2=74%)により作動するLC Star Nebulizer Set(Model 22F51,PARI Respiratory Equipment,Inc.Midlothian,VA)を使用し、水溶液から生じさせた。この作動圧においてネブライザーを通した気体流は、約3L/分であった。この生じたエアゾールを、陽圧によりチャンバ内に押し流した。エアゾール化した溶液の送達の間、希釈用空気は使用されなかった。10分の噴霧療法の間、約1.8mLの溶液を噴霧した。この値を、充填したネブライザーの噴霧前および噴霧後の重量を比較することにより、重量分析的に測定した。
【0174】
吸引により投与されるテスト化合物の気管支保護効果を、投与後1.5時間、24時間、48時間、および72時間に、全身プレチスモグラフ法を使用して評価した。
肺評価の開始45分前に、各モルモットをケタミン(43.75mg/kg)、キシラジン(3.50mg/kg)、およびアセプロマジン(1.05mg/kg)の筋肉内注射で麻酔した。手術部位を剃り、70%アルコールで清潔にした後、頸部腹側面の2〜3cmの正中切開を実施した。次いで、頚静脈を分離し、生理食塩水中のアセチルコリン(Ach)またはヒスタミンの静脈内注入を考慮して、生理食塩水を充填したポリエチレンカテーテル(PE−50,Becton Dickinson,Sparks,MD)と共にカニューレを挿入した。次いで、この気管を切開解放(dissected free)し、14Gテフロン(登録商標)チューブ(#NE−014,Small Parts,Miami Lakes,FL)と共にカニューレを挿入した。必要な場合は、前記した麻酔混合物のさらなる筋肉内注射によって知覚麻痺を維持した。知覚麻痺の程度をモニターし、動物が足をつまんで(pinch)反応する場合、または呼吸数が100呼吸/分を超えた場合に調節した。
【0175】
カニューレの挿入が完了すると、動物をプレチスモグラフ(#PLY3114,Buxco Electronics,Inc.,Sharon,CT)に置き、食道圧力カニューレ(PE−160,Becton Dickinson,Sparks,MD)を、肺の駆動圧(プレッシャー)を測定するために挿入した。テフロン(登録商標)導管チューブをプレチスモグラフの開口部に取り付け、モルモットにチャンバの外からの大気を吸わせた。次いで、チャンバを密閉した。体温を維持するために加熱灯を使用し、モルモットの肺を、10mL較正シリンジ(calibration syringe)(#5520 Series,Hans Rudolph,Kansas City,MO)を使用して、4mLの空気で3回膨張させ、気道下部が無空気状態にならないことを、そして動物が過呼吸に陥らないことを確実にした。
基準線の値がコンプライアンスについて0.3〜0.9mL/cm H2Oの範囲内であると決定し、抵抗について0.1〜0.199cm H2O/mL毎秒の範囲内であると決定すると、肺評価を開始した。Buxco肺測定コンピュータープログラムにより、肺の値の収集および導出を実施した。
【0176】
このプログラムをスタートさせると、実験プロトコルおよびデータ収集を開始した。各呼吸に関するプレチスモグラフ中で起こる、容量の経時変化を、Buxco圧力変換器により測定した。この経時シグナルを取り入れることにより、各呼吸についてのフローの測定値を算定した。このシグナルを、Sensym圧力変換器(#TRD4100)を使用して回収された肺の駆動圧力の変化と共に、Buxco(MAX 2270)前置増幅器によりデータ収集インターフェース(#SFT3400および#SFT3813)と結びつけた。全ての他の肺のパラメーターをこれら2つのインプットから引き出した。
【0177】
基準線値を5分間で収集し、その後、モルモットをAchまたはヒスタミンでチャレンジした。ムスカリンアンタゴニストの効果を評価する場合、プロパノロール(5mg/Kg、iv)(Sigma−Aldrich,St.Louis,MO)を、Achでチャレンジする15分前に投与した。Ach(Sigma−Aldrich,St.Louis,MO)(0.1mg/mL)を、シリンジポンプ(sp210iw、World Precision Instruments,Inc.,Sarasota,FL)から1分間、次の用量および指定された実験の開始からの時間で静脈内に注入した:5分の時点で1.9μg/分、10分の時点で3.8μg/分、15分の時点で7.5μg/分、20分の時点で15.0μg、25分の時点で30μg/分、および30分の時点で60μg/分。あるいは、テスト化合物の気管支保護を、β遮断化合物での前処理なしのアセチルコリンチャレンジモデルで評価した。
【0178】
テスト化合物のβ2アドレナリンレセプターアゴニスト効果を評価する場合、ヒスタミン(25μg/mL)(Sigma−Aldrich,St.Louis,MO)を、シリンジポンプから1分間、次の用量および指定された実験の開始からの時間で静脈内に注入した:5分の時点で0.5μg/分、10分の時点で0.9μg/分、15分の時点で1.9μg/分、20分の時点で3.8μg、25分の時点で7.5μg/分、および30分の時点で15μg/分。抵抗またはコンプライアンスが、Achまたはヒスタミンのそれぞれの投薬に続く3分の時点で基準線値に戻らない場合は、モルモットの肺を、10mL較正シリングから4mLの空気で3回膨張させた。記録された肺のパラメーターには、呼吸回数(呼吸/分)、コンプライアンス(mL/cm H2O)、および肺抵抗(cm H2O/mL毎秒)が含まれる。このプロトコルの35分の時点で肺機能測定が完了すると、モルモットをプレチスモグラフから取り出し、二酸化炭素の窒息により安楽死させた。
【0179】
これらのデータを2つの方法のうちの一方で評価した:
(a)肺抵抗(RL、cm H2O/mL毎秒)を、「圧力の変化」対「流量の変化」の比から計算した。AChへのRL応答(60μg/分、IH)をビヒクルおよびテスト化合物群について、コンピュータで算定した。ビヒクル−処置動物において、各前処理時点の平均ACh応答を計算し、各テスト化合物用量にて対応する前処理時点のACh応答の阻害%をコンピュータで算定するために使用した。「RL」についての阻害用量−応答曲線を、気管支保護ID50(ACh(60μg/分)気管支収縮物質(bronchocontrictor)応答を50%阻害するのに必要とされる用量)を推定するために、Windows(登録商標)用GraphPad Prism,version 3.00(GraphPad Software,San Diego,California)を使用して、4パラメーター論理方程式を用いて調節した。使用された方程式は次のとおりである:
Y=Min+(Max−Min)/(1+10((logID50−X)*Hillslope))
ここでXは用量の対数であり、Yは応答(Achが誘発するRL増加の阻害%)である。YはMinで始まり、漸近的にMaxへとS字状(sigmoidal shape)を描く。
【0180】
(b)基準線の肺抵抗を倍にするために必要とされるAchまたはヒスタミンの量、として規定される量PD2は、次の方程式(クリニックでPC20値を計算するために使用される方程式から導かれる)を使用して、Achまたはヒスタミンのチャレンジの範囲にわたるフローおよびプレッシャーから導かれる肺の抵抗値を使用して算定される(Am.Thoracic Soc,2000を参照)。
【0181】
【数1】
ここで:
C1=C2に先行するAchまたはヒスタミンの濃度
C2=肺抵抗(RL)の少なくとも2倍の増加をもたらすAchまたはヒスタミンの濃度
R0=基準線RL値
R1=C1に従うRL値
R2=C2に従うRL値
データの統計分析を、両側スチューデントt検定を使用して実施した。P値<0.05を有意とみなした。
【0182】
このアッセイにおいてテストされる場合、式Iの化合物は、MCh誘発気管支収縮およびHis誘発気管支収縮に対する、用量依存性気管支保護効果をもたらした。さらに、式Iの化合物は、このアッセイにおいて少なくとも約24時間の気管支保護活性の持続時間(PD T1/2)を有した。
【0183】
本発明は、その特異的な局面または実施形態を参照して記載されているが、本発明の精神および範囲から逸脱することなしに、種々の変更がなされ得、等価物を代わりに使用し得ることが、当業者には理解される。さらに、適用し得る特許法および規制により認可される範囲まで、本明細書中で引用するすべての刊行物、特許および特許出願を、各書類が個々に本明細書中に参考として援用された場合と同程度に、それらの全体が参考として本明細書中で援用される。
【図面の簡単な説明】
【0184】
本発明の種々の局面が、添付の図の参照により説明される。
【図1】図1は、示差走査熱量測定(DSC)の記録および熱重量分析(TGA)の記録を示している。
【図2】図2は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについてのDSCの記録を示す。
【図3】図3は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの粉末エックス線回析(PXRD)パターンを示している。
【図4】図4は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの粉末エックス線回析(PXRD)パターンを示している。
【図5】図5は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩についての赤外線(IR)吸収スペクトルを示している。
【図6】図6は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩についての力学的水分吸着(DMS)の記録を示している。
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、肺障害を処置するための治療因子として有用であると期待されるビフェニル化合物の新規結晶性1,2−エタンジスルホン酸塩に関する。本発明はまた、そのような結晶性化合物を含む薬学的組成物、もしくはそのような結晶性化合物から調製される薬学的組成物、そのような結晶性化合物を調製するためのプロセスおよび中間体、ならびにそのような結晶性化合物を使用して肺障害を処置する方法に関する。
【背景技術】
【0002】
(技術的現状)
同一出願人による2004年2月13日出願の米国特許出願第10/779,157号は、慢性閉塞性肺疾患(COPD)および喘息などの肺障害を処置するための治療因子として有用である新規ビフェニル化合物を開示している。特に、この化合物、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルは、ムスカリンアンタゴニストおよびβ2アドレナリンレセプターアゴニスト活性の両方を有するとして、これらの出願に特異的に開示される。ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの化学構造は、式I:
【0003】
【化2】
によって表される。
【0004】
肺障害を処置するために有用な治療因子は、吸入により、好都合に直接気道へと投与される。そのため、いくつかの種類の薬学的吸入デバイスが、吸入により治療因子を投与するために開発されており、その中には、乾燥粉末吸入器(DPI)、定量吸入器(MDI)、およびネブライザー吸入器が含まれる。そのようなデバイス中での使用のための薬学的組成物および調合物を調製する場合、吸湿性でも潮解性でもなく、比較的高い融点(すなわち、約150℃より高い融点)を有し、それにより結晶性の重大な分壊または喪失なくこの物質を微粉化し得る結晶性形態の治療因子を有することが非常に望ましい。
【発明の開示】
【発明が解決しようとする課題】
【0005】
式Iの化合物の結晶性塩形態は、以前に報告されていない。したがって、受容可能な水準の吸湿性および比較的高い融点を有する式Iの化合物の、安定した、非潮解性の結晶性塩形態に対する必要性が存在する。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を提供する。
【0007】
驚くことに、式Iの化合物のそのような結晶性1,2−エタンジスルホン酸塩は、大気中の湿気に曝露される場合でさえも、潮解性ではないことが見出された。さらに、そのような結晶性塩は、受容可能な水準の吸湿性および非常に高い融点(例えば、約215℃より高い融点)を有する。特定の実施形態において、本発明の結晶性塩は、約230℃より高い融点を有する。
【0008】
いくつかある使用の中で、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、肺障害を処置するために有用であると期待される薬学的組成物を調製するために有用である。したがって、その組成物の別の局面おいて、本発明は、薬学的に受容可能なキャリアおよびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物を提供する。
【0009】
特定の実施形態において、本発明の薬学的組成物は、さらにステロイド抗炎症因子(たとえば、コルチコステロイド(cortecosteroid));もしくはホスホジエステラーゼ−4インヒビター;またはそれらの組み合わせを含む。
【0010】
別の実施形態において、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩を含み、約4〜約6の範囲におけるpHを有する等張生理食塩水溶液を含む薬学的組成物を提供する。
【0011】
さらに別の実施形態において、本発明は:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子
を含む組み合わせを提供する。
【0012】
式Iの化合物は、ムスカリンアンタゴニスト活性およびβ2アドレナリンレセプターアゴニスト活性の両方を有する。したがって、本発明の1,2−エタンジスルホン酸塩は、肺障害(例えば、喘息、および慢性閉塞性肺疾患)を治療するための治療因子として有用であると期待される。
【0013】
さらに、方法の局面のうちの1つにおいて、本発明は、肺障害を処置するための方法を提供し、この方法は、処置を必要としている患者に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を投与する工程を包含する。
【0014】
さらに、方法の別の局面において、本発明は、患者において気管支拡張(bronchodilation)を生じさせる方法を提供し、この方法は、気管支拡張を生じさせる量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を、吸入により患者に投与する工程を包含する。
【0015】
本発明はまた、慢性閉塞性肺疾患または喘息を処置する方法を提供し、この方法は、処置を必要としている患者に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を投与する工程を包含する。
【0016】
本発明はまた、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスに向けられる。したがって、その方法の別の局面において、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を調製するためのプロセス;ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを1,2−エタンジスルホン酸と接触させる工程を包含するプロセスを提供する。
【0017】
その方法のさらに別の局面において、本発明は、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスを提供し、このプロセスは:
(a)式IIの化合物:
【0018】
【化3】
をフッ化物イオンと接触させる工程であって、ここで
R1a、R1b、およびR1cが、C1〜4アルキル、フェニル、−C1〜4アルキル−(フェニル)から独立して選択されるか、またはR1a、R1b、およびR1cのうちの1つが−O−(C1〜4アルキル)である工程;および
(b)工程(a)からの生成物を、1,2−エタンジスルホン酸もしくはその水和物と接触させて式Iの化合物の結晶性1,2−エタンジスルホン酸塩を形成させる工程であって、
工程(a)および(b)が、同じ反応容器中で、工程(a)の生成物を分離せずに実施される工程
を包含する。
【0019】
その方法の別の局面において、本発明は、約230℃より高い融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するプロセスを提供し、このプロセスは、不活性希釈剤に溶解させた式Iの化合物の1,2−エタンジスルホン酸塩を含む溶液に、式Iの化合物の結晶性1,2−エタンジスルホン酸塩の種結晶を加える工程を包含し、ここでこの種結晶は、約230℃より高い融点を有する。
【0020】
このプロセスはまた、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を再結晶化し、約230℃より高い融点を有する結晶性形態を提供するために使用され得る。したがって、本発明は、約230℃より高い融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製するためのプロセスをさらに提供し、このプロセスは:
(a)式Iの化合物の結晶性1,2−エタンジスルホン酸塩を、第一の温度にて不活性な希釈剤に溶解させる工程;
(b)工程(a)の生成物を第二の温度に冷却する工程;および
(c)式Iの化合物の1,2−エタンジスルホン酸塩の種結晶を加える工程
を包含し、ここで、種結晶は約230℃より高い融点を有し、第一の温度は、1,2−エタンジスルホン酸塩を溶解させるのに十分な温度であり、そして第二の温度は、種結晶が工程(b)の生成物に加えられる場合に完全に溶解する温度より低い。
【0021】
さらに、本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを精製するためのプロセス;ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩を形成させる工程を包含するプロセスに向けられる。本発明はまた、本明細書中に記載されるプロセスによって調製される生成物にも向けられる。
【0022】
本発明はまた、治療における使用または薬としての使用のためのビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物に向けられる。
【0023】
さらに、本発明は、薬の製造;特に肺障害の処置のための薬の製造のためのビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の使用に向けられる。
【0024】
本発明はまた、肺障害の処置のための薬の製造における:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子
の使用に向けられる。
【0025】
本発明はまた、微粉形態のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;ならびに、微粉形態の薬学的に受容可能なキャリアおよびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物に向けられる。
【発明を実施するための最良の形態】
【0026】
(発明の詳細な説明)
本発明は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩またはその溶媒和物を提供する。これらの塩(すなわち、式Iの化合物)中の活性な治療因子は、(R)配置を有する1つのキラル中心を含んでいる。しかしながら、全体として組成物の任意の有用性がそのような異性体の存在によって排除されないならば、他に示さない限り、少量の(S)立体異性体が本発明の組成物中に存在し得ることが、当業者には理解される。
【0027】
式Iの化合物は、市販のAutoNomソフトウェア(MDL,San Leandro,California)を使用して命名された。加えて、1,2−エタンジスルホン酸塩はまた、時にエジシラート(edisylateまたはedisilate)塩として述べられることがある。
【0028】
(定義)
本発明の化合物、組成物、方法、およびプロセスを記載する場合、次の用語は、他に示さない限り、次の意味を有する。
【0029】
用語「融点」は、本明細書中で使用される場合、最大吸熱熱流が、示差走査熱量測定により得られる温度を意味する。
【0030】
用語「微粉形態」は、少なくとも約90%の粒子が約10μmより小さな直径を有する粒子の形態を意味する。
【0031】
用語「溶媒和物」は、溶質の1つ以上の分子によって形成される複合体または凝集物(すなわち、式Iの化合物の1,2−エタンジスルホン酸塩)、および溶媒の1つ以上の分子を意味する。そのような溶媒和物は、典型的に、実質的に固定された溶質と溶媒とのモル比を有する。この用語はまた、水を有する包接化合物を含む包接化合物を含む。代表的な溶媒は、例として、水、メタノール、エタノール、イソプロパノール、酢酸などが挙げられる。溶媒が水の場合、その形成される溶媒は水和物である。
【0032】
用語「治療有効量」は、処置を必要としている患者に投与される場合、処置を達成するのに十分な量を意味する。
【0033】
用語「処置する」または「処置」は、本明細書中で使用される場合:
(a)疾患または医学的状態(medical condition)が生じるのを防ぐ(すなわち、患者の予防的処置)こと;
(b)疾患または医学的状態を改善する(すなわち、患者において、疾患または医学的状態の排除または軽減をもたらす)こと;
(c)疾患または医学的状態を抑制する(すなわち、患者において、疾患または医学的状態の発達を減速させるかまたは阻止する)こと;あるいは
(d)患者において、疾患の症状または医学的状態を緩和すること
を含み、患者(例えば、哺乳動物(特にヒト)において疾患もしくは医学的状態(例えば、COPD)を処置することまたはその処置を意味する。
【0034】
用語「単位投薬形態」は、患者に投薬するのに適した物理的に分離した単位、すなわち、本発明の塩の予め決定された量を含む各単位を表す。この予め決定された量は、単独で、または1つ以上の付加的な単位との組み合わせにおいて所望の治療効果を生むように算定される。例えば、そのような単位投薬形態は、乾燥粉末吸入カプセル、定量吸入器からの一定用量、カプセル、錠剤、丸剤などであり得る。
【0035】
(本発明の1,2−エタンジスルホン酸塩)
本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩は、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステル、および1,2−エタンジスルホン酸塩、またはその水和物から調製され得る。
【0036】
本発明の1,2−エタンジスルホン酸塩は、典型的に、式Iの化合物1モル当量当たり約0.90と約1.10モル当量との間の1,2−エタンジスルホン酸を含み;式Iの化合物1モル当量当たり約0.95と約1.05モル当量との間の1,2−エタンジスルホン酸を含む。特定の実施形態において、本発明の1,2−エタンジスルホン酸塩は、式Iの化合物1モル当量当たり約1モル当量の1,2−エタンジスルホン酸を含む。
【0037】
1,2−エタンジスルホン酸のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルに対するモル比率は、当業者が利用し得る種々の方法により、容易に決定され得る。例えば、そのようなモル比率は、1H NMRにより容易に決定され得る。あるいは、元素分析およびHPLC法が、モル比率を決定するために使用され得る。
【0038】
本発明で使用されるビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルは、市販の出発物質および試薬から、以下の実施例に記載される手順を使用するか;または本出願の背景部分に記載される同一出願人による米国特許出願に記載される手順を使用して、容易に調製され得る。
【0039】
1,2−エタンジスルホン酸は、例えば、Alfa Chemicals Ltd.,Berkshire,UKから市販されている。1つの実施形態において、本発明の塩を調製するのに使用される1,2−エタンジスルホン酸は、二水和物である。特定の実施形態において、1,2−エタンジスルホン酸二水和物は、97%以上の純度(HPLCによって決定される場合)を有する。望ましい場合は、本発明に使用される1,2−エタンジスルホン酸二水和物を、使用する前に、例えば酢酸および無水酢酸から再結晶化し得る。
【0040】
本発明の結晶性塩を調製するために、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを、典型的には、約0.75〜約1.3モル当量の1,2−エタンジスルホン酸、またはその水和物と接触させる。一般的に、この反応は、約0℃〜約60℃;約20℃〜約55℃(例えば、約25℃〜約50℃)を含む範囲の温度にて、不活性な希釈剤中で実施される。この反応に適した不活性な希釈剤としては、これらに限定されないが、メタノール、エタノール、イソプロパノール、イソブタノール、酢酸エチル、ジクロロメタンなどが挙げられ、必要に応じて水を含む。特定の実施形態において、エタノール中の1,2−エタンジスルホン酸二水和物の溶液を、イソプロパノールとジクロロメタン(64:1)との混合物中のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの体積の約5倍まで加える。他の特定の実施形態において、1,2−エタンジスルホン酸二水和物の溶液は、希釈剤として水またはエタノールを含み、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステル溶液は、希釈剤としてイソプロパノールまたはエタノールを含む。
【0041】
あるいは、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、式Iの化合物のシリルで保護された誘導体(すなわち、式IIの化合物)を、フッ化物イオンの供給源と接触させ、次いで同じ反応容器中で、その生成物を1,2−エタンジスルホン酸またはその水和物と接触させることにより調製し得る。特定の実施形態において、シリル保護基は、tert−ブチルジメチルシリル基である。他の適切なシリル保護基としては、tert−ブチルジフェニルシリル、ジフェニルメチルシリル、ジ−tert−ブチルメチルシリル(buylmethylsilyl)、tert−ブトキシジフェニルシリルなどが挙げられる。このプロセスで使用されるフッ化物イオンの供給源は、フッ化物イオンまたはフッ化水素を含んでいるか、または包含している任意の試薬であり得る。特定の実施形態において、フッ化物イオンの供給源は、三フッ化水素トリエチルアミンである。フッ化物イオンの他の適切な供給源は、フッ化テトラブチルアンモニウム、18−クラウン−6を有するフッ化カリウム、フッ化水素、ピリジンフッ化水素塩などが挙げられる。
【0042】
一般的に、このプロセスは、約0℃〜約50℃;約20℃〜約35℃(例えば、約25℃〜約30℃)を含む範囲の温度において不活性な希釈剤中で実施される。この反応に適切した不活性な希釈剤としては、これらに限定されないが、ジクロロメタン、メタノール、およびそれらの混合物が挙げられる。特定の実施形態において、ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−tert−ブチルジメチルシラニルオキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロ−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの溶液を、ジクロロメタン中の約2.5〜約3.0モル当量のトリエチルアミン三フッ化水素塩と、周囲温度にて約12〜24時間、またはシリル基の除去が実質的に完了するまで接触させる。得られた溶液を、反応生成物を分離せず、メタノール中の約0.9〜約1.1モル当量の1,2−エタンジスルホン酸二水和物を加え、そしてこの混合物を約25℃〜約35℃にて約2〜約6時間加熱する。この反応の完了の際に、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩は、任意の従来の方法(例えば、沈殿、濃縮、遠心分離など)により、反応混合物から分離される。
【0043】
必要に応じて、本発明の結晶性1,2−エタンジスルホン酸塩は、容量で約15%〜約25%(約20%を含む)の水を含むイソプロパノール中で、塩を攪拌または懸濁(slurring)させることによりさらに精製され得る。特定の実施形態において、1,2−エタンジスルホン酸塩1g当たり約10mLのイソプロパノール/水混合物が使用される。
【0044】
本発明の結晶性1,2−エタンジスルホン酸塩を調製するプロセスは、必要に応じて、特定の結晶性塩を優位に生産するための種結晶の使用を含む。例えば、高温で(すなわち、約230℃より高い温度で)融解する結晶性塩の種結晶を使用することにより、種結晶と本質的に同じ融点を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を調製し得る。そのような種結晶は、最初、結晶性塩を形成する場合に使用され得るか、または結晶性もしくは一部が結晶性の塩を再結晶化するために使用され得る。
【0045】
典型的に、種結晶は、攪拌せず、冷却を適用しないゆっくりとした結晶化により、小規模で調製される。種結晶を得るための例として、典型的に、溶解を与えるのに十分な温度で不活性な希釈剤に結晶性塩を溶解させる。一般的に、種結晶を得る最初のプロセスにおいて、少量の、典型的に10g未満(5g未満(例えば1g未満)を含む)の結晶性塩が使用される。特定の実施形態において、希釈剤として約12%〜約20%の水(約13%〜約15%の水を含む)が、約60℃〜約70℃(例えば、約60℃〜約65℃)の範囲の温度にて使用される。この溶液を、室温に冷却させる。約1日〜約3日後、得られた結晶を濾過、または他の慣例的な方法により単離する。あるいは、以前の結晶性物質の調製から、種結晶が得られ得る。
【0046】
種結晶を使用した再結晶化プロセスにおいて、本発明の結晶性1,2−エタンジスルホン酸塩を、種結晶を得るプロセスの場合のように、不活性な希釈剤、典型的には15%の水を含むメタノールに、約60℃〜約65℃の範囲の温度にて溶解させる。この溶液を種結晶が溶解しない温度、例えば、約30℃〜約40℃の範囲における温度に冷却させ、次いで、種結晶を加える。典型的に、この溶液中の結晶性塩の重量に対する種結晶の重量の比は、約1:5と約1:35との間である。この溶液を結晶化が起こる温度、例えば約20度に冷却し、約2時間〜約24時間、攪拌する。得られた結晶を、慣例的な方法により単離する。大量の物質を調製するのに十分な種結晶を得るために、最初の再結晶化によって得られた結晶を、次の再結晶化工程のための種結晶として使用し、この再結晶化プロセスを連続的に実施し得る。再結晶化プロセスの工程が実施される特異的な温度は、希釈剤の性質および希釈剤中の結晶性塩の濃度に従い選択されることが理解される。さらに、冷却する代わりにエバポレーションかまたは抗溶媒を、結晶化を促進するために使用して、この再結晶化プロセスを実施し得る。
【0047】
いくつかある利点の中で、式Iの化合物の結晶性1,2−エタンジスルホン酸塩を形成することが、式Iの化合物を精製するために有用であることが見出された。一般的に、本発明の結晶性1,2−エタンジスルホン酸塩は、高速液体クロマトグラフィーにより決定される場合、95%より高い純度;典型的に98%より高い純度を有する。
【0048】
本発明の結晶性1,2−エタンジスルホン酸塩は、約215℃〜約240℃の範囲で吸熱熱流におけるピークを示す示差走査熱量測定(DSC)の記録によって明示されるように、非常に高い融点を特徴とする。結晶性塩の融点温度は、その結晶性塩が形成されたプロセスに従うことが認められた。攪拌せず、冷却を適用しないゆっくりとした結晶化によって形成される種結晶は、約230℃より高い融点を示す。そのような種結晶を用いた再結晶化を含むプロセスによって形成される結晶性塩は、典型的に、例えば図1に示されるように、約230℃〜約245℃の範囲における融点を示す。約230℃より高い融点を有する種結晶を用いずに形成された結晶性塩は、典型的に、例えば図2に示されるように、約215℃〜約229℃の範囲における融点を示す。したがって、特定の実施形態において、本発明は、実質的に図1または図2に示される記録に従う約200℃より高い温度範囲にDSCの記録を有する式Iの化合物の結晶性1,2−エタンジスルホン酸塩を提供する。
【0049】
別の実施形態において、本発明の結晶性1,2−エタンジスルホン酸塩は、5.0±0.3および15.0±0.3の2θの値において有意な回析ピークを有する粉末エックス線回析(PXRD)パターンを特徴とする。図3に示されるような融点が高い種結晶からの再結晶化により調製した結晶化塩のPXRDスペクトルにおけるピーク位置と、図4に示されるようなそのような種結晶を使用せずに調製した塩のピーク位置との間に、微妙な違いが認められた。従って、別の実施形態において、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、ピーク位置が実質的に図3または図4に示されるPXRDスペクトルにおけるピーク位置と一致する粉末エックス線回析パターンを特徴とする。
【0050】
別の実施形態において、式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、図5に図示されるように、約704cm−1、約748cm−1、約768cm−1、約841cm−1、約900cm−1、約1055cm−1、約1104cm−1、約1166cm−1、約1218cm−1、約1294cm−1、約1408cm−1、約1522cm−1、約1609cm−1、約1655cm−1、および約1701cm−1において有意な吸収帯を示す赤外線(IR)吸収スペクトルを特徴とする。
【0051】
式Iの化合物の結晶性1,2−エタンジスルホン酸塩は、受容可能な適度な水準の吸湿性(すなわち、相対湿度40%〜相対湿度75%の湿度範囲において約2.5%未満重量増)を有する可逆的吸着/脱着プロフィールを有することが示された。
【0052】
本発明の塩のこれらの特性について、以下の実施例においてさらに説明する。
【0053】
(薬学的組成物および調合物)
式Iの化合物の1,2−エタンジスルホン酸塩を、典型的には、薬学的組成物または調合物の形態で患者に投与する。そのような薬学的組成物を、投与の任意の受容可能な経路(これらに限定されないが、吸引、経口、鼻腔、局所(経皮を含む)を含む)、および投与の非経口形態によって患者に投与し得る。しかしながら、本発明の結晶性塩は調合されてしまうと結晶性形態ではなくなり得る(すなわち、この塩を適切なキャリアに溶解し得る)ということが当業者には理解される。
【0054】
したがって、組成物の局面のうちの1つにおいて、本発明は、薬学的に受容可能なキャリアもしくは賦形剤、およびビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む薬学的組成物に向けられる。必要に応じ、そのような薬学的組成物は、望ましい場合、他の治療剤および/または調合剤を含み得る。
【0055】
本発明の薬学的組成物は、典型的に、治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む。典型的に、そのような薬学的組成物は、約0.01〜約95重量%;約0.01〜約30重量%;例えば、約0.01〜約10%の活性因子を含む。
【0056】
任意の従来のキャリアもしくは賦形剤を、本発明の薬学的組成物に使用し得る。特定のキャリアもしくは賦形剤の選択、またはキャリアもしくは賦形剤の組み合わせの選択は、特定の患者を治療するために使用されている投与の形態、または医学的状態もしくは病状(disease state)の種類に従う。この点で、特定の形態の投与に適した薬学的組成物の調製は、まさに薬学分野の当業者の範囲内である。さらに、そのような組成物の成分は、例えば、Sigma,P.O.Box 14508、St.Louis, MO 63178から市販されている。さらなる例として、慣例的な調合技術は、Remington:The Science and Pracice of Pharmacy,20th Edition,Lippincott Williams & White,Baltimore,Maryland(2000);およびH.C.Ansel et al.,Pharmaceutical Dosage Forms and Drug Delivery Systems,7th Edition, Lippincott Williams & White,Baltimore,Maryland(1999)に記載されている。
【0057】
薬学的に受容可能なキャリアとなり得る物質の代表的な例としては、これらに限定されないが、次の:(1)糖(例えば、ラクトース、ブドウ糖、およびスクロース);(2)デンプン(例えば、コーンスターチ、およびジャガイモデンプン);(3)セルロース、およびその誘導体(例えば、カルボキシメチルセルロースナトリウム、エチルセルロース、および酢酸セルロース);(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤(例えば、ココアバター、および座剤用ワックス;(9)油(例えば、ピーナツ油、綿実油、ベニバナ油、ゴマ油、オリーブ油、コーン油、およびダイズ油;(10)グリコール(例えば、プロピレングリコール);(11)多価アルコール(例えば、グリセリン、ソルビトール、マンニトール、およびポリエチレングリコール);(12)エステル(例えば、オレイン酸エチル、およびラウリン酸エチル);(13)寒天;(14)緩衝剤(例えば、水酸化マグネシウム、および水酸化アルミニウム);(15)アルギン酸;(16)発熱因子非含有水(pyrogen−free water);(17)等張食塩水;(18)リンゲル溶液;(19)エチルアルコール;(20)リン酸緩衝溶液;(21)圧縮噴霧ガス(例えば、クロロフルオロカーボン、およびハイドロフルオロカーボン);そして(22)薬学的組成物に使用される他の無毒性適合性物質が挙げられる。
【0058】
本発明の薬学的組成物は、典型的に、薬学的に受容可能なキャリアおよび1つ以上の任意の成分と本発明の塩とを混合するかまたは組み合わせて調製される。必要な場合、または望ましい場合は、得られた均質に混和した混合物を、慣例的な手順および装置を使用して形作られるか、あるいは錠剤、カプセル剤、丸剤、キャニスター、カートリッジ、ディペンサーなどに詰め込み得る。
【0059】
1つの実施形態において、本発明の薬学的組成物は、吸入投与に適している。吸入投与に適した薬学的組成物は、典型的に、エアゾール、または粉末の形態である。そのような組成物は、一般的に、周知の送達デバイス(例えば、ネブライザー吸入器、定量吸入器(MDI)、乾燥粉末吸入器(DPI)、または類似した送達デバイスを使用して投与される。
【0060】
本発明の特異的な実施形態において、活性因子を含む薬学的組成物は、ネブライザー吸入器を使用して吸入により投与される。そのようなネブライザーデバイスは、典型的に、高速の気流を生成し、活性因子を含む薬学的組成物がミストとしてスプレーされ、患者の気道に運ばれる。したがって、ネブライザー吸入器中での使用のために調合される場合、典型的に、適切なキャリアにこの活性因子を溶解させて溶液を形成する。適切なネブライザーデバイスは、例えば、PARI GmbH(Starnberg,German)により市販されている。他のネブライザーデバイスとしては、Respimat(Boehringer Ingelheim)、ならびに例えば、米国特許第6,123,068号および国際公開第97/12687号で開示されているものが挙げられる。
【0061】
ネブライザー吸入器中での使用のための代表的な薬学的組成物は、約0.05μg/mL〜約10mg/mLの式Iの化合物の1,2−エタンジスルホン酸塩、またはその溶媒和物を含む水溶液を含む。1つの実施形態において、このネブライザー調合水は等張性である。1つの実施形態において、このネブライザー調合水は、約4〜約6の範囲におけるpHを有する。特定の実施形態において、このネブライザー調合水は、pHが約5までのクエン酸緩衝液で緩衝される。別の特定の実施形態において、このネブライザー調合水は約0.1mg/mL〜約1.0mg/mLの遊離塩基当量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルを含む。
【0062】
本発明の別の特定の実施形態において、活性因子を含む薬学的組成物は、乾燥粉末吸入器を使用して、吸入により投与される。そのような乾燥粉末吸入器は、典型的に、吸息中に患者の気流中に分散される自由流れ粉末(free−flowing powder)として活性因子を投与する。自由流れ粉末を達成するために、活性因子は、典型的に、適切な賦形剤(例えば、ラクトース、デンプン、マンニトール、ブドウ糖、ポリ乳酸(PLA)、ポリ乳酸・グリコリド共重合体(PLGA)、またはそれらの組み合わせ)と共に調合される。典型的に、この活性因子は、微粉化され、適切なキャリアと組み合わせて呼吸に適した大きさの微粉粒の混合物を形成する。ここで「微粉粒」または「微粉形態」は、少なくとも約90%の粒子が約10μm未満の直径を有することを意味する。
【0063】
乾燥粉末吸入器中での使用のための代表的な薬学的組成物は、約1μmと約100μmとの間の粒径を有するラクトース、および式Iの化合物の1,2−エタンジスルホン酸塩、またはその溶媒和物の微粉粒子を含む。
【0064】
そのような乾燥粉末調合物を、例えば、ラクトースを活性因子と組み合わせ、次いでその成分を乾式混合することにより作製し得る。あるいは、望ましい場合、この活性因子を、賦形剤を用いずに調合し得る。次いで、薬学的組成物を、典型的に、乾燥粉末ディスペンサー、または乾燥粉末送達デバイスと共に使用するための吸入カートリッジ、もしくはカプセル剤に詰める。
【0065】
乾燥粉末吸入器送達デバイスの例としては、Diskhaler(GlaxoSmithKline,Research Triangle Park,NC)(例えば、米国特許第5,035,237号を参照);Diskus(GlaxoSmithKline)(例えば、米国特許第6,378,519号を参照);Turbuhaler(AstraZeneca,Wilmington,DE)(例えば、米国特許第4,524,769号を参照);Rotahaler(GlaxoSmithKline)(例えば、米国特許第4,353,365号を参照)、およびHandihaler(Boehringer Ingelheim)が挙げられる。適切なDPIデバイスのさらなる例には、米国特許第5,415,162号、米国特許第5,239,993号、および米国特許第5,715,810号に記載されており、これらの文献は、本明細書中で引用される。
【0066】
本発明のさらに別の特異的な実施形態において、活性因子を含む薬学的組成物は、定量吸入器を使用して吸入により投与される。そのような定量吸入器は、典型的に、圧縮噴霧ガスを使用して、一定量の活性因子またはその薬学的に受容可能な塩を放出する。したがって、定量吸入器を使用して投与される薬学的組成物は、典型的に、液化された噴霧剤中に、活性因子の溶液または懸濁液を含む。クロロフルオロカーボン(例えば、CCl3F)、およびハイドロフルオロアルカン(HFA)(例えば、1,1,1,2−テトラフルオロエタン(HFA 134a)、および1,1,1,2,3,3,3−へプタフルオロ−n−プロパン(HFA 227))を含む任意の適した液化噴霧剤を使用し得る。オゾン層に影響を及ぼすクロロフルオロカーボンについての懸念から、HFAを含む調合物が、一般的に好ましい。HFA調合物のさらなる任意の成分としては、共溶媒(例えば、エタノールまたはペンタン)および表面活性剤(例えば、ソルビタントリオレエイト(trioleate)、オレイン酸、レシチン、およびグリセリン)が挙げられる。例えば、米国特許第5,225,183号、欧州特許第0717987A2号、および国際公開92/22286号を参照されたい。
【0067】
定量吸入器中における使用のための代表的な薬学的組成物は、約0.01重量%〜約5重量%の式Iの化合物の1,2−エタンジスルホン酸塩またはその溶媒和物;約0重量%〜20重量%のエタノール;そして約0重量%〜約5重量%の表面活性剤を含み、その残りがHFA噴霧剤である。
【0068】
そのような組成物を、典型的に、冷却もしくは加圧ヒドロフルオロアルカンを、活性因子、エタノール(あれば)、および表面活性剤(あれば)を含む適切な容器に加えることにより調製する。懸濁液を調製するために、活性因子を微粉化し、次いで、噴霧剤と組み合わせる。次いで、この調合物をエアゾールキャニスターに詰めて、定量吸入デバイスの一部とする。特異的にHFA噴霧剤と共に使用するために開発された定量吸入器デバイスの例が、米国特許第6,006,745号、および第6,143,277号に提供されている。あるいは、懸濁調合物を、活性因子の微粉粒子上の表面活性剤のコーティングをスプレー乾燥させることにより調製し得る。例えば、国際公開第99/53901号、および国際公開第00/61108号を参照されたい。
【0069】
呼吸に適した粒子を調製するプロセス、ならびに吸入投薬に適した調合物およびデバイスのさらなる例については、米国特許第6,268,533号、第5,983,956号、第5,874,063号、および6,221,398号、ならびに国際公開第99/55319号、および国際公開第00/30614号を参照されたい。
【0070】
別の実施形態において、本発明の薬学的組成物は、経口投与に適している。経口投与に適した薬学的組成物は、カプセル剤、錠剤、丸剤、ドロップ剤、カシェ剤、糖衣剤、粉剤、顆粒剤の形態であるか;あるいは溶液または水性もしくは非水性の液体中の懸濁液として;あるいは水中油もしくは油中水液体乳剤として;あるいはエリキシル剤またはシロップ剤として;などであり得、それぞれ活性成分として所定量の本発明の塩を含む。
【0071】
固形の投薬形態(すなわち、カプセル剤、錠剤、丸剤など)での経口投与に向けられる場合、本発明の薬学的組成物は、典型的に、活性成分として本発明の塩、および1種以上の薬学的に受容可能なキャリア(例えば、クエン酸ナトリウムまたはリン酸二カルシウム)を含む。必要に応じ、または代替的に、そのような固形の投薬形態はまた:(1)充填剤または増量剤(例えば、デンプン、ラクトース、スクロース、グルコース、マンニトール、および/またはケイ酸);(2)結合剤(例えば、カルボキシメチルセルロース、アルギナート、ゼラチン、ポリビニルピロリドン、スクロース、および/またはアカシア);(3)湿潤剤(例えば、グリセリン);(4)崩壊剤(例えば、寒天、炭酸カルシウム、ジャガイモもしくはタピオカデンプン、アルギン酸、特定のシリケート、および/または炭酸ナトリウム);(5)溶液緩染剤(例えば、パラフィン);(6)吸収促進剤(例えば、第四アンモニウム化合物);(7)加湿剤(例えば、セチルアルコール、および/またはグリセロールモノステアレート);(8)吸収剤(例えば、カロリン、および/またはベントナイトクレー);(9)潤滑剤(例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、および/またはそれらの混合物);(10)着色剤;そして(11)緩衝剤を含む。
【0072】
離型剤、加湿剤、コーティング剤、甘味剤、香味剤および芳香剤、保存剤、ならびに酸化防止剤もまた、本発明の薬学的組成物中に存在し得る。薬学的に受容可能な酸化防止剤の例としては:(1)水溶性酸化防止剤(例えば、アスコルビン酸、塩酸システイン、硫酸水素ナトリウム、メタ重亜硫酸ナトリウム、亜硫酸ナトリウムなど);(2)油溶性酸化防止剤(例えば、アスコルビン酸パルミテート、ブチル化ヒドロキシアニソール(BHA)、ブチル化ヒドロキシトルエン(BHT)、レシチン、没食子酸プロピル、α−トコフェロールなど);そして(3)金属キレート因子(例えば、クエン酸、エチレンジアミン四酢酸(EDTA)、ソルビトール、酒石酸、リン酸、など)が挙げられる。錠剤、カプセル剤、丸剤などのための被膜因子としては、腸溶コーティング用に使用される被膜因子(例えば、酢酸フタル酸セルロース(CAP)、ポリビニルアセテートフタレート(PVAP)、フタル酸ヒドロキシプロピルメチルセルロース、メタクリル酸−メタクリル酸エステルコポリマー、セルソースアセテートトリメリテート(trimellitate)(CAT)、カルボキシメチルエチルセルロース(CMEC),ヒドロキシプロピルメチルセルロースアセテートスクシナート(HPMCAS)など)が挙げられる。
【0073】
望ましい場合、本発明の薬学的組成物は、例として、様々な割合のヒドロキシプロピルメチルセルロース;あるいは他のポリマーマトリックス(例えば、ポリ乳酸(PLA)もしくはポリ乳酸・グリコリド共重合体(PLGA)、リポソームおよび/または小球体を有するようなポリマーマトリクス)を使用して、活性成分のゆっくりとした、もしくは制御された放出を提供するように調合され得る。
【0074】
さらに、本発明の薬学的組成物は、必要に応じて不透明化因子を含み得、胃腸管の特定の部分において、必要に応じ、遅延させる様式で、活性成分のみを、または活性成分を優先的に、放出するように調合され得る。使用し得る包埋組成物(embedding composition)の例としては、高分子物質、およびワックスが挙げられる。この活性成分はまた、適切である場合、1種以上の上記賦形剤を有するマイクロカプセル形態であり得る。
【0075】
経口投与に適した液体投薬形態としては、例として、薬学的に受容可能な乳剤、ミクロエマルジョン、液剤、懸濁剤、シロップ剤、およびエリキシル剤が挙げられる。そのような液体投与形態は、典型的に、活性成分および不活性な希釈剤(例えば、水または他の溶媒など)、可溶化剤、および乳化剤(例えば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセリン、テトラヒドロフリルアルコール、ポリエチレングリコール、およびソルビタン脂肪酸エステル、ならびにこれらの混合物を含む。活性成分に加えて懸濁剤は、懸濁化剤(例えば、エトキシ化したイソステアリールアルコール、ポリオキシエチレンソルビトール、およびソルビタンエステルなど)、微結晶性セルロース、アルミニウムメタヒドロキシド、ベントナイト、寒天、およびトラガカント、ならびにこれらの混合物を含み得る。
【0076】
経口投与に向けられる場合、本発明の薬学的組成物は、好ましくは、単位投薬形態にまとめられる。例えば、そのような単位投薬形態は、カプセル、錠剤、丸剤などであり得る。
【0077】
本発明の塩はまた、公知の経皮送達システムおよび賦形剤を使用して、経皮投与され得る。例えば、本発明の化合物を、浸透促進剤(permeation enhancer)(例えば、プロピレングリコール、モノラウリン酸ポリエチレングリコルム(polyethylene glycolm monolaurate)、アザシクロアルカン−2−オンなど)と混合し、貼付剤もしくは類似の送達システムに組み込み得る。ゲル化剤、乳白剤、および緩衝剤を含むさらなる賦形剤を、望ましい場合、そのような経皮組成物中に使用し得る。
【0078】
本発明の薬学的組成物はまた、他の治療因子を含んで式Iの化合物の1,2−エタンジスルホン酸塩またはその溶媒和物と同時投与し得る。例えば、本発明の薬学的組成物は、抗炎症剤(例えば、ステロイド抗炎症剤(コルチコステロイドなど);および非ステロイド抗炎症剤(NSAID)、ホスホジエステラーゼIV阻害物質、抗感染剤(例えば、抗生物質または抗ウィルス薬)、抗ヒスタミン剤、β2アドレナリンレセプターアゴニスト、ムスカリンレセプターアンタゴニスト(すなわち、抗コリン作用薬)などから選択される1種以上の治療因子をさらに含み得る。他の治療因子は、薬学的に受容可能な塩もしくは溶媒和物の形態で使用され得る。さらに、適切である場合、他の治療因子は、光学的に純粋な立体異性体として使用され得る。
【0079】
望ましい場合、本発明の塩はまた、別の治療因子または本明細書中に記載される因子と組み合せて投与され得る。この実施形態において、成分が物理的に混合しているのではなく、同時にまたは連続的に、別個の組成物として投与される。例えば本発明の塩は、各治療因子に対し別個のコンパートメントを使用する吸入薬送達デバイス(例えば、ブリスターパック)を使用して、ステロイド抗炎症剤(例えば、コルチコステロイド)と共に、吸入により同時にまたは連続的に投与され得る。あるいは、この組み合せは、多数の送達デバイス(すなわち各治療因子に対し1つの送達デバイス)から投与され得る。
【0080】
本発明の化合物と組み合わせて使用し得る代表的なβ2アドレナリンレセプターアゴニストとしては、これらに限定されないが、サルメテロール(salmeterol)、サルブタモール、フォルモテロール、サルメファモール(salmefamol)、フェノテロール、テルブタリン、アルブテロール、イソエタリン、メタプロテレノール、ビトルテロール、ピルブテロール、レバルブテロール(levalbuterol)など、またはそれらの薬学的に受容可能な塩が挙げられる。本発明の化合物と組み合わせて使用し得る他のβ2アドレナリンレセプターアゴニストとしては、これらに限定されないが、3−(4−{[6−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、3−(−3−{[7−({(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル}アミノ)へプチル]オキシ}プロピル)ベンゼンスルホンアミド、および2002年8月29日公開の国際公開第02/066422号に開示されている関連化合物;3−[3−(4−{[6−([(2R)−2−ヒドロキシ−2−[4−ヒドロキシ−3−(ヒドロキシメチル)フェニル]エチル]アミノ)ヘキシル]オキシ}ブチル)フェニル]イミダゾリジン−2,4−ジオン、および2002年9月12日公開の国際公開第02/070490号に開示されている関連化合物;3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)−ベンゼンスルホンアミド、3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、N−(tert−ブチル)−3−(4−{[6−({(2R/S)−2−[3−(ホルミルアミノ)−4−ヒドロキシフェニル]−2−ヒドロキシエチル}アミノ)ヘキシル]オキシ}ブチル)ベンゼンスルホンアミド、および2002年10月3日公開の国際公開第02/076933号に開示されている関連化合物;4−{(1R)−2−[(6−{2−[(2,6−ジクロロベンジル)オキシ]エトキシ}ヘキシル)アミノ]−1−ヒドロキシエチル}−2−(ヒドロキシメチル)フェノール、および2003年3月27日公開の国際公開第03/024439号に開示されている関連化合物;N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミン、および2003年6月10日発行の米国特許第6,576,793号B1に開示されている関連化合物;N−{2−[4−(3−フェニル−4−メトキシフェニル)アミノフェニル]エチル}−(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2(1H)−キノリノン−5−イル)エチルアミン、および2003年11月25日発行の米国特許第6,653,323号B2に開示されている関連化合物;ならびにそれらの薬学的に受容可能な塩が挙げられる。特定の実施形態において、β2アドレナリンレセプターアゴニストは、N−{2−[4−((R)−2−ヒドロキシ−2−フェニルエチルアミノ)フェニル]エチル}−(R)−2−ヒドロキシ−2−(3−ホルムアミド−4−ヒドロキシフェニル)エチルアミンの結晶性モノヒドロクロリド塩である。使用される場合、β2アドレナリンレセプターアゴニストは、治療有効量で薬学的組成物中に存在する。典型的に、このβ2アドレナリンレセプターアゴニストは、1用量あたり約0.05μg〜約500μgを提供するのに十分な量で存在する。
【0081】
本発明の化合物と組み合わせて使用し得る代表的なステロイド抗炎症剤としては、これらに限定されないが、メチルプレドニゾロン、プレドニゾロン、デキサメタゾン、フルチカソンプロピオナート(fluticasone propionate)、6,9−ジフルオロ−17−[(2−フラニルカルボニル)オキシ]−11−ヒドロキシ−16−メチル−3−オキソアンドロスタ−1,4−ジエン−17−カルボチオ酸S−フルオロメチルエステル、6,9−ジフルオロ−11−ヒドロキシ−16−メチル−3−オキソ−17−プロピオニルオキシ−アンドロスタ−1,4−ジエン−17−カルボチオ酸S−(2−オキソテトラヒドロフラン−3S−イル)エステル、ベクロメタゾンエステル(例えば、17−プロピオン酸エステル、または17,21−ジプロピオン酸エステル)、ブデソニド、フルニソリド、モメタソンエステル(mometasone ester)(例えば、フロン酸エステル)、トリアムシノロンアセトニド、ロフレポニド(rofleponide)、シクルソニド(ciclesonide)、ブチキソコルトプロピオナート(butixocort propionate)、RPR−106541、ST−126など、またはそれらの薬学的に受容可能な塩が挙げられる。特定の実施形態において、ステロイド抗炎症剤は、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその薬学的に受容可能な塩もしくは溶媒和物である。使用される場合、ステロイド抗炎症剤は、治療有効量で薬学的組成物中に存在する。典型的に、ステロイド抗炎症剤は、1用量あたり約0.05μg〜約500μgを提供するのに十分な量で存在する。
【0082】
他の適切な組み合わせとしては、例として、他の抗炎症剤、例えば、NSAID(クロモグリク酸ナトリウム;ネドクロミルナトリウム(nedocromil sodium);ホスホジエステラーゼ(PDE)阻害物質(例えば、テオフィリン、PDE4阻害物質、または混合PDE3/PDE4阻害物質);ロイコトリエンアンタゴニスト(例えば、モンテルカスト);ロイコトリエン合成の阻害物質;iNOS阻害物質;プロテアーゼ阻害物質(例えば、トリプターゼ阻害物質、およびエラスターゼ阻害物質);β−2インテグリンアンタゴニスト、およびアデノシンレセプターアゴニストもしくはアンタゴニスト(例えば、アデノシン2aアゴニスト);サイトカインアンタゴニスト(例えば、ケモカインアンタゴニスト(インターロイキン抗体(IL抗体)、特にIL−4療法、IL−13療法、またはそれらの組み合わせなど));あるいはサイトカイン合成の阻害物質が挙げられる。
【0083】
例えば、本発明の化合物と組み合わせて使用し得る代表的なホスホジエステラーゼ−4(PDE4)阻害物質、または混合PDE3/PDE4阻害物質としては、これらに限定されないが、シス4−シアノ−4−(3−シクロペンチルオキシ−4−メトキシフェニル)シクロヘキサン−1−カルボン酸、2−カルボメトキシ−4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オン;シス−[4−シアノ−4−(3−シクロプロピルメトキシ−4−ジフルオロメトキシフェニル)シクロヘキサン−1−オール];シス−4−シアノ−4−[3−(シクロペンチルオキシ)−4−メトキシフェニル]シクロヘキサン−1−カルボン酸など、またはそれらの薬学的に受容可能な塩が挙げられる。他の代表的なPDE4または混合PDE4/PDE3阻害物質としては、AWD−12−281(elbion);NCS−613(INSERM);D−4418(Chiroscience and Schering−Plough);CI−1018、またはPD−168787(Pfizer);国際公開第99/16766号で開示されているベンゾジオキソール化合物(Kyowa Hakko);K−34(Kyowa Hakko);V−11294A(Napp);roflumilast(Byk−Gulden);国際公開第99/47505号で開示されているフタラジノン(pthalazinone)化合物(Byk−Gulden);Pumafentrine(Byk−Gulden、現在はAltana);arofylline(Almirall−Prodesfarma);VM554/UM565(Vernalis);T−440(Tanabe Seiyaku);およびT2585(Tanabe Seiyaku)が挙げられる。
【0084】
本発明の化合物と組み合わせて、そして本発明の化合物に加えて、使用し得る代表的なムスカリンアンタゴニスト(すなわち、抗コリン作用薬)としては、これらに限定されないが、アトロピン、硫酸アトロピン、酸化アトロピン、硝酸メチルアトロピン、臭化水素酸ホマトロピン、臭化水素酸ヒオスシアミン(d,l)、臭化水素酸スコポラミン、臭化イプラトロピウム、臭化オキシトロピウム、臭化チオトロピウム(tiotropium)、メタンテリン、臭化プロパンテリン、臭化メチルアニソトロピン、臭化クリジニウム、copyrrolate(Robinul)、ヨウ化イソプロパミド、臭化メペンゾラート、塩化トリジヘキセチル(Pathilone)、ヘキソシクリウム硫酸メチル(hexocyclium methylsulfate)、塩酸シクロペントレート、トロピカミド、塩酸トリヘキシフェニジル、ピレンゼピン、テレンゼピン(telenzepine)、AF−DX 116、およびメトクトラミン(methoctramine)など、またはそれらの薬学的に受容可能な塩;もしくは、塩として列挙された化合物の代わりに、それらの薬学的に受容可能な塩が挙げられる。
【0085】
本発明の化合物と組み合わせて使用し得る代表的な抗ヒスタミン剤(すなわち、H1−レセプターアンタゴニスト)としては、これらに限定されないが、エタノールアミン(例えば、マレイン酸カルビノキサミン、フマル酸クレマスチン、塩酸ジフェニルヒドラミン、およびジメンヒドリナート;エチレンジアミン(例えば、ピリラミンアムリエート(pyrilamine amleate))、塩酸トリペレナミン(tripelennamine)、クエン酸トリペレンアミン;アルキルアミン(例えば、クロルフェニラミン、およびアクリバスチン(acrivastine);ピペラジン(例えば、塩酸ヒドロキシジン、ヒドロキシジンパモエート、塩酸シクリジン、乳酸シクリジン、塩酸メクリジン、および塩酸セチリジン(cetirizine);ピペリジン(例えば、アステミゾール、塩酸レボカバスチン(levocabastine)、ロラタジン(loratadine)もしくはそのデスカルボエトキシアナログ(descarboethoxy analogue)、テルフェナジン、および塩酸フェキソフェナジン(fexofenadine);塩酸アゼラスチン;など、またはそれらの薬学的に受容可能な塩;もしくは、塩として列挙された化合物の代わりとして、それらの薬学的に受容可能な塩が挙げられる。
【0086】
本発明の化合物と組み合わせて投与される他の治療因子に適した用量は、約0.05mg/日〜約100mg/日の範囲である。
【0087】
次の調合物は、本発明の代表的な薬学的組成物を示している:
(調合物実施例A)
吸入による投与のための乾燥粉末を次のように調製する:
【0088】
【化4】
代表的な手順:本発明の化合物を微粉化し、次いでラクトースと混和する。次いで、この混和した混合物を、ゼラチン吸入カートリッジに詰める。このカートリッジの内容物を、粉末吸入器を使用して投与する。
【0089】
(調合物実施例B)
乾燥粉末吸入デバイス中における使用のための乾燥粉末調合物を次のように調製する:
(代表的な手順)本発明の微粉化した塩とラクトースとの調合容積比(bulk formulation ratio)が1:200である薬学的組成物を調製する。この組成物を1用量当たり約10μgと約100μgとの間の本発明の化合物を送達し得る乾燥粉末吸入デバイスに詰め込む。
【0090】
(調合物実施例C)
定量吸入器での吸入による投与のための乾燥粉末を次のように調製する:
代表的な手順:5重量%の本発明の塩、および0.1重量%のレシチンを含む懸濁液を、0.2gのレシチンを200mLの脱塩水に溶解させて生成した溶液に、10μm未満の平均粒径を有する微粉粒子として10gの本発明の化合物を分散させることにより調製する。この懸濁液を噴霧乾燥(spray dry)させ、得られた物質を1.5μm未満の平均直径を有する粒子に微粉化する。この粒子を加圧した1,1,1,2−テトラフルオロエタンと共にカートリッジに詰める。
【0091】
(調合物実施例D)
定量吸入器中における使用のための薬学的組成物を次のように調製する:
代表的な手順:5%の本発明の塩、0.5%のレシチン、および0.5%のトレハロースを含む懸濁液を、0.5gのトレハロース、および0.5gのレシチンを100mLの脱塩水に溶解させて生成したコロイド溶液に、10μm未満の平均粒径を有する微粉粒子として5gの活性成分を分散させることにより調製する。この懸濁液を噴霧乾燥させ、得られた物質を1.5μm未満の平均直径を有する粒子に微粉化する。この粒子を加圧した1,1,1,2−テトラフルオロエタンと共にキャニスターに詰める。
【0092】
(調合物実施例E)
ネブライザー吸入器中における使用のための薬学的組成物を次のように調製する:
代表的な手順:ネブライザー中における使用のためのエアゾール調合水を、クエン酸を用いて酸性化した1mLの0.9%塩化ナトリム溶液に、0.5mgの本発明の塩を溶解させることにより調製する。この混合物を攪拌し、活性成分が溶解するまで超音波処理する。この溶液のpHを、NaOHのゆっくりとした添加により約5の値に調節する。
【0093】
(調合物実施例F)
経口投与のための硬ゼラチンカプセル剤を、次のように調製する:
【0094】
【化5】
代表的な手順:これらの成分を完全に混和させ、次いで、硬カプセルに詰める(1カプセル当たり460mgの組成物)。
【0095】
(調合物実施例G)
経口投与のための懸濁液を次のように調製する:
【0096】
【化6】
代表的な手順:これらの成分を混合し、10mLの懸濁液につき100mgの活性成分を含む懸濁液を生成する。
【0097】
(調合物実施例H)
注射可能な調合物を次のように調製する:
【0098】
【化7】
代表的な手順:上記成分を混和させ、0.5N HCl、または0.5N NaOHを使用して、pHを4±0.5に調節する。
【0099】
(効用)
式Iの化合物は、β2アドレナリンレセプターアゴニスト、およびムスカリンレセプターアンタゴニスト活性の両方を有するため、本発明の式Iの化合物の1,2−エタンジスルホン酸塩は、β2アドレナリンレセプターもしくはムスカリンレセプターが介在する医学的状態(すなわち、β2アドレナリンレセプターアゴニストもしくはムスカリンレセプターアンタゴニストを用いた処置により改善される医学的状態)を処置するための治療因子として有用であると期待される。そのような医学的状態としては、例として、肺障害、または可逆性気道閉塞と関連する疾患(例えば、慢性閉塞性肺疾患(例えば、慢性的で息苦しい(wheezy)気管支炎、および肺気腫)、喘息、肺線維症、アレルギー性鼻炎、鼻漏など))を含む疾患が挙げられる。処置し得る他の状態としては、早期分娩、鬱病、鬱血性心不全、皮膚病(例えば、炎症性、アレルギー性、乾癬性、および増殖性皮膚疾患)、ペプシンの酸性を低下させることが望ましい状態(例えば、消化性および胃潰瘍)、および筋肉消耗性疾患が挙げられる。
【0100】
したがって、1つの実施形態において本発明は、肺障害を処置するための方法に向けられ、この方法は、処置を必要とする患者に治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を投与する工程を包含する。肺障害を処置するために使用される場合、本発明の塩は、典型的に、1日当たり複数回分の用量、1回で1日分の用量、または1回で1週間分の用量で、吸入により投与される。一般的に、肺障害を処置するための用量は、約10μg/日〜約200μg/日の範囲である。
【0101】
吸入により投与される場合、本発明の化合物は、典型的に気管支拡張を提供する効果を有する。したがって、その方法の別の局面において、本発明は、気管支拡張を必要としている患者に気管支拡張を提供する方法に向けられ、この方法は、気管支拡張を生じる量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を患者に投与する工程を包含する。一般的に、気管支拡張を提供するための用量は、約10μg/日〜約200μg/日の範囲である。
【0102】
1つの実施形態において、本発明は、慢性閉塞性肺疾患または喘息を処置する方法に向けられ、この方法は、処置を必要とする患者に治療有効量のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩またはその溶媒和物を投与する工程を包含する。COPDまたは喘息を処置するために使用される場合、本発明の塩は、典型的に、1日当たり複数回分の用量、または1回で1日分の用量で、吸入により投与される。一般的に、COPDまたは喘息を処置するための用量は、約10μg/日〜約200μg/日の範囲である。本明細書中で使用される場合、COPDは、慢性閉塞性気管支炎および肺気腫を含む(例えば、Barnes,Chronic Obstructive Pulmonary Disease,N Engl J Med 2000:343:269−78を参照)。
【0103】
肺障害を処置するために使用される場合、本発明の塩は、必要に応じ、他の治療因子と組み合せて投与される。したがって、特定の実施形態において、本発明の薬学的組成物および方法は、治療有効量のステロイド抗炎症剤をさらに含む。本発明の1,2−エタンジスルホン酸塩の特性および効用は、当該技術分野で周知の種々のインビトロおよびインビボアッセイを使用して明らかにされ得る。例えば代表的なアッセイは、次の実施例においてさらに詳細に記載されている。
【実施例】
【0104】
次の(調製)および(実施例)は、本発明の具体的な実施例を説明するために提供される。しかしながら、これらの具体的な実施例は、他に示さない限り、決して本発明の範囲を限定することを意図していない。
【0105】
次の略語は、他に示さない限り次の意味を有し、本明細書中で使用され規定されていない任意の他の略語は、それらの標準的な意味を有する:
AC アデニリルシクラーゼ
Ach アセチルコリン
ATCC アメリカン・タイプ・カルチャー・コレクション
BSA ウシ血清アルブミン
cAMP 3’−5’環状アデノシン一リン酸
CHO チャイニーズハムスター卵巣
cM5 クローンチンパンジーのM5レセプター
DCM ジクロロメタン(すなわち、塩化メチレン)
DIPEA N,N−ジイソプロピルエチルアミン
dPBS ダルベッコリン酸緩衝生理食塩水
DMEM ダルベッコ改変イーグル培地
DMSO ジメチルスルホキシド
EDTA エチレンジアミン四酢酸
Emax 最大効果
EtOAc 酢酸エチル
EtOH エタノール
FBS 胎仔ウシ血清
FLIPR 蛍光定量的イメージングプレートリーダー
Gly グリシン
HATU O−(7−アザベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート
HBSS ハンクス緩衝食塩水
HEK ヒト胚腎臓細胞
HEPES 4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸
hM1 クローンヒトM1レセプター
hM2 クローンヒトM2レセプター
hM3 クローンヒトM3レセプター
hM4 クローンヒトM4レセプター
hM5 クローンヒトM5レセプター
HPLC 高速液体クロマトグラフィー
IBMX 3−イソブチル−1−メチルキサンチン
%Eff %効果
PBS リン酸緩衝生理食塩水
PyBOP ベンゾトリアゾール−1−イルオキシトリピロリジノホスホニウムヘキサフルオロホスフェート
rpm 1分当たりの回転数
TFA トリフルオロ酢酸
THF テトラヒドロフラン
Tris トリス(ヒドロキシメチル)アミノメタン
他に断りのない限り、試薬、出発物質、および溶媒は、供給業者(例えば、Aldrich、Fluka、Sigmaなど)から購入され、さらなる精製なしに使用された。
【0106】
下記の実施例において、HPLC分析を、Agilentが提供する3.5ミクロンの粒径を有するZorbax Bonus RP2.1×50mmのカラム(C14カラム)を用いたAgilent(Palo Alto,CA)1100シリーズ機器を使用して実施した。検出は、214nmにおけるUV吸収度による。HPLC10〜70データを、10%〜70%のB0.5mL/分の流速で6分間にわたり入手した。流動相Aは、2%−98%−0.1%のACN−H2O−TFA:そして流動相Bは、90%−10%−0.1%のACN−H2O−TFAであった。上記流動相AおよびBを使用して、HPLC5−35のデータ、およびHPLC10−90のデータを、5分間の勾配で入手した。
【0107】
液体クロマトグラフィー質量分析(LCMS)データを、Applied Biosystems(Foster City,CA)のAPI−150EX型の機器を使用して入手した。LCMS10−90のデータを、10%〜90%の流動相Bを用い、5分の勾配で入手した。
【0108】
小規模な精製を、Applied BiosystemsのAPI 150EX Prep Workstation systemを使用して入手した。流動相は、A:水+0.05容量(v/v)%TFA;そしてB:アセトニトリル+0.05容量(v/v)%TFAであった。アレイ(典型的に約3〜50mgの回収サンプルサイズ)に対し、次の条件が使用された:流速20mL/分;勾配15分、および5ミクロンの粒子を有する20mm×50mmのPrism RPカラム(Thermo Hypersil−Keystone,Bellefonete,PA)。より大規模な精製(典型的に100mgより大きなの粗サンプル)に対しては、次の条件を使用した:流速60mL/分;勾配30分、および10ミクロンの粒子を有する41.4mm×250mmのMicrosorb BDSカラム(Varian,Palo Alto,CA)。
【0109】
タングステンハロゲン光源、および589nmフィルターを用いたJasco Polarimeter(P−1010型)を使用して、20℃にてキラル化合物([α]20Dと示す)についての比旋光度を測定した。テスト化合物のサンプルは、典型的に、1mg/mLの水にて測定された。
【0110】
(調製1)
(4−アミノ−5−クロロ−2−メトキシ安息香酸メチル)
トルエン(9mL)とメタノール(1mL)との混合物中の4−アミノ−5−クロロ−2−メトキシ安息香酸(1.008g、5.0mmol)の溶液に、(トリメチルシリル)ジアゾメタン(ヘキサン中2.0M、3.0mL、6.0mmol)を、0℃にて滴下した。次いで、この反応混合物を室温に温め、16時間、攪拌した。過剰な(トリメチルシリル)ジアゾメタンを、反応混合物の鮮明な黄色が消えるまで、酢酸を加えることによってクエンチした。次いで、この混合物を真空下で濃縮することにより、オフホワイトの固体として表題の化合物を得、これをさらなる精製なしに使用した。
【0111】
(調製2)
(4−アクリロイルアミノ−5−クロロ−2−メトキシ安息香酸メチル)
調製2の粗生成物に、ジクロロメタン(10mL、0.5M)およびトリエチルアミン(2.1mL、15mmol)を加えた。この混合物を0℃に冷却し、塩化アクリロイル(812μL、10mmol)を攪拌しながら滴下した。2時間後、この反応物をメタノール(約2mL)を0℃にて加えることによりクエンチし、得られた混合物を室温にて15分間攪拌し、次いで、真空下で濃縮した。ジクロロメタン(30mL)および水(30mL)をこの残留物に加え、この混合物を十分に混合した。この層を分離し、水層をジクロロメタン(20mL)を用いて抽出した。有機層を合わせて、乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去することにより、褐色の泡沫状固体として表題の化合物を得、これをさらなる精製なしに使用した。
【0112】
(調製3)
(ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル)
ビフェニル−2−イソシアネート(97.5g、521mmol)および4−ヒドロキシ−1−ベンジルピペリジン(105g、549mmol)(共にAldrich、Milwaukee、WIから市販されている)を、12時間、70℃にて一緒に加熱し、その間、ビフェニル−2−イルカルバミン酸1−ベンジルピペリジン−4−イルエステルの形成をLCMSによりモニターした。次いで、この反応混合物を50℃に冷却し、エタノール(1L)を加え、次いで、6M 塩酸(191mL)をゆっくりと加えた。次いで、この反応混合物を周囲温度に冷却し、蟻酸アンモニウム(98.5g、1.56mol)を加え、窒素ガスを20分間溶液に通して活発に泡立てた。次いで、パラジウム(活性炭担持10重量%(乾燥ベース))(20g)を加えた。この反応混合物を、12時間、40℃にて加熱し、次いで、セライトパッドに通して濾過した。次いで、溶媒を減圧下で除去し、1M 塩酸(40mL)を粗残留物に加えた。次いで、水酸化ナトリウム(10N)を加えてpHを12に調節した。水層を酢酸エチル(2×150mL)を用いて抽出し、乾燥させ(硫酸マグネシウム)、次いで、溶媒を減圧下で除去することにより、表題の化合物(155g、100%)を得た。HPLC(10−70)Rt=2.52;MS m/z:[M+H+]C18H20N2O2についての計算値297.15;測定値297.3。
【0113】
(調製4)
(4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ}−5−クロロ−2−メトキシ安息香酸メチル)
調製2からの粗生成物に、調製3の生成物(1.33g、4.5mmol)、およびTHF(22.5mL)とメタノール(2.5mL)との混合物を加えた。この混合物を50℃にて16時間、攪拌しながら加熱し、次いで溶媒を、真空下で除去した。残留物をクロマトグラフィー法(シリカゲル;EtOAc)で分離することにより、オフホワイトの泡沫状固体として、表題の化合物(0.82g;Rf=0.4、3工程にわたる収率29%)を得た。MS m/z566.4(M+H、C30H32ClN3O6についての期待値565.20)。
【0114】
(調製5)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ヒドロキシメチル−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
THF(4.5mL)とメタノール(0.5mL)との混合物中の調製4の生成物(0.82mg、1.45mmol)の溶液に、0℃にてホウ化水素リチウム(32mg、1.45mmol)を加えた。この反応混合物を、室温に温めさせ、41時間、攪拌した。次いで、この反応物を、さらなる泡立ちが観察されなくなるまで、1N塩酸水を0℃にて加えることによりクエンチし、この混合物を10分間、攪拌した。溶媒を真空下で除去し、残留物をアセトニトリル(約2mL)に溶解させた。この溶液を、prep−RP−HPLC(勾配:0.05%のTFAを有する水中に2〜50%のアセトニトリル)により精製した。適切な分画を回収して合わせ、凍結乾燥することにより、トリフルオロ酢酸塩として表題の化合物を得た。この塩を酢酸イソプロピル(10mL)、および1N 水酸化ナトリウム水(10mL)を用いて処理し、有機層を回収し、乾燥させ(Na2SO4)、濾過し、そして溶媒を真空下で除去することにより、白色泡沫状固体として表題の化合物(161mg、収率21%)を得た。MS m/z538.4(M+H、C29H32ClN3O5についての期待値537.20)。
【0115】
(調製6)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイル)エチル]ピペリジン−4−イルエステル)
ジクロロメタン(3mL)中の調製5の生成物(161mg、0.3mmol)の溶液に、ジメチルスルホキシド(213μL、3.0mmol)、およびジイソプロピルエチルアミン(261μL、1.5mmol)を加えた。この混合物を−20℃に冷却し、三酸化硫黄ピリジン錯体(238mg、1.5mmol)をゆっくりと加えた。30分後、この反応混合物を水(約3mL)を加えることによりクエンチした。層を分離して、有機層を乾燥させ(Na2SO4)、濾過し、溶媒を真空下で除去することにより、薄黄色の固体として表題の化合物を得た。MS m/z536.3(M+H、C29H30ClN3O5についての期待値535.19)。
【0116】
(調製7)
(8−ベンジルオキシ−5−(2−ブロモアセチル)−1H−キノリン−2−オン)
((a)8−アセトキシ−1H−キノリン−2−オン)
8−ヒドロキシキノリン−N−オキシド(160.0g、1.0mol)(Aldrich、Milwaukee、WIから市販されている)、および無水酢酸(800mL、8.4mol)を、3時間、100℃にて加熱し、次いで、氷中で冷却した。この生成物を、ブフナー漏斗で回収し、無水酢酸(2×100mL)を用いて洗浄し、減圧下で乾燥させることにより、固体として8−アセトキシ−1H−キノリン−2−オン(144g)を得た。
【0117】
((b)5−アセチル−8−ヒドロキシ−1H−キノリン−2−オン)
1,2−ジクロロエタン(280mL)中の塩化アルミニウム(85.7g、640mmol)のスラリーを氷中で冷却し、工程(a)からの生成物(56.8g、280mmol)を加えた。この混合物を室温に温め、次いで、85℃にて加熱した。30分後、塩化アセチル(1.5mL、21mmol)を加え、この混合物をさらに60分間、加熱した。次いで、この反応混合物を冷却し、0℃にてよく攪拌しながら1N 塩酸(3L)に加えた。2時間攪拌後、固体をブフナー漏斗で回収し、水(3×250mL)を用いて洗浄し、減圧下で乾燥させた。いくつかのバッチから分離させた粗生成物(135g)を合わせ、6時間、ジクロロメタン(4L)を用いて粉砕した。得られた固体をブフナー漏斗で回収し、減圧下で乾燥させることにより、表題の化合物(121g)を得た。
【0118】
((c)5−アセチル−8−ベンジルオキシ−1H−キノリン−2−オン)
工程(b)からの生成物(37.7g、186mmol)に、N,N−ジメチルホルムアミド(200mL)、および炭酸カリウム(34.5g、250mmol)を加え、続いて臭化ベンジル(31.8g、186mmol)を加えた。この混合物を2.25時間、室温にて攪拌し、次いで、飽和塩化ナトリウム(3.5L)に0℃にて注ぎ入れ、1時間、攪拌した。この生成物を回収し、1時間、ブフナー漏斗で乾燥させ、得られた固体をジクロロメタン(2L)に溶解させ、この混合物を硫酸ナトリウムで乾燥させた。溶液をセライトパッドに通して濾過し、次いで、ジクロロメタン(5×200mL)を用いて洗浄した。次いで、合わせた濾液を濃縮乾固し、得られた固体を2時間、エーテル(500mL)を用いて粉砕した。この生成物をブフナー漏斗で回収し、エーテル(2×250mL)を用いて洗浄し、減圧下で乾燥させ、粉末として表題の化合物(44g)を得た。
【0119】
((d)8−ベンジルオキシ−5−(2−ブロモアセチル)−1H−キノリン−2−オン)
工程(c)からの生成物(20.0g、68.2mmol)を、ジクロロメタン(200mL)に溶解させ、0℃にて冷却した。三フッ化ホウ素ジエチルエーテラート(10.4mL、82.0mmol)をシリンジによって加え、この混合物を室温に温めることにより、濃厚な懸濁液を得た。この懸濁液を45℃(油浴)にて加熱し、ジクロロメタン(100mL)中の臭素(11.5g、72.0mmol)の溶液を、40分間にわたり加えた。この混合物をさらに15分間、45℃にて保ち、次いで、室温に冷却した。この混合物を減圧下で濃縮し、次いで1時間、10%炭酸ナトリウム水(200mL)を用いて粉砕した。固体をブフナー漏斗で回収し、水(4×100mL)を用いて洗浄し、減圧下で乾燥させた。2処理分の生成物を精製用に合わせた。この粗生成物(52g)を、クロロホルム(500mL)中の50%メタノールを用いて1時間、粉砕した。この生成物をブフナー漏斗で回収し、クロロホルム(2×50mL)およびメタノール(2×50mL)中の50%メタノールを用いて洗浄した。固体を減圧下で乾燥させることにより、粉末として表題の化合物(34.1g)を得た。
【0120】
(調製8)
(8−ベンゾイルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ(butyldimethylsilanyloxy))エチル]−1H−キノリン−2−オン)
((a)8−ベンジルオキシ−5−((R)−2−ブロモ−1−ヒドロキシエチル)−1H−キノリン−2−オン)
(R)−(+)−α,α−ジフェニルプロリノール(30.0g、117mmol)とトリメチルボロキシン(11.1mL、78mmol)とをトルエン(300mL)中で合わせ、30分間、室温にて攪拌した。この混合物を、150℃の油浴中に置き、液体を蒸留により取り除いた。トルエンを20mLのアリコートに加え、4時間、蒸留を続けた。総量で300mLのトルエンを加えた。次いで、この混合物を室温に冷却した。500μLのアリコートをエバポレートして乾固し、計量し(246mg)、この触媒濃度を1.8Mと決定した。
【0121】
8−ベンジルオキシ5−(2−ブロモアセチル)−1H−キノリン−2−オン(90.0g、243mmol)を、窒素下に置き、テトラヒドロフラン(900mL)を加え、続いて上記の触媒(トルエン中1.8M、15mL、27mmol)を加えた。この懸濁液を氷/イソプロパノール浴中で、−10±5℃に冷却した。ボラン(THF中1.0M、294mL、294mmol)を、4時間にわたり加えた。次いでこの反応物を、さらに45分間、−10℃にて攪拌し、次いで、メタノール(250mL)をゆっくりと加えた。この混合物を真空下で濃縮し、残留物を沸騰アセトニトリル(1.3L)に溶解させ、熱いうちに濾過し、次いで、室温に冷却した。結晶を濾過し、アセトニトリルを用いて洗浄し、真空下で乾燥させることにより、表題の化合物を得た(72.5g、196mmol、収率81%、95%ee、HPLCによる純度95%)。
【0122】
((b)8−ベンジルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
工程(b)の生成物(70.2g、189mmol)に、N,N−ジメチルホルムアミド(260mL)を加え、窒素下にてこの混合物を氷浴中で冷却した。2,6−ルチジン(40.3g、376mmol)を、5分間にわたり加え、次いで、tert−ブチルジメチルシリルトリフルオロメタンスルホナート(99.8g、378mmol)を、20℃未満の温度を維持しながらゆっくりと加えた。この混合物を45分間、室温に温めさせた。メタノール(45mL)を混合物に10分にわたって滴下し、この混合物を、酢酸エチル/クロロヘキサン(1:1、500mL)と水/ブライン(1:1、500mL)との間で分割した。有機物(organics)をさらに2度、水/ブライン(1:1、各回500mL)を用いて洗浄した。合わせた有機物を減圧下でエバポレートすることにより、薄黄色の油状体を得た。シクロヘキサン(400mL)を、2度に分けてこの油状体に加え、濃厚な白色のスラリーが生成するまで蒸留を続けた。シクロヘキサン(300mL)をスラリーに加え、得られた白色結晶を濾過し、シクロヘキサン(300mL)を用いて洗浄し、減圧下で乾燥させ、表題の化合物(75.4g、151mmol、収率80%、98.6%ee)を得た。
【0123】
(調製9A)
(8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
DMSO(1.7mL)中の調製8の生成物(1.00g、2.05mmol)とベンジルアミン(493μL、4.51mmol)との攪拌溶液を、105℃にて4時間、加熱した。この反応混合物を冷却させ、次いでEtOAc(10mL)を用いて希釈し、有機層を飽和塩化アンモニア水溶液(5mL)および1N 水酸化ナトリウム(5mL)を用いて洗浄し、乾燥させ(MgSO4)、溶媒を減圧下で除去した。この粗残留物を、カラムクロマトグラフィー(50% EtOAc/ヘキサン)により精製し、表題の化合物(700mg、67%)を得た。MS m/z:[M+H+]C31H38N2O3Siについての計算値515.27;測定値515.5。
【0124】
(調製9B)
(8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン)
500mLの三ッ口丸底フラスコに、8−ベンジルオキシ−5−[(R)−2−ブロモ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン(43g、0.124mol、キラル純度約95%)、1−メチル−2−ピロリジノン(210mL)、およびベンジルアミン(28.3g、0.37mol)を加えた。得られた混合物を窒素を用いて洗い流し、次いで、90℃にて6時間、攪拌した。次いで、この混合物を室温に冷却し、水(300mL)および酢酸エチル(300mL)を加えた。層を分離し、有機層を水(200mL)、水と飽和塩化ナトリウム水溶液との1:1混合物(200mL)、および水(200mL)を用いて洗浄した。次いで、有機層を硫酸マグネシウムで乾燥させ、濾過し、減圧下で濃縮することにより、橙色の油状体として表題の化合物を得た。
【0125】
この橙色の油状体にヘプタン(200mL)および酢酸エチル(200mL)を加え、得られた混合物を65℃に加熱することにより、透明な溶液を得た。この溶液を室温に冷却し、一晩(約16時間)おいた時点で沈殿物を生成した。この沈殿物を濾過により回収し、立体化学的には不純な表題の化合物(8.85g、79.6%ee)を得た。濾液を減圧下で濃縮することにより、表題の化合物(38.6g、99.4%)を得た。この物質を先の物質(19.2g、99.5%ee)のバッチと合わせ、そしてヘプタン(250mL)および酢酸エチル(100mL)を加えた。この混合物を80℃に加熱し(濁りを帯びた〜透明な溶液)、次いで、室温に冷却し、一晩おいた。得られた沈殿物を濾過により回収し、白色固体(36.8g、98.4%ee、化学的純度99.9%)として表題の化合物を得た。濾液を減圧下で濃縮し、残渣をヘプタン(100mL)に溶解させた。得られた固体を回収することにより、黄褐色の固体(24g、キラル純度100%、化学的純度95%)として表題の化合物を得た。
【0126】
(調製10A)
(5−[(R)−2−アミノ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−8−ヒドロキシー1H−キノリン−2−オン)
エタノール(62mL)中の調製9Aの生成物(3.16g、6.15mmol)とパラジウム(活性炭担持10重量%(乾燥ベース))(1.58g)との攪拌溶液を、24時間、水素雰囲気下に置いた。この反応混合物をセライトに通して濾過し、メタノール(15mL)を用いて洗浄し、次いで、溶媒を減圧下で除去することにより、固体として表題の化合物(1.52g、4.55mmol、74%)を得た。
【0127】
(調製10B)
(5−[(R)−2−アミノ−1−(tert−ブチルジメチルシラニルオキシ)エチル]−8−ヒドロキシ−1H−キノリン−2−オン酢酸塩)
8−ベンジルオキシ−5−[(R)−2−(N−ベンジルアミノ)−1−(tert−ブチルジメチルシラニルオキシ)エチル]−1H−キノリン−2−オン(100g、194mmol)および酢酸(17.5mL、291mmol)を、メタノール(1L)に溶解させた。この透明な溶液を窒素を用いてパージし、次いで、炭素担持水酸化パラジウム(20g、20重量%のPd(乾燥ベース)、湿気(約50%の水分))を加えた。室温にて6時間、水素ガスを攪拌溶液に通じて泡立て、その間に、濃厚なスラリーを生じさせた。次いで、この反応混合物を窒素を用いてパージし、メタノール(1L)を加えた。約30分間、得られた混合物を(生成物を溶解させるために)攪拌し、次いで、この混合物をセライトパッドに通して濾過した。濾液を約500mLの容量まで減圧下で濃縮し、得られたスラリーにエタノール(500mL)を加えた。得られた混合物を再度、減圧下で約500mLの容量まで濃縮し、得られた沈殿物を濾過により回収し、乾燥させることにより、黄白色の固体として表題の化合物を得た(65g、収率85%、純度>98%)。
【0128】
(調製11)
(ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−(tert−ブチルジメチルシラニルオキシ)−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロー5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
ジクロロメタン(0.5mL)とメタノール(0.5mL)との混合物中の調製6からの生成物に、調製10Aの生成物(124.1mg、3.1mmol)を加え、得られた混合物を、室温にて1.5時間、攪拌した。トリアセトキシホウ化水素ナトリウム(190.7mg、0.9mmol)を加え、得られた混合物を、室温にて15時間、攪拌した。この反応物を、水(約0.2mL)を加えることによりクエンチし、この混合物を真空下で濃縮することにより、表題の化合物を得、これをさらなる精製なしに使用した。MS m/z854.5(M+H、C46H56ClN5O7Siについての期待値853.36)。
【0129】
(調製12)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル)
ジクロロメタン(1.0mL、0.3M)中の調製11の生成物の懸濁液に、トリエチルアミン三フッ化水素塩(245μL、1.5mmol)を加えた。この混合物を、室温にて45時間、攪拌し、次いで、この混合物を真空下で濃縮した。残留物を、DMF(0.5mL)と、アセトニトリル/水(1:1、0.1%TFAを含む、0.6mL)と、TFA(0.3mL)と、アセトニトリル(約1mL)との混合物に溶解させ、この混合物を、prep−RP−HPLC(勾配:0.05%TFAを含む水中のアセトニトリル2〜50%)によって精製した。適切な分画を回収して合わせ、凍結乾燥させることにより、オフホワイトの固体として、表題の化合物のジトリフルオロ酢酸塩(100mg、収率34%、HPLCによる純度98.7%)を得た。MS m/z740.5(M+H、C40H42ClN5O7についての期待値739.28)。
【0130】
(実施例1)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
エタノール(0.2mL)中の1,2−エタンジスルホン酸二水和物(3.8mg、0.02mmol)の溶液を、イソプロパノールとジクロロメタン(1mL)との64:1v/v混合物中の調製12の生成物(14.3mg、0.02mmol)の溶液にゆっくりと加えた。得られた溶液を、45℃〜50℃にて約30分間、加熱した。次いで、この混合物をゆっくりと室温に冷却し、室温になった時点で溶液は僅かに濁った。この溶液を、一晩、窒素の緩流下で周囲温度においた。得られた沈殿物を濾過により回収し、乾燥させることにより、白色結晶性固体として表題の化合物(13mg、収率72%)を得た。
【0131】
(実施例2)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル(26.8mg、0.0362mmol)の溶液を、エタノール(5.36mL)中に調製し、室温にて、完全な溶解が得られるまで(5分)攪拌した。エタノール(0.2mL)中の1,2エタンジスルホン酸二水和物(8.2mg、0.0362mmol)の溶液を、約1分かけて最初の溶液にゆっくりと加えた。5分間、得られた懸濁液を攪拌し、次いで、窒素下で濾過により分離した。得られた沈殿物を乾燥させることにより、白色固体として表題の化合物(28.5mg、収率85%)を得た。
【0132】
(実施例3)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル(5g、6.75mmol、純度>99%)を、イソプロパノール(100mL)に溶解させ、水(20mL)に溶解させたエタンジスルホン酸二水和物(1.525mg、6.75mmol)を添加した。得られたスラリーを、室温にて1時間攪拌し、次いで約30℃にて一晩攪拌した。表題の化合物(6.0g)を、白色粉末として分離した。この生成物を、30℃にて48時間、イソプロパノール中の20%の水(100mL)中で加熱した。室温に冷却後、得られた沈殿物を濾過により単離し、2時間、風乾させることにより、表題の化合物(5.4g)を得た。
【0133】
(調製13)
(4−アクリロイルアミノ−5−クロロ−2−メトキシ安息香酸メチル)
オーバーヘッドスターラー、温度調節および追加漏斗(addition funnel)を備えた1リットルの3ツ口丸底フラスコに、4−アミノ−5−クロロ−2−メトキシ安息香酸メチル(44.2g、200mmol)、ジクロロメタン(500ml)、およびジイソプロピルエチルアミン(104.5mL、600mmol)を加えた。この得られた混合物を、室温にて成分が溶解するまで攪拌し、次いでこの混合物を0℃に冷却した。次いで、内部の反応混合物温度を10℃未満に維持しながら、塩化アクリロイル(16.25mL、200mmol)を滴下した。添加の合計時間は約30分であった。次いでこの反応混合物を、約2時間かけて0℃〜室温にゆっくりと温めた。次いで、飽和炭酸水素ナトリウム水溶液(200mL)、およびジクロロメタン(200mL)を加え、この混合物を15分間攪拌し、次いで層を分離した。ジクロロメタン層を1M 塩酸(200mL)を用いて洗浄し、次いで、減圧下で元の容量の約3分の1に濃縮し、濃厚なスラリーを得た。このスラリーを濾過し、濾塊をジクロロメタン(100mL)で洗浄し、乾燥させることにより、オフホワイトの固体として表題の化合物(36g、収率67%、HPLCによる純度>98%)を得た。
(調製14)
(4−{3−[4−(ビフェニル−2−イルカルバモイルオキシ)ピペリジン−1−イル]プロピオニルアミノ}−5−クロロ−2−メトキシ安息香酸メチル)
オーバーヘッドスターラー、温度調節および還流凝縮器を備えた1リットルの3ツ口丸底フラスコに、ビフェニル−2−イルカルバミン酸ピペリジン−4−イルエステル(36.3g、122mmol)、ジクロロメタン(500mL)、およびイソプロパノール(100mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いで、調製10からの生成物(30g、111.5mmol)を加えた。攪拌を、成分が溶解するまで室温で続け、次いでこの混合物を、還流下(50℃〜55℃)で18時間、加熱した。次いで、この反応混合物を室温に冷却し、エタノール(200mL)を加えた。この混合物を減圧下で約150mLの容量に濃縮し、濃厚なスラリーを得た。このスラリーを濾過し、濾塊をエタノール(50mL)で洗浄し、乾燥させることにより、白色固体として表題の化合物(58g、収率92%、HPLCによる純度99.5%)を得た。
【0134】
(調製15)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ヒドロキシメチル−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
2リットルの丸底フラスコに、調製14からの生成物(40g、70.8mmol)およびTHF(400mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いでこのフラスコを、窒素で5分間パージした。次いでこの混合物を0℃(内部温度)に冷却し、THF(106mL、106mmol)中の水素化リチウムアルミニウムの1M溶液を、内部の反応混合物温度を10℃未満に維持しながら、追加漏斗を通じて滴下した。合計添加時間は、約40分であった。次いでこの反応混合物を、0℃にて1時間、攪拌し、次いで内部の反応混合物温度を15℃未満に維持しながら、1M水酸化ナトリウム(200mL)を加えた。次いで、層を分離し、THF層を飽和塩化ナトリウム水溶液(100mL)で洗浄し、硫化ナトリウムで乾燥させ、濾過し、減圧下で濃縮することにより、白色固体として表題の化合物(38g、収率100%、HPLCによる純度94%)を得た。
【0135】
(調製16)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−ホルミル−5−メトキシフェニル−カルバモイル)エチル]ピペリジン−4−イルエステル)
1リットルの丸底フラスコに、調製15からの生成物(28g、52mmol)、およびジクロロメタン(500mL)を加えた。得られた混合物を、成分が溶解するまで室温にて攪拌し、次いで、活性酸化マンガン(IV)(45g、520mmol)を加えた。この反応混合物を、窒素下で室温にて12時間、攪拌し、次いで、セライトパッドに通して濾過した。次いで、この混合物を減圧下で濃縮し、残留物を真空下で一晩乾燥させることにより、黄色固体として表題の化合物(26g、収率93%、HPLCによる純度約93%)を得た。
【0136】
(調製17)
(ビフェニル−2−イルカルバミン酸1−[2−(4−{[(R)−2−(tert−ブチルジメチルシラニルオキシ)−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−2−クロロ−5−メトキシ−フェニルカルバモイル)エチル]ピペリジン−4−イルエステル)
500mLの丸底フラスコに、調製16からの生成物(6g、11.2mmol)、およびジクロロメタン(50mL)を加えた。得られた混合物を成分が溶解するまで室温にて攪拌し、次いで、調製7の生成物(6g、15.0mmol)および乾燥メタノール(50mL)を加えた。この混合物を窒素下で室温にて2時間、攪拌し(透明な黄色から橙色の溶液)、次いで、この混合物を0℃〜5℃に冷却した。固体のナトリウムトリアセトキシボロヒドリド(7.2g、34mmol)を10分間かけて少しずつ加え、次いで、この反応混合物を、約2時間かけて、0℃〜室温にゆっくりと温めた。次いで、この混合物を0℃に冷却し、1M 水酸化ナトリウム水溶液(50mL)およびジクロロメタン(150mL)を加えた。この混合物を十分に攪拌し、次いで、層を分離した。有機層を飽和塩化ナトリウム水溶液(50mL)で洗浄し、濾過し、硫化ナトリウムで乾燥させ、濾過し、減圧下で濃縮することにより、黄色固体として表題の化合物(10.1g、収率100%、HPLCによる純度87%)を得た。
【0137】
(実施例4)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩)
50mLの丸底フラスコに、調製17からの生成物(2.0g、2.5mmol)およびジクロロメタン(10mL)を加えた。得られた混合物を、成分を溶解するまで室温にて攪拌し、次いで、三フッ化水素トリエチルアミン(1.2mL、7.5mmol)を加え、得られた混合物を、25℃にて20時間、攪拌した。次いで、メタノール(10mL)中の1,2−エタンジスルホン酸二水和物(0.56g、2.5mmol)の溶液を加え、この混合物を30℃にて2時間攪拌した時点で、濃厚な白色スラリーが形成された。このスラリーをゆっくりと濾過し、濾塊をメタノール(10mL)で洗浄し、2時間、風乾し、次いで、真空下で一晩乾燥させることにより、微細な白色粉末として表題の化合物(1.5g、HPLCによる純度>98%)を得た。
【0138】
(実施例5)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の精製)
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(80g)に、イソプロパノール(800mL)中の容量で20%の水の溶液を加えた。得られたスラリーを室温にて一晩おき、次いで濾過することにより、改善した結晶性および純度を有する表題の化合物(74g)を得た。
【0139】
(調製18)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の種結晶)
(工程(a))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(100mg)を、メタノール(20mL)中の13%の水に、約60℃にて溶解させた。得られた透明溶液を、密閉容器中で室温に冷却させた。48時間後、得られた板様結晶を、濾過により分離した。
【0140】
(工程(b))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(1.0g)を、メタノール(100mL)中の15%の水に、60〜65℃にて溶解させた。この透明な攪拌溶液を、30℃に冷却させ、次いで、工程(a)の結晶性生成物(4.2mg)を加えた。この溶液を20℃に冷却し、2時間攪拌した。得られた沈殿物を濾過により分離し、空気中で1時間、乾燥させることにより、表題の化合物(680mg)を得た。
【0141】
(工程(c))
工程(b)の手順を、工程(a)の生成物の代わりに工程(b)の生成物(20mg)を用いて繰り返し、表題の化合物(690mg)を得た。
【0142】
(工程(d))
実施例4のとおり調製したビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩(10g)を、メタノール(1L)中の15%の水に、60〜65℃にて溶解させた。この透明な攪拌溶液を、30℃に冷却させ、次いで、工程(c)の結晶性生成物(4.2mg)を加えた。この溶液を20℃に冷却し、18時間攪拌した。得られた沈殿物を濾過により分離し、空気中で2時間、乾燥させることにより、表題の化合物(5.5g)を得た。
【0143】
(実施例6)
(ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステル1,2−エタンジスルホン酸塩の種結晶を使用した再結晶化)
12Lの丸底フラスコに、実施例5からの生成物(60g、64.5mmol)、水(0.9L)、およびメタノール(5.1L)を加えた。得られた混合物を、成分が溶解するまで攪拌しながら25℃から61〜65℃に加熱し、さらに20分間、60〜65℃にて攪拌した。この混合物を30℃に冷却させ、次いで、調製18の生成物(2g、2.15mmol)を加えた。この混合物を20℃にゆっくりと冷却し、得られたスラリーを、30℃にてさらに2時間、攪拌した。この生成物をメタノール(500mL)と共に濾過し、2時間空気中で乾燥させ、次いで真空中で25〜30℃にて18時間、乾燥させることにより、表題の化合物(43g、収率72%、純度99.2%)を得た。
【0144】
(実施例7)
(熱分析)
示差走査熱量測定(DSC)を、Thermal Analystコントローラーを備えたTA InstrumentsのQ−10型モジュールを使用して実施した。データを回収し、TA InstrumentsのThermal Solutionsソフトウェアを使用して分析した。約1mgのサンプルを正確に計量して蓋付きのアルミパンに入れた。このサンプルを、周囲温度から約300℃まで5℃/分の一次加熱傾斜(linear heating ramp)を使用して評価した。使用中、DSCセルを、乾燥窒素を用いてパージした。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なDSCの記録を図1に、実施例2のサンプルについての代表的なDSCの記録を図2に示す。
【0145】
熱重量分析(TGA)を、高分解能を備えたTA InstrumentsのQ−50型モジュールを使用して実施した。データを回収し、TA InstrumentsのThermal Solutionsソフトウェアを使用して分析した。約10mgのサンプルをプラチナパンに置き、周囲温度〜300℃の高分解能加熱速度(high resolution−heating rate)でスキャンした。使用中、天秤および加熱炉チャンバーを、窒素フローでパージした。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なTGAの記録を図1に示す。
【0146】
DSCの記録は、本発明の1,2−エタンジスルホン酸塩が、それぞれ約239℃、および約219℃の融点を有して熱安定性に優れており、約200℃未満で熱分解しないことを示している。
【0147】
(実施例8)
(粉末エックス線回析)
粉末エックス線回析パターンを、Si(Li)半導体検出器を備え、1.542Å(45kV、40mA)におけるCuのKα線を使用したThermo ARL X−Ray DiffractometerのX’TRA型(Thermo ARL SA、Switzerland)を用いて得た。この分析を、典型的に、2θ角で2〜30°の範囲にかけて1点当たり0.03°の刻み幅を用い、2°/分のスキャン速度で実施した。入手したままかまたは微細な粉末に磨り潰したサンプルを、機器の分析用のトップローディングカップに合わせて設計された特別注文の小容量インサートに優しく詰め込んだ。機器を週に一度、±0.02°の2θ角内のシリコン金属基準に対応させる。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なPXRDパターンを図3に示し、実施例2のサンプルについてのパターンを図4に示す。
【0148】
(実施例9)
(赤外分析法)
赤外(IR)吸収スペクトルを、Nicolet減衰全反射(ATR)サンプルホルダーを備えたAvatar 360 FT−IR分光計を使用して、4000〜675cm−1の周波数範囲にわたり決定した。実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについての代表的なIR吸収スペクトルは、図5に示されるように、704±1、748±1、768±1、841±1、900±1、1055±1、1104±1、1166±1、1218±1、1294±1、1408±1、1522±1、1609±1、1655±1、および1701±1において有意な吸収帯を有する。
【0149】
(実施例10)
(力学的水分吸着アセスメント)
力学的水分吸着(DMS:dynamic moisture sorption)アセスメント(水分吸着−脱着プロフィールとしても知られる)を、VTI大気マイクロバランス(atmospheric microbalance)、SGA−100システム(VTI Corp.,Hialeah,FL 33016)を使用して、手で砕いた調製18のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについて実施した。約10mgのサンプルサイズを使用し、湿度を分析の開始時に周囲値に設定した。典型的なDMS分析は、3つのスキャンからなる:周囲〜相対湿度(RH)2%、2%RH〜90%RH、90%RH〜5%RH(スキャン速度:5%RH/ステップ)。質量を2分毎に測定し、サンプルの質量が5連続点の間、0.01%以内に安定したときに、RHを次の値(+/−5%RH)に変更した。代表的なDMSの記録を、図6に示す。
【0150】
このDMSの記録は、本発明の1,2−エタンジスルホン酸塩が、中程度(<9%)の吸湿性と共に、可逆的な吸着/脱着プロフィールを有することを示している。この塩は、40%RH〜75%RHの湿度範囲において2.5%未満の増量がある。この可逆的な湿気吸着/脱着プロフィールは、本発明の結晶性塩が、受容可能な吸湿性を有し、潮解性ではないことを示している。
【0151】
(実施例11)
(元素分析、および対イオン比率)
実施例6のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの、炭素、水素、窒素、および硫黄の次の元素百分比を、Flash EA 1112 Elemental Analyzer(CE Elantech、Lakewood、NJ)を使用した燃焼分析により決定した:炭素52.95%、水素5.43%、窒素6.83%、および硫黄6.87%。硫黄の測定重量百分比から計算した結晶性サンプル中の1,2−エタンジスルホン酸の重量百分比は、20.4%で理論上の重量百分比20.4%と等しく、対イオン比率1:1が得られた。
【0152】
(調製A)
(細胞培養およびヒトβ1、β2またはβ3アドレナリンレセプターを発現する細胞からの細胞膜の調製)
クローニングしたヒトβ1、β2、またはβ3アドレナリンレセプターをそれぞれ安定的に発現するチャイニーズハムスター卵巣(CHO)細胞株を、500μg/mLのジェネテシンの存在下で10%FBSを用いたHams F−12培地において、ほぼコンフルエンシーまで培養した。細胞単層をPBS中の2mM EDTAで浮かせた。細胞を1,000rpmにおける遠心分離によりペレットにし、この細胞ペレットを、−80℃にて冷凍保存するか、または使用の直前に、細胞膜を調製した。β1およびβ2レセプターを発現する細胞膜の調製に関し、細胞ペレットを溶解緩衝液(10mM HEPES/HCl、10mM EDTA、4℃にてpH7.4)中に再懸濁させ、ぴったりと適合したDounceガラス製ホモジナイザー(30ストローク)を氷上で使用して均質化した。さらなるプロテアーゼ感受性のβ3レセプターを発現する細胞膜に関しては、細胞ペレットを、「Complete Protease Inhibitor Cocktail Tablets with 2mM EDTA」の錠剤(Rocheカタログ番号1697498、Roche Molecular Biochemicals,Indianapolis,IN)を緩衝液50mLにつき1つで補った溶解緩衝液(10mM Tris/HCl、pH7.4)中で均質化した。このホモジェネートを20,000×gにて遠心分離し、得られたペレットを、上記のような再懸濁および遠心分離により、溶解緩衝液を用いて1度、洗浄した。次いでこの最終ペレットを、氷冷の結合アッセイ緩衝液(75mM Tris/HCl pH7.4、12.5mM MgCl2、1mM EDTA)に再懸濁させた。この細胞膜懸濁液のタンパク質濃度を、Lowry et al.,1951,Journal of Biological Chemistry,193,265;およびBradford,Analytical Biochemistry,1976,72,248−54に記載される方法により決定した。全ての細胞膜を、−80℃にて分割して保存するか、または直ちに使用した。
【0153】
(調製B)
(細胞培養およびヒトM1、M2、M3およびM4ムスカリンレセプターを発現する細胞からの細胞膜の調製)
クローニングしたヒトhM1、hM2、hM3、およびhM4ムスカリンレセプターサブタイプをそれぞれ安定的に発現するCHO細胞株を、10%FBS、および250μg/mLのジェネテシンで補ったHams F−12培地において、ほぼコンフルエンシーまで培養した。これらの細胞を、CO25%、37℃のインキュベータ中で増殖させ、dPBS中の2mM EDTAで浮かせた。細胞を650×gにおける5分の遠心分離により回収し、細胞ペレットを、−80℃にて冷凍保存するか、または使用の直前に、細胞膜を調製した。細胞膜の調製に関し、細胞ペレットを溶解緩衝液に再懸濁させ、Polytron PT−2100組織粉砕器(tissue disrupter)(Kinematica AG;20秒×2バースト)を用いて均質化した。粗細胞膜を、40,000×gで4℃にて15分間、遠心分離した。次いでこの細胞膜ペレットを、再懸濁緩衝液を用いて再懸濁させ、Polytron組織粉砕器を用いて再度均質化した。この細胞膜懸濁液のタンパク質濃度を、Lowry et al.,1951,Journal of Biochemistry,193,265に記載される方法により決定した。全ての細胞膜を、分割して−80℃にて保存するか、または直ちに使用した。調製されたhM5レセプター細胞膜のアリコートを、Perkin Elmerから直接購入し、使用まで−80℃にて保存した。
【0154】
(アッセイテスト手順A)
(ヒトβ1、β2およびβ3アドレナリンレセプターについての放射性リガンド結合アッセイ)
結合アッセイを、96ウェルマイクロタイタープレートにおいて、アッセイ緩衝液(75mM Tris/HCl 25℃にてpH7.4、12.5mM MgCl2、1mM EDTA,0.2% BSA)中のヒトβ1、β2、またはβ3アドレナリンレセプターを含む10〜15μgの細胞膜タンパク質を用い、総アッセイ容量100μLで実施した。放射性リガンドのKd値の決定のための飽和結合調査を、β1およびβ2レセプターについては[3H]−ジヒドロアルプレノロール(NET−720、100 Ci/mmol、PerkinElmer Life Sciences Inc.,Boston,MA)を、そして[125I]−(−)−ヨードシアノピンドロール(NEX−189、220 Ci/mmol、PerkinElmer Life Sciences Inc.,Boston,MA)を、0.01nM〜20nMの範囲の10もしくは11の異なる濃度で使用して実施した。テスト化合物のKi値の決定のための置換アッセイを、[3H]−ジヒドロアルプレノロールを1nMにて、そして[125I]−(−)−ヨードシアノピンドロールを0.5nMにて用い、10pM〜10μMの範囲における10もしくは11の異なる濃度のテスト化合物について実施した。非特異的な結合を、10μMのプロプラノロールの存在下で決定した。分析物を37℃にて1時間インキュベートし、次いで、β1およびβ2レセプターについてはGF/B、β3レセプターについてはGF/Cガラス繊維フィルタープレート(Packard BioScience Co.,Meriden,CT)(0.3%のポリエチレンイミンに予浸しておく)を用いた急速濾過により、結合反応を終わらせた。フィルタープレートを濾過緩衝液(75mM Tris/HCl 4℃にてpH7.4、12.5mM MgCl2、1mM EDTA)を用いて3回洗浄し、結合していない放射能を除去した。次いでこのプレートを乾燥させ、50μLのMicroscint−20液体シンチレーション液(Packard BioScience Co.,Meriden,CT)を加え、プレートをPackard Topcount液体シンチレーションカウンター(Packard BioScience Co.,Meriden,CT)で計数した。結合データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、ワンサイト・コンペティション(one−site competition)についての3パラメーターモデルを使用して、非線形回帰分析により分析した。曲線の最小値(curve minimum)を、10μMのプロプラノロールの存在下で決定された非特異的な結合についての値に固定した。テスト化合物についてのKi値を、観測されたIC50値、および放射性リガンドのKd値から、Cheng−Prusoff方程式(Cheng Y,and Prusoff WH.,Biochemical Pharmacology,1973,22,23,3099−108)を使用して計算した。
【0155】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターに対するより高い結合親和力を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトβ2アドレナリンレセプターに対し、10nM未満のKi値を有することが見出された。
【0156】
(アッセイテスト手順B)
(ムスカリンレセプターについての放射性リガンド結合アッセイ)
クローニングしたヒトムスカリンレセプターについての放射性リガンド結合アッセイを、総アッセイ容量100μLで、96ウェルマイクロタイタープレートにて実施した。hM1、hM2、hM3、hM4、またはhM5のいずれかのムスカリンサブタイプを安定的に発現するCHO細胞の細胞膜を、類似したシグナル(cpm)を得るために、次の特異的なターゲットタンパク質濃度(μg/ウェル)までアッセイ緩衝液で希釈した:hM1については10μg、hM2については10〜15μg、hM3については10〜20μg、hM4については10〜20μg、そしてhM5については10〜12μg。これらの細胞膜を、アッセイプレートへの添加前に、Polytron組織粉砕器を使用して(10秒)簡単に均質化した。放射性リガンドのKD値を決定するための飽和結合調査を、0.001nM〜20nMの範囲の濃度でL−[N−メチル−3H]スコポラミンメチルクロリド([3H]−NMS)(TRK666、84.0 Ci/mmol、Amersham Pharmacia Biotech,Buckinghamshire,England)を使用して実施した。テスト化合物のKi値の決定のための置換アッセイを、1nMの[3H]−NMS、および11の異なるテスト化合物濃度を用いて実施した。これらのテスト化合物を、初めに希釈緩衝液に溶解して400μMの濃度にし、次いで、連続的に希釈緩衝液で5×に希釈して、最終濃度を10pM〜100μMの範囲にした。アッセイプレートへの添加順序および容量は次のとおりである:25μLの放射性リガンド、25μLの希釈したテスト化合物、そして50μLの細胞膜。アッセイプレートを、37℃にて60分間、インキュベートした。結合反応を、1%BSA中で予め処理したGF/Bガラス繊維フィルタープレート(PerkinElmer Inc.,Wellesley,MA)を用いた急速濾過により終わらせた。フィルタープレートを、洗浄緩衝液(10mM HEPES)で3回すすぎ、結合していない放射能を除去した。次いで、これらのプレートを風乾させ、50μLのMicroscint−20液体シンチレーション液(PerkinElmer Inc.,Wellesley,MA)を各ウェルに加えた。次いでこれらのプレートを、PerkinElmer Topcount液体シンチレーションカウンター(PerkinElmer Inc.,Wellesley,MA)で計数した。結合データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、ワンサイト・コンペティションモデルを使用して、非線形回帰分析により分析した。テスト化合物についてのKi値を、観測されたIC50値、および放射性リガンドのKD値から、Cheng−Prusoff方程式(Cheng Y;Prusoff WH.(1973)Biochemical Pharmacology,22(23):3099−108)を使用して計算した。Ki値をpKi値に変換し、幾何平均および95%信頼区間を決定した。次いで、これらの要約統計を、データ報告のためにKi値に変換し直した。
【0157】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターに対するより高い結合親和力を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトM2およびM3ムスカリンレセプターに対し、10nM未満のKi値を有することが見出された。
【0158】
(アッセイテスト手順C)
(非相同的にヒトβ1、β2またはβ3アドレナリンレセプターを発現するCHO細胞株における細胞全体cAMPフラッシュアッセイ)
cAMPアッセイを、製造業者の説明に従い、[125I]−cAMPを用いたFlashplate Adenylyl Cyclase Activation Assay System(NEN SMP004,PerkinElmer Life Sciences Inc.,Boston,MA)を使用して、放射線免疫アッセイ形式で実施した。βレセプターアゴニストの効力(EC50)の決定のために、クローニングしたヒトβ1、β2またはβ3アドレナリンレセプターを安定的に発現するCHO−K1細胞株を、10%FBS、およびジェネテシン(250μg/mL)で補ったHams F−12培地において、ほぼコンフルエンシーまで培養した。細胞をPBSですすぎ、2mM EDTAまたはトリプシン−EDTA溶液(0.05%トリプシン/0.53mM EDTA)を含むdPBS(ダルベッコリン酸緩衝生理食塩水、CaCl2およびMgCl2を含まない)で分離した。Coulter細胞カウンターで細胞を計数後、細胞を1,000rpmにおける遠心分離によってペレットにし、予め室温に温めたIBMX(PerkinElmer Kit)を含む刺激緩衝液に再懸濁させ、1.6×106〜2.8×106細胞/mLの濃度にした。1ウェル当たり約60,000〜80,000細胞を、このアッセイで使用した。テスト化合物(DMSO中10mM)をBeckman Biomek−2000中の0.1%BSAを含むPBS中で希釈し、100μM〜1pMの範囲の11の異なる濃度にてテストした。反応物を37℃にて10分間インキュベートし、[125I]−cAMP(NEN SMP004、PerkinElmer Life Sciences,Boston,MA)を含む100μLの冷たい検出緩衝液を加えることにより止めた。生成されたcAMPの量(pmol/ウェル)を、サンプルについて観測された数、および製造業者の使用者マニュアルに記載されるcAMP標準に基づき計算した。データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド方程式(sigmoidal equation)を用いて、非線形回帰分析により分析した。Cheng−Prusoff方程式(Cheng Y,and Prusoff WH.,Biochemical Pharmacology,1973,22,23:3099−108)を使用して、EC50値を計算した。
【0159】
このアッセイにおいて、より低いEC50値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、ヒトβ2アドレナリンレセプターに対し、10nM未満のEC50値を有することが見出された。
【0160】
(アッセイテスト手順D)
(ムスカリンレセプターサブタイプについての拮抗作用の機能アッセイ)
(A.アゴニストが媒介する[35S]GTPγS結合の遮断)
テスト化合物の機能的効力を、hM2レセプターを発現するCHO−K1細胞に結合するオキソトレモリンが刺激する[35S]GTPγS結合を遮断する化合物の能力を計測することにより決定した。
使用のときに、冷凍細胞膜を解凍し、次いで、最終ターゲット組織濃度1ウェル当たり5〜10μgのタンパク質を用いてアッセイ緩衝液中で希釈した。この細胞膜を、Polytron PT−2100組織粉砕器を使用して簡単に均質化し、次いで、アッセイプレートに加えた。
【0161】
アゴニストオキソトレモリンによる[35S]GTPγS結合の刺激に対するEC90値(90%の最大応答に対する有効濃度)をそれぞれの実験において決定した。
【0162】
オキソトレモリンが刺激する[35S]GTPγS結合を阻害するテスト化合物の能力を決定するために、次の:[35S]GTPγS(0.4nM)を有する25μLのアッセイ緩衝液、25μLのオキソトレモリン(EC90)およびGDP(3μM)、25μLの希釈したテスト化合物、ならびにhM2レセプターを発現する25μLのCHO細胞の細胞膜を、96ウェルプレートの各ウェルに加えた。次いで、アッセイプレートを37℃にて60分間インキュベートした。これらのアッセイプレートを、PerkinElmer 96ウェルハーベスターを使用し、1%BSAで予め処理したGF/Bフィルターを用いて濾過した。これらのプレートを、3回×3秒間、氷冷の洗浄緩衝液を用いてすすぎ、次いで、風乾または真空乾燥させた。Microscint−20シンチレーション液(50μL)を、各ウェルに加え、各プレートを密封し、放射能をTopcounter(PerkinElmer)で算出した。データを、GraphPad Prism Software package(GraphPad Software,Inc.,San Diego,CA)を用い、非線形回帰分析であるワンサイト・コンペティションを使用して分析した。Cheng−Prusoff方程式を、Ki値を計算するために使用し、KDおよび[L]のリガンド濃度として、それぞれアッセイにおけるテスト化合物およびオキソトレモリン濃度についての濃度−応答曲線のIC50値を使用した。
【0163】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、hM2レセプターを発現するCHO−K1細胞におけるオキソトレモリンが刺激する[35S]GTPγS結合の遮断に対して、約10nM未満のKi値を有することが見出された。
【0164】
(B.FLIPRアッセイによるアゴニストが媒介するカルシウム放出の遮断)
Gqタンパク質に結合するムスカリンレセプターサブタイプ(M1、M3、およびM5レセプター)は、アゴニストがレセプターに結合すると、ホスホリパーゼC(PLC)経路を活性化する。その結果、活性化されたPLCはホスファチジル(phosphatyl)イノシトール二リン酸(PIP2)を、ジアシルグリセロール(DAG)とホスファチジル−1,4,5−三リン酸(IP3)とに加水分解し、次いで、細胞内貯蔵(すなわち、小胞体および筋小胞体)からカルシウム放出を生じさせる。FLIPR(Molecular Devices,Sunnyvale,CA)アッセイは、遊離カルシウムが結合するときに蛍光を発するカルシウム感受性色素(Fluo−4AM,Molecular Probes,Eugene,OR)を使用することにより、この細胞内カルシウムの増加を利用する(capitalize)。この蛍光イベントを、ヒトM1およびM3レセプター、ならびにチンパンジーM5レセプターを有するクローンされた細胞の単層から蛍光の変化を検出するFLIPRにより、実時間で計測する。アンタゴニストの効力を、アゴニストが媒介する細胞内カルシウムの増加を阻害するアンタゴニストの能力によって決定し得る。
【0165】
FLIPRカルシウム刺激アッセイに関し、hM1、hM3、およびcM5レセプターを安定的に発現するCHO細胞を、アッセイを実施する前夜に、96ウェルFLIPRプレートに播種した。播種した細胞をFLIPR緩衝液(10mM HEPES、pH7.4、2mM 塩化カルシウム、カルシウムおよびマグネシウムを含まないハンクス緩衝食塩水中(HBSS)の2.5mM プロベネシド)を用いて、Cellwash(MTX Labsystems,Inc.)により2回洗浄し、増殖培地、そしてFLIPR緩衝液50μL/ウェルを残した。次いでこれらの細胞を、二酸化炭素5%で37℃にて40分間、50μL/ウェルの4μM FLUO−4AM(2X溶液が生成された)を用いてインキュベートした。染色インキュベーションに続き、FLIPR緩衝液で2回洗浄し、最終用量50μL/ウェルを残した。
アンタゴニストの効力を決定するために、後でアンタゴニストの効力をEC90濃度におけるオキソトレモリン刺激に対して計測し得るように、まず、オキソトレモリンについての細胞内Ca2+放出の用量依存性刺激を決定した。細胞をまず20分間、化合物希釈緩衝液を用いてインキュベートし、続くアゴニストの添加はFLIPRによって実施された。オキソトレモリンについてのEC90値を、以下のFLIPR測定およびデータ整理の節にて、式ECF=((F/100−F)^1/H)*EC50とともに詳述される方法に従って得た。オキソトレモリンのEC90濃度がアンタゴニスト阻害アッセイプレート中の各ウェルに加えられるように、3×ECFのオキソトレモリン濃度を刺激プレート中に調製した。
【0166】
FLIPRに使用されるパラメーターは:0.4秒の曝露時間、0.5ワットのレーザー強度、488nmの励起波長、550nmの発光波長であった。アゴニストの添加前に、10秒間の蛍光の変化を測定することにより、基準線(baseline)を決定した。アゴニストの刺激に続き、FLIPRは、0.5秒から1秒毎の蛍光の変化を1.5分間、最大蛍光変化を得るために、継続的に計測した。
発光の変化を各ウェルについて、最大蛍光マイナス基準線蛍光として表した。生データを、GraphPad Prism(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド用量反応についてのビルトインモデルを使用して、非線形回帰分析により、薬物濃度の対数に対して分析した。アンタゴニストのK1値を、KDとしてオキソトレモリンEC50値、およびCheng−Prusoff方程式(Cheng&Prusoff,1973)に従うリガンド濃度についてのオキソトレモリンEC90を使用し、Prismにより決定した。
【0167】
このアッセイにおいて、より低いKi値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、hM1、hM3、およびcM5レセプターを安定的に発現するCHO細胞におけるアゴニストが媒介するカルシウム放出の遮断に対し、約10nM未満のKi値を有することが見出された。
【0168】
(アッセイテスト手順E)
(内因的にヒトβ2アドレナリンレセプターを発現する肺上皮細胞株を用いた細胞全体cAMPフラッシュアッセイ)
内因的濃度のβ2アドレナリンレセプターを発現している細胞株におけるアゴニストの効力および効能(内因性の活性)の決定に関し、ヒト肺上皮細胞株(BEAS−2B)を使用した(ATCC CRL−9609、American Type Culture Collection,Manassas,VA)(January B,et al.,British Journal of Pharmacology,1998,123,4,701−11)。細胞を、完全な無血清培地(エピネフリンおよびレチノイン酸を含むLHC−9 MEDIUM、カタログ番号181−500、Biosource International,Camarillo,CA)において、75〜90%のコンフルエンシーまで培養した。アッセイ前日に、培地をLHC−8(エピネフリンおよびレチノイン酸を含まない、カタログ番号141−500、Biosource International,Camarillo,CA)に変更した。
cAMPアッセイを、製造業者の説明に従い、[125I]−cAMPを用いたFlashplate Adenylyl Cyclase Activation Assay System(NEN SMP004,PerkinElmer Life Sciences Inc.,Boston,MA)を使用して、放射線免疫アッセイ形式で実施した。
【0169】
アッセイの当日、細胞をPBSですすぎ、PBS中の5mM EDTAで擦る(scrape)ことによって浮かせ、計数した。細胞を1,000rpmにて遠心分離によりペレットにし、37℃に予め温めておいた刺激緩衝液に、600,000細胞/mLの最終濃度で再懸濁させた。このアッセイでは、100,000〜120,000細胞/ウェルの最終濃度で、細胞を使用した。テスト化合物を、Beckman Biomek−2000で、アッセイ緩衝液(75mM Tris/HCl 25℃にてpH7.4、12.5mM MgCl2、1mM EDTA、0.2%BSA)で、連続的に希釈した。このアッセイでは、10μM〜10pMの範囲における11の異なる濃度で、テスト化合物をテストした。反応物を37℃にて10分間、インキュベートし、100μLの氷冷の検出緩衝液の添加により止めた。プレートを密閉し、4℃にて一晩中インキュベートし、翌朝、Topcountシンチレーションカウンター(Packard BioScience Co.,Meriden,CT)で計数した。反応物1mL当たりに生成されたcAMP量を、製造業者の使用者マニュアルに記載のとおり、サンプルについて観測された数、およびcAMP標準に基づき算定した。データを、GraphPad Prism Softwareパッケージ(GraphPad Software,Inc.,San Diego,CA)を用い、シグモイド用量反応に関する4−パラメーターモデルを使用して、非線形回帰分析により分析した。
【0170】
このアッセイにおいて、より低いEC50値は、テスト化合物が、テストされたレセプターにおいてより高い機能活性を有することを示す。このアッセイにおいてテストされる場合、式Iの化合物は、β2アドレナリンレセプターに対し、約10nM未満のEC50値を有することが見出された。
【0171】
(アッセイテスト手順F)
(アセチルコリン誘発またはヒスタミン誘発気管支収縮のモルモットモデルにおける気管支保護(bronchoprotection)の持続時間)
これらのインビボアッセイを使用して、ムスカリンレセプターアンタゴニストおよびβ2アドレナリンレセプターアゴニスト活性の両方を示すテスト化合物の気管支保護効果を評価した。アセチルコリン誘発気管支収縮モデルにおけるムスカリンアンタゴニスト活性を分離するために、これらの動物にプロパノロール(βレセプター活性を遮断する化合物)を、アセチルコリンの投与の前に投与した。ヒスタミン誘発気管支収縮モデルにおける気管支保護の持続時間は、β2アドレナリンレセプターアゴニスト活性を反映する。
【0172】
250gと350gとの間の重さの6匹の雄のモルモット(Duncan−Hartley(HsdPoc:DH)Harlan,Madison,WI)群を、それぞれケージカードにより同定した。調査の間中、動物に自由に食料および水を入手させた。
【0173】
テスト化合物を、全身曝露投薬チャンバ(whole−body exposure dosing chamber)(R&S Molds,San Carlos,CA)で10分間にわたり、吸引により投与した。この投薬チャンバを、エアゾールが中央の多岐管から6つの個々のチャンバに同時に送達されるように設計した。モルモットをテスト化合物またはビヒクル(WFI)のエアゾールに曝露させた。これらのエアゾールを、22psiの圧力にて気体の混合物(CO2=5%、O2=21%、およびN2=74%)により作動するLC Star Nebulizer Set(Model 22F51,PARI Respiratory Equipment,Inc.Midlothian,VA)を使用し、水溶液から生じさせた。この作動圧においてネブライザーを通した気体流は、約3L/分であった。この生じたエアゾールを、陽圧によりチャンバ内に押し流した。エアゾール化した溶液の送達の間、希釈用空気は使用されなかった。10分の噴霧療法の間、約1.8mLの溶液を噴霧した。この値を、充填したネブライザーの噴霧前および噴霧後の重量を比較することにより、重量分析的に測定した。
【0174】
吸引により投与されるテスト化合物の気管支保護効果を、投与後1.5時間、24時間、48時間、および72時間に、全身プレチスモグラフ法を使用して評価した。
肺評価の開始45分前に、各モルモットをケタミン(43.75mg/kg)、キシラジン(3.50mg/kg)、およびアセプロマジン(1.05mg/kg)の筋肉内注射で麻酔した。手術部位を剃り、70%アルコールで清潔にした後、頸部腹側面の2〜3cmの正中切開を実施した。次いで、頚静脈を分離し、生理食塩水中のアセチルコリン(Ach)またはヒスタミンの静脈内注入を考慮して、生理食塩水を充填したポリエチレンカテーテル(PE−50,Becton Dickinson,Sparks,MD)と共にカニューレを挿入した。次いで、この気管を切開解放(dissected free)し、14Gテフロン(登録商標)チューブ(#NE−014,Small Parts,Miami Lakes,FL)と共にカニューレを挿入した。必要な場合は、前記した麻酔混合物のさらなる筋肉内注射によって知覚麻痺を維持した。知覚麻痺の程度をモニターし、動物が足をつまんで(pinch)反応する場合、または呼吸数が100呼吸/分を超えた場合に調節した。
【0175】
カニューレの挿入が完了すると、動物をプレチスモグラフ(#PLY3114,Buxco Electronics,Inc.,Sharon,CT)に置き、食道圧力カニューレ(PE−160,Becton Dickinson,Sparks,MD)を、肺の駆動圧(プレッシャー)を測定するために挿入した。テフロン(登録商標)導管チューブをプレチスモグラフの開口部に取り付け、モルモットにチャンバの外からの大気を吸わせた。次いで、チャンバを密閉した。体温を維持するために加熱灯を使用し、モルモットの肺を、10mL較正シリンジ(calibration syringe)(#5520 Series,Hans Rudolph,Kansas City,MO)を使用して、4mLの空気で3回膨張させ、気道下部が無空気状態にならないことを、そして動物が過呼吸に陥らないことを確実にした。
基準線の値がコンプライアンスについて0.3〜0.9mL/cm H2Oの範囲内であると決定し、抵抗について0.1〜0.199cm H2O/mL毎秒の範囲内であると決定すると、肺評価を開始した。Buxco肺測定コンピュータープログラムにより、肺の値の収集および導出を実施した。
【0176】
このプログラムをスタートさせると、実験プロトコルおよびデータ収集を開始した。各呼吸に関するプレチスモグラフ中で起こる、容量の経時変化を、Buxco圧力変換器により測定した。この経時シグナルを取り入れることにより、各呼吸についてのフローの測定値を算定した。このシグナルを、Sensym圧力変換器(#TRD4100)を使用して回収された肺の駆動圧力の変化と共に、Buxco(MAX 2270)前置増幅器によりデータ収集インターフェース(#SFT3400および#SFT3813)と結びつけた。全ての他の肺のパラメーターをこれら2つのインプットから引き出した。
【0177】
基準線値を5分間で収集し、その後、モルモットをAchまたはヒスタミンでチャレンジした。ムスカリンアンタゴニストの効果を評価する場合、プロパノロール(5mg/Kg、iv)(Sigma−Aldrich,St.Louis,MO)を、Achでチャレンジする15分前に投与した。Ach(Sigma−Aldrich,St.Louis,MO)(0.1mg/mL)を、シリンジポンプ(sp210iw、World Precision Instruments,Inc.,Sarasota,FL)から1分間、次の用量および指定された実験の開始からの時間で静脈内に注入した:5分の時点で1.9μg/分、10分の時点で3.8μg/分、15分の時点で7.5μg/分、20分の時点で15.0μg、25分の時点で30μg/分、および30分の時点で60μg/分。あるいは、テスト化合物の気管支保護を、β遮断化合物での前処理なしのアセチルコリンチャレンジモデルで評価した。
【0178】
テスト化合物のβ2アドレナリンレセプターアゴニスト効果を評価する場合、ヒスタミン(25μg/mL)(Sigma−Aldrich,St.Louis,MO)を、シリンジポンプから1分間、次の用量および指定された実験の開始からの時間で静脈内に注入した:5分の時点で0.5μg/分、10分の時点で0.9μg/分、15分の時点で1.9μg/分、20分の時点で3.8μg、25分の時点で7.5μg/分、および30分の時点で15μg/分。抵抗またはコンプライアンスが、Achまたはヒスタミンのそれぞれの投薬に続く3分の時点で基準線値に戻らない場合は、モルモットの肺を、10mL較正シリングから4mLの空気で3回膨張させた。記録された肺のパラメーターには、呼吸回数(呼吸/分)、コンプライアンス(mL/cm H2O)、および肺抵抗(cm H2O/mL毎秒)が含まれる。このプロトコルの35分の時点で肺機能測定が完了すると、モルモットをプレチスモグラフから取り出し、二酸化炭素の窒息により安楽死させた。
【0179】
これらのデータを2つの方法のうちの一方で評価した:
(a)肺抵抗(RL、cm H2O/mL毎秒)を、「圧力の変化」対「流量の変化」の比から計算した。AChへのRL応答(60μg/分、IH)をビヒクルおよびテスト化合物群について、コンピュータで算定した。ビヒクル−処置動物において、各前処理時点の平均ACh応答を計算し、各テスト化合物用量にて対応する前処理時点のACh応答の阻害%をコンピュータで算定するために使用した。「RL」についての阻害用量−応答曲線を、気管支保護ID50(ACh(60μg/分)気管支収縮物質(bronchocontrictor)応答を50%阻害するのに必要とされる用量)を推定するために、Windows(登録商標)用GraphPad Prism,version 3.00(GraphPad Software,San Diego,California)を使用して、4パラメーター論理方程式を用いて調節した。使用された方程式は次のとおりである:
Y=Min+(Max−Min)/(1+10((logID50−X)*Hillslope))
ここでXは用量の対数であり、Yは応答(Achが誘発するRL増加の阻害%)である。YはMinで始まり、漸近的にMaxへとS字状(sigmoidal shape)を描く。
【0180】
(b)基準線の肺抵抗を倍にするために必要とされるAchまたはヒスタミンの量、として規定される量PD2は、次の方程式(クリニックでPC20値を計算するために使用される方程式から導かれる)を使用して、Achまたはヒスタミンのチャレンジの範囲にわたるフローおよびプレッシャーから導かれる肺の抵抗値を使用して算定される(Am.Thoracic Soc,2000を参照)。
【0181】
【数1】
ここで:
C1=C2に先行するAchまたはヒスタミンの濃度
C2=肺抵抗(RL)の少なくとも2倍の増加をもたらすAchまたはヒスタミンの濃度
R0=基準線RL値
R1=C1に従うRL値
R2=C2に従うRL値
データの統計分析を、両側スチューデントt検定を使用して実施した。P値<0.05を有意とみなした。
【0182】
このアッセイにおいてテストされる場合、式Iの化合物は、MCh誘発気管支収縮およびHis誘発気管支収縮に対する、用量依存性気管支保護効果をもたらした。さらに、式Iの化合物は、このアッセイにおいて少なくとも約24時間の気管支保護活性の持続時間(PD T1/2)を有した。
【0183】
本発明は、その特異的な局面または実施形態を参照して記載されているが、本発明の精神および範囲から逸脱することなしに、種々の変更がなされ得、等価物を代わりに使用し得ることが、当業者には理解される。さらに、適用し得る特許法および規制により認可される範囲まで、本明細書中で引用するすべての刊行物、特許および特許出願を、各書類が個々に本明細書中に参考として援用された場合と同程度に、それらの全体が参考として本明細書中で援用される。
【図面の簡単な説明】
【0184】
本発明の種々の局面が、添付の図の参照により説明される。
【図1】図1は、示差走査熱量測定(DSC)の記録および熱重量分析(TGA)の記録を示している。
【図2】図2は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルについてのDSCの記録を示す。
【図3】図3は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの粉末エックス線回析(PXRD)パターンを示している。
【図4】図4は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩のサンプルの粉末エックス線回析(PXRD)パターンを示している。
【図5】図5は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩についての赤外線(IR)吸収スペクトルを示している。
【図6】図6は、本発明のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩についての力学的水分吸着(DMS)の記録を示している。
【特許請求の範囲】
【請求項1】
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物。
【請求項2】
請求項1に記載の化合物であって、約215℃〜約240℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項3】
請求項1に記載の化合物であって、約215℃〜約229℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項4】
請求項1に記載の化合物であって、約230℃〜約240℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項5】
請求項1に記載の化合物であって、200℃を超える温度範囲において図1に示される記録に実質的に従う示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項6】
請求項1に記載の化合物であって、200℃を超える温度範囲において図2に示される記録に実質的に従う示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項7】
請求項1に記載の化合物であって、5.0±0.3および15.0±0.3の2θの値における回析ピークを有する粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項8】
請求項1に記載の化合物であって、そのピーク位置が、図3に示されるパターンのピーク位置に実質的に従う粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項9】
請求項1に記載の化合物であって、そのピーク位置が、図4に示されるパターンのピーク位置に実質的に従う粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項10】
請求項1に記載の化合物であって、約704cm−1、約748cm−1、約768cm−1、約841cm−1、約900cm−1、約1055cm−1、約1104cm−1、約1166cm−1、約1218cm−1、約1294cm−1、約1408cm−1、約1522cm−1、約1609cm−1、約1655cm−1、および約1701cm−1において有意な吸収帯を有する赤外線吸収スペクトルを有する、化合物。
【請求項11】
請求項1に記載の化合物であって、図5に示されるスペクトルに実質的に従う赤外線吸収スペクトルによって特徴付けられる、化合物。
【請求項12】
請求項1に記載の化合物であって、微粉形態である、化合物。
【請求項13】
薬学的に受容可能なキャリアおよび請求項1〜12のいずれか1項に記載の化合物を含む、薬学的組成物。
【請求項14】
請求項13に記載の薬学的組成物であって、ステロイド抗炎症因子をさらに含む、薬学的組成物。
【請求項15】
請求項14に記載の薬学的組成物であって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、薬学的組成物。
【請求項16】
請求項13に記載の薬学的組成物であって、ホスホジエステラーゼ−4インヒビターをさらに含む、薬学的組成物。
【請求項17】
請求項13に記載の薬学的組成物であって、吸入による投与のために処方される、薬学的組成物。
【請求項18】
請求項13に記載の薬学的組成物であって、微粉形態である、薬学的組成物。
【請求項19】
請求項17に記載の薬学的組成物であって、前記キャリアが、ラクトース、デンプン、マンニトール、ブドウ糖、ポリ乳酸、ポリラクチド−コ−グリコリド、またはそれらの組み合わせである、薬学的組成物。
【請求項20】
組み合わせであって、
(a)請求項1〜12のうちいずれか1項に記載の化合物;および
(b)ステロイド抗炎症因子
を含む、組み合わせ。
【請求項21】
請求項20に記載の組み合わせであって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、組み合わせ。
【請求項22】
肺障害を処置するための方法であって、処置を必要としている患者に、治療有効量の請求項1〜12のうちいずれか1項に記載の化合物を投与する工程を包含する、方法。
【請求項23】
患者において気管支拡張を生じさせる方法であって、該患者に、気管支拡張を生じさせる量の請求項1〜12のうちいずれか1項に記載の化合物を吸引により投与する工程を包含する、方法。
【請求項24】
慢性閉塞性肺疾患または喘息を処置する方法であって、処置を必要としている患者に、治療有効量の請求項1〜12のうちいずれか1項に記載の化合物を投与する工程を包含する、方法。
【請求項25】
請求項1に記載の化合物を調製するプロセスであって、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルを、1,2−エタンジスルホン酸またはその水和物と接触させる工程を包含する、プロセス。
【請求項26】
請求項1に記載の化合物を調製するためのプロセスであって:
(a)式II:
【化1】
の化合物を、フッ化物イオンと接触させる工程であって、ここで、R1a、R1b、およびR1cが、C1〜4アルキル、フェニル、−C1〜4アルキル−(フェニル)から独立して選択されるか、またはR1a、R1b、およびR1cのうちの1つが−O−(C1〜4アルキル)である、工程;そして
(b)工程(a)からの生成物を、1,2−エタンジスルホン酸塩、またはその溶媒和物と接触させてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩またはその溶媒和物を形成させる工程
を包含し、ここで、工程(a)および(b)が、同じ反応容器中で、該工程(a)の生成物を分離せずに実施される、プロセス。
【請求項27】
約230℃よりも高い融点を有する請求項1に記載の化合物を調製するためのプロセスであって、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の種結晶を、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、または不活性な希釈剤に溶解させたその溶媒和物を含む溶液に加える工程を包含し、ここで該種結晶は、約230℃より高い融点を有する、プロセス。
【請求項28】
約230℃より高い融点を有する請求項1に記載の化合物を調製するための工程であって:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩を、第一の温度にて、不活性な希釈剤に溶解させる工程;
(b)工程(a)の生成物を第二の温度に冷却する工程;そして
(c)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩の種結晶を加える工程、
を包含し、ここで種結晶が、約230℃より高い融点を有し、第一の温度が、1,2−エタンジスルホン酸塩を溶解させるのに十分な温度であり、そして、第二の温度が、種結晶が工程(b)の生成物に加えられる場合に完全に溶解する温度より低い、
プロセス。
【請求項29】
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルを精製するためのプロセスであって、請求項1に記載の化合物を形成させる工程を包含する、プロセス。
【請求項30】
治療における使用のため、または薬としての、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物。
【請求項31】
薬の製造のための、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の、使用。
【請求項32】
請求項31に記載の使用であって、前記薬が、肺障害を処置するための薬である、使用。
【請求項33】
請求項32に記載の使用であって、前記肺障害が、慢性閉塞性肺疾患または喘息である、使用。
【請求項34】
結晶性のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む、薬。
【請求項35】
肺障害の処置用薬の製造における:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子;
の、使用。
【請求項36】
請求項35に記載の使用であって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、使用。
【請求項1】
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)−エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物。
【請求項2】
請求項1に記載の化合物であって、約215℃〜約240℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項3】
請求項1に記載の化合物であって、約215℃〜約229℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項4】
請求項1に記載の化合物であって、約230℃〜約240℃の範囲において最大吸熱熱流を示す示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項5】
請求項1に記載の化合物であって、200℃を超える温度範囲において図1に示される記録に実質的に従う示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項6】
請求項1に記載の化合物であって、200℃を超える温度範囲において図2に示される記録に実質的に従う示差走査熱量測定の記録によって特徴付けられる、化合物。
【請求項7】
請求項1に記載の化合物であって、5.0±0.3および15.0±0.3の2θの値における回析ピークを有する粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項8】
請求項1に記載の化合物であって、そのピーク位置が、図3に示されるパターンのピーク位置に実質的に従う粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項9】
請求項1に記載の化合物であって、そのピーク位置が、図4に示されるパターンのピーク位置に実質的に従う粉末エックス線回析パターンによって特徴付けられる、化合物。
【請求項10】
請求項1に記載の化合物であって、約704cm−1、約748cm−1、約768cm−1、約841cm−1、約900cm−1、約1055cm−1、約1104cm−1、約1166cm−1、約1218cm−1、約1294cm−1、約1408cm−1、約1522cm−1、約1609cm−1、約1655cm−1、および約1701cm−1において有意な吸収帯を有する赤外線吸収スペクトルを有する、化合物。
【請求項11】
請求項1に記載の化合物であって、図5に示されるスペクトルに実質的に従う赤外線吸収スペクトルによって特徴付けられる、化合物。
【請求項12】
請求項1に記載の化合物であって、微粉形態である、化合物。
【請求項13】
薬学的に受容可能なキャリアおよび請求項1〜12のいずれか1項に記載の化合物を含む、薬学的組成物。
【請求項14】
請求項13に記載の薬学的組成物であって、ステロイド抗炎症因子をさらに含む、薬学的組成物。
【請求項15】
請求項14に記載の薬学的組成物であって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、薬学的組成物。
【請求項16】
請求項13に記載の薬学的組成物であって、ホスホジエステラーゼ−4インヒビターをさらに含む、薬学的組成物。
【請求項17】
請求項13に記載の薬学的組成物であって、吸入による投与のために処方される、薬学的組成物。
【請求項18】
請求項13に記載の薬学的組成物であって、微粉形態である、薬学的組成物。
【請求項19】
請求項17に記載の薬学的組成物であって、前記キャリアが、ラクトース、デンプン、マンニトール、ブドウ糖、ポリ乳酸、ポリラクチド−コ−グリコリド、またはそれらの組み合わせである、薬学的組成物。
【請求項20】
組み合わせであって、
(a)請求項1〜12のうちいずれか1項に記載の化合物;および
(b)ステロイド抗炎症因子
を含む、組み合わせ。
【請求項21】
請求項20に記載の組み合わせであって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、組み合わせ。
【請求項22】
肺障害を処置するための方法であって、処置を必要としている患者に、治療有効量の請求項1〜12のうちいずれか1項に記載の化合物を投与する工程を包含する、方法。
【請求項23】
患者において気管支拡張を生じさせる方法であって、該患者に、気管支拡張を生じさせる量の請求項1〜12のうちいずれか1項に記載の化合物を吸引により投与する工程を包含する、方法。
【請求項24】
慢性閉塞性肺疾患または喘息を処置する方法であって、処置を必要としている患者に、治療有効量の請求項1〜12のうちいずれか1項に記載の化合物を投与する工程を包含する、方法。
【請求項25】
請求項1に記載の化合物を調製するプロセスであって、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルを、1,2−エタンジスルホン酸またはその水和物と接触させる工程を包含する、プロセス。
【請求項26】
請求項1に記載の化合物を調製するためのプロセスであって:
(a)式II:
【化1】
の化合物を、フッ化物イオンと接触させる工程であって、ここで、R1a、R1b、およびR1cが、C1〜4アルキル、フェニル、−C1〜4アルキル−(フェニル)から独立して選択されるか、またはR1a、R1b、およびR1cのうちの1つが−O−(C1〜4アルキル)である、工程;そして
(b)工程(a)からの生成物を、1,2−エタンジスルホン酸塩、またはその溶媒和物と接触させてビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩またはその溶媒和物を形成させる工程
を包含し、ここで、工程(a)および(b)が、同じ反応容器中で、該工程(a)の生成物を分離せずに実施される、プロセス。
【請求項27】
約230℃よりも高い融点を有する請求項1に記載の化合物を調製するためのプロセスであって、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の種結晶を、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、または不活性な希釈剤に溶解させたその溶媒和物を含む溶液に加える工程を包含し、ここで該種結晶は、約230℃より高い融点を有する、プロセス。
【請求項28】
約230℃より高い融点を有する請求項1に記載の化合物を調製するための工程であって:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩を、第一の温度にて、不活性な希釈剤に溶解させる工程;
(b)工程(a)の生成物を第二の温度に冷却する工程;そして
(c)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩の種結晶を加える工程、
を包含し、ここで種結晶が、約230℃より高い融点を有し、第一の温度が、1,2−エタンジスルホン酸塩を溶解させるのに十分な温度であり、そして、第二の温度が、種結晶が工程(b)の生成物に加えられる場合に完全に溶解する温度より低い、
プロセス。
【請求項29】
ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルを精製するためのプロセスであって、請求項1に記載の化合物を形成させる工程を包含する、プロセス。
【請求項30】
治療における使用のため、または薬としての、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物。
【請求項31】
薬の製造のための、ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]−ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物の、使用。
【請求項32】
請求項31に記載の使用であって、前記薬が、肺障害を処置するための薬である、使用。
【請求項33】
請求項32に記載の使用であって、前記肺障害が、慢性閉塞性肺疾患または喘息である、使用。
【請求項34】
結晶性のビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの1,2−エタンジスルホン酸塩、またはその溶媒和物を含む、薬。
【請求項35】
肺障害の処置用薬の製造における:
(a)ビフェニル−2−イルカルバミン酸1−[2−(2−クロロ−4−{[(R)−2−ヒドロキシ−2−(8−ヒドロキシ−2−オキソ−1,2−ジヒドロキノリン−5−イル)エチルアミノ]メチル}−5−メトキシフェニルカルバモイル)エチル]ピペリジン−4−イルエステルの結晶性1,2−エタンジスルホン酸塩、またはその溶媒和物;および
(b)ステロイド抗炎症因子;
の、使用。
【請求項36】
請求項35に記載の使用であって、前記ステロイド抗炎症因子が、6α,9α−ジフルオロ−17α−[(2−フラニルカルボニル)オキシ]−11β−ヒドロキシ−16α−メチル−3−オキソアンドロスタ−1,4−ジエン−17β−カルボチオ酸S−フルオロメチルエステル、またはその溶媒和物である、使用。
【図1】

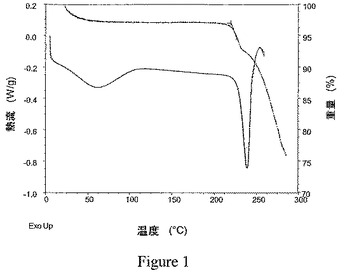
【図2】
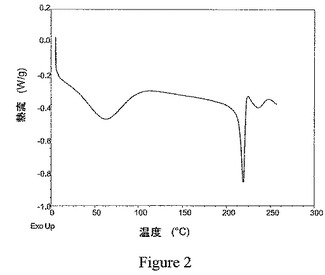
【図3】
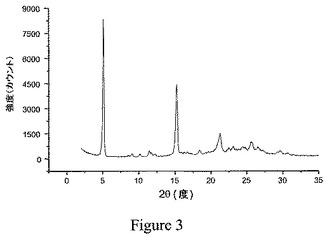
【図4】
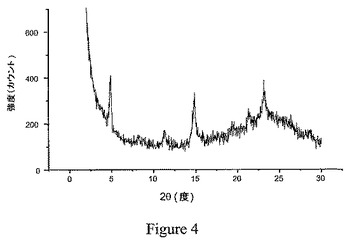
【図5】
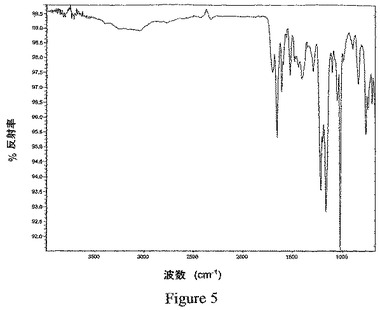
【図6】
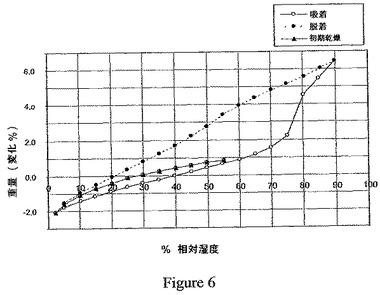
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2008−513358(P2008−513358A)
【公表日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願番号】特願2007−527919(P2007−527919)
【出願日】平成17年8月15日(2005.8.15)
【国際出願番号】PCT/US2005/029013
【国際公開番号】WO2006/023454
【国際公開日】平成18年3月2日(2006.3.2)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
【公表日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願日】平成17年8月15日(2005.8.15)
【国際出願番号】PCT/US2005/029013
【国際公開番号】WO2006/023454
【国際公開日】平成18年3月2日(2006.3.2)
【出願人】(500154711)セラヴァンス, インコーポレーテッド (129)
【Fターム(参考)】
[ Back to top ]