
結晶性糖組成物および作製方法
新規な結晶性ピバロイルフラノースおよびピバロイルフラノースを結晶化する方法が記載される。これらの化合物は、デオキシノジリマイシンおよびノジリマイシンなどの化合物の合成で中間体として有用であり、数kg規模での製造のために中間体として特に有用である。詳細な結晶性化合物としては、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース、1,2,3,6−テトラピバロイル−α−L−アルトロフラノース、および5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースが挙げられる。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本出願は2005年6月8日に出願された米国仮特許出願第60/689,119号からの優先権を主張し、その開示は参照としてその全体が本明細書に組み入れられている。
【0002】
[発明の分野]
本発明は、結晶性ピバロイルフラノースおよびビバロイルフラノースの結晶化方法に関するものである。これらの化合物は、D−1−デオキシガラクトノジリマイシン(DGJ)などの糖の合成での中間体として有用である。
【0003】
[発明の背景]
DGJはまた、(2R,3S,4R,5S)−2−ヒドロキシメチル−3,4,5−トリヒドロキシピペリジン、1−デオキシ−ガラクトスタチンとしても、そしてD−1−デオキシガラクトノジリマイシンとしても記載される。DGJは、D−ガラクトースのイミノ糖(5−アミノ−5−デオキシ−D−グルコピラノース)類似体であり、α−およびβ−D−ガラクトシダーゼの両方の強力な阻害物質である。ガラクトシダーゼは、グリコシド結合の加水分解を触媒し、複合糖質の代謝において重要である。DGJなどのガラクトシダーゼ阻害物質は、糖尿病(例えば米国特許第4,634,765号)、癌(例えば米国特許第5,250,545号)、ヘルペス(例えば米国特許第4,957,926号)、Hivおよびファブリ病(Fanら,Nat.Med.1999 5:1,112−5)を含む、多くの疾患および状態の治療に使用されうる。
【0004】
デオキシノジリマイシンなどのノジリマイシン誘導体の発表された化学合成は一般に、市販用途には適切でない複数のステップを有する。中間体の多くは安定でなく、中間体および最終生成物の両方の精製は数キログラム規模で扱いにくい。Grabnerによって特許取得された化学微生物学的方法(米国特許第5,695,969号;米国特許第5,610,039号)は、グルコースの細菌による酸化で得られた5−ケトアルドースの還元的アミノ化によって、糖をそのイミノ誘導体に変換する方法を提供する。しかしながら該方法は、D−ガラクトノジリマイシン誘導体には適用できない。他の関連特許(米国特許第5,227,479号、米国特許第4,908,439号および米国特許第4,634,765号)は、保護グリコシルハライドを使用するホモノジリマイシンの製剤、D−グルクロノラクトンの水素化物還元について議論している。米国特許第4,908,439号は、アジドに水素化アルミニウムリチウムなどのヒドリド還元剤を反応させることによって、グルコースジリマイシン誘導体である5−アミノ−5−デオキシ−1,2−O−イソプロピリデン−D−グルコンウロラクトン(DNJ誘導体)を調製するプロセスを教示する。
【0005】
米国特許第6,740,780号、同第6,683,185号、同第6,653,482号、同第6,653,480号、同第6,649,766号、同第6,605,724号、同第6,590,121号、および同第6,462,197号は、D−ジデオキシガラクトノジリマイシンの製剤において中間体として有用であるイミノ糖の製剤のプロセスについて記載している。これらの化合物は、ヘキソース糖の1,5−ジデオキシ−1,5−イミノヘキシトールであり、ヒドロキシル保護オキシム中間体から調製される。これらのイミノ糖を作製するプロセスは、ヘキシトールに還元されるラクタムの生成を含む。しかしながら本プロセスは、安全性、アップスケール、取り扱いおよび合成の複雑さに関する、数キログラム規模での産生に関してある欠点を有する。例えば該プロセスは、大規模では実際的でない手順である、フラッシュクロマトグラフィーを精製に使用する。
【0006】
文献で発表されたD−1−デオキシガラクトノジリマイシン(DGJ)の複数の製剤があり、製剤の大半は、予備規模手順(>100g)での産業研究所における反復には適切でない。これらの合成のいくつかとしては、D−グルコース(Legler G,ら,Carbohydr Res.1986 Nov 1;155:119−29);D−ガラクトース(Uriel,C,Santoyo−Gonzalez,F.,ら,Synlett 1999 593−595;Synthesis 1998 1787−1792(ピバロイル化中間体を開示));ガラクトピラノース(Bernotas RC,ら,Carbohydr Res.1987 Sep 15;167:305−ll);L−酒石酸(Aoyagiら,J.Org.Chem.1991,56,815);ケブラコイトール(Chidaら,J.Chem.Soc,Chem Commun.1994,1247);ガラクトフラノース(Paulsenら,Chem.Ber.1980,113,2601);ベンゼン(Johnsonら,Tetrahedron Lett.1995,36,653);アラビノ−ヘキソース−5−ウロース(Bariliら,tetrahedron 1997,3407);5−アジド−1,4−ラクトン(Shilvockら,Synlett,1998,554);ドキシノジリマイシン(Takahashiら,J.Carbohydr.Chem.1998,17,117);アセチルグルコサミン(Heightmanら,Helv.Chim.Acta 1995,78,514);ミオ−イノシトール(Chida N,ら,Carbohydr Res.1992 Dec 31;237:185−94);ジオキサニルピペリデン(Takahataら,Org.Lett.2003;5(14);2527−2529);および(E)−2,4−ペンタジエノール(Martin R,ら,Org Lett.2000 Jan;2(1):93−5)(Hughes AB,ら,Nat Prod Rep.1994 Apr;11(2):135−62)からの合成が挙げられる。N,N−メチル−1−デオキシノジリマイシン含有オリゴサッカライドの合成は、Kisoによって記載されている(Bioorg Med Chem.1994 Nov;2(11):1295−308)。Kisoは、保護1−デオキシノジリマイシン誘導体に、D−ガラクトースのメチル−1−チオグリコシド(グリコシル供与体)をグリコシルプロモータとして使用されたトリフラートと共に結合させた。
【0007】
精製のためのカラムクロマトグラフィーの使用は、上で開示された参考文献によって教示された反応で産生されるような小規模合成には可能であるが、数kg規模での使用には不十分である。必要なカラムのサイズはもちろんのこと、要求される溶媒の量もこの手順を非現実的にしている。文献で報告されているDGJ製剤の最大規模は13.3gであり(Fred−Robert Heiker,Alfred Matthias Schueller,Carbohydrate Research,1989,203 314−318)、13.3gは治療薬としての使用のためのプラント規模合成で要求されるよりもはるかに少ない。Heikerらは、イオン交換樹脂Lewatit MP 400(OH−)およびエタノールからの結晶化を使用してDGJを精製した。しかしながらこのプロセスも数キログラム量には容易に拡大できない。
【0008】
したがって、クロマトグラフィーまたはイオン交換樹脂を利用しない合成が好ましい。化学製造において化合物を単離する最も容易な方法は、結晶化である。結晶化は、他の方法よりも一般に高速、安全、経費節約的であり、スケールアップが容易である。しかしながら炭水化物は通常、油の形であり、油は結晶化させるのが困難である。いくつかの例外がある。例えば米国特許第6,620,921号は、あるグルコフラノシドの製剤に有用な化合物である、結晶性1,2,3,5,6−ペンタ−O−プロパノイル−β−D−グルコフラノースを開示している。多くのグルコフラノース誘導体は常温常圧にて油であるが、‘921特許は一部のフラノースがこれらの条件下で結晶性であることを開示している。これらのフラノースとしては:フェニルβ−D−グルコフラノシド、4−ニトロフェニルα−D−グルコフラノシド、メチル2,3,5,6−テトラ−O−プロパノイル−1−チオ−β−D−グルコフラノシド、および1−β D−グルコフラノシルウラシルが挙げられる。
【0009】
しかしながら、他の結晶性中間体に対する、そしてDGJなどのデオキシジリマイシンの合成に有用であり、大規模合成にとって実際的である、結晶化による中間体の容易で拡張可能な精製工程(合成において中間体を精製することを含む)に対する要求がなお存在する。
【0010】
[発明の概要]
フラノースの結晶形およびこれらのフラノースを結晶化する方法が開示される。結晶性フラノースは、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基を有する。
【0011】
フラノースの分子量は300g/mol〜1000g/molである。好ましくは、分子量は少なくとも350g/mol、少なくとも400g/mol、またはさらに好ましくは少なくとも450g/molである。別の実施態様において、分子量は900g/mol未満または800g/mol未満である。
【0012】
別の実施形態において、少なくとも3個のトリメチルアセチル保護基がある。フラノースは、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース、1,2,3,6−テトラピバロイル−α−L−アルトロフラノース、または5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースなどのテトラピバロイルフラノースでありうる。
【0013】
フラノースを溶媒に添加する、またはフラノースを溶媒中で生成するステップと;溶媒から該フラノースを結晶化させるステップとを備える、結晶性フラノースを産生する方法も提供される。結晶化は好ましくは、第2の溶媒を添加する工程と、周囲圧にて冷却する工程とによって実施される。
【0014】
結晶性テトラピバロイルフラノースを含み、該テトラピバロイルフラノースに加えてモノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースの少なくとも1つが生成される本発明の一態様において;本モノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースは、テトラピバロイルフラノースが結晶化されるときには結晶化されない。同様に、トリピバロイル(あるいは例えばペンタピバロイルまたは他の保護糖)が目的の生成物である場合、そして追加の望ましくない保護糖が反応で生成される場合に、該トリピバロイル(あるいはペンタピバロイルまたは他の保護糖)は溶媒から結晶化され、該望ましくない保護糖は結晶化されない。
【0015】
好ましい溶媒は、ヘプタンおよびメタノールである。なお本発明の別の態様において、結晶化させるステップは、フラノースおよび溶媒を溶媒の沸点付近の温度まで加熱する工程と、0℃以下、またはさらに好ましくは−20℃〜−10℃の温度まで冷却する工程と、フラノースが沈殿するまで待機する工程とを含む;一態様において、この時間は少なくとも36時間である。
【0016】
なお別の実施形態において、結晶性フラノースを産生する方法は、フラノースおよび第1の溶媒を含む溶液を調製するステップと、第2の溶媒であって、第1の溶媒と混和性であり、フラノースを溶解させることができる第2の溶媒を添加するステップと、フラノースの上記結晶形を得るために、溶液に結晶化処理を受けさせるステップと、を備える。結晶化処理は、溶媒系を冷却する工程と、外部冷却源を用いずに溶媒を冷却させる工程と、室温にて溶液と共にある期間にわたって待機する工程と、種結晶を添加する工程と、および/またはフラノースを溶液から析出させるために追加の溶媒または溶媒系を添加する工程とを含みうる。
【0017】
なお別の実施形態において、結晶性フラノースを産生する方法は、フラノースおよび1つ以上の溶媒を含む溶液を調製するステップと、追加の溶媒の過剰量であって、第1の溶媒と混和性であり、フラノースの上記溶解形を得るためにフラノースを溶解させない追加の溶媒の過剰量をゆっくりと添加するステップとを含む。
【0018】
なお別の実施形態において、本発明は、DGJなどのノジリマイシン誘導体を作製する方法における改良を提供する。そのような方法は例えば、Santoyo−Gonzalez,F.,ら,Synlett 1999 593−595に見出される。該改良は、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または他の保護基を有するフラノースを結晶化させるステップと、ノジリマイシン誘導体の産生において、該フラノースを精製するためにクロマトグラフィーまたはイオン交換樹脂を含む精製ステップを用いずにフラノースを使用するステップとを備える。
【0019】
本発明の他の特徴、利点および実施形態は、次の説明、付随するデータおよび添付請求項から当業者に明らかとなるであろう。
【0020】
[好ましい実施形態の詳細な説明]
次の図面は、本明細書の一部を形成し、本発明のある態様をさらに説明するために含まれている。本発明は、本明細書に示す具体的な実施形態の詳細な説明と併せた、1つ以上のこれらの図面の参照によって、さらに良好に理解されうる。
【0021】
「アルキル」という用語は、炭素および水素原子のみから成り、不飽和を含有せず、単結合によって分子の残りに結合された、直鎖または分岐C1〜C20炭化水素基、例えばメチル、エチル、n−プロピル、1−メチルエチル(イソプロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)を指す。本明細書で使用するアルキルは、好ましくはC1〜C8アルキルである。
【0022】
「アルケニル」という用語は、少なくとも1個の炭素間二重結合を含有し、直鎖または分岐鎖でありうるC2〜C20脂肪族炭化水素基、例えばエテニル、1−プロペニル、2−プロペニル(アリル)、イソプロペニル、2−メチル−1−プロペニル、1−ブテニル、2−ブテニルを指す。
【0023】
「シクロアルキル」という用語は、不飽和、非芳香族単環または多環式炭化水素環系、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルを示す。多環式シクロアルキル基の例としては、パーヒドロナフチル、アダマンチルおよびノルボルニル基、架橋環式基またはスピロ二環式基、例えばスピロ(4,4)ノン−2−イルが挙げられる。
【0024】
「シクロアルカルキル」という用語は、上で定義したようなアルキル基に直接結合した、上で定義したようなシクロアルキルを指し、該シクロアルキルはシクロプロピルメチル、シクロブチルエチル、シクロペンチルエチルなどの安定した構造の生成をもたらす。
【0025】
「アルキルエーテル」という用語は、アルキル鎖内に包含された少なくとも1個の炭素を有する、上で定義したようなアルキル基またはシクロアルキル基、例えばメチルエチルエーテル、ジエチルエーテル、テトラヒドロフランを指す。
【0026】
「アルキルアミン」という用語は、少なくとも1個の窒素原子を有する、上で定義したようなアルキル基またはシクロアルキル基、例えばn−ブチルアミンおよびテトラヒドロオキサジンを指す。
【0027】
「アリール」という用語は、約6〜約14個の範囲の炭素原子を有する芳香族ラジカル、例えばフェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニルを指す。
【0028】
「アリールアルキル」という用語は、上で定義したようなアルキル基に直接結合した、上で定義したようなアリール基、例えばCH2C6H5、および−C2H4C6H5を指す。
【0029】
「複素環式」という用語は、炭素原子および窒素、リン、酸素および硫黄から成る群より選択される1〜5個のヘテロ原子より成る、安定な3〜15員環ラジカルを指す。本発明の目的では、複素環式環は、縮合、架橋またはスピロ環系を含みうる単環式、二環式または三環式環系であり、複素環式環ラジカル中の窒素、リン、炭素または硫黄原子が各種の酸化状態へ場合により酸化されうる。加えて、窒素原子は場合により4級化されうる;環ラジカルは、一部または完全に飽和されうる(すなわちヘテロ芳香族またはヘテロアリール芳香族)。そのような複素環式環ラジカルの例としては、これに限定されるわけではないが、アゼチジニル、アクリジニル、ベンゾジオキソリル、ベンゾジオキサニル、ベンゾフラニル、カルバゾリル、シンノリニル、ジオキソラニル、インドリジニル、ナフチリジニル、パーヒドロアゼピニル、フェナジニル、フェノチアジニル、フェノキサジニル、フタラジニル、ピリジル、プテリジニル、プリニル、キナゾリニル、キノキサリニル、キノリニル、イソキノリニル、テトラゾリル、イミダゾリル、テトラヒドロイソキノリル、ピペリジニル、ピペラジニル、2−オキソピペラジニル、2−オキソピペリジニル、2−オキソピロリジニル、2−オキソアゼピニル、アゼピニル、ピロリル、4−ピペリドニル、ピロリジニル、ピラジニル、ピリミジニル、ピリダジニル、オキサゾリル、オキサゾリニル、オキサソリジニル、トリアゾリル、インダニル、イソキサゾリル、イソキサソリジニル、モルホリニル、チアゾリル、チアゾリニル、チアゾリジニル、イソチアゾリル、キヌクリジニル、イソチアゾリジニル、インドリル、イソインドリル、インドリニル、イソインドリニル、オクタヒドロインドリル、オクタヒドロイソインドリル、キノリル、イソキノリル、デカヒドロイソキノリル、ベンズイミダゾリル、チアジアゾリル、ベンゾピラニル、ベンゾチアゾリル、ベンゾオキサゾリル、フリル、テトラヒドロフリル、テトラヒドロピラニル、チエニル、ベンゾチエニル、チアモルホリニル、チアモルホリニルスルホキシド、チアモルホリニルスルホン、ジオキサホスホラニル、オキサジアゾリル、クロマニル、イソクロマニルが挙げられる。
【0030】
複素環式環ラジカルは、安定な構造の生成をもたらす主構造にいずれかのヘテロ原子または炭素原子にて結合されうる。
【0031】
「ヘテロアリール」という用語は、環が芳香族である複素環式環を指す。
【0032】
「ヘテロアリールアルキル」という用語は、アルキル基に直接結合した、上で定義したようなヘテロアリール環ラジカルを指す。ヘテロアリールアルキルラジカルは、安定な構造の生成をもたらす主構造に、アルキル基からのいずれかの炭素原子にて結合されうる。
【0033】
「ヘテロシクリル」という用語は、上で定義したような複素環式環ラジカルを指す。ヘテロシクリル環ラジカルは、安定な構造の生成をもたらす主構造にいずれかのヘテロ原子または炭素原子にて結合されうる。
【0034】
「ヘテロシクリルアルキル」という用語は、アルキル基に直接結合した、上で定義したような複素環式環ラジカルを指す。ヘテロシクリルアルキルラジカルは、安定な構造の生成をもたらす主構造に、アルキル基内のいずれかの炭素原子にて結合されうる。
【0035】
「置換アルキル」、「置換アルケニル」、「置換アルキニル」、「置換シクロアルキル」、「置換シクロアルカルキル」、「置換シクロアルケニル」、「置換アリールアルキル」、「置換アリール」、「置換複素環式環」、「置換へテロアリール環」、「置換へテロアリールアルキル」、または「置換ヘテロシクリルアルキル環」における置換基は、水素、ヒドロキシル、ハロゲン、カルボキシル、シアノ、アミノ、ニトロ、オキソ(=O)、チオ(=S)の基より選択される1個以上、あるいはアルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アリール、ヘテロアリール、ヘテロアリールアルキル、複素環式環、−COORx、−C(O)Rx、−C(S)Rx、−C(O)NRxRy、−C(O)ONRxRy、−NRxCONRyRz、−N(Rx)SORy、−N(Rx)SO2Ry、−(=N−N(Rx)Ry)、−NRxC(O)ORy、−NRxRy、−NRxC(O)Ry−、−NRxC(S)Ry、−NRxC(S)NRyRz、−SONRxRy−、−SO2NRxRy−、−ORx、−ORxC(O)NRyRz、−ORxC(O)ORy−、−OC(O)Rx、−OC(O)NRxRy、−RxNRyRz、−RxRyRz、−RxCF3、−RxNRyC(O)Rz、−RxORy、−RxC(O)ORy、−RxC(O)NRyRz、−RxC(O)Rx、−RxOC(O)Ry、−SRx、−SORx、−SO2Rx、−ONO2より選択される場合により置換された基と同じまたは異なっており、上の基のそれぞれにおける、Rx、RyおよびRzは、水素原子、置換または非置換アルキル、ハロアルキル、置換または非置換アリールアルキル、置換または非置換アリール、置換または非置換シクロアルキル、置換または非置換アルカルキル、置換または非置換複素環式環、置換または非置換ヘテロシクリルアルキル、置換または非置換へテロアリールまたは置換または非置換へテロアリールアルキルでありうる。
【0036】
「ハロゲン」という用語は、フッ素、塩素、臭素およびヨウ素のラジカルを指す。
【0037】
ピバロイルフラノース化合物が結晶形でただちに得られることが見出されている。これらの化合物の結晶化による精製は、特にクロマトグラフィーによる精製が実現不可能である大規模合成において、非結晶性生成物の精製と比較して単純化されている。クロマトグラフィーは有用なツールでありうるが、クロマトグラフィーは数キログラム規模の合成では無効である。本発明の方法によって産生されたピバロイルフラノースは、糖の合成で有用であり、クロマトグラフィーによる精製が不適切である合成工程に特に適している。多岐にわたる糖および糖の誘導体は、保護フラノース化合物が立体選択的に合成され、結晶化により単離されうるために、本明細書で述べる結晶化方法によって生成されうる。例えばL−アルトロースなどの糖は、最初に炭素C−5における配置の選択的にピバロイル化された中間体転化を生成する工程と、結晶化による中間体を精製する工程と、次に糖を生成するために脱保護する工程によって、D−ガラクトース糖などのより安価な糖より生成されうる。D−1−デオキシガラクトノジリマイシン(DGJ)などの化合物は、本発明のピバロイルフラノースを使用して生成されうる。結晶性ピバロイルフラノースは、クロマトグラフィー分離を使用せずに結晶化によって精製可能であり、高い純度および良好な収率での数キログラム規模での合成を可能にする、DGJの合成における有用な中間体である。
【0038】
糖
本発明は、溶液を反応中に生成された固体の結晶性生成物からデカンテーションすることにより、粗保護フラノースの単離を可能にする。本発明は、カラムクロマトグラフィーおよび他の方法と比較した容易さおよび費用削減のために、文献に見出される単離方法よりも好ましい。本発明は、ピバロイルフラノースが結晶化され、固体として単離されうるという驚くべき発見のために可能である。
【0039】
本明細書で述べる方法によって精製されうるフラノース化合物は、式:
【化1】

を有する、300g/molを超える分子量の保護フラノース化合物を含み、式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRsが、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択される。好ましい実施形態において、各Rはトリメチルアセチル(ピバロイル)である。別の態様において、糖は3個のピバロイル基を有する。
【0040】
R1およびR2は、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHである。各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシルである。一実施形態において、各Rはピバロイルであり、R1およびR2の一方はトリメチルアセトキシ(ペンタピバロイル)である。
【0041】
好ましいアリールおよびアリールアルキルは、フェニル、ベンジルまたはC7〜C12アルキルフェニル、特にC1〜C4アルキルフェニルまたはアルキルベンジルである。好ましいアシルは、C2〜C8アシル、例えばアセチル、プロパノイル、ブタノイル、ペンタノイル、ヘキサノイルおよびベンゾイルである。好ましいアルキルは、C1〜C6アルキルである。
【0042】
OR部分を有する位置のいずれも保護されうるか、またはOHとして放置されうる。遊離ヒドロキシル基の位置は、実施された反応の位置選択性によって定義される。
【0043】
R基は、フラノースの分子量が少なくとも300g/molであるように選択される。好ましくは、分子量は少なくとも325g/mol、または少なくとも350g/mol、または少なくとも375g/mol、または少なくとも400g/mol、または少なくとも425g/molである。最も好ましくは、分子量は少なくとも500g/molである。ある実施形態において、分子量は少なくとも525g/molまたは550g/mol、または575g/mol、または600g/molであろう。分子量は、1000g/mol未満、好ましくは800g/mol未満であろう。
【0044】
好ましい保護基は、ピバロイル基である。本保護基は大型であり、85g/molの分子量を有し、例えば他の非常に大型の基であるトリフェニルメチル基のような結晶メーカーと見なされうる。大きいサイズは、より小型の化合物ではそうであるように糖部分を油のままにしておく代わりに、糖部分を結晶化させる。あるいはジメチルアセチルは、保護基として使用されうる。アセチル基およびメチルアセチルはどちらも小さすぎて結晶メーカー保護基にはなれないが、化合物が少なくとも300g/molの分子量を有する場合に、1または2個のR基がアセチルまたはメチルアセチルであり、残りのR基がジメチルアセチルまたはトリメチルアセチル基でありうることが考慮される。
【0045】
4個のピバロイル基(それぞれ85g/mol)を有する化合物では、R1またはR2位置にヒドロキシル基を有する糖は、516g/molの分子量を有するであろう;R1またはR2の一方がアジドである場合、分子量は541g/molである。これらの化合物のそれぞれが、適切な溶媒から結晶化するであろう。少なくとも300g/molの分子量の化合物も結晶化するであろう。したがって4個のピバロイル基の代わりに4個のジメチルアセチル基(アゾ糖では460および485g/molの比較分子量の場合)を使用して糖が保護される場合、糖は本明細書で述べるように結晶化されうる。
【0046】
同様に、3個のピバロイル基を有する化合物では、R1またはR2位置および分子内の別の箇所に2つのヒドロキシル基を有する糖は、432g/molの分子量を有するであろう;R1またはR2の一方がアジドである場合、分子量は457g/molである。これらの化合物のそれぞれが、適切な溶媒から結晶化するであろう。
【0047】
好ましい実施形態において、テトラピバロイルフラノースが結晶化される。保護反応はモノ、ジ、トリおよびペンタピバロイル誘導体はもちろんのこと、所望のテトラピバロイル誘導体も潜在的に生成する(または代わりにテトラピバロイルが、好ましいペンタピバロイルまたはトリピバロイルに加えて生成される)ので、所望の生成物のみの結晶化がこれらの副生成物/不純物の分離を可能にする。使用される溶媒または溶媒系は、化合物の分子量および極性に基づいて結晶化される特定のテトラピバロイルフラノースに合せて「調整」されうる。
【0048】
保護基の1個以上がピバロイルまたは関連アルキルアセチル基でないことが考慮される。本発明のピバロイルフラノースの一部として含有されうる他の保護基は、糖のヒドロキシル基を誘導する着脱可能な保護基を含む。例えばフラノースは、ピバロイル基4個および他の保護基1個、またはピバロイル基3個および他の保護基2個、またはピバロイル基2個および他の保護基2個、またはピバロイル基3個および他の保護基1個およびヒドロキシル基1個を含有しうる。この種の保護基および誘導体を生成する工程は一般に糖化学において既知であり、これに限定されるわけではないが、直鎖または分岐C1〜C8アルキル、特にC1〜C4アルキル、例えばメチル、エチル、n−プロピル、イソプロピルまたはn−、イソ−およびt−ブチル;C7〜C12アリールアルキル、例えばベンジル、3〜20個の、特に3〜10個の炭素原子を有するトリアルキルシリル、例えばトリメチルシリル、トリエチルシリル、トリ−n−プロピルシリル、イソプロピルジメチルシリル、t−ブチルジメチルシリル、n−オクチルジメチルシリルまたは(1,1,2,2−テトラメチルエチル)−ジメチルシリル;アルデヒドおよびケトンによって糖または糖誘導体の隣接するOHからアセタールまたはケタールを生成することによって得られ、好ましくはそれぞれ2〜12個の、または3〜12個のC原子を含有する置換メチリデン基、例えばC1〜C12アルキリデン、好ましくはC1〜C6アルキリデンおよび特にC1〜C4アルキリデン、またはベンジリデン(エチリデン、1,1−プロピリデン、2,2−プロピリデン、1,1−ブチリデンまたは2,2−ブチリデン);C2〜C12アシル、特にC2〜C8アシル、例えばアセチル、プロパノイル、ブタノイル、ペンタノイル、ヘキサノイルおよびベンゾイル;R5−SO−(式中、R5は、C1〜C12アルキル、特にC1〜C6アルキル、C5シクロアルキル、C6シクロアルキル、フェニル、ベンジルあるいはC7〜C12アルキルフェニル、特にC1〜C4アルキルフェニル、あるいはC1〜C12アルキルベンジル、特にC1〜C4アルキルスルホニルまたはアリールスルホニル、例えばメチルスルホニル、エチルスルホニル、プロピルスルホニル、ブチルスルホニル、フェニルスルホニル、ベンジスルスルホニル、およびp−メチルフェニルスルホニルである)を含む(例えば米国特許第5,218,097号を参照)。1個以上のピバロイラート基に加えて使用される好ましい保護基は、アセチル、ベンジル、シリルまたはトリチルである。
【0049】
本明細書で示した糖構造は、ヘキソフラノース形である。しかしながら結晶性糖は別の形、例えば5員(フラノースにおいてのように)または6員(ピラノースにおいてのように)環形のどちらかおよび開鎖形の環式ヘミアセタールなどの、別の形にも適合しうる。
【0050】
他のピバロイルフラノースは、本発明の結晶化方法によって精製されうる。フラノシドは、異なる開始物質を使用することによる本明細書に記載した方法によっても産生されうる。例えば、糖:アロース、アルトロース、グルコース、マンノース、グロース、イドースおよびタロースのいずれか1つが、結晶性ピバロイルフラノースを産生するための開始物質として使用されうる。本明細書に記載したフラノース化合物のDおよびL系列のどちらも考慮される;最も好ましい立体化学は、D系列を含む。
【0051】
結晶化
本発明の化合物は、結晶性であることが見出されており、糖は一般に粘性液体の形であり、結晶化できないので、糖合成中に普通、必要とされるようなカラムクロマトグラフィーまたはイオン交換樹脂などの文献に記載された精製手順の使用を必要としない。
【0052】
フラノース糖は当分野で周知の方法によって結晶化されうる。溶媒は、極性および糖との反応性がないことに基づいて選択される。結晶化に理想的な溶媒は、糖と反応してはならず、高温時に適度に大量のフラノースを、低温時にはごく少量のフラノースを溶解させねばならない。該溶媒は、糖の融点以下の温度にて沸騰すべきである。使用されうる多数の溶媒がある。一般に、ガラクトースおよびアルトロース糖などのより極性の糖は、C6〜C9アルカンおよびシクロアルカンなどの、より非極性の溶媒から結晶化するであろう。他の糖、例えばアジドによって置換される糖はより極性が低く、結晶化にはメタノールなどのより極性の高い溶媒を使用すべきである。
【0053】
本発明で使用されうる溶媒としては、これに限定されるわけではないが、エタノール、メタノール、プロパノール、n−ヘキサン、シクロヘキサン、ヘプタン、オクタン、テトラヒドロフラン、ジエチルエーテル、酢酸エチル、ジブチルエーテル、ジメチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、塩化メチレン、クロロホルム、四塩化炭素、ジクロロメタン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、ジオキサン、アセトニトリル、ペンタノール、イソプロパノール、ベンゼン、トルエン、キシレン、アセトン、エチレングリコール、およびこれらの溶媒の2つ以上の組合せが挙げられる。
【0054】
溶媒が高温であるか、または沸騰しているときに溶媒に溶媒和される糖の量は、好ましくは5〜60重量%である。さらに好ましくは、溶媒中に重量で10〜50%の、または20〜40%の、または最も好ましくは25〜35%の糖がある。糖を溶媒和させた後に、温度を低下させる。温度は結晶化のために、好ましくは0℃以下に、さらに好ましくは−10℃または−20℃まで低下させる。好ましい場合、種添加が使用されうる。フラノースの結晶化は低速で進行し、低速での進行は、結晶格子中の分子が溶液中の分子と平衡にあるために、結晶構造が成長するときに不純物の排除を可能にする。一実施形態において、結晶化を発生させるために、溶液は−10℃〜−20℃にて約2日間維持される。
【0055】
本発明は、カラムクロマトグラフィーまたはイオン交換樹脂による精製の追加ステップに頼ることなく、フラノース糖を生成する他の方法によって達成されうるよりも高い、好ましくは著しく高い純度レベルで生成される、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基を有するフラノース糖を提供する。これらの結晶性フラノース糖は実質的により純粋である。本明細書に記載したような結晶化工程は、該結晶化工程が、追加のピバロイラート部分を有する反応副生成物、または未保護基を含む汚染物質からのフラノースの分離を可能にするので好都合である。
【0056】
一実施形態において、粗ピバロイルフラノースは、DMF水溶液などの溶液からの結晶化によって単離される。本溶液は、保護フラノースの生成中に使用可能であり、保護反応を停止させた後に入手取得されるので有用である。DMF溶液からの結晶化は最大約2日間を要しうる。粗生成物がいったん回収されると、粗生成物はヘプタン/酢酸エチルなどの溶液に溶解される。粗生成物は次に、洗浄、乾燥、濃縮および例えばヘプタンからの再結晶化によって精製されうる。この再結晶化工程は、反応で母液中に生成された汚染物質および副生成物(例えばテトラピバロイラートが所望であるとき、ペンタピバロイラートなど)を残すと同時に、所望のピバロイルフラノースが結晶化される。この結晶化は低速でもあり、最大2日間を要しうる。所望ならば本反応で種添加が使用されうる。
【0057】
好ましい実施形態において、テトラピバロイルフラノースの1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)または1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)は、C6〜C9アルカン、例えばヘキサンまたはヘプタンからの結晶化によって単離される。これらのフラノシド生成物は、高い純度で産生されうる。別の好ましい実施形態において、アジドテトラピバロイルフラノースの5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)がメタノールからの結晶化によって単離される。このことは、テトラピバロイルフラノース化合物(II)および(III)で実施されるように、ヘプタンからの結晶化よりも高い純度の生成物を与える。同様のアジド糖、例えば5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−アルトロフラノース、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−ガラクトフラノースも考慮される。
【0058】
DGJの合成
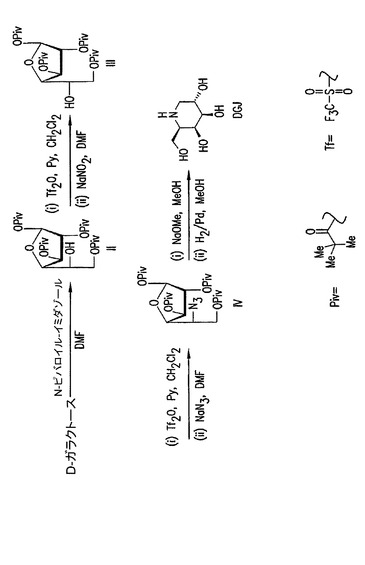
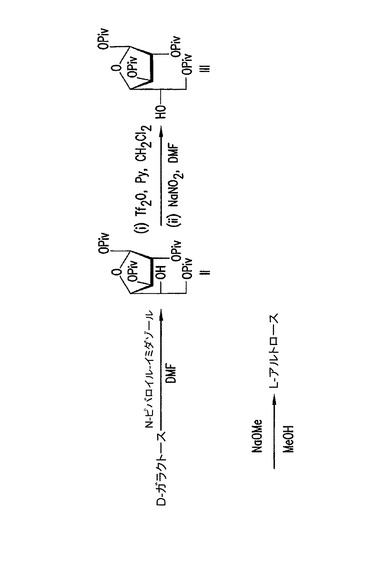
DGJの合成方法において、D−ガラクトースは、参照により本明細書に組み入れられているSantoyo−Gonzalez(1999)に記載されているように、開始物質として使用されうる。本合成での方法としては、N,N−ジメチルホルムアミド(DMF)中でD−ガラクトースに1−(トリメチルアセチル)イミダゾール(ピバロイルイミダゾール)と砂糖を反応させることによって、主生成物としての該保護フラノシド誘導体:1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)と、微量生成物としての1,2,3,5,6−ペンタ−O−ピバロイル−D−ガラクトフラノースのα,β−アノマーの混合物を生成する、D−ガラクトースのヒドロキシル基の、ピバロイル基による保護が挙げられる。ガラクトフラノシドは次に、アルトロフラノシドの1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)に変換される。次にヒドロキシルは保護されて、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)を得るためにアジド基によって置換される。脱保護の後、DGJを得るためにガラクトフラノシド誘導体が還元される。Santoyo−Gonzalezは、3つのフラノシド中間体はもちろんのこと、DJG生成物も精製するためにカラムクロマトグラフィーを使用した。本参考文献に記載されたDGJの合成は、全収率が約20%の最終生成物約200mgの規模でのみ有用である。
【0059】
3つのフラノシド中間体、1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)、1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)、および5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)は共通の溶媒からそれぞれ結晶化されうるので、本発明はDGJの合成の改良された方法を提供する(図1)。中間ステップのそれぞれの間に精製のためにカラムクロマトグラフィーを使用する代わりに、フラノシド中間体は結晶化によって精製されうる。ガラクトフラノシド(IV)は、Santoyo−Gonzalezによって記載された方法などによってDGJを生成するのに使用されうる。
【0060】
アルトロース誘導体の合成
L−アルトロースは、低い全収率で一連の化学反応によって、または細菌Butyrivibrio fibrisolvensから培養された細胞外ポリサッカライドから合成されうる非栄養性甘味料である(米国特許第4,966,845号)。しかしながら、これらの方法は高価である。本発明の結晶性ピバロイルフラノースの使用は、D−ガラクトース誘導体のさらに高価なL−アルトロース誘導体への簡単な変換を可能にする。この変換はクロマトグラフィーによる分離および精製の必要なしに達成されうる(図2)。結晶性1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースは、安価なD−ガラクトースから開始して上記の方法で調製されうる。クリスタリン1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースは後で、ピバロイル保護基(例えばメタノール中でナトリウムメトキシド)を除去するために脱保護反応を受けることができ、純粋なα−L−アルトロ−フラノシドが単離されうる。
【0061】
他の糖の合成
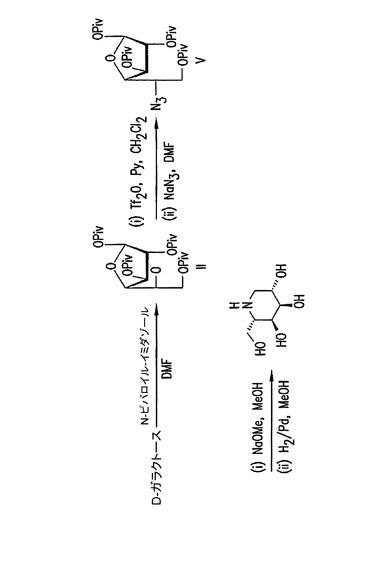
本発明のピバロイルフラノースは、多数の糖および糖誘導体の合成において有用な中間体である。例えばDGJの合成と類似して、D−ガラクトースは、参照により本明細書に組み入れられているSantoyo−Gonzalez(1999)によって記載されているように、(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオールを調製するための開始物質として使用されうる(図3)。結晶性1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)は、上述のように調製されうる。次にヒドロキシルは保護されて、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(V)を得るためにアジド基によって置換される。脱保護の後、イミノ糖を得るために5−アジドアルトロフラノシド誘導体が還元される。フラノシド中間体の1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)および5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(V)はそれぞれ共通の溶媒から結晶化されうるので、本発明はDGJの(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオール異性体の合成の改良された方法を提供する。
【0062】
チオヘキソース、例えばWhistlerによって述べられたチオヘキソースも、本明細書に記載した方法によってピバロイル化および結晶化されうる。(Whistler,J.Org.Chem.,1968,396−8)。
【0063】
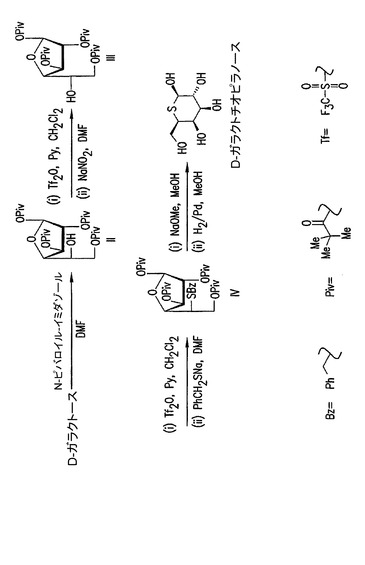
D−ガラクトースは、DGJの合成と類似して、(2R,3R,4S,5R,6R)−6−ヒドロキシメチル−テトラヒドロ−チオピラン−2,3,4,5−テトラオール(D−ガラクトチオピラノース)を調製するための開始物質として使用されうる(図4)。1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)は上述のように調製されうる。ヒドロキシルは保護されて、5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)を得るためにベンジルチオ基によって置換される。このテトラピバロイルは、この中間体を精製するために結晶化されうる。脱保護の後、D−ガラクトチオピラノースを得るためにガラクトフラノシド中間体が還元される。
【0064】
本明細書で使用するように、「数キログラム」「数kg」および「予備規模」という用語は、生成物が、1回の合成で生成物1kgを超える、あるいは10kgすら超える量である合成規模を示す。
【0065】
[実施例]
本発明は、本発明の範囲を限定すると解釈されるべきではない次の実施例でさらに例示される。
【0066】
[実施例1]
結晶性1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(II)の製剤およびキャラクタリゼーション
【0067】
1−(トリメチルアセチル)イミダゾール(ピバロイルイミダゾール)(42.2kg、5倍過剰)をDMF(90kg)およびヘプタン(3.4kg)に溶解させて、溶液を60℃まで加温した。D−ガラクトース(10kg)を溶液に注入し、混合物を75℃まで加熱した。反応物を90〜100℃まで発熱させて、発熱が収まった後に反応が完了するまで80〜100℃に維持した。反応の進行はTLC(ヘキサン:酢酸エチル=4:1)によって監視した。進行を描出するために、TLCを後で希硫酸によって染色および加熱した;TLC上での生成物のスポット(Rf=0.5)が主な成分となったときに反応が完了したと見なした。反応が完了した後に、水(200kg)および氷(82kg)を含有する混合物中に反応生成物をただちに移した。粗生成物を本混合物から結晶化によって単離した。本結晶化は低速で、一般に2日間を要する。粗生成物を収集して、ヘプタン/酢酸エチルに溶解させ、水で洗浄して、硫酸マグネシウムによって乾燥させ、濃縮して、2〜3倍量のヘプタン(〜25kg)から−20℃にて再び結晶化させた;本工程は母液にペンタピバロイラートを残した。本ステップの収率は、数kg規模で実施したときに25〜35%(7.2〜10kg)であった。1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(II)は、高純度を有する白色結晶性粉末であった。融点は、105〜108℃の範囲内であった。IR(KBr,cm−1):3432(OH,s),2974(C−H stretch,s),1740(ester of pivaloylate,vs),1284(C−O,weak),1143(C−O,vs),1031(C−O,weak);1HNMR(CDCl3,400MHz,TMS):δ=1.18(s,3H),1.19(s,3H),1.20(s,3H),1.23(s,3H),2.45(d,J=7.9Hz,1H);3.90−3.96(m,1H),4.07(dd,J=3.4Hz,J=6.5Hz,1H),4.13(dd,J=11.6Hz,J=5.3Hz,1H),4.19(dd,J=11.6Hz,J=6.1Hz,1H),5.44(dd,J=7.9Hz,J=4.6Hz,1H),5.62(dd,J=7.9Hz,J=7.0Hz,1H),6.37(d,J=4.6Hz,1H).
【0068】
[実施例2]
結晶性1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)の製剤およびキャラクタリゼーション
【0069】
ピリジン(3.82kg)の塩化メチレン(15L)溶液を窒素雰囲気下で0℃まで冷却した。トリフルオロメタンスルホン酸無水物(3.28kg)を0℃にて滴加し、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド(5kg)の塩化メチレン(10L)溶液の滴加を続けた。反応混合物を0℃にて2時間撹拌して、TLC(ヘキサン:酢酸エチル=4:1)によって反応の完了を確認した。反応がこの時点で完了していない場合、トリフルオロメタンスルホン酸無水物の追加量(0.1kg)を添加した。トリフラート化化合物の5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシドが、反応の本段階でガラクトフラノシドから生成された。反応混合物を次に冷6%塩酸(3回30L)、塩水(30L)および7.5%重炭酸ナトリウム溶液(30L)によって洗浄した。N,N−ジイソプロピルエチルアミン(230mL)を次に添加して、反応物を炭酸ナトリウム(1.5kg)上で1時間撹拌した。反応物を濾過して、乾燥まで濃縮した。本質的に純粋な5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースを結晶性固体として単離した。
【0070】
5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースをDMF9.5Lに溶解させ、亜硝酸ナトリウム5当量(1.67kg)と12時間にわたって反応させた。反応物をヘプタン(24L)および酢酸エチル(12L)によって希釈し、濾過して、2%重炭酸塩溶液(40L)に注入した。生成物をヘプタン/酢酸エチルによって抽出し、実施例1で行ったようにヘプタンから結晶化した。1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)の収率は35〜45%であった((II)の5kgより2kg)。HPLCは、転化アルコールへの完全な変換を証明した。生成物は、オフホワイト結晶性固体であった。融点109〜112℃,IR(KBr,cm−1):3444(OH,s),2977(C−H stretch,s),1732(ester of pivalate,vs),1481(weak),1284(C−O,weak),1156(C−O,vs),1028(C−O,weak);1H NMR(CDCl3,400MHz,TMS):δ=1.18(s,3H),1.20(s,3H),1.21(s,3H),1.22(s,3H),3.01(d,J=2.45,1H),3.99−4.02m,2H),4.11−4.07(m,1H),4.26(dd,J=12.4Hz,J=2.7Hz,1H),5.43(dd,J=7.3Hz,J=4.6Hz,1H),5.58(dd,J=7.3Hz,J=5.2Hz,1H),6.37(d,J=4.8Hz,1H).
【0071】
[実施例3]
結晶性5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)の製剤およびキャラクタリゼーション
【0072】
トリフラート化化合物の5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノースは、実施例2に記載したような手順で実施例2のアルトロフラノースIII(5kg)から生成した。本化合物にアジ化ナトリウム(1.6kg)をDMF(9.5L)中で反応させた。反応は転化反応中に観察された最適条件を使用して実施した。粗生成物はメタノール(1.3〜1.7mL/g)から2回結晶化させた。5kg規模では、IIIからの5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)の収率は通常、50〜70%(〜3.3kg)であった。生成物は、オフホワイト結晶性固体であった。融点103〜104℃。IR(KBr,cm−1):2090(azide,s),1740(ester of pivalate,vs),1480(weak),1280(C−O,s),1160(C−O,vs),1042(C−O,weak);1HNMR(CDCl3,400MHz,TMS):δ=1.19(s,3H),1.20(s,3H),1.22(s,3H),1.25(s,3H),3.83−3.79(m,1H),4.05(dd,J=6.7,J=4.8Hz 1H),4.15(dd,J=11.7Hz,J=8.0Hz,1H),4.30(dd,J=11.7Hz,J=4.2Hz,1H),5.41(dd,J=7.9Hz,J=4.6Hz,1H),5.59(t,J=7.5Hz,1H),6.33(d,J=4.5Hz,1H).
【0073】
[実施例4]
結晶性5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)
【0074】
実施例3から生成された粗生成物を、上述の結晶化手順を使用してEtOAc:MeOH 1:6およびメタノールから結晶化させた。本結晶性の収率は、50〜60%の5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)であった。
【0075】
[実施例5]
結晶性5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシドの製剤
【0076】
5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースは、アジ化ナトリウムをナトリウムα−トルエンチオキシドに代えて、実施例3に記載したようにサンプルを結晶化することによって、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースと同様の方法で調製する。
【0077】
本発明の多くの変形は、上述の説明に照らして当業者にそれ自体を示唆するであろう。例えば、糖の結晶化は、各種の溶媒から実施されうる。そのようなすべての明らかな変形は、添付請求項の十分に意図された範囲内である。当業者は、本開示に照らして、本明細書で開示される具体的な実施形態で変更を実施して、本発明の精神および範囲から逸脱せずに同じまたは同様の結果が得られることを認識すべきである。
【0078】
上記の特許、出願、試験方法、刊行物は、参照によりその全体が本明細書に組み入れられている。
【図面の簡単な説明】
【0079】
【図1】結晶性誘導体II、IIIおよびIVを使用するDGJの合成。
【図2】結晶性誘導体IIおよびIIIを使用するL−アルトロースの合成。
【図3】結晶性誘導体IIおよびVを使用するD−ガラクトーゼからの(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオールの合成。
【図4】(2R,3R,4S,5R,6R)−6−ヒドロキシメチル−テトラヒドロ−チオピラン−2,3,4,5−テトラオール(D−ガラクトチオピラノース)の合成。
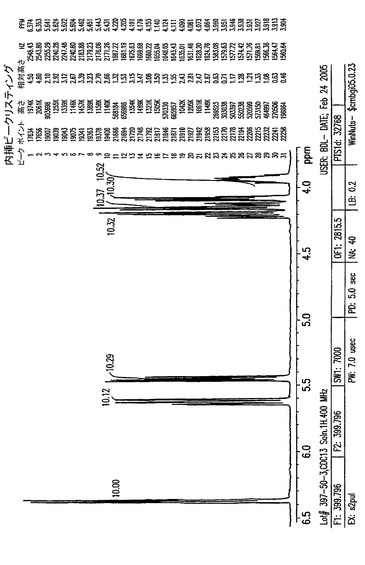
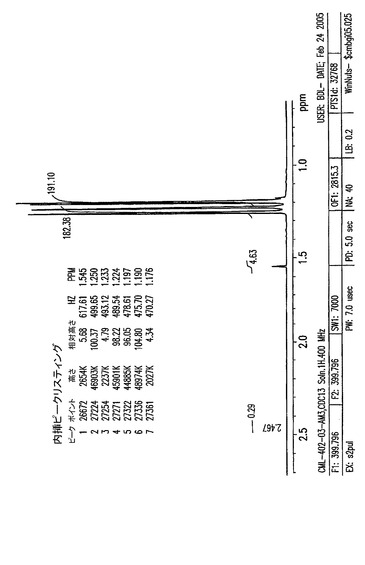
【図5A】0〜14ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
【図5B】0.7〜2.6ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
【図5C】3.8〜6.5ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
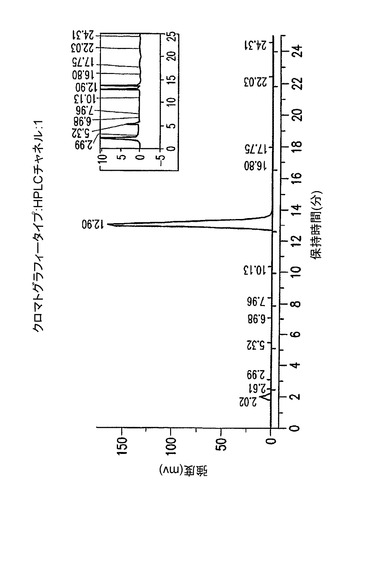
【図6】他の異性体(II)からの完全な除去を示す、結晶化1,2,3,6−テトラピバロイル−α−L−アルトロ−フラノース(III)のHPLC。化合物(III)は約27.5分にて溶離するのに対して、関連する異性体(II)は29.0分にて溶離する。
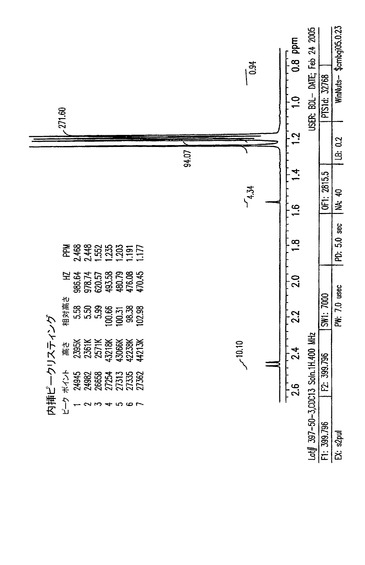
【図7A】0〜14ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図7B】3.8〜6.6ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
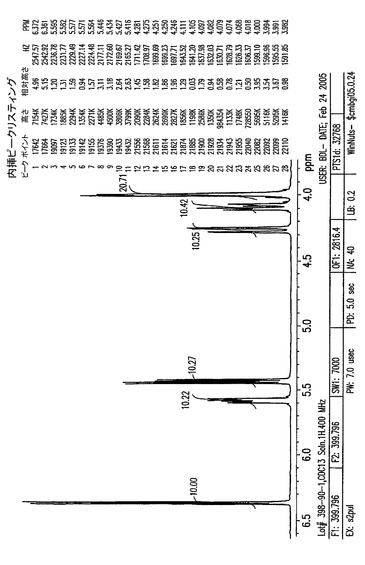
【図7C】0.7〜3.2ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
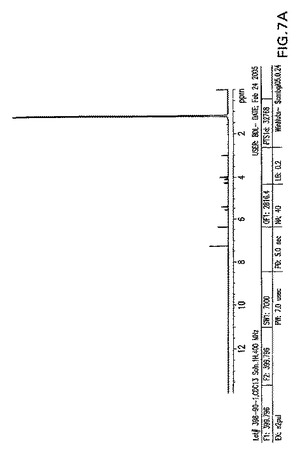
【図8A】0〜14ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
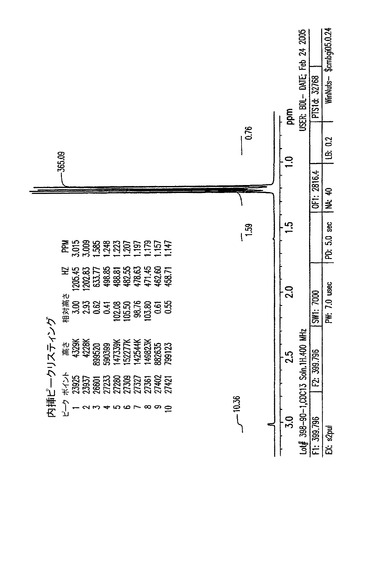
【図8B】3.7〜6.6ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
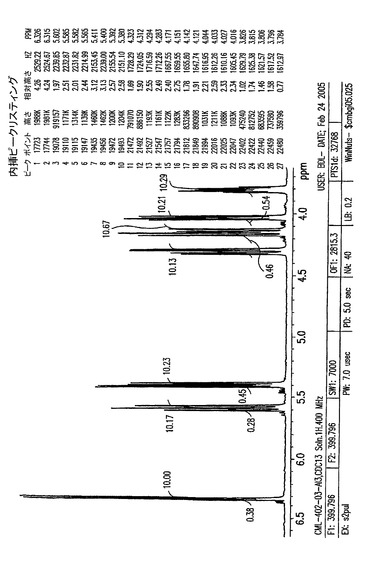
【図8C】0.7〜2.7ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【発明の詳細な説明】
【0001】
本出願は2005年6月8日に出願された米国仮特許出願第60/689,119号からの優先権を主張し、その開示は参照としてその全体が本明細書に組み入れられている。
【0002】
[発明の分野]
本発明は、結晶性ピバロイルフラノースおよびビバロイルフラノースの結晶化方法に関するものである。これらの化合物は、D−1−デオキシガラクトノジリマイシン(DGJ)などの糖の合成での中間体として有用である。
【0003】
[発明の背景]
DGJはまた、(2R,3S,4R,5S)−2−ヒドロキシメチル−3,4,5−トリヒドロキシピペリジン、1−デオキシ−ガラクトスタチンとしても、そしてD−1−デオキシガラクトノジリマイシンとしても記載される。DGJは、D−ガラクトースのイミノ糖(5−アミノ−5−デオキシ−D−グルコピラノース)類似体であり、α−およびβ−D−ガラクトシダーゼの両方の強力な阻害物質である。ガラクトシダーゼは、グリコシド結合の加水分解を触媒し、複合糖質の代謝において重要である。DGJなどのガラクトシダーゼ阻害物質は、糖尿病(例えば米国特許第4,634,765号)、癌(例えば米国特許第5,250,545号)、ヘルペス(例えば米国特許第4,957,926号)、Hivおよびファブリ病(Fanら,Nat.Med.1999 5:1,112−5)を含む、多くの疾患および状態の治療に使用されうる。
【0004】
デオキシノジリマイシンなどのノジリマイシン誘導体の発表された化学合成は一般に、市販用途には適切でない複数のステップを有する。中間体の多くは安定でなく、中間体および最終生成物の両方の精製は数キログラム規模で扱いにくい。Grabnerによって特許取得された化学微生物学的方法(米国特許第5,695,969号;米国特許第5,610,039号)は、グルコースの細菌による酸化で得られた5−ケトアルドースの還元的アミノ化によって、糖をそのイミノ誘導体に変換する方法を提供する。しかしながら該方法は、D−ガラクトノジリマイシン誘導体には適用できない。他の関連特許(米国特許第5,227,479号、米国特許第4,908,439号および米国特許第4,634,765号)は、保護グリコシルハライドを使用するホモノジリマイシンの製剤、D−グルクロノラクトンの水素化物還元について議論している。米国特許第4,908,439号は、アジドに水素化アルミニウムリチウムなどのヒドリド還元剤を反応させることによって、グルコースジリマイシン誘導体である5−アミノ−5−デオキシ−1,2−O−イソプロピリデン−D−グルコンウロラクトン(DNJ誘導体)を調製するプロセスを教示する。
【0005】
米国特許第6,740,780号、同第6,683,185号、同第6,653,482号、同第6,653,480号、同第6,649,766号、同第6,605,724号、同第6,590,121号、および同第6,462,197号は、D−ジデオキシガラクトノジリマイシンの製剤において中間体として有用であるイミノ糖の製剤のプロセスについて記載している。これらの化合物は、ヘキソース糖の1,5−ジデオキシ−1,5−イミノヘキシトールであり、ヒドロキシル保護オキシム中間体から調製される。これらのイミノ糖を作製するプロセスは、ヘキシトールに還元されるラクタムの生成を含む。しかしながら本プロセスは、安全性、アップスケール、取り扱いおよび合成の複雑さに関する、数キログラム規模での産生に関してある欠点を有する。例えば該プロセスは、大規模では実際的でない手順である、フラッシュクロマトグラフィーを精製に使用する。
【0006】
文献で発表されたD−1−デオキシガラクトノジリマイシン(DGJ)の複数の製剤があり、製剤の大半は、予備規模手順(>100g)での産業研究所における反復には適切でない。これらの合成のいくつかとしては、D−グルコース(Legler G,ら,Carbohydr Res.1986 Nov 1;155:119−29);D−ガラクトース(Uriel,C,Santoyo−Gonzalez,F.,ら,Synlett 1999 593−595;Synthesis 1998 1787−1792(ピバロイル化中間体を開示));ガラクトピラノース(Bernotas RC,ら,Carbohydr Res.1987 Sep 15;167:305−ll);L−酒石酸(Aoyagiら,J.Org.Chem.1991,56,815);ケブラコイトール(Chidaら,J.Chem.Soc,Chem Commun.1994,1247);ガラクトフラノース(Paulsenら,Chem.Ber.1980,113,2601);ベンゼン(Johnsonら,Tetrahedron Lett.1995,36,653);アラビノ−ヘキソース−5−ウロース(Bariliら,tetrahedron 1997,3407);5−アジド−1,4−ラクトン(Shilvockら,Synlett,1998,554);ドキシノジリマイシン(Takahashiら,J.Carbohydr.Chem.1998,17,117);アセチルグルコサミン(Heightmanら,Helv.Chim.Acta 1995,78,514);ミオ−イノシトール(Chida N,ら,Carbohydr Res.1992 Dec 31;237:185−94);ジオキサニルピペリデン(Takahataら,Org.Lett.2003;5(14);2527−2529);および(E)−2,4−ペンタジエノール(Martin R,ら,Org Lett.2000 Jan;2(1):93−5)(Hughes AB,ら,Nat Prod Rep.1994 Apr;11(2):135−62)からの合成が挙げられる。N,N−メチル−1−デオキシノジリマイシン含有オリゴサッカライドの合成は、Kisoによって記載されている(Bioorg Med Chem.1994 Nov;2(11):1295−308)。Kisoは、保護1−デオキシノジリマイシン誘導体に、D−ガラクトースのメチル−1−チオグリコシド(グリコシル供与体)をグリコシルプロモータとして使用されたトリフラートと共に結合させた。
【0007】
精製のためのカラムクロマトグラフィーの使用は、上で開示された参考文献によって教示された反応で産生されるような小規模合成には可能であるが、数kg規模での使用には不十分である。必要なカラムのサイズはもちろんのこと、要求される溶媒の量もこの手順を非現実的にしている。文献で報告されているDGJ製剤の最大規模は13.3gであり(Fred−Robert Heiker,Alfred Matthias Schueller,Carbohydrate Research,1989,203 314−318)、13.3gは治療薬としての使用のためのプラント規模合成で要求されるよりもはるかに少ない。Heikerらは、イオン交換樹脂Lewatit MP 400(OH−)およびエタノールからの結晶化を使用してDGJを精製した。しかしながらこのプロセスも数キログラム量には容易に拡大できない。
【0008】
したがって、クロマトグラフィーまたはイオン交換樹脂を利用しない合成が好ましい。化学製造において化合物を単離する最も容易な方法は、結晶化である。結晶化は、他の方法よりも一般に高速、安全、経費節約的であり、スケールアップが容易である。しかしながら炭水化物は通常、油の形であり、油は結晶化させるのが困難である。いくつかの例外がある。例えば米国特許第6,620,921号は、あるグルコフラノシドの製剤に有用な化合物である、結晶性1,2,3,5,6−ペンタ−O−プロパノイル−β−D−グルコフラノースを開示している。多くのグルコフラノース誘導体は常温常圧にて油であるが、‘921特許は一部のフラノースがこれらの条件下で結晶性であることを開示している。これらのフラノースとしては:フェニルβ−D−グルコフラノシド、4−ニトロフェニルα−D−グルコフラノシド、メチル2,3,5,6−テトラ−O−プロパノイル−1−チオ−β−D−グルコフラノシド、および1−β D−グルコフラノシルウラシルが挙げられる。
【0009】
しかしながら、他の結晶性中間体に対する、そしてDGJなどのデオキシジリマイシンの合成に有用であり、大規模合成にとって実際的である、結晶化による中間体の容易で拡張可能な精製工程(合成において中間体を精製することを含む)に対する要求がなお存在する。
【0010】
[発明の概要]
フラノースの結晶形およびこれらのフラノースを結晶化する方法が開示される。結晶性フラノースは、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基を有する。
【0011】
フラノースの分子量は300g/mol〜1000g/molである。好ましくは、分子量は少なくとも350g/mol、少なくとも400g/mol、またはさらに好ましくは少なくとも450g/molである。別の実施態様において、分子量は900g/mol未満または800g/mol未満である。
【0012】
別の実施形態において、少なくとも3個のトリメチルアセチル保護基がある。フラノースは、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース、1,2,3,6−テトラピバロイル−α−L−アルトロフラノース、または5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースなどのテトラピバロイルフラノースでありうる。
【0013】
フラノースを溶媒に添加する、またはフラノースを溶媒中で生成するステップと;溶媒から該フラノースを結晶化させるステップとを備える、結晶性フラノースを産生する方法も提供される。結晶化は好ましくは、第2の溶媒を添加する工程と、周囲圧にて冷却する工程とによって実施される。
【0014】
結晶性テトラピバロイルフラノースを含み、該テトラピバロイルフラノースに加えてモノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースの少なくとも1つが生成される本発明の一態様において;本モノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースは、テトラピバロイルフラノースが結晶化されるときには結晶化されない。同様に、トリピバロイル(あるいは例えばペンタピバロイルまたは他の保護糖)が目的の生成物である場合、そして追加の望ましくない保護糖が反応で生成される場合に、該トリピバロイル(あるいはペンタピバロイルまたは他の保護糖)は溶媒から結晶化され、該望ましくない保護糖は結晶化されない。
【0015】
好ましい溶媒は、ヘプタンおよびメタノールである。なお本発明の別の態様において、結晶化させるステップは、フラノースおよび溶媒を溶媒の沸点付近の温度まで加熱する工程と、0℃以下、またはさらに好ましくは−20℃〜−10℃の温度まで冷却する工程と、フラノースが沈殿するまで待機する工程とを含む;一態様において、この時間は少なくとも36時間である。
【0016】
なお別の実施形態において、結晶性フラノースを産生する方法は、フラノースおよび第1の溶媒を含む溶液を調製するステップと、第2の溶媒であって、第1の溶媒と混和性であり、フラノースを溶解させることができる第2の溶媒を添加するステップと、フラノースの上記結晶形を得るために、溶液に結晶化処理を受けさせるステップと、を備える。結晶化処理は、溶媒系を冷却する工程と、外部冷却源を用いずに溶媒を冷却させる工程と、室温にて溶液と共にある期間にわたって待機する工程と、種結晶を添加する工程と、および/またはフラノースを溶液から析出させるために追加の溶媒または溶媒系を添加する工程とを含みうる。
【0017】
なお別の実施形態において、結晶性フラノースを産生する方法は、フラノースおよび1つ以上の溶媒を含む溶液を調製するステップと、追加の溶媒の過剰量であって、第1の溶媒と混和性であり、フラノースの上記溶解形を得るためにフラノースを溶解させない追加の溶媒の過剰量をゆっくりと添加するステップとを含む。
【0018】
なお別の実施形態において、本発明は、DGJなどのノジリマイシン誘導体を作製する方法における改良を提供する。そのような方法は例えば、Santoyo−Gonzalez,F.,ら,Synlett 1999 593−595に見出される。該改良は、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または他の保護基を有するフラノースを結晶化させるステップと、ノジリマイシン誘導体の産生において、該フラノースを精製するためにクロマトグラフィーまたはイオン交換樹脂を含む精製ステップを用いずにフラノースを使用するステップとを備える。
【0019】
本発明の他の特徴、利点および実施形態は、次の説明、付随するデータおよび添付請求項から当業者に明らかとなるであろう。
【0020】
[好ましい実施形態の詳細な説明]
次の図面は、本明細書の一部を形成し、本発明のある態様をさらに説明するために含まれている。本発明は、本明細書に示す具体的な実施形態の詳細な説明と併せた、1つ以上のこれらの図面の参照によって、さらに良好に理解されうる。
【0021】
「アルキル」という用語は、炭素および水素原子のみから成り、不飽和を含有せず、単結合によって分子の残りに結合された、直鎖または分岐C1〜C20炭化水素基、例えばメチル、エチル、n−プロピル、1−メチルエチル(イソプロピル)、n−ブチル、n−ペンチル、1,1−ジメチルエチル(t−ブチル)を指す。本明細書で使用するアルキルは、好ましくはC1〜C8アルキルである。
【0022】
「アルケニル」という用語は、少なくとも1個の炭素間二重結合を含有し、直鎖または分岐鎖でありうるC2〜C20脂肪族炭化水素基、例えばエテニル、1−プロペニル、2−プロペニル(アリル)、イソプロペニル、2−メチル−1−プロペニル、1−ブテニル、2−ブテニルを指す。
【0023】
「シクロアルキル」という用語は、不飽和、非芳香族単環または多環式炭化水素環系、例えばシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルを示す。多環式シクロアルキル基の例としては、パーヒドロナフチル、アダマンチルおよびノルボルニル基、架橋環式基またはスピロ二環式基、例えばスピロ(4,4)ノン−2−イルが挙げられる。
【0024】
「シクロアルカルキル」という用語は、上で定義したようなアルキル基に直接結合した、上で定義したようなシクロアルキルを指し、該シクロアルキルはシクロプロピルメチル、シクロブチルエチル、シクロペンチルエチルなどの安定した構造の生成をもたらす。
【0025】
「アルキルエーテル」という用語は、アルキル鎖内に包含された少なくとも1個の炭素を有する、上で定義したようなアルキル基またはシクロアルキル基、例えばメチルエチルエーテル、ジエチルエーテル、テトラヒドロフランを指す。
【0026】
「アルキルアミン」という用語は、少なくとも1個の窒素原子を有する、上で定義したようなアルキル基またはシクロアルキル基、例えばn−ブチルアミンおよびテトラヒドロオキサジンを指す。
【0027】
「アリール」という用語は、約6〜約14個の範囲の炭素原子を有する芳香族ラジカル、例えばフェニル、ナフチル、テトラヒドロナフチル、インダニル、ビフェニルを指す。
【0028】
「アリールアルキル」という用語は、上で定義したようなアルキル基に直接結合した、上で定義したようなアリール基、例えばCH2C6H5、および−C2H4C6H5を指す。
【0029】
「複素環式」という用語は、炭素原子および窒素、リン、酸素および硫黄から成る群より選択される1〜5個のヘテロ原子より成る、安定な3〜15員環ラジカルを指す。本発明の目的では、複素環式環は、縮合、架橋またはスピロ環系を含みうる単環式、二環式または三環式環系であり、複素環式環ラジカル中の窒素、リン、炭素または硫黄原子が各種の酸化状態へ場合により酸化されうる。加えて、窒素原子は場合により4級化されうる;環ラジカルは、一部または完全に飽和されうる(すなわちヘテロ芳香族またはヘテロアリール芳香族)。そのような複素環式環ラジカルの例としては、これに限定されるわけではないが、アゼチジニル、アクリジニル、ベンゾジオキソリル、ベンゾジオキサニル、ベンゾフラニル、カルバゾリル、シンノリニル、ジオキソラニル、インドリジニル、ナフチリジニル、パーヒドロアゼピニル、フェナジニル、フェノチアジニル、フェノキサジニル、フタラジニル、ピリジル、プテリジニル、プリニル、キナゾリニル、キノキサリニル、キノリニル、イソキノリニル、テトラゾリル、イミダゾリル、テトラヒドロイソキノリル、ピペリジニル、ピペラジニル、2−オキソピペラジニル、2−オキソピペリジニル、2−オキソピロリジニル、2−オキソアゼピニル、アゼピニル、ピロリル、4−ピペリドニル、ピロリジニル、ピラジニル、ピリミジニル、ピリダジニル、オキサゾリル、オキサゾリニル、オキサソリジニル、トリアゾリル、インダニル、イソキサゾリル、イソキサソリジニル、モルホリニル、チアゾリル、チアゾリニル、チアゾリジニル、イソチアゾリル、キヌクリジニル、イソチアゾリジニル、インドリル、イソインドリル、インドリニル、イソインドリニル、オクタヒドロインドリル、オクタヒドロイソインドリル、キノリル、イソキノリル、デカヒドロイソキノリル、ベンズイミダゾリル、チアジアゾリル、ベンゾピラニル、ベンゾチアゾリル、ベンゾオキサゾリル、フリル、テトラヒドロフリル、テトラヒドロピラニル、チエニル、ベンゾチエニル、チアモルホリニル、チアモルホリニルスルホキシド、チアモルホリニルスルホン、ジオキサホスホラニル、オキサジアゾリル、クロマニル、イソクロマニルが挙げられる。
【0030】
複素環式環ラジカルは、安定な構造の生成をもたらす主構造にいずれかのヘテロ原子または炭素原子にて結合されうる。
【0031】
「ヘテロアリール」という用語は、環が芳香族である複素環式環を指す。
【0032】
「ヘテロアリールアルキル」という用語は、アルキル基に直接結合した、上で定義したようなヘテロアリール環ラジカルを指す。ヘテロアリールアルキルラジカルは、安定な構造の生成をもたらす主構造に、アルキル基からのいずれかの炭素原子にて結合されうる。
【0033】
「ヘテロシクリル」という用語は、上で定義したような複素環式環ラジカルを指す。ヘテロシクリル環ラジカルは、安定な構造の生成をもたらす主構造にいずれかのヘテロ原子または炭素原子にて結合されうる。
【0034】
「ヘテロシクリルアルキル」という用語は、アルキル基に直接結合した、上で定義したような複素環式環ラジカルを指す。ヘテロシクリルアルキルラジカルは、安定な構造の生成をもたらす主構造に、アルキル基内のいずれかの炭素原子にて結合されうる。
【0035】
「置換アルキル」、「置換アルケニル」、「置換アルキニル」、「置換シクロアルキル」、「置換シクロアルカルキル」、「置換シクロアルケニル」、「置換アリールアルキル」、「置換アリール」、「置換複素環式環」、「置換へテロアリール環」、「置換へテロアリールアルキル」、または「置換ヘテロシクリルアルキル環」における置換基は、水素、ヒドロキシル、ハロゲン、カルボキシル、シアノ、アミノ、ニトロ、オキソ(=O)、チオ(=S)の基より選択される1個以上、あるいはアルキル、アルコキシ、アルケニル、アルキニル、アリール、アリールアルキル、シクロアルキル、アリール、ヘテロアリール、ヘテロアリールアルキル、複素環式環、−COORx、−C(O)Rx、−C(S)Rx、−C(O)NRxRy、−C(O)ONRxRy、−NRxCONRyRz、−N(Rx)SORy、−N(Rx)SO2Ry、−(=N−N(Rx)Ry)、−NRxC(O)ORy、−NRxRy、−NRxC(O)Ry−、−NRxC(S)Ry、−NRxC(S)NRyRz、−SONRxRy−、−SO2NRxRy−、−ORx、−ORxC(O)NRyRz、−ORxC(O)ORy−、−OC(O)Rx、−OC(O)NRxRy、−RxNRyRz、−RxRyRz、−RxCF3、−RxNRyC(O)Rz、−RxORy、−RxC(O)ORy、−RxC(O)NRyRz、−RxC(O)Rx、−RxOC(O)Ry、−SRx、−SORx、−SO2Rx、−ONO2より選択される場合により置換された基と同じまたは異なっており、上の基のそれぞれにおける、Rx、RyおよびRzは、水素原子、置換または非置換アルキル、ハロアルキル、置換または非置換アリールアルキル、置換または非置換アリール、置換または非置換シクロアルキル、置換または非置換アルカルキル、置換または非置換複素環式環、置換または非置換ヘテロシクリルアルキル、置換または非置換へテロアリールまたは置換または非置換へテロアリールアルキルでありうる。
【0036】
「ハロゲン」という用語は、フッ素、塩素、臭素およびヨウ素のラジカルを指す。
【0037】
ピバロイルフラノース化合物が結晶形でただちに得られることが見出されている。これらの化合物の結晶化による精製は、特にクロマトグラフィーによる精製が実現不可能である大規模合成において、非結晶性生成物の精製と比較して単純化されている。クロマトグラフィーは有用なツールでありうるが、クロマトグラフィーは数キログラム規模の合成では無効である。本発明の方法によって産生されたピバロイルフラノースは、糖の合成で有用であり、クロマトグラフィーによる精製が不適切である合成工程に特に適している。多岐にわたる糖および糖の誘導体は、保護フラノース化合物が立体選択的に合成され、結晶化により単離されうるために、本明細書で述べる結晶化方法によって生成されうる。例えばL−アルトロースなどの糖は、最初に炭素C−5における配置の選択的にピバロイル化された中間体転化を生成する工程と、結晶化による中間体を精製する工程と、次に糖を生成するために脱保護する工程によって、D−ガラクトース糖などのより安価な糖より生成されうる。D−1−デオキシガラクトノジリマイシン(DGJ)などの化合物は、本発明のピバロイルフラノースを使用して生成されうる。結晶性ピバロイルフラノースは、クロマトグラフィー分離を使用せずに結晶化によって精製可能であり、高い純度および良好な収率での数キログラム規模での合成を可能にする、DGJの合成における有用な中間体である。
【0038】
糖
本発明は、溶液を反応中に生成された固体の結晶性生成物からデカンテーションすることにより、粗保護フラノースの単離を可能にする。本発明は、カラムクロマトグラフィーおよび他の方法と比較した容易さおよび費用削減のために、文献に見出される単離方法よりも好ましい。本発明は、ピバロイルフラノースが結晶化され、固体として単離されうるという驚くべき発見のために可能である。
【0039】
本明細書で述べる方法によって精製されうるフラノース化合物は、式:
【化1】
を有する、300g/molを超える分子量の保護フラノース化合物を含み、式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRsが、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択される。好ましい実施形態において、各Rはトリメチルアセチル(ピバロイル)である。別の態様において、糖は3個のピバロイル基を有する。
【0040】
R1およびR2は、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHである。各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシルである。一実施形態において、各Rはピバロイルであり、R1およびR2の一方はトリメチルアセトキシ(ペンタピバロイル)である。
【0041】
好ましいアリールおよびアリールアルキルは、フェニル、ベンジルまたはC7〜C12アルキルフェニル、特にC1〜C4アルキルフェニルまたはアルキルベンジルである。好ましいアシルは、C2〜C8アシル、例えばアセチル、プロパノイル、ブタノイル、ペンタノイル、ヘキサノイルおよびベンゾイルである。好ましいアルキルは、C1〜C6アルキルである。
【0042】
OR部分を有する位置のいずれも保護されうるか、またはOHとして放置されうる。遊離ヒドロキシル基の位置は、実施された反応の位置選択性によって定義される。
【0043】
R基は、フラノースの分子量が少なくとも300g/molであるように選択される。好ましくは、分子量は少なくとも325g/mol、または少なくとも350g/mol、または少なくとも375g/mol、または少なくとも400g/mol、または少なくとも425g/molである。最も好ましくは、分子量は少なくとも500g/molである。ある実施形態において、分子量は少なくとも525g/molまたは550g/mol、または575g/mol、または600g/molであろう。分子量は、1000g/mol未満、好ましくは800g/mol未満であろう。
【0044】
好ましい保護基は、ピバロイル基である。本保護基は大型であり、85g/molの分子量を有し、例えば他の非常に大型の基であるトリフェニルメチル基のような結晶メーカーと見なされうる。大きいサイズは、より小型の化合物ではそうであるように糖部分を油のままにしておく代わりに、糖部分を結晶化させる。あるいはジメチルアセチルは、保護基として使用されうる。アセチル基およびメチルアセチルはどちらも小さすぎて結晶メーカー保護基にはなれないが、化合物が少なくとも300g/molの分子量を有する場合に、1または2個のR基がアセチルまたはメチルアセチルであり、残りのR基がジメチルアセチルまたはトリメチルアセチル基でありうることが考慮される。
【0045】
4個のピバロイル基(それぞれ85g/mol)を有する化合物では、R1またはR2位置にヒドロキシル基を有する糖は、516g/molの分子量を有するであろう;R1またはR2の一方がアジドである場合、分子量は541g/molである。これらの化合物のそれぞれが、適切な溶媒から結晶化するであろう。少なくとも300g/molの分子量の化合物も結晶化するであろう。したがって4個のピバロイル基の代わりに4個のジメチルアセチル基(アゾ糖では460および485g/molの比較分子量の場合)を使用して糖が保護される場合、糖は本明細書で述べるように結晶化されうる。
【0046】
同様に、3個のピバロイル基を有する化合物では、R1またはR2位置および分子内の別の箇所に2つのヒドロキシル基を有する糖は、432g/molの分子量を有するであろう;R1またはR2の一方がアジドである場合、分子量は457g/molである。これらの化合物のそれぞれが、適切な溶媒から結晶化するであろう。
【0047】
好ましい実施形態において、テトラピバロイルフラノースが結晶化される。保護反応はモノ、ジ、トリおよびペンタピバロイル誘導体はもちろんのこと、所望のテトラピバロイル誘導体も潜在的に生成する(または代わりにテトラピバロイルが、好ましいペンタピバロイルまたはトリピバロイルに加えて生成される)ので、所望の生成物のみの結晶化がこれらの副生成物/不純物の分離を可能にする。使用される溶媒または溶媒系は、化合物の分子量および極性に基づいて結晶化される特定のテトラピバロイルフラノースに合せて「調整」されうる。
【0048】
保護基の1個以上がピバロイルまたは関連アルキルアセチル基でないことが考慮される。本発明のピバロイルフラノースの一部として含有されうる他の保護基は、糖のヒドロキシル基を誘導する着脱可能な保護基を含む。例えばフラノースは、ピバロイル基4個および他の保護基1個、またはピバロイル基3個および他の保護基2個、またはピバロイル基2個および他の保護基2個、またはピバロイル基3個および他の保護基1個およびヒドロキシル基1個を含有しうる。この種の保護基および誘導体を生成する工程は一般に糖化学において既知であり、これに限定されるわけではないが、直鎖または分岐C1〜C8アルキル、特にC1〜C4アルキル、例えばメチル、エチル、n−プロピル、イソプロピルまたはn−、イソ−およびt−ブチル;C7〜C12アリールアルキル、例えばベンジル、3〜20個の、特に3〜10個の炭素原子を有するトリアルキルシリル、例えばトリメチルシリル、トリエチルシリル、トリ−n−プロピルシリル、イソプロピルジメチルシリル、t−ブチルジメチルシリル、n−オクチルジメチルシリルまたは(1,1,2,2−テトラメチルエチル)−ジメチルシリル;アルデヒドおよびケトンによって糖または糖誘導体の隣接するOHからアセタールまたはケタールを生成することによって得られ、好ましくはそれぞれ2〜12個の、または3〜12個のC原子を含有する置換メチリデン基、例えばC1〜C12アルキリデン、好ましくはC1〜C6アルキリデンおよび特にC1〜C4アルキリデン、またはベンジリデン(エチリデン、1,1−プロピリデン、2,2−プロピリデン、1,1−ブチリデンまたは2,2−ブチリデン);C2〜C12アシル、特にC2〜C8アシル、例えばアセチル、プロパノイル、ブタノイル、ペンタノイル、ヘキサノイルおよびベンゾイル;R5−SO−(式中、R5は、C1〜C12アルキル、特にC1〜C6アルキル、C5シクロアルキル、C6シクロアルキル、フェニル、ベンジルあるいはC7〜C12アルキルフェニル、特にC1〜C4アルキルフェニル、あるいはC1〜C12アルキルベンジル、特にC1〜C4アルキルスルホニルまたはアリールスルホニル、例えばメチルスルホニル、エチルスルホニル、プロピルスルホニル、ブチルスルホニル、フェニルスルホニル、ベンジスルスルホニル、およびp−メチルフェニルスルホニルである)を含む(例えば米国特許第5,218,097号を参照)。1個以上のピバロイラート基に加えて使用される好ましい保護基は、アセチル、ベンジル、シリルまたはトリチルである。
【0049】
本明細書で示した糖構造は、ヘキソフラノース形である。しかしながら結晶性糖は別の形、例えば5員(フラノースにおいてのように)または6員(ピラノースにおいてのように)環形のどちらかおよび開鎖形の環式ヘミアセタールなどの、別の形にも適合しうる。
【0050】
他のピバロイルフラノースは、本発明の結晶化方法によって精製されうる。フラノシドは、異なる開始物質を使用することによる本明細書に記載した方法によっても産生されうる。例えば、糖:アロース、アルトロース、グルコース、マンノース、グロース、イドースおよびタロースのいずれか1つが、結晶性ピバロイルフラノースを産生するための開始物質として使用されうる。本明細書に記載したフラノース化合物のDおよびL系列のどちらも考慮される;最も好ましい立体化学は、D系列を含む。
【0051】
結晶化
本発明の化合物は、結晶性であることが見出されており、糖は一般に粘性液体の形であり、結晶化できないので、糖合成中に普通、必要とされるようなカラムクロマトグラフィーまたはイオン交換樹脂などの文献に記載された精製手順の使用を必要としない。
【0052】
フラノース糖は当分野で周知の方法によって結晶化されうる。溶媒は、極性および糖との反応性がないことに基づいて選択される。結晶化に理想的な溶媒は、糖と反応してはならず、高温時に適度に大量のフラノースを、低温時にはごく少量のフラノースを溶解させねばならない。該溶媒は、糖の融点以下の温度にて沸騰すべきである。使用されうる多数の溶媒がある。一般に、ガラクトースおよびアルトロース糖などのより極性の糖は、C6〜C9アルカンおよびシクロアルカンなどの、より非極性の溶媒から結晶化するであろう。他の糖、例えばアジドによって置換される糖はより極性が低く、結晶化にはメタノールなどのより極性の高い溶媒を使用すべきである。
【0053】
本発明で使用されうる溶媒としては、これに限定されるわけではないが、エタノール、メタノール、プロパノール、n−ヘキサン、シクロヘキサン、ヘプタン、オクタン、テトラヒドロフラン、ジエチルエーテル、酢酸エチル、ジブチルエーテル、ジメチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、塩化メチレン、クロロホルム、四塩化炭素、ジクロロメタン、1,2−ジクロロエタン、1,1,2,2−テトラクロロエタン、ジオキサン、アセトニトリル、ペンタノール、イソプロパノール、ベンゼン、トルエン、キシレン、アセトン、エチレングリコール、およびこれらの溶媒の2つ以上の組合せが挙げられる。
【0054】
溶媒が高温であるか、または沸騰しているときに溶媒に溶媒和される糖の量は、好ましくは5〜60重量%である。さらに好ましくは、溶媒中に重量で10〜50%の、または20〜40%の、または最も好ましくは25〜35%の糖がある。糖を溶媒和させた後に、温度を低下させる。温度は結晶化のために、好ましくは0℃以下に、さらに好ましくは−10℃または−20℃まで低下させる。好ましい場合、種添加が使用されうる。フラノースの結晶化は低速で進行し、低速での進行は、結晶格子中の分子が溶液中の分子と平衡にあるために、結晶構造が成長するときに不純物の排除を可能にする。一実施形態において、結晶化を発生させるために、溶液は−10℃〜−20℃にて約2日間維持される。
【0055】
本発明は、カラムクロマトグラフィーまたはイオン交換樹脂による精製の追加ステップに頼ることなく、フラノース糖を生成する他の方法によって達成されうるよりも高い、好ましくは著しく高い純度レベルで生成される、少なくとも1個のメチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基を有するフラノース糖を提供する。これらの結晶性フラノース糖は実質的により純粋である。本明細書に記載したような結晶化工程は、該結晶化工程が、追加のピバロイラート部分を有する反応副生成物、または未保護基を含む汚染物質からのフラノースの分離を可能にするので好都合である。
【0056】
一実施形態において、粗ピバロイルフラノースは、DMF水溶液などの溶液からの結晶化によって単離される。本溶液は、保護フラノースの生成中に使用可能であり、保護反応を停止させた後に入手取得されるので有用である。DMF溶液からの結晶化は最大約2日間を要しうる。粗生成物がいったん回収されると、粗生成物はヘプタン/酢酸エチルなどの溶液に溶解される。粗生成物は次に、洗浄、乾燥、濃縮および例えばヘプタンからの再結晶化によって精製されうる。この再結晶化工程は、反応で母液中に生成された汚染物質および副生成物(例えばテトラピバロイラートが所望であるとき、ペンタピバロイラートなど)を残すと同時に、所望のピバロイルフラノースが結晶化される。この結晶化は低速でもあり、最大2日間を要しうる。所望ならば本反応で種添加が使用されうる。
【0057】
好ましい実施形態において、テトラピバロイルフラノースの1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)または1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)は、C6〜C9アルカン、例えばヘキサンまたはヘプタンからの結晶化によって単離される。これらのフラノシド生成物は、高い純度で産生されうる。別の好ましい実施形態において、アジドテトラピバロイルフラノースの5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)がメタノールからの結晶化によって単離される。このことは、テトラピバロイルフラノース化合物(II)および(III)で実施されるように、ヘプタンからの結晶化よりも高い純度の生成物を与える。同様のアジド糖、例えば5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−アルトロフラノース、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−ガラクトフラノースも考慮される。
【0058】
DGJの合成
DGJの合成方法において、D−ガラクトースは、参照により本明細書に組み入れられているSantoyo−Gonzalez(1999)に記載されているように、開始物質として使用されうる。本合成での方法としては、N,N−ジメチルホルムアミド(DMF)中でD−ガラクトースに1−(トリメチルアセチル)イミダゾール(ピバロイルイミダゾール)と砂糖を反応させることによって、主生成物としての該保護フラノシド誘導体:1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)と、微量生成物としての1,2,3,5,6−ペンタ−O−ピバロイル−D−ガラクトフラノースのα,β−アノマーの混合物を生成する、D−ガラクトースのヒドロキシル基の、ピバロイル基による保護が挙げられる。ガラクトフラノシドは次に、アルトロフラノシドの1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)に変換される。次にヒドロキシルは保護されて、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)を得るためにアジド基によって置換される。脱保護の後、DGJを得るためにガラクトフラノシド誘導体が還元される。Santoyo−Gonzalezは、3つのフラノシド中間体はもちろんのこと、DJG生成物も精製するためにカラムクロマトグラフィーを使用した。本参考文献に記載されたDGJの合成は、全収率が約20%の最終生成物約200mgの規模でのみ有用である。
【0059】
3つのフラノシド中間体、1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)、1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)、および5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)は共通の溶媒からそれぞれ結晶化されうるので、本発明はDGJの合成の改良された方法を提供する(図1)。中間ステップのそれぞれの間に精製のためにカラムクロマトグラフィーを使用する代わりに、フラノシド中間体は結晶化によって精製されうる。ガラクトフラノシド(IV)は、Santoyo−Gonzalezによって記載された方法などによってDGJを生成するのに使用されうる。
【0060】
アルトロース誘導体の合成
L−アルトロースは、低い全収率で一連の化学反応によって、または細菌Butyrivibrio fibrisolvensから培養された細胞外ポリサッカライドから合成されうる非栄養性甘味料である(米国特許第4,966,845号)。しかしながら、これらの方法は高価である。本発明の結晶性ピバロイルフラノースの使用は、D−ガラクトース誘導体のさらに高価なL−アルトロース誘導体への簡単な変換を可能にする。この変換はクロマトグラフィーによる分離および精製の必要なしに達成されうる(図2)。結晶性1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースは、安価なD−ガラクトースから開始して上記の方法で調製されうる。クリスタリン1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースは後で、ピバロイル保護基(例えばメタノール中でナトリウムメトキシド)を除去するために脱保護反応を受けることができ、純粋なα−L−アルトロ−フラノシドが単離されうる。
【0061】
他の糖の合成
本発明のピバロイルフラノースは、多数の糖および糖誘導体の合成において有用な中間体である。例えばDGJの合成と類似して、D−ガラクトースは、参照により本明細書に組み入れられているSantoyo−Gonzalez(1999)によって記載されているように、(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオールを調製するための開始物質として使用されうる(図3)。結晶性1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)は、上述のように調製されうる。次にヒドロキシルは保護されて、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(V)を得るためにアジド基によって置換される。脱保護の後、イミノ糖を得るために5−アジドアルトロフラノシド誘導体が還元される。フラノシド中間体の1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)および5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(V)はそれぞれ共通の溶媒から結晶化されうるので、本発明はDGJの(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオール異性体の合成の改良された方法を提供する。
【0062】
チオヘキソース、例えばWhistlerによって述べられたチオヘキソースも、本明細書に記載した方法によってピバロイル化および結晶化されうる。(Whistler,J.Org.Chem.,1968,396−8)。
【0063】
D−ガラクトースは、DGJの合成と類似して、(2R,3R,4S,5R,6R)−6−ヒドロキシメチル−テトラヒドロ−チオピラン−2,3,4,5−テトラオール(D−ガラクトチオピラノース)を調製するための開始物質として使用されうる(図4)。1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)は上述のように調製されうる。ヒドロキシルは保護されて、5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)を得るためにベンジルチオ基によって置換される。このテトラピバロイルは、この中間体を精製するために結晶化されうる。脱保護の後、D−ガラクトチオピラノースを得るためにガラクトフラノシド中間体が還元される。
【0064】
本明細書で使用するように、「数キログラム」「数kg」および「予備規模」という用語は、生成物が、1回の合成で生成物1kgを超える、あるいは10kgすら超える量である合成規模を示す。
【0065】
[実施例]
本発明は、本発明の範囲を限定すると解釈されるべきではない次の実施例でさらに例示される。
【0066】
[実施例1]
結晶性1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(II)の製剤およびキャラクタリゼーション
【0067】
1−(トリメチルアセチル)イミダゾール(ピバロイルイミダゾール)(42.2kg、5倍過剰)をDMF(90kg)およびヘプタン(3.4kg)に溶解させて、溶液を60℃まで加温した。D−ガラクトース(10kg)を溶液に注入し、混合物を75℃まで加熱した。反応物を90〜100℃まで発熱させて、発熱が収まった後に反応が完了するまで80〜100℃に維持した。反応の進行はTLC(ヘキサン:酢酸エチル=4:1)によって監視した。進行を描出するために、TLCを後で希硫酸によって染色および加熱した;TLC上での生成物のスポット(Rf=0.5)が主な成分となったときに反応が完了したと見なした。反応が完了した後に、水(200kg)および氷(82kg)を含有する混合物中に反応生成物をただちに移した。粗生成物を本混合物から結晶化によって単離した。本結晶化は低速で、一般に2日間を要する。粗生成物を収集して、ヘプタン/酢酸エチルに溶解させ、水で洗浄して、硫酸マグネシウムによって乾燥させ、濃縮して、2〜3倍量のヘプタン(〜25kg)から−20℃にて再び結晶化させた;本工程は母液にペンタピバロイラートを残した。本ステップの収率は、数kg規模で実施したときに25〜35%(7.2〜10kg)であった。1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(II)は、高純度を有する白色結晶性粉末であった。融点は、105〜108℃の範囲内であった。IR(KBr,cm−1):3432(OH,s),2974(C−H stretch,s),1740(ester of pivaloylate,vs),1284(C−O,weak),1143(C−O,vs),1031(C−O,weak);1HNMR(CDCl3,400MHz,TMS):δ=1.18(s,3H),1.19(s,3H),1.20(s,3H),1.23(s,3H),2.45(d,J=7.9Hz,1H);3.90−3.96(m,1H),4.07(dd,J=3.4Hz,J=6.5Hz,1H),4.13(dd,J=11.6Hz,J=5.3Hz,1H),4.19(dd,J=11.6Hz,J=6.1Hz,1H),5.44(dd,J=7.9Hz,J=4.6Hz,1H),5.62(dd,J=7.9Hz,J=7.0Hz,1H),6.37(d,J=4.6Hz,1H).
【0068】
[実施例2]
結晶性1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)の製剤およびキャラクタリゼーション
【0069】
ピリジン(3.82kg)の塩化メチレン(15L)溶液を窒素雰囲気下で0℃まで冷却した。トリフルオロメタンスルホン酸無水物(3.28kg)を0℃にて滴加し、1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシド(5kg)の塩化メチレン(10L)溶液の滴加を続けた。反応混合物を0℃にて2時間撹拌して、TLC(ヘキサン:酢酸エチル=4:1)によって反応の完了を確認した。反応がこの時点で完了していない場合、トリフルオロメタンスルホン酸無水物の追加量(0.1kg)を添加した。トリフラート化化合物の5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシドが、反応の本段階でガラクトフラノシドから生成された。反応混合物を次に冷6%塩酸(3回30L)、塩水(30L)および7.5%重炭酸ナトリウム溶液(30L)によって洗浄した。N,N−ジイソプロピルエチルアミン(230mL)を次に添加して、反応物を炭酸ナトリウム(1.5kg)上で1時間撹拌した。反応物を濾過して、乾燥まで濃縮した。本質的に純粋な5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースを結晶性固体として単離した。
【0070】
5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースをDMF9.5Lに溶解させ、亜硝酸ナトリウム5当量(1.67kg)と12時間にわたって反応させた。反応物をヘプタン(24L)および酢酸エチル(12L)によって希釈し、濾過して、2%重炭酸塩溶液(40L)に注入した。生成物をヘプタン/酢酸エチルによって抽出し、実施例1で行ったようにヘプタンから結晶化した。1,2,3,6−テトラピバロイル−α−L−アルトロフラノース(III)の収率は35〜45%であった((II)の5kgより2kg)。HPLCは、転化アルコールへの完全な変換を証明した。生成物は、オフホワイト結晶性固体であった。融点109〜112℃,IR(KBr,cm−1):3444(OH,s),2977(C−H stretch,s),1732(ester of pivalate,vs),1481(weak),1284(C−O,weak),1156(C−O,vs),1028(C−O,weak);1H NMR(CDCl3,400MHz,TMS):δ=1.18(s,3H),1.20(s,3H),1.21(s,3H),1.22(s,3H),3.01(d,J=2.45,1H),3.99−4.02m,2H),4.11−4.07(m,1H),4.26(dd,J=12.4Hz,J=2.7Hz,1H),5.43(dd,J=7.3Hz,J=4.6Hz,1H),5.58(dd,J=7.3Hz,J=5.2Hz,1H),6.37(d,J=4.8Hz,1H).
【0071】
[実施例3]
結晶性5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)の製剤およびキャラクタリゼーション
【0072】
トリフラート化化合物の5−トリフルオロメタンスルホニルオキシ−5−デオキシ−1,2,3,6−テトラピバロイル−α−L−アルトロフラノースは、実施例2に記載したような手順で実施例2のアルトロフラノースIII(5kg)から生成した。本化合物にアジ化ナトリウム(1.6kg)をDMF(9.5L)中で反応させた。反応は転化反応中に観察された最適条件を使用して実施した。粗生成物はメタノール(1.3〜1.7mL/g)から2回結晶化させた。5kg規模では、IIIからの5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)の収率は通常、50〜70%(〜3.3kg)であった。生成物は、オフホワイト結晶性固体であった。融点103〜104℃。IR(KBr,cm−1):2090(azide,s),1740(ester of pivalate,vs),1480(weak),1280(C−O,s),1160(C−O,vs),1042(C−O,weak);1HNMR(CDCl3,400MHz,TMS):δ=1.19(s,3H),1.20(s,3H),1.22(s,3H),1.25(s,3H),3.83−3.79(m,1H),4.05(dd,J=6.7,J=4.8Hz 1H),4.15(dd,J=11.7Hz,J=8.0Hz,1H),4.30(dd,J=11.7Hz,J=4.2Hz,1H),5.41(dd,J=7.9Hz,J=4.6Hz,1H),5.59(t,J=7.5Hz,1H),6.33(d,J=4.5Hz,1H).
【0073】
[実施例4]
結晶性5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)
【0074】
実施例3から生成された粗生成物を、上述の結晶化手順を使用してEtOAc:MeOH 1:6およびメタノールから結晶化させた。本結晶性の収率は、50〜60%の5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノース(IV)であった。
【0075】
[実施例5]
結晶性5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノシドの製剤
【0076】
5−ベンジルチオ−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースは、アジ化ナトリウムをナトリウムα−トルエンチオキシドに代えて、実施例3に記載したようにサンプルを結晶化することによって、5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースと同様の方法で調製する。
【0077】
本発明の多くの変形は、上述の説明に照らして当業者にそれ自体を示唆するであろう。例えば、糖の結晶化は、各種の溶媒から実施されうる。そのようなすべての明らかな変形は、添付請求項の十分に意図された範囲内である。当業者は、本開示に照らして、本明細書で開示される具体的な実施形態で変更を実施して、本発明の精神および範囲から逸脱せずに同じまたは同様の結果が得られることを認識すべきである。
【0078】
上記の特許、出願、試験方法、刊行物は、参照によりその全体が本明細書に組み入れられている。
【図面の簡単な説明】
【0079】
【図1】結晶性誘導体II、IIIおよびIVを使用するDGJの合成。
【図2】結晶性誘導体IIおよびIIIを使用するL−アルトロースの合成。
【図3】結晶性誘導体IIおよびVを使用するD−ガラクトーゼからの(2S,3S,4R,5S)−2−ヒドロキシメチル−ピペリジン−3,4,5−トリオールの合成。
【図4】(2R,3R,4S,5R,6R)−6−ヒドロキシメチル−テトラヒドロ−チオピラン−2,3,4,5−テトラオール(D−ガラクトチオピラノース)の合成。
【図5A】0〜14ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
【図5B】0.7〜2.6ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
【図5C】3.8〜6.5ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−D−ガラクトフラノース(II)のプロトンNMR。
【図6】他の異性体(II)からの完全な除去を示す、結晶化1,2,3,6−テトラピバロイル−α−L−アルトロ−フラノース(III)のHPLC。化合物(III)は約27.5分にて溶離するのに対して、関連する異性体(II)は29.0分にて溶離する。
【図7A】0〜14ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図7B】3.8〜6.6ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図7C】0.7〜3.2ppmの、結晶化1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図8A】0〜14ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図8B】3.7〜6.6ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【図8C】0.7〜2.7ppmの、結晶化5−アジド−5−デオキシ−1,2,3,6−テトラ−O−ピバロイル−α−L−アルトロフラノースのプロトンNMR。
【特許請求の範囲】
【請求項1】
式:
【化1】
(式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRは、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択され;
R1およびR2は独立して、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHであり;
各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシル、またはその組合せであり;
ここでフラノースの分子量が少なくとも300g/mol〜1000g/molである)
の結晶性フラノース。
【請求項2】
前記フラノースが少なくとも350g/molの分子量を有する、請求項1に記載の結晶性フラノース。
【請求項3】
前記フラノースが少なくとも400g/molの分子量を有する、請求項2に記載の結晶性フラノース。
【請求項4】
前記フラノースが少なくとも450g/molの分子量を有する、請求項3に記載の結晶性フラノース。
【請求項5】
少なくとも3個のR基がトリメチルアセチルである、請求項1に記載の結晶性フラノース。
【請求項6】
前記フラノースがテトラピバロイルフラノースである、請求項5に記載の結晶性フラノース。
【請求項7】
R1がOHまたはN3であり、R2がHである、請求項1に記載の結晶性フラノース。
【請求項8】
前記フラノースが1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースまたは1,2,3,6−テトラピバロイル−α−L−アルトロフラノースである、請求項1に記載の結晶性フラノース。
【請求項9】
前記フラノースが5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースである、請求項1に記載の結晶性フラノース。
【請求項10】
式:
【化2】
(式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRは、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択され;
R1およびR2は、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHであり;
各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシル、またはその組合せであり;
ここでフラノースの分子量が少なくとも300g/mol〜1000g/molである)によって表される結晶性フラノースを産生する方法であって、
フラノースを溶媒に添加する、またはフラノースを溶媒中で生成するステップと、前記溶媒から前記フラノースを結晶化させるステップと、
を備える方法。
【請求項11】
前記フラノースが少なくとも350g/molの分子量を有する、請求項10に記載の方法。
【請求項12】
前記フラノースが少なくとも400g/molの分子量を有する、請求項11に記載の方法。
【請求項13】
前記フラノースが少なくとも450g/molの分子量を有する、請求項12に記載の方法。
【請求項14】
少なくとも3個のR基がトリメチルアセチルである、請求項10に記載の方法。
【請求項15】
前記フラノースがテトラピバロイルフラノースである、請求項16に記載の方法。
【請求項16】
前記テトラピバロイルフラノースに加えてモノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースの少なくとも1つが生成され、前記テトラピバロイルフラノースが結晶化されるときに前記モノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースが結晶化されない、請求項15に記載の方法。
【請求項17】
R1がOHであり、R2がHである、請求項10に記載の方法。
【請求項18】
前記溶媒がヘプタンを含む、請求項17に記載の方法。
【請求項19】
R1がN3であり、R2がHである、請求項10に記載の方法。
【請求項20】
前記溶媒がメタノールを含む、請求項19に記載の方法。
【請求項21】
前記結晶化させるステップが、前記溶媒系を冷却する工程、外部冷却源を用いずに前記溶媒を冷却させる工程、種結晶を添加する工程、前記フラノースを溶液から析出させるために追加の溶媒または溶媒系を添加する工程、またはその組合せを備える、請求項10に記載の方法。
【請求項22】
前記結晶化させるステップが最初に前記フラノースおよび前記溶媒を前記溶媒の沸点付近の温度まで加熱する工程と、次に−20℃〜−10℃の温度まで冷却する工程と、少なくとも36時間待機する工程とを備える、請求項21に記載の方法。
【請求項23】
第2の溶媒であって、前記溶媒と混和性であり、前記フラノースを溶解させることができる第2の溶媒を添加するステップと、
前記フラノースの前記結晶形を得るために、溶液に結晶化処理を受けさせるステップと、
をさらに備える、請求項10に記載の方法。
【請求項1】
式:
【化1】
(式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRは、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択され;
R1およびR2は独立して、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHであり;
各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシル、またはその組合せであり;
ここでフラノースの分子量が少なくとも300g/mol〜1000g/molである)
の結晶性フラノース。
【請求項2】
前記フラノースが少なくとも350g/molの分子量を有する、請求項1に記載の結晶性フラノース。
【請求項3】
前記フラノースが少なくとも400g/molの分子量を有する、請求項2に記載の結晶性フラノース。
【請求項4】
前記フラノースが少なくとも450g/molの分子量を有する、請求項3に記載の結晶性フラノース。
【請求項5】
少なくとも3個のR基がトリメチルアセチルである、請求項1に記載の結晶性フラノース。
【請求項6】
前記フラノースがテトラピバロイルフラノースである、請求項5に記載の結晶性フラノース。
【請求項7】
R1がOHまたはN3であり、R2がHである、請求項1に記載の結晶性フラノース。
【請求項8】
前記フラノースが1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースまたは1,2,3,6−テトラピバロイル−α−L−アルトロフラノースである、請求項1に記載の結晶性フラノース。
【請求項9】
前記フラノースが5−アジド−5−デオキシ−1,2,3,6−テトラピバロイル−α−D−ガラクトフラノースである、請求項1に記載の結晶性フラノース。
【請求項10】
式:
【化2】
(式中、各Rは独立して、H、アセチル、メチルアセチル、ジメチルアセチル、トリメチルアセチル、または保護基であり、少なくとも2個のRは、メチルアセチル、ジメチルアセチル、およびトリメチルアセチルから成る群より選択され;
R1およびR2は、H、OH、OR3、N3、NH2、NHR3、NR32、SH、SR3、OS(=O)2R3、C(=O)R3、メチルアセトキシ、ジメチルアセトキシ、トリメチルアセトキシ、アセトキシ、クロロアセトキシ、ジクロロアセトキシ、トリクロロアセトキシまたはO保護基であり、ここでR1およびR2の少なくとも1つがHであり;
各R3は独立して、Hあるいは置換または非置換C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C5〜C6シクロアルキル、C5〜C12シクロアルケニル、C5〜C12アリール、C4〜C12ヘテロアリール、C6〜C12アリールアルキル、C4〜C12複素環、C6〜C12複素環アルキル、C5〜C12ヘテロアリールアルキルまたはC2〜C12アシル、またはその組合せであり;
ここでフラノースの分子量が少なくとも300g/mol〜1000g/molである)によって表される結晶性フラノースを産生する方法であって、
フラノースを溶媒に添加する、またはフラノースを溶媒中で生成するステップと、前記溶媒から前記フラノースを結晶化させるステップと、
を備える方法。
【請求項11】
前記フラノースが少なくとも350g/molの分子量を有する、請求項10に記載の方法。
【請求項12】
前記フラノースが少なくとも400g/molの分子量を有する、請求項11に記載の方法。
【請求項13】
前記フラノースが少なくとも450g/molの分子量を有する、請求項12に記載の方法。
【請求項14】
少なくとも3個のR基がトリメチルアセチルである、請求項10に記載の方法。
【請求項15】
前記フラノースがテトラピバロイルフラノースである、請求項16に記載の方法。
【請求項16】
前記テトラピバロイルフラノースに加えてモノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースの少なくとも1つが生成され、前記テトラピバロイルフラノースが結晶化されるときに前記モノピバロイル、ジピバロイル、トリピバロイル、またはペンタピバロイルフラノースが結晶化されない、請求項15に記載の方法。
【請求項17】
R1がOHであり、R2がHである、請求項10に記載の方法。
【請求項18】
前記溶媒がヘプタンを含む、請求項17に記載の方法。
【請求項19】
R1がN3であり、R2がHである、請求項10に記載の方法。
【請求項20】
前記溶媒がメタノールを含む、請求項19に記載の方法。
【請求項21】
前記結晶化させるステップが、前記溶媒系を冷却する工程、外部冷却源を用いずに前記溶媒を冷却させる工程、種結晶を添加する工程、前記フラノースを溶液から析出させるために追加の溶媒または溶媒系を添加する工程、またはその組合せを備える、請求項10に記載の方法。
【請求項22】
前記結晶化させるステップが最初に前記フラノースおよび前記溶媒を前記溶媒の沸点付近の温度まで加熱する工程と、次に−20℃〜−10℃の温度まで冷却する工程と、少なくとも36時間待機する工程とを備える、請求項21に記載の方法。
【請求項23】
第2の溶媒であって、前記溶媒と混和性であり、前記フラノースを溶解させることができる第2の溶媒を添加するステップと、
前記フラノースの前記結晶形を得るために、溶液に結晶化処理を受けさせるステップと、
をさらに備える、請求項10に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5A】

【図5b】
【図5c】
【図6】
【図7A】
【図7b】
【図7c】
【図8A】
【図8b】
【図8c】
【図2】
【図3】
【図4】
【図5A】
【図5b】
【図5c】
【図6】
【図7A】
【図7b】
【図7c】
【図8A】
【図8b】
【図8c】
【公表番号】特表2008−543784(P2008−543784A)
【公表日】平成20年12月4日(2008.12.4)
【国際特許分類】
【出願番号】特願2008−516015(P2008−516015)
【出願日】平成18年6月8日(2006.6.8)
【国際出願番号】PCT/US2006/022755
【国際公開番号】WO2006/133447
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(507170099)アミカス セラピューティックス インコーポレイテッド (21)
【Fターム(参考)】
【公表日】平成20年12月4日(2008.12.4)
【国際特許分類】
【出願日】平成18年6月8日(2006.6.8)
【国際出願番号】PCT/US2006/022755
【国際公開番号】WO2006/133447
【国際公開日】平成18年12月14日(2006.12.14)
【出願人】(507170099)アミカス セラピューティックス インコーポレイテッド (21)
【Fターム(参考)】
[ Back to top ]