
結核菌感染の予防または治療のための新規方法
【課題】活発なまたは潜伏中の結核菌(M. tuberculosis)感染の再活性化を防止する方法、および結核菌感染に対する化学療法レジメンの期間を短縮する方法の提供。
【解決手段】Mtb72f融合タンパク質をコードする核酸、またはMtb72f融合タンパク質もしくはその免疫原性断片を、例えばアジュバントと共に含んでなる医薬組成物の投与。
【解決手段】Mtb72f融合タンパク質をコードする核酸、またはMtb72f融合タンパク質もしくはその免疫原性断片を、例えばアジュバントと共に含んでなる医薬組成物の投与。
【発明の詳細な説明】
【技術分野】
【0001】
発明の属する分野
本発明は、哺乳動物における結核菌(M. tuberculosis)感染の再活性化を予防または治療する方法および結核菌感染に対する化学療法の期間を短縮する方法に関する。
【背景技術】
【0002】
発明の背景
結核は、結核菌および他のマイコバクテリア属(Mycobacterium)の種の感染によって引き起こされる慢性感染症である。それは開発途上国における主要な疾患であり、そしてもちろん世界の先進地域においても高まりつつある問題であり、毎年約8百万の新規症例および3百万の死がもたらされている。該感染は、かなりの期間、無症候性であることがあるが、最も一般的には、該疾患は急性肺炎として現れ、発熱および乾性咳嗽を生じる。治療しないと、典型的に重大な合併症および死にいたる。
【0003】
結核は、一般に、長期の抗生物質治療を使用して抑制できるが、そのような治療は該疾患の伝播を防ぐために十分でない。感染個体は、ある期間、無症候性であるが伝染性であることがある。さらに、治療レジメンのコンプライアンスが重要であるが、患者の行動をモニターすることが困難である。治療過程を完了しない患者があり、その結果、治療が無効になり、薬物耐性が発生しうる。全治療過程を完了した場合でさえ、結核菌の感染は感染個体から根絶されず、潜伏感染として残り、それは再活性化しうる。
【0004】
結核の伝播を抑制するために、有効なワクチン接種および疾患の正確な早期診断が最も重要である。現在、生細菌でのワクチン接種が、防御免疫を誘発するための最も有効な方法である。この目的で使用される最も一般的なマイコバクテリウムは、M. bovisの弱毒性株であるカルメット・ゲラン桿菌(BCG)である。しかし、BCGの安全性および効力は論議の種であり、いくつかの国、例えば米国は、一般の人々にこの物質をワクチン接種しない。
【0005】
結核の診断は、一般に、皮膚検査を使用して達成され、その検査は、ツベルクリンPPD(タンパク質精製誘導体)に対する皮内曝露を伴う。抗原特異的T細胞応答によって、注射後48〜72時間までに注射部位で測定可能な硬化が生じ、マイコバクテリア抗原に対する曝露が示される。しかし、この検査に関して、感度および特異性が問題となっており、BCGでワクチン接種された個体は感染個体と識別することができない。
【0006】
マクロファージがマイコバクテリウム免疫の主要なエフェクターとして機能することが示されている一方、T細胞は該免疫の最も重要な誘導因子である。マイコバクテリウム感染に対する防御におけるT細胞の必須の役割は、マイコバクテリウム感染がAIDS患者において頻繁に出現することによって説明される。それは、ヒト免疫不全ウイルス(HIV)感染と関連するCD4+T細胞の枯渇に起因する。マイコバクテリウム反応性CD4+T細胞は、γ−インターフェロン(IFN−γ)の強力な生産者であることが示されていて、γ−インターフェロンは、次いで、マウスにおけるマクロファージの抗マイコバクテリア効果を惹起することが示されている。ヒトにおけるIFN−γの役割は明らかではないが、研究では、1,25−ジヒドロキシ−ビタミンD3が、単独で、あるいはIFN−γまたは腫瘍壊死因子−αとの組み合わせで、ヒトマクロファージを活性化させて、結核菌感染を阻害することが示されている。さらにまた、IFN−γがヒトマクロファージを刺激して、1,25−ジヒドロキシ−ビタミンD3を生成させることが知られている。同様に、インターロイキン−12(IL−12)は、結核菌感染に対する抵抗性を刺激することに役割を果たすことが示されている。結核菌感染の免疫学についての総説としては、Chan & Kaufmann, Tuberculosis: Pathogenesis, Protection and Control (Bloom ed., 1994), Tuberculosis (2nd ed., Rom and Garay, eds., 2003), およびHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Chan & Kaufmann, Tuberculosis: Pathogenesis, Protection and Control (Bloom ed., 1994)
【非特許文献2】Tuberculosis (2nd ed., Rom and Garay, eds., 2003)
【非特許文献3】Harrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
活発な感染および潜伏感染の両者に由来する結核菌(Mycobacterium tuberculosis)感染の再活性化を防止するための有効な治療ストラテジーに関する必要性が依然として存在する。本発明は、この必要性および他の必要性を満たす。
【課題を解決するための手段】
【0009】
列挙される配列の説明
配列番号1:N末端の6Hisタグを有するMtb72f(DNA)。
【0010】
配列番号2:N末端の6Hisタグを有するMtb72f(タンパク質)。
【0011】
配列番号3:N末端の2His挿入物を有するM72(Mtb72fの変異体)(DNA)。
【0012】
配列番号4:N末端の2His挿入物を有するM72(Mtb72fの変異体)(タンパク質)。
【0013】
配列番号5:N末端のHis挿入物を有さないMtb72f(DNA)。
【0014】
配列番号6:N末端のHis挿入物を有さないMtb72f(タンパク質)。
【0015】
本発明の概要
本発明は、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片を、例えば1以上のアジュバントと共に含んでなる医薬組成物を提供する。該アジュバントには、AS01BおよびAS02Aが含まれる。
【0016】
本発明は、Mtb72f融合タンパク質またはその免疫原性断片を、例えば1以上のアジュバントと共に投与するか、あるいはMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を投与すると、活性または不活性な結核菌感染の再活性化を予防または治療できるという発明者らの発見に部分的に基づく。好ましい実施形態では、Mtb72f融合タンパク質または核酸を、結核菌感染に対して有効な1以上の化学療法剤と共に投与する。
【0017】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を予防または治療する。
【0018】
別の態様では、被験体での結核の再活性化を防止するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、発現されたMtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を予防または治療する。
【0019】
別の態様では、結核菌感染に対する化学療法の期間を減少させるための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤および、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質またはその免疫原性断片は結核菌に対する免疫応答を誘導することにより、結核菌感染に対する化学療法の期間を減少させることが可能である。結核菌感染に対する化学療法の期間を短縮することによって、本方法は、結核菌感染に関する治療対象の個体が全治療過程を完了するコンプライアンスの向上に有効でもある。
【図面の簡単な説明】
【0020】
【図1】図1は、Swiss Websterマウス(SWR/J)における結核菌の再活性化モデルを図示する。この図面では、感染、化学療法処置(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)、免疫化、および菌数(bacterial load)/コロニー形成単位(CFU)の算出の時点を示す。
【図2】図2は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIgG1およびIgG2a抗体応答免疫応答を示す。マウスは、無処置のままか、化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)で処置するか、あるいは化学療法で処置し、かつアジュバントを伴わずに製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。最後の免疫化の10日後にマウスから採血し、血清を、IgG1(赤色)およびIgG2a(黒色)同位体の両者に関する抗Mtb72f抗体応答に関してELISAによって試験した。
【図3】図3は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIgG1およびIgG2a抗体応答免疫応答を示す。マウスは、無処置のままか、化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)で処置するか、あるいは化学療法で処置し、かつアジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。最後の免疫化の10日後にマウスから採血し、血清を、IgG1(赤色)およびIgG2a(黒色)同位体の両者に関する抗Mtb72f抗体応答に関してELISAによって試験した。
【図4】図4は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスにおけるインターフェロン−γ(IFN−γ)応答を示す。種々の時点でマウスから脾臓細胞を取得し、記載のようにin vitroで10μg/mlのrMtb72fまたは構成要素(Mtb32CおよびMtb39)で3日間刺激した。対照として、PPD(3μg/ml)、BCG溶解物(10μg/ml)、conA(3μg/ml)または培地のみでも脾細胞培養物を刺激した。次いでELISAによってIFN−γ生産を測定した。
【図5】図5は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIFN−γ応答を示す。種々の時点でマウスから脾臓細胞を取得し、記載のようにin vitroで10μg/mlのrMtb72fまたは構成要素(Mtb32CおよびMtb39)で3日間刺激した。対照として、PPD(3μg/ml)、BCG溶解物(10μg/ml)、conA(3μg/ml)または培地のみでも脾細胞培養物を刺激した。次いでELISAによってIFN−γ生産を測定した。
【図6A】図6は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるCD4+T細胞およびIFN−γサイトカイン応答を示す。種々の時点でマウスから脾臓細胞を取得し、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4およびIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色(ICS)によって測定した。
【図6B】図6は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるCD4+T細胞およびIFN−γサイトカイン応答を示す。種々の時点でマウスから脾臓細胞を取得し、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4およびIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色(ICS)によって測定した。
【図7】図7は、Mtb感染後120日の時点でのCD4+およびCD8+T細胞特異的IFN−γ+生産の値についての表形式の概要を示す。無処置のままか、30日間、60日間もしくは90日間の併用化学療法で処置、またはMtb72fワクチンの補助療法として併用化学療法で処置されたマウスの群から脾臓細胞を取得した。脾細胞を、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4、CD8またはIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+およびCD8+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色によって測定した。
【図8】図8は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスの生存率を示す。50〜100CFUのMtbH37Rvを含むエアロゾルでマウスを感染させ、30日後、一部のマウスにおいて化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)を開始した。化学療法は60日間継続した。化学療法を受けたマウスの半数を、アジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。
【図9】図9は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスの生存率を示す。50〜100CFUのMtbH37Rvを含むエアロゾルでマウスを感染させ、30日後、一部のマウスにおいて化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)を開始した。別々の群のマウスにおいて30日間、60日間または90日間、化学療法を継続した。化学療法を受けたマウスの半数を、アジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。
【発明を実施するための形態】
【0021】
具体的な実施形態の詳細な説明
本発明は、活性または不活性な(すなわち潜伏性の)マイコバクテリウム感染の再活性化を治療するか、予防するか、あるいは遅延させるために有用なMtb72f核酸または融合タンパク質およびアジュバントを含んでなる組成物、ならびにそれらの使用のための方法に関する。より具体的には、本発明の組成物は、結核菌群のマイコバクテリア属の種、例えばM. tuberculosis、M. bovis、またはM. africanum等の種、または環境性または日和見性であり、かつ免疫不全宿主(例えばAIDS患者)において日和見感染、例えば肺感染を引き起こすマイコバクテリア属の種、例えばBCG、M. avium、M. intracellulare、M. celatum、M. genavense、M. haemophilum、M. kansasii、M. simiae、M. vaccae、M. fortuitum、およびM. scrofulaceumに由来する成分を有するMtb72f融合ポリペプチドもしくはその免疫原性断片またはMtb72f融合ポリペプチドもしくはその免疫原性断片をコードする核酸を含む(例えばHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと)。本出願の発明者らは、驚くべきことに、Mtb72f融合ポリペプチドまたはMtb72f融合ポリペプチドをコードする核酸、またはその免疫原性断片を含んでなる組成物が、結核菌感染の再活性化を治療し、予防し、あるいは遅延させるのに有用であることを発見した。好ましい実施形態では、Mtb72f融合ポリペプチドまたは核酸を1以上の化学療法剤と共に投与する。したがって、これらの組成物、ポリペプチド、およびそれらをコードする核酸は、哺乳動物において、疾患症状の再活性化に対して防御性である免疫応答を誘発するために有用である。
【0022】
本発明のMtb72f核酸および融合ポリペプチドは、それらの抗原性を増強し、あるいは他の態様におけるこれらの抗原を改善するように設計された他の成分をさらに含みうる。例えば、該抗原の一端に対して一続きのヒスチジン残基を付加することによって、該融合ポリペプチド抗原の単離の改善を促してよい。本発明の組成物、ポリペプチド、および核酸は、追加コピーの抗原、すなわちマイコバクテリア属の種に由来する追加の異種性ポリペプチド、例えばMTB8.4抗原、MTB9.8抗原、MTB9.9抗原、MTB40抗原、MTB41抗原、ESAT−6抗原、MTB85複合抗原、α−結晶性抗原、またはNS1抗原を含みうる。あるいは、またはさらに、本発明の組成物、ポリペプチド、および核酸は、マイコバクテリア属の種に由来する他の抗原の追加のコピー、例えばAg85BまたはMTCC#2を含みうる。本発明の組成物、ポリペプチド、および核酸は、また、他の供給源に由来する追加のポリペプチドを含みうる。例えば、本発明の組成物および融合タンパク質には、ポリペプチドまたはポリペプチドをコードする核酸が含まれうる。該ポリペプチドは、抗原、例えばNS1、インフルエンザウイルスタンパク質の発現を増強するものである(例えばWO99/40188およびWO93/04175を参照のこと)。本発明の核酸は、選択された生物種、例えばヒトにおけるコドン優先度に基づいて遺伝子操作することができる。
【0023】
Mtb72f融合タンパク質組成物は、通常、1以上のアジュバント、例えばAS01B(リポソーム製剤中のモノホスホリル脂質A(MPL)およびQS21;米国特許公開第2003/0143240号を参照のこと);AS02A(3D−MPLおよびQS21および水中油型エマルジョン;Bojang, et al., Lancet (2001) 358:1927を参照のこと);ENHANZYN(Detox);3D−MPL;サポニン、例えばQuil Aおよびその成分、例えばQS21およびサポニン模倣物;CWS;TDM;AGP;免疫賦活性オリゴヌクレオチド(oligonucleoptides)、例えばCPG;Leif;およびその誘導体を含む。好ましい実施形態では、リポソーム製剤中の3D−MPLおよびQS21、例えばAS01BおよびMPLおよびQS21および水中油型エマルジョン(例えばAS02A)からなる群から選択される1以上のアジュバントと共にMtb72f融合ポリペプチドを投与する。アジュバントAS01BおよびAS02Aは、Pichyangkul, et al., Vaccine (2004) 22:3831-40にさらに記載されている。
【0024】
Mtb72f抗原を核酸として送達する場合、例えばウイルスベクター(すなわちアデノウイルスベクター)中で、または突然変異体細菌宿主細胞(すなわち突然変異体、弱毒性マイコバクテリウム、ラクトバシラス属(Lactobacillus)またはバシラス属(Bacillus)宿主細胞、例えばカルメット・ゲラン桿菌(BCG)およびLactococcus lactis)中で送達することができる。
【0025】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止する。本発明の方法を実施することによって、結核菌感染の再活性化を遅延させることができる(例えば数か月、数年の期間または無期限に)。
【0026】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、発現されたMtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止する。
【0027】
一実施形態では、活発な結核菌感染を有する個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、不活性すなわち潜伏性の結核菌感染を有する個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、結核菌の多剤耐性株に感染している個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、以前にカルメット・ゲラン桿菌(BCG)で免疫化されている個体にMtb72f核酸または融合タンパク質を投与する。
【0028】
いくつかの実施形態では、Mtb72f核酸または融合タンパク質を、結核菌感染に対して有効な1以上の化学療法剤と共に投与する。そのような化学療法剤の例には、非限定的に、アミカシン、アミノサリチル酸、カプレオマイシン、サイクロセリン、エタンブトール、エチオナミド、イソニアジド、カナマイシン、ピラチナミド、リファマイシン(すなわちリファンピン、リファペンチンおよびリファブチン)、ストレプトマイシン、オフロキサシン、シプロフロキサシン、クラリスロマイシン、アジスロマイシンおよびフルオロキノロンが含まれる。そのような化学療法は、好ましい薬物の組み合わせを使用するよう担当医の判断によって決定される。薬剤耐性でない結核菌感染を治療するために使用される「第一選択」の化学療法剤には、イソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミドが含まれる。1以上の「第一選択」の薬物に対して薬剤耐性を示している結核菌感染を治療するために使用される「第二選択」の化学療法剤には、オフロキサシン、シプロフロキサシン、エチオナミド、アミノサリチル酸、サイクロセリン、アミカシン、カナマイシンおよびカプレオマイシンが含まれる。
【0029】
Mtb72f核酸または融合タンパク質は、結核菌感染に対して有効な1以上の化学療法剤の投与前に、その投与と同時に、あるいはその投与後に投与することができる。一実施形態では、1以上の化学療法剤の投与を開始した約2週間後にMtb72f核酸または融合タンパク質を投与する。1以上の化学療法剤を、一般に、例えば約1週間、2週間、3週間、または4週間、2ヵ月間、3ヵ月間、4ヵ月間、5ヵ月間、6ヵ月間、または8ヵ月間、1年間またはそれ以上の期間にわたって投与する。
【0030】
特定の実施形態では、カルメット・ゲラン桿菌(BCG)を投与することによってMtb72f核酸または融合タンパク質の効果を増強する。
【0031】
いくつかの実施形態では、Mtb72f核酸または融合ポリペプチドの初回免疫すなわち最初の投与に続いて、Mtb72f核酸または融合ポリペプチドの1回以上の「追加免疫」すなわち後の投与を行う(「初回免疫および追加免疫」法)。例えば、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f核酸または融合タンパク質の1回以上の後の投与を行う。一実施形態では、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f融合ポリペプチドの1回以上の後の投与を行う。一実施形態では、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f核酸の1回以上の後の投与を行う。通常、最初の、すなわち「初回免疫」の投与および次の、すなわち「追加免疫」の投与は、約2〜12週間をあけて、あるいは4〜6か月までの期間をあけて行う。その後の「追加免疫」投与は約6か月をあけて、あるいは1、2、3、4または5年もの長期間をあけて行う。慣用の追加免疫処置(例えばタンパク質初回免疫投与、その後のタンパク質追加免疫投与)は、また、結核菌の再活性化に対する予防または治療において有用である。
【0032】
別の態様では、結核菌感染に対する化学療法の期間を減少させるか、あるいは短縮するための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤、ならびに結核菌群のマイコバクテリア属の種に由来するMtb72f融合ポリペプチドまたはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合ポリペプチドは結核菌に対する免疫応答を誘導することにより、結核菌感染に対する化学療法の期間を減少させるか、あるいは短縮することが可能である。通常、Mtb72f核酸または融合ポリペプチドの投与により、6か月、5か月、4か月、3か月、またはそれ未満以内に結核菌感染に対する有効な化学療法処置が可能になる。
【0033】
該Mtb72f組成物は、通常、ヒトに投与するが、他の哺乳動物においても有効であり、該哺乳動物には、家畜哺乳動物(すなわちイヌ、ネコ、ウサギ、ラット、マウス、モルモット、ハムスター、チンチラ)および農業用哺乳動物(すなわちウシ、ブタ、ヒツジ、ヤギ、ウマ)が含まれる。
【0034】
その最も一般的な関連において、本発明のMtb72f融合タンパク質は、3抗原Ra12−TbH9−Ra35のそれぞれの少なくとも免疫原性断片を含むタンパク質である。
【0035】
本出願の命名法では、Ra35とは、Mtb32A(Ra35FL)のN末端を表し、それは少なくとも結核菌に由来するMtb32Aの最初の約205アミノ酸を含み、そのヌクレオチドおよびアミノ酸配列は米国特許出願第09/597,796号の図4に開示され、あるいは別のマイコバクテリア属の種に由来する対応する領域である。最も典型的には、Ra35とは、本出願で開示される配列番号2の残基535−729に対応する部分を表す。あるいは、それは、配列番号2の710位に対応するアミノ酸SerがAlaに置換されているRa35に関する変異体を表す。
【0036】
Ra12とは、Mtb32A(Ra35FL)のC末端を表し、それは少なくとも結核菌に由来するMTB32Aの最後の約132アミノ酸を含み、その配列は米国特許出願第09/072,967号の配列番号4(DNA)および配列番号66(予測されるアミノ酸配列)として開示され、あるいは別のマイコバクテリア属の種に由来する対応する領域である。最も典型的には、Ra12とは、本出願で開示される配列番号2の残基8−139に対応する部分を表す。
【0037】
Mtb39(TbH9)とは、基本的には、米国特許出願第08/658,800号、第08/659,683号、同第08/818,112号、および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号106(完全長cDNA)および配列番号107(完全長タンパク質)として開示されている配列を表す。該配列は、米国特許出願第09/056,559号の配列番号33(DNA)および配列番号91(アミノ酸)としても開示されている。最も典型的には、TbH9とは、本出願で開示される配列番号2の残基143−532に対応する部分を表す。
【0038】
以下に、本発明の組成物および融合タンパク質中で使用されるいくつかの個別の抗原の配列を提供する。
【0039】
Mtb32A(TbRa35FLまたはRa35FL)。その配列は米国特許出願第08/523,436号、同第08/523,435号、同第08/658,800号、同第08/659,683号、同第08/818,112号、同第09/056,556号、および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号17(cDNA)および配列番号79(タンパク質)として開示されている。さらにSkeiky et al., Infection and Immunity 67:3998-4007 (1999)を参照のこと。
【0040】
以下に、本発明のいくつかの融合タンパク質の配列を提供する。
【0041】
TbH9−Ra35(Mtb59F)。その配列は米国特許出願第09/287,849号およびPCT/US99/07717に配列番号23(cDNA)および配列番号24(タンパク質)として開示されている。
【0042】
Ra12−TbH9−Ra35(Mtb72f)。その配列は、本出願の配列番号1または配列番号5(DNA)および配列番号2または配列番号6(タンパク質)として、ならびに米国特許出願第09/223,040号、およびPCT/US99/07717に開示されている。配列番号1および配列番号2の配列には、6His残基のHisタグが含まれる。
【0043】
Mtb72fの突然変異体であるM72は、配列番号2の710位に対応するアミノ酸におけるセリン残基がAlaに変更され(ならびに4His残基がN末端のHisタグから除去され)ていて、その配列は本出願において配列番号3(DNA)および配列番号4(タンパク質)として開示されている。タンパク質が6His残基のHisタグを有するこれらの配列に関する変異体が、米国特許出願第09/597,796号およびPCT/US01/19959に開示されている。Ser710がAlaに置換されているために、M72は自己融解に対してMtb72fより抵抗性であると考えられる。
【0044】
以下に、本発明の組成物および融合タンパク質中で使用されるいくつかの追加の抗原の配列を提供する。
【0045】
Mtb8.4(DPV)。その配列は米国特許出願第08/658,800号、同第08/659,683号、同第08/818,112号および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号101(cDNA)および配列番号102(タンパク質)として開示されている。
【0046】
Mtb9.8(MSL)。その配列は米国特許出願第08/859,381号、同第08/858,998号、同第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号12(DNA)、配列番号109(予測されるアミノ酸配列)および配列番号110〜124(ペプチド)として開示されている。
【0047】
Mtb9.9A(MTI(MTI−Aとしても知られている))。その配列は米国特許出願第08/859,381号、同第08/858,998号、同第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号3および配列番号4(DNA)および配列番号29および配列番号51〜66(MTIのORFペプチド)として開示されている。さらに、2種の他のMTI変異体が存在し、MTI−BおよびMTI−Cと称される。
【0048】
Mtb40(HTCC#1)。その配列は米国特許出願第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号137(cDNA)および138(予測されるアミノ酸配列)として開示されている。
【0049】
Mtb41(MTCC#2)。その配列は米国特許出願第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514出願に配列番号140(cDNA)および配列番号142(予測されるアミノ酸配列)として開示されている。
【0050】
ESAT−6。その配列は米国特許出願第09/072,967号の配列番号103(DNA)および配列番号104(予測されるアミノ酸配列)として開示されている。ESAT−6の配列は米国特許第5,955,077号にも開示されている。
【0051】
α−結晶性抗原。その配列はVerbon et al., J. Bact. 174:1352-1359 (1992)に開示されている。
【0052】
85複合抗原。その配列はContent et al., Infect. & Immunol. 59:3205-3212 (1991)に開示されている。
【0053】
上記各配列はCole et al. Nature 393:537 (1998)にも開示されていて、例えばhttp://www.sanger.ac.ukおよびhttp:/www.pasteur.fr/mycdb/で見出すことができる。
【0054】
上記配列は米国特許出願第08/523,435号、同第08/523,436号、同第08/658,800号、同第08/659,683号、同第08/818,111号、同第08/818,112号、同第08/942,341号、同第08/942,578号、同第08/858,998号、同第08/859,381号、同第09/056,556号、同第09/072,596号、同第09/072,967号、同第09/073,009号、同第09/073,010号、同第09/223,040号、同第09/287,849号およびPCT特許出願PCT/US98/10407、PCT/US98/10514、PCT/US99/03265、PCT/US99/03268、PCT/US99/07717、WO97/09428およびWO97/09429、WO98/16645、WO98/16646に開示されている。各文献は参照により本明細書中に組み入れられる。
【0055】
本明細書中に記載の抗原には、多型変異体および保存的に改変された変異、ならびに株間および種間マイコバクテリウムホモログが含まれる。さらに、本明細書中に記載の抗原には、サブシークエンスまたはトランケート配列が含まれる。融合タンパク質は、追加のポリペプチド、場合によりマイコバクテリウムまたは他の供給源に由来する異種性ペプチドを含有してもよい。これらの抗原は、例えば以下に記載のリンカーペプチド配列を付加することによって改変してよい。これらのリンカーペプチドは、融合タンパク質の各々を構成する1以上の成分の間に挿入してよい。
【0056】
定義
用語「結核の再活性化」とは、ツベルクリン検査で陽性と出るが見かけの疾患症状を有さない個体における疾患症状の後の発現を表す。該個体は結核菌に感染していて、結核が不活性すなわち潜伏期に入るように十分に治療されている活性な疾患症状を以前に示していてもいなくてもよい。しかし、結核の再活性化を予防または治療するための方法は、疾患の活発な症状を示す個体において開始することができる。
【0057】
「一次性結核」とは、結核菌感染に直接続く臨床疾病(疾患症状の発現)を表す。Harrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと。
【0058】
「二次性結核」または「一次性感染後結核」とは、休眠性、不活性すなわち潜伏性の結核菌感染の再活性化を表す。Harrison's Principles of Internal Medicine(上記)を参照のこと。
【0059】
「結核菌の活発(活性)な感染」とは、顕性疾患症状を伴う結核菌感染を表す。
【0060】
「結核菌の不活性、休眠性すなわち潜伏性の感染」とは、顕性疾患症状を伴わない結核菌感染を表す。
【0061】
「薬剤耐性」結核菌感染とは、感染株が、結核菌感染の治療に有効な1以上のいわゆる「第一選択」の化学療法剤(例えばイソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミド)によって、静的な状態にされたり、死滅させられたりしない(それらに耐性の)結核菌感染を表す。
【0062】
「多剤耐性」結核菌感染とは、感染株が結核菌感染の治療に有効な2以上の「第一選択」の化学療法剤に耐性である結核菌感染を表す。
【0063】
「結核菌感染の治療に有効な化学療法剤」とは、当技術分野において公知でかつ結核菌感染の治療に使用されている薬理学的物質を表す。結核菌感染を治療するために使用される例示的薬理学的物質には、非限定的に、アミカシン、アミノサリチル酸、カプレオマイシン、サイクロセリン、エタンブトール、エチオナミド、イソニアジド、カナマイシン、ピラチナミド、リファマイシン(すなわちリファンピン、リファペンチンおよびリファブチン)、ストレプトマイシン、オフロキサシン、シプロフロキサシン、クラリスロマイシン、アジスロマイシンおよびフルオロキノロンが含まれる。薬剤耐性でない結核菌感染を治療するために使用される「第一選択」の化学療法剤には、イソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミドが含まれる。1以上の「第一選択」の薬物に対して薬剤耐性を示している結核菌感染を治療するために使用される「第二選択」の化学療法剤には、オフロキサシン、シプロフロキサシン、エチオナミド、アミノサリチル酸、サイクロセリン、アミカシン、カナマイシンおよびカプレオマイシンが含まれる。そのような薬理学的物質はGoodman and Gilman's The Pharmacological Basis of Therapeutics, Hardman and Limbird eds., 2001のChapter 48で概説されている。
【0064】
「FL」とは、完全長、すなわち野生型ポリペプチドと同じ長さのポリペプチドを表す。
【0065】
「Hisタグ」とは、N末端の、通常、開始Met残基の直後に挿入されているか、あるいはC末端に挿入されている一連のHis残基、典型的に6残基を表す。それらは、通常、天然配列に対して異種性であるが、それらは、固定化金属アフィニティークロマトグラフィー樹脂(IMAC)に対するタンパク質結合を向上させることによって単離を容易にするために組み込まれる。一般的に言えば、Hisタグの存在または不存在は、誘発対象の抗原タンパク質に対する有用な免疫応答を引き起こす観点からは重要でない。Hisタグ自体に対する有害な免疫反応が誘発されるといけないので、Hisタグの長さを最小に、例えば4以下の残基、特に2残基にすることが最良であると見なされる。
【0066】
用語「その免疫原性断片」とは、細胞障害性Tリンパ球、ヘルパーTリンパ球またはB細胞によって認識されるエピトープを含むポリペプチドを表す。典型的に、Mtb72fの免疫原性断片は、500以上のアミノ酸、例えば600以上のアミノ酸、例えば700以上のアミノ酸を含有するポリペプチドである。本発明は、また、Mtb72F融合タンパク質の配列の全体または実質的に全体(例えば500以上のアミノ酸、例えば600以上のアミノ酸、例えば700以上のアミノ酸)を一緒になってカバーする複数の断片、例えば重複断片を包含する。
【0067】
用語「結核菌群のマイコバクテリア属の種」には、結核という疾患を引き起こすと従来より見なされている種、ならびに免疫無防備状態の患者、例えばAIDS患者において結核および肺疾患を引き起こすマイコバクテリウムの環境性および日和見感染性の種が含まれ、例えばM. tuberculosis、M. bovis、またはM. africanum、BCG、M. avium、M. intracellular、M. celatum、M. genavense、M. haemophilum、M. kansasii、M. simiae、M. vaccae、M. fortuitum、およびM. scrofulaceumである(例えばHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005を参照のこと)。
【0068】
アジュバントとは、抗原に対する特定の免疫応答を高めるワクチンまたは治療用組成物中の成分を表す(例えばEdelman, AIDS Res. Hum Retroviruses 8:1409-1411 (1992)を参照のこと)。アジュバントはTh1型およびTh2型応答の免疫応答を誘発する。Th1型サイトカイン(例えばIFN−γ、IL−2、およびIL−12)は投与抗原に対する細胞性免疫応答の誘発に好都合である傾向があり、一方、Th2型サイトカイン(例えばIL−4、IL−5、IL−6、IL−10およびTNF−β)は体液性免疫応答の誘発に好都合である傾向がある。Th1細胞性免疫応答の優先的刺激が可能なアジュバントはWO94/00153およびWO95/17209に記載されている。
【0069】
「核酸」とは、一本鎖または二本鎖型のデオキシリボヌクレオチドまたはリボヌクレオチドおよびそのポリマーを表す。該用語は、公知のヌクレオチドアナログまたは改変された主鎖残基または結合を含有する核酸であって、合成の、天然に存在する、および非天然に存在するものであり、参照核酸と同様の結合特性を有し、かつ参照ヌクレオチドと同様の様式で代謝される核酸を包含する。そのようなアナログの例には、非限定的に、ホスホロチオアート、ホスホルアミダート、メチルホスホナート、キラルメチルホスホナート、2−O−メチルリボヌクレオチド、ペプチド核酸(PNA)が含まれる。
【0070】
特に指定しない限り、具体的核酸配列は、さらに、保存的に改変されたその変異体(例えば縮重コドン置換物)および相補的配列、ならびに明示的に記載されている配列を暗黙のうちに包含する。特に、縮重コドン置換物は、1以上の選択(またはすべての)コドンの第3位が混合塩基および/またはデオキシイノシン残基で置換されている配列を作成することによって達成してよい(Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); Rossolini et al., Mol. Cell. Probes 8:91-98 (1994))。核酸という用語は、遺伝子、cDNA、mRNA、オリゴヌクレオチド、およびポリヌクレオチドと交換可能に使用される。
【0071】
用語「ポリペプチド」、「ペプチド」および「タンパク質」は本明細書中で交換可能に使用され、アミノ酸残基のポリマーを表す。該用語は、1以上のアミノ酸残基が、対応する天然に存在するアミノ酸の人工の化学的模倣物であるアミノ酸ポリマー、ならびに天然に存在するアミノ酸ポリマーおよび非天然に存在するアミノ酸ポリマーに適用される。
【0072】
用語「アミノ酸」とは、天然に存在するアミノ酸および合成アミノ酸、ならびに天然に存在するアミノ酸と同様の様式で機能するアミノ酸アナログおよびアミノ酸模倣物を表す。天然に存在するアミノ酸とは、遺伝暗号によってコードされるアミノ酸、ならびに後に改変されたそれらのアミノ酸、例えばヒドロキシプロリン、γ−カルボキシグルタマート、およびO−ホスホセリンである。アミノ酸アナログとは、天然に存在するアミノ酸と同じ基本化学構造、すなわち水素、カルボキシル基、アミノ基、およびR基に結合しているα炭素を有する化合物を表し、例えばホモセリン、ノルロイシン、メチオニンスルホキシド、メチオニンメチルスルホニウムである。そのようなアナログは改変されたR基(例えばノルロイシン)または改変されたペプチド主鎖を有するが、天然に存在するアミノ酸と同じ基本化学構造を保持する。アミノ酸模倣物とは、アミノ酸の一般的化学構造と異なる構造を有するが、天然に存在するアミノ酸と同様の様式で機能する化学物質を表す。
【0073】
本明細書中で、アミノ酸は、IUPAC-IUB Biochemical Nomenclature Commissionによって推奨されるそれらの一般に知られている3文字記号または1文字記号によって言及されることがある。同様に、ヌクレオチドは、それらの一般に許容される1文字コードによって言及されることがある。
【0074】
「保存的に改変された変異体」は、アミノ酸および核酸配列の両者に適用される。特定の核酸配列に関して、保存的に改変された変異体とは、同一または本質的に同一のアミノ酸配列をコードする核酸を表すか、あるいは該核酸がアミノ酸配列をコードしない場合には、本質的に同一の配列を表す。遺伝暗号の縮重のせいで、多数の機能的に同一の核酸が任意の所定のタンパク質をコードする。例えば、コドンGCA、GCC、GCGおよびGCUはすべてアミノ酸アラニンをコードする。ゆえに、コドンによってアラニンが指定されるすべての位置で、該コドンは、対応する上記コドンのいずれかに改変することができ、コード対象のポリペプチドは変更されない。そのような核酸変異は「サイレント変異」であり、保存的に改変された変異の1種である。ポリペプチドをコードする本明細書中のすべての核酸配列は、該核酸のすべてのサイレント変異候補を同時に記載している。当業者は、核酸中の各コドン(通常メチオニンに関する唯一のコドンであるAUG、および通常トリプトファンに関する唯一のコドンであるTGGを除く)を改変して機能的に同一の分子を得ることができることを認識する。したがって、ポリペプチドをコードする核酸の各サイレント変異は、記載される各配列に暗黙のうちに含まれる。
【0075】
アミノ酸配列に関して、当業者は、コード配列中の単一のアミノ酸または少ないパーセンテージのアミノ酸を変更、付加または欠失する、核酸、ペプチド、ポリペプチド、またはタンパク質配列の個々の置換、欠失または付加が「保存的に改変された変異体」であり、その場合、該改変は、化学的に類似のアミノ酸によるアミノ酸の置換を生じさせることを認識する。機能的に類似のアミノ酸を提供する同類置換表は当技術分野において周知である。そのような保存的に改変された変異体は、本発明の多型変異体、種間ホモログ、および対立遺伝子に追加されるものであり、それらを排除しない。
【0076】
以下の8群は、それぞれ、互いに同類置換であるアミノ酸を含有する:
1)アラニン(A)、グリシン(G);
2)アスパラギン酸(D)、グルタミン酸(E);
3)アスパラギン(N)、グルタミン(Q);
4)アルギニン(R)、リシン(K);
5)イソロイシン(I)、ロイシン(L)、メチオニン(M)、バリン(V);
6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W);
7)セリン(S)、スレオニン(T);および
8)システイン(C)、メチオニン(M)
(例えばCreighton, Proteins (1984)を参照のこと)。
【0077】
用語「異種性」とは、核酸の部分に関して使用される場合、該核酸が、自然界で、互いに同一の関連性において見出されない2以上の部分配列を含むことを示す。例えば、該核酸は典型的に組換え生産され、新規の機能的核酸を作成するように配置された無関係の遺伝子に由来する2以上の配列、例えば1つの供給源に由来するプロモーターおよび別の供給源に由来するコード領域を有する。同様に、異種性タンパク質とは、該タンパク質が、自然界で、互いに同一の関連性において見出されない2以上の部分配列を含むこと(例えば融合タンパク質)を示す。
【0078】
「融合ポリペプチド」または「融合タンパク質」とは、直接またはアミノ酸リンカーを介して、共有結合によって連結された少なくとも2個の異種性のマイコバクテリア属の種のポリペプチドを有するタンパク質を表す。融合タンパク質を形成するポリペプチドは、典型的に、C末端からN末端に連結されるが、それらをC末端からC末端、N末端からN末端、またはN末端からC末端に連結することもできる。融合タンパク質のポリペプチドは任意の順序であってよい。この用語は、また、該融合タンパク質を構成する抗原の保存的に改変された変異体、多型変異体、対立遺伝子、突然変異体、部分配列、および種間ホモログを表す。結核菌抗原はCole et al., Nature 393:537 (1998)に記載されている。該文献は結核菌ゲノム全体を開示している。結核菌の完全配列はhttp://www.sanger.ac.ukおよびhttp://www.pasteur.fr/mycdb/(MycDB)で見出すこともできる。結核菌抗原に対応する他のマイコバクテリア属の種に由来する抗原は、例えば本明細書中に記載の配列比較アルゴリズムまたは当業者に公知の他の方法、例えばハイブリダイゼーションアッセイおよび抗体結合アッセイを使用して特定することができる。
【0079】
本発明において有用な典型的なMtb72f融合タンパク質には以下のものが含まれる:
配列番号2の配列の残基8−729を含むタンパク質;
場合により該配列のHisタグ形成残基2−7を有さないか、あるいは異なる長さのHisタグを有する、配列番号2(=Mtb72f)の配列を含むか、あるいはそれからなるタンパク質;
場合により該配列のHisタグ形成残基2−7を有さないか、あるいは異なる長さのHisタグを有する配列番号2の配列(例えば配列番号2の配列の残基8−729を含むタンパク質)を1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質;
配列番号4(=M72)の配列の残基4−725を含むタンパク質;
場合により該配列のHisタグ形成残基2−3を有さないか、あるいは異なる長さのHisタグを有する、配列番号4(=M72)の配列を含むか、あるいはそれからなるタンパク質;および
場合により該配列のHisタグ形成残基2−3を有さないか、あるいは異なる長さのHisタグを有する配列番号4の配列(例えば配列番号4の配列の残基4−725を含むタンパク質)を1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0080】
本発明において有用な、Mtb72f融合タンパク質の典型的な免疫原性断片には以下のものが含まれる:
TbH9−Ra35(Mtb59F);またはTbH9;またはRa35;またはRa12の配列を含むか、あるいはそれからなるタンパク質;および
該配列を、1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0081】
本発明において有用な、Mtb72f融合タンパク質の追加の典型的な免疫原性断片には以下のものが含まれる:
配列番号2のSer710に対応する位置がAlaに変更されているTbH9−Ra35(Mtb59F)またはRa35の配列を含むか、あるいはそれからなるタンパク質;および
該配列を、1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0082】
より具体的には、Mtb72fは以下のものである:
配列番号2の残基8−729を含むポリペプチド;または
場合により開始Met残基の後ろにHisタグが挿入されている配列番号2の残基1および8−729からなるポリペプチド;または
配列番号2のポリペプチド;または
配列番号4の残基4−725を含むポリペプチド;または
場合により開始Met残基の後ろにHisタグが挿入されている配列番号4の残基1および4−725からなるポリペプチド;または
配列番号4のポリペプチド;または
配列番号6のポリペプチド。
【0083】
追加の典型的なMtb72f融合タンパク質およびその免疫原性断片には、N末端および/またはC末端が例えば5または4または3または2または1アミノ酸残基短縮されている上述のタンパク質が含まれる。
【0084】
追加の典型的なMtb72f融合タンパク質およびその免疫原性断片には、10%までのアミノ酸、例えば5%までのアミノ酸(例えば10まで、例えば5までの)アミノ酸が、本明細書中で規定される同類置換によって置き換えられている上述のタンパク質が含まれる。
【0085】
本発明において用いられる典型的なMtb72f核酸には、上述の典型的なMtb72f融合タンパク質およびその免疫原性断片をコードする核酸(例えばDNA分子)が含まれる。言及することができる1セットの具体的なDNA分子は、配列番号1のヌクレオチド63−2228を含むものである。言及することができる別のセットの具体的なDNA分子は、配列番号3のヌクレオチド10−2175を含むものである。言及することができる具体的なDNA分子は、配列番号1または配列番号3または配列番号5を含むか、あるいはそれからなるものである。
【0086】
用語「融合(fused)」とは、融合タンパク質中の2ポリペプチド間の共有結合による連結を表す。該ポリペプチドは、典型的に、互いに直接的にまたはアミノ酸リンカーを介して、ペプチド結合を介して連結されている。場合により、該ペプチドは、当業者に公知の非ペプチド共有結合性の連結を介して連結することができる。
【0087】
「〜に選択的に(または特異的に)ハイブリダイズする」との言い回しは、該配列が複合混合物(例えば細胞の全DNAもしくはRNA、またはライブラリーのDNAもしくはRNA)中に存在する場合に、ストリンジェントなハイブリダイゼーション条件下で、分子が特定のヌクレオチド配列に対してのみ結合、二重鎖形成、またはハイブリダイズすることを表す。
【0088】
「ストリンジェントなハイブリダイゼーション条件」との言い回しは、プローブが、典型的に核酸の複合混合物中で、その標的部分配列にハイブリダイズするが、他の配列にはハイブリダイズしない条件を表す。ストリンジェントな条件は配列依存的であり、状況ごとに異なる。配列が長いほど、より高い温度で特異的にハイブリダイズする。広範囲にわたる、核酸のハイブリダイゼーションについての指針はTijssen, Techniques in Biochemistry and Molecular Biology-Hybridization with Nucleic Probes, "Overview of principles of hybridization and the strategy of nucleic acid assays" (1993)に見出せる。概して、ストリンジェントな条件は、所定のイオン強度pHで、具体的配列に関する熱融点(Tm)より約5〜10℃低いように選択される。Tmは、平衡状態で、標的に相補的なプローブの50%が標的配列にハイブリダイズする(所定のイオン強度、pH、および核酸濃度条件下での)温度である(標的配列が過剰に存在する場合、Tmでは、平衡状態で50%のプローブが占有される)。ストリンジェントな条件は、塩濃度が、pH7.0〜8.3で約1.0Mナトリウムイオン未満、典型的に約0.01〜1.0Mナトリウムイオン濃度(または他の塩)であり、かつ温度が、短いプローブ(例えば10〜50ヌクレオチド)では少なくとも約30℃および長いプローブ(例えば50ヌクレオチド以上)では少なくとも約60℃である条件である。ストリンジェントな条件は、不安定化物質、例えばホルムアミドを加えることによって達成してもよい。
【0089】
選択的または特異的ハイブリダイゼーションでは、陽性シグナルはバックグラウンドの少なくとも2倍であり、場合によりバックグラウンドハイブリダイゼーションの10倍である。典型的なストリンジェントなハイブリダイゼーション条件は以下の通りであってよい:50%ホルムアミド、5×SSC、および1%SDS、42℃でインキュベート、または、5×SSC、1%SDS、65℃でインキュベートし、0.2×SSC、および0.1%SDS中で65℃で洗浄。
【0090】
ストリンジェントな条件下で互いにハイブリダイズしない核酸は、それらがコードするポリペプチドが実質的に同一であれば、依然として実質的に同一である。これは、例えば遺伝暗号によって許容される最大コドン縮重を使用して1コピーの核酸が作成された場合に生じる。そのような場合、該核酸は、典型的に、中程度にストリンジェントなハイブリダイゼーション条件下でハイブリダイズする。典型的な「中程度にストリンジェントなハイブリダイゼーション条件」には、40%ホルムアミド、1M NaCl、1%SDSのバッファー中で37℃でのハイブリダイゼーション、および1×SSC中で45℃での洗浄が含まれる。陽性ハイブリダイゼーションはバックグラウンドの少なくとも2倍である。当業者は、代替のハイブリダイゼーション条件および洗浄条件を利用して同様にストリンジェントな条件を提供できることを容易に認識する。
【0091】
「抗体」とは、抗原に特異的に結合し、それを認識する、免疫グロブリン遺伝子に由来するフレームワーク領域またはそのフラグメントを含むポリペプチドを表す。一般に認識されている免疫グロブリン遺伝子には、κ、λ、α、γ、δ、ε、およびμ定常領域遺伝子、ならびに種々の免疫グロブリン可変領域遺伝子が含まれる。軽鎖はκまたはλとして分類される。重鎖はγ、μ、α、δ、またはεとして分類され、そしてそれらは、それぞれ、免疫グロブリンクラス、IgG、IgM、IgA、IgDおよびIgEを規定する。
【0092】
典型的な免疫グロブリン(抗体)の構造単位は四量体を含む。各四量体は2個の同一ペアのポリペプチド鎖から構成され、各ペアは1つの「軽」鎖(約25kDa)および1つの「重」鎖(約50〜70kDa)を有する。各鎖のN末端は、抗原認識を主に担う約100〜110またはそれ以上のアミノ酸からなる可変領域を規定する。可変軽鎖(VL)および可変重鎖(VH)という用語は、それぞれ、これらの軽鎖および重鎖を表す。
【0093】
抗体は、例えばインタクトの免疫グロブリンとして、あるいは種々のペプチダーゼでの消化によって生産されるいくつかの十分に特徴付けされているフラグメントとして存在する。ゆえに、例えば、ペプシンは、ヒンジ領域のジスルフィド結合の下で抗体を消化して、それ自体がジスルフィド結合によってVH−CH1に連結された軽鎖であるFabの二量体であるF(ab)’2を生じさせる。F(ab)’2を穏やかな条件下で還元して、ヒンジ領域中のジスルフィド結合を分解することにより、F(ab)’2二量体をFab’モノマーに変換してよい。Fab’モノマーは、本質的に、ヒンジ領域の部分を有するFabである(Fundamental Immunology (Paul ed., 3d ed. 1993)を参照のこと)。種々の抗体フラグメントはインタクトの抗体の消化の観点から定義されるが、当業者は、そのようなフラグメントを、化学的に、あるいは組換えDNA方法論を使用して新たに合成してよいことを認識する。ゆえに、本明細書中で使用される抗体という用語には、完全抗体の改変によって生産される抗体フラグメント、または組換えDNA方法論を使用して新たに合成されるもの(例えば単鎖Fv)またはファージディスプレイライブラリーを使用して特定されるもの(例えばMcCafferty et al., Nature 348:552-554 (1990)を参照のこと)がさらに含まれる。
【0094】
モノクローナル抗体またはポリクローナル抗体の調製では、当技術分野において公知の任意の技術を使用することができる(例えばKohler & Milstein, Nature 256:495-497 (1975); Kozbor et al., Immunology Today 4: 72 (1983); Cole et al., pp. 77-96 in Monoclonal Antibodies and Cancer Therapy (1985)を参照のこと)。単鎖抗体を生産するための技術(米国特許第4,946,778号)を適用して、本発明のポリペプチドに対する抗体を生産することができる。また、トランスジェニックマウス、または他の生物、例えば他の哺乳動物を使用して、ヒト化抗体を発現させてよい。あるいは、ファージディスプレイテクノロジーを使用して、選択抗原に特異的に結合する抗体およびヘテロメリック(heteromeric)Fabフラグメントを特定することができる(例えばMcCafferty et al., Nature 348:552-554 (1990); Marks et al., Biotechnology 10:779-783 (1992)を参照のこと)。
【0095】
抗体に「特異的に(または選択的に)結合する」または、それと「特異的に(または選択的に)免疫反応性である」との言い回しは、タンパク質またはペプチドに言及する場合、タンパク質の混成集団および他の生物学的物質(biologics)中のタンパク質の存在を決定する結合反応を表す。ゆえに、指定の免疫アッセイ条件下で、指定の抗体は、特定のタンパク質に対して、バックグラウンドの少なくとも2倍結合し、サンプル中に存在する他のタンパク質に対して実質的に顕著な量では結合しない。そのような条件下での抗体に対する特異的結合には、特定のタンパク質に関するその特異性に関して選択された抗体が必要とされる。例えば、融合タンパク質に対して産生されるポリクローナル抗体を選択して、融合タンパク質と特異的に免疫反応性であり、かつ該融合タンパク質の個別の成分とはそうでないポリクローナル抗体のみを取得することができる。この選択は、個別の抗原と交差反応する抗体を差し引くことによって達成してよい。種々の免疫アッセイ形式を使用して、特定のタンパク質と特異的に免疫反応性の抗体を選択してよい。例えば、固相ELISA免疫アッセイを通常通り使用して、タンパク質と特異的に免疫反応性の抗体を選択する(特異的免疫反応性を決定するために使用できる免疫アッセイ形式および条件の説明に関しては、例えばHarlow & Lane, Antibodies, A Laboratory Manual (1988)およびUsing Antibodies: A Laboratory Manual (1998)を参照のこと)。典型的には、特異的または選択的反応は、バックグラウンドシグナルすなわちノイズの少なくとも2倍であり、より典型的には、バックグラウンドの10〜100倍を超える。
【0096】
ポリヌクレオチドは、ネイティブの配列(すなわち、個々の抗原またはその部分をコードする内因性配列)を含んでよく、あるいはそのような配列の変異体を含んでよい。ポリヌクレオチド変異体に1以上の置換、付加、欠失および/または挿入を含有させて、コード対象の融合ポリペプチドの生物学的活性が、ネイティブの抗原を含む融合ポリペプチドと比べて減少しないようにしてよい。変異体は、好ましくは、ネイティブのポリペプチドまたはその部分をコードするポリヌクレオチド配列に対して、少なくとも約70%同一性、より好ましくは少なくとも約80%同一性、最も好ましくは少なくとも約90%同一性を示す。
【0097】
2以上の核酸またはポリペプチド配列の関連で、用語「同一の」または「同一性」パーセントとは、比較ウインドウ、すなわち、以下の配列比較アルゴリズムの1つを使用するか、あるいはマニュアルアライメントおよび視覚的点検によって測定される指定領域にわたる最大一致に関して比較およびアライメントされた場合に、同一であるか、あるいは指定のパーセンテージの同一のアミノ酸残基またはヌクレオチド(すなわち、指定領域にわたって70%同一性、場合により75%、80%、85%、90%、または95%同一性)を有する2以上の配列または部分配列を表す。そしてそのような配列は「実質的に同一」であると称される。この定義は試験配列の相補物(compliment)をも表す。場合により、同一性は、少なくとも約25〜約50アミノ酸またはヌクレオチド長の領域にわたって、あるいは場合により75〜100アミノ酸またはヌクレオチド長の領域にわたって存在する。
【0098】
配列比較では、典型的に、1つの配列が参照配列として働き、それに対して試験配列を比較する。配列比較アルゴリズムを使用する場合、試験配列および参照配列をコンピュータに入力し、必要であれば部分配列座標を指定し、配列アルゴリズムプログラムパラメータを指定する。デフォルトプログラムパラメータを使用することができ、あるいは代替パラメータを指定することができる。そして配列比較アルゴリズムは、プログラムパラメータに基づいて、参照配列に対する試験配列の配列同一性パーセントを算出する。
【0099】
本明細書中で使用される「比較ウインドウ」には、25〜500、通常には約50〜約200、より通常には約100〜約150からなる群から選択される数の連続位置のいずれか1つのセグメントへの言及が含まれ、そのセグメントにおいて、2つの配列を最適にアライメントした後に、配列を同一数の連続位置の参照配列と比較してよい。比較のために配列をアライメントする方法は当技術分野において周知である。比較のための最適な配列のアライメントは、例えばSmith & Watermanの局所的相同性アルゴリズム(Adv. Appl. Math. 2:482 (1981))、Needleman & Wunschの相同性アライメントアルゴリズム(J. Mol. Biol. 48:443 (1970))、Pearson & Lipmanの類似度検索法(the search for similarity method)(Proc. Nat'l. Acad. Sci. USA 85:2444 (1988))、これらのアルゴリズムのコンピュータによる実装(GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, WI)、またはマニュアルアライメントおよび視覚的点検(例えばCurrent Protocols in Molecular Biology (Ausubel et al., eds. 1995 supplement)を参照のこと)によって行うことができる。
【0100】
有用なアルゴリズムの一例はPILEUPである。PILEUPは、累進ペアワイズアライメント(progressive, pairwise alignments)を使用して関連配列の群に由来する多重配列アライメントを作成し、関連性および配列同一性パーセントを示す。さらに該アライメントの作成に使用されるクラスタリング関連性を示すツリーまたはデンドログラム(dendogram)をプロットする。PILEUPは、Feng & Doolittleの累進アライメント法(J. Mol. Evol. 35:351-360 (1987))を簡素化したものを使用する。使用される方法は、Higgins & Sharp, CABIOS 5:151-153 (1989)に記載の方法に類似の方法である。このプログラムは、それぞれ最大5,000ヌクレオチドまたはアミノ酸長の300までの配列をアライメントすることができる。マルチプルアライメント手順は、2個の最も類似する配列のペアワイズアライメントで開始され、2個のアライメント済み配列のクラスターが得られる。そしてこのクラスターを、次に最も関連している配列または、アライメント済み配列のクラスターに対してアライメントする。配列の2個のクラスターは、2個の個別の配列のペアワイズアライメントを単純に拡張することによってアライメントされる。一連の累進ペアワイズアライメントによって最終のアライメントを達成する。このプログラムは、特定の配列および、配列比較の領域に関するそれらのアミノ酸またはヌクレオチド座標を指定し、プログラムパラメータを指定することによって実行される。PILEUPを使用して、参照配列を他の試験配列と比較して、配列同一性パーセントの関連性を決定する。その場合、次のパラメータ:デフォルトギャップ重み付け(gap weight)(3.00)、デフォルトギャップ長重み付け(gap length weight)(0.10)、および重み付けエンドギャップ(weighted end gaps)を使用する。PILEUPはGCG配列解析ソフトウェアパッケージ、例えばバージョン7.0(Devereaux et al., Nuc. Acids Res. 12:387-395 (1984))から入手することができる。
【0101】
配列同一性および配列類似性パーセントの決定に適しているアルゴリズムの別の例はBLASTおよびBLAST2.0アルゴリズムであり、それぞれAltschul et al., Nuc. Acids Res. 25:3389-3402 (1977)およびAltschul et al., J. Mol. Biol. 215:403-410 (1990)に記載されている。BLAST解析を実施するためのソフトウェアはNational Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/)から一般に利用可能である。このアルゴリズムは、データベース配列の同じ長さのワードとアライメントされた場合にいくつかの正値の閾値スコアTに適合するか、あるいはそれを満たすクエリ配列の長さWの短いワードを特定することによって高スコアの配列ペア(HSP)を最初に特定することを含む。Tは近縁ワードスコア閾値と称される(Altschul et al., 上記)。これらの最初の近縁ワードヒットは、それらを含むより長いHSPを発見するための検索開始のシードとして機能する。そのワードヒットを各配列に沿って両方向に、累積のアラインメントスコアが増加しうる限り伸長する。ヌクレオチド配列の場合、パラメータM(適合残基ペアに対する褒賞スコア;常に>0)およびN(非適合残基に対する罰則スコア;常に<0)を使用して累積スコアを算出する。アミノ酸配列の場合は、スコアリングマトリックスを使用して累積スコアを算出する。各方向におけるワードヒットの伸長は以下の時点で停止させる:累積のアラインメントスコアがその最大達成値から量Xだけ減少したとき;累積スコアが、1個以上の負のスコアを有する残基のアラインメントの蓄積のために0以下になったとき;またはいずれかの配列の末端に達したとき。BLASTアルゴリズムパラメータW、T、およびXによってアライメントの感度および速度が決定される。BLASTNプログラム(ヌクレオチド配列用)では、デフォルトとして、ワード長(W)11、期待値(E)10、M=5、N=−4および両鎖の比較を使用する。アミノ酸配列用のBLASTPプログラムでは、デフォルトとして、ワード長(W)3、および期待値(E)10、およびBLOSUM62スコアリ
ングマトリックス(Henikoff & Henikoff, Proc. Natl. Acad. Sci. USA 89:10915 (1989)を参照のこと)アライメント(B)50、期待値(E)10、M=5、N=−4、および両鎖の比較を使用する。
【0102】
BLASTアルゴリズムでは、2つの配列間の類似性の統計解析をさらに実施する(例えばKarlin & Altschul, Proc. Nat'l. Acad. Sci. USA 90:5873-5787 (1993)を参照のこと)。BLASTアルゴリズムによって提供される類似性の1つの測定値は、最小合計確率(smallest sum probability)(P(N))であり、それは2つのヌクレオチドまたはアミノ酸配列間の適合が偶然生じる確率の指標を提供する。例えば、参照核酸と試験核酸の比較において最小合計確率が約0.2未満、より好ましくは約0.01未満、最も好ましくは約0.001未満であれば、該核酸は参照配列と類似であると見なされる。
【0103】
ポリヌクレオチド組成物
本明細書中で使用される用語「DNAセグメント」および「ポリヌクレオチド」とは、特定の生物種のトータルゲノムDNAからは単離されたDNA分子を表す。したがって、ポリペプチドをコードするDNAセグメントとは、1以上のコード配列を含有するが、該DNAセグメントの取得元の生物種のトータルゲノムDNAから実質的に単離されているか、あるいは精製されているDNAセグメントを表す。DNAセグメントおよびそのようなセグメントの小断片、ならびに組換えベクター、例えばプラスミド、コスミド、ファージミド、ファージ、ウイルス等も、用語「DNAセグメント」および「ポリヌクレオチド」の範囲内に含まれる。
【0104】
当業者に理解されるように、本発明のDNAセグメントには、タンパク質、ポリペプチド、ペプチド等を発現するか、あるいはそれらを発現するように作り変えてよいゲノム配列、ゲノム外およびプラスミドによってコードされる配列および遺伝子操作された小さい遺伝子セグメントが含まれうる。そのようなセグメントは、天然に単離されるか、あるいは人の手によって合成的に改変してよい。
【0105】
本明細書中で使用される「単離された」とは、ポリヌクレオチドが他のコード配列から実質的に離れていること、および該DNAセグメントが、大部分の無関係のコードDNA、例えば大きい染色体断片または他の機能的遺伝子またはポリペプチドコード領域を含有しないことを意味する。当然ながら、これは元々単離されているDNAセグメントを表すが、人の手によって該セグメントに後に付加された遺伝子またはコード領域を排除しない。
【0106】
当業者に認識されるように、ポリヌクレオチドは一本鎖(コードまたはアンチセンス)または二本鎖であってよく、DNA(ゲノム、cDNAまたは合成)またはRNA分子であってよい。RNA分子には、イントロンを含有し、DNA分子と1対1様式で対応するHnRNA分子、およびイントロンを含有しないmRNA分子が含まれる。追加のコードまたは非コード配列は、その必要はないが、本発明のポリヌクレオチド内に存在してよく、ポリヌクレオチドは、その必要はないが、他の分子および/または支持材料に連結してよい。
【0107】
ポリヌクレオチドは、ネイティブの配列(すなわち、マイコバクテリウム抗原またはその一部分をコードする内因性配列)を含んでよく、あるいはそのような配列の変異体、または生物学的もしくは抗原機能的等価物を含んでよい。以下にさらに記載されるように、ポリヌクレオチド変異体に1以上の置換、付加、欠失および/または挿入を含有させて、好ましくは、コード対象のポリペプチドの免疫原性が、ネイティブの腫瘍タンパク質と比べて減少しないようにしてよい。コード対象のポリペプチドの免疫原性に対する効果は、一般に、本明細書中に記載のように評価してよい。用語「変異体」は異種起源の相同遺伝子をも包含する。
【0108】
追加の実施形態では、本発明は、本明細書中で開示される1以上の配列と同一または相補的な種々の長さの一連の連続する配列を含む、単離されたポリヌクレオチドおよびポリペプチドを提供する。例えば、本明細書中で開示される1以上の配列の少なくとも約15、20、30、40、50、75、100、150、200、300、400、500または1000またはそれ以上の連続ヌクレオチド、ならびにそれらの間のすべての中間の長さを含むポリヌクレオチドが本発明によって提供される。この関連で「中間の長さ」とは、引用された値の間の任意の長さ、例えば16、17、18、19、等;21、22、23、等;30、31、32、等;50、51、52、53、等;100、101、102、103、等;150、151、152、153、等;例えば200〜500;500〜1,000の間のすべての整数、等を意味することが容易に理解される。
【0109】
本発明のポリヌクレオチド、またはその断片を、コード配列自体の長さにかかわらず、他のDNA配列、例えばプロモーター、ポリアデニル化シグナル、追加の制限酵素部位、マルチクローニング部位、他のコードセグメント、等と組み合わせて、それらの全体の長さがかなり変動するようにしてよい。したがって、ほとんど任意の長さの核酸断片を使用してよく、トータルの長さは、好ましくは、意図される組換えDNAプロトコルにおける調製および使用の容易さによって制限されることが考慮される。例えば、トータルの長さが約10,000、約5,000、約3,000、約2,000、約1,000、約500、約200、約100、約50塩基対長、等(すべての中間の長さを含む)である例示的DNAセグメントは、本発明の多数の実行において有用であると予測される。
【0110】
さらに、遺伝暗号の縮重の結果として、本明細書中に記載のポリペプチドをコードする多数のヌクレオチド配列が存在することが当業者に理解される。これらのポリヌクレオチドのいくつかは、いずれかのネイティブの遺伝子のヌクレオチド配列に対して最小の相同性しか有さない。それにもかかわらず、コドン使用の差異に起因して変動するポリヌクレオチド、例えばヒトおよび/または霊長類のコドン選択に関して最適化されたポリヌクレオチドが本発明によって具体的に考慮される。さらに、本明細書中で提供されるポリヌクレオチド配列を含む遺伝子の対立遺伝子は本発明の範囲内である。対立遺伝子は、ヌクレオチドの1以上の突然変異、例えば欠失、付加および/または置換の結果として変更される内因性遺伝子である。その結果得られるmRNAおよびタンパク質は、その必要はないが、改変された構造または機能を有してよい。対立遺伝子は標準的技術(例えばハイブリダイゼーション、増幅および/またはデータベース配列比較)を使用して特定することができる。
【0111】
ポリヌクレオチドの特定および特徴付け
ポリヌクレオチドは、任意の種々の十分に確立されている技術を使用して、特定、調製および/または操作してよい。例えば、ポリヌクレオチドは、以下にさらに詳細に記載されるように、cDNAのマイクロアレイを腫瘍関連発現(すなわち、本明細書中で提供される代表的なアッセイを使用して測定された場合の、腫瘍における、正常組織より少なくとも2倍高い発現)に関してスクリーニングすることによって特定してよい。そのようなスクリーニングは、例えばSynteniマイクロアレイ(Palo Alto, CA)を製造元の指示書にしたがって使用して(かつ本質的にSchena et al., Proc. Natl. Acad. Sci. USA 93:10614-10619 (1996)およびHeller et al., Proc. Natl. Acad. Sci. USA 94:2150-2155 (1997)に記載されるように)実施してよい。あるいは、本明細書中に記載のタンパク質を発現する細胞、例えば結核菌細胞から調製されたcDNAからポリヌクレオチドを増幅してよい。そのようなポリヌクレオチドは、ポリメラーゼ連鎖反応(PCR)によって増幅してよい。このアプローチでは、本明細書中で提供される配列に基づいて配列特異的プライマーを設計してよく、購入または合成してよい。
【0112】
本発明のポリヌクレオチドの増幅された部分を使用し、周知の技術を使用して好適なライブラリー(例えば結核菌cDNAライブラリー)から全長遺伝子を単離してよい。そのような技術には、増幅に適した1以上のポリヌクレオチドプローブまたはプライマーを使用してライブラリー(cDNAまたはゲノム)をスクリーニングすることが含まれる。好ましくは、ライブラリーをサイズで選択して大きい分子を含ませる。遺伝子の5’および上流領域を特定するためには、ランダムプライムドライブラリー(Random primed libraries)も好ましいかもしれない。ゲノムライブラリーはイントロンの取得および5’配列の伸長のために好ましい。
【0113】
ハイブリダイゼーション技術では、周知の技術を使用して(例えばニックトランスレーションまたは32Pでの末端標識によって)部分配列を標識してよい。そして、一般に、変性細菌コロニーを含有するフィルター(またはファージプラークを含有するローン(lawns))を標識プローブとハイブリダイズさせることによって細菌またはバクテリオファージライブラリーをスクリーニングする(Sambrook et al., Molecular Cloning: A Laboratory Manual (2000)を参照のこと)。ハイブリダイズするコロニーまたはプラークを選択し、増殖させ、追加の分析のためにDNAを単離する。cDNAクローンを解析して、例えば、該部分配列に由来するプライマーおよびベクターに由来するプライマーを使用するPCRによって追加の配列の量を決定してよい。制限酵素地図および部分配列を作成して、1以上の重複クローンを特定してよい。そして、標準的技術を使用して完全配列を決定してよい。その技術は一連の欠失クローンを作成するステップを含んでよい。そして、得られた重複配列を組み立てて、単一の連続配列にすることができる。周知の技術を使用して好適な断片をライゲートすることによって全長cDNA分子を作成することができる。
【0114】
一方、部分cDNA配列から全長コード配列を取得するための多数の増幅技術が存在する。そのような技術では、概してPCRによって増幅を実施する。種々の市販のキットのいずれかを使用して増幅ステップを実施してよい。例えば当技術分野において周知のソフトウェアを使用してプライマーを設計してよい。プライマーは、好ましくは22〜30ヌクレオチド長であり、少なくとも50%のGC含量を有し、約68℃〜72℃の温度で標的配列にアニーリングする。増幅された領域は、上記のようにシークエンシングしてよく、重複配列を組み立てて連続配列にしてよい。
【0115】
そのような増幅技術の1つはインバースPCR(inverse PCR)であり(Triglia et al., Nucl. Acids Res. 16:8186 (1988)を参照のこと)、その場合、制限酵素を使用して、遺伝子の既知領域の断片を作成する。そして該断片を分子内ライゲーションによって環化し、該既知領域に由来する種々のプライマーを用いるPCRの鋳型として使用する。代替のアプローチでは、リンカー配列に対するプライマーおよび既知領域に特異的なプライマーを用いる増幅によって部分配列に隣接する配列を回収してよい。増幅された配列を、典型的には、同リンカープライマーおよび該既知領域に特異的な第2のプライマーを用いる第2ラウンドの増幅に付する。該既知配列からの反対方向の伸長を開始させる2種のプライマーを使用するこの手順のバリエーションがWO96/38591に記載されている。別のそのような技術は「cDNA末端の高速増幅」またはRACEとして知られる。この技術には、内部プライマーおよび外部プライマーの使用が含まれ、外部プライマーはポリA領域またはベクター配列にハイブリダイズして、既知配列の5’および3’である配列を特定する。追加の技術には、捕捉PCR(capture PCR)(Lagerstrom et al., PCR Methods Applic. 1 :111-19 (1991))およびウォーキングPCR(walking PCR)(Parker et al., Nucl. Acids. Res. 19:3055-60 (1991))が含まれる。増幅を使用する他の方法を同様に使用して、全長cDNA配列を取得してよい。
【0116】
特定の場合、発現配列タグ(EST)データベースにおいて提供される、例えばGenBankから入手可能な配列の解析によって全長cDNA配列を取得することが可能である。重複ESTの検索は、一般に、周知のプログラム(例えばNCBI BLAST検索)を使用して実施してよく、そのようなESTを使用して連続の全長配列を作成してよい。全長DNA配列はゲノム断片の解析によって取得してもよい。
【0117】
宿主細胞でのポリヌクレオチド発現
本発明の他の実施形態では、本発明のポリペプチド、または融合タンパク質もしくはその機能的等価物をコードするポリヌクレオチド配列またはその断片を組換えDNA分子中で使用して、適切な宿主細胞におけるポリペプチドの発現を導いてよい。遺伝暗号に特有の縮重に基づいて、実質的に同一または機能的に等価なアミノ酸配列をコードする他のDNA配列を生産してよく、これらの配列を使用して、所定のポリペプチドをクローニングし、発現させてよい。
【0118】
当業者に理解されるように、いくつかの場合には、非天然に存在するコドンを有する、ポリペプチドをコードするヌクレオチド配列を生産することが有益であるかもしれない。例えば、特定の原核生物または真核生物の宿主が好むコドンを選択して、タンパク質発現の割合を増加させるか、あるいは、所望の特性、例えば天然に存在する配列から作成された転写産物の半減期より長い半減期を有する組換えRNA転写産物を生産することができる。
【0119】
さらに、本発明のポリヌクレオチド配列は、当技術分野において一般に知られている方法を使用して遺伝子操作することができる。その目的は、ポリペプチドをコードする配列を種々の理由で変化させることであり、それには、非限定的に、クローニング、プロセシング、および/または遺伝子産物の発現を改変する変化が含まれる。例えば、ランダムな断片化によるDNAシャッフリングおよび遺伝子断片のPCRによる組み立ておよび合成オリゴヌクレオチドを使用して、該ヌクレオチド配列を遺伝子操作してよい。さらに、部位特異的突然変異誘発を使用して、新規制限部位を挿入し、グリコシル化パターンを変化させ、コドン優先度を変更し、スプライシング変異体を生成し、あるいは突然変異を導入すること、等を行ってよい。
【0120】
本発明の別の実施形態では、天然、改変、または組換え核酸配列を異種性配列にライゲーションして、融合タンパク質をコードするようにしてよい。例えば、ポリペプチド活性のインヒビターに関してペプチドライブラリーをスクリーニングするために、市販の抗体によって認識できるキメラタンパク質をコードするようにすることが有用であるかもしれない。融合タンパク質を遺伝子操作して、ポリペプチドをコードする配列と異種性タンパク質配列の間に位置する切断部位を含有させて、該ポリペプチドが切断され、異種性部分から精製されるようにしてもよい。
【0121】
所望のポリペプチドをコードする配列は、完全にまたは部分的に、当技術分野において周知の化学的方法を使用して合成してよい(Caruthers, M. H. et al., Nucl. Acids Res. Symp. Ser. pp. 215-223 (1980), Horn et al., Nucl. Acids Res. Symp. Ser. pp. 225-232 (1980)を参照のこと)。あるいは、ポリペプチドのアミノ酸配列、またはその部分を合成する化学的方法を使用してタンパク質自体を生産してよい。例えば、ペプチド合成は種々の固相技術を使用して実施することができ(Roberge et al., Science 269:202-204 (1995))、例えばABI 431A Peptide Synthesizer(Perkin Elmer, Palo Alto, CA)を使用して自動合成を達成してよい。
【0122】
新たに合成されたペプチドを調製用高性能液体クロマトグラフィー(例えばCreighton, Proteins, Structures and Molecular Principles (1983))または当技術分野において利用可能な他の同等の技術によって実質的に精製してよい。アミノ酸解析またはシークエンシング(例えばエドマン分解手順)によって合成ペプチドの組成を確認してよい。さらに、ポリペプチド、またはその任意の部分のアミノ酸配列を、直接合成中に改変し、ならびに/あるいは、化学的方法を使用して他のタンパク質、またはその任意の部分に由来する配列と組み合わせて、変異体ポリペプチドを作成してよい。
【0123】
所望のポリペプチドを発現させるために、該ポリペプチド、または機能的等価物をコードするヌクレオチド配列を適切な発現ベクター、すなわち挿入されたコード配列の転写および翻訳に必要なエレメントを含有するベクターに挿入してよい。当業者に周知の方法を使用して、目的のポリペプチドをコードする配列ならびに適切な転写および翻訳調節エレメントを含有する発現ベクターを構築してよい。これらの方法には、in vitro組換えDNA技術、合成技術、およびin vivo遺伝子組換えが含まれる。そのような技術は、Sambrook et al., Molecular Cloning, A Laboratory Manual (2000), およびAusubel et al., Current Protocols in Molecular Biology (毎年更新される)に記載されている。
【0124】
種々の発現ベクター/宿主系を利用して、ポリヌクレオチド配列を含有および発現させてよい。これらには、非限定的に、組換えバクテリオファージ、プラスミド、またはコスミドDNA発現ベクターで形質転換された微生物、例えば細菌;酵母発現ベクターで形質転換された酵母;ウイルス発現ベクター(例えばバキュロウイルス)で感染させた昆虫細胞系;ウイルス発現ベクター(例えばカリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)または細菌発現ベクター(例えばTiまたはpBR322プラスミド)で形質転換された植物細胞系;または動物細胞系が含まれる。
【0125】
発現ベクター中に存在する「制御エレメント(control elements)」または「調節配列(regulatory sequences)」は、宿主細胞のタンパク質と相互作用して転写および翻訳を実行するベクターの非翻訳領域:エンハンサー、プロモーター、5’および3’非翻訳領域である。そのようなエレメントは強度および特異性において様々である。利用されるベクター系および宿主に応じて、任意の数の好適な転写および翻訳エレメント、例えば構成的および誘導性プロモーターを使用してよい。例えば、細菌系でのクローニングの場合、誘導性プロモーター、例えばPBLUESCRIPTファージミド(Stratagene, La Jolla, Calif.)またはPSPORT1プラスミド(Gibco BRL, Gaithersburg, MD)のハイブリッドlacZプロモーター等を使用してよい。哺乳動物の細胞系では、哺乳動物の遺伝子または哺乳動物ウイルスに由来するプロモーターが概して好ましい。ポリペプチドをコードする複数コピーの配列を含有する細胞株を作製することが必要である場合、SV40またはEBVに基づくベクターを適切な選択マーカーと共に好都合に使用してよい。
【0126】
細菌系では、発現対象のポリペプチドに関して意図される用途に応じて、いくつかの発現ベクターを選択してよい。例として、例えば抗体を誘導するために大規模な量が必要である場合、容易に精製可能な融合タンパク質の大量発現を導くベクターを使用してよい。そのようなベクターには、非限定的に、多機能性の大腸菌(E. coli)クローニングおよび発現ベクター、例えばBLUESCRIPT(Stratagene)(その場合、目的のポリペプチドをコードする配列をアミノ末端Metおよびそれに続くβ−ガラクトシダーゼの7残基の配列とインフレームで該ベクターにライゲーションして、ハイブリッドタンパク質が生産されるようにしてよい);pINベクター(Van Heeke & Schuster, J. Biol. Chem. 264:5503-5509 (1989));等が含まれる。pGEXベクター(Promega, Madison, Wis.)を使用して、グルタチオンS−トランスフェラーゼ(GST)との融合タンパク質として外来性ポリペプチドを発現させてもよい。一般に、そのような融合タンパク質は可溶性であり、グルタチオン−アガロースビーズへの吸着、その後の遊離グルタチオンの存在下での溶出によって溶解細胞から容易に精製することができる。そのような系で作成されるタンパク質が、ヘパリン、トロンビン、または第XA因子プロテアーゼ切断部位を含むように設計して、目的のクローニング対象ポリペプチドをGST部分から自在に遊離できるようにしてよい。
【0127】
酵母(Saccharomyces cerevisiae)では、構成的または誘導性プロモーター、例えばα因子、アルコールオキシダーゼ、およびPGHを含有するいくつかのベクターを使用してよい。総説としては、Ausubel et al. (上記)およびGrant et al., Methods Enzymol. 153:516-544 (1987)を参照のこと。
【0128】
植物の発現ベクターが使用される場合、多数のプロモーターのいずれかによってポリペプチドをコードする配列の発現を誘導してよい。例えば、ウイルスプロモーター、例えばCaMVの35Sおよび19Sプロモーターを、単独で、あるいはTMVに由来するオメガリーダー配列と組み合わせて使用してよい(Takamatsu, EMBO J. 6:307-311 (1987))。あるいは、植物プロモーター、例えばRUBISCOの小サブユニットまたは熱ショックプロモーターを使用してよい(Coruzzi et al., EMBO J. 3:1671-1680 (1984); Broglie et al., Science 224:838-843 (1984); およびWinter et al., Results Probl. Cell Differ. 17:85-105 (1991))。これらの構築物は、直接DNA形質転換または病原体媒介性トランスフェクションによって植物細胞に導入することができる。そのような技術は、いくつかの一般に入手可能な総説に記載されている(例えばHobbs in McGraw Hill Yearbook of Science and Technology pp. 191-196 (1992)を参照のこと)。
【0129】
昆虫系を使用して目的のポリペプチドを発現させてもよい。例えば、そのような系の1つでは、Autographa californica核多核体病ウイルス(AcNPV)をベクターとして使用して、Spodoptera frugiperda細胞またはTrichoplusia larvaeにおいて外来遺伝子を発現させる。該ポリペプチドをコードする配列を、ウイルスの非必須領域、例えばポリヘドリン遺伝子内にクローニングし、ポリヘドリンプロモーターの制御下に配置してよい。該ポリペプチドをコードする配列が首尾良く挿入されるとポリヘドリン遺伝子が不活性になり、コートタンパク質を欠く組換えウイルスが生産される。そして該組換えウイルスを使用して、例えばS. frugiperda細胞またはTrichoplusia larvaeを感染させてよく、それらにおいて目的のポリペプチドを発現させてよい(Engelhard et al., Proc. Natl. Acad. Sci. U.S.A. 91:3224-3227 (1994))。
【0130】
哺乳動物の宿主細胞では、いくつかのウイルスベースの発現系が一般に利用可能である。例えば、アデノウイルスが発現ベクターとして使用される場合、目的のポリペプチドをコードする配列を、後期プロモーターおよび3成分(tripartite)リーダー配列からなるアデノウイルス転写/翻訳複合体内にライゲーションしてよい。ウイルスゲノムの非必須E1またはE3領域への挿入を使用して、感染宿主細胞において該ポリペプチドを発現可能である生存可能なウイルスを取得してよい(Logan & Shenk, Proc. Natl. Acad. Sci. U.S.A. 81:3655-3659 (1984))。さらに、転写エンハンサー、例えばラウス肉腫ウイルス(RSV)エンハンサーを使用して、哺乳動物の宿主細胞における発現を増加させてよい。アデノウイルスベクターを用いる作業に関する方法およびプロトコルは、Wold, Adenovirus Methods and Protocols, 1998で概説されている。アデノウイルスベクターの使用に関する追加の参考文献は、Adenovirus: A Medical Dictionary, Bibliography, and Annotated Research Guide to Internet References, 2004に見出すことができる。
【0131】
特定の開始シグナルを使用して、目的のポリペプチドをコードする配列の、より効率的な翻訳を達成してもよい。そのようなシグナルには、ATG開始コドンおよび隣接配列が含まれる。ポリペプチドをコードする配列、その開始コドン、および上流配列が適切な発現ベクター中に挿入される場合、追加の転写または翻訳調節シグナルが必要とされないかもしれない。しかし、コード配列、またはその一部分しか挿入されない場合、ATG開始コドンを含めた外因性翻訳調節シグナルが提供されるべきであろう。さらにまた、インサート全体の翻訳を保証するために、該開始コドンは正しいリーディングフレームであるべきであろう。外因性翻訳エレメントおよび開始コドンは、種々の起源の、天然物および合成物の両者であってよい。使用される具体的な細胞系に適切なエンハンサーを含ませることによって発現効率を高めてよい。そのようなエンハンサーは、例えば文献に記載されているエンハンサーである(Scharf. et al., Results Probl. Cell Differ. 20:125-162 (1994))。
【0132】
さらに、宿主細胞株を、所望の様式で、挿入配列の発現をモジュレートするか、あるいは発現タンパク質をプロセシングするその能力に関して選択してよい。そのようなポリペプチドの改変には、非限定的に、アセチル化、カルボキシル化、グリコシル化、リン酸化、脂質化(lipidation)、およびアシル化が含まれる。「プレプロ(prepro)」形式のタンパク質を切断する翻訳後プロセシングを使用して、正しい挿入、折りたたみおよび/または機能を促進してもよい。そのような翻訳後の作業に特有の細胞機構および特徴的機構を有する、異なる宿主細胞、例えばCHO、HeLa、MDCK、HEK293、およびWI38を選択して、外来タンパク質の正しい改変およびプロセシングを保証してよい。
【0133】
組換えタンパク質の長期の、高収量の生産のためには、概して、安定発現が好ましい。例えば、ウイルスの複製起点および/または内因性発現エレメントおよび、同一または別のベクター上の選択マーカー遺伝子を含有してよい発現ベクターを使用して、目的のポリヌクレオチドを安定に発現する細胞株を形質転換してよい。該ベクターの導入後、細胞を強化培地中で1〜2日間成長させてよく、その後、それらを選択培地に切り換える。選択マーカーの目的は選択のための耐性を付与することであり、その存在により、導入配列を首尾良く発現する細胞の培養および回収が可能になる。細胞タイプに適切な組織培養技術を使用して、安定に形質転換された細胞の耐性クローンを増殖させてよい。
【0134】
任意の数の選択系を使用して形質転換細胞株を回収してよい。これらには、非限定的に、単純ヘルペスウイルスチミジンキナーゼ(Wigler et al., Cell 11 :223-32 (1977))およびアデニンホスホリボシルトランスフェラーゼ(Lowy et al., Cell 22:817-23 (1990))遺伝子が含まれる。それらは、それぞれ、tk.sup.−またはaprt.sup.−細胞において使用することができる。また、代謝拮抗剤、抗生物質または除草剤抵抗性を選択の基礎として使用することができる;例えば、メトトレキセートに対する耐性を付与するdhfr(Wigler et al., Proc. Natl. Acad. Sci. U.S.A. 77:3567-70 (1980));アミノグリコシド、ネオマイシンおよびG−418に対する耐性を付与するnpt(Colbere-Garapin et al., J. Mol. Biol. 150:1-14 (1981));および、それぞれクロルスルフロン(chlorsulfuron)およびホスフィノトリシン(phosphinotricin)アセチルトランスフェラーゼに対する耐性を付与するalsまたはpat(Murry, 上記)である。追加の選択可能遺伝子が報告されている。例えば、細胞がトリプトファンの代わりにインドールを利用することを可能にするtrpB、または細胞がヒスチジンの代わりにヒスチノール(histinol)を利用することを可能にするhisD(Hartman & Mulligan, Proc. Natl. Acad. Sci. U.S.A. 85:8047-51 (1988))である。最近、可視マーカーの使用が人気を獲得している。アントシアニン、β−グルクロニダーゼおよびその基質GUS、ならびにルシフェラーゼおよびその基質ルシフェリン等のマーカーが、特定のベクター系に起因する、形質転換体を特定するためだけでなく、一時的または安定なタンパク質発現の量を定量するためにも広く使用されている(Rhodes et al., Methods Mol. Biol. 55:121-131 (1995))。
【0135】
マーカー遺伝子発現の存在/不存在は目的の遺伝子も存在することを示唆するが、その存在および発現を確認する必要があるかもしれない。例えば、ポリペプチドをコードする配列がマーカー遺伝子配列内に挿入されれば、マーカー遺伝子機能の不存在によって配列を含有する組換え細胞を特定することができる。あるいは、単一のプロモーターの制御下でポリペプチドをコードする配列と直列にマーカー遺伝子を配置することができる。誘導または選択に応答して該マーカー遺伝子が発現されると、通常、直列型遺伝子の発現も示される。
【0136】
あるいは、当業者に公知の種々の手順によって、所望のポリヌクレオチド配列を含有および発現する宿主細胞を特定してよい。これらの手順としては、非限定的に、DNA−DNAまたはDNA−RNAハイブリダイゼーションおよびタンパク質バイオアッセイまたは免疫アッセイ技術(核酸またはタンパク質を検出および/または定量するためのメンブレン、溶液、またはチップベースのテクノロジーが含まれる)が挙げられる。
【0137】
ポリヌクレオチドによってコードされる生成物の発現を検出および測定するための種々のプロトコルであって、該生成物に特異的なポリクローナル抗体またはモノクローナル抗体を使用するプロトコルは当技術分野において公知である。その例には、酵素結合免疫吸着検定(ELISA)、ラジオイムノアッセイ(RIA)、および蛍光標示式細胞分取(FACS)が含まれる。いくつかの適用では、所定のポリペプチド上の2種の不干渉エピトープと反応性のモノクローナル抗体を利用する2部位のモノクローナルベースの免疫アッセイが好ましいかもしれないが、競合的結合アッセイを使用してもよい。これらのアッセイおよび他のアッセイは、特にHampton et al., Serological Methods, a Laboratory Manual (1990)およびMaddox et al., J. Exp. Med. 158:1211-1216 (1983)に記載されている。
【0138】
多種多様な標識およびコンジュゲーション技術が当業者に公知であり、種々の核酸およびアミノ酸アッセイにおいて使用してよい。ポリヌクレオチドに関連する配列を検出するための標識ハイブリダイゼーションまたはPCRプローブの作成手段には、オリゴラベリング(oligolabeling)、ニックトランスレーション、末端標識化またはPCR増幅(標識ヌクレオチドを使用する)が含まれる。あるいは、配列、またはその任意の部分を、mRNAプローブを生産するためのベクターにクローニングしてよい。そのようなベクターは当技術分野において公知であり、市販されていて、適切なRNAポリメラーゼ、例えばT7、T3、またはSP6および標識ヌクレオチドを加えることによってin vitroでRNAプローブを合成するために使用してよい。種々の市販のキットを使用してこれらの手順を実行してよい。使用することができる好適なレポーター分子または標識には、放射性核種、酵素、蛍光、化学発光、または発色物質ならびに基質、補助因子、インヒビター、磁気粒子、等が含まれる。
【0139】
目的のポリヌクレオチド配列で形質転換された宿主細胞を、細胞培養から該タンパク質を発現および回収するために適した条件下で培養してよい。組換え細胞によって生産されたタンパク質を分泌させるか、あるいは細胞内に含有させてよい。それは使用される配列および/またはベクターに依存する。当業者に理解されるように、本発明のポリヌクレオチドを含有する発現ベクターは、原核細胞膜または真核細胞膜を通過するコード対象のポリペプチドの分泌を導くシグナル配列を含有するように設計してよい。他の組換え構築物を使用して、目的のポリペプチドをコードする配列を、可溶性タンパク質の精製を容易にするポリペプチドドメインをコードするヌクレオチド配列と連結してもよい。そのような精製を容易にするドメインには、非限定的に、金属キレートペプチド、例えば固定金属上での精製を可能にするヒスチジン−トリプトファンモジュール、固定免疫グロブリン上での精製を可能にするプロテインAドメイン、およびFLAGS伸長/親和性精製系(Immunex Corp., Seattle, Wash.)で利用されるドメインが含まれる。切断可能なリンカー配列、例えば第XA因子またはエンテロキナーゼ(Invitrogen. San Diego, Calif.)に特異的な配列を精製ドメインおよびコード対象のポリペプチドの間に含ませることによって精製を容易にしてよい。そのような発現ベクターの1つは、目的のポリペプチドを含有する融合タンパク質および、チオレドキシンまたはエンテロキナーゼ切断部位に先行する6ヒスチジン残基をコードする核酸の発現を提供する。該ヒスチジン残基は、Porath et al., Prot. Exp. Purif. 3:263-281 (1992)に記載のようにIMIAC(固定金属イオンアフィニティークロマトグラフィー)上での精製を容易にし、一方、エンテロキナーゼ切断部位は該融合タンパク質に由来する所望のポリペプチドを精製するための手段を提供する。融合タンパク質を含有するベクターについての考察はKroll et al., DNA Cell Biol. 12:441-453 (1993)に提供されている。
【0140】
組換え生産方法に加えて、固相技術を使用する直接ペプチド合成(Merrifield, J. Am. Chem. Soc. 85:2149-2154 (1963))によって本発明のポリペプチド、およびその断片を生産してよい。マニュアル技術を使用するか、あるいは自動化によってタンパク質合成を実施してよい。自動合成は、例えばApplied Biosystems 431A Peptide Synthesizer(Perkin Elmer)を使用して達成してよい。あるいは、種々の断片を別々に化学合成し、化学的方法を使用して組み合わせて、全長分子を生産してよい。
【0141】
in vivoポリヌクレオチド送達技術
追加の実施形態では、1以上の本発明のポリヌクレオチドを含む遺伝子構築物をin vivoで細胞に導入する。これは多様な周知のアプローチのいずれかを使用して達成してよく、説明のためにそのアプローチのいつくかを以下に概説する。
【0142】
1.アデノウイルス
1以上の核酸配列をin vivoで送達するための好ましい方法の1つにはアデノウイルス発現ベクターの使用が含まれる。「アデノウイルス発現ベクター」とは、(a)構築物のパッケージングをサポートし、かつ(b)センスまたはアンチセンス方向で構築物にクローニングされているポリヌクレオチドを発現するために十分なアデノウイルス配列を含有する構築物を含むものとする。当然、アンチセンス構築物の関連では、発現には、その遺伝子産物が合成されることは必要とされない。
【0143】
該発現ベクターは遺伝子改変型のアデノウイルスを含む。36kbで、線状の二本鎖DNAウイルスであるアデノウイルスの遺伝子構成の知識によって、アデノウイルスDNAの大きな部分を7kbまでの外来性配列で置き換えることが可能になっている(Grunhaus & Horwitz, 1992)。レトロウイルスとは対照的に、宿主細胞のアデノウイルス感染では染色体への組込みが生じない。その理由は、アデノウイルスDNAがエピソーム様式で複製できるからであり、潜在的な遺伝毒性を有さない。また、アデノウイルスは構造的に安定であり、大規模な増幅後にゲノムの再編成は検出されていない。アデノウイルスは、実質的にすべての上皮細胞に、それらの細胞周期段階にかかわらずに感染することができる。現在までのところ、アデノウイルス感染は、軽度の疾患、例えばヒトの急性呼吸器疾患にしか関連しないようである。
【0144】
アデノウイルスは、その中程度のゲノムサイズ、操作の容易さ、高い力価、広い標的細胞の範囲および高い感染力のために遺伝子導入ベクターとしての使用に特に適している。該ウイルスゲノムの両端は100〜200塩基対の逆位反復(ITR)を含有する。それはウイルスDNAの複製およびパッケージングに必要なシスエレメントである。該ゲノムの初期(E)および後期(L)領域は、ウイルスDNA複製の開始によって分割される異なる転写単位を含有する。E1領域(E1AおよびE1B)は、ウイルスゲノムおよび少数の細胞遺伝子の転写の調節を担うタンパク質をコードする。E2領域(E2AおよびE2B)の発現によって、ウイルスDNA複製のためのタンパク質の合成が生じる。これらのタンパク質は、DNA複製、後期遺伝子発現および宿主細胞停止(host cell shut-off)に関与する(Renan, 1990)。大多数のウイルスキャプシドタンパク質を含む後期遺伝子の生成物は、主要後期プロモーター(MLP)によって放出される単一の初期転写産物の重大なプロセシング後にしか発現されない。該MLP(16.8m.u.に位置する)は感染の後期に特に効率的であり、このプロモーターから放出されるすべてのmRNAは5’の3成分リーダー(TPL)配列を有し、その配列はそれらを翻訳に好ましいmRNAにする。
【0145】
現行の系では、シャトルベクターおよびプロウイルスベクター間の相同組換えの結果、組換えアデノウイルスを作成する。2つのプロウイルスベクター間で起こりうる組換えのために、このプロセスから野生型アデノウイルスが生じる可能性がある。したがって、個々のプラークから単一クローンのウイルスを単離し、そのゲノム構造を検査することが重要である。
【0146】
複製欠損である現行のアデノウイルスベクターの作成および増殖は、Ad5 DNA断片によってヒト胚性腎細胞から形質転換され、E1タンパク質を構成的に発現する、293と称される特有のヘルパー細胞株に依存する(Graham et al., 1977)。アデノウイルスゲノムに由来するE3領域は不必要である(Jones & Shenk, 1978)ため、現行のアデノウイルスベクターは、293細胞の支援を受けて、E1、D3または両領域中に外来DNAを保持する(Graham & Prevec, 1991)。自然界では、アデノウイルスは約105%の野生型ゲノム(Ghosh-Choudhury et al., 1987)をパッケージングすることができ、追加の約2kBのDNAの受容能を提供する。E1およびE3領域中で取り替え可能な約5.5kBのDNAと合わせて、現行のアデノウイルスベクターの最大容量は7.5kB未満であり、すなわち該ベクターの全長の約15%である。アデノウイルスのウイルスゲノムの80%を超える部分はベクター主鎖中に残存し、それはベクターが有する細胞障害性の源である。また、E1欠失ウイルスの複製欠損は不完全である。例えば、現在利用可能なベクターを高い感染多重度(MOI)で用いた場合、ウイルス遺伝子発現の漏出が観察されている(Mulligan, 1993)。
【0147】
ヘルパー細胞株は、ヒト細胞、例えばヒト胚性腎細胞、筋細胞、造血細胞または他のヒトの胚性の間葉細胞または上皮細胞に由来してよい。あるいは、該ヘルパー細胞は、ヒトアデノウイルスに関して許容される他の哺乳動物種の細胞に由来してよい。そのような細胞には、例えばVero細胞または他のサルの胚性の間葉細胞または上皮細胞が含まれる。上記のように、現在好ましいヘルパー細胞株は293である。
【0148】
最近、Racher et al.(1995)は、293細胞を培養してアデノウイルスを増殖させるための改善された方法を開示した。一形式では、100〜200mlの培地を含有する1リットルのシリコン処理スピナーフラスコ(Techne, Cambridge, UK)中に個々の細胞を接種することによって天然細胞凝集物を培養する。40rpmで撹拌した後、トリパンブルーで細胞生存度を見積もる。別の形式では、Fibra-Celマイクロキャリア(Bibby Sterlin, Stone, UK)(5g/l)を以下のように使用する。5mlの培地に再懸濁した接種用細胞を250ml三角フラスコ中の担体(50ml)に加え、1〜4時間、時折撹拌しながら静置する。そしてその培地を50mlの新鮮な培地と取り替え、振とうを開始する。ウイルス生産では、細胞を約80%集密まで成長させ、その後、培地を取り除き(最終容量の25%にする)、アデノウイルスをMOI0.05で加える。培養を一晩静置し、その後、容量を増加させて100%にし、さらに72時間の振とうを開始する。
【0149】
アデノウイルスベクターが複製欠損、または少なくとも条件的に欠損であるという必要条件以外に、アデノウイルスベクターの性質は発明の実施を成功させるために重要ではないと考えられる。アデノウイルスは、42種の異なる公知の血清型または亜群A〜Fのいずれかであってよい。亜群Cの5型アデノウイルスは、本発明で使用するための条件的複製欠損アデノウイルスベクターを取得するために好ましい出発材料である。その理由は、5型アデノウイルスがヒトアデノウイルスであり、それについて非常に多くの生化学的情報および遺伝情報が公知であり、かつ歴史的に、ベクターとしてアデノウイルスを使用するほとんどの構築物に使用されているからである。
【0150】
上記のように、本発明の典型的ベクターは複製欠損であり、アデノウイルスE1領域を有さない。ゆえに、E1コード配列が除去されている位置で目的の遺伝子をコードするポリヌクレオチドを導入することが最も好都合である。しかし、アデノウイルス配列内の構築物を挿入する位置は本発明にとって重要ではない。目的の遺伝子をコードするポリヌクレオチドは、Karlsson et al. (1986)によって報告されているようにE3置換ベクター中の欠失E3領域の代わりに挿入するか、あるいはヘルパー細胞株またはヘルパーウイルスがE4欠損を補完する場合のE4領域中に挿入してもよい。
【0151】
アデノウイルスは容易に培養および操作することができ、in vitroおよびin vivoで広い宿主範囲を示す。この群のウイルスは高い力価、例えば109〜1011プラーク形成単位/mlで取得することができ、それらは非常に感染性である。アデノウイルスの生活環は宿主細胞ゲノム内への組込みを必要としない。アデノウイルスベクターによって送達される外来遺伝子はエピソーム性であり、したがって宿主細胞に対する低い遺伝毒性しか有さない。野生型アデノウイルスでのワクチン接種の研究において副作用は報告されていない(Couch et al., 1963; Top et al., 1971)。このことはそれらの安全性およびin vivo遺伝子導入ベクターとしての治療的潜在能力を示す。
【0152】
アデノウイルスベクターは、真核生物の遺伝子の発現(Levrero et al., 1991; Gomez-Foix et al., 1992)およびワクチン開発(Grunhaus & Horwitz, 1992; Graham & Prevec, 1992)において使用されている。最近、動物試験では、組換えアデノウイルスを遺伝子治療に使用できることが示唆された(Stratford-Perricaudet & Perricaudet, 1991; Stratford-Perricaudet et al., 1990; Rich et al., 1993)。組換えアデノウイルスを種々の組織に投与する研究には、気管点滴注入(Rosenfeld et al., 1991; Rosenfeld et al., 1992)、筋肉注射(Ragot et al., 1993)、末梢静脈内注射(Herz & Gerard, 1993)および脳内への定位接種(Le Gal La Salle et al., 1993)が含まれる。
【0153】
アデノウイルスベクターはヒトアデノウイルスに由来してよい。あるいは、それらは他の動物種、例えばチンパンジーのアデノウイルスに由来してよく、それは、多数のヒト被験体中で循環している、ヒトアデノウイルスに対する抗体によってウイルスベクターが中和されないという利点を有するかもしれない(例えば:Tatsis N et al (2005) Gene Ther. Dec 1; [Epub ahead of print]を参照のこと)。
【0154】
2.レトロウイルス
レトロウイルスは、感染細胞中で逆転写のプロセスによってそれらのRNAを二本鎖DNAに変換する能力を特徴とする一本鎖RNAウイルスの一群である(Coffin, 1990)。そのように生じたDNAはプロウイルスとして細胞の染色体内に安定に組込まれ、ウイルスタンパク質の合成を導く。組込みの結果、レシピエント細胞およびその子孫においてウイルス遺伝子の配列が保持される。レトロウイルスのゲノムは、それぞれキャプシドタンパク質、ポリメラーゼ酵素、およびエンベロープ成分をコードするgag、pol、およびenvの3遺伝子を含有する。gag遺伝子の上流に見出される配列は、ゲノムをビリオン内へパッケージングするためのシグナルを含有する。ウイルスゲノムの5’および3’末端に2個の長末端反復(LTR)配列が存在する。これらは強いプロモーターおよびエンハンサー配列を含有し、宿主細胞ゲノムへの組込みにも必要とされる(Coffin, 1990)。
【0155】
レトロウイルスベクターを構築するために、ウイルスゲノムの特定のウイルス配列の代わりに1以上の目的のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸を挿入し、複製欠損であるウイルスを得る。ビリオンを生産するために、gag、pol、およびenv遺伝子を含有するが、LTRおよびパッケージング成分を有さないパッケージング細胞株を構築する(Mann et al., 1983)。レトロウイルスのLTRおよびパッケージング配列と共にcDNAを含有する組換えプラスミドをこの細胞株に(例えばリン酸カルシウム沈殿によって)導入すると、該パッケージング配列によって、該組換えプラスミドのRNA転写産物をウイルス粒子中にパッケージングさせることが可能になり、そしてそれは培養培地中に分泌される(Nicolas & Rubenstein, 1988; Temin, 1986; Mann et al., 1983)。そして該組換えレトロウイルスを含有する培地を収集し、場合により濃縮し、遺伝子導入に使用する。レトロウイルスベクターは多種多様な細胞タイプに感染することができる。しかし、組込みおよび安定な発現には宿主細胞の分裂が必要とされる(Paskind et al., 1975)。
【0156】
ウイルスエンベロープにラクトース残基を化学付加することによるレトロウイルスの化学修飾に基づいて、レトロウイルスベクターの特異的ターゲティングを可能にするように設計された新規アプローチが最近開発された。この改変は、シアロ糖タンパク質受容体を介する肝細胞の特異的感染を可能にしうる。
【0157】
レトロウイルスエンベロープタンパク質および特異的細胞受容体に対するビオチン化抗体を使用する、組換えレトロウイルスをターゲティングするための異なるアプローチが設計された。ストレプトアビジンを使用し、ビオチン成分を介して該抗体をカップリングした(Roux et al., 1989)。主要組織適合複合体クラスIおよびクラスII抗原に対する抗体を使用して、それらは、in vitroで、エコトロピックウイルスを用いた、該表面抗原を有する種々のヒト細胞の感染を実証した(Roux et al., 1989)。
【0158】
3.アデノ随伴ウイルス
AAV(Ridgeway, 1988; Hermonat & Muzycska, 1984)はパルボウイルス(parovirus)であり、アデノウイルスストックの混入物として発見された。それは遍在性ウイルス(米国のヒト集団の85%に抗体が存在する)であり、いかなる疾患とも関連付けられていない。それは、また、その複製がヘルパーウイルス、例えばアデノウイルスの存在に依存することから、ディペンドウイルス属として分類される。5血清型が特定されていて、そのうちAAV−2が最もよく特徴付けされている。AAVは、直径20〜24nmの正二十面体ビリオンを形成するキャプシドタンパク質VP1、VP2およびVP3内に包まれている一本鎖線状DNAを有する(Muzyczka & McLaughlin, 1988)。
【0159】
AAVのDNAは約4.7キロベース長である。それは2種のオープンリーディングフレームを含有し、2個のITRが隣接している。AAVゲノムには、2個の主要遺伝子:repおよびcapが存在する。rep遺伝子はウイルス複製を担うタンパク質をコードし、一方capはキャプシドタンパク質VP1〜3をコードする。各ITRはT字型のヘアピン構造を形成する。これらの末端反復は、染色体組込みのためのAAVの唯一の必須シス成分である。したがって、すべてのウイルスコード配列が除去され、送達用の遺伝子カセットで置き換えられているベクターとしてAAVを使用することができる。3個のウイルスプロモーターが特定されていて、その地図上の位置にしたがって、p5、p19、およびp40と称される。p5およびp19からの転写の結果、repタンパク質が生産され、p40からの転写の結果、キャプシドタンパク質が生産される(Hermonat & Muzyczka, 1984)。
【0160】
研究者がrAAVを発現ベクターとして使用する可能性を研究することを促すいくつかの要因が存在する。その1つは、遺伝子を送達して宿主染色体中に組込むための必要条件が、驚くべきことに、わずかしかないことである。145bpのITRを有することが必要であり、それはAAVゲノムのほんの6%である。これにより、該ベクター中には、4.5kbのDNA挿入を組み立てるための余地が残る。この積載能力はAAVが大きい遺伝子を送達することを妨げるかもしれないが、本発明のアンチセンス構築物の送達には十分に適している。
【0161】
AAVは、また、その安全性のために送達ビヒクルの良好な選択肢である。比較的複雑な救済機構が存在し:AAV遺伝子は野生型アデノウイルスだけでなくrAAVも動員しなければならない。同様に、AAVは病原性でなく、いかなる疾患とも関連していない。ウイルスコード配列を除去するとウイルス遺伝子の発現に対する免疫反応が最小になり、したがってrAAVは炎症反応を誘発しない。
【0162】
4.発現構築物としての他のウイルスベクター
オリゴヌクレオチドまたはポリヌクレオチド配列を宿主細胞に送達するために、本発明の発現構築物として他のウイルスベクターを使用してよい。ウイルス、例えばワクシニアウイルス(Ridgeway, 1988; Coupar et al., 1988)、レンチウイルス、ポリオウイルスおよびヘルペスウイルスに由来するベクターを使用してよい。それらは種々の哺乳動物細胞に関するいくつかの魅力的な特徴を提供する(Friedmann, 1989; Ridgeway, 1988; Coupar et al., 1988; Horwich et al., 1990)。
【0163】
最近、欠損B型肝炎ウイルスが認識され、種々のウイルス配列の構造と機能の関連性への新規洞察が得られた。in vitro研究では、該ウイルスは、80%までのゲノムの欠失にもかかわらず、ヘルパー依存的パッケージングおよび逆転写の能力を保持できることが示された(Horwich et al., 1990)。このことは、ゲノムの大部分を外来性遺伝物質で置き換えることができることを示唆した。肝親和性(hepatotropism)および持続性(組込み)は、肝臓を標的にする遺伝子導入に関して特に魅力的な特性であった。Chang et al.(1991)は、クロラムフェニコールアセチルトランスフェラーゼ(CAT)遺伝子をアヒルB型肝炎ウイルスゲノムのポリメラーゼ、表面、およびプレ表面コード配列の代わりに導入した。それはトリ肝細胞腫細胞株に野生型ウイルスと同時にトランスフェクトされた。高い力価の組換えウイルスを含有する培養培地を使用して初代子ガモ肝細胞を感染させた。トランスフェクション後、少なくとも24日間、安定なCAT遺伝子発現が検出された(Chang et al., 1991)。
【0164】
5.非ウイルスベクター
本発明のオリゴヌクレオチドまたはポリヌクレオチド配列の発現を実行するためには、発現構築物を細胞内に送達しなければならない。この送達は、細胞株を形質転換するための実験室手順の場合のようにin vitroで、あるいは特定の疾患状態の治療の場合のようにin vivoまたはex vivoで達成してよい。上記のように、好ましい送達機構の1つは、発現構築物が感染性ウイルス粒子中に封入されているウイルスの感染を介するものである。
【0165】
該発現構築物が細胞内に送達されると、所望のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は種々の部位に配置され発現される。特定の実施形態では、該構築物をコードする核酸は細胞のゲノム内に安定に組込まれる。この組込みは相同組換え(遺伝子置換)を介する特異的な位置および方向での組込みであってよく、ランダムな非特異的な位置(遺伝子付加(gene augmentation))で組込まれてもよい。さらに別の実施形態では、該核酸は、分離したエピソーム性DNAセグメントとして細胞中で安定に維持される。そのような核酸セグメントまたは「エピソーム」は、宿主細胞周期と無関係の、あるいはそれと同期した維持および複製を可能にするために十分な配列をコードする。該発現構築物がどのように細胞に送達されるか、および該核酸が細胞内のどこにとどまるかは、使用される発現構築物のタイプに依存する。
【0166】
本発明の特定の実施形態では、1以上のオリゴヌクレオチドまたはポリヌクレオチド配列を含む発現構築物は、単に裸の組換えDNAまたはプラスミドからなる。該構築物の導入は、細胞膜を物理的または化学的に透過性にする上記方法のいずれかによって実施してよい。このことは、in vitroでの導入に特に適用可能であるが、in vivoでの使用にも同様に適用してよい。Dubensky et al.(1984)は、リン酸カルシウム沈殿の形式のポリオーマウイルスDNAを成体および新生児マウスの肝臓および脾臓に首尾良く注射し、活発なウイルス複製および急性感染を実証した。Benvenisty & Reshef(1986)は、また、リン酸カルシウム沈殿済みプラスミドを直接腹腔内注射すると、トランスフェクトされた遺伝子の発現が生じることを実証した。また、目的の遺伝子をコードするDNAをin vivoで同様の様式で導入し、該遺伝子産物を発現させることが想定される。
【0167】
裸のDNA発現構築物を細胞内に導入するための本発明の別の実施形態には、微粒子銃が含まれる。この方法は、DNAでコーティングされた微粒子を高速に加速して、それらが細胞膜を貫通して、細胞を死滅させることなく細胞に入ることを可能にする能力に依存する(Klein et al., 1987)。小粒子を加速するためのいくつかの装置が開発されている。そのような装置の1つは、電流を生成するための高電圧の放電に依存し、そしてその電流が輸送力を提供する(Yang et al., 1990)。使用される微粒子は、生物学的に不活性な物質、例えばタングステンまたは金ビーズから構成されている。
【0168】
ラットおよびマウスの選択された器官、例えば肝臓、皮膚、および筋組織にin vivoで粒子を衝突させている(Yang et al., 1990; Zelenin et al., 1991)。これは、銃と標的器官の間の任意の介在組織を排除するために組織または細胞の外科的露出、すなわちex vivo処理を必要とするかもしれない。やはり、特定の遺伝子をコードするDNAをこの方法によって送達してよく、依然として本発明に包含される。
【0169】
ポリペプチド組成物
本発明は、他の態様では、ポリペプチド組成物を提供する。一般に、本発明のポリペプチドは、哺乳動物種に由来する単離されたポリペプチド(またはエピトープ、変異体、またはその活性断片)である。好ましくは、該ポリペプチドは、本明細書中で開示されるポリヌクレオチド配列または本明細書中で開示されるポリヌクレオチド配列に中程度にストリンジェントな条件下でハイブリダイズする配列によってコードされる。あるいは、該ポリペプチドは、本明細書中で開示されるアミノ酸配列に由来する連続アミノ酸配列を含むポリペプチド、または本明細書中で開示されるアミノ酸配列全体を含むポリペプチドとして規定される。
【0170】
免疫原性部分は、概して、周知の技術、例えばPaul, Fundamental Immunology, 3rd ed., 243-247 (1993)およびその引用文献で概説されている技術を使用して特定してよい。そのような技術には、抗原特異的抗体、抗血清および/またはT細胞株またはクローンと反応する能力に関してポリペプチドをスクリーニングすることが含まれる。本明細書中で使用されるように、抗血清および抗体は、それらが抗原に特異的に結合する(すなわちそれらがELISAまたは他の免疫アッセイにおいてタンパク質と反応し、無関係のタンパク質と検出可能な程度に反応することはない)場合に「抗原特異的」である。そのような抗血清および抗体は、本明細書中に記載のように、周知の技術を使用して調製してよい。マイコバクテリア属の種のタンパク質の免疫原性部分は、全長ポリペプチドの反応性に実質的に劣らないレベルで前記抗血清および/またはT細胞と反応する部分である(例えばELISAおよび/またはT細胞反応性アッセイにおいて)。そのような免疫原性部分は、前記アッセイ内で全長ポリペプチドの反応性と同様のレベルまたはそれを超えるレベルで反応する。そのようなスクリーニングは、概して、当業者に周知の方法、例えばHarlow & Lane, Antibodies: A Laboratory Manual (1988)およびUsing Antibodies: A Laboratory Manual (1998)に記載の方法を使用して実施してよい。例えば、ポリペプチドを固形支持体上に固定し、患者の血清と接触させて、固定ポリペプチドに対する該血清内の抗体の結合を可能にしてよい。次いで未結合血清を除去し、例えば125I標識プロテインAを使用して結合抗体を検出してよい。
【0171】
ポリペプチドは種々の周知の技術のいずれかを使用して調製してよい。上記のようなDNA配列によってコードされる組換えポリペプチドは、当業者に公知の種々の発現ベクターのいずれかを使用してDNA配列から容易に調製してよい。発現は、組換えポリペプチドをコードするDNA分子を含有する発現ベクターで形質転換またはトランスフェクトされている任意の適切な宿主細胞において達成してよい。好適な宿主細胞には、原核生物、酵母、および高等真核細胞、例えば哺乳動物細胞および植物細胞が含まれる。好ましくは、使用される宿主細胞は、大腸菌、酵母または哺乳動物細胞株、例えばCOSまたはCHOである。組換えタンパク質またはポリペプチドを培養培地に分泌する好適な宿主/ベクター系から得られた上清を、まず、市販のフィルターを使用して濃縮してよい。濃縮後、その濃縮物を好適な精製マトリックス、例えば親和性マトリックスまたはイオン交換樹脂に適用してよい。最後に、1以上の逆相HPLCステップを使用して、組換えポリペプチドをさらに精製することができる。
【0172】
約100未満のアミノ酸、概して約50未満のアミノ酸を有する本発明のポリペプチド、その免疫原性断片、および他の変異体は、当業者に周知の技術を使用する合成手段によって作成してもよい。例えば、そのようなポリペプチドは、市販の固相技術のいずれか、例えばメリフィールド固相合成法を使用して合成してよく、その場合、成長中のアミノ酸鎖にアミノ酸が順次付加される。Merrifield, J. Am. Chem. Soc. 85:2149-2146 (1963)を参照のこと。ポリペプチドの自動合成のための設備がPerkin Elmer/Applied BioSystems Division(Foster City, CA)のような供給元から市販されており、それを製造元の指示書にしたがって操作してよい。
【0173】
ある具体的な実施形態では、ポリペプチドは、本明細書中に記載の複数のポリペプチドを含むか、あるいは本明細書中に記載の少なくとも1つのポリペプチドおよび無関係の配列、例えば公知の腫瘍タンパク質を含む融合タンパク質であってよい。融合パートナーは、例えば、Tヘルパーエピトープ、好ましくはヒトによって認識されるTヘルパーエピトープの提供を支援(免疫学的融合パートナー)してよく、あるいはネイティブの組換えタンパク質より高い収率での該タンパク質の発現を支援(発現エンハンサー)してよい。特定の好ましい融合パートナーは免疫学的かつ発現増強性の融合パートナーである。該タンパク質の溶解度を増加させるか、あるいは該タンパク質を所望の細胞内区画にターゲティングさせることが可能であるように、他の融合パートナーを選択してよい。さらに別の融合パートナーには親和性タグが含まれ、それは該タンパク質の精製を容易にする。
【0174】
融合タンパク質は、一般的に、標準的技術、例えば化学的コンジュゲーションを使用して調製してよい。好ましくは、発現系において、融合タンパク質を組換えタンパク質として発現させ、非融合タンパク質と比べて高レベルの生産を可能にする。簡潔に言えば、前記ポリペプチド成分をコードするDNA配列を別々に組み立てて、適切な発現ベクター中にライゲーションしてよい。1つのポリペプチド成分をコードするDNA配列の3’末端を、ペプチドリンカーを用いるか、あるいは用いずに、第二のポリペプチド成分をコードするDNA配列の5’末端にライゲーションし、該配列のリーディングフレームが一致するようにする。これにより、両成分ポリペプチドの生物学的活性を保持する単一融合タンパク質への翻訳が可能になる。
【0175】
ペプチドリンカー配列を使用して、第一および第二のポリペプチド成分を、各ポリペプチドが折りたたまれてその二次および三次構造になることを保証するために十分な距離で分離してよい。そのようなペプチドリンカー配列は、当技術分野において周知の標準的技術を使用して融合タンパク質に組み入れられる。好適なペプチドリンカー配列は以下の要因に基づいて選択してよい:(1)可動性の伸張したコンフォメーションをとることができること;(2)第一および第二のポリペプチド上の機能的エピトープと相互作用しうる二次構造をとることができないこと;および(3)該ポリペプチドの機能的エピトープと反応する可能性がある疎水性または荷電残基を欠いていること。好ましいペプチドリンカー配列は、Gly、AsnおよびSer残基を含有する。他のほぼ中性のアミノ酸、例えばThrおよびAlaをリンカー配列中で使用してもよい。リンカーとして有用に使用されるアミノ酸配列には、Maratea et al., Gene 40:39-46 (1985); Murphy et al., Proc. Natl. Acad. Sci. USA 83:8258-8262 (1986); 米国特許第4,935,233号および同第4,751,180号に開示される配列が含まれる。該リンカー配列は、概して、1〜約50アミノ酸長であってよい。第一および第二のポリペプチドが、機能的ドメインを分離して立体的干渉を妨げるために使用できる必須でないN末端アミノ酸領域を有する場合には、リンカー配列は必要とされない。
【0176】
ライゲーションされたDNA配列は好適な転写または翻訳調節エレメントに作動可能に連結される。DNAの発現を担う調節エレメントは、第一のポリペプチドをコードするDNA配列の5’側にのみ位置する。同様に、翻訳を終わらせるために必要とされる停止コドンおよび転写終結シグナルは第二のポリペプチドをコードするDNA配列の3’側にのみ存在する。
【0177】
融合タンパク質もまた提供される。そのようなタンパク質は、無関係の免疫原性タンパク質と共に本明細書中に記載のポリペプチドを含む。好ましくは免疫原性タンパク質はリコール応答(recall response)を誘発可能なものである。そのようなタンパク質の例には、テタヌス、結核および肝炎タンパク質が含まれる(例えばStoute et al., New Engl. J. Med. 336:86-91 (1997)を参照のこと)。
【0178】
好ましい実施形態では、免疫学的融合パートナーは、グラム陰性菌であるインフルエンザ菌B(Haemophilus influenza B)の表面タンパク質であるプロテインDに由来する(WO91/18926)。好ましくは、プロテインD誘導体は該タンパク質の最初の約3分の1(例えば最初のN末端100〜110アミノ酸)を含み、プロテインD誘導体は脂質化してよい。特定の好ましい実施形態では、リポプロテインD融合パートナーの最初の109残基をN末端に包含させて、追加の外因性T細胞エピトープを有するポリペプチドを提供し、大腸菌での発現レベルを増加させる(ゆえに発現エンハンサーとして機能する)。脂質の尾部によって、抗原提示細胞への該抗原の最適な提示が保証される。他の融合パートナーには、インフルエンザウイルスに由来する非構造タンパク質、NS1(赤血球凝集素(hemaglutinin))が含まれる。典型的にN末端81アミノ酸が使用されるが、Tヘルパーエピトープを含む異なる断片を使用してよい。
【0179】
別の実施形態では、免疫学的融合パートナーはLYTAとして知られるタンパク質、またはその一部分(好ましくはC末端部分)である。LYTAは肺炎連鎖球菌(Streptococcus pneumoniae)に由来し、それはアミダーゼLYTAとして知られるN−アセチル−L−アラニンアミダーゼを合成する(LytA遺伝子によってコードされる; Gene 43:265-292 (1986))。LYTAは、ペプチドグリカン主鎖中の特定の結合を特異的に分解する自己溶解素である。LYTAタンパク質のC末端ドメインはコリンまたはいくつかのコリンアナログ、例えばDEAEに対する親和性を担う。この特性は、融合タンパク質の発現に有用な大腸菌C−LYTA発現プラスミドの開発に活用されている。アミノ末端にC−LYTA断片を含有するハイブリッドタンパク質の精製が報告されている(Biotechnology 10:795-798 (1992)を参照のこと)。好ましい実施形態では、LYTAの反復部分を融合タンパク質中に組み入れてよい。反復部分は、残基178で出発するC末端領域中に見出せる。特に好ましい反復部分は残基188〜305を含んでいる。
【0180】
一般に、本明細書中に記載のポリペプチド(融合タンパク質を含む)およびポリヌクレオチドは単離型である。「単離された」ポリペプチドまたはポリヌクレオチドとは、その元の環境から取り出されたものである。例えば、天然に存在するタンパク質は、自然系におけるいくつかまたはすべての共存物質から分離されている場合に単離されている。好ましくは、そのようなポリペプチドは少なくとも約90%の純度を有し、より好ましくは少なくとも約95%純度、最も好ましくは少なくとも約99%の純度を有する。ポリヌクレオチドは、例えば、天然環境の一部分ではないベクターにクローニングされている場合に単離されていると見なされる。
【0181】
T細胞
免疫療法組成物は、さらに、またはあるいは、マイコバクテリウム抗原に特異的なT細胞を含んでよい。そのような細胞は、概して、標準的手順を使用してin vitroまたはex vivoで調製してよい。例えば、市販の細胞分離系、例えばNexell Therapeutics, Inc.(Irvine, CA)から入手可能なIsolexTM System(さらに米国特許第5,240,856号;同第5,215,926号;WO89/06280;WO91/16116およびWO92/07243を参照のこと)を使用して、患者の骨髄、末梢血、または骨髄もしくは末梢血の一部分からT細胞を単離してよい。あるいは、T細胞は、関連または無関連のヒト、非ヒト哺乳動物、細胞株または培養に由来してよい。
【0182】
T細胞は、本発明のポリペプチド、該ポリペプチドをコードするポリヌクレオチド、および/または該ポリペプチドを発現している抗原提示細胞(APC)で刺激してよい。該ポリペプチドに特異的なT細胞の産生を可能にするために十分な条件下でかつ十分な時間、そのような刺激を実施する。好ましくは、該ポリペプチドまたはポリヌクレオチドを送達ビヒクル、例えばミクロスフェア内に存在させて、特異的T細胞の産生を促進する。
【0183】
T細胞が特異的に増殖し、サイトカインを分泌するか、あるいは、該ポリペプチドでコーティングされている標的細胞または該ポリペプチドをコードする遺伝子を発現している標的細胞を死滅させれば、該T細胞は本発明のポリペプチドに特異的であると見なされる。T細胞の特異性は種々の標準的技術のいずれかを使用して評価してよい。例えば、クロム放出アッセイまたは増殖アッセイでは、溶解および/または増殖が陰性対照と比較して2倍を超える増加を示す刺激指数がT細胞の特異性を示す。そのようなアッセイは、例えばChen et al., Cancer Res. 54:1065-1070 (1994)に記載のように実施してよい。あるいは、種々の公知の技術によってT細胞増殖の検出を達成してよい。例えば、T細胞の増殖は、DNA合成の割合の増加を測定することによって(例えば、トリチウム標識チミジンでT細胞培養をパルス標識し、DNA中に取り込まれたトリチウム標識チミジンの量を測定することによって)検出することができる。本発明のポリペプチド(100ng/ml〜100μg/ml、好ましくは200ng/ml〜25μg/ml)を3〜7日間接触させると、T細胞の増殖が少なくとも2倍増加するべきであろう。2〜3時間の上記接触により、T細胞の活性化が生じるはずであろう。その活性化は標準サイトカインアッセイを使用して測定され、その場合、サイトカイン放出(例えばTNFまたはIFN−γ)のレベルの2倍増加がT細胞の活性化を示す(Coligan et al., Current Protocols in Immunology, vol. 1 (1998)を参照のこと)。ポリペプチド、ポリヌクレオチドまたはポリペプチド発現APCに応答して活性化されたT細胞はCD4+および/またはCD8+であるかもしれない。タンパク質特異的T細胞は標準的技術を使用して増殖させてよい。好ましい実施形態では、T細胞は、患者、血縁関係ドナーまたは非血縁関係のドナーに由来し、刺激および増殖後に該患者に投与される。
【0184】
治療目的では、ポリペプチド、ポリヌクレオチドまたはAPCに応答して増殖するCD4+またはCD8+T細胞の数をin vitroまたはin vivoで増やすことができる。そのようなT細胞のin vitroでの増殖は種々の方法で達成してよい。例えば、ポリペプチド、または該ポリペプチドの免疫原性部分に相当する短いペプチドにT細胞を再度曝露することができ、その場合、T細胞成長因子、例えばインターロイキン−2、および/またはポリペプチドを合成する刺激細胞を加えるか、あるいは加えない。あるいは、タンパク質の存在下で増殖する1種以上のT細胞の数を、クローニングによって増やすことができる。細胞をクローニングするための方法は当技術分野において周知であり、それには限界希釈が含まれる。
【0185】
医薬組成物
追加の実施形態では、本発明は、単独で、あるいは1以上の他の様式の治療と組み合わせて、細胞または動物に投与するための、製薬上許容される溶液中の本明細書中で開示される1以上のポリヌクレオチド、ポリペプチド、T細胞、抗体、および化学療法組成物の製剤に関する。
【0186】
所望であれば、本明細書中で開示されるポリペプチドを発現する核酸セグメント(例えばRNAまたはDNA)を、さらに他の物質、例えば他のタンパク質もしくはポリペプチドまたは種々の医薬活性物質、例えば結核菌感染に対して有効な化学療法剤と組み合わせて投与してよいことがさらに理解される。実際、標的細胞または宿主組織との接触時に追加の物質が重大な有害作用を生じさせないならば、同時に含ませてよい他の成分に関する制限は実質的にない。ゆえに、該組成物は、具体的な事例において必要とされる種々の他の物質と共に送達してよい。そのような組成物は、宿主細胞または他の生物学的供給源から精製するか、あるいは本明細書中に記載のように化学合成してよい。同様に、そのような組成物は置換または誘導体化RNAまたはDNA組成物をさらに含んでよい。
【0187】
製薬上許容される賦形剤および担体溶液の製剤化は当業者に周知であり、それは種々の治療レジメン、例えば経口、非経口、静脈内、鼻腔内、および筋肉内投与および製剤において本明細書中に記載の具体的な組成物を使用するために好適な投薬および治療レジメンの開発も同様である。典型的には、治療有効量を含む製剤は、約2μg〜約50μgのMtb72fポリペプチド/投与、より典型的には約5μg〜約40μgのMtb72fポリペプチド/投与を送達する。
【0188】
1.経口送達
特定の適用では、本明細書中で開示される医薬組成物は経口投与によって動物に送達してよい。そのような場合、これらの組成物は不活性希釈剤または吸収可能な食用担体と共に製剤化してよく、あるいはゼラチン硬カプセルまたはゼラチン軟カプセル(hard- or soft-shell gelatin capsule)に封入してよく、あるいは圧縮して錠剤にしてよく、あるいは食事の食物中に直接組み入れてよい。
【0189】
活性化合物は、賦形剤と共に組み入れてよく、経口摂取可能な錠剤、口腔錠(buccal tables)、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、ウェーハ剤、等の剤形で使用してもよい(Mathiowitz et al., 1997; Hwang et al., 1998; 米国特許第5,641,515号;同第5,580,579号および同第5,792,451号(各文献は参照によりその全体が本明細書中に具体的に組み入れられる))。該錠剤、トローチ剤、丸剤、カプセル剤等は以下のものを含有してもよい:結合剤、例えばトラガカントガム、アカシア、コーンスターチ、またはゼラチン;賦形剤、例えば第二リン酸カルシウム;崩壊剤、例えばコーンスターチ、ジャガイモデンプン、アルギン酸等;滑沢剤、例えばステアリン酸マグネシウム;および甘味剤、例えばショ糖、ラクトースまたはサッカリン、あるいは香味物質、例えばペパーミント、冬緑油、またはサクランボ香味物質を加えてよい。単位投与剤形がカプセル剤である場合、上記タイプの物質に加えて、液体担体を含有してよい。種々の他の物質を、コーティングとして、あるいは該投与単位の物理的形状を別の様式で修飾するために存在させてよい。例えば、錠剤、丸剤、またはカプセル剤は、シェラック、糖、または両者でコーティングしてよい。シロップ剤またはエリキシル剤は、活性化合物、甘味剤であるショ糖、保存剤であるメチルおよびプロピルパラベン、色素および香味物質、例えばサクランボまたはオレンジ香料を含有してよい。当然、すべての単位投与剤形の調製に使用されるすべての物質は、製薬的に純粋であり、かつ使用される量において実質的に無毒であるべきであろう。さらに、該活性化合物を徐放性調製物および製剤中に組み入れてよい。
【0190】
典型的に、これらの製剤は、通常、2μg〜50μgの範囲のMtb72fポリペプチドを含有する。当然、調製することができる治療上有用な各組成物中の活性化合物(群)の量は、該化合物の任意の所定の単位容量において好適な用量が達成されるような量である。そのような医薬製剤を調製する技術分野の当業者には、溶解度、バイオアベイラビリティ、生物学的半減期、投与経路、製品保存期間、ならびに他の薬理学的考察等の要因が考慮され、それゆえに、種々の用量および治療レジメンが望ましいかもしれない。
【0191】
経口投与では、別法として、本発明の組成物を、洗口剤、歯磨剤、口腔錠、経口スプレー、または舌下経口投与用製剤の剤形中に1以上の賦形剤と共に組み入れてよい。例えば、ホウ酸ナトリウム溶液(ドーベル液)等の適切な溶媒中に必要量の活性成分を包含する洗口剤を調製してよい。あるいは、活性成分を経口溶液、例えばホウ酸ナトリウム、グリセリンおよび炭酸水素カリウムを含有する溶液に組み入れるか、あるいは歯磨剤中に分散させるか、あるいは、水、結合剤、研磨剤、香味物質、発泡剤、および湿潤剤を含んでよい治療有効量の組成物に加えてよい。あるいは、該組成物は、舌下に投与されるか、あるいは別の様式で口内で溶解させてよい錠剤または溶液剤の剤形で作成してよい。
【0192】
2.注射送達
特定の状況では、本明細書中で開示される医薬組成物を、非経口、静脈内、筋肉内、または、さらには腹腔内で送達することが望ましい。それは、米国特許第5,543,158号;同第5,641,515号および同第5,399,363号に記載される通りである(各文献は参照によりその全体が本明細書中に具体的に組み入れられる)。遊離塩基である活性化合物または製薬上許容されるその塩の溶液剤を、界面活性剤、例えばヒドロキシプロピルセルロースと好適に混合された水中で調製してよい。さらに、グリセロール、液状ポリエチレングリコール、およびその混合物および油中で分散剤を調製してよい。通常の保存および使用条件下では、これらの調製物は、微生物の生育を妨げるための保存剤を含有する。
【0193】
注入使用に適した医薬剤形には、滅菌水性溶液剤または分散剤および、滅菌注射用溶液剤または分散剤の即席調製のための滅菌粉末剤が含まれる(米国特許第5,466,468号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。全事例で、その剤形は無菌である必要があり、容易な注射針通過性(syringability)が存在する程度に液状である必要がある。それは、製造および保存条件下で安定である必要があり、微生物、例えば細菌および真菌の混入作用に対して保護される必要がある。該担体は、溶媒または分散媒でありうる。それには、例えば水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール、および液状ポリエチレングリコール、等)、好適なその混合物、および/または植物油が含有される。例えば、コーティング、例えばレシチンの使用によって、分散剤の場合、必要な粒子サイズの維持によって、ならびに界面活性剤の使用によって、適正な流動性を維持してよい。微生物の作用の防止は、種々の抗菌剤および抗真菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール、等によって促進することができる。多数の場合、等張性物質、例えば糖または塩化ナトリウムを含ませることが好ましい。注射用組成物の長期吸収は、吸収を遅延させる物質、例えばモノステアリン酸アルミニウムおよびゼラチンを組成物中で使用することによって達成することができる。
【0194】
水性溶液剤中での非経口投与では、例えば、必要であれば該溶液剤を好適に緩衝化するべきであろう。まず、十分な生理食塩水またはグルコースを用いて該液状希釈剤を等張性にするべきであろう。これらの特定の水性溶液剤は、静脈内、筋肉内、皮下および腹腔内投与に特に適している。この関連で、使用できる滅菌水性媒体は、本開示内容を踏まえて当業者に公知である。例えば、一用量を1mlの等張性NaCl溶液に溶解し、1000mlの皮下注入用の液体に加えるか、あるいは注入の予定部位に注射してよい(例えばRemington's Pharmaceutical Sciences, 15th Edition, pp. 1035-1038および1570-1580を参照のこと)。治療対象の被験体の症状に応じていくらかの用量バリエーションが必然的に存在する。いずれにしても、処方担当者が個々の被験体に適切な用量を決定する。さらに、ヒトへの投与では、調製物は、FDA Office of Biologics standardsが要求する滅菌性、発熱原性、および全般的安全性および純度基準を満たすべきであろう。
【0195】
滅菌注射用溶液剤は、必要量の活性化合物を、必要に応じて上に列挙される種々の他の成分と共に、適切な溶媒中に組み入れた後、ろ過滅菌することによって調製される。概して、分散剤は、ベースとなる分散媒および、上に列挙されるものから選択される必要な他の成分を含有する滅菌ビヒクル中に種々の滅菌済み活性成分を組み入れることによって調製される。滅菌注射用溶液剤の調製のための滅菌粉末剤の場合、好ましい調製方法は真空乾燥および凍結乾燥技術であり、それにより、活性成分と任意の所望の追加成分からなる粉末剤が、あらかじめ滅菌ろ過されたその溶液から得られる。
【0196】
本明細書中で開示される組成物は中性または塩形式で製剤化してよい。製薬上許容される塩には、酸付加塩(タンパク質の遊離アミノ基と共に形成される)であって、無機酸、例えば塩酸もしくはリン酸、または酢酸、シュウ酸、酒石酸、マンデル酸、等の有機酸と共に形成されるものが含まれる。また、遊離カルボキシル基と共に形成される塩を、無機塩基、例えばナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第二鉄、およびイソプロピルアミン、トリメチルアミン、ヒスチジン、プロカイン等の有機塩基から生成させることができる。製剤化された溶液剤は、投与製剤に適合する様式でかつ治療上有効な量で投与される。該製剤は、注射用溶液剤、薬物放出カプセル、等の種々の剤形で容易に投与することができる。
【0197】
本明細書中で使用される「担体」には、任意およびすべての溶媒、分散媒、ビヒクル、コーティング、希釈剤、抗菌剤および抗真菌剤、等張性および吸収遅延物質、バッファー、担体溶液、懸濁液、コロイド、等が含まれる。医薬活性物質に関するそのような媒体および物質の使用は当技術分野において周知である。任意の慣用の媒体または物質が活性成分と不適合性である場合を除いて、治療用組成物中でのその使用が想定される。該組成物に補充活性成分を組み入れることもできる。
【0198】
「製薬上許容される」との言い回しは、ヒトへの投与時に、アレルギー性または同様の有害反応を生じさせない分子的実体および組成物を表す。活性成分としてタンパク質を含有する水性組成物の調製は当技術分野において十分に理解されている。典型的に、そのような組成物は注射剤として、液状溶液剤または懸濁剤として調製され;注射前の液体中の溶解または懸濁に適した固形剤形を調製することもできる。該調製物は乳化することもできる。
【0199】
3.鼻腔および口腔送達
特定の実施形態では、鼻内スプレー、口腔スプレー、吸入、および/または他のエアロゾル送達ビヒクルによって医薬組成物を送達してよい。遺伝子、核酸、およびペプチド組成物を、例えば鼻腔および口腔エアロゾルスプレーによって直接に肺に送達するための方法は、例えば米国特許第5,756,353号および同第5,804,212号に記載されている(各文献は参照によりその全体が本明細書中に具体的に組み入れられる)。同様に、鼻腔内微粒子樹脂(Takenaga et al., 1998)およびリゾホスファチジル−グリセロール化合物(米国特許第5,725,871号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))を使用する薬物送達もまた、製薬技術分野において周知である。同様に、ポリテトラフルオロエチレン支持マトリックス(polytetrafluoroetheylene support matrix)の形式での経粘膜薬物送達が米国特許第5,780,045号に記載されている(該文献は参照によりその全体が本明細書中に具体的に組み入れられる)。
【0200】
4.リポソーム媒介性、ナノカプセル媒介性、および微粒子媒介性送達
特定の実施形態では、本発明者らは、本発明の組成物を好適な宿主細胞内に導入するための、リポソーム、ナノカプセル、微粒子、ミクロスフェア、脂質粒子、小胞、等の使用を意図する。特に、本発明の組成物は、脂質粒子、リポソーム、小胞、ナノスフェア、またはナノ粒子等に封入して送達するために製剤化してよい。
【0201】
そのような製剤は、本明細書中で開示される核酸または構築物の製薬上許容される製剤の導入に好ましいかもしれない。リポソームの形成および使用は、当業者に一般に公知である(例えばCouvreur et al., 1977; Couvreur, 1988; Lasic, 1998を参照のこと(該文献には、細胞内細菌感染および疾患に関する標的化された抗生物質治療におけるリポソームおよびナノカプセルの使用が記載されている))。最近、改善された血清安定性および循環半減期を有するリポソームが開発された(Gabizon & Papahadjopoulos, 1988; Allen and Choun, 1987; 米国特許第5,741,516号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。さらに、潜在的薬物担体としてのリポソームおよびリポソーム様調製物を調製する種々の方法が概説されている(Takakura, 1998; Chandran et al., 1997; Margalit, 1995; 米国特許第5,567,434号;同第5,552,157号;同第5,565,213号;同第5,738,868号および同第5,795,587号(各文献は参照によりその全体が本明細書中に具体的に組み入れられる))。
【0202】
リポソームは、通常、他の手順によるトランスフェクションに抵抗性のいくつかの細胞タイプ、例えばT細胞懸濁物、初代肝細胞培養およびPC12細胞で首尾良く使用されている(Renneisen et al., 1990; Muller et al., 1990)。さらに、リポソームは、ウイルスベースの送達系に典型的なDNAの長さの制約を有さない。リポソームは、遺伝子、薬物(Heath & Martin, 1986; Heath et al., 1986; Balazsovits et al., 1989; Fresta & Puglisi, 1996)、放射線治療薬(Pikul et al., 1987)、酵素(Imaizumi et al., 1990a; Imaizumi et al., 1990b)、ウイルス(Faller & Baltimore, 1984)、転写因子およびアロステリックエフェクター(Nicolau & Gersonde, 1979)を種々の培養細胞株および動物に導入するために効果的に使用されている。さらに、リポソーム媒介性薬物送達の有効性を調査するいくつかの臨床試験の成功が達成されている(Lopez-Berestein et al., 1985a; 1985b; Coune, 1988; Sculier et al., 1988)。さらにまた、いくつかの研究で、リポソームの使用が、自己免疫性応答、毒性または全身性送達後の生殖腺局在化を伴わないことが示唆されている(Mori & Fukatsu, 1992)。
【0203】
リポソームは、水性媒体中に分散され、自発的に多重膜同心性二分子膜小胞(multilamellar concentric bilayer vesicles)(多重膜小胞(MLV)とも称される)を形成するリン脂質から形成される。MLVは一般的には25nm〜4μmの直径を有する。MLVを超音波処理すると、200〜500Åの範囲の直径で、コア中に水性溶液を含有する小さい単層の小胞(SUV)が形成される。
【0204】
リポソームは細胞膜と類似点を有し、ペプチド組成物の担体として本発明に関連した使用が意図される。それらは、水溶性および脂溶性の両物質を、すなわち、それぞれ水性間隙中および二重層自体の中にトラップできるために、広く適している。薬物保持リポソームを、リポソーム製剤を選択的に改変することによって活性物質の部位特異的送達に使用することさえ可能である。
【0205】
Couvreur et al. (1977; 1988)の教示内容に加えて、以下の情報をリポソーム製剤の作成に利用してよい。リン脂質は、水に分散させると、水に対する脂質のモル比に応じてリポソーム以外の種々の構造を形成しうる。低い割合で、リポソームは好ましい構造になる。リポソームの物理的特性は、pH、イオン強度および2価カチオンの存在に依存する。リポソームは、イオン性および極性物質に対して低い透過性を示しうるが、高い温度では、それらの透過性を顕著に変化させる相転移を受ける。該相転移には、ゲル状態として知られる密接に充填された秩序構造から、液体状態として知られる緩く充填された低秩序構造への変化が関与する。これは特徴的な相転移温度で生じ、その結果、イオン、糖および薬物に対する透過性が増加する。
【0206】
温度に加えて、タンパク質への曝露によりリポソームの透過性が変化しうる。特定の可溶性タンパク質、例えばシトクロムcは、二重層に結合し、それを変形させ、それを貫通することによって、透過性の変化を生じさせる。コレステロールは、おそらくリン脂質をより密接にパッキングすることによってこのタンパク質の貫通を阻害する。抗生物質およびインヒビター送達のための最も有用なリポソーム構成はコレステロールを含有することが想定される。
【0207】
溶質をトラップする能力は、異なるタイプのリポソーム間で様々である。例えば、溶質のトラップ時にMLVは中程度に効率的であるが、SUVは非常に非効率的である。しかし、SUVはサイズ分布の均質性および再現性の利点を提供し、大きい単層の小胞(LUV)によってサイズおよびトラップ効率の間の妥協点が提供される。これらはエーテル蒸発によって調製され、溶質トラップ時にMLVより3〜4倍効率的である。
【0208】
リポソーム特性に加えて、化合物のトラップに関する重要な決定要因は該化合物自体の物理化学的特性である。極性化合物は水性間隙中にトラップされ、無極性化合物は小胞の脂質二重層に結合する。極性化合物は、浸透によって、あるいは二重層が崩壊した時に、放出されるが、無極性化合物は、温度またはリポタンパク質への曝露によってそれが崩壊しない限り、二重層に付属したままである。両タイプは相転移温度で最大流出率を示す。
【0209】
リポソームは以下の4種の異なる機構で細胞と相互作用する:細網内皮系の食細胞、例えばマクロファージおよび好中球によるエンドサイトーシス;非特異的な弱い疎水性または静電力によるか、あるいは細胞表面成分との特異的相互作用による細胞表面への吸着;リポソームの脂質二重層が原形質膜内へ挿入されることによる形質細胞膜との融合、およびリポソーム内容物の細胞質内への同時放出;および、リポソーム内容物の結合を全く伴わない、細胞膜またはオルガネラ膜(subcellular membranes)へのリポソーム脂質の移入またはその逆に基づく機構。どの機構が作動するかを決定することは困難であることがよくあり、2以上の機構が同時に作動するかもしれない。
【0210】
静脈内に注射されたリポソームの末路および傾向は、それらの物性、例えばサイズ、流動性、および表面電荷に依存する。それらは、その組成、および血液中での分〜数時間の範囲の半減期に依存して、数時間または数日間、組織中に残存するかもしれない。大きいリポソーム、例えばMLVおよびLUVは、細網内皮系の食細胞によって急速に摂取されるが、循環系の生理学はほとんどの部位でそのような大きい分子種が外に出ることを抑制する。それらは、毛細血管内皮中の大きな開口部または細孔が存在する場所、例えば肝臓または脾臓のシヌソイドでしか外に出ることができない。ゆえに、これらの器官は取り込みの顕著な部位(predominate site)である。一方、SUVはより広い組織分布を示すが、依然として肝臓および脾臓に高度に隔離される。概して、このin vivo挙動により、リポソームの潜在的ターゲティングは、大きいサイズのリポソームがアクセスできる器官および組織のみに制限される。これらには、血液、肝臓、脾臓、骨髄、およびリンパ器官が含まれる。
【0211】
ターゲティングは、概して、本発明に関する制約ではない。しかし、万一、特異的ターゲティングが所望であれば、それを達成するための方法が利用可能である。リポソーム表面に結合する抗体を使用し、該抗体およびその薬物内容物を、特定の細胞タイプの表面上に位置する特異的抗原性受容体に向けてよい。炭水化物決定基(細胞−細胞認識、相互作用および接着に役割を果たす糖タンパク質または糖脂質細胞表面成分)を認識部位として使用してもよい。それは、それらがリポソームを特定の細胞タイプに向ける潜在能力を有する場合である。ほとんどの場合、リポソーム調製物の静脈内注射を使用することが想定されるが、他の投与経路も考えられる。
【0212】
あるいは、本発明は、本発明の組成物の製薬上許容されるナノカプセル製剤を提供する。ナノカプセルは、概して、安定かつ再現可能な様式で化合物をトラップすることができる(Henry-Michelland et al., 1987; Quintanar-Guerrero et al., 1998; Douglas et al., 1987)。細胞内ポリマーの過剰詰め込みに起因する副作用を回避するために、in vivoで分解可能なポリマーを使用してそのような超微粒子(約0.1μmのサイズ)を設計すべきであろう。本発明での使用には、これらの必要条件を満たす生分解性ポリアルキル−シアノアクリラートナノ粒子が想定される。そのような粒子は文献に記載されるように容易に作成してよい(Couvreur et al., 1980; 1988; zur Muhlen et al., 1998; Zambaux et al. 1998; Pinto-Alphandry et al., 1995および米国特許第5,145,684号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。
【0213】
ワクチン
本発明の特定の好ましい実施形態では、ワクチンを提供する。該ワクチンは、一般的に、1以上の上記のような医薬組成物を、免疫賦活剤と組み合わせて含む。免疫賦活剤は、外因性抗原に対する免疫応答(抗体および/または細胞媒介性)を高めるか、あるいは増強する任意の物質であってよい。免疫賦活剤の例には、アジュバント、生分解性ミクロスフェア(例えばポリラクティックガラクチド(polylactic galactide))およびリポソーム(内部に化合物が組み入れられている;例えばFullerton, 米国特許第4,235,877号を参照のこと)が含まれる。ワクチン調製物は、概して、例えばPowell & Newman, eds., Vaccine Design (the subunit and adjuvant approach) (1995)に記載されている。本発明の範囲内の医薬組成物およびワクチンは、他の化合物を含有してもよく、該化合物は生物活性または不活性であってよい。例えば、1以上の、他の腫瘍抗原の免疫原性部分を、該組成物またはワクチン内で、融合ポリペプチドに組み入れるか、あるいは分離した化合物として存在させてよい。
【0214】
例示的なワクチンには、1以上の上記ポリペプチドをコードするDNAを含有させて、該ポリペプチドがin situで生成されるようにしてよい。上記のように、当業者に公知の任意の種々の送達系内に該DNAを存在させてよく、該送達系には、核酸発現系、細菌およびウイルス発現系が含まれる。多数の遺伝子送達技術が当技術分野において周知であり、例えばRolland, Crit. Rev. Therap. Drug Carrier Systems 15:143-198 (1998)、およびその引用文献に記載の技術がある。適切な核酸発現系は、患者での発現に必要なDNA配列(例えば好適なプロモーターおよび終結シグナル)を含有する。細菌送達系には、該ポリペプチドの免疫原性部分を細胞表面上で発現するか、あるいはそのようなエピトープを分泌する細菌宿主細胞(例えばマイコバクテリウム、バシラスまたはラクトバシラス株、例えばカルメット・ゲラン桿菌またはLactococcus lactis)の投与が関与する(例えばFerreira, et al., An Acad Bras Cienc (2005) 77:113-124; およびRaha, et al., Appl Microbiol Biotechnol (2005) PubMedID 15635459を参照のこと)。好ましい実施形態では、ウイルス発現系(例えばワクシニアまたは他のポックスウイルス、レトロウイルス、またはアデノウイルス)を使用して該DNAを導入してよく、それには、非病原性(欠損)複製可能ウイルスの使用が関与してよい。好適な系は、例えば以下の文献に開示されている:Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA 86:317-321 (1989); Flexner et al., Ann. N.Y. Acad. Sci. 569:86-103 (1989); Flexner et al., Vaccine 8:17-21 (1990); 米国特許第4,603,112号、同第4,769,330号、および同第5,017,487号;WO89/01973;米国特許第4,777,127号;GB2,200,651;EP0,345,242;WO91/02805; Berkner, Biotechniques 6:616-627 (1988); Rosenfeld et al., Science 252:431-434 (1991); Kolls et al., Proc. Natl. Acad. Sci. USA 91:215-219 (1994); Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90:11498-11502 (1993); Guzman et al., Circulation 88:2838-2848 (1993); およびGuzman et al., Cir. Res. 73:1202-1207 (1993)。そのような発現系にDNAを組み入れるための技術は当業者に周知である。該DNAは「裸の」DNAであってよく、それは、例えばUlmer et al., Science 259:1745-1749 (1993)に記載され、Cohen, Science 259:1691-1692 (1993)に概説されている。裸のDNAの取り込みは、該DNAを生分解性ビーズ上にコーティングすることによって高めてよく、該ビーズは細胞内に効率的に輸送される。ワクチンはポリヌクレオチドおよびポリペプチドの両成分を含んでよいことが明らかである。そのようなワクチンは向上した免疫応答を提供するかもしれない。
【0215】
ワクチンは、本明細書中で提供されるポリヌクレオチドおよびポリペプチドの製薬上許容される塩を含有してよいことが明らかである。そのような塩は、製薬上許容される無毒の塩基、例えば有機塩基(例えば第1級、第2級および第3級アミンおよび塩基性アミノ酸の塩)および無機塩基(例えばナトリウム、カリウム、リチウム、アンモニウム、カルシウムおよびマグネシウム塩)から調製してよい。
【0216】
本発明のワクチン組成物中では当業者に公知の任意の適切な担体を使用してよいが、担体のタイプは投与様式に応じて様々である。本発明の組成物は任意の適切な投与様式、例えば局所、経口、鼻腔、静脈内、頭蓋内、腹腔内、皮下または筋肉内投与のために製剤化してよい。非経口投与、例えば皮下注射では、担体は、好ましくは、水、生理食塩水、アルコール、脂肪、ワックスまたはバッファーを含む。経口投与では、上記担体のいずれかまたは固形担体、例えばマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、滑石、セルロース、グルコース、ショ糖、および炭酸マグネシウムを使用してよい。本発明の医薬組成物用の担体として、生分解性ミクロスフェア(例えばポリラクタートポリグリコラート)を使用してもよい。好適な生分解性ミクロスフェアは、例えば米国特許第4,897,268号;同第5,075,109号;同第5,928,647号;同第5,811,128号;同第5,820,883号;同第5,853,763号;同第5,814,344号および同第5,942,252号で開示されている。米国特許第5,928,647号に記載の微粒子−タンパク質複合体を含む担体を使用してもよく、該複合体は宿主においてクラスI限定の細胞障害性Tリンパ球応答を誘発可能である。
【0217】
そのような組成物は、バッファー(例えば中性緩衝生理食塩水またはリン酸緩衝食塩水)、炭水化物(例えばグルコース、マンノース、ショ糖またはデキストラン)、マンニトール、タンパク質、ポリペプチドまたはアミノ酸、例えばグリシン、酸化防止剤、静菌剤、キレート剤、例えばEDTAまたはグルタチオン、アジュバント(例えば水酸化アルミニウム)、製剤をレシピエントの血液と等張、低張または弱い高張性にする溶質、懸濁化剤、増粘剤および/または保存剤を含んでもよい。あるいは、本発明の組成物は凍結乾燥物(lyophilizate)として製剤化してよい。周知のテクノロジーを使用してリポソーム内に化合物を封入してもよい。
【0218】
本発明のワクチン中で任意の種々の免疫賦活剤を使用してよい。例えば、アジュバントを含ませてよい。ほとんどのアジュバントは、急速な異化作用から抗原を保護するように設計された物質、例えば水酸化アルミニウムまたは鉱油、および免疫応答の刺激因子、例えばリピドA、Bortadella pertussisまたはマイコバクテリア属の種またはマイコバクテリウム由来タンパク質を含有する。例えば、脱脂(delipidated)、脱糖脂質化(deglycolipidated)M. vaccae(「pVac」)を使用することができる。好適なアジュバントは、例えばフロイント不完全アジュバントおよび完全アジュバント(Difco Laboratories, Detroit, MI);Merck Adjuvant 65(Merck and Company, Inc., Rahway, NJ);AS01B、AS02A、AS15、AS−2およびその誘導体(GlaxoSmithKline, Philadelphia, PA);CWS、TDM、Leif、アルミニウム塩、例えば水酸化アルミニウムゲル(alum)またはリン酸アルミニウム;カルシウム、鉄または亜鉛の塩;アシル化チロシンの不溶性懸濁物;アシル化糖;カチオンまたはアニオンによって誘導体化された多糖;ポリホスファゼン(polyphosphazenes);生分解性ミクロスフェア;モノホスホリル脂質Aおよびquil Aとして市販されている。サイトカイン、例えばGM−CSFまたはインターロイキン−2、−7、または−12をアジュバントとして使用してもよい。
【0219】
本明細書中で提供されるワクチンでは、該アジュバント組成物は、好ましくは、主にTh1タイプの免疫応答を誘発するように設計される。高レベルのTh1タイプサイトカイン(例えばIFN−γ、TNFα、IL−2およびIL−12)は、投与された抗原に対する細胞性免疫応答の誘発に好都合である傾向がある。対照的に、高レベルのTh2タイプサイトカイン(例えばIL−4、IL−5、IL−6およびIL−10)は体液性免疫応答の誘発に好都合である傾向がある。本明細書中で提供されるワクチンの適用後、患者は、Th1−およびTh2−タイプ応答を含む免疫応答を支持する。応答が主にTh1タイプである好ましい実施形態では、Th1タイプサイトカインのレベルがTh2タイプサイトカインのレベルより高い程度にまで増加する。これらのサイトカインのレベルは、標準的アッセイを使用して容易に評価してよい。サイトカインのファミリーについての総説としては、Janeway, et al., Immunobiology, 5th Edition, 2001を参照のこと。
【0220】
主にTh1タイプ応答の誘発に使用するための好ましいアジュバントには、例えば、場合によりアルミニウム塩を伴う、モノホスホリル脂質A、好ましくは3−O−脱アシル化モノホスホリル脂質A(3D−MPL)の組み合わせが含まれる(例えばRibi, et al., 1986, Immunology and Immunopharmacology of Bacterial Endotoxins, Plenum Publ. Corp., NY, pp. 407-419; GB2122204B;GB2220211;およびUS4,912,094を参照のこと)。好ましい形式の3D−MPLは、直径0.2mm未満の小さい粒子サイズを有するエマルジョンの形式であり、その製造方法はWO94/21292に開示されている。モノホスホリル脂質Aおよび界面活性剤を含む水性製剤はWO98/43670に記載されている。例示的な好ましいアジュバントには、AS01B(リポソーム製剤中のMPLおよびQS21)、リポソーム製剤中の3D−MPLおよびQS21、AS02A(MPLおよびQS21および水中油型エマルジョン)、3D−MPLおよびQS21および水中油型エマルジョン、およびAS15(GlaxoSmithKlineから入手可能)が含まれる。MPLアジュバントはGlaxoSmithKline, Seattle, WAから入手可能である(米国特許第4,436,727号;同第4,877,611号;同第4,866,034号および同第4,912,094号を参照のこと)。
【0221】
CpG含有オリゴヌクレオチド(該オリゴヌクレオチドではCpGジヌクレオチドがメチル化されていない)もまた、主にTh1応答を誘発する。CpGとは、DNA中に存在するシトシン−グアノシンジヌクレオチドモチーフの略語である。そのようなオリゴヌクレオチドは周知であり、例えばWO96/02555、WO99/33488および米国特許第6,008,200号および同第5,856,462号に記載されている。また、免疫賦活性DNA配列は、例えばSato et al., Science 273:352 (1996)に記載されている。CpGは、ワクチンに製剤化されると、概して、遊離溶液中で遊離抗原と共に投与される(WO 96/02555; McCluskie and Davis, 上記)か、あるいは共有結合によって抗原にコンジュゲートされる(WO98/16247)か、あるいは水酸化アルミニウム等の担体と共に製剤化される((肝炎表面抗原))Davis et al. 上記; Brazolot-Millan et al., Proc.Natl.Acad.Sci., USA, 1998, 95(26), 15553-8)。CpGは、全身および粘膜の両経路によって投与できるアジュバントであることが当技術分野において知られている(WO96/02555、EP468520、Davis et al., J.Immunol, 1998, 160(2):870-876; McCluskie and Davis, J.Immunol., 1998, 161(9):4463-6)。
【0222】
別の好ましいアジュバントはサポニンまたはサポニン模倣物もしくは誘導体、好ましくはQS21(Aquila Biopharmaceuticals Inc., Framingham, MA)であり、それは単独で、あるいは他のアジュバントと組み合わせて使用してよい。例えば、強化された系には、モノホスホリル脂質Aおよびサポニン誘導体の組み合わせ、例えばWO94/00153に記載のQS21および3D−MPLの組み合わせ、またはWO96/33739に記載のQS21がコレステロールでクエンチされる低反応原性組成物が含まれる。他の好ましい製剤には、水中油型エマルジョンおよびトコフェロールが含まれる。水中油型エマルジョン中にQS21、3D−MPLおよびトコフェロールを含む特に強力なアジュバント製剤がWO95/17210に記載されている。本発明で有用な追加のサポニンアジュバントには、QS7(WO96/33739およびWO96/11711に記載)およびQS17(米国特許第5,057,540号およびEP0 362 279B1に記載)が含まれる。
【0223】
他の好ましいアジュバントには、Montanide ISA 720(Seppic, France)、SAF(Chiron, California, United States)、ISCOMS(CSL)、MF-59(Chiron)、SBASシリーズのアジュバント(例えばSBAS-2、AS2'、AS2''、SBAS-4、またはSBAS6(GlaxoSmithKline, Rixensart, Belgiumから入手可能))、Detox(Corixa, Hamilton, MT)、RC-529(Corixa, Hamilton, MT)および他のアミノアルキルグルコサミニド4−ホスファート(AGP)、例えば係属中の米国特許出願第08/853,826号および同第09/074,720号(その開示内容は参照によりその全体が本明細書中に組み入れられる)に記載のものが含まれる。
【0224】
アジュバントの別の例には、合成MPLおよび志賀毒素Bサブユニットベースのアジュバントが含まれる(WO2005/112991を参照のこと)。
【0225】
本明細書中で提供される任意のワクチンは、抗原、免疫応答エンハンサーおよび好適な担体または賦形剤の組み合わせに結実する周知の方法を使用して調製してよい。本明細書中に記載の組成物は、徐放性製剤(すなわち、投与後に化合物の持続放出を生じさせる製剤、例えばカプセル、スポンジまたはゲル(例えば多糖から構成される))の部分として投与してよい。そのような製剤は、概して、周知のテクノロジーを使用して調製してよく(例えばCoombes et al., Vaccine 14:1429-1438 (1996)を参照のこと)、例えば経口、経直腸または皮下移植によって、あるいは所望の標的部位での移植によって投与してよい。徐放性製剤は、ポリペプチド、ポリヌクレオチドまたは抗体を含有してよく、それらは担体マトリックス中に分散され、ならびに/あるいは律速メンブレン(rate controlling membrane)によって包囲されているリザーバ内に含有されていてよい。
【0226】
そのような製剤内で使用するための担体は生体適合性であり、生分解性であってもよく;好ましくは該製剤は比較的一定レベルの活性成分放出を提供する。そのような担体には、ポリ(ラクチドコグリコリド)、ポリアクリラート、ラテックス、デンプン、セルロース、デキストラン等の微粒子が含まれる。他の遅延放出担体には、非液状親水性コア(例えば架橋された多糖またはオリゴ糖)および、場合により、両親媒性化合物、例えばリン脂質を含む外層を含む超分子バイオベクター(supramolecular biovectors)が含まれる(例えば米国特許第5,151,254号およびPCT出願WO94/20078、WO94/23701およびWO96/06638を参照のこと)。徐放性製剤内に含有される活性化合物の量は、移植部位、放出の速度および予測される持続時間および治療または予防対象の症状の性質に依存する。
【0227】
任意の種々の送達ビヒクルを医薬組成物およびワクチン中で使用して、腫瘍細胞を標的にする抗原特異的免疫応答の発生を促進してよい。送達ビヒクルには、抗原提示細胞(APC)、例えば樹状細胞、マクロファージ、B細胞、単球および、遺伝子操作によって有効なAPCにしてよい他の細胞が含まれる。そのような細胞は、その必要はないが、遺伝子改変して、抗原を提示する能力を高め、T細胞応答の活性化および/または維持を改善し、それ自体で抗腫瘍効果を有するようにし、ならびに/あるいは、受け手と免疫適合性(すなわち適合型HLAハプロタイプ)にしてよい。APCは、概して、腫瘍および腫瘍周囲の組織を含む任意の種々の生体液および器官から単離してよく、自己、同種異系、同系または異種細胞であってよい。
【0228】
本発明の特定の好ましい実施形態では、樹状細胞またはその前駆体を抗原提示細胞として使用する。樹状細胞は非常に強力なAPCであり(Banchereau & Steinman, Nature 392:245-251 (1998))、予防的または治療的な抗腫瘍性免疫を誘発するための生理学的アジュバントとして有効であることが示されている(Timmerman & Levy, Ann. Rev. Med. 50:507-529 (1999)を参照のこと)。一般に、樹状細胞は、その典型的な形状(in situでは星状であり、in vitroで可視の顕著な細胞質の突起(樹状突起)を有する)、高い効率で抗原を取り込み、プロセシングし、提示するその能力、およびナイーブT細胞応答を活性化するその能力に基づいて特定してよい。当然、樹状細胞は、in vivoまたはex vivoで樹状細胞上には一般に認められない特異的細胞表面受容体またはリガンドを発現するように遺伝子操作してよく、そのような改変樹状細胞は本発明によって想定される。樹状細胞の代替物として、分泌小胞である抗原積載樹状細胞(エキソソームと称される)をワクチン内で使用してよい(Zitvogel et al., Nature Med. 4:594-600 (1998)を参照のこと)。
【0229】
樹状細胞および前駆体は、末梢血、骨髄、腫瘍に浸潤している細胞、腫瘍周囲の組織に浸潤している細胞、リンパ節、脾臓、皮膚、臍帯血または任意の他の好適な組織もしくは液体から取得してよい。例えば、末梢血から回収された単球の培養にサイトカイン、例えばGM−CSF、IL−4、IL−13および/またはTNFαの組み合わせを加えることによって、ex vivoで樹状細胞を分化させてよい。あるいは、GM−CSF、IL−3、TNFα、CD40リガンド、LPS、flt3リガンドおよび/または、樹状細胞の分化、成熟および増殖を誘発する他の化合物(群)の組み合わせを培養培地に加えることによって、末梢血、臍帯血または骨髄から収集されたCD34陽性細胞を樹状細胞に分化させてよい。
【0230】
樹状細胞は、好都合には、「未成熟」および「成熟」細胞として分類され、それにより、2種の十分に特徴付けられている表現型を識別する単純な様式が提供される。しかし、この命名法は、すべての起こりうる、分化の中間段階を排除すると解釈されるべきではない。未成熟樹状細胞は、抗原の取り込みおよびプロセシングの高い能力を有するAPCとして特徴付けられ、それはFcγ受容体およびマンノース受容体の高い発現と相関している。成熟表現型は、典型的に、これらのマーカーの低い発現、反面、T細胞活性化を担う細胞表面分子、例えばクラスIおよびクラスII MHC、接着分子(例えばCD54およびCD11)および共刺激分子(例えばCD40、CD80、CD86および4−1BB)の高い発現によって特徴付けられる。
【0231】
一般に、タンパク質(またはその一部分もしくは他の変異体)をコードするポリヌクレオチドでAPCをトランスフェクトして、該ポリペプチド、またはその免疫原性部分が細胞表面上に発現されるようにしてよい。そのようなトランスフェクションはex vivoで行ってよく、そして、本明細書中で記載されるように、該トランスフェクト済み細胞を含む組成物またはワクチンを治療目的で使用してよい。あるいは、樹状または他の抗原提示細胞を標的にする遺伝子送達ビヒクルを患者に投与してよく、その結果、トランスフェクションがin vivoで生じる。例えば、樹状細胞のin vivoおよびex vivoトランスフェクションは、概して、当技術分野において公知の任意の方法、例えばWO97/24447に記載の方法、またはMahvi et al., Immunology and Cell Biology 75:456-460 (1997)に記載の遺伝子銃アプローチを使用して実施してよい。樹状細胞の抗原積載は、樹状細胞または前駆細胞を、ポリペプチド、DNA(裸のDNAまたはプラスミドベクター内のDNA)もしくはRNA;または抗原を発現する組換え細菌またはウイルス(例えばワクシニア、鶏痘、アデノウイルスまたはレンチウイルスベクター)とインキュベートすることによって達成してよい。積載前に、該ポリペプチドを免疫学的パートナーに共有結合によってコンジュゲートして、T細胞支援(例えば担体分子)を提供してよい。あるいは、非コンジュゲート型の免疫学的パートナーを、別途、あるいは該ポリペプチドの存在下で樹状細胞にパルスしてよい。
【0232】
ワクチンおよび医薬組成物は、単位用量または複数回用量容器、例えば密封アンプルまたはバイアル中で提供してよい。好ましくは、使用時まで該製剤の無菌性を保つために該容器を密封する。一般に、製剤は、油性または水性ビヒクル中の懸濁液、溶液またはエマルジョンとして保存してよい。あるいは、ワクチンまたは医薬組成物は凍結乾燥条件で保存してよい。その場合、使用直前に滅菌液状担体を加えることのみが必要とされる。
【0233】
本明細書中で引用されるすべての刊行物および特許出願は、個々の各刊行物または特許出願が参照により組み入れられることが具体的かつ個別に示されているかのように、参照により本明細書中に組み入れられる。
【0234】
前記発明について、理解を明確にするために、説明および例を用いていくらか詳細に説明してきたが、特許請求の範囲の思想または範囲から逸脱することなく、それに対して特定の変更および改変を施してよいことが、本発明の教授内容を踏まえて当業者には直ちに自明である。
【実施例】
【0235】
実施例
限定目的ではなく単に説明目的で以下に実施例を挙げる。当業者は、本質的に同様の結果を生じさせるよう変更または改変することができる種々の重要でないパラメータを容易に認識するであろう。
【0236】
実施例1: Mtb72f(Hisタグなし)(配列番号6)の調製
Mtb72f発現ベクターの構築
Mtb72fは、2種の結核菌タンパク質Mtb32およびMtb39から形成される融合タンパク質である。Mtb39と、Mtb32のN末端およびC末端部分を、Mtb32のC末端−Mtb39−Mtb32のN末端のように融合させることによって、Mtb72fを構築する。具体的には、Mtb32の約14kDaのC末端断片(残基192−323;132アミノ酸)をコードするオープンリーディングフレーム(ORF)にMtb39の全長ORFを直列に連続して連結し、続いてMtb32の約20kDaのN末端部分(残基1−195)をC末端に連結することによって、Mtb72fタンパク質を作成した。結核菌株H37Rvに由来するゲノムDNAからのポリメラーゼ連鎖反応(PCR)のために、ユニークな制限部位(EcoRIおよびEcoRV)を含有し、C末端の停止コドンを欠いている(Mtb32−CおよびMtb39の場合)配列特異的オリゴヌクレオチドを使用することによって、それを達成した。該プロセスの詳細は以下の通りである。
【0237】
まず、以下のオリゴヌクレオチドを用いるPCRを使用して、H37RvからMtb32のC末端部分(Mtb32C)をコードするDNAをクローニングした:5’(5’−CAA−TTA−CAT−ATG−CAT−CAC−CAT−CAC−CAT−CAC−ACG−GCC−GCG−TCC−GAT−AAC−TTC−3’)および3’(5’−CTA−ATC−GAA−TCC−GGC−CGG−GGG−TCC−CTC−GGC−CAA−3’)。5’オリゴヌクレオチドは、ATG開始コドンを包含するNdeI制限部位(下線部)を含有した。3’オリゴヌクレオチドはEcoRI制限部位(下線部)を含有した。これらのオリゴヌクレオチドを使用して、Mtb32の396ヌクレオチド部分であるMtb32Cを増幅し、得られた産物を発現ベクターのNdeIおよびEcoRI部位にサブクローニングした。次いでEcoRIおよびEcoRVで消化して、Mtb32Cプラスミドを直線状にした。
【0238】
Mtb39に関しては、PCR増幅およびクローニングに以下のオリゴヌクレオチドを使用した:5’−(5’−CTA−ATC−GAA−TTC−ATG−GTG−GAT−TTC−GGG−GCG−TTA−3’)および3’(5’−CTA−ATC−GAT−ATC−GCC−GGC−TGC−CGG−AGA−ATG−CGG−3’)。5’オリゴヌクレオチドはEcoRI制限部位(下線部)を含有し、一方、3’オリゴヌクレオチドはEcoRV制限部位(下線部)を含有した。Mtb39の全長コード配列を増幅し、消化し、第1ステップであらかじめ消化されたプラスミドを使用してMtb32cの下流にインフレームでサブクローニングした。
【0239】
Mtb32のN末端断片の5’および3’オリゴヌクレオチドを以下のように設計した:5’−(5’−CTA−ATC−GAT−ATC−GCC−CCG−CCG−GCC−TTG−TCG−CAG−GAC−3’)および3’(5’−CTA−ATC−GAT−ATC−cta−GGA−CGC−GGC−CGT−GTT−CAT−AC−3’)。両セットのオリゴヌクレオチドはEcoRV制限部位(下線部)を含有し、一方、3’オリゴヌクレオチドは停止コドン(小文字)をさらに含んだ。このタンパク質の予測されるN末端ドメインをコードするMtb32の585bp部分を増幅するように該オリゴヌクレオチドを設計した。得られたPCR産物をMtb32c−Mtb39融合プラスミドにサブクローニングした。次いで、適正な挿入方向および突然変異の不存在をDNAシークエンシングによって確認した。
【0240】
Master Cell BankおよびManufacturer's Working Cell Bankを作成するために使用される最終構築物では、6×His親和性タグをPCRによって除去し、Mtb72fのオープンリーディングフレーム(ORF)を、pET由来の発現ベクターであるpPDMにサブクローニングした。該ORFは約72kDaのポリタンパク質(Mtb72f)をコードし、直列順序:Mtb32C−Mtb39−Mtb32Nで構成されたドメインを有する。次いでこのDNAを大腸菌のHMS174 pLysS株に形質転換し、試験、細胞バンク作成、および製造に使用した。
【0241】
Mtb72fバルク原体(Bulk Drug Substance)の生産
Mtb72fを生産するための製造プロセスを以下にまとめる:
・発酵、次いで遠心分離による細胞収集、細胞破壊(microfluidizer)および遠心分離による封入体ペレットの取得;
・8M尿素中での抽出による封入体ペレットの精製、次いでQ Sepharose Fast Flow(QFF)クロマトグラフィー、Ceramic Hydroxyapatite(CHT)クロマトグラフィー、ダイアフィルトレーション(diafilitration)、および滅菌ろ過による精製バルク原体の取得。
【0242】
発酵
発酵は10Lの作業容量で実施する。37℃で一晩培養された作業用種細胞の300mLの振とうフラスコ培養を発酵槽に接種する。接種および発酵ではともに、主要炭素源である植物由来グリセロールを含む半合成培地(semidefined medium)を使用する。培地の組成は以下の表に示される通りである。すべての培地成分を121℃で20分間加熱するか、あるいは滅菌ろ過によって滅菌する。発酵中、発酵槽を37℃の温度で維持する。5標準リットル/分(SLPM)の割合で空気を混入する。酸(H2SO4)または塩基(NaOH)の自動添加によって培地のpHを7.0で維持する。200rpmの最小撹拌を維持しつつ撹拌を自動調節することによって、溶存酸素を30%に制御するように発酵槽をプログラムする。1.05%SAG−471シリコーン消泡剤(Witco Corp.)の自動添加によって発酵槽内の発泡抑制を達成する。細胞密度が約3.5の光学濃度(600nm)に達したら、イソプロピル−β−D−チオガラクトピラノシド(IPTG)を1.0mMの濃度まで発酵槽に加える。IPTGは、Mtb72fタンパク質をコードする組換え遺伝子の発現を誘導する。誘導後3.0時間の時点で、発酵槽を冷却し、1L遠心分離ボトル(centrifuge bottles)中で遠心分離することによって細胞を収集する。
【表1】

【0243】
封入体の単離
細胞ペレットを、2.3Lの溶解バッファー(50mM NaCl、10mM Tris pH8.0)中に再懸濁してプールし、M-l 10Y Microfluidizer(登録商標)を使用して細胞を破壊する。細胞は、11,000±1,000psiの圧力で5回、Microfluidizerを通過させる。500mLボトル中で8000×gで懸濁液を遠心分離する。これらの条件下で、Mtb72fタンパク質を含有する封入体(IB)はペレットになり、ほとんどの細胞片は上清中に残る。IBペレットを洗浄バッファー(2M尿素、50mM NaCl、10mM Tris pH8.0)に再懸濁した後、8,000gで遠心分離する。上清画分を廃棄し、追加の精製に必要とされるまで−70℃〜−80℃でIBペレットを保存する。
【0244】
ポリタンパク質の精製
凍結IB調製物を37℃で15分間解凍した後、穏やかな機械的撹拌を使用して、8M尿素、50mM NaCl、20mM Bis−trisプロパン、pH7.0(バッファーA)に再懸濁する。そして再懸濁されたIBを、マグネチックスターラーバーで300rpmで2時間室温で撹拌する。次いでIB抽出物を高速で遠心分離し、得られた上清画分を0.45uMフィルター(Pall, Supor)を通してろ過した後、クロマトグラフィーによって分画する。
【0245】
あらかじめ1N NaOHで衛生化され、次いでバッファーAで平衡化されたQ Sepharose Fast Flow(QFF)アニオン交換樹脂(10×12.5cm、Amersham/Pharmacia BPG; 1L充填床)を含有するカラムにIB抽出物をアプライする。バッファーAで60cm/時間の線流速でカラムを展開し、主に低質量の混入物を含有するフロースルーを参照用に回収する。8M尿素、90mM NaCl、20mM Bis−trisプロパン、pH7.0を使用してMtb72fのバルクを1ステップで溶出し、吸光度に基づいて単一のバルクピークとして回収する。
【0246】
QFF樹脂は、精製時に使用される条件下で正に帯電する第4級アミン官能基を有する高度に架橋されたアガロース樹脂である。荷電マトリックスは、種々のアニオンの結合を可能にし、そして該アニオンは、塩勾配を使用して選択的に溶出させることができる。このアニオン交換クロマトグラフィーを使用して、該タンパク質から、樹脂に密接に結合している核酸および内毒素を分離する。該タンパク質は弱く結合し、これらの混入物より前に溶出される。さらに、このステップでは、無電荷の混入物および大部分のタンパク質不純物が除去される。
【0247】
QFFカラムからの90mM NaCl溶出液ピークを、MacroPrep(登録商標)セラミックヒドロキシアパタイト(CHT)(タイプI、40uM、BioRad)を含有するカラム(2.6×12cm、Amersham/Pharmacia XK26/20; 63mL充填床)にアプライする。該カラムは、1N NaOHを使用してあらかじめ衛生化され、次いでバッファーC(8M尿素、250mM NaCl、および20mM Bis−trisプロパン、pH7.0)で平衡化されている。大部分のMtb72fを含有し、混入物を含まないフロースルー材料(FT1)を回収する。カラムをバッファーCで洗浄し、残ったすべてのUV吸収材料を回収する。最後に、バッファーD(8M尿素、200mMリン酸ナトリウム、pH7.4)中でカラムを溶出させる。
【0248】
MacroPrep(登録商標)CHTは、球状の、マクロ細孔状のヒドロキシアパタイト[Ca5(PO4)3OH]2である。CHTクロマトグラフィーは、適正な結合条件および溶出条件を見出せば、非常に選択的な精製方法でありうる。結合様式には、荷電カルシウムおよびリン酸イオンとのイオン交換タイプの結合ならびに分子のキレート化が含まれる。DNAはこの樹脂に結合し、個別タンパク質に関する高い選択性を達成することができる。Mtb72fの精製に使用される条件は洗練ステップとして機能し、検出可能な宿主細胞混入物の実質的に完全な除去を可能にする。
【0249】
クロマトグラフィーによる分離中に、紫外(UV)吸光度、伝導率、圧力、pH、流速、および室温をモニターし、記録する。初期CHTフロースルー材料(FT1)をさらなる下流の処理に使用する。
【0250】
ダイアフィルトレーションおよび滅菌ろ過
CHT FT1プールに対してダイアフィルトレーションを実施して、尿素を除去し、バッファーを20mM Tris pH7.5で置き換える。ダイアフィルトレーションは、30kDa分子量カットオフ(MWCO)の限外ろ過メンブレンを用いて、LV-CentramateTMタンジェンシャルフローろ過デバイスを含むPall MinimTMシステムを使用して実施する。20mM Tris pH7.5中のMtb72f溶液を、0.2um滅菌フィルター(Millipak 40)を使用してろ過滅菌する。50mLの溶液を滅菌60mL PETG(ポリエチレンテレフタラートコポリマー)培地ボトル中に分配した後、−70℃で凍結および保存する。この材料がMtb72f精製済みバルク原体である。
【0251】
実施例2: Mtb72f(6Hisタグ)(配列番号2)の調製
Hisタグを除去するためのpPDMへのサブクローニングのステップを省略することを除き、実施例1の方法にしたがってよい。
【0252】
実施例3: M72(2Hisタグ)(配列番号4)の調製
M72発現ベクターの構築
M72抗原の構築用の出発材料は組換えプラスミド6His−Mtb72fmutであった。6His−Mtb72f組換えプラスミド(実施例1を参照のこと)を鋳型として使用する部位特異的突然変異誘発によって、6His−Mtb72fmutを調製した。部位特異的突然変異誘発には、配列番号1の710位のSerに対するコドンをAlaに対するコドンで置き変えることが含まれた。
【0253】
「Gene Tailor Site-Directed Mutagenesis System」(Invitrogen)を用いて、6His−Mtb72fmut構築物(Corixaプラスミド)上に存在する4個のN末端ヒスチジンの欠失を達成し、予測される2His−Mtb72Fmut構築物を得た。配列確認後、該プラスミドから(酵素による制限処理によって)2His−Mtb72fmutコード配列を切り出し、ゲル精製し、pET29a発現ベクターにライゲーションして、最終的な組換えプラスミドpET29a/2His−Mtb72fmutを得た。配列確認後、その組換えプラスミドに正式名称pRIT15497を付与し、HMS174(DE3)宿主細胞の形質転換に使用した。pRIT15497は、M72と称される725アミノ酸のタンパク質をコードする。
【0254】
M72タンパク質の生産
M72の生産に関しては、発酵培地中にクロラムフェニコールが存在しないことを除き、Mtb72fに関する記載(実施例1を参照のこと)と同一の生産プロセスを使用してよい。
【0255】
生物学的実施例1: 不活性/潜伏期の結核菌感染のマウスモデル
潜伏性結核菌感染のマウスモデルを確立するために、SWR系統を使用した。SWRマウスは免疫無防備状態ではないが、補体成分C5の分泌欠損である(Ooi and Colten, Nature (1979) 282:207-8を参照のこと)。SWRマウスは慢性状態のMtb感染を確立できないが、大型類上皮細胞(epitheloid)および泡沫状マクロファージ(クリスタロイド封入体を伴う)(貪食された好中球または好酸球由来顆粒)、多発性壊死、好中球蓄積およびリンパ球欠乏を特徴とするびまん性肉芽腫性肺炎(diffuse granulomatous pneumonia)を発症する(Turner, et al., J Submicrosc Cytol Pathol. (2001) 33(l-2):217-9; およびTurner, et al., Infect Immun. (2003) 71(9):5266-72を参照のこと)。潜伏性結核菌感染のモデルにおけるAS01Bアジュバントと共に製剤化されたMtb72f(配列番号6)の治療効力を評価するために、Swiss Webster(SWR/J)マウス系統を使用するプロトコルを以下に挙げる。コレステロール(25μg)(WO96/33739)およびモノホスホリル脂質A(MPL)(5μg)を膜中に含有するジオレオイルホスファチジルコリン(100μg)の小型単層小胞(SUV)にQS21(5μg)を加えることによって、2倍強度AS01Bを調製する(米国特許公開第2003/0143240号を参照のこと)。バッファー(PBS pH6.8)中の4μgのタンパク質を50μlの2倍強度AS01Bと混合することによって、注射用アリコート(50μl)を調製する。各マウスに2回の50μl注射(すなわち8μgのタンパク質)を施した。
【0256】
潜伏性結核菌感染のモデルを確立するための代表的時間軸は図1に模式的に示される通りである。
【0257】
1日目:50〜100コロニー形成単位(CFU)の結核菌生物体をエアロゾルによって感染させる
30〜90日目:飲料水1リットルあたり50mgリファンピン/85mgイソニアジドで一部のマウスを処置する
61日目:ワクチン候補5を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
82日目:ワクチン候補を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
103日目:ワクチン候補を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
113日目:IgGアッセイのために採血する
諸時点:CFUの数え上げおよび免疫原性のために脾臓および肺を採取する。
【0258】
バリエーション1→60日間の化学療法での処置。30日目に開始→3、4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0259】
バリエーション2→90日間の化学療法での処置。30日目に開始→4、5か月間の休止→各時点で2マウスのCFUおよび7マウスを生存研究のために残す。
【0260】
バリエーション3→4、5、6か月間の休止→各時点で2マウスのCFUおよび4マウスを生存研究のために残す。
【0261】
バリエーション4→60日間の化学療法での処置。30日目に開始→60日目に開始される筋肉内(i.m.)r72F+AS01Bでの3回の免疫化→3、4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0262】
バリエーション5→90日間の化学療法での処置。30日目に開始→60日目に開始されるr72F+AS01B(i.m.)での3回の免疫化→4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0263】
ELISAプレートをコーティングするためにrMtb72fを使用する感染後の抗体応答の分析では、化学療法およびMtb72f+AS01B免疫化の組み合わせを施された群が、無処置または化学療法処置のみを施されたマウス(0.5未満のOD)より高い抗体応答(2.0までのOD)を有することが示された(図2)。Mtb72fで免疫化されたマウスは、60日間の化学療法を施されたか90日間の化学療法を施されたかにかかわらず、かなりのMtb72f特異的抗体応答(1.5〜2.5の範囲のOD)に達した(図3)。
【0264】
マウスに結核菌を感染させた後、種々の間隔でマウスから脾臓細胞を収集した。その脾細胞をin vitroで組換え抗原で再刺激し、IFN−γ分泌を測定した。これらの細胞によって産生されるIFN−γレベルは、Mtb39を除き、60日目の群1(無処置)および群2(化学療法のみ)では一様に無視できた。conA、PPDおよびBCG溶解物での陽性対照刺激では、細胞が他の刺激分子に応答してIFN−γを合成および分泌可能であることが実証された(図4)。IFN−γレベルは、Mtb72f+AS01Bを施された群で高かったが、Mtb72f+AS01Bで免疫化されなかった群では、化学療法が施されたかどうかにかかわらず、それらは低いか、あるいは無視できた(図5)。
【0265】
結核感染および以後の処置経過中に、特異的T細胞が応答する。IFN−γに対する細胞内サイトカイン染色を使用して、Mtb72Fに対する特異的CD4+細胞応答のパーセンテージを測定した(図6)。このアッセイによって測定した場合、化学療法単独の経過中にはいずれの時点でも、Mtb72F特異的CD4+ IFNγ+ T細胞応答は変化しないようであった(図7)。Mtb感染後120日目の時点で、Mtb72f+AS01Bワクチンを施された群において、Mtb72Fに対するCD4+ IFNγ+ 応答の傾向は、化学療法中の期間の長さに伴って増加するように見えた(図7)。
【0266】
発明者らの実験の結果は、SWRマウスが結核菌の感染に対して感受性であることを実証する。無処置のままにすると、Mtb感染後115日目までにSWRマウスは死にいたる(図8および図9)。60日間の併用化学療法を施されたマウスの平均生存期間は170日間であった(図8および図9)。60日間の併用化学療法およびMtb72f/AS01Bでの3回の免疫化を施されたマウスの平均生存期間は215日であった(図8および図9)。化学療法を施されたマウスの群に関する生存率は、化学療法およびMtb72f/AS01Bワクチンを施された群と有意に異なる(95%信頼区間(p=0.0067))。
【技術分野】
【0001】
発明の属する分野
本発明は、哺乳動物における結核菌(M. tuberculosis)感染の再活性化を予防または治療する方法および結核菌感染に対する化学療法の期間を短縮する方法に関する。
【背景技術】
【0002】
発明の背景
結核は、結核菌および他のマイコバクテリア属(Mycobacterium)の種の感染によって引き起こされる慢性感染症である。それは開発途上国における主要な疾患であり、そしてもちろん世界の先進地域においても高まりつつある問題であり、毎年約8百万の新規症例および3百万の死がもたらされている。該感染は、かなりの期間、無症候性であることがあるが、最も一般的には、該疾患は急性肺炎として現れ、発熱および乾性咳嗽を生じる。治療しないと、典型的に重大な合併症および死にいたる。
【0003】
結核は、一般に、長期の抗生物質治療を使用して抑制できるが、そのような治療は該疾患の伝播を防ぐために十分でない。感染個体は、ある期間、無症候性であるが伝染性であることがある。さらに、治療レジメンのコンプライアンスが重要であるが、患者の行動をモニターすることが困難である。治療過程を完了しない患者があり、その結果、治療が無効になり、薬物耐性が発生しうる。全治療過程を完了した場合でさえ、結核菌の感染は感染個体から根絶されず、潜伏感染として残り、それは再活性化しうる。
【0004】
結核の伝播を抑制するために、有効なワクチン接種および疾患の正確な早期診断が最も重要である。現在、生細菌でのワクチン接種が、防御免疫を誘発するための最も有効な方法である。この目的で使用される最も一般的なマイコバクテリウムは、M. bovisの弱毒性株であるカルメット・ゲラン桿菌(BCG)である。しかし、BCGの安全性および効力は論議の種であり、いくつかの国、例えば米国は、一般の人々にこの物質をワクチン接種しない。
【0005】
結核の診断は、一般に、皮膚検査を使用して達成され、その検査は、ツベルクリンPPD(タンパク質精製誘導体)に対する皮内曝露を伴う。抗原特異的T細胞応答によって、注射後48〜72時間までに注射部位で測定可能な硬化が生じ、マイコバクテリア抗原に対する曝露が示される。しかし、この検査に関して、感度および特異性が問題となっており、BCGでワクチン接種された個体は感染個体と識別することができない。
【0006】
マクロファージがマイコバクテリウム免疫の主要なエフェクターとして機能することが示されている一方、T細胞は該免疫の最も重要な誘導因子である。マイコバクテリウム感染に対する防御におけるT細胞の必須の役割は、マイコバクテリウム感染がAIDS患者において頻繁に出現することによって説明される。それは、ヒト免疫不全ウイルス(HIV)感染と関連するCD4+T細胞の枯渇に起因する。マイコバクテリウム反応性CD4+T細胞は、γ−インターフェロン(IFN−γ)の強力な生産者であることが示されていて、γ−インターフェロンは、次いで、マウスにおけるマクロファージの抗マイコバクテリア効果を惹起することが示されている。ヒトにおけるIFN−γの役割は明らかではないが、研究では、1,25−ジヒドロキシ−ビタミンD3が、単独で、あるいはIFN−γまたは腫瘍壊死因子−αとの組み合わせで、ヒトマクロファージを活性化させて、結核菌感染を阻害することが示されている。さらにまた、IFN−γがヒトマクロファージを刺激して、1,25−ジヒドロキシ−ビタミンD3を生成させることが知られている。同様に、インターロイキン−12(IL−12)は、結核菌感染に対する抵抗性を刺激することに役割を果たすことが示されている。結核菌感染の免疫学についての総説としては、Chan & Kaufmann, Tuberculosis: Pathogenesis, Protection and Control (Bloom ed., 1994), Tuberculosis (2nd ed., Rom and Garay, eds., 2003), およびHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Chan & Kaufmann, Tuberculosis: Pathogenesis, Protection and Control (Bloom ed., 1994)
【非特許文献2】Tuberculosis (2nd ed., Rom and Garay, eds., 2003)
【非特許文献3】Harrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)
【発明の概要】
【発明が解決しようとする課題】
【0008】
活発な感染および潜伏感染の両者に由来する結核菌(Mycobacterium tuberculosis)感染の再活性化を防止するための有効な治療ストラテジーに関する必要性が依然として存在する。本発明は、この必要性および他の必要性を満たす。
【課題を解決するための手段】
【0009】
列挙される配列の説明
配列番号1:N末端の6Hisタグを有するMtb72f(DNA)。
【0010】
配列番号2:N末端の6Hisタグを有するMtb72f(タンパク質)。
【0011】
配列番号3:N末端の2His挿入物を有するM72(Mtb72fの変異体)(DNA)。
【0012】
配列番号4:N末端の2His挿入物を有するM72(Mtb72fの変異体)(タンパク質)。
【0013】
配列番号5:N末端のHis挿入物を有さないMtb72f(DNA)。
【0014】
配列番号6:N末端のHis挿入物を有さないMtb72f(タンパク質)。
【0015】
本発明の概要
本発明は、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片を、例えば1以上のアジュバントと共に含んでなる医薬組成物を提供する。該アジュバントには、AS01BおよびAS02Aが含まれる。
【0016】
本発明は、Mtb72f融合タンパク質またはその免疫原性断片を、例えば1以上のアジュバントと共に投与するか、あるいはMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を投与すると、活性または不活性な結核菌感染の再活性化を予防または治療できるという発明者らの発見に部分的に基づく。好ましい実施形態では、Mtb72f融合タンパク質または核酸を、結核菌感染に対して有効な1以上の化学療法剤と共に投与する。
【0017】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を予防または治療する。
【0018】
別の態様では、被験体での結核の再活性化を防止するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、発現されたMtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を予防または治療する。
【0019】
別の態様では、結核菌感染に対する化学療法の期間を減少させるための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤および、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質またはその免疫原性断片は結核菌に対する免疫応答を誘導することにより、結核菌感染に対する化学療法の期間を減少させることが可能である。結核菌感染に対する化学療法の期間を短縮することによって、本方法は、結核菌感染に関する治療対象の個体が全治療過程を完了するコンプライアンスの向上に有効でもある。
【図面の簡単な説明】
【0020】
【図1】図1は、Swiss Websterマウス(SWR/J)における結核菌の再活性化モデルを図示する。この図面では、感染、化学療法処置(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)、免疫化、および菌数(bacterial load)/コロニー形成単位(CFU)の算出の時点を示す。
【図2】図2は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIgG1およびIgG2a抗体応答免疫応答を示す。マウスは、無処置のままか、化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)で処置するか、あるいは化学療法で処置し、かつアジュバントを伴わずに製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。最後の免疫化の10日後にマウスから採血し、血清を、IgG1(赤色)およびIgG2a(黒色)同位体の両者に関する抗Mtb72f抗体応答に関してELISAによって試験した。
【図3】図3は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIgG1およびIgG2a抗体応答免疫応答を示す。マウスは、無処置のままか、化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)で処置するか、あるいは化学療法で処置し、かつアジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。最後の免疫化の10日後にマウスから採血し、血清を、IgG1(赤色)およびIgG2a(黒色)同位体の両者に関する抗Mtb72f抗体応答に関してELISAによって試験した。
【図4】図4は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスにおけるインターフェロン−γ(IFN−γ)応答を示す。種々の時点でマウスから脾臓細胞を取得し、記載のようにin vitroで10μg/mlのrMtb72fまたは構成要素(Mtb32CおよびMtb39)で3日間刺激した。対照として、PPD(3μg/ml)、BCG溶解物(10μg/ml)、conA(3μg/ml)または培地のみでも脾細胞培養物を刺激した。次いでELISAによってIFN−γ生産を測定した。
【図5】図5は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるIFN−γ応答を示す。種々の時点でマウスから脾臓細胞を取得し、記載のようにin vitroで10μg/mlのrMtb72fまたは構成要素(Mtb32CおよびMtb39)で3日間刺激した。対照として、PPD(3μg/ml)、BCG溶解物(10μg/ml)、conA(3μg/ml)または培地のみでも脾細胞培養物を刺激した。次いでELISAによってIFN−γ生産を測定した。
【図6A】図6は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるCD4+T細胞およびIFN−γサイトカイン応答を示す。種々の時点でマウスから脾臓細胞を取得し、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4およびIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色(ICS)によって測定した。
【図6B】図6は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/JマウスにおけるCD4+T細胞およびIFN−γサイトカイン応答を示す。種々の時点でマウスから脾臓細胞を取得し、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4およびIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色(ICS)によって測定した。
【図7】図7は、Mtb感染後120日の時点でのCD4+およびCD8+T細胞特異的IFN−γ+生産の値についての表形式の概要を示す。無処置のままか、30日間、60日間もしくは90日間の併用化学療法で処置、またはMtb72fワクチンの補助療法として併用化学療法で処置されたマウスの群から脾臓細胞を取得した。脾細胞を、in vitroで一晩、10μg/mlのrMtb72fで刺激した。次いでその細胞をCD4、CD8またはIFN−γに関して染色した。対照として、脾細胞培養物を培地のみでも刺激した。次いでCD4+およびCD8+T細胞特異的IFN−γ+生産を細胞内サイトカイン染色によって測定した。
【図8】図8は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスの生存率を示す。50〜100CFUのMtbH37Rvを含むエアロゾルでマウスを感染させ、30日後、一部のマウスにおいて化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)を開始した。化学療法は60日間継続した。化学療法を受けたマウスの半数を、アジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。
【図9】図9は、化学療法で処置された後、Mtb72fで免疫化された結核菌感染SWR/Jマウスの生存率を示す。50〜100CFUのMtbH37Rvを含むエアロゾルでマウスを感染させ、30日後、一部のマウスにおいて化学療法(飲料水1リットルあたりリファンピン50mg/イソニアジド85mg)を開始した。別々の群のマウスにおいて30日間、60日間または90日間、化学療法を継続した。化学療法を受けたマウスの半数を、アジュバントAS01Bと共に製剤化された用量あたり8μgのMtb72fで筋肉内に3回免疫化した。
【発明を実施するための形態】
【0021】
具体的な実施形態の詳細な説明
本発明は、活性または不活性な(すなわち潜伏性の)マイコバクテリウム感染の再活性化を治療するか、予防するか、あるいは遅延させるために有用なMtb72f核酸または融合タンパク質およびアジュバントを含んでなる組成物、ならびにそれらの使用のための方法に関する。より具体的には、本発明の組成物は、結核菌群のマイコバクテリア属の種、例えばM. tuberculosis、M. bovis、またはM. africanum等の種、または環境性または日和見性であり、かつ免疫不全宿主(例えばAIDS患者)において日和見感染、例えば肺感染を引き起こすマイコバクテリア属の種、例えばBCG、M. avium、M. intracellulare、M. celatum、M. genavense、M. haemophilum、M. kansasii、M. simiae、M. vaccae、M. fortuitum、およびM. scrofulaceumに由来する成分を有するMtb72f融合ポリペプチドもしくはその免疫原性断片またはMtb72f融合ポリペプチドもしくはその免疫原性断片をコードする核酸を含む(例えばHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと)。本出願の発明者らは、驚くべきことに、Mtb72f融合ポリペプチドまたはMtb72f融合ポリペプチドをコードする核酸、またはその免疫原性断片を含んでなる組成物が、結核菌感染の再活性化を治療し、予防し、あるいは遅延させるのに有用であることを発見した。好ましい実施形態では、Mtb72f融合ポリペプチドまたは核酸を1以上の化学療法剤と共に投与する。したがって、これらの組成物、ポリペプチド、およびそれらをコードする核酸は、哺乳動物において、疾患症状の再活性化に対して防御性である免疫応答を誘発するために有用である。
【0022】
本発明のMtb72f核酸および融合ポリペプチドは、それらの抗原性を増強し、あるいは他の態様におけるこれらの抗原を改善するように設計された他の成分をさらに含みうる。例えば、該抗原の一端に対して一続きのヒスチジン残基を付加することによって、該融合ポリペプチド抗原の単離の改善を促してよい。本発明の組成物、ポリペプチド、および核酸は、追加コピーの抗原、すなわちマイコバクテリア属の種に由来する追加の異種性ポリペプチド、例えばMTB8.4抗原、MTB9.8抗原、MTB9.9抗原、MTB40抗原、MTB41抗原、ESAT−6抗原、MTB85複合抗原、α−結晶性抗原、またはNS1抗原を含みうる。あるいは、またはさらに、本発明の組成物、ポリペプチド、および核酸は、マイコバクテリア属の種に由来する他の抗原の追加のコピー、例えばAg85BまたはMTCC#2を含みうる。本発明の組成物、ポリペプチド、および核酸は、また、他の供給源に由来する追加のポリペプチドを含みうる。例えば、本発明の組成物および融合タンパク質には、ポリペプチドまたはポリペプチドをコードする核酸が含まれうる。該ポリペプチドは、抗原、例えばNS1、インフルエンザウイルスタンパク質の発現を増強するものである(例えばWO99/40188およびWO93/04175を参照のこと)。本発明の核酸は、選択された生物種、例えばヒトにおけるコドン優先度に基づいて遺伝子操作することができる。
【0023】
Mtb72f融合タンパク質組成物は、通常、1以上のアジュバント、例えばAS01B(リポソーム製剤中のモノホスホリル脂質A(MPL)およびQS21;米国特許公開第2003/0143240号を参照のこと);AS02A(3D−MPLおよびQS21および水中油型エマルジョン;Bojang, et al., Lancet (2001) 358:1927を参照のこと);ENHANZYN(Detox);3D−MPL;サポニン、例えばQuil Aおよびその成分、例えばQS21およびサポニン模倣物;CWS;TDM;AGP;免疫賦活性オリゴヌクレオチド(oligonucleoptides)、例えばCPG;Leif;およびその誘導体を含む。好ましい実施形態では、リポソーム製剤中の3D−MPLおよびQS21、例えばAS01BおよびMPLおよびQS21および水中油型エマルジョン(例えばAS02A)からなる群から選択される1以上のアジュバントと共にMtb72f融合ポリペプチドを投与する。アジュバントAS01BおよびAS02Aは、Pichyangkul, et al., Vaccine (2004) 22:3831-40にさらに記載されている。
【0024】
Mtb72f抗原を核酸として送達する場合、例えばウイルスベクター(すなわちアデノウイルスベクター)中で、または突然変異体細菌宿主細胞(すなわち突然変異体、弱毒性マイコバクテリウム、ラクトバシラス属(Lactobacillus)またはバシラス属(Bacillus)宿主細胞、例えばカルメット・ゲラン桿菌(BCG)およびLactococcus lactis)中で送達することができる。
【0025】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止する。本発明の方法を実施することによって、結核菌感染の再活性化を遅延させることができる(例えば数か月、数年の期間または無期限に)。
【0026】
一態様では、被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌に感染している哺乳動物に、結核菌群のマイコバクテリア属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、発現されたMtb72f融合タンパク質は結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止する。
【0027】
一実施形態では、活発な結核菌感染を有する個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、不活性すなわち潜伏性の結核菌感染を有する個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、結核菌の多剤耐性株に感染している個体にMtb72f核酸または融合タンパク質を投与する。一実施形態では、以前にカルメット・ゲラン桿菌(BCG)で免疫化されている個体にMtb72f核酸または融合タンパク質を投与する。
【0028】
いくつかの実施形態では、Mtb72f核酸または融合タンパク質を、結核菌感染に対して有効な1以上の化学療法剤と共に投与する。そのような化学療法剤の例には、非限定的に、アミカシン、アミノサリチル酸、カプレオマイシン、サイクロセリン、エタンブトール、エチオナミド、イソニアジド、カナマイシン、ピラチナミド、リファマイシン(すなわちリファンピン、リファペンチンおよびリファブチン)、ストレプトマイシン、オフロキサシン、シプロフロキサシン、クラリスロマイシン、アジスロマイシンおよびフルオロキノロンが含まれる。そのような化学療法は、好ましい薬物の組み合わせを使用するよう担当医の判断によって決定される。薬剤耐性でない結核菌感染を治療するために使用される「第一選択」の化学療法剤には、イソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミドが含まれる。1以上の「第一選択」の薬物に対して薬剤耐性を示している結核菌感染を治療するために使用される「第二選択」の化学療法剤には、オフロキサシン、シプロフロキサシン、エチオナミド、アミノサリチル酸、サイクロセリン、アミカシン、カナマイシンおよびカプレオマイシンが含まれる。
【0029】
Mtb72f核酸または融合タンパク質は、結核菌感染に対して有効な1以上の化学療法剤の投与前に、その投与と同時に、あるいはその投与後に投与することができる。一実施形態では、1以上の化学療法剤の投与を開始した約2週間後にMtb72f核酸または融合タンパク質を投与する。1以上の化学療法剤を、一般に、例えば約1週間、2週間、3週間、または4週間、2ヵ月間、3ヵ月間、4ヵ月間、5ヵ月間、6ヵ月間、または8ヵ月間、1年間またはそれ以上の期間にわたって投与する。
【0030】
特定の実施形態では、カルメット・ゲラン桿菌(BCG)を投与することによってMtb72f核酸または融合タンパク質の効果を増強する。
【0031】
いくつかの実施形態では、Mtb72f核酸または融合ポリペプチドの初回免疫すなわち最初の投与に続いて、Mtb72f核酸または融合ポリペプチドの1回以上の「追加免疫」すなわち後の投与を行う(「初回免疫および追加免疫」法)。例えば、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f核酸または融合タンパク質の1回以上の後の投与を行う。一実施形態では、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f融合ポリペプチドの1回以上の後の投与を行う。一実施形態では、Mtb72f核酸または融合ポリペプチドでの最初の投与に続いて、Mtb72f核酸の1回以上の後の投与を行う。通常、最初の、すなわち「初回免疫」の投与および次の、すなわち「追加免疫」の投与は、約2〜12週間をあけて、あるいは4〜6か月までの期間をあけて行う。その後の「追加免疫」投与は約6か月をあけて、あるいは1、2、3、4または5年もの長期間をあけて行う。慣用の追加免疫処置(例えばタンパク質初回免疫投与、その後のタンパク質追加免疫投与)は、また、結核菌の再活性化に対する予防または治療において有用である。
【0032】
別の態様では、結核菌感染に対する化学療法の期間を減少させるか、あるいは短縮するための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤、ならびに結核菌群のマイコバクテリア属の種に由来するMtb72f融合ポリペプチドまたはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含む方法において前記組成物を使用し、その場合、該Mtb72f融合ポリペプチドは結核菌に対する免疫応答を誘導することにより、結核菌感染に対する化学療法の期間を減少させるか、あるいは短縮することが可能である。通常、Mtb72f核酸または融合ポリペプチドの投与により、6か月、5か月、4か月、3か月、またはそれ未満以内に結核菌感染に対する有効な化学療法処置が可能になる。
【0033】
該Mtb72f組成物は、通常、ヒトに投与するが、他の哺乳動物においても有効であり、該哺乳動物には、家畜哺乳動物(すなわちイヌ、ネコ、ウサギ、ラット、マウス、モルモット、ハムスター、チンチラ)および農業用哺乳動物(すなわちウシ、ブタ、ヒツジ、ヤギ、ウマ)が含まれる。
【0034】
その最も一般的な関連において、本発明のMtb72f融合タンパク質は、3抗原Ra12−TbH9−Ra35のそれぞれの少なくとも免疫原性断片を含むタンパク質である。
【0035】
本出願の命名法では、Ra35とは、Mtb32A(Ra35FL)のN末端を表し、それは少なくとも結核菌に由来するMtb32Aの最初の約205アミノ酸を含み、そのヌクレオチドおよびアミノ酸配列は米国特許出願第09/597,796号の図4に開示され、あるいは別のマイコバクテリア属の種に由来する対応する領域である。最も典型的には、Ra35とは、本出願で開示される配列番号2の残基535−729に対応する部分を表す。あるいは、それは、配列番号2の710位に対応するアミノ酸SerがAlaに置換されているRa35に関する変異体を表す。
【0036】
Ra12とは、Mtb32A(Ra35FL)のC末端を表し、それは少なくとも結核菌に由来するMTB32Aの最後の約132アミノ酸を含み、その配列は米国特許出願第09/072,967号の配列番号4(DNA)および配列番号66(予測されるアミノ酸配列)として開示され、あるいは別のマイコバクテリア属の種に由来する対応する領域である。最も典型的には、Ra12とは、本出願で開示される配列番号2の残基8−139に対応する部分を表す。
【0037】
Mtb39(TbH9)とは、基本的には、米国特許出願第08/658,800号、第08/659,683号、同第08/818,112号、および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号106(完全長cDNA)および配列番号107(完全長タンパク質)として開示されている配列を表す。該配列は、米国特許出願第09/056,559号の配列番号33(DNA)および配列番号91(アミノ酸)としても開示されている。最も典型的には、TbH9とは、本出願で開示される配列番号2の残基143−532に対応する部分を表す。
【0038】
以下に、本発明の組成物および融合タンパク質中で使用されるいくつかの個別の抗原の配列を提供する。
【0039】
Mtb32A(TbRa35FLまたはRa35FL)。その配列は米国特許出願第08/523,436号、同第08/523,435号、同第08/658,800号、同第08/659,683号、同第08/818,112号、同第09/056,556号、および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号17(cDNA)および配列番号79(タンパク質)として開示されている。さらにSkeiky et al., Infection and Immunity 67:3998-4007 (1999)を参照のこと。
【0040】
以下に、本発明のいくつかの融合タンパク質の配列を提供する。
【0041】
TbH9−Ra35(Mtb59F)。その配列は米国特許出願第09/287,849号およびPCT/US99/07717に配列番号23(cDNA)および配列番号24(タンパク質)として開示されている。
【0042】
Ra12−TbH9−Ra35(Mtb72f)。その配列は、本出願の配列番号1または配列番号5(DNA)および配列番号2または配列番号6(タンパク質)として、ならびに米国特許出願第09/223,040号、およびPCT/US99/07717に開示されている。配列番号1および配列番号2の配列には、6His残基のHisタグが含まれる。
【0043】
Mtb72fの突然変異体であるM72は、配列番号2の710位に対応するアミノ酸におけるセリン残基がAlaに変更され(ならびに4His残基がN末端のHisタグから除去され)ていて、その配列は本出願において配列番号3(DNA)および配列番号4(タンパク質)として開示されている。タンパク質が6His残基のHisタグを有するこれらの配列に関する変異体が、米国特許出願第09/597,796号およびPCT/US01/19959に開示されている。Ser710がAlaに置換されているために、M72は自己融解に対してMtb72fより抵抗性であると考えられる。
【0044】
以下に、本発明の組成物および融合タンパク質中で使用されるいくつかの追加の抗原の配列を提供する。
【0045】
Mtb8.4(DPV)。その配列は米国特許出願第08/658,800号、同第08/659,683号、同第08/818,112号および同第08/818,111号ならびにWO97/09428およびWO97/09429に配列番号101(cDNA)および配列番号102(タンパク質)として開示されている。
【0046】
Mtb9.8(MSL)。その配列は米国特許出願第08/859,381号、同第08/858,998号、同第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号12(DNA)、配列番号109(予測されるアミノ酸配列)および配列番号110〜124(ペプチド)として開示されている。
【0047】
Mtb9.9A(MTI(MTI−Aとしても知られている))。その配列は米国特許出願第08/859,381号、同第08/858,998号、同第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号3および配列番号4(DNA)および配列番号29および配列番号51〜66(MTIのORFペプチド)として開示されている。さらに、2種の他のMTI変異体が存在し、MTI−BおよびMTI−Cと称される。
【0048】
Mtb40(HTCC#1)。その配列は米国特許出願第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514に配列番号137(cDNA)および138(予測されるアミノ酸配列)として開示されている。
【0049】
Mtb41(MTCC#2)。その配列は米国特許出願第09/073,009号および同第09/073,010号ならびにPCT/US98/10407およびPCT/US98/10514出願に配列番号140(cDNA)および配列番号142(予測されるアミノ酸配列)として開示されている。
【0050】
ESAT−6。その配列は米国特許出願第09/072,967号の配列番号103(DNA)および配列番号104(予測されるアミノ酸配列)として開示されている。ESAT−6の配列は米国特許第5,955,077号にも開示されている。
【0051】
α−結晶性抗原。その配列はVerbon et al., J. Bact. 174:1352-1359 (1992)に開示されている。
【0052】
85複合抗原。その配列はContent et al., Infect. & Immunol. 59:3205-3212 (1991)に開示されている。
【0053】
上記各配列はCole et al. Nature 393:537 (1998)にも開示されていて、例えばhttp://www.sanger.ac.ukおよびhttp:/www.pasteur.fr/mycdb/で見出すことができる。
【0054】
上記配列は米国特許出願第08/523,435号、同第08/523,436号、同第08/658,800号、同第08/659,683号、同第08/818,111号、同第08/818,112号、同第08/942,341号、同第08/942,578号、同第08/858,998号、同第08/859,381号、同第09/056,556号、同第09/072,596号、同第09/072,967号、同第09/073,009号、同第09/073,010号、同第09/223,040号、同第09/287,849号およびPCT特許出願PCT/US98/10407、PCT/US98/10514、PCT/US99/03265、PCT/US99/03268、PCT/US99/07717、WO97/09428およびWO97/09429、WO98/16645、WO98/16646に開示されている。各文献は参照により本明細書中に組み入れられる。
【0055】
本明細書中に記載の抗原には、多型変異体および保存的に改変された変異、ならびに株間および種間マイコバクテリウムホモログが含まれる。さらに、本明細書中に記載の抗原には、サブシークエンスまたはトランケート配列が含まれる。融合タンパク質は、追加のポリペプチド、場合によりマイコバクテリウムまたは他の供給源に由来する異種性ペプチドを含有してもよい。これらの抗原は、例えば以下に記載のリンカーペプチド配列を付加することによって改変してよい。これらのリンカーペプチドは、融合タンパク質の各々を構成する1以上の成分の間に挿入してよい。
【0056】
定義
用語「結核の再活性化」とは、ツベルクリン検査で陽性と出るが見かけの疾患症状を有さない個体における疾患症状の後の発現を表す。該個体は結核菌に感染していて、結核が不活性すなわち潜伏期に入るように十分に治療されている活性な疾患症状を以前に示していてもいなくてもよい。しかし、結核の再活性化を予防または治療するための方法は、疾患の活発な症状を示す個体において開始することができる。
【0057】
「一次性結核」とは、結核菌感染に直接続く臨床疾病(疾患症状の発現)を表す。Harrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005)を参照のこと。
【0058】
「二次性結核」または「一次性感染後結核」とは、休眠性、不活性すなわち潜伏性の結核菌感染の再活性化を表す。Harrison's Principles of Internal Medicine(上記)を参照のこと。
【0059】
「結核菌の活発(活性)な感染」とは、顕性疾患症状を伴う結核菌感染を表す。
【0060】
「結核菌の不活性、休眠性すなわち潜伏性の感染」とは、顕性疾患症状を伴わない結核菌感染を表す。
【0061】
「薬剤耐性」結核菌感染とは、感染株が、結核菌感染の治療に有効な1以上のいわゆる「第一選択」の化学療法剤(例えばイソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミド)によって、静的な状態にされたり、死滅させられたりしない(それらに耐性の)結核菌感染を表す。
【0062】
「多剤耐性」結核菌感染とは、感染株が結核菌感染の治療に有効な2以上の「第一選択」の化学療法剤に耐性である結核菌感染を表す。
【0063】
「結核菌感染の治療に有効な化学療法剤」とは、当技術分野において公知でかつ結核菌感染の治療に使用されている薬理学的物質を表す。結核菌感染を治療するために使用される例示的薬理学的物質には、非限定的に、アミカシン、アミノサリチル酸、カプレオマイシン、サイクロセリン、エタンブトール、エチオナミド、イソニアジド、カナマイシン、ピラチナミド、リファマイシン(すなわちリファンピン、リファペンチンおよびリファブチン)、ストレプトマイシン、オフロキサシン、シプロフロキサシン、クラリスロマイシン、アジスロマイシンおよびフルオロキノロンが含まれる。薬剤耐性でない結核菌感染を治療するために使用される「第一選択」の化学療法剤には、イソニアジド、リファンピン、エタンブトール、ストレプトマイシンおよびピラチナミドが含まれる。1以上の「第一選択」の薬物に対して薬剤耐性を示している結核菌感染を治療するために使用される「第二選択」の化学療法剤には、オフロキサシン、シプロフロキサシン、エチオナミド、アミノサリチル酸、サイクロセリン、アミカシン、カナマイシンおよびカプレオマイシンが含まれる。そのような薬理学的物質はGoodman and Gilman's The Pharmacological Basis of Therapeutics, Hardman and Limbird eds., 2001のChapter 48で概説されている。
【0064】
「FL」とは、完全長、すなわち野生型ポリペプチドと同じ長さのポリペプチドを表す。
【0065】
「Hisタグ」とは、N末端の、通常、開始Met残基の直後に挿入されているか、あるいはC末端に挿入されている一連のHis残基、典型的に6残基を表す。それらは、通常、天然配列に対して異種性であるが、それらは、固定化金属アフィニティークロマトグラフィー樹脂(IMAC)に対するタンパク質結合を向上させることによって単離を容易にするために組み込まれる。一般的に言えば、Hisタグの存在または不存在は、誘発対象の抗原タンパク質に対する有用な免疫応答を引き起こす観点からは重要でない。Hisタグ自体に対する有害な免疫反応が誘発されるといけないので、Hisタグの長さを最小に、例えば4以下の残基、特に2残基にすることが最良であると見なされる。
【0066】
用語「その免疫原性断片」とは、細胞障害性Tリンパ球、ヘルパーTリンパ球またはB細胞によって認識されるエピトープを含むポリペプチドを表す。典型的に、Mtb72fの免疫原性断片は、500以上のアミノ酸、例えば600以上のアミノ酸、例えば700以上のアミノ酸を含有するポリペプチドである。本発明は、また、Mtb72F融合タンパク質の配列の全体または実質的に全体(例えば500以上のアミノ酸、例えば600以上のアミノ酸、例えば700以上のアミノ酸)を一緒になってカバーする複数の断片、例えば重複断片を包含する。
【0067】
用語「結核菌群のマイコバクテリア属の種」には、結核という疾患を引き起こすと従来より見なされている種、ならびに免疫無防備状態の患者、例えばAIDS患者において結核および肺疾患を引き起こすマイコバクテリウムの環境性および日和見感染性の種が含まれ、例えばM. tuberculosis、M. bovis、またはM. africanum、BCG、M. avium、M. intracellular、M. celatum、M. genavense、M. haemophilum、M. kansasii、M. simiae、M. vaccae、M. fortuitum、およびM. scrofulaceumである(例えばHarrison's Principles of Internal Medicine, Chapter 150, pp. 953-966 (16th ed., Braunwald, et al., eds., 2005を参照のこと)。
【0068】
アジュバントとは、抗原に対する特定の免疫応答を高めるワクチンまたは治療用組成物中の成分を表す(例えばEdelman, AIDS Res. Hum Retroviruses 8:1409-1411 (1992)を参照のこと)。アジュバントはTh1型およびTh2型応答の免疫応答を誘発する。Th1型サイトカイン(例えばIFN−γ、IL−2、およびIL−12)は投与抗原に対する細胞性免疫応答の誘発に好都合である傾向があり、一方、Th2型サイトカイン(例えばIL−4、IL−5、IL−6、IL−10およびTNF−β)は体液性免疫応答の誘発に好都合である傾向がある。Th1細胞性免疫応答の優先的刺激が可能なアジュバントはWO94/00153およびWO95/17209に記載されている。
【0069】
「核酸」とは、一本鎖または二本鎖型のデオキシリボヌクレオチドまたはリボヌクレオチドおよびそのポリマーを表す。該用語は、公知のヌクレオチドアナログまたは改変された主鎖残基または結合を含有する核酸であって、合成の、天然に存在する、および非天然に存在するものであり、参照核酸と同様の結合特性を有し、かつ参照ヌクレオチドと同様の様式で代謝される核酸を包含する。そのようなアナログの例には、非限定的に、ホスホロチオアート、ホスホルアミダート、メチルホスホナート、キラルメチルホスホナート、2−O−メチルリボヌクレオチド、ペプチド核酸(PNA)が含まれる。
【0070】
特に指定しない限り、具体的核酸配列は、さらに、保存的に改変されたその変異体(例えば縮重コドン置換物)および相補的配列、ならびに明示的に記載されている配列を暗黙のうちに包含する。特に、縮重コドン置換物は、1以上の選択(またはすべての)コドンの第3位が混合塩基および/またはデオキシイノシン残基で置換されている配列を作成することによって達成してよい(Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); Rossolini et al., Mol. Cell. Probes 8:91-98 (1994))。核酸という用語は、遺伝子、cDNA、mRNA、オリゴヌクレオチド、およびポリヌクレオチドと交換可能に使用される。
【0071】
用語「ポリペプチド」、「ペプチド」および「タンパク質」は本明細書中で交換可能に使用され、アミノ酸残基のポリマーを表す。該用語は、1以上のアミノ酸残基が、対応する天然に存在するアミノ酸の人工の化学的模倣物であるアミノ酸ポリマー、ならびに天然に存在するアミノ酸ポリマーおよび非天然に存在するアミノ酸ポリマーに適用される。
【0072】
用語「アミノ酸」とは、天然に存在するアミノ酸および合成アミノ酸、ならびに天然に存在するアミノ酸と同様の様式で機能するアミノ酸アナログおよびアミノ酸模倣物を表す。天然に存在するアミノ酸とは、遺伝暗号によってコードされるアミノ酸、ならびに後に改変されたそれらのアミノ酸、例えばヒドロキシプロリン、γ−カルボキシグルタマート、およびO−ホスホセリンである。アミノ酸アナログとは、天然に存在するアミノ酸と同じ基本化学構造、すなわち水素、カルボキシル基、アミノ基、およびR基に結合しているα炭素を有する化合物を表し、例えばホモセリン、ノルロイシン、メチオニンスルホキシド、メチオニンメチルスルホニウムである。そのようなアナログは改変されたR基(例えばノルロイシン)または改変されたペプチド主鎖を有するが、天然に存在するアミノ酸と同じ基本化学構造を保持する。アミノ酸模倣物とは、アミノ酸の一般的化学構造と異なる構造を有するが、天然に存在するアミノ酸と同様の様式で機能する化学物質を表す。
【0073】
本明細書中で、アミノ酸は、IUPAC-IUB Biochemical Nomenclature Commissionによって推奨されるそれらの一般に知られている3文字記号または1文字記号によって言及されることがある。同様に、ヌクレオチドは、それらの一般に許容される1文字コードによって言及されることがある。
【0074】
「保存的に改変された変異体」は、アミノ酸および核酸配列の両者に適用される。特定の核酸配列に関して、保存的に改変された変異体とは、同一または本質的に同一のアミノ酸配列をコードする核酸を表すか、あるいは該核酸がアミノ酸配列をコードしない場合には、本質的に同一の配列を表す。遺伝暗号の縮重のせいで、多数の機能的に同一の核酸が任意の所定のタンパク質をコードする。例えば、コドンGCA、GCC、GCGおよびGCUはすべてアミノ酸アラニンをコードする。ゆえに、コドンによってアラニンが指定されるすべての位置で、該コドンは、対応する上記コドンのいずれかに改変することができ、コード対象のポリペプチドは変更されない。そのような核酸変異は「サイレント変異」であり、保存的に改変された変異の1種である。ポリペプチドをコードする本明細書中のすべての核酸配列は、該核酸のすべてのサイレント変異候補を同時に記載している。当業者は、核酸中の各コドン(通常メチオニンに関する唯一のコドンであるAUG、および通常トリプトファンに関する唯一のコドンであるTGGを除く)を改変して機能的に同一の分子を得ることができることを認識する。したがって、ポリペプチドをコードする核酸の各サイレント変異は、記載される各配列に暗黙のうちに含まれる。
【0075】
アミノ酸配列に関して、当業者は、コード配列中の単一のアミノ酸または少ないパーセンテージのアミノ酸を変更、付加または欠失する、核酸、ペプチド、ポリペプチド、またはタンパク質配列の個々の置換、欠失または付加が「保存的に改変された変異体」であり、その場合、該改変は、化学的に類似のアミノ酸によるアミノ酸の置換を生じさせることを認識する。機能的に類似のアミノ酸を提供する同類置換表は当技術分野において周知である。そのような保存的に改変された変異体は、本発明の多型変異体、種間ホモログ、および対立遺伝子に追加されるものであり、それらを排除しない。
【0076】
以下の8群は、それぞれ、互いに同類置換であるアミノ酸を含有する:
1)アラニン(A)、グリシン(G);
2)アスパラギン酸(D)、グルタミン酸(E);
3)アスパラギン(N)、グルタミン(Q);
4)アルギニン(R)、リシン(K);
5)イソロイシン(I)、ロイシン(L)、メチオニン(M)、バリン(V);
6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W);
7)セリン(S)、スレオニン(T);および
8)システイン(C)、メチオニン(M)
(例えばCreighton, Proteins (1984)を参照のこと)。
【0077】
用語「異種性」とは、核酸の部分に関して使用される場合、該核酸が、自然界で、互いに同一の関連性において見出されない2以上の部分配列を含むことを示す。例えば、該核酸は典型的に組換え生産され、新規の機能的核酸を作成するように配置された無関係の遺伝子に由来する2以上の配列、例えば1つの供給源に由来するプロモーターおよび別の供給源に由来するコード領域を有する。同様に、異種性タンパク質とは、該タンパク質が、自然界で、互いに同一の関連性において見出されない2以上の部分配列を含むこと(例えば融合タンパク質)を示す。
【0078】
「融合ポリペプチド」または「融合タンパク質」とは、直接またはアミノ酸リンカーを介して、共有結合によって連結された少なくとも2個の異種性のマイコバクテリア属の種のポリペプチドを有するタンパク質を表す。融合タンパク質を形成するポリペプチドは、典型的に、C末端からN末端に連結されるが、それらをC末端からC末端、N末端からN末端、またはN末端からC末端に連結することもできる。融合タンパク質のポリペプチドは任意の順序であってよい。この用語は、また、該融合タンパク質を構成する抗原の保存的に改変された変異体、多型変異体、対立遺伝子、突然変異体、部分配列、および種間ホモログを表す。結核菌抗原はCole et al., Nature 393:537 (1998)に記載されている。該文献は結核菌ゲノム全体を開示している。結核菌の完全配列はhttp://www.sanger.ac.ukおよびhttp://www.pasteur.fr/mycdb/(MycDB)で見出すこともできる。結核菌抗原に対応する他のマイコバクテリア属の種に由来する抗原は、例えば本明細書中に記載の配列比較アルゴリズムまたは当業者に公知の他の方法、例えばハイブリダイゼーションアッセイおよび抗体結合アッセイを使用して特定することができる。
【0079】
本発明において有用な典型的なMtb72f融合タンパク質には以下のものが含まれる:
配列番号2の配列の残基8−729を含むタンパク質;
場合により該配列のHisタグ形成残基2−7を有さないか、あるいは異なる長さのHisタグを有する、配列番号2(=Mtb72f)の配列を含むか、あるいはそれからなるタンパク質;
場合により該配列のHisタグ形成残基2−7を有さないか、あるいは異なる長さのHisタグを有する配列番号2の配列(例えば配列番号2の配列の残基8−729を含むタンパク質)を1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質;
配列番号4(=M72)の配列の残基4−725を含むタンパク質;
場合により該配列のHisタグ形成残基2−3を有さないか、あるいは異なる長さのHisタグを有する、配列番号4(=M72)の配列を含むか、あるいはそれからなるタンパク質;および
場合により該配列のHisタグ形成残基2−3を有さないか、あるいは異なる長さのHisタグを有する配列番号4の配列(例えば配列番号4の配列の残基4−725を含むタンパク質)を1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0080】
本発明において有用な、Mtb72f融合タンパク質の典型的な免疫原性断片には以下のものが含まれる:
TbH9−Ra35(Mtb59F);またはTbH9;またはRa35;またはRa12の配列を含むか、あるいはそれからなるタンパク質;および
該配列を、1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0081】
本発明において有用な、Mtb72f融合タンパク質の追加の典型的な免疫原性断片には以下のものが含まれる:
配列番号2のSer710に対応する位置がAlaに変更されているTbH9−Ra35(Mtb59F)またはRa35の配列を含むか、あるいはそれからなるタンパク質;および
該配列を、1以上の結核菌抗原、例えば上記段落[0045]〜[0052]に列挙される1以上のタンパク質、またはそれらのいずれかの免疫原性断片と共に含む融合タンパク質。
【0082】
より具体的には、Mtb72fは以下のものである:
配列番号2の残基8−729を含むポリペプチド;または
場合により開始Met残基の後ろにHisタグが挿入されている配列番号2の残基1および8−729からなるポリペプチド;または
配列番号2のポリペプチド;または
配列番号4の残基4−725を含むポリペプチド;または
場合により開始Met残基の後ろにHisタグが挿入されている配列番号4の残基1および4−725からなるポリペプチド;または
配列番号4のポリペプチド;または
配列番号6のポリペプチド。
【0083】
追加の典型的なMtb72f融合タンパク質およびその免疫原性断片には、N末端および/またはC末端が例えば5または4または3または2または1アミノ酸残基短縮されている上述のタンパク質が含まれる。
【0084】
追加の典型的なMtb72f融合タンパク質およびその免疫原性断片には、10%までのアミノ酸、例えば5%までのアミノ酸(例えば10まで、例えば5までの)アミノ酸が、本明細書中で規定される同類置換によって置き換えられている上述のタンパク質が含まれる。
【0085】
本発明において用いられる典型的なMtb72f核酸には、上述の典型的なMtb72f融合タンパク質およびその免疫原性断片をコードする核酸(例えばDNA分子)が含まれる。言及することができる1セットの具体的なDNA分子は、配列番号1のヌクレオチド63−2228を含むものである。言及することができる別のセットの具体的なDNA分子は、配列番号3のヌクレオチド10−2175を含むものである。言及することができる具体的なDNA分子は、配列番号1または配列番号3または配列番号5を含むか、あるいはそれからなるものである。
【0086】
用語「融合(fused)」とは、融合タンパク質中の2ポリペプチド間の共有結合による連結を表す。該ポリペプチドは、典型的に、互いに直接的にまたはアミノ酸リンカーを介して、ペプチド結合を介して連結されている。場合により、該ペプチドは、当業者に公知の非ペプチド共有結合性の連結を介して連結することができる。
【0087】
「〜に選択的に(または特異的に)ハイブリダイズする」との言い回しは、該配列が複合混合物(例えば細胞の全DNAもしくはRNA、またはライブラリーのDNAもしくはRNA)中に存在する場合に、ストリンジェントなハイブリダイゼーション条件下で、分子が特定のヌクレオチド配列に対してのみ結合、二重鎖形成、またはハイブリダイズすることを表す。
【0088】
「ストリンジェントなハイブリダイゼーション条件」との言い回しは、プローブが、典型的に核酸の複合混合物中で、その標的部分配列にハイブリダイズするが、他の配列にはハイブリダイズしない条件を表す。ストリンジェントな条件は配列依存的であり、状況ごとに異なる。配列が長いほど、より高い温度で特異的にハイブリダイズする。広範囲にわたる、核酸のハイブリダイゼーションについての指針はTijssen, Techniques in Biochemistry and Molecular Biology-Hybridization with Nucleic Probes, "Overview of principles of hybridization and the strategy of nucleic acid assays" (1993)に見出せる。概して、ストリンジェントな条件は、所定のイオン強度pHで、具体的配列に関する熱融点(Tm)より約5〜10℃低いように選択される。Tmは、平衡状態で、標的に相補的なプローブの50%が標的配列にハイブリダイズする(所定のイオン強度、pH、および核酸濃度条件下での)温度である(標的配列が過剰に存在する場合、Tmでは、平衡状態で50%のプローブが占有される)。ストリンジェントな条件は、塩濃度が、pH7.0〜8.3で約1.0Mナトリウムイオン未満、典型的に約0.01〜1.0Mナトリウムイオン濃度(または他の塩)であり、かつ温度が、短いプローブ(例えば10〜50ヌクレオチド)では少なくとも約30℃および長いプローブ(例えば50ヌクレオチド以上)では少なくとも約60℃である条件である。ストリンジェントな条件は、不安定化物質、例えばホルムアミドを加えることによって達成してもよい。
【0089】
選択的または特異的ハイブリダイゼーションでは、陽性シグナルはバックグラウンドの少なくとも2倍であり、場合によりバックグラウンドハイブリダイゼーションの10倍である。典型的なストリンジェントなハイブリダイゼーション条件は以下の通りであってよい:50%ホルムアミド、5×SSC、および1%SDS、42℃でインキュベート、または、5×SSC、1%SDS、65℃でインキュベートし、0.2×SSC、および0.1%SDS中で65℃で洗浄。
【0090】
ストリンジェントな条件下で互いにハイブリダイズしない核酸は、それらがコードするポリペプチドが実質的に同一であれば、依然として実質的に同一である。これは、例えば遺伝暗号によって許容される最大コドン縮重を使用して1コピーの核酸が作成された場合に生じる。そのような場合、該核酸は、典型的に、中程度にストリンジェントなハイブリダイゼーション条件下でハイブリダイズする。典型的な「中程度にストリンジェントなハイブリダイゼーション条件」には、40%ホルムアミド、1M NaCl、1%SDSのバッファー中で37℃でのハイブリダイゼーション、および1×SSC中で45℃での洗浄が含まれる。陽性ハイブリダイゼーションはバックグラウンドの少なくとも2倍である。当業者は、代替のハイブリダイゼーション条件および洗浄条件を利用して同様にストリンジェントな条件を提供できることを容易に認識する。
【0091】
「抗体」とは、抗原に特異的に結合し、それを認識する、免疫グロブリン遺伝子に由来するフレームワーク領域またはそのフラグメントを含むポリペプチドを表す。一般に認識されている免疫グロブリン遺伝子には、κ、λ、α、γ、δ、ε、およびμ定常領域遺伝子、ならびに種々の免疫グロブリン可変領域遺伝子が含まれる。軽鎖はκまたはλとして分類される。重鎖はγ、μ、α、δ、またはεとして分類され、そしてそれらは、それぞれ、免疫グロブリンクラス、IgG、IgM、IgA、IgDおよびIgEを規定する。
【0092】
典型的な免疫グロブリン(抗体)の構造単位は四量体を含む。各四量体は2個の同一ペアのポリペプチド鎖から構成され、各ペアは1つの「軽」鎖(約25kDa)および1つの「重」鎖(約50〜70kDa)を有する。各鎖のN末端は、抗原認識を主に担う約100〜110またはそれ以上のアミノ酸からなる可変領域を規定する。可変軽鎖(VL)および可変重鎖(VH)という用語は、それぞれ、これらの軽鎖および重鎖を表す。
【0093】
抗体は、例えばインタクトの免疫グロブリンとして、あるいは種々のペプチダーゼでの消化によって生産されるいくつかの十分に特徴付けされているフラグメントとして存在する。ゆえに、例えば、ペプシンは、ヒンジ領域のジスルフィド結合の下で抗体を消化して、それ自体がジスルフィド結合によってVH−CH1に連結された軽鎖であるFabの二量体であるF(ab)’2を生じさせる。F(ab)’2を穏やかな条件下で還元して、ヒンジ領域中のジスルフィド結合を分解することにより、F(ab)’2二量体をFab’モノマーに変換してよい。Fab’モノマーは、本質的に、ヒンジ領域の部分を有するFabである(Fundamental Immunology (Paul ed., 3d ed. 1993)を参照のこと)。種々の抗体フラグメントはインタクトの抗体の消化の観点から定義されるが、当業者は、そのようなフラグメントを、化学的に、あるいは組換えDNA方法論を使用して新たに合成してよいことを認識する。ゆえに、本明細書中で使用される抗体という用語には、完全抗体の改変によって生産される抗体フラグメント、または組換えDNA方法論を使用して新たに合成されるもの(例えば単鎖Fv)またはファージディスプレイライブラリーを使用して特定されるもの(例えばMcCafferty et al., Nature 348:552-554 (1990)を参照のこと)がさらに含まれる。
【0094】
モノクローナル抗体またはポリクローナル抗体の調製では、当技術分野において公知の任意の技術を使用することができる(例えばKohler & Milstein, Nature 256:495-497 (1975); Kozbor et al., Immunology Today 4: 72 (1983); Cole et al., pp. 77-96 in Monoclonal Antibodies and Cancer Therapy (1985)を参照のこと)。単鎖抗体を生産するための技術(米国特許第4,946,778号)を適用して、本発明のポリペプチドに対する抗体を生産することができる。また、トランスジェニックマウス、または他の生物、例えば他の哺乳動物を使用して、ヒト化抗体を発現させてよい。あるいは、ファージディスプレイテクノロジーを使用して、選択抗原に特異的に結合する抗体およびヘテロメリック(heteromeric)Fabフラグメントを特定することができる(例えばMcCafferty et al., Nature 348:552-554 (1990); Marks et al., Biotechnology 10:779-783 (1992)を参照のこと)。
【0095】
抗体に「特異的に(または選択的に)結合する」または、それと「特異的に(または選択的に)免疫反応性である」との言い回しは、タンパク質またはペプチドに言及する場合、タンパク質の混成集団および他の生物学的物質(biologics)中のタンパク質の存在を決定する結合反応を表す。ゆえに、指定の免疫アッセイ条件下で、指定の抗体は、特定のタンパク質に対して、バックグラウンドの少なくとも2倍結合し、サンプル中に存在する他のタンパク質に対して実質的に顕著な量では結合しない。そのような条件下での抗体に対する特異的結合には、特定のタンパク質に関するその特異性に関して選択された抗体が必要とされる。例えば、融合タンパク質に対して産生されるポリクローナル抗体を選択して、融合タンパク質と特異的に免疫反応性であり、かつ該融合タンパク質の個別の成分とはそうでないポリクローナル抗体のみを取得することができる。この選択は、個別の抗原と交差反応する抗体を差し引くことによって達成してよい。種々の免疫アッセイ形式を使用して、特定のタンパク質と特異的に免疫反応性の抗体を選択してよい。例えば、固相ELISA免疫アッセイを通常通り使用して、タンパク質と特異的に免疫反応性の抗体を選択する(特異的免疫反応性を決定するために使用できる免疫アッセイ形式および条件の説明に関しては、例えばHarlow & Lane, Antibodies, A Laboratory Manual (1988)およびUsing Antibodies: A Laboratory Manual (1998)を参照のこと)。典型的には、特異的または選択的反応は、バックグラウンドシグナルすなわちノイズの少なくとも2倍であり、より典型的には、バックグラウンドの10〜100倍を超える。
【0096】
ポリヌクレオチドは、ネイティブの配列(すなわち、個々の抗原またはその部分をコードする内因性配列)を含んでよく、あるいはそのような配列の変異体を含んでよい。ポリヌクレオチド変異体に1以上の置換、付加、欠失および/または挿入を含有させて、コード対象の融合ポリペプチドの生物学的活性が、ネイティブの抗原を含む融合ポリペプチドと比べて減少しないようにしてよい。変異体は、好ましくは、ネイティブのポリペプチドまたはその部分をコードするポリヌクレオチド配列に対して、少なくとも約70%同一性、より好ましくは少なくとも約80%同一性、最も好ましくは少なくとも約90%同一性を示す。
【0097】
2以上の核酸またはポリペプチド配列の関連で、用語「同一の」または「同一性」パーセントとは、比較ウインドウ、すなわち、以下の配列比較アルゴリズムの1つを使用するか、あるいはマニュアルアライメントおよび視覚的点検によって測定される指定領域にわたる最大一致に関して比較およびアライメントされた場合に、同一であるか、あるいは指定のパーセンテージの同一のアミノ酸残基またはヌクレオチド(すなわち、指定領域にわたって70%同一性、場合により75%、80%、85%、90%、または95%同一性)を有する2以上の配列または部分配列を表す。そしてそのような配列は「実質的に同一」であると称される。この定義は試験配列の相補物(compliment)をも表す。場合により、同一性は、少なくとも約25〜約50アミノ酸またはヌクレオチド長の領域にわたって、あるいは場合により75〜100アミノ酸またはヌクレオチド長の領域にわたって存在する。
【0098】
配列比較では、典型的に、1つの配列が参照配列として働き、それに対して試験配列を比較する。配列比較アルゴリズムを使用する場合、試験配列および参照配列をコンピュータに入力し、必要であれば部分配列座標を指定し、配列アルゴリズムプログラムパラメータを指定する。デフォルトプログラムパラメータを使用することができ、あるいは代替パラメータを指定することができる。そして配列比較アルゴリズムは、プログラムパラメータに基づいて、参照配列に対する試験配列の配列同一性パーセントを算出する。
【0099】
本明細書中で使用される「比較ウインドウ」には、25〜500、通常には約50〜約200、より通常には約100〜約150からなる群から選択される数の連続位置のいずれか1つのセグメントへの言及が含まれ、そのセグメントにおいて、2つの配列を最適にアライメントした後に、配列を同一数の連続位置の参照配列と比較してよい。比較のために配列をアライメントする方法は当技術分野において周知である。比較のための最適な配列のアライメントは、例えばSmith & Watermanの局所的相同性アルゴリズム(Adv. Appl. Math. 2:482 (1981))、Needleman & Wunschの相同性アライメントアルゴリズム(J. Mol. Biol. 48:443 (1970))、Pearson & Lipmanの類似度検索法(the search for similarity method)(Proc. Nat'l. Acad. Sci. USA 85:2444 (1988))、これらのアルゴリズムのコンピュータによる実装(GAP, BESTFIT, FASTA, and TFASTA in the Wisconsin Genetics Software Package, Genetics Computer Group, 575 Science Dr., Madison, WI)、またはマニュアルアライメントおよび視覚的点検(例えばCurrent Protocols in Molecular Biology (Ausubel et al., eds. 1995 supplement)を参照のこと)によって行うことができる。
【0100】
有用なアルゴリズムの一例はPILEUPである。PILEUPは、累進ペアワイズアライメント(progressive, pairwise alignments)を使用して関連配列の群に由来する多重配列アライメントを作成し、関連性および配列同一性パーセントを示す。さらに該アライメントの作成に使用されるクラスタリング関連性を示すツリーまたはデンドログラム(dendogram)をプロットする。PILEUPは、Feng & Doolittleの累進アライメント法(J. Mol. Evol. 35:351-360 (1987))を簡素化したものを使用する。使用される方法は、Higgins & Sharp, CABIOS 5:151-153 (1989)に記載の方法に類似の方法である。このプログラムは、それぞれ最大5,000ヌクレオチドまたはアミノ酸長の300までの配列をアライメントすることができる。マルチプルアライメント手順は、2個の最も類似する配列のペアワイズアライメントで開始され、2個のアライメント済み配列のクラスターが得られる。そしてこのクラスターを、次に最も関連している配列または、アライメント済み配列のクラスターに対してアライメントする。配列の2個のクラスターは、2個の個別の配列のペアワイズアライメントを単純に拡張することによってアライメントされる。一連の累進ペアワイズアライメントによって最終のアライメントを達成する。このプログラムは、特定の配列および、配列比較の領域に関するそれらのアミノ酸またはヌクレオチド座標を指定し、プログラムパラメータを指定することによって実行される。PILEUPを使用して、参照配列を他の試験配列と比較して、配列同一性パーセントの関連性を決定する。その場合、次のパラメータ:デフォルトギャップ重み付け(gap weight)(3.00)、デフォルトギャップ長重み付け(gap length weight)(0.10)、および重み付けエンドギャップ(weighted end gaps)を使用する。PILEUPはGCG配列解析ソフトウェアパッケージ、例えばバージョン7.0(Devereaux et al., Nuc. Acids Res. 12:387-395 (1984))から入手することができる。
【0101】
配列同一性および配列類似性パーセントの決定に適しているアルゴリズムの別の例はBLASTおよびBLAST2.0アルゴリズムであり、それぞれAltschul et al., Nuc. Acids Res. 25:3389-3402 (1977)およびAltschul et al., J. Mol. Biol. 215:403-410 (1990)に記載されている。BLAST解析を実施するためのソフトウェアはNational Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/)から一般に利用可能である。このアルゴリズムは、データベース配列の同じ長さのワードとアライメントされた場合にいくつかの正値の閾値スコアTに適合するか、あるいはそれを満たすクエリ配列の長さWの短いワードを特定することによって高スコアの配列ペア(HSP)を最初に特定することを含む。Tは近縁ワードスコア閾値と称される(Altschul et al., 上記)。これらの最初の近縁ワードヒットは、それらを含むより長いHSPを発見するための検索開始のシードとして機能する。そのワードヒットを各配列に沿って両方向に、累積のアラインメントスコアが増加しうる限り伸長する。ヌクレオチド配列の場合、パラメータM(適合残基ペアに対する褒賞スコア;常に>0)およびN(非適合残基に対する罰則スコア;常に<0)を使用して累積スコアを算出する。アミノ酸配列の場合は、スコアリングマトリックスを使用して累積スコアを算出する。各方向におけるワードヒットの伸長は以下の時点で停止させる:累積のアラインメントスコアがその最大達成値から量Xだけ減少したとき;累積スコアが、1個以上の負のスコアを有する残基のアラインメントの蓄積のために0以下になったとき;またはいずれかの配列の末端に達したとき。BLASTアルゴリズムパラメータW、T、およびXによってアライメントの感度および速度が決定される。BLASTNプログラム(ヌクレオチド配列用)では、デフォルトとして、ワード長(W)11、期待値(E)10、M=5、N=−4および両鎖の比較を使用する。アミノ酸配列用のBLASTPプログラムでは、デフォルトとして、ワード長(W)3、および期待値(E)10、およびBLOSUM62スコアリ
ングマトリックス(Henikoff & Henikoff, Proc. Natl. Acad. Sci. USA 89:10915 (1989)を参照のこと)アライメント(B)50、期待値(E)10、M=5、N=−4、および両鎖の比較を使用する。
【0102】
BLASTアルゴリズムでは、2つの配列間の類似性の統計解析をさらに実施する(例えばKarlin & Altschul, Proc. Nat'l. Acad. Sci. USA 90:5873-5787 (1993)を参照のこと)。BLASTアルゴリズムによって提供される類似性の1つの測定値は、最小合計確率(smallest sum probability)(P(N))であり、それは2つのヌクレオチドまたはアミノ酸配列間の適合が偶然生じる確率の指標を提供する。例えば、参照核酸と試験核酸の比較において最小合計確率が約0.2未満、より好ましくは約0.01未満、最も好ましくは約0.001未満であれば、該核酸は参照配列と類似であると見なされる。
【0103】
ポリヌクレオチド組成物
本明細書中で使用される用語「DNAセグメント」および「ポリヌクレオチド」とは、特定の生物種のトータルゲノムDNAからは単離されたDNA分子を表す。したがって、ポリペプチドをコードするDNAセグメントとは、1以上のコード配列を含有するが、該DNAセグメントの取得元の生物種のトータルゲノムDNAから実質的に単離されているか、あるいは精製されているDNAセグメントを表す。DNAセグメントおよびそのようなセグメントの小断片、ならびに組換えベクター、例えばプラスミド、コスミド、ファージミド、ファージ、ウイルス等も、用語「DNAセグメント」および「ポリヌクレオチド」の範囲内に含まれる。
【0104】
当業者に理解されるように、本発明のDNAセグメントには、タンパク質、ポリペプチド、ペプチド等を発現するか、あるいはそれらを発現するように作り変えてよいゲノム配列、ゲノム外およびプラスミドによってコードされる配列および遺伝子操作された小さい遺伝子セグメントが含まれうる。そのようなセグメントは、天然に単離されるか、あるいは人の手によって合成的に改変してよい。
【0105】
本明細書中で使用される「単離された」とは、ポリヌクレオチドが他のコード配列から実質的に離れていること、および該DNAセグメントが、大部分の無関係のコードDNA、例えば大きい染色体断片または他の機能的遺伝子またはポリペプチドコード領域を含有しないことを意味する。当然ながら、これは元々単離されているDNAセグメントを表すが、人の手によって該セグメントに後に付加された遺伝子またはコード領域を排除しない。
【0106】
当業者に認識されるように、ポリヌクレオチドは一本鎖(コードまたはアンチセンス)または二本鎖であってよく、DNA(ゲノム、cDNAまたは合成)またはRNA分子であってよい。RNA分子には、イントロンを含有し、DNA分子と1対1様式で対応するHnRNA分子、およびイントロンを含有しないmRNA分子が含まれる。追加のコードまたは非コード配列は、その必要はないが、本発明のポリヌクレオチド内に存在してよく、ポリヌクレオチドは、その必要はないが、他の分子および/または支持材料に連結してよい。
【0107】
ポリヌクレオチドは、ネイティブの配列(すなわち、マイコバクテリウム抗原またはその一部分をコードする内因性配列)を含んでよく、あるいはそのような配列の変異体、または生物学的もしくは抗原機能的等価物を含んでよい。以下にさらに記載されるように、ポリヌクレオチド変異体に1以上の置換、付加、欠失および/または挿入を含有させて、好ましくは、コード対象のポリペプチドの免疫原性が、ネイティブの腫瘍タンパク質と比べて減少しないようにしてよい。コード対象のポリペプチドの免疫原性に対する効果は、一般に、本明細書中に記載のように評価してよい。用語「変異体」は異種起源の相同遺伝子をも包含する。
【0108】
追加の実施形態では、本発明は、本明細書中で開示される1以上の配列と同一または相補的な種々の長さの一連の連続する配列を含む、単離されたポリヌクレオチドおよびポリペプチドを提供する。例えば、本明細書中で開示される1以上の配列の少なくとも約15、20、30、40、50、75、100、150、200、300、400、500または1000またはそれ以上の連続ヌクレオチド、ならびにそれらの間のすべての中間の長さを含むポリヌクレオチドが本発明によって提供される。この関連で「中間の長さ」とは、引用された値の間の任意の長さ、例えば16、17、18、19、等;21、22、23、等;30、31、32、等;50、51、52、53、等;100、101、102、103、等;150、151、152、153、等;例えば200〜500;500〜1,000の間のすべての整数、等を意味することが容易に理解される。
【0109】
本発明のポリヌクレオチド、またはその断片を、コード配列自体の長さにかかわらず、他のDNA配列、例えばプロモーター、ポリアデニル化シグナル、追加の制限酵素部位、マルチクローニング部位、他のコードセグメント、等と組み合わせて、それらの全体の長さがかなり変動するようにしてよい。したがって、ほとんど任意の長さの核酸断片を使用してよく、トータルの長さは、好ましくは、意図される組換えDNAプロトコルにおける調製および使用の容易さによって制限されることが考慮される。例えば、トータルの長さが約10,000、約5,000、約3,000、約2,000、約1,000、約500、約200、約100、約50塩基対長、等(すべての中間の長さを含む)である例示的DNAセグメントは、本発明の多数の実行において有用であると予測される。
【0110】
さらに、遺伝暗号の縮重の結果として、本明細書中に記載のポリペプチドをコードする多数のヌクレオチド配列が存在することが当業者に理解される。これらのポリヌクレオチドのいくつかは、いずれかのネイティブの遺伝子のヌクレオチド配列に対して最小の相同性しか有さない。それにもかかわらず、コドン使用の差異に起因して変動するポリヌクレオチド、例えばヒトおよび/または霊長類のコドン選択に関して最適化されたポリヌクレオチドが本発明によって具体的に考慮される。さらに、本明細書中で提供されるポリヌクレオチド配列を含む遺伝子の対立遺伝子は本発明の範囲内である。対立遺伝子は、ヌクレオチドの1以上の突然変異、例えば欠失、付加および/または置換の結果として変更される内因性遺伝子である。その結果得られるmRNAおよびタンパク質は、その必要はないが、改変された構造または機能を有してよい。対立遺伝子は標準的技術(例えばハイブリダイゼーション、増幅および/またはデータベース配列比較)を使用して特定することができる。
【0111】
ポリヌクレオチドの特定および特徴付け
ポリヌクレオチドは、任意の種々の十分に確立されている技術を使用して、特定、調製および/または操作してよい。例えば、ポリヌクレオチドは、以下にさらに詳細に記載されるように、cDNAのマイクロアレイを腫瘍関連発現(すなわち、本明細書中で提供される代表的なアッセイを使用して測定された場合の、腫瘍における、正常組織より少なくとも2倍高い発現)に関してスクリーニングすることによって特定してよい。そのようなスクリーニングは、例えばSynteniマイクロアレイ(Palo Alto, CA)を製造元の指示書にしたがって使用して(かつ本質的にSchena et al., Proc. Natl. Acad. Sci. USA 93:10614-10619 (1996)およびHeller et al., Proc. Natl. Acad. Sci. USA 94:2150-2155 (1997)に記載されるように)実施してよい。あるいは、本明細書中に記載のタンパク質を発現する細胞、例えば結核菌細胞から調製されたcDNAからポリヌクレオチドを増幅してよい。そのようなポリヌクレオチドは、ポリメラーゼ連鎖反応(PCR)によって増幅してよい。このアプローチでは、本明細書中で提供される配列に基づいて配列特異的プライマーを設計してよく、購入または合成してよい。
【0112】
本発明のポリヌクレオチドの増幅された部分を使用し、周知の技術を使用して好適なライブラリー(例えば結核菌cDNAライブラリー)から全長遺伝子を単離してよい。そのような技術には、増幅に適した1以上のポリヌクレオチドプローブまたはプライマーを使用してライブラリー(cDNAまたはゲノム)をスクリーニングすることが含まれる。好ましくは、ライブラリーをサイズで選択して大きい分子を含ませる。遺伝子の5’および上流領域を特定するためには、ランダムプライムドライブラリー(Random primed libraries)も好ましいかもしれない。ゲノムライブラリーはイントロンの取得および5’配列の伸長のために好ましい。
【0113】
ハイブリダイゼーション技術では、周知の技術を使用して(例えばニックトランスレーションまたは32Pでの末端標識によって)部分配列を標識してよい。そして、一般に、変性細菌コロニーを含有するフィルター(またはファージプラークを含有するローン(lawns))を標識プローブとハイブリダイズさせることによって細菌またはバクテリオファージライブラリーをスクリーニングする(Sambrook et al., Molecular Cloning: A Laboratory Manual (2000)を参照のこと)。ハイブリダイズするコロニーまたはプラークを選択し、増殖させ、追加の分析のためにDNAを単離する。cDNAクローンを解析して、例えば、該部分配列に由来するプライマーおよびベクターに由来するプライマーを使用するPCRによって追加の配列の量を決定してよい。制限酵素地図および部分配列を作成して、1以上の重複クローンを特定してよい。そして、標準的技術を使用して完全配列を決定してよい。その技術は一連の欠失クローンを作成するステップを含んでよい。そして、得られた重複配列を組み立てて、単一の連続配列にすることができる。周知の技術を使用して好適な断片をライゲートすることによって全長cDNA分子を作成することができる。
【0114】
一方、部分cDNA配列から全長コード配列を取得するための多数の増幅技術が存在する。そのような技術では、概してPCRによって増幅を実施する。種々の市販のキットのいずれかを使用して増幅ステップを実施してよい。例えば当技術分野において周知のソフトウェアを使用してプライマーを設計してよい。プライマーは、好ましくは22〜30ヌクレオチド長であり、少なくとも50%のGC含量を有し、約68℃〜72℃の温度で標的配列にアニーリングする。増幅された領域は、上記のようにシークエンシングしてよく、重複配列を組み立てて連続配列にしてよい。
【0115】
そのような増幅技術の1つはインバースPCR(inverse PCR)であり(Triglia et al., Nucl. Acids Res. 16:8186 (1988)を参照のこと)、その場合、制限酵素を使用して、遺伝子の既知領域の断片を作成する。そして該断片を分子内ライゲーションによって環化し、該既知領域に由来する種々のプライマーを用いるPCRの鋳型として使用する。代替のアプローチでは、リンカー配列に対するプライマーおよび既知領域に特異的なプライマーを用いる増幅によって部分配列に隣接する配列を回収してよい。増幅された配列を、典型的には、同リンカープライマーおよび該既知領域に特異的な第2のプライマーを用いる第2ラウンドの増幅に付する。該既知配列からの反対方向の伸長を開始させる2種のプライマーを使用するこの手順のバリエーションがWO96/38591に記載されている。別のそのような技術は「cDNA末端の高速増幅」またはRACEとして知られる。この技術には、内部プライマーおよび外部プライマーの使用が含まれ、外部プライマーはポリA領域またはベクター配列にハイブリダイズして、既知配列の5’および3’である配列を特定する。追加の技術には、捕捉PCR(capture PCR)(Lagerstrom et al., PCR Methods Applic. 1 :111-19 (1991))およびウォーキングPCR(walking PCR)(Parker et al., Nucl. Acids. Res. 19:3055-60 (1991))が含まれる。増幅を使用する他の方法を同様に使用して、全長cDNA配列を取得してよい。
【0116】
特定の場合、発現配列タグ(EST)データベースにおいて提供される、例えばGenBankから入手可能な配列の解析によって全長cDNA配列を取得することが可能である。重複ESTの検索は、一般に、周知のプログラム(例えばNCBI BLAST検索)を使用して実施してよく、そのようなESTを使用して連続の全長配列を作成してよい。全長DNA配列はゲノム断片の解析によって取得してもよい。
【0117】
宿主細胞でのポリヌクレオチド発現
本発明の他の実施形態では、本発明のポリペプチド、または融合タンパク質もしくはその機能的等価物をコードするポリヌクレオチド配列またはその断片を組換えDNA分子中で使用して、適切な宿主細胞におけるポリペプチドの発現を導いてよい。遺伝暗号に特有の縮重に基づいて、実質的に同一または機能的に等価なアミノ酸配列をコードする他のDNA配列を生産してよく、これらの配列を使用して、所定のポリペプチドをクローニングし、発現させてよい。
【0118】
当業者に理解されるように、いくつかの場合には、非天然に存在するコドンを有する、ポリペプチドをコードするヌクレオチド配列を生産することが有益であるかもしれない。例えば、特定の原核生物または真核生物の宿主が好むコドンを選択して、タンパク質発現の割合を増加させるか、あるいは、所望の特性、例えば天然に存在する配列から作成された転写産物の半減期より長い半減期を有する組換えRNA転写産物を生産することができる。
【0119】
さらに、本発明のポリヌクレオチド配列は、当技術分野において一般に知られている方法を使用して遺伝子操作することができる。その目的は、ポリペプチドをコードする配列を種々の理由で変化させることであり、それには、非限定的に、クローニング、プロセシング、および/または遺伝子産物の発現を改変する変化が含まれる。例えば、ランダムな断片化によるDNAシャッフリングおよび遺伝子断片のPCRによる組み立ておよび合成オリゴヌクレオチドを使用して、該ヌクレオチド配列を遺伝子操作してよい。さらに、部位特異的突然変異誘発を使用して、新規制限部位を挿入し、グリコシル化パターンを変化させ、コドン優先度を変更し、スプライシング変異体を生成し、あるいは突然変異を導入すること、等を行ってよい。
【0120】
本発明の別の実施形態では、天然、改変、または組換え核酸配列を異種性配列にライゲーションして、融合タンパク質をコードするようにしてよい。例えば、ポリペプチド活性のインヒビターに関してペプチドライブラリーをスクリーニングするために、市販の抗体によって認識できるキメラタンパク質をコードするようにすることが有用であるかもしれない。融合タンパク質を遺伝子操作して、ポリペプチドをコードする配列と異種性タンパク質配列の間に位置する切断部位を含有させて、該ポリペプチドが切断され、異種性部分から精製されるようにしてもよい。
【0121】
所望のポリペプチドをコードする配列は、完全にまたは部分的に、当技術分野において周知の化学的方法を使用して合成してよい(Caruthers, M. H. et al., Nucl. Acids Res. Symp. Ser. pp. 215-223 (1980), Horn et al., Nucl. Acids Res. Symp. Ser. pp. 225-232 (1980)を参照のこと)。あるいは、ポリペプチドのアミノ酸配列、またはその部分を合成する化学的方法を使用してタンパク質自体を生産してよい。例えば、ペプチド合成は種々の固相技術を使用して実施することができ(Roberge et al., Science 269:202-204 (1995))、例えばABI 431A Peptide Synthesizer(Perkin Elmer, Palo Alto, CA)を使用して自動合成を達成してよい。
【0122】
新たに合成されたペプチドを調製用高性能液体クロマトグラフィー(例えばCreighton, Proteins, Structures and Molecular Principles (1983))または当技術分野において利用可能な他の同等の技術によって実質的に精製してよい。アミノ酸解析またはシークエンシング(例えばエドマン分解手順)によって合成ペプチドの組成を確認してよい。さらに、ポリペプチド、またはその任意の部分のアミノ酸配列を、直接合成中に改変し、ならびに/あるいは、化学的方法を使用して他のタンパク質、またはその任意の部分に由来する配列と組み合わせて、変異体ポリペプチドを作成してよい。
【0123】
所望のポリペプチドを発現させるために、該ポリペプチド、または機能的等価物をコードするヌクレオチド配列を適切な発現ベクター、すなわち挿入されたコード配列の転写および翻訳に必要なエレメントを含有するベクターに挿入してよい。当業者に周知の方法を使用して、目的のポリペプチドをコードする配列ならびに適切な転写および翻訳調節エレメントを含有する発現ベクターを構築してよい。これらの方法には、in vitro組換えDNA技術、合成技術、およびin vivo遺伝子組換えが含まれる。そのような技術は、Sambrook et al., Molecular Cloning, A Laboratory Manual (2000), およびAusubel et al., Current Protocols in Molecular Biology (毎年更新される)に記載されている。
【0124】
種々の発現ベクター/宿主系を利用して、ポリヌクレオチド配列を含有および発現させてよい。これらには、非限定的に、組換えバクテリオファージ、プラスミド、またはコスミドDNA発現ベクターで形質転換された微生物、例えば細菌;酵母発現ベクターで形質転換された酵母;ウイルス発現ベクター(例えばバキュロウイルス)で感染させた昆虫細胞系;ウイルス発現ベクター(例えばカリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)または細菌発現ベクター(例えばTiまたはpBR322プラスミド)で形質転換された植物細胞系;または動物細胞系が含まれる。
【0125】
発現ベクター中に存在する「制御エレメント(control elements)」または「調節配列(regulatory sequences)」は、宿主細胞のタンパク質と相互作用して転写および翻訳を実行するベクターの非翻訳領域:エンハンサー、プロモーター、5’および3’非翻訳領域である。そのようなエレメントは強度および特異性において様々である。利用されるベクター系および宿主に応じて、任意の数の好適な転写および翻訳エレメント、例えば構成的および誘導性プロモーターを使用してよい。例えば、細菌系でのクローニングの場合、誘導性プロモーター、例えばPBLUESCRIPTファージミド(Stratagene, La Jolla, Calif.)またはPSPORT1プラスミド(Gibco BRL, Gaithersburg, MD)のハイブリッドlacZプロモーター等を使用してよい。哺乳動物の細胞系では、哺乳動物の遺伝子または哺乳動物ウイルスに由来するプロモーターが概して好ましい。ポリペプチドをコードする複数コピーの配列を含有する細胞株を作製することが必要である場合、SV40またはEBVに基づくベクターを適切な選択マーカーと共に好都合に使用してよい。
【0126】
細菌系では、発現対象のポリペプチドに関して意図される用途に応じて、いくつかの発現ベクターを選択してよい。例として、例えば抗体を誘導するために大規模な量が必要である場合、容易に精製可能な融合タンパク質の大量発現を導くベクターを使用してよい。そのようなベクターには、非限定的に、多機能性の大腸菌(E. coli)クローニングおよび発現ベクター、例えばBLUESCRIPT(Stratagene)(その場合、目的のポリペプチドをコードする配列をアミノ末端Metおよびそれに続くβ−ガラクトシダーゼの7残基の配列とインフレームで該ベクターにライゲーションして、ハイブリッドタンパク質が生産されるようにしてよい);pINベクター(Van Heeke & Schuster, J. Biol. Chem. 264:5503-5509 (1989));等が含まれる。pGEXベクター(Promega, Madison, Wis.)を使用して、グルタチオンS−トランスフェラーゼ(GST)との融合タンパク質として外来性ポリペプチドを発現させてもよい。一般に、そのような融合タンパク質は可溶性であり、グルタチオン−アガロースビーズへの吸着、その後の遊離グルタチオンの存在下での溶出によって溶解細胞から容易に精製することができる。そのような系で作成されるタンパク質が、ヘパリン、トロンビン、または第XA因子プロテアーゼ切断部位を含むように設計して、目的のクローニング対象ポリペプチドをGST部分から自在に遊離できるようにしてよい。
【0127】
酵母(Saccharomyces cerevisiae)では、構成的または誘導性プロモーター、例えばα因子、アルコールオキシダーゼ、およびPGHを含有するいくつかのベクターを使用してよい。総説としては、Ausubel et al. (上記)およびGrant et al., Methods Enzymol. 153:516-544 (1987)を参照のこと。
【0128】
植物の発現ベクターが使用される場合、多数のプロモーターのいずれかによってポリペプチドをコードする配列の発現を誘導してよい。例えば、ウイルスプロモーター、例えばCaMVの35Sおよび19Sプロモーターを、単独で、あるいはTMVに由来するオメガリーダー配列と組み合わせて使用してよい(Takamatsu, EMBO J. 6:307-311 (1987))。あるいは、植物プロモーター、例えばRUBISCOの小サブユニットまたは熱ショックプロモーターを使用してよい(Coruzzi et al., EMBO J. 3:1671-1680 (1984); Broglie et al., Science 224:838-843 (1984); およびWinter et al., Results Probl. Cell Differ. 17:85-105 (1991))。これらの構築物は、直接DNA形質転換または病原体媒介性トランスフェクションによって植物細胞に導入することができる。そのような技術は、いくつかの一般に入手可能な総説に記載されている(例えばHobbs in McGraw Hill Yearbook of Science and Technology pp. 191-196 (1992)を参照のこと)。
【0129】
昆虫系を使用して目的のポリペプチドを発現させてもよい。例えば、そのような系の1つでは、Autographa californica核多核体病ウイルス(AcNPV)をベクターとして使用して、Spodoptera frugiperda細胞またはTrichoplusia larvaeにおいて外来遺伝子を発現させる。該ポリペプチドをコードする配列を、ウイルスの非必須領域、例えばポリヘドリン遺伝子内にクローニングし、ポリヘドリンプロモーターの制御下に配置してよい。該ポリペプチドをコードする配列が首尾良く挿入されるとポリヘドリン遺伝子が不活性になり、コートタンパク質を欠く組換えウイルスが生産される。そして該組換えウイルスを使用して、例えばS. frugiperda細胞またはTrichoplusia larvaeを感染させてよく、それらにおいて目的のポリペプチドを発現させてよい(Engelhard et al., Proc. Natl. Acad. Sci. U.S.A. 91:3224-3227 (1994))。
【0130】
哺乳動物の宿主細胞では、いくつかのウイルスベースの発現系が一般に利用可能である。例えば、アデノウイルスが発現ベクターとして使用される場合、目的のポリペプチドをコードする配列を、後期プロモーターおよび3成分(tripartite)リーダー配列からなるアデノウイルス転写/翻訳複合体内にライゲーションしてよい。ウイルスゲノムの非必須E1またはE3領域への挿入を使用して、感染宿主細胞において該ポリペプチドを発現可能である生存可能なウイルスを取得してよい(Logan & Shenk, Proc. Natl. Acad. Sci. U.S.A. 81:3655-3659 (1984))。さらに、転写エンハンサー、例えばラウス肉腫ウイルス(RSV)エンハンサーを使用して、哺乳動物の宿主細胞における発現を増加させてよい。アデノウイルスベクターを用いる作業に関する方法およびプロトコルは、Wold, Adenovirus Methods and Protocols, 1998で概説されている。アデノウイルスベクターの使用に関する追加の参考文献は、Adenovirus: A Medical Dictionary, Bibliography, and Annotated Research Guide to Internet References, 2004に見出すことができる。
【0131】
特定の開始シグナルを使用して、目的のポリペプチドをコードする配列の、より効率的な翻訳を達成してもよい。そのようなシグナルには、ATG開始コドンおよび隣接配列が含まれる。ポリペプチドをコードする配列、その開始コドン、および上流配列が適切な発現ベクター中に挿入される場合、追加の転写または翻訳調節シグナルが必要とされないかもしれない。しかし、コード配列、またはその一部分しか挿入されない場合、ATG開始コドンを含めた外因性翻訳調節シグナルが提供されるべきであろう。さらにまた、インサート全体の翻訳を保証するために、該開始コドンは正しいリーディングフレームであるべきであろう。外因性翻訳エレメントおよび開始コドンは、種々の起源の、天然物および合成物の両者であってよい。使用される具体的な細胞系に適切なエンハンサーを含ませることによって発現効率を高めてよい。そのようなエンハンサーは、例えば文献に記載されているエンハンサーである(Scharf. et al., Results Probl. Cell Differ. 20:125-162 (1994))。
【0132】
さらに、宿主細胞株を、所望の様式で、挿入配列の発現をモジュレートするか、あるいは発現タンパク質をプロセシングするその能力に関して選択してよい。そのようなポリペプチドの改変には、非限定的に、アセチル化、カルボキシル化、グリコシル化、リン酸化、脂質化(lipidation)、およびアシル化が含まれる。「プレプロ(prepro)」形式のタンパク質を切断する翻訳後プロセシングを使用して、正しい挿入、折りたたみおよび/または機能を促進してもよい。そのような翻訳後の作業に特有の細胞機構および特徴的機構を有する、異なる宿主細胞、例えばCHO、HeLa、MDCK、HEK293、およびWI38を選択して、外来タンパク質の正しい改変およびプロセシングを保証してよい。
【0133】
組換えタンパク質の長期の、高収量の生産のためには、概して、安定発現が好ましい。例えば、ウイルスの複製起点および/または内因性発現エレメントおよび、同一または別のベクター上の選択マーカー遺伝子を含有してよい発現ベクターを使用して、目的のポリヌクレオチドを安定に発現する細胞株を形質転換してよい。該ベクターの導入後、細胞を強化培地中で1〜2日間成長させてよく、その後、それらを選択培地に切り換える。選択マーカーの目的は選択のための耐性を付与することであり、その存在により、導入配列を首尾良く発現する細胞の培養および回収が可能になる。細胞タイプに適切な組織培養技術を使用して、安定に形質転換された細胞の耐性クローンを増殖させてよい。
【0134】
任意の数の選択系を使用して形質転換細胞株を回収してよい。これらには、非限定的に、単純ヘルペスウイルスチミジンキナーゼ(Wigler et al., Cell 11 :223-32 (1977))およびアデニンホスホリボシルトランスフェラーゼ(Lowy et al., Cell 22:817-23 (1990))遺伝子が含まれる。それらは、それぞれ、tk.sup.−またはaprt.sup.−細胞において使用することができる。また、代謝拮抗剤、抗生物質または除草剤抵抗性を選択の基礎として使用することができる;例えば、メトトレキセートに対する耐性を付与するdhfr(Wigler et al., Proc. Natl. Acad. Sci. U.S.A. 77:3567-70 (1980));アミノグリコシド、ネオマイシンおよびG−418に対する耐性を付与するnpt(Colbere-Garapin et al., J. Mol. Biol. 150:1-14 (1981));および、それぞれクロルスルフロン(chlorsulfuron)およびホスフィノトリシン(phosphinotricin)アセチルトランスフェラーゼに対する耐性を付与するalsまたはpat(Murry, 上記)である。追加の選択可能遺伝子が報告されている。例えば、細胞がトリプトファンの代わりにインドールを利用することを可能にするtrpB、または細胞がヒスチジンの代わりにヒスチノール(histinol)を利用することを可能にするhisD(Hartman & Mulligan, Proc. Natl. Acad. Sci. U.S.A. 85:8047-51 (1988))である。最近、可視マーカーの使用が人気を獲得している。アントシアニン、β−グルクロニダーゼおよびその基質GUS、ならびにルシフェラーゼおよびその基質ルシフェリン等のマーカーが、特定のベクター系に起因する、形質転換体を特定するためだけでなく、一時的または安定なタンパク質発現の量を定量するためにも広く使用されている(Rhodes et al., Methods Mol. Biol. 55:121-131 (1995))。
【0135】
マーカー遺伝子発現の存在/不存在は目的の遺伝子も存在することを示唆するが、その存在および発現を確認する必要があるかもしれない。例えば、ポリペプチドをコードする配列がマーカー遺伝子配列内に挿入されれば、マーカー遺伝子機能の不存在によって配列を含有する組換え細胞を特定することができる。あるいは、単一のプロモーターの制御下でポリペプチドをコードする配列と直列にマーカー遺伝子を配置することができる。誘導または選択に応答して該マーカー遺伝子が発現されると、通常、直列型遺伝子の発現も示される。
【0136】
あるいは、当業者に公知の種々の手順によって、所望のポリヌクレオチド配列を含有および発現する宿主細胞を特定してよい。これらの手順としては、非限定的に、DNA−DNAまたはDNA−RNAハイブリダイゼーションおよびタンパク質バイオアッセイまたは免疫アッセイ技術(核酸またはタンパク質を検出および/または定量するためのメンブレン、溶液、またはチップベースのテクノロジーが含まれる)が挙げられる。
【0137】
ポリヌクレオチドによってコードされる生成物の発現を検出および測定するための種々のプロトコルであって、該生成物に特異的なポリクローナル抗体またはモノクローナル抗体を使用するプロトコルは当技術分野において公知である。その例には、酵素結合免疫吸着検定(ELISA)、ラジオイムノアッセイ(RIA)、および蛍光標示式細胞分取(FACS)が含まれる。いくつかの適用では、所定のポリペプチド上の2種の不干渉エピトープと反応性のモノクローナル抗体を利用する2部位のモノクローナルベースの免疫アッセイが好ましいかもしれないが、競合的結合アッセイを使用してもよい。これらのアッセイおよび他のアッセイは、特にHampton et al., Serological Methods, a Laboratory Manual (1990)およびMaddox et al., J. Exp. Med. 158:1211-1216 (1983)に記載されている。
【0138】
多種多様な標識およびコンジュゲーション技術が当業者に公知であり、種々の核酸およびアミノ酸アッセイにおいて使用してよい。ポリヌクレオチドに関連する配列を検出するための標識ハイブリダイゼーションまたはPCRプローブの作成手段には、オリゴラベリング(oligolabeling)、ニックトランスレーション、末端標識化またはPCR増幅(標識ヌクレオチドを使用する)が含まれる。あるいは、配列、またはその任意の部分を、mRNAプローブを生産するためのベクターにクローニングしてよい。そのようなベクターは当技術分野において公知であり、市販されていて、適切なRNAポリメラーゼ、例えばT7、T3、またはSP6および標識ヌクレオチドを加えることによってin vitroでRNAプローブを合成するために使用してよい。種々の市販のキットを使用してこれらの手順を実行してよい。使用することができる好適なレポーター分子または標識には、放射性核種、酵素、蛍光、化学発光、または発色物質ならびに基質、補助因子、インヒビター、磁気粒子、等が含まれる。
【0139】
目的のポリヌクレオチド配列で形質転換された宿主細胞を、細胞培養から該タンパク質を発現および回収するために適した条件下で培養してよい。組換え細胞によって生産されたタンパク質を分泌させるか、あるいは細胞内に含有させてよい。それは使用される配列および/またはベクターに依存する。当業者に理解されるように、本発明のポリヌクレオチドを含有する発現ベクターは、原核細胞膜または真核細胞膜を通過するコード対象のポリペプチドの分泌を導くシグナル配列を含有するように設計してよい。他の組換え構築物を使用して、目的のポリペプチドをコードする配列を、可溶性タンパク質の精製を容易にするポリペプチドドメインをコードするヌクレオチド配列と連結してもよい。そのような精製を容易にするドメインには、非限定的に、金属キレートペプチド、例えば固定金属上での精製を可能にするヒスチジン−トリプトファンモジュール、固定免疫グロブリン上での精製を可能にするプロテインAドメイン、およびFLAGS伸長/親和性精製系(Immunex Corp., Seattle, Wash.)で利用されるドメインが含まれる。切断可能なリンカー配列、例えば第XA因子またはエンテロキナーゼ(Invitrogen. San Diego, Calif.)に特異的な配列を精製ドメインおよびコード対象のポリペプチドの間に含ませることによって精製を容易にしてよい。そのような発現ベクターの1つは、目的のポリペプチドを含有する融合タンパク質および、チオレドキシンまたはエンテロキナーゼ切断部位に先行する6ヒスチジン残基をコードする核酸の発現を提供する。該ヒスチジン残基は、Porath et al., Prot. Exp. Purif. 3:263-281 (1992)に記載のようにIMIAC(固定金属イオンアフィニティークロマトグラフィー)上での精製を容易にし、一方、エンテロキナーゼ切断部位は該融合タンパク質に由来する所望のポリペプチドを精製するための手段を提供する。融合タンパク質を含有するベクターについての考察はKroll et al., DNA Cell Biol. 12:441-453 (1993)に提供されている。
【0140】
組換え生産方法に加えて、固相技術を使用する直接ペプチド合成(Merrifield, J. Am. Chem. Soc. 85:2149-2154 (1963))によって本発明のポリペプチド、およびその断片を生産してよい。マニュアル技術を使用するか、あるいは自動化によってタンパク質合成を実施してよい。自動合成は、例えばApplied Biosystems 431A Peptide Synthesizer(Perkin Elmer)を使用して達成してよい。あるいは、種々の断片を別々に化学合成し、化学的方法を使用して組み合わせて、全長分子を生産してよい。
【0141】
in vivoポリヌクレオチド送達技術
追加の実施形態では、1以上の本発明のポリヌクレオチドを含む遺伝子構築物をin vivoで細胞に導入する。これは多様な周知のアプローチのいずれかを使用して達成してよく、説明のためにそのアプローチのいつくかを以下に概説する。
【0142】
1.アデノウイルス
1以上の核酸配列をin vivoで送達するための好ましい方法の1つにはアデノウイルス発現ベクターの使用が含まれる。「アデノウイルス発現ベクター」とは、(a)構築物のパッケージングをサポートし、かつ(b)センスまたはアンチセンス方向で構築物にクローニングされているポリヌクレオチドを発現するために十分なアデノウイルス配列を含有する構築物を含むものとする。当然、アンチセンス構築物の関連では、発現には、その遺伝子産物が合成されることは必要とされない。
【0143】
該発現ベクターは遺伝子改変型のアデノウイルスを含む。36kbで、線状の二本鎖DNAウイルスであるアデノウイルスの遺伝子構成の知識によって、アデノウイルスDNAの大きな部分を7kbまでの外来性配列で置き換えることが可能になっている(Grunhaus & Horwitz, 1992)。レトロウイルスとは対照的に、宿主細胞のアデノウイルス感染では染色体への組込みが生じない。その理由は、アデノウイルスDNAがエピソーム様式で複製できるからであり、潜在的な遺伝毒性を有さない。また、アデノウイルスは構造的に安定であり、大規模な増幅後にゲノムの再編成は検出されていない。アデノウイルスは、実質的にすべての上皮細胞に、それらの細胞周期段階にかかわらずに感染することができる。現在までのところ、アデノウイルス感染は、軽度の疾患、例えばヒトの急性呼吸器疾患にしか関連しないようである。
【0144】
アデノウイルスは、その中程度のゲノムサイズ、操作の容易さ、高い力価、広い標的細胞の範囲および高い感染力のために遺伝子導入ベクターとしての使用に特に適している。該ウイルスゲノムの両端は100〜200塩基対の逆位反復(ITR)を含有する。それはウイルスDNAの複製およびパッケージングに必要なシスエレメントである。該ゲノムの初期(E)および後期(L)領域は、ウイルスDNA複製の開始によって分割される異なる転写単位を含有する。E1領域(E1AおよびE1B)は、ウイルスゲノムおよび少数の細胞遺伝子の転写の調節を担うタンパク質をコードする。E2領域(E2AおよびE2B)の発現によって、ウイルスDNA複製のためのタンパク質の合成が生じる。これらのタンパク質は、DNA複製、後期遺伝子発現および宿主細胞停止(host cell shut-off)に関与する(Renan, 1990)。大多数のウイルスキャプシドタンパク質を含む後期遺伝子の生成物は、主要後期プロモーター(MLP)によって放出される単一の初期転写産物の重大なプロセシング後にしか発現されない。該MLP(16.8m.u.に位置する)は感染の後期に特に効率的であり、このプロモーターから放出されるすべてのmRNAは5’の3成分リーダー(TPL)配列を有し、その配列はそれらを翻訳に好ましいmRNAにする。
【0145】
現行の系では、シャトルベクターおよびプロウイルスベクター間の相同組換えの結果、組換えアデノウイルスを作成する。2つのプロウイルスベクター間で起こりうる組換えのために、このプロセスから野生型アデノウイルスが生じる可能性がある。したがって、個々のプラークから単一クローンのウイルスを単離し、そのゲノム構造を検査することが重要である。
【0146】
複製欠損である現行のアデノウイルスベクターの作成および増殖は、Ad5 DNA断片によってヒト胚性腎細胞から形質転換され、E1タンパク質を構成的に発現する、293と称される特有のヘルパー細胞株に依存する(Graham et al., 1977)。アデノウイルスゲノムに由来するE3領域は不必要である(Jones & Shenk, 1978)ため、現行のアデノウイルスベクターは、293細胞の支援を受けて、E1、D3または両領域中に外来DNAを保持する(Graham & Prevec, 1991)。自然界では、アデノウイルスは約105%の野生型ゲノム(Ghosh-Choudhury et al., 1987)をパッケージングすることができ、追加の約2kBのDNAの受容能を提供する。E1およびE3領域中で取り替え可能な約5.5kBのDNAと合わせて、現行のアデノウイルスベクターの最大容量は7.5kB未満であり、すなわち該ベクターの全長の約15%である。アデノウイルスのウイルスゲノムの80%を超える部分はベクター主鎖中に残存し、それはベクターが有する細胞障害性の源である。また、E1欠失ウイルスの複製欠損は不完全である。例えば、現在利用可能なベクターを高い感染多重度(MOI)で用いた場合、ウイルス遺伝子発現の漏出が観察されている(Mulligan, 1993)。
【0147】
ヘルパー細胞株は、ヒト細胞、例えばヒト胚性腎細胞、筋細胞、造血細胞または他のヒトの胚性の間葉細胞または上皮細胞に由来してよい。あるいは、該ヘルパー細胞は、ヒトアデノウイルスに関して許容される他の哺乳動物種の細胞に由来してよい。そのような細胞には、例えばVero細胞または他のサルの胚性の間葉細胞または上皮細胞が含まれる。上記のように、現在好ましいヘルパー細胞株は293である。
【0148】
最近、Racher et al.(1995)は、293細胞を培養してアデノウイルスを増殖させるための改善された方法を開示した。一形式では、100〜200mlの培地を含有する1リットルのシリコン処理スピナーフラスコ(Techne, Cambridge, UK)中に個々の細胞を接種することによって天然細胞凝集物を培養する。40rpmで撹拌した後、トリパンブルーで細胞生存度を見積もる。別の形式では、Fibra-Celマイクロキャリア(Bibby Sterlin, Stone, UK)(5g/l)を以下のように使用する。5mlの培地に再懸濁した接種用細胞を250ml三角フラスコ中の担体(50ml)に加え、1〜4時間、時折撹拌しながら静置する。そしてその培地を50mlの新鮮な培地と取り替え、振とうを開始する。ウイルス生産では、細胞を約80%集密まで成長させ、その後、培地を取り除き(最終容量の25%にする)、アデノウイルスをMOI0.05で加える。培養を一晩静置し、その後、容量を増加させて100%にし、さらに72時間の振とうを開始する。
【0149】
アデノウイルスベクターが複製欠損、または少なくとも条件的に欠損であるという必要条件以外に、アデノウイルスベクターの性質は発明の実施を成功させるために重要ではないと考えられる。アデノウイルスは、42種の異なる公知の血清型または亜群A〜Fのいずれかであってよい。亜群Cの5型アデノウイルスは、本発明で使用するための条件的複製欠損アデノウイルスベクターを取得するために好ましい出発材料である。その理由は、5型アデノウイルスがヒトアデノウイルスであり、それについて非常に多くの生化学的情報および遺伝情報が公知であり、かつ歴史的に、ベクターとしてアデノウイルスを使用するほとんどの構築物に使用されているからである。
【0150】
上記のように、本発明の典型的ベクターは複製欠損であり、アデノウイルスE1領域を有さない。ゆえに、E1コード配列が除去されている位置で目的の遺伝子をコードするポリヌクレオチドを導入することが最も好都合である。しかし、アデノウイルス配列内の構築物を挿入する位置は本発明にとって重要ではない。目的の遺伝子をコードするポリヌクレオチドは、Karlsson et al. (1986)によって報告されているようにE3置換ベクター中の欠失E3領域の代わりに挿入するか、あるいはヘルパー細胞株またはヘルパーウイルスがE4欠損を補完する場合のE4領域中に挿入してもよい。
【0151】
アデノウイルスは容易に培養および操作することができ、in vitroおよびin vivoで広い宿主範囲を示す。この群のウイルスは高い力価、例えば109〜1011プラーク形成単位/mlで取得することができ、それらは非常に感染性である。アデノウイルスの生活環は宿主細胞ゲノム内への組込みを必要としない。アデノウイルスベクターによって送達される外来遺伝子はエピソーム性であり、したがって宿主細胞に対する低い遺伝毒性しか有さない。野生型アデノウイルスでのワクチン接種の研究において副作用は報告されていない(Couch et al., 1963; Top et al., 1971)。このことはそれらの安全性およびin vivo遺伝子導入ベクターとしての治療的潜在能力を示す。
【0152】
アデノウイルスベクターは、真核生物の遺伝子の発現(Levrero et al., 1991; Gomez-Foix et al., 1992)およびワクチン開発(Grunhaus & Horwitz, 1992; Graham & Prevec, 1992)において使用されている。最近、動物試験では、組換えアデノウイルスを遺伝子治療に使用できることが示唆された(Stratford-Perricaudet & Perricaudet, 1991; Stratford-Perricaudet et al., 1990; Rich et al., 1993)。組換えアデノウイルスを種々の組織に投与する研究には、気管点滴注入(Rosenfeld et al., 1991; Rosenfeld et al., 1992)、筋肉注射(Ragot et al., 1993)、末梢静脈内注射(Herz & Gerard, 1993)および脳内への定位接種(Le Gal La Salle et al., 1993)が含まれる。
【0153】
アデノウイルスベクターはヒトアデノウイルスに由来してよい。あるいは、それらは他の動物種、例えばチンパンジーのアデノウイルスに由来してよく、それは、多数のヒト被験体中で循環している、ヒトアデノウイルスに対する抗体によってウイルスベクターが中和されないという利点を有するかもしれない(例えば:Tatsis N et al (2005) Gene Ther. Dec 1; [Epub ahead of print]を参照のこと)。
【0154】
2.レトロウイルス
レトロウイルスは、感染細胞中で逆転写のプロセスによってそれらのRNAを二本鎖DNAに変換する能力を特徴とする一本鎖RNAウイルスの一群である(Coffin, 1990)。そのように生じたDNAはプロウイルスとして細胞の染色体内に安定に組込まれ、ウイルスタンパク質の合成を導く。組込みの結果、レシピエント細胞およびその子孫においてウイルス遺伝子の配列が保持される。レトロウイルスのゲノムは、それぞれキャプシドタンパク質、ポリメラーゼ酵素、およびエンベロープ成分をコードするgag、pol、およびenvの3遺伝子を含有する。gag遺伝子の上流に見出される配列は、ゲノムをビリオン内へパッケージングするためのシグナルを含有する。ウイルスゲノムの5’および3’末端に2個の長末端反復(LTR)配列が存在する。これらは強いプロモーターおよびエンハンサー配列を含有し、宿主細胞ゲノムへの組込みにも必要とされる(Coffin, 1990)。
【0155】
レトロウイルスベクターを構築するために、ウイルスゲノムの特定のウイルス配列の代わりに1以上の目的のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸を挿入し、複製欠損であるウイルスを得る。ビリオンを生産するために、gag、pol、およびenv遺伝子を含有するが、LTRおよびパッケージング成分を有さないパッケージング細胞株を構築する(Mann et al., 1983)。レトロウイルスのLTRおよびパッケージング配列と共にcDNAを含有する組換えプラスミドをこの細胞株に(例えばリン酸カルシウム沈殿によって)導入すると、該パッケージング配列によって、該組換えプラスミドのRNA転写産物をウイルス粒子中にパッケージングさせることが可能になり、そしてそれは培養培地中に分泌される(Nicolas & Rubenstein, 1988; Temin, 1986; Mann et al., 1983)。そして該組換えレトロウイルスを含有する培地を収集し、場合により濃縮し、遺伝子導入に使用する。レトロウイルスベクターは多種多様な細胞タイプに感染することができる。しかし、組込みおよび安定な発現には宿主細胞の分裂が必要とされる(Paskind et al., 1975)。
【0156】
ウイルスエンベロープにラクトース残基を化学付加することによるレトロウイルスの化学修飾に基づいて、レトロウイルスベクターの特異的ターゲティングを可能にするように設計された新規アプローチが最近開発された。この改変は、シアロ糖タンパク質受容体を介する肝細胞の特異的感染を可能にしうる。
【0157】
レトロウイルスエンベロープタンパク質および特異的細胞受容体に対するビオチン化抗体を使用する、組換えレトロウイルスをターゲティングするための異なるアプローチが設計された。ストレプトアビジンを使用し、ビオチン成分を介して該抗体をカップリングした(Roux et al., 1989)。主要組織適合複合体クラスIおよびクラスII抗原に対する抗体を使用して、それらは、in vitroで、エコトロピックウイルスを用いた、該表面抗原を有する種々のヒト細胞の感染を実証した(Roux et al., 1989)。
【0158】
3.アデノ随伴ウイルス
AAV(Ridgeway, 1988; Hermonat & Muzycska, 1984)はパルボウイルス(parovirus)であり、アデノウイルスストックの混入物として発見された。それは遍在性ウイルス(米国のヒト集団の85%に抗体が存在する)であり、いかなる疾患とも関連付けられていない。それは、また、その複製がヘルパーウイルス、例えばアデノウイルスの存在に依存することから、ディペンドウイルス属として分類される。5血清型が特定されていて、そのうちAAV−2が最もよく特徴付けされている。AAVは、直径20〜24nmの正二十面体ビリオンを形成するキャプシドタンパク質VP1、VP2およびVP3内に包まれている一本鎖線状DNAを有する(Muzyczka & McLaughlin, 1988)。
【0159】
AAVのDNAは約4.7キロベース長である。それは2種のオープンリーディングフレームを含有し、2個のITRが隣接している。AAVゲノムには、2個の主要遺伝子:repおよびcapが存在する。rep遺伝子はウイルス複製を担うタンパク質をコードし、一方capはキャプシドタンパク質VP1〜3をコードする。各ITRはT字型のヘアピン構造を形成する。これらの末端反復は、染色体組込みのためのAAVの唯一の必須シス成分である。したがって、すべてのウイルスコード配列が除去され、送達用の遺伝子カセットで置き換えられているベクターとしてAAVを使用することができる。3個のウイルスプロモーターが特定されていて、その地図上の位置にしたがって、p5、p19、およびp40と称される。p5およびp19からの転写の結果、repタンパク質が生産され、p40からの転写の結果、キャプシドタンパク質が生産される(Hermonat & Muzyczka, 1984)。
【0160】
研究者がrAAVを発現ベクターとして使用する可能性を研究することを促すいくつかの要因が存在する。その1つは、遺伝子を送達して宿主染色体中に組込むための必要条件が、驚くべきことに、わずかしかないことである。145bpのITRを有することが必要であり、それはAAVゲノムのほんの6%である。これにより、該ベクター中には、4.5kbのDNA挿入を組み立てるための余地が残る。この積載能力はAAVが大きい遺伝子を送達することを妨げるかもしれないが、本発明のアンチセンス構築物の送達には十分に適している。
【0161】
AAVは、また、その安全性のために送達ビヒクルの良好な選択肢である。比較的複雑な救済機構が存在し:AAV遺伝子は野生型アデノウイルスだけでなくrAAVも動員しなければならない。同様に、AAVは病原性でなく、いかなる疾患とも関連していない。ウイルスコード配列を除去するとウイルス遺伝子の発現に対する免疫反応が最小になり、したがってrAAVは炎症反応を誘発しない。
【0162】
4.発現構築物としての他のウイルスベクター
オリゴヌクレオチドまたはポリヌクレオチド配列を宿主細胞に送達するために、本発明の発現構築物として他のウイルスベクターを使用してよい。ウイルス、例えばワクシニアウイルス(Ridgeway, 1988; Coupar et al., 1988)、レンチウイルス、ポリオウイルスおよびヘルペスウイルスに由来するベクターを使用してよい。それらは種々の哺乳動物細胞に関するいくつかの魅力的な特徴を提供する(Friedmann, 1989; Ridgeway, 1988; Coupar et al., 1988; Horwich et al., 1990)。
【0163】
最近、欠損B型肝炎ウイルスが認識され、種々のウイルス配列の構造と機能の関連性への新規洞察が得られた。in vitro研究では、該ウイルスは、80%までのゲノムの欠失にもかかわらず、ヘルパー依存的パッケージングおよび逆転写の能力を保持できることが示された(Horwich et al., 1990)。このことは、ゲノムの大部分を外来性遺伝物質で置き換えることができることを示唆した。肝親和性(hepatotropism)および持続性(組込み)は、肝臓を標的にする遺伝子導入に関して特に魅力的な特性であった。Chang et al.(1991)は、クロラムフェニコールアセチルトランスフェラーゼ(CAT)遺伝子をアヒルB型肝炎ウイルスゲノムのポリメラーゼ、表面、およびプレ表面コード配列の代わりに導入した。それはトリ肝細胞腫細胞株に野生型ウイルスと同時にトランスフェクトされた。高い力価の組換えウイルスを含有する培養培地を使用して初代子ガモ肝細胞を感染させた。トランスフェクション後、少なくとも24日間、安定なCAT遺伝子発現が検出された(Chang et al., 1991)。
【0164】
5.非ウイルスベクター
本発明のオリゴヌクレオチドまたはポリヌクレオチド配列の発現を実行するためには、発現構築物を細胞内に送達しなければならない。この送達は、細胞株を形質転換するための実験室手順の場合のようにin vitroで、あるいは特定の疾患状態の治療の場合のようにin vivoまたはex vivoで達成してよい。上記のように、好ましい送達機構の1つは、発現構築物が感染性ウイルス粒子中に封入されているウイルスの感染を介するものである。
【0165】
該発現構築物が細胞内に送達されると、所望のオリゴヌクレオチドまたはポリヌクレオチド配列をコードする核酸は種々の部位に配置され発現される。特定の実施形態では、該構築物をコードする核酸は細胞のゲノム内に安定に組込まれる。この組込みは相同組換え(遺伝子置換)を介する特異的な位置および方向での組込みであってよく、ランダムな非特異的な位置(遺伝子付加(gene augmentation))で組込まれてもよい。さらに別の実施形態では、該核酸は、分離したエピソーム性DNAセグメントとして細胞中で安定に維持される。そのような核酸セグメントまたは「エピソーム」は、宿主細胞周期と無関係の、あるいはそれと同期した維持および複製を可能にするために十分な配列をコードする。該発現構築物がどのように細胞に送達されるか、および該核酸が細胞内のどこにとどまるかは、使用される発現構築物のタイプに依存する。
【0166】
本発明の特定の実施形態では、1以上のオリゴヌクレオチドまたはポリヌクレオチド配列を含む発現構築物は、単に裸の組換えDNAまたはプラスミドからなる。該構築物の導入は、細胞膜を物理的または化学的に透過性にする上記方法のいずれかによって実施してよい。このことは、in vitroでの導入に特に適用可能であるが、in vivoでの使用にも同様に適用してよい。Dubensky et al.(1984)は、リン酸カルシウム沈殿の形式のポリオーマウイルスDNAを成体および新生児マウスの肝臓および脾臓に首尾良く注射し、活発なウイルス複製および急性感染を実証した。Benvenisty & Reshef(1986)は、また、リン酸カルシウム沈殿済みプラスミドを直接腹腔内注射すると、トランスフェクトされた遺伝子の発現が生じることを実証した。また、目的の遺伝子をコードするDNAをin vivoで同様の様式で導入し、該遺伝子産物を発現させることが想定される。
【0167】
裸のDNA発現構築物を細胞内に導入するための本発明の別の実施形態には、微粒子銃が含まれる。この方法は、DNAでコーティングされた微粒子を高速に加速して、それらが細胞膜を貫通して、細胞を死滅させることなく細胞に入ることを可能にする能力に依存する(Klein et al., 1987)。小粒子を加速するためのいくつかの装置が開発されている。そのような装置の1つは、電流を生成するための高電圧の放電に依存し、そしてその電流が輸送力を提供する(Yang et al., 1990)。使用される微粒子は、生物学的に不活性な物質、例えばタングステンまたは金ビーズから構成されている。
【0168】
ラットおよびマウスの選択された器官、例えば肝臓、皮膚、および筋組織にin vivoで粒子を衝突させている(Yang et al., 1990; Zelenin et al., 1991)。これは、銃と標的器官の間の任意の介在組織を排除するために組織または細胞の外科的露出、すなわちex vivo処理を必要とするかもしれない。やはり、特定の遺伝子をコードするDNAをこの方法によって送達してよく、依然として本発明に包含される。
【0169】
ポリペプチド組成物
本発明は、他の態様では、ポリペプチド組成物を提供する。一般に、本発明のポリペプチドは、哺乳動物種に由来する単離されたポリペプチド(またはエピトープ、変異体、またはその活性断片)である。好ましくは、該ポリペプチドは、本明細書中で開示されるポリヌクレオチド配列または本明細書中で開示されるポリヌクレオチド配列に中程度にストリンジェントな条件下でハイブリダイズする配列によってコードされる。あるいは、該ポリペプチドは、本明細書中で開示されるアミノ酸配列に由来する連続アミノ酸配列を含むポリペプチド、または本明細書中で開示されるアミノ酸配列全体を含むポリペプチドとして規定される。
【0170】
免疫原性部分は、概して、周知の技術、例えばPaul, Fundamental Immunology, 3rd ed., 243-247 (1993)およびその引用文献で概説されている技術を使用して特定してよい。そのような技術には、抗原特異的抗体、抗血清および/またはT細胞株またはクローンと反応する能力に関してポリペプチドをスクリーニングすることが含まれる。本明細書中で使用されるように、抗血清および抗体は、それらが抗原に特異的に結合する(すなわちそれらがELISAまたは他の免疫アッセイにおいてタンパク質と反応し、無関係のタンパク質と検出可能な程度に反応することはない)場合に「抗原特異的」である。そのような抗血清および抗体は、本明細書中に記載のように、周知の技術を使用して調製してよい。マイコバクテリア属の種のタンパク質の免疫原性部分は、全長ポリペプチドの反応性に実質的に劣らないレベルで前記抗血清および/またはT細胞と反応する部分である(例えばELISAおよび/またはT細胞反応性アッセイにおいて)。そのような免疫原性部分は、前記アッセイ内で全長ポリペプチドの反応性と同様のレベルまたはそれを超えるレベルで反応する。そのようなスクリーニングは、概して、当業者に周知の方法、例えばHarlow & Lane, Antibodies: A Laboratory Manual (1988)およびUsing Antibodies: A Laboratory Manual (1998)に記載の方法を使用して実施してよい。例えば、ポリペプチドを固形支持体上に固定し、患者の血清と接触させて、固定ポリペプチドに対する該血清内の抗体の結合を可能にしてよい。次いで未結合血清を除去し、例えば125I標識プロテインAを使用して結合抗体を検出してよい。
【0171】
ポリペプチドは種々の周知の技術のいずれかを使用して調製してよい。上記のようなDNA配列によってコードされる組換えポリペプチドは、当業者に公知の種々の発現ベクターのいずれかを使用してDNA配列から容易に調製してよい。発現は、組換えポリペプチドをコードするDNA分子を含有する発現ベクターで形質転換またはトランスフェクトされている任意の適切な宿主細胞において達成してよい。好適な宿主細胞には、原核生物、酵母、および高等真核細胞、例えば哺乳動物細胞および植物細胞が含まれる。好ましくは、使用される宿主細胞は、大腸菌、酵母または哺乳動物細胞株、例えばCOSまたはCHOである。組換えタンパク質またはポリペプチドを培養培地に分泌する好適な宿主/ベクター系から得られた上清を、まず、市販のフィルターを使用して濃縮してよい。濃縮後、その濃縮物を好適な精製マトリックス、例えば親和性マトリックスまたはイオン交換樹脂に適用してよい。最後に、1以上の逆相HPLCステップを使用して、組換えポリペプチドをさらに精製することができる。
【0172】
約100未満のアミノ酸、概して約50未満のアミノ酸を有する本発明のポリペプチド、その免疫原性断片、および他の変異体は、当業者に周知の技術を使用する合成手段によって作成してもよい。例えば、そのようなポリペプチドは、市販の固相技術のいずれか、例えばメリフィールド固相合成法を使用して合成してよく、その場合、成長中のアミノ酸鎖にアミノ酸が順次付加される。Merrifield, J. Am. Chem. Soc. 85:2149-2146 (1963)を参照のこと。ポリペプチドの自動合成のための設備がPerkin Elmer/Applied BioSystems Division(Foster City, CA)のような供給元から市販されており、それを製造元の指示書にしたがって操作してよい。
【0173】
ある具体的な実施形態では、ポリペプチドは、本明細書中に記載の複数のポリペプチドを含むか、あるいは本明細書中に記載の少なくとも1つのポリペプチドおよび無関係の配列、例えば公知の腫瘍タンパク質を含む融合タンパク質であってよい。融合パートナーは、例えば、Tヘルパーエピトープ、好ましくはヒトによって認識されるTヘルパーエピトープの提供を支援(免疫学的融合パートナー)してよく、あるいはネイティブの組換えタンパク質より高い収率での該タンパク質の発現を支援(発現エンハンサー)してよい。特定の好ましい融合パートナーは免疫学的かつ発現増強性の融合パートナーである。該タンパク質の溶解度を増加させるか、あるいは該タンパク質を所望の細胞内区画にターゲティングさせることが可能であるように、他の融合パートナーを選択してよい。さらに別の融合パートナーには親和性タグが含まれ、それは該タンパク質の精製を容易にする。
【0174】
融合タンパク質は、一般的に、標準的技術、例えば化学的コンジュゲーションを使用して調製してよい。好ましくは、発現系において、融合タンパク質を組換えタンパク質として発現させ、非融合タンパク質と比べて高レベルの生産を可能にする。簡潔に言えば、前記ポリペプチド成分をコードするDNA配列を別々に組み立てて、適切な発現ベクター中にライゲーションしてよい。1つのポリペプチド成分をコードするDNA配列の3’末端を、ペプチドリンカーを用いるか、あるいは用いずに、第二のポリペプチド成分をコードするDNA配列の5’末端にライゲーションし、該配列のリーディングフレームが一致するようにする。これにより、両成分ポリペプチドの生物学的活性を保持する単一融合タンパク質への翻訳が可能になる。
【0175】
ペプチドリンカー配列を使用して、第一および第二のポリペプチド成分を、各ポリペプチドが折りたたまれてその二次および三次構造になることを保証するために十分な距離で分離してよい。そのようなペプチドリンカー配列は、当技術分野において周知の標準的技術を使用して融合タンパク質に組み入れられる。好適なペプチドリンカー配列は以下の要因に基づいて選択してよい:(1)可動性の伸張したコンフォメーションをとることができること;(2)第一および第二のポリペプチド上の機能的エピトープと相互作用しうる二次構造をとることができないこと;および(3)該ポリペプチドの機能的エピトープと反応する可能性がある疎水性または荷電残基を欠いていること。好ましいペプチドリンカー配列は、Gly、AsnおよびSer残基を含有する。他のほぼ中性のアミノ酸、例えばThrおよびAlaをリンカー配列中で使用してもよい。リンカーとして有用に使用されるアミノ酸配列には、Maratea et al., Gene 40:39-46 (1985); Murphy et al., Proc. Natl. Acad. Sci. USA 83:8258-8262 (1986); 米国特許第4,935,233号および同第4,751,180号に開示される配列が含まれる。該リンカー配列は、概して、1〜約50アミノ酸長であってよい。第一および第二のポリペプチドが、機能的ドメインを分離して立体的干渉を妨げるために使用できる必須でないN末端アミノ酸領域を有する場合には、リンカー配列は必要とされない。
【0176】
ライゲーションされたDNA配列は好適な転写または翻訳調節エレメントに作動可能に連結される。DNAの発現を担う調節エレメントは、第一のポリペプチドをコードするDNA配列の5’側にのみ位置する。同様に、翻訳を終わらせるために必要とされる停止コドンおよび転写終結シグナルは第二のポリペプチドをコードするDNA配列の3’側にのみ存在する。
【0177】
融合タンパク質もまた提供される。そのようなタンパク質は、無関係の免疫原性タンパク質と共に本明細書中に記載のポリペプチドを含む。好ましくは免疫原性タンパク質はリコール応答(recall response)を誘発可能なものである。そのようなタンパク質の例には、テタヌス、結核および肝炎タンパク質が含まれる(例えばStoute et al., New Engl. J. Med. 336:86-91 (1997)を参照のこと)。
【0178】
好ましい実施形態では、免疫学的融合パートナーは、グラム陰性菌であるインフルエンザ菌B(Haemophilus influenza B)の表面タンパク質であるプロテインDに由来する(WO91/18926)。好ましくは、プロテインD誘導体は該タンパク質の最初の約3分の1(例えば最初のN末端100〜110アミノ酸)を含み、プロテインD誘導体は脂質化してよい。特定の好ましい実施形態では、リポプロテインD融合パートナーの最初の109残基をN末端に包含させて、追加の外因性T細胞エピトープを有するポリペプチドを提供し、大腸菌での発現レベルを増加させる(ゆえに発現エンハンサーとして機能する)。脂質の尾部によって、抗原提示細胞への該抗原の最適な提示が保証される。他の融合パートナーには、インフルエンザウイルスに由来する非構造タンパク質、NS1(赤血球凝集素(hemaglutinin))が含まれる。典型的にN末端81アミノ酸が使用されるが、Tヘルパーエピトープを含む異なる断片を使用してよい。
【0179】
別の実施形態では、免疫学的融合パートナーはLYTAとして知られるタンパク質、またはその一部分(好ましくはC末端部分)である。LYTAは肺炎連鎖球菌(Streptococcus pneumoniae)に由来し、それはアミダーゼLYTAとして知られるN−アセチル−L−アラニンアミダーゼを合成する(LytA遺伝子によってコードされる; Gene 43:265-292 (1986))。LYTAは、ペプチドグリカン主鎖中の特定の結合を特異的に分解する自己溶解素である。LYTAタンパク質のC末端ドメインはコリンまたはいくつかのコリンアナログ、例えばDEAEに対する親和性を担う。この特性は、融合タンパク質の発現に有用な大腸菌C−LYTA発現プラスミドの開発に活用されている。アミノ末端にC−LYTA断片を含有するハイブリッドタンパク質の精製が報告されている(Biotechnology 10:795-798 (1992)を参照のこと)。好ましい実施形態では、LYTAの反復部分を融合タンパク質中に組み入れてよい。反復部分は、残基178で出発するC末端領域中に見出せる。特に好ましい反復部分は残基188〜305を含んでいる。
【0180】
一般に、本明細書中に記載のポリペプチド(融合タンパク質を含む)およびポリヌクレオチドは単離型である。「単離された」ポリペプチドまたはポリヌクレオチドとは、その元の環境から取り出されたものである。例えば、天然に存在するタンパク質は、自然系におけるいくつかまたはすべての共存物質から分離されている場合に単離されている。好ましくは、そのようなポリペプチドは少なくとも約90%の純度を有し、より好ましくは少なくとも約95%純度、最も好ましくは少なくとも約99%の純度を有する。ポリヌクレオチドは、例えば、天然環境の一部分ではないベクターにクローニングされている場合に単離されていると見なされる。
【0181】
T細胞
免疫療法組成物は、さらに、またはあるいは、マイコバクテリウム抗原に特異的なT細胞を含んでよい。そのような細胞は、概して、標準的手順を使用してin vitroまたはex vivoで調製してよい。例えば、市販の細胞分離系、例えばNexell Therapeutics, Inc.(Irvine, CA)から入手可能なIsolexTM System(さらに米国特許第5,240,856号;同第5,215,926号;WO89/06280;WO91/16116およびWO92/07243を参照のこと)を使用して、患者の骨髄、末梢血、または骨髄もしくは末梢血の一部分からT細胞を単離してよい。あるいは、T細胞は、関連または無関連のヒト、非ヒト哺乳動物、細胞株または培養に由来してよい。
【0182】
T細胞は、本発明のポリペプチド、該ポリペプチドをコードするポリヌクレオチド、および/または該ポリペプチドを発現している抗原提示細胞(APC)で刺激してよい。該ポリペプチドに特異的なT細胞の産生を可能にするために十分な条件下でかつ十分な時間、そのような刺激を実施する。好ましくは、該ポリペプチドまたはポリヌクレオチドを送達ビヒクル、例えばミクロスフェア内に存在させて、特異的T細胞の産生を促進する。
【0183】
T細胞が特異的に増殖し、サイトカインを分泌するか、あるいは、該ポリペプチドでコーティングされている標的細胞または該ポリペプチドをコードする遺伝子を発現している標的細胞を死滅させれば、該T細胞は本発明のポリペプチドに特異的であると見なされる。T細胞の特異性は種々の標準的技術のいずれかを使用して評価してよい。例えば、クロム放出アッセイまたは増殖アッセイでは、溶解および/または増殖が陰性対照と比較して2倍を超える増加を示す刺激指数がT細胞の特異性を示す。そのようなアッセイは、例えばChen et al., Cancer Res. 54:1065-1070 (1994)に記載のように実施してよい。あるいは、種々の公知の技術によってT細胞増殖の検出を達成してよい。例えば、T細胞の増殖は、DNA合成の割合の増加を測定することによって(例えば、トリチウム標識チミジンでT細胞培養をパルス標識し、DNA中に取り込まれたトリチウム標識チミジンの量を測定することによって)検出することができる。本発明のポリペプチド(100ng/ml〜100μg/ml、好ましくは200ng/ml〜25μg/ml)を3〜7日間接触させると、T細胞の増殖が少なくとも2倍増加するべきであろう。2〜3時間の上記接触により、T細胞の活性化が生じるはずであろう。その活性化は標準サイトカインアッセイを使用して測定され、その場合、サイトカイン放出(例えばTNFまたはIFN−γ)のレベルの2倍増加がT細胞の活性化を示す(Coligan et al., Current Protocols in Immunology, vol. 1 (1998)を参照のこと)。ポリペプチド、ポリヌクレオチドまたはポリペプチド発現APCに応答して活性化されたT細胞はCD4+および/またはCD8+であるかもしれない。タンパク質特異的T細胞は標準的技術を使用して増殖させてよい。好ましい実施形態では、T細胞は、患者、血縁関係ドナーまたは非血縁関係のドナーに由来し、刺激および増殖後に該患者に投与される。
【0184】
治療目的では、ポリペプチド、ポリヌクレオチドまたはAPCに応答して増殖するCD4+またはCD8+T細胞の数をin vitroまたはin vivoで増やすことができる。そのようなT細胞のin vitroでの増殖は種々の方法で達成してよい。例えば、ポリペプチド、または該ポリペプチドの免疫原性部分に相当する短いペプチドにT細胞を再度曝露することができ、その場合、T細胞成長因子、例えばインターロイキン−2、および/またはポリペプチドを合成する刺激細胞を加えるか、あるいは加えない。あるいは、タンパク質の存在下で増殖する1種以上のT細胞の数を、クローニングによって増やすことができる。細胞をクローニングするための方法は当技術分野において周知であり、それには限界希釈が含まれる。
【0185】
医薬組成物
追加の実施形態では、本発明は、単独で、あるいは1以上の他の様式の治療と組み合わせて、細胞または動物に投与するための、製薬上許容される溶液中の本明細書中で開示される1以上のポリヌクレオチド、ポリペプチド、T細胞、抗体、および化学療法組成物の製剤に関する。
【0186】
所望であれば、本明細書中で開示されるポリペプチドを発現する核酸セグメント(例えばRNAまたはDNA)を、さらに他の物質、例えば他のタンパク質もしくはポリペプチドまたは種々の医薬活性物質、例えば結核菌感染に対して有効な化学療法剤と組み合わせて投与してよいことがさらに理解される。実際、標的細胞または宿主組織との接触時に追加の物質が重大な有害作用を生じさせないならば、同時に含ませてよい他の成分に関する制限は実質的にない。ゆえに、該組成物は、具体的な事例において必要とされる種々の他の物質と共に送達してよい。そのような組成物は、宿主細胞または他の生物学的供給源から精製するか、あるいは本明細書中に記載のように化学合成してよい。同様に、そのような組成物は置換または誘導体化RNAまたはDNA組成物をさらに含んでよい。
【0187】
製薬上許容される賦形剤および担体溶液の製剤化は当業者に周知であり、それは種々の治療レジメン、例えば経口、非経口、静脈内、鼻腔内、および筋肉内投与および製剤において本明細書中に記載の具体的な組成物を使用するために好適な投薬および治療レジメンの開発も同様である。典型的には、治療有効量を含む製剤は、約2μg〜約50μgのMtb72fポリペプチド/投与、より典型的には約5μg〜約40μgのMtb72fポリペプチド/投与を送達する。
【0188】
1.経口送達
特定の適用では、本明細書中で開示される医薬組成物は経口投与によって動物に送達してよい。そのような場合、これらの組成物は不活性希釈剤または吸収可能な食用担体と共に製剤化してよく、あるいはゼラチン硬カプセルまたはゼラチン軟カプセル(hard- or soft-shell gelatin capsule)に封入してよく、あるいは圧縮して錠剤にしてよく、あるいは食事の食物中に直接組み入れてよい。
【0189】
活性化合物は、賦形剤と共に組み入れてよく、経口摂取可能な錠剤、口腔錠(buccal tables)、トローチ剤、カプセル剤、エリキシル剤、懸濁剤、シロップ剤、ウェーハ剤、等の剤形で使用してもよい(Mathiowitz et al., 1997; Hwang et al., 1998; 米国特許第5,641,515号;同第5,580,579号および同第5,792,451号(各文献は参照によりその全体が本明細書中に具体的に組み入れられる))。該錠剤、トローチ剤、丸剤、カプセル剤等は以下のものを含有してもよい:結合剤、例えばトラガカントガム、アカシア、コーンスターチ、またはゼラチン;賦形剤、例えば第二リン酸カルシウム;崩壊剤、例えばコーンスターチ、ジャガイモデンプン、アルギン酸等;滑沢剤、例えばステアリン酸マグネシウム;および甘味剤、例えばショ糖、ラクトースまたはサッカリン、あるいは香味物質、例えばペパーミント、冬緑油、またはサクランボ香味物質を加えてよい。単位投与剤形がカプセル剤である場合、上記タイプの物質に加えて、液体担体を含有してよい。種々の他の物質を、コーティングとして、あるいは該投与単位の物理的形状を別の様式で修飾するために存在させてよい。例えば、錠剤、丸剤、またはカプセル剤は、シェラック、糖、または両者でコーティングしてよい。シロップ剤またはエリキシル剤は、活性化合物、甘味剤であるショ糖、保存剤であるメチルおよびプロピルパラベン、色素および香味物質、例えばサクランボまたはオレンジ香料を含有してよい。当然、すべての単位投与剤形の調製に使用されるすべての物質は、製薬的に純粋であり、かつ使用される量において実質的に無毒であるべきであろう。さらに、該活性化合物を徐放性調製物および製剤中に組み入れてよい。
【0190】
典型的に、これらの製剤は、通常、2μg〜50μgの範囲のMtb72fポリペプチドを含有する。当然、調製することができる治療上有用な各組成物中の活性化合物(群)の量は、該化合物の任意の所定の単位容量において好適な用量が達成されるような量である。そのような医薬製剤を調製する技術分野の当業者には、溶解度、バイオアベイラビリティ、生物学的半減期、投与経路、製品保存期間、ならびに他の薬理学的考察等の要因が考慮され、それゆえに、種々の用量および治療レジメンが望ましいかもしれない。
【0191】
経口投与では、別法として、本発明の組成物を、洗口剤、歯磨剤、口腔錠、経口スプレー、または舌下経口投与用製剤の剤形中に1以上の賦形剤と共に組み入れてよい。例えば、ホウ酸ナトリウム溶液(ドーベル液)等の適切な溶媒中に必要量の活性成分を包含する洗口剤を調製してよい。あるいは、活性成分を経口溶液、例えばホウ酸ナトリウム、グリセリンおよび炭酸水素カリウムを含有する溶液に組み入れるか、あるいは歯磨剤中に分散させるか、あるいは、水、結合剤、研磨剤、香味物質、発泡剤、および湿潤剤を含んでよい治療有効量の組成物に加えてよい。あるいは、該組成物は、舌下に投与されるか、あるいは別の様式で口内で溶解させてよい錠剤または溶液剤の剤形で作成してよい。
【0192】
2.注射送達
特定の状況では、本明細書中で開示される医薬組成物を、非経口、静脈内、筋肉内、または、さらには腹腔内で送達することが望ましい。それは、米国特許第5,543,158号;同第5,641,515号および同第5,399,363号に記載される通りである(各文献は参照によりその全体が本明細書中に具体的に組み入れられる)。遊離塩基である活性化合物または製薬上許容されるその塩の溶液剤を、界面活性剤、例えばヒドロキシプロピルセルロースと好適に混合された水中で調製してよい。さらに、グリセロール、液状ポリエチレングリコール、およびその混合物および油中で分散剤を調製してよい。通常の保存および使用条件下では、これらの調製物は、微生物の生育を妨げるための保存剤を含有する。
【0193】
注入使用に適した医薬剤形には、滅菌水性溶液剤または分散剤および、滅菌注射用溶液剤または分散剤の即席調製のための滅菌粉末剤が含まれる(米国特許第5,466,468号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。全事例で、その剤形は無菌である必要があり、容易な注射針通過性(syringability)が存在する程度に液状である必要がある。それは、製造および保存条件下で安定である必要があり、微生物、例えば細菌および真菌の混入作用に対して保護される必要がある。該担体は、溶媒または分散媒でありうる。それには、例えば水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール、および液状ポリエチレングリコール、等)、好適なその混合物、および/または植物油が含有される。例えば、コーティング、例えばレシチンの使用によって、分散剤の場合、必要な粒子サイズの維持によって、ならびに界面活性剤の使用によって、適正な流動性を維持してよい。微生物の作用の防止は、種々の抗菌剤および抗真菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール、等によって促進することができる。多数の場合、等張性物質、例えば糖または塩化ナトリウムを含ませることが好ましい。注射用組成物の長期吸収は、吸収を遅延させる物質、例えばモノステアリン酸アルミニウムおよびゼラチンを組成物中で使用することによって達成することができる。
【0194】
水性溶液剤中での非経口投与では、例えば、必要であれば該溶液剤を好適に緩衝化するべきであろう。まず、十分な生理食塩水またはグルコースを用いて該液状希釈剤を等張性にするべきであろう。これらの特定の水性溶液剤は、静脈内、筋肉内、皮下および腹腔内投与に特に適している。この関連で、使用できる滅菌水性媒体は、本開示内容を踏まえて当業者に公知である。例えば、一用量を1mlの等張性NaCl溶液に溶解し、1000mlの皮下注入用の液体に加えるか、あるいは注入の予定部位に注射してよい(例えばRemington's Pharmaceutical Sciences, 15th Edition, pp. 1035-1038および1570-1580を参照のこと)。治療対象の被験体の症状に応じていくらかの用量バリエーションが必然的に存在する。いずれにしても、処方担当者が個々の被験体に適切な用量を決定する。さらに、ヒトへの投与では、調製物は、FDA Office of Biologics standardsが要求する滅菌性、発熱原性、および全般的安全性および純度基準を満たすべきであろう。
【0195】
滅菌注射用溶液剤は、必要量の活性化合物を、必要に応じて上に列挙される種々の他の成分と共に、適切な溶媒中に組み入れた後、ろ過滅菌することによって調製される。概して、分散剤は、ベースとなる分散媒および、上に列挙されるものから選択される必要な他の成分を含有する滅菌ビヒクル中に種々の滅菌済み活性成分を組み入れることによって調製される。滅菌注射用溶液剤の調製のための滅菌粉末剤の場合、好ましい調製方法は真空乾燥および凍結乾燥技術であり、それにより、活性成分と任意の所望の追加成分からなる粉末剤が、あらかじめ滅菌ろ過されたその溶液から得られる。
【0196】
本明細書中で開示される組成物は中性または塩形式で製剤化してよい。製薬上許容される塩には、酸付加塩(タンパク質の遊離アミノ基と共に形成される)であって、無機酸、例えば塩酸もしくはリン酸、または酢酸、シュウ酸、酒石酸、マンデル酸、等の有機酸と共に形成されるものが含まれる。また、遊離カルボキシル基と共に形成される塩を、無機塩基、例えばナトリウム、カリウム、アンモニウム、カルシウム、または水酸化第二鉄、およびイソプロピルアミン、トリメチルアミン、ヒスチジン、プロカイン等の有機塩基から生成させることができる。製剤化された溶液剤は、投与製剤に適合する様式でかつ治療上有効な量で投与される。該製剤は、注射用溶液剤、薬物放出カプセル、等の種々の剤形で容易に投与することができる。
【0197】
本明細書中で使用される「担体」には、任意およびすべての溶媒、分散媒、ビヒクル、コーティング、希釈剤、抗菌剤および抗真菌剤、等張性および吸収遅延物質、バッファー、担体溶液、懸濁液、コロイド、等が含まれる。医薬活性物質に関するそのような媒体および物質の使用は当技術分野において周知である。任意の慣用の媒体または物質が活性成分と不適合性である場合を除いて、治療用組成物中でのその使用が想定される。該組成物に補充活性成分を組み入れることもできる。
【0198】
「製薬上許容される」との言い回しは、ヒトへの投与時に、アレルギー性または同様の有害反応を生じさせない分子的実体および組成物を表す。活性成分としてタンパク質を含有する水性組成物の調製は当技術分野において十分に理解されている。典型的に、そのような組成物は注射剤として、液状溶液剤または懸濁剤として調製され;注射前の液体中の溶解または懸濁に適した固形剤形を調製することもできる。該調製物は乳化することもできる。
【0199】
3.鼻腔および口腔送達
特定の実施形態では、鼻内スプレー、口腔スプレー、吸入、および/または他のエアロゾル送達ビヒクルによって医薬組成物を送達してよい。遺伝子、核酸、およびペプチド組成物を、例えば鼻腔および口腔エアロゾルスプレーによって直接に肺に送達するための方法は、例えば米国特許第5,756,353号および同第5,804,212号に記載されている(各文献は参照によりその全体が本明細書中に具体的に組み入れられる)。同様に、鼻腔内微粒子樹脂(Takenaga et al., 1998)およびリゾホスファチジル−グリセロール化合物(米国特許第5,725,871号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))を使用する薬物送達もまた、製薬技術分野において周知である。同様に、ポリテトラフルオロエチレン支持マトリックス(polytetrafluoroetheylene support matrix)の形式での経粘膜薬物送達が米国特許第5,780,045号に記載されている(該文献は参照によりその全体が本明細書中に具体的に組み入れられる)。
【0200】
4.リポソーム媒介性、ナノカプセル媒介性、および微粒子媒介性送達
特定の実施形態では、本発明者らは、本発明の組成物を好適な宿主細胞内に導入するための、リポソーム、ナノカプセル、微粒子、ミクロスフェア、脂質粒子、小胞、等の使用を意図する。特に、本発明の組成物は、脂質粒子、リポソーム、小胞、ナノスフェア、またはナノ粒子等に封入して送達するために製剤化してよい。
【0201】
そのような製剤は、本明細書中で開示される核酸または構築物の製薬上許容される製剤の導入に好ましいかもしれない。リポソームの形成および使用は、当業者に一般に公知である(例えばCouvreur et al., 1977; Couvreur, 1988; Lasic, 1998を参照のこと(該文献には、細胞内細菌感染および疾患に関する標的化された抗生物質治療におけるリポソームおよびナノカプセルの使用が記載されている))。最近、改善された血清安定性および循環半減期を有するリポソームが開発された(Gabizon & Papahadjopoulos, 1988; Allen and Choun, 1987; 米国特許第5,741,516号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。さらに、潜在的薬物担体としてのリポソームおよびリポソーム様調製物を調製する種々の方法が概説されている(Takakura, 1998; Chandran et al., 1997; Margalit, 1995; 米国特許第5,567,434号;同第5,552,157号;同第5,565,213号;同第5,738,868号および同第5,795,587号(各文献は参照によりその全体が本明細書中に具体的に組み入れられる))。
【0202】
リポソームは、通常、他の手順によるトランスフェクションに抵抗性のいくつかの細胞タイプ、例えばT細胞懸濁物、初代肝細胞培養およびPC12細胞で首尾良く使用されている(Renneisen et al., 1990; Muller et al., 1990)。さらに、リポソームは、ウイルスベースの送達系に典型的なDNAの長さの制約を有さない。リポソームは、遺伝子、薬物(Heath & Martin, 1986; Heath et al., 1986; Balazsovits et al., 1989; Fresta & Puglisi, 1996)、放射線治療薬(Pikul et al., 1987)、酵素(Imaizumi et al., 1990a; Imaizumi et al., 1990b)、ウイルス(Faller & Baltimore, 1984)、転写因子およびアロステリックエフェクター(Nicolau & Gersonde, 1979)を種々の培養細胞株および動物に導入するために効果的に使用されている。さらに、リポソーム媒介性薬物送達の有効性を調査するいくつかの臨床試験の成功が達成されている(Lopez-Berestein et al., 1985a; 1985b; Coune, 1988; Sculier et al., 1988)。さらにまた、いくつかの研究で、リポソームの使用が、自己免疫性応答、毒性または全身性送達後の生殖腺局在化を伴わないことが示唆されている(Mori & Fukatsu, 1992)。
【0203】
リポソームは、水性媒体中に分散され、自発的に多重膜同心性二分子膜小胞(multilamellar concentric bilayer vesicles)(多重膜小胞(MLV)とも称される)を形成するリン脂質から形成される。MLVは一般的には25nm〜4μmの直径を有する。MLVを超音波処理すると、200〜500Åの範囲の直径で、コア中に水性溶液を含有する小さい単層の小胞(SUV)が形成される。
【0204】
リポソームは細胞膜と類似点を有し、ペプチド組成物の担体として本発明に関連した使用が意図される。それらは、水溶性および脂溶性の両物質を、すなわち、それぞれ水性間隙中および二重層自体の中にトラップできるために、広く適している。薬物保持リポソームを、リポソーム製剤を選択的に改変することによって活性物質の部位特異的送達に使用することさえ可能である。
【0205】
Couvreur et al. (1977; 1988)の教示内容に加えて、以下の情報をリポソーム製剤の作成に利用してよい。リン脂質は、水に分散させると、水に対する脂質のモル比に応じてリポソーム以外の種々の構造を形成しうる。低い割合で、リポソームは好ましい構造になる。リポソームの物理的特性は、pH、イオン強度および2価カチオンの存在に依存する。リポソームは、イオン性および極性物質に対して低い透過性を示しうるが、高い温度では、それらの透過性を顕著に変化させる相転移を受ける。該相転移には、ゲル状態として知られる密接に充填された秩序構造から、液体状態として知られる緩く充填された低秩序構造への変化が関与する。これは特徴的な相転移温度で生じ、その結果、イオン、糖および薬物に対する透過性が増加する。
【0206】
温度に加えて、タンパク質への曝露によりリポソームの透過性が変化しうる。特定の可溶性タンパク質、例えばシトクロムcは、二重層に結合し、それを変形させ、それを貫通することによって、透過性の変化を生じさせる。コレステロールは、おそらくリン脂質をより密接にパッキングすることによってこのタンパク質の貫通を阻害する。抗生物質およびインヒビター送達のための最も有用なリポソーム構成はコレステロールを含有することが想定される。
【0207】
溶質をトラップする能力は、異なるタイプのリポソーム間で様々である。例えば、溶質のトラップ時にMLVは中程度に効率的であるが、SUVは非常に非効率的である。しかし、SUVはサイズ分布の均質性および再現性の利点を提供し、大きい単層の小胞(LUV)によってサイズおよびトラップ効率の間の妥協点が提供される。これらはエーテル蒸発によって調製され、溶質トラップ時にMLVより3〜4倍効率的である。
【0208】
リポソーム特性に加えて、化合物のトラップに関する重要な決定要因は該化合物自体の物理化学的特性である。極性化合物は水性間隙中にトラップされ、無極性化合物は小胞の脂質二重層に結合する。極性化合物は、浸透によって、あるいは二重層が崩壊した時に、放出されるが、無極性化合物は、温度またはリポタンパク質への曝露によってそれが崩壊しない限り、二重層に付属したままである。両タイプは相転移温度で最大流出率を示す。
【0209】
リポソームは以下の4種の異なる機構で細胞と相互作用する:細網内皮系の食細胞、例えばマクロファージおよび好中球によるエンドサイトーシス;非特異的な弱い疎水性または静電力によるか、あるいは細胞表面成分との特異的相互作用による細胞表面への吸着;リポソームの脂質二重層が原形質膜内へ挿入されることによる形質細胞膜との融合、およびリポソーム内容物の細胞質内への同時放出;および、リポソーム内容物の結合を全く伴わない、細胞膜またはオルガネラ膜(subcellular membranes)へのリポソーム脂質の移入またはその逆に基づく機構。どの機構が作動するかを決定することは困難であることがよくあり、2以上の機構が同時に作動するかもしれない。
【0210】
静脈内に注射されたリポソームの末路および傾向は、それらの物性、例えばサイズ、流動性、および表面電荷に依存する。それらは、その組成、および血液中での分〜数時間の範囲の半減期に依存して、数時間または数日間、組織中に残存するかもしれない。大きいリポソーム、例えばMLVおよびLUVは、細網内皮系の食細胞によって急速に摂取されるが、循環系の生理学はほとんどの部位でそのような大きい分子種が外に出ることを抑制する。それらは、毛細血管内皮中の大きな開口部または細孔が存在する場所、例えば肝臓または脾臓のシヌソイドでしか外に出ることができない。ゆえに、これらの器官は取り込みの顕著な部位(predominate site)である。一方、SUVはより広い組織分布を示すが、依然として肝臓および脾臓に高度に隔離される。概して、このin vivo挙動により、リポソームの潜在的ターゲティングは、大きいサイズのリポソームがアクセスできる器官および組織のみに制限される。これらには、血液、肝臓、脾臓、骨髄、およびリンパ器官が含まれる。
【0211】
ターゲティングは、概して、本発明に関する制約ではない。しかし、万一、特異的ターゲティングが所望であれば、それを達成するための方法が利用可能である。リポソーム表面に結合する抗体を使用し、該抗体およびその薬物内容物を、特定の細胞タイプの表面上に位置する特異的抗原性受容体に向けてよい。炭水化物決定基(細胞−細胞認識、相互作用および接着に役割を果たす糖タンパク質または糖脂質細胞表面成分)を認識部位として使用してもよい。それは、それらがリポソームを特定の細胞タイプに向ける潜在能力を有する場合である。ほとんどの場合、リポソーム調製物の静脈内注射を使用することが想定されるが、他の投与経路も考えられる。
【0212】
あるいは、本発明は、本発明の組成物の製薬上許容されるナノカプセル製剤を提供する。ナノカプセルは、概して、安定かつ再現可能な様式で化合物をトラップすることができる(Henry-Michelland et al., 1987; Quintanar-Guerrero et al., 1998; Douglas et al., 1987)。細胞内ポリマーの過剰詰め込みに起因する副作用を回避するために、in vivoで分解可能なポリマーを使用してそのような超微粒子(約0.1μmのサイズ)を設計すべきであろう。本発明での使用には、これらの必要条件を満たす生分解性ポリアルキル−シアノアクリラートナノ粒子が想定される。そのような粒子は文献に記載されるように容易に作成してよい(Couvreur et al., 1980; 1988; zur Muhlen et al., 1998; Zambaux et al. 1998; Pinto-Alphandry et al., 1995および米国特許第5,145,684号(該文献は参照によりその全体が本明細書中に具体的に組み入れられる))。
【0213】
ワクチン
本発明の特定の好ましい実施形態では、ワクチンを提供する。該ワクチンは、一般的に、1以上の上記のような医薬組成物を、免疫賦活剤と組み合わせて含む。免疫賦活剤は、外因性抗原に対する免疫応答(抗体および/または細胞媒介性)を高めるか、あるいは増強する任意の物質であってよい。免疫賦活剤の例には、アジュバント、生分解性ミクロスフェア(例えばポリラクティックガラクチド(polylactic galactide))およびリポソーム(内部に化合物が組み入れられている;例えばFullerton, 米国特許第4,235,877号を参照のこと)が含まれる。ワクチン調製物は、概して、例えばPowell & Newman, eds., Vaccine Design (the subunit and adjuvant approach) (1995)に記載されている。本発明の範囲内の医薬組成物およびワクチンは、他の化合物を含有してもよく、該化合物は生物活性または不活性であってよい。例えば、1以上の、他の腫瘍抗原の免疫原性部分を、該組成物またはワクチン内で、融合ポリペプチドに組み入れるか、あるいは分離した化合物として存在させてよい。
【0214】
例示的なワクチンには、1以上の上記ポリペプチドをコードするDNAを含有させて、該ポリペプチドがin situで生成されるようにしてよい。上記のように、当業者に公知の任意の種々の送達系内に該DNAを存在させてよく、該送達系には、核酸発現系、細菌およびウイルス発現系が含まれる。多数の遺伝子送達技術が当技術分野において周知であり、例えばRolland, Crit. Rev. Therap. Drug Carrier Systems 15:143-198 (1998)、およびその引用文献に記載の技術がある。適切な核酸発現系は、患者での発現に必要なDNA配列(例えば好適なプロモーターおよび終結シグナル)を含有する。細菌送達系には、該ポリペプチドの免疫原性部分を細胞表面上で発現するか、あるいはそのようなエピトープを分泌する細菌宿主細胞(例えばマイコバクテリウム、バシラスまたはラクトバシラス株、例えばカルメット・ゲラン桿菌またはLactococcus lactis)の投与が関与する(例えばFerreira, et al., An Acad Bras Cienc (2005) 77:113-124; およびRaha, et al., Appl Microbiol Biotechnol (2005) PubMedID 15635459を参照のこと)。好ましい実施形態では、ウイルス発現系(例えばワクシニアまたは他のポックスウイルス、レトロウイルス、またはアデノウイルス)を使用して該DNAを導入してよく、それには、非病原性(欠損)複製可能ウイルスの使用が関与してよい。好適な系は、例えば以下の文献に開示されている:Fisher-Hoch et al., Proc. Natl. Acad. Sci. USA 86:317-321 (1989); Flexner et al., Ann. N.Y. Acad. Sci. 569:86-103 (1989); Flexner et al., Vaccine 8:17-21 (1990); 米国特許第4,603,112号、同第4,769,330号、および同第5,017,487号;WO89/01973;米国特許第4,777,127号;GB2,200,651;EP0,345,242;WO91/02805; Berkner, Biotechniques 6:616-627 (1988); Rosenfeld et al., Science 252:431-434 (1991); Kolls et al., Proc. Natl. Acad. Sci. USA 91:215-219 (1994); Kass-Eisler et al., Proc. Natl. Acad. Sci. USA 90:11498-11502 (1993); Guzman et al., Circulation 88:2838-2848 (1993); およびGuzman et al., Cir. Res. 73:1202-1207 (1993)。そのような発現系にDNAを組み入れるための技術は当業者に周知である。該DNAは「裸の」DNAであってよく、それは、例えばUlmer et al., Science 259:1745-1749 (1993)に記載され、Cohen, Science 259:1691-1692 (1993)に概説されている。裸のDNAの取り込みは、該DNAを生分解性ビーズ上にコーティングすることによって高めてよく、該ビーズは細胞内に効率的に輸送される。ワクチンはポリヌクレオチドおよびポリペプチドの両成分を含んでよいことが明らかである。そのようなワクチンは向上した免疫応答を提供するかもしれない。
【0215】
ワクチンは、本明細書中で提供されるポリヌクレオチドおよびポリペプチドの製薬上許容される塩を含有してよいことが明らかである。そのような塩は、製薬上許容される無毒の塩基、例えば有機塩基(例えば第1級、第2級および第3級アミンおよび塩基性アミノ酸の塩)および無機塩基(例えばナトリウム、カリウム、リチウム、アンモニウム、カルシウムおよびマグネシウム塩)から調製してよい。
【0216】
本発明のワクチン組成物中では当業者に公知の任意の適切な担体を使用してよいが、担体のタイプは投与様式に応じて様々である。本発明の組成物は任意の適切な投与様式、例えば局所、経口、鼻腔、静脈内、頭蓋内、腹腔内、皮下または筋肉内投与のために製剤化してよい。非経口投与、例えば皮下注射では、担体は、好ましくは、水、生理食塩水、アルコール、脂肪、ワックスまたはバッファーを含む。経口投与では、上記担体のいずれかまたは固形担体、例えばマンニトール、ラクトース、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、滑石、セルロース、グルコース、ショ糖、および炭酸マグネシウムを使用してよい。本発明の医薬組成物用の担体として、生分解性ミクロスフェア(例えばポリラクタートポリグリコラート)を使用してもよい。好適な生分解性ミクロスフェアは、例えば米国特許第4,897,268号;同第5,075,109号;同第5,928,647号;同第5,811,128号;同第5,820,883号;同第5,853,763号;同第5,814,344号および同第5,942,252号で開示されている。米国特許第5,928,647号に記載の微粒子−タンパク質複合体を含む担体を使用してもよく、該複合体は宿主においてクラスI限定の細胞障害性Tリンパ球応答を誘発可能である。
【0217】
そのような組成物は、バッファー(例えば中性緩衝生理食塩水またはリン酸緩衝食塩水)、炭水化物(例えばグルコース、マンノース、ショ糖またはデキストラン)、マンニトール、タンパク質、ポリペプチドまたはアミノ酸、例えばグリシン、酸化防止剤、静菌剤、キレート剤、例えばEDTAまたはグルタチオン、アジュバント(例えば水酸化アルミニウム)、製剤をレシピエントの血液と等張、低張または弱い高張性にする溶質、懸濁化剤、増粘剤および/または保存剤を含んでもよい。あるいは、本発明の組成物は凍結乾燥物(lyophilizate)として製剤化してよい。周知のテクノロジーを使用してリポソーム内に化合物を封入してもよい。
【0218】
本発明のワクチン中で任意の種々の免疫賦活剤を使用してよい。例えば、アジュバントを含ませてよい。ほとんどのアジュバントは、急速な異化作用から抗原を保護するように設計された物質、例えば水酸化アルミニウムまたは鉱油、および免疫応答の刺激因子、例えばリピドA、Bortadella pertussisまたはマイコバクテリア属の種またはマイコバクテリウム由来タンパク質を含有する。例えば、脱脂(delipidated)、脱糖脂質化(deglycolipidated)M. vaccae(「pVac」)を使用することができる。好適なアジュバントは、例えばフロイント不完全アジュバントおよび完全アジュバント(Difco Laboratories, Detroit, MI);Merck Adjuvant 65(Merck and Company, Inc., Rahway, NJ);AS01B、AS02A、AS15、AS−2およびその誘導体(GlaxoSmithKline, Philadelphia, PA);CWS、TDM、Leif、アルミニウム塩、例えば水酸化アルミニウムゲル(alum)またはリン酸アルミニウム;カルシウム、鉄または亜鉛の塩;アシル化チロシンの不溶性懸濁物;アシル化糖;カチオンまたはアニオンによって誘導体化された多糖;ポリホスファゼン(polyphosphazenes);生分解性ミクロスフェア;モノホスホリル脂質Aおよびquil Aとして市販されている。サイトカイン、例えばGM−CSFまたはインターロイキン−2、−7、または−12をアジュバントとして使用してもよい。
【0219】
本明細書中で提供されるワクチンでは、該アジュバント組成物は、好ましくは、主にTh1タイプの免疫応答を誘発するように設計される。高レベルのTh1タイプサイトカイン(例えばIFN−γ、TNFα、IL−2およびIL−12)は、投与された抗原に対する細胞性免疫応答の誘発に好都合である傾向がある。対照的に、高レベルのTh2タイプサイトカイン(例えばIL−4、IL−5、IL−6およびIL−10)は体液性免疫応答の誘発に好都合である傾向がある。本明細書中で提供されるワクチンの適用後、患者は、Th1−およびTh2−タイプ応答を含む免疫応答を支持する。応答が主にTh1タイプである好ましい実施形態では、Th1タイプサイトカインのレベルがTh2タイプサイトカインのレベルより高い程度にまで増加する。これらのサイトカインのレベルは、標準的アッセイを使用して容易に評価してよい。サイトカインのファミリーについての総説としては、Janeway, et al., Immunobiology, 5th Edition, 2001を参照のこと。
【0220】
主にTh1タイプ応答の誘発に使用するための好ましいアジュバントには、例えば、場合によりアルミニウム塩を伴う、モノホスホリル脂質A、好ましくは3−O−脱アシル化モノホスホリル脂質A(3D−MPL)の組み合わせが含まれる(例えばRibi, et al., 1986, Immunology and Immunopharmacology of Bacterial Endotoxins, Plenum Publ. Corp., NY, pp. 407-419; GB2122204B;GB2220211;およびUS4,912,094を参照のこと)。好ましい形式の3D−MPLは、直径0.2mm未満の小さい粒子サイズを有するエマルジョンの形式であり、その製造方法はWO94/21292に開示されている。モノホスホリル脂質Aおよび界面活性剤を含む水性製剤はWO98/43670に記載されている。例示的な好ましいアジュバントには、AS01B(リポソーム製剤中のMPLおよびQS21)、リポソーム製剤中の3D−MPLおよびQS21、AS02A(MPLおよびQS21および水中油型エマルジョン)、3D−MPLおよびQS21および水中油型エマルジョン、およびAS15(GlaxoSmithKlineから入手可能)が含まれる。MPLアジュバントはGlaxoSmithKline, Seattle, WAから入手可能である(米国特許第4,436,727号;同第4,877,611号;同第4,866,034号および同第4,912,094号を参照のこと)。
【0221】
CpG含有オリゴヌクレオチド(該オリゴヌクレオチドではCpGジヌクレオチドがメチル化されていない)もまた、主にTh1応答を誘発する。CpGとは、DNA中に存在するシトシン−グアノシンジヌクレオチドモチーフの略語である。そのようなオリゴヌクレオチドは周知であり、例えばWO96/02555、WO99/33488および米国特許第6,008,200号および同第5,856,462号に記載されている。また、免疫賦活性DNA配列は、例えばSato et al., Science 273:352 (1996)に記載されている。CpGは、ワクチンに製剤化されると、概して、遊離溶液中で遊離抗原と共に投与される(WO 96/02555; McCluskie and Davis, 上記)か、あるいは共有結合によって抗原にコンジュゲートされる(WO98/16247)か、あるいは水酸化アルミニウム等の担体と共に製剤化される((肝炎表面抗原))Davis et al. 上記; Brazolot-Millan et al., Proc.Natl.Acad.Sci., USA, 1998, 95(26), 15553-8)。CpGは、全身および粘膜の両経路によって投与できるアジュバントであることが当技術分野において知られている(WO96/02555、EP468520、Davis et al., J.Immunol, 1998, 160(2):870-876; McCluskie and Davis, J.Immunol., 1998, 161(9):4463-6)。
【0222】
別の好ましいアジュバントはサポニンまたはサポニン模倣物もしくは誘導体、好ましくはQS21(Aquila Biopharmaceuticals Inc., Framingham, MA)であり、それは単独で、あるいは他のアジュバントと組み合わせて使用してよい。例えば、強化された系には、モノホスホリル脂質Aおよびサポニン誘導体の組み合わせ、例えばWO94/00153に記載のQS21および3D−MPLの組み合わせ、またはWO96/33739に記載のQS21がコレステロールでクエンチされる低反応原性組成物が含まれる。他の好ましい製剤には、水中油型エマルジョンおよびトコフェロールが含まれる。水中油型エマルジョン中にQS21、3D−MPLおよびトコフェロールを含む特に強力なアジュバント製剤がWO95/17210に記載されている。本発明で有用な追加のサポニンアジュバントには、QS7(WO96/33739およびWO96/11711に記載)およびQS17(米国特許第5,057,540号およびEP0 362 279B1に記載)が含まれる。
【0223】
他の好ましいアジュバントには、Montanide ISA 720(Seppic, France)、SAF(Chiron, California, United States)、ISCOMS(CSL)、MF-59(Chiron)、SBASシリーズのアジュバント(例えばSBAS-2、AS2'、AS2''、SBAS-4、またはSBAS6(GlaxoSmithKline, Rixensart, Belgiumから入手可能))、Detox(Corixa, Hamilton, MT)、RC-529(Corixa, Hamilton, MT)および他のアミノアルキルグルコサミニド4−ホスファート(AGP)、例えば係属中の米国特許出願第08/853,826号および同第09/074,720号(その開示内容は参照によりその全体が本明細書中に組み入れられる)に記載のものが含まれる。
【0224】
アジュバントの別の例には、合成MPLおよび志賀毒素Bサブユニットベースのアジュバントが含まれる(WO2005/112991を参照のこと)。
【0225】
本明細書中で提供される任意のワクチンは、抗原、免疫応答エンハンサーおよび好適な担体または賦形剤の組み合わせに結実する周知の方法を使用して調製してよい。本明細書中に記載の組成物は、徐放性製剤(すなわち、投与後に化合物の持続放出を生じさせる製剤、例えばカプセル、スポンジまたはゲル(例えば多糖から構成される))の部分として投与してよい。そのような製剤は、概して、周知のテクノロジーを使用して調製してよく(例えばCoombes et al., Vaccine 14:1429-1438 (1996)を参照のこと)、例えば経口、経直腸または皮下移植によって、あるいは所望の標的部位での移植によって投与してよい。徐放性製剤は、ポリペプチド、ポリヌクレオチドまたは抗体を含有してよく、それらは担体マトリックス中に分散され、ならびに/あるいは律速メンブレン(rate controlling membrane)によって包囲されているリザーバ内に含有されていてよい。
【0226】
そのような製剤内で使用するための担体は生体適合性であり、生分解性であってもよく;好ましくは該製剤は比較的一定レベルの活性成分放出を提供する。そのような担体には、ポリ(ラクチドコグリコリド)、ポリアクリラート、ラテックス、デンプン、セルロース、デキストラン等の微粒子が含まれる。他の遅延放出担体には、非液状親水性コア(例えば架橋された多糖またはオリゴ糖)および、場合により、両親媒性化合物、例えばリン脂質を含む外層を含む超分子バイオベクター(supramolecular biovectors)が含まれる(例えば米国特許第5,151,254号およびPCT出願WO94/20078、WO94/23701およびWO96/06638を参照のこと)。徐放性製剤内に含有される活性化合物の量は、移植部位、放出の速度および予測される持続時間および治療または予防対象の症状の性質に依存する。
【0227】
任意の種々の送達ビヒクルを医薬組成物およびワクチン中で使用して、腫瘍細胞を標的にする抗原特異的免疫応答の発生を促進してよい。送達ビヒクルには、抗原提示細胞(APC)、例えば樹状細胞、マクロファージ、B細胞、単球および、遺伝子操作によって有効なAPCにしてよい他の細胞が含まれる。そのような細胞は、その必要はないが、遺伝子改変して、抗原を提示する能力を高め、T細胞応答の活性化および/または維持を改善し、それ自体で抗腫瘍効果を有するようにし、ならびに/あるいは、受け手と免疫適合性(すなわち適合型HLAハプロタイプ)にしてよい。APCは、概して、腫瘍および腫瘍周囲の組織を含む任意の種々の生体液および器官から単離してよく、自己、同種異系、同系または異種細胞であってよい。
【0228】
本発明の特定の好ましい実施形態では、樹状細胞またはその前駆体を抗原提示細胞として使用する。樹状細胞は非常に強力なAPCであり(Banchereau & Steinman, Nature 392:245-251 (1998))、予防的または治療的な抗腫瘍性免疫を誘発するための生理学的アジュバントとして有効であることが示されている(Timmerman & Levy, Ann. Rev. Med. 50:507-529 (1999)を参照のこと)。一般に、樹状細胞は、その典型的な形状(in situでは星状であり、in vitroで可視の顕著な細胞質の突起(樹状突起)を有する)、高い効率で抗原を取り込み、プロセシングし、提示するその能力、およびナイーブT細胞応答を活性化するその能力に基づいて特定してよい。当然、樹状細胞は、in vivoまたはex vivoで樹状細胞上には一般に認められない特異的細胞表面受容体またはリガンドを発現するように遺伝子操作してよく、そのような改変樹状細胞は本発明によって想定される。樹状細胞の代替物として、分泌小胞である抗原積載樹状細胞(エキソソームと称される)をワクチン内で使用してよい(Zitvogel et al., Nature Med. 4:594-600 (1998)を参照のこと)。
【0229】
樹状細胞および前駆体は、末梢血、骨髄、腫瘍に浸潤している細胞、腫瘍周囲の組織に浸潤している細胞、リンパ節、脾臓、皮膚、臍帯血または任意の他の好適な組織もしくは液体から取得してよい。例えば、末梢血から回収された単球の培養にサイトカイン、例えばGM−CSF、IL−4、IL−13および/またはTNFαの組み合わせを加えることによって、ex vivoで樹状細胞を分化させてよい。あるいは、GM−CSF、IL−3、TNFα、CD40リガンド、LPS、flt3リガンドおよび/または、樹状細胞の分化、成熟および増殖を誘発する他の化合物(群)の組み合わせを培養培地に加えることによって、末梢血、臍帯血または骨髄から収集されたCD34陽性細胞を樹状細胞に分化させてよい。
【0230】
樹状細胞は、好都合には、「未成熟」および「成熟」細胞として分類され、それにより、2種の十分に特徴付けられている表現型を識別する単純な様式が提供される。しかし、この命名法は、すべての起こりうる、分化の中間段階を排除すると解釈されるべきではない。未成熟樹状細胞は、抗原の取り込みおよびプロセシングの高い能力を有するAPCとして特徴付けられ、それはFcγ受容体およびマンノース受容体の高い発現と相関している。成熟表現型は、典型的に、これらのマーカーの低い発現、反面、T細胞活性化を担う細胞表面分子、例えばクラスIおよびクラスII MHC、接着分子(例えばCD54およびCD11)および共刺激分子(例えばCD40、CD80、CD86および4−1BB)の高い発現によって特徴付けられる。
【0231】
一般に、タンパク質(またはその一部分もしくは他の変異体)をコードするポリヌクレオチドでAPCをトランスフェクトして、該ポリペプチド、またはその免疫原性部分が細胞表面上に発現されるようにしてよい。そのようなトランスフェクションはex vivoで行ってよく、そして、本明細書中で記載されるように、該トランスフェクト済み細胞を含む組成物またはワクチンを治療目的で使用してよい。あるいは、樹状または他の抗原提示細胞を標的にする遺伝子送達ビヒクルを患者に投与してよく、その結果、トランスフェクションがin vivoで生じる。例えば、樹状細胞のin vivoおよびex vivoトランスフェクションは、概して、当技術分野において公知の任意の方法、例えばWO97/24447に記載の方法、またはMahvi et al., Immunology and Cell Biology 75:456-460 (1997)に記載の遺伝子銃アプローチを使用して実施してよい。樹状細胞の抗原積載は、樹状細胞または前駆細胞を、ポリペプチド、DNA(裸のDNAまたはプラスミドベクター内のDNA)もしくはRNA;または抗原を発現する組換え細菌またはウイルス(例えばワクシニア、鶏痘、アデノウイルスまたはレンチウイルスベクター)とインキュベートすることによって達成してよい。積載前に、該ポリペプチドを免疫学的パートナーに共有結合によってコンジュゲートして、T細胞支援(例えば担体分子)を提供してよい。あるいは、非コンジュゲート型の免疫学的パートナーを、別途、あるいは該ポリペプチドの存在下で樹状細胞にパルスしてよい。
【0232】
ワクチンおよび医薬組成物は、単位用量または複数回用量容器、例えば密封アンプルまたはバイアル中で提供してよい。好ましくは、使用時まで該製剤の無菌性を保つために該容器を密封する。一般に、製剤は、油性または水性ビヒクル中の懸濁液、溶液またはエマルジョンとして保存してよい。あるいは、ワクチンまたは医薬組成物は凍結乾燥条件で保存してよい。その場合、使用直前に滅菌液状担体を加えることのみが必要とされる。
【0233】
本明細書中で引用されるすべての刊行物および特許出願は、個々の各刊行物または特許出願が参照により組み入れられることが具体的かつ個別に示されているかのように、参照により本明細書中に組み入れられる。
【0234】
前記発明について、理解を明確にするために、説明および例を用いていくらか詳細に説明してきたが、特許請求の範囲の思想または範囲から逸脱することなく、それに対して特定の変更および改変を施してよいことが、本発明の教授内容を踏まえて当業者には直ちに自明である。
【実施例】
【0235】
実施例
限定目的ではなく単に説明目的で以下に実施例を挙げる。当業者は、本質的に同様の結果を生じさせるよう変更または改変することができる種々の重要でないパラメータを容易に認識するであろう。
【0236】
実施例1: Mtb72f(Hisタグなし)(配列番号6)の調製
Mtb72f発現ベクターの構築
Mtb72fは、2種の結核菌タンパク質Mtb32およびMtb39から形成される融合タンパク質である。Mtb39と、Mtb32のN末端およびC末端部分を、Mtb32のC末端−Mtb39−Mtb32のN末端のように融合させることによって、Mtb72fを構築する。具体的には、Mtb32の約14kDaのC末端断片(残基192−323;132アミノ酸)をコードするオープンリーディングフレーム(ORF)にMtb39の全長ORFを直列に連続して連結し、続いてMtb32の約20kDaのN末端部分(残基1−195)をC末端に連結することによって、Mtb72fタンパク質を作成した。結核菌株H37Rvに由来するゲノムDNAからのポリメラーゼ連鎖反応(PCR)のために、ユニークな制限部位(EcoRIおよびEcoRV)を含有し、C末端の停止コドンを欠いている(Mtb32−CおよびMtb39の場合)配列特異的オリゴヌクレオチドを使用することによって、それを達成した。該プロセスの詳細は以下の通りである。
【0237】
まず、以下のオリゴヌクレオチドを用いるPCRを使用して、H37RvからMtb32のC末端部分(Mtb32C)をコードするDNAをクローニングした:5’(5’−CAA−TTA−CAT−ATG−CAT−CAC−CAT−CAC−CAT−CAC−ACG−GCC−GCG−TCC−GAT−AAC−TTC−3’)および3’(5’−CTA−ATC−GAA−TCC−GGC−CGG−GGG−TCC−CTC−GGC−CAA−3’)。5’オリゴヌクレオチドは、ATG開始コドンを包含するNdeI制限部位(下線部)を含有した。3’オリゴヌクレオチドはEcoRI制限部位(下線部)を含有した。これらのオリゴヌクレオチドを使用して、Mtb32の396ヌクレオチド部分であるMtb32Cを増幅し、得られた産物を発現ベクターのNdeIおよびEcoRI部位にサブクローニングした。次いでEcoRIおよびEcoRVで消化して、Mtb32Cプラスミドを直線状にした。
【0238】
Mtb39に関しては、PCR増幅およびクローニングに以下のオリゴヌクレオチドを使用した:5’−(5’−CTA−ATC−GAA−TTC−ATG−GTG−GAT−TTC−GGG−GCG−TTA−3’)および3’(5’−CTA−ATC−GAT−ATC−GCC−GGC−TGC−CGG−AGA−ATG−CGG−3’)。5’オリゴヌクレオチドはEcoRI制限部位(下線部)を含有し、一方、3’オリゴヌクレオチドはEcoRV制限部位(下線部)を含有した。Mtb39の全長コード配列を増幅し、消化し、第1ステップであらかじめ消化されたプラスミドを使用してMtb32cの下流にインフレームでサブクローニングした。
【0239】
Mtb32のN末端断片の5’および3’オリゴヌクレオチドを以下のように設計した:5’−(5’−CTA−ATC−GAT−ATC−GCC−CCG−CCG−GCC−TTG−TCG−CAG−GAC−3’)および3’(5’−CTA−ATC−GAT−ATC−cta−GGA−CGC−GGC−CGT−GTT−CAT−AC−3’)。両セットのオリゴヌクレオチドはEcoRV制限部位(下線部)を含有し、一方、3’オリゴヌクレオチドは停止コドン(小文字)をさらに含んだ。このタンパク質の予測されるN末端ドメインをコードするMtb32の585bp部分を増幅するように該オリゴヌクレオチドを設計した。得られたPCR産物をMtb32c−Mtb39融合プラスミドにサブクローニングした。次いで、適正な挿入方向および突然変異の不存在をDNAシークエンシングによって確認した。
【0240】
Master Cell BankおよびManufacturer's Working Cell Bankを作成するために使用される最終構築物では、6×His親和性タグをPCRによって除去し、Mtb72fのオープンリーディングフレーム(ORF)を、pET由来の発現ベクターであるpPDMにサブクローニングした。該ORFは約72kDaのポリタンパク質(Mtb72f)をコードし、直列順序:Mtb32C−Mtb39−Mtb32Nで構成されたドメインを有する。次いでこのDNAを大腸菌のHMS174 pLysS株に形質転換し、試験、細胞バンク作成、および製造に使用した。
【0241】
Mtb72fバルク原体(Bulk Drug Substance)の生産
Mtb72fを生産するための製造プロセスを以下にまとめる:
・発酵、次いで遠心分離による細胞収集、細胞破壊(microfluidizer)および遠心分離による封入体ペレットの取得;
・8M尿素中での抽出による封入体ペレットの精製、次いでQ Sepharose Fast Flow(QFF)クロマトグラフィー、Ceramic Hydroxyapatite(CHT)クロマトグラフィー、ダイアフィルトレーション(diafilitration)、および滅菌ろ過による精製バルク原体の取得。
【0242】
発酵
発酵は10Lの作業容量で実施する。37℃で一晩培養された作業用種細胞の300mLの振とうフラスコ培養を発酵槽に接種する。接種および発酵ではともに、主要炭素源である植物由来グリセロールを含む半合成培地(semidefined medium)を使用する。培地の組成は以下の表に示される通りである。すべての培地成分を121℃で20分間加熱するか、あるいは滅菌ろ過によって滅菌する。発酵中、発酵槽を37℃の温度で維持する。5標準リットル/分(SLPM)の割合で空気を混入する。酸(H2SO4)または塩基(NaOH)の自動添加によって培地のpHを7.0で維持する。200rpmの最小撹拌を維持しつつ撹拌を自動調節することによって、溶存酸素を30%に制御するように発酵槽をプログラムする。1.05%SAG−471シリコーン消泡剤(Witco Corp.)の自動添加によって発酵槽内の発泡抑制を達成する。細胞密度が約3.5の光学濃度(600nm)に達したら、イソプロピル−β−D−チオガラクトピラノシド(IPTG)を1.0mMの濃度まで発酵槽に加える。IPTGは、Mtb72fタンパク質をコードする組換え遺伝子の発現を誘導する。誘導後3.0時間の時点で、発酵槽を冷却し、1L遠心分離ボトル(centrifuge bottles)中で遠心分離することによって細胞を収集する。
【表1】
【0243】
封入体の単離
細胞ペレットを、2.3Lの溶解バッファー(50mM NaCl、10mM Tris pH8.0)中に再懸濁してプールし、M-l 10Y Microfluidizer(登録商標)を使用して細胞を破壊する。細胞は、11,000±1,000psiの圧力で5回、Microfluidizerを通過させる。500mLボトル中で8000×gで懸濁液を遠心分離する。これらの条件下で、Mtb72fタンパク質を含有する封入体(IB)はペレットになり、ほとんどの細胞片は上清中に残る。IBペレットを洗浄バッファー(2M尿素、50mM NaCl、10mM Tris pH8.0)に再懸濁した後、8,000gで遠心分離する。上清画分を廃棄し、追加の精製に必要とされるまで−70℃〜−80℃でIBペレットを保存する。
【0244】
ポリタンパク質の精製
凍結IB調製物を37℃で15分間解凍した後、穏やかな機械的撹拌を使用して、8M尿素、50mM NaCl、20mM Bis−trisプロパン、pH7.0(バッファーA)に再懸濁する。そして再懸濁されたIBを、マグネチックスターラーバーで300rpmで2時間室温で撹拌する。次いでIB抽出物を高速で遠心分離し、得られた上清画分を0.45uMフィルター(Pall, Supor)を通してろ過した後、クロマトグラフィーによって分画する。
【0245】
あらかじめ1N NaOHで衛生化され、次いでバッファーAで平衡化されたQ Sepharose Fast Flow(QFF)アニオン交換樹脂(10×12.5cm、Amersham/Pharmacia BPG; 1L充填床)を含有するカラムにIB抽出物をアプライする。バッファーAで60cm/時間の線流速でカラムを展開し、主に低質量の混入物を含有するフロースルーを参照用に回収する。8M尿素、90mM NaCl、20mM Bis−trisプロパン、pH7.0を使用してMtb72fのバルクを1ステップで溶出し、吸光度に基づいて単一のバルクピークとして回収する。
【0246】
QFF樹脂は、精製時に使用される条件下で正に帯電する第4級アミン官能基を有する高度に架橋されたアガロース樹脂である。荷電マトリックスは、種々のアニオンの結合を可能にし、そして該アニオンは、塩勾配を使用して選択的に溶出させることができる。このアニオン交換クロマトグラフィーを使用して、該タンパク質から、樹脂に密接に結合している核酸および内毒素を分離する。該タンパク質は弱く結合し、これらの混入物より前に溶出される。さらに、このステップでは、無電荷の混入物および大部分のタンパク質不純物が除去される。
【0247】
QFFカラムからの90mM NaCl溶出液ピークを、MacroPrep(登録商標)セラミックヒドロキシアパタイト(CHT)(タイプI、40uM、BioRad)を含有するカラム(2.6×12cm、Amersham/Pharmacia XK26/20; 63mL充填床)にアプライする。該カラムは、1N NaOHを使用してあらかじめ衛生化され、次いでバッファーC(8M尿素、250mM NaCl、および20mM Bis−trisプロパン、pH7.0)で平衡化されている。大部分のMtb72fを含有し、混入物を含まないフロースルー材料(FT1)を回収する。カラムをバッファーCで洗浄し、残ったすべてのUV吸収材料を回収する。最後に、バッファーD(8M尿素、200mMリン酸ナトリウム、pH7.4)中でカラムを溶出させる。
【0248】
MacroPrep(登録商標)CHTは、球状の、マクロ細孔状のヒドロキシアパタイト[Ca5(PO4)3OH]2である。CHTクロマトグラフィーは、適正な結合条件および溶出条件を見出せば、非常に選択的な精製方法でありうる。結合様式には、荷電カルシウムおよびリン酸イオンとのイオン交換タイプの結合ならびに分子のキレート化が含まれる。DNAはこの樹脂に結合し、個別タンパク質に関する高い選択性を達成することができる。Mtb72fの精製に使用される条件は洗練ステップとして機能し、検出可能な宿主細胞混入物の実質的に完全な除去を可能にする。
【0249】
クロマトグラフィーによる分離中に、紫外(UV)吸光度、伝導率、圧力、pH、流速、および室温をモニターし、記録する。初期CHTフロースルー材料(FT1)をさらなる下流の処理に使用する。
【0250】
ダイアフィルトレーションおよび滅菌ろ過
CHT FT1プールに対してダイアフィルトレーションを実施して、尿素を除去し、バッファーを20mM Tris pH7.5で置き換える。ダイアフィルトレーションは、30kDa分子量カットオフ(MWCO)の限外ろ過メンブレンを用いて、LV-CentramateTMタンジェンシャルフローろ過デバイスを含むPall MinimTMシステムを使用して実施する。20mM Tris pH7.5中のMtb72f溶液を、0.2um滅菌フィルター(Millipak 40)を使用してろ過滅菌する。50mLの溶液を滅菌60mL PETG(ポリエチレンテレフタラートコポリマー)培地ボトル中に分配した後、−70℃で凍結および保存する。この材料がMtb72f精製済みバルク原体である。
【0251】
実施例2: Mtb72f(6Hisタグ)(配列番号2)の調製
Hisタグを除去するためのpPDMへのサブクローニングのステップを省略することを除き、実施例1の方法にしたがってよい。
【0252】
実施例3: M72(2Hisタグ)(配列番号4)の調製
M72発現ベクターの構築
M72抗原の構築用の出発材料は組換えプラスミド6His−Mtb72fmutであった。6His−Mtb72f組換えプラスミド(実施例1を参照のこと)を鋳型として使用する部位特異的突然変異誘発によって、6His−Mtb72fmutを調製した。部位特異的突然変異誘発には、配列番号1の710位のSerに対するコドンをAlaに対するコドンで置き変えることが含まれた。
【0253】
「Gene Tailor Site-Directed Mutagenesis System」(Invitrogen)を用いて、6His−Mtb72fmut構築物(Corixaプラスミド)上に存在する4個のN末端ヒスチジンの欠失を達成し、予測される2His−Mtb72Fmut構築物を得た。配列確認後、該プラスミドから(酵素による制限処理によって)2His−Mtb72fmutコード配列を切り出し、ゲル精製し、pET29a発現ベクターにライゲーションして、最終的な組換えプラスミドpET29a/2His−Mtb72fmutを得た。配列確認後、その組換えプラスミドに正式名称pRIT15497を付与し、HMS174(DE3)宿主細胞の形質転換に使用した。pRIT15497は、M72と称される725アミノ酸のタンパク質をコードする。
【0254】
M72タンパク質の生産
M72の生産に関しては、発酵培地中にクロラムフェニコールが存在しないことを除き、Mtb72fに関する記載(実施例1を参照のこと)と同一の生産プロセスを使用してよい。
【0255】
生物学的実施例1: 不活性/潜伏期の結核菌感染のマウスモデル
潜伏性結核菌感染のマウスモデルを確立するために、SWR系統を使用した。SWRマウスは免疫無防備状態ではないが、補体成分C5の分泌欠損である(Ooi and Colten, Nature (1979) 282:207-8を参照のこと)。SWRマウスは慢性状態のMtb感染を確立できないが、大型類上皮細胞(epitheloid)および泡沫状マクロファージ(クリスタロイド封入体を伴う)(貪食された好中球または好酸球由来顆粒)、多発性壊死、好中球蓄積およびリンパ球欠乏を特徴とするびまん性肉芽腫性肺炎(diffuse granulomatous pneumonia)を発症する(Turner, et al., J Submicrosc Cytol Pathol. (2001) 33(l-2):217-9; およびTurner, et al., Infect Immun. (2003) 71(9):5266-72を参照のこと)。潜伏性結核菌感染のモデルにおけるAS01Bアジュバントと共に製剤化されたMtb72f(配列番号6)の治療効力を評価するために、Swiss Webster(SWR/J)マウス系統を使用するプロトコルを以下に挙げる。コレステロール(25μg)(WO96/33739)およびモノホスホリル脂質A(MPL)(5μg)を膜中に含有するジオレオイルホスファチジルコリン(100μg)の小型単層小胞(SUV)にQS21(5μg)を加えることによって、2倍強度AS01Bを調製する(米国特許公開第2003/0143240号を参照のこと)。バッファー(PBS pH6.8)中の4μgのタンパク質を50μlの2倍強度AS01Bと混合することによって、注射用アリコート(50μl)を調製する。各マウスに2回の50μl注射(すなわち8μgのタンパク質)を施した。
【0256】
潜伏性結核菌感染のモデルを確立するための代表的時間軸は図1に模式的に示される通りである。
【0257】
1日目:50〜100コロニー形成単位(CFU)の結核菌生物体をエアロゾルによって感染させる
30〜90日目:飲料水1リットルあたり50mgリファンピン/85mgイソニアジドで一部のマウスを処置する
61日目:ワクチン候補5を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
82日目:ワクチン候補を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
103日目:ワクチン候補を施されたすべてのマウスをrMtb72f+AS01Bで免疫化しなければならない
113日目:IgGアッセイのために採血する
諸時点:CFUの数え上げおよび免疫原性のために脾臓および肺を採取する。
【0258】
バリエーション1→60日間の化学療法での処置。30日目に開始→3、4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0259】
バリエーション2→90日間の化学療法での処置。30日目に開始→4、5か月間の休止→各時点で2マウスのCFUおよび7マウスを生存研究のために残す。
【0260】
バリエーション3→4、5、6か月間の休止→各時点で2マウスのCFUおよび4マウスを生存研究のために残す。
【0261】
バリエーション4→60日間の化学療法での処置。30日目に開始→60日目に開始される筋肉内(i.m.)r72F+AS01Bでの3回の免疫化→3、4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0262】
バリエーション5→90日間の化学療法での処置。30日目に開始→60日目に開始されるr72F+AS01B(i.m.)での3回の免疫化→4、5か月間の休止→各時点で2マウスのCFUおよび4〜7マウスを生存研究のために残す。
【0263】
ELISAプレートをコーティングするためにrMtb72fを使用する感染後の抗体応答の分析では、化学療法およびMtb72f+AS01B免疫化の組み合わせを施された群が、無処置または化学療法処置のみを施されたマウス(0.5未満のOD)より高い抗体応答(2.0までのOD)を有することが示された(図2)。Mtb72fで免疫化されたマウスは、60日間の化学療法を施されたか90日間の化学療法を施されたかにかかわらず、かなりのMtb72f特異的抗体応答(1.5〜2.5の範囲のOD)に達した(図3)。
【0264】
マウスに結核菌を感染させた後、種々の間隔でマウスから脾臓細胞を収集した。その脾細胞をin vitroで組換え抗原で再刺激し、IFN−γ分泌を測定した。これらの細胞によって産生されるIFN−γレベルは、Mtb39を除き、60日目の群1(無処置)および群2(化学療法のみ)では一様に無視できた。conA、PPDおよびBCG溶解物での陽性対照刺激では、細胞が他の刺激分子に応答してIFN−γを合成および分泌可能であることが実証された(図4)。IFN−γレベルは、Mtb72f+AS01Bを施された群で高かったが、Mtb72f+AS01Bで免疫化されなかった群では、化学療法が施されたかどうかにかかわらず、それらは低いか、あるいは無視できた(図5)。
【0265】
結核感染および以後の処置経過中に、特異的T細胞が応答する。IFN−γに対する細胞内サイトカイン染色を使用して、Mtb72Fに対する特異的CD4+細胞応答のパーセンテージを測定した(図6)。このアッセイによって測定した場合、化学療法単独の経過中にはいずれの時点でも、Mtb72F特異的CD4+ IFNγ+ T細胞応答は変化しないようであった(図7)。Mtb感染後120日目の時点で、Mtb72f+AS01Bワクチンを施された群において、Mtb72Fに対するCD4+ IFNγ+ 応答の傾向は、化学療法中の期間の長さに伴って増加するように見えた(図7)。
【0266】
発明者らの実験の結果は、SWRマウスが結核菌の感染に対して感受性であることを実証する。無処置のままにすると、Mtb感染後115日目までにSWRマウスは死にいたる(図8および図9)。60日間の併用化学療法を施されたマウスの平均生存期間は170日間であった(図8および図9)。60日間の併用化学療法およびMtb72f/AS01Bでの3回の免疫化を施されたマウスの平均生存期間は215日であった(図8および図9)。化学療法を施されたマウスの群に関する生存率は、化学療法およびMtb72f/AS01Bワクチンを施された群と有意に異なる(95%信頼区間(p=0.0067))。
【特許請求の範囲】
【請求項1】
被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌(Mycobacterium tuberculosis)に感染している哺乳動物に、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含み、該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止するものである、上記方法。
【請求項2】
前記哺乳動物が結核菌の活発な感染を有している、請求項1に記載の方法。
【請求項3】
前記哺乳動物が結核菌の潜伏感染を有している、請求項1に記載の方法。
【請求項4】
前記哺乳動物が結核菌の多剤耐性株に感染している、請求項1に記載の方法。
【請求項5】
前記哺乳動物が以前にカルメット・ゲラン桿菌(BCG)を用いて免疫化されている、請求項1に記載の方法。
【請求項6】
Mtb72fが結核菌(Mycobacterium tuberculosis)に由来するものである、請求項1に記載の方法。
【請求項7】
Mtb72fが配列番号2の残基8−729を含むポリペプチドである、請求項1に記載の方法。
【請求項8】
Mtb72fが配列番号2の残基1および8−729からなり、開始Met残基の後にHisタグが挿入されていてもよい、請求項7に記載の方法。
【請求項9】
Mtb72fが配列番号2に示されるポリペプチドである、請求項7に記載の方法。
【請求項10】
Mtb72fが配列番号6に示されるポリペプチドである、請求項7に記載の方法。
【請求項11】
Mtb72fが配列番号4の残基4−725を含むポリペプチドである、請求項1に記載の方法。
【請求項12】
Mtb72fが配列番号4の残基1および4−725からなり、開始Met残基の後にHisタグが挿入されていてもよい、請求項11に記載の方法。
【請求項13】
Mtb72fが配列番号4に示されるポリペプチドである、請求項11に記載の方法。
【請求項14】
前記哺乳動物がヒトである、請求項1に記載の方法。
【請求項15】
前記アジュバントが、リポソーム製剤中の3D−MPLおよびQS21ならびに水中油型エマルジョン中の3D−MPLおよびQS21からなる群より選択される、請求項1に記載の方法。
【請求項16】
結核菌感染の治療に有効な1以上の化学治療剤の投与をさらに含む、請求項1に記載の方法。
【請求項17】
前記1以上の化学治療剤がイソニアジドおよびリファンピンより選択される、請求項16に記載の方法。
【請求項18】
前記哺乳動物が、まず一定期間にわたって1以上の化学療法剤を投与され、次に請求項1に記載の医薬組成物を投与される、請求項16に記載の方法。
【請求項19】
前記哺乳動物が、まず請求項1に記載の医薬組成物を投与され、次に一定期間にわたって1以上の化学療法剤を投与される、請求項16に記載の方法。
【請求項20】
1以上の化学療法剤の投与と、請求項1に記載の医薬組成物の投与とを同時に開始する、請求項16に記載の方法。
【請求項21】
続いて1回以上、請求項1に記載の医薬組成物を投与するステップをさらに含む、請求項1に記載の方法。
【請求項22】
初回免疫と、続いて結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を投与することによる追加免疫とを含む方法をさらに含む、請求項1に記載の方法。
【請求項23】
被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌(Mycobacterium tuberculosis)に感染している哺乳動物に、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる医薬組成物を免疫学的に有効な量で投与するステップを含み、発現される該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止するものである、上記方法。
【請求項24】
前記核酸が配列番号1である、請求項23に記載の方法。
【請求項25】
前記核酸が配列番号1のヌクレオチド63−2222を含む、請求項23に記載の方法。
【請求項26】
前記核酸が配列番号3である、請求項23に記載の方法。
【請求項27】
前記核酸が配列番号3のヌクレオチド10−2175を含む、請求項23に記載の方法。
【請求項28】
前記核酸をアデノウイルスベクター中で送達する、請求項23に記載の方法。
【請求項29】
前記核酸を、突然変異体マイコバクテリウム属またはバシラス属宿主細胞ベクター中で送達する、請求項23に記載の方法。
【請求項30】
初回免疫と、続いて結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片を投与することによる追加免疫とを含む方法をさらに含む、請求項23に記載の方法。
【請求項31】
結核菌(M. tuberculosis)感染に対する化学療法の期間を短縮するための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤と、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含み、該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導し、それにより結核菌感染に対する化学療法の期間を短縮することが可能になるものである、上記方法。
【請求項1】
被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌(Mycobacterium tuberculosis)に感染している哺乳動物に、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含み、該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止するものである、上記方法。
【請求項2】
前記哺乳動物が結核菌の活発な感染を有している、請求項1に記載の方法。
【請求項3】
前記哺乳動物が結核菌の潜伏感染を有している、請求項1に記載の方法。
【請求項4】
前記哺乳動物が結核菌の多剤耐性株に感染している、請求項1に記載の方法。
【請求項5】
前記哺乳動物が以前にカルメット・ゲラン桿菌(BCG)を用いて免疫化されている、請求項1に記載の方法。
【請求項6】
Mtb72fが結核菌(Mycobacterium tuberculosis)に由来するものである、請求項1に記載の方法。
【請求項7】
Mtb72fが配列番号2の残基8−729を含むポリペプチドである、請求項1に記載の方法。
【請求項8】
Mtb72fが配列番号2の残基1および8−729からなり、開始Met残基の後にHisタグが挿入されていてもよい、請求項7に記載の方法。
【請求項9】
Mtb72fが配列番号2に示されるポリペプチドである、請求項7に記載の方法。
【請求項10】
Mtb72fが配列番号6に示されるポリペプチドである、請求項7に記載の方法。
【請求項11】
Mtb72fが配列番号4の残基4−725を含むポリペプチドである、請求項1に記載の方法。
【請求項12】
Mtb72fが配列番号4の残基1および4−725からなり、開始Met残基の後にHisタグが挿入されていてもよい、請求項11に記載の方法。
【請求項13】
Mtb72fが配列番号4に示されるポリペプチドである、請求項11に記載の方法。
【請求項14】
前記哺乳動物がヒトである、請求項1に記載の方法。
【請求項15】
前記アジュバントが、リポソーム製剤中の3D−MPLおよびQS21ならびに水中油型エマルジョン中の3D−MPLおよびQS21からなる群より選択される、請求項1に記載の方法。
【請求項16】
結核菌感染の治療に有効な1以上の化学治療剤の投与をさらに含む、請求項1に記載の方法。
【請求項17】
前記1以上の化学治療剤がイソニアジドおよびリファンピンより選択される、請求項16に記載の方法。
【請求項18】
前記哺乳動物が、まず一定期間にわたって1以上の化学療法剤を投与され、次に請求項1に記載の医薬組成物を投与される、請求項16に記載の方法。
【請求項19】
前記哺乳動物が、まず請求項1に記載の医薬組成物を投与され、次に一定期間にわたって1以上の化学療法剤を投与される、請求項16に記載の方法。
【請求項20】
1以上の化学療法剤の投与と、請求項1に記載の医薬組成物の投与とを同時に開始する、請求項16に記載の方法。
【請求項21】
続いて1回以上、請求項1に記載の医薬組成物を投与するステップをさらに含む、請求項1に記載の方法。
【請求項22】
初回免疫と、続いて結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を投与することによる追加免疫とを含む方法をさらに含む、請求項1に記載の方法。
【請求項23】
被験体での結核の再活性化を予防または治療するための方法であって、既に結核菌(Mycobacterium tuberculosis)に感染している哺乳動物に、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片をコードする核酸を含んでなる医薬組成物を免疫学的に有効な量で投与するステップを含み、発現される該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導することにより、結核の再活性化を防止するものである、上記方法。
【請求項24】
前記核酸が配列番号1である、請求項23に記載の方法。
【請求項25】
前記核酸が配列番号1のヌクレオチド63−2222を含む、請求項23に記載の方法。
【請求項26】
前記核酸が配列番号3である、請求項23に記載の方法。
【請求項27】
前記核酸が配列番号3のヌクレオチド10−2175を含む、請求項23に記載の方法。
【請求項28】
前記核酸をアデノウイルスベクター中で送達する、請求項23に記載の方法。
【請求項29】
前記核酸を、突然変異体マイコバクテリウム属またはバシラス属宿主細胞ベクター中で送達する、請求項23に記載の方法。
【請求項30】
初回免疫と、続いて結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片を投与することによる追加免疫とを含む方法をさらに含む、請求項23に記載の方法。
【請求項31】
結核菌(M. tuberculosis)感染に対する化学療法の期間を短縮するための方法であって、既に結核菌に感染している哺乳動物に、結核菌感染に対して有効な1以上の化学療法剤と、結核菌群のマイコバクテリウム属の種に由来するMtb72f融合タンパク質またはその免疫原性断片およびアジュバントを含んでなる免疫学的に有効な量の医薬組成物を投与するステップを含み、該Mtb72f融合タンパク質が結核菌に対する免疫応答を誘導し、それにより結核菌感染に対する化学療法の期間を短縮することが可能になるものである、上記方法。
【図1】

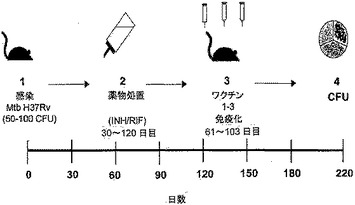
【図2】
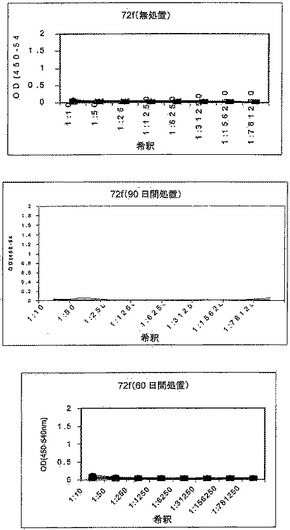
【図3】
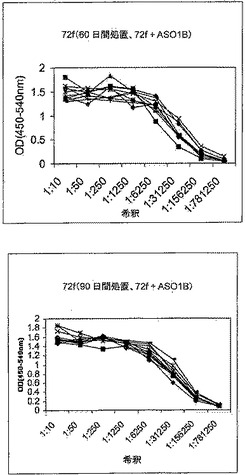
【図4】
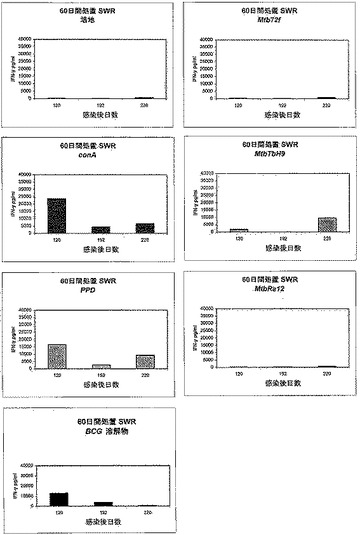
【図5】
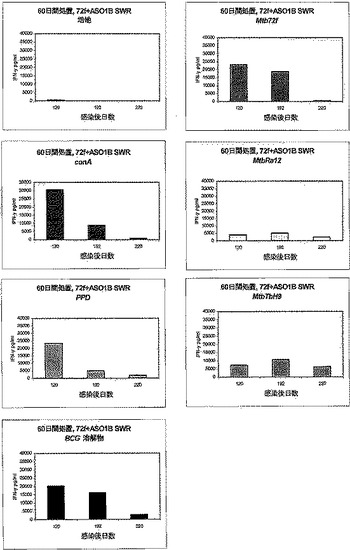
【図6A】
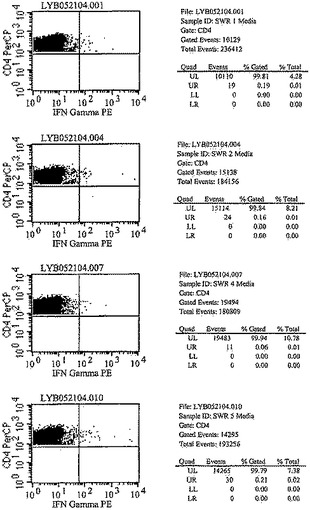
【図6B】
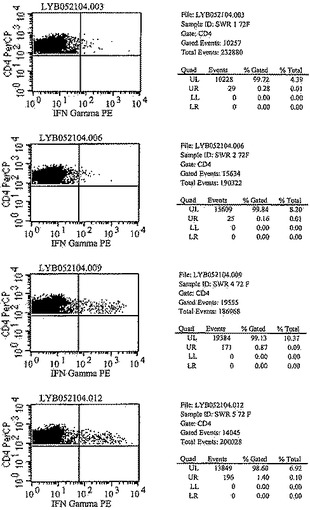
【図7】
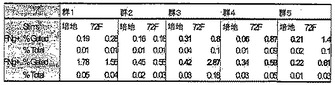
【図8】
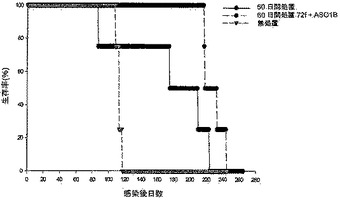
【図9】
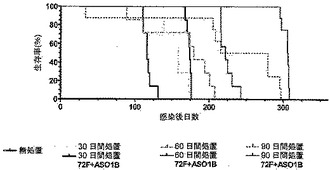
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【図8】
【図9】
【公開番号】特開2013−40190(P2013−40190A)
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願番号】特願2012−227192(P2012−227192)
【出願日】平成24年10月12日(2012.10.12)
【分割の表示】特願2008−508169(P2008−508169)の分割
【原出願日】平成18年4月27日(2006.4.27)
【出願人】(305060279)グラクソスミスクライン バイオロジカルズ ソシエテ アノニム (169)
【出願人】(507355113)インフェクティアス ディジーズ リサーチ インスティチュート(アイディーアールアイ) (3)
【Fターム(参考)】
【公開日】平成25年2月28日(2013.2.28)
【国際特許分類】
【出願日】平成24年10月12日(2012.10.12)
【分割の表示】特願2008−508169(P2008−508169)の分割
【原出願日】平成18年4月27日(2006.4.27)
【出願人】(305060279)グラクソスミスクライン バイオロジカルズ ソシエテ アノニム (169)
【出願人】(507355113)インフェクティアス ディジーズ リサーチ インスティチュート(アイディーアールアイ) (3)
【Fターム(参考)】
[ Back to top ]