
絨毛癌の治療剤
【課題】絨毛癌に対して治療効果を有する、絨毛癌に対する新規な治療剤を提供すること。
【解決手段】絨毛癌の治療剤脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する。この抑制剤の例としては、チロシンキナーゼ抑制剤、遊離のTrkB若しくはBDNFとの結合性を有するその断片又はTrkB若しくは前記断片を細胞内で生産する組換えベクター、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、BDNF又はTrkBに対する抗体、及びBDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクター等が挙げられる。
【解決手段】絨毛癌の治療剤脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する。この抑制剤の例としては、チロシンキナーゼ抑制剤、遊離のTrkB若しくはBDNFとの結合性を有するその断片又はTrkB若しくは前記断片を細胞内で生産する組換えベクター、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、BDNF又はTrkBに対する抗体、及びBDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクター等が挙げられる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、絨毛癌の治療剤に関する。
【背景技術】
【0002】
絨毛癌は、栄養芽細胞から誘導される高度に未分化で浸潤性の悪性腫瘍である。絨毛癌の治療は、現在、メトトレキセート、アクチノマイシン−D、エトポシド等の抗癌剤を用いる化学療法により行われている(非特許文献31)。確立された化学療法により多くの絨毛癌患者は寛解に至るが、それでも絨毛癌患者の5〜25%は抗癌剤により十分に治療されず、あるいは、一旦寛解しても再発する。
【0003】
一方、脳由来神経栄養因子(brain-derived neurotrophic factor (BDNF))は、高親和性チロシンキナーゼ(Trk)B受容体を全ニューロトロフィン低親和性共受容体p75(p75NTR)と共に活性化するタンパク質であるニューロトロフィンファミリーの1つである(非特許文献1)。BDNFが結合後、TrkB受容体シグナルは、フォスファチジルイノシトール 3-キナーゼ(PI3K)、MAPK/ERK, フォスフォリパーゼC-γ、及びプロテインCキナーゼカスケードを包含する複数のシグナル経路を活性化し、異なる種類の細胞の細胞増殖、分化及び生存において重要な役割を果たしている(非特許文献1、2)。ニューロトロフィンは、中枢神経系で広く発現し、ニューロンの生存と分化にとって重要である(非特許文献3)が、それらはまた、非ニューロン組織においても重要な役割を果たす(非特許文献4)。正常組織中での生理作用に加え、TrkBを介するニューロトロフィンシグナルが変更されることにより悪性腫瘍の形成及び転移が促進されることを示すデータが蓄積されている(非特許文献5)。TrkBは、ときどきそのリガンドであるBDNFと共に、神経芽細胞腫、ウィルムス腫瘍並びに前立腺及び膵臓腺癌を包含する種々のヒト癌、並びに多発性骨髄腫及び非固形癌において過剰発現している(非特許文献5)。これらのデータは、腫瘍の発達のためのBDNF/TrkBシグナルの潜在的なオートクリン役割を示唆している。しかしながら、腫瘍形成中における内発性BDNF/TrkBシグナルの分子機構を利用した研究はほとんどない。
【0004】
本願発明者らは、浸潤性栄養芽細胞に分化することができる、胚盤胞段階の胚の栄養外胚葉細胞中でのTrkB及びそのリガンドであるBDNFの発現を最近見出した。本願発明者らはまた、着床前の栄養外胚葉細胞の増殖及び生存におけるBDNFの促進効果を示した(非特許文献6)。着床後も胎盤栄養芽細胞中でTrkBとそのリガンドの発現は持続する。本願発明者は、妊娠中の胎盤発達における栄養芽細胞細胞の成長と生存におけるTrkBシグナル系のオートクリン/パラクリン調節役割を示した(非特許文献7)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Barbacid M 1994 The Trk family of neurotrophin receptors. J Neurobiol 25:1386-1403
【非特許文献2】Huang EJ, Reichardt LF 2003 Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609-642
【非特許文献3】Jones KR, Farinas I, Backus C, Reichardt LF 1994 Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989-999
【非特許文献4】Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD 1993 Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron 10:137-149
【非特許文献5】Desmet CJ, Peeper DS 2006 The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cell Mol Life Sci 63:755-759
【非特許文献6】Kawamura K, Kawamura N, Fukuda J, Kumagai J, Hsueh AJ, Tanaka T 2007 Regulation of preimplantation embryo development by brain-derived neurotrophic factor. Dev Biol 311:147-158
【非特許文献7】Kawamura K, Kawamura N, Sato W, Fukuda J, Kumagai J, Tanaka T 2009 Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology 150:3774-3782
【非特許文献8】Kawamura K, Kawamura N, Mulders SM, Sollewijn Gelpke MD, Hsueh AJ 2005 Ovarian brain-derived neurotrophic factor (BDNF) promotes the development of oocytes into preimplantation embryos. Proc Natl Acad Sci USA 102:9206-9211
【非特許文献9】Kawamura K, Sato N, Fukuda J, Kodama H, Kumagai J, Tanikawa H, Nakamura A, Honda Y, Sato T, Tanaka T 2003 Ghrelin inhibits the development of mouse preimplantation embryos in vitro. Endocrinology 144:2623-2633
【非特許文献10】Klein R, Conway D, Parada LF, BarbacidM1990 The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61:647-656
【非特許文献11】Tapley P, Lamballe F, BarbacidM1992 K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene 7:371-381
【非特許文献12】Ross AH, McKinnon CA, Daou MC, Ratliff K, Wolf DE 1995 Differential biological effects of K252 kinase inhibitors are related to membrane solubility but not to permeability. J Neurochem 65:2748-2756
【非特許文献13】Liu D, Li C, Chen Y, Burnett C, Liu XY, Downs S, Collins RD, Hawiger J 2004 Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem 279:48434-48442
【非特許文献14】Kawamura K, Fukuda J, Shimizu Y, Kodama H, Tanaka T 2005 Survivin contributes to the anti-apoptotic activities of transforming growth factor α in mouse blastocysts through phosphatidylinositol 3' kinase pathway. Biol Reprod 73:1094-1101
【非特許文献15】Kawamura K, Fukuda J, Kumagai J, Shimizu Y, Kodama H, Nakamura A, Tanaka T 2005 Gonadotropin-releasing hormone I analog acts as an antiapoptotic factor in mouse blastocysts. Endocrinology 146:4105-4116
【非特許文献16】Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, Nagashima Y, Kubota Y 2006 Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res 66:3699-3705
【非特許文献17】Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G 1999 Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441-446
【非特許文献18】BrodeurGM2003 Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3:203-216
【非特許文献19】McAllister AK, Katz LC, Lo DC 1999 Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22:295-318
【非特許文献20】Hamada AL, Nakabayashi K, Sato A, Kiyoshi K, Takamatsu Y, Laoag-Fernandez JB, Ohara N, Maruo T 2005 Transfection of antisense chorionic gonadotropin β gene into choriocarcinoma cells suppresses the cell proliferation and induces apoptosis. J Clin Endocrinol Metab 90:4873-4879
【非特許文献21】RaffMC1992 Social controls on cell survival and cell death. Nature 356:397-400
【非特許文献22】Wang X 2001 The expanding role of mitochondria in apoptosis. Genes Dev 15:2922-2933
【非特許文献23】KaplanDR,MillerFD2000 Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10:381-391
【非特許文献24】Yuan J, Yankner BA 2000 Apoptosis in the nervous system. Nature 407:802-809
【非特許文献25】Simpson PJ, Moon C, Kleman AM, Connolly E, Ronnett GV 2007 Progressive and inhibitory cell cycle proteins act simultaneously to regulate neurotrophin-mediated proliferation and maturation of neuronal precursors. Cell Cycle 6:1077-1089
【非特許文献26】Li J, Melvin WS, Tsai MD, Muscarella P 2004 The nuclear protein p34SEI-1 regulates the kinase activity of cyclin-dependent kinase 4 in a concentration-dependent manner. Biochemistry 43:4394-4399
【非特許文献27】Jin M, Udagawa K, Miyagi E, Nakazawa T, Hirahara F, Yasumitsu H, Miyazaki K, Nagashima Y, Aoki I, Miyagi Y 2001 Expression of serine proteinase inhibitor PP5/TFPI-2/MSPI decreases the invasive potential of human choriocarcinoma cells in vitro and in vivo. Gynecol Oncol 83:325-333
【非特許文献28】Krause DS, Van Etten RA 2005 Tyrosine kinases as targets for cancer therapy. N Engl J Med 353:172-187
【非特許文献29】Kawamura K, Kawamura N, Mulders SM, Sollewijn Gelpke MD, Hsueh AJ 2005 Ovarianbrain-derived neurotrophic factor (BDNF) promotes the development of oocytes intopreimplantation embryos. Proc Natl Acad Sci U S A 102:9206-9211
【非特許文献30】Hamada AL, Nakabayashi K, Sato A, Kiyoshi K, Takamatsu Y, Laoag-Fernandez JB,Ohara N, Maruo T 2005 Transfection of antisense chorionic gonadotropin beta gene intochoriocarcinoma cells suppresses the cell proliferation and induces apoptosis. J Clin Endocrinol Metab 90:4873-4879
【非特許文献31】石川 哲也ら、症例報告 日産婦関東連会報44:9-14, 2007
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、絨毛癌に対して治療効果を有する、絨毛癌に対する新規な治療剤を提供することである。
【課題を解決するための手段】
【0007】
本願発明者らは鋭意研究の結果、BDNF及び/又はTrkBの作用を抑制することにより絨毛癌細胞の増殖を抑制することができ、絨毛癌が治療可能であることを見出し本発明を完成した。
【0008】
すなわち、本発明は、脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する絨毛癌の治療剤を提供する。
【発明の効果】
【0009】
本発明により、絨毛癌に対して優れた治療効果を有する新規な絨毛癌治療剤が提供された。
【図面の簡単な説明】
【0010】
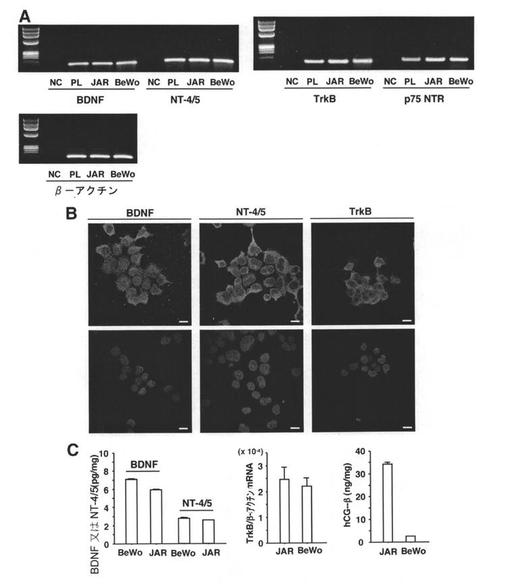
【図1】絨毛癌細胞におけるBDNF、NT−4/5、TrkBの発現を示す図である。Aでは、TrkBリガンドおよび受容体のmRNAの発現をRT−PCRにより検出した。β−アクチンのレベルをローディングコントロールとして使用した。ヒト胎盤cDNAをポジティブコントロールとして使用し、ネガティブコントロールには鋳型DNAを含めなかった。PLは胎盤の略、NCはネガティブコントロールの略である。Bは、BDNFタンパク質、NT−4/5タンパク質、TrkBタンパク質を免疫組織化学的に検出したものである。JAR細胞において、BDNF、NT−4/5、TrkBの特異的なシグナルが認められる。下段はネガティブコントロールである。細胞の核は、ヘキスト33342を用いて青色に染色した。スケールバーは20μmを表す。BeWo細胞においても同様の染色パターンが観察された(データ非掲載)。Cは、BDNFタンパク質とNT−4/5タンパク質、TrkB転写物、またはhCG−βタンパク質のレベルを、それぞれ、ELISA(左)、リアルタイムRT−PCR(真ん中)、またはRIA(右)を用いて定量化したものである。サンプルは異なる培養液中で取得した(n=6)。TrkBのmRNAレベルは、同一サンプル中のβ−アクチンの転写レベルを用いて正規化した。柱が平均を、バーが標準誤差を示している。
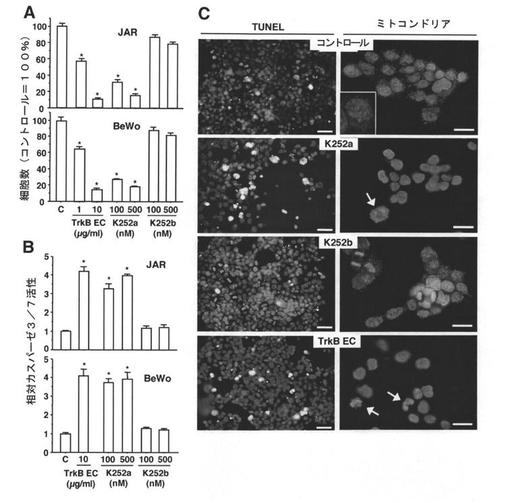
【図2】内在性TrkBリガンドのin vitroの絨毛癌細胞成長における役割を示す図である。内在性TrkBリガンドの抑制の、細胞増殖への影響(A)および生存への影響(B)。JAR細胞およびBeWo細胞を、培地のみ[コントロール(C)]で、種々の用量のTrkBの細胞外ドメイン(TrkB EC)とともに、K252aまたはK252bとともに培養した。細胞数はトリパンブルー排除試験を用いて測定し、細胞のアポトーシスはカスパーゼ3/7アッセイを用いて定量化した(n=5)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。Cは、DNA断片化および機能的なミトコンドリアの減少を、それぞれin situ TUNEL染色およびMitoTracker Orange CMTMRosを用いて検出したものである。掲載されている画像は、TrkBの細胞外ドメイン(10μg/ml)、K252a(500nM)、またはK252b(500nM)で処理した、または処理しなかったJAR細胞から得た。TUNELアッセイでは、細胞の核酸を、ヨウ化プロピジウムを用いて赤色に染色した。陽性アポトーシスシグナル数(緑色蛍光)は、TrkBの細胞外ドメインまたはK252a処理をした細胞において増加した。スケールバーは50μmを表す。ミトコンドリアアッセイでは、細胞の核をヘキスト33342で青色に染色した。コントロールグループまたはK252bグループの細胞は点状の染色パターン(赤色)を示し、TrkBの細胞外ドメインおよびK252aで処理した細胞は不鮮明な染色パターンを示す。挿入図は、より高い倍率に拡大した画像である。TrkBの細胞外ドメインまたはK252aで処理した細胞のいくつかは、クロマチン凝縮と核断片化とで特徴付けられるアポトーシスの形態学的特徴(矢印)を示す。スケールバーは25μmを表す。同様の結果がBeWo細胞でも観察された(データ非掲載)。
【図3】PI3KおよびERKシグナル伝達経路を介した絨毛癌細胞の細胞増殖のBDNFによる促進。細胞を、培地のみで、もしくは種々の用量のBDNFとともに培養し、またはBDNFなし[コントロール(C)]もしくはBDNF(30ng/ml)とともに、PI3KインヒビターであるLY294002(LY2)、もしくはその不活性類似体であるLY303511(LY3)の存在下または非存在下で培養した。いくつかの実験においては、細胞を、MEK1/2のインヒビターであるU0126、もしくはその不活性類似体であるU0124、またはLY294002とU0126の両方か、LY303511とU0124の両方の存在下または非存在下において、BDNFで処理した。培養後48時間で、トリパンブルー排除試験を用いて細胞数を測定し、BDNF処理の細胞増殖への影響(A)と、BDNFによる細胞増殖刺激におけるPI3KおよびERK経路の関与(B)を評価した(n≧3)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。
【図4】絨毛癌細胞において内在性TrkBシグナル伝達の抑制によって制御される細胞周期関連遺伝子およびアポトーシス関連遺伝子の同定結果を示す図である。JAR細胞をTrkBの細胞外ドメイン(500nM)の存在下または非存在下で培養した。培養後8時間で、細胞周期について(A)およびアポトーシスについて(B)のPCRに基づく遺伝子アレイ解析を行った。柱は、TrkBの細胞外ドメインでの処理をしなかったサンプルと比較した、3つの別のサンプルにおける個々の遺伝子発現の平均倍率(fold difference)を表している。すべての差が統計的に有意(P>0.05)であった。
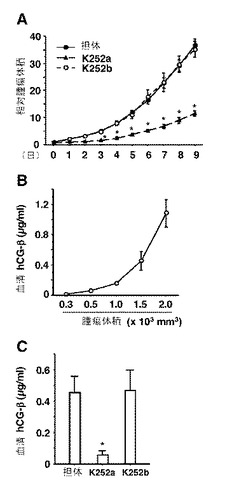
【図5】ヌードマウス中のJAR細胞異種移植片の、in vivoでの腫瘍成長に対するTrk受容体の効果を示す図である。A:K252aを投与したマウスにおける腫瘍成長の経時変化を示す。K252a又はK252b(500μg/kg)を3日毎に胸腺欠損マウス(n=9)に投与した。各マウスが投与を受けた最初の日における各腫瘍の体積を相対的腫瘍体積1と表した。点は平均、バーは標準誤差。*:P<0.05 vs.コントロール群。B:腫瘍成長の間における血清hCG-β濃度。異なる体積(n=4)の腫瘍を担持するマウスから血清試料を得た。血清hCG-β濃度はRIAにより測定した。点は平均、バーは標準誤差。C:治療9日目におけるマウス中の血清hCG-β濃度。カラムは平均、バーは標準誤差。*:P<0.05 vs.コントロール群。
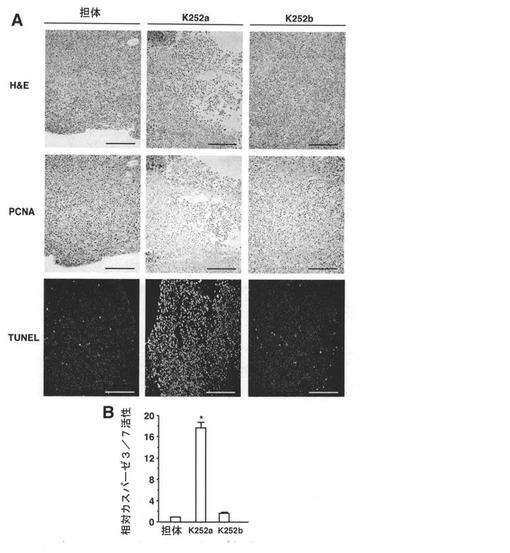
【図6】Trk受容体インヒビターによるin vivoでの腫瘍増殖の抑制及びヌードマウス中でのJAR細胞異種移植片の生存を示す図である。A:細胞増殖の組織学的特徴付け及び腫瘍におけるアポトーシスを示す。細胞増殖及びDNA断片化の特異的検出は、それぞれ、PCNA免疫染色及びin situ TUNEL染色を用いて行った。腫瘍を担持する動物に、K252a又はK252b(500μg/kg)を3日毎に投与し又は投与しなかった。図示の画像は、治療9日目の切除した腫瘍から得た。H&E染色は、K252a投与後の有糸分裂細胞数の減少及びクロマチンの凝縮を示している。K252a投与により、PCNAシグナル(茶色)は減少し、一方、TUNEL染色核(緑色蛍光)は増大した。スケールバー:200μm。B:K252a投与による、腫瘍中のcaspase-3/7活性の増大を示す。試料は、投与9日目のマウスから得た(n=9)。データは、タンパク濃度により正規化し、コントロール(担体のみ)に対する増加倍率で示し、1に正規化した。カラムは平均、バーは標準誤差。*:P<0.05 vs.コントロール群。
【図7】JAR細胞における、siRNAによって媒介されるBDNFおよびTrkBのノックダウンによる細胞成長の阻害を示す図である。細胞を、siRNAでトランスフェクションしなかった(コントロール、C)か、またはBDNF、TrkB、またはネガティブコントロールsiRNA(ネガティブコントロール、NC)でトランスフェクションした。Aは、siRNAによって媒介されたBDNFまたはTrkBの転写レベルの減少を示している。siRNAのトランスフェクション後72時間で、リアルタイムRT−PCRによりBDNFおよびTrkBのmRNAの発現を解析した。サンプルは異なる培養液中の細胞から取得した(n=3)。BDNFおよびTrkBのmRNAレベルは、同一サンプルのβ−アクチンの転写レベルを用いて正規化した。Bは、siRNAによって媒介されるBDNFおよびTrkBのノックダウンを経た、BDNF/TrkBシグナル伝達の抑制による細胞増殖への影響を示している。siRNAのトランスフェクション後72時間で培養液を無血清培地に交換し、次いで、インキュベーション後48時間で細胞数を、トリパンブルー排除試験を用いて測定した(n=4)。Cは、BDNF siRNAをトランスフェクションした細胞の、BDNFによる増殖促進を示している。siRNAをトランスフェクションした細胞を無血清培地において、BDNF非存在下(コントロール、C)またはBDNF(30ng/ml)存在下で培養した。培養後48時間で細胞数を、トリパンブルー排除試験により測定した(n=4)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。
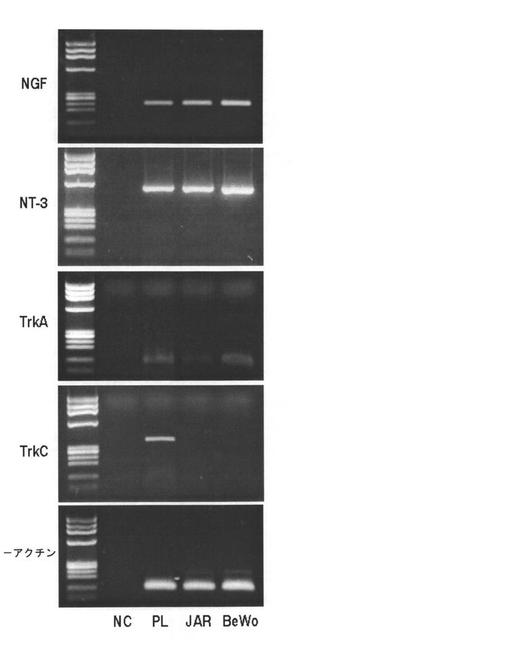
【図8】絨毛癌細胞におけるTrkリガンド(NGFおよびNT−3)と受容体(TrkAおよびTrkC)の発現を示す図である。JAR細胞およびBeWo細胞中のTrkBリガンドおよび受容体のmRNAの発現をRT−PCRにより検出した。β−アクチンのレベルをローディングコントロールとして使用する。ヒト胎盤cDNAをポジティブコントロールとして使用し、ネガティブコントロールには鋳型DNAを含めなかった。PLは胎盤の略、NCはネガティブコントロールの略である。
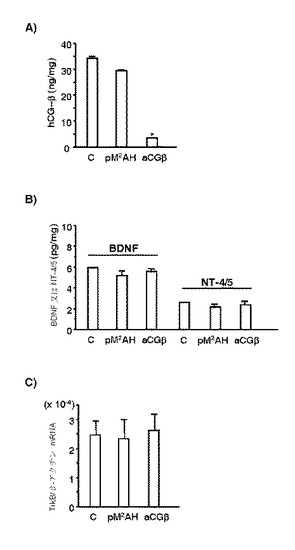
【図9】JAR細胞における、アンチセンスhCG−βのトランスフェクションによる、hCG−βタンパク質(A)、BDNFタンパク質およびNT−4/5タンパク質(B)、ならびにTrkB転写物(A)の発現レベルへの影響を示す図である。細胞を、トランスフェクションしなかった(コントロール、C)か、または、アンチセンスhCG−β(aCGβ)もしくは空ベクター(pM2AH)でトランスフェクションした。hCG−β、BDNFおよびNT−4/5、ならびにTrkBの転写レベルに対するタンパク質レベルを、それぞれRIA/ELISA、ならびにリアルタイムRT−PCRを用いて定量化した。サンプルは異なる培養液からの細胞から取得した(n=3)。TrkBのmRNAレベルは、同一サンプル中のβ−アクチンの転写レベルを用いて正規化した。柱が平均を、バーが標準誤差を示している。
【発明を実施するための形態】
【0011】
上記の通り、本発明の絨毛癌治療剤は、BDNF及び/又はTrkBの抑制剤を有効成分として含有する。ここで、「BDNF及び/又はTrkBの抑制剤」とは、(1)BDNF及びTrkBの少なくとも一方の生理作用を抑制する物質、(2) BDNFとTrkBの結合を抑制する物質、(3) BDNF及びTrkBの少なくとも一方の、細胞内における生産を抑制する物質を意味する。(1)の例として、チロシンキナーゼ抑制剤を挙げることができる。(2)の例として、(i)遊離のTrkB又はBDNFとの結合性を有するTrkB断片、及び、(ii)BDNF又はTrkBに対する抗体を挙げることができる。(3)の例として、(i)BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、及び(ii) BDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクターを挙げることができる。以下、これらについて説明する。
【0012】
TrkBは、チロシンキナーゼ活性を有しており、下記実施例に具体的に記載されるように、チロシンキナーゼ活性を抑制することにより絨毛癌治療効果が発揮される。従って、チロシンキナーゼ抑制剤(阻害剤)を、本発明の絨毛癌治療剤の有効成分として用いることができる。チロシンキナーゼ抑制剤は、既に種々のものが公知であり、市販されているものも少なくない。市販品を好ましく用いることができる。公知のチロシンキナーゼ抑制剤の例として、K252a、AZ-23(Wang et al. J Med Chem 2008, 51, 4672-84)、CEP-701(Cephalon Inc., West Chester, PA)、CEP-751(Kyowa Hakko Kogyo, Tokyo, Japan)、CEP-2563(Cephalon Inc.)及びCEP-7801(Somaiah et al. J Thorac Oncol,2009,4, S1045-83)等を挙げることができるがこれらに限定されるものではない。
【0013】
これらのうち、下記実施例において採用したK252aは、下記の化学構造を有する、土壌真菌により生産される物質であり、チロシンキナーゼ抑制剤として広く用いられており、市販されているので市販品を好都合に用いることができる。
【0014】
【化1】

【0015】
チロシンキナーゼ抑制剤を本発明の絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。投与量は、用いるチロシンキナーゼ抑制剤の種類や患者の状態等に応じて適宜設定されるが、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。
【0016】
チロシンキナーゼ抑制剤を本発明の絨毛癌治療剤の有効成分として用いる場合、本発明の絨毛癌治療剤は、上記チロシンキナーゼ抑制剤のみから成っていてもよいし、また、各投与形態に適した、薬剤的に許容される担体及び/又は希釈剤を用いて製剤することもできる。製剤方法及びそのための各種担体は、医薬製剤の分野において周知である。薬剤的に許容される担体又は希釈剤は、例えば、生理緩衝液のような緩衝液や、賦形剤(砂糖、乳糖、コーンスターチ、リン酸カルシウム、ソルビトール、グリシン等)であってよく、結合剤(シロップ、ゼラチン、アラビアゴム、ソルビトール、ポリビニルクロリド、トラガント等)、滑沢剤(ステアリン酸マグネシウム、ポリエチレングリコール、タルク、シリカ等)等が適宜混合されていてもよい。投与形態としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などによる経口剤、吸入剤、注射剤、座剤、液剤などによる非経口剤などを挙げることができる。これらの製剤は一般的に知られている製法によって作ることができる。
【0017】
上記の通り、BDNFとTrkBの結合を抑制する物質も本発明の絨毛癌治療剤の有効成分として用いることができる。このような物質としては、まず、遊離のTrkB又はBDNFとの結合性を有するTrkB断片を挙げることができる。遊離のTrkBは、BDNFと結合するので、遊離のTrkBを投与すると、投与したTrkBは、細胞膜上の本来のTrkBと競合してBDNFと結合するので、細胞膜上の本来のTrkBと結合するBDNFの量が減少する。すなわち、遊離のTrkBは、細胞自体のTrkBとBDNFとの結合を競合的に抑制する。また、細胞膜上のTrkBがBDNFと結合する部分は、TrkBの細胞外ドメインである。従って、下記実施例に具体的に記載するように、TrkBの細胞外ドメイン、又は該細胞外ドメインを含むTrkBの断片も全長TrkBと同様、BDNFとTrkBの結合を競合的に抑制するので、本発明の絨毛癌治療剤の有効成分として用いることができる。ヒトTrkB遺伝子のcDNAの塩基配列を、それがコードするアミノ酸配列と共に配列番号1に示し、アミノ酸配列のみを取り出したものを配列番号2に示す。なお、ヒトTrkB遺伝子のcDNA及びそれがコードするアミノ酸配列は公知であり、GenBank Accession No.NM_006180として登録されている。配列番号2に示すアミノ酸配列(すなわちTrkB全長のアミノ酸配列)のうち、細胞外ドメインは、N末端から-31番目のアミノ酸(以下、「-31aa」のように記載)〜397aaまでである。この細胞外ドメインから成るTrkB断片も本発明の絨毛癌治療剤の有効成分として用いることができる。一般に、ポリペプチドはサイズが小さい方が製造が容易で細胞に取り込まれやすいので、上記TrkB断片はこれらの観点から好ましい。
【0018】
一般に、生理活性を有するポリペプチドにおいて、少数のアミノ酸が置換し、欠失し又は挿入された場合であってもその生理活性が維持される場合があることは当業者にとって周知である。従って、上記したTrkB又はその断片に加え、アミノ酸配列が、配列番号2で示されるアミノ酸配列、及び該アミノ酸配列のうち-31aa〜397aaである細胞外ドメイン領域のアミノ酸配列と90%以上、好ましくは95%以上、さらに好ましくは99%以上の配列同一性を有するポリペプチドであって、BDNFと結合して絨毛癌の治療効果を発揮するポリペプチドもそれぞれ遊離のTrkBやその細胞外ドメイン断片と同様に本発明の絨毛癌治療剤の有効成分として用いることができる。ここで、アミノ酸配列の配列同一性とは、一致するアミノ酸残基の数が最大となるように(必要に応じてギャップを挿入する)、2つのアミノ酸配列を並べ、一致したアミノ酸残基の数を完全長の配列のアミノ酸残基(2つの配列間で全アミノ酸残基の数が異なる場合、長い方のアミノ酸残基)の数で除することよって求めた値を意味する。そのような相同性の計算は、BLASTのような周知のソフトウェアによって容易に入手し得る。特に、アミノ酸配列が、配列番号2で示されるアミノ酸配列、又は該アミノ酸配列のうち-31aa〜397aaである細胞外ドメイン領域のアミノ酸配列において、1個ないし数個のアミノ酸が置換し若しくは欠失し、又は1個ないし数個のアミノ酸が挿入され若しくは付加されたアミノ酸配列であるポリペプチドであって、BDNFとの結合性、ひいては絨毛癌の治療効果を有するポリペプチドも本発明の絨毛癌治療剤の有効成分として用いることができる。なお、天然のタンパク質を構成する20種類のアミノ酸は、低極性側鎖を有する中性アミノ酸(Gly, Ile, Val, Leu, Ala, Met, Pro)、親水性側鎖を有する中性アミノ酸(Asn, Gln, Thr, Ser, Tyr, Cys)、酸性アミノ酸(Asp, Glu)、塩基性アミノ酸(Arg, Lys, His)、芳香族アミノ酸(Phe, Tyr, Trp)のように類似の性質を有するものにグループ分けでき、これらの間での置換であればペプチドの性質が変化しないことが多いことが知られている。従って、配列番号2で示されるアミノ酸配列又はその細胞外ドメイン領域のアミノ酸配列から成るポリペプチド中のアミノ酸残基を置換する場合には、これらの各グループの間で置換することにより、当該ポリペプチドのBDNF結合能が維持される可能性が高くなる。
【0019】
また、生理活性を有する2種類のポリペプチドが連結された融合ポリペプチドが、各ポリペプチドの生理活性を維持する場合があることからも明らかなように、生理活性を有するポリペプチドをそっくり含み、その一端又は両端に他のアミノ酸配列が連結されたポリペプチドであってもその生理活性が維持される場合があることは当業者にとって周知である。従って、上記したBDNFとの結合能を有するポリペプチドを含み、BDNFとの結合能を有するポリペプチドを本発明の絨毛癌治療剤の有効成分として用いることも可能である。この場合、上記したBDNFとの結合能を有するポリペプチドの一端又は両端に付加されるアミノ酸の数は、最終的なポリペプチドがBDNFとの結合能、ひいては絨毛癌の治療効果を発揮する限り特に限定されないが、合成の容易さ及び単位重量当たりの活性を高くする観点から、1個〜数個であることが好ましい。
【0020】
なお、一般に、ポリペプチド製剤においては、生体内でのプロテアーゼによる分解を受けにくくするためにポリペプチドの一端にポリエチレングリコール(PEG)鎖等を結合したものが広く用いられている。本発明の絨毛癌治療剤においても、同様に、上記したポリペプチドをそっくり含み、その一端にPEG鎖等の安定化構造を付加したものを有効成分として用いることができる。なお、PEG化によりペプチドを安定化する場合には、PEGのサイズは分子量数千〜5万、好ましくは1万〜5万程度である。また、ポリペプチドの一端にPEGを結合する方法は周知である。
【0021】
なお、本明細書及び特許請求の範囲において、遊離のTrkB若しくはBDNFとの結合性を有するその断片の、絨毛癌の治療効果を有する「修飾体」とは、配列番号2で表されるアミノ酸配列又はその細胞外ドメイン領域から成るアミノ酸配列とは異なるアミノ酸配列を持ち、BDNFとの結合性ひいては絨毛癌の治療効果を有する、上記したポリペプチド並びにそれらにPEG鎖等の安定化構造を付加したものを意味する。
【0022】
上記した、遊離のTrkB、その細胞外ドメイン断片やそれらの上記修飾体(以下、便宜的に「BDNF結合性TrkB断片等」ということがある)を絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。もっとも、体内への吸収性や消化酵素による分解を避ける観点から非経口投与が好ましい。投与量は、用いるチロシンキナーゼ抑制剤の種類や患者の状態等に応じて適宜設定されるが、非経口投与の場合、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。また、BDNF結合性TrkB断片等を有効成分として用いる場合も、上記と同様、常法に基づいて製剤することができる。
【0023】
BDNF結合性TrkB断片等は、それ自体を有効成分として用いることができるが、BDNF結合性TrkB断片等をコードする核酸を組み込んだ組換えベクターであって、細胞中でBDNF結合性TrkB断片等を発現することができる組換えベクターを有効成分として用いることもできる。哺乳動物の遺伝子治療用のベクターは、種々のものが公知であり、市販されているものも少なくないので、市販の遺伝子治療用ベクターのクローニング部位にBDNF結合性TrkB断片等をコードするDNAを挿入した組換えベクターを好ましく用いることができる。なお、所望の遺伝子をベクターに挿入して遺伝子治療用組換えベクターを作製する有料サービスも行われており、このような有料サービスを利用することも可能である。
【0024】
哺乳動物への組換えベクターの投与自体は、周知の方法により行うことができる。すなわち、好ましくは、治療すべき絨毛癌細胞の近傍の組織に注射等の非経口投与により投与することができる。組換えベクターをリン酸緩衝液(PBS)等の緩衝液に懸濁したものを投与することができる。投与に際し、細胞内への遺伝子ワクチンの侵入を容易にするために、注射部位に電界パルスを与えてもよい。この場合、電界の強さは、特に限定されないが、通常、10V/cm〜60V/cm程度、好ましくは25V/cm〜35V/cm程度、パルスの持続時間は、通常、20ミリ秒〜100ミリ秒、好ましくは、40ミリ秒〜60ミリ秒程度であり、パルスを通常、1回〜6回、好ましくは2回〜4回程度当てることができる。組換えベクターの投与量は、症状や神経損傷部位の状態等に応じて適宜選択することができるが、通常、組換えベクターの重量で1ng〜10mg程度、特に100ng〜1mg程度である。
【0025】
BDNFとTrkBとの結合を抑制する物質として、BDNFに対する抗体又はBDNF結合性TrkB断片等に対する抗体を用いることもできる。BDNF及びBDNF結合性TrkB断片等は容易に入手可能であるので、これらに対する抗体は、BDNF又はTrkBを免疫原として動物(ヒトを除く)に投与するして抗体を誘導することを含む常法により得ることができる。抗体は、ポリクローナル抗体でもモノクローナル抗体でもよく、モノクローナル抗体も常法であるハイブリドーマ法により作製することができる。抗体は、BDNFとTrkBとの結合を抑制できるものである必要があるので、モノクローナル抗体の場合には、得られたモノクローナル抗体のうち、BDNFとTrkBとの結合を抑制するモノクローナル抗体をスクリーニングする。ポリクローナル抗体の場合には、免疫原の全エピトープに体する種々の抗体が含まれるので、このようなスクリーニングを行わなくてもBDNFとTrkBとの結合を抑制するが得られる。
【0026】
上記抗体を絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。もっとも、体内への吸収性や消化酵素による分解を避ける観点から非経口投与が好ましい。投与量は、用いる抗体の力価や患者の状態等に応じて適宜設定されるが、非経口投与の場合、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。また、上記抗体を有効成分として用いる場合も、上記と同様、常法に基づいて製剤することができる。
【0027】
BDNF又はTrkBの、細胞内における生産を抑制する物質を本発明の絨毛癌治療剤の有効成分として用いることもできる。このような物質として、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA(iRNA)を挙げることができる。BDNF遺伝子又はTrkB遺伝子の発現を抑制する抑制剤としては、BDNF遺伝子又はTrkB遺伝子のmRNAを標的とするiRNA、好ましくはsiRNAを挙げることができる。iRNAは、標的となるmRNAと相補的な鎖を含む二本鎖RNAであり、標的となるmRNAと結合してこれを切断するものである。siRNAは、サイズが21〜23塩基程度の短い(small)iRNAである。siRNAは、サイズが小さいので合成が容易で、それによるmRNAの切断部位を設定し易いので好ましい。siRNAによる遺伝子発現の抑制技術は、既に周知であり、mRNAの配列(cDNA配列)さえ提示すれば、それを標的とするsiRNAを設計し、そのsiRNAを発現ベクターに組み込んだ組換えベクターを作製するサービスを行なっている業者が多数存在するほどである。上記の通り、TrkB遺伝子のcDNAの配列は配列番号1に記載したとおりであり、また、BDNF遺伝子のcDNAの塩基配列(GenBank Accession No. NM_170735)は配列番号3に示す通りであるので、これらに対するsiRNAは当業者であれば容易に設定することができる。簡単に説明すると、siRNAは標的とするmRNAと相補的な鎖を含む二本鎖RNAで、そのサイズは通常、21〜23塩基であり、通常、二本鎖RNAの両端にそれぞれハングオーバーを有する。ハングオーバーのサイズは、それぞれ1塩基〜2塩基であり、ハングオーバー部分はデオキシヌクレオチドでもよい。また、mRNAとの相補性は、完全な相補性が好ましいが、1〜2塩基程度のミスマッチがあっても十分な切断作用を発揮する場合も多い。また、ハングオーバー部分は相補的でなくてもよい。siRNAは、mRNAの塩基配列中のaaに続く19〜21塩基として設定することが好ましい場合が多く、gc含量が50%前後(通常45〜55%程度)のものが好ましい。また、成熟タンパク質で切断される部分に設定されないように、5'末端から50塩基以上離れた部位に設定することが多い。
【0028】
siRNAはそのまま投与することもできるが、該siRNAを発現するDNAを哺乳動物細胞用の発現ベクターに組み込み、得られた組換えベクターを投与することにより、細胞内でsiRNAを生産させBDNF遺伝子又はTrkB遺伝子の発現を抑制してもよい。哺乳動物細胞用の発現ベクターは種々市販されており、それらのマルチクローニング部位に上記DNAを挿入することができる。なお、上記の通り、siRNAを発現するDNAを組み込んだ発現ベクターを作製する業者のサービスも利用できる。
【0029】
投与量は、絨毛癌の程度、患者の状態や体重等に応じて適宜選択されるが、抑制剤がsiRNAの場合、その投与量は、成人(体重60kg)1日当たり通常、0.01mg/kg〜10mg/kg程度、特に0.1mg/kg〜5mg/kg程度、siRNAを発現する組換えベクターの場合、治療全体を通して成人1日当たり0.01mg/kg〜10mg/kg程度、特に0.1mg/kg〜5mg/kg程度であるが、投与量はもちろんこれらに限定されるものではない。
【0030】
さらに、本発明の絨毛癌治療剤の有効成分として、BDNF遺伝子又はTrkB遺伝子のアンチセンスRNAを用いることもできる。アンチセンスRNAは、標的遺伝子のmRNAの全長又はその一部と相補的な塩基配列を有し、該mRNAとハイブリダイズして、mRNAが翻訳されることを抑制し、ひいては標的遺伝子の遺伝子産物が生産されることを抑制するものである。TrkB遺伝子及びBDNF遺伝子のcDNAの塩基配列はそれぞれ配列番号1及び配列番号3に記載されているので、これらのアンチセンスRNAも容易に調製することができる。アンチセンスRNAのサイズは、標的遺伝子のmRNAと特異的にハイブリダイズすることが可能で該mRNAの翻訳を抑制できるサイズであれば特に限定されないが、通常、20塩基〜mRNAのコード領域の全長程度である。
【0031】
iRNAの場合と同様、アンチセンスRNAもそのまま投与することもできるが、該アンチセンスRNAを発現するDNAを哺乳動物細胞用の発現ベクターに組み込み、得られた組換えベクターを投与することにより、細胞内でアンチセンスRNAを生産させBDNF遺伝子又はTrkB遺伝子の発現を抑制してもよい。哺乳動物細胞用の発現ベクターは種々市販されており、それらのマルチクローニング部位に上記DNAを挿入することができる。
【0032】
アンチセンスRNAの投与量は、絨毛癌の程度、患者の状態や体重等に応じて適宜選択されるが、上記したiRNAの投与量と同程度であってよい。
【0033】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【実施例】
【0034】
実施例1
材料および方法
細胞
絨毛癌細胞株であるJARおよびBeWoはATCC(バージニア州マナッサス)から購入した。JAR細胞は、10%ウシ胎仔血清(インビトロジェン社)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)を添加したDMEM/F12(インビトロジェン社、カリフォルニア州カールズバッド)で保持し、BeWo細胞は、15%ウシ胎仔血清と抗菌剤とを添加したF−12培地で、37℃、5%CO2環境で保持した。
【0035】
RT−PCR
JAR細胞およびBeWo細胞におけるTrkBリガンド(BDNFおよびNT−4/5)と受容体(TrkBおよびp75NTR)の発現を調査するため、従来のRT−PCRで、非特許文献7〜9のプライマーを用いた。ネガティブコントロールには、mRNAを含めなかった。
【0036】
JAR細胞およびBeWo細胞中のTrkBの転写レベルについての定量的リアルタイムRT−PCRを、非特許文献8のTrkBおよびβ−アクチンに対するプライマーおよびハイブリダイゼーションプローブにより、SmartCycler(タカラ、東京、日本)を用いて行った。TrkBに対するプライマーは受容体の触媒キナーゼドメインに合致し、切断されたアイソフォームの増幅を回避する(非特許文献10)。データはβ−アクチンの転写レベルに基づいて正規化した。
【0037】
PCRアレイ実験には、ヒトの細胞周期用およびアポトーシス用のRT2 Profiler PCRアレイ(SAバイオサイエンス社、メリーランド州フレデリック)を用い、メーカーのプロトコルに従ってそれぞれの96ウェルPCRアレイプレート中の84個の遺伝子のmRNAレベルを同時に検討した。RNeasy Mini kit(キアゲン、東京、日本)を用いて、全RNAを細胞から抽出した。RT2 First Strandキット(SAバイオサイエンス社)を用いて1μgの全RNAを逆転写し、次いで、LightCycler480(ロシュ・アプライド・サイエンス社、インディアナ州インディアナポリス)でリアルタイムPCRを行った。ΔΔサイクル閾値法によりデータを解析し、発現における変化倍率(fold change)を測定した。
【0038】
免疫測定
JAR細胞およびBeWo細胞を用いて、非特許文献6、8のように、BDNF、NT−4/5、およびTrkBの免疫蛍光測定を行った。
【0039】
ELISAには、JAR細胞およびBeWo細胞を、137mM NaCl、20mM Tris−HCl、1%ノニデットP-40、10%グリセロール、プロテアーゼインヒビターカクテル(ロシュ・アプライド・サイエンス社)を含有するバッファー中でホモジナイズし、8000×g、4℃で5分間、遠心した。上清中のBDNFおよびNT−4/5の定量を、非特許文献6、8のように行った。結果は、タンパク質濃度により正規化し、細胞(mg)当たりのBDNF(pg)またはNT−4/5(pg)として表した。
【0040】
血清中のヒト絨毛性ゴナドトロピン(hCG)−βタンパク質のレベルと、細胞および組織ホモジネートとを、RIA(三菱BCL社、東京、日本)を用いて測定した。このアッセイでの最小検出可能レベルは0.1ng/mlであり、アッセイ内およびアッセイ間の変動係数はそれぞれ4.5%および6.3%である。
【0041】
細胞培養および細胞の増殖とアポトーシスの測定
内在性TrkBリガンドの細胞増殖およびアポトーシスにおける役割を調べるため、JAR細胞およびBeWo細胞を96ウェル培養プレートに1×104個/ウェルの密度で播種した。細胞を一晩インキュベートし、種々のインヒビターで処理した。プレインキュベーション後、種々の用量のTrkBの可溶性細胞外ドメイン(R&Dシステムズ社、ミネソタ州ミネアポリス)、汎特異的(pan-specific)Trk受容体インヒビターであるK252a(カルバイオケム社、カリフォルニア州ラ・ホーヤ)(非特許文献11)、または不活性細胞膜非透過性K252b(カルバイオケム社)(非特許文献12)を用いて、または用いずに、48時間、無血清培地中で、細胞を培養した。さらに、一晩のプレインキュベーション後、96ウェル培養プレートに0.2×104個/ウェルの密度のJAR細胞およびBeWo細胞を、48時間、無血清培地中で、種々の用量のBDNF(R&Dシステムズ社)で処理するか、または処理しないことによりBDNFの細胞増殖およびアポトーシスへの影響を測定した。BDNFによる細胞増殖促進におけるPI3KおよびERK経路の関与を解析するため、JAR細胞およびBeWo細胞を、BDNF(30ng/ml)とともに、PI3KインヒビターであるLY294002(0.1〜1.0μM;シグマ社、ミズーリ州セントルイス)またはその不活性類似体であるLY303511(1.0μM;カルバイオケム社)の存在下または非存在下で培養した。いくつかの実験においては、細胞を、BDNFとともに、MAPK/ERKキナーゼ1/2インヒビターであるU0126(0.1〜10μM;カルバイオケム社)もしくはその不活性類似体であるU0124(10μM;カルバイオケム社)、またはLY294002とU0126の両方か、LY303511とU0124の両方の存在下または非存在下で培養した。
【0042】
トリパンブルー排除試験(インビトロジェン社)を用いて、細胞増殖を測定した。生存可能な細胞の数を、生存可能な細胞による色素排除に基づいて血球計算板上で測定した。アポトーシスの進行を測定するため、一部の細胞を、培養8時間において、定量的カスパーゼ3/7酵素アッセイにかけた。アポトーシス測定のための培養期間は、カスパーゼ3/7の活性化がTrkBの細胞外ドメインまたはK252aで12時間処理した細胞において明らかな最終的細胞死よりも早くに起こる、との仮説に基づいて選択した。カスパーゼ3/7活性を、非特許文献13のように、Caspase-Glo 3/7アッセイ(プロメガ社、ウィスコンシン州マディソン)によって測定した。細胞のアポトーシスについても、培養8時間においてin situ 末端デオキシヌクレオチジルトランスフェラーゼ媒介2’−デオキシウリジン 5’−二リン酸ニック末端標識法(TUNEL法)を用いてDNAの断片化を検出することにより分析した非特許文献14。さらに、非特許文献15のように、アポトーシスで誘導されたミトコンドリアの膜電位の変化をMitoTracker Orange CMTMRos(インビトロジェン社)を用いて検討した。いくつかの実験においては、培養8時間で、細胞をPCRアレイ実験に供した。
【0043】
In vivo試験
絨毛癌の腫瘍形成における内在性TrkBリガンドのin vivoでの役割を探るため、JAR腫瘍を有する胸腺欠損ヌードマウス(BALB/c nu/nu)(日本クレア、東京、日本)で、K252aの抗腫瘍活性を測定した。動物の世話および使用は秋田大学医学部動物実験委員会により承認を受けた。JAR細胞(0.1ml、5×106個)を4週齢のメスのヌードマウスの右脇腹の皮下に移植した。腫瘍容積がおよそ60mm3になったときに、動物への処理を開始した。動物の体重は処理の日において17〜21gであった。生理食塩水(500μg/kg)に溶かしたK252aの腹腔内投与を3日ごとに行った。同じ用量の投与も毎日継続して行った。ネガティブコントロールとして、K252b(500μg/kg)での処理または賦形剤のみの処置を行った。これらの実験のために選択したK252aおよびK252bの用量は先の調査(非特許文献16)に基づいた。式:腫瘍容積(mm3)=長さ×(幅)2×0.5を用いて、腫瘍容積を毎日測定した。処理の9日後にマウスを殺し、試験したすべてのマウスから血液サンプルを回収し、血清hCG−βレベルを測定した。ヘマトキシリン・エオシン(H&E)染色に加え、in vivoでの細胞増殖およびアポトーシスを、それぞれ、増殖性細胞核抗原(PCNA)の免疫染色、およびTUNELアッセイにより評価した。PCNAの免疫染色は、マウス抗PCNAモノクローナル抗体(Cell Signaling Technology社、マサチューセッツ州ダンバーズ)を1:4000希釈で用い、メーカーのプロトコルに従って行った。切除した腫瘍サンプル中のカスパーゼ3/7活性をCaspase-Glo 3/7アッセイにより測定した。データは、タンパク質濃度により正規化した。このデータはコントロールに対する増加倍率を示した。
【0044】
統計解析
マン・ホイットニーのU検定を行い、絨毛癌細胞株中のBDNF、NT−4/5、TrkB、およびhCG−βのレベルを比較した。薬剤処理による腫瘍容積への影響はスチューデンドt検定を用いて解析した。PCRアレイからのデータを、独立スチューデンドt検定により、有意差について試験した。一元配置ANOVA、次いでフィッシャーの保護最小有意差検定を用いて、その他の差を評価した。データは平均±標準誤差である。
【0045】
結果
絨毛癌細胞におけるニューロトロフィンとTrk受容体の発現
絨毛癌細胞におけるBDNF、NT−4/5、TrkBの発現について、PCRおよび免疫測定により検討した。これら3種すべての転写物が、JAR細胞とBeWo細胞のいずれにおいても認められた(図1A)。絨毛癌細胞におけるこれらの発現を、さらに、免疫蛍光染色(JAR細胞)により確認した(図1B)。加えて、p75NTRのmRNAも両細胞株で検出した(図1A)。ELISA分析により、両絨毛癌細胞において、BDNFのタンパク質レベルがNT−4/5よりも2.3倍高いことが示された(図1C)。hCG−βのタンパク質レベルはBeWo細胞よりもJAR細胞での方が高いが、ELISA分析および定量的リアルタイムRT−PCR分析によると、BDNFタンパク質、NT−4/5タンパク質、およびTrkB転写物のレベルは、JAR細胞とBeWo細胞とで差はなかった(図1C)。
【0046】
絨毛癌細胞の成長に対する内在性TrkBシグナル伝達のin vitro抑制
異なる絨毛癌細胞株におけるTrkBリガンドと受容体の両方の発現は、TrkBシグナル伝達系が、絨毛癌の成長において、オートクリンの役割を果たしている可能性があることを示唆する。内在性TrkBリガンドが絨毛癌細胞にとって生存因子として作用するかどうかを判定するため、我々は、TrkBの細胞外ドメインとK252aで処理した培養JAR細胞および培養BeWo細胞の増殖とアポトーシスとを評価した。図2Aに示されるように、コントロールの細胞数は、培養後に増加(13.5±0.9,×104個/ウェル)し、不活性のK252bで処理した場合ではなく、TrkBの細胞外ドメインまたはK252aのどちらかで処理した場合に、生存可能な細胞の数は、両細胞株で同じような効率で減少した。我々はさらに、種々のインヒビターで処理することにより、両細胞株においてカスパーゼ3/7活性の増大を検出した(図2B)。TrkBの細胞外ドメインおよびK252aでの処理によりTUNEL陽性の核の割合も増加した(図2C)。これはアポトーシスが誘導されたことを示す。絨毛癌細胞における内在性TrkBシグナル伝達の抑制によって誘導されるアポトーシスをさらに特徴づけるため、我々は、ミトコンドリアの膜電位感受性色素であるMitoTracker Orange CMTMRosを用いてミトコンドリアの機能の低下を検討し、アポトーシス中のミトコンドリアの膜電位の状態を評価した(非特許文献17)。機能的なミトコンドリアは色素を取り込み、オレンジ色の蛍光で点状の染色パターンを示すが、アポトーシスを起こした細胞のミトコンドリアは染色されない。我々は、コントロール細胞でのMitoTracker色素染色が、それらのミトコンドリアの局在として予想される通りに点状に現れる(図2C)ことを見出した。不活性のK252bでの処理ではなく、rkBの細胞外ドメインまたはK252aのどちらかでの処理により、MitoTracker色素染色は減少した(図2C)。
【0047】
BDNF処理の絨毛癌細胞増殖への影響とin vitroのPI3KおよびERKの媒介的役割
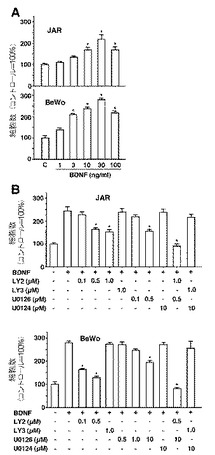
BDNFの細胞増殖への影響を検討するため、細胞を、種々の用量のBDNFの存在下または非存在下で培養した。BDNFの細胞増殖への影響は、細胞をより高い密度で培養した場合、おそらくオートクリン因子の蓄積により、より目立たなくなった(データ非掲載)。従って、我々は、低い密度で細胞を培養し、BDNFを含む細胞由来因子の影響を排除した。この条件下では、BDNF処理は、両絨毛癌細胞において用量依存的に細胞増殖を増加させた(図3A)。
【0048】
我々はさらに、2種類の異なる絨毛癌細胞株において、BDNFによって誘導された細胞増殖の下流メディエーターとしてのPI3KおよびERKシグナル伝達経路の役割を解析した。低密度で細胞数を増加させるBDNFの能力は、不活性類似体であるLY303511で処理するのではなくPI3KインヒビターであるLY294002とともに処理するか、または不活性類似体であるU0124で処理するのではなくMAPKキナーゼ(MEK)1/2インヒビターであるU0126とともに処理するかのどちらかによって抑制され(図3B)、また、PI3KインヒビターおよびMEK1/2インヒビターの両方とともに処理することによって、完全に抑制され、これはこれらの経路のみが内在性TrkBシグナル伝達の媒介に関与しているとのことを示している(図3B)。しかしながら、PI3KインヒビターおよびMEK1/2インヒビターへの感受性はJAR細胞とBeWo細胞とで異なっていた。より低用量のU0126(0.5μMおよび1.0μM)は、BeWo細胞ではBDNFによる細胞増殖刺激を抑制するには効果がなかった。
【0049】
BDNFおよびTrkBに対するsiRNAを用いて、我々はさらに、内在性BDNF/TrkBシグナル伝達の抑制後の絨毛癌細胞の成長阻害について検証した。BDNFまたはTrkBのノックダウンは、細胞増殖を、それぞれ50.5%、47.8%阻害し、細胞成長における内在性BDNF/TrkBシグナル伝達の特異的役割を支援した。しかしながら、両siRNAの細胞成長への阻害効果は、TrkBの細胞外ドメインおよびTrk受容体インヒビターの場合(>80%阻害)よりも効力が少なかった。加えて、BDNF siRNAをトランスフェクションした細胞におけるBDNFによる細胞増殖刺激は、おそらく、siRNAによって媒介されたノックダウンの後のBDNFまたはTrkBの残存発現(図5C)のために、そのsiRNAをトランスフェクションしなかった細胞の場合(コントロールの223%)よりも弱かった(コントロールの177%)。
【0050】
内在性TrkBシグナル伝達抑制後の培養絨毛癌細胞における細胞周期関連遺伝子およびアポトーシス関連遺伝子のmRNA発現プロファイル変化
TrkBシグナル伝達インヒビターによる細胞増殖阻害および生存阻害の根底にある分子機構を探るため、TrkBの細胞外ドメイン(500nM)で処理した、または処理しなかったJAR細胞から全RNAを精製し、PCRアレイ解析に使用して、細胞周期関連遺伝子およびアポトーシス関連遺伝子の発現をプロファイルした。TrkBの細胞外ドメインで処理することにより、アレイ中でスクリーニングされた84種類の細胞周期関連mRNAのパネル全体にわたって、3つの刺激性遺伝子(CCND1、DDX11、DNM2)の発現が2倍以上減少し、2つの阻害遺伝子(CDKN2B、SERTAD1)の発現が2倍以上増加した(図4A)。アポトーシス関連遺伝子の解析においては、異なるアポトーシス促進性の遺伝子の転写レベルが、内在性TrkBシグナル伝達の抑制に応答して増加した(図4B)。発現が増加したアポトーシス促進性遺伝子のうち、TNFにおいて、もっとも有意な増加(25倍)が認められた(図4B)。対照的に、抗アポトーシス性の遺伝子の発現への有意な影響は、TrkBの細胞外ドメインで処理した細胞では観察されなかった。
【0051】
絨毛癌成長へのTrk受容体インヒビターのIn vivoでの影響
観察されたTrkBインヒビターによる絨毛癌細胞成長のin vitro阻害が、in vivoでの抗腫瘍活性となるのかどうかを判定するため、K252aを、JAR細胞の腫瘍異種移植片を有する胸腺欠損ヌードマウスに投与した。これらの調査にはJAR細胞を用いた。これは、この細胞では、腫瘍マーカーであるhCG−βの産生レベルが高かった(図1C)ためである。腫瘍容積がおよそ60mm3に達したときに、動物に、K252aまたはK252bを、500μg/kgで3日ごとに与えた。K252aで処理した動物の腫瘍は、コントロールに比べて有意に小さくなり、これは処理開始から3日後程度の早期に始まり、9日目の実験終了まで続いた(図5A)。処理9日目で、最大の腫瘍成長阻害(68%)が観察された。図5Bに示すように、JAR腫瘍を有するマウスにおける血清hCG−βレベルは腫瘍容積に相関し、腫瘍マーカーとしてのその役割と一致した。K252aで処理した動物は、処理9日目において、コントロールと比べて87%の血清hCG−βレベルの減少を示した(図5C)。試験されたすべての動物において、実験期間中、腫瘍の転移は観察されなかった。K252aを9日目まで毎日投与した場合に、成長抑制効率が比較可能であった(67%の腫瘍成長阻害、82%の血清hCG−βレベル増加)。試験したすべての動物において、実験中、重大な副作用は観察されず、また、K252aで処理したグループでは、調査中、有意な体重減少はなかった(賦形剤の場合には、22.08±0.54g、K252aの場合には、21.15±0.47g、K252bの場合には、21.13±0.45g)。
【0052】
切除した腫瘍をさらに検査し、K252aのin vivoでの影響を測定した。H&E染色による組織病理学的検査では、K252aで処理したマウスの腫瘍中に、細胞増殖抑制を示唆する有糸分裂活性の減少、および生存抑制を示唆するクロマチン凝縮を有する細胞数の増加が検出された(図6A)。PCNA染色によりK252a処理の細胞増殖抑制への影響を確認し、アポトーシスの誘導をTUNELアッセイにより確認した(図6A)。我々はさらに、カスパーゼ活性を定量化することにより、腫瘍中でK252aにより誘導されるアポトーシスを特徴づけ、またK252aで処理したマウスの腫瘍中でカスパーゼ3/7の活性が17.7倍減少することを観察した(図6B)。重要なのは、不活性細胞膜非透過性K252bが、試験されたすべてのパラメーターで効果がなかったことである。
【0053】
実施例2
siRNAのトランスフェクション
アプライドバイオシステムズ社(カリフォルニア州フォスターシティ)製の、BDNFを標的(s1962;センスとしてuuacuaugguuauuucauatt(配列番号5)、アンチセンスとしてuaugaaauaaccauaguaagg(配列番号6)、および、センスとしてcccuuaccauggauagcaatt(配列番号7)、アンチセンスとしてuugcuauccaugguaagggcc(配列番号8))、またはTrkBを標的(s9750;センスとしてgguuagaaaucaucaacgatt(配列番号9)、アンチセンスとしてucguugaugauuucuaacctt(配列番号10)、および、s9749;センスとしてgaauugacgauggugcaaatt(配列番号11)、アンチセンスとしてuuugcaccaucgucaauucca(配列番号12))とした2種類の特異的なsiRNA二本鎖のセット(Silencer(登録商標) Select Pre-designed siRNA)を用いてBDNFまたはTrkBのノックダウンを行った。Select Negative Control #1 siRNAおよびGAPDH Positive Control siRNAを、それぞれネガティブコントロールおよびポジティブコントロールとして使用した。siPORT(商品名) NeoFX(商品名)Transfection Agent(アプライドバイオシステムズ社)を、メーカーの説明書に従ってリバーストランスフェクション法に使用した。このポジティブコントロールとKDalert(商品名)GAPDH Assay Kit(アプライドバイオシステムズ社)とを用い、siRNAのトランスフェクション条件をJAR細胞において最適化した。BDNFおよびTrkBのノックダウンのために、細胞を終濃度5nMの各siRNA二本鎖セットでトランスフェクションした。この細胞を24時間インキュベートし、次いで無血清培地を10%FBS含有完全培地に交換した。siRNAのトランスフェクションの72時間後に、リアルタイムRT−PCR解析用に細胞を回収し、BDNFおよびTrkBのmRNAの発現のノックダウンを確認した。培養液中に蓄積した内在性BDNFを除去するために、siRNAのトランスフェクション後72時間で、完全培地を無血清培地に交換した。48時間のインキュベーションの後、細胞増殖をトリパンブルー排除試験を用いて測定した。いくつかの実験においては、siRNAのトランスフェクション後72時間に、BDNF siRNAをトランスフェクションした細胞をBDNF(30ng/ml)(R&Dシステムズ社、ミネソタ州ミネアポリス)培地で処理し、BDNFによる細胞成長への影響を評価した。
【0054】
RT−PCR
絨毛癌細胞におけるニューロトロフィン(神経成長因子、NGF、およびニューロトロフィン−3、NT−3)およびTrk受容体(TrkA、およびTrkC)の発現を調査するための従来のRT−PCRに用いる、NGF、NT−3、TrkA、およびTrkCに対するプライマーは以下に示す通り:NGFには、センスとして5'-tcatcatcccatcccatcttcc-3'(配列番号13)、アンチセンスとして5'-tccagtgctttgagtcaatgcc-3'(配列番号14);NT−3には、センスとして5'-gccagaataacacagactcagc-3'(配列番号15)、アンチセンスとして5'-tcggtgactcttatgctccg-3'(配列番号16);TrkAには、センスとして5'-catcgtgaagagtggtctccg-3'(配列番号17)、アンチセンスとして5'-gagagagactccagagcgttgaa-3'(配列番号18);TrkCには、センスとして5'-agcgtctggctggactatgt-3'(配列番号19)、アンチセンスとして5'-gtgtggtgagccggttactt-3'(配列番号20)。β−アクチンに対するプライマーは既に記載されている(非特許文献1)。PCR反応は、94℃で30秒の変性、57℃(TrkA)、60℃(TrkCおよびβ−アクチン)、または62℃(NGFおよびNT−3)で30秒のアニーリング、および72℃で30秒の伸長、による増幅を35サイクルとした。ネガティブコントロールにはmRNAを含めなかった。
【0055】
細胞培養
hCG刺激によるTrkBリガンドおよび受容体の産生調節に取り組むため、アンチセンスの絨毛性ゴナドトロピンβ(aCGβ)(Koji Nakabayashi博士から分与(非特許文献29))を有する発現ベクターpM2HAでトランスフェクションしたJAR細胞を使用した。JAR−pM2HA細胞とともに、トランスフェクションしていないJAR細胞をコントロールとして実験において使用した。これらの細胞株を、成長曲線およびマイコプラズマ解析を用いるとともに、これらの細胞の形態により検証した。JAR−aCG細胞およびJAR−pM2HA細胞は、10%FBS、ジェネティシン(62.5μg/ml)(インビトロジェン社)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)を添加したDMEM/F12で保持した。トランスフェクションした細胞におけるhCG−β、BDNF、およびNT−4/5の測定のために、細胞をサブコンフルエントな状態まで増殖させ、PBSで洗浄し、次いで無血清培地に交換した。一晩培養した後、その細胞のホモジネートをRIAまたはELISAに供し、本文中に記載したように、それらのhCG−βタンパク質、BDNFタンパク質またはNT−4/5タンパク質のレベルを測定した。定量的リアルタイムRT−PCRを行い、非特許文献30のように、これらのトランスフェクションした細胞におけるTrkBの転写レベルを測定した。
【0056】
結果を図7〜図9に示す。
【技術分野】
【0001】
本発明は、絨毛癌の治療剤に関する。
【背景技術】
【0002】
絨毛癌は、栄養芽細胞から誘導される高度に未分化で浸潤性の悪性腫瘍である。絨毛癌の治療は、現在、メトトレキセート、アクチノマイシン−D、エトポシド等の抗癌剤を用いる化学療法により行われている(非特許文献31)。確立された化学療法により多くの絨毛癌患者は寛解に至るが、それでも絨毛癌患者の5〜25%は抗癌剤により十分に治療されず、あるいは、一旦寛解しても再発する。
【0003】
一方、脳由来神経栄養因子(brain-derived neurotrophic factor (BDNF))は、高親和性チロシンキナーゼ(Trk)B受容体を全ニューロトロフィン低親和性共受容体p75(p75NTR)と共に活性化するタンパク質であるニューロトロフィンファミリーの1つである(非特許文献1)。BDNFが結合後、TrkB受容体シグナルは、フォスファチジルイノシトール 3-キナーゼ(PI3K)、MAPK/ERK, フォスフォリパーゼC-γ、及びプロテインCキナーゼカスケードを包含する複数のシグナル経路を活性化し、異なる種類の細胞の細胞増殖、分化及び生存において重要な役割を果たしている(非特許文献1、2)。ニューロトロフィンは、中枢神経系で広く発現し、ニューロンの生存と分化にとって重要である(非特許文献3)が、それらはまた、非ニューロン組織においても重要な役割を果たす(非特許文献4)。正常組織中での生理作用に加え、TrkBを介するニューロトロフィンシグナルが変更されることにより悪性腫瘍の形成及び転移が促進されることを示すデータが蓄積されている(非特許文献5)。TrkBは、ときどきそのリガンドであるBDNFと共に、神経芽細胞腫、ウィルムス腫瘍並びに前立腺及び膵臓腺癌を包含する種々のヒト癌、並びに多発性骨髄腫及び非固形癌において過剰発現している(非特許文献5)。これらのデータは、腫瘍の発達のためのBDNF/TrkBシグナルの潜在的なオートクリン役割を示唆している。しかしながら、腫瘍形成中における内発性BDNF/TrkBシグナルの分子機構を利用した研究はほとんどない。
【0004】
本願発明者らは、浸潤性栄養芽細胞に分化することができる、胚盤胞段階の胚の栄養外胚葉細胞中でのTrkB及びそのリガンドであるBDNFの発現を最近見出した。本願発明者らはまた、着床前の栄養外胚葉細胞の増殖及び生存におけるBDNFの促進効果を示した(非特許文献6)。着床後も胎盤栄養芽細胞中でTrkBとそのリガンドの発現は持続する。本願発明者は、妊娠中の胎盤発達における栄養芽細胞細胞の成長と生存におけるTrkBシグナル系のオートクリン/パラクリン調節役割を示した(非特許文献7)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Barbacid M 1994 The Trk family of neurotrophin receptors. J Neurobiol 25:1386-1403
【非特許文献2】Huang EJ, Reichardt LF 2003 Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609-642
【非特許文献3】Jones KR, Farinas I, Backus C, Reichardt LF 1994 Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989-999
【非特許文献4】Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD 1993 Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron 10:137-149
【非特許文献5】Desmet CJ, Peeper DS 2006 The neurotrophic receptor TrkB: a drug target in anti-cancer therapy? Cell Mol Life Sci 63:755-759
【非特許文献6】Kawamura K, Kawamura N, Fukuda J, Kumagai J, Hsueh AJ, Tanaka T 2007 Regulation of preimplantation embryo development by brain-derived neurotrophic factor. Dev Biol 311:147-158
【非特許文献7】Kawamura K, Kawamura N, Sato W, Fukuda J, Kumagai J, Tanaka T 2009 Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology 150:3774-3782
【非特許文献8】Kawamura K, Kawamura N, Mulders SM, Sollewijn Gelpke MD, Hsueh AJ 2005 Ovarian brain-derived neurotrophic factor (BDNF) promotes the development of oocytes into preimplantation embryos. Proc Natl Acad Sci USA 102:9206-9211
【非特許文献9】Kawamura K, Sato N, Fukuda J, Kodama H, Kumagai J, Tanikawa H, Nakamura A, Honda Y, Sato T, Tanaka T 2003 Ghrelin inhibits the development of mouse preimplantation embryos in vitro. Endocrinology 144:2623-2633
【非特許文献10】Klein R, Conway D, Parada LF, BarbacidM1990 The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 61:647-656
【非特許文献11】Tapley P, Lamballe F, BarbacidM1992 K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene 7:371-381
【非特許文献12】Ross AH, McKinnon CA, Daou MC, Ratliff K, Wolf DE 1995 Differential biological effects of K252 kinase inhibitors are related to membrane solubility but not to permeability. J Neurochem 65:2748-2756
【非特許文献13】Liu D, Li C, Chen Y, Burnett C, Liu XY, Downs S, Collins RD, Hawiger J 2004 Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide. J Biol Chem 279:48434-48442
【非特許文献14】Kawamura K, Fukuda J, Shimizu Y, Kodama H, Tanaka T 2005 Survivin contributes to the anti-apoptotic activities of transforming growth factor α in mouse blastocysts through phosphatidylinositol 3' kinase pathway. Biol Reprod 73:1094-1101
【非特許文献15】Kawamura K, Fukuda J, Kumagai J, Shimizu Y, Kodama H, Nakamura A, Tanaka T 2005 Gonadotropin-releasing hormone I analog acts as an antiapoptotic factor in mouse blastocysts. Endocrinology 146:4105-4116
【非特許文献16】Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, Nagashima Y, Kubota Y 2006 Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res 66:3699-3705
【非特許文献17】Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G 1999 Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441-446
【非特許文献18】BrodeurGM2003 Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 3:203-216
【非特許文献19】McAllister AK, Katz LC, Lo DC 1999 Neurotrophins and synaptic plasticity. Annu Rev Neurosci 22:295-318
【非特許文献20】Hamada AL, Nakabayashi K, Sato A, Kiyoshi K, Takamatsu Y, Laoag-Fernandez JB, Ohara N, Maruo T 2005 Transfection of antisense chorionic gonadotropin β gene into choriocarcinoma cells suppresses the cell proliferation and induces apoptosis. J Clin Endocrinol Metab 90:4873-4879
【非特許文献21】RaffMC1992 Social controls on cell survival and cell death. Nature 356:397-400
【非特許文献22】Wang X 2001 The expanding role of mitochondria in apoptosis. Genes Dev 15:2922-2933
【非特許文献23】KaplanDR,MillerFD2000 Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10:381-391
【非特許文献24】Yuan J, Yankner BA 2000 Apoptosis in the nervous system. Nature 407:802-809
【非特許文献25】Simpson PJ, Moon C, Kleman AM, Connolly E, Ronnett GV 2007 Progressive and inhibitory cell cycle proteins act simultaneously to regulate neurotrophin-mediated proliferation and maturation of neuronal precursors. Cell Cycle 6:1077-1089
【非特許文献26】Li J, Melvin WS, Tsai MD, Muscarella P 2004 The nuclear protein p34SEI-1 regulates the kinase activity of cyclin-dependent kinase 4 in a concentration-dependent manner. Biochemistry 43:4394-4399
【非特許文献27】Jin M, Udagawa K, Miyagi E, Nakazawa T, Hirahara F, Yasumitsu H, Miyazaki K, Nagashima Y, Aoki I, Miyagi Y 2001 Expression of serine proteinase inhibitor PP5/TFPI-2/MSPI decreases the invasive potential of human choriocarcinoma cells in vitro and in vivo. Gynecol Oncol 83:325-333
【非特許文献28】Krause DS, Van Etten RA 2005 Tyrosine kinases as targets for cancer therapy. N Engl J Med 353:172-187
【非特許文献29】Kawamura K, Kawamura N, Mulders SM, Sollewijn Gelpke MD, Hsueh AJ 2005 Ovarianbrain-derived neurotrophic factor (BDNF) promotes the development of oocytes intopreimplantation embryos. Proc Natl Acad Sci U S A 102:9206-9211
【非特許文献30】Hamada AL, Nakabayashi K, Sato A, Kiyoshi K, Takamatsu Y, Laoag-Fernandez JB,Ohara N, Maruo T 2005 Transfection of antisense chorionic gonadotropin beta gene intochoriocarcinoma cells suppresses the cell proliferation and induces apoptosis. J Clin Endocrinol Metab 90:4873-4879
【非特許文献31】石川 哲也ら、症例報告 日産婦関東連会報44:9-14, 2007
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、絨毛癌に対して治療効果を有する、絨毛癌に対する新規な治療剤を提供することである。
【課題を解決するための手段】
【0007】
本願発明者らは鋭意研究の結果、BDNF及び/又はTrkBの作用を抑制することにより絨毛癌細胞の増殖を抑制することができ、絨毛癌が治療可能であることを見出し本発明を完成した。
【0008】
すなわち、本発明は、脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する絨毛癌の治療剤を提供する。
【発明の効果】
【0009】
本発明により、絨毛癌に対して優れた治療効果を有する新規な絨毛癌治療剤が提供された。
【図面の簡単な説明】
【0010】
【図1】絨毛癌細胞におけるBDNF、NT−4/5、TrkBの発現を示す図である。Aでは、TrkBリガンドおよび受容体のmRNAの発現をRT−PCRにより検出した。β−アクチンのレベルをローディングコントロールとして使用した。ヒト胎盤cDNAをポジティブコントロールとして使用し、ネガティブコントロールには鋳型DNAを含めなかった。PLは胎盤の略、NCはネガティブコントロールの略である。Bは、BDNFタンパク質、NT−4/5タンパク質、TrkBタンパク質を免疫組織化学的に検出したものである。JAR細胞において、BDNF、NT−4/5、TrkBの特異的なシグナルが認められる。下段はネガティブコントロールである。細胞の核は、ヘキスト33342を用いて青色に染色した。スケールバーは20μmを表す。BeWo細胞においても同様の染色パターンが観察された(データ非掲載)。Cは、BDNFタンパク質とNT−4/5タンパク質、TrkB転写物、またはhCG−βタンパク質のレベルを、それぞれ、ELISA(左)、リアルタイムRT−PCR(真ん中)、またはRIA(右)を用いて定量化したものである。サンプルは異なる培養液中で取得した(n=6)。TrkBのmRNAレベルは、同一サンプル中のβ−アクチンの転写レベルを用いて正規化した。柱が平均を、バーが標準誤差を示している。
【図2】内在性TrkBリガンドのin vitroの絨毛癌細胞成長における役割を示す図である。内在性TrkBリガンドの抑制の、細胞増殖への影響(A)および生存への影響(B)。JAR細胞およびBeWo細胞を、培地のみ[コントロール(C)]で、種々の用量のTrkBの細胞外ドメイン(TrkB EC)とともに、K252aまたはK252bとともに培養した。細胞数はトリパンブルー排除試験を用いて測定し、細胞のアポトーシスはカスパーゼ3/7アッセイを用いて定量化した(n=5)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。Cは、DNA断片化および機能的なミトコンドリアの減少を、それぞれin situ TUNEL染色およびMitoTracker Orange CMTMRosを用いて検出したものである。掲載されている画像は、TrkBの細胞外ドメイン(10μg/ml)、K252a(500nM)、またはK252b(500nM)で処理した、または処理しなかったJAR細胞から得た。TUNELアッセイでは、細胞の核酸を、ヨウ化プロピジウムを用いて赤色に染色した。陽性アポトーシスシグナル数(緑色蛍光)は、TrkBの細胞外ドメインまたはK252a処理をした細胞において増加した。スケールバーは50μmを表す。ミトコンドリアアッセイでは、細胞の核をヘキスト33342で青色に染色した。コントロールグループまたはK252bグループの細胞は点状の染色パターン(赤色)を示し、TrkBの細胞外ドメインおよびK252aで処理した細胞は不鮮明な染色パターンを示す。挿入図は、より高い倍率に拡大した画像である。TrkBの細胞外ドメインまたはK252aで処理した細胞のいくつかは、クロマチン凝縮と核断片化とで特徴付けられるアポトーシスの形態学的特徴(矢印)を示す。スケールバーは25μmを表す。同様の結果がBeWo細胞でも観察された(データ非掲載)。
【図3】PI3KおよびERKシグナル伝達経路を介した絨毛癌細胞の細胞増殖のBDNFによる促進。細胞を、培地のみで、もしくは種々の用量のBDNFとともに培養し、またはBDNFなし[コントロール(C)]もしくはBDNF(30ng/ml)とともに、PI3KインヒビターであるLY294002(LY2)、もしくはその不活性類似体であるLY303511(LY3)の存在下または非存在下で培養した。いくつかの実験においては、細胞を、MEK1/2のインヒビターであるU0126、もしくはその不活性類似体であるU0124、またはLY294002とU0126の両方か、LY303511とU0124の両方の存在下または非存在下において、BDNFで処理した。培養後48時間で、トリパンブルー排除試験を用いて細胞数を測定し、BDNF処理の細胞増殖への影響(A)と、BDNFによる細胞増殖刺激におけるPI3KおよびERK経路の関与(B)を評価した(n≧3)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。
【図4】絨毛癌細胞において内在性TrkBシグナル伝達の抑制によって制御される細胞周期関連遺伝子およびアポトーシス関連遺伝子の同定結果を示す図である。JAR細胞をTrkBの細胞外ドメイン(500nM)の存在下または非存在下で培養した。培養後8時間で、細胞周期について(A)およびアポトーシスについて(B)のPCRに基づく遺伝子アレイ解析を行った。柱は、TrkBの細胞外ドメインでの処理をしなかったサンプルと比較した、3つの別のサンプルにおける個々の遺伝子発現の平均倍率(fold difference)を表している。すべての差が統計的に有意(P>0.05)であった。
【図5】ヌードマウス中のJAR細胞異種移植片の、in vivoでの腫瘍成長に対するTrk受容体の効果を示す図である。A:K252aを投与したマウスにおける腫瘍成長の経時変化を示す。K252a又はK252b(500μg/kg)を3日毎に胸腺欠損マウス(n=9)に投与した。各マウスが投与を受けた最初の日における各腫瘍の体積を相対的腫瘍体積1と表した。点は平均、バーは標準誤差。*:P<0.05 vs.コントロール群。B:腫瘍成長の間における血清hCG-β濃度。異なる体積(n=4)の腫瘍を担持するマウスから血清試料を得た。血清hCG-β濃度はRIAにより測定した。点は平均、バーは標準誤差。C:治療9日目におけるマウス中の血清hCG-β濃度。カラムは平均、バーは標準誤差。*:P<0.05 vs.コントロール群。
【図6】Trk受容体インヒビターによるin vivoでの腫瘍増殖の抑制及びヌードマウス中でのJAR細胞異種移植片の生存を示す図である。A:細胞増殖の組織学的特徴付け及び腫瘍におけるアポトーシスを示す。細胞増殖及びDNA断片化の特異的検出は、それぞれ、PCNA免疫染色及びin situ TUNEL染色を用いて行った。腫瘍を担持する動物に、K252a又はK252b(500μg/kg)を3日毎に投与し又は投与しなかった。図示の画像は、治療9日目の切除した腫瘍から得た。H&E染色は、K252a投与後の有糸分裂細胞数の減少及びクロマチンの凝縮を示している。K252a投与により、PCNAシグナル(茶色)は減少し、一方、TUNEL染色核(緑色蛍光)は増大した。スケールバー:200μm。B:K252a投与による、腫瘍中のcaspase-3/7活性の増大を示す。試料は、投与9日目のマウスから得た(n=9)。データは、タンパク濃度により正規化し、コントロール(担体のみ)に対する増加倍率で示し、1に正規化した。カラムは平均、バーは標準誤差。*:P<0.05 vs.コントロール群。
【図7】JAR細胞における、siRNAによって媒介されるBDNFおよびTrkBのノックダウンによる細胞成長の阻害を示す図である。細胞を、siRNAでトランスフェクションしなかった(コントロール、C)か、またはBDNF、TrkB、またはネガティブコントロールsiRNA(ネガティブコントロール、NC)でトランスフェクションした。Aは、siRNAによって媒介されたBDNFまたはTrkBの転写レベルの減少を示している。siRNAのトランスフェクション後72時間で、リアルタイムRT−PCRによりBDNFおよびTrkBのmRNAの発現を解析した。サンプルは異なる培養液中の細胞から取得した(n=3)。BDNFおよびTrkBのmRNAレベルは、同一サンプルのβ−アクチンの転写レベルを用いて正規化した。Bは、siRNAによって媒介されるBDNFおよびTrkBのノックダウンを経た、BDNF/TrkBシグナル伝達の抑制による細胞増殖への影響を示している。siRNAのトランスフェクション後72時間で培養液を無血清培地に交換し、次いで、インキュベーション後48時間で細胞数を、トリパンブルー排除試験を用いて測定した(n=4)。Cは、BDNF siRNAをトランスフェクションした細胞の、BDNFによる増殖促進を示している。siRNAをトランスフェクションした細胞を無血清培地において、BDNF非存在下(コントロール、C)またはBDNF(30ng/ml)存在下で培養した。培養後48時間で細胞数を、トリパンブルー排除試験により測定した(n=4)。柱が平均を、バーが標準誤差を示している。*は、コントロールグループに対してP<0.05。
【図8】絨毛癌細胞におけるTrkリガンド(NGFおよびNT−3)と受容体(TrkAおよびTrkC)の発現を示す図である。JAR細胞およびBeWo細胞中のTrkBリガンドおよび受容体のmRNAの発現をRT−PCRにより検出した。β−アクチンのレベルをローディングコントロールとして使用する。ヒト胎盤cDNAをポジティブコントロールとして使用し、ネガティブコントロールには鋳型DNAを含めなかった。PLは胎盤の略、NCはネガティブコントロールの略である。
【図9】JAR細胞における、アンチセンスhCG−βのトランスフェクションによる、hCG−βタンパク質(A)、BDNFタンパク質およびNT−4/5タンパク質(B)、ならびにTrkB転写物(A)の発現レベルへの影響を示す図である。細胞を、トランスフェクションしなかった(コントロール、C)か、または、アンチセンスhCG−β(aCGβ)もしくは空ベクター(pM2AH)でトランスフェクションした。hCG−β、BDNFおよびNT−4/5、ならびにTrkBの転写レベルに対するタンパク質レベルを、それぞれRIA/ELISA、ならびにリアルタイムRT−PCRを用いて定量化した。サンプルは異なる培養液からの細胞から取得した(n=3)。TrkBのmRNAレベルは、同一サンプル中のβ−アクチンの転写レベルを用いて正規化した。柱が平均を、バーが標準誤差を示している。
【発明を実施するための形態】
【0011】
上記の通り、本発明の絨毛癌治療剤は、BDNF及び/又はTrkBの抑制剤を有効成分として含有する。ここで、「BDNF及び/又はTrkBの抑制剤」とは、(1)BDNF及びTrkBの少なくとも一方の生理作用を抑制する物質、(2) BDNFとTrkBの結合を抑制する物質、(3) BDNF及びTrkBの少なくとも一方の、細胞内における生産を抑制する物質を意味する。(1)の例として、チロシンキナーゼ抑制剤を挙げることができる。(2)の例として、(i)遊離のTrkB又はBDNFとの結合性を有するTrkB断片、及び、(ii)BDNF又はTrkBに対する抗体を挙げることができる。(3)の例として、(i)BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、及び(ii) BDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクターを挙げることができる。以下、これらについて説明する。
【0012】
TrkBは、チロシンキナーゼ活性を有しており、下記実施例に具体的に記載されるように、チロシンキナーゼ活性を抑制することにより絨毛癌治療効果が発揮される。従って、チロシンキナーゼ抑制剤(阻害剤)を、本発明の絨毛癌治療剤の有効成分として用いることができる。チロシンキナーゼ抑制剤は、既に種々のものが公知であり、市販されているものも少なくない。市販品を好ましく用いることができる。公知のチロシンキナーゼ抑制剤の例として、K252a、AZ-23(Wang et al. J Med Chem 2008, 51, 4672-84)、CEP-701(Cephalon Inc., West Chester, PA)、CEP-751(Kyowa Hakko Kogyo, Tokyo, Japan)、CEP-2563(Cephalon Inc.)及びCEP-7801(Somaiah et al. J Thorac Oncol,2009,4, S1045-83)等を挙げることができるがこれらに限定されるものではない。
【0013】
これらのうち、下記実施例において採用したK252aは、下記の化学構造を有する、土壌真菌により生産される物質であり、チロシンキナーゼ抑制剤として広く用いられており、市販されているので市販品を好都合に用いることができる。
【0014】
【化1】
【0015】
チロシンキナーゼ抑制剤を本発明の絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。投与量は、用いるチロシンキナーゼ抑制剤の種類や患者の状態等に応じて適宜設定されるが、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。
【0016】
チロシンキナーゼ抑制剤を本発明の絨毛癌治療剤の有効成分として用いる場合、本発明の絨毛癌治療剤は、上記チロシンキナーゼ抑制剤のみから成っていてもよいし、また、各投与形態に適した、薬剤的に許容される担体及び/又は希釈剤を用いて製剤することもできる。製剤方法及びそのための各種担体は、医薬製剤の分野において周知である。薬剤的に許容される担体又は希釈剤は、例えば、生理緩衝液のような緩衝液や、賦形剤(砂糖、乳糖、コーンスターチ、リン酸カルシウム、ソルビトール、グリシン等)であってよく、結合剤(シロップ、ゼラチン、アラビアゴム、ソルビトール、ポリビニルクロリド、トラガント等)、滑沢剤(ステアリン酸マグネシウム、ポリエチレングリコール、タルク、シリカ等)等が適宜混合されていてもよい。投与形態としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などによる経口剤、吸入剤、注射剤、座剤、液剤などによる非経口剤などを挙げることができる。これらの製剤は一般的に知られている製法によって作ることができる。
【0017】
上記の通り、BDNFとTrkBの結合を抑制する物質も本発明の絨毛癌治療剤の有効成分として用いることができる。このような物質としては、まず、遊離のTrkB又はBDNFとの結合性を有するTrkB断片を挙げることができる。遊離のTrkBは、BDNFと結合するので、遊離のTrkBを投与すると、投与したTrkBは、細胞膜上の本来のTrkBと競合してBDNFと結合するので、細胞膜上の本来のTrkBと結合するBDNFの量が減少する。すなわち、遊離のTrkBは、細胞自体のTrkBとBDNFとの結合を競合的に抑制する。また、細胞膜上のTrkBがBDNFと結合する部分は、TrkBの細胞外ドメインである。従って、下記実施例に具体的に記載するように、TrkBの細胞外ドメイン、又は該細胞外ドメインを含むTrkBの断片も全長TrkBと同様、BDNFとTrkBの結合を競合的に抑制するので、本発明の絨毛癌治療剤の有効成分として用いることができる。ヒトTrkB遺伝子のcDNAの塩基配列を、それがコードするアミノ酸配列と共に配列番号1に示し、アミノ酸配列のみを取り出したものを配列番号2に示す。なお、ヒトTrkB遺伝子のcDNA及びそれがコードするアミノ酸配列は公知であり、GenBank Accession No.NM_006180として登録されている。配列番号2に示すアミノ酸配列(すなわちTrkB全長のアミノ酸配列)のうち、細胞外ドメインは、N末端から-31番目のアミノ酸(以下、「-31aa」のように記載)〜397aaまでである。この細胞外ドメインから成るTrkB断片も本発明の絨毛癌治療剤の有効成分として用いることができる。一般に、ポリペプチドはサイズが小さい方が製造が容易で細胞に取り込まれやすいので、上記TrkB断片はこれらの観点から好ましい。
【0018】
一般に、生理活性を有するポリペプチドにおいて、少数のアミノ酸が置換し、欠失し又は挿入された場合であってもその生理活性が維持される場合があることは当業者にとって周知である。従って、上記したTrkB又はその断片に加え、アミノ酸配列が、配列番号2で示されるアミノ酸配列、及び該アミノ酸配列のうち-31aa〜397aaである細胞外ドメイン領域のアミノ酸配列と90%以上、好ましくは95%以上、さらに好ましくは99%以上の配列同一性を有するポリペプチドであって、BDNFと結合して絨毛癌の治療効果を発揮するポリペプチドもそれぞれ遊離のTrkBやその細胞外ドメイン断片と同様に本発明の絨毛癌治療剤の有効成分として用いることができる。ここで、アミノ酸配列の配列同一性とは、一致するアミノ酸残基の数が最大となるように(必要に応じてギャップを挿入する)、2つのアミノ酸配列を並べ、一致したアミノ酸残基の数を完全長の配列のアミノ酸残基(2つの配列間で全アミノ酸残基の数が異なる場合、長い方のアミノ酸残基)の数で除することよって求めた値を意味する。そのような相同性の計算は、BLASTのような周知のソフトウェアによって容易に入手し得る。特に、アミノ酸配列が、配列番号2で示されるアミノ酸配列、又は該アミノ酸配列のうち-31aa〜397aaである細胞外ドメイン領域のアミノ酸配列において、1個ないし数個のアミノ酸が置換し若しくは欠失し、又は1個ないし数個のアミノ酸が挿入され若しくは付加されたアミノ酸配列であるポリペプチドであって、BDNFとの結合性、ひいては絨毛癌の治療効果を有するポリペプチドも本発明の絨毛癌治療剤の有効成分として用いることができる。なお、天然のタンパク質を構成する20種類のアミノ酸は、低極性側鎖を有する中性アミノ酸(Gly, Ile, Val, Leu, Ala, Met, Pro)、親水性側鎖を有する中性アミノ酸(Asn, Gln, Thr, Ser, Tyr, Cys)、酸性アミノ酸(Asp, Glu)、塩基性アミノ酸(Arg, Lys, His)、芳香族アミノ酸(Phe, Tyr, Trp)のように類似の性質を有するものにグループ分けでき、これらの間での置換であればペプチドの性質が変化しないことが多いことが知られている。従って、配列番号2で示されるアミノ酸配列又はその細胞外ドメイン領域のアミノ酸配列から成るポリペプチド中のアミノ酸残基を置換する場合には、これらの各グループの間で置換することにより、当該ポリペプチドのBDNF結合能が維持される可能性が高くなる。
【0019】
また、生理活性を有する2種類のポリペプチドが連結された融合ポリペプチドが、各ポリペプチドの生理活性を維持する場合があることからも明らかなように、生理活性を有するポリペプチドをそっくり含み、その一端又は両端に他のアミノ酸配列が連結されたポリペプチドであってもその生理活性が維持される場合があることは当業者にとって周知である。従って、上記したBDNFとの結合能を有するポリペプチドを含み、BDNFとの結合能を有するポリペプチドを本発明の絨毛癌治療剤の有効成分として用いることも可能である。この場合、上記したBDNFとの結合能を有するポリペプチドの一端又は両端に付加されるアミノ酸の数は、最終的なポリペプチドがBDNFとの結合能、ひいては絨毛癌の治療効果を発揮する限り特に限定されないが、合成の容易さ及び単位重量当たりの活性を高くする観点から、1個〜数個であることが好ましい。
【0020】
なお、一般に、ポリペプチド製剤においては、生体内でのプロテアーゼによる分解を受けにくくするためにポリペプチドの一端にポリエチレングリコール(PEG)鎖等を結合したものが広く用いられている。本発明の絨毛癌治療剤においても、同様に、上記したポリペプチドをそっくり含み、その一端にPEG鎖等の安定化構造を付加したものを有効成分として用いることができる。なお、PEG化によりペプチドを安定化する場合には、PEGのサイズは分子量数千〜5万、好ましくは1万〜5万程度である。また、ポリペプチドの一端にPEGを結合する方法は周知である。
【0021】
なお、本明細書及び特許請求の範囲において、遊離のTrkB若しくはBDNFとの結合性を有するその断片の、絨毛癌の治療効果を有する「修飾体」とは、配列番号2で表されるアミノ酸配列又はその細胞外ドメイン領域から成るアミノ酸配列とは異なるアミノ酸配列を持ち、BDNFとの結合性ひいては絨毛癌の治療効果を有する、上記したポリペプチド並びにそれらにPEG鎖等の安定化構造を付加したものを意味する。
【0022】
上記した、遊離のTrkB、その細胞外ドメイン断片やそれらの上記修飾体(以下、便宜的に「BDNF結合性TrkB断片等」ということがある)を絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。もっとも、体内への吸収性や消化酵素による分解を避ける観点から非経口投与が好ましい。投与量は、用いるチロシンキナーゼ抑制剤の種類や患者の状態等に応じて適宜設定されるが、非経口投与の場合、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。また、BDNF結合性TrkB断片等を有効成分として用いる場合も、上記と同様、常法に基づいて製剤することができる。
【0023】
BDNF結合性TrkB断片等は、それ自体を有効成分として用いることができるが、BDNF結合性TrkB断片等をコードする核酸を組み込んだ組換えベクターであって、細胞中でBDNF結合性TrkB断片等を発現することができる組換えベクターを有効成分として用いることもできる。哺乳動物の遺伝子治療用のベクターは、種々のものが公知であり、市販されているものも少なくないので、市販の遺伝子治療用ベクターのクローニング部位にBDNF結合性TrkB断片等をコードするDNAを挿入した組換えベクターを好ましく用いることができる。なお、所望の遺伝子をベクターに挿入して遺伝子治療用組換えベクターを作製する有料サービスも行われており、このような有料サービスを利用することも可能である。
【0024】
哺乳動物への組換えベクターの投与自体は、周知の方法により行うことができる。すなわち、好ましくは、治療すべき絨毛癌細胞の近傍の組織に注射等の非経口投与により投与することができる。組換えベクターをリン酸緩衝液(PBS)等の緩衝液に懸濁したものを投与することができる。投与に際し、細胞内への遺伝子ワクチンの侵入を容易にするために、注射部位に電界パルスを与えてもよい。この場合、電界の強さは、特に限定されないが、通常、10V/cm〜60V/cm程度、好ましくは25V/cm〜35V/cm程度、パルスの持続時間は、通常、20ミリ秒〜100ミリ秒、好ましくは、40ミリ秒〜60ミリ秒程度であり、パルスを通常、1回〜6回、好ましくは2回〜4回程度当てることができる。組換えベクターの投与量は、症状や神経損傷部位の状態等に応じて適宜選択することができるが、通常、組換えベクターの重量で1ng〜10mg程度、特に100ng〜1mg程度である。
【0025】
BDNFとTrkBとの結合を抑制する物質として、BDNFに対する抗体又はBDNF結合性TrkB断片等に対する抗体を用いることもできる。BDNF及びBDNF結合性TrkB断片等は容易に入手可能であるので、これらに対する抗体は、BDNF又はTrkBを免疫原として動物(ヒトを除く)に投与するして抗体を誘導することを含む常法により得ることができる。抗体は、ポリクローナル抗体でもモノクローナル抗体でもよく、モノクローナル抗体も常法であるハイブリドーマ法により作製することができる。抗体は、BDNFとTrkBとの結合を抑制できるものである必要があるので、モノクローナル抗体の場合には、得られたモノクローナル抗体のうち、BDNFとTrkBとの結合を抑制するモノクローナル抗体をスクリーニングする。ポリクローナル抗体の場合には、免疫原の全エピトープに体する種々の抗体が含まれるので、このようなスクリーニングを行わなくてもBDNFとTrkBとの結合を抑制するが得られる。
【0026】
上記抗体を絨毛癌治療剤の有効成分として用いる場合、投与経路は、経口でも非経口でもよく、非経口の場合、絨毛癌組織への直接投与、静脈内、筋肉内、皮下、皮内、経皮、直腸内、点眼等、通常の各種投与経路で投与可能である。もっとも、体内への吸収性や消化酵素による分解を避ける観点から非経口投与が好ましい。投与量は、用いる抗体の力価や患者の状態等に応じて適宜設定されるが、非経口投与の場合、通常、成人1日当たり、1 mg〜100,000 mg、好ましくは1 mg〜1,000 mg程度であるがもちろんこの範囲に限定されるものではない。また、上記抗体を有効成分として用いる場合も、上記と同様、常法に基づいて製剤することができる。
【0027】
BDNF又はTrkBの、細胞内における生産を抑制する物質を本発明の絨毛癌治療剤の有効成分として用いることもできる。このような物質として、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA(iRNA)を挙げることができる。BDNF遺伝子又はTrkB遺伝子の発現を抑制する抑制剤としては、BDNF遺伝子又はTrkB遺伝子のmRNAを標的とするiRNA、好ましくはsiRNAを挙げることができる。iRNAは、標的となるmRNAと相補的な鎖を含む二本鎖RNAであり、標的となるmRNAと結合してこれを切断するものである。siRNAは、サイズが21〜23塩基程度の短い(small)iRNAである。siRNAは、サイズが小さいので合成が容易で、それによるmRNAの切断部位を設定し易いので好ましい。siRNAによる遺伝子発現の抑制技術は、既に周知であり、mRNAの配列(cDNA配列)さえ提示すれば、それを標的とするsiRNAを設計し、そのsiRNAを発現ベクターに組み込んだ組換えベクターを作製するサービスを行なっている業者が多数存在するほどである。上記の通り、TrkB遺伝子のcDNAの配列は配列番号1に記載したとおりであり、また、BDNF遺伝子のcDNAの塩基配列(GenBank Accession No. NM_170735)は配列番号3に示す通りであるので、これらに対するsiRNAは当業者であれば容易に設定することができる。簡単に説明すると、siRNAは標的とするmRNAと相補的な鎖を含む二本鎖RNAで、そのサイズは通常、21〜23塩基であり、通常、二本鎖RNAの両端にそれぞれハングオーバーを有する。ハングオーバーのサイズは、それぞれ1塩基〜2塩基であり、ハングオーバー部分はデオキシヌクレオチドでもよい。また、mRNAとの相補性は、完全な相補性が好ましいが、1〜2塩基程度のミスマッチがあっても十分な切断作用を発揮する場合も多い。また、ハングオーバー部分は相補的でなくてもよい。siRNAは、mRNAの塩基配列中のaaに続く19〜21塩基として設定することが好ましい場合が多く、gc含量が50%前後(通常45〜55%程度)のものが好ましい。また、成熟タンパク質で切断される部分に設定されないように、5'末端から50塩基以上離れた部位に設定することが多い。
【0028】
siRNAはそのまま投与することもできるが、該siRNAを発現するDNAを哺乳動物細胞用の発現ベクターに組み込み、得られた組換えベクターを投与することにより、細胞内でsiRNAを生産させBDNF遺伝子又はTrkB遺伝子の発現を抑制してもよい。哺乳動物細胞用の発現ベクターは種々市販されており、それらのマルチクローニング部位に上記DNAを挿入することができる。なお、上記の通り、siRNAを発現するDNAを組み込んだ発現ベクターを作製する業者のサービスも利用できる。
【0029】
投与量は、絨毛癌の程度、患者の状態や体重等に応じて適宜選択されるが、抑制剤がsiRNAの場合、その投与量は、成人(体重60kg)1日当たり通常、0.01mg/kg〜10mg/kg程度、特に0.1mg/kg〜5mg/kg程度、siRNAを発現する組換えベクターの場合、治療全体を通して成人1日当たり0.01mg/kg〜10mg/kg程度、特に0.1mg/kg〜5mg/kg程度であるが、投与量はもちろんこれらに限定されるものではない。
【0030】
さらに、本発明の絨毛癌治療剤の有効成分として、BDNF遺伝子又はTrkB遺伝子のアンチセンスRNAを用いることもできる。アンチセンスRNAは、標的遺伝子のmRNAの全長又はその一部と相補的な塩基配列を有し、該mRNAとハイブリダイズして、mRNAが翻訳されることを抑制し、ひいては標的遺伝子の遺伝子産物が生産されることを抑制するものである。TrkB遺伝子及びBDNF遺伝子のcDNAの塩基配列はそれぞれ配列番号1及び配列番号3に記載されているので、これらのアンチセンスRNAも容易に調製することができる。アンチセンスRNAのサイズは、標的遺伝子のmRNAと特異的にハイブリダイズすることが可能で該mRNAの翻訳を抑制できるサイズであれば特に限定されないが、通常、20塩基〜mRNAのコード領域の全長程度である。
【0031】
iRNAの場合と同様、アンチセンスRNAもそのまま投与することもできるが、該アンチセンスRNAを発現するDNAを哺乳動物細胞用の発現ベクターに組み込み、得られた組換えベクターを投与することにより、細胞内でアンチセンスRNAを生産させBDNF遺伝子又はTrkB遺伝子の発現を抑制してもよい。哺乳動物細胞用の発現ベクターは種々市販されており、それらのマルチクローニング部位に上記DNAを挿入することができる。
【0032】
アンチセンスRNAの投与量は、絨毛癌の程度、患者の状態や体重等に応じて適宜選択されるが、上記したiRNAの投与量と同程度であってよい。
【0033】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【実施例】
【0034】
実施例1
材料および方法
細胞
絨毛癌細胞株であるJARおよびBeWoはATCC(バージニア州マナッサス)から購入した。JAR細胞は、10%ウシ胎仔血清(インビトロジェン社)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)を添加したDMEM/F12(インビトロジェン社、カリフォルニア州カールズバッド)で保持し、BeWo細胞は、15%ウシ胎仔血清と抗菌剤とを添加したF−12培地で、37℃、5%CO2環境で保持した。
【0035】
RT−PCR
JAR細胞およびBeWo細胞におけるTrkBリガンド(BDNFおよびNT−4/5)と受容体(TrkBおよびp75NTR)の発現を調査するため、従来のRT−PCRで、非特許文献7〜9のプライマーを用いた。ネガティブコントロールには、mRNAを含めなかった。
【0036】
JAR細胞およびBeWo細胞中のTrkBの転写レベルについての定量的リアルタイムRT−PCRを、非特許文献8のTrkBおよびβ−アクチンに対するプライマーおよびハイブリダイゼーションプローブにより、SmartCycler(タカラ、東京、日本)を用いて行った。TrkBに対するプライマーは受容体の触媒キナーゼドメインに合致し、切断されたアイソフォームの増幅を回避する(非特許文献10)。データはβ−アクチンの転写レベルに基づいて正規化した。
【0037】
PCRアレイ実験には、ヒトの細胞周期用およびアポトーシス用のRT2 Profiler PCRアレイ(SAバイオサイエンス社、メリーランド州フレデリック)を用い、メーカーのプロトコルに従ってそれぞれの96ウェルPCRアレイプレート中の84個の遺伝子のmRNAレベルを同時に検討した。RNeasy Mini kit(キアゲン、東京、日本)を用いて、全RNAを細胞から抽出した。RT2 First Strandキット(SAバイオサイエンス社)を用いて1μgの全RNAを逆転写し、次いで、LightCycler480(ロシュ・アプライド・サイエンス社、インディアナ州インディアナポリス)でリアルタイムPCRを行った。ΔΔサイクル閾値法によりデータを解析し、発現における変化倍率(fold change)を測定した。
【0038】
免疫測定
JAR細胞およびBeWo細胞を用いて、非特許文献6、8のように、BDNF、NT−4/5、およびTrkBの免疫蛍光測定を行った。
【0039】
ELISAには、JAR細胞およびBeWo細胞を、137mM NaCl、20mM Tris−HCl、1%ノニデットP-40、10%グリセロール、プロテアーゼインヒビターカクテル(ロシュ・アプライド・サイエンス社)を含有するバッファー中でホモジナイズし、8000×g、4℃で5分間、遠心した。上清中のBDNFおよびNT−4/5の定量を、非特許文献6、8のように行った。結果は、タンパク質濃度により正規化し、細胞(mg)当たりのBDNF(pg)またはNT−4/5(pg)として表した。
【0040】
血清中のヒト絨毛性ゴナドトロピン(hCG)−βタンパク質のレベルと、細胞および組織ホモジネートとを、RIA(三菱BCL社、東京、日本)を用いて測定した。このアッセイでの最小検出可能レベルは0.1ng/mlであり、アッセイ内およびアッセイ間の変動係数はそれぞれ4.5%および6.3%である。
【0041】
細胞培養および細胞の増殖とアポトーシスの測定
内在性TrkBリガンドの細胞増殖およびアポトーシスにおける役割を調べるため、JAR細胞およびBeWo細胞を96ウェル培養プレートに1×104個/ウェルの密度で播種した。細胞を一晩インキュベートし、種々のインヒビターで処理した。プレインキュベーション後、種々の用量のTrkBの可溶性細胞外ドメイン(R&Dシステムズ社、ミネソタ州ミネアポリス)、汎特異的(pan-specific)Trk受容体インヒビターであるK252a(カルバイオケム社、カリフォルニア州ラ・ホーヤ)(非特許文献11)、または不活性細胞膜非透過性K252b(カルバイオケム社)(非特許文献12)を用いて、または用いずに、48時間、無血清培地中で、細胞を培養した。さらに、一晩のプレインキュベーション後、96ウェル培養プレートに0.2×104個/ウェルの密度のJAR細胞およびBeWo細胞を、48時間、無血清培地中で、種々の用量のBDNF(R&Dシステムズ社)で処理するか、または処理しないことによりBDNFの細胞増殖およびアポトーシスへの影響を測定した。BDNFによる細胞増殖促進におけるPI3KおよびERK経路の関与を解析するため、JAR細胞およびBeWo細胞を、BDNF(30ng/ml)とともに、PI3KインヒビターであるLY294002(0.1〜1.0μM;シグマ社、ミズーリ州セントルイス)またはその不活性類似体であるLY303511(1.0μM;カルバイオケム社)の存在下または非存在下で培養した。いくつかの実験においては、細胞を、BDNFとともに、MAPK/ERKキナーゼ1/2インヒビターであるU0126(0.1〜10μM;カルバイオケム社)もしくはその不活性類似体であるU0124(10μM;カルバイオケム社)、またはLY294002とU0126の両方か、LY303511とU0124の両方の存在下または非存在下で培養した。
【0042】
トリパンブルー排除試験(インビトロジェン社)を用いて、細胞増殖を測定した。生存可能な細胞の数を、生存可能な細胞による色素排除に基づいて血球計算板上で測定した。アポトーシスの進行を測定するため、一部の細胞を、培養8時間において、定量的カスパーゼ3/7酵素アッセイにかけた。アポトーシス測定のための培養期間は、カスパーゼ3/7の活性化がTrkBの細胞外ドメインまたはK252aで12時間処理した細胞において明らかな最終的細胞死よりも早くに起こる、との仮説に基づいて選択した。カスパーゼ3/7活性を、非特許文献13のように、Caspase-Glo 3/7アッセイ(プロメガ社、ウィスコンシン州マディソン)によって測定した。細胞のアポトーシスについても、培養8時間においてin situ 末端デオキシヌクレオチジルトランスフェラーゼ媒介2’−デオキシウリジン 5’−二リン酸ニック末端標識法(TUNEL法)を用いてDNAの断片化を検出することにより分析した非特許文献14。さらに、非特許文献15のように、アポトーシスで誘導されたミトコンドリアの膜電位の変化をMitoTracker Orange CMTMRos(インビトロジェン社)を用いて検討した。いくつかの実験においては、培養8時間で、細胞をPCRアレイ実験に供した。
【0043】
In vivo試験
絨毛癌の腫瘍形成における内在性TrkBリガンドのin vivoでの役割を探るため、JAR腫瘍を有する胸腺欠損ヌードマウス(BALB/c nu/nu)(日本クレア、東京、日本)で、K252aの抗腫瘍活性を測定した。動物の世話および使用は秋田大学医学部動物実験委員会により承認を受けた。JAR細胞(0.1ml、5×106個)を4週齢のメスのヌードマウスの右脇腹の皮下に移植した。腫瘍容積がおよそ60mm3になったときに、動物への処理を開始した。動物の体重は処理の日において17〜21gであった。生理食塩水(500μg/kg)に溶かしたK252aの腹腔内投与を3日ごとに行った。同じ用量の投与も毎日継続して行った。ネガティブコントロールとして、K252b(500μg/kg)での処理または賦形剤のみの処置を行った。これらの実験のために選択したK252aおよびK252bの用量は先の調査(非特許文献16)に基づいた。式:腫瘍容積(mm3)=長さ×(幅)2×0.5を用いて、腫瘍容積を毎日測定した。処理の9日後にマウスを殺し、試験したすべてのマウスから血液サンプルを回収し、血清hCG−βレベルを測定した。ヘマトキシリン・エオシン(H&E)染色に加え、in vivoでの細胞増殖およびアポトーシスを、それぞれ、増殖性細胞核抗原(PCNA)の免疫染色、およびTUNELアッセイにより評価した。PCNAの免疫染色は、マウス抗PCNAモノクローナル抗体(Cell Signaling Technology社、マサチューセッツ州ダンバーズ)を1:4000希釈で用い、メーカーのプロトコルに従って行った。切除した腫瘍サンプル中のカスパーゼ3/7活性をCaspase-Glo 3/7アッセイにより測定した。データは、タンパク質濃度により正規化した。このデータはコントロールに対する増加倍率を示した。
【0044】
統計解析
マン・ホイットニーのU検定を行い、絨毛癌細胞株中のBDNF、NT−4/5、TrkB、およびhCG−βのレベルを比較した。薬剤処理による腫瘍容積への影響はスチューデンドt検定を用いて解析した。PCRアレイからのデータを、独立スチューデンドt検定により、有意差について試験した。一元配置ANOVA、次いでフィッシャーの保護最小有意差検定を用いて、その他の差を評価した。データは平均±標準誤差である。
【0045】
結果
絨毛癌細胞におけるニューロトロフィンとTrk受容体の発現
絨毛癌細胞におけるBDNF、NT−4/5、TrkBの発現について、PCRおよび免疫測定により検討した。これら3種すべての転写物が、JAR細胞とBeWo細胞のいずれにおいても認められた(図1A)。絨毛癌細胞におけるこれらの発現を、さらに、免疫蛍光染色(JAR細胞)により確認した(図1B)。加えて、p75NTRのmRNAも両細胞株で検出した(図1A)。ELISA分析により、両絨毛癌細胞において、BDNFのタンパク質レベルがNT−4/5よりも2.3倍高いことが示された(図1C)。hCG−βのタンパク質レベルはBeWo細胞よりもJAR細胞での方が高いが、ELISA分析および定量的リアルタイムRT−PCR分析によると、BDNFタンパク質、NT−4/5タンパク質、およびTrkB転写物のレベルは、JAR細胞とBeWo細胞とで差はなかった(図1C)。
【0046】
絨毛癌細胞の成長に対する内在性TrkBシグナル伝達のin vitro抑制
異なる絨毛癌細胞株におけるTrkBリガンドと受容体の両方の発現は、TrkBシグナル伝達系が、絨毛癌の成長において、オートクリンの役割を果たしている可能性があることを示唆する。内在性TrkBリガンドが絨毛癌細胞にとって生存因子として作用するかどうかを判定するため、我々は、TrkBの細胞外ドメインとK252aで処理した培養JAR細胞および培養BeWo細胞の増殖とアポトーシスとを評価した。図2Aに示されるように、コントロールの細胞数は、培養後に増加(13.5±0.9,×104個/ウェル)し、不活性のK252bで処理した場合ではなく、TrkBの細胞外ドメインまたはK252aのどちらかで処理した場合に、生存可能な細胞の数は、両細胞株で同じような効率で減少した。我々はさらに、種々のインヒビターで処理することにより、両細胞株においてカスパーゼ3/7活性の増大を検出した(図2B)。TrkBの細胞外ドメインおよびK252aでの処理によりTUNEL陽性の核の割合も増加した(図2C)。これはアポトーシスが誘導されたことを示す。絨毛癌細胞における内在性TrkBシグナル伝達の抑制によって誘導されるアポトーシスをさらに特徴づけるため、我々は、ミトコンドリアの膜電位感受性色素であるMitoTracker Orange CMTMRosを用いてミトコンドリアの機能の低下を検討し、アポトーシス中のミトコンドリアの膜電位の状態を評価した(非特許文献17)。機能的なミトコンドリアは色素を取り込み、オレンジ色の蛍光で点状の染色パターンを示すが、アポトーシスを起こした細胞のミトコンドリアは染色されない。我々は、コントロール細胞でのMitoTracker色素染色が、それらのミトコンドリアの局在として予想される通りに点状に現れる(図2C)ことを見出した。不活性のK252bでの処理ではなく、rkBの細胞外ドメインまたはK252aのどちらかでの処理により、MitoTracker色素染色は減少した(図2C)。
【0047】
BDNF処理の絨毛癌細胞増殖への影響とin vitroのPI3KおよびERKの媒介的役割
BDNFの細胞増殖への影響を検討するため、細胞を、種々の用量のBDNFの存在下または非存在下で培養した。BDNFの細胞増殖への影響は、細胞をより高い密度で培養した場合、おそらくオートクリン因子の蓄積により、より目立たなくなった(データ非掲載)。従って、我々は、低い密度で細胞を培養し、BDNFを含む細胞由来因子の影響を排除した。この条件下では、BDNF処理は、両絨毛癌細胞において用量依存的に細胞増殖を増加させた(図3A)。
【0048】
我々はさらに、2種類の異なる絨毛癌細胞株において、BDNFによって誘導された細胞増殖の下流メディエーターとしてのPI3KおよびERKシグナル伝達経路の役割を解析した。低密度で細胞数を増加させるBDNFの能力は、不活性類似体であるLY303511で処理するのではなくPI3KインヒビターであるLY294002とともに処理するか、または不活性類似体であるU0124で処理するのではなくMAPKキナーゼ(MEK)1/2インヒビターであるU0126とともに処理するかのどちらかによって抑制され(図3B)、また、PI3KインヒビターおよびMEK1/2インヒビターの両方とともに処理することによって、完全に抑制され、これはこれらの経路のみが内在性TrkBシグナル伝達の媒介に関与しているとのことを示している(図3B)。しかしながら、PI3KインヒビターおよびMEK1/2インヒビターへの感受性はJAR細胞とBeWo細胞とで異なっていた。より低用量のU0126(0.5μMおよび1.0μM)は、BeWo細胞ではBDNFによる細胞増殖刺激を抑制するには効果がなかった。
【0049】
BDNFおよびTrkBに対するsiRNAを用いて、我々はさらに、内在性BDNF/TrkBシグナル伝達の抑制後の絨毛癌細胞の成長阻害について検証した。BDNFまたはTrkBのノックダウンは、細胞増殖を、それぞれ50.5%、47.8%阻害し、細胞成長における内在性BDNF/TrkBシグナル伝達の特異的役割を支援した。しかしながら、両siRNAの細胞成長への阻害効果は、TrkBの細胞外ドメインおよびTrk受容体インヒビターの場合(>80%阻害)よりも効力が少なかった。加えて、BDNF siRNAをトランスフェクションした細胞におけるBDNFによる細胞増殖刺激は、おそらく、siRNAによって媒介されたノックダウンの後のBDNFまたはTrkBの残存発現(図5C)のために、そのsiRNAをトランスフェクションしなかった細胞の場合(コントロールの223%)よりも弱かった(コントロールの177%)。
【0050】
内在性TrkBシグナル伝達抑制後の培養絨毛癌細胞における細胞周期関連遺伝子およびアポトーシス関連遺伝子のmRNA発現プロファイル変化
TrkBシグナル伝達インヒビターによる細胞増殖阻害および生存阻害の根底にある分子機構を探るため、TrkBの細胞外ドメイン(500nM)で処理した、または処理しなかったJAR細胞から全RNAを精製し、PCRアレイ解析に使用して、細胞周期関連遺伝子およびアポトーシス関連遺伝子の発現をプロファイルした。TrkBの細胞外ドメインで処理することにより、アレイ中でスクリーニングされた84種類の細胞周期関連mRNAのパネル全体にわたって、3つの刺激性遺伝子(CCND1、DDX11、DNM2)の発現が2倍以上減少し、2つの阻害遺伝子(CDKN2B、SERTAD1)の発現が2倍以上増加した(図4A)。アポトーシス関連遺伝子の解析においては、異なるアポトーシス促進性の遺伝子の転写レベルが、内在性TrkBシグナル伝達の抑制に応答して増加した(図4B)。発現が増加したアポトーシス促進性遺伝子のうち、TNFにおいて、もっとも有意な増加(25倍)が認められた(図4B)。対照的に、抗アポトーシス性の遺伝子の発現への有意な影響は、TrkBの細胞外ドメインで処理した細胞では観察されなかった。
【0051】
絨毛癌成長へのTrk受容体インヒビターのIn vivoでの影響
観察されたTrkBインヒビターによる絨毛癌細胞成長のin vitro阻害が、in vivoでの抗腫瘍活性となるのかどうかを判定するため、K252aを、JAR細胞の腫瘍異種移植片を有する胸腺欠損ヌードマウスに投与した。これらの調査にはJAR細胞を用いた。これは、この細胞では、腫瘍マーカーであるhCG−βの産生レベルが高かった(図1C)ためである。腫瘍容積がおよそ60mm3に達したときに、動物に、K252aまたはK252bを、500μg/kgで3日ごとに与えた。K252aで処理した動物の腫瘍は、コントロールに比べて有意に小さくなり、これは処理開始から3日後程度の早期に始まり、9日目の実験終了まで続いた(図5A)。処理9日目で、最大の腫瘍成長阻害(68%)が観察された。図5Bに示すように、JAR腫瘍を有するマウスにおける血清hCG−βレベルは腫瘍容積に相関し、腫瘍マーカーとしてのその役割と一致した。K252aで処理した動物は、処理9日目において、コントロールと比べて87%の血清hCG−βレベルの減少を示した(図5C)。試験されたすべての動物において、実験期間中、腫瘍の転移は観察されなかった。K252aを9日目まで毎日投与した場合に、成長抑制効率が比較可能であった(67%の腫瘍成長阻害、82%の血清hCG−βレベル増加)。試験したすべての動物において、実験中、重大な副作用は観察されず、また、K252aで処理したグループでは、調査中、有意な体重減少はなかった(賦形剤の場合には、22.08±0.54g、K252aの場合には、21.15±0.47g、K252bの場合には、21.13±0.45g)。
【0052】
切除した腫瘍をさらに検査し、K252aのin vivoでの影響を測定した。H&E染色による組織病理学的検査では、K252aで処理したマウスの腫瘍中に、細胞増殖抑制を示唆する有糸分裂活性の減少、および生存抑制を示唆するクロマチン凝縮を有する細胞数の増加が検出された(図6A)。PCNA染色によりK252a処理の細胞増殖抑制への影響を確認し、アポトーシスの誘導をTUNELアッセイにより確認した(図6A)。我々はさらに、カスパーゼ活性を定量化することにより、腫瘍中でK252aにより誘導されるアポトーシスを特徴づけ、またK252aで処理したマウスの腫瘍中でカスパーゼ3/7の活性が17.7倍減少することを観察した(図6B)。重要なのは、不活性細胞膜非透過性K252bが、試験されたすべてのパラメーターで効果がなかったことである。
【0053】
実施例2
siRNAのトランスフェクション
アプライドバイオシステムズ社(カリフォルニア州フォスターシティ)製の、BDNFを標的(s1962;センスとしてuuacuaugguuauuucauatt(配列番号5)、アンチセンスとしてuaugaaauaaccauaguaagg(配列番号6)、および、センスとしてcccuuaccauggauagcaatt(配列番号7)、アンチセンスとしてuugcuauccaugguaagggcc(配列番号8))、またはTrkBを標的(s9750;センスとしてgguuagaaaucaucaacgatt(配列番号9)、アンチセンスとしてucguugaugauuucuaacctt(配列番号10)、および、s9749;センスとしてgaauugacgauggugcaaatt(配列番号11)、アンチセンスとしてuuugcaccaucgucaauucca(配列番号12))とした2種類の特異的なsiRNA二本鎖のセット(Silencer(登録商標) Select Pre-designed siRNA)を用いてBDNFまたはTrkBのノックダウンを行った。Select Negative Control #1 siRNAおよびGAPDH Positive Control siRNAを、それぞれネガティブコントロールおよびポジティブコントロールとして使用した。siPORT(商品名) NeoFX(商品名)Transfection Agent(アプライドバイオシステムズ社)を、メーカーの説明書に従ってリバーストランスフェクション法に使用した。このポジティブコントロールとKDalert(商品名)GAPDH Assay Kit(アプライドバイオシステムズ社)とを用い、siRNAのトランスフェクション条件をJAR細胞において最適化した。BDNFおよびTrkBのノックダウンのために、細胞を終濃度5nMの各siRNA二本鎖セットでトランスフェクションした。この細胞を24時間インキュベートし、次いで無血清培地を10%FBS含有完全培地に交換した。siRNAのトランスフェクションの72時間後に、リアルタイムRT−PCR解析用に細胞を回収し、BDNFおよびTrkBのmRNAの発現のノックダウンを確認した。培養液中に蓄積した内在性BDNFを除去するために、siRNAのトランスフェクション後72時間で、完全培地を無血清培地に交換した。48時間のインキュベーションの後、細胞増殖をトリパンブルー排除試験を用いて測定した。いくつかの実験においては、siRNAのトランスフェクション後72時間に、BDNF siRNAをトランスフェクションした細胞をBDNF(30ng/ml)(R&Dシステムズ社、ミネソタ州ミネアポリス)培地で処理し、BDNFによる細胞成長への影響を評価した。
【0054】
RT−PCR
絨毛癌細胞におけるニューロトロフィン(神経成長因子、NGF、およびニューロトロフィン−3、NT−3)およびTrk受容体(TrkA、およびTrkC)の発現を調査するための従来のRT−PCRに用いる、NGF、NT−3、TrkA、およびTrkCに対するプライマーは以下に示す通り:NGFには、センスとして5'-tcatcatcccatcccatcttcc-3'(配列番号13)、アンチセンスとして5'-tccagtgctttgagtcaatgcc-3'(配列番号14);NT−3には、センスとして5'-gccagaataacacagactcagc-3'(配列番号15)、アンチセンスとして5'-tcggtgactcttatgctccg-3'(配列番号16);TrkAには、センスとして5'-catcgtgaagagtggtctccg-3'(配列番号17)、アンチセンスとして5'-gagagagactccagagcgttgaa-3'(配列番号18);TrkCには、センスとして5'-agcgtctggctggactatgt-3'(配列番号19)、アンチセンスとして5'-gtgtggtgagccggttactt-3'(配列番号20)。β−アクチンに対するプライマーは既に記載されている(非特許文献1)。PCR反応は、94℃で30秒の変性、57℃(TrkA)、60℃(TrkCおよびβ−アクチン)、または62℃(NGFおよびNT−3)で30秒のアニーリング、および72℃で30秒の伸長、による増幅を35サイクルとした。ネガティブコントロールにはmRNAを含めなかった。
【0055】
細胞培養
hCG刺激によるTrkBリガンドおよび受容体の産生調節に取り組むため、アンチセンスの絨毛性ゴナドトロピンβ(aCGβ)(Koji Nakabayashi博士から分与(非特許文献29))を有する発現ベクターpM2HAでトランスフェクションしたJAR細胞を使用した。JAR−pM2HA細胞とともに、トランスフェクションしていないJAR細胞をコントロールとして実験において使用した。これらの細胞株を、成長曲線およびマイコプラズマ解析を用いるとともに、これらの細胞の形態により検証した。JAR−aCG細胞およびJAR−pM2HA細胞は、10%FBS、ジェネティシン(62.5μg/ml)(インビトロジェン社)、ペニシリン(100U/ml)、ストレプトマイシン(100μg/ml)を添加したDMEM/F12で保持した。トランスフェクションした細胞におけるhCG−β、BDNF、およびNT−4/5の測定のために、細胞をサブコンフルエントな状態まで増殖させ、PBSで洗浄し、次いで無血清培地に交換した。一晩培養した後、その細胞のホモジネートをRIAまたはELISAに供し、本文中に記載したように、それらのhCG−βタンパク質、BDNFタンパク質またはNT−4/5タンパク質のレベルを測定した。定量的リアルタイムRT−PCRを行い、非特許文献30のように、これらのトランスフェクションした細胞におけるTrkBの転写レベルを測定した。
【0056】
結果を図7〜図9に示す。
【特許請求の範囲】
【請求項1】
脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する絨毛癌の治療剤。
【請求項2】
チロシンキナーゼ抑制剤、遊離のTrkB若しくはBDNFとの結合性を有するその断片又は絨毛癌の治療効果を有するそれらの修飾体又はTrkB若しくは前記断片又は前記修飾体を細胞内で生産する組換えベクター、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、BDNF又はTrkBに対する抗体、及びBDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクターから成る群より選ばれる少なくとも1種を有効成分として含有する請求項1記載の治療剤。
【請求項3】
チロシンキナーゼ抑制剤、遊離のTrkB又はBDNFとの結合性を有するTrkB断片、及びBDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクターから成る群より選ばれる少なくとも1種を有効成分として含有する請求項2記載の治療剤。
【請求項4】
チロシンキナーゼ抑制剤が、K252aである請求項3記載の治療剤。
【請求項5】
遊離のTrkB又はBDNFとの結合性を有するその断片が、BDNFと結合するTrkBの細胞外ドメインを含むTrkB断片である請求項3記載の治療剤。
【請求項6】
BDNF遺伝子又はTrkB遺伝子に対する干渉RNAが、BDNF遺伝子又はTrkB遺伝子に対するsiRNAである請求項3記載の治療剤。
【請求項1】
脳由来神経栄養因子(BDNF)及び/又は脳由来神経栄養因子受容体(TrkB)の抑制剤を有効成分として含有する絨毛癌の治療剤。
【請求項2】
チロシンキナーゼ抑制剤、遊離のTrkB若しくはBDNFとの結合性を有するその断片又は絨毛癌の治療効果を有するそれらの修飾体又はTrkB若しくは前記断片又は前記修飾体を細胞内で生産する組換えベクター、BDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクター、BDNF又はTrkBに対する抗体、及びBDNF遺伝子若しくはTrkB遺伝子に対するアンチセンス核酸又は該アンチセンス核酸を細胞内で生産する組換えベクターから成る群より選ばれる少なくとも1種を有効成分として含有する請求項1記載の治療剤。
【請求項3】
チロシンキナーゼ抑制剤、遊離のTrkB又はBDNFとの結合性を有するTrkB断片、及びBDNF遺伝子若しくはTrkB遺伝子に対する干渉RNA又は該干渉RNAを細胞内で生産する組換えベクターから成る群より選ばれる少なくとも1種を有効成分として含有する請求項2記載の治療剤。
【請求項4】
チロシンキナーゼ抑制剤が、K252aである請求項3記載の治療剤。
【請求項5】
遊離のTrkB又はBDNFとの結合性を有するその断片が、BDNFと結合するTrkBの細胞外ドメインを含むTrkB断片である請求項3記載の治療剤。
【請求項6】
BDNF遺伝子又はTrkB遺伝子に対する干渉RNAが、BDNF遺伝子又はTrkB遺伝子に対するsiRNAである請求項3記載の治療剤。
【図3】

【図4】

【図5】

【図7】

【図9】
【図1】
【図2】
【図6】
【図8】
【図4】
【図5】
【図7】
【図9】
【図1】
【図2】
【図6】
【図8】
【公開番号】特開2012−41311(P2012−41311A)
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願番号】特願2010−185384(P2010−185384)
【出願日】平成22年8月20日(2010.8.20)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り Endocrinology,July 2010,Volume 151,Issue 7,pp.3006−3014(2010年5月12日にdoi:10.1210/en.2009−1378としてwebにて先行開示)
【出願人】(504409543)国立大学法人秋田大学 (210)
【Fターム(参考)】
【公開日】平成24年3月1日(2012.3.1)
【国際特許分類】
【出願日】平成22年8月20日(2010.8.20)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り Endocrinology,July 2010,Volume 151,Issue 7,pp.3006−3014(2010年5月12日にdoi:10.1210/en.2009−1378としてwebにて先行開示)
【出願人】(504409543)国立大学法人秋田大学 (210)
【Fターム(参考)】
[ Back to top ]