
統合失調症を処置するためのビフェプルノックス用量
本開示は、統合失調症を有する患者の処置のためのビフェプルノックスの日用量に関する。そのような用量は、少なくとも1種のビフェプルノックス化合物の有効量を含んでなる製薬学的組成物を患者に投与することを含んでなる統合失調症を処置する方法において効果的に使用され、他の効果の中でも、体重増加なし、非空腹時トリグリセリドレベルおよび/もしくは総コレステロールレベルの改善のような統合失調症処置と関連する副作用の軽減をもたらす。処置効果は、例えば患者におけるPANSS合計スコアの減少および統合失調症の増悪までの時間の増加および精神病症状の改善である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、統合失調症を有する患者の処置のためのビフェプルノックスの日用量、安定した統合失調症を有する患者のそして統合失調症の急性増悪を有する患者のビフェプルノックスでの処置に、そして少なくとも1種のビフェプルノックス化合物の用量を含んでなる製薬学的組成物に関する。
【0002】
統合失調症は、陽性および陰性症状、認知障害および鬱病を包含する重篤なそして変わりやすい症状を特徴とする生涯にわたる日常生活に支障をきたす精神障害である。疾患の経過は4つの主要な時期:発病前、急性、安定/維持および後期経過に分けられることができる。発病前期は、陽性症状の発症前に起こる症状をさす。急性期中に患者は妄想および幻覚のような明白な陽性症状を経験する。安定/維持期は2つの亜期(subphase)に分けることができる。疾患の最初の5〜10年は陽性症状の多数の増悪を特徴とすることが多く、より安定な期間が急性エピソードの間に散在する。この亜期の後に症状の安定化および増悪の数の減少を特徴とするプラトー期が続く。維持期中の重要な処置目標は、地域社会への患者の復帰を促進しそして長期の維持計画を定めることである。疾患の後期経過期において、陽性症状は年齢とともに減少する傾向があり、そして長期的な障害を有する多数の患者はある程度の社会的および職業的能力を取り戻し、しかしながら、長年の機能障害の影響はめったに克服されない。
【0003】
統合失調症の維持および後期経過期において、患者の大部分は、再発の防止;認知機能の維持;体重増加、高血糖および脂質異常症の予防;ならびに生活の質の改善を包含する、さらなる症状改善および精神病症状の長期制御の必要性のような多数の問題に直面し続ける。さらに、陽性症状は、各々の続いて起こるエピソードで処置に対してより耐性になり得る。この概念と一致して、統合失調症を有する患者の85%〜90%は臨床的増悪を経験する。さらに、抗精神病薬を与えられた患者の少なくとも半分は処方された処置処方計画を順守せず、従って、再発の危険がある。従って、処置を改善することを目指す熱心な努力にもかからず、統合失調症を有する患者の大部分は重篤な障害を有し;再発することが多く、そして入院を必要とし得る。
【0004】
統合失調症を処置するために現在使用される化合物は、いくつかの望ましくない副作用と関連している。これらの副作用には体重増加、高プロラクチン血症、上昇したトリグリセリドレベル、メタボリック症候群(マーカー:糖尿病、高脂血症、高血圧および肥満)、延長したQTc間隔、グルコース異常ならびに錐体外路症状の表出が包含される。例えば、延長したQTc間隔、すなわち、心電図における補正QT間隔は、問題のある心律動もしくは心臓不整脈をもたらし得る。同様に、リスペリドンおよびオランザピンのような従来の非定型抗精神病薬で認められる体重増加は、心臓血管疾患および糖尿病の増加した危険性と関連している。
【0005】
さらに、薬剤を用いる統合失調症処置は長期間にわたってであり得る。そのようなものとして、これらの望ましくない副作用は日常的にならびに彼らの長期の健康に寄与して患者に影響を及ぼす。これらの副作用はまた、患者の処置処方計画の不順守にもつながり得る。処置が限られたそして/もしくは短い期間にわたってである場合でさえ、副作用は処置処方計画を順守しようとする患者の意欲に影響を及ぼす。
【0006】
従って、これらの望ましくない副作用を防ぎそして/もしくは軽減するならびに統合失調症処置を受けている患者のベースライン症状を維持し、軽減しそして/もしくは改善する方法の必要性がある。
【発明の概要】
【0007】
少なくとも1種のビフェプルノックス化合物の用量、特に20〜30mgの日用量を含んでなる(製薬学的組成物)で統合失調症患者を処置することは、これらの副作用の1つもしくはそれ以上および統合失調症の1つもしくはそれ以上の症状を軽減しそして/もしくは防ぐことを可能にすることを本発明者等は見出した。本発明の態様において、用量は毎日1回投与される。特定の用量の態様は、20mg用量および30mg用量である。特に好ましいのは、20mgの日用量である。
【0008】
この処置の好ましい効果には、患者における陽性および陰性症状評価尺度(PANSS)合計スコアの減少、体重の維持、トリグリセリドレベルおよび/もしくは総コレステロールレベルの維持および/もしくは改善、特に慢性の安定した統合失調症を有する患者における統合失調症の臨床的安定の維持(処置効果は、例えば増悪までの時間の増加である)、1つもしくはそれ以上の精神病症状の改善または投与前のベースライン測定と同様の錐体外路兆候および症状(EPS)プロフィールの維持および/もしくは減少が包含されるがこれらに限定されるものではない。他の好ましい効果は、高血糖および/または1つもしくはそれ以上の糖尿病関連有害事象の発生の減少である。
【0009】
本発明の態様において、ビフェプルノックスは、特に20〜30mgの日用量において、統合失調症を有する患者の長期処置に使用される。「長期処置」という用語は、例えば少なくとも3ヶ月もしくは少なくとも6ヶ月の処置をさす。
【0010】
本明細書に開示される態様は、本開示の様々な態様のいくつかの一般的な概説を提供するが、いかなる方法によっても本開示の範囲を限定するものではない。
【0011】
ビフェプルノックス化合物は米国特許第6,225,312号および米国特許第7,030,241号に記述され、これらの内容は引用することにより本明細書に組み込まれる。この化合物(7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン(ビフェプルノックス)の塩酸塩はWO97/36893に記述されそして請求され、そしてモノメタンスルホン酸塩はWO02/066449に記述されそして請求される。これらの特許公開の第二のものにおいて、N,N,N−ビス(2−エタノール)−m−フェニルベンジルアミンの反応性メシル酸エステルと7−アミノ−2(3H)−ベンゾキサゾロンとの間の反応によるモノメタンスルホン酸塩の直接形成が開示される。ビフェプルノックスモノメタンスルホン酸塩の安定な多形は、WO2005/016898に開示されそして請求される。また「ビフェプルノックス化合物」という用語に包含されるのは、ビフェプルノックスN−オキシドである。ビフェプルノックスN−オキシドは、WO2007/023141に開示されそして請求される。
【0012】
ビフェプルノックス化合物は、統合失調症、他の精神病性障害(特に精神病)およびパーキンソン病を包含するCNS(中枢神経系)障害の処置に適応される。本発明の枠組みにおいて、投薬量強度(もしくは用量)はビフェプルノックス塩基(bifeprunox base)に相当する量で表される。本明細書において用いる場合、「ビフェプルノックス塩基」という用語は以下の式:
【0013】
【化1】

【0014】
を有する化合物7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン(INNビフェプルノックス)をさす。
【0015】
本明細書において用いる場合、「ビフェプルノックス化合物」という用語は活性化合物7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン、そのN−オキシドおよび製薬学的に許容しうる塩、その溶媒和物および水和物をさす。N−オキシドがビフェプルノックス化合物として用いられる場合、ミリグラム単位の量は、当業者が酸化物なしのビフェプルノックス化合物に選択する量と同じ量である。さらに、ビフェプルノックスもしくはそのN−オキシドの製薬学的に許容しうる塩は、当該技術分野において周知である標準的な方法を用いて、例えば、本発明の化合物を適当な酸、例えば無機酸もしくは有機酸と混合することにより得ることができる。
【0016】
本開示は、少なくとも1種のビフェプルノックス化合物の用量を含んでなる製薬学的組成物を処置を必要とする患者に投与することにより統合失調症の処置と関連する症状を維持し、軽減しそして/もしくは改善するための統合失調症の処置におけるビフェプルノックスの使用に関する。例えば、製薬学的組成物は、10mg〜40mgもしくはさらに例えば20mg〜30mgのビフェプルノックス化合物のような5mg〜40mgの間の量の少なくとも1種のビフェプルノックス化合物を含んでなる。1つの態様において、ビフェプルノックスは体重の問題を有するかもしくは体重の問題を起こしやすい統合失調症を有する患者の処置において用いられる。
【0017】
本発明の態様は、用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、統合失調症を有する患者の処置のためのビフェプルノックスの日用量に関する。特に、該用量は、安定した統合失調症およびさらに特に慢性の安定した統合失調症を有する患者において臨床的安定を維持するために有用である。その態様は、それぞれ20mgの用量および30mgの用量である。
【0018】
さらなる態様において、本発明は、用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、急性増悪統合失調症を有する患者の処置のためのビフェプルノックスの日用量に関する。その態様は、好ましい副作用で該処置において用いられる、それぞれ20mgの用量および30mgの用量である。
【0019】
本発明の態様は、ビフェプルノックス(すなわち、少なくとも1種のビフェプルノックス化合物)が気分安定剤リチウムと組み合わせて投与される、精神病および気分障害を有する(特に統合失調症を有する)患者の処置における使用のためのビフェプルノックスおよび該使用のためのキットに関する。
【0020】
本発明のさらなる態様は、少なくとも1種のビフェプルノックス化合物が抗鬱剤(特にSSRI、特にパロキセチン)と組み合わせて投与される、CNS障害を有する(特に統合失調症を有する)患者の処置における使用のためのビフェプルノックスおよび該使用のためのキットに関する。
【0021】
本発明のさらなる態様は、それぞれ、CYP2C9阻害剤(例えばフルコナゾール)と、CYP3A4阻害剤(例えばケトコナゾールおよびカルバマゼピン)と、CYP2D6阻害剤(例えばパロキセチン)とそしてH2−アンタゴニスト(例えばファモチジン)とビフェプルノックスとの共投与(のための方法)および該処置のためのキットに関する。
【0022】
本開示の態様において、少なくとも1種のビフェプルノックス化合物はメシル酸ビフェプルノックスを含んでなる。好ましくは、少なくとも1種のビフェプルノックス化合物はメシル酸ビフェプルノックスである。メシル酸ビフェプルノックスは、α、γもしくはδ結晶多形形態およびその混合物から選択することができる。例えば、少なくとも1種のビフェプルノックス化合物は、αおよびγ多形形態から選択される少なくとも1種の多形形態を含んでなる。
【0023】
本開示によるαメシル酸ビフェプルノックスの結晶多形形態は、WO2005/016898に開示されるような少なくとも物理化学パラメーターにより定義される。
【0024】
別の態様において、本開示は、少なくとも約50重量パーセント(wt.%)、少なくとも約60wt.%、少なくとも約70wt.%、少なくとも約80wt.%、少なくとも約90wt.%もしくは少なくとも約95wt.%のメシル酸ビフェプルノックスが多形α形態であるメシル酸ビフェプルノックスを提供する。別の態様において、製薬学的組成物は、メシル酸ビフェプルノックスのあらゆるγもしくはδ多形形態を実質的に欠いている。別の態様において、本開示により提供されるメシル酸ビフェプルノックスは10wt.%未満、5wt.%未満、2.5wt.%未満のγもしくはδ多形形態のメシル酸ビフェプルノックスを含んでなる。別の態様において、少なくとも約99wt.%のメシル酸ビフェプルノックスは多形α形態である。
【0025】
多形形態αの製造は、WO2005/016898に記述される方法に従って実施することができる。
【0026】
本開示の少なくとも1種のビフェプルノックス化合物は、活性物質が当該技術分野において既知である方法により固形形態で存在する投与形態物に調合することができる。該投与形態物の例は(場合によりコーティングされていてもよい)錠剤、カプセル剤、粒状エアロゾル、座薬および懸濁剤である。そのような投与形態物は、少なくとも1種のビフェプルノックス化合物を不活性の製薬学的に許容しうる賦形剤および担体と混合することにより製造することができる。
【0027】
本開示の製薬学的組成物は少なくとも1種の製薬学的賦形剤を含んでなることができる。適当な賦形剤の限定されない例には、沈殿防止剤(例えば、ゴム、キサンタン、セルロース誘導体および糖)、保湿剤(例えば、ソルビトール)、可溶化剤(例えば、エタノール、水、PEGおよびプロピレングリコール)、界面活性剤(例えば、ラウリル硫酸ナトリウム、スパン(Span)、ツィーン(Tween)およびセチルピリジン)、防腐剤、酸化防止剤(例えば、パラベン、ならびにビタミンEおよびC)、固化防止剤(anti−caking agent)、コーティング剤、キレート剤(例えば、EDTA)、安定剤、抗微生物剤、抗真菌もしくは抗菌剤(例えば、パラベン、クロロブタノール、フェノール、ソルビン酸)、等張剤(例えば、糖、塩化ナトリウム)、増粘剤(例えば、メチルセルロース)、香料(例えば、チョコレート、ザルマンチン(thalmantin
)、アスパルテーム、ルートビアもしくはスイカまたはpH7〜9で安定な他の香料)、消泡剤(例えば、シメチコン、Mylicon(R))、崩壊剤、流動助剤(flow aid)、潤滑剤、添加剤、着色剤、希釈剤、湿潤剤、防腐剤、担体、結合剤(例えば、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、他のセルロース系材料および澱粉)、希釈剤(例えば、ラクトースおよび他の糖、澱粉、第二リン酸カルシウムならびにセルロース系材料)、崩壊剤(例えば、澱粉ポリマーおよびセルロース系材料)、流動促進剤および不水溶性もしくは水溶性潤滑剤もしくは平滑剤が包含される。
【0028】
1つの実例となる投与形態物は、活性物質(本明細書に記述されるようなビフェプルノックス)の粉砕しそしてふるいにかけた用量のほかに、ラクトース1水和物、微晶質セルロース、澱粉グリコール酸ナトリウム(例えばタイプA)、ステアリルフマル酸ナトリウムおよび場合によりコロイド状無水シリカを含んでなる。1つの態様において、ラクトースは、錠剤コアの総重量に基づいて約20重量%〜約90重量%、約70重量%〜約90重量%もしくは約75重量%〜約85重量%の量で存在する。微晶質セルロースは、錠剤コアの総重量に基づいて約5重量%〜約90重量%、約10重量%〜約15重量%もしくは約11重量%〜約12重量%の量で存在する。澱粉グリコール酸ナトリウム(例えばタイプA)は、錠剤コアの総重量に基づいて約0.1重量%〜約2.5重量%、約0.3重量%〜約0.7重量%もしくは約0.5重量%の量で存在する。ステアリルフマル酸ナトリウムは、錠剤コアの総重量に基づいて約0.1重量%〜約1.5重量%、約0.6重量%〜約1.3重量%もしくは約1.0重量%の量で存在する。コロイド状無水シリカは、粉末の流動特性を向上するために場合により製剤に加えてもよい。所望に応じて、コロイド状無水シリカは、錠剤コアの総重量に基づいて約0.05重量%〜約0.5重量%もしくは約0.4重量%の量で典型的に存在する。任意のコーティングの量は、錠剤コアの総重量に基づいて約2.0重量%〜約5.0重量%、約3.0重量%〜約4.0重量%もしくは約3.5重量%である。
【0029】
少なくとも1種の態様において、本開示の少なくとも1種のビフェプルノックス化合物を含んでなる製薬学的系組成物は、それを必要とする患者、例えばヒト患者に投与することができる。
【0030】
本開示はまた、患者におけるPANSS合計スコアを減らすこと、体重を維持すること、トリグリセリドレベルおよび/もしくは総コレステロールレベルを維持することおよび/もしくは改善すること、統合失調症の臨床的安定を維持すること、1つもしくはそれ以上の精神病症状を改善することまたは投与前のベースライン測定と同様のEPSプロフィールを維持することにも関するが、これらに限定されるものではない。本開示はまた、高血糖および/もしくは糖尿病関連有害事象の発生を減らす方法にも関する。これらの方法は、以下に提供される下記の臨床実施例において例示される。
【0031】
米国特許出願第10/920,361号、第10/920,386号および第11/354,652号は、それらの全部が本明細書に引用することにより本明細書に組み込まれる。本明細書における全てのデータは概算であり、そして例えば使用した装置ならびにピーク位置およびピーク強度に影響を及ぼす他のパラメーターにより通常の測定誤差を受けることが理解される。他に特に定義されないかもしくは他に文脈が要求しない限り、「約」という用語は本明細書において用いる場合に列挙した値の±5%を一般に意味する。
【図面の簡単な説明】
【0032】
【図1】パネル34 PANSS陽性スコア(FAS、LOCF)を示す。
【図2】パネル35 PANSS陰性スコア(FAS、LOCF)を示す。
【図3】パネル36 PANSS総合精神病理スコア(FAS、LOCF)を示す。
【図4】パネル42 PANSS合計スコアの少なくとも25%の減少を有する患者の割合を示す(FAS、LOCF)。
【図5】パネル43 2以下のCGI−Iスコアを有する患者の割合を示す(FAS、LOCF)。
【実施例】
【0033】
以下の実施例は、より詳細に本開示をさらに説明することを意図するだけであり、従って、これらの実施例は決して本開示の範囲を限定すると見なされない。
【0034】
実施例1.統合失調症の処置におけるビフェプルノックスの効能
統合失調症の処置におけるビフェプルノックスの固定用量の効能および安全性を評価するために6週の無作為化二重盲検プラセボ対照およびリスペリドン参照研究を用いた。合計599人の患者を無作為抽出した。
【0035】
処置は少なくとも3日の単純盲検プラセボ導入期間から開始し、その後にビフェプルノックス処置患者には0.25mgから30mg/日もしくは40mg/日までのビフェプルノックスの滴定を続けた。リスペリドン処置患者は3日の期間にわたって毎日2mgから6mgまで滴定され、そして処置期間の残りの間6mg/日で維持された。
【0036】
PANSS合計スコアのベースラインからエンドポイントまでの変化を測定するために、評定尺度評価は5週目を除いて毎週行った。他の評価には:PANSS陽性症状下位尺度スコア、PANSS陰性症状下位尺度スコア、PANSS総合精神病理下位尺度スコア、BPRS合計スコア、BPRS精神病スコア、CGI−Sスコア、CGI−Iスコア、PANSS合計スコアに基づくレスポンダー率および統合失調症のためのカルガリー鬱病評価尺度(Calgary Depression Scale for Schizophrenia)(CDSS)が包含された。
【0037】
安全性および耐容性測定には、身体所見(physical examinations)、体重、ウエスト囲、バイタルサイン、12誘導心電図(ECG)、臨床検査評価(血液学、生化学、検尿、プロラクチン、IGF−1、IGFBP−3、甲状腺機能の特定の検査評価)、二重盲検処置期間の間の抗コリン作用薬処置の必要性、併用薬使用、有害事象モニタリングおよび正常な動き(normal movement)の評価が包含された。
【0038】
20mgビフェプルノックス処置群は、PANSS合計スコアにおけるベースラインからエンドポイントまでの変化の主要評価項目についてプラセボとの統計的に有意な差を示した。PANSS合計スコアにおけるベースラインからエンドポイントまでの平均的変化(標準偏差)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった。
【0039】
30mgビフェプルノックス群は、CGI−Sスコア、PANSS陰性症状下位尺度スコアおよびPANSS陽性症状下位尺度スコアについてプラセボとの顕著な差を示した。顕著な差はまた、PANSS総合精神病理下位尺度スコア、BPRS合計スコア、BPRS精神病クラスター(cluster)スコア、PANSSレスポンダー率およびCGI−Iレスポンダー率におけるベースラインからエンドポイントまでの変化についても30mgビフェプルノックス群とプラセボとの間で認められた。本研究において、PANSSレスポンダーは、そのPANSS合計スコアがベースラインからエンドポイントまで20%以上減少した患者をさす。CGI−Iレスポンダーは、エンドポイントでCGI全般改善度評価尺度において「著明改善」もしくは「中等度改善」と分類された患者をさす。
【0040】
40mgビフェプルノックス群は、PANSS陽性症状下位尺度スコアおよびBPRS
精神病クラスタースコアにおける変化についてプラセボとの顕著な差を示した。
【0041】
プラセボおよびリスペリドン処置群における増加と対照的にビフェプルノックス処置群において体重の減少が見られた。
【0042】
ビフェプルノックス群は、プラセボおよびリスペリドン群と比較してトリグリセリド、VLDLおよびLDLにおけるN→Hシフト(N to H shifts)のより低い発生率ならびに総コレステロールおよびVLDLにおけるN→Lシフトのより高い発生率を有した。
【0043】
実施例2.統合失調症の処置におけるビフェプルノックスに関する臨床研究
実施例2a−臨床研究1
目的:本臨床研究の主要目的は、主要アウトカム評価尺度として陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、5mg、10mgもしくは20mgのビフェプルノックスでの6週の処置が統合失調症を有する成人患者においてプラセボでの処置より優れているかどうかを調べることであった。二次的目的は、PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、ならびにPANSS合計スコアに基づくレスポンダー率およびCGI−Iレスポンダー率を用いて統合失調症を処置することにおけるビフェプルノックスの効能を評価することであった。身体所見、体重、バイタルサイン(脈拍数および最高/最低血圧(BP)−横たわって5分後そして立って2分後の両方(both lying after five minutes and standing after two minutes)、ならびに口腔温を包含する)、12誘導心電図(ECG)、血液学、生化学および検尿を包含する安全性検査評価、二重盲検処置期間中の抗コリン作用薬処置の必要性、併用薬使用、有害事象モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価することもまた本研究の目的であった。
【0044】
方法論:これは、統合失調症を有する成人患者における無作為化二重盲検固定用量プラセボ対照リスペリドン参照並行群多施設研究であった。本研究には5つの処置群があった。処置群は下記のとおりであった:ビフェプルノックス5mg、ビフェプルノックス10mg、ビフェプルノックス20mg、リスペリドン6mgおよびプラセボ。研究薬剤は、毎日1回投与した。ベースライン測定を行った後に、滴定期を開始した。ビフェプルノックス処置患者は、標準化滴定スケジュール(1日:0.125mg、2日:0.25mg、3日:0.5mg、4日:1.0mg、5日:2.0mg、6日:5.0mg、7日:10.0mg、8日:20mg)に従って5mg、10mgもしくは20mgまで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。リスペリドン処置患者は、毎日1回の処方計画を用いて3日にわたって6mgまで滴定された(1日:2mg、2日:4mg、3日:6mg)。
【0045】
患者数(計画し、スクリーニングし、無作為抽出し、そして分析した):統合失調症を有する合計575人の患者を研究に包含することを計画した。合計836人の患者を40の施設でスクリーニングし、そして合計589人の患者(5mgビフェプルノックス:115人の患者;10mgビフェプルノックス:120人の患者;20mgビフェプルノックス:115人の患者;プラセボ:119人の患者;リスペリドン:120人の患者)を37の施設で無作為抽出した。
【0046】
試験製品、用量および投与形態:ビフェプルノックス錠剤、総日用量0.125mg〜20mg、毎日1回の投与処方計画を用いて経口投与する。
【0047】
参照治療、用量および投与形態:プラセボおよびリスペリドン、2mg〜6mg、毎日1回の投与処方計画を用いて経口投与する。
【0048】
効能結果:20mgのビフェプルノックス用量は、PANSS合計および下位尺度スコアの分析からの統計的に有意な比較により示されるようにプラセボと比較して統合失調症の陽性および陰性症状の両方を軽減することならびに総合精神病理を減少させることにおいて有効であった。20mgビフェプルノックス群における患者は、プラセボ患者と比較してPANSS合計スコアにおいてベースラインからの5.8ポイント大きい改善を有した。ビフェプルノックスのより低い用量は有効ではなかった。10mg用量のビフェプルノックスは、いずれの効能評価項目についても統計的により大きい改善を示さなかった。5mg用量は、プラセボと比較していくつかの副次的効能評価尺度に関してより大きい改善を示したが、主要効能評価項目(PANSS合計スコア)についてプラセボに対する優位性を示さなかった。リスペリドン6mgは本研究における活性対照(active reference)として用いられ、そしてプラセボ群からの明らかな分離を示した。一般に、20mgビフェプルノックス群において見られる改善の大きさは、大部分の効能評価項目についてリスペリドン群において見られるものより小さかった。
【0049】
安全性結果:少なくとも1種の処置下で発現した有害事象(TEAE)を有する患者の割合は処置群にわたって同様であった:5mgビフェプルノックス群において89%(102人の患者)、10mgビフェプルノックス群において87%(104人の患者)、20mgビフェプルノックス群において83%(95人の患者)、プラセボ群において85%(101人の患者)、そしてリスペリドン群において89%(107人の患者)。全体で、ビフェプルノックスで処置した349人の患者から、最も頻繁に報告されるTEAEは頭痛、消化不良、不眠、悪心、嘔吐NOS、便秘および激越であった。プラセボ群(N=119人の患者)と比較して20mgビフェプルノックス群(N=114人の患者)においてより高い発生率(>5%の差)を有するTEAEには、便秘、消化不良および嘔吐NOSが包含された。プラセボと比較して20mgビフェプルノックス群においてより高い発生率(>5%の差)を有する関連TEAEは、便秘、嘔吐NOSおよび頭痛NOSであった。少なくとも1種の重篤なTEAEを有する患者の割合は、20mgビフェプルノックス群(11人の患者、10%)において最も低く、続いてプラセボ群(15人の患者、13%)であった。残りの群について、少なくとも1種の重篤なTEAEを有する患者の割合は16%〜18%の間であった。重篤であると考えられるTEAEの発生率は、全てのTEAEについて20mgビフェプルノックス群とプラセボ群間で同様であった(<5%の差)。任意の事象について認められる全発生率、関連TEAEの発生率もしくは重篤なTEAEの発生率においてビフェプルノックス群における用量依存的傾向はなかった。
【0050】
少なくとも1種のSAEを有する患者の総数は、プラセボ群(9%)と比較して積極的処置群において高かった(ビフェプルノックス群:12〜15%、リスペリドン群:16%)。最も一般的に報告されるSAE(任意の処置群において>5%)は、増悪精神病および増悪統合失調症NOSであった。プラセボおよびリスペリドン(各々5人の患者、4%)と比較して20mgビフェプルノックス群(8人の患者、7%)において増悪統合失調症NOSのわずかに高い発生があった。自殺未遂は10mgビフェプルノックス群における1人の患者(<1%)、20mgビフェプルノックス群における2人の患者(2%)、プラセボ群における0人の患者およびリスペリドン群における0人の患者について報告された。自殺念慮の重篤な有害事象は、10mgビフェプルノックスおよびリスペリドン
群における各々1人の患者(<1%)について報告された(20mgビフェプルノックス群における1人のさらなる患者は、自殺念慮の非重篤な有害事象を経験した)。任意のSAEの発生率における用量依存的増加の傾向は、ビフェプルノックス群について認められなかった。ビフェプルノックスは、全ての用量レベルで安全でありそして良好な耐容性を示した。中止につながる少なくとも1種のAEを有する患者の総数は、処置群間で同様であった(5mgビフェプルノックス:13人の患者、11%;10mgビフェプルノックス:17人の患者、14%;20mgビフェプルノックス:11人の患者、10%;プラセボ:15人の患者、13%;リスペリドン:17人の患者、14%)。中止につながる最も一般的な(任意の処置群における患者の>2%により報告される)AEは、激越、増悪精神病および増悪統合失調症NOSであった。20mgビフェプルノックス群とプラセボ群との間で研究薬剤の中止につながるAEの発生率の処置群差はなかった。ビフェプルノックス群において中止につながるAEの発生率の用量依存的増加の傾向はなかった。臨床検査、バイタルサイン、ECGおよび身体所見結果の評価は、いかなる予想外の安全性懸念も引き起こさなかった。ビフェプルノックス患者は、プラセボ群と比較してプロラクチンの減少を示した。軽度の体重減少はビフェプルノックス群において認められたが、プラセボもしくはリスペリドン群においては認められなかった。
【0051】
BAS、SASもしくはAIMSスコアにおけるベースラインからエンドポイントまでの変化の処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であり、そしてリスペリドンでの患者におけるよりも少なかった。
【0052】
結論:本研究の1つの結論は、6週間毎日1回与えるビフェプルノックスの20mg用量が統合失調症の陽性および陰性症状の両方を軽減することにおいて有効であったことである。全体で、ビフェプルノックスの全ての用量は安全でありそして統合失調症患者により良好な耐容性が示された。安全性/耐容性の用量反応関係は見られなかった。
【0053】
実施例2b−臨床研究2
主要目的:主要アウトカムとして陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、ビフェプルノックスの固定用量(30mg/日もしくは40mg/日)での6週の処置が統合失調症を有する成人患者においてプラセボと比較して優れた効能を示すことができるかどうかを調べること。
【0054】
二次的目的:PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病スコア、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、PANSS合計スコアに基づくレスポンダー率、統合失調症のカルガリー鬱病評価尺度(CDSS)および患者満足度を用いて統合失調症を処置することにおけるビフェプルノックスの効能を評価すること。統合失調症患者におけるビフェプルノックスの薬物動態(PK)データもまた評価し、そして別個の報告において提示する(研究S1543003からのデータと合わせて)。身体所見、体重、ウエスト囲、バイタルサイン(脈拍数および最高/最低血圧[BP]−横たわって5分後そして立って2分後の両方、ならびに口腔温)、12誘導心電図(ECG)、臨床検査評価(血液学、生化学、検尿、プロラクチン、IGF−1、IGFBP−3および甲状腺機能についての特定の検査評価)、二重盲検処置期間の間の抗コリン作用薬処置の必要性、併用薬使用、有害事象(AE)モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価すること。
【0055】
方法論:これは、統合失調症を有する成人患者におけるビフェプルノックスの効能、耐容性および安全性の第III相6週無作為化二重盲検プラセボ対照リスペリドン参照並行群多施設研究であった。本研究を完了した患者は、長期で継続する選択肢を有した。処置群は:ビフェプルノックス30mg/日、ビフェプルノックス40mg/日、リスペリドン6mg/日およびプラセボであった。少なくとも3日の単純盲検プラセボ導入期間を完了した後に、ビフェプルノックス処置患者は8日の期間にわたって標準化滴定スキームに従って0.25mgから30mg/日もしくは40mg/日まで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。リスペリドン処置患者は、3日の期間にわたって毎日2mgから6mgまで滴定され、そして次に処置期間の残りの間6mg/日で維持された。効能および異常運動障害の評定尺度評価は、5週目を除いて毎週行われた。安全性評価はスクリーニングで、処置の間にそして研究の最後で行われた。研究薬剤での患者満足度は6週で評価された。血液のサンプルは、血漿中のビフェプルノックスの測定用に2、4および6週で得られた。血液サンプルはまた、臨床検査評価用にスクリーニング/ベースライン、3週および6週でも得られた。
【0056】
患者数(計画し、承認し、任意抽出し、そして分析した):統合失調症を有する合計576人の患者を研究に包含することを計画した。783人のスクリーニングした患者のうち、合計599人の患者を無作為抽出した(30mgビフェプルノックス:148人の患者、40mgビフェプルノックス:148人の患者、プラセボ:149人の患者;リスペリドン:154人の患者)。
【0057】
診断および包含のための主な基準:(DSM−IV−TR基準に従って)統合失調症を有する18〜75歳の男性もしくは女性患者。患者は、70〜120の間のPANSSでの合計スコアを有していなければならず;4つのPANSS項目(概念の統合障害、幻覚による行動、猜疑心、不自然な思考内容)の少なくとも2つがスコア>4を有していなければならず;そしてCGI−Sでのスコアが少なくとも4でなければならなかった。
【0058】
参照治療、用量および投与形態:プラセボおよびリスペリドン、6mg、毎日1回経口投与する。
【0059】
効能結果:30mgビフェプルノックス処置群は、Hochberg補正したp値(補正したp=0.020)に基づいてPANSS合計スコア(LOCF)におけるベースラインからエンドポイントまでの変化の主要評価項目についてプラセボとの統計的に有意な差を示した。40mgビフェプルノックス処置群は、主要効能評価項目についてプラセボ群と有意に異ならなかった(補正したp=0.156)。PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:30mgビフェプルノックス群について−5.9、そして40mgビフェプルノックス群について−3.2であった。計画されたステップダウン法に基づく統計的に有意な差は、プラセボと比較して30mgビフェプルノックス処置群についてCGI−Sのベースラインからエンドポイントまでの変化において見られなかった。従って、PANSS陰性症状およびPANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの変化に関するプラセボと30mgビフェプルノックス群との間の差は、プラセボと比較した統計的有意性についてステップダウン法を用いて評価されなかった。30mgビフェプルノックス群は、CGI−Sスコア(公称(nominal)p=0.028)、PANSS陰性症状下位尺度スコア(公称p=0.027)およびPANSS陽性症状下位尺度スコア(公称p=0.010)についてプラセボとの顕著な差を示した。
【0060】
顕著な差は、PANSS総合精神病理下位尺度スコア(p=0.025)、BPRS合計スコア(p=0.019)、BPRS精神病クラスタースコア(p=0.002)、PANSS(30%)レスポンダー率(p=0.019)およびCGI−Iレスポンダー率(p=0.039)におけるベースラインからエンドポイントまでの変化について30mgビフェプルノックス群とプラセボとの間で認められた。エンドポイントでの顕著な差は、患者満足度(p=0.051)、CGI改善度スコアもしくはCDSSスコアについて30mgビフェプルノックス群とプラセボとの間で認められなかった。40mgビフェプルノックス群について、プラセボとの統計的に有意な差は主要効能パラメーターについて見られなかった。プラセボとの顕著な差は、PANSS陽性症状下位尺度スコア(p=0.020)およびBPRS精神病クラスタースコア(p=0.031)を除いて副次的効能パラメーターのいずれについても40mgビフェプルノックス群では認められなかった。
【0061】
安全性結果:少なくとも1種のTEAEを有する患者の割合はビフェプルノックス用量群において同程度であり(74%〜76%)、そしてプラセボ群(64%)より高かったが、リスペリドン群(78%)におけるよりもわずかに低かった。プラセボ群と比較してビフェプルノックス群においてより高い(>5%の差)発生率を有する処置下で発現したAEには、悪心、嘔吐、便秘、消化不良、下痢および眩暈が包含された。ビフェプルノックス処置群間の差は、ビフェプルノックス群における個々のTEAEの発生率において一般に認められなかった。任意の処置群における患者の少なくとも5%において起こるTEAEのうち、30mgビフェプルノックス群と比較して40mgビフェプルノックス群においてより高い(>2%の差)発生率を有するものには、悪心、嘔吐、歯痛、食欲不振、静座不能、眩暈、頭痛および不眠が包含された。対照的に、口渇、唾液分泌過多、食欲減退、鎮静、傾眠、不安および膣炎は、40mgビフェプルノックス群と比較して30mgビフェプルノックス群においてより高い(>2%の差)発生率で起こった。重篤なTEAEの発生率は、精神病性障害(<6%)および統合失調症(ビフェプルノックスおよびプラセボ群において各々3%そしてリスペリドン群において2%)のTEAEを除いて一般に低かった(<1%の発生率)。重篤なTEAEの発生率は処置群にわたって一般に同程度であり、そして任意の事象について重篤なTEAEの発生率におけるビフェプルノックス処置群間の差はなかった。特定の興味深いTEAEはデータベースロックの前に定義され、そして自殺、自殺未遂、性機能障害、失神、血管迷走神経性発作および起立性低血圧に関連する事象が包含された。全体で合計76人の患者(13%)が研究中に少なくとも1種の特定の興味深いTEAEを報告した。少なくとも1種の特定の興味深いTEAEを有する患者の割合は、プラセボ群(6%)と比較してビフェプルノックスおよびリスペリドン処置群(12%〜16%)において高かった。特定の興味深いTEAEの大部分は、任意の処置群における患者の<1%において起こった。眩暈は最も一般的に報告され、そして他の2つの群と比較してビフェプルノックス処置群においてわずかに高い発生率で起こった。
【0062】
全体で合計60人の患者(10%)は81のSAE(死亡を包含する)を経験した。大部分のSAEは、患者の<1%において起こった。例外は、精神病性障害(<5%)および統合失調症(4つの処置群の各々において3%)であった。SAEの発生率は、ビフェプルノックス群およびプラセボ間で同程度であった。異常運動の評価尺度(BAS、SASもしくはAIMSスコア)で処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボで処置した患者のものと同様であった。全体で、ビフェプルノックスで臨床検査パラメーター、身体所見結果もしくはECG読み取りにおける臨床的に有意な変化はなかった。ビフェプルノックス群は、プラセボおよびリスペリドン群と比較してトリグリセリド、VLDLおよびLDLにおけるN→Hシフトのより低い発生率ならびに総コレステロールおよびVLDLにおけるN→Lシフト
のより高い発生率を有した。著しく異常な総コレステロール値の発生率は、処置群にわたって同様であった(1%〜2%)。著しく異常なトリグリセリド値は、他の群と比較して40mgビフェプルノックス群においてわずかに少ない患者により報告された。プラセボ(4%)および40mgビフェプルノックス(5%)群と比較して30mgビフェプルノックスおよびリスペリドン群(各々6%)におけるトリヨウ素サイロニンのN→Hシフトのわずかに大きい発生率ならびにプラセボ(2%)およびリスペリドン(<1%)群と比較して30mgビフェプルノックス(4%)および40mgビフェプルノックス(5%)におけるTSHのN→Lシフトのわずかに大きい発生率があった。プラセボおよびリスペリドン処置群における増加と対照的にビフェプルノックス処置群において体重のわずかな同程度の減少が見られた。体重の著しく異常な減少の発生率はプラセボおよびリスペリドン群に対してビフェプルノックス処置群の両方においてわずかに高く、一方、体重の著しく異常な増加の発生率はビフェプルノックスおよびプラセボ群に対してリスペリドン処置群において高かった。
【0063】
結論:本研究の1つの結論は、6週間毎日1回与えるビフェプルノックスの30mg用量がPANSS合計スコアの分析からの統計的に有意な比較により示されるように統合失調症の症状を軽減することにおいて有効であったことである。30mgビフェプルノックス群における患者は、LOCFデータに基づいてプラセボ患者と比較してPANSS合計スコアにおけるベースラインからエンドポイントまでの5.9ポイント大きい改善を有した。主要および副次的効能パラメーターについて実対照薬、リスペリドンとプラセボ処置との間で認められる統計的に有意な差は、これが有効な研究であることを示す。
【0064】
30mgビフェプルノックス処置群は、3つのキーとなる副次的効能評価項目、CGI−S、PANSS陰性および陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの変化についてプラセボとの顕著な差を示し(公称p値に基づいて);これら3つのキーとなる副次的効能評価項目についてステップダウン法に基づいて統計的有意性は得られなかった。顕著な差は、他の副次的効能パラメーター(PANSS総合精神病理下位尺度スコア、BPRS合計スコアおよびBPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの変化)の大部分についてエンドポイントで30mgビフェプルノックス群とプラセボとの間で認められた。30mgビフェプルノックス用量とプラセボとの間の顕著な差はまた、PANSSおよびCGI−Iレスポンダー率についてもエンドポイントで示された。40mgビフェプルノックス処置群は、主要効能パラメーターについてプラセボとの統計的に有意な差を示さなかった。PANSS陽性症状下位尺度およびBPRSクラスタースコアを除いて副次的効能パラメーターのいずれについても40mgビフェプルノックスとプラセボ群との間で顕著な差はなかった。全体で、ビフェプルノックスの30mgおよび40mg用量は安全であり、そして統合失調症患者により良好な耐容性が示された。
【0065】
実施例2c:臨床研究3
主要目的:主要アウトカムとして陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、ビフェプルノックスの固定用量(20mg/日もしくは30mg/日)での6週の処置が統合失調症を有する成人患者においてプラセボと比較して優れた効能を示すことができるかどうかを調べること。
【0066】
二次的目的:PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病スコア、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、CGI−Iレスポンダー率、PANSS合計スコアに基づくPANSSレスポンダー率、統合失調症のカルガリー鬱病評価尺度(CDSS)および患者満足度を用いて統合失調症を処置することにお
けるビフェプルノックスの効能を評価すること。身体所見、体重、ウエスト囲、バイタルサイン(脈拍数および最高/最低血圧[BP]ならびに口腔温)、12誘導心電図(ECG)、臨床検査評価(血液学、空腹時インシュリンレベル、空腹時グルコース、空腹時脂質プロフィールを包含する生化学、検尿、プロラクチン、IGF−1、IGFBP−3および甲状腺機能の特定の検査評価)、二重盲検処置期間中の抗コリン作用薬処置の必要性、併用薬使用、有害事象(AE)モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価すること。
【0067】
方法論:これは、統合失調症を有する成人患者におけるビフェプルノックスの効能、耐容性および安全性の第III相6週無作為化二重盲検プラセボ対照オランザピン参照並行群多施設研究であった。本研究には4つの処置群があった、処置群当たり約144人の患者。処置群は:ビフェプルノックス20mg/日、ビフェプルノックス30mg/日、オランザピン15mg/日およびプラセボであった。少なくとも3日の単純盲検プラセボ導入期間を完了した後に、ビフェプルノックス処置患者はそれぞれ7もしくは8日の期間にわたって標準化滴定スキームに従って0.25mgから20mg/日もしくは30mg/日まで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。オランザピン処置患者は最初の7日の期間にわたって10mg/日で投薬を開始し、そして次に処置期間の残りの間15mg/日で維持された。スクリーニング来院(Screening Visit)から開始してベースライン後少なくとも10日まで、研究参加資格が確認された後に患者を入院させた(すでに入院患者ではない場合)。患者は、研究者により医学上必要と見なされる場合には10日より長く入院させることができた。効能および異常運動障害の評定尺度評価は、5週目を除いて毎週行われた。安全性評価はスクリーニングで、処置の間にそして研究の最後で行われた。研究薬剤での患者満足度は6週で評価された。全血のサンプルは、血漿におけるビフェプルノックスの測定用に2、4および6週で得られた。
【0068】
患者数(計画し、承認し、任意抽出し、そして分析した):統合失調症を有する合計576人の患者を研究に包含することを計画した。合計814人の患者を32の施設でスクリーニングし、そして合計604人の患者(20mgビフェプルノックス:154人の患者、30mgビフェプルノックス:150人の患者;プラセボ:150人の患者;オランザピン:150人の患者)を32の施設で無作為抽出した。
【0069】
診断および包含のための主な基準:(DSM−IV−TR基準に従って)統合失調症を有する18〜75歳の男性もしくは女性患者。患者は、70〜120の間のPANSSでの合計スコアを有していなければならず;4つのPANSS項目(概念の統合障害、幻覚による行動、猜疑心、不自然な思考内容)の少なくとも2つがスコア>4を有していなければならず;CGI−Sでのスコアが少なくとも4でなければならない。
【0070】
試験製品、用量および投与形態:ビフェプルノックス錠剤、総日用量20mgもしくは30mg(1錠の20mg錠剤および1錠の10mg錠剤)、毎日1回の投与処方計画を用いて経口投与する。
【0071】
参照治療、用量および投与形態:プラセボおよびオランザピン、5mgおよび15mg、毎日1回の投与処方計画を用いて経口投与する。
【0072】
効能結果:20mgおよび30mg処置群は両方ともエンドポイントでベースラインに対して改善を示したが、主要およびキーとなる副次的効能パラメーターに関してプラセボ群と比較した場合に効能を示さなかった。しかしながら、20mgビフェプルノックス群
は、副次的効能パラメーター、CGI改善度スコア(公称p=0.027)におけるベースラインからエンドポイントまでの変化についてプラセボ群に対して顕著な改善を示した。さらに1つの他の副次的効能パラメーターにおいて、20mgビフェプルノックス群とアプローチしたプラセボ群との間の差は、PANSS−20%レスポンダー率について顕著である(p=0.061)。しかしながら、副次的効能パラメーター間の顕著なそしてほぼ顕著な差のこれらの発生は、偶然、すなわち、処置比較の5%において起こると予想されるものを上回らない。全ての他の副次的およびキーとなる副次的パラメーターにおいて、ビフェプルノックス処置群はいずれも任意の効能評価項目についてプラセボ群に対して顕著な改善を示さなかった。15mgの用量のオランザピンは、本研究における活性対照として用いられた。オランザピンとプラセボとの間のPANSS合計スコアの差は、感度分析に従って分析された。これらの結果は、オランザピンがプラセボと顕著に異なることを示した(p<0.001)。一般に、ビフェプルノックス用量群において見られる改善の大きさはプラセボ群において見られるものより高かったが、大部分の効能評価項目についてオランザピン群において見られるものより低かった。
【0073】
安全性結果:少なくとも1種の処置下で発現した有害事象(TEAE)を有する患者の割合は、20mgビフェプルノックス群(126人の患者、82%)において最も高く、続いて30mgビフェプルノックス(115人の患者、77%)、オランザピン(110人の患者、73%)、そしてプラセボ(107人の患者、72%)群であった。全体で、ビフェプルノックスで処置した304人の患者から、最も頻繁に報告されるTEAEは頭痛、悪心、嘔吐、消化不良および不眠であった。プラセボ群と比較してビフェプルノックス群においてより高い発生率(>5%の差)を有するTEAEには、悪心、嘔吐および便秘が包含された。一般に、個々のTEAEの発生率の明らかな用量依存的増加はビフェプルノックス群において認められなかった。20mgビフェプルノックス群と比較して30mgビフェプルノックス群においてわずかに高い(>2%の差)発生率を有するTEAEには、疲労、眩暈および鎮静が包含された。少なくとも1種の重篤なTEAEを有する患者の割合は、ビフェプルノックス処置群およびプラセボ群(9%〜12%)において同程度であり、そしてオランザピン処置群(6%)においてわずかに少なかった。任意の事象についてビフェプルノックス群における重篤なTEAEの発生率の用量依存的増加の明示はなかった。特定の興味深いTEAEはデータベースロックの前に定義され、そして以下の事象:自殺、自殺未遂、性機能障害、失神、血管迷走神経性発作および起立性低血圧に関連する事象が包含された。全体で合計58人の患者(10%)が研究中に少なくとも1種の特定の興味深いTEAEを報告した。少なくとも1種の特定の興味深いTEAEを有する患者の総数は、プラセボ(11人の患者、7%)もしくはオランザピン(10人の患者、7%)群と比較してビフェプルノックス処置群(30mgビフェプルノックス;20人の患者、13%;20mgビフェプルノックス;17人の患者、11%)において高かった。特定の興味深いTEAEの大部分は、任意の処置群における患者の<1%において起こった。最も一般的に報告される特定の興味深いTEAEは眩暈であり、それは他の2つの群と比較してビフェプルノックス処置群においてわずかに高い発生率を有した(30mgビフェプルノックス:15人の患者、10%;20mgビフェプルノックス:13人の患者、8%;プラセボ:9人の患者、6%;オランザピン:8人の患者、5%)。処置群内の少なくとも2人の患者において起こる他の特定の興味深いTEAEには、血管迷走神経性失神(30mgビフェプルノックス:2人の患者)および起立性低血圧(30mgビフェプルノックス:3人の患者)が包含された。
【0074】
少なくとも1種のSAEを有する患者の割合は、オランザピン処置群(6人の患者、4%)において最も少なく、続いてビフェプルノックス群(20mg:15人の患者、10%;30mg:12人の患者、8%)であり、SAEの最も高い発生率はプラセボ群(20人の患者、13%)において認められた。最も一般的に報告されるSAEは、精神病性障害(全体で患者の4%)および統合失調症(全体で2%)であった。これらのSAEの発生率は、プラセボ群と比較してビフェプルノックス群において同様であるかもしくは低かった。他の処置群における0人の患者と比較して30mgビフェプルノックス群における2人の患者は、血管迷走神経性失神のSAEを有した。この起こり得る例外は別として、ビフェプルノックス群について認められる任意の他のSAEの発生率の用量依存的増加の他の顕著な表示はなかった。AEのために研究薬剤を中止した患者の割合はプラセボ群(11%)において最も大きく、続いてビフェプルノックス群(各々8%)およびオランザピン処置群(6%)であった。研究終了につながる少なくとも1種のAEを有する患者の割合はプラセボ群(12%)において最も大きく、同程度の割合の患者が20mgビフェプルノックス(8%)、30mgビフェプルノックス(7%)およびオランザピン(6%)処置群において研究終了につながる少なくとも1種のAEを有した。研究薬剤の中止につながる最も一般的な(任意の処置群における患者の>2%により報告される)AEは、精神病性障害および統合失調症であった。ビフェプルノックス群において研究薬剤の中止につながるAEの発生率の用量依存的増加の明らかな傾向はなかった。検査、バイタルサインおよびECG結果の評価は、いかなる予想されない安全性の懸念も引き起こさなかった。ビフェプルノックス群における患者は、プラセボおよびオランザピン群における患者と比較してプロラクチンの減少を示した。体重の著しく異常な減少の発生率は、ビフェプルノックス処置およびプラセボ群間で同程度であり(5%〜6%)、そしてオランザピン処置群において低かった(<1%)。体重の著しく異常な増加の発生率は、ビフェプルノックスおよびプラセボ群において同程度であり(1%〜3%)、そしてオランザピン群においてはるかに高かった(19%)。BAS、SASもしくはAIMSスコアにおいてベースラインからエンドポイントまでの変化における処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であった。
【0075】
説明および結論:これは、統合失調症を有する604人の患者の処置におけるオランザピンを活性参照とするビフェプルノックスの効能、耐容性および安全性の6週無作為化二重盲検固定用量プラセボ対照並行群多施設研究であった。本研究は米国(26)、コロンビア(3)およびインド(5)における32の施設で実施された。
【0076】
これは、主要効能パラメーターについて実対照薬オランザピンとプラセボ処置間で統計的に有意な差を示す結果により証明される有効な研究であった。
【0077】
20mgおよび30mg処置群は両方ともエンドポイントでベースラインに対して改善を示したが、主要およびキーとなる副次的効能パラメーターに関してプラセボ群と比較した場合に効能を示さなかった。しかしながら、20mgビフェプルノックス群は副次的効能パラメーター、CGI改善度スコアにおけるベースラインからエンドポイントまでの変化についてプラセボ群に対して顕著な改善を示した(公称p=0.027)。さらに1つの他の副次的効能パラメーターにおいて、20mgビフェプルノックス群とアプローチしたプラセボ群との間の差はPANSS−20%レスポンダー率について顕著である(p=0.061)。しかしながら、副次的効能パラメーター間の顕著なそしてほぼ顕著な差のこれらの発生は、偶然、すなわち、処置比較の5%において起こると予想されるものを上回らない。全ての他の副次的およびキーとなる副次的パラメーターにおいて、ビフェプルノックス処置群はいずれも任意の効能評価項目についてプラセボ群に対して顕著な改善を示さなかった。
【0078】
15mgの用量のオランザピンは、本研究における活性参照として用いられた。一般に、ビフェプルノックス用量群において見られる改善の大きさはプラセボ群において見られるものより高かったが、大部分の効能評価項目についてオランザピン群において見られるものより低かった。
【0079】
対照的に、臨床研究1における大規模研究において、20mgビフェプルノックス用量は、PANSS合計および下位尺度スコアの分析からの統計的に有意な比較により示されるようにプラセボと比較して統合失調症の陽性および陰性症状の両方を軽減することならびに総合精神病理を減少することにおいて有効であった。本研究において、PANSS合計スコアにおけるベースラインから6週までの変化の観察値(observed values)分析を用いて、結果は20mgビフェプルノックス(−24.42[15.6])、30mgビフェプルノックス(−24.56[17.02])およびオランザピン(−29.11[16.88])群間でさらに同様であり、しかしながら、プラセボ群もまた同様の結果(−22.29[19.14])を示した。これは、6週間研究にとどまった患者がそれらの処置処方計画によく反応したことを示す。
【0080】
ビフェプルノックスは、両方の用量レベルで良好な耐容性を示した。有害事象による離脱率は、プラセボ群と比較してビフェプルノックス群において低かった。プラセボ患者におけるよりもビフェプルノックス処置患者において頻繁に生じる有害事象は主に胃腸性のものであり、そして軽度〜中等度の重症度であった。2人の患者のみが(20mgビフェプルノックス群における)胃腸のAEのために研究薬剤を中止した(1人の患者は悪心のために中止し、1人の患者は悪心および嘔吐のために中止した)。少なくとも1種のSAEを有する患者の割合は、プラセボ群(13%)と比較してビフェプルノックス群(7%〜8%)において低かった。最も一般的に報告されるSAEは精神病性障害(全体で4%)および統合失調症(全体で1%)であった。これらのSAEの発生率は、プラセボ群と比較してビフェプルノックス群において同様であるかもしくは低かった。他の処置群における0人の患者と比較して30mgビフェプルノックス群における2人の患者は、血管迷走神経性失神のSAEを有した。この起こり得る例外は別として、ビフェプルノックス群について任意の他のSAEの発生率の用量依存的増加の他の顕著な表示はなかった。検査、バイタルサインおよびECG結果の評価は、いかなる予想されない安全性の懸念も引き起こさなかった。ビフェプルノックスは、以前の研究において見られておりそして薬剤の部分ドーパミンアゴニストプロフィールに基づいて予想されているものと一致するプロラクチンの減少と関連し、そしていずれのAEとも関連しなかった。対照的に、プロラクチンの増加がプラセボおよびオランザピン群において認められた。平均体重のわずかな減少がビフェプルノックス群において認められ、一方、体重の増加がオランザピン群に認められた。体重の著しく異常な減少の発生率はビフェプルノックス処置およびプラセボ群間で同程度であり(5%〜6%)、そしてオランザピン処置群において低かった(1%)。体重の著しく異常な増加の発生率はビフェプルノックスおよびプラセボ群において同程度であり(1%〜3%)、そしてオランザピン(19%)群においてはるかに高かった。これらの結果は他のビフェプルノックス研究において認められているものと一致し、そして体重増加は非定型抗精神病薬にとって問題でありそして心臓血管疾患および糖尿病の増加した危険性と関連することを考えれば注目に値する。
【0081】
異常運動の評価尺度(BAS、SASもしくはAIMSスコア)に関して処置群間で顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であった。錐体外路障害の発生率は、全ての群において低かった(ビフェプルノックス群:<1%〜3%;オランザピン:1%;プラセボ:3%)。ビフェプルノックスの20mgおよび30mg用量は、大部分の効能評価項目についてプラセボと比較して統計的に有意に大きい改善を示さなかった。より良いCGI改善度スコアは、プラセボ群と比較して20mg(しかし30mgではそうでない)ビフェプルノックス群において見られた。
【0082】
悪心、嘔吐および便秘は、プラセボに対してより顕著なAEであった。全体で、ビフェプルノックスの20mgおよび30mg用量は安全であり、そして統合失調症を有する患者により良好な耐容性が示された。
【0083】
実施例2d−臨床研究4
主要目的:主要アウトカム評価尺度として無作為抽出から増悪までの時間を用いて、6ヶ月のビフェプルノックス処置が慢性統合失調症を有する患者においてプラセボでの処置より優れているかどうかを調べること。
【0084】
二次的目的:アウトカム評価尺度として陽性および陰性症候群評価尺度(PANSS)合計スコアにおけるベースラインからの変化を用いて、6週のビフェプルノックス処置後の急性効果がプラセボでの処置より優れているかどうかを調べること。
【0085】
他の二次的目的:プラセボに対するビフェプルノックスの長期安全性および耐容性を評価すること。
【0086】
方法論:本研究は、多国籍多施設無作為化二重盲検並行群プラセボ対照固定用量研究であった。研究は3〜6日の抗精神病薬を使わない導入期間からなり、その後に患者は固定用量のビフェプルノックス(20mg/日(BX20)もしくは30mg/日(BX30))もしくはプラセボ(PBO)での6ヶ月の二重盲検処置に無作為抽出された。BX群に割り当てられた患者は、7日(BX20)もしくは8日(BX30)にわたって0.25mg/日から漸増され、そして次に研究の残りの間これらの用量で続けられた。効能評価はベースラインで(CGI−Iを除く)、そして1、2、4、6および9週で、そして3、4、5および6ヶ月で行われた。安全性評価はスクリーニングで、処置の間に、そして研究の最後で行われた。既定の時間点で、血液サンプルがBXおよびその主要代謝物(BXの3’−および4’−硫酸抱合体)の薬剤濃度分析用に得られ、そして医薬品経済性評価が行われた。
【0087】
計画しそして分析した患者数:参加が計画された合計495人の患者があった:各処置群において165人。
【0088】
診断および主な包含基準:スクリーニングおよびベースラインでPANSS合計スコア>60およびCGI−Sスコア>4(中等症)を有し;スクリーニングおよびベースラインでPANSS項目P7(敵意)およびG8(非協調性)スコア<4(中等度)を有し;18〜65歳の間であり(極値を包含する);入院患者であるか、部分入院しているか、もしくは90日以内にデイケアプログラムにおいて追跡される外来患者であり;そしてスクリーニングの前にスクリーニングの前の1ヶ月以内に抗精神病薬の改変を有さなかった、2年より多くにわたって、DSM−IV−TR基準に従って、統合失調症の一次診断を有する患者。
【0089】
治験製品、用量および投与形態、バッチ番号:ビフェプルノックス−毎日1回20もしくは30mgまで7(20mg)もしくは8日(30mg)にわたって漸増;カプセル封入錠剤、経口的。
【0090】
参照治療、用量および投与形態:プラセボ−カプセル封入錠剤、経口的。
【0091】
効能結果:主要効能変数は増悪までの時間であり、そして分析はFASに基づいた。主要効能分析は、3つの処置群における統合失調症の増悪までの等しい時間の仮説を退けた(Coxモデル、p=0.008)。続いてBX群の各々とPBO群の対比較により、BX群における患者はPBO群における患者よりも統合失調症の増悪までの統計的に有意に長い期間を有することが示された(BX20:p=0.008およびBX30:p=0.006)。増悪した患者の割合はPBO群において59%、BX20群において41%、そしてBX30群において38%であった。Cox比例ハザードモデルは、PBOと比較して0.66(BX20)および0.65(BX30)の概算ハザード比を与え;すなわち、増悪の危険性は、BX20もしくはBX30群における患者に対してよりもPBO群における患者に対して約1.5倍高かった。ビフェプルノックスはまた、PPSに基づく増悪までの時間の分析においてもPBOより統計的に有意に優れていた。PPSにおける患者の大部分は研究の大部分に参加したので、得られる概算ハザード比およびp値の両方について、結果は主要分析の結果に非常に近かった。これは、主要効能分析の結論のロバスト性を説明する。副次的効能変数は、6週でのPANSS合計スコアであった。各BX群のPANSS合計スコア(FAS、LOCF)におけるベースラインから6週までの補正した平均変化(BX20:−4.0;BX30:−2.7)は、Hochbergのステップアップ法によればPBO群のもの(1.1)より統計的に有意に大きかった(BX20:p=0.002;BX30:p=0.017)。
【0092】
PANSS合計スコア、PANSS陽性および総合精神病理下位尺度スコア、BPRS合計スコアならびにBPRS精神病クラスタースコア(全てFAS、LOCF)における経時的な進展について、同じ一般的パターンが見られ:最初に(6もしくは9週まで)、平均スコアは減少し、そして次にそれらは両方のBX群において安定し、一方、PBO群において、平均スコアは2もしくは4週まで減少し、その後にスコアは着実に増加した。これらの変数の各々について、LOCF(ANCOVA)を用いたFASについてのPBO群とBX群の各々との対比較により、BX(いずれかの用量)での処置は6もしくは9週以降PBOでの処置より一般に統計的に有意に優れていることが示された。
【0093】
平均PANSS陰性下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後にスコアは安定した。PBO群において、スコアは4週まで減少し、その後にスコアは増加する傾向があった。平均PANSS陰性下位尺度スコアの来院ごとの(per−visit)LOCF分析(FAS、ANCOVA)において、両方のBX用量での処置は、BX30群についての2および4週を除いて、全ての時間点でPBOでのものより統計的に有意に優れていた。
【0094】
全ての処置群について、平均CDSS合計スコア(FAS、LOCF)は最初に減少し(ベースライン〜2週)、その後にスコアは安定なままであるか(両方のBX群)もしくは経時的に増加した(PBO群)。平均CDSS合計スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、任意の時間点でBX群のいずれかとPBO群との間に統計的に有意な差はなかった。この結果は、低レベルの鬱病症状を反映して、ベースラインCDSSスコアが低かったこととの関連において見られるべきである。
【0095】
平均CGI−Sスコア(FAS、LOCF)は両方のBX群において6週まで減少し、その後にスコアは安定した。PBO群において、平均CGI−Sスコアは4週まで減少し、その後にスコアは増加する傾向があった。平均CGI−Sスコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX群はBX20については3、4、5および6ヶ月そしてBX30については5および6ヶ月でPBO群より統計的に有意に優れていた。
【0096】
平均CGI−Iスコア(FAS、LOCF)は両方のBX群において6週まで減少し、その後にスコアは安定した。PBO群において、スコアは2週までごくわずか減少し、その後にスコアは着実に増加した。平均CGI−Iスコアの来院ごとのLOCF分析(FAS、ANCOVA)において、両方のBX用量は6週以降PBOより統計的に有意に優れていた。
【0097】
主に陰性の症状の基準を満たした少数の患者(PBO:25人の患者;BX20:32人の患者;BX30:36人の患者)について、PANSS合計スコア(全ての群)にお
けるそしてPANSS陽性下位尺度スコア(PBOおよびBX20)におけるベースラインからの補正した平均変化は一般に同じパターンをたどり、そして全集団のものと同じ範囲内であった。対照的に、BX30で処置した患者のPANSS陽性下位尺度スコアにおけるベースラインからの補正した平均変化は全集団のと経時的に同じパターンをたどったが、変化はより大きかった。PANSS陰性下位尺度スコア(全ての群)におけるベースラインからの補正した平均変化は全集団と経時的に同じパターンをたどり;しかしながら、平均変化は全集団と比較してこの集団において一般に2倍大きかった。PANSS合計スコア、PANSS陽性下位尺度スコアおよびPANSS陰性下位尺度スコアの来院ごとのLOCF分析(ANCOVA)において、BX20は任意の時間点でPBOと統計的に有意に異ならなかった。BX30は6週以降にPANSS合計スコアにおいてそして2週以降にPANSS陽性下位尺度スコアにおいてPBOより統計的に有意に優れており、一方、BX30はPANSS陰性下位尺度スコアにおいて2週以降PBOと統計的に有意に異ならなかった。
【0098】
主に鬱病の症状の基準を満たした少数の患者(PBO:18人の患者;BX20:15人の患者;BX30:22人の患者)について、PANSS合計スコア、PANSS陽性下位尺度スコア、PANSS陰性下位尺度スコアおよびCDSS合計スコアは全集団のものと一般的に同様の全体的パターンをたどった。PANSS合計スコア、PANSS陽性下位尺度スコア、PANSS陰性下位尺度スコアおよびCDSS合計スコアの来院ごとのLOCF分析(ANCOVA)において、おそらく少数の患者およびベースラインで存在する鬱病症状の低いレベルのために、この亜集団における任意の時間点でBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0099】
BX20群におけるPANSS合計スコア(FAS、LOCF)の少なくとも25%の減少を有する患者の割合は、9週以降PBO群におけるものより統計的に有意に大きく、一方、BX30群におけるレスポンダーの割合は5ヶ月でのみPBO群におけるものより統計的に有意に大きかった。
【0100】
わずかな割合(0%〜8%)の患者(処置にかかわらず)は、PANSS合計スコアの減少>35%、>45%もしくは>55%を有した。特定の減少を有する患者の割合における分布に関して処置群間もしくは内で傾向はなかった。
【0101】
6週および6ヶ月でCGI−Iスコア<2(FAS、LOCF)を有する患者の割合は、PBO群(6週:11%;6ヶ月:9%)におけるよりもBX群(6週:BX20 17%;BX30 19%;6ヶ月:BX20 22%;BX30 20%)において大きかった。BX群とPBO群との間の差は9週以降(BX20)そして3ヶ月を除いて9週以降(BX30)統計的に有意であった(p<0.05)。
【0102】
計算される平均総コレステロールおよび平均LDLは、処置および空腹時/非空腹時条件にかかわらず(非空腹時PBO患者を除いて)全ての群においてベースラインから6ヶ月まで減少した。計算される平均VLDLおよびトリグリセリドは、処置および空腹時/非空腹時条件にかかわらず(非空腹時BX30患者を除いて)全ての群においてベースラインから6ヶ月まで減少した。平均HDLは、処置および空腹時/非空腹時条件にかかわらず全ての群においてベースラインから6ヶ月まで増加した。補正した平均HDLコレステロール値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで増加した(PBO:0.04/0.06(空腹時/非空腹時);BX20:0.07/0.08;BX30:0.07/0.08mmol/L)。BX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0103】
補正した平均トリグリセリド値は、空腹時/非空腹時条件にかかわらず全ての3つの処
置群においてベースラインから6ヶ月まで減少した(PBO:−0.06/−0.22(空腹時/非空腹時);BX20:−0.16/−0.21;BX30:−0.37/−0.03mmol/L)。BX群のいずれか(BX30(空腹時)を除いて)とPBO群との間で統計的に有意な差はなかった。
【0104】
補正した平均空腹時グルコース値は全ての3つの処置群においてベースラインから6ヶ月まで増加し(PBO:0.10;BX20:0.13;BX30:0.09mmol/L)、そしてBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0105】
ベースラインから6ヶ月までの補正した平均体重変化(APTS、OC、ANCOVA)はPBO群において−0.8kg、BX20群において−0.3kg、そしてBX30群において−0.5kgであった。BX20およびBX30群における補正した平均体重減少は、PBO群におけるものと統計的に有意に異ならなかった。
【0106】
全ての処置群において、悪心および/もしくは嘔吐も有した患者はより大きい体重減少を有したが(PBO:−0.6対−1.9kg;BX20:−1.0対−1.9kg;BX30:−1.1対−2.3kg)、患者は悪心および/もしくは嘔吐を有するかどうかにかかわらず体重を減らした。
【0107】
研究中にメタボリック症候群の患者の状態へのBX(いずれかの用量)の処置効果はなかった。約75%(範囲:70%〜80%)の患者は、ベースラインでもしくは研究の最後でメタボリック症候群を有さなかった。メタボリック症候群の患者の状態における統計的に有意な処置差はなかった。
【0108】
臨床検査値、バイタルサイン、メタボリック症候群もしくはECGパラメーターにおける処置群内の臨床的に関連する変化もしくは処置群間の差はなかった。
【0109】
結論:本研究の1つの結論は、ビフェプルノックスの両方の用量(20mg/日および30mg/日)がプラセボより統計的に有意に良く統合失調症の増悪を防いだことである。慢性の安定した統合失調症を有する患者について、ベースライン症状は6週の処置後もしくは長期処置後のいずれかでPBOでよりもBX(両方の用量)で有意に良く維持された。ビフェプルノックスとプラセボとの安全性プロフィールの比較により、プラセボ群に対してビフェプルノックス群における胃腸系および眩暈と関連する有害事象のより高い発生率が示された。研究からの離脱につながる悪心および嘔吐ならびに異常運動の発生率は、BX20群におけるよりもBX30群において高かった。好ましい代謝プロフィール(体重変化、血液脂質およびメタボリック症候群の存在/不在に基づく)は、ビフェプルノックスについて見られた。
【0110】
臨床研究1〜4に関連する結果
PANSS合計スコア
臨床研究1:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(S.D.)は、ビフェプルノックス5mg群について−9.7(17.5)、ビフェプルノックス10mg群について−5.0(18.3)、ビフェプルノックス20mg群について−11.3(17.0)、プラセボ群について−5.3(16.3)、そしてリスペリドン群について−15.7(14.9)であった。エンドポイント(LOCF)でのベースラインからのビフェプルノックスとプラセボ平均変化間の差に対応する処置効果値は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−4.1、0.6および−5.8であった。対比較から、エンドポイント(LOCF)での統計的に有意に大きい減少がプラセボに対して20mgビフェプルノックス群について見られた(多重比較のために補正したp値、p=0.031)。有意な処置群差は、それぞれプラセボと比較した5mgビフェプルノックスおよび10mgビフェプルノックス処置群について見られなかった。
【0111】
【表1】
【0112】
臨床研究2:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった(以下の表に示されるとおり)。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:30mgビフェプルノックス群について−5.9、そして40mgビフェプルノックス群について−3.2であった。統計的に有意な処置群差は、Hochberg補正したp値(p=0.020)に基づいてエンドポイントでプラセボと比較して30mgビフェプルノックス処置群について見られた。30mgビフェプルノックスとプラセボ処置群間の差はまた、公称p値(2週〜4週の各々でp<0.004)から2週〜4週でも認められた。さらに、処置効果は6週の研究にわたって一貫して増加した(効果の範囲:2週で−5.6そして4週で−6.3;表3.0.1)。40mgビフェプルノックス群とプラセボ群との間の統計的に有意な差は、Hochberg補正したp値に基づいてエンドポイントで明らかではなかった。
【0113】
【表2】
【0114】
臨床研究3:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−13.8(19.9)、30mgビフェプルノックス群について−13.1(20.2)、プラセボ群について−10.7(19.4)、そしてオランザピン群について−22.0(18.2)であった(表15)。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:20mgビフェプルノックス群について−3.5、そして30mgビフェプルノックス群について−2.2であった。統計的に有意な処置群差は、Hochberg補正したp値に基づいてプラセボ群と比較した20mgもしくは30mgビフェプルノックス処置群について見られなかった。同様に、研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。各来院について同じ評価者を有する患者についてのみデータを分析した場合、PANSS合計スコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)の差は一次解析において認められるものと同様であった。オランザピンとプラセボとの間のPANSS合計スコアの差は、感度分析に従って分析した。これらの結果は、オランザピンがプラセボと顕著に異なることを示した(p<0.001)。
【0115】
【表3】
【0116】
臨床研究4:各BX群のPANSS合計スコアにおけるベースラインから6週までの補正した平均変化(BX20:−4.0およびBX30:−2.7)は、PBO(1.1)群のものより統計的に有意に大きかった(BX20:p=0.002;BX30:p=0.017)。
【0117】
【表4】
【0118】
PANSS陽性
臨床研究1:以下の表は、ITT集団についてベースラインでの平均PANSS陽性下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−1.1、0.7、−1.5であった。エンドポイント(LOCF)での統計的に有意に大きい減少(未補正p=0.037)は、ビフェプルノックス20mg群およびプラセボの処置効果概算の比較についてPANSS陽性下位尺度スコアにおいて見られた。
【0119】
【表5】
【0120】
ベースラインからの変化の観察値分析について、PANSS合計スコアと同様の傾向が経時的なビフェプルノックス処置群におけるPANSS陽性下位尺度スコアのベースラインからの変化において見られた。ビフェプルノックスとプラセボ群との間の対比較は、統計的に有意ではなかった。
【0121】
臨床研究2:以下の表は、ITT集団についてベースラインでのPANSS陽性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−4.5(6.6)、40mgビフェプルノックス群について−4.2(6.9)、プラセボ群について−2.5(6.0)、そしてリスペリドン群について−7.2(6.6)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群
について−1.9、そして40mgビフェプルノックス群について−1.7であった。SEP1は有意な結果をもたらさず、従って、SEP2およびSEP3は評価されなかった(7.4.1.1節を参照)。しかしながら、ビフェプルノックス30mg処置群とプラセボとの間の顕著な差がエンドポイントで認められた(公称p=0.01)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.006)。
【0122】
同様に、ビフェプルノックス40mg処置群とプラセボとの間の顕著な差がエンドポイントで認められた(p=0.020)。プラセボに対する差はまた、1週〜3週でも認められた(p<0.013)。
【0123】
【表6】
【0124】
臨床研究3:以下の表は、ITT集団についてベースラインでのPANSS陽性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−4.3(6.1)、30mgビ
フェプルノックス群について−4.2(6.8)、プラセボ群について−3.5(6.2)、そしてオランザピン群について−7.0(5.8)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−1.1、そして30mgビフェプルノックス群について−0.7であった。公称p値(<0.05)での差は、6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で認められなかった。
【0125】
【表7】
【0126】
臨床研究4:平均PANSS陽性下位尺度スコア(FAS、LOCF)は、9週まで両方のBX群において経時的に減少し、その後にスコアはごくわずか増加した。PBO群において、平均PANSS合計スコアは2週まで減少し、その後にスコアは着実に増加した(図1;パネル34)。PANSS陽性下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20でもしくはBX30での処置は6週以降PBOでのものより統計的に有意に優れていた。
【0127】
PANSS陰性
臨床研究1:以下の表は、ITT集団についてベースラインでの平均PANSS陰性下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。ビフェプルノックス5mg、20mgおよびリスペリドン6mg群は、経時的に最も大きい改善を示した。エンドポイントLOCFでの処置効果(ビフェプルノックス−プラセボ)の概算は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−1.0、−0.3、−1.4であった。PANSS陰性下位尺度スコアにおける統計的に有意に大きい減少は、プラセボに対してビフェプルノックス20mg群について3週(未補正p=0.013)でそしてエンドポイント(LOCF)(未補正p=0.026)で見られた。
【0128】
【表8】
【0129】
PANSS陰性下位尺度スコアにおけるベースラインからの変化の観察値分析について、経時的なスコアの減少は、ビフェプルノックス処置群の各々における改善を示した。ビフェプルノックスの統計的に有意な処置効果は、プラセボと比較してビフェプルノックス5mg群について2週で認められた(p=0.032)。他の有意な差は認められなかった。
【0130】
臨床研究2:以下の表は、ITT集団についてベースラインでのPANSS陰性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陰性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−3.1(5.6)、40mgビフェプルノックス群について−2.2(5.4)、プラセボ群について−1.8(5.6)、そしてリスペリドン群について−3.8(5.5)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−1.4、そして40mgビフェプルノックス群について−0.9であった。SEP1は有意な結果をもたらさず、従って、SEP2およびSEP3は評価されなかった。
【0131】
30mgビフェプルノックス処置群は、エンドポイントでプラセボとの顕著な差を示した(公称p=0.027)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.021)。研究中の任意の時間点(1週〜6週)でプラセボと40mgビフェプルノックス用量群との間で差は認められなかった。
【0132】
【表9】
【0133】
臨床研究3:以下の表は、ITT集団についてベースラインでのPANSS陰性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陰性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−3.3(5.0)、30mgビフェプルノックス群について−3.1(5.2)、プラセボ群について−2.4(5.1)、そしてオランザピン群について−4.6(5.2)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.9、そして30mgビフェプルノックス群について−0.6であった。公称p値(<0.05)での差は、6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で認められなかった。
【0134】
【表10】
【0135】
臨床研究4:平均PANSS陰性下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後でスコアは安定した(図2;パネル35)。PBO群において、スコアは4週まで減少し、その後でスコアは増加する傾向があった(パネル35)。
【0136】
平均PANSS陰性下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20でもしくはBX30での処置は、BX30について2および4週を除いて、全ての時間点でPBOでのものより統計的に有意に優れていた。
【0137】
臨床研究4のPANSSデータ
平均PANSS合計スコア(FAS、LOCF)は9週まで両方のBX群において経時的に減少し、その後でスコアは安定した(パネル28)。PBO群において、平均PANSS合計スコアは2週まで減少し、その後でスコアは着実に増加した(パネル28)。
【0138】
PANSS合計スコア(パネル28)のLOCF分析により、最終観察値で補完すると、BX群におけるPANSS合計スコアは安定なままであったことが示される。両方のBX群における変化は、6週以降PBO群におけるものより統計的に有意に優れていた。さらに、BX20は4週以降PBOより統計的に有意に優れていた。PANSS合計スコアのOC分析により、研究を続行した患者は経時的に改善したことが示される(パネル29)。両方のBX群における変化は6週を包含するが6ヶ月を除く多数の時間点でPBO群におけるものより統計的に有意に優れていた。
【0139】
LOCFとPOCF分析との間の差は、離脱した多数の患者がそれらのPANSS合計スコアにおける増悪を有しそして増悪が見られた最初の来院で離脱したことを示す。これは、少なくともごくわずかでも悪くなった場合に研究から離脱するように患者に求める研究設計と一致する。
【0140】
他の効能変数について、効能スコアの経時的な進展の傾向は、研究に含まれる患者が鬱状態でなかったことをベースラインレベルが示すCDSSを除いて、同様であった。
【0141】
【表11】
【0142】
【表12】
【0143】
【表13】
【0144】
【表14】
【0145】
【表15】
【0146】
総合精神病理スコア
臨床研究1:総合精神病理スコアは、以下の表に示されるようにビフェプルノックス処置群の各々についてベースラインから6週まで減少した。LOCFを用いたビフェプルノックスの処置効果の概算は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−2.2、0.4および−2.8であった。ビフェプルノックス20mg群は、2週(未補正p=0.029)、3週(未補正p=0.032)そしてエンドポイント(LOCF)(未補正p=0.016)でプラセボより統計的に有意に大きい減少を有した。
【0147】
【表16】
【0148】
PANSS総合精神病理下位尺度スコアにおけるベースラインからの変化の観察値分析について、経時的なスコアの減少はビフェプルノックス処置群の各々における改善を示した。プラセボに対するビフェプルノックスの統計的に有意な処置効果は、プラセボに対してビフェプルノックス20mg群について2週で(p=0.038)そしてビフェプルノックス5mg群について6週で(p=0.017)認められた。他の有意な差は認められなかった。
【0149】
臨床研究2:ITT集団(LOCF)についてのベースラインでの平均PANSS総合精神病理下位尺度スコアおよび各ベースライン後来院でのPANSS総合精神病理下位尺度におけるベースラインからの変化を以下の表に提示する。PANSS総合精神病理下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−6.0(10.2)、40mgビフェプルノックス群について−3.8(10.1)、プラセボ群について−3.5(9.7)、そしてリスペリドン群について−8.6(9.8)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−2.5、そして40mgビフェプルノックス群について−0.7であった。2週〜エンドポイン
トで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.025)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕著な差はなかった。
【0150】
【表17】
【0151】
臨床研究3:ITT集団(LOCF)についてのベースラインでの平均PANSS総合精神病理下位尺度スコアおよび各ベースライン後来院でのPANSS総合精神病理下位尺度スコアにおけるベースラインからの変化を以下の表に提示する。PANSS総合精神病理下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−6.1(10.7)、30mgビフェプルノックス群について−5.8(10.3)、プラセボ群について−4.8(10.0)、そしてオランザピン群について−10.5(9.4)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−1.5、そして30mgビフェプルノックス群について−0.9であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0152】
【表18】
【0153】
臨床研究4:平均PANSS総合精神病理下位尺度スコアは、PANSS合計スコアと同じパターンを一般にたどった。平均PANSS総合精神病理下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後でスコアは安定した(図3;パネル36)。PBO群において、平均PANSS総合精神病理下位尺度スコアは2週まで減少し、その後でスコアは着実に増加した(パネル36)。
【0154】
PANSS総合精神病理下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20およびBX30は両方とも9週以降PBOより統計的に有意に優れていた。さらに、BX20は6週でPBOより統計的に有意に優れていた。
【0155】
BPRS合計スコア
臨床研究1:以下の表に示されるように、ビフェプルノックス20mg群はプラセボ群よりBPRS合計スコアにおける大きい変化を示した。2週(未補正p=0.042)から始まるあらゆる時間点;3週(未補正p=0.020);4週(未補正p=0.024);エンドポイント(LOCF)(未補正p=0.012)でプラセボに対してビフェプルノックス20mgについて統計的に有意な処置群差が見られた。
【0156】
【表19】
【0157】
LOCF分析において認められる同様の傾向は、観察値を用いたBPRS合計スコア分析におけるベースラインからの変化において認められた。プラセボに対するビフェプルノックス処置効果の比較は、5mg群のみについて6週で統計的に有意であった(p=0.024)。他の有意な比較は認められなかった。
【0158】
臨床研究2:以下の表は、ITT集団(LOCF)についてベースラインでの平均BPRS合計スコア(PANSS由来)および各ベースライン後来院でのBPRS合計スコアにおけるベースラインからの変化を提示する。BPRS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−8.1(12.3)、40mgビフェプルノックス群について−6.5(11.7)、プラセボ群について−4.9(11.5)、そしてリスペリドン群について−12.2(11.7)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−3.2、そして40mgビフェプルノックス群について−1.9であった。2週〜エンドポイントで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.019)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕
著な差は見られなかった。
【0159】
【表20】
【0160】
臨床研究3:以下の表は、ITT集団(LOCF)についてのベースラインでの平均BPRS合計スコア(PANSS由来)および各ベースライン後来院でのBPRS合計スコアにおけるベースラインからの変化を提示する。BPRS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−8.5(11.9)、30mgビフェプルノックス群について−7.9(12.0)、プラセボ群について−6.7(11.7)、そしてオランザピン群について−13.2(10.7)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−2.1、そして30mgビフェプルノックス群について−1.1であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0161】
【表21】
【0162】
BPRS精神病クラスタースコア
臨床研究1:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−0.9、0.4、−1.1であった。ビフェプルノックスとプラセボのエンドポイント(LOCF)でのベースラインからの平均変化間の差における統計的に有意に大きい減少(未補正p=0.044)は、プラセボに対するビフェプルノックス20mg群のBPRS精神病クラスタースコアにおいて見られた。
【0163】
【表22】
【0164】
BPRS精神病クラスタースコアにおけるベースラインからの変化の観察値分析について、プラセボと比較してスコアにおけるより大きい減少が、4週から始まり3つのビフェプルノックス処置群の各々について見られた。統計的に有意な差は認められなかった。
【0165】
臨床研究2:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。BPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−3.2(4.2)、40mgビフェプルノックス群について−2.6(4.4)、プラセボ群について−1.8(3.5)、そしてリスペリドン群について−4.9(4.0)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−1.4、そして40mgビフェプルノックス群について−1.0であった。2週〜エンドポイントで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.002)。エンドポイントで(p=0.031)そして2週および3週で(p<0.036)で40mgビフェプルノックス群とプラセボとの間で顕著な差が見られた。
【0166】
【表23】
【0167】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。BPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−3.1(3.9)、30mgビフェプルノックス群について−3.2(4.4)、プラセボ群について−2.6(4.0)、そしてオランザピン群について−4.9(4.0)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.6、そして30mgビフェプルノックス群について−0.5であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0168】
【表24】
【0169】
CGI−S
臨床研究1:以下の表は、ITT集団についてのベースラインでの平均CGI疾患重症度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−0.31、0.12、−0.19であった。対処置群比較により、2週(p=0.008)、3週(p=0.020)および4週(p=0.032)でプラセボに対してビフェプルノックス20mgにおいて統計的に有意に大きい減少が示されたが、エンドポイント(LOCF)ではそうでなかった。ビフェプルノックス5mg対プラセボの処置群比較は、3週(p=0.049)、4週(p=0.033)およびエンドポイント(LOCF)(p=0.013)で有意であった。
【0170】
【表25】
【0171】
臨床研究2:以下の表は、ITT集団(LOCF)についてのベースラインでの平均CGI−SスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CGI疾患重症度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−0.69(1.19)、40mgビフェプルノックス群について−0.54(1.12)、プラセボ群について−0.37(1.07)、そしてリスペリドン群について−1.06(1.20)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−0.28、そして40mgビフェプルノックス群について−0.18であった。CGI−Sにおけるベースラインからエンドポイントまでの変化の統計的に有意な差は、ステップダウン法に基づいてプラセボと比較して30mgビフェプルノックス処置群について見られなかった(補正したp=0.056)。30mgビフェプルノックス群は、エンドポイントでプラセボとの顕著な差を示した(公称p=0.028)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.015)。研究中の任意の時間点(1週〜4週)でプラセボと40mgビフェプルノックス用量群との間で差は認められなかった。
【0172】
【表26】
【0173】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均CGI疾患重症度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CGI疾患重症度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−0.63(0.98)、30mgビフェプルノックス群について−0.62(1.03)、プラセボ群について−0.49(1.06)、そしてオランザピン群について−1.03(1.00)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.13、そして30mgビフェプルノックス群について−0.09であった。6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で公称p値(p<0.05)では差は顕著でなかった。
【0174】
【表27】
【0175】
CGI−I
臨床研究1:表24に示されるように20mgビフェプルノックス処置群のCGI改善度スコアにおいて改善が認められた。処置群差は、LOCF分析においてプラセボに対してビフェプルノックス20mg群について1週(p=0.040)および2週(p=0.016)で統計的に有意であったが、エンドポイントではそうでなかった。他の有意な差は認められなかった。
【0176】
【表28】
【0177】
臨床研究2:ベースラインからエンドポイント(LOCF)まで中等度もしくは著明改善を報告した患者の割合は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において28%、プラセボ群において25%、そしてリスペリドン群において52%であった(以下の表)。エンドポイントで、プラセボと2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められず;しかしながら、30mgビフェプルノックス群とプラセボとの間の顕著な差は2週〜4週で見られた(p<0.024)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕著な差は見られなかった。2週〜エンドポイントのデータを以下の表に要約する。
【0178】
【表29】
【0179】
臨床研究3:以下の表は、ITT集団についてのLOCFを用いた来院ごとのCGI改善度評価の7つのカテゴリーの頻度を提示する。平均CGI改善度スコアに関する処置群間の統計的差は、修正リジットスコアを用いて7つのCGI改善度カテゴリーの頻度に適用したプールした施設により階層化されるCMH検定を用いて評価された。ベースラインからエンドポイントまで(表23における対応する個々の割合を合計することにより得られる)、合わせた中等度もしくは著名改善を報告した患者の割合は:20mgビフェプルノックス群において38%、30mgビフェプルノックス群において34%、プラセボ群において32%、そしてオランザピン群において46%であった。エンドポイントでのプラセボ群に対する20mgビフェプルノックス群の比較は、顕著であることが見出された(p=0.027)。プラセボ群に対する30mgビフェプルノックス群の処置比較は、顕著ではなかった(p=0.162)。研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0180】
【表30】
【0181】
PANSSレスポンダー率
臨床研究1:PANSSレスポンダー率は、プラセボ群と比較してビフェプルノックス処置群の各々について高かった。PANSSレスポンダー率は、研究プロトコルから20%定義を用いてそれぞれ3つの用量群について28%、24%および34%であった。全ての4つの定義を用いるPANSSのレスポンダー率は、以下の表に提示される。
【0182】
【表31】
【0183】
LOCFを用いたPANSSレスポンダー率の有意な差は、プラセボに対するビフェプルノックス5mgの25%定義を用いてそしてプラセボに対するビフェプルノックス20mg群の25%、30%および35%定義について見られた。
【0184】
臨床研究2:PANSSレスポンダーは、そのPANSS合計スコアがLOCFデータに基づいてベースラインからエンドポイントまで20%以上減少した患者として定義された。探索的解析として、レスポンダー率はまたレスポンダーの30%、35%、40%および50%定義も用いて分析された。エンドポイント(LOCF)でのPANSSレスポンダー率の要約は、以下の表25に提示される。
【0185】
PANSSレスポンダー率は、プラセボ群と比較して2つのビフェプルノックス処置群についてわずかに高かった。PANSS20%レスポンダー率は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において30%、プラセボ群において26%、そしてリスペリドン群において54%であった。しかしながら、30mgビフェプルノックス群とプラセボとの間の差は顕著ではなかった(p=0.052)。PANSS 30%レスポンダー率は:30mgビフェプルノックス群において24%、40mgビフェプルノックス群において22%、プラセボ群において14%、そしてリスペリドン群において31%であった。30mgビフェプルノックス群とプラセボとの間の差は顕著であった(p=0.019)。30mgビフェプルノックス群とプラセボとの間の顕著な差はまた、35%および50%レスポンダー率についても見られた。
【0186】
【表32】
【0187】
臨床研究3:PANSSレスポンダーは、そのPANSS合計スコアがベースラインからエンドポイントまで20%以上減少した患者として定義された。探索的解析として、レスポンダー率はまたレスポンダーの30%、35%、40%および50%定義も用いて分析された。エンドポイント(LOCF)でのPANSSレスポンダー率の要約を以下の表に提示する。20%PANSSレスポンダー率は、プラセボ群と比較して2つのビフェプルノックス処置群についてわずかに高く:20mgビフェプルノックス群における患者の42%、30mgビフェプルノックス群における患者の39%、プラセボ群における患者の32%、そしてオランザピン群における患者の54%は、PANSS合計スコアにおいてベースラインからエンドポイントまで20%以上改善した。20mgビフェプルノックス群とアプローチしたプラセボ群との間の差は、顕著である(p=0.061)。しかしながら、レスポンダーの定義のさらに厳格な基準(30%〜50%)を用いた場合、20mgもしくは30mgビフェプルノックス群とプラセボ群との間の差は顕著ではなかった(全てp>0.118)。
【0188】
【表33】
【0189】
臨床研究4:各来院でベースラインに対してPANSS合計スコアの>25%、>35%、>45%もしくは>55%の減少を有する患者(PANSSレスポンダー)の割合は、LOCFにより示される(図4;パネル42)。
【0190】
CGIレスポンダー率
臨床研究1:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。ビフェプルノックス5mgおよび20mg群のCGIレスポンダー率は、プラセボ群のレスポンダー率より高かった。プラセボと比較した場合に3つのビフェプルノックス用量群のいずれについても統計的に有意な差は見られなかった。
【0191】
【表34】
【0192】
臨床研究2:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。エンドポイント(LOCF)でのCGI−Iレスポンダー率の要約を以下の表に提示する。CGI−Iレスポンダー率は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において28%、プラセボ群において25%、そしてリスペリドン群において52%であった。30mgビフェプルノックス群とプラセボとの間の顕著な差が見られた(p=0.039)。
【0193】
【表35】
【0194】
臨床研究3:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。エンドポイント(LOCF)でのCGI−Iレスポンダー率の要約を表3.10.0(LOCF)および表26に提示する。CGI−Iレスポンダー率は:20mgビフェプルノックス群において38%、30mgビフェプルノックス群において34%、プラセボ群において32%、そしてオランザピン群において46%であった。プラセボ群と2つのビフェプルノックス用量群のいずれかとの
間で顕著な差は認められなかった。
【0195】
【表36】
【0196】
CDSS
臨床研究2:統合失調症のカルガリー鬱病評価尺度スコアを以下の表に提示する。この表は、ITT集団についてのベースラインでの平均CDSSスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CDSSスコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−0.66(3.64)、40mgビフェプルノックス群について0.01(3.66)、プラセボ群について−0.39(3.45)、そしてリスペリドン群について−0.89(3.42)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス、プラセボ)は:30mgビフェプルノックス群について−0.38、そして40mgビフェプルノックス群について0.29であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボと2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0197】
【表37】
【0198】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均CDSSスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CDSSスコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−1.30(3.85)、30mgビフェプルノックス群について−0.79(3.02)、プラセボ群について−0.59(4.20)、そしてオランザピン群について−1.47(3.95)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス、プラセボ)は:20mgビフェプルノックス群について−0.54、そして20mgビフェプルノックス群について−0.34であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0199】
【表38】
【0200】
臨床研究4:各来院でCGI−Iスコア<2を有する患者(CGI−Iレスポンダー;FAS、LOCF)の割合を表(図5;パネル43)において以下に示す。
【0201】
EPS
EPSは、静座不能、ジスキネジー、パーキンソン症候群などのような処置下で発現した有害事象ならびに/またはSAS、BASおよび/もしくはAIMSのような公式評価尺度を用いて評価された。
【0202】
シンプソン・アンガス評価尺度16(SAS)
SASは、抗精神病薬を投与された患者におけるパーキンソン症候群タイプ症状を測定するために用いられる。評価尺度は、各々0(症状が全くない)〜4(極端な形の症状の存在)の間の5点評価尺度で評価される10項目からなる。SAS合計スコアは全ての項目スコアの合計として定義され、そして範囲は0〜40である。3までのSAS合計スコアは、正常と見なされる。
【0203】
バーンズ・アカシジア評価尺度17(BAS)
BASは、薬剤誘発性静座不能の観察可能な落ち着きのない動きならびに落ち着きのなさの自覚および静座不能と関連する任意の苦痛を評価するために用いられる。評価尺度は、0(静座不能の症状なし)から3(重篤な静座不能)まで評価される3項目からなる。
BAS合計スコアはこれら3つのBAS項目スコアの合計として定義され、そして範囲は0〜9である。さらに、静座不能の包括的(global)臨床評価は0(静座不能の症状なし)から5(重篤な静座不能)まで評価される。
【0204】
異常不随意運動評価尺度18(AIMS)
ジスキネジー運動の発生を記録するために考案されたAIMSは、12項目からなる。項目1〜7は、0(なし)〜4(重篤)の評価尺度で特定の不随意運動を測定する。項目8〜10は、0(自覚なし)〜4(自覚する、重篤な苦痛)の評価尺度で異常運動の包括的評価を測定する。項目11および12は、イエス/ノーで答える、患者の歯の症状に関する質問である。合計スコアはAIMS項目1〜10を合計することにより計算され、そして0〜40の間であり;非包括的合計スコアは項目1〜7を合計することにより計算される。
【0205】
臨床研究1:
【0206】
【表39】
【0207】
【表40】
【0208】
【表41】
【0209】
臨床研究2:
【0210】
【表42】
【0211】
【表43】
【0212】
臨床研究3:
【0213】
【表44】
【0214】
【表45】
【0215】
【表46】
【0216】
臨床研究4:
EPSに関連するTEAEを有する患者の割合は、BX群のいずれか(BX20:10%;BX30:15%)におけるよりもPBO群(4%)において低かった。PBO群に対していずれかのBX群において>3人の患者がいたEPSに関連するTEAEは:BX20−静座不能;BX30−ジスキネジー、静座不能、錐体外路障害を含んでなった。全体で、15人の患者が離脱につながるEPSに関連するTEAEを有した(PBO群において2人;BX20群において4人;BX30群において9人)。
【0217】
【表47】
【0218】
SAS、BASおよびAIMS
臨床研究1:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。各時間点で正常および異常スコアを有する患者の割合。ベースラインで、異常SASスコアを有する患者の割合は8%〜12%の間であった。エンドポイントで、異常SASスコアを有する患者の割合は3%〜10%の間であった。異常SASスコアを有する患者の割合は、全ての処置群においてベースラインとエンドポイントとの間で減少した。ベースラインからエンドポイントまでカテゴリー
間でシフトする患者の割合において処置群間で統計的に有意な差はなかった(p=0.800)。
【0219】
BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛スコア、3項目合計スコアもしくは包括的臨床評価スコアについてベースラインもしくはエンドポイントで処置群間で統計的に有意な差はなかった。
【0220】
BAS合計スコアもしくはBAS包括的臨床評価スコアにおいてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった。ベースラインもしくはエンドポイントで処置群間で統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群間で統計的に有意な差はなかった。
【0221】
臨床研究2:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。ベースラインで、異常SASスコアを有する患者の割合は:30mgビフェプルノックス群において14%、40mgビフェプルノックス群において11%、プラセボ群において7%、そしてリスペリドン群において6%であった。ベースラインとエンドポイントとの間で異常SASスコアを有する患者の割合は、ビフェプルノックス群において減少し、プラセボ群において変化せず、そしてリスペリドン群において増加した。エンドポイントで、異常SASスコアを有する患者の割合は、40mgビフェプルノックスおよびプラセボ群における7%からリスペリドン群における14%までの間であった。
【0222】
ベースラインで正常からエンドポイントで異常へのシフトは、他の3つの処置群(30mgビフェプルノックス:1人の患者、<1%;40mgビフェプルノックス:5人の患者、4%;プラセボ:6人の患者、4%)と比較してリスペリドン群(19人の患者、12%)において最も高かった。処置群間での統計的に有意な差(p<0.001)が認められた。
【0223】
BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア(p=0.093)、落ち着きのなさの自覚スコア(p=0.368)、落ち着きのなさと関連する苦痛スコア(p=0.779)、3項目合計スコア(p=0.433)もしくは静座不能の包括的臨床評価スコア(p=0.541)についてエンドポイントで処置群にわたる統計的に有意な差はなかった。また、上記の評価尺度についてベースラインでも処置群にわたる統計的に有意な差はなかった(p>0.168)。
【0224】
BAS合計スコア(p=0.482)もしくはBAS包括的臨床評価スコア(p=0.911)においてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合における処置群にわたる統計的に有意な差はなかった。
【0225】
ベースライン(平均範囲=1.0〜1.2;p=0.923)もしくはエンドポイント(平均範囲=0.9〜1.6;p=0.138)で処置群にわたる統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群にわたる統計的に有意な差はなかった(平均範囲=−0.3〜0.4;p=0.138)。
【0226】
臨床研究3:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。ベースラインで、異常SASスコアを有
する患者の割合は6%〜10%の間であった。エンドポイントで、異常SASスコアを有する患者の割合は3%〜8%の間であった。異常SASスコアを有する患者の割合は、全ての処置群においてベースラインとエンドポイントとの間で減少した。ベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった(p=0.463)。BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア(p=0.421)、落ち着きのなさの自覚スコア(p=0.584)、落ち着きのなさと関連する苦痛スコア(p=0.254)、3項目合計スコア(p=0.865)もしくは包括的臨床評価スコア(p=0.518)についてエンドポイントで処置群間で統計的に有意な差はなかった。落ち着きのなさと関連する主観的苦痛スコアについてベースラインで処置群間で統計的に有意な差があったが(p=0.029)、任意の他のスコアについてはそうでなかった。
【0227】
BAS合計スコア(p=0.808)もしくはBAS包括的臨床評価スコア(p=0.525)においてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった。
【0228】
ベースライン(p=0.362)もしくはエンドポイント(p=0.187)で処置群間で統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群間で統計的に有意な差はなかった(p=0.187)。
【0229】
臨床研究4:
SAS
ベースラインで、全ての3つの処置群における平均SAS合計スコアは2.17〜2.33の間であった。
【0230】
研究中に全ての3つの処置群における平均SAS合計スコアのわずかな変動があり、そして6ヶ月で、平均SAS合計スコアは0.57〜1.16の間であり(APTS、OC);従って、ベースラインでそして6ヶ月で平均合計スコアは正常範囲内であった。SAS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は、各処置群において<1であった(APTS、OC、ANCOVA)。SAS合計スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった。
【0231】
ベースラインで、患者の大部分(PBO:74%;BX20:71%;BX30:72%)はSAS状態に関して正常であり、そして正常患者の割合は6ヶ月で増加していた(PBO:85%;BX20:89%;BX30:78%)。SAS状態のシフトにおいて処置群のいずれかの間で統計的に有意な差はなかった(カテゴリー:不変;異常→正常;正常→異常)。
【0232】
BAS
ベースラインで、全ての3つの処置群における平均BAS合計スコアは0.44〜0.46の間であった。研究中に全ての3つの処置群における平均BAS合計スコアのわずかな変動があり、そして6ヶ月で、平均BAS合計スコアは0.05〜0.17の間であった(APTS、OC)。BAS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は、各処置群において<0.6であった(APTS、OC、ANCOVA)。BAS合計スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった。
【0233】
BAS包括的評価スコアについて、傾向は同様であり:研究中に全ての3つの処置群において平均BAS包括的評価スコアのわずかな変動があり;平均スコアはベースラインで0.22〜0.26、そして6ヶ月で0.07〜0.14の間であった(APTS、OC
)。6ヶ月でBAS包括的評価スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった(APTS、OC)。BAS項目1、2もしくは3の分布において処置群のいずれかの間で統計的に有意な差はなかった。
【0234】
AIMS
全ての3つの処置群における平均AIMS合計スコアは低く、そしてベースラインで1.04〜1.35の間であった。全ての処置群について補正した平均スコアの初期増加があり(BX30群において最大)、その後で平均スコアは経時的に減少し;6ヶ月で、スコアは全ての3つの処置群においてベースラインレベルまで戻っていた(PBO:0.11;BX20:1.08;BX30:0.86(APTS、OC))。BX20(全ての時間点)およびBX30(5および6ヶ月)群におけるAIMS合計スコアの補正した平均変化はPBO群におけるものと統計的に有意に異ならず、一方、BX30(1週〜4ヶ月)群の補正した平均変化はそうでなかった。AIMS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は小さかった(PBO:0.39;BX20:1.21;BX30:2.45、APTS、OC、ANCOVA)。差はいずれも臨床的に有意と見なされなかった。
【0235】
【表48】
【0236】
体重
臨床研究1:
体重のわずかな減少がビフェプルノックス処置群において認められたが、プラセボ処置群においてはそうでなかった。エンドポイントで、ビフェプルノックス処置群における患者の平均体重減少は約1 lb(5mgビフェプルノックス、−1.0 lb;10mgビフェプルノックス、−1.3 lb;20mgビフェプルノックス、−0.6 lb)であった。プラセボ処置群は、1.9 lbの平均体重増加を有した。
【0237】
体重におけるベースラインからの変化の統計的検定は、副次的効能パラメーターに使用するものと同様の方法論を用いて行われた(処置およびベースラインでの体重の因子を有するANCOVAモデル)。プラセボに対するビフェプルノックス処置群の対比較は、ビフェプルノックスとプラセボのエンドポイントでの体重のベースラインからの平均変化間
の差について全て統計的に有意であった(ビフェプルノックス5mg[p=0.025]、ビフェプルノックス10mg[p=0.009]およびビフェプルノックス20mg[p=0.031])。
【0238】
リスペリドン処置群における患者の平均体重変化は、4.8 lbの増加であった。
【0239】
体重の増加:ビフェプルノックス処置群におけるその体重が7%より多く増加した患者の割合(2〜4%)は、プラセボ処置群において認められる割合(5%)より少ないかもしくは同様であった。リスペリドン処置群において、患者の13%はそれらの体重を>7%増加した。
【0240】
体重の減少:その体重が7%より多く減少した患者の割合は、プラセボ処置群(3%)におけるよりもビフェプルノックス処置群(5mg、6%;10mg、7%;20mg、8%)において高かった。リスペリドン処置群にはそれらの体重を>7%減少した患者はいなかった。
【0241】
臨床研究2:
平均体重のわずかな減少がエンドポイントでビフェプルノックス処置群およびプラセボ群において認められ、一方、リスペリドン処置群について増加が認められた(以下の表)。エンドポイントで、平均体重変化は:30mgビフェプルノックス群において−2.2 lb;40mgビフェプルノックス群において−1.9 lb;プラセボ群において0.5 lb;そしてリスペリドン群において3.2 lbであった。ビフェプルノックス処置群において、6週での平均変化はエンドポイントでのものより高かった。
【0242】
【表49】
【0243】
臨床研究3:平均体重のわずかな減少がビフェプルノックス処置群およびプラセボ群において認められ、一方、オランザピン処置群について増加が認められた。エンドポイントで、平均体重変化は:20mgビフェプルノックス群において−2.3 lb;30mgビフェプルノックス群において−1.1 lb;プラセボ群において−1.3 lb、そしてオランザピン群において5.2 lbであった。6週での平均変化は、エンドポイントでのものと同程度であった(以下の表)。
【0244】
体重の著しく異常な(>7%)減少の発生率は、ビフェプルノックス処置およびプラセボ群間で同程度であり(20mgビフェプルノックス:6%;30mgビフェプルノックス:5%;プラセボ:5%)、そしてオランザピン処置群において低かった(<1%)。体重の著しく異常な(>7%)増加の発生率においては、これを経験した30mgビフェプルノックスにおける5%、20mgビフェプルノックスにおける1%、そしてプラセボ群における5%と比較してオランザピン処置群では19%で逆のことが見られた。
【0245】
【表50】
【0246】
臨床研究4:
体重およびBMIならびにベースラインに対するその変化を以下の表に要約する。全ての群において、補正した平均体重およびBMIはベースラインから6ヶ月まで減少した。ベースラインから6ヶ月までの補正した平均体重変化(APTS、OC、ANCOVA)は、PBO群において−0.8kg、BX20群において−0.3kg、そしてBX30群において−0.5kgであった。BX20およびBX30群における補正した平均体重減少は、PBO群におけるものと統計的に有意に異ならなかった。
【0247】
減少した体重は、全ての処置群における患者についてTEAEとして報告された(PBO:5%;BX20:8%;BX30:8%)(パネル45)。増加した体重は、PBO群(1.8%)におけるそしてBX30群(1.2%)における患者についてTEAEとして報告された。
【0248】
悪心および/もしくは嘔吐を有する/有さない患者におけるベースラインから6ヶ月までの補正した平均体重変化(APTS、LOCF、ANCOVA)は、悪心および/もしくは嘔吐も有した患者はより大きい体重減少を有したが、全ての群における患者が、悪心および/もしくは嘔吐を有するかどうかにかかわらず体重を減少することを示した(PBO:−0.6対−1.9kg;BX20:−1.0対−1.9kg;BX30:−1.1対−2.3kg)。
【0249】
【表51】
【0250】
【表52】
【0251】
増悪までの時間
臨床研究4:
主要効能変数は増悪までの時間であり、そして分析はFASに基づいた。主要効能分析は、3つの処置群における統合失調症の増悪までの等しい時間の仮説を退けた(p=0.008)。増悪した患者の割合はPBO群において59%、BX20群において41%、そしてBX30群において38%であった。Cox比例ハザードモデルは、PBOに対して0.66(BX20)および0.65(BX30)の概算ハザード比を与え;すなわち、増悪の危険性はBX20もしくはBX30群における患者についてよりもPBO群における患者について約1.5倍高かった。
【0252】
次にBX群の各々とPBO群との対比較により、BX群における患者はPBO群におけ
る患者よりも統合失調症の増悪までの統計的に有意に長い時間を有することが示された(BX20:p=0.008およびBX30:p=0.006)。主要効能分析をPPSについて繰り返した。PPSにおける患者の大部分は研究の大部分に参加したので、得られる概算ハザード比およびp値の両方について、結果は主要分析の結果に非常に近かった。これは、主要効能分析の結論のロバスト性を説明する。
【0253】
【表53】
【0254】
脂質プロフィール
臨床研究4:補正した平均HDLコレステロール値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで増加した(PBO:0.04/0.06(空腹時/非空腹時);BX20:0.07/0.08;BX30:0.07/0.08mmol/L)。BX群のいずれかとPBO群の間で統計的に有意な差はなかった。補正した平均トリグリセリド値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで減少した(PBO:−0.06/−0.22(空腹時/非空腹時);BX20:−0.16/−0.21;BX30:−0.37/−0.03mmol/L)。BX群のいずれか(BX30(空腹時)を除く)とPBO群との間で統計的に有意な差はなかった。
【0255】
補正した平均空腹時グルコース値は全ての3つの処置群においてベースラインから6ヶ月まで増加し(PBO:0.10;BX20:0.13;BX30:0.09mmol/L)、そしてBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0256】
以下のパネル50は、空腹時および非空腹時患者におけるベースラインでの(平均値)そして6ヶ月での(ベースラインからの平均変化)脂質プロフィールを要約する。計算される平均総コレステロールおよび平均LDLは、処置および空腹時/非空腹時条件にかかわらず全ての群(非空腹時PBO患者を除く)においてベースラインから6ヶ月まで減少した。計算される平均VLDLおよび平均トリグリセリドは、処置および空腹時/非空腹時条件にかかわらず全ての群(非空腹時BX30患者を除く)においてベースラインから6ヶ月まで減少した。平均HDLは、処置および空腹時/非空腹時条件にかかわらず全ての群においてベースラインから6ヶ月まで増加した。
【0257】
【表54】
【0258】
高血糖および糖尿病
ある場合に著明なそしてケトアシドーシスもしくは高浸透圧性昏睡もしくは死亡と関連する高血糖は、非定型抗精神病薬で処置した患者において報告されている。ビフェプルノックスで処置した患者における高血糖および糖尿病関連有害事象(高血糖、上昇した血糖、耐糖能異常、糖尿病、不十分に制御される糖尿病のような)の発生率は、6週のプラセボ対照試験において0.5%(5/1050)であり、そしてプラセボ処置患者においては0.6%(3/469)であった。26週のプラセボ対照試験において、高血糖もしくは糖尿病関連有害事象を報告した患者はいなかった。非定型抗精神病薬使用とグルコール異常との間の関係の評価は、統合失調症を有する患者における糖尿病の増加した背景リスクの可能性および一般集団における糖尿病の増加する発生率により複雑化される。これらの交絡因子を考えると、非定型抗精神病薬使用と高血糖関連有害事象との間の関係は完全に理解されるわけではない。しかしながら、ビフェプルノックスを含まなかった疫学的研究は、これらの研究に含まれる非定型抗精神病薬で処置した患者における処置下で発現した高血糖関連有害事象の増加した危険性を示唆する。ビフェプルノックスはこれらの研究が行われた時点で市販されていなかったので、ビフェプルノックスがこの増加した危険性と関連するかどうかは知られていない。非定型抗精神病薬で処置した患者における高血糖関連有害事象の正確な危険性概算は利用可能でない。
【0259】
PR、QT、QTc、QRS
臨床研究1:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平
均値の変化はほとんどなかった。これらの間隔の平均変化は、約−2msec〜4msecの間であった。処置群による平均変化の傾向はなかった。ベースラインとエンドポイントとの間の平均心拍数の変化は、−1.5bpm〜0.9bpmの間であった。処置群による平均変化の傾向はなかった。
【0260】
臨床研究2:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平均値の変化はほとんどなかった。処置群にわたる平均変化の範囲は:PR=−2.5msec〜0.2msec;QT=−2.8msec〜7msec;QTc=0.4msec〜3.9msec;QRS=−0.4msec〜1.4msecであった。処置群による平均変化の傾向はなかった。処置群にわたるベースラインとエンドポイントとの間の平均心拍数の変化は、おおよそ−1.3bpm〜1.7bpmの間であった。処置群による平均変化の傾向はなかった。
【0261】
臨床研究3:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。これらの要約について、Bazett補正したQT間隔はQTcとして提示された。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平均値の変化はほとんどなかった。これらの間隔の平均変化は、約3msec〜4msecの間であった。処置群による平均変化の傾向はなかった。ベースラインとエンドポイントとの間の平均心拍数の変化は、−1bpm〜1bpmの間であった。処置群による平均変化の傾向はなかった。
【0262】
実施例3a:ビフェプルノックスの薬物動態
本研究の目的は、ビフェプルノックスの薬物動態(PK)を評価することである。健常被験者におけるビフェプルノックスのPKを21の臨床薬理学研究からのPKパラメーターのプールした分析に基づいて調べた。プールした分析には、それぞれ132および399人の被験者への単回および複数回投与後のPKプロフィールが包含され、そして年齢、性別、体重および人種の潜在的影響を調べた。さらに、第II相研究中の376人の患者および第III相研究中の434人の患者からのサンプルに基づく母集団PK法を用いて統合失調症を有する患者におけるPKを調べた。ビフェプルノックスは、経口投与後に迅速に吸収された(全ての用量レベルで1.5〜2時間のtmax)。ビフェプルノックス複数回投与PKは、20〜40mg/日の範囲で用量に比例した。定常状態の平均の見かけのクリアランスおよび見かけの分布容積は、それぞれ、62.2L/hおよび1300Lであった。ビフェプルノックスは、14.4時間の平均血漿定常状態半減期で排出された。標準的な高脂肪食での40mg用量の投与は、tmax(1.5時間)のわずかな遅延ならびにCmax(10%)およびAUC(29%)のわずかな増加と関連した。ビフェプルノックスは、血清タンパク質に約99%結合している。ビフェプルノックスは、CYP2C9、CYP3A4そしてより少ない程度にCYP2D6により代謝される。ビフェプルノックス暴露は、フルコナゾール(CYP2C9阻害剤)そしてわずかな程度にケトコナゾール(CYP3A4阻害剤)との共投与により増加されたが、パロキセチン(CYP2D6阻害剤)およびファモチジン(H2アンタゴニスト)との共投与により増加されなかった。ビフェプルノックス暴露は、カルバマゼピン(CYP3A4誘導剤)の共投与により軽減された。ビフェプルノックスと狭い治療指数化合物ワーファリンおよびリチウムとの共投与(実施例3bを参照)は、いかなる関連する程度にもこれらの化合物のPKに影響を及ぼさなかった。CYP2C9遅い/中間代謝個体において、正常酵素活性を有する被験者におけるよりも高い血漿レベルのビフェプルノックスが認められた。[14C]で標識したビフェプルノックスの単回経口投与後に、放射活性の13%および74%はそれぞれ尿および便において排出された。ビフェプルノックスPKへの臨床的に有意な
年齢、性別、体重もしくは人種に関連する効果は認められなかった。統合失調症を有する患者におけるPKは、健常被験者においてみられるものと同様であった。
【0263】
本研究の1つの結論は、ビフェプルノックスが経口投与後に迅速に吸収され;平均排出半減期が約14時間であることである。複数回投与PKは、20〜40mgの範囲で用量に比例した。ビフェプルノックスは、低い相互作用ポテンシャルを有する。
【0264】
実施例3b:健常男性被験者におけるリチウムおよびビフェプルノックスの薬物動態学的相互作用
目的:ドーパミンD2および5−HT1A受容体の部分アゴニスト、ビフェプルノックスは、統合失調症の処置用に開発されている。ビフェプルノックスは、精神病および気分障害を有する患者の処置のために気分安定剤リチウムと組み合わせて用いることができるので、リチウムの薬物動態(PK)プロフィールへのビフェプルノックスの複数回投与の影響を評価した。リチウムは、治療を複雑にし得る狭い治療指数を有し、そして1.5mmol/Lより大きい血清レベルは、より低いレベルよりリチウム毒性のより大きい危険性を有する。方法:これは、48人の健常男性被験者における単一施設二重盲検無作為化プラセボ対照並列設計研究であった。全ての被検者は、1〜8日にそして血清レベルが5日〜7日に安定であっったならば9〜20日に再び、毎日2回非盲検でリチウム(450mg)を服用する予定であった。血清レベルが5〜7日に安定であった被験者は、毎日2回リチウム450mgに加えて、9〜17日に毎日1回プラセボもしくは上昇する用量のビフェプルノックス(0.025〜40mg)を、そして18〜21日にプラセボもしくはビフェプルノックス40mgを服用するように無作為抽出されたものに含まれた。リチウム450mgの単回朝用量のみを21日に投与した。投与間隔(0〜t)にわたるリチウム、定常状態Cmax、AUCおよび腎クリアランス(CL−R)値を8日に測定したリチウムのベースライン値を共変量としてANCOVAを用いてビフェプルノックスとプラセボ群との間で比較した。結果:ビフェプルノックス群においてリチウムの平均CmaxおよびAUC(0〜t)のわずかな増加があったが、AUC、CmaxおよびCL−Rの2つの処置の幾何最小二乗平均の比および90%信頼区間(CI)は0.80〜1.25の所定範囲内であった。Cmax、AUC(0〜t)およびCL−Rの処置比および90%CIは、それぞれ1.11(90%CI:1.01〜1.20)、1.13(90%CI:1.06〜1.21)および0.94(90%CI:0.87〜1.02)であった。1日あたり40mgまでのビフェプルノックスおよび毎日2回リチウム450mgの組み合わせた投与は、良好な耐容性を示した。結論:リチウム定常状態薬物動態へのビフェプルノックスの複数回投与の共投与の臨床的に関連する影響はなかった。結果は、ビフェプルノックスとの同時投与中にリチウムの投薬量調整は必要とされないことを示唆する。
【0265】
実施例4:急性増悪統合失調症を有する患者の処置におけるビフェプルノックスの効能および安全性
目的:統合失調症を有する急性疾患患者の処置におけるビフェプルノックスの効能および安全性を評価すること。
【0266】
評価:6週の無作為化プラセボ対照リスペリドン参照用量設定研究には、統合失調症(DSM−IV−TR)の急性増悪を有する589人の無作為抽出患者が含まれた。患者は、ビフェプルノックス5mg(n=115)、ビフェプルノックス10mg(n=120)、ビフェプルノックス20mg(n=115)、プラセボ(n=119)もしくはリスペリドン6mg(n=120)に無作為に割り当てられた。全てのビフェプルノックス用量での処置は目標用量、1日に0.125mgの用量から開始して、2日に0.25mg、3日に0.5mg、4日に1mg、5日に2mgそして6日に5mg、7日に10mgもしくは8日に20mgまで滴定され、一方、リスペリドンでの処置は3日にわたって滴定された。ベースラインからエンドポイントまでの陽性および陰性症状評価尺度(PAN
SS)合計スコアの変化は、主要アウトカム評価尺度であった。副次的効能評価尺度には:PANSS陽性、PANSS陰性、PANSS総合精神病理(GPP)スコア、PANSS由来の簡易精神症状評価尺度(BPRS)スコア、臨床全般印象疾患重症度(CGI−S)、CGI−I改善度(CGI−I)スコアならびにレスポンダー率が包含された。安全性および耐容性評価には、錐体外路症状(EPS)、体重増加、脂質プロフィールおよび血清プロラクチンが包含された。リスペリドンは、アッセイ感度用に含まれた。
【0267】
結果:ビフェプルノックスのPANSS合計スコアの減少は、20mg用量について3週および6週/エンドポイントでプラセボと比較して統計的に有意であった(P<0.05)。ビフェプルノックス20mgのプラス効果はまた、副次的効能評価尺度PANSS陽性、陰性、GPP下位尺度、BPRSおよびレスポンダー率に対しても認められた。リスペリドン6mgは、全ての効能評価尺度についてプラセボと比較してエンドポイントで統計的に有意であった。最も一般的な有害事象(発生率>5%およびプラセボに対して2倍)には:消化不良、悪心、嘔吐および便秘が包含された。用量関係は、最も頻繁な有害事象のいずれについても明らかではなかった。ビフェプルノックスは、減少したプロラクチンレベルおよびプラセボと同程度であるEPSの割合と関連した。さらに、ビフェプルノックスを服用する患者は、統計的に有意な(P<0.05)体重減少を経験し、そしてプラセボと比較して非空腹時トリグリセリド(P<0.005)および総コレステロール(P<0.005)の統計的に有意な改善を示した。
【0268】
結論:本研究において、ビフェプルノックス20mgは急性統合失調症の処置において有効であることが示された。ビフェプルノックスは、体重の減少および脂質プロフィールの改善に起因する安全性利点を有し得る。
【技術分野】
【0001】
本発明は、統合失調症を有する患者の処置のためのビフェプルノックスの日用量、安定した統合失調症を有する患者のそして統合失調症の急性増悪を有する患者のビフェプルノックスでの処置に、そして少なくとも1種のビフェプルノックス化合物の用量を含んでなる製薬学的組成物に関する。
【0002】
統合失調症は、陽性および陰性症状、認知障害および鬱病を包含する重篤なそして変わりやすい症状を特徴とする生涯にわたる日常生活に支障をきたす精神障害である。疾患の経過は4つの主要な時期:発病前、急性、安定/維持および後期経過に分けられることができる。発病前期は、陽性症状の発症前に起こる症状をさす。急性期中に患者は妄想および幻覚のような明白な陽性症状を経験する。安定/維持期は2つの亜期(subphase)に分けることができる。疾患の最初の5〜10年は陽性症状の多数の増悪を特徴とすることが多く、より安定な期間が急性エピソードの間に散在する。この亜期の後に症状の安定化および増悪の数の減少を特徴とするプラトー期が続く。維持期中の重要な処置目標は、地域社会への患者の復帰を促進しそして長期の維持計画を定めることである。疾患の後期経過期において、陽性症状は年齢とともに減少する傾向があり、そして長期的な障害を有する多数の患者はある程度の社会的および職業的能力を取り戻し、しかしながら、長年の機能障害の影響はめったに克服されない。
【0003】
統合失調症の維持および後期経過期において、患者の大部分は、再発の防止;認知機能の維持;体重増加、高血糖および脂質異常症の予防;ならびに生活の質の改善を包含する、さらなる症状改善および精神病症状の長期制御の必要性のような多数の問題に直面し続ける。さらに、陽性症状は、各々の続いて起こるエピソードで処置に対してより耐性になり得る。この概念と一致して、統合失調症を有する患者の85%〜90%は臨床的増悪を経験する。さらに、抗精神病薬を与えられた患者の少なくとも半分は処方された処置処方計画を順守せず、従って、再発の危険がある。従って、処置を改善することを目指す熱心な努力にもかからず、統合失調症を有する患者の大部分は重篤な障害を有し;再発することが多く、そして入院を必要とし得る。
【0004】
統合失調症を処置するために現在使用される化合物は、いくつかの望ましくない副作用と関連している。これらの副作用には体重増加、高プロラクチン血症、上昇したトリグリセリドレベル、メタボリック症候群(マーカー:糖尿病、高脂血症、高血圧および肥満)、延長したQTc間隔、グルコース異常ならびに錐体外路症状の表出が包含される。例えば、延長したQTc間隔、すなわち、心電図における補正QT間隔は、問題のある心律動もしくは心臓不整脈をもたらし得る。同様に、リスペリドンおよびオランザピンのような従来の非定型抗精神病薬で認められる体重増加は、心臓血管疾患および糖尿病の増加した危険性と関連している。
【0005】
さらに、薬剤を用いる統合失調症処置は長期間にわたってであり得る。そのようなものとして、これらの望ましくない副作用は日常的にならびに彼らの長期の健康に寄与して患者に影響を及ぼす。これらの副作用はまた、患者の処置処方計画の不順守にもつながり得る。処置が限られたそして/もしくは短い期間にわたってである場合でさえ、副作用は処置処方計画を順守しようとする患者の意欲に影響を及ぼす。
【0006】
従って、これらの望ましくない副作用を防ぎそして/もしくは軽減するならびに統合失調症処置を受けている患者のベースライン症状を維持し、軽減しそして/もしくは改善する方法の必要性がある。
【発明の概要】
【0007】
少なくとも1種のビフェプルノックス化合物の用量、特に20〜30mgの日用量を含んでなる(製薬学的組成物)で統合失調症患者を処置することは、これらの副作用の1つもしくはそれ以上および統合失調症の1つもしくはそれ以上の症状を軽減しそして/もしくは防ぐことを可能にすることを本発明者等は見出した。本発明の態様において、用量は毎日1回投与される。特定の用量の態様は、20mg用量および30mg用量である。特に好ましいのは、20mgの日用量である。
【0008】
この処置の好ましい効果には、患者における陽性および陰性症状評価尺度(PANSS)合計スコアの減少、体重の維持、トリグリセリドレベルおよび/もしくは総コレステロールレベルの維持および/もしくは改善、特に慢性の安定した統合失調症を有する患者における統合失調症の臨床的安定の維持(処置効果は、例えば増悪までの時間の増加である)、1つもしくはそれ以上の精神病症状の改善または投与前のベースライン測定と同様の錐体外路兆候および症状(EPS)プロフィールの維持および/もしくは減少が包含されるがこれらに限定されるものではない。他の好ましい効果は、高血糖および/または1つもしくはそれ以上の糖尿病関連有害事象の発生の減少である。
【0009】
本発明の態様において、ビフェプルノックスは、特に20〜30mgの日用量において、統合失調症を有する患者の長期処置に使用される。「長期処置」という用語は、例えば少なくとも3ヶ月もしくは少なくとも6ヶ月の処置をさす。
【0010】
本明細書に開示される態様は、本開示の様々な態様のいくつかの一般的な概説を提供するが、いかなる方法によっても本開示の範囲を限定するものではない。
【0011】
ビフェプルノックス化合物は米国特許第6,225,312号および米国特許第7,030,241号に記述され、これらの内容は引用することにより本明細書に組み込まれる。この化合物(7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン(ビフェプルノックス)の塩酸塩はWO97/36893に記述されそして請求され、そしてモノメタンスルホン酸塩はWO02/066449に記述されそして請求される。これらの特許公開の第二のものにおいて、N,N,N−ビス(2−エタノール)−m−フェニルベンジルアミンの反応性メシル酸エステルと7−アミノ−2(3H)−ベンゾキサゾロンとの間の反応によるモノメタンスルホン酸塩の直接形成が開示される。ビフェプルノックスモノメタンスルホン酸塩の安定な多形は、WO2005/016898に開示されそして請求される。また「ビフェプルノックス化合物」という用語に包含されるのは、ビフェプルノックスN−オキシドである。ビフェプルノックスN−オキシドは、WO2007/023141に開示されそして請求される。
【0012】
ビフェプルノックス化合物は、統合失調症、他の精神病性障害(特に精神病)およびパーキンソン病を包含するCNS(中枢神経系)障害の処置に適応される。本発明の枠組みにおいて、投薬量強度(もしくは用量)はビフェプルノックス塩基(bifeprunox base)に相当する量で表される。本明細書において用いる場合、「ビフェプルノックス塩基」という用語は以下の式:
【0013】
【化1】
【0014】
を有する化合物7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン(INNビフェプルノックス)をさす。
【0015】
本明細書において用いる場合、「ビフェプルノックス化合物」という用語は活性化合物7−[4−([1,1’−ビフェニル]−3−イルメチル)−1−ピペラジニル]−2(3H)−ベンゾキサゾロン、そのN−オキシドおよび製薬学的に許容しうる塩、その溶媒和物および水和物をさす。N−オキシドがビフェプルノックス化合物として用いられる場合、ミリグラム単位の量は、当業者が酸化物なしのビフェプルノックス化合物に選択する量と同じ量である。さらに、ビフェプルノックスもしくはそのN−オキシドの製薬学的に許容しうる塩は、当該技術分野において周知である標準的な方法を用いて、例えば、本発明の化合物を適当な酸、例えば無機酸もしくは有機酸と混合することにより得ることができる。
【0016】
本開示は、少なくとも1種のビフェプルノックス化合物の用量を含んでなる製薬学的組成物を処置を必要とする患者に投与することにより統合失調症の処置と関連する症状を維持し、軽減しそして/もしくは改善するための統合失調症の処置におけるビフェプルノックスの使用に関する。例えば、製薬学的組成物は、10mg〜40mgもしくはさらに例えば20mg〜30mgのビフェプルノックス化合物のような5mg〜40mgの間の量の少なくとも1種のビフェプルノックス化合物を含んでなる。1つの態様において、ビフェプルノックスは体重の問題を有するかもしくは体重の問題を起こしやすい統合失調症を有する患者の処置において用いられる。
【0017】
本発明の態様は、用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、統合失調症を有する患者の処置のためのビフェプルノックスの日用量に関する。特に、該用量は、安定した統合失調症およびさらに特に慢性の安定した統合失調症を有する患者において臨床的安定を維持するために有用である。その態様は、それぞれ20mgの用量および30mgの用量である。
【0018】
さらなる態様において、本発明は、用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、急性増悪統合失調症を有する患者の処置のためのビフェプルノックスの日用量に関する。その態様は、好ましい副作用で該処置において用いられる、それぞれ20mgの用量および30mgの用量である。
【0019】
本発明の態様は、ビフェプルノックス(すなわち、少なくとも1種のビフェプルノックス化合物)が気分安定剤リチウムと組み合わせて投与される、精神病および気分障害を有する(特に統合失調症を有する)患者の処置における使用のためのビフェプルノックスおよび該使用のためのキットに関する。
【0020】
本発明のさらなる態様は、少なくとも1種のビフェプルノックス化合物が抗鬱剤(特にSSRI、特にパロキセチン)と組み合わせて投与される、CNS障害を有する(特に統合失調症を有する)患者の処置における使用のためのビフェプルノックスおよび該使用のためのキットに関する。
【0021】
本発明のさらなる態様は、それぞれ、CYP2C9阻害剤(例えばフルコナゾール)と、CYP3A4阻害剤(例えばケトコナゾールおよびカルバマゼピン)と、CYP2D6阻害剤(例えばパロキセチン)とそしてH2−アンタゴニスト(例えばファモチジン)とビフェプルノックスとの共投与(のための方法)および該処置のためのキットに関する。
【0022】
本開示の態様において、少なくとも1種のビフェプルノックス化合物はメシル酸ビフェプルノックスを含んでなる。好ましくは、少なくとも1種のビフェプルノックス化合物はメシル酸ビフェプルノックスである。メシル酸ビフェプルノックスは、α、γもしくはδ結晶多形形態およびその混合物から選択することができる。例えば、少なくとも1種のビフェプルノックス化合物は、αおよびγ多形形態から選択される少なくとも1種の多形形態を含んでなる。
【0023】
本開示によるαメシル酸ビフェプルノックスの結晶多形形態は、WO2005/016898に開示されるような少なくとも物理化学パラメーターにより定義される。
【0024】
別の態様において、本開示は、少なくとも約50重量パーセント(wt.%)、少なくとも約60wt.%、少なくとも約70wt.%、少なくとも約80wt.%、少なくとも約90wt.%もしくは少なくとも約95wt.%のメシル酸ビフェプルノックスが多形α形態であるメシル酸ビフェプルノックスを提供する。別の態様において、製薬学的組成物は、メシル酸ビフェプルノックスのあらゆるγもしくはδ多形形態を実質的に欠いている。別の態様において、本開示により提供されるメシル酸ビフェプルノックスは10wt.%未満、5wt.%未満、2.5wt.%未満のγもしくはδ多形形態のメシル酸ビフェプルノックスを含んでなる。別の態様において、少なくとも約99wt.%のメシル酸ビフェプルノックスは多形α形態である。
【0025】
多形形態αの製造は、WO2005/016898に記述される方法に従って実施することができる。
【0026】
本開示の少なくとも1種のビフェプルノックス化合物は、活性物質が当該技術分野において既知である方法により固形形態で存在する投与形態物に調合することができる。該投与形態物の例は(場合によりコーティングされていてもよい)錠剤、カプセル剤、粒状エアロゾル、座薬および懸濁剤である。そのような投与形態物は、少なくとも1種のビフェプルノックス化合物を不活性の製薬学的に許容しうる賦形剤および担体と混合することにより製造することができる。
【0027】
本開示の製薬学的組成物は少なくとも1種の製薬学的賦形剤を含んでなることができる。適当な賦形剤の限定されない例には、沈殿防止剤(例えば、ゴム、キサンタン、セルロース誘導体および糖)、保湿剤(例えば、ソルビトール)、可溶化剤(例えば、エタノール、水、PEGおよびプロピレングリコール)、界面活性剤(例えば、ラウリル硫酸ナトリウム、スパン(Span)、ツィーン(Tween)およびセチルピリジン)、防腐剤、酸化防止剤(例えば、パラベン、ならびにビタミンEおよびC)、固化防止剤(anti−caking agent)、コーティング剤、キレート剤(例えば、EDTA)、安定剤、抗微生物剤、抗真菌もしくは抗菌剤(例えば、パラベン、クロロブタノール、フェノール、ソルビン酸)、等張剤(例えば、糖、塩化ナトリウム)、増粘剤(例えば、メチルセルロース)、香料(例えば、チョコレート、ザルマンチン(thalmantin
)、アスパルテーム、ルートビアもしくはスイカまたはpH7〜9で安定な他の香料)、消泡剤(例えば、シメチコン、Mylicon(R))、崩壊剤、流動助剤(flow aid)、潤滑剤、添加剤、着色剤、希釈剤、湿潤剤、防腐剤、担体、結合剤(例えば、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、他のセルロース系材料および澱粉)、希釈剤(例えば、ラクトースおよび他の糖、澱粉、第二リン酸カルシウムならびにセルロース系材料)、崩壊剤(例えば、澱粉ポリマーおよびセルロース系材料)、流動促進剤および不水溶性もしくは水溶性潤滑剤もしくは平滑剤が包含される。
【0028】
1つの実例となる投与形態物は、活性物質(本明細書に記述されるようなビフェプルノックス)の粉砕しそしてふるいにかけた用量のほかに、ラクトース1水和物、微晶質セルロース、澱粉グリコール酸ナトリウム(例えばタイプA)、ステアリルフマル酸ナトリウムおよび場合によりコロイド状無水シリカを含んでなる。1つの態様において、ラクトースは、錠剤コアの総重量に基づいて約20重量%〜約90重量%、約70重量%〜約90重量%もしくは約75重量%〜約85重量%の量で存在する。微晶質セルロースは、錠剤コアの総重量に基づいて約5重量%〜約90重量%、約10重量%〜約15重量%もしくは約11重量%〜約12重量%の量で存在する。澱粉グリコール酸ナトリウム(例えばタイプA)は、錠剤コアの総重量に基づいて約0.1重量%〜約2.5重量%、約0.3重量%〜約0.7重量%もしくは約0.5重量%の量で存在する。ステアリルフマル酸ナトリウムは、錠剤コアの総重量に基づいて約0.1重量%〜約1.5重量%、約0.6重量%〜約1.3重量%もしくは約1.0重量%の量で存在する。コロイド状無水シリカは、粉末の流動特性を向上するために場合により製剤に加えてもよい。所望に応じて、コロイド状無水シリカは、錠剤コアの総重量に基づいて約0.05重量%〜約0.5重量%もしくは約0.4重量%の量で典型的に存在する。任意のコーティングの量は、錠剤コアの総重量に基づいて約2.0重量%〜約5.0重量%、約3.0重量%〜約4.0重量%もしくは約3.5重量%である。
【0029】
少なくとも1種の態様において、本開示の少なくとも1種のビフェプルノックス化合物を含んでなる製薬学的系組成物は、それを必要とする患者、例えばヒト患者に投与することができる。
【0030】
本開示はまた、患者におけるPANSS合計スコアを減らすこと、体重を維持すること、トリグリセリドレベルおよび/もしくは総コレステロールレベルを維持することおよび/もしくは改善すること、統合失調症の臨床的安定を維持すること、1つもしくはそれ以上の精神病症状を改善することまたは投与前のベースライン測定と同様のEPSプロフィールを維持することにも関するが、これらに限定されるものではない。本開示はまた、高血糖および/もしくは糖尿病関連有害事象の発生を減らす方法にも関する。これらの方法は、以下に提供される下記の臨床実施例において例示される。
【0031】
米国特許出願第10/920,361号、第10/920,386号および第11/354,652号は、それらの全部が本明細書に引用することにより本明細書に組み込まれる。本明細書における全てのデータは概算であり、そして例えば使用した装置ならびにピーク位置およびピーク強度に影響を及ぼす他のパラメーターにより通常の測定誤差を受けることが理解される。他に特に定義されないかもしくは他に文脈が要求しない限り、「約」という用語は本明細書において用いる場合に列挙した値の±5%を一般に意味する。
【図面の簡単な説明】
【0032】
【図1】パネル34 PANSS陽性スコア(FAS、LOCF)を示す。
【図2】パネル35 PANSS陰性スコア(FAS、LOCF)を示す。
【図3】パネル36 PANSS総合精神病理スコア(FAS、LOCF)を示す。
【図4】パネル42 PANSS合計スコアの少なくとも25%の減少を有する患者の割合を示す(FAS、LOCF)。
【図5】パネル43 2以下のCGI−Iスコアを有する患者の割合を示す(FAS、LOCF)。
【実施例】
【0033】
以下の実施例は、より詳細に本開示をさらに説明することを意図するだけであり、従って、これらの実施例は決して本開示の範囲を限定すると見なされない。
【0034】
実施例1.統合失調症の処置におけるビフェプルノックスの効能
統合失調症の処置におけるビフェプルノックスの固定用量の効能および安全性を評価するために6週の無作為化二重盲検プラセボ対照およびリスペリドン参照研究を用いた。合計599人の患者を無作為抽出した。
【0035】
処置は少なくとも3日の単純盲検プラセボ導入期間から開始し、その後にビフェプルノックス処置患者には0.25mgから30mg/日もしくは40mg/日までのビフェプルノックスの滴定を続けた。リスペリドン処置患者は3日の期間にわたって毎日2mgから6mgまで滴定され、そして処置期間の残りの間6mg/日で維持された。
【0036】
PANSS合計スコアのベースラインからエンドポイントまでの変化を測定するために、評定尺度評価は5週目を除いて毎週行った。他の評価には:PANSS陽性症状下位尺度スコア、PANSS陰性症状下位尺度スコア、PANSS総合精神病理下位尺度スコア、BPRS合計スコア、BPRS精神病スコア、CGI−Sスコア、CGI−Iスコア、PANSS合計スコアに基づくレスポンダー率および統合失調症のためのカルガリー鬱病評価尺度(Calgary Depression Scale for Schizophrenia)(CDSS)が包含された。
【0037】
安全性および耐容性測定には、身体所見(physical examinations)、体重、ウエスト囲、バイタルサイン、12誘導心電図(ECG)、臨床検査評価(血液学、生化学、検尿、プロラクチン、IGF−1、IGFBP−3、甲状腺機能の特定の検査評価)、二重盲検処置期間の間の抗コリン作用薬処置の必要性、併用薬使用、有害事象モニタリングおよび正常な動き(normal movement)の評価が包含された。
【0038】
20mgビフェプルノックス処置群は、PANSS合計スコアにおけるベースラインからエンドポイントまでの変化の主要評価項目についてプラセボとの統計的に有意な差を示した。PANSS合計スコアにおけるベースラインからエンドポイントまでの平均的変化(標準偏差)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった。
【0039】
30mgビフェプルノックス群は、CGI−Sスコア、PANSS陰性症状下位尺度スコアおよびPANSS陽性症状下位尺度スコアについてプラセボとの顕著な差を示した。顕著な差はまた、PANSS総合精神病理下位尺度スコア、BPRS合計スコア、BPRS精神病クラスター(cluster)スコア、PANSSレスポンダー率およびCGI−Iレスポンダー率におけるベースラインからエンドポイントまでの変化についても30mgビフェプルノックス群とプラセボとの間で認められた。本研究において、PANSSレスポンダーは、そのPANSS合計スコアがベースラインからエンドポイントまで20%以上減少した患者をさす。CGI−Iレスポンダーは、エンドポイントでCGI全般改善度評価尺度において「著明改善」もしくは「中等度改善」と分類された患者をさす。
【0040】
40mgビフェプルノックス群は、PANSS陽性症状下位尺度スコアおよびBPRS
精神病クラスタースコアにおける変化についてプラセボとの顕著な差を示した。
【0041】
プラセボおよびリスペリドン処置群における増加と対照的にビフェプルノックス処置群において体重の減少が見られた。
【0042】
ビフェプルノックス群は、プラセボおよびリスペリドン群と比較してトリグリセリド、VLDLおよびLDLにおけるN→Hシフト(N to H shifts)のより低い発生率ならびに総コレステロールおよびVLDLにおけるN→Lシフトのより高い発生率を有した。
【0043】
実施例2.統合失調症の処置におけるビフェプルノックスに関する臨床研究
実施例2a−臨床研究1
目的:本臨床研究の主要目的は、主要アウトカム評価尺度として陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、5mg、10mgもしくは20mgのビフェプルノックスでの6週の処置が統合失調症を有する成人患者においてプラセボでの処置より優れているかどうかを調べることであった。二次的目的は、PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、ならびにPANSS合計スコアに基づくレスポンダー率およびCGI−Iレスポンダー率を用いて統合失調症を処置することにおけるビフェプルノックスの効能を評価することであった。身体所見、体重、バイタルサイン(脈拍数および最高/最低血圧(BP)−横たわって5分後そして立って2分後の両方(both lying after five minutes and standing after two minutes)、ならびに口腔温を包含する)、12誘導心電図(ECG)、血液学、生化学および検尿を包含する安全性検査評価、二重盲検処置期間中の抗コリン作用薬処置の必要性、併用薬使用、有害事象モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価することもまた本研究の目的であった。
【0044】
方法論:これは、統合失調症を有する成人患者における無作為化二重盲検固定用量プラセボ対照リスペリドン参照並行群多施設研究であった。本研究には5つの処置群があった。処置群は下記のとおりであった:ビフェプルノックス5mg、ビフェプルノックス10mg、ビフェプルノックス20mg、リスペリドン6mgおよびプラセボ。研究薬剤は、毎日1回投与した。ベースライン測定を行った後に、滴定期を開始した。ビフェプルノックス処置患者は、標準化滴定スケジュール(1日:0.125mg、2日:0.25mg、3日:0.5mg、4日:1.0mg、5日:2.0mg、6日:5.0mg、7日:10.0mg、8日:20mg)に従って5mg、10mgもしくは20mgまで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。リスペリドン処置患者は、毎日1回の処方計画を用いて3日にわたって6mgまで滴定された(1日:2mg、2日:4mg、3日:6mg)。
【0045】
患者数(計画し、スクリーニングし、無作為抽出し、そして分析した):統合失調症を有する合計575人の患者を研究に包含することを計画した。合計836人の患者を40の施設でスクリーニングし、そして合計589人の患者(5mgビフェプルノックス:115人の患者;10mgビフェプルノックス:120人の患者;20mgビフェプルノックス:115人の患者;プラセボ:119人の患者;リスペリドン:120人の患者)を37の施設で無作為抽出した。
【0046】
試験製品、用量および投与形態:ビフェプルノックス錠剤、総日用量0.125mg〜20mg、毎日1回の投与処方計画を用いて経口投与する。
【0047】
参照治療、用量および投与形態:プラセボおよびリスペリドン、2mg〜6mg、毎日1回の投与処方計画を用いて経口投与する。
【0048】
効能結果:20mgのビフェプルノックス用量は、PANSS合計および下位尺度スコアの分析からの統計的に有意な比較により示されるようにプラセボと比較して統合失調症の陽性および陰性症状の両方を軽減することならびに総合精神病理を減少させることにおいて有効であった。20mgビフェプルノックス群における患者は、プラセボ患者と比較してPANSS合計スコアにおいてベースラインからの5.8ポイント大きい改善を有した。ビフェプルノックスのより低い用量は有効ではなかった。10mg用量のビフェプルノックスは、いずれの効能評価項目についても統計的により大きい改善を示さなかった。5mg用量は、プラセボと比較していくつかの副次的効能評価尺度に関してより大きい改善を示したが、主要効能評価項目(PANSS合計スコア)についてプラセボに対する優位性を示さなかった。リスペリドン6mgは本研究における活性対照(active reference)として用いられ、そしてプラセボ群からの明らかな分離を示した。一般に、20mgビフェプルノックス群において見られる改善の大きさは、大部分の効能評価項目についてリスペリドン群において見られるものより小さかった。
【0049】
安全性結果:少なくとも1種の処置下で発現した有害事象(TEAE)を有する患者の割合は処置群にわたって同様であった:5mgビフェプルノックス群において89%(102人の患者)、10mgビフェプルノックス群において87%(104人の患者)、20mgビフェプルノックス群において83%(95人の患者)、プラセボ群において85%(101人の患者)、そしてリスペリドン群において89%(107人の患者)。全体で、ビフェプルノックスで処置した349人の患者から、最も頻繁に報告されるTEAEは頭痛、消化不良、不眠、悪心、嘔吐NOS、便秘および激越であった。プラセボ群(N=119人の患者)と比較して20mgビフェプルノックス群(N=114人の患者)においてより高い発生率(>5%の差)を有するTEAEには、便秘、消化不良および嘔吐NOSが包含された。プラセボと比較して20mgビフェプルノックス群においてより高い発生率(>5%の差)を有する関連TEAEは、便秘、嘔吐NOSおよび頭痛NOSであった。少なくとも1種の重篤なTEAEを有する患者の割合は、20mgビフェプルノックス群(11人の患者、10%)において最も低く、続いてプラセボ群(15人の患者、13%)であった。残りの群について、少なくとも1種の重篤なTEAEを有する患者の割合は16%〜18%の間であった。重篤であると考えられるTEAEの発生率は、全てのTEAEについて20mgビフェプルノックス群とプラセボ群間で同様であった(<5%の差)。任意の事象について認められる全発生率、関連TEAEの発生率もしくは重篤なTEAEの発生率においてビフェプルノックス群における用量依存的傾向はなかった。
【0050】
少なくとも1種のSAEを有する患者の総数は、プラセボ群(9%)と比較して積極的処置群において高かった(ビフェプルノックス群:12〜15%、リスペリドン群:16%)。最も一般的に報告されるSAE(任意の処置群において>5%)は、増悪精神病および増悪統合失調症NOSであった。プラセボおよびリスペリドン(各々5人の患者、4%)と比較して20mgビフェプルノックス群(8人の患者、7%)において増悪統合失調症NOSのわずかに高い発生があった。自殺未遂は10mgビフェプルノックス群における1人の患者(<1%)、20mgビフェプルノックス群における2人の患者(2%)、プラセボ群における0人の患者およびリスペリドン群における0人の患者について報告された。自殺念慮の重篤な有害事象は、10mgビフェプルノックスおよびリスペリドン
群における各々1人の患者(<1%)について報告された(20mgビフェプルノックス群における1人のさらなる患者は、自殺念慮の非重篤な有害事象を経験した)。任意のSAEの発生率における用量依存的増加の傾向は、ビフェプルノックス群について認められなかった。ビフェプルノックスは、全ての用量レベルで安全でありそして良好な耐容性を示した。中止につながる少なくとも1種のAEを有する患者の総数は、処置群間で同様であった(5mgビフェプルノックス:13人の患者、11%;10mgビフェプルノックス:17人の患者、14%;20mgビフェプルノックス:11人の患者、10%;プラセボ:15人の患者、13%;リスペリドン:17人の患者、14%)。中止につながる最も一般的な(任意の処置群における患者の>2%により報告される)AEは、激越、増悪精神病および増悪統合失調症NOSであった。20mgビフェプルノックス群とプラセボ群との間で研究薬剤の中止につながるAEの発生率の処置群差はなかった。ビフェプルノックス群において中止につながるAEの発生率の用量依存的増加の傾向はなかった。臨床検査、バイタルサイン、ECGおよび身体所見結果の評価は、いかなる予想外の安全性懸念も引き起こさなかった。ビフェプルノックス患者は、プラセボ群と比較してプロラクチンの減少を示した。軽度の体重減少はビフェプルノックス群において認められたが、プラセボもしくはリスペリドン群においては認められなかった。
【0051】
BAS、SASもしくはAIMSスコアにおけるベースラインからエンドポイントまでの変化の処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であり、そしてリスペリドンでの患者におけるよりも少なかった。
【0052】
結論:本研究の1つの結論は、6週間毎日1回与えるビフェプルノックスの20mg用量が統合失調症の陽性および陰性症状の両方を軽減することにおいて有効であったことである。全体で、ビフェプルノックスの全ての用量は安全でありそして統合失調症患者により良好な耐容性が示された。安全性/耐容性の用量反応関係は見られなかった。
【0053】
実施例2b−臨床研究2
主要目的:主要アウトカムとして陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、ビフェプルノックスの固定用量(30mg/日もしくは40mg/日)での6週の処置が統合失調症を有する成人患者においてプラセボと比較して優れた効能を示すことができるかどうかを調べること。
【0054】
二次的目的:PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病スコア、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、PANSS合計スコアに基づくレスポンダー率、統合失調症のカルガリー鬱病評価尺度(CDSS)および患者満足度を用いて統合失調症を処置することにおけるビフェプルノックスの効能を評価すること。統合失調症患者におけるビフェプルノックスの薬物動態(PK)データもまた評価し、そして別個の報告において提示する(研究S1543003からのデータと合わせて)。身体所見、体重、ウエスト囲、バイタルサイン(脈拍数および最高/最低血圧[BP]−横たわって5分後そして立って2分後の両方、ならびに口腔温)、12誘導心電図(ECG)、臨床検査評価(血液学、生化学、検尿、プロラクチン、IGF−1、IGFBP−3および甲状腺機能についての特定の検査評価)、二重盲検処置期間の間の抗コリン作用薬処置の必要性、併用薬使用、有害事象(AE)モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価すること。
【0055】
方法論:これは、統合失調症を有する成人患者におけるビフェプルノックスの効能、耐容性および安全性の第III相6週無作為化二重盲検プラセボ対照リスペリドン参照並行群多施設研究であった。本研究を完了した患者は、長期で継続する選択肢を有した。処置群は:ビフェプルノックス30mg/日、ビフェプルノックス40mg/日、リスペリドン6mg/日およびプラセボであった。少なくとも3日の単純盲検プラセボ導入期間を完了した後に、ビフェプルノックス処置患者は8日の期間にわたって標準化滴定スキームに従って0.25mgから30mg/日もしくは40mg/日まで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。リスペリドン処置患者は、3日の期間にわたって毎日2mgから6mgまで滴定され、そして次に処置期間の残りの間6mg/日で維持された。効能および異常運動障害の評定尺度評価は、5週目を除いて毎週行われた。安全性評価はスクリーニングで、処置の間にそして研究の最後で行われた。研究薬剤での患者満足度は6週で評価された。血液のサンプルは、血漿中のビフェプルノックスの測定用に2、4および6週で得られた。血液サンプルはまた、臨床検査評価用にスクリーニング/ベースライン、3週および6週でも得られた。
【0056】
患者数(計画し、承認し、任意抽出し、そして分析した):統合失調症を有する合計576人の患者を研究に包含することを計画した。783人のスクリーニングした患者のうち、合計599人の患者を無作為抽出した(30mgビフェプルノックス:148人の患者、40mgビフェプルノックス:148人の患者、プラセボ:149人の患者;リスペリドン:154人の患者)。
【0057】
診断および包含のための主な基準:(DSM−IV−TR基準に従って)統合失調症を有する18〜75歳の男性もしくは女性患者。患者は、70〜120の間のPANSSでの合計スコアを有していなければならず;4つのPANSS項目(概念の統合障害、幻覚による行動、猜疑心、不自然な思考内容)の少なくとも2つがスコア>4を有していなければならず;そしてCGI−Sでのスコアが少なくとも4でなければならなかった。
【0058】
参照治療、用量および投与形態:プラセボおよびリスペリドン、6mg、毎日1回経口投与する。
【0059】
効能結果:30mgビフェプルノックス処置群は、Hochberg補正したp値(補正したp=0.020)に基づいてPANSS合計スコア(LOCF)におけるベースラインからエンドポイントまでの変化の主要評価項目についてプラセボとの統計的に有意な差を示した。40mgビフェプルノックス処置群は、主要効能評価項目についてプラセボ群と有意に異ならなかった(補正したp=0.156)。PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:30mgビフェプルノックス群について−5.9、そして40mgビフェプルノックス群について−3.2であった。計画されたステップダウン法に基づく統計的に有意な差は、プラセボと比較して30mgビフェプルノックス処置群についてCGI−Sのベースラインからエンドポイントまでの変化において見られなかった。従って、PANSS陰性症状およびPANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの変化に関するプラセボと30mgビフェプルノックス群との間の差は、プラセボと比較した統計的有意性についてステップダウン法を用いて評価されなかった。30mgビフェプルノックス群は、CGI−Sスコア(公称(nominal)p=0.028)、PANSS陰性症状下位尺度スコア(公称p=0.027)およびPANSS陽性症状下位尺度スコア(公称p=0.010)についてプラセボとの顕著な差を示した。
【0060】
顕著な差は、PANSS総合精神病理下位尺度スコア(p=0.025)、BPRS合計スコア(p=0.019)、BPRS精神病クラスタースコア(p=0.002)、PANSS(30%)レスポンダー率(p=0.019)およびCGI−Iレスポンダー率(p=0.039)におけるベースラインからエンドポイントまでの変化について30mgビフェプルノックス群とプラセボとの間で認められた。エンドポイントでの顕著な差は、患者満足度(p=0.051)、CGI改善度スコアもしくはCDSSスコアについて30mgビフェプルノックス群とプラセボとの間で認められなかった。40mgビフェプルノックス群について、プラセボとの統計的に有意な差は主要効能パラメーターについて見られなかった。プラセボとの顕著な差は、PANSS陽性症状下位尺度スコア(p=0.020)およびBPRS精神病クラスタースコア(p=0.031)を除いて副次的効能パラメーターのいずれについても40mgビフェプルノックス群では認められなかった。
【0061】
安全性結果:少なくとも1種のTEAEを有する患者の割合はビフェプルノックス用量群において同程度であり(74%〜76%)、そしてプラセボ群(64%)より高かったが、リスペリドン群(78%)におけるよりもわずかに低かった。プラセボ群と比較してビフェプルノックス群においてより高い(>5%の差)発生率を有する処置下で発現したAEには、悪心、嘔吐、便秘、消化不良、下痢および眩暈が包含された。ビフェプルノックス処置群間の差は、ビフェプルノックス群における個々のTEAEの発生率において一般に認められなかった。任意の処置群における患者の少なくとも5%において起こるTEAEのうち、30mgビフェプルノックス群と比較して40mgビフェプルノックス群においてより高い(>2%の差)発生率を有するものには、悪心、嘔吐、歯痛、食欲不振、静座不能、眩暈、頭痛および不眠が包含された。対照的に、口渇、唾液分泌過多、食欲減退、鎮静、傾眠、不安および膣炎は、40mgビフェプルノックス群と比較して30mgビフェプルノックス群においてより高い(>2%の差)発生率で起こった。重篤なTEAEの発生率は、精神病性障害(<6%)および統合失調症(ビフェプルノックスおよびプラセボ群において各々3%そしてリスペリドン群において2%)のTEAEを除いて一般に低かった(<1%の発生率)。重篤なTEAEの発生率は処置群にわたって一般に同程度であり、そして任意の事象について重篤なTEAEの発生率におけるビフェプルノックス処置群間の差はなかった。特定の興味深いTEAEはデータベースロックの前に定義され、そして自殺、自殺未遂、性機能障害、失神、血管迷走神経性発作および起立性低血圧に関連する事象が包含された。全体で合計76人の患者(13%)が研究中に少なくとも1種の特定の興味深いTEAEを報告した。少なくとも1種の特定の興味深いTEAEを有する患者の割合は、プラセボ群(6%)と比較してビフェプルノックスおよびリスペリドン処置群(12%〜16%)において高かった。特定の興味深いTEAEの大部分は、任意の処置群における患者の<1%において起こった。眩暈は最も一般的に報告され、そして他の2つの群と比較してビフェプルノックス処置群においてわずかに高い発生率で起こった。
【0062】
全体で合計60人の患者(10%)は81のSAE(死亡を包含する)を経験した。大部分のSAEは、患者の<1%において起こった。例外は、精神病性障害(<5%)および統合失調症(4つの処置群の各々において3%)であった。SAEの発生率は、ビフェプルノックス群およびプラセボ間で同程度であった。異常運動の評価尺度(BAS、SASもしくはAIMSスコア)で処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボで処置した患者のものと同様であった。全体で、ビフェプルノックスで臨床検査パラメーター、身体所見結果もしくはECG読み取りにおける臨床的に有意な変化はなかった。ビフェプルノックス群は、プラセボおよびリスペリドン群と比較してトリグリセリド、VLDLおよびLDLにおけるN→Hシフトのより低い発生率ならびに総コレステロールおよびVLDLにおけるN→Lシフト
のより高い発生率を有した。著しく異常な総コレステロール値の発生率は、処置群にわたって同様であった(1%〜2%)。著しく異常なトリグリセリド値は、他の群と比較して40mgビフェプルノックス群においてわずかに少ない患者により報告された。プラセボ(4%)および40mgビフェプルノックス(5%)群と比較して30mgビフェプルノックスおよびリスペリドン群(各々6%)におけるトリヨウ素サイロニンのN→Hシフトのわずかに大きい発生率ならびにプラセボ(2%)およびリスペリドン(<1%)群と比較して30mgビフェプルノックス(4%)および40mgビフェプルノックス(5%)におけるTSHのN→Lシフトのわずかに大きい発生率があった。プラセボおよびリスペリドン処置群における増加と対照的にビフェプルノックス処置群において体重のわずかな同程度の減少が見られた。体重の著しく異常な減少の発生率はプラセボおよびリスペリドン群に対してビフェプルノックス処置群の両方においてわずかに高く、一方、体重の著しく異常な増加の発生率はビフェプルノックスおよびプラセボ群に対してリスペリドン処置群において高かった。
【0063】
結論:本研究の1つの結論は、6週間毎日1回与えるビフェプルノックスの30mg用量がPANSS合計スコアの分析からの統計的に有意な比較により示されるように統合失調症の症状を軽減することにおいて有効であったことである。30mgビフェプルノックス群における患者は、LOCFデータに基づいてプラセボ患者と比較してPANSS合計スコアにおけるベースラインからエンドポイントまでの5.9ポイント大きい改善を有した。主要および副次的効能パラメーターについて実対照薬、リスペリドンとプラセボ処置との間で認められる統計的に有意な差は、これが有効な研究であることを示す。
【0064】
30mgビフェプルノックス処置群は、3つのキーとなる副次的効能評価項目、CGI−S、PANSS陰性および陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの変化についてプラセボとの顕著な差を示し(公称p値に基づいて);これら3つのキーとなる副次的効能評価項目についてステップダウン法に基づいて統計的有意性は得られなかった。顕著な差は、他の副次的効能パラメーター(PANSS総合精神病理下位尺度スコア、BPRS合計スコアおよびBPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの変化)の大部分についてエンドポイントで30mgビフェプルノックス群とプラセボとの間で認められた。30mgビフェプルノックス用量とプラセボとの間の顕著な差はまた、PANSSおよびCGI−Iレスポンダー率についてもエンドポイントで示された。40mgビフェプルノックス処置群は、主要効能パラメーターについてプラセボとの統計的に有意な差を示さなかった。PANSS陽性症状下位尺度およびBPRSクラスタースコアを除いて副次的効能パラメーターのいずれについても40mgビフェプルノックスとプラセボ群との間で顕著な差はなかった。全体で、ビフェプルノックスの30mgおよび40mg用量は安全であり、そして統合失調症患者により良好な耐容性が示された。
【0065】
実施例2c:臨床研究3
主要目的:主要アウトカムとして陽性および陰性症状評価尺度(PANSS)合計スコアのベースラインからエンドポイントまでの変化を用いて、ビフェプルノックスの固定用量(20mg/日もしくは30mg/日)での6週の処置が統合失調症を有する成人患者においてプラセボと比較して優れた効能を示すことができるかどうかを調べること。
【0066】
二次的目的:PANSSの陽性症状下位尺度スコア、PANSSの陰性症状下位尺度スコア、PANSSの総合精神病理下位尺度、PANSS由来の簡易精神症状評価尺度(BPRS)合計スコア、PANSS由来のBPRS精神病スコア、臨床全般印象疾患重症度スコア(CGI−S)、臨床全般印象改善度スコア(CGI−I)、CGI−Iレスポンダー率、PANSS合計スコアに基づくPANSSレスポンダー率、統合失調症のカルガリー鬱病評価尺度(CDSS)および患者満足度を用いて統合失調症を処置することにお
けるビフェプルノックスの効能を評価すること。身体所見、体重、ウエスト囲、バイタルサイン(脈拍数および最高/最低血圧[BP]ならびに口腔温)、12誘導心電図(ECG)、臨床検査評価(血液学、空腹時インシュリンレベル、空腹時グルコース、空腹時脂質プロフィールを包含する生化学、検尿、プロラクチン、IGF−1、IGFBP−3および甲状腺機能の特定の検査評価)、二重盲検処置期間中の抗コリン作用薬処置の必要性、併用薬使用、有害事象(AE)モニタリング、ならびにシンプソン・アンガス評価尺度(SAS)、バーンズ・アカシジア評価尺度(BAS)および異常不随意運動評価尺度(AIMS)を包含する異常運動の評価を用いてビフェプルノックスの安全性および耐容性を評価すること。
【0067】
方法論:これは、統合失調症を有する成人患者におけるビフェプルノックスの効能、耐容性および安全性の第III相6週無作為化二重盲検プラセボ対照オランザピン参照並行群多施設研究であった。本研究には4つの処置群があった、処置群当たり約144人の患者。処置群は:ビフェプルノックス20mg/日、ビフェプルノックス30mg/日、オランザピン15mg/日およびプラセボであった。少なくとも3日の単純盲検プラセボ導入期間を完了した後に、ビフェプルノックス処置患者はそれぞれ7もしくは8日の期間にわたって標準化滴定スキームに従って0.25mgから20mg/日もしくは30mg/日まで滴定された。指定用量に到達すると、患者は6週の処置期間の残りの間その用量で維持された。オランザピン処置患者は最初の7日の期間にわたって10mg/日で投薬を開始し、そして次に処置期間の残りの間15mg/日で維持された。スクリーニング来院(Screening Visit)から開始してベースライン後少なくとも10日まで、研究参加資格が確認された後に患者を入院させた(すでに入院患者ではない場合)。患者は、研究者により医学上必要と見なされる場合には10日より長く入院させることができた。効能および異常運動障害の評定尺度評価は、5週目を除いて毎週行われた。安全性評価はスクリーニングで、処置の間にそして研究の最後で行われた。研究薬剤での患者満足度は6週で評価された。全血のサンプルは、血漿におけるビフェプルノックスの測定用に2、4および6週で得られた。
【0068】
患者数(計画し、承認し、任意抽出し、そして分析した):統合失調症を有する合計576人の患者を研究に包含することを計画した。合計814人の患者を32の施設でスクリーニングし、そして合計604人の患者(20mgビフェプルノックス:154人の患者、30mgビフェプルノックス:150人の患者;プラセボ:150人の患者;オランザピン:150人の患者)を32の施設で無作為抽出した。
【0069】
診断および包含のための主な基準:(DSM−IV−TR基準に従って)統合失調症を有する18〜75歳の男性もしくは女性患者。患者は、70〜120の間のPANSSでの合計スコアを有していなければならず;4つのPANSS項目(概念の統合障害、幻覚による行動、猜疑心、不自然な思考内容)の少なくとも2つがスコア>4を有していなければならず;CGI−Sでのスコアが少なくとも4でなければならない。
【0070】
試験製品、用量および投与形態:ビフェプルノックス錠剤、総日用量20mgもしくは30mg(1錠の20mg錠剤および1錠の10mg錠剤)、毎日1回の投与処方計画を用いて経口投与する。
【0071】
参照治療、用量および投与形態:プラセボおよびオランザピン、5mgおよび15mg、毎日1回の投与処方計画を用いて経口投与する。
【0072】
効能結果:20mgおよび30mg処置群は両方ともエンドポイントでベースラインに対して改善を示したが、主要およびキーとなる副次的効能パラメーターに関してプラセボ群と比較した場合に効能を示さなかった。しかしながら、20mgビフェプルノックス群
は、副次的効能パラメーター、CGI改善度スコア(公称p=0.027)におけるベースラインからエンドポイントまでの変化についてプラセボ群に対して顕著な改善を示した。さらに1つの他の副次的効能パラメーターにおいて、20mgビフェプルノックス群とアプローチしたプラセボ群との間の差は、PANSS−20%レスポンダー率について顕著である(p=0.061)。しかしながら、副次的効能パラメーター間の顕著なそしてほぼ顕著な差のこれらの発生は、偶然、すなわち、処置比較の5%において起こると予想されるものを上回らない。全ての他の副次的およびキーとなる副次的パラメーターにおいて、ビフェプルノックス処置群はいずれも任意の効能評価項目についてプラセボ群に対して顕著な改善を示さなかった。15mgの用量のオランザピンは、本研究における活性対照として用いられた。オランザピンとプラセボとの間のPANSS合計スコアの差は、感度分析に従って分析された。これらの結果は、オランザピンがプラセボと顕著に異なることを示した(p<0.001)。一般に、ビフェプルノックス用量群において見られる改善の大きさはプラセボ群において見られるものより高かったが、大部分の効能評価項目についてオランザピン群において見られるものより低かった。
【0073】
安全性結果:少なくとも1種の処置下で発現した有害事象(TEAE)を有する患者の割合は、20mgビフェプルノックス群(126人の患者、82%)において最も高く、続いて30mgビフェプルノックス(115人の患者、77%)、オランザピン(110人の患者、73%)、そしてプラセボ(107人の患者、72%)群であった。全体で、ビフェプルノックスで処置した304人の患者から、最も頻繁に報告されるTEAEは頭痛、悪心、嘔吐、消化不良および不眠であった。プラセボ群と比較してビフェプルノックス群においてより高い発生率(>5%の差)を有するTEAEには、悪心、嘔吐および便秘が包含された。一般に、個々のTEAEの発生率の明らかな用量依存的増加はビフェプルノックス群において認められなかった。20mgビフェプルノックス群と比較して30mgビフェプルノックス群においてわずかに高い(>2%の差)発生率を有するTEAEには、疲労、眩暈および鎮静が包含された。少なくとも1種の重篤なTEAEを有する患者の割合は、ビフェプルノックス処置群およびプラセボ群(9%〜12%)において同程度であり、そしてオランザピン処置群(6%)においてわずかに少なかった。任意の事象についてビフェプルノックス群における重篤なTEAEの発生率の用量依存的増加の明示はなかった。特定の興味深いTEAEはデータベースロックの前に定義され、そして以下の事象:自殺、自殺未遂、性機能障害、失神、血管迷走神経性発作および起立性低血圧に関連する事象が包含された。全体で合計58人の患者(10%)が研究中に少なくとも1種の特定の興味深いTEAEを報告した。少なくとも1種の特定の興味深いTEAEを有する患者の総数は、プラセボ(11人の患者、7%)もしくはオランザピン(10人の患者、7%)群と比較してビフェプルノックス処置群(30mgビフェプルノックス;20人の患者、13%;20mgビフェプルノックス;17人の患者、11%)において高かった。特定の興味深いTEAEの大部分は、任意の処置群における患者の<1%において起こった。最も一般的に報告される特定の興味深いTEAEは眩暈であり、それは他の2つの群と比較してビフェプルノックス処置群においてわずかに高い発生率を有した(30mgビフェプルノックス:15人の患者、10%;20mgビフェプルノックス:13人の患者、8%;プラセボ:9人の患者、6%;オランザピン:8人の患者、5%)。処置群内の少なくとも2人の患者において起こる他の特定の興味深いTEAEには、血管迷走神経性失神(30mgビフェプルノックス:2人の患者)および起立性低血圧(30mgビフェプルノックス:3人の患者)が包含された。
【0074】
少なくとも1種のSAEを有する患者の割合は、オランザピン処置群(6人の患者、4%)において最も少なく、続いてビフェプルノックス群(20mg:15人の患者、10%;30mg:12人の患者、8%)であり、SAEの最も高い発生率はプラセボ群(20人の患者、13%)において認められた。最も一般的に報告されるSAEは、精神病性障害(全体で患者の4%)および統合失調症(全体で2%)であった。これらのSAEの発生率は、プラセボ群と比較してビフェプルノックス群において同様であるかもしくは低かった。他の処置群における0人の患者と比較して30mgビフェプルノックス群における2人の患者は、血管迷走神経性失神のSAEを有した。この起こり得る例外は別として、ビフェプルノックス群について認められる任意の他のSAEの発生率の用量依存的増加の他の顕著な表示はなかった。AEのために研究薬剤を中止した患者の割合はプラセボ群(11%)において最も大きく、続いてビフェプルノックス群(各々8%)およびオランザピン処置群(6%)であった。研究終了につながる少なくとも1種のAEを有する患者の割合はプラセボ群(12%)において最も大きく、同程度の割合の患者が20mgビフェプルノックス(8%)、30mgビフェプルノックス(7%)およびオランザピン(6%)処置群において研究終了につながる少なくとも1種のAEを有した。研究薬剤の中止につながる最も一般的な(任意の処置群における患者の>2%により報告される)AEは、精神病性障害および統合失調症であった。ビフェプルノックス群において研究薬剤の中止につながるAEの発生率の用量依存的増加の明らかな傾向はなかった。検査、バイタルサインおよびECG結果の評価は、いかなる予想されない安全性の懸念も引き起こさなかった。ビフェプルノックス群における患者は、プラセボおよびオランザピン群における患者と比較してプロラクチンの減少を示した。体重の著しく異常な減少の発生率は、ビフェプルノックス処置およびプラセボ群間で同程度であり(5%〜6%)、そしてオランザピン処置群において低かった(<1%)。体重の著しく異常な増加の発生率は、ビフェプルノックスおよびプラセボ群において同程度であり(1%〜3%)、そしてオランザピン群においてはるかに高かった(19%)。BAS、SASもしくはAIMSスコアにおいてベースラインからエンドポイントまでの変化における処置群間の顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であった。
【0075】
説明および結論:これは、統合失調症を有する604人の患者の処置におけるオランザピンを活性参照とするビフェプルノックスの効能、耐容性および安全性の6週無作為化二重盲検固定用量プラセボ対照並行群多施設研究であった。本研究は米国(26)、コロンビア(3)およびインド(5)における32の施設で実施された。
【0076】
これは、主要効能パラメーターについて実対照薬オランザピンとプラセボ処置間で統計的に有意な差を示す結果により証明される有効な研究であった。
【0077】
20mgおよび30mg処置群は両方ともエンドポイントでベースラインに対して改善を示したが、主要およびキーとなる副次的効能パラメーターに関してプラセボ群と比較した場合に効能を示さなかった。しかしながら、20mgビフェプルノックス群は副次的効能パラメーター、CGI改善度スコアにおけるベースラインからエンドポイントまでの変化についてプラセボ群に対して顕著な改善を示した(公称p=0.027)。さらに1つの他の副次的効能パラメーターにおいて、20mgビフェプルノックス群とアプローチしたプラセボ群との間の差はPANSS−20%レスポンダー率について顕著である(p=0.061)。しかしながら、副次的効能パラメーター間の顕著なそしてほぼ顕著な差のこれらの発生は、偶然、すなわち、処置比較の5%において起こると予想されるものを上回らない。全ての他の副次的およびキーとなる副次的パラメーターにおいて、ビフェプルノックス処置群はいずれも任意の効能評価項目についてプラセボ群に対して顕著な改善を示さなかった。
【0078】
15mgの用量のオランザピンは、本研究における活性参照として用いられた。一般に、ビフェプルノックス用量群において見られる改善の大きさはプラセボ群において見られるものより高かったが、大部分の効能評価項目についてオランザピン群において見られるものより低かった。
【0079】
対照的に、臨床研究1における大規模研究において、20mgビフェプルノックス用量は、PANSS合計および下位尺度スコアの分析からの統計的に有意な比較により示されるようにプラセボと比較して統合失調症の陽性および陰性症状の両方を軽減することならびに総合精神病理を減少することにおいて有効であった。本研究において、PANSS合計スコアにおけるベースラインから6週までの変化の観察値(observed values)分析を用いて、結果は20mgビフェプルノックス(−24.42[15.6])、30mgビフェプルノックス(−24.56[17.02])およびオランザピン(−29.11[16.88])群間でさらに同様であり、しかしながら、プラセボ群もまた同様の結果(−22.29[19.14])を示した。これは、6週間研究にとどまった患者がそれらの処置処方計画によく反応したことを示す。
【0080】
ビフェプルノックスは、両方の用量レベルで良好な耐容性を示した。有害事象による離脱率は、プラセボ群と比較してビフェプルノックス群において低かった。プラセボ患者におけるよりもビフェプルノックス処置患者において頻繁に生じる有害事象は主に胃腸性のものであり、そして軽度〜中等度の重症度であった。2人の患者のみが(20mgビフェプルノックス群における)胃腸のAEのために研究薬剤を中止した(1人の患者は悪心のために中止し、1人の患者は悪心および嘔吐のために中止した)。少なくとも1種のSAEを有する患者の割合は、プラセボ群(13%)と比較してビフェプルノックス群(7%〜8%)において低かった。最も一般的に報告されるSAEは精神病性障害(全体で4%)および統合失調症(全体で1%)であった。これらのSAEの発生率は、プラセボ群と比較してビフェプルノックス群において同様であるかもしくは低かった。他の処置群における0人の患者と比較して30mgビフェプルノックス群における2人の患者は、血管迷走神経性失神のSAEを有した。この起こり得る例外は別として、ビフェプルノックス群について任意の他のSAEの発生率の用量依存的増加の他の顕著な表示はなかった。検査、バイタルサインおよびECG結果の評価は、いかなる予想されない安全性の懸念も引き起こさなかった。ビフェプルノックスは、以前の研究において見られておりそして薬剤の部分ドーパミンアゴニストプロフィールに基づいて予想されているものと一致するプロラクチンの減少と関連し、そしていずれのAEとも関連しなかった。対照的に、プロラクチンの増加がプラセボおよびオランザピン群において認められた。平均体重のわずかな減少がビフェプルノックス群において認められ、一方、体重の増加がオランザピン群に認められた。体重の著しく異常な減少の発生率はビフェプルノックス処置およびプラセボ群間で同程度であり(5%〜6%)、そしてオランザピン処置群において低かった(1%)。体重の著しく異常な増加の発生率はビフェプルノックスおよびプラセボ群において同程度であり(1%〜3%)、そしてオランザピン(19%)群においてはるかに高かった。これらの結果は他のビフェプルノックス研究において認められているものと一致し、そして体重増加は非定型抗精神病薬にとって問題でありそして心臓血管疾患および糖尿病の増加した危険性と関連することを考えれば注目に値する。
【0081】
異常運動の評価尺度(BAS、SASもしくはAIMSスコア)に関して処置群間で顕著な差はなかった。ビフェプルノックスで処置した患者に対する抗コリン作用薬の使用は、プラセボでの患者のものと同様であった。錐体外路障害の発生率は、全ての群において低かった(ビフェプルノックス群:<1%〜3%;オランザピン:1%;プラセボ:3%)。ビフェプルノックスの20mgおよび30mg用量は、大部分の効能評価項目についてプラセボと比較して統計的に有意に大きい改善を示さなかった。より良いCGI改善度スコアは、プラセボ群と比較して20mg(しかし30mgではそうでない)ビフェプルノックス群において見られた。
【0082】
悪心、嘔吐および便秘は、プラセボに対してより顕著なAEであった。全体で、ビフェプルノックスの20mgおよび30mg用量は安全であり、そして統合失調症を有する患者により良好な耐容性が示された。
【0083】
実施例2d−臨床研究4
主要目的:主要アウトカム評価尺度として無作為抽出から増悪までの時間を用いて、6ヶ月のビフェプルノックス処置が慢性統合失調症を有する患者においてプラセボでの処置より優れているかどうかを調べること。
【0084】
二次的目的:アウトカム評価尺度として陽性および陰性症候群評価尺度(PANSS)合計スコアにおけるベースラインからの変化を用いて、6週のビフェプルノックス処置後の急性効果がプラセボでの処置より優れているかどうかを調べること。
【0085】
他の二次的目的:プラセボに対するビフェプルノックスの長期安全性および耐容性を評価すること。
【0086】
方法論:本研究は、多国籍多施設無作為化二重盲検並行群プラセボ対照固定用量研究であった。研究は3〜6日の抗精神病薬を使わない導入期間からなり、その後に患者は固定用量のビフェプルノックス(20mg/日(BX20)もしくは30mg/日(BX30))もしくはプラセボ(PBO)での6ヶ月の二重盲検処置に無作為抽出された。BX群に割り当てられた患者は、7日(BX20)もしくは8日(BX30)にわたって0.25mg/日から漸増され、そして次に研究の残りの間これらの用量で続けられた。効能評価はベースラインで(CGI−Iを除く)、そして1、2、4、6および9週で、そして3、4、5および6ヶ月で行われた。安全性評価はスクリーニングで、処置の間に、そして研究の最後で行われた。既定の時間点で、血液サンプルがBXおよびその主要代謝物(BXの3’−および4’−硫酸抱合体)の薬剤濃度分析用に得られ、そして医薬品経済性評価が行われた。
【0087】
計画しそして分析した患者数:参加が計画された合計495人の患者があった:各処置群において165人。
【0088】
診断および主な包含基準:スクリーニングおよびベースラインでPANSS合計スコア>60およびCGI−Sスコア>4(中等症)を有し;スクリーニングおよびベースラインでPANSS項目P7(敵意)およびG8(非協調性)スコア<4(中等度)を有し;18〜65歳の間であり(極値を包含する);入院患者であるか、部分入院しているか、もしくは90日以内にデイケアプログラムにおいて追跡される外来患者であり;そしてスクリーニングの前にスクリーニングの前の1ヶ月以内に抗精神病薬の改変を有さなかった、2年より多くにわたって、DSM−IV−TR基準に従って、統合失調症の一次診断を有する患者。
【0089】
治験製品、用量および投与形態、バッチ番号:ビフェプルノックス−毎日1回20もしくは30mgまで7(20mg)もしくは8日(30mg)にわたって漸増;カプセル封入錠剤、経口的。
【0090】
参照治療、用量および投与形態:プラセボ−カプセル封入錠剤、経口的。
【0091】
効能結果:主要効能変数は増悪までの時間であり、そして分析はFASに基づいた。主要効能分析は、3つの処置群における統合失調症の増悪までの等しい時間の仮説を退けた(Coxモデル、p=0.008)。続いてBX群の各々とPBO群の対比較により、BX群における患者はPBO群における患者よりも統合失調症の増悪までの統計的に有意に長い期間を有することが示された(BX20:p=0.008およびBX30:p=0.006)。増悪した患者の割合はPBO群において59%、BX20群において41%、そしてBX30群において38%であった。Cox比例ハザードモデルは、PBOと比較して0.66(BX20)および0.65(BX30)の概算ハザード比を与え;すなわち、増悪の危険性は、BX20もしくはBX30群における患者に対してよりもPBO群における患者に対して約1.5倍高かった。ビフェプルノックスはまた、PPSに基づく増悪までの時間の分析においてもPBOより統計的に有意に優れていた。PPSにおける患者の大部分は研究の大部分に参加したので、得られる概算ハザード比およびp値の両方について、結果は主要分析の結果に非常に近かった。これは、主要効能分析の結論のロバスト性を説明する。副次的効能変数は、6週でのPANSS合計スコアであった。各BX群のPANSS合計スコア(FAS、LOCF)におけるベースラインから6週までの補正した平均変化(BX20:−4.0;BX30:−2.7)は、Hochbergのステップアップ法によればPBO群のもの(1.1)より統計的に有意に大きかった(BX20:p=0.002;BX30:p=0.017)。
【0092】
PANSS合計スコア、PANSS陽性および総合精神病理下位尺度スコア、BPRS合計スコアならびにBPRS精神病クラスタースコア(全てFAS、LOCF)における経時的な進展について、同じ一般的パターンが見られ:最初に(6もしくは9週まで)、平均スコアは減少し、そして次にそれらは両方のBX群において安定し、一方、PBO群において、平均スコアは2もしくは4週まで減少し、その後にスコアは着実に増加した。これらの変数の各々について、LOCF(ANCOVA)を用いたFASについてのPBO群とBX群の各々との対比較により、BX(いずれかの用量)での処置は6もしくは9週以降PBOでの処置より一般に統計的に有意に優れていることが示された。
【0093】
平均PANSS陰性下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後にスコアは安定した。PBO群において、スコアは4週まで減少し、その後にスコアは増加する傾向があった。平均PANSS陰性下位尺度スコアの来院ごとの(per−visit)LOCF分析(FAS、ANCOVA)において、両方のBX用量での処置は、BX30群についての2および4週を除いて、全ての時間点でPBOでのものより統計的に有意に優れていた。
【0094】
全ての処置群について、平均CDSS合計スコア(FAS、LOCF)は最初に減少し(ベースライン〜2週)、その後にスコアは安定なままであるか(両方のBX群)もしくは経時的に増加した(PBO群)。平均CDSS合計スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、任意の時間点でBX群のいずれかとPBO群との間に統計的に有意な差はなかった。この結果は、低レベルの鬱病症状を反映して、ベースラインCDSSスコアが低かったこととの関連において見られるべきである。
【0095】
平均CGI−Sスコア(FAS、LOCF)は両方のBX群において6週まで減少し、その後にスコアは安定した。PBO群において、平均CGI−Sスコアは4週まで減少し、その後にスコアは増加する傾向があった。平均CGI−Sスコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX群はBX20については3、4、5および6ヶ月そしてBX30については5および6ヶ月でPBO群より統計的に有意に優れていた。
【0096】
平均CGI−Iスコア(FAS、LOCF)は両方のBX群において6週まで減少し、その後にスコアは安定した。PBO群において、スコアは2週までごくわずか減少し、その後にスコアは着実に増加した。平均CGI−Iスコアの来院ごとのLOCF分析(FAS、ANCOVA)において、両方のBX用量は6週以降PBOより統計的に有意に優れていた。
【0097】
主に陰性の症状の基準を満たした少数の患者(PBO:25人の患者;BX20:32人の患者;BX30:36人の患者)について、PANSS合計スコア(全ての群)にお
けるそしてPANSS陽性下位尺度スコア(PBOおよびBX20)におけるベースラインからの補正した平均変化は一般に同じパターンをたどり、そして全集団のものと同じ範囲内であった。対照的に、BX30で処置した患者のPANSS陽性下位尺度スコアにおけるベースラインからの補正した平均変化は全集団のと経時的に同じパターンをたどったが、変化はより大きかった。PANSS陰性下位尺度スコア(全ての群)におけるベースラインからの補正した平均変化は全集団と経時的に同じパターンをたどり;しかしながら、平均変化は全集団と比較してこの集団において一般に2倍大きかった。PANSS合計スコア、PANSS陽性下位尺度スコアおよびPANSS陰性下位尺度スコアの来院ごとのLOCF分析(ANCOVA)において、BX20は任意の時間点でPBOと統計的に有意に異ならなかった。BX30は6週以降にPANSS合計スコアにおいてそして2週以降にPANSS陽性下位尺度スコアにおいてPBOより統計的に有意に優れており、一方、BX30はPANSS陰性下位尺度スコアにおいて2週以降PBOと統計的に有意に異ならなかった。
【0098】
主に鬱病の症状の基準を満たした少数の患者(PBO:18人の患者;BX20:15人の患者;BX30:22人の患者)について、PANSS合計スコア、PANSS陽性下位尺度スコア、PANSS陰性下位尺度スコアおよびCDSS合計スコアは全集団のものと一般的に同様の全体的パターンをたどった。PANSS合計スコア、PANSS陽性下位尺度スコア、PANSS陰性下位尺度スコアおよびCDSS合計スコアの来院ごとのLOCF分析(ANCOVA)において、おそらく少数の患者およびベースラインで存在する鬱病症状の低いレベルのために、この亜集団における任意の時間点でBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0099】
BX20群におけるPANSS合計スコア(FAS、LOCF)の少なくとも25%の減少を有する患者の割合は、9週以降PBO群におけるものより統計的に有意に大きく、一方、BX30群におけるレスポンダーの割合は5ヶ月でのみPBO群におけるものより統計的に有意に大きかった。
【0100】
わずかな割合(0%〜8%)の患者(処置にかかわらず)は、PANSS合計スコアの減少>35%、>45%もしくは>55%を有した。特定の減少を有する患者の割合における分布に関して処置群間もしくは内で傾向はなかった。
【0101】
6週および6ヶ月でCGI−Iスコア<2(FAS、LOCF)を有する患者の割合は、PBO群(6週:11%;6ヶ月:9%)におけるよりもBX群(6週:BX20 17%;BX30 19%;6ヶ月:BX20 22%;BX30 20%)において大きかった。BX群とPBO群との間の差は9週以降(BX20)そして3ヶ月を除いて9週以降(BX30)統計的に有意であった(p<0.05)。
【0102】
計算される平均総コレステロールおよび平均LDLは、処置および空腹時/非空腹時条件にかかわらず(非空腹時PBO患者を除いて)全ての群においてベースラインから6ヶ月まで減少した。計算される平均VLDLおよびトリグリセリドは、処置および空腹時/非空腹時条件にかかわらず(非空腹時BX30患者を除いて)全ての群においてベースラインから6ヶ月まで減少した。平均HDLは、処置および空腹時/非空腹時条件にかかわらず全ての群においてベースラインから6ヶ月まで増加した。補正した平均HDLコレステロール値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで増加した(PBO:0.04/0.06(空腹時/非空腹時);BX20:0.07/0.08;BX30:0.07/0.08mmol/L)。BX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0103】
補正した平均トリグリセリド値は、空腹時/非空腹時条件にかかわらず全ての3つの処
置群においてベースラインから6ヶ月まで減少した(PBO:−0.06/−0.22(空腹時/非空腹時);BX20:−0.16/−0.21;BX30:−0.37/−0.03mmol/L)。BX群のいずれか(BX30(空腹時)を除いて)とPBO群との間で統計的に有意な差はなかった。
【0104】
補正した平均空腹時グルコース値は全ての3つの処置群においてベースラインから6ヶ月まで増加し(PBO:0.10;BX20:0.13;BX30:0.09mmol/L)、そしてBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0105】
ベースラインから6ヶ月までの補正した平均体重変化(APTS、OC、ANCOVA)はPBO群において−0.8kg、BX20群において−0.3kg、そしてBX30群において−0.5kgであった。BX20およびBX30群における補正した平均体重減少は、PBO群におけるものと統計的に有意に異ならなかった。
【0106】
全ての処置群において、悪心および/もしくは嘔吐も有した患者はより大きい体重減少を有したが(PBO:−0.6対−1.9kg;BX20:−1.0対−1.9kg;BX30:−1.1対−2.3kg)、患者は悪心および/もしくは嘔吐を有するかどうかにかかわらず体重を減らした。
【0107】
研究中にメタボリック症候群の患者の状態へのBX(いずれかの用量)の処置効果はなかった。約75%(範囲:70%〜80%)の患者は、ベースラインでもしくは研究の最後でメタボリック症候群を有さなかった。メタボリック症候群の患者の状態における統計的に有意な処置差はなかった。
【0108】
臨床検査値、バイタルサイン、メタボリック症候群もしくはECGパラメーターにおける処置群内の臨床的に関連する変化もしくは処置群間の差はなかった。
【0109】
結論:本研究の1つの結論は、ビフェプルノックスの両方の用量(20mg/日および30mg/日)がプラセボより統計的に有意に良く統合失調症の増悪を防いだことである。慢性の安定した統合失調症を有する患者について、ベースライン症状は6週の処置後もしくは長期処置後のいずれかでPBOでよりもBX(両方の用量)で有意に良く維持された。ビフェプルノックスとプラセボとの安全性プロフィールの比較により、プラセボ群に対してビフェプルノックス群における胃腸系および眩暈と関連する有害事象のより高い発生率が示された。研究からの離脱につながる悪心および嘔吐ならびに異常運動の発生率は、BX20群におけるよりもBX30群において高かった。好ましい代謝プロフィール(体重変化、血液脂質およびメタボリック症候群の存在/不在に基づく)は、ビフェプルノックスについて見られた。
【0110】
臨床研究1〜4に関連する結果
PANSS合計スコア
臨床研究1:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(S.D.)は、ビフェプルノックス5mg群について−9.7(17.5)、ビフェプルノックス10mg群について−5.0(18.3)、ビフェプルノックス20mg群について−11.3(17.0)、プラセボ群について−5.3(16.3)、そしてリスペリドン群について−15.7(14.9)であった。エンドポイント(LOCF)でのベースラインからのビフェプルノックスとプラセボ平均変化間の差に対応する処置効果値は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−4.1、0.6および−5.8であった。対比較から、エンドポイント(LOCF)での統計的に有意に大きい減少がプラセボに対して20mgビフェプルノックス群について見られた(多重比較のために補正したp値、p=0.031)。有意な処置群差は、それぞれプラセボと比較した5mgビフェプルノックスおよび10mgビフェプルノックス処置群について見られなかった。
【0111】
【表1】
【0112】
臨床研究2:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−13.5(20.1)、40mgビフェプルノックス群について−10.3(20.5)、プラセボ群について−7.7(19.2)、そしてリスペリドン群について−19.7(19.3)であった(以下の表に示されるとおり)。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:30mgビフェプルノックス群について−5.9、そして40mgビフェプルノックス群について−3.2であった。統計的に有意な処置群差は、Hochberg補正したp値(p=0.020)に基づいてエンドポイントでプラセボと比較して30mgビフェプルノックス処置群について見られた。30mgビフェプルノックスとプラセボ処置群間の差はまた、公称p値(2週〜4週の各々でp<0.004)から2週〜4週でも認められた。さらに、処置効果は6週の研究にわたって一貫して増加した(効果の範囲:2週で−5.6そして4週で−6.3;表3.0.1)。40mgビフェプルノックス群とプラセボ群との間の統計的に有意な差は、Hochberg補正したp値に基づいてエンドポイントで明らかではなかった。
【0113】
【表2】
【0114】
臨床研究3:PANSS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−13.8(19.9)、30mgビフェプルノックス群について−13.1(20.2)、プラセボ群について−10.7(19.4)、そしてオランザピン群について−22.0(18.2)であった(表15)。ビフェプルノックスとプラセボとの間の差に対応する処置効果値(エンドポイント[LOCF]でのベースラインからの平均変化について)は:20mgビフェプルノックス群について−3.5、そして30mgビフェプルノックス群について−2.2であった。統計的に有意な処置群差は、Hochberg補正したp値に基づいてプラセボ群と比較した20mgもしくは30mgビフェプルノックス処置群について見られなかった。同様に、研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。各来院について同じ評価者を有する患者についてのみデータを分析した場合、PANSS合計スコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)の差は一次解析において認められるものと同様であった。オランザピンとプラセボとの間のPANSS合計スコアの差は、感度分析に従って分析した。これらの結果は、オランザピンがプラセボと顕著に異なることを示した(p<0.001)。
【0115】
【表3】
【0116】
臨床研究4:各BX群のPANSS合計スコアにおけるベースラインから6週までの補正した平均変化(BX20:−4.0およびBX30:−2.7)は、PBO(1.1)群のものより統計的に有意に大きかった(BX20:p=0.002;BX30:p=0.017)。
【0117】
【表4】
【0118】
PANSS陽性
臨床研究1:以下の表は、ITT集団についてベースラインでの平均PANSS陽性下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−1.1、0.7、−1.5であった。エンドポイント(LOCF)での統計的に有意に大きい減少(未補正p=0.037)は、ビフェプルノックス20mg群およびプラセボの処置効果概算の比較についてPANSS陽性下位尺度スコアにおいて見られた。
【0119】
【表5】
【0120】
ベースラインからの変化の観察値分析について、PANSS合計スコアと同様の傾向が経時的なビフェプルノックス処置群におけるPANSS陽性下位尺度スコアのベースラインからの変化において見られた。ビフェプルノックスとプラセボ群との間の対比較は、統計的に有意ではなかった。
【0121】
臨床研究2:以下の表は、ITT集団についてベースラインでのPANSS陽性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−4.5(6.6)、40mgビフェプルノックス群について−4.2(6.9)、プラセボ群について−2.5(6.0)、そしてリスペリドン群について−7.2(6.6)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群
について−1.9、そして40mgビフェプルノックス群について−1.7であった。SEP1は有意な結果をもたらさず、従って、SEP2およびSEP3は評価されなかった(7.4.1.1節を参照)。しかしながら、ビフェプルノックス30mg処置群とプラセボとの間の顕著な差がエンドポイントで認められた(公称p=0.01)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.006)。
【0122】
同様に、ビフェプルノックス40mg処置群とプラセボとの間の顕著な差がエンドポイントで認められた(p=0.020)。プラセボに対する差はまた、1週〜3週でも認められた(p<0.013)。
【0123】
【表6】
【0124】
臨床研究3:以下の表は、ITT集団についてベースラインでのPANSS陽性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陽性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−4.3(6.1)、30mgビ
フェプルノックス群について−4.2(6.8)、プラセボ群について−3.5(6.2)、そしてオランザピン群について−7.0(5.8)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−1.1、そして30mgビフェプルノックス群について−0.7であった。公称p値(<0.05)での差は、6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で認められなかった。
【0125】
【表7】
【0126】
臨床研究4:平均PANSS陽性下位尺度スコア(FAS、LOCF)は、9週まで両方のBX群において経時的に減少し、その後にスコアはごくわずか増加した。PBO群において、平均PANSS合計スコアは2週まで減少し、その後にスコアは着実に増加した(図1;パネル34)。PANSS陽性下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20でもしくはBX30での処置は6週以降PBOでのものより統計的に有意に優れていた。
【0127】
PANSS陰性
臨床研究1:以下の表は、ITT集団についてベースラインでの平均PANSS陰性下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。ビフェプルノックス5mg、20mgおよびリスペリドン6mg群は、経時的に最も大きい改善を示した。エンドポイントLOCFでの処置効果(ビフェプルノックス−プラセボ)の概算は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−1.0、−0.3、−1.4であった。PANSS陰性下位尺度スコアにおける統計的に有意に大きい減少は、プラセボに対してビフェプルノックス20mg群について3週(未補正p=0.013)でそしてエンドポイント(LOCF)(未補正p=0.026)で見られた。
【0128】
【表8】
【0129】
PANSS陰性下位尺度スコアにおけるベースラインからの変化の観察値分析について、経時的なスコアの減少は、ビフェプルノックス処置群の各々における改善を示した。ビフェプルノックスの統計的に有意な処置効果は、プラセボと比較してビフェプルノックス5mg群について2週で認められた(p=0.032)。他の有意な差は認められなかった。
【0130】
臨床研究2:以下の表は、ITT集団についてベースラインでのPANSS陰性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陰性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−3.1(5.6)、40mgビフェプルノックス群について−2.2(5.4)、プラセボ群について−1.8(5.6)、そしてリスペリドン群について−3.8(5.5)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−1.4、そして40mgビフェプルノックス群について−0.9であった。SEP1は有意な結果をもたらさず、従って、SEP2およびSEP3は評価されなかった。
【0131】
30mgビフェプルノックス処置群は、エンドポイントでプラセボとの顕著な差を示した(公称p=0.027)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.021)。研究中の任意の時間点(1週〜6週)でプラセボと40mgビフェプルノックス用量群との間で差は認められなかった。
【0132】
【表9】
【0133】
臨床研究3:以下の表は、ITT集団についてベースラインでのPANSS陰性症状下位尺度スコアおよびLOCFを用いた来院ごとのベースラインからの変化を提示する。PANSS陰性症状下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−3.3(5.0)、30mgビフェプルノックス群について−3.1(5.2)、プラセボ群について−2.4(5.1)、そしてオランザピン群について−4.6(5.2)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.9、そして30mgビフェプルノックス群について−0.6であった。公称p値(<0.05)での差は、6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で認められなかった。
【0134】
【表10】
【0135】
臨床研究4:平均PANSS陰性下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後でスコアは安定した(図2;パネル35)。PBO群において、スコアは4週まで減少し、その後でスコアは増加する傾向があった(パネル35)。
【0136】
平均PANSS陰性下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20でもしくはBX30での処置は、BX30について2および4週を除いて、全ての時間点でPBOでのものより統計的に有意に優れていた。
【0137】
臨床研究4のPANSSデータ
平均PANSS合計スコア(FAS、LOCF)は9週まで両方のBX群において経時的に減少し、その後でスコアは安定した(パネル28)。PBO群において、平均PANSS合計スコアは2週まで減少し、その後でスコアは着実に増加した(パネル28)。
【0138】
PANSS合計スコア(パネル28)のLOCF分析により、最終観察値で補完すると、BX群におけるPANSS合計スコアは安定なままであったことが示される。両方のBX群における変化は、6週以降PBO群におけるものより統計的に有意に優れていた。さらに、BX20は4週以降PBOより統計的に有意に優れていた。PANSS合計スコアのOC分析により、研究を続行した患者は経時的に改善したことが示される(パネル29)。両方のBX群における変化は6週を包含するが6ヶ月を除く多数の時間点でPBO群におけるものより統計的に有意に優れていた。
【0139】
LOCFとPOCF分析との間の差は、離脱した多数の患者がそれらのPANSS合計スコアにおける増悪を有しそして増悪が見られた最初の来院で離脱したことを示す。これは、少なくともごくわずかでも悪くなった場合に研究から離脱するように患者に求める研究設計と一致する。
【0140】
他の効能変数について、効能スコアの経時的な進展の傾向は、研究に含まれる患者が鬱状態でなかったことをベースラインレベルが示すCDSSを除いて、同様であった。
【0141】
【表11】
【0142】
【表12】
【0143】
【表13】
【0144】
【表14】
【0145】
【表15】
【0146】
総合精神病理スコア
臨床研究1:総合精神病理スコアは、以下の表に示されるようにビフェプルノックス処置群の各々についてベースラインから6週まで減少した。LOCFを用いたビフェプルノックスの処置効果の概算は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−2.2、0.4および−2.8であった。ビフェプルノックス20mg群は、2週(未補正p=0.029)、3週(未補正p=0.032)そしてエンドポイント(LOCF)(未補正p=0.016)でプラセボより統計的に有意に大きい減少を有した。
【0147】
【表16】
【0148】
PANSS総合精神病理下位尺度スコアにおけるベースラインからの変化の観察値分析について、経時的なスコアの減少はビフェプルノックス処置群の各々における改善を示した。プラセボに対するビフェプルノックスの統計的に有意な処置効果は、プラセボに対してビフェプルノックス20mg群について2週で(p=0.038)そしてビフェプルノックス5mg群について6週で(p=0.017)認められた。他の有意な差は認められなかった。
【0149】
臨床研究2:ITT集団(LOCF)についてのベースラインでの平均PANSS総合精神病理下位尺度スコアおよび各ベースライン後来院でのPANSS総合精神病理下位尺度におけるベースラインからの変化を以下の表に提示する。PANSS総合精神病理下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−6.0(10.2)、40mgビフェプルノックス群について−3.8(10.1)、プラセボ群について−3.5(9.7)、そしてリスペリドン群について−8.6(9.8)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−2.5、そして40mgビフェプルノックス群について−0.7であった。2週〜エンドポイン
トで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.025)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕著な差はなかった。
【0150】
【表17】
【0151】
臨床研究3:ITT集団(LOCF)についてのベースラインでの平均PANSS総合精神病理下位尺度スコアおよび各ベースライン後来院でのPANSS総合精神病理下位尺度スコアにおけるベースラインからの変化を以下の表に提示する。PANSS総合精神病理下位尺度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−6.1(10.7)、30mgビフェプルノックス群について−5.8(10.3)、プラセボ群について−4.8(10.0)、そしてオランザピン群について−10.5(9.4)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−1.5、そして30mgビフェプルノックス群について−0.9であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0152】
【表18】
【0153】
臨床研究4:平均PANSS総合精神病理下位尺度スコアは、PANSS合計スコアと同じパターンを一般にたどった。平均PANSS総合精神病理下位尺度スコア(FAS、LOCF)は6週まで両方のBX群において減少し、その後でスコアは安定した(図3;パネル36)。PBO群において、平均PANSS総合精神病理下位尺度スコアは2週まで減少し、その後でスコアは着実に増加した(パネル36)。
【0154】
PANSS総合精神病理下位尺度スコアの来院ごとのLOCF分析(FAS、ANCOVA)において、BX20およびBX30は両方とも9週以降PBOより統計的に有意に優れていた。さらに、BX20は6週でPBOより統計的に有意に優れていた。
【0155】
BPRS合計スコア
臨床研究1:以下の表に示されるように、ビフェプルノックス20mg群はプラセボ群よりBPRS合計スコアにおける大きい変化を示した。2週(未補正p=0.042)から始まるあらゆる時間点;3週(未補正p=0.020);4週(未補正p=0.024);エンドポイント(LOCF)(未補正p=0.012)でプラセボに対してビフェプルノックス20mgについて統計的に有意な処置群差が見られた。
【0156】
【表19】
【0157】
LOCF分析において認められる同様の傾向は、観察値を用いたBPRS合計スコア分析におけるベースラインからの変化において認められた。プラセボに対するビフェプルノックス処置効果の比較は、5mg群のみについて6週で統計的に有意であった(p=0.024)。他の有意な比較は認められなかった。
【0158】
臨床研究2:以下の表は、ITT集団(LOCF)についてベースラインでの平均BPRS合計スコア(PANSS由来)および各ベースライン後来院でのBPRS合計スコアにおけるベースラインからの変化を提示する。BPRS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−8.1(12.3)、40mgビフェプルノックス群について−6.5(11.7)、プラセボ群について−4.9(11.5)、そしてリスペリドン群について−12.2(11.7)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−3.2、そして40mgビフェプルノックス群について−1.9であった。2週〜エンドポイントで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.019)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕
著な差は見られなかった。
【0159】
【表20】
【0160】
臨床研究3:以下の表は、ITT集団(LOCF)についてのベースラインでの平均BPRS合計スコア(PANSS由来)および各ベースライン後来院でのBPRS合計スコアにおけるベースラインからの変化を提示する。BPRS合計スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−8.5(11.9)、30mgビフェプルノックス群について−7.9(12.0)、プラセボ群について−6.7(11.7)、そしてオランザピン群について−13.2(10.7)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−2.1、そして30mgビフェプルノックス群について−1.1であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0161】
【表21】
【0162】
BPRS精神病クラスタースコア
臨床研究1:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−0.9、0.4、−1.1であった。ビフェプルノックスとプラセボのエンドポイント(LOCF)でのベースラインからの平均変化間の差における統計的に有意に大きい減少(未補正p=0.044)は、プラセボに対するビフェプルノックス20mg群のBPRS精神病クラスタースコアにおいて見られた。
【0163】
【表22】
【0164】
BPRS精神病クラスタースコアにおけるベースラインからの変化の観察値分析について、プラセボと比較してスコアにおけるより大きい減少が、4週から始まり3つのビフェプルノックス処置群の各々について見られた。統計的に有意な差は認められなかった。
【0165】
臨床研究2:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。BPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−3.2(4.2)、40mgビフェプルノックス群について−2.6(4.4)、プラセボ群について−1.8(3.5)、そしてリスペリドン群について−4.9(4.0)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−1.4、そして40mgビフェプルノックス群について−1.0であった。2週〜エンドポイントで30mgビフェプルノックス群とプラセボとの間で顕著な差が認められた(p<0.002)。エンドポイントで(p=0.031)そして2週および3週で(p<0.036)で40mgビフェプルノックス群とプラセボとの間で顕著な差が見られた。
【0166】
【表23】
【0167】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均BPRS精神病クラスタースコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。BPRS精神病クラスタースコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−3.1(3.9)、30mgビフェプルノックス群について−3.2(4.4)、プラセボ群について−2.6(4.0)、そしてオランザピン群について−4.9(4.0)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.6、そして30mgビフェプルノックス群について−0.5であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0168】
【表24】
【0169】
CGI−S
臨床研究1:以下の表は、ITT集団についてのベースラインでの平均CGI疾患重症度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は、それぞれビフェプルノックス5mg、10mgおよび20mg群について−0.31、0.12、−0.19であった。対処置群比較により、2週(p=0.008)、3週(p=0.020)および4週(p=0.032)でプラセボに対してビフェプルノックス20mgにおいて統計的に有意に大きい減少が示されたが、エンドポイント(LOCF)ではそうでなかった。ビフェプルノックス5mg対プラセボの処置群比較は、3週(p=0.049)、4週(p=0.033)およびエンドポイント(LOCF)(p=0.013)で有意であった。
【0170】
【表25】
【0171】
臨床研究2:以下の表は、ITT集団(LOCF)についてのベースラインでの平均CGI−SスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CGI疾患重症度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−0.69(1.19)、40mgビフェプルノックス群について−0.54(1.12)、プラセボ群について−0.37(1.07)、そしてリスペリドン群について−1.06(1.20)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:30mgビフェプルノックス群について−0.28、そして40mgビフェプルノックス群について−0.18であった。CGI−Sにおけるベースラインからエンドポイントまでの変化の統計的に有意な差は、ステップダウン法に基づいてプラセボと比較して30mgビフェプルノックス処置群について見られなかった(補正したp=0.056)。30mgビフェプルノックス群は、エンドポイントでプラセボとの顕著な差を示した(公称p=0.028)。30mgビフェプルノックスとプラセボ処置群との間の顕著な差はまた、2週〜4週でも認められた(p<0.015)。研究中の任意の時間点(1週〜4週)でプラセボと40mgビフェプルノックス用量群との間で差は認められなかった。
【0172】
【表26】
【0173】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均CGI疾患重症度スコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CGI疾患重症度スコアにおけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−0.63(0.98)、30mgビフェプルノックス群について−0.62(1.03)、プラセボ群について−0.49(1.06)、そしてオランザピン群について−1.03(1.00)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス−プラセボ)は:20mgビフェプルノックス群について−0.13、そして30mgビフェプルノックス群について−0.09であった。6週/エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群とビフェプルノックス用量群のいずれかとの間で公称p値(p<0.05)では差は顕著でなかった。
【0174】
【表27】
【0175】
CGI−I
臨床研究1:表24に示されるように20mgビフェプルノックス処置群のCGI改善度スコアにおいて改善が認められた。処置群差は、LOCF分析においてプラセボに対してビフェプルノックス20mg群について1週(p=0.040)および2週(p=0.016)で統計的に有意であったが、エンドポイントではそうでなかった。他の有意な差は認められなかった。
【0176】
【表28】
【0177】
臨床研究2:ベースラインからエンドポイント(LOCF)まで中等度もしくは著明改善を報告した患者の割合は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において28%、プラセボ群において25%、そしてリスペリドン群において52%であった(以下の表)。エンドポイントで、プラセボと2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められず;しかしながら、30mgビフェプルノックス群とプラセボとの間の顕著な差は2週〜4週で見られた(p<0.024)。研究中の任意の時間点(1週〜エンドポイント)で40mgビフェプルノックス群とプラセボとの間で顕著な差は見られなかった。2週〜エンドポイントのデータを以下の表に要約する。
【0178】
【表29】
【0179】
臨床研究3:以下の表は、ITT集団についてのLOCFを用いた来院ごとのCGI改善度評価の7つのカテゴリーの頻度を提示する。平均CGI改善度スコアに関する処置群間の統計的差は、修正リジットスコアを用いて7つのCGI改善度カテゴリーの頻度に適用したプールした施設により階層化されるCMH検定を用いて評価された。ベースラインからエンドポイントまで(表23における対応する個々の割合を合計することにより得られる)、合わせた中等度もしくは著名改善を報告した患者の割合は:20mgビフェプルノックス群において38%、30mgビフェプルノックス群において34%、プラセボ群において32%、そしてオランザピン群において46%であった。エンドポイントでのプラセボ群に対する20mgビフェプルノックス群の比較は、顕著であることが見出された(p=0.027)。プラセボ群に対する30mgビフェプルノックス群の処置比較は、顕著ではなかった(p=0.162)。研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0180】
【表30】
【0181】
PANSSレスポンダー率
臨床研究1:PANSSレスポンダー率は、プラセボ群と比較してビフェプルノックス処置群の各々について高かった。PANSSレスポンダー率は、研究プロトコルから20%定義を用いてそれぞれ3つの用量群について28%、24%および34%であった。全ての4つの定義を用いるPANSSのレスポンダー率は、以下の表に提示される。
【0182】
【表31】
【0183】
LOCFを用いたPANSSレスポンダー率の有意な差は、プラセボに対するビフェプルノックス5mgの25%定義を用いてそしてプラセボに対するビフェプルノックス20mg群の25%、30%および35%定義について見られた。
【0184】
臨床研究2:PANSSレスポンダーは、そのPANSS合計スコアがLOCFデータに基づいてベースラインからエンドポイントまで20%以上減少した患者として定義された。探索的解析として、レスポンダー率はまたレスポンダーの30%、35%、40%および50%定義も用いて分析された。エンドポイント(LOCF)でのPANSSレスポンダー率の要約は、以下の表25に提示される。
【0185】
PANSSレスポンダー率は、プラセボ群と比較して2つのビフェプルノックス処置群についてわずかに高かった。PANSS20%レスポンダー率は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において30%、プラセボ群において26%、そしてリスペリドン群において54%であった。しかしながら、30mgビフェプルノックス群とプラセボとの間の差は顕著ではなかった(p=0.052)。PANSS 30%レスポンダー率は:30mgビフェプルノックス群において24%、40mgビフェプルノックス群において22%、プラセボ群において14%、そしてリスペリドン群において31%であった。30mgビフェプルノックス群とプラセボとの間の差は顕著であった(p=0.019)。30mgビフェプルノックス群とプラセボとの間の顕著な差はまた、35%および50%レスポンダー率についても見られた。
【0186】
【表32】
【0187】
臨床研究3:PANSSレスポンダーは、そのPANSS合計スコアがベースラインからエンドポイントまで20%以上減少した患者として定義された。探索的解析として、レスポンダー率はまたレスポンダーの30%、35%、40%および50%定義も用いて分析された。エンドポイント(LOCF)でのPANSSレスポンダー率の要約を以下の表に提示する。20%PANSSレスポンダー率は、プラセボ群と比較して2つのビフェプルノックス処置群についてわずかに高く:20mgビフェプルノックス群における患者の42%、30mgビフェプルノックス群における患者の39%、プラセボ群における患者の32%、そしてオランザピン群における患者の54%は、PANSS合計スコアにおいてベースラインからエンドポイントまで20%以上改善した。20mgビフェプルノックス群とアプローチしたプラセボ群との間の差は、顕著である(p=0.061)。しかしながら、レスポンダーの定義のさらに厳格な基準(30%〜50%)を用いた場合、20mgもしくは30mgビフェプルノックス群とプラセボ群との間の差は顕著ではなかった(全てp>0.118)。
【0188】
【表33】
【0189】
臨床研究4:各来院でベースラインに対してPANSS合計スコアの>25%、>35%、>45%もしくは>55%の減少を有する患者(PANSSレスポンダー)の割合は、LOCFにより示される(図4;パネル42)。
【0190】
CGIレスポンダー率
臨床研究1:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。ビフェプルノックス5mgおよび20mg群のCGIレスポンダー率は、プラセボ群のレスポンダー率より高かった。プラセボと比較した場合に3つのビフェプルノックス用量群のいずれについても統計的に有意な差は見られなかった。
【0191】
【表34】
【0192】
臨床研究2:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。エンドポイント(LOCF)でのCGI−Iレスポンダー率の要約を以下の表に提示する。CGI−Iレスポンダー率は:30mgビフェプルノックス群において36%、40mgビフェプルノックス群において28%、プラセボ群において25%、そしてリスペリドン群において52%であった。30mgビフェプルノックス群とプラセボとの間の顕著な差が見られた(p=0.039)。
【0193】
【表35】
【0194】
臨床研究3:CGIレスポンダーは、CGI改善度評価尺度で「著明改善」もしくは「中等度改善」と分類される患者として定義される。エンドポイント(LOCF)でのCGI−Iレスポンダー率の要約を表3.10.0(LOCF)および表26に提示する。CGI−Iレスポンダー率は:20mgビフェプルノックス群において38%、30mgビフェプルノックス群において34%、プラセボ群において32%、そしてオランザピン群において46%であった。プラセボ群と2つのビフェプルノックス用量群のいずれかとの
間で顕著な差は認められなかった。
【0195】
【表36】
【0196】
CDSS
臨床研究2:統合失調症のカルガリー鬱病評価尺度スコアを以下の表に提示する。この表は、ITT集団についてのベースラインでの平均CDSSスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CDSSスコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)は、30mgビフェプルノックス群について−0.66(3.64)、40mgビフェプルノックス群について0.01(3.66)、プラセボ群について−0.39(3.45)、そしてリスペリドン群について−0.89(3.42)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス、プラセボ)は:30mgビフェプルノックス群について−0.38、そして40mgビフェプルノックス群について0.29であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボと2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0197】
【表37】
【0198】
臨床研究3:以下の表は、ITT集団についてのベースラインでの平均CDSSスコアおよびLOCFを用いた来院ごとのベースラインからの平均変化を提示する。CDSSスコア(LOCF)におけるベースラインからエンドポイントまでの平均変化(SD)は、20mgビフェプルノックス群について−1.30(3.85)、30mgビフェプルノックス群について−0.79(3.02)、プラセボ群について−0.59(4.20)、そしてオランザピン群について−1.47(3.95)であった。エンドポイントLOCFでの処置効果値(ビフェプルノックス、プラセボ)は:20mgビフェプルノックス群について−0.54、そして20mgビフェプルノックス群について−0.34であった。エンドポイントでもしくは研究中の任意の他の時間点(1週〜4週)でプラセボ群と2つのビフェプルノックス用量群のいずれかとの間で顕著な差は認められなかった。
【0199】
【表38】
【0200】
臨床研究4:各来院でCGI−Iスコア<2を有する患者(CGI−Iレスポンダー;FAS、LOCF)の割合を表(図5;パネル43)において以下に示す。
【0201】
EPS
EPSは、静座不能、ジスキネジー、パーキンソン症候群などのような処置下で発現した有害事象ならびに/またはSAS、BASおよび/もしくはAIMSのような公式評価尺度を用いて評価された。
【0202】
シンプソン・アンガス評価尺度16(SAS)
SASは、抗精神病薬を投与された患者におけるパーキンソン症候群タイプ症状を測定するために用いられる。評価尺度は、各々0(症状が全くない)〜4(極端な形の症状の存在)の間の5点評価尺度で評価される10項目からなる。SAS合計スコアは全ての項目スコアの合計として定義され、そして範囲は0〜40である。3までのSAS合計スコアは、正常と見なされる。
【0203】
バーンズ・アカシジア評価尺度17(BAS)
BASは、薬剤誘発性静座不能の観察可能な落ち着きのない動きならびに落ち着きのなさの自覚および静座不能と関連する任意の苦痛を評価するために用いられる。評価尺度は、0(静座不能の症状なし)から3(重篤な静座不能)まで評価される3項目からなる。
BAS合計スコアはこれら3つのBAS項目スコアの合計として定義され、そして範囲は0〜9である。さらに、静座不能の包括的(global)臨床評価は0(静座不能の症状なし)から5(重篤な静座不能)まで評価される。
【0204】
異常不随意運動評価尺度18(AIMS)
ジスキネジー運動の発生を記録するために考案されたAIMSは、12項目からなる。項目1〜7は、0(なし)〜4(重篤)の評価尺度で特定の不随意運動を測定する。項目8〜10は、0(自覚なし)〜4(自覚する、重篤な苦痛)の評価尺度で異常運動の包括的評価を測定する。項目11および12は、イエス/ノーで答える、患者の歯の症状に関する質問である。合計スコアはAIMS項目1〜10を合計することにより計算され、そして0〜40の間であり;非包括的合計スコアは項目1〜7を合計することにより計算される。
【0205】
臨床研究1:
【0206】
【表39】
【0207】
【表40】
【0208】
【表41】
【0209】
臨床研究2:
【0210】
【表42】
【0211】
【表43】
【0212】
臨床研究3:
【0213】
【表44】
【0214】
【表45】
【0215】
【表46】
【0216】
臨床研究4:
EPSに関連するTEAEを有する患者の割合は、BX群のいずれか(BX20:10%;BX30:15%)におけるよりもPBO群(4%)において低かった。PBO群に対していずれかのBX群において>3人の患者がいたEPSに関連するTEAEは:BX20−静座不能;BX30−ジスキネジー、静座不能、錐体外路障害を含んでなった。全体で、15人の患者が離脱につながるEPSに関連するTEAEを有した(PBO群において2人;BX20群において4人;BX30群において9人)。
【0217】
【表47】
【0218】
SAS、BASおよびAIMS
臨床研究1:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。各時間点で正常および異常スコアを有する患者の割合。ベースラインで、異常SASスコアを有する患者の割合は8%〜12%の間であった。エンドポイントで、異常SASスコアを有する患者の割合は3%〜10%の間であった。異常SASスコアを有する患者の割合は、全ての処置群においてベースラインとエンドポイントとの間で減少した。ベースラインからエンドポイントまでカテゴリー
間でシフトする患者の割合において処置群間で統計的に有意な差はなかった(p=0.800)。
【0219】
BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛スコア、3項目合計スコアもしくは包括的臨床評価スコアについてベースラインもしくはエンドポイントで処置群間で統計的に有意な差はなかった。
【0220】
BAS合計スコアもしくはBAS包括的臨床評価スコアにおいてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった。ベースラインもしくはエンドポイントで処置群間で統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群間で統計的に有意な差はなかった。
【0221】
臨床研究2:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。ベースラインで、異常SASスコアを有する患者の割合は:30mgビフェプルノックス群において14%、40mgビフェプルノックス群において11%、プラセボ群において7%、そしてリスペリドン群において6%であった。ベースラインとエンドポイントとの間で異常SASスコアを有する患者の割合は、ビフェプルノックス群において減少し、プラセボ群において変化せず、そしてリスペリドン群において増加した。エンドポイントで、異常SASスコアを有する患者の割合は、40mgビフェプルノックスおよびプラセボ群における7%からリスペリドン群における14%までの間であった。
【0222】
ベースラインで正常からエンドポイントで異常へのシフトは、他の3つの処置群(30mgビフェプルノックス:1人の患者、<1%;40mgビフェプルノックス:5人の患者、4%;プラセボ:6人の患者、4%)と比較してリスペリドン群(19人の患者、12%)において最も高かった。処置群間での統計的に有意な差(p<0.001)が認められた。
【0223】
BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア(p=0.093)、落ち着きのなさの自覚スコア(p=0.368)、落ち着きのなさと関連する苦痛スコア(p=0.779)、3項目合計スコア(p=0.433)もしくは静座不能の包括的臨床評価スコア(p=0.541)についてエンドポイントで処置群にわたる統計的に有意な差はなかった。また、上記の評価尺度についてベースラインでも処置群にわたる統計的に有意な差はなかった(p>0.168)。
【0224】
BAS合計スコア(p=0.482)もしくはBAS包括的臨床評価スコア(p=0.911)においてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合における処置群にわたる統計的に有意な差はなかった。
【0225】
ベースライン(平均範囲=1.0〜1.2;p=0.923)もしくはエンドポイント(平均範囲=0.9〜1.6;p=0.138)で処置群にわたる統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群にわたる統計的に有意な差はなかった(平均範囲=−0.3〜0.4;p=0.138)。
【0226】
臨床研究3:シンプソン・アンガス評価尺度総合スコアは、ベースラインおよびエンドポイントで正常もしくは異常として評価された。ベースラインで、異常SASスコアを有
する患者の割合は6%〜10%の間であった。エンドポイントで、異常SASスコアを有する患者の割合は3%〜8%の間であった。異常SASスコアを有する患者の割合は、全ての処置群においてベースラインとエンドポイントとの間で減少した。ベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった(p=0.463)。BASスコアは客観的スコア、落ち着きのなさの自覚スコア、落ち着きのなさと関連する苦痛のスコア、3項目合計スコアおよび包括的臨床評価スコアからなる。客観的スコア(p=0.421)、落ち着きのなさの自覚スコア(p=0.584)、落ち着きのなさと関連する苦痛スコア(p=0.254)、3項目合計スコア(p=0.865)もしくは包括的臨床評価スコア(p=0.518)についてエンドポイントで処置群間で統計的に有意な差はなかった。落ち着きのなさと関連する主観的苦痛スコアについてベースラインで処置群間で統計的に有意な差があったが(p=0.029)、任意の他のスコアについてはそうでなかった。
【0227】
BAS合計スコア(p=0.808)もしくはBAS包括的臨床評価スコア(p=0.525)においてベースラインからエンドポイントまでカテゴリー間でシフトする患者の割合において処置群間で統計的に有意な差はなかった。
【0228】
ベースライン(p=0.362)もしくはエンドポイント(p=0.187)で処置群間で統計的に有意な差はなかった。ベースラインからエンドポイントまでの変化の値において処置群間で統計的に有意な差はなかった(p=0.187)。
【0229】
臨床研究4:
SAS
ベースラインで、全ての3つの処置群における平均SAS合計スコアは2.17〜2.33の間であった。
【0230】
研究中に全ての3つの処置群における平均SAS合計スコアのわずかな変動があり、そして6ヶ月で、平均SAS合計スコアは0.57〜1.16の間であり(APTS、OC);従って、ベースラインでそして6ヶ月で平均合計スコアは正常範囲内であった。SAS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は、各処置群において<1であった(APTS、OC、ANCOVA)。SAS合計スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった。
【0231】
ベースラインで、患者の大部分(PBO:74%;BX20:71%;BX30:72%)はSAS状態に関して正常であり、そして正常患者の割合は6ヶ月で増加していた(PBO:85%;BX20:89%;BX30:78%)。SAS状態のシフトにおいて処置群のいずれかの間で統計的に有意な差はなかった(カテゴリー:不変;異常→正常;正常→異常)。
【0232】
BAS
ベースラインで、全ての3つの処置群における平均BAS合計スコアは0.44〜0.46の間であった。研究中に全ての3つの処置群における平均BAS合計スコアのわずかな変動があり、そして6ヶ月で、平均BAS合計スコアは0.05〜0.17の間であった(APTS、OC)。BAS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は、各処置群において<0.6であった(APTS、OC、ANCOVA)。BAS合計スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった。
【0233】
BAS包括的評価スコアについて、傾向は同様であり:研究中に全ての3つの処置群において平均BAS包括的評価スコアのわずかな変動があり;平均スコアはベースラインで0.22〜0.26、そして6ヶ月で0.07〜0.14の間であった(APTS、OC
)。6ヶ月でBAS包括的評価スコアにおいて処置群のいずれかの間で臨床的に関連する差はなかった(APTS、OC)。BAS項目1、2もしくは3の分布において処置群のいずれかの間で統計的に有意な差はなかった。
【0234】
AIMS
全ての3つの処置群における平均AIMS合計スコアは低く、そしてベースラインで1.04〜1.35の間であった。全ての処置群について補正した平均スコアの初期増加があり(BX30群において最大)、その後で平均スコアは経時的に減少し;6ヶ月で、スコアは全ての3つの処置群においてベースラインレベルまで戻っていた(PBO:0.11;BX20:1.08;BX30:0.86(APTS、OC))。BX20(全ての時間点)およびBX30(5および6ヶ月)群におけるAIMS合計スコアの補正した平均変化はPBO群におけるものと統計的に有意に異ならず、一方、BX30(1週〜4ヶ月)群の補正した平均変化はそうでなかった。AIMS合計スコアにおけるベースラインから6ヶ月までの補正した平均最大変化は小さかった(PBO:0.39;BX20:1.21;BX30:2.45、APTS、OC、ANCOVA)。差はいずれも臨床的に有意と見なされなかった。
【0235】
【表48】
【0236】
体重
臨床研究1:
体重のわずかな減少がビフェプルノックス処置群において認められたが、プラセボ処置群においてはそうでなかった。エンドポイントで、ビフェプルノックス処置群における患者の平均体重減少は約1 lb(5mgビフェプルノックス、−1.0 lb;10mgビフェプルノックス、−1.3 lb;20mgビフェプルノックス、−0.6 lb)であった。プラセボ処置群は、1.9 lbの平均体重増加を有した。
【0237】
体重におけるベースラインからの変化の統計的検定は、副次的効能パラメーターに使用するものと同様の方法論を用いて行われた(処置およびベースラインでの体重の因子を有するANCOVAモデル)。プラセボに対するビフェプルノックス処置群の対比較は、ビフェプルノックスとプラセボのエンドポイントでの体重のベースラインからの平均変化間
の差について全て統計的に有意であった(ビフェプルノックス5mg[p=0.025]、ビフェプルノックス10mg[p=0.009]およびビフェプルノックス20mg[p=0.031])。
【0238】
リスペリドン処置群における患者の平均体重変化は、4.8 lbの増加であった。
【0239】
体重の増加:ビフェプルノックス処置群におけるその体重が7%より多く増加した患者の割合(2〜4%)は、プラセボ処置群において認められる割合(5%)より少ないかもしくは同様であった。リスペリドン処置群において、患者の13%はそれらの体重を>7%増加した。
【0240】
体重の減少:その体重が7%より多く減少した患者の割合は、プラセボ処置群(3%)におけるよりもビフェプルノックス処置群(5mg、6%;10mg、7%;20mg、8%)において高かった。リスペリドン処置群にはそれらの体重を>7%減少した患者はいなかった。
【0241】
臨床研究2:
平均体重のわずかな減少がエンドポイントでビフェプルノックス処置群およびプラセボ群において認められ、一方、リスペリドン処置群について増加が認められた(以下の表)。エンドポイントで、平均体重変化は:30mgビフェプルノックス群において−2.2 lb;40mgビフェプルノックス群において−1.9 lb;プラセボ群において0.5 lb;そしてリスペリドン群において3.2 lbであった。ビフェプルノックス処置群において、6週での平均変化はエンドポイントでのものより高かった。
【0242】
【表49】
【0243】
臨床研究3:平均体重のわずかな減少がビフェプルノックス処置群およびプラセボ群において認められ、一方、オランザピン処置群について増加が認められた。エンドポイントで、平均体重変化は:20mgビフェプルノックス群において−2.3 lb;30mgビフェプルノックス群において−1.1 lb;プラセボ群において−1.3 lb、そしてオランザピン群において5.2 lbであった。6週での平均変化は、エンドポイントでのものと同程度であった(以下の表)。
【0244】
体重の著しく異常な(>7%)減少の発生率は、ビフェプルノックス処置およびプラセボ群間で同程度であり(20mgビフェプルノックス:6%;30mgビフェプルノックス:5%;プラセボ:5%)、そしてオランザピン処置群において低かった(<1%)。体重の著しく異常な(>7%)増加の発生率においては、これを経験した30mgビフェプルノックスにおける5%、20mgビフェプルノックスにおける1%、そしてプラセボ群における5%と比較してオランザピン処置群では19%で逆のことが見られた。
【0245】
【表50】
【0246】
臨床研究4:
体重およびBMIならびにベースラインに対するその変化を以下の表に要約する。全ての群において、補正した平均体重およびBMIはベースラインから6ヶ月まで減少した。ベースラインから6ヶ月までの補正した平均体重変化(APTS、OC、ANCOVA)は、PBO群において−0.8kg、BX20群において−0.3kg、そしてBX30群において−0.5kgであった。BX20およびBX30群における補正した平均体重減少は、PBO群におけるものと統計的に有意に異ならなかった。
【0247】
減少した体重は、全ての処置群における患者についてTEAEとして報告された(PBO:5%;BX20:8%;BX30:8%)(パネル45)。増加した体重は、PBO群(1.8%)におけるそしてBX30群(1.2%)における患者についてTEAEとして報告された。
【0248】
悪心および/もしくは嘔吐を有する/有さない患者におけるベースラインから6ヶ月までの補正した平均体重変化(APTS、LOCF、ANCOVA)は、悪心および/もしくは嘔吐も有した患者はより大きい体重減少を有したが、全ての群における患者が、悪心および/もしくは嘔吐を有するかどうかにかかわらず体重を減少することを示した(PBO:−0.6対−1.9kg;BX20:−1.0対−1.9kg;BX30:−1.1対−2.3kg)。
【0249】
【表51】
【0250】
【表52】
【0251】
増悪までの時間
臨床研究4:
主要効能変数は増悪までの時間であり、そして分析はFASに基づいた。主要効能分析は、3つの処置群における統合失調症の増悪までの等しい時間の仮説を退けた(p=0.008)。増悪した患者の割合はPBO群において59%、BX20群において41%、そしてBX30群において38%であった。Cox比例ハザードモデルは、PBOに対して0.66(BX20)および0.65(BX30)の概算ハザード比を与え;すなわち、増悪の危険性はBX20もしくはBX30群における患者についてよりもPBO群における患者について約1.5倍高かった。
【0252】
次にBX群の各々とPBO群との対比較により、BX群における患者はPBO群におけ
る患者よりも統合失調症の増悪までの統計的に有意に長い時間を有することが示された(BX20:p=0.008およびBX30:p=0.006)。主要効能分析をPPSについて繰り返した。PPSにおける患者の大部分は研究の大部分に参加したので、得られる概算ハザード比およびp値の両方について、結果は主要分析の結果に非常に近かった。これは、主要効能分析の結論のロバスト性を説明する。
【0253】
【表53】
【0254】
脂質プロフィール
臨床研究4:補正した平均HDLコレステロール値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで増加した(PBO:0.04/0.06(空腹時/非空腹時);BX20:0.07/0.08;BX30:0.07/0.08mmol/L)。BX群のいずれかとPBO群の間で統計的に有意な差はなかった。補正した平均トリグリセリド値は、空腹時/非空腹時条件にかかわらず全ての3つの処置群においてベースラインから6ヶ月まで減少した(PBO:−0.06/−0.22(空腹時/非空腹時);BX20:−0.16/−0.21;BX30:−0.37/−0.03mmol/L)。BX群のいずれか(BX30(空腹時)を除く)とPBO群との間で統計的に有意な差はなかった。
【0255】
補正した平均空腹時グルコース値は全ての3つの処置群においてベースラインから6ヶ月まで増加し(PBO:0.10;BX20:0.13;BX30:0.09mmol/L)、そしてBX群のいずれかとPBO群との間で統計的に有意な差はなかった。
【0256】
以下のパネル50は、空腹時および非空腹時患者におけるベースラインでの(平均値)そして6ヶ月での(ベースラインからの平均変化)脂質プロフィールを要約する。計算される平均総コレステロールおよび平均LDLは、処置および空腹時/非空腹時条件にかかわらず全ての群(非空腹時PBO患者を除く)においてベースラインから6ヶ月まで減少した。計算される平均VLDLおよび平均トリグリセリドは、処置および空腹時/非空腹時条件にかかわらず全ての群(非空腹時BX30患者を除く)においてベースラインから6ヶ月まで減少した。平均HDLは、処置および空腹時/非空腹時条件にかかわらず全ての群においてベースラインから6ヶ月まで増加した。
【0257】
【表54】
【0258】
高血糖および糖尿病
ある場合に著明なそしてケトアシドーシスもしくは高浸透圧性昏睡もしくは死亡と関連する高血糖は、非定型抗精神病薬で処置した患者において報告されている。ビフェプルノックスで処置した患者における高血糖および糖尿病関連有害事象(高血糖、上昇した血糖、耐糖能異常、糖尿病、不十分に制御される糖尿病のような)の発生率は、6週のプラセボ対照試験において0.5%(5/1050)であり、そしてプラセボ処置患者においては0.6%(3/469)であった。26週のプラセボ対照試験において、高血糖もしくは糖尿病関連有害事象を報告した患者はいなかった。非定型抗精神病薬使用とグルコール異常との間の関係の評価は、統合失調症を有する患者における糖尿病の増加した背景リスクの可能性および一般集団における糖尿病の増加する発生率により複雑化される。これらの交絡因子を考えると、非定型抗精神病薬使用と高血糖関連有害事象との間の関係は完全に理解されるわけではない。しかしながら、ビフェプルノックスを含まなかった疫学的研究は、これらの研究に含まれる非定型抗精神病薬で処置した患者における処置下で発現した高血糖関連有害事象の増加した危険性を示唆する。ビフェプルノックスはこれらの研究が行われた時点で市販されていなかったので、ビフェプルノックスがこの増加した危険性と関連するかどうかは知られていない。非定型抗精神病薬で処置した患者における高血糖関連有害事象の正確な危険性概算は利用可能でない。
【0259】
PR、QT、QTc、QRS
臨床研究1:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平
均値の変化はほとんどなかった。これらの間隔の平均変化は、約−2msec〜4msecの間であった。処置群による平均変化の傾向はなかった。ベースラインとエンドポイントとの間の平均心拍数の変化は、−1.5bpm〜0.9bpmの間であった。処置群による平均変化の傾向はなかった。
【0260】
臨床研究2:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平均値の変化はほとんどなかった。処置群にわたる平均変化の範囲は:PR=−2.5msec〜0.2msec;QT=−2.8msec〜7msec;QTc=0.4msec〜3.9msec;QRS=−0.4msec〜1.4msecであった。処置群による平均変化の傾向はなかった。処置群にわたるベースラインとエンドポイントとの間の平均心拍数の変化は、おおよそ−1.3bpm〜1.7bpmの間であった。処置群による平均変化の傾向はなかった。
【0261】
臨床研究3:ベースラインとエンドポイントとの間の平均値の変化を評価することにより経時的なPR、QT、QTc、QRS間隔および心拍数の変化を評価した。これらの要約について、Bazett補正したQT間隔はQTcとして提示された。任意の処置群においてベースラインとエンドポイントとの間のPR、QTcもしくはQRS間隔の平均値の変化はほとんどなかった。これらの間隔の平均変化は、約3msec〜4msecの間であった。処置群による平均変化の傾向はなかった。ベースラインとエンドポイントとの間の平均心拍数の変化は、−1bpm〜1bpmの間であった。処置群による平均変化の傾向はなかった。
【0262】
実施例3a:ビフェプルノックスの薬物動態
本研究の目的は、ビフェプルノックスの薬物動態(PK)を評価することである。健常被験者におけるビフェプルノックスのPKを21の臨床薬理学研究からのPKパラメーターのプールした分析に基づいて調べた。プールした分析には、それぞれ132および399人の被験者への単回および複数回投与後のPKプロフィールが包含され、そして年齢、性別、体重および人種の潜在的影響を調べた。さらに、第II相研究中の376人の患者および第III相研究中の434人の患者からのサンプルに基づく母集団PK法を用いて統合失調症を有する患者におけるPKを調べた。ビフェプルノックスは、経口投与後に迅速に吸収された(全ての用量レベルで1.5〜2時間のtmax)。ビフェプルノックス複数回投与PKは、20〜40mg/日の範囲で用量に比例した。定常状態の平均の見かけのクリアランスおよび見かけの分布容積は、それぞれ、62.2L/hおよび1300Lであった。ビフェプルノックスは、14.4時間の平均血漿定常状態半減期で排出された。標準的な高脂肪食での40mg用量の投与は、tmax(1.5時間)のわずかな遅延ならびにCmax(10%)およびAUC(29%)のわずかな増加と関連した。ビフェプルノックスは、血清タンパク質に約99%結合している。ビフェプルノックスは、CYP2C9、CYP3A4そしてより少ない程度にCYP2D6により代謝される。ビフェプルノックス暴露は、フルコナゾール(CYP2C9阻害剤)そしてわずかな程度にケトコナゾール(CYP3A4阻害剤)との共投与により増加されたが、パロキセチン(CYP2D6阻害剤)およびファモチジン(H2アンタゴニスト)との共投与により増加されなかった。ビフェプルノックス暴露は、カルバマゼピン(CYP3A4誘導剤)の共投与により軽減された。ビフェプルノックスと狭い治療指数化合物ワーファリンおよびリチウムとの共投与(実施例3bを参照)は、いかなる関連する程度にもこれらの化合物のPKに影響を及ぼさなかった。CYP2C9遅い/中間代謝個体において、正常酵素活性を有する被験者におけるよりも高い血漿レベルのビフェプルノックスが認められた。[14C]で標識したビフェプルノックスの単回経口投与後に、放射活性の13%および74%はそれぞれ尿および便において排出された。ビフェプルノックスPKへの臨床的に有意な
年齢、性別、体重もしくは人種に関連する効果は認められなかった。統合失調症を有する患者におけるPKは、健常被験者においてみられるものと同様であった。
【0263】
本研究の1つの結論は、ビフェプルノックスが経口投与後に迅速に吸収され;平均排出半減期が約14時間であることである。複数回投与PKは、20〜40mgの範囲で用量に比例した。ビフェプルノックスは、低い相互作用ポテンシャルを有する。
【0264】
実施例3b:健常男性被験者におけるリチウムおよびビフェプルノックスの薬物動態学的相互作用
目的:ドーパミンD2および5−HT1A受容体の部分アゴニスト、ビフェプルノックスは、統合失調症の処置用に開発されている。ビフェプルノックスは、精神病および気分障害を有する患者の処置のために気分安定剤リチウムと組み合わせて用いることができるので、リチウムの薬物動態(PK)プロフィールへのビフェプルノックスの複数回投与の影響を評価した。リチウムは、治療を複雑にし得る狭い治療指数を有し、そして1.5mmol/Lより大きい血清レベルは、より低いレベルよりリチウム毒性のより大きい危険性を有する。方法:これは、48人の健常男性被験者における単一施設二重盲検無作為化プラセボ対照並列設計研究であった。全ての被検者は、1〜8日にそして血清レベルが5日〜7日に安定であっったならば9〜20日に再び、毎日2回非盲検でリチウム(450mg)を服用する予定であった。血清レベルが5〜7日に安定であった被験者は、毎日2回リチウム450mgに加えて、9〜17日に毎日1回プラセボもしくは上昇する用量のビフェプルノックス(0.025〜40mg)を、そして18〜21日にプラセボもしくはビフェプルノックス40mgを服用するように無作為抽出されたものに含まれた。リチウム450mgの単回朝用量のみを21日に投与した。投与間隔(0〜t)にわたるリチウム、定常状態Cmax、AUCおよび腎クリアランス(CL−R)値を8日に測定したリチウムのベースライン値を共変量としてANCOVAを用いてビフェプルノックスとプラセボ群との間で比較した。結果:ビフェプルノックス群においてリチウムの平均CmaxおよびAUC(0〜t)のわずかな増加があったが、AUC、CmaxおよびCL−Rの2つの処置の幾何最小二乗平均の比および90%信頼区間(CI)は0.80〜1.25の所定範囲内であった。Cmax、AUC(0〜t)およびCL−Rの処置比および90%CIは、それぞれ1.11(90%CI:1.01〜1.20)、1.13(90%CI:1.06〜1.21)および0.94(90%CI:0.87〜1.02)であった。1日あたり40mgまでのビフェプルノックスおよび毎日2回リチウム450mgの組み合わせた投与は、良好な耐容性を示した。結論:リチウム定常状態薬物動態へのビフェプルノックスの複数回投与の共投与の臨床的に関連する影響はなかった。結果は、ビフェプルノックスとの同時投与中にリチウムの投薬量調整は必要とされないことを示唆する。
【0265】
実施例4:急性増悪統合失調症を有する患者の処置におけるビフェプルノックスの効能および安全性
目的:統合失調症を有する急性疾患患者の処置におけるビフェプルノックスの効能および安全性を評価すること。
【0266】
評価:6週の無作為化プラセボ対照リスペリドン参照用量設定研究には、統合失調症(DSM−IV−TR)の急性増悪を有する589人の無作為抽出患者が含まれた。患者は、ビフェプルノックス5mg(n=115)、ビフェプルノックス10mg(n=120)、ビフェプルノックス20mg(n=115)、プラセボ(n=119)もしくはリスペリドン6mg(n=120)に無作為に割り当てられた。全てのビフェプルノックス用量での処置は目標用量、1日に0.125mgの用量から開始して、2日に0.25mg、3日に0.5mg、4日に1mg、5日に2mgそして6日に5mg、7日に10mgもしくは8日に20mgまで滴定され、一方、リスペリドンでの処置は3日にわたって滴定された。ベースラインからエンドポイントまでの陽性および陰性症状評価尺度(PAN
SS)合計スコアの変化は、主要アウトカム評価尺度であった。副次的効能評価尺度には:PANSS陽性、PANSS陰性、PANSS総合精神病理(GPP)スコア、PANSS由来の簡易精神症状評価尺度(BPRS)スコア、臨床全般印象疾患重症度(CGI−S)、CGI−I改善度(CGI−I)スコアならびにレスポンダー率が包含された。安全性および耐容性評価には、錐体外路症状(EPS)、体重増加、脂質プロフィールおよび血清プロラクチンが包含された。リスペリドンは、アッセイ感度用に含まれた。
【0267】
結果:ビフェプルノックスのPANSS合計スコアの減少は、20mg用量について3週および6週/エンドポイントでプラセボと比較して統計的に有意であった(P<0.05)。ビフェプルノックス20mgのプラス効果はまた、副次的効能評価尺度PANSS陽性、陰性、GPP下位尺度、BPRSおよびレスポンダー率に対しても認められた。リスペリドン6mgは、全ての効能評価尺度についてプラセボと比較してエンドポイントで統計的に有意であった。最も一般的な有害事象(発生率>5%およびプラセボに対して2倍)には:消化不良、悪心、嘔吐および便秘が包含された。用量関係は、最も頻繁な有害事象のいずれについても明らかではなかった。ビフェプルノックスは、減少したプロラクチンレベルおよびプラセボと同程度であるEPSの割合と関連した。さらに、ビフェプルノックスを服用する患者は、統計的に有意な(P<0.05)体重減少を経験し、そしてプラセボと比較して非空腹時トリグリセリド(P<0.005)および総コレステロール(P<0.005)の統計的に有意な改善を示した。
【0268】
結論:本研究において、ビフェプルノックス20mgは急性統合失調症の処置において有効であることが示された。ビフェプルノックスは、体重の減少および脂質プロフィールの改善に起因する安全性利点を有し得る。
【特許請求の範囲】
【請求項1】
用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、CNS障害を有する患者の処置のためのビフェプルノックスの日用量。
【請求項2】
統合失調症を有する患者の処置のための請求項1の用量。
【請求項3】
用量が毎日1回の用量である請求項1もしくは2の用量。
【請求項4】
統合失調症を有する患者の長期処置のための請求項1〜3のいずれか1項の用量。
【請求項5】
統合失調症を有する患者における臨床的安定を維持するための請求項1〜4のいずれか1項の用量。
【請求項6】
用量が20mgの少なくとも1種のビフェプルノックス化合物である請求項5の用量。
【請求項7】
用量が30mgの少なくとも1種のビフェプルノックス化合物である請求項5の用量。
【請求項8】
急性増悪統合失調症を有する患者の処置のための請求項1〜3のいずれか1項の用量。
【請求項9】
用量が20mgの少なくとも1種のビフェプルノックス化合物である請求項8の用量。
【請求項10】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項1〜9のいずれか1項の用量。
【請求項11】
ビフェプルノックス化合物がメシル酸ビフェプスノックスのアルファ多形である請求項10の用量。
【請求項12】
統合失調症を有する患者の処置における使用のための30mgの少なくとも1種のビフェプルノックス化合物および少なくとも1種の製薬学的に許容しうる担体を含んでなる製薬学的組成物。
【請求項13】
統合失調症を有する患者の処置における使用のための20mgの少なくとも1種のビフェプルノックス化合物および少なくとも1種の製薬学的に許容しうる担体を含んでなる製薬学的組成物。
【請求項14】
統合失調症を有する患者の長期処置における使用のための請求項12もしくは13の製薬学的組成物。
【請求項15】
統合失調症を有する患者における臨床的安定を維持するために使用する請求項12〜14のいずれか1項の製薬学的組成物。
【請求項16】
急性増悪統合失調症を有する患者の処置のために使用する請求項12もしくは13の製薬学的組成物。
【請求項17】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項12〜16のいずれか1項の製薬学的組成物。
【請求項18】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項17の製薬学的組成物。
【請求項19】
統合失調症を有する患者の長期処置における使用のためのビフェプルノックス。
【請求項20】
安定した統合失調症を有する患者の処置における請求項19に記載の使用のためのビフェプルノックス。
【請求項21】
安定した統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【請求項22】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項19〜21のいずれか1項の使用のためのビフェプルノックス。
【請求項23】
20mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項22の使用のためのビフェプルノックス。
【請求項24】
30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項22の使用のためのビフェプルノックス。
【請求項25】
急性増悪統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【請求項26】
20mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項25の使用のためのビフェプルノックス。
【請求項27】
30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項25の使用のためのビフェプルノックス。
【請求項28】
少なくとも1種のビフェプルノックス化合物が気分安定剤リチウムと組み合わせて投与される請求項19〜27のいずれか1項の使用のためのビフェプルノックス。
【請求項29】
少なくとも1種のビフェプルノックス化合物が気分安定剤リチウムと組み合わせて投与される精神病および気分障害を有する患者の処置における使用のためのビフェプルノックス。
【請求項30】
少なくとも1種のビフェプルノックス化合物が抗欝剤と組み合わせて投与される請求項19〜27のいずれか1項の使用のためのビフェプルノックス。
【請求項31】
抗欝剤がパロキセチンである請求項30の使用のためのビフェプルノックス。
【請求項32】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項19〜31のいずれか1項のビフェプルノックス。
【請求項33】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項32のビフェプルノックス。
【請求項34】
少なくとも1種のビフェプルノックス化合物を含んでなる組成物および気分安定剤リチウムを含んでなる組成物を含んでなる精神病および気分障害を有する患者の処置のためのキット。
【請求項35】
少なくとも1種のビフェプルノックス化合物を含んでなる組成物および抗鬱剤を含んでなる組成物を含んでなるCNS障害を有する患者の処置のためのキット。
【請求項36】
抗欝剤がパロキセチンである請求項35のキット。
【請求項37】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項34〜36のいずれか1項のキット。
【請求項38】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項37のキット。
【請求項39】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与され、そして該処置が好ましい代謝プロフィールをもたらす、統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項40】
統合失調症を有する患者のビフェプルノックスでの処置が、体重増加、トリグリセリドレベルおよび/もしくは総コレステロールレベルの障害、高血糖および/または1つもしくはそれ以上の糖尿病関連有害事象の発生から選択される副作用を防ぎそして/もしくは軽減する請求項40の使用。
【請求項41】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される、安定した統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項42】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される、急性増悪統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項43】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項39〜42のいずれか1項の使用。
【請求項44】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項43の使用。
【請求項45】
体重の問題を有するかもしくは体重の問題を起こしやすい統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【請求項1】
用量が20〜30mgの少なくとも1種のビフェプルノックス化合物である、CNS障害を有する患者の処置のためのビフェプルノックスの日用量。
【請求項2】
統合失調症を有する患者の処置のための請求項1の用量。
【請求項3】
用量が毎日1回の用量である請求項1もしくは2の用量。
【請求項4】
統合失調症を有する患者の長期処置のための請求項1〜3のいずれか1項の用量。
【請求項5】
統合失調症を有する患者における臨床的安定を維持するための請求項1〜4のいずれか1項の用量。
【請求項6】
用量が20mgの少なくとも1種のビフェプルノックス化合物である請求項5の用量。
【請求項7】
用量が30mgの少なくとも1種のビフェプルノックス化合物である請求項5の用量。
【請求項8】
急性増悪統合失調症を有する患者の処置のための請求項1〜3のいずれか1項の用量。
【請求項9】
用量が20mgの少なくとも1種のビフェプルノックス化合物である請求項8の用量。
【請求項10】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項1〜9のいずれか1項の用量。
【請求項11】
ビフェプルノックス化合物がメシル酸ビフェプスノックスのアルファ多形である請求項10の用量。
【請求項12】
統合失調症を有する患者の処置における使用のための30mgの少なくとも1種のビフェプルノックス化合物および少なくとも1種の製薬学的に許容しうる担体を含んでなる製薬学的組成物。
【請求項13】
統合失調症を有する患者の処置における使用のための20mgの少なくとも1種のビフェプルノックス化合物および少なくとも1種の製薬学的に許容しうる担体を含んでなる製薬学的組成物。
【請求項14】
統合失調症を有する患者の長期処置における使用のための請求項12もしくは13の製薬学的組成物。
【請求項15】
統合失調症を有する患者における臨床的安定を維持するために使用する請求項12〜14のいずれか1項の製薬学的組成物。
【請求項16】
急性増悪統合失調症を有する患者の処置のために使用する請求項12もしくは13の製薬学的組成物。
【請求項17】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項12〜16のいずれか1項の製薬学的組成物。
【請求項18】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項17の製薬学的組成物。
【請求項19】
統合失調症を有する患者の長期処置における使用のためのビフェプルノックス。
【請求項20】
安定した統合失調症を有する患者の処置における請求項19に記載の使用のためのビフェプルノックス。
【請求項21】
安定した統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【請求項22】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項19〜21のいずれか1項の使用のためのビフェプルノックス。
【請求項23】
20mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項22の使用のためのビフェプルノックス。
【請求項24】
30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項22の使用のためのビフェプルノックス。
【請求項25】
急性増悪統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【請求項26】
20mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項25の使用のためのビフェプルノックス。
【請求項27】
30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される請求項25の使用のためのビフェプルノックス。
【請求項28】
少なくとも1種のビフェプルノックス化合物が気分安定剤リチウムと組み合わせて投与される請求項19〜27のいずれか1項の使用のためのビフェプルノックス。
【請求項29】
少なくとも1種のビフェプルノックス化合物が気分安定剤リチウムと組み合わせて投与される精神病および気分障害を有する患者の処置における使用のためのビフェプルノックス。
【請求項30】
少なくとも1種のビフェプルノックス化合物が抗欝剤と組み合わせて投与される請求項19〜27のいずれか1項の使用のためのビフェプルノックス。
【請求項31】
抗欝剤がパロキセチンである請求項30の使用のためのビフェプルノックス。
【請求項32】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項19〜31のいずれか1項のビフェプルノックス。
【請求項33】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項32のビフェプルノックス。
【請求項34】
少なくとも1種のビフェプルノックス化合物を含んでなる組成物および気分安定剤リチウムを含んでなる組成物を含んでなる精神病および気分障害を有する患者の処置のためのキット。
【請求項35】
少なくとも1種のビフェプルノックス化合物を含んでなる組成物および抗鬱剤を含んでなる組成物を含んでなるCNS障害を有する患者の処置のためのキット。
【請求項36】
抗欝剤がパロキセチンである請求項35のキット。
【請求項37】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項34〜36のいずれか1項のキット。
【請求項38】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項37のキット。
【請求項39】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与され、そして該処置が好ましい代謝プロフィールをもたらす、統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項40】
統合失調症を有する患者のビフェプルノックスでの処置が、体重増加、トリグリセリドレベルおよび/もしくは総コレステロールレベルの障害、高血糖および/または1つもしくはそれ以上の糖尿病関連有害事象の発生から選択される副作用を防ぎそして/もしくは軽減する請求項40の使用。
【請求項41】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される、安定した統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項42】
20〜30mgの少なくとも1種のビフェプルノックス化合物の日用量が患者に投与される、急性増悪統合失調症を有する患者の処置用の薬剤の製造のためのビフェプルノックスの使用。
【請求項43】
ビフェプルノックス化合物がメシル酸ビフェプルノックスである請求項39〜42のいずれか1項の使用。
【請求項44】
ビフェプルノックス化合物がメシル酸ビフェプルノックスのアルファ多形である請求項43の使用。
【請求項45】
体重の問題を有するかもしくは体重の問題を起こしやすい統合失調症を有する患者の処置における使用のためのビフェプルノックス。
【図1】

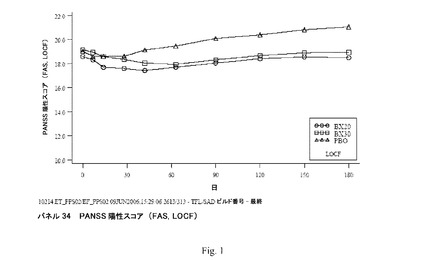
【図2】
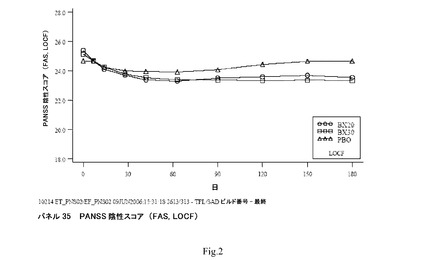
【図3】
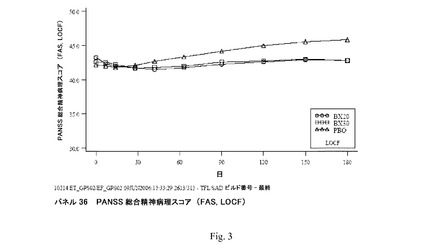
【図4】
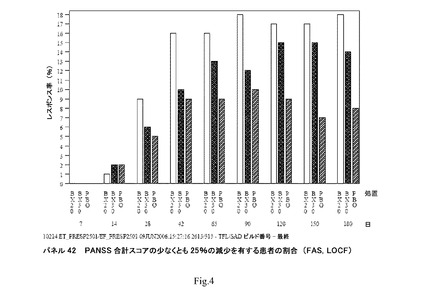
【図5】
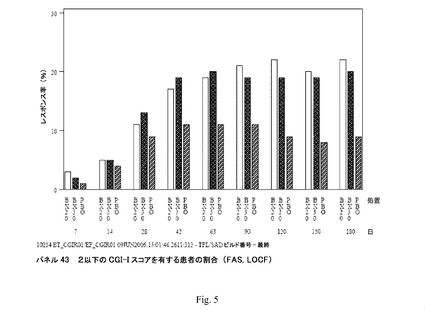
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2010−501626(P2010−501626A)
【公表日】平成22年1月21日(2010.1.21)
【国際特許分類】
【出願番号】特願2009−526089(P2009−526089)
【出願日】平成19年8月29日(2007.8.29)
【国際出願番号】PCT/EP2007/058958
【国際公開番号】WO2008/025781
【国際公開日】平成20年3月6日(2008.3.6)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【出願人】(509058449)
【Fターム(参考)】
【公表日】平成22年1月21日(2010.1.21)
【国際特許分類】
【出願日】平成19年8月29日(2007.8.29)
【国際出願番号】PCT/EP2007/058958
【国際公開番号】WO2008/025781
【国際公開日】平成20年3月6日(2008.3.6)
【出願人】(501439149)ソルベイ・フアーマシユーチカルズ・ベー・ブイ (71)
【出願人】(509058449)
【Fターム(参考)】
[ Back to top ]