
縮合多環化合物及びこれを用いた有機発光素子
【課題】発光効率が高く駆動電圧の低い有機発光素子を提供する。
【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層とから構成され、前記有機化合物層に下式で示される縮合多環化合物のαの位置に3−(ジベンゾチオフェン−1−イル)フェニル基等が置換した縮合多環誘導体が含まれることを特徴とする、有機発光素子。

【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層とから構成され、前記有機化合物層に下式で示される縮合多環化合物のαの位置に3−(ジベンゾチオフェン−1−イル)フェニル基等が置換した縮合多環誘導体が含まれることを特徴とする、有機発光素子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、縮合多環化合物及びこれを用いた有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、陽極と陰極と、これら両電極間に配置される有機化合物層とを有する電子素子である。各電極からそれぞれ注入させる正孔(ホール)及び電子が、有機化合物層、特に発光層内で再結合することで生成する励起子が基底状態に戻る際に、有機発光素子は光を放出する。
【0003】
有機発光素子の最近の進歩は著しく、その特徴として、低駆動電圧、多様な発光波長、高速応答性、発光デバイスの薄型化・軽量化が可能であることが挙げられる。
【0004】
ところで有機発光素子は、発光に関与する励起子の種類によって蛍光発光素子と燐光発光素子とに大別される。そのうち燐光発光素子では三重項励起子が発光に関与し、有機発光素子を構成する有機化合物層、具体的には発光層中に燐光発光材料を有する電子素子である。ここで燐光発光材料は、正孔及び電子の再結合によって三重項状態に励起され、基底状態に戻る際に燐光を放出する。このため燐光発光素子は、この三重項励起子由来の発光が得られる有機発光素子である。
【0005】
ところで、燐光発光素子の内部量子収率は、理論上蛍光発光素子の内部量子収率の4倍になるので、近年では燐光発光素子に注目が集まっている。しかし燐光発光素子において、発光効率についてはさらなる改善の余地がある。
【0006】
一方、燐光発光素子で使用される材料について様々な提案がなされている。例えば、非特許文献1及び特許文献1に示される下記に示される部分構造を有する化合物が提案されている。
【0007】
【化1】
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−290991号公報
【非特許文献】
【0009】
【非特許文献1】Journal of Organic Chemistry 2006,71,6822−6828
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、上記課題を解決するためになされたものであり、その目的は、発光効率が高く駆動電圧の低い有機発光素子を提供することにある。
【課題を解決するための手段】
【0011】
本発明の縮合多環化合物は、下記一般式[1]及至[4]のいずれかで示されることを特徴とする。
【0012】
【化2】
(式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。)
【発明の効果】
【0013】
本発明によれば、発光効率が高く駆動電圧の低い有機発光素子を提供することができる。
【図面の簡単な説明】
【0014】
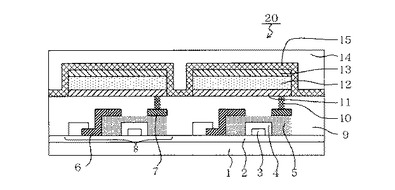
【図1】本発明の有機発光素子と、この有機発光素子に電気接続されるスイッチング素子の一例であるTFT素子とを有する表示装置の例を示す断面模式図である。
【発明を実施するための形態】
【0015】
まず本発明の縮合多環化合物について説明する。本発明の縮合多環化合物は、下記一般式[1]及至[4]のいずれかで示される化合物である。
【0016】
【化3】
【0017】
式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。
【0018】
上記フェニル基、ジベンゾチオフェニル基、フェナンスリル基、フルオレニル基、トリフェニレニル基及びナフチル基が有してもよい置換基として、メチル基、エチル基、プロピル基等のアルキル基、フェニル基、フルオレニル基、フェナンスリル基、トリフェニレニル基、ナフチル基等のアリール基等が挙げられる。
【0019】
式[1]及至[4]において、R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。
【0020】
R1及至R6で表されるアルキル基として、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基及びtert−ブチル基が挙げられる。
【0021】
式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。
【0022】
式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。
【0023】
式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。
【0024】
本発明の縮合多環化合物は、例えば、以下に示す合成ルートに従って合成することができる。
【0025】
【化4】
【0026】
上記合成スキームにおいて、中間体の1つである化合物d−8は、本発明の縮合多環化合物の母骨格を有する化合物である。ここで化合物d−8は、例えば、トリフェニレン(化合物d−1)を出発原料として、下記(i)乃至(v)のプロセスにより合成される。
(i)トリフェニレンの臭素化(化合物d−2の合成)
(ii)臭化トリフェニレンのピナコールボロン化(化合物d−4の合成)
(iii)(ii)にて合成されるトリフェニレニルボロン酸エステル(化合物d−4)と、ブロモクロロ安息香酸メチル(化合物d−5)との鈴木−宮浦カップリング反応(化合物d−6の合成)
(iv)グリニャール反応(化合物d−7の合成)
(v)ポリリン酸による脱水環化反応(化合物d−8の合成)
【0027】
一方、化合物d−5は、式[1]及至[4]の化合物をそれぞれ合成する際の原料として有効なクロロ体(化合物d−8)を合成する際に重要な化合物である。尚、上記合成スキームにて表される中間体d−5はベンゼン環の4位の位置に塩素原子を有しているが、この塩素原子が他のハロゲン原子に置換されていてもよいし、この塩素原子がトリフラート基又はピナコールボロン酸基に置換されていてもよい。
【0028】
次に、本発明の縮合多環化合物の特性について説明する。下記に示される(a−1)は、本発明の縮合多環化合物の母骨格となる化合物である。
【0029】
【化5】
上記化合物(a−1)に類似する化合物として、下記に示される化合物(a−2)がある。
【0030】
【化6】
【0031】
ここで化合物(a−1)は、化合物(a−2)と比べると、分子会合性が低いという性質を有する。化合物(a−1)及び化合物(a−2)は、いずれもトリフェニレン骨格とジメチルインデン骨格とが縮合することで形成される骨格であるが、フルオレン環に縮合化する方向が異なることによりこの特徴が生まれる。即ち、本発明の縮合多環化合物の母骨格となる化合物(a−1)は、分子会合性の強いトリフェニレン骨格と2つのメチル基との距離が化合物(a−2)よりも近い。このため化合物(a−1)同士が会合しようとすると、所定の分子が有するメチル基が別の分子が有するトリフェニレン骨格同士のスタッキングを抑制するため分子会合性が低くなるといえる。
【0032】
分子会合性が低いことは、分子会合による濃度消光やエキサイマー発光を抑えることにつながるため、化合物の発光特性に関して有利に働く。
【0033】
次に、化合物(a−1)と化合物(a−2)に導入される置換基の置換位置の違いについて述べる。
【0034】
本発明の縮合多環化合物は、下記(a−1)で示される化合物を母骨格として、置換基が当該母骨格のα位に導入されるという特徴を有する。
【0035】
【化7】
【0036】
この特徴を有することにより、トリフェニレン環とベンゼン環とで形成される共役が置換基を介してさらに延伸されることがない、即ち、母骨格と置換基との間で母骨格が有する共役が切れるという特徴を有する。これにより、本発明の縮合多環化合物の最低三重項励起状態のエネルギー(T1エネルギー)は、化合物の母骨格である(a−1)で決定されるものであり、高いT1エネルギーが維持される。
【0037】
一方、下記構造(a−2)で示される化合物を母骨格として、置換基が当該母骨格のβ位に置換基を有する化合物は、トリフェニレン環とベンゼン環とで形成される共役が置換基を介してさらに延伸されることに特徴を有する。
【0038】
【化8】
【0039】
この特徴を有することにより、a−2の最低三重項励起状態のエネルギー(T1エネルギー)は、a−2とβ位で置換する置換基との相互作用(共役の延伸)で決定されるものとなり、本発明の縮合多環化合物に比べて低いT1エネルギーになる。
【0040】
ここで発明者らは、下記に示される化合物についてトルエン希薄溶液中でのT1エネルギーを測定した。尚、T1の測定はトルエン溶液(1×10-4mol/l)を77Kに冷却した上で、励起波長350nmにて燐光発光スペクトルを測定し、第一発光ピークをT1として用いた。装置は日立製分光光度計U−3010を用いた。
【0041】
【化9】
【0042】
【表1】
【0043】
表1より、本発明の縮合多環化合物である化合物D−1のT1は、自身の部分骨格であるa−1と同じである。このことから2つのa−1骨格同士の共役が切れていることが示される。
【0044】
一方、比較化合物に相当する化合物F−1のT1は、自身の部分骨格であるa−2よりも大きく長波長化している。このことから2つのa−2骨格同士の共役がつながっていることが示される。
【0045】
ところで、式[1]及至[4]で示されるArは、T1が高いアリール基が好ましく、530nm以下のアリール基より選ばれる。具体的には、ベンゼン、ジベンゾチオフェン、フェナンスレン、フルオレン、トリフェニレン及びナフタレンから選ばれる。尚、Arで表されるアリール基は、さらに置換基を有してもよい。
【0046】
以上により、高いT1を持つ母骨格a−1と、Ar基の所定の位置の置換により、本発明の縮合多環化合物はそのT1が470nm以上500nm以下の範囲になっている。
【0047】
以上の作用効果により、本発明の縮合多環化合物は、有機発光素子用材料、特に、発光材料として用いた場合、高効率の発光素子を提供することができる。
【0048】
ところで、緑色に燐光発光する燐光発光材料のT1は490nm以上530nm以下であり、本発明の縮合多環化合物はそれよりも高いT1エネルギーを有する。従って、本発明の縮合多環化合物は、緑色の燐光を発する有機発光素子の発光層のホストもしくは電子輸送材料として用いると、素子の発光効率を向上させることができる。この場合、燐光発光する化合物は、発光層のゲスト(燐光発光材料)である。
【0049】
本発明の縮合多環化合物は、母骨格であるa−1にArで示されるアリール基あるいはa−1が所定の位置に結合されていることを特徴とする。ここで母骨格であるa−1にArが結合することにより、分子全体の平面性が崩れることで安定なアモルファス膜を形成することに効果がある。
【0050】
従って、本発明の縮合多環化合物を有機発光素子用材料として用いた場合、耐久性が向上された発光素子を提供することができる。
【0051】
本発明の縮合多環化合物の具体例を以下に示す。ただし、本発明はこれらに限られるものではない。
【0052】
【化10】
【0053】
【化11】
【0054】
上記具体例において、A群に属する化合物は、式[1]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがフェニレン基で連結されている化合物である。ここでA群に属する化合物は分子量が小さいので、蒸着によって化合物の薄膜を成膜する際に、蒸着温度を低くすることができる。
【0055】
上記具体例において、B群に属する化合物は、式[2]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがビフェニレン基で連結されている化合物である。ここでB群に属する化合物は分子中での回転を可能にする結合が多いため、アモルファス膜を形成したときの膜の安定性が高い。
【0056】
上記具体例において、C群に属する化合物は、式[3]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがフルオレニレン基で連結されている化合物である。ここで母骨格(a−1)とアリール基とを連結するフルオレニレン基は剛直であるため、アモルファス膜を成膜した際にその膜の電子及び正孔の移動度が高くなる。
【0057】
上記具体例において、D群に属する化合物は、式[4]で示される化合物群である。即ち、母骨格(a−1)のダイマー体である。ここでD群に属する化合物は、分子の対称性が高いため、アモルファス膜を成膜した際にその膜の電子及び正孔の移動度が高くなる。
【0058】
ここで上記具体例にて示される化合物のうち、正孔注入・輸送性が高いという観点から、式[1]乃至[4]にて示されるArがジベンゾチオフェンである化合物、具体的にはA−8、B−2及びC−4は好ましい材料であるといえる。
【0059】
次に、本発明の有機発光素子について説明する。
【0060】
本発明の有機発光素子は、一対の電極である陽極と陰極と、これら陽極と陰極との間に配置される有機化合物層と、から構成される。
【0061】
本発明において、有機発光素子の構成部材である有機化合物層は、発光層又は発光機能を有する層が含まれていれば、単一の層であってもよいし、複数の層からなる積層体であってもよい。
【0062】
有機化合物層が複数の層からなる場合、発光層(又は発光機能を有する層)以外の層であって、かつ有機化合物層に含まれる層として、ホール注入層、ホール輸送層、発光層、ホールブロック層、電子輸送層、電子注入層、エキシトンブロック層等が挙げられる。もちろん、上記群の中から1又は複数を選択して、かつそれらを組み合わせて用いることができる。
【0063】
尚、本発明の有機発光素子の構成はこれらに限定されるものではない。例えば、電極と有機化合物層界面に絶縁性層、接着層あるいは干渉層を設ける、電子輸送層もしくはホール輸送層をイオン化ポテンシャルの異なる二層から構成される等の多様な層構成をとることができる。
【0064】
本発明の有機発光素子において、素子形態としては、基板とは反対側の電極から光を取り出すいわゆるトップエミッション方式でもよいし、基板側から光を取り出すいわゆるボトムエミッション方式でもよい。また基板や電極を透明な材料にして両面から光を取り出す構成にしてもよい。
【0065】
本発明の有機発光素子において、本発明の縮合多環化合物は、有機化合物層に含まれている。本発明の有機発光素子において、本発明の縮合多環化合物を有している有機化合物層は、特に限定されないが、好ましくは、発光層に含まれる。本発明の有機発光素子において、発光層は、本発明の縮合多環化合物のみからなる層であってもよいが、好ましくは、ホストとゲストとからなる層である。
【0066】
ここで本発明の縮合多環化合物は、発光層のホストとして使用してもよいし、ゲストとして使用してもよいが、好ましくは、発光層のホストとして使用する。ここで本発明の縮合多環化合物を燐光発光するゲストと組み合わせて使用されるホストとして使用すると、発光効率の観点から好ましい。特に、本発明の縮合多環化合物は、490nm乃至660nmの領域に発光ピークを持つ発光色が緑乃至赤であるゲストと組み合わせると、三重項エネルギーのロスが少ないため、発光素子の効率が高くなる。
【0067】
尚、本発明の縮合多環化合物をゲストとして使用する場合、ホストに対するゲストの濃度は、発光層の全体量に対して0.1重量%以上30重量%以下であることが好ましく、0.5重量%以上10重量%以下であることがより好ましい。
【0068】
本発明の有機発光素子は、本発明の縮合多環化合物以外にも、必要に応じて他の化合物を構成材料として使用することができる。具体的には、従来公知の低分子系及び高分子系の正孔注入・輸送性材料、ホスト、ゲストあるいは電子注入・輸送性材料等を一緒に使用することができる。
【0069】
以下にこれらの化合物例を挙げる。
【0070】
正孔注入・輸送性材料としては、正孔移動度が高い材料であることが好ましい。正孔注入性能あるいは正孔輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられるが、もちろんこれらに限定されるものではない。
【0071】
ホストとしては、トリアリールアミン誘導体、フェニレン誘導体、縮合環芳香族化合物(例えばナフタレン誘導体、フェナントレン誘導体、フルオレン誘導体、クリセン誘導体、等)、有機金属錯体(例えば、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、有機イリジウム錯体、有機プラチナ錯体等)及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体、ポリ(チエニレンビニレン)誘導体、ポリ(アセチレン)誘導体等の高分子
誘導体が挙げられるが、もちろんこれらに限定されるものではない。
【0072】
ゲストとしては、燐光発光材料が好ましい。具体的には、以下に示す、燐光発光性のIr錯体や、プラチナ錯体等が挙げられる。
【0073】
【化12】
【0074】
また、蛍光発光性のドーパントを用いることもでき、縮環化合物(例えばフルオレン誘導体、ナフタレン誘導体、ピレン誘導体、ペリレン誘導体、テトラセン誘導体、アントラセン誘導体、ルブレン等)、キナクリドン誘導体、クマリン誘導体、スチルベン誘導体、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体等の高分子誘導体が挙げられる。
【0075】
電子注入・輸送性材料としては、ホール注入性材料あるいはホール輸送性材料のホール移動度とのバランス等を考慮し選択される。電子注入性能あるいは電子輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられるが、もちろんこれらに限定されるものではない。
【0076】
陽極の構成材料としては、仕事関数がなるべく大きなものがよい。例えば、金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれら金属単体を複数種組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物である。また、ポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーでもよい。これらの電極物質は一種類を単独で使用してもよいし複数種類を併用して使用してもよい。また、陽極は一層構成でもよいし、多層構成でもよい。
【0077】
一方、陰極を構成する材料としては、仕事関数の小さなものがよい。例えば、リチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えば、マグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、複数種類を併用して使用してもよい。また、陰極は一層構成でもよいし、多層構成でもよい。
【0078】
本発明の有機発光素子において、本発明の縮合多環化合物が含まれている層及びその他の層については、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により層を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で形成する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0079】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコーン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として1種単独で使用してもよいし、2種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0080】
本発明の有機発光素子は、表示装置や照明装置に用いることができる。他にも電子写真方式の画像形成装置の露光光源や液晶表示装置のバックライト等がある。
【0081】
表示装置は本発明の有機発光素子を表示部に有する。この表示部は複数の画素を有する。この画素は本実施形態に係る有機発光素子と発光輝度を制御するためのスイッチング素子の一例としてTFT素子とを有し、この有機発光素子の陽極又は陰極とTFT素子のドレイン電極又はソース電極とが接続されている。表示装置はPC等の画像表示装置として用いることができる。
【0082】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの情報を入力する画像入力部を有し、入力された画像を表示部に出力する画像出力装置でもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報に基づいて画像を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。また表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0083】
次に、本実施形態に係る有機発光素子を使用した表示装置について図1を用いて説明する。
【0084】
図1は、本発明の有機発光素子と、この有機発光素子に電気接続されるスイッチング素子の一例であるTFT素子とを有する表示装置の例を示す断面模式図である。図1の表示装置20には有機発光素子とTFT素子との組が2組図示されている。構造の詳細を以下に説明する。
【0085】
図1の表示装置20は、ガラス等の基板1とその上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属のゲート電極3である。符号4はゲート絶縁膜4であり、5は半導体層である。
【0086】
TFT素子8は半導体層5とドレイン電極6とソース電極7とを有している。TFT素子8の上部には絶縁膜9が設けられている。コンタクトホール10を介して有機発光素子の陽極11とソース電極7とが接続されている。表示装置はこの構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子ソース電極又はドレイン電極のいずれか一方とが接続されていればよい。
【0087】
尚、図1の表示装置20において、有機化合物層12は、多層あるいは単層の有機化合物層を1つの層の如く図示をしている。陰極13の上には有機発光素子の劣化を抑制するための第一の保護層14や第二の保護層15が設けられている。
【0088】
本実施形態に係る表示装置においてスイッチング素子に特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0089】
以下、実施例において本発明を詳細に説明する。ただし本発明はこれらに限定されるものではない。
【0090】
[実施例1]例示化合物A−8の合成
以下に示す合成スキームにより合成した。
【0091】
【化13】
【0092】
(1)化合物d−2の合成
500mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−1:9.99g(43.8mmol)
ジクロロメタン:300ml
【0093】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、臭素7.7g(48.2mmol)とジクロロメタン7.0mlとの混合溶液を滴下した。当該混合溶液を滴下した後、反応溶液を室温で12時間攪拌した。反応終了後、チオ硫酸ナトリウム溶液中に反応溶液をあけた後、有機層をクロロホルムで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、化合物d−2を白色固体として11.6g(収率86.3%)得た。
【0094】
(2)化合物d−4の合成
300mlの三ツ口フラスコ内を窒素雰囲気にした後、下記に示す試薬、溶媒を投入した。
化合物d−2:11.5g(37.4mmol)
化合物d−3:11.4g(44.9mmol)
酢酸カリウム:6.61g(67.4mmol)
ジオキサン:100ml
【0095】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、ビス(ジフェニルフォスフィノ)フェロセンパラジウム(II)ジクロライド・ジクロロメタン1.53g(1.87mmol)を添加した。次に、反応溶液を100℃に昇温し、この温度(100℃)で4時間攪拌した。反応終了後、反応溶液中の溶媒を減圧留去して粗生成物を得た。次に、この粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−ヘプタン混合溶媒)で精製することにより、化合物d−4を白色固体として10.42g(収率68.6%)得た。
【0096】
(3)化合物d−6の合成
200mlの三ツ口フラスコ内を窒素雰囲気にした後、下記に示す試薬、溶媒を投入した。
化合物d−4:7.08g(20.0mmol)
化合物d−5:5.46g(22.0mmol)
炭酸ナトリウム:10.6g(100mmol)
トルエン:100ml
エタノール:20ml
水:100ml
【0097】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、テトラキス(トリフェニルホスフィン)パラジウム(0)を1.16g添加した。次に、反応溶液を80℃に昇温し、この温度(80℃)で12時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−酢酸エチル混合溶媒)で精製することにより、化合物d−6を白色固体として4.92g(収率62%)得た。
【0098】
(4)化合物d−7の合成
100mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−6:3.46g(9.06mmol)
THF:80ml
【0099】
次に、窒素雰囲気中、氷冷下で反応溶液を攪拌しながら、メチルマグネシウムブロマイド22.6mlをゆっくり滴下した。滴下終了後、反応溶液を室温まで昇温し、この温度(室温)で15時間攪拌した。次に、反応溶液を水100ml中にあけた後、トルエンで有機層を抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン)で精製することにより、化合物d−7を白色固体として2.16g(収率60.2%)得た。
【0100】
(5)化合物d−8の合成
50mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−7:2.10g(5.30mmol)
ポリリン酸:30ml
クロロホルム:20ml
【0101】
次に、反応溶液を60℃に昇温した後、この温度(60℃)で3時間攪拌した。次に、反応溶液を、水30ml中にあけた後、トルエンで有機層を抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製した後、ゲルろ過クロマトグラフィーにより、異性体を分離除去した。以上に示す方法により、化合物d−8を白色固体として1.65g(収率82.2%)得た。
【0102】
(6)例示化合物A−8の合成
50mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−8:0.378g(1.00mmol)
化合物d−11:0.425g(1.10mmol)
リン酸カリウム:1.06g
トルエン:5ml
水:0.1ml
【0103】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、下記に示す試薬を添加した。
酢酸パラジウム:22mg
化合物d−12:82mg
【0104】
次に、反応溶液を90℃に昇温した後、この温度(90℃)で5時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、例示化合物A−8を白色固体として0.440g(収率73.1%)得た。
【0105】
質量分析法により、例示化合物A−8のM+である602を確認した。
【0106】
次に、例示化合物A−8についてトルエン希薄溶液中でのT1を測定した。具体的には、トルエン溶液(1×10-4mol/l)を77Kに冷却し、このトルエン溶液に励起波長350nmの光を照射したときの燐光発光スペクトルを測定し、測定により得られた第一発光ピークをT1として用いた。尚、測定に当たっては、装置は日立製分光光度計U−3010を用いた。測定の結果、例示化合物A−8のT1は482nmであった。また例示化合物A−8についてイオン化ポテンシャルを測定した。具体的には、真空蒸着法によりガラス基板上に成膜された厚さ20nmの蒸着膜について、大気光電子分光装置(理研計器社製AC−3)を用いてイオン化ポテンシャルの測定を行った。測定の結果、イオン化ポテンシャルは6.16eVであった。
【0107】
[実施例2]例示化合物A−1の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−1を使用したことを除いては、実施例1と同様の方法により例示化合物A−1を合成した。
【0108】
【化14】
【0109】
質量分析法により、例示化合物A−1のM+である648を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物A−1のT1を測定したところ、T1は481nmであった。
【0110】
[実施例3]例示化合物A−5の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−2を使用したことを除いては、実施例1と同様の方法により例示化合物A−5を合成した。
【0111】
【化15】
【0112】
質量分析法により、例示化合物A−5のM+である612を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物A−5のT1を測定したところ、T1は482nmであった。
【0113】
[実施例4]例示化合物B−2の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−3を使用したことを除いては、実施例1と同様の方法により例示化合物B−2を合成した。
【0114】
【化16】
【0115】
質量分析法により、例示化合物B−2のM+である678を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−2のT1を測定したところ、T1は481nmであった。
【0116】
[実施例5]例示化合物B−5の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−4を使用したことを除いては、実施例1と同様の方法により例示化合物B−5を合成した。
【0117】
【化17】
【0118】
質量分析法により、例示化合物B−5のM+である688を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−5のT1を測定したところ、T1は482nmであった。
【0119】
[実施例6]例示化合物B−6の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−5を使用したことを除いては、実施例1と同様の方法により例示化合物B−6を合成した。
【0120】
【化18】
【0121】
質量分析法により、例示化合物B−6のM+である722を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−6のT1を測定したところ、T1は482nmであった。
【0122】
[実施例7]例示化合物C−3の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−6を使用したことを除いては、実施例1と同様の方法により例示化合物C−3を合成した。
【0123】
【化19】
【0124】
質量分析法により、例示化合物C−3のM+である728を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物C−3のT1を測定したところ、T1は483nmであった。
【0125】
[実施例8]例示化合物D−1の合成
以下に示す合成スキームにより、例示化合物D−1を合成した。
【0126】
【化20】
【0127】
(1)化合物d−13の合成
100mlの三ツ口フラスコ内を窒素雰囲気にした後、以下に示す試薬、溶媒を投入した。
化合物d−8:0.378g(1.00mmol)
化合物d−3:0.305g(1.20mmol)
酢酸カリウム:0.294g(3.00mmol)
ジオキサン:30ml
【0128】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、以下に示し試薬を添加した。
酢酸パラジウム:22mg
トリシクロヘキシルフォスフィン:56mg
【0129】
次に、反応溶液を100℃に昇温した後、この温度(100℃)で6時間攪拌した。反応終了後、反応溶液中の溶媒を減圧留去して粗生成物を得た。次に、この粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−ヘプタン混合溶媒)で精製することにより、化合物d−13を白色固体として0.446g(収率65.0%)得た。
【0130】
(2)例示化合物D−1の合成
50mlの三ツ口フラスコに、以下に示す試薬、溶媒を投入した。
化合物d−13:0.400g(0.85mmol)
化合物d−8:0.302g(0.80mmol)
リン酸カリウム:1.0g
トルエン:5ml
水:0.1ml
【0131】
次に、反応溶液を、窒素雰囲気中、室温で攪拌しながら、以下に示す試薬、溶媒を添加した。
酢酸パラジウム:22mg
化合物d−12:82mg
【0132】
次に、反応溶液を90℃に昇温した後、この温度(90℃)で5時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、例示化合物D−1を白色固体として0.390g(収率72.0%)得た。
【0133】
質量分析法により、例示化合物D−1のM+である686を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物D−1のT1を測定したところ、T1は482nmであった。
【0134】
(比較例1)比較化合物F−1の合成
実施例8(1)及び(2)において、化合物d−8の代わりに下記に示すe−15を使用したことを除いては、実施例8と同様の方法により、下記に示す比較化合物F−1を合成した。
【0135】
【化21】
【0136】
尚、化合物e−15は、例えば、実施例1(3)において、化合物d−5に代わりに下記に示す化合物e−14を使用して、実施例1(1)乃至(5)と同じ方法で合成を行うことにより得られる。
【0137】
【化22】
【0138】
質量分析法により、比較化合物F−1のM+である686を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における比較化合物F−1のT1を測定したところ、T1は529nmであった。
【0139】
[実施例9]
ガラス基板(基板)上に、陽極/ホール輸送層/発光層/電子輸送層/陰極が順次設けられた構成の有機発光素子を以下に示す方法で作製した。以下に、本実施例で使用した材料の一部を示す。
【0140】
【化23】
【0141】
スパッタ法により、ガラス基板上に、ITOを成膜して陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO電極付基板)として以下の工程で使用した。
【0142】
次に、1×10-5Paの真空チャンバー内における抵抗加熱による真空蒸着によって、ITO電極付基板上に、以下の表2に示す有機化合物層及び電極層を連続的に成膜した。このとき対向する電極面積は3mm2になるように作製した。
【0143】
【表2】
【0144】
得られた有機発光素子について、ITO電極を正極、Al電極を負極にして、4.0Vの印加電圧をかけたところ、電流密度3.40mA/cm2であった。また素子の発光輝度を4000cd/m2にした時の電圧は4.2Vであり、発光効率は66cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【0145】
さらに、本実施例の有機発光素子について、窒素雰囲気下で電流密度を40mA/cm2に保ちながら、連続して素子を駆動させた。その結果、初期輝度に対して輝度が半減するまでの時間は80時間以上であった。
【0146】
[実施例10]
実施例9において、発光層に含まれるホストを、例示化合物A−8に代えて例示化合物A−1とした以外は、実施例9と同様の方法により有機発光素子を作製した。
【0147】
本実施例で作製した有機発光素子について、ITO電極を正極、Al電極を負極にして、印加電圧をかけたところ、発光輝度が4000cd/m2の時の電圧は4.3Vであった。またこの素子において発光効率は63cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【0148】
[実施例11]
実施例9において、発光層に含まれるホストを、例示化合物A−8に代えて例示化合物A−5とした以外は、実施例9と同様の方法により有機発光素子を作製した。
【0149】
本実施例で作製した有機発光素子について、ITO電極を正極、Al電極を負極にして、印加電圧をかけたところ、発光輝度が4000cd/m2の時の電圧は4.3Vであった。またこの素子において発光効率は60cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【符号の説明】
【0150】
1:基板、2:防湿膜、3:ゲート電極、4:ゲート絶縁膜、5:半導体層、6:ドレイン電極、7:ソース電極、8:TFT素子、9:絶縁膜、10:コンタクトホール、11:陽極、12:有機化合物層、13:陰極、14:第一の保護層、15:第二の保護層、20:表示装置
【技術分野】
【0001】
本発明は、縮合多環化合物及びこれを用いた有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、陽極と陰極と、これら両電極間に配置される有機化合物層とを有する電子素子である。各電極からそれぞれ注入させる正孔(ホール)及び電子が、有機化合物層、特に発光層内で再結合することで生成する励起子が基底状態に戻る際に、有機発光素子は光を放出する。
【0003】
有機発光素子の最近の進歩は著しく、その特徴として、低駆動電圧、多様な発光波長、高速応答性、発光デバイスの薄型化・軽量化が可能であることが挙げられる。
【0004】
ところで有機発光素子は、発光に関与する励起子の種類によって蛍光発光素子と燐光発光素子とに大別される。そのうち燐光発光素子では三重項励起子が発光に関与し、有機発光素子を構成する有機化合物層、具体的には発光層中に燐光発光材料を有する電子素子である。ここで燐光発光材料は、正孔及び電子の再結合によって三重項状態に励起され、基底状態に戻る際に燐光を放出する。このため燐光発光素子は、この三重項励起子由来の発光が得られる有機発光素子である。
【0005】
ところで、燐光発光素子の内部量子収率は、理論上蛍光発光素子の内部量子収率の4倍になるので、近年では燐光発光素子に注目が集まっている。しかし燐光発光素子において、発光効率についてはさらなる改善の余地がある。
【0006】
一方、燐光発光素子で使用される材料について様々な提案がなされている。例えば、非特許文献1及び特許文献1に示される下記に示される部分構造を有する化合物が提案されている。
【0007】
【化1】
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−290991号公報
【非特許文献】
【0009】
【非特許文献1】Journal of Organic Chemistry 2006,71,6822−6828
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、上記課題を解決するためになされたものであり、その目的は、発光効率が高く駆動電圧の低い有機発光素子を提供することにある。
【課題を解決するための手段】
【0011】
本発明の縮合多環化合物は、下記一般式[1]及至[4]のいずれかで示されることを特徴とする。
【0012】
【化2】
(式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。)
【発明の効果】
【0013】
本発明によれば、発光効率が高く駆動電圧の低い有機発光素子を提供することができる。
【図面の簡単な説明】
【0014】
【図1】本発明の有機発光素子と、この有機発光素子に電気接続されるスイッチング素子の一例であるTFT素子とを有する表示装置の例を示す断面模式図である。
【発明を実施するための形態】
【0015】
まず本発明の縮合多環化合物について説明する。本発明の縮合多環化合物は、下記一般式[1]及至[4]のいずれかで示される化合物である。
【0016】
【化3】
【0017】
式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。
【0018】
上記フェニル基、ジベンゾチオフェニル基、フェナンスリル基、フルオレニル基、トリフェニレニル基及びナフチル基が有してもよい置換基として、メチル基、エチル基、プロピル基等のアルキル基、フェニル基、フルオレニル基、フェナンスリル基、トリフェニレニル基、ナフチル基等のアリール基等が挙げられる。
【0019】
式[1]及至[4]において、R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。
【0020】
R1及至R6で表されるアルキル基として、メチル基、エチル基、n−プロピル基、iso−プロピル基、n−ブチル基、iso−ブチル基、sec−ブチル基及びtert−ブチル基が挙げられる。
【0021】
式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。
【0022】
式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。
【0023】
式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。
【0024】
本発明の縮合多環化合物は、例えば、以下に示す合成ルートに従って合成することができる。
【0025】
【化4】
【0026】
上記合成スキームにおいて、中間体の1つである化合物d−8は、本発明の縮合多環化合物の母骨格を有する化合物である。ここで化合物d−8は、例えば、トリフェニレン(化合物d−1)を出発原料として、下記(i)乃至(v)のプロセスにより合成される。
(i)トリフェニレンの臭素化(化合物d−2の合成)
(ii)臭化トリフェニレンのピナコールボロン化(化合物d−4の合成)
(iii)(ii)にて合成されるトリフェニレニルボロン酸エステル(化合物d−4)と、ブロモクロロ安息香酸メチル(化合物d−5)との鈴木−宮浦カップリング反応(化合物d−6の合成)
(iv)グリニャール反応(化合物d−7の合成)
(v)ポリリン酸による脱水環化反応(化合物d−8の合成)
【0027】
一方、化合物d−5は、式[1]及至[4]の化合物をそれぞれ合成する際の原料として有効なクロロ体(化合物d−8)を合成する際に重要な化合物である。尚、上記合成スキームにて表される中間体d−5はベンゼン環の4位の位置に塩素原子を有しているが、この塩素原子が他のハロゲン原子に置換されていてもよいし、この塩素原子がトリフラート基又はピナコールボロン酸基に置換されていてもよい。
【0028】
次に、本発明の縮合多環化合物の特性について説明する。下記に示される(a−1)は、本発明の縮合多環化合物の母骨格となる化合物である。
【0029】
【化5】
上記化合物(a−1)に類似する化合物として、下記に示される化合物(a−2)がある。
【0030】
【化6】
【0031】
ここで化合物(a−1)は、化合物(a−2)と比べると、分子会合性が低いという性質を有する。化合物(a−1)及び化合物(a−2)は、いずれもトリフェニレン骨格とジメチルインデン骨格とが縮合することで形成される骨格であるが、フルオレン環に縮合化する方向が異なることによりこの特徴が生まれる。即ち、本発明の縮合多環化合物の母骨格となる化合物(a−1)は、分子会合性の強いトリフェニレン骨格と2つのメチル基との距離が化合物(a−2)よりも近い。このため化合物(a−1)同士が会合しようとすると、所定の分子が有するメチル基が別の分子が有するトリフェニレン骨格同士のスタッキングを抑制するため分子会合性が低くなるといえる。
【0032】
分子会合性が低いことは、分子会合による濃度消光やエキサイマー発光を抑えることにつながるため、化合物の発光特性に関して有利に働く。
【0033】
次に、化合物(a−1)と化合物(a−2)に導入される置換基の置換位置の違いについて述べる。
【0034】
本発明の縮合多環化合物は、下記(a−1)で示される化合物を母骨格として、置換基が当該母骨格のα位に導入されるという特徴を有する。
【0035】
【化7】
【0036】
この特徴を有することにより、トリフェニレン環とベンゼン環とで形成される共役が置換基を介してさらに延伸されることがない、即ち、母骨格と置換基との間で母骨格が有する共役が切れるという特徴を有する。これにより、本発明の縮合多環化合物の最低三重項励起状態のエネルギー(T1エネルギー)は、化合物の母骨格である(a−1)で決定されるものであり、高いT1エネルギーが維持される。
【0037】
一方、下記構造(a−2)で示される化合物を母骨格として、置換基が当該母骨格のβ位に置換基を有する化合物は、トリフェニレン環とベンゼン環とで形成される共役が置換基を介してさらに延伸されることに特徴を有する。
【0038】
【化8】
【0039】
この特徴を有することにより、a−2の最低三重項励起状態のエネルギー(T1エネルギー)は、a−2とβ位で置換する置換基との相互作用(共役の延伸)で決定されるものとなり、本発明の縮合多環化合物に比べて低いT1エネルギーになる。
【0040】
ここで発明者らは、下記に示される化合物についてトルエン希薄溶液中でのT1エネルギーを測定した。尚、T1の測定はトルエン溶液(1×10-4mol/l)を77Kに冷却した上で、励起波長350nmにて燐光発光スペクトルを測定し、第一発光ピークをT1として用いた。装置は日立製分光光度計U−3010を用いた。
【0041】
【化9】
【0042】
【表1】
【0043】
表1より、本発明の縮合多環化合物である化合物D−1のT1は、自身の部分骨格であるa−1と同じである。このことから2つのa−1骨格同士の共役が切れていることが示される。
【0044】
一方、比較化合物に相当する化合物F−1のT1は、自身の部分骨格であるa−2よりも大きく長波長化している。このことから2つのa−2骨格同士の共役がつながっていることが示される。
【0045】
ところで、式[1]及至[4]で示されるArは、T1が高いアリール基が好ましく、530nm以下のアリール基より選ばれる。具体的には、ベンゼン、ジベンゾチオフェン、フェナンスレン、フルオレン、トリフェニレン及びナフタレンから選ばれる。尚、Arで表されるアリール基は、さらに置換基を有してもよい。
【0046】
以上により、高いT1を持つ母骨格a−1と、Ar基の所定の位置の置換により、本発明の縮合多環化合物はそのT1が470nm以上500nm以下の範囲になっている。
【0047】
以上の作用効果により、本発明の縮合多環化合物は、有機発光素子用材料、特に、発光材料として用いた場合、高効率の発光素子を提供することができる。
【0048】
ところで、緑色に燐光発光する燐光発光材料のT1は490nm以上530nm以下であり、本発明の縮合多環化合物はそれよりも高いT1エネルギーを有する。従って、本発明の縮合多環化合物は、緑色の燐光を発する有機発光素子の発光層のホストもしくは電子輸送材料として用いると、素子の発光効率を向上させることができる。この場合、燐光発光する化合物は、発光層のゲスト(燐光発光材料)である。
【0049】
本発明の縮合多環化合物は、母骨格であるa−1にArで示されるアリール基あるいはa−1が所定の位置に結合されていることを特徴とする。ここで母骨格であるa−1にArが結合することにより、分子全体の平面性が崩れることで安定なアモルファス膜を形成することに効果がある。
【0050】
従って、本発明の縮合多環化合物を有機発光素子用材料として用いた場合、耐久性が向上された発光素子を提供することができる。
【0051】
本発明の縮合多環化合物の具体例を以下に示す。ただし、本発明はこれらに限られるものではない。
【0052】
【化10】
【0053】
【化11】
【0054】
上記具体例において、A群に属する化合物は、式[1]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがフェニレン基で連結されている化合物である。ここでA群に属する化合物は分子量が小さいので、蒸着によって化合物の薄膜を成膜する際に、蒸着温度を低くすることができる。
【0055】
上記具体例において、B群に属する化合物は、式[2]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがビフェニレン基で連結されている化合物である。ここでB群に属する化合物は分子中での回転を可能にする結合が多いため、アモルファス膜を形成したときの膜の安定性が高い。
【0056】
上記具体例において、C群に属する化合物は、式[3]で示される化合物群である。即ち、母骨格(a−1)とアリール基とがフルオレニレン基で連結されている化合物である。ここで母骨格(a−1)とアリール基とを連結するフルオレニレン基は剛直であるため、アモルファス膜を成膜した際にその膜の電子及び正孔の移動度が高くなる。
【0057】
上記具体例において、D群に属する化合物は、式[4]で示される化合物群である。即ち、母骨格(a−1)のダイマー体である。ここでD群に属する化合物は、分子の対称性が高いため、アモルファス膜を成膜した際にその膜の電子及び正孔の移動度が高くなる。
【0058】
ここで上記具体例にて示される化合物のうち、正孔注入・輸送性が高いという観点から、式[1]乃至[4]にて示されるArがジベンゾチオフェンである化合物、具体的にはA−8、B−2及びC−4は好ましい材料であるといえる。
【0059】
次に、本発明の有機発光素子について説明する。
【0060】
本発明の有機発光素子は、一対の電極である陽極と陰極と、これら陽極と陰極との間に配置される有機化合物層と、から構成される。
【0061】
本発明において、有機発光素子の構成部材である有機化合物層は、発光層又は発光機能を有する層が含まれていれば、単一の層であってもよいし、複数の層からなる積層体であってもよい。
【0062】
有機化合物層が複数の層からなる場合、発光層(又は発光機能を有する層)以外の層であって、かつ有機化合物層に含まれる層として、ホール注入層、ホール輸送層、発光層、ホールブロック層、電子輸送層、電子注入層、エキシトンブロック層等が挙げられる。もちろん、上記群の中から1又は複数を選択して、かつそれらを組み合わせて用いることができる。
【0063】
尚、本発明の有機発光素子の構成はこれらに限定されるものではない。例えば、電極と有機化合物層界面に絶縁性層、接着層あるいは干渉層を設ける、電子輸送層もしくはホール輸送層をイオン化ポテンシャルの異なる二層から構成される等の多様な層構成をとることができる。
【0064】
本発明の有機発光素子において、素子形態としては、基板とは反対側の電極から光を取り出すいわゆるトップエミッション方式でもよいし、基板側から光を取り出すいわゆるボトムエミッション方式でもよい。また基板や電極を透明な材料にして両面から光を取り出す構成にしてもよい。
【0065】
本発明の有機発光素子において、本発明の縮合多環化合物は、有機化合物層に含まれている。本発明の有機発光素子において、本発明の縮合多環化合物を有している有機化合物層は、特に限定されないが、好ましくは、発光層に含まれる。本発明の有機発光素子において、発光層は、本発明の縮合多環化合物のみからなる層であってもよいが、好ましくは、ホストとゲストとからなる層である。
【0066】
ここで本発明の縮合多環化合物は、発光層のホストとして使用してもよいし、ゲストとして使用してもよいが、好ましくは、発光層のホストとして使用する。ここで本発明の縮合多環化合物を燐光発光するゲストと組み合わせて使用されるホストとして使用すると、発光効率の観点から好ましい。特に、本発明の縮合多環化合物は、490nm乃至660nmの領域に発光ピークを持つ発光色が緑乃至赤であるゲストと組み合わせると、三重項エネルギーのロスが少ないため、発光素子の効率が高くなる。
【0067】
尚、本発明の縮合多環化合物をゲストとして使用する場合、ホストに対するゲストの濃度は、発光層の全体量に対して0.1重量%以上30重量%以下であることが好ましく、0.5重量%以上10重量%以下であることがより好ましい。
【0068】
本発明の有機発光素子は、本発明の縮合多環化合物以外にも、必要に応じて他の化合物を構成材料として使用することができる。具体的には、従来公知の低分子系及び高分子系の正孔注入・輸送性材料、ホスト、ゲストあるいは電子注入・輸送性材料等を一緒に使用することができる。
【0069】
以下にこれらの化合物例を挙げる。
【0070】
正孔注入・輸送性材料としては、正孔移動度が高い材料であることが好ましい。正孔注入性能あるいは正孔輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられるが、もちろんこれらに限定されるものではない。
【0071】
ホストとしては、トリアリールアミン誘導体、フェニレン誘導体、縮合環芳香族化合物(例えばナフタレン誘導体、フェナントレン誘導体、フルオレン誘導体、クリセン誘導体、等)、有機金属錯体(例えば、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、有機イリジウム錯体、有機プラチナ錯体等)及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体、ポリ(チエニレンビニレン)誘導体、ポリ(アセチレン)誘導体等の高分子
誘導体が挙げられるが、もちろんこれらに限定されるものではない。
【0072】
ゲストとしては、燐光発光材料が好ましい。具体的には、以下に示す、燐光発光性のIr錯体や、プラチナ錯体等が挙げられる。
【0073】
【化12】
【0074】
また、蛍光発光性のドーパントを用いることもでき、縮環化合物(例えばフルオレン誘導体、ナフタレン誘導体、ピレン誘導体、ペリレン誘導体、テトラセン誘導体、アントラセン誘導体、ルブレン等)、キナクリドン誘導体、クマリン誘導体、スチルベン誘導体、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体等の高分子誘導体が挙げられる。
【0075】
電子注入・輸送性材料としては、ホール注入性材料あるいはホール輸送性材料のホール移動度とのバランス等を考慮し選択される。電子注入性能あるいは電子輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられるが、もちろんこれらに限定されるものではない。
【0076】
陽極の構成材料としては、仕事関数がなるべく大きなものがよい。例えば、金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれら金属単体を複数種組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物である。また、ポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーでもよい。これらの電極物質は一種類を単独で使用してもよいし複数種類を併用して使用してもよい。また、陽極は一層構成でもよいし、多層構成でもよい。
【0077】
一方、陰極を構成する材料としては、仕事関数の小さなものがよい。例えば、リチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えば、マグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、複数種類を併用して使用してもよい。また、陰極は一層構成でもよいし、多層構成でもよい。
【0078】
本発明の有機発光素子において、本発明の縮合多環化合物が含まれている層及びその他の層については、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により層を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で形成する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0079】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコーン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として1種単独で使用してもよいし、2種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0080】
本発明の有機発光素子は、表示装置や照明装置に用いることができる。他にも電子写真方式の画像形成装置の露光光源や液晶表示装置のバックライト等がある。
【0081】
表示装置は本発明の有機発光素子を表示部に有する。この表示部は複数の画素を有する。この画素は本実施形態に係る有機発光素子と発光輝度を制御するためのスイッチング素子の一例としてTFT素子とを有し、この有機発光素子の陽極又は陰極とTFT素子のドレイン電極又はソース電極とが接続されている。表示装置はPC等の画像表示装置として用いることができる。
【0082】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの情報を入力する画像入力部を有し、入力された画像を表示部に出力する画像出力装置でもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報に基づいて画像を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。また表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0083】
次に、本実施形態に係る有機発光素子を使用した表示装置について図1を用いて説明する。
【0084】
図1は、本発明の有機発光素子と、この有機発光素子に電気接続されるスイッチング素子の一例であるTFT素子とを有する表示装置の例を示す断面模式図である。図1の表示装置20には有機発光素子とTFT素子との組が2組図示されている。構造の詳細を以下に説明する。
【0085】
図1の表示装置20は、ガラス等の基板1とその上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属のゲート電極3である。符号4はゲート絶縁膜4であり、5は半導体層である。
【0086】
TFT素子8は半導体層5とドレイン電極6とソース電極7とを有している。TFT素子8の上部には絶縁膜9が設けられている。コンタクトホール10を介して有機発光素子の陽極11とソース電極7とが接続されている。表示装置はこの構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子ソース電極又はドレイン電極のいずれか一方とが接続されていればよい。
【0087】
尚、図1の表示装置20において、有機化合物層12は、多層あるいは単層の有機化合物層を1つの層の如く図示をしている。陰極13の上には有機発光素子の劣化を抑制するための第一の保護層14や第二の保護層15が設けられている。
【0088】
本実施形態に係る表示装置においてスイッチング素子に特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0089】
以下、実施例において本発明を詳細に説明する。ただし本発明はこれらに限定されるものではない。
【0090】
[実施例1]例示化合物A−8の合成
以下に示す合成スキームにより合成した。
【0091】
【化13】
【0092】
(1)化合物d−2の合成
500mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−1:9.99g(43.8mmol)
ジクロロメタン:300ml
【0093】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、臭素7.7g(48.2mmol)とジクロロメタン7.0mlとの混合溶液を滴下した。当該混合溶液を滴下した後、反応溶液を室温で12時間攪拌した。反応終了後、チオ硫酸ナトリウム溶液中に反応溶液をあけた後、有機層をクロロホルムで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、化合物d−2を白色固体として11.6g(収率86.3%)得た。
【0094】
(2)化合物d−4の合成
300mlの三ツ口フラスコ内を窒素雰囲気にした後、下記に示す試薬、溶媒を投入した。
化合物d−2:11.5g(37.4mmol)
化合物d−3:11.4g(44.9mmol)
酢酸カリウム:6.61g(67.4mmol)
ジオキサン:100ml
【0095】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、ビス(ジフェニルフォスフィノ)フェロセンパラジウム(II)ジクロライド・ジクロロメタン1.53g(1.87mmol)を添加した。次に、反応溶液を100℃に昇温し、この温度(100℃)で4時間攪拌した。反応終了後、反応溶液中の溶媒を減圧留去して粗生成物を得た。次に、この粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−ヘプタン混合溶媒)で精製することにより、化合物d−4を白色固体として10.42g(収率68.6%)得た。
【0096】
(3)化合物d−6の合成
200mlの三ツ口フラスコ内を窒素雰囲気にした後、下記に示す試薬、溶媒を投入した。
化合物d−4:7.08g(20.0mmol)
化合物d−5:5.46g(22.0mmol)
炭酸ナトリウム:10.6g(100mmol)
トルエン:100ml
エタノール:20ml
水:100ml
【0097】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、テトラキス(トリフェニルホスフィン)パラジウム(0)を1.16g添加した。次に、反応溶液を80℃に昇温し、この温度(80℃)で12時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−酢酸エチル混合溶媒)で精製することにより、化合物d−6を白色固体として4.92g(収率62%)得た。
【0098】
(4)化合物d−7の合成
100mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−6:3.46g(9.06mmol)
THF:80ml
【0099】
次に、窒素雰囲気中、氷冷下で反応溶液を攪拌しながら、メチルマグネシウムブロマイド22.6mlをゆっくり滴下した。滴下終了後、反応溶液を室温まで昇温し、この温度(室温)で15時間攪拌した。次に、反応溶液を水100ml中にあけた後、トルエンで有機層を抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン)で精製することにより、化合物d−7を白色固体として2.16g(収率60.2%)得た。
【0100】
(5)化合物d−8の合成
50mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−7:2.10g(5.30mmol)
ポリリン酸:30ml
クロロホルム:20ml
【0101】
次に、反応溶液を60℃に昇温した後、この温度(60℃)で3時間攪拌した。次に、反応溶液を、水30ml中にあけた後、トルエンで有機層を抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製した後、ゲルろ過クロマトグラフィーにより、異性体を分離除去した。以上に示す方法により、化合物d−8を白色固体として1.65g(収率82.2%)得た。
【0102】
(6)例示化合物A−8の合成
50mlの三ツ口フラスコに、下記に示す試薬、溶媒を投入した。
化合物d−8:0.378g(1.00mmol)
化合物d−11:0.425g(1.10mmol)
リン酸カリウム:1.06g
トルエン:5ml
水:0.1ml
【0103】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、下記に示す試薬を添加した。
酢酸パラジウム:22mg
化合物d−12:82mg
【0104】
次に、反応溶液を90℃に昇温した後、この温度(90℃)で5時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、例示化合物A−8を白色固体として0.440g(収率73.1%)得た。
【0105】
質量分析法により、例示化合物A−8のM+である602を確認した。
【0106】
次に、例示化合物A−8についてトルエン希薄溶液中でのT1を測定した。具体的には、トルエン溶液(1×10-4mol/l)を77Kに冷却し、このトルエン溶液に励起波長350nmの光を照射したときの燐光発光スペクトルを測定し、測定により得られた第一発光ピークをT1として用いた。尚、測定に当たっては、装置は日立製分光光度計U−3010を用いた。測定の結果、例示化合物A−8のT1は482nmであった。また例示化合物A−8についてイオン化ポテンシャルを測定した。具体的には、真空蒸着法によりガラス基板上に成膜された厚さ20nmの蒸着膜について、大気光電子分光装置(理研計器社製AC−3)を用いてイオン化ポテンシャルの測定を行った。測定の結果、イオン化ポテンシャルは6.16eVであった。
【0107】
[実施例2]例示化合物A−1の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−1を使用したことを除いては、実施例1と同様の方法により例示化合物A−1を合成した。
【0108】
【化14】
【0109】
質量分析法により、例示化合物A−1のM+である648を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物A−1のT1を測定したところ、T1は481nmであった。
【0110】
[実施例3]例示化合物A−5の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−2を使用したことを除いては、実施例1と同様の方法により例示化合物A−5を合成した。
【0111】
【化15】
【0112】
質量分析法により、例示化合物A−5のM+である612を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物A−5のT1を測定したところ、T1は482nmであった。
【0113】
[実施例4]例示化合物B−2の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−3を使用したことを除いては、実施例1と同様の方法により例示化合物B−2を合成した。
【0114】
【化16】
【0115】
質量分析法により、例示化合物B−2のM+である678を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−2のT1を測定したところ、T1は481nmであった。
【0116】
[実施例5]例示化合物B−5の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−4を使用したことを除いては、実施例1と同様の方法により例示化合物B−5を合成した。
【0117】
【化17】
【0118】
質量分析法により、例示化合物B−5のM+である688を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−5のT1を測定したところ、T1は482nmであった。
【0119】
[実施例6]例示化合物B−6の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−5を使用したことを除いては、実施例1と同様の方法により例示化合物B−6を合成した。
【0120】
【化18】
【0121】
質量分析法により、例示化合物B−6のM+である722を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物B−6のT1を測定したところ、T1は482nmであった。
【0122】
[実施例7]例示化合物C−3の合成
実施例1(6)において、化合物d−11の代わりに下記に示す化合物e−6を使用したことを除いては、実施例1と同様の方法により例示化合物C−3を合成した。
【0123】
【化19】
【0124】
質量分析法により、例示化合物C−3のM+である728を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物C−3のT1を測定したところ、T1は483nmであった。
【0125】
[実施例8]例示化合物D−1の合成
以下に示す合成スキームにより、例示化合物D−1を合成した。
【0126】
【化20】
【0127】
(1)化合物d−13の合成
100mlの三ツ口フラスコ内を窒素雰囲気にした後、以下に示す試薬、溶媒を投入した。
化合物d−8:0.378g(1.00mmol)
化合物d−3:0.305g(1.20mmol)
酢酸カリウム:0.294g(3.00mmol)
ジオキサン:30ml
【0128】
次に、反応溶液を、窒素雰囲気下、室温で攪拌しながら、以下に示し試薬を添加した。
酢酸パラジウム:22mg
トリシクロヘキシルフォスフィン:56mg
【0129】
次に、反応溶液を100℃に昇温した後、この温度(100℃)で6時間攪拌した。反応終了後、反応溶液中の溶媒を減圧留去して粗生成物を得た。次に、この粗生成物をシリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−ヘプタン混合溶媒)で精製することにより、化合物d−13を白色固体として0.446g(収率65.0%)得た。
【0130】
(2)例示化合物D−1の合成
50mlの三ツ口フラスコに、以下に示す試薬、溶媒を投入した。
化合物d−13:0.400g(0.85mmol)
化合物d−8:0.302g(0.80mmol)
リン酸カリウム:1.0g
トルエン:5ml
水:0.1ml
【0131】
次に、反応溶液を、窒素雰囲気中、室温で攪拌しながら、以下に示す試薬、溶媒を添加した。
酢酸パラジウム:22mg
化合物d−12:82mg
【0132】
次に、反応溶液を90℃に昇温した後、この温度(90℃)で5時間攪拌した。反応終了後、有機層をトルエンで抽出してこの有機層を無水硫酸ナトリウムで乾燥させた。次に、この有機層を減圧濃縮することで粗生成物を得た。次に、この粗生成物を、シリカゲルカラムクロマトグラフィー(展開溶媒:トルエン−ヘプタン混合溶媒)で精製することにより、例示化合物D−1を白色固体として0.390g(収率72.0%)得た。
【0133】
質量分析法により、例示化合物D−1のM+である686を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における例示化合物D−1のT1を測定したところ、T1は482nmであった。
【0134】
(比較例1)比較化合物F−1の合成
実施例8(1)及び(2)において、化合物d−8の代わりに下記に示すe−15を使用したことを除いては、実施例8と同様の方法により、下記に示す比較化合物F−1を合成した。
【0135】
【化21】
【0136】
尚、化合物e−15は、例えば、実施例1(3)において、化合物d−5に代わりに下記に示す化合物e−14を使用して、実施例1(1)乃至(5)と同じ方法で合成を行うことにより得られる。
【0137】
【化22】
【0138】
質量分析法により、比較化合物F−1のM+である686を確認した。また、実施例1と同様の方法により、トルエン希薄溶液中における比較化合物F−1のT1を測定したところ、T1は529nmであった。
【0139】
[実施例9]
ガラス基板(基板)上に、陽極/ホール輸送層/発光層/電子輸送層/陰極が順次設けられた構成の有機発光素子を以下に示す方法で作製した。以下に、本実施例で使用した材料の一部を示す。
【0140】
【化23】
【0141】
スパッタ法により、ガラス基板上に、ITOを成膜して陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO電極付基板)として以下の工程で使用した。
【0142】
次に、1×10-5Paの真空チャンバー内における抵抗加熱による真空蒸着によって、ITO電極付基板上に、以下の表2に示す有機化合物層及び電極層を連続的に成膜した。このとき対向する電極面積は3mm2になるように作製した。
【0143】
【表2】
【0144】
得られた有機発光素子について、ITO電極を正極、Al電極を負極にして、4.0Vの印加電圧をかけたところ、電流密度3.40mA/cm2であった。また素子の発光輝度を4000cd/m2にした時の電圧は4.2Vであり、発光効率は66cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【0145】
さらに、本実施例の有機発光素子について、窒素雰囲気下で電流密度を40mA/cm2に保ちながら、連続して素子を駆動させた。その結果、初期輝度に対して輝度が半減するまでの時間は80時間以上であった。
【0146】
[実施例10]
実施例9において、発光層に含まれるホストを、例示化合物A−8に代えて例示化合物A−1とした以外は、実施例9と同様の方法により有機発光素子を作製した。
【0147】
本実施例で作製した有機発光素子について、ITO電極を正極、Al電極を負極にして、印加電圧をかけたところ、発光輝度が4000cd/m2の時の電圧は4.3Vであった。またこの素子において発光効率は63cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【0148】
[実施例11]
実施例9において、発光層に含まれるホストを、例示化合物A−8に代えて例示化合物A−5とした以外は、実施例9と同様の方法により有機発光素子を作製した。
【0149】
本実施例で作製した有機発光素子について、ITO電極を正極、Al電極を負極にして、印加電圧をかけたところ、発光輝度が4000cd/m2の時の電圧は4.3Vであった。またこの素子において発光効率は60cd/Aであり、CIE色度座標(0.35,0.62)の緑色発光が観測された。
【符号の説明】
【0150】
1:基板、2:防湿膜、3:ゲート電極、4:ゲート絶縁膜、5:半導体層、6:ドレイン電極、7:ソース電極、8:TFT素子、9:絶縁膜、10:コンタクトホール、11:陽極、12:有機化合物層、13:陰極、14:第一の保護層、15:第二の保護層、20:表示装置
【特許請求の範囲】
【請求項1】
下記一般式[1]及至[4]のいずれかで示されることを特徴とする、縮合多環化合物。
【化1】
(式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。)
【請求項2】
前記Arが置換あるいは無置換のジベンゾチオフェンであることを特徴とする、請求項1に記載の縮合多環化合物。
【請求項3】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、から構成され、
前記有機化合物層に請求項1又は2に記載の縮合多環化合物が含まれることを特徴とする、有機発光素子。
【請求項4】
前記縮合多環化合物が、発光層に含まれており、
前記発光層が、ホストとゲストとを有し、
前記ホストが、前記縮合多環化合物であることを特徴とする、請求項3に記載の有機発光素子。
【請求項5】
前記ゲストが、燐光発光材料であることを特徴とする、請求項4に記載の有機発光素子。
【請求項6】
請求項3及至5のいずれか一項に記載の有機発光素子と、前記有機発光素子に電気接続されるスイッチング素子と、を有することを特徴とする、表示装置。
【請求項1】
下記一般式[1]及至[4]のいずれかで示されることを特徴とする、縮合多環化合物。
【化1】
(式[1]及至[4]において、Arは、置換あるいは無置換のフェニル基、置換あるいは無置換のジベンゾチオフェニル基、置換あるいは無置換のフェナンスリル基、置換あるいは無置換のフルオレニル基、置換あるいは無置換のトリフェニレニル基又は置換あるいは無置換のナフチル基である。R1及至R6は、それぞれ水素原子又は炭素数1以上4以下のアルキル基である。式[1]及至[4]において、R1及びR2は、同じであってもよいし異なっていてもよい。式[3]において、R3及びR4は、同じであってもよいし異なっていてもよい。式[4]において、R5及びR6は、同じであってもよいし異なっていてもよい。)
【請求項2】
前記Arが置換あるいは無置換のジベンゾチオフェンであることを特徴とする、請求項1に記載の縮合多環化合物。
【請求項3】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、から構成され、
前記有機化合物層に請求項1又は2に記載の縮合多環化合物が含まれることを特徴とする、有機発光素子。
【請求項4】
前記縮合多環化合物が、発光層に含まれており、
前記発光層が、ホストとゲストとを有し、
前記ホストが、前記縮合多環化合物であることを特徴とする、請求項3に記載の有機発光素子。
【請求項5】
前記ゲストが、燐光発光材料であることを特徴とする、請求項4に記載の有機発光素子。
【請求項6】
請求項3及至5のいずれか一項に記載の有機発光素子と、前記有機発光素子に電気接続されるスイッチング素子と、を有することを特徴とする、表示装置。
【図1】

【公開番号】特開2012−131752(P2012−131752A)
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願番号】特願2010−286970(P2010−286970)
【出願日】平成22年12月24日(2010.12.24)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願日】平成22年12月24日(2010.12.24)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]