
縮合多環化合物及びこれを用いた有機発光素子
【課題】主として青色発光素子の構成材料として用いるのに適した縮合多環化合物及びこれを用いた有機発光素子を提供する。
【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層と、を有する有機発光素子において、前記有機化合物層のうち少なくとも一層が、下記に記載の縮合多環化合物のいずれかを有することを特徴とする、有機発光素子。

(式[1]、[2]において、Rは、水素原子又はメチル基を表す。Qは、芳香環を含む電子吸引性の特定の置換基である。
【解決手段】陽極と陰極と、前記陽極と前記陰極との間に配置される有機化合物層と、を有する有機発光素子において、前記有機化合物層のうち少なくとも一層が、下記に記載の縮合多環化合物のいずれかを有することを特徴とする、有機発光素子。
(式[1]、[2]において、Rは、水素原子又はメチル基を表す。Qは、芳香環を含む電子吸引性の特定の置換基である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、縮合多環化合物及びこれを用いた有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、一対の電極間に蛍光性又は燐光性有機化合物を含む薄膜が挟持されている電子素子である。各電極から電子及びホール(正孔)を注入することにより、蛍光性又は燐光性化合物の励起子が生成され、この励起子が基底状態に戻る際に、有機発光素子は光を放出する。
【0003】
有機発光素子における最近の進歩は著しく、その特徴として、低印加電圧で高輝度、発光波長の多様性、高速応答性、薄型、軽量の発光デバイス化の可能性であることが挙げられる。このことから、有機発光素子は広汎な用途への可能性を示唆している。
【0004】
ところで、有機発光素子の構成材料として用いられる蛍光性又は燐光性有機化合物についても盛んに研究がなされている。その中でも青色発光材料については、例えば、特許文献1や2に示されている有機化合物が提案されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−254610号公報
【特許文献2】特開2010−270103号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかし特許文献1や2にて提案されている有機化合物及びこれを用いた有機発光素子は、実用化という観点からさらなる改善の余地がある。
【0007】
具体的には、実用化のためにはさらなる高輝度の光出力あるいは高変換効率が必要であること、長時間の使用による経時変化や酸素を含む雰囲気気体や湿気等による劣化等の耐久面において改善が必要であること、が挙げられる。さらにはフルカラーディスプレイ等への応用を考えた場合、使用される青色発光する有機発光素子には色純度が良く高効率の青色発光が要求されるが、これらの問題に関してもまだ十分に解決されたとはいえない。
【0008】
従って、青色発光する有機発光素子に関して言えば、特に、色純度、発光効率及び耐久性が高い有機発光素子及びこれを実現するための材料が要求されている。
【0009】
本発明は、上述した従来技術の問題点を解決するためになされたものである。即ち、本発明の目的は、主として青色発光素子の構成材料として用いるのに適した縮合多環化合物及びこれを用いた有機発光素子を提供することにある。
【課題を解決するための手段】
【0010】
本発明の縮合多環化合物は、下記一般式[1]、[2]、[8]又は[9]で示される化合物であることを特徴とする。
【0011】
【化1】
(式[1]、[2]、[8]及び[9]において、Rは、水素原子又はメチル基を表す。式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【0012】
【化2】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【発明の効果】
【0013】
本発明の縮合多環化合物は、発光ピークが460nm以下の純度のよい青色発光色相を呈する。また本発明の縮合多環化合物を用いた有機発光素子は、低い印加電圧で高輝度な発光が得られ、耐久性にも優れる。従って、本発明によれば、主として青色発光素子の構成材料として用いるのに適した縮合多環化合物及びこれを用いた有機発光素子を提供することができる。
【図面の簡単な説明】
【0014】
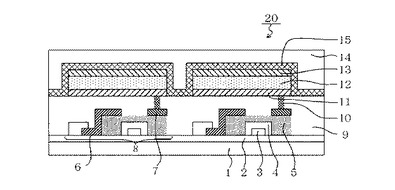
【図1】本発明の有機発光素子と、この有機発光素子に電気接続するスイッチング素子の一例であるTFT素子と、を有する表示装置の例を示す断面模式図である。
【発明を実施するための形態】
【0015】
本発明の縮合多環化合物であるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、下記一般式[1]、[2]、[8]又は[9]で示される化合物である。
【0016】
【化3】
【0017】
式[1]、[2]、[8]及び[9]において、Rは、水素原子又はメチル基を表す。ここで式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。
【0018】
式[1]、[2]、[8]及び[9]において、Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【0019】
【化4】
【0020】
式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。
【0021】
R1乃至R4で表されるアルキル基として、メチル基、エチル基、n−プロピル基、n−ブチル基、iso−プロピル基、iso−ブチル基、sec−ブチル基、tert−ブチル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基、パーフルオロエチル基、3−フルオロプロピル基、パーフルオロプロピル基、4−フルオロブチル基、パーフルオロブチル基、シクロプロピル基、シクロブチル基等が挙げられるが、もちろんこれらに限定されるものではない。
【0022】
尚、R1乃至R4は、それぞれ同じであってもよいし異なっていてもよい。
【0023】
式[7]において、R5は、水素原子又はメチル基である。
【0024】
ここで本発明の縮合多環化合物が式[1]又は[2]で示される化合物である場合、好ましくは、下記一般式[10]、[11]、[30]又は[31]で示される化合物である。
【0025】
【化5】
【0026】
式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。
【0027】
R1乃至R4で表されるアルキル基として、メチル基、エチル基、n−プロピル基、n−ブチル基、iso−プロピル基、iso−ブチル基、sec−ブチル基、tert−ブチル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基、パーフルオロエチル基、3−フルオロプロピル基、パーフルオロプロピル基、4−フルオロブチル基、パーフルオロブチル基、シクロプロピル基、シクロブチル基等が挙げられるが、もちろんこれらに限定されるものではない。
【0028】
式[30]及び[31]において、Rは、水素原子又はメチル基を表す。ここで式[30]にて示されるRは同一の置換基である。式[31]にて示されるRは同一の置換基である。
【0029】
一方、本発明の縮合多環化合物が式[8]又は[9]で示される化合物である場合、好ましくは、下記一般式[20]又は[21]で示される化合物である。
【0030】
【化6】
【0031】
式[20]及び[21]において、Rは、水素原子又はメチル基を表す。
【0032】
[合成ルートの説明]
本発明の縮合多環化合物、具体的には、一般式[1]、[2]、[8]又は[9]に示される化合物は、例えば、以下に示す合成ルートに従って合成することができる。
【0033】
【化7】
【0034】
この合成ルートでは、基本骨格が共通するが置換基Arの置換位置が異なる二種類の化合物が同時に得られる。具体的には、式[1]に示される化合物と式[2]に示される化合物との混合物、あるいは式[8]に示される化合物と式[9]に示される化合物との混合物が得られる。ただし式[1]に示される化合物と式[2]に示される化合物との関係、あるいは式[8]に示される化合物と式[9]に示される化合物との関係においては、Arが同じであれば、Arの置換位置によっては発光特性にほとんど差がない。このため、再結晶等で単離して使用しても良いし、混合物の状態でそのまま用いてもよい。また混合物をそのまま使用したとしても単体と比べて発光特性が特段低下することはないため、混合比は特に限定されない。また、混合物の状態で用いた際は、結晶性を抑えることができるため、濃度消光の抑制等の効果も期待できる。
【0035】
[化合物に関する考察]
以下、本発明の縮合多環化合物についてさらに詳細を説明する。
【0036】
一般的に有機発光素子の発光効率を高めるためには、発光中心そのものの発光量子収率が大きいことが望まれる。そのためには、下記(A)及び(B)が要求されている。
(A)振動子強度が高いこと
(B)発光にかかわる骨格の振動部分が少ないこと
【0037】
(A)に関しては、分子の発光に関わる骨格の対称性を高くすることが重要である。ただし、高対称性分子特有の禁制遷移条件によっては全く発光しなくなる場合もある。ところで振動子強度を高くする方法として、最も共役面の長い方向を軸として、さらに共役を伸ばす方法がある。この方法を採用することによって分子の双極子モーメントが大きくなり振動子強度が向上する。この点では、本発明の縮合多環化合物、特に、一般式[1]又は[2]で表わされる化合物は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の最も共役面が長い方向を軸とした上で、さらに共役長が伸びる位置に特定の置換基を有している。このように分子設計を行うことで分子全体の振動子強度が向上した構造になっている。
【0038】
(B)に関しては、発光に関わる骨格に回転構造を有さないことで、回転振動による量子収率の低下を抑制することができる。ここで本発明の縮合多環化合物の基本骨格は回転構造を有していない。従って回転振動による量子収率の低下を抑制することができる。
【0039】
一方、有機ELディスプレイとして青色発光に適した材料に求められる物性値の1つであるが、材料自体の発光ピークが430nm〜480nmの範囲にあることは重要である。特に、青色発光材料に対して色純度の良として要求される発光ピークの存在範囲としては440nm〜460nmであることがより好ましい。
【0040】
本発明の縮合多環化合物が有する基本骨格、即ち、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格は、これ自体で最大発光波長が青色領域に存在する。このため、この基本骨格に置換基を導入する位置及び置換基の種類を適切に選ぶことによって460nm以下の発光ピークを有する青色発光材料になる化合物を提供することができる。具体的には、一般式[1]、[2]、[8]又は[9]で表わされる縮合多環化合物は、上述のように基本骨格内で最も共役面が長い方向、即ち、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位或いは13位に特定の置換基を導入してなる化合物である。12位或いは13位に特定の置換基、具体的には、式[3]乃至[7]のうちのいずれかを導入することにより、共役長が伸びて振動子強度が高まる。またこれと同時に、青色発光に適した発光波長として、特に、460nm以下の良好な色純度を満たすために設計された分子構造である。
【0041】
ここで460nm以下の良好な青色発光を高い量子収率で出力させるために、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位置或いは13位に導入する置換基としては、式[3]乃至[7]から選ばれる置換基が望ましい。特に、式[3]又は[4]で表わされる置換基が好ましい。
【0042】
また、本発明の縮合多環化合物は、骨格内に二つの5員環構造を有するため、HOMO−LUMOのエネルギーレベルが低くなる。ここで、本発明の縮合多環化合物の基本骨格に、電子吸引性の置換基、具体的には、式[3]乃至[7]から選ばれる置換基のいずれかを導入することで、さらに分子全体のHOMO−LUMOのエネルギーレベルが低くなる。一方、酸化電位が低くなるということは、酸化されるのにより必要なエネルギーがより多くなることを意味する。従って、本発明の縮合多環化合物は酸化に対して安定である。
【0043】
本発明の縮合多環化合物は、有機発光素子の構成材料として使用できる。その中でも、発光層の構成材料として使用すると、層内においてはドーパント(ゲスト)として使用できる。このため、本発明の縮合多環化合物を発光層の構成材料とすることで発光効率が高くなり、長い期間高輝度を保ち、通電劣化が小さい素子を得ることができる。
【0044】
また発光層が、キャリア輸送性のホストとゲストからなる場合、発光にいたる主な過程は、以下に示される過程(a)乃至(d)からなる。
(a)発光層内での電子・ホールの輸送。
(b)ホストの励起子生成。
(c)ホスト分子間の励起エネルギー伝達。
(d)ホストからゲストへの励起エネルギー移動。
【0045】
それぞれの過程における所望のエネルギー移動や、発光はさまざまな失活過程と競争でおこる。
【0046】
有機発光素子の発光効率を高めるためには、発光中心材料そのものの発光量子収率を大きくすることは言うまでもない。しかしながら、ホスト−ホスト間、あるいはホスト−ゲスト間のエネルギー移動が如何に効率的にできるかも大きな問題となる。また、通電による発光劣化は今のところ原因は明らかではないが、少なくとも発光中心材料そのもの、又は、その周辺分子による発光材料の環境変化に関連したものと想定される。
【0047】
そこで本発明者らは種々の検討を行い、一般式[1]、[2]、[8]及び[9]で表されるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体に着目した。そして、式[1]、[2]、[8]及び[9]で表される化合物が、特に発光層のゲストに用いた素子が高効率発光し、長い期間高輝度を保ち、通電劣化が小さいことを見出した。
【0048】
本発明に係る縮合多環化合物は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレンの基本骨格の12位置或いは13位にフェニル基を介して一般式[3]乃至[7]から選ばれる電子吸引性の置換基が導入されている化合物である。そしてこの化合物は、還元電位が高く電子受容性の大きいことに特徴がある材料である。ここで一般式[3]乃至[7]にて示される電子吸引性の置換基を適切に選択し還元電位をコントロールすることで電子移動度を調節することが可能となる。これにより、本発明者らは、種々のホストとの組み合わせにより、駆動電圧が低く長い期間高輝度を保ち、通電劣化の減少を可能にすることを見出した。
【0049】
本発明は、以上の考察のもとに分子設計が行われ発明がなされたものである。
【0050】
以下、本発明の縮合多環化合物であるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体の具体例を示す。但し、これらはあくまでも具体例を示すものであり、本発明は、これに限定されるものではない。
【0051】
(1)化合物例1:下記一般式[10]又は[11]で示される化合物群
【0052】
【化8】
(式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0053】
【化9】
【0054】
【化10】
【0055】
(2)化合物例2:下記一般式[12]又は[13]で示される化合物群
【0056】
【化11】
(式[12]及び[13]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0057】
【化12】
【0058】
【化13】
【0059】
(3)化合物例3:下記一般式[14]又は[15]で示される化合物群
【0060】
【化14】
(式[14]及び[15]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0061】
【化15】
【0062】
【化16】
【0063】
(4)化合物例4:下記一般式[16]又は[17]で示される化合物群
【0064】
【化17】
(式[16]及び[17]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0065】
【化18】
【0066】
【化19】
【0067】
(5)化合物例5:下記一般式[18]又は[19]で示される化合物群
【0068】
【化20】
(式[18]及び[19]において、Rは、水素原子又はメチル基である。)
【0069】
【化21】
【0070】
(6)化合物例6:下記一般式[20]又は[21]で示される化合物群
【0071】
【化22】
(式[20]及び[21]において、Rは、水素原子又はメチル基である。)
【0072】
【化23】
【0073】
(7)化合物例7:下記一般式[1]又は[2]で示され、式[1]、[2]中のQが下記一般式[5]又は[6]で示される化合物群
【0074】
【化24】
(式[1]、[2]、[5]及び[6]において、Rは、水素原子又はメチル基である。)
【0075】
【化25】
【0076】
(8)化合物例8:下記一般式[22]又は[23]で示され、式[22]、[23]中のQが下記一般式[5]又は[6]で示される化合物群
【0077】
【化26】
【0078】
【化27】
【0079】
[有機発光素子の説明]
次に、本発明の有機発光素子について説明する。
【0080】
本発明の有機発光素子は、一対の電極である陽極と陰極と、陽極と陰極との間に配置される有機化合物層とを有している。ここで有機化合物層は単層又は複数の層からなる積層体であり、本発明においては、この有機化合物層のうち少なくとも一層が本発明の縮合多環化合物を有している。
【0081】
本発明の有機発光素子を構成する有機化合物層は、少なくとも発光層を含んでなる単層又は複数の層からなる積層体である。有機化合物層が複数層から構成される積層体である場合、有機化合物層は、発光層の他に、ホール注入層、ホール輸送層、ホール・エキシトンブロッキング層、電子輸送層、電子注入層等のうちいずれかを有している積層体である。
【0082】
以下に本発明の有機発光素子の具体例を示す。
(i)(陽極/)発光層(/陰極)
(ii)(陽極/)正孔輸送層/電子輸送層(/陰極)
(iii)(陽極/)正孔輸送層/発光層/電子輸送層(/陰極)
(iv)(陽極/)正孔注入層/正孔輸送層/発光層/電子輸送層(/陰極)
(v)(陽極/)正孔輸送層/発光層/正孔・エキシトンブロッキング層/電子輸送層(/陰極)
【0083】
ただし上記(i)乃至(v)の構成は、あくまでもごく基本的な素子構成の具体例を示すものであり、本発明の有機発光素子における有機化合物層の構成はこれらに限定されるものではない。
【0084】
本発明の有機発光素子において、本発明の縮合多環化合物は、有機化合物層を構成する層のいずれかに含まれる。具体的には、正孔注入層、正孔輸送層、発光層、正孔・エキシトンブロッキング層、電子輸送層等に含まれる。好ましくは、発光層に含まれる。
【0085】
ここで本発明の縮合多環化合物が発光層に含まれる場合、発光層は、本発明の縮合多環化合物のみで構成されてもよいし、ホストとゲストとで構成されていてもよい。また発光層がホストとゲストとで構成される場合、本発明の縮合多環化合物はゲストとして用いることができる。
【0086】
ここで、本発明の縮合多環化合物をゲストとして用いる場合、ホストに対するゲストの濃度は0.1重量%以上30重量%以下であることが好ましく、0.5重量%以上10重量%以下であることがより好ましい。
【0087】
本発明の有機発光素子は、本発明の縮合多環化合物以外にも、必要に応じて従来公知の低分子系及び高分子系の材料を使用することができる。具体的には、正孔注入性材料、正孔輸送性材料、ホスト、電子注入性材料、電子輸送性材料等を構成材料として一緒に使用することができる。
【0088】
以下に、これらの具体例を挙げる。
【0089】
正孔注入性材料あるいは正孔輸送性材料としては、正孔移動度が高い材料であることが好ましい。正孔注入性能あるいは正孔輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられるが、もちろんこれらに限定されるものではない。
【0090】
ホストとしては、トリアリールアミン誘導体、フェニレン誘導体、縮合環芳香族化合物(例えばナフタレン誘導体、フェナントレン誘導体、フルオレン誘導体、クリセン誘導体、アントラセン誘導体、ピレン誘導体等)、有機金属錯体(例えば、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、有機イリジウム錯体、有機プラチナ錯体等)及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体、ポリ(チエニレンビニレン)誘導体、ポリ(アセチレン)誘導体等の高分子誘導体が挙げられるが、もちろんこれらに限定されるものではない。
【0091】
電子注入性材料あるいは電子輸送性材料としては、ホール注入性材料あるいはホール輸送性材料のホール移動度とのバランス等を考慮し選択される。電子注入性能あるいは電子輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられるが、もちろんこれらに限定されるものではない。
【0092】
次に、有機化合物層以外の部材の構成材料について説明する。
【0093】
陽極の構成材料としては、仕事関数がなるべく大きなものがよい。例えば、金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれら金属単体を複数組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物である。また、ポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーでもよい。これらの電極物質は一種類を単独で使用してもよいし二種類以上を併用して使用してもよい。また、陽極は一層構成でもよく、多層構成でもよい。
【0094】
一方、陰極の構成材料としては、仕事関数の小さなものがよい。例えば、リチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えば、マグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また、陰極は一層構成でもよく、多層構成でもよい。
【0095】
本発明の有機発光素子において、本発明の縮合多環化合物を含有する層及びその他の有機化合物からなる層は、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により層を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で形成する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0096】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコーン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として1種単独で使用してもよいし、2種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0097】
[有機発光素子の用途]
本発明に係る有機発光素子は、表示装置や照明装置の構成部材として用いることができる。他にも電子写真方式の画像形成装置の露光光源や、液晶表示装置のバックライト等としての用途がある。
【0098】
ここで表示装置とは、本発明の有機発光素子を表示部に有する装置である。この表示部は複数の画素を有しており、各画素はそれぞれ本発明の有機発光素子と、本発明の有機発光素子の発光輝度を制御するためのTFT素子等のスイッチング素子と、を有している。ここでスイッチング素子としてTFT素子が使用されている場合、本発明の有機発光素子の構成部材である陽極又は陰極とTFT素子のドレイン電極又はソース電極とが電気的に接続されている。尚、表示装置は、例えば、PC等の画像表示装置として用いることができる。
【0099】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの情報を入力する画像入力部を有し、入力された画像を表示部に出力する画像出力装置であってもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報に基づいて画像を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。さらに表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0100】
次に、本発明の有機発光素子を使用した表示装置について、図面を参照しながら説明する。
【0101】
図1は、本発明の有機発光素子と、この有機発光素子に電気接続するスイッチング素子の一例であるTFT素子と、を有する表示装置の例を示す断面模式図である。構造の詳細を以下に説明する。
【0102】
図1の表示装置20は、ガラス等の基板1とその上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属のゲート電極3である。符号4はゲート絶縁膜4であり、5は半導体層である。
【0103】
TFT素子8は半導体層5とドレイン電極6とソース電極7とを有している。TFT素子8の上部には絶縁膜9が設けられている。コンタクトホール(スルーホール)10を介して有機発光素子の陽極11とソース電極7とが接続されている。尚、本発明の表示装置は、図1の構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子ソース電極又はドレイン電極のいずれか一方とが接続されていればよい。
【0104】
図1の表示装置20において、単層あるいは多層構造である有機化合物層12は、1つの層の如き図示がなされている。陰極13の上には有機発光素子の劣化を抑制するための第一の保護層14や第二の保護層15が設けられている。
【0105】
尚、本発明の表示装置において、表示装置を構成するスイッチング素子は、特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0106】
以下、本発明について実施例を用いて詳細に説明する。ただし本発明はこれらに限定されるものではない。
【0107】
[実施例1]例示化合物D1、D2の合成
以下に説明する合成方法に従い、例示化合物D1、D2を合成した。
【0108】
(1)中間体化合物M1の合成
【0109】
【化28】
【0110】
アルゴン雰囲気下、2Lの四つ口フラスコに、下記に示す試薬、溶媒を仕込んだ。
3−アミノフルオランテン:45.4g(209mmol)
DMF:910mL
【0111】
次に、反応系内を遮光した状態で、氷浴を用いて反応溶液を0℃まで冷却した。次に、NBS(37.2g、209mmol)を加えた後、氷浴を外して反応溶液を室温に戻した。次に、反応溶液を室温で1時間攪拌した。次に、反応溶液を氷水2L中に注いだ時に析出した結晶(黄緑色結晶)をろ取した。次に、ろ取した結晶を酢酸エチル4L中に溶解し、硫酸マグネシウム50g、フロリジル500gに順次通液して原料物質を除去した。次に、ろ液を減圧濃縮したときに生じたスラリー状の物質をろ過して結晶を得た。次に、得られた結晶を減圧乾燥することにより、2−ブロモフルオランテン−3−アミンを54.0g(収率87.1%)得た。
【0112】
続いてアルゴン雰囲気下、5Lの四つ口フラスコに、下記に示す試薬、溶媒を仕込んだ。
2−ブロモフルオランテン−3−アミン:55.6g(188mmol)
特級シクロペンチルメチルエーテル:1.12L
【0113】
次に、反応溶液を攪拌しながら氷浴を用いて反応溶液を内温0℃に冷却した。次に、n−BuLiの1.65M溶液596mL(939mmol、5eq)を30分かけて滴下した。次に、反応溶液を室温に戻した後、この温度(室温)で3.5時間攪拌した。次に、反応系内に炭酸ガスを吹き込んだ後(この炭酸ガスの吹込みにより反応溶液の内温が18℃から48℃に上昇)、反応系にガスバックを取り付けてから反応溶液を一晩攪拌した。次に、反応溶液中に5mol/L塩酸水溶液を滴下した。ここで塩酸水溶液の滴下の際に析出した結晶をろ過したがそのほとんどが目的物以外の化合物であった。そこでろ過の際に得られたろ液について分液操作を行うことで得られた有機層をフロリジル500gに通液した。次に、通液したろ液を濃縮乾固することで得られた残渣をTHF/CHCl3混合溶媒で再沈殿を行うことで、粗結晶を10.5g得た(HPLC純度92%)。次に、得られた粗結晶を再度THF/CHCl3で再沈殿を行うことで、結晶Aを8.50gを得た(HPLC純度98%)。次に、2回目の再沈殿の際に得たろ液をカラムクロマトグラフィー(ゲル1kg、展開溶媒:ヘプタン/酢酸エチル=1/1)で精製することで、結晶Bを900mg得た(HPLC純度98%)。次に、結晶A及び結晶Bを合わせた上でTHFに溶解して均一な溶液にした後減圧濃縮することでスラリー状物質を得た。次に、このスラリー状の物質にクロロホルムを加えることで結晶Cを取り出した。次に、減圧下で結晶Cを乾燥することにより、中間体化合物M1を9.23g(収率18.6%)得た。
【0114】
(2)中間体化合物M2の合成
【0115】
【化29】
【0116】
窒素雰囲気下、500mLの三口反応容器に、下記に示す試薬、溶媒を仕込んだ。
5−ブロモ−アセナフチレン−1,2−ジオン:15.0g(57.5mmmol)
1,3−ジフェニルプロパン−2−オン:12.1g(57.5mmol)
エタノール:300mL
【0117】
次に、室温にて、反応溶液中に6M水酸化ナトリウム水溶液20mLを滴下した。滴下終了後反応溶液を60℃まで加熱して、この温度(60℃)で反応溶液を約2時間撹拌した。次に、反応溶液を室温まで冷却した際に生成した析出物をろ過し、この析出物を水、エタノールで順次洗浄した。次に、この析出物を80℃で減圧加熱乾燥を行うことにより、中間体化合物M2を濃緑色の固体として22.0g(収率88%)得た。
【0118】
(3)中間体化合物M3(M3−1、M3−2)の合成
【0119】
【化30】
【0120】
窒素雰囲気下かつ室温下において、1Lの三口反応容器に、下記に示す試薬、溶媒を入れた。
中間体化合物M2:18.0g(41.3mmol)
中間体化合物M1:14.0g(53.8mmol)
亜硝酸イソアミル:8.72g(74.4mmol)
トルエン:720mL
【0121】
次に、反応溶液を95℃に加熱して、この温度(95℃)で反応溶液を1.5時間撹拌した。次に、反応溶液を冷却した後、反応溶液を水1Lで3回洗浄した。次に、水による洗浄により得られた有機層を飽和食塩水でさらに洗浄した後、硫酸マグネシウムで乾燥した。次に、この有機層をろ過することで得られたろ液を減圧濃縮することで茶褐色の液体を得た。次に、得られた茶褐色の液体をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/3)にて精製した。次に、この精製の際に得られた溶液を減圧濃縮して結晶を得た。次に、得られた結晶にメタノール345mLを添加し加熱スラリー洗浄を行った後、室温まで放冷した時に生成した結晶をろ過することにより、黄色結晶の中間体化合物M3を、異性体の混合物(M3−1、M3−2)として21.9g(収率:87%)得た。
【0122】
(4)中間体化合物M4(M4−1、M4−2)の合成
【0123】
【化31】
【0124】
窒素雰囲気下、1Lの三口容器に、下記に示す試薬、溶媒を入れた。
中間体化合物M3(M3−1とM3−2との混合物):19.4g(31.9mmol)
ビス−ジオキサボロラン:16.2g(63.9mmol)
酢酸カリウム:6.27g(63.9mmol)
テトラキストリフェニルフォスフィンパラジウム:1.107g(0.96mmol)
DMSO:388mL
【0125】
次に、反応溶液を100℃まで加熱した後、この温度(100℃)下で反応溶液を1.5時間撹拌を行った。次に、反応溶液を30℃まで冷却した後に、メタノール400mLを加えた。次に、反応溶液を10℃まで冷却してからこの温度(10℃)下で反応溶液を1時間撹拌を行った後、ろ過を行った。次に、ろ過によって得られたろ物についてメタノール800mLを加え、溶媒(メタノール)を還流させながら加熱スラリー洗浄を1時間行った後、室温まで冷却しろ過を行った。これにより、中間体化合物M4の粗結晶を18.3g(収率:87.5%)得た。次に、得られた粗結晶をトルエン265mLに溶解させて得た溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/2→トルエン/ヘプタン=1/1)にて精製した。次に、トルエン/メタノールで加熱スリラー洗浄を行うことにより、黄色結晶の中間体化合物M4を、異性体の混合物(M4−1、M4−2)として9.75g(収率:46.6%)得た。
【0126】
(5)中間体化合物M5の合成
【0127】
【化32】
【0128】
窒素雰囲気下、200mLの三口容器に、下記に示す試薬、溶媒を入れた。
4−ブロモ−3,5−ジメチルフェノール:3.19g(16mmol)
ピリジン:5.65g(71mmol)
トルエン:65mL
【0129】
次に、反応溶液を撹拌しながら0℃まで冷却した。次に、反応溶液を撹拌しながらトリフルオロメタンスルホン酸無水物13.43g(48mmol)を約15分かけてゆっくり滴下した。滴下終了後、反応溶液を室温まで昇温し、室温下でさらに反応溶液を1時間撹拌した。次に、水100mLを添加して反応を停止し、続いてトルエン40mLを添加した後、分液操作を行い有機層を分離した。次に、得られた有機層を減圧濃縮した後、カラムクロマトグラフィー(展開溶媒:トルエン)にて精製した。このときに得られた溶液を減圧濃縮することにより、無色透明液体の4−ブロモ−3,5−ジメチルフェニル−トルフルオロメタンスルホン酸エステルを5.94g(収率:97.1%)得た。
【0130】
続いて窒素雰囲気下、500mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
4−ブロモ−3,5−ジメチルフェニル−トルフルオロメタンスルホン酸エステル:4.90g(14.7mmol)
4−シアノフェニルボロン酸:2.05g(14.0mmol)
ジオキサン:245mL
炭酸カリウム:4.07g(29.4mmol)
テトラキストリフェニルホスフィンパラジウム:0.17g(0.15mmol)
【0131】
次に、反応溶液を80℃に加熱し、この温度(80℃)下で反応溶液を1.5時間撹拌した。次に、反応溶液を室温まで放冷した後、トルエン100mL及び水100mLを添加し分液操作を行い、有機層を分離した。次に、得られた有機層を硫酸マグネシウムで乾燥した後、この有機層をろ過することで得られたろ液を減圧濃縮することで中間体化合物5の粗結晶を4.64g得た。次に、得られた粗結晶とトルエンとを混合することで得た溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/5→トルエン/ヘプタン=1/4)にて精製した後、濃縮乾固化した。以上により、白色結晶の中間体化合物5を1.54g(収率:37%)得た。
【0132】
(6)例示化合物D1、D2の合成
【0133】
【化33】
【0134】
窒素雰囲気下、500mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):4.40g(6.72mmol)
中間体化合物M5:1.54g(5.38mmol)
トルエン:220mL
エタノール:110mL
炭酸ナトリウム:1.42g(13.4mmol)
水:100mL
テトラキストリフェニルホスフィンパラジウム:0.39g(0.34mmol)
【0135】
次に、反応溶液を70℃に加熱して、この温度(70℃)下にて反応溶液を3時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで2回洗浄することで有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、溶液をろ過することで得られたろ液を減圧濃縮することで黄色粗結晶を得た。次に、この粗結晶をトルエン60mlとヘプタン60mLとを混合して調製した溶媒に溶解させた後、この溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/1→トルエン/ヘプタン=2/1)にて精製した。次に、溶液を減圧濃縮した後、メタノールを添加して加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D1及びD2の混合物を3.26g(収率:66%)得た。尚、例示化合物D1は、中間化合物M4−1から合成されるものであり、例示化合物D2は、中間化合物M4−2から合成されるものである。
【0136】
質量分析法により、この化合物のM+である733を確認した。また、NMR測定により、例示化合物D1とD2の混合比が1:1であることを確認した。
【0137】
1H−NMR(CDCl3):δ(ppm)=8.20(s,2H),7.89−7.97(m,4H),7.83−7.65(m,30H),7.40−7.37(m,6H),7.34−7.28(m,6H),7.25−7.20(m,4H),7.12(d,1H,J=7.99Hz),7.09(d,1H,J=7.99Hz),6.61(d,1H,J=7.99Hz),6.53(dt,1H,J1=7.99Hz,J2=3.99Hz),6.38(d,1H,J=7.99Hz),6.30(dd,1H,J1=7.99Hz,J2=3.99Hz),1.97(s,12H)
【0138】
さらに例示化合物D1及びD2の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク450nmに最大強度を有する青色発光スペクトルが確認された。
【0139】
[実施例2]例示化合物D87、D88の合成
実施例1(6)において、中間体化合物M5の代わりに下記に示される中間体化合物M6を用いたことを除いては、実施例1と同様の方法で合成を行い、例示化合物D87、D88(の混合物)を得た。以下に、中間体化合物M6、並びに例示化合物D87及びD88の具体的な合成方法について説明する。
【0140】
(1)中間体化合物M6の合成
【0141】
【化34】
【0142】
Chemistry of Materials,2009,21(12),2452に記載の方法で、1−フルオロ−2−ニトロベンゼンと1−ブロモ−4−アミノベンゼンとを出発物質として、中間体化合物M6を30g合成した。
【0143】
(2)例示化合物D87、D88の合成
【0144】
【化35】
【0145】
窒素雰囲気下、200mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):1.30g(1.99mmol)
中間体化合物M6:1.54g(1.39mmol)
トルエン:39mL
エタノール:13mL
炭酸ナトリウム:0.42g(3.97mmol)
水:13mL
テトラキストリフェニルホスフィンパラジウム:0.12g(0.11mmol)
【0146】
次に、反応溶液を70℃に加熱し、この温度(70℃)下で反応溶液を5時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで3回洗浄し有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、ろ過により得られたろ液を減圧濃縮することで黄色の粗結晶を得た。次に、得られた粗結晶をトルエン19mと混合して得られた溶液について、カラムクロマトグラフィー(展開溶媒:トルエン/酢酸エチル=9/1→トルエン/酢酸エチル=5/1)にて精製した。次に、溶液を濃縮した後メタノールを添加し加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D87及びD88の混合物を0.96g(収率:58%)得た。尚、例示化合物D87は、中間化合物M4−1から合成されるものであり、例示化合物D88は、中間化合物M4−2から合成されるものである。
【0147】
質量分析法により、この化合物のM+である796を確認した。また、NMR測定により、例示化合物D87とD88との混合比が1:1であることを確認した。
【0148】
1H−NMR(CDCl3):δ(ppm)=8.21(s,1H),8.20(s,1H),7.93−7.63(m,38H),7.43−7.29(m,28H),6.59(dd,2H,J1=7.99Hz,J2=3.99Hz),6.35(d,2H,J=7.99Hz)
【0149】
さらに例示化合物D87及びD88の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク455nmに最大強度を有する青色発光スペクトルが確認された。
【0150】
[実施例3]例示化合物D83、D84の合成
実施例1(6)において、中間体化合物M5の代わりに下記に示される中間体化合物M7を用いたことを除いては、実施例1と同様の方法で合成を行い、例示化合物D83、D84(の混合物)を得た。以下に、中間体化合物M7、並びに例示化合物D83及びD84の具体的な合成方法について説明する。
【0151】
(1)中間体化合物M7の合成
【0152】
【化36】
【0153】
Heterocycles,2001,55,1329に記載の方法で、3−ヒドロキシ−2−アミノピリジンと3−ヨード−1−カルボキシルベンゼンとを出発物質として、中間体化合物M7を22g合成した。
【0154】
(2)例示化合物D83、D84の合成
【0155】
【化37】
【0156】
窒素雰囲気下、200mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):1.40g(2.10mmol)
中間体化合物M7:0.48g(1.39mmol)
トルエン:70mL
エタノール:30mL
炭酸ナトリウム:0.17g(3.97mmol)
水:30mL
テトラキストリフェニルホスフィンパラジウム:0.12g(0.11mmol)
【0157】
次に、反応溶液を70℃に加熱し、この温度(70℃)下で反応溶液を2.5時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで3回洗浄し有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、ろ過により得られたろ液を減圧濃縮することで黄色の粗結晶を得た。次に、得られた粗結晶をトルエン20mと混合して得られた溶液について、カラムクロマトグラフィー(展開溶媒:トルエン/酢酸エチル=9/1)にて精製した。次に、溶液を濃縮した後メタノールを添加し加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D83及びD84の混合物を0.93g(収率:56%)得た。尚、例示化合物D83は、中間化合物M4−1から合成されるものであり、例示化合物D84は、中間化合物M4−2から合成されるものである。
【0158】
質量分析法により、この化合物のM+である722を確認した。
【0159】
また、NMR測定により、例示化合物D83とD84との混合比が1:1であることを確認した。
【0160】
1H−NMR(CDCl3):δ(ppm)=8.59(dd,2H,J1=3.99Hz,J2=1.33Hz),8.48(s,2H),8.38(d,2H,J=7.99Hz),8.22(s,1H),8.21(s,1H),7.88−7.64(m,34H),7.42−7.28(m,14H),6.59(dd,2H,J1=9.99Hz,J2=7.99Hz),6.35(dd,2H,J1=7.99Hz,J2=7.99Hz)
【0161】
さらに例示化合物D83及びD84の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク453nmに最大強度を有する青色発光スペクトルが確認された。
【0162】
[比較例1]比較化合物R1、R2の合成
【0163】
【化38】
【0164】
実施例1(6)において、中間体化合物M4(M4−1とM4−2との混合物)の代わりに中間体化合物M3(M3−1とM3−2との混合物)を用いた。また中間体化合物M5の代わりに2,6−ジメチルフェニルボロン酸を用いた。これらを除いては、実施例1と同様の方法で合成を行い、比較化合物R1及びR2を、R1とR2との混合物として0.95g(収率:75%)得た。
【0165】
質量分析法により、この化合物のM+である632を確認した。
【0166】
また比較化合物R1及びR2の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長414nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク447nmに最大強度を有する青色発光スペクトルが確認された。
【0167】
[比較例2]比較化合物R3、R4の合成
【0168】
【化39】
【0169】
実施例1(6)において、中間体化合物M4(M4−1とM4−2との混合物)の代わりに中間体化合物M3(M3−1とM3−2との混合物)を用いた。また中間体化合物M5の代わりに4−シアノフェニルボロン酸を用いた。これらを除いては、実施例1と同様の方法で合成を行い、比較化合物R3及びR4を、R3とR4との混合物として0.8g(収率:70%)得た。
【0170】
質量分析法により、この化合物のM+である629を確認した。
【0171】
また比較化合物R3及びR4の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長414nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク458nmに最大強度を有する青色発光スペクトルが確認された。
【0172】
[蛍光量子収率及び酸化還元電位の比較]
以下に説明する方法で、実施例1乃至3にてそれぞれ得られた化合物(例示化合物D1、D2、D83、D84、D87、D88)及び比較例1及び2にて得られた化合物(比較化合物R1、R2、R3、R4)について量子収率及び酸化還元電位を評価した。
【0173】
(1)蛍光量子収率
まず濃度10-6mol/lのトルエン溶液を調製した。次に、調製した溶液について、絶対PL量子収率測定装置(浜松ホトニクス社製、商品名:C9920)を用い、励起波長を414nmとしたときの値を測定した。そして、比較化合物R1及びR2の混合物(混合比=1:1)を1とした場合の発光量子収率の相対値を求めた。結果を表1に示す。
【0174】
(2)酸化還元電位
サイクリックボルタンメトリー法により酸化還元電位を以下のように測定した。具体的には、0.1mol/Lのテトラブチルアンモニウムパークロレイトを支持電解物質とし、以下の条件で、酸化電位及び還元電位を測定した。酸化電位は、各化合物の1×10-4mol/L以上1×10-6mol/L以下の塩化メチレン溶液中で測定した。還元電位は、各化合物の1×10-4mol/L以上1×10-6mol/L以下のN,N−ジメチルホルムアミド溶液中で測定した。尚、測定にあたり測定温度を各々25℃に設定し、Ag/AgNO3を参照電極とし、白金電極を対向電極とし、グラシックカーボンを作用電極としてそれぞれ用いて測定を行った。結果を表1に示す。
【0175】
【表1】
【0176】
表1より、本発明のアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、比較化合物R1,R2よりも蛍光量子収率が1.1倍以上向上していることがわかった。
【0177】
また、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位或いは13位に直接電子吸引性の4−シアノフェニル基が導入された比較化合物R3,R4は、還元電位が最も深くなり4−シアノフェニル基が導入された効果が大きい。しかしその一方で、比較化合物R1,R2よりも蛍光量子収率が低下していることがわかった。
【0178】
以上より、本発明のアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の特定の位置にフェニル基を介して特定の電子吸引性の置換基を導入することで、深い還元電位を有するのと同時に高い蛍光量子収率を有する材料であることが示された。
【0179】
[実施例4]
本実施例では、基板上に、陽極、正孔輸送層、発光層、正孔・エキシトンブロッキング層、電子輸送層及び陰極が順次形成されている有機発光素子を作製した。以下に、本実施例で使用した材料の一部を示す。
【0180】
【化40】
【0181】
有機発光素子の具体的な製造方法を以下に説明する。
【0182】
スパッタ法により、ガラス基板上に、ITOを成膜して陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO基板)として以下の工程で使用した。次に、このITO基板上に、下記表2に示される有機化合物層及び電極層を、1×10-5Paの真空チャンバー内で抵抗加熱による真空蒸着によって連続的に成膜した。このとき対向する電極(金属電極層、陰極)の面積が3mm2になるように作製した。
【0183】
【表2】
【0184】
得られた素子について、素子の特性を測定・評価した。具体的には、電流電圧特性をヒューレッドパッカード社製・微小電流計4140Bで測定し、発光輝度は、トプコン社製BM7で測定した。測定の結果を表3に示す。
【0185】
[実施例5,6]
実施例4において、ゲストを、表3に示される化合物に適宜変更する以外は、実施例4と同様の方法により有機発光素子を作製した。得られた素子について実施例4と同様に素子の特性を測定・評価した。測定の結果を表3に示す。
【0186】
[比較例3]
実施例4において、ゲストを、比較化合物R1、R2(R1とR2との重量混合比は、1:1である。)に適宜変更する以外は、実施例4と同様の方法により有機発光素子を作製した。得られた素子について実施例4と同様に素子の特性を測定・評価した。測定の結果を表3に示す。
【0187】
【表3】
【0188】
[結果と考察]
本発明の縮合多環化合物は、高い量子収率と青色発光を呈する有機化合物であり、有機発光素子の構成材料に用いた場合、良好な色度と高効率を示すと同時に特に耐久特性が良好な発光素子を提供することができる。
【符号の説明】
【0189】
8:TFT素子、11:陽極、12:有機化合物層、13:陰極
【技術分野】
【0001】
本発明は、縮合多環化合物及びこれを用いた有機発光素子に関する。
【背景技術】
【0002】
有機発光素子は、一対の電極間に蛍光性又は燐光性有機化合物を含む薄膜が挟持されている電子素子である。各電極から電子及びホール(正孔)を注入することにより、蛍光性又は燐光性化合物の励起子が生成され、この励起子が基底状態に戻る際に、有機発光素子は光を放出する。
【0003】
有機発光素子における最近の進歩は著しく、その特徴として、低印加電圧で高輝度、発光波長の多様性、高速応答性、薄型、軽量の発光デバイス化の可能性であることが挙げられる。このことから、有機発光素子は広汎な用途への可能性を示唆している。
【0004】
ところで、有機発光素子の構成材料として用いられる蛍光性又は燐光性有機化合物についても盛んに研究がなされている。その中でも青色発光材料については、例えば、特許文献1や2に示されている有機化合物が提案されている。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−254610号公報
【特許文献2】特開2010−270103号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
しかし特許文献1や2にて提案されている有機化合物及びこれを用いた有機発光素子は、実用化という観点からさらなる改善の余地がある。
【0007】
具体的には、実用化のためにはさらなる高輝度の光出力あるいは高変換効率が必要であること、長時間の使用による経時変化や酸素を含む雰囲気気体や湿気等による劣化等の耐久面において改善が必要であること、が挙げられる。さらにはフルカラーディスプレイ等への応用を考えた場合、使用される青色発光する有機発光素子には色純度が良く高効率の青色発光が要求されるが、これらの問題に関してもまだ十分に解決されたとはいえない。
【0008】
従って、青色発光する有機発光素子に関して言えば、特に、色純度、発光効率及び耐久性が高い有機発光素子及びこれを実現するための材料が要求されている。
【0009】
本発明は、上述した従来技術の問題点を解決するためになされたものである。即ち、本発明の目的は、主として青色発光素子の構成材料として用いるのに適した縮合多環化合物及びこれを用いた有機発光素子を提供することにある。
【課題を解決するための手段】
【0010】
本発明の縮合多環化合物は、下記一般式[1]、[2]、[8]又は[9]で示される化合物であることを特徴とする。
【0011】
【化1】
(式[1]、[2]、[8]及び[9]において、Rは、水素原子又はメチル基を表す。式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【0012】
【化2】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【発明の効果】
【0013】
本発明の縮合多環化合物は、発光ピークが460nm以下の純度のよい青色発光色相を呈する。また本発明の縮合多環化合物を用いた有機発光素子は、低い印加電圧で高輝度な発光が得られ、耐久性にも優れる。従って、本発明によれば、主として青色発光素子の構成材料として用いるのに適した縮合多環化合物及びこれを用いた有機発光素子を提供することができる。
【図面の簡単な説明】
【0014】
【図1】本発明の有機発光素子と、この有機発光素子に電気接続するスイッチング素子の一例であるTFT素子と、を有する表示装置の例を示す断面模式図である。
【発明を実施するための形態】
【0015】
本発明の縮合多環化合物であるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、下記一般式[1]、[2]、[8]又は[9]で示される化合物である。
【0016】
【化3】
【0017】
式[1]、[2]、[8]及び[9]において、Rは、水素原子又はメチル基を表す。ここで式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。
【0018】
式[1]、[2]、[8]及び[9]において、Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【0019】
【化4】
【0020】
式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。
【0021】
R1乃至R4で表されるアルキル基として、メチル基、エチル基、n−プロピル基、n−ブチル基、iso−プロピル基、iso−ブチル基、sec−ブチル基、tert−ブチル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基、パーフルオロエチル基、3−フルオロプロピル基、パーフルオロプロピル基、4−フルオロブチル基、パーフルオロブチル基、シクロプロピル基、シクロブチル基等が挙げられるが、もちろんこれらに限定されるものではない。
【0022】
尚、R1乃至R4は、それぞれ同じであってもよいし異なっていてもよい。
【0023】
式[7]において、R5は、水素原子又はメチル基である。
【0024】
ここで本発明の縮合多環化合物が式[1]又は[2]で示される化合物である場合、好ましくは、下記一般式[10]、[11]、[30]又は[31]で示される化合物である。
【0025】
【化5】
【0026】
式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。
【0027】
R1乃至R4で表されるアルキル基として、メチル基、エチル基、n−プロピル基、n−ブチル基、iso−プロピル基、iso−ブチル基、sec−ブチル基、tert−ブチル基、フルオロメチル基、ジフルオロメチル基、トリフルオロメチル基、2−フルオロエチル基、2,2,2−トリフルオロエチル基、パーフルオロエチル基、3−フルオロプロピル基、パーフルオロプロピル基、4−フルオロブチル基、パーフルオロブチル基、シクロプロピル基、シクロブチル基等が挙げられるが、もちろんこれらに限定されるものではない。
【0028】
式[30]及び[31]において、Rは、水素原子又はメチル基を表す。ここで式[30]にて示されるRは同一の置換基である。式[31]にて示されるRは同一の置換基である。
【0029】
一方、本発明の縮合多環化合物が式[8]又は[9]で示される化合物である場合、好ましくは、下記一般式[20]又は[21]で示される化合物である。
【0030】
【化6】
【0031】
式[20]及び[21]において、Rは、水素原子又はメチル基を表す。
【0032】
[合成ルートの説明]
本発明の縮合多環化合物、具体的には、一般式[1]、[2]、[8]又は[9]に示される化合物は、例えば、以下に示す合成ルートに従って合成することができる。
【0033】
【化7】
【0034】
この合成ルートでは、基本骨格が共通するが置換基Arの置換位置が異なる二種類の化合物が同時に得られる。具体的には、式[1]に示される化合物と式[2]に示される化合物との混合物、あるいは式[8]に示される化合物と式[9]に示される化合物との混合物が得られる。ただし式[1]に示される化合物と式[2]に示される化合物との関係、あるいは式[8]に示される化合物と式[9]に示される化合物との関係においては、Arが同じであれば、Arの置換位置によっては発光特性にほとんど差がない。このため、再結晶等で単離して使用しても良いし、混合物の状態でそのまま用いてもよい。また混合物をそのまま使用したとしても単体と比べて発光特性が特段低下することはないため、混合比は特に限定されない。また、混合物の状態で用いた際は、結晶性を抑えることができるため、濃度消光の抑制等の効果も期待できる。
【0035】
[化合物に関する考察]
以下、本発明の縮合多環化合物についてさらに詳細を説明する。
【0036】
一般的に有機発光素子の発光効率を高めるためには、発光中心そのものの発光量子収率が大きいことが望まれる。そのためには、下記(A)及び(B)が要求されている。
(A)振動子強度が高いこと
(B)発光にかかわる骨格の振動部分が少ないこと
【0037】
(A)に関しては、分子の発光に関わる骨格の対称性を高くすることが重要である。ただし、高対称性分子特有の禁制遷移条件によっては全く発光しなくなる場合もある。ところで振動子強度を高くする方法として、最も共役面の長い方向を軸として、さらに共役を伸ばす方法がある。この方法を採用することによって分子の双極子モーメントが大きくなり振動子強度が向上する。この点では、本発明の縮合多環化合物、特に、一般式[1]又は[2]で表わされる化合物は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の最も共役面が長い方向を軸とした上で、さらに共役長が伸びる位置に特定の置換基を有している。このように分子設計を行うことで分子全体の振動子強度が向上した構造になっている。
【0038】
(B)に関しては、発光に関わる骨格に回転構造を有さないことで、回転振動による量子収率の低下を抑制することができる。ここで本発明の縮合多環化合物の基本骨格は回転構造を有していない。従って回転振動による量子収率の低下を抑制することができる。
【0039】
一方、有機ELディスプレイとして青色発光に適した材料に求められる物性値の1つであるが、材料自体の発光ピークが430nm〜480nmの範囲にあることは重要である。特に、青色発光材料に対して色純度の良として要求される発光ピークの存在範囲としては440nm〜460nmであることがより好ましい。
【0040】
本発明の縮合多環化合物が有する基本骨格、即ち、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格は、これ自体で最大発光波長が青色領域に存在する。このため、この基本骨格に置換基を導入する位置及び置換基の種類を適切に選ぶことによって460nm以下の発光ピークを有する青色発光材料になる化合物を提供することができる。具体的には、一般式[1]、[2]、[8]又は[9]で表わされる縮合多環化合物は、上述のように基本骨格内で最も共役面が長い方向、即ち、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位或いは13位に特定の置換基を導入してなる化合物である。12位或いは13位に特定の置換基、具体的には、式[3]乃至[7]のうちのいずれかを導入することにより、共役長が伸びて振動子強度が高まる。またこれと同時に、青色発光に適した発光波長として、特に、460nm以下の良好な色純度を満たすために設計された分子構造である。
【0041】
ここで460nm以下の良好な青色発光を高い量子収率で出力させるために、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位置或いは13位に導入する置換基としては、式[3]乃至[7]から選ばれる置換基が望ましい。特に、式[3]又は[4]で表わされる置換基が好ましい。
【0042】
また、本発明の縮合多環化合物は、骨格内に二つの5員環構造を有するため、HOMO−LUMOのエネルギーレベルが低くなる。ここで、本発明の縮合多環化合物の基本骨格に、電子吸引性の置換基、具体的には、式[3]乃至[7]から選ばれる置換基のいずれかを導入することで、さらに分子全体のHOMO−LUMOのエネルギーレベルが低くなる。一方、酸化電位が低くなるということは、酸化されるのにより必要なエネルギーがより多くなることを意味する。従って、本発明の縮合多環化合物は酸化に対して安定である。
【0043】
本発明の縮合多環化合物は、有機発光素子の構成材料として使用できる。その中でも、発光層の構成材料として使用すると、層内においてはドーパント(ゲスト)として使用できる。このため、本発明の縮合多環化合物を発光層の構成材料とすることで発光効率が高くなり、長い期間高輝度を保ち、通電劣化が小さい素子を得ることができる。
【0044】
また発光層が、キャリア輸送性のホストとゲストからなる場合、発光にいたる主な過程は、以下に示される過程(a)乃至(d)からなる。
(a)発光層内での電子・ホールの輸送。
(b)ホストの励起子生成。
(c)ホスト分子間の励起エネルギー伝達。
(d)ホストからゲストへの励起エネルギー移動。
【0045】
それぞれの過程における所望のエネルギー移動や、発光はさまざまな失活過程と競争でおこる。
【0046】
有機発光素子の発光効率を高めるためには、発光中心材料そのものの発光量子収率を大きくすることは言うまでもない。しかしながら、ホスト−ホスト間、あるいはホスト−ゲスト間のエネルギー移動が如何に効率的にできるかも大きな問題となる。また、通電による発光劣化は今のところ原因は明らかではないが、少なくとも発光中心材料そのもの、又は、その周辺分子による発光材料の環境変化に関連したものと想定される。
【0047】
そこで本発明者らは種々の検討を行い、一般式[1]、[2]、[8]及び[9]で表されるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体に着目した。そして、式[1]、[2]、[8]及び[9]で表される化合物が、特に発光層のゲストに用いた素子が高効率発光し、長い期間高輝度を保ち、通電劣化が小さいことを見出した。
【0048】
本発明に係る縮合多環化合物は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレンの基本骨格の12位置或いは13位にフェニル基を介して一般式[3]乃至[7]から選ばれる電子吸引性の置換基が導入されている化合物である。そしてこの化合物は、還元電位が高く電子受容性の大きいことに特徴がある材料である。ここで一般式[3]乃至[7]にて示される電子吸引性の置換基を適切に選択し還元電位をコントロールすることで電子移動度を調節することが可能となる。これにより、本発明者らは、種々のホストとの組み合わせにより、駆動電圧が低く長い期間高輝度を保ち、通電劣化の減少を可能にすることを見出した。
【0049】
本発明は、以上の考察のもとに分子設計が行われ発明がなされたものである。
【0050】
以下、本発明の縮合多環化合物であるアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体の具体例を示す。但し、これらはあくまでも具体例を示すものであり、本発明は、これに限定されるものではない。
【0051】
(1)化合物例1:下記一般式[10]又は[11]で示される化合物群
【0052】
【化8】
(式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0053】
【化9】
【0054】
【化10】
【0055】
(2)化合物例2:下記一般式[12]又は[13]で示される化合物群
【0056】
【化11】
(式[12]及び[13]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0057】
【化12】
【0058】
【化13】
【0059】
(3)化合物例3:下記一般式[14]又は[15]で示される化合物群
【0060】
【化14】
(式[14]及び[15]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0061】
【化15】
【0062】
【化16】
【0063】
(4)化合物例4:下記一般式[16]又は[17]で示される化合物群
【0064】
【化17】
(式[16]及び[17]において、R1乃至R4は、それぞれ水素原子、メチル基、トリフルオロメチル基等の炭素数1以上4以下のアルキル基又はシアノ基である。)
【0065】
【化18】
【0066】
【化19】
【0067】
(5)化合物例5:下記一般式[18]又は[19]で示される化合物群
【0068】
【化20】
(式[18]及び[19]において、Rは、水素原子又はメチル基である。)
【0069】
【化21】
【0070】
(6)化合物例6:下記一般式[20]又は[21]で示される化合物群
【0071】
【化22】
(式[20]及び[21]において、Rは、水素原子又はメチル基である。)
【0072】
【化23】
【0073】
(7)化合物例7:下記一般式[1]又は[2]で示され、式[1]、[2]中のQが下記一般式[5]又は[6]で示される化合物群
【0074】
【化24】
(式[1]、[2]、[5]及び[6]において、Rは、水素原子又はメチル基である。)
【0075】
【化25】
【0076】
(8)化合物例8:下記一般式[22]又は[23]で示され、式[22]、[23]中のQが下記一般式[5]又は[6]で示される化合物群
【0077】
【化26】
【0078】
【化27】
【0079】
[有機発光素子の説明]
次に、本発明の有機発光素子について説明する。
【0080】
本発明の有機発光素子は、一対の電極である陽極と陰極と、陽極と陰極との間に配置される有機化合物層とを有している。ここで有機化合物層は単層又は複数の層からなる積層体であり、本発明においては、この有機化合物層のうち少なくとも一層が本発明の縮合多環化合物を有している。
【0081】
本発明の有機発光素子を構成する有機化合物層は、少なくとも発光層を含んでなる単層又は複数の層からなる積層体である。有機化合物層が複数層から構成される積層体である場合、有機化合物層は、発光層の他に、ホール注入層、ホール輸送層、ホール・エキシトンブロッキング層、電子輸送層、電子注入層等のうちいずれかを有している積層体である。
【0082】
以下に本発明の有機発光素子の具体例を示す。
(i)(陽極/)発光層(/陰極)
(ii)(陽極/)正孔輸送層/電子輸送層(/陰極)
(iii)(陽極/)正孔輸送層/発光層/電子輸送層(/陰極)
(iv)(陽極/)正孔注入層/正孔輸送層/発光層/電子輸送層(/陰極)
(v)(陽極/)正孔輸送層/発光層/正孔・エキシトンブロッキング層/電子輸送層(/陰極)
【0083】
ただし上記(i)乃至(v)の構成は、あくまでもごく基本的な素子構成の具体例を示すものであり、本発明の有機発光素子における有機化合物層の構成はこれらに限定されるものではない。
【0084】
本発明の有機発光素子において、本発明の縮合多環化合物は、有機化合物層を構成する層のいずれかに含まれる。具体的には、正孔注入層、正孔輸送層、発光層、正孔・エキシトンブロッキング層、電子輸送層等に含まれる。好ましくは、発光層に含まれる。
【0085】
ここで本発明の縮合多環化合物が発光層に含まれる場合、発光層は、本発明の縮合多環化合物のみで構成されてもよいし、ホストとゲストとで構成されていてもよい。また発光層がホストとゲストとで構成される場合、本発明の縮合多環化合物はゲストとして用いることができる。
【0086】
ここで、本発明の縮合多環化合物をゲストとして用いる場合、ホストに対するゲストの濃度は0.1重量%以上30重量%以下であることが好ましく、0.5重量%以上10重量%以下であることがより好ましい。
【0087】
本発明の有機発光素子は、本発明の縮合多環化合物以外にも、必要に応じて従来公知の低分子系及び高分子系の材料を使用することができる。具体的には、正孔注入性材料、正孔輸送性材料、ホスト、電子注入性材料、電子輸送性材料等を構成材料として一緒に使用することができる。
【0088】
以下に、これらの具体例を挙げる。
【0089】
正孔注入性材料あるいは正孔輸送性材料としては、正孔移動度が高い材料であることが好ましい。正孔注入性能あるいは正孔輸送性能を有する低分子及び高分子系材料としては、トリアリールアミン誘導体、フェニレンジアミン誘導体、スチルベン誘導体、フタロシアニン誘導体、ポルフィリン誘導体、ポリ(ビニルカルバゾール)、ポリ(チオフェン)、その他導電性高分子が挙げられるが、もちろんこれらに限定されるものではない。
【0090】
ホストとしては、トリアリールアミン誘導体、フェニレン誘導体、縮合環芳香族化合物(例えばナフタレン誘導体、フェナントレン誘導体、フルオレン誘導体、クリセン誘導体、アントラセン誘導体、ピレン誘導体等)、有機金属錯体(例えば、トリス(8−キノリノラート)アルミニウム等の有機アルミニウム錯体、有機ベリリウム錯体、有機イリジウム錯体、有機プラチナ錯体等)及びポリ(フェニレンビニレン)誘導体、ポリ(フルオレン)誘導体、ポリ(フェニレン)誘導体、ポリ(チエニレンビニレン)誘導体、ポリ(アセチレン)誘導体等の高分子誘導体が挙げられるが、もちろんこれらに限定されるものではない。
【0091】
電子注入性材料あるいは電子輸送性材料としては、ホール注入性材料あるいはホール輸送性材料のホール移動度とのバランス等を考慮し選択される。電子注入性能あるいは電子輸送性能を有する材料としては、オキサジアゾール誘導体、オキサゾール誘導体、ピラジン誘導体、トリアゾール誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、有機アルミニウム錯体等が挙げられるが、もちろんこれらに限定されるものではない。
【0092】
次に、有機化合物層以外の部材の構成材料について説明する。
【0093】
陽極の構成材料としては、仕事関数がなるべく大きなものがよい。例えば、金、白金、銀、銅、ニッケル、パラジウム、コバルト、セレン、バナジウム、タングステン等の金属単体あるいはこれら金属単体を複数組み合わせた合金、酸化錫、酸化亜鉛、酸化インジウム、酸化錫インジウム(ITO)、酸化亜鉛インジウム等の金属酸化物である。また、ポリアニリン、ポリピロール、ポリチオフェン等の導電性ポリマーでもよい。これらの電極物質は一種類を単独で使用してもよいし二種類以上を併用して使用してもよい。また、陽極は一層構成でもよく、多層構成でもよい。
【0094】
一方、陰極の構成材料としては、仕事関数の小さなものがよい。例えば、リチウム等のアルカリ金属、カルシウム等のアルカリ土類金属、アルミニウム、チタニウム、マンガン、銀、鉛、クロム等の金属単体が挙げられる。あるいはこれら金属単体を複数種組み合わせた合金も使用することができる。例えば、マグネシウム−銀、アルミニウム−リチウム、アルミニウム−マグネシウム等が使用できる。酸化錫インジウム(ITO)等の金属酸化物の利用も可能である。これらの電極物質は一種類を単独で使用してもよいし、二種類以上を併用して使用してもよい。また、陰極は一層構成でもよく、多層構成でもよい。
【0095】
本発明の有機発光素子において、本発明の縮合多環化合物を含有する層及びその他の有機化合物からなる層は、以下に示す方法により形成される。一般には真空蒸着法、イオン化蒸着法、スパッタリング法、プラズマあるいは、適当な溶媒に溶解させて公知の塗布法(例えば、スピンコーティング、ディッピング、キャスト法、LB法、インクジェット法等)により層を形成する。ここで真空蒸着法や溶液塗布法等によって層を形成すると、結晶化等が起こりにくく経時安定性に優れる。また塗布法で形成する場合は、適当なバインダー樹脂と組み合わせて膜を形成することもできる。
【0096】
上記バインダー樹脂としては、ポリビニルカルバゾール樹脂、ポリカーボネート樹脂、ポリエステル樹脂、ABS樹脂、アクリル樹脂、ポリイミド樹脂、フェノール樹脂、エポキシ樹脂、シリコーン樹脂、尿素樹脂等が挙げられるが、これらに限定されるものではない。また、これらバインダー樹脂は、ホモポリマー又は共重合体として1種単独で使用してもよいし、2種以上を混合して使用してもよい。さらに必要に応じて、公知の可塑剤、酸化防止剤、紫外線吸収剤等の添加剤を併用してもよい。
【0097】
[有機発光素子の用途]
本発明に係る有機発光素子は、表示装置や照明装置の構成部材として用いることができる。他にも電子写真方式の画像形成装置の露光光源や、液晶表示装置のバックライト等としての用途がある。
【0098】
ここで表示装置とは、本発明の有機発光素子を表示部に有する装置である。この表示部は複数の画素を有しており、各画素はそれぞれ本発明の有機発光素子と、本発明の有機発光素子の発光輝度を制御するためのTFT素子等のスイッチング素子と、を有している。ここでスイッチング素子としてTFT素子が使用されている場合、本発明の有機発光素子の構成部材である陽極又は陰極とTFT素子のドレイン電極又はソース電極とが電気的に接続されている。尚、表示装置は、例えば、PC等の画像表示装置として用いることができる。
【0099】
表示装置は、エリアCCD、リニアCCD、メモリーカード等からの情報を入力する画像入力部を有し、入力された画像を表示部に出力する画像出力装置であってもよい。また、撮像装置やインクジェットプリンタが有する表示部として、外部から入力された画像情報に基づいて画像を表示する画像出力機能と操作パネルとして画像への加工情報を入力する入力機能との両方を有していてもよい。さらに表示装置はマルチファンクションプリンタの表示部に用いられてもよい。
【0100】
次に、本発明の有機発光素子を使用した表示装置について、図面を参照しながら説明する。
【0101】
図1は、本発明の有機発光素子と、この有機発光素子に電気接続するスイッチング素子の一例であるTFT素子と、を有する表示装置の例を示す断面模式図である。構造の詳細を以下に説明する。
【0102】
図1の表示装置20は、ガラス等の基板1とその上部にTFT素子又は有機化合物層を保護するための防湿膜2が設けられている。また符号3は金属のゲート電極3である。符号4はゲート絶縁膜4であり、5は半導体層である。
【0103】
TFT素子8は半導体層5とドレイン電極6とソース電極7とを有している。TFT素子8の上部には絶縁膜9が設けられている。コンタクトホール(スルーホール)10を介して有機発光素子の陽極11とソース電極7とが接続されている。尚、本発明の表示装置は、図1の構成に限られず、陽極又は陰極のうちいずれか一方とTFT素子ソース電極又はドレイン電極のいずれか一方とが接続されていればよい。
【0104】
図1の表示装置20において、単層あるいは多層構造である有機化合物層12は、1つの層の如き図示がなされている。陰極13の上には有機発光素子の劣化を抑制するための第一の保護層14や第二の保護層15が設けられている。
【0105】
尚、本発明の表示装置において、表示装置を構成するスイッチング素子は、特に制限はなく、単結晶シリコン基板やMIM素子、a−Si型の素子等を用いてもよい。
【実施例】
【0106】
以下、本発明について実施例を用いて詳細に説明する。ただし本発明はこれらに限定されるものではない。
【0107】
[実施例1]例示化合物D1、D2の合成
以下に説明する合成方法に従い、例示化合物D1、D2を合成した。
【0108】
(1)中間体化合物M1の合成
【0109】
【化28】
【0110】
アルゴン雰囲気下、2Lの四つ口フラスコに、下記に示す試薬、溶媒を仕込んだ。
3−アミノフルオランテン:45.4g(209mmol)
DMF:910mL
【0111】
次に、反応系内を遮光した状態で、氷浴を用いて反応溶液を0℃まで冷却した。次に、NBS(37.2g、209mmol)を加えた後、氷浴を外して反応溶液を室温に戻した。次に、反応溶液を室温で1時間攪拌した。次に、反応溶液を氷水2L中に注いだ時に析出した結晶(黄緑色結晶)をろ取した。次に、ろ取した結晶を酢酸エチル4L中に溶解し、硫酸マグネシウム50g、フロリジル500gに順次通液して原料物質を除去した。次に、ろ液を減圧濃縮したときに生じたスラリー状の物質をろ過して結晶を得た。次に、得られた結晶を減圧乾燥することにより、2−ブロモフルオランテン−3−アミンを54.0g(収率87.1%)得た。
【0112】
続いてアルゴン雰囲気下、5Lの四つ口フラスコに、下記に示す試薬、溶媒を仕込んだ。
2−ブロモフルオランテン−3−アミン:55.6g(188mmol)
特級シクロペンチルメチルエーテル:1.12L
【0113】
次に、反応溶液を攪拌しながら氷浴を用いて反応溶液を内温0℃に冷却した。次に、n−BuLiの1.65M溶液596mL(939mmol、5eq)を30分かけて滴下した。次に、反応溶液を室温に戻した後、この温度(室温)で3.5時間攪拌した。次に、反応系内に炭酸ガスを吹き込んだ後(この炭酸ガスの吹込みにより反応溶液の内温が18℃から48℃に上昇)、反応系にガスバックを取り付けてから反応溶液を一晩攪拌した。次に、反応溶液中に5mol/L塩酸水溶液を滴下した。ここで塩酸水溶液の滴下の際に析出した結晶をろ過したがそのほとんどが目的物以外の化合物であった。そこでろ過の際に得られたろ液について分液操作を行うことで得られた有機層をフロリジル500gに通液した。次に、通液したろ液を濃縮乾固することで得られた残渣をTHF/CHCl3混合溶媒で再沈殿を行うことで、粗結晶を10.5g得た(HPLC純度92%)。次に、得られた粗結晶を再度THF/CHCl3で再沈殿を行うことで、結晶Aを8.50gを得た(HPLC純度98%)。次に、2回目の再沈殿の際に得たろ液をカラムクロマトグラフィー(ゲル1kg、展開溶媒:ヘプタン/酢酸エチル=1/1)で精製することで、結晶Bを900mg得た(HPLC純度98%)。次に、結晶A及び結晶Bを合わせた上でTHFに溶解して均一な溶液にした後減圧濃縮することでスラリー状物質を得た。次に、このスラリー状の物質にクロロホルムを加えることで結晶Cを取り出した。次に、減圧下で結晶Cを乾燥することにより、中間体化合物M1を9.23g(収率18.6%)得た。
【0114】
(2)中間体化合物M2の合成
【0115】
【化29】
【0116】
窒素雰囲気下、500mLの三口反応容器に、下記に示す試薬、溶媒を仕込んだ。
5−ブロモ−アセナフチレン−1,2−ジオン:15.0g(57.5mmmol)
1,3−ジフェニルプロパン−2−オン:12.1g(57.5mmol)
エタノール:300mL
【0117】
次に、室温にて、反応溶液中に6M水酸化ナトリウム水溶液20mLを滴下した。滴下終了後反応溶液を60℃まで加熱して、この温度(60℃)で反応溶液を約2時間撹拌した。次に、反応溶液を室温まで冷却した際に生成した析出物をろ過し、この析出物を水、エタノールで順次洗浄した。次に、この析出物を80℃で減圧加熱乾燥を行うことにより、中間体化合物M2を濃緑色の固体として22.0g(収率88%)得た。
【0118】
(3)中間体化合物M3(M3−1、M3−2)の合成
【0119】
【化30】
【0120】
窒素雰囲気下かつ室温下において、1Lの三口反応容器に、下記に示す試薬、溶媒を入れた。
中間体化合物M2:18.0g(41.3mmol)
中間体化合物M1:14.0g(53.8mmol)
亜硝酸イソアミル:8.72g(74.4mmol)
トルエン:720mL
【0121】
次に、反応溶液を95℃に加熱して、この温度(95℃)で反応溶液を1.5時間撹拌した。次に、反応溶液を冷却した後、反応溶液を水1Lで3回洗浄した。次に、水による洗浄により得られた有機層を飽和食塩水でさらに洗浄した後、硫酸マグネシウムで乾燥した。次に、この有機層をろ過することで得られたろ液を減圧濃縮することで茶褐色の液体を得た。次に、得られた茶褐色の液体をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/3)にて精製した。次に、この精製の際に得られた溶液を減圧濃縮して結晶を得た。次に、得られた結晶にメタノール345mLを添加し加熱スラリー洗浄を行った後、室温まで放冷した時に生成した結晶をろ過することにより、黄色結晶の中間体化合物M3を、異性体の混合物(M3−1、M3−2)として21.9g(収率:87%)得た。
【0122】
(4)中間体化合物M4(M4−1、M4−2)の合成
【0123】
【化31】
【0124】
窒素雰囲気下、1Lの三口容器に、下記に示す試薬、溶媒を入れた。
中間体化合物M3(M3−1とM3−2との混合物):19.4g(31.9mmol)
ビス−ジオキサボロラン:16.2g(63.9mmol)
酢酸カリウム:6.27g(63.9mmol)
テトラキストリフェニルフォスフィンパラジウム:1.107g(0.96mmol)
DMSO:388mL
【0125】
次に、反応溶液を100℃まで加熱した後、この温度(100℃)下で反応溶液を1.5時間撹拌を行った。次に、反応溶液を30℃まで冷却した後に、メタノール400mLを加えた。次に、反応溶液を10℃まで冷却してからこの温度(10℃)下で反応溶液を1時間撹拌を行った後、ろ過を行った。次に、ろ過によって得られたろ物についてメタノール800mLを加え、溶媒(メタノール)を還流させながら加熱スラリー洗浄を1時間行った後、室温まで冷却しろ過を行った。これにより、中間体化合物M4の粗結晶を18.3g(収率:87.5%)得た。次に、得られた粗結晶をトルエン265mLに溶解させて得た溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/2→トルエン/ヘプタン=1/1)にて精製した。次に、トルエン/メタノールで加熱スリラー洗浄を行うことにより、黄色結晶の中間体化合物M4を、異性体の混合物(M4−1、M4−2)として9.75g(収率:46.6%)得た。
【0126】
(5)中間体化合物M5の合成
【0127】
【化32】
【0128】
窒素雰囲気下、200mLの三口容器に、下記に示す試薬、溶媒を入れた。
4−ブロモ−3,5−ジメチルフェノール:3.19g(16mmol)
ピリジン:5.65g(71mmol)
トルエン:65mL
【0129】
次に、反応溶液を撹拌しながら0℃まで冷却した。次に、反応溶液を撹拌しながらトリフルオロメタンスルホン酸無水物13.43g(48mmol)を約15分かけてゆっくり滴下した。滴下終了後、反応溶液を室温まで昇温し、室温下でさらに反応溶液を1時間撹拌した。次に、水100mLを添加して反応を停止し、続いてトルエン40mLを添加した後、分液操作を行い有機層を分離した。次に、得られた有機層を減圧濃縮した後、カラムクロマトグラフィー(展開溶媒:トルエン)にて精製した。このときに得られた溶液を減圧濃縮することにより、無色透明液体の4−ブロモ−3,5−ジメチルフェニル−トルフルオロメタンスルホン酸エステルを5.94g(収率:97.1%)得た。
【0130】
続いて窒素雰囲気下、500mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
4−ブロモ−3,5−ジメチルフェニル−トルフルオロメタンスルホン酸エステル:4.90g(14.7mmol)
4−シアノフェニルボロン酸:2.05g(14.0mmol)
ジオキサン:245mL
炭酸カリウム:4.07g(29.4mmol)
テトラキストリフェニルホスフィンパラジウム:0.17g(0.15mmol)
【0131】
次に、反応溶液を80℃に加熱し、この温度(80℃)下で反応溶液を1.5時間撹拌した。次に、反応溶液を室温まで放冷した後、トルエン100mL及び水100mLを添加し分液操作を行い、有機層を分離した。次に、得られた有機層を硫酸マグネシウムで乾燥した後、この有機層をろ過することで得られたろ液を減圧濃縮することで中間体化合物5の粗結晶を4.64g得た。次に、得られた粗結晶とトルエンとを混合することで得た溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/5→トルエン/ヘプタン=1/4)にて精製した後、濃縮乾固化した。以上により、白色結晶の中間体化合物5を1.54g(収率:37%)得た。
【0132】
(6)例示化合物D1、D2の合成
【0133】
【化33】
【0134】
窒素雰囲気下、500mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):4.40g(6.72mmol)
中間体化合物M5:1.54g(5.38mmol)
トルエン:220mL
エタノール:110mL
炭酸ナトリウム:1.42g(13.4mmol)
水:100mL
テトラキストリフェニルホスフィンパラジウム:0.39g(0.34mmol)
【0135】
次に、反応溶液を70℃に加熱して、この温度(70℃)下にて反応溶液を3時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで2回洗浄することで有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、溶液をろ過することで得られたろ液を減圧濃縮することで黄色粗結晶を得た。次に、この粗結晶をトルエン60mlとヘプタン60mLとを混合して調製した溶媒に溶解させた後、この溶液をカラムクロマトグラフィー(展開溶媒:トルエン/ヘプタン=1/1→トルエン/ヘプタン=2/1)にて精製した。次に、溶液を減圧濃縮した後、メタノールを添加して加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D1及びD2の混合物を3.26g(収率:66%)得た。尚、例示化合物D1は、中間化合物M4−1から合成されるものであり、例示化合物D2は、中間化合物M4−2から合成されるものである。
【0136】
質量分析法により、この化合物のM+である733を確認した。また、NMR測定により、例示化合物D1とD2の混合比が1:1であることを確認した。
【0137】
1H−NMR(CDCl3):δ(ppm)=8.20(s,2H),7.89−7.97(m,4H),7.83−7.65(m,30H),7.40−7.37(m,6H),7.34−7.28(m,6H),7.25−7.20(m,4H),7.12(d,1H,J=7.99Hz),7.09(d,1H,J=7.99Hz),6.61(d,1H,J=7.99Hz),6.53(dt,1H,J1=7.99Hz,J2=3.99Hz),6.38(d,1H,J=7.99Hz),6.30(dd,1H,J1=7.99Hz,J2=3.99Hz),1.97(s,12H)
【0138】
さらに例示化合物D1及びD2の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク450nmに最大強度を有する青色発光スペクトルが確認された。
【0139】
[実施例2]例示化合物D87、D88の合成
実施例1(6)において、中間体化合物M5の代わりに下記に示される中間体化合物M6を用いたことを除いては、実施例1と同様の方法で合成を行い、例示化合物D87、D88(の混合物)を得た。以下に、中間体化合物M6、並びに例示化合物D87及びD88の具体的な合成方法について説明する。
【0140】
(1)中間体化合物M6の合成
【0141】
【化34】
【0142】
Chemistry of Materials,2009,21(12),2452に記載の方法で、1−フルオロ−2−ニトロベンゼンと1−ブロモ−4−アミノベンゼンとを出発物質として、中間体化合物M6を30g合成した。
【0143】
(2)例示化合物D87、D88の合成
【0144】
【化35】
【0145】
窒素雰囲気下、200mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):1.30g(1.99mmol)
中間体化合物M6:1.54g(1.39mmol)
トルエン:39mL
エタノール:13mL
炭酸ナトリウム:0.42g(3.97mmol)
水:13mL
テトラキストリフェニルホスフィンパラジウム:0.12g(0.11mmol)
【0146】
次に、反応溶液を70℃に加熱し、この温度(70℃)下で反応溶液を5時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで3回洗浄し有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、ろ過により得られたろ液を減圧濃縮することで黄色の粗結晶を得た。次に、得られた粗結晶をトルエン19mと混合して得られた溶液について、カラムクロマトグラフィー(展開溶媒:トルエン/酢酸エチル=9/1→トルエン/酢酸エチル=5/1)にて精製した。次に、溶液を濃縮した後メタノールを添加し加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D87及びD88の混合物を0.96g(収率:58%)得た。尚、例示化合物D87は、中間化合物M4−1から合成されるものであり、例示化合物D88は、中間化合物M4−2から合成されるものである。
【0147】
質量分析法により、この化合物のM+である796を確認した。また、NMR測定により、例示化合物D87とD88との混合比が1:1であることを確認した。
【0148】
1H−NMR(CDCl3):δ(ppm)=8.21(s,1H),8.20(s,1H),7.93−7.63(m,38H),7.43−7.29(m,28H),6.59(dd,2H,J1=7.99Hz,J2=3.99Hz),6.35(d,2H,J=7.99Hz)
【0149】
さらに例示化合物D87及びD88の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク455nmに最大強度を有する青色発光スペクトルが確認された。
【0150】
[実施例3]例示化合物D83、D84の合成
実施例1(6)において、中間体化合物M5の代わりに下記に示される中間体化合物M7を用いたことを除いては、実施例1と同様の方法で合成を行い、例示化合物D83、D84(の混合物)を得た。以下に、中間体化合物M7、並びに例示化合物D83及びD84の具体的な合成方法について説明する。
【0151】
(1)中間体化合物M7の合成
【0152】
【化36】
【0153】
Heterocycles,2001,55,1329に記載の方法で、3−ヒドロキシ−2−アミノピリジンと3−ヨード−1−カルボキシルベンゼンとを出発物質として、中間体化合物M7を22g合成した。
【0154】
(2)例示化合物D83、D84の合成
【0155】
【化37】
【0156】
窒素雰囲気下、200mLの三口フラスコに、下記に示す試薬、溶媒を入れた。
中間体化合物M4(M4−1とM4−2との混合物):1.40g(2.10mmol)
中間体化合物M7:0.48g(1.39mmol)
トルエン:70mL
エタノール:30mL
炭酸ナトリウム:0.17g(3.97mmol)
水:30mL
テトラキストリフェニルホスフィンパラジウム:0.12g(0.11mmol)
【0157】
次に、反応溶液を70℃に加熱し、この温度(70℃)下で反応溶液を2.5時間撹拌した。次に、反応溶液を室温まで放冷した後、反応溶液を水100mLで3回洗浄し有機層を得た。次に、この有機層を硫酸マグネシウムで乾燥した後、ろ過により得られたろ液を減圧濃縮することで黄色の粗結晶を得た。次に、得られた粗結晶をトルエン20mと混合して得られた溶液について、カラムクロマトグラフィー(展開溶媒:トルエン/酢酸エチル=9/1)にて精製した。次に、溶液を濃縮した後メタノールを添加し加熱スラリー洗浄を行うことにより、黄色結晶の例示化合物D83及びD84の混合物を0.93g(収率:56%)得た。尚、例示化合物D83は、中間化合物M4−1から合成されるものであり、例示化合物D84は、中間化合物M4−2から合成されるものである。
【0158】
質量分析法により、この化合物のM+である722を確認した。
【0159】
また、NMR測定により、例示化合物D83とD84との混合比が1:1であることを確認した。
【0160】
1H−NMR(CDCl3):δ(ppm)=8.59(dd,2H,J1=3.99Hz,J2=1.33Hz),8.48(s,2H),8.38(d,2H,J=7.99Hz),8.22(s,1H),8.21(s,1H),7.88−7.64(m,34H),7.42−7.28(m,14H),6.59(dd,2H,J1=9.99Hz,J2=7.99Hz),6.35(dd,2H,J1=7.99Hz,J2=7.99Hz)
【0161】
さらに例示化合物D83及びD84の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長418nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク453nmに最大強度を有する青色発光スペクトルが確認された。
【0162】
[比較例1]比較化合物R1、R2の合成
【0163】
【化38】
【0164】
実施例1(6)において、中間体化合物M4(M4−1とM4−2との混合物)の代わりに中間体化合物M3(M3−1とM3−2との混合物)を用いた。また中間体化合物M5の代わりに2,6−ジメチルフェニルボロン酸を用いた。これらを除いては、実施例1と同様の方法で合成を行い、比較化合物R1及びR2を、R1とR2との混合物として0.95g(収率:75%)得た。
【0165】
質量分析法により、この化合物のM+である632を確認した。
【0166】
また比較化合物R1及びR2の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長414nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク447nmに最大強度を有する青色発光スペクトルが確認された。
【0167】
[比較例2]比較化合物R3、R4の合成
【0168】
【化39】
【0169】
実施例1(6)において、中間体化合物M4(M4−1とM4−2との混合物)の代わりに中間体化合物M3(M3−1とM3−2との混合物)を用いた。また中間体化合物M5の代わりに4−シアノフェニルボロン酸を用いた。これらを除いては、実施例1と同様の方法で合成を行い、比較化合物R3及びR4を、R3とR4との混合物として0.8g(収率:70%)得た。
【0170】
質量分析法により、この化合物のM+である629を確認した。
【0171】
また比較化合物R3及びR4の混合物について、濃度1.0×10-6mol/Lのトルエン溶液における発光スペクトルを測定した。具体的には、日立製F−5400を用いて、励起波長414nmにおいてフォトルミネッセンスの測定を行った。その結果、発光ピーク458nmに最大強度を有する青色発光スペクトルが確認された。
【0172】
[蛍光量子収率及び酸化還元電位の比較]
以下に説明する方法で、実施例1乃至3にてそれぞれ得られた化合物(例示化合物D1、D2、D83、D84、D87、D88)及び比較例1及び2にて得られた化合物(比較化合物R1、R2、R3、R4)について量子収率及び酸化還元電位を評価した。
【0173】
(1)蛍光量子収率
まず濃度10-6mol/lのトルエン溶液を調製した。次に、調製した溶液について、絶対PL量子収率測定装置(浜松ホトニクス社製、商品名:C9920)を用い、励起波長を414nmとしたときの値を測定した。そして、比較化合物R1及びR2の混合物(混合比=1:1)を1とした場合の発光量子収率の相対値を求めた。結果を表1に示す。
【0174】
(2)酸化還元電位
サイクリックボルタンメトリー法により酸化還元電位を以下のように測定した。具体的には、0.1mol/Lのテトラブチルアンモニウムパークロレイトを支持電解物質とし、以下の条件で、酸化電位及び還元電位を測定した。酸化電位は、各化合物の1×10-4mol/L以上1×10-6mol/L以下の塩化メチレン溶液中で測定した。還元電位は、各化合物の1×10-4mol/L以上1×10-6mol/L以下のN,N−ジメチルホルムアミド溶液中で測定した。尚、測定にあたり測定温度を各々25℃に設定し、Ag/AgNO3を参照電極とし、白金電極を対向電極とし、グラシックカーボンを作用電極としてそれぞれ用いて測定を行った。結果を表1に示す。
【0175】
【表1】
【0176】
表1より、本発明のアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、比較化合物R1,R2よりも蛍光量子収率が1.1倍以上向上していることがわかった。
【0177】
また、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の12位或いは13位に直接電子吸引性の4−シアノフェニル基が導入された比較化合物R3,R4は、還元電位が最も深くなり4−シアノフェニル基が導入された効果が大きい。しかしその一方で、比較化合物R1,R2よりも蛍光量子収率が低下していることがわかった。
【0178】
以上より、本発明のアセナフト[1,2−k]ベンゾ[e]アセフェナンスレン誘導体は、アセナフト[1,2−k]ベンゾ[e]アセフェナンスレン骨格の特定の位置にフェニル基を介して特定の電子吸引性の置換基を導入することで、深い還元電位を有するのと同時に高い蛍光量子収率を有する材料であることが示された。
【0179】
[実施例4]
本実施例では、基板上に、陽極、正孔輸送層、発光層、正孔・エキシトンブロッキング層、電子輸送層及び陰極が順次形成されている有機発光素子を作製した。以下に、本実施例で使用した材料の一部を示す。
【0180】
【化40】
【0181】
有機発光素子の具体的な製造方法を以下に説明する。
【0182】
スパッタ法により、ガラス基板上に、ITOを成膜して陽極を形成した。このとき陽極の膜厚を120nmとした。このようにITO電極が形成されている基板を透明導電性支持基板(ITO基板)として以下の工程で使用した。次に、このITO基板上に、下記表2に示される有機化合物層及び電極層を、1×10-5Paの真空チャンバー内で抵抗加熱による真空蒸着によって連続的に成膜した。このとき対向する電極(金属電極層、陰極)の面積が3mm2になるように作製した。
【0183】
【表2】
【0184】
得られた素子について、素子の特性を測定・評価した。具体的には、電流電圧特性をヒューレッドパッカード社製・微小電流計4140Bで測定し、発光輝度は、トプコン社製BM7で測定した。測定の結果を表3に示す。
【0185】
[実施例5,6]
実施例4において、ゲストを、表3に示される化合物に適宜変更する以外は、実施例4と同様の方法により有機発光素子を作製した。得られた素子について実施例4と同様に素子の特性を測定・評価した。測定の結果を表3に示す。
【0186】
[比較例3]
実施例4において、ゲストを、比較化合物R1、R2(R1とR2との重量混合比は、1:1である。)に適宜変更する以外は、実施例4と同様の方法により有機発光素子を作製した。得られた素子について実施例4と同様に素子の特性を測定・評価した。測定の結果を表3に示す。
【0187】
【表3】
【0188】
[結果と考察]
本発明の縮合多環化合物は、高い量子収率と青色発光を呈する有機化合物であり、有機発光素子の構成材料に用いた場合、良好な色度と高効率を示すと同時に特に耐久特性が良好な発光素子を提供することができる。
【符号の説明】
【0189】
8:TFT素子、11:陽極、12:有機化合物層、13:陰極
【特許請求の範囲】
【請求項1】
下記一般式[1]又は[2]で示される化合物であることを特徴とする、縮合多環化合物。
【化1】
(式[1]及び[2]において、Rは、水素原子又はメチル基を表す。式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【化2】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【請求項2】
下記一般式[8]又は[9]で示される化合物であることを特徴とする、縮合多環化合物。
【化3】
(式[8]及び[9]において、Rは、水素原子又はメチル基を表す。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【化4】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基、シアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【請求項3】
下記一般式[10]又は[11]で示される化合物であることを特徴とする、請求項1に記載の縮合多環化合物。
【化5】
(式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。)
【請求項4】
下記一般式[20]又は[21]で示される化合物であることを特徴とする、請求項2に記載の縮合多環化合物。
【化6】
(式[20]及び[21]において、Rは、水素原子又はメチル基である。)
【請求項5】
下記一般式[30]及び[31]で示される化合物であることを特徴とする、請求項1に記載の縮合多環化合物。
【化7】
(式[30]及び[31]において、Rは、水素原子又はメチル基である。式[30]にて示されるRは同一の置換基である。式[31]にて示されるRは同一の置換基である。)
【請求項6】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、を有する有機発光素子において、
前記有機化合物層のうち少なくとも一層が、請求項1乃至5のいずれか一項に記載の縮合多環化合物を有することを特徴とする、有機発光素子。
【請求項7】
前記縮合多環化合物が発光層に含まれることを特徴とする、請求項6に記載の有機発光素子。
【請求項8】
前記発光層が、ホストとゲストとから構成されることを特徴とする、請求項7に記載の有機発光素子。
【請求項1】
下記一般式[1]又は[2]で示される化合物であることを特徴とする、縮合多環化合物。
【化1】
(式[1]及び[2]において、Rは、水素原子又はメチル基を表す。式[1]にて示されるRは同一の置換基である。式[2]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【化2】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【請求項2】
下記一般式[8]又は[9]で示される化合物であることを特徴とする、縮合多環化合物。
【化3】
(式[8]及び[9]において、Rは、水素原子又はメチル基を表す。式[8]にて示されるRは同一の置換基である。式[9]にて示されるRは同一の置換基である。Qは、下記一般式[3]乃至[7]から選ばれる電子吸引性の置換基である。
【化4】
(式[3]及び[4]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基、シアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。式[7]において、R5は、水素原子又はメチル基である。))
【請求項3】
下記一般式[10]又は[11]で示される化合物であることを特徴とする、請求項1に記載の縮合多環化合物。
【化5】
(式[10]及び[11]において、R1乃至R4は、それぞれ水素原子、炭素数1以上4以下のアルキル基及びシアノ基から選ばれる置換基である。R1乃至R4は、同じであってもよいし異なっていてもよい。)
【請求項4】
下記一般式[20]又は[21]で示される化合物であることを特徴とする、請求項2に記載の縮合多環化合物。
【化6】
(式[20]及び[21]において、Rは、水素原子又はメチル基である。)
【請求項5】
下記一般式[30]及び[31]で示される化合物であることを特徴とする、請求項1に記載の縮合多環化合物。
【化7】
(式[30]及び[31]において、Rは、水素原子又はメチル基である。式[30]にて示されるRは同一の置換基である。式[31]にて示されるRは同一の置換基である。)
【請求項6】
陽極と陰極と、
前記陽極と前記陰極との間に配置される有機化合物層と、を有する有機発光素子において、
前記有機化合物層のうち少なくとも一層が、請求項1乃至5のいずれか一項に記載の縮合多環化合物を有することを特徴とする、有機発光素子。
【請求項7】
前記縮合多環化合物が発光層に含まれることを特徴とする、請求項6に記載の有機発光素子。
【請求項8】
前記発光層が、ホストとゲストとから構成されることを特徴とする、請求項7に記載の有機発光素子。
【図1】

【公開番号】特開2012−246258(P2012−246258A)
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願番号】特願2011−120164(P2011−120164)
【出願日】平成23年5月30日(2011.5.30)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
【公開日】平成24年12月13日(2012.12.13)
【国際特許分類】
【出願日】平成23年5月30日(2011.5.30)
【出願人】(000001007)キヤノン株式会社 (59,756)
【Fターム(参考)】
[ Back to top ]