
繊毛虫類の宿主細胞中におけるウイルスタンパク質の異種発現系。
本発明は、ウイルスタンパク質またはそのフラグメントの異種タンパク質発現系に関する。この発現系は、(a)繊毛虫類宿主細胞と、(b)ウイルスタンパク質またはそのフラグメントをコードする少なくとも1つのcDNAと、(c)cDNAに機能しうるように連結されたプロモーターとを備える。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、繊毛虫類の宿主細胞中におけるウイルスタンパク質の異種発現系に関連する。
【背景技術】
【0002】
多くのウイルスは、ヒト病原体または動物病原体として重要な役割を果たしているので、ウイルス病原体に対するワクチンとして、ウイルスタンパク質を使用することが注目されている。ウイルス(特に、RNAウイルス)は、非常に多様性があり、ウイルス感染は、様々な血清型を有するウイルス(例えば、インフルエンザウイルス、FMDウイルス)に起因する。結果として、既存のウイルス性ワクチンの多くは、しばしば、現場で流行している菌株に対処することができない。また、新しいウイルス性ワクチンは、新たな感染が発生したときに、現場の菌株から製造する必要がある。
【0003】
現代のバイオテクノロジー技術は、ほとんど全ての考えられるタンパク質の異種発現を可能にしているが、ウイルスタンパク質の発現は、あまり見られないのが現状である。多くの場合、ウイルスタンパク質は構造タンパク質(すなわち、酵素でもなく、タンパク質ホルモンでもなく、抗体でもない。)であり、製薬目的または工業目的で大して関心をひかないからである。
【0004】
しかしながら、多くのウイルスは、ヒト病原体または動物病原体として、重要な役割を果たしているので、ウイルス病原体に対するワクチンとして、ウイルスタンパク質を使用することが注目されている。
【0005】
近年、ウイルス病原体に対するワクチンを製造する標準手順は、特定の系において、それぞれのウイルスを培養し、そのようにして得られたウイルス粒子を収集および不活性化し、不活性化された粒子をワクチンとして含む医薬組成物を製造する。
【0006】
上記原理は、例えば、インフルエンザワクチンの製造において実施される。インフルエンザワクチンは、受精鶏卵中で製造されている。受精11日後、それぞれの卵のアルブミン中に、インフルエンザウイルス株が注入され、発生中の胚の肺を感染させる。数日間培養された後、ウイルスが集菌および精製され、化学的に不活性化され、ワクチンの製造に用いられる。1ダースのワクチンの製造には、平均して、約1〜2個の卵が必要になる。全製造工程は、少なくとも6ヶ月続く。
【発明の概要】
【0007】
このワクチン製造方法は、確立されており、コスト効率がよい。しかし、このワクチン製造方法には、数百万個もの卵の調達、長いタイムライン、卵のハンドリングの退屈さ、需要の急増または新しいウイルス株の突然の出現に対する適応性のなさといったデメリットがある。さらに、受精鶏卵は、いくつかの病原性ウイルス、中でも鳥インフルエンザA型ウイルス(H5N1)に対するワクチンの製造に使用することができない。
【0008】
受精鶏卵は、少なくとも、ホストとしてニワトリ胚と相性のよい全てのウイルスの生産のために、または、前者の成分(element)を含むワクチン生産のために、主に使用することができる。
【0009】
ウイルスまたは前者の成分を含むワクチンを生産する他の方法は、細胞培養または組織培養に基づく。ある特定のアプローチよれば、哺乳類の腎臓細胞中に、ウイルスが注入される。ウイルスの増殖後、細胞を溶解させて、ウイルス粒子を集菌し、精製し、不活性化させる。しかしながら、これらの系では、複合増殖培地を使用する必要があり、それらの取り扱いは、面倒で、時間がかかる。さらに、このような感染アプローチは、実施が難しく、再現性が十分でない。
【0010】
国際公開第03/048348号に開示されている別のアプローチによれば、ヒト細胞株PER.C6(登録商標)に関する不活性化されたウイルスワクチンの製造方法が開示されている。その表面にレセプタを有するSia2-6GalおよびSia2-3Galが存在するので、この細胞株は、様々なウイルス(例えば、インフルエンザウイルス、パラインフルエンザウイルス、アデノ随伴ウイルスまたはポリオーマウイルス・タイプ(poliomavirus type))に感染しやすい。それにより、生産プロセスに関する安全性の問題を引き起こす。
【0011】
一方、哺乳類の細胞培養を利用した系(mammalian cell culture based system)は、上述の卵を利用した系(egg based system)とは対照的に、急速に規模を拡大することができ、緊急時にスケールアップすることができる。一方、そのような製造設備(巨大なバイオリアクターを有する。)の操作上の即応性に対して事前に必要な費用(up-front cost)は、卵を利用した系に関する費用と比較して、はるかに高額であり、収率は、わずかに劣る。さらに、これらの系は、複合増殖培地を使用する必要があり、その取り扱いは、面倒で、時間がかかる。
【0012】
それゆえ、ワクチンとして用いることができるウイルスタンパク質を生産する代替手法を開発することは、最優先の課題である。
【0013】
ある特定のアプローチによれば、組み換えDNA技術を用いて、これらのタンパク質を生産する。明らかな利点の1つは、大きく改善されたワクチンの安全性である。卵を用いたワクチンの生産方法と比較して、組換え発現のウイルスタンパク質を精製するため条件が有利だからである。さらに、適応性が大きく向上して、様々な季節性の亜型ウイルスに適応することができる。
【0014】
しかしながら、ウイルスタンパク質は、多くの場合、構造タンパク質であるので、製薬目的および工業目的での生産は、複雑で面倒である。当初、大腸菌中で、ウイルスタンパク質に対応するポリペプチド(例えば、血球凝集素)を発現させることが試みられた。しかしながら、後者は、翻訳後修飾装置(posttranslational modification apparatus)を有しないので、ワクチンとして使用されうるタンパク質の原核生物中における異種発現が意味をなさないことは、注目に値する。それゆえ、原核生物(例えば、大腸菌)中において発現させられたタンパク質は、翻訳後修飾(例えば、糖鎖付加パターン)が十分ではなく、安定性に影響を与え、抗原の免疫原性に大きく貢献するように思われる(Nayakら、1984年)。
【0015】
真核生物のサッカロマイセス・セレヴィシエ(血球凝集素に関する米国特許第4,752,473号明細書)中におけるウイルスタンパク質の発現は、高分子量の外鎖マンナン(outer chain mannan)の広範囲に及ぶ付加を伴う組み換えタンパク質のハイパーグリコシル化によって問題を引き起こしたが(JabbarおよびNayak、1987年。Jabbarら、1985年)、十分には立証されていない。
【0016】
米国特許第5,858,368号明細書は、組み換えバキュロウイルスに感染した昆虫細胞株中において、インフルエンザA型ウイルスの血球凝集素(HA)を発現させる方法を開示している。タンパク質は、非変性、非イオン洗剤を用いて感染細胞の表在性膜から抽出された後、クロマトグラフにより精製される。しかしながら、精製プロセスは、必要以上に時間がかかる(rather time-consuming)。さらに、バキュロウイルス/昆虫細胞発現系において生産されたタンパク質が、それらの糖鎖付加パターンのおかげで、哺乳類の治療用途に適しているのかどうかは、疑わしい(Kulakoskyら、1998年)。また、昆虫細胞中で発現させられたタンパク質のN−グリカン中の特定の残基(certain residue)が、アレルギー性のエピトープである可能性を示すヒントもある(Tomiyaら、2004年)。
【0017】
(定義)
本明細書において、「異種発現(heterologous expression)」という用語は、その中で発現が生じている有機体(organism)にとって異質である遺伝子、核酸またはcDNAのタンパク質の発現を示す。
【0018】
本明細書において、「繊毛虫類(ciliate)」という用語は、繊毛虫(Ciliophora)の系統だった門(scientific phylum)を示す。繊毛虫は、単細胞の真核生物(原虫(protozoa)または原生生物(protist))であり、特に、その比較的大きなサイズ(種によっては、その長さが2mmに達する。)、その繊毛細胞表面、2つの異なる種類の核(すなわち、小さな2倍体の小核と、大きな倍数体の大核(転写活性)とである。)によって特徴付けられる。後者は、ゲノムの増幅およびヘビー・エディティング(heavy editing)によって、小核から生成される。
【0019】
本明細書において、「cDNA」という用語は、発現させられるべきタンパク質をコードするDNA分子であって、エンコードしていない部分(non-encoding part)(例えば、イントロン)を有していないDNA分子を示す。多くの場合、cDNAは、逆転写酵素およびオリゴdTプライマー(oligo dT-primer)を用いて、RNAテンプレートから直接合成される。しかしながら、当該用語には、他の方法で得られた合成遺伝子およびエンコーディングDNA(encoding DNA)も同様に含まれる。
【0020】
本明細書において、「プロモーター遺伝子(promoter)」という用語は、DNAの調節領域を示す。当該領域は、通常、遺伝子(gene)またはcDNAの上流(センス鎖の5'領域に向かって)に配され、当該遺伝子または当該cDNAの転写を可能にするまたは増進する本質的な遺伝因子(essential genetic element)を含む。
【0021】
本明細書において、「フラグメント(fragment)」という用語は、ある部分もしくはドメインが欠落しており、酵素活性、免疫原性、標的結合もしくはそれらに類似する特性に関して、いくらかの活性を維持している天然タンパク質もしくは野生型タンパク質の一部、または、それらに類似するものを示す。
【0022】
本明細書において、「シグナル配列(signal sequence)」という用語は、オリゴペプチド(「シグナル・ペプチド」または「輸送ペプチド」)をコードする核酸配列を示す。オリゴペプチドは、タンパク質の特定の細胞小器官(例えば、核、糸粒体基質、小胞体、葉緑体、アポプラスト、ペルオキシソーム)の輸送を指示する。小胞体(endoplasmatic reticulum)へと輸送されたほとんど全てのタンパク質は、N末端に、5−10個の疎水性アミノ酸を含む配列を有する。これらのシグナル・ペプチドは、ERの内腔へのタンパク質の翻訳時挿入(cotranslational insertion)の後、シグナル・ぺプチダーゼによって、たんぱく質から分割されるその後、多くのタンパク質は、ゴルジ体(golgi apparatus)を介して、分泌経路の下流へと輸送される。
【0023】
本明細書において、「機能しうるように連結される(operably linked)」という用語は、プロモーター遺伝子が適切な条件下で遺伝子産物の発現を制御するように、遺伝子産物をコードし得るヌクレオチド配列が、当該プロモーター遺伝子に連結されることを意味する。
【0024】
本明細書において、「疎水性ドメイン(hydrophobic domain)」という用語は、「親油性ドメイン(lipophilic domain)」および/または「非極性ドメイン(non-polar domain)」という用語と同意語として用いられる用語である。本明細書において、これらの用語は、少なくともその末端において、親油性/非極性のアミノ酸残基の存在によって主に制御される疎水性(「ヒドロパシー(hydropathy)」と呼ばれる場合もある。)を有し、そのため、例えば、細胞膜中に見られるリン脂質二重層のように、脂質画分に対してある程度の親和性を有するタンパク質ドメインを示す。
【0025】
上記の疎水性ドメインは、例えば、いわゆる「膜貫通ドメインtransmembrane domain)」(すなわち、膜の中で熱力学的に安定したタンパク質の3次元構造)とみなすこともできる。このような膜貫通ドメインには、単一のαへリックス(single alpha helix)、いくつかの膜貫通αへリックスの安定な複合体(complex)、膜貫通βバレル、βヘリックス、潜在的に(potentially)疎水性アミノ酸の残基を取り囲む疎水性アミノ酸の外側の環(outer ring)、および、それらに類似するものが含まれてよい。
【0026】
本発明者らは、疎水性ドメインが、タンパク質の環境中への分泌を妨げることを見出した。表1に、疎水性タンパク質の一例を示す。
【表1】

【0027】
本明細書において、「核酸分子(nucleic acid molecule)」という用語は、一本鎖核酸分子または二本鎖核酸分子を示すことを意図しており、例えば、DNA(cDNAおよび/もしくはゲノムDNA)、RNA(好ましくは、mRNA)、PNA、LNA、ならびに/または、モルホリノが含まれる。
【0028】
本明細書において、「ストリンジェントな条件(stringent conditions)」という用語は、その条件の下において、プローブが、他の配列よりも、当該プローブの標的部分配列(target subsequence)と分子交雑(hybridize)しやすい条件に関連する。ストリンジェントな条件は、配列依存性(sequence-dependent)であり、環境が異なれば当該条件も異なるであろう。特に、配列が長いほど、高い温度で分子交雑する。一般的に、ストリンジェントな条件は、一定の(defined)イオン強度およびpHにおける特定配列の融点(thermal melting point)(Tm)よりも約5℃低くなるように選択される。Tmは、その温度において、平衡状態で、標的配列に相補的なプローブの50%が標的配列に分子交雑する(一定のイオン強度、pHおよび核酸濃度の下での)温度である(一般に、標的配列は過剰に存在するので、Tmにおいて、平衡状態では、プローブの50%は占有される。)。通常、ストリンジェントな条件における塩濃度は、Naイオン濃度が約1.0mol/L(Mと表記する場合がある。)より小さく、典型的には、pHが7.0から8.3であり、Naイオン濃度(またはその他の塩)濃度が約0.01から1.0Mである。また、ストリンジェントな条件における温度は、短いプローブ(例えば、10から50個のヌクレオチドを含む。)の場合は少なくとも約30℃であり、それより長いプローブの場合には少なくとも約60℃である。ストリンジェントな条件は、不安定化剤(destabilizing agent)(例えば、ホルムアミドおよびそれに類似するもの)を添加することで調整されてもよい。
【0029】
本明細書において、「PDBコード(PDB code)」という用語は、特有の4種類の文字表記(unique four letter code)に関連する。その文字表記は、基本識別子としての役割を果たし、それによって、生体高分子の構造データの世界的なレポジトリであるタンパク質構造データベース(Protein Data Bank)を用いてエントリーを検索することができる。
【0030】
本明細書において、「核酸分子のフラグメント(fragment of the nucleic acid molecule)」という用語は、特許請求の範囲に記載された配列の1つに従う核酸分子のサブセットを含む核酸を示すことを意図している。
【0031】
「核酸分子のフラクション(fraction of the nucleic acid molecule)」という用語についても同様である。
【0032】
本明細書において、「核酸分子の変異体(variant of the nucleic acid molecule)」という用語は、構造および特許請求の範囲に記載された配列の1つに従う核酸分子に対する生物活性において、実質的に同様である核酸分子を示す。
【0033】
本明細書において、「核酸分子のホモログ(homologue of the nucleic acid molecule)」という用語は、特許請求の範囲に記載された配列の1つに従う、核酸分子と比較して、その配列が、付加、削除、置換またはその他の化学修飾を施された1以上のヌクレオチドを有する核酸分子に関連し、このホモログは、特許請求の範囲に記載された配列の1つに従う核酸分子と実質的に同一の特性を保持することが常に要求される。
【0034】
本明細書において、「コドン最適化された(codon optimized)」という用語は、発現させられるべき異種タンパク質をコードするcDNAを、普遍的遺伝子コード表(universal genetic code scheme)から派生した宿主特異的な(host specific)コドン使用頻度(codon usage)に適応させるプロセスを示す。繊毛虫類は、AとTに富むゲノム(AT-rich genome)を有し、テトラヒメナのDNAにおいては、その約75%がAおよびTからなる。ここでのコドン使用頻度は、他の有機体と、特に、特定のアミノ酸をコードする目的におけるコドンの使用頻度(「コドン・バイアス(codon bias)」)において異なる。異種タンパク質をコードするcDNAが最適化されていない場合、当該cDNAが、繊毛虫においてほとんど使用されないコドンを使用すると、タンパク質の発現効率に大きく影響する可能性がある。このことは、一方で、研究中の遺伝子のコドン使用頻度(codon frequency)が、繊毛虫類の発現系のコドン使用頻度に適合した場合には、異種タンパク質の発現が劇的に改善される可能性があることを意味する。さらに、多くの繊毛虫類、中でもテトラヒメナは、グルタミンをコードするUAAトリプレットおよびUAGトリプレットを有する標準的でないヌクレオチド・コードを利用する。しかし、他の多くの有機体において、これらのコドンは、翻訳を終了させる終止コドンとして利用される。このことから、UAAトリプレットおよびUAGトリプレットを終止コドンとして有する外来遺伝子(繊毛虫類の遺伝子ではない遺伝子)は、正確に発現しないということを導き出すことができるかもしれない。この目的のために、繊毛虫類の宿主細胞を形質転換する前に、UAAトリプレットおよびUAGトリプレットをUAAに修正するような方法で、異種タンパク質をコードするcDNAのコード最適化が実施されるべきである。コード最適化(code optimization)は、例えば、部位特異的突然変異誘発法(site directed mutagenesis)またはcDNAデノボ合成(de novo cDNA synthesis)によって実施することができる。
【0035】
本明細書において、「誘導体(derivative)」という用語は、特許請求の範囲に記載された配列の1つに従う核酸分子のように、標的核酸配列と似た特性を有する、関連した核酸分子(related nucleic acid molecule)を示す。
【0036】
本明細書において、「少なくともX%の配列同一性(sequence identity of at least X %)」という用語は、BLASTアルゴリズムの系統(family)のアルゴリズム(特に、メガブラスト、不連続メガブラスト、blastn、blastp、PSI−BLAST、PHI−BLAST、blastx、blastn、または、tblastx)を用いて配列アラインメントを実施した後に決定される配列同一性を示す。これらのアルゴリズムは、例えば、NCBIによって提供される、それぞれのインターネット・ドメインからアクセスすることができる。
【0037】
本明細書において、「宿主細胞(host cell)」という用語は、2つの異なる意味を有する。これらは、それぞれの文脈から区別することができる。異種タンパク質発現の文脈において、「宿主細胞」という用語は、発現宿主として用いられる遺伝子組み換え細胞(transgenic cell)を示す。このように、この細胞またはその前駆細胞(progenitor)には、発現させられるべきタンパク質のcDNAを含む適切なベクターを用いて、核酸が導入されている。ウイルスのライフサイクルの文脈において、「宿主細胞」という用語は、ウイルスに感染された細胞を示す。ウイルスは、その細胞を、複製のために利用する。
【0038】
本明細書において、「ベクター(vector)」という用語は、外来遺伝子を別の細胞に導入させるために用いられる分子ベヒクル(molecular vehicle)を示す。一般的に、ベクター自身は、インサート(目的の配列)と、ベクターの「バックボーン(backbone)」の役割を果たす、より大きな配列とを含むDNA配列である。遺伝情報を別の細胞に導入するベクターの目的は、通常、標的細胞中のインサートの単離、複製または発現である。
【0039】
本明細書において、「プラスミド(plasmid)」という用語は、プラスミド・ベクターを示す。すなわち、複製起点(ORI)に起因して、適切な宿主の内部で自己複製することができる環状DNAを示す。さらに、上記のマーカーは、形質転換(transformation)の成功、または、外来DNAを細胞もしくは多重クローニング部位に導入するように意図されているその他のプロシージャの成功を示唆する目的で、プラスミドが、選択マーカー(selectable marker)を含んでもよい。クローニング・ベクターまたはドナー・ベクターと呼ばれるプラスミド・ベクターは、クローニングを容易にして、目的の配列を増殖する目的で用いられる。発現ベクターまたはアクセプター・ベクターと呼ばれるプラスミド・ベクターは、特に、一定の(defined)標的細胞における目的の遺伝子の発現に用いられる。これらのプラスミド・ベクターは、一般に、プロモーター、導入遺伝子および終了配列を含む発現カセットを与える。発現プラスミドは、異なる宿主細胞における増殖(propagation)および淘汰を可能にする要素を含むシャトル・プラスミドであってもよい。
【0040】
本明細書において、「ウイルスタンパク質(viral protein)」という用語は、ウイルスによって生産されたタンパク質を示す。このタンパク質は、ウイルスのエンベロープおよび/またはカプシドを形成してもよく、非構造性のタンパク質、調節タンパク質または付属タンパク質であってもよい。
【0041】
本明細書において、「表面タンパク質(surface protein)」という用語は、ウイルスの外層を形成するウイルスタンパク質を示す。このようなタンパク質としては、(i)エンベロープを有するウイルス(エンベロープウイルスと称する場合がある。)の脂質二重層コーティングに固定された(anchored)タンパク質、(ii)エンベロープウイルスおよびエンベロープを有しないウイルス中のカプシドタンパク質、および/または、(iii)多くのウイルス(例えば、バクテリオファージ、または、ヒトおよび鳥類を含む哺乳類を含むに感染するウイルス)中に見られるようなスパイクタンパク質を例示することができる。多くの場合、エンベロープウイルスの脂質二重層コーティングは、ウイルスの複製サイクル中(すなわち、開口分泌時または細胞溶解時)に、宿主細胞中の細胞内膜(例えば、核の内膜もしくはゴルジ膜)または宿主細胞の外膜から、ウイルスによって獲得される。
【0042】
本明細書において、「ウイルス融合タンパク質(viral fusion protein)」という用語は、エンベロープウイルスの糖タンパク質を示す。この糖タンパク質は、宿主細胞の感染(ウイルスの遺伝物質の宿主細胞中への挿入)を促進する。その構造の一体部分(integral part)として脂質二重層を有するエンベロープウイルスは、ウイルス膜と宿主細胞膜との融合により、当該ウイルスが感染した細胞の一員となる。ウイルス融合タンパク質は、2つの主要な特徴を兼ね備える。つまり、ウイルス融合タンパク質は、レセプタ結合機能(receptor binding function)を有する。レセプタ結合機能は、ウイルスを宿主細胞に付着させる機能である。また、ウイルス融合タンパク質は、融合機能(fusion function)を有する。融合機能は、ウイルス膜と宿主細胞膜との融合を仲介すべく、活性化されてよい。ウイルス融合タンパク質としては、後述するような、クラスIのウイルス融合タンパク質が、よく知られている。
【0043】
本明細書において、「融合ペプチド(fusion peptide)」という用語は、融合プロセスの間に宿主細胞膜の中に挿入されたウイルス融合タンパク質の明確に保存された疎水性領域(distinct conserved hydrophobic region)を示す。融合ペプチドは、どちらかといえば、無極性の領域であり、比較的、グリシン残基およびアラニン残基に富み、いくつかの大きな疎水性残基を含む。クラスIのウイルス融合タンパク質に関して、融合ペプチドは、タンパク質の膜貫領域のN末端に配される。
【0044】
(本発明の目的)
本発明の目的は、上述の問題点を打開する方法および/または実施態様を提供することにある。この目的は、独立項に記載された実施態様によって解決される。
【図面の簡単な説明】
【0045】
【図1A】様々な分類群のN−結合型グリコシル化パターン概要を示す。
【図1B】いくつかの種類の繊毛虫類におけるパターンの可能性のあるバリエーション(potential variation)を示す。
【図2A】合成HA遺伝子をコードするドナー・ベクターを示す。
【図2B】テトラヒメナ好熱菌において用いられる発現ベクターを示す。
【図3A】血球凝集素を、そのサブユニットであるHA1およびHA2とともに示す。
【図3B】血球凝集素の概要を、そのサブユニットであるHA1およびHA2とともに示した概略図である。
【図4A】クラスIのウイルス融合タンパク質の一般的な構造を示す。
【図4B】インフルエンザAウイルス株の血球凝集素のサブユニットが例示的に説明される。
【図5】テトラヒメナ好熱菌と、ホモ・サピエンスとの間におけるコドン出現頻度の比較を示す。
【図6】繊毛虫類において用いられている遺伝子コードを示す。
【図7】ウイルス血球凝集素配列、テトラヒメナ好熱菌における発現に関してコドン最適化された血球凝集素の配列との間におけるコドン出現頻度の比較を示す。
【図8】組み換えHAを発現している形質転換された繊毛虫類の免疫ブロットを示す。
【図9】血球凝集素タンパク質フラグメントの免疫ブロットを示す。
【図10A】HAロングに関連するタンパク質の疎水性プロットを示す。
【図10B】主要な繊毛虫類タンパク質の疎水性プロットを示す。
【発明を実施するための形態】
【0046】
本発明によれば、ウイルスタンパク質の異種タンパク質発現系が提供される。この発現系は、繊毛虫類の宿主細胞と、ウイルスタンパク質またはそのフラグメントをコードする少なくとも1つの核酸と、当該核酸に機能しうるように連結されたプロモーターとを備える。
【0047】
好ましい実施形態において、上記の核酸はcDNAである。
【0048】
多くの場合、ウイルスタンパク質は構造タンパク質(すなわち、酵素でもなく、タンパク質ホルモンでもなく、抗体でもない。)であり、製薬目的または工業目的で大して関心をひかないので、基本的には、現在、ウイルスタンパク質の異種タンパク質発現は一般的ではない。
【0049】
さらに、繊毛虫類におけるウイルスタンパク質の発現は、上述の参考文献において、いまだに示唆されていない。本発明者らは、繊毛虫類に関しては、バクテリアまたは後生動物に関するものとは異なり、特異的なウイルスがあまり多くは知られていないことに気がついた。このことは、繊毛虫類に共通する核の二形性に起因する可能性がある。他の原因としては、繊毛虫類の独特なコドン使用頻度と、AおよびTに富むゲノムとが考えられる。そこで、本発明者らは、高等生物の病原性ウイルスは、大部分の繊毛虫類(例えば、テトラヒメナ)の中では増殖することができないと仮定した。
【0050】
上記の議論は、今までのところ、繊毛虫類中でウイルスタンパク質を生産する試みが知られていないという事実の理由を説明する。
【0051】
今までに知られていたような、繊毛虫類は、ウイルスに感染しにくいという事実は、驚くべき利点をもたらす。このことは、繊毛虫類を用いた生産プロセスにおいて、付随的な(adventitious)ウイルスの増幅または増殖が起こらないことを意味する。このことは、さらに、タンパク質が治療上の使用目的で生産される場合、工業プロセスにおいて、必要に応じて、ヒト細胞培養または動物細胞培養を用いた高価なウイルス枯渇プロシージャ(virus depletion procedure)を省略することができることをも意味する。
【0052】
さらに、繊毛虫類中のウイルスタンパク質発現に適したプロモーターは、例えば、本発明の出願人に関連して登録された国際公開第2007/006812号に開示されており、その内容は、参照によって本願に組み込まれる。添付の配列表の配列ID番号12および13は、本発明との関連で特に好ましい、繊毛虫類に特異的な2つのプロモーターの配列を与える。これらは、すなわち、熱誘発性プロモーター(heat-inducible promoter)と、メタロチオネイン・プロモーター(metallothionein-promoter)である。
【0053】
Gaertigら(1999年、欧州特許出願公開第1151118号明細書を参照。)は、原生動物(テトラヒメナ好熱菌(Tetrahymena thermophila))を、他の原生動物(例えば、ウオノカイセンチュウ(Ichthyophthirius multifiliis))の表面抗原タンパク質(surface antigen protein)のタンパク質発現系として用いたことを開示している。彼らは、このようにして生産されたタンパク質は、形質転換されたテトラヒメナ細胞の表面上に提示されていたと報告している。また、そのタンパク質は採取され、ウオノカイセンチュウに対するワクチンの製造に用いられたと報告している。他のアイデアとしては、形質変換され、その表面に抗原を提示しているテトラヒメナ細胞を、生ワクチンとして直接使用することが考えられる。
【0054】
しかしながら、その著者らは、テトラヒメナは、AおよびTに富んだゲノムを有する有機体(例えば、マラリア原虫のような原生動物、マイコプラズマ、リシューマニアなどであり、これらは、同様にヒト病原体である。)からの遺伝子のクローニングおよび発現用としてのみ利用することができると提言している。従来の系(例えば、大腸菌)のATトラクトは、本来的に不安定だったので、これらの有機体から遺伝子をクローニングすることは難しいことが判明していた。例えば、テトラヒメナDNAは、ほぼ75%がATからなる。
【0055】
さらに、コドン使用頻度の問題によって、テトラヒメナ中で外来遺伝子を発現させることが妨げられていた。テトラヒメナ好熱菌(T. thermophila)は、グルタミン用に、UAAトリプレットおよびUAGトリプレットを利用する。しかし、他の多くの有機体において、これらのコドンは、翻訳を終了させる終止コドンとして利用される。このことから、UAAトリプレットおよびUAGトリプレットを終止コドンとして有する外来遺伝子(繊毛虫類の遺伝子ではない遺伝子)は、正確に発現しないということを導き出すことができるかもしれない。
【0056】
本発明の好ましい実施態様において、発現系は、さらに、上記の核酸に機能しうるように連結されたシグナル配列を含む。シグナル配列は、上記の核酸によってコードされたウイルスタンパク質またはそのフラグメントの細胞外培地(extracellular medium)への分泌(secretion)の原因となる。
【0057】
ウイルスタンパク質が自然に発現した場合(つまり、バクテリオファージに感染されたバクテリアの宿主細胞によって、または、ウイルスに感染された後生動物の宿主細胞によって出現した場合)には、ウイルスは、その宿主の機構および代謝作用を利用して、単に自分自身のタンパク質を生産するだけなので、ウイルスタンパク質は分泌の対象にはならない。なお、生産されたタンパク質は、その後、宿主細胞が溶解する前に、第2世代のウイルスへと組み立てられる。このことは、一方で、ウイルスタンパク質が、いまだ、分泌される能力を最適化しようとする進化的圧力にさらされていないことを意味する。
【0058】
これは、遺伝子組み換え技術による可溶性のウイルスタンパク質の生産と、その後の工程におけるこれらの可溶性のウイルスタンパク質の分泌とが複雑な問題であるという事実についての原因の1つである。例えば、適切なシグナル配列は、本発明の出願人に関連して登録された国際公開第03/078566号に開示されており、その内容は、参照によって本願に組み込まれる。
【0059】
添付の配列表の配列ID番号8および10は、本発明との関連で特に好ましい2つのシグナル・ペプチドの核酸配列を与える。これらは、すなわち、HA遺伝子の内因性のシグナル・ペプチドと、繊毛虫類のホスホリパーゼのA1シグナル・ペプチドである。
【0060】
本発明のさらに他の好ましい実施形態によれば、ウイルスタンパク質は、上述のウイルスの表面タンパク質である。
【0061】
宿主の感染に関して、宿主が免疫応答を許す場合(例えば、よく発達した免疫システムを有する哺乳類の場合)、ウイルスの表面タンパク質は、宿主と接触して、免疫応答を引き起こす場合がある。このように、これらのタンパク質は、単離された形態で、または、アジュバント(不活性化されたウイルスもしくはそのフラグメント)に関連して、ワクチンとして機能する可能性がある。
【0062】
本発明のさらに別の好ましい実施態様において、遺伝子組み換えの繊毛虫類は、テトラヒメナ科(family Tetrahymenidae)のメンバーであってよい。
【0063】
特に好ましい実施形態において、遺伝子組み換えの繊毛虫類は、テトラヒメナ(Tetrahymena sp)(特に、テトラヒメナ好熱菌)であってよい。テトラヒメナは、非病原性の単細胞真核微生物(nonpathogenic unicellular eukaryotic microorganism)である。テトラヒメナは、数箇所の研究所において、発現宿主として確立されている。テトラヒメナは、異種タンパク質発現に適した多くの利点を有することを特徴とする。テトラヒメナは、広く調べられたモデル生物であり、50年以上にわたる基礎研究において、ウイルスまたは内部寄生生物(endoparasite)が観測されていない。指標細胞系を用いた試験では、ウイルス、マイコプラズマなどの内因性の感染性病原体(endogenous infectious agen)が発見されていない。
【0064】
第一に、繊毛虫類におけるコドン使用頻度に関する上記の考察は、テトラヒメナにも同様に当てはまる。さらに、ハイコピープラスミド(high copy number plasmid)は、テトラヒメナに使用可能であり、ミニ染色体のrDNAからの複製起点(ORI)を含む。このミニ染色体のrDNAは、1つの細胞あたり最大9.000のコピー数の中に含まれる。その上、安定した統合は、大核DNA中で発生しうる。大核DNAにおいて、全ての遺伝子は、45個のコピー数の中に含まれる。高遺伝子量(high gene dose)は、効率的なタンパク質生合成および高い生産性の理想的な前提条件である。酵母菌およびバクテリアとは対照的に、テトラヒメナ属(genus Tetrahymena)の繊毛虫類は、発酵上澄み(fermentation supernatant)に、生物タンパク質(biologically protein)を効率よく分泌する。
【0065】
テトラヒメナは、タンパク質に翻訳後修飾(例えば、ジスルフィド架橋、GPIアンカー、リン酸化反応、アセチル化、グリコシル化)を施す(attach)ことができる。これらは、酵母菌またはその他の真核生物発現系において検出されるものよりも、哺乳類細胞中のものに似ている。
【0066】
哺乳類細胞とは異なり、テトラヒメナは、合成培地(chemically defined media)を使用することができ、増殖因子のように、ペプチドまたは血清成分を必要としないので、短い世代時間(1.5−3時間)による成長の容易さと、コスト削減とを兼ね備える。
【0067】
最大2×107細胞/mLの細胞密度と、最大80g/Lの乾燥質量の条件で、回分式、供給回文式および連続式のテトラヒメナの発酵が実施され、最大1000Lまでの生産規模の拡大(スケールアップ)が問題なく実施できることが実証された。レポーター・タンパク質を用いたフィージビリティ・スタディにおいて、1日当たり500−90pg/細胞の空時収量(space-time yield)が既に得られた。相同的発現を用いた最初の実験では、分泌されたタンパク質に関して、1日当たり200mg/L以上の収率(yield)が得られた。テトラヒメナは、従来型の微生物学的な(microbiological)発現系(バクテリアまたは酵母菌)用の生産設備中で発酵させることができる。このことは、既存の生産プラントの改修に費用をかけたり、生産設備を新たに建設したりする必要のないことを意味する。
【0068】
このような利点にも関わらず、繊毛虫類発現系(特に、テトラヒメナ)については、いまだにあまり知られていない。異種タンパク質発現系となる見込みのある系について尋ねられた当業者は、むしろ、大腸菌、酵母菌もしくはバキュロウイルスを用いた系、または、不死化哺乳類細胞株に想到するであろう。
【0069】
繊毛虫類発現系(特に、テトラヒメナ)の利用には、上述の状況を背景としても予測することができない別の大きな利点がある。哺乳類の免疫系の自己識別/非自己識別は、糖タンパク質の糖質組成(carbohydrate composition)に基づいて実現されるので、抗原のグリコシル化パターンは、かなりの程度、その免疫原性に寄与する。
【0070】
Readingら(2000年)は、哺乳類細胞において、マンノース受容体が、インフルエンザウイルス(およびマクロファージ中の他のエンベロープウイルス)の感染性侵入における主要なエンドサイトーシス受容体として機能することを報告している。
【0071】
マンノース受容体は、マンノース、フコースまたはN−アセチルグルコサミン(acytylglucosamin)および、保存されている(conserved)糖認識ドメイン(CRD)を含むC型レクチン中で終了する(terminate)糖タンパク質の取り込みを仲介する、膜結合型のレクチンタンパク質(「マンノース結合レクチン」、「MBL」とも呼ばれる。)として機能する能力がある。
【0072】
マンノース結合レクチン(MBL)のCRDは、ヘキソース(例えば、ピラノース環のC3およびC4の位置に、エクアトリアル配位の(equatorial)ヒドロキシル基を有するマンノースおよびN−アセチルグルコサミン)と結合する。それらは、哺乳類の糖タンパク質に一般的に見られるオリゴ糖と親和性がない。
【0073】
マクロファージは、哺乳類の免疫系において重要な役割を果たす。マクロファージは、エンドサイトーシスによって病原体を取り上げて、それらを消化した後、病原体に関連する抗原を対応するヘルパーT細胞に提示する。抗原を細胞膜中に一体化し(integrate)、他の白血球細胞に対しては、当該マクロファージは、表面に抗原を有するけれども、病原体ではないことを示しながら、対応するヘルパーT細胞に対して、MHCクラスII分子に結合した抗原を提示することで、抗原の提示が実施される。
【0074】
Readingら(2000年)の研究から、本発明者らは、以下の通り推測した。つまり、ワクチンの免疫原性を強化する目的で、ワクチン中に含まれるタンパク質のグリコシル化パターン中のマンノース量を増加させることが有用なのではないかと推測した。しかしながら、タンパク質のグリコシル化は、真核生物にしかない翻訳後修飾装置(posttranslational modification apparatus)中で実行されるので、意図的に修飾することが難しい。
【0075】
しかしながら、真核生物分類群は、それらのグリコシル化スキームの相違点を示している。グリコシル化は、主に、真核生物中および古細菌中で起こり、バクテリア中では起こらない。一般的に、N−結合型グリコシル化(N-glycosylation)という用語は、アスパラギン(N)アミノ酸残基のグリコシル化を示す。オリゴ糖鎖は、オリゴサッカリルトランスフェラーゼ酵素によって、トリペプチド配列Asn−X−SerまたはAsn−X−Thrにおいて生じるようなアスパラギン残基に取り付けられる。ここで、Xは、任意のアミノ酸であってよい。なお、ProおよびAspは、まれにしかみられない。
【0076】
驚いたことに、本発明の発明者らは、繊毛虫類が独特のN−結合型グリコシル化パターンを有し、高度(high degree)なマンノース残基を有することを見出した(図1Aおよび図1Bを参照。)。本発明の発明者らは、さらに、このことから、哺乳類のワクチン接種に用いられるであろうタンパク質は、末端のマンノース残基から利益を得ている可能性があることを見出した。すなわち、マンノース受容体を介したマクロファージ中におけるこれらのタンパク質の取り込みが増加した結果、免疫応答が増加した可能性があることを見出した。
【0077】
これらのことから、本発明者らは、繊毛虫類(特に、テトラヒメナ)において発現させられたタンパク質は、高度なマンノース残基に起因した高度の免疫原性を有し、遺伝子組み換えの繊毛虫類は、哺乳類用のワクチン(特に、ウイルスワクチン)を生産するための有望な候補であることを導き出した。
【0078】
さらに、本発明者らは、マクロファージが、マンノースに富むグリコシル化パターンを有する病原体の食作用を増加させることを見出した。このことから、このようにして生産されたワクチンが対象となる哺乳類動物に投薬(administer)されると、マンノースに富むグリコシル化パターンに起因する繊毛虫類におけるワクチンの発現が免疫反応を増加させることが証明される。
【0079】
この発見は、上述のGaertigら(1999年)の研究では予測されていない。Gaertigらは、ただ、魚のワクチンとして使用する目的で、遺伝子組み換えのテトラヒメナ中で生産された繊毛虫類のタンパク質を使用することを記載しているだけである。しかしながら、上述したような、マクロファージにおけるマンノース受容体の効果は、現在の知識によれば、魚用ではなく、哺乳類にのみ適用することができる。このことから、魚のワクチンが、ワクチンのグリコシル化パターン中のマンノース量の増加によって、同様の効果をあげることができるとは、まったく予測不能であった。
【0080】
特に好ましい実施形態において、ウイルスの表面タンパク質は、ウイルス融合タンパク質(上述の定義を参照。)である。
【0081】
別の好ましい実施形態において、ウイルスの表面タンパク質は、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質からなる群から選ばれるウイルス融合タンパク質の少なくとも1つであってよい。
【0082】
これらのウイルス融合タンパク質は、構造的に全く異なるが、これらの融合サブユニットは、折りたたまれて、最終的にはヘアピン構造の三量体(trimer-of-hairpins)になる。ヘアピン構造の三量体において、3つのC末端領域は、中心のN末端三量体コア(central N-terminal trimeric core.)の外側を包む(pack)。
【0083】
上述のとおり、ウイルス融合タンパク質は、クラスI、クラスIIおよびクラスIIIのウイルス融合タンパク質に細分される。表2に、上述のとおり定義されたエンベロープウイルスに由来するウイルス融合タンパク質のいくつかの概要を示す。
【0084】
上記の表において、「−S−S−」は、ジスルフィド架橋を示す。「/」は、当該記号によって示されたサブユニットが、ジスルフィド結合以外の方法でお互いに関連していることを示す。「GPX」は、「糖タンパク質 X」を示し、「FX」は、「融合タンパク質 X」を示す。
【表2】
【0085】
多くの場合、クラスIのウイルス融合タンパク質の2以上のサブユニットは、ジスルフィド架橋またはその他の手段によって結合される。そのような二量体として、HA1−S−S−HA2、SU−S−S−TM、HA1−S−S−HA2、SU−S−S−TM、SU/TM、F2−S−S−F1、S1/S2、GP1−S−S−GP2、GP1/GP2/SSPを例示することができる。ここで、サブユニットの少なくとも1つは、疎水性ドメイン(例えば、HA1−S−S−HA2構造体におけるHA2サブユニット)によって特徴付けられる。
【0086】
好ましい実施形態において、タンパク質は、クラスIのウイルス融合タンパク質であってよい。図4Aに、クラスIのウイルス融合タンパク質の概要を示す。
【0087】
特に好ましいウイルスタンパク質は、血球凝集素(hemagglutinin)である。血球凝集素(HA)は、クラス1の膜融合タンパク質サブユニットを2つ含む(すなわち、HA2およびHA1)、抗原性の糖タンパク質(antigenic glycoprotein)である。血球凝集素は、多くの他のバクテリアおよびウイルスと同様、インフルエンザウイルス(オルソミクソウイルス科)の表面に見られる。
【0088】
血球凝集素は、ウイルスと、当該ウイルスに感染されようとしている細胞との結合に関与する。今日では、少なくとも16種類の異なるHA亜類型(subtype)が知られており、H1−H16に分類される。最初の3つの血球凝集素(すなわち、H1、H2およびH3)は、ヒト・インフルエンザウイルス中に見出された。他方で、例えば、鳥インフルエンザウイルスは、H5血球凝集素を有する。
【0089】
HAの主な機能には、標的となる脊椎動物細胞の認識がある。この認識は、標的細胞膜上のシアル酸を含む受容体を介して達成される。HAの主な機能には、標的細胞膜とウイルス細胞膜との融合を引き起こすことで、標的細胞へのウイルスゲノムの侵入がある。この文脈において、オルソミクソウイルス科が、ヌクレオカプシドを包み込むリン脂質膜を有し、当該リン脂質膜が、宿主細胞から放出されて獲得されたものであることは、言及する価値がある。
【0090】
HAは、ホモ三量体の、内在性膜糖タンパク質(homotrimeric integral membrane glycoprotein)である。その形状は略円筒形状であり、その長さはおよそ135Åである。HAを構成する3つの同一の(identical)モノマーが組み立てられて、中心のαヘリックス・コイル(central α helix coil)と、サイトに結合するシアル酸を含む3つの球状のヘッド部とを形成する。HAモノマーは、前駆体として合成される。この前駆体は、後に、グリコシル化され、2つのより小さなポリペプチド(すなわち、HA1サブユニットとHA2サブユニット)に開裂される。それぞれのHAモノマーは、HA2によって膜に固定され、大きなHA1小球が先端に配された(topped)、長いらせん鎖を含む(図3Aおよび図3Bを参照。)。
【0091】
結合機構は、以下の通りである。HAは、標的細胞の表面に存在する単糖類シアル酸(monosaccharide sialic acid)に結合する。これにより、ウイルス粒子が細胞の表面にとどまる。次に、細胞膜がウイルスを飲み込んで、細胞内でウイルスをピンチオフして、飲み込んだウイルスを含み、エンドソームと呼ばれる新しい膜結合コンパートメントを形成する。その後、細胞は、エンドソームの内部を酸性化して、リソソームに送り込むことでエンドソームの中身を消化する。しかし、pHが低くなると(pH<6.0)、HA分子の立体構造変化が引き起こされ、HA2からHA1が分離した後、再び折りたたまれて、全く異なる形状になる。このプロセスの末期には、いわゆる「融合ペプチド」が分子のフック(molecular hook)のように機能する。つまり、融合ペプチドを、鉤のようにエンドソーム膜に挿入して固定する。HA分子の残りの部分が新たな構造(より低いpHでも安定な構造)に折りたたみ直された後、「掴み鉤」を引っ込めて、エンドソーム膜をウイルス粒子自身の膜のすぐ近くに引き寄せる。これにより、この2つがお互いに融合するようになる。これらの工程が起こると、ウイルスの中身(そのRNAを含む。)は、標的細胞の細胞質の中へと、自由に流出することができる。
【0092】
本発明者らは、可溶性タンパク質としての発現(細胞内発現)も、テトラヒメナにおける全ての(full)血球凝集素の分泌(細胞外発現)も、困難であることを見出した。予備実験において、細胞膜内へのタンパク質発現それ自体は成功したけれども、可溶性タンパク質の発現およびその分泌は、問題を引き起こした。つまり、血球凝集素分子は、テトラヒメナ内で首尾よく発現し、シグナル・ペプチドを備えていたが、血球凝集素分子は、細胞内膜の構造体および宿主細胞の細胞膜に付着したままのようであった。
【0093】
他のタンパク質の発現および分泌において有用であることが分かっていた(例えば、国際公開第03/078566号を参照。)シグナル・ペプチドを使用したにもかかわらず、上述の問題が見出された。国際公開第03/078566号は、参照によって本願に組み込まれる。
【0094】
本発明者らは、この現象の原因は、疎水性の強い領域(特に、HA2の当該領域)にあり、それらが、首尾よく発現したタンパク質を細胞膜中に保持することに関与している可能性があると考えた。
【0095】
この現象は、感染宿主細胞中における、ウイルス粒子の自然の複製プロセスに似ていると考えられる。自然の複製プロセスにおいて、感染宿主によって発現させられたウイルスタンパク質の多くは、細胞質の内部でビリオンを組み立てるのに用いられる。一方、血球凝集素は、小胞体(endoplasmatic reticulum)およびゴルジ体によって、細胞表面に運ばれる。血球凝集素は、成熟ウイルスが出芽して、宿主のリン脂質膜(かつての宿主の細胞膜)に取り囲まれた状態で細胞から離れるまで、細胞膜に固定されて、細胞表面にとどまる。このようにして、血球凝集素がその外被を獲得する。
【0096】
本発明の別の好ましい実施形態において、ウイルス融合タンパク質またはそのフラグメントが提供される。これらは、核酸によってコードされており、少なくとも1つの疎水性ドメインまたはそのフラクションが欠けている。
【0097】
驚くべきことに、本発明の場面において、本発明者らは、少なくとも疎水性の融合ペプチドドメインまたはそのフラクションを欠くウイルスタンパク質が、タンパク質の分泌を促進することを見出した。
【0098】
上述のとおり、異種タンパク質発現において、タンパク質の分泌はシグナル配列によってサポートされる。大きな疎水性ドメインは、タンパク質の分泌の場面において、問題を引き起こす可能性がある。大きな疎水性ドメインは、発現宿主の細胞膜または細胞内膜との間で親和力を発揮する可能性があるからである。基本的に、遺伝子組み換えの繊毛虫類(例えば、テトラヒメナ)におけるタンパク質の分泌は、容易に達成される。遺伝子組み換えの繊毛虫類は、食物の細胞外消化のために、大量の加水分解酵素を分泌するからである。しかしながら、繊毛虫類におけるウイルス表面タンパク質のフルスケールの分泌が厄介になる可能性のあることがわかった。
【0099】
本発明者らは、そのような場合には、疎水性ドメインの除去が、タンパク質の分泌を促進して、そのように修飾されたタンパク質の免疫原性に影響を与えることなく、分泌されたタンパク質の収率を向上させるのに有用であることを見出した。
【0100】
ウイルスタンパク質において、抗原ドメインは親水性であることが多い。抗原ドメインは、水性媒体(唾液、血液およびその他の体液、汚水など)であることの多い周囲の媒体(medium)中にむかって、ウイルスの表面から伸長するからである。このことは、そのような場合において、疎水性ドメインの除去は、タンパク質の免疫原性に影響を与えないことを意味する。
【0101】
以下において、タンパク質の免疫原性を維持しながら、タンパク質の分泌または細胞溶解後の精製を促進する目的で、ウイルスタンパク質の疎水性ドメインを除去するという基本原理が、血球凝集素(HA)を用いた場合について説明されるであろう。
【0102】
上述のHAタンパク質発現において、HA0は、前駆体タンパク質(precursor protein)であり、それぞれのcDNAによってコードされたHA1およびHA2という2つのサブユニットからなる。HA0は、タンパク質分解酵素(例えば、多くの脊椎動物の肺組織中に見られるようなトリプターゼ)によって開裂されて、そのサブユニットに分割される。分割されたサブユニットは、ジスルフィド架橋によって、お互いに結合された状態を維持する(HA1−HA2−タンパク質。図3A、図3B、図4Aおよび図4Bを参照。)。
【0103】
HA1は、受容体の認識に関与しており、特に強い免疫原性を有するドメイン(特に、球状ドメインの「ヘッド部」)を備える。このドメインは、宿主細胞膜のグリコカリックスにあるシアル酸との結合サイトを含む。一方、HA2(完全なHA1−HA2複合体をウイルス膜にとどめておく(anchor)役割もある。)は、膜融合に関与する。HA2は、比較的疎水性の配列(表1を参照。)からなるN末端融合ペプチド(FP)を含む。
【0104】
この融合ペプチドは、約24個の高度保存されたN末端残基(N-terminal, highly conserved residue)からなる。これら残基のうちの少なくとも6個は、グリシンである(図4Aおよび図4Bを参照。同図において、融合ペプチドは8個のグリシンを含む。)。次に、3個のα−へリックス(α−H1、α−H2およびα−H3)が続く。その次には、疎水性の強い膜貫通ドメイン(TMD)が続く。TMDは、タンパク質を、ウイルスエンベロープの脂質二重層にとどめる(図3Aおよび図3Bを参照。)。
【0105】
次表は、本発明に関連して、少なくとも1つの疎水性ドメインを欠く、修飾された血球凝集素の候補の概要を示す。この表において、「TMD」は膜貫通ドメインを示し、「FP」は融合ペプチドを示す。上述の記載から、他のクラスI、クラスIIおよび/クラスIIIのウイルス融合ペプチドにも、この原理を適用できることが明らかである。
【表3】
【0106】
本発明者らが、タンパク質の分泌の代替手段として、細胞質中に可溶な形態でのウイルスタンパク質の細胞内発現が有望なオプションであることを見出したことは、特筆に価する。本発明者らは、細胞内に発現させられたフル装備の(full-featured)クラスIのウイルス融合タンパク質は、細胞膜だけでなく、繊毛虫類宿主の細胞内膜(小胞体、ゴルジ、核膜、ミトコンドリアなどたくさんある。)にも付着しやすいであろうことを見出し、また、このような場合には、宿主細胞が溶解した後で単離することができないことを見出した。さらに、本発明者らは、ウイルス融合タンパク質の少なくとも1つの疎水性ドメインが欠けていれば、ウイルス融合タンパク質は、細胞内膜に付着しにくくなるであろうことを見出した。したがって、そのように修飾されたタンパク質(例えば、表3に示されるようなタンパク質)は、全細胞溶解液からより容易に単離することができる。
【0107】
さらに、本発明によれば、少なくとも1つの核酸分子が提供される。核酸分子は、以下の(a)〜(j)の少なくとも1つからなる群から選択されてよい。
(a)配列ID番号2、4および/または6として表されたヌクレオチド配列を含む核酸分子。
(b)配列ID番号3、5および/または7として表されたアミノ酸配列を含むポリペプチドをコードする核酸分子。ここで、上記のポリペプチドは、ウイルスタンパク質またはそのフラグメントである。
(c)少なくとも1つの疎水性ドメインを欠いたウイルスタンパク質の切断型(truncated form)をコードする核酸分子。ここで、上記のウイルスタンパク質は、好ましくはウイルス融合タンパク質であり、さらに好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質である。
(d)膜貫通ドメイン(TMD)、融合ペプチド(FP)および/またはHA2サブユニットを欠いたクラスIのウイルス融合タンパク質の切断型をコードする核酸分子。
(e)(a)〜(d)の核酸分子のフラクション、変異体(variant)、ホモログまたは誘導体(derivative)である核酸分子。
(f)(a)〜(e)のいずれかの核酸分子の相補体である核酸分子。
(g)ストリンジェントな条件下で、(a)〜(f)のいずれかの核酸分子と分子交雑することができる核酸分子。
(h)(a)〜(g)のいずれかの核酸分子と比較して、少なくとも1つの無症状の単一ヌクレオチドの置換(silent single nucleotide substitution)を含む核酸分子。
(i)原生動物の発現宿主に最適化するようコードされた、(a)および(c)〜(h)核酸分子に関連する核酸分子。
(j)(a)〜(i)のいずれかの核酸分子と、少なくとも70%、好ましくは少なくとも95%の配列同一性を有する核酸分子。
【0108】
これらの核酸分子の特に好ましい代替策(fallback position)は、(a)〜(i)のいずれかの核酸分子と、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98または99%の配列同一性を有する。
【0109】
さらに、繊毛虫類の宿主細胞のトランスフェクションに用いられるベクターが提供される。このベクターは、(a)ウイルスタンパク質またはそのフラグメントをコードする核酸と、(b)当該核酸に機能しうるように連結されたプロモーターとを備える。
【0110】
これらの核酸は、cDNAであることが好ましい。
【0111】
さらに、本発明によれば、本発明に係るベクターを用いて核酸を導入された繊毛虫類の宿主細胞が提供される。この繊毛虫類は、テトラヒメナ科のメンバーであることが好ましい。
【0112】
さらに、本発明によれば、少なくとも1つのウイルスタンパク質またはそのフラグメントを生産するプロセスが提供される。この方法は、(a)本発明に係るベクターを用いて、繊毛虫類の宿主細胞に核酸を導入する段階と、(b)タンパク質の発現が可能な条件下で、宿主細胞を培養する段階とを有する。
【0113】
好ましい実施態様において、このプロセスは、(c)ある特定のタンパク質について発現させられたタンパク質の分泌が可能な条件下で、宿主細胞を培養する段階を、さらに有してよい。
【0114】
さらに、本発明によれば、以下の(a)〜(d)の少なくとも1つからなる群から選択されるタンパク質が提供される。
(a)本発明に係る核酸によってコードされたタンパク質。
(b)本発明に係るアミノ酸配列を含むタンパク質。
(c)少なくとも1つの保存アミノ酸置換(conservative amino acid substitution)を含む、(a)または(b)に関連するタンパク質。
(d)本発明に係るプロセスによって得られたタンパク質。
【0115】
さらに、本発明によれば、医薬組成物を生産する方法が提供される。この方法は、(a)本発明に係る繊毛虫類発現系において本発明に係るタンパク質を発現させる段階と、(b)そのようにして得られたタンパク質を単離または精製する段階とを有する。
【0116】
他の実施態様において、本発明によれば、本発明に係るプロセスを含む医薬組成物を生産する方法が提供される。
【0117】
さらに、本発明によれば、本発明に係るタンパク質または本発明に係る方法を用いて生産されたタンパク質を含む医薬組成物が提供される。好ましい実施形態において、この組成物はワクチンである。
【0118】
(ディスクレーマー)
明細書を過度に長くすることなく、包括的に開示する目的で、出願人は、以上で参照した特許および特許出願のそれぞれを参照により組み込む。
【0119】
上述の詳細な実施態様における要素および特徴の特定の組合せは、単なる例示に過ぎない。これらの内容(teaching)を、本明細書または参照によって組み込まれた特許もしくは出願に記載されている他の内容と、置換または代用できることも、明白である。当業者がそう認識するように、当業者であれば、特許請求の範囲に記載された本発明の趣旨および範囲から逸脱することなく、本明細書に記載されている事項の変更、改良およびその他の実施に想到することができる。したがって、前述の開示は、ほんの一例であり、本発明の範囲を限定することは意図されていない。本発明の範囲は、後述の特許請求の範囲において定義され、それに加えて、その均等物も含まれる。さらに、特許請求の範囲に記載された本発明の範囲は、本明細書および特許請求の範囲において用いられる参照符号によって限定されない。
【実施例】
【0120】
(実施例および図面の簡単な説明)
本発明の目的(object)の付加的な詳細、特徴、特色および利点は、下位クレームおよびそれぞれの図面および実施例に関する記載において開示される。それらは、本発明の好ましい実施態様を例示的に説明する。しかしながら、これらの図面は、本発明の範囲を限定するものとして理解されるべきではない。
【0121】
(実施例および図面)
(1.発現ベクターの作製)
ドナー・ベクターの中に、様々な血球凝集素のフラグメント(配列ID番号2、4および6を参照。)のための合成遺伝子のクローンを作製した(図2Aを参照。)。Cre依存性リコンビナーゼ系(Cre dependent recombinase system)を用いて、全てのドナー・ベクターに由来する発現カセットを、アクセプター・ベクターに移動させた(図2Bを参照。)。
【0122】
(2.野生型テトラヒメナの培養および発現プラスミドの形質転換(transformation))
スキムミルク培地中、プロテオースペプトンを添加した(supplemented proteose peptone)(SPP)培地中、または、既知組成培地(CDM)中で、野生型のテトラヒメナ好熱菌株(例えば、B 1868/4、B 1868/7およびB 2068/1)を培養した。テトラヒメナ好熱菌(T. thermophila)細胞の形質変換は、前述のCassidy-Hanleyら(1997年)と同様の方法で実施した。
【0123】
(3.組み換え血球凝集素の検出)
80rpmの攪拌器中で、30℃における淘汰圧下において、形質変換されたテトラヒメナ細胞をSPP培地中で培養した。標的遺伝子発現は、41℃での熱ショック(HSP−P)または対数増殖培養への20nMのCd2+(MTT1−P)の添加により誘導された。
【0124】
標的遺伝子発現の誘導工程の後、形質変換された細胞の部分標本(aliquot)と、細胞の添加されていないSPP(cell free SPP)の上澄液の部分標本とを、24時間培養した。収集した細胞を、氷冷したRIPAバッファー中で可溶化した(150mM NaCl、10mM TrisHCl、5mM EDTA、0.1% SDS、0.1% DOC、1% Triton X100、2.5μg/mL E64の混合溶液中で、5000細胞/μL)。その後、超音波処理器中で15分間粉砕(disrupt)した。通常の方法に従って、SDS−ポリアクリルアミドゲル電気泳動法(SDS−PAGE)およびウエスタンブロット解析を実施した。粉砕された細胞の部分標本(つまり、1000個の細胞)または細胞の添加されていない上澄液を、Laemmliサンプルバッファー(125mM TrisHCl、pH6.8、10%Glycerol、4%SDS)中に再懸濁させて、12%SDS−ポリアクリルアミドゲル電気泳動法により分離した。ニトロセルロース膜にゲルを転写して、0.05%Tween20と、5%スキムミルクまたは3%ウシ血清アルブミンを含むPBSを用いてブロッキングした。ウイルス株に特異的な一次抗体(virus strain specific primary antibody)を用いて、形質転換された繊毛虫類における組み換え血球凝集素の発現を検出した。PBS/Tを用いて洗浄し、HRP標識二次抗体(secondary HRP-conjugated antibody)を適用した後、スーパーシグナル・ウエスト・ピコ・ケミルミネッセント基質(Perbio、Fischer Scientific社製)と、通常のX線フィルム現像とを組み合わせて用いて、ブロットを展開させた。図8、図9のA、図9のBおよび図9のCは、標的遺伝子発現が誘導された後の、形質転換されたテトラヒメナ細胞の細胞溶解物(cell lysate)および上清(supernatant)の代表的なウエスタンブロット(representative Western blot)を示す。野生型の対照群(図9のB。レーン3は対照群の細胞ペレットであり、レーン4は対照群の上清である。)は、全て空白である。図8から、発酵プロセス中に、HA抗原が上清に分泌されたことがわかる。図9から、形質転換された繊毛虫類の上清に関して、先端を切り取られた(truncated)血球凝集素が検出された(細胞内ドメインが除去された)こと(図9のAの「HAロング」)、さらに膜貫通領域が除去されたこと(図9のBの「HAショート」)、および、HA2サブユニット全体が除去されたこと(図9のCの「HA1」)が分かる。
【0125】
(4.血球凝集素の生産)
発酵には、Braun社のUD50(50リットル)および標準ラッシュトン・インペラ(standard Rushton impeller)を備えたInfors社のSixfors(0.5リットル)を使用した攪拌速度は、300rpmおよび400rpmに設定した。pO2は20%に設定し、pHは7.0に設定した。発酵は、標準培地中で実施した。
【0126】
(5.組み換え血球凝集素の精製)
中空糸モジュール(0.3m2、3L/min、管径11mm)を用いて、発酵もろみ液(fermentation broth)から細胞を分離することで、50L発酵プロセスから細胞を採取した。氷冷したpH7.4のリン酸ナトリウムバッファー(10mM、PB)を用いて、細胞ペレットを3〜4回洗浄した。その後、ムチン分泌胞体の中身を除去すべく、10℃、2400xgの条件で8分間の遠心分離処理(Sorvall(登録商標)evolution(商標)SLA−1500ローター)を実施して、小粒状にした。得られた細胞ペレットを、システインプロテアーゼ阻害剤E−64(70μM)および3%Tween20(登録商標)を補充したPB中に再懸濁させた。Ultra Turrax(IKA社製、UT T25+S25N−25G)を用いて、氷上で10,000rpmの条件で、5分間処理することで、細胞を粉砕した。PB(pH7.4)を用いて、溶解物の容積を1.8Lとし、4℃で17時間攪拌することで、可溶化した。中空糸モジュール(0.45μm、表面積850cm2)を用いて、細胞溶解物をろ過した。2.3LのPB(pH7.4)を用いて、中空糸モジュールを3回洗浄した。
【0127】
3段階クロマトグラフィー精製プロセスを用いて、クロマトグラフィー精製を実施した。その結果、高度に純化された組み換えHA抗原が得られた。この組み換えHA抗原は、変性しておらず、ワクチンの成分として適している。
【0128】
全てのクロマトグラフィーは、4℃で実施した。第1の精製ステップでは、上述のように準備したろ液を含むHAを、5%グリセロールおよび3%Tween20(登録商標)を含みpHが7.4のPBとの平衡を保たせながら、アンモニウム基が導入されたアニオン交換カラム(ammonium anion exchange column)(Capto(商標)Qカラム、強い第四級アンモニウムを有するアガロースビーズ・カラム)に、15mL/minの流量で供給した。最初に、3%Tween20(登録商標)を含むローディングバッファーを用いて、カラムを洗浄した。その後、Tween20(登録商標)を含まないローディングバッファーを用いた洗浄工程を実施して、洗浄剤を除去した。5%グリセロールと、NaClとを含むPBを用いて、部分的に精製されたHAを溶出させた。NaClの濃度は、第1ステップでは150mMであり、第2ステップ(pH7.4)では、1Mであった。収集したフラクションの全てのサンプルを、SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析によって解析した。HAが陽性のフラクション(HA positive fraction)は、次のクロマトグラフィー工程に備えて貯蔵した。
【0129】
組み換えHAは、セラミック・ヒドロキシアパタイト・カラム(CHT)とは結合しないので、セラミック・ヒドロキシアパタイト・カラムを用いて、夾雑タンパク質を除去した。Capto(商標)Qカラムから得て、貯蔵しておいたフラクションを濃縮した。NaClを除去すべく、5%グリセロールを含むPBとともに実験室規模のTFFモジュール(30kDa)を用いて、緩衝液交換を実施した。サンプルのpHは、CHTカラムに供給する前の段階で7.5に設定した。供給流量は、7mL/minに設定した。その後、5%グリセロールを含むPBを用いて、カラムを洗浄した。5%グリセロールを含み、pHが7.5である150mMのPBを用いて、段階溶離法による溶出工程を実施した。SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析により、溶出液およびフロースルー(flow through)を分析した。200mLのフロースルーを含むHAを採取して、第3のカラムに供給した。
【0130】
第3の精製ステップでは、Con A Sepharose 4Bカラム(臭化シアン法によってセファロース4Bと結合したコンカナバリンAと親和性のある媒体)を使用した。ヒドロキシアパタイト・カラムのフロースルーに150mMのNaClを補充して、5mL/minの流量でカラムに供給した。リン酸緩衝生理食塩水(PBS、pH7.4)を用いてカラムを洗浄し、0.5Mのメチルα‐D‐マンノピラノシド(pH7.4)を含むPBSを用いて、精製された組み換えHAを溶出させた。SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析により、溶出画分(elution fraction)のサンプルを分析した。
【0131】
(図面)
図1Aは、様々な分類群のN−結合型グリコシル化パターン概要を示す。一般的に、「N−結合型グリコシル化」という用語は、アミノ酸残基アスパラギン(N)のグリコシル化を示す。そして、オリゴサッカリルトランスフェラーゼによって、そのようなアスパラギン残基にオリゴ糖鎖が取り付けられる。この反応は、トリペプチド配列Asn−X−SerまたはAsn−X−Thrにおいて生じる。なお、Xは、Pro以外であればどのようなアミノ酸であってもよい。
【0132】
原核生物が全くN−結合型グリコシル化を有しない一方で、繊毛虫類は、マンノースに富み、タンパク質の免疫原性を強化するN−結合型グリコシル化パターンを有することを特徴としている(本文を参照。)。図1Bは、いくつかの種類の繊毛虫類におけるパターンの可能性のあるバリエーション(potential variation)を示す。
【0133】
図2Aは、合成HA遺伝子をコードするドナー・ベクターを示す。ドナー・ベクターは、大腸菌中で増幅するためのバクテリアのバックボーン(pUC_ori)と、カナマイシン(kanR)と、大腸菌内での淘汰のためのクロラムフェニコール(cmR)耐性遺伝子カセットおよびスクラーゼ遺伝子(sacB)と、誘導プロモーターによってコントロールされ、その後にテトラヒメナ好熱菌のβチューブリン2終了配列(btu2)が続く、標的遺伝子のオープン・リーディング・フレーム(HA遺伝子)とを含む。
【0134】
図2Bは、テトラヒメナ好熱菌において用いられる発現ベクターを示す。このベクターは、アンピシリン(ampR)と、大腸菌内での淘汰のためのクロラムフェニコール(cmR)耐性遺伝子と、テトラヒメナ好熱菌内でのプラスミド増殖のためのテトラヒメナ好熱菌に特異的な複製起点(rDNA_ori)と、形質転換された繊毛虫類を識別するためのネオマイシンを用いた耐性遺伝子カセット(neoR)と、誘導プロモーターによってコントロールされ、その後にテトラヒメナ好熱菌のβチューブリン2終了配列(btu2)が続く、標的遺伝子のオープン・リーディング・フレーム(HA遺伝子)とを含む。
【0135】
図3Aは、血球凝集素を、そのサブユニットであるHA1(黒色)およびHA2(灰色)とともに示す。αヘリックスおよびβシートが示される。
【0136】
図3Bは、血球凝集素の概要を、そのサブユニットであるHA1(黒色)およびHA2とともに示した概略図である。融合ペプチド(FP、上記の記載を参照。)と、タンパク質をウイルスエンペロープ膜に固定する膜貫通ドメイン(TMD)とが記載されている。HA1およびHA2は、ジスルフィド架橋(DSB)によって結合されている。HA2において2つのαヘリックス(α−H1およびα−H2)と連結する部分は、「ループドメイン(LD)」と呼ばれる。HA2のC末端細胞質尾部(C-terminal cytoplasmatic tail)は図示されていない。
【0137】
HA1は、図示されていないジスルフィド架橋によって安定化された球状ドメイン(「ヘッド部」)を有する。このヘッド部は、宿主細胞膜のグリコカリックスにあるシアル酸との結合サイトを含む。同様に、ヘッド部は、強い免疫原性を有し、そのため、免疫付与後にHAに対して作製された多くの抗原の標的となる。
【0138】
ウイルスエンベロープ二重層の宿主細胞膜との融合を引き起こすべく、pHの低下をトリガーとして、HA1およびHA2は、お互いに切り離される。その結果、HAホモ三量体中において、HA1サブユニットは正電荷を有する。
【0139】
これにより反発力が発生して、HA1サブユニットがお互いから離れて、それにより、HA2サブユニットとの連結が解ける。その結果、HA2サブユニットが活性化される。活性化されたHA2は、膜の融合の主な原因となる。HA2は、1度だけ活性化されることができ、その後は不活性なままである。そのため、HA1はあまりにも早く連結を解放しないことが重要である。そうしなければ、ウイルスは、その感染力を喪失してしまうからである。
【0140】
HA2は、大きく引き伸ばせば(over large stretches)、αらせん構造を有し、大きなループ領域を含む。加えて、HA2は、膜貫通ドメインおよび融合ペプチドを含む。融合ペプチドは、HA1サブユニットの分離によって、解放される。
【0141】
融合を解除する目的で、HA1の球状ドメイン(「ヘッド部」)がHA2から分離せざるを得ない。そのため、HA2は、HA2が展開して、融合ペプチドが宿主細胞膜を貫通するように、その構造を変化させることができる。このようにして、ウイルスが、宿主細胞膜に直接結合する。
【0142】
展開プロセスによって、HA2は、一方の側に引き伸ばされる。他の領域は、タンパク質の正味の大きさが増大しないように、巻き込まれる。その後、HAは、その構造を変化させて、膜融合が生じるように、ウイルスを宿主細胞膜の近くに持っていく。
【0143】
図4Aは、血球凝集素を例として、本文中で定義されたクラスIのウイルス融合タンパク質の一般的な構造を示す。HA1およびHA2は、インフルエンザHA0クラスIタンパク質のサブユニットである。ラベルは、シグナル・ペプチド(SP)、融合ペプチド(FP)、加工部位(矢印)、膜貫通ドメイン(TMD)の位置を特定する。これらは、一般的なクラスIウイルス融合ペプチドのサブユニットである。
【0144】
図4B(配列ID番号3も参照。)では、インフルエンザAウイルス株、A/ニューカレドニア/20/1999 H1N1(ジェンバンク信託番号:AAP34324.1)、の血球凝集素のサブユニットが例示的に説明される。アンダーラインを引いた一節は、シグナル・ペプチド(SP)、HA2融合ペプチド(FP)およびHA2膜貫通ドメイン(TMD)を示す。
【0145】
図示されているタンパク質は、565個のアミノ酸残基(「AA」)からなる。特定配列の特徴を、表4に示す。ここで、HA2融合ペプチドおよびHA2膜貫通ドメインでは、疎水性アミノ酸の占める割合が大きく(表1のヒドロパシーのデータに基づいて計算されたデータ)、例えば、融合ペプチドは、8個のグリシン残基を有することに留意すべきである。本発明者らは、繊毛虫類における異種タンパク質発現を目的として、本発明によって提供されるように、これらのドメインを切断することで、タンパク質の分泌および/または細胞溶解後の精製を促進することができることを示した。
【表4】
【0146】
表4は、HA2膜貫通ドメインおよびHA2融合ペプチドにおいて、疎水性アミノ酸の占める割合が非常に大きいことを明確に示す。本発明者らは、この発見から、これらの2つのドメインの少なくとも1つを切断することで、媒体または培地(medium)中へのタンパク質の分泌が促進されるであろうと推測した。
【0147】
図4Aおよび図4Bのタンパク質に関連して説明された特徴は、他の血球凝集素にも、表2に示されたクラスIウイルス融合タンパク質の多くにも適用することができることに注目すべきである。
【0148】
図5は、テトラヒメナ好熱菌と、ホモ・サピエンスとの間におけるコドン出現頻度の比較を示す。ホモ・サピエンスのコドン出現頻度は、ヒト宿主細胞から発現させられた血球凝集素と同様に、ヒト宿主において発現させられるウイルスタンパク質に適用することができる。さらなる説明については、本文を参照されたい。
【0149】
図6は、繊毛虫類(特に、テトラヒメナ)において用いられている遺伝子コードを示す。標準的でないヌクレオチド・コード(non-canonical nucleotide code)であり、グルタミンをコードするUAAおよびUAGは、太字で印刷されている。しかしながら、一般的な遺伝子コードによれば、これらのトリプレットは、終止コドン(削除されたトリプレットを参照。)である。「1LC」は、「1文字コード(one letter code)」を表し、「3LC」は、「3文字コード(three letter code)」を表す。
【0150】
図7は、一例として、ウイルス血球凝集素配列(配列ID番号1を参照。)と、テトラヒメナ好熱菌における発現に関してコドン最適化された血球凝集素の配列(配列ID番号2を参照。)との間におけるコドン出現頻度の比較を示す。コドン出現頻度における相違点は、灰色の四角で示される。
【0151】
図8は、供給回分式の発酵プロセス(0.5L)を用いて培養され、組み換えHAを発現している形質転換された繊毛虫類の免疫ブロットを示す。集菌時期の異なる上清のサンプル(SN1は47.5時間、SN2は66時間、SN3は70時間、SN4は90時間)に対して、非還元条件下でSDS−PAGEが適用され、その後、ウエスタンブロット分析が適用された。染色には、特定のインフルエンザ・アンチ−B/フロリダ/4/2006血清(Influenza anti-B/Florida/4/2006 serum)を用いた。適用されたNIBSC−HA抗原(陽性対照)は、約90kDa(A、NIBSC、45ng)に相当するシグナルを示した。47.5時間の培養の後、上清に関して、約72kDaおよび約90kDaにおいて、HA特異沈降線(HA specific band)が見られた。このタンパク質は、配列ID番号9に対応する。
【0152】
図9は、テトラヒメナ中で遺伝子組み換え技術によって発現させられた、長さの異なる(本発明の内容に従って切断された)血球凝集素タンパク質フラグメントの免疫ブロットを示す。染色には、特定のギニア・ブタ−アンチ−H1N1抗体(Guinea Pig-anti-H1N1 antibody)を用いた。Pは、テトラヒメナ細胞ペレットであり、SNは、細胞培養上清である。レーン1および2は、テトラヒメナのクローンを表し、レーン3および4は、野生型の対照群を表す。
【0153】
図9のAは、「HAロング」と呼ばれるタンパク質を示す。HAロングは、HA2の細胞内ドメイン(CT、図4Aを参照。)を欠いた、HA1−HA2の完全な二量体(full HA1-HA2-dimer)である(予測分子量は61.4kDaである。)。このタンパク質は、配列ID番号3に対応する。
【0154】
図9のBは、「HAショート」と呼ばれるタンパク質を示す。HAショートは、HA2の細胞内ドメイン(CT)および疎水性の膜貫通ドメイン(TMD)を欠いた、HA1−HA2二量体である(予測分子量は57.6kDaである。)。このタンパク質は、配列ID番号5に対応する。
【0155】
図9のCは、単なるHA1(「HA_1」と称され、予測分子量は34.2kDaである。)を示す。すなわち、少なくとも2つの疎水性ドメイン(融合ペプチドおよび膜貫通ドメイン)を有するHA2が切断されている。このタンパク質は、配列ID番号7に対応する。
【0156】
免疫ブロットから、HAショートおよびHA_1(これらは、少なくとも1つの疎水性ドメインを欠く。)は、上清中に、HAロングよりも豊富に含まれることが明らかである。このことは、ウイルス融合タンパク質の繊毛虫類におけるタンパク質分泌は、少なくとも1つの疎水性ドメインを除去することにより促進されるという結論をサポートする。細胞膜への付着しやすさが低減するからである。さらに、ウイルス融合タンパク質(特に、血球凝集素)の免疫原性ドメインは、HA1サブユニット中に配されるので、HA1サブユニットのみを含むワクチンまたはHA1サブユニットと切断されたHA2サブユニットとを含むワクチンが、依然として免疫応答を引き起こすことが予想される。したがって、ワクチンの生産にとって有用であることが予想される。
【0157】
図10Aは、本発明に係る配列ID番号3(すなわち、「HAロング」)に関連するタンパク質の疎水性プロットを示す。疎水性プロットは、カイト−ドリトルの指標(Kyte-Doolittle scale)に従って作図されている。融合ペプチド(AA344−368)および膜貫通ドメイン(AA530−550)が、タンパク質の主要な疎水性領域の間に配されており(矢印を参照。)、それらが、タンパク質の媒体または培地(medium)への効率的な分泌を妨げていることを、容易に判定することができる。
【0158】
図10Bは、分泌された主要な繊毛虫類タンパク質(a ciliate major secreted protein)の疎水性プロット(本発明の発明者らによる国際公開第03/078566号の配列ID番号2)を示す。このタンパク質は、自然な環境においてテトラヒメナによって大量に分泌される。このタンパク質は、強い疎水性ドメインを有しておらず、平均して、HAロングよりも疎水性が小さいことが明白である。本発明者らは、この現象を、HAロングとは対照的に、このタンパク質の分泌の容易化に関与させた。
【0159】
(配列表)
別表において、完全な配列表が添付される。配列表には、以下の配列を示す(NAは核酸であり、AAはアミノ酸である。)。
【表5】
【0160】
(参照文献)
Cassidy-Hanley, D.、Bowen, J.、Lee, J.、Cole, E.、VerPlank, L.、Gaertig, J.、Gorovsky, M.およびBruns, P.(1997年);粒子衝撃によるテトラヒメナ好熱菌を交配する生殖細胞系列および体細胞形質転換(Germline and somatic transformation of mating Tetrahymena thermophila by particle bombardment);Genetics 146巻:135−47頁。
Gaertig, J.、Gao, Y.、Tishgarten, T.、Clark, T.およびDickerson, H.(1999年);テトラヒメナ好熱菌におけるパラサイト抗原の表面提示(Surface display of a parasite antigen in the ciliate Tetrahymena thermophila);Nat Biotechnol 17巻:462−5頁。
Kulakosky, P.、Hughes, P.およびWood, H.(1998年);昆虫の幼虫および組織培養細胞におけるバキュロ・ウイルス発現による組み換え糖タンパク質のN結合型グリコシル化(N-Linked glycosylation of a baculovirus-expressed recombinant glycoprotein in insect larvae and tissue culture cells);Glycobiology 8巻:741−5頁。
Reading, P.、Miller, J.およびAnders, E.(2000);インフルエンザウイルスによるマクロファージの感染中におけるマンノース受容体の取り込み(Involvement of the mannose receptor in infection of macrophages by influenza virus);J Virol 74巻:5190−7頁。
Tomiya, N.、Narang, S.、Lee, Y.C.およびBetenbaugh, M.J.(2004年);哺乳類細胞株における、自然のおよび改変された鱗翅目の昆虫細胞株のN−グリカン・プロセッシング(Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines);Glycoconj J 21巻:343−60頁。
Kyte, J.およびDoolittle RF.(1982年);タンパク質の疎水性特性を表示するシンプルな方法(A simple method for displaying the hydropathic character of a protein);J Mol Biol 157巻:105頁。
【技術分野】
【0001】
本発明は、繊毛虫類の宿主細胞中におけるウイルスタンパク質の異種発現系に関連する。
【背景技術】
【0002】
多くのウイルスは、ヒト病原体または動物病原体として重要な役割を果たしているので、ウイルス病原体に対するワクチンとして、ウイルスタンパク質を使用することが注目されている。ウイルス(特に、RNAウイルス)は、非常に多様性があり、ウイルス感染は、様々な血清型を有するウイルス(例えば、インフルエンザウイルス、FMDウイルス)に起因する。結果として、既存のウイルス性ワクチンの多くは、しばしば、現場で流行している菌株に対処することができない。また、新しいウイルス性ワクチンは、新たな感染が発生したときに、現場の菌株から製造する必要がある。
【0003】
現代のバイオテクノロジー技術は、ほとんど全ての考えられるタンパク質の異種発現を可能にしているが、ウイルスタンパク質の発現は、あまり見られないのが現状である。多くの場合、ウイルスタンパク質は構造タンパク質(すなわち、酵素でもなく、タンパク質ホルモンでもなく、抗体でもない。)であり、製薬目的または工業目的で大して関心をひかないからである。
【0004】
しかしながら、多くのウイルスは、ヒト病原体または動物病原体として、重要な役割を果たしているので、ウイルス病原体に対するワクチンとして、ウイルスタンパク質を使用することが注目されている。
【0005】
近年、ウイルス病原体に対するワクチンを製造する標準手順は、特定の系において、それぞれのウイルスを培養し、そのようにして得られたウイルス粒子を収集および不活性化し、不活性化された粒子をワクチンとして含む医薬組成物を製造する。
【0006】
上記原理は、例えば、インフルエンザワクチンの製造において実施される。インフルエンザワクチンは、受精鶏卵中で製造されている。受精11日後、それぞれの卵のアルブミン中に、インフルエンザウイルス株が注入され、発生中の胚の肺を感染させる。数日間培養された後、ウイルスが集菌および精製され、化学的に不活性化され、ワクチンの製造に用いられる。1ダースのワクチンの製造には、平均して、約1〜2個の卵が必要になる。全製造工程は、少なくとも6ヶ月続く。
【発明の概要】
【0007】
このワクチン製造方法は、確立されており、コスト効率がよい。しかし、このワクチン製造方法には、数百万個もの卵の調達、長いタイムライン、卵のハンドリングの退屈さ、需要の急増または新しいウイルス株の突然の出現に対する適応性のなさといったデメリットがある。さらに、受精鶏卵は、いくつかの病原性ウイルス、中でも鳥インフルエンザA型ウイルス(H5N1)に対するワクチンの製造に使用することができない。
【0008】
受精鶏卵は、少なくとも、ホストとしてニワトリ胚と相性のよい全てのウイルスの生産のために、または、前者の成分(element)を含むワクチン生産のために、主に使用することができる。
【0009】
ウイルスまたは前者の成分を含むワクチンを生産する他の方法は、細胞培養または組織培養に基づく。ある特定のアプローチよれば、哺乳類の腎臓細胞中に、ウイルスが注入される。ウイルスの増殖後、細胞を溶解させて、ウイルス粒子を集菌し、精製し、不活性化させる。しかしながら、これらの系では、複合増殖培地を使用する必要があり、それらの取り扱いは、面倒で、時間がかかる。さらに、このような感染アプローチは、実施が難しく、再現性が十分でない。
【0010】
国際公開第03/048348号に開示されている別のアプローチによれば、ヒト細胞株PER.C6(登録商標)に関する不活性化されたウイルスワクチンの製造方法が開示されている。その表面にレセプタを有するSia2-6GalおよびSia2-3Galが存在するので、この細胞株は、様々なウイルス(例えば、インフルエンザウイルス、パラインフルエンザウイルス、アデノ随伴ウイルスまたはポリオーマウイルス・タイプ(poliomavirus type))に感染しやすい。それにより、生産プロセスに関する安全性の問題を引き起こす。
【0011】
一方、哺乳類の細胞培養を利用した系(mammalian cell culture based system)は、上述の卵を利用した系(egg based system)とは対照的に、急速に規模を拡大することができ、緊急時にスケールアップすることができる。一方、そのような製造設備(巨大なバイオリアクターを有する。)の操作上の即応性に対して事前に必要な費用(up-front cost)は、卵を利用した系に関する費用と比較して、はるかに高額であり、収率は、わずかに劣る。さらに、これらの系は、複合増殖培地を使用する必要があり、その取り扱いは、面倒で、時間がかかる。
【0012】
それゆえ、ワクチンとして用いることができるウイルスタンパク質を生産する代替手法を開発することは、最優先の課題である。
【0013】
ある特定のアプローチによれば、組み換えDNA技術を用いて、これらのタンパク質を生産する。明らかな利点の1つは、大きく改善されたワクチンの安全性である。卵を用いたワクチンの生産方法と比較して、組換え発現のウイルスタンパク質を精製するため条件が有利だからである。さらに、適応性が大きく向上して、様々な季節性の亜型ウイルスに適応することができる。
【0014】
しかしながら、ウイルスタンパク質は、多くの場合、構造タンパク質であるので、製薬目的および工業目的での生産は、複雑で面倒である。当初、大腸菌中で、ウイルスタンパク質に対応するポリペプチド(例えば、血球凝集素)を発現させることが試みられた。しかしながら、後者は、翻訳後修飾装置(posttranslational modification apparatus)を有しないので、ワクチンとして使用されうるタンパク質の原核生物中における異種発現が意味をなさないことは、注目に値する。それゆえ、原核生物(例えば、大腸菌)中において発現させられたタンパク質は、翻訳後修飾(例えば、糖鎖付加パターン)が十分ではなく、安定性に影響を与え、抗原の免疫原性に大きく貢献するように思われる(Nayakら、1984年)。
【0015】
真核生物のサッカロマイセス・セレヴィシエ(血球凝集素に関する米国特許第4,752,473号明細書)中におけるウイルスタンパク質の発現は、高分子量の外鎖マンナン(outer chain mannan)の広範囲に及ぶ付加を伴う組み換えタンパク質のハイパーグリコシル化によって問題を引き起こしたが(JabbarおよびNayak、1987年。Jabbarら、1985年)、十分には立証されていない。
【0016】
米国特許第5,858,368号明細書は、組み換えバキュロウイルスに感染した昆虫細胞株中において、インフルエンザA型ウイルスの血球凝集素(HA)を発現させる方法を開示している。タンパク質は、非変性、非イオン洗剤を用いて感染細胞の表在性膜から抽出された後、クロマトグラフにより精製される。しかしながら、精製プロセスは、必要以上に時間がかかる(rather time-consuming)。さらに、バキュロウイルス/昆虫細胞発現系において生産されたタンパク質が、それらの糖鎖付加パターンのおかげで、哺乳類の治療用途に適しているのかどうかは、疑わしい(Kulakoskyら、1998年)。また、昆虫細胞中で発現させられたタンパク質のN−グリカン中の特定の残基(certain residue)が、アレルギー性のエピトープである可能性を示すヒントもある(Tomiyaら、2004年)。
【0017】
(定義)
本明細書において、「異種発現(heterologous expression)」という用語は、その中で発現が生じている有機体(organism)にとって異質である遺伝子、核酸またはcDNAのタンパク質の発現を示す。
【0018】
本明細書において、「繊毛虫類(ciliate)」という用語は、繊毛虫(Ciliophora)の系統だった門(scientific phylum)を示す。繊毛虫は、単細胞の真核生物(原虫(protozoa)または原生生物(protist))であり、特に、その比較的大きなサイズ(種によっては、その長さが2mmに達する。)、その繊毛細胞表面、2つの異なる種類の核(すなわち、小さな2倍体の小核と、大きな倍数体の大核(転写活性)とである。)によって特徴付けられる。後者は、ゲノムの増幅およびヘビー・エディティング(heavy editing)によって、小核から生成される。
【0019】
本明細書において、「cDNA」という用語は、発現させられるべきタンパク質をコードするDNA分子であって、エンコードしていない部分(non-encoding part)(例えば、イントロン)を有していないDNA分子を示す。多くの場合、cDNAは、逆転写酵素およびオリゴdTプライマー(oligo dT-primer)を用いて、RNAテンプレートから直接合成される。しかしながら、当該用語には、他の方法で得られた合成遺伝子およびエンコーディングDNA(encoding DNA)も同様に含まれる。
【0020】
本明細書において、「プロモーター遺伝子(promoter)」という用語は、DNAの調節領域を示す。当該領域は、通常、遺伝子(gene)またはcDNAの上流(センス鎖の5'領域に向かって)に配され、当該遺伝子または当該cDNAの転写を可能にするまたは増進する本質的な遺伝因子(essential genetic element)を含む。
【0021】
本明細書において、「フラグメント(fragment)」という用語は、ある部分もしくはドメインが欠落しており、酵素活性、免疫原性、標的結合もしくはそれらに類似する特性に関して、いくらかの活性を維持している天然タンパク質もしくは野生型タンパク質の一部、または、それらに類似するものを示す。
【0022】
本明細書において、「シグナル配列(signal sequence)」という用語は、オリゴペプチド(「シグナル・ペプチド」または「輸送ペプチド」)をコードする核酸配列を示す。オリゴペプチドは、タンパク質の特定の細胞小器官(例えば、核、糸粒体基質、小胞体、葉緑体、アポプラスト、ペルオキシソーム)の輸送を指示する。小胞体(endoplasmatic reticulum)へと輸送されたほとんど全てのタンパク質は、N末端に、5−10個の疎水性アミノ酸を含む配列を有する。これらのシグナル・ペプチドは、ERの内腔へのタンパク質の翻訳時挿入(cotranslational insertion)の後、シグナル・ぺプチダーゼによって、たんぱく質から分割されるその後、多くのタンパク質は、ゴルジ体(golgi apparatus)を介して、分泌経路の下流へと輸送される。
【0023】
本明細書において、「機能しうるように連結される(operably linked)」という用語は、プロモーター遺伝子が適切な条件下で遺伝子産物の発現を制御するように、遺伝子産物をコードし得るヌクレオチド配列が、当該プロモーター遺伝子に連結されることを意味する。
【0024】
本明細書において、「疎水性ドメイン(hydrophobic domain)」という用語は、「親油性ドメイン(lipophilic domain)」および/または「非極性ドメイン(non-polar domain)」という用語と同意語として用いられる用語である。本明細書において、これらの用語は、少なくともその末端において、親油性/非極性のアミノ酸残基の存在によって主に制御される疎水性(「ヒドロパシー(hydropathy)」と呼ばれる場合もある。)を有し、そのため、例えば、細胞膜中に見られるリン脂質二重層のように、脂質画分に対してある程度の親和性を有するタンパク質ドメインを示す。
【0025】
上記の疎水性ドメインは、例えば、いわゆる「膜貫通ドメインtransmembrane domain)」(すなわち、膜の中で熱力学的に安定したタンパク質の3次元構造)とみなすこともできる。このような膜貫通ドメインには、単一のαへリックス(single alpha helix)、いくつかの膜貫通αへリックスの安定な複合体(complex)、膜貫通βバレル、βヘリックス、潜在的に(potentially)疎水性アミノ酸の残基を取り囲む疎水性アミノ酸の外側の環(outer ring)、および、それらに類似するものが含まれてよい。
【0026】
本発明者らは、疎水性ドメインが、タンパク質の環境中への分泌を妨げることを見出した。表1に、疎水性タンパク質の一例を示す。
【表1】
【0027】
本明細書において、「核酸分子(nucleic acid molecule)」という用語は、一本鎖核酸分子または二本鎖核酸分子を示すことを意図しており、例えば、DNA(cDNAおよび/もしくはゲノムDNA)、RNA(好ましくは、mRNA)、PNA、LNA、ならびに/または、モルホリノが含まれる。
【0028】
本明細書において、「ストリンジェントな条件(stringent conditions)」という用語は、その条件の下において、プローブが、他の配列よりも、当該プローブの標的部分配列(target subsequence)と分子交雑(hybridize)しやすい条件に関連する。ストリンジェントな条件は、配列依存性(sequence-dependent)であり、環境が異なれば当該条件も異なるであろう。特に、配列が長いほど、高い温度で分子交雑する。一般的に、ストリンジェントな条件は、一定の(defined)イオン強度およびpHにおける特定配列の融点(thermal melting point)(Tm)よりも約5℃低くなるように選択される。Tmは、その温度において、平衡状態で、標的配列に相補的なプローブの50%が標的配列に分子交雑する(一定のイオン強度、pHおよび核酸濃度の下での)温度である(一般に、標的配列は過剰に存在するので、Tmにおいて、平衡状態では、プローブの50%は占有される。)。通常、ストリンジェントな条件における塩濃度は、Naイオン濃度が約1.0mol/L(Mと表記する場合がある。)より小さく、典型的には、pHが7.0から8.3であり、Naイオン濃度(またはその他の塩)濃度が約0.01から1.0Mである。また、ストリンジェントな条件における温度は、短いプローブ(例えば、10から50個のヌクレオチドを含む。)の場合は少なくとも約30℃であり、それより長いプローブの場合には少なくとも約60℃である。ストリンジェントな条件は、不安定化剤(destabilizing agent)(例えば、ホルムアミドおよびそれに類似するもの)を添加することで調整されてもよい。
【0029】
本明細書において、「PDBコード(PDB code)」という用語は、特有の4種類の文字表記(unique four letter code)に関連する。その文字表記は、基本識別子としての役割を果たし、それによって、生体高分子の構造データの世界的なレポジトリであるタンパク質構造データベース(Protein Data Bank)を用いてエントリーを検索することができる。
【0030】
本明細書において、「核酸分子のフラグメント(fragment of the nucleic acid molecule)」という用語は、特許請求の範囲に記載された配列の1つに従う核酸分子のサブセットを含む核酸を示すことを意図している。
【0031】
「核酸分子のフラクション(fraction of the nucleic acid molecule)」という用語についても同様である。
【0032】
本明細書において、「核酸分子の変異体(variant of the nucleic acid molecule)」という用語は、構造および特許請求の範囲に記載された配列の1つに従う核酸分子に対する生物活性において、実質的に同様である核酸分子を示す。
【0033】
本明細書において、「核酸分子のホモログ(homologue of the nucleic acid molecule)」という用語は、特許請求の範囲に記載された配列の1つに従う、核酸分子と比較して、その配列が、付加、削除、置換またはその他の化学修飾を施された1以上のヌクレオチドを有する核酸分子に関連し、このホモログは、特許請求の範囲に記載された配列の1つに従う核酸分子と実質的に同一の特性を保持することが常に要求される。
【0034】
本明細書において、「コドン最適化された(codon optimized)」という用語は、発現させられるべき異種タンパク質をコードするcDNAを、普遍的遺伝子コード表(universal genetic code scheme)から派生した宿主特異的な(host specific)コドン使用頻度(codon usage)に適応させるプロセスを示す。繊毛虫類は、AとTに富むゲノム(AT-rich genome)を有し、テトラヒメナのDNAにおいては、その約75%がAおよびTからなる。ここでのコドン使用頻度は、他の有機体と、特に、特定のアミノ酸をコードする目的におけるコドンの使用頻度(「コドン・バイアス(codon bias)」)において異なる。異種タンパク質をコードするcDNAが最適化されていない場合、当該cDNAが、繊毛虫においてほとんど使用されないコドンを使用すると、タンパク質の発現効率に大きく影響する可能性がある。このことは、一方で、研究中の遺伝子のコドン使用頻度(codon frequency)が、繊毛虫類の発現系のコドン使用頻度に適合した場合には、異種タンパク質の発現が劇的に改善される可能性があることを意味する。さらに、多くの繊毛虫類、中でもテトラヒメナは、グルタミンをコードするUAAトリプレットおよびUAGトリプレットを有する標準的でないヌクレオチド・コードを利用する。しかし、他の多くの有機体において、これらのコドンは、翻訳を終了させる終止コドンとして利用される。このことから、UAAトリプレットおよびUAGトリプレットを終止コドンとして有する外来遺伝子(繊毛虫類の遺伝子ではない遺伝子)は、正確に発現しないということを導き出すことができるかもしれない。この目的のために、繊毛虫類の宿主細胞を形質転換する前に、UAAトリプレットおよびUAGトリプレットをUAAに修正するような方法で、異種タンパク質をコードするcDNAのコード最適化が実施されるべきである。コード最適化(code optimization)は、例えば、部位特異的突然変異誘発法(site directed mutagenesis)またはcDNAデノボ合成(de novo cDNA synthesis)によって実施することができる。
【0035】
本明細書において、「誘導体(derivative)」という用語は、特許請求の範囲に記載された配列の1つに従う核酸分子のように、標的核酸配列と似た特性を有する、関連した核酸分子(related nucleic acid molecule)を示す。
【0036】
本明細書において、「少なくともX%の配列同一性(sequence identity of at least X %)」という用語は、BLASTアルゴリズムの系統(family)のアルゴリズム(特に、メガブラスト、不連続メガブラスト、blastn、blastp、PSI−BLAST、PHI−BLAST、blastx、blastn、または、tblastx)を用いて配列アラインメントを実施した後に決定される配列同一性を示す。これらのアルゴリズムは、例えば、NCBIによって提供される、それぞれのインターネット・ドメインからアクセスすることができる。
【0037】
本明細書において、「宿主細胞(host cell)」という用語は、2つの異なる意味を有する。これらは、それぞれの文脈から区別することができる。異種タンパク質発現の文脈において、「宿主細胞」という用語は、発現宿主として用いられる遺伝子組み換え細胞(transgenic cell)を示す。このように、この細胞またはその前駆細胞(progenitor)には、発現させられるべきタンパク質のcDNAを含む適切なベクターを用いて、核酸が導入されている。ウイルスのライフサイクルの文脈において、「宿主細胞」という用語は、ウイルスに感染された細胞を示す。ウイルスは、その細胞を、複製のために利用する。
【0038】
本明細書において、「ベクター(vector)」という用語は、外来遺伝子を別の細胞に導入させるために用いられる分子ベヒクル(molecular vehicle)を示す。一般的に、ベクター自身は、インサート(目的の配列)と、ベクターの「バックボーン(backbone)」の役割を果たす、より大きな配列とを含むDNA配列である。遺伝情報を別の細胞に導入するベクターの目的は、通常、標的細胞中のインサートの単離、複製または発現である。
【0039】
本明細書において、「プラスミド(plasmid)」という用語は、プラスミド・ベクターを示す。すなわち、複製起点(ORI)に起因して、適切な宿主の内部で自己複製することができる環状DNAを示す。さらに、上記のマーカーは、形質転換(transformation)の成功、または、外来DNAを細胞もしくは多重クローニング部位に導入するように意図されているその他のプロシージャの成功を示唆する目的で、プラスミドが、選択マーカー(selectable marker)を含んでもよい。クローニング・ベクターまたはドナー・ベクターと呼ばれるプラスミド・ベクターは、クローニングを容易にして、目的の配列を増殖する目的で用いられる。発現ベクターまたはアクセプター・ベクターと呼ばれるプラスミド・ベクターは、特に、一定の(defined)標的細胞における目的の遺伝子の発現に用いられる。これらのプラスミド・ベクターは、一般に、プロモーター、導入遺伝子および終了配列を含む発現カセットを与える。発現プラスミドは、異なる宿主細胞における増殖(propagation)および淘汰を可能にする要素を含むシャトル・プラスミドであってもよい。
【0040】
本明細書において、「ウイルスタンパク質(viral protein)」という用語は、ウイルスによって生産されたタンパク質を示す。このタンパク質は、ウイルスのエンベロープおよび/またはカプシドを形成してもよく、非構造性のタンパク質、調節タンパク質または付属タンパク質であってもよい。
【0041】
本明細書において、「表面タンパク質(surface protein)」という用語は、ウイルスの外層を形成するウイルスタンパク質を示す。このようなタンパク質としては、(i)エンベロープを有するウイルス(エンベロープウイルスと称する場合がある。)の脂質二重層コーティングに固定された(anchored)タンパク質、(ii)エンベロープウイルスおよびエンベロープを有しないウイルス中のカプシドタンパク質、および/または、(iii)多くのウイルス(例えば、バクテリオファージ、または、ヒトおよび鳥類を含む哺乳類を含むに感染するウイルス)中に見られるようなスパイクタンパク質を例示することができる。多くの場合、エンベロープウイルスの脂質二重層コーティングは、ウイルスの複製サイクル中(すなわち、開口分泌時または細胞溶解時)に、宿主細胞中の細胞内膜(例えば、核の内膜もしくはゴルジ膜)または宿主細胞の外膜から、ウイルスによって獲得される。
【0042】
本明細書において、「ウイルス融合タンパク質(viral fusion protein)」という用語は、エンベロープウイルスの糖タンパク質を示す。この糖タンパク質は、宿主細胞の感染(ウイルスの遺伝物質の宿主細胞中への挿入)を促進する。その構造の一体部分(integral part)として脂質二重層を有するエンベロープウイルスは、ウイルス膜と宿主細胞膜との融合により、当該ウイルスが感染した細胞の一員となる。ウイルス融合タンパク質は、2つの主要な特徴を兼ね備える。つまり、ウイルス融合タンパク質は、レセプタ結合機能(receptor binding function)を有する。レセプタ結合機能は、ウイルスを宿主細胞に付着させる機能である。また、ウイルス融合タンパク質は、融合機能(fusion function)を有する。融合機能は、ウイルス膜と宿主細胞膜との融合を仲介すべく、活性化されてよい。ウイルス融合タンパク質としては、後述するような、クラスIのウイルス融合タンパク質が、よく知られている。
【0043】
本明細書において、「融合ペプチド(fusion peptide)」という用語は、融合プロセスの間に宿主細胞膜の中に挿入されたウイルス融合タンパク質の明確に保存された疎水性領域(distinct conserved hydrophobic region)を示す。融合ペプチドは、どちらかといえば、無極性の領域であり、比較的、グリシン残基およびアラニン残基に富み、いくつかの大きな疎水性残基を含む。クラスIのウイルス融合タンパク質に関して、融合ペプチドは、タンパク質の膜貫領域のN末端に配される。
【0044】
(本発明の目的)
本発明の目的は、上述の問題点を打開する方法および/または実施態様を提供することにある。この目的は、独立項に記載された実施態様によって解決される。
【図面の簡単な説明】
【0045】
【図1A】様々な分類群のN−結合型グリコシル化パターン概要を示す。
【図1B】いくつかの種類の繊毛虫類におけるパターンの可能性のあるバリエーション(potential variation)を示す。
【図2A】合成HA遺伝子をコードするドナー・ベクターを示す。
【図2B】テトラヒメナ好熱菌において用いられる発現ベクターを示す。
【図3A】血球凝集素を、そのサブユニットであるHA1およびHA2とともに示す。
【図3B】血球凝集素の概要を、そのサブユニットであるHA1およびHA2とともに示した概略図である。
【図4A】クラスIのウイルス融合タンパク質の一般的な構造を示す。
【図4B】インフルエンザAウイルス株の血球凝集素のサブユニットが例示的に説明される。
【図5】テトラヒメナ好熱菌と、ホモ・サピエンスとの間におけるコドン出現頻度の比較を示す。
【図6】繊毛虫類において用いられている遺伝子コードを示す。
【図7】ウイルス血球凝集素配列、テトラヒメナ好熱菌における発現に関してコドン最適化された血球凝集素の配列との間におけるコドン出現頻度の比較を示す。
【図8】組み換えHAを発現している形質転換された繊毛虫類の免疫ブロットを示す。
【図9】血球凝集素タンパク質フラグメントの免疫ブロットを示す。
【図10A】HAロングに関連するタンパク質の疎水性プロットを示す。
【図10B】主要な繊毛虫類タンパク質の疎水性プロットを示す。
【発明を実施するための形態】
【0046】
本発明によれば、ウイルスタンパク質の異種タンパク質発現系が提供される。この発現系は、繊毛虫類の宿主細胞と、ウイルスタンパク質またはそのフラグメントをコードする少なくとも1つの核酸と、当該核酸に機能しうるように連結されたプロモーターとを備える。
【0047】
好ましい実施形態において、上記の核酸はcDNAである。
【0048】
多くの場合、ウイルスタンパク質は構造タンパク質(すなわち、酵素でもなく、タンパク質ホルモンでもなく、抗体でもない。)であり、製薬目的または工業目的で大して関心をひかないので、基本的には、現在、ウイルスタンパク質の異種タンパク質発現は一般的ではない。
【0049】
さらに、繊毛虫類におけるウイルスタンパク質の発現は、上述の参考文献において、いまだに示唆されていない。本発明者らは、繊毛虫類に関しては、バクテリアまたは後生動物に関するものとは異なり、特異的なウイルスがあまり多くは知られていないことに気がついた。このことは、繊毛虫類に共通する核の二形性に起因する可能性がある。他の原因としては、繊毛虫類の独特なコドン使用頻度と、AおよびTに富むゲノムとが考えられる。そこで、本発明者らは、高等生物の病原性ウイルスは、大部分の繊毛虫類(例えば、テトラヒメナ)の中では増殖することができないと仮定した。
【0050】
上記の議論は、今までのところ、繊毛虫類中でウイルスタンパク質を生産する試みが知られていないという事実の理由を説明する。
【0051】
今までに知られていたような、繊毛虫類は、ウイルスに感染しにくいという事実は、驚くべき利点をもたらす。このことは、繊毛虫類を用いた生産プロセスにおいて、付随的な(adventitious)ウイルスの増幅または増殖が起こらないことを意味する。このことは、さらに、タンパク質が治療上の使用目的で生産される場合、工業プロセスにおいて、必要に応じて、ヒト細胞培養または動物細胞培養を用いた高価なウイルス枯渇プロシージャ(virus depletion procedure)を省略することができることをも意味する。
【0052】
さらに、繊毛虫類中のウイルスタンパク質発現に適したプロモーターは、例えば、本発明の出願人に関連して登録された国際公開第2007/006812号に開示されており、その内容は、参照によって本願に組み込まれる。添付の配列表の配列ID番号12および13は、本発明との関連で特に好ましい、繊毛虫類に特異的な2つのプロモーターの配列を与える。これらは、すなわち、熱誘発性プロモーター(heat-inducible promoter)と、メタロチオネイン・プロモーター(metallothionein-promoter)である。
【0053】
Gaertigら(1999年、欧州特許出願公開第1151118号明細書を参照。)は、原生動物(テトラヒメナ好熱菌(Tetrahymena thermophila))を、他の原生動物(例えば、ウオノカイセンチュウ(Ichthyophthirius multifiliis))の表面抗原タンパク質(surface antigen protein)のタンパク質発現系として用いたことを開示している。彼らは、このようにして生産されたタンパク質は、形質転換されたテトラヒメナ細胞の表面上に提示されていたと報告している。また、そのタンパク質は採取され、ウオノカイセンチュウに対するワクチンの製造に用いられたと報告している。他のアイデアとしては、形質変換され、その表面に抗原を提示しているテトラヒメナ細胞を、生ワクチンとして直接使用することが考えられる。
【0054】
しかしながら、その著者らは、テトラヒメナは、AおよびTに富んだゲノムを有する有機体(例えば、マラリア原虫のような原生動物、マイコプラズマ、リシューマニアなどであり、これらは、同様にヒト病原体である。)からの遺伝子のクローニングおよび発現用としてのみ利用することができると提言している。従来の系(例えば、大腸菌)のATトラクトは、本来的に不安定だったので、これらの有機体から遺伝子をクローニングすることは難しいことが判明していた。例えば、テトラヒメナDNAは、ほぼ75%がATからなる。
【0055】
さらに、コドン使用頻度の問題によって、テトラヒメナ中で外来遺伝子を発現させることが妨げられていた。テトラヒメナ好熱菌(T. thermophila)は、グルタミン用に、UAAトリプレットおよびUAGトリプレットを利用する。しかし、他の多くの有機体において、これらのコドンは、翻訳を終了させる終止コドンとして利用される。このことから、UAAトリプレットおよびUAGトリプレットを終止コドンとして有する外来遺伝子(繊毛虫類の遺伝子ではない遺伝子)は、正確に発現しないということを導き出すことができるかもしれない。
【0056】
本発明の好ましい実施態様において、発現系は、さらに、上記の核酸に機能しうるように連結されたシグナル配列を含む。シグナル配列は、上記の核酸によってコードされたウイルスタンパク質またはそのフラグメントの細胞外培地(extracellular medium)への分泌(secretion)の原因となる。
【0057】
ウイルスタンパク質が自然に発現した場合(つまり、バクテリオファージに感染されたバクテリアの宿主細胞によって、または、ウイルスに感染された後生動物の宿主細胞によって出現した場合)には、ウイルスは、その宿主の機構および代謝作用を利用して、単に自分自身のタンパク質を生産するだけなので、ウイルスタンパク質は分泌の対象にはならない。なお、生産されたタンパク質は、その後、宿主細胞が溶解する前に、第2世代のウイルスへと組み立てられる。このことは、一方で、ウイルスタンパク質が、いまだ、分泌される能力を最適化しようとする進化的圧力にさらされていないことを意味する。
【0058】
これは、遺伝子組み換え技術による可溶性のウイルスタンパク質の生産と、その後の工程におけるこれらの可溶性のウイルスタンパク質の分泌とが複雑な問題であるという事実についての原因の1つである。例えば、適切なシグナル配列は、本発明の出願人に関連して登録された国際公開第03/078566号に開示されており、その内容は、参照によって本願に組み込まれる。
【0059】
添付の配列表の配列ID番号8および10は、本発明との関連で特に好ましい2つのシグナル・ペプチドの核酸配列を与える。これらは、すなわち、HA遺伝子の内因性のシグナル・ペプチドと、繊毛虫類のホスホリパーゼのA1シグナル・ペプチドである。
【0060】
本発明のさらに他の好ましい実施形態によれば、ウイルスタンパク質は、上述のウイルスの表面タンパク質である。
【0061】
宿主の感染に関して、宿主が免疫応答を許す場合(例えば、よく発達した免疫システムを有する哺乳類の場合)、ウイルスの表面タンパク質は、宿主と接触して、免疫応答を引き起こす場合がある。このように、これらのタンパク質は、単離された形態で、または、アジュバント(不活性化されたウイルスもしくはそのフラグメント)に関連して、ワクチンとして機能する可能性がある。
【0062】
本発明のさらに別の好ましい実施態様において、遺伝子組み換えの繊毛虫類は、テトラヒメナ科(family Tetrahymenidae)のメンバーであってよい。
【0063】
特に好ましい実施形態において、遺伝子組み換えの繊毛虫類は、テトラヒメナ(Tetrahymena sp)(特に、テトラヒメナ好熱菌)であってよい。テトラヒメナは、非病原性の単細胞真核微生物(nonpathogenic unicellular eukaryotic microorganism)である。テトラヒメナは、数箇所の研究所において、発現宿主として確立されている。テトラヒメナは、異種タンパク質発現に適した多くの利点を有することを特徴とする。テトラヒメナは、広く調べられたモデル生物であり、50年以上にわたる基礎研究において、ウイルスまたは内部寄生生物(endoparasite)が観測されていない。指標細胞系を用いた試験では、ウイルス、マイコプラズマなどの内因性の感染性病原体(endogenous infectious agen)が発見されていない。
【0064】
第一に、繊毛虫類におけるコドン使用頻度に関する上記の考察は、テトラヒメナにも同様に当てはまる。さらに、ハイコピープラスミド(high copy number plasmid)は、テトラヒメナに使用可能であり、ミニ染色体のrDNAからの複製起点(ORI)を含む。このミニ染色体のrDNAは、1つの細胞あたり最大9.000のコピー数の中に含まれる。その上、安定した統合は、大核DNA中で発生しうる。大核DNAにおいて、全ての遺伝子は、45個のコピー数の中に含まれる。高遺伝子量(high gene dose)は、効率的なタンパク質生合成および高い生産性の理想的な前提条件である。酵母菌およびバクテリアとは対照的に、テトラヒメナ属(genus Tetrahymena)の繊毛虫類は、発酵上澄み(fermentation supernatant)に、生物タンパク質(biologically protein)を効率よく分泌する。
【0065】
テトラヒメナは、タンパク質に翻訳後修飾(例えば、ジスルフィド架橋、GPIアンカー、リン酸化反応、アセチル化、グリコシル化)を施す(attach)ことができる。これらは、酵母菌またはその他の真核生物発現系において検出されるものよりも、哺乳類細胞中のものに似ている。
【0066】
哺乳類細胞とは異なり、テトラヒメナは、合成培地(chemically defined media)を使用することができ、増殖因子のように、ペプチドまたは血清成分を必要としないので、短い世代時間(1.5−3時間)による成長の容易さと、コスト削減とを兼ね備える。
【0067】
最大2×107細胞/mLの細胞密度と、最大80g/Lの乾燥質量の条件で、回分式、供給回文式および連続式のテトラヒメナの発酵が実施され、最大1000Lまでの生産規模の拡大(スケールアップ)が問題なく実施できることが実証された。レポーター・タンパク質を用いたフィージビリティ・スタディにおいて、1日当たり500−90pg/細胞の空時収量(space-time yield)が既に得られた。相同的発現を用いた最初の実験では、分泌されたタンパク質に関して、1日当たり200mg/L以上の収率(yield)が得られた。テトラヒメナは、従来型の微生物学的な(microbiological)発現系(バクテリアまたは酵母菌)用の生産設備中で発酵させることができる。このことは、既存の生産プラントの改修に費用をかけたり、生産設備を新たに建設したりする必要のないことを意味する。
【0068】
このような利点にも関わらず、繊毛虫類発現系(特に、テトラヒメナ)については、いまだにあまり知られていない。異種タンパク質発現系となる見込みのある系について尋ねられた当業者は、むしろ、大腸菌、酵母菌もしくはバキュロウイルスを用いた系、または、不死化哺乳類細胞株に想到するであろう。
【0069】
繊毛虫類発現系(特に、テトラヒメナ)の利用には、上述の状況を背景としても予測することができない別の大きな利点がある。哺乳類の免疫系の自己識別/非自己識別は、糖タンパク質の糖質組成(carbohydrate composition)に基づいて実現されるので、抗原のグリコシル化パターンは、かなりの程度、その免疫原性に寄与する。
【0070】
Readingら(2000年)は、哺乳類細胞において、マンノース受容体が、インフルエンザウイルス(およびマクロファージ中の他のエンベロープウイルス)の感染性侵入における主要なエンドサイトーシス受容体として機能することを報告している。
【0071】
マンノース受容体は、マンノース、フコースまたはN−アセチルグルコサミン(acytylglucosamin)および、保存されている(conserved)糖認識ドメイン(CRD)を含むC型レクチン中で終了する(terminate)糖タンパク質の取り込みを仲介する、膜結合型のレクチンタンパク質(「マンノース結合レクチン」、「MBL」とも呼ばれる。)として機能する能力がある。
【0072】
マンノース結合レクチン(MBL)のCRDは、ヘキソース(例えば、ピラノース環のC3およびC4の位置に、エクアトリアル配位の(equatorial)ヒドロキシル基を有するマンノースおよびN−アセチルグルコサミン)と結合する。それらは、哺乳類の糖タンパク質に一般的に見られるオリゴ糖と親和性がない。
【0073】
マクロファージは、哺乳類の免疫系において重要な役割を果たす。マクロファージは、エンドサイトーシスによって病原体を取り上げて、それらを消化した後、病原体に関連する抗原を対応するヘルパーT細胞に提示する。抗原を細胞膜中に一体化し(integrate)、他の白血球細胞に対しては、当該マクロファージは、表面に抗原を有するけれども、病原体ではないことを示しながら、対応するヘルパーT細胞に対して、MHCクラスII分子に結合した抗原を提示することで、抗原の提示が実施される。
【0074】
Readingら(2000年)の研究から、本発明者らは、以下の通り推測した。つまり、ワクチンの免疫原性を強化する目的で、ワクチン中に含まれるタンパク質のグリコシル化パターン中のマンノース量を増加させることが有用なのではないかと推測した。しかしながら、タンパク質のグリコシル化は、真核生物にしかない翻訳後修飾装置(posttranslational modification apparatus)中で実行されるので、意図的に修飾することが難しい。
【0075】
しかしながら、真核生物分類群は、それらのグリコシル化スキームの相違点を示している。グリコシル化は、主に、真核生物中および古細菌中で起こり、バクテリア中では起こらない。一般的に、N−結合型グリコシル化(N-glycosylation)という用語は、アスパラギン(N)アミノ酸残基のグリコシル化を示す。オリゴ糖鎖は、オリゴサッカリルトランスフェラーゼ酵素によって、トリペプチド配列Asn−X−SerまたはAsn−X−Thrにおいて生じるようなアスパラギン残基に取り付けられる。ここで、Xは、任意のアミノ酸であってよい。なお、ProおよびAspは、まれにしかみられない。
【0076】
驚いたことに、本発明の発明者らは、繊毛虫類が独特のN−結合型グリコシル化パターンを有し、高度(high degree)なマンノース残基を有することを見出した(図1Aおよび図1Bを参照。)。本発明の発明者らは、さらに、このことから、哺乳類のワクチン接種に用いられるであろうタンパク質は、末端のマンノース残基から利益を得ている可能性があることを見出した。すなわち、マンノース受容体を介したマクロファージ中におけるこれらのタンパク質の取り込みが増加した結果、免疫応答が増加した可能性があることを見出した。
【0077】
これらのことから、本発明者らは、繊毛虫類(特に、テトラヒメナ)において発現させられたタンパク質は、高度なマンノース残基に起因した高度の免疫原性を有し、遺伝子組み換えの繊毛虫類は、哺乳類用のワクチン(特に、ウイルスワクチン)を生産するための有望な候補であることを導き出した。
【0078】
さらに、本発明者らは、マクロファージが、マンノースに富むグリコシル化パターンを有する病原体の食作用を増加させることを見出した。このことから、このようにして生産されたワクチンが対象となる哺乳類動物に投薬(administer)されると、マンノースに富むグリコシル化パターンに起因する繊毛虫類におけるワクチンの発現が免疫反応を増加させることが証明される。
【0079】
この発見は、上述のGaertigら(1999年)の研究では予測されていない。Gaertigらは、ただ、魚のワクチンとして使用する目的で、遺伝子組み換えのテトラヒメナ中で生産された繊毛虫類のタンパク質を使用することを記載しているだけである。しかしながら、上述したような、マクロファージにおけるマンノース受容体の効果は、現在の知識によれば、魚用ではなく、哺乳類にのみ適用することができる。このことから、魚のワクチンが、ワクチンのグリコシル化パターン中のマンノース量の増加によって、同様の効果をあげることができるとは、まったく予測不能であった。
【0080】
特に好ましい実施形態において、ウイルスの表面タンパク質は、ウイルス融合タンパク質(上述の定義を参照。)である。
【0081】
別の好ましい実施形態において、ウイルスの表面タンパク質は、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質からなる群から選ばれるウイルス融合タンパク質の少なくとも1つであってよい。
【0082】
これらのウイルス融合タンパク質は、構造的に全く異なるが、これらの融合サブユニットは、折りたたまれて、最終的にはヘアピン構造の三量体(trimer-of-hairpins)になる。ヘアピン構造の三量体において、3つのC末端領域は、中心のN末端三量体コア(central N-terminal trimeric core.)の外側を包む(pack)。
【0083】
上述のとおり、ウイルス融合タンパク質は、クラスI、クラスIIおよびクラスIIIのウイルス融合タンパク質に細分される。表2に、上述のとおり定義されたエンベロープウイルスに由来するウイルス融合タンパク質のいくつかの概要を示す。
【0084】
上記の表において、「−S−S−」は、ジスルフィド架橋を示す。「/」は、当該記号によって示されたサブユニットが、ジスルフィド結合以外の方法でお互いに関連していることを示す。「GPX」は、「糖タンパク質 X」を示し、「FX」は、「融合タンパク質 X」を示す。
【表2】
【0085】
多くの場合、クラスIのウイルス融合タンパク質の2以上のサブユニットは、ジスルフィド架橋またはその他の手段によって結合される。そのような二量体として、HA1−S−S−HA2、SU−S−S−TM、HA1−S−S−HA2、SU−S−S−TM、SU/TM、F2−S−S−F1、S1/S2、GP1−S−S−GP2、GP1/GP2/SSPを例示することができる。ここで、サブユニットの少なくとも1つは、疎水性ドメイン(例えば、HA1−S−S−HA2構造体におけるHA2サブユニット)によって特徴付けられる。
【0086】
好ましい実施形態において、タンパク質は、クラスIのウイルス融合タンパク質であってよい。図4Aに、クラスIのウイルス融合タンパク質の概要を示す。
【0087】
特に好ましいウイルスタンパク質は、血球凝集素(hemagglutinin)である。血球凝集素(HA)は、クラス1の膜融合タンパク質サブユニットを2つ含む(すなわち、HA2およびHA1)、抗原性の糖タンパク質(antigenic glycoprotein)である。血球凝集素は、多くの他のバクテリアおよびウイルスと同様、インフルエンザウイルス(オルソミクソウイルス科)の表面に見られる。
【0088】
血球凝集素は、ウイルスと、当該ウイルスに感染されようとしている細胞との結合に関与する。今日では、少なくとも16種類の異なるHA亜類型(subtype)が知られており、H1−H16に分類される。最初の3つの血球凝集素(すなわち、H1、H2およびH3)は、ヒト・インフルエンザウイルス中に見出された。他方で、例えば、鳥インフルエンザウイルスは、H5血球凝集素を有する。
【0089】
HAの主な機能には、標的となる脊椎動物細胞の認識がある。この認識は、標的細胞膜上のシアル酸を含む受容体を介して達成される。HAの主な機能には、標的細胞膜とウイルス細胞膜との融合を引き起こすことで、標的細胞へのウイルスゲノムの侵入がある。この文脈において、オルソミクソウイルス科が、ヌクレオカプシドを包み込むリン脂質膜を有し、当該リン脂質膜が、宿主細胞から放出されて獲得されたものであることは、言及する価値がある。
【0090】
HAは、ホモ三量体の、内在性膜糖タンパク質(homotrimeric integral membrane glycoprotein)である。その形状は略円筒形状であり、その長さはおよそ135Åである。HAを構成する3つの同一の(identical)モノマーが組み立てられて、中心のαヘリックス・コイル(central α helix coil)と、サイトに結合するシアル酸を含む3つの球状のヘッド部とを形成する。HAモノマーは、前駆体として合成される。この前駆体は、後に、グリコシル化され、2つのより小さなポリペプチド(すなわち、HA1サブユニットとHA2サブユニット)に開裂される。それぞれのHAモノマーは、HA2によって膜に固定され、大きなHA1小球が先端に配された(topped)、長いらせん鎖を含む(図3Aおよび図3Bを参照。)。
【0091】
結合機構は、以下の通りである。HAは、標的細胞の表面に存在する単糖類シアル酸(monosaccharide sialic acid)に結合する。これにより、ウイルス粒子が細胞の表面にとどまる。次に、細胞膜がウイルスを飲み込んで、細胞内でウイルスをピンチオフして、飲み込んだウイルスを含み、エンドソームと呼ばれる新しい膜結合コンパートメントを形成する。その後、細胞は、エンドソームの内部を酸性化して、リソソームに送り込むことでエンドソームの中身を消化する。しかし、pHが低くなると(pH<6.0)、HA分子の立体構造変化が引き起こされ、HA2からHA1が分離した後、再び折りたたまれて、全く異なる形状になる。このプロセスの末期には、いわゆる「融合ペプチド」が分子のフック(molecular hook)のように機能する。つまり、融合ペプチドを、鉤のようにエンドソーム膜に挿入して固定する。HA分子の残りの部分が新たな構造(より低いpHでも安定な構造)に折りたたみ直された後、「掴み鉤」を引っ込めて、エンドソーム膜をウイルス粒子自身の膜のすぐ近くに引き寄せる。これにより、この2つがお互いに融合するようになる。これらの工程が起こると、ウイルスの中身(そのRNAを含む。)は、標的細胞の細胞質の中へと、自由に流出することができる。
【0092】
本発明者らは、可溶性タンパク質としての発現(細胞内発現)も、テトラヒメナにおける全ての(full)血球凝集素の分泌(細胞外発現)も、困難であることを見出した。予備実験において、細胞膜内へのタンパク質発現それ自体は成功したけれども、可溶性タンパク質の発現およびその分泌は、問題を引き起こした。つまり、血球凝集素分子は、テトラヒメナ内で首尾よく発現し、シグナル・ペプチドを備えていたが、血球凝集素分子は、細胞内膜の構造体および宿主細胞の細胞膜に付着したままのようであった。
【0093】
他のタンパク質の発現および分泌において有用であることが分かっていた(例えば、国際公開第03/078566号を参照。)シグナル・ペプチドを使用したにもかかわらず、上述の問題が見出された。国際公開第03/078566号は、参照によって本願に組み込まれる。
【0094】
本発明者らは、この現象の原因は、疎水性の強い領域(特に、HA2の当該領域)にあり、それらが、首尾よく発現したタンパク質を細胞膜中に保持することに関与している可能性があると考えた。
【0095】
この現象は、感染宿主細胞中における、ウイルス粒子の自然の複製プロセスに似ていると考えられる。自然の複製プロセスにおいて、感染宿主によって発現させられたウイルスタンパク質の多くは、細胞質の内部でビリオンを組み立てるのに用いられる。一方、血球凝集素は、小胞体(endoplasmatic reticulum)およびゴルジ体によって、細胞表面に運ばれる。血球凝集素は、成熟ウイルスが出芽して、宿主のリン脂質膜(かつての宿主の細胞膜)に取り囲まれた状態で細胞から離れるまで、細胞膜に固定されて、細胞表面にとどまる。このようにして、血球凝集素がその外被を獲得する。
【0096】
本発明の別の好ましい実施形態において、ウイルス融合タンパク質またはそのフラグメントが提供される。これらは、核酸によってコードされており、少なくとも1つの疎水性ドメインまたはそのフラクションが欠けている。
【0097】
驚くべきことに、本発明の場面において、本発明者らは、少なくとも疎水性の融合ペプチドドメインまたはそのフラクションを欠くウイルスタンパク質が、タンパク質の分泌を促進することを見出した。
【0098】
上述のとおり、異種タンパク質発現において、タンパク質の分泌はシグナル配列によってサポートされる。大きな疎水性ドメインは、タンパク質の分泌の場面において、問題を引き起こす可能性がある。大きな疎水性ドメインは、発現宿主の細胞膜または細胞内膜との間で親和力を発揮する可能性があるからである。基本的に、遺伝子組み換えの繊毛虫類(例えば、テトラヒメナ)におけるタンパク質の分泌は、容易に達成される。遺伝子組み換えの繊毛虫類は、食物の細胞外消化のために、大量の加水分解酵素を分泌するからである。しかしながら、繊毛虫類におけるウイルス表面タンパク質のフルスケールの分泌が厄介になる可能性のあることがわかった。
【0099】
本発明者らは、そのような場合には、疎水性ドメインの除去が、タンパク質の分泌を促進して、そのように修飾されたタンパク質の免疫原性に影響を与えることなく、分泌されたタンパク質の収率を向上させるのに有用であることを見出した。
【0100】
ウイルスタンパク質において、抗原ドメインは親水性であることが多い。抗原ドメインは、水性媒体(唾液、血液およびその他の体液、汚水など)であることの多い周囲の媒体(medium)中にむかって、ウイルスの表面から伸長するからである。このことは、そのような場合において、疎水性ドメインの除去は、タンパク質の免疫原性に影響を与えないことを意味する。
【0101】
以下において、タンパク質の免疫原性を維持しながら、タンパク質の分泌または細胞溶解後の精製を促進する目的で、ウイルスタンパク質の疎水性ドメインを除去するという基本原理が、血球凝集素(HA)を用いた場合について説明されるであろう。
【0102】
上述のHAタンパク質発現において、HA0は、前駆体タンパク質(precursor protein)であり、それぞれのcDNAによってコードされたHA1およびHA2という2つのサブユニットからなる。HA0は、タンパク質分解酵素(例えば、多くの脊椎動物の肺組織中に見られるようなトリプターゼ)によって開裂されて、そのサブユニットに分割される。分割されたサブユニットは、ジスルフィド架橋によって、お互いに結合された状態を維持する(HA1−HA2−タンパク質。図3A、図3B、図4Aおよび図4Bを参照。)。
【0103】
HA1は、受容体の認識に関与しており、特に強い免疫原性を有するドメイン(特に、球状ドメインの「ヘッド部」)を備える。このドメインは、宿主細胞膜のグリコカリックスにあるシアル酸との結合サイトを含む。一方、HA2(完全なHA1−HA2複合体をウイルス膜にとどめておく(anchor)役割もある。)は、膜融合に関与する。HA2は、比較的疎水性の配列(表1を参照。)からなるN末端融合ペプチド(FP)を含む。
【0104】
この融合ペプチドは、約24個の高度保存されたN末端残基(N-terminal, highly conserved residue)からなる。これら残基のうちの少なくとも6個は、グリシンである(図4Aおよび図4Bを参照。同図において、融合ペプチドは8個のグリシンを含む。)。次に、3個のα−へリックス(α−H1、α−H2およびα−H3)が続く。その次には、疎水性の強い膜貫通ドメイン(TMD)が続く。TMDは、タンパク質を、ウイルスエンベロープの脂質二重層にとどめる(図3Aおよび図3Bを参照。)。
【0105】
次表は、本発明に関連して、少なくとも1つの疎水性ドメインを欠く、修飾された血球凝集素の候補の概要を示す。この表において、「TMD」は膜貫通ドメインを示し、「FP」は融合ペプチドを示す。上述の記載から、他のクラスI、クラスIIおよび/クラスIIIのウイルス融合ペプチドにも、この原理を適用できることが明らかである。
【表3】
【0106】
本発明者らが、タンパク質の分泌の代替手段として、細胞質中に可溶な形態でのウイルスタンパク質の細胞内発現が有望なオプションであることを見出したことは、特筆に価する。本発明者らは、細胞内に発現させられたフル装備の(full-featured)クラスIのウイルス融合タンパク質は、細胞膜だけでなく、繊毛虫類宿主の細胞内膜(小胞体、ゴルジ、核膜、ミトコンドリアなどたくさんある。)にも付着しやすいであろうことを見出し、また、このような場合には、宿主細胞が溶解した後で単離することができないことを見出した。さらに、本発明者らは、ウイルス融合タンパク質の少なくとも1つの疎水性ドメインが欠けていれば、ウイルス融合タンパク質は、細胞内膜に付着しにくくなるであろうことを見出した。したがって、そのように修飾されたタンパク質(例えば、表3に示されるようなタンパク質)は、全細胞溶解液からより容易に単離することができる。
【0107】
さらに、本発明によれば、少なくとも1つの核酸分子が提供される。核酸分子は、以下の(a)〜(j)の少なくとも1つからなる群から選択されてよい。
(a)配列ID番号2、4および/または6として表されたヌクレオチド配列を含む核酸分子。
(b)配列ID番号3、5および/または7として表されたアミノ酸配列を含むポリペプチドをコードする核酸分子。ここで、上記のポリペプチドは、ウイルスタンパク質またはそのフラグメントである。
(c)少なくとも1つの疎水性ドメインを欠いたウイルスタンパク質の切断型(truncated form)をコードする核酸分子。ここで、上記のウイルスタンパク質は、好ましくはウイルス融合タンパク質であり、さらに好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質である。
(d)膜貫通ドメイン(TMD)、融合ペプチド(FP)および/またはHA2サブユニットを欠いたクラスIのウイルス融合タンパク質の切断型をコードする核酸分子。
(e)(a)〜(d)の核酸分子のフラクション、変異体(variant)、ホモログまたは誘導体(derivative)である核酸分子。
(f)(a)〜(e)のいずれかの核酸分子の相補体である核酸分子。
(g)ストリンジェントな条件下で、(a)〜(f)のいずれかの核酸分子と分子交雑することができる核酸分子。
(h)(a)〜(g)のいずれかの核酸分子と比較して、少なくとも1つの無症状の単一ヌクレオチドの置換(silent single nucleotide substitution)を含む核酸分子。
(i)原生動物の発現宿主に最適化するようコードされた、(a)および(c)〜(h)核酸分子に関連する核酸分子。
(j)(a)〜(i)のいずれかの核酸分子と、少なくとも70%、好ましくは少なくとも95%の配列同一性を有する核酸分子。
【0108】
これらの核酸分子の特に好ましい代替策(fallback position)は、(a)〜(i)のいずれかの核酸分子と、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98または99%の配列同一性を有する。
【0109】
さらに、繊毛虫類の宿主細胞のトランスフェクションに用いられるベクターが提供される。このベクターは、(a)ウイルスタンパク質またはそのフラグメントをコードする核酸と、(b)当該核酸に機能しうるように連結されたプロモーターとを備える。
【0110】
これらの核酸は、cDNAであることが好ましい。
【0111】
さらに、本発明によれば、本発明に係るベクターを用いて核酸を導入された繊毛虫類の宿主細胞が提供される。この繊毛虫類は、テトラヒメナ科のメンバーであることが好ましい。
【0112】
さらに、本発明によれば、少なくとも1つのウイルスタンパク質またはそのフラグメントを生産するプロセスが提供される。この方法は、(a)本発明に係るベクターを用いて、繊毛虫類の宿主細胞に核酸を導入する段階と、(b)タンパク質の発現が可能な条件下で、宿主細胞を培養する段階とを有する。
【0113】
好ましい実施態様において、このプロセスは、(c)ある特定のタンパク質について発現させられたタンパク質の分泌が可能な条件下で、宿主細胞を培養する段階を、さらに有してよい。
【0114】
さらに、本発明によれば、以下の(a)〜(d)の少なくとも1つからなる群から選択されるタンパク質が提供される。
(a)本発明に係る核酸によってコードされたタンパク質。
(b)本発明に係るアミノ酸配列を含むタンパク質。
(c)少なくとも1つの保存アミノ酸置換(conservative amino acid substitution)を含む、(a)または(b)に関連するタンパク質。
(d)本発明に係るプロセスによって得られたタンパク質。
【0115】
さらに、本発明によれば、医薬組成物を生産する方法が提供される。この方法は、(a)本発明に係る繊毛虫類発現系において本発明に係るタンパク質を発現させる段階と、(b)そのようにして得られたタンパク質を単離または精製する段階とを有する。
【0116】
他の実施態様において、本発明によれば、本発明に係るプロセスを含む医薬組成物を生産する方法が提供される。
【0117】
さらに、本発明によれば、本発明に係るタンパク質または本発明に係る方法を用いて生産されたタンパク質を含む医薬組成物が提供される。好ましい実施形態において、この組成物はワクチンである。
【0118】
(ディスクレーマー)
明細書を過度に長くすることなく、包括的に開示する目的で、出願人は、以上で参照した特許および特許出願のそれぞれを参照により組み込む。
【0119】
上述の詳細な実施態様における要素および特徴の特定の組合せは、単なる例示に過ぎない。これらの内容(teaching)を、本明細書または参照によって組み込まれた特許もしくは出願に記載されている他の内容と、置換または代用できることも、明白である。当業者がそう認識するように、当業者であれば、特許請求の範囲に記載された本発明の趣旨および範囲から逸脱することなく、本明細書に記載されている事項の変更、改良およびその他の実施に想到することができる。したがって、前述の開示は、ほんの一例であり、本発明の範囲を限定することは意図されていない。本発明の範囲は、後述の特許請求の範囲において定義され、それに加えて、その均等物も含まれる。さらに、特許請求の範囲に記載された本発明の範囲は、本明細書および特許請求の範囲において用いられる参照符号によって限定されない。
【実施例】
【0120】
(実施例および図面の簡単な説明)
本発明の目的(object)の付加的な詳細、特徴、特色および利点は、下位クレームおよびそれぞれの図面および実施例に関する記載において開示される。それらは、本発明の好ましい実施態様を例示的に説明する。しかしながら、これらの図面は、本発明の範囲を限定するものとして理解されるべきではない。
【0121】
(実施例および図面)
(1.発現ベクターの作製)
ドナー・ベクターの中に、様々な血球凝集素のフラグメント(配列ID番号2、4および6を参照。)のための合成遺伝子のクローンを作製した(図2Aを参照。)。Cre依存性リコンビナーゼ系(Cre dependent recombinase system)を用いて、全てのドナー・ベクターに由来する発現カセットを、アクセプター・ベクターに移動させた(図2Bを参照。)。
【0122】
(2.野生型テトラヒメナの培養および発現プラスミドの形質転換(transformation))
スキムミルク培地中、プロテオースペプトンを添加した(supplemented proteose peptone)(SPP)培地中、または、既知組成培地(CDM)中で、野生型のテトラヒメナ好熱菌株(例えば、B 1868/4、B 1868/7およびB 2068/1)を培養した。テトラヒメナ好熱菌(T. thermophila)細胞の形質変換は、前述のCassidy-Hanleyら(1997年)と同様の方法で実施した。
【0123】
(3.組み換え血球凝集素の検出)
80rpmの攪拌器中で、30℃における淘汰圧下において、形質変換されたテトラヒメナ細胞をSPP培地中で培養した。標的遺伝子発現は、41℃での熱ショック(HSP−P)または対数増殖培養への20nMのCd2+(MTT1−P)の添加により誘導された。
【0124】
標的遺伝子発現の誘導工程の後、形質変換された細胞の部分標本(aliquot)と、細胞の添加されていないSPP(cell free SPP)の上澄液の部分標本とを、24時間培養した。収集した細胞を、氷冷したRIPAバッファー中で可溶化した(150mM NaCl、10mM TrisHCl、5mM EDTA、0.1% SDS、0.1% DOC、1% Triton X100、2.5μg/mL E64の混合溶液中で、5000細胞/μL)。その後、超音波処理器中で15分間粉砕(disrupt)した。通常の方法に従って、SDS−ポリアクリルアミドゲル電気泳動法(SDS−PAGE)およびウエスタンブロット解析を実施した。粉砕された細胞の部分標本(つまり、1000個の細胞)または細胞の添加されていない上澄液を、Laemmliサンプルバッファー(125mM TrisHCl、pH6.8、10%Glycerol、4%SDS)中に再懸濁させて、12%SDS−ポリアクリルアミドゲル電気泳動法により分離した。ニトロセルロース膜にゲルを転写して、0.05%Tween20と、5%スキムミルクまたは3%ウシ血清アルブミンを含むPBSを用いてブロッキングした。ウイルス株に特異的な一次抗体(virus strain specific primary antibody)を用いて、形質転換された繊毛虫類における組み換え血球凝集素の発現を検出した。PBS/Tを用いて洗浄し、HRP標識二次抗体(secondary HRP-conjugated antibody)を適用した後、スーパーシグナル・ウエスト・ピコ・ケミルミネッセント基質(Perbio、Fischer Scientific社製)と、通常のX線フィルム現像とを組み合わせて用いて、ブロットを展開させた。図8、図9のA、図9のBおよび図9のCは、標的遺伝子発現が誘導された後の、形質転換されたテトラヒメナ細胞の細胞溶解物(cell lysate)および上清(supernatant)の代表的なウエスタンブロット(representative Western blot)を示す。野生型の対照群(図9のB。レーン3は対照群の細胞ペレットであり、レーン4は対照群の上清である。)は、全て空白である。図8から、発酵プロセス中に、HA抗原が上清に分泌されたことがわかる。図9から、形質転換された繊毛虫類の上清に関して、先端を切り取られた(truncated)血球凝集素が検出された(細胞内ドメインが除去された)こと(図9のAの「HAロング」)、さらに膜貫通領域が除去されたこと(図9のBの「HAショート」)、および、HA2サブユニット全体が除去されたこと(図9のCの「HA1」)が分かる。
【0125】
(4.血球凝集素の生産)
発酵には、Braun社のUD50(50リットル)および標準ラッシュトン・インペラ(standard Rushton impeller)を備えたInfors社のSixfors(0.5リットル)を使用した攪拌速度は、300rpmおよび400rpmに設定した。pO2は20%に設定し、pHは7.0に設定した。発酵は、標準培地中で実施した。
【0126】
(5.組み換え血球凝集素の精製)
中空糸モジュール(0.3m2、3L/min、管径11mm)を用いて、発酵もろみ液(fermentation broth)から細胞を分離することで、50L発酵プロセスから細胞を採取した。氷冷したpH7.4のリン酸ナトリウムバッファー(10mM、PB)を用いて、細胞ペレットを3〜4回洗浄した。その後、ムチン分泌胞体の中身を除去すべく、10℃、2400xgの条件で8分間の遠心分離処理(Sorvall(登録商標)evolution(商標)SLA−1500ローター)を実施して、小粒状にした。得られた細胞ペレットを、システインプロテアーゼ阻害剤E−64(70μM)および3%Tween20(登録商標)を補充したPB中に再懸濁させた。Ultra Turrax(IKA社製、UT T25+S25N−25G)を用いて、氷上で10,000rpmの条件で、5分間処理することで、細胞を粉砕した。PB(pH7.4)を用いて、溶解物の容積を1.8Lとし、4℃で17時間攪拌することで、可溶化した。中空糸モジュール(0.45μm、表面積850cm2)を用いて、細胞溶解物をろ過した。2.3LのPB(pH7.4)を用いて、中空糸モジュールを3回洗浄した。
【0127】
3段階クロマトグラフィー精製プロセスを用いて、クロマトグラフィー精製を実施した。その結果、高度に純化された組み換えHA抗原が得られた。この組み換えHA抗原は、変性しておらず、ワクチンの成分として適している。
【0128】
全てのクロマトグラフィーは、4℃で実施した。第1の精製ステップでは、上述のように準備したろ液を含むHAを、5%グリセロールおよび3%Tween20(登録商標)を含みpHが7.4のPBとの平衡を保たせながら、アンモニウム基が導入されたアニオン交換カラム(ammonium anion exchange column)(Capto(商標)Qカラム、強い第四級アンモニウムを有するアガロースビーズ・カラム)に、15mL/minの流量で供給した。最初に、3%Tween20(登録商標)を含むローディングバッファーを用いて、カラムを洗浄した。その後、Tween20(登録商標)を含まないローディングバッファーを用いた洗浄工程を実施して、洗浄剤を除去した。5%グリセロールと、NaClとを含むPBを用いて、部分的に精製されたHAを溶出させた。NaClの濃度は、第1ステップでは150mMであり、第2ステップ(pH7.4)では、1Mであった。収集したフラクションの全てのサンプルを、SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析によって解析した。HAが陽性のフラクション(HA positive fraction)は、次のクロマトグラフィー工程に備えて貯蔵した。
【0129】
組み換えHAは、セラミック・ヒドロキシアパタイト・カラム(CHT)とは結合しないので、セラミック・ヒドロキシアパタイト・カラムを用いて、夾雑タンパク質を除去した。Capto(商標)Qカラムから得て、貯蔵しておいたフラクションを濃縮した。NaClを除去すべく、5%グリセロールを含むPBとともに実験室規模のTFFモジュール(30kDa)を用いて、緩衝液交換を実施した。サンプルのpHは、CHTカラムに供給する前の段階で7.5に設定した。供給流量は、7mL/minに設定した。その後、5%グリセロールを含むPBを用いて、カラムを洗浄した。5%グリセロールを含み、pHが7.5である150mMのPBを用いて、段階溶離法による溶出工程を実施した。SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析により、溶出液およびフロースルー(flow through)を分析した。200mLのフロースルーを含むHAを採取して、第3のカラムに供給した。
【0130】
第3の精製ステップでは、Con A Sepharose 4Bカラム(臭化シアン法によってセファロース4Bと結合したコンカナバリンAと親和性のある媒体)を使用した。ヒドロキシアパタイト・カラムのフロースルーに150mMのNaClを補充して、5mL/minの流量でカラムに供給した。リン酸緩衝生理食塩水(PBS、pH7.4)を用いてカラムを洗浄し、0.5Mのメチルα‐D‐マンノピラノシド(pH7.4)を含むPBSを用いて、精製された組み換えHAを溶出させた。SDS−PAGE法、ウエスタンブロット分析およびブラッドフォード分析により、溶出画分(elution fraction)のサンプルを分析した。
【0131】
(図面)
図1Aは、様々な分類群のN−結合型グリコシル化パターン概要を示す。一般的に、「N−結合型グリコシル化」という用語は、アミノ酸残基アスパラギン(N)のグリコシル化を示す。そして、オリゴサッカリルトランスフェラーゼによって、そのようなアスパラギン残基にオリゴ糖鎖が取り付けられる。この反応は、トリペプチド配列Asn−X−SerまたはAsn−X−Thrにおいて生じる。なお、Xは、Pro以外であればどのようなアミノ酸であってもよい。
【0132】
原核生物が全くN−結合型グリコシル化を有しない一方で、繊毛虫類は、マンノースに富み、タンパク質の免疫原性を強化するN−結合型グリコシル化パターンを有することを特徴としている(本文を参照。)。図1Bは、いくつかの種類の繊毛虫類におけるパターンの可能性のあるバリエーション(potential variation)を示す。
【0133】
図2Aは、合成HA遺伝子をコードするドナー・ベクターを示す。ドナー・ベクターは、大腸菌中で増幅するためのバクテリアのバックボーン(pUC_ori)と、カナマイシン(kanR)と、大腸菌内での淘汰のためのクロラムフェニコール(cmR)耐性遺伝子カセットおよびスクラーゼ遺伝子(sacB)と、誘導プロモーターによってコントロールされ、その後にテトラヒメナ好熱菌のβチューブリン2終了配列(btu2)が続く、標的遺伝子のオープン・リーディング・フレーム(HA遺伝子)とを含む。
【0134】
図2Bは、テトラヒメナ好熱菌において用いられる発現ベクターを示す。このベクターは、アンピシリン(ampR)と、大腸菌内での淘汰のためのクロラムフェニコール(cmR)耐性遺伝子と、テトラヒメナ好熱菌内でのプラスミド増殖のためのテトラヒメナ好熱菌に特異的な複製起点(rDNA_ori)と、形質転換された繊毛虫類を識別するためのネオマイシンを用いた耐性遺伝子カセット(neoR)と、誘導プロモーターによってコントロールされ、その後にテトラヒメナ好熱菌のβチューブリン2終了配列(btu2)が続く、標的遺伝子のオープン・リーディング・フレーム(HA遺伝子)とを含む。
【0135】
図3Aは、血球凝集素を、そのサブユニットであるHA1(黒色)およびHA2(灰色)とともに示す。αヘリックスおよびβシートが示される。
【0136】
図3Bは、血球凝集素の概要を、そのサブユニットであるHA1(黒色)およびHA2とともに示した概略図である。融合ペプチド(FP、上記の記載を参照。)と、タンパク質をウイルスエンペロープ膜に固定する膜貫通ドメイン(TMD)とが記載されている。HA1およびHA2は、ジスルフィド架橋(DSB)によって結合されている。HA2において2つのαヘリックス(α−H1およびα−H2)と連結する部分は、「ループドメイン(LD)」と呼ばれる。HA2のC末端細胞質尾部(C-terminal cytoplasmatic tail)は図示されていない。
【0137】
HA1は、図示されていないジスルフィド架橋によって安定化された球状ドメイン(「ヘッド部」)を有する。このヘッド部は、宿主細胞膜のグリコカリックスにあるシアル酸との結合サイトを含む。同様に、ヘッド部は、強い免疫原性を有し、そのため、免疫付与後にHAに対して作製された多くの抗原の標的となる。
【0138】
ウイルスエンベロープ二重層の宿主細胞膜との融合を引き起こすべく、pHの低下をトリガーとして、HA1およびHA2は、お互いに切り離される。その結果、HAホモ三量体中において、HA1サブユニットは正電荷を有する。
【0139】
これにより反発力が発生して、HA1サブユニットがお互いから離れて、それにより、HA2サブユニットとの連結が解ける。その結果、HA2サブユニットが活性化される。活性化されたHA2は、膜の融合の主な原因となる。HA2は、1度だけ活性化されることができ、その後は不活性なままである。そのため、HA1はあまりにも早く連結を解放しないことが重要である。そうしなければ、ウイルスは、その感染力を喪失してしまうからである。
【0140】
HA2は、大きく引き伸ばせば(over large stretches)、αらせん構造を有し、大きなループ領域を含む。加えて、HA2は、膜貫通ドメインおよび融合ペプチドを含む。融合ペプチドは、HA1サブユニットの分離によって、解放される。
【0141】
融合を解除する目的で、HA1の球状ドメイン(「ヘッド部」)がHA2から分離せざるを得ない。そのため、HA2は、HA2が展開して、融合ペプチドが宿主細胞膜を貫通するように、その構造を変化させることができる。このようにして、ウイルスが、宿主細胞膜に直接結合する。
【0142】
展開プロセスによって、HA2は、一方の側に引き伸ばされる。他の領域は、タンパク質の正味の大きさが増大しないように、巻き込まれる。その後、HAは、その構造を変化させて、膜融合が生じるように、ウイルスを宿主細胞膜の近くに持っていく。
【0143】
図4Aは、血球凝集素を例として、本文中で定義されたクラスIのウイルス融合タンパク質の一般的な構造を示す。HA1およびHA2は、インフルエンザHA0クラスIタンパク質のサブユニットである。ラベルは、シグナル・ペプチド(SP)、融合ペプチド(FP)、加工部位(矢印)、膜貫通ドメイン(TMD)の位置を特定する。これらは、一般的なクラスIウイルス融合ペプチドのサブユニットである。
【0144】
図4B(配列ID番号3も参照。)では、インフルエンザAウイルス株、A/ニューカレドニア/20/1999 H1N1(ジェンバンク信託番号:AAP34324.1)、の血球凝集素のサブユニットが例示的に説明される。アンダーラインを引いた一節は、シグナル・ペプチド(SP)、HA2融合ペプチド(FP)およびHA2膜貫通ドメイン(TMD)を示す。
【0145】
図示されているタンパク質は、565個のアミノ酸残基(「AA」)からなる。特定配列の特徴を、表4に示す。ここで、HA2融合ペプチドおよびHA2膜貫通ドメインでは、疎水性アミノ酸の占める割合が大きく(表1のヒドロパシーのデータに基づいて計算されたデータ)、例えば、融合ペプチドは、8個のグリシン残基を有することに留意すべきである。本発明者らは、繊毛虫類における異種タンパク質発現を目的として、本発明によって提供されるように、これらのドメインを切断することで、タンパク質の分泌および/または細胞溶解後の精製を促進することができることを示した。
【表4】
【0146】
表4は、HA2膜貫通ドメインおよびHA2融合ペプチドにおいて、疎水性アミノ酸の占める割合が非常に大きいことを明確に示す。本発明者らは、この発見から、これらの2つのドメインの少なくとも1つを切断することで、媒体または培地(medium)中へのタンパク質の分泌が促進されるであろうと推測した。
【0147】
図4Aおよび図4Bのタンパク質に関連して説明された特徴は、他の血球凝集素にも、表2に示されたクラスIウイルス融合タンパク質の多くにも適用することができることに注目すべきである。
【0148】
図5は、テトラヒメナ好熱菌と、ホモ・サピエンスとの間におけるコドン出現頻度の比較を示す。ホモ・サピエンスのコドン出現頻度は、ヒト宿主細胞から発現させられた血球凝集素と同様に、ヒト宿主において発現させられるウイルスタンパク質に適用することができる。さらなる説明については、本文を参照されたい。
【0149】
図6は、繊毛虫類(特に、テトラヒメナ)において用いられている遺伝子コードを示す。標準的でないヌクレオチド・コード(non-canonical nucleotide code)であり、グルタミンをコードするUAAおよびUAGは、太字で印刷されている。しかしながら、一般的な遺伝子コードによれば、これらのトリプレットは、終止コドン(削除されたトリプレットを参照。)である。「1LC」は、「1文字コード(one letter code)」を表し、「3LC」は、「3文字コード(three letter code)」を表す。
【0150】
図7は、一例として、ウイルス血球凝集素配列(配列ID番号1を参照。)と、テトラヒメナ好熱菌における発現に関してコドン最適化された血球凝集素の配列(配列ID番号2を参照。)との間におけるコドン出現頻度の比較を示す。コドン出現頻度における相違点は、灰色の四角で示される。
【0151】
図8は、供給回分式の発酵プロセス(0.5L)を用いて培養され、組み換えHAを発現している形質転換された繊毛虫類の免疫ブロットを示す。集菌時期の異なる上清のサンプル(SN1は47.5時間、SN2は66時間、SN3は70時間、SN4は90時間)に対して、非還元条件下でSDS−PAGEが適用され、その後、ウエスタンブロット分析が適用された。染色には、特定のインフルエンザ・アンチ−B/フロリダ/4/2006血清(Influenza anti-B/Florida/4/2006 serum)を用いた。適用されたNIBSC−HA抗原(陽性対照)は、約90kDa(A、NIBSC、45ng)に相当するシグナルを示した。47.5時間の培養の後、上清に関して、約72kDaおよび約90kDaにおいて、HA特異沈降線(HA specific band)が見られた。このタンパク質は、配列ID番号9に対応する。
【0152】
図9は、テトラヒメナ中で遺伝子組み換え技術によって発現させられた、長さの異なる(本発明の内容に従って切断された)血球凝集素タンパク質フラグメントの免疫ブロットを示す。染色には、特定のギニア・ブタ−アンチ−H1N1抗体(Guinea Pig-anti-H1N1 antibody)を用いた。Pは、テトラヒメナ細胞ペレットであり、SNは、細胞培養上清である。レーン1および2は、テトラヒメナのクローンを表し、レーン3および4は、野生型の対照群を表す。
【0153】
図9のAは、「HAロング」と呼ばれるタンパク質を示す。HAロングは、HA2の細胞内ドメイン(CT、図4Aを参照。)を欠いた、HA1−HA2の完全な二量体(full HA1-HA2-dimer)である(予測分子量は61.4kDaである。)。このタンパク質は、配列ID番号3に対応する。
【0154】
図9のBは、「HAショート」と呼ばれるタンパク質を示す。HAショートは、HA2の細胞内ドメイン(CT)および疎水性の膜貫通ドメイン(TMD)を欠いた、HA1−HA2二量体である(予測分子量は57.6kDaである。)。このタンパク質は、配列ID番号5に対応する。
【0155】
図9のCは、単なるHA1(「HA_1」と称され、予測分子量は34.2kDaである。)を示す。すなわち、少なくとも2つの疎水性ドメイン(融合ペプチドおよび膜貫通ドメイン)を有するHA2が切断されている。このタンパク質は、配列ID番号7に対応する。
【0156】
免疫ブロットから、HAショートおよびHA_1(これらは、少なくとも1つの疎水性ドメインを欠く。)は、上清中に、HAロングよりも豊富に含まれることが明らかである。このことは、ウイルス融合タンパク質の繊毛虫類におけるタンパク質分泌は、少なくとも1つの疎水性ドメインを除去することにより促進されるという結論をサポートする。細胞膜への付着しやすさが低減するからである。さらに、ウイルス融合タンパク質(特に、血球凝集素)の免疫原性ドメインは、HA1サブユニット中に配されるので、HA1サブユニットのみを含むワクチンまたはHA1サブユニットと切断されたHA2サブユニットとを含むワクチンが、依然として免疫応答を引き起こすことが予想される。したがって、ワクチンの生産にとって有用であることが予想される。
【0157】
図10Aは、本発明に係る配列ID番号3(すなわち、「HAロング」)に関連するタンパク質の疎水性プロットを示す。疎水性プロットは、カイト−ドリトルの指標(Kyte-Doolittle scale)に従って作図されている。融合ペプチド(AA344−368)および膜貫通ドメイン(AA530−550)が、タンパク質の主要な疎水性領域の間に配されており(矢印を参照。)、それらが、タンパク質の媒体または培地(medium)への効率的な分泌を妨げていることを、容易に判定することができる。
【0158】
図10Bは、分泌された主要な繊毛虫類タンパク質(a ciliate major secreted protein)の疎水性プロット(本発明の発明者らによる国際公開第03/078566号の配列ID番号2)を示す。このタンパク質は、自然な環境においてテトラヒメナによって大量に分泌される。このタンパク質は、強い疎水性ドメインを有しておらず、平均して、HAロングよりも疎水性が小さいことが明白である。本発明者らは、この現象を、HAロングとは対照的に、このタンパク質の分泌の容易化に関与させた。
【0159】
(配列表)
別表において、完全な配列表が添付される。配列表には、以下の配列を示す(NAは核酸であり、AAはアミノ酸である。)。
【表5】
【0160】
(参照文献)
Cassidy-Hanley, D.、Bowen, J.、Lee, J.、Cole, E.、VerPlank, L.、Gaertig, J.、Gorovsky, M.およびBruns, P.(1997年);粒子衝撃によるテトラヒメナ好熱菌を交配する生殖細胞系列および体細胞形質転換(Germline and somatic transformation of mating Tetrahymena thermophila by particle bombardment);Genetics 146巻:135−47頁。
Gaertig, J.、Gao, Y.、Tishgarten, T.、Clark, T.およびDickerson, H.(1999年);テトラヒメナ好熱菌におけるパラサイト抗原の表面提示(Surface display of a parasite antigen in the ciliate Tetrahymena thermophila);Nat Biotechnol 17巻:462−5頁。
Kulakosky, P.、Hughes, P.およびWood, H.(1998年);昆虫の幼虫および組織培養細胞におけるバキュロ・ウイルス発現による組み換え糖タンパク質のN結合型グリコシル化(N-Linked glycosylation of a baculovirus-expressed recombinant glycoprotein in insect larvae and tissue culture cells);Glycobiology 8巻:741−5頁。
Reading, P.、Miller, J.およびAnders, E.(2000);インフルエンザウイルスによるマクロファージの感染中におけるマンノース受容体の取り込み(Involvement of the mannose receptor in infection of macrophages by influenza virus);J Virol 74巻:5190−7頁。
Tomiya, N.、Narang, S.、Lee, Y.C.およびBetenbaugh, M.J.(2004年);哺乳類細胞株における、自然のおよび改変された鱗翅目の昆虫細胞株のN−グリカン・プロセッシング(Comparing N-glycan processing in mammalian cell lines to native and engineered lepidopteran insect cell lines);Glycoconj J 21巻:343−60頁。
Kyte, J.およびDoolittle RF.(1982年);タンパク質の疎水性特性を表示するシンプルな方法(A simple method for displaying the hydropathic character of a protein);J Mol Biol 157巻:105頁。
【特許請求の範囲】
【請求項1】
ウイルスタンパク質またはそのフラグメントの異種タンパク質発現系であって、
(a)繊毛虫類宿主細胞と、
(b)ウイルスタンパク質またはそのフラグメントをコードする少なくとも1つの核酸と、
(c)前記核酸に機能しうるように連結されたプロモーターと、
を備える、異種タンパク質発現系。
【請求項2】
(d)前記核酸に機能しうるように連結され、前記核酸によってコードされる、前記ウイルスタンパク質または前記フラグメントの細胞外培地への分泌の原因となるシグナル配列をさらに備える、
請求項1に記載の異種タンパク質発現系。
【請求項3】
前記ウイルスタンパク質は、ウイルス表面タンパク質である、
請求項1または請求項2に記載の異種タンパク質発現系。
【請求項4】
遺伝子組み換えの前記繊毛虫類は、テトラヒメナ科(family Tetrahymenidae)のメンバーである、
請求項1から請求項3までのいずれか一項に記載の異種タンパク質発現系。
【請求項5】
前記ウイルスタンパク質は、ウイルス融合タンパク質であり、好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質からなる群の少なくとも1つである、
請求項1から請求項4までのいずれか一項に記載の異種タンパク質発現系。
【請求項6】
前記ウイルスタンパク質またはそのフラグメントは、
前記核酸によってコードされ、
疎水性ドメインまたはそのフラクションの少なくとも1つを欠いている、
請求項1から請求項5までのいずれか一項に記載の異種タンパク質発現系。
【請求項7】
(a)配列ID番号2、4、6および/または14として表されたヌクレオチド配列を含む核酸分子、
(b)配列ID番号3、5、7および/または15として表されたアミノ酸配列を含み、ウイルスタンパク質またはそのフラグメントであるポリペプチドをコードする核酸分子、
(c)少なくとも1つの疎水性ドメインを欠いたウイルスタンパク質(好ましくはウイルス融合タンパク質であり、さらに好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質である。)の切断型(truncated form)をコードする核酸分子、
(d)膜貫通ドメイン(TMD)、融合ペプチド(FP)および/またはHA2サブユニットを欠いたクラスIのウイルス融合タンパク質の切断型をコードする核酸分子、
(e)(a)〜(d)の核酸分子のフラクション、変異体(variant)、ホモログまたは誘導体(derivative)である核酸分子、
(f)(a)〜(e)のいずれかの核酸分子の相補体である核酸分子、
(g)ストリンジェントな条件下で、(a)〜(f)のいずれかの核酸分子と分子交雑することができる核酸分子、
(h)(a)〜(g)のいずれかの核酸分子と比較して、少なくとも1つの無症状の単一ヌクレオチドの置換(silent single nucleotide substitution)を含む核酸分子、
(i)原生動物の発現宿主に最適化するようコードされた、(a)および(c)〜(h)核酸分子に関連する核酸分子、
(j)(a)〜(i)のいずれかの核酸分子と、少なくとも70%、好ましくは少なくとも95%の配列同一性を有する核酸分子、
のうちの少なくとも1つの核酸分子からなる群から選択される核酸分子。
【請求項8】
繊毛虫類の宿主細胞のトランスフェクションに用いられるベクターであって、
(a)ウイルスタンパク質またはそのフラグメントをコードする核酸と、
(b)当該核酸に機能しうるように連結されたプロモーターと、
を備えるベクター。
【請求項9】
請求項8に記載のベクターを用いて核酸を導入された繊毛虫類の宿主細胞。
【請求項10】
繊毛虫類の宿主細胞において、少なくとも1つのウイルスタンパク質またはそのフラグメントを生産する方法であって、
(a)請求項8に記載のベクターを用いて、前記繊毛虫類の宿主細胞に核酸をトランスフェクトする段階と、
(b)タンパク質の発現が可能な条件下で、前記宿主細胞を培養する段階と、
を有する方法。
【請求項11】
(c)発現させられたタンパク質の分泌が可能な条件下で、宿主細胞を培養する段階をさらに有する、
請求項10に記載の方法。
【請求項12】
(a)請求項7に記載の核酸によってコードされたタンパク質、
(b)請求項7に記載のアミノ酸配列を含むタンパク質、
(c)少なくとも1つの保存アミノ酸置換を含む、(a)または(b)のタンパク質に関連するタンパク質、
(d)請求項10または請求項11に記載の方法によって得られたタンパク質、
のうちの少なくとも1つのタンパク質からなる群から選択されるタンパク質。
【請求項13】
医薬組成物を生産する方法であって、
(a)請求項1に記載の繊毛虫類の発現系において、請求項12に記載のタンパク質を発現させる段階と、
(b)得られたタンパク質を単離または精製する段階と、
を有する方法。
【請求項14】
医薬組成物を生産する方法であって、
請求項10に記載の方法を含む、方法。
【請求項15】
請求項12に記載のタンパク質、ならびに、請求項13および/または請求項14に記載の方法を用いて生産されたタンパク質の少なくとも一方を含む、
医薬組成物。
【請求項16】
前記医薬組成物がワクチンである、
請求項15に記載の医薬組成物。
【請求項1】
ウイルスタンパク質またはそのフラグメントの異種タンパク質発現系であって、
(a)繊毛虫類宿主細胞と、
(b)ウイルスタンパク質またはそのフラグメントをコードする少なくとも1つの核酸と、
(c)前記核酸に機能しうるように連結されたプロモーターと、
を備える、異種タンパク質発現系。
【請求項2】
(d)前記核酸に機能しうるように連結され、前記核酸によってコードされる、前記ウイルスタンパク質または前記フラグメントの細胞外培地への分泌の原因となるシグナル配列をさらに備える、
請求項1に記載の異種タンパク質発現系。
【請求項3】
前記ウイルスタンパク質は、ウイルス表面タンパク質である、
請求項1または請求項2に記載の異種タンパク質発現系。
【請求項4】
遺伝子組み換えの前記繊毛虫類は、テトラヒメナ科(family Tetrahymenidae)のメンバーである、
請求項1から請求項3までのいずれか一項に記載の異種タンパク質発現系。
【請求項5】
前記ウイルスタンパク質は、ウイルス融合タンパク質であり、好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質からなる群の少なくとも1つである、
請求項1から請求項4までのいずれか一項に記載の異種タンパク質発現系。
【請求項6】
前記ウイルスタンパク質またはそのフラグメントは、
前記核酸によってコードされ、
疎水性ドメインまたはそのフラクションの少なくとも1つを欠いている、
請求項1から請求項5までのいずれか一項に記載の異種タンパク質発現系。
【請求項7】
(a)配列ID番号2、4、6および/または14として表されたヌクレオチド配列を含む核酸分子、
(b)配列ID番号3、5、7および/または15として表されたアミノ酸配列を含み、ウイルスタンパク質またはそのフラグメントであるポリペプチドをコードする核酸分子、
(c)少なくとも1つの疎水性ドメインを欠いたウイルスタンパク質(好ましくはウイルス融合タンパク質であり、さらに好ましくは、クラスI、クラスIIおよび/またはクラスIIIのウイルス融合タンパク質である。)の切断型(truncated form)をコードする核酸分子、
(d)膜貫通ドメイン(TMD)、融合ペプチド(FP)および/またはHA2サブユニットを欠いたクラスIのウイルス融合タンパク質の切断型をコードする核酸分子、
(e)(a)〜(d)の核酸分子のフラクション、変異体(variant)、ホモログまたは誘導体(derivative)である核酸分子、
(f)(a)〜(e)のいずれかの核酸分子の相補体である核酸分子、
(g)ストリンジェントな条件下で、(a)〜(f)のいずれかの核酸分子と分子交雑することができる核酸分子、
(h)(a)〜(g)のいずれかの核酸分子と比較して、少なくとも1つの無症状の単一ヌクレオチドの置換(silent single nucleotide substitution)を含む核酸分子、
(i)原生動物の発現宿主に最適化するようコードされた、(a)および(c)〜(h)核酸分子に関連する核酸分子、
(j)(a)〜(i)のいずれかの核酸分子と、少なくとも70%、好ましくは少なくとも95%の配列同一性を有する核酸分子、
のうちの少なくとも1つの核酸分子からなる群から選択される核酸分子。
【請求項8】
繊毛虫類の宿主細胞のトランスフェクションに用いられるベクターであって、
(a)ウイルスタンパク質またはそのフラグメントをコードする核酸と、
(b)当該核酸に機能しうるように連結されたプロモーターと、
を備えるベクター。
【請求項9】
請求項8に記載のベクターを用いて核酸を導入された繊毛虫類の宿主細胞。
【請求項10】
繊毛虫類の宿主細胞において、少なくとも1つのウイルスタンパク質またはそのフラグメントを生産する方法であって、
(a)請求項8に記載のベクターを用いて、前記繊毛虫類の宿主細胞に核酸をトランスフェクトする段階と、
(b)タンパク質の発現が可能な条件下で、前記宿主細胞を培養する段階と、
を有する方法。
【請求項11】
(c)発現させられたタンパク質の分泌が可能な条件下で、宿主細胞を培養する段階をさらに有する、
請求項10に記載の方法。
【請求項12】
(a)請求項7に記載の核酸によってコードされたタンパク質、
(b)請求項7に記載のアミノ酸配列を含むタンパク質、
(c)少なくとも1つの保存アミノ酸置換を含む、(a)または(b)のタンパク質に関連するタンパク質、
(d)請求項10または請求項11に記載の方法によって得られたタンパク質、
のうちの少なくとも1つのタンパク質からなる群から選択されるタンパク質。
【請求項13】
医薬組成物を生産する方法であって、
(a)請求項1に記載の繊毛虫類の発現系において、請求項12に記載のタンパク質を発現させる段階と、
(b)得られたタンパク質を単離または精製する段階と、
を有する方法。
【請求項14】
医薬組成物を生産する方法であって、
請求項10に記載の方法を含む、方法。
【請求項15】
請求項12に記載のタンパク質、ならびに、請求項13および/または請求項14に記載の方法を用いて生産されたタンパク質の少なくとも一方を含む、
医薬組成物。
【請求項16】
前記医薬組成物がワクチンである、
請求項15に記載の医薬組成物。
【図1A】

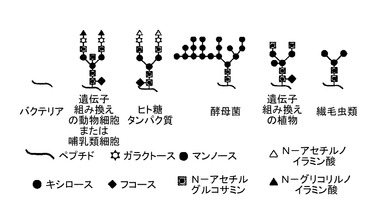
【図1B】
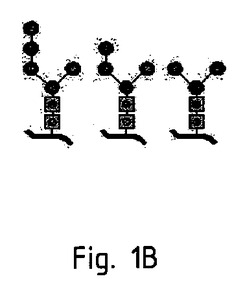
【図2A】
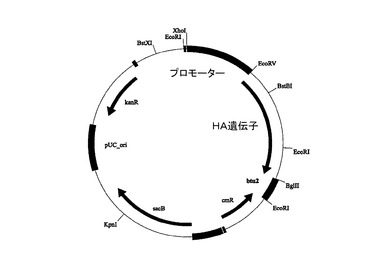
【図2B】
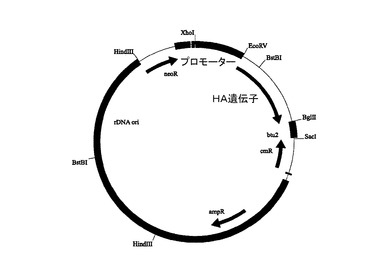
【図3A】

【図3B】
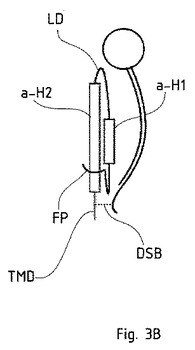
【図4A】
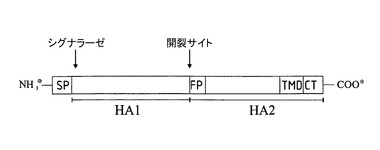
【図4B】
【図5】

【図6】
【図7】

【図8】
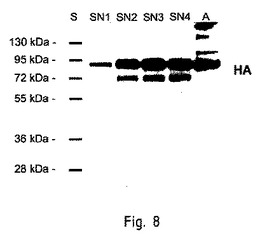
【図9】
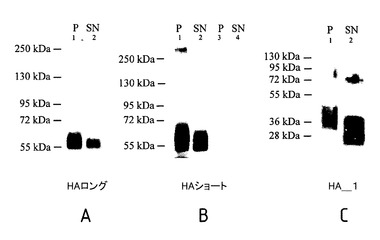
【図10A】
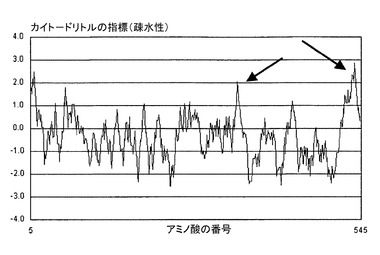
【図10B】
【図1B】
【図2A】
【図2B】
【図3A】
【図3B】
【図4A】
【図4B】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【公表番号】特表2012−529899(P2012−529899A)
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2012−515458(P2012−515458)
【出願日】平成22年6月15日(2010.6.15)
【国際出願番号】PCT/EP2010/058360
【国際公開番号】WO2010/146043
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(511306239)
【Fターム(参考)】
【公表日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成22年6月15日(2010.6.15)
【国際出願番号】PCT/EP2010/058360
【国際公開番号】WO2010/146043
【国際公開日】平成22年12月23日(2010.12.23)
【出願人】(511306239)
【Fターム(参考)】
[ Back to top ]