
繊維状ファージ上でのジスルフィド結合二量体タンパク質の提示
バクテリオファージ粒子の表面上での複合ホモ二量体タンパク質、及び繊維状ファージpIXコートタンパク質との融合ポリペプチドとして提示される、そのようなタンパク質の組み合わせ合成ライブラリの提示のための方法が提供される。本発明の方法を用いて、ヘテロ二量体又はジスルフィド結合した多量体タンパク質のような、更に複合の鎖間結合構造が提示され得る。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2009年11月17日に出願された米国出願第61/261,767号に対する優先権を請求し、この出願は、参照により全体的に組み込まれる。
【0002】
(発明の分野)
本発明は、二量体抗体断片、全抗体、又は他のジスルフィド結合多量体構築物を産生するために、pIXファージディスプレイライブラリを生成し、用いるための、組成物及び方法に関する。
【背景技術】
【0003】
繊維状ファージディスプレイは、各ファージ粒子が、選択プロセスにおいて一緒にそのコートタンパク質のN末端と融合したポリペプチドをコードする核酸を結合させるときの、タンパク質の親和性に基づく選択のために広範に使用される技術である。M13バクテリオファージは、5つのコートタンパク質をコードし、このうちマイナーコートタンパク質pIII及びpVIのおよそ5つのコピーは、ファージの一方の末端部にあり、同じ数のpVII及びpIXが、ファージの他方の末端部にある。ファージDNAは、メジャーコートタンパク質、pVIIIのおよそ3000個のコピーによって封入される。異物ポリペプチドの提示はM13の各コートタンパク質によって達成されてきたが、このうち最も一般的な融合パートナーはpIII及びpVIIIである。この技術を用いて、ペプチド、Fab、scFv、及び他のタンパク質バインダーのライブラリが構築されており、多様な用途において使用が見出され、かつ高い商業的価値を有する。
【0004】
pIIIコートタンパク質は、そのサイズ、立体配座、少ないコピー数に起因して、pVIIIタンパク質よりも好まれている。pIIIマイナーコートタンパク質は、柔軟な可動性ヒンジセグメントによって接続される3つのドメインを含む、大腸菌中へのファージ感染に関与する404アミノ酸、42kDタンパク質である。pIII N末端への融合は、ファージ表面から離れて提示されるタンパク質を繋ぎ止め、小さく、高いコピー数のpVIIIコートタンパク質への融合よりも、リガンド結合に対して潜在的により高いアクセスを提供する。pIIIタンパク質は、感染の最初の工程に必須であり、ほとんどの小さいペプチド及びタンパク質の融合は、このプロセスを干渉し得る。この問題は、例えば、野生型pIIIタンパク質の第2のコピー又はヘルパーファージを用いるファージミド系を含有する、ウイルスベクターの使用によって回避される。pIIIとは対照的に、かつpVIIIと同様に、pVII及びpIXは、それぞれ、ファージ表面上で密接に詰められた33及び32aaの短いヘリックスタンパク質である。それにもかかわらず、scFv(Gao,C.ら、Proc Natl Acad Sci U S A 99,12612〜12616,2002)及びFab(Shi,Lら、J Mol Biol 397,385〜396,2010、Tornetta,Mら、J Immunol Meth 360,39〜46,2010)ライブラリは、pIX上で提示及び選択されている。Fv及びペプチドのヘテロ二量体ディスプレイは、異なるポリペプチドをpVII及び密接に隣接したpIXの両方に融合することによって記載されている(Gaoら、1999 Proc Nat Acad Sci 96:6025〜6030及びJanda米国特許第7078166号)。加えて、単一特異的なscFvのpVIIディスプレイが報告されている(Kwasnikowskiら、2005.J Immunol Methods 307:135)。ファージ又はファージミドベクターによってコードされる細胞外タンパク質がコートタンパク質に融合されず、むしろ再操作されたコートタンパク質pIII及びpIXにジスルフィド結合を通じて共有結合する、代替的なアプローチもまた記載されている(米国特許第6753136号)。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ファージ粒子の表面上で二量体タンパク質並びにヘテロ二量体タンパク質を提示する能力は、組み合わせライブラリフォーマットにおけるより複合のタンパク質構造を模倣する際に有利である。分子間ジスルフィド結合を介して接続される重鎖及び軽鎖対(ヘテロ二量体)のホモ二量体である、ヒトIgGのタンパク質のような、複合タンパク質の変異型をスクリーニングする高処理方法を生成するために、当該技術を前進させる要求が引き続き存在する。現在まで、繊維状ファージ上の完全な抗体重鎖の正確なアセンブリ及び提示を実証することは可能でなかった。本発明のライブラリ及び方法は、総合設計、アセンブリ技術、及びファージpIX Fabライブラリを組み合わせることにより、これらの要求を満たす。
【課題を解決するための手段】
【0006】
本発明は、M13コートタンパク質、pIXを用いた、繊維状ファージ上での二量体ジスルフィド結合タンパク質及びより複合の構造の提示のための簡易な手段を提供する。本発明において、提示されるタンパク質は、pIXコートタンパク質を含む融合タンパク質であり、CH2ドメインのような、折り畳まれたドメインは、ヒンジドメインのような、システイン残基を含む多量体化ドメインに結合される。具体的な実施形態では、二量体タンパク質は、ホモ二量体であり、ここでメンバーは、ジスルフィド結合され、タンパク質は抗体Fcを含む。別の実施形態では、ホモ二量体ジスルフィド結合タンパク質は、ヒト抗体タンパク質を含み、ここで少なくともヒンジドメイン及び定常ドメインは、ホモ二量体を含むポリペプチドの各々において存在し、かつ任意に、ホモ二量体構造は更に、ジスルフィド結合形成によって独立して発現される抗体軽鎖と会合する。
【0007】
本発明は、pIXコートタンパク質をコードする配列と融合した外因性ポリペプチドをコードする配列を有する、少なくとも1つの融合タンパク質をコード化する複製可能なベクターを提供し、ここで外因性非ファージタンパク質部分は、ホモ二量体形成ポリペプチド鎖である。一実施形態では、融合したホモ二量体形成ポリペプチドは、Fc融合タンパク質を形成する。別の実施形態では、ホモ二量体構造は更に、ヘテロポリペプチドと会合して、より複合の構造を形成し得る。一態様では、単一のファージ分子における抗体重鎖ポリペプチド及び軽鎖ポリペプチドの両方の提示は、完全なIgG分子等であるが、これらに限定されない、ファージ粒子の表面における機能的抗体分子のアセンブリをもたらす。二量体ジスルフィド結合タンパク質として、ファージの表面上で融合ポリペプチドを提示することができる複製可能なベクター及びファージ粒子を含有する宿主細胞が、本発明に含まれる。ベクターは任意に、提示されたポリペプチド−コートタンパク質融合をコードするポリヌクレオチド配列に操作可能に融合した、分泌シグナルをコードするポリヌクレオチドを含む。
【0008】
抗体組成物の選択及び改善のためのアセンブリ、スクリーニング、並びに当該技術分野において実施されるような他の尋問技術に有用な、二量体ジスルフィド結合タンパク質のpIXファージディスプレイのデノボライブラリを構築するための方法及びベクターもまた提供される。一実施形態では、ファージ粒子表面上で多量体構造を形成することができる複数個の異なる融合ポリペプチドを提示するファージ粒子を含有する宿主細胞のライブラリは、pIXタンパク質に結合される。一態様では、ライブラリは、ファージミド系においてコードされる。
【0009】
一実施形態では、本発明のライブラリは、重鎖可変領域のライブラリを含み得、それは更に、軽鎖可変領域のライブラリを含み得、それは更に、変異型Fc領域のライブラリを含み得る。本発明のライブラリは、標的リガンドへの変化した結合、又はエフェクター分子(例えば、FcγRs及び/又はC1q)に対する変化した結合を有するような、所望の、増強された、又は軽減された特性を有するタンパク質をコードするライブラリから、ポリヌクレオチドを特定及び単離するために、パンニング、分類、又は他の選択手順に供されてもよい。
【図面の簡単な説明】
【0010】
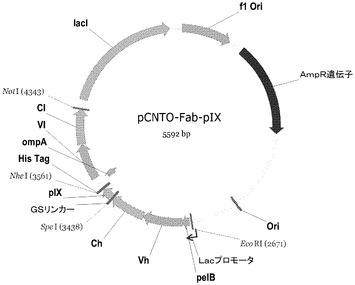
【図1】pIXで繋ぎ止められたFabを発現するために使用される出発ベクターのダイアグラム。
【図2】A〜Bは、lacZプロモータ;細菌シグナルペプチド、pelBの上流に付加されたリボソーム結合部位(RIBS);Fcポリペプチドをコードするポリヌクレオチド配列、及びファージマイナーコートタンパク質pIX又はpVIIを接続する、可動性リンカー(G4S)の位置;並びにpIX上の完全IgG構造の発現のためのジシストロニックなファージミドベクター(B)の相対的な位置を示す、Fc形成構築物(A)のためのpIXファージミドベクターの概略図。
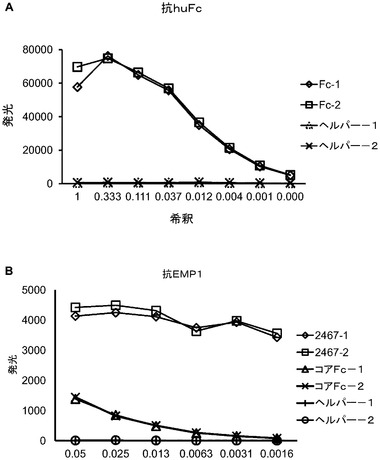
【図3】A〜Bは、実施例1に記載されるように構築された組み換えファージ粒子に対する、ELISAの結果を示すグラフであり、組み換えファージ粒子が、抗Fc Mab(A)で又はCNTO 3443、抗EMP1 mAb(B)でコーティングされたプレート上に捕捉され、捕捉されたファージが、HRP共役した抗pVIII mAbを用いて検出された、Fc融合タンパク質(A)又はEMP−1−Fc(B)の増加を実証している。ヘルパーファージは、BにおけるFcファージと同様に陰性対照であった。この実施例において、ファージの2つの個々の調製物が使用された。
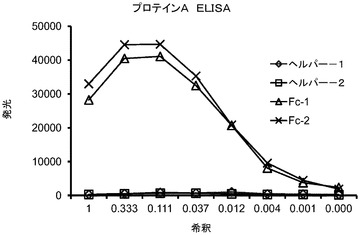
【図4】ファージ上に提示されたFcドメインが、プロテインAに結合できることを示す結合アッセイからのグラフ。
【図5】A〜Bは、最適な結合酸性度、pH 6.0(上部)で、及び非特異的な結合条件下、pH 7.5(下部)で行われた、FcRn結合アッセイからのグラフ。
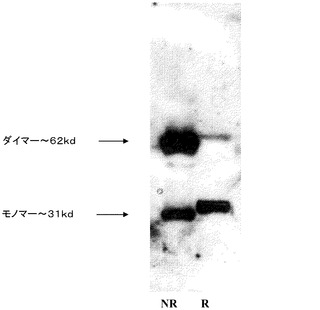
【図6】検出のために抗ヒトFc抗体を用いたウェスタンブロットを示し、非還元条件、レーン1、NR;及び還元条件、レーン2、Rの下で、単離及び電気泳動されたタンパク質の二量体的性質を実証し、非還元下でメジャーバンドが、還元条件下でのメジャーバンドのおよそ2倍の分子量であることを示している。
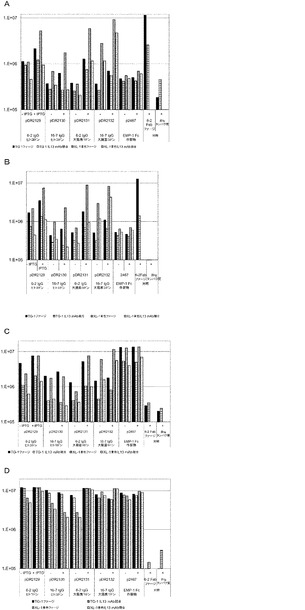
【図7】A〜Dは、ファージ上で発現された抗体ドメイン、発現されたEMP−1−Fc構築物、又はファージ自体のいずれかに特異的な種々のリガンドを用いて指定の調製物から捕捉され、lAc誘導物質IPTGである(A)抗FD(CH1)抗体捕捉;(B)抗カッパ抗体;(C)抗CH2抗体;及び(D)抗CH3抗体を用いて又は用いずに培養された、ファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。pIX上で6−2 Fab又は非免疫グロブリンタンパク質を提示するファージが陰性対照として含まれる。
【図8】抗IL13 IgG pIXと同じ特異性で競合する可溶性抗IL13 mAb(格子縞棒)の存在又は不在下で、商業用の抗IL13抗体を用いて指定の調製物から捕捉されたファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。EMP−1−Fc構築物は、陰性対照であり、IL13に結合せず、IL13特異的6−2 Fabは、pIX融合物が陽性対照として含まれるため、ファージ上でpIXを提示した。
【図9】A〜Bは、商業用の抗IL13抗体によってプレート中に捕捉され、続いてファージ上で漸増量の競合する抗IL13抗体(6−2完全IgG)、又はIL13(抗EMMPRIN)(A)に特異的でない対照抗体を付加され、またIL13によって、商業用の抗IL13抗体によってプレート中に捕捉され、続いてファージ上の6−2 Fabを付加されたファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。漸増量の抗IL13 mAb又は抗EMMPRIN mAbのいずれかが付加された(B)。
【図10】ビオチン化IL13又はIL17A抗原を使用してIL13又はIL17Aの完全IgG構築物を提示するファージを捕捉した後に、ドメイン特異的な抗体抗Fd、抗カッパ、抗CH2、及び抗CH3のいずれかによって、及び競合する可溶性抗IL13 mAb又は抗IL17A mAbの存在又は不在下で捕捉されたファージからのシグナルを示す。ファージは、抗M13抗体(y軸)で検出された。
【0011】
【表1】
【発明を実施するための形態】
【0012】
略語
ADCC=抗体依存性細胞媒介型細胞傷害性、ADMC=抗体依存性単核細胞媒介型細胞傷害性、c1q=補体因子1q、EPO=組み換えエリスロポエチン、FcR=Fc受容体、Ig=免疫グロブリン、Hc=重鎖、Lc=軽鎖、IPTG=イソプロピルチオ−β−ガラクトシド。
【0013】
定義
本明細書で使用されるとき、別段の指示がない限り又は文脈から明白でない限り、抗体ドメイン、領域、及び断片は、当該技術分野において周知であるような標準的な定義に従う。本発明のタンパク質は、1つ以上の免疫グロブリンクラスの抗体に由来するか、又はその部分を組み込む。免疫グロブリンクラスには、IgG、IgM、IgA、IgD、及びIgEアイソタイプ、またIgG及びIgAの場合は、それらのサブタイプ、例えば、IgG1、IgG2、IgG3、及びIgG4。
【0014】
が含まれる。「シストロン」とは、アミノ酸配列をコード化し、上流及び下流DNA発現制御要素を含む、DNA分子におけるヌクレオチドの配列を意味する。
【0015】
「外因性ポリペプチド」又は「外因性タンパク質」又は「細胞外タンパク質」とは、通常は野生型繊維状ファージゲノムによってコードされず、むしろ通常のファージタンパク質にとって異質であるタンパク質を意味する。典型的な外因性ポリペプチドは、それらがとりわけ、CH3、CH2、ヒンジ領域、及び/若しくはCH1ドメイン又はそれらの断片を含み得るFcドメインとして、天然に発生するように、抗体免疫グロブリン重鎖(Hc)ドメイン若しくは免疫グロブリン軽鎖(Lc)ドメイン、免疫グロブリン重鎖可変ドメイン(VH)、免疫グロブリン軽鎖可変ドメイン(VL)、天然若しくは合成ポリペプチド、単鎖抗体(scFv)、又は免疫グロブリンドメインの配列若しくは組み合わせを含む、目的とする任意のポリペプチドである。
【0016】
「Fc」により、消化されたIgGの結晶化可能な開裂断片に与えられる標識であり、抗体定常ドメインに由来し、かつジスルフィド結合の鎖間結合を有するポリペプチド鎖の二量体構造を含む、抗体の機能的断片を意味する。ヒトIgG1において、パパインは、Cys226に対する断片C末端を作り出す(参照により本明細書に明示的に組み込まれる、Kabatら、Sequences of Proteins of Immunological Interest,5th Ed.Public Health Service,National Institutes of Health,Bethesda,Md.(1991)にあるようなEUインデックスを用いて付番される)。「KabatにあるようなEUインデックス」は、ヒトIgG1 EU抗体の残基付番を指す。FcのN末端残基の定義は、異なり得るが、第1の鎖間結合システイン(Kabat系におけるC226)に対してN末端の第3残基である、Kabat付番系における少なくとも残基223を含むことが概して理解される。分子のFc部分は、抗体のその特異的な標的抗原との接触に直接に関与しないが、エフェクター機能を媒介する。これらの機能は、次の2つの種類からなる:(1)C1q結合及び/又はIgGに対するFc受容体γ−タイプ結合、IgEに対するFc受容体ε結合、及びIgAに対するFc受容体α結合に続く、補体依存性細胞傷害性(CDC)活性若しくはADCC及びADMCのような、抗体の抗原への結合を要求する機能、並びに(2)FcRnに結合し、細胞及び組織障壁(腸のような)を越えて経細胞輸送される能力による循環における持続性のような、抗原結合から独立した機能。特に、Fcの融合を介して、抗体分子又は他の分子の血清半減期を有意に増加させる能力は、非常に有利である。より長命の分子は、臨床治療に必要とされる量を低減させ、それによって投与の頻度を低減させ得る。
【0017】
用語「Fc受容体」又は「FcR」は、抗体のFc領域に結合する受容体を説明するために使用される。FcRには、FcγRI、FcγRII、及びFcγRIIIサブクラスが含まれ、これにはこれらの受容体の対立遺伝子変異型、あるいはスプライスされた形態を含む。FcγRII受容体には、FcγRIIA(「活性化受容体」)及びFcγRIIB(「阻害受容体」)が含まれ、それは主にその細胞質ドメインにおいて異なる類似したアミノ酸配列を有する。活性化受容体FcγRIIAは、その細胞質ドメインにおいて免疫受容活性化チロシンモチーフ(FAM)を含有する。阻害受容体FcγRIIBは、その細胞質ドメインにおいて免疫受容抑制性チロシンモチーフ(ITIM)を含有する(Daeron、Annu.Rev.Immunol.,1997,15:203〜234における概説を参照されたく、FcRsは、Ravetch及びKinet、Annu.Rev.Immunol.,1991,9:457〜92、Capelら、Immunomethods,1994,4:25〜34、及びde Haasら、J.Lab.Clin.Med.,1995,126:330〜41において概説され、これらの各々は参照により本明細書に組み込まれる)。
【0018】
「融合ポリペプチド」又は「融合タンパク質」とは、操作可能に結合される、それぞれ第1及び第2の核酸配列によってコードされる第1及び第2のポリペプチドを含む、融合ポリペプチド(タンパク質)を意味する。本明細書で使用されるとき、融合タンパク質は、「操作可能に結合」される構成成分及びドメインを含有し、例えば、ポリペプチド又はポリヌクレオチドの融合した要素が、各々が意図されるように働く又は機能するように結合されることを意味することが理解される。例えば、プロモータ、オペレータ、又はエンハンサのような、発現を調節する要素は、その発現が調節されるべきヌクレオチド配列に操作可能に結合され得る。要素間の及び要素の間の結合は、直接的であっても、又はリンカーを介するような間接的であってもよい。これらの要素は、必ずしも隣接していない。
【0019】
用語「ライブラリ」は、変異型である、すなわち、ある種の領域が同じであるか又は類似しており、他の領域が異なる、コードされたタンパク質の集積を意味する。変異領域は、指向性又は無作為の変異(確率的又は非確率的変化)によるものであってもよい。ライブラリ又は変異型は、異なる変異型の数又はライブラリの「サイズ」の観点から説明することができる。有用なデノボ抗体ライブラリは、高い多様性(>1010)を有し、変化に応じやすく、組立が容易であり、所望でない配列のバックグラウンドが低い。次の方法を組み合わせることにより、ライブラリアセンブリが加速され、低バックグラウンドをもたらされる:(a)Kunkel系単鎖突然変異誘発、(b)制限部位を有するパリンドロームループ、及び(c)メガプライマーアプローチの使用。
【0020】
「ファージミド」又は「ファージベクター」は、プラスミドに由来するもののような、ファージ染色体及び外因性DNAの両方に由来する構成成分を含有するクローニング及び発現ベクターである。ファージミドは、ファージゲノムの部分を含有するため、宿主のヘルパーファージとの共感染時に、それはファージ粒子中にパッケージ化され得る。本発明のファージミドは、ファージM13粒子中にパッケージ化され得る。ファージミド又はファージベクターは、本明細書における融合タンパク質をコードする核酸又は本明細書で提供される発現カセットのような、非相同のDNAの挿入又は組み込みによって操られている。そのような発現ベクターは、典型的には、宿主細胞中の挿入された核酸の効率的な転写のためのプロモータ配列を含有する。
【0021】
概論
二価抗原結合タンパク質である天然抗体は、重鎖の適切な会合のために、Fc定常ドメイン及びヒンジ領域に依存する。CH2及びCH3ドメインは、好ましくは、国際公開第2005005604号に記載されるもののような、又は天然又は操作された抗体配列の配列を含む検索データベースによって見出され得るもののような、ヒト生殖細胞系配列に由来する。広くは、本発明のタンパク質構築物は、1つ以上の定常ドメイン又はその部分に結合されたヒンジ領域を含む。能力受容体に結合し、体内の持続性を増加させる能力のようなすべての関連する機能を保持するために、Fcにおいて通常存在するすべての定常ドメイン:1つ以上のシステイン残基又は他のスルフィド若しくはセレノスルフィド結合形成残基を含有する、配列番号1〜4に示されるヒンジ又はその部分;配列番号5〜8によって例示されるCH2又はその変異型、及び配列番号9〜12によって例示されるCH3又はその変異型を組み込むことが通常は所望される。本明細書で提供されるものによって表される配列は、非限定的であり、天然及び変異型抗体ドメイン配列は、インターネット上の種々のデータベースにおいて、又は本発明の実践において有用であり得る多数の公報において見出され得ることが当業者によって理解されよう。加えて、構築物は任意に、CH1ドメインの幾つか若しくは、すべてを含んでもよく、又は配列番号13のもののように、抗体可変ドメインの幾つか若しくはすべてがまた存在してもよい。当然のことながら、これらのドメインは、完全IgG構築物において存在するであろう。抗体ドメインの適切な発現及び折り畳みに対する必要に応じて、配列番号14(pelB)及び15(ompA)のアミノ酸配列をコードするもののような、他の抗体配列及び非抗体配列が含まれてもよい。しかしながら、本発明は、ある種の定常ドメインのみを含み、その他を含まない構造、並びに非抗体由来のドメインが存在し得る構造を企図する。
【0022】
種々のFc機能は、Fcの異なる部分に依存するため、完全未満の機能性が所望される場合、より少ないCHドメインを重鎖に組み込むことができる。例えば、補体の有意な活性化は、IgGのCH2又はIgMのCH3を要求する。本発明はまた、修飾されたヒンジ及び重鎖が安定な複合で会合し得る限り、置換、欠失、挿入、又は修飾されたアミノ酸を有し得るFc重鎖ドメインの使用を企図する。
【0023】
加えて、典型的にはジスルフィド結合構造として形成されるであろう二量体共有結合構造もまた、セレノシステイン結合、ホモシステイン結合、又は混合スルフィド−セレニド結合によって形成され得る。鎖間共有結合残基を含む抗体ヒンジに加えて、他の多量体化ドメインを置換して、二量体又はより高秩序の構造を形成してもよい。これらの多量体化ドメインは、ファージ粒子の表面上での細胞外タンパク質−コートタンパク質融合タンパク質のポリペプチドの会合を補助するために、単独のシステイン若しくはセレノシステイン残基のように天然若しくは人工であってもよく、又はロイシンジッパーモチーフのようにモチーフを含んでもよい。
【0024】
完全抗体タンパク質の場合は、重鎖−軽鎖ヘテロ二量体は、特定の重鎖定常ドメインを介して会合して、より高秩序の構造を形成する。例えば、IgG型抗体は、四量体構造における共有結合によって接合される2つの重鎖−軽鎖ヘテロ二量体を含む。ある種の他の抗体型は、例えば、2個の四量体(IgA)又は10個の四量体(IgM)を含む、より高秩序の構造中に組み込まれる類似した四量体構造を含む。
【0025】
ファージコートタンパク質を用いて大型の細胞外タンパク質分子を提示する際に、提示されるタンパク質は、コートタンパク質の全コピーに結合される場合、組み換えファージ粒子のアセンブリを干渉する可能性がある。アセンブリ干渉を回避するために、pIXディスプレイについて(Gaoら、Proc Natl Acad Sci USA,99:12612〜12616,2002)によって記載されるようなファージミド系を使用してもよく、それによって野生型及び細胞外タンパク質結合コード配列の両方がベクターにおいて存在し、両方のタンパク質が組み換えファージ粒子中に組み込まれる。
【0026】
本発明の適用は、予想外に、本明細書に記載されるような抗体のFc部分を形成する抗体構成成分が、繊維状ファージ粒子の表面上のpIX又はpVIIコートタンパク質に対する融合タンパク質として、Fc受容体結合のような、天然抗体のFcドメインの既知の生物学的活性を提示するホモ二量体ジスルフィド結合タンパク質として、提示され得、また二価抗原結合タンパク質の形態であるとき、抗原結合の能力があることを見出している。故に、ファージ粒子上での抗体結合断片の単量体一価の提示とは対照的に、多価のタンパク質提示の多量体の提示が企図される。故に、本発明は、天然抗体の機能的特徴のより完全なスペクトルの間の操縦及び選択のための体系を提供する。そのような特徴には、細胞表面上で目的とする特定の抗原を提示する、細胞に対して向けられる免疫反応を促進し、製造される抗体様治療薬の生物活性の重要な構成成分である、Fc機能が含まれる。免疫系エフェクター細胞には、細胞免疫反応を活性化するT細胞のような抗原特異的細胞、並びに細胞免疫反応を媒介するマクロファージ、好中球、及びナチュラルキラー(NK)細胞のような非特異的細胞が含まれる。
【0027】
本発明を作製する方法
繊維状ファージ粒子上に提示された融合タンパク質において、外因性ポリペプチドと繊維状ファージpVII又はpIXタンパク質との間の「融合」は、アミド結合によって直接に結合されてもよく、又はリンカーポリペプチド(すなわち、「リンカー」)を含んでもよい。典型的に一続きの長さ約5〜50のアミノ酸である様々なリンカーのいずれを使用してもよい。特に好ましいリンカーは、そのリンカーの位置で融合タンパク質に対して大きな可動度を付与する。リピートの数が典型的には1〜12であるG4S(Gly−Gly−Gly−Gly−Ser)リピート又はG3S(Gly−Gly−Gly−Ser)を有するもののような、支配的にグリシン(G、Gly)残基からなるもののような、二次構造が欠けているリンカーを、この目的のために使用してもよい。
【0028】
第1のポリペプチドは、外因性タンパク質であり、第2のポリペプチドは、繊維状ファージpVII又はpIXタンパク質であり、それによって外因性タンパク質は、繊維状ファージタンパク質のアミノ末端に融合される。更に、融合タンパク質が未成熟形態であるとき、すなわち、リーダー配列がプロセス(除去)されていないとき、融合タンパク質はまた、本明細書に記載されるような野生型又は突然変異体pelB又はompA配列(それぞれ配列番号14及び15)等のような、アミノ末端原核分泌シグナルを含有し得る。
【0029】
天然抗体において、軽鎖ポリペプチド及び重鎖ポリペプチド鎖は、別個にコードされ、発現される。分子の典型的なヘテロの二量体構造IgGクラスは、分子の4つのポリペプチド鎖、2つの重鎖及び2つの軽鎖の中で及びそれらの間での、ジスルフィド結合の適切なアセンブリ及びその形成に依存している。故に、本発明において、抗体の二量体Fc部分のアセンブリ及び/又は軽鎖の会合は、存在するとき、タンパク質の個々のドメインが自己会合し、それらの間にジスルフィド結合を形成する限りにおいて、抗体形成の天然プロセスを再現する。
【0030】
一実施形態では、繊維状ファージ粒子の表面上に提示されるべきFc含有タンパク質は、天然抗体であり、ジシストロニックなベクターは、Fc構築物−pIX融合タンパク質並びに自己会合するであろう、別個にコードされ、発現された抗体Lc又は抗原結合ドメインの発現のために構築される。本発明の抗原結合タンパク質は、任意のエピトープ、抗原部位、又はタンパク質のための結合部位を有することができる。好ましい抗原結合タンパク質は、受容体への直接結合によって、又はそれらの同族のリガンド(複数可)への結合によって、受容体タンパク質の活性化を中和する。概して、抗原結合ドメインは、Fcドメインを含む天然抗体Hc配列と融合した抗体Lc及び抗体Hc可変ドメインから形成されるであろう。別の実施形態では、pIX融合タンパク質は、Fcドメインに結合されたscFvを含む。本発明の別の態様では、sscFvを含む重鎖及び軽鎖の抗原結合部位は、2つの異なる結合特異性を提供し、それによってファージ表面に提示された自己組立されたジスルフィド結合構築物タンパク質を、二特異的かつ二価の分子にするように異なってもよい。例えば、IgG分子のVL及びVHドメインの代わりに置換されるのは、異なる特異性のscFvドメインであり、これにより結果として生じる分子が2つの異なるエピトープに同時に結合することができるようになる。本発明の方法を用いてファージ粒子上に提示され得る、多数の可変ドメイン対を有する二特異的な抗体分子を作り出す他の方法は、参照により本明細書に組み込まれる米国特許第20020103345A1号に教示される。
【0031】
一実施形態では抗原結合又は受容体結合ドメインは、抗体ドメインに由来しないが、Fcドメインに融合される既知の又は無作為ペプチド配列である。Fcの代替鎖と結合されて、所望によりその間にリンカー部分を有する生物活性ペプチド類は、同一であっても異なってもよい。生物活性ペプチド類は、最終共役物が所望の生物活性を示すのであれば、介在するリンカーと又はペプチド上のいずれかの残基から生じるFcと結合していてもよい。生物活性は、例えば、結合活性についてはインビトロアッセイにより、又は動物の疾病モデルでのようにインビボ活性により、あるいは共役物を投与した後の被験者の反応により、測定することができる。
【0032】
同時係属中の出願、国際公開第04/002417号、同第04/002424号、同第05/081687号、及び同第05/032460号の出願者は、本明細書でMIMETIBODY(商標)構造と称される構造を説明し、これらの参考文献の各々は、参照により本明細書に全体的に組み込まれ、これらの構造は、pIX又はpVIIファージコートタンパク質に融合され、ファージ粒子の外表面上に提示され得る、本発明の二量体ジスルフィド結合構造として含まれる。
【0033】
一実施形態では、MIMETIBODYは、生物活性ペプチドリンカー−ヒンジ−CH2−CH3ポリペプチドの対を含み、この対は、会合又は共有結合、具体的には、Cys−Cysジスルフィド結合によって結合される。生物活性ペプチドは、任意の長さの上に(長さで?)あってもよく、任意の種に由来する天然産の配列であっても、人工配列であってもよい。ペプチドは概して、ファージミドベクターによってコードされ、ファージ粒子上での提示のために構築物のFc部分に融合されるであろう。そのような組成物の一例としては、生物活性ペプチドとしてのEPO模倣ペプチドが挙げられる。故に、EPO模倣のCH1欠失MIMETIBODYは、治療用ペプチド及びその本来の又は後天性の生体外、生体内、又はその場での特性又は活性を提示しながら、その本来の特性及び機能により抗体構造を模倣する。ペプチドが既知の生物活性を有さないが、マーカー、タグ、抗原としての機能を呈示するか、又はレポーター基、キレート基等を提供する、類似した構造の他の構築物もまた、本発明によって包含される。
【0034】
典型的な実施形態では、Fc含有融合タンパク質又は「MIMETIBODY(商標)」は、幾つかの又は全体的な免疫グロブリンCH1ドメインが不在である式(I):
V1o−Pepa−Flexn−V2m−ヒンジ−CH2−CH3 (I)を含み、
式中、Pepは、標的を特異的に認識することができる生物活性ペプチド又はポリペプチドを表し、Flexは、MIMETIBODYが代替的な配向及び結合特性を有することを可能にすることによって、構造的な可動性を提供する任意の可動性リンカーポリペプチドであり、V1及びV2は、ブラケティング配列であり、ヒンジは、免疫グロブリンヒンジ領域の少なくとも一部分、例えば配列番号1〜4であり、CH2は、免疫グロブリンCH2定常領域の少なくとも一部分、例えば配列番号5〜8であり、CH3は、免疫グロブリンCH3定常領域の少なくとも一部分、例えば配列番号9〜12であり、m、N、及びoは、ゼロであり得るか、又は1〜10の間の整数であり得、かつ1〜10までの整数であり得る。Pep配列は、任意に、目的又は安定化、又は任意の数の生物物理学的な機能のための配列を含むことができる。典型的な実施形態では、ブラケティング配列は、Vhフレームワークのような抗体可変(V)ドメインに由来し、V1は、配列QIQであり、V2は、免疫グロブリンJ遺伝子ドメインに由来する配列を表し、GTLVTVSS(配列番号13)である。結果として生じるポリペプチドは、Cys−Cysジスルフィド結合等であるが、これらに限定されない、会合又は共有結合によって他のポリペプチドに結合することができる。
【0035】
pIX融合タンパク質の発現のレベルは、転写レベルにおいて追加的に制御することができる。融合タンパク質は、Lac Zプロモータ/オペレータ系の誘導可能な制御下にある(図1を参照されたい)。他の誘導可能なプロモータも同様に作動することができ、それらは当業者に既知である。高レベルの表面発現については、サプレッサーライブラリが、イソプロピルチオ−β−ガラクトシド(IPTG)のようなLac Zプロモータの誘導物質中で培養される。誘導可能な制御は、非発現条件下でライブラリを培養することによって、非機能的pIX融合タンパク質に対する生物学的選択が最小化され得るため、有益である。次いで発現は、ライブラリ内の抗体の全体的な集団がファージ表面上に正確に表されていることを確実にするためのスクリーニングの時にのみ誘導することができる。
【0036】
二量体化ポリペプチドファージコートタンパク質融合タンパク質をコードするベクターは、細胞外タンパク質及びファージコートタンパク質コード領域の接合部において、翻訳終止コドンを含み得る。対応する翻訳終止サプレッサーを担持する細菌細胞中で発現されるとき、融合タンパク質が産生される。対応する翻訳終止サプレッサーのない細菌細胞中で発現されるとき、遊離細胞外タンパク質は、産生されない。
【0037】
本発明の使用方法
本明細書に出発点として例示されたファージベクターを用いて、タンパク質は、分子のライブラリを生成するための指向性突然変異誘発を用いて特異的な個別的な残基位置で、又はNXT配列と一般的に称されるN結合グリコシル化配列のような領域で、多様であってもよい。各々が異なる細胞外タンパク質配列を保有する数十億の大腸菌コロニーを生成するために使用され得る、修飾されたKunkel突然変異誘発法が、特に有用である。効率的である一方で、高度に複合の配列ライブラリを生成するとき、突然変異誘発されていない親DNAの割合は増加する。加えて、長いオリゴヌクレオチドの合成の技術的制限は、遠位領域において配列多様性を含有するライブラリを作製するために使用されるとき、本方法の有効性を低下させる。これらの制限を克服するために、350塩基を超えるオリゴヌクレオチドを生成する追加的な技術を使用することができる。これらの技術には、米国特許第20050048617号に記載されるような、標準的なKunkel突然変異誘発法(Kunkelら、1987 Methods Enzymol 154:367〜382)と組み合わせた、メガプライマーの使用及び突然変異誘発テンプレートにおいて制限酵素認識部位を含有するステム−ループ配列の作製が含まれる。制限クローニング(Marksら、1991 J.Mol.Biol.222:581〜597、Griffithsら、1994 EMBO J.13,3245〜3260、Hoetら、2005 Nature Biotechnol 23,344〜348)、ファージ組み換え(Gigapack,Invitrogen)、及び配列特異的組み換えのような他のライブラリ技術と比較して、改善されたKunkelに基づく方法は、配列多様なライブラリ(109を超える)を生成する際に、有意により有効であり、標的化されたDNA中の任意の場所に配列多様性を導入するのにより万能である。
【0038】
Fc含有タンパク質の繊維状ファージ上での提示は、所望の結合特徴のために、そのような分子の大集団をスクリーニングすることが所望される場合に特に有用である。一実施形態では、Fc構築物−pIXタンパク質融合を発現する細菌細胞は、Fc構築物−pIX融合遺伝子を担持するベクターDNAの、ファージ粒子中への優先的なパッケージングを可能にするM13変異型に感染させられる。各々結果として生じるファージ粒子は、特定のFc構築物−pIX融合タンパク質を提示し、Fc構築物−pIX融合物をコードするベクターを含有する。そのようなファージ粒子の集団は、パンニング手順によって、所望の結合特徴のために強化することができる。典型的には、所望の粒子は、所望のファージ粒子がそこに結合し得る抗原によりコーティングされた固体表面上に固定化される。結合粒子は、収集され、細菌細胞を更に感染させるために使用される。パンニング手順は、所望の結合特徴のために更に強化するために反復される。
【0039】
一実施形態では、ファージライブラリは、FcRgammaIII(CD16)、FcRgammaII(CD32)、及びFcRgammaI(CD64)のような、天然又は組み換えFc受容体への増強された、減少された、又は変化した結合のために、分子のFc部分の変異型をスクリーニングするために使用される。
【0040】
ファージ及び他の抗体ディスプレイ方法は、生体外の抗原又は受容体標的に対する選択を操る機会を利用可能にする。生体外選択法の1つの特定の利点は、標的タンパク質上の多様な部位への抗体結合を得るように選択手順を操る能力である。あるいは、全細胞を使用して、バインダーを選択してもよい。
【0041】
ファージライブラリは、機能的属性に関連する遺伝物質の検索を単純化するが、ライブラリから最良の候補を単離するために多重ステップのパンニング戦略が要求される。ドメイン又はエピトープ指向性パンニングは、標的タンパク質に結合する抗体を選択する日常的な方法となっている。そのような選択は、選択的パンニング、脱選択的(de-selective)パンニング、リガンド捕捉、サブトラクティブパンニング又は道しるべ(pathfinder)選択として多様に知られる方法を利用する抗体のステップによる選択を用いることによって、主に達成されてきた。
【0042】
サブトラクティブパンニングにおいては、重複するが、完全に同一でない結合部位を有する標的(複数可)を使用して、不必要なバインダーを脱選択することができる。この戦略は、癌細胞に対するバインダーを脱選択するために通常の細胞を使用する際と同様に、未知の抗原さえにも対応するバインダーを特定するために使用されている。あるいは、関連する抗原の間で異なる又は共通の部位への抗体結合を得るために、幾つかの共通のドメイン又は構造を有する天然産のタンパク質が、連続的な又は競合選択において使用される。場合によっては、関連するケモカイン又はタンパク質の突然変異バージョンのような天然産のタンパク質が、サブトラクティブパンニングにおいて使用され得る。
【0043】
リガンド捕捉指向性パンニングは、無関係の及び隣接していないエピトープに固定化された抗体が、ファージパンニングのために好ましい標的リガンドの結合面を捕捉し、呈示するために使用されるという点で、ELISAサンドイッチアッセイに類似している(米国特許第6376170号)。その他のものは、所望の標的ドメイン以外における抗原を選択的にマスクするために競合抗体を使用している(Tsui,P.ら、2002.J.Immunol.Meth.263:123〜132)。道しるべ技術は、モノクローナル及びポリクローナル抗体、並びに西洋ワサビビペルオキシダーゼ(HRP)に直接に又は関節に共役させられた天然リガンドを使用する。ビオチンチラミンの存在下で、これらの分子は、標的抗原に極めて接近したファージ結合のビオチン化を触媒し、ストレプトアビジンを用いて全集団からの「タグされた」ファージの特異的な回収を可能にする。このようにして、標的自体に又はそのすぐ近くに結合するファージは、選択的に回収される(Osborn,J.K.ら、1998.Immunotechnol.3:293〜302)。これらの方法、方法の変形、及び当業者に既知の他の方法を用いて、本発明のpIX細胞外タンパク質のライブラリを問い合わせることができる。
【0044】
本発明は一般論として記述されてきているが、本発明の実施形態は、特許請求の範囲を限定するように解釈されるべきではない以下の実施例で更に開示される。
【実施例】
【0045】
実施例1.pIX上でのFc融合タンパク質の提示
A.ファージミドベクター構築
ファージミドベクター、pCGMT9(Gaoら、Proc.Natl.Acad.Sci.96:6025〜6030,1999,米国特許第6472147号)は、pIX融合を介したファージディスプレイのために重鎖定常ドメインを挿入することができるファージミドpIXディスプレイベクターの発達のための主鎖としての機能を果たした。このファージミドにおいて、アンピシリンに対する耐性を付与するβ−ラクタマーゼ遺伝子と共に、大腸菌(colE1)及び繊維状ファージ(f1)のための複製の源が存在する。
【0046】
MIMETIBODY(商標)分子を含む、Fc含有タンパク質を提示するためのpIXファージミドベクターを、国際公開第2009/085462号及び図1に開示させるように、2シストロン性の発現、pCNTO−Fab−pIXのために適合させられたGaoベクターに基づいて構築した。それにより可溶性軽鎖が同じ細胞中で発現され、繋ぎ止められたポリペプチドと会合する、Fabファージディスプレイのために使用された戦略とは異なり、可溶性Fcは、全く発現されなかった(図2A)。
【0047】
ベクターにおけるFab軽鎖配列は欠失された。ベクターにおけるFab重鎖配列を、Fc又は構築物又はMIMETIBODY(商標)構築物のいずれかと置き換えた。コアヒンジを含有するシステイン対を含有するファージミドベクターFc構築を次のように達成した。ヒトIgG1のコアヒンジ、CH2、及びCH3をコードするFc遺伝子セグメントを、PCRによってFc含有プラスミドから増幅した。NcoI制限部位を、5’プライマー末端部中に、及びSacII制限エンドヌクレアーゼ部位を3’プライマー末端部において組み入れた。PCR増幅したDNA断片及びファージミドベクター(pCNTO−Fc−pIXコアHg)を、NcoI及びSacII制限エンドヌクレアーゼで消化した。消化産物を生成し、迅速ライゲーションキットを用いてライゲートし、DH10B大腸菌に形質転換させた。形質転換したクローンを、DNA塩基配列決定法を用いてスクリーニングし、正確な配列を示したものを次いで、ファージ調製のために、TG−1大腸菌に形質転換した。
【0048】
制限酵素クローニングを介して、Fcをコードする配列を、完全なCNTO530融合タンパク質をコードする配列と置き換えることによって、米国特許第7393662号及びその中の配列番号88に記載され、「EPO MIMETIBODY(商標)」又はCNTO530と称される、EMP−1(配列番号16)Fc(配列番号17)構築物をコードするpIXファージミドベクター(p2467)を構築した。CNTO530コード配列を、PCRによってプラスミドp2467から増幅した。制限エンドヌクレアーゼ部位NcoI及びSpeIを、それぞれ5’末端及び3’末端プライマーに含めた。PCR産物及びファージミドベクター、pCNTO−Fc−pIXコアHgをNcoI及びSpeIで消化し、精製し、迅速ライゲーションキットを用いてライゲートし、DH10B大腸菌に形質転換した。形質転換クローンを、DNA塩基配列決定法によってスクリーニングし、正確な配列を有するものをファージディスプレイのためにTG−1大腸菌に形質転換した。
【0049】
B.組み換えファージの調製及び特徴付け
ファージミドベクターでトランスフェクトしたTG−1大腸菌を、液体培養でOD600=0.5〜0.6になるまで増殖させた。VCSM13ヘルパーファージストックを培養物に添加し、感染は、静的インキュベーションとして、37℃で45分間進行させた。培養物を遠心分離にかけて、細菌をペレット化し、カルベニシリン、カナマイシン、及びIPTGを補充した培地中に再懸濁させ、250RPMで振盪しながら、30℃で12〜16時間インキュベートした。一晩の培養物を遠心分離にかけ、ファージ含有上清を未使用の管に移し、そこに10分の1の容量の塩化ナトリウム/PEG溶液(NaCl及びPEGの濃度は?又は単に標準的な方法を用いて沈殿させたPEGと述べる(Ref))を添加した。各管を混合し、時折混合しながら氷上でおよそ3時間インキュベートし、その後、この管を遠心分離にかけてファージをペレット化した。ファージペレットを、PBS中に注意深く再懸濁させ、新たな管に移し、2回目の遠心分離にかけてあらゆる残存する細胞残屑を除去した。精製しファージを、−80℃のアリコート中で保管した。スポット滴定を行って、ミリリットル当たりのコロニー形成単位(cfu)としてファージ力価を推定した。
【0050】
C.提示されたタンパク質の特徴付け
Fc及びペプチド−Fc構築物の提示の確認
この実施例において、ファージの2つの個々の調製物が使用された。Fc支持又はCNTO530支持ファージを検出するために、黒色ELISAプレートを、抗ヒトFcγ特異的ポリクローナル抗体又は抗EMP1ペプチドモノクローナル抗体(CNTO 3443)のいずれかでコーティングした。コーティングしたプレートを、TBST中の5%ミルクでブロックし、TBSTで洗浄した。ヘルパーファージ、Fc−又はCNTO530組み換えファージをプレートに添加し、室温で1時間インキュベートし、洗浄して非結合ファージを除去した。結合ファージを、HRP共役した抗M13 mAb及び化学発光基質で検出した。捕捉されたファージを、HRP共役した抗pVIII mAbを用いて検出した。ヘルパーファージ、及びCNTO530支持ファージの場合は、EMP1ペプチドを有さないFc組み換えファージを、陰性対照として使用した。
【0051】
プロテインA結合。精製した組み換えプロテイン−Aを、黒色ウェルのELISAプレート上に一晩、4℃でコーティングした。コーティングしたプレートを、TBST中の5%ミルクでブロックし、TBSTで洗浄した。ヘルパーファージ又はFc提示ファージの適切な希釈物をプレートに添加した。プレートを室温で1時間インキュベートし、洗浄して非結合ファージを除去した。コーティングしたプロテイン−A上のあらゆる残存する非占有のFc結合部位をブロックするために、ヒト抗体由来のFcを飽和濃度でプレートに添加した。30分のインキュベーション後、結合ファージをHRP共役した抗M13 mAb及び化学発光基質で検出した。
【0052】
FcRn結合。FcRn(新生児型Fc受容体)は、抗体再取り込み、区画転座、及び再循環を可能にし、故に、抗体の循環半減期を持続させる。FcRnに結合するFcは、pH依存性であり、ELISA結合アッセイをそれに応じて行った。FcRn結合ファージを、ニュートラアビジンでコーティングした96ウェルのプレート上に捕捉し、HRP共役した抗pVIII mAbで検出した。簡潔に述べると、黒色ウェルのELISAプレートを、ニュートラアビジンでコーティングし、SuperBlock T20(TBS)及びケミブロッカーの50/50混合物でブロックした。プレートをTBSTで洗浄し、ビオチン化FcRnを1時間捕捉した。ヘルパーファージ又はFc提示ファージの適切な希釈物を、TBST中、pH=6又はpH=7.5で調製した。非結合FcRnを、プレートから洗浄し、ファージを添加し、1時間インキュベートした。あるいは、プレートへの添加前に、ビオチン化FcRnをファージと室温で1時間混合した。コーティングしたFcRn上の残存する非占有のFc結合部位をブロックするために、ヒト抗体由来のFcを飽和濃度でプレートに添加した。結合ファージを、HRP共役した抗M13 mAb及び化学発光基質で検出した。
【0053】
D.結果
Fcを提示するファージは、抗Fc抗体を用いて捕捉し、抗pVIII抗体を用いて検出したため、ELISAアッセイは、Fcを提示するファージの比率を示すように設計した(図3A)。Fc組み換えファージについて観察された強力なシグナル及びヘルパーファージについて観察されたシグナルの欠如は、Fcが効率的にファージ表面上に提示されたことを実証する。EMP1融合タンパク質構築物、CNTO530、提示を、図3Bに示される捕捉リガンドとしてEMP−1特異的抗体を用いて確認した。Fc領域が適切な生物学的活性を保持し、故に、二量体であることを確認するために、特異的な結合アッセイを行った:プロテインA結合、及びFcRn結合。図4に示されるように、その表面上に提示されたFcを有するファージは、プロテイン−Aに結合する一方で、Fcを欠く対照ヘルパーファージは、それに結合しない。プロテインA結合についての化学発光シグナルは、ヒト免疫グロブリンγ特異的なポリクローナル抗体で捕捉されたFc提示ファージのそれに類似しており、ファージ提示されたFcの大部分が、プロテインAへの結合に適格な立体配座へと折り畳まれることを示唆する。
【0054】
Fcは、pH 6.0でFcRnに結合するが、pH 7.5で数桁の結合親和性を失う。ファージを、pH 6.0(図5A)又はpH 7.5(図5B)のいずれかで、ビオチン化FcRnでインキュベートした。pH 6.0で観察された強力なシグナルによって実証されるように、Fc組み換えファージは、pH 6.0でFcRnに効率的に結合した。対照的に、同じファージは、試験したすべての濃度で、それよりもはるかに低いシグナルを示した。したがって、pH依存性結合は、pIXファージミド系を用いて提示されたFcについて保持された。
【0055】
IgG及び他のFc含有分子は、CH3ドメインの相互作用を介してホモ二量体を形成する。ホモ二量体は、そのコアヒンジ領域における2つのジスルフィド結合によって安定化される。Fcファージミドディスプレイベクターは、Fc遺伝子の単独コピーのみをコードするため、ウェスタンブロットを介して提示されたFcの凝集状態を検査した。濃縮されたファージ粒子を、還元又は非還元条件下でSDSゲル上に直接充填した。図4に示されるように、非還元条件下で、タンパク質の大部分は、二量体Fc−pIX融合タンパク質について予想されたように、約62kDの分子量を有する二量体として移動した。逆に、還元条件下で、Fc−pIXタンパク質の大部分は、31kDの単量体として移動した。故に、ファージ表面上に提示されたFc分子の大部分は、ホモ二量体であり、IgG又は他のFc含有分子と同じ様態で、ジスルフィド結合で共有結合される。
【0056】
E.発明の概要
組み換えファージについて観察された強力なシグナルは、ヘルパーファージについてのシグナルの欠如と一緒に、EPO受容体作用物質(EMP−1)Fc構築物が、ペプチド並びにファージ粒子上のFcの検出によって実証されるように、効率的に提示されることを実証する。データは、Fc含有タンパク質が、天然リガンドへの結合を可能にする特徴的な立体配座特徴を有するホモ二量体として、ファージ上で効率的に提示されたことを示す。
【0057】
実施例2:ペプチド−Fc融合ライブラリ
ペプチド−Fc融合ライブラリを生成するために、無作為アミノ酸配列の部位においてヘアピンループを含有する、テンプレートファージミドを生成した。ヘアピンが二本鎖DNAを形成する場所に固有の制限部位、XbaIが配置されるような方法で、ヘアピンを設計した。これは後に、XbaIでの制限消化を介してテンプレートDNAを除去し、それによって最終構築されたライブラリにおいてテンプレートファージミドが密接に詰められたファージを還元するために使用される予定であった。この細胞株を通じる継代は、ウラシルのssDNA中への組み込みを引き起こすため、二本鎖テンプレートプラスミドを、Dut−/uNg−大腸菌宿主株、CJ236に形質転換した。次いでウラシル含有ssDNAテンプレートを、最終ライブラリ宿主細胞の酵素によって分解した。プラスミドを保有する単独コロニーを、液体培養で増殖させ、それをその後、VCS−M13ヘルパーファージで感染させた。ファージをPEGと生理食塩水で沈殿させ、一本鎖DNAの精製のために使用した。
【0058】
DNAライブラリを、修飾されたKunkel突然変異誘発プロトコルを用いて生成した。無作為化ライブラリヌクレオチドをコードするオリゴマー、並びに5’及び3’隣接配列を、T4キナーゼを用いて酵素的にリン酸化した。リン酸化オリゴを、3ステップの温度低下プログラムを用いて、それらのそれぞれのssDNAテンプレートにアニールした。第2のストランド合成を、T7 DNAポリメラーゼ及びT4 DNAリガーゼを反応混合物に添加して、共有結合閉環状DNA(CCC−DNA)を形成することによって行った。CCC−DNAを精製し、次いでヘアピン配列においてXbaIで消化して、バックグラウンドを低減するためにテンプレートDNAを開裂した。消化前及び後の両方で、CCC−DNA産物をアガロースゲル電気泳動によって検査して、細胞中へのその導入の前にライブラリ調製物の質を評価した。次いでライゲーション混合物を、MC1061F’宿主細胞株(大腸菌)に形質転換した。
【0059】
4つのpIX提示されたライブラリを構築し、ここで7つ(A1及びA2)又は8つ(B3及びB4)の無作為アミノ酸ループは、各々2つのFc含有MIMETIBODY(商標)構築物(上記の式1を参照されたい)においてジスルフィド結合で拘束され、ここでリンカーは、GGSG又はGSであり、V領域J−ピース(配列番号13)は、存在するか又は不在であり、ヒンジは、隣接した配列を有するか又は有さないCPPCのコアアミノ酸、IgG1タイプのヒンジのいずれかを含む。これらの2つの変異型Fc領域は、下記に示すように表され、それは配列番号17及び18によって表され、ここで天然産のIgG4と異なる残基は下線を引かれている。2つの更なる無作為アミノ酸を、拘束ループの各末端部に付加した。
【0060】
A.7NNKライブラリ(XXCXXXXXXXCXX)
1)Fc=V領域及び完全ヒンジを有する突然変異体IgG4(配列番号18)。2)Fc=ヒンジコアを有する突然変異体IgG4(配列番号17)
B.8NNKライブラリ(XXCXXXXXXXXCXX)
3)Fc=V領域及び完全ヒンジを有する突然変異体IgG4(配列番号18)
4)Fc=ヒンジコアを有する突然変異体IgG4(配列番号17)
【0061】
生成された各ライブラリについて、総計31回の電気穿孔を行った。形質転換効率性を滴定するために小アリコートを除去した後、増殖培養物を即座に、1リットルの培養容量にまでスケールアップし、それを1.0のOD600にまで増殖させた。この時点で、培養物を次のように分割した:培養物の10分の1を、VCSM13ヘルパーファージに感染させてファージライブラリを生成した一方で、培養物の大半を使用して、細菌ライブラリのグリセロールストックを確立した。ファージ感染された培養物を増加したスケールにまで再び拡張し、一晩増殖させた。ファージライブラリを、氷上のPEG/NACl沈殿を用いて培養上清から精製した。結果として生じたファージ力価を、ミリリットル当たりのコロニー形成単位(cfu/mL)の数を測定するためにスポット滴定を用いて推定した。スポット滴定調製物からの1×10-9及び1×10-10希釈物のアリコートを、単独のコロニーを単離するためにグルコース及びカルベニシリンを補充したLB培地プレート上に広げた。各ライブラリについて、最終ファージライブラリの多様性及び機能性を評価するために、96個の単独のコロニーの配列を決定した。また、これを使用して、提供されたバックグラウンド汚染残留テンプレートの量も決定した。
【0062】
発明の概要
一方は短い可動性グリシン−セリンリンカー(GS)、コアヒンジ、CH2、及びCH3を有し(配列番号17によって表される)、他方は可動性グリシン−セリンリンカー(GGGS)、Vhドメインの部分、突然変異IgG4ヒンジ、CH2、及びCH3(配列番号18によって表される)を有する、2つのFc足場は、約1〜3×109の複雑性を有するライブラリを産生した。各ライブラリからの96個のクローンの配列決定は、クローンのどの配列も同一でないことを示し、ライブラリの多様性が良好であることを示した。
【0063】
実施例3:ファージ粒子上の完全IgGディスプレイ
A.ベクター設計。
完全IgGディスプレイファージミド(vDR47、図2B)を、図1に示される、また国際公開第2009/085462号に記載されるような、pCNTO Fab IX構築物を用いて構築し、それは重鎖のVh及びCH1(配列番号19)ドメインを含んだ。ヒトIgG1(配列番号20)のヒンジ、CH2、及びCH3ドメインをコードする配列、並びに野生型配列、P6S(配列番号14)からの単独突然変異を有する変異型pelBシグナル配列を付加して、pVIIマイナーコートタンパク質でのペプチドディスプレイ及びタンパク質分泌における有意な改善を引き起こし(出願人は出願を同時係属中)、このベクターは、lacI遺伝子を有さないが、lacプロモータを有する。
【0064】
B.完全IgGディスプレイのために使用される構築物の特徴付け。
pIX上での完全IgGの提示を査定するために、試験構築物のパネルを作製した。6〜2及び16〜7と表記されるIL13に対する抗体、並びに抗サイトカイン抗体9〜4を、新たな完全IgG分子を構築するためのプロトタイプとして選択した。異なるコドン利用の効果を決定するために、2つの構築物を抗IL13抗体の各々に対して作製し、このうち1つはヒトコドン最適化を伴い、もう1つは大腸菌コドン最適化を伴った。表1は、5つの完全IgG試験構築物についてのベクター表記を列挙する。最適化された遺伝子を合成し、米国特許第6,670,127号及び同第6,521,427号に記載されるような二本鎖DNAに組み立てた。加えて、pIXと融合したEMP−1 Fc(実施例1)を、それがIgGヒンジ、CH2、及びCH3ドメインを含有するが、軽鎖を含有しないため、対照として含めた。
【0065】
【表2】
【0066】
C.ファージ産生
上記のB節に記載される完全IgGディスプレイ構築物を、標準的なプロトコルに従って2つの異なるF’大腸菌株、TG−1、及びXL−1ブルーに形質転換した。これらの2つの株を試験する理由は、仮説上、完全IgG pIX融合タンパク質のパッケージング及びディスプレイに影響を及ぼし得る、増殖率におけるそれらの差異である。個々の形質転換体を採取し、カルベニシリンを補充した(常に100μg/mLで使用される)2XYT培地中で一晩増殖させた。次いで一晩の培養物(500μL)を使用して、25mLの2XYT/カルベニシリンを接種し、培養物を37℃、250rpmで、OD(600nm)が0.5に到達するまで増殖させた。振盪を行わずに37℃で30分間インキュベーションする間に、細菌を1011pfu/mLのVCSM13ヘルパーファージ(Stratagene,La Jolla,CA)に感染させ、続いて3,000rpmで15分間、遠心分離工程を行った。この工程において、標準的なプロトコルにより、2XYT/カルベニシリン/IPTG(1mM)での細菌培養が求められた。しかしながら、我々は、この体系の漏出性が、その後のファージパッケージングを伴う融合タンパク質を産生するために十分であろうとの仮説により、培養物を2つに分割し、1mM IPTGを一方に添加し、他方には添加しなかった。要約すると、各々の構築物について、4つの異なるファージ調製を作製した:(i)IPTGを有するTG−1(ii)IPTGを有さないTG−1(iii)IPTGを有するXL−1ブルー(iv)IPTGを有さないXL−1ブルー。培養を30℃、250rpmで一晩増殖させ、翌日、3,000rpmで15分間遠沈させ、続いてPEG/NACl中でファージ上清の沈殿を行った。氷上で2時間後、沈殿したファージを10,000rpmで、15分間遠沈させ、ファージペレットを2mLのPBS中に再懸濁させた。ファージ調製物を更に、10,000rpmで10分間スピンすることによって、あらゆる残存する細菌ペレットから清澄化し、2mL管中、4℃で保管した。
【0067】
D.ファージ力価
標準的なプロトコルに従ってファージ力価を決定した。簡潔に述べると、OD(600nm)が0.5に到達するまで、TG−1細胞を2XYT中で増殖させた。ファージ調製物を、96ウェルプレート中のPBS中に連続的に希釈し、TG−1細胞をファージに添加し、37℃でインキュベートして感染可能にした。30分後、1%グルコース及びカルベニシリンを含有するLB寒天プレートに、2μLの各ウェルを分与することによって、スポット滴定を行った。プレートを37℃で一晩インキュベートし、ファージ濃度を、mL当たりのコロニー形成単位(cfu)の観点から決定した。表2は、すべての構築物及び培養条件についてのファージ滴定からの結果を示す。すべてのクローンは、10^11〜10^13cfu/mLの間の高いファージ力価を産生し、それは予測範囲内であり、ファージが効率的に産生されることを示した。
【0068】
【表3】
【0069】
E.機能的提示を査定するためのIgGドメイン−特異的なサンドイッチELISA
ファージpIX上での完全IgG分子の提示を査定するために、一連のサンドイッチELISAを設定した。黒色マキシソーププレートを、1μg/mLの、TBS中に希釈した捕捉抗体;ヒツジ抗ヒトIgG(FD、CH1)抗体(The Binding Site,Birmingham,UK)、マウス抗ヒトカッパ軽鎖(Southern Biotech,Birmingham,AL)、マウス抗ヒトIgG(CH2ドメイン)抗体(AbD Serotec,Raleigh,NC)、及びマウス抗ヒトIgG(CH3ドメイン)抗体(AbD Serotec)のうちの1つでコーティングした。プレートをケミブロッカー(Chemicon/Millipore,Billerica,MA)でブロックした後、プレートを洗浄し、ファージを2×1011cfu/mLの濃度(10%ケミブロッカー/TBST中に希釈)で添加し、1時間インキュベートした。プレートを洗浄し、HRP共役したマウス抗M13抗体をプレートに添加した。30分のインキュベーション後、プレートを洗浄し、化学発光基質をウェルに添加し、プレートをEnvisionプレートリーダーにおいて読み取った。図7A〜Dは、それぞれCH1(図7A)、カッパ(図7B)、CH2(図7C)、及びCH3(図7D)サンドイッチELISAからの結果を示す。ELISAで使用した対照は、vDR10中のクローン6−2のFab−pIX融合物(ヒトコドン最適化、TG−1細胞中で作製、IPTG誘導を伴う)、非特異的な足場タンパク質−pIX融合物、又はCNTO530−pIX融合物を提示するファージであった。CH1及びカッパELISAにおいて、6−2 Fabは、陽性対照としての機能を果たす一方で、EMP−1構築物(CNTO530)分子は、陰性対照としての機能を果たす。CH2及びCH3 ELISAにおいて、6−2 Fabは、陰性対照としての機能を果たし、CNTO530分子は、陽性対照としての機能を果たす。足場タンパク質ファージは、それがいかなる抗体ドメインも担持しないため、すべてのELISAにおいて陰性対照としての機能を果たす。ファージの異なる捕捉抗体への結合を防止するために、可溶性競合物質として、5μg/mLの濃度での抗IL13完全IgG1抗体の添加によるELISAアッセイもまた行った。
【0070】
図7A〜Dに示されるように、サンドイッチELISAのすべてにおいてファージを検出し、ファージが実際には、表面上で異なる抗体ドメインを提示しているという証拠を提供した。XL−1ブルー細胞中で産生されたファージは、最も高いシグナルを有し、IPTGの添加は、結合シグナルに対する明白な効果を有した。ファージの結合は、特異的な相互作用を示す、可溶性抗IL13抗体の添加によって阻害され得る。しかしながら、可溶性抗IL13抗体は、ファージとCH3ドメインとの間の相互作用に完全に勝る(compete off)ことはできなかった(図7D)。これは、完全IgG−pIX融合物の両方について、並びにEMP−1−Fc−pIX融合物(CNTO530)について観察された。
【0071】
F.IL13への完全IgG pIXファージ結合
IgG分子のすべてのドメインを、ELISAによってファージ粒子上で検出できることを実証した後、構築物が、それらのそれぞれの抗原に結合する能力も保持するかどうかを決定することが必要であった。IL13結合ELISAを、黒色マキシソーププレートを、1μg/mLの商業用の抗IL13抗体(マウス抗ヒトIL13、MAB213、R&D Systems)でコーティングすることによって設定した。MAB213は、IL13への結合をめぐって6−2又は16−7と競合せず、故に、サンドイッチELISA捕捉抗体として理想的である。洗浄及びブロッキング後、ビオチン化ヒトIL13R130Qヒト(Peprotech)を100nMで添加し、1時間インキュベートした。プレートを洗浄し、pIX上で6−2及び16−7の完全IgGバージョンを提示するファージを2×1011cfu/mLで、単独で又は競合のための可溶性抗IL13抗体と一緒に添加した。結合ファージを、HRP共役したマウス抗M13抗体で検出し、化学発光をEnvision機器において読み取った。図8は、IL13ファージELISAの結果を示す。結合は、ほとんどの条件において検出され、このうち1mM IPTGを有するXL−1ブルー細胞中で産生されたファージが最も高いシグナルを示した。ペプチド−Fc−pIX及び代替的な足場分子−pIX融合物は、予測通り陰性であり、6−2 Fab pIX対照は、陽性であった。結合は、可溶性抗IL13抗体を添加することによって阻害され、相互作用が特異的であることを示した。IL13結合を更に検査するために、ELISAを設定し、そこで可溶性競合抗体を50μg/mL〜0.01μg/mLまで連続的に希釈した。対照抗体もまた含めた。図9A及びBは、それぞれ6〜2 IgG pIX及び6〜2Fab pIXのIL13結合に及ぼす可溶性抗体競合の効果を示す。結合の阻害は、およそ0.1μg/mLのIC50により、両方の構築物について見られた。しかしながら、完全IgG pIX構築物については、阻害は、非常に高い競合物質濃度においてさえ不完全であり、あるレベルの非特異的な相互作用が存在することを示唆する。
【0072】
G.IL13及びIL17への完全IgG pIXファージ結合
第2の確証的な実験を行った。これは、抗IL17A抗体の完全IgGバージョンをクローニングすることによって行った。構築物をXL−1ブルー細胞に形質転換し、ファージを、上述のように産生した。ELISAを行って、図6に示されるように、pIX上でのIL17 IgGの提示、並びにヒトIL17Amut6抗原へのその結合を確認した。各ELISA(FD捕捉、カッパ捕捉、CH2捕捉、CH3捕捉、IL13捕捉、及びIL17捕捉)について、ファージを単独で又は可溶性抗IL13 mAb若しくはA可溶性抗IL17A mAbと一緒にのいずれかで添加した。競合物質mAbの添加は、ELISAの特異性を示す。図10において明白なように、IL17 IgGは、IL13 IgGよりも低いレベルではあるが、pIX上に提示された。これは、これらの構築物の間のFab発現レベルにおける差異と一致する(データは示されず)。ファージ上の抗IL13 IgGは、IL17に結合せず、ファージ上の抗IL17 IgGは、IL13に結合しないことから、抗原結合の特異性を見ることができる。加えて、ファージの2つのタイプの各々の結合は、それらの可溶性mAb対応物によって阻害され得る。
【0073】
実施例4:pVIIと融合したFc含有タンパク質の提示
追加的に、pVIIファージミド系を用いて、Fc及びMIMETIBODY(商標)タンパク質がファージ表面上に提示され得ることを実証した。
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2009年11月17日に出願された米国出願第61/261,767号に対する優先権を請求し、この出願は、参照により全体的に組み込まれる。
【0002】
(発明の分野)
本発明は、二量体抗体断片、全抗体、又は他のジスルフィド結合多量体構築物を産生するために、pIXファージディスプレイライブラリを生成し、用いるための、組成物及び方法に関する。
【背景技術】
【0003】
繊維状ファージディスプレイは、各ファージ粒子が、選択プロセスにおいて一緒にそのコートタンパク質のN末端と融合したポリペプチドをコードする核酸を結合させるときの、タンパク質の親和性に基づく選択のために広範に使用される技術である。M13バクテリオファージは、5つのコートタンパク質をコードし、このうちマイナーコートタンパク質pIII及びpVIのおよそ5つのコピーは、ファージの一方の末端部にあり、同じ数のpVII及びpIXが、ファージの他方の末端部にある。ファージDNAは、メジャーコートタンパク質、pVIIIのおよそ3000個のコピーによって封入される。異物ポリペプチドの提示はM13の各コートタンパク質によって達成されてきたが、このうち最も一般的な融合パートナーはpIII及びpVIIIである。この技術を用いて、ペプチド、Fab、scFv、及び他のタンパク質バインダーのライブラリが構築されており、多様な用途において使用が見出され、かつ高い商業的価値を有する。
【0004】
pIIIコートタンパク質は、そのサイズ、立体配座、少ないコピー数に起因して、pVIIIタンパク質よりも好まれている。pIIIマイナーコートタンパク質は、柔軟な可動性ヒンジセグメントによって接続される3つのドメインを含む、大腸菌中へのファージ感染に関与する404アミノ酸、42kDタンパク質である。pIII N末端への融合は、ファージ表面から離れて提示されるタンパク質を繋ぎ止め、小さく、高いコピー数のpVIIIコートタンパク質への融合よりも、リガンド結合に対して潜在的により高いアクセスを提供する。pIIIタンパク質は、感染の最初の工程に必須であり、ほとんどの小さいペプチド及びタンパク質の融合は、このプロセスを干渉し得る。この問題は、例えば、野生型pIIIタンパク質の第2のコピー又はヘルパーファージを用いるファージミド系を含有する、ウイルスベクターの使用によって回避される。pIIIとは対照的に、かつpVIIIと同様に、pVII及びpIXは、それぞれ、ファージ表面上で密接に詰められた33及び32aaの短いヘリックスタンパク質である。それにもかかわらず、scFv(Gao,C.ら、Proc Natl Acad Sci U S A 99,12612〜12616,2002)及びFab(Shi,Lら、J Mol Biol 397,385〜396,2010、Tornetta,Mら、J Immunol Meth 360,39〜46,2010)ライブラリは、pIX上で提示及び選択されている。Fv及びペプチドのヘテロ二量体ディスプレイは、異なるポリペプチドをpVII及び密接に隣接したpIXの両方に融合することによって記載されている(Gaoら、1999 Proc Nat Acad Sci 96:6025〜6030及びJanda米国特許第7078166号)。加えて、単一特異的なscFvのpVIIディスプレイが報告されている(Kwasnikowskiら、2005.J Immunol Methods 307:135)。ファージ又はファージミドベクターによってコードされる細胞外タンパク質がコートタンパク質に融合されず、むしろ再操作されたコートタンパク質pIII及びpIXにジスルフィド結合を通じて共有結合する、代替的なアプローチもまた記載されている(米国特許第6753136号)。
【発明の概要】
【発明が解決しようとする課題】
【0005】
ファージ粒子の表面上で二量体タンパク質並びにヘテロ二量体タンパク質を提示する能力は、組み合わせライブラリフォーマットにおけるより複合のタンパク質構造を模倣する際に有利である。分子間ジスルフィド結合を介して接続される重鎖及び軽鎖対(ヘテロ二量体)のホモ二量体である、ヒトIgGのタンパク質のような、複合タンパク質の変異型をスクリーニングする高処理方法を生成するために、当該技術を前進させる要求が引き続き存在する。現在まで、繊維状ファージ上の完全な抗体重鎖の正確なアセンブリ及び提示を実証することは可能でなかった。本発明のライブラリ及び方法は、総合設計、アセンブリ技術、及びファージpIX Fabライブラリを組み合わせることにより、これらの要求を満たす。
【課題を解決するための手段】
【0006】
本発明は、M13コートタンパク質、pIXを用いた、繊維状ファージ上での二量体ジスルフィド結合タンパク質及びより複合の構造の提示のための簡易な手段を提供する。本発明において、提示されるタンパク質は、pIXコートタンパク質を含む融合タンパク質であり、CH2ドメインのような、折り畳まれたドメインは、ヒンジドメインのような、システイン残基を含む多量体化ドメインに結合される。具体的な実施形態では、二量体タンパク質は、ホモ二量体であり、ここでメンバーは、ジスルフィド結合され、タンパク質は抗体Fcを含む。別の実施形態では、ホモ二量体ジスルフィド結合タンパク質は、ヒト抗体タンパク質を含み、ここで少なくともヒンジドメイン及び定常ドメインは、ホモ二量体を含むポリペプチドの各々において存在し、かつ任意に、ホモ二量体構造は更に、ジスルフィド結合形成によって独立して発現される抗体軽鎖と会合する。
【0007】
本発明は、pIXコートタンパク質をコードする配列と融合した外因性ポリペプチドをコードする配列を有する、少なくとも1つの融合タンパク質をコード化する複製可能なベクターを提供し、ここで外因性非ファージタンパク質部分は、ホモ二量体形成ポリペプチド鎖である。一実施形態では、融合したホモ二量体形成ポリペプチドは、Fc融合タンパク質を形成する。別の実施形態では、ホモ二量体構造は更に、ヘテロポリペプチドと会合して、より複合の構造を形成し得る。一態様では、単一のファージ分子における抗体重鎖ポリペプチド及び軽鎖ポリペプチドの両方の提示は、完全なIgG分子等であるが、これらに限定されない、ファージ粒子の表面における機能的抗体分子のアセンブリをもたらす。二量体ジスルフィド結合タンパク質として、ファージの表面上で融合ポリペプチドを提示することができる複製可能なベクター及びファージ粒子を含有する宿主細胞が、本発明に含まれる。ベクターは任意に、提示されたポリペプチド−コートタンパク質融合をコードするポリヌクレオチド配列に操作可能に融合した、分泌シグナルをコードするポリヌクレオチドを含む。
【0008】
抗体組成物の選択及び改善のためのアセンブリ、スクリーニング、並びに当該技術分野において実施されるような他の尋問技術に有用な、二量体ジスルフィド結合タンパク質のpIXファージディスプレイのデノボライブラリを構築するための方法及びベクターもまた提供される。一実施形態では、ファージ粒子表面上で多量体構造を形成することができる複数個の異なる融合ポリペプチドを提示するファージ粒子を含有する宿主細胞のライブラリは、pIXタンパク質に結合される。一態様では、ライブラリは、ファージミド系においてコードされる。
【0009】
一実施形態では、本発明のライブラリは、重鎖可変領域のライブラリを含み得、それは更に、軽鎖可変領域のライブラリを含み得、それは更に、変異型Fc領域のライブラリを含み得る。本発明のライブラリは、標的リガンドへの変化した結合、又はエフェクター分子(例えば、FcγRs及び/又はC1q)に対する変化した結合を有するような、所望の、増強された、又は軽減された特性を有するタンパク質をコードするライブラリから、ポリヌクレオチドを特定及び単離するために、パンニング、分類、又は他の選択手順に供されてもよい。
【図面の簡単な説明】
【0010】
【図1】pIXで繋ぎ止められたFabを発現するために使用される出発ベクターのダイアグラム。
【図2】A〜Bは、lacZプロモータ;細菌シグナルペプチド、pelBの上流に付加されたリボソーム結合部位(RIBS);Fcポリペプチドをコードするポリヌクレオチド配列、及びファージマイナーコートタンパク質pIX又はpVIIを接続する、可動性リンカー(G4S)の位置;並びにpIX上の完全IgG構造の発現のためのジシストロニックなファージミドベクター(B)の相対的な位置を示す、Fc形成構築物(A)のためのpIXファージミドベクターの概略図。
【図3】A〜Bは、実施例1に記載されるように構築された組み換えファージ粒子に対する、ELISAの結果を示すグラフであり、組み換えファージ粒子が、抗Fc Mab(A)で又はCNTO 3443、抗EMP1 mAb(B)でコーティングされたプレート上に捕捉され、捕捉されたファージが、HRP共役した抗pVIII mAbを用いて検出された、Fc融合タンパク質(A)又はEMP−1−Fc(B)の増加を実証している。ヘルパーファージは、BにおけるFcファージと同様に陰性対照であった。この実施例において、ファージの2つの個々の調製物が使用された。
【図4】ファージ上に提示されたFcドメインが、プロテインAに結合できることを示す結合アッセイからのグラフ。
【図5】A〜Bは、最適な結合酸性度、pH 6.0(上部)で、及び非特異的な結合条件下、pH 7.5(下部)で行われた、FcRn結合アッセイからのグラフ。
【図6】検出のために抗ヒトFc抗体を用いたウェスタンブロットを示し、非還元条件、レーン1、NR;及び還元条件、レーン2、Rの下で、単離及び電気泳動されたタンパク質の二量体的性質を実証し、非還元下でメジャーバンドが、還元条件下でのメジャーバンドのおよそ2倍の分子量であることを示している。
【図7】A〜Dは、ファージ上で発現された抗体ドメイン、発現されたEMP−1−Fc構築物、又はファージ自体のいずれかに特異的な種々のリガンドを用いて指定の調製物から捕捉され、lAc誘導物質IPTGである(A)抗FD(CH1)抗体捕捉;(B)抗カッパ抗体;(C)抗CH2抗体;及び(D)抗CH3抗体を用いて又は用いずに培養された、ファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。pIX上で6−2 Fab又は非免疫グロブリンタンパク質を提示するファージが陰性対照として含まれる。
【図8】抗IL13 IgG pIXと同じ特異性で競合する可溶性抗IL13 mAb(格子縞棒)の存在又は不在下で、商業用の抗IL13抗体を用いて指定の調製物から捕捉されたファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。EMP−1−Fc構築物は、陰性対照であり、IL13に結合せず、IL13特異的6−2 Fabは、pIX融合物が陽性対照として含まれるため、ファージ上でpIXを提示した。
【図9】A〜Bは、商業用の抗IL13抗体によってプレート中に捕捉され、続いてファージ上で漸増量の競合する抗IL13抗体(6−2完全IgG)、又はIL13(抗EMMPRIN)(A)に特異的でない対照抗体を付加され、またIL13によって、商業用の抗IL13抗体によってプレート中に捕捉され、続いてファージ上の6−2 Fabを付加されたファージについての、ELISAフォーマットで産生されたシグナルを示す棒グラフである。漸増量の抗IL13 mAb又は抗EMMPRIN mAbのいずれかが付加された(B)。
【図10】ビオチン化IL13又はIL17A抗原を使用してIL13又はIL17Aの完全IgG構築物を提示するファージを捕捉した後に、ドメイン特異的な抗体抗Fd、抗カッパ、抗CH2、及び抗CH3のいずれかによって、及び競合する可溶性抗IL13 mAb又は抗IL17A mAbの存在又は不在下で捕捉されたファージからのシグナルを示す。ファージは、抗M13抗体(y軸)で検出された。
【0011】
【表1】
【発明を実施するための形態】
【0012】
略語
ADCC=抗体依存性細胞媒介型細胞傷害性、ADMC=抗体依存性単核細胞媒介型細胞傷害性、c1q=補体因子1q、EPO=組み換えエリスロポエチン、FcR=Fc受容体、Ig=免疫グロブリン、Hc=重鎖、Lc=軽鎖、IPTG=イソプロピルチオ−β−ガラクトシド。
【0013】
定義
本明細書で使用されるとき、別段の指示がない限り又は文脈から明白でない限り、抗体ドメイン、領域、及び断片は、当該技術分野において周知であるような標準的な定義に従う。本発明のタンパク質は、1つ以上の免疫グロブリンクラスの抗体に由来するか、又はその部分を組み込む。免疫グロブリンクラスには、IgG、IgM、IgA、IgD、及びIgEアイソタイプ、またIgG及びIgAの場合は、それらのサブタイプ、例えば、IgG1、IgG2、IgG3、及びIgG4。
【0014】
が含まれる。「シストロン」とは、アミノ酸配列をコード化し、上流及び下流DNA発現制御要素を含む、DNA分子におけるヌクレオチドの配列を意味する。
【0015】
「外因性ポリペプチド」又は「外因性タンパク質」又は「細胞外タンパク質」とは、通常は野生型繊維状ファージゲノムによってコードされず、むしろ通常のファージタンパク質にとって異質であるタンパク質を意味する。典型的な外因性ポリペプチドは、それらがとりわけ、CH3、CH2、ヒンジ領域、及び/若しくはCH1ドメイン又はそれらの断片を含み得るFcドメインとして、天然に発生するように、抗体免疫グロブリン重鎖(Hc)ドメイン若しくは免疫グロブリン軽鎖(Lc)ドメイン、免疫グロブリン重鎖可変ドメイン(VH)、免疫グロブリン軽鎖可変ドメイン(VL)、天然若しくは合成ポリペプチド、単鎖抗体(scFv)、又は免疫グロブリンドメインの配列若しくは組み合わせを含む、目的とする任意のポリペプチドである。
【0016】
「Fc」により、消化されたIgGの結晶化可能な開裂断片に与えられる標識であり、抗体定常ドメインに由来し、かつジスルフィド結合の鎖間結合を有するポリペプチド鎖の二量体構造を含む、抗体の機能的断片を意味する。ヒトIgG1において、パパインは、Cys226に対する断片C末端を作り出す(参照により本明細書に明示的に組み込まれる、Kabatら、Sequences of Proteins of Immunological Interest,5th Ed.Public Health Service,National Institutes of Health,Bethesda,Md.(1991)にあるようなEUインデックスを用いて付番される)。「KabatにあるようなEUインデックス」は、ヒトIgG1 EU抗体の残基付番を指す。FcのN末端残基の定義は、異なり得るが、第1の鎖間結合システイン(Kabat系におけるC226)に対してN末端の第3残基である、Kabat付番系における少なくとも残基223を含むことが概して理解される。分子のFc部分は、抗体のその特異的な標的抗原との接触に直接に関与しないが、エフェクター機能を媒介する。これらの機能は、次の2つの種類からなる:(1)C1q結合及び/又はIgGに対するFc受容体γ−タイプ結合、IgEに対するFc受容体ε結合、及びIgAに対するFc受容体α結合に続く、補体依存性細胞傷害性(CDC)活性若しくはADCC及びADMCのような、抗体の抗原への結合を要求する機能、並びに(2)FcRnに結合し、細胞及び組織障壁(腸のような)を越えて経細胞輸送される能力による循環における持続性のような、抗原結合から独立した機能。特に、Fcの融合を介して、抗体分子又は他の分子の血清半減期を有意に増加させる能力は、非常に有利である。より長命の分子は、臨床治療に必要とされる量を低減させ、それによって投与の頻度を低減させ得る。
【0017】
用語「Fc受容体」又は「FcR」は、抗体のFc領域に結合する受容体を説明するために使用される。FcRには、FcγRI、FcγRII、及びFcγRIIIサブクラスが含まれ、これにはこれらの受容体の対立遺伝子変異型、あるいはスプライスされた形態を含む。FcγRII受容体には、FcγRIIA(「活性化受容体」)及びFcγRIIB(「阻害受容体」)が含まれ、それは主にその細胞質ドメインにおいて異なる類似したアミノ酸配列を有する。活性化受容体FcγRIIAは、その細胞質ドメインにおいて免疫受容活性化チロシンモチーフ(FAM)を含有する。阻害受容体FcγRIIBは、その細胞質ドメインにおいて免疫受容抑制性チロシンモチーフ(ITIM)を含有する(Daeron、Annu.Rev.Immunol.,1997,15:203〜234における概説を参照されたく、FcRsは、Ravetch及びKinet、Annu.Rev.Immunol.,1991,9:457〜92、Capelら、Immunomethods,1994,4:25〜34、及びde Haasら、J.Lab.Clin.Med.,1995,126:330〜41において概説され、これらの各々は参照により本明細書に組み込まれる)。
【0018】
「融合ポリペプチド」又は「融合タンパク質」とは、操作可能に結合される、それぞれ第1及び第2の核酸配列によってコードされる第1及び第2のポリペプチドを含む、融合ポリペプチド(タンパク質)を意味する。本明細書で使用されるとき、融合タンパク質は、「操作可能に結合」される構成成分及びドメインを含有し、例えば、ポリペプチド又はポリヌクレオチドの融合した要素が、各々が意図されるように働く又は機能するように結合されることを意味することが理解される。例えば、プロモータ、オペレータ、又はエンハンサのような、発現を調節する要素は、その発現が調節されるべきヌクレオチド配列に操作可能に結合され得る。要素間の及び要素の間の結合は、直接的であっても、又はリンカーを介するような間接的であってもよい。これらの要素は、必ずしも隣接していない。
【0019】
用語「ライブラリ」は、変異型である、すなわち、ある種の領域が同じであるか又は類似しており、他の領域が異なる、コードされたタンパク質の集積を意味する。変異領域は、指向性又は無作為の変異(確率的又は非確率的変化)によるものであってもよい。ライブラリ又は変異型は、異なる変異型の数又はライブラリの「サイズ」の観点から説明することができる。有用なデノボ抗体ライブラリは、高い多様性(>1010)を有し、変化に応じやすく、組立が容易であり、所望でない配列のバックグラウンドが低い。次の方法を組み合わせることにより、ライブラリアセンブリが加速され、低バックグラウンドをもたらされる:(a)Kunkel系単鎖突然変異誘発、(b)制限部位を有するパリンドロームループ、及び(c)メガプライマーアプローチの使用。
【0020】
「ファージミド」又は「ファージベクター」は、プラスミドに由来するもののような、ファージ染色体及び外因性DNAの両方に由来する構成成分を含有するクローニング及び発現ベクターである。ファージミドは、ファージゲノムの部分を含有するため、宿主のヘルパーファージとの共感染時に、それはファージ粒子中にパッケージ化され得る。本発明のファージミドは、ファージM13粒子中にパッケージ化され得る。ファージミド又はファージベクターは、本明細書における融合タンパク質をコードする核酸又は本明細書で提供される発現カセットのような、非相同のDNAの挿入又は組み込みによって操られている。そのような発現ベクターは、典型的には、宿主細胞中の挿入された核酸の効率的な転写のためのプロモータ配列を含有する。
【0021】
概論
二価抗原結合タンパク質である天然抗体は、重鎖の適切な会合のために、Fc定常ドメイン及びヒンジ領域に依存する。CH2及びCH3ドメインは、好ましくは、国際公開第2005005604号に記載されるもののような、又は天然又は操作された抗体配列の配列を含む検索データベースによって見出され得るもののような、ヒト生殖細胞系配列に由来する。広くは、本発明のタンパク質構築物は、1つ以上の定常ドメイン又はその部分に結合されたヒンジ領域を含む。能力受容体に結合し、体内の持続性を増加させる能力のようなすべての関連する機能を保持するために、Fcにおいて通常存在するすべての定常ドメイン:1つ以上のシステイン残基又は他のスルフィド若しくはセレノスルフィド結合形成残基を含有する、配列番号1〜4に示されるヒンジ又はその部分;配列番号5〜8によって例示されるCH2又はその変異型、及び配列番号9〜12によって例示されるCH3又はその変異型を組み込むことが通常は所望される。本明細書で提供されるものによって表される配列は、非限定的であり、天然及び変異型抗体ドメイン配列は、インターネット上の種々のデータベースにおいて、又は本発明の実践において有用であり得る多数の公報において見出され得ることが当業者によって理解されよう。加えて、構築物は任意に、CH1ドメインの幾つか若しくは、すべてを含んでもよく、又は配列番号13のもののように、抗体可変ドメインの幾つか若しくはすべてがまた存在してもよい。当然のことながら、これらのドメインは、完全IgG構築物において存在するであろう。抗体ドメインの適切な発現及び折り畳みに対する必要に応じて、配列番号14(pelB)及び15(ompA)のアミノ酸配列をコードするもののような、他の抗体配列及び非抗体配列が含まれてもよい。しかしながら、本発明は、ある種の定常ドメインのみを含み、その他を含まない構造、並びに非抗体由来のドメインが存在し得る構造を企図する。
【0022】
種々のFc機能は、Fcの異なる部分に依存するため、完全未満の機能性が所望される場合、より少ないCHドメインを重鎖に組み込むことができる。例えば、補体の有意な活性化は、IgGのCH2又はIgMのCH3を要求する。本発明はまた、修飾されたヒンジ及び重鎖が安定な複合で会合し得る限り、置換、欠失、挿入、又は修飾されたアミノ酸を有し得るFc重鎖ドメインの使用を企図する。
【0023】
加えて、典型的にはジスルフィド結合構造として形成されるであろう二量体共有結合構造もまた、セレノシステイン結合、ホモシステイン結合、又は混合スルフィド−セレニド結合によって形成され得る。鎖間共有結合残基を含む抗体ヒンジに加えて、他の多量体化ドメインを置換して、二量体又はより高秩序の構造を形成してもよい。これらの多量体化ドメインは、ファージ粒子の表面上での細胞外タンパク質−コートタンパク質融合タンパク質のポリペプチドの会合を補助するために、単独のシステイン若しくはセレノシステイン残基のように天然若しくは人工であってもよく、又はロイシンジッパーモチーフのようにモチーフを含んでもよい。
【0024】
完全抗体タンパク質の場合は、重鎖−軽鎖ヘテロ二量体は、特定の重鎖定常ドメインを介して会合して、より高秩序の構造を形成する。例えば、IgG型抗体は、四量体構造における共有結合によって接合される2つの重鎖−軽鎖ヘテロ二量体を含む。ある種の他の抗体型は、例えば、2個の四量体(IgA)又は10個の四量体(IgM)を含む、より高秩序の構造中に組み込まれる類似した四量体構造を含む。
【0025】
ファージコートタンパク質を用いて大型の細胞外タンパク質分子を提示する際に、提示されるタンパク質は、コートタンパク質の全コピーに結合される場合、組み換えファージ粒子のアセンブリを干渉する可能性がある。アセンブリ干渉を回避するために、pIXディスプレイについて(Gaoら、Proc Natl Acad Sci USA,99:12612〜12616,2002)によって記載されるようなファージミド系を使用してもよく、それによって野生型及び細胞外タンパク質結合コード配列の両方がベクターにおいて存在し、両方のタンパク質が組み換えファージ粒子中に組み込まれる。
【0026】
本発明の適用は、予想外に、本明細書に記載されるような抗体のFc部分を形成する抗体構成成分が、繊維状ファージ粒子の表面上のpIX又はpVIIコートタンパク質に対する融合タンパク質として、Fc受容体結合のような、天然抗体のFcドメインの既知の生物学的活性を提示するホモ二量体ジスルフィド結合タンパク質として、提示され得、また二価抗原結合タンパク質の形態であるとき、抗原結合の能力があることを見出している。故に、ファージ粒子上での抗体結合断片の単量体一価の提示とは対照的に、多価のタンパク質提示の多量体の提示が企図される。故に、本発明は、天然抗体の機能的特徴のより完全なスペクトルの間の操縦及び選択のための体系を提供する。そのような特徴には、細胞表面上で目的とする特定の抗原を提示する、細胞に対して向けられる免疫反応を促進し、製造される抗体様治療薬の生物活性の重要な構成成分である、Fc機能が含まれる。免疫系エフェクター細胞には、細胞免疫反応を活性化するT細胞のような抗原特異的細胞、並びに細胞免疫反応を媒介するマクロファージ、好中球、及びナチュラルキラー(NK)細胞のような非特異的細胞が含まれる。
【0027】
本発明を作製する方法
繊維状ファージ粒子上に提示された融合タンパク質において、外因性ポリペプチドと繊維状ファージpVII又はpIXタンパク質との間の「融合」は、アミド結合によって直接に結合されてもよく、又はリンカーポリペプチド(すなわち、「リンカー」)を含んでもよい。典型的に一続きの長さ約5〜50のアミノ酸である様々なリンカーのいずれを使用してもよい。特に好ましいリンカーは、そのリンカーの位置で融合タンパク質に対して大きな可動度を付与する。リピートの数が典型的には1〜12であるG4S(Gly−Gly−Gly−Gly−Ser)リピート又はG3S(Gly−Gly−Gly−Ser)を有するもののような、支配的にグリシン(G、Gly)残基からなるもののような、二次構造が欠けているリンカーを、この目的のために使用してもよい。
【0028】
第1のポリペプチドは、外因性タンパク質であり、第2のポリペプチドは、繊維状ファージpVII又はpIXタンパク質であり、それによって外因性タンパク質は、繊維状ファージタンパク質のアミノ末端に融合される。更に、融合タンパク質が未成熟形態であるとき、すなわち、リーダー配列がプロセス(除去)されていないとき、融合タンパク質はまた、本明細書に記載されるような野生型又は突然変異体pelB又はompA配列(それぞれ配列番号14及び15)等のような、アミノ末端原核分泌シグナルを含有し得る。
【0029】
天然抗体において、軽鎖ポリペプチド及び重鎖ポリペプチド鎖は、別個にコードされ、発現される。分子の典型的なヘテロの二量体構造IgGクラスは、分子の4つのポリペプチド鎖、2つの重鎖及び2つの軽鎖の中で及びそれらの間での、ジスルフィド結合の適切なアセンブリ及びその形成に依存している。故に、本発明において、抗体の二量体Fc部分のアセンブリ及び/又は軽鎖の会合は、存在するとき、タンパク質の個々のドメインが自己会合し、それらの間にジスルフィド結合を形成する限りにおいて、抗体形成の天然プロセスを再現する。
【0030】
一実施形態では、繊維状ファージ粒子の表面上に提示されるべきFc含有タンパク質は、天然抗体であり、ジシストロニックなベクターは、Fc構築物−pIX融合タンパク質並びに自己会合するであろう、別個にコードされ、発現された抗体Lc又は抗原結合ドメインの発現のために構築される。本発明の抗原結合タンパク質は、任意のエピトープ、抗原部位、又はタンパク質のための結合部位を有することができる。好ましい抗原結合タンパク質は、受容体への直接結合によって、又はそれらの同族のリガンド(複数可)への結合によって、受容体タンパク質の活性化を中和する。概して、抗原結合ドメインは、Fcドメインを含む天然抗体Hc配列と融合した抗体Lc及び抗体Hc可変ドメインから形成されるであろう。別の実施形態では、pIX融合タンパク質は、Fcドメインに結合されたscFvを含む。本発明の別の態様では、sscFvを含む重鎖及び軽鎖の抗原結合部位は、2つの異なる結合特異性を提供し、それによってファージ表面に提示された自己組立されたジスルフィド結合構築物タンパク質を、二特異的かつ二価の分子にするように異なってもよい。例えば、IgG分子のVL及びVHドメインの代わりに置換されるのは、異なる特異性のscFvドメインであり、これにより結果として生じる分子が2つの異なるエピトープに同時に結合することができるようになる。本発明の方法を用いてファージ粒子上に提示され得る、多数の可変ドメイン対を有する二特異的な抗体分子を作り出す他の方法は、参照により本明細書に組み込まれる米国特許第20020103345A1号に教示される。
【0031】
一実施形態では抗原結合又は受容体結合ドメインは、抗体ドメインに由来しないが、Fcドメインに融合される既知の又は無作為ペプチド配列である。Fcの代替鎖と結合されて、所望によりその間にリンカー部分を有する生物活性ペプチド類は、同一であっても異なってもよい。生物活性ペプチド類は、最終共役物が所望の生物活性を示すのであれば、介在するリンカーと又はペプチド上のいずれかの残基から生じるFcと結合していてもよい。生物活性は、例えば、結合活性についてはインビトロアッセイにより、又は動物の疾病モデルでのようにインビボ活性により、あるいは共役物を投与した後の被験者の反応により、測定することができる。
【0032】
同時係属中の出願、国際公開第04/002417号、同第04/002424号、同第05/081687号、及び同第05/032460号の出願者は、本明細書でMIMETIBODY(商標)構造と称される構造を説明し、これらの参考文献の各々は、参照により本明細書に全体的に組み込まれ、これらの構造は、pIX又はpVIIファージコートタンパク質に融合され、ファージ粒子の外表面上に提示され得る、本発明の二量体ジスルフィド結合構造として含まれる。
【0033】
一実施形態では、MIMETIBODYは、生物活性ペプチドリンカー−ヒンジ−CH2−CH3ポリペプチドの対を含み、この対は、会合又は共有結合、具体的には、Cys−Cysジスルフィド結合によって結合される。生物活性ペプチドは、任意の長さの上に(長さで?)あってもよく、任意の種に由来する天然産の配列であっても、人工配列であってもよい。ペプチドは概して、ファージミドベクターによってコードされ、ファージ粒子上での提示のために構築物のFc部分に融合されるであろう。そのような組成物の一例としては、生物活性ペプチドとしてのEPO模倣ペプチドが挙げられる。故に、EPO模倣のCH1欠失MIMETIBODYは、治療用ペプチド及びその本来の又は後天性の生体外、生体内、又はその場での特性又は活性を提示しながら、その本来の特性及び機能により抗体構造を模倣する。ペプチドが既知の生物活性を有さないが、マーカー、タグ、抗原としての機能を呈示するか、又はレポーター基、キレート基等を提供する、類似した構造の他の構築物もまた、本発明によって包含される。
【0034】
典型的な実施形態では、Fc含有融合タンパク質又は「MIMETIBODY(商標)」は、幾つかの又は全体的な免疫グロブリンCH1ドメインが不在である式(I):
V1o−Pepa−Flexn−V2m−ヒンジ−CH2−CH3 (I)を含み、
式中、Pepは、標的を特異的に認識することができる生物活性ペプチド又はポリペプチドを表し、Flexは、MIMETIBODYが代替的な配向及び結合特性を有することを可能にすることによって、構造的な可動性を提供する任意の可動性リンカーポリペプチドであり、V1及びV2は、ブラケティング配列であり、ヒンジは、免疫グロブリンヒンジ領域の少なくとも一部分、例えば配列番号1〜4であり、CH2は、免疫グロブリンCH2定常領域の少なくとも一部分、例えば配列番号5〜8であり、CH3は、免疫グロブリンCH3定常領域の少なくとも一部分、例えば配列番号9〜12であり、m、N、及びoは、ゼロであり得るか、又は1〜10の間の整数であり得、かつ1〜10までの整数であり得る。Pep配列は、任意に、目的又は安定化、又は任意の数の生物物理学的な機能のための配列を含むことができる。典型的な実施形態では、ブラケティング配列は、Vhフレームワークのような抗体可変(V)ドメインに由来し、V1は、配列QIQであり、V2は、免疫グロブリンJ遺伝子ドメインに由来する配列を表し、GTLVTVSS(配列番号13)である。結果として生じるポリペプチドは、Cys−Cysジスルフィド結合等であるが、これらに限定されない、会合又は共有結合によって他のポリペプチドに結合することができる。
【0035】
pIX融合タンパク質の発現のレベルは、転写レベルにおいて追加的に制御することができる。融合タンパク質は、Lac Zプロモータ/オペレータ系の誘導可能な制御下にある(図1を参照されたい)。他の誘導可能なプロモータも同様に作動することができ、それらは当業者に既知である。高レベルの表面発現については、サプレッサーライブラリが、イソプロピルチオ−β−ガラクトシド(IPTG)のようなLac Zプロモータの誘導物質中で培養される。誘導可能な制御は、非発現条件下でライブラリを培養することによって、非機能的pIX融合タンパク質に対する生物学的選択が最小化され得るため、有益である。次いで発現は、ライブラリ内の抗体の全体的な集団がファージ表面上に正確に表されていることを確実にするためのスクリーニングの時にのみ誘導することができる。
【0036】
二量体化ポリペプチドファージコートタンパク質融合タンパク質をコードするベクターは、細胞外タンパク質及びファージコートタンパク質コード領域の接合部において、翻訳終止コドンを含み得る。対応する翻訳終止サプレッサーを担持する細菌細胞中で発現されるとき、融合タンパク質が産生される。対応する翻訳終止サプレッサーのない細菌細胞中で発現されるとき、遊離細胞外タンパク質は、産生されない。
【0037】
本発明の使用方法
本明細書に出発点として例示されたファージベクターを用いて、タンパク質は、分子のライブラリを生成するための指向性突然変異誘発を用いて特異的な個別的な残基位置で、又はNXT配列と一般的に称されるN結合グリコシル化配列のような領域で、多様であってもよい。各々が異なる細胞外タンパク質配列を保有する数十億の大腸菌コロニーを生成するために使用され得る、修飾されたKunkel突然変異誘発法が、特に有用である。効率的である一方で、高度に複合の配列ライブラリを生成するとき、突然変異誘発されていない親DNAの割合は増加する。加えて、長いオリゴヌクレオチドの合成の技術的制限は、遠位領域において配列多様性を含有するライブラリを作製するために使用されるとき、本方法の有効性を低下させる。これらの制限を克服するために、350塩基を超えるオリゴヌクレオチドを生成する追加的な技術を使用することができる。これらの技術には、米国特許第20050048617号に記載されるような、標準的なKunkel突然変異誘発法(Kunkelら、1987 Methods Enzymol 154:367〜382)と組み合わせた、メガプライマーの使用及び突然変異誘発テンプレートにおいて制限酵素認識部位を含有するステム−ループ配列の作製が含まれる。制限クローニング(Marksら、1991 J.Mol.Biol.222:581〜597、Griffithsら、1994 EMBO J.13,3245〜3260、Hoetら、2005 Nature Biotechnol 23,344〜348)、ファージ組み換え(Gigapack,Invitrogen)、及び配列特異的組み換えのような他のライブラリ技術と比較して、改善されたKunkelに基づく方法は、配列多様なライブラリ(109を超える)を生成する際に、有意により有効であり、標的化されたDNA中の任意の場所に配列多様性を導入するのにより万能である。
【0038】
Fc含有タンパク質の繊維状ファージ上での提示は、所望の結合特徴のために、そのような分子の大集団をスクリーニングすることが所望される場合に特に有用である。一実施形態では、Fc構築物−pIXタンパク質融合を発現する細菌細胞は、Fc構築物−pIX融合遺伝子を担持するベクターDNAの、ファージ粒子中への優先的なパッケージングを可能にするM13変異型に感染させられる。各々結果として生じるファージ粒子は、特定のFc構築物−pIX融合タンパク質を提示し、Fc構築物−pIX融合物をコードするベクターを含有する。そのようなファージ粒子の集団は、パンニング手順によって、所望の結合特徴のために強化することができる。典型的には、所望の粒子は、所望のファージ粒子がそこに結合し得る抗原によりコーティングされた固体表面上に固定化される。結合粒子は、収集され、細菌細胞を更に感染させるために使用される。パンニング手順は、所望の結合特徴のために更に強化するために反復される。
【0039】
一実施形態では、ファージライブラリは、FcRgammaIII(CD16)、FcRgammaII(CD32)、及びFcRgammaI(CD64)のような、天然又は組み換えFc受容体への増強された、減少された、又は変化した結合のために、分子のFc部分の変異型をスクリーニングするために使用される。
【0040】
ファージ及び他の抗体ディスプレイ方法は、生体外の抗原又は受容体標的に対する選択を操る機会を利用可能にする。生体外選択法の1つの特定の利点は、標的タンパク質上の多様な部位への抗体結合を得るように選択手順を操る能力である。あるいは、全細胞を使用して、バインダーを選択してもよい。
【0041】
ファージライブラリは、機能的属性に関連する遺伝物質の検索を単純化するが、ライブラリから最良の候補を単離するために多重ステップのパンニング戦略が要求される。ドメイン又はエピトープ指向性パンニングは、標的タンパク質に結合する抗体を選択する日常的な方法となっている。そのような選択は、選択的パンニング、脱選択的(de-selective)パンニング、リガンド捕捉、サブトラクティブパンニング又は道しるべ(pathfinder)選択として多様に知られる方法を利用する抗体のステップによる選択を用いることによって、主に達成されてきた。
【0042】
サブトラクティブパンニングにおいては、重複するが、完全に同一でない結合部位を有する標的(複数可)を使用して、不必要なバインダーを脱選択することができる。この戦略は、癌細胞に対するバインダーを脱選択するために通常の細胞を使用する際と同様に、未知の抗原さえにも対応するバインダーを特定するために使用されている。あるいは、関連する抗原の間で異なる又は共通の部位への抗体結合を得るために、幾つかの共通のドメイン又は構造を有する天然産のタンパク質が、連続的な又は競合選択において使用される。場合によっては、関連するケモカイン又はタンパク質の突然変異バージョンのような天然産のタンパク質が、サブトラクティブパンニングにおいて使用され得る。
【0043】
リガンド捕捉指向性パンニングは、無関係の及び隣接していないエピトープに固定化された抗体が、ファージパンニングのために好ましい標的リガンドの結合面を捕捉し、呈示するために使用されるという点で、ELISAサンドイッチアッセイに類似している(米国特許第6376170号)。その他のものは、所望の標的ドメイン以外における抗原を選択的にマスクするために競合抗体を使用している(Tsui,P.ら、2002.J.Immunol.Meth.263:123〜132)。道しるべ技術は、モノクローナル及びポリクローナル抗体、並びに西洋ワサビビペルオキシダーゼ(HRP)に直接に又は関節に共役させられた天然リガンドを使用する。ビオチンチラミンの存在下で、これらの分子は、標的抗原に極めて接近したファージ結合のビオチン化を触媒し、ストレプトアビジンを用いて全集団からの「タグされた」ファージの特異的な回収を可能にする。このようにして、標的自体に又はそのすぐ近くに結合するファージは、選択的に回収される(Osborn,J.K.ら、1998.Immunotechnol.3:293〜302)。これらの方法、方法の変形、及び当業者に既知の他の方法を用いて、本発明のpIX細胞外タンパク質のライブラリを問い合わせることができる。
【0044】
本発明は一般論として記述されてきているが、本発明の実施形態は、特許請求の範囲を限定するように解釈されるべきではない以下の実施例で更に開示される。
【実施例】
【0045】
実施例1.pIX上でのFc融合タンパク質の提示
A.ファージミドベクター構築
ファージミドベクター、pCGMT9(Gaoら、Proc.Natl.Acad.Sci.96:6025〜6030,1999,米国特許第6472147号)は、pIX融合を介したファージディスプレイのために重鎖定常ドメインを挿入することができるファージミドpIXディスプレイベクターの発達のための主鎖としての機能を果たした。このファージミドにおいて、アンピシリンに対する耐性を付与するβ−ラクタマーゼ遺伝子と共に、大腸菌(colE1)及び繊維状ファージ(f1)のための複製の源が存在する。
【0046】
MIMETIBODY(商標)分子を含む、Fc含有タンパク質を提示するためのpIXファージミドベクターを、国際公開第2009/085462号及び図1に開示させるように、2シストロン性の発現、pCNTO−Fab−pIXのために適合させられたGaoベクターに基づいて構築した。それにより可溶性軽鎖が同じ細胞中で発現され、繋ぎ止められたポリペプチドと会合する、Fabファージディスプレイのために使用された戦略とは異なり、可溶性Fcは、全く発現されなかった(図2A)。
【0047】
ベクターにおけるFab軽鎖配列は欠失された。ベクターにおけるFab重鎖配列を、Fc又は構築物又はMIMETIBODY(商標)構築物のいずれかと置き換えた。コアヒンジを含有するシステイン対を含有するファージミドベクターFc構築を次のように達成した。ヒトIgG1のコアヒンジ、CH2、及びCH3をコードするFc遺伝子セグメントを、PCRによってFc含有プラスミドから増幅した。NcoI制限部位を、5’プライマー末端部中に、及びSacII制限エンドヌクレアーゼ部位を3’プライマー末端部において組み入れた。PCR増幅したDNA断片及びファージミドベクター(pCNTO−Fc−pIXコアHg)を、NcoI及びSacII制限エンドヌクレアーゼで消化した。消化産物を生成し、迅速ライゲーションキットを用いてライゲートし、DH10B大腸菌に形質転換させた。形質転換したクローンを、DNA塩基配列決定法を用いてスクリーニングし、正確な配列を示したものを次いで、ファージ調製のために、TG−1大腸菌に形質転換した。
【0048】
制限酵素クローニングを介して、Fcをコードする配列を、完全なCNTO530融合タンパク質をコードする配列と置き換えることによって、米国特許第7393662号及びその中の配列番号88に記載され、「EPO MIMETIBODY(商標)」又はCNTO530と称される、EMP−1(配列番号16)Fc(配列番号17)構築物をコードするpIXファージミドベクター(p2467)を構築した。CNTO530コード配列を、PCRによってプラスミドp2467から増幅した。制限エンドヌクレアーゼ部位NcoI及びSpeIを、それぞれ5’末端及び3’末端プライマーに含めた。PCR産物及びファージミドベクター、pCNTO−Fc−pIXコアHgをNcoI及びSpeIで消化し、精製し、迅速ライゲーションキットを用いてライゲートし、DH10B大腸菌に形質転換した。形質転換クローンを、DNA塩基配列決定法によってスクリーニングし、正確な配列を有するものをファージディスプレイのためにTG−1大腸菌に形質転換した。
【0049】
B.組み換えファージの調製及び特徴付け
ファージミドベクターでトランスフェクトしたTG−1大腸菌を、液体培養でOD600=0.5〜0.6になるまで増殖させた。VCSM13ヘルパーファージストックを培養物に添加し、感染は、静的インキュベーションとして、37℃で45分間進行させた。培養物を遠心分離にかけて、細菌をペレット化し、カルベニシリン、カナマイシン、及びIPTGを補充した培地中に再懸濁させ、250RPMで振盪しながら、30℃で12〜16時間インキュベートした。一晩の培養物を遠心分離にかけ、ファージ含有上清を未使用の管に移し、そこに10分の1の容量の塩化ナトリウム/PEG溶液(NaCl及びPEGの濃度は?又は単に標準的な方法を用いて沈殿させたPEGと述べる(Ref))を添加した。各管を混合し、時折混合しながら氷上でおよそ3時間インキュベートし、その後、この管を遠心分離にかけてファージをペレット化した。ファージペレットを、PBS中に注意深く再懸濁させ、新たな管に移し、2回目の遠心分離にかけてあらゆる残存する細胞残屑を除去した。精製しファージを、−80℃のアリコート中で保管した。スポット滴定を行って、ミリリットル当たりのコロニー形成単位(cfu)としてファージ力価を推定した。
【0050】
C.提示されたタンパク質の特徴付け
Fc及びペプチド−Fc構築物の提示の確認
この実施例において、ファージの2つの個々の調製物が使用された。Fc支持又はCNTO530支持ファージを検出するために、黒色ELISAプレートを、抗ヒトFcγ特異的ポリクローナル抗体又は抗EMP1ペプチドモノクローナル抗体(CNTO 3443)のいずれかでコーティングした。コーティングしたプレートを、TBST中の5%ミルクでブロックし、TBSTで洗浄した。ヘルパーファージ、Fc−又はCNTO530組み換えファージをプレートに添加し、室温で1時間インキュベートし、洗浄して非結合ファージを除去した。結合ファージを、HRP共役した抗M13 mAb及び化学発光基質で検出した。捕捉されたファージを、HRP共役した抗pVIII mAbを用いて検出した。ヘルパーファージ、及びCNTO530支持ファージの場合は、EMP1ペプチドを有さないFc組み換えファージを、陰性対照として使用した。
【0051】
プロテインA結合。精製した組み換えプロテイン−Aを、黒色ウェルのELISAプレート上に一晩、4℃でコーティングした。コーティングしたプレートを、TBST中の5%ミルクでブロックし、TBSTで洗浄した。ヘルパーファージ又はFc提示ファージの適切な希釈物をプレートに添加した。プレートを室温で1時間インキュベートし、洗浄して非結合ファージを除去した。コーティングしたプロテイン−A上のあらゆる残存する非占有のFc結合部位をブロックするために、ヒト抗体由来のFcを飽和濃度でプレートに添加した。30分のインキュベーション後、結合ファージをHRP共役した抗M13 mAb及び化学発光基質で検出した。
【0052】
FcRn結合。FcRn(新生児型Fc受容体)は、抗体再取り込み、区画転座、及び再循環を可能にし、故に、抗体の循環半減期を持続させる。FcRnに結合するFcは、pH依存性であり、ELISA結合アッセイをそれに応じて行った。FcRn結合ファージを、ニュートラアビジンでコーティングした96ウェルのプレート上に捕捉し、HRP共役した抗pVIII mAbで検出した。簡潔に述べると、黒色ウェルのELISAプレートを、ニュートラアビジンでコーティングし、SuperBlock T20(TBS)及びケミブロッカーの50/50混合物でブロックした。プレートをTBSTで洗浄し、ビオチン化FcRnを1時間捕捉した。ヘルパーファージ又はFc提示ファージの適切な希釈物を、TBST中、pH=6又はpH=7.5で調製した。非結合FcRnを、プレートから洗浄し、ファージを添加し、1時間インキュベートした。あるいは、プレートへの添加前に、ビオチン化FcRnをファージと室温で1時間混合した。コーティングしたFcRn上の残存する非占有のFc結合部位をブロックするために、ヒト抗体由来のFcを飽和濃度でプレートに添加した。結合ファージを、HRP共役した抗M13 mAb及び化学発光基質で検出した。
【0053】
D.結果
Fcを提示するファージは、抗Fc抗体を用いて捕捉し、抗pVIII抗体を用いて検出したため、ELISAアッセイは、Fcを提示するファージの比率を示すように設計した(図3A)。Fc組み換えファージについて観察された強力なシグナル及びヘルパーファージについて観察されたシグナルの欠如は、Fcが効率的にファージ表面上に提示されたことを実証する。EMP1融合タンパク質構築物、CNTO530、提示を、図3Bに示される捕捉リガンドとしてEMP−1特異的抗体を用いて確認した。Fc領域が適切な生物学的活性を保持し、故に、二量体であることを確認するために、特異的な結合アッセイを行った:プロテインA結合、及びFcRn結合。図4に示されるように、その表面上に提示されたFcを有するファージは、プロテイン−Aに結合する一方で、Fcを欠く対照ヘルパーファージは、それに結合しない。プロテインA結合についての化学発光シグナルは、ヒト免疫グロブリンγ特異的なポリクローナル抗体で捕捉されたFc提示ファージのそれに類似しており、ファージ提示されたFcの大部分が、プロテインAへの結合に適格な立体配座へと折り畳まれることを示唆する。
【0054】
Fcは、pH 6.0でFcRnに結合するが、pH 7.5で数桁の結合親和性を失う。ファージを、pH 6.0(図5A)又はpH 7.5(図5B)のいずれかで、ビオチン化FcRnでインキュベートした。pH 6.0で観察された強力なシグナルによって実証されるように、Fc組み換えファージは、pH 6.0でFcRnに効率的に結合した。対照的に、同じファージは、試験したすべての濃度で、それよりもはるかに低いシグナルを示した。したがって、pH依存性結合は、pIXファージミド系を用いて提示されたFcについて保持された。
【0055】
IgG及び他のFc含有分子は、CH3ドメインの相互作用を介してホモ二量体を形成する。ホモ二量体は、そのコアヒンジ領域における2つのジスルフィド結合によって安定化される。Fcファージミドディスプレイベクターは、Fc遺伝子の単独コピーのみをコードするため、ウェスタンブロットを介して提示されたFcの凝集状態を検査した。濃縮されたファージ粒子を、還元又は非還元条件下でSDSゲル上に直接充填した。図4に示されるように、非還元条件下で、タンパク質の大部分は、二量体Fc−pIX融合タンパク質について予想されたように、約62kDの分子量を有する二量体として移動した。逆に、還元条件下で、Fc−pIXタンパク質の大部分は、31kDの単量体として移動した。故に、ファージ表面上に提示されたFc分子の大部分は、ホモ二量体であり、IgG又は他のFc含有分子と同じ様態で、ジスルフィド結合で共有結合される。
【0056】
E.発明の概要
組み換えファージについて観察された強力なシグナルは、ヘルパーファージについてのシグナルの欠如と一緒に、EPO受容体作用物質(EMP−1)Fc構築物が、ペプチド並びにファージ粒子上のFcの検出によって実証されるように、効率的に提示されることを実証する。データは、Fc含有タンパク質が、天然リガンドへの結合を可能にする特徴的な立体配座特徴を有するホモ二量体として、ファージ上で効率的に提示されたことを示す。
【0057】
実施例2:ペプチド−Fc融合ライブラリ
ペプチド−Fc融合ライブラリを生成するために、無作為アミノ酸配列の部位においてヘアピンループを含有する、テンプレートファージミドを生成した。ヘアピンが二本鎖DNAを形成する場所に固有の制限部位、XbaIが配置されるような方法で、ヘアピンを設計した。これは後に、XbaIでの制限消化を介してテンプレートDNAを除去し、それによって最終構築されたライブラリにおいてテンプレートファージミドが密接に詰められたファージを還元するために使用される予定であった。この細胞株を通じる継代は、ウラシルのssDNA中への組み込みを引き起こすため、二本鎖テンプレートプラスミドを、Dut−/uNg−大腸菌宿主株、CJ236に形質転換した。次いでウラシル含有ssDNAテンプレートを、最終ライブラリ宿主細胞の酵素によって分解した。プラスミドを保有する単独コロニーを、液体培養で増殖させ、それをその後、VCS−M13ヘルパーファージで感染させた。ファージをPEGと生理食塩水で沈殿させ、一本鎖DNAの精製のために使用した。
【0058】
DNAライブラリを、修飾されたKunkel突然変異誘発プロトコルを用いて生成した。無作為化ライブラリヌクレオチドをコードするオリゴマー、並びに5’及び3’隣接配列を、T4キナーゼを用いて酵素的にリン酸化した。リン酸化オリゴを、3ステップの温度低下プログラムを用いて、それらのそれぞれのssDNAテンプレートにアニールした。第2のストランド合成を、T7 DNAポリメラーゼ及びT4 DNAリガーゼを反応混合物に添加して、共有結合閉環状DNA(CCC−DNA)を形成することによって行った。CCC−DNAを精製し、次いでヘアピン配列においてXbaIで消化して、バックグラウンドを低減するためにテンプレートDNAを開裂した。消化前及び後の両方で、CCC−DNA産物をアガロースゲル電気泳動によって検査して、細胞中へのその導入の前にライブラリ調製物の質を評価した。次いでライゲーション混合物を、MC1061F’宿主細胞株(大腸菌)に形質転換した。
【0059】
4つのpIX提示されたライブラリを構築し、ここで7つ(A1及びA2)又は8つ(B3及びB4)の無作為アミノ酸ループは、各々2つのFc含有MIMETIBODY(商標)構築物(上記の式1を参照されたい)においてジスルフィド結合で拘束され、ここでリンカーは、GGSG又はGSであり、V領域J−ピース(配列番号13)は、存在するか又は不在であり、ヒンジは、隣接した配列を有するか又は有さないCPPCのコアアミノ酸、IgG1タイプのヒンジのいずれかを含む。これらの2つの変異型Fc領域は、下記に示すように表され、それは配列番号17及び18によって表され、ここで天然産のIgG4と異なる残基は下線を引かれている。2つの更なる無作為アミノ酸を、拘束ループの各末端部に付加した。
【0060】
A.7NNKライブラリ(XXCXXXXXXXCXX)
1)Fc=V領域及び完全ヒンジを有する突然変異体IgG4(配列番号18)。2)Fc=ヒンジコアを有する突然変異体IgG4(配列番号17)
B.8NNKライブラリ(XXCXXXXXXXXCXX)
3)Fc=V領域及び完全ヒンジを有する突然変異体IgG4(配列番号18)
4)Fc=ヒンジコアを有する突然変異体IgG4(配列番号17)
【0061】
生成された各ライブラリについて、総計31回の電気穿孔を行った。形質転換効率性を滴定するために小アリコートを除去した後、増殖培養物を即座に、1リットルの培養容量にまでスケールアップし、それを1.0のOD600にまで増殖させた。この時点で、培養物を次のように分割した:培養物の10分の1を、VCSM13ヘルパーファージに感染させてファージライブラリを生成した一方で、培養物の大半を使用して、細菌ライブラリのグリセロールストックを確立した。ファージ感染された培養物を増加したスケールにまで再び拡張し、一晩増殖させた。ファージライブラリを、氷上のPEG/NACl沈殿を用いて培養上清から精製した。結果として生じたファージ力価を、ミリリットル当たりのコロニー形成単位(cfu/mL)の数を測定するためにスポット滴定を用いて推定した。スポット滴定調製物からの1×10-9及び1×10-10希釈物のアリコートを、単独のコロニーを単離するためにグルコース及びカルベニシリンを補充したLB培地プレート上に広げた。各ライブラリについて、最終ファージライブラリの多様性及び機能性を評価するために、96個の単独のコロニーの配列を決定した。また、これを使用して、提供されたバックグラウンド汚染残留テンプレートの量も決定した。
【0062】
発明の概要
一方は短い可動性グリシン−セリンリンカー(GS)、コアヒンジ、CH2、及びCH3を有し(配列番号17によって表される)、他方は可動性グリシン−セリンリンカー(GGGS)、Vhドメインの部分、突然変異IgG4ヒンジ、CH2、及びCH3(配列番号18によって表される)を有する、2つのFc足場は、約1〜3×109の複雑性を有するライブラリを産生した。各ライブラリからの96個のクローンの配列決定は、クローンのどの配列も同一でないことを示し、ライブラリの多様性が良好であることを示した。
【0063】
実施例3:ファージ粒子上の完全IgGディスプレイ
A.ベクター設計。
完全IgGディスプレイファージミド(vDR47、図2B)を、図1に示される、また国際公開第2009/085462号に記載されるような、pCNTO Fab IX構築物を用いて構築し、それは重鎖のVh及びCH1(配列番号19)ドメインを含んだ。ヒトIgG1(配列番号20)のヒンジ、CH2、及びCH3ドメインをコードする配列、並びに野生型配列、P6S(配列番号14)からの単独突然変異を有する変異型pelBシグナル配列を付加して、pVIIマイナーコートタンパク質でのペプチドディスプレイ及びタンパク質分泌における有意な改善を引き起こし(出願人は出願を同時係属中)、このベクターは、lacI遺伝子を有さないが、lacプロモータを有する。
【0064】
B.完全IgGディスプレイのために使用される構築物の特徴付け。
pIX上での完全IgGの提示を査定するために、試験構築物のパネルを作製した。6〜2及び16〜7と表記されるIL13に対する抗体、並びに抗サイトカイン抗体9〜4を、新たな完全IgG分子を構築するためのプロトタイプとして選択した。異なるコドン利用の効果を決定するために、2つの構築物を抗IL13抗体の各々に対して作製し、このうち1つはヒトコドン最適化を伴い、もう1つは大腸菌コドン最適化を伴った。表1は、5つの完全IgG試験構築物についてのベクター表記を列挙する。最適化された遺伝子を合成し、米国特許第6,670,127号及び同第6,521,427号に記載されるような二本鎖DNAに組み立てた。加えて、pIXと融合したEMP−1 Fc(実施例1)を、それがIgGヒンジ、CH2、及びCH3ドメインを含有するが、軽鎖を含有しないため、対照として含めた。
【0065】
【表2】
【0066】
C.ファージ産生
上記のB節に記載される完全IgGディスプレイ構築物を、標準的なプロトコルに従って2つの異なるF’大腸菌株、TG−1、及びXL−1ブルーに形質転換した。これらの2つの株を試験する理由は、仮説上、完全IgG pIX融合タンパク質のパッケージング及びディスプレイに影響を及ぼし得る、増殖率におけるそれらの差異である。個々の形質転換体を採取し、カルベニシリンを補充した(常に100μg/mLで使用される)2XYT培地中で一晩増殖させた。次いで一晩の培養物(500μL)を使用して、25mLの2XYT/カルベニシリンを接種し、培養物を37℃、250rpmで、OD(600nm)が0.5に到達するまで増殖させた。振盪を行わずに37℃で30分間インキュベーションする間に、細菌を1011pfu/mLのVCSM13ヘルパーファージ(Stratagene,La Jolla,CA)に感染させ、続いて3,000rpmで15分間、遠心分離工程を行った。この工程において、標準的なプロトコルにより、2XYT/カルベニシリン/IPTG(1mM)での細菌培養が求められた。しかしながら、我々は、この体系の漏出性が、その後のファージパッケージングを伴う融合タンパク質を産生するために十分であろうとの仮説により、培養物を2つに分割し、1mM IPTGを一方に添加し、他方には添加しなかった。要約すると、各々の構築物について、4つの異なるファージ調製を作製した:(i)IPTGを有するTG−1(ii)IPTGを有さないTG−1(iii)IPTGを有するXL−1ブルー(iv)IPTGを有さないXL−1ブルー。培養を30℃、250rpmで一晩増殖させ、翌日、3,000rpmで15分間遠沈させ、続いてPEG/NACl中でファージ上清の沈殿を行った。氷上で2時間後、沈殿したファージを10,000rpmで、15分間遠沈させ、ファージペレットを2mLのPBS中に再懸濁させた。ファージ調製物を更に、10,000rpmで10分間スピンすることによって、あらゆる残存する細菌ペレットから清澄化し、2mL管中、4℃で保管した。
【0067】
D.ファージ力価
標準的なプロトコルに従ってファージ力価を決定した。簡潔に述べると、OD(600nm)が0.5に到達するまで、TG−1細胞を2XYT中で増殖させた。ファージ調製物を、96ウェルプレート中のPBS中に連続的に希釈し、TG−1細胞をファージに添加し、37℃でインキュベートして感染可能にした。30分後、1%グルコース及びカルベニシリンを含有するLB寒天プレートに、2μLの各ウェルを分与することによって、スポット滴定を行った。プレートを37℃で一晩インキュベートし、ファージ濃度を、mL当たりのコロニー形成単位(cfu)の観点から決定した。表2は、すべての構築物及び培養条件についてのファージ滴定からの結果を示す。すべてのクローンは、10^11〜10^13cfu/mLの間の高いファージ力価を産生し、それは予測範囲内であり、ファージが効率的に産生されることを示した。
【0068】
【表3】
【0069】
E.機能的提示を査定するためのIgGドメイン−特異的なサンドイッチELISA
ファージpIX上での完全IgG分子の提示を査定するために、一連のサンドイッチELISAを設定した。黒色マキシソーププレートを、1μg/mLの、TBS中に希釈した捕捉抗体;ヒツジ抗ヒトIgG(FD、CH1)抗体(The Binding Site,Birmingham,UK)、マウス抗ヒトカッパ軽鎖(Southern Biotech,Birmingham,AL)、マウス抗ヒトIgG(CH2ドメイン)抗体(AbD Serotec,Raleigh,NC)、及びマウス抗ヒトIgG(CH3ドメイン)抗体(AbD Serotec)のうちの1つでコーティングした。プレートをケミブロッカー(Chemicon/Millipore,Billerica,MA)でブロックした後、プレートを洗浄し、ファージを2×1011cfu/mLの濃度(10%ケミブロッカー/TBST中に希釈)で添加し、1時間インキュベートした。プレートを洗浄し、HRP共役したマウス抗M13抗体をプレートに添加した。30分のインキュベーション後、プレートを洗浄し、化学発光基質をウェルに添加し、プレートをEnvisionプレートリーダーにおいて読み取った。図7A〜Dは、それぞれCH1(図7A)、カッパ(図7B)、CH2(図7C)、及びCH3(図7D)サンドイッチELISAからの結果を示す。ELISAで使用した対照は、vDR10中のクローン6−2のFab−pIX融合物(ヒトコドン最適化、TG−1細胞中で作製、IPTG誘導を伴う)、非特異的な足場タンパク質−pIX融合物、又はCNTO530−pIX融合物を提示するファージであった。CH1及びカッパELISAにおいて、6−2 Fabは、陽性対照としての機能を果たす一方で、EMP−1構築物(CNTO530)分子は、陰性対照としての機能を果たす。CH2及びCH3 ELISAにおいて、6−2 Fabは、陰性対照としての機能を果たし、CNTO530分子は、陽性対照としての機能を果たす。足場タンパク質ファージは、それがいかなる抗体ドメインも担持しないため、すべてのELISAにおいて陰性対照としての機能を果たす。ファージの異なる捕捉抗体への結合を防止するために、可溶性競合物質として、5μg/mLの濃度での抗IL13完全IgG1抗体の添加によるELISAアッセイもまた行った。
【0070】
図7A〜Dに示されるように、サンドイッチELISAのすべてにおいてファージを検出し、ファージが実際には、表面上で異なる抗体ドメインを提示しているという証拠を提供した。XL−1ブルー細胞中で産生されたファージは、最も高いシグナルを有し、IPTGの添加は、結合シグナルに対する明白な効果を有した。ファージの結合は、特異的な相互作用を示す、可溶性抗IL13抗体の添加によって阻害され得る。しかしながら、可溶性抗IL13抗体は、ファージとCH3ドメインとの間の相互作用に完全に勝る(compete off)ことはできなかった(図7D)。これは、完全IgG−pIX融合物の両方について、並びにEMP−1−Fc−pIX融合物(CNTO530)について観察された。
【0071】
F.IL13への完全IgG pIXファージ結合
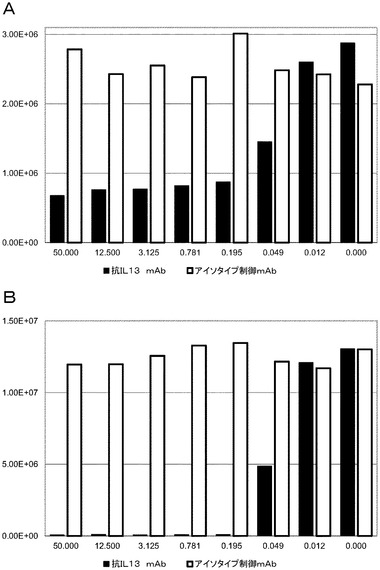
IgG分子のすべてのドメインを、ELISAによってファージ粒子上で検出できることを実証した後、構築物が、それらのそれぞれの抗原に結合する能力も保持するかどうかを決定することが必要であった。IL13結合ELISAを、黒色マキシソーププレートを、1μg/mLの商業用の抗IL13抗体(マウス抗ヒトIL13、MAB213、R&D Systems)でコーティングすることによって設定した。MAB213は、IL13への結合をめぐって6−2又は16−7と競合せず、故に、サンドイッチELISA捕捉抗体として理想的である。洗浄及びブロッキング後、ビオチン化ヒトIL13R130Qヒト(Peprotech)を100nMで添加し、1時間インキュベートした。プレートを洗浄し、pIX上で6−2及び16−7の完全IgGバージョンを提示するファージを2×1011cfu/mLで、単独で又は競合のための可溶性抗IL13抗体と一緒に添加した。結合ファージを、HRP共役したマウス抗M13抗体で検出し、化学発光をEnvision機器において読み取った。図8は、IL13ファージELISAの結果を示す。結合は、ほとんどの条件において検出され、このうち1mM IPTGを有するXL−1ブルー細胞中で産生されたファージが最も高いシグナルを示した。ペプチド−Fc−pIX及び代替的な足場分子−pIX融合物は、予測通り陰性であり、6−2 Fab pIX対照は、陽性であった。結合は、可溶性抗IL13抗体を添加することによって阻害され、相互作用が特異的であることを示した。IL13結合を更に検査するために、ELISAを設定し、そこで可溶性競合抗体を50μg/mL〜0.01μg/mLまで連続的に希釈した。対照抗体もまた含めた。図9A及びBは、それぞれ6〜2 IgG pIX及び6〜2Fab pIXのIL13結合に及ぼす可溶性抗体競合の効果を示す。結合の阻害は、およそ0.1μg/mLのIC50により、両方の構築物について見られた。しかしながら、完全IgG pIX構築物については、阻害は、非常に高い競合物質濃度においてさえ不完全であり、あるレベルの非特異的な相互作用が存在することを示唆する。
【0072】
G.IL13及びIL17への完全IgG pIXファージ結合
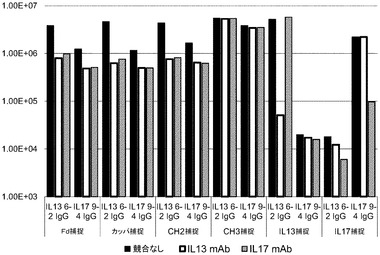
第2の確証的な実験を行った。これは、抗IL17A抗体の完全IgGバージョンをクローニングすることによって行った。構築物をXL−1ブルー細胞に形質転換し、ファージを、上述のように産生した。ELISAを行って、図6に示されるように、pIX上でのIL17 IgGの提示、並びにヒトIL17Amut6抗原へのその結合を確認した。各ELISA(FD捕捉、カッパ捕捉、CH2捕捉、CH3捕捉、IL13捕捉、及びIL17捕捉)について、ファージを単独で又は可溶性抗IL13 mAb若しくはA可溶性抗IL17A mAbと一緒にのいずれかで添加した。競合物質mAbの添加は、ELISAの特異性を示す。図10において明白なように、IL17 IgGは、IL13 IgGよりも低いレベルではあるが、pIX上に提示された。これは、これらの構築物の間のFab発現レベルにおける差異と一致する(データは示されず)。ファージ上の抗IL13 IgGは、IL17に結合せず、ファージ上の抗IL17 IgGは、IL13に結合しないことから、抗原結合の特異性を見ることができる。加えて、ファージの2つのタイプの各々の結合は、それらの可溶性mAb対応物によって阻害され得る。
【0073】
実施例4:pVIIと融合したFc含有タンパク質の提示
追加的に、pVIIファージミド系を用いて、Fc及びMIMETIBODY(商標)タンパク質がファージ表面上に提示され得ることを実証した。
【特許請求の範囲】
【請求項1】
繊維状ファージ粒子の表面上での機能的な多量体鎖間ジスルフィド結合タンパク質の表示のための複製可能なファージベクターであって、細胞外タンパク質に由来する第1のアミノ酸配列をコードするポリヌクレオチド配列と融合したファージpIX又はpVIIタンパク質をコードする核酸配列を含み、前記コードされた細胞外タンパク質のアミノ酸配列が、少なくとも1つのシステイン残基を含み、前記システイン残基が、pIX又はpVIIとの細胞外タンパク質融合として発現される第2のポリペプチド鎖上のシステイン残基に酸化的に結合されるようになることができ、前記第2のアミノ酸配列が、前記第1のアミノ酸配列と同じ若しくはその変異型であるか、又は異なるタンパク質であり、それによって、そのように形成された前記システイン結合が、前記維状ファージ粒子の表面上に提示されている前記機能的多量体構造の鎖間ジスルフィドである、前記ファージベクター。
【請求項2】
前記タンパク質構造の機能的活性が、プロテインA結合及びFcRn結合から選択される、請求項1に記載のファージベクター。
【請求項3】
前記コードされたアミノ酸配列が、配列番号1〜3及び4からなる群から選択される抗体ヒンジドメイン、並びに配列番号1〜3、及び4のコア残基を含む、請求項1に記載のファージベクター。
【請求項4】
前記鎖間ジスルフィドが、前記アミノ酸配列の前記抗体ヒンジドメイン、又はコアヒンジ残基内に位置する、請求項3に記載のファージベクター。
【請求項5】
配列番号5〜16、及び17〜22から選択される配列、又はその中の少なくとも1つの残基が置換されるそれらの変異型を更に含む、請求項4に記載のファージベクター。
【請求項6】
前記核酸が更に、前記細胞外タンパク質コード配列のうちの1つに操作可能に融合された細菌分泌シグナルをコードする、請求項1に記載のファージベクター。
【請求項7】
前記細菌分泌シグナルが、配列番号14又は15に示される、pelB配列若しくは変異型pelB配列から選択されるか、又はompA若しくはompAの変異型である、請求項6に記載のファージベクター。
【請求項8】
誘導可能なプロモータを更に含む、請求項1に記載のファージベクター。
【請求項9】
前記誘導可能なプロモータが、lacプロモータ又はlacの突然変異体である、請求項8に記載のファージベクター。
【請求項10】
融合ポリペプチドをコードするベクターを封入し、表面上に機能的な多量体鎖間ジスルフィド結合タンパク質を有する、繊維状ファージ粒子であって、前記タンパク質が、繊維状ファージpVII又はpIXタンパク質のアミノ末端と融合したシステイン残基を有する第1の外因性ポリペプチドを含み、その上で、前記タンパク質が繊維状ファージタンパク質の表面で発現されるとき、前記システイン残基が、pIX又はpVIIと融合した第2のポリペプチド鎖上のシステイン残基に酸化的に結合されるようになり、前記第2のアミノ酸配列が、前記第1のアミノ酸配列と同じ若しくはその変異型であるか、又は異なるタンパク質であり、それによって、そのように形成された前記システイン結合が、前記繊維状ファージ粒子の表面上に提示されている前記機能的多量体構造の鎖間ジスルフィドである、前記繊維状ファージ粒子。
【請求項11】
前記タンパク質構造の機能的活性が、プロテインA結合及びFcRn結合から選択される、請求項10に記載の繊維状ファージ粒子。
【請求項12】
請求項10に記載の繊維状ファージを含む、細菌宿主細胞。
【請求項13】
前記機能的タンパク質が、原核宿主細胞の培養培地から回収されることができる、請求項12に記載の宿主細胞。
【請求項14】
前記タンパク質が、繊維状ファージ粒子の表面上に表示される、請求項13に記載の宿主細胞。
【請求項15】
測定可能な機能的タンパク質結合活性を有する、請求項12に記載の細菌宿主細胞によって発現される二量体又は多量体融合タンパク質。
【請求項16】
前記測定可能なタンパク質結合活性が、プロテインA結合又はFcRn受容体結合である、請求項15に記載の融合タンパク質。
【請求項17】
請求項16に記載の多量体タンパク質構造の、生物学的に活性な細胞外タンパク質変異型。
【請求項18】
請求項4に記載の核酸ファージベクターを含み、前記抗体配列内の個別的な残基又はドメインをコードする前記ベクター内の特異的位置が、多様である、細菌宿主細胞のファージライブラリ。
【請求項19】
前記核酸が、アミノ酸置換を有するFc形成配列をコードする、請求項18に記載のファージライブラリ。
【請求項20】
前記Fc形成配列が、更に、リガンド結合ドメインを含む、請求項19に記載のファージライブラリ。
【請求項21】
前記リガンド結合ドメインが、受容体結合リガンド、受容体細胞外ドメイン、可変ドメイン及び定常ドメインを含む抗体Fabドメイン、並びに単鎖Fv構築物からなる群から選択される、請求項20に記載のファージライブラリ。
【請求項22】
前記リガンド結合ドメインが、前記結合ドメイン内の特異的残基において互いに異なる配列を含む、請求項21に記載のファージライブラリ。
【請求項23】
リガンド結合ドメインが、CDR1、CDR2、及びCDR3からなる群から選択される生殖細胞系重鎖相補性決定領域において複数個の多様な変異を含む、請求項22に記載のファージライブラリ。
【請求項24】
求項12に記載の細菌宿主細胞中で、抗体又はFc断片のライブラリを産生するための方法であって、
a.集団宿主細胞を、ファージコートタンパク質と融合した組み換え抗体又はその断片をコードする、請求項1に記載のポリヌクレオチドを含むベクターのライブラリに感染させること、
b.前記細胞集団をファージ表面上での前記抗体又はそのFc含有断片の発現を可能にする条件下で培養することと、及び
c.所望の又は増強された特性を有する前記抗体又はその断片を提示するファージを選択することを含む、前記方法。
【請求項25】
改善された特性を有する変異型の選択のために、請求項20に記載のファージライブラリを使用する方法。
【請求項26】
前記特性が、改善された標的結合、特異的な標的エピトープへの結合、Fc受容体結合親和性、減少されたグリコシル化部位、増加されたグリコシル化部位、及び増強された熱安定性からなる群から選択される、請求項23に記載の変異型を選択する方法。
【請求項27】
所望の生物活性について選択するために、請求項23に記載のファージライブラリを使用する方法であって、前記選択が、(a)抗体Fc断片を、請求項13に記載のファージライブラリから発現させること、及び(b)前記所望の生物活性を有するファージ粒子を選択することを含む、前記方法。
【請求項28】
請求項22に記載の方法から得られる、核酸をコードするFc含有抗体又は抗体断片。
【請求項29】
IgA、IgE、IgM、IgD、IgY、及びIgGからなる群から選択される、請求項23に記載の抗体又は抗体断片。
【請求項30】
前記抗体又はその断片が、マウス抗体、キメラ抗体、ヒト化抗体、又はヒト抗体である、請求項23に記載の抗体又は抗体断片。
【請求項31】
請求項24又は25に記載の方法を用いて選択されたFc含有抗体又は抗体断片を含む、医薬組成物。
【請求項1】
繊維状ファージ粒子の表面上での機能的な多量体鎖間ジスルフィド結合タンパク質の表示のための複製可能なファージベクターであって、細胞外タンパク質に由来する第1のアミノ酸配列をコードするポリヌクレオチド配列と融合したファージpIX又はpVIIタンパク質をコードする核酸配列を含み、前記コードされた細胞外タンパク質のアミノ酸配列が、少なくとも1つのシステイン残基を含み、前記システイン残基が、pIX又はpVIIとの細胞外タンパク質融合として発現される第2のポリペプチド鎖上のシステイン残基に酸化的に結合されるようになることができ、前記第2のアミノ酸配列が、前記第1のアミノ酸配列と同じ若しくはその変異型であるか、又は異なるタンパク質であり、それによって、そのように形成された前記システイン結合が、前記維状ファージ粒子の表面上に提示されている前記機能的多量体構造の鎖間ジスルフィドである、前記ファージベクター。
【請求項2】
前記タンパク質構造の機能的活性が、プロテインA結合及びFcRn結合から選択される、請求項1に記載のファージベクター。
【請求項3】
前記コードされたアミノ酸配列が、配列番号1〜3及び4からなる群から選択される抗体ヒンジドメイン、並びに配列番号1〜3、及び4のコア残基を含む、請求項1に記載のファージベクター。
【請求項4】
前記鎖間ジスルフィドが、前記アミノ酸配列の前記抗体ヒンジドメイン、又はコアヒンジ残基内に位置する、請求項3に記載のファージベクター。
【請求項5】
配列番号5〜16、及び17〜22から選択される配列、又はその中の少なくとも1つの残基が置換されるそれらの変異型を更に含む、請求項4に記載のファージベクター。
【請求項6】
前記核酸が更に、前記細胞外タンパク質コード配列のうちの1つに操作可能に融合された細菌分泌シグナルをコードする、請求項1に記載のファージベクター。
【請求項7】
前記細菌分泌シグナルが、配列番号14又は15に示される、pelB配列若しくは変異型pelB配列から選択されるか、又はompA若しくはompAの変異型である、請求項6に記載のファージベクター。
【請求項8】
誘導可能なプロモータを更に含む、請求項1に記載のファージベクター。
【請求項9】
前記誘導可能なプロモータが、lacプロモータ又はlacの突然変異体である、請求項8に記載のファージベクター。
【請求項10】
融合ポリペプチドをコードするベクターを封入し、表面上に機能的な多量体鎖間ジスルフィド結合タンパク質を有する、繊維状ファージ粒子であって、前記タンパク質が、繊維状ファージpVII又はpIXタンパク質のアミノ末端と融合したシステイン残基を有する第1の外因性ポリペプチドを含み、その上で、前記タンパク質が繊維状ファージタンパク質の表面で発現されるとき、前記システイン残基が、pIX又はpVIIと融合した第2のポリペプチド鎖上のシステイン残基に酸化的に結合されるようになり、前記第2のアミノ酸配列が、前記第1のアミノ酸配列と同じ若しくはその変異型であるか、又は異なるタンパク質であり、それによって、そのように形成された前記システイン結合が、前記繊維状ファージ粒子の表面上に提示されている前記機能的多量体構造の鎖間ジスルフィドである、前記繊維状ファージ粒子。
【請求項11】
前記タンパク質構造の機能的活性が、プロテインA結合及びFcRn結合から選択される、請求項10に記載の繊維状ファージ粒子。
【請求項12】
請求項10に記載の繊維状ファージを含む、細菌宿主細胞。
【請求項13】
前記機能的タンパク質が、原核宿主細胞の培養培地から回収されることができる、請求項12に記載の宿主細胞。
【請求項14】
前記タンパク質が、繊維状ファージ粒子の表面上に表示される、請求項13に記載の宿主細胞。
【請求項15】
測定可能な機能的タンパク質結合活性を有する、請求項12に記載の細菌宿主細胞によって発現される二量体又は多量体融合タンパク質。
【請求項16】
前記測定可能なタンパク質結合活性が、プロテインA結合又はFcRn受容体結合である、請求項15に記載の融合タンパク質。
【請求項17】
請求項16に記載の多量体タンパク質構造の、生物学的に活性な細胞外タンパク質変異型。
【請求項18】
請求項4に記載の核酸ファージベクターを含み、前記抗体配列内の個別的な残基又はドメインをコードする前記ベクター内の特異的位置が、多様である、細菌宿主細胞のファージライブラリ。
【請求項19】
前記核酸が、アミノ酸置換を有するFc形成配列をコードする、請求項18に記載のファージライブラリ。
【請求項20】
前記Fc形成配列が、更に、リガンド結合ドメインを含む、請求項19に記載のファージライブラリ。
【請求項21】
前記リガンド結合ドメインが、受容体結合リガンド、受容体細胞外ドメイン、可変ドメイン及び定常ドメインを含む抗体Fabドメイン、並びに単鎖Fv構築物からなる群から選択される、請求項20に記載のファージライブラリ。
【請求項22】
前記リガンド結合ドメインが、前記結合ドメイン内の特異的残基において互いに異なる配列を含む、請求項21に記載のファージライブラリ。
【請求項23】
リガンド結合ドメインが、CDR1、CDR2、及びCDR3からなる群から選択される生殖細胞系重鎖相補性決定領域において複数個の多様な変異を含む、請求項22に記載のファージライブラリ。
【請求項24】
求項12に記載の細菌宿主細胞中で、抗体又はFc断片のライブラリを産生するための方法であって、
a.集団宿主細胞を、ファージコートタンパク質と融合した組み換え抗体又はその断片をコードする、請求項1に記載のポリヌクレオチドを含むベクターのライブラリに感染させること、
b.前記細胞集団をファージ表面上での前記抗体又はそのFc含有断片の発現を可能にする条件下で培養することと、及び
c.所望の又は増強された特性を有する前記抗体又はその断片を提示するファージを選択することを含む、前記方法。
【請求項25】
改善された特性を有する変異型の選択のために、請求項20に記載のファージライブラリを使用する方法。
【請求項26】
前記特性が、改善された標的結合、特異的な標的エピトープへの結合、Fc受容体結合親和性、減少されたグリコシル化部位、増加されたグリコシル化部位、及び増強された熱安定性からなる群から選択される、請求項23に記載の変異型を選択する方法。
【請求項27】
所望の生物活性について選択するために、請求項23に記載のファージライブラリを使用する方法であって、前記選択が、(a)抗体Fc断片を、請求項13に記載のファージライブラリから発現させること、及び(b)前記所望の生物活性を有するファージ粒子を選択することを含む、前記方法。
【請求項28】
請求項22に記載の方法から得られる、核酸をコードするFc含有抗体又は抗体断片。
【請求項29】
IgA、IgE、IgM、IgD、IgY、及びIgGからなる群から選択される、請求項23に記載の抗体又は抗体断片。
【請求項30】
前記抗体又はその断片が、マウス抗体、キメラ抗体、ヒト化抗体、又はヒト抗体である、請求項23に記載の抗体又は抗体断片。
【請求項31】
請求項24又は25に記載の方法を用いて選択されたFc含有抗体又は抗体断片を含む、医薬組成物。
【図1】

【図2】

【図3】
【図4】
【図5】

【図6】
【図7】
【図8】

【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2013−510591(P2013−510591A)
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−539959(P2012−539959)
【出願日】平成22年11月15日(2010.11.15)
【国際出願番号】PCT/US2010/056675
【国際公開番号】WO2011/062859
【国際公開日】平成23年5月26日(2011.5.26)
【出願人】(509087759)ヤンセン バイオテツク,インコーポレーテツド (77)
【Fターム(参考)】
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願日】平成22年11月15日(2010.11.15)
【国際出願番号】PCT/US2010/056675
【国際公開番号】WO2011/062859
【国際公開日】平成23年5月26日(2011.5.26)
【出願人】(509087759)ヤンセン バイオテツク,インコーポレーテツド (77)
【Fターム(参考)】
[ Back to top ]