
繊維
【課題】 従来とは異なり、マトリックスポリマーと分子鎖の絡み合いが抑制され、紡糸過程での伸長変形を阻害し難い樹状ポリエステルを用いることにより、紡糸過程での流動性を向上させ紡糸温度の低温化を図るとともに、糸斑が小さく、さらに繊維の複屈折あたりの弾性率が向上、すなわち高弾性率でありながら分子配向の低い繊維を提供するものである。
【解決手段】 芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
【解決手段】 芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、マトリックスポリマーに樹状ポリエステルをブレンドしたポリマーブレンドから成る繊維に関するものである。
【背景技術】
【0002】
ポリエステルやポリアミドなどの熱可塑性ポリマーを用いた繊維は幅広く利用されており、産業上の価値は極めて高い。そして、これらの製造方法は、ポリマーを融解した後、細い口金孔から押し出す溶融紡糸が採用されている。この時、溶融紡糸における紡糸温度はポリマーの融点や溶融粘度により決定されるが、融点が同じであっても高粘度ポリマーを紡糸する際は低粘度ポリマーの場合に比べ高い紡糸温度を設定する必要がある。しかしながら、過度に高温にするとポリマーそのものの分解が促進されるなど不具合があった。また、繊維に機能性を付加するため機能付加成分を同時に紡糸することが行われているが、この機能付加成分の耐熱性が低い場合には分解やそれに伴うガスが発生する場合がある。このため、特に高粘度ポリマーの紡糸や耐熱性の低い機能性付加成分を同時に紡糸する場合に於いてはポリマーの流動性を向上させ、紡糸温度をなるべく低温下する技術が重要である。
【0003】
このため、種々の減粘剤が利用される場合があるが、現在まで成功した例は少ない。従来の技術の中でも特に興味深いものとしては、超分岐ポリマー(ハイパーブランチポリマー)をナイロンに添加した例を挙げることができる(特許文献1)。ここでは、マトリックスポリマーと非反応性の超分岐ポリマーをマトリックスポリマーに添加することで、未添加の場合に比べ分子量減少が7%未満で、かつ流動性が向上することが記載されている。しかしながら、実際には超分岐ポリマーの主鎖である枝構造部分(D)が脂肪族(実施例ではε−カプロラクタムから誘導された物)となるため、柔軟性が著しく高い。このため、超分岐ポリマーとマトリックスポリマーが非反応性であるとしても、超分岐ポリマー主鎖部分とマトリックスポリマー主鎖部分でいわゆる分子鎖の絡み合いが多く発生してしまう可能性があった。これは、樹脂の押し出し加工などでは変形量が小さく、さらに剪断変形が支配的であるため大きな問題とはならないが、特に紡糸などの大きな伸長変形を伴う場合には深刻な問題を引き起こしてしまう場合があった。すなわち、分子鎖の絡み合いの程度が大きくなることで、マトリックスポリマー分子鎖のスムーズな伸長変形が阻害され、紡糸性を著しく損ない、場合よってはポリマーの弾性的振る舞いが顕著となり紡糸不能に陥る場合があった。また、紡糸不能に至らないまでも紡糸線での伸長変形に大規模な経時変動(斑)が発生し、糸の太細斑が過大となり、実用的な繊維が得られない場合があった。特に、マトリックスポリマーとして高粘度ポリマーや高分子量ポリマーを用いた場合、また細繊度紡糸や紡糸速度を高速化した場合にこの傾向が顕著であった。
【0004】
このため、紡糸や繊維に適したハイパーブランチポリマーが、またこれが添加された糸斑の小さな繊維が求められていた。
【特許文献1】特表2005−513186号公報(6〜12ページ)
【発明の開示】
【発明が解決しようとする課題】
【0005】
従来とは異なり、マトリックスポリマーと分子鎖の絡み合いが抑制され、紡糸過程での伸長変形を阻害し難い樹状ポリエステルを用いることにより、紡糸過程での流動性を向上させ紡糸温度の低温化を図るとともに、糸斑が小さく、さらに繊維の複屈折あたりの弾性率が向上、すなわち高弾性率でありながら分子配向の低い繊維を提供するものである。
【課題を解決するための手段】
【0006】
上記目的は、以下の手段により達成される。
(1)芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
(2)熱可塑性のマトリックスポリマーがポリエステルである(1)記載の繊維。
(3)弾性率が110〜1000cN/dtexである請求項1または2記載の繊維。
(4)弾性率が50cN/dtex以上、110cN/dtex未満である(1)または(2)記載の繊維。
(5)樹状ポリエステルをマトリックスポリマーにブレンドしたポリマーブレンド全体に対して、エステル交換反応抑制剤を0.01〜1wt%含有する(1)〜(4)のいずれか1項記載の繊維。
(6)繊維がモノフィラメントである(1)〜(3)および(5)のいずれか1項記載の繊維。
(7)請求項(1)〜(2)および(4)のいずれか1項記載の繊維が芯糸として用いられた混繊糸。
(8)(1)〜(7)のいずれか1項記載の繊維を少なくとも一部に使用した繊維製品。
【発明の効果】
【0007】
本発明の繊維により、糸斑が小さく、さらに繊維の複屈折あたりの弾性率を向上することができる。すなわち同じ複屈折であれば高弾性率に、同じ弾性率であれば低複屈折とできるため、高弾性率でありながら分子配向を低下させることで耐摩耗性を向上させることができ、産業資材、小さな糸斑ともあいまって、特にスクリーン紗などに好適に使用することができる。また、紡糸混繊の一成分に利用することで、低分子配向であっても芯糸を形成し易い繊維を得ることができ、高級衣料用として最適な混繊糸を提供することができる。
【発明を実施するための最良の形態】
【0008】
本発明で言う樹状ポリエステルとは、ハイパーブランチポリマーの一種であるが、本発明で用いられる樹状ポリエステルは、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含んでいる。本発明で用いられる樹状ポリエステルは、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルである。
【0009】
ここで、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および、芳香族ジカルボニル単位(R)は、それぞれ下式(1)で表される構造単位であることが好ましい。
【0010】
【化1】

【0011】
ここで、R1、R3は、それぞれ芳香族残基であり、R2は芳香族および脂肪族を両方含む残基である。またR1、R2、およびR3は、それぞれ複数の構造単位を含んでも良い。
【0012】
上記の芳香族残基としては、置換または非置換のフェニレン基、ナフチレン基、ビフェニレン基などが挙げられ、脂肪族残基としてはエチレン、プロピレン、ブチレンなどが挙げられる。R1、R2およびR3は、好ましくは、それぞれ下式で表される構造単位から選ばれる少なくとも1種以上の構造単位であり、R2は芳香族残基である(R2−1)と脂肪族残基である(R2−2)を両方含む。
【0013】
【化2】
【0014】
ただし、式中nは2〜8の整数である。
【0015】
本発明の樹状ポリエステルは、3官能以上の有機残基(B)が、互いにエステル結合および/またはアミド結合により直接、あるいは、枝構造部分(D)を構成するP、QおよびRから選ばれる構造単位を介して結合した、3分岐以上の分岐構造を基本骨格としている。分岐構造は、3分岐、4分岐など単一の基本骨格で形成されていてもよいし、3分岐と4分岐など、複数の基本骨格が共存していてもよい。ポリマーの全てが該基本骨格からなる必要はなく、たとえば末端封鎖のために末端に他の構造が含まれても良い。また、Bが3官能性の有機残基である場合には、樹状ポリエステル中には、Bの3つの官能基が全て反応している構造、2つだけが反応している構造、および1つだけしか反応していない構造が混在していてもよい。好ましくはBの3つの官能基が全て反応した構造が、B全体に対して15モル%以上であることが好ましく、より好ましくは20モル%以上であり、さらに好ましくは30モル%以上である。また、Bが4官能性の有機残基である場合には、樹状ポリエステル中には、Bの4つの官能基が全て反応している構造、3つだけが反応している構造、2つだけが反応している構造、および1つしか反応していない構造が混在していてもよい。好ましくはBの4つの官能基が全て反応した構造がB全体に対して10モル%以上かつ3つの官能基が反応した構造が20モル%以上であることが好ましく、より好ましくは4つの官能基が反応した構造がB全体に対して20モル%以上かつ3つの官能基が反応した構造がB全体に対して30モル%以上であり、さらに好ましくは4つの官能基が反応した構造がB全体に対して25モル%以上かつ3つの官能基が反応した構造がB全体に対して35モル%以上である。
【0016】
Bは3官能化合物および/または4官能化合物の有機残基であることが好ましく、3官能化合物の有機残基であることが最も好ましい。
【0017】
上記3分岐の基本骨格を模式的に示すと、式(2)で示される。また上記4分岐の基本骨格を模式的に示すと、式(3)で示される。
【0018】
【化3】
【0019】
【化4】
【0020】
本発明の樹状ポリエステルは、溶融液晶性を示すことが好ましい。ここで溶融液晶性を示すとは、室温(25℃)から昇温していった際に、ある温度域で液晶状態を示すことである。液晶状態とは、剪断下において光学的異方性を示す状態である。
【0021】
溶融液晶性を示すために、3分岐の場合の基本骨格は、下式(4)で示されるように、Bが、P、QおよびRから選ばれる構造単位により構成される枝構造部分(D)を介して結合していることが好ましい。
【0022】
【化5】
【0023】
同様に、4分岐の場合の基本骨格は、下式(5)で示される構造が好ましい。
【0024】
【化6】
【0025】
3官能の有機残基Bとしては、カルボキシル基、ヒドロキシル基およびアミノ基から選ばれる官能基を含有する化合物の有機残基であることが好ましい。例えばグリセロール、1,2,3−トリカルボキシプロパン、ジアミノプロパノール、ジアミノプロピオン酸などの脂肪族化合物や、トリメシン酸、トリメリット酸、4−ヒドロキシ−1,2−ベンゼンジカルボン酸、フロログルシノール、α−レゾルシン酸、β−レゾルシン酸、γ−レゾルシン酸、トリカルボキシナフタレン、ジヒドロキシナフトエ酸、アミノフタル酸、5−アミノイソフタル酸、アミノテレフタル酸、ジアミノ安息香酸、メラミンなどの芳香族化合物の残基が好ましく用いられる。下式で表される芳香族化合物の残基がさらに好ましい。
【0026】
【化7】
【0027】
上記の3官能の有機残基の具体例としては、フロログルシノール、トリメシン酸、トリメリット酸、無水トリメリット酸、α−レゾルシル酸、4−ヒドロキシ−1,2−ベンゼンジカルボン酸などの残基が好ましく、さらに好ましくは、トリメシン酸、α−レゾルシル酸の残基であり、最も好ましくはトリメシン酸の残基である。
【0028】
また、4官能以上の有機残基Bとしては、カルボキシル基、ヒドロキシル基およびアミノ基から選ばれる官能基を含有する化合物の有機残基であることが好ましい。例えば、エリスリトール、ペンタエリスリトール、スレイトール、キシリトール、グルシトール、マンニトール、1,2,3,4−ブタンテトラカルボン酸、1,2,4,5−シクロヘキサンテトラオール、1,2,3,4,5−シクロヘキサンペンタンオール、1,2,3,4,5,6−シクロヘキサンヘキサンオール、1,2,4,5−シクロヘキサンテトラカルボン酸、1,2,3,4,5−シクロヘキサンペンタカルボン酸、1,2,3,4,5,6−シクロヘキサンヘキサカルボン酸、クエン酸、酒石酸などの脂肪族化合物の残基や1,2,4,5−ベンゼンテトラオ−ル、1,2,3,4−ベンゼンテトラオ−ル、1,2,3,5−ベンゼンテトラオ−ル、1,2,3,4,5−ベンゼンペンタンオ−ル、1,2,3,4,5,6−ベンゼンヘキサンオ−ル、2,2’,3,3’−テトラヒドロキシビフェニル、2,2’,4,4’−テトラヒドロキシビフェニル、3,3’,4,4’−テトラヒドロキシビフェニル、3,3’,5,5’−テトラヒドロキシビフェニル、2,3,6,7−ナフタレンテトラオール、1,4,5,8−ナフタレンテトラオール、ピロメリット酸、メロファン酸、プレーニト酸、メリット酸、2,2’,3,3’−ビフェニルテトラカルボン酸、2,2’,4,4’−ビフェニルテトラカルボン酸、3,3’,4,4’−ビフェニルテトラカルボン酸、3,3’,5,5’−ビフェニルテトラカルボン酸、2,3,6,7−ナフタレンテトラカルボン酸、1,4,5,8−ナフタレンテトラカルボン酸、2,3,6,7−ナフタレンテトラオール、1,4,5,8−ナフタレンテトラオール、1,2,4,5,6,8−ナフタレンヘキサオール、1,2,4,5,6,8−ナフタレンヘキサカルボン酸、没食子酸、などの芳香族化合物の残基が挙げられる。下式で表される芳香族化合物の残基がさらに好ましい。
【0029】
【化8】
【0030】
上式の4官能の有機残基の具体例としては、1,2,4,5−ベンゼンテトラオ−ル、1,2,3,4−ベンゼンテトラオ−ル、1,2,3,5−ベンゼンテトラオ−ル、ピロメリット酸、メロファン酸、プレーニト酸、没食子酸などの残基が好ましく、没食子酸の残基が特に好ましい。
【0031】
また、樹状ポリエステルの芳香族オキシカルボニル単位(P)、芳香族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)は、樹状ポリエステルの分岐間の枝構造部分(D)を構成する単位である。p、qおよびrはそれぞれ構造単位P、QおよびRの平均含有量(モル比)であり、Bの含有量bの1モルに対して、p+q+r=1〜10モルの範囲であることが好ましい。p+q+rは、より好ましくは、2〜6モルの範囲である。枝構造長が長すぎると、剛直で綿密な樹状構造に基づく剪断応答性などの効果やマトリックスポリマー分子鎖との絡み合い抑制効果が低減するため好ましくない。
【0032】
このp、qおよびrの値は、例えば、樹状ポリエステルをペンタフルオロフェノール50重量%:重クロロホルム50重量%の混合溶媒に溶解し、40℃でプロトン核の核磁気共鳴スペクトル分析を行い、それぞれの構造単位に由来するピーク強度比から求めることができる。各構造単位のピーク面積強度比から、平均含有率を算出し、小数点3桁は四捨五入する。Bの含有量bにあたるピークとの面積強度比から、枝構造部分(D)の平均鎖長を算出し、p+q+rの値とする。この場合にも小数点3桁は四捨五入する。
【0033】
pとqの比率およびpとrの比率(p/q、p/r)は、いずれも5/95〜95/5の範囲が好ましく、より好ましくは10/90〜90/10であり、さらに好ましくは20/80〜80/20である。この範囲であれば、液晶性が発現しやすく好ましい。p/qおよびp/rの比率を95/5以下とすることで、樹状ポリエステルの融点を適当な範囲とすることができるため好ましい。また、p/qおよびp/rを5/95以上とすることで樹状ポリエステルの溶融液晶性を発現することができるため好ましい。
【0034】
qとrは、実質的に等モルであることが好ましいが、末端基を制御するためにどちらかの成分を過剰に加えることもできる。q/rの比率としては0.7〜1.5の範囲であることが好ましく、より好ましくは0.9〜1.1である。ここでいう等モルとは、繰り返し単位内でのモル量が等しいことを意味し、末端構造は含めない。ここで、末端構造とは、枝構造部分(D)の末端を意味し、末端が封鎖されている場合などには、最も末端に近い枝構造部分(D)の末端を意味する。
【0035】
前記式(1)において、R1は芳香族オキシカルボニル単位由来の構造単位であり、具体例としては、p−ヒドロキシ安息香酸、6−ヒドロキシ−2−ナフトエ酸から生成した構造単位などが挙げられる。好ましくはp−ヒドロキシ安息香酸由来の構造単位であり、6−ヒドロキシ−2−ナフトエ酸由来の構造単位部併用することも可能である。また本発明の効果を損なわない範囲でグリコール酸、乳酸、ヒドロキシプロピオン酸、ヒドロキシ酪酸、ヒドロキシ吉草酸、ヒドロキシカプロン酸などの脂肪族ヒドロキシカルボン酸由来の構造単位を含有しても良い。
【0036】
R2は芳香族および脂肪族由来の構造を両方含む構造単位であり、芳香族単位としては、例えば、4,4’−ジヒドロキシビフェニル、ハイドロキノン、3,3’,5,5’−テトラメチル−4,4’−ジヒドロキシビフェニル、t−ブチルハイドロキノン、フェニルハイドロキノン、メチルハイドロキノン、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、2,2−ビス(4−ヒドロキシフェニル)プロパンおよび4,4’−ジヒドロキシジフェニルエーテル、など由来の構造単位が挙げられ、脂肪族単位としてはエチレングリコール、プロピレングリコール、1,4−ブタンジオールなど脂肪族ジオール由来の構造単位が挙げられる。好ましくは、4,4’−ジヒドロキシビフェニル、エチレングリコール由来の構造単位であり、4,4’−ジヒドロキシビフェニルとエチレングリコール由来の構造単位が含まれることが液晶性の制御の点から好ましい。
【0037】
本発明で用いる樹状ポリエステルとしては、このR2単位を芳香族と脂肪族の両方を含む構造単位とすることが特に重要である。まず、芳香族を含有させることにより枝構造に剛直性を与え(好ましくは液晶性を与え)、マトリックスポリマー分子鎖との絡み合いを抑制し、紡糸などの伸長大変形の場でも伸長変形を阻害することを抑制できるのである。これにより、工業的に充分な紡糸性(曳糸性、安定性など)が得られ、かつ糸斑も大きく改善できるのである。また、完全に芳香族だけにせず脂肪族も含有させるのは、マトリックスポリマーの伸長変形を大きく阻害しない範囲で適度な絡み合いを持たせることで、繊維の複屈折あたりの弾性率を向上させることができるからである。本発明では、このように芳香族と脂肪族を混合することで分子鎖の絡み合いを制御し、紡糸性・糸斑改善と複屈折あたりの弾性率向上という、従来、二律背反の効果を両立させているのである。ここで、複屈折あたりの弾性率が向上すると、同じ複屈折であれば高弾性率に、同じ弾性率であれば低複屈折とできるため、高弾性率でありながら分子配向を低下させることができ、やはり従来、二律背反であった繊維の弾性率と耐摩耗性を両立させることができるのである。このため、産業資材、小さな糸斑ともあいまって、特にスクリーン紗などに好適に使用することができる。
【0038】
R3は芳香族ジカルボニル単位由来の構造単位であり、例えば、テレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、4,4’−ジフェニルジカルボン酸、1,2−ビス(フェノキシ)エタン−4,4’−ジカルボン酸、1,2−ビス(2−クロロフェノキシ)エタン−4,4’−ジカルボン酸および4,4’−ジフェニルエーテルジカルボン酸など由来の構造単位が挙げられる。好ましくはテレフタル酸またはイソフタル酸由来の構造単位であり、特に両者を併用した場合に融点調節がしやすく好ましい。また、本発明の効果に影響を及ぼさない範囲で、セバシン酸やアジピン酸などの脂肪族ジカルボン酸由来の構造単位が一部含まれていてもよい。
【0039】
本発明の樹状ポリエステルの枝構造部分(D)は、主としてポリエステル骨格からなることが好ましいが、カーボネート構造やアミド構造、ウレタン構造などを、特性に大きな影響を与えない程度に導入することも可能である。中でもアミド構造を導入することが好ましい。このような別の結合を導入することで、多種多様な熱可塑性樹脂に対する相溶性を調整することが可能であり、好ましい。アミド結合の導入の方法としては、p−アミノ安息香酸、m−アミノ安息香酸、p−アミノフェノール、m−アミノフェノール、p−フェニレンジアミン、m−フェニレンジアミン、テトラメチレンジアミンペンタメチレンジアミン、ヘキサメチレンジアミン、2−メチルペンタメチレンジアミン、ノナメチレンジアミン、ウンデカメチレンジアミン、ドデカメチレンジアミン、2,2,4−/2,4,4−トリメチルヘキサメチレンジアミン、5−メチルノナメチレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン、1,3−ビス(アミノメチル)シクロヘキサン、1,4−ビス(アミノメチル)シクロヘキサン、1−アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン、ビス(4−アミノシクロヘキシル)メタン、ビス(3−メチル−4−アミノシクロヘキシル)メタン、2,2−ビス(4−アミノシクロヘキシル)プロパン、ビス(アミノプロピル)ピペラジン、アミノエチルピペラジンなどの脂肪族、脂環族、あるいは芳香族のアミン化合物などを共重合することが好ましい。中でもp−アミノフェノールまたはp−アミノ安息香酸の共重合が好ましい。
【0040】
樹状ポリエステルの枝構造部分(D)の具体例としては、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位およびテレフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位およびテレフタル酸由来の構造単位からなるものなどが挙げられる。
【0041】
特に好ましいのは、枝構造部分(D)が、下記構造単位(I)、(II)、(III)および(IV)から構成されることである。ここで、(I)は構造単位(P)の、(II)、(III)は構造単位(Q)の、(IV)は構造単位(R)の特に好ましい様態である。
【0042】
【化9】
【0043】
枝構造部分(D)が、上記構造単位(I)、(II)、(III)および(IV)から構成される場合には、上記構造単位(I)の含有量pは、p+q+rに対して30〜90モル%が好ましく、40〜80モル%がより好ましい。また、構造単位(VI)の含有量q(III)は、(II)と(III)の合計含有量qに対して70〜5モル%が好ましく、60〜8モル%がより好ましい。前記のように、構造単位(IV) の含有量rは、構造単位(II)および(III)の合計含有量qと実質的に等モルであることが好ましいが、いずれかの成分を過剰に加えてもよい。
【0044】
また、本発明の樹状ポリエステルの末端は、カルボキシル基、水酸基、アミノ基、またはそれらの誘導体が好ましい。水酸基の誘導体もしくは、カルボン酸の誘導体としては、メチルエステルなどのアルキルエステルやフェニルエステルやベンジルエステルなどの芳香族エステルが挙げられる。また、単官能エポキシ化合物、オキサゾリン化合物、オルトエステル、酸無水物化合物などを用いて末端封鎖することも可能である。末端封鎖の方法としては、樹状ポリエステルを合成する際に、あらかじめ単官能性の有機化合物を添加する方法や、ある程度樹状ポリステルの骨格が形成された段階で単官能性の有機化合物を添加する方法などが挙げられる。
【0045】
具体的には、水酸基末端やアセトキシ末端を封鎖する場合には、安息香酸、4−t−ブチル安息香酸、3−t−ブチル安息香酸、4−クロロ安息香酸、3−クロロ安息香酸、4−メチル安息香酸、3−メチル安息香酸、3,5−ジメチル安息香酸などを添加することで可能である。
【0046】
また、カルボキシル基末端の封鎖は、カルボン酸反応性単官能化合物を反応することにより行うことができる。ここで、カルボン酸反応性単官能化合物とは、常温または加熱時にカルボン酸と反応し、エステル、アミド、ウレタン、ウレア結合を形成しうる官能基を分子内に1つ有する化合物をいう。樹状ポリエステルの分子末端に存在するカルボン酸基に、カルボン酸反応性単官能化合物を反応させ、分子末端に単官能化合物を導入することにより、樹状ポリエステルの滞留安定性や耐加水分解性を向上させ、さらに他の熱可塑性ポリマーと混練した際には、熱可塑性ポリマーの分解を抑制でき、また樹状ポリエステルの分散性が向上することによって、流動性や物性の改良が期待できる。
【0047】
本発明の樹状ポリエステルに用いることのできるカルボン酸反応性単官能化合物としては、オキサゾリン、エポキシド、オルトエステル、イソシアネート、カルボジイミド、ジアゾ化合物から選ばれる1種類以上の化合物である。カルボン酸との反応性およびハンドリング性の観点から、オキサゾリン、エポキシド、オルトエステル、イソシアネートが好ましく用いることができる。カルボン酸反応性単官能化合物は、単独で使用または2種類以上のカルボン酸反応性単官能化合物を併用しても構わない。
【0048】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちオキサゾリン化合物としては、例えば、2−メトキシ−2−オキサゾリン、2−エトキシ−2−オキサゾリン、2−プロポキシ−2−オキサゾリン、2−ブトキシ−2−オキサゾリン、2−ペンチルオキシ−2−オキサゾリン、2−ヘキシルオキシ−2−オキサゾリン、2−ヘプチルオキシ−2−オキサゾリン、2−オクチルオキシ−2−オキサゾリン、2−デシルオキシ−2−オキサゾリン、2−シクロペンチルオキシ−2−オキサゾリン、2−シクロヘキシル−2−オキサゾリン、2−アリルオキシ−2−オキサゾリン、2−メタアリルオキシ−2−オキサゾリン、2−フェノキシ−2−オキサゾリン、2−クレジル−2−オキサゾリン、2−p−フェニルフェノキシ−2−オキサゾリン、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−ペンチル−2−オキサゾリン、2−ヘキシル−2−オキサゾリン、2−ヘプチル−2−オキサゾリン、2−オクチル−2−オキサゾリン、2−ノニル−2−オキサゾリン、2−デシル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−シクロペンチル−2−オキサゾリン、2−シクロヘキシル−2−オキサゾリン、2−アリル−2−オキサゾリン、2−メタアリル−2−オキサゾリン、2−クロチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性、および耐熱性の観点から、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンが好ましく、特に好ましくは2−フェニル−2−オキサゾリンである。
【0049】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちエポキシ化合物としては、例えば、N−グリシジルフタルイミド、N−グリシジル−4−メチルフタルイミド、N−グリシジル−4,5−ジメチルフタルイミド、N−グリシジル−3−メチルフタルイミド、N−グリシジル−3,6−ジメチルフタルイミド、N−グリシジル−4−エトキシフタルイミド、N−グリシジル−4−クロルフタルイミド、N−グリシジル−4,5−ジクロルフタルイミド、N−グリシジルサクシンイミド、N−グリシジルヘキサヒドロフタルイミド、N−グリシジルマレインイミド、N−グリシジルベンズアミド、N−グリシジル−p−メチルベンズアミド、N−グリシジルナフトアミド、N−グリシジルステラアミド、o−フェニルフェニルグリシジルエーテル、2−メチルオクチルグリシジルエーテル、フェニルグリシジルエーテル、3−(2−キセニルオキシ)−1,2−エポキシプロパン、アリルグリシジルエーテル、ブチルグリシジルエーテル、ラウリルグリシジルエーテル、ベンジルグリシジルエーテル、シクロヘキシルグリシジルエーテル、α−クレシルグリシジルエーテル、p−tert−ブチルフェニルグリシジルエーテル、メタクリル酸グリシジルエーテル、エチレンオキサイド、プロピレンオキサイド、スチレンオキサイド、オクトイレンオキサイド、酢酸グリシジルエステル、プロピオン酸グリシジルエステル、ブタン酸グリシジルエステル、ペンタン酸グリシジルエステル、ヘキサン酸グリシジルエステル、オクタン酸グリシジルエステル、デカン酸グリシジルエステル、ネオデカン酸グリシジルエステル、安息香酸グリシジルエステルなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、エチレンオキサイド、プロピレンオキサイド、ブチルグリシジルエーテル、フェニルグリシジルエーテル、安息香酸グリシジルエステルが好ましく、特に好ましくは安息香酸グリシジルエステルである。
【0050】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちオルトエステル化合物としては、例えば、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト酢酸トリプロピル、オルト酢酸トリブチル、オルト酢酸トリベンジル、オルト蟻酸トリメチル、オルト蟻酸トリエチル、オルト蟻酸トリプロピル、オルト蟻酸トリブチル、オルト蟻酸トリベンジル、オルトプロピオン酸トリメチル、オルトプロピオン酸トリエチル、オルトプロピオン酸トリプロピル、オルトプロピオン酸トリブチル、オルトプロピオン酸トリベンジル、オルト安息香酸トリメチル、オルト安息香酸トリエチル、オルト安息香酸トリプロピル、オルト安息香酸トリブチル、オルト安息香酸トリベンジルなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性およびハンドリング性の観点から、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト蟻酸トリメチル、オルト蟻酸トリエチルが好ましく、特に好ましくはオルト酢酸トリメチルまたはオルト酢酸トリエチルである。
【0051】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちイソシアネート化合物としては、例えば、メチルイソシアネート、エチルイソシアネート、プロピルイソシアネート、ブチルイソシアネート、ペンチルイソシアネート、ヘキシルイソシアネート、ヘプチルイソシアネート、オクチルイソシアネート、ノニルイソシアネート、デシルイソシアネート、ドデシルイソシアネート、オクタデシルイソシアネート、ベンジルイソシアネート、シクロへキシルイソシアネート、フェニルイソシアネート、p−クロロフェニルイソシアネート、p−ニトロフェニルイソシアネート、2−クロロエチルイソシアネート、ステアロイルイソシアネート、p−トルオルスルフォニルイソシアネートが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、メチルイソシアネート、エチルイソシアネート、プロピルイソシアネート、ブチルイソシアネート、フェニルイソシアネートが好ましく、特に好ましくはフェニルイソシアネートである。
【0052】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちジアゾ化合物としては、例えば、ジアゾメタン、ジアゾエタン、ジアゾプロパン、ジアゾブタン、トリメチルシリルジアゾメタンが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、ジアゾメタンおよびトリメチルシリルジアゾメタンが好ましく用いられる。
【0053】
理論的には、上記末端の封鎖に用いる有機化合物を、封鎖したい末端基に相当する量添加することで末端封鎖が可能である。封鎖したい末端基相当量に対して、末端封鎖に用いる有機化合物を、1.005倍当量以上用いることが好ましく、より好ましくは1.008倍当量以上である。また、末端封鎖に用いる有機化合物の添加量は2.5倍当量以下であることが好ましい。末端封鎖に用いる化合物の添加量が少なすぎると、末端封鎖が充分ではない。一方、添加量が多すぎると、過剰に添加した化合物が系中に残存して、ガスを発生したりするため好ましくない。
【0054】
また、Bの含有量は、枝構造部分(D)の連鎖長が、樹状ポリエステルが樹状の形態をとるのに適した長さとなる観点から決定されるが、充分な樹状を形成させるためには、樹状ポリエステルを構成する全単量体の含有量に対して7.5モル%以上とすることが重要であり、10モル%以上がより好ましく、さらに好ましくは20モル%以上である。一方、枝構造部が過度に密に混み合わないようにするためには、Bの含有量の上限としては、50モル%以下とすることが重要であり、45モル%以下が好ましく、40モル%以下がより好ましい。
【0055】
また本発明の樹状ポリエステルは特性に影響が出ない範囲で、部分的に架橋構造を有していてもよい。
【0056】
本発明において、樹状ポリエステルの製造方法は、公知のポリエステルの重縮合法に準じて製造できる。前記R1で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、R2で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能以上の有機残基(B)を形成する3官能以上の多官能単量体を反応させる方法であって、該多官能単量体の添加量(モル)が、樹状ポリエステルを構成する全単量体(モル)に対して7.5モル%以上として製造する方法が好ましい。多官能単量体の添加量は、より好ましくは10モル%以上、より好ましくは15モル%以上、さらに好ましくは20モル%以上である。また、添加量の上限としては、50モル%以下が好ましく、より好ましくは33モル%以下、さらに好ましくは25モル%以下である。
【0057】
また、上記反応に際して、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体をアシル化した後、3官能以上の多官能単量体を反応させる態様も好ましい。また、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能以上の多官能単量体をアシル化した後、重合反応させる態様も好ましい。
【0058】
前記構造単位(I)、(II)、(III)および(IV)とトリメシン酸残基から構成される樹状ポリエステルを製造する場合を例に挙げて、好ましい製造方法を説明する。
(1)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、テレフタル酸およびポリエチレンテレフタレートポリマーから脱酢酸縮重合反応によって液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(2)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよびトリメシン酸から脱酢酸縮重合反応によって製造する方法。
(3)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸およびポリエチレンテレフタレートポリマーに無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって液晶性ポリエステルオリゴマーを合成し、さらにトリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(4)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよびトリメシン酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって製造する方法。
(5)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、テレフタル酸ジフェニルエステルおよびポリエチレンテレフタレートポリマーから脱フェノール重縮合反応により液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱フェノール重縮合反応によって製造する方法。
(6)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、テレフタル酸ジフェニルエステル、ポリエチレンテレフタレートポリマーおよびトリメシン酸のフェニルエステルから脱フェノール重縮合反応によって製造する方法。
(7)p−ヒドロキシ安息香酸、テレフタル酸、トリメシン酸にジフェニルカーボネートを反応させて、それぞれフェニルエステルとした後、4,4’−ジヒドロキシビフェニル、ポリエチレンテレフタレートポリマーを加え、脱フェノール重縮合反応によって製造する方法。
【0059】
なかでも(1)〜(4)の製造方法が好ましく、(3)および(4)の方法がより好ましい。
【0060】
(3)および(4)の製造方法において、無水酢酸の使用量は、鎖長制御の点からフェノール性水酸基の合計の0.95当量以上1.10当量以下であることが好ましく、1.00当量以上1.08当量以下であることがより好ましく、最も好ましくは1.02当量以上1.05当量以下である。無水酢酸量を制御すること、ジヒドロキシモノマーおよびジカルボン酸モノマーのいずれかを過剰に添加すること等により、末端基を制御することが可能である。
【0061】
分子量を上げるためには、トリメシン酸のカルボン酸量に相当する分だけ、4,4’−ジヒドロキシビフェニルなどのジヒドロキシモノマーを、ジカルボン酸モノマーに対して過剰に加え、全単量体におけるカルボン酸と水酸基当量を合わせることが好ましい。一方、カルボン酸を意図的に末端基に残す場合には、前記のようなジヒドロキシモノマーの過剰添加を行わないことが好ましい。さらに、水酸基を意図的に末端に残す場合には、ジヒドロキシモノマーをトリメシン酸のカルボン酸当量以上に過剰に添加し、かつ無水酢酸の使用量をフェノール性水酸基の1.00当量未満で行うことが好ましい。
【0062】
これらの方法により、本発明の樹状ポリエステルには、種々の熱可塑性ポリマーとの反応性に富む末端基構造を選択的に設けることが可能である。ただし、マトリックスポリマーとなる熱可塑性ポリマーによっては、過剰な反応性を抑制するために、単官能エポキシ化合物などを用いて末端を封鎖した方が分散状態を制御しやすい場合もある。
【0063】
脱酢酸重縮合反応を行う場合には、樹状ポリエステルが溶融する温度で、場合によっては減圧下で反応させ、所定量の酢酸を留出させ、重縮合反応を完了させる溶融重合法が好ましい。例えば、所定量のp−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよび無水酢酸を、攪拌翼および留出管を備え、下部に吐出口を備えた反応容器中に仕込む。混合物を、窒素ガス雰囲気下で攪拌しながら加熱して、水酸基をアセチル化させた後、200〜350℃まで昇温して脱酢酸重縮合反応を行い、酢酸を留出させる。酢酸が、理論留出量の50%まで留出した段階で、トリメシン酸を所定量加えて、さらに理論留出量の91%まで酢酸を留出させ、反応を完了させる。
【0064】
アセチル化させる条件としては、反応温度は、130〜170℃の範囲が好ましく、より好ましくは135〜155℃の範囲である。反応時間は、0.5〜6時間が好ましく、より好ましくは1〜2時間である。
【0065】
重縮合させる温度は、樹状ポリエステルが溶融する温度であり、好ましくは樹状ポリエステルの融点+10℃以上の温度である。具体的には、例えば、200〜350℃の範囲であり、240〜280℃が好ましい。重縮合させるときの雰囲気は、常圧窒素下でも問題ないが、減圧すると反応が早く進み、系内の残留酢酸が少なくなるため好ましい。減圧度は、0.1mmHg(13.3Pa)〜200mmHg(26600Pa)が好ましく、より好ましくは10mmHg(1330Pa)〜100mmHg(13300Pa)である。なお、アセチル化と重縮合は同一の反応容器で連続して行っても良いし、アセチル化と重縮合を異なる反応容器で行っても良い。
【0066】
重縮合反応が完了した後、反応容器内を樹状ポリエステルが溶融する温度に保ち、例えば、0.01〜1.0kg/cm2(0.001〜0.1MPa)に加圧し、反応容器下部に設けられた吐出口より、樹状ポリエステルをストランド状に吐出する。吐出口には断続的に開閉する機構を設け、液滴状に吐出することも可能である。吐出した樹状ポリエステルは、空気中もしくは水中を通過して冷却された後、必要に応じて、カッティングもしくは粉砕される。
【0067】
得られたペレット状、粒状または粉状の樹状ポリエステルは、さらに必要に応じて、熱乾燥や真空乾燥により水、酢酸などを除く。また、重合度の微調整、あるいは、さらに重合度を上げるために、固相重合をすることも可能である。固相重合は、例えば、上記により得られた樹状ポリエステルを、窒素気流下、または、減圧下、樹状ポリエステルの融点−50℃〜融点−5℃(例えば、200〜300℃)の温度範囲で1〜50時間加熱する方法が挙げられる。
【0068】
樹状ポリエステルの重縮合反応は無触媒でも進行するが、酢酸第一錫、テトラブチルチタネート、酢酸カリウムおよび酢酸ナトリウム、三酸化アンチモン、金属マグネシウムなどの金属化合物を使用することもできる。
【0069】
本発明の樹状ポリエステルは、数平均分子量は1,000〜40,000であることが好ましく、より好ましくは1,000〜20,000、さらに好ましくは1,000〜10,000であり、最も好ましくは1,000〜5,000の範囲である。なお、この数平均分子量は、樹状ポリエステルが可溶な溶媒を使用して、GPC−LS(ゲル浸透クロマトグラフ−光散乱)法により絶対分子量として測定した値である。
【0070】
また、本発明における樹状ポリエステルの溶融粘度は、0.01〜30Pa・sが好ましく、0.5〜20Pa・sがより好ましく、1〜10Pa・sが特に好ましい。なお、この溶融粘度は、樹状ポリエステルの液晶開始温度+10℃の条件で、ずり速度100/sの条件下で高化式フローテスターによって測定した値である。
【0071】
本発明では、上記した樹状ポリエステルをマトリックスポリマーにブレンドすることで、流動性向上効果が得られ、紡糸温度を低下させることができるが、このメカニズムは以下のように推定している。すなわち、J.Polym.Sci.PartB:Polum.Phys.,vol.34,2433(1996).によると、非相溶系ポリマーブレンドにおけるポリマーの粘度は分子量項+分散相互作用項+スリップ効果項で記述されるが、本発明で用いる樹状ポリエステルは、分子量項については低分子量化、分散相互作用項については樹状構造、スリップ効果項について液晶性の効果により、ポリマーブレンドの流動性を向上させていると考えられる。すなわち、単に低粘度ポリマーあるいは液晶ポリマー、脂肪族ハイパーブランチポリマーをブレンドさせただけでは得られない、より高度な流動性向上効果を得ることができるのである。また、Macromolecule,vol.38,10571(2005).ではハイパーブランチポリマーと通常の直鎖状のポリマーのサイズを同一分子量で比較したところ、ハイパーブランチポリマーは1/5以下の分子サイズとなること、さらにPolymer,vol.45,7491(2004)によると、ハイパーブランチポリマーと通常の直鎖状ポリマーで分子間の絡み合いを第2ビリアル係数A2で評価したところ、ハイパーブランチポリマーではA2が2桁小さく、自己分子での分子間絡み合いが極めて少ないことが報告されている。以上より、本発明で用いる樹状ポリエステルは、マトリックスポリマー中で有機ナノ粒子的に振る舞い、流動性を向上させていると考えられる。さらに、前記したようにR2部分を芳香族と脂肪族の混合とすることで、分子鎖の絡み合いを制御し、紡糸性・糸斑改善と複屈折あたりの弾性率向上という、従来、二律背反の効果を両立させているのである。
【0072】
本発明では上記した樹状ポリエステルを0.1〜10wt%、熱可塑性ポリマーから成るマトリックスポリマーにブレンドすることが重要である。
【0073】
マトリックスポリマーは、ポリエステル、ポリアミド、ポリフェニレンスルフィド、ポリオレフィン、ポリカーボネート、ポリエステルカーボネート、ポリイミド、ポリアミドイミド、ポリエーテルケトン、ポリエーテルエーテルケトン、ポリフッ化ビニリデンなどを挙げることができる。中でも、汎用性が高く、かつ弾性率が比較的高いポリエステルや、耐熱性・薬品性に優れるポリフェニレンスルフィドが好ましい。特に、極限粘度が1.0dL/g以上の高分子量ポリエステルは融点はそれほど高くないものの超高粘度のため、紡糸温度を衣料用ポリエステルの場合に比べ10〜20℃程度高くする場合も有り、加水分解や熱分解抑制による高分子量保持の観点から有用である。また、汎用的ではないが、ポリエーテルケトン、ポリエーテルエーテルケトン、ポリイミド、ポリフッ化ビニリデンなど通常、紡糸温度がかなりの高温になるポリマー種も流動性向上効果により紡糸温度を低下させる効果からすると、好ましいポリマーである。特にポリフッ化ビニリデンなど腐食性物質や有害物質が発生しやすいポリマーでは、紡糸温度低下効果は有用である。また、分子量が10万以上の高分子量ポリマーの場合には、分子鎖の絡み合い抑制効果は有用である。特に、ポリ乳酸のように高分子量であるが熱分解温度が低いポリマーの場合には、紡糸温度を低下させることができると、加水分解や熱分解による分子量低下を押さえられるだけでなく、分解ガスの発生を抑制できるため非常に有用である。本発明においては、汎用性の観点から極限粘度1.0dL/g以上の高分子量ポリエチレンテレフタレート(PET)やポリエチレンナフタレート(PEN)をマトリックスポリマーとすることが好ましい。また、紡糸温度が高くなり熱分解しやすい、重量平均分子量15万以上のポリ乳酸、また融点が向上したステレオコンプレックスポリ乳酸をマトリックスポリマーとすることも有用性が高い。
【0074】
また別の観点では、ポリ乳酸とPETやポリブチレンテレフタレート(PBT)、ポリトリメチレンテレフタレート(PTT)、ナイロン6、ナイロン66などを複合紡糸する際に、ポリ乳酸の相手となるポリマーに本発明の樹状ポリエステルを添加すると、流動性を向上させることで複合紡糸の紡糸温度を低下させることができるため、これも有用である。
【0075】
さらに別の観点では、紡糸温度を低下できるということは紡糸口金付近での温度も低下できるため、口金汚れを低減し経時的な紡糸安定性を向上できるとともに、口金修正周期を延長できるため生産効率の向上も図ることができる。特に、PBTやPTTに酸化チタンを含有させた場合には、PETの場合とは比べものにならないほど口金孔外周に酸化チタンを核とする汚れが堆積しやすいという問題があるが、本発明ではこれの抑制にも寄与することが可能である。
【0076】
また、樹状ポリエステルのブレンド率は0.1wt%以上であれば流動性向上による紡糸温度低下効果や弾性率向上効果が認められ、0.7wt%以上であればより効果が上がり好ましい。一方、ブレンド率を10wt%以下とすることで、伸長変形を大きく阻害することなく良好な紡糸性と糸斑を達成することができる。好ましくは2wt%以下である。
【0077】
本発明の繊維では糸の太細斑の指標であるウースター斑(U%)は5%以下であることが重要である。これにより、糸の強度や弾性率の長手方向斑が小さくなり、特に産業資材用途に用いる場合に、材料設計し易くなるのである。また、衣料用繊維の観点からは染色斑を抑制するためにU%は2%以下であることが好ましく、1.2%以下であることがより好ましい。U%は小さいほど好ましいが、現実的な下限値としては0.1%である。紡糸性を向上させ、糸斑を小さくするためには、樹状ポリエステルとマトリックスポリマーの反応を抑制することが有用であり、樹状ポリエステルの末端封鎖が有効である。特にカルボキシ末端は封鎖しておくことが好ましい。
【0078】
また、エステル交換反応抑制剤の添加も効果的であり、ポリマーブレンド全体に対して0.01〜1wt%添加することが好ましい。多量添加の方が反応抑制には好ましいが、過度に添加すると紡糸性や糸斑が低下する場合があるので、0.05〜0.2wt%添加とすることがより好ましい。
【0079】
本発明で言うエステル交換反応抑制剤とは、樹状ポリエステルとマトリックスポリマーとの反応を抑制するものであり、特にマトリックスポリマーがポリエステルおよび/またはポリアミドの時に効果が大きい。これは、残留金属イオンなどに対して単座配位子または多座配位子として配位し、金属イオンの触媒能を失活させる作用を有する。例えば、トリアゾール系化合物、多価アミン化合物、ヒドラジン誘導体系化合物、リン系化合物、イオウ系化合物などが挙げられる。以下に具体例をあげる。
【0080】
トリアゾール系化合物の具体例としては、ベンゾトリアゾール、3−(N−サリシロイル)アミノ−1,2,4−トリアゾールなどが挙げられる。
【0081】
多価アミンの具体例としては、3,9−ビス[2−(3,5−ジアミノ−2,4,6−トリアザフェニル)エチル]−2,4,8,10−テトラオキサスピロ[5.5]ウンデカン、エチレンジアミン−テトラアセチックアシッド、エチレンジアミン−テトラアセチックアシッドのアルカリ金属塩(Li,Na,K)塩、N,N’−ジサリシリデン−エチレンジアミン、N,N’−ジサリシリデン−1,2−プロピレンジアミン、N,N’’−ジサリシリデン−N’−メチル−ジプロピレントリアミン、3−サリシロイルアミノ−1,2,4−トリアゾールなどが挙げられる。
【0082】
ヒドラジン誘導体系化合物の具体例としては、デカメチレンジカルボキシリックアシッド−ビス(N’−サリシロイルヒドラジド)、イソフタル酸ビス(2−フェノキシプロピオニルヒドラジド)、N−ホルミル−N’−サリシロイルヒドラジン、2,2−オキザミドビス[エチル−3−(3,5−ジ−t−ブチル−4−ハイドロオキシフェニル)プロピオネート]、オギザリル−ビス−ベンジリデン−ヒドラジド、ニッケル−ビス(1−フェニル−3−メチル−4−デカノイル−5−ピラゾレート)、2−エトキシ−2’−エチルオキサニリド、5−t−ブチル−2−エトキシ−2’−エチルオキサニリド、N,N−ジエチル−N’,N’−ジフェニルオキサミド、N,N’−ジエチル−N,N’−ジフェニルオキサミド、オキサリックアシッド−ビス(ベンジリデンヒドラジド)、チオジプロピオニックアシッド−ビス(ベンジリデンヒドラジド)、イソフタリックアシッド−ビス(2−フェノキシプロピオニルヒドラジド)、ビス(サリシロイルヒドラジン)、N−サリシリデン−N’−サリシロイルヒドラゾン、N,N’−ビス[3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオニル]ヒドラジン、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,6−ヘキサメチレン−ビス(N −ヒドロキシエチル−N−メチルセミカルバジド)−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,10−デカメチレン−ジ−カルボキシリックアシッド−ジ−ヒドロキシエチルカルボニルヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,10−デカメチレン−ジ−カルボキシリックアシッド−ジ−サリシロイルヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−ジ(ヒドロキシエチルカルボニル)ヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−N,N’−ビス(ヒドロキシエチル)オキサミド−ジホスファイト、N,N’−ビス[2−〔3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオニルオキシ〕エチル]オキサミドなどが挙げられる。
【0083】
リン系化合物としては、ホスフェート系化合物やホスファイト系化合物など、分子中にリン原子を含む化合物が挙げられる。
【0084】
ホスフェート系化合物の具体例としては、脂肪族リン酸エステル(トリメチルホスフェート、トリエチルホスフェート、トリブチルホスフェート、トリ(2−エチルヘキシル)ホスフェート、トリブトキシエチルホスフェート、トリオレイルホスフェート、ジブチルホスフェート、モノブチルホスフェート、ジ(2−エチルヘキシル)ホスフェート、モノイソデシルホスフェート、2−アクリロイルオキシエチルアシッドホスフェート、2−メタクリロイルオキシエチルアシッドホスフェート、ジシクロペンチルハイポジホスフェートなど)、芳香族リン酸エステル(トリフェニルホスフェート、トリクレジルホスフェート、トリキシレニルホスフェート、トリス(イソプロピルフェニル)ホスフェート、トリス(o−フェニルフェニル)ホスフェート、トリス(p−フェニルフェニル)ホスフェート、トリナフチルホスフェート、クレジルジフェニルホスフェート、キシレニルジフェニルホスフェート、ジ(イソプロピルフェニル)フェニルホスフェート、o−フェニルフェニルジクレジルホスフェート、ジピロカテコールハイポジホスフェートなど)、脂肪族−芳香族リン酸エステル(ジフェニル(2−エチルヘキシル)ホスフェート、ジフェニル−2−アクリロイルオキシエチルホスフェート、ジフェニル−2−メタクリロイルオキシエチルホスフェート、フェニルネオペンチルホスフェート、ペンタエリスリトールジフェニルジホスフェート、エチルピロカテコールホスフェートなど)などの正リン酸エステル及びこれらの縮合物が挙げられる。
【0085】
ホスファイト系化合物の具体例としては、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト、テトラキス(2,4−ジ−t−ブチルフェニル)4,4’−ビフェニレンホスフォナイト、ビス(2,4−ジ−t−ブチルフェニル)ペンタエリスリトール−ジ−ホスファイト、ビス(2,6−ジ−t−ブチル−4−メチルフェニル)ペンタエリスリトール−ジ−ホスファイト、2,2−メチレンビス(4,6−ジ−t−ブチルフェニル)オクチルホスファイト、4,4’−ブチリデン−ビス(3−メチル−6−t−ブチルフェニル−ジ−トリデシル)ホスファイト、1,1,3−トリス(2−メチル−4−ジトリデシルホスファイト−5−t−ブチル−フェニル)ブタン、トリス(ミックスドモノおよびジ−ノニルフェニル)ホスファイト、トリス(ノニルフェニル)ホスファイト、4,4’−イソプロピリデンビス(フェニル−ジアルキルホスファイト)などが挙げられる。
【0086】
イオウ系化合物としては、分子中に硫黄原子を含んでいれば良く、具体的には、ジラウリル3,3′−チオジプロピオネート、ジミリスチル3,3′−チオジプロピオネート、ジステアリル3,3′−チオジプロピオネート、ラウリルステアリル3,3′−チオジプロピオネート、ペンタエリスリトール−テトラキス−(β−ラウリル−チオ−プロピオネート)、3,9−ビス(2−ドデシルチオエチル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなどが挙げられる。
【0087】
これらのエステル交換反応抑制剤は、ポリマーに含まれる残留金属イオンに強く配位して錯体形成しやすい物を選ぶことで、金属イオンの触媒能を失活させやすい。
【0088】
また、ポリマーをブレンド、紡糸する際にはポリマーの融点以上で処理されることから、エステル交換反応抑制剤自体の耐熱性も重要となる。本発明のエステル交換反応抑制剤の中で、ヒドラジン誘導体系化合物やイオウ系化合物と比較し、リン系化合物は耐熱性が高く着色しにくいため好ましく、この中でも脂肪族リン酸エステル系化合物が特に好ましい。
【0089】
本発明の繊維においては、複屈折あたりの弾性率が樹状ポリエステル未添加の場合に比べ高くすることができるが、これは以下のように推定される。すなわち、前記したように枝構造を完全に芳香族だけにせず脂肪族も含有させることで、マトリックスポリマーの伸長変形を大きく阻害しない範囲で分子鎖の適度な絡み合いを持たせることで、局所的に分子鎖の拘束性が強い部分を誘起させ、これを伸長変形過程でさらに拘束性を強くする。これにより、全体の伸長度(延伸度)としてはそれほど高くなくても(複屈折が低くとも)、局所的に分子鎖の拘束性が強い部分が存在するため、弾性率のような微小変形特性には大きな影響を与えると考えられる。弾性率の向上という観点からは前記した樹状ポリエステルのブレンド率は多いほど好ましいが、紡糸性や糸斑との兼ね合いから上限が定められる。極限粘度1.0dL/g以上の高分子量PETであっても1wt%添加で十分な効果が得られる。
【0090】
本発明の繊維においては、弾性率は110cN/dtex以上であれば産業資材用途に好適であり、好ましい。この場合、弾性率は高い方が好ましいが、弾性率の現実的な上限は2000cN/dtexである。また、比較的低複屈折であるため、屈曲疲労や摩耗特性も従来品よりも向上し、タイヤコードやタイヤのキャップレイヤー材などのゴム補強用途のみならず漁網や農業資材の他、スクリーン紗などにも好適に用いることができる。特にスクリーン紗は従来の捺染から、より薄くて精度が求められるフラットディスプレイの電極印刷などにも利用されるようになっており、さらなるモノフィラメントの細繊度化と高弾性率化が求められるようになってきている。しかしながら、これを達成するため単純に繊維を高倍率延伸しただけでは分子配向が高くなり過ぎ(複屈折が高くなり過ぎ)、耐摩耗性が低下し製織時の毛羽などが多発する問題があった。しかしながら、本発明では複屈折が低くとも弾性率が高いため、このような課題を解決しうるのである。なお、特にスクリーン用途に用いる場合にはモノフィラメントとすることが好ましい。また、スクリーン紗に適用する場合には、弾性率は110cN/dtex以上、複屈折は0.180以下であることが好ましい。
【0091】
また、本発明の繊維において、弾性率が50cN/dtex以上、110cN/dtex未満であれば衣料用途などに好適に用いることができる。特に、混繊糸の芯糸として有用である。ここで、混繊糸とは、異なる2種類の繊維を混合した物であり、混合方法としては、紡糸混繊、エア交絡や合撚、複合仮撚りなどを用いることができる。特に、紡糸混繊では同一の口金から紡糸し、引き取り速度が同速度であることがほとんどであるので、分子配向の調整が難しかった。例えば、単糸繊度が細い物と太い物を紡糸混繊する場合には、太繊度糸を芯糸として混繊糸の比較的内層部に位置させ、細繊度糸を鞘糸として比較的外層部に位置させたいのであるが、現実には細繊度糸の方が分子配向が進む(複屈折が大きい)ため、延伸時の張力バランスにより期待とは逆に細繊度糸が芯糸を形成してしまうことが普通であった。しかしながら、太繊度糸側に前記した樹状ポリエステルをブレンドすると、糸の複屈折は低くとも弾性率が高いため比較的芯糸を形成しやすくなり、期待通りの糸の配置とすることが可能となるのである。
【0092】
本発明の繊維では、強度は2cN/dtex以上が好ましく、現実的な上限としては20cN/dtexである。また、伸度は延伸糸で2〜60%、特に高弾性率が必要とされる産業資材分野では2〜15%、衣料用では25〜60%とすることが好ましい。繊維の断面形状は、丸断面、三葉などの多葉断面、中空、星形、不定形など適宜選択可能である。
【0093】
上記した繊維は、繊維巻き取りパッケージやトウ、カットファイバー、わた、ファイバーボール、コード、パイル、織編、不織布、紙、液体分散体など多用な繊維製品とすることができる。
【0094】
本発明の繊維を得る方法の一例について説明する。樹状ポリエステルについては前記した通りであるが、ブレンド、繊維化については例えば以下のような方法を用いることができる。すなわち、前記した構造単位(I)〜(IV)とトリメシン酸残基から構成される樹状ポリエステルにおいて末端アセチル基が安息香酸で封鎖された絶対分子量2000〜5000の範囲の樹状ポリエステルとマトリックスポリマーを必要に応じ乾燥し、二軸押し出し混練機に導入する。この時、ブレンド装置としてはブレンド斑を低減するために二軸押し出し混練機とすることが好ましい。ここで、作製したポリマーブレンドをそのまま紡糸機に導いても、マスターペレットとして一旦ペレット化しても良い。省力化のためには混練直結紡糸が好ましいが、樹状ポリエステルのブレンド率やポリエステル分子量が異なる品種をいくつかつくるなど汎用性を持たせるためにはマスターペレット化が好ましい。また、混練直結紡糸の場合には、二軸押し出し混練機では一軸押し出し混練機の場合とは異なり、混練機中で誘起された発泡が仕込み側に抜け難いため、発泡が繊維にまで混入し糸切れが頻発する場合がある。このため、特に高分子量ポリエステルなど高粘度ポリマーをマトリックスポリマーとする場合には、二軸押し出し混練機の吐出側でベントを行い、泡を抜く操作を行うことが好ましい。なお、マスターペレット化場合にもガット切れが頻発する時はベントを行うことが好ましい。また、本発明においては樹状ポリエステル添加による良流動化効果により、未添加の場合に比べ同一温度であればスクリュートルクが小さくなるため、混練温度の低温化が可能である。これにより、ポリマーの熱分解や熱変性、また加水分解などを抑制することができ、バージンポリマーが本来持っていた高分子量や易加工性などを利用し易くできるのである。
【0095】
なお、樹脂加工の場合にはガラス繊維などの無機フィラーを多量に混合させることで機械的特性(弾性率向上など)やガスバリア性を向上させることも多いが、繊維化の場合に無機フィラーを混合させると紡糸機内のフィルターで詰まりが発生し濾圧が急上昇したり、また紡糸口金孔に無機フィラーが詰まり紡糸不能に陥る場合がある。また、紡糸不能に至らずとも、紡糸口金孔からのポリマーの吐出が安定せず糸切れの頻発や糸斑の悪化などの問題が発生する場合がある。このため、繊維化の場合には樹脂加工とは異なり、無機フィラーは混合しない方が良く、混合したとしてもブレンドポリマー全体に対し0.5wt%未満である。無機フィラーを0.5wt%以上混合した場合には、U%を5%以下とすることはできない。ここで言う無機フィラーとは、80wt%以上が無機物から構成され、円換算の平均直径が10nm以上かつ平均長さが100nm以上のものである。
【0096】
先の樹状ポリエステルブレンドでマスターペレット化した場合には、紡糸過程でバージンポリマーで希釈されるわけであるが、この時も二軸押し出し混練機を用いる方がブンレンドの均一性の観点から好ましい。というのは、本発明では樹状ポリエステルブレンド率で良流動化効果の程度が異なるため、ポリマーブレンド中でブレンドが不均一であるとスクリュートルクや先端圧、濾圧、口金背面圧、ひいては紡糸応力などの斑が発生し、安定した紡糸が不能となる場合があるからである。やむを得ず一軸押し出し混練機を用いる場合には、ダルメージなどの混練機能を付加するとともに、一軸押し出し混練機吐出付近や紡糸機あるいは紡糸パック内に静止混練器を設け、充分にブレンドを均一化することが好ましい。
【0097】
前記したように、樹状ポリエステルブレンドによる良流動化効果のため、未添加の場合に比べ混練機温度を低温化できるのであるが、紡糸機についても設定温度を低下させることが可能であり、例えば高分子量ポリマーによる高粘度のため通常では紡糸温度を融点より大幅に高温化せざるを得ない場合であっても、樹状ポリエステル添加により5℃以上の低温化も可能である。この効果は、高粘度ポリマーほど大きく発現する。また、この良流動化効果により紡糸口金孔からのポリマーの吐出が安定し、本来紡糸が不安定で糸斑が発生しやすい高粘度ポリマー紡糸であっても糸斑を小さくすることができる。
【0098】
また、本発明で用いる樹状ポリマーは枝構造に芳香族成分を導入し剛直性を向上、好ましくは液晶性を発現させるため、マトリックスポリマー分子鎖と絡み合い難いため、伸長変形がスムーズになるため、紡糸性が向上し糸斑が低減されるのみならず、枝構造が脂肪族のハイパーブランチポリマー添加では難しかった高速紡糸も可能となるのである。これも紡糸という伸長変形場特有の効果である。一方、本発明で用いる樹状ポリマーは枝構造に脂肪族成分も混合しているため、適度な分子鎖の絡み合いがあり複屈折あたりの弾性率が向上するが、この効果も高速紡糸や高倍率延伸など極限的な伸長変形場で強く発現する効果である。
【0099】
本発明において、紡糸温度は、樹状ポリエステル未添加の場合に比較して5〜20℃低下させることが好ましい。より好ましくは、7〜15℃低下である。また、紡糸速度はマトリックスポリマーの物性や繊維の目的によって異なるが、500〜6000m/分程度とすることができる。特に、産業資材用途で高弾性率が必要な場合には、高分子量ポリマーを用い、500〜1500m/分とし、その後高倍率延伸することが好ましい。
【0100】
延伸に際しては、特に予熱温度を適切に設定することが好ましい。というのは本発明で用いる樹状ポリエステルはガラス転移温度などの軟化温度が70℃より高い場合があり、例えばPETの通常の予熱温度である85〜95℃程度では、樹状ポリエステルが延伸過程で異物として振る舞い結果として延伸糸のタフネスの低下を招く場合がある。この影響は、特に高倍率延伸時ほど顕著に現れる。このため、樹状ポリエステルの添加量が微量であっても予熱温度は樹状ポリエステルのガラス転移温度や軟化温度以上に設定することが好ましい。予熱温度の上限としては、予熱過程で繊維の自発伸長により糸道乱れが発生しない温度とすることが好ましい。この延伸時の予熱温度設定も糸斑低減に寄与することができる。
【0101】
なお、スクリーン紗に用いる高弾性率細繊度モノフィラメントでは、高度に分子配向させて弾性率を向上させるのであるが、これだけでは耐摩耗性が低下し、製織過程で毛羽などが多発する場合が有り、この時は弛緩熱処理を行い分子配向を低下させこれを回避する場合がある。しかし、同時に弾性率も低下するため、従来技術では弾性率と耐摩耗性は二律背反であったが、本発明では複屈折あたりの弾性率が高いため、弛緩熱処理による低配向化が不要であり、この二律背反を解決できるのである。また、細繊度モノフィラメント紡糸であるが故、低吐出量であり紡糸機内でのポリマーの滞留時間が長く、さらに異常滞留によるポリマーの熱劣化によりゲル化などが発生しやすく、これがモノフィラメントの節を誘発する場合がある。この節とは繊維の異常太部であり、製織欠点となるのである。このため、この節はゼロにすべきものであるが、紡糸機や紡糸パック・口金構造の工夫の他にはあまり良い手段が無かった。しかしながら、本発明では溶融部である混練機温度や紡糸温度を低温化できるため、低吐出量であっても前記問題を緩和でき、節の発生を抑制できるため、産業上の価値は高い。
【0102】
また、ポリエステルやポリアミドで行われる機能付加成分を同時に紡糸する場合、機能付加成分の耐熱性が低くともこれの分解や変性・劣化を抑制することができるのである。
【実施例】
【0103】
以下、本発明を実施例を用いて詳細に説明する。なお、実施例中の測定方法は以下の方法を用いた。
【0104】
A.絶対分子量
樹状ポリエステルの絶対分子量は樹状ポリエステルが可溶な溶媒であるペンタフルオロフェノールを使用して、GPC−MALLS(ゲル浸透クロマトグラフ(ShodexGPC−101)−光散乱検出器(Wyatt製DAWN HELEOS))により、試料濃度0.04%、測定温度23℃で測定した。
【0105】
B.重量平均分子量
本発明の熱可塑性マトリックスポリマーの重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)により測定した標準PMMA換算の値である。GPC測定は、検出器にWATERS社示差屈折計WATERS410を用い、ポンプにMODEL510高速液体クロマトグラフィーを用いて、溶媒にヘキサフルオロイソプロパノールを用いて測定した。
【0106】
ただし、ポリ乳酸の重量平均分子量は以下のようにして求めた。試料のクロロホルム溶液にTHF(テトロヒドロフラン)を混合し測定溶液とした。これをWATERS社製GPC WATERS2690を用いて25℃で測定し、ポリスチレン換算で重量平均分子量を求めた。
【0107】
C.ポリエステルの極限粘度
ポリエステルの極限粘度はo−クロロフェノールに溶解してオストワルド式粘度計を用いて25℃で測定した。
【0108】
D.ナイロンの相対粘度
98%硫酸水溶液にナイロンを溶解し0.01g/mLの濃度に調整した後、オストワルド式粘度計を用いて25℃で測定した。
【0109】
E.樹状ポリエステルの化学組成比
樹状ポリエステルの化学組成比は核磁気共鳴装置(日本電子製JNM−AL400)を用いて、ペンタフルオロフェノール/重水素化クロロホルム(50/50)混合溶媒に溶解して、40℃で1H−NMR測定を行い、ピーク強度比から各成分の化学組成比を算出した。
【0110】
F.融点およびガラス転移点
TA Instruments社製DSC2920 Modulated DSCを用いて2nd runでポリマーの融解を示すピークトップ温度をポリマーの融点とした。この時の昇温速度は16℃/分、サンプル量は10mgとした。
また、同じく2nd runでの階段状の吸熱を示す領域の中点をガラス転移点とした。
【0111】
G.液晶開始温度
剪断応力加熱装置(CSS−450)により、剪断速度1.0(1/秒)、昇温速度5.0℃/分、対物レンズ60倍において測定し、視野全体が流動開始する温度とした。
【0112】
H.繊維のウースター斑(U%)
ツェルベガーウスター株式会社製USTER TESTER 4を用い、給糸速度200m/分でノーマルモードで測定を行った。
【0113】
I.繊維の力学特性(強度、伸度、弾性率)
室温(25℃)で、初期試料長=200mm、引っ張り速度=200mm/分とし、JIS L1013に示される条件で荷重−伸長曲線を求めた。次に破断時の荷重値を初期の繊度で割り、それを強度とし、破断時の伸びを初期試料長で割り伸度として強伸度曲線を求めた。また、弾性率は荷重−伸長曲線の初期立ち上がり部分を直線近似し、その傾きから求めた。
【0114】
J.繊維の複屈折
OLIMPUS BH−2偏光顕微鏡により単糸のレターデーションと路長を測定し、複屈折を求めた。
【0115】
K.耐摩耗性の評価
耐摩耗性の評価は特開2004−232182号公報に準じ、φ3mmの梨地金属棒に接触角35°で糸を掛け、金属棒から340mmの所で糸張力1.5cN/dtexとして把持し、ストローク長30mm、速度100回/分の往復運動を与え、毛羽(剥離、フィブリル化)の発生した往復回数を測定した。測定は5回行いそれの平均値とし、毛羽発生まで1000回以上を合格とした。
【0116】
参考例1(樹状ポリエステルA−1の合成)
攪拌翼、留出管を備えた反応容器にp−ヒドロキシ安息香酸48.0g(0.35モル)、4,4’−ジヒドロキシビフェニル30.9g(0.17モル)、テレフタル酸5.41g(0.033モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレート10.4g(0.054モル)、トリメシン酸42.0g(0.20モル)、および無水酢酸76.3g(フェノール性水酸基合計の1.1当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で1.5時間反応させた後、250℃まで昇温して脱酢酸縮合反応を行った。反応器内温が250℃に達した後、安息香酸14.7g(0.12モル)を加えて280℃まで昇温させた。酢酸の理論留出量の100%が留出したところで加熱、攪拌を停止し、内容物を冷水中に吐出し、樹状ポリエステル樹脂(A−1)を得た。
【0117】
この樹状ポリエステル樹脂(A−1)は、核磁気共鳴スペクトル解析の結果、R部分の構造が、p−オキシベンゾエート単位の含量pが2.0、4,4’−ジオキシビフェニル単位とエチレンオキシド単位の含量qが0.5、テレフタレート単位の含量rが0.5であり、p+q+r=3であり、分岐点、すなわちBの含有率は樹状ポリエステルを構成する全単量体に対して25モル%であった。また末端構造はカルボン酸と安息香酸エステルであった。
【0118】
得られた樹状ポリエステル樹脂の融点Tmは182℃、液晶開始温度は163℃で、絶対分子量は5500であった。
【0119】
参考例2(樹状ポリエステルA−2の合成)
攪拌翼、留出管を備えた反応容器にp−ヒドロキシ安息香酸66.30g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.40g(0.075モル)、α−レゾルシル酸42.72g(0.28モル)および無水酢酸78.26g(フェノール性水酸基合計の1.08当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、260℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで、安息香酸25.6g(0.21モル;理論アセトキシ末端に対して1.000倍)を添加し、酢酸を100%まで留出させたところで、加熱および攪拌を停止し、内容物を冷水中に吐出して樹状ポリエステル樹脂(A−2)を得た。
【0120】
この樹状ポリエステル樹脂(A−2)は、核磁気共鳴スペクトル解析の結果、R部分の構造が、p−オキシベンゾエート単位の含量pが1.32、4,4’−ジオキシビフェニル単位とエチレンオキシド単位の含量qが0.33、テレフタレート単位の含量rが0.33であり、p+q+r=2であり、分岐点含有率は30モル%であった。また末端構造はカルボン酸とアセチル基であった。
【0121】
得られた樹状ポリエステル樹脂の融点Tmは182℃、液晶開始温度は152℃で、絶対分子量は6500であった。
【0122】
参考例3(脂肪族系超分岐ポリマーA−3の合成)
特表2005−513186号公報に準じ脂肪族系超分岐ポリマーを合成した。
【0123】
実施例1〜5、比較例1
参考例1で合成した樹状ポリエステル(A−1)と、極限粘度1.11dL/g(重量平均分子量=25,500)の高分子量PETを乾燥した後、別々に計量し、独立に二軸押し出し混練機に仕込んだ。この時、樹状ポリエステルの添加量はポリマーブレンドに対し1wt%とした。二軸押し出し混練機の吐出側でベントを行い、泡を消した。混練機および紡糸温度は表1のように設定した。吐出量は42.3g/分で絶対濾過径10μの金属不織布で濾過した後、丸孔24ホール(φ=0.6mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し巻き取った。
【0124】
まず、紡糸速度を500m/分として、樹状ポリエステル添加による流動性向上効果を確かめた。実施例1〜5と比較例1の比較から明らかなように、樹状ポリエステルを添加することで顕著な紡糸パック圧力の低下が起こり流動性が高分子量PETの流動性が向上していることが確かめられた。また、図1に紡糸温度と紡糸パック圧力の関係をプロットしたが、同一紡糸パック圧力で比較すると、樹状ポリエステル1wt%添加(実施例1〜5)では、8℃の紡糸温度低下効果が見込めることがわかった。また、念のため実施例2(樹状ポリエステル1wt%添加、紡糸温度295℃)で、PETの重量平均分子量を測定したが、18,600であり、樹状ポリエステル無添加(比較例1)と同じ値であり、樹状ポリエステル添加による加水分解や熱分解の促進は見られなかった。得られた繊維はほとんどがU%が1%以下と実用的に充分小さいレベルの糸斑であった。
【0125】
【表1】
【0126】
実施例6
さらに、脂肪族リン酸エステル系のエステル交換反応抑制剤であるアデカ社製アデカスタブAX−71を乾燥した後、ブレンドポリマーに対し0.1wt%添加し、実施例4(紡糸温度291℃)と同様に紡糸を行ったところ、さらに顕著な紡糸パック圧力低下が起こり、流動性向上に非常に有効であった(表1、図1)。
【0127】
実施例7〜9
次に、樹状ポリエステル(A−1)の添加量を種々変更し実施例3と同様に紡糸を行ったところ、添加量に応じた流動性向上効果が得られた。U%については、実施例9では1.41%となったが、実用上大きな問題のないレベルである。実施例7,8では1%以下であり優れたU%であった。
【0128】
実施例10、11
次に紡糸速度を変更して実施例3と同様に紡糸を行ったが、紡糸速度を高速化しても紡糸性は良好でありU%も1%以下であった。
【0129】
実施例12〜18、比較例2〜8
実施例2,4,5,6および比較例1で得られた未延伸糸の延伸・熱処理実験を行った。この時、延伸機としては3ホットローラー型延伸機を用い、フィードローラー(非加熱)、第1ホットローラー、第2ホットローラー、第3ホットローラー、デリバリーローラー(非加熱)と糸を通して延伸・熱処理を行った。第1ホットローラー温度(予熱温度)を90℃、第2ホットローラー温度を140℃、第3ホットローラー温度を230℃とし、第1ホットローラーと第2ホットローラー間の延伸倍率を3.85に固定し、第2ホットローラーと第3ホットローラー間の延伸倍率を変化させた。表2にはフィードローラーからデリバリーローラーまでのトータル延伸倍率を示した。
【0130】
いずれも延伸性に問題はなく糸切れ、毛羽等は発生せず、U%も1%以下であった。また、複屈折と弾性率の関係を図2にプロットしたが、樹状ポリエステルを添加した実施例12〜18は未添加の比較例2〜8に比べ同一複屈折での弾性率は1割以上高いものであった。また、特に複屈折が0.185付近よりも小さい領域は効果が高いものであった。
【0131】
【表2】
【0132】
実施例19
樹状ポリエステル(A−1)の熱流動開始温度が160℃と高温であるため、第2ホットローラー温度をその温度以上に変更して、延伸性と延伸糸の物性向上について調べた。未延伸糸としては実施例2で得た物を用い、延伸倍率を5.6倍とし、実施例12と同様に延伸・熱処理を行った。
【0133】
まず第2ホットローラー温度が140℃の時にはトータル延伸倍率5.7倍では糸切れのため延伸不能であったが、第2ホットローラー温度を165℃にしたところ、糸切れは無く延伸性良好であった。また得られた延伸糸物性としては強度7.1cN/dtex、伸度7.5%、弾性率125cN/dtex、複屈折0.186、U%=0.78%と実施例12に比べ、より高倍率延伸できた結果、力学特性の向上が認められた。また、糸斑も改善した。
【0134】
実施例20
極限粘度0.63dL/gのPETと参考例1で合成した樹状ポリエステル(A−1)を乾燥した後、別々に軽量し、独立に二軸押し出し混練機に仕込みブレンドを行った。この時、樹状ポリエステルはポリマーブレンド全体に対し1wt%とした。また、混練機温度は285℃とて、複合紡糸機にブレンドポリマーを導いた。一方、極限粘度0.63dL/gのPETを一軸押し出し混練機で290℃で溶融し、やはり同じ複合紡糸機に導いた。そして、紡糸温度を287℃として、ポリマーブレンドは単糸繊度6dtex(12フィラメント)、PETは単糸繊度2dtex(72フィラメント)となるように紡糸速度3000m/分で巻き取った。そして、この混繊未延伸糸にベルトニップ型仮撚り機を用い、ヒーター温度190℃、延伸倍率1.8倍で常法に従い仮撚りを施した。得られた仮撚り加工糸断面に於いて、ポリマーブレンドから成る太繊度糸が混繊糸の比較的内側に位置し芯糸を形成し、PETから成る細繊度糸が混繊糸の比較的外側に位置し鞘糸を形成していた。なお、紡糸過程でポリマーブレンドから成る太繊度糸のみを巻き取り、U%を測定したところ、U%=0.98%と良好であった。さらにこれに、前記したのと同様の仮撚り加工を加え、仮撚り加工糸のU%を測定したが1.00%とやはり糸斑は良好であった。また、この糸の弾性率は60cN/dtexであった。
【0135】
そして、この混繊糸から成る仮撚り加工糸に700T/mの実撚りを加え、S撚り/Z撚りを交互配列し経糸および緯糸に用いて2/2ツイル織物を作製した。さらにこの織物を80℃で精練を施した後170℃で中間セットした後、常法により染色し、170℃で仕上げセットを行った。得られた織物は嵩高さとソフトさ、またハリ腰を備えた風合いに優れる物であった。また、染色斑も無く審美性にも優れる物であった。
【0136】
比較例9
樹状ポリエステルを添加せず実施例20と同様に紡糸、仮撚り加工を行ったところ、太繊度糸が鞘糸を細繊度糸が芯糸を形成していた。この混繊糸を経糸および緯糸に用い、実施例20と同様に織物を作製し仕上げセットまで行ったところ、芯糸と鞘糸が逆転しているため、粗硬感はあるがハリ腰の無い風合いに劣る物であった。
【0137】
実施例21
実施例20で得た未延伸混繊糸を予熱温度90℃、延伸倍率1.85倍、熱セット温度130℃で延伸・熱処理した。得られた延伸糸断面に於いて、ポリマーブレンドから成る太繊度糸が混繊糸の比較的内側に位置し芯糸を形成し、PETから成る細繊度糸が混繊糸の比較的外側に位置し鞘糸を形成していた。なお、紡糸過程でポリマーブレンドから成る太繊度糸のみを巻き取り、U%を測定したところ、U%=0.98%と良好であった。さらにこれに、前記したのと延伸・熱処理を加え、延伸糸のU%を測定したが0.82%とやはり糸斑は良好であった。また、この糸の弾性率は85cN/dtexであった。
【0138】
この混繊糸を用いて丸編みを作製し、80℃で精練を施した後170℃で中間セットした後、常法により染色し、170℃で仕上げセットを行った。得られた編物は嵩高さとソフトさ、また適度なハリ腰を備えた風合いに優れる物であった。
【0139】
実施例22
参考例1で合成した樹状ポリエステル(A−1)と、極限粘度1.11dL/gの高分子量PETおよびエステル交換反応抑制剤であるアデカ社製アデカスタブAX−71を乾燥した後、別々に計量し、二軸押し出し混練機に仕込んだ。この時、樹状ポリエステルとエステル交換反応抑制剤は計量した物をドライブレンドし、仕込んだ。添加量はポリマーブレンドに対し、樹状ポリエステルは1wt%、エステル交換反応抑制剤は0.1wt%とした。また、二軸押し出し混練機の吐出側でベントを行い、泡を消した。混練機および紡糸温度は287℃に設定し、丸孔4ホールの口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、モノフィラメントに分割後給油し、それぞれモノフィラメントとして巻き取った。そして、トータル延伸倍率4.72倍、第2ホットローラー温度165℃として、実施例16と同様に延伸熱処理を行い、単糸繊度8dtexのモノフィラメントを得た。得られた繊維は、U%=0.78%、弾性率=111cN/dtex、複屈折=0.178、強度=5.5cN/dtex、伸度=16.5%であった。
【0140】
これの耐摩耗性の評価を行ったところ、毛羽発生までの往復回数は1000回以上であり、良好な耐摩耗性を示した。これは、充分な弾性率と耐摩耗特性を有したモノフィラメントであり、IT用などの薄くてかつ高精度が求められるスクリーン紗に最適な物である。
【0141】
比較例10
樹状ポリエステルとエステル交換反応抑制剤を添加しないで、混練機温度および紡糸温度を300℃として実施例22と同様に紡糸を行った。なお吐出量は延伸糸繊度に応じて調整した。得られたモノフィラメントをトータル延伸倍率5.70倍として実施例22と同様に延伸・熱処理し、8dtexのモノフィラメントを得た。これの弾性率は111cN/dtex、複屈折は0.186であった。続いて耐摩耗性を実施例22と同様に測定したが、毛羽発生まで500回と耐摩耗性の低い物であった。
【0142】
実施例23
参考例2で合成した樹状ポリエステル(A−2)を用い、実施例2と同様に紡糸を行ったところ、紡糸パック圧力は5.4Pa・sと良流動化効果を確認できた。また得られた未延伸糸のU%は0.94%と充分糸斑の小さな物であった。
これを第2ホットローラー温度を165℃、トータル延伸倍率5.20倍として実施例12と同様に延伸・熱処理し、U%=0.77%、弾性率=110cN/dtex、複屈折=0.180、強度=5.8cN/dtex、伸度=18%の繊維を得た。A−1を使用した場合に比べると若干複屈折あたりの弾性率が低いが、未添加に比べると十分な効果であった。
【0143】
比較例11
参考例3で合成した脂肪族系超分岐ポリマー(A−3)を用い、実施例11と同様に紡糸を行ったところ、紡糸パック圧力は8.0Pa・sと本発明のA−1、A−2を用いた場合に比べ良流動化効果は小さいものであった。また、紡糸が安定せず、糸切れが多発するなど紡糸性も実施例11には及ばなかった。
【0144】
実施例24
光学純度99%のポリL乳酸(重量平均分子量19万)を乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は220℃および紡糸温度は230℃とし、吐出量は30g/分、口金として孔径0.30mm、18ホールの物を用い、紡糸速度は5000m/分とした。この時の紡糸パック圧力は16MPaであった。紡糸温度をポリ乳酸の分解が著しくなる温度(240℃)よりも下げることができたため、分解ガスの発生を抑制することができた。得られた高速紡糸繊維のU%は0.81%であった。
【0145】
比較例12
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度240℃、紡糸温度245℃として実施例24と同様に溶融紡糸を行った。この時の紡糸パック圧力は17MPaであった。紡糸温度がポリ乳酸に対して高いため、分解ガスの発生が著しく作業環境が悪化した。
【0146】
実施例25
極限粘度0.75dL/gのPENを乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は290℃および紡糸温度は290℃とし、吐出量は20g/分、紡糸口金としてはφ0.60mm、10ホールの物を用い、紡糸速度は1350m/分とした。この時の紡糸パック圧力は5.8MPaであり、流動性向上効果を確かめることができた。得られた高速紡糸繊維のU%は0.95%であった
比較例13
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度295℃、紡糸温度300℃として実施例25と同様に溶融紡糸を行った。この時の紡糸パック圧力は6.8MPaであった。
【0147】
実施例26
相対粘度3.8のナイロン66を乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は290℃および紡糸温度は290℃とし、吐出量は33.5g/分、紡糸口金としてはφ0.30mm、18ホールの物を用い、紡糸速度は1500m/分とした。この時の紡糸パック圧力は12.5MPaであり、流動性向上効果を確かめることができた。得られた繊維のU%は0.95%であった
比較例14
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度295℃、紡糸温度295℃として実施例26と同様に溶融紡糸を行った。この時の紡糸パック圧力は16.5MPaであった。
【図面の簡単な説明】
【0148】
【図1】紡糸温度と紡糸パック圧力の関係を示す図
【図2】複屈折と弾性率の関係を示す図
【技術分野】
【0001】
本発明は、マトリックスポリマーに樹状ポリエステルをブレンドしたポリマーブレンドから成る繊維に関するものである。
【背景技術】
【0002】
ポリエステルやポリアミドなどの熱可塑性ポリマーを用いた繊維は幅広く利用されており、産業上の価値は極めて高い。そして、これらの製造方法は、ポリマーを融解した後、細い口金孔から押し出す溶融紡糸が採用されている。この時、溶融紡糸における紡糸温度はポリマーの融点や溶融粘度により決定されるが、融点が同じであっても高粘度ポリマーを紡糸する際は低粘度ポリマーの場合に比べ高い紡糸温度を設定する必要がある。しかしながら、過度に高温にするとポリマーそのものの分解が促進されるなど不具合があった。また、繊維に機能性を付加するため機能付加成分を同時に紡糸することが行われているが、この機能付加成分の耐熱性が低い場合には分解やそれに伴うガスが発生する場合がある。このため、特に高粘度ポリマーの紡糸や耐熱性の低い機能性付加成分を同時に紡糸する場合に於いてはポリマーの流動性を向上させ、紡糸温度をなるべく低温下する技術が重要である。
【0003】
このため、種々の減粘剤が利用される場合があるが、現在まで成功した例は少ない。従来の技術の中でも特に興味深いものとしては、超分岐ポリマー(ハイパーブランチポリマー)をナイロンに添加した例を挙げることができる(特許文献1)。ここでは、マトリックスポリマーと非反応性の超分岐ポリマーをマトリックスポリマーに添加することで、未添加の場合に比べ分子量減少が7%未満で、かつ流動性が向上することが記載されている。しかしながら、実際には超分岐ポリマーの主鎖である枝構造部分(D)が脂肪族(実施例ではε−カプロラクタムから誘導された物)となるため、柔軟性が著しく高い。このため、超分岐ポリマーとマトリックスポリマーが非反応性であるとしても、超分岐ポリマー主鎖部分とマトリックスポリマー主鎖部分でいわゆる分子鎖の絡み合いが多く発生してしまう可能性があった。これは、樹脂の押し出し加工などでは変形量が小さく、さらに剪断変形が支配的であるため大きな問題とはならないが、特に紡糸などの大きな伸長変形を伴う場合には深刻な問題を引き起こしてしまう場合があった。すなわち、分子鎖の絡み合いの程度が大きくなることで、マトリックスポリマー分子鎖のスムーズな伸長変形が阻害され、紡糸性を著しく損ない、場合よってはポリマーの弾性的振る舞いが顕著となり紡糸不能に陥る場合があった。また、紡糸不能に至らないまでも紡糸線での伸長変形に大規模な経時変動(斑)が発生し、糸の太細斑が過大となり、実用的な繊維が得られない場合があった。特に、マトリックスポリマーとして高粘度ポリマーや高分子量ポリマーを用いた場合、また細繊度紡糸や紡糸速度を高速化した場合にこの傾向が顕著であった。
【0004】
このため、紡糸や繊維に適したハイパーブランチポリマーが、またこれが添加された糸斑の小さな繊維が求められていた。
【特許文献1】特表2005−513186号公報(6〜12ページ)
【発明の開示】
【発明が解決しようとする課題】
【0005】
従来とは異なり、マトリックスポリマーと分子鎖の絡み合いが抑制され、紡糸過程での伸長変形を阻害し難い樹状ポリエステルを用いることにより、紡糸過程での流動性を向上させ紡糸温度の低温化を図るとともに、糸斑が小さく、さらに繊維の複屈折あたりの弾性率が向上、すなわち高弾性率でありながら分子配向の低い繊維を提供するものである。
【課題を解決するための手段】
【0006】
上記目的は、以下の手段により達成される。
(1)芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
(2)熱可塑性のマトリックスポリマーがポリエステルである(1)記載の繊維。
(3)弾性率が110〜1000cN/dtexである請求項1または2記載の繊維。
(4)弾性率が50cN/dtex以上、110cN/dtex未満である(1)または(2)記載の繊維。
(5)樹状ポリエステルをマトリックスポリマーにブレンドしたポリマーブレンド全体に対して、エステル交換反応抑制剤を0.01〜1wt%含有する(1)〜(4)のいずれか1項記載の繊維。
(6)繊維がモノフィラメントである(1)〜(3)および(5)のいずれか1項記載の繊維。
(7)請求項(1)〜(2)および(4)のいずれか1項記載の繊維が芯糸として用いられた混繊糸。
(8)(1)〜(7)のいずれか1項記載の繊維を少なくとも一部に使用した繊維製品。
【発明の効果】
【0007】
本発明の繊維により、糸斑が小さく、さらに繊維の複屈折あたりの弾性率を向上することができる。すなわち同じ複屈折であれば高弾性率に、同じ弾性率であれば低複屈折とできるため、高弾性率でありながら分子配向を低下させることで耐摩耗性を向上させることができ、産業資材、小さな糸斑ともあいまって、特にスクリーン紗などに好適に使用することができる。また、紡糸混繊の一成分に利用することで、低分子配向であっても芯糸を形成し易い繊維を得ることができ、高級衣料用として最適な混繊糸を提供することができる。
【発明を実施するための最良の形態】
【0008】
本発明で言う樹状ポリエステルとは、ハイパーブランチポリマーの一種であるが、本発明で用いられる樹状ポリエステルは、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含んでいる。本発明で用いられる樹状ポリエステルは、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルである。
【0009】
ここで、芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および、芳香族ジカルボニル単位(R)は、それぞれ下式(1)で表される構造単位であることが好ましい。
【0010】
【化1】
【0011】
ここで、R1、R3は、それぞれ芳香族残基であり、R2は芳香族および脂肪族を両方含む残基である。またR1、R2、およびR3は、それぞれ複数の構造単位を含んでも良い。
【0012】
上記の芳香族残基としては、置換または非置換のフェニレン基、ナフチレン基、ビフェニレン基などが挙げられ、脂肪族残基としてはエチレン、プロピレン、ブチレンなどが挙げられる。R1、R2およびR3は、好ましくは、それぞれ下式で表される構造単位から選ばれる少なくとも1種以上の構造単位であり、R2は芳香族残基である(R2−1)と脂肪族残基である(R2−2)を両方含む。
【0013】
【化2】
【0014】
ただし、式中nは2〜8の整数である。
【0015】
本発明の樹状ポリエステルは、3官能以上の有機残基(B)が、互いにエステル結合および/またはアミド結合により直接、あるいは、枝構造部分(D)を構成するP、QおよびRから選ばれる構造単位を介して結合した、3分岐以上の分岐構造を基本骨格としている。分岐構造は、3分岐、4分岐など単一の基本骨格で形成されていてもよいし、3分岐と4分岐など、複数の基本骨格が共存していてもよい。ポリマーの全てが該基本骨格からなる必要はなく、たとえば末端封鎖のために末端に他の構造が含まれても良い。また、Bが3官能性の有機残基である場合には、樹状ポリエステル中には、Bの3つの官能基が全て反応している構造、2つだけが反応している構造、および1つだけしか反応していない構造が混在していてもよい。好ましくはBの3つの官能基が全て反応した構造が、B全体に対して15モル%以上であることが好ましく、より好ましくは20モル%以上であり、さらに好ましくは30モル%以上である。また、Bが4官能性の有機残基である場合には、樹状ポリエステル中には、Bの4つの官能基が全て反応している構造、3つだけが反応している構造、2つだけが反応している構造、および1つしか反応していない構造が混在していてもよい。好ましくはBの4つの官能基が全て反応した構造がB全体に対して10モル%以上かつ3つの官能基が反応した構造が20モル%以上であることが好ましく、より好ましくは4つの官能基が反応した構造がB全体に対して20モル%以上かつ3つの官能基が反応した構造がB全体に対して30モル%以上であり、さらに好ましくは4つの官能基が反応した構造がB全体に対して25モル%以上かつ3つの官能基が反応した構造がB全体に対して35モル%以上である。
【0016】
Bは3官能化合物および/または4官能化合物の有機残基であることが好ましく、3官能化合物の有機残基であることが最も好ましい。
【0017】
上記3分岐の基本骨格を模式的に示すと、式(2)で示される。また上記4分岐の基本骨格を模式的に示すと、式(3)で示される。
【0018】
【化3】
【0019】
【化4】
【0020】
本発明の樹状ポリエステルは、溶融液晶性を示すことが好ましい。ここで溶融液晶性を示すとは、室温(25℃)から昇温していった際に、ある温度域で液晶状態を示すことである。液晶状態とは、剪断下において光学的異方性を示す状態である。
【0021】
溶融液晶性を示すために、3分岐の場合の基本骨格は、下式(4)で示されるように、Bが、P、QおよびRから選ばれる構造単位により構成される枝構造部分(D)を介して結合していることが好ましい。
【0022】
【化5】
【0023】
同様に、4分岐の場合の基本骨格は、下式(5)で示される構造が好ましい。
【0024】
【化6】
【0025】
3官能の有機残基Bとしては、カルボキシル基、ヒドロキシル基およびアミノ基から選ばれる官能基を含有する化合物の有機残基であることが好ましい。例えばグリセロール、1,2,3−トリカルボキシプロパン、ジアミノプロパノール、ジアミノプロピオン酸などの脂肪族化合物や、トリメシン酸、トリメリット酸、4−ヒドロキシ−1,2−ベンゼンジカルボン酸、フロログルシノール、α−レゾルシン酸、β−レゾルシン酸、γ−レゾルシン酸、トリカルボキシナフタレン、ジヒドロキシナフトエ酸、アミノフタル酸、5−アミノイソフタル酸、アミノテレフタル酸、ジアミノ安息香酸、メラミンなどの芳香族化合物の残基が好ましく用いられる。下式で表される芳香族化合物の残基がさらに好ましい。
【0026】
【化7】
【0027】
上記の3官能の有機残基の具体例としては、フロログルシノール、トリメシン酸、トリメリット酸、無水トリメリット酸、α−レゾルシル酸、4−ヒドロキシ−1,2−ベンゼンジカルボン酸などの残基が好ましく、さらに好ましくは、トリメシン酸、α−レゾルシル酸の残基であり、最も好ましくはトリメシン酸の残基である。
【0028】
また、4官能以上の有機残基Bとしては、カルボキシル基、ヒドロキシル基およびアミノ基から選ばれる官能基を含有する化合物の有機残基であることが好ましい。例えば、エリスリトール、ペンタエリスリトール、スレイトール、キシリトール、グルシトール、マンニトール、1,2,3,4−ブタンテトラカルボン酸、1,2,4,5−シクロヘキサンテトラオール、1,2,3,4,5−シクロヘキサンペンタンオール、1,2,3,4,5,6−シクロヘキサンヘキサンオール、1,2,4,5−シクロヘキサンテトラカルボン酸、1,2,3,4,5−シクロヘキサンペンタカルボン酸、1,2,3,4,5,6−シクロヘキサンヘキサカルボン酸、クエン酸、酒石酸などの脂肪族化合物の残基や1,2,4,5−ベンゼンテトラオ−ル、1,2,3,4−ベンゼンテトラオ−ル、1,2,3,5−ベンゼンテトラオ−ル、1,2,3,4,5−ベンゼンペンタンオ−ル、1,2,3,4,5,6−ベンゼンヘキサンオ−ル、2,2’,3,3’−テトラヒドロキシビフェニル、2,2’,4,4’−テトラヒドロキシビフェニル、3,3’,4,4’−テトラヒドロキシビフェニル、3,3’,5,5’−テトラヒドロキシビフェニル、2,3,6,7−ナフタレンテトラオール、1,4,5,8−ナフタレンテトラオール、ピロメリット酸、メロファン酸、プレーニト酸、メリット酸、2,2’,3,3’−ビフェニルテトラカルボン酸、2,2’,4,4’−ビフェニルテトラカルボン酸、3,3’,4,4’−ビフェニルテトラカルボン酸、3,3’,5,5’−ビフェニルテトラカルボン酸、2,3,6,7−ナフタレンテトラカルボン酸、1,4,5,8−ナフタレンテトラカルボン酸、2,3,6,7−ナフタレンテトラオール、1,4,5,8−ナフタレンテトラオール、1,2,4,5,6,8−ナフタレンヘキサオール、1,2,4,5,6,8−ナフタレンヘキサカルボン酸、没食子酸、などの芳香族化合物の残基が挙げられる。下式で表される芳香族化合物の残基がさらに好ましい。
【0029】
【化8】
【0030】
上式の4官能の有機残基の具体例としては、1,2,4,5−ベンゼンテトラオ−ル、1,2,3,4−ベンゼンテトラオ−ル、1,2,3,5−ベンゼンテトラオ−ル、ピロメリット酸、メロファン酸、プレーニト酸、没食子酸などの残基が好ましく、没食子酸の残基が特に好ましい。
【0031】
また、樹状ポリエステルの芳香族オキシカルボニル単位(P)、芳香族ジオキシ単位(Q)、芳香族ジカルボニル単位(R)は、樹状ポリエステルの分岐間の枝構造部分(D)を構成する単位である。p、qおよびrはそれぞれ構造単位P、QおよびRの平均含有量(モル比)であり、Bの含有量bの1モルに対して、p+q+r=1〜10モルの範囲であることが好ましい。p+q+rは、より好ましくは、2〜6モルの範囲である。枝構造長が長すぎると、剛直で綿密な樹状構造に基づく剪断応答性などの効果やマトリックスポリマー分子鎖との絡み合い抑制効果が低減するため好ましくない。
【0032】
このp、qおよびrの値は、例えば、樹状ポリエステルをペンタフルオロフェノール50重量%:重クロロホルム50重量%の混合溶媒に溶解し、40℃でプロトン核の核磁気共鳴スペクトル分析を行い、それぞれの構造単位に由来するピーク強度比から求めることができる。各構造単位のピーク面積強度比から、平均含有率を算出し、小数点3桁は四捨五入する。Bの含有量bにあたるピークとの面積強度比から、枝構造部分(D)の平均鎖長を算出し、p+q+rの値とする。この場合にも小数点3桁は四捨五入する。
【0033】
pとqの比率およびpとrの比率(p/q、p/r)は、いずれも5/95〜95/5の範囲が好ましく、より好ましくは10/90〜90/10であり、さらに好ましくは20/80〜80/20である。この範囲であれば、液晶性が発現しやすく好ましい。p/qおよびp/rの比率を95/5以下とすることで、樹状ポリエステルの融点を適当な範囲とすることができるため好ましい。また、p/qおよびp/rを5/95以上とすることで樹状ポリエステルの溶融液晶性を発現することができるため好ましい。
【0034】
qとrは、実質的に等モルであることが好ましいが、末端基を制御するためにどちらかの成分を過剰に加えることもできる。q/rの比率としては0.7〜1.5の範囲であることが好ましく、より好ましくは0.9〜1.1である。ここでいう等モルとは、繰り返し単位内でのモル量が等しいことを意味し、末端構造は含めない。ここで、末端構造とは、枝構造部分(D)の末端を意味し、末端が封鎖されている場合などには、最も末端に近い枝構造部分(D)の末端を意味する。
【0035】
前記式(1)において、R1は芳香族オキシカルボニル単位由来の構造単位であり、具体例としては、p−ヒドロキシ安息香酸、6−ヒドロキシ−2−ナフトエ酸から生成した構造単位などが挙げられる。好ましくはp−ヒドロキシ安息香酸由来の構造単位であり、6−ヒドロキシ−2−ナフトエ酸由来の構造単位部併用することも可能である。また本発明の効果を損なわない範囲でグリコール酸、乳酸、ヒドロキシプロピオン酸、ヒドロキシ酪酸、ヒドロキシ吉草酸、ヒドロキシカプロン酸などの脂肪族ヒドロキシカルボン酸由来の構造単位を含有しても良い。
【0036】
R2は芳香族および脂肪族由来の構造を両方含む構造単位であり、芳香族単位としては、例えば、4,4’−ジヒドロキシビフェニル、ハイドロキノン、3,3’,5,5’−テトラメチル−4,4’−ジヒドロキシビフェニル、t−ブチルハイドロキノン、フェニルハイドロキノン、メチルハイドロキノン、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、2,2−ビス(4−ヒドロキシフェニル)プロパンおよび4,4’−ジヒドロキシジフェニルエーテル、など由来の構造単位が挙げられ、脂肪族単位としてはエチレングリコール、プロピレングリコール、1,4−ブタンジオールなど脂肪族ジオール由来の構造単位が挙げられる。好ましくは、4,4’−ジヒドロキシビフェニル、エチレングリコール由来の構造単位であり、4,4’−ジヒドロキシビフェニルとエチレングリコール由来の構造単位が含まれることが液晶性の制御の点から好ましい。
【0037】
本発明で用いる樹状ポリエステルとしては、このR2単位を芳香族と脂肪族の両方を含む構造単位とすることが特に重要である。まず、芳香族を含有させることにより枝構造に剛直性を与え(好ましくは液晶性を与え)、マトリックスポリマー分子鎖との絡み合いを抑制し、紡糸などの伸長大変形の場でも伸長変形を阻害することを抑制できるのである。これにより、工業的に充分な紡糸性(曳糸性、安定性など)が得られ、かつ糸斑も大きく改善できるのである。また、完全に芳香族だけにせず脂肪族も含有させるのは、マトリックスポリマーの伸長変形を大きく阻害しない範囲で適度な絡み合いを持たせることで、繊維の複屈折あたりの弾性率を向上させることができるからである。本発明では、このように芳香族と脂肪族を混合することで分子鎖の絡み合いを制御し、紡糸性・糸斑改善と複屈折あたりの弾性率向上という、従来、二律背反の効果を両立させているのである。ここで、複屈折あたりの弾性率が向上すると、同じ複屈折であれば高弾性率に、同じ弾性率であれば低複屈折とできるため、高弾性率でありながら分子配向を低下させることができ、やはり従来、二律背反であった繊維の弾性率と耐摩耗性を両立させることができるのである。このため、産業資材、小さな糸斑ともあいまって、特にスクリーン紗などに好適に使用することができる。
【0038】
R3は芳香族ジカルボニル単位由来の構造単位であり、例えば、テレフタル酸、イソフタル酸、2,6−ナフタレンジカルボン酸、4,4’−ジフェニルジカルボン酸、1,2−ビス(フェノキシ)エタン−4,4’−ジカルボン酸、1,2−ビス(2−クロロフェノキシ)エタン−4,4’−ジカルボン酸および4,4’−ジフェニルエーテルジカルボン酸など由来の構造単位が挙げられる。好ましくはテレフタル酸またはイソフタル酸由来の構造単位であり、特に両者を併用した場合に融点調節がしやすく好ましい。また、本発明の効果に影響を及ぼさない範囲で、セバシン酸やアジピン酸などの脂肪族ジカルボン酸由来の構造単位が一部含まれていてもよい。
【0039】
本発明の樹状ポリエステルの枝構造部分(D)は、主としてポリエステル骨格からなることが好ましいが、カーボネート構造やアミド構造、ウレタン構造などを、特性に大きな影響を与えない程度に導入することも可能である。中でもアミド構造を導入することが好ましい。このような別の結合を導入することで、多種多様な熱可塑性樹脂に対する相溶性を調整することが可能であり、好ましい。アミド結合の導入の方法としては、p−アミノ安息香酸、m−アミノ安息香酸、p−アミノフェノール、m−アミノフェノール、p−フェニレンジアミン、m−フェニレンジアミン、テトラメチレンジアミンペンタメチレンジアミン、ヘキサメチレンジアミン、2−メチルペンタメチレンジアミン、ノナメチレンジアミン、ウンデカメチレンジアミン、ドデカメチレンジアミン、2,2,4−/2,4,4−トリメチルヘキサメチレンジアミン、5−メチルノナメチレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン、1,3−ビス(アミノメチル)シクロヘキサン、1,4−ビス(アミノメチル)シクロヘキサン、1−アミノ−3−アミノメチル−3,5,5−トリメチルシクロヘキサン、ビス(4−アミノシクロヘキシル)メタン、ビス(3−メチル−4−アミノシクロヘキシル)メタン、2,2−ビス(4−アミノシクロヘキシル)プロパン、ビス(アミノプロピル)ピペラジン、アミノエチルピペラジンなどの脂肪族、脂環族、あるいは芳香族のアミン化合物などを共重合することが好ましい。中でもp−アミノフェノールまたはp−アミノ安息香酸の共重合が好ましい。
【0040】
樹状ポリエステルの枝構造部分(D)の具体例としては、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位およびテレフタル酸由来の構造単位からなるもの、p−ヒドロキシ安息香酸由来の構造単位、エチレングリコール由来の構造単位、4,4’−ジヒドロキシビフェニル由来の構造単位およびテレフタル酸由来の構造単位からなるものなどが挙げられる。
【0041】
特に好ましいのは、枝構造部分(D)が、下記構造単位(I)、(II)、(III)および(IV)から構成されることである。ここで、(I)は構造単位(P)の、(II)、(III)は構造単位(Q)の、(IV)は構造単位(R)の特に好ましい様態である。
【0042】
【化9】
【0043】
枝構造部分(D)が、上記構造単位(I)、(II)、(III)および(IV)から構成される場合には、上記構造単位(I)の含有量pは、p+q+rに対して30〜90モル%が好ましく、40〜80モル%がより好ましい。また、構造単位(VI)の含有量q(III)は、(II)と(III)の合計含有量qに対して70〜5モル%が好ましく、60〜8モル%がより好ましい。前記のように、構造単位(IV) の含有量rは、構造単位(II)および(III)の合計含有量qと実質的に等モルであることが好ましいが、いずれかの成分を過剰に加えてもよい。
【0044】
また、本発明の樹状ポリエステルの末端は、カルボキシル基、水酸基、アミノ基、またはそれらの誘導体が好ましい。水酸基の誘導体もしくは、カルボン酸の誘導体としては、メチルエステルなどのアルキルエステルやフェニルエステルやベンジルエステルなどの芳香族エステルが挙げられる。また、単官能エポキシ化合物、オキサゾリン化合物、オルトエステル、酸無水物化合物などを用いて末端封鎖することも可能である。末端封鎖の方法としては、樹状ポリエステルを合成する際に、あらかじめ単官能性の有機化合物を添加する方法や、ある程度樹状ポリステルの骨格が形成された段階で単官能性の有機化合物を添加する方法などが挙げられる。
【0045】
具体的には、水酸基末端やアセトキシ末端を封鎖する場合には、安息香酸、4−t−ブチル安息香酸、3−t−ブチル安息香酸、4−クロロ安息香酸、3−クロロ安息香酸、4−メチル安息香酸、3−メチル安息香酸、3,5−ジメチル安息香酸などを添加することで可能である。
【0046】
また、カルボキシル基末端の封鎖は、カルボン酸反応性単官能化合物を反応することにより行うことができる。ここで、カルボン酸反応性単官能化合物とは、常温または加熱時にカルボン酸と反応し、エステル、アミド、ウレタン、ウレア結合を形成しうる官能基を分子内に1つ有する化合物をいう。樹状ポリエステルの分子末端に存在するカルボン酸基に、カルボン酸反応性単官能化合物を反応させ、分子末端に単官能化合物を導入することにより、樹状ポリエステルの滞留安定性や耐加水分解性を向上させ、さらに他の熱可塑性ポリマーと混練した際には、熱可塑性ポリマーの分解を抑制でき、また樹状ポリエステルの分散性が向上することによって、流動性や物性の改良が期待できる。
【0047】
本発明の樹状ポリエステルに用いることのできるカルボン酸反応性単官能化合物としては、オキサゾリン、エポキシド、オルトエステル、イソシアネート、カルボジイミド、ジアゾ化合物から選ばれる1種類以上の化合物である。カルボン酸との反応性およびハンドリング性の観点から、オキサゾリン、エポキシド、オルトエステル、イソシアネートが好ましく用いることができる。カルボン酸反応性単官能化合物は、単独で使用または2種類以上のカルボン酸反応性単官能化合物を併用しても構わない。
【0048】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちオキサゾリン化合物としては、例えば、2−メトキシ−2−オキサゾリン、2−エトキシ−2−オキサゾリン、2−プロポキシ−2−オキサゾリン、2−ブトキシ−2−オキサゾリン、2−ペンチルオキシ−2−オキサゾリン、2−ヘキシルオキシ−2−オキサゾリン、2−ヘプチルオキシ−2−オキサゾリン、2−オクチルオキシ−2−オキサゾリン、2−デシルオキシ−2−オキサゾリン、2−シクロペンチルオキシ−2−オキサゾリン、2−シクロヘキシル−2−オキサゾリン、2−アリルオキシ−2−オキサゾリン、2−メタアリルオキシ−2−オキサゾリン、2−フェノキシ−2−オキサゾリン、2−クレジル−2−オキサゾリン、2−p−フェニルフェノキシ−2−オキサゾリン、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−ペンチル−2−オキサゾリン、2−ヘキシル−2−オキサゾリン、2−ヘプチル−2−オキサゾリン、2−オクチル−2−オキサゾリン、2−ノニル−2−オキサゾリン、2−デシル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−シクロペンチル−2−オキサゾリン、2−シクロヘキシル−2−オキサゾリン、2−アリル−2−オキサゾリン、2−メタアリル−2−オキサゾリン、2−クロチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性、および耐熱性の観点から、2−メチル−2−オキサゾリン、2−エチル−2−オキサゾリン、2−プロピル−2−オキサゾリン、2−ブチル−2−オキサゾリン、2−イソプロピル−2−オキサゾリン、2−イソブチル−2−オキサゾリン、2−sec−ブチル−2−オキサゾリン、2−tert−ブチル−2−オキサゾリン、2−フェニル−2−オキサゾリン、2−ビフェニル−2−オキサゾリンが好ましく、特に好ましくは2−フェニル−2−オキサゾリンである。
【0049】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちエポキシ化合物としては、例えば、N−グリシジルフタルイミド、N−グリシジル−4−メチルフタルイミド、N−グリシジル−4,5−ジメチルフタルイミド、N−グリシジル−3−メチルフタルイミド、N−グリシジル−3,6−ジメチルフタルイミド、N−グリシジル−4−エトキシフタルイミド、N−グリシジル−4−クロルフタルイミド、N−グリシジル−4,5−ジクロルフタルイミド、N−グリシジルサクシンイミド、N−グリシジルヘキサヒドロフタルイミド、N−グリシジルマレインイミド、N−グリシジルベンズアミド、N−グリシジル−p−メチルベンズアミド、N−グリシジルナフトアミド、N−グリシジルステラアミド、o−フェニルフェニルグリシジルエーテル、2−メチルオクチルグリシジルエーテル、フェニルグリシジルエーテル、3−(2−キセニルオキシ)−1,2−エポキシプロパン、アリルグリシジルエーテル、ブチルグリシジルエーテル、ラウリルグリシジルエーテル、ベンジルグリシジルエーテル、シクロヘキシルグリシジルエーテル、α−クレシルグリシジルエーテル、p−tert−ブチルフェニルグリシジルエーテル、メタクリル酸グリシジルエーテル、エチレンオキサイド、プロピレンオキサイド、スチレンオキサイド、オクトイレンオキサイド、酢酸グリシジルエステル、プロピオン酸グリシジルエステル、ブタン酸グリシジルエステル、ペンタン酸グリシジルエステル、ヘキサン酸グリシジルエステル、オクタン酸グリシジルエステル、デカン酸グリシジルエステル、ネオデカン酸グリシジルエステル、安息香酸グリシジルエステルなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、エチレンオキサイド、プロピレンオキサイド、ブチルグリシジルエーテル、フェニルグリシジルエーテル、安息香酸グリシジルエステルが好ましく、特に好ましくは安息香酸グリシジルエステルである。
【0050】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちオルトエステル化合物としては、例えば、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト酢酸トリプロピル、オルト酢酸トリブチル、オルト酢酸トリベンジル、オルト蟻酸トリメチル、オルト蟻酸トリエチル、オルト蟻酸トリプロピル、オルト蟻酸トリブチル、オルト蟻酸トリベンジル、オルトプロピオン酸トリメチル、オルトプロピオン酸トリエチル、オルトプロピオン酸トリプロピル、オルトプロピオン酸トリブチル、オルトプロピオン酸トリベンジル、オルト安息香酸トリメチル、オルト安息香酸トリエチル、オルト安息香酸トリプロピル、オルト安息香酸トリブチル、オルト安息香酸トリベンジルなどが挙げられる。このうち、樹状ポリエステルとの反応性や親和性およびハンドリング性の観点から、オルト酢酸トリメチル、オルト酢酸トリエチル、オルト蟻酸トリメチル、オルト蟻酸トリエチルが好ましく、特に好ましくはオルト酢酸トリメチルまたはオルト酢酸トリエチルである。
【0051】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちイソシアネート化合物としては、例えば、メチルイソシアネート、エチルイソシアネート、プロピルイソシアネート、ブチルイソシアネート、ペンチルイソシアネート、ヘキシルイソシアネート、ヘプチルイソシアネート、オクチルイソシアネート、ノニルイソシアネート、デシルイソシアネート、ドデシルイソシアネート、オクタデシルイソシアネート、ベンジルイソシアネート、シクロへキシルイソシアネート、フェニルイソシアネート、p−クロロフェニルイソシアネート、p−ニトロフェニルイソシアネート、2−クロロエチルイソシアネート、ステアロイルイソシアネート、p−トルオルスルフォニルイソシアネートが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、メチルイソシアネート、エチルイソシアネート、プロピルイソシアネート、ブチルイソシアネート、フェニルイソシアネートが好ましく、特に好ましくはフェニルイソシアネートである。
【0052】
本発明に用いることのできるカルボン酸反応性単官能化合物のうちジアゾ化合物としては、例えば、ジアゾメタン、ジアゾエタン、ジアゾプロパン、ジアゾブタン、トリメチルシリルジアゾメタンが挙げられる。このうち、樹状ポリエステルとの反応性や親和性の観点から、ジアゾメタンおよびトリメチルシリルジアゾメタンが好ましく用いられる。
【0053】
理論的には、上記末端の封鎖に用いる有機化合物を、封鎖したい末端基に相当する量添加することで末端封鎖が可能である。封鎖したい末端基相当量に対して、末端封鎖に用いる有機化合物を、1.005倍当量以上用いることが好ましく、より好ましくは1.008倍当量以上である。また、末端封鎖に用いる有機化合物の添加量は2.5倍当量以下であることが好ましい。末端封鎖に用いる化合物の添加量が少なすぎると、末端封鎖が充分ではない。一方、添加量が多すぎると、過剰に添加した化合物が系中に残存して、ガスを発生したりするため好ましくない。
【0054】
また、Bの含有量は、枝構造部分(D)の連鎖長が、樹状ポリエステルが樹状の形態をとるのに適した長さとなる観点から決定されるが、充分な樹状を形成させるためには、樹状ポリエステルを構成する全単量体の含有量に対して7.5モル%以上とすることが重要であり、10モル%以上がより好ましく、さらに好ましくは20モル%以上である。一方、枝構造部が過度に密に混み合わないようにするためには、Bの含有量の上限としては、50モル%以下とすることが重要であり、45モル%以下が好ましく、40モル%以下がより好ましい。
【0055】
また本発明の樹状ポリエステルは特性に影響が出ない範囲で、部分的に架橋構造を有していてもよい。
【0056】
本発明において、樹状ポリエステルの製造方法は、公知のポリエステルの重縮合法に準じて製造できる。前記R1で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、R2で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能以上の有機残基(B)を形成する3官能以上の多官能単量体を反応させる方法であって、該多官能単量体の添加量(モル)が、樹状ポリエステルを構成する全単量体(モル)に対して7.5モル%以上として製造する方法が好ましい。多官能単量体の添加量は、より好ましくは10モル%以上、より好ましくは15モル%以上、さらに好ましくは20モル%以上である。また、添加量の上限としては、50モル%以下が好ましく、より好ましくは33モル%以下、さらに好ましくは25モル%以下である。
【0057】
また、上記反応に際して、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体をアシル化した後、3官能以上の多官能単量体を反応させる態様も好ましい。また、R1、R2およびR3で表される構造単位から選ばれる少なくとも1種の構造単位を含む単量体、および、3官能以上の多官能単量体をアシル化した後、重合反応させる態様も好ましい。
【0058】
前記構造単位(I)、(II)、(III)および(IV)とトリメシン酸残基から構成される樹状ポリエステルを製造する場合を例に挙げて、好ましい製造方法を説明する。
(1)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、テレフタル酸およびポリエチレンテレフタレートポリマーから脱酢酸縮重合反応によって液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(2)p−アセトキシ安息香酸、4,4’−ジアセトキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよびトリメシン酸から脱酢酸縮重合反応によって製造する方法。
(3)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸およびポリエチレンテレフタレートポリマーに無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって液晶性ポリエステルオリゴマーを合成し、さらにトリメシン酸を加えて脱酢酸重合反応させて製造する方法。
(4)p−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよびトリメシン酸に無水酢酸を反応させて、フェノール性水酸基をアシル化した後、脱酢酸重縮合反応によって製造する方法。
(5)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、テレフタル酸ジフェニルエステルおよびポリエチレンテレフタレートポリマーから脱フェノール重縮合反応により液晶性ポリエステルオリゴマーを合成した後、トリメシン酸を加えて脱フェノール重縮合反応によって製造する方法。
(6)p−ヒドロキシ安息香酸のフェニルエステル、4,4’−ジヒドロキシビフェニル、テレフタル酸ジフェニルエステル、ポリエチレンテレフタレートポリマーおよびトリメシン酸のフェニルエステルから脱フェノール重縮合反応によって製造する方法。
(7)p−ヒドロキシ安息香酸、テレフタル酸、トリメシン酸にジフェニルカーボネートを反応させて、それぞれフェニルエステルとした後、4,4’−ジヒドロキシビフェニル、ポリエチレンテレフタレートポリマーを加え、脱フェノール重縮合反応によって製造する方法。
【0059】
なかでも(1)〜(4)の製造方法が好ましく、(3)および(4)の方法がより好ましい。
【0060】
(3)および(4)の製造方法において、無水酢酸の使用量は、鎖長制御の点からフェノール性水酸基の合計の0.95当量以上1.10当量以下であることが好ましく、1.00当量以上1.08当量以下であることがより好ましく、最も好ましくは1.02当量以上1.05当量以下である。無水酢酸量を制御すること、ジヒドロキシモノマーおよびジカルボン酸モノマーのいずれかを過剰に添加すること等により、末端基を制御することが可能である。
【0061】
分子量を上げるためには、トリメシン酸のカルボン酸量に相当する分だけ、4,4’−ジヒドロキシビフェニルなどのジヒドロキシモノマーを、ジカルボン酸モノマーに対して過剰に加え、全単量体におけるカルボン酸と水酸基当量を合わせることが好ましい。一方、カルボン酸を意図的に末端基に残す場合には、前記のようなジヒドロキシモノマーの過剰添加を行わないことが好ましい。さらに、水酸基を意図的に末端に残す場合には、ジヒドロキシモノマーをトリメシン酸のカルボン酸当量以上に過剰に添加し、かつ無水酢酸の使用量をフェノール性水酸基の1.00当量未満で行うことが好ましい。
【0062】
これらの方法により、本発明の樹状ポリエステルには、種々の熱可塑性ポリマーとの反応性に富む末端基構造を選択的に設けることが可能である。ただし、マトリックスポリマーとなる熱可塑性ポリマーによっては、過剰な反応性を抑制するために、単官能エポキシ化合物などを用いて末端を封鎖した方が分散状態を制御しやすい場合もある。
【0063】
脱酢酸重縮合反応を行う場合には、樹状ポリエステルが溶融する温度で、場合によっては減圧下で反応させ、所定量の酢酸を留出させ、重縮合反応を完了させる溶融重合法が好ましい。例えば、所定量のp−ヒドロキシ安息香酸、4,4’−ジヒドロキシビフェニル、テレフタル酸、ポリエチレンテレフタレートポリマーおよび無水酢酸を、攪拌翼および留出管を備え、下部に吐出口を備えた反応容器中に仕込む。混合物を、窒素ガス雰囲気下で攪拌しながら加熱して、水酸基をアセチル化させた後、200〜350℃まで昇温して脱酢酸重縮合反応を行い、酢酸を留出させる。酢酸が、理論留出量の50%まで留出した段階で、トリメシン酸を所定量加えて、さらに理論留出量の91%まで酢酸を留出させ、反応を完了させる。
【0064】
アセチル化させる条件としては、反応温度は、130〜170℃の範囲が好ましく、より好ましくは135〜155℃の範囲である。反応時間は、0.5〜6時間が好ましく、より好ましくは1〜2時間である。
【0065】
重縮合させる温度は、樹状ポリエステルが溶融する温度であり、好ましくは樹状ポリエステルの融点+10℃以上の温度である。具体的には、例えば、200〜350℃の範囲であり、240〜280℃が好ましい。重縮合させるときの雰囲気は、常圧窒素下でも問題ないが、減圧すると反応が早く進み、系内の残留酢酸が少なくなるため好ましい。減圧度は、0.1mmHg(13.3Pa)〜200mmHg(26600Pa)が好ましく、より好ましくは10mmHg(1330Pa)〜100mmHg(13300Pa)である。なお、アセチル化と重縮合は同一の反応容器で連続して行っても良いし、アセチル化と重縮合を異なる反応容器で行っても良い。
【0066】
重縮合反応が完了した後、反応容器内を樹状ポリエステルが溶融する温度に保ち、例えば、0.01〜1.0kg/cm2(0.001〜0.1MPa)に加圧し、反応容器下部に設けられた吐出口より、樹状ポリエステルをストランド状に吐出する。吐出口には断続的に開閉する機構を設け、液滴状に吐出することも可能である。吐出した樹状ポリエステルは、空気中もしくは水中を通過して冷却された後、必要に応じて、カッティングもしくは粉砕される。
【0067】
得られたペレット状、粒状または粉状の樹状ポリエステルは、さらに必要に応じて、熱乾燥や真空乾燥により水、酢酸などを除く。また、重合度の微調整、あるいは、さらに重合度を上げるために、固相重合をすることも可能である。固相重合は、例えば、上記により得られた樹状ポリエステルを、窒素気流下、または、減圧下、樹状ポリエステルの融点−50℃〜融点−5℃(例えば、200〜300℃)の温度範囲で1〜50時間加熱する方法が挙げられる。
【0068】
樹状ポリエステルの重縮合反応は無触媒でも進行するが、酢酸第一錫、テトラブチルチタネート、酢酸カリウムおよび酢酸ナトリウム、三酸化アンチモン、金属マグネシウムなどの金属化合物を使用することもできる。
【0069】
本発明の樹状ポリエステルは、数平均分子量は1,000〜40,000であることが好ましく、より好ましくは1,000〜20,000、さらに好ましくは1,000〜10,000であり、最も好ましくは1,000〜5,000の範囲である。なお、この数平均分子量は、樹状ポリエステルが可溶な溶媒を使用して、GPC−LS(ゲル浸透クロマトグラフ−光散乱)法により絶対分子量として測定した値である。
【0070】
また、本発明における樹状ポリエステルの溶融粘度は、0.01〜30Pa・sが好ましく、0.5〜20Pa・sがより好ましく、1〜10Pa・sが特に好ましい。なお、この溶融粘度は、樹状ポリエステルの液晶開始温度+10℃の条件で、ずり速度100/sの条件下で高化式フローテスターによって測定した値である。
【0071】
本発明では、上記した樹状ポリエステルをマトリックスポリマーにブレンドすることで、流動性向上効果が得られ、紡糸温度を低下させることができるが、このメカニズムは以下のように推定している。すなわち、J.Polym.Sci.PartB:Polum.Phys.,vol.34,2433(1996).によると、非相溶系ポリマーブレンドにおけるポリマーの粘度は分子量項+分散相互作用項+スリップ効果項で記述されるが、本発明で用いる樹状ポリエステルは、分子量項については低分子量化、分散相互作用項については樹状構造、スリップ効果項について液晶性の効果により、ポリマーブレンドの流動性を向上させていると考えられる。すなわち、単に低粘度ポリマーあるいは液晶ポリマー、脂肪族ハイパーブランチポリマーをブレンドさせただけでは得られない、より高度な流動性向上効果を得ることができるのである。また、Macromolecule,vol.38,10571(2005).ではハイパーブランチポリマーと通常の直鎖状のポリマーのサイズを同一分子量で比較したところ、ハイパーブランチポリマーは1/5以下の分子サイズとなること、さらにPolymer,vol.45,7491(2004)によると、ハイパーブランチポリマーと通常の直鎖状ポリマーで分子間の絡み合いを第2ビリアル係数A2で評価したところ、ハイパーブランチポリマーではA2が2桁小さく、自己分子での分子間絡み合いが極めて少ないことが報告されている。以上より、本発明で用いる樹状ポリエステルは、マトリックスポリマー中で有機ナノ粒子的に振る舞い、流動性を向上させていると考えられる。さらに、前記したようにR2部分を芳香族と脂肪族の混合とすることで、分子鎖の絡み合いを制御し、紡糸性・糸斑改善と複屈折あたりの弾性率向上という、従来、二律背反の効果を両立させているのである。
【0072】
本発明では上記した樹状ポリエステルを0.1〜10wt%、熱可塑性ポリマーから成るマトリックスポリマーにブレンドすることが重要である。
【0073】
マトリックスポリマーは、ポリエステル、ポリアミド、ポリフェニレンスルフィド、ポリオレフィン、ポリカーボネート、ポリエステルカーボネート、ポリイミド、ポリアミドイミド、ポリエーテルケトン、ポリエーテルエーテルケトン、ポリフッ化ビニリデンなどを挙げることができる。中でも、汎用性が高く、かつ弾性率が比較的高いポリエステルや、耐熱性・薬品性に優れるポリフェニレンスルフィドが好ましい。特に、極限粘度が1.0dL/g以上の高分子量ポリエステルは融点はそれほど高くないものの超高粘度のため、紡糸温度を衣料用ポリエステルの場合に比べ10〜20℃程度高くする場合も有り、加水分解や熱分解抑制による高分子量保持の観点から有用である。また、汎用的ではないが、ポリエーテルケトン、ポリエーテルエーテルケトン、ポリイミド、ポリフッ化ビニリデンなど通常、紡糸温度がかなりの高温になるポリマー種も流動性向上効果により紡糸温度を低下させる効果からすると、好ましいポリマーである。特にポリフッ化ビニリデンなど腐食性物質や有害物質が発生しやすいポリマーでは、紡糸温度低下効果は有用である。また、分子量が10万以上の高分子量ポリマーの場合には、分子鎖の絡み合い抑制効果は有用である。特に、ポリ乳酸のように高分子量であるが熱分解温度が低いポリマーの場合には、紡糸温度を低下させることができると、加水分解や熱分解による分子量低下を押さえられるだけでなく、分解ガスの発生を抑制できるため非常に有用である。本発明においては、汎用性の観点から極限粘度1.0dL/g以上の高分子量ポリエチレンテレフタレート(PET)やポリエチレンナフタレート(PEN)をマトリックスポリマーとすることが好ましい。また、紡糸温度が高くなり熱分解しやすい、重量平均分子量15万以上のポリ乳酸、また融点が向上したステレオコンプレックスポリ乳酸をマトリックスポリマーとすることも有用性が高い。
【0074】
また別の観点では、ポリ乳酸とPETやポリブチレンテレフタレート(PBT)、ポリトリメチレンテレフタレート(PTT)、ナイロン6、ナイロン66などを複合紡糸する際に、ポリ乳酸の相手となるポリマーに本発明の樹状ポリエステルを添加すると、流動性を向上させることで複合紡糸の紡糸温度を低下させることができるため、これも有用である。
【0075】
さらに別の観点では、紡糸温度を低下できるということは紡糸口金付近での温度も低下できるため、口金汚れを低減し経時的な紡糸安定性を向上できるとともに、口金修正周期を延長できるため生産効率の向上も図ることができる。特に、PBTやPTTに酸化チタンを含有させた場合には、PETの場合とは比べものにならないほど口金孔外周に酸化チタンを核とする汚れが堆積しやすいという問題があるが、本発明ではこれの抑制にも寄与することが可能である。
【0076】
また、樹状ポリエステルのブレンド率は0.1wt%以上であれば流動性向上による紡糸温度低下効果や弾性率向上効果が認められ、0.7wt%以上であればより効果が上がり好ましい。一方、ブレンド率を10wt%以下とすることで、伸長変形を大きく阻害することなく良好な紡糸性と糸斑を達成することができる。好ましくは2wt%以下である。
【0077】
本発明の繊維では糸の太細斑の指標であるウースター斑(U%)は5%以下であることが重要である。これにより、糸の強度や弾性率の長手方向斑が小さくなり、特に産業資材用途に用いる場合に、材料設計し易くなるのである。また、衣料用繊維の観点からは染色斑を抑制するためにU%は2%以下であることが好ましく、1.2%以下であることがより好ましい。U%は小さいほど好ましいが、現実的な下限値としては0.1%である。紡糸性を向上させ、糸斑を小さくするためには、樹状ポリエステルとマトリックスポリマーの反応を抑制することが有用であり、樹状ポリエステルの末端封鎖が有効である。特にカルボキシ末端は封鎖しておくことが好ましい。
【0078】
また、エステル交換反応抑制剤の添加も効果的であり、ポリマーブレンド全体に対して0.01〜1wt%添加することが好ましい。多量添加の方が反応抑制には好ましいが、過度に添加すると紡糸性や糸斑が低下する場合があるので、0.05〜0.2wt%添加とすることがより好ましい。
【0079】
本発明で言うエステル交換反応抑制剤とは、樹状ポリエステルとマトリックスポリマーとの反応を抑制するものであり、特にマトリックスポリマーがポリエステルおよび/またはポリアミドの時に効果が大きい。これは、残留金属イオンなどに対して単座配位子または多座配位子として配位し、金属イオンの触媒能を失活させる作用を有する。例えば、トリアゾール系化合物、多価アミン化合物、ヒドラジン誘導体系化合物、リン系化合物、イオウ系化合物などが挙げられる。以下に具体例をあげる。
【0080】
トリアゾール系化合物の具体例としては、ベンゾトリアゾール、3−(N−サリシロイル)アミノ−1,2,4−トリアゾールなどが挙げられる。
【0081】
多価アミンの具体例としては、3,9−ビス[2−(3,5−ジアミノ−2,4,6−トリアザフェニル)エチル]−2,4,8,10−テトラオキサスピロ[5.5]ウンデカン、エチレンジアミン−テトラアセチックアシッド、エチレンジアミン−テトラアセチックアシッドのアルカリ金属塩(Li,Na,K)塩、N,N’−ジサリシリデン−エチレンジアミン、N,N’−ジサリシリデン−1,2−プロピレンジアミン、N,N’’−ジサリシリデン−N’−メチル−ジプロピレントリアミン、3−サリシロイルアミノ−1,2,4−トリアゾールなどが挙げられる。
【0082】
ヒドラジン誘導体系化合物の具体例としては、デカメチレンジカルボキシリックアシッド−ビス(N’−サリシロイルヒドラジド)、イソフタル酸ビス(2−フェノキシプロピオニルヒドラジド)、N−ホルミル−N’−サリシロイルヒドラジン、2,2−オキザミドビス[エチル−3−(3,5−ジ−t−ブチル−4−ハイドロオキシフェニル)プロピオネート]、オギザリル−ビス−ベンジリデン−ヒドラジド、ニッケル−ビス(1−フェニル−3−メチル−4−デカノイル−5−ピラゾレート)、2−エトキシ−2’−エチルオキサニリド、5−t−ブチル−2−エトキシ−2’−エチルオキサニリド、N,N−ジエチル−N’,N’−ジフェニルオキサミド、N,N’−ジエチル−N,N’−ジフェニルオキサミド、オキサリックアシッド−ビス(ベンジリデンヒドラジド)、チオジプロピオニックアシッド−ビス(ベンジリデンヒドラジド)、イソフタリックアシッド−ビス(2−フェノキシプロピオニルヒドラジド)、ビス(サリシロイルヒドラジン)、N−サリシリデン−N’−サリシロイルヒドラゾン、N,N’−ビス[3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオニル]ヒドラジン、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,6−ヘキサメチレン−ビス(N −ヒドロキシエチル−N−メチルセミカルバジド)−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,10−デカメチレン−ジ−カルボキシリックアシッド−ジ−ヒドロキシエチルカルボニルヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−1,10−デカメチレン−ジ−カルボキシリックアシッド−ジ−サリシロイルヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−ジ(ヒドロキシエチルカルボニル)ヒドラジド−ジホスファイト、テトラキス[2−t−ブチル−4−チオ(2’−メチル−4’−ヒドロキシ−5’−t−ブチルフェニル)−5−メチルフェニル]−N,N’−ビス(ヒドロキシエチル)オキサミド−ジホスファイト、N,N’−ビス[2−〔3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオニルオキシ〕エチル]オキサミドなどが挙げられる。
【0083】
リン系化合物としては、ホスフェート系化合物やホスファイト系化合物など、分子中にリン原子を含む化合物が挙げられる。
【0084】
ホスフェート系化合物の具体例としては、脂肪族リン酸エステル(トリメチルホスフェート、トリエチルホスフェート、トリブチルホスフェート、トリ(2−エチルヘキシル)ホスフェート、トリブトキシエチルホスフェート、トリオレイルホスフェート、ジブチルホスフェート、モノブチルホスフェート、ジ(2−エチルヘキシル)ホスフェート、モノイソデシルホスフェート、2−アクリロイルオキシエチルアシッドホスフェート、2−メタクリロイルオキシエチルアシッドホスフェート、ジシクロペンチルハイポジホスフェートなど)、芳香族リン酸エステル(トリフェニルホスフェート、トリクレジルホスフェート、トリキシレニルホスフェート、トリス(イソプロピルフェニル)ホスフェート、トリス(o−フェニルフェニル)ホスフェート、トリス(p−フェニルフェニル)ホスフェート、トリナフチルホスフェート、クレジルジフェニルホスフェート、キシレニルジフェニルホスフェート、ジ(イソプロピルフェニル)フェニルホスフェート、o−フェニルフェニルジクレジルホスフェート、ジピロカテコールハイポジホスフェートなど)、脂肪族−芳香族リン酸エステル(ジフェニル(2−エチルヘキシル)ホスフェート、ジフェニル−2−アクリロイルオキシエチルホスフェート、ジフェニル−2−メタクリロイルオキシエチルホスフェート、フェニルネオペンチルホスフェート、ペンタエリスリトールジフェニルジホスフェート、エチルピロカテコールホスフェートなど)などの正リン酸エステル及びこれらの縮合物が挙げられる。
【0085】
ホスファイト系化合物の具体例としては、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト、テトラキス(2,4−ジ−t−ブチルフェニル)4,4’−ビフェニレンホスフォナイト、ビス(2,4−ジ−t−ブチルフェニル)ペンタエリスリトール−ジ−ホスファイト、ビス(2,6−ジ−t−ブチル−4−メチルフェニル)ペンタエリスリトール−ジ−ホスファイト、2,2−メチレンビス(4,6−ジ−t−ブチルフェニル)オクチルホスファイト、4,4’−ブチリデン−ビス(3−メチル−6−t−ブチルフェニル−ジ−トリデシル)ホスファイト、1,1,3−トリス(2−メチル−4−ジトリデシルホスファイト−5−t−ブチル−フェニル)ブタン、トリス(ミックスドモノおよびジ−ノニルフェニル)ホスファイト、トリス(ノニルフェニル)ホスファイト、4,4’−イソプロピリデンビス(フェニル−ジアルキルホスファイト)などが挙げられる。
【0086】
イオウ系化合物としては、分子中に硫黄原子を含んでいれば良く、具体的には、ジラウリル3,3′−チオジプロピオネート、ジミリスチル3,3′−チオジプロピオネート、ジステアリル3,3′−チオジプロピオネート、ラウリルステアリル3,3′−チオジプロピオネート、ペンタエリスリトール−テトラキス−(β−ラウリル−チオ−プロピオネート)、3,9−ビス(2−ドデシルチオエチル)−2,4,8,10−テトラオキサスピロ[5,5]ウンデカンなどが挙げられる。
【0087】
これらのエステル交換反応抑制剤は、ポリマーに含まれる残留金属イオンに強く配位して錯体形成しやすい物を選ぶことで、金属イオンの触媒能を失活させやすい。
【0088】
また、ポリマーをブレンド、紡糸する際にはポリマーの融点以上で処理されることから、エステル交換反応抑制剤自体の耐熱性も重要となる。本発明のエステル交換反応抑制剤の中で、ヒドラジン誘導体系化合物やイオウ系化合物と比較し、リン系化合物は耐熱性が高く着色しにくいため好ましく、この中でも脂肪族リン酸エステル系化合物が特に好ましい。
【0089】
本発明の繊維においては、複屈折あたりの弾性率が樹状ポリエステル未添加の場合に比べ高くすることができるが、これは以下のように推定される。すなわち、前記したように枝構造を完全に芳香族だけにせず脂肪族も含有させることで、マトリックスポリマーの伸長変形を大きく阻害しない範囲で分子鎖の適度な絡み合いを持たせることで、局所的に分子鎖の拘束性が強い部分を誘起させ、これを伸長変形過程でさらに拘束性を強くする。これにより、全体の伸長度(延伸度)としてはそれほど高くなくても(複屈折が低くとも)、局所的に分子鎖の拘束性が強い部分が存在するため、弾性率のような微小変形特性には大きな影響を与えると考えられる。弾性率の向上という観点からは前記した樹状ポリエステルのブレンド率は多いほど好ましいが、紡糸性や糸斑との兼ね合いから上限が定められる。極限粘度1.0dL/g以上の高分子量PETであっても1wt%添加で十分な効果が得られる。
【0090】
本発明の繊維においては、弾性率は110cN/dtex以上であれば産業資材用途に好適であり、好ましい。この場合、弾性率は高い方が好ましいが、弾性率の現実的な上限は2000cN/dtexである。また、比較的低複屈折であるため、屈曲疲労や摩耗特性も従来品よりも向上し、タイヤコードやタイヤのキャップレイヤー材などのゴム補強用途のみならず漁網や農業資材の他、スクリーン紗などにも好適に用いることができる。特にスクリーン紗は従来の捺染から、より薄くて精度が求められるフラットディスプレイの電極印刷などにも利用されるようになっており、さらなるモノフィラメントの細繊度化と高弾性率化が求められるようになってきている。しかしながら、これを達成するため単純に繊維を高倍率延伸しただけでは分子配向が高くなり過ぎ(複屈折が高くなり過ぎ)、耐摩耗性が低下し製織時の毛羽などが多発する問題があった。しかしながら、本発明では複屈折が低くとも弾性率が高いため、このような課題を解決しうるのである。なお、特にスクリーン用途に用いる場合にはモノフィラメントとすることが好ましい。また、スクリーン紗に適用する場合には、弾性率は110cN/dtex以上、複屈折は0.180以下であることが好ましい。
【0091】
また、本発明の繊維において、弾性率が50cN/dtex以上、110cN/dtex未満であれば衣料用途などに好適に用いることができる。特に、混繊糸の芯糸として有用である。ここで、混繊糸とは、異なる2種類の繊維を混合した物であり、混合方法としては、紡糸混繊、エア交絡や合撚、複合仮撚りなどを用いることができる。特に、紡糸混繊では同一の口金から紡糸し、引き取り速度が同速度であることがほとんどであるので、分子配向の調整が難しかった。例えば、単糸繊度が細い物と太い物を紡糸混繊する場合には、太繊度糸を芯糸として混繊糸の比較的内層部に位置させ、細繊度糸を鞘糸として比較的外層部に位置させたいのであるが、現実には細繊度糸の方が分子配向が進む(複屈折が大きい)ため、延伸時の張力バランスにより期待とは逆に細繊度糸が芯糸を形成してしまうことが普通であった。しかしながら、太繊度糸側に前記した樹状ポリエステルをブレンドすると、糸の複屈折は低くとも弾性率が高いため比較的芯糸を形成しやすくなり、期待通りの糸の配置とすることが可能となるのである。
【0092】
本発明の繊維では、強度は2cN/dtex以上が好ましく、現実的な上限としては20cN/dtexである。また、伸度は延伸糸で2〜60%、特に高弾性率が必要とされる産業資材分野では2〜15%、衣料用では25〜60%とすることが好ましい。繊維の断面形状は、丸断面、三葉などの多葉断面、中空、星形、不定形など適宜選択可能である。
【0093】
上記した繊維は、繊維巻き取りパッケージやトウ、カットファイバー、わた、ファイバーボール、コード、パイル、織編、不織布、紙、液体分散体など多用な繊維製品とすることができる。
【0094】
本発明の繊維を得る方法の一例について説明する。樹状ポリエステルについては前記した通りであるが、ブレンド、繊維化については例えば以下のような方法を用いることができる。すなわち、前記した構造単位(I)〜(IV)とトリメシン酸残基から構成される樹状ポリエステルにおいて末端アセチル基が安息香酸で封鎖された絶対分子量2000〜5000の範囲の樹状ポリエステルとマトリックスポリマーを必要に応じ乾燥し、二軸押し出し混練機に導入する。この時、ブレンド装置としてはブレンド斑を低減するために二軸押し出し混練機とすることが好ましい。ここで、作製したポリマーブレンドをそのまま紡糸機に導いても、マスターペレットとして一旦ペレット化しても良い。省力化のためには混練直結紡糸が好ましいが、樹状ポリエステルのブレンド率やポリエステル分子量が異なる品種をいくつかつくるなど汎用性を持たせるためにはマスターペレット化が好ましい。また、混練直結紡糸の場合には、二軸押し出し混練機では一軸押し出し混練機の場合とは異なり、混練機中で誘起された発泡が仕込み側に抜け難いため、発泡が繊維にまで混入し糸切れが頻発する場合がある。このため、特に高分子量ポリエステルなど高粘度ポリマーをマトリックスポリマーとする場合には、二軸押し出し混練機の吐出側でベントを行い、泡を抜く操作を行うことが好ましい。なお、マスターペレット化場合にもガット切れが頻発する時はベントを行うことが好ましい。また、本発明においては樹状ポリエステル添加による良流動化効果により、未添加の場合に比べ同一温度であればスクリュートルクが小さくなるため、混練温度の低温化が可能である。これにより、ポリマーの熱分解や熱変性、また加水分解などを抑制することができ、バージンポリマーが本来持っていた高分子量や易加工性などを利用し易くできるのである。
【0095】
なお、樹脂加工の場合にはガラス繊維などの無機フィラーを多量に混合させることで機械的特性(弾性率向上など)やガスバリア性を向上させることも多いが、繊維化の場合に無機フィラーを混合させると紡糸機内のフィルターで詰まりが発生し濾圧が急上昇したり、また紡糸口金孔に無機フィラーが詰まり紡糸不能に陥る場合がある。また、紡糸不能に至らずとも、紡糸口金孔からのポリマーの吐出が安定せず糸切れの頻発や糸斑の悪化などの問題が発生する場合がある。このため、繊維化の場合には樹脂加工とは異なり、無機フィラーは混合しない方が良く、混合したとしてもブレンドポリマー全体に対し0.5wt%未満である。無機フィラーを0.5wt%以上混合した場合には、U%を5%以下とすることはできない。ここで言う無機フィラーとは、80wt%以上が無機物から構成され、円換算の平均直径が10nm以上かつ平均長さが100nm以上のものである。
【0096】
先の樹状ポリエステルブレンドでマスターペレット化した場合には、紡糸過程でバージンポリマーで希釈されるわけであるが、この時も二軸押し出し混練機を用いる方がブンレンドの均一性の観点から好ましい。というのは、本発明では樹状ポリエステルブレンド率で良流動化効果の程度が異なるため、ポリマーブレンド中でブレンドが不均一であるとスクリュートルクや先端圧、濾圧、口金背面圧、ひいては紡糸応力などの斑が発生し、安定した紡糸が不能となる場合があるからである。やむを得ず一軸押し出し混練機を用いる場合には、ダルメージなどの混練機能を付加するとともに、一軸押し出し混練機吐出付近や紡糸機あるいは紡糸パック内に静止混練器を設け、充分にブレンドを均一化することが好ましい。
【0097】
前記したように、樹状ポリエステルブレンドによる良流動化効果のため、未添加の場合に比べ混練機温度を低温化できるのであるが、紡糸機についても設定温度を低下させることが可能であり、例えば高分子量ポリマーによる高粘度のため通常では紡糸温度を融点より大幅に高温化せざるを得ない場合であっても、樹状ポリエステル添加により5℃以上の低温化も可能である。この効果は、高粘度ポリマーほど大きく発現する。また、この良流動化効果により紡糸口金孔からのポリマーの吐出が安定し、本来紡糸が不安定で糸斑が発生しやすい高粘度ポリマー紡糸であっても糸斑を小さくすることができる。
【0098】
また、本発明で用いる樹状ポリマーは枝構造に芳香族成分を導入し剛直性を向上、好ましくは液晶性を発現させるため、マトリックスポリマー分子鎖と絡み合い難いため、伸長変形がスムーズになるため、紡糸性が向上し糸斑が低減されるのみならず、枝構造が脂肪族のハイパーブランチポリマー添加では難しかった高速紡糸も可能となるのである。これも紡糸という伸長変形場特有の効果である。一方、本発明で用いる樹状ポリマーは枝構造に脂肪族成分も混合しているため、適度な分子鎖の絡み合いがあり複屈折あたりの弾性率が向上するが、この効果も高速紡糸や高倍率延伸など極限的な伸長変形場で強く発現する効果である。
【0099】
本発明において、紡糸温度は、樹状ポリエステル未添加の場合に比較して5〜20℃低下させることが好ましい。より好ましくは、7〜15℃低下である。また、紡糸速度はマトリックスポリマーの物性や繊維の目的によって異なるが、500〜6000m/分程度とすることができる。特に、産業資材用途で高弾性率が必要な場合には、高分子量ポリマーを用い、500〜1500m/分とし、その後高倍率延伸することが好ましい。
【0100】
延伸に際しては、特に予熱温度を適切に設定することが好ましい。というのは本発明で用いる樹状ポリエステルはガラス転移温度などの軟化温度が70℃より高い場合があり、例えばPETの通常の予熱温度である85〜95℃程度では、樹状ポリエステルが延伸過程で異物として振る舞い結果として延伸糸のタフネスの低下を招く場合がある。この影響は、特に高倍率延伸時ほど顕著に現れる。このため、樹状ポリエステルの添加量が微量であっても予熱温度は樹状ポリエステルのガラス転移温度や軟化温度以上に設定することが好ましい。予熱温度の上限としては、予熱過程で繊維の自発伸長により糸道乱れが発生しない温度とすることが好ましい。この延伸時の予熱温度設定も糸斑低減に寄与することができる。
【0101】
なお、スクリーン紗に用いる高弾性率細繊度モノフィラメントでは、高度に分子配向させて弾性率を向上させるのであるが、これだけでは耐摩耗性が低下し、製織過程で毛羽などが多発する場合が有り、この時は弛緩熱処理を行い分子配向を低下させこれを回避する場合がある。しかし、同時に弾性率も低下するため、従来技術では弾性率と耐摩耗性は二律背反であったが、本発明では複屈折あたりの弾性率が高いため、弛緩熱処理による低配向化が不要であり、この二律背反を解決できるのである。また、細繊度モノフィラメント紡糸であるが故、低吐出量であり紡糸機内でのポリマーの滞留時間が長く、さらに異常滞留によるポリマーの熱劣化によりゲル化などが発生しやすく、これがモノフィラメントの節を誘発する場合がある。この節とは繊維の異常太部であり、製織欠点となるのである。このため、この節はゼロにすべきものであるが、紡糸機や紡糸パック・口金構造の工夫の他にはあまり良い手段が無かった。しかしながら、本発明では溶融部である混練機温度や紡糸温度を低温化できるため、低吐出量であっても前記問題を緩和でき、節の発生を抑制できるため、産業上の価値は高い。
【0102】
また、ポリエステルやポリアミドで行われる機能付加成分を同時に紡糸する場合、機能付加成分の耐熱性が低くともこれの分解や変性・劣化を抑制することができるのである。
【実施例】
【0103】
以下、本発明を実施例を用いて詳細に説明する。なお、実施例中の測定方法は以下の方法を用いた。
【0104】
A.絶対分子量
樹状ポリエステルの絶対分子量は樹状ポリエステルが可溶な溶媒であるペンタフルオロフェノールを使用して、GPC−MALLS(ゲル浸透クロマトグラフ(ShodexGPC−101)−光散乱検出器(Wyatt製DAWN HELEOS))により、試料濃度0.04%、測定温度23℃で測定した。
【0105】
B.重量平均分子量
本発明の熱可塑性マトリックスポリマーの重量平均分子量は、ゲルパーミエーションクロマトグラフィー(GPC)により測定した標準PMMA換算の値である。GPC測定は、検出器にWATERS社示差屈折計WATERS410を用い、ポンプにMODEL510高速液体クロマトグラフィーを用いて、溶媒にヘキサフルオロイソプロパノールを用いて測定した。
【0106】
ただし、ポリ乳酸の重量平均分子量は以下のようにして求めた。試料のクロロホルム溶液にTHF(テトロヒドロフラン)を混合し測定溶液とした。これをWATERS社製GPC WATERS2690を用いて25℃で測定し、ポリスチレン換算で重量平均分子量を求めた。
【0107】
C.ポリエステルの極限粘度
ポリエステルの極限粘度はo−クロロフェノールに溶解してオストワルド式粘度計を用いて25℃で測定した。
【0108】
D.ナイロンの相対粘度
98%硫酸水溶液にナイロンを溶解し0.01g/mLの濃度に調整した後、オストワルド式粘度計を用いて25℃で測定した。
【0109】
E.樹状ポリエステルの化学組成比
樹状ポリエステルの化学組成比は核磁気共鳴装置(日本電子製JNM−AL400)を用いて、ペンタフルオロフェノール/重水素化クロロホルム(50/50)混合溶媒に溶解して、40℃で1H−NMR測定を行い、ピーク強度比から各成分の化学組成比を算出した。
【0110】
F.融点およびガラス転移点
TA Instruments社製DSC2920 Modulated DSCを用いて2nd runでポリマーの融解を示すピークトップ温度をポリマーの融点とした。この時の昇温速度は16℃/分、サンプル量は10mgとした。
また、同じく2nd runでの階段状の吸熱を示す領域の中点をガラス転移点とした。
【0111】
G.液晶開始温度
剪断応力加熱装置(CSS−450)により、剪断速度1.0(1/秒)、昇温速度5.0℃/分、対物レンズ60倍において測定し、視野全体が流動開始する温度とした。
【0112】
H.繊維のウースター斑(U%)
ツェルベガーウスター株式会社製USTER TESTER 4を用い、給糸速度200m/分でノーマルモードで測定を行った。
【0113】
I.繊維の力学特性(強度、伸度、弾性率)
室温(25℃)で、初期試料長=200mm、引っ張り速度=200mm/分とし、JIS L1013に示される条件で荷重−伸長曲線を求めた。次に破断時の荷重値を初期の繊度で割り、それを強度とし、破断時の伸びを初期試料長で割り伸度として強伸度曲線を求めた。また、弾性率は荷重−伸長曲線の初期立ち上がり部分を直線近似し、その傾きから求めた。
【0114】
J.繊維の複屈折
OLIMPUS BH−2偏光顕微鏡により単糸のレターデーションと路長を測定し、複屈折を求めた。
【0115】
K.耐摩耗性の評価
耐摩耗性の評価は特開2004−232182号公報に準じ、φ3mmの梨地金属棒に接触角35°で糸を掛け、金属棒から340mmの所で糸張力1.5cN/dtexとして把持し、ストローク長30mm、速度100回/分の往復運動を与え、毛羽(剥離、フィブリル化)の発生した往復回数を測定した。測定は5回行いそれの平均値とし、毛羽発生まで1000回以上を合格とした。
【0116】
参考例1(樹状ポリエステルA−1の合成)
攪拌翼、留出管を備えた反応容器にp−ヒドロキシ安息香酸48.0g(0.35モル)、4,4’−ジヒドロキシビフェニル30.9g(0.17モル)、テレフタル酸5.41g(0.033モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレート10.4g(0.054モル)、トリメシン酸42.0g(0.20モル)、および無水酢酸76.3g(フェノール性水酸基合計の1.1当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で1.5時間反応させた後、250℃まで昇温して脱酢酸縮合反応を行った。反応器内温が250℃に達した後、安息香酸14.7g(0.12モル)を加えて280℃まで昇温させた。酢酸の理論留出量の100%が留出したところで加熱、攪拌を停止し、内容物を冷水中に吐出し、樹状ポリエステル樹脂(A−1)を得た。
【0117】
この樹状ポリエステル樹脂(A−1)は、核磁気共鳴スペクトル解析の結果、R部分の構造が、p−オキシベンゾエート単位の含量pが2.0、4,4’−ジオキシビフェニル単位とエチレンオキシド単位の含量qが0.5、テレフタレート単位の含量rが0.5であり、p+q+r=3であり、分岐点、すなわちBの含有率は樹状ポリエステルを構成する全単量体に対して25モル%であった。また末端構造はカルボン酸と安息香酸エステルであった。
【0118】
得られた樹状ポリエステル樹脂の融点Tmは182℃、液晶開始温度は163℃で、絶対分子量は5500であった。
【0119】
参考例2(樹状ポリエステルA−2の合成)
攪拌翼、留出管を備えた反応容器にp−ヒドロキシ安息香酸66.30g(0.48モル)、4,4’−ジヒドロキシビフェニル8.38g(0.045モル)、テレフタル酸7.48g(0.045モル)、固有粘度が約0.6dl/gのポリエチレンテレフタレ−ト14.40g(0.075モル)、α−レゾルシル酸42.72g(0.28モル)および無水酢酸78.26g(フェノール性水酸基合計の1.08当量)を仕込み、窒素ガス雰囲気下で攪拌しながら145℃で2時間反応させた。その後、260℃まで昇温し、3時間攪拌し、理論留出量の91%の酢酸が留出したところで、安息香酸25.6g(0.21モル;理論アセトキシ末端に対して1.000倍)を添加し、酢酸を100%まで留出させたところで、加熱および攪拌を停止し、内容物を冷水中に吐出して樹状ポリエステル樹脂(A−2)を得た。
【0120】
この樹状ポリエステル樹脂(A−2)は、核磁気共鳴スペクトル解析の結果、R部分の構造が、p−オキシベンゾエート単位の含量pが1.32、4,4’−ジオキシビフェニル単位とエチレンオキシド単位の含量qが0.33、テレフタレート単位の含量rが0.33であり、p+q+r=2であり、分岐点含有率は30モル%であった。また末端構造はカルボン酸とアセチル基であった。
【0121】
得られた樹状ポリエステル樹脂の融点Tmは182℃、液晶開始温度は152℃で、絶対分子量は6500であった。
【0122】
参考例3(脂肪族系超分岐ポリマーA−3の合成)
特表2005−513186号公報に準じ脂肪族系超分岐ポリマーを合成した。
【0123】
実施例1〜5、比較例1
参考例1で合成した樹状ポリエステル(A−1)と、極限粘度1.11dL/g(重量平均分子量=25,500)の高分子量PETを乾燥した後、別々に計量し、独立に二軸押し出し混練機に仕込んだ。この時、樹状ポリエステルの添加量はポリマーブレンドに対し1wt%とした。二軸押し出し混練機の吐出側でベントを行い、泡を消した。混練機および紡糸温度は表1のように設定した。吐出量は42.3g/分で絶対濾過径10μの金属不織布で濾過した後、丸孔24ホール(φ=0.6mm)の口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、給油し巻き取った。
【0124】
まず、紡糸速度を500m/分として、樹状ポリエステル添加による流動性向上効果を確かめた。実施例1〜5と比較例1の比較から明らかなように、樹状ポリエステルを添加することで顕著な紡糸パック圧力の低下が起こり流動性が高分子量PETの流動性が向上していることが確かめられた。また、図1に紡糸温度と紡糸パック圧力の関係をプロットしたが、同一紡糸パック圧力で比較すると、樹状ポリエステル1wt%添加(実施例1〜5)では、8℃の紡糸温度低下効果が見込めることがわかった。また、念のため実施例2(樹状ポリエステル1wt%添加、紡糸温度295℃)で、PETの重量平均分子量を測定したが、18,600であり、樹状ポリエステル無添加(比較例1)と同じ値であり、樹状ポリエステル添加による加水分解や熱分解の促進は見られなかった。得られた繊維はほとんどがU%が1%以下と実用的に充分小さいレベルの糸斑であった。
【0125】
【表1】
【0126】
実施例6
さらに、脂肪族リン酸エステル系のエステル交換反応抑制剤であるアデカ社製アデカスタブAX−71を乾燥した後、ブレンドポリマーに対し0.1wt%添加し、実施例4(紡糸温度291℃)と同様に紡糸を行ったところ、さらに顕著な紡糸パック圧力低下が起こり、流動性向上に非常に有効であった(表1、図1)。
【0127】
実施例7〜9
次に、樹状ポリエステル(A−1)の添加量を種々変更し実施例3と同様に紡糸を行ったところ、添加量に応じた流動性向上効果が得られた。U%については、実施例9では1.41%となったが、実用上大きな問題のないレベルである。実施例7,8では1%以下であり優れたU%であった。
【0128】
実施例10、11
次に紡糸速度を変更して実施例3と同様に紡糸を行ったが、紡糸速度を高速化しても紡糸性は良好でありU%も1%以下であった。
【0129】
実施例12〜18、比較例2〜8
実施例2,4,5,6および比較例1で得られた未延伸糸の延伸・熱処理実験を行った。この時、延伸機としては3ホットローラー型延伸機を用い、フィードローラー(非加熱)、第1ホットローラー、第2ホットローラー、第3ホットローラー、デリバリーローラー(非加熱)と糸を通して延伸・熱処理を行った。第1ホットローラー温度(予熱温度)を90℃、第2ホットローラー温度を140℃、第3ホットローラー温度を230℃とし、第1ホットローラーと第2ホットローラー間の延伸倍率を3.85に固定し、第2ホットローラーと第3ホットローラー間の延伸倍率を変化させた。表2にはフィードローラーからデリバリーローラーまでのトータル延伸倍率を示した。
【0130】
いずれも延伸性に問題はなく糸切れ、毛羽等は発生せず、U%も1%以下であった。また、複屈折と弾性率の関係を図2にプロットしたが、樹状ポリエステルを添加した実施例12〜18は未添加の比較例2〜8に比べ同一複屈折での弾性率は1割以上高いものであった。また、特に複屈折が0.185付近よりも小さい領域は効果が高いものであった。
【0131】
【表2】
【0132】
実施例19
樹状ポリエステル(A−1)の熱流動開始温度が160℃と高温であるため、第2ホットローラー温度をその温度以上に変更して、延伸性と延伸糸の物性向上について調べた。未延伸糸としては実施例2で得た物を用い、延伸倍率を5.6倍とし、実施例12と同様に延伸・熱処理を行った。
【0133】
まず第2ホットローラー温度が140℃の時にはトータル延伸倍率5.7倍では糸切れのため延伸不能であったが、第2ホットローラー温度を165℃にしたところ、糸切れは無く延伸性良好であった。また得られた延伸糸物性としては強度7.1cN/dtex、伸度7.5%、弾性率125cN/dtex、複屈折0.186、U%=0.78%と実施例12に比べ、より高倍率延伸できた結果、力学特性の向上が認められた。また、糸斑も改善した。
【0134】
実施例20
極限粘度0.63dL/gのPETと参考例1で合成した樹状ポリエステル(A−1)を乾燥した後、別々に軽量し、独立に二軸押し出し混練機に仕込みブレンドを行った。この時、樹状ポリエステルはポリマーブレンド全体に対し1wt%とした。また、混練機温度は285℃とて、複合紡糸機にブレンドポリマーを導いた。一方、極限粘度0.63dL/gのPETを一軸押し出し混練機で290℃で溶融し、やはり同じ複合紡糸機に導いた。そして、紡糸温度を287℃として、ポリマーブレンドは単糸繊度6dtex(12フィラメント)、PETは単糸繊度2dtex(72フィラメント)となるように紡糸速度3000m/分で巻き取った。そして、この混繊未延伸糸にベルトニップ型仮撚り機を用い、ヒーター温度190℃、延伸倍率1.8倍で常法に従い仮撚りを施した。得られた仮撚り加工糸断面に於いて、ポリマーブレンドから成る太繊度糸が混繊糸の比較的内側に位置し芯糸を形成し、PETから成る細繊度糸が混繊糸の比較的外側に位置し鞘糸を形成していた。なお、紡糸過程でポリマーブレンドから成る太繊度糸のみを巻き取り、U%を測定したところ、U%=0.98%と良好であった。さらにこれに、前記したのと同様の仮撚り加工を加え、仮撚り加工糸のU%を測定したが1.00%とやはり糸斑は良好であった。また、この糸の弾性率は60cN/dtexであった。
【0135】
そして、この混繊糸から成る仮撚り加工糸に700T/mの実撚りを加え、S撚り/Z撚りを交互配列し経糸および緯糸に用いて2/2ツイル織物を作製した。さらにこの織物を80℃で精練を施した後170℃で中間セットした後、常法により染色し、170℃で仕上げセットを行った。得られた織物は嵩高さとソフトさ、またハリ腰を備えた風合いに優れる物であった。また、染色斑も無く審美性にも優れる物であった。
【0136】
比較例9
樹状ポリエステルを添加せず実施例20と同様に紡糸、仮撚り加工を行ったところ、太繊度糸が鞘糸を細繊度糸が芯糸を形成していた。この混繊糸を経糸および緯糸に用い、実施例20と同様に織物を作製し仕上げセットまで行ったところ、芯糸と鞘糸が逆転しているため、粗硬感はあるがハリ腰の無い風合いに劣る物であった。
【0137】
実施例21
実施例20で得た未延伸混繊糸を予熱温度90℃、延伸倍率1.85倍、熱セット温度130℃で延伸・熱処理した。得られた延伸糸断面に於いて、ポリマーブレンドから成る太繊度糸が混繊糸の比較的内側に位置し芯糸を形成し、PETから成る細繊度糸が混繊糸の比較的外側に位置し鞘糸を形成していた。なお、紡糸過程でポリマーブレンドから成る太繊度糸のみを巻き取り、U%を測定したところ、U%=0.98%と良好であった。さらにこれに、前記したのと延伸・熱処理を加え、延伸糸のU%を測定したが0.82%とやはり糸斑は良好であった。また、この糸の弾性率は85cN/dtexであった。
【0138】
この混繊糸を用いて丸編みを作製し、80℃で精練を施した後170℃で中間セットした後、常法により染色し、170℃で仕上げセットを行った。得られた編物は嵩高さとソフトさ、また適度なハリ腰を備えた風合いに優れる物であった。
【0139】
実施例22
参考例1で合成した樹状ポリエステル(A−1)と、極限粘度1.11dL/gの高分子量PETおよびエステル交換反応抑制剤であるアデカ社製アデカスタブAX−71を乾燥した後、別々に計量し、二軸押し出し混練機に仕込んだ。この時、樹状ポリエステルとエステル交換反応抑制剤は計量した物をドライブレンドし、仕込んだ。添加量はポリマーブレンドに対し、樹状ポリエステルは1wt%、エステル交換反応抑制剤は0.1wt%とした。また、二軸押し出し混練機の吐出側でベントを行い、泡を消した。混練機および紡糸温度は287℃に設定し、丸孔4ホールの口金から紡糸を行い、ユニフローの冷却風帯域を通過させた後、モノフィラメントに分割後給油し、それぞれモノフィラメントとして巻き取った。そして、トータル延伸倍率4.72倍、第2ホットローラー温度165℃として、実施例16と同様に延伸熱処理を行い、単糸繊度8dtexのモノフィラメントを得た。得られた繊維は、U%=0.78%、弾性率=111cN/dtex、複屈折=0.178、強度=5.5cN/dtex、伸度=16.5%であった。
【0140】
これの耐摩耗性の評価を行ったところ、毛羽発生までの往復回数は1000回以上であり、良好な耐摩耗性を示した。これは、充分な弾性率と耐摩耗特性を有したモノフィラメントであり、IT用などの薄くてかつ高精度が求められるスクリーン紗に最適な物である。
【0141】
比較例10
樹状ポリエステルとエステル交換反応抑制剤を添加しないで、混練機温度および紡糸温度を300℃として実施例22と同様に紡糸を行った。なお吐出量は延伸糸繊度に応じて調整した。得られたモノフィラメントをトータル延伸倍率5.70倍として実施例22と同様に延伸・熱処理し、8dtexのモノフィラメントを得た。これの弾性率は111cN/dtex、複屈折は0.186であった。続いて耐摩耗性を実施例22と同様に測定したが、毛羽発生まで500回と耐摩耗性の低い物であった。
【0142】
実施例23
参考例2で合成した樹状ポリエステル(A−2)を用い、実施例2と同様に紡糸を行ったところ、紡糸パック圧力は5.4Pa・sと良流動化効果を確認できた。また得られた未延伸糸のU%は0.94%と充分糸斑の小さな物であった。
これを第2ホットローラー温度を165℃、トータル延伸倍率5.20倍として実施例12と同様に延伸・熱処理し、U%=0.77%、弾性率=110cN/dtex、複屈折=0.180、強度=5.8cN/dtex、伸度=18%の繊維を得た。A−1を使用した場合に比べると若干複屈折あたりの弾性率が低いが、未添加に比べると十分な効果であった。
【0143】
比較例11
参考例3で合成した脂肪族系超分岐ポリマー(A−3)を用い、実施例11と同様に紡糸を行ったところ、紡糸パック圧力は8.0Pa・sと本発明のA−1、A−2を用いた場合に比べ良流動化効果は小さいものであった。また、紡糸が安定せず、糸切れが多発するなど紡糸性も実施例11には及ばなかった。
【0144】
実施例24
光学純度99%のポリL乳酸(重量平均分子量19万)を乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は220℃および紡糸温度は230℃とし、吐出量は30g/分、口金として孔径0.30mm、18ホールの物を用い、紡糸速度は5000m/分とした。この時の紡糸パック圧力は16MPaであった。紡糸温度をポリ乳酸の分解が著しくなる温度(240℃)よりも下げることができたため、分解ガスの発生を抑制することができた。得られた高速紡糸繊維のU%は0.81%であった。
【0145】
比較例12
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度240℃、紡糸温度245℃として実施例24と同様に溶融紡糸を行った。この時の紡糸パック圧力は17MPaであった。紡糸温度がポリ乳酸に対して高いため、分解ガスの発生が著しく作業環境が悪化した。
【0146】
実施例25
極限粘度0.75dL/gのPENを乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は290℃および紡糸温度は290℃とし、吐出量は20g/分、紡糸口金としてはφ0.60mm、10ホールの物を用い、紡糸速度は1350m/分とした。この時の紡糸パック圧力は5.8MPaであり、流動性向上効果を確かめることができた。得られた高速紡糸繊維のU%は0.95%であった
比較例13
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度295℃、紡糸温度300℃として実施例25と同様に溶融紡糸を行った。この時の紡糸パック圧力は6.8MPaであった。
【0147】
実施例26
相対粘度3.8のナイロン66を乾燥し、実施例6と同様にブレンド、溶融紡糸を行った。この時、混練機温度は290℃および紡糸温度は290℃とし、吐出量は33.5g/分、紡糸口金としてはφ0.30mm、18ホールの物を用い、紡糸速度は1500m/分とした。この時の紡糸パック圧力は12.5MPaであり、流動性向上効果を確かめることができた。得られた繊維のU%は0.95%であった
比較例14
樹状ポリエステルおよびエステル交換抑制剤を添加せず混練機温度295℃、紡糸温度295℃として実施例26と同様に溶融紡糸を行った。この時の紡糸パック圧力は16.5MPaであった。
【図面の簡単な説明】
【0148】
【図1】紡糸温度と紡糸パック圧力の関係を示す図
【図2】複屈折と弾性率の関係を示す図
【特許請求の範囲】
【請求項1】
芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
【請求項2】
熱可塑性のマトリックスポリマーがポリエステルである請求項1記載の繊維。
【請求項3】
弾性率が110〜2000cN/dtexである請求項1または2記載の繊維。
【請求項4】
弾性率が50cN/dtex以上、110cN/dtex未満である請求項1または2記載の繊維。
【請求項5】
樹状ポリエステルをマトリックスポリマーにブレンドしたポリマーブレンド全体に対して、エステル交換反応抑制剤を0.01〜1wt%含有する請求項1〜4のいずれか1項記載の繊維。
【請求項6】
繊維がモノフィラメントである請求項1〜3および5のいずれか1項記載の繊維。
【請求項7】
請求項1〜2および4のいずれか1項記載の繊維が芯糸として用いられた混繊糸。
【請求項8】
請求項1〜7のいずれか1項記載の繊維を少なくとも一部に使用した繊維製品。
【請求項1】
芳香族オキシカルボニル単位(P)、芳香族および脂肪族ジオキシ単位(Q)、および芳香族ジカルボニル単位(R)からなる構造単位と3官能以上の有機残基(B)とを含み、かつ、Bの含有量が樹状ポリエステルを構成する全単量体に対して7.5〜50モル%の範囲にある樹状ポリエステルを熱可塑性のマトリックスポリマーに0.1〜10wt%ブレンドしたポリマーブレンドから成る繊維であって、繊維のウースター斑が0.1〜5%である繊維。
【請求項2】
熱可塑性のマトリックスポリマーがポリエステルである請求項1記載の繊維。
【請求項3】
弾性率が110〜2000cN/dtexである請求項1または2記載の繊維。
【請求項4】
弾性率が50cN/dtex以上、110cN/dtex未満である請求項1または2記載の繊維。
【請求項5】
樹状ポリエステルをマトリックスポリマーにブレンドしたポリマーブレンド全体に対して、エステル交換反応抑制剤を0.01〜1wt%含有する請求項1〜4のいずれか1項記載の繊維。
【請求項6】
繊維がモノフィラメントである請求項1〜3および5のいずれか1項記載の繊維。
【請求項7】
請求項1〜2および4のいずれか1項記載の繊維が芯糸として用いられた混繊糸。
【請求項8】
請求項1〜7のいずれか1項記載の繊維を少なくとも一部に使用した繊維製品。
【図1】

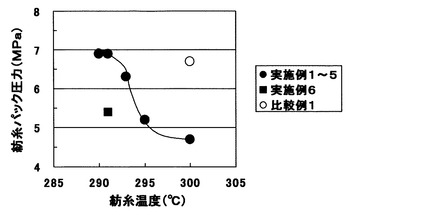
【図2】
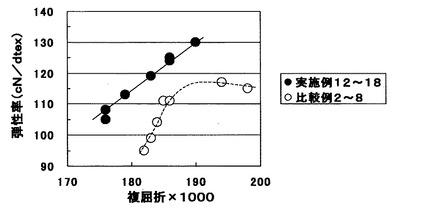
【図2】
【公開番号】特開2009−52150(P2009−52150A)
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願番号】特願2007−216971(P2007−216971)
【出願日】平成19年8月23日(2007.8.23)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成21年3月12日(2009.3.12)
【国際特許分類】
【出願日】平成19年8月23日(2007.8.23)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]