
置換トリアゾロ−ピリダジン誘導体
【課題】α1−GABA−A受容体アンタゴニスト、ならびに/または、α2、α3、およびα5 GABA−A受容体アゴニストの投与によって有益な形で治療される疾患および状態を治療する方法の提供。
【解決手段】式1の置換トリアゾロ−ピリダジン、その誘導体、およびその薬学的に許容可能な塩。

(式中、R1はCH3、CDH2、CD2H、またはCD3であり、R2は、0〜9個の重水素原子を有するt−ブチル基であり、各Yは独立して、水素または重水素であり、R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
【解決手段】式1の置換トリアゾロ−ピリダジン、その誘導体、およびその薬学的に許容可能な塩。
(式中、R1はCH3、CDH2、CD2H、またはCD3であり、R2は、0〜9個の重水素原子を有するt−ブチル基であり、各Yは独立して、水素または重水素であり、R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
【発明の詳細な説明】
【関連出願の相互参照】
【0001】
本出願は、2008年8月29日に提出された米国特許仮出願第61/093,293号の優先権を主張するものであり、その内容は、参照により本明細書に組み込まれる。
【技術分野】
【0002】
本発明は、新規な置換トリアゾロ−ピリダジン、その誘導体、およびそれらの薬学的に許容可能な塩に関する。本発明は、本発明の化合物を含む組成物、ならびに、α1−GABA−A受容体アンタゴニストの投与によって有益な形で治療される疾患および状態を治療する方法において、前記組成物を使用することも提供する。
【背景技術】
【0003】
L−838417(7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−(1−メチル−1H−1,2,4−トリアゾール−5−イルメトキシ)[1,2,4]トリアゾロ[4,3−b]ピリダジンとしても知られている)は、GABA−A受容体のベンゾジアゼピン部分で、サブタイプα1のアンタゴニストとして、ならびに、サブタイプα2、α3、およびα5の機能選択的アロステリックアゴニストとしての役割を果たす。
【0004】
L−838417は現在のところ、中枢神経系障害に対する前臨床段階の候補である。
L−838417の有益な活性にもかかわらず、α1−GABA−A受容体アンタゴニストである新たな化合物に対するニーズが依然として存在する。
【図面の簡単な説明】
【0005】
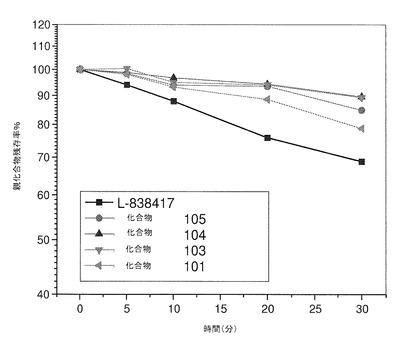
【図1】ヒト肝ミクロソームにおける本発明の化合物の経時安定性を示している。
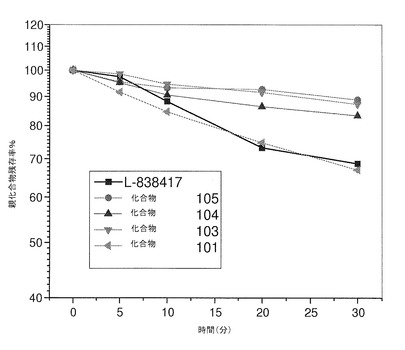
【図2】ラット肝ミクロソームにおける本発明の化合物の経時安定性を示している。
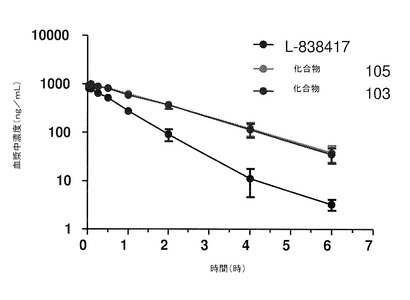
【図3】ラットに静脈内投与した後の本発明の化合物の血漿中濃度の変化を示している。
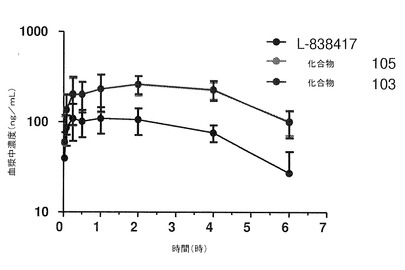
【図4】ラットに経口投与した後の本発明の化合物の血漿中濃度の変化を示している。
【発明を実施するための形態】
【0006】
定義
「治療する」という用語は、疾患(例えば、本明細書に示されている疾患または障害)の発生または進行を減少、抑制、減衰、縮小、停止、または安定させることを意味する。
【0007】
「疾患」とは、細胞、組織、または器官の正常機能を損傷させるか、またはそれに干渉するいずれかの状態または障害を意味する。
【0008】
合成化合物においては、その合成で用いた化学物質の起源に応じて、天然同位体存在度の多少の変動が生じることが認められる。したがって、L−838417の調製物は本質的に、少量の重水素化アイソトポローグを含有することになる。上記の変動にもかかわらず、天然に豊富な安定水素および炭素の同位体の濃度は、本発明の化合物の安定同位体置換の程度と比較すると低く、重要ではない。例えば、Wada,E et al.,Seikagaku,1994,66:15、Gannes,LZ et al.,Comp Biochem Physiol Mol Integr Physiol,1998,119:725を参照されたい。本発明の化合物では、特定の位置が、重水素を有する位置として指定されている場合には、その位置の重水素の存在度が、重水素の天然の存在度(0.015%)よりも実質的に大きいことが理解される。重水素を有する位置として指定されている位置では典型的に、前記化合物において重水素として指定されている各原子における最小同位体濃縮係数は少なくとも3000(重水素導入度45%)である。
【0009】
「同位体濃縮係数」という用語は、本明細書で使用する場合、特定の同位体の同位体存在度と天然存在度との間の比率を意味する。
【0010】
別の実施形態では、本発明の化合物では、指定されている各重水素原子の同位体濃縮係数は、少なくとも3500(指定されている各重水素原子の重水素導入度52.5%)、少なくとも4000(重水素導入度60%)、少なくとも4500(重水素導入度67.5%)、少なくとも5000(重水素75%)、少なくとも5500(重水素導入度82.5%)、少なくとも6000(重水素導入度90%)、少なくとも6333.3(重水素導入度95%)、少なくとも6466.7(重水素導入度97%)、少なくとも6600(重水素導入度99%)、または少なくとも6633.3(重水素導入度99.5%)である。
【0011】
本発明の化合物では、特定の同位体として具体的に指定されていないいずれの原子も、その原子のいずれかの安定同位体を表すことを意味する。別段の記載のない限り、ある位置が、「H」または「水素」として具体的に指定されている場合には、その位置は、その天然存在度の同位体組成で水素を有するものと理解される。また、別段の記載のない限り、ある位置が、「D」または「重水素」として具体的に指定されている場合には、その位置は、0.015%という重水素の天然存在度(すなわち、少なくとも50.1%の重水素導入度)よりも少なくとも3340倍大きい存在度で重水素を有するものと理解される。
【0012】
「アイソトポローグ」という用語は、本発明の特定の化合物と同位体組成のみが異なる種を指す。
【0013】
「化合物」という用語は、本明細書で使用する場合、同一の化学構造を有する分子の一群を指す。ただし、それらの分子の構成原子間に同位体変化があってもよい。したがって、指定の重水素原子を含む特定の化学構造によって表される化合物が、その構造内の指定の重水素位置の1つ以上に水素原子を有するアイソトポローグを、より少ない量で含むことは当業者には明らかであろう。本発明の化合物におけるこのようなアイソトポローグの相対量は、本発明の化合物を作製する目的で用いる重水素化試薬の同位体純度、および、本発明の化合物を調製する目的で用いる各種の合成工程における重水素の導入効率といった多くの要因に左右されることになる。しかし、上に示したように、上記のアイソトポローグの相対量は、本発明の化合物の49.9%未満となる。
【0014】
本発明には、本明細書に開示されている化合物の塩も含まれる。
【0015】
本発明の化合物の塩は、酸と、本発明の化合物の塩基性基、例えばアミノ官能基との間、または、塩基と、本発明の化合物の酸性基、例えばカルボキシル官能基との間で形成される。別の実施形態によれば、本発明の化合物は、薬学的に許容可能な酸付加塩である。
【0016】
「薬学的に許容可能な」という用語は、本明細書で使用する場合、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などなしに、ヒトおよびその他の哺乳類の組織と接触させて用いるのに適していると共に、合理的なベネフィット/リスク比に見合う成分を指す。「薬学的に許容可能な塩」は、受容者への投与において、本発明の化合物を直接的または間接的に与えることができるいずれかの無毒性の塩を意味する。「薬学的に許容可能な対イオン」は、受容者への投与において、塩から放出された際に毒性でない塩のイオン部分である。
【0017】
薬学的に許容可能な塩を形成するのによく用いられる酸としては、二硫化水素、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸のような無機酸、ならびに、p−トルエンスルホン酸、サリチル酸、酒石酸、重酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、p−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸のような有機酸、さらには、関連する無機酸および有機酸が挙げられる。すなわち、このような薬学的に許容可能な塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオル酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−二酸塩、ヘキシン−1,6−二酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スフホン酸塩、マンデル酸塩、およびその他の塩が挙げられる。1つの実施形態では、薬学的に許容可能な酸付加塩としては、塩酸および臭化水素酸のような鉱酸によって形成されるもの、特に、マレイン酸のような有機酸によって形成されるものが挙げられる。
【0018】
本発明の化合物(例えば式Iの化合物)は、例えば重水素置換などの結果として、不斉炭素原子を含んでもよい。それゆえ、本発明の化合物は、個別の鏡像異性体、または、2つの鏡像異性体の混合物のいずれかとして存在することができる。したがって、本発明の化合物は、ラセミ混合物もしくはスケールミック混合物のいずれかとして、または、別の考え得る立体異性体を実質的に含まない個別の立体異性体として存在してもよい。「他の立体異性体を実質的に含まない」という用語は、本明細書で使用する場合、他の立体異性体が25%未満存在すること、好ましくは、他の立体異性体が10%未満存在すること、より好ましくは、他の立体異性体が5%未満存在すること、最も好ましくは、他の立体異性体が2%未満存在すること、または他の立体異性体が「X」%未満存在すること(Xは0以上、100以下の数字である)を意味する。所与の化合物に対する個別の鏡像異性体を得たりまたは合成したりする方法は、当該技術分野において既知であり、最終化合物または出発材料もしくは中間体に実際に使用可能な方法として適用できる。
【0019】
別段の指定がない限り、開示されている化合物が、その立体化学の特定なしに、構造によって命名または表示されていると共に、1つ以上のキラル中心を有する場合には、その化合物のすべての考え得る立体異性体を表すものと理解する。
【0020】
「安定化合物」という用語は、本明細書で使用する場合、その化合物を製造可能にするほど十分な安定性を有すると共に、本願明細書に詳述されている目的(例えば、治療用製品への製剤化、治療用化合物の作製で用いられる中間体、単離可能または保存可能な中間化合物、治療剤に応答する疾患または状態の治療)において有用であるほど、十分な期間にわたって、その化合物の完全性を保持する化合物を指す。
【0021】
「D」は重水素を指す。「立体異性体」は、鏡像異性体とジアステレオマーの両方を指す。「tert」、「t」、および「t−」はそれぞれ三級を指す。「US」は米国を指す。
【0022】
本明細書全体を通じて、可変基については、一般的に示されている場合もあれば(例えば「各R」)、具体的に示されている場合もある(例えば、R1、R2、R3など)。別段の指定がない限り、可変基について一般的に示されている場合には、その特定の可変基のすべての具体的な実施形態を含むことを意味する。
治療用化合物
【0023】
本発明は、式I
【化1】
(式中、R1はCH3、CDH2、CD2H、またはCD3であり、
R2は、0〜9個の重水素原子を有するt−ブチル基であり、
各Yは独立して、水素または重水素であり、
R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
の化合物、またはその薬学的に許容可能な塩を提供する。
【0024】
本発明の1つの実施形態は、式Iの化合物であって、式中のR1がCH3またはCD3である化合物を提供する。この実施形態の1つの態様では、Y1aとY1bは水素である。別の態様では、Y1aとY1bは重水素である。別の態様では、Y2は水素である。別の態様では、Y2は重水素である。別の態様では、R2は−C(CH3)3または−C(CD3)3である。この態様の一例としては、R2は−C(CD3)3である。
【0025】
別の実施形態は、式中のR2が−C(CH3)3または−C(CD3)3である化合物を提供する。一例としては、R2は−C(CD3)3である。この実施形態の1つの態様では、Y1aとY1bは水素である。別の態様では、Y1aとY1bは重水素である。別の態様では、Y2は水素である。別の態様では、Y2は重水素である。
【0026】
別の実施形態は、式中のY1aとY1bが同じである化合物を提供する。この実施形態の1つの態様では、Y1aとY1bは重水素である。この実施形態の1つの態様では、R2は−C(CD3)3である。
【0027】
さらに別の実施形態では、本発明の化合物は、下記の表1に示されている化合物のいずれか1つから選択する。
【表1】
【0028】
特定の実施形態では、本発明の化合物は、化合物102、103、104、105、109、110、111、および112、またはこれらの薬学的に許容可能な塩のいずれか1つから選択する。他の実施形態では、本発明の化合物は、化合物102、103、および105、またはこれらの薬学的に許容可能な塩のいずれか1つから選択する。1つの態様では、本発明の化合物は、化合物103もしくは化合物105、またはこれらの薬学的に許容可能な塩から選択する。
【0029】
別の実施形態群では、上記の実施形態のいずれかで重水素として指定されていないいずれかの原子も、その天然同位体存在度で存在する。
【0030】
通常の技能を持つ合成化学者は、本明細書に開示されている代表的合成法と実施例に従って、式Iの化合物の合成を容易に行うことができる。他の関連する手順および中間体は、例えば、国際公開第WO98/04559号および同第WO00/44752号に開示されている。
【0031】
このような方法は、本明細書に示されている化合物を合成するために、対応する重水素化試薬および/または中間体、ならびに任意で、他の同位体を含有する試薬および/または中間体を用いるか、あるいは、同位体原子を化学構造に導入するためのものとして当該技術分野において知られている標準的な合成プロトコールを用いて行うことができる。特定の中間体は、精製(例えば、ろ過、蒸留、昇華、結晶化、粉砕、固相抽出、およびクロマトグラフィー)を行って、または精製を行わずに用いることができる。
代表的合成法
【0032】
式Iの化合物は、下記のスキームに従って調製してよい。
スキーム1.式Iの化合物の一般的なルート
【化2】
【0033】
式Iの化合物の合成は、一般的にスキーム1に示されているように行うことができる。中間体12は、3,6−ジクロロピリダジン11を、適切に重水素化したピバル酸10によってラジカルアルキル化することによって調製する。D9−ピバル酸は、これらの化合物の調整用として市販されており、R2は−C(CD3)3である。続いて、適切に重水素化した3,6−ジクロロ−4−t−ブチルピリダジン12を2,5−ジフルオロベンゾヒドラジド13と縮合して、14をもたらす。適切に重水素化した(2−メチル−2H−1,2,4−トリアゾール−3−イル)メタノール15とNaHから発生するアニオンによって塩化物を置換して、式Iの化合物をもたらす。あるいは、THF中のn−BuLiもしくはDMSO中の炭酸セシウムを用いることによって、または、当業者に既知の他の同様の条件下で、14を式Iの化合物に変換する。
スキーム2.化合物15の合成
【化3】
【0034】
スキーム2は、15の重水素化類似体の調製法を示している。Dallacker F et al,Chemiker−Zeitung 1986,110:101−108、およびDallacker F et al,Chemiker−Zeitung 1986,110,p.275−281に記載されているように、1,2,4−トリアゾール(16)をR1−Iと反応させて、適切に重水素化したメチルトリアゾール17をもたらしてから、ホルムアルデヒドまたは重水素化ホルムアルデヒドで処理して15をもたらす。当業者であれば、重水素交換が潜在的にこれらの条件下で生じて、Y2が重水素である化合物をもたらし得ることが分かるであろう。
【0035】
上記の具体的なアプローチおよび化合物は、限定することを意図するものではない。本明細書のスキームの化学構造は、同じ可変基名(すなわち、R1、R2、R3など)によって同定されるか否かに関わらず、本明細書の式の化合物の対応する位置の化学基の定義(部分、原子など)に相応して定義される可変基を示している。別の化合物を合成するのに用いる化学構造内の化学基の適合性は、当業者の知識の範囲内である。
【0036】
式Iの化合物とその合成前駆体(本明細書のスキームに明示的に示されていないルート内の前駆体を含む)を合成する追加的な方法は、当該技術分野の通常の技能を持つ化学者の手段の範囲内である。適用可能な化合物を合成するのに有用な合成化学変換および保護基の方法論(保護および脱保護)は、当該技術分野において既知であり、それらの方法論としては例えば、Larock R,Comprehensive Organic Transformations,VCH Publishers(1989)、Greene TW et al.,Protective Groups in Organic Synthesis,3rd Ed.,John Wiley and Sons(1999)、Fieser L et al.,Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994)、Paquette L,ed.,Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、およびこれらのその後の版に記載されているものが挙げられる。
【0037】
本発明によって想定される置換基と可変基との組み合わせは、安定化合物を形成させる組み合わせのみである。
組成物
【0038】
本発明は、発熱物質を含まない組成物であって、式I(例えば、本明細書内の式のいずれかを含む)の化合物、または前記化合物の薬学的に許容可能な塩の有効量と、許容可能な担体とを含む組成物も提供する。本発明の組成物は、医薬用途用に製剤化するのが好ましく(「医薬組成物」)、この場合の担体は、薬学的に許容可能な担体である。この担体(単数または複数)は、その製剤の他の成分と混合可能であるという意味で「許容可能」であり、薬学的に許容可能な担体の場合には、医薬中に用いられる量では、その受容者に対して有毒ではない。
【0039】
薬学的に許容可能な担体としては、本発明の医薬組成物中で用いてよいアジュバントとビヒクルが挙げられる。薬学的に許容可能な担体としては、1種以上の塩、電解質、可溶化剤、溶媒、緩衝剤、乳化剤、矯味矯臭剤、着色剤、甘味剤、充填剤、潤沢剤、賦形剤、懸濁化剤、増粘剤、分散剤、湿潤剤、バイオアベイラビリティ促進剤、および吸収促進剤が挙げられる。具体的な薬学的に許容可能な担体としては、1,3−ブタンジオール、2−オクチルドデカノール、アカシア、アルミナ、ステアリン酸アルミニウム、蜜ろう、ベンジルアルコール、リン酸塩、セルロース系物質、セテアリルアルコール、セチルエステルワックス、ココアバター、コロイドシリカ、コーンスターチ、リン酸水素二ナトリウム、乳化ろう、エチレンオキシド−プロピレンオキシドブロックコポリマー、ゼラチン、グリセリン、グリシン、ヒト血清アルブミン、イオン交換剤、等張食塩水、ラクトース、レシチン、液化石油、長鎖アルコール、LUTROL(登録商標)、ステアリン酸マグネシウム、三ケイ酸マグネシウム、マンニトール、鉱油、オレイン酸およびそのグリセリド誘導体、オリーブ油または特にポリオキシエチル化された型のヒマシ油、飽和植物脂肪酸の部分グリセリド混合物、PLURONIC(登録商標)、ポリアクリレート、ポリエチレングリコール、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリソルベート60、ポリビニルピロリドン、リン酸水素カリウム、ソルビン酸カリウム、プロピレングリコール、硫酸プロタミン、リンゲル液、血清タンパク、カルボキシメチルセルロースナトリウム、塩化ナトリウム、ソルビン酸、モノステアリン酸ソルビタン、スクロース、トラガカント、Tween80、水、ろう、白色ワセリン、羊毛脂、および亜鉛塩が挙げられるが、これらに限らない。
【0040】
本発明の医薬組成物としては、経口投与、直腸投与、経鼻投与、局所投与(口腔投与および舌下投与を含む)、膣内投与、非経口投与(皮下投与、筋内投与、静脈内投与、および皮内投与を含む)、ならびに、経皮投与に適したものが挙げられる。各タイプの組成物と共に用いるのに適切な薬学的に許容可能な担体の選択は、当該技術分野において周知である。同様に、有効成分(単数または複数)と担体とを併せて、本発明の各種医薬組成物の単位投与形態を作製するための方法も、当該技術分野において周知である。例えば、Remington:The Science and Practice of Pharmacy,Lippincott Williams&Wilkins,Baltimore,MD(20th ed.2000)を参照されたい。
【0041】
別の実施形態では、本発明の組成物は、第2の治療剤をさらに含む。この第2の治療剤は、L−838417と同じ作用メカニズムを有する化合物と共に投与すると、有益な特性を有するかまたは示すことが知られているいずれかの化合物または治療剤から選択してよい。
【0042】
好ましくは、第2の治療剤は、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態の治療または予防に有用な薬剤である。
【0043】
別の実施形態では、本発明は、別々の投与形態の本発明の化合物と、上記の第2の治療剤のいずれかの1つ以上とを提供し、この際、本発明の化合物と第2の治療剤は相互に付随している。「相互に付随している」という用語は、本明細書で使用する場合、上記の別々の投与形態が一緒に販売および(一方の薬剤の24時間未満以内に連続してまたは同時に)投与されるように意図されていることが容易に分かるように、別々の投与形態が一緒に包装されているか、または別の形で相互に添付されていることを意味する。
【0044】
本発明の医薬組成物中には、本発明の化合物が有効量存在する。本明細書で使用する場合、「有効量」という用語は、適切な投与レジメンで投与した場合に、治療対象の障害の重症度、持続期間、もしくは進行を軽減もしくは改善するか、治療対象の障害の進展を予防するか、治療対象の障害を退行させるか、または、別の療法の予防効果もしくは治療効果(単数もしくは複数)を促進もしくは向上させるのに十分な量を指す。
【0045】
動物およびヒトに対する用量の相互関係(体表面1平方メートル当たりのミリグラムベース)については、Freireich et al.,(1996)Cancer Chemother.Rep 50:219に記載されている。体表面積は、患者の身長および体重から適切に割り出すことができる。例えば、Scientific Tables, Geigy Pharmaceuticals,Ardsley,N.Y.,1970,537を参照されたい。
【0046】
1つの実施形態では、本発明の化合物の有効量は、1回の治療当たり約0.01〜約5000mgの範囲であることができる。さらに具体的な実施形態では、この範囲は約0.1〜2500mg、または0.2〜1000mg、または最も具体的には約1〜500mgである。治療薬は典型的には1日に1〜3回投与する。
【0047】
当業者であれば分かるように、有効用量も、治療する疾患、疾患の重症度、投与経路、患者の性別、年齢、および全身の健康状態、賦形剤の使用、他の治療処置との併用(他の薬剤の使用など)の可能性、ならびに、治療を行う医師の判断に応じて変化することになる。例えば、有効用量を選択するための指針は、L−838417の処方に関する情報を参照することによって決定することができる。
【0048】
第2の治療剤を含む医薬組成物では、その第2の治療剤の有効量は、その治療剤のみを用いる単剤療法レジームで通常用いられる用量の約20%〜100%である。好ましくは、有効量は、単剤療法での通常の用量の約70%〜100%である。これらの第2の治療剤の単剤療法での通常の用量は、当該技術分野において周知である。例えば、Wells et al.,eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000)、PDR Pharmacopia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)を参照されたい。これらの各参考文献は、参照によりその全体が本明細書に組み込まれる。
【0049】
上に参照として示されている第2の治療剤のいくつかは、本発明の化合物と相乗的に作用すると予想される。相乗的に作用すると、第2の治療剤および/または本発明の化合物の有効用量を単剤療法で必要な有効用量よりも減らすことができるようになる。これには、本発明の化合物のいずれかの第2の治療剤の有毒な副作用を最小限に抑えるという利点、効能を相乗的に向上させる利点、投与もしくは使用のしやすさを向上させる利点、および/または、化合物を調製もしくは製剤化する際の全体的コストが低下するという利点がある。
治療法
【0050】
別の実施形態では、本発明は、細胞内のGABA−A受容体のサブタイプα1を阻害する方法であって、本明細書の式Iの化合物1種以上と細胞を接触させることを含む方法を提供する。別の実施形態では、本発明は、細胞内のGABA−A受容体のサブタイプα2、α3、およびα5のうちの1つ以上を活性化させる方法を提供する。
【0051】
別の実施形態によれば、本発明は、L−838417によって有益な形で治療される疾患を罹患しているか、または罹患しやすい患者を治療する方法であって、式Iの化合物もしくはその塩、または本発明の組成物を有効量、前記患者に投与する工程を含む方法を提供する。このような疾患は、当該技術分野において周知であり、国際公開第WO1998004559号、同第WO2000044752号、同第WO2006061428号に開示されているが、これらの特許および特許出願に限らない。上記のような疾患としては、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛が挙げられるが、これらに限らない。
【0052】
本明細書に示されている方法は、特定的に示されている治療が必要な患者として認定する方法も含む。このような治療が必要な患者としての認定は、患者または医療の専門家の判断で行うことができると共に、主観的(例えば意見)であることも、客観的(例えば、試験または診断法によって測定可能)であることもできる。
【0053】
別の実施形態では、上記の治療法のいずれかが、1種以上の第2の治療剤を前記患者に同時投与するさらなる工程を含む。この第2の治療剤は、L−838417との同時投与用として有用であることが知られているいずれかの第2の治療剤から選択してよい。第2の治療剤の選択は、治療対象の特定の疾患または状態によっても左右される。本発明の方法で用いてよい第2の治療剤の例は、本発明の化合物と第2の治療剤を含む複合組成物で用いるものとして上に示したものである。
【0054】
「同時投与」という用語は、本明細書で使用する場合、単回投与形態(本発明の化合物と、上記のような第2の治療剤とを含む本発明の組成物など)、または別々の複数回投与形態の一部として、第2の治療剤を本発明の化合物と共に投与してよいことを意味する。あるいは、本発明の化合物の投与前、投与に連続的に、または投与の後に、追加の薬剤を投与してもよい。このような併用療法の治療では、本発明の化合物も、第2の治療剤(単数または複数)も、従来の方法によって投与する。本発明の化合物と第2の治療剤の両方を含む本発明の組成物を患者に投与することは、治療過程中の別の時期に、同じ治療剤、いずれかの他の第2の治療剤、または本発明のいずれかの化合物を前記患者に別々に投与することを妨げない。
【0055】
これらの第2の治療剤の有効量は当業者に周知であり、投与指針は、本明細書に参照として記載されている特許および公開特許出願、ならびに、Wells et al.,eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000)、PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)、およびその他の医学書で見ることができる。しかし、第2の医薬剤の最適な有効量範囲を割り出すことは、十分に当業者の視野の範囲内である。
【0056】
第2の治療剤を対象に投与する本発明の1つの実施形態では、本発明の化合物の有効量は、第2の治療剤を投与しない場合の有効量よりも少ない。別の実施形態では、第2の治療剤の有効量は、本発明の化合物を投与しない場合の有効量よりも少ない。このように、高用量のいずれかの薬剤に付随する望ましくない副作用を最小限に抑えることができる。他の潜在的な利点(投与レジメンの改善および/または薬剤コストの削減が挙げられるが、これらに限らない)は、当業者には明らかであろう。
【0057】
さらに別の態様では、本発明は、上記の疾患、障害、または症状の患者における治療または予防用の単一組成物または別々の投与形態のいずれかとして医薬を製造する際に、式Iの化合物を単独で、または、上記の第2の治療剤の1種以上と共に用いることを提供する。本発明の別の態様は、本明細書に示されている疾患、障害、または症状の患者における治療または予防で用いる式Iの化合物、または式Iの化合物を含む医薬組成物である。
医薬キット
【0058】
本発明は、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛の治療で用いるキットも提供する。これらのキットは、(a)式Iの化合物またはその塩を含む医薬組成物であって、入れ物に入っている医薬組成物と、(b)この医薬組成物を用いて、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛を治療する方法を説明している説明書とを含む。
【0059】
上記の入れ物は、前記医薬組成物を保持できるいずれかの容器、または密閉されているか、もしくは密閉可能なその他の器具であってよい。例としては、ボトル、アンプル、分画もしくは多チャンバー型ホルダー、ボトル(その各分画部もしくはチャンバーは、単回用量の前記組成物を含む)、分画型のホイル製小包(その各分画部は、単回用量の前記組成物を含む)、または、単回用量の前記組成物を分注する分注器が挙げられる。上記の入れ物は、当該技術分野において既知のようないずれかの従来の形または形態のものであって、薬学的に許容可能な材料で作られているもの、例えば、紙箱、段ボール箱、ガラスもしくはプラスチック製ボトルもしくは広口瓶、再密閉可能なバッグ(例えば、錠剤の「リフィル」を異なる入れ物に入れて保持するためのもの)、または、治療スケジュールに従ってパックから押し出すように、個別の用量が入ったブリスターパックであることができる。用いる入れ物は、関連する正確な投与形態に応じて決めることができ、例えば、従来の段ボール箱は一般的には、液体懸濁剤を保持するのには用いられない。単一のパッケージの中で2つ以上の入れ物を併せて用いて、単一の投与形態を市販することも可能である。例えば、錠剤をボトルに入れてから、そのボトルを箱に入れてもよい。1つの実施形態では、上記の入れ物はブリスターパックである。
【0060】
本発明のキットは、単位用量の医薬組成物を投与または測り分ける器具も含んでもよい。このような器具としては、吸入器(前記組成物が吸入可能な組成物である場合)、注射器および注射針(前記組成物が注射可能な組成物の場合)、注射器、スプーン、ポンプ、もしくは容器(目盛りの有無は問わない)(前記組成物が経口用液体組成物である場合)、または、本発明のキット内に含まれる組成物の投与剤形に適している他のいずれかの測定用もしくは送達用器具を挙げてよい。
【0061】
特定の実施形態では、本発明のキットは、本発明の化合物との同時投与用に用いる上記のもののうちの1つのような第2の治療剤を含む医薬組成物を、入れ物の別個の容器内に含んでもよい。
【実施例】
【0062】
実施例1.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−メチル−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物103)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物103を調製した。
【化4】
【0063】
工程1.4−(tert−ブチル−d9)−3,6−ジクロロピリダジン(12a) 新たに精製した3,6−ジクロロ−ピリダジン、11(5.4g、33.5mmol)を蒸留水(130mL)に懸濁させた懸濁液に、濃硫酸(5.7mL、108mmol)を加えた。この混合物を65℃まで加温し、トリメチル酢酸−d9、10a(6.0g、54mmol、CDNアイソトープス、99atom%D)を加えてから、硝酸銀(1.1g、7mmol)を加えた。反応温度を65〜75℃に保ちながら、10〜15分にわたって、アンモニウムペルオキシ二硫酸(12.3g、54mmol)を蒸留水(35mL)に溶解させた溶液を上記の混合物に加えた。この混合物を30分間攪拌し、室温まで冷却した。この混合物を氷(100g)の上に注いで、その混合物を濃水酸化アンモニウムによってpH=9〜10に調節した。この水性混合物をジクロロメタン(2×30mL)で抽出した。合わせた抽出物を1Nの水酸化ナトリウム(10mL)で洗浄し、Na2SO4で乾燥させ、ろ過し、減圧下で濃縮した。この粗生成物をシリカゲルカラムクロマトグラフィー(10%酢酸エチル/ヘプタンで溶出)によって精製し、6.1g(80%)の12aを無色の油として得た。
【0064】
工程2.7−(tert−ブチル−d9)−6−クロロ−3−(2,5−ジフルオロフェニル)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(14a) キシレン(30mL)中の12a(6g、28mmol)、13(7.2g、42mmol、市販品)、およびトリエチルアミン塩酸塩(5.8g、42mmol)の混合物を攪拌しながら150℃で36時間加熱した。室温まで冷却後、この混合物を減圧下で濃縮した。その残渣をジクロロメタン(40mL)で粉砕し、ろ過し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルクロマトグラフィー(20〜50%酢酸エチル/ヘプタンで溶出)によって精製し、5.6g(60%)の14aをオフホワイトの固体として得た。
【0065】
工程3.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−メチル−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物103) (1−メチル−1H−1,2,4−トリアゾール−5−イル)メタノール15a(0.45g、4.0mmol、市販品)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水酸化ナトリウム(0.17、4.3mmol)を加えた。この混合物を15分間攪拌し、14a(1.2g、3.6mmol)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過によって回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル−ヘプタン(1:1)からの再結晶によってさらに精製し、1.25g(78%)の化合物103を白色固体として得た。1H−NMR(300MHz、CDCl3):δ3.91(s,3H)、5.55(s、2H)、7.23−7.28(m、2H)、7.62−7.68(m、1H)、7.93(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):δ34.55、35.66、59.37、115.58(dd、J1=16.6、J2=9.2)、117.63(dd、J1=25.8、J2=6.6)、117.72(dd、J1=24.5、J2=12.2)、118.72(dd、J1=24.0、J2=8.5)、121.74、137.85、143.47、145.00、149.49,151.13、155.70(d、J=160.9)、159.01(d、J=155.9)、158.70。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.41分、純度99.3%。MS(M+H):409.2。元素分析(C19H10D9F2N7O):計算値:C=55.88、H=4.69、N=24.01、実測値:C=55.98、H=4.53、N=23.98。
【0066】
実施例2.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メタノール(15b)の合成 上記のスキーム2に一般的に概要が示されているように、適切に重水素化した中間体から中間体15bを調製した。
【化5】
【0067】
工程1.1−(メチル−d3)−1H−1,2,4−トリアゾール(17a) 窒素雰囲気下の、機械的スターラーを備えたフラスコ内で、無水THF(60mL)に1,2,4−トリアゾール16(6.0g、87mol)を加えてから、ヨードメタン−d3(6.5mL、1.05mol、ケンブリッジアイソトープス、99atom%D)を加えた。この濁った混合物を0℃まで冷却し、20分にわたって、1,8−ジアザビシクロ[5.4.0]ウンデス−7−エン「DBU」(13.2mL、0.87mol)を加えた。この混合物を室温までゆっくり加温し、オーバーナイトで攪拌した。続いて、この混合物をセライトパッドに通してろ過し、そのろ液を減圧下で濃縮して、7.3g(>100%)の粗生成物17aを黄色の油として得た。GCMSによれば、純度は90%である。位置異性体の比率は12:1であった。
【0068】
工程2.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メタノール(15b) 17a(5g、58mmol)とパラホルムアルデヒド−d2(10g、333mmol、ケンブリッジアイソトープス、99atom%D)との混合物を密閉チューブ内で170℃にて5時間加熱した。この混合物を室温まで冷却し、ジクロロメタン(20mL)で希釈した。その固体をろ過して除去し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルショートカラムクロマトグラフィー(75%THF/ヘプタンで溶出)によって精製し、4.8g(71%)の15bをオフホワイトの固体として得た。
【0069】
実施例3.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物105)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水化した中間体から化合物105を調製した。
【化6】
7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物105) 15b(0.24g、2.0mmol)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.08、2.1mmol)を加えた。この混合物を15分間攪拌し、14a(0.6g、1.8mmol、実施例1参照)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、0.52g(70%)の化合物105を白色固体として得た。1H−NMR(300MHz、CDCl3):δ7.23−7.28(m、2H)、7.63−7.67(m、1H)、7.92(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):35.66および59.37でピークが観測されず。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.40分、純度99.0%。MS(M+H):414.3。元素分析(C19H5D14F2N7O):計算値:C=55.20、H=4.63、N=23.72、F=9.19、実測値:C=54.88、H=4.45、N=23.46、F=9.59。
【0070】
実施例4.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−メタノール(15c)の合成 上記のスキーム2に一般的に概要が示されているように、適切に重水素化した中間体から中間体15cを調製した。
【化7】
(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−メタノール(15c) 17a(5g、58mmol、実施例2参照)とパラホルムアルデヒド(10g、333mmol)との混合物を密閉チューブ内で170℃にて5時間加熱した。この混合物を室温まで冷却し、ジクロロメタン(20mL)で希釈した。その固体をろ過して除去し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルショートカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製し、5.0g(75%)の15cをオフホワイトの固体として得た。
【0071】
実施例5.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物104)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物104を調製した。
【化8】
【0072】
7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物104) 15c(0.46g、4.0mmol)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.17、4.3mmol)を加えた。この混合物を15分間攪拌し、14a(1.2g、3.6mmol、実施例1参照)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、1.31g(88%)の化合物104を白色固体として得た。
1H−NMR(300MHz、CDCl3):δ5.55(s、2H)、7.23−7.28(m、2H)、7.62−7.67(m、1H)、7.93(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):δ34.55、59.36、115.53(dd、J1=16.6、J2=8.8)、117.63(dd、J1=24.4、J2=12.8)、117.71(dd、J1=24.1、J2=8.0)、118.77(dd、J1=23.9、J2=8.5)、121.75、137.85、143.48、145.00、149.50、151.15、155.76(d、J=163.5)、159.08(d、J=156.9)、158.71。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.40分、純度99.6%。MS(M+H):412.2。元素分析(C19H7D12F2N7O):計算値:C=55.47、H=4.67、N=23.83、実測値:C=55.49、H=4.76、N=23.87。
【0073】
実施例6.7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物101)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物101を調製した。
【化9】
7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物101) 15b(0.24g、4.0mmol、実施例2参照)をDMF(10mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.08、2.1mmol)を加えた。この混合物を15分間攪拌し、既知の化合物7−tert−ブチル−6−クロロ−3−(2,5−ジフルオロフェニル)−[1,2,4]トリアゾロ[4,3−b]ピリダジン、14b(0.58g、1.8mmol、国際公開第WO1998004559号に記載されているように調製)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(75%THF/ヘプタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、0.53g(72%)の化合物101を白色固体として得た。1H−NMR(300MHz、CDCl3):δ1.41(s、9H)、7.23−7.28(m、2H)、7.62−7.68(m、1H)、7.92(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):28.96でピークが観察され、35.66および59.36ではピークは観察されず。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.42分、純度99.7%。MS(M+H):405.3。元素分析(C19H14D5F2N7O):計算値:C=56.43、H=4.74、N=24.25、F=9.40、実測値:C=56.22、H=4.73、N=23.87、F=9.35。
【0074】
実施例7.代謝安定性の評価 ヒト肝ミクロソーム(20mg/mL)とラット肝ミクロソーム(20mg/mL)をゼノテック社(カンザス州レネクサ)から入手した。β−ニコチンアミドアデニンジヌクレオチドリン酸還元体(NADPH)、塩化マグネシウム(MgCl2)、およびジメチスルホキシド(DMSO)をシグマ−アルドリッチから購入した。
【0075】
代謝安定性の測定:試験化合物(L−838417、化合物101、化合物103、化合物104、および化合物105)の7.5mMストック溶液をDMSO中で調製した。これらの7.5mMストック溶液をアセトニトリル(ACN)中で12.5μMまで希釈した。3mMのMgCl2を含有するpH7.4の0.1Mリン酸カリウム緩衝液中で上記の20mg/mLの肝ミクロソーム(ヒトまたは肝臓のいずれか)を2.5mg/mLまで希釈した。この希釈ミクロソーム(375μL)を96ウェルのポリプロピレン製ディープウェルプレートのウェルに3連で加えた。このミクロソームに12.5μMの試験化合物10μLを加え、その混合物を10分間予熱した。予熱したNADPH溶液125μLを加えることによって、反応を開始させた。最終反応体積は0.5mLであり、pH7.4の0.1Mリン酸カリウム緩衝液および3mMのMgCl2中に、0.5mg/mLのヒト肝ミクロソーム、0.25μMの試験化合物、および2mMのNADPHを含有していた。この反応混合物を37℃でインキュベートし、50μLのアリコートを0分、5分、10分、20分、および30分の時点で除去して、内部標準を有する50μLの氷冷ACNの入った96ウェルのシャローウェルプレートに加えて、反応を停止させた。このプレートを4℃で20分間保存した後、そのプレートのウェルに100μLの水を加えてから遠心分離して、ペレット沈殿タンパクを得た。上清を別の96ウェルプレートに移し、アプライドバイオシステムのAPI 4000質量分析計を用いて、LC−MS/MSによって、残存の親化合物の量について分析した。7−エトキシクマリン(1μM)をポジティブコントロールとして用いた。
【0076】
データ解析:下記の数式を用いて、親化合物の残存率(%)(ln)とインキュベーション時間との関係の直線回帰の傾斜から、試験化合物のインビトロt1/2を算出した。
インビトロt1/2=0.693/k(式中、k=−[親化合物の残存率(%)(ln)とインキュベーション時間との関係の直線回帰の傾斜]
データ解析は、マイクロソフトのソフトウェアExcelを用いて行った。
【0077】
これらの実験の結果は、図1(ヒト肝ミクロソーム)および図2(ラット肝ミクロソーム)に示されている。図1に示されているように、ヒト肝ミクロソームと共に30分インキュベーションした後、約70%のL−838417が元の状態を保っていた。L−838417の半減期を算出したところ、54.4分であった。これに対し、化合物101、103、104、および105はそれぞれ、ヒト肝ミクロソーム中で安定していた(30分のインキュベーション後、80%超の親化合物が元の状態を保っていた)。
【0078】
ラット肝ミクロソームでは、L−838417も化合物101も、30分のインキュベーション後、約70%が元の状態を保っていた(図2)。半減期の計算値は、L−838417では50.5分、化合物101では52.7分であった。化合物103、104、および105はいずれも、30分のインキュベーション後、80%超が元の状態を保っており、いずれもラットミクロソームにおいて安定的であるとみなされた。
【0079】
実施例8.ラットに経口および静脈内投与した後の化合物103および105の薬物動態およびバイオアベイラビリティ解析 3匹の雄スプラーグドーリーラット(それぞれ200〜250g)の頸静脈にカニューレを挿入し、頸静脈のカニューレを通じて、L−838417、化合物103、および化合物105それぞれを1mg/kg含む単回用量分を(10%ジメチルスルホキシド(DMSO)、10%N,N−ジメチルアセトアミド(DMA)、および60%ポリエチレングリコール(PG)中に、これらの3つの化合物を1mg/mL含む1:1:1混合物として)投与した。3匹の追加の雄スプラーグドーリーラット(それぞれ200〜250g)に対して、L−838417、化合物103、および化合物105をそれぞれ1mg/kg含む単回用量分を(10%ジメチルスルホキシド(DMSO)、10%N,N−ジメチルアセトアミド(DMA)、および60%ポリエチレングリコール(PG)中に、これらの3つの化合物をそれぞれ1mg/mL含む1:1:1混合物として)強制経口投与した。
【0080】
静脈処置した試験ラットの血液(0.25mL)を、投与から2分、5分、15分、30分、1時間、2時間、4時間、および6時間の時点に眼窩後穿刺で採取した。経口処置した試験ラットの血液(0.25mL)を、投与から5分、15分、30分、45分、1時間、2時間、4時間、および6時間の時点に眼窩後穿刺で採取した。血液は、上記の時点に、抗血液凝固剤としてK2EDTAを含むチューブの中に集めた。血液サンプルを氷上で保存してから、遠心分離して血漿を得た。この血漿(約0.125μL)を96ウェルのディーププレートに分注し、アプライドバイオシステムのAPI 4000質量分析計を用いたLC−MS/MSによる解析まで、−80℃で保存した。
【0081】
この研究の静脈投与の部分の結果は、図3および下記の表2に示されている。
【表2】
【0082】
これらの結果によって、ラットに静脈内投与した後は、非重水素化L−838417よりも、化合物103および105はそれぞれ半減期が約80%長くなり、またAUC0−6が2倍超となることが示されている。加えて、化合物103および105の両方が、L−838417よりもそれぞれ約60%以上ゆっくりと消失する。
【0083】
この研究の経口投与部分の結果は、図4および下記の表3に示されている。
【表3】
【0084】
これらの結果によって、ラットに経口投与した後は、L−838417よりも、化合物103および105のCmaxが2倍超高いことが示されている。加えて、上記の結果によって、経口投与後は、非重水素化L−838417よりも、化合物103および105のAUC0−6が2.5倍超となることが示されている。
【0085】
更なる説明がなくても、当業者であれば、上記の説明および実例となる実施例を用いて、本発明の化合物を作製および利用できると共に、請求されている方法を実施できると考えられる。上記の論議および実施例は、特定の好ましい実施形態の詳細な説明を示しているに過ぎないと理解されたい。本発明の趣旨および範囲から逸脱することなく、各種の変形形態および均等形態を作製できることは当業者には明らかであろう。上で論じられているか、または引用されているすべての特許、学術論文、およびその他の文書は、参照により本明細書に組み込まれる。
【関連出願の相互参照】
【0001】
本出願は、2008年8月29日に提出された米国特許仮出願第61/093,293号の優先権を主張するものであり、その内容は、参照により本明細書に組み込まれる。
【技術分野】
【0002】
本発明は、新規な置換トリアゾロ−ピリダジン、その誘導体、およびそれらの薬学的に許容可能な塩に関する。本発明は、本発明の化合物を含む組成物、ならびに、α1−GABA−A受容体アンタゴニストの投与によって有益な形で治療される疾患および状態を治療する方法において、前記組成物を使用することも提供する。
【背景技術】
【0003】
L−838417(7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−(1−メチル−1H−1,2,4−トリアゾール−5−イルメトキシ)[1,2,4]トリアゾロ[4,3−b]ピリダジンとしても知られている)は、GABA−A受容体のベンゾジアゼピン部分で、サブタイプα1のアンタゴニストとして、ならびに、サブタイプα2、α3、およびα5の機能選択的アロステリックアゴニストとしての役割を果たす。
【0004】
L−838417は現在のところ、中枢神経系障害に対する前臨床段階の候補である。
L−838417の有益な活性にもかかわらず、α1−GABA−A受容体アンタゴニストである新たな化合物に対するニーズが依然として存在する。
【図面の簡単な説明】
【0005】
【図1】ヒト肝ミクロソームにおける本発明の化合物の経時安定性を示している。
【図2】ラット肝ミクロソームにおける本発明の化合物の経時安定性を示している。
【図3】ラットに静脈内投与した後の本発明の化合物の血漿中濃度の変化を示している。
【図4】ラットに経口投与した後の本発明の化合物の血漿中濃度の変化を示している。
【発明を実施するための形態】
【0006】
定義
「治療する」という用語は、疾患(例えば、本明細書に示されている疾患または障害)の発生または進行を減少、抑制、減衰、縮小、停止、または安定させることを意味する。
【0007】
「疾患」とは、細胞、組織、または器官の正常機能を損傷させるか、またはそれに干渉するいずれかの状態または障害を意味する。
【0008】
合成化合物においては、その合成で用いた化学物質の起源に応じて、天然同位体存在度の多少の変動が生じることが認められる。したがって、L−838417の調製物は本質的に、少量の重水素化アイソトポローグを含有することになる。上記の変動にもかかわらず、天然に豊富な安定水素および炭素の同位体の濃度は、本発明の化合物の安定同位体置換の程度と比較すると低く、重要ではない。例えば、Wada,E et al.,Seikagaku,1994,66:15、Gannes,LZ et al.,Comp Biochem Physiol Mol Integr Physiol,1998,119:725を参照されたい。本発明の化合物では、特定の位置が、重水素を有する位置として指定されている場合には、その位置の重水素の存在度が、重水素の天然の存在度(0.015%)よりも実質的に大きいことが理解される。重水素を有する位置として指定されている位置では典型的に、前記化合物において重水素として指定されている各原子における最小同位体濃縮係数は少なくとも3000(重水素導入度45%)である。
【0009】
「同位体濃縮係数」という用語は、本明細書で使用する場合、特定の同位体の同位体存在度と天然存在度との間の比率を意味する。
【0010】
別の実施形態では、本発明の化合物では、指定されている各重水素原子の同位体濃縮係数は、少なくとも3500(指定されている各重水素原子の重水素導入度52.5%)、少なくとも4000(重水素導入度60%)、少なくとも4500(重水素導入度67.5%)、少なくとも5000(重水素75%)、少なくとも5500(重水素導入度82.5%)、少なくとも6000(重水素導入度90%)、少なくとも6333.3(重水素導入度95%)、少なくとも6466.7(重水素導入度97%)、少なくとも6600(重水素導入度99%)、または少なくとも6633.3(重水素導入度99.5%)である。
【0011】
本発明の化合物では、特定の同位体として具体的に指定されていないいずれの原子も、その原子のいずれかの安定同位体を表すことを意味する。別段の記載のない限り、ある位置が、「H」または「水素」として具体的に指定されている場合には、その位置は、その天然存在度の同位体組成で水素を有するものと理解される。また、別段の記載のない限り、ある位置が、「D」または「重水素」として具体的に指定されている場合には、その位置は、0.015%という重水素の天然存在度(すなわち、少なくとも50.1%の重水素導入度)よりも少なくとも3340倍大きい存在度で重水素を有するものと理解される。
【0012】
「アイソトポローグ」という用語は、本発明の特定の化合物と同位体組成のみが異なる種を指す。
【0013】
「化合物」という用語は、本明細書で使用する場合、同一の化学構造を有する分子の一群を指す。ただし、それらの分子の構成原子間に同位体変化があってもよい。したがって、指定の重水素原子を含む特定の化学構造によって表される化合物が、その構造内の指定の重水素位置の1つ以上に水素原子を有するアイソトポローグを、より少ない量で含むことは当業者には明らかであろう。本発明の化合物におけるこのようなアイソトポローグの相対量は、本発明の化合物を作製する目的で用いる重水素化試薬の同位体純度、および、本発明の化合物を調製する目的で用いる各種の合成工程における重水素の導入効率といった多くの要因に左右されることになる。しかし、上に示したように、上記のアイソトポローグの相対量は、本発明の化合物の49.9%未満となる。
【0014】
本発明には、本明細書に開示されている化合物の塩も含まれる。
【0015】
本発明の化合物の塩は、酸と、本発明の化合物の塩基性基、例えばアミノ官能基との間、または、塩基と、本発明の化合物の酸性基、例えばカルボキシル官能基との間で形成される。別の実施形態によれば、本発明の化合物は、薬学的に許容可能な酸付加塩である。
【0016】
「薬学的に許容可能な」という用語は、本明細書で使用する場合、適切な医学的判断の範囲内で、過度の毒性、刺激、アレルギー反応などなしに、ヒトおよびその他の哺乳類の組織と接触させて用いるのに適していると共に、合理的なベネフィット/リスク比に見合う成分を指す。「薬学的に許容可能な塩」は、受容者への投与において、本発明の化合物を直接的または間接的に与えることができるいずれかの無毒性の塩を意味する。「薬学的に許容可能な対イオン」は、受容者への投与において、塩から放出された際に毒性でない塩のイオン部分である。
【0017】
薬学的に許容可能な塩を形成するのによく用いられる酸としては、二硫化水素、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、およびリン酸のような無機酸、ならびに、p−トルエンスルホン酸、サリチル酸、酒石酸、重酒石酸、アスコルビン酸、マレイン酸、ベシル酸、フマル酸、グルコン酸、グルクロン酸、ギ酸、グルタミン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、乳酸、シュウ酸、p−ブロモフェニルスルホン酸、炭酸、コハク酸、クエン酸、安息香酸、および酢酸のような有機酸、さらには、関連する無機酸および有機酸が挙げられる。すなわち、このような薬学的に許容可能な塩としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプリン酸塩、ヘプタン酸塩、プロピオル酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−二酸塩、ヘキシン−1,6−二酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、ヒドロキシ安息香酸塩、メトキシ安息香酸塩、フタル酸塩、テレフタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、β−ヒドロキシ酪酸塩、グリコール酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン−1−スルホン酸塩、ナフタレン−2−スフホン酸塩、マンデル酸塩、およびその他の塩が挙げられる。1つの実施形態では、薬学的に許容可能な酸付加塩としては、塩酸および臭化水素酸のような鉱酸によって形成されるもの、特に、マレイン酸のような有機酸によって形成されるものが挙げられる。
【0018】
本発明の化合物(例えば式Iの化合物)は、例えば重水素置換などの結果として、不斉炭素原子を含んでもよい。それゆえ、本発明の化合物は、個別の鏡像異性体、または、2つの鏡像異性体の混合物のいずれかとして存在することができる。したがって、本発明の化合物は、ラセミ混合物もしくはスケールミック混合物のいずれかとして、または、別の考え得る立体異性体を実質的に含まない個別の立体異性体として存在してもよい。「他の立体異性体を実質的に含まない」という用語は、本明細書で使用する場合、他の立体異性体が25%未満存在すること、好ましくは、他の立体異性体が10%未満存在すること、より好ましくは、他の立体異性体が5%未満存在すること、最も好ましくは、他の立体異性体が2%未満存在すること、または他の立体異性体が「X」%未満存在すること(Xは0以上、100以下の数字である)を意味する。所与の化合物に対する個別の鏡像異性体を得たりまたは合成したりする方法は、当該技術分野において既知であり、最終化合物または出発材料もしくは中間体に実際に使用可能な方法として適用できる。
【0019】
別段の指定がない限り、開示されている化合物が、その立体化学の特定なしに、構造によって命名または表示されていると共に、1つ以上のキラル中心を有する場合には、その化合物のすべての考え得る立体異性体を表すものと理解する。
【0020】
「安定化合物」という用語は、本明細書で使用する場合、その化合物を製造可能にするほど十分な安定性を有すると共に、本願明細書に詳述されている目的(例えば、治療用製品への製剤化、治療用化合物の作製で用いられる中間体、単離可能または保存可能な中間化合物、治療剤に応答する疾患または状態の治療)において有用であるほど、十分な期間にわたって、その化合物の完全性を保持する化合物を指す。
【0021】
「D」は重水素を指す。「立体異性体」は、鏡像異性体とジアステレオマーの両方を指す。「tert」、「t」、および「t−」はそれぞれ三級を指す。「US」は米国を指す。
【0022】
本明細書全体を通じて、可変基については、一般的に示されている場合もあれば(例えば「各R」)、具体的に示されている場合もある(例えば、R1、R2、R3など)。別段の指定がない限り、可変基について一般的に示されている場合には、その特定の可変基のすべての具体的な実施形態を含むことを意味する。
治療用化合物
【0023】
本発明は、式I
【化1】
(式中、R1はCH3、CDH2、CD2H、またはCD3であり、
R2は、0〜9個の重水素原子を有するt−ブチル基であり、
各Yは独立して、水素または重水素であり、
R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
の化合物、またはその薬学的に許容可能な塩を提供する。
【0024】
本発明の1つの実施形態は、式Iの化合物であって、式中のR1がCH3またはCD3である化合物を提供する。この実施形態の1つの態様では、Y1aとY1bは水素である。別の態様では、Y1aとY1bは重水素である。別の態様では、Y2は水素である。別の態様では、Y2は重水素である。別の態様では、R2は−C(CH3)3または−C(CD3)3である。この態様の一例としては、R2は−C(CD3)3である。
【0025】
別の実施形態は、式中のR2が−C(CH3)3または−C(CD3)3である化合物を提供する。一例としては、R2は−C(CD3)3である。この実施形態の1つの態様では、Y1aとY1bは水素である。別の態様では、Y1aとY1bは重水素である。別の態様では、Y2は水素である。別の態様では、Y2は重水素である。
【0026】
別の実施形態は、式中のY1aとY1bが同じである化合物を提供する。この実施形態の1つの態様では、Y1aとY1bは重水素である。この実施形態の1つの態様では、R2は−C(CD3)3である。
【0027】
さらに別の実施形態では、本発明の化合物は、下記の表1に示されている化合物のいずれか1つから選択する。
【表1】
【0028】
特定の実施形態では、本発明の化合物は、化合物102、103、104、105、109、110、111、および112、またはこれらの薬学的に許容可能な塩のいずれか1つから選択する。他の実施形態では、本発明の化合物は、化合物102、103、および105、またはこれらの薬学的に許容可能な塩のいずれか1つから選択する。1つの態様では、本発明の化合物は、化合物103もしくは化合物105、またはこれらの薬学的に許容可能な塩から選択する。
【0029】
別の実施形態群では、上記の実施形態のいずれかで重水素として指定されていないいずれかの原子も、その天然同位体存在度で存在する。
【0030】
通常の技能を持つ合成化学者は、本明細書に開示されている代表的合成法と実施例に従って、式Iの化合物の合成を容易に行うことができる。他の関連する手順および中間体は、例えば、国際公開第WO98/04559号および同第WO00/44752号に開示されている。
【0031】
このような方法は、本明細書に示されている化合物を合成するために、対応する重水素化試薬および/または中間体、ならびに任意で、他の同位体を含有する試薬および/または中間体を用いるか、あるいは、同位体原子を化学構造に導入するためのものとして当該技術分野において知られている標準的な合成プロトコールを用いて行うことができる。特定の中間体は、精製(例えば、ろ過、蒸留、昇華、結晶化、粉砕、固相抽出、およびクロマトグラフィー)を行って、または精製を行わずに用いることができる。
代表的合成法
【0032】
式Iの化合物は、下記のスキームに従って調製してよい。
スキーム1.式Iの化合物の一般的なルート
【化2】
【0033】
式Iの化合物の合成は、一般的にスキーム1に示されているように行うことができる。中間体12は、3,6−ジクロロピリダジン11を、適切に重水素化したピバル酸10によってラジカルアルキル化することによって調製する。D9−ピバル酸は、これらの化合物の調整用として市販されており、R2は−C(CD3)3である。続いて、適切に重水素化した3,6−ジクロロ−4−t−ブチルピリダジン12を2,5−ジフルオロベンゾヒドラジド13と縮合して、14をもたらす。適切に重水素化した(2−メチル−2H−1,2,4−トリアゾール−3−イル)メタノール15とNaHから発生するアニオンによって塩化物を置換して、式Iの化合物をもたらす。あるいは、THF中のn−BuLiもしくはDMSO中の炭酸セシウムを用いることによって、または、当業者に既知の他の同様の条件下で、14を式Iの化合物に変換する。
スキーム2.化合物15の合成
【化3】
【0034】
スキーム2は、15の重水素化類似体の調製法を示している。Dallacker F et al,Chemiker−Zeitung 1986,110:101−108、およびDallacker F et al,Chemiker−Zeitung 1986,110,p.275−281に記載されているように、1,2,4−トリアゾール(16)をR1−Iと反応させて、適切に重水素化したメチルトリアゾール17をもたらしてから、ホルムアルデヒドまたは重水素化ホルムアルデヒドで処理して15をもたらす。当業者であれば、重水素交換が潜在的にこれらの条件下で生じて、Y2が重水素である化合物をもたらし得ることが分かるであろう。
【0035】
上記の具体的なアプローチおよび化合物は、限定することを意図するものではない。本明細書のスキームの化学構造は、同じ可変基名(すなわち、R1、R2、R3など)によって同定されるか否かに関わらず、本明細書の式の化合物の対応する位置の化学基の定義(部分、原子など)に相応して定義される可変基を示している。別の化合物を合成するのに用いる化学構造内の化学基の適合性は、当業者の知識の範囲内である。
【0036】
式Iの化合物とその合成前駆体(本明細書のスキームに明示的に示されていないルート内の前駆体を含む)を合成する追加的な方法は、当該技術分野の通常の技能を持つ化学者の手段の範囲内である。適用可能な化合物を合成するのに有用な合成化学変換および保護基の方法論(保護および脱保護)は、当該技術分野において既知であり、それらの方法論としては例えば、Larock R,Comprehensive Organic Transformations,VCH Publishers(1989)、Greene TW et al.,Protective Groups in Organic Synthesis,3rd Ed.,John Wiley and Sons(1999)、Fieser L et al.,Fieser and Fieser’s Reagents for Organic Synthesis,John Wiley and Sons(1994)、Paquette L,ed.,Encyclopedia of Reagents for Organic Synthesis,John Wiley and Sons(1995)、およびこれらのその後の版に記載されているものが挙げられる。
【0037】
本発明によって想定される置換基と可変基との組み合わせは、安定化合物を形成させる組み合わせのみである。
組成物
【0038】
本発明は、発熱物質を含まない組成物であって、式I(例えば、本明細書内の式のいずれかを含む)の化合物、または前記化合物の薬学的に許容可能な塩の有効量と、許容可能な担体とを含む組成物も提供する。本発明の組成物は、医薬用途用に製剤化するのが好ましく(「医薬組成物」)、この場合の担体は、薬学的に許容可能な担体である。この担体(単数または複数)は、その製剤の他の成分と混合可能であるという意味で「許容可能」であり、薬学的に許容可能な担体の場合には、医薬中に用いられる量では、その受容者に対して有毒ではない。
【0039】
薬学的に許容可能な担体としては、本発明の医薬組成物中で用いてよいアジュバントとビヒクルが挙げられる。薬学的に許容可能な担体としては、1種以上の塩、電解質、可溶化剤、溶媒、緩衝剤、乳化剤、矯味矯臭剤、着色剤、甘味剤、充填剤、潤沢剤、賦形剤、懸濁化剤、増粘剤、分散剤、湿潤剤、バイオアベイラビリティ促進剤、および吸収促進剤が挙げられる。具体的な薬学的に許容可能な担体としては、1,3−ブタンジオール、2−オクチルドデカノール、アカシア、アルミナ、ステアリン酸アルミニウム、蜜ろう、ベンジルアルコール、リン酸塩、セルロース系物質、セテアリルアルコール、セチルエステルワックス、ココアバター、コロイドシリカ、コーンスターチ、リン酸水素二ナトリウム、乳化ろう、エチレンオキシド−プロピレンオキシドブロックコポリマー、ゼラチン、グリセリン、グリシン、ヒト血清アルブミン、イオン交換剤、等張食塩水、ラクトース、レシチン、液化石油、長鎖アルコール、LUTROL(登録商標)、ステアリン酸マグネシウム、三ケイ酸マグネシウム、マンニトール、鉱油、オレイン酸およびそのグリセリド誘導体、オリーブ油または特にポリオキシエチル化された型のヒマシ油、飽和植物脂肪酸の部分グリセリド混合物、PLURONIC(登録商標)、ポリアクリレート、ポリエチレングリコール、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリソルベート60、ポリビニルピロリドン、リン酸水素カリウム、ソルビン酸カリウム、プロピレングリコール、硫酸プロタミン、リンゲル液、血清タンパク、カルボキシメチルセルロースナトリウム、塩化ナトリウム、ソルビン酸、モノステアリン酸ソルビタン、スクロース、トラガカント、Tween80、水、ろう、白色ワセリン、羊毛脂、および亜鉛塩が挙げられるが、これらに限らない。
【0040】
本発明の医薬組成物としては、経口投与、直腸投与、経鼻投与、局所投与(口腔投与および舌下投与を含む)、膣内投与、非経口投与(皮下投与、筋内投与、静脈内投与、および皮内投与を含む)、ならびに、経皮投与に適したものが挙げられる。各タイプの組成物と共に用いるのに適切な薬学的に許容可能な担体の選択は、当該技術分野において周知である。同様に、有効成分(単数または複数)と担体とを併せて、本発明の各種医薬組成物の単位投与形態を作製するための方法も、当該技術分野において周知である。例えば、Remington:The Science and Practice of Pharmacy,Lippincott Williams&Wilkins,Baltimore,MD(20th ed.2000)を参照されたい。
【0041】
別の実施形態では、本発明の組成物は、第2の治療剤をさらに含む。この第2の治療剤は、L−838417と同じ作用メカニズムを有する化合物と共に投与すると、有益な特性を有するかまたは示すことが知られているいずれかの化合物または治療剤から選択してよい。
【0042】
好ましくは、第2の治療剤は、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態の治療または予防に有用な薬剤である。
【0043】
別の実施形態では、本発明は、別々の投与形態の本発明の化合物と、上記の第2の治療剤のいずれかの1つ以上とを提供し、この際、本発明の化合物と第2の治療剤は相互に付随している。「相互に付随している」という用語は、本明細書で使用する場合、上記の別々の投与形態が一緒に販売および(一方の薬剤の24時間未満以内に連続してまたは同時に)投与されるように意図されていることが容易に分かるように、別々の投与形態が一緒に包装されているか、または別の形で相互に添付されていることを意味する。
【0044】
本発明の医薬組成物中には、本発明の化合物が有効量存在する。本明細書で使用する場合、「有効量」という用語は、適切な投与レジメンで投与した場合に、治療対象の障害の重症度、持続期間、もしくは進行を軽減もしくは改善するか、治療対象の障害の進展を予防するか、治療対象の障害を退行させるか、または、別の療法の予防効果もしくは治療効果(単数もしくは複数)を促進もしくは向上させるのに十分な量を指す。
【0045】
動物およびヒトに対する用量の相互関係(体表面1平方メートル当たりのミリグラムベース)については、Freireich et al.,(1996)Cancer Chemother.Rep 50:219に記載されている。体表面積は、患者の身長および体重から適切に割り出すことができる。例えば、Scientific Tables, Geigy Pharmaceuticals,Ardsley,N.Y.,1970,537を参照されたい。
【0046】
1つの実施形態では、本発明の化合物の有効量は、1回の治療当たり約0.01〜約5000mgの範囲であることができる。さらに具体的な実施形態では、この範囲は約0.1〜2500mg、または0.2〜1000mg、または最も具体的には約1〜500mgである。治療薬は典型的には1日に1〜3回投与する。
【0047】
当業者であれば分かるように、有効用量も、治療する疾患、疾患の重症度、投与経路、患者の性別、年齢、および全身の健康状態、賦形剤の使用、他の治療処置との併用(他の薬剤の使用など)の可能性、ならびに、治療を行う医師の判断に応じて変化することになる。例えば、有効用量を選択するための指針は、L−838417の処方に関する情報を参照することによって決定することができる。
【0048】
第2の治療剤を含む医薬組成物では、その第2の治療剤の有効量は、その治療剤のみを用いる単剤療法レジームで通常用いられる用量の約20%〜100%である。好ましくは、有効量は、単剤療法での通常の用量の約70%〜100%である。これらの第2の治療剤の単剤療法での通常の用量は、当該技術分野において周知である。例えば、Wells et al.,eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000)、PDR Pharmacopia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)を参照されたい。これらの各参考文献は、参照によりその全体が本明細書に組み込まれる。
【0049】
上に参照として示されている第2の治療剤のいくつかは、本発明の化合物と相乗的に作用すると予想される。相乗的に作用すると、第2の治療剤および/または本発明の化合物の有効用量を単剤療法で必要な有効用量よりも減らすことができるようになる。これには、本発明の化合物のいずれかの第2の治療剤の有毒な副作用を最小限に抑えるという利点、効能を相乗的に向上させる利点、投与もしくは使用のしやすさを向上させる利点、および/または、化合物を調製もしくは製剤化する際の全体的コストが低下するという利点がある。
治療法
【0050】
別の実施形態では、本発明は、細胞内のGABA−A受容体のサブタイプα1を阻害する方法であって、本明細書の式Iの化合物1種以上と細胞を接触させることを含む方法を提供する。別の実施形態では、本発明は、細胞内のGABA−A受容体のサブタイプα2、α3、およびα5のうちの1つ以上を活性化させる方法を提供する。
【0051】
別の実施形態によれば、本発明は、L−838417によって有益な形で治療される疾患を罹患しているか、または罹患しやすい患者を治療する方法であって、式Iの化合物もしくはその塩、または本発明の組成物を有効量、前記患者に投与する工程を含む方法を提供する。このような疾患は、当該技術分野において周知であり、国際公開第WO1998004559号、同第WO2000044752号、同第WO2006061428号に開示されているが、これらの特許および特許出願に限らない。上記のような疾患としては、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛が挙げられるが、これらに限らない。
【0052】
本明細書に示されている方法は、特定的に示されている治療が必要な患者として認定する方法も含む。このような治療が必要な患者としての認定は、患者または医療の専門家の判断で行うことができると共に、主観的(例えば意見)であることも、客観的(例えば、試験または診断法によって測定可能)であることもできる。
【0053】
別の実施形態では、上記の治療法のいずれかが、1種以上の第2の治療剤を前記患者に同時投与するさらなる工程を含む。この第2の治療剤は、L−838417との同時投与用として有用であることが知られているいずれかの第2の治療剤から選択してよい。第2の治療剤の選択は、治療対象の特定の疾患または状態によっても左右される。本発明の方法で用いてよい第2の治療剤の例は、本発明の化合物と第2の治療剤を含む複合組成物で用いるものとして上に示したものである。
【0054】
「同時投与」という用語は、本明細書で使用する場合、単回投与形態(本発明の化合物と、上記のような第2の治療剤とを含む本発明の組成物など)、または別々の複数回投与形態の一部として、第2の治療剤を本発明の化合物と共に投与してよいことを意味する。あるいは、本発明の化合物の投与前、投与に連続的に、または投与の後に、追加の薬剤を投与してもよい。このような併用療法の治療では、本発明の化合物も、第2の治療剤(単数または複数)も、従来の方法によって投与する。本発明の化合物と第2の治療剤の両方を含む本発明の組成物を患者に投与することは、治療過程中の別の時期に、同じ治療剤、いずれかの他の第2の治療剤、または本発明のいずれかの化合物を前記患者に別々に投与することを妨げない。
【0055】
これらの第2の治療剤の有効量は当業者に周知であり、投与指針は、本明細書に参照として記載されている特許および公開特許出願、ならびに、Wells et al.,eds.,Pharmacotherapy Handbook,2nd Edition,Appleton and Lange,Stamford,Conn.(2000)、PDR Pharmacopoeia,Tarascon Pocket Pharmacopoeia 2000,Deluxe Edition,Tarascon Publishing,Loma Linda,Calif.(2000)、およびその他の医学書で見ることができる。しかし、第2の医薬剤の最適な有効量範囲を割り出すことは、十分に当業者の視野の範囲内である。
【0056】
第2の治療剤を対象に投与する本発明の1つの実施形態では、本発明の化合物の有効量は、第2の治療剤を投与しない場合の有効量よりも少ない。別の実施形態では、第2の治療剤の有効量は、本発明の化合物を投与しない場合の有効量よりも少ない。このように、高用量のいずれかの薬剤に付随する望ましくない副作用を最小限に抑えることができる。他の潜在的な利点(投与レジメンの改善および/または薬剤コストの削減が挙げられるが、これらに限らない)は、当業者には明らかであろう。
【0057】
さらに別の態様では、本発明は、上記の疾患、障害、または症状の患者における治療または予防用の単一組成物または別々の投与形態のいずれかとして医薬を製造する際に、式Iの化合物を単独で、または、上記の第2の治療剤の1種以上と共に用いることを提供する。本発明の別の態様は、本明細書に示されている疾患、障害、または症状の患者における治療または予防で用いる式Iの化合物、または式Iの化合物を含む医薬組成物である。
医薬キット
【0058】
本発明は、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛の治療で用いるキットも提供する。これらのキットは、(a)式Iの化合物またはその塩を含む医薬組成物であって、入れ物に入っている医薬組成物と、(b)この医薬組成物を用いて、不安および痙攣を含む中枢神経系障害、ならびに、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛を治療する方法を説明している説明書とを含む。
【0059】
上記の入れ物は、前記医薬組成物を保持できるいずれかの容器、または密閉されているか、もしくは密閉可能なその他の器具であってよい。例としては、ボトル、アンプル、分画もしくは多チャンバー型ホルダー、ボトル(その各分画部もしくはチャンバーは、単回用量の前記組成物を含む)、分画型のホイル製小包(その各分画部は、単回用量の前記組成物を含む)、または、単回用量の前記組成物を分注する分注器が挙げられる。上記の入れ物は、当該技術分野において既知のようないずれかの従来の形または形態のものであって、薬学的に許容可能な材料で作られているもの、例えば、紙箱、段ボール箱、ガラスもしくはプラスチック製ボトルもしくは広口瓶、再密閉可能なバッグ(例えば、錠剤の「リフィル」を異なる入れ物に入れて保持するためのもの)、または、治療スケジュールに従ってパックから押し出すように、個別の用量が入ったブリスターパックであることができる。用いる入れ物は、関連する正確な投与形態に応じて決めることができ、例えば、従来の段ボール箱は一般的には、液体懸濁剤を保持するのには用いられない。単一のパッケージの中で2つ以上の入れ物を併せて用いて、単一の投与形態を市販することも可能である。例えば、錠剤をボトルに入れてから、そのボトルを箱に入れてもよい。1つの実施形態では、上記の入れ物はブリスターパックである。
【0060】
本発明のキットは、単位用量の医薬組成物を投与または測り分ける器具も含んでもよい。このような器具としては、吸入器(前記組成物が吸入可能な組成物である場合)、注射器および注射針(前記組成物が注射可能な組成物の場合)、注射器、スプーン、ポンプ、もしくは容器(目盛りの有無は問わない)(前記組成物が経口用液体組成物である場合)、または、本発明のキット内に含まれる組成物の投与剤形に適している他のいずれかの測定用もしくは送達用器具を挙げてよい。
【0061】
特定の実施形態では、本発明のキットは、本発明の化合物との同時投与用に用いる上記のもののうちの1つのような第2の治療剤を含む医薬組成物を、入れ物の別個の容器内に含んでもよい。
【実施例】
【0062】
実施例1.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−メチル−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物103)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物103を調製した。
【化4】
【0063】
工程1.4−(tert−ブチル−d9)−3,6−ジクロロピリダジン(12a) 新たに精製した3,6−ジクロロ−ピリダジン、11(5.4g、33.5mmol)を蒸留水(130mL)に懸濁させた懸濁液に、濃硫酸(5.7mL、108mmol)を加えた。この混合物を65℃まで加温し、トリメチル酢酸−d9、10a(6.0g、54mmol、CDNアイソトープス、99atom%D)を加えてから、硝酸銀(1.1g、7mmol)を加えた。反応温度を65〜75℃に保ちながら、10〜15分にわたって、アンモニウムペルオキシ二硫酸(12.3g、54mmol)を蒸留水(35mL)に溶解させた溶液を上記の混合物に加えた。この混合物を30分間攪拌し、室温まで冷却した。この混合物を氷(100g)の上に注いで、その混合物を濃水酸化アンモニウムによってpH=9〜10に調節した。この水性混合物をジクロロメタン(2×30mL)で抽出した。合わせた抽出物を1Nの水酸化ナトリウム(10mL)で洗浄し、Na2SO4で乾燥させ、ろ過し、減圧下で濃縮した。この粗生成物をシリカゲルカラムクロマトグラフィー(10%酢酸エチル/ヘプタンで溶出)によって精製し、6.1g(80%)の12aを無色の油として得た。
【0064】
工程2.7−(tert−ブチル−d9)−6−クロロ−3−(2,5−ジフルオロフェニル)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(14a) キシレン(30mL)中の12a(6g、28mmol)、13(7.2g、42mmol、市販品)、およびトリエチルアミン塩酸塩(5.8g、42mmol)の混合物を攪拌しながら150℃で36時間加熱した。室温まで冷却後、この混合物を減圧下で濃縮した。その残渣をジクロロメタン(40mL)で粉砕し、ろ過し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルクロマトグラフィー(20〜50%酢酸エチル/ヘプタンで溶出)によって精製し、5.6g(60%)の14aをオフホワイトの固体として得た。
【0065】
工程3.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−メチル−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物103) (1−メチル−1H−1,2,4−トリアゾール−5−イル)メタノール15a(0.45g、4.0mmol、市販品)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水酸化ナトリウム(0.17、4.3mmol)を加えた。この混合物を15分間攪拌し、14a(1.2g、3.6mmol)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過によって回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル−ヘプタン(1:1)からの再結晶によってさらに精製し、1.25g(78%)の化合物103を白色固体として得た。1H−NMR(300MHz、CDCl3):δ3.91(s,3H)、5.55(s、2H)、7.23−7.28(m、2H)、7.62−7.68(m、1H)、7.93(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):δ34.55、35.66、59.37、115.58(dd、J1=16.6、J2=9.2)、117.63(dd、J1=25.8、J2=6.6)、117.72(dd、J1=24.5、J2=12.2)、118.72(dd、J1=24.0、J2=8.5)、121.74、137.85、143.47、145.00、149.49,151.13、155.70(d、J=160.9)、159.01(d、J=155.9)、158.70。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.41分、純度99.3%。MS(M+H):409.2。元素分析(C19H10D9F2N7O):計算値:C=55.88、H=4.69、N=24.01、実測値:C=55.98、H=4.53、N=23.98。
【0066】
実施例2.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メタノール(15b)の合成 上記のスキーム2に一般的に概要が示されているように、適切に重水素化した中間体から中間体15bを調製した。
【化5】
【0067】
工程1.1−(メチル−d3)−1H−1,2,4−トリアゾール(17a) 窒素雰囲気下の、機械的スターラーを備えたフラスコ内で、無水THF(60mL)に1,2,4−トリアゾール16(6.0g、87mol)を加えてから、ヨードメタン−d3(6.5mL、1.05mol、ケンブリッジアイソトープス、99atom%D)を加えた。この濁った混合物を0℃まで冷却し、20分にわたって、1,8−ジアザビシクロ[5.4.0]ウンデス−7−エン「DBU」(13.2mL、0.87mol)を加えた。この混合物を室温までゆっくり加温し、オーバーナイトで攪拌した。続いて、この混合物をセライトパッドに通してろ過し、そのろ液を減圧下で濃縮して、7.3g(>100%)の粗生成物17aを黄色の油として得た。GCMSによれば、純度は90%である。位置異性体の比率は12:1であった。
【0068】
工程2.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メタノール(15b) 17a(5g、58mmol)とパラホルムアルデヒド−d2(10g、333mmol、ケンブリッジアイソトープス、99atom%D)との混合物を密閉チューブ内で170℃にて5時間加熱した。この混合物を室温まで冷却し、ジクロロメタン(20mL)で希釈した。その固体をろ過して除去し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルショートカラムクロマトグラフィー(75%THF/ヘプタンで溶出)によって精製し、4.8g(71%)の15bをオフホワイトの固体として得た。
【0069】
実施例3.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物105)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水化した中間体から化合物105を調製した。
【化6】
7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物105) 15b(0.24g、2.0mmol)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.08、2.1mmol)を加えた。この混合物を15分間攪拌し、14a(0.6g、1.8mmol、実施例1参照)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、0.52g(70%)の化合物105を白色固体として得た。1H−NMR(300MHz、CDCl3):δ7.23−7.28(m、2H)、7.63−7.67(m、1H)、7.92(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):35.66および59.37でピークが観測されず。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.40分、純度99.0%。MS(M+H):414.3。元素分析(C19H5D14F2N7O):計算値:C=55.20、H=4.63、N=23.72、F=9.19、実測値:C=54.88、H=4.45、N=23.46、F=9.59。
【0070】
実施例4.(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−メタノール(15c)の合成 上記のスキーム2に一般的に概要が示されているように、適切に重水素化した中間体から中間体15cを調製した。
【化7】
(1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−メタノール(15c) 17a(5g、58mmol、実施例2参照)とパラホルムアルデヒド(10g、333mmol)との混合物を密閉チューブ内で170℃にて5時間加熱した。この混合物を室温まで冷却し、ジクロロメタン(20mL)で希釈した。その固体をろ過して除去し、そのろ液を減圧下で濃縮した。この粗生成物をシリカゲルショートカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製し、5.0g(75%)の15cをオフホワイトの固体として得た。
【0071】
実施例5.7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物104)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物104を調製した。
【化8】
【0072】
7−(tert−ブチル−d9)−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物104) 15c(0.46g、4.0mmol)をDMF(20mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.17、4.3mmol)を加えた。この混合物を15分間攪拌し、14a(1.2g、3.6mmol、実施例1参照)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(5%メタノール/ジクロロメタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、1.31g(88%)の化合物104を白色固体として得た。
1H−NMR(300MHz、CDCl3):δ5.55(s、2H)、7.23−7.28(m、2H)、7.62−7.67(m、1H)、7.93(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):δ34.55、59.36、115.53(dd、J1=16.6、J2=8.8)、117.63(dd、J1=24.4、J2=12.8)、117.71(dd、J1=24.1、J2=8.0)、118.77(dd、J1=23.9、J2=8.5)、121.75、137.85、143.48、145.00、149.50、151.15、155.76(d、J=163.5)、159.08(d、J=156.9)、158.71。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.40分、純度99.6%。MS(M+H):412.2。元素分析(C19H7D12F2N7O):計算値:C=55.47、H=4.67、N=23.83、実測値:C=55.49、H=4.76、N=23.87。
【0073】
実施例6.7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物101)の合成 上記のスキーム1に一般的に概要が示されているように、適切に重水素化した中間体から化合物101を調製した。
【化9】
7−tert−ブチル−3−(2,5−ジフルオロフェニル)−6−((1−(メチル−d3)−1H−1,2,4−トリアゾール−5−イル)−1,1−d2−メトキシ)−[1,2,4]トリアゾロ[4,3−b]ピリダジン(化合物101) 15b(0.24g、4.0mmol、実施例2参照)をDMF(10mL)に溶解させた溶液に、鉱油中の60%水素化ナトリウム(0.08、2.1mmol)を加えた。この混合物を15分間攪拌し、既知の化合物7−tert−ブチル−6−クロロ−3−(2,5−ジフルオロフェニル)−[1,2,4]トリアゾロ[4,3−b]ピリダジン、14b(0.58g、1.8mmol、国際公開第WO1998004559号に記載されているように調製)を加えた。この混合物を3時間、室温で攪拌してから、水(100mL)で希釈した。その沈殿物をろ過して回収し、水で数回洗浄した。この粗生成物をシリカゲルカラムクロマトグラフィー(75%THF/ヘプタンで溶出)によって精製した。この生成物を、酢酸エチル/ヘプタン(1:1)からの再結晶によってさらに精製し、0.53g(72%)の化合物101を白色固体として得た。1H−NMR(300MHz、CDCl3):δ1.41(s、9H)、7.23−7.28(m、2H)、7.62−7.68(m、1H)、7.92(s、1H)、8.00(s、1H)。13C−NMR(75MHz、CDCl3):28.96でピークが観察され、35.66および59.36ではピークは観察されず。HPLC(方法:ウォーターズ製Atlantis T3 2.1×50mm 3μm C18−RPカラム−勾配法:14分で5〜95%ACN+0.1%ギ酸(1.0mL/分)、95%ACNで4分保持、波長:254nm):保持時間:5.42分、純度99.7%。MS(M+H):405.3。元素分析(C19H14D5F2N7O):計算値:C=56.43、H=4.74、N=24.25、F=9.40、実測値:C=56.22、H=4.73、N=23.87、F=9.35。
【0074】
実施例7.代謝安定性の評価 ヒト肝ミクロソーム(20mg/mL)とラット肝ミクロソーム(20mg/mL)をゼノテック社(カンザス州レネクサ)から入手した。β−ニコチンアミドアデニンジヌクレオチドリン酸還元体(NADPH)、塩化マグネシウム(MgCl2)、およびジメチスルホキシド(DMSO)をシグマ−アルドリッチから購入した。
【0075】
代謝安定性の測定:試験化合物(L−838417、化合物101、化合物103、化合物104、および化合物105)の7.5mMストック溶液をDMSO中で調製した。これらの7.5mMストック溶液をアセトニトリル(ACN)中で12.5μMまで希釈した。3mMのMgCl2を含有するpH7.4の0.1Mリン酸カリウム緩衝液中で上記の20mg/mLの肝ミクロソーム(ヒトまたは肝臓のいずれか)を2.5mg/mLまで希釈した。この希釈ミクロソーム(375μL)を96ウェルのポリプロピレン製ディープウェルプレートのウェルに3連で加えた。このミクロソームに12.5μMの試験化合物10μLを加え、その混合物を10分間予熱した。予熱したNADPH溶液125μLを加えることによって、反応を開始させた。最終反応体積は0.5mLであり、pH7.4の0.1Mリン酸カリウム緩衝液および3mMのMgCl2中に、0.5mg/mLのヒト肝ミクロソーム、0.25μMの試験化合物、および2mMのNADPHを含有していた。この反応混合物を37℃でインキュベートし、50μLのアリコートを0分、5分、10分、20分、および30分の時点で除去して、内部標準を有する50μLの氷冷ACNの入った96ウェルのシャローウェルプレートに加えて、反応を停止させた。このプレートを4℃で20分間保存した後、そのプレートのウェルに100μLの水を加えてから遠心分離して、ペレット沈殿タンパクを得た。上清を別の96ウェルプレートに移し、アプライドバイオシステムのAPI 4000質量分析計を用いて、LC−MS/MSによって、残存の親化合物の量について分析した。7−エトキシクマリン(1μM)をポジティブコントロールとして用いた。
【0076】
データ解析:下記の数式を用いて、親化合物の残存率(%)(ln)とインキュベーション時間との関係の直線回帰の傾斜から、試験化合物のインビトロt1/2を算出した。
インビトロt1/2=0.693/k(式中、k=−[親化合物の残存率(%)(ln)とインキュベーション時間との関係の直線回帰の傾斜]
データ解析は、マイクロソフトのソフトウェアExcelを用いて行った。
【0077】
これらの実験の結果は、図1(ヒト肝ミクロソーム)および図2(ラット肝ミクロソーム)に示されている。図1に示されているように、ヒト肝ミクロソームと共に30分インキュベーションした後、約70%のL−838417が元の状態を保っていた。L−838417の半減期を算出したところ、54.4分であった。これに対し、化合物101、103、104、および105はそれぞれ、ヒト肝ミクロソーム中で安定していた(30分のインキュベーション後、80%超の親化合物が元の状態を保っていた)。
【0078】
ラット肝ミクロソームでは、L−838417も化合物101も、30分のインキュベーション後、約70%が元の状態を保っていた(図2)。半減期の計算値は、L−838417では50.5分、化合物101では52.7分であった。化合物103、104、および105はいずれも、30分のインキュベーション後、80%超が元の状態を保っており、いずれもラットミクロソームにおいて安定的であるとみなされた。
【0079】
実施例8.ラットに経口および静脈内投与した後の化合物103および105の薬物動態およびバイオアベイラビリティ解析 3匹の雄スプラーグドーリーラット(それぞれ200〜250g)の頸静脈にカニューレを挿入し、頸静脈のカニューレを通じて、L−838417、化合物103、および化合物105それぞれを1mg/kg含む単回用量分を(10%ジメチルスルホキシド(DMSO)、10%N,N−ジメチルアセトアミド(DMA)、および60%ポリエチレングリコール(PG)中に、これらの3つの化合物を1mg/mL含む1:1:1混合物として)投与した。3匹の追加の雄スプラーグドーリーラット(それぞれ200〜250g)に対して、L−838417、化合物103、および化合物105をそれぞれ1mg/kg含む単回用量分を(10%ジメチルスルホキシド(DMSO)、10%N,N−ジメチルアセトアミド(DMA)、および60%ポリエチレングリコール(PG)中に、これらの3つの化合物をそれぞれ1mg/mL含む1:1:1混合物として)強制経口投与した。
【0080】
静脈処置した試験ラットの血液(0.25mL)を、投与から2分、5分、15分、30分、1時間、2時間、4時間、および6時間の時点に眼窩後穿刺で採取した。経口処置した試験ラットの血液(0.25mL)を、投与から5分、15分、30分、45分、1時間、2時間、4時間、および6時間の時点に眼窩後穿刺で採取した。血液は、上記の時点に、抗血液凝固剤としてK2EDTAを含むチューブの中に集めた。血液サンプルを氷上で保存してから、遠心分離して血漿を得た。この血漿(約0.125μL)を96ウェルのディーププレートに分注し、アプライドバイオシステムのAPI 4000質量分析計を用いたLC−MS/MSによる解析まで、−80℃で保存した。
【0081】
この研究の静脈投与の部分の結果は、図3および下記の表2に示されている。
【表2】
【0082】
これらの結果によって、ラットに静脈内投与した後は、非重水素化L−838417よりも、化合物103および105はそれぞれ半減期が約80%長くなり、またAUC0−6が2倍超となることが示されている。加えて、化合物103および105の両方が、L−838417よりもそれぞれ約60%以上ゆっくりと消失する。
【0083】
この研究の経口投与部分の結果は、図4および下記の表3に示されている。
【表3】
【0084】
これらの結果によって、ラットに経口投与した後は、L−838417よりも、化合物103および105のCmaxが2倍超高いことが示されている。加えて、上記の結果によって、経口投与後は、非重水素化L−838417よりも、化合物103および105のAUC0−6が2.5倍超となることが示されている。
【0085】
更なる説明がなくても、当業者であれば、上記の説明および実例となる実施例を用いて、本発明の化合物を作製および利用できると共に、請求されている方法を実施できると考えられる。上記の論議および実施例は、特定の好ましい実施形態の詳細な説明を示しているに過ぎないと理解されたい。本発明の趣旨および範囲から逸脱することなく、各種の変形形態および均等形態を作製できることは当業者には明らかであろう。上で論じられているか、または引用されているすべての特許、学術論文、およびその他の文書は、参照により本明細書に組み込まれる。
【特許請求の範囲】
【請求項1】
式I
【化1】
(式中、R1はCH3、CDH2、CD2H、またはCD3であり、
R2は、0〜9個の重水素原子を有するt−ブチル基であり、
各Yは独立して、水素または重水素であり、
R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
の化合物、またはその薬学的に許容可能な塩。
【請求項2】
式中のR1がCH3またはCD3である、請求項1に記載の化合物。
【請求項3】
式中のR2が−C(CH3)3または−C(CD3)3である、請求項1または2に記載の化合物。
【請求項4】
式中のY1aとY1bが同じである、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
式中のR2が−C(CD3)3である、請求項1〜4のいずれか一項に記載の化合物。
【請求項6】
下記の表に示されている化合物のいずれか1つ、またはその薬学的に許容可能な塩から選択される、請求項1に記載の化合物。
【表1】
【請求項7】
上記の実施形態のいずれかで重水素として指定されていないいずれの原子も、その天然同位体存在度で存在する、請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
発熱物質を含まない組成物であって、請求項1の化合物と、許容可能な担体とを含む組成物。
【請求項9】
医薬投与用に製剤化された請求項8に記載の組成物であって、その担体が、薬学的に許容可能な担体である組成物。
【請求項10】
中枢神経系障害、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態の治療または予防に有用な第2の治療剤を追加的に含む、請求項9に記載の組成物。
【請求項11】
a.細胞内のGABA−A受容体のサブタイプα1を阻害するのに、または、
b.細胞内のGABA−A受容体のサブタイプα2、α3、およびα5のうちの1つ以上を活性化させるのに
用いられる、請求項1〜7のいずれか一項に記載の化合物。
【請求項12】
中枢神経系障害、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態を治療するのに用いられる、請求項8または9に記載の組成物。
【請求項13】
不安または痙攣から選択される疾患または状態を治療するのに用いられる、請求項8または9に記載の組成物。
【請求項1】
式I
【化1】
(式中、R1はCH3、CDH2、CD2H、またはCD3であり、
R2は、0〜9個の重水素原子を有するt−ブチル基であり、
各Yは独立して、水素または重水素であり、
R1がCH3であり、各Yが水素である場合、R2は1〜9個の重水素原子を有する)
の化合物、またはその薬学的に許容可能な塩。
【請求項2】
式中のR1がCH3またはCD3である、請求項1に記載の化合物。
【請求項3】
式中のR2が−C(CH3)3または−C(CD3)3である、請求項1または2に記載の化合物。
【請求項4】
式中のY1aとY1bが同じである、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
式中のR2が−C(CD3)3である、請求項1〜4のいずれか一項に記載の化合物。
【請求項6】
下記の表に示されている化合物のいずれか1つ、またはその薬学的に許容可能な塩から選択される、請求項1に記載の化合物。
【表1】
【請求項7】
上記の実施形態のいずれかで重水素として指定されていないいずれの原子も、その天然同位体存在度で存在する、請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
発熱物質を含まない組成物であって、請求項1の化合物と、許容可能な担体とを含む組成物。
【請求項9】
医薬投与用に製剤化された請求項8に記載の組成物であって、その担体が、薬学的に許容可能な担体である組成物。
【請求項10】
中枢神経系障害、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態の治療または予防に有用な第2の治療剤を追加的に含む、請求項9に記載の組成物。
【請求項11】
a.細胞内のGABA−A受容体のサブタイプα1を阻害するのに、または、
b.細胞内のGABA−A受容体のサブタイプα2、α3、およびα5のうちの1つ以上を活性化させるのに
用いられる、請求項1〜7のいずれか一項に記載の化合物。
【請求項12】
中枢神経系障害、神経病性疼痛、炎症性疼痛、および片頭痛関連疼痛から選択される疾患または状態を治療するのに用いられる、請求項8または9に記載の組成物。
【請求項13】
不安または痙攣から選択される疾患または状態を治療するのに用いられる、請求項8または9に記載の組成物。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2013−100322(P2013−100322A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2013−4449(P2013−4449)
【出願日】平成25年1月15日(2013.1.15)
【分割の表示】特願2011−525250(P2011−525250)の分割
【原出願日】平成21年8月28日(2009.8.28)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成25年1月15日(2013.1.15)
【分割の表示】特願2011−525250(P2011−525250)の分割
【原出願日】平成21年8月28日(2009.8.28)
【出願人】(509049012)コンサート ファーマシューティカルズ インコーポレイテッド (24)
【Fターム(参考)】
[ Back to top ]