
置換プロパルギルエトキシアミドヌクレオシド
【課題】標識したポリヌクレオチドの提供。
【解決手段】以下の構造を有する置換プロパルギルエトキシアミドヌクレオシドが開示されている:ここで、Xは、アミノアルカン酸、アルキルアミノ安息香酸、α−アミノ酸、及び4−アミノ−2−ブチン酸からなる群から選択される。R1及びR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;R3は、−H及び低級アルキルからなる群から選択される。Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である。さらに、プライマー伸長方法が提供され、これは、上記X置換プロパルギルエトキシアミドヌクレオシドを使用し、そして、上記X置換プロパルギルエトキシアミドヌクレオシドを含有するポリヌクレオチドが提供される。

【解決手段】以下の構造を有する置換プロパルギルエトキシアミドヌクレオシドが開示されている:ここで、Xは、アミノアルカン酸、アルキルアミノ安息香酸、α−アミノ酸、及び4−アミノ−2−ブチン酸からなる群から選択される。R1及びR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;R3は、−H及び低級アルキルからなる群から選択される。Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である。さらに、プライマー伸長方法が提供され、これは、上記X置換プロパルギルエトキシアミドヌクレオシドを使用し、そして、上記X置換プロパルギルエトキシアミドヌクレオシドを含有するポリヌクレオチドが提供される。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、一般に、ポリメラーゼ酵素の基質として有用なヌクレオチド化合物、プライマー伸長反応でこのようなヌクレオチド化合物を使用する方法、およびこのようなヌクレオチド化合物を含有するポリヌクレオチドに関する。
【0002】
(参考文献)
【0003】
【数1】
【0004】
【数2】
【背景技術】
【0005】
(背景)
核酸配列決定は、現代の生物学および生物工学において必須の重要な技術となっており、基本的な生物研究から薬剤の発見や臨床医学までの範囲の分野に関連した情報を提供する。大容量のDNA配列データが収集されているので、核酸配列決定法のスループットを高めると共にコストを下げるために、自動化技術が開発されている(Smith;Connell;Trainor)。
【0006】
好ましい自動化核酸配列決定法は、Sangerにより開発された酵素複製技術に基づいている(Sanger)。Sangerの方法では、一本鎖テンプレート核酸の配列は、一組のポリヌクレオチドフラグメントを合成するための核酸ポリメラーゼを用いて決定され、ここで、このフラグメントは、(i)このテンプレート配列に対する配列相補性を有し、(ii)単一ヌクレオチドにより長さが変わり、そして(iii)公知のヌクレオチド(例えば、A、C、GまたはT)での5’−末端終止を有する。この方法では、オリゴヌクレオチドプライマーは、配列決定されるテンプレート核酸の3’−末端にアニールされ、このプライマーの3’−末端は、相補的ポリヌクレオチドフラグメントのポリメラーゼ媒介重合に対する開始部位として、役立つ。この酵素的重合工程は、このテンプレート−プライマーハイブリッドと4個の天然デオキシヌクレオチド(「dNTP」)、核酸ポリメラーゼ酵素および2’,3’−ジデオキシヌクレオチド三リン酸(「ddNTP」)「ターミネーター」とを組み合わせることにより、行われる。このターミネーターの組み込みは、この3’−末端の水酸基を欠いたフラグメントを形成し、それゆえ、このポリメラーゼによって、さらに伸長され得ない(すなわち、このフラグメントは、「終止されている」)。組込みのためのddNTPおよびその対応dNTPの間の競合の結果、異なるサイズのフラグメントが分配され、各フラグメントは、この反応で使用される特定のターミネーターで終止される。このテンプレート核酸の完全な配列を発見するために、4個の並発反応が行われ、各反応は、異なるddNTPターミネーターを使用する。これらのフラグメントのサイズ分配を決定するために、これらのフラグメントは、サイズが異なるフラグメントを単一ヌクレオチドによって分割するように、電気泳動により分離される。
【0007】
古典的なSanger法の現代版変形法では、このヌクレオチドターミネーターは、蛍光色素で標識され(Prober;Hobbs)、熱安定性のDNAポリメラーゼ酵素が使用される(Murray)。色素標識したターミネーターを使用することにより、以下のいくつかの利点が得られる:(i)放射性同位体の貯蔵、使用および廃棄に付随した問題がなくなる;(ii)色素標識したプライマーを合成する必要性がなくなる;および(iii)各A、G、CまたはTヌクレオチド用に異なる色素標識を使用するとき、単一の管にて、4個の反応の全てを同時に行うことができる。熱安定性のポリメラーゼ酵素を使用することにより、(i)この重合反応を高温で行うことが可能になり、それにより、このテンプレートのいずれかの二次構造が分断され、その結果、より少ない配列依存性アーチファクトが得られ、そして、(ii)この配列決定反応が熱サイクルでき、それにより、生成する伸長生成物の量を線形的に増幅させるのに役立ち、それゆえ、配列を得るのに必要なDNAテンプレートの量が低減される。
【0008】
Sanger配列決定法のこれらの現代版変形法は、効果的であることが証明されているものの、それらの性能および経済性を最適化することに関して、いくつかの問題点が残っている。Sanger型核酸配列決定法で熱安定性のポリメラーゼ酵素と組み合わせて現在入手できる色素標識ターミネーターを使用するとき、特に、フルオレセイン型色素標識の場合に遭遇する1つの問題点には、50:1の比まで、非標識dNTPよりも大過剰の色素標識ターミネーターが必要なことがある。この大過剰の標識ターミネーターにより、この電気泳動分離工程を行う前に、この配列決定反応生成物を精製する必要が生じる。この清浄化(clean−up)工程は、組み込まれていない標識化ターミネーター種および真正の配列決定フラグメントの同時移入により起こる干渉を回避するために、必要である。典型的な清浄化方法には、エタノール沈殿またはクロマトグラフィー分離が含まれる(ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Protocol)。このような清浄化工程は、完全に自動化した配列決定系(ここで、その配列決定反応生成物は、電気泳動分離プロセスに直接移される)を開発する仕事を著しく複雑にする。
【0009】
Sanger型核酸配列決定法で熱安定性のポリメラーゼと組み合わせて現在入手できる色素標識ターミネーターを使用するとき遭遇する第二の問題点には、これらの反応生成物を電気泳動により分離し蛍光検出を用いて検出するとき、ピーク高さの不均一な分布が得られることがある。このような不均一なピーク高さは、自動化した配列測定およびヘテロ接合体の検出を実質的に信頼できないものとするために、不利である。
【発明の概要】
【課題を解決するための手段】
【0010】
(要旨)
本発明は、プライマー伸長反応(例えば、Sanger型DNA配列決定反応またはPCR反応)において、鎖終止ヌクレオチドおよび鎖伸長ヌクレオチドとして有用な、新規なクラスの置換プロパルギルエトキシアミドヌクレオシドの本発明者らによる発見に関する。
【0011】
本発明の目的は、標識した鎖終止ヌクレオチドを形成するのに使用できるヌクレオチドを提供することにある。
【0012】
本発明のさらなる目的は、標識を含む鎖終止ヌクレオチドを提供することにある。
【0013】
本発明のなおさらなる目的は、蛍光標識を含む鎖終止ヌクレオチドであって、Sanger型DNA配列決定法において、非標識の鎖終止ヌクレオチドよりも低い過剰濃度のこのような標識した鎖終止ヌクレオチドを必要とするものを提供することにある。
【0014】
本発明の別の目的は、Sanger型DNA配列決定法において、ピーク高さのより均一な分布が得られる標識した鎖終止ヌクレオチドを提供することにある。
【0015】
本発明の目的は、標識した鎖伸長ヌクレオチドを形成するのに使用できるヌクレオチドを提供することにある。
【0016】
本発明のさらなる目的は、標識を含む鎖伸長ヌクレオチドを提供することにある。
【0017】
本発明の別の目的は、標識したポリヌクレオチドを提供することにある。
【0018】
本発明のさらなる目的は、本発明の置換プロパルギルエトキシアミドヌクレオチドを使用するプライマー伸長反応を包含する方法を提供することにある。
【0019】
第一の局面では、本発明の上述の目的および他の目的は、以下の構造を有するヌクレオシド化合物により、達成される:
【0020】
【化1】
ここで、Xは、
【0021】
【化2】
ここで、nは、1〜5の範囲である、
【0022】
【化3】
ここで、nは、1〜5の範囲である、
【0023】
【化4】
および
【0024】
【化5】
からなる群から選択される;そしてR1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される。R3は、−Hおよび低級アルキルからなる群から選択される。Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基であり、ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、このプリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、このピリミジンのN1位にて結合しており、Bがプリンのとき、その隣接する三重結合炭素は、このプリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、この7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、このピリミジンの5位に結合している。W1は、−Hおよび−OHからなる群から選択される;W2は、−OH、またはこのヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そしてW3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
【0025】
本発明の第一の局面の第一の好ましい実施態様では、R1およびR2の1個は、標識であり、より好ましくは、この標識は、フルオレセイン型色素、ローダミン型色素またはFLAN型色素である。
【0026】
本発明の第一の局面の第二の好ましい実施態様では、W1は、−Hである;W2は、−OH、またはその3’位でホスホジエステル結合を形成できなくする部分である;そしてW3は、−P3O10である。より好ましくは、W2は、−OH、−H、アジド、アミノ、フルオロおよびメトキシからなる群から選択される。
【0027】
本発明の第一の局面の第三の好ましい実施態様では、Bは、ウラシル、シトシン、7−デアザアデニンおよび7−デアザグアノシンからなる群から選択される。
【0028】
本発明の第一の局面の第四の好ましい実施態様では、Xは、
【0029】
【化6】
であり、n=1であり、そしてΦ基は、パラ立体配置にある。
【0030】
第二の局面では、本発明の上述の目的および他の目的は、プライマー伸長反応を行う方法により達成され、この方法は、テンプレート核酸を提供する工程;このテンプレート核酸の一部にオリゴヌクレオチドプライマーをアニールする工程;およびこのプライマーを伸長するために、このプライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加する工程を包含し、このプライマー伸長試薬は、上記の本発明の第一局面のヌクレオシド化合物を含有する。
【0031】
第三の局面では、本発明の上述の目的および他の目的は、上記の本発明の第一の局面のヌクレオシド化合物を含有するポリヌクレオチドにより、達成される。
【0032】
本発明は、例えば以下の項目を提供する。
(項目1) 以下の構造を有するヌクレオシド化合物:
【0033】
【化18】
ここで、Xは、
【0034】
【化19】
ここで、nは、1〜5の範囲である、
【0035】
【化20】
ここで、nは、1〜5の範囲である、
【0036】
【化21】
および
【0037】
【化22】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
(項目2) R1およびR2の1個が、標識である、項目1に記載のヌクレオシド化合物。
(項目3) 前記標識が、フルオレセイン型色素である、項目2に記載のヌクレオシド化合物。
(項目4) 前記標識が、ローダミン型色素およびFLAN型色素からなる群から選択される、項目2に記載のヌクレオシド化合物。
(項目5) W1が、−Hであり;
W2が、−OH、または前記ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分であり;そして
W3が、−P3O10である、
項目1に記載のヌクレオシド化合物。
(項目6) W2が、−OH、−H、アジド、アミノ、ハロ、およびメトキシからなる群から選択される、項目1に記載のヌクレオシド化合物。
(項目7) W2が、−Hおよびフルオロからなる群から選択される、項目1に記載のヌクレオシド化合物。
(項目8) Bが、ウラシル、シトシン、7−デアザアデニン、および7−デアザグアノシンからなる群から選択される、項目1に記載のヌクレオシド。
(項目9) Xが、パラ立体配置を有するアルキルアミノ安息香酸であり、そしてn=1である、項目1に記載のヌクレオシド化合物。
(項目10) プライマー伸長反応を行う方法であって、該方法は、以下の工程を包含する:
テンプレート核酸を提供すること;
該テンプレート核酸の一部にオリゴヌクレオチドプライマーをアニールすること;および
該プライマーを伸長するために、該プライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加することであって、該プライマー伸長試薬は、以下の構造を有するヌクレオシド化合物を含有する:
【0038】
【化23】
ここで、Xは、
【0039】
【化24】
ここで、nは、1〜5の範囲である、
【0040】
【化25】
ここで、nは、1〜5の範囲である、
【0041】
【化26】
および
【0042】
【化27】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
(項目11) R1およびR2が、標識であり、そして他が、−Hである、項目10に記載の方法。
(項目12) 以下の構造を有するヌクレオチド化合物を含有するポリヌクレオチド:
【0043】
【化28】
ここで、Xは、
【0044】
【化29】
ここで、nは、1〜5の範囲である、
【0045】
【化30】
ここで、nは、1〜5の範囲である、
【0046】
【化31】
および
【0047】
【化32】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、ホスフェートアナログ、および−OHからなる群から選択される。
【図面の簡単な説明】
【0048】
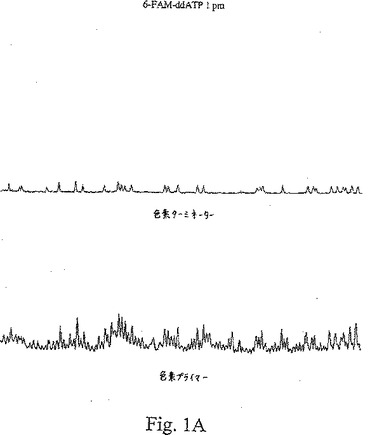
【図1A】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
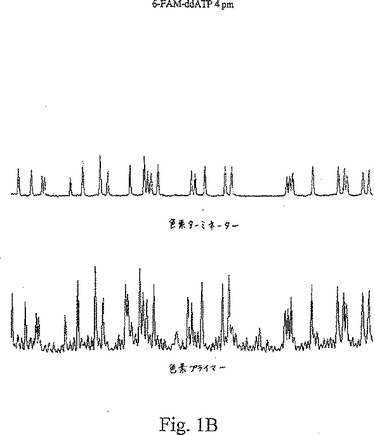
【図1B】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
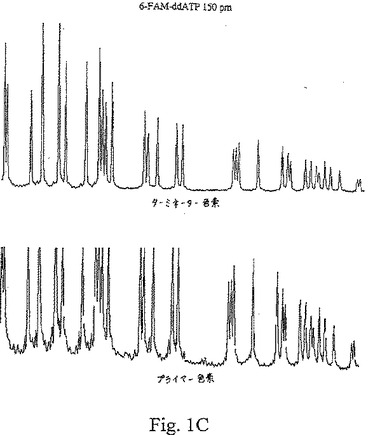
【図1C】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
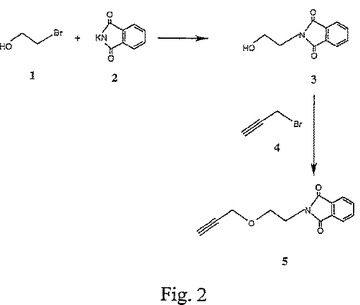
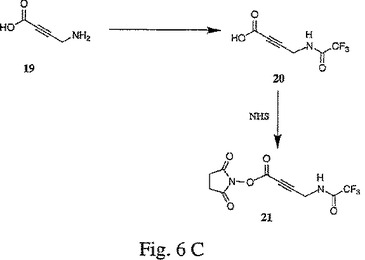
【図2】図2は、2−フタルイミドエタノール(3)および3−(2−フタルイミドエトキシ)プロピン(5)の合成を示す。
【図3】図3は、5−{3−(2−フタルアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン(7)および5−{3−(2−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン(8)の合成を示す。
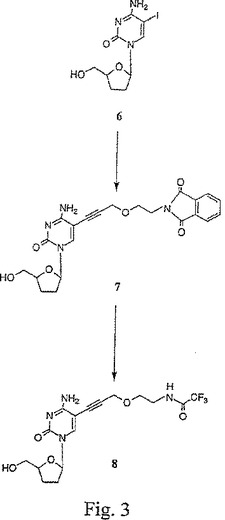
【図4】図4は、5−{3−(2’−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン一リン酸(10)の合成を示す。
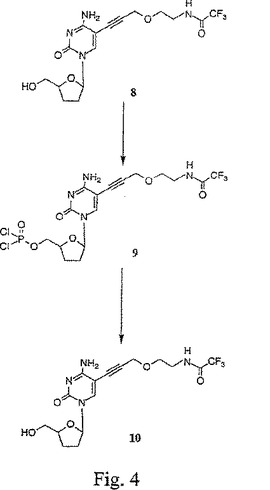
【図5】図5は、5−{3−(2−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(12)および5−{3−(2−アミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成を示す。
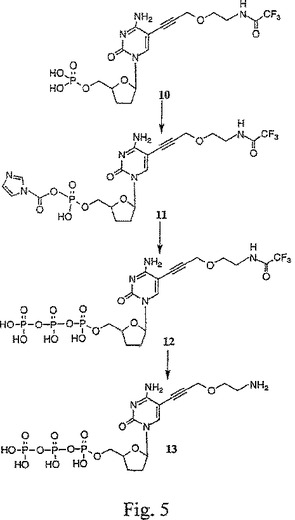
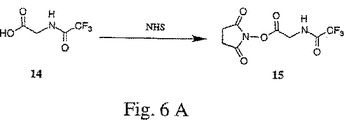
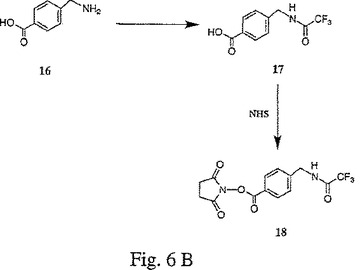
【図6A】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図6B】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図6C】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図7】図7は、5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)の合成を示す。
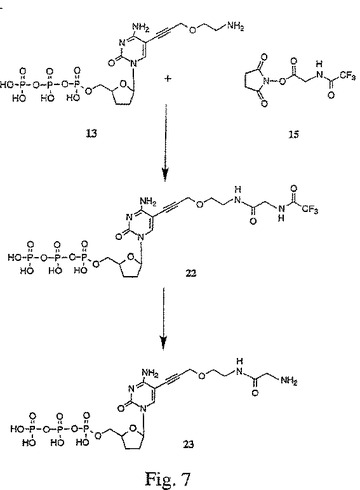
【図8】図8は、5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)の合成を示す。
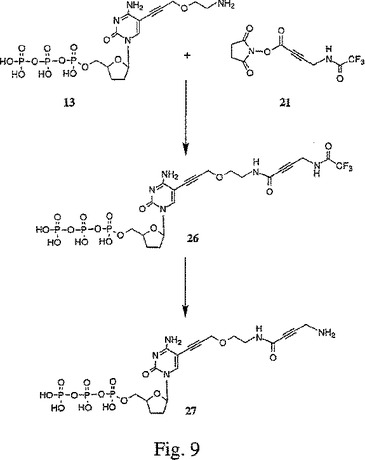
【図9】図9は、5−[3−{2−[N−(2−アミノ−ブト−2−イノイック)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(27)の合成を示す。
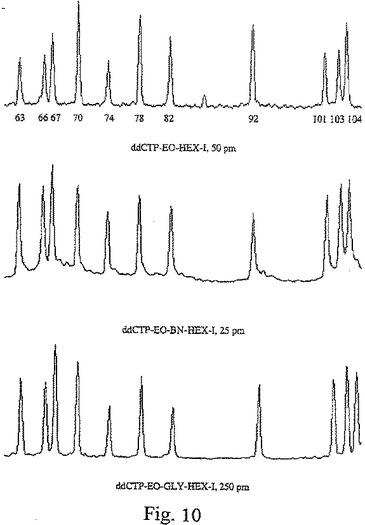
【図10】図10は、3個の異なる色素ターミネーター配列決定反応から得た結果を示し、ここで、各反応は、色素をターミネーターに結合する異なるリンカーを使用した。
【発明を実施するための形態】
【0049】
(好ましい実施態様の詳細な説明)
ここで、本発明の好ましい実施態様に対して、詳細な参照がなされ、その実施例は、添付の図面に例示されている。本発明は、その好ましい実施態様に関連して記述されているものの、それらは、本発明をこれらの実施態様に限定する意図はないことが分かる。反対に、本発明は、その代替、改変および等価物を含むことを意図しており、これらは、添付の請求の範囲で規定される本発明の範囲内に含まれ得る。
【0050】
一般に、本発明は、ポリメラーゼ酵素に対して基質として有用な新規な種類の置換プロパルギルエトキシアミドヌクレオシド化合物、このようなヌクレオシド化合物を含むポリヌクレオチド、およびプライマー伸長反応においてこのようなヌクレオシド化合物を使用する方法を包含する。本発明の化合物は、Sanger型DNA配列決定法で使用する色素標識ヌクレオチド鎖終止試薬の調製、およびプライマー伸長反応(例えば、PCR)を含む方法で使用する色素標識ヌクレオチド鎖伸長試薬の調製において、特定の用途が見出されている。
【0051】
本発明は、一部には、目的の置換プロパルギルエトキシアミドヌクレオチドが、熱安定性DNAポリメラーゼ酵素用の特に良好な基質であること、すなわち、(i)Sanger型DNA配列決定反応にて、現在入手できる標識ターミネーターを使用したときに必要なものと比較して、著しく低いモル過剰しか必要としないこと、および(ii)Sanger型DNA配列決定プロセスにて、現在入手できる標識ターミネーターを使用したときに見られるものと比較して、ピーク高さのより均一な分布が見られることの発見に基づいている。
【0052】
I.定義
他に述べられていなければ、本明細書中で使用する以下の用語および語句は、以下の意味を有することを意図している:
「低級アルキル」との用語は、1個〜8個の炭素原子を含有する直鎖および分枝炭化水素部分(すなわち、メチル、エチル、プロピル、イソプロピル、tert−ブチル、イソブチル、sec−ブチル、ネオペンチル、tert−ペンチルなど)を表わす。
【0053】
「標識」との用語は、本発明のヌクレオシドに結合したとき、このようなヌクレオシドおよびこのようなヌクレオチドを含有するポリヌクレオチドを、公知の検出手段を用いて検出可能にする部分を意味する。例示の標識には、蛍光体、発色団、放射性同位元素、スピン標識、酵素標識、化学発光標識など(これらは、適切な検出器による標識化合物の直接的な検出を可能にする)、またはリガンド、例えば、抗原またはビオチン(これは、検出可能な抗リガンド(例えば、標識抗体またはアビジン)に、高い親和性で特異的に結合できる)が挙げられる。好ましくは、これらの標識は、蛍光色素(例えば、フルオレセイン型色素またはローダミン型色素(Lee;Menchen))である。
【0054】
「ヌクレオシド」との用語は、2’−デオキシ形状および2’−ヒドロキシル形状を含めて、その1’位でペントースに結合したプリン、デアザプリンまたはピリミジンヌクレオシド塩基(例えば、アデニン、グアニン、シトシン、ウラシル、チミン、デアザアデニン、デアザグアノシンなど)からなる化合物を意味する(Stryer)。本明細書中で使用する「ヌクレオチド」との用語は、ヌクレオシドのリン酸エステル(例えば、三リン酸エステル)を意味し、ここで、最も一般的なエステル化部位は、このペントースのC−5位に結合した水酸基である。多くの場合、本発明の開示では、ヌクレオシドとの用語は、ヌクレオシドおよびヌクレオチドの両方を包含することを意図している。ヌクレオシドに関する「アナログ」には、例えば、他の文献で記述のように(Scheit;Eckstein 1991)、変性塩基部分、変性糖部分および/または変性リン酸エステル部分を有する合成アナログが挙げられる。
【0055】
本明細書中で使用する「ポリヌクレオチド」または「オリゴヌクレオチド」との用語は、天然ヌクレオチドモノマーまたはそれらのアナログの線状ポリマーを意味し、これには、二本鎖および一本鎖のデオキシリボヌクレオチド、リボヌクレオチド、それらのα−アノマー形状などが含まれる。通常、このヌクレオシドモノマーは、ホスホジエステル連結により連結されており、本明細書中で使用する「ホスホジエステル連結」との用語は、ホスホジエステル結合またはそれらのホスフェートアナログを含む結合を意味し、これらには、会合した対イオンが存在するなら、このような対イオン(例えば、H、NH4、Naなど)を含む。このポリヌクレオチドの大きさは、典型的には、数個(例えば、8個〜40個)のモノマー単位から、数千個のモノマー単位までに及ぶ。ポリヌクレオチドが「ATGCCTG」のような一連の文字で表わされるときは、このヌクレオチドは、他に指示がなければ、5’→3’の順序で左から右に配列されており、「A」は、デオキシアデノシンを表わし、「C」は、デオキシシチジンを表わし、「G」は、デオキシグアノシンを表わし、そして「T」は、チミジンを表わすことが分かる。
【0056】
「ホスフェートアナログ」との用語は、ホスフェートのアナログであって、ここでそのリン原子が+5酸化状態であり、そしてその酸素原子の1個またはそれ以上が非酸素部分で置き換えられているものを意味し、例示のアナログには、ホスホロチオエート、ホスホロジチオエート、ホスホロセレノエート、ホスホロジセレノエート、ホスホロアニロチオエート、ホスホロアニリデート、ホスホロアミデート、ボロノホスフェートなど(会合した対イオン(例えば、H、NH4、Naなど)があれば、このような対イオンを含めて)が挙げられる。
【0057】
本明細書中で使用される「プロパルギルアミドリンカー」との用語は、以下の構造を有するリンカーを意味する:
【0058】
【化7】
また、「プロパルギルエトキシアミドリンカー」との用語は、以下の構造を有するリンカーを意味する:
【0059】
【化8】
ここで、上記構造のそれぞれについて、そのアセチレンの終止末端は、ヌクレオチド塩基に結合しており、そしてそのアミド窒素は、標識との好都合な連結を介して、結合される。
【0060】
本発明の「X」部分に関連して、「アミノアルカン酸」との用語は、以下の構造を意味する:
【0061】
【化9】
ここで、nは、1〜5の範囲である;「アルキルアミノ安息香酸」との用語は、以下の構造を意味する:
【0062】
【化10】
ここで、nは、1〜5の範囲である;「α−アミノ酸」との用語は、以下の構造を意味する:
【0063】
【化11】
そして「4−アミノ−2−ブチン酸」との用語は、以下の構造を意味する:
【0064】
【化12】
ここで、上記構造では、R1およびR2は、別個に、−H、低級アルキル、保護基または標識であり、そしてR4は、アミノ酸側鎖(天然(Stryer)または合成のいずれか)である。
【0065】
「プロパルギルエトキシアミドヌクレオチド」との用語は、ヌクレオチドのヌクレオ塩基に結合したプロパルギルエトキシアミドリンカーを含むヌクレオチドを意味する。
【0066】
「フルオレセイン型色素」との用語は、以下の縮合三環系を含有する種類のキサンテン色素分子を意味する:
【0067】
【化13】
ここで、各デオキシ環位置では、広範囲の置換が可能である。フルオレセイン型色素の特に好ましいサブセットには、4,7−ジクロロフルオレセイン(Menchen)が挙げられる。DNA配列決定法で蛍光標識として使用されるフルオレセイン型色素の例には、6−カルボキシフルオレセイン(6−FAM)、5−カルボキシフルオレセイン(5−FAM)、6−カルボキシ−4,7,2’,7’−テトラクロロフルオレセイン(TET)、6−カルボキシ−4,7,2’,4’,5’,7’−ヘキサクロロフルオレセイン(HEX)、5−(および6)カルボキシ−4’,5’−ジクロロ−2’,7’−ジメトキシフルオレセイン(JOE)、および5−カルボキシ−2’,4’,5’,7’−テトラクロロフルオレセイン(ZOE)が包含される。多くの場合、−1または−2との名称は、特定の色素(例えば、HEX−1)の略語の後で、付けられる。この「−1」および「−2」との名称は、使用する特定の色素異性体を意味する。これらの1および2異性体は、C−8カラムおよび溶出勾配(15%アセトニトリル/85% 0.1 M酢酸トリエチルアンモニウム〜35%アセトニトリル/65% 0.1 M酢酸トリエチルアンモニウム)を使用する逆相クロマトグラフィー分離系にて、遊離色素の溶出順序(この1異性体が最初に溶出する)により、定義される。
【0068】
「ローダミン型色素」との用語は、以下の縮合三環系を含有する種類のキサンテン色素分子を意味する:
【0069】
【化14】
ここで、好ましくは、Y1〜Y4は、別個に、水素または低級アルキルであり、または一緒になって、Y1およびR2は、プロパノであり、そしてY2およびR1は、プロパノであるか、または一緒になって、Y3およびR3は、プロパノであり、そしてY4およびR4は、プロパノである。R1〜R4位置を含めた各デオキシ環位置にて、広範囲の置換が可能である。ヌクレオシド標識として有用な例示のローダミン型色素には、テトラメチルローダミン(TAMRA)、4,7−ジクロロテトラメチルローダミン(DTAMRA)、ローダミンX(ROX)、ローダミン6G(R6G)、ローダミン110(R110)などが挙げられる(Bergot;Lee)。
【0070】
本明細書中で使用する「FLAN色素」との用語は、次式を有する非対称ベンゾキサンテン色素化合物を意味する:
【0071】
【化15】
ここで、Y1およびY2は、別個に、ヒドロキシル、酸素、イミニウムまたはアミンである。R1〜R8は、別個に、水素、フッ素、塩素、低級アルキル、低級アルケン、低級アルキン、スルホネート、アミノ、アンモニウム、アミド、ニトリル、アルコキシ、連結基、またはそれらの組合せである。そして、R9は、アセチレン、アルカン、アルケン、シアノ、置換フェニル、またはそれらの組合せ、以下の構造を有する置換フェニルである:
【0072】
【化16】
ここで、X1は、カルボン酸またはスルホン酸である;X2およびX5は、別個に、水素、塩素、フッ素または低級アルキルである;そしてX3およびX4は、別個に、水素、塩素、フッ素、低級アルキル、カルボン酸、スルホン酸または連結基である(Benson)。
【0073】
本明細書中で使用する「プライマー伸長試薬」との用語は、オリゴヌクレオチドプライマーの酵素的テンプレート媒介の伸長を起こすのに必要な成分を含有する試薬を意味する。プライマー伸長試薬には、以下が挙げられる:(i)ポリメラーゼ酵素、例えば、熱安定性ポリメラーゼ酵素(例えば、Taqポリメラーゼ);(ii)緩衝液;(iii)鎖伸長ヌクレオチド、例えば、デオキシヌクレオチド三リン酸(例えば、ジオキシグアノシン5’−三リン酸、7−デアザデオキシグアノシン5’−三リン酸、デオキシアデノシン5’−三リン酸、デオキシチミジン5’−三リン酸、デオキシシチジン5’−三リン酸);および必要に応じて、Sanger型DNA配列決定反応の場合に、(iv)1個またはそれ以上の鎖終止ヌクレオチド、例えば、ジデオキシヌクレオチド三リン酸(例えば、ジデオキシグアノシン5’−三リン酸、7−デアザジデオキシグアノシン5’−三リン酸、ジデオキシアデノシン5’−三リン酸、ジデオキチミジン5’−三リン酸、およびジデオキシシチジン5’−三リン酸)。
【0074】
「テンプレート核酸」とは、単一鎖形状で提示できる任意の核酸であって、プライマーオリゴヌクレオチドでアニールできるものを意味する。例示のテンプレート核酸には、DNA、RNAが挙げられ、このDNAまたはRNAは、一本鎖または二本鎖であり得る。さらに特定すると、テンプレート核酸は、ゲノムDNA、メッセンジャーRNA、cDNA、PCR反応に由来のDNA増幅生成物などであり得る。テンプレートDNAの調製方法は、他の文献に見られ得る(ABI PRISMTM Dye Primer Cycle Sequencing Core Kit)。
【0075】
II.置換プロパルギルエトキシアミドヌクレオチド化合物
第一の局面では、本発明は、すぐ下の式Iで示す一般構造を有する新規なクラスの置換プロパルギルエトキシアミドヌクレオチド化合物を包含する。(本開示全体で提供された全ての分子構造は、提示した正確な電子構造だけでなく、それらの全ての共鳴構造およびプロトン化状態を含むことを意図することに留意のこと)。
【0076】
【化17】
式Iの構造では、Bは、7−デアザプリン、プリンまたはピリミジンヌクレオチド塩基であり、好ましい実施態様では、Bは、ウラシル、シトシン、7−デアザアデニンおよび7−デアザグアノシンからなる群から選択される。Bがプリンまたは7−デアザプリンのとき、このヌクレオチドの糖部分は、このプリンまたはデアザプリンのN9位にて結合され、Bがピリミジンのとき、その糖部分は、このピリミジンのN1位にて結合される。Bがプリンのとき、隣接三重結合炭素は、このプリンの8位に結合され、Bが7−デアザプリンのとき、隣接三重結合炭素は、7−デアザプリンの7位に結合され、Bがピリミジンのとき、隣接三重結合炭素は、このピリミジンの5位に結合される。
【0077】
W1は、−Hおよび−OHから選択される。W1が−OHであるとき、このヌクレオシドは、リボヌクレオチドであり、そしてW1が−Hであるとき、このヌクレオシドは、デオキシリボヌクレオチドである。
【0078】
W2は、−OH、またはこのヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である。この機能に有用な好ましい部分には、−H、アジド、アミノ、ハロ、メトキシなどが挙げられる。特に好ましい実施態様では、W2は、−Hまたはフルオロである。
【0079】
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。ポリヌクレオチドの酵素的合成に有用な好ましい実施態様では、W3は−P3O10である。
【0080】
Xは、アミノアルカン酸、アルキルアミノ安息香酸、α−アミノ酸、および4−アミノ−2−ブチン酸からなる群から選択される。好ましくは、Xは、アルキルアミノ安息香酸である。Xがアミノアルカン酸またはアルキルアミノ安息香酸のとき、nは、1〜5の範囲であり、さらに好ましくは、nは、1〜3の範囲である。Xがアルキルアミノ安息香酸のとき、そのフェニル基は、その隣接するカルボニル基およびメチレン基に関して、メタ、パラまたはオルト配置であり得る。好ましい実施態様では、フェニル基は、パラ配置である。Xがα−アミノ酸のとき、アミノ酸側鎖は、任意の適切な天然または合成のアミノ酸側鎖であり得る。
【0081】
上記X部分のうちの任意のものについて、R1およびR2は、−H、低級アルキル、保護基または標識から選択される。好ましくは、標識は、蛍光色素である。より好ましくは、標識は、フルオレセイン型蛍光色素、ローダミン型蛍光色素またはFLAN型蛍光色素である。好ましくは、R1およびR2の一方が標識のとき、他方は、−Hまたは低級アルキルのいずれかである。好ましい保護基には、ハロアセチル、アシル、アルコキシカルボニルまたはスルホニルが挙げられる。より好ましくは、この保護基は、トリフルオロアセチルである。
【0082】
この標識は、置換プロパルギルエトキシアミドヌクレオシドの第一級または第二級アミノ部分と、標識上に位置している「相補的官能性」との反応により、典型的に形成される「連結」を介して、このヌクレオシドに結合される。好ましくは、この相補的官能性は、イソチオシアネート、イソシアネート、アシルアジド、N−ヒドロキシスクシンイミド(NHS)エステル、スルホニルクロライド、アルデヒドまたはグリオキサール、エポキシド、カーボネート、ハロゲン化アリール、イミドエステル、カルボジイミド、無水物、4,6−ジクロロトリアジニルアミンまたは他の活性カルボキシレートである(Hermanson)。特に好ましい実施態様では、この相補的官能性は、活性化NHSエステルであり、これは、本発明の置換プロパルギルエトキシアミドヌクレオシドのアミンと反応し、この場合、活性化NHSエステルを形成するには、カルボキシレート相補的官能性を含む標識は、ジシクロヘキシルカルボジイミドおよびN−ヒドロキシスクシンイミドと反応されて、NHSエステルを形成する(Khanna;Kasai)。以下の表1は、代表的な相補的官能性およびこの相補的官能性とこの置換プロパルギルエトキシアミドヌクレオシドのアミンとの反応により形成されて得られた連結のサンプリングを示す。
【0083】
【表1】
R3は、−Hおよび低級アルキルからなる群から選択される。
【0084】
III.置換プロパルギルエトキシアミドヌクレオチドおよび標識ヌクレオチドの合成
図2〜9は、本発明の置換プロパルギルエトキシアミドヌクレオシドのいくつかの代表的な合成を示す。この合成では、ブロモエタノール1は、フタルイミドカリウム2と反応して、フタルイミド誘導体3が得られる。このフタルイミド誘導体は、次いで、NaHの存在下にて、プロパルギルブロマイド4でO−アルキル化されて、保護した3−(2−フタルイミドエトキシ)プロピン連結アーム5が得られる。図2を参照せよ。次いで、ヨードヌクレオシド6は、ジメチルホルムアミド中、ヨウ化第一銅、テトラキス(トリフェニルホスフィン)パラジウム、およびトリエチルアミンの存在下にて、室温でおよそ12時間またはこの反応がTLCで決定されるように完結するまで、保護連結アームと反応されて、それにより、化合物7が形成される。この溶液は、次いで、真空下で濃縮され、生成物は、シリカゲルフラッシュクロマトグラフィーにより精製され、そして同定および純度測定のために、プロトンNMRおよび分析用逆相HPLCにより分析される。エチレンジアミンでの処理に続いて、トリフルオロ酢酸エチルでのアセチル化により、ヌクレオシド連結アーム化合物8が得られる。図3を参照せよ。リン酸トリメチル中にて、−30℃で、このヌクレオシド連結アーム化合物8に、新たに蒸留したオキシ塩化リンを添加して、対応するジクロロモノホスフェート9を形成する。反応混合物を2M重炭酸テトラエチルアンモニウム(TEAB)(pH 8.0)でクエンチして、モノホスフェート10を得、これは、次いで、分取逆相HPLCにより精製される。図4を参照せよ。モノホスフェート10は、カルボニルジイミダゾール(CDI)で活性化され、過剰のCDIは、MeOHでクエンチされて、活性化モノホスフェート11が得られる。この活性化モノホスフェートは、室温で、ピロリン酸トリブチルアンモニウムと反応される。この反応は、完結したとき、0.2M TEABでクエンチされ、そして逆相HPLCにより精製されて、保護トリホスフェート12が得られる。精製した保護トリホスフェートは、乾燥するまでエバポレートされ、そして濃NH4OH水溶液中に再懸濁されて、TFA基が除去され、プロパルギルエトキシアミドヌクレオチド13が得られる。図5を参照せよ。
【0085】
一般に、X部分のプロパルギルエトキシアミドヌクレオチド(例えば、13)への結合は、以下のようにして行われる。X部分のアミン官能性を保護するために、X部分とアミン保護試薬(例えば、トリフルオロアセテート、例えば、図6A〜Cの化合物14、17または20)とを反応させることにより、アミン保護X部分が形成される。アミン保護X部分の酸官能性を活性化するために、アミン保護X部分は、酸活性化剤(例えば、N−ヒドロキシスクシンイミド、例えば、図6A〜Cの化合物15、18または21)と反応される。次に、酸活性化されたアミン保護X部分は、塩基条件下(例えば、250 mMの重炭酸塩緩衝液、pH 9)で、プロパルギルエトキシアミドヌクレオチドと反応されて、アミン保護された置換プロパルギルエトキシアミドヌクレオチド(例えば、図7の化合物22、図8の化合物24、または図9の化合物26を参照のこと)を生じる。最後に、アミン保護基は、強塩基(例えば、NH4OH)との反応により、アミン保護された置換プロパルギルエトキシアミドヌクレオチドから除去されて、置換プロパルギルエトキシアミドヌクレオチド(例えば、図7の化合物23、図8の化合物25、または図9の化合物27を参照のこと)が得られる。
【0086】
一般に、好ましい方法では、本発明の標識した置換プロパルギルエトキシアミドヌクレオシドは、以下のようにして調製される。置換プロパルギルエトキシアミドヌクレオシド(例えば、23、25または27)は、中性緩衝溶液(例えば、100 mM TEAB(pH 7.0))に溶解され、溶液を乾燥状態までエバポレートされ、そして、ヌクレオシドを塩基性緩衝溶液(例えば、250 mM重炭酸ナトリウム緩衝液、pH=9)に再懸濁する。この溶液に、標識−NHSエステル(DMSO中)が添加され、そして撹拌しながら、一晩反応される。完結すると、この反応混合物は、イオン交換および逆相HPLCにより精製されて、標識した置換プロパルギルエトキシアミドヌクレオシドが得られる。
【0087】
IV.プロパルギルエトキシアミド化合物を使用する方法
本発明のプロパルギルエトキシアミド化合物は、以下の工程を包含するタイプのテンプレート媒介プライマー伸長反応を含めた方法での使用によく適合する:(i)テンプレート核酸を提供する工程;(ii)このテンプレート核酸の一部に、オリゴヌクレオチドプライマーをアニーリングし、それにより、プライマー−テンプレートハイブリッドを形成する工程;および(iii)このプライマーを伸長するために、このプライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加する工程。特に、本発明の化合物は、プライマー伸長生成物に標識を直接含入させる手段を提供する。
【0088】
プライマー伸長反応を使用する第一の好ましい種類の方法では、この伸長生成物は、本発明の標識した置換プロパルギルエトキシアミドヌクレオチドを、プライマー伸長反応物に含有させ、それにより、この伸長生成物全体にわたって、標識をランダムに含入させることにより、標識される([F]dNTP試薬プロトコル)。このような方法は、PCR単位複製配列、および単一のプライマー誘導伸長生成物を標識するのに使用できる。このようにして伸長生成物を標識するために、プライマー伸長反応は、確立したプロトコルを用いて行われるが、この反応系には、標識した置換プロパルギルエトキシアミドヌクレオチドが添加される。一般に、PCRの状況で、プライマー伸長反応を行うためには、テンプレート核酸は、各プライマー20 pmolと、プライマー伸長試薬(これは、20 mMの緩衝液(pH 8)、1.5 mMのMgCl2、50 mMの各デオキシヌクレオチド三リン酸(dNTP)、および2単位のTaqポリメラーゼまたは他の適切な熱安定性ポリメラーゼを含有する)と混合される。次いで、反応混合物を熱サイクルにかけるが、典型的な熱サイクルプロフィールは、変性工程(例えば、96℃で15秒間)、プライマーアニーリング工程(例えば、55℃で30秒間)、およびプライマー伸長工程(例えば、72℃で90秒間)を包含する。典型的には、この熱サイクルは、約10〜40サイクル繰り返される。PCR増幅のためには、標識デオキシヌクレオチド三リン酸と非標識デオキシヌクレオチド三リン酸との典型的な比は、所望のシグナル量に依存して、100:1〜1000:1の間である。増幅効率に悪影響を起こすことなくPCR反応混合物中にて使用できる標識デオキシヌクレオチド三リン酸と非標識デオキシヌクレオチド三リン酸との最大比は、およそ1:4である。
【0089】
プライマー伸長反応を使用する第二の好ましい種類の方法では、この伸長生成物は、プライマー伸長反応物に本発明の置換プロパルギルエトキシアミドヌクレオチドを含有させて、それにより、伸長生成物の3’−末端ヌクレオチドにて、検出可能な標識をランダムに含入させることにより標識される(例えば、Sanger型DNA配列決定)。一般に、本発明の標識ジデオキシヌクレオチド三リン酸を用いたSanger型DNA配列決定の状況でプライマー伸長反応を行うためには、1μlのテンプレート溶液(水5μlで希釈したPCR反応物1μl)および2μlのプライマー(0.4 pmol/μl)は、プライマー伸長試薬(これは、2μlの緩衝液(400 mM Tris−HCl、10 mM MgCl2、pH 9.0)、2μlのデオキシヌクレオチド/標識ジデオキシヌクレオチド混合物(T−終止反応、1250μM ddTTP、250μM dATP、250μM dCTP、180μM 7−デアザ−dGTP、および250μM dTTP)、および2μlのポリメラーゼ酵素(5単位/μlであって、この場合、1単位は、Lawyerのように定義される)を含有する)と混合される。次いで、反応物は、以下の代表的なプログラムを用いて、熱サイクルにかけられる:98℃で5秒間の変性に続いて、96℃で5秒間;55℃で40秒間;60℃で1分間のサイクルの繰り返し、このサイクルは、およそ15回繰り返される。
【0090】
本発明の置換プロパルギルエトキシアミドヌクレオシド化合物はまた、プライマー伸長生成物の塩基特異的な開裂に依存したSanger型配列決定の変形法(例えば、標識ヌクレオチド使用する方法)の状況にて、よく適合する(Eckstein 1988;Shaw)。
【実施例】
【0091】
本発明は、以下の実施例を考慮することにより、さらに明らかにされるが、これらの実施例は、本発明の純粋な例示とすることを意図しており、いずれの様式でも、その範囲を限定することを意図していない。
【0092】
(実施例1)
配列決定反応に必要なターミネーター過剰を決定するためのターミネーター滴定アッセイ
完全な配列決定ラダー(ladder)(すなわち、約20ヌクレオチドと約600ヌクレオチドの間の長さを有する特定の塩基で終止する全てのフラグメントを含有する配列決定ラダー)を作るのに必要な最小量の色素標識ターミネーターを決定するために、ターミネーター滴定アッセイを使用した。このターミネーター滴定アッセイの不可欠な成分は、(i)第一色素で標識したプライマー、および(ii)この第一色素とスペクトル分解可能な第二色素で標識したターミネーターであった。このアッセイでは、この配列決定反応物に、不充分な濃度の色素ターミネーターを添加したとき、ジデオキシ終止フラグメントは形成されず、配列決定ゲル上では、第一色素のみで標識した「偽スポット」により形成された生成物だけが見られた。本明細書中で使用する「偽スポット」との用語は、ジデオキシターミネーターで終止していないプライマー伸長生成物を意味し、このような生成物は、おそらく、ポリメラーゼ酵素がテンプレート核酸鎖から自発的に分離するときに形成される。多すぎるターミネーターを使用したときには、短い終止生成物(すなわち、長さが約50個未満のヌクレオチド)しか形成されず、このような生成物は、第一色素および第二色素の両方を含有していた。適切な量のターミネーターを使用したとき、完全な配列決定ラダーが生成し、ラダーの各フラグメントは、第一色素および第二色素の両方で標識されていた。
【0093】
色素−ターミネーター反応は、ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Manual(PE Applied Biosystems p/n 402116)で提供されたプロトコルに従ったAmpliTaq DNA Polymerase、FSを用いて、行った。(このFS酵素は、2点変異−−G46DおよびF667Yを有する組み換えThermus水生DNAポリメラーゼである)。dNTPミックス、色素標識プライマーおよび色素標識ターミネーター以外の全ての試薬は、ABI PRISMTM Dye Terminator Core Kit(PE Applied Biosystems p/n 402117)から得た。dNTPミックスは、各2 mMのdATP、dCTP、7−デアザ−dGTPおよびdTTPからなっていた。反応成分のプレミックスを、以下の表に示すように調製し、ここで、全ての量は、反応ベースあたりで示されている:
【0094】
【表2】
反応物を、Perkin−Elmer 480 DNA Thermal Cycler(PE Applied Biosystems p/n N801−100)に適合した0.5 ml微量遠心分離管に組み込んだ。反応物の容量は20μLであり、これには、上記反応プレミックス15μL、可変量の色素標識ターミネーター、および全反応容量を20μLにするのに充分な量の水を含む。各反応物に、1〜500 pmolの色素標識ターミネーターを添加した。蒸発を防止するために、各反応物の頂部に、鉱油30μLを添加した。これらの反応物を、以下のようにして熱サイクルにかけた:96℃で30秒間、50℃で15秒間、および60℃で4分間を25サイクルに続いて、4℃で保持するサイクル。
【0095】
全ての反応物を、製造業者の指示(Princeton Separations p/n CS−901)に従って、Centri−Sepスピンカラム上のスピン−カラム精製により精製した。このカラム内のゲル物質は、室温で少なくとも30分間にわたり、脱イオン水0.8 mLで水和させた。カラムが水和され、ゲル物質に気泡が取り込まれていないことを確認した後、カラムの上部および下部の末端キャップを取り除き、カラムを、重力により排水した。次いで、カラムを、キットに備え付けた洗浄管に挿入し、そして可変速度微量遠心分離機で、1300xgで2分間にわたり遠心分離し、洗浄管を取り除き、そして試料収集管に挿入した。この反応混合物を、オイルの下部から注意深く取り出し、そしてゲル物質上に充填した。カラムを、可変速度微量遠心分離で、1300xgで2分間遠心分離した。次いで、溶出した試料を、真空遠心分離で乾燥した。
【0096】
配列決定ゲル上に充填する前に、乾燥した試料を、25μLのTemplate Suppression Reagent(PE Applied Biosystems p/n 401674)中に再懸濁させ、ボルテックスし、2分間にわたって95℃まで加熱し、氷上で冷却し、再びボルテックスし、そして遠心分離した(13,000xg)。再懸濁した試料10μLを、PE ABI PRISMTM 310 Genetic Analyzer(PE Applied Biosystems p/n 310−00−100/120)に適合させた試料バイアル(PE Applied Biosystems p/n 401957)に等分した。310 Genetic Analyzerでの電気泳動は、DNA配列決定分析に特別に適合させたふるい分けポリマーおよび毛細管(PE Applied Biosystems p/n 402837(ポリマー)およびp/n 402840(毛細管))を用いて行った。各場合では、ふるい分けポリマーは、核酸変性物を含んでいた。試料は、2.5 kVで30秒間、毛細管に動電的に注入し、そしてこの毛細管の外壁を50℃に維持しつつ、12.2 kVで2時間走査した。
【0097】
図1A〜Cは、Terminator Titration Assayから得た典型的な結果を示し、この場合、プライマーはTAMRA色素で標識され、ddATPターミネーターは、6−FAM色素で標識され、TAMRA色素をこのジデオキシヌクレオチドターミネーターに連結するために、伝統的なプロパルギルアミドリンカーを使用した。これらのトレースは、ヌクレオチド71〜175に対する電気泳動走査時間の関数として、一定波長での蛍光強度を示す。プライマー伸長反応物に添加した色素−ターミネーターの量は、可変であった:図1Aでは、1 pmolのターミネーターを使用し、図1Bでは、4 pmolのターミネーターを使用し、そして図1Cでは、150 pmolのターミネーターを使用した。各パネルの上のトレースは、色素標識ターミネーターで発光し、535〜545 nmで集めた蛍光であり、各パネルの下のトレースは、色素標識プライマーで発光し、575〜585 nmで集めた蛍光である。色素プライマートレース(下)は、適切に終止したフラグメントでけでなく、偽ストップ(false stop)(すなわち、色素標識ターミネーターで終止していないフラグメント)も示している。色素ターミネータートレース(上)は、色素標識ターミネーターの特異的な取り込みを示している。
【0098】
図1Aは、1 pmolの6−FAM−ddATPターミネーターを用いる反応についてのデータを示す。この色素ターミネータートレースでは、小さなピークにより明らかなように、極く僅かな特異的混入が検出された。この色素−プライマートレースで示された偽ストップは、任意の特異的に終止したピークとも実質的に同じ大きさであった。このパターンから、この色素−ターミネーター濃度が非常に低いことが明らかとなった。図1Bは、4 pmolの6−FAM−ddATPターミネーターを用いた反応についてのデータを示す。このターミネータートレースでは、この配列決定ラダー全体を通じて、比較的に一定のピーク高さで、良好な特異的ターミネーター混入が認められた。この色素プライマートレースでは、偽ストップノイズの上部に容易に識別できるピークが存在しており、これらのピークは、色素ターミネータートレースのピークと共に移動していた。このパターンは、この色素ターミネーター濃度は、使用可能な範囲内であったことを示す。図1Cは、150 pmolの6−FAM−ddATPターミネーターを用いた反応についてのデータを示す。「トップヘビー(top heavy)」パターンが見られ、色素ターミネータートレースの初期のピークが、非常に高レベルの色素ターミネーター混入を示し、その後のピークは、ずっと低いレベルの混入を示していた。このパターンは、この色素−ターミネーター濃度が高すぎたことを示している。
【0099】
(実施例2)
リンカー型の関数として、完全配列決定ラダーを形成するのに必要なHEX−1−標識したC−ターミネーターの量
以下の表は、実施例1にて上で記述のターミネーター滴定アッセイに従って、完全配列決定ラダーを形成するのに必要な色素標識したC−ターミネーターの相対モル過剰(molar excess)を示す。この相対モル過剰は、完全配列決定ラダーを形成するのに必要な非標識ジデオキシターミネーターの量が1の値となるように、定義される。各場合では、C−ターミネーターは、HEX−1色素に連結した。
【0100】
【表3】
a この相対モル過剰は、完全配列決定ラダーを形成するのに必要な非標識ジデオキシターミネーターの量が1.0の値となるように定義される。
【0101】
(実施例3)
5−{3−(2−アミノエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成
A.物質および方法
薄層クロマトグラフィー(TLC)を、250μm層のシリカゲル60−F254で予め塗装したガラス板上で行った。蛍光のクエンチおよび/または5%硫酸での炭化により展開した後、化合物をTLC板上に配置した。SIPブランドシリカゲル60Å、230〜400 Mesh ASTM(Baxter Scientific p/n C4582−87)上で、フラッシュカラムクロマトグラフィーを行った。以下のようにして、NMRスペクトルを得た:1H NMRスペクトルを、室温で、CDCl3(内部Me4Si、δ0)またはD2O(外部Me4Si、δ0)に溶解した溶液で300 MHzにて記録した;13C NMRスペクトルを、CDCl3(内部Me4Si、δ0)に溶解した溶液で、75.5 MHzにて記録した;19F NMRスペクトルを、CDCl3またはD2O(外部CFCl3、δ0)に溶解した溶液で、282.23 MHzにて記録した;そして31P NMRスペクトルを、D2O中の溶液で、121.44 MHzにて記録した。全ての場合において、NMRデータは、提案した構造に一致していた。他に指示がなければ、全ての反応は室温で行い、後処理において、有機溶媒の溶液を、等容量の水溶液で洗浄した。有機溶液を、40〜50℃の浴温を用いた真空下でのロータリーエバポレーターでの濃縮の前に、無水Na2SO4で一般的に乾燥した。分析および分取目的で使用したHPLC系は以下の通りであった:
分析用逆相HPLC:カラム:Spheri−5 RP−C18、5μmの粒子サイズ、220×4.6 mm(PE Applied Biosystems p/n 0711−0017);勾配:0〜50%アセトニトリルで1.5 ml/分にて20分間に続いて、50%アセトニトリル〜100%アセトニトリルで1.5 ml/分にて10分間。
【0102】
分析用イオン対HPLC:カラム:AquaporeTM OD−300、7μmの粒子サイズ、220×4.6 mm(PE Applied Biosystems p/n 0711−0331);勾配:0〜40%アセトニトリルで1.5 ml/分にて30分間に続いて、40%アセトニトリル〜60%アセトニトリルで1.5 ml/分にて5分間。
【0103】
分取アニオン交換HPLC:カラム:AquaporeTM Anion、20μmの粒子サイズ、250×10 mm(PE Applied Biosystems p/n 0711−0172);勾配:40%アセトニトリル:60% 100 mM TEAB、pH 7.0〜40%アセトニトリル:60% 1.5 mM TEAB pH 8で4.5 ml/分にて20分間に続いて、アイソクラチック(isocratic)溶出。
【0104】
分取逆相HPLC:カラム:Prep Nova Pak HR−C18、6μmの粒子サイズ、60Åの細孔サイズ、300×40 mm(Waters Division of the Millipore Corporation p/n WAT037704);勾配(モノホスフェートおよびトリホスフェートについて):100% 100 mM TEAB、pH 7〜20%アセトニトリル:80% 100 mM TEAB pH 7で50 ml/分にて30分間に続いて、20%アセトニトリル:80% 100 mM TEAB pH 7〜50%アセトニトリル:50% 100 mM TEAB pH 7で10分間;勾配(色素標識三リン酸について):100% 100 mM TEAB pH 7〜10% 100 mM TEAB pH 7:90%アセトニトリル。
【0105】
B.2−フタルイミドエタノール(3)の合成
フタルイミドカリウム2(2.7g、14.6 mmol)を、ブロモエタノール1のN,N−ジメチルホルムアミド溶液(12 mL、14.1 mmol)に添加した。70℃で12時間撹拌した後、この混合物を濃縮し、次いで、ジクロロメタン(100 mL)で希釈した。濾過により固形分を除去した後、その有機層を水で洗浄し、乾燥し、そして濃縮した。この濃縮物をフラッシュカラムクロマトグラフィー(3:2〜2:3ヘキサン−酢酸エチル)により精製して、0.22のRF(3:2のヘキサン−酢酸エチル)を有する白色固形分(1.19g、44.12%)として、化合物3を得た。図2を参照せよ。
【0106】
C.3−(2−フタルイミドエトキシ)プロピン(5)の合成
化合物3(1.14g、5.96 mmol)のN,N−ジメチルホルムアミド(20 mL)撹拌溶液に、NaH(0.36g、80%)を滴下した。NaHを完全に添加した後、撹拌を室温で0.5時間継続し、その反応系を0℃まで冷却した。プロパルギルブロマイド4(1.5 mL、13.47 mmol)を添加し、この撹拌を、0℃でさらに0.5時間、次いで、室温で2時間継続した。メタノールを注意深く添加して過剰のNaHを分解した後、溶媒を蒸発させ、その粗生成物をフラッシュカラムクロマトグラフィー(3:2〜1:1〜2:3ヘキサン−酢酸エチル)により精製して、0.22のRF(3:2のヘキサン−酢酸エチル)を有する固形物(495 mg、36.2%)として、化合物5を得た。図2を参照せよ。
【0107】
D.5−{3−(2−フタルアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン(7)の合成
5−ヨード−2’,3’−ジデオキシシチジン6(100 mg、0.3 mmol)を、N,N−ジメチルホルムアミド(1 mL)中、ヨウ化第一銅(11.4 mg、0.06 mmol)、テトラキス(トリフェニルホスフィン)パラジウム(69 mg、0.06 mmol)、およびトリエチルアミン(84μL、0.6 mmol)の存在下にて、アルゴン雰囲気下にて、室温で12時間にわたり、化合物5(158 mg、0.69 mmol)と反応させた。次いで、この反応物を、メタノール中の重炭酸塩形態のDowex−1アニオン交換樹脂2gで希釈した。室温で1時間撹拌した後、この反応混合物を濾過し、そして濃縮した。この生成物を、フラッシュカラムクロマトグラフィー(13:1のジクロロメタン−メタノール)により精製して、0.23のRF(溶媒は9:1のジクロロメタン−メタノール)を有する化合物7(75 mg、57.66%)を得た。図3を参照せよ。
【0108】
E.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン(8)の合成
化合物7(73 mg、0.17 mmol)およびエチレンジアミン(400μL)の混合物を、エタノール(4 mL)中にて、80℃で1時間加熱した。次いで、この反応系を乾燥状態までエバポレートし、その残留物を、N,N−ジメチルホルムアミド(2 mL)に溶解し、トリフルオロ酢酸メチル(6.5 mL)を添加した。80℃で1時間撹拌した後、この溶媒をエバポレートし、その残留物を、フラッシュカラムクロマトグラフィー(溶媒は9:1のジクロロメタン−メタノール)により精製して、0.24のRF(9:1のジクロロメタン−メタノール)を有する化合物8(36 mg、50.7%)を得た。図3を参照せよ。
【0109】
F.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン一リン酸(10)の合成
新たに蒸留したオキシ塩化リン(16.2μL、0.17 mmol)を、−30℃で、リン酸トリメチル(150μL)中のヌクレオシド8(18.8 mg、0.046 mmol)に添加して、対応するジクロロモノホスフェート9を形成した。この反応混合物を、80分間にわたって−5℃まで暖めて、撹拌を、室温でさらに1時間継続した。この反応物を、2 M TEAB緩衝液(pH 8.0)でクエンチし、そして上記のような分取逆相HPLCにより精製した。生成物に対応する画分を濃縮して、モノホスフェート10(12.3 mg、54.56%)を得た。図4を参照せよ。
【0110】
G.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(12)の合成
N,N−ジメチルホルムアミド(200μl)に溶解したモノホスフェート10(7.4 mg、15.3 mmol)を、室温で1時間にわたって、カルボニルジイミダゾール(CDI)(4.2 mg、25.9 mmol)と共に撹拌した。過剰のCDIを、乾燥メタノール(40μL)の添加によりクエンチした。活性化モノホスフェート11を、室温で24時間にわたって、n−トリブチルアミン(16μL)を含有するN,N−ジメチルホルムアミド(160μL)中のピロリン酸トリブチルアンモニウムの溶液と共に撹拌した。この反応物を、2 M TEAB(pH 8.0)でクエンチし、そして上記のような分取逆相HPLCにより精製した。生成物に対応する画分を濃縮して、トリホスフェート12を得た。図5を参照せよ。
【0111】
H.5−{3−(2−アミノエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成
精製し保護したトリホスフェート12を、濃NH4OH水(4 mL)に取り出し、そして室温で2.5時間撹拌した。この反応混合物を濃縮して、プロパルギルエトキシアミドヌクレオチド13を得た。次いで、この濃縮した化合物を、2.6 mMの濃度まで、0.1 M TEAB(pH 7.0)で、バルクとして調合した。調合したバルクの濃度および純度は、それぞれ、UV/Vis分光分析および上記の分析用イオン対HPLCにより、確認した。図5を参照せよ。
【0112】
(実施例4)
5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)[グリシン置換プロパルギルエトキシアミドヌクレオチド]の合成
A.トリフルオロアセトアミドグリシンNHSエステル(15)の合成−−図6A
トリフルオロアセトアミドグリシン14(0.96g、5.6 mmoles)を、室温で1時間にわたって、乾燥DMF(10 mL)中にて、N−ヒドロキシスクシンイミド(0.65g、5.7 mmoles)および1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドメチオジド(methiodide)(1.68g、5.7 mmoles)と反応させた。この反応溶液を酢酸エチル(200 mL)で希釈し、そして0.1 N HCl(2×100 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮して、このトリフルオロアセトアミドグリシンNHSエステル15(1.43g、100%)を得た。
【0113】
B.5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)の合成−−図7
100 mM TEA−重炭酸塩(pH 7.0)中のプロパルギルエトキシアミドヌクレオチド13(200μl、15.2 mM)を、乾燥状態までエバポレートした。次いで、それを、250 mMの重炭酸塩緩衝液(pH 9.0)100μlに再懸濁させた。このトリフルオロアセトアミドグリシンNHSエステル15の溶液(50μl、DMSO中で23% w/w)を添加し、そして室温で一晩撹拌した。次いで、この反応混合物を、以下のようにして、分取逆相HPLC(C−18逆相)により精製した:カラム:Aquapore ODS、20μmの粒子サイズ、250×10 mm(PE Applied Biosystems p/n 0711−0163);勾配:0〜50%アセトニトリルで4.5 ml/分にて20分間に続いて、40%アセトニトリル〜100%アセトニトリルで、4.5 ml/分にて10分間。生成物に対応する画分をプールし、そして等容量の100 mM TEA−重炭酸塩(pH 7.0)で希釈し、次いで、真空中で濃縮して、精製し保護したトリホスフェート22を得た。
【0114】
精製し保護したトリホスフェート22を、濃NH4OH水(2 mL)に取り出し、そして室温で一晩撹拌した。この反応混合物を濃縮して、グリシン置換プロパルギルアミノヌクレオチド23を得た。このグリシン置換プロパルギルアミノヌクレオチド23のバルクを、4.84 mMの濃度まで、0.1 M TEAB(pH 7.0)中へのこの反応混合物の溶解により調合した。調合したバルク溶液の濃度および純度は、UV/Vis分光分析および上記実施例3の分析用逆相HPLCにより、確認した。
【0115】
(実施例5)
5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)[メチルアミノ安息香酸(パラ)置換プロパルギルエトキシアミドヌクレオチド]の合成
A.4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)の合成−−図6B
4−アミノメチル安息香酸16(4g、26.5 mmol)を、DMSO(60 mL)およびトリフルオロ酢酸メチル(3 mL)に溶解し、この溶液に、トリエチルアミン(3 mL)を添加した。60℃で5時間撹拌した後、この反応溶液を酢酸エチル(500 mL)で希釈し、そして水(6×200 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮して、白色の固形物として、トリフルオロアセトアミドメチル安息香酸17(4.6g、70.3%)を得た。
【0116】
このトリフルオロアセトアミドメチル安息香酸17(4.49g、18.2 mmol)を、室温で2時間にわたり、酢酸エチル100 ml中にて、N−ヒドロキシスクシンイミド(2.09g、18.2 mmol)および1,3−ジシクロヘキシルカルボジイミド(3.74g、18.2 mmol)と反応させた。この反応溶液を濾過して、白色沈殿物を除去し、その有機層を1N HCl(100 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮した。この濃縮物を、以下のステップ勾配を用いるシリカゲル固定相を用いた順相カラムクロマトグラフィーにより、クロマトグラフにかけて、白色の固形物(3.5g、56%)として、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル18を得た:40:1のジクロロメタン:酢酸エチルに続いて、10:1のジクロロメタン:酢酸エチル。
【0117】
B.5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)の合成−−図8
100 mM TEA−重炭酸塩(pH 7.0)中のプロパルギルエトキシアミドヌクレオシド13(200μl、15.2 mM)を、乾燥状態までエバポレートした。次いで、それを、250 mM重炭酸塩緩衝液(pH 9.0)150μlに再懸濁させた。4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル18の溶液(100μl、100μlのDMSO中で25 mg)を添加し、そして室温で一晩撹拌した。この反応混合物を、実施例4にて上で記述した分取逆相HPLCにより精製した。生成物に対応する画分をプールし、そして等容量の100 mM TEA−重炭酸塩(pH 7.0)で希釈し、次いで、真空中で濃縮して、精製し保護したトリホスフェート22を得た。
【0118】
この精製し保護したトリホスフェート22を、濃NH4OH水(2 mL)に取り出し、そして室温で一晩撹拌した。この反応混合物を濃縮して、メチルアミノ安息香酸(パラ)置換プロパルギルアミノヌクレオチド25を得た。次いで、この置換プロパルギルアミノヌクレオチド25のバルク溶液を、0.1 M TEAB(pH 7.0)へのこの反応混合物の溶解により調合した。調合したバルク溶液の濃度および純度は、それぞれ、UV/Vis分光分析および上記実施例3の分析用逆相HPLCにより確認した。
【0119】
(実施例6)
置換プロパルギルエトキシアミドヌクレオチドへの色素の結合
100 mM TEA−重炭酸塩(pH 7.0)中の置換プロパルギルエトキシアミドヌクレオチドを、乾燥状態まで蒸発させた。次いで、それを、250 mM重炭酸塩緩衝液(pH 9.0)に再懸濁させた。所望の色素−NHS溶液(DMSO中)を添加し、そして暗所中にて、室温で一晩撹拌した。この反応混合物を、上記の分取アニオン交換HPLCにより精製した。生成物に対応する画分を濃縮し、そして上記の分取逆相HPLCにより再精製した。最終生成物を真空中で乾燥し、そして1 mMの濃度まで、50 mM CAPSO(pH 9.6)で希釈した。調合したバルクの濃度および純度は、それぞれ、UV/VIS分光分析および上記の分析用イオン対HPLCにより確認した。
【0120】
(実施例7)
本発明の置換プロパルギルエトキシアミドジデオキシヌクレオチドを用いて改良したピーク高さ均等性
図10は、HEX−1−標識ddCTPを用いた単色配列決定反応の電気泳動図であり、この場合、このHEX−1色素は、3個の異なるタイプのリンカーのそれぞれを用いて、ターミネーターに結合している。上記ターミネーター滴定アッセイに基づき、各リンカータイプについて、その最適なターミネーター濃度を使用した。上のパネルは、50 pmでのプロパルギルエトキシアミドリンカーを用いた結果を示し、中央のパネルは、25 pmでのメチルアミノ安息香酸(パラ)置換プロパルギルエトキシアミドリンカーを用いた結果を示し、下のパネルは、250 pmでのグリシン置換プロパルギルエトキシアミドリンカーを用いた結果を示す。図10の上の電気泳動図における番号により示されるように、図10で示した配列決定ラダー部分は、−21 M13配列決定プライマー(前方)を用いたpGEM−3Zf(+)の配列の塩基63から塩基104までに及んでいる。各実験の相対誤差は、以下のようであり、この場合、本明細書中で使用する用語「相対誤差」は、この図で示した電気泳動図部分にわたるピーク高さの比または標準偏差および中間ピーク高さ(mean peak height)を意味する。
【0121】
【表4】
全ての文献および特許出願の内容は、個々の文献または特許出願のそれぞれの内容が具体的かつ個別的に本明細書中で参考として援用されているかのように、本明細書中で参考として援用されている。
【0122】
少数の実施態様だけを上で詳細に記述しているものの、化学分野の当業者は、その好ましい実施態様において、それらの教示から逸脱することなく、多くの改良が可能であることを、充分に理解している。このような改良の全ては、以下の請求の範囲の範囲内に包含されることを意図している。
【技術分野】
【0001】
(発明の分野)
本発明は、一般に、ポリメラーゼ酵素の基質として有用なヌクレオチド化合物、プライマー伸長反応でこのようなヌクレオチド化合物を使用する方法、およびこのようなヌクレオチド化合物を含有するポリヌクレオチドに関する。
【0002】
(参考文献)
【0003】
【数1】
【0004】
【数2】
【背景技術】
【0005】
(背景)
核酸配列決定は、現代の生物学および生物工学において必須の重要な技術となっており、基本的な生物研究から薬剤の発見や臨床医学までの範囲の分野に関連した情報を提供する。大容量のDNA配列データが収集されているので、核酸配列決定法のスループットを高めると共にコストを下げるために、自動化技術が開発されている(Smith;Connell;Trainor)。
【0006】
好ましい自動化核酸配列決定法は、Sangerにより開発された酵素複製技術に基づいている(Sanger)。Sangerの方法では、一本鎖テンプレート核酸の配列は、一組のポリヌクレオチドフラグメントを合成するための核酸ポリメラーゼを用いて決定され、ここで、このフラグメントは、(i)このテンプレート配列に対する配列相補性を有し、(ii)単一ヌクレオチドにより長さが変わり、そして(iii)公知のヌクレオチド(例えば、A、C、GまたはT)での5’−末端終止を有する。この方法では、オリゴヌクレオチドプライマーは、配列決定されるテンプレート核酸の3’−末端にアニールされ、このプライマーの3’−末端は、相補的ポリヌクレオチドフラグメントのポリメラーゼ媒介重合に対する開始部位として、役立つ。この酵素的重合工程は、このテンプレート−プライマーハイブリッドと4個の天然デオキシヌクレオチド(「dNTP」)、核酸ポリメラーゼ酵素および2’,3’−ジデオキシヌクレオチド三リン酸(「ddNTP」)「ターミネーター」とを組み合わせることにより、行われる。このターミネーターの組み込みは、この3’−末端の水酸基を欠いたフラグメントを形成し、それゆえ、このポリメラーゼによって、さらに伸長され得ない(すなわち、このフラグメントは、「終止されている」)。組込みのためのddNTPおよびその対応dNTPの間の競合の結果、異なるサイズのフラグメントが分配され、各フラグメントは、この反応で使用される特定のターミネーターで終止される。このテンプレート核酸の完全な配列を発見するために、4個の並発反応が行われ、各反応は、異なるddNTPターミネーターを使用する。これらのフラグメントのサイズ分配を決定するために、これらのフラグメントは、サイズが異なるフラグメントを単一ヌクレオチドによって分割するように、電気泳動により分離される。
【0007】
古典的なSanger法の現代版変形法では、このヌクレオチドターミネーターは、蛍光色素で標識され(Prober;Hobbs)、熱安定性のDNAポリメラーゼ酵素が使用される(Murray)。色素標識したターミネーターを使用することにより、以下のいくつかの利点が得られる:(i)放射性同位体の貯蔵、使用および廃棄に付随した問題がなくなる;(ii)色素標識したプライマーを合成する必要性がなくなる;および(iii)各A、G、CまたはTヌクレオチド用に異なる色素標識を使用するとき、単一の管にて、4個の反応の全てを同時に行うことができる。熱安定性のポリメラーゼ酵素を使用することにより、(i)この重合反応を高温で行うことが可能になり、それにより、このテンプレートのいずれかの二次構造が分断され、その結果、より少ない配列依存性アーチファクトが得られ、そして、(ii)この配列決定反応が熱サイクルでき、それにより、生成する伸長生成物の量を線形的に増幅させるのに役立ち、それゆえ、配列を得るのに必要なDNAテンプレートの量が低減される。
【0008】
Sanger配列決定法のこれらの現代版変形法は、効果的であることが証明されているものの、それらの性能および経済性を最適化することに関して、いくつかの問題点が残っている。Sanger型核酸配列決定法で熱安定性のポリメラーゼ酵素と組み合わせて現在入手できる色素標識ターミネーターを使用するとき、特に、フルオレセイン型色素標識の場合に遭遇する1つの問題点には、50:1の比まで、非標識dNTPよりも大過剰の色素標識ターミネーターが必要なことがある。この大過剰の標識ターミネーターにより、この電気泳動分離工程を行う前に、この配列決定反応生成物を精製する必要が生じる。この清浄化(clean−up)工程は、組み込まれていない標識化ターミネーター種および真正の配列決定フラグメントの同時移入により起こる干渉を回避するために、必要である。典型的な清浄化方法には、エタノール沈殿またはクロマトグラフィー分離が含まれる(ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Protocol)。このような清浄化工程は、完全に自動化した配列決定系(ここで、その配列決定反応生成物は、電気泳動分離プロセスに直接移される)を開発する仕事を著しく複雑にする。
【0009】
Sanger型核酸配列決定法で熱安定性のポリメラーゼと組み合わせて現在入手できる色素標識ターミネーターを使用するとき遭遇する第二の問題点には、これらの反応生成物を電気泳動により分離し蛍光検出を用いて検出するとき、ピーク高さの不均一な分布が得られることがある。このような不均一なピーク高さは、自動化した配列測定およびヘテロ接合体の検出を実質的に信頼できないものとするために、不利である。
【発明の概要】
【課題を解決するための手段】
【0010】
(要旨)
本発明は、プライマー伸長反応(例えば、Sanger型DNA配列決定反応またはPCR反応)において、鎖終止ヌクレオチドおよび鎖伸長ヌクレオチドとして有用な、新規なクラスの置換プロパルギルエトキシアミドヌクレオシドの本発明者らによる発見に関する。
【0011】
本発明の目的は、標識した鎖終止ヌクレオチドを形成するのに使用できるヌクレオチドを提供することにある。
【0012】
本発明のさらなる目的は、標識を含む鎖終止ヌクレオチドを提供することにある。
【0013】
本発明のなおさらなる目的は、蛍光標識を含む鎖終止ヌクレオチドであって、Sanger型DNA配列決定法において、非標識の鎖終止ヌクレオチドよりも低い過剰濃度のこのような標識した鎖終止ヌクレオチドを必要とするものを提供することにある。
【0014】
本発明の別の目的は、Sanger型DNA配列決定法において、ピーク高さのより均一な分布が得られる標識した鎖終止ヌクレオチドを提供することにある。
【0015】
本発明の目的は、標識した鎖伸長ヌクレオチドを形成するのに使用できるヌクレオチドを提供することにある。
【0016】
本発明のさらなる目的は、標識を含む鎖伸長ヌクレオチドを提供することにある。
【0017】
本発明の別の目的は、標識したポリヌクレオチドを提供することにある。
【0018】
本発明のさらなる目的は、本発明の置換プロパルギルエトキシアミドヌクレオチドを使用するプライマー伸長反応を包含する方法を提供することにある。
【0019】
第一の局面では、本発明の上述の目的および他の目的は、以下の構造を有するヌクレオシド化合物により、達成される:
【0020】
【化1】
ここで、Xは、
【0021】
【化2】
ここで、nは、1〜5の範囲である、
【0022】
【化3】
ここで、nは、1〜5の範囲である、
【0023】
【化4】
および
【0024】
【化5】
からなる群から選択される;そしてR1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される。R3は、−Hおよび低級アルキルからなる群から選択される。Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基であり、ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、このプリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、このピリミジンのN1位にて結合しており、Bがプリンのとき、その隣接する三重結合炭素は、このプリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、この7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、このピリミジンの5位に結合している。W1は、−Hおよび−OHからなる群から選択される;W2は、−OH、またはこのヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そしてW3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
【0025】
本発明の第一の局面の第一の好ましい実施態様では、R1およびR2の1個は、標識であり、より好ましくは、この標識は、フルオレセイン型色素、ローダミン型色素またはFLAN型色素である。
【0026】
本発明の第一の局面の第二の好ましい実施態様では、W1は、−Hである;W2は、−OH、またはその3’位でホスホジエステル結合を形成できなくする部分である;そしてW3は、−P3O10である。より好ましくは、W2は、−OH、−H、アジド、アミノ、フルオロおよびメトキシからなる群から選択される。
【0027】
本発明の第一の局面の第三の好ましい実施態様では、Bは、ウラシル、シトシン、7−デアザアデニンおよび7−デアザグアノシンからなる群から選択される。
【0028】
本発明の第一の局面の第四の好ましい実施態様では、Xは、
【0029】
【化6】
であり、n=1であり、そしてΦ基は、パラ立体配置にある。
【0030】
第二の局面では、本発明の上述の目的および他の目的は、プライマー伸長反応を行う方法により達成され、この方法は、テンプレート核酸を提供する工程;このテンプレート核酸の一部にオリゴヌクレオチドプライマーをアニールする工程;およびこのプライマーを伸長するために、このプライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加する工程を包含し、このプライマー伸長試薬は、上記の本発明の第一局面のヌクレオシド化合物を含有する。
【0031】
第三の局面では、本発明の上述の目的および他の目的は、上記の本発明の第一の局面のヌクレオシド化合物を含有するポリヌクレオチドにより、達成される。
【0032】
本発明は、例えば以下の項目を提供する。
(項目1) 以下の構造を有するヌクレオシド化合物:
【0033】
【化18】
ここで、Xは、
【0034】
【化19】
ここで、nは、1〜5の範囲である、
【0035】
【化20】
ここで、nは、1〜5の範囲である、
【0036】
【化21】
および
【0037】
【化22】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
(項目2) R1およびR2の1個が、標識である、項目1に記載のヌクレオシド化合物。
(項目3) 前記標識が、フルオレセイン型色素である、項目2に記載のヌクレオシド化合物。
(項目4) 前記標識が、ローダミン型色素およびFLAN型色素からなる群から選択される、項目2に記載のヌクレオシド化合物。
(項目5) W1が、−Hであり;
W2が、−OH、または前記ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分であり;そして
W3が、−P3O10である、
項目1に記載のヌクレオシド化合物。
(項目6) W2が、−OH、−H、アジド、アミノ、ハロ、およびメトキシからなる群から選択される、項目1に記載のヌクレオシド化合物。
(項目7) W2が、−Hおよびフルオロからなる群から選択される、項目1に記載のヌクレオシド化合物。
(項目8) Bが、ウラシル、シトシン、7−デアザアデニン、および7−デアザグアノシンからなる群から選択される、項目1に記載のヌクレオシド。
(項目9) Xが、パラ立体配置を有するアルキルアミノ安息香酸であり、そしてn=1である、項目1に記載のヌクレオシド化合物。
(項目10) プライマー伸長反応を行う方法であって、該方法は、以下の工程を包含する:
テンプレート核酸を提供すること;
該テンプレート核酸の一部にオリゴヌクレオチドプライマーをアニールすること;および
該プライマーを伸長するために、該プライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加することであって、該プライマー伸長試薬は、以下の構造を有するヌクレオシド化合物を含有する:
【0038】
【化23】
ここで、Xは、
【0039】
【化24】
ここで、nは、1〜5の範囲である、
【0040】
【化25】
ここで、nは、1〜5の範囲である、
【0041】
【化26】
および
【0042】
【化27】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。
(項目11) R1およびR2が、標識であり、そして他が、−Hである、項目10に記載の方法。
(項目12) 以下の構造を有するヌクレオチド化合物を含有するポリヌクレオチド:
【0043】
【化28】
ここで、Xは、
【0044】
【化29】
ここで、nは、1〜5の範囲である、
【0045】
【化30】
ここで、nは、1〜5の範囲である、
【0046】
【化31】
および
【0047】
【化32】
からなる群から選択される;
R1およびR2は、別個に、−H、低級アルキル、保護基、および標識からなる群から選択される;
R3は、−Hおよび低級アルキルからなる群から選択される;
Bは、7−デアザプリン、プリン、またはピリミジンヌクレオシド塩基である;
ここで、Bがプリンまたは7−デアザプリンのとき、その糖部分は、該プリンまたはデアザプリンのN9位にて結合しており、Bがピリミジンのとき、その糖部分は、該ピリミジンのN1位にて結合している;そして
ここで、Bがプリンのとき、その隣接する三重結合炭素は、該プリンの8位に結合しており、Bが7−デアザプリンのとき、その隣接する三重結合炭素は、該7−デアザプリンの7位に結合しており、Bがピリミジンのとき、その隣接する三重結合炭素は、該ピリミジンの5位に結合している;
W1は、−Hおよび−OHからなる群から選択される;
W2は、−OH、または該ヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である;そして
W3は、−PO4、ホスフェートアナログ、および−OHからなる群から選択される。
【図面の簡単な説明】
【0048】
【図1A】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
【図1B】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
【図1C】図1A〜1Cは、種々の濃度の色素標識ターミネーターを用いたターミネーター滴定アッセイから得た結果を示す。
【図2】図2は、2−フタルイミドエタノール(3)および3−(2−フタルイミドエトキシ)プロピン(5)の合成を示す。
【図3】図3は、5−{3−(2−フタルアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン(7)および5−{3−(2−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン(8)の合成を示す。
【図4】図4は、5−{3−(2’−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン一リン酸(10)の合成を示す。
【図5】図5は、5−{3−(2−トリフルオロアセトアミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(12)および5−{3−(2−アミドエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成を示す。
【図6A】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図6B】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図6C】図6A〜Cは、トリフルオロアセトアミドグリシンNHSエステル(15)、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)、およびトリフルオロアセトアミドブト−2−イノイックNHSエステル(21)の合成を示す。
【図7】図7は、5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)の合成を示す。
【図8】図8は、5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)の合成を示す。
【図9】図9は、5−[3−{2−[N−(2−アミノ−ブト−2−イノイック)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(27)の合成を示す。
【図10】図10は、3個の異なる色素ターミネーター配列決定反応から得た結果を示し、ここで、各反応は、色素をターミネーターに結合する異なるリンカーを使用した。
【発明を実施するための形態】
【0049】
(好ましい実施態様の詳細な説明)
ここで、本発明の好ましい実施態様に対して、詳細な参照がなされ、その実施例は、添付の図面に例示されている。本発明は、その好ましい実施態様に関連して記述されているものの、それらは、本発明をこれらの実施態様に限定する意図はないことが分かる。反対に、本発明は、その代替、改変および等価物を含むことを意図しており、これらは、添付の請求の範囲で規定される本発明の範囲内に含まれ得る。
【0050】
一般に、本発明は、ポリメラーゼ酵素に対して基質として有用な新規な種類の置換プロパルギルエトキシアミドヌクレオシド化合物、このようなヌクレオシド化合物を含むポリヌクレオチド、およびプライマー伸長反応においてこのようなヌクレオシド化合物を使用する方法を包含する。本発明の化合物は、Sanger型DNA配列決定法で使用する色素標識ヌクレオチド鎖終止試薬の調製、およびプライマー伸長反応(例えば、PCR)を含む方法で使用する色素標識ヌクレオチド鎖伸長試薬の調製において、特定の用途が見出されている。
【0051】
本発明は、一部には、目的の置換プロパルギルエトキシアミドヌクレオチドが、熱安定性DNAポリメラーゼ酵素用の特に良好な基質であること、すなわち、(i)Sanger型DNA配列決定反応にて、現在入手できる標識ターミネーターを使用したときに必要なものと比較して、著しく低いモル過剰しか必要としないこと、および(ii)Sanger型DNA配列決定プロセスにて、現在入手できる標識ターミネーターを使用したときに見られるものと比較して、ピーク高さのより均一な分布が見られることの発見に基づいている。
【0052】
I.定義
他に述べられていなければ、本明細書中で使用する以下の用語および語句は、以下の意味を有することを意図している:
「低級アルキル」との用語は、1個〜8個の炭素原子を含有する直鎖および分枝炭化水素部分(すなわち、メチル、エチル、プロピル、イソプロピル、tert−ブチル、イソブチル、sec−ブチル、ネオペンチル、tert−ペンチルなど)を表わす。
【0053】
「標識」との用語は、本発明のヌクレオシドに結合したとき、このようなヌクレオシドおよびこのようなヌクレオチドを含有するポリヌクレオチドを、公知の検出手段を用いて検出可能にする部分を意味する。例示の標識には、蛍光体、発色団、放射性同位元素、スピン標識、酵素標識、化学発光標識など(これらは、適切な検出器による標識化合物の直接的な検出を可能にする)、またはリガンド、例えば、抗原またはビオチン(これは、検出可能な抗リガンド(例えば、標識抗体またはアビジン)に、高い親和性で特異的に結合できる)が挙げられる。好ましくは、これらの標識は、蛍光色素(例えば、フルオレセイン型色素またはローダミン型色素(Lee;Menchen))である。
【0054】
「ヌクレオシド」との用語は、2’−デオキシ形状および2’−ヒドロキシル形状を含めて、その1’位でペントースに結合したプリン、デアザプリンまたはピリミジンヌクレオシド塩基(例えば、アデニン、グアニン、シトシン、ウラシル、チミン、デアザアデニン、デアザグアノシンなど)からなる化合物を意味する(Stryer)。本明細書中で使用する「ヌクレオチド」との用語は、ヌクレオシドのリン酸エステル(例えば、三リン酸エステル)を意味し、ここで、最も一般的なエステル化部位は、このペントースのC−5位に結合した水酸基である。多くの場合、本発明の開示では、ヌクレオシドとの用語は、ヌクレオシドおよびヌクレオチドの両方を包含することを意図している。ヌクレオシドに関する「アナログ」には、例えば、他の文献で記述のように(Scheit;Eckstein 1991)、変性塩基部分、変性糖部分および/または変性リン酸エステル部分を有する合成アナログが挙げられる。
【0055】
本明細書中で使用する「ポリヌクレオチド」または「オリゴヌクレオチド」との用語は、天然ヌクレオチドモノマーまたはそれらのアナログの線状ポリマーを意味し、これには、二本鎖および一本鎖のデオキシリボヌクレオチド、リボヌクレオチド、それらのα−アノマー形状などが含まれる。通常、このヌクレオシドモノマーは、ホスホジエステル連結により連結されており、本明細書中で使用する「ホスホジエステル連結」との用語は、ホスホジエステル結合またはそれらのホスフェートアナログを含む結合を意味し、これらには、会合した対イオンが存在するなら、このような対イオン(例えば、H、NH4、Naなど)を含む。このポリヌクレオチドの大きさは、典型的には、数個(例えば、8個〜40個)のモノマー単位から、数千個のモノマー単位までに及ぶ。ポリヌクレオチドが「ATGCCTG」のような一連の文字で表わされるときは、このヌクレオチドは、他に指示がなければ、5’→3’の順序で左から右に配列されており、「A」は、デオキシアデノシンを表わし、「C」は、デオキシシチジンを表わし、「G」は、デオキシグアノシンを表わし、そして「T」は、チミジンを表わすことが分かる。
【0056】
「ホスフェートアナログ」との用語は、ホスフェートのアナログであって、ここでそのリン原子が+5酸化状態であり、そしてその酸素原子の1個またはそれ以上が非酸素部分で置き換えられているものを意味し、例示のアナログには、ホスホロチオエート、ホスホロジチオエート、ホスホロセレノエート、ホスホロジセレノエート、ホスホロアニロチオエート、ホスホロアニリデート、ホスホロアミデート、ボロノホスフェートなど(会合した対イオン(例えば、H、NH4、Naなど)があれば、このような対イオンを含めて)が挙げられる。
【0057】
本明細書中で使用される「プロパルギルアミドリンカー」との用語は、以下の構造を有するリンカーを意味する:
【0058】
【化7】
また、「プロパルギルエトキシアミドリンカー」との用語は、以下の構造を有するリンカーを意味する:
【0059】
【化8】
ここで、上記構造のそれぞれについて、そのアセチレンの終止末端は、ヌクレオチド塩基に結合しており、そしてそのアミド窒素は、標識との好都合な連結を介して、結合される。
【0060】
本発明の「X」部分に関連して、「アミノアルカン酸」との用語は、以下の構造を意味する:
【0061】
【化9】
ここで、nは、1〜5の範囲である;「アルキルアミノ安息香酸」との用語は、以下の構造を意味する:
【0062】
【化10】
ここで、nは、1〜5の範囲である;「α−アミノ酸」との用語は、以下の構造を意味する:
【0063】
【化11】
そして「4−アミノ−2−ブチン酸」との用語は、以下の構造を意味する:
【0064】
【化12】
ここで、上記構造では、R1およびR2は、別個に、−H、低級アルキル、保護基または標識であり、そしてR4は、アミノ酸側鎖(天然(Stryer)または合成のいずれか)である。
【0065】
「プロパルギルエトキシアミドヌクレオチド」との用語は、ヌクレオチドのヌクレオ塩基に結合したプロパルギルエトキシアミドリンカーを含むヌクレオチドを意味する。
【0066】
「フルオレセイン型色素」との用語は、以下の縮合三環系を含有する種類のキサンテン色素分子を意味する:
【0067】
【化13】
ここで、各デオキシ環位置では、広範囲の置換が可能である。フルオレセイン型色素の特に好ましいサブセットには、4,7−ジクロロフルオレセイン(Menchen)が挙げられる。DNA配列決定法で蛍光標識として使用されるフルオレセイン型色素の例には、6−カルボキシフルオレセイン(6−FAM)、5−カルボキシフルオレセイン(5−FAM)、6−カルボキシ−4,7,2’,7’−テトラクロロフルオレセイン(TET)、6−カルボキシ−4,7,2’,4’,5’,7’−ヘキサクロロフルオレセイン(HEX)、5−(および6)カルボキシ−4’,5’−ジクロロ−2’,7’−ジメトキシフルオレセイン(JOE)、および5−カルボキシ−2’,4’,5’,7’−テトラクロロフルオレセイン(ZOE)が包含される。多くの場合、−1または−2との名称は、特定の色素(例えば、HEX−1)の略語の後で、付けられる。この「−1」および「−2」との名称は、使用する特定の色素異性体を意味する。これらの1および2異性体は、C−8カラムおよび溶出勾配(15%アセトニトリル/85% 0.1 M酢酸トリエチルアンモニウム〜35%アセトニトリル/65% 0.1 M酢酸トリエチルアンモニウム)を使用する逆相クロマトグラフィー分離系にて、遊離色素の溶出順序(この1異性体が最初に溶出する)により、定義される。
【0068】
「ローダミン型色素」との用語は、以下の縮合三環系を含有する種類のキサンテン色素分子を意味する:
【0069】
【化14】
ここで、好ましくは、Y1〜Y4は、別個に、水素または低級アルキルであり、または一緒になって、Y1およびR2は、プロパノであり、そしてY2およびR1は、プロパノであるか、または一緒になって、Y3およびR3は、プロパノであり、そしてY4およびR4は、プロパノである。R1〜R4位置を含めた各デオキシ環位置にて、広範囲の置換が可能である。ヌクレオシド標識として有用な例示のローダミン型色素には、テトラメチルローダミン(TAMRA)、4,7−ジクロロテトラメチルローダミン(DTAMRA)、ローダミンX(ROX)、ローダミン6G(R6G)、ローダミン110(R110)などが挙げられる(Bergot;Lee)。
【0070】
本明細書中で使用する「FLAN色素」との用語は、次式を有する非対称ベンゾキサンテン色素化合物を意味する:
【0071】
【化15】
ここで、Y1およびY2は、別個に、ヒドロキシル、酸素、イミニウムまたはアミンである。R1〜R8は、別個に、水素、フッ素、塩素、低級アルキル、低級アルケン、低級アルキン、スルホネート、アミノ、アンモニウム、アミド、ニトリル、アルコキシ、連結基、またはそれらの組合せである。そして、R9は、アセチレン、アルカン、アルケン、シアノ、置換フェニル、またはそれらの組合せ、以下の構造を有する置換フェニルである:
【0072】
【化16】
ここで、X1は、カルボン酸またはスルホン酸である;X2およびX5は、別個に、水素、塩素、フッ素または低級アルキルである;そしてX3およびX4は、別個に、水素、塩素、フッ素、低級アルキル、カルボン酸、スルホン酸または連結基である(Benson)。
【0073】
本明細書中で使用する「プライマー伸長試薬」との用語は、オリゴヌクレオチドプライマーの酵素的テンプレート媒介の伸長を起こすのに必要な成分を含有する試薬を意味する。プライマー伸長試薬には、以下が挙げられる:(i)ポリメラーゼ酵素、例えば、熱安定性ポリメラーゼ酵素(例えば、Taqポリメラーゼ);(ii)緩衝液;(iii)鎖伸長ヌクレオチド、例えば、デオキシヌクレオチド三リン酸(例えば、ジオキシグアノシン5’−三リン酸、7−デアザデオキシグアノシン5’−三リン酸、デオキシアデノシン5’−三リン酸、デオキシチミジン5’−三リン酸、デオキシシチジン5’−三リン酸);および必要に応じて、Sanger型DNA配列決定反応の場合に、(iv)1個またはそれ以上の鎖終止ヌクレオチド、例えば、ジデオキシヌクレオチド三リン酸(例えば、ジデオキシグアノシン5’−三リン酸、7−デアザジデオキシグアノシン5’−三リン酸、ジデオキシアデノシン5’−三リン酸、ジデオキチミジン5’−三リン酸、およびジデオキシシチジン5’−三リン酸)。
【0074】
「テンプレート核酸」とは、単一鎖形状で提示できる任意の核酸であって、プライマーオリゴヌクレオチドでアニールできるものを意味する。例示のテンプレート核酸には、DNA、RNAが挙げられ、このDNAまたはRNAは、一本鎖または二本鎖であり得る。さらに特定すると、テンプレート核酸は、ゲノムDNA、メッセンジャーRNA、cDNA、PCR反応に由来のDNA増幅生成物などであり得る。テンプレートDNAの調製方法は、他の文献に見られ得る(ABI PRISMTM Dye Primer Cycle Sequencing Core Kit)。
【0075】
II.置換プロパルギルエトキシアミドヌクレオチド化合物
第一の局面では、本発明は、すぐ下の式Iで示す一般構造を有する新規なクラスの置換プロパルギルエトキシアミドヌクレオチド化合物を包含する。(本開示全体で提供された全ての分子構造は、提示した正確な電子構造だけでなく、それらの全ての共鳴構造およびプロトン化状態を含むことを意図することに留意のこと)。
【0076】
【化17】
式Iの構造では、Bは、7−デアザプリン、プリンまたはピリミジンヌクレオチド塩基であり、好ましい実施態様では、Bは、ウラシル、シトシン、7−デアザアデニンおよび7−デアザグアノシンからなる群から選択される。Bがプリンまたは7−デアザプリンのとき、このヌクレオチドの糖部分は、このプリンまたはデアザプリンのN9位にて結合され、Bがピリミジンのとき、その糖部分は、このピリミジンのN1位にて結合される。Bがプリンのとき、隣接三重結合炭素は、このプリンの8位に結合され、Bが7−デアザプリンのとき、隣接三重結合炭素は、7−デアザプリンの7位に結合され、Bがピリミジンのとき、隣接三重結合炭素は、このピリミジンの5位に結合される。
【0077】
W1は、−Hおよび−OHから選択される。W1が−OHであるとき、このヌクレオシドは、リボヌクレオチドであり、そしてW1が−Hであるとき、このヌクレオシドは、デオキシリボヌクレオチドである。
【0078】
W2は、−OH、またはこのヌクレオシドがその3’位でホスホジエステル結合を形成できなくする部分である。この機能に有用な好ましい部分には、−H、アジド、アミノ、ハロ、メトキシなどが挙げられる。特に好ましい実施態様では、W2は、−Hまたはフルオロである。
【0079】
W3は、−PO4、−P2O7、−P3O10、ホスフェートアナログ、および−OHからなる群から選択される。ポリヌクレオチドの酵素的合成に有用な好ましい実施態様では、W3は−P3O10である。
【0080】
Xは、アミノアルカン酸、アルキルアミノ安息香酸、α−アミノ酸、および4−アミノ−2−ブチン酸からなる群から選択される。好ましくは、Xは、アルキルアミノ安息香酸である。Xがアミノアルカン酸またはアルキルアミノ安息香酸のとき、nは、1〜5の範囲であり、さらに好ましくは、nは、1〜3の範囲である。Xがアルキルアミノ安息香酸のとき、そのフェニル基は、その隣接するカルボニル基およびメチレン基に関して、メタ、パラまたはオルト配置であり得る。好ましい実施態様では、フェニル基は、パラ配置である。Xがα−アミノ酸のとき、アミノ酸側鎖は、任意の適切な天然または合成のアミノ酸側鎖であり得る。
【0081】
上記X部分のうちの任意のものについて、R1およびR2は、−H、低級アルキル、保護基または標識から選択される。好ましくは、標識は、蛍光色素である。より好ましくは、標識は、フルオレセイン型蛍光色素、ローダミン型蛍光色素またはFLAN型蛍光色素である。好ましくは、R1およびR2の一方が標識のとき、他方は、−Hまたは低級アルキルのいずれかである。好ましい保護基には、ハロアセチル、アシル、アルコキシカルボニルまたはスルホニルが挙げられる。より好ましくは、この保護基は、トリフルオロアセチルである。
【0082】
この標識は、置換プロパルギルエトキシアミドヌクレオシドの第一級または第二級アミノ部分と、標識上に位置している「相補的官能性」との反応により、典型的に形成される「連結」を介して、このヌクレオシドに結合される。好ましくは、この相補的官能性は、イソチオシアネート、イソシアネート、アシルアジド、N−ヒドロキシスクシンイミド(NHS)エステル、スルホニルクロライド、アルデヒドまたはグリオキサール、エポキシド、カーボネート、ハロゲン化アリール、イミドエステル、カルボジイミド、無水物、4,6−ジクロロトリアジニルアミンまたは他の活性カルボキシレートである(Hermanson)。特に好ましい実施態様では、この相補的官能性は、活性化NHSエステルであり、これは、本発明の置換プロパルギルエトキシアミドヌクレオシドのアミンと反応し、この場合、活性化NHSエステルを形成するには、カルボキシレート相補的官能性を含む標識は、ジシクロヘキシルカルボジイミドおよびN−ヒドロキシスクシンイミドと反応されて、NHSエステルを形成する(Khanna;Kasai)。以下の表1は、代表的な相補的官能性およびこの相補的官能性とこの置換プロパルギルエトキシアミドヌクレオシドのアミンとの反応により形成されて得られた連結のサンプリングを示す。
【0083】
【表1】
R3は、−Hおよび低級アルキルからなる群から選択される。
【0084】
III.置換プロパルギルエトキシアミドヌクレオチドおよび標識ヌクレオチドの合成
図2〜9は、本発明の置換プロパルギルエトキシアミドヌクレオシドのいくつかの代表的な合成を示す。この合成では、ブロモエタノール1は、フタルイミドカリウム2と反応して、フタルイミド誘導体3が得られる。このフタルイミド誘導体は、次いで、NaHの存在下にて、プロパルギルブロマイド4でO−アルキル化されて、保護した3−(2−フタルイミドエトキシ)プロピン連結アーム5が得られる。図2を参照せよ。次いで、ヨードヌクレオシド6は、ジメチルホルムアミド中、ヨウ化第一銅、テトラキス(トリフェニルホスフィン)パラジウム、およびトリエチルアミンの存在下にて、室温でおよそ12時間またはこの反応がTLCで決定されるように完結するまで、保護連結アームと反応されて、それにより、化合物7が形成される。この溶液は、次いで、真空下で濃縮され、生成物は、シリカゲルフラッシュクロマトグラフィーにより精製され、そして同定および純度測定のために、プロトンNMRおよび分析用逆相HPLCにより分析される。エチレンジアミンでの処理に続いて、トリフルオロ酢酸エチルでのアセチル化により、ヌクレオシド連結アーム化合物8が得られる。図3を参照せよ。リン酸トリメチル中にて、−30℃で、このヌクレオシド連結アーム化合物8に、新たに蒸留したオキシ塩化リンを添加して、対応するジクロロモノホスフェート9を形成する。反応混合物を2M重炭酸テトラエチルアンモニウム(TEAB)(pH 8.0)でクエンチして、モノホスフェート10を得、これは、次いで、分取逆相HPLCにより精製される。図4を参照せよ。モノホスフェート10は、カルボニルジイミダゾール(CDI)で活性化され、過剰のCDIは、MeOHでクエンチされて、活性化モノホスフェート11が得られる。この活性化モノホスフェートは、室温で、ピロリン酸トリブチルアンモニウムと反応される。この反応は、完結したとき、0.2M TEABでクエンチされ、そして逆相HPLCにより精製されて、保護トリホスフェート12が得られる。精製した保護トリホスフェートは、乾燥するまでエバポレートされ、そして濃NH4OH水溶液中に再懸濁されて、TFA基が除去され、プロパルギルエトキシアミドヌクレオチド13が得られる。図5を参照せよ。
【0085】
一般に、X部分のプロパルギルエトキシアミドヌクレオチド(例えば、13)への結合は、以下のようにして行われる。X部分のアミン官能性を保護するために、X部分とアミン保護試薬(例えば、トリフルオロアセテート、例えば、図6A〜Cの化合物14、17または20)とを反応させることにより、アミン保護X部分が形成される。アミン保護X部分の酸官能性を活性化するために、アミン保護X部分は、酸活性化剤(例えば、N−ヒドロキシスクシンイミド、例えば、図6A〜Cの化合物15、18または21)と反応される。次に、酸活性化されたアミン保護X部分は、塩基条件下(例えば、250 mMの重炭酸塩緩衝液、pH 9)で、プロパルギルエトキシアミドヌクレオチドと反応されて、アミン保護された置換プロパルギルエトキシアミドヌクレオチド(例えば、図7の化合物22、図8の化合物24、または図9の化合物26を参照のこと)を生じる。最後に、アミン保護基は、強塩基(例えば、NH4OH)との反応により、アミン保護された置換プロパルギルエトキシアミドヌクレオチドから除去されて、置換プロパルギルエトキシアミドヌクレオチド(例えば、図7の化合物23、図8の化合物25、または図9の化合物27を参照のこと)が得られる。
【0086】
一般に、好ましい方法では、本発明の標識した置換プロパルギルエトキシアミドヌクレオシドは、以下のようにして調製される。置換プロパルギルエトキシアミドヌクレオシド(例えば、23、25または27)は、中性緩衝溶液(例えば、100 mM TEAB(pH 7.0))に溶解され、溶液を乾燥状態までエバポレートされ、そして、ヌクレオシドを塩基性緩衝溶液(例えば、250 mM重炭酸ナトリウム緩衝液、pH=9)に再懸濁する。この溶液に、標識−NHSエステル(DMSO中)が添加され、そして撹拌しながら、一晩反応される。完結すると、この反応混合物は、イオン交換および逆相HPLCにより精製されて、標識した置換プロパルギルエトキシアミドヌクレオシドが得られる。
【0087】
IV.プロパルギルエトキシアミド化合物を使用する方法
本発明のプロパルギルエトキシアミド化合物は、以下の工程を包含するタイプのテンプレート媒介プライマー伸長反応を含めた方法での使用によく適合する:(i)テンプレート核酸を提供する工程;(ii)このテンプレート核酸の一部に、オリゴヌクレオチドプライマーをアニーリングし、それにより、プライマー−テンプレートハイブリッドを形成する工程;および(iii)このプライマーを伸長するために、このプライマー−テンプレートハイブリッドに、プライマー伸長試薬を添加する工程。特に、本発明の化合物は、プライマー伸長生成物に標識を直接含入させる手段を提供する。
【0088】
プライマー伸長反応を使用する第一の好ましい種類の方法では、この伸長生成物は、本発明の標識した置換プロパルギルエトキシアミドヌクレオチドを、プライマー伸長反応物に含有させ、それにより、この伸長生成物全体にわたって、標識をランダムに含入させることにより、標識される([F]dNTP試薬プロトコル)。このような方法は、PCR単位複製配列、および単一のプライマー誘導伸長生成物を標識するのに使用できる。このようにして伸長生成物を標識するために、プライマー伸長反応は、確立したプロトコルを用いて行われるが、この反応系には、標識した置換プロパルギルエトキシアミドヌクレオチドが添加される。一般に、PCRの状況で、プライマー伸長反応を行うためには、テンプレート核酸は、各プライマー20 pmolと、プライマー伸長試薬(これは、20 mMの緩衝液(pH 8)、1.5 mMのMgCl2、50 mMの各デオキシヌクレオチド三リン酸(dNTP)、および2単位のTaqポリメラーゼまたは他の適切な熱安定性ポリメラーゼを含有する)と混合される。次いで、反応混合物を熱サイクルにかけるが、典型的な熱サイクルプロフィールは、変性工程(例えば、96℃で15秒間)、プライマーアニーリング工程(例えば、55℃で30秒間)、およびプライマー伸長工程(例えば、72℃で90秒間)を包含する。典型的には、この熱サイクルは、約10〜40サイクル繰り返される。PCR増幅のためには、標識デオキシヌクレオチド三リン酸と非標識デオキシヌクレオチド三リン酸との典型的な比は、所望のシグナル量に依存して、100:1〜1000:1の間である。増幅効率に悪影響を起こすことなくPCR反応混合物中にて使用できる標識デオキシヌクレオチド三リン酸と非標識デオキシヌクレオチド三リン酸との最大比は、およそ1:4である。
【0089】
プライマー伸長反応を使用する第二の好ましい種類の方法では、この伸長生成物は、プライマー伸長反応物に本発明の置換プロパルギルエトキシアミドヌクレオチドを含有させて、それにより、伸長生成物の3’−末端ヌクレオチドにて、検出可能な標識をランダムに含入させることにより標識される(例えば、Sanger型DNA配列決定)。一般に、本発明の標識ジデオキシヌクレオチド三リン酸を用いたSanger型DNA配列決定の状況でプライマー伸長反応を行うためには、1μlのテンプレート溶液(水5μlで希釈したPCR反応物1μl)および2μlのプライマー(0.4 pmol/μl)は、プライマー伸長試薬(これは、2μlの緩衝液(400 mM Tris−HCl、10 mM MgCl2、pH 9.0)、2μlのデオキシヌクレオチド/標識ジデオキシヌクレオチド混合物(T−終止反応、1250μM ddTTP、250μM dATP、250μM dCTP、180μM 7−デアザ−dGTP、および250μM dTTP)、および2μlのポリメラーゼ酵素(5単位/μlであって、この場合、1単位は、Lawyerのように定義される)を含有する)と混合される。次いで、反応物は、以下の代表的なプログラムを用いて、熱サイクルにかけられる:98℃で5秒間の変性に続いて、96℃で5秒間;55℃で40秒間;60℃で1分間のサイクルの繰り返し、このサイクルは、およそ15回繰り返される。
【0090】
本発明の置換プロパルギルエトキシアミドヌクレオシド化合物はまた、プライマー伸長生成物の塩基特異的な開裂に依存したSanger型配列決定の変形法(例えば、標識ヌクレオチド使用する方法)の状況にて、よく適合する(Eckstein 1988;Shaw)。
【実施例】
【0091】
本発明は、以下の実施例を考慮することにより、さらに明らかにされるが、これらの実施例は、本発明の純粋な例示とすることを意図しており、いずれの様式でも、その範囲を限定することを意図していない。
【0092】
(実施例1)
配列決定反応に必要なターミネーター過剰を決定するためのターミネーター滴定アッセイ
完全な配列決定ラダー(ladder)(すなわち、約20ヌクレオチドと約600ヌクレオチドの間の長さを有する特定の塩基で終止する全てのフラグメントを含有する配列決定ラダー)を作るのに必要な最小量の色素標識ターミネーターを決定するために、ターミネーター滴定アッセイを使用した。このターミネーター滴定アッセイの不可欠な成分は、(i)第一色素で標識したプライマー、および(ii)この第一色素とスペクトル分解可能な第二色素で標識したターミネーターであった。このアッセイでは、この配列決定反応物に、不充分な濃度の色素ターミネーターを添加したとき、ジデオキシ終止フラグメントは形成されず、配列決定ゲル上では、第一色素のみで標識した「偽スポット」により形成された生成物だけが見られた。本明細書中で使用する「偽スポット」との用語は、ジデオキシターミネーターで終止していないプライマー伸長生成物を意味し、このような生成物は、おそらく、ポリメラーゼ酵素がテンプレート核酸鎖から自発的に分離するときに形成される。多すぎるターミネーターを使用したときには、短い終止生成物(すなわち、長さが約50個未満のヌクレオチド)しか形成されず、このような生成物は、第一色素および第二色素の両方を含有していた。適切な量のターミネーターを使用したとき、完全な配列決定ラダーが生成し、ラダーの各フラグメントは、第一色素および第二色素の両方で標識されていた。
【0093】
色素−ターミネーター反応は、ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Manual(PE Applied Biosystems p/n 402116)で提供されたプロトコルに従ったAmpliTaq DNA Polymerase、FSを用いて、行った。(このFS酵素は、2点変異−−G46DおよびF667Yを有する組み換えThermus水生DNAポリメラーゼである)。dNTPミックス、色素標識プライマーおよび色素標識ターミネーター以外の全ての試薬は、ABI PRISMTM Dye Terminator Core Kit(PE Applied Biosystems p/n 402117)から得た。dNTPミックスは、各2 mMのdATP、dCTP、7−デアザ−dGTPおよびdTTPからなっていた。反応成分のプレミックスを、以下の表に示すように調製し、ここで、全ての量は、反応ベースあたりで示されている:
【0094】
【表2】
反応物を、Perkin−Elmer 480 DNA Thermal Cycler(PE Applied Biosystems p/n N801−100)に適合した0.5 ml微量遠心分離管に組み込んだ。反応物の容量は20μLであり、これには、上記反応プレミックス15μL、可変量の色素標識ターミネーター、および全反応容量を20μLにするのに充分な量の水を含む。各反応物に、1〜500 pmolの色素標識ターミネーターを添加した。蒸発を防止するために、各反応物の頂部に、鉱油30μLを添加した。これらの反応物を、以下のようにして熱サイクルにかけた:96℃で30秒間、50℃で15秒間、および60℃で4分間を25サイクルに続いて、4℃で保持するサイクル。
【0095】
全ての反応物を、製造業者の指示(Princeton Separations p/n CS−901)に従って、Centri−Sepスピンカラム上のスピン−カラム精製により精製した。このカラム内のゲル物質は、室温で少なくとも30分間にわたり、脱イオン水0.8 mLで水和させた。カラムが水和され、ゲル物質に気泡が取り込まれていないことを確認した後、カラムの上部および下部の末端キャップを取り除き、カラムを、重力により排水した。次いで、カラムを、キットに備え付けた洗浄管に挿入し、そして可変速度微量遠心分離機で、1300xgで2分間にわたり遠心分離し、洗浄管を取り除き、そして試料収集管に挿入した。この反応混合物を、オイルの下部から注意深く取り出し、そしてゲル物質上に充填した。カラムを、可変速度微量遠心分離で、1300xgで2分間遠心分離した。次いで、溶出した試料を、真空遠心分離で乾燥した。
【0096】
配列決定ゲル上に充填する前に、乾燥した試料を、25μLのTemplate Suppression Reagent(PE Applied Biosystems p/n 401674)中に再懸濁させ、ボルテックスし、2分間にわたって95℃まで加熱し、氷上で冷却し、再びボルテックスし、そして遠心分離した(13,000xg)。再懸濁した試料10μLを、PE ABI PRISMTM 310 Genetic Analyzer(PE Applied Biosystems p/n 310−00−100/120)に適合させた試料バイアル(PE Applied Biosystems p/n 401957)に等分した。310 Genetic Analyzerでの電気泳動は、DNA配列決定分析に特別に適合させたふるい分けポリマーおよび毛細管(PE Applied Biosystems p/n 402837(ポリマー)およびp/n 402840(毛細管))を用いて行った。各場合では、ふるい分けポリマーは、核酸変性物を含んでいた。試料は、2.5 kVで30秒間、毛細管に動電的に注入し、そしてこの毛細管の外壁を50℃に維持しつつ、12.2 kVで2時間走査した。
【0097】
図1A〜Cは、Terminator Titration Assayから得た典型的な結果を示し、この場合、プライマーはTAMRA色素で標識され、ddATPターミネーターは、6−FAM色素で標識され、TAMRA色素をこのジデオキシヌクレオチドターミネーターに連結するために、伝統的なプロパルギルアミドリンカーを使用した。これらのトレースは、ヌクレオチド71〜175に対する電気泳動走査時間の関数として、一定波長での蛍光強度を示す。プライマー伸長反応物に添加した色素−ターミネーターの量は、可変であった:図1Aでは、1 pmolのターミネーターを使用し、図1Bでは、4 pmolのターミネーターを使用し、そして図1Cでは、150 pmolのターミネーターを使用した。各パネルの上のトレースは、色素標識ターミネーターで発光し、535〜545 nmで集めた蛍光であり、各パネルの下のトレースは、色素標識プライマーで発光し、575〜585 nmで集めた蛍光である。色素プライマートレース(下)は、適切に終止したフラグメントでけでなく、偽ストップ(false stop)(すなわち、色素標識ターミネーターで終止していないフラグメント)も示している。色素ターミネータートレース(上)は、色素標識ターミネーターの特異的な取り込みを示している。
【0098】
図1Aは、1 pmolの6−FAM−ddATPターミネーターを用いる反応についてのデータを示す。この色素ターミネータートレースでは、小さなピークにより明らかなように、極く僅かな特異的混入が検出された。この色素−プライマートレースで示された偽ストップは、任意の特異的に終止したピークとも実質的に同じ大きさであった。このパターンから、この色素−ターミネーター濃度が非常に低いことが明らかとなった。図1Bは、4 pmolの6−FAM−ddATPターミネーターを用いた反応についてのデータを示す。このターミネータートレースでは、この配列決定ラダー全体を通じて、比較的に一定のピーク高さで、良好な特異的ターミネーター混入が認められた。この色素プライマートレースでは、偽ストップノイズの上部に容易に識別できるピークが存在しており、これらのピークは、色素ターミネータートレースのピークと共に移動していた。このパターンは、この色素ターミネーター濃度は、使用可能な範囲内であったことを示す。図1Cは、150 pmolの6−FAM−ddATPターミネーターを用いた反応についてのデータを示す。「トップヘビー(top heavy)」パターンが見られ、色素ターミネータートレースの初期のピークが、非常に高レベルの色素ターミネーター混入を示し、その後のピークは、ずっと低いレベルの混入を示していた。このパターンは、この色素−ターミネーター濃度が高すぎたことを示している。
【0099】
(実施例2)
リンカー型の関数として、完全配列決定ラダーを形成するのに必要なHEX−1−標識したC−ターミネーターの量
以下の表は、実施例1にて上で記述のターミネーター滴定アッセイに従って、完全配列決定ラダーを形成するのに必要な色素標識したC−ターミネーターの相対モル過剰(molar excess)を示す。この相対モル過剰は、完全配列決定ラダーを形成するのに必要な非標識ジデオキシターミネーターの量が1の値となるように、定義される。各場合では、C−ターミネーターは、HEX−1色素に連結した。
【0100】
【表3】
a この相対モル過剰は、完全配列決定ラダーを形成するのに必要な非標識ジデオキシターミネーターの量が1.0の値となるように定義される。
【0101】
(実施例3)
5−{3−(2−アミノエトキシ)プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成
A.物質および方法
薄層クロマトグラフィー(TLC)を、250μm層のシリカゲル60−F254で予め塗装したガラス板上で行った。蛍光のクエンチおよび/または5%硫酸での炭化により展開した後、化合物をTLC板上に配置した。SIPブランドシリカゲル60Å、230〜400 Mesh ASTM(Baxter Scientific p/n C4582−87)上で、フラッシュカラムクロマトグラフィーを行った。以下のようにして、NMRスペクトルを得た:1H NMRスペクトルを、室温で、CDCl3(内部Me4Si、δ0)またはD2O(外部Me4Si、δ0)に溶解した溶液で300 MHzにて記録した;13C NMRスペクトルを、CDCl3(内部Me4Si、δ0)に溶解した溶液で、75.5 MHzにて記録した;19F NMRスペクトルを、CDCl3またはD2O(外部CFCl3、δ0)に溶解した溶液で、282.23 MHzにて記録した;そして31P NMRスペクトルを、D2O中の溶液で、121.44 MHzにて記録した。全ての場合において、NMRデータは、提案した構造に一致していた。他に指示がなければ、全ての反応は室温で行い、後処理において、有機溶媒の溶液を、等容量の水溶液で洗浄した。有機溶液を、40〜50℃の浴温を用いた真空下でのロータリーエバポレーターでの濃縮の前に、無水Na2SO4で一般的に乾燥した。分析および分取目的で使用したHPLC系は以下の通りであった:
分析用逆相HPLC:カラム:Spheri−5 RP−C18、5μmの粒子サイズ、220×4.6 mm(PE Applied Biosystems p/n 0711−0017);勾配:0〜50%アセトニトリルで1.5 ml/分にて20分間に続いて、50%アセトニトリル〜100%アセトニトリルで1.5 ml/分にて10分間。
【0102】
分析用イオン対HPLC:カラム:AquaporeTM OD−300、7μmの粒子サイズ、220×4.6 mm(PE Applied Biosystems p/n 0711−0331);勾配:0〜40%アセトニトリルで1.5 ml/分にて30分間に続いて、40%アセトニトリル〜60%アセトニトリルで1.5 ml/分にて5分間。
【0103】
分取アニオン交換HPLC:カラム:AquaporeTM Anion、20μmの粒子サイズ、250×10 mm(PE Applied Biosystems p/n 0711−0172);勾配:40%アセトニトリル:60% 100 mM TEAB、pH 7.0〜40%アセトニトリル:60% 1.5 mM TEAB pH 8で4.5 ml/分にて20分間に続いて、アイソクラチック(isocratic)溶出。
【0104】
分取逆相HPLC:カラム:Prep Nova Pak HR−C18、6μmの粒子サイズ、60Åの細孔サイズ、300×40 mm(Waters Division of the Millipore Corporation p/n WAT037704);勾配(モノホスフェートおよびトリホスフェートについて):100% 100 mM TEAB、pH 7〜20%アセトニトリル:80% 100 mM TEAB pH 7で50 ml/分にて30分間に続いて、20%アセトニトリル:80% 100 mM TEAB pH 7〜50%アセトニトリル:50% 100 mM TEAB pH 7で10分間;勾配(色素標識三リン酸について):100% 100 mM TEAB pH 7〜10% 100 mM TEAB pH 7:90%アセトニトリル。
【0105】
B.2−フタルイミドエタノール(3)の合成
フタルイミドカリウム2(2.7g、14.6 mmol)を、ブロモエタノール1のN,N−ジメチルホルムアミド溶液(12 mL、14.1 mmol)に添加した。70℃で12時間撹拌した後、この混合物を濃縮し、次いで、ジクロロメタン(100 mL)で希釈した。濾過により固形分を除去した後、その有機層を水で洗浄し、乾燥し、そして濃縮した。この濃縮物をフラッシュカラムクロマトグラフィー(3:2〜2:3ヘキサン−酢酸エチル)により精製して、0.22のRF(3:2のヘキサン−酢酸エチル)を有する白色固形分(1.19g、44.12%)として、化合物3を得た。図2を参照せよ。
【0106】
C.3−(2−フタルイミドエトキシ)プロピン(5)の合成
化合物3(1.14g、5.96 mmol)のN,N−ジメチルホルムアミド(20 mL)撹拌溶液に、NaH(0.36g、80%)を滴下した。NaHを完全に添加した後、撹拌を室温で0.5時間継続し、その反応系を0℃まで冷却した。プロパルギルブロマイド4(1.5 mL、13.47 mmol)を添加し、この撹拌を、0℃でさらに0.5時間、次いで、室温で2時間継続した。メタノールを注意深く添加して過剰のNaHを分解した後、溶媒を蒸発させ、その粗生成物をフラッシュカラムクロマトグラフィー(3:2〜1:1〜2:3ヘキサン−酢酸エチル)により精製して、0.22のRF(3:2のヘキサン−酢酸エチル)を有する固形物(495 mg、36.2%)として、化合物5を得た。図2を参照せよ。
【0107】
D.5−{3−(2−フタルアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン(7)の合成
5−ヨード−2’,3’−ジデオキシシチジン6(100 mg、0.3 mmol)を、N,N−ジメチルホルムアミド(1 mL)中、ヨウ化第一銅(11.4 mg、0.06 mmol)、テトラキス(トリフェニルホスフィン)パラジウム(69 mg、0.06 mmol)、およびトリエチルアミン(84μL、0.6 mmol)の存在下にて、アルゴン雰囲気下にて、室温で12時間にわたり、化合物5(158 mg、0.69 mmol)と反応させた。次いで、この反応物を、メタノール中の重炭酸塩形態のDowex−1アニオン交換樹脂2gで希釈した。室温で1時間撹拌した後、この反応混合物を濾過し、そして濃縮した。この生成物を、フラッシュカラムクロマトグラフィー(13:1のジクロロメタン−メタノール)により精製して、0.23のRF(溶媒は9:1のジクロロメタン−メタノール)を有する化合物7(75 mg、57.66%)を得た。図3を参照せよ。
【0108】
E.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン(8)の合成
化合物7(73 mg、0.17 mmol)およびエチレンジアミン(400μL)の混合物を、エタノール(4 mL)中にて、80℃で1時間加熱した。次いで、この反応系を乾燥状態までエバポレートし、その残留物を、N,N−ジメチルホルムアミド(2 mL)に溶解し、トリフルオロ酢酸メチル(6.5 mL)を添加した。80℃で1時間撹拌した後、この溶媒をエバポレートし、その残留物を、フラッシュカラムクロマトグラフィー(溶媒は9:1のジクロロメタン−メタノール)により精製して、0.24のRF(9:1のジクロロメタン−メタノール)を有する化合物8(36 mg、50.7%)を得た。図3を参照せよ。
【0109】
F.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン一リン酸(10)の合成
新たに蒸留したオキシ塩化リン(16.2μL、0.17 mmol)を、−30℃で、リン酸トリメチル(150μL)中のヌクレオシド8(18.8 mg、0.046 mmol)に添加して、対応するジクロロモノホスフェート9を形成した。この反応混合物を、80分間にわたって−5℃まで暖めて、撹拌を、室温でさらに1時間継続した。この反応物を、2 M TEAB緩衝液(pH 8.0)でクエンチし、そして上記のような分取逆相HPLCにより精製した。生成物に対応する画分を濃縮して、モノホスフェート10(12.3 mg、54.56%)を得た。図4を参照せよ。
【0110】
G.5−{3−(2−トリフルオロアセトアミドエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(12)の合成
N,N−ジメチルホルムアミド(200μl)に溶解したモノホスフェート10(7.4 mg、15.3 mmol)を、室温で1時間にわたって、カルボニルジイミダゾール(CDI)(4.2 mg、25.9 mmol)と共に撹拌した。過剰のCDIを、乾燥メタノール(40μL)の添加によりクエンチした。活性化モノホスフェート11を、室温で24時間にわたって、n−トリブチルアミン(16μL)を含有するN,N−ジメチルホルムアミド(160μL)中のピロリン酸トリブチルアンモニウムの溶液と共に撹拌した。この反応物を、2 M TEAB(pH 8.0)でクエンチし、そして上記のような分取逆相HPLCにより精製した。生成物に対応する画分を濃縮して、トリホスフェート12を得た。図5を参照せよ。
【0111】
H.5−{3−(2−アミノエトキシ)−プロピン−1−イル}−2’,3’−ジデオキシシチジン三リン酸(13)の合成
精製し保護したトリホスフェート12を、濃NH4OH水(4 mL)に取り出し、そして室温で2.5時間撹拌した。この反応混合物を濃縮して、プロパルギルエトキシアミドヌクレオチド13を得た。次いで、この濃縮した化合物を、2.6 mMの濃度まで、0.1 M TEAB(pH 7.0)で、バルクとして調合した。調合したバルクの濃度および純度は、それぞれ、UV/Vis分光分析および上記の分析用イオン対HPLCにより、確認した。図5を参照せよ。
【0112】
(実施例4)
5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)[グリシン置換プロパルギルエトキシアミドヌクレオチド]の合成
A.トリフルオロアセトアミドグリシンNHSエステル(15)の合成−−図6A
トリフルオロアセトアミドグリシン14(0.96g、5.6 mmoles)を、室温で1時間にわたって、乾燥DMF(10 mL)中にて、N−ヒドロキシスクシンイミド(0.65g、5.7 mmoles)および1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミドメチオジド(methiodide)(1.68g、5.7 mmoles)と反応させた。この反応溶液を酢酸エチル(200 mL)で希釈し、そして0.1 N HCl(2×100 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮して、このトリフルオロアセトアミドグリシンNHSエステル15(1.43g、100%)を得た。
【0113】
B.5−[3−{2−[N−(2−アミノアセチル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(23)の合成−−図7
100 mM TEA−重炭酸塩(pH 7.0)中のプロパルギルエトキシアミドヌクレオチド13(200μl、15.2 mM)を、乾燥状態までエバポレートした。次いで、それを、250 mMの重炭酸塩緩衝液(pH 9.0)100μlに再懸濁させた。このトリフルオロアセトアミドグリシンNHSエステル15の溶液(50μl、DMSO中で23% w/w)を添加し、そして室温で一晩撹拌した。次いで、この反応混合物を、以下のようにして、分取逆相HPLC(C−18逆相)により精製した:カラム:Aquapore ODS、20μmの粒子サイズ、250×10 mm(PE Applied Biosystems p/n 0711−0163);勾配:0〜50%アセトニトリルで4.5 ml/分にて20分間に続いて、40%アセトニトリル〜100%アセトニトリルで、4.5 ml/分にて10分間。生成物に対応する画分をプールし、そして等容量の100 mM TEA−重炭酸塩(pH 7.0)で希釈し、次いで、真空中で濃縮して、精製し保護したトリホスフェート22を得た。
【0114】
精製し保護したトリホスフェート22を、濃NH4OH水(2 mL)に取り出し、そして室温で一晩撹拌した。この反応混合物を濃縮して、グリシン置換プロパルギルアミノヌクレオチド23を得た。このグリシン置換プロパルギルアミノヌクレオチド23のバルクを、4.84 mMの濃度まで、0.1 M TEAB(pH 7.0)中へのこの反応混合物の溶解により調合した。調合したバルク溶液の濃度および純度は、UV/Vis分光分析および上記実施例3の分析用逆相HPLCにより、確認した。
【0115】
(実施例5)
5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)[メチルアミノ安息香酸(パラ)置換プロパルギルエトキシアミドヌクレオチド]の合成
A.4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル(18)の合成−−図6B
4−アミノメチル安息香酸16(4g、26.5 mmol)を、DMSO(60 mL)およびトリフルオロ酢酸メチル(3 mL)に溶解し、この溶液に、トリエチルアミン(3 mL)を添加した。60℃で5時間撹拌した後、この反応溶液を酢酸エチル(500 mL)で希釈し、そして水(6×200 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮して、白色の固形物として、トリフルオロアセトアミドメチル安息香酸17(4.6g、70.3%)を得た。
【0116】
このトリフルオロアセトアミドメチル安息香酸17(4.49g、18.2 mmol)を、室温で2時間にわたり、酢酸エチル100 ml中にて、N−ヒドロキシスクシンイミド(2.09g、18.2 mmol)および1,3−ジシクロヘキシルカルボジイミド(3.74g、18.2 mmol)と反応させた。この反応溶液を濾過して、白色沈殿物を除去し、その有機層を1N HCl(100 mL)で洗浄した。次いで、この酢酸エチル抽出物を硫酸ナトリウムで乾燥し、そして濃縮した。この濃縮物を、以下のステップ勾配を用いるシリカゲル固定相を用いた順相カラムクロマトグラフィーにより、クロマトグラフにかけて、白色の固形物(3.5g、56%)として、4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル18を得た:40:1のジクロロメタン:酢酸エチルに続いて、10:1のジクロロメタン:酢酸エチル。
【0117】
B.5−[3−{2−[N−(4−アミノメチルベンゾイル)]アミノエトキシ}プロピン−1−イル]−2’,3’−ジデオキシシチジン三リン酸(25)の合成−−図8
100 mM TEA−重炭酸塩(pH 7.0)中のプロパルギルエトキシアミドヌクレオシド13(200μl、15.2 mM)を、乾燥状態までエバポレートした。次いで、それを、250 mM重炭酸塩緩衝液(pH 9.0)150μlに再懸濁させた。4−(トリフルオロアセトアミドメチル)安息香酸NHSエステル18の溶液(100μl、100μlのDMSO中で25 mg)を添加し、そして室温で一晩撹拌した。この反応混合物を、実施例4にて上で記述した分取逆相HPLCにより精製した。生成物に対応する画分をプールし、そして等容量の100 mM TEA−重炭酸塩(pH 7.0)で希釈し、次いで、真空中で濃縮して、精製し保護したトリホスフェート22を得た。
【0118】
この精製し保護したトリホスフェート22を、濃NH4OH水(2 mL)に取り出し、そして室温で一晩撹拌した。この反応混合物を濃縮して、メチルアミノ安息香酸(パラ)置換プロパルギルアミノヌクレオチド25を得た。次いで、この置換プロパルギルアミノヌクレオチド25のバルク溶液を、0.1 M TEAB(pH 7.0)へのこの反応混合物の溶解により調合した。調合したバルク溶液の濃度および純度は、それぞれ、UV/Vis分光分析および上記実施例3の分析用逆相HPLCにより確認した。
【0119】
(実施例6)
置換プロパルギルエトキシアミドヌクレオチドへの色素の結合
100 mM TEA−重炭酸塩(pH 7.0)中の置換プロパルギルエトキシアミドヌクレオチドを、乾燥状態まで蒸発させた。次いで、それを、250 mM重炭酸塩緩衝液(pH 9.0)に再懸濁させた。所望の色素−NHS溶液(DMSO中)を添加し、そして暗所中にて、室温で一晩撹拌した。この反応混合物を、上記の分取アニオン交換HPLCにより精製した。生成物に対応する画分を濃縮し、そして上記の分取逆相HPLCにより再精製した。最終生成物を真空中で乾燥し、そして1 mMの濃度まで、50 mM CAPSO(pH 9.6)で希釈した。調合したバルクの濃度および純度は、それぞれ、UV/VIS分光分析および上記の分析用イオン対HPLCにより確認した。
【0120】
(実施例7)
本発明の置換プロパルギルエトキシアミドジデオキシヌクレオチドを用いて改良したピーク高さ均等性
図10は、HEX−1−標識ddCTPを用いた単色配列決定反応の電気泳動図であり、この場合、このHEX−1色素は、3個の異なるタイプのリンカーのそれぞれを用いて、ターミネーターに結合している。上記ターミネーター滴定アッセイに基づき、各リンカータイプについて、その最適なターミネーター濃度を使用した。上のパネルは、50 pmでのプロパルギルエトキシアミドリンカーを用いた結果を示し、中央のパネルは、25 pmでのメチルアミノ安息香酸(パラ)置換プロパルギルエトキシアミドリンカーを用いた結果を示し、下のパネルは、250 pmでのグリシン置換プロパルギルエトキシアミドリンカーを用いた結果を示す。図10の上の電気泳動図における番号により示されるように、図10で示した配列決定ラダー部分は、−21 M13配列決定プライマー(前方)を用いたpGEM−3Zf(+)の配列の塩基63から塩基104までに及んでいる。各実験の相対誤差は、以下のようであり、この場合、本明細書中で使用する用語「相対誤差」は、この図で示した電気泳動図部分にわたるピーク高さの比または標準偏差および中間ピーク高さ(mean peak height)を意味する。
【0121】
【表4】
全ての文献および特許出願の内容は、個々の文献または特許出願のそれぞれの内容が具体的かつ個別的に本明細書中で参考として援用されているかのように、本明細書中で参考として援用されている。
【0122】
少数の実施態様だけを上で詳細に記述しているものの、化学分野の当業者は、その好ましい実施態様において、それらの教示から逸脱することなく、多くの改良が可能であることを、充分に理解している。このような改良の全ては、以下の請求の範囲の範囲内に包含されることを意図している。
【特許請求の範囲】
【請求項1】
本明細書に記載の発明。
【請求項1】
本明細書に記載の発明。
【図1A】

【図1B】
【図1C】
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7】
【図8】
【図9】
【図10】
【図1B】
【図1C】
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2009−143946(P2009−143946A)
【公開日】平成21年7月2日(2009.7.2)
【国際特許分類】
【出願番号】特願2009−26772(P2009−26772)
【出願日】平成21年2月6日(2009.2.6)
【分割の表示】特願平10−542803の分割
【原出願日】平成10年3月23日(1998.3.23)
【出願人】(500069057)アプライド バイオシステムズ インコーポレイテッド (120)
【Fターム(参考)】
【公開日】平成21年7月2日(2009.7.2)
【国際特許分類】
【出願日】平成21年2月6日(2009.2.6)
【分割の表示】特願平10−542803の分割
【原出願日】平成10年3月23日(1998.3.23)
【出願人】(500069057)アプライド バイオシステムズ インコーポレイテッド (120)
【Fターム(参考)】
[ Back to top ]