
置換5−オキサゾール−2−イル−キノリン化合物のキシナホ酸塩
【課題】置換5−オキサゾール−2−イル−キノリン化合物のキシナホ酸塩の提供。
【解決手段】下記の式の化合物、上部閉塞性気道疾患および下部閉塞性気道疾患を、上記化合物を使用して処置する方法、上記化合物を含む組成物、ならびに、多形体、および、多形形態を合成するプロセス。

【解決手段】下記の式の化合物、上部閉塞性気道疾患および下部閉塞性気道疾患を、上記化合物を使用して処置する方法、上記化合物を含む組成物、ならびに、多形体、および、多形形態を合成するプロセス。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、1−[[5−(1(S)−アミノエチル)−2−[8−メトキシ−2−(トリフルオロメチル)−5−キノリル]−4−オキサゾリル]カルボニル]−4(R)−[(シクロプロピルカルボニル)アミノ]−L−プロリン、エチルエステルのキシナホ酸塩、上記塩を含む医薬組成物、ならびに、気道の上部閉塞性疾患および下部閉塞性疾患を上記塩の吸入によって処置する方法に関連する。
【背景技術】
【0002】
(発明の背景)
様々なホスホジエステラーゼが、環状AMPを調節することが知られており、また、ホスホジエステラーゼ4(PDE4)が、気道平滑筋および炎症性細胞における環状AMPの主要な調節因子であることが示されている。PDE4の阻害剤は、アレルギー性疾患および炎症性疾患、糖尿病、中枢神経系疾患、疼痛、ならびに、TNFを産生するウイルスを含む様々な疾患の処置に有用である。
【0003】
アミノ置換キノリルのPDE4阻害剤が特許文献1に開示され、スルホンアミド置換キノリルのPDE4阻害剤が特許文献2に開示され、また、(ベンゼン縮合)ヘテロアリール置換のPDE4阻害剤が特許文献3に開示される。オキサゾリル置換のPDE4阻害剤がPCT/US2005/017134に開示される。
【0004】
本明細書中において化合物Aとして示される化合物が、特許文献4において、95頁、実施例26〜実施例347および228頁(請求項19)に、その遊離塩基および医薬的に許容され得る塩形態として記載される(それらの記載は本明細書に参考として組み込まれる)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第5,804,588号明細書
【特許文献2】米国特許第5,834,485号明細書
【特許文献3】米国特許第6,069,151号明細書
【特許文献4】国際公開第2005/116009号パンフレット
【発明の概要】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、1−[[5−(1(S)−アミノエチル)−2−[8−メトキシ−2−(トリフルオロメチル)−5−キノリル]−4−オキサゾリル]カルボニル]−4(R)−[(シクロプロピルカルボニル)アミノ]−L−プロリン、エチルエステルのキシナホ酸塩(すなわち、下記式Iの化合物:
【0007】
【化1】
)を提供する。
【0008】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法に関し、有効量の式1の化合物を吸入によって上記患者に投与することを含む。
【0009】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法に関し、式1の化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを吸入によって上記患者に投与することを含む。好ましいさらなる薬剤は、β−アゴニスト、ムスカリンアンタゴニストまたはコルチコステロイドである。
【0010】
本発明はさらに、有効量の式Iの化合物を含む吸入可能な医薬組成物に関する。
【0011】
本発明はさらに、式1の化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを含む吸入可能な医薬組成物に関する。
【0012】
本発明はまた、式Iの化合物の結晶性多形体および偽多形体(水和物)に関し、上記多形体は、
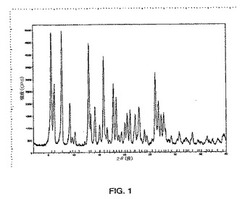
図1に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態1、
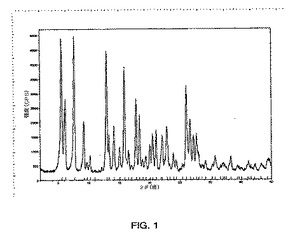
図2に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態2、および
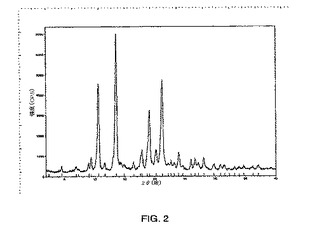
図3に示されるパターンと実質的に同じである粉末x線回折パターンを示す二水和物形態1、
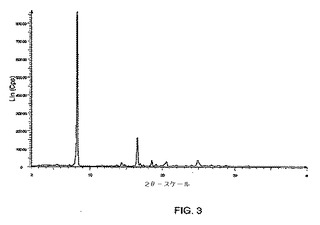
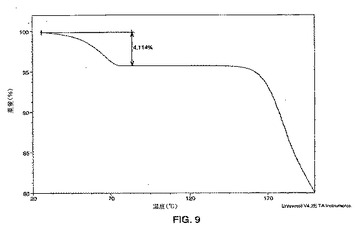
図10に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態3、からなる群から選択される。
【0013】
本発明はさらに、6.1度、7.7度、13.0度および15.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態1を提供する。
【0014】
別の実施形態において、式Iの結晶性多形体形態1は、5.6度、6.1度、7.7度、13.0度、15.9度、17.8度、18.4度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0015】
別の実施形態において、式Iの結晶性多形体形態1は、5.6度、6.1度、7.7度、9.2度、13.0度、14.2度、15.9度、17.8度、18.4度、20.5度、22.9度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0016】
本発明はさらに、10.6度、13.6度、19.1度および21.2度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態2を提供する。
【0017】
別の実施形態において、式Iの結晶性多形体形態2は、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度および23.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0018】
別の実施形態において、式Iの結晶性多形体形態2は、9.4度、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度、23.9度、26.0度、26.6度および28.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0019】
本発明はさらに、8.2度、16.5度、18.5度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性二水和物形態1を提供する。
【0020】
別の実施形態において、式Iの結晶性二水和物形態1は、5.5度、8.2度、14.3度、16.5度、16.9度、18.5度、20.6度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0021】
別の実施形態において、式Iの結晶性二水和物形態1は、5.5度、7.2度、8.2度、14.3度、14.7度、16.5度、16.9度、18.5度、20.6度、24.1度、24.9度および26.8度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0022】
本発明はさらに、4.6度、7.9度、12.1度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態3を提供する。
【0023】
別の実施形態において、式Iの結晶性多形体形態3は、4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0024】
別の実施形態において、式Iの結晶性多形体形態3は、4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度、18.9度、20.0度、23.9度、24.3度および25.7度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0025】
本発明はさらに、形態1の多形体キシナホ酸塩を化合物Aから調製するための2つのプロセスを提供する。
【0026】
第1の方法:
【0027】
【化2】
この方法は下記の工程を含む。
【0028】
a)化合物Aを熱エタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)さらなるエタノールおよび水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、混合物を、形態1の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態1の結晶をろ過する工程。
【0029】
第2の方法:
【0030】
【化3】
この方法は下記の工程を含む。
【0031】
a)トルエンおよびメタノールを化合物Aおよびキシナホ酸に加え、混合し、これにより、スラリーを形成する工程、
b)上記スラリーを混合しながら約62℃に加熱し、これにより、均一な混合物を得る工程、
c)上記均一な混合物を大気圧で蒸留し、蒸留された混合物を約50℃に冷却し、上記蒸留された混合物に化合物Aの形態1の種結晶を接種し、これにより、スラリー状の結晶を生じさせる工程、
d)上記スラリーを約50℃で約30分間撹拌し、スラリーを約10℃に冷却する工程、
e)さらなるトルエンを上記冷却したスラリーに加え、真空蒸留し、その後、さらなるトルエンを加え、約20℃で約20分間撹拌し、これにより、固体物質を形成させる工程、
f)得られた固体を、撹拌型乾燥機を真空下で使用して収集し、これにより、湿潤ケーキを形成し、次いで、上記湿潤ケーキをトルエンにより洗浄し、撹拌を伴うことなく約3時間の約50℃での乾燥、その後、約20R.P.M.の撹拌を伴う約12時間の約80℃での乾燥、その後、約60R.P.M.の撹拌を伴う約12時間の約80℃での乾燥をすべて真空下で行う工程。
【0032】
第3の方法:
【0033】
【化4】
この方法は下記の工程を含む。
【0034】
a)化合物Aおよびキシナホ酸を熱メタノールに別々に溶解する工程、
b)熱溶液の両方をろ過し、これら2つの溶液を混合する工程、
c)混合物を還流し、過剰なメタノールを留去する工程、および
d)混合物を0℃に冷却し、これにより、沈殿物を形成させ、形態1の結晶をろ過する工程。
【0035】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性多形体形態1を提供する。
【0036】
本発明はさらに、式2の多形体キシナホ酸塩を、化合物A:
【0037】
【化5】
から調製するためのプロセスを提供し、このプロセスは、
a)化合物Aを熱メタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、混合物を、形態2の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態2の結晶をろ過する工程
を含む。
【0038】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性形態2の多形体キシナホ酸塩を提供する。
【0039】
本発明はさらに、二水和物形態1を、化合物A:
【0040】
【化6】
の形態1の多形体キシナホ酸塩から調製するためのプロセスを提供し、このプロセスは、下記の工程を含む:
a)キシナホ酸塩形成時における水の添加が、結晶性形態を得るために必要である。化合物Aの形態1の多形体キシナホ酸塩を、水およびメタノールの混合物に懸濁させる、
b)懸濁物を21時間撹拌し、懸濁物を遠心分離して、その後、上清をデカンテーションにより除くことによって固体を単離した、
c)固体を真空下において室温で乾燥した。
【0041】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性二水和物形態2のキシナホ酸塩を提供する。
【0042】
本発明はさらに、形態3の多形体キシナホ酸塩を、化合物A:
【0043】
【化7】
から調製するためのプロセスを提供し、このプロセスは、
a)化合物Aおよびキシナホ酸の混合物を2−プロパノール中で合わせる工程、
b)混合物を加熱して還流させ、さらに2−プロパノールを加え、次いで、混合物を還流状態で約1時間保ち、その後、室温まで冷却する工程、
c)混合物をろ過し、固体を2−プロパノールにより洗浄し、真空下で乾燥する工程
を含む。
【0044】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性形態3の多形体キシナホ酸塩を提供する。
【0045】
本発明はさらに、式Iの化合物の形態1の多形体の精製された形態を提供する。
【0046】
本発明はさらに、式Iの化合物の形態2の多形体の精製された形態を提供する。
【0047】
本発明はさらに、式Iの化合物の二水和物形態1の精製された形態を提供する。
【0048】
本発明はさらに、式Iの化合物の形態3の多形体の精製された形態を提供する。
【0049】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態1の多形体、ならびに有効量の式Iの化合物の形態1の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0050】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態2の多形体、ならびに有効量の式Iの化合物の形態2の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0051】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の二水和物形態1、ならびに、有効量の式Iの化合物の二水和物形態1と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0052】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態3の多形体、ならびに有効量の式Iの化合物の形態3の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
例えば、本願発明は以下の項目を提供する。
(項目1)
下記の構造式:
【化23】
を有する化合物。
(項目2)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目1に記載される化合物を吸入によって該患者に投与することを含む、方法。
(項目3)
前記処置される疾患が喘息または慢性閉塞性肺疾患である、項目2に記載の方法。
(項目4)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、項目1に記載される化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを吸入によって該患者に投与することを含む、方法。
(項目5)
前記処置される疾患が喘息または慢性閉塞性肺疾患である、項目4に記載の方法。
(項目6)
前記さらなる薬剤が、β−アゴニスト、ムスカリンアンタゴニストおよびコルチコステロイドからなる群から選択される、項目4に記載の方法。
(項目7)
有効量の項目1に記載される化合物を含む吸入可能な医薬組成物。
(項目8)
項目1に記載される化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを含む吸入可能な医薬組成物。
(項目9)
前記さらなる薬剤が、β−アゴニスト、ムスカリンアンタゴニストおよびコルチコステロイドからなる群から選択される、項目8に記載の組成物。
(項目10)
下記式の化合物:
【化24】
の結晶性多形体であって、該多形体が:
図1に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態1、
図2に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態2、および
図3に示されるパターンと実質的に同じである粉末x線回折パターンを示す二水和物形態1、
図10に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態3、からなる群から選択される、結晶性多形体。
(項目11)
6.1度、7.7度、13.0度および15.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態1。
(項目12)
5.6度、6.1度、7.7度、13.0度、15.9度、17.8度、18.4度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目11に記載の結晶性多形体。
(項目13)
5.6度、6.1度、7.7度、9.2度、13.0度、14.2度、15.9度、17.8度、18.4度、20.5度、22.9度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目11に記載の結晶性多形体。
(項目14)
項目10に記載の結晶性多形体形態1。
(項目15)
10.6度、13.6度、19.1度および21.2度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態2。
(項目16)
10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度および23.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目15に記載の結晶性多形体。
(項目17)
9.4度、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度、23.9度、26.0度、26.6度および28.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目15に記載の結晶性多形体。
(項目18)
項目10に記載の結晶性多形体形態2。
(項目19)
8.2度、16.5度、18.5度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性二水和物形態1。
(項目20)
5.5度、8.2度、14.3度、16.5度、16.9度、18.5度、20.6度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目19に記載の結晶性二水和物。
(項目21)
5.5度、7.2度、8.2度、14.3度、14.7度、16.5度、16.9度、18.5度、20.6度、24.1度、24.9度および26.8度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目19に記載の結晶性二水和物。
(項目22)
項目10に記載の結晶性二水和物形態1。
(項目23)
4.6度、7.9度、12.1度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態3。
(項目24)
4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目23に記載の結晶性多形体。
(項目25)
4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度、18.9度、20.0度、23.9度、24.3度および25.7度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目23に記載の結晶性多形体。
(項目26)
項目10に記載の結晶性多形体形態3。
(項目27)
項目10に記載される多形体形態1を、化合物A:
【化25】
から調製するためのプロセスであって、
a)化合物Aを熱エタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)さらなるエタノールおよび水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、該混合物を、形態1の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態1の結晶をろ過する工程
を含む、プロセス。
(項目28)
項目10に記載される多形体形態1を、化合物A:
【化26】
から調製するためのプロセスであって、
e)トルエンおよびメタノールを化合物Aおよびキシナホ酸に加え、混合し、これにより、スラリーを形成する工程、
f)該スラリーを混合しながら約62℃に加熱し、これにより、均一な混合物を得る工程、
g)該均一な混合物を大気圧で蒸留し、蒸留された混合物を約50℃に冷却し、該蒸留された混合物に化合物Aの形態1の種結晶を接種し、これにより、スラリー状の結晶を生じさせる工程、
h)該スラリーを約50℃で約30分間撹拌し、スラリーを約10℃に冷却する工程、
i)さらなるトルエンを該冷却したスラリーに加え、真空蒸留し、その後、さらなるトルエンを加え、約20℃で約20分間撹拌し、これにより、固体物質を形成させる工程、
j)得られた固体を、撹拌型乾燥機を真空下で使用して収集し、これにより、湿潤ケーキを形成させ、次いで、該湿潤ケーキをトルエンにより洗浄し、撹拌を伴うことなく約3時間の約50℃での乾燥、その後、約20R.P.M.の撹拌を伴う約12時間の約80℃での乾燥、その後、約60R.P.M.の撹拌を伴う約12時間の約80℃での乾燥をすべて真空下で行う工程
を含む、プロセス。
(項目29)
項目10に記載される多形体形態1を、化合物A:
【化27】
から調製するためのプロセスであって、
k)化合物Aおよびキシナホ酸を熱メタノールに別々に溶解する工程、
l)両方の熱溶液をろ過し、これら2つの溶液を混合する工程、
m)混合物を還流し、過剰なメタノールを留去する工程、および
n)混合物を0℃に冷却し、これにより、沈殿物を形成させ、形態1の結晶をろ過する工程
を含む、プロセス。
(項目30)
項目10に記載される形態1の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目31)
項目10に記載される形態1の多形体の精製された形態。
(項目32)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態1の多形体を吸入によって該患者に投与することを含む、方法。
(項目33)
項目10に記載される多形体形態2を、化合物A:
【化28】
から調製するためのプロセスであって、
o)化合物Aを熱メタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
p)水を加え、混合物を沸騰近くまで加熱する工程、
q)熱混合物をろ過し、その後、室温までゆっくり冷却し、該混合物を、形態2の結晶が沈殿するまで室温で一晩放置する工程、および
r)ろ液を0℃に冷却し、形態2の結晶をろ過する工程
を含む、プロセス。
(項目34)
項目10に記載される形態2の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目35)
項目10に記載される形態2の多形体の精製された形態。
(項目36)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態2の多形体を吸入によって該患者に投与することを含む、方法。
(項目37)
項目10に記載される二水和物形態1を、化合物A:
【化29】
のキシナホ酸塩多形体形態1から調製するためのプロセスであって、
r)化合物Aの形態1の多形体キシナホ酸塩を水およびメタノールの混合物に懸濁させる工程、
s)懸濁物を21時間撹拌し、懸濁物を遠心分離して、その後、上清をデカンテーションにより除くことによって固体を単離する工程、
t)固体を真空下において室温で乾燥させる工程
を含む、プロセス。
(項目38)
項目10に記載される二水和物形態1の結晶と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目39)
項目10に記載される結晶性二水和物形態1の精製された形態。
(項目40)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される二水和物形態1を吸入によって該患者に投与することを含む、方法。
(項目41)
項目10に記載される多形体形態3を、化合物A:
【化30】
から調製するためのプロセスであって、
u)化合物Aおよびキシナホ酸の混合物を2−プロパノール中で合わせる工程、
v)混合物を加熱して還流させ、さらに2−プロパノールを加え、次いで、混合物を還流状態で1時間保ち、その後、室温まで冷却する工程、
w)混合物をろ過し、固体を2−プロパノールで洗浄し、真空下で乾燥させる工程
を含む、プロセス。
(項目42)
項目10に記載される形態3の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目43)
項目10に記載される形態3の多形体の精製された形態。
(項目44)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態3の多形体を吸入によって該患者に投与することを含む、方法。
【図面の簡単な説明】
【0053】
【図1】X線回折計を使用して得られる、式Iの化合物の形態1の粉末x線回折(PXRD)パターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図2】X線回折計を使用して得られる、式Iの化合物の形態2のPXRDパターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図3】X線回折計を使用して得られる、式Iの化合物の二水和物形態1のPXRDパターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図4】化合物10(工程8の生成物)のNMRスペクトルの写しである。
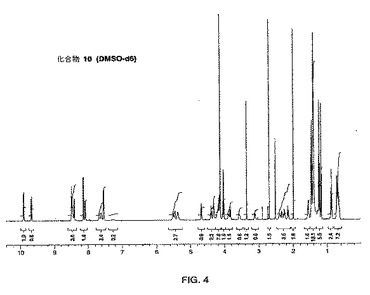
【図5】化合物11(化合物Aとも呼ばれる)のNMRスペクトルの写しである。
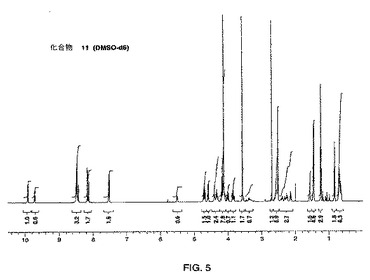
【図6】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態1の熱分析のプロットである。
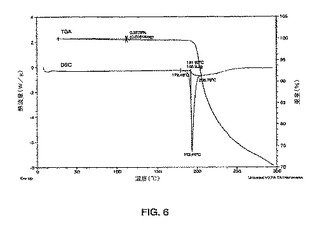
【図7】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態2の熱分析のプロットである。
【図8】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の二水和物形態1の熱分析のプロットである。
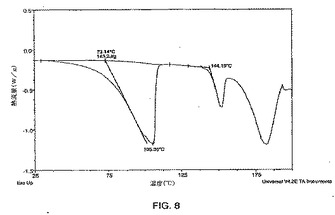
【図9】熱重量分析(TGA)によって得られる、式Iの化合物の二水和物形態1の熱重量分析のプロットである。
【図10】X線回折計を使用して得られる、式Iの化合物の形態3のPXRDパターンのグラフである。グラフでは、カウント/秒によって定義されるようなピークの強度が、度での回折角度2θに対してプロットされる。
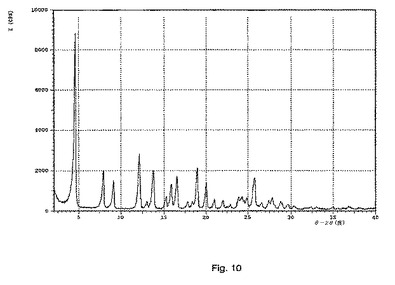
【図11】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態3の熱分析のプロットである。
【発明を実施するための形態】
【0054】
(詳細な説明)
式Iの遊離塩基(これは本明細書中下記では化合物Aとして示され、下記の構造:
【0055】
【化8】
を有する)が、PCT/US2005/017134(これは参考として本明細書中に組み込まれる)において実施例26〜実施例381として開示される。
【0056】
式Iの化合物、すなわち、化合物Aのキシナホ酸塩は非吸湿性の結晶性塩であり、3つの多形体および1つの水和物を示す。
【0057】
式Iの化合物は、化合物Aまたは化合物Aの他の塩と比較して、吸入によって投与される場合、気道の上部閉塞性疾患および下部閉塞性疾患を処置するための予想外に優れた物理的プロフィルおよび薬物動態学プロフィルを有する。リン酸塩、マレイン酸塩およびコハク酸塩は非晶質である。酒石酸塩は結晶性であるが吸湿性である。フマル酸塩は結晶性の形態を有しているが、それらの形態は不安定な水和物である。従って、キシナホ酸塩は、他の塩と比較して、吸入処方物における使用について、他の塩よりも予想外に優れている。そのうえ、キシナホ酸塩は、経口投与と比較して、気管内投与により、25倍高い炎症性細胞の阻害を示す。
【0058】
式Iの化合物には3つの異なった結晶性多形体および1つの水和物が存在することが見出された。これら4つの形態は本明細書中では、形態1、形態2、形態3および二水和物形態1として示される。この化合物は治療活性医薬剤としての使用が意図されているので、式Iの化合物の最も安定な医薬的に許容され得る形態は非常に注目される。
【0059】
形態1が、本発明の方法における使用のための好ましい形態である。
【0060】
多形は、化合物が、同じ化学式を維持しながら、異なる結晶形態に結晶化し得る能力として特徴づけることができる。所与の薬物物質の結晶性多形体は、同じ様式で互いに結合する同じ原子を含むという点で、この薬物物質の任意の他の結晶性多形体と化学的に同一であるが、1つまたは複数の物理的性質(例えば、安定性、溶解性、融点、嵩密度、流動特性、バイオアベイラビリティなど)に影響を及ぼし得るその結晶形態において異なる。
【0061】
本明細書を通して使用される下記の用語は、別途指示されない限り、下記の意味を有することが理解される。
【0062】
「患者」はヒトおよび他の動物の両方を包含する。
【0063】
「哺乳動物」はヒトおよび他の哺乳類動物を包含する。
【0064】
「多形体」は、別の結晶性形態とは異なるが、同じ化学式をともに有する、物質の結晶性形態を意味する。
【0065】
「本発明の多形体」は式Iの化合物の結晶性多形体を意味する。
【0066】
「アルコール」は、ヒドロキシル基(−OH)を含む有機化合物を意味する。
【0067】
「賦形剤」は、希釈剤として使用されるか、あるいは、形態またはコンシステンシーを処方物に与えるために使用される本質的に不活性な物質を意味する。
【0068】
「有効」または「治療有効」は、PDE4阻害剤として効果的であり、従って、所望される治療効果、改善効果、阻害効果または防止効果をもたらす、本発明の化合物の多形体または組成物を表すことが意味される。「有効量」または「治療有効量」は、PDE4阻害剤として効果的であり、従って、所望される治療効果、改善効果、阻害効果または防止効果をもたらす、本発明の多形体または組成物の量を表すことが意味される。
【0069】
式Iの化合物によって処置される上部気道閉塞性疾患および下部気道閉塞性疾患には、喘息、COPD(慢性閉塞性肺疾患)、慢性気管支炎、嚢胞性線維症、アレルギー性鼻炎、非アレルギー性鼻炎、鼻副鼻腔炎、成人呼吸疾患、急性呼吸窮迫症候群、呼吸器系ウイルス、咳、間質性肺炎、慢性副鼻腔炎、気流閉塞、気道過応答(すなわち、気道過敏性)、気管支拡張症、細気管支炎、閉塞性細気管支炎(すなわち、閉塞性細気管支炎症候群)、呼吸困難、気腫、高炭酸ガス症、過膨張、低酸素血症、高酸素症誘発性炎症、肺線維症、肺高血圧症、小気道疾患、喘鳴および風邪が含まれる。
【0070】
式Iの化合物は好ましくは、喘息、COPD、咳、気流閉塞、気道過応答(すなわち、気道過敏性)、細気管支炎、慢性気管支炎、気腫、肺線維症、肺高血圧症、小気道疾患、喘鳴およびアレルギー性鼻炎の処置に有用である。
【0071】
より好ましくは、式Iの化合物は、COPDおよび喘息を処置するために有用である。
【0072】
式Iの化合物と組合せて使用するための、閉塞性気道疾患(例えば、COPDまたは喘息)を処置するための他の薬剤は、ステロイド剤(例えば、グルココルチコイド剤)、5−リポキシゲナーゼ阻害剤、β−2アドレナリン受容体アゴニスト、α−アドレナリン作動性受容体アゴニスト、ムスカリンM1アンタゴニスト、ムスカリンM3アンタゴニスト、ムスカリンM2アンタゴニスト、LTB4アンタゴニスト、システイニルロイコトリエンアンタゴニスト、気管支拡張剤、PDE4阻害剤、エラスターゼ阻害剤、MMP阻害剤、ホスホリパーゼA2阻害剤、ホスホリパーゼD阻害剤、ヒスタミンH1アンタゴニスト、ヒスタミンH3アンタゴニスト、ドーパミンアゴニスト、アデノシンA2アゴニスト、NK1アンタゴニスト、NK2アンタゴニスト、NK3アンタゴニスト、GABA−bアゴニスト、ノシセプチンアゴニスト、去痰剤、粘液溶解剤、うっ血除去剤、マスト細胞安定化剤、抗酸化剤、抗IL−8抗体、抗IL−5抗体、抗IgE抗体、抗TNF抗体、IL−10、接着分子阻害剤、成長ホルモンおよび他のPDE4阻害剤からなる群から選択される。
【0073】
式Iの化合物と組合せて使用するための、抗ヒスタミン剤の限定されない例には、アステミゾール、アザタジン、アゼラスチン、アクリバスチン、ブロムフェニルアミン、セルチリジン(certirizine)、クロルフェニラミン、クレマスチン、シクリジン、カレバスチン(carebastine)、シプロヘプタジン、カルビノキサミン、デスカルボエトキシロラタジン(descarboethoxyloratadine)、ドキシラミン、ジメチンデン、エバスチン、エピナスチン、エフレチリジン(efletirizine)、フェキソフェナジン、ヒドロキシジン、ケトチフェン、ロラタジン、レボカバスチン、ミゾラスチン、エキタジン(equitazine)、ミアンセリン、ノベラスチン(noberastine)、メクリジン、ノルアステミゾール(norastemizole)、ピクマスト(picumast)、ピリラミン、プロメタジン、テルフェナジン、トリペレンアミン、テメラスチン(temelastine)、トリメプラジンおよびトリプロリジンが含まれる。
【0074】
ヒスタミンH3受容体アンタゴニストの限定されない例には、チオペラミド、インプロミジン、ブリマミド、クロベンプロピト(clobenpropit)、インペンタミン(impentamine)、ミフェチジン(mifetidine)、S−ソプロミジン(sopromidine)、R−ソプロミジン、SKF−91486、GR−175737、GT−2016、UCL−1199およびクロザピンが含まれる。他の化合物を、H3受容体への活性を明らかにするために種々の公知の方法で容易に評価することができ、そのような方法には、モルモット脳膜アッセイおよびモルモットニューロン性回腸収縮アッセイが含まれる(これらはともに米国特許第5,352,707号に記載される)。別の有用なアッセイでは、ラットの脳膜が利用され、このアッセイが、Westら、「Identification of Two−H3−Histamine Receptor Subtypes」、Molecular Pharmacology、第38巻、610頁〜613頁(1990)によって記載される。
【0075】
用語「ロイコトリエン阻害剤」には、ロイコトリエンの作用または活性を阻害するか、あるいは抑制するか、あるいは阻止するか、あるいは、そうでない場合には、ロイコトリエンの作用または活性と相互作用する任意の薬剤または化合物が含まれる。ロイコトリエン阻害剤の限定されない例には、モンテルカストおよびそのナトリウム塩、1−(((R)−(3−(2−(6,7−ジフルオロ−2−キノリニル)エテニル)フェニル)−3−(2−(2−ヒドロキシ−2−プロピル)フェニル)チオ)メチルシクロプロパン酢酸およびそのナトリウム塩(これらは米国特許第5,270,324号に記載される)、1−(((1(R)−3(3−(2−(2,3−ジクロロチエノ[3,2−b]ピリジン−5−イル)−(E)−エテニル)フェニル)−3−(2−(1−ヒドロキシ−1−メチルエチル)フェニル)プロピル)チオ)メチル)シクロプロパン酢酸およびそのナトリウム塩(これらは米国特許第5,472,964号に記載される)、プランルカスト;ザフィルルカスト、ならびに[2−[[2(4−tert−ブチル−2−チアゾリル)−5−ベンゾフラニル]オキシメチル]フェニル]酢酸(これは米国特許第5,296,495号に記載される)が含まれる。
【0076】
β−アドレナリン作動性受容体アゴニストの限定されない例には、アルブテロール、ビトルテロール、イソエタリン、マタプロテレノール(mataproterenol)、ペルブテロール(perbuterol)、サルメテロール、テルブタリン、イソプロテレノール、エフェドリンおよびエピネフリンが含まれる。α−アドレナリン作動性受容体アゴニストの限定されない例には、アリールアルキルアミン(例えば、フェニルプロパノールアミンおよびプソイドエフェドリン)、イミダゾール(例えば、ナファゾリン、オキシメタゾリン、テトラヒドロゾリンおよびキシロメタゾリン)およびシクロアルキルアミン(例えば、プロピルヘキセドリン)が含まれる。
【0077】
マスト細胞安定化剤の限定されない一例がネドクロミルナトリウムである。去痰剤の限定されない一例がグアイフェネシンである。うっ血除去剤の限定されない例には、プソイドエフェドリン、フェニルプロパノールアミンおよびフェニレフリンがある。
【0078】
他のPDE4阻害剤の限定されない例には、ロフルミラスト、テオフィリン、ロリプラム、ピクラミスト(piclamist)、シロミラストおよびCDP−840が含まれる。ステロイド剤の例には、プレドニゾロン、フルチカゾン、トリアムシノロン、ベクロメタゾン、モメタゾン、ブジサミド(budisamide)、ベタメタゾン、デキサメタゾン、プレドニゾン、フルニソリドおよびコルチゾンが含まれる。
【0079】
NK1タキキニン受容体アンタゴニスト、NK2タキキニン受容体アンタゴニストおよびNK3タキキニン受容体アンタゴニストの限定されない例には、CP−99,994およびSR48968が含まれる。ムスカリンアンタゴニストの限定されない例には、臭化イプラトロピウムおよび臭化チアトロピウムが含まれる。
【0080】
GABABアゴニストの限定されない例には、バクロフェンおよび3−アミノプロピル−ホスフィン酸が含まれる。ドーパミンアゴニストには、キンピロール、ロピニロール、プラミペキソール、ペルゴリドおよびブロモクリプチンが含まれる。
【0081】
「5−リポキシゲナーゼ阻害剤」には、5−リポキシゲナーゼの酵素作用を阻害するか、または抑制するか、または阻止するか、または、そうでない場合には、5−リポキシゲナーゼの酵素作用と相互作用する任意の薬剤または化合物が含まれる。5−リポキシゲナーゼ阻害剤の限定されない例には、ジロートン、ドセベノン(docebenone)、ピリポスト(piripost)、ICI−D2318およびABT761が含まれる。
【0082】
式Iの化合物は、スキーム1またはスキーム2に概説され、また、下記の実施例1または実施例2に詳しく記載される手順によって調製された。本出願明細書における実施例1および他の箇所において、Etはエチルを意味し、Meはメチルを意味し、THFはテトラヒドロフランであり、DMFはN,N−ジメチルホルムアミドであり、t−BOCおよびBOCはt−ブトキシカルボニルを意味し、RTは室温であり、HATUはN−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシドである。
【0083】
【化9】
【実施例】
【0084】
(実施例1)
工程1:
化合物1(100.6g、0.767mol)をEtOH(1000ml)に懸濁し、0℃に冷却した機械的に撹拌されているこの懸濁液に、SOCl2(136.9g、1.15mol、84.0ml)を、内部温度が15℃未満であるように滴下漏斗により滴下した。反応混合物を2.5時間にわたって加熱還流し、その後、0℃に冷却した。エーテル(1000ml)を加え、白色の固体が沈殿した。固体を真空ろ過によって単離し、エーテルにより洗浄した。生成物2(HCl塩)を真空オーブンで乾燥して、146.3g(97%)の白色の固体を得た。MS(M+1):m/e 160。
【0085】
【化10】
工程2:
化合物2(HCl塩、146.2g、0.747mol)をCH2Cl2(1600ml)およびEtOH(100ml)に溶解し、0℃に冷却したこの溶液に、Et3N(113.4g、1.12mol、156.2ml)を加えた。t−BOC無水物(195.6、0.90mol)を少量ずつ加えた。反応混合物を0℃で15分間撹拌し、その後、RTで16時間撹拌した。得られた混合物を約800mlの体積まで濃縮し、水により洗浄した。有機溶液を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:20%EtOAc−CH2Cl2)により、生成物3(193.7g、100%)を黄色の油状物として得た。MS(M+Na):m/e 282。
【0086】
【化11】
工程3:
化合物3(36.5g、0.141mol)およびトリフェニルホスフィン(46.2g、0.176mol)を乾燥THF(1000ml)に溶解し、0℃に冷却したこの溶液に、ジエチルアゾジカルボキシラート(30.7g、0.176mol)を滴下漏斗により滴下した。反応混合物を0℃で5分間撹拌し、その後、LiBr(61.1g、0.704mol)を一度に加えた。得られた混合物をRTで16時間撹拌した。溶媒をエバポレーションし、水(1500ml)を加え、水溶液をCH2Cl2により抽出した。あわせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:2%EtOAc−CH2Cl2から5%EtOAc−CH2Cl2)により、生成物4(31.8g、70%)を黄色の油状物として得た。MS(M+1):m/e 322および324。
【0087】
【化12】
工程4:
化合物4(41.2g、0.128mol)を乾燥DMSO(300ml)に溶解した溶液に、NaN3(9.15g、0.141mol)を加えた。反応混合物をRTで16時間撹拌した。水(300ml)を加え、水溶液をエーテルにより抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮して、生成物5(36.4g、100%)を油状物として得た。MS(M+Na):m/e 307。
【0088】
【化13】
工程5:
化合物5(36.4g、0.128mol)をTHF(800ml)に溶解した溶液に、10%パラジウム炭素触媒(10.0g)を加えた。反応混合物を40psiの水素圧下、Parr振とう機で16時間振とうした。触媒をろ過によって除き、イソプロパノールにより洗浄した。ろ液を濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:CH2Cl2、次いでNH3−CH2Cl2を含む10%MeOH)により、生成物6(24.2g、73%)を明灰色の固体として得た。MS(M+1):m/e 259。
【0089】
【化14】
工程6:
化合物6(12.0g、0.0464mol)を乾燥CH2Cl2(300ml)に溶解した溶液に、Et3N(9.4g、0.093mol、13.0ml)を加え、その後、シクロプロパンカルボニルクロリド(5.3g、0.051mol、4.64ml)を加えた。反応混合物をRTで16時間撹拌した。水(200ml)を加え、水溶液をCH2Cl2により抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:NH3−CH2Cl2を含む5%MeOH)により、生成物7(14.3g、94%)を油状物として得た。MS(M+Na):m/e 349。
【0090】
【化15】
工程7:
化合物7(40.0g、0.123mol)をCH2Cl2(550ml)に溶解した溶液に、4N HClのジオキサン溶液(153ml、0.613mol)を加えた。反応混合物をRTで4時間撹拌し、その後、濃縮して、生成物8(32.2g、100%)を無色の泡状物として得た。MS(M+1):m/e 227。
【0091】
【化16】
工程8:
乾燥DMF(300ml)中の化合物8(5.5g、20.8mmol)およびカルボン酸9(10.0g、20.8mmol)の混合物に、3Aシーブス(10.0g)、Et3N(6.3g、62.3mmol、8.7ml)、次いでHATU(15.8g、41.6mmol)を加えた。反応混合物をRTで21時間撹拌し、その後、溶媒を濃縮した。水(400ml)を加え、水溶液をCH2Cl2により抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:20%EtOAc−CH2Cl2から60%EtOAc−CH2Cl2に)により、生成物10(14.0g、98%)を無色の泡状物として得た。MS(M+1):m/e 690。NMRスペクトルについては図3を参照のこと。
【0092】
工程9:
化合物10(42.1g、0.061mol)をCH2Cl2(600ml)に溶解し、0℃に冷却したこの溶液に、4N HClのジオキサン溶液(76ml、0.305mol)を加えた。その後、反応混合物をRTで5時間撹拌し、その後、濃縮した。粗生成物を1:1のEtOH:H2O(120ml)に溶解し、25%NaOH水溶液により塩基性にした(pH=9〜10)。CH2Cl2(700ml)を加え、反応混合物を、すべての固体が溶解するまで撹拌した。層を分離し、水溶液をCH2Cl2により抽出した。合わせた有機抽出物をブラインにより洗浄し、乾燥させ(MgSO4)、ろ過し、濃縮した。さらなるCH2Cl2を加え、混合物を再び濃縮した。エーテルを加え、混合物を濃縮して、化合物11(化合物A)(34.4g、96%)を明黄色の固体として得た。MS(M+1):m/e 590。NMRスペクトルについては図4を参照のこと。
【0093】
本出願明細書における実施例2および他の箇所において、Etはエチルを意味し、Meはメチルを意味し、EtOHはエタノールを意味し、NMRは核磁気共鳴を意味し、THFはテトラヒドロフランであり、DMFはN,N−ジメチルホルムアミドであり、t−BOCおよびBOCはt−ブトキシカルボニルを意味し、RTは室温であり、DMSOはジメチルスルホキシドを意味し、Et3Nはトリエチルアミンを意味し、NaHMDSはナトリウムビス(トリメチルシリル)アミドであり、HOBTはヒドロキシベンゾトリアゾールであり、EDCl HClは1−エチル−3−[3−ジメチルアミノ]プロピル]カルボジイミド塩酸塩であり、NMPはN−メチルピロリジノンであり、caはcirca(約)であり、KFはカールフィッシャーであり、EtOAcは酢酸エチルである。
【0094】
【化17】
(実施例2)
工程1:
(S)−2−tert−ブトキシカルボニルアミノプロピオン酸(8.8kg(46.5モル、2当量))を、熱電対、N2導入管および供給タンクを備える50LのHastelloy反応装置に加えた。乾燥テトラヒドロフラン(90リットル)(THF、KF<0.05%)をバッチに加え、溶解するように装入した。ジシクロヘキシルアミン(8.5kg(46.9モル、2当量))をバッチに加え、−5℃〜5℃の間の温度範囲で約30分かけてゆっくり装入した。バッチを−5℃〜5℃の間の温度範囲で約15分間撹拌した。トリメチルアセチルクロリド(5.7kg(47.3モル、2当量))をバッチに加え、−5℃〜5℃の間の温度範囲で約30分かけてゆっくり装入した。バッチを−5℃〜5℃の間の温度範囲で約3時間撹拌した。ヘプタン(27リットル)をバッチに加え、装入し、続いて、4.5kgのセライトを加えた。バッチをN2下でろ過し、フィルターケーキを30%(v/v)THF/ヘプタンにより洗浄した。ろ液を濃縮した。ろ液および洗浄液は、真空下でバッチを約36リットルのバッチ容量にした。THF(27リットル)をバッチに加え、装入した。バッチの温度を約20℃〜30℃に調節した。バッチをKFのためにサンプリングした(0.06ppm未満)。バッチは混合無水物のTHF溶液であり、これを、さらに精製することなく次工程において使用した。
【0095】
化合物(1A)(9.0kg(23.3モル、1当量))を、熱電対、N2導入管および供給タンクを備える50ガロンのガラス内張り反応装置に加えた。乾燥テトラヒドロフランをバッチに加え(126リットル(THF、KF<0.05%))、溶解するように装入した。バッチを1気圧で濃縮して約81リットルのバッチ容量にした。温度を約−60℃〜−70℃に調節した。NaHMDS(THF中2M、2.70kg、5.9モル、0.25当量)を加え、−60℃〜−70℃の間の温度範囲で約15分かけて装入した。バッチを−60℃〜−70℃の間の温度範囲で約5分間撹拌した。上記混合無水物のTHF溶液(0.83kg活性、3.2モル、0.14当量)を加え、−60℃〜−70℃の間の温度範囲で約15分かけて装入した。バッチを−60℃〜−70℃の間の温度範囲で約10分間撹拌した。連続する2回の装入(NaHMDS、THF中2M)および混合無水物を、合計で8セットの装入のためにさらに7回、または、転換率が70%以上になるまで繰り返した。転換率が94%以上になるまで、NaHMDS(THF中2M)を装入し、続いて、混合無水物を、残留する出発物質の量に基づいて同じ比率で装入することを続けた。ゆっくりと約15分かけて、バッチの温度を30℃未満で維持しながら、バッチを90リットルのH2Oに溶解した13.5kgのKH2PO4の水溶液に移した。酢酸エチル(59リットル)を加え、充填し、その後、約15分間撹拌し、層を静置させた。水層を45リットルの酢酸エチルにより抽出した。合わせた有機層を約32リットルの10%(w/v)NaCl水溶液により2回洗浄した。有機層を、バッチとして1気圧で約45リットルのバッチ容量まで濃縮した。メチルtertブチルエーテル(MTBE)(90リットル)をバッチに加え、装入した。バッチを約54リットルのバッチ容量まで1気圧で濃縮した。メチルtertブチルエーテル(45リットル)を55℃〜65℃の間の温度で装入した。ヘプタン(108リットル)をバッチに加え、55℃〜65℃の間の温度で装入した。温度を約45℃〜55℃に調節し、約30分間撹拌した。その後、温度を約1時間かけて約−5℃〜5℃に調節した。バッチを−5℃〜5℃の間の温度で約30分間撹拌した。バッチをろ過し、これにより、フィルターケーキを形成させ、33%(v/v)メチルtertブチルエーテル/ヘプタンにより洗浄した。バッチを真空オーブン中、45℃〜55℃で少なくとも約12時間乾燥し、これにより、8.4kg(72.2%)の化合物(2A)を、eeが99.0%を越える固体として得た。
【0096】
【化18】
工程2:
化合物(2A)(20g(39.3mmol、1当量))を、機械的撹拌装置、滴下漏斗および熱電対が取り付けられた500mLの三口丸底フラスコに加え、装入した。THF(60ml)、EtOH(20mL)および水(100mL)をフラスコに加え、反応混合物を装入した。次いで、8mLの25%水酸化ナトリウム溶液を反応混合物に加え、装入した。反応混合物を40℃で約4時間撹拌した。反応の完了をHPLCアッセイによって判断すると、水(100ml)を混合物に加え、バッチを装入し、50℃に加熱した。50℃に達すると、1N HCl溶液(30ml)をバッチに加え、30分間かけて装入した。バッチをこの温度でさらに30分間撹拌し、その後、さらに24mlの1N HCl溶液をバッチに加え、バッチを30分かけて装入した。水(60ml)をバッチに加え、バッチを50℃で30分かけて装入し、これにより、スラリーを形成させた。得られたスラリーを1時間以上にわたって室温に冷却し、これにより、生成物を形成させ、この生成物を吸引ろ過によって集め、これにより、湿潤ケーキを形成させた。湿潤ケーキをエタノールおよび水(1/5、v/v)の40mlの溶媒混合物により洗浄した。得られた固体を真空下60℃で12時間乾燥し、これにより、16.8g(90%)の化合物(3A)を灰白色の固体として得た。
【0097】
【化19】
工程3:
パートA
(2R,4S)−4(シクロプロパンカルボニルアミノ)ピロリジン−1,2−ジカルボン酸−1−tert−ブチルエステル2−エチルエステル(BP)(60g、184mmol、1当量)をEtOAc(1.2L)に溶解し、サンプルを100%のHPLC標準物として採取した。バッチを20℃〜35℃に冷却し、HCl(g)(36g、980mmol、5.3当量)をバッチに加え、反応温度を20℃〜35℃の間で維持しながら装入した。生成物のHCl塩が、反応が進行するにつれ沈殿した。HCl装入が終了したとき、バッチを20℃〜30℃に加熱し、1時間撹拌した。1時間後、反応混合物をサンプリングし、反応物のHPLC面積応答を上記標準物と比較することによって反応の完了について調べた。反応液を、標準物に対するBPの量が0.5%以下の面積になるまでサンプリングした。バッチを真空下、35℃〜45℃で600mLまで濃縮し、これは濃厚なスラリーを形成した。その後、NMP(280mL)をバッチに加えた。バッチを真空下、35℃〜45℃で約560mLの容量まで濃縮し、これは透明な溶液を形成した。この透明な溶液をパートBにおけるカップリング工程においてそのまま使用した。
【0098】
パートB:
化合物(3)を、1Lの三口丸底フラスコ中のHOBT・H2O(28g、182mmol、1.1当量)およびEDC・HCl(48g、250mmol、1.4当量)をNMP(320mL)およびEtOAc(320mL)の溶液中に溶解した(80g、166mmol、1当量)。バッチを25℃で40分間撹拌した。(パートAからの)BPの溶液をバッチに加え、10分間撹拌した。N−メチルモルホリン(80mL、724mmol、4.4当量)を、温度を35℃未満に維持する速度で反応液に加えた。反応が完了したと判断すると、EtOAc(320mL)および水(800mL)をバッチに加えた。得られたバッチを15分間撹拌し、層を分離した。有機層を1M HCl(400mL)により洗浄し、次いで、10%K2CO3(400mL)および水(400mL)により洗浄した。有機層を約160mLまで濃縮し、アセトン(800mL)を有機層に加えた。バッチを減圧下において約40℃〜50℃で約240mLまで再び濃縮した。反応液をさらに800mLのアセトンにより希釈し、バッチを減圧下において40℃〜50℃で約240mLまで濃縮した。バッチの温度を約40℃で維持し、800mLのヘプタンをバッチにゆっくり加え、その結果、固体が一部形成した。固体生成物をろ過によって収集し、真空下50℃で12時間乾燥させて、103g(90%)の化合物(4A)を灰白色の固体として得た。
【0099】
【化20】
注意:回転異性体の存在のために、測定されたピークは、測定された通りに列挙される。
【0100】
工程4:
化合物(4A)(20g、29mmol、1当量)をフラスコに加え、THF(60ml)に溶解するように装入し、溶液を0℃〜10℃に冷却した。濃HCl(20ml)を、温度を0℃〜20℃に維持するようにゆっくり加えた。装入が終了したとき、溶液を20℃〜30℃に加温し、約4時間撹拌した。約4時間で、HPLC分析によって、反応が完了していることが明らかにされた。バッチを2−Me−THF(120ml)およびTHF(40ml)により希釈し、反応を、8〜8.5のpHを達成するため20%K2CO3(110ml)により停止させた。pHを調節した後、さらに水(80ml)を加え、バッチを約30℃に加熱して、明瞭な相分離を達成した。バッチを約15分間静置し、下層を分離し、有機層を水(80ml)により洗浄した。有機相を2−Me−THF(200ml)により希釈し、その後、大気圧において還流下で約100mlまで濃縮した。固体生成物がこの体積で観測された。その後、バッチを0℃〜10℃に冷却し、ろ過し、これにより、湿ったケーキが残留した。湿ったケーキを2−Me−THFにより2回洗浄した(それぞれ、40ml)。洗浄された湿ったケーキを真空下において60℃で少なくとも12時間乾燥し、これにより、13.50g(79%)の化合物(5A)(これは本明細書中では化合物Aとも呼ばれる)を白色の固体として得た。
1H−NMR(スペクトルは回転異性体を示し、化学シフトのみが報告され、積分またはピーク多重度は報告されない;
【0101】
【化21】
キシナホ酸塩の形成:
多形体形態1:方法1:
化合物A(34.4g、0.0583mol)を熱EtOH(800ml)に溶解した溶液に、キシナホ酸(10.98g、0.0583mol)を、EtOH溶液を加熱し続けながら少量ずつ注意深く加えた。さらなるEtOH(200ml)および水(6ml)を加えた。反応混合物を沸騰近くまで加熱して、すべての固体を溶解し、その後、ろ過した。ろ液をRTにゆっくり冷却した。このとき、結晶化が生じた。混合物をRTで一晩放置した。ろ液を0℃に冷却し、固体のキシナホ酸塩を真空ろ過によって単離した。固体のキシナホ酸塩をイソプロパノールにより洗浄し、次いで、エーテルにより洗浄し、高真空下において60℃で乾燥させて、36.8g(81%)の白色固体を得た。
【0102】
多形体形態1:方法2
窒素導入管および還流冷却器を備えた500mLの三口丸底フラスコに、スキーム2(実施例2)の化合物(5A)(30g、50.89mmol、1当量)および1−ヒドロキシ−2−ナフトエ酸(10.5g、55.80mmol、1.1当量)を加えた。その後、このフラスコにトルエン(154mL)およびメタノール(103mL)を加え、得られたスラリーを約62℃に加熱した。このとき、内容物が均一になった。15分間撹拌した後、内容物を大気圧で蒸留して210mLにし、その後、約50℃に冷却し、その後、形態1の結晶(10mLのトルエン中3g、10重量%)を接種し、これにより、生成物の塩を結晶化させ、スラリーを形成した。このスラリーを50℃で30分間撹拌した後、内容物を約10℃に冷却した。そのとき、トルエン(90mL)を加え、スラリーを真空蒸留して約210mLにした。トルエン(90mL)の再度の添加を行い、内容物を約20℃で20分間撹拌した。得られた固体を、真空下で撹拌型乾燥機を使用して収集し、湿ったケーキをトルエン(60mL)により洗浄した。これらの固体を、下記のプロトコルを使用して乾燥した。(a)Tj=50℃、圧力=0.1bar、撹拌なし、時間=3h;(b)Tj=80℃、圧力=0.1bar、20rpm、時間=12h;(c)Tj=80℃、圧力=0.1bar、60rpm、時間=12h。合計で35g(81%)の、スキーム2(実施例2)の化合物(6A)を固体として回収した。
1H−NMR(スペクトルは回転異性体を示し、化学シフトのみが報告され、積分またはピーク多重度は報告されない;
【0103】
【化22】
多形体形態1、方法3:化合物A(5.0g、0.00848mol)をMeOH(75ml)に溶解した溶液を50℃に加熱し、ろ過し、MeOH(10ml)によりリンスした。キシナホ酸(1.76g、0.00933mol)をMeOH(35ml)に溶解した溶液を50℃に加熱し、ろ過し、化合物Aの溶液に加えた。混合物を約10分間にわたって加熱還流し、大気圧で蒸留して約50mLにし、約1時間かけて0℃に冷却し、約30分間撹拌した。混合物をろ過し、冷却したMeOH(20ml)により洗浄し、真空下において室温で約12時間乾燥させて、灰白色の固体を得た(5.63g(85.4%))。
【0104】
多形体形態2:化合物A(34.4g、0.0583mol)を熱CH3OH(800ml)に溶解した溶液に、キシナホ酸(10.98g、0.0583mol)を、CH3OH溶液を加熱し続けながら少量ずつ注意深く加えた。水(6ml)を加えた。反応混合物を沸騰近くまで加熱して、すべての固体を溶解し、その後、ろ過した。ろ液をRTにゆっくり冷却し、これにより結晶化がもたらされた。混合物をRTで一晩放置した。ろ液を0℃に冷却し、固体のキシナホ酸塩を真空ろ過によって単離した。固体のキシナホ酸塩をイソプロパノールにより洗浄し、次いで、エーテルにより洗浄し、高真空下において60℃で乾燥させて、36.8g(81%)の白色固体を得た。
【0105】
二水和物形態1:キシナホ酸塩形成時における水の添加が、結晶性形態を得るために必要である。二水和物形態1を、形態1(504.83mg、0.65mmol)を水(0.9mL)およびメタノール(3.1mL)の混合物に懸濁させることによって調製した。懸濁物を21時間撹拌した。懸濁物を遠心分離し、その後、上清をデカンテーションにより除くことによって固体を単離した。固体を真空下において室温で乾燥させた。
【0106】
多形体形態3:形態3を、遊離塩基の化合物A(3.0g、5.1mmol)と、キシナホ酸(0.96g、5.1mmol)との混合物を2−プロパノール(90mL)中で合わせることによって調製した。混合物を加熱還流し、さらに2−プロパノール(30mL)を加えた。混合物を1時間にわたって還流状態で保ち、その後、室温に冷却した。混合物をろ過し、固体を2−プロパノール(6mL)により洗浄し、その後、真空下で乾燥させて、3.24gの生成物を得た。
【0107】
粉末X線回折用サンプルの調製
キシナホ酸塩の、形態1、形態2、形態3、および形態1の二水和物を、粉末x線回折(「PXRD」)分析のための乾燥粉末として分析した。形態1を、下記の手順を使用して、PXRD分析の前にジェットミルで微粉化した。
【0108】
ジェットミル粉砕による微粉化
微粉化された粉末の粒子サイズ分布が、ジェット圧およびジェットミル内への供給速度を調節することによって制御される。粒子が、加圧窒素によるベンチュリシステムを介して、MC ONE JETMILL(Jetpharma Group、South Plainfield、NJ)の粉砕チャンバーに1g/分の速度で供給される。ベンチュリでの圧力低下を5barに設定する。粒子は、粉砕チャンバーの外周に置かれた4つのノズルによって粉砕チャンバーの内部においてらせん運動で加速される。ノズルでの圧力低下を4barに設定する。微粉化作用が、より遅い進入粒子と、渦巻き流において既に加速された粒子との間における衝突によって生じる。遠心力により、より大きい粒子が粉砕チャンバーの外周部に保持され、一方、より小さい粒子が静的分級機によってチャンバーの中心から排出ガスとともに出ていき、ジェットミルのすぐ下方にある捕集容器に回収される。
【0109】
サンプルを、任意の形態変化を防止するために最小限の調製物を用いて分析した。サンプルを、粒子が凝集していないことを確かめるために軽くこすった。溶媒、乾燥工程または他の調製工程はいずれも、これらの分析のために使用しなかった。PXRDデータにより、水和物形態および多形形態を一意的に特定することができる。
【0110】
粉末X線回折
形態1および形態2のX線粉末回折パターンを、30kv、15mAでのCuKα放射線(λ=1.54056Å)、および、半導体検出器を備えるRigaku Miniflex回折計(Rigaku MSC、The Woodlands、TX)で収集した。連続走査を0.02°の2θのステップサイズおよび2°/分の走査速度ですべてのサンプルについて記録した。
【0111】
二水和物形態1のX線粉末回折パターンを、40kvおよび40mAでのCuKα1線源(λ=1.5406Å)を用いてBrukar D8回折計で集めた。連続走査を0.032°の2θのステップサイズおよび0.5秒のステップ時間により記録した。
【0112】
形態3のX線粉末回折パターンをKratos XRD6000で集めた。材料をサンプルホルダーに軽く詰め、静かに滑らかにして、平坦なサンプル表面を作製することによってサンプルを調製した。サンプルを、0.02度のステップサイズおよび0.6秒のステップ持続時間で、2度から40度までの2θで分析した。データ分析を、Kratosによって供給されるBasic Processソフトウエア(バージョン2.6)を使用して行った。データを、ソフトウエア内の自動平滑化プロセスを使用して平滑化した。
【0113】
上記で記載された方法および設備を使用して、化合物Aの形態1、形態2および形態3の多形体キシナホ酸塩、ならびに、二水和物形態をPXRD分析に供した。PXRDパターンが得られた。PXRDパターンが図1〜図3および図10に示される。ピークの強度(y軸はカウント/秒の単位である)が2θの角度(x軸は度(2θ)の単位である)に対してプロットされる。加えて、データを、2θの角度に対するステップあたりの収集時間について正規化された検出器カウントでプロットした。これらのプロフィルと一致するピーク存在位置(2θのX軸における存在位置)が表1に示される。これらのPXRDピークの存在位置は、式Iの化合物の形態1、形態2、形態3の結晶性多形体および結晶性二水和物形態1に特徴的である。
【0114】
【表1−1】
【0115】
【表1−2】
【0116】
【表1−3】
表1に示されるようなPXRDピーク存在位置をはじめとして、それぞれの多形体または水和物の最も特徴的なピーク存在位置を、結晶性構造を他の構造から都合よく識別するために選択し、相対強度によってグループ化することができる。
【0117】
特徴的なピークのそのような選択が表2に示される。従って、例えば、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群1(これは、4個の特徴的なPXRDピーク存在位置からなる)によって特定することができる。代替として、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群2(これは、群1の4個の特徴的なPXRDピーク存在位置、およびさらなる4個のピーク存在位置からなる)によって特定することができる。代替として、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群3(これは、群2の8個の特徴的なPXRDピーク存在位置、およびさらなる4個のピーク存在位置からなる)によって特定することができる。このスキームは、4つの多形形態のそれぞれを特定するために、また、4つの多形形態のそれぞれを他の形態から識別するために、4つの多形形態のそれぞれに適用される。
【0118】
【表2】
当業者は、同じ化合物の所与の結晶性形態についてのPXRDピーク存在位置の測定値が誤差限界内で変動することを認識する。そのような変動は、とりわけ、サンプル調製、装置使用または分析技術における様々な違いによって持ち込まれ得る。個々のピーク存在位置の測定値はわずかに変動することがあり、しかし、ピークプロフィル全体が、例えば、充填サンプルの密度における変動に起因して、より大規模に変動することがある。
【0119】
多形体の純度
好ましくは、式Iの化合物の結晶性多形体の形態1、形態2、形態3および二水和物形態1は、化学的不純物(例えば、多形体の調製時に生じる副生成物)および他の多形性結晶性形態を実質的に含まない。化学的不純物を「実質的に含まない」とは、本発明の目的のためには、約5%(w/w)以下の化学的不純物、好ましくは、約3%(w/w)以下の化学的不純物、より好ましくは、約2%(w/w)以下の化学的不純物、さらに一層好ましくは、約1%(w/w)以下の化学的不純物を含むことを意味する。多形体についての用語「精製された」または用語「精製された形態で」は、本明細書中に記載されるか、または、当業者に広く知られている標準的な分析技術によって特徴づけ可能であるための十分な純度で、本明細書中に記載されるか、または、当業者に広く知られている精製プロセスから得られた後の上記多形体の物理的状態を示す。式Iの化合物の結晶性多形体の形態1、形態2、形態3および二水和物形態1の精製された形態は、化学的不純物を実質的に含まない。
【0120】
示差走査熱量測定
多形体形態1および多形体形態2のサンプルを試験するために使用したDSC装置は、冷却された冷却系を備えた、TA Instruments(登録商標)モデル2920(2001年製造)であった。DSCセル/サンプルチャンバーが40ml/分の超高純度窒素ガスによりパージされた。装置は高純度インジウムにより校正された。この方法による測定されたサンプル温度の正確度は約+/−1℃の範囲内であり、融解熱を約+/−5%の相対的誤差の範囲内で測定することができる。サンプルを、圧力放出を可能にするための2つのピンホールを有するふたを伴う標準的なアルミニウムDSC皿に入れた。約2mgのサンプル粉末を皿の底に入れ、皿と接触させるために軽くたたいた。サンプルの重量を正確に測定し、ミリグラムの1/100まで記録した。この装置では、空の参照皿を使用された。DSC分析を10℃/分の加熱速度で行った。
【0121】
二水和物形態1および多形体形態3のサンプルを試験するために使用したDSC装置は、Q100 TAInstruments(登録商標)であった。再度、サンプルを気密型アルミニウム皿に密封し、2つのピンホールをサンプル皿のふたに開けた。分析を、窒素パージ下、10℃/分の加熱速度で行った。
【0122】
熱流量(これはサンプル重量によって正規化された)が、測定されたサンプル温度に対してプロットされた。データがワット/グラム(「W/g」)の単位で報告された。プロットを作製した(吸熱ピークは下向きである)。吸熱の融解ピークを、この分析における外挿された開始温度および終了(発生)温度、ピーク温度、ならびに、融解熱について評価した。
【0123】
式Iの形態1のDSCプロフィルが図6に示される。式Iの化合物の形態1について、単独の吸熱が観測され、開始温度が192℃であり、ピーク温度が193℃であった。
【0124】
式Iの形態2のDSCプロフィルが図7に示される。式Iの化合物の形態2について、2つの重なった吸熱が観測され、開始温度が152℃であり、ピーク温度が161℃および181℃であった。
【0125】
式Iの二水和物形態1のDSCプロフィルが図8に示される。式Iの化合物の二水和物形態1については、10℃/分で、二水和物形態1は脱水を受け、室温で準安定性の無水形態に変わる。この事象は、73℃での開始温度および143J/gの熱を有する幅広い吸熱としてDSCサーモグラム(図8)に反映される。加熱期間中に失われた水和物の水の量は総重量の4.1%を占め、これが、二水和物の化学的量論を示す、TGAデータ(図9)における階段様の重量減少として示される。室温で準安定性の形態は、144℃の開始温度で融解する。融解熱は、TGAデータ(図9)における150℃以降の重量減少に対応する分解が、融解事象が完了する前に始まるために、求めることができない。
【0126】
式Iの形態3のDSCプロフィルが図11に示される。式Iの化合物の形態3について、182℃の開始温度および186℃のピーク温度を伴う単独の吸熱が観測された。
【0127】
吸入による処置のための前提は、全身の副作用を最小限に抑えながら、薬物を作用部位(肺)に直接送達することである。従って、吸入された化合物は、経口バイオアベイラビリティの低さに起因する低い血中濃度(AUC)、および/または、吸入もしくは経口による服用経路によって与えられる場合の高いクリアランスを有する薬物動態学プロフィルを示さなければならない。吸入期間中の何らかの飲み込まれた薬物の影響を最小限に抑えるために、経口AUCが低いことは重要である。多くの場合、低いAUCレベルは、測定することが困難である。従って、再現可能なAUCデータが好ましい。
【0128】
アレルギー性Brown−Norwayラットのためのアッセイプロトコル:
体重が150g〜200gの同系交配されたオスのBNラットをCharles River Laboratory(Wilmington、MA)から得た。使用前、動物に、食物および水を自由に摂取させた。以下の「試験化合物の送達」の節に記載されるように、試験化合物を経口経路または吸入経路のいずれかによって抗原攻撃の5時間前に投与した。
【0129】
感作および抗原気管支誘発
動物を2つの主要な群(すなわち、ミョウバン群および抗原群)に分けた。抗原群では、動物を、0.9%の生理的食塩水ビヒクルに懸濁させた20μgの卵白アルブミン(OVA、グレードIII;Sigma chemical Co.、St Louis、MO)および8mgのAl(OH)3を含む1mlのミョウバン沈殿抗原の腹腔内(i.p.)注射によって感作させた。このミョウバン−OVA混合物の追加抗原刺激注射を7日後に再び行った。ミョウバン群に属する動物には、ミョウバンのみを含有する注射を受けさせた。2回目の注射の7日後、動物を、エアロゾル化抗原気管支誘発にさらし、これは、ラットを密閉型プレキシガラスチャンバー(21リットル)に置き、ラットをエアロゾル化OVA(1%)に30分間暴露させることによって行われた。エアロゾル化OVAは、超音波ネブライザー(DeVilbiss、Somerset、PA、米国;モデルUltra−Neb99)によっておよそ8リットル/分の流速で作製した。エアロゾル化OVAによる攻撃の24時間後、動物を過量のペントバルビタールナトリウムにより安楽死させた。気管を取り出し、挿管し、肺を3mlの生理食塩水により2回洗浄した。このようにして集められた気管支肺胞洗浄液(BALF)を細胞計数に供した。10マイクロリットルのBALFを利用して、総白血球を、血球計を使用して手作業により計数した。100マイクロリットルのBALFを使用して、細胞遠心分離物を調製し、この細胞遠心分離物をHema3(商標)染色システム(Fisher Scientific、Springfield、NJ)により染色して、種々の白血球(例えば、好酸球、好中球、単核細胞および上皮細胞など)を特定し、計数した。合計で200個の細胞をそれぞれの細胞遠心分離物から計数した。炎症性細胞を気道内に動員することを阻害する化合物の能力が報告される。
【0130】
試験化合物の送達:
経口投与:化合物を0.4%メチルセルロースに溶解し、動物に3ml/kgで経口送達した。等体積の0.4%メチルセルロースを陰性コントロール群(ミョウバン群)および陽性コントロール群(抗原源)の両方に与えた。
【0131】
気管内投与:適切な用量の化合物をラクトース粉末と混合して、3mgの最終量を達成し、これを、麻酔した動物に、先の細い微量噴霧器を使用して気管内送達した。動物を、3分間〜4分間、直立姿勢で保ち、麻酔から回復させ、その後、そのケージに戻した。
【0132】
上記の試験手順を使用して、下記の結果が得られた:
酒石酸塩:0.02mpkで炎症性細胞の52%阻害(気管内投与)
キシナホ酸塩:0.02mpkで炎症性細胞の69%阻害(気管内投与)
サルPKアッセイのためのアッセイプロトコル:
2匹の絶食させたサルに、0.4%のHPMCビヒクル中の試験化合物を3mpkで経口投与した。投与容量は2ml/kgであった。血漿を、0.5時間、1時間、2時間、4時間、8時間および24時間後に収集した。血液サンプルをヘパリンとともに収集し、血漿をEDTAとともに保存した。それぞれの個々の動物についての血液サンプルをMS/MS分析によって特徴決定した。
【0133】
上記の試験手順を使用して、下記の結果が得られた:
酒石酸塩:サルAUC=10mpk(po)で30ng.h/mL
キシナホ酸塩:サルAUC=10mpk(po)で0ng.h/mL
ラットPKアッセイのためのアッセイプロトコル:
2匹の絶食させたSprague Dawleyラットに、0.4%のHPMCビヒクル中の化合物を10mpkで経口投与した。投与容量は5ml/kgであった。血漿を、0.5時間、1時間、2時間、3時間、4時間および6時間後に収集した。血液サンプルをヘパリンとともに収集した、血漿をEDTAとともに保存した。それぞれの時点での2つの血液サンプルをMS/MS分析によって特徴決定した。
【0134】
酒石酸塩:AUC=30mpk(po)で0〜1350ng.h/mL(変動)
キシナホ酸塩:AUC=30mpk(po)で350ng.h/mL
肺機能アッセイのためのアッセイプロトコル:
肺機能を、強制呼気技術を使用して測定した。この手法では、ラットを麻酔し、気管カテーテルを挿入した。ラットを、肺の膨張および収縮を分離することができる呼吸弁を含有する全身プレチスモグラフの中に置いた。その後、肺を、総肺気量に対する肺の強制膨張に供し、その後、残気量に対する急速収縮に供した。努力肺活量および最高呼気流量の測定値を使用して、抗原攻撃の影響を測定し、式Iの化合物の阻害効果を評価した。薬物を気管内送達のためにラクトースと混合させ、抗原攻撃の5時間前に微細微量噴霧器により気管内に直接に与えた。経口送達される化合物は抗原攻撃の5時間前に0.4%メチルセルロースビヒクル中で与えた。コントロール動物は気管内ラクトースまたは気管内メチルセルロースをそれぞれ受けた。抗原攻撃は、1%卵白アルブミンへの30分間のエアロゾル暴露からなった。強制呼気による肺機能を抗原(卵白アルブミン)暴露後24時間で測定した。
【0135】
式Iの化合物は0.02mpk(it(気管内))で努力肺活量(FVC)の54%阻害を示し、3mpk(po)でFVCの31%阻害を示す。
【0136】
医薬組成物
医薬組成物を本発明により記載される多形体から調製するために、不活性な医薬的に許容され得るキャリアは固体または液体のいずれかであり得る。様々な組成物のための医薬的に許容され得るキャリアおよび製造方法の例を、A.Gennaro(編)、Remington’s Pharmaceutical Sciences(第18版(1990)、Mack Publishing Co.、Easton、Pennsylvania)に見出すことができる。
【0137】
液体形態の調製物には、鼻腔内投与のための溶液、懸濁物およびエマルジョンが含まれる。
【0138】
吸入のための好適なエアロゾル調製物には、溶液、および、粉末形態の固体が含まれ得る。この場合、これらは、医薬的に許容され得るキャリア(例えば、不活性な圧縮ガス(例えば、窒素)など)との組合せであり得る。
【0139】
投薬量
単位服用量の調製物における活性な化合物の量は、特定の適用に従って、約0.01μgから約100mgまで、好ましくは約0.01μgから約75mgまで、より好ましくは約0.01μgから約50mgまで、最も好ましくは約0.01μgから約25mgまで変化させるかまたは調節することができる。
【0140】
用いられる正確な投薬量は、患者の要件、および、処置されている状態の重篤度に依存して変化させることができる。具体的な状況のための適正な投薬計画の決定は当業者の技術の範囲内である。便宜上、総投薬量は分割することができ、必要に応じて、1日のうちに何回かに分けて投与することができる。
【0141】
本発明の化合物および/またはその医薬的に許容され得る塩の投与量および投与頻度は、患者の年齢、状態およびサイズ、ならびに、処置されている症状の重篤度のような要因を考慮して、主治医の判断に従って調節される。吸入のために推奨される典型的な1日投薬量は、1回〜4回に分割した投与において、約0.04μg/日から約400mg/日の範囲であり得る。
【0142】
操作実施例に示される場合、または別途示される場合を除き、成分の量および反応条件などを表す、本明細書および請求項において使用されるすべての数字は、あらゆる場合、用語「約」によって修飾されているとして理解される。上の記述は、本発明のすべての改変および変化を詳しく記載することを意図するものではない。様々な変化が、本発明の概念から逸脱することなく、上記で記載された実施形態に対して行われ得ることが当業者によって理解されるであろう。従って、本発明は、上述した特定の実施形態に限定されず、しかし、下記の請求項の範囲によって定義されるような本発明の趣旨および範囲内にある改変を包含するように意図されることが理解される。
【技術分野】
【0001】
(発明の分野)
本発明は、1−[[5−(1(S)−アミノエチル)−2−[8−メトキシ−2−(トリフルオロメチル)−5−キノリル]−4−オキサゾリル]カルボニル]−4(R)−[(シクロプロピルカルボニル)アミノ]−L−プロリン、エチルエステルのキシナホ酸塩、上記塩を含む医薬組成物、ならびに、気道の上部閉塞性疾患および下部閉塞性疾患を上記塩の吸入によって処置する方法に関連する。
【背景技術】
【0002】
(発明の背景)
様々なホスホジエステラーゼが、環状AMPを調節することが知られており、また、ホスホジエステラーゼ4(PDE4)が、気道平滑筋および炎症性細胞における環状AMPの主要な調節因子であることが示されている。PDE4の阻害剤は、アレルギー性疾患および炎症性疾患、糖尿病、中枢神経系疾患、疼痛、ならびに、TNFを産生するウイルスを含む様々な疾患の処置に有用である。
【0003】
アミノ置換キノリルのPDE4阻害剤が特許文献1に開示され、スルホンアミド置換キノリルのPDE4阻害剤が特許文献2に開示され、また、(ベンゼン縮合)ヘテロアリール置換のPDE4阻害剤が特許文献3に開示される。オキサゾリル置換のPDE4阻害剤がPCT/US2005/017134に開示される。
【0004】
本明細書中において化合物Aとして示される化合物が、特許文献4において、95頁、実施例26〜実施例347および228頁(請求項19)に、その遊離塩基および医薬的に許容され得る塩形態として記載される(それらの記載は本明細書に参考として組み込まれる)。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許第5,804,588号明細書
【特許文献2】米国特許第5,834,485号明細書
【特許文献3】米国特許第6,069,151号明細書
【特許文献4】国際公開第2005/116009号パンフレット
【発明の概要】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、1−[[5−(1(S)−アミノエチル)−2−[8−メトキシ−2−(トリフルオロメチル)−5−キノリル]−4−オキサゾリル]カルボニル]−4(R)−[(シクロプロピルカルボニル)アミノ]−L−プロリン、エチルエステルのキシナホ酸塩(すなわち、下記式Iの化合物:
【0007】
【化1】
)を提供する。
【0008】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法に関し、有効量の式1の化合物を吸入によって上記患者に投与することを含む。
【0009】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法に関し、式1の化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを吸入によって上記患者に投与することを含む。好ましいさらなる薬剤は、β−アゴニスト、ムスカリンアンタゴニストまたはコルチコステロイドである。
【0010】
本発明はさらに、有効量の式Iの化合物を含む吸入可能な医薬組成物に関する。
【0011】
本発明はさらに、式1の化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを含む吸入可能な医薬組成物に関する。
【0012】
本発明はまた、式Iの化合物の結晶性多形体および偽多形体(水和物)に関し、上記多形体は、
図1に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態1、
図2に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態2、および
図3に示されるパターンと実質的に同じである粉末x線回折パターンを示す二水和物形態1、
図10に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態3、からなる群から選択される。
【0013】
本発明はさらに、6.1度、7.7度、13.0度および15.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態1を提供する。
【0014】
別の実施形態において、式Iの結晶性多形体形態1は、5.6度、6.1度、7.7度、13.0度、15.9度、17.8度、18.4度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0015】
別の実施形態において、式Iの結晶性多形体形態1は、5.6度、6.1度、7.7度、9.2度、13.0度、14.2度、15.9度、17.8度、18.4度、20.5度、22.9度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0016】
本発明はさらに、10.6度、13.6度、19.1度および21.2度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態2を提供する。
【0017】
別の実施形態において、式Iの結晶性多形体形態2は、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度および23.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0018】
別の実施形態において、式Iの結晶性多形体形態2は、9.4度、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度、23.9度、26.0度、26.6度および28.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0019】
本発明はさらに、8.2度、16.5度、18.5度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性二水和物形態1を提供する。
【0020】
別の実施形態において、式Iの結晶性二水和物形態1は、5.5度、8.2度、14.3度、16.5度、16.9度、18.5度、20.6度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0021】
別の実施形態において、式Iの結晶性二水和物形態1は、5.5度、7.2度、8.2度、14.3度、14.7度、16.5度、16.9度、18.5度、20.6度、24.1度、24.9度および26.8度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0022】
本発明はさらに、4.6度、7.9度、12.1度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す式Iの結晶性多形体形態3を提供する。
【0023】
別の実施形態において、式Iの結晶性多形体形態3は、4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0024】
別の実施形態において、式Iの結晶性多形体形態3は、4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度、18.9度、20.0度、23.9度、24.3度および25.7度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す。
【0025】
本発明はさらに、形態1の多形体キシナホ酸塩を化合物Aから調製するための2つのプロセスを提供する。
【0026】
第1の方法:
【0027】
【化2】
この方法は下記の工程を含む。
【0028】
a)化合物Aを熱エタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)さらなるエタノールおよび水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、混合物を、形態1の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態1の結晶をろ過する工程。
【0029】
第2の方法:
【0030】
【化3】
この方法は下記の工程を含む。
【0031】
a)トルエンおよびメタノールを化合物Aおよびキシナホ酸に加え、混合し、これにより、スラリーを形成する工程、
b)上記スラリーを混合しながら約62℃に加熱し、これにより、均一な混合物を得る工程、
c)上記均一な混合物を大気圧で蒸留し、蒸留された混合物を約50℃に冷却し、上記蒸留された混合物に化合物Aの形態1の種結晶を接種し、これにより、スラリー状の結晶を生じさせる工程、
d)上記スラリーを約50℃で約30分間撹拌し、スラリーを約10℃に冷却する工程、
e)さらなるトルエンを上記冷却したスラリーに加え、真空蒸留し、その後、さらなるトルエンを加え、約20℃で約20分間撹拌し、これにより、固体物質を形成させる工程、
f)得られた固体を、撹拌型乾燥機を真空下で使用して収集し、これにより、湿潤ケーキを形成し、次いで、上記湿潤ケーキをトルエンにより洗浄し、撹拌を伴うことなく約3時間の約50℃での乾燥、その後、約20R.P.M.の撹拌を伴う約12時間の約80℃での乾燥、その後、約60R.P.M.の撹拌を伴う約12時間の約80℃での乾燥をすべて真空下で行う工程。
【0032】
第3の方法:
【0033】
【化4】
この方法は下記の工程を含む。
【0034】
a)化合物Aおよびキシナホ酸を熱メタノールに別々に溶解する工程、
b)熱溶液の両方をろ過し、これら2つの溶液を混合する工程、
c)混合物を還流し、過剰なメタノールを留去する工程、および
d)混合物を0℃に冷却し、これにより、沈殿物を形成させ、形態1の結晶をろ過する工程。
【0035】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性多形体形態1を提供する。
【0036】
本発明はさらに、式2の多形体キシナホ酸塩を、化合物A:
【0037】
【化5】
から調製するためのプロセスを提供し、このプロセスは、
a)化合物Aを熱メタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、混合物を、形態2の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態2の結晶をろ過する工程
を含む。
【0038】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性形態2の多形体キシナホ酸塩を提供する。
【0039】
本発明はさらに、二水和物形態1を、化合物A:
【0040】
【化6】
の形態1の多形体キシナホ酸塩から調製するためのプロセスを提供し、このプロセスは、下記の工程を含む:
a)キシナホ酸塩形成時における水の添加が、結晶性形態を得るために必要である。化合物Aの形態1の多形体キシナホ酸塩を、水およびメタノールの混合物に懸濁させる、
b)懸濁物を21時間撹拌し、懸濁物を遠心分離して、その後、上清をデカンテーションにより除くことによって固体を単離した、
c)固体を真空下において室温で乾燥した。
【0041】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性二水和物形態2のキシナホ酸塩を提供する。
【0042】
本発明はさらに、形態3の多形体キシナホ酸塩を、化合物A:
【0043】
【化7】
から調製するためのプロセスを提供し、このプロセスは、
a)化合物Aおよびキシナホ酸の混合物を2−プロパノール中で合わせる工程、
b)混合物を加熱して還流させ、さらに2−プロパノールを加え、次いで、混合物を還流状態で約1時間保ち、その後、室温まで冷却する工程、
c)混合物をろ過し、固体を2−プロパノールにより洗浄し、真空下で乾燥する工程
を含む。
【0044】
本発明はさらに、上記プロセスの生成物である化合物Aの結晶性形態3の多形体キシナホ酸塩を提供する。
【0045】
本発明はさらに、式Iの化合物の形態1の多形体の精製された形態を提供する。
【0046】
本発明はさらに、式Iの化合物の形態2の多形体の精製された形態を提供する。
【0047】
本発明はさらに、式Iの化合物の二水和物形態1の精製された形態を提供する。
【0048】
本発明はさらに、式Iの化合物の形態3の多形体の精製された形態を提供する。
【0049】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態1の多形体、ならびに有効量の式Iの化合物の形態1の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0050】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態2の多形体、ならびに有効量の式Iの化合物の形態2の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0051】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の二水和物形態1、ならびに、有効量の式Iの化合物の二水和物形態1と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
【0052】
本発明はまた、気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者においてこの疾患を処置する方法を特許請求し、この方法は、有効量の式Iの化合物の形態3の多形体、ならびに有効量の式Iの化合物の形態3の多形体と、医薬的に許容され得るキャリアとを含む吸入可能な医薬組成物を吸入によって上記患者に投与することを含む。
例えば、本願発明は以下の項目を提供する。
(項目1)
下記の構造式:
【化23】
を有する化合物。
(項目2)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目1に記載される化合物を吸入によって該患者に投与することを含む、方法。
(項目3)
前記処置される疾患が喘息または慢性閉塞性肺疾患である、項目2に記載の方法。
(項目4)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、項目1に記載される化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを吸入によって該患者に投与することを含む、方法。
(項目5)
前記処置される疾患が喘息または慢性閉塞性肺疾患である、項目4に記載の方法。
(項目6)
前記さらなる薬剤が、β−アゴニスト、ムスカリンアンタゴニストおよびコルチコステロイドからなる群から選択される、項目4に記載の方法。
(項目7)
有効量の項目1に記載される化合物を含む吸入可能な医薬組成物。
(項目8)
項目1に記載される化合物と、気道の上部閉塞性疾患または下部閉塞性疾患を処置するために有用な少なくとも1つのさらなる薬剤との有効量の組合せを含む吸入可能な医薬組成物。
(項目9)
前記さらなる薬剤が、β−アゴニスト、ムスカリンアンタゴニストおよびコルチコステロイドからなる群から選択される、項目8に記載の組成物。
(項目10)
下記式の化合物:
【化24】
の結晶性多形体であって、該多形体が:
図1に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態1、
図2に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態2、および
図3に示されるパターンと実質的に同じである粉末x線回折パターンを示す二水和物形態1、
図10に示されるパターンと実質的に同じである粉末x線回折パターンを示す形態3、からなる群から選択される、結晶性多形体。
(項目11)
6.1度、7.7度、13.0度および15.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態1。
(項目12)
5.6度、6.1度、7.7度、13.0度、15.9度、17.8度、18.4度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目11に記載の結晶性多形体。
(項目13)
5.6度、6.1度、7.7度、9.2度、13.0度、14.2度、15.9度、17.8度、18.4度、20.5度、22.9度および26.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目11に記載の結晶性多形体。
(項目14)
項目10に記載の結晶性多形体形態1。
(項目15)
10.6度、13.6度、19.1度および21.2度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態2。
(項目16)
10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度および23.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目15に記載の結晶性多形体。
(項目17)
9.4度、10.6度、13.6度、17.9度、18.8度、19.1度、20.2度、21.2度、23.9度、26.0度、26.6度および28.1度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目15に記載の結晶性多形体。
(項目18)
項目10に記載の結晶性多形体形態2。
(項目19)
8.2度、16.5度、18.5度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性二水和物形態1。
(項目20)
5.5度、8.2度、14.3度、16.5度、16.9度、18.5度、20.6度および24.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目19に記載の結晶性二水和物。
(項目21)
5.5度、7.2度、8.2度、14.3度、14.7度、16.5度、16.9度、18.5度、20.6度、24.1度、24.9度および26.8度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目19に記載の結晶性二水和物。
(項目22)
項目10に記載の結晶性二水和物形態1。
(項目23)
4.6度、7.9度、12.1度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目10に記載の化合物の結晶性多形体形態3。
(項目24)
4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度および18.9度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目23に記載の結晶性多形体。
(項目25)
4.6度、7.9度、9.1度、12.1度、13.7度、15.8度、16.5度、18.9度、20.0度、23.9度、24.3度および25.7度の2θの特徴的なピーク存在位置を有する粉末x線回折パターンを示す、項目23に記載の結晶性多形体。
(項目26)
項目10に記載の結晶性多形体形態3。
(項目27)
項目10に記載される多形体形態1を、化合物A:
【化25】
から調製するためのプロセスであって、
a)化合物Aを熱エタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
b)さらなるエタノールおよび水を加え、混合物を沸騰近くまで加熱する工程、
c)熱混合物をろ過し、その後、室温までゆっくり冷却し、該混合物を、形態1の結晶が沈殿するまで室温で一晩放置する工程、および
d)ろ液を0℃に冷却し、形態1の結晶をろ過する工程
を含む、プロセス。
(項目28)
項目10に記載される多形体形態1を、化合物A:
【化26】
から調製するためのプロセスであって、
e)トルエンおよびメタノールを化合物Aおよびキシナホ酸に加え、混合し、これにより、スラリーを形成する工程、
f)該スラリーを混合しながら約62℃に加熱し、これにより、均一な混合物を得る工程、
g)該均一な混合物を大気圧で蒸留し、蒸留された混合物を約50℃に冷却し、該蒸留された混合物に化合物Aの形態1の種結晶を接種し、これにより、スラリー状の結晶を生じさせる工程、
h)該スラリーを約50℃で約30分間撹拌し、スラリーを約10℃に冷却する工程、
i)さらなるトルエンを該冷却したスラリーに加え、真空蒸留し、その後、さらなるトルエンを加え、約20℃で約20分間撹拌し、これにより、固体物質を形成させる工程、
j)得られた固体を、撹拌型乾燥機を真空下で使用して収集し、これにより、湿潤ケーキを形成させ、次いで、該湿潤ケーキをトルエンにより洗浄し、撹拌を伴うことなく約3時間の約50℃での乾燥、その後、約20R.P.M.の撹拌を伴う約12時間の約80℃での乾燥、その後、約60R.P.M.の撹拌を伴う約12時間の約80℃での乾燥をすべて真空下で行う工程
を含む、プロセス。
(項目29)
項目10に記載される多形体形態1を、化合物A:
【化27】
から調製するためのプロセスであって、
k)化合物Aおよびキシナホ酸を熱メタノールに別々に溶解する工程、
l)両方の熱溶液をろ過し、これら2つの溶液を混合する工程、
m)混合物を還流し、過剰なメタノールを留去する工程、および
n)混合物を0℃に冷却し、これにより、沈殿物を形成させ、形態1の結晶をろ過する工程
を含む、プロセス。
(項目30)
項目10に記載される形態1の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目31)
項目10に記載される形態1の多形体の精製された形態。
(項目32)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態1の多形体を吸入によって該患者に投与することを含む、方法。
(項目33)
項目10に記載される多形体形態2を、化合物A:
【化28】
から調製するためのプロセスであって、
o)化合物Aを熱メタノールに溶解し、キシナホ酸を、混合物を加熱し続けながら加える工程、
p)水を加え、混合物を沸騰近くまで加熱する工程、
q)熱混合物をろ過し、その後、室温までゆっくり冷却し、該混合物を、形態2の結晶が沈殿するまで室温で一晩放置する工程、および
r)ろ液を0℃に冷却し、形態2の結晶をろ過する工程
を含む、プロセス。
(項目34)
項目10に記載される形態2の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目35)
項目10に記載される形態2の多形体の精製された形態。
(項目36)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態2の多形体を吸入によって該患者に投与することを含む、方法。
(項目37)
項目10に記載される二水和物形態1を、化合物A:
【化29】
のキシナホ酸塩多形体形態1から調製するためのプロセスであって、
r)化合物Aの形態1の多形体キシナホ酸塩を水およびメタノールの混合物に懸濁させる工程、
s)懸濁物を21時間撹拌し、懸濁物を遠心分離して、その後、上清をデカンテーションにより除くことによって固体を単離する工程、
t)固体を真空下において室温で乾燥させる工程
を含む、プロセス。
(項目38)
項目10に記載される二水和物形態1の結晶と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目39)
項目10に記載される結晶性二水和物形態1の精製された形態。
(項目40)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される二水和物形態1を吸入によって該患者に投与することを含む、方法。
(項目41)
項目10に記載される多形体形態3を、化合物A:
【化30】
から調製するためのプロセスであって、
u)化合物Aおよびキシナホ酸の混合物を2−プロパノール中で合わせる工程、
v)混合物を加熱して還流させ、さらに2−プロパノールを加え、次いで、混合物を還流状態で1時間保ち、その後、室温まで冷却する工程、
w)混合物をろ過し、固体を2−プロパノールで洗浄し、真空下で乾燥させる工程
を含む、プロセス。
(項目42)
項目10に記載される形態3の結晶性多形体と、少なくとも1つの医薬的に許容され得る賦形剤またはキャリアとを含む吸入可能な医薬組成物。
(項目43)
項目10に記載される形態3の多形体の精製された形態。
(項目44)
気道の上部閉塞性疾患または下部閉塞性疾患の処置を必要としている患者において該疾患を処置する方法であって、有効量の項目10に記載される形態3の多形体を吸入によって該患者に投与することを含む、方法。
【図面の簡単な説明】
【0053】
【図1】X線回折計を使用して得られる、式Iの化合物の形態1の粉末x線回折(PXRD)パターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図2】X線回折計を使用して得られる、式Iの化合物の形態2のPXRDパターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図3】X線回折計を使用して得られる、式Iの化合物の二水和物形態1のPXRDパターンのグラフである。グラフでは、カウント/秒によって規定されるピークの強度が、度での回折角度2θに対してプロットされる。
【図4】化合物10(工程8の生成物)のNMRスペクトルの写しである。
【図5】化合物11(化合物Aとも呼ばれる)のNMRスペクトルの写しである。
【図6】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態1の熱分析のプロットである。
【図7】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態2の熱分析のプロットである。
【図8】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の二水和物形態1の熱分析のプロットである。
【図9】熱重量分析(TGA)によって得られる、式Iの化合物の二水和物形態1の熱重量分析のプロットである。
【図10】X線回折計を使用して得られる、式Iの化合物の形態3のPXRDパターンのグラフである。グラフでは、カウント/秒によって定義されるようなピークの強度が、度での回折角度2θに対してプロットされる。
【図11】示差走査熱量測定法(DSC)によって得られる、式Iの化合物の形態3の熱分析のプロットである。
【発明を実施するための形態】
【0054】
(詳細な説明)
式Iの遊離塩基(これは本明細書中下記では化合物Aとして示され、下記の構造:
【0055】
【化8】
を有する)が、PCT/US2005/017134(これは参考として本明細書中に組み込まれる)において実施例26〜実施例381として開示される。
【0056】
式Iの化合物、すなわち、化合物Aのキシナホ酸塩は非吸湿性の結晶性塩であり、3つの多形体および1つの水和物を示す。
【0057】
式Iの化合物は、化合物Aまたは化合物Aの他の塩と比較して、吸入によって投与される場合、気道の上部閉塞性疾患および下部閉塞性疾患を処置するための予想外に優れた物理的プロフィルおよび薬物動態学プロフィルを有する。リン酸塩、マレイン酸塩およびコハク酸塩は非晶質である。酒石酸塩は結晶性であるが吸湿性である。フマル酸塩は結晶性の形態を有しているが、それらの形態は不安定な水和物である。従って、キシナホ酸塩は、他の塩と比較して、吸入処方物における使用について、他の塩よりも予想外に優れている。そのうえ、キシナホ酸塩は、経口投与と比較して、気管内投与により、25倍高い炎症性細胞の阻害を示す。
【0058】
式Iの化合物には3つの異なった結晶性多形体および1つの水和物が存在することが見出された。これら4つの形態は本明細書中では、形態1、形態2、形態3および二水和物形態1として示される。この化合物は治療活性医薬剤としての使用が意図されているので、式Iの化合物の最も安定な医薬的に許容され得る形態は非常に注目される。
【0059】
形態1が、本発明の方法における使用のための好ましい形態である。
【0060】
多形は、化合物が、同じ化学式を維持しながら、異なる結晶形態に結晶化し得る能力として特徴づけることができる。所与の薬物物質の結晶性多形体は、同じ様式で互いに結合する同じ原子を含むという点で、この薬物物質の任意の他の結晶性多形体と化学的に同一であるが、1つまたは複数の物理的性質(例えば、安定性、溶解性、融点、嵩密度、流動特性、バイオアベイラビリティなど)に影響を及ぼし得るその結晶形態において異なる。
【0061】
本明細書を通して使用される下記の用語は、別途指示されない限り、下記の意味を有することが理解される。
【0062】
「患者」はヒトおよび他の動物の両方を包含する。
【0063】
「哺乳動物」はヒトおよび他の哺乳類動物を包含する。
【0064】
「多形体」は、別の結晶性形態とは異なるが、同じ化学式をともに有する、物質の結晶性形態を意味する。
【0065】
「本発明の多形体」は式Iの化合物の結晶性多形体を意味する。
【0066】
「アルコール」は、ヒドロキシル基(−OH)を含む有機化合物を意味する。
【0067】
「賦形剤」は、希釈剤として使用されるか、あるいは、形態またはコンシステンシーを処方物に与えるために使用される本質的に不活性な物質を意味する。
【0068】
「有効」または「治療有効」は、PDE4阻害剤として効果的であり、従って、所望される治療効果、改善効果、阻害効果または防止効果をもたらす、本発明の化合物の多形体または組成物を表すことが意味される。「有効量」または「治療有効量」は、PDE4阻害剤として効果的であり、従って、所望される治療効果、改善効果、阻害効果または防止効果をもたらす、本発明の多形体または組成物の量を表すことが意味される。
【0069】
式Iの化合物によって処置される上部気道閉塞性疾患および下部気道閉塞性疾患には、喘息、COPD(慢性閉塞性肺疾患)、慢性気管支炎、嚢胞性線維症、アレルギー性鼻炎、非アレルギー性鼻炎、鼻副鼻腔炎、成人呼吸疾患、急性呼吸窮迫症候群、呼吸器系ウイルス、咳、間質性肺炎、慢性副鼻腔炎、気流閉塞、気道過応答(すなわち、気道過敏性)、気管支拡張症、細気管支炎、閉塞性細気管支炎(すなわち、閉塞性細気管支炎症候群)、呼吸困難、気腫、高炭酸ガス症、過膨張、低酸素血症、高酸素症誘発性炎症、肺線維症、肺高血圧症、小気道疾患、喘鳴および風邪が含まれる。
【0070】
式Iの化合物は好ましくは、喘息、COPD、咳、気流閉塞、気道過応答(すなわち、気道過敏性)、細気管支炎、慢性気管支炎、気腫、肺線維症、肺高血圧症、小気道疾患、喘鳴およびアレルギー性鼻炎の処置に有用である。
【0071】
より好ましくは、式Iの化合物は、COPDおよび喘息を処置するために有用である。
【0072】
式Iの化合物と組合せて使用するための、閉塞性気道疾患(例えば、COPDまたは喘息)を処置するための他の薬剤は、ステロイド剤(例えば、グルココルチコイド剤)、5−リポキシゲナーゼ阻害剤、β−2アドレナリン受容体アゴニスト、α−アドレナリン作動性受容体アゴニスト、ムスカリンM1アンタゴニスト、ムスカリンM3アンタゴニスト、ムスカリンM2アンタゴニスト、LTB4アンタゴニスト、システイニルロイコトリエンアンタゴニスト、気管支拡張剤、PDE4阻害剤、エラスターゼ阻害剤、MMP阻害剤、ホスホリパーゼA2阻害剤、ホスホリパーゼD阻害剤、ヒスタミンH1アンタゴニスト、ヒスタミンH3アンタゴニスト、ドーパミンアゴニスト、アデノシンA2アゴニスト、NK1アンタゴニスト、NK2アンタゴニスト、NK3アンタゴニスト、GABA−bアゴニスト、ノシセプチンアゴニスト、去痰剤、粘液溶解剤、うっ血除去剤、マスト細胞安定化剤、抗酸化剤、抗IL−8抗体、抗IL−5抗体、抗IgE抗体、抗TNF抗体、IL−10、接着分子阻害剤、成長ホルモンおよび他のPDE4阻害剤からなる群から選択される。
【0073】
式Iの化合物と組合せて使用するための、抗ヒスタミン剤の限定されない例には、アステミゾール、アザタジン、アゼラスチン、アクリバスチン、ブロムフェニルアミン、セルチリジン(certirizine)、クロルフェニラミン、クレマスチン、シクリジン、カレバスチン(carebastine)、シプロヘプタジン、カルビノキサミン、デスカルボエトキシロラタジン(descarboethoxyloratadine)、ドキシラミン、ジメチンデン、エバスチン、エピナスチン、エフレチリジン(efletirizine)、フェキソフェナジン、ヒドロキシジン、ケトチフェン、ロラタジン、レボカバスチン、ミゾラスチン、エキタジン(equitazine)、ミアンセリン、ノベラスチン(noberastine)、メクリジン、ノルアステミゾール(norastemizole)、ピクマスト(picumast)、ピリラミン、プロメタジン、テルフェナジン、トリペレンアミン、テメラスチン(temelastine)、トリメプラジンおよびトリプロリジンが含まれる。
【0074】
ヒスタミンH3受容体アンタゴニストの限定されない例には、チオペラミド、インプロミジン、ブリマミド、クロベンプロピト(clobenpropit)、インペンタミン(impentamine)、ミフェチジン(mifetidine)、S−ソプロミジン(sopromidine)、R−ソプロミジン、SKF−91486、GR−175737、GT−2016、UCL−1199およびクロザピンが含まれる。他の化合物を、H3受容体への活性を明らかにするために種々の公知の方法で容易に評価することができ、そのような方法には、モルモット脳膜アッセイおよびモルモットニューロン性回腸収縮アッセイが含まれる(これらはともに米国特許第5,352,707号に記載される)。別の有用なアッセイでは、ラットの脳膜が利用され、このアッセイが、Westら、「Identification of Two−H3−Histamine Receptor Subtypes」、Molecular Pharmacology、第38巻、610頁〜613頁(1990)によって記載される。
【0075】
用語「ロイコトリエン阻害剤」には、ロイコトリエンの作用または活性を阻害するか、あるいは抑制するか、あるいは阻止するか、あるいは、そうでない場合には、ロイコトリエンの作用または活性と相互作用する任意の薬剤または化合物が含まれる。ロイコトリエン阻害剤の限定されない例には、モンテルカストおよびそのナトリウム塩、1−(((R)−(3−(2−(6,7−ジフルオロ−2−キノリニル)エテニル)フェニル)−3−(2−(2−ヒドロキシ−2−プロピル)フェニル)チオ)メチルシクロプロパン酢酸およびそのナトリウム塩(これらは米国特許第5,270,324号に記載される)、1−(((1(R)−3(3−(2−(2,3−ジクロロチエノ[3,2−b]ピリジン−5−イル)−(E)−エテニル)フェニル)−3−(2−(1−ヒドロキシ−1−メチルエチル)フェニル)プロピル)チオ)メチル)シクロプロパン酢酸およびそのナトリウム塩(これらは米国特許第5,472,964号に記載される)、プランルカスト;ザフィルルカスト、ならびに[2−[[2(4−tert−ブチル−2−チアゾリル)−5−ベンゾフラニル]オキシメチル]フェニル]酢酸(これは米国特許第5,296,495号に記載される)が含まれる。
【0076】
β−アドレナリン作動性受容体アゴニストの限定されない例には、アルブテロール、ビトルテロール、イソエタリン、マタプロテレノール(mataproterenol)、ペルブテロール(perbuterol)、サルメテロール、テルブタリン、イソプロテレノール、エフェドリンおよびエピネフリンが含まれる。α−アドレナリン作動性受容体アゴニストの限定されない例には、アリールアルキルアミン(例えば、フェニルプロパノールアミンおよびプソイドエフェドリン)、イミダゾール(例えば、ナファゾリン、オキシメタゾリン、テトラヒドロゾリンおよびキシロメタゾリン)およびシクロアルキルアミン(例えば、プロピルヘキセドリン)が含まれる。
【0077】
マスト細胞安定化剤の限定されない一例がネドクロミルナトリウムである。去痰剤の限定されない一例がグアイフェネシンである。うっ血除去剤の限定されない例には、プソイドエフェドリン、フェニルプロパノールアミンおよびフェニレフリンがある。
【0078】
他のPDE4阻害剤の限定されない例には、ロフルミラスト、テオフィリン、ロリプラム、ピクラミスト(piclamist)、シロミラストおよびCDP−840が含まれる。ステロイド剤の例には、プレドニゾロン、フルチカゾン、トリアムシノロン、ベクロメタゾン、モメタゾン、ブジサミド(budisamide)、ベタメタゾン、デキサメタゾン、プレドニゾン、フルニソリドおよびコルチゾンが含まれる。
【0079】
NK1タキキニン受容体アンタゴニスト、NK2タキキニン受容体アンタゴニストおよびNK3タキキニン受容体アンタゴニストの限定されない例には、CP−99,994およびSR48968が含まれる。ムスカリンアンタゴニストの限定されない例には、臭化イプラトロピウムおよび臭化チアトロピウムが含まれる。
【0080】
GABABアゴニストの限定されない例には、バクロフェンおよび3−アミノプロピル−ホスフィン酸が含まれる。ドーパミンアゴニストには、キンピロール、ロピニロール、プラミペキソール、ペルゴリドおよびブロモクリプチンが含まれる。
【0081】
「5−リポキシゲナーゼ阻害剤」には、5−リポキシゲナーゼの酵素作用を阻害するか、または抑制するか、または阻止するか、または、そうでない場合には、5−リポキシゲナーゼの酵素作用と相互作用する任意の薬剤または化合物が含まれる。5−リポキシゲナーゼ阻害剤の限定されない例には、ジロートン、ドセベノン(docebenone)、ピリポスト(piripost)、ICI−D2318およびABT761が含まれる。
【0082】
式Iの化合物は、スキーム1またはスキーム2に概説され、また、下記の実施例1または実施例2に詳しく記載される手順によって調製された。本出願明細書における実施例1および他の箇所において、Etはエチルを意味し、Meはメチルを意味し、THFはテトラヒドロフランであり、DMFはN,N−ジメチルホルムアミドであり、t−BOCおよびBOCはt−ブトキシカルボニルを意味し、RTは室温であり、HATUはN−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシドである。
【0083】
【化9】
【実施例】
【0084】
(実施例1)
工程1:
化合物1(100.6g、0.767mol)をEtOH(1000ml)に懸濁し、0℃に冷却した機械的に撹拌されているこの懸濁液に、SOCl2(136.9g、1.15mol、84.0ml)を、内部温度が15℃未満であるように滴下漏斗により滴下した。反応混合物を2.5時間にわたって加熱還流し、その後、0℃に冷却した。エーテル(1000ml)を加え、白色の固体が沈殿した。固体を真空ろ過によって単離し、エーテルにより洗浄した。生成物2(HCl塩)を真空オーブンで乾燥して、146.3g(97%)の白色の固体を得た。MS(M+1):m/e 160。
【0085】
【化10】
工程2:
化合物2(HCl塩、146.2g、0.747mol)をCH2Cl2(1600ml)およびEtOH(100ml)に溶解し、0℃に冷却したこの溶液に、Et3N(113.4g、1.12mol、156.2ml)を加えた。t−BOC無水物(195.6、0.90mol)を少量ずつ加えた。反応混合物を0℃で15分間撹拌し、その後、RTで16時間撹拌した。得られた混合物を約800mlの体積まで濃縮し、水により洗浄した。有機溶液を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:20%EtOAc−CH2Cl2)により、生成物3(193.7g、100%)を黄色の油状物として得た。MS(M+Na):m/e 282。
【0086】
【化11】
工程3:
化合物3(36.5g、0.141mol)およびトリフェニルホスフィン(46.2g、0.176mol)を乾燥THF(1000ml)に溶解し、0℃に冷却したこの溶液に、ジエチルアゾジカルボキシラート(30.7g、0.176mol)を滴下漏斗により滴下した。反応混合物を0℃で5分間撹拌し、その後、LiBr(61.1g、0.704mol)を一度に加えた。得られた混合物をRTで16時間撹拌した。溶媒をエバポレーションし、水(1500ml)を加え、水溶液をCH2Cl2により抽出した。あわせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:2%EtOAc−CH2Cl2から5%EtOAc−CH2Cl2)により、生成物4(31.8g、70%)を黄色の油状物として得た。MS(M+1):m/e 322および324。
【0087】
【化12】
工程4:
化合物4(41.2g、0.128mol)を乾燥DMSO(300ml)に溶解した溶液に、NaN3(9.15g、0.141mol)を加えた。反応混合物をRTで16時間撹拌した。水(300ml)を加え、水溶液をエーテルにより抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮して、生成物5(36.4g、100%)を油状物として得た。MS(M+Na):m/e 307。
【0088】
【化13】
工程5:
化合物5(36.4g、0.128mol)をTHF(800ml)に溶解した溶液に、10%パラジウム炭素触媒(10.0g)を加えた。反応混合物を40psiの水素圧下、Parr振とう機で16時間振とうした。触媒をろ過によって除き、イソプロパノールにより洗浄した。ろ液を濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:CH2Cl2、次いでNH3−CH2Cl2を含む10%MeOH)により、生成物6(24.2g、73%)を明灰色の固体として得た。MS(M+1):m/e 259。
【0089】
【化14】
工程6:
化合物6(12.0g、0.0464mol)を乾燥CH2Cl2(300ml)に溶解した溶液に、Et3N(9.4g、0.093mol、13.0ml)を加え、その後、シクロプロパンカルボニルクロリド(5.3g、0.051mol、4.64ml)を加えた。反応混合物をRTで16時間撹拌した。水(200ml)を加え、水溶液をCH2Cl2により抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:NH3−CH2Cl2を含む5%MeOH)により、生成物7(14.3g、94%)を油状物として得た。MS(M+Na):m/e 349。
【0090】
【化15】
工程7:
化合物7(40.0g、0.123mol)をCH2Cl2(550ml)に溶解した溶液に、4N HClのジオキサン溶液(153ml、0.613mol)を加えた。反応混合物をRTで4時間撹拌し、その後、濃縮して、生成物8(32.2g、100%)を無色の泡状物として得た。MS(M+1):m/e 227。
【0091】
【化16】
工程8:
乾燥DMF(300ml)中の化合物8(5.5g、20.8mmol)およびカルボン酸9(10.0g、20.8mmol)の混合物に、3Aシーブス(10.0g)、Et3N(6.3g、62.3mmol、8.7ml)、次いでHATU(15.8g、41.6mmol)を加えた。反応混合物をRTで21時間撹拌し、その後、溶媒を濃縮した。水(400ml)を加え、水溶液をCH2Cl2により抽出した。合わせた有機抽出物を乾燥させ(MgSO4)、ろ過し、濃縮した。シリカゲルクロマトグラフィーによる精製(溶出液:20%EtOAc−CH2Cl2から60%EtOAc−CH2Cl2に)により、生成物10(14.0g、98%)を無色の泡状物として得た。MS(M+1):m/e 690。NMRスペクトルについては図3を参照のこと。
【0092】
工程9:
化合物10(42.1g、0.061mol)をCH2Cl2(600ml)に溶解し、0℃に冷却したこの溶液に、4N HClのジオキサン溶液(76ml、0.305mol)を加えた。その後、反応混合物をRTで5時間撹拌し、その後、濃縮した。粗生成物を1:1のEtOH:H2O(120ml)に溶解し、25%NaOH水溶液により塩基性にした(pH=9〜10)。CH2Cl2(700ml)を加え、反応混合物を、すべての固体が溶解するまで撹拌した。層を分離し、水溶液をCH2Cl2により抽出した。合わせた有機抽出物をブラインにより洗浄し、乾燥させ(MgSO4)、ろ過し、濃縮した。さらなるCH2Cl2を加え、混合物を再び濃縮した。エーテルを加え、混合物を濃縮して、化合物11(化合物A)(34.4g、96%)を明黄色の固体として得た。MS(M+1):m/e 590。NMRスペクトルについては図4を参照のこと。
【0093】
本出願明細書における実施例2および他の箇所において、Etはエチルを意味し、Meはメチルを意味し、EtOHはエタノールを意味し、NMRは核磁気共鳴を意味し、THFはテトラヒドロフランであり、DMFはN,N−ジメチルホルムアミドであり、t−BOCおよびBOCはt−ブトキシカルボニルを意味し、RTは室温であり、DMSOはジメチルスルホキシドを意味し、Et3Nはトリエチルアミンを意味し、NaHMDSはナトリウムビス(トリメチルシリル)アミドであり、HOBTはヒドロキシベンゾトリアゾールであり、EDCl HClは1−エチル−3−[3−ジメチルアミノ]プロピル]カルボジイミド塩酸塩であり、NMPはN−メチルピロリジノンであり、caはcirca(約)であり、KFはカールフィッシャーであり、EtOAcは酢酸エチルである。
【0094】
【化17】
(実施例2)
工程1:
(S)−2−tert−ブトキシカルボニルアミノプロピオン酸(8.8kg(46.5モル、2当量))を、熱電対、N2導入管および供給タンクを備える50LのHastelloy反応装置に加えた。乾燥テトラヒドロフラン(90リットル)(THF、KF<0.05%)をバッチに加え、溶解するように装入した。ジシクロヘキシルアミン(8.5kg(46.9モル、2当量))をバッチに加え、−5℃〜5℃の間の温度範囲で約30分かけてゆっくり装入した。バッチを−5℃〜5℃の間の温度範囲で約15分間撹拌した。トリメチルアセチルクロリド(5.7kg(47.3モル、2当量))をバッチに加え、−5℃〜5℃の間の温度範囲で約30分かけてゆっくり装入した。バッチを−5℃〜5℃の間の温度範囲で約3時間撹拌した。ヘプタン(27リットル)をバッチに加え、装入し、続いて、4.5kgのセライトを加えた。バッチをN2下でろ過し、フィルターケーキを30%(v/v)THF/ヘプタンにより洗浄した。ろ液を濃縮した。ろ液および洗浄液は、真空下でバッチを約36リットルのバッチ容量にした。THF(27リットル)をバッチに加え、装入した。バッチの温度を約20℃〜30℃に調節した。バッチをKFのためにサンプリングした(0.06ppm未満)。バッチは混合無水物のTHF溶液であり、これを、さらに精製することなく次工程において使用した。
【0095】
化合物(1A)(9.0kg(23.3モル、1当量))を、熱電対、N2導入管および供給タンクを備える50ガロンのガラス内張り反応装置に加えた。乾燥テトラヒドロフランをバッチに加え(126リットル(THF、KF<0.05%))、溶解するように装入した。バッチを1気圧で濃縮して約81リットルのバッチ容量にした。温度を約−60℃〜−70℃に調節した。NaHMDS(THF中2M、2.70kg、5.9モル、0.25当量)を加え、−60℃〜−70℃の間の温度範囲で約15分かけて装入した。バッチを−60℃〜−70℃の間の温度範囲で約5分間撹拌した。上記混合無水物のTHF溶液(0.83kg活性、3.2モル、0.14当量)を加え、−60℃〜−70℃の間の温度範囲で約15分かけて装入した。バッチを−60℃〜−70℃の間の温度範囲で約10分間撹拌した。連続する2回の装入(NaHMDS、THF中2M)および混合無水物を、合計で8セットの装入のためにさらに7回、または、転換率が70%以上になるまで繰り返した。転換率が94%以上になるまで、NaHMDS(THF中2M)を装入し、続いて、混合無水物を、残留する出発物質の量に基づいて同じ比率で装入することを続けた。ゆっくりと約15分かけて、バッチの温度を30℃未満で維持しながら、バッチを90リットルのH2Oに溶解した13.5kgのKH2PO4の水溶液に移した。酢酸エチル(59リットル)を加え、充填し、その後、約15分間撹拌し、層を静置させた。水層を45リットルの酢酸エチルにより抽出した。合わせた有機層を約32リットルの10%(w/v)NaCl水溶液により2回洗浄した。有機層を、バッチとして1気圧で約45リットルのバッチ容量まで濃縮した。メチルtertブチルエーテル(MTBE)(90リットル)をバッチに加え、装入した。バッチを約54リットルのバッチ容量まで1気圧で濃縮した。メチルtertブチルエーテル(45リットル)を55℃〜65℃の間の温度で装入した。ヘプタン(108リットル)をバッチに加え、55℃〜65℃の間の温度で装入した。温度を約45℃〜55℃に調節し、約30分間撹拌した。その後、温度を約1時間かけて約−5℃〜5℃に調節した。バッチを−5℃〜5℃の間の温度で約30分間撹拌した。バッチをろ過し、これにより、フィルターケーキを形成させ、33%(v/v)メチルtertブチルエーテル/ヘプタンにより洗浄した。バッチを真空オーブン中、45℃〜55℃で少なくとも約12時間乾燥し、これにより、8.4kg(72.2%)の化合物(2A)を、eeが99.0%を越える固体として得た。
【0096】
【化18】
工程2:
化合物(2A)(20g(39.3mmol、1当量))を、機械的撹拌装置、滴下漏斗および熱電対が取り付けられた500mLの三口丸底フラスコに加え、装入した。THF(60ml)、EtOH(20mL)および水(100mL)をフラスコに加え、反応混合物を装入した。次いで、8mLの25%水酸化ナトリウム溶液を反応混合物に加え、装入した。反応混合物を40℃で約4時間撹拌した。反応の完了をHPLCアッセイによって判断すると、水(100ml)を混合物に加え、バッチを装入し、50℃に加熱した。50℃に達すると、1N HCl溶液(30ml)をバッチに加え、30分間かけて装入した。バッチをこの温度でさらに30分間撹拌し、その後、さらに24mlの1N HCl溶液をバッチに加え、バッチを30分かけて装入した。水(60ml)をバッチに加え、バッチを50℃で30分かけて装入し、これにより、スラリーを形成させた。得られたスラリーを1時間以上にわたって室温に冷却し、これにより、生成物を形成させ、この生成物を吸引ろ過によって集め、これにより、湿潤ケーキを形成させた。湿潤ケーキをエタノールおよび水(1/5、v/v)の40mlの溶媒混合物により洗浄した。得られた固体を真空下60℃で12時間乾燥し、これにより、16.8g(90%)の化合物(3A)を灰白色の固体として得た。
【0097】
【化19】
工程3:
パートA
(2R,4S)−4(シクロプロパンカルボニルアミノ)ピロリジン−1,2−ジカルボン酸−1−tert−ブチルエステル2−エチルエステル(BP)(60g、184mmol、1当量)をEtOAc(1.2L)に溶解し、サンプルを100%のHPLC標準物として採取した。バッチを20℃〜35℃に冷却し、HCl(g)(36g、980mmol、5.3当量)をバッチに加え、反応温度を20℃〜35℃の間で維持しながら装入した。生成物のHCl塩が、反応が進行するにつれ沈殿した。HCl装入が終了したとき、バッチを20℃〜30℃に加熱し、1時間撹拌した。1時間後、反応混合物をサンプリングし、反応物のHPLC面積応答を上記標準物と比較することによって反応の完了について調べた。反応液を、標準物に対するBPの量が0.5%以下の面積になるまでサンプリングした。バッチを真空下、35℃〜45℃で600mLまで濃縮し、これは濃厚なスラリーを形成した。その後、NMP(280mL)をバッチに加えた。バッチを真空下、35℃〜45℃で約560mLの容量まで濃縮し、これは透明な溶液を形成した。この透明な溶液をパートBにおけるカップリング工程においてそのまま使用した。
【0098】
パートB:
化合物(3)を、1Lの三口丸底フラスコ中のHOBT・H2O(28g、182mmol、1.1当量)およびEDC・HCl(48g、250mmol、1.4当量)をNMP(320mL)およびEtOAc(320mL)の溶液中に溶解した(80g、166mmol、1当量)。バッチを25℃で40分間撹拌した。(パートAからの)BPの溶液をバッチに加え、10分間撹拌した。N−メチルモルホリン(80mL、724mmol、4.4当量)を、温度を35℃未満に維持する速度で反応液に加えた。反応が完了したと判断すると、EtOAc(320mL)および水(800mL)をバッチに加えた。得られたバッチを15分間撹拌し、層を分離した。有機層を1M HCl(400mL)により洗浄し、次いで、10%K2CO3(400mL)および水(400mL)により洗浄した。有機層を約160mLまで濃縮し、アセトン(800mL)を有機層に加えた。バッチを減圧下において約40℃〜50℃で約240mLまで再び濃縮した。反応液をさらに800mLのアセトンにより希釈し、バッチを減圧下において40℃〜50℃で約240mLまで濃縮した。バッチの温度を約40℃で維持し、800mLのヘプタンをバッチにゆっくり加え、その結果、固体が一部形成した。固体生成物をろ過によって収集し、真空下50℃で12時間乾燥させて、103g(90%)の化合物(4A)を灰白色の固体として得た。
【0099】
【化20】
注意:回転異性体の存在のために、測定されたピークは、測定された通りに列挙される。
【0100】
工程4:
化合物(4A)(20g、29mmol、1当量)をフラスコに加え、THF(60ml)に溶解するように装入し、溶液を0℃〜10℃に冷却した。濃HCl(20ml)を、温度を0℃〜20℃に維持するようにゆっくり加えた。装入が終了したとき、溶液を20℃〜30℃に加温し、約4時間撹拌した。約4時間で、HPLC分析によって、反応が完了していることが明らかにされた。バッチを2−Me−THF(120ml)およびTHF(40ml)により希釈し、反応を、8〜8.5のpHを達成するため20%K2CO3(110ml)により停止させた。pHを調節した後、さらに水(80ml)を加え、バッチを約30℃に加熱して、明瞭な相分離を達成した。バッチを約15分間静置し、下層を分離し、有機層を水(80ml)により洗浄した。有機相を2−Me−THF(200ml)により希釈し、その後、大気圧において還流下で約100mlまで濃縮した。固体生成物がこの体積で観測された。その後、バッチを0℃〜10℃に冷却し、ろ過し、これにより、湿ったケーキが残留した。湿ったケーキを2−Me−THFにより2回洗浄した(それぞれ、40ml)。洗浄された湿ったケーキを真空下において60℃で少なくとも12時間乾燥し、これにより、13.50g(79%)の化合物(5A)(これは本明細書中では化合物Aとも呼ばれる)を白色の固体として得た。
1H−NMR(スペクトルは回転異性体を示し、化学シフトのみが報告され、積分またはピーク多重度は報告されない;
【0101】
【化21】
キシナホ酸塩の形成:
多形体形態1:方法1:
化合物A(34.4g、0.0583mol)を熱EtOH(800ml)に溶解した溶液に、キシナホ酸(10.98g、0.0583mol)を、EtOH溶液を加熱し続けながら少量ずつ注意深く加えた。さらなるEtOH(200ml)および水(6ml)を加えた。反応混合物を沸騰近くまで加熱して、すべての固体を溶解し、その後、ろ過した。ろ液をRTにゆっくり冷却した。このとき、結晶化が生じた。混合物をRTで一晩放置した。ろ液を0℃に冷却し、固体のキシナホ酸塩を真空ろ過によって単離した。固体のキシナホ酸塩をイソプロパノールにより洗浄し、次いで、エーテルにより洗浄し、高真空下において60℃で乾燥させて、36.8g(81%)の白色固体を得た。
【0102】
多形体形態1:方法2
窒素導入管および還流冷却器を備えた500mLの三口丸底フラスコに、スキーム2(実施例2)の化合物(5A)(30g、50.89mmol、1当量)および1−ヒドロキシ−2−ナフトエ酸(10.5g、55.80mmol、1.1当量)を加えた。その後、このフラスコにトルエン(154mL)およびメタノール(103mL)を加え、得られたスラリーを約62℃に加熱した。このとき、内容物が均一になった。15分間撹拌した後、内容物を大気圧で蒸留して210mLにし、その後、約50℃に冷却し、その後、形態1の結晶(10mLのトルエン中3g、10重量%)を接種し、これにより、生成物の塩を結晶化させ、スラリーを形成した。このスラリーを50℃で30分間撹拌した後、内容物を約10℃に冷却した。そのとき、トルエン(90mL)を加え、スラリーを真空蒸留して約210mLにした。トルエン(90mL)の再度の添加を行い、内容物を約20℃で20分間撹拌した。得られた固体を、真空下で撹拌型乾燥機を使用して収集し、湿ったケーキをトルエン(60mL)により洗浄した。これらの固体を、下記のプロトコルを使用して乾燥した。(a)Tj=50℃、圧力=0.1bar、撹拌なし、時間=3h;(b)Tj=80℃、圧力=0.1bar、20rpm、時間=12h;(c)Tj=80℃、圧力=0.1bar、60rpm、時間=12h。合計で35g(81%)の、スキーム2(実施例2)の化合物(6A)を固体として回収した。
1H−NMR(スペクトルは回転異性体を示し、化学シフトのみが報告され、積分またはピーク多重度は報告されない;
【0103】
【化22】
多形体形態1、方法3:化合物A(5.0g、0.00848mol)をMeOH(75ml)に溶解した溶液を50℃に加熱し、ろ過し、MeOH(10ml)によりリンスした。キシナホ酸(1.76g、0.00933mol)をMeOH(35ml)に溶解した溶液を50℃に加熱し、ろ過し、化合物Aの溶液に加えた。混合物を約10分間にわたって加熱還流し、大気圧で蒸留して約50mLにし、約1時間かけて0℃に冷却し、約30分間撹拌した。混合物をろ過し、冷却したMeOH(20ml)により洗浄し、真空下において室温で約12時間乾燥させて、灰白色の固体を得た(5.63g(85.4%))。
【0104】
多形体形態2:化合物A(34.4g、0.0583mol)を熱CH3OH(800ml)に溶解した溶液に、キシナホ酸(10.98g、0.0583mol)を、CH3OH溶液を加熱し続けながら少量ずつ注意深く加えた。水(6ml)を加えた。反応混合物を沸騰近くまで加熱して、すべての固体を溶解し、その後、ろ過した。ろ液をRTにゆっくり冷却し、これにより結晶化がもたらされた。混合物をRTで一晩放置した。ろ液を0℃に冷却し、固体のキシナホ酸塩を真空ろ過によって単離した。固体のキシナホ酸塩をイソプロパノールにより洗浄し、次いで、エーテルにより洗浄し、高真空下において60℃で乾燥させて、36.8g(81%)の白色固体を得た。
【0105】
二水和物形態1:キシナホ酸塩形成時における水の添加が、結晶性形態を得るために必要である。二水和物形態1を、形態1(504.83mg、0.65mmol)を水(0.9mL)およびメタノール(3.1mL)の混合物に懸濁させることによって調製した。懸濁物を21時間撹拌した。懸濁物を遠心分離し、その後、上清をデカンテーションにより除くことによって固体を単離した。固体を真空下において室温で乾燥させた。
【0106】
多形体形態3:形態3を、遊離塩基の化合物A(3.0g、5.1mmol)と、キシナホ酸(0.96g、5.1mmol)との混合物を2−プロパノール(90mL)中で合わせることによって調製した。混合物を加熱還流し、さらに2−プロパノール(30mL)を加えた。混合物を1時間にわたって還流状態で保ち、その後、室温に冷却した。混合物をろ過し、固体を2−プロパノール(6mL)により洗浄し、その後、真空下で乾燥させて、3.24gの生成物を得た。
【0107】
粉末X線回折用サンプルの調製
キシナホ酸塩の、形態1、形態2、形態3、および形態1の二水和物を、粉末x線回折(「PXRD」)分析のための乾燥粉末として分析した。形態1を、下記の手順を使用して、PXRD分析の前にジェットミルで微粉化した。
【0108】
ジェットミル粉砕による微粉化
微粉化された粉末の粒子サイズ分布が、ジェット圧およびジェットミル内への供給速度を調節することによって制御される。粒子が、加圧窒素によるベンチュリシステムを介して、MC ONE JETMILL(Jetpharma Group、South Plainfield、NJ)の粉砕チャンバーに1g/分の速度で供給される。ベンチュリでの圧力低下を5barに設定する。粒子は、粉砕チャンバーの外周に置かれた4つのノズルによって粉砕チャンバーの内部においてらせん運動で加速される。ノズルでの圧力低下を4barに設定する。微粉化作用が、より遅い進入粒子と、渦巻き流において既に加速された粒子との間における衝突によって生じる。遠心力により、より大きい粒子が粉砕チャンバーの外周部に保持され、一方、より小さい粒子が静的分級機によってチャンバーの中心から排出ガスとともに出ていき、ジェットミルのすぐ下方にある捕集容器に回収される。
【0109】
サンプルを、任意の形態変化を防止するために最小限の調製物を用いて分析した。サンプルを、粒子が凝集していないことを確かめるために軽くこすった。溶媒、乾燥工程または他の調製工程はいずれも、これらの分析のために使用しなかった。PXRDデータにより、水和物形態および多形形態を一意的に特定することができる。
【0110】
粉末X線回折
形態1および形態2のX線粉末回折パターンを、30kv、15mAでのCuKα放射線(λ=1.54056Å)、および、半導体検出器を備えるRigaku Miniflex回折計(Rigaku MSC、The Woodlands、TX)で収集した。連続走査を0.02°の2θのステップサイズおよび2°/分の走査速度ですべてのサンプルについて記録した。
【0111】
二水和物形態1のX線粉末回折パターンを、40kvおよび40mAでのCuKα1線源(λ=1.5406Å)を用いてBrukar D8回折計で集めた。連続走査を0.032°の2θのステップサイズおよび0.5秒のステップ時間により記録した。
【0112】
形態3のX線粉末回折パターンをKratos XRD6000で集めた。材料をサンプルホルダーに軽く詰め、静かに滑らかにして、平坦なサンプル表面を作製することによってサンプルを調製した。サンプルを、0.02度のステップサイズおよび0.6秒のステップ持続時間で、2度から40度までの2θで分析した。データ分析を、Kratosによって供給されるBasic Processソフトウエア(バージョン2.6)を使用して行った。データを、ソフトウエア内の自動平滑化プロセスを使用して平滑化した。
【0113】
上記で記載された方法および設備を使用して、化合物Aの形態1、形態2および形態3の多形体キシナホ酸塩、ならびに、二水和物形態をPXRD分析に供した。PXRDパターンが得られた。PXRDパターンが図1〜図3および図10に示される。ピークの強度(y軸はカウント/秒の単位である)が2θの角度(x軸は度(2θ)の単位である)に対してプロットされる。加えて、データを、2θの角度に対するステップあたりの収集時間について正規化された検出器カウントでプロットした。これらのプロフィルと一致するピーク存在位置(2θのX軸における存在位置)が表1に示される。これらのPXRDピークの存在位置は、式Iの化合物の形態1、形態2、形態3の結晶性多形体および結晶性二水和物形態1に特徴的である。
【0114】
【表1−1】
【0115】
【表1−2】
【0116】
【表1−3】
表1に示されるようなPXRDピーク存在位置をはじめとして、それぞれの多形体または水和物の最も特徴的なピーク存在位置を、結晶性構造を他の構造から都合よく識別するために選択し、相対強度によってグループ化することができる。
【0117】
特徴的なピークのそのような選択が表2に示される。従って、例えば、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群1(これは、4個の特徴的なPXRDピーク存在位置からなる)によって特定することができる。代替として、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群2(これは、群1の4個の特徴的なPXRDピーク存在位置、およびさらなる4個のピーク存在位置からなる)によって特定することができる。代替として、式Iの化合物の形態1の結晶性構造を、ピーク存在位置群3(これは、群2の8個の特徴的なPXRDピーク存在位置、およびさらなる4個のピーク存在位置からなる)によって特定することができる。このスキームは、4つの多形形態のそれぞれを特定するために、また、4つの多形形態のそれぞれを他の形態から識別するために、4つの多形形態のそれぞれに適用される。
【0118】
【表2】
当業者は、同じ化合物の所与の結晶性形態についてのPXRDピーク存在位置の測定値が誤差限界内で変動することを認識する。そのような変動は、とりわけ、サンプル調製、装置使用または分析技術における様々な違いによって持ち込まれ得る。個々のピーク存在位置の測定値はわずかに変動することがあり、しかし、ピークプロフィル全体が、例えば、充填サンプルの密度における変動に起因して、より大規模に変動することがある。
【0119】
多形体の純度
好ましくは、式Iの化合物の結晶性多形体の形態1、形態2、形態3および二水和物形態1は、化学的不純物(例えば、多形体の調製時に生じる副生成物)および他の多形性結晶性形態を実質的に含まない。化学的不純物を「実質的に含まない」とは、本発明の目的のためには、約5%(w/w)以下の化学的不純物、好ましくは、約3%(w/w)以下の化学的不純物、より好ましくは、約2%(w/w)以下の化学的不純物、さらに一層好ましくは、約1%(w/w)以下の化学的不純物を含むことを意味する。多形体についての用語「精製された」または用語「精製された形態で」は、本明細書中に記載されるか、または、当業者に広く知られている標準的な分析技術によって特徴づけ可能であるための十分な純度で、本明細書中に記載されるか、または、当業者に広く知られている精製プロセスから得られた後の上記多形体の物理的状態を示す。式Iの化合物の結晶性多形体の形態1、形態2、形態3および二水和物形態1の精製された形態は、化学的不純物を実質的に含まない。
【0120】
示差走査熱量測定
多形体形態1および多形体形態2のサンプルを試験するために使用したDSC装置は、冷却された冷却系を備えた、TA Instruments(登録商標)モデル2920(2001年製造)であった。DSCセル/サンプルチャンバーが40ml/分の超高純度窒素ガスによりパージされた。装置は高純度インジウムにより校正された。この方法による測定されたサンプル温度の正確度は約+/−1℃の範囲内であり、融解熱を約+/−5%の相対的誤差の範囲内で測定することができる。サンプルを、圧力放出を可能にするための2つのピンホールを有するふたを伴う標準的なアルミニウムDSC皿に入れた。約2mgのサンプル粉末を皿の底に入れ、皿と接触させるために軽くたたいた。サンプルの重量を正確に測定し、ミリグラムの1/100まで記録した。この装置では、空の参照皿を使用された。DSC分析を10℃/分の加熱速度で行った。
【0121】
二水和物形態1および多形体形態3のサンプルを試験するために使用したDSC装置は、Q100 TAInstruments(登録商標)であった。再度、サンプルを気密型アルミニウム皿に密封し、2つのピンホールをサンプル皿のふたに開けた。分析を、窒素パージ下、10℃/分の加熱速度で行った。
【0122】
熱流量(これはサンプル重量によって正規化された)が、測定されたサンプル温度に対してプロットされた。データがワット/グラム(「W/g」)の単位で報告された。プロットを作製した(吸熱ピークは下向きである)。吸熱の融解ピークを、この分析における外挿された開始温度および終了(発生)温度、ピーク温度、ならびに、融解熱について評価した。
【0123】
式Iの形態1のDSCプロフィルが図6に示される。式Iの化合物の形態1について、単独の吸熱が観測され、開始温度が192℃であり、ピーク温度が193℃であった。
【0124】
式Iの形態2のDSCプロフィルが図7に示される。式Iの化合物の形態2について、2つの重なった吸熱が観測され、開始温度が152℃であり、ピーク温度が161℃および181℃であった。
【0125】
式Iの二水和物形態1のDSCプロフィルが図8に示される。式Iの化合物の二水和物形態1については、10℃/分で、二水和物形態1は脱水を受け、室温で準安定性の無水形態に変わる。この事象は、73℃での開始温度および143J/gの熱を有する幅広い吸熱としてDSCサーモグラム(図8)に反映される。加熱期間中に失われた水和物の水の量は総重量の4.1%を占め、これが、二水和物の化学的量論を示す、TGAデータ(図9)における階段様の重量減少として示される。室温で準安定性の形態は、144℃の開始温度で融解する。融解熱は、TGAデータ(図9)における150℃以降の重量減少に対応する分解が、融解事象が完了する前に始まるために、求めることができない。
【0126】
式Iの形態3のDSCプロフィルが図11に示される。式Iの化合物の形態3について、182℃の開始温度および186℃のピーク温度を伴う単独の吸熱が観測された。
【0127】
吸入による処置のための前提は、全身の副作用を最小限に抑えながら、薬物を作用部位(肺)に直接送達することである。従って、吸入された化合物は、経口バイオアベイラビリティの低さに起因する低い血中濃度(AUC)、および/または、吸入もしくは経口による服用経路によって与えられる場合の高いクリアランスを有する薬物動態学プロフィルを示さなければならない。吸入期間中の何らかの飲み込まれた薬物の影響を最小限に抑えるために、経口AUCが低いことは重要である。多くの場合、低いAUCレベルは、測定することが困難である。従って、再現可能なAUCデータが好ましい。
【0128】
アレルギー性Brown−Norwayラットのためのアッセイプロトコル:
体重が150g〜200gの同系交配されたオスのBNラットをCharles River Laboratory(Wilmington、MA)から得た。使用前、動物に、食物および水を自由に摂取させた。以下の「試験化合物の送達」の節に記載されるように、試験化合物を経口経路または吸入経路のいずれかによって抗原攻撃の5時間前に投与した。
【0129】
感作および抗原気管支誘発
動物を2つの主要な群(すなわち、ミョウバン群および抗原群)に分けた。抗原群では、動物を、0.9%の生理的食塩水ビヒクルに懸濁させた20μgの卵白アルブミン(OVA、グレードIII;Sigma chemical Co.、St Louis、MO)および8mgのAl(OH)3を含む1mlのミョウバン沈殿抗原の腹腔内(i.p.)注射によって感作させた。このミョウバン−OVA混合物の追加抗原刺激注射を7日後に再び行った。ミョウバン群に属する動物には、ミョウバンのみを含有する注射を受けさせた。2回目の注射の7日後、動物を、エアロゾル化抗原気管支誘発にさらし、これは、ラットを密閉型プレキシガラスチャンバー(21リットル)に置き、ラットをエアロゾル化OVA(1%)に30分間暴露させることによって行われた。エアロゾル化OVAは、超音波ネブライザー(DeVilbiss、Somerset、PA、米国;モデルUltra−Neb99)によっておよそ8リットル/分の流速で作製した。エアロゾル化OVAによる攻撃の24時間後、動物を過量のペントバルビタールナトリウムにより安楽死させた。気管を取り出し、挿管し、肺を3mlの生理食塩水により2回洗浄した。このようにして集められた気管支肺胞洗浄液(BALF)を細胞計数に供した。10マイクロリットルのBALFを利用して、総白血球を、血球計を使用して手作業により計数した。100マイクロリットルのBALFを使用して、細胞遠心分離物を調製し、この細胞遠心分離物をHema3(商標)染色システム(Fisher Scientific、Springfield、NJ)により染色して、種々の白血球(例えば、好酸球、好中球、単核細胞および上皮細胞など)を特定し、計数した。合計で200個の細胞をそれぞれの細胞遠心分離物から計数した。炎症性細胞を気道内に動員することを阻害する化合物の能力が報告される。
【0130】
試験化合物の送達:
経口投与:化合物を0.4%メチルセルロースに溶解し、動物に3ml/kgで経口送達した。等体積の0.4%メチルセルロースを陰性コントロール群(ミョウバン群)および陽性コントロール群(抗原源)の両方に与えた。
【0131】
気管内投与:適切な用量の化合物をラクトース粉末と混合して、3mgの最終量を達成し、これを、麻酔した動物に、先の細い微量噴霧器を使用して気管内送達した。動物を、3分間〜4分間、直立姿勢で保ち、麻酔から回復させ、その後、そのケージに戻した。
【0132】
上記の試験手順を使用して、下記の結果が得られた:
酒石酸塩:0.02mpkで炎症性細胞の52%阻害(気管内投与)
キシナホ酸塩:0.02mpkで炎症性細胞の69%阻害(気管内投与)
サルPKアッセイのためのアッセイプロトコル:
2匹の絶食させたサルに、0.4%のHPMCビヒクル中の試験化合物を3mpkで経口投与した。投与容量は2ml/kgであった。血漿を、0.5時間、1時間、2時間、4時間、8時間および24時間後に収集した。血液サンプルをヘパリンとともに収集し、血漿をEDTAとともに保存した。それぞれの個々の動物についての血液サンプルをMS/MS分析によって特徴決定した。
【0133】
上記の試験手順を使用して、下記の結果が得られた:
酒石酸塩:サルAUC=10mpk(po)で30ng.h/mL
キシナホ酸塩:サルAUC=10mpk(po)で0ng.h/mL
ラットPKアッセイのためのアッセイプロトコル:
2匹の絶食させたSprague Dawleyラットに、0.4%のHPMCビヒクル中の化合物を10mpkで経口投与した。投与容量は5ml/kgであった。血漿を、0.5時間、1時間、2時間、3時間、4時間および6時間後に収集した。血液サンプルをヘパリンとともに収集した、血漿をEDTAとともに保存した。それぞれの時点での2つの血液サンプルをMS/MS分析によって特徴決定した。
【0134】
酒石酸塩:AUC=30mpk(po)で0〜1350ng.h/mL(変動)
キシナホ酸塩:AUC=30mpk(po)で350ng.h/mL
肺機能アッセイのためのアッセイプロトコル:
肺機能を、強制呼気技術を使用して測定した。この手法では、ラットを麻酔し、気管カテーテルを挿入した。ラットを、肺の膨張および収縮を分離することができる呼吸弁を含有する全身プレチスモグラフの中に置いた。その後、肺を、総肺気量に対する肺の強制膨張に供し、その後、残気量に対する急速収縮に供した。努力肺活量および最高呼気流量の測定値を使用して、抗原攻撃の影響を測定し、式Iの化合物の阻害効果を評価した。薬物を気管内送達のためにラクトースと混合させ、抗原攻撃の5時間前に微細微量噴霧器により気管内に直接に与えた。経口送達される化合物は抗原攻撃の5時間前に0.4%メチルセルロースビヒクル中で与えた。コントロール動物は気管内ラクトースまたは気管内メチルセルロースをそれぞれ受けた。抗原攻撃は、1%卵白アルブミンへの30分間のエアロゾル暴露からなった。強制呼気による肺機能を抗原(卵白アルブミン)暴露後24時間で測定した。
【0135】
式Iの化合物は0.02mpk(it(気管内))で努力肺活量(FVC)の54%阻害を示し、3mpk(po)でFVCの31%阻害を示す。
【0136】
医薬組成物
医薬組成物を本発明により記載される多形体から調製するために、不活性な医薬的に許容され得るキャリアは固体または液体のいずれかであり得る。様々な組成物のための医薬的に許容され得るキャリアおよび製造方法の例を、A.Gennaro(編)、Remington’s Pharmaceutical Sciences(第18版(1990)、Mack Publishing Co.、Easton、Pennsylvania)に見出すことができる。
【0137】
液体形態の調製物には、鼻腔内投与のための溶液、懸濁物およびエマルジョンが含まれる。
【0138】
吸入のための好適なエアロゾル調製物には、溶液、および、粉末形態の固体が含まれ得る。この場合、これらは、医薬的に許容され得るキャリア(例えば、不活性な圧縮ガス(例えば、窒素)など)との組合せであり得る。
【0139】
投薬量
単位服用量の調製物における活性な化合物の量は、特定の適用に従って、約0.01μgから約100mgまで、好ましくは約0.01μgから約75mgまで、より好ましくは約0.01μgから約50mgまで、最も好ましくは約0.01μgから約25mgまで変化させるかまたは調節することができる。
【0140】
用いられる正確な投薬量は、患者の要件、および、処置されている状態の重篤度に依存して変化させることができる。具体的な状況のための適正な投薬計画の決定は当業者の技術の範囲内である。便宜上、総投薬量は分割することができ、必要に応じて、1日のうちに何回かに分けて投与することができる。
【0141】
本発明の化合物および/またはその医薬的に許容され得る塩の投与量および投与頻度は、患者の年齢、状態およびサイズ、ならびに、処置されている症状の重篤度のような要因を考慮して、主治医の判断に従って調節される。吸入のために推奨される典型的な1日投薬量は、1回〜4回に分割した投与において、約0.04μg/日から約400mg/日の範囲であり得る。
【0142】
操作実施例に示される場合、または別途示される場合を除き、成分の量および反応条件などを表す、本明細書および請求項において使用されるすべての数字は、あらゆる場合、用語「約」によって修飾されているとして理解される。上の記述は、本発明のすべての改変および変化を詳しく記載することを意図するものではない。様々な変化が、本発明の概念から逸脱することなく、上記で記載された実施形態に対して行われ得ることが当業者によって理解されるであろう。従って、本発明は、上述した特定の実施形態に限定されず、しかし、下記の請求項の範囲によって定義されるような本発明の趣旨および範囲内にある改変を包含するように意図されることが理解される。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2012−214482(P2012−214482A)
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−133753(P2012−133753)
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2009−519488(P2009−519488)の分割
【原出願日】平成19年7月10日(2007.7.10)
【出願人】(596129215)メルク・シャープ・アンド・ドーム・コーポレーション (785)
【氏名又は名称原語表記】Merck Sharp & Dohme Corp.
【住所又は居所原語表記】One Merck Drive,Whitehouse Station,New Jersey 08889,U.S.A.
【Fターム(参考)】
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2012−133753(P2012−133753)
【出願日】平成24年6月13日(2012.6.13)
【分割の表示】特願2009−519488(P2009−519488)の分割
【原出願日】平成19年7月10日(2007.7.10)
【出願人】(596129215)メルク・シャープ・アンド・ドーム・コーポレーション (785)
【氏名又は名称原語表記】Merck Sharp & Dohme Corp.
【住所又は居所原語表記】One Merck Drive,Whitehouse Station,New Jersey 08889,U.S.A.
【Fターム(参考)】
[ Back to top ]