
耐熱性に優れる重合体の製造方法
【課題】耐熱性に優れるスチレン系重合体を、汎用の重合開始剤などを用いて重合を行なうことにより、簡単に製造できる方法の提供。
【解決手段】下記の一般式(I);

(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)で表されるスチレン系誘導体(I)の単独および共重合体を製造する方法。
【解決手段】下記の一般式(I);
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)で表されるスチレン系誘導体(I)の単独および共重合体を製造する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、耐熱性に優れる重合体の製造方法に関する。より詳細には、本発明は、多環構造を有する脂環式炭化水素基からなる置換基をベンゼン環に有するスチレン系誘導体に由来する構造単位を分子中に有する耐熱性に優れる重合体の製造方法に関する。本発明により得られる重合体はその分子構造に応じて熱可塑性樹脂または熱可塑性エラストマーとしての性質を有し、そのいずれもが耐熱性および力学的特性に優れ、エラストマー構造を有するものは弾性特性においても優れている。そのため、本発明により得られる重合体は、それらの特性を活かして、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することができる。
【背景技術】
【0002】
スチレン系樹脂は、その優れた成形加工性、物性などにより、電気製品、家庭用部品、産業用資材などの広範な分野で従来から汎用されているが、近年、用途の拡大などに伴って一層高い性能、特に耐熱性の向上が強く求められるようになっている。
【0003】
耐熱性の向上したスチレン系樹脂としては、立体規則性が主としてシンジオタクチック構造であるスチレン系重合体が知られている(特許文献1及び2を参照)。これらのスチレン系重合体は、シンジオタクティシティーが高く、結晶性であり高い耐熱性を示す。しかしながら、該スチレン系重合体の製造に当たって、有機アルミニウム化合物と縮合剤とを接触させて得られる有機アルミノキサンにチタン化合物を組み合わせた触媒などを使用する必要がある。そのため、触媒を特別に調製しなければならず、汎用の触媒を用いる従来のスチレン系樹脂の製造法に比べて製造工程が複雑で手間や時間を要する。
【0004】
また、スチレン系重合体ブロックと共役ジエン系重合体ブロックを有するブロック共重合体またはその水素添加物からなる熱可塑性エラストマー[以下「(水添)スチレン系熱可塑性エラストマー」ということがある]は、熱可塑性エラストマーの1種として成形材料、粘接着剤、樹脂の改質をはじめとして、種々の用途に広く用いられるようになっている。
【0005】
近年、工業技術の進歩に伴って、熱可塑性エラストマーを高温で使用する必要が生じており、かかる点から高温特性に優れる熱可塑性エラストマーの開発が強く望まれているが、そのことは(水添)スチレン系熱可塑性エラストマーにおいても例外ではない。
高温特性が要求される用途分野としては、例えば、自動車部品、電気・電子部品、フィルム、シート、医療材料などを挙げることができ、熱可塑性エラストマーに求められる高温特性としては、高温に曝されても劣化や分解などの生じにくい耐熱特性を有すること、高温時に引張り強度、引張り伸び、応力緩和特性などの機械的特性に優れること、高温時に加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを示すことなどが挙げられる。
しかしながら、(水添)スチレン系熱可塑性エラストマーは、拘束相をなすスチレン重合体ブロックのガラス転移温度が一般に100℃前後であるため、前記温度を超える高温条件下では弾性が著しく低下し、熱可塑性エラストマーとしての性能を十分に維持できないという問題があった。
【0006】
(水添)スチレン系熱可塑性エラストマーの耐熱性を向上させる方法として、(水添)スチレン系熱可塑性エラストマーにポリα−メチルスチレン樹脂、ポリオレフィン、ポリフェニレンエーテルなどを配合することが知られているが、耐熱性の改善の程度は市場の要求を満たすには至っていない。
【0007】
また、(水添)スチレン系熱可塑性エラストマーにおいて、スチレン系重合体ブロックとして結晶性を有するシンジオタクチックポリスチレンを導入したブロック共重合体が知られている(特許文献3を参照)。このブロック共重合体では、拘束相であるシンジオタクチックポリスチレンブロックが260〜270℃という高い融点を有するため、ブロック共重合体は高い耐熱性が示す。しかしながら、このブロック共重合体の製造に当たっては、まず二官能性アニオン重合開始剤を調製して共役ジエンを重合した後、そのアニオン重合活性末端を配位重合活性末端に変換し、次いでスチレンの重合を行う必要があり、製造工程が極めて複雑である。
【0008】
さらに、(水添)スチレン系熱可塑性エラストマーにおけるスチレン系重合体ブロックを、ガラス転移温度の高いα−メチルスチレン重合体ブロックから形成したブロック共重合体が知られている(特許文献4を参照)。このブロック共重合体は、α−メチルスチレン重合体ブロックを導入したことにより高い耐熱使用温度を有しているが、α−メチルスチレンの重合をスチレンの重合に比べて低温で行う必要があるため、温度管理が複雑であり、改良の余地がある。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭62−104818号公報
【特許文献2】特開昭63−241009号公報
【特許文献3】特開平5−295056公報
【特許文献4】特開2001−172324公報
【非特許文献】
【0010】
【非特許文献1】“Synthesis”,1998年、第2号、148−152頁
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、耐熱性に優れていて、高温に曝されても劣化や分解が生じにくく、高温において良好な力学的特性を有し、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することのできる、熱可塑性樹脂状または熱可塑性エラストマー状のスチレン系重合体を提供することである。
また、本発明の目的は、特別に調製した触媒を使用する必要がなく、更には複雑で手間および時間のかかる重合工程や複雑な温度管理を要せず、汎用の触媒を用いて、簡単な重合工程および単純な温度管理の元で簡単に且つ円滑に製造することのできる耐熱性スチレン系重合体およびその製造方法を提供することである。
さらに、本発明の目的は、安価で、入手が容易なスチレン系重合体を基質として用いて、フリーデル・クラフツ反応などの芳香族求電子置換反応を行うことにより、耐熱性スチレン系重合体を簡単に且つ円滑に製造する方法を提供することである。
【課題を解決するための手段】
【0012】
上記の目的を達成すべく本発明者らは検討を重ねてきた。そして、多環構造を有する脂環式炭化水素基よりなる置換基をベンゼン環に有する特定のスチレン系誘導体を用いて重合を行って耐熱性に優れるスチレン系重合体を得ることができた。
それを踏まえて更に研究を続けたところ、多環構造を有する脂環式炭化水素基からなる置換基をベンゼン環に有する前記スチレン系誘導体を用いると、単独重合体、ランダム共重合体、ブロック共重合体などの種々の耐熱性に優れる重合体が、汎用の重合開始剤を用いて簡単な工程で、しかも複雑な温度管理を要せずに円滑に製造できることを見出した。
さらに、本発明者らは、耐熱性に優れる前記スチレン系重合体は、汎用のスチレン系樹脂を基質として用い、フリーデル・クラフツ反応などの芳香族求電子置換反応によって該基質(スチレン系樹脂)のベンゼン環に多環構造を有する脂環式炭化水素基を導入する方法によっても、簡単に製造できることを見出した。
そして、それにより得られた重合体は、高温に曝されても劣化や分解などが生じにくく、高温下で良好な機械的特性を有することを見出した。
また、本発明者らは、前記したスチレン系誘導体に由来する構造単位を有する重合体ブロックとそれよりもガラス転移温度の低い重合体ブロックを有するブロック共重合体は、熱可塑性エラストマーとしての特性を有し、引張り強度、引張り伸び、応力緩和特性などに優れ、しかも高温下において加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを有し、優れたエラストマー特性を有することを見出し、それらの知見に基づいて本発明を完成した。
【0013】
すなわち、本発明は、
(1) 下記の一般式(I);
【0014】
【化1】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または前記スチレン系誘導体(I)および他の重合性単量体を用いて重合を行うことを特徴とする、下記の一般式(I−A);
【0015】
【化2】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法である。
【0016】
そして、本発明は、
(2) 一般式(I)および一般式(I−A)におけるR1が多環構造を有する橋架け脂環式炭化水素基である前記(1)の重合体の製造方法;
(3) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)を10質量%以上の割合で有する重合体を製造するものである前記(1)または(2)の重合体の製造方法;および、
(4) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)の単独からなるか、または該構造単位(I−A)および他の重合性単量体に由来する構造単位を有する重合体を製造するものである前記(1)〜(3)のいずれかの重合体の製造方法;
である。
【0017】
さらに、本発明は、
(5) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)よりなる重合体ブロックAと、他の重合性単量体に由来する構造単位よりなる重合体ブロックBを有するブロック共重合体を製造するものである前記(1)〜(4)のいずれかの重合体の製造方法;
(6) 重合体ブロックAの含有割合が、ブロック共重合体の質量に基づいて10質量%以上であるブロック共重合体を製造するものである前記(5)の重合体の製造方法;
(7) 重合体ブロックBのガラス転移温度が重合体ブロックAのガラス転移温度よりも低いブロック共重合体を製造するものである前記(5)または(6)の重合体の製造方法;および、
(8) 重合体ブロックBが共役ジエンに由来する構造単位を主体とする共役ジエン系重合体または該共役ジエン系重合体の水素添加物よりなる重合体ブロックであるブロック共重合体を製造するものである前記(5)〜(7)のいずれかの製造方法;
である。
【0018】
そして、本発明は、
(9) 一般式(I)で表されるスチレン系誘導体(I)として、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンの少なくとも1種を用いる前記(1)〜(8)のいずれかの重合体の製造方法;および、
(10) 有機リチウム化合物を重合開始剤として用いてアニオン重合を行う前記(1)〜(9)のいずれかの重合体の製造方法;
である。
【0019】
さらに、本発明は、
(11) 下記の一般式(II);
【0020】
【化3】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるか、または当該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多環構造を有する脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のスチレン由来のベンゼン環に基R1を導入する工程を含むことを特徴とする、下記の一般式(I−A);
【0021】
【化4】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法;および、
(12) 芳香族求電子置換反応が、フリーデル・クラフツ反応である前記(11)の製造方法;
である。
【発明の効果】
【0022】
本発明の製造方法により得られる重合体は、耐熱性に優れており、高温に曝されても劣化や分解などが生じにくく、高温下においても良好な機械的特性および物理的特性を有する。
本発明の製造方法により得られる重合体のうち、一般式(I)に由来する構造単位を有する重合体ブロックとそれよりもガラス転移温度の低い重合体ブロックを有する熱可塑性エラストマーは、引張り強度、引張り伸び、応力緩和特性などに優れ、しかも高温下において加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを有し、良好な熱可塑性エラストマー特性を備えている。
本発明の製造方法による場合は、特別の触媒(重合開始剤)などを別途調製する必要がなく、スチレン系単量体などの重合において従来から用いられている汎用の重合開始剤を使用して、しかも重合時に複雑な温度管理などを要せずに、目的とする耐熱性スチレン系重合体を簡単に且つ円滑に製造することができる。
さらに、本発明では、安価で入手が容易な汎用のスチレン系重合体を基質として用いて、該スチレン系重合体中のベンゼン環に、フリーデル・クラフツ反応などの芳香族求電子置換反応によって多環構造を有する脂環式炭化水素基を導入することによっても、耐熱性スチレン系重合体を簡単に且つ円滑に製造することができる。
本発明の製造方法により得られる重合体は、前記した優れた特性を活かして、例えば、家電製品の筐体、OA機器、食品容器、包装材用発泡体、食品トレー、自動車部品、電気・電子部品、フィルム・シート、医療材料、伸縮部材などの広範な用途に有効に使用することができる。
【図面の簡単な説明】
【0023】
【図1】図1は引張試験に用いた試験片の形状および寸法を示す図である。
【図2】図2は参考例1で得られた1−(4−ビニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図3】図3は参考例2で得られた3−(4−ビニルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図4】図4は参考例3で得られた1−(4−イソプロペニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図5】図5は実施例11、実施例12および比較例2の水添トリブロック共重合体(熱可塑性エラストマー)の貯蔵弾性率E’、損失弾性率E’’および損失正接tanδの測定結果を示すグラフである。
【図6】図6は実施例11、実施例12および比較例2の水添トリブロック共重合体(熱可塑性エラストマー)の応力緩和特性の測定結果を示すグラフである。
【発明を実施するための最良の形態】
【0024】
以下に本発明について詳細に説明する。
本発明の製造方法により得られる重合体(以下、本発明の製造方法により得られる重合体を、「本発明の重合体」ということがある)は、スチレン系誘導体由来の下記の一般式(I−A)で表される構造単位(I−A)を重合体分子中に有する。
【0025】
【化5】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
【0026】
上記の構造単位(I−A)においてR1は多環構造を有する脂環式炭化水素基である。
ここで、「多環構造を有する脂環式炭化水素基」とは、2個以上の脂肪族環(脂環)を有する炭化水素基を意味する。多環構造を有する脂環式炭化水素基(以下「多脂環式炭化水素基」ということがある)R1は、多環構造を有する橋架け脂環式炭化水素基、すなわち隣り合う2つの脂環が2個以上の炭素原子を互いに共有している多脂環式炭化水素基であることが、重合体の耐熱性が良好になる点から好ましい。多脂環式炭化水素基R1は、場合によりR1を構成している2個以上の脂環のうちの1個または2個以上に脂肪族不飽和結合を有していてもよい。但し、脂環中に存在する脂肪族不飽和結合は、重合に関与しないか又はスチレン系誘導体(I)中のビニル結合よりも重合性が低いことが必要である。そうでないと、スチレン系誘導体(I)の円滑な重合が阻害されて、上記の構造単位(I−A)を有する本発明の重合体を円滑に得ることが困難になる。
【0027】
多脂環式炭化水素基R1の具体例としては、アダマンチル基(トリシクロ〔3.3.1.13,7〕デシル基)、ビアダマンチル基、ノルボルニル基、イソボルニル基、ビシクロノニル基、ビシクロ〔2.1.0〕ペンチル基、ビシクロ〔3.2.1〕オクチル基、トリクロ〔2.2.1.02,6〕ペンチル基などを挙げることができる。これらの多脂環式炭化水素基は、場合によりアルキル基、ハロゲン、アルコキシル基などにより置換されていてもよい。
【0028】
構造単位(I−A)における多脂環式炭化水素基R1のベンゼン環での結合位置は、ベンゼン環における重合体主鎖への結合部位[スチレン系誘導体(I)におけるビニル結合:−C(R2)=CH2の結合部位]に対してオルト位、メタ位またはパラ位のいずれであってもよく、そのうちでも、重合体の製造原料であるスチレン系誘導体(I)の重合反応性などの点から、多脂環式炭化水素基R1はパラ位に結合していることが好ましい。
【0029】
上記の一般式(I−A)で表される構造単位(I−A)において、R2は水素原子、炭素数1〜10のアルキル基またはアリール基のいずれであってもよい。
R2が炭素数1〜10のアルキル基である場合は、直鎖状または分岐状のいずれであってもよく、具体的にはメチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基などの直鎖状アルキル基、イソプロピル基、イソブチル基、sec−ブチル基、tert−ブチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、1−メチルヘプチル基などの分岐状アルキル基などを挙げることができる。
また、R2がアリール基である場合の具体例としては、フェニル基、ナフチル基などを挙げることができ、これらのアリール基は、アルキル基、ハロゲン、アルコキシル基などの置換基を1個または2個以上有していてもよい。
上記したうちでも、R2は水素原子または炭素数1〜3の直鎖状アルキル基、そのうちでも水素原子またはメチル基、特に水素原子であることが、重合体の製造原料であるスチレン系誘導体(I)の製造容易性、重合反応性などの点から好ましい。
【0030】
構造単位(I−A)を有する本発明の重合体は、上記の一般式(I)で表されるスチレン系誘導体(I)を少なくとも用いて重合を行うことによって製造することができる。
本発明の重合体の製造に好ましく用い得るスチレン誘導体(I)の具体例としては、下記の化学式で示すスチレン系誘導体を挙げることができる。
【0031】
【化6】
【0032】
上記したスチレン系誘導体(I)のうちでも、R1がアダマンチル基である上記の化学式(Ia)で表される1−(4−ビニルフェニル)アダマンタン[別称:4−(1−アダマンチル)スチレン]およびR1がアダマンチル基でR2がメチル基である上記の化学式(Ic)で表される1−(4−イソプロペニルフェニル)アダマンタン[別称:4−(1−アダマンチル)−α−メチルスチレン]、並びにR1がビアダマンチル基である上記の化学式(Ib)で表される3−(4−ビニルフェニル)−1,1’−ビアダマンタンのうちの1つ以上、特に1−(4−ビニルフェニル)アダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンのうちの一方または両方に由来する構造単位を有する本発明の重合体は、耐熱性に優れ、しかも単量体の製造が容易であることから好ましい。
【0033】
本発明の重合体は、スチレン系誘導体由来の構造単位(I−A)のみから構成される単独重合体であってもよいし、または構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体であってもいずれでもよい。
本発明の重合体が、構造単位(I−A)のみから構成される単独重合体である場合は、重合体のガラス転移温度が一般に200℃を超えるようになり、使用可能温度域が広がり、より高温での使用に耐えることができる。
また、本発明の重合体が、構造単位(I−A)および他の重合性単量体に由来する構造単位を有する共重合体である場合は、構造単位(I−A)のみからなる単独重合体に比べてガラス転移温度は低いものの、構造単位(I−A)の存在によりそのガラス転移温度は汎用のスチレン系重合体に比べて大幅に高く、やはり汎用のスチレン系重合体に比べて高温での使用に耐えることができる。
構造単位(I−A)において、R2がメチル基である場合は、R2が水素原子である場合に比べて、重合体のガラス転移温度が高くなり、より高温での使用に耐えることができるようになるため、重合体の使用可能温度域が広がる。
【0034】
本発明の重合体がスチレン系誘導体由来の構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体である場合は、他の重合性単量体の種類は特に制限されず、一般に重合可能な化合物であればよい。
本発明の重合体(共重合体)を形成するのに用い得る他の重合性単量体の具体例としては、エチレン、プロピレン、1−ブテン、1−オクテン、イソブチレンなどのオレフィン類;塩化ビニル、塩化ビニリデン、フッ化ビニル、フッ化ビニリデン、テトラフルオロエチレンなどの不飽和ハロゲン化合物;酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、ピバリン酸ビニルなどのビニルエステル化合物;アクリル酸、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、アクリル酸アダマンチル、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸2−エチルヘキシル、メタクリル酸2−ヒドロキシエチル、メタクリル酸アダマンチルなどの(メタ)アクリル酸エステル化合物;アクリロニトリル、メタクリロニトリルなどのニトリル化合物;アクリルアミド、N−メチルアクリルアミド、N,N−ジメチルアクリルアミド、N−イソプロピルアクリルアミド、N,N−ジイソプロピルアクリルアミド、メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−イソプロピルメタクリルアミド、N,N−ジイソプロピルメタクリルアミドなどの(メタ)アクリルアミド系化合物;無水マレイン酸、フマル酸ジエチルなどの二重結合含有ジカルボン酸誘導体;スチレン、α−メチルスチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、2,4−ジメチルスチレン、・−メチル−p−メチルスチレン、o−メトキシスチレン、m−メトキシスチレン、p−メトキシスチレン、1,1−ジフェニルエチレン、ビニルナフタレン、ビニルアントラセン、2−ビニルピリジン、4−ビニルピリジンなどの芳香族ビニル化合物;1,3−ブタジエン、イソプレン、2,3−ジメチル−1,3−ブタジエン、3,4−ジメチル−1,3−ペンタジエン、1,3−シクロヘキサジエン等の共役ジエン化合物;エチレンオキシド、プロピレンオキシド、テトラヒドロフランなどの環状エーテル化合物;・−カプロラクトンなどのラクトン化合物;ヘキサメチルシクロトリシロキサンなどの含ケイ素へテロ環状化合物などを挙げることができる。
本発明の重合体は、構造単位(I−A)と共に、前記した重合性単量体の1種または2種以上に由来する構造単位を有することができる。
【0035】
本発明の重合体が構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体である場合は、構造単位(I−A)の含有割合が重合体(共重合体)の質量に基づいて10質量%以上であることが好ましく、20質量%以上であることがより好ましい。構造単位(I−A)の含有割合が少ないと、構造単位(I−A)による耐熱性の向上効果が発揮されにくくなる。
【0036】
本発明の重合体が共重合体である場合は、構造単位(I−A)と共に存在する他の重合性単量体に由来する構造単位の結合様式は特に制限されず、ブロック状、テーパード状、ランダム状のいずれであってもよい。
【0037】
本発明の重合体がブロック共重合体である場合は、ブロック共重合体は、スチレン系誘導体由来の構造単位(I−A)を有する重合体ブロックAの少なくとも1個と、他の重合性単量体に由来する重合体ブロックBの少なくとも1個が結合したブロック共重合体であればいずれでもよい。なお、重合体ブロックBは、重合体ブロックAと明確に区別でき且つ重合体ブロックB本来の特性を損なわい限りは、構造単位(I−A)を微量であれば有していても差し支えない。
構造単位(I−A)を有する重合体ブロックAをAで表し、重合体ブロックBをBで表すと、本発明の重合体(ブロック共重合体)の具体例としては、A−B型のジブロック共重合体、A−B−A型またはB−A−B型のトリブロック共重合体、A−B−A−B型のテトラブロック共重合体、A−B−A−B−A型またはB−A−B−A−B型のペンタブロック共重合体などを挙げることができる。前記したブロック共重合体のうちでも、製造の容易性、力学的物性などの点から、A−B型のジブロック共重合体、A−B−A型のトリブロック共重合体、A−B−A−B型のテトラブロック共重合体またはA−B−A−B−A型のペンタブロック共重合体が好ましい。
【0038】
重合体ブロックAは、構造単位(I−A)のみから構成される単独重合体であってもよいし、または構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体であってもよい。また、重合体ブロックBは、スチレン系誘導体(I)以外の重合性単量体(他の重合性単量体)のうちの1種類のみから形成された単独重合体であってもよいし、または2種類以上の他の重合性単量体から形成された共重合体であってもよい。前記したように、重合体ブロックBは、スチレン系誘導体(I)以外の重合性単量体から実質的になっていて、重合体ブロックAと明確に区別でき且つ重合体ブロックB本来の特性に影響を及ぼさない限りは、構造単位(I−A)を微量[例えば重合体ブロックB中に構造単位(I−A)を1〜3ユニットというような微量]で有していても差し支えない。
【0039】
本発明の重合体がブロック共重合体であって、該ブロック共重合体が2個以上の重合体ブロックAを有する場合を有する場合は、2個以上の重合体ブロックAは、構造単位(I−A)を有する重合体ブロックである限りは、構造や分子量などが互いに同じであっても又は異なっていてもよい。また、該ブロック共重合体が2個以上の重合体ブロックBを有する場合は、2個以上の重合体ブロックBは、構造単位(I−A)以外の構造単位から実質的に構成される重合体ブロックである限りはその構造や分子量などが互いに同じであっても又は異なっていてもよい。
【0040】
重合体ブロックBを形成する他の重合性単量体、また重合体ブロックAが共重合体である場合に構造単位(I−A)と共に重合体ブロックAを形成する他の重合性単量体としては、本発明の重合体が共重合体である場合に該共重合体を形成するのに用い得る他の重合性単量体の具体例として前記で挙げた種々の重合性単量体、すなわち上記したオレフィン類、不飽和ハロゲン化合物、ビニルエステル化合物、(メタ)アクリル酸エステル化合物、不飽和ニトリル化合物、(メタ)アクリルアミド系化合物、二重結合含有ジカルボン酸誘導体、芳香族ビニル化合物、共役ジエン化合物、環状エーテル化合物、ラクトン化合物、含ケイ素へテロ環状化合物などの重合性単量体のうちの1種または2種以上を用いることができる。
重合体ブロックAがスチレン誘導体由来の構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体から形成されている場合は、重合体ブロックAを形成する他の重合性単量体が、上記したスチレン系誘導体(I)以外の芳香族ビニル化合物の1種または2種以上であることが、スチレン系誘導体(I)との共重合の容易性などの点から好ましい。特に、重合体ブロックAが共重合体である場合は、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび4−(1−イソプロペニルフェニル)アダマンタンのうちの1種または2種以上と、スチレンおよび/またはα−メチルスチレンとの共重合体であることがより好ましい。
【0041】
本発明の重合体がブロック共重合体である場合は、構造単位(I−A)を有する重合体ブロックAの割合が、ブロック共重合体の質量に基づいて、10質量%以上であることが好ましく、13〜70質量%であることがより好ましい。また、ブロック共重合体の全質量に対する構造単位(I−A)の割合は、5質量%以上であることが好ましく、10質量%以上であることがより好ましく、15〜70質量%であることが更に好ましい。構造単位(I−A)の割合が少ないと構造単位(I−A)による耐熱性の向上効果が発揮されにくくなる。
【0042】
本発明の重合体がブロック共重合体である場合に、重合体ブロックBを、構造単位(I−A)を有する重合体ブロックAよりもガラス転移温度の低い重合体から形成すると、熱可塑性で且つ弾性を有するブロック共重合体、すなわち熱可塑性エラストマーにすることができる。
重合体ブロックAよりもガラス転移温度の低い重合体ブロックBを形成するための重合性単量体の種類は特に制限されず、ガラス転移温度の低い重合体ブロックBを形成し得る重合性単量体であればいずれでもよく、そのうちでも共役ジエン、特に1,3−ブタジエン、イソプレンまたはこれらの混合物が、耐寒性および柔軟性に優れるブロック共重合体を形成し得ることから好ましく用いられる。
【0043】
前記したブロック共重合体を含めて、本発明の重合体(共重合体)が、共役ジエンに由来する構造単位を有し、それに伴って分子中に共役ジエンに由来する炭素−炭素不飽和二重結合が存在している場合には、耐候性、耐熱劣化性、耐オゾン性の向上などの観点から、該炭素−炭素不飽和二重結合の少なくとも一部が水素添加されていることが好ましく、その際の水素添加率は50%以上であることがより好ましく、90%以上であることが更に好ましい(以下、水素添加を「水添」ということがある)。
水素添加は一般的に利用される方法で行うことができ、例えば、アルキルアルミニウム化合物とコバルト、ニッケルなどからなるチーグラー触媒等の水添触媒の存在下に水素を供給する方法、p−トルエンスルホン酸ヒドラジドのようなジイミドを系内で発生する化合物を使用する方法などを採用することができる。
【0044】
本発明の重合体の分子量は特に制限されないが、重合体の強度および成形性の点から、単独重合体および共重合体のいずれであっても、重合体全体の数平均分子量が3000〜1000000であることが好ましく、5000〜500000であることがより好ましい。
また、本発明の重合体がブロック共重合体である場合は、構造単位(I−A)を有する重合体ブロックAの数平均分子量が3000〜100000、特に4500〜50000で、重合体ブロックBの数平均分子量が10000〜400000、特に30000〜350000であることが、力学物性、成形性の点から好ましい。
なお、本明細書における数平均分子量(Mn)は、ゲル浸透クロマトグラフィー(GPC)を用いて測定した標準ポリスチレン換算による数平均分子量をいう。
また、本明細書における分子量分布(Mw/Mn)は、前記と同様にして測定した重量平均分子量(Mw)と前記した方法で測定した数平均分子量(Mn)との比(Mw/Mn)をいう。
【0045】
本発明の重合体は、本発明の効果を損なわない限り、分子鎖中および/または分子末端に、場合によりカルボキシル基、水酸基、酸無水物基、アミノ基、エポキシ基の官能基を有していてもよい。
【0046】
本発明の重合体の製法は特に制限されず、スチレン系誘導体由来の構造単位(I−A)を分子中に有する重合体を製造し得る方法であれば、いずれの方法を採用してもよい。
そのうちでも、本発明の重合体は、
(i) 下記の一般式(I);
【0047】
【化7】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または該スチレン系誘導体(I)および他の重合性単量体を用いて重合を行う方法[以下これを「製造法(i)」ということがある];または、
(ii) 下記の一般式(II)
【0048】
【化8】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるかまたは該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のベンゼン環に基R1を導入する方法[以下これを「製造法(ii)」ということがある];
によって、円滑に製造することができ、本発明は前記した製造法(i)および製造法(ii)を包含する。
【0049】
[製造法(i)]
まず、製造法(i)により本発明の重合体を製造する場合について説明する。
製造法(i)では、スチレン系誘導体(I)として、例えば上記した化学式(Ia)〜(Ih2)に示したスチレン系誘導体や、それ以外のスチレン系誘導体(I)の1種または2種以上を用いて重合を行なう。
製造法(i)では、スチレン系誘導体(I)を単独で用いるか、またはスチレン系誘導体(I)と他の重合性単量体を用いて、アニオン重合、ラジカル重合、カチオン重合、配位重合などを行うことによって本発明の重合体を製造することができる。
また、本発明の重合体がブロック共重合体である場合は、例えば、スチレン系誘導体(I)と他の重合性単量体を用いて、リビングアニオン重合、リビングラジカル重合、リビングカチオン重合などの、いわゆるリビング重合を行ってブロック共重合体を製造する方法を採用することができる。
【0050】
そのうちでも、本発明の重合体はアニオン重合またはリビングアニオン重合により円滑に製造することができる。
本発明の重合体をアニオン重合によって製造するに当たっては、スチレン系誘導体(I)を単独で有機溶媒中に溶解するか、またはスチレン系誘導体(I)と他の重合性単量体を有機溶媒中に溶解し、そこにアニオン重合開始剤を添加し、窒素、アルゴンなどの不活性ガス雰囲気下、常圧ないし加圧条件下で、−100℃〜100℃、特には−78℃〜70℃の温度で重合を行う方法が好ましく採用される。
その際のアニオン重合開始剤としては、ブチルリチウム、エチルリチウム、メチルリチウムなどのアルキルリチウム等の従来から知られているアニオン重合開始剤を用いることができ、これらのアニオン重合開始剤は単独で使用しても又は2種以上を併用してもよい。そのうちでも、ブチルリチウム、特にsec−ブチルリチウムが、重合体の収率、重合開始速度などの点から好ましく用いられる。アニオン重合開始剤の使用量は、一般に、単量体の合計質量に基づいて、0.005〜3質量%、特に0.01〜1.5質量%であることが、得られる重合体の分子量などの点から好ましい。
また、アニオン重合時に用い得る有機溶媒としては、シクロヘキサン、トルエン、ベンゼンなどの炭化水素溶媒、テトラヒドロフランなどを挙げることができ、これらの有機溶媒は単独で使用しても、または2種以上を併用してもよい。そのうちでもシクロヘキサン、トルエンなどの炭化水素が、副反応が少ない点から好ましく用いられる。有機溶媒の使用量は、単量体1gに対して、0.1〜20ml、特に1〜5ml程度であることが、撹拌の容易性、製造コストなどの点から好ましい。
【0051】
また、リビングアニオン重合によってブロック共重合体を製造する場合は、構造単位(I−A)を有する重合体ブロックAを製造するためにスチレン系誘導体(I)を単独で有機溶媒中に溶解するかまたはスチレン系誘導体(I)と他の重合性単量体を有機溶媒中に溶解し、そこにリビングアニオン重合を開始させるアニオン重合開始剤を添加して重合を行って重合体ブロックAを構成する重合体(リビングポリマー)を製造した後、重合体ブロックBを製造するための他の重合性単量体を添加して重合を引き続いて行う方法などを採用することによって、重合体ブロックAと重合体ブロックBを有するブロック共重合体を製造することができる。その際に、重合体ブロックBを構成する重合体(リビングポリマー)を先に製造した後にそこに重合体ブロックAを形成するスチレン系誘導体(I)またはスチレン系誘導体(I)と他の単量体を添加して重合を行って重合体ブロックAと重合体ブロックBを有するブロック共重合体を製造してもよい。トリブロック以上のマルチブロック共重合体を製造する場合は、従来と同様にして、上記した重合操作を順次繰り返すことによって目的とするマルチブロック共重合体を製造することができる。
【0052】
ブロック共重合体の製造に当たっては、リビングアニオン重合開始剤として、ブチルリチウム、メチルリチウムなどのリビングアニオン重合開始剤が好ましく用いられ、特にブチルリチウムが好ましく用いられる。リビングアニオン重合開始剤の使用量は、ブロック共重合体の製造に用いる単量体の全質量に基づいて0.000001〜0.1質量%であることが好ましく、0.000001〜0.01質量%であることがより好ましい。
また、有機溶媒としては、上記で挙げた有機溶媒の1種または2種以上、特にシクロヘキサン、ベンゼンなどの炭化水素溶媒が好ましく用いられる。重合温度は一般に−100℃〜100℃、特に−78℃〜70℃が好ましく採用され、重合圧力は一般に0.1〜3MPa、特に0.1〜1MPaが好ましい。
【0053】
単独重合体、共重合体のいずれの場合も、重合が終了した時点で、重合混合物(反応液)中にメタノール、エタノールなどの重合停止剤を添加するか、または重合混合物を前記したメタノールなどの重合停止剤中に投入することによって、重合を停止することができる。
【0054】
製造法(i)に用いるスチレン系誘導体(I)は、公知の方法、またはそれに準じた方法で製造することができる。
上記の化学式(Ia)で表される1−(4−ビニルフェニル)アダマンタンは、例えば非特許文献1に記載されている方法により製造することができる。
具体的には、下記の反応式(III)に示すように、1−ブロモアダマンタンとベンゼンを炭酸カリウムの存在下にパラジウム/炭素触媒(Pd/C)を用いてカップリング反応させて1−フェニルアダマンタンを生成させ、その1−フェニルアダマンタンにジクロロメタン中でTiCl4の存在下で1,1−ジクロロメチルエーテルを反応させてホルミル化を行い、次いで前記で生成したホルミル化物にテトラヒドロフラン(THF)中でトリフェニルホスフィンメチレン(別名:メチレントリフェニルホスホラン)(Ph3P=CH2)を反応させること(Wittigオレフィン反応)により製造することができる。
【0055】
【化9】
【0056】
また、上記とは別に、下記の反応式(IV)に示すように、1−ブロモアダマンタンとフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて1−フェニルアダマンタンを生成させ、生成した1−フェニルアダマンタンに四塩化炭素中で臭素を反応させて1−(4−ブロモフェニル)アダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−アダマンチル−フェニルマグネシウムブロミド(中間体)を生成させ、そこに窒素雰囲気下でジメチルホルムアミド(DMF)を加えて反応させて、1−(4−ホルミルフェニル)アダマンタン[別称:4−(1−アダマンチル)ベンズアルデヒド]を生成させ、生成した1−(4−ホルミルフェニル)アダマンタンにメチルトリフェニルホスホニウムブロミドとカリウムtert−ブトキシドを加えて脱水THF中で反応させて1−(4−ビニルフェニル)アダマンタン[別称:4−(1−アダマンチル)スチレン]を製造する方法を挙げることができる。
【0057】
【化10】
【0058】
また、上記の化学式(Ib)で表される3−(4−ビニルフェニル)−1,1’−ビアダマンタンは、例えば、下記の反応式(V)に示すように、1−ブロモアダマンタンをn−オクタン中でナトリウムを用いて反応させてビアダマンタンを生成させ、それに四塩化炭素中で臭素を反応させて3−ブロモ−1,1’−ビアダマンタンを生成させ、それにフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて3−フェニル1,1’−ビアダマンタンを生成させ、それに四塩化炭素中で臭素を反応させて3−(4−ブロモフェニル)−1,1’−ビアダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−ビアダマンチル−フェニルマグネシウムブロミド(中間生成物)を生成させ、そこに窒素雰囲気下でジメチルホルムアミド(DMF)を加えて反応させて、3−(4−ホルミルフェニル)−1,1’−ビアダマンタンを生成させ、それにメチルトリフェニルホスホニウムブロミドとカリウムtert−ブトキシドを加えて脱水THF中で反応させて、3−(4−ビニルフェニル)−1,1’−ビアダマンタンを生成させる方法により製造することができる。
【0059】
【化11】
【0060】
また、上記の化学式(Ic)で表される1−(4−イソプロペニルフェニル)アダマンタンは、例えば、下記の反応式(VI)に示すように、1−ブロモアダマンタンとフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて1−フェニルアダマンタンを生成させ、生成した1−フェニルアダマンタンに四塩化炭素中で臭素を反応させて1−(4−ブロモフェニル)アダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−アダマンチル−フェニルマグネシウムブロマイド(中間体)を生成させ、そこに窒素雰囲気下で脱水アセトンを加えて反応させて4−(1−アダマンチル)クミルアルコールを生成させ、生成した4−(1−アダマンチル)クミルアルコールをトルエンスルホン酸の存在下にトルエン中で脱水反応させて、4−(1−イソプロペニルフェニル)アダマンタンを生成させる方法により製造することができる。
【0061】
【化12】
【0062】
1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタン以外の、上記した一般式(Id)〜(Ih2)などに示したスチレン系誘導体(I)およびそれら以外のスチレン系誘導体(I)も、前記した反応式(III)〜(VI)に準じた方法で製造することができる。
【0063】
[製造法(ii)]
次に、製造法(ii)により本発明の重合体を製造する場合について説明する。
上記の一般式(II)で表されるスチレン系化合物(II)を単独で用いるかまたは該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体としては、従来既知のスチレン系重合体を用いることができる。限定されるものではないが、製造法(ii)で用いることのできる、スチレン系化合物(II)の重合体の代表例としては、ポリスチレン、スチレンと他の重合性単量体との共重合体、α−メチルスチレンの単独重合体、スチレンとα−メチルスチレンの共重合体、スチレンとα−メチルスチレンと他の重合性単量体との共重合体などを挙げることができる。
スチレン系重合体が共重合体である場合は、ブロック共重合体、テーパード共重合体、ランダム共重合体のいずれであってもよい。また、その際の他の重合性単量体としては、本発明の重合体がスチレン誘導体由来の構造単位(I−A)と他の重合性単量体由来の構造単位を有する共重合体である場合について上記で例示した種々の単量体を用いることができる。
【0064】
また、スチレン系化合物(II)に芳香族求電子置換反応させるのに用いる上記の一般式(III)で表される化合物(III)(R1−X)における多脂環式炭化水素基R1の具体例としては、アダマンチル基(トリシクロ〔3.3.1.13,7〕デシル基)、ビアダマンチル基、ノルボルニル基、イソボルニル基、ビシクロノニル基、ビシクロ〔2.1.0〕ペンチル基、ビシクロ〔3.2.1〕オクチル基、トリクロ〔2.2.1.02,6〕ペンチル基などを挙げることができる。これらの多脂環式炭化水素基は、場合によりアルキル基、ハロゲン、アルコキシル基などにより置換されていてもよい。
【0065】
化合物(III)における基Xとしては、化合物(III)をスチレン系化合物(II)に対して芳香族求電子置換反応させたときに、スチレン系重合体中のスチレン由来のベンゼン環に多脂環式炭化水素基R1を結合させる作用を有する基であればいずれでもよい。基Xの具体例としては、ハロゲン原子、アルコール性水酸基、エステル基、トリフレート基、トシル基、オレフィン置換基などを挙げることができる。これらの中でも、化合物(III)の入手容易性、反応性などの観点から、化合物(III)としては、基Xがハロゲン原子またはトリフレート基である化合物(多脂環式炭化水素化合物)を用いることが好ましい。
本発明の重合体の製造に当たっては、1種類の化合物(III)のみを用いても、または2種類以上の化合物(III)を用いてもよい。
スチレン系重合体に対する化合物(III)(R1−X)の使用割合は、スチレン系重合体中に存在するスチレン系化合物(II)に由来するベンゼン環の量、スチレン系重合体中に導入すべき多脂環式炭化水素基R1の割合、芳香族求電子置換反応時の反応条件などに応じて異なり得るが、一般的には、スチレン系重合体中に存在するスチレン系化合物(II)に由来するベンゼン環1個に対して、化合物(III)(R1−X)を0.001〜10モル、特に0.1〜3モルの割合で用いることが、分子中に構造単位(I−A)を有する耐熱性に優れる本発明の重合体が円滑に得られる点から好ましい。その際に、化合物(III)の使用割合を、芳香族求電子置換反応によって得られる重合体における構造単位(I−A)の割合が10質量%以上になるようにして前記範囲から選択することが好ましい。
【0066】
スチレン系重合体に化合物(III)(R1−X)を芳香族求電子置換反応させる際の芳香族求電子置換反応の種類は特に限定されず、スチレン系重合体中のベンゼン環に多脂環式炭化水素基R1を円滑に結合させ得る芳香族求電子置換反応であればいずれでもよく、例えばフリーデル・クラフツ反応などを挙げることができる。
芳香族求電子置換反応は、触媒や溶媒などを用いずに、スチレン系重合体に化合物(III)をそのまま直接反応させて行なってもよいが、スチレン系重合体および化合物(III)を有機溶媒に溶解させて触媒の存在下に行なうことが、芳香族求電子置換反応を速やかに且つ円滑に行わせる点から好ましい。
【0067】
芳香族求電子置換反応に用いる有機溶媒の種類は特に制限されず、例えば、ヘキサン、シクロヘキサン、ヘプタン、オクタンなどの脂肪族炭化水素、ベンゼン、トルエン、キシレンなどの芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、ジオキサンなどのエーテル化合物、クロロホルム、ジクロロメタン、ジクロロエタンなどのハロゲン化炭化水素などを挙げることができ、これら1種または2種以上を用いることができる。そのうちでも、反応制御の容易性の点から、脂肪族炭化水素および/またはハロゲン化炭化水素が好ましく用いられる。
有機溶媒の使用量は、有機溶媒の種類などに応じて調節できるが、芳香族求電子置換反応の円滑な進行、温度管理の容易性などの点から、一般にスチレン系重合体100質量部に対して、0.1〜10000質量部、特に1〜500質量部であることが好ましい。
【0068】
芳香族求電子置換反応に用い得る触媒の例としては、ルイス酸、ブレンステッド酸などが挙げられる。
ルイス酸としては、特に一般に知られるものであれば特に制限はなく、例えば塩化アルミニウム、臭化アルミニウム、アルキルアルミニウムジクロライド、アルキルアルミニウムクロライド、塩化鉄(III)、三フッ化ホウ素、三塩化ホウ素、三臭化ホウ素、塩化チタンなどを挙げることができ、これらは単独で用いてもまたは2種類以上を組み合わせて用いてもよい。そのうちでも、入手容易性などの点から、塩化アルミニウム、塩化鉄(III)、三フッ化ホウ素が好ましく用いられる。
ブレンステッド酸としては、リン酸、硫酸、フッ化水素などの強酸を挙げることができ、これらは単独で用いてもまたは2種類以上を組み合わせて用いてもよい。
また、上記したルイス酸の1種または2種以上と、ブレンステッド酸の1種または2種以上を組み合わせて使用してもよい。
触媒の使用量は、触媒の種類、化合物(III)(R1−X)の種類などに応じて異なり得るが、一般的には、化合物(III)1モルに対して、0.0001〜100モル、特に0.001〜10モルであることが、芳香族求電子置換反応の円滑な進行などの点から好ましい。
【0069】
芳香族求電子置換反応を実施する際の温度には特に制限はないが、熱効率、反応の円滑な進行などの点から、0〜100℃、特に15〜70℃の範囲内で行うことが好ましい。
【0070】
本発明の重合体は単独で使用してもよいし、または必要に応じて他の熱可塑性樹脂、熱硬化性樹脂、ゴム、熱可塑性エラストマーなどとブレンドして使用してもよい。その際に、本発明の趣旨を損なわない範囲で、さらに酸化防止剤、軟化剤、可塑剤、帯電防止剤、滑剤、紫外線吸収剤、難燃剤、顔料、無機充填剤などを配合してもよい。
【実施例】
【0071】
以下に、実施例、比較例および参考例により本発明をさらに具体的に説明するが、本発明は以下の例により何ら限定されるものではない。なお、以下の例で用いた薬品(化合物など)は入手しうる限りの最高の純度のものを用いた。また、溶媒は十分に脱気したものを使用した。
以下の例において、参考例1または参考例2で得られた生成物(中間化合物、最終化合物)の構造の確認、重合の進行度、各実施例で得られた重合体の数平均分子量および分子量分布の測定、重合体のガラス転移温度の測定、熱質量分析、引張試験、動的粘弾性試験および応力緩和特性の測定は次のようにして行った。
【0072】
(1)生成物の構造の確認:
以下の参考例1または参考例2、参考例3で生成した生成物(中間化合物、最終化合物)を重クロロホルムに溶解し、核磁気共鳴装置(ブルカー社製「BRUCKER DPX300」)を使用して、プロトン核(1H)、炭素核(13C)を27℃で測定して、その構造の確認を行った。また、参考例2で生成した一部の生成物については、構造の確認に当たって、日本分光社製の赤外線分析装置「FT/IR−460」を使用してKBr法により測定した。
【0073】
(2)重合の進行度の測定:
以下の実施例で生成した重合反応液または重合体を重クロロホルムに溶解させ、核磁気共鳴分光装置(日本電子データム社製「JNMLA400」)を使用して、プロトン核(1H)を50℃で測定して重合の進行度を測定した。
【0074】
(3)数平均分子量(Mn)および分子量分布(Mw/Mn)の測定:
ピーク分子量が既知の標準ポリスチレンを用い、該標準で校正したゲル浸透クロマトグラフィー(GPC)(東ソー社製「HLC−8020」)を使用して、重合体の数平均分子量(Mn)および重量平均分子量(Mw)を測定した(溶媒:テトラヒドロフラン、温度:40℃)。分子量分布は重量平均分子量(Mw)と数平均分子量(Mn)の比(Mw/Mn)として求めた。
【0075】
(4)重合体のガラス転移温度(Tg):
DSC装置(メトラー社製「DSC822e」)を用いて測定した。測定は、一旦280℃まで試料を加熱し、同温度で5分間アニールを施した後、降温速度を毎分10℃として0℃まで冷却した。この後再び昇温速度を毎分10℃として昇温して、ガラス転移温度(Tg)を測定した。
【0076】
(5)重合体の熱重量分析(10%質量減少温度の測定):
熱重量(TGA)装置(メトラー社製「TG50」)を使用して、重合体試料を窒素雰囲気下に置き、雰囲気温度を50℃から500℃まで毎分10℃の速度で昇温しながら、重合体の質量が当初よりも10%減少した時点の温度を測定した。
【0077】
(6)引張試験:
以下の実施例11、実施例12または比較例2で得られた重合体を用いて、図1に示す形状および寸法の試験片(厚さ1mm)を作製し、該試験片を使用して、万能材料試験機(インストロンジャパン社製「TM−MS−134」)を用いて、JIS K6251に準拠して、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)、破断伸度(EB)を測定した。なお、図1における各部の寸法の単位はmmである。
【0078】
(7)動的粘弾性試験:
以下の実施例11、実施例12または比較例2で得られた重合体から形成したシート(厚さ1mm)から、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用い、広域動的粘弾性測定装置(レオロジ社製「DVE−V4FTレオスペクトラー」)を使用して、引張りモード(周波数 11Hz)で、昇温速度を毎分3℃として、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。
【0079】
(8)応力緩和特性:
以下の実施例11、実施例12または比較例2で得られた重合体から形成したシート(厚さ1mm)から、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用い、広域動的粘弾性測定装置(レオロジ社製「DVE−V4FTレオスペクトラー」)を使用して、40℃の温度下で引張りひずみを100%与え、経過時間に対して応力の保持率を測定した。保持率が1(100)に近いほど応力緩和特性は良好であることを表す。
【0080】
《参考例1》[1−(4−ビニルフェニル)アダマンタンの製造]
上記した反応式(III)にしたがって1−(4−ビニルフェニル)アダマンタンを以下のようにして製造した。
(1)1−フェニルアダマンタンの製造:
窒素雰囲気下で、マグネシウム3.36g(138.0ミリモル)に脱水エーテル20mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、ブロモベンゼン10ml(84ミリモル)の脱水エーテル溶液100mlを滴下して室温で3時間撹拌した後、エーテルを減圧留去することにより、フェニルマグネシウムブロミドの灰色固体を得た。この灰色固体に、1−ブロモアダマンタン5.00g(23.2ミリモル)と脱水塩化メチレン70mlを加えて、GCとNMRで反応を追跡しながら1−ブロモアダマンタンの消費が完全に確認されるまで還流した(約24時間後)ところで、系を0℃に冷却し、水を加えて反応を停止させ、更に2N塩酸を加えた。塩化メチレンで抽出を3回、有機層を水で3回洗浄した。有機層を無水硫酸マグネシウムを加えて乾燥した後、硫酸マグネシウムを濾別し、有機溶媒を減圧留去して、4.52gの淡黄色固体を得た。この淡黄色固体を、ヘキサンを展開溶媒に用いたシリカゲルカラムクロマトグラフィーにて精製して8.45gの白色固体を得た後、該白色固体をメタノールから再結晶して、1−フェニルアダマンタン6.30g(収率64%)の白色固体(mp84−85℃)を得た。
これにより得られた1−フェニルアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.73−1.82(m,6H,C(2)H2),1.92(s,6H,C(4)H2),2.10(s,3H,C(3)H),7.15−7.20(m,1H,C(d)H),7.29−7.39(m,4H,C(b)H,C(c)H)
・13C−NMR(CDCl3):δ 29.0(C−3),36.2(C−1),36.9(C−2),43.2(C−4),124.9(C−c),125.6(C−d),128.2(C−b),151.4(C−a)
【0081】
(2)1−(4−ブロモフェニル)アダマンタンの製造:
上記(1)で得られた1−フェニルアダマンタン3.60g(17.0ミリモル)に四塩化炭素34mlを加えた後、臭素17ml(330ミリモル)を加えて室温で4時間撹拌した。反応溶液を氷水中に注ぎ込み、過剰の臭素を亜硫酸水素ナトリウムで処理した後、塩化メチレンで3回抽出し、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して、1−(4−ブロモフェニル)アダマンタン4.95g(収率100%、白色固体、mp101−102℃)を得た。
これにより得られた1−(4−ブロモフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.71−1.81(m,6H,C(2)H2),1.87(s,6H,C(4)H2),2.09(s,3H,C(3)H),7.21−7.24(d,2H,C(c)H,J=8.6Hz),7.40−7.43(d,2H,C(b)H,J=8.6Hz)
・13C−NMR(CDCl3):δ 28.9(C−3),36.1(C−1),36.7(C−2),43.1(C−4),119.3(C−d),126.9(C−c),131.1(C−b),150.4(C−a)
【0082】
(3)1−(4−ホルミルフェニル)アダマンタンの製造:
窒素雰囲気下に、マグネシウム0.84g(35ミリモル)に脱水THF20mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。これに、上記(2)で得られた1−(4−ブロモフェニル)アダマンタン4.95g(17.0ミリモル)の脱水THF溶液40mlを室温で滴下した後、還流した。還流中に、ガスクロマトグラフィーで反応を追跡し、1−(4−ブロモフェニル)アダマンタンが完全に消費されて4−アダマンチル−フェニルマグネシウムブロミドに由来する1−フェニルアダマンタンの生成が確認された時点(約2時間後)で、系を0℃に冷却し、脱水DMF25ml(323ミリモル)を滴下したところ、滴下と同時に白濁した。その後、室温で一晩撹拌した後、系に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体4.87gを得た。これを、ヘキサンを展開溶媒に用いてクロマトグラフィーにて処理して、副生した1−フェニルアダマンタン1.01g(4.76ミリモル、回収率28%)を除去した後、ヘキサン/酢酸エチル=20/1,10/1(v/v)を展開溶媒に用いて3.25gの白色固体を得た。これを昇華精製(120〜130℃/0.2mmHg)して、1−(4−ホルミルフェニル)アダマンタン[別称:4−(1−アダマンチル)ベンズアルデヒド]2.88g(収率70%、白色粉末、mp253−255℃)を得た。
これにより得られた1−(4−ホルミルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.74−1.84(m,6H,C(2)H2),1.94(s,6H,C(4)H2),2.13(s,3H,C(3)H),7.51−7.54(d,2H,C(c)H,J=8.3Hz),9.98(s,1H,CHO)
・13C−NMR(CDCl3):δ 28.8(C−3),36.7(C−2),37.0(C−1),42.9(C−4),125.7(C−c),129.8(C−b),134.2(C−d),158.6(C−a),192.2(CHO)
・IR(KBr):2903 and 2848cm-1(C−H伸縮振動),1698,1686 and 1654cm-1(C=O伸縮振動),1604 cm-1(芳香環C−C振動),1448,1167,802cm-1(C−H変角振動)
【0083】
(4)1−(4−ビニルフェニル)アダマンタンの製造:
窒素雰囲気下に、メチルトリフェニルホスホニウムブロミド4.59g(12.5ミリモル)にカリウムtert−ブトキシド1.97g(17.6ミリモル)を加えた後、脱水THF95mlを加えて室温で約20分間撹拌した。系を0℃に冷却した後、上記(3)で得られた1−(4−ホルミルフェニル)アダマンタン2.61g(10.9ミリモル)の脱水THF溶液45mlを滴下し、室温で一晩撹拌した。ガスクロマトグラフィーにて1−(4−ホルミルフェニル)アダマンタンの消費が完全に確認された後、系に水を加えて反応を停止させた。エバポレートによりTHFを除去した後、エーテルで抽出を3回、有機層を水で2回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して白色固体を得た。これを少量のTHFに溶解し、大量のヘキサン中に注ぎ込むことにより、反応により生成したトリフェニルホスフィンオキシドを沈殿させ、濾別によりトリフェニルホスフィンオキシドを除去後、有機溶媒を減圧留去して3.94gの白色固体を得た。この白色固体をヘキサンを展開溶媒
に用いたシリカゲルグロマトグラフィーにより精製して3.45gの白色粉末を得た。この白色粉末をメタノールから再結晶して、1−(4−ビニルフェニル)アダマンタン2.08g(8.72ミリモル、収率80%、白色針状結晶、mp98−99℃)を得た。
これにより得られた1−(4−ビニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図2に示すとおりであり、NMRスペクトルおよびIR分析のピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.72−1.82(m,6H,C(2)H2),1.91(s,6H,C(4)H2),2.10(s,3H,C(3)H),5.17−5.21(d,1H,CH2=trans,J=11.1Hz),5.68−5.74(d,1H,CH2= cis,J=17.7Hz),6.65−6.75(d,1H,=CH,J=11.1Hz and 17.7Hz),7.31−7.39(m,4H,C(b)H and C(c)H).
・13C−NMR(CDCl3):δ 29.0(C−3),36.2(C−1),36.9(C−2),43.2(C−4),113.1(CH2=),125.1(C−c),126.1(C−b),135.0(C−d),136.7(=CH),151.2(C−a)
・IR(KBr):2917,2903 and 2847cm-1(C−H伸縮振動),1629 cm-1(C=C伸縮振動),1510,1446,1345,988,897,838 and 808cm-1(C−H変角振動)
(5) 上記の結果から、この参考例1で最終的に得られた生成物は、1−(4−ビニルフェニル)アダマンタンであることが確認された。
【0084】
《参考例2》[3−(4−ビニルフェニル)−1,1’−ビアダマンタンの製造]
上記した反応式(IV)に示す方法により、3−(4−ビニルフェニル)−1,1’−ビアダマンタンを次のようにして製造した。
(1)1,1’−ビアダマンタンの製造:
窒素雰囲気下、n−オクタン50mlに1−ブロモアダマンタン25.12g(118.3ミリモル)を溶かし、ナトリウム2.73gを加えた後、140℃に加熱して還流した。ナトリウムが融解すると系は青色を呈した。ガスクロマトグラフィー(GC)で反応を追跡し、1−ブロモアダマンタンの消費が完全に確認された時点(8時間半)で放冷し、ナトリウムをメタノールと水で処理した後、塩酸を加えた。クロロホルムで3回抽出し、水で2回洗浄した後、有機層の溶媒を減圧留去し、それにより得られた白色固体14.1gを180℃、26.7Pa(0.2mmHg)で昇華精製して、1,1’−ビアダマンタン6.96g(収率44%)を得た。
これにより得られた1,1’−ビアダマンタンの1H−NMRスペクトルおよび13CNMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.54−1.69(s,24H,CH2),1.95(s,6H,CH)
・13C−NMR(CDCl3):δ 29.1(C−3),35.3(C−2),36.4(C−1),37.7(C−4)
【0085】
(2)3−ブロモ−1,1’−ビアダマンタンの製造:
上記(1)で得られた1,1’−ビアダマンタン6.23g(23.1ミリモル)に四塩化炭素160mlと臭素80ml(1.56mol)を加え、室温で72時間撹拌した。反応溶液を氷浴中に注ぎ込み、過剰に用いた臭素を亜硫酸水素ナトリウムで処理した後、クロロホルムで3回抽出し、有機層を水で2回洗浄した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムを濾別し、有機溶媒を減圧留去して淡黄色粉末7.96gを得た。これを最初はヘキサンのみで、次にヘキサン/酢酸=20/1、10/1(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて精製して、3−ブロモ−1,1’−ビアダマンタン4.42g(収率56%、白色粉末)を得た。
これにより得られた3−ブロモ−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.53−1.70(m,18H,C(4)H2,C(6)H2,C(2')H2,C(4')H2),1.98(s,3H,C(3')H),2.16−2.33(m,8H,C(2)H2,C(5)H,C(7)H2)
・13C−NMR(CDCl3):δ 29.0(C−3'),32.9(C−5),33.5(C−4),35.5(C−6,C−2'),36.7(C−1'),37.5(C−4'),42.8(C−1'),47.7(C−2),49.3(C−7),69.6(C−3)
【0086】
(3)3−フェニル−1,1’−ビアダマンタンの製造:
窒素雰囲気下で、マグネシウム1.65g(67.8ミリモル)に脱水エーテル15mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、ブロモベンゼン4.9ml(41ミリモル)の脱水エーテル溶液45mlを滴下して室温で3時間撹拌した後、エーテルを減圧留去することにより、フェニルマグネシウムブロミドの灰色固体を得た。この灰色固体に、上記(2)で得られた3−ブロモ−1,1’−ビアダマンタン3.96g(11.3ミリモル)と脱水塩化メチレン35mlを加えて、GCとNMRで反応を追跡しながら3−ブロモ−1,1’−ビアダマンタンの消費が完全に確認されるまで還流(約40時間後)したところで、系を0℃に冷却し、水を加えて反応を停止させ、更に2N塩酸を加えた。塩化メチレンで抽出を3回、有機層を水で3回洗浄し、更に塩化メチレンで2回抽出した。有機溶媒を減圧留去し、4.53gの淡黄色固体を得た。これを40〜60℃、26.7Pa(0.2mmHg)で昇華精製して反応により副生したビフェニルを除去した後、ヘキサンから再結晶して、3−フェニル−1,1’−ビアダマンタン3.43g(収率67%)の白色針状結晶(mp204−20
6℃)を得た。
これにより得られた3−フェニル−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.64−1.70(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.85(s,4H,C(4)H2),1.98(s,3H,C(3')H),2.20(s,2H,C(5)H),7.16−7.21(t,1H,C(d)H),7.30−7.40(m,4H,C(b)H,C(c)H)
・13C−NMR(CDCl3):δ 29.0(C−3'),29.5(C−5),34.5(C−7),35.4(C−2'),36.5(C−1'),36.7(C−2),37.0(C−3),37.6(C−4'),37.6(C−1),41.7(C−6),42.7(C−4)
【0087】
(4)3−(4−ブロモフェニル)−1,1’−ビアダマンタンの製造:
上記(3)で得られた3−フェニル−1,1’−ビアダマンタン2.17g(6.26ミリモル)に四塩化炭素70mlを加えた後、臭素20ml(390ミリモル)を加えて室温で2.5時間撹拌した。反応溶液を氷水中に注ぎ込み、過剰の臭素を亜硫酸水素ナトリウムで処理した後、塩化メチレンで3回抽出し、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体2.69gを得た。この一連の操作を再度繰り返して、合計で4.48gの淡黄色固体(収率100%)を得た。これをヘキサンから再結晶して、3−(4−ブロモフェニル)−1,1’−ビアダマンタン3.07g(白色針状結晶、mp184−186℃)を得た。
これにより得られた3−(4−ブロモフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.63−1.72(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.81(s,4H,C(4)H2),1.99(s,3H,C(3')H),2.21(s,2H,C(5)H),7.26−7.29(d,2H,C(c)H),7.44−7.47(d,2H,C(b)H)
・13C−NMR(CDCl3):δ 29.0(C−3'),29.4(C−5),34.4(C−7),35.4(C−2'),36.5(C−1'),36.5(C−2),36.9(C−3),37.6(C−4'),37.6(C−1),41.6(C−6),42.6(C−4)
・IR(KBr):2904,2847cm-1(C−H伸縮振動),1490cm-1(C−Br伸縮振動),1449,1396,1343,803,730cm-1(C−H変角振動)
【0088】
(5)3−(4−ホルミルフェニル)−1,1’−ビアダマンタンの製造:
窒素雰囲気下に、マグネシウム0.34g(14.0ミリモル)に脱水THF10mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。これに、上記(4)で得られた3−(4−ブロモフェニル)−1,1’−ビアダマンタン2.86g(6.73ミリモル)の脱水THF溶液70mlを室温で滴下した後、還流した。還流中に、一部をサンプリングしDMFを加えることでホルミル体の生成と3−(4−ブロモフェニル)−1,1’−ビアダマンタンの消費をNMRで確認することで反応を追跡しながら、3−(4−ブロモフェニル)−1,1’−ビアダマンタンが完全に消費されるまで還流を続けた(約6時間)。次いで、系を0℃に冷却し、脱水DMF3ml(38.8ミリモル)を滴下したところ、滴下と同時に白濁して系は灰色になった。その後、室温で一晩撹拌した後、系に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させ
た。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体2.29gを得た。これを、初めはヘキサンのみで、その後ヘキサン/酢酸エチル=10/1,10/3,1/1(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて副生した3−フェニル−1,1’−ビアダマンタンを除去し、3−(4−ホルミルフェニル)−1,1’−ビアダマンタン1.49g(収率59%、白色粉末、mp205−207℃)を得た。
これにより得られた3−(4−ホルミルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.54−1.69(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.85(s,4H,C(4)H2),1.97(s,3H,C(3')H),2.21(s,2H,C(5)H),7.52−7.55(d,2H,C(c)H),7.82−7.85(d,2H,C(b)H),9.98(s,1H,CHO)
・13C−NMR(CDCl3):δ 29.3(C−3'),29.7(C−5),34.7(C−7),35.7(C−2'),36.8(C−1'),36.8(C−2),37.9(C−4'),37.9(C−1),38.2(C−3),41.7(C−6),42.8(C−4)
・IR(KBr):2927,2902,2849cm-1(C−H伸縮振動),1707cm-1(C=O伸縮振動),1447,1346,827,730cm-1(C−H変角振動)
【0089】
(6)3−(4−ビニルフェニル)−1,1’−ビアダマンタンの製造:
窒素雰囲気下に、メチルトリフェニルホスホニウムブロミド1.00g(2.80ミリモル)にカリウムtert−ブトキシド0.43g(3.88ミリモル)を加えた後、脱水THF20mlを加えて室温で約20分間撹拌した。系を0℃に冷却した後、上記(5)で得られた3−(4−ホルミルフェニル)−1,1’−ビアダマンタン0.88g(2.34ミリモル)の脱水THF溶液を60ml滴下し、室温で一晩撹拌した。TLC板(メルク社製「TLCアルミニウムシート シリカゲル60F254」)およびNMRで3−(4−ホルミルフェニル)−1,1’−ビアダマンタンが完全に消費されたのを確認した後、系に水を加えて反応を停止させた。エバポレートによりTHFを除去した後、エーテルで抽出を3回、有機層を水で2回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して白色固体を得た。これを、初めはヘキサンのみで、その後ヘキサン/酢酸エチル=10/1,10/3(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて、トリフェニルホスフィンオキシドを除去した。続いてエタノール/ヘキサン混合溶媒から再結晶し、3−(4−ビニルフェニル)−1,1’−ビアダマンタン0.61g(1.64ミリモル、収率68%、白色針状結晶、mp170−172℃)を得た。
これにより得られた3−(4−ビニルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図3に示すとおりであり、NMRスペクトルおよびIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.63−1.71(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.84(s,4H,C(4)H2),1.98(s,3H,C(3')H),2.20(s,2H,C(5)H),5.19−5.23(d,1H,CH2=trans),5.70−5.76(d,1H,CH2= cis),6.68−6.77(d,1H,=CH),7.34−7.41(m,4H,C(b)H,C(c)H).
・13C−NMR(CDCl3):δ 29.0(C−3'),29.5(C−5),34.5(C−7),35.4(C−2'),36.5(C−1'),36.7(C−2),37.0(C−3),37.6(C−4'),37.6(C−1),41.7(C−6),42.7(C−4)
・IR(KBr):2902,2848cm-1(C−H伸縮振動),1629,1510 cm-1(C=C伸縮振動),1446,1343,830cm-1(C−H変角振動)
【0090】
《参考例3》[1−(4−イソプロペニルフェニル)アダマンタンの製造]
(1)4−(1−アダマンチル)クミルアルコールの製造;
窒素雰囲気下、マグネシウム3.32g(137ミリモル)に脱水THF15mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、参考例1の中間体として得られた1−(4−ブロモフェニルアダマンタン)21.8g(75ミリモル)の脱水THF溶液130mlを室温で滴下した後、還流した。還流中にガスクロマトグラフィーで反応を追跡し、1−(4−ブロモフェニル)アダマンタンが完全に消費されて4−アダマンチル−フェニルマグネシウムブロミドに由来する1−フェニルアダマンタンの生成が確認された時点(約2時間後)で、系を0℃に冷却し、脱水アセトン100ml(1.36モル)を滴下したところ、滴下と同時に白濁した。その後、室温で1時間攪拌した後、系内に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFおよび過剰なアセトンを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して23.7gの淡黄色固体を得た。
(2)1−(4−イソプロペニルフェニル)アダマンタンの製造;
上記(1)で得られた4−(1−アダマンチル)クミルアルコール23.7gと触媒量のトルエンスルホン酸(1g)にトルエン200mlを加えて溶解させた。溶解させた後に、系内に発生する水を除去しながら還流し、ガスクロマトグラフィーにより4−(1−アダマンチル)クミルアルコールが完全に消費されたことを確認し、系を室温まで冷却した。その後、系に水および炭酸水素ナトリウム水溶液を順に加えて反応を停止させ、有機層を水で3回洗浄した。有機層に無水硫酸マグネシウムを加えて乾燥し、硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して18.3gの白色固体を得た。該白色固体を、ヘキサンを展開溶媒に用いたシリカゲルクロマトグラフィーにて精製したのち、メタノール/エーテル混合溶媒から再結晶し、1−(4−イソプロペニルフェニル)アダマンタン9.81g(白色固体)を得た。
これにより得られた1−(4−イソプロペニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図4に示すとおりであり、それらのピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.72−1.82(m,6H,C(2)H2),1.91(s,6H,C(4)H2), 2.10(s,3H,C(3)H),2.15(s,3H,α−CH3),5.04(s,1H,CH2=trans),5.35(s,1H,CH2=cis),7.25−7.45(m,4H,C(b)H and C(c)H)
・13C−NMR(CDCl3):δ 21.9(α−CH3),29.0(C−3),36.1(C−1),36.9 (C−2),43.2(C−4),111.7(CH2=),124.8(C−b),125.3(C−c),138.4(C−a), 143.1(=C(Me)(Ph)),150.8(C−d)
【0091】
《実施例1》[1−(4−ビニルフェニル)アダマンタン単独重合体の製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に参考例1で製造した1−(4−ビニルフェニル)アダマンタン0.3g(1.26ミリモル)を入れ、次に溶媒としてシクロヘキサン20mlを加えて1−(4−ビニルフェニル)アダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.02ml(sec−ブチルリチウムとして0.026ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、1時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)8500、分子量分布(Mw/Mn)1.29であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークは観測されず、1−(4−ビニルフェニル)アダマンタンの重合反応率は99.9%以上であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は237℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は408℃であった。この結果を下記の表2に示す。
【0092】
《実施例2》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(1)の製造]
(1) 3本の乾燥したガラス製反応容器を用意し、各反応容器内の気体を窒素ガスで置換した後、それぞれの反応容器に、参考例1で製造した1−(4−ビニルフェニル)アダマンタン217mg(0.912ミリモル)およびスチレン95mg(0.912ミリモル)を入れ、次に溶媒としてシクロヘキサンを2ml加えて、1−(4−ビニルフェニル)アダマンタンおよびスチレンの溶液を3系列調製した。
(2) 上記(1)で調製した各溶液を攪拌しながら、各反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.028ml(sec−ブチルリチウムとして0.0365ミリモル)を各反応溶液に加えた。sec−ブチルリチウムの添加後、即座に溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、第1の反応溶液にはsec−ブチルリチウムの添加10分後に少量の脱気したメタノールを添加して重合反応を停止させ、第2の反応溶液にはsec−ブチルリチウムの添加20分後に少量の脱気したメタノールを添加して重合反応を停止させ、第3の反応溶液にはsec−ブチルリチウムの添加30分後に少量の脱気したメタノールを添加して重合反応を停止させた。
【0093】
(3) 各反応容器から、反応液の一部をサンプリングし、サンプリングした反応液の一部を乾燥し、それにより得られた半固形状物をテトラヒドロフランに溶解させてGPC測定を行い、またサンプリングした反応液の一部は重クロロホルムで希釈して50℃で1H−NMR測定を行って、生成した共重合体の数平均分子量(Mw)、分子量分布(Mw/Mn)および1−(4−ビニルフェニル)アダマンタンとスチレンの重合反応率を求めた。なお、1−(4−ビニルフェニル)アダマンタンのβ−炭素上のプロトン(trans側)のピークは5.17ppmおよび5.20ppmにダブレットピークとして現れるのに対し、スチレンのβ−炭素上のプロトン(trans側)のピークは5.23ppmおよび5.26ppmにダブレットピークとして現れるため、1H−NMRスペクトル上においてそれぞれを区別することが可能であり、それに基づいて各単量体の重合反応率を測定することができる。結果を下記の表1に示す。
【0094】
【表1】
【0095】
上記の表1の結果から、1−(4−ビニルフェニル)アダマンタンとスチレンの重合反応速度はほぼ同等であることが確認された。
【0096】
(4) 上記(2)において、30分間重合反応を行った第3の反応容器内の反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体[ランダム共重合体(1)]のガラス転移温度(Tg)は180℃であり、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は404℃であった。この結果を下記の表2に示す。
【0097】
《実施例3》[3−(4−ビニルフェニル)−1,1’−ビアダマンタンの単独重合体の製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に参考例2で製造した3−(4−ビニルフェニル)−1,1’−ビアダマンタン0.6g(1.61ミリモル)を入れ、次に溶媒としてシクロヘキサン40mlを加えて3−(4−ビニルフェニル)−1,1’−ビアダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.025ml(sec−ブチルリチウムとして0.033ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、2時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)7900、分子量分布(Mw/Mn)1.34であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークは観測されず、3−(4−ビニルフェニル)−1,1’−ビアダマンタンの重合反応率は99.9%以上であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は245℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は426℃であった。この結果を下記の表2に示す。
【0098】
《実施例4》[1−(4−イソプロペニルフェニル)アダマンタンの単独重合体の製造]
(1) 乾燥したガラス製反応容器内を窒素ガスで置換した後、反応容器内に参考例3で製造した1−(4−イソプロペニルフェニル)アダマンタン0.79g(3.12ミリモル)を入れ、次に溶媒として脱水THF30mlを加えて1−(4−イソプロペニルフェニル)アダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を−78℃に冷却した後、sec−ブチルリチウムのシクロヘキサン溶液0.05ml(sec−ブチルリチウムとして0.065ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を−78℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、2時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)9200、分子量分布(Mw/Mn)1.04であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークが観測され、芳香族由来のピークとの積分値の比較から、1−(4−イソプロペニルフェニル)アダマンタンの反応率は81%であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノールに注ぎ込み、精製した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は273℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は360℃であった。この結果を下記の表2に示す。
【0099】
《比較例1》[ポリスチレンの製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に溶媒としてシクロヘキサン40mlを加え、更にsec−ブチルリチウムのシクロヘキサン溶液0.14ml(sec−ブチルリチウムとして0.18ミリモル)を加えた。得られた溶液を撹拌しながら反応容器内の温度を50℃に加温した後、スチレン3g(28.8ミルモル)を加えた。スチレンを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、スチレンを添加した後、1時間後に少量のメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)17000、分子量分布(Mw/Mn)1.01であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体(ポリスチレン)について上記した方法でDSC測定を行ったところ、ポリスチレンのガラス転移温度(Tg)は98℃と低く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は397℃であった。この結果を下記の表2に示す。
【0100】
【表2】
【0101】
上記の表2にみるように、重合体のガラス転移温度(Tg)が、比較例1のポリスチレン(通常のポリスチレン)では98℃であるのに対して、実施例1の1−(4−ビニルフェニル)アダマンタン単独重合体では237℃、実施例2の1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(1)では180℃、実施例3の3−(4−ビニルフェニル)−1,1’−ビアダマンタン単独重合体では245℃、実施例4の1−(4−イソプロペニルフェニル)アダマンタン単独重合体では273℃であって、実施例1〜4の重合体はポリスチレンに比べてガラス転移温度(Tg)が大幅に高い。
また、10%質量減量温度が、比較例1のポリスチレンでは397℃であるのに対して、実施例1の1−(4−ビニルフェニル)アダマンタン単独重合体では408℃、実施例2の1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体では404℃、実施例3の3−(4−ビニルフェニル)−1,1’−ビアダマンタン単独重合体では426℃であり、比較例1のポリスチレンに比べて10%質量減少温度が高い。
かかる点から、上記の一般式(I)で表されるスチレン系誘導体(I)に由来する構造単位を有する本発明の重合体が、耐熱性に優れていることが裏付けられる。
【0102】
《実施例5》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(2)の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、通常のアニオン重合法により調製したポリスチレン[数平均分子量(Mn)5730、分子量分布(Mw/Mn)1.03]0.4g(スチレンユニットとして3.85ミリモル)および1−ブロモアダマンタン1g(4.64ミリモル)を入れ、次に溶媒として塩化メチレン10ml、触媒として塩化アルミニウム20mg(0.15ミリモル)を加えた。得られた溶液を16時間還流したのち、系を室温まで冷却し、水を加えて反応を停止させた。反応後、クロロホルムで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して1.13gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノール中に注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して、白色粉末0.46gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、ポリスチレンにおける14%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0103】
《実施例6》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(3)の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、1−ブロモアダマンタン83mg(0.39ミリモル)を入れ、次に溶媒として塩化メチレン5mLを加えて溶解させた。アルミホイルで遮光し、0℃に冷却したのちトリフルオロメタンスルホン酸銀0.12g(0.47ミリモル)を加え3時間反応させ、ガスクロマトグラフィーにより1−ブロモアダマンタンが完全に消費され、トリフルオロメタンスルホン酸(1−アダマンチル)が生成したことを確認した。ここに通常のアニオン重合法により調製したポリスチレン[数平均分子量(Mn)5730、分子量分布(Mw/Mn)1.03]0.2g(スチレンユニットとして1.92ミリモル)の塩化メチレン溶液2mlを加え、系を室温に戻し3時間攪拌を行い、水を加えて反応を停止させた。反応後、クロロホルムで3回抽出、有機層をチオ硫酸ナトリウム水溶液で3回洗浄、次いで水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥した。硫酸マグネシウムを濾別後、有機溶媒を減圧留去して0.24gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノールに注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して、白色粉末0.15gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、10%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0104】
《実施例7》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(4)の製造]
1−ブロモアダマンタン165mg(0.77ミリモル)、トリフルオロメタンスルホン酸銀0.23g(0.90ミリモル)を用いたこと以外は、実施例6と同様にして白色粉末を得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、22%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0105】
《実施例8》[1−(4−イソプロペニル)アダマンタン/α−メチルスチレンランダム共重合体の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、1−ブロモアダマンタン73mg(0.34ミリモル)を入れ、次に溶媒として塩化メチレン5mlを加えて溶解させた。アルミホイルで遮光し、0℃に冷却したのちトリフルオロメタンスルホン酸銀0.1g(0.39ミリモル)を加えて3時間反応させ、ガスクロマトグラフィーにより1−ブロモアダマンタンが完全に消費され、トリフルオロメタンスルホン酸(1−アダマンチル)が生成したことを確認した。ここに通常のアニオン重合法により調製したポリ(α−メチルスチレン)[数平均分子量(Mn)9300、分子量分布(Mw/Mn)1.04]0.2g(α−メチルスチレンユニットとして1.69ミリモル)の塩化メチレン溶液2mlを加え、系を室温に戻し1時間攪拌を行い、水を加えて反応を停止させた。反応後、クロロホルムで3回抽出、有機層をチオ硫酸ナトリウム水溶液で3回洗浄、次いで水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥した。硫酸マグネシウムを濾別後、有機溶媒を減圧留去して0.18gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノールに注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して白色粉末0.15gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、20%のα−メチルスチレンユニットにアダマンチル基が導入されたことを確認した。
【0106】
上記した実施例5〜8の結果が示すように、入手が容易なポリスチレン、ポリ(α−メチルスチレン)を基質として用いた芳香族求電子置換反応によっても本発明の重合体を得ることが可能である。
【0107】
《実施例9》[{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製容器内の気体を窒素ガスで置換した後、1−(4−ビニルフェニル)アダマンタン2.087g(8.76ミリモル)およびスチレン0.912g(8.76ミリモル)を入れ、次に溶媒としてシクロヘキサン25mlを加えて1−(4−ビニルフェニル)アダマンタンおよびスチレンを溶解させて単量体混合物溶液を調製した。この単量体混合物溶液の密度が0.7804g/mlであり、かかる点から、溶液中の1−(4−ビニルフェニル)アダマンタンおよびスチレンの濃度はいずれも0.304モル/リットルであった。
(2) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、シクロヘキサン50mlを入れて50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.081ml(sec−ブチルリチウムとして0.105ミリモル)を加え、ここに上記(1)で調製した単量体混合物溶液の8.65ml[1−(4−ビニルフェニル)アダマンタン2.63ミリモル、スチレン2.63ミリモルを含有]を加えて、50℃で30分間重合した後、反応液の一部をサンプリングし、GPC測定を行ったところ数平均分子量(Mn)6900、分子量分布(Mw/Mn)1.07の重合体が生成していた。
(3) 続いて、前記(2)の反応容器に、イソプレン4.2g(61.7ミリモル)を加えて50℃で1.5時間重合を行った。反応液の一部をサンプリングし、GPC測定を行ったところ、数平均分子量(Mn)107000、平均分子量1.02の重合体が生成していた。続いて反応容器に、上記(1)で調製した混合単量体溶液の8.65ml[1−(4−ビニルフェニル)アダマンタン2.63ミリモル、スチレン2.63ミリモルを含有]を加えて50℃で1時間重合反応を行った後、脱気したメタノールを少量添加して重合を停止した。
【0108】
(4) 上記(3)で得られた反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)114000、分子量分布(Mw/Mn)1.03のブロック共重合体が生成していた。得られたブロック共重合体の溶液(反応液)を水で洗浄した後、メタノール/アセトン混合溶媒で沈殿させて回収した。これにより得られたブロック共重合体の1H−NMR測定を行ったところ、得られたブロック共重合体は、{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体であり、ポリイソプレンブロックには3,4−結合が5.5%含まれており、ブロック共重合体の質量に基づく1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロックの含有割合は28.7質量%であった。
【0109】
《実施例10》[{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製容器内の気体を窒素ガスで置換した後、1−(4−ビニルフェニル)アダマンタン2g(8.39ミリモル)を入れ、次に溶媒としてシクロヘキサン15mlを加えて1−(4−ビニルフェニル)アダマンタンを溶解させて単量体溶液を調製した。この単量体溶液の密度は0.795g/mlであり、これより単量体溶液中での1−(4−ビニルフェニル)アダマンタンの濃度は0.504モル/リットルであった。(2) 乾燥したガラス製反応容器内の気体を窒素ガスで置換し、シクロヘキサン50mlを入れて50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.058ml(sec−ブチルリチウムとして0.076ミリモル)を加え、ここに上記(1)で調製した単量体溶液の7.5ml[1−(4−ビニルフェニル)アダマンタンとして0.9g(3.78ミリモル)]を加えて50℃で30分間重合した後、反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)8500、分子量分布(Mw/Mn)1.29の重合体が生成していた。
(3) 続いて反応容器に、イソプレン4.2g(61.7ミリモル)を加えて50℃で1.5時間重合を行った。反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)100000、分子量分布(Mw/Mn)1.05の重合体が生成していた。続いて反応容器に、上記(1)で調製した単量体溶液の7.5ml[1−(4−ビニルフェニル)アダマンタンとして0.9g(3.78ミリモル)]を加えて50℃で2時間重合反応を行った後、脱気したメタノールを少量添加して重合を停止させた。
【0110】
(4) 上記(3)で得られた反応液の一部をサンプリングしGPC測定を行ったところ、数平均分子量(Mn)110000、分子量分布(Mw/Mn)1.03のブロック共重合体が生成していた。得られたブロック共重合体の溶液(反応液)を水で洗浄した後、メタノール/アセトン混合溶媒で沈殿させてブロック共重合体を回収した。これにより得られたブロック共重合体の1H−NMR測定を行ったところ、{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体であり、ポリイソプレンブロックには3,4−結合が5.7%含まれており、ブロック共重合体の質量に基づく1−(4−ビニルフェニル)アダマンタン重合体ブロックの含有割合は24.7質量%であった。
【0111】
《実施例11》[水添{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製反応容器に、実施例9で製造したトリブロック共重合体[{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体]3.6g、酸化防止剤(日本チバ・ガイギー社製「Irganox 1010」)0.36gおよびp−トルエンスルホン酸ヒドラジド20gを入れて30分間真空脱気を行った。その後、反応容器内に窒素ガスを導入し、キシレン250mlを加えて攪拌して120℃に加熱した。溶液は当初懸濁状態であったが、120℃まで加熱すると均一の溶液となるともに、窒素ガス、水素ガスの発生による気泡の発生が確認された。反応液の温度をそのまま120℃に保って15時間反応を継続した。反応終了後、反応液を室温まで冷却した後、大過剰のメタノールに投入して、水添トリブロック共重合体を沈殿させて回収した。回収した水添トリブロック共重合体をトルエンに溶解させた後、メタノールで再沈する作業をさらに3回繰り返して精製し、それにより得られた水添トリブロック共重合体を50℃で一晩真空乾燥した。
【0112】
(2) 上記(1)で得られた水添トリブロック共重合体について1H−NMR測定を行った結果、イソプレン中の二重結合に由来するするピークの大部分が消失し、水添率は98%であった。また水添トリブロック共重合体のGPC測定を行ったところ、ゲル化や分解を示すピークは観測されず、数平均分子量(Mn)122000、分子量分布(Mw/Mn)1.03であった。
(3)(i) 上記(1)で得られた水添トリブロック共重合体をトルエンに溶解し、平らな台上に設置したテフロン(登録商標)シート上でキャストした後、乾燥して厚さ1mmのシートを製造した。
(ii) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。その結果を図5に示す。
(iii) 上記(i)で得られたシートから、図1に示す形状および寸法のダンベル試験片を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iv) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0113】
《実施例12》[水添{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 実施例10で得られたトリブロック共重合体[{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体]を3.6g用いた以外は実施例11と同様にして水素添加処理、水添トリブロック共重合体の回収および精製を行って、水添{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体を製造した。
(2) 上記(1)で得られた水添トリブロック共重合体について1H−NMR測定を行った結果、イソプレン中の二重結合に由来するするピークの大部分が消失し、水添率は98%であった。また水添トリブロック共重合体のGPC測定を行ったところ、ゲル化や分解を示すピークは観測されず、数平均分子量(Mn)125000、分子量分布(Mw/Mn)1.03であった。
【0114】
(3)(i) 上記(1)で得られた水添トリブロック共重合体をトルエンに溶解し、平らな台上に設置したテフロン(登録商標)シート上でキャストした後、乾燥して厚さ1mmのシートを製造した。
(ii) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。その結果を図5に示す。
(iii) 上記(i)で得られたシートから、図1に示す形状および寸法のダンベル試験片を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iv) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0115】
《比較例2》[ポリスチレン−水添ポリイソプレン−ポリスチレンよりなるトリブロック共重合体の製造]
(1) 実施例11の水添{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の代りに、ポリスチレン−水添ポリイソプレン−ポリスチレンよりなるトリブロック共重合体(株式会社クラレ製「セプトン2002」、スチレン単位含量30質量%)を用いて、実施例11と同様にして厚さ1mmのシートをつくった。
(2)(i) 上記(1)で得られたシートから、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’及び損失正接tanδを測定した。その結果を図5に示す。
(ii) 上記(1)で得られたシートから、図1に示す形状および寸法のダンベル試験片
を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iii) 上記(1)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0116】
【表3】
【0117】
上記の表3の結果にみるように、実施例11および実施例12の水添トリブロック共重合体(熱可塑性エラストマー)は、スチレン系誘導体由来の構造単位(I−A)を有する重合体ブロックを有していることにより、構造単位(I−A)を持たない比較例2の水添トリブロック共重合体(熱可塑性エラストマー)に比べて、100%モジュラス(M100)および300モジュラス(M300)が小さくて伸縮性に優れ、応力緩和特性(Eb)にも優れている。しかも、実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて破断強度TB(引張強度)が高く、引張強度にも優れている。
また、図5のグラフにみるように、実施例11および実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて、高い温度まで貯蔵弾性率E’が保持されており、比較例2の水添トリブロック共重合体が使用不可能な温度域においてもエラストマーとしての使用が可能である。
さらに、図6のグラフにみるように、実施例11および実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて、応力緩和性に優れている。
【産業上の利用可能性】
【0118】
本発明の製造方法により得られる重合体は、耐熱性に極めているため、その特性を活かして、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することができる。
【技術分野】
【0001】
本発明は、耐熱性に優れる重合体の製造方法に関する。より詳細には、本発明は、多環構造を有する脂環式炭化水素基からなる置換基をベンゼン環に有するスチレン系誘導体に由来する構造単位を分子中に有する耐熱性に優れる重合体の製造方法に関する。本発明により得られる重合体はその分子構造に応じて熱可塑性樹脂または熱可塑性エラストマーとしての性質を有し、そのいずれもが耐熱性および力学的特性に優れ、エラストマー構造を有するものは弾性特性においても優れている。そのため、本発明により得られる重合体は、それらの特性を活かして、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することができる。
【背景技術】
【0002】
スチレン系樹脂は、その優れた成形加工性、物性などにより、電気製品、家庭用部品、産業用資材などの広範な分野で従来から汎用されているが、近年、用途の拡大などに伴って一層高い性能、特に耐熱性の向上が強く求められるようになっている。
【0003】
耐熱性の向上したスチレン系樹脂としては、立体規則性が主としてシンジオタクチック構造であるスチレン系重合体が知られている(特許文献1及び2を参照)。これらのスチレン系重合体は、シンジオタクティシティーが高く、結晶性であり高い耐熱性を示す。しかしながら、該スチレン系重合体の製造に当たって、有機アルミニウム化合物と縮合剤とを接触させて得られる有機アルミノキサンにチタン化合物を組み合わせた触媒などを使用する必要がある。そのため、触媒を特別に調製しなければならず、汎用の触媒を用いる従来のスチレン系樹脂の製造法に比べて製造工程が複雑で手間や時間を要する。
【0004】
また、スチレン系重合体ブロックと共役ジエン系重合体ブロックを有するブロック共重合体またはその水素添加物からなる熱可塑性エラストマー[以下「(水添)スチレン系熱可塑性エラストマー」ということがある]は、熱可塑性エラストマーの1種として成形材料、粘接着剤、樹脂の改質をはじめとして、種々の用途に広く用いられるようになっている。
【0005】
近年、工業技術の進歩に伴って、熱可塑性エラストマーを高温で使用する必要が生じており、かかる点から高温特性に優れる熱可塑性エラストマーの開発が強く望まれているが、そのことは(水添)スチレン系熱可塑性エラストマーにおいても例外ではない。
高温特性が要求される用途分野としては、例えば、自動車部品、電気・電子部品、フィルム、シート、医療材料などを挙げることができ、熱可塑性エラストマーに求められる高温特性としては、高温に曝されても劣化や分解などの生じにくい耐熱特性を有すること、高温時に引張り強度、引張り伸び、応力緩和特性などの機械的特性に優れること、高温時に加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを示すことなどが挙げられる。
しかしながら、(水添)スチレン系熱可塑性エラストマーは、拘束相をなすスチレン重合体ブロックのガラス転移温度が一般に100℃前後であるため、前記温度を超える高温条件下では弾性が著しく低下し、熱可塑性エラストマーとしての性能を十分に維持できないという問題があった。
【0006】
(水添)スチレン系熱可塑性エラストマーの耐熱性を向上させる方法として、(水添)スチレン系熱可塑性エラストマーにポリα−メチルスチレン樹脂、ポリオレフィン、ポリフェニレンエーテルなどを配合することが知られているが、耐熱性の改善の程度は市場の要求を満たすには至っていない。
【0007】
また、(水添)スチレン系熱可塑性エラストマーにおいて、スチレン系重合体ブロックとして結晶性を有するシンジオタクチックポリスチレンを導入したブロック共重合体が知られている(特許文献3を参照)。このブロック共重合体では、拘束相であるシンジオタクチックポリスチレンブロックが260〜270℃という高い融点を有するため、ブロック共重合体は高い耐熱性が示す。しかしながら、このブロック共重合体の製造に当たっては、まず二官能性アニオン重合開始剤を調製して共役ジエンを重合した後、そのアニオン重合活性末端を配位重合活性末端に変換し、次いでスチレンの重合を行う必要があり、製造工程が極めて複雑である。
【0008】
さらに、(水添)スチレン系熱可塑性エラストマーにおけるスチレン系重合体ブロックを、ガラス転移温度の高いα−メチルスチレン重合体ブロックから形成したブロック共重合体が知られている(特許文献4を参照)。このブロック共重合体は、α−メチルスチレン重合体ブロックを導入したことにより高い耐熱使用温度を有しているが、α−メチルスチレンの重合をスチレンの重合に比べて低温で行う必要があるため、温度管理が複雑であり、改良の余地がある。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特開昭62−104818号公報
【特許文献2】特開昭63−241009号公報
【特許文献3】特開平5−295056公報
【特許文献4】特開2001−172324公報
【非特許文献】
【0010】
【非特許文献1】“Synthesis”,1998年、第2号、148−152頁
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、耐熱性に優れていて、高温に曝されても劣化や分解が生じにくく、高温において良好な力学的特性を有し、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することのできる、熱可塑性樹脂状または熱可塑性エラストマー状のスチレン系重合体を提供することである。
また、本発明の目的は、特別に調製した触媒を使用する必要がなく、更には複雑で手間および時間のかかる重合工程や複雑な温度管理を要せず、汎用の触媒を用いて、簡単な重合工程および単純な温度管理の元で簡単に且つ円滑に製造することのできる耐熱性スチレン系重合体およびその製造方法を提供することである。
さらに、本発明の目的は、安価で、入手が容易なスチレン系重合体を基質として用いて、フリーデル・クラフツ反応などの芳香族求電子置換反応を行うことにより、耐熱性スチレン系重合体を簡単に且つ円滑に製造する方法を提供することである。
【課題を解決するための手段】
【0012】
上記の目的を達成すべく本発明者らは検討を重ねてきた。そして、多環構造を有する脂環式炭化水素基よりなる置換基をベンゼン環に有する特定のスチレン系誘導体を用いて重合を行って耐熱性に優れるスチレン系重合体を得ることができた。
それを踏まえて更に研究を続けたところ、多環構造を有する脂環式炭化水素基からなる置換基をベンゼン環に有する前記スチレン系誘導体を用いると、単独重合体、ランダム共重合体、ブロック共重合体などの種々の耐熱性に優れる重合体が、汎用の重合開始剤を用いて簡単な工程で、しかも複雑な温度管理を要せずに円滑に製造できることを見出した。
さらに、本発明者らは、耐熱性に優れる前記スチレン系重合体は、汎用のスチレン系樹脂を基質として用い、フリーデル・クラフツ反応などの芳香族求電子置換反応によって該基質(スチレン系樹脂)のベンゼン環に多環構造を有する脂環式炭化水素基を導入する方法によっても、簡単に製造できることを見出した。
そして、それにより得られた重合体は、高温に曝されても劣化や分解などが生じにくく、高温下で良好な機械的特性を有することを見出した。
また、本発明者らは、前記したスチレン系誘導体に由来する構造単位を有する重合体ブロックとそれよりもガラス転移温度の低い重合体ブロックを有するブロック共重合体は、熱可塑性エラストマーとしての特性を有し、引張り強度、引張り伸び、応力緩和特性などに優れ、しかも高温下において加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを有し、優れたエラストマー特性を有することを見出し、それらの知見に基づいて本発明を完成した。
【0013】
すなわち、本発明は、
(1) 下記の一般式(I);
【0014】
【化1】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または前記スチレン系誘導体(I)および他の重合性単量体を用いて重合を行うことを特徴とする、下記の一般式(I−A);
【0015】
【化2】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法である。
【0016】
そして、本発明は、
(2) 一般式(I)および一般式(I−A)におけるR1が多環構造を有する橋架け脂環式炭化水素基である前記(1)の重合体の製造方法;
(3) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)を10質量%以上の割合で有する重合体を製造するものである前記(1)または(2)の重合体の製造方法;および、
(4) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)の単独からなるか、または該構造単位(I−A)および他の重合性単量体に由来する構造単位を有する重合体を製造するものである前記(1)〜(3)のいずれかの重合体の製造方法;
である。
【0017】
さらに、本発明は、
(5) 一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)よりなる重合体ブロックAと、他の重合性単量体に由来する構造単位よりなる重合体ブロックBを有するブロック共重合体を製造するものである前記(1)〜(4)のいずれかの重合体の製造方法;
(6) 重合体ブロックAの含有割合が、ブロック共重合体の質量に基づいて10質量%以上であるブロック共重合体を製造するものである前記(5)の重合体の製造方法;
(7) 重合体ブロックBのガラス転移温度が重合体ブロックAのガラス転移温度よりも低いブロック共重合体を製造するものである前記(5)または(6)の重合体の製造方法;および、
(8) 重合体ブロックBが共役ジエンに由来する構造単位を主体とする共役ジエン系重合体または該共役ジエン系重合体の水素添加物よりなる重合体ブロックであるブロック共重合体を製造するものである前記(5)〜(7)のいずれかの製造方法;
である。
【0018】
そして、本発明は、
(9) 一般式(I)で表されるスチレン系誘導体(I)として、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンの少なくとも1種を用いる前記(1)〜(8)のいずれかの重合体の製造方法;および、
(10) 有機リチウム化合物を重合開始剤として用いてアニオン重合を行う前記(1)〜(9)のいずれかの重合体の製造方法;
である。
【0019】
さらに、本発明は、
(11) 下記の一般式(II);
【0020】
【化3】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるか、または当該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多環構造を有する脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のスチレン由来のベンゼン環に基R1を導入する工程を含むことを特徴とする、下記の一般式(I−A);
【0021】
【化4】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法;および、
(12) 芳香族求電子置換反応が、フリーデル・クラフツ反応である前記(11)の製造方法;
である。
【発明の効果】
【0022】
本発明の製造方法により得られる重合体は、耐熱性に優れており、高温に曝されても劣化や分解などが生じにくく、高温下においても良好な機械的特性および物理的特性を有する。
本発明の製造方法により得られる重合体のうち、一般式(I)に由来する構造単位を有する重合体ブロックとそれよりもガラス転移温度の低い重合体ブロックを有する熱可塑性エラストマーは、引張り強度、引張り伸び、応力緩和特性などに優れ、しかも高温下において加硫ゴムにより近い圧縮永久ひずみや動的ヒステリシスなどを有し、良好な熱可塑性エラストマー特性を備えている。
本発明の製造方法による場合は、特別の触媒(重合開始剤)などを別途調製する必要がなく、スチレン系単量体などの重合において従来から用いられている汎用の重合開始剤を使用して、しかも重合時に複雑な温度管理などを要せずに、目的とする耐熱性スチレン系重合体を簡単に且つ円滑に製造することができる。
さらに、本発明では、安価で入手が容易な汎用のスチレン系重合体を基質として用いて、該スチレン系重合体中のベンゼン環に、フリーデル・クラフツ反応などの芳香族求電子置換反応によって多環構造を有する脂環式炭化水素基を導入することによっても、耐熱性スチレン系重合体を簡単に且つ円滑に製造することができる。
本発明の製造方法により得られる重合体は、前記した優れた特性を活かして、例えば、家電製品の筐体、OA機器、食品容器、包装材用発泡体、食品トレー、自動車部品、電気・電子部品、フィルム・シート、医療材料、伸縮部材などの広範な用途に有効に使用することができる。
【図面の簡単な説明】
【0023】
【図1】図1は引張試験に用いた試験片の形状および寸法を示す図である。
【図2】図2は参考例1で得られた1−(4−ビニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図3】図3は参考例2で得られた3−(4−ビニルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図4】図4は参考例3で得られた1−(4−イソプロペニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルを示す図である。
【図5】図5は実施例11、実施例12および比較例2の水添トリブロック共重合体(熱可塑性エラストマー)の貯蔵弾性率E’、損失弾性率E’’および損失正接tanδの測定結果を示すグラフである。
【図6】図6は実施例11、実施例12および比較例2の水添トリブロック共重合体(熱可塑性エラストマー)の応力緩和特性の測定結果を示すグラフである。
【発明を実施するための最良の形態】
【0024】
以下に本発明について詳細に説明する。
本発明の製造方法により得られる重合体(以下、本発明の製造方法により得られる重合体を、「本発明の重合体」ということがある)は、スチレン系誘導体由来の下記の一般式(I−A)で表される構造単位(I−A)を重合体分子中に有する。
【0025】
【化5】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
【0026】
上記の構造単位(I−A)においてR1は多環構造を有する脂環式炭化水素基である。
ここで、「多環構造を有する脂環式炭化水素基」とは、2個以上の脂肪族環(脂環)を有する炭化水素基を意味する。多環構造を有する脂環式炭化水素基(以下「多脂環式炭化水素基」ということがある)R1は、多環構造を有する橋架け脂環式炭化水素基、すなわち隣り合う2つの脂環が2個以上の炭素原子を互いに共有している多脂環式炭化水素基であることが、重合体の耐熱性が良好になる点から好ましい。多脂環式炭化水素基R1は、場合によりR1を構成している2個以上の脂環のうちの1個または2個以上に脂肪族不飽和結合を有していてもよい。但し、脂環中に存在する脂肪族不飽和結合は、重合に関与しないか又はスチレン系誘導体(I)中のビニル結合よりも重合性が低いことが必要である。そうでないと、スチレン系誘導体(I)の円滑な重合が阻害されて、上記の構造単位(I−A)を有する本発明の重合体を円滑に得ることが困難になる。
【0027】
多脂環式炭化水素基R1の具体例としては、アダマンチル基(トリシクロ〔3.3.1.13,7〕デシル基)、ビアダマンチル基、ノルボルニル基、イソボルニル基、ビシクロノニル基、ビシクロ〔2.1.0〕ペンチル基、ビシクロ〔3.2.1〕オクチル基、トリクロ〔2.2.1.02,6〕ペンチル基などを挙げることができる。これらの多脂環式炭化水素基は、場合によりアルキル基、ハロゲン、アルコキシル基などにより置換されていてもよい。
【0028】
構造単位(I−A)における多脂環式炭化水素基R1のベンゼン環での結合位置は、ベンゼン環における重合体主鎖への結合部位[スチレン系誘導体(I)におけるビニル結合:−C(R2)=CH2の結合部位]に対してオルト位、メタ位またはパラ位のいずれであってもよく、そのうちでも、重合体の製造原料であるスチレン系誘導体(I)の重合反応性などの点から、多脂環式炭化水素基R1はパラ位に結合していることが好ましい。
【0029】
上記の一般式(I−A)で表される構造単位(I−A)において、R2は水素原子、炭素数1〜10のアルキル基またはアリール基のいずれであってもよい。
R2が炭素数1〜10のアルキル基である場合は、直鎖状または分岐状のいずれであってもよく、具体的にはメチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基などの直鎖状アルキル基、イソプロピル基、イソブチル基、sec−ブチル基、tert−ブチル基、イソペンチル基、ネオペンチル基、tert−ペンチル基、1−メチルヘプチル基などの分岐状アルキル基などを挙げることができる。
また、R2がアリール基である場合の具体例としては、フェニル基、ナフチル基などを挙げることができ、これらのアリール基は、アルキル基、ハロゲン、アルコキシル基などの置換基を1個または2個以上有していてもよい。
上記したうちでも、R2は水素原子または炭素数1〜3の直鎖状アルキル基、そのうちでも水素原子またはメチル基、特に水素原子であることが、重合体の製造原料であるスチレン系誘導体(I)の製造容易性、重合反応性などの点から好ましい。
【0030】
構造単位(I−A)を有する本発明の重合体は、上記の一般式(I)で表されるスチレン系誘導体(I)を少なくとも用いて重合を行うことによって製造することができる。
本発明の重合体の製造に好ましく用い得るスチレン誘導体(I)の具体例としては、下記の化学式で示すスチレン系誘導体を挙げることができる。
【0031】
【化6】
【0032】
上記したスチレン系誘導体(I)のうちでも、R1がアダマンチル基である上記の化学式(Ia)で表される1−(4−ビニルフェニル)アダマンタン[別称:4−(1−アダマンチル)スチレン]およびR1がアダマンチル基でR2がメチル基である上記の化学式(Ic)で表される1−(4−イソプロペニルフェニル)アダマンタン[別称:4−(1−アダマンチル)−α−メチルスチレン]、並びにR1がビアダマンチル基である上記の化学式(Ib)で表される3−(4−ビニルフェニル)−1,1’−ビアダマンタンのうちの1つ以上、特に1−(4−ビニルフェニル)アダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンのうちの一方または両方に由来する構造単位を有する本発明の重合体は、耐熱性に優れ、しかも単量体の製造が容易であることから好ましい。
【0033】
本発明の重合体は、スチレン系誘導体由来の構造単位(I−A)のみから構成される単独重合体であってもよいし、または構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体であってもいずれでもよい。
本発明の重合体が、構造単位(I−A)のみから構成される単独重合体である場合は、重合体のガラス転移温度が一般に200℃を超えるようになり、使用可能温度域が広がり、より高温での使用に耐えることができる。
また、本発明の重合体が、構造単位(I−A)および他の重合性単量体に由来する構造単位を有する共重合体である場合は、構造単位(I−A)のみからなる単独重合体に比べてガラス転移温度は低いものの、構造単位(I−A)の存在によりそのガラス転移温度は汎用のスチレン系重合体に比べて大幅に高く、やはり汎用のスチレン系重合体に比べて高温での使用に耐えることができる。
構造単位(I−A)において、R2がメチル基である場合は、R2が水素原子である場合に比べて、重合体のガラス転移温度が高くなり、より高温での使用に耐えることができるようになるため、重合体の使用可能温度域が広がる。
【0034】
本発明の重合体がスチレン系誘導体由来の構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体である場合は、他の重合性単量体の種類は特に制限されず、一般に重合可能な化合物であればよい。
本発明の重合体(共重合体)を形成するのに用い得る他の重合性単量体の具体例としては、エチレン、プロピレン、1−ブテン、1−オクテン、イソブチレンなどのオレフィン類;塩化ビニル、塩化ビニリデン、フッ化ビニル、フッ化ビニリデン、テトラフルオロエチレンなどの不飽和ハロゲン化合物;酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、ピバリン酸ビニルなどのビニルエステル化合物;アクリル酸、アクリル酸メチル、アクリル酸エチル、アクリル酸ブチル、アクリル酸アダマンチル、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸ブチル、メタクリル酸2−エチルヘキシル、メタクリル酸2−ヒドロキシエチル、メタクリル酸アダマンチルなどの(メタ)アクリル酸エステル化合物;アクリロニトリル、メタクリロニトリルなどのニトリル化合物;アクリルアミド、N−メチルアクリルアミド、N,N−ジメチルアクリルアミド、N−イソプロピルアクリルアミド、N,N−ジイソプロピルアクリルアミド、メタクリルアミド、N−メチルメタクリルアミド、N,N−ジメチルメタクリルアミド、N−イソプロピルメタクリルアミド、N,N−ジイソプロピルメタクリルアミドなどの(メタ)アクリルアミド系化合物;無水マレイン酸、フマル酸ジエチルなどの二重結合含有ジカルボン酸誘導体;スチレン、α−メチルスチレン、o−メチルスチレン、m−メチルスチレン、p−メチルスチレン、2,4−ジメチルスチレン、・−メチル−p−メチルスチレン、o−メトキシスチレン、m−メトキシスチレン、p−メトキシスチレン、1,1−ジフェニルエチレン、ビニルナフタレン、ビニルアントラセン、2−ビニルピリジン、4−ビニルピリジンなどの芳香族ビニル化合物;1,3−ブタジエン、イソプレン、2,3−ジメチル−1,3−ブタジエン、3,4−ジメチル−1,3−ペンタジエン、1,3−シクロヘキサジエン等の共役ジエン化合物;エチレンオキシド、プロピレンオキシド、テトラヒドロフランなどの環状エーテル化合物;・−カプロラクトンなどのラクトン化合物;ヘキサメチルシクロトリシロキサンなどの含ケイ素へテロ環状化合物などを挙げることができる。
本発明の重合体は、構造単位(I−A)と共に、前記した重合性単量体の1種または2種以上に由来する構造単位を有することができる。
【0035】
本発明の重合体が構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体である場合は、構造単位(I−A)の含有割合が重合体(共重合体)の質量に基づいて10質量%以上であることが好ましく、20質量%以上であることがより好ましい。構造単位(I−A)の含有割合が少ないと、構造単位(I−A)による耐熱性の向上効果が発揮されにくくなる。
【0036】
本発明の重合体が共重合体である場合は、構造単位(I−A)と共に存在する他の重合性単量体に由来する構造単位の結合様式は特に制限されず、ブロック状、テーパード状、ランダム状のいずれであってもよい。
【0037】
本発明の重合体がブロック共重合体である場合は、ブロック共重合体は、スチレン系誘導体由来の構造単位(I−A)を有する重合体ブロックAの少なくとも1個と、他の重合性単量体に由来する重合体ブロックBの少なくとも1個が結合したブロック共重合体であればいずれでもよい。なお、重合体ブロックBは、重合体ブロックAと明確に区別でき且つ重合体ブロックB本来の特性を損なわい限りは、構造単位(I−A)を微量であれば有していても差し支えない。
構造単位(I−A)を有する重合体ブロックAをAで表し、重合体ブロックBをBで表すと、本発明の重合体(ブロック共重合体)の具体例としては、A−B型のジブロック共重合体、A−B−A型またはB−A−B型のトリブロック共重合体、A−B−A−B型のテトラブロック共重合体、A−B−A−B−A型またはB−A−B−A−B型のペンタブロック共重合体などを挙げることができる。前記したブロック共重合体のうちでも、製造の容易性、力学的物性などの点から、A−B型のジブロック共重合体、A−B−A型のトリブロック共重合体、A−B−A−B型のテトラブロック共重合体またはA−B−A−B−A型のペンタブロック共重合体が好ましい。
【0038】
重合体ブロックAは、構造単位(I−A)のみから構成される単独重合体であってもよいし、または構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体であってもよい。また、重合体ブロックBは、スチレン系誘導体(I)以外の重合性単量体(他の重合性単量体)のうちの1種類のみから形成された単独重合体であってもよいし、または2種類以上の他の重合性単量体から形成された共重合体であってもよい。前記したように、重合体ブロックBは、スチレン系誘導体(I)以外の重合性単量体から実質的になっていて、重合体ブロックAと明確に区別でき且つ重合体ブロックB本来の特性に影響を及ぼさない限りは、構造単位(I−A)を微量[例えば重合体ブロックB中に構造単位(I−A)を1〜3ユニットというような微量]で有していても差し支えない。
【0039】
本発明の重合体がブロック共重合体であって、該ブロック共重合体が2個以上の重合体ブロックAを有する場合を有する場合は、2個以上の重合体ブロックAは、構造単位(I−A)を有する重合体ブロックである限りは、構造や分子量などが互いに同じであっても又は異なっていてもよい。また、該ブロック共重合体が2個以上の重合体ブロックBを有する場合は、2個以上の重合体ブロックBは、構造単位(I−A)以外の構造単位から実質的に構成される重合体ブロックである限りはその構造や分子量などが互いに同じであっても又は異なっていてもよい。
【0040】
重合体ブロックBを形成する他の重合性単量体、また重合体ブロックAが共重合体である場合に構造単位(I−A)と共に重合体ブロックAを形成する他の重合性単量体としては、本発明の重合体が共重合体である場合に該共重合体を形成するのに用い得る他の重合性単量体の具体例として前記で挙げた種々の重合性単量体、すなわち上記したオレフィン類、不飽和ハロゲン化合物、ビニルエステル化合物、(メタ)アクリル酸エステル化合物、不飽和ニトリル化合物、(メタ)アクリルアミド系化合物、二重結合含有ジカルボン酸誘導体、芳香族ビニル化合物、共役ジエン化合物、環状エーテル化合物、ラクトン化合物、含ケイ素へテロ環状化合物などの重合性単量体のうちの1種または2種以上を用いることができる。
重合体ブロックAがスチレン誘導体由来の構造単位(I−A)と共に他の重合性単量体に由来する構造単位を有する共重合体から形成されている場合は、重合体ブロックAを形成する他の重合性単量体が、上記したスチレン系誘導体(I)以外の芳香族ビニル化合物の1種または2種以上であることが、スチレン系誘導体(I)との共重合の容易性などの点から好ましい。特に、重合体ブロックAが共重合体である場合は、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび4−(1−イソプロペニルフェニル)アダマンタンのうちの1種または2種以上と、スチレンおよび/またはα−メチルスチレンとの共重合体であることがより好ましい。
【0041】
本発明の重合体がブロック共重合体である場合は、構造単位(I−A)を有する重合体ブロックAの割合が、ブロック共重合体の質量に基づいて、10質量%以上であることが好ましく、13〜70質量%であることがより好ましい。また、ブロック共重合体の全質量に対する構造単位(I−A)の割合は、5質量%以上であることが好ましく、10質量%以上であることがより好ましく、15〜70質量%であることが更に好ましい。構造単位(I−A)の割合が少ないと構造単位(I−A)による耐熱性の向上効果が発揮されにくくなる。
【0042】
本発明の重合体がブロック共重合体である場合に、重合体ブロックBを、構造単位(I−A)を有する重合体ブロックAよりもガラス転移温度の低い重合体から形成すると、熱可塑性で且つ弾性を有するブロック共重合体、すなわち熱可塑性エラストマーにすることができる。
重合体ブロックAよりもガラス転移温度の低い重合体ブロックBを形成するための重合性単量体の種類は特に制限されず、ガラス転移温度の低い重合体ブロックBを形成し得る重合性単量体であればいずれでもよく、そのうちでも共役ジエン、特に1,3−ブタジエン、イソプレンまたはこれらの混合物が、耐寒性および柔軟性に優れるブロック共重合体を形成し得ることから好ましく用いられる。
【0043】
前記したブロック共重合体を含めて、本発明の重合体(共重合体)が、共役ジエンに由来する構造単位を有し、それに伴って分子中に共役ジエンに由来する炭素−炭素不飽和二重結合が存在している場合には、耐候性、耐熱劣化性、耐オゾン性の向上などの観点から、該炭素−炭素不飽和二重結合の少なくとも一部が水素添加されていることが好ましく、その際の水素添加率は50%以上であることがより好ましく、90%以上であることが更に好ましい(以下、水素添加を「水添」ということがある)。
水素添加は一般的に利用される方法で行うことができ、例えば、アルキルアルミニウム化合物とコバルト、ニッケルなどからなるチーグラー触媒等の水添触媒の存在下に水素を供給する方法、p−トルエンスルホン酸ヒドラジドのようなジイミドを系内で発生する化合物を使用する方法などを採用することができる。
【0044】
本発明の重合体の分子量は特に制限されないが、重合体の強度および成形性の点から、単独重合体および共重合体のいずれであっても、重合体全体の数平均分子量が3000〜1000000であることが好ましく、5000〜500000であることがより好ましい。
また、本発明の重合体がブロック共重合体である場合は、構造単位(I−A)を有する重合体ブロックAの数平均分子量が3000〜100000、特に4500〜50000で、重合体ブロックBの数平均分子量が10000〜400000、特に30000〜350000であることが、力学物性、成形性の点から好ましい。
なお、本明細書における数平均分子量(Mn)は、ゲル浸透クロマトグラフィー(GPC)を用いて測定した標準ポリスチレン換算による数平均分子量をいう。
また、本明細書における分子量分布(Mw/Mn)は、前記と同様にして測定した重量平均分子量(Mw)と前記した方法で測定した数平均分子量(Mn)との比(Mw/Mn)をいう。
【0045】
本発明の重合体は、本発明の効果を損なわない限り、分子鎖中および/または分子末端に、場合によりカルボキシル基、水酸基、酸無水物基、アミノ基、エポキシ基の官能基を有していてもよい。
【0046】
本発明の重合体の製法は特に制限されず、スチレン系誘導体由来の構造単位(I−A)を分子中に有する重合体を製造し得る方法であれば、いずれの方法を採用してもよい。
そのうちでも、本発明の重合体は、
(i) 下記の一般式(I);
【0047】
【化7】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または該スチレン系誘導体(I)および他の重合性単量体を用いて重合を行う方法[以下これを「製造法(i)」ということがある];または、
(ii) 下記の一般式(II)
【0048】
【化8】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるかまたは該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のベンゼン環に基R1を導入する方法[以下これを「製造法(ii)」ということがある];
によって、円滑に製造することができ、本発明は前記した製造法(i)および製造法(ii)を包含する。
【0049】
[製造法(i)]
まず、製造法(i)により本発明の重合体を製造する場合について説明する。
製造法(i)では、スチレン系誘導体(I)として、例えば上記した化学式(Ia)〜(Ih2)に示したスチレン系誘導体や、それ以外のスチレン系誘導体(I)の1種または2種以上を用いて重合を行なう。
製造法(i)では、スチレン系誘導体(I)を単独で用いるか、またはスチレン系誘導体(I)と他の重合性単量体を用いて、アニオン重合、ラジカル重合、カチオン重合、配位重合などを行うことによって本発明の重合体を製造することができる。
また、本発明の重合体がブロック共重合体である場合は、例えば、スチレン系誘導体(I)と他の重合性単量体を用いて、リビングアニオン重合、リビングラジカル重合、リビングカチオン重合などの、いわゆるリビング重合を行ってブロック共重合体を製造する方法を採用することができる。
【0050】
そのうちでも、本発明の重合体はアニオン重合またはリビングアニオン重合により円滑に製造することができる。
本発明の重合体をアニオン重合によって製造するに当たっては、スチレン系誘導体(I)を単独で有機溶媒中に溶解するか、またはスチレン系誘導体(I)と他の重合性単量体を有機溶媒中に溶解し、そこにアニオン重合開始剤を添加し、窒素、アルゴンなどの不活性ガス雰囲気下、常圧ないし加圧条件下で、−100℃〜100℃、特には−78℃〜70℃の温度で重合を行う方法が好ましく採用される。
その際のアニオン重合開始剤としては、ブチルリチウム、エチルリチウム、メチルリチウムなどのアルキルリチウム等の従来から知られているアニオン重合開始剤を用いることができ、これらのアニオン重合開始剤は単独で使用しても又は2種以上を併用してもよい。そのうちでも、ブチルリチウム、特にsec−ブチルリチウムが、重合体の収率、重合開始速度などの点から好ましく用いられる。アニオン重合開始剤の使用量は、一般に、単量体の合計質量に基づいて、0.005〜3質量%、特に0.01〜1.5質量%であることが、得られる重合体の分子量などの点から好ましい。
また、アニオン重合時に用い得る有機溶媒としては、シクロヘキサン、トルエン、ベンゼンなどの炭化水素溶媒、テトラヒドロフランなどを挙げることができ、これらの有機溶媒は単独で使用しても、または2種以上を併用してもよい。そのうちでもシクロヘキサン、トルエンなどの炭化水素が、副反応が少ない点から好ましく用いられる。有機溶媒の使用量は、単量体1gに対して、0.1〜20ml、特に1〜5ml程度であることが、撹拌の容易性、製造コストなどの点から好ましい。
【0051】
また、リビングアニオン重合によってブロック共重合体を製造する場合は、構造単位(I−A)を有する重合体ブロックAを製造するためにスチレン系誘導体(I)を単独で有機溶媒中に溶解するかまたはスチレン系誘導体(I)と他の重合性単量体を有機溶媒中に溶解し、そこにリビングアニオン重合を開始させるアニオン重合開始剤を添加して重合を行って重合体ブロックAを構成する重合体(リビングポリマー)を製造した後、重合体ブロックBを製造するための他の重合性単量体を添加して重合を引き続いて行う方法などを採用することによって、重合体ブロックAと重合体ブロックBを有するブロック共重合体を製造することができる。その際に、重合体ブロックBを構成する重合体(リビングポリマー)を先に製造した後にそこに重合体ブロックAを形成するスチレン系誘導体(I)またはスチレン系誘導体(I)と他の単量体を添加して重合を行って重合体ブロックAと重合体ブロックBを有するブロック共重合体を製造してもよい。トリブロック以上のマルチブロック共重合体を製造する場合は、従来と同様にして、上記した重合操作を順次繰り返すことによって目的とするマルチブロック共重合体を製造することができる。
【0052】
ブロック共重合体の製造に当たっては、リビングアニオン重合開始剤として、ブチルリチウム、メチルリチウムなどのリビングアニオン重合開始剤が好ましく用いられ、特にブチルリチウムが好ましく用いられる。リビングアニオン重合開始剤の使用量は、ブロック共重合体の製造に用いる単量体の全質量に基づいて0.000001〜0.1質量%であることが好ましく、0.000001〜0.01質量%であることがより好ましい。
また、有機溶媒としては、上記で挙げた有機溶媒の1種または2種以上、特にシクロヘキサン、ベンゼンなどの炭化水素溶媒が好ましく用いられる。重合温度は一般に−100℃〜100℃、特に−78℃〜70℃が好ましく採用され、重合圧力は一般に0.1〜3MPa、特に0.1〜1MPaが好ましい。
【0053】
単独重合体、共重合体のいずれの場合も、重合が終了した時点で、重合混合物(反応液)中にメタノール、エタノールなどの重合停止剤を添加するか、または重合混合物を前記したメタノールなどの重合停止剤中に投入することによって、重合を停止することができる。
【0054】
製造法(i)に用いるスチレン系誘導体(I)は、公知の方法、またはそれに準じた方法で製造することができる。
上記の化学式(Ia)で表される1−(4−ビニルフェニル)アダマンタンは、例えば非特許文献1に記載されている方法により製造することができる。
具体的には、下記の反応式(III)に示すように、1−ブロモアダマンタンとベンゼンを炭酸カリウムの存在下にパラジウム/炭素触媒(Pd/C)を用いてカップリング反応させて1−フェニルアダマンタンを生成させ、その1−フェニルアダマンタンにジクロロメタン中でTiCl4の存在下で1,1−ジクロロメチルエーテルを反応させてホルミル化を行い、次いで前記で生成したホルミル化物にテトラヒドロフラン(THF)中でトリフェニルホスフィンメチレン(別名:メチレントリフェニルホスホラン)(Ph3P=CH2)を反応させること(Wittigオレフィン反応)により製造することができる。
【0055】
【化9】
【0056】
また、上記とは別に、下記の反応式(IV)に示すように、1−ブロモアダマンタンとフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて1−フェニルアダマンタンを生成させ、生成した1−フェニルアダマンタンに四塩化炭素中で臭素を反応させて1−(4−ブロモフェニル)アダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−アダマンチル−フェニルマグネシウムブロミド(中間体)を生成させ、そこに窒素雰囲気下でジメチルホルムアミド(DMF)を加えて反応させて、1−(4−ホルミルフェニル)アダマンタン[別称:4−(1−アダマンチル)ベンズアルデヒド]を生成させ、生成した1−(4−ホルミルフェニル)アダマンタンにメチルトリフェニルホスホニウムブロミドとカリウムtert−ブトキシドを加えて脱水THF中で反応させて1−(4−ビニルフェニル)アダマンタン[別称:4−(1−アダマンチル)スチレン]を製造する方法を挙げることができる。
【0057】
【化10】
【0058】
また、上記の化学式(Ib)で表される3−(4−ビニルフェニル)−1,1’−ビアダマンタンは、例えば、下記の反応式(V)に示すように、1−ブロモアダマンタンをn−オクタン中でナトリウムを用いて反応させてビアダマンタンを生成させ、それに四塩化炭素中で臭素を反応させて3−ブロモ−1,1’−ビアダマンタンを生成させ、それにフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて3−フェニル1,1’−ビアダマンタンを生成させ、それに四塩化炭素中で臭素を反応させて3−(4−ブロモフェニル)−1,1’−ビアダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−ビアダマンチル−フェニルマグネシウムブロミド(中間生成物)を生成させ、そこに窒素雰囲気下でジメチルホルムアミド(DMF)を加えて反応させて、3−(4−ホルミルフェニル)−1,1’−ビアダマンタンを生成させ、それにメチルトリフェニルホスホニウムブロミドとカリウムtert−ブトキシドを加えて脱水THF中で反応させて、3−(4−ビニルフェニル)−1,1’−ビアダマンタンを生成させる方法により製造することができる。
【0059】
【化11】
【0060】
また、上記の化学式(Ic)で表される1−(4−イソプロペニルフェニル)アダマンタンは、例えば、下記の反応式(VI)に示すように、1−ブロモアダマンタンとフェニルマグネシウムブロミドを脱水塩化メチレン中で反応させて1−フェニルアダマンタンを生成させ、生成した1−フェニルアダマンタンに四塩化炭素中で臭素を反応させて1−(4−ブロモフェニル)アダマンタンを生成させ、それにTHF中、窒素雰囲気下にマグネシウムを加えて還流して反応系中に4−アダマンチル−フェニルマグネシウムブロマイド(中間体)を生成させ、そこに窒素雰囲気下で脱水アセトンを加えて反応させて4−(1−アダマンチル)クミルアルコールを生成させ、生成した4−(1−アダマンチル)クミルアルコールをトルエンスルホン酸の存在下にトルエン中で脱水反応させて、4−(1−イソプロペニルフェニル)アダマンタンを生成させる方法により製造することができる。
【0061】
【化12】
【0062】
1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタン以外の、上記した一般式(Id)〜(Ih2)などに示したスチレン系誘導体(I)およびそれら以外のスチレン系誘導体(I)も、前記した反応式(III)〜(VI)に準じた方法で製造することができる。
【0063】
[製造法(ii)]
次に、製造法(ii)により本発明の重合体を製造する場合について説明する。
上記の一般式(II)で表されるスチレン系化合物(II)を単独で用いるかまたは該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体としては、従来既知のスチレン系重合体を用いることができる。限定されるものではないが、製造法(ii)で用いることのできる、スチレン系化合物(II)の重合体の代表例としては、ポリスチレン、スチレンと他の重合性単量体との共重合体、α−メチルスチレンの単独重合体、スチレンとα−メチルスチレンの共重合体、スチレンとα−メチルスチレンと他の重合性単量体との共重合体などを挙げることができる。
スチレン系重合体が共重合体である場合は、ブロック共重合体、テーパード共重合体、ランダム共重合体のいずれであってもよい。また、その際の他の重合性単量体としては、本発明の重合体がスチレン誘導体由来の構造単位(I−A)と他の重合性単量体由来の構造単位を有する共重合体である場合について上記で例示した種々の単量体を用いることができる。
【0064】
また、スチレン系化合物(II)に芳香族求電子置換反応させるのに用いる上記の一般式(III)で表される化合物(III)(R1−X)における多脂環式炭化水素基R1の具体例としては、アダマンチル基(トリシクロ〔3.3.1.13,7〕デシル基)、ビアダマンチル基、ノルボルニル基、イソボルニル基、ビシクロノニル基、ビシクロ〔2.1.0〕ペンチル基、ビシクロ〔3.2.1〕オクチル基、トリクロ〔2.2.1.02,6〕ペンチル基などを挙げることができる。これらの多脂環式炭化水素基は、場合によりアルキル基、ハロゲン、アルコキシル基などにより置換されていてもよい。
【0065】
化合物(III)における基Xとしては、化合物(III)をスチレン系化合物(II)に対して芳香族求電子置換反応させたときに、スチレン系重合体中のスチレン由来のベンゼン環に多脂環式炭化水素基R1を結合させる作用を有する基であればいずれでもよい。基Xの具体例としては、ハロゲン原子、アルコール性水酸基、エステル基、トリフレート基、トシル基、オレフィン置換基などを挙げることができる。これらの中でも、化合物(III)の入手容易性、反応性などの観点から、化合物(III)としては、基Xがハロゲン原子またはトリフレート基である化合物(多脂環式炭化水素化合物)を用いることが好ましい。
本発明の重合体の製造に当たっては、1種類の化合物(III)のみを用いても、または2種類以上の化合物(III)を用いてもよい。
スチレン系重合体に対する化合物(III)(R1−X)の使用割合は、スチレン系重合体中に存在するスチレン系化合物(II)に由来するベンゼン環の量、スチレン系重合体中に導入すべき多脂環式炭化水素基R1の割合、芳香族求電子置換反応時の反応条件などに応じて異なり得るが、一般的には、スチレン系重合体中に存在するスチレン系化合物(II)に由来するベンゼン環1個に対して、化合物(III)(R1−X)を0.001〜10モル、特に0.1〜3モルの割合で用いることが、分子中に構造単位(I−A)を有する耐熱性に優れる本発明の重合体が円滑に得られる点から好ましい。その際に、化合物(III)の使用割合を、芳香族求電子置換反応によって得られる重合体における構造単位(I−A)の割合が10質量%以上になるようにして前記範囲から選択することが好ましい。
【0066】
スチレン系重合体に化合物(III)(R1−X)を芳香族求電子置換反応させる際の芳香族求電子置換反応の種類は特に限定されず、スチレン系重合体中のベンゼン環に多脂環式炭化水素基R1を円滑に結合させ得る芳香族求電子置換反応であればいずれでもよく、例えばフリーデル・クラフツ反応などを挙げることができる。
芳香族求電子置換反応は、触媒や溶媒などを用いずに、スチレン系重合体に化合物(III)をそのまま直接反応させて行なってもよいが、スチレン系重合体および化合物(III)を有機溶媒に溶解させて触媒の存在下に行なうことが、芳香族求電子置換反応を速やかに且つ円滑に行わせる点から好ましい。
【0067】
芳香族求電子置換反応に用いる有機溶媒の種類は特に制限されず、例えば、ヘキサン、シクロヘキサン、ヘプタン、オクタンなどの脂肪族炭化水素、ベンゼン、トルエン、キシレンなどの芳香族炭化水素、ジエチルエーテル、テトラヒドロフラン、ジオキサンなどのエーテル化合物、クロロホルム、ジクロロメタン、ジクロロエタンなどのハロゲン化炭化水素などを挙げることができ、これら1種または2種以上を用いることができる。そのうちでも、反応制御の容易性の点から、脂肪族炭化水素および/またはハロゲン化炭化水素が好ましく用いられる。
有機溶媒の使用量は、有機溶媒の種類などに応じて調節できるが、芳香族求電子置換反応の円滑な進行、温度管理の容易性などの点から、一般にスチレン系重合体100質量部に対して、0.1〜10000質量部、特に1〜500質量部であることが好ましい。
【0068】
芳香族求電子置換反応に用い得る触媒の例としては、ルイス酸、ブレンステッド酸などが挙げられる。
ルイス酸としては、特に一般に知られるものであれば特に制限はなく、例えば塩化アルミニウム、臭化アルミニウム、アルキルアルミニウムジクロライド、アルキルアルミニウムクロライド、塩化鉄(III)、三フッ化ホウ素、三塩化ホウ素、三臭化ホウ素、塩化チタンなどを挙げることができ、これらは単独で用いてもまたは2種類以上を組み合わせて用いてもよい。そのうちでも、入手容易性などの点から、塩化アルミニウム、塩化鉄(III)、三フッ化ホウ素が好ましく用いられる。
ブレンステッド酸としては、リン酸、硫酸、フッ化水素などの強酸を挙げることができ、これらは単独で用いてもまたは2種類以上を組み合わせて用いてもよい。
また、上記したルイス酸の1種または2種以上と、ブレンステッド酸の1種または2種以上を組み合わせて使用してもよい。
触媒の使用量は、触媒の種類、化合物(III)(R1−X)の種類などに応じて異なり得るが、一般的には、化合物(III)1モルに対して、0.0001〜100モル、特に0.001〜10モルであることが、芳香族求電子置換反応の円滑な進行などの点から好ましい。
【0069】
芳香族求電子置換反応を実施する際の温度には特に制限はないが、熱効率、反応の円滑な進行などの点から、0〜100℃、特に15〜70℃の範囲内で行うことが好ましい。
【0070】
本発明の重合体は単独で使用してもよいし、または必要に応じて他の熱可塑性樹脂、熱硬化性樹脂、ゴム、熱可塑性エラストマーなどとブレンドして使用してもよい。その際に、本発明の趣旨を損なわない範囲で、さらに酸化防止剤、軟化剤、可塑剤、帯電防止剤、滑剤、紫外線吸収剤、難燃剤、顔料、無機充填剤などを配合してもよい。
【実施例】
【0071】
以下に、実施例、比較例および参考例により本発明をさらに具体的に説明するが、本発明は以下の例により何ら限定されるものではない。なお、以下の例で用いた薬品(化合物など)は入手しうる限りの最高の純度のものを用いた。また、溶媒は十分に脱気したものを使用した。
以下の例において、参考例1または参考例2で得られた生成物(中間化合物、最終化合物)の構造の確認、重合の進行度、各実施例で得られた重合体の数平均分子量および分子量分布の測定、重合体のガラス転移温度の測定、熱質量分析、引張試験、動的粘弾性試験および応力緩和特性の測定は次のようにして行った。
【0072】
(1)生成物の構造の確認:
以下の参考例1または参考例2、参考例3で生成した生成物(中間化合物、最終化合物)を重クロロホルムに溶解し、核磁気共鳴装置(ブルカー社製「BRUCKER DPX300」)を使用して、プロトン核(1H)、炭素核(13C)を27℃で測定して、その構造の確認を行った。また、参考例2で生成した一部の生成物については、構造の確認に当たって、日本分光社製の赤外線分析装置「FT/IR−460」を使用してKBr法により測定した。
【0073】
(2)重合の進行度の測定:
以下の実施例で生成した重合反応液または重合体を重クロロホルムに溶解させ、核磁気共鳴分光装置(日本電子データム社製「JNMLA400」)を使用して、プロトン核(1H)を50℃で測定して重合の進行度を測定した。
【0074】
(3)数平均分子量(Mn)および分子量分布(Mw/Mn)の測定:
ピーク分子量が既知の標準ポリスチレンを用い、該標準で校正したゲル浸透クロマトグラフィー(GPC)(東ソー社製「HLC−8020」)を使用して、重合体の数平均分子量(Mn)および重量平均分子量(Mw)を測定した(溶媒:テトラヒドロフラン、温度:40℃)。分子量分布は重量平均分子量(Mw)と数平均分子量(Mn)の比(Mw/Mn)として求めた。
【0075】
(4)重合体のガラス転移温度(Tg):
DSC装置(メトラー社製「DSC822e」)を用いて測定した。測定は、一旦280℃まで試料を加熱し、同温度で5分間アニールを施した後、降温速度を毎分10℃として0℃まで冷却した。この後再び昇温速度を毎分10℃として昇温して、ガラス転移温度(Tg)を測定した。
【0076】
(5)重合体の熱重量分析(10%質量減少温度の測定):
熱重量(TGA)装置(メトラー社製「TG50」)を使用して、重合体試料を窒素雰囲気下に置き、雰囲気温度を50℃から500℃まで毎分10℃の速度で昇温しながら、重合体の質量が当初よりも10%減少した時点の温度を測定した。
【0077】
(6)引張試験:
以下の実施例11、実施例12または比較例2で得られた重合体を用いて、図1に示す形状および寸法の試験片(厚さ1mm)を作製し、該試験片を使用して、万能材料試験機(インストロンジャパン社製「TM−MS−134」)を用いて、JIS K6251に準拠して、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)、破断伸度(EB)を測定した。なお、図1における各部の寸法の単位はmmである。
【0078】
(7)動的粘弾性試験:
以下の実施例11、実施例12または比較例2で得られた重合体から形成したシート(厚さ1mm)から、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用い、広域動的粘弾性測定装置(レオロジ社製「DVE−V4FTレオスペクトラー」)を使用して、引張りモード(周波数 11Hz)で、昇温速度を毎分3℃として、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。
【0079】
(8)応力緩和特性:
以下の実施例11、実施例12または比較例2で得られた重合体から形成したシート(厚さ1mm)から、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用い、広域動的粘弾性測定装置(レオロジ社製「DVE−V4FTレオスペクトラー」)を使用して、40℃の温度下で引張りひずみを100%与え、経過時間に対して応力の保持率を測定した。保持率が1(100)に近いほど応力緩和特性は良好であることを表す。
【0080】
《参考例1》[1−(4−ビニルフェニル)アダマンタンの製造]
上記した反応式(III)にしたがって1−(4−ビニルフェニル)アダマンタンを以下のようにして製造した。
(1)1−フェニルアダマンタンの製造:
窒素雰囲気下で、マグネシウム3.36g(138.0ミリモル)に脱水エーテル20mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、ブロモベンゼン10ml(84ミリモル)の脱水エーテル溶液100mlを滴下して室温で3時間撹拌した後、エーテルを減圧留去することにより、フェニルマグネシウムブロミドの灰色固体を得た。この灰色固体に、1−ブロモアダマンタン5.00g(23.2ミリモル)と脱水塩化メチレン70mlを加えて、GCとNMRで反応を追跡しながら1−ブロモアダマンタンの消費が完全に確認されるまで還流した(約24時間後)ところで、系を0℃に冷却し、水を加えて反応を停止させ、更に2N塩酸を加えた。塩化メチレンで抽出を3回、有機層を水で3回洗浄した。有機層を無水硫酸マグネシウムを加えて乾燥した後、硫酸マグネシウムを濾別し、有機溶媒を減圧留去して、4.52gの淡黄色固体を得た。この淡黄色固体を、ヘキサンを展開溶媒に用いたシリカゲルカラムクロマトグラフィーにて精製して8.45gの白色固体を得た後、該白色固体をメタノールから再結晶して、1−フェニルアダマンタン6.30g(収率64%)の白色固体(mp84−85℃)を得た。
これにより得られた1−フェニルアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.73−1.82(m,6H,C(2)H2),1.92(s,6H,C(4)H2),2.10(s,3H,C(3)H),7.15−7.20(m,1H,C(d)H),7.29−7.39(m,4H,C(b)H,C(c)H)
・13C−NMR(CDCl3):δ 29.0(C−3),36.2(C−1),36.9(C−2),43.2(C−4),124.9(C−c),125.6(C−d),128.2(C−b),151.4(C−a)
【0081】
(2)1−(4−ブロモフェニル)アダマンタンの製造:
上記(1)で得られた1−フェニルアダマンタン3.60g(17.0ミリモル)に四塩化炭素34mlを加えた後、臭素17ml(330ミリモル)を加えて室温で4時間撹拌した。反応溶液を氷水中に注ぎ込み、過剰の臭素を亜硫酸水素ナトリウムで処理した後、塩化メチレンで3回抽出し、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して、1−(4−ブロモフェニル)アダマンタン4.95g(収率100%、白色固体、mp101−102℃)を得た。
これにより得られた1−(4−ブロモフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.71−1.81(m,6H,C(2)H2),1.87(s,6H,C(4)H2),2.09(s,3H,C(3)H),7.21−7.24(d,2H,C(c)H,J=8.6Hz),7.40−7.43(d,2H,C(b)H,J=8.6Hz)
・13C−NMR(CDCl3):δ 28.9(C−3),36.1(C−1),36.7(C−2),43.1(C−4),119.3(C−d),126.9(C−c),131.1(C−b),150.4(C−a)
【0082】
(3)1−(4−ホルミルフェニル)アダマンタンの製造:
窒素雰囲気下に、マグネシウム0.84g(35ミリモル)に脱水THF20mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。これに、上記(2)で得られた1−(4−ブロモフェニル)アダマンタン4.95g(17.0ミリモル)の脱水THF溶液40mlを室温で滴下した後、還流した。還流中に、ガスクロマトグラフィーで反応を追跡し、1−(4−ブロモフェニル)アダマンタンが完全に消費されて4−アダマンチル−フェニルマグネシウムブロミドに由来する1−フェニルアダマンタンの生成が確認された時点(約2時間後)で、系を0℃に冷却し、脱水DMF25ml(323ミリモル)を滴下したところ、滴下と同時に白濁した。その後、室温で一晩撹拌した後、系に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体4.87gを得た。これを、ヘキサンを展開溶媒に用いてクロマトグラフィーにて処理して、副生した1−フェニルアダマンタン1.01g(4.76ミリモル、回収率28%)を除去した後、ヘキサン/酢酸エチル=20/1,10/1(v/v)を展開溶媒に用いて3.25gの白色固体を得た。これを昇華精製(120〜130℃/0.2mmHg)して、1−(4−ホルミルフェニル)アダマンタン[別称:4−(1−アダマンチル)ベンズアルデヒド]2.88g(収率70%、白色粉末、mp253−255℃)を得た。
これにより得られた1−(4−ホルミルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.74−1.84(m,6H,C(2)H2),1.94(s,6H,C(4)H2),2.13(s,3H,C(3)H),7.51−7.54(d,2H,C(c)H,J=8.3Hz),9.98(s,1H,CHO)
・13C−NMR(CDCl3):δ 28.8(C−3),36.7(C−2),37.0(C−1),42.9(C−4),125.7(C−c),129.8(C−b),134.2(C−d),158.6(C−a),192.2(CHO)
・IR(KBr):2903 and 2848cm-1(C−H伸縮振動),1698,1686 and 1654cm-1(C=O伸縮振動),1604 cm-1(芳香環C−C振動),1448,1167,802cm-1(C−H変角振動)
【0083】
(4)1−(4−ビニルフェニル)アダマンタンの製造:
窒素雰囲気下に、メチルトリフェニルホスホニウムブロミド4.59g(12.5ミリモル)にカリウムtert−ブトキシド1.97g(17.6ミリモル)を加えた後、脱水THF95mlを加えて室温で約20分間撹拌した。系を0℃に冷却した後、上記(3)で得られた1−(4−ホルミルフェニル)アダマンタン2.61g(10.9ミリモル)の脱水THF溶液45mlを滴下し、室温で一晩撹拌した。ガスクロマトグラフィーにて1−(4−ホルミルフェニル)アダマンタンの消費が完全に確認された後、系に水を加えて反応を停止させた。エバポレートによりTHFを除去した後、エーテルで抽出を3回、有機層を水で2回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して白色固体を得た。これを少量のTHFに溶解し、大量のヘキサン中に注ぎ込むことにより、反応により生成したトリフェニルホスフィンオキシドを沈殿させ、濾別によりトリフェニルホスフィンオキシドを除去後、有機溶媒を減圧留去して3.94gの白色固体を得た。この白色固体をヘキサンを展開溶媒
に用いたシリカゲルグロマトグラフィーにより精製して3.45gの白色粉末を得た。この白色粉末をメタノールから再結晶して、1−(4−ビニルフェニル)アダマンタン2.08g(8.72ミリモル、収率80%、白色針状結晶、mp98−99℃)を得た。
これにより得られた1−(4−ビニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図2に示すとおりであり、NMRスペクトルおよびIR分析のピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.72−1.82(m,6H,C(2)H2),1.91(s,6H,C(4)H2),2.10(s,3H,C(3)H),5.17−5.21(d,1H,CH2=trans,J=11.1Hz),5.68−5.74(d,1H,CH2= cis,J=17.7Hz),6.65−6.75(d,1H,=CH,J=11.1Hz and 17.7Hz),7.31−7.39(m,4H,C(b)H and C(c)H).
・13C−NMR(CDCl3):δ 29.0(C−3),36.2(C−1),36.9(C−2),43.2(C−4),113.1(CH2=),125.1(C−c),126.1(C−b),135.0(C−d),136.7(=CH),151.2(C−a)
・IR(KBr):2917,2903 and 2847cm-1(C−H伸縮振動),1629 cm-1(C=C伸縮振動),1510,1446,1345,988,897,838 and 808cm-1(C−H変角振動)
(5) 上記の結果から、この参考例1で最終的に得られた生成物は、1−(4−ビニルフェニル)アダマンタンであることが確認された。
【0084】
《参考例2》[3−(4−ビニルフェニル)−1,1’−ビアダマンタンの製造]
上記した反応式(IV)に示す方法により、3−(4−ビニルフェニル)−1,1’−ビアダマンタンを次のようにして製造した。
(1)1,1’−ビアダマンタンの製造:
窒素雰囲気下、n−オクタン50mlに1−ブロモアダマンタン25.12g(118.3ミリモル)を溶かし、ナトリウム2.73gを加えた後、140℃に加熱して還流した。ナトリウムが融解すると系は青色を呈した。ガスクロマトグラフィー(GC)で反応を追跡し、1−ブロモアダマンタンの消費が完全に確認された時点(8時間半)で放冷し、ナトリウムをメタノールと水で処理した後、塩酸を加えた。クロロホルムで3回抽出し、水で2回洗浄した後、有機層の溶媒を減圧留去し、それにより得られた白色固体14.1gを180℃、26.7Pa(0.2mmHg)で昇華精製して、1,1’−ビアダマンタン6.96g(収率44%)を得た。
これにより得られた1,1’−ビアダマンタンの1H−NMRスペクトルおよび13CNMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.54−1.69(s,24H,CH2),1.95(s,6H,CH)
・13C−NMR(CDCl3):δ 29.1(C−3),35.3(C−2),36.4(C−1),37.7(C−4)
【0085】
(2)3−ブロモ−1,1’−ビアダマンタンの製造:
上記(1)で得られた1,1’−ビアダマンタン6.23g(23.1ミリモル)に四塩化炭素160mlと臭素80ml(1.56mol)を加え、室温で72時間撹拌した。反応溶液を氷浴中に注ぎ込み、過剰に用いた臭素を亜硫酸水素ナトリウムで処理した後、クロロホルムで3回抽出し、有機層を水で2回洗浄した。有機層を無水硫酸マグネシウムで乾燥した後、硫酸マグネシウムを濾別し、有機溶媒を減圧留去して淡黄色粉末7.96gを得た。これを最初はヘキサンのみで、次にヘキサン/酢酸=20/1、10/1(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて精製して、3−ブロモ−1,1’−ビアダマンタン4.42g(収率56%、白色粉末)を得た。
これにより得られた3−ブロモ−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.53−1.70(m,18H,C(4)H2,C(6)H2,C(2')H2,C(4')H2),1.98(s,3H,C(3')H),2.16−2.33(m,8H,C(2)H2,C(5)H,C(7)H2)
・13C−NMR(CDCl3):δ 29.0(C−3'),32.9(C−5),33.5(C−4),35.5(C−6,C−2'),36.7(C−1'),37.5(C−4'),42.8(C−1'),47.7(C−2),49.3(C−7),69.6(C−3)
【0086】
(3)3−フェニル−1,1’−ビアダマンタンの製造:
窒素雰囲気下で、マグネシウム1.65g(67.8ミリモル)に脱水エーテル15mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、ブロモベンゼン4.9ml(41ミリモル)の脱水エーテル溶液45mlを滴下して室温で3時間撹拌した後、エーテルを減圧留去することにより、フェニルマグネシウムブロミドの灰色固体を得た。この灰色固体に、上記(2)で得られた3−ブロモ−1,1’−ビアダマンタン3.96g(11.3ミリモル)と脱水塩化メチレン35mlを加えて、GCとNMRで反応を追跡しながら3−ブロモ−1,1’−ビアダマンタンの消費が完全に確認されるまで還流(約40時間後)したところで、系を0℃に冷却し、水を加えて反応を停止させ、更に2N塩酸を加えた。塩化メチレンで抽出を3回、有機層を水で3回洗浄し、更に塩化メチレンで2回抽出した。有機溶媒を減圧留去し、4.53gの淡黄色固体を得た。これを40〜60℃、26.7Pa(0.2mmHg)で昇華精製して反応により副生したビフェニルを除去した後、ヘキサンから再結晶して、3−フェニル−1,1’−ビアダマンタン3.43g(収率67%)の白色針状結晶(mp204−20
6℃)を得た。
これにより得られた3−フェニル−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルにおけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.64−1.70(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.85(s,4H,C(4)H2),1.98(s,3H,C(3')H),2.20(s,2H,C(5)H),7.16−7.21(t,1H,C(d)H),7.30−7.40(m,4H,C(b)H,C(c)H)
・13C−NMR(CDCl3):δ 29.0(C−3'),29.5(C−5),34.5(C−7),35.4(C−2'),36.5(C−1'),36.7(C−2),37.0(C−3),37.6(C−4'),37.6(C−1),41.7(C−6),42.7(C−4)
【0087】
(4)3−(4−ブロモフェニル)−1,1’−ビアダマンタンの製造:
上記(3)で得られた3−フェニル−1,1’−ビアダマンタン2.17g(6.26ミリモル)に四塩化炭素70mlを加えた後、臭素20ml(390ミリモル)を加えて室温で2.5時間撹拌した。反応溶液を氷水中に注ぎ込み、過剰の臭素を亜硫酸水素ナトリウムで処理した後、塩化メチレンで3回抽出し、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体2.69gを得た。この一連の操作を再度繰り返して、合計で4.48gの淡黄色固体(収率100%)を得た。これをヘキサンから再結晶して、3−(4−ブロモフェニル)−1,1’−ビアダマンタン3.07g(白色針状結晶、mp184−186℃)を得た。
これにより得られた3−(4−ブロモフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.63−1.72(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.81(s,4H,C(4)H2),1.99(s,3H,C(3')H),2.21(s,2H,C(5)H),7.26−7.29(d,2H,C(c)H),7.44−7.47(d,2H,C(b)H)
・13C−NMR(CDCl3):δ 29.0(C−3'),29.4(C−5),34.4(C−7),35.4(C−2'),36.5(C−1'),36.5(C−2),36.9(C−3),37.6(C−4'),37.6(C−1),41.6(C−6),42.6(C−4)
・IR(KBr):2904,2847cm-1(C−H伸縮振動),1490cm-1(C−Br伸縮振動),1449,1396,1343,803,730cm-1(C−H変角振動)
【0088】
(5)3−(4−ホルミルフェニル)−1,1’−ビアダマンタンの製造:
窒素雰囲気下に、マグネシウム0.34g(14.0ミリモル)に脱水THF10mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。これに、上記(4)で得られた3−(4−ブロモフェニル)−1,1’−ビアダマンタン2.86g(6.73ミリモル)の脱水THF溶液70mlを室温で滴下した後、還流した。還流中に、一部をサンプリングしDMFを加えることでホルミル体の生成と3−(4−ブロモフェニル)−1,1’−ビアダマンタンの消費をNMRで確認することで反応を追跡しながら、3−(4−ブロモフェニル)−1,1’−ビアダマンタンが完全に消費されるまで還流を続けた(約6時間)。次いで、系を0℃に冷却し、脱水DMF3ml(38.8ミリモル)を滴下したところ、滴下と同時に白濁して系は灰色になった。その後、室温で一晩撹拌した後、系に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させ
た。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して淡黄色固体2.29gを得た。これを、初めはヘキサンのみで、その後ヘキサン/酢酸エチル=10/1,10/3,1/1(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて副生した3−フェニル−1,1’−ビアダマンタンを除去し、3−(4−ホルミルフェニル)−1,1’−ビアダマンタン1.49g(収率59%、白色粉末、mp205−207℃)を得た。
これにより得られた3−(4−ホルミルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトル並びにIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.54−1.69(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.85(s,4H,C(4)H2),1.97(s,3H,C(3')H),2.21(s,2H,C(5)H),7.52−7.55(d,2H,C(c)H),7.82−7.85(d,2H,C(b)H),9.98(s,1H,CHO)
・13C−NMR(CDCl3):δ 29.3(C−3'),29.7(C−5),34.7(C−7),35.7(C−2'),36.8(C−1'),36.8(C−2),37.9(C−4'),37.9(C−1),38.2(C−3),41.7(C−6),42.8(C−4)
・IR(KBr):2927,2902,2849cm-1(C−H伸縮振動),1707cm-1(C=O伸縮振動),1447,1346,827,730cm-1(C−H変角振動)
【0089】
(6)3−(4−ビニルフェニル)−1,1’−ビアダマンタンの製造:
窒素雰囲気下に、メチルトリフェニルホスホニウムブロミド1.00g(2.80ミリモル)にカリウムtert−ブトキシド0.43g(3.88ミリモル)を加えた後、脱水THF20mlを加えて室温で約20分間撹拌した。系を0℃に冷却した後、上記(5)で得られた3−(4−ホルミルフェニル)−1,1’−ビアダマンタン0.88g(2.34ミリモル)の脱水THF溶液を60ml滴下し、室温で一晩撹拌した。TLC板(メルク社製「TLCアルミニウムシート シリカゲル60F254」)およびNMRで3−(4−ホルミルフェニル)−1,1’−ビアダマンタンが完全に消費されたのを確認した後、系に水を加えて反応を停止させた。エバポレートによりTHFを除去した後、エーテルで抽出を3回、有機層を水で2回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して白色固体を得た。これを、初めはヘキサンのみで、その後ヘキサン/酢酸エチル=10/1,10/3(v/v)を展開溶媒に用いたシリカゲルクロマトグラフィーにて、トリフェニルホスフィンオキシドを除去した。続いてエタノール/ヘキサン混合溶媒から再結晶し、3−(4−ビニルフェニル)−1,1’−ビアダマンタン0.61g(1.64ミリモル、収率68%、白色針状結晶、mp170−172℃)を得た。
これにより得られた3−(4−ビニルフェニル)−1,1’−ビアダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図3に示すとおりであり、NMRスペクトルおよびIR分析におけるピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.63−1.71(m,20H,C(2)H2,C(6)H2,C(7)H2,C(2')H2,C(4')H2),1.84(s,4H,C(4)H2),1.98(s,3H,C(3')H),2.20(s,2H,C(5)H),5.19−5.23(d,1H,CH2=trans),5.70−5.76(d,1H,CH2= cis),6.68−6.77(d,1H,=CH),7.34−7.41(m,4H,C(b)H,C(c)H).
・13C−NMR(CDCl3):δ 29.0(C−3'),29.5(C−5),34.5(C−7),35.4(C−2'),36.5(C−1'),36.7(C−2),37.0(C−3),37.6(C−4'),37.6(C−1),41.7(C−6),42.7(C−4)
・IR(KBr):2902,2848cm-1(C−H伸縮振動),1629,1510 cm-1(C=C伸縮振動),1446,1343,830cm-1(C−H変角振動)
【0090】
《参考例3》[1−(4−イソプロペニルフェニル)アダマンタンの製造]
(1)4−(1−アダマンチル)クミルアルコールの製造;
窒素雰囲気下、マグネシウム3.32g(137ミリモル)に脱水THF15mlを加えた後、1,2−ジブロモエタンを数滴加えてマグネシウムの表面を活性化した。次いで、参考例1の中間体として得られた1−(4−ブロモフェニルアダマンタン)21.8g(75ミリモル)の脱水THF溶液130mlを室温で滴下した後、還流した。還流中にガスクロマトグラフィーで反応を追跡し、1−(4−ブロモフェニル)アダマンタンが完全に消費されて4−アダマンチル−フェニルマグネシウムブロミドに由来する1−フェニルアダマンタンの生成が確認された時点(約2時間後)で、系を0℃に冷却し、脱水アセトン100ml(1.36モル)を滴下したところ、滴下と同時に白濁した。その後、室温で1時間攪拌した後、系内に水を加えて反応を停止させ、さらに2N塩酸を加えて過剰に用いたマグネシウムを処理した。エバポレートによりTHFおよび過剰なアセトンを除去した後、塩化メチレンで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して23.7gの淡黄色固体を得た。
(2)1−(4−イソプロペニルフェニル)アダマンタンの製造;
上記(1)で得られた4−(1−アダマンチル)クミルアルコール23.7gと触媒量のトルエンスルホン酸(1g)にトルエン200mlを加えて溶解させた。溶解させた後に、系内に発生する水を除去しながら還流し、ガスクロマトグラフィーにより4−(1−アダマンチル)クミルアルコールが完全に消費されたことを確認し、系を室温まで冷却した。その後、系に水および炭酸水素ナトリウム水溶液を順に加えて反応を停止させ、有機層を水で3回洗浄した。有機層に無水硫酸マグネシウムを加えて乾燥し、硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して18.3gの白色固体を得た。該白色固体を、ヘキサンを展開溶媒に用いたシリカゲルクロマトグラフィーにて精製したのち、メタノール/エーテル混合溶媒から再結晶し、1−(4−イソプロペニルフェニル)アダマンタン9.81g(白色固体)を得た。
これにより得られた1−(4−イソプロペニルフェニル)アダマンタンの1H−NMRスペクトルおよび13C−NMRスペクトルは、図4に示すとおりであり、それらのピークは次のとおりであった。
・1H−NMR(CDCl3):δ 1.72−1.82(m,6H,C(2)H2),1.91(s,6H,C(4)H2), 2.10(s,3H,C(3)H),2.15(s,3H,α−CH3),5.04(s,1H,CH2=trans),5.35(s,1H,CH2=cis),7.25−7.45(m,4H,C(b)H and C(c)H)
・13C−NMR(CDCl3):δ 21.9(α−CH3),29.0(C−3),36.1(C−1),36.9 (C−2),43.2(C−4),111.7(CH2=),124.8(C−b),125.3(C−c),138.4(C−a), 143.1(=C(Me)(Ph)),150.8(C−d)
【0091】
《実施例1》[1−(4−ビニルフェニル)アダマンタン単独重合体の製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に参考例1で製造した1−(4−ビニルフェニル)アダマンタン0.3g(1.26ミリモル)を入れ、次に溶媒としてシクロヘキサン20mlを加えて1−(4−ビニルフェニル)アダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.02ml(sec−ブチルリチウムとして0.026ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、1時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)8500、分子量分布(Mw/Mn)1.29であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークは観測されず、1−(4−ビニルフェニル)アダマンタンの重合反応率は99.9%以上であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は237℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は408℃であった。この結果を下記の表2に示す。
【0092】
《実施例2》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(1)の製造]
(1) 3本の乾燥したガラス製反応容器を用意し、各反応容器内の気体を窒素ガスで置換した後、それぞれの反応容器に、参考例1で製造した1−(4−ビニルフェニル)アダマンタン217mg(0.912ミリモル)およびスチレン95mg(0.912ミリモル)を入れ、次に溶媒としてシクロヘキサンを2ml加えて、1−(4−ビニルフェニル)アダマンタンおよびスチレンの溶液を3系列調製した。
(2) 上記(1)で調製した各溶液を攪拌しながら、各反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.028ml(sec−ブチルリチウムとして0.0365ミリモル)を各反応溶液に加えた。sec−ブチルリチウムの添加後、即座に溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、第1の反応溶液にはsec−ブチルリチウムの添加10分後に少量の脱気したメタノールを添加して重合反応を停止させ、第2の反応溶液にはsec−ブチルリチウムの添加20分後に少量の脱気したメタノールを添加して重合反応を停止させ、第3の反応溶液にはsec−ブチルリチウムの添加30分後に少量の脱気したメタノールを添加して重合反応を停止させた。
【0093】
(3) 各反応容器から、反応液の一部をサンプリングし、サンプリングした反応液の一部を乾燥し、それにより得られた半固形状物をテトラヒドロフランに溶解させてGPC測定を行い、またサンプリングした反応液の一部は重クロロホルムで希釈して50℃で1H−NMR測定を行って、生成した共重合体の数平均分子量(Mw)、分子量分布(Mw/Mn)および1−(4−ビニルフェニル)アダマンタンとスチレンの重合反応率を求めた。なお、1−(4−ビニルフェニル)アダマンタンのβ−炭素上のプロトン(trans側)のピークは5.17ppmおよび5.20ppmにダブレットピークとして現れるのに対し、スチレンのβ−炭素上のプロトン(trans側)のピークは5.23ppmおよび5.26ppmにダブレットピークとして現れるため、1H−NMRスペクトル上においてそれぞれを区別することが可能であり、それに基づいて各単量体の重合反応率を測定することができる。結果を下記の表1に示す。
【0094】
【表1】
【0095】
上記の表1の結果から、1−(4−ビニルフェニル)アダマンタンとスチレンの重合反応速度はほぼ同等であることが確認された。
【0096】
(4) 上記(2)において、30分間重合反応を行った第3の反応容器内の反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体[ランダム共重合体(1)]のガラス転移温度(Tg)は180℃であり、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は404℃であった。この結果を下記の表2に示す。
【0097】
《実施例3》[3−(4−ビニルフェニル)−1,1’−ビアダマンタンの単独重合体の製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に参考例2で製造した3−(4−ビニルフェニル)−1,1’−ビアダマンタン0.6g(1.61ミリモル)を入れ、次に溶媒としてシクロヘキサン40mlを加えて3−(4−ビニルフェニル)−1,1’−ビアダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.025ml(sec−ブチルリチウムとして0.033ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、2時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)7900、分子量分布(Mw/Mn)1.34であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークは観測されず、3−(4−ビニルフェニル)−1,1’−ビアダマンタンの重合反応率は99.9%以上であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は245℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は426℃であった。この結果を下記の表2に示す。
【0098】
《実施例4》[1−(4−イソプロペニルフェニル)アダマンタンの単独重合体の製造]
(1) 乾燥したガラス製反応容器内を窒素ガスで置換した後、反応容器内に参考例3で製造した1−(4−イソプロペニルフェニル)アダマンタン0.79g(3.12ミリモル)を入れ、次に溶媒として脱水THF30mlを加えて1−(4−イソプロペニルフェニル)アダマンタンを溶解させた。得られた溶液を攪拌しながら反応容器内の温度を−78℃に冷却した後、sec−ブチルリチウムのシクロヘキサン溶液0.05ml(sec−ブチルリチウムとして0.065ミリモル)を加えた。sec−ブチルリチウムを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を−78℃に保ちながら反応を継続し、sec−ブチルリチウムを添加した後、2時間後に少量の脱気したメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)9200、分子量分布(Mw/Mn)1.04であった。サンプリングした反応液の一部を重クロロホルムで希釈し、50℃で1H−NMR測定を行ったところ、二重結合の存在を示すピークが観測され、芳香族由来のピークとの積分値の比較から、1−(4−イソプロペニルフェニル)アダマンタンの反応率は81%であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノールに注ぎ込み、精製した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体について上記した方法でDSC測定を行ったところ、重合体のガラス転移温度(Tg)は273℃と極めて高く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は360℃であった。この結果を下記の表2に示す。
【0099】
《比較例1》[ポリスチレンの製造]
(1) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、反応容器内に溶媒としてシクロヘキサン40mlを加え、更にsec−ブチルリチウムのシクロヘキサン溶液0.14ml(sec−ブチルリチウムとして0.18ミリモル)を加えた。得られた溶液を撹拌しながら反応容器内の温度を50℃に加温した後、スチレン3g(28.8ミルモル)を加えた。スチレンを添加後、反応溶液は赤色を呈し、アニオン重合が開始されたことを示した。反応容器内の温度を50℃に保ちながら反応を継続し、スチレンを添加した後、1時間後に少量のメタノールを添加し重合反応を停止させた。
(2) 上記(1)で得られた反応液の一部をサンプリングした。サンプリングした反応液の一部を乾燥し、得られた半固形状物をテトラヒドロフランに溶解させ、GPC測定を行ったところ、数平均分子量(Mn)17000、分子量分布(Mw/Mn)1.01であった。
(3) 上記(1)で得られた反応液の残部を大過剰のメタノール中に注ぎ込み、生成した重合体を回収して減圧下に50℃で一晩乾燥した。得られた重合体(ポリスチレン)について上記した方法でDSC測定を行ったところ、ポリスチレンのガラス転移温度(Tg)は98℃と低く、また上記した方法で熱質量分析を行ったところ、10%質量減少温度は397℃であった。この結果を下記の表2に示す。
【0100】
【表2】
【0101】
上記の表2にみるように、重合体のガラス転移温度(Tg)が、比較例1のポリスチレン(通常のポリスチレン)では98℃であるのに対して、実施例1の1−(4−ビニルフェニル)アダマンタン単独重合体では237℃、実施例2の1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(1)では180℃、実施例3の3−(4−ビニルフェニル)−1,1’−ビアダマンタン単独重合体では245℃、実施例4の1−(4−イソプロペニルフェニル)アダマンタン単独重合体では273℃であって、実施例1〜4の重合体はポリスチレンに比べてガラス転移温度(Tg)が大幅に高い。
また、10%質量減量温度が、比較例1のポリスチレンでは397℃であるのに対して、実施例1の1−(4−ビニルフェニル)アダマンタン単独重合体では408℃、実施例2の1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体では404℃、実施例3の3−(4−ビニルフェニル)−1,1’−ビアダマンタン単独重合体では426℃であり、比較例1のポリスチレンに比べて10%質量減少温度が高い。
かかる点から、上記の一般式(I)で表されるスチレン系誘導体(I)に由来する構造単位を有する本発明の重合体が、耐熱性に優れていることが裏付けられる。
【0102】
《実施例5》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(2)の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、通常のアニオン重合法により調製したポリスチレン[数平均分子量(Mn)5730、分子量分布(Mw/Mn)1.03]0.4g(スチレンユニットとして3.85ミリモル)および1−ブロモアダマンタン1g(4.64ミリモル)を入れ、次に溶媒として塩化メチレン10ml、触媒として塩化アルミニウム20mg(0.15ミリモル)を加えた。得られた溶液を16時間還流したのち、系を室温まで冷却し、水を加えて反応を停止させた。反応後、クロロホルムで抽出を3回、有機層を水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥させた。硫酸マグネシウムを濾別した後、有機溶媒を減圧留去して1.13gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノール中に注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して、白色粉末0.46gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、ポリスチレンにおける14%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0103】
《実施例6》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(3)の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、1−ブロモアダマンタン83mg(0.39ミリモル)を入れ、次に溶媒として塩化メチレン5mLを加えて溶解させた。アルミホイルで遮光し、0℃に冷却したのちトリフルオロメタンスルホン酸銀0.12g(0.47ミリモル)を加え3時間反応させ、ガスクロマトグラフィーにより1−ブロモアダマンタンが完全に消費され、トリフルオロメタンスルホン酸(1−アダマンチル)が生成したことを確認した。ここに通常のアニオン重合法により調製したポリスチレン[数平均分子量(Mn)5730、分子量分布(Mw/Mn)1.03]0.2g(スチレンユニットとして1.92ミリモル)の塩化メチレン溶液2mlを加え、系を室温に戻し3時間攪拌を行い、水を加えて反応を停止させた。反応後、クロロホルムで3回抽出、有機層をチオ硫酸ナトリウム水溶液で3回洗浄、次いで水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥した。硫酸マグネシウムを濾別後、有機溶媒を減圧留去して0.24gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノールに注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して、白色粉末0.15gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、10%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0104】
《実施例7》[1−(4−ビニルフェニル)アダマンタン/スチレンランダム共重合体(4)の製造]
1−ブロモアダマンタン165mg(0.77ミリモル)、トリフルオロメタンスルホン酸銀0.23g(0.90ミリモル)を用いたこと以外は、実施例6と同様にして白色粉末を得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、22%のスチレンユニットにアダマンチル基が導入されたことを確認した。
【0105】
《実施例8》[1−(4−イソプロペニル)アダマンタン/α−メチルスチレンランダム共重合体の製造]
乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、1−ブロモアダマンタン73mg(0.34ミリモル)を入れ、次に溶媒として塩化メチレン5mlを加えて溶解させた。アルミホイルで遮光し、0℃に冷却したのちトリフルオロメタンスルホン酸銀0.1g(0.39ミリモル)を加えて3時間反応させ、ガスクロマトグラフィーにより1−ブロモアダマンタンが完全に消費され、トリフルオロメタンスルホン酸(1−アダマンチル)が生成したことを確認した。ここに通常のアニオン重合法により調製したポリ(α−メチルスチレン)[数平均分子量(Mn)9300、分子量分布(Mw/Mn)1.04]0.2g(α−メチルスチレンユニットとして1.69ミリモル)の塩化メチレン溶液2mlを加え、系を室温に戻し1時間攪拌を行い、水を加えて反応を停止させた。反応後、クロロホルムで3回抽出、有機層をチオ硫酸ナトリウム水溶液で3回洗浄、次いで水で3回洗浄し、有機層に無水硫酸マグネシウムを加えて乾燥した。硫酸マグネシウムを濾別後、有機溶媒を減圧留去して0.18gの白色粉末を得た。この白色粉末を少量のTHFに溶解させた後、大過剰のメタノールに注ぎ込み、生成した重合体を回収して50℃で一晩乾燥して白色粉末0.15gを得た。得られた白色粉末の1H−NMR測定を行い、芳香族由来の1H核(6−8ppm)の積分値と、環構造、主鎖構造に由来する1H核(1−3ppm)の積分値の比から、20%のα−メチルスチレンユニットにアダマンチル基が導入されたことを確認した。
【0106】
上記した実施例5〜8の結果が示すように、入手が容易なポリスチレン、ポリ(α−メチルスチレン)を基質として用いた芳香族求電子置換反応によっても本発明の重合体を得ることが可能である。
【0107】
《実施例9》[{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製容器内の気体を窒素ガスで置換した後、1−(4−ビニルフェニル)アダマンタン2.087g(8.76ミリモル)およびスチレン0.912g(8.76ミリモル)を入れ、次に溶媒としてシクロヘキサン25mlを加えて1−(4−ビニルフェニル)アダマンタンおよびスチレンを溶解させて単量体混合物溶液を調製した。この単量体混合物溶液の密度が0.7804g/mlであり、かかる点から、溶液中の1−(4−ビニルフェニル)アダマンタンおよびスチレンの濃度はいずれも0.304モル/リットルであった。
(2) 乾燥したガラス製反応容器内の気体を窒素ガスで置換した後、シクロヘキサン50mlを入れて50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.081ml(sec−ブチルリチウムとして0.105ミリモル)を加え、ここに上記(1)で調製した単量体混合物溶液の8.65ml[1−(4−ビニルフェニル)アダマンタン2.63ミリモル、スチレン2.63ミリモルを含有]を加えて、50℃で30分間重合した後、反応液の一部をサンプリングし、GPC測定を行ったところ数平均分子量(Mn)6900、分子量分布(Mw/Mn)1.07の重合体が生成していた。
(3) 続いて、前記(2)の反応容器に、イソプレン4.2g(61.7ミリモル)を加えて50℃で1.5時間重合を行った。反応液の一部をサンプリングし、GPC測定を行ったところ、数平均分子量(Mn)107000、平均分子量1.02の重合体が生成していた。続いて反応容器に、上記(1)で調製した混合単量体溶液の8.65ml[1−(4−ビニルフェニル)アダマンタン2.63ミリモル、スチレン2.63ミリモルを含有]を加えて50℃で1時間重合反応を行った後、脱気したメタノールを少量添加して重合を停止した。
【0108】
(4) 上記(3)で得られた反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)114000、分子量分布(Mw/Mn)1.03のブロック共重合体が生成していた。得られたブロック共重合体の溶液(反応液)を水で洗浄した後、メタノール/アセトン混合溶媒で沈殿させて回収した。これにより得られたブロック共重合体の1H−NMR測定を行ったところ、得られたブロック共重合体は、{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体であり、ポリイソプレンブロックには3,4−結合が5.5%含まれており、ブロック共重合体の質量に基づく1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロックの含有割合は28.7質量%であった。
【0109】
《実施例10》[{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製容器内の気体を窒素ガスで置換した後、1−(4−ビニルフェニル)アダマンタン2g(8.39ミリモル)を入れ、次に溶媒としてシクロヘキサン15mlを加えて1−(4−ビニルフェニル)アダマンタンを溶解させて単量体溶液を調製した。この単量体溶液の密度は0.795g/mlであり、これより単量体溶液中での1−(4−ビニルフェニル)アダマンタンの濃度は0.504モル/リットルであった。(2) 乾燥したガラス製反応容器内の気体を窒素ガスで置換し、シクロヘキサン50mlを入れて50℃に加温した後、sec−ブチルリチウムのシクロヘキサン溶液0.058ml(sec−ブチルリチウムとして0.076ミリモル)を加え、ここに上記(1)で調製した単量体溶液の7.5ml[1−(4−ビニルフェニル)アダマンタンとして0.9g(3.78ミリモル)]を加えて50℃で30分間重合した後、反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)8500、分子量分布(Mw/Mn)1.29の重合体が生成していた。
(3) 続いて反応容器に、イソプレン4.2g(61.7ミリモル)を加えて50℃で1.5時間重合を行った。反応液の一部をサンプリングしてGPC測定を行ったところ、数平均分子量(Mn)100000、分子量分布(Mw/Mn)1.05の重合体が生成していた。続いて反応容器に、上記(1)で調製した単量体溶液の7.5ml[1−(4−ビニルフェニル)アダマンタンとして0.9g(3.78ミリモル)]を加えて50℃で2時間重合反応を行った後、脱気したメタノールを少量添加して重合を停止させた。
【0110】
(4) 上記(3)で得られた反応液の一部をサンプリングしGPC測定を行ったところ、数平均分子量(Mn)110000、分子量分布(Mw/Mn)1.03のブロック共重合体が生成していた。得られたブロック共重合体の溶液(反応液)を水で洗浄した後、メタノール/アセトン混合溶媒で沈殿させてブロック共重合体を回収した。これにより得られたブロック共重合体の1H−NMR測定を行ったところ、{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体であり、ポリイソプレンブロックには3,4−結合が5.7%含まれており、ブロック共重合体の質量に基づく1−(4−ビニルフェニル)アダマンタン重合体ブロックの含有割合は24.7質量%であった。
【0111】
《実施例11》[水添{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 乾燥したガラス製反応容器に、実施例9で製造したトリブロック共重合体[{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体]3.6g、酸化防止剤(日本チバ・ガイギー社製「Irganox 1010」)0.36gおよびp−トルエンスルホン酸ヒドラジド20gを入れて30分間真空脱気を行った。その後、反応容器内に窒素ガスを導入し、キシレン250mlを加えて攪拌して120℃に加熱した。溶液は当初懸濁状態であったが、120℃まで加熱すると均一の溶液となるともに、窒素ガス、水素ガスの発生による気泡の発生が確認された。反応液の温度をそのまま120℃に保って15時間反応を継続した。反応終了後、反応液を室温まで冷却した後、大過剰のメタノールに投入して、水添トリブロック共重合体を沈殿させて回収した。回収した水添トリブロック共重合体をトルエンに溶解させた後、メタノールで再沈する作業をさらに3回繰り返して精製し、それにより得られた水添トリブロック共重合体を50℃で一晩真空乾燥した。
【0112】
(2) 上記(1)で得られた水添トリブロック共重合体について1H−NMR測定を行った結果、イソプレン中の二重結合に由来するするピークの大部分が消失し、水添率は98%であった。また水添トリブロック共重合体のGPC測定を行ったところ、ゲル化や分解を示すピークは観測されず、数平均分子量(Mn)122000、分子量分布(Mw/Mn)1.03であった。
(3)(i) 上記(1)で得られた水添トリブロック共重合体をトルエンに溶解し、平らな台上に設置したテフロン(登録商標)シート上でキャストした後、乾燥して厚さ1mmのシートを製造した。
(ii) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。その結果を図5に示す。
(iii) 上記(i)で得られたシートから、図1に示す形状および寸法のダンベル試験片を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iv) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0113】
《実施例12》[水添{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体の製造]
(1) 実施例10で得られたトリブロック共重合体[{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体]を3.6g用いた以外は実施例11と同様にして水素添加処理、水添トリブロック共重合体の回収および精製を行って、水添{1−(4−ビニルフェニル)アダマンタン重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン重合体ブロック}よりなるトリブロック共重合体を製造した。
(2) 上記(1)で得られた水添トリブロック共重合体について1H−NMR測定を行った結果、イソプレン中の二重結合に由来するするピークの大部分が消失し、水添率は98%であった。また水添トリブロック共重合体のGPC測定を行ったところ、ゲル化や分解を示すピークは観測されず、数平均分子量(Mn)125000、分子量分布(Mw/Mn)1.03であった。
【0114】
(3)(i) 上記(1)で得られた水添トリブロック共重合体をトルエンに溶解し、平らな台上に設置したテフロン(登録商標)シート上でキャストした後、乾燥して厚さ1mmのシートを製造した。
(ii) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’および損失正接tanδを測定した。その結果を図5に示す。
(iii) 上記(i)で得られたシートから、図1に示す形状および寸法のダンベル試験片を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iv) 上記(i)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0115】
《比較例2》[ポリスチレン−水添ポリイソプレン−ポリスチレンよりなるトリブロック共重合体の製造]
(1) 実施例11の水添{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}−{ポリイソプレンブロック}−{1−(4−ビニルフェニル)アダマンタン/スチレン共重合体ブロック}よりなるトリブロック共重合体の代りに、ポリスチレン−水添ポリイソプレン−ポリスチレンよりなるトリブロック共重合体(株式会社クラレ製「セプトン2002」、スチレン単位含量30質量%)を用いて、実施例11と同様にして厚さ1mmのシートをつくった。
(2)(i) 上記(1)で得られたシートから、縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で動的粘弾性試験を行って、貯蔵弾性率E’、損失弾性率E’’及び損失正接tanδを測定した。その結果を図5に示す。
(ii) 上記(1)で得られたシートから、図1に示す形状および寸法のダンベル試験片
を採取し、該試験片を用いて上記した方法で引張試験を行って、100%モジュラス(M100)、300%モジュラス(M300)、破断強度(TB)および破断伸度(EB)を測定した。その結果を下記の表3に示す。
(iii) 上記(1)で得られたシートから縦×横×厚さ=20mm×5mm×1mmの試験片を採取し、該試験片を用いて上記した方法で応力緩和特性の測定を行った。結果を図6に示す。
【0116】
【表3】
【0117】
上記の表3の結果にみるように、実施例11および実施例12の水添トリブロック共重合体(熱可塑性エラストマー)は、スチレン系誘導体由来の構造単位(I−A)を有する重合体ブロックを有していることにより、構造単位(I−A)を持たない比較例2の水添トリブロック共重合体(熱可塑性エラストマー)に比べて、100%モジュラス(M100)および300モジュラス(M300)が小さくて伸縮性に優れ、応力緩和特性(Eb)にも優れている。しかも、実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて破断強度TB(引張強度)が高く、引張強度にも優れている。
また、図5のグラフにみるように、実施例11および実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて、高い温度まで貯蔵弾性率E’が保持されており、比較例2の水添トリブロック共重合体が使用不可能な温度域においてもエラストマーとしての使用が可能である。
さらに、図6のグラフにみるように、実施例11および実施例12の水添トリブロック共重合体は、比較例2の水添トリブロック共重合体に比べて、応力緩和性に優れている。
【産業上の利用可能性】
【0118】
本発明の製造方法により得られる重合体は、耐熱性に極めているため、その特性を活かして、家電製品の筐体、OA機器、食品容器、梱包材、食品トレー、発泡体、自動車部品、電気・電子部品、フィルム、シート、医療材料、伸縮部材などの種々の用途に好適に使用することができる。
【特許請求の範囲】
【請求項1】
下記の一般式(I);
【化1】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または前記スチレン系誘導体(I)および他の重合性単量体を用いて重合を行うことを特徴とする、下記の一般式(I−A);
【化2】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法。
【請求項2】
一般式(I)および一般式(I−A)におけるR1が多環構造を有する橋架け脂環式炭化水素基である請求項1に記載の重合体の製造方法。
【請求項3】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)を10質量%以上の割合で有する重合体を製造するものである請求項1または2に記載の重合体の製造方法。
【請求項4】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)の単独からなるか、または該構造単位(I−A)および他の重合性単量体に由来する構造単位を有する重合体を製造するものである請求項1〜3のいずれか1項に記載の重合体の製造方法。
【請求項5】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)よりなる重合体ブロックAと、他の重合性単量体に由来する構造単位よりなる重合体ブロックBを有するブロック共重合体を製造するものである請求項1〜4のいずれか1項に記載の重合体の製造方法。
【請求項6】
重合体ブロックAの含有割合が、ブロック共重合体の質量に基づいて10質量%以上であるブロック共重合体を製造するものである請求項5に記載の重合体の製造方法。
【請求項7】
重合体ブロックBのガラス転移温度が重合体ブロックAのガラス転移温度よりも低いブロック共重合体を製造するものである請求項5または6に記載の重合体の製造方法。
【請求項8】
重合体ブロックBが共役ジエンに由来する構造単位を主体とする共役ジエン系重合体または該共役ジエン系重合体の水素添加物よりなる重合体ブロックであるブロック共重合体を製造するものである請求項5〜7のいずれか1項に記載の重合体の製造方法。
【請求項9】
一般式(I)で表されるスチレン系誘導体(I)として、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンの少なくとも1種を用いる請求項1〜8のいずれか1項に記載の重合体の製造方法。
【請求項10】
有機リチウム化合物を重合開始剤として用いてアニオン重合を行う請求項1〜9のいずれか1項に記載の重合体の製造方法。
【請求項11】
下記の一般式(II);
【化3】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるか、または当該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多環構造を有する脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のスチレン由来のベンゼン環に基R1を導入する工程を含むことを特徴とする、下記の一般式(I−A);
【化4】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法。
【請求項12】
芳香族求電子置換反応が、フリーデル・クラフツ反応である請求項11に記載の製造方法。
【請求項1】
下記の一般式(I);
【化1】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系誘導体(I)を単独で用いるか、または前記スチレン系誘導体(I)および他の重合性単量体を用いて重合を行うことを特徴とする、下記の一般式(I−A);
【化2】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法。
【請求項2】
一般式(I)および一般式(I−A)におけるR1が多環構造を有する橋架け脂環式炭化水素基である請求項1に記載の重合体の製造方法。
【請求項3】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)を10質量%以上の割合で有する重合体を製造するものである請求項1または2に記載の重合体の製造方法。
【請求項4】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)の単独からなるか、または該構造単位(I−A)および他の重合性単量体に由来する構造単位を有する重合体を製造するものである請求項1〜3のいずれか1項に記載の重合体の製造方法。
【請求項5】
一般式(I−A)で表されるスチレン系誘導体由来の構造単位(I−A)よりなる重合体ブロックAと、他の重合性単量体に由来する構造単位よりなる重合体ブロックBを有するブロック共重合体を製造するものである請求項1〜4のいずれか1項に記載の重合体の製造方法。
【請求項6】
重合体ブロックAの含有割合が、ブロック共重合体の質量に基づいて10質量%以上であるブロック共重合体を製造するものである請求項5に記載の重合体の製造方法。
【請求項7】
重合体ブロックBのガラス転移温度が重合体ブロックAのガラス転移温度よりも低いブロック共重合体を製造するものである請求項5または6に記載の重合体の製造方法。
【請求項8】
重合体ブロックBが共役ジエンに由来する構造単位を主体とする共役ジエン系重合体または該共役ジエン系重合体の水素添加物よりなる重合体ブロックであるブロック共重合体を製造するものである請求項5〜7のいずれか1項に記載の重合体の製造方法。
【請求項9】
一般式(I)で表されるスチレン系誘導体(I)として、1−(4−ビニルフェニル)アダマンタン、3−(4−ビニルフェニル)−1,1’−ビアダマンタンおよび1−(4−イソプロペニルフェニル)アダマンタンの少なくとも1種を用いる請求項1〜8のいずれか1項に記載の重合体の製造方法。
【請求項10】
有機リチウム化合物を重合開始剤として用いてアニオン重合を行う請求項1〜9のいずれか1項に記載の重合体の製造方法。
【請求項11】
下記の一般式(II);
【化3】
(式中、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表されるスチレン系化合物(II)を単独で用いるか、または当該スチレン系化合物(II)および他の重合性単量体を用いて得られるスチレン系重合体に、下記の一般式(III);
R1−X (III)
(式中、R1は多環構造を有する脂環式炭化水素基、Xは基R1を芳香族求電子置換反応によってスチレン系重合体中のベンゼン環に結合させる作用を有する基を示す。)
で表される化合物(III)を、芳香族求電子置換反応させて、スチレン系重合体中のスチレン由来のベンゼン環に基R1を導入する工程を含むことを特徴とする、下記の一般式(I−A);
【化4】
(式中、R1は多環構造を有する脂環式炭化水素基、R2は水素原子、炭素数1〜10のアルキル基またはアリール基を示す。)
で表される、スチレン系誘導体由来の構造単位(I−A)を有する重合体の製造方法。
【請求項12】
芳香族求電子置換反応が、フリーデル・クラフツ反応である請求項11に記載の製造方法。
【図1】

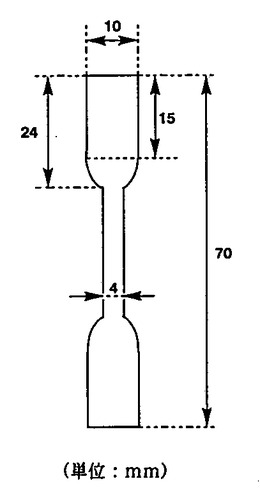
【図2】
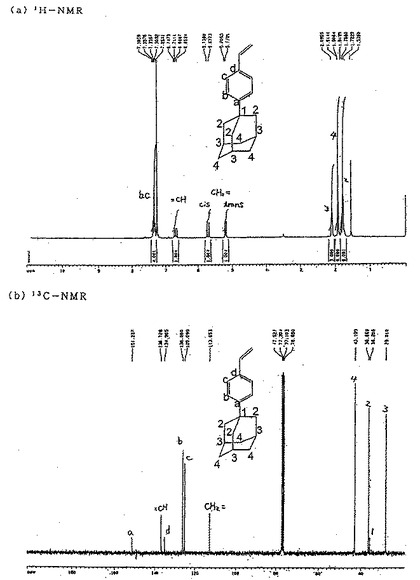
【図3】
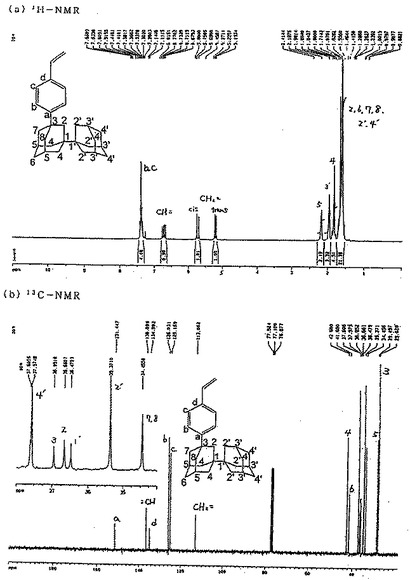
【図4】
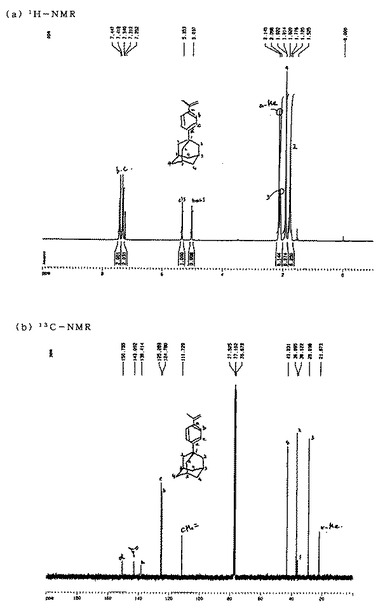
【図5】
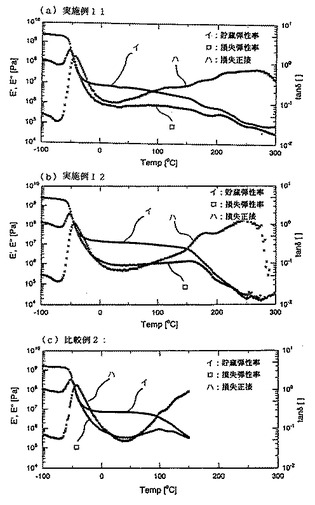
【図6】
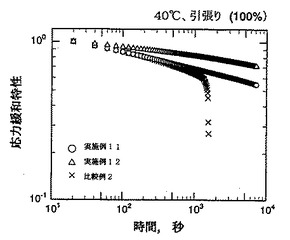
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2011−252164(P2011−252164A)
【公開日】平成23年12月15日(2011.12.15)
【国際特許分類】
【出願番号】特願2011−174585(P2011−174585)
【出願日】平成23年8月10日(2011.8.10)
【分割の表示】特願2005−244039(P2005−244039)の分割
【原出願日】平成17年8月25日(2005.8.25)
【出願人】(000001085)株式会社クラレ (1,607)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
【公開日】平成23年12月15日(2011.12.15)
【国際特許分類】
【出願日】平成23年8月10日(2011.8.10)
【分割の表示】特願2005−244039(P2005−244039)の分割
【原出願日】平成17年8月25日(2005.8.25)
【出願人】(000001085)株式会社クラレ (1,607)
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【Fターム(参考)】
[ Back to top ]