
肥満および糖尿病の有望な治療薬としてのグレリンO−アシルトランスフェラーゼ阻害剤の合成方法および使用方法
【課題】本発明は、グレリンO−アシルトランスフェラーゼの阻害剤およびその製造方法および使用方法を提供する。
【解決手段】いくつかの実施形態では、本発明は、例えば、肥満および糖尿病の治療に有効であり得るグレリンO−アシルトランスフェラーゼの2基質類似体阻害剤を提供する。
【解決手段】いくつかの実施形態では、本発明は、例えば、肥満および糖尿病の治療に有効であり得るグレリンO−アシルトランスフェラーゼの2基質類似体阻害剤を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願で記載した研究は、国立衛生研究所の助成金U54 RR020839およびR01 AG19186によって支援された。合衆国政府は、本出願に一定の権利を有する。
【0002】
本出願は、全ての目的のために参照により全体を本明細書に組み込まれる2008年10月3日出願の米国仮出願第61/102,549号に対する優先権を主張する。
【0003】
本発明は、グレリンO−アシルトランスフェラーゼの阻害剤およびその製造方法および使用方法に関する。
【背景技術】
【0004】
過去30年にわたり西欧社会において体重過剰者の割合が増加し続けていることは、罹病率のかなりの超過と関連があり、重大な公衆衛生問題として広く認識されている。この問題に取り組むために、体重増加に神経内分泌が寄与していることを明らかにするたゆみない努力を重ねている。レプチン(1)の単離を始めとして、中枢および末梢に作用して体重に影響を及ぼす一連のホルモンが発見されてきた。中でも、胃ペプチドホルモン、アシル−グレリンは、体重増加の重要な刺激因子(2〜5)およびグルコース恒常性の調節因子(6〜8)として非常に興味を持たれてきた。治療の発展における様々な戦略の中で、アシル−グレリンの拮抗作用が記載されているが(9)、未だに臨床的に有益性なものは出現していない。
【0005】
アシル−グレリンの生合成には、グレリンペプチドの3位のセリンの通常にはない翻訳後オクタノイル化が関与する。このオクタノイル化は、成長ホルモン分泌促進物質受容体(GHSR)との相互作用を介して行われる生理活性に必要である。このエステル化を担う酵素、グレリンO−アシルトランスフェラーゼ(GOAT)が最近クローニングされた(10、11)。
【発明の概要】
【発明が解決しようとする課題】
【0006】
当業界では依然として、肥満および糖尿病の治療に使用するための改善された治療薬、特に神経内分泌経路を標的とした治療薬が必要とされている。毎週推定10,000例の早死の原因となっている世界的な肥満の流行に対処する中で、効果的な食欲抑制薬物療法は大規模に命を救う可能性があり得る。
【課題を解決するための手段】
【0007】
本発明者らは、GOATの阻害剤が肥満患者の満腹感および体重減少を誘導できることを発見した。さらに、アシル化グレリンを遮断すると膵島のグルコース応答性を改善することができ、したがってGOAT阻害剤はまた、II型糖尿病患者集団で使用するための有効な治療薬として期待される。
【0008】
これらの目的およびその他の目的は、2基質類似体グレリンO−アシルトランスフェラーゼ(GOAT)阻害剤、例えば、インビトロ、培養細胞およびマウスにおいて強力、選択的かつ効果的であるGO−CoA−Tatなどを提供することによって実現される。GO−CoA−Tatなどの化合物の投与によって、グルコース負荷に対するインスリン応答が増強され、高脂肪食を与えられた対象において統計学的に有意な体重減少が導かれる。例えば、GO−CoA−Tatによる治療はまた、除脂肪量よりも体脂肪量に対して選択的な減少を導く。
【0009】
一態様では、本発明は、膜結合O−アシルトランスフェラーゼ(mBOAT)膜タンパク質、例えば、グレリンO−アシルトランスフェラーゼを阻害することができる化合物を提供する。いくつかの実施形態では、化合物は下式を有する。
【化1】

【0010】
式中、Rは存在しないか、あるいは置換もしくは非置換のアルキル基、置換もしくは非置換のペプチド鎖、置換もしくは非置換の多糖、またはペプトイド残基(例えば、N−メチルもしくはその他のN−アルキル置換グリシンなど)からなる群から選択される有機部分である。Rは、ペプチド鎖の場合、例えば、1〜20個のアミノ酸残基の鎖であってもよい。いくつかの実施形態では、Rはグレリン残基4〜28、または4〜15、または4〜10または4〜5に対応するペプチド鎖である。
【0011】
本明細書では、「アミノ酸」は、天然アミノ酸および合成アミノ酸の両方を含む。これらには、特に、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン酸、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、トレオニン、トリプトファン、チロシン、バリン、5−ヒドロキシリシン、4−ヒドロキシプロリン、チロキシン、3−メチルヒスチジン、ε−N−メチルリシン、ε−N,N,N−トリメチルリシン、アミノアジピン酸、γ−カルボキシグルタミン酸、ホスホセリン、ホスホトレオニン、ホスホチロシン、N−メチルアルギニンおよびN−アセチルリシンが含まれる。アルファアミノ酸のR型およびL型の両方ならびにR型およびL型の混合物が企図される。さらに可能性のある置換基には、アミノ基とカルボキシル基の間に2個以上の−CH2−基(通常2〜4個)が存在するアミノ酸およびそれらの誘導体が含まれる。したがって、アミノ酸残基は、アルファ、ベータ、ガンマまたはさらに高級な(higher order)アミノ酸の残基であることができ、好ましくはその他に関しては天然に生じるアミノ酸に対応する残基であることができる。本明細書では、「アミノ酸残基」には、例えば、このような鎖が直鎖であろうと枝分かれ鎖であろうと、アミノ酸配列またはその他の鎖構造に組み込まれた、前記に挙げた任意のアミノ酸が包含される。
【0012】
別の態様では、本発明は組成物を提供する。例えば、組成物は本発明の化合物および薬学的に許容される賦形剤または担体を含むことができる。
【0013】
さらに別の態様では、本発明は本発明の化合物の調製方法を提供する。例えば、この方法は、3〜15個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、D4-Tatがペプチド配列の一末端に結合している工程と、ブロモオクタノイル化中間体を得るために、1または複数個の反応容器中でグレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、前記ブロモオクタノイル化中間体と補酵素Aを一緒にする工程とを含むことができる。
【0014】
さらに別の態様では、本発明は本発明の化合物および組成物を使用して、疾患、障害または症状を治療する方法を提供する。いくつかの実施形態では、この方法は肥満を治療するために使用される。これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。本発明は、例えば、対象の除脂肪量(lean mass)よりも大きく脂肪量減少をもたらすことができるか、または対象におけるアシル−グレリンに対するグレリンの比の増加をもたらすことができる。いくつかの実施形態では、この方法は糖尿病、例えば、II型糖尿病を治療するために使用することができる。これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。これらの方法は、例えば、対象におけるインスリン産生増加、または対象におけるグルコース曝露に対する応答性増加、または対象における脱共役タンパク質2(UCP−2)mRNAレベルの減少をもたらすことができる。いくつかの実施形態では、この方法は過敏性腸症候群を治療するために使用することができる。これらの方法は、本発明の化合物の1または複数種の治療有効量を含む組成物を対象に投与することを含むことができる。
【0015】
別の態様では、本発明はmBOAT関連疾患の治療方法を提供する。いくつかの実施形態では、これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。
【0016】
さらに別の態様では、本発明はキットを提供する。例えば、このキットは、本明細書で開示する化合物および組成物の治療有効量の少なくとも1用量を含む組成物を含むことができる。いくつかの実施形態では、このキットはまた、さらなる体重減少治療薬の少なくとも1用量を含む。
【図面の簡単な説明】
【0017】
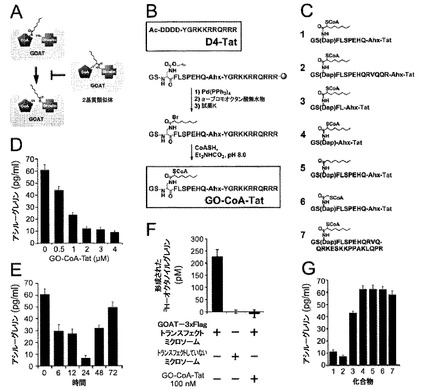
【図1】GO−CoA−Tatは、GOATを阻害し、アシルグレリンレベルを低下させる2基質阻害剤である。(A)2基質類似体阻害剤が、2種類の基質を共有結合的に一緒にし(covalently combine)、それによって効力および特異性を高めることを示す図である。(B)D4-Tatの構造ならびに2基質阻害剤の合成スキームを示した図であり、2基質阻害剤は、3つの成分またはそれらの実質的な同等物:補酵素A;オクタノイル化部分、例えば、置換または非置換のグレリンペプチドおよび;Tatペプチドからなる。(C)本発明による化合物の構造を示した図である。(D)24時間インキュベートした後の、HEL細胞溶解物(2.5×106細胞)における、GO−CoA−Tatによるアシル−グレリンレベルの用量応答性減少を示した図である。(E)HEL細胞溶解物(2.5×106細胞)中におけるGO−CoA−Tat 2μMの経時的(temporal)阻害を示した図である。(F)インビトロアッセイで、グレリン−O−アシルトランスフェラーゼ(GOAT)はGO−CoA−Tat 100nMによって阻害されることを示した図である。(G)図1Cに挙げた7種類の化合物の1つと24時間インキュベートした後の、HEL細胞溶解物(2.5×106細胞)におけるアシル−グレリンレベル(pg/ml)を示した図である。
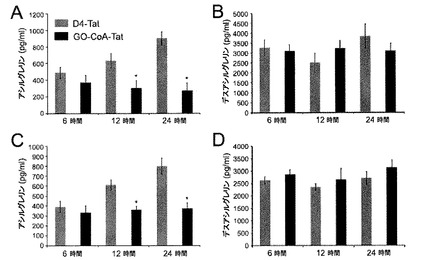
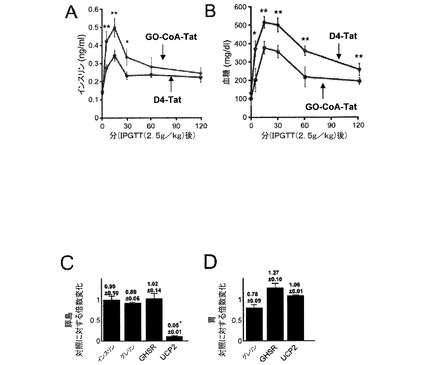
【図2】GO−CoA−Tatは、マウスにおいて、循環アシル−グレリンレベルを低下させるが、デスアシル−グレリンレベルは低下させないことを示した図である。(A)GO−CoA−Tat 29mg/kgで処理したマウス(n=3)は、D4-Tatで処理した対照マウスと比較して、アシル−グレリンの統計学的に有意な減少を示した。(B)12時間後および24時間後のデスアシル−グレリンレベルは、GO−CoA−Tatで処理したマウスでは同等モルのD4-Tatで処理した対照マウス(n=3)に対して統計学的に有意な差を示さなかったことを示す図である。GO−CoA−Tat 15mg/kgでは、阻害はより小さいが類似の結果が認められた(C,D)。
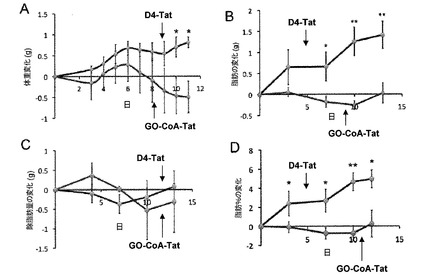
【図3】[マウスの体重および体組成に対するGO−CoA−Tatの効果を示す図である。GO−CoA−Tat 29mg/kgで処理したマウス(n=3)は、D4-Tatで処理したマウスに対して、(A)体重の統計学的に有意な減少を示し(*<0.05、3日間の標準誤差)、(B)体脂肪のグラムの統計学的に有意な減少を示し、(C)除脂肪量の統計学的に有意な差を示さず、(D)体脂肪の割合の統計学的に有意な減少を示した(*<0.05、**<0.005)。
【図4】GO−CoA−Tatはインスリンを増加させ、グルコースレベルを減少させ、UCP2mRNAを下方制御することを示した図である。経口的にグルコースを負荷する24時間前に化合物を投与(1g/kg)した場合、腹腔内グルコース負荷試験(IPGTT)(2.5g/kg)後30分毎に血糖値およびインスリン値を測定したとき、GO−CoA−Tat 29mg/kgで処理したマウス(n=4)は、D4-Tatで処理した対照マウス(n=6)と比較して、(A)インスリン分泌の統計学的に有意な増加を示し、(B)血糖値の統計学的に有意な減少を示した。分離の24時間前に阻害剤で処理したマウスから分離した、(C)膵島のQRT−PCRおよび(D)胃底部のQRT−PCRを示す;mRNA発現を単離し、対照(n=3)と比較して測定した。
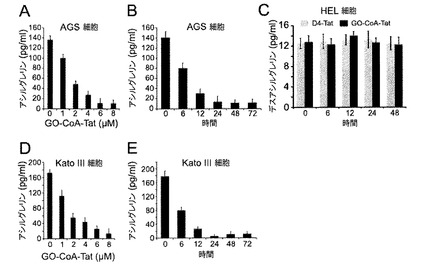
【図5】GO−CoA−Tatは、AGSおよびKatoIII細胞においてアシル−グレリンレベルを低下させるが、デスアシル−グレリンレベルは低下させない。(A)AGS細胞溶解物(2.5×106細胞)において、様々な濃度のGO−CoA−Tatで24時間インキュベートした後のアシル−グレリン(pg/ml)の用量応答性阻害を示した図である。(B)AGS細胞溶解物(2.5×106細胞)におけるGO−CoA−Tat 6μMの経時的阻害を示した図である。(C)HEL細胞溶解物(2.5×106細胞)中において、様々な濃度のGO−CoA−TatおよびD4-Tatと24時間インキュベートした後のデスアシル−グレリン(pg/ml)の用量応答性応阻害を示した図である。KatoIII細胞において、(D)GO−CoA−Tatで24時間インキュベートした後の用量応答性阻害および(E)GO−CoA−Tat 6μMの経時的阻害を示した図である。
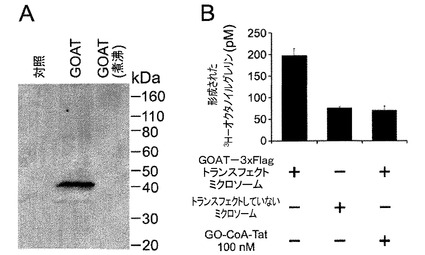
【図6】GOATのインビトロアッセイの確立。GOATはHEK293TGnTI(−)細胞から単離されたミクロソーム内に存在した。HEK293T GnTI(−)細胞に、GOAT−3xFlagまたは空のベクター(対照)をトランスフェクトし、ミクロソームは実施例で説明したように調製した。(A)トランスフェクトした細胞およびトランスフェクトしていない細胞のミクロソーム(50μgのタンパク質)を抗FLAGモノクローナル抗体10μg/mLでイムノブロット分析した図である。50℃でインキュベートしたGOATトランスフェクトミクロソームではSDS−PAGEにおいて5分で色素負荷したが、同じ時間100℃で煮沸したGOATトランスフェクトミクロソームではせず、42kDaのバンドがGOATの存在を示すことが明らかになった。(B)GOAT活性はトランスフェクトした細胞のミクロソーム中には存在しているが、トランスフェクトしていない細胞のミクロソームには存在しておらず、GO−CoA−Tat 100nMによって完全に阻害されることを示した図である。
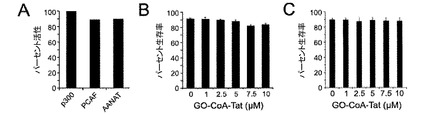
【図7】GOCoA−Tatの非特異的阻害および毒性のアッセイ。(A)p300、PCAFおよびAANATにおけるGO−CoA−Tat 10μMの存在下でのパーセント活性。GO−CoA−Tatの濃度を変化させて24時間インキュベートした後の(B)HepG2細胞の生存率および(C)HEL細胞の生存率を示した図である。
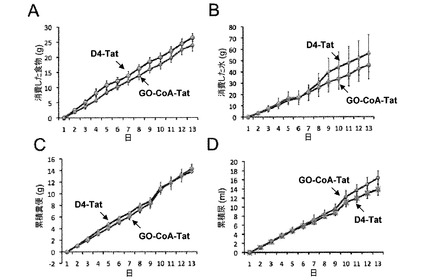
【図8】摂取および排泄に対するGO−CoA−Tatの効果。GO−CoA−Tat 29mg/kg(n=3)およびD4-Tat(n=3)で処理したマウスは(A)食物の累積消費および(B)水の累積消費に統計学的に有意な差を示さなかった。これらのマウスの(C)累積糞便生成および(D)尿生成も、統計学的に有意な程度の差を示さなかった。
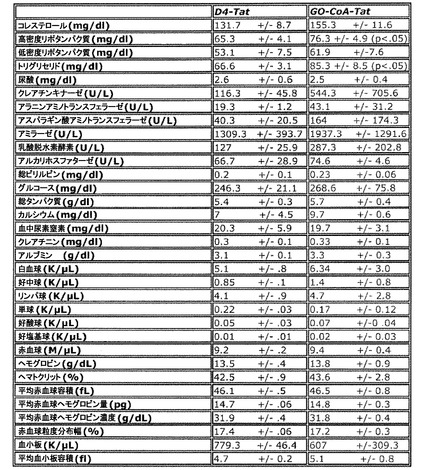
【図9】D4−TatおよびGOCoA−Tatで処理したマウスの血液パネルを示した図である。毎日化合物を投与して12日後の午後2時に採取した試料について、GO−CoA−TatマウスではD4-Tat処理マウスと比較して高密度リポタンパク質(p=0.04)およびトリグリセリド(p=0.02)の統計学的に有意な増加が示された。2つの群の間のその他の値には統計学的に有意な差は認められなかった。
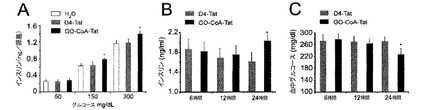
【図10】膵島およびマウスにおける、インスリンおよびグルコースに対するGO−CoA−Tatの効果。(A)化合物GO−CoA−Tat 5μMとインキュベートすることによって、ヒト膵島をグルコース150mg/dlおよび300mg/dl中でインキュベートしたときに(n=9)、インスリン産生の統計学的に有意な増加が引き起こされる。GO−CoA−Tat 15mg/kgで処理したマウス(n=6)は、(B)インスリン分泌の統計学的に有意な増加を示し、(C)血糖の統計学的に有意な減少を示した。
【図11】GOAT拮抗剤を含む、または含まない中鎖トリグリセリド食による、野生型またはグレリンノックアウトマウスにおける体重増加を示した図である。GOAT阻害剤GO−CoA−Tatは野生型の体重に影響を及ぼすが、遺伝子改変した(グレリンノックアウト)マウスには及ぼさず、グレリン経路を標的としていることが裏付けられる。
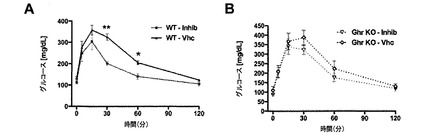
【図12】GOAT拮抗剤を投与された野生型およびグレリンノックアウトマウスにおける、グルコース負荷試験を示した図である。GOAT阻害剤GO−CoA−Tatは野生型のグルコースレベルに影響を及ぼすが、遺伝子改変した(グレリンノックアウト)マウスには及ぼさず、グレリン経路を標的としていることが裏付けられる。
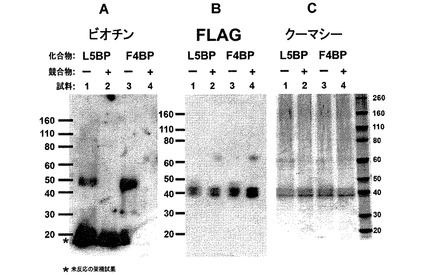
【図13】光活性化可能なGO−CoA−Tat類似体は、可溶性GOATを架橋結合することができる。GO−CoA−Tatの2つの光活性化可能変種は、GOATに直接結合し、化学的に架橋結合することができる。
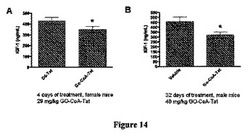
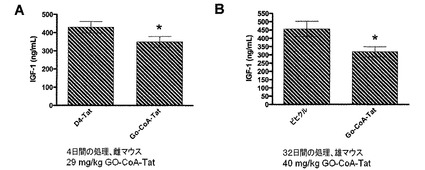
【図14】GOAT阻害剤は、雌雄マウスの血液IGF1レベルを抑制することを示した図である。これは、アシル−グレリンに対する影響と矛盾しない。
【発明を実施するための形態】
【0018】
本発明の実施形態を以下に詳細に論じる。記載する実施形態では、明瞭にするために特定の用語を使用する。しかし、本発明はそのように選択した特定の用語に限定されるものではない。当業者であれば、本発明の精神および範囲を逸脱することなく、その他の同等の部分を使用でき、その他の方法を開発できることを理解するであろう。本明細書で引用した参考文献は全て、それぞれが個々に組み込まれるように、全体があらゆる目的のために参考として組み込まれる。
【0019】
化合物
胃によって分泌される強力な食欲刺激ホルモン、グレリンは、動物およびヒトの体重増加を刺激する重要なペプチドホルモンであるとされてきた。その効果を発揮するために、グレリンは、アシル化、すなわち3位のセリン残基の通常ではない翻訳後オクタノイル化を必要とする。グレリンのオクタノイル化型は、通常アシル−グレリンと呼ばれる。
【0020】
グレリンのオクタノイル化は、グレリンO−アシルトランスフェラーゼ(GOAT)によって媒介される。GOATは膜タンパク質の膜結合O−アシルトランスフェラーゼ(mBOAT)ファミリーのメンバーである。本明細書で開示するのは、インビトロ、培養細胞およびマウスにおいて、強力、選択的かつ効果的な2基質類似体GOAT阻害剤である、GO−CoA−Tatである。
【0021】
いくつかの実施形態では、本発明は、例えば、肥満および/または糖尿病の治療で使用するための化合物を提供する。この化合物は一般式(I)を有することができる。
【0022】
【化2】
【0023】
式Iでは、本明細書で開示するように、Rは存在しなくてもよく、またはRは化合物の適切な機能と矛盾しない任意の有機部分であることができる。Rは、例えば、アルキル基、ならびにペプトイド残基(例えば、N−アルキルGly)、ベータアミノ酸、アミノラク酸およびフェニルGlyの組み合わせであることができる。いくつかの実施形態では、Rはアミノ酸残基で構成されるペプチド鎖、例えば、グレリンペプチドのフラグメントを含むペプチド鎖などである。グレリンペプチドのフラグメントは、例えば、残基4で開始してもよい。Rは、例えば、グレリンペプチドの残基4〜5または4〜10または4〜15または4〜28で構成されてもよい。実際に、本発明は、Rが本明細書で開示するように本発明による機能を可能とする任意の数の残基であってもよいことを企図する。式Iの化合物は「GO−CoA−Tat」とも呼ばれる。さらに、式Iによって定義された属に含まれる化合物の特定の例も、「GO−CoA−Tat」と呼ぶことができる。GO−CoA−Tatは、グレリンのオクタノイル化を媒介してアシル−グレリンを生成するGOATの阻害剤である。
【0024】
式Iにおいて、「GS」で示した部分は、従来の一文字命名法にしたがって、グリシンおよびセリンそれぞれに対応するアミノ酸残基で構成される。グリシンおよびセリンは、本明細書で開示するように本発明にしたがって機能することができる、任意の長さのペプチド鎖を含む任意の有機部分で置換することができる。セリン残基の適切な置換基の例には、例えば、D−AlaまたはD−SerなどのD−アミノ酸が含まれ、これらはまた、アセチル基またはプロピオニル基に切断されていてもよく(truncated to)、ヒドロキシアセチル置換基であってもよい。グリシン残基の適切な置換基の例には、セリンについて挙げたものならびにペプトイド残基(例えば、N−アルキルGly)、ベータアミノ酸、アミノラク酸およびフェニルGlyが含まれる。本明細書では、「ペプチド鎖」は、ペプトイド残基およびアミノ酸のその他の変種または誘導体を含む部分を包含し、天然に生じるアミノ酸、ペプトイド残基およびその他のアミノ酸変種または誘導体あるいはそれらの任意の部分集団の組み合わせからなる部分も包含する。
【0025】
「Dap」とは、1,2−ジアミノプロピオン酸を意味する。GS−Dap−RがグレリンペプチドのN末端に由来する場合、通常3位に存在するセリンをDapが置換する。Dapは、側鎖の酸素が窒素に置換されたセリンアイソスターで、アミド結合を導く。「Ahx」とは、RとTat配列を結合するのに用いられるアミノ−ヘキサノイルリンカーを意味する。Ahxは、例えば、アミノヘキサン酸、グリシン、アミノプロピオン酸、アミノ酪酸、アミノペンタン酸およびエチレングリコールをベースにした置換基であることができる。
【0026】
「転写のトランス作用活性化剤」の短縮形である「Tat」は、HIVウイルスで通常見出されるペプチド配列を意味する。例えば、「Tat」は、アミノ酸YGRKKRRQRRRを含む11残基のペプチドであることができる。Tatタンパク質は、分子、例えば、タンパク質またはその他の部分の細胞膜通過輸送を媒介することができる。このような媒介は、分子の大きさには関係ないものと考えられ、細胞膜のいかなる破壊も伴わないようである。いかなる特定の理論にも結びつけることを望まないが、GO−CoA−TatのTat部分は化合物の細胞内部への輸送の助けとなると仮定される。したがって、同様にこのような輸送の助けになる任意の部分が、本発明の化合物のTatと置換していてもよい。Tatの適切な置換基の例には、例えば、オリゴArg、オリゴアルキルグアニジウムおよびTatのd−もしくはl−立体異性体のいずれかが含まれる。「D4−Tat」は、本明細書では、Tat配列に結合した4個のアスパラギン酸残基のペプチドを含む。D4-Tatは、Fmoc方法またはそれらの変法を使用して調製した。
【0027】
いかなる特定の理論にも結びつけることを望まないが、GO−CoA−Tatの設計は、オクタノイル−CoAおよびグレリンペプチドを鋳型とする3要素複合体機構をGOATが使用するならば、切断可能でない架橋により2個の基質を結合すると、3要素複合体の形成に関連したエントロピーを損失せずに個々のリガンドの結合エネルギーを一緒にすることができるという理論に基づいている(図1A)。関連のある戦略が、ヒストンアセチルトランスフェラーゼ(HAT)およびタンパク質キナーゼを含むその他のペプチド修飾酵素にうまく使用されてきた。グレリンペプチドを式IのR基として使用する場合に、GOATによる認識に必要なグレリンペプチド長について我々は不確かであったので、高度に保存されたグレリン残基を最大限に含めるため、オクタノイル−CoAへのカップリングにアミノ酸1〜10を選択した。Tat由来ペプチド配列、例えば、11マー HIV Tatも、細胞透過を高めるために、アミノヘキサノイルリンカーを介してC末端に結合することができる。
【0028】
この研究に着手したとき、インビトロGOATアッセイはまだ開発されておらず、したがって3種類の細胞においてアシル−グレリン産生に対するGO−CoA−Tatの影響を調べた。D4-Tat(Tatペプチドに同じように結合する、負のCoA電荷を模倣したテトラ−アスパラギン酸、またはDDDD)を対照として使用した。試験した3種類の細胞系それぞれにおいて、アシル−グレリンのレベルは、D4-Tat対照と比べてGO−CoA−TatによってIC50が1〜2μMの範囲で有意に減少したが、デスアシル−グレリンは減少しなかった。
【0029】
GO−CoA−TatはGOATの選択的拮抗剤であると考えられ、その理由は、GO−CoA−Tatが10μMで、インビトロにおいて、p300 HAT、PCAF HATおよびセロトニンN−アセチルトランスフェラーゼを含む、アセチル−CoAを利用する3種類のその他の酵素を15%未満阻害することが示されたからである(図7)。さらに、GO−CoA−Tat 10μMはHEL細胞およびHEPG2細胞の生存率に非毒性であると考えられる(図7)。さらに、GO−CoA−TatはGOAT活性の拮抗において真正な2基質類似体として作用するようである(図1G)。Tat配列または細胞内移行を助けるその他の部分を含めると阻害活性が増加するという事実によって、細胞透過が関与しており、この化合物は細胞表面受容体には作用しないことが示唆される。
【0030】
本発明の一態様では、GO−CoA−Tatは、グルコース負荷に対するインスリン応答を高め、哺乳類、例えば、高脂肪食を与えられたマウスにおいて相対的な体重減少を導くために、例えば、腹腔内に投与する。定量的MR分光法によって、GO−CoA−Tat処理が除脂肪量よりも脂肪量に対して選択的な減少を導くことが立証された。したがって、GOATは肥満およびグルコース不耐性の治療の標的となることができ、GO−CoA−Tatは有効なGOAT阻害剤となる。
【0031】
組成物
いくつかの実施形態では、本発明は式Iの化合物および薬学的に許容される賦形剤を含む組成物を提供する。本明細書では、「薬学的に許容される」は、健全な医学的判断の範囲内で、妥当な便益/リスク比に見合って、過剰な毒性、刺激、アレルギー応答またはその他の問題となる合併症がなくヒトおよび動物の組織との接触に適した化合物、物質、組成物および/または剤形を包含する。いくつかの実施形態では、用語「薬学的に許容される」は、連邦政府または合衆国政府の規制当局によって承認されているか、あるいは合衆国薬局方または動物、より特定すればヒトで使用するために一般的に認識されているその他の国際的薬局方に挙げられていることを意味する。本明細書では、「賦形剤」とは、医薬組成物の製剤で使用される物質を意味し、それ自体は治療価値の無いものであってもよい。参考として組み込んだRemington: The Science and Practice of Pharmacy, 21st Ed. (2006)において記載されたものを含む様々な賦形剤を本発明で使用することができる。賦形剤には、限定はしないが、抗酸化剤、(治療効果を示すものとは対照的に)製剤自体の崩壊を防ぐ抗菌剤、保存剤、キレート化剤、緩衝剤、毒性を調節するための薬剤、着色剤、芳香剤および希釈剤、乳化剤および懸濁剤ならびに薬学的に適用されるその他の物質が含まれる。
【0032】
本発明の組成物は、肥満治療のその他の形態、例えば、食事および運動または肥満外科手術と併用して投与することができる。このような併用療法では、本発明の組成物はその他の形態の肥満治療の前、同時または後に投与することができる。
【0033】
本発明に従って使用するための医薬組成物は、薬剤として使用することができる調製物への活性成分のプロセシングを容易にする賦形剤および補助剤を含む一または複数種の生理学的に許容される担体を使用して従来の方法で製剤化することができる。適切な製剤は、選択した投与経路に左右される。
【0034】
適切な投与経路は、例えば、経口、直腸、経粘膜、特に経鼻、局所、腸内または非経口送達を含んでもよく、筋肉内、皮下および脊髄内ならびにクモ膜下腔内、直接脳室内、静脈内、腹腔内、鼻腔内または眼内注射を含む。医薬組成物は、局所的または全身的に投与することができる。例えば、組成物は、患者の体の特定の領域に直接調製物を注射することによって局所的に投与することができる。
【0035】
注射のために、本発明の活性成分は水性溶液、好ましくはハンクス液、リンゲル液または生理学的塩緩衝液などの生理学的に適合した緩衝液で製剤化してもよい。経粘膜投与のために、浸透させるバリアに適した浸透剤が製剤中で使用される。このような浸透剤は当業界では一般的に公知である。
【0036】
経口投与のために、化合物は当業界で周知の薬学的に許容される担体と活性化合物を一緒にすることによって容易に製剤化することができる。このような担体は、患者による経口摂取のために、本発明の化合物の錠剤、丸剤、糖衣錠、カプセル、液剤、ジェル、シロップ、スラリー、懸濁物などへの製剤化を可能にする。経口使用のための薬理学的調製物は、所望により、錠剤または糖衣錠の核を得るために適切な補助剤を添加した後、固形賦形剤を使用し、得られた混合物を場合により粉砕し、顆粒の混合物を加工して形成することができる。適切な賦形剤は、特に:乳糖、スクロース、マンニトールまたはソルビトールを含む糖などの充填剤;例えば、トウモロコシ澱粉、コムギ澱粉、米澱粉、ジャガイモ澱粉、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチル−セルロース、カルボキシメチルセルロースナトリウムなどのセルロース調製物;および/またはポリビニルピロリドン(PVP)などの生理学的に許容されるポリマーである。所望により、例えば、架橋結合ポリビニルピロリドン、寒天またはアルギン酸もしくはアルギン酸ナトリウムなどのそれらの塩のような崩壊剤を使用してもよい。
【0037】
糖衣錠核は適切なコーティングを施す。このために、場合によりアラビアゴム、タルク、ポリビニルピロリドン、カーボポールゲル、ポリエチレングリコール、二酸化チタン、塗料溶液および適切な有機溶媒または溶媒混合物を含有してもよい、濃縮糖溶液を使用することができる。色素または顔料は、活性化合物用量の様々な組み合わせを識別し、または特徴付けるために、錠剤または糖衣錠コーティングに添加することができる。
【0038】
経口使用できる医薬組成物には、ゼラチンで作られた押し込み型(push-fit)カプセル、ゼラチンおよび可塑剤、例えば、グリセロールまたはソルビトールで作られた密封軟カプセルが含まれる。押し込み型カプセルは、乳糖などの充填剤、澱粉などの結合剤、タルクまたはステアリン酸マグネシウムなどの潤滑剤および場合により安定化剤と混合して活性成分を含有することができる。軟カプセルでは、活性成分は適切な液体、例えば、脂肪油、液体パラフィンまたは液体ポリエチレングリコールに溶解または懸濁することができる。さらに、安定化剤を添加することができる。経口投与のための製剤は全て、選択した投与経路に適した投薬量であるべきである。
【0039】
頬側投与のためには、組成物は従来の方法で製剤化された錠剤またはトローチ剤の形態をとることができる。
【0040】
鼻吸入による投与のためには、本発明によって使用するための活性成分は、適切な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロ−テトラフルオロメタンまたは二酸化炭素を使用して加圧容器または噴霧器からエアロゾルスプレー状の形態で適宜送達させる。加圧エアロゾルの場合では、投薬単位は計量した量を送達するために弁を形成することによって測定することができる。ディスペンサーで使用するためのゼラチンなどのカプセルまたはカートリッジは、化合物および乳糖または澱粉などの適切な粉末基剤の粉末ミックスを含有して製剤化することができる。
【0041】
本明細書で記載した調製物は、例えばボーラス注射または連続注入によって、非経口投与用に製剤化することができる。注射用製剤は、例えば、アンプルまたは反復投与容器に入れて、場合により保存剤を添加して、単位投与形態にすることができる。組成物は、油性または水性媒体に溶かした懸濁液、溶液またはエマルジョンであってもよく、懸濁剤、安定化剤および/または分散剤などの製剤化剤を含有してもよい。
【0042】
非経口投与用の医薬組成物には、水溶性形態の活性調製物の水溶液が含まれる。さらに、活性成分の懸濁液は適当な油または水をベースにした注射用懸濁液として調製することができる。適切な親油性溶剤またはビヒクルは、ゴマ油などの脂肪油、またはオレイン酸エチル、トリグリセリドまたはリポソームなどの合成脂肪酸エステルを含む。水性注射用懸濁液は、カルボキシメチルセルロースナトリウム、ソルビトールまたはデキストランなどの懸濁液の粘度を高める物質を含有することができる。任意選択により、懸濁液はまた、適切な安定化剤または高濃度溶液の調製を可能にするために活性成分の溶解度を高める薬剤を含有することができる。
【0043】
あるいは、活性成分は、使用前に適当なビヒクル、例えば、発熱性物質を含まない無菌水をベースにした溶液によって構成するための粉末形態であってもよい。活性成分はまた、当業界で公知の方法によってペグ化を行うことができる。
【0044】
本発明の調製物はまた、カカオ脂またはその他のグリセリドなどの従来の座薬基剤を使用して、座薬または停留浣腸剤などの直腸用組成物として製剤化することができる。
【0045】
本発明の状況で使用するために適した医薬組成物には、活性成分を企図した目的を実現するために有効な量で含有する組成物が含まれる。より特定すれば、治療有効量は、疾患の症状の予防、軽減または回復あるいは治療した対象の生存を延長させるために有効な1または複数種の活性成分の量である。
【0046】
本明細書で記載した活性成分の毒性および治療効果は、インビトロ、細胞培養物または実験動物において、標準的な薬学的手法によって測定することができる。これらのインビトロおよび細胞培養物アッセイならびに動物研究から得られたデータは、ヒトにおいて使用するために用量の範囲の製剤設計において使用することができる。投薬量は、使用する剤形および利用する投与経路に応じて変化させることができる。正確な処方、投与経路および投薬量は、患者の状態を考慮して個々の医師が選択することができる。
【0047】
治療する状態の重症度および応答性に応じて、投薬は1回または複数回投与であることができ、治療過程は、数日から数週間まで持続するか、または治癒が達成されるか、または疾患状態の軽減が実現されるまでであることができる。
【0048】
投与する組成物の量は、もちろん、治療する対象、苦痛の重症度、投与方法、処方する医師の判断などに応じて左右される。
【0049】
適合した医薬担体中に製剤化された本発明の調製物を含む組成物も、調製し、適切な容器に入れ、適応する症状の治療を表示することができる。
【0050】
本発明の医薬組成物は、当業界で周知の工程によって、例えば、従来の混合、溶解、顆粒化、糖衣錠形成、微粒子化、エマルジョン化、カプセル化、封入または凍結乾燥工程によって製造することができる。
【0051】
調製方法
別の態様では、本発明は式Iの化合物の調製方法を提供する。いくつかの実施形態では、この方法は、3〜15個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、Tat配列がペプチド配列の一末端に結合している工程と、ブロモ−オクタノイル化中間体を得るために、1または複数個の反応容器中で、グレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、前記ブロモオクタノイル化中間体と補酵素Aを一緒にする工程とを含むことができる。
【0052】
Tatは、例えば、式IのR配列の一末端に結合させることができる。例えば、Rがペプチド配列の場合、Tatはペプチド配列のC末端に結合させることができる。いくつかの実施形態では、Tatは、連結部分、例えば、アミノ−ヘキサノイルリンカーなどによって結合するが、GO−CoA−Tatの適切な機能を妨害しない任意の連結部分は本発明の範囲内である。
【0053】
「試薬K」は、トリフルオロ酢酸、水、チオアニソール、フェノールおよびエタンジチオールの標準的混合物を意味する。保護されたアミノ酸を脱保護し、樹脂から切断するために使用される。
【0054】
GO−CoA−Tat(例えば、図1Cで挙げたような化合物1)の合成は、例えば、固相方法を使用して実施することができる(図1B)。ペプチド長の長さが異なり、CoA、オクチルおよびTatそれぞれが個々に欠損している一連の関連化合物(化合物2〜7、図1C)も合成して(図1C)、本発明にしたがって使用することができる。
【0055】
予防方法および治療方法
別の態様では、本発明は、治療有効量の式Iの化合物を含む組成物を使用して、様々な疾患、障害または症状を予防および/または治療する方法を提供する。
【0056】
[0070] 「治療」とは、本明細書で記載した疾患、障害または状態の症状または有害な影響の少なくとも1つを治癒、逆戻り、軽減、緩和、最小化、抑制または停止させることを意味する。「治療」とは、疾患、障害または症状を有する対象の治療的処置のいずれも意味する。「予防」とは、障害の徴候または症状を発症していない対象における予防的または防御的処置を意味する。「治療」とは、例えば、症状の軽減;症状、障害または疾患の範囲の減少;症状、障害または疾患の状態の安定化(すなわち、悪化阻止);症状、障害または疾患進行の緩徐化;症状、障害または疾患状態の回復;部分的または全体的な寛解;あるいは症状、障害または疾患の改善もしくは改良を包含する。治療を必要とするものには、既に障害を有するものが含まれる。防御または予防を必要とするものには、障害を防御できるものが含まれる。したがって、本明細書で開示する化合物を投与する患者は、障害を有すると診断されていてもよく、または障害に罹患しやすいか、もしくは感受性であってもよい。「治療すること」には、測定可能な応答、好ましくは過剰なレベルの副作用無しに臨床的に重要な応答を惹起することが含まれる。「治療すること」にはまた、治療を受けていない場合に予測される生存と比較して、生存を延長させることが含まれる。
【0057】
本発明による対象は、本明細書で記載した疾患、障害または症状の1つであると診断された、あるいは本明細書で記載した疾患、障害または症状の少なくとも1種類に罹患している哺乳類、例えば、ヒトである。本発明の組成物は、本発明の化合物の有益な効果を経験することができる、組成物を必要とするあらゆる哺乳類に投与することができる。このような任意の哺乳類は、「対象」と考えられる。このような対象には、ヒトおよび非ヒト、例えば、ヒト、飼育動物および家畜、動物園用、スポーツ用もしくは愛玩用動物、例えば、イヌ、ウマ、ネコ、ウシなどが含まれる。対象は男性または女性であることができる。
【0058】
治療有効量を構成するために必要な活性剤の量は、治療する障害の重症度;患者のアイデンティティ、年齢、体重、一般的な健康状態、性別、食事および化学的組成(chemical make-up);実現する細胞応答の種類および程度;使用する特定の薬剤または組成物およびその活性;投与時間、投与経路および薬剤の排泄速度;治療期間;特定の薬剤と組み合わせてまたは同時に使用する薬剤;ならびに医薬品業界で周知の類似の要素を含むいくつかの要素に基づいて変化する。一般的に、治療有効量は、疾患または障害に関連した1または複数種の症状の測定可能な軽減を生じる量である。
【0059】
例えば、肥満の治療又は予防のための治療有効量は、最大でまたは少なくとも1日あたり、約1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、5mg/kg、7.5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kgまたはそれ以上の活性薬剤であることができる。70kgの人では、治療有効用量は、最大でまたは少なくとも1日あたり、約50mg、60mg、70mg、80mg、90mg、100mg、120mg、140mg、160mg、180mg、200mg、210mg、250mg、300mg、350mg、400mg、450mg、500mg、600mg、700mg、800mg、900mg、1000mg、1100mg、1200mg、1300mg、1400mg、1500mg、1600mg、1700mg、1800mg、1900mg、2000mgまたはそれ以上の活性薬剤であることができる。本明細書中において、「約」は、参照数字の10%以下から10%以上までの範囲を指し得る。例えば、「約50」は、45〜55を意味し得る。
【0060】
例えば、所望の治療効果を実現するため、および所望の効果が実現するまで投薬量を徐々に増加するために、必要なレベルより低いレベルの薬剤用量で開始することは当業者の技量の範囲内である。これらの状況下での活性薬剤の治療有効量は、動物および臨床試験および医学的観察を用いて医師が容易に決定することができる。
【0061】
いくつかの実施形態では、本発明は、肥満の治療方法または予防方法を提供する。これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与することを含むことができる。
【0062】
本発明では、組成物の投与は、例えば、対象において除脂肪量よりも大きい脂肪量減少をもたらすことができる。例えば、対象においてアシル−グレリンに対するグレリンの比の増加ももたらすことができる。
【0063】
いくつかの実施形態では、本発明は、II型糖尿病の治療または予防方法を提供する。これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与することを含むことができる。組成物の投与は、例えば、対象においてインスリン産生の増加をもたらすことができる。例えば、対象においてグルコース曝露に対する応答性増加、および/または対象において脱共役タンパク質2(UCP−2)mRNAレベルの減少ももたらすことができる。
【0064】
糖尿病を治療または予防するための式Iの化合物の治療有効量は、当業者が決定することができるが、例えば、肥満について前述した量と類似であってもよい。
【0065】
例えば、糖尿病の治療又は予防のための治療有効量は、最大でまたは少なくとも1日あたり、約1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、5mg/kg、7.5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kgまたはそれ以上の活性薬剤であることができる。70kgの人では、治療有効用量は、最大でまたは少なくとも1日あたり、約50mg、60mg、70mg、80mg、90mg、100mg、120mg、140mg、160mg、180mg、200mg、210mg、250mg、300mg、350mg、400mg、450mg、500mg、600mg、700mg、800mg、900mg、1000mg、1100mg、1200mg、1300mg、1400mg、1500mg、1600mg、1700mg、1800mg、1900mg、2000mgまたはそれ以上の活性薬剤であることができる。
【0066】
特定の理論に結びつけることは望まないが、GOAT阻害剤は膵臓のランゲルハンス島細胞における脱共役タンパク質−2(UCP−2)のレベルを減少させることによって糖尿病を治療するものと考えられる。GOAT阻害剤がUCP2レベルを劇的に調節することはさらに、アシル−グレリンと肥満およびII型糖尿病との関連を実証する。アシルグレリンは、ニューロペプチドYおよびアグーチ関連タンパク質を共発現する弓状核ニューロンにおけるUCP2レベルの増加を引き起こすことによって、摂食行動を誘導する。UCP2は、肥満誘導性糖尿病モデルであるob/obマウスの膵島で著しく上方制御され得る。UCP2を欠如したob/obマウスは、インスリン初期分泌の回復、血清インスリンレベルの増加および血糖値の著しい減少を示すことは重要である。
【0067】
さらに、ヒトUCP2多形のA55Vは肥満と関係があり、インスリンおよびレプチン濃度を著しく増加させる。さらに、−866A UCP2プロモータ多形の同型接合型のII型糖尿病患者はUCP2発現が上昇しており、より高頻度のインスリン治療を必要とする。これらの患者はまた、グルコース誘導性インスリン分泌(GIIS)が有意に低下している。さらに、−866 A/Aドナーから単離された膵島では、−866 G/Aヘテロ接合型およびG/G同型接合型の個体から単離された膵島よりもGIISが有意に低かった。
【0068】
いくつかの実施形態では、本発明は、mBOAT関連疾患の治療方法を提供する。例えば、これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与し、それによってmBOAT関連疾患を治療することを含むことができる。
【0069】
「mBOAT関連疾患」は、mBOATペプチドが関与する経路の調節異常を特徴とする疾患である。mBOAT遺伝子は少なくとも16個存在する。例えば、mBOATタンパク質に類似した構造を有する「porcupine」として知られる遺伝子産物は、癌に関連したプロセスである、Wntシグナル伝達に不可欠である。Wntはporcupineアシルトランスフェラーゼ酵素によるアシル修飾を必要とする。Wntシグナル伝達は多くの癌に寄与する可能性があり、porcupine阻害剤によって遮断することができるかもしれない。mBOAT関連疾患には、脂質代謝調節異常の様々な形態が含まれる。脂質代謝変化は癌に関与しており、mBOATの遮断は癌の治療となり得る。
【0070】
いくつかの実施形態では、本発明は、胃運動の調節異常に関連した障害、例えば、過敏性腸症候群などの治療または予防方法を提供する。グレリン投与は、胃内容排出を増加させ、胃不全麻痺を減少させる。したがって、当業者によって理解されるように、GO−CoA−Tatの投与は、GOAT阻害および付随したアシル−グレリンレベルの低下によって胃運動を阻害し、過敏性腸症候群および胃運動調節異常に関連したその他の障害の患者における緩和効果を発揮するために使用することができる。
【0071】
キット
別の態様では、本発明はキットを提供する。いくつかの実施形態では、これらのキットは、式Iの化合物の治療有効量の少なくとも1用量を含む組成物を含む。
【0072】
これらのキットはさらに、さらなるの体重減少治療薬(treatment)の少なくとも1用量を含む。このようなさらなる治療薬の例には、例えば、アシル−グレリンに対する抗体ならびにGHSRに対する拮抗剤が含まれる。
【0073】
活性アシル−グレリンホルモンの生合成の直接的な標的化には、受容体拮抗剤を上回るいくつかの利点がある。第1に、作用に重要な部位の多くが脳内に存在するアシル−グレリン受容体遮断薬とは対照的に、これらの酵素阻害剤は血液脳関門を通過する必要がない。第2に、酵素阻害剤の効果は範囲がより広範囲である可能性がある。例えば、まだ同定されておらず、公知の受容体を標的とする受容体拮抗剤によってはおそらく影響を受けない、GHSR以外のクラスのアシル−グレリン受容体が存在する可能性がある。しかし、これらの未だに知られていない受容体の活性は、当然のことながら、ホルモンレベルの減少によって影響を受けるだろう。第3に、受容体遮断薬には、動物が受容体遮断に応答してより多くのアシル−グレリン形成を引き起こすフィードバック機構を有する可能性があり、それが受容体遮断薬を上回るかもしれないという欠点がある。しかし、生合成経路を標的とし、したがってフィードバック機構がアシル−グレリンレベルを増加させるのを妨害する酵素阻害剤によって、このようなフィードバック機構は効果的に相殺されるだろう。第4に、GOAT酵素は触媒的に作用するので、所与の用量のGOAT阻害剤は、大体1対1でのみ効果的であることができるアシル−グレリン受容体遮断薬の類似用量よりも、比例的に有効である可能性がある。第5に、グレリンのアシル−グレリンに対する比は、体重増加の調節に重要な因子であり、比較的高いグレリン濃度が体重減少には都合がよいといういくつかの示唆がなされており、これがまさにGOAT阻害剤によって実現する効果である。要約すると、本発明は、体重およびグルコース制御の薬理学的管理のための新たなアプローチを示す。
【0074】
本発明の他の目的、利点および新たな特性は、本発明の範囲を決して制限するものではない以下の実施例を考慮することによって当業者には明らかとなろう。
【実施例】
【0075】
(実施例1)
3種類の細胞系におけるアシル−グレリン産生に対するGO−CoA−Tatの効果
この研究に着手したとき、インビトロGOATアッセイはまだ開発されていなかったため、HELヒト赤白血病細胞(図1D〜E)、KatoIIIヒト胃細胞(図5)およびAGSヒト胃上皮細胞(図5)を含む3種類の細胞においてアシル−グレリン産生に対するGO−CoA−Tatの効果を調べた(3種類の細胞系は全てATCC、Manassas、VAから入手した)。D4-Tat(テトラ−アスパラギン酸は、負のCoA電荷を模倣し、同様にTatペプチドに結合する)を対照として使用した。細胞系それぞれにおいて、アシル−グレリンのレベルは、GO−CoA−Tatによって、IC50が1〜2μMの範囲でD4-Tat対照と比べて有意に減少したが、デスアシル−グレリンは減少しなかった。興味深いことに、最大阻害は、化合物に24時間曝露した後においてのみ実現し、遅い反応速度は非定型酵素特性または予め形成されたアシル−グレリン貯蔵のいずれかから生じているかもしれない。この遅延をさらに調べるために、放射活性アッセイを使用し、組換えミクロソームGOATを用いてインビトロでGO−CoA−Tatを試験した(14)(図1Fおよび6)。完全なGOAT阻害はGO−CoA−Tat 100nMで5分以内に実現し、細胞アシル−グレリンレベルのゆっくりした見かけ上の減少は大きな既存の細胞内貯蔵を反映している可能性があることが示唆された。
【0076】
GO−CoA−Tatは選択的GOAT拮抗剤であると考えられ、その理由は、GO−CoA−Tatは10μMで、インビトロにおいてp300 HAT、PCAF HATおよびセロトニンN−アセチルトランスフェラーゼを含む3種類のアセチル−CoA利用酵素を15%未満阻害することが示されたからである(図7)。さらに、GO−CoA−Tat 10μMはHEL細胞およびHEPG2細胞(ATCC, Manassas, VA)の生存率に対して非毒性であると考えられる(図7)。GO−CoA−Tatおよび類似体の広範な分析によって、アシル−グレリン産生の強力な細胞阻害のためには、少なくとも10個のグレリン残基、ならびにCoA、オクタノイルおよびTat成分を使用して最適な結果が実現することが示唆される。これらの結果は、GO−CoA−TatがGOAT活性の拮抗において、真正な2基質類似体として作用するようであることを示唆している(図1G)。さらに、最良の結果はTat配列などの細胞内移行を助ける部分を含めることによって実現するという事実によって、細胞透過が関与しており、この化合物は細胞表面受容体には作用しないことが示唆される。
【0077】
(実施例2)
マウスにおけるアシル−グレリン産生に対するGO−CoA−Tat投与の効果
次に、正常マウスにおいてGO−CoA−Tatがアシル−グレリン産生を阻害するかどうかを調べた。化合物を投与する6時間前に絶食したマウスにおいて、2種類の腹腔内(IP)GO−CoA−Tat用量、15mg/kgおよび29mg/kgを試験した。分布容積が動物の体重と等しいと仮定すると、これらの用量はそれぞれ、約4μMおよび8μMの最大濃度を実現すると予測される。培養細胞で観察されたように、GO−CoA−Tatによる処理は血清アシル−グレリンの用量依存的阻害を引き起こし(図2)、29mg/kg用量で24時間後に最大の70%阻害が得られたが、D4-Tat対照では阻害は引き起こされなかった(図2)。また、デスアシルグレリンの血清レベルにあまり影響はなかったが、少し増加傾向があった。これらの結果は、GO−CoA−Tatがインビボにおいて選択的にアシル−グレリン産生を標的としていることを示している。
【0078】
(実施例3)
マウスにおける体重増加に対するGO−CoA−Tat投与の効果
次に、正常マウスに12日間にわたって高脂肪食を与えることによって、体重増加に対するGO−CoA−Tatの効果を調べた。24時間毎にGO−CoA−Tat(29mg/kg IP)でこれらのマウスを処理し、代謝ケージに入れて、毎日摂取および排出ならびに体重のモニターを可能にした。さらに、動物の脂肪量および除脂肪量を評価するために、マウスに3日毎に定量的核磁気共鳴(QMR)分光法を実施した(15)。これらの実験によって、GO−CoA−Tatで処理したマウスは、D4-Tatで処理したマウスと比較して統計学的に有意な体重減少を表すことが示された(図3A)。さらに、QMR測定によって、対象と比較して、GO−CoA−Tat処理動物では脂肪量の約5%の相対的減少を表し、6日後も維持されたが、除脂肪量の減少は表れないことが示された(図3B〜D)。食事または水の摂取および糞尿排出には統計学的有意差はなかったが(図8)、中程度の試料数ではこれらの値には比較的大きな変動があり、そのため摂取減少の関与を除外することはできなった。以前の遺伝子研究では、グレリンの欠如は、食欲ならびに代謝率に影響を及ぼすことによって体重に影響を与え得ることが示唆されている(2〜8)。
【0079】
GO−CoA−Tatが誘導する、体重減少の原因となる全身毒性または臓器障害の可能性について調べるために、12日間GO−CoA−Tat処理した動物の血液化学および血液細胞数を評価した。これらの分析によって、体重減少の原因となり得る肝臓、腎臓、膵臓または骨髄毒性の証拠は示されなかった(図9)。興味深いことに、これらの血液測定では、GO−CoA−Tatは、少しではあるが有意なHDLならびにトリグリセリドの増加を誘導した。別の研究では、3日間の全糞便脂肪をQMRによって測定すると、GO−CoA−Tat処理マウス(4.97±0.10g)とD4-Tat処理マウス(5.01±0.07g)の間には統計学的有意差は示されず、GOAT阻害は腸内脂肪吸収に影響を及ぼさないことを示唆している。総合すると、これらのデータは、GO−CoA−Tatはアシル−グレリンを低下させることによって体重減少を引き起こすことを示唆している。
【0080】
(実施例4)
グルコース曝露に対するインスリン応答へのGO−CoA−Tatの効果
グルコース曝露に対するインスリン応答へのGO−CoA−Tatの効果を調べるために、ヒト膵島細胞をGO−CoA−Tatで24時間まで予め処理して、これらの細胞が、24時間GO−CoA−Tatに曝露したとき、グルコース曝露に対するインスリン応答の統計学的有意な増加を示すことを示した(図10)。これらの結果は、アシル化グレリンはインスリン応答鈍化に直接的役割を担うことを示唆している。このことをインビボで調べるために、GO−CoA−Tatで予め処理した後、経口的にグルコース曝露したマウスを研究した。膵島細胞研究と矛盾することなく、経口的グルコース曝露(1mg/kg)の前に24時間GO−CoA−Tat 15mg/kgを与えられた動物は、インスリン応答の統計学的有意な増加を示した(図10)。これらの結果は、2.5g/kgの腹腔内グルコース曝露と共にGO−CoA−Tat 29mg/kgを投与されたマウスにおいてさらによりはっきりしていた(図4A)。このインスリン応答の増加には、グルコースの統計学的有意な減少が伴い、機能的に重要な効果が示唆された(図4B)。
【0081】
GOAT阻害とインスリン調節との間の関係をさらに調べるために、GO−CoA−Tatで処理したマウスから単離した膵臓のランゲルハンス島を研究した。インスリン産生β細胞はGHSRが陽性に染色し、膵島ではβ細胞とは区別されるグレリン発現細胞がわずかな割合で示され、インスリンおよびグレリン受容体は別の細胞で産生されるようであることが示唆された。GO−CoA−Tatで処理したマウスから単離された膵島のQRT−PCRでは、UCP2 mRNAレベルはD4−tat処理マウスと比較して20倍減少していたが(図4C)、インスリン、グレリンまたはGHSRのレベルには変化がないことが示された。比較すると、QRT−PCRは胃底部においてUCP2に対して非統計学的に有意な効果を示した(図4D)。総合すると、これらのデータは、UCP2レベルの抑制およびグルコースに応答したインスリン放出の増強における、GOAT阻害の組織特異的役割を示唆している。
【0082】
GOAT阻害剤がUCP2レベルを劇的に調節することはさらに、アシル−グレリンと肥満およびII型糖尿病との関連を実証している(19〜21)。アシル−グレリン誘導性摂食行動は、ニューロペプチドYおよびアグーチ関連タンパク質を共発現する弓状核ニューロンにおいてUCP2レベルを増加させることよって制御することができる(21)。UCP2は、肥満誘導性糖尿病モデルであるob/obマウスの膵島で著しく上方制御される。UCP2を欠如したob/obマウスは、インスリン初期分泌の回復、血清インスリンレベルの増加および糖血症レベルの著しい減少を示すことは重要である(19)。さらに、ヒトUCP2多形のA55Vは肥満と関係があり、インスリンおよびレプチン濃度を著しく増加させる(22)。さらに、−866A UCP2プロモータ多形の同型接合型のII型糖尿病患者はUCP2発現が上昇しており、より高頻度のインスリン治療を必要とする。これらの患者はまた、グルコース誘導性インスリン分泌(GIIS)が有意に低下している(23)。さらに、−866 A/Aドナーから単離された膵島では、−866 G/Aヘテロ接合型およびG/G同型接合型の個体から単離された膵島よりもGIISが有意に低かった(24)。
【0083】
(実施例5)
化学合成(化合物1〜7、D4-Tat、グレリン−27−ビオチン)
化学合成用の試薬は全て、AldrichまたはAcros Organicsから購入した。市販の試薬は全て、購入時のままさらに精製することなく使用した。2基質類似体阻害剤、ペプチドおよび対照化合物の調製用HPLC単離は、逆相C−18カラム(25×2.14cm、Microsorbtm-100, Rainin)を使用して実施し、水(トリフルオロ酢酸(TFA)0.05%、移動相A)およびアセトニトリル(TFA0.05%、移動相B)の勾配で溶出した。グレリン−27−ビオチン(GSSFLSPEHQRVQQRKESKKPPAKLQPK(ビオチン)G)およびD4−Tatは、Fmoc法を使用して調製した。GO−CoA−Tatおよび関連誘導体の合成は、以前に記載された方法(33、34)と類似しており、グレリンから開始して、Fmoc法を使用した。様々な長さのグレリンペプチド(3から15個のC末端配列)を使用し、グレリンのSer3をAlloc(アリルオキシカルボニル)保護1,2−ジアミノプロピオン酸で置換した。遊離アミノ基を得るために、CHCl3:AcOH:N−メチルモルホリン(37:2:1)中においてテトラキス(トリフェニルホスフィン)パラジウム(0)を使用したAllocの直交脱保護を実施し、次いでラセミ化α−ブロモオクタン酸無水物(化合物5ではオクタン酸無水物および化合物6ではブロモ酢酸無水物)と反応させた(図1C参照)。補酵素Aとの結合は、α−ブロモ−アミドをCoASHと反応させることによって実施した。ブロモ−アミドペプチドの撹拌溶液(濃度2〜10mMのddH2O溶液)に室温で重炭酸トリエチルアンモニウムの水性緩衝液(pH8.0)1.0Mに溶かしたCoASHの溶液(2.0〜3.0当量)を滴下した。48〜72時間後、混合物を一晩凍結乾燥し、所望の生成物を単離するために残渣を調製用HPLCに添加した。HPLCカラムは水(TFA0.05%、移動相A)およびアセトニトリル(TFA0.05%、移動相B)(0分、5%B;5〜65分、60%Bまで直線的に増加;10mL/分)の勾配で溶出し、214nmでモニターした。収集した画分を減圧下で濃縮し、凍結乾燥して最終生成物を白色固形物として得た。マトリックス支援レーザー脱離イオン化(MALDI)およびエレクトロスプレー(ES)質量分析データは、計算値と矛盾せず、アッセイ用の水性溶液中の化合物最終濃度はアミノ酸分析によって測定した。
【0084】
(実施例6)
細胞培養、細胞溶解調製物およびEIA測定
HEL細胞およびAGS細胞(ATCC, Manassas VA)は、不活性化牛胎児血清10%、ストレプトマイシン−ペニシリン100IU/mlおよびグルタミン4mMを補給したRPMI1640培地(Sigma)で増殖させ、週に2回常法通り継代した。KatoIII細胞(ATCC, Manaasas VA)は、不活性化牛胎児血清10%、ストレプトマイシン−ペニシリン100IU/mlおよびグルタミン4mMを補給したDMEM培地(Sigma)で増殖させ、週に2回常法通り継代した。いくつかの実施形態では、アシル−グレリン産生の阻害を評価するために、HEL細胞は7種類の化合物の1種で処理した。3種類の細胞種全ての細胞溶解物を以下のようにRIPA緩衝液中で調製した。細胞(7.5×106細胞)は、卓上型遠心機で1000rpmで5分間遠心分離した。次に細胞ペレットを洗浄し、これに含有する氷冷した改変RIPA緩衝液0.25mlを添加し、激しくピペット操作し、ボルテックスして、その後完全に細胞が溶解するまで4℃で10分間静置させた。次に、溶解物を予め冷却した遠心機で2000rpmで15分間遠心した。遠心直後、上清を−80℃で保存した。細胞溶解物および血清中のアシル−およびデスアシル−グレリンのレベルならびに培養培地および血清試料中のインスリンレベルを評価するために、2抗体サンドイッチをベースにした市販の酵素免疫アッセイ法(Alpco Diagnostics)で分析した。
【0085】
(実施例7)
分子クローニング
GOATは、マウス胃から2段階ネステッドRT−PCR法によってクローニングした。全RNAは、組織ホモジェナイザーおよびRNeasy(登録商標)キット(Qiagen)を使用して、マウス胃の約半分から抽出した。cDNAは、Invitrogen SuperScript(商標)II RTで逆転写することによって調製した。cDNAの2μlをプライマー、TTTACAAGGGCACCGCTTAG/CAAGGCATCTTCTGGCATTTで増幅した。次いで、反応混合物1μlをネステッドプライマーGCCACCATGGATTGGCTCCAGCTC/GAGATGAAGGGCAGGGAAAでさらに増幅した。約1.3kBのバンドをゲルから切り出し、pCR(登録商標)2.1−TOPO(登録商標)(Invitrogen)に連結した。
【0086】
[00108] GOATは5’平滑末端連結および3’EcoRI切断のために設計されたプライマーを使用してこのベクターから増幅し、次にpHLsecから得られたpαHベクター(Aricescu et al. Acta Cryst.(2006). D62, 1243-1250)にKpnI(平滑末端化)およびEcoRIを使用してクローニングした。QuikChange(登録商標)変異誘発(Stratagene)を使用して、製造元の方法をWang and Malcolm(35)によって開発された2段階法に置き換えて、3xFlag(登録商標)タグ(Sigma)をC末端に付加した。
【0087】
(実施例8)
GOATトランスフェクションおよびミクロソーム単離
GnTI欠損HEK293T細胞(Reeves et al., Proc Natl Acad Sci U S A. 2002 Oct 15 ;99(21): 13419-24)は、L−グルタミン2mMおよびFBS1%を補給したFreestyle293T培地(Invitrogen)中でCO28%を含有する雰囲気中で増殖させた。トランスフェクションの日に、細胞400mlを2×106/mlの濃度に設定した。細胞に、Aricescuらによって記載されたように(Acta Cryst. (2006). D62, 1243-1250)、ポリエチレンイミン「Max」強力直鎖状PEI(Polysciences)を使用してプラスミドをトランスフェクトした。簡単に説明すると、最終培養量の20分の1と等しい未補給ハイブリドーマSFM培地(Invitrogen)中で、DNA1μg/mlをトランスフェクション試薬3μg/mlと混合し、室温で20分間インキュベートし、細胞に添加した。4日後、細胞を実験毎に収集し、PBSで1回洗浄した。
【0088】
HEK293T GnTI細胞の各ペレットを緩衝液D(Tris−HCl 50mM、pH7.4、NaCl 150mM、ナトリウムEGTA 1mM、フェニルメチルスルホニルフルオリド30μg/ml、ペプスタチンA 3μg/mlおよび完全プロテアーゼ阻害剤カクテル錠、EDTAなし(Roche、1錠/緩衝液40ml))20mlに再懸濁した。細胞は、加圧型ホモジェナイザー(B型内筒、排出30回)を使用して溶解した。溶解物を4℃で2135×gで10分間遠心分離し、上清を新たな管に移し、同条件下で再度遠心分離した。上清を4℃で100000×gで30分間遠心分離し、上清を捨てた。ミクロソームペレットをTris緩衝生理食塩水(TBS)(Tris-HCl 50mM、pH7.4、NaCl 150mM)に再懸濁し、不溶性物質を1000×gで1分間遠心分離することによって除去した。イムノブロット分析または液体窒素中での急速冷凍のために一定量を準備し、アッセイ時間まで−80℃で保存した。タンパク質濃度は、Triton X−100 0.5%を補給したBCAアッセイ(Thermo)を使用して測定した。
【0089】
(実施例9)
GOATアシルトランスフェラーゼアッセイ
アッセイ方法は、Yangら(36)を改変した。アッセイ混合物は、HEPES 50mM、pH7.0、オクタノイル−CoA(33dpm/fMol、American Radiolabeled Chemicals)1μM、グレリン27−ビオチン 10μM、ミクロソームタンパク質50μgおよびパルミトイル−CoA(Avanti Polar Lipids)50μMを含有した。反応は、膜タンパク質を添加することによって開始し、37℃で5分間インキュベートした。指示がある場合、GO−CoA−Tatは膜タンパク質と共に5分間予めインキュベートした。ストレプトアビジンプラスウルトラリンク樹脂(Pierce)150μlを予め混合した反応停止緩衝液(TBS+SDS 2%(w/v))850μlを添加し、回転ミキサーで15分間インキュベートすることによって反応を停止した。この混合物をPoly−Prep(登録商標)クロマトグラフィーカラム(Bio-Rad)に移し、洗浄緩衝液(TBS+SDS 0.1%)30mlで洗浄した。混合物は反応停止緩衝液1mlを含有する1.5ml微量遠心管に移し、98℃で5分間煮沸し、シンチレーション測定を行った。
【0090】
(実施例10)
細胞生存率アッセイ
様々な濃度のGO−CoA−Tatで処置したHEL細胞系およびヒト不死化肝細胞系HepG2(ATCC, Manassas, VA)の生存率は、LIVE/DEAD生存率アッセイキット(Molecular Probes)を使用して測定した。ラット骨髄間質細胞(RBMSC)は、2種類のプローブ、細胞内エステラーゼ活性についてはカルセイン−AM(緑色)2μMで30分間、細胞膜強度についてはエチジウムホモダイマー−1(EtdD-1、淡赤色)4μMで10分間インキュベートした。カルセイン−AMは、500nmレーザー光線を使用して励起し、放出した蛍光は535nmロングパスフィルターによって検出した。EtdD−1は、514nmレーザー光線を使用して励起し、放出した蛍光は550nmロングパスフィルターによって検出した。赤色蛍光(EtdD-1)を放つ細胞は死細胞として計数し、緑色蛍光(カルセイン-AM)は生細胞として計数した。部分的に赤および緑で2重に染色している場合は、死細胞として計数した。
【0091】
(実施例11)
ヒト膵島の実験
新鮮なヒト死体膵島は、National Islet Cell Resource centerから提供された。平均純度は70%、生存率は85%であった。ヒト膵島実験用に、膵島をGO−CoA−Tat 5μMを含む無血清RPMI培地で24時間インキュベートし、その後グルコース50、150および300mg/dlを添加した無グルコースRPMI培地中で30分間静的インキュベーションアッセイを行い、培地中に分泌されたインスリンをELISA(Alpco Diagnostics)で評価した。
【0092】
(実施例12)
アセチルトランスフェラーゼアッセイ
GO−CoA−Tatのアセチルトランスフェラーゼp300、PCAFおよびAANATとの特異性は、α−ケトグルタル酸脱水素酵素(α-KGDH)関連分光測定法(37)を使用して測定した。組換えアセチルトランスフェラーゼドメインおよびヒストン尾部ペプチドは、以前に記載されたように調製した(38〜40)。アセチルトランスフェラーゼによるCoASHの産生は、NADHの形成と共役しており、Beckman DU−640分光光度計によって340nm(ε340=6230M−1cm−1)でモニターした。反応物は全て、チアミンピロリン酸200μM、MgCl2 5mM、DTT 1mM、BSA 50μg/mL、NAD 200μM、α−ケトグルタル酸2.4mM、GO−CoA−Tat 10μMおよびアセチル−CoA 50μMを含有する。p300との反応は、HEPES 100mM、pH7.9(α−KGDH 0.1単位およびH4〜15 200μM、ヒストンH4尾部の配列に基づく15マーペプチド基質を含有する)中で実施した。p300反応は、30℃でインキュベートした後、p300 100nMを添加して開始して30℃で行う。PCAFとの反応は、HEPES 100mM、pH7.9(α−KGDH 0.037単位およびH3〜20 100μM、ヒストンH3尾部の配列に基づく20マーペプチド基質を含有する)中で実施する。PCAF反応は、30℃で10分間インキュベートした後、PCAF 100nMを添加して開始して30℃で行う。AANATとの反応は、NH4OAc 100mM、pH6.8(α−KGDH 0.1単位およびトリプタミン200μMを含有する)中で実施する。AANAT反応は、25℃で10分間インキュベートした後、AANAT 10.83nMを添加して開始して25℃で行う。反応は全て反応進行曲線の直線部分にあり、直線回帰によって初速度が得られる。
【0093】
(実施例13)
マウスにおける実験
24時間の実験では全て、C57BL/6マウスはGO−CoA−TatまたはD4−TatをIP注射する前に6時間絶食させた。各時点で、経心的穿刺によって血液を採取した。血液試料は、EDTAおよびp−ヒドロキシメルクリ安息香酸(PHMB)1mMを含有する試験管に収集した。試料を+4℃で3500rpmで5分間遠心分離して、次に上清を別の試験管に移した。次いですぐに細胞溶解物1mL当たり1N HCl 100μlを添加し、試験管を+4℃で3500rpmで5分間遠心分離した。上清をクライオバイアルに移し、−80℃で保存した。デスアシル−およびアシル−グレリンの血清レベルは、ELISA(Alpco Diagnostics)によって測定した。糖尿病の成果については、経口的グルコース曝露(1g/kg)を血液試料採取の30分前に行うこと以外は前記の方法にしたがった。糖尿病の成果のために、化合物を絶食していないマウスに18時間の絶食の6時間前に投与し、その時点で2.5g/kg腹腔内グルコース負荷試験(IP-GTT)を意識のあるマウスに実施した。血液は、IP−GTT後0、15、30、60および120分に尾静脈から採取した。グルコースは、グルコメータ(LifeScan OneTouch)で測定し、インスリン値はELISA(Alpco Diagnostics)によって評価した。毎日のGO−CoA−TatのIP投与の体組成に対する効果を評価するために、同腹仔の8週齢C57BL6Jマウス(Jackson Laboratories, Bar Harbor, ME)の体重を2群23.2±1.2g(n=3)および22.6±2.2(n=3)に一致させ、処置前に7日間高脂肪食を与えた。8日目に、マウスには高脂肪食を継続させたが、GO−CoA−Tat 29mg/kg用量(n=3)または等モル用量のD4−Tatペプチド(n=3)の投与を開始した。マウスは、食物および水の摂取ならびに糞尿の生成を毎日測定する直接熱量計測ケージ(Nalgene)中で2週間モニターした。さらに、マウスの身体プロファイルを3日毎にQNMR(EchoMRI)で評価した。身体測定それぞれについて、3つの別個の測定の平均を得た。糞便分析では、糞便を3日間収集し、1.5mL超遠心管に保存した。糞便は再水和して、GO−CoA−Tat(n=4)およびD4−Tat(n=4)の全糞便を4つの別個の測定と比較した。
【0094】
(実施例14)
QRT−PCR
膵島は膵臓をコラゲナーゼおよびDNアーゼIで消化した後、Songら(41)で記載されたように単離した。全RNAは、トリゾールを使用して抽出した。RT−PCRは、one−tube RT−PCR Sybr greenミックス(BioRad)を使用して標準的方法にしたがって実施した。発現レベルの倍数変化は、DDCT法を使用して算出した。2連の結果をステューデントt検定を使用して分析した。RT−PCRで使用したプライマー:インスリンFw:CGAGGCTTCTTCTACACACC;インスリンRv:GAGGGAGCAGATGCTGGT;グルカゴンFw:CCACTCACAGGGCACATTCA;グルカゴンRv:GTCCCTGGTGGCAAGATTGT GHSR FW:ACCTGCTCTGCAAACTCTTCCAGT;GHSR RV:CAAACACCACCACAGCAAGCATCT グレリンFw:ACTCAGCATGCTCTGGATGGACA;グレリンRv:ATGCCAACATCGAAGGGAGCATTG UCP2−001 Fw:TGGTTGGTTTCAAGGCCACAGATG;UCP2−001 Rv:TCTCGTGCAATGGTCTTGTAGGCT 36B4 Fw:TGTTTGACAACGGCAGCATTT;36B4 Rv:CCGAGGCAACAGTTGGGTA
【0095】
(実施例15)
免疫組織化学
マウスの膵臓およびヒト膵島を10%緩衝ホルマリンまたはブアン固定液で固定し、パラフィンに包埋し、5μmの切片を作製した。脱脂し、再水和してクエン酸緩衝液中で抗原を回復した後、免疫染色をモルモット抗インスリン(Abcam)、ウサギ抗グレリン(Abcam)およびニワトリ抗GHSR(Chemicon)で実施した。適切な蛍光タグ第2抗体(ロバ抗モルモットCy3、ロバ抗ウサギFITC、Jackson Immunoresearch)を抗原分布のために使用した。核は、封入媒体に含めたDAPI(Vector Biolabs)を使用して対比染色した。画像作製のために擬似カラー化用CCDデジタルカメラを装着したZeiss Axioskopおよびデジタル画像処理装置を連結したApotome光学切片装置(optical sectioning device)を使用した。
【0096】
(実施例16)
MCT食でのグレリン欠損(グレリンKO)および野生型(WT)マウスの長期調査
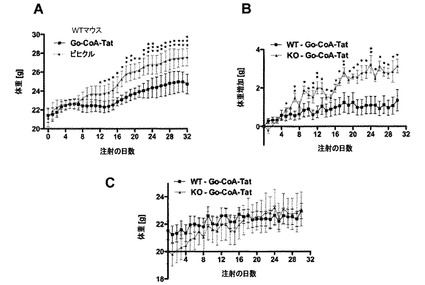
グレリン欠損マウスは、前述されたように、ハイスループットVelociGene遺伝子標的化システムを使用して作製した(42)。全動物は、22℃で12:12時間の明暗サイクルで維持し、中鎖トリグリセリドの豊富な食餌(MCT)−(MCT10%、スクロース40%、Teklad TD 08622)を与え、食餌および水は自由に摂取させた。GO−CoA−Tat 40mg/kgはWTおよびKOマウスに投与し、ビヒクルはWTマウスに投与した。全体の体組成(脂肪および除脂肪)は3〜4日毎にNMR技術(EchoMRI, Houston, TX)を使用して測定し、体重測定は毎日行った。結果を図11に示す。
【0097】
(実施例17)
グレリンKOおよびWTマウスにおけるグルコース負荷試験
糖尿病の成果のために、GO−CoA−Tat 40mg/kg(図12の「inhib」)を絶食していないwtマウス(n=5)およびグレリン−koマウス(n=5)に投与し、ビヒクル(2回蒸留水、図12の「Vhc」)を月齢を一致させたwtマウス(n=5)およびグレリン−koマウス(n=5)に投与し、6時間経ってから18時間絶食させた。18時間絶食した後、2.5g/kg腹腔内グルコース負荷試験(IP-GTT)を意識のあるマウスに実施した。血液は、IP−GTT後0、15、30、60および120分に尾静脈から採取した。グルコースはグルコメータ(Lifescan OneTouch)で測定した。
【0098】
(実施例18)
光活性化可能な(photoactivatable)GO−CoA−Tat類似体および可溶性GOATの架橋結合
FLAG−GOAT酵素を、SF9細胞で過剰産生し、界面活性剤で可溶化し、GO−CoA−Tatのベンゾイル−フェニルアラニン含有類似体およびビオチン含有類似体(5μM)の存在下でUV光で処理した。これらはビオチンブロットで示したように架橋結合を誘導し(図13A)、標準GO−CoA−Tat(100uM)で阻害することができた。FLAG(図13B)およびクーマシー(図13C)ゲルでは、これらのレーンへの負荷が等しいことを示している。
【0099】
(実施例19)
GOAT阻害剤はIGF−1に対して影響を及ぼす
血液は、EDTAコーティングMicrovette管を使用して経心的穿刺によって収集し、すぐに氷で冷却した。4℃で3000gで15分間遠心した後、血漿を−80℃で保存した。血漿IGF−1レベルを定量するために、市販のアッセイを使用した(R&D Systems, Minneapolis, MN)。このアッセイは、酸性化していない血漿試料におけるIGF−1の測定に有効で、タンパク質抽出を必要としない。このアッセイはアッセイ製造元の指示に従って実施した。結果を図14に示す。
【0100】
本明細書で例示し検討した実施形態は、本発明者が知っている本発明の作製および使用の最良の方法を当業者に教示するためのみのものである。本明細書において本発明の範囲の制限を意味するものはない。提示した実施例は全て代表的なものであって制限はしない。本発明の前述の実施形態は、前記の教示を考慮して当業者が理解するように、本発明から逸脱することなく改変または変化させることができる。したがって、特許請求の範囲およびその同等物の範囲内であれば、本発明は具体的に記載したものとは別に実施することができるものと理解される。
【0101】
(参考文献)
【化3】
【技術分野】
【0001】
本出願で記載した研究は、国立衛生研究所の助成金U54 RR020839およびR01 AG19186によって支援された。合衆国政府は、本出願に一定の権利を有する。
【0002】
本出願は、全ての目的のために参照により全体を本明細書に組み込まれる2008年10月3日出願の米国仮出願第61/102,549号に対する優先権を主張する。
【0003】
本発明は、グレリンO−アシルトランスフェラーゼの阻害剤およびその製造方法および使用方法に関する。
【背景技術】
【0004】
過去30年にわたり西欧社会において体重過剰者の割合が増加し続けていることは、罹病率のかなりの超過と関連があり、重大な公衆衛生問題として広く認識されている。この問題に取り組むために、体重増加に神経内分泌が寄与していることを明らかにするたゆみない努力を重ねている。レプチン(1)の単離を始めとして、中枢および末梢に作用して体重に影響を及ぼす一連のホルモンが発見されてきた。中でも、胃ペプチドホルモン、アシル−グレリンは、体重増加の重要な刺激因子(2〜5)およびグルコース恒常性の調節因子(6〜8)として非常に興味を持たれてきた。治療の発展における様々な戦略の中で、アシル−グレリンの拮抗作用が記載されているが(9)、未だに臨床的に有益性なものは出現していない。
【0005】
アシル−グレリンの生合成には、グレリンペプチドの3位のセリンの通常にはない翻訳後オクタノイル化が関与する。このオクタノイル化は、成長ホルモン分泌促進物質受容体(GHSR)との相互作用を介して行われる生理活性に必要である。このエステル化を担う酵素、グレリンO−アシルトランスフェラーゼ(GOAT)が最近クローニングされた(10、11)。
【発明の概要】
【発明が解決しようとする課題】
【0006】
当業界では依然として、肥満および糖尿病の治療に使用するための改善された治療薬、特に神経内分泌経路を標的とした治療薬が必要とされている。毎週推定10,000例の早死の原因となっている世界的な肥満の流行に対処する中で、効果的な食欲抑制薬物療法は大規模に命を救う可能性があり得る。
【課題を解決するための手段】
【0007】
本発明者らは、GOATの阻害剤が肥満患者の満腹感および体重減少を誘導できることを発見した。さらに、アシル化グレリンを遮断すると膵島のグルコース応答性を改善することができ、したがってGOAT阻害剤はまた、II型糖尿病患者集団で使用するための有効な治療薬として期待される。
【0008】
これらの目的およびその他の目的は、2基質類似体グレリンO−アシルトランスフェラーゼ(GOAT)阻害剤、例えば、インビトロ、培養細胞およびマウスにおいて強力、選択的かつ効果的であるGO−CoA−Tatなどを提供することによって実現される。GO−CoA−Tatなどの化合物の投与によって、グルコース負荷に対するインスリン応答が増強され、高脂肪食を与えられた対象において統計学的に有意な体重減少が導かれる。例えば、GO−CoA−Tatによる治療はまた、除脂肪量よりも体脂肪量に対して選択的な減少を導く。
【0009】
一態様では、本発明は、膜結合O−アシルトランスフェラーゼ(mBOAT)膜タンパク質、例えば、グレリンO−アシルトランスフェラーゼを阻害することができる化合物を提供する。いくつかの実施形態では、化合物は下式を有する。
【化1】
【0010】
式中、Rは存在しないか、あるいは置換もしくは非置換のアルキル基、置換もしくは非置換のペプチド鎖、置換もしくは非置換の多糖、またはペプトイド残基(例えば、N−メチルもしくはその他のN−アルキル置換グリシンなど)からなる群から選択される有機部分である。Rは、ペプチド鎖の場合、例えば、1〜20個のアミノ酸残基の鎖であってもよい。いくつかの実施形態では、Rはグレリン残基4〜28、または4〜15、または4〜10または4〜5に対応するペプチド鎖である。
【0011】
本明細書では、「アミノ酸」は、天然アミノ酸および合成アミノ酸の両方を含む。これらには、特に、アラニン、アルギニン、アスパラギン、アスパラギン酸、システイン、グルタミン酸、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、プロリン、セリン、トレオニン、トリプトファン、チロシン、バリン、5−ヒドロキシリシン、4−ヒドロキシプロリン、チロキシン、3−メチルヒスチジン、ε−N−メチルリシン、ε−N,N,N−トリメチルリシン、アミノアジピン酸、γ−カルボキシグルタミン酸、ホスホセリン、ホスホトレオニン、ホスホチロシン、N−メチルアルギニンおよびN−アセチルリシンが含まれる。アルファアミノ酸のR型およびL型の両方ならびにR型およびL型の混合物が企図される。さらに可能性のある置換基には、アミノ基とカルボキシル基の間に2個以上の−CH2−基(通常2〜4個)が存在するアミノ酸およびそれらの誘導体が含まれる。したがって、アミノ酸残基は、アルファ、ベータ、ガンマまたはさらに高級な(higher order)アミノ酸の残基であることができ、好ましくはその他に関しては天然に生じるアミノ酸に対応する残基であることができる。本明細書では、「アミノ酸残基」には、例えば、このような鎖が直鎖であろうと枝分かれ鎖であろうと、アミノ酸配列またはその他の鎖構造に組み込まれた、前記に挙げた任意のアミノ酸が包含される。
【0012】
別の態様では、本発明は組成物を提供する。例えば、組成物は本発明の化合物および薬学的に許容される賦形剤または担体を含むことができる。
【0013】
さらに別の態様では、本発明は本発明の化合物の調製方法を提供する。例えば、この方法は、3〜15個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、D4-Tatがペプチド配列の一末端に結合している工程と、ブロモオクタノイル化中間体を得るために、1または複数個の反応容器中でグレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、前記ブロモオクタノイル化中間体と補酵素Aを一緒にする工程とを含むことができる。
【0014】
さらに別の態様では、本発明は本発明の化合物および組成物を使用して、疾患、障害または症状を治療する方法を提供する。いくつかの実施形態では、この方法は肥満を治療するために使用される。これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。本発明は、例えば、対象の除脂肪量(lean mass)よりも大きく脂肪量減少をもたらすことができるか、または対象におけるアシル−グレリンに対するグレリンの比の増加をもたらすことができる。いくつかの実施形態では、この方法は糖尿病、例えば、II型糖尿病を治療するために使用することができる。これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。これらの方法は、例えば、対象におけるインスリン産生増加、または対象におけるグルコース曝露に対する応答性増加、または対象における脱共役タンパク質2(UCP−2)mRNAレベルの減少をもたらすことができる。いくつかの実施形態では、この方法は過敏性腸症候群を治療するために使用することができる。これらの方法は、本発明の化合物の1または複数種の治療有効量を含む組成物を対象に投与することを含むことができる。
【0015】
別の態様では、本発明はmBOAT関連疾患の治療方法を提供する。いくつかの実施形態では、これらの方法は、本発明の1または複数種の化合物の治療有効量を含む組成物を対象に投与することを含むことができる。
【0016】
さらに別の態様では、本発明はキットを提供する。例えば、このキットは、本明細書で開示する化合物および組成物の治療有効量の少なくとも1用量を含む組成物を含むことができる。いくつかの実施形態では、このキットはまた、さらなる体重減少治療薬の少なくとも1用量を含む。
【図面の簡単な説明】
【0017】
【図1】GO−CoA−Tatは、GOATを阻害し、アシルグレリンレベルを低下させる2基質阻害剤である。(A)2基質類似体阻害剤が、2種類の基質を共有結合的に一緒にし(covalently combine)、それによって効力および特異性を高めることを示す図である。(B)D4-Tatの構造ならびに2基質阻害剤の合成スキームを示した図であり、2基質阻害剤は、3つの成分またはそれらの実質的な同等物:補酵素A;オクタノイル化部分、例えば、置換または非置換のグレリンペプチドおよび;Tatペプチドからなる。(C)本発明による化合物の構造を示した図である。(D)24時間インキュベートした後の、HEL細胞溶解物(2.5×106細胞)における、GO−CoA−Tatによるアシル−グレリンレベルの用量応答性減少を示した図である。(E)HEL細胞溶解物(2.5×106細胞)中におけるGO−CoA−Tat 2μMの経時的(temporal)阻害を示した図である。(F)インビトロアッセイで、グレリン−O−アシルトランスフェラーゼ(GOAT)はGO−CoA−Tat 100nMによって阻害されることを示した図である。(G)図1Cに挙げた7種類の化合物の1つと24時間インキュベートした後の、HEL細胞溶解物(2.5×106細胞)におけるアシル−グレリンレベル(pg/ml)を示した図である。
【図2】GO−CoA−Tatは、マウスにおいて、循環アシル−グレリンレベルを低下させるが、デスアシル−グレリンレベルは低下させないことを示した図である。(A)GO−CoA−Tat 29mg/kgで処理したマウス(n=3)は、D4-Tatで処理した対照マウスと比較して、アシル−グレリンの統計学的に有意な減少を示した。(B)12時間後および24時間後のデスアシル−グレリンレベルは、GO−CoA−Tatで処理したマウスでは同等モルのD4-Tatで処理した対照マウス(n=3)に対して統計学的に有意な差を示さなかったことを示す図である。GO−CoA−Tat 15mg/kgでは、阻害はより小さいが類似の結果が認められた(C,D)。
【図3】[マウスの体重および体組成に対するGO−CoA−Tatの効果を示す図である。GO−CoA−Tat 29mg/kgで処理したマウス(n=3)は、D4-Tatで処理したマウスに対して、(A)体重の統計学的に有意な減少を示し(*<0.05、3日間の標準誤差)、(B)体脂肪のグラムの統計学的に有意な減少を示し、(C)除脂肪量の統計学的に有意な差を示さず、(D)体脂肪の割合の統計学的に有意な減少を示した(*<0.05、**<0.005)。
【図4】GO−CoA−Tatはインスリンを増加させ、グルコースレベルを減少させ、UCP2mRNAを下方制御することを示した図である。経口的にグルコースを負荷する24時間前に化合物を投与(1g/kg)した場合、腹腔内グルコース負荷試験(IPGTT)(2.5g/kg)後30分毎に血糖値およびインスリン値を測定したとき、GO−CoA−Tat 29mg/kgで処理したマウス(n=4)は、D4-Tatで処理した対照マウス(n=6)と比較して、(A)インスリン分泌の統計学的に有意な増加を示し、(B)血糖値の統計学的に有意な減少を示した。分離の24時間前に阻害剤で処理したマウスから分離した、(C)膵島のQRT−PCRおよび(D)胃底部のQRT−PCRを示す;mRNA発現を単離し、対照(n=3)と比較して測定した。
【図5】GO−CoA−Tatは、AGSおよびKatoIII細胞においてアシル−グレリンレベルを低下させるが、デスアシル−グレリンレベルは低下させない。(A)AGS細胞溶解物(2.5×106細胞)において、様々な濃度のGO−CoA−Tatで24時間インキュベートした後のアシル−グレリン(pg/ml)の用量応答性阻害を示した図である。(B)AGS細胞溶解物(2.5×106細胞)におけるGO−CoA−Tat 6μMの経時的阻害を示した図である。(C)HEL細胞溶解物(2.5×106細胞)中において、様々な濃度のGO−CoA−TatおよびD4-Tatと24時間インキュベートした後のデスアシル−グレリン(pg/ml)の用量応答性応阻害を示した図である。KatoIII細胞において、(D)GO−CoA−Tatで24時間インキュベートした後の用量応答性阻害および(E)GO−CoA−Tat 6μMの経時的阻害を示した図である。
【図6】GOATのインビトロアッセイの確立。GOATはHEK293TGnTI(−)細胞から単離されたミクロソーム内に存在した。HEK293T GnTI(−)細胞に、GOAT−3xFlagまたは空のベクター(対照)をトランスフェクトし、ミクロソームは実施例で説明したように調製した。(A)トランスフェクトした細胞およびトランスフェクトしていない細胞のミクロソーム(50μgのタンパク質)を抗FLAGモノクローナル抗体10μg/mLでイムノブロット分析した図である。50℃でインキュベートしたGOATトランスフェクトミクロソームではSDS−PAGEにおいて5分で色素負荷したが、同じ時間100℃で煮沸したGOATトランスフェクトミクロソームではせず、42kDaのバンドがGOATの存在を示すことが明らかになった。(B)GOAT活性はトランスフェクトした細胞のミクロソーム中には存在しているが、トランスフェクトしていない細胞のミクロソームには存在しておらず、GO−CoA−Tat 100nMによって完全に阻害されることを示した図である。
【図7】GOCoA−Tatの非特異的阻害および毒性のアッセイ。(A)p300、PCAFおよびAANATにおけるGO−CoA−Tat 10μMの存在下でのパーセント活性。GO−CoA−Tatの濃度を変化させて24時間インキュベートした後の(B)HepG2細胞の生存率および(C)HEL細胞の生存率を示した図である。
【図8】摂取および排泄に対するGO−CoA−Tatの効果。GO−CoA−Tat 29mg/kg(n=3)およびD4-Tat(n=3)で処理したマウスは(A)食物の累積消費および(B)水の累積消費に統計学的に有意な差を示さなかった。これらのマウスの(C)累積糞便生成および(D)尿生成も、統計学的に有意な程度の差を示さなかった。
【図9】D4−TatおよびGOCoA−Tatで処理したマウスの血液パネルを示した図である。毎日化合物を投与して12日後の午後2時に採取した試料について、GO−CoA−TatマウスではD4-Tat処理マウスと比較して高密度リポタンパク質(p=0.04)およびトリグリセリド(p=0.02)の統計学的に有意な増加が示された。2つの群の間のその他の値には統計学的に有意な差は認められなかった。
【図10】膵島およびマウスにおける、インスリンおよびグルコースに対するGO−CoA−Tatの効果。(A)化合物GO−CoA−Tat 5μMとインキュベートすることによって、ヒト膵島をグルコース150mg/dlおよび300mg/dl中でインキュベートしたときに(n=9)、インスリン産生の統計学的に有意な増加が引き起こされる。GO−CoA−Tat 15mg/kgで処理したマウス(n=6)は、(B)インスリン分泌の統計学的に有意な増加を示し、(C)血糖の統計学的に有意な減少を示した。
【図11】GOAT拮抗剤を含む、または含まない中鎖トリグリセリド食による、野生型またはグレリンノックアウトマウスにおける体重増加を示した図である。GOAT阻害剤GO−CoA−Tatは野生型の体重に影響を及ぼすが、遺伝子改変した(グレリンノックアウト)マウスには及ぼさず、グレリン経路を標的としていることが裏付けられる。
【図12】GOAT拮抗剤を投与された野生型およびグレリンノックアウトマウスにおける、グルコース負荷試験を示した図である。GOAT阻害剤GO−CoA−Tatは野生型のグルコースレベルに影響を及ぼすが、遺伝子改変した(グレリンノックアウト)マウスには及ぼさず、グレリン経路を標的としていることが裏付けられる。
【図13】光活性化可能なGO−CoA−Tat類似体は、可溶性GOATを架橋結合することができる。GO−CoA−Tatの2つの光活性化可能変種は、GOATに直接結合し、化学的に架橋結合することができる。
【図14】GOAT阻害剤は、雌雄マウスの血液IGF1レベルを抑制することを示した図である。これは、アシル−グレリンに対する影響と矛盾しない。
【発明を実施するための形態】
【0018】
本発明の実施形態を以下に詳細に論じる。記載する実施形態では、明瞭にするために特定の用語を使用する。しかし、本発明はそのように選択した特定の用語に限定されるものではない。当業者であれば、本発明の精神および範囲を逸脱することなく、その他の同等の部分を使用でき、その他の方法を開発できることを理解するであろう。本明細書で引用した参考文献は全て、それぞれが個々に組み込まれるように、全体があらゆる目的のために参考として組み込まれる。
【0019】
化合物
胃によって分泌される強力な食欲刺激ホルモン、グレリンは、動物およびヒトの体重増加を刺激する重要なペプチドホルモンであるとされてきた。その効果を発揮するために、グレリンは、アシル化、すなわち3位のセリン残基の通常ではない翻訳後オクタノイル化を必要とする。グレリンのオクタノイル化型は、通常アシル−グレリンと呼ばれる。
【0020】
グレリンのオクタノイル化は、グレリンO−アシルトランスフェラーゼ(GOAT)によって媒介される。GOATは膜タンパク質の膜結合O−アシルトランスフェラーゼ(mBOAT)ファミリーのメンバーである。本明細書で開示するのは、インビトロ、培養細胞およびマウスにおいて、強力、選択的かつ効果的な2基質類似体GOAT阻害剤である、GO−CoA−Tatである。
【0021】
いくつかの実施形態では、本発明は、例えば、肥満および/または糖尿病の治療で使用するための化合物を提供する。この化合物は一般式(I)を有することができる。
【0022】
【化2】
【0023】
式Iでは、本明細書で開示するように、Rは存在しなくてもよく、またはRは化合物の適切な機能と矛盾しない任意の有機部分であることができる。Rは、例えば、アルキル基、ならびにペプトイド残基(例えば、N−アルキルGly)、ベータアミノ酸、アミノラク酸およびフェニルGlyの組み合わせであることができる。いくつかの実施形態では、Rはアミノ酸残基で構成されるペプチド鎖、例えば、グレリンペプチドのフラグメントを含むペプチド鎖などである。グレリンペプチドのフラグメントは、例えば、残基4で開始してもよい。Rは、例えば、グレリンペプチドの残基4〜5または4〜10または4〜15または4〜28で構成されてもよい。実際に、本発明は、Rが本明細書で開示するように本発明による機能を可能とする任意の数の残基であってもよいことを企図する。式Iの化合物は「GO−CoA−Tat」とも呼ばれる。さらに、式Iによって定義された属に含まれる化合物の特定の例も、「GO−CoA−Tat」と呼ぶことができる。GO−CoA−Tatは、グレリンのオクタノイル化を媒介してアシル−グレリンを生成するGOATの阻害剤である。
【0024】
式Iにおいて、「GS」で示した部分は、従来の一文字命名法にしたがって、グリシンおよびセリンそれぞれに対応するアミノ酸残基で構成される。グリシンおよびセリンは、本明細書で開示するように本発明にしたがって機能することができる、任意の長さのペプチド鎖を含む任意の有機部分で置換することができる。セリン残基の適切な置換基の例には、例えば、D−AlaまたはD−SerなどのD−アミノ酸が含まれ、これらはまた、アセチル基またはプロピオニル基に切断されていてもよく(truncated to)、ヒドロキシアセチル置換基であってもよい。グリシン残基の適切な置換基の例には、セリンについて挙げたものならびにペプトイド残基(例えば、N−アルキルGly)、ベータアミノ酸、アミノラク酸およびフェニルGlyが含まれる。本明細書では、「ペプチド鎖」は、ペプトイド残基およびアミノ酸のその他の変種または誘導体を含む部分を包含し、天然に生じるアミノ酸、ペプトイド残基およびその他のアミノ酸変種または誘導体あるいはそれらの任意の部分集団の組み合わせからなる部分も包含する。
【0025】
「Dap」とは、1,2−ジアミノプロピオン酸を意味する。GS−Dap−RがグレリンペプチドのN末端に由来する場合、通常3位に存在するセリンをDapが置換する。Dapは、側鎖の酸素が窒素に置換されたセリンアイソスターで、アミド結合を導く。「Ahx」とは、RとTat配列を結合するのに用いられるアミノ−ヘキサノイルリンカーを意味する。Ahxは、例えば、アミノヘキサン酸、グリシン、アミノプロピオン酸、アミノ酪酸、アミノペンタン酸およびエチレングリコールをベースにした置換基であることができる。
【0026】
「転写のトランス作用活性化剤」の短縮形である「Tat」は、HIVウイルスで通常見出されるペプチド配列を意味する。例えば、「Tat」は、アミノ酸YGRKKRRQRRRを含む11残基のペプチドであることができる。Tatタンパク質は、分子、例えば、タンパク質またはその他の部分の細胞膜通過輸送を媒介することができる。このような媒介は、分子の大きさには関係ないものと考えられ、細胞膜のいかなる破壊も伴わないようである。いかなる特定の理論にも結びつけることを望まないが、GO−CoA−TatのTat部分は化合物の細胞内部への輸送の助けとなると仮定される。したがって、同様にこのような輸送の助けになる任意の部分が、本発明の化合物のTatと置換していてもよい。Tatの適切な置換基の例には、例えば、オリゴArg、オリゴアルキルグアニジウムおよびTatのd−もしくはl−立体異性体のいずれかが含まれる。「D4−Tat」は、本明細書では、Tat配列に結合した4個のアスパラギン酸残基のペプチドを含む。D4-Tatは、Fmoc方法またはそれらの変法を使用して調製した。
【0027】
いかなる特定の理論にも結びつけることを望まないが、GO−CoA−Tatの設計は、オクタノイル−CoAおよびグレリンペプチドを鋳型とする3要素複合体機構をGOATが使用するならば、切断可能でない架橋により2個の基質を結合すると、3要素複合体の形成に関連したエントロピーを損失せずに個々のリガンドの結合エネルギーを一緒にすることができるという理論に基づいている(図1A)。関連のある戦略が、ヒストンアセチルトランスフェラーゼ(HAT)およびタンパク質キナーゼを含むその他のペプチド修飾酵素にうまく使用されてきた。グレリンペプチドを式IのR基として使用する場合に、GOATによる認識に必要なグレリンペプチド長について我々は不確かであったので、高度に保存されたグレリン残基を最大限に含めるため、オクタノイル−CoAへのカップリングにアミノ酸1〜10を選択した。Tat由来ペプチド配列、例えば、11マー HIV Tatも、細胞透過を高めるために、アミノヘキサノイルリンカーを介してC末端に結合することができる。
【0028】
この研究に着手したとき、インビトロGOATアッセイはまだ開発されておらず、したがって3種類の細胞においてアシル−グレリン産生に対するGO−CoA−Tatの影響を調べた。D4-Tat(Tatペプチドに同じように結合する、負のCoA電荷を模倣したテトラ−アスパラギン酸、またはDDDD)を対照として使用した。試験した3種類の細胞系それぞれにおいて、アシル−グレリンのレベルは、D4-Tat対照と比べてGO−CoA−TatによってIC50が1〜2μMの範囲で有意に減少したが、デスアシル−グレリンは減少しなかった。
【0029】
GO−CoA−TatはGOATの選択的拮抗剤であると考えられ、その理由は、GO−CoA−Tatが10μMで、インビトロにおいて、p300 HAT、PCAF HATおよびセロトニンN−アセチルトランスフェラーゼを含む、アセチル−CoAを利用する3種類のその他の酵素を15%未満阻害することが示されたからである(図7)。さらに、GO−CoA−Tat 10μMはHEL細胞およびHEPG2細胞の生存率に非毒性であると考えられる(図7)。さらに、GO−CoA−TatはGOAT活性の拮抗において真正な2基質類似体として作用するようである(図1G)。Tat配列または細胞内移行を助けるその他の部分を含めると阻害活性が増加するという事実によって、細胞透過が関与しており、この化合物は細胞表面受容体には作用しないことが示唆される。
【0030】
本発明の一態様では、GO−CoA−Tatは、グルコース負荷に対するインスリン応答を高め、哺乳類、例えば、高脂肪食を与えられたマウスにおいて相対的な体重減少を導くために、例えば、腹腔内に投与する。定量的MR分光法によって、GO−CoA−Tat処理が除脂肪量よりも脂肪量に対して選択的な減少を導くことが立証された。したがって、GOATは肥満およびグルコース不耐性の治療の標的となることができ、GO−CoA−Tatは有効なGOAT阻害剤となる。
【0031】
組成物
いくつかの実施形態では、本発明は式Iの化合物および薬学的に許容される賦形剤を含む組成物を提供する。本明細書では、「薬学的に許容される」は、健全な医学的判断の範囲内で、妥当な便益/リスク比に見合って、過剰な毒性、刺激、アレルギー応答またはその他の問題となる合併症がなくヒトおよび動物の組織との接触に適した化合物、物質、組成物および/または剤形を包含する。いくつかの実施形態では、用語「薬学的に許容される」は、連邦政府または合衆国政府の規制当局によって承認されているか、あるいは合衆国薬局方または動物、より特定すればヒトで使用するために一般的に認識されているその他の国際的薬局方に挙げられていることを意味する。本明細書では、「賦形剤」とは、医薬組成物の製剤で使用される物質を意味し、それ自体は治療価値の無いものであってもよい。参考として組み込んだRemington: The Science and Practice of Pharmacy, 21st Ed. (2006)において記載されたものを含む様々な賦形剤を本発明で使用することができる。賦形剤には、限定はしないが、抗酸化剤、(治療効果を示すものとは対照的に)製剤自体の崩壊を防ぐ抗菌剤、保存剤、キレート化剤、緩衝剤、毒性を調節するための薬剤、着色剤、芳香剤および希釈剤、乳化剤および懸濁剤ならびに薬学的に適用されるその他の物質が含まれる。
【0032】
本発明の組成物は、肥満治療のその他の形態、例えば、食事および運動または肥満外科手術と併用して投与することができる。このような併用療法では、本発明の組成物はその他の形態の肥満治療の前、同時または後に投与することができる。
【0033】
本発明に従って使用するための医薬組成物は、薬剤として使用することができる調製物への活性成分のプロセシングを容易にする賦形剤および補助剤を含む一または複数種の生理学的に許容される担体を使用して従来の方法で製剤化することができる。適切な製剤は、選択した投与経路に左右される。
【0034】
適切な投与経路は、例えば、経口、直腸、経粘膜、特に経鼻、局所、腸内または非経口送達を含んでもよく、筋肉内、皮下および脊髄内ならびにクモ膜下腔内、直接脳室内、静脈内、腹腔内、鼻腔内または眼内注射を含む。医薬組成物は、局所的または全身的に投与することができる。例えば、組成物は、患者の体の特定の領域に直接調製物を注射することによって局所的に投与することができる。
【0035】
注射のために、本発明の活性成分は水性溶液、好ましくはハンクス液、リンゲル液または生理学的塩緩衝液などの生理学的に適合した緩衝液で製剤化してもよい。経粘膜投与のために、浸透させるバリアに適した浸透剤が製剤中で使用される。このような浸透剤は当業界では一般的に公知である。
【0036】
経口投与のために、化合物は当業界で周知の薬学的に許容される担体と活性化合物を一緒にすることによって容易に製剤化することができる。このような担体は、患者による経口摂取のために、本発明の化合物の錠剤、丸剤、糖衣錠、カプセル、液剤、ジェル、シロップ、スラリー、懸濁物などへの製剤化を可能にする。経口使用のための薬理学的調製物は、所望により、錠剤または糖衣錠の核を得るために適切な補助剤を添加した後、固形賦形剤を使用し、得られた混合物を場合により粉砕し、顆粒の混合物を加工して形成することができる。適切な賦形剤は、特に:乳糖、スクロース、マンニトールまたはソルビトールを含む糖などの充填剤;例えば、トウモロコシ澱粉、コムギ澱粉、米澱粉、ジャガイモ澱粉、ゼラチン、トラガカントガム、メチルセルロース、ヒドロキシプロピルメチル−セルロース、カルボキシメチルセルロースナトリウムなどのセルロース調製物;および/またはポリビニルピロリドン(PVP)などの生理学的に許容されるポリマーである。所望により、例えば、架橋結合ポリビニルピロリドン、寒天またはアルギン酸もしくはアルギン酸ナトリウムなどのそれらの塩のような崩壊剤を使用してもよい。
【0037】
糖衣錠核は適切なコーティングを施す。このために、場合によりアラビアゴム、タルク、ポリビニルピロリドン、カーボポールゲル、ポリエチレングリコール、二酸化チタン、塗料溶液および適切な有機溶媒または溶媒混合物を含有してもよい、濃縮糖溶液を使用することができる。色素または顔料は、活性化合物用量の様々な組み合わせを識別し、または特徴付けるために、錠剤または糖衣錠コーティングに添加することができる。
【0038】
経口使用できる医薬組成物には、ゼラチンで作られた押し込み型(push-fit)カプセル、ゼラチンおよび可塑剤、例えば、グリセロールまたはソルビトールで作られた密封軟カプセルが含まれる。押し込み型カプセルは、乳糖などの充填剤、澱粉などの結合剤、タルクまたはステアリン酸マグネシウムなどの潤滑剤および場合により安定化剤と混合して活性成分を含有することができる。軟カプセルでは、活性成分は適切な液体、例えば、脂肪油、液体パラフィンまたは液体ポリエチレングリコールに溶解または懸濁することができる。さらに、安定化剤を添加することができる。経口投与のための製剤は全て、選択した投与経路に適した投薬量であるべきである。
【0039】
頬側投与のためには、組成物は従来の方法で製剤化された錠剤またはトローチ剤の形態をとることができる。
【0040】
鼻吸入による投与のためには、本発明によって使用するための活性成分は、適切な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロ−テトラフルオロメタンまたは二酸化炭素を使用して加圧容器または噴霧器からエアロゾルスプレー状の形態で適宜送達させる。加圧エアロゾルの場合では、投薬単位は計量した量を送達するために弁を形成することによって測定することができる。ディスペンサーで使用するためのゼラチンなどのカプセルまたはカートリッジは、化合物および乳糖または澱粉などの適切な粉末基剤の粉末ミックスを含有して製剤化することができる。
【0041】
本明細書で記載した調製物は、例えばボーラス注射または連続注入によって、非経口投与用に製剤化することができる。注射用製剤は、例えば、アンプルまたは反復投与容器に入れて、場合により保存剤を添加して、単位投与形態にすることができる。組成物は、油性または水性媒体に溶かした懸濁液、溶液またはエマルジョンであってもよく、懸濁剤、安定化剤および/または分散剤などの製剤化剤を含有してもよい。
【0042】
非経口投与用の医薬組成物には、水溶性形態の活性調製物の水溶液が含まれる。さらに、活性成分の懸濁液は適当な油または水をベースにした注射用懸濁液として調製することができる。適切な親油性溶剤またはビヒクルは、ゴマ油などの脂肪油、またはオレイン酸エチル、トリグリセリドまたはリポソームなどの合成脂肪酸エステルを含む。水性注射用懸濁液は、カルボキシメチルセルロースナトリウム、ソルビトールまたはデキストランなどの懸濁液の粘度を高める物質を含有することができる。任意選択により、懸濁液はまた、適切な安定化剤または高濃度溶液の調製を可能にするために活性成分の溶解度を高める薬剤を含有することができる。
【0043】
あるいは、活性成分は、使用前に適当なビヒクル、例えば、発熱性物質を含まない無菌水をベースにした溶液によって構成するための粉末形態であってもよい。活性成分はまた、当業界で公知の方法によってペグ化を行うことができる。
【0044】
本発明の調製物はまた、カカオ脂またはその他のグリセリドなどの従来の座薬基剤を使用して、座薬または停留浣腸剤などの直腸用組成物として製剤化することができる。
【0045】
本発明の状況で使用するために適した医薬組成物には、活性成分を企図した目的を実現するために有効な量で含有する組成物が含まれる。より特定すれば、治療有効量は、疾患の症状の予防、軽減または回復あるいは治療した対象の生存を延長させるために有効な1または複数種の活性成分の量である。
【0046】
本明細書で記載した活性成分の毒性および治療効果は、インビトロ、細胞培養物または実験動物において、標準的な薬学的手法によって測定することができる。これらのインビトロおよび細胞培養物アッセイならびに動物研究から得られたデータは、ヒトにおいて使用するために用量の範囲の製剤設計において使用することができる。投薬量は、使用する剤形および利用する投与経路に応じて変化させることができる。正確な処方、投与経路および投薬量は、患者の状態を考慮して個々の医師が選択することができる。
【0047】
治療する状態の重症度および応答性に応じて、投薬は1回または複数回投与であることができ、治療過程は、数日から数週間まで持続するか、または治癒が達成されるか、または疾患状態の軽減が実現されるまでであることができる。
【0048】
投与する組成物の量は、もちろん、治療する対象、苦痛の重症度、投与方法、処方する医師の判断などに応じて左右される。
【0049】
適合した医薬担体中に製剤化された本発明の調製物を含む組成物も、調製し、適切な容器に入れ、適応する症状の治療を表示することができる。
【0050】
本発明の医薬組成物は、当業界で周知の工程によって、例えば、従来の混合、溶解、顆粒化、糖衣錠形成、微粒子化、エマルジョン化、カプセル化、封入または凍結乾燥工程によって製造することができる。
【0051】
調製方法
別の態様では、本発明は式Iの化合物の調製方法を提供する。いくつかの実施形態では、この方法は、3〜15個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、Tat配列がペプチド配列の一末端に結合している工程と、ブロモ−オクタノイル化中間体を得るために、1または複数個の反応容器中で、グレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、前記ブロモオクタノイル化中間体と補酵素Aを一緒にする工程とを含むことができる。
【0052】
Tatは、例えば、式IのR配列の一末端に結合させることができる。例えば、Rがペプチド配列の場合、Tatはペプチド配列のC末端に結合させることができる。いくつかの実施形態では、Tatは、連結部分、例えば、アミノ−ヘキサノイルリンカーなどによって結合するが、GO−CoA−Tatの適切な機能を妨害しない任意の連結部分は本発明の範囲内である。
【0053】
「試薬K」は、トリフルオロ酢酸、水、チオアニソール、フェノールおよびエタンジチオールの標準的混合物を意味する。保護されたアミノ酸を脱保護し、樹脂から切断するために使用される。
【0054】
GO−CoA−Tat(例えば、図1Cで挙げたような化合物1)の合成は、例えば、固相方法を使用して実施することができる(図1B)。ペプチド長の長さが異なり、CoA、オクチルおよびTatそれぞれが個々に欠損している一連の関連化合物(化合物2〜7、図1C)も合成して(図1C)、本発明にしたがって使用することができる。
【0055】
予防方法および治療方法
別の態様では、本発明は、治療有効量の式Iの化合物を含む組成物を使用して、様々な疾患、障害または症状を予防および/または治療する方法を提供する。
【0056】
[0070] 「治療」とは、本明細書で記載した疾患、障害または状態の症状または有害な影響の少なくとも1つを治癒、逆戻り、軽減、緩和、最小化、抑制または停止させることを意味する。「治療」とは、疾患、障害または症状を有する対象の治療的処置のいずれも意味する。「予防」とは、障害の徴候または症状を発症していない対象における予防的または防御的処置を意味する。「治療」とは、例えば、症状の軽減;症状、障害または疾患の範囲の減少;症状、障害または疾患の状態の安定化(すなわち、悪化阻止);症状、障害または疾患進行の緩徐化;症状、障害または疾患状態の回復;部分的または全体的な寛解;あるいは症状、障害または疾患の改善もしくは改良を包含する。治療を必要とするものには、既に障害を有するものが含まれる。防御または予防を必要とするものには、障害を防御できるものが含まれる。したがって、本明細書で開示する化合物を投与する患者は、障害を有すると診断されていてもよく、または障害に罹患しやすいか、もしくは感受性であってもよい。「治療すること」には、測定可能な応答、好ましくは過剰なレベルの副作用無しに臨床的に重要な応答を惹起することが含まれる。「治療すること」にはまた、治療を受けていない場合に予測される生存と比較して、生存を延長させることが含まれる。
【0057】
本発明による対象は、本明細書で記載した疾患、障害または症状の1つであると診断された、あるいは本明細書で記載した疾患、障害または症状の少なくとも1種類に罹患している哺乳類、例えば、ヒトである。本発明の組成物は、本発明の化合物の有益な効果を経験することができる、組成物を必要とするあらゆる哺乳類に投与することができる。このような任意の哺乳類は、「対象」と考えられる。このような対象には、ヒトおよび非ヒト、例えば、ヒト、飼育動物および家畜、動物園用、スポーツ用もしくは愛玩用動物、例えば、イヌ、ウマ、ネコ、ウシなどが含まれる。対象は男性または女性であることができる。
【0058】
治療有効量を構成するために必要な活性剤の量は、治療する障害の重症度;患者のアイデンティティ、年齢、体重、一般的な健康状態、性別、食事および化学的組成(chemical make-up);実現する細胞応答の種類および程度;使用する特定の薬剤または組成物およびその活性;投与時間、投与経路および薬剤の排泄速度;治療期間;特定の薬剤と組み合わせてまたは同時に使用する薬剤;ならびに医薬品業界で周知の類似の要素を含むいくつかの要素に基づいて変化する。一般的に、治療有効量は、疾患または障害に関連した1または複数種の症状の測定可能な軽減を生じる量である。
【0059】
例えば、肥満の治療又は予防のための治療有効量は、最大でまたは少なくとも1日あたり、約1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、5mg/kg、7.5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kgまたはそれ以上の活性薬剤であることができる。70kgの人では、治療有効用量は、最大でまたは少なくとも1日あたり、約50mg、60mg、70mg、80mg、90mg、100mg、120mg、140mg、160mg、180mg、200mg、210mg、250mg、300mg、350mg、400mg、450mg、500mg、600mg、700mg、800mg、900mg、1000mg、1100mg、1200mg、1300mg、1400mg、1500mg、1600mg、1700mg、1800mg、1900mg、2000mgまたはそれ以上の活性薬剤であることができる。本明細書中において、「約」は、参照数字の10%以下から10%以上までの範囲を指し得る。例えば、「約50」は、45〜55を意味し得る。
【0060】
例えば、所望の治療効果を実現するため、および所望の効果が実現するまで投薬量を徐々に増加するために、必要なレベルより低いレベルの薬剤用量で開始することは当業者の技量の範囲内である。これらの状況下での活性薬剤の治療有効量は、動物および臨床試験および医学的観察を用いて医師が容易に決定することができる。
【0061】
いくつかの実施形態では、本発明は、肥満の治療方法または予防方法を提供する。これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与することを含むことができる。
【0062】
本発明では、組成物の投与は、例えば、対象において除脂肪量よりも大きい脂肪量減少をもたらすことができる。例えば、対象においてアシル−グレリンに対するグレリンの比の増加ももたらすことができる。
【0063】
いくつかの実施形態では、本発明は、II型糖尿病の治療または予防方法を提供する。これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与することを含むことができる。組成物の投与は、例えば、対象においてインスリン産生の増加をもたらすことができる。例えば、対象においてグルコース曝露に対する応答性増加、および/または対象において脱共役タンパク質2(UCP−2)mRNAレベルの減少ももたらすことができる。
【0064】
糖尿病を治療または予防するための式Iの化合物の治療有効量は、当業者が決定することができるが、例えば、肥満について前述した量と類似であってもよい。
【0065】
例えば、糖尿病の治療又は予防のための治療有効量は、最大でまたは少なくとも1日あたり、約1mg/kg、1.5mg/kg、2mg/kg、2.5mg/kg、3mg/kg、5mg/kg、7.5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kgまたはそれ以上の活性薬剤であることができる。70kgの人では、治療有効用量は、最大でまたは少なくとも1日あたり、約50mg、60mg、70mg、80mg、90mg、100mg、120mg、140mg、160mg、180mg、200mg、210mg、250mg、300mg、350mg、400mg、450mg、500mg、600mg、700mg、800mg、900mg、1000mg、1100mg、1200mg、1300mg、1400mg、1500mg、1600mg、1700mg、1800mg、1900mg、2000mgまたはそれ以上の活性薬剤であることができる。
【0066】
特定の理論に結びつけることは望まないが、GOAT阻害剤は膵臓のランゲルハンス島細胞における脱共役タンパク質−2(UCP−2)のレベルを減少させることによって糖尿病を治療するものと考えられる。GOAT阻害剤がUCP2レベルを劇的に調節することはさらに、アシル−グレリンと肥満およびII型糖尿病との関連を実証する。アシルグレリンは、ニューロペプチドYおよびアグーチ関連タンパク質を共発現する弓状核ニューロンにおけるUCP2レベルの増加を引き起こすことによって、摂食行動を誘導する。UCP2は、肥満誘導性糖尿病モデルであるob/obマウスの膵島で著しく上方制御され得る。UCP2を欠如したob/obマウスは、インスリン初期分泌の回復、血清インスリンレベルの増加および血糖値の著しい減少を示すことは重要である。
【0067】
さらに、ヒトUCP2多形のA55Vは肥満と関係があり、インスリンおよびレプチン濃度を著しく増加させる。さらに、−866A UCP2プロモータ多形の同型接合型のII型糖尿病患者はUCP2発現が上昇しており、より高頻度のインスリン治療を必要とする。これらの患者はまた、グルコース誘導性インスリン分泌(GIIS)が有意に低下している。さらに、−866 A/Aドナーから単離された膵島では、−866 G/Aヘテロ接合型およびG/G同型接合型の個体から単離された膵島よりもGIISが有意に低かった。
【0068】
いくつかの実施形態では、本発明は、mBOAT関連疾患の治療方法を提供する。例えば、これらの方法は、式Iの化合物の治療有効量を含む組成物を対象に投与し、それによってmBOAT関連疾患を治療することを含むことができる。
【0069】
「mBOAT関連疾患」は、mBOATペプチドが関与する経路の調節異常を特徴とする疾患である。mBOAT遺伝子は少なくとも16個存在する。例えば、mBOATタンパク質に類似した構造を有する「porcupine」として知られる遺伝子産物は、癌に関連したプロセスである、Wntシグナル伝達に不可欠である。Wntはporcupineアシルトランスフェラーゼ酵素によるアシル修飾を必要とする。Wntシグナル伝達は多くの癌に寄与する可能性があり、porcupine阻害剤によって遮断することができるかもしれない。mBOAT関連疾患には、脂質代謝調節異常の様々な形態が含まれる。脂質代謝変化は癌に関与しており、mBOATの遮断は癌の治療となり得る。
【0070】
いくつかの実施形態では、本発明は、胃運動の調節異常に関連した障害、例えば、過敏性腸症候群などの治療または予防方法を提供する。グレリン投与は、胃内容排出を増加させ、胃不全麻痺を減少させる。したがって、当業者によって理解されるように、GO−CoA−Tatの投与は、GOAT阻害および付随したアシル−グレリンレベルの低下によって胃運動を阻害し、過敏性腸症候群および胃運動調節異常に関連したその他の障害の患者における緩和効果を発揮するために使用することができる。
【0071】
キット
別の態様では、本発明はキットを提供する。いくつかの実施形態では、これらのキットは、式Iの化合物の治療有効量の少なくとも1用量を含む組成物を含む。
【0072】
これらのキットはさらに、さらなるの体重減少治療薬(treatment)の少なくとも1用量を含む。このようなさらなる治療薬の例には、例えば、アシル−グレリンに対する抗体ならびにGHSRに対する拮抗剤が含まれる。
【0073】
活性アシル−グレリンホルモンの生合成の直接的な標的化には、受容体拮抗剤を上回るいくつかの利点がある。第1に、作用に重要な部位の多くが脳内に存在するアシル−グレリン受容体遮断薬とは対照的に、これらの酵素阻害剤は血液脳関門を通過する必要がない。第2に、酵素阻害剤の効果は範囲がより広範囲である可能性がある。例えば、まだ同定されておらず、公知の受容体を標的とする受容体拮抗剤によってはおそらく影響を受けない、GHSR以外のクラスのアシル−グレリン受容体が存在する可能性がある。しかし、これらの未だに知られていない受容体の活性は、当然のことながら、ホルモンレベルの減少によって影響を受けるだろう。第3に、受容体遮断薬には、動物が受容体遮断に応答してより多くのアシル−グレリン形成を引き起こすフィードバック機構を有する可能性があり、それが受容体遮断薬を上回るかもしれないという欠点がある。しかし、生合成経路を標的とし、したがってフィードバック機構がアシル−グレリンレベルを増加させるのを妨害する酵素阻害剤によって、このようなフィードバック機構は効果的に相殺されるだろう。第4に、GOAT酵素は触媒的に作用するので、所与の用量のGOAT阻害剤は、大体1対1でのみ効果的であることができるアシル−グレリン受容体遮断薬の類似用量よりも、比例的に有効である可能性がある。第5に、グレリンのアシル−グレリンに対する比は、体重増加の調節に重要な因子であり、比較的高いグレリン濃度が体重減少には都合がよいといういくつかの示唆がなされており、これがまさにGOAT阻害剤によって実現する効果である。要約すると、本発明は、体重およびグルコース制御の薬理学的管理のための新たなアプローチを示す。
【0074】
本発明の他の目的、利点および新たな特性は、本発明の範囲を決して制限するものではない以下の実施例を考慮することによって当業者には明らかとなろう。
【実施例】
【0075】
(実施例1)
3種類の細胞系におけるアシル−グレリン産生に対するGO−CoA−Tatの効果
この研究に着手したとき、インビトロGOATアッセイはまだ開発されていなかったため、HELヒト赤白血病細胞(図1D〜E)、KatoIIIヒト胃細胞(図5)およびAGSヒト胃上皮細胞(図5)を含む3種類の細胞においてアシル−グレリン産生に対するGO−CoA−Tatの効果を調べた(3種類の細胞系は全てATCC、Manassas、VAから入手した)。D4-Tat(テトラ−アスパラギン酸は、負のCoA電荷を模倣し、同様にTatペプチドに結合する)を対照として使用した。細胞系それぞれにおいて、アシル−グレリンのレベルは、GO−CoA−Tatによって、IC50が1〜2μMの範囲でD4-Tat対照と比べて有意に減少したが、デスアシル−グレリンは減少しなかった。興味深いことに、最大阻害は、化合物に24時間曝露した後においてのみ実現し、遅い反応速度は非定型酵素特性または予め形成されたアシル−グレリン貯蔵のいずれかから生じているかもしれない。この遅延をさらに調べるために、放射活性アッセイを使用し、組換えミクロソームGOATを用いてインビトロでGO−CoA−Tatを試験した(14)(図1Fおよび6)。完全なGOAT阻害はGO−CoA−Tat 100nMで5分以内に実現し、細胞アシル−グレリンレベルのゆっくりした見かけ上の減少は大きな既存の細胞内貯蔵を反映している可能性があることが示唆された。
【0076】
GO−CoA−Tatは選択的GOAT拮抗剤であると考えられ、その理由は、GO−CoA−Tatは10μMで、インビトロにおいてp300 HAT、PCAF HATおよびセロトニンN−アセチルトランスフェラーゼを含む3種類のアセチル−CoA利用酵素を15%未満阻害することが示されたからである(図7)。さらに、GO−CoA−Tat 10μMはHEL細胞およびHEPG2細胞(ATCC, Manassas, VA)の生存率に対して非毒性であると考えられる(図7)。GO−CoA−Tatおよび類似体の広範な分析によって、アシル−グレリン産生の強力な細胞阻害のためには、少なくとも10個のグレリン残基、ならびにCoA、オクタノイルおよびTat成分を使用して最適な結果が実現することが示唆される。これらの結果は、GO−CoA−TatがGOAT活性の拮抗において、真正な2基質類似体として作用するようであることを示唆している(図1G)。さらに、最良の結果はTat配列などの細胞内移行を助ける部分を含めることによって実現するという事実によって、細胞透過が関与しており、この化合物は細胞表面受容体には作用しないことが示唆される。
【0077】
(実施例2)
マウスにおけるアシル−グレリン産生に対するGO−CoA−Tat投与の効果
次に、正常マウスにおいてGO−CoA−Tatがアシル−グレリン産生を阻害するかどうかを調べた。化合物を投与する6時間前に絶食したマウスにおいて、2種類の腹腔内(IP)GO−CoA−Tat用量、15mg/kgおよび29mg/kgを試験した。分布容積が動物の体重と等しいと仮定すると、これらの用量はそれぞれ、約4μMおよび8μMの最大濃度を実現すると予測される。培養細胞で観察されたように、GO−CoA−Tatによる処理は血清アシル−グレリンの用量依存的阻害を引き起こし(図2)、29mg/kg用量で24時間後に最大の70%阻害が得られたが、D4-Tat対照では阻害は引き起こされなかった(図2)。また、デスアシルグレリンの血清レベルにあまり影響はなかったが、少し増加傾向があった。これらの結果は、GO−CoA−Tatがインビボにおいて選択的にアシル−グレリン産生を標的としていることを示している。
【0078】
(実施例3)
マウスにおける体重増加に対するGO−CoA−Tat投与の効果
次に、正常マウスに12日間にわたって高脂肪食を与えることによって、体重増加に対するGO−CoA−Tatの効果を調べた。24時間毎にGO−CoA−Tat(29mg/kg IP)でこれらのマウスを処理し、代謝ケージに入れて、毎日摂取および排出ならびに体重のモニターを可能にした。さらに、動物の脂肪量および除脂肪量を評価するために、マウスに3日毎に定量的核磁気共鳴(QMR)分光法を実施した(15)。これらの実験によって、GO−CoA−Tatで処理したマウスは、D4-Tatで処理したマウスと比較して統計学的に有意な体重減少を表すことが示された(図3A)。さらに、QMR測定によって、対象と比較して、GO−CoA−Tat処理動物では脂肪量の約5%の相対的減少を表し、6日後も維持されたが、除脂肪量の減少は表れないことが示された(図3B〜D)。食事または水の摂取および糞尿排出には統計学的有意差はなかったが(図8)、中程度の試料数ではこれらの値には比較的大きな変動があり、そのため摂取減少の関与を除外することはできなった。以前の遺伝子研究では、グレリンの欠如は、食欲ならびに代謝率に影響を及ぼすことによって体重に影響を与え得ることが示唆されている(2〜8)。
【0079】
GO−CoA−Tatが誘導する、体重減少の原因となる全身毒性または臓器障害の可能性について調べるために、12日間GO−CoA−Tat処理した動物の血液化学および血液細胞数を評価した。これらの分析によって、体重減少の原因となり得る肝臓、腎臓、膵臓または骨髄毒性の証拠は示されなかった(図9)。興味深いことに、これらの血液測定では、GO−CoA−Tatは、少しではあるが有意なHDLならびにトリグリセリドの増加を誘導した。別の研究では、3日間の全糞便脂肪をQMRによって測定すると、GO−CoA−Tat処理マウス(4.97±0.10g)とD4-Tat処理マウス(5.01±0.07g)の間には統計学的有意差は示されず、GOAT阻害は腸内脂肪吸収に影響を及ぼさないことを示唆している。総合すると、これらのデータは、GO−CoA−Tatはアシル−グレリンを低下させることによって体重減少を引き起こすことを示唆している。
【0080】
(実施例4)
グルコース曝露に対するインスリン応答へのGO−CoA−Tatの効果
グルコース曝露に対するインスリン応答へのGO−CoA−Tatの効果を調べるために、ヒト膵島細胞をGO−CoA−Tatで24時間まで予め処理して、これらの細胞が、24時間GO−CoA−Tatに曝露したとき、グルコース曝露に対するインスリン応答の統計学的有意な増加を示すことを示した(図10)。これらの結果は、アシル化グレリンはインスリン応答鈍化に直接的役割を担うことを示唆している。このことをインビボで調べるために、GO−CoA−Tatで予め処理した後、経口的にグルコース曝露したマウスを研究した。膵島細胞研究と矛盾することなく、経口的グルコース曝露(1mg/kg)の前に24時間GO−CoA−Tat 15mg/kgを与えられた動物は、インスリン応答の統計学的有意な増加を示した(図10)。これらの結果は、2.5g/kgの腹腔内グルコース曝露と共にGO−CoA−Tat 29mg/kgを投与されたマウスにおいてさらによりはっきりしていた(図4A)。このインスリン応答の増加には、グルコースの統計学的有意な減少が伴い、機能的に重要な効果が示唆された(図4B)。
【0081】
GOAT阻害とインスリン調節との間の関係をさらに調べるために、GO−CoA−Tatで処理したマウスから単離した膵臓のランゲルハンス島を研究した。インスリン産生β細胞はGHSRが陽性に染色し、膵島ではβ細胞とは区別されるグレリン発現細胞がわずかな割合で示され、インスリンおよびグレリン受容体は別の細胞で産生されるようであることが示唆された。GO−CoA−Tatで処理したマウスから単離された膵島のQRT−PCRでは、UCP2 mRNAレベルはD4−tat処理マウスと比較して20倍減少していたが(図4C)、インスリン、グレリンまたはGHSRのレベルには変化がないことが示された。比較すると、QRT−PCRは胃底部においてUCP2に対して非統計学的に有意な効果を示した(図4D)。総合すると、これらのデータは、UCP2レベルの抑制およびグルコースに応答したインスリン放出の増強における、GOAT阻害の組織特異的役割を示唆している。
【0082】
GOAT阻害剤がUCP2レベルを劇的に調節することはさらに、アシル−グレリンと肥満およびII型糖尿病との関連を実証している(19〜21)。アシル−グレリン誘導性摂食行動は、ニューロペプチドYおよびアグーチ関連タンパク質を共発現する弓状核ニューロンにおいてUCP2レベルを増加させることよって制御することができる(21)。UCP2は、肥満誘導性糖尿病モデルであるob/obマウスの膵島で著しく上方制御される。UCP2を欠如したob/obマウスは、インスリン初期分泌の回復、血清インスリンレベルの増加および糖血症レベルの著しい減少を示すことは重要である(19)。さらに、ヒトUCP2多形のA55Vは肥満と関係があり、インスリンおよびレプチン濃度を著しく増加させる(22)。さらに、−866A UCP2プロモータ多形の同型接合型のII型糖尿病患者はUCP2発現が上昇しており、より高頻度のインスリン治療を必要とする。これらの患者はまた、グルコース誘導性インスリン分泌(GIIS)が有意に低下している(23)。さらに、−866 A/Aドナーから単離された膵島では、−866 G/Aヘテロ接合型およびG/G同型接合型の個体から単離された膵島よりもGIISが有意に低かった(24)。
【0083】
(実施例5)
化学合成(化合物1〜7、D4-Tat、グレリン−27−ビオチン)
化学合成用の試薬は全て、AldrichまたはAcros Organicsから購入した。市販の試薬は全て、購入時のままさらに精製することなく使用した。2基質類似体阻害剤、ペプチドおよび対照化合物の調製用HPLC単離は、逆相C−18カラム(25×2.14cm、Microsorbtm-100, Rainin)を使用して実施し、水(トリフルオロ酢酸(TFA)0.05%、移動相A)およびアセトニトリル(TFA0.05%、移動相B)の勾配で溶出した。グレリン−27−ビオチン(GSSFLSPEHQRVQQRKESKKPPAKLQPK(ビオチン)G)およびD4−Tatは、Fmoc法を使用して調製した。GO−CoA−Tatおよび関連誘導体の合成は、以前に記載された方法(33、34)と類似しており、グレリンから開始して、Fmoc法を使用した。様々な長さのグレリンペプチド(3から15個のC末端配列)を使用し、グレリンのSer3をAlloc(アリルオキシカルボニル)保護1,2−ジアミノプロピオン酸で置換した。遊離アミノ基を得るために、CHCl3:AcOH:N−メチルモルホリン(37:2:1)中においてテトラキス(トリフェニルホスフィン)パラジウム(0)を使用したAllocの直交脱保護を実施し、次いでラセミ化α−ブロモオクタン酸無水物(化合物5ではオクタン酸無水物および化合物6ではブロモ酢酸無水物)と反応させた(図1C参照)。補酵素Aとの結合は、α−ブロモ−アミドをCoASHと反応させることによって実施した。ブロモ−アミドペプチドの撹拌溶液(濃度2〜10mMのddH2O溶液)に室温で重炭酸トリエチルアンモニウムの水性緩衝液(pH8.0)1.0Mに溶かしたCoASHの溶液(2.0〜3.0当量)を滴下した。48〜72時間後、混合物を一晩凍結乾燥し、所望の生成物を単離するために残渣を調製用HPLCに添加した。HPLCカラムは水(TFA0.05%、移動相A)およびアセトニトリル(TFA0.05%、移動相B)(0分、5%B;5〜65分、60%Bまで直線的に増加;10mL/分)の勾配で溶出し、214nmでモニターした。収集した画分を減圧下で濃縮し、凍結乾燥して最終生成物を白色固形物として得た。マトリックス支援レーザー脱離イオン化(MALDI)およびエレクトロスプレー(ES)質量分析データは、計算値と矛盾せず、アッセイ用の水性溶液中の化合物最終濃度はアミノ酸分析によって測定した。
【0084】
(実施例6)
細胞培養、細胞溶解調製物およびEIA測定
HEL細胞およびAGS細胞(ATCC, Manassas VA)は、不活性化牛胎児血清10%、ストレプトマイシン−ペニシリン100IU/mlおよびグルタミン4mMを補給したRPMI1640培地(Sigma)で増殖させ、週に2回常法通り継代した。KatoIII細胞(ATCC, Manaasas VA)は、不活性化牛胎児血清10%、ストレプトマイシン−ペニシリン100IU/mlおよびグルタミン4mMを補給したDMEM培地(Sigma)で増殖させ、週に2回常法通り継代した。いくつかの実施形態では、アシル−グレリン産生の阻害を評価するために、HEL細胞は7種類の化合物の1種で処理した。3種類の細胞種全ての細胞溶解物を以下のようにRIPA緩衝液中で調製した。細胞(7.5×106細胞)は、卓上型遠心機で1000rpmで5分間遠心分離した。次に細胞ペレットを洗浄し、これに含有する氷冷した改変RIPA緩衝液0.25mlを添加し、激しくピペット操作し、ボルテックスして、その後完全に細胞が溶解するまで4℃で10分間静置させた。次に、溶解物を予め冷却した遠心機で2000rpmで15分間遠心した。遠心直後、上清を−80℃で保存した。細胞溶解物および血清中のアシル−およびデスアシル−グレリンのレベルならびに培養培地および血清試料中のインスリンレベルを評価するために、2抗体サンドイッチをベースにした市販の酵素免疫アッセイ法(Alpco Diagnostics)で分析した。
【0085】
(実施例7)
分子クローニング
GOATは、マウス胃から2段階ネステッドRT−PCR法によってクローニングした。全RNAは、組織ホモジェナイザーおよびRNeasy(登録商標)キット(Qiagen)を使用して、マウス胃の約半分から抽出した。cDNAは、Invitrogen SuperScript(商標)II RTで逆転写することによって調製した。cDNAの2μlをプライマー、TTTACAAGGGCACCGCTTAG/CAAGGCATCTTCTGGCATTTで増幅した。次いで、反応混合物1μlをネステッドプライマーGCCACCATGGATTGGCTCCAGCTC/GAGATGAAGGGCAGGGAAAでさらに増幅した。約1.3kBのバンドをゲルから切り出し、pCR(登録商標)2.1−TOPO(登録商標)(Invitrogen)に連結した。
【0086】
[00108] GOATは5’平滑末端連結および3’EcoRI切断のために設計されたプライマーを使用してこのベクターから増幅し、次にpHLsecから得られたpαHベクター(Aricescu et al. Acta Cryst.(2006). D62, 1243-1250)にKpnI(平滑末端化)およびEcoRIを使用してクローニングした。QuikChange(登録商標)変異誘発(Stratagene)を使用して、製造元の方法をWang and Malcolm(35)によって開発された2段階法に置き換えて、3xFlag(登録商標)タグ(Sigma)をC末端に付加した。
【0087】
(実施例8)
GOATトランスフェクションおよびミクロソーム単離
GnTI欠損HEK293T細胞(Reeves et al., Proc Natl Acad Sci U S A. 2002 Oct 15 ;99(21): 13419-24)は、L−グルタミン2mMおよびFBS1%を補給したFreestyle293T培地(Invitrogen)中でCO28%を含有する雰囲気中で増殖させた。トランスフェクションの日に、細胞400mlを2×106/mlの濃度に設定した。細胞に、Aricescuらによって記載されたように(Acta Cryst. (2006). D62, 1243-1250)、ポリエチレンイミン「Max」強力直鎖状PEI(Polysciences)を使用してプラスミドをトランスフェクトした。簡単に説明すると、最終培養量の20分の1と等しい未補給ハイブリドーマSFM培地(Invitrogen)中で、DNA1μg/mlをトランスフェクション試薬3μg/mlと混合し、室温で20分間インキュベートし、細胞に添加した。4日後、細胞を実験毎に収集し、PBSで1回洗浄した。
【0088】
HEK293T GnTI細胞の各ペレットを緩衝液D(Tris−HCl 50mM、pH7.4、NaCl 150mM、ナトリウムEGTA 1mM、フェニルメチルスルホニルフルオリド30μg/ml、ペプスタチンA 3μg/mlおよび完全プロテアーゼ阻害剤カクテル錠、EDTAなし(Roche、1錠/緩衝液40ml))20mlに再懸濁した。細胞は、加圧型ホモジェナイザー(B型内筒、排出30回)を使用して溶解した。溶解物を4℃で2135×gで10分間遠心分離し、上清を新たな管に移し、同条件下で再度遠心分離した。上清を4℃で100000×gで30分間遠心分離し、上清を捨てた。ミクロソームペレットをTris緩衝生理食塩水(TBS)(Tris-HCl 50mM、pH7.4、NaCl 150mM)に再懸濁し、不溶性物質を1000×gで1分間遠心分離することによって除去した。イムノブロット分析または液体窒素中での急速冷凍のために一定量を準備し、アッセイ時間まで−80℃で保存した。タンパク質濃度は、Triton X−100 0.5%を補給したBCAアッセイ(Thermo)を使用して測定した。
【0089】
(実施例9)
GOATアシルトランスフェラーゼアッセイ
アッセイ方法は、Yangら(36)を改変した。アッセイ混合物は、HEPES 50mM、pH7.0、オクタノイル−CoA(33dpm/fMol、American Radiolabeled Chemicals)1μM、グレリン27−ビオチン 10μM、ミクロソームタンパク質50μgおよびパルミトイル−CoA(Avanti Polar Lipids)50μMを含有した。反応は、膜タンパク質を添加することによって開始し、37℃で5分間インキュベートした。指示がある場合、GO−CoA−Tatは膜タンパク質と共に5分間予めインキュベートした。ストレプトアビジンプラスウルトラリンク樹脂(Pierce)150μlを予め混合した反応停止緩衝液(TBS+SDS 2%(w/v))850μlを添加し、回転ミキサーで15分間インキュベートすることによって反応を停止した。この混合物をPoly−Prep(登録商標)クロマトグラフィーカラム(Bio-Rad)に移し、洗浄緩衝液(TBS+SDS 0.1%)30mlで洗浄した。混合物は反応停止緩衝液1mlを含有する1.5ml微量遠心管に移し、98℃で5分間煮沸し、シンチレーション測定を行った。
【0090】
(実施例10)
細胞生存率アッセイ
様々な濃度のGO−CoA−Tatで処置したHEL細胞系およびヒト不死化肝細胞系HepG2(ATCC, Manassas, VA)の生存率は、LIVE/DEAD生存率アッセイキット(Molecular Probes)を使用して測定した。ラット骨髄間質細胞(RBMSC)は、2種類のプローブ、細胞内エステラーゼ活性についてはカルセイン−AM(緑色)2μMで30分間、細胞膜強度についてはエチジウムホモダイマー−1(EtdD-1、淡赤色)4μMで10分間インキュベートした。カルセイン−AMは、500nmレーザー光線を使用して励起し、放出した蛍光は535nmロングパスフィルターによって検出した。EtdD−1は、514nmレーザー光線を使用して励起し、放出した蛍光は550nmロングパスフィルターによって検出した。赤色蛍光(EtdD-1)を放つ細胞は死細胞として計数し、緑色蛍光(カルセイン-AM)は生細胞として計数した。部分的に赤および緑で2重に染色している場合は、死細胞として計数した。
【0091】
(実施例11)
ヒト膵島の実験
新鮮なヒト死体膵島は、National Islet Cell Resource centerから提供された。平均純度は70%、生存率は85%であった。ヒト膵島実験用に、膵島をGO−CoA−Tat 5μMを含む無血清RPMI培地で24時間インキュベートし、その後グルコース50、150および300mg/dlを添加した無グルコースRPMI培地中で30分間静的インキュベーションアッセイを行い、培地中に分泌されたインスリンをELISA(Alpco Diagnostics)で評価した。
【0092】
(実施例12)
アセチルトランスフェラーゼアッセイ
GO−CoA−Tatのアセチルトランスフェラーゼp300、PCAFおよびAANATとの特異性は、α−ケトグルタル酸脱水素酵素(α-KGDH)関連分光測定法(37)を使用して測定した。組換えアセチルトランスフェラーゼドメインおよびヒストン尾部ペプチドは、以前に記載されたように調製した(38〜40)。アセチルトランスフェラーゼによるCoASHの産生は、NADHの形成と共役しており、Beckman DU−640分光光度計によって340nm(ε340=6230M−1cm−1)でモニターした。反応物は全て、チアミンピロリン酸200μM、MgCl2 5mM、DTT 1mM、BSA 50μg/mL、NAD 200μM、α−ケトグルタル酸2.4mM、GO−CoA−Tat 10μMおよびアセチル−CoA 50μMを含有する。p300との反応は、HEPES 100mM、pH7.9(α−KGDH 0.1単位およびH4〜15 200μM、ヒストンH4尾部の配列に基づく15マーペプチド基質を含有する)中で実施した。p300反応は、30℃でインキュベートした後、p300 100nMを添加して開始して30℃で行う。PCAFとの反応は、HEPES 100mM、pH7.9(α−KGDH 0.037単位およびH3〜20 100μM、ヒストンH3尾部の配列に基づく20マーペプチド基質を含有する)中で実施する。PCAF反応は、30℃で10分間インキュベートした後、PCAF 100nMを添加して開始して30℃で行う。AANATとの反応は、NH4OAc 100mM、pH6.8(α−KGDH 0.1単位およびトリプタミン200μMを含有する)中で実施する。AANAT反応は、25℃で10分間インキュベートした後、AANAT 10.83nMを添加して開始して25℃で行う。反応は全て反応進行曲線の直線部分にあり、直線回帰によって初速度が得られる。
【0093】
(実施例13)
マウスにおける実験
24時間の実験では全て、C57BL/6マウスはGO−CoA−TatまたはD4−TatをIP注射する前に6時間絶食させた。各時点で、経心的穿刺によって血液を採取した。血液試料は、EDTAおよびp−ヒドロキシメルクリ安息香酸(PHMB)1mMを含有する試験管に収集した。試料を+4℃で3500rpmで5分間遠心分離して、次に上清を別の試験管に移した。次いですぐに細胞溶解物1mL当たり1N HCl 100μlを添加し、試験管を+4℃で3500rpmで5分間遠心分離した。上清をクライオバイアルに移し、−80℃で保存した。デスアシル−およびアシル−グレリンの血清レベルは、ELISA(Alpco Diagnostics)によって測定した。糖尿病の成果については、経口的グルコース曝露(1g/kg)を血液試料採取の30分前に行うこと以外は前記の方法にしたがった。糖尿病の成果のために、化合物を絶食していないマウスに18時間の絶食の6時間前に投与し、その時点で2.5g/kg腹腔内グルコース負荷試験(IP-GTT)を意識のあるマウスに実施した。血液は、IP−GTT後0、15、30、60および120分に尾静脈から採取した。グルコースは、グルコメータ(LifeScan OneTouch)で測定し、インスリン値はELISA(Alpco Diagnostics)によって評価した。毎日のGO−CoA−TatのIP投与の体組成に対する効果を評価するために、同腹仔の8週齢C57BL6Jマウス(Jackson Laboratories, Bar Harbor, ME)の体重を2群23.2±1.2g(n=3)および22.6±2.2(n=3)に一致させ、処置前に7日間高脂肪食を与えた。8日目に、マウスには高脂肪食を継続させたが、GO−CoA−Tat 29mg/kg用量(n=3)または等モル用量のD4−Tatペプチド(n=3)の投与を開始した。マウスは、食物および水の摂取ならびに糞尿の生成を毎日測定する直接熱量計測ケージ(Nalgene)中で2週間モニターした。さらに、マウスの身体プロファイルを3日毎にQNMR(EchoMRI)で評価した。身体測定それぞれについて、3つの別個の測定の平均を得た。糞便分析では、糞便を3日間収集し、1.5mL超遠心管に保存した。糞便は再水和して、GO−CoA−Tat(n=4)およびD4−Tat(n=4)の全糞便を4つの別個の測定と比較した。
【0094】
(実施例14)
QRT−PCR
膵島は膵臓をコラゲナーゼおよびDNアーゼIで消化した後、Songら(41)で記載されたように単離した。全RNAは、トリゾールを使用して抽出した。RT−PCRは、one−tube RT−PCR Sybr greenミックス(BioRad)を使用して標準的方法にしたがって実施した。発現レベルの倍数変化は、DDCT法を使用して算出した。2連の結果をステューデントt検定を使用して分析した。RT−PCRで使用したプライマー:インスリンFw:CGAGGCTTCTTCTACACACC;インスリンRv:GAGGGAGCAGATGCTGGT;グルカゴンFw:CCACTCACAGGGCACATTCA;グルカゴンRv:GTCCCTGGTGGCAAGATTGT GHSR FW:ACCTGCTCTGCAAACTCTTCCAGT;GHSR RV:CAAACACCACCACAGCAAGCATCT グレリンFw:ACTCAGCATGCTCTGGATGGACA;グレリンRv:ATGCCAACATCGAAGGGAGCATTG UCP2−001 Fw:TGGTTGGTTTCAAGGCCACAGATG;UCP2−001 Rv:TCTCGTGCAATGGTCTTGTAGGCT 36B4 Fw:TGTTTGACAACGGCAGCATTT;36B4 Rv:CCGAGGCAACAGTTGGGTA
【0095】
(実施例15)
免疫組織化学
マウスの膵臓およびヒト膵島を10%緩衝ホルマリンまたはブアン固定液で固定し、パラフィンに包埋し、5μmの切片を作製した。脱脂し、再水和してクエン酸緩衝液中で抗原を回復した後、免疫染色をモルモット抗インスリン(Abcam)、ウサギ抗グレリン(Abcam)およびニワトリ抗GHSR(Chemicon)で実施した。適切な蛍光タグ第2抗体(ロバ抗モルモットCy3、ロバ抗ウサギFITC、Jackson Immunoresearch)を抗原分布のために使用した。核は、封入媒体に含めたDAPI(Vector Biolabs)を使用して対比染色した。画像作製のために擬似カラー化用CCDデジタルカメラを装着したZeiss Axioskopおよびデジタル画像処理装置を連結したApotome光学切片装置(optical sectioning device)を使用した。
【0096】
(実施例16)
MCT食でのグレリン欠損(グレリンKO)および野生型(WT)マウスの長期調査
グレリン欠損マウスは、前述されたように、ハイスループットVelociGene遺伝子標的化システムを使用して作製した(42)。全動物は、22℃で12:12時間の明暗サイクルで維持し、中鎖トリグリセリドの豊富な食餌(MCT)−(MCT10%、スクロース40%、Teklad TD 08622)を与え、食餌および水は自由に摂取させた。GO−CoA−Tat 40mg/kgはWTおよびKOマウスに投与し、ビヒクルはWTマウスに投与した。全体の体組成(脂肪および除脂肪)は3〜4日毎にNMR技術(EchoMRI, Houston, TX)を使用して測定し、体重測定は毎日行った。結果を図11に示す。
【0097】
(実施例17)
グレリンKOおよびWTマウスにおけるグルコース負荷試験
糖尿病の成果のために、GO−CoA−Tat 40mg/kg(図12の「inhib」)を絶食していないwtマウス(n=5)およびグレリン−koマウス(n=5)に投与し、ビヒクル(2回蒸留水、図12の「Vhc」)を月齢を一致させたwtマウス(n=5)およびグレリン−koマウス(n=5)に投与し、6時間経ってから18時間絶食させた。18時間絶食した後、2.5g/kg腹腔内グルコース負荷試験(IP-GTT)を意識のあるマウスに実施した。血液は、IP−GTT後0、15、30、60および120分に尾静脈から採取した。グルコースはグルコメータ(Lifescan OneTouch)で測定した。
【0098】
(実施例18)
光活性化可能な(photoactivatable)GO−CoA−Tat類似体および可溶性GOATの架橋結合
FLAG−GOAT酵素を、SF9細胞で過剰産生し、界面活性剤で可溶化し、GO−CoA−Tatのベンゾイル−フェニルアラニン含有類似体およびビオチン含有類似体(5μM)の存在下でUV光で処理した。これらはビオチンブロットで示したように架橋結合を誘導し(図13A)、標準GO−CoA−Tat(100uM)で阻害することができた。FLAG(図13B)およびクーマシー(図13C)ゲルでは、これらのレーンへの負荷が等しいことを示している。
【0099】
(実施例19)
GOAT阻害剤はIGF−1に対して影響を及ぼす
血液は、EDTAコーティングMicrovette管を使用して経心的穿刺によって収集し、すぐに氷で冷却した。4℃で3000gで15分間遠心した後、血漿を−80℃で保存した。血漿IGF−1レベルを定量するために、市販のアッセイを使用した(R&D Systems, Minneapolis, MN)。このアッセイは、酸性化していない血漿試料におけるIGF−1の測定に有効で、タンパク質抽出を必要としない。このアッセイはアッセイ製造元の指示に従って実施した。結果を図14に示す。
【0100】
本明細書で例示し検討した実施形態は、本発明者が知っている本発明の作製および使用の最良の方法を当業者に教示するためのみのものである。本明細書において本発明の範囲の制限を意味するものはない。提示した実施例は全て代表的なものであって制限はしない。本発明の前述の実施形態は、前記の教示を考慮して当業者が理解するように、本発明から逸脱することなく改変または変化させることができる。したがって、特許請求の範囲およびその同等物の範囲内であれば、本発明は具体的に記載したものとは別に実施することができるものと理解される。
【0101】
(参考文献)
【化3】
【特許請求の範囲】
【請求項1】
式(I)の化合物
【化1】
(式中、Rは存在しないか、あるいは置換または非置換のアルキル基、置換または非置換のペプチド鎖および置換または非置換の多糖からなる群から選択される有機部分である)。
【請求項2】
Rが1〜12個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項3】
Rがグレリン残基4から10までに対応する7個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項4】
Rがグレリン残基4から15までに対応する12個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項5】
Rがグレリン残基4から5までに対応する2個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項6】
Rがグレリン残基4から28までに対応する25個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項7】
請求項1〜6のいずれか1項に記載の化合物および薬学的に許容される賦形剤または担体を含む組成物。
【請求項8】
請求項1〜6のいずれか1項に記載の化合物を調製する方法であって、
3〜5個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、D4−Tatがペプチド配列の一末端に結合している、工程と、
ブロモオクタノイル化中間体を得るために、1または複数個の反応容器中で、グレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、
前記ブロモオクタノイル化中間体を補酵素Aと一緒にする工程と
を含む方法。
【請求項9】
請求項1〜6のいずれか1項に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、肥満の治療方法。
【請求項10】
前記組成物の投与が、対象において除脂肪量よりも大きい脂肪量減少をもたらす、請求項9に記載の方法。
【請求項11】
前記組成物の投与が、対象においてアシル−グレリンに対するグレリンの比の増加をもたらす、請求項9に記載の方法。
【請求項12】
前記治療有効量が少なくとも約5mg/kg対象の体重である、請求項9に記載の方法。
【請求項13】
前記治療有効量が少なくとも約15mg/kg対象の体重である、請求項9に記載の方法。
【請求項14】
前記治療有効量が少なくとも約25mg/kg対象の体重である、請求項9に記載の方法。
【請求項15】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、II型糖尿病の治療方法。
【請求項16】
前記組成物の投与が、対象においてインスリン産生増加をもたらす、請求項15に記載の方法。
【請求項17】
前記組成物の投与が、対象においてグルコース曝露に対する応答性増加をもたらす、請求項15に記載の方法。
【請求項18】
前記組成物の投与が対象において脱共役タンパク質2(UCP−2)mRNAレベルの減少をもたらす、請求項15に記載の方法。
【請求項19】
前記治療有効量が少なくとも約5mg/kg対象の体重である、請求項15に記載の方法。
【請求項20】
前記治療有効量が少なくとも約15mg/kg対象の体重である、請求項15に記載の方法。
【請求項21】
前記治療有効量が少なくとも約25mg/kg対象の体重である、請求項15に記載の方法。
【請求項22】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、mBOAT関連疾患の治療方法。
【請求項23】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、過敏性腸症候群の治療方法。
【請求項24】
請求項1に記載の化合物の治療有効量の少なくとも1用量を含む組成物を含むキット。
【請求項25】
さらなる体重減少治療薬の少なくとも1用量をさらに含む、請求項24に記載のキット。
【請求項26】
前記対象がヒトである、請求項9〜23のいずれか1項に記載の方法。
【請求項1】
式(I)の化合物
【化1】
(式中、Rは存在しないか、あるいは置換または非置換のアルキル基、置換または非置換のペプチド鎖および置換または非置換の多糖からなる群から選択される有機部分である)。
【請求項2】
Rが1〜12個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項3】
Rがグレリン残基4から10までに対応する7個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項4】
Rがグレリン残基4から15までに対応する12個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項5】
Rがグレリン残基4から5までに対応する2個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項6】
Rがグレリン残基4から28までに対応する25個のアミノ酸残基のペプチドである、請求項1に記載の化合物。
【請求項7】
請求項1〜6のいずれか1項に記載の化合物および薬学的に許容される賦形剤または担体を含む組成物。
【請求項8】
請求項1〜6のいずれか1項に記載の化合物を調製する方法であって、
3〜5個のN末端アミノ酸残基を含むグレリンペプチド配列を提供する工程であって、3位のセリン残基がアロック保護1,2−ジアミノプロピオン酸に置換されており、D4−Tatがペプチド配列の一末端に結合している、工程と、
ブロモオクタノイル化中間体を得るために、1または複数個の反応容器中で、グレリンペプチド配列を、Pd(PPh3)4パラジウム試薬、n−ブロモオクタン酸無水物および試薬Kと一緒にする工程と、
前記ブロモオクタノイル化中間体を補酵素Aと一緒にする工程と
を含む方法。
【請求項9】
請求項1〜6のいずれか1項に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、肥満の治療方法。
【請求項10】
前記組成物の投与が、対象において除脂肪量よりも大きい脂肪量減少をもたらす、請求項9に記載の方法。
【請求項11】
前記組成物の投与が、対象においてアシル−グレリンに対するグレリンの比の増加をもたらす、請求項9に記載の方法。
【請求項12】
前記治療有効量が少なくとも約5mg/kg対象の体重である、請求項9に記載の方法。
【請求項13】
前記治療有効量が少なくとも約15mg/kg対象の体重である、請求項9に記載の方法。
【請求項14】
前記治療有効量が少なくとも約25mg/kg対象の体重である、請求項9に記載の方法。
【請求項15】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、II型糖尿病の治療方法。
【請求項16】
前記組成物の投与が、対象においてインスリン産生増加をもたらす、請求項15に記載の方法。
【請求項17】
前記組成物の投与が、対象においてグルコース曝露に対する応答性増加をもたらす、請求項15に記載の方法。
【請求項18】
前記組成物の投与が対象において脱共役タンパク質2(UCP−2)mRNAレベルの減少をもたらす、請求項15に記載の方法。
【請求項19】
前記治療有効量が少なくとも約5mg/kg対象の体重である、請求項15に記載の方法。
【請求項20】
前記治療有効量が少なくとも約15mg/kg対象の体重である、請求項15に記載の方法。
【請求項21】
前記治療有効量が少なくとも約25mg/kg対象の体重である、請求項15に記載の方法。
【請求項22】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、mBOAT関連疾患の治療方法。
【請求項23】
請求項1に記載の化合物の治療有効量を含む組成物を対象に投与することを含む、過敏性腸症候群の治療方法。
【請求項24】
請求項1に記載の化合物の治療有効量の少なくとも1用量を含む組成物を含むキット。
【請求項25】
さらなる体重減少治療薬の少なくとも1用量をさらに含む、請求項24に記載のキット。
【請求項26】
前記対象がヒトである、請求項9〜23のいずれか1項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2012−504619(P2012−504619A)
【公表日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願番号】特願2011−530103(P2011−530103)
【出願日】平成21年9月18日(2009.9.18)
【国際出願番号】PCT/US2009/057512
【国際公開番号】WO2010/039461
【国際公開日】平成22年4月8日(2010.4.8)
【出願人】(505005016)ザ ジョンズ ホプキンス ユニバーシティ (4)
【Fターム(参考)】
【公表日】平成24年2月23日(2012.2.23)
【国際特許分類】
【出願日】平成21年9月18日(2009.9.18)
【国際出願番号】PCT/US2009/057512
【国際公開番号】WO2010/039461
【国際公開日】平成22年4月8日(2010.4.8)
【出願人】(505005016)ザ ジョンズ ホプキンス ユニバーシティ (4)
【Fターム(参考)】
[ Back to top ]