
肥満細胞トリプターゼインヒビターとしての[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン
本発明は、トリプターゼインヒビターとして有用なインドールベンジルアミン化合物に関する。さらに、本発明はトリプターゼの阻害により改善の必要がある生理学的症状を患っているか、または罹りやすい患者を治療するための化合物の使用に関し、治療有効量の化合物をその患者に投与することを包含する。
さらに、本発明は治療有効量の式Iの化合物および薬学的に受容可能な担体を含む医薬組成物に関する。
【化1】

さらに、本発明は治療有効量の式Iの化合物および薬学的に受容可能な担体を含む医薬組成物に関する。
【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、置換されたインドールベンジルアミン化合物、その製造、化合物を含有する医薬組成物、その用途およびそれらの中間体に関する。
【背景技術】
【0002】
肥満細胞介在性炎症症状、特に喘息は、深刻な公衆の健康問題である。喘息は、慢性炎症の発症をもたらす免疫特異的アレルゲンおよび全身性の化学的もしくは物理的刺激の両方に対する気管および気管支の過敏症(hyper−responsiveness)の進行性発生によって特徴付けられる。IgE受容体を含有する白血球、特に肥満細胞および好塩基球は、気管支の上皮およびその下部の平滑筋組織に存在する。これらの白血球が、先ずIgE受容体への特異的な吸入抗原の結合によって活性化され、次いで、多数の化学的媒介物を放出する。例えば、肥満細胞の脱顆粒により、プロテオグリカン、ペルオキシダーゼ、アリールスルファターゼB、キマーゼおよびトリプターゼの放出がもたらされ、細気管支の収縮が起こる。
【0003】
トリプターゼは、肥満細胞分泌性顆粒内に貯留され、そしてヒト肥満細胞の主要な分泌性プロテアーゼである。トリプターゼは、血管拡張性および気管支弛緩(bronchorelaxing)の神経ペプチドの分解(非特許文献1;非特許文献2;および非特許文献3)およびヒスタミンへの気管支応答性の変調(非特許文献4)を含む種々の生物学的プロセスに関与している。
【0004】
その結果、トリプターゼインヒビターは抗炎症薬(非特許文献5)として、特に慢性喘息の治療(非特許文献6)において有用であり得、そしてまたアレルギー性鼻炎(非特許文献7)、炎症性腸疾患(非特許文献8)、乾癬(非特許文献9)、結膜炎(非特許文献10)、アトピー性皮膚炎(非特許文献11)、関節リウマチ(非特許文献12)、変形性関節症(非特許文献13)、痛風性関節炎、リウマチ様脊椎炎および関節軟骨崩壊の疾患の治療または予防にも有用であり得る。
【0005】
さらに、トリプターゼは、線維芽細胞の強力な有糸分裂促進物質であることがわかっており、喘息および間質性肺炎における肺線維症に関与していることが示唆されている(非特許文献14)。
【0006】
従って、トリプターゼインヒビターは、線維性症状(非特許文献15)、例えば、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫および肥厚性瘢痕の治療または予防に有用であり得る。
【0007】
さらに、トリプターゼインヒビターは、心筋梗塞、発作、狭心症およびアテローム性動脈硬化性プラーク破壊のその他の結果の治療または予防に有用であり得る(非特許文献16)。
【0008】
トリプターゼはまた、コラゲナーゼを活性化するプロストロメリシンを活性化し、これにより軟骨および歯周の結合組織の破壊をそれぞれ開始させることがわかっている。
【0009】
従って、トリプターゼインヒビターは、関節炎、歯周病、糖尿病性網膜症および腫瘍増殖の治療または予防に有用であり得る(非特許文献17)。また、トリプターゼインヒビターは、過敏症(非特許文献18)、多発性硬化症(非特許文献19)、消化性潰瘍および合胞体ウイルス感染症の治療にも有用であり得る。
【0010】
式(A):
【化1】
の化合物として表される置換アリールメチルアミン、それらの製造、それらの化合物を含有する医薬組成物およびトリプターゼの阻害によって調節され得る疾患状態の治療におけるそれらの薬学的使用は、特許文献1に記載されている。特許文献1に具体的に開示されている化合物は、以下の式のものである:
【化2】
【0011】
しかしながら、特許文献1は、前述の[(アミノメチル−フェニル)−ピペリジン−1−イル]−[インドリル]−メタノン種(そのフェニル部分におけるアミノメチル基に対するパラ位はまた、フルオロ基で置換されている)のいずれも開示していない。さらに、特許文献1は、そのインドール部分における芳香族炭素(カルボニルに結合されているもの以外)が置換されている:より具体的にはインドールの5位がメトキシで置換されている、[(アミノメチル−フェニル)−ピペリジン−1−イル]−[インドリル]−メタノンのみを開示している。
【0012】
非特許文献20は、トリプターゼインヒビターとして3種類の [(アミノメチル−フェニル)−ピペリジン−1−イル]−[1H−インドリル−3−イル]−メタノンを開示している。第1のタイプのインヒビターは、式Bの化合物:
【化3】
であり、式中、インドール部分の芳香族炭素(カルボニルに結合しているもの以外)はいずれも置換されていないのに対し、インドール窒素は、水素、メチル、エチル、イソプロピル、プロピル、イソブチル、ブチル、ヘキシル、2−メトキシエチル、シクロヘキシルメチル、シクロプロピルメチル、3−ピリジル、2−チアゾール、アセチル、チオフェン−2−カルボニル、ベンゼンスルホニル,またはメタンスルホニルのようなR1で置換されている。第2のタイプのインヒビターは、式Cの化合物:
【化4】
であり、式中、インドール窒素は水素によってのみ置換され、そのインドール部分の単一の芳香族炭素(カルボニルに結合されているもの以外)は、4位、5位、6位、もしくは7位のメチル、または7位のフルオロのようなRで置換されている。第3のタイプのインヒビターは、式Dの化合物:
【化5】
であり、式中そのインドール部分中の単一の芳香族炭素(カルボニルに結合されているもの以外)が、7位においてメチルで置換されており、インドール窒素はメチル、エチル、プロピル、ブチル、または2−メトキシエチルのようなR1で置換されている。非特許文献20もまた、5位または7位においてインドール中の芳香族炭素における置換は耐性があるが、4位または6位の置換はほとんど不活性な化合物をもたらすことを開示している。
【0013】
特許文献1または非特許文献20には、(1)そのフェニル部分上のアミノメチル基に対するパラ位もまたフルオロ基で置換されている;(2)インドール窒素が2−メトキシエチルで置換されている;または(3)そのインドール部分中の2つもしくはそれ以上の芳香族炭素(カルボニルに結合しているもの以外)が置換されている、トリプターゼインヒビターとして特に価値のある薬学的特性を有するトリプターゼインヒビターを含むインドールが開示されていない。このような化合物は、トリプターゼインヒビターの投与によって改善され得る症状、例えば肥満細胞介在性炎症症状、炎症、および血管拡張性および気管支拡張性の神経ペプチドの分解に関連する疾患または状態を患っている患者の治療において容易に有用であり、セミカルバジド感受性アミン酸化酵素(SSAO)代謝についての障害を減少させる。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第6977263号
【非特許文献】
【0015】
【非特許文献1】Caughey,et al.,J.Pharmacol.Exp.Ther.,1988,244,133−137頁
【非特許文献2】Franconi,et al.,J.Pharmacol.Exp.Ther.,1988,248,947−951頁
【非特許文献3】Tam,et al.,Am.J.Respir.Cellmol.Biol.,1990,3,27−32頁
【非特許文献4】Sekizawa,et al.,J.Clin.Invest.,1989,83,175−179頁
【非特許文献5】K.Rice,P.A.Sprengler,Current Opinion in Drug Discovery and Development,1999,2(5),463−474頁
【非特許文献6】M.Q.Zhang,H.Timmerman,Mediators Inflamm.,1997,112,311−317頁
【非特許文献7】S.J.Wilson,et al.,Clin.Exp.Allergy,1998,28,220−227頁
【非特許文献8】S.C.Bischoff,et al.,Histopathology,1996,28,1−13頁
【非特許文献9】A.Naukkarinen,et al.,Arch.Dermatol.Res.,1993,285,341−346頁
【非特許文献10】A.A.Irani,et al.,Allergy Clin.Immunol.,1990,86,34−40頁
【非特許文献11】A.Jarvikallio,et al.,Br.J.Dermatol.,1997,136,871−877頁
【非特許文献12】L.C.Tetlow,et al.,Ann.Rheum.Dis,1998,54,549−555頁
【非特許文献13】M.G.Buckley,et al.,J.Pathol.,1998,186,67−74頁
【非特許文献14】Rouss et al.,J.Clin.Invest.,1991,88,493−499頁
【非特許文献15】J.A.CairnsおよびA.F.Walls,J.Clin.Invest.,1997,99,1313−1321頁
【非特許文献16】M.Jeziorska et al,J.Pathol.,1997,182,115−122頁
【非特許文献17】W.J.Beil et al,Exp.Hematol.,(1998)26,158−169頁
【非特許文献18】L.B.Schwarz et al,J.Clin.Invest.,1995,96,2702−2710頁
【非特許文献19】M.Steinhoff et al,Nat.Med.(N.Y.),2000,6(2),151−158頁
【非特許文献20】Bioorg.Med.Chem.Lett.15、2734(2005)
【発明の概要】
【課題を解決するための手段】
【0016】
本発明は、式I:
【化6】
の化合物または前記化合物のプロドラッグ、薬学的に受容可能な塩もしくは溶媒和物に及ぶ。
【0017】
さらに、本発明は、薬学的有効量の式Iの化合物および薬学的に受容可能な担体を含有する医薬組成物に関する。
【0018】
さらに、本発明は、トリプターゼインヒビターとしての式Iの化合物の使用に関し、トリプターゼインヒビター受容体を含む組成物中に化合物を導入することを包含する。さらに、本発明は、トリプターゼインヒビターで改善の必要がある生理学的症状を患っているか、または被っている患者を治療するための式Iの化合物の使用に関し、治療有効量の請求項1に記載の化合物を患者に投与することを包含する。
【0019】
本発明はまた、式Iの化合物およびその中で有用な中間体の製造に関する。
【0020】
本発明の態様、特性および利点は、以下の詳細な説明からより理解され、これらは、例としてのみとして与えられ、そして本発明を制限しない。
【0021】
略語のリスト
上記および本発明の説明を通して用いられる場合、他に示されない限り以下の略号は以下の意味を有すると理解される:
n−BuOAc n−酢酸ブチル
n−BuLi n−ブチルリチウム
sec−BuLi sec−ブチルリチウム
t−Bu tert−ブチル
t−BuOH tert−ブタノール
CuI ヨウ化銅
DCM ジクロロメタン、CH2Cl2または塩化メチレン
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
dppf 1,1’−ビス(ジフェニルホスフィノ)フェロセン
DSC 示差走査熱量測定
EDCI 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド HCl
eq 当量
Et エチル
Et2O ジエチルエーテル
TEA トリエチルアミン
EtOH エタノール
EtOAc 酢酸エチル
EtOC(O)Cl クロロギ酸エチル
HPLC 高速液体クロマトグラフィー
MgSO4 硫酸マグネシウム
Me メチル
MeOH メタノール
MS 質量分析
MTBE メチル t−ブチルエーテル
NaHCO3 重炭酸ナトリウム
Na2SO3 亜硫酸ナトリウム
Na2SO4 硫酸ナトリウム
NMR 核磁気共鳴
Pd(PPh3)2Cl2 ビストリフェニルホスフィンパラジウム(II)二塩化物
PdCl2dppf 1,1’−ビス(ジフェニルホスフィノ)フェロセン パラジウム(II)二塩化物
Pd(dtbpf)Cl2 (1,1’ビス(ジ−t−ブチルホスフィノ)フェロセン
パラジウム 二塩化物
Pd2(dba)3 トリス(ジベンジリデンアセトン)ジパラジウム(0)
Pd(OAc)2 酢酸パラジウム(II)
P(Cy)3 トリシクロヘキシルホスフィン
t−Bu3P トリ−t−ブチルホスフィン
PPh3 トリフェニルホスフィン
PrOH プロパノール
iPrOH イソ−プロパノール
i−PrOAc イソ−酢酸プロピル
t−BuOK カリウム tert−ブトキシド
PPSE ポリ−リン酸トリメチルシリルエステル
K2CO3 炭酸カリウム
K2SO4 硫酸カリウム
LC 液体クロマトグラフィー
Na2SO4 硫酸ナトリウム
rt 室温
Rt 保持時間
TFA トリフルオロ酢酸
TFAA トリフルオロ酢酸無水物
TGA 熱重量分析
THF テトラヒドロフラン
TLC 薄層クロマトグラフィー
TMS−アセチレン トリメチルシリル−アセチレン
【0022】
定義
上記ならびに本明細書および添付の特許請求の範囲の全体を通して使用される場合、他に示されない限り、以下の用語は以下の意味を有することが理解されるべきである。
【0023】
本明細書中で使用される場合、用語「本発明の化合物」および同等の表現とは、前記の式Iの化合物を包含し、そしてその表現は、プロドラッグ、薬学的に受容可能な塩および溶媒和物(例えば、水和物)を包含することが意味される。同様に、中間体を言及する場合、それ自体が請求項に記載されていてもいなくても、可能な限りその塩および溶媒和物を包含することが意味される。明確化のために、可能な限り特定の例を本文中に時々示すが、これらの例は単なる説明であり、可能な限りの他の例を排除することを意図しない。
【0024】
本明細書中で使用される場合、用語「治療(treatment)」または「治療(treating)」は、患者の症状の改善のためのような、予防的治療ならびに確立された症状の治療を包含する。このような改善としては、疾患の進行を遅らせることや患者の症状の有益な緩和を含む。
【0025】
「患者」とは、ヒトまたは他の哺乳動物を意味する。
【0026】
「有効量」とは、所望の治療効果を与えるのに効果的な化合物の量を記載することを意味する。
【0027】
「プロドラッグ」とは、望ましくない毒性、刺激性、アレルギー応答などを伴うことなく患者に投与するのに適切であり、そして、代謝的手段により(例えば、加水分解により)インビボで本発明の化合物に転換可能である化合物を意味する。プロドラッグの十分な議論は、T.HiguchiおよびV.Stella,Pro−drugs as Novel Delivery Systems,Vol.14 of the A.C.S.Symposium SeriesおよびEdward B.Roche編,Bioreversible Carriers in Drug Design,American Pharmaceutical Association and Pergamon Press,1987に提供され、これらの両方は参照によって本明細書中に援用される。
【0028】
「薬学的に受容可能な塩」は、望ましくない毒性または副作用の生じることのない酸を用いたこれらの活性成分の任意の塩を意味する。これらの酸は、薬学の専門家に十分公知である。適切な塩の非限定的な例としては以下が挙げられる:塩化物;臭化物;ヨウ化物;アスパラギン酸塩、特に酸性のアスパラギン酸塩;安息香酸塩、特に酸性の安息香酸塩;クエン酸塩、特に酸性のクエン酸塩;酒石酸塩;リン酸塩、特に酸性のリン酸塩;フマル酸塩、特に酸性のフマル酸塩;グリセロリン酸塩;グルコースリン酸塩;乳酸塩;マレイン酸塩、特に酸性のマレイン酸塩;オロト酸塩;シュウ酸塩、特に酸性のシュウ酸塩;硫酸塩、特に酸性の硫酸塩;トリクロロ酢酸塩;トリフルオロ酢酸塩;ベシル酸塩;トシラートおよびメタンスルホン酸塩。FDA公認の薬理学的に受容可能な塩のリストは、以下:Philip L.Gould、「Salt Selection for Basic Drugs」33 Int’l J.Pharm.201、202、214−216(1986);Stephen M.Berge et al.「Pharmaceutical Salts」、Journal of Pharmaceutical Sciences Vol.66、No.1、January 1977、pages 1−19のさらなる情報;およびHandbook of Pharmaceutical Salts、P.Heinrich Stahl、Camille G.Wermuth(Eds.)、IUPAC Wiley−VCH、2002より当該分野で公知の塩を製造するための方法;に与えられ、これらの文献は参照により本明細書中で援用される。
【0029】
「溶媒和物」は、本発明の化合物と1つ又はそれ以上の溶媒分子との物理的会合を意味する。この物理的会合には、水素結合が含まれる。いくつかの例では、例えば1つ又はそれ以上の溶媒分子が結晶性固体の結晶格子中に組み込まれる場合、溶媒和物は単離可能である。「溶媒和物」は、溶液相及び単離可能な溶媒和物の両方を包含する。典型的な溶媒和物には、水和物、エタノラート、メタノラート、及び同種のものが含まれる。
【0030】
「鈴木カップリング条件」は、鈴木カップリング溶媒、鈴木カップリング触媒および鈴木カップリング反応を用いた条件を意味する。
【0031】
「鈴木カップリング溶媒」は、イソプロピルアルコール以上の沸点を有するアルコール溶媒:例えばn−プロピルアルコール、n−ブチルアルコールなど;極性非プロトン性溶媒:例えばジメチルホルムアミド、1−メチル−2−ピロリドン、ジメチルスルホキシドなど;エーテル溶媒:例えばTHF、2−メチルTHF、ジメトキシエタンなど;または前出の溶媒と水もしくはトルエンの任意の混合物を意味する。
【0032】
「鈴木カップリング触媒」は、Pd(PPh3)4、Pd(PPh3)2Cl2、Pd2(dba)3、Pd(dtbpf)Cl2、のようなPd触媒;またはPPh3、dppf、t−Bu3P、P(Cy)3などのホスフィンリガンドと結合する、Pd(OAc)2、Pd2(dba)3のようなPd触媒を意味する。
【0033】
「鈴木カップリング温度」は、約60℃〜鈴木カップリング反応混合物の沸点温度の温度を意味する。
【0034】
「トリフルオロアセチル化条件」は、トリフルオロアセチル化試薬、トリフルオロアセチル化溶媒、およびトリフルオロアセチル化反応温度を用いた条件を意味する。
【0035】
「トリフルオロアセチル化試薬」は、トリフルオロ酢酸無水物、1,1,1−トリクロロ−3,3,3−トリフルオロアセトン、トリフルオロ酢酸およびポリ−リン酸トリメチルシリルエステル(PPSE)、塩化トリフルオロアセチル、フッ化トリフルオロアセチル、ペンタフルオロフェニルトリフルオロアセテートのなどを意味する。
【0036】
「トリフルオロアセチル化溶媒」は、酢酸エチル、酢酸イソプロピル、n−ブチルアセテートなどのようなエステル溶媒;トルエンなどのような芳香族炭化水素溶媒;塩化メチレン、1,2−ジクロロエタンなどのような塩素化炭化水素などの溶媒を意味する。
【0037】
トリフルオロアセチル化反応温度」は約−20℃〜約30℃を意味する。
【0038】
「水素化条件」は、水素化触媒、水素化溶媒、水素化反応温度および水素化圧力を用いる条件を意味する。
【0039】
「水素化反応溶媒」は、メタノール、エタノール、イソプロピルアルコールなどのようなアルコール溶媒;または酢酸;またはアルコール溶媒もしくは酢酸と水との混合物を意味する。
【0040】
「水素化触媒」は、HClなどのような無機酸または酢酸などのような有機酸を加えるかまたは加えない、PtO2、Pd/C、Pd(OH)2、Rh/Cなどを意味する。
【0041】
「水素化反応温度」は、約10〜約60℃を意味する。
【0042】
「水素化圧力」は、約10〜約1000psi水素(装置能力によって指示される上限)を意味する。
【0043】
特定または好ましい実施態様
さらに、本発明は、治療有効量の式Iの化合物を患者に投与することによって改善され得る生理学的症状を患っている患者を治療するための式Iの化合物の使用に関する。本発明の化合物を用いて治療され得る生理学的症状の特定の実施態様としては、以下が挙げられるが、特にこれらに限定されない:炎症性疾患、例えば、関節の炎症、関節炎、関節リウマチ、リウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎および他の慢性炎症性関節疾患および全国および他の炎症性呼吸状態。本発明によって治療され得る生理学的症状の他の実施態様としては、慢性閉塞性肺疾患(COPD)、COPDの悪化、関節軟骨崩壊(joint cartilage destruction)、眼結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺炎、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫、肥厚性瘢痕、種々の皮膚科学的症状(例えば、アトピー性皮膚炎および乾癬)、心筋梗塞、発作、狭心症およびアテローム硬化性プラーク破壊(atherosclerotic plaque rapture)のその他の結果、ならびに歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍および合胞体ウイルス感染症(syncytial viral infection)のような生理学的症状が挙げられる。
【0044】
特定の実施態様において、本発明は喘息および他の炎症性呼吸状態を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0045】
別の特定の実施態様において、本発明はCOPDを患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0046】
別の特定の実施態様において、本発明はCOPDの悪化を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0047】
別の特定の実施態様において、本発明はアレルギー性鼻炎を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0048】
別の特定の実施態様において、本発明は関節の炎症を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0049】
別の特定の実施態様において、本発明は炎症性腸疾患を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0050】
さらに、本発明は、式Iの化合物、βアドレナリン作用性アゴニスト(beta andrenergic agonist)、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬からなる群から選択される第二の化合物ならびにそれらの薬学的に受容可能な担体を含有する医薬組成物にまで及ぶ。このような組成物において、式Iの化合物および第二の化合物は、治療的に有効な活性、すなわち、相加効果または相乗効果を提供するような量で存在する。このような医薬組成物を用いて治療され得る特定の炎症性疾患または障害としては、喘息が挙げられるが、これに限定されない。
【0051】
さらに、本発明は、炎症性障害を患っている患者を治療するための方法に関し、式Iの化合物ならびにβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬からなる群から選択される第二の化合物を患者に投与することを包含する。このような方法において、式Iの化合物および第二の化合物は、治療的に有効な活性、すなわち、相加効果または相乗効果を提供するような量で存在する。このような本発明の方法において、本発明の化合物は、第二の化合物の前に患者に投与され得るか、第二の化合物は、本発明の化合物の前に患者に投与され得るか、または本発明の化合物および第二の化合物は同時に投与され得る。本方法に従って適用されるアドレナリン作用性アゴニスト(andrenergic agonist)、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬の特定の例は、以下に記載される。本発明で用いられることが企図される抗コリン作用薬は、臭化イプラトロピウムおよびチオトロピウムを含む。本発明で用いられることが企図される抗炎症性コルチコステロイドは、ジプロピオン酸ベクロメタゾン、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンおよびデキサメタゾンを含む。
【0052】
本発明はまた、式Iの化合物を製造するための、式2〜9の中間化合物にも向けられる:
【化7】
【0053】
医薬組成物
上に説明される通り、本発明の化合物は有用な薬理学的活性を示し、従って医薬組成物中に組み込まれ、そして特定の内科的疾患を患っている患者の治療に使用され得る。このように、さらなる態様に従って、本発明は、本発明の化合物およびその薬学的に受容可能な担体を含有する医薬組成物を提供する。本明細書中で使用される場合、用語「薬学的に受容可能」とは、好ましくは政府、特に連邦政府または州政府の規制当局により認可されているか、または米国薬局方もしくは動物、特にヒトにおける使用に関する別の一般的に認知されている薬局方に記載されていることを意味する。適切な薬学的担体は、E.W.Martinによる「Remington’s Pharmaceutical Sciences」に記載されている。
【0054】
本発明に従う医薬組成物は、1種またはそれ以上の薬学的に受容可能な補助剤または賦形剤を使用して慣用的な方法に従って製造され得る。とりわけ、補助剤としては、希釈剤、増量剤、結合剤、錠剤分解物質、流動促進剤、滑沢剤、界面活性剤、滅菌水性媒体および種々の非毒性の有機溶媒が挙げられる。組成物は、錠剤、カプセル剤、丸薬、徐放製剤、顆粒、粉末、水性溶液または水性懸濁液、注射用溶液、エリキシル剤またはシロップ剤の形態で製造されてもよく、そして、甘味料、着香料、着色料または安定化剤からなる群から選択される1種またはそれ以上の薬剤を含有することにより薬学的に受容可能な製剤を得ることができる。ビヒクルの選択およびビヒクル中の活性物質の量は、活性化合物の溶解度および化学的特性、特定の投与様式ならびに薬務において守られるべき条件に従って一般的に決定される。例えば、ラクトース、微結晶性セルロース、α化デンプン、未変性デンプン、ケイ化微結晶性セルロース、マンニトール、ソルビトール、キシリトール、デキストラート、フルクトース、クエン酸ナトリウム、炭酸カルシウム、第二リン酸カルシウム二水和物、無水第二リン酸カルシウム、硫酸カルシウムのような賦形剤は、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシルメチルセルロースナトリウム、α化デンプン、デンプン、ポリエチレングリコール、ポリエチレンオキシド、ポリカルボフィル、ゼラチンおよびアラビアゴムのような結合剤ならびにクロスカルメロース、グリコール酸ナトリウムデンプン、クロスポビドン、デンプン、微結晶性セルロース、アルギン酸および特定の複合ケイ酸塩のような錠剤崩壊剤と共に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、硬化植物油、鉱油、ポリエチレングリコール、脂肪酸のグリセリルエステル、ラウリル硫酸ナトリウムのような滑沢剤および二酸化ケイ素、タルク、デンプンのような流動促進剤と組み合わせて、ラウリル硫酸ナトリウム、ソルビタンエステル、ポリオキシエチレン脂肪酸エステル、ポロキサマー、ポリオキシエチレンエーテル、ドキュセートナトリウム、ポリエトキシ化ヒマシ油および塩化ベンザルコニウムのようないくつかの適切な湿潤剤と共に、錠剤を製造するために使用され得る。カプセルを製造するために、ラクトース、微晶性セルロース、α化デンプン、未変性デンプン、ケイ化微結晶性セルロースのような増量剤を、単独もしくは2種またはそれ以上の増量剤の混合物で、上記のバインダーを含むか含まずに、上記の適切な湿潤剤、錠剤崩壊剤、流動促進剤、滑沢剤などと共に使用するのに都合がよい。水性懸濁液を使用する場合、それらは乳化剤または懸濁を促進する薬剤を含有し得る。スクロース、エタノール、ポリエチレングリコール、プロピレングリコール、グリセロールおよびクロロホルムまたはこれらの混合物のような希釈剤をもまた使用してもよい。このような薬学的に受容可能な担体はまた、滅菌水および滅菌オイルであり得、これらとしては、ピーナツ油、大豆油、鉱油、ゴマ油などのような石油、動物、植物または合成起源のものが挙げられる。医薬組成物を静脈内投与する場合、水が好ましい担体である。生理食塩水および水性デキストロースとグリセロールとの溶液もまた、特に注射用溶液のための液体担体として利用され得る。適切な薬学的賦形剤としては、マンニトール、ヒト血清アルブミン(HSA)、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、米、小麦、チョーク、シリカゲル、炭酸マグネシウム、ステアリン酸マグネシウム、ステアリン酸ナトリウム、グリセロールモノステアレート、タルク、塩化ナトリウム、乾燥脱脂乳、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。これらの組成物は、溶液、懸濁液、錠剤、丸薬、カプセル、粉末、徐放製剤などの形態をとり得る。
【0055】
当然ながら、本発明の医薬組成物は、治療有効量の活性化合物を適切な量の担体と共に含有することによって、患者への適切な投与のための形態を提供する。静脈内注射が極めて有効な投与形態であるが、以下に議論される注射または経口、経鼻もしくは非経口の投与による他の様式が利用され得る。
【0056】
治療方法
式Iの化合物は、文献に記載され、そして本明細書中以下に記載されている試験によると、トリプターゼ阻害活性を有し、この試験結果はヒトおよび他の哺乳動物における薬理学的活性と相関すると考えられている。従って、さらなる実施態様において、本発明は、トリプターゼインヒビターの投与によって改善され得る症状を患っているか、または罹りやすい患者を治療するための式Iまたは式Iを含有する組成物の使用に関する。例えば、式Iの化合物は、炎症性疾患、例えば関節の炎症(関節炎、関節リュウマチおよびリウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎、変形性関節症もしくは他の慢性炎症性関節疾患のような他の関節炎状態または関節軟骨崩壊の疾患を含む)、結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺疾患、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経腺維腫、肥大性瘢痕、種々の皮膚科的症状(例えば、アトピー性皮膚炎および乾癬)、心筋梗塞、発作、狭心症もしくはアテローム性動脈硬化性プラーク破壊のその他の結果、ならびに歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍または合胞体ウイルス感染症を治療するのに有用である。
【0057】
本発明のさらなる特性によれば、トリプターゼインヒビターの投与によって改善され得る症状、例えば、前述したような症状を患っているか、または罹りやすいヒト患者または動物罹患体の治療方法を提供し、有効量の本発明の化合物または本発明の化合物を含有する組成物を患者に投与することを包含する。
【0058】
組合せ療法
上で説明した通り、他の薬学的活性剤は、治療されるべき疾患によって、式Iの化合物と組合せて利用され得る。例えば、喘息の治療において、アルブテロール、ターブタリン、フォルモテロール、フェノテロールまたはプレナリン(prenaline)のようなβアドレナリン作用性アゴニストを包含し得、同様に、臭化イプラトロピウムのような抗コリン作用薬;ジプロピオン酸ベクロメタゾン、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンまたはプレジノーズ(predinose)のような抗炎症性コルチコステロイド;ならびにクロモグリク酸ナトリウムおよびネドクロミルナトリウムのような抗炎症剤を包含し得る。従って、本発明は、式Iの化合物およびβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症剤からなる群から選択される第二の化合物;ならびにそれらの薬学的に受容可能な担体を含有する医薬組成物にまで及ぶ。ロイコトリエンアンタゴニストとして本発明で使用することが特に意図されるのはモンテルカストである。またホスホジエステラーゼ−4 インヒビターとして本発明で用いることが特に意図されるのは、シフルモラスト(ciflumolast)である。本医薬組成物に適用される特定の薬学的担体は、本明細書中に記載される。
【0059】
さらに、本発明は、喘息を患っている患者を治療する方法に及び、本発明の化合物およびβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症剤からなる群から選択される第二の化合物を患者に投与する工程を包含する。このような組合せ方法において、本発明の化合物を第二の化合物の投与前に投与し得るか、本発明の化合物を第二の化合物の投与後に投与し得るか、または本発明の化合物と第二の化合物を同時に投与し得る。
【0060】
送達様式
本発明に従って、式Iの化合物または本化合物を含有する医薬組成物は、非経口的、経粘膜的(例えば、経口、経鼻、肺内もしくは直腸内)または経皮的に患者に導入されてもよい。
【0061】
経口送達
一般的に、Remington’s Pharmaceutical Sciences,第18編、1990(Mack Publishing Co.Easton PA 18042)の第89章(参照により本明細書中に援用される)に記載されている経口固体投薬形態が、本明細書中で使用するために意図される。固体投薬形態としては、錠剤、カプセル、丸薬、口中錠またはトローチ剤(lozenge)、カシェ剤またはペレット剤が挙げられる。また、リポソーム被包またはプロテイノイドカプセル化を使用して、本発明の組成物を製剤し得る(例えば、米国特許第4,925,673号に記載されているプロテイノイドミクロスフィア)。リポソーム被包を使用してもよく、そしてリポソームは種々のポリマーで誘導体化されていてもよい(例えば、米国特許第5,013,556号)。治療のための可能な固体投薬形態の説明は、Marshall,K.,In:Modern Pharmaceutics,Edited by G.S.Banker and C.T.Rhodes、第10章、1979に記載されており、これは参照により本明細書中に援用される。一般的に、製剤は本発明の化合物および胃環境に対して保護を与え、そして腸内において生物学的に活性な物質(即ち、本発明の化合物)を放出する不活性成分を含有する。
【0062】
本発明の化合物の経口投薬形態もまた特に意図される。このような化合物は、経口送達がより効率的になるように化学的に修飾されてもよい。一般的に、意図される化学的修飾は、成分分子自体への少なくとも1つの部分の連結であり、前記部分は、(a)タンパク質分解の阻害;および(b)胃または腸から血流への取りこみを可能にする。本発明の化合物の全体的な安定性の向上および体内循環時間の延長もまた所望される。このような部分の例としては、ポリエチレングリコール、エチレングリコールとプロピレングリコールのコポリマー、カルボキシメチルセルロース、デキストラン、ポリビニルアルコール、ポリビニルピロリドンおよびポリプロリンが挙げられる。Abuchowski and Davis,1981,「Soluble Polymer−Enzyme Adducts」In:Enzymes as Drugs,Hocenberg and Roberts編、Wiley−Interscience,New York,NY,367−383頁;Newmark et al.,1982,J.Appl.Biochem.4:185−189。使用され得る他のポリマーは、ポリ−1,3−ジオキソランおよびポリ−1,3,6−チオキソカンである。上で示される通り、薬学的用法はポリエチレングリコール部分であることが好ましい。
【0063】
本発明の化合物について、放出場所は、胃、小腸(十二指腸、空腸もしくは回腸)または大腸であり得る。胃内では溶解しないが十二指腸または腸内のどこかで物質を放出する利用可能な製剤を当業者は承知している。発明の化合物を保護することによってか、または腸のような胃環境の先で化合物を放出することによって、放出による胃環境への悪影響を回避することが好ましい。
【0064】
完全に胃耐性とするために、少なくともpH5.0で不浸透性であるコーティングが必須である。腸溶性コーティングとして使用される、より一般的な不活性成分の例としては、セルロースアセテートトリメリテート(CAT)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、HPMCP50、HPMCP55、ポリビニルアセテートフタレート(PVAP)、Eudragit L30D、Aquateric、セルロースアセテートフタレート(CAP)、Eudragit L、Eudragit Sおよびセラックが挙げられる。これらのコーティングは混合フイルムとして使用してもよい。
【0065】
コーティングまたはコーティング混合物はまた、胃に対する保護を意図しない錠剤に使用され得る。これには、糖コーティングまたは錠剤を嚥下しやすくするコーティングが包含され得る。カプセルは、ドライ治療薬(即ち、粉末)の送達のためのハードシェル(例えば、ゼラチン)からなり;液体形態について、ソフトゼラチンシェル使用されてもよい。カシェ剤のシェル物質は、濃厚なデンプンまたは他の食用紙であり得る。丸薬、トローチ剤、湿製錠剤またはすりこみ錠剤について、湿潤塊化技術(moist massing technique)を使用し得る。
【0066】
治療薬は、約1mmの粒径の顆粒またはペレットの形態の微細マルチ粒子としての製剤中に含有され得る。カプセル投与のための物質の製剤はまた、粉末、軽度に圧縮されたプラグまたは錠剤としても存在し得る。治療薬は圧縮によって製造され得る。
【0067】
着色料および着香料は全て含有してもよい。例えば、本発明の化合物を製剤し(例えば、リポソームカプセル化または微小球カプセル化によって)、次いで着色料および着香料を含有する冷蔵飲料のような食品中にさらに含有してもよい。
【0068】
不活性物質を用いて治療薬の容量を希釈させるか、または増大させてもよい。これらの希釈剤としては、炭水化物、特にマンニトール、α−ラクトース、無水ラクトース、セルロース、スクロース、変性デキストランおよびデンプンが挙げられ得る。特定の無機塩もまた増量剤として使用されてもよく、これらとしては、第三リン酸カルシウム、炭酸マグネシウムおよび塩化ナトリウムが挙げられる。いくつかの市販の希釈剤としては、Fast−Flo、Emdex、STA−Rx1500、EmcompressおよびAvicellが挙げられる。
【0069】
治療薬を固体投薬形態に製剤するのに錠剤崩壊剤を含有してもよい。錠剤崩壊剤として使用される物質としては、デンプンをベースにした市販の錠剤崩壊剤であるExplotabが挙げられるが、デンプンに限定されない。デンプングリコール酸ナトリウム、Amberlite、カルボキシメチルセルロースナトリウム、ウルトラミロペクチン、アルギン酸ナトリウム、ゼラチン、オレンジ果皮、酸性カルボキシメチルセルロース、天然の海綿およびベントナイトを全て使用してもよい。錠剤崩壊剤の別の形態は、不溶性カチオン交換樹脂である。粉末化ガムを錠剤崩壊剤および結合剤として使用してもよく、そしてこれらは、寒天、カラヤ(Karaya)またはトラガカントのような粉末化ガムが挙げられ得る。アルギン酸およびそのナトリウム塩もまた、錠剤崩壊剤として有用である。
【0070】
結合剤を使用して治療薬を保持し、同時に硬質錠剤を形成し、そしてこれらとしては、アラビアゴム、トラガカント、デンプンおよびゼラチンのような天然の産物由来の物質が挙げられる。他のものとしては、メチルセルロース(MC)、エチルセルロース(EC)およびカルボキシメチルセルロース(CMC)が挙げられる。ポリビニルピロリドン(PVP)およびヒドロキシプロピルメチルセルロース(HPMC)の両方が、治療薬を顆粒化するためにアルコール溶液中で使用され得る。
【0071】
製剤プロセスの過程で、付着を防止するために治療薬の製剤において抗摩擦剤を含有させてもよい。滑沢剤は治療薬とダイ壁との間の層として使用されてもよく、そしてこれらとしては、以下が挙げられるが、これらに限定されない;ステアリン酸(そのマグネシウム塩およびカルシウム塩を含む)、ポリテトラフルオロエチレン(PTFE)、流動パラフィン、植物油およびワックス。ラウリル硫酸ナトリウム、ラウリル硫酸マグネシウム、種々の分子量のポリエチレングリコール、Carbowax4000および6000のような可溶性滑沢剤も使用してもよい。
【0072】
製剤化の過程で、薬物の流動特性を改善し、圧縮の過程で、再配列を助け得る流動促進剤を添加してもよい。流動促進剤はデンプン、タルク、熱分解法シリカおよび水和シリコアルミネートが挙げられ得る。
【0073】
水性環境中への治療薬の溶解を助けるために、界面活性剤を湿潤剤として添加してもよい。界面活性剤としては、ラウリル硫酸ナトリウム、スルホコハク酸ジオクチルナトリウムおよび硫酸ジオクチルナトリウムのような陰イオン清浄剤が挙げられ得る。陽イオン清浄剤が使用されてもよく、これとしては、塩化ベンザルコニウムまたは塩化ベンゼトニウムが挙げられる。界面活性剤として製剤中に含有し得る、可能性のある非イオン性洗剤のリストは、ラウロマクロゴール400、ポリオキシ40ステアレート、ポリオキシエチレン水素化ヒマシ油10、50および60、グリセロールモノステアレート、ポリソルベート40、60、65および80、スクロース脂肪酸エステル、メチルセルロースならびにカルボキシメチルセルロースが挙げられる。これらの界面活性剤は、本発明の化合物の製剤中に単独または異なった比率の混合物として存在し得る。
【0074】
本発明の化合物の取りこみを増強する可能性のある添加剤は、例えば、脂肪酸のオレイン酸、リノール酸およびリノレン酸である。徐放性経口製剤が望ましくあり得る。拡散または浸出の機構のいずれかによって放出を可能にする不活性マトリクス(例えば、ガム)中に薬物を組み込み得る。徐々に崩壊するマトリクスをもまた製剤中に組み込んでもよい。いくつかの腸溶性コーティングはまた、遅延放出効果を有する。本治療薬の徐放の別の形態は、Oros治療薬システム(Alza Corp.)に基づく方法によるものであり、即ち、水を侵入させて、そして浸透圧作用によって単一の小型開口部より薬剤を押出す半透膜中に薬物を封入する。
【0075】
他のコーティングが製剤に使用されてもよい。これらとしては、コーティングパンに適用し得る種々の糖が挙げられる。治療薬はまた、フィルムコート化錠剤中にあってもよく、そしてこの場合に使用される物質は、2グループに分けられる。第一グループは非腸溶性物質であり、そしてこれらとしては、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、メチルヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウム、ポビドン(providone)およびポリエチレングリコールが挙げられる。第二グループは、一般的にフタル酸のエステルである腸溶性物質からなる。
【0076】
物質の混合物を使用して、最適なフィルムコーティングを提供し得る。フィルムコーティングは、パンコーター中もしくは流動床中、または圧縮コーティングによって行なわれ得る。
【0077】
肺送達
単独または医薬組成物中の本発明の化合物の肺送達がまた、本明細書において意図される。吸入の間、化合物は哺乳動物の肺に送達され、そして肺上皮を通過して血流に満たされる。この他の報告はAdjei et al.,1990,Pharmaceutical Research,7:565−569;Adjei et al.,1990,International Journal of Pharmaceutics,63:135−144(酢酸ロイプロリド);Braquet et al.,1989,Journal of Cardiovascular Pharmacology,13(補遺5):143−146(エンドセリン−1);Hubbard et al.,1989,Annals of Internal Medicine,Vol.III,206−212頁(a1−アンチトリプシン);Smith et al.,1989,J.Clin.Invest.84:1145−1146(a−1−プロテイナーゼ);Oswein et al.,1990,「Aerosolization of Proteins」,Proceedings of Symposium on Respiratory Drug Delivery II,Keystone,Colorado,March(組換えヒト成長ホルモン);Debs et al.,1988,J.Immunol.140:3482−3488(インターフェロン−γおよび腫瘍壊死因子α)およびPlatz et al.,米国特許第5,284,656号(顆粒球コロニー刺激因子)が挙げられる。全身作用のための薬物の肺送達のための方法および組成物は、Wong et al.に対して1995年9月19日に発行された米国特許第5,451,569号に記載されている。
【0078】
治療薬の肺送達のために設計された広範な機械的デバイスが、本発明の実施における使用について意図され、このデバイスとしては、ネブライザー、定量吸入器および粉末吸入器が挙げられるが、これらに限定されず、これらの全てを当業者が承知している。
【0079】
本発明の実施に適切な市販のデバイスのいくつかの特定の例としては、ほんの一部の名前について、Mallinckrodt,Inc.,St Louis,Missouri製のUltraventネブライザー;Marquest Medical Product,Englewood,Colorado製のAcorn IIネブライザー;Glaxo Inc.,Research Triangle Park,North Carolina製のVentolin定量吸入器;およびFisons Corp.,Bedford,massachusetts製のSpinhaler粉末吸入器が挙げられる。全てのこのようなデバイスは、本発明の化合物を投与するために適切な製剤の使用を必要とする。典型的に、各製剤は、利用されるデバイスの種類に特異的であり、そして治療に有用な通常の希釈剤、補助剤および/または担体に加えて、適切な高圧ガス物質の使用を包含し得る。リポソーム、マイクロカプセルもしくは微小球、封入複合体または他の種類の担体の使用もまた意図される。本発明の化学修飾された化合物もまた、化学修飾の種類または利用されるデバイスの種類に応じて、異なる製剤に製造され得る。
【0080】
ジェット型または超音波型のいずれかのネブライザーと共に使用するのに適切な製剤は、典型的に、溶液1mLあたり約0.1〜25mgの化合物濃度で水中に溶解された本発明の化合物を含有する。製剤はまた、緩衝化剤および単純な糖(例えば、浸透圧の安定化および調節のため)を含有してもよい。ネブライザー製剤はまた、エアロゾルを形成するのに溶液の噴霧により起こる化合物の表面誘導凝集を低減または防止するために界面活性剤を含有してもよい。
【0081】
定量吸入器デバイスと共に使用するための製剤は、一般的に、界面活性剤の助けで高圧ガス中に懸濁された本発明の化合物を含有する微細分割粉末を含有する。クロロフルオロカーボン、ヒドロクロロフルオロカーボン、ヒドロフルオロカーボンまたはヒドロカーボン(トリクロロフルオロメタン、ジクロロジフルオロメタン、ジクロロテトラフルオロエタノールおよび1,1,1,2−テトラヒドロエタンまたはこれらの組合せを含む)のような高圧ガスは、この目的のために利用される任意の慣用的な物質であり得る。適切な界面活性剤としては、ソルビタントリオレエートおよび大豆レシチンが挙げられる。オレイン酸もまた界面活性剤として有用であり得る。
【0082】
粉末吸入器デバイスから投与するための製剤は、本発明の化合物を含有する微細分割乾燥粉末を含有し、そしてまた、ラクトース、ソルビトール、スクロースまたはマンニトールのような充填剤を、デバイスからの粉末の分散を促進するような量(例えば、製剤の50〜90質量%)で含有し得る。本発明の化合物は、肺の末端にまで最も効果的に送達するために、平均粒径10mm(またはミクロン)未満、最も好ましくは0.5〜5mmを有する粒子形態に製造することが最も好都合である。
【0083】
経鼻送達
本発明の化合物の経鼻送達もまた意図される。経鼻送達により、鼻への治療薬の投与後に直接血流に化合物が移動するようになり、肺中に治療薬を付着させる必要が無い。経鼻送達のための製剤としては、デキストランまたはシクロデキストランを含有するものが挙げられる。
【0084】
経皮送達
薬物の経皮投与の分野において種々の多数の方法(例えば、経皮パッチを介する)が公知であり本発明において適用される。経皮パッチは、例えば、米国特許第5,407,713号、第5,352,456号、第5,332,213号、第5,336,168号、第5,290,561号、第5,254,346号、第5,164,189号、第5,163,899号、第5,088,977号および第5,087,240号、第5,008,110号、および第4,921,475号に記載され、これらの開示の各々は、その全体が参照により本明細書中に援用される。
【0085】
経皮投与経路は、経皮浸透増強剤(例えば、米国特許第5,164,189号、第5,008,110号、および第4,879,119号に記載された増強剤)の使用によって増強され得ることが容易に理解され、これらの開示の各々は、その全体が参照により本明細書中に援用される。
【0086】
局所投与
局所投与のために、本発明の化合物を含有するゲル(水またはアルコールベース)、クリームまたは軟膏が使用されてもよい。本発明の化合物はまた、パッチ適用のためのゲルまたはマトリクス基剤中に組み込まれてもよく、これにより経皮バリアを通して化合物の制御放出が可能になる。
【0087】
直腸投与
直腸投与用の固体組成物としては、公知の方法で製剤され、そして本発明の化合物を含有する坐剤が挙げられる。
【0088】
投薬量
本発明の組成物中の活性成分の比率は変更されてもよく、適切な投薬量が得られるような比率を構成することが必要である。明らかに、いくつかの単位投薬形態を、ほとんど同時に投与してもよい。利用される用量は、医師により決定され、そして所望の治療効果、投与経路および治療期間、ならびに患者の症状に依存する。成人において、用量は一般的に、吸入では約0.001〜約50、好ましくは約0.001〜約5mg/kg体重/日であり、経口投与では約0.01〜約100、好ましくは0.1〜70、より特に0.5〜10mg/kg体重/日であり、そして静脈内投与では約0.001〜約10、好ましくは0.01〜1mg/kg体重/日である。いずれの場合にも、用量は治療されるべき対象に対する年齢、体重、一般的健康状態および医薬品の効果に影響し得る他の特性のような固有の要因に従って決定される。
【0089】
さらに、本発明に従う化合物は、所望の治療効果を得るために必要な頻度で投与され得る。一部の患者は、より高用量または低用量に急速に応答し、そしてはるかに低用量の維持量で十分であることが見出され得る。別の患者について、各特定の患者の生理学的必要性に従って、1〜4用量/日の比率で長期間の治療を要する必要があり得る。一般的に、活性成分は1〜4回/日で経口投与され得る。もちろん、一部の患者については、多くて1または2用量/日で処方される必要がある。
【0090】
当然ながら、本発明の化合物の投与が有効な治療計画となる患者は、好ましくはヒトであるが、いずれの動物でもあり得る。従って、当業者によって容易に理解され得る通り、本発明の方法および医薬組成物は、いずれの動物、特に哺乳動物への投与(すなわち、獣医用途)に特に相応し、これらの動物としては、以下が挙げられるがこれらに限定されない:ネコ科またはイヌ科の動物の罹患体のような家畜(domestic animal);限定されないが、ウシ科、ウマ科、ヤギ、ヒツジおよびブタの罹患体のような家畜(farm animal);野生動物(野外または動物園内にかかわらず);マウス、ラット、ウサギ、ヤギ、ヒツジ、ブタ、イヌ、ネコなどのような実験動物;ニワトリ、シチメンチョウ、鳴き鳥などのようなトリ種。
【0091】
製造の詳細
式Iの化合物は、本明細書中でこれまでに使用されてるかまたは文献に記載される方法(例えば、R.C.LarockのComprehensive Organic Transformations,VCH publishes,1989によって記載されるもの)または本明細書中に記載されるような方法を意味する、公知の方法を適用または適合させることによって製造され得る。
【0092】
本明細書中の以下に記載される反応において、反応におけるそれらの望まれていない関与を避けるために、反応性官能基(例えば、アミノ基)を保護する必要があり得る。標準的技法に従って慣用的な保護基を使用し得る;例えば、T.W.GreeneおよびP.G.M.Wutsの「Protective Groups in Organic Chemistry」,John Wiley and Sons,1991を参照のこと。
【0093】
特に、式Iの化合物はスキーム1−2によって示されるように製造することができる。
【0094】
例えば、本発明の化合物はそれらの製造が収束性の合成からなるアキラルな化合物である。安息香酸塩としての本発明の化合物は、以下のスキームに示されるとおりに製造される。
【0095】
スキーム1
【化8】
【0096】
(i)クロロギ酸エチル、ピリジン、THF、0℃、100%;(ii)a:sec−BuLi、THF、−78℃、b:I2、THF、−78℃、52−68%;(iii)TMS−アセチレン、TEA、CuI、Pd(PPh3)2Cl2、脱気THF、60℃、93%;(iv)KOH、t−BuOH、70℃、91%;(v)KOH粉末、2−メトキシエチル臭化物、DMSO、室温、95%;(vi)TFAA、DMF、40℃、89%;(vii)5M NaOH、MeOH、85℃、96%;(viii)2,2,2−トリフルオロ−N−(フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩、EDCI、TEA、CH2Cl2(DCM)、室温、99%;(ix)a:K2CO3、MeOH/H2O、b:1M HCl(Et2O中、90%)
【0097】
化合物1は、適切な塩基(例えばピリジン)の存在下においてアミノ基保護剤(例えばクロロギ酸エチル)でアミノ基を保護することにより、化合物2に転換され、保護された化合物2が得られる。
【0098】
化合物2は、3段階の工程で化合物5に転換される。化合物2は、2と強塩基(例えば第2級ブチルリチウム)とを反応させることによってカルバミン酸エステルの隣の位置でヨウ素化されて、分子ヨウ素のようなヨウ素源と反応するアニオンを形成し、化合物3を得る。その後化合物3を、トリメチルシリルアセチレンおよび塩基(例えばトリエチルアミン)の存在下で、ヨウ化銅(I)およびビストリフェニルホスフィンパラジウム(II)二塩化物のような触媒条件を用いてアセチレン化合物4に転換する。化合物4を、水酸化カリウムのような強塩基を用いて環化し、加熱してインドール化合物5を得る。
【0099】
化合物5は、室温にて水酸化カリウムのような強塩基の存在下、ジメチルスルホキシドのような二極性非プロトン性溶媒中で、そのインドール窒素をアルキルハライドを用いてアルキル化することによって化合物6に転換され、化合物6を得られる。
【0100】
化合物6は、2工程で化合物8に転換される。最初に、化合物6をN,N−ジメチルホルムアミドのような溶媒の存在下でトリフルオロ酢酸無水物を用いて処理し、加熱することにより化合物6を化合物7に転換する。化合物7を水酸化ナトリウムのような強塩基で処理してその3位に酸性官能基を持つ化合物8を得る。
【0101】
化合物8は、ジクロロメタンのような不活性溶媒中、酸性カップリング試薬(EDCIなど)および有機塩基(トリエチルアミンなど)の存在下で酸性の8を2,2,2−トリフルオロ−N−(フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(化合物14)と反応させることにより、アミド9に転換される。
【0102】
化合物9は、メタノール/水のような溶媒混合物中で炭酸カリウムのような弱塩基での処理でN−ベンジル トリフルオロアセトアミドを脱保護することにより、化合物10に転換される。この塩酸塩は、エーテルのような極性有機溶媒の存在下で形成することができ、式Iの([4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−メチル−1H−インドール−3−イル]−メタノン)の塩酸塩である化合物10を得る。
【0103】
このスキームの反応は以下のとおりである。
【0104】
工程A:(2−フルオロ−5−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(2)の製造
【化9】
THF(500mL)に溶解させた1(50.72g、0.26mol)およびピリジン(27.3mL、0.34mol)の溶液に、0℃でクロロギ酸エチル(32.2mL、0.39mol)を30分間にわたって滴下した。1時間後、LC/MSとTLCの両方が、反応が完了したことを示した。その反応混合物をH2OとEtOAcに分配した。2層を分離し、その有機層を1M HCl、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(95/5〜70/30)を用いてシリカゲル上で精製し、69.23g(99%)の生成物2を透明な無色の液体として得た。
1H NMR(CDCl3)δ8.11(br s,1H)、7.07(dd,J=9.1、9.3Hz,1H)、7.00−6.80(m,2H)、4.27(q,J=7.1Hz,2H)、1.33(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−57.84(s,3F)、−134.01(br s,1F); MS 309(M+CH3CN+1、100%)、268(M+1).
【0105】
工程B:(6−フルオロ−2−ヨード−3−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(3)の製造
【化10】
THF(180mL)に溶解させた2(31.34g、117.2mmol)の溶液に−78℃でsec−BuLi(シクロヘキサン中、1.4M、200mL、280mmol)を1時間にわたり滴下した。20分後、THF(150mL)に溶解させたI2(44.6g、175.8mmol)の溶液を30分間にわたって滴下した。次いでこの混合物を−78℃で30分間攪拌した。飽和NH4Clを加え、冷却浴を取り除いた。反応混合物をH2OとEtOAcに分配した。2層を分離し、その有機層を10% Na2SO3、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM(50mL)に懸濁させ、ヘプタン(300mL)を加えた。得られた懸濁液からの白色粉末3(18.1g、39%)をサクションフィルターで回収し、空気乾燥させた。ろ液を減圧下で濃縮し、残留物をヘプタン(200mL)中に懸濁させた。別バッチの3(3.8g、8%)をサクションフィルターで回収し、空気乾燥させた。シリカゲルクロマトグラフィーによって精製することにより、追加生成物を得ることができた。
1H NMR(CDCl3)δ7.30−17.10(m,2H)、6.16(br s,1H)、4.26(q,J=7.1Hz,2H)、1.32(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−56.90(s,3F)、−114.35(d、J=8.5Hz,1F);MS 394(M+1、100%)、374、364、321、267.
【0106】
工程C:(6−フルオロ−3−トリフルオロメトキシ−2−トリメチルシラニルエチニル−フェニル)−カルバミン酸エチルエステル(4)の製造
【化11】
脱気したTHF(180mL)に溶解させた3(18.1g、45.9mmol)、Et3N(12.8mL、91.9mmol)、Pd(PPh)2Cl2(1.6g、5%mol)、CuI(0.7g、8%mol)、およびTMS−アセチレン(19.6mL、137.8mmol)の混合物を60℃で一晩加熱した。その混合物を室温まで冷却し、その後H2OとEtOAcに分配した。この混合物をセライトを通して濾過し、不溶性物質を除去した。ろ液の2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAcを用いてシリカゲル上で精製し、15.6g(93%)の生成物4をベージュの固形物として得た。
1H NMR(CDCl3)δ7.15−7.00(m,2H)、6.41(br s,
1H)、4.26(q,J=7.1Hz,2H)、1.31(t,J=7.1Hz,3H)、0.27(s,9H);19F NMR(CDCl3)δ−57.59(s,3F)、−118.15(s,1F);MS 364(M+1、100%).
【0107】
工程D:7−フルオロ−4−トリフルオロメトキシ−1H−インドール(5)の製造
【化12】
脱気したt−BuOH(300mL)に溶解させた4(28.9g、79.6mmol)およびKOH(35.7g、636.7mmol)の混合物を70℃で一晩加熱した。LC/MSが反応の完了を示した。この混合物を室温まで冷却し、その後H2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜60/40)を用いてシリカゲル上で精製し、16g(91%)の5を黄色液体として得た。
1H NMR(CDCl3)δ8.47(br s,1H)、7.35−7.20(m,1H)、6.95−6.80(m,2H)、6.68(d、J=2.5Hz,1H);19F NMR(CDCl3)δ−57.63(s,3F)、−136.10(d、J=8.5Hz,1F);MS 220(M+1、100%)、200.
【0108】
工程E:P7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール(6)の製造
【化13】
DMSO(150mL)に溶解した5(16g、72.8mmol)およびKOH粉末(20.4g、364.2mmol)の混合物を室温で10分間攪拌した。2−メトキシエチル臭化物(10.3mL、109.2mmol)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。この混合物をH2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、19.3g(95%)の6を黄色液体として得た。
1H NMR(CDCl3)δ7.15(d、J=2.1Hz,1H)、6.90−6.75(m,2H)、6.56(t,J=2.5Hz,1 H)、3.72(t,J=5.2Hz,2H)、3.72(t,J=5.2Hz,2H)、3.31(s,3H);19F NMR(CDCl3)δ−57.54(s,3F)、−137.00(d、J=11.3Hz,1F);MS 278(M+1、100%).
【0109】
工程F:2,2,2−トリフルオロ−1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−エタノン(7)の製造
【化14】
DMF(135mL)に溶解させた6(19.3g、69.7mmol)の混合物にTFAA(26.2mL、188.2mmol)を加えた。この混合物を40℃で一晩加熱した。TLCが、反応が完了したことを示した。混合物を室温まで冷却し、その後H2OとEt2Oの間に分配した。2層を分離し、有機層を飽和NaHCO3(2X)、H2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製して、23.4g(89%)の7をわずかに緑色の固形物として得た。
1H NMR(CDCl3)δ8.03(d、J=1.4Hz,1H)、7.20−6.95(m,2H)、4.54(t,J=4.9Hz,2H)、3.76(t,J=4.8Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−71.10(s,3F)、−134.95(d、J=11.5Hz,1F);MS 374(M+1、100%).
【0110】
工程G:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボン酸(8)の製造
【化15】
MeOH(100mL)および5M NaOH(100mL)に溶解させた7(23.4g、62.6mmol)の混合物を80℃で一晩加熱した。LC/MSが、反応が完了したことを示した。反応混合物を室温まで冷却し、その後減圧下で濃縮して大部分のMeOHを除去した。残留物をH2Oに溶解し、その後Et2Oで一度洗浄した。水層を濃HClによってゆっくりとpH2まで酸性化した。酸性化した懸濁液をEt2Oで抽出し、有機抽出物をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM/ヘプタン(10/90)に懸濁させた。懸濁液中の白色粉末8(19.4g、96%)をサクションフィルターで回収し、空気乾燥させた。
1H NMR(CDCl3)δ8.02(s,1H)、7.15−7.05(m,1H)、7.00−6.90(m,1H)、4.49(t,J=5.0Hz,2H)、3.75(t,J=4.9Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−135.65(d、J=11.3Hz,1F);MS 363(M+CH3CN+1)、322(M+1、100%).
【0111】
工程H:2,2,2−トリフルオロ−N−(4−フルオロ−3−{1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボニル]−ピペリジン−4−イル}−ベンジル)−アセトアミド(9)の製造
【化16】
CH2Cl2に溶解させた8(19.1g、59.6mmol)、Et3N(24.8mL、177.9mmol)、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(11、26.4g、77.5mmol)(14)、およびEDCI(17.1g、89.3mmol)の混合物を室温で一晩攪拌した。TLCとLC/MSの両方が、反応が完了したことを示した。この混合物をH2OとCH2Cl2の間に分配した。2層を分離し、有機層をブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(40/60〜0/100)を用いてシリカゲル上で精製し、9(36g、99%)を白色泡沫として得た。
1H NMR(CDCl3)δ7.37(s,1H)、7.20−7.10(m,2H)、7.10−6.85(m,4H)、4.95(br s,1H)、4.60−4.35(m,4H)、3.90(br s,1 H)、3.73(t,J=5.0Hz,2H)、3.32(s,3H)、3.25−2.70(m,3H)、2.05−1.50(m,4H);19F NMR(CDCl3)δ−57.54(s,3F)、−75.39(s,3F)、−119.31(s,1F)、−134.96(d、J=11.3Hz,1F);MS 608(M+1、100%).
【0112】
工程I:[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン塩酸塩(10)の製造
【化17】
MeOH(400mL)に溶解させた9(36g、59.3mmol)の混合物に、K2CO3水溶液(65.5g、474mmol、120mL H2Oに溶解)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。反応混合物を減圧下で濃縮してほとんどのメタノールを除去した。残留物をH2OとEtOAcの間に分配した。2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮して、27.5g(90%)の10を透明な無色の粘着性ゴムとして得た。
1H NMR(CDCl3)δ7.42(s,1H)、7.25−7.10(m,2H)、7.05−6.85(m,3H)、4.92(br s,1H)、4.46(t,J=5.2Hz,2H)、3.86(br s,3 H)、3.74(t,J=5.1Hz,2H)、3.32(s,3H)、3.30−2.75(m,3H)、2.24(br s,2H)、2.05−1.55(m,4H);19F NMR(CDCl3)δ−57.52(s,3F)、−121.64(s,1F)、−136.03(d、J=11.3Hz,1F);MS 512(M+1、100%).
【0113】
Et2O(30mL)に溶解させた上記物質 (2.856g、5.59mmol)の溶液に、2N HCl/Et2O(3mL、6mmol)を滴下した。固体沈殿物が形成し、エーテル溶液をデカントした。その固形物をさらにEt2Oで洗浄し、その後デカントした。残りの淡黄色固形物を温めたMeOH(10mL)に溶解し、その後Et2O(50mL)を、その溶液がわずかに濁るまで加えた。約2時間後、固体沈殿物が出現した。さらにEt2O(5−10mL)を加え、その後懸濁液を冷蔵庫の中に一晩置いた。白色の結晶生成物(2.475g、4.52mmol)を回収し、高減圧下で4時間乾燥した。
1H NMR(DMSO−d6)δ8.32(br s,2H)、7.71(s,1H)、7.43(d、1H、J=7.2Hz)、7.36(m,1H)、7.26−7.20(m,1H)、7.12−7.08(m,2H)、4.49(t,J=5.1Hz、2H)、4.00(s,2H)、3.71(t,J= 5.1Hz、2H)、3.32(s,3H)、3.21−3.07(m,3H)、2.99(br s,2H)、1.80−1.62(m,4H);19F NMR(DMSO−d6)δ−56.79(s,3F)、−119.34(s,1F)、−134.53(d、J=9.6Hz,1F);MS 512(M+1、100%). CHN:理論値:C 53.06%、H 5.16%、N 7.42%(1.0 H2Oとして算出). 実測値:C 53.03%、H 4.82%、N 7.22、Cl 6.64%.
【0114】
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(10 安息香酸塩)
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(1320g、2.58mol)を含んでいると思われるトルエン溶液をすでに入れてある20Lのガラスジャケット反応器を攪拌し、61℃まで加熱した。安息香酸(316g、2.58mol)を加え、全ての安息香酸が溶解した後、シクロヘキサン(6.04L)を加えた。上記のバッチからの[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(0.100g)で結晶が生じる77℃まで反応物を加熱した。結晶化を77℃で進行させ、15分後、その反応物を−10℃/hのランプで冷却した。反応が61℃まで到達したときに攪拌も冷却も停止させ、反応物を室温まで冷却した。一晩放置した後、攪拌を再開し、生成物を濾過により回収した。濾過ケークをトルエン(3L)およびシクロヘキサン(1.5L)より調製した溶媒混合物で洗浄した。サクションにより部分的に乾燥した後、生成物を40℃で乾燥させる乾燥オーブンに移し、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエートを無色の固形物として得た。:1408.8g(86%)、mp=156−159℃.元素分析:C25H26F5N3O3.C7H6O2についての算出値:C、60.66;H、5.09;N、6.63. 実測値:C、60.44;H、5.01;N、6.87。 赤外線スペクトル項目 (cm−1):1612、1526、1511、1501、1394、1362、1256、1232、1211、1158、1117、999、826.
【0115】
スキーム2
【化18】
【0116】
3−ブロモ−4−フルオロベンジルアミン塩酸塩(Wychem)を、少なくともイソプロピルアルコールの沸点を持つアルコール溶媒(例えばn−プロピルアルコール、n−ブチルアルコールなど);極性非プロトン性溶媒(例えばジメチルホルムアミド、1−メチル−2−ピロリドン、ジメチルスルホキシド)、およびエーテル溶媒(例えば2−メチルテトラヒドロフラン、ジメトキシエタンなど)中でピリジン−4−ボロン酸(ClariantまたはBoronmolecular)と反応させた。1,1’−ビス(ジフェニルホスフィノ)フェロセン−パラジウム(II)二塩化物 ジクロロメタン複合体(PdCl2dppf−CH2Cl2)、Pd(PPh3)4、PdCl2(PPh3)2、Pd(dtbpf)Cl2、などのような適切な触媒の存在下で、上述の溶媒と水との任意の混合物中の化合物12および化合物13を約70℃から鈴木カップリング反応混合物の沸点温度まで十分加熱してピリジンを得た。
【0117】
このピリジンを、トリフルオロ酢酸無水物、フッ化トリフルオロアセチル、ペンタフルオロフェニル、トリフルオロ酢酸のようなトリフルオロアセチル化剤を用いたトリフルオロアセチル化条件下で、エステル溶媒(例えば酢酸エチル、酢酸イソプロピル、など);芳香族炭化水素溶媒(トルエンなど);塩素化炭化水素溶媒(例えば塩化メチレン、1,2−ジクロロエタンなど)などのトリフルオロアセチル化溶媒中で、約−20〜約30℃のトリフルオロアセチル化反応温度においてトリフルオロアセトアミド化合物である2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩に転換し、その後塩酸で処理した。
【0118】
2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩を、水素化触媒(PtO2、Pd/C、Pd(OH)2Rh/Cなど)の存在下で、HClなどの無機酸もしくは酢酸などの有機酸を加えるかもしくは加えていないアルコール溶媒(例えばエタノール、イソプロピルアルコールなど);または酢酸;またはアルコール溶媒もしくは酢酸と水の混合物中のような水素化反応溶媒中で約10〜約60℃の水素化反応温度で、かつ約20〜約1000psiの水素化圧力下で水素で処理することにより、水素化条件下で化合物14に還元した。
【0119】
本発明の化合物は塩基性であり、そしてこのような化合物は、遊離の塩基の形態またはそれらの薬学的に受容可能な酸付加塩の形態で有用である。
【0120】
酸付加塩は、より慣用的な使用形態であり得;そして実際に塩形態の使用は、遊離塩基の形態での使用量に匹敵する。酸付加塩を製造するために使用され得る酸は、好ましくは、遊離の塩基と組合せた場合、薬学的に受容可能な塩、すなわち、そのアニオンが薬学的用量の塩で患者に対して非毒性であり、遊離塩基に固有の有益な阻害作用がアニオンに起因する副作用によって低下しないような塩を製造するものを包含する。前記塩基性化合物の薬学的に受容可能な塩が好ましいが、特定の塩自体が、例えば、塩が単に精製および同定の目的のために形成される場合、またはそれイオン交換法によって薬学的に受容可能な塩を製造する際の中間体として使用される場合、単に中間体生成物としてのみ所望されるとしても、全ての酸付加塩が遊離塩基の形態の供給源として有用である。本発明の範囲に包含される薬学的に受容可能な塩は、無機酸および有機酸から誘導されるものが挙げられ、そしてこれらとしては、ハロゲン化水素酸塩(例えば、塩酸塩および臭化水素酸塩)、硫酸塩、リン酸塩、硝酸塩、スルファミン酸塩、酢酸塩、クエン酸塩、乳酸塩、酒石酸塩、マロン酸塩、シュウ酸塩、サリチル酸塩、プロピオン酸塩、コハク酸塩、フマル酸塩、マレイン酸塩、メチレン−ビス−b−ヒドロキシナフトエ酸塩、安息香酸塩、トシレート、ゲンチジン酸塩、イセチオン酸塩、ジ−p−トルオイル酒石酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、シクロヘキシルスルファミン酸塩およびキナ酸塩が挙げられる。より特定の塩は、式Iの化合物の塩が、塩酸塩であるものである。本発明の別の特定の塩は、式Iの化合物のフマル酸塩である。本発明の好ましい薬学的に許容され得る塩は、式Iの化合物の安息香酸塩である。
【0121】
それ自体が活性化合物として有用であると同時に、本発明の化合物の塩は、例えば、当業者に周知の技術による塩と親化合物、副生成物、および/または出発物質との溶解度の差を利用することによって化合物の精製目的のために有用である。
【0122】
本発明のさらなる特徴に従って、公知の方法の応用または適用によって遊離塩基を適切な酸と反応させることによって、本発明の化合物の酸付加塩を製造し得る。例えば、本発明の化合物の酸付加塩は、遊離塩基を適切な酸を含む水または水性アルコール溶液または他の適切な溶媒中に溶解し、そして溶液を蒸発させることによって塩を単離するか、または有機溶媒中で遊離塩基を酸と反応させる(この場合は塩を直接分離するか、または溶液を濃縮することによって得ることができる)ことによって製造し得る。
【0123】
公知方法の応用または適用によって、本発明の化合物の酸付加塩を塩から再生し得る。例えば、アルカリ(例えば、重炭酸ナトリウム水溶液またはアンモニア水)を用いる処理によってその酸付加塩から本発明の親化合物を再生し得る。
【0124】
公知の方法(例えば、参照の実施例に記載される方法またはそれらの明らかな化学同等物)の応用もしくは適用によって、出発物質および中間体を製造し得る。
【0125】
本発明はまた、上記のスキーム1中のいくつかの中間体に関し、そしてまたそれらの製造についての本明細書中に記載されている方法は本発明のさらなる特徴を構成する。
【実施例】
【0126】
本発明は、本発明の例示として提供される以下の非限定的な実施例を参照することにより良く理解され得る。以下の実施例は、本発明の特定の実施形態をより十分に説明するために提示される。しかし、これらは本発明の範囲を限定するものと見なすべきではない。以下の参照実施例は、式Iの化合物を作製するために用いられる中間体を作製する方法を開示するために提供される。
【0127】
以下に報告される核磁気共鳴スペクトル(NMR)において、化学シフトは、テトラメチルシランに対するppmで表示する。略語は以下の意味を有する:br=ブロード、dd=ダブル二重項、s=一重項;m=多重項。
【0128】
参考実施例1
工程A:2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩(13)の製造
【化19】
フラスコにNaHCO3(126g、1.5mol)、3−ブロモ−4−フルオロベンジルアミン塩酸塩(11、120g、0.5mole)、ピリジン−4−ボロン酸(13、67.6g、0.55mmol)、iPrOH(750mL)および水(375mL)を室温で充填した。この懸濁液をN2で10℃にて1時間脱気した。この混合物中に1,1’−ビス(ジフェニルホスフィノ)フェロセン−パラジウム(II)二塩化物 ジクロロメタン複合体(PdCl2dppf−CH2Cl2、16.4g、20mmol)を加えた。この反応混合物を、内部温度が80℃に到達するまでいくらかの部分が蒸散するが、80℃まで加熱し、10時間攪拌した。この反応が完了(HPLC分析)した後、混合物を室温まで冷却し、2N HCl水溶液(750mL)を加え、0.5時間攪拌した。その溶液をDCM(750mL及び500mL)で洗浄した。水層に50% NaOH水溶液(100mL)をpH>13に調整されるまで加えた。n−BuOAc(2L)を加えた後、活性炭(50g)を有機層に加えた。この混合物をセライトパッド(50g)を通して濾過した。共沸蒸留を行った。追加のn−BuOAc(1L)を加えた後、反応物を5℃まで冷却した。TFAA(157g、0.6mol)をその溶液中にゆっくりと5℃で加えた。その反応が完了(HPLC分析)した後、反応混合物を10% Na2CO3水溶液(1L)で洗浄した。iPrOH(120mL)に溶解した5−6N HClの溶液を、粗製の有機層中に10℃で加えた。その後追加のn−BuOAc(1L)を加え、懸濁液を室温で一晩放置した。得られた固形物を10℃で濾過し、50℃のオーブンで乾燥させて124g(75%)の化合物15を白色固体として得た:mp=220℃. 分析 C14H10F4N2O−HClについての算出値:C、50.24;H、3.31;N、8.37.実測値:C、50.16;H、3.08;N、8.38. MS(ESI)m/z 299(M+H).1H NMR(300MHz、D2O)δ8.70(d、J=6.9Hz,2 H)、8.14(d、J=6.9Hz,2H)、7.56−7.20(m,3H)、4.51(s,2H).
【0129】
工程B:2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(14)の製造
【化20】
パーフラスコ(Parr flask)に、室温で化合物13(123g、0.37mol)およびMeOH(740mL)を入れ、その後5% Pt/C(36.9g、30
w/w%)を加えた。その反応フラスコをパー(Parr)水素化システム内に置き、H2により50−60psiで満たした。H2を充填する間、圧力が安定状態(H2を日中は2−3時間ごとに50−60psiまで補充したが、一晩超えた後は、さらなる補充なしに10−20psiが観察された)に達するまで、その混合物を>48時間振盪した。HPLC分析が反応の完了を示すと、反応混合物をセライトのパッドを通して濾過した。そのろ液を40−50℃で蒸留する一方、n−BuOAc(1.25L)を加えた。MeOHの蒸留が完了した後、追加のn−BuOAc(1L)を加えた。得られた懸濁液を一晩室温まで冷却させた。懸濁液を10℃まで冷却し、濾過し、50℃のオーブンで乾燥させ、112g(89%)の化合物14を白色固形物として得た:mp=134℃.分析 C14H10F4N2O−HClについての算出値:C、50.24;H、3.31;N、8.37.実測値:C、50.16;H、3.08;N、8.38. MS(ESI)m/z 305.4(M+H).
1H NMR(300MHz、D2O)δ 7.16−6.98(m,3 H)、4.34(s,2H)、3.42(d、J=12.9Hz,2H)、3.14−2.99(m,3H)、1.98−1.81(m,4H).
【0130】
参考実施例2
工程A:(2−フルオロ−5−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(2)の製造
【化21】
THF(500mL)に溶解させた1(50.72g、0.26mol)およびピリジン(27.3mL、0.34mol)の溶液に0℃でクロロギ酸エチル(32.2mL、0.39mol)を30分間にわたり滴下した、1時間後 LC/MSとTLCの両方が、反応が完了したことを示した。反応混合物をH2OとEtOAcに分配した。2層を分離し、有機層を1M HCl、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(95/5〜70/30)を用いてシリカゲル上で精製して69.23g(99%)の生成物2を透明な無色の液体として得た。
1H NMR(CDCl3)δ8.11(br s,1H)、7.07(dd,J=9.1、9.3Hz,1H)、7.00−6.80(m,2H)、4.27(q,J=7.1Hz,2H)、1.33(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−57.84(s,3F)、−134.01(br s,1F); MS 309(M+CH3CN+1、100%)、268(M+1).
【0131】
工程B:(6−フルオロ−2−ヨード−3−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(3)の製造
【化22】
THF(180mL)に溶解させた2(31.34g、117.2mmol)の溶液に−78℃でsec−BuLi(1.4M シクロヘキサン中、200mL、280mmol)を1時間にわたって滴下した。20分後、THF(150mL)に溶解させたI2(44.6g、175.8mmol)の溶液を30分にわたって滴下した。その後この混合物を−78℃で30分間攪拌した。飽和NH4Clを加え、冷却浴を取り除いた。反応混合物をH2OとEtOAcとの間に分配した。2層を分離し、有機層を10% Na2SO3、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM(50mL)に懸濁させ、ヘプタン(300mL)を加えた。残りの懸濁液から白色粉末3(18.1g、39%)をサクションフィルターによって回収し、空気乾燥させた。ろ液を減圧下で濃縮し、残留物をヘプタン(200mL)に懸濁させた。別バッチの3(3.8g、8%)をサクション濾過によって回収し、空気乾燥させた。シリカゲルクロマトグラフィーによるろ液の精製により、追加の生成物を得ることができた。
1H NMR(CDCl3)δ7.30−17.10(m,2H)、6.16(br s,1H)、4.26(q,J=7.1Hz,2H)、1.32(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−56.90(s,3F)、−114.35(d、J=8.5Hz,1F);MS 394(M+1、100%)、374、364、321、267.
【0132】
工程C:(6−フルオロ−3−トリフルオロメトキシ−2−トリメチルシラニルエチニル−フェニル)−カルバミン酸エチルエステル(4)の製造
【化23】
脱気したTHF(180mL)中に溶解した3(18.1g、45.9mmol)、Et3N(12.8mL、91.9mmol)、Pd(PPh)2Cl2(1.6g、5%mol)、CuI(0.7g、8%mol)、およびTMS−アセチレン(19.6mL、137.8mmol)混合物を60℃で一晩加熱した。混合物を室温まで冷却し、その後H2OとEtOAcの間に分配した。混合物をセライトを通して濾過し、不溶性物質を除去した。2層のろ液を分離し、有機層をH2O およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAcを用いてシリカゲル上で精製し、15.6g(93%)の生成物4をベージュの固形物として得た。
1H NMR(CDCl3)δ7.15−7.00(m,2H)、6.41(br s,1H)、4.26(q,J=7.1Hz,2H)、1.31(t,J=7.1Hz,3H)、0.27(s,9H);19F NMR(CDCl3)δ−57.59(s,3F)、−118.15(s,1F);MS 364(M+1、100%).
【0133】
工程D:7−フルオロ−4−トリフルオロメトキシ−1H−インドール(5)の製造
【化24】
脱気したt−BuOH(300mL)に溶解した4(28.9g、79.6mmol)およびKOH(35.7g、636.7mmol)の混合物を70℃で一晩加熱した。LC/MSが、反応が完了したことを示した。その混合物を室温まで冷却し、H2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜60/40)を用いてシリカゲル上で精製し、16g(91%)の5を黄色液体として得た。
1H NMR(CDCl3)δ8.47(br s,1H)、7.35−7.20(m,1H)、6.95−6.80(m,2H)、6.68(d、J=2.5Hz,1H);19F NMR(CDCl3)δ−57.63(s,3F)、−136.10(d、J=8.5Hz,1F);MS 220(M+1、100%)、200.
【0134】
工程E:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール(6)の製造
【化25】
DMSO(150mL)に溶解させた5(16g、72.8mmol)およびKOH粉末(20.4g、364.2mmol)の混合物を室温で10分間攪拌した。2−メトキシエチル臭化物(10.3mL、109.2mmol)を加えた。その混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。この混合物をH2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、19.3g(95%)の6を黄色液体として得た。
1H NMR(CDCl3)δ7.15(d、J=2.1Hz,1H)、6.90−6.75(m,2H)、6.56(t,J=2.5Hz,1 H)、3.72(t,J=5.2Hz,2H)、3.72(t,J=5.2Hz,2H)、3.31(s,3H);19F NMR(CDCl3)δ−57.54(s,3F)、−137.00(d、J=11.3Hz,1F);MS 278(M+1、100%).
【0135】
工程F:2,2,2−トリフルオロ−1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−エタノン(7)の製造
【化26】
DMF(135mL)に溶解させた6(19.3g、69.7mmol)の混合物にTFAA(26.2mL、188.2mmol)を加えた。この混合物を40℃で一晩加熱した。TLCが、反応が完了したことを示した。混合物を室温まで冷却し、その後H2O
とEt2Oの間に分配した。2層を分離し、有機層を飽和NaHCO3(2X)、H2O およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、23.4g(89%)の7をわずかに緑色の固形物として得た。
1H NMR(CDCl3)δ8.03(d、J=1.4Hz,1H)、7.20−6.95(m,2H)、4.54(t,J=4.9Hz,2H)、3.76(t,J=4.8Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−71.10(s,3F)、−134.95(d、J=11.5Hz,1F);MS 374(M+1、100%).
【0136】
工程G:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボン酸(8)の製造
【化27】
MeOH(100mL)および5M NaOH(100mL)に7(23.4g、62.6mmol)を溶解させた混合物を80℃で一晩加熱した。LC/MSが、反応が完了したことを示した。その反応混合物を室温まで冷却し、その後減圧下で濃縮して大部分のMeOHを除去した。残留物をH2Oに溶解させて、その後Et2Oで一度洗浄した。水層を濃HClを用いてpH2までゆっくりと酸性化させた。酸性化させた懸濁液をEt2Oで抽出し、有機抽出物をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM/ヘプタン(10/90)に懸濁させた。懸濁液中の白色粉末8(19.4g、96%)をサクションフィルターで回収し、空気乾燥させた。
1H NMR(CDCl3)δ8.02(s,1H)、7.15−7.05(m,1H)、7.00−6.90(m,1H)、4.49(t,J=5.0Hz,2H)、3.75(t,J=4.9Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−135.65(d、J=11.3Hz,1F);MS 363(M+CH3CN+1)、322(M+1、100%).
【0137】
工程H:2,2,2−トリフルオロ−N−(4−フルオロ−3−{1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボニル]−ピペリジン−4−イル}−ベンジル)−アセトアミド(9)の製造
【化28】
CH2Cl2に8(19.1g、59.6mmol)、Et3N(24.8mL、177.9mmol)、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(11、26.4g、77.5mmol)(14)、およびEDCI(17.1g、89.3mmol)を溶解させた混合物を室温で一晩攪拌した。TLCとLC/MSの両方が、反応が完了したことを示した。混合物をH2OとCH2Cl2の間に分配した。2層を分離し、有機層をブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(40/60〜0/100)を用いてシリカゲル上で精製し、9(36g、99%)を白色泡沫として得た。
1H NMR(CDCl3)δ7.37(s,1H)、7.20−7.10(m,2H)、7.10−6.85(m,4H)、4.95(br s,1H)、4.60−4.35(m,4H)、3.90(br s,1 H)、3.73(t,J=5.0Hz,2H)、3.32(s,3H)、3.25−2.70(m,3H)、2.05−1.50(m,4H);19F NMR(CDCl3)δ−57.54(s,3F)、−75.39(s,3F)、−119.31(s,1F)、−134.96(d、J=11.3Hz,1F);MS 608(M+1、100%).
【0138】
工程I:[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン塩酸塩(10)の製造
【化29】
MeOH(400mL)に9(36g、59.3mmol)を溶解した混合物にK2CO3水溶液(65.5g、474mmol、120mL H2Oに溶解)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。反応混合物を減圧下で濃縮し、大部分のメタノールを除去した。残留物をH2OとEtOAcの間に分配した。2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮して27.5g(90%)の10を透明な無色の粘着性ゴムとして得た。
1H NMR(CDCl3)δ7.42(s,1H)、7.25−7.10(m,2H)、7.05−6.85(m,3H)、4.92(br s,1H)、4.46(t,J=5.2Hz,2H)、3.86(br s,3 H)、3.74(t,J=5.1Hz,2H)、3.32(s,3H)、3.30−2.75(m,3H)、2.24(br s,2H)、2.05−1.55(m,4H);19F NMR(CDCl3)δ−57.52(s,3F)、−121.64(s,1F)、−136.03(d、J=11.3Hz,1F);MS 512(M+1、100%).
【0139】
Et2O(30mL)に溶解させた上記の物質(2.856g、5.59mmol)の溶液に2N HCl/Et2O(3mL、6mmol)を滴下した。固体沈殿物が形成し、エーテル溶液をデカントした。固体をさらなるEt2Oで洗浄してその後デカントした。残りの淡黄色固形物を温MeOH(10mL)に溶解し、その後Et2O(50mL)をその溶液がわずかににごるまで加えた。約2時間後、固体沈殿物が出現した。さらなるEt2O(5−10mL)を加え、その後懸濁液を冷蔵庫に一晩置いておいた。白色の結晶生成物(2.475g、4.52mmol)を回収し、高減圧下で4時間乾燥させた。
1H NMR(DMSO−d6)δ8.32(br s,2H)、7.71(s,1H)、7.43(d、1H、J=7.2Hz)、7.36(m,1H)、7.26−7.20(m,1H)、7.12−7.08(m,2H)、4.49(t,J=5.1Hz、2H)、4.00(s,2H)、3.71(t,J= 5.1Hz、2H)、3.32(s,3H)、3.21−3.07(m,3H)、2.99(br s,2H)、1.80−1.62(m,4H);19F NMR(DMSO−d6)δ−56.79(s,3F)、−119.34(s,1F)、−134.53(d、J=9.6Hz,1F);MS 512(M+1、100%). CHN:理論値:C 53.06%、H 5.16%、N 7.42%(calc’d as 1.0 H2O). 実測値:C 53.03%、H
4.82%、N 7.22、Cl 6.64%.
【0140】
参考実施例3
式Iの化合物の安息香酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(1320g、2.58mol)を含んでいると思われるトルエン溶液をすでに入れてある20Lのガラスジャケット反応器を攪拌し、61℃まで加熱した。安息香酸(316g、2.58mol)を加え、全ての安息香酸が溶解した後、シクロヘキサン(6.04L)を加えた。上記のバッチからの[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(0.100g)で結晶が生じる77℃まで反応物を加熱した。結晶化を77℃で進行させ、15分後、その反応物を−10℃/hのランプで冷却した。反応が61℃まで到達したときに攪拌も冷却も停止させ、反応を室温まで冷却した。一晩放置した後、攪拌を再開し、生成物を濾過により回収した。濾過ケークをトルエン(3L)およびシクロヘキサン(1.5L)より調製した溶媒混合物で洗浄した。サクションにより部分的に乾燥した後、生成物を40℃で乾燥させる乾燥オーブンに移し、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエートを無色の固形物として得た。:1408.8g(86%)、mp=156−159℃.
【0141】
参考実施例4
式Iの化合物のベシル酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゼンスルホン酸塩
アセトニトリル(12mL)にベンゼンスルホン酸一水和物(698mg、3.84mmol)を溶解した溶液を、攪拌したアセトニトリル(5mL)に溶解させた[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(2.0g、3.91mmol)の懸濁液に滴下した。溶解した遊離塩基の最期として混合物からベンゼンスルホン酸塩が結晶化し始めた。2時間後、生成物を濾過により回収し、アセトニトリルで洗浄した。フィルターケークを一晩乾燥させた。固形物を砕き、減圧オーブン中(43〜44℃、6.8−7.3Hg)で7.5時間窒素を流しながら乾燥させ、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノンベンゼンスルホン酸塩を無色の固体として得た。:2.27g 1(86.7%)、mp=215−218℃.C25H26F5N3O3.C6H6O3Sについての分析算出値:C、55.60;H、4.82;N、6.27. 実測値:C、55.65;H、4.65;N、6.27. Karl Fischer:<0.10.赤外線スペクトルの項目(cm−1):1587、1545、1445、1210、1167、1125、1036、1018.
【0142】
参考実施例5
式Iの化合物のセスキフマル酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン セスキフマル酸塩 一水和物
丸底フラスコを、[4−(5−アミノメチル−2−フルオロフェニル)−ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(10.4g、20.4mmol)およびフマル酸(4.74g、40.7mmol)で満たした。イソプロパノール(IPA、62mL)を加え、得られた混合物をスチームバスで加熱した。塩の結晶化が起こる前に、ほとんどの物質が溶解した。スチームバスで加熱している間、さらなるIPAを30mLのポーションで加えた。合計152mLのIPAを加えた後、完全な溶液に到達した。得られた溶液を濾過し、そのろ液を室温まで冷却した。ろ液をさらに氷浴で1.5時間冷却し、その後生成物を濾過により回収した。回収した生成物を冷IPA(50mL)で洗浄し、サクションにより部分的に乾燥させ、45℃で乾燥させる乾燥オーブンに移した。一晩乾燥させた後、所望の生成物を無色の固体として得た。:11.8g(84%).IR(cm-1):3122−2700、2920、2824、1698、1584、1512、1443、1397−1368、1293−1217、822、794、639.1H NMR(300MHz、DMSO−d6):δ 10.07(br、3H)、7.71(s,1H)、7.43(dd,J=2.4、7.1、1H)、7.36(ddd、J=2.4、4.9、8.4、1H)、7.19(d、J=8.4、10.7、1H)、7.10(d、J=8.7、11.7、1H)、7.05(ddd、J=1.4、3.3、8.7、1H)、6.50(s,3H)、4.69(br、1H)、4.48(t,J=5.3、2H)、3.97(s,2H)、3.69(t,J=5.4、2H)、3.24(s,3H)、3.08(dddd、J= 3.5、3.5、12.1、12.1、1H)、2.91(br、2H)、1.75(br、2H)、1.63(br、2H).C25H26F5N3O3−1.5C4H4O4についての分析算出値:C、54.31;H、4.70;N、6.13.実測値:C、54.30;H、4.62;N、6.04. MS(ESI)m/z 512.2(M+H).
【0143】
参考実施例6
式Iの化合物のトシル化塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノンp−トルエンスルホン酸
アセトニトリル(3mL)に[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(488mg、0.95mmol)を溶解させた混合物に、p−トルエンスルホン酸一水和物(181mg、0.95mmol)を加えた、この混合物を冷蔵庫に一晩貯蔵した。得られたベージュの結晶をサクションフィルターで回収し、トルエンで洗浄し、減圧下の50℃で一晩乾燥させた。453mg(69%)得られた。
1H NMR(DMSO−d6)δ8.08(bs、3H)、7.70(s,1H)、7.80−6.95(m,9H)、5.00−4.30(m,3H)、4.20−3.90(m,2H)、3.80−3.60(m,3H)、3.23(s,3H)、3.25−2.80(m,3H)、2.28(s,3H)、1.95−1.45(m,4H);19F NMR(DMSO−d6)δ−55.61(s,3F)、−118.98(s,1F)、−134.33(d、J=9.3Hz,1F);LC 2.627 min;MS 512(M+1、100%).Mp 219℃.赤外線スペクトルの項目(cm−1):1583、1548、1511、1501、1250、1200、1169、1123、1115.
【0144】
参考実施例7
式Iの化合物の硫酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(423mg、0.827mmol)を20mLのガラスバイアル中に秤量した。この固体に硫酸(1.0N 試薬、1.5当量、1.30mmol、2.60mL)および1.7mL 水の溶液を加えた。室温で2時間攪拌した後、結晶性生成物が沈殿した。濾過および乾燥後、固体がアモルファスとして発見された。数滴の水で処理すると、そのアモルファス固体は結晶形態に戻った。Mp 62℃.赤外線スペクトルの項目(cm−1):1574、1545、1511、1483、1362、1267、1219、1212、1162、1096、1051.
【0145】
参考実施例8
式Iの化合物のクエン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(265mg、0.52mmol)を20mLのガラスバイアル中に秤量した。この溶液に2:1(v/v)アセトニトリル/水に溶解させたクエン酸溶液(0.158mmol/mlクエン酸3.30mL)を加えた。全ての固体が素早く溶解し、得られた無色の溶液を室温で1時間置いておいた。溶液を窒素ガス流の元でエバポレートし、その後室温の減圧下で乾燥させた。固体を最小限の量の水を加えた温アセトニトリル中で再結晶化させて無色の溶液を得た。冷却するとその溶液に、室温に置いておくとプレート状にに変形するような非常に長い繊維の粒子状の生成物が沈殿した。Mp 112℃.赤外線スペクトルの項目(cm−1):1721、1590、1553、1369、1245、1174、1155、1119。
【0146】
参考実施例9
式Iの化合物のメタンスルホン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(0.250g、0.489mmol)を20mLのガラスバイアル中に秤量した。水に溶解させたメタンスルホン酸(0.50mmol/ml溶液0.98mL)を加え、その混合物を攪拌しながら60℃まで加熱した。全ての固形物が溶解したわけではないが、さらに25mlのメタンスルホン酸溶液を加えて透明な溶液を得た。室温で1時間攪拌した後、溶液を、ロータリーエバポレーターにより減圧下でエバポレートして非常に粘性のオイルを得た。このオイルを、アセトニトリル中で正方形プレートに再結晶化させた。赤外線スペクトルの項目(cm−1):1596、1540、1214、1159、1112、1040、1020.
【0147】
参考実施例10
式Iの化合物の酒石酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(0.250g、0.554mmol)を、20mLのガラスバイアル中に秤量した。5:1(v/v)アセトニトリル/水に溶解させたL−(+)−酒石酸溶液2.66mmol/mlを調製し、0.2084mLのこの溶液を秤量した固体に加え、攪拌しながら60℃まで加熱して透明の溶液を得た。その後この溶液を、ロータリーエバポレーターを用いて減圧下でエバポレートして温酢酸イソプロピル中で再結晶化したガラス状の固形物を分離し、そこに最小限の量のイソプロパノールを加えて透明な溶液を得た。濾過によって単離した結晶生成物を冷却し、室温にて減圧下で乾燥させた。
【0148】
参考実施例11
式Iの化合物のリン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(133.9mg、0.262mmol)に、リン酸溶液(1mmole/mL イソプロパノールに溶解、1.1当量)を加えた。この混合物を、マグネットスターラーを用いて攪拌しながら500μLのイソプロパノールに室温で溶解させた。この物質をエバポレートして室温で乾燥させ、結晶性の物質は単離されなかった。この物質を500μLのアセトン、500μLの酢酸エチル、および1mLのヘプタンに再溶解させた。この物質をオイルとして分離した。この混合物をエバポレートして窒素流下で乾燥させた。一旦乾燥させ、酢酸エチル(500μL)およびトルエン(500μL)を加え、その物質をオイルとして分離した。この混合物をエバポレートして一晩室温で乾燥させた。メチルイソブチルケトン(1mL)およびトルエン(500μL)を加え、その物質を溶解させた。混合物を室温で一晩エバポレートした。結晶が出現し、室温での減圧濾過により、回収した。物質を室温での減圧オーブン(300mbar)中で一晩乾燥させた。
【0149】
参考実施例12
式Iの化合物のグルタミン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(138.8mg、0.271mmol)、グルタミン酸溶液(162.4mg/20mL(水に溶解)、1.1当量)を加えた。メタノール(2mL)を加えて物質を溶解させた。その混合物を室温で一晩エバポレートし、白色のアモルファス物質が沈殿した。その物質にイソプロパノール(600μL)をく加えた。結晶が出現し、それを室温にて減圧濾過により回収した。物質を減圧オーブン(約300mbar)中で一晩乾燥させた。
【0150】
参考実施例13
式Iの化合物の安息香酸塩の結晶形態
サンプル準備:材料を上記の参考実施例3のように準備した。安息香酸塩の懸濁液をナノピュアウオーター中に50mg/mLの遊離塩基当量(1mLの水中に63.6mgの塩)として調製した。サンプルを500rpmで一晩攪拌し、4時間放置したのち遠心分離した(懸濁として計29時間)。13000rpmで8分間遠心分離し、回収した固体を、湿潤サンプルとしてXRPD(x−ray power diffraction)で分析し、顕微鏡で評価した。その後この湿潤固体を周囲室温で一晩空気乾燥し、乾燥サンプルとしてXRPDおよび熱分析により分析した。そのままの薬物物質を初期の物質と比較した。遊離塩基薬物物質のXRPDもまた比較として用いた。XRPDが水量の異なる複数のピークを示しており、出現した安息香酸塩は可変の水和物であると考えられる。
【0151】
装置パラメータ
XRPD法
Cu対陰極を備えるジーメンスモデル D5000
プログラム:1.0 Sec.dql
範囲:2°〜40°.2−θスケール
工程サイズ:0.02°
雰囲気:周囲条件の温度および湿度
標準的なトップロード、低容量のキャビティー試験片マウントを用いた
【0152】
DSC−TGA:
TA Instruments Model Q−600 Simultaneous
DSC−TGA
パージガス:ヘリウム100mL/分
温度プログラム:10℃/分 線形加熱速度
サンプル準備:約3−5mgの粉末を開口アルミニウムパンに移し、TGA中にロードした。空のアルミニウムパンを参照として用いた。
【0153】
結果:
XRPDおよび熱分析を湿潤および乾燥サンプルで実施した。湿潤サンプルのXRPDは、ベースラインのいくらかのシフトと上昇を示した。しかし乾燥(一晩)サンプルでは、XRPDは、初期の物質と比較してピークの向上した分解を示した。乾燥サンプルの熱分析は、初期と同じTGAプロフィールを示した。XRPDおよび熱分析に基づいて、遊離塩基または水和物形態への転換は報告されなかった。遊離塩基のXRPDより、以下を得た:
【0154】
図1は、式Iの化合物の安息香酸塩の結晶形態AについてのXRPD結果を示す。この図は、サンプルについて角度に対する相対強度(%)を示している。ピークは、以下の角度で見られた:7.75、10.13、17.03、17.16、17.99、18.39、20.51、21.33、21.88、23.19、23.43、および27.59.
【0155】
図2は、式Iの化合物の安息香酸塩の結晶形態AについてのDSC結果を示している。この図は、160.29℃で溶解が開始し、162℃でその形態が溶解することを示している。
【0156】
生物活性
本発明の化合物の特性は、1)そのβ−トリプターゼ阻害能力(IC50およびKi値)によって証明される。
【0157】
インビトロ試験手順
背景技術の章に記載されたように、トリプターゼの全ての作用がその触媒活性に依存しているので、その触媒活性を阻害する化合物は、トリプターゼの作用を強力に阻害する。この触媒活性の阻害は、インビトロの酵素アッセイおよび細胞アッセイにより測定される。
【0158】
単離ヒト肺トリプターゼまたは酵母細胞内に発現させた組換えヒトβトリプターゼのいずれかを使用して、トリプターゼの阻害活性を確認する。単離天然酵素または発現酵素を使用して、本質的に同等の結果が得られる。アッセイ手順は、基質としてL−ピログルタミル−L−プロリル−L−アルギニン−パラ−ニトロアニリド(S2366:Quadratech)を使用する、96ウェルのマイクロプレート(Costar3590)を利用する(本質的にはMcEuen et al.Biochem Pharm.1996,52,331−340頁に記載されている)。アッセイは0.5mM基質(2×Km)を用いて50mM Tris(pH8.2)、100nM NaCl、0.05% Tween20、50μg/mlヘパリン中で実施され、そしてマイクロプレートは405nmの波長でマイクロプレートリーダー(Beckman Biomek Plate reader)上で読み取られる。
【0159】
プロトコル(IC50およびKiの測定)
化合物は2連で、以下の最終濃度:0.01、0.03、0.1、0.3、1、3、10μMで添加することを除いて、プロトコルは本質的に上記と同様である(全ての希釈は手作業で行なう)。各アッセイについて、一点測定またはIC50測定のいずれにおいても、標準化合物を使用して、比較のためのIC50を導く。IC50値から、式:Ki=IC50/(1+[基質]/Km)を使用して、Kiを計算し得る。
【0160】
式Iの化合物についてのβ−トリプターゼ阻害能力は、26±5nMのKi値であった。
【0161】
抗原誘導性気道過敏症アッセイのプロトコル
抗原感作およびチャレンジ:雄のHartleyモルモット(225〜250g)をオボアルブミン(1%溶液0.5mL、i.p.およびs.c.)で1日目(8/25/08)に感作した。4日目(8/28/08)に、動物は1%オボアルブミン(0.5mL)のブースタ注射を受けた(i.p.)。21日目(09/16/08)に、抗原チャレンジの24時間前に、動物にビヒクル(0.5%メチルセルロース/0.2%Tween 80)または試験化合物のいずれかを経口投薬した(2mL/kg)。また、抗原チャレンジの30分前に、動物にメピラミン(10mg/kg、i.p.)を注射し、アナフィラキシー性の衰弱(anaphylactic collapse)を予防した。次いで、動物を、DeVilbiss Ultranebネブライザーを使用して、1%オボアルブミンのエアロゾルに20分間暴露させた。ネガティブコントロールの動物はチャレンジしなかった。感作溶液:鶏卵白(Sigma A55031G;lot#087K7004)由来のアルブミン1gを100mLの生理食塩水に加え、溶液中に入れた。
【0162】
気道抵抗測定:チャレンジの18〜25時間後、動物に麻酔(ケタミン(62mg/kg)、キシラジン(30mg/kg)およびプロメース(promace)(1.5mg/kg)を含む反応混液0.5ml用量(i.m.))をかけ、外科的準備をしてその後全身を体積変動記録計に置いた。動物を、気管カニューレを介して50呼吸/分の速度で1mL/100gの1回呼吸量を送達するUgo−Basile人工呼吸器につないだ。ヒスタミンチャレンジのために頚静脈にもカニューレを挿入した。肺圧差を記録できるように、水を満たした食道カニューレを取り付けた。異なる圧力のトランスデューサを使用して、気管と食道カニューレとの間の差異として肺圧差を測定した。容量、気流および肺圧差のシグナルを、肺分析システム(Buxco XAソフトウェア)を使用してモニターし、そしてこれらを使用して肺抵抗(cm H2O/mL/s)および動的コンプライアンス(mL/cm H2O)を計算した。気道抵抗および動的コンプライアンスを呼吸基礎(breath basis)による呼吸数で計算した。ヒスタミンを静脈内に投与し、そして増大する濃度(1〜20μg/kg)についての反応性を評価した。
【0163】
式Iの化合物のフマル酸塩についてのこのアッセイ結果を以下の表に示す。このアッセイは、喘息を潜在的に治療するための化合物の有効性に関する。式Iの化合物のフマル酸塩は、これらの表によって示されるように、アレルゲンチャレンジの24時間前に投与された場合、抗原誘導性気道過敏症の用量関連阻害を示した。
【0164】
【表1】
【0165】
【表2】
【0166】
本発明は本明細書中に記載した特定の実施態様によって範囲は制限されない。実際、本明細書中に記載されたものに加えて、本発明の種々の変更が、上記の説明および添付の図面から当業者に明らかとなる。このような変更は、添付の特許請求の範囲内に属することが意図される。
【0167】
種々の文献を本明細書中で引用し、この開示はその内容が参照によって援用される。
【図面の簡単な説明】
【0168】
【図1】式Iの化合物の安息香酸塩の結晶形態AについてのXRPD結果を示す。
【図2】式Iの化合物の安息香酸塩の結晶形態AについてのDSC結果を示している。
【技術分野】
【0001】
本発明は、置換されたインドールベンジルアミン化合物、その製造、化合物を含有する医薬組成物、その用途およびそれらの中間体に関する。
【背景技術】
【0002】
肥満細胞介在性炎症症状、特に喘息は、深刻な公衆の健康問題である。喘息は、慢性炎症の発症をもたらす免疫特異的アレルゲンおよび全身性の化学的もしくは物理的刺激の両方に対する気管および気管支の過敏症(hyper−responsiveness)の進行性発生によって特徴付けられる。IgE受容体を含有する白血球、特に肥満細胞および好塩基球は、気管支の上皮およびその下部の平滑筋組織に存在する。これらの白血球が、先ずIgE受容体への特異的な吸入抗原の結合によって活性化され、次いで、多数の化学的媒介物を放出する。例えば、肥満細胞の脱顆粒により、プロテオグリカン、ペルオキシダーゼ、アリールスルファターゼB、キマーゼおよびトリプターゼの放出がもたらされ、細気管支の収縮が起こる。
【0003】
トリプターゼは、肥満細胞分泌性顆粒内に貯留され、そしてヒト肥満細胞の主要な分泌性プロテアーゼである。トリプターゼは、血管拡張性および気管支弛緩(bronchorelaxing)の神経ペプチドの分解(非特許文献1;非特許文献2;および非特許文献3)およびヒスタミンへの気管支応答性の変調(非特許文献4)を含む種々の生物学的プロセスに関与している。
【0004】
その結果、トリプターゼインヒビターは抗炎症薬(非特許文献5)として、特に慢性喘息の治療(非特許文献6)において有用であり得、そしてまたアレルギー性鼻炎(非特許文献7)、炎症性腸疾患(非特許文献8)、乾癬(非特許文献9)、結膜炎(非特許文献10)、アトピー性皮膚炎(非特許文献11)、関節リウマチ(非特許文献12)、変形性関節症(非特許文献13)、痛風性関節炎、リウマチ様脊椎炎および関節軟骨崩壊の疾患の治療または予防にも有用であり得る。
【0005】
さらに、トリプターゼは、線維芽細胞の強力な有糸分裂促進物質であることがわかっており、喘息および間質性肺炎における肺線維症に関与していることが示唆されている(非特許文献14)。
【0006】
従って、トリプターゼインヒビターは、線維性症状(非特許文献15)、例えば、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫および肥厚性瘢痕の治療または予防に有用であり得る。
【0007】
さらに、トリプターゼインヒビターは、心筋梗塞、発作、狭心症およびアテローム性動脈硬化性プラーク破壊のその他の結果の治療または予防に有用であり得る(非特許文献16)。
【0008】
トリプターゼはまた、コラゲナーゼを活性化するプロストロメリシンを活性化し、これにより軟骨および歯周の結合組織の破壊をそれぞれ開始させることがわかっている。
【0009】
従って、トリプターゼインヒビターは、関節炎、歯周病、糖尿病性網膜症および腫瘍増殖の治療または予防に有用であり得る(非特許文献17)。また、トリプターゼインヒビターは、過敏症(非特許文献18)、多発性硬化症(非特許文献19)、消化性潰瘍および合胞体ウイルス感染症の治療にも有用であり得る。
【0010】
式(A):
【化1】
の化合物として表される置換アリールメチルアミン、それらの製造、それらの化合物を含有する医薬組成物およびトリプターゼの阻害によって調節され得る疾患状態の治療におけるそれらの薬学的使用は、特許文献1に記載されている。特許文献1に具体的に開示されている化合物は、以下の式のものである:
【化2】
【0011】
しかしながら、特許文献1は、前述の[(アミノメチル−フェニル)−ピペリジン−1−イル]−[インドリル]−メタノン種(そのフェニル部分におけるアミノメチル基に対するパラ位はまた、フルオロ基で置換されている)のいずれも開示していない。さらに、特許文献1は、そのインドール部分における芳香族炭素(カルボニルに結合されているもの以外)が置換されている:より具体的にはインドールの5位がメトキシで置換されている、[(アミノメチル−フェニル)−ピペリジン−1−イル]−[インドリル]−メタノンのみを開示している。
【0012】
非特許文献20は、トリプターゼインヒビターとして3種類の [(アミノメチル−フェニル)−ピペリジン−1−イル]−[1H−インドリル−3−イル]−メタノンを開示している。第1のタイプのインヒビターは、式Bの化合物:
【化3】
であり、式中、インドール部分の芳香族炭素(カルボニルに結合しているもの以外)はいずれも置換されていないのに対し、インドール窒素は、水素、メチル、エチル、イソプロピル、プロピル、イソブチル、ブチル、ヘキシル、2−メトキシエチル、シクロヘキシルメチル、シクロプロピルメチル、3−ピリジル、2−チアゾール、アセチル、チオフェン−2−カルボニル、ベンゼンスルホニル,またはメタンスルホニルのようなR1で置換されている。第2のタイプのインヒビターは、式Cの化合物:
【化4】
であり、式中、インドール窒素は水素によってのみ置換され、そのインドール部分の単一の芳香族炭素(カルボニルに結合されているもの以外)は、4位、5位、6位、もしくは7位のメチル、または7位のフルオロのようなRで置換されている。第3のタイプのインヒビターは、式Dの化合物:
【化5】
であり、式中そのインドール部分中の単一の芳香族炭素(カルボニルに結合されているもの以外)が、7位においてメチルで置換されており、インドール窒素はメチル、エチル、プロピル、ブチル、または2−メトキシエチルのようなR1で置換されている。非特許文献20もまた、5位または7位においてインドール中の芳香族炭素における置換は耐性があるが、4位または6位の置換はほとんど不活性な化合物をもたらすことを開示している。
【0013】
特許文献1または非特許文献20には、(1)そのフェニル部分上のアミノメチル基に対するパラ位もまたフルオロ基で置換されている;(2)インドール窒素が2−メトキシエチルで置換されている;または(3)そのインドール部分中の2つもしくはそれ以上の芳香族炭素(カルボニルに結合しているもの以外)が置換されている、トリプターゼインヒビターとして特に価値のある薬学的特性を有するトリプターゼインヒビターを含むインドールが開示されていない。このような化合物は、トリプターゼインヒビターの投与によって改善され得る症状、例えば肥満細胞介在性炎症症状、炎症、および血管拡張性および気管支拡張性の神経ペプチドの分解に関連する疾患または状態を患っている患者の治療において容易に有用であり、セミカルバジド感受性アミン酸化酵素(SSAO)代謝についての障害を減少させる。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第6977263号
【非特許文献】
【0015】
【非特許文献1】Caughey,et al.,J.Pharmacol.Exp.Ther.,1988,244,133−137頁
【非特許文献2】Franconi,et al.,J.Pharmacol.Exp.Ther.,1988,248,947−951頁
【非特許文献3】Tam,et al.,Am.J.Respir.Cellmol.Biol.,1990,3,27−32頁
【非特許文献4】Sekizawa,et al.,J.Clin.Invest.,1989,83,175−179頁
【非特許文献5】K.Rice,P.A.Sprengler,Current Opinion in Drug Discovery and Development,1999,2(5),463−474頁
【非特許文献6】M.Q.Zhang,H.Timmerman,Mediators Inflamm.,1997,112,311−317頁
【非特許文献7】S.J.Wilson,et al.,Clin.Exp.Allergy,1998,28,220−227頁
【非特許文献8】S.C.Bischoff,et al.,Histopathology,1996,28,1−13頁
【非特許文献9】A.Naukkarinen,et al.,Arch.Dermatol.Res.,1993,285,341−346頁
【非特許文献10】A.A.Irani,et al.,Allergy Clin.Immunol.,1990,86,34−40頁
【非特許文献11】A.Jarvikallio,et al.,Br.J.Dermatol.,1997,136,871−877頁
【非特許文献12】L.C.Tetlow,et al.,Ann.Rheum.Dis,1998,54,549−555頁
【非特許文献13】M.G.Buckley,et al.,J.Pathol.,1998,186,67−74頁
【非特許文献14】Rouss et al.,J.Clin.Invest.,1991,88,493−499頁
【非特許文献15】J.A.CairnsおよびA.F.Walls,J.Clin.Invest.,1997,99,1313−1321頁
【非特許文献16】M.Jeziorska et al,J.Pathol.,1997,182,115−122頁
【非特許文献17】W.J.Beil et al,Exp.Hematol.,(1998)26,158−169頁
【非特許文献18】L.B.Schwarz et al,J.Clin.Invest.,1995,96,2702−2710頁
【非特許文献19】M.Steinhoff et al,Nat.Med.(N.Y.),2000,6(2),151−158頁
【非特許文献20】Bioorg.Med.Chem.Lett.15、2734(2005)
【発明の概要】
【課題を解決するための手段】
【0016】
本発明は、式I:
【化6】
の化合物または前記化合物のプロドラッグ、薬学的に受容可能な塩もしくは溶媒和物に及ぶ。
【0017】
さらに、本発明は、薬学的有効量の式Iの化合物および薬学的に受容可能な担体を含有する医薬組成物に関する。
【0018】
さらに、本発明は、トリプターゼインヒビターとしての式Iの化合物の使用に関し、トリプターゼインヒビター受容体を含む組成物中に化合物を導入することを包含する。さらに、本発明は、トリプターゼインヒビターで改善の必要がある生理学的症状を患っているか、または被っている患者を治療するための式Iの化合物の使用に関し、治療有効量の請求項1に記載の化合物を患者に投与することを包含する。
【0019】
本発明はまた、式Iの化合物およびその中で有用な中間体の製造に関する。
【0020】
本発明の態様、特性および利点は、以下の詳細な説明からより理解され、これらは、例としてのみとして与えられ、そして本発明を制限しない。
【0021】
略語のリスト
上記および本発明の説明を通して用いられる場合、他に示されない限り以下の略号は以下の意味を有すると理解される:
n−BuOAc n−酢酸ブチル
n−BuLi n−ブチルリチウム
sec−BuLi sec−ブチルリチウム
t−Bu tert−ブチル
t−BuOH tert−ブタノール
CuI ヨウ化銅
DCM ジクロロメタン、CH2Cl2または塩化メチレン
DMF ジメチルホルムアミド
DMSO ジメチルスルホキシド
dppf 1,1’−ビス(ジフェニルホスフィノ)フェロセン
DSC 示差走査熱量測定
EDCI 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド HCl
eq 当量
Et エチル
Et2O ジエチルエーテル
TEA トリエチルアミン
EtOH エタノール
EtOAc 酢酸エチル
EtOC(O)Cl クロロギ酸エチル
HPLC 高速液体クロマトグラフィー
MgSO4 硫酸マグネシウム
Me メチル
MeOH メタノール
MS 質量分析
MTBE メチル t−ブチルエーテル
NaHCO3 重炭酸ナトリウム
Na2SO3 亜硫酸ナトリウム
Na2SO4 硫酸ナトリウム
NMR 核磁気共鳴
Pd(PPh3)2Cl2 ビストリフェニルホスフィンパラジウム(II)二塩化物
PdCl2dppf 1,1’−ビス(ジフェニルホスフィノ)フェロセン パラジウム(II)二塩化物
Pd(dtbpf)Cl2 (1,1’ビス(ジ−t−ブチルホスフィノ)フェロセン
パラジウム 二塩化物
Pd2(dba)3 トリス(ジベンジリデンアセトン)ジパラジウム(0)
Pd(OAc)2 酢酸パラジウム(II)
P(Cy)3 トリシクロヘキシルホスフィン
t−Bu3P トリ−t−ブチルホスフィン
PPh3 トリフェニルホスフィン
PrOH プロパノール
iPrOH イソ−プロパノール
i−PrOAc イソ−酢酸プロピル
t−BuOK カリウム tert−ブトキシド
PPSE ポリ−リン酸トリメチルシリルエステル
K2CO3 炭酸カリウム
K2SO4 硫酸カリウム
LC 液体クロマトグラフィー
Na2SO4 硫酸ナトリウム
rt 室温
Rt 保持時間
TFA トリフルオロ酢酸
TFAA トリフルオロ酢酸無水物
TGA 熱重量分析
THF テトラヒドロフラン
TLC 薄層クロマトグラフィー
TMS−アセチレン トリメチルシリル−アセチレン
【0022】
定義
上記ならびに本明細書および添付の特許請求の範囲の全体を通して使用される場合、他に示されない限り、以下の用語は以下の意味を有することが理解されるべきである。
【0023】
本明細書中で使用される場合、用語「本発明の化合物」および同等の表現とは、前記の式Iの化合物を包含し、そしてその表現は、プロドラッグ、薬学的に受容可能な塩および溶媒和物(例えば、水和物)を包含することが意味される。同様に、中間体を言及する場合、それ自体が請求項に記載されていてもいなくても、可能な限りその塩および溶媒和物を包含することが意味される。明確化のために、可能な限り特定の例を本文中に時々示すが、これらの例は単なる説明であり、可能な限りの他の例を排除することを意図しない。
【0024】
本明細書中で使用される場合、用語「治療(treatment)」または「治療(treating)」は、患者の症状の改善のためのような、予防的治療ならびに確立された症状の治療を包含する。このような改善としては、疾患の進行を遅らせることや患者の症状の有益な緩和を含む。
【0025】
「患者」とは、ヒトまたは他の哺乳動物を意味する。
【0026】
「有効量」とは、所望の治療効果を与えるのに効果的な化合物の量を記載することを意味する。
【0027】
「プロドラッグ」とは、望ましくない毒性、刺激性、アレルギー応答などを伴うことなく患者に投与するのに適切であり、そして、代謝的手段により(例えば、加水分解により)インビボで本発明の化合物に転換可能である化合物を意味する。プロドラッグの十分な議論は、T.HiguchiおよびV.Stella,Pro−drugs as Novel Delivery Systems,Vol.14 of the A.C.S.Symposium SeriesおよびEdward B.Roche編,Bioreversible Carriers in Drug Design,American Pharmaceutical Association and Pergamon Press,1987に提供され、これらの両方は参照によって本明細書中に援用される。
【0028】
「薬学的に受容可能な塩」は、望ましくない毒性または副作用の生じることのない酸を用いたこれらの活性成分の任意の塩を意味する。これらの酸は、薬学の専門家に十分公知である。適切な塩の非限定的な例としては以下が挙げられる:塩化物;臭化物;ヨウ化物;アスパラギン酸塩、特に酸性のアスパラギン酸塩;安息香酸塩、特に酸性の安息香酸塩;クエン酸塩、特に酸性のクエン酸塩;酒石酸塩;リン酸塩、特に酸性のリン酸塩;フマル酸塩、特に酸性のフマル酸塩;グリセロリン酸塩;グルコースリン酸塩;乳酸塩;マレイン酸塩、特に酸性のマレイン酸塩;オロト酸塩;シュウ酸塩、特に酸性のシュウ酸塩;硫酸塩、特に酸性の硫酸塩;トリクロロ酢酸塩;トリフルオロ酢酸塩;ベシル酸塩;トシラートおよびメタンスルホン酸塩。FDA公認の薬理学的に受容可能な塩のリストは、以下:Philip L.Gould、「Salt Selection for Basic Drugs」33 Int’l J.Pharm.201、202、214−216(1986);Stephen M.Berge et al.「Pharmaceutical Salts」、Journal of Pharmaceutical Sciences Vol.66、No.1、January 1977、pages 1−19のさらなる情報;およびHandbook of Pharmaceutical Salts、P.Heinrich Stahl、Camille G.Wermuth(Eds.)、IUPAC Wiley−VCH、2002より当該分野で公知の塩を製造するための方法;に与えられ、これらの文献は参照により本明細書中で援用される。
【0029】
「溶媒和物」は、本発明の化合物と1つ又はそれ以上の溶媒分子との物理的会合を意味する。この物理的会合には、水素結合が含まれる。いくつかの例では、例えば1つ又はそれ以上の溶媒分子が結晶性固体の結晶格子中に組み込まれる場合、溶媒和物は単離可能である。「溶媒和物」は、溶液相及び単離可能な溶媒和物の両方を包含する。典型的な溶媒和物には、水和物、エタノラート、メタノラート、及び同種のものが含まれる。
【0030】
「鈴木カップリング条件」は、鈴木カップリング溶媒、鈴木カップリング触媒および鈴木カップリング反応を用いた条件を意味する。
【0031】
「鈴木カップリング溶媒」は、イソプロピルアルコール以上の沸点を有するアルコール溶媒:例えばn−プロピルアルコール、n−ブチルアルコールなど;極性非プロトン性溶媒:例えばジメチルホルムアミド、1−メチル−2−ピロリドン、ジメチルスルホキシドなど;エーテル溶媒:例えばTHF、2−メチルTHF、ジメトキシエタンなど;または前出の溶媒と水もしくはトルエンの任意の混合物を意味する。
【0032】
「鈴木カップリング触媒」は、Pd(PPh3)4、Pd(PPh3)2Cl2、Pd2(dba)3、Pd(dtbpf)Cl2、のようなPd触媒;またはPPh3、dppf、t−Bu3P、P(Cy)3などのホスフィンリガンドと結合する、Pd(OAc)2、Pd2(dba)3のようなPd触媒を意味する。
【0033】
「鈴木カップリング温度」は、約60℃〜鈴木カップリング反応混合物の沸点温度の温度を意味する。
【0034】
「トリフルオロアセチル化条件」は、トリフルオロアセチル化試薬、トリフルオロアセチル化溶媒、およびトリフルオロアセチル化反応温度を用いた条件を意味する。
【0035】
「トリフルオロアセチル化試薬」は、トリフルオロ酢酸無水物、1,1,1−トリクロロ−3,3,3−トリフルオロアセトン、トリフルオロ酢酸およびポリ−リン酸トリメチルシリルエステル(PPSE)、塩化トリフルオロアセチル、フッ化トリフルオロアセチル、ペンタフルオロフェニルトリフルオロアセテートのなどを意味する。
【0036】
「トリフルオロアセチル化溶媒」は、酢酸エチル、酢酸イソプロピル、n−ブチルアセテートなどのようなエステル溶媒;トルエンなどのような芳香族炭化水素溶媒;塩化メチレン、1,2−ジクロロエタンなどのような塩素化炭化水素などの溶媒を意味する。
【0037】
トリフルオロアセチル化反応温度」は約−20℃〜約30℃を意味する。
【0038】
「水素化条件」は、水素化触媒、水素化溶媒、水素化反応温度および水素化圧力を用いる条件を意味する。
【0039】
「水素化反応溶媒」は、メタノール、エタノール、イソプロピルアルコールなどのようなアルコール溶媒;または酢酸;またはアルコール溶媒もしくは酢酸と水との混合物を意味する。
【0040】
「水素化触媒」は、HClなどのような無機酸または酢酸などのような有機酸を加えるかまたは加えない、PtO2、Pd/C、Pd(OH)2、Rh/Cなどを意味する。
【0041】
「水素化反応温度」は、約10〜約60℃を意味する。
【0042】
「水素化圧力」は、約10〜約1000psi水素(装置能力によって指示される上限)を意味する。
【0043】
特定または好ましい実施態様
さらに、本発明は、治療有効量の式Iの化合物を患者に投与することによって改善され得る生理学的症状を患っている患者を治療するための式Iの化合物の使用に関する。本発明の化合物を用いて治療され得る生理学的症状の特定の実施態様としては、以下が挙げられるが、特にこれらに限定されない:炎症性疾患、例えば、関節の炎症、関節炎、関節リウマチ、リウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎および他の慢性炎症性関節疾患および全国および他の炎症性呼吸状態。本発明によって治療され得る生理学的症状の他の実施態様としては、慢性閉塞性肺疾患(COPD)、COPDの悪化、関節軟骨崩壊(joint cartilage destruction)、眼結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺炎、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫、肥厚性瘢痕、種々の皮膚科学的症状(例えば、アトピー性皮膚炎および乾癬)、心筋梗塞、発作、狭心症およびアテローム硬化性プラーク破壊(atherosclerotic plaque rapture)のその他の結果、ならびに歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍および合胞体ウイルス感染症(syncytial viral infection)のような生理学的症状が挙げられる。
【0044】
特定の実施態様において、本発明は喘息および他の炎症性呼吸状態を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0045】
別の特定の実施態様において、本発明はCOPDを患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0046】
別の特定の実施態様において、本発明はCOPDの悪化を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0047】
別の特定の実施態様において、本発明はアレルギー性鼻炎を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0048】
別の特定の実施態様において、本発明は関節の炎症を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0049】
別の特定の実施態様において、本発明は炎症性腸疾患を患っている患者を治療するための式Iの化合物の使用に関し、生理学的有効量の化合物を患者に投与することを包含する。
【0050】
さらに、本発明は、式Iの化合物、βアドレナリン作用性アゴニスト(beta andrenergic agonist)、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬からなる群から選択される第二の化合物ならびにそれらの薬学的に受容可能な担体を含有する医薬組成物にまで及ぶ。このような組成物において、式Iの化合物および第二の化合物は、治療的に有効な活性、すなわち、相加効果または相乗効果を提供するような量で存在する。このような医薬組成物を用いて治療され得る特定の炎症性疾患または障害としては、喘息が挙げられるが、これに限定されない。
【0051】
さらに、本発明は、炎症性障害を患っている患者を治療するための方法に関し、式Iの化合物ならびにβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬からなる群から選択される第二の化合物を患者に投与することを包含する。このような方法において、式Iの化合物および第二の化合物は、治療的に有効な活性、すなわち、相加効果または相乗効果を提供するような量で存在する。このような本発明の方法において、本発明の化合物は、第二の化合物の前に患者に投与され得るか、第二の化合物は、本発明の化合物の前に患者に投与され得るか、または本発明の化合物および第二の化合物は同時に投与され得る。本方法に従って適用されるアドレナリン作用性アゴニスト(andrenergic agonist)、抗コリン作用薬、抗炎症性コルチコステロイドおよび抗炎症薬の特定の例は、以下に記載される。本発明で用いられることが企図される抗コリン作用薬は、臭化イプラトロピウムおよびチオトロピウムを含む。本発明で用いられることが企図される抗炎症性コルチコステロイドは、ジプロピオン酸ベクロメタゾン、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンおよびデキサメタゾンを含む。
【0052】
本発明はまた、式Iの化合物を製造するための、式2〜9の中間化合物にも向けられる:
【化7】
【0053】
医薬組成物
上に説明される通り、本発明の化合物は有用な薬理学的活性を示し、従って医薬組成物中に組み込まれ、そして特定の内科的疾患を患っている患者の治療に使用され得る。このように、さらなる態様に従って、本発明は、本発明の化合物およびその薬学的に受容可能な担体を含有する医薬組成物を提供する。本明細書中で使用される場合、用語「薬学的に受容可能」とは、好ましくは政府、特に連邦政府または州政府の規制当局により認可されているか、または米国薬局方もしくは動物、特にヒトにおける使用に関する別の一般的に認知されている薬局方に記載されていることを意味する。適切な薬学的担体は、E.W.Martinによる「Remington’s Pharmaceutical Sciences」に記載されている。
【0054】
本発明に従う医薬組成物は、1種またはそれ以上の薬学的に受容可能な補助剤または賦形剤を使用して慣用的な方法に従って製造され得る。とりわけ、補助剤としては、希釈剤、増量剤、結合剤、錠剤分解物質、流動促進剤、滑沢剤、界面活性剤、滅菌水性媒体および種々の非毒性の有機溶媒が挙げられる。組成物は、錠剤、カプセル剤、丸薬、徐放製剤、顆粒、粉末、水性溶液または水性懸濁液、注射用溶液、エリキシル剤またはシロップ剤の形態で製造されてもよく、そして、甘味料、着香料、着色料または安定化剤からなる群から選択される1種またはそれ以上の薬剤を含有することにより薬学的に受容可能な製剤を得ることができる。ビヒクルの選択およびビヒクル中の活性物質の量は、活性化合物の溶解度および化学的特性、特定の投与様式ならびに薬務において守られるべき条件に従って一般的に決定される。例えば、ラクトース、微結晶性セルロース、α化デンプン、未変性デンプン、ケイ化微結晶性セルロース、マンニトール、ソルビトール、キシリトール、デキストラート、フルクトース、クエン酸ナトリウム、炭酸カルシウム、第二リン酸カルシウム二水和物、無水第二リン酸カルシウム、硫酸カルシウムのような賦形剤は、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、メチルセルロース、カルボキシルメチルセルロースナトリウム、α化デンプン、デンプン、ポリエチレングリコール、ポリエチレンオキシド、ポリカルボフィル、ゼラチンおよびアラビアゴムのような結合剤ならびにクロスカルメロース、グリコール酸ナトリウムデンプン、クロスポビドン、デンプン、微結晶性セルロース、アルギン酸および特定の複合ケイ酸塩のような錠剤崩壊剤と共に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、硬化植物油、鉱油、ポリエチレングリコール、脂肪酸のグリセリルエステル、ラウリル硫酸ナトリウムのような滑沢剤および二酸化ケイ素、タルク、デンプンのような流動促進剤と組み合わせて、ラウリル硫酸ナトリウム、ソルビタンエステル、ポリオキシエチレン脂肪酸エステル、ポロキサマー、ポリオキシエチレンエーテル、ドキュセートナトリウム、ポリエトキシ化ヒマシ油および塩化ベンザルコニウムのようないくつかの適切な湿潤剤と共に、錠剤を製造するために使用され得る。カプセルを製造するために、ラクトース、微晶性セルロース、α化デンプン、未変性デンプン、ケイ化微結晶性セルロースのような増量剤を、単独もしくは2種またはそれ以上の増量剤の混合物で、上記のバインダーを含むか含まずに、上記の適切な湿潤剤、錠剤崩壊剤、流動促進剤、滑沢剤などと共に使用するのに都合がよい。水性懸濁液を使用する場合、それらは乳化剤または懸濁を促進する薬剤を含有し得る。スクロース、エタノール、ポリエチレングリコール、プロピレングリコール、グリセロールおよびクロロホルムまたはこれらの混合物のような希釈剤をもまた使用してもよい。このような薬学的に受容可能な担体はまた、滅菌水および滅菌オイルであり得、これらとしては、ピーナツ油、大豆油、鉱油、ゴマ油などのような石油、動物、植物または合成起源のものが挙げられる。医薬組成物を静脈内投与する場合、水が好ましい担体である。生理食塩水および水性デキストロースとグリセロールとの溶液もまた、特に注射用溶液のための液体担体として利用され得る。適切な薬学的賦形剤としては、マンニトール、ヒト血清アルブミン(HSA)、デンプン、グルコース、ラクトース、スクロース、ゼラチン、麦芽、米、小麦、チョーク、シリカゲル、炭酸マグネシウム、ステアリン酸マグネシウム、ステアリン酸ナトリウム、グリセロールモノステアレート、タルク、塩化ナトリウム、乾燥脱脂乳、グリセロール、プロピレン、グリコール、水、エタノールなどが挙げられる。これらの組成物は、溶液、懸濁液、錠剤、丸薬、カプセル、粉末、徐放製剤などの形態をとり得る。
【0055】
当然ながら、本発明の医薬組成物は、治療有効量の活性化合物を適切な量の担体と共に含有することによって、患者への適切な投与のための形態を提供する。静脈内注射が極めて有効な投与形態であるが、以下に議論される注射または経口、経鼻もしくは非経口の投与による他の様式が利用され得る。
【0056】
治療方法
式Iの化合物は、文献に記載され、そして本明細書中以下に記載されている試験によると、トリプターゼ阻害活性を有し、この試験結果はヒトおよび他の哺乳動物における薬理学的活性と相関すると考えられている。従って、さらなる実施態様において、本発明は、トリプターゼインヒビターの投与によって改善され得る症状を患っているか、または罹りやすい患者を治療するための式Iまたは式Iを含有する組成物の使用に関する。例えば、式Iの化合物は、炎症性疾患、例えば関節の炎症(関節炎、関節リュウマチおよびリウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎、変形性関節症もしくは他の慢性炎症性関節疾患のような他の関節炎状態または関節軟骨崩壊の疾患を含む)、結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺疾患、線維症、強皮症、肺線維症、肝硬変、心筋線維症、神経腺維腫、肥大性瘢痕、種々の皮膚科的症状(例えば、アトピー性皮膚炎および乾癬)、心筋梗塞、発作、狭心症もしくはアテローム性動脈硬化性プラーク破壊のその他の結果、ならびに歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍または合胞体ウイルス感染症を治療するのに有用である。
【0057】
本発明のさらなる特性によれば、トリプターゼインヒビターの投与によって改善され得る症状、例えば、前述したような症状を患っているか、または罹りやすいヒト患者または動物罹患体の治療方法を提供し、有効量の本発明の化合物または本発明の化合物を含有する組成物を患者に投与することを包含する。
【0058】
組合せ療法
上で説明した通り、他の薬学的活性剤は、治療されるべき疾患によって、式Iの化合物と組合せて利用され得る。例えば、喘息の治療において、アルブテロール、ターブタリン、フォルモテロール、フェノテロールまたはプレナリン(prenaline)のようなβアドレナリン作用性アゴニストを包含し得、同様に、臭化イプラトロピウムのような抗コリン作用薬;ジプロピオン酸ベクロメタゾン、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンまたはプレジノーズ(predinose)のような抗炎症性コルチコステロイド;ならびにクロモグリク酸ナトリウムおよびネドクロミルナトリウムのような抗炎症剤を包含し得る。従って、本発明は、式Iの化合物およびβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症剤からなる群から選択される第二の化合物;ならびにそれらの薬学的に受容可能な担体を含有する医薬組成物にまで及ぶ。ロイコトリエンアンタゴニストとして本発明で使用することが特に意図されるのはモンテルカストである。またホスホジエステラーゼ−4 インヒビターとして本発明で用いることが特に意図されるのは、シフルモラスト(ciflumolast)である。本医薬組成物に適用される特定の薬学的担体は、本明細書中に記載される。
【0059】
さらに、本発明は、喘息を患っている患者を治療する方法に及び、本発明の化合物およびβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症剤からなる群から選択される第二の化合物を患者に投与する工程を包含する。このような組合せ方法において、本発明の化合物を第二の化合物の投与前に投与し得るか、本発明の化合物を第二の化合物の投与後に投与し得るか、または本発明の化合物と第二の化合物を同時に投与し得る。
【0060】
送達様式
本発明に従って、式Iの化合物または本化合物を含有する医薬組成物は、非経口的、経粘膜的(例えば、経口、経鼻、肺内もしくは直腸内)または経皮的に患者に導入されてもよい。
【0061】
経口送達
一般的に、Remington’s Pharmaceutical Sciences,第18編、1990(Mack Publishing Co.Easton PA 18042)の第89章(参照により本明細書中に援用される)に記載されている経口固体投薬形態が、本明細書中で使用するために意図される。固体投薬形態としては、錠剤、カプセル、丸薬、口中錠またはトローチ剤(lozenge)、カシェ剤またはペレット剤が挙げられる。また、リポソーム被包またはプロテイノイドカプセル化を使用して、本発明の組成物を製剤し得る(例えば、米国特許第4,925,673号に記載されているプロテイノイドミクロスフィア)。リポソーム被包を使用してもよく、そしてリポソームは種々のポリマーで誘導体化されていてもよい(例えば、米国特許第5,013,556号)。治療のための可能な固体投薬形態の説明は、Marshall,K.,In:Modern Pharmaceutics,Edited by G.S.Banker and C.T.Rhodes、第10章、1979に記載されており、これは参照により本明細書中に援用される。一般的に、製剤は本発明の化合物および胃環境に対して保護を与え、そして腸内において生物学的に活性な物質(即ち、本発明の化合物)を放出する不活性成分を含有する。
【0062】
本発明の化合物の経口投薬形態もまた特に意図される。このような化合物は、経口送達がより効率的になるように化学的に修飾されてもよい。一般的に、意図される化学的修飾は、成分分子自体への少なくとも1つの部分の連結であり、前記部分は、(a)タンパク質分解の阻害;および(b)胃または腸から血流への取りこみを可能にする。本発明の化合物の全体的な安定性の向上および体内循環時間の延長もまた所望される。このような部分の例としては、ポリエチレングリコール、エチレングリコールとプロピレングリコールのコポリマー、カルボキシメチルセルロース、デキストラン、ポリビニルアルコール、ポリビニルピロリドンおよびポリプロリンが挙げられる。Abuchowski and Davis,1981,「Soluble Polymer−Enzyme Adducts」In:Enzymes as Drugs,Hocenberg and Roberts編、Wiley−Interscience,New York,NY,367−383頁;Newmark et al.,1982,J.Appl.Biochem.4:185−189。使用され得る他のポリマーは、ポリ−1,3−ジオキソランおよびポリ−1,3,6−チオキソカンである。上で示される通り、薬学的用法はポリエチレングリコール部分であることが好ましい。
【0063】
本発明の化合物について、放出場所は、胃、小腸(十二指腸、空腸もしくは回腸)または大腸であり得る。胃内では溶解しないが十二指腸または腸内のどこかで物質を放出する利用可能な製剤を当業者は承知している。発明の化合物を保護することによってか、または腸のような胃環境の先で化合物を放出することによって、放出による胃環境への悪影響を回避することが好ましい。
【0064】
完全に胃耐性とするために、少なくともpH5.0で不浸透性であるコーティングが必須である。腸溶性コーティングとして使用される、より一般的な不活性成分の例としては、セルロースアセテートトリメリテート(CAT)、ヒドロキシプロピルメチルセルロースフタレート(HPMCP)、HPMCP50、HPMCP55、ポリビニルアセテートフタレート(PVAP)、Eudragit L30D、Aquateric、セルロースアセテートフタレート(CAP)、Eudragit L、Eudragit Sおよびセラックが挙げられる。これらのコーティングは混合フイルムとして使用してもよい。
【0065】
コーティングまたはコーティング混合物はまた、胃に対する保護を意図しない錠剤に使用され得る。これには、糖コーティングまたは錠剤を嚥下しやすくするコーティングが包含され得る。カプセルは、ドライ治療薬(即ち、粉末)の送達のためのハードシェル(例えば、ゼラチン)からなり;液体形態について、ソフトゼラチンシェル使用されてもよい。カシェ剤のシェル物質は、濃厚なデンプンまたは他の食用紙であり得る。丸薬、トローチ剤、湿製錠剤またはすりこみ錠剤について、湿潤塊化技術(moist massing technique)を使用し得る。
【0066】
治療薬は、約1mmの粒径の顆粒またはペレットの形態の微細マルチ粒子としての製剤中に含有され得る。カプセル投与のための物質の製剤はまた、粉末、軽度に圧縮されたプラグまたは錠剤としても存在し得る。治療薬は圧縮によって製造され得る。
【0067】
着色料および着香料は全て含有してもよい。例えば、本発明の化合物を製剤し(例えば、リポソームカプセル化または微小球カプセル化によって)、次いで着色料および着香料を含有する冷蔵飲料のような食品中にさらに含有してもよい。
【0068】
不活性物質を用いて治療薬の容量を希釈させるか、または増大させてもよい。これらの希釈剤としては、炭水化物、特にマンニトール、α−ラクトース、無水ラクトース、セルロース、スクロース、変性デキストランおよびデンプンが挙げられ得る。特定の無機塩もまた増量剤として使用されてもよく、これらとしては、第三リン酸カルシウム、炭酸マグネシウムおよび塩化ナトリウムが挙げられる。いくつかの市販の希釈剤としては、Fast−Flo、Emdex、STA−Rx1500、EmcompressおよびAvicellが挙げられる。
【0069】
治療薬を固体投薬形態に製剤するのに錠剤崩壊剤を含有してもよい。錠剤崩壊剤として使用される物質としては、デンプンをベースにした市販の錠剤崩壊剤であるExplotabが挙げられるが、デンプンに限定されない。デンプングリコール酸ナトリウム、Amberlite、カルボキシメチルセルロースナトリウム、ウルトラミロペクチン、アルギン酸ナトリウム、ゼラチン、オレンジ果皮、酸性カルボキシメチルセルロース、天然の海綿およびベントナイトを全て使用してもよい。錠剤崩壊剤の別の形態は、不溶性カチオン交換樹脂である。粉末化ガムを錠剤崩壊剤および結合剤として使用してもよく、そしてこれらは、寒天、カラヤ(Karaya)またはトラガカントのような粉末化ガムが挙げられ得る。アルギン酸およびそのナトリウム塩もまた、錠剤崩壊剤として有用である。
【0070】
結合剤を使用して治療薬を保持し、同時に硬質錠剤を形成し、そしてこれらとしては、アラビアゴム、トラガカント、デンプンおよびゼラチンのような天然の産物由来の物質が挙げられる。他のものとしては、メチルセルロース(MC)、エチルセルロース(EC)およびカルボキシメチルセルロース(CMC)が挙げられる。ポリビニルピロリドン(PVP)およびヒドロキシプロピルメチルセルロース(HPMC)の両方が、治療薬を顆粒化するためにアルコール溶液中で使用され得る。
【0071】
製剤プロセスの過程で、付着を防止するために治療薬の製剤において抗摩擦剤を含有させてもよい。滑沢剤は治療薬とダイ壁との間の層として使用されてもよく、そしてこれらとしては、以下が挙げられるが、これらに限定されない;ステアリン酸(そのマグネシウム塩およびカルシウム塩を含む)、ポリテトラフルオロエチレン(PTFE)、流動パラフィン、植物油およびワックス。ラウリル硫酸ナトリウム、ラウリル硫酸マグネシウム、種々の分子量のポリエチレングリコール、Carbowax4000および6000のような可溶性滑沢剤も使用してもよい。
【0072】
製剤化の過程で、薬物の流動特性を改善し、圧縮の過程で、再配列を助け得る流動促進剤を添加してもよい。流動促進剤はデンプン、タルク、熱分解法シリカおよび水和シリコアルミネートが挙げられ得る。
【0073】
水性環境中への治療薬の溶解を助けるために、界面活性剤を湿潤剤として添加してもよい。界面活性剤としては、ラウリル硫酸ナトリウム、スルホコハク酸ジオクチルナトリウムおよび硫酸ジオクチルナトリウムのような陰イオン清浄剤が挙げられ得る。陽イオン清浄剤が使用されてもよく、これとしては、塩化ベンザルコニウムまたは塩化ベンゼトニウムが挙げられる。界面活性剤として製剤中に含有し得る、可能性のある非イオン性洗剤のリストは、ラウロマクロゴール400、ポリオキシ40ステアレート、ポリオキシエチレン水素化ヒマシ油10、50および60、グリセロールモノステアレート、ポリソルベート40、60、65および80、スクロース脂肪酸エステル、メチルセルロースならびにカルボキシメチルセルロースが挙げられる。これらの界面活性剤は、本発明の化合物の製剤中に単独または異なった比率の混合物として存在し得る。
【0074】
本発明の化合物の取りこみを増強する可能性のある添加剤は、例えば、脂肪酸のオレイン酸、リノール酸およびリノレン酸である。徐放性経口製剤が望ましくあり得る。拡散または浸出の機構のいずれかによって放出を可能にする不活性マトリクス(例えば、ガム)中に薬物を組み込み得る。徐々に崩壊するマトリクスをもまた製剤中に組み込んでもよい。いくつかの腸溶性コーティングはまた、遅延放出効果を有する。本治療薬の徐放の別の形態は、Oros治療薬システム(Alza Corp.)に基づく方法によるものであり、即ち、水を侵入させて、そして浸透圧作用によって単一の小型開口部より薬剤を押出す半透膜中に薬物を封入する。
【0075】
他のコーティングが製剤に使用されてもよい。これらとしては、コーティングパンに適用し得る種々の糖が挙げられる。治療薬はまた、フィルムコート化錠剤中にあってもよく、そしてこの場合に使用される物質は、2グループに分けられる。第一グループは非腸溶性物質であり、そしてこれらとしては、メチルセルロース、エチルセルロース、ヒドロキシエチルセルロース、メチルヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウム、ポビドン(providone)およびポリエチレングリコールが挙げられる。第二グループは、一般的にフタル酸のエステルである腸溶性物質からなる。
【0076】
物質の混合物を使用して、最適なフィルムコーティングを提供し得る。フィルムコーティングは、パンコーター中もしくは流動床中、または圧縮コーティングによって行なわれ得る。
【0077】
肺送達
単独または医薬組成物中の本発明の化合物の肺送達がまた、本明細書において意図される。吸入の間、化合物は哺乳動物の肺に送達され、そして肺上皮を通過して血流に満たされる。この他の報告はAdjei et al.,1990,Pharmaceutical Research,7:565−569;Adjei et al.,1990,International Journal of Pharmaceutics,63:135−144(酢酸ロイプロリド);Braquet et al.,1989,Journal of Cardiovascular Pharmacology,13(補遺5):143−146(エンドセリン−1);Hubbard et al.,1989,Annals of Internal Medicine,Vol.III,206−212頁(a1−アンチトリプシン);Smith et al.,1989,J.Clin.Invest.84:1145−1146(a−1−プロテイナーゼ);Oswein et al.,1990,「Aerosolization of Proteins」,Proceedings of Symposium on Respiratory Drug Delivery II,Keystone,Colorado,March(組換えヒト成長ホルモン);Debs et al.,1988,J.Immunol.140:3482−3488(インターフェロン−γおよび腫瘍壊死因子α)およびPlatz et al.,米国特許第5,284,656号(顆粒球コロニー刺激因子)が挙げられる。全身作用のための薬物の肺送達のための方法および組成物は、Wong et al.に対して1995年9月19日に発行された米国特許第5,451,569号に記載されている。
【0078】
治療薬の肺送達のために設計された広範な機械的デバイスが、本発明の実施における使用について意図され、このデバイスとしては、ネブライザー、定量吸入器および粉末吸入器が挙げられるが、これらに限定されず、これらの全てを当業者が承知している。
【0079】
本発明の実施に適切な市販のデバイスのいくつかの特定の例としては、ほんの一部の名前について、Mallinckrodt,Inc.,St Louis,Missouri製のUltraventネブライザー;Marquest Medical Product,Englewood,Colorado製のAcorn IIネブライザー;Glaxo Inc.,Research Triangle Park,North Carolina製のVentolin定量吸入器;およびFisons Corp.,Bedford,massachusetts製のSpinhaler粉末吸入器が挙げられる。全てのこのようなデバイスは、本発明の化合物を投与するために適切な製剤の使用を必要とする。典型的に、各製剤は、利用されるデバイスの種類に特異的であり、そして治療に有用な通常の希釈剤、補助剤および/または担体に加えて、適切な高圧ガス物質の使用を包含し得る。リポソーム、マイクロカプセルもしくは微小球、封入複合体または他の種類の担体の使用もまた意図される。本発明の化学修飾された化合物もまた、化学修飾の種類または利用されるデバイスの種類に応じて、異なる製剤に製造され得る。
【0080】
ジェット型または超音波型のいずれかのネブライザーと共に使用するのに適切な製剤は、典型的に、溶液1mLあたり約0.1〜25mgの化合物濃度で水中に溶解された本発明の化合物を含有する。製剤はまた、緩衝化剤および単純な糖(例えば、浸透圧の安定化および調節のため)を含有してもよい。ネブライザー製剤はまた、エアロゾルを形成するのに溶液の噴霧により起こる化合物の表面誘導凝集を低減または防止するために界面活性剤を含有してもよい。
【0081】
定量吸入器デバイスと共に使用するための製剤は、一般的に、界面活性剤の助けで高圧ガス中に懸濁された本発明の化合物を含有する微細分割粉末を含有する。クロロフルオロカーボン、ヒドロクロロフルオロカーボン、ヒドロフルオロカーボンまたはヒドロカーボン(トリクロロフルオロメタン、ジクロロジフルオロメタン、ジクロロテトラフルオロエタノールおよび1,1,1,2−テトラヒドロエタンまたはこれらの組合せを含む)のような高圧ガスは、この目的のために利用される任意の慣用的な物質であり得る。適切な界面活性剤としては、ソルビタントリオレエートおよび大豆レシチンが挙げられる。オレイン酸もまた界面活性剤として有用であり得る。
【0082】
粉末吸入器デバイスから投与するための製剤は、本発明の化合物を含有する微細分割乾燥粉末を含有し、そしてまた、ラクトース、ソルビトール、スクロースまたはマンニトールのような充填剤を、デバイスからの粉末の分散を促進するような量(例えば、製剤の50〜90質量%)で含有し得る。本発明の化合物は、肺の末端にまで最も効果的に送達するために、平均粒径10mm(またはミクロン)未満、最も好ましくは0.5〜5mmを有する粒子形態に製造することが最も好都合である。
【0083】
経鼻送達
本発明の化合物の経鼻送達もまた意図される。経鼻送達により、鼻への治療薬の投与後に直接血流に化合物が移動するようになり、肺中に治療薬を付着させる必要が無い。経鼻送達のための製剤としては、デキストランまたはシクロデキストランを含有するものが挙げられる。
【0084】
経皮送達
薬物の経皮投与の分野において種々の多数の方法(例えば、経皮パッチを介する)が公知であり本発明において適用される。経皮パッチは、例えば、米国特許第5,407,713号、第5,352,456号、第5,332,213号、第5,336,168号、第5,290,561号、第5,254,346号、第5,164,189号、第5,163,899号、第5,088,977号および第5,087,240号、第5,008,110号、および第4,921,475号に記載され、これらの開示の各々は、その全体が参照により本明細書中に援用される。
【0085】
経皮投与経路は、経皮浸透増強剤(例えば、米国特許第5,164,189号、第5,008,110号、および第4,879,119号に記載された増強剤)の使用によって増強され得ることが容易に理解され、これらの開示の各々は、その全体が参照により本明細書中に援用される。
【0086】
局所投与
局所投与のために、本発明の化合物を含有するゲル(水またはアルコールベース)、クリームまたは軟膏が使用されてもよい。本発明の化合物はまた、パッチ適用のためのゲルまたはマトリクス基剤中に組み込まれてもよく、これにより経皮バリアを通して化合物の制御放出が可能になる。
【0087】
直腸投与
直腸投与用の固体組成物としては、公知の方法で製剤され、そして本発明の化合物を含有する坐剤が挙げられる。
【0088】
投薬量
本発明の組成物中の活性成分の比率は変更されてもよく、適切な投薬量が得られるような比率を構成することが必要である。明らかに、いくつかの単位投薬形態を、ほとんど同時に投与してもよい。利用される用量は、医師により決定され、そして所望の治療効果、投与経路および治療期間、ならびに患者の症状に依存する。成人において、用量は一般的に、吸入では約0.001〜約50、好ましくは約0.001〜約5mg/kg体重/日であり、経口投与では約0.01〜約100、好ましくは0.1〜70、より特に0.5〜10mg/kg体重/日であり、そして静脈内投与では約0.001〜約10、好ましくは0.01〜1mg/kg体重/日である。いずれの場合にも、用量は治療されるべき対象に対する年齢、体重、一般的健康状態および医薬品の効果に影響し得る他の特性のような固有の要因に従って決定される。
【0089】
さらに、本発明に従う化合物は、所望の治療効果を得るために必要な頻度で投与され得る。一部の患者は、より高用量または低用量に急速に応答し、そしてはるかに低用量の維持量で十分であることが見出され得る。別の患者について、各特定の患者の生理学的必要性に従って、1〜4用量/日の比率で長期間の治療を要する必要があり得る。一般的に、活性成分は1〜4回/日で経口投与され得る。もちろん、一部の患者については、多くて1または2用量/日で処方される必要がある。
【0090】
当然ながら、本発明の化合物の投与が有効な治療計画となる患者は、好ましくはヒトであるが、いずれの動物でもあり得る。従って、当業者によって容易に理解され得る通り、本発明の方法および医薬組成物は、いずれの動物、特に哺乳動物への投与(すなわち、獣医用途)に特に相応し、これらの動物としては、以下が挙げられるがこれらに限定されない:ネコ科またはイヌ科の動物の罹患体のような家畜(domestic animal);限定されないが、ウシ科、ウマ科、ヤギ、ヒツジおよびブタの罹患体のような家畜(farm animal);野生動物(野外または動物園内にかかわらず);マウス、ラット、ウサギ、ヤギ、ヒツジ、ブタ、イヌ、ネコなどのような実験動物;ニワトリ、シチメンチョウ、鳴き鳥などのようなトリ種。
【0091】
製造の詳細
式Iの化合物は、本明細書中でこれまでに使用されてるかまたは文献に記載される方法(例えば、R.C.LarockのComprehensive Organic Transformations,VCH publishes,1989によって記載されるもの)または本明細書中に記載されるような方法を意味する、公知の方法を適用または適合させることによって製造され得る。
【0092】
本明細書中の以下に記載される反応において、反応におけるそれらの望まれていない関与を避けるために、反応性官能基(例えば、アミノ基)を保護する必要があり得る。標準的技法に従って慣用的な保護基を使用し得る;例えば、T.W.GreeneおよびP.G.M.Wutsの「Protective Groups in Organic Chemistry」,John Wiley and Sons,1991を参照のこと。
【0093】
特に、式Iの化合物はスキーム1−2によって示されるように製造することができる。
【0094】
例えば、本発明の化合物はそれらの製造が収束性の合成からなるアキラルな化合物である。安息香酸塩としての本発明の化合物は、以下のスキームに示されるとおりに製造される。
【0095】
スキーム1
【化8】
【0096】
(i)クロロギ酸エチル、ピリジン、THF、0℃、100%;(ii)a:sec−BuLi、THF、−78℃、b:I2、THF、−78℃、52−68%;(iii)TMS−アセチレン、TEA、CuI、Pd(PPh3)2Cl2、脱気THF、60℃、93%;(iv)KOH、t−BuOH、70℃、91%;(v)KOH粉末、2−メトキシエチル臭化物、DMSO、室温、95%;(vi)TFAA、DMF、40℃、89%;(vii)5M NaOH、MeOH、85℃、96%;(viii)2,2,2−トリフルオロ−N−(フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩、EDCI、TEA、CH2Cl2(DCM)、室温、99%;(ix)a:K2CO3、MeOH/H2O、b:1M HCl(Et2O中、90%)
【0097】
化合物1は、適切な塩基(例えばピリジン)の存在下においてアミノ基保護剤(例えばクロロギ酸エチル)でアミノ基を保護することにより、化合物2に転換され、保護された化合物2が得られる。
【0098】
化合物2は、3段階の工程で化合物5に転換される。化合物2は、2と強塩基(例えば第2級ブチルリチウム)とを反応させることによってカルバミン酸エステルの隣の位置でヨウ素化されて、分子ヨウ素のようなヨウ素源と反応するアニオンを形成し、化合物3を得る。その後化合物3を、トリメチルシリルアセチレンおよび塩基(例えばトリエチルアミン)の存在下で、ヨウ化銅(I)およびビストリフェニルホスフィンパラジウム(II)二塩化物のような触媒条件を用いてアセチレン化合物4に転換する。化合物4を、水酸化カリウムのような強塩基を用いて環化し、加熱してインドール化合物5を得る。
【0099】
化合物5は、室温にて水酸化カリウムのような強塩基の存在下、ジメチルスルホキシドのような二極性非プロトン性溶媒中で、そのインドール窒素をアルキルハライドを用いてアルキル化することによって化合物6に転換され、化合物6を得られる。
【0100】
化合物6は、2工程で化合物8に転換される。最初に、化合物6をN,N−ジメチルホルムアミドのような溶媒の存在下でトリフルオロ酢酸無水物を用いて処理し、加熱することにより化合物6を化合物7に転換する。化合物7を水酸化ナトリウムのような強塩基で処理してその3位に酸性官能基を持つ化合物8を得る。
【0101】
化合物8は、ジクロロメタンのような不活性溶媒中、酸性カップリング試薬(EDCIなど)および有機塩基(トリエチルアミンなど)の存在下で酸性の8を2,2,2−トリフルオロ−N−(フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(化合物14)と反応させることにより、アミド9に転換される。
【0102】
化合物9は、メタノール/水のような溶媒混合物中で炭酸カリウムのような弱塩基での処理でN−ベンジル トリフルオロアセトアミドを脱保護することにより、化合物10に転換される。この塩酸塩は、エーテルのような極性有機溶媒の存在下で形成することができ、式Iの([4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−メチル−1H−インドール−3−イル]−メタノン)の塩酸塩である化合物10を得る。
【0103】
このスキームの反応は以下のとおりである。
【0104】
工程A:(2−フルオロ−5−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(2)の製造
【化9】
THF(500mL)に溶解させた1(50.72g、0.26mol)およびピリジン(27.3mL、0.34mol)の溶液に、0℃でクロロギ酸エチル(32.2mL、0.39mol)を30分間にわたって滴下した。1時間後、LC/MSとTLCの両方が、反応が完了したことを示した。その反応混合物をH2OとEtOAcに分配した。2層を分離し、その有機層を1M HCl、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(95/5〜70/30)を用いてシリカゲル上で精製し、69.23g(99%)の生成物2を透明な無色の液体として得た。
1H NMR(CDCl3)δ8.11(br s,1H)、7.07(dd,J=9.1、9.3Hz,1H)、7.00−6.80(m,2H)、4.27(q,J=7.1Hz,2H)、1.33(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−57.84(s,3F)、−134.01(br s,1F); MS 309(M+CH3CN+1、100%)、268(M+1).
【0105】
工程B:(6−フルオロ−2−ヨード−3−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(3)の製造
【化10】
THF(180mL)に溶解させた2(31.34g、117.2mmol)の溶液に−78℃でsec−BuLi(シクロヘキサン中、1.4M、200mL、280mmol)を1時間にわたり滴下した。20分後、THF(150mL)に溶解させたI2(44.6g、175.8mmol)の溶液を30分間にわたって滴下した。次いでこの混合物を−78℃で30分間攪拌した。飽和NH4Clを加え、冷却浴を取り除いた。反応混合物をH2OとEtOAcに分配した。2層を分離し、その有機層を10% Na2SO3、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM(50mL)に懸濁させ、ヘプタン(300mL)を加えた。得られた懸濁液からの白色粉末3(18.1g、39%)をサクションフィルターで回収し、空気乾燥させた。ろ液を減圧下で濃縮し、残留物をヘプタン(200mL)中に懸濁させた。別バッチの3(3.8g、8%)をサクションフィルターで回収し、空気乾燥させた。シリカゲルクロマトグラフィーによって精製することにより、追加生成物を得ることができた。
1H NMR(CDCl3)δ7.30−17.10(m,2H)、6.16(br s,1H)、4.26(q,J=7.1Hz,2H)、1.32(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−56.90(s,3F)、−114.35(d、J=8.5Hz,1F);MS 394(M+1、100%)、374、364、321、267.
【0106】
工程C:(6−フルオロ−3−トリフルオロメトキシ−2−トリメチルシラニルエチニル−フェニル)−カルバミン酸エチルエステル(4)の製造
【化11】
脱気したTHF(180mL)に溶解させた3(18.1g、45.9mmol)、Et3N(12.8mL、91.9mmol)、Pd(PPh)2Cl2(1.6g、5%mol)、CuI(0.7g、8%mol)、およびTMS−アセチレン(19.6mL、137.8mmol)の混合物を60℃で一晩加熱した。その混合物を室温まで冷却し、その後H2OとEtOAcに分配した。この混合物をセライトを通して濾過し、不溶性物質を除去した。ろ液の2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAcを用いてシリカゲル上で精製し、15.6g(93%)の生成物4をベージュの固形物として得た。
1H NMR(CDCl3)δ7.15−7.00(m,2H)、6.41(br s,
1H)、4.26(q,J=7.1Hz,2H)、1.31(t,J=7.1Hz,3H)、0.27(s,9H);19F NMR(CDCl3)δ−57.59(s,3F)、−118.15(s,1F);MS 364(M+1、100%).
【0107】
工程D:7−フルオロ−4−トリフルオロメトキシ−1H−インドール(5)の製造
【化12】
脱気したt−BuOH(300mL)に溶解させた4(28.9g、79.6mmol)およびKOH(35.7g、636.7mmol)の混合物を70℃で一晩加熱した。LC/MSが反応の完了を示した。この混合物を室温まで冷却し、その後H2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜60/40)を用いてシリカゲル上で精製し、16g(91%)の5を黄色液体として得た。
1H NMR(CDCl3)δ8.47(br s,1H)、7.35−7.20(m,1H)、6.95−6.80(m,2H)、6.68(d、J=2.5Hz,1H);19F NMR(CDCl3)δ−57.63(s,3F)、−136.10(d、J=8.5Hz,1F);MS 220(M+1、100%)、200.
【0108】
工程E:P7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール(6)の製造
【化13】
DMSO(150mL)に溶解した5(16g、72.8mmol)およびKOH粉末(20.4g、364.2mmol)の混合物を室温で10分間攪拌した。2−メトキシエチル臭化物(10.3mL、109.2mmol)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。この混合物をH2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、19.3g(95%)の6を黄色液体として得た。
1H NMR(CDCl3)δ7.15(d、J=2.1Hz,1H)、6.90−6.75(m,2H)、6.56(t,J=2.5Hz,1 H)、3.72(t,J=5.2Hz,2H)、3.72(t,J=5.2Hz,2H)、3.31(s,3H);19F NMR(CDCl3)δ−57.54(s,3F)、−137.00(d、J=11.3Hz,1F);MS 278(M+1、100%).
【0109】
工程F:2,2,2−トリフルオロ−1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−エタノン(7)の製造
【化14】
DMF(135mL)に溶解させた6(19.3g、69.7mmol)の混合物にTFAA(26.2mL、188.2mmol)を加えた。この混合物を40℃で一晩加熱した。TLCが、反応が完了したことを示した。混合物を室温まで冷却し、その後H2OとEt2Oの間に分配した。2層を分離し、有機層を飽和NaHCO3(2X)、H2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製して、23.4g(89%)の7をわずかに緑色の固形物として得た。
1H NMR(CDCl3)δ8.03(d、J=1.4Hz,1H)、7.20−6.95(m,2H)、4.54(t,J=4.9Hz,2H)、3.76(t,J=4.8Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−71.10(s,3F)、−134.95(d、J=11.5Hz,1F);MS 374(M+1、100%).
【0110】
工程G:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボン酸(8)の製造
【化15】
MeOH(100mL)および5M NaOH(100mL)に溶解させた7(23.4g、62.6mmol)の混合物を80℃で一晩加熱した。LC/MSが、反応が完了したことを示した。反応混合物を室温まで冷却し、その後減圧下で濃縮して大部分のMeOHを除去した。残留物をH2Oに溶解し、その後Et2Oで一度洗浄した。水層を濃HClによってゆっくりとpH2まで酸性化した。酸性化した懸濁液をEt2Oで抽出し、有機抽出物をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM/ヘプタン(10/90)に懸濁させた。懸濁液中の白色粉末8(19.4g、96%)をサクションフィルターで回収し、空気乾燥させた。
1H NMR(CDCl3)δ8.02(s,1H)、7.15−7.05(m,1H)、7.00−6.90(m,1H)、4.49(t,J=5.0Hz,2H)、3.75(t,J=4.9Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−135.65(d、J=11.3Hz,1F);MS 363(M+CH3CN+1)、322(M+1、100%).
【0111】
工程H:2,2,2−トリフルオロ−N−(4−フルオロ−3−{1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボニル]−ピペリジン−4−イル}−ベンジル)−アセトアミド(9)の製造
【化16】
CH2Cl2に溶解させた8(19.1g、59.6mmol)、Et3N(24.8mL、177.9mmol)、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(11、26.4g、77.5mmol)(14)、およびEDCI(17.1g、89.3mmol)の混合物を室温で一晩攪拌した。TLCとLC/MSの両方が、反応が完了したことを示した。この混合物をH2OとCH2Cl2の間に分配した。2層を分離し、有機層をブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(40/60〜0/100)を用いてシリカゲル上で精製し、9(36g、99%)を白色泡沫として得た。
1H NMR(CDCl3)δ7.37(s,1H)、7.20−7.10(m,2H)、7.10−6.85(m,4H)、4.95(br s,1H)、4.60−4.35(m,4H)、3.90(br s,1 H)、3.73(t,J=5.0Hz,2H)、3.32(s,3H)、3.25−2.70(m,3H)、2.05−1.50(m,4H);19F NMR(CDCl3)δ−57.54(s,3F)、−75.39(s,3F)、−119.31(s,1F)、−134.96(d、J=11.3Hz,1F);MS 608(M+1、100%).
【0112】
工程I:[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン塩酸塩(10)の製造
【化17】
MeOH(400mL)に溶解させた9(36g、59.3mmol)の混合物に、K2CO3水溶液(65.5g、474mmol、120mL H2Oに溶解)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。反応混合物を減圧下で濃縮してほとんどのメタノールを除去した。残留物をH2OとEtOAcの間に分配した。2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮して、27.5g(90%)の10を透明な無色の粘着性ゴムとして得た。
1H NMR(CDCl3)δ7.42(s,1H)、7.25−7.10(m,2H)、7.05−6.85(m,3H)、4.92(br s,1H)、4.46(t,J=5.2Hz,2H)、3.86(br s,3 H)、3.74(t,J=5.1Hz,2H)、3.32(s,3H)、3.30−2.75(m,3H)、2.24(br s,2H)、2.05−1.55(m,4H);19F NMR(CDCl3)δ−57.52(s,3F)、−121.64(s,1F)、−136.03(d、J=11.3Hz,1F);MS 512(M+1、100%).
【0113】
Et2O(30mL)に溶解させた上記物質 (2.856g、5.59mmol)の溶液に、2N HCl/Et2O(3mL、6mmol)を滴下した。固体沈殿物が形成し、エーテル溶液をデカントした。その固形物をさらにEt2Oで洗浄し、その後デカントした。残りの淡黄色固形物を温めたMeOH(10mL)に溶解し、その後Et2O(50mL)を、その溶液がわずかに濁るまで加えた。約2時間後、固体沈殿物が出現した。さらにEt2O(5−10mL)を加え、その後懸濁液を冷蔵庫の中に一晩置いた。白色の結晶生成物(2.475g、4.52mmol)を回収し、高減圧下で4時間乾燥した。
1H NMR(DMSO−d6)δ8.32(br s,2H)、7.71(s,1H)、7.43(d、1H、J=7.2Hz)、7.36(m,1H)、7.26−7.20(m,1H)、7.12−7.08(m,2H)、4.49(t,J=5.1Hz、2H)、4.00(s,2H)、3.71(t,J= 5.1Hz、2H)、3.32(s,3H)、3.21−3.07(m,3H)、2.99(br s,2H)、1.80−1.62(m,4H);19F NMR(DMSO−d6)δ−56.79(s,3F)、−119.34(s,1F)、−134.53(d、J=9.6Hz,1F);MS 512(M+1、100%). CHN:理論値:C 53.06%、H 5.16%、N 7.42%(1.0 H2Oとして算出). 実測値:C 53.03%、H 4.82%、N 7.22、Cl 6.64%.
【0114】
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(10 安息香酸塩)
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(1320g、2.58mol)を含んでいると思われるトルエン溶液をすでに入れてある20Lのガラスジャケット反応器を攪拌し、61℃まで加熱した。安息香酸(316g、2.58mol)を加え、全ての安息香酸が溶解した後、シクロヘキサン(6.04L)を加えた。上記のバッチからの[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(0.100g)で結晶が生じる77℃まで反応物を加熱した。結晶化を77℃で進行させ、15分後、その反応物を−10℃/hのランプで冷却した。反応が61℃まで到達したときに攪拌も冷却も停止させ、反応物を室温まで冷却した。一晩放置した後、攪拌を再開し、生成物を濾過により回収した。濾過ケークをトルエン(3L)およびシクロヘキサン(1.5L)より調製した溶媒混合物で洗浄した。サクションにより部分的に乾燥した後、生成物を40℃で乾燥させる乾燥オーブンに移し、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエートを無色の固形物として得た。:1408.8g(86%)、mp=156−159℃.元素分析:C25H26F5N3O3.C7H6O2についての算出値:C、60.66;H、5.09;N、6.63. 実測値:C、60.44;H、5.01;N、6.87。 赤外線スペクトル項目 (cm−1):1612、1526、1511、1501、1394、1362、1256、1232、1211、1158、1117、999、826.
【0115】
スキーム2
【化18】
【0116】
3−ブロモ−4−フルオロベンジルアミン塩酸塩(Wychem)を、少なくともイソプロピルアルコールの沸点を持つアルコール溶媒(例えばn−プロピルアルコール、n−ブチルアルコールなど);極性非プロトン性溶媒(例えばジメチルホルムアミド、1−メチル−2−ピロリドン、ジメチルスルホキシド)、およびエーテル溶媒(例えば2−メチルテトラヒドロフラン、ジメトキシエタンなど)中でピリジン−4−ボロン酸(ClariantまたはBoronmolecular)と反応させた。1,1’−ビス(ジフェニルホスフィノ)フェロセン−パラジウム(II)二塩化物 ジクロロメタン複合体(PdCl2dppf−CH2Cl2)、Pd(PPh3)4、PdCl2(PPh3)2、Pd(dtbpf)Cl2、などのような適切な触媒の存在下で、上述の溶媒と水との任意の混合物中の化合物12および化合物13を約70℃から鈴木カップリング反応混合物の沸点温度まで十分加熱してピリジンを得た。
【0117】
このピリジンを、トリフルオロ酢酸無水物、フッ化トリフルオロアセチル、ペンタフルオロフェニル、トリフルオロ酢酸のようなトリフルオロアセチル化剤を用いたトリフルオロアセチル化条件下で、エステル溶媒(例えば酢酸エチル、酢酸イソプロピル、など);芳香族炭化水素溶媒(トルエンなど);塩素化炭化水素溶媒(例えば塩化メチレン、1,2−ジクロロエタンなど)などのトリフルオロアセチル化溶媒中で、約−20〜約30℃のトリフルオロアセチル化反応温度においてトリフルオロアセトアミド化合物である2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩に転換し、その後塩酸で処理した。
【0118】
2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩を、水素化触媒(PtO2、Pd/C、Pd(OH)2Rh/Cなど)の存在下で、HClなどの無機酸もしくは酢酸などの有機酸を加えるかもしくは加えていないアルコール溶媒(例えばエタノール、イソプロピルアルコールなど);または酢酸;またはアルコール溶媒もしくは酢酸と水の混合物中のような水素化反応溶媒中で約10〜約60℃の水素化反応温度で、かつ約20〜約1000psiの水素化圧力下で水素で処理することにより、水素化条件下で化合物14に還元した。
【0119】
本発明の化合物は塩基性であり、そしてこのような化合物は、遊離の塩基の形態またはそれらの薬学的に受容可能な酸付加塩の形態で有用である。
【0120】
酸付加塩は、より慣用的な使用形態であり得;そして実際に塩形態の使用は、遊離塩基の形態での使用量に匹敵する。酸付加塩を製造するために使用され得る酸は、好ましくは、遊離の塩基と組合せた場合、薬学的に受容可能な塩、すなわち、そのアニオンが薬学的用量の塩で患者に対して非毒性であり、遊離塩基に固有の有益な阻害作用がアニオンに起因する副作用によって低下しないような塩を製造するものを包含する。前記塩基性化合物の薬学的に受容可能な塩が好ましいが、特定の塩自体が、例えば、塩が単に精製および同定の目的のために形成される場合、またはそれイオン交換法によって薬学的に受容可能な塩を製造する際の中間体として使用される場合、単に中間体生成物としてのみ所望されるとしても、全ての酸付加塩が遊離塩基の形態の供給源として有用である。本発明の範囲に包含される薬学的に受容可能な塩は、無機酸および有機酸から誘導されるものが挙げられ、そしてこれらとしては、ハロゲン化水素酸塩(例えば、塩酸塩および臭化水素酸塩)、硫酸塩、リン酸塩、硝酸塩、スルファミン酸塩、酢酸塩、クエン酸塩、乳酸塩、酒石酸塩、マロン酸塩、シュウ酸塩、サリチル酸塩、プロピオン酸塩、コハク酸塩、フマル酸塩、マレイン酸塩、メチレン−ビス−b−ヒドロキシナフトエ酸塩、安息香酸塩、トシレート、ゲンチジン酸塩、イセチオン酸塩、ジ−p−トルオイル酒石酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、シクロヘキシルスルファミン酸塩およびキナ酸塩が挙げられる。より特定の塩は、式Iの化合物の塩が、塩酸塩であるものである。本発明の別の特定の塩は、式Iの化合物のフマル酸塩である。本発明の好ましい薬学的に許容され得る塩は、式Iの化合物の安息香酸塩である。
【0121】
それ自体が活性化合物として有用であると同時に、本発明の化合物の塩は、例えば、当業者に周知の技術による塩と親化合物、副生成物、および/または出発物質との溶解度の差を利用することによって化合物の精製目的のために有用である。
【0122】
本発明のさらなる特徴に従って、公知の方法の応用または適用によって遊離塩基を適切な酸と反応させることによって、本発明の化合物の酸付加塩を製造し得る。例えば、本発明の化合物の酸付加塩は、遊離塩基を適切な酸を含む水または水性アルコール溶液または他の適切な溶媒中に溶解し、そして溶液を蒸発させることによって塩を単離するか、または有機溶媒中で遊離塩基を酸と反応させる(この場合は塩を直接分離するか、または溶液を濃縮することによって得ることができる)ことによって製造し得る。
【0123】
公知方法の応用または適用によって、本発明の化合物の酸付加塩を塩から再生し得る。例えば、アルカリ(例えば、重炭酸ナトリウム水溶液またはアンモニア水)を用いる処理によってその酸付加塩から本発明の親化合物を再生し得る。
【0124】
公知の方法(例えば、参照の実施例に記載される方法またはそれらの明らかな化学同等物)の応用もしくは適用によって、出発物質および中間体を製造し得る。
【0125】
本発明はまた、上記のスキーム1中のいくつかの中間体に関し、そしてまたそれらの製造についての本明細書中に記載されている方法は本発明のさらなる特徴を構成する。
【実施例】
【0126】
本発明は、本発明の例示として提供される以下の非限定的な実施例を参照することにより良く理解され得る。以下の実施例は、本発明の特定の実施形態をより十分に説明するために提示される。しかし、これらは本発明の範囲を限定するものと見なすべきではない。以下の参照実施例は、式Iの化合物を作製するために用いられる中間体を作製する方法を開示するために提供される。
【0127】
以下に報告される核磁気共鳴スペクトル(NMR)において、化学シフトは、テトラメチルシランに対するppmで表示する。略語は以下の意味を有する:br=ブロード、dd=ダブル二重項、s=一重項;m=多重項。
【0128】
参考実施例1
工程A:2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩(13)の製造
【化19】
フラスコにNaHCO3(126g、1.5mol)、3−ブロモ−4−フルオロベンジルアミン塩酸塩(11、120g、0.5mole)、ピリジン−4−ボロン酸(13、67.6g、0.55mmol)、iPrOH(750mL)および水(375mL)を室温で充填した。この懸濁液をN2で10℃にて1時間脱気した。この混合物中に1,1’−ビス(ジフェニルホスフィノ)フェロセン−パラジウム(II)二塩化物 ジクロロメタン複合体(PdCl2dppf−CH2Cl2、16.4g、20mmol)を加えた。この反応混合物を、内部温度が80℃に到達するまでいくらかの部分が蒸散するが、80℃まで加熱し、10時間攪拌した。この反応が完了(HPLC分析)した後、混合物を室温まで冷却し、2N HCl水溶液(750mL)を加え、0.5時間攪拌した。その溶液をDCM(750mL及び500mL)で洗浄した。水層に50% NaOH水溶液(100mL)をpH>13に調整されるまで加えた。n−BuOAc(2L)を加えた後、活性炭(50g)を有機層に加えた。この混合物をセライトパッド(50g)を通して濾過した。共沸蒸留を行った。追加のn−BuOAc(1L)を加えた後、反応物を5℃まで冷却した。TFAA(157g、0.6mol)をその溶液中にゆっくりと5℃で加えた。その反応が完了(HPLC分析)した後、反応混合物を10% Na2CO3水溶液(1L)で洗浄した。iPrOH(120mL)に溶解した5−6N HClの溶液を、粗製の有機層中に10℃で加えた。その後追加のn−BuOAc(1L)を加え、懸濁液を室温で一晩放置した。得られた固形物を10℃で濾過し、50℃のオーブンで乾燥させて124g(75%)の化合物15を白色固体として得た:mp=220℃. 分析 C14H10F4N2O−HClについての算出値:C、50.24;H、3.31;N、8.37.実測値:C、50.16;H、3.08;N、8.38. MS(ESI)m/z 299(M+H).1H NMR(300MHz、D2O)δ8.70(d、J=6.9Hz,2 H)、8.14(d、J=6.9Hz,2H)、7.56−7.20(m,3H)、4.51(s,2H).
【0129】
工程B:2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(14)の製造
【化20】
パーフラスコ(Parr flask)に、室温で化合物13(123g、0.37mol)およびMeOH(740mL)を入れ、その後5% Pt/C(36.9g、30
w/w%)を加えた。その反応フラスコをパー(Parr)水素化システム内に置き、H2により50−60psiで満たした。H2を充填する間、圧力が安定状態(H2を日中は2−3時間ごとに50−60psiまで補充したが、一晩超えた後は、さらなる補充なしに10−20psiが観察された)に達するまで、その混合物を>48時間振盪した。HPLC分析が反応の完了を示すと、反応混合物をセライトのパッドを通して濾過した。そのろ液を40−50℃で蒸留する一方、n−BuOAc(1.25L)を加えた。MeOHの蒸留が完了した後、追加のn−BuOAc(1L)を加えた。得られた懸濁液を一晩室温まで冷却させた。懸濁液を10℃まで冷却し、濾過し、50℃のオーブンで乾燥させ、112g(89%)の化合物14を白色固形物として得た:mp=134℃.分析 C14H10F4N2O−HClについての算出値:C、50.24;H、3.31;N、8.37.実測値:C、50.16;H、3.08;N、8.38. MS(ESI)m/z 305.4(M+H).
1H NMR(300MHz、D2O)δ 7.16−6.98(m,3 H)、4.34(s,2H)、3.42(d、J=12.9Hz,2H)、3.14−2.99(m,3H)、1.98−1.81(m,4H).
【0130】
参考実施例2
工程A:(2−フルオロ−5−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(2)の製造
【化21】
THF(500mL)に溶解させた1(50.72g、0.26mol)およびピリジン(27.3mL、0.34mol)の溶液に0℃でクロロギ酸エチル(32.2mL、0.39mol)を30分間にわたり滴下した、1時間後 LC/MSとTLCの両方が、反応が完了したことを示した。反応混合物をH2OとEtOAcに分配した。2層を分離し、有機層を1M HCl、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(95/5〜70/30)を用いてシリカゲル上で精製して69.23g(99%)の生成物2を透明な無色の液体として得た。
1H NMR(CDCl3)δ8.11(br s,1H)、7.07(dd,J=9.1、9.3Hz,1H)、7.00−6.80(m,2H)、4.27(q,J=7.1Hz,2H)、1.33(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−57.84(s,3F)、−134.01(br s,1F); MS 309(M+CH3CN+1、100%)、268(M+1).
【0131】
工程B:(6−フルオロ−2−ヨード−3−トリフルオロメトキシ−フェニル)−カルバミン酸エチルエステル(3)の製造
【化22】
THF(180mL)に溶解させた2(31.34g、117.2mmol)の溶液に−78℃でsec−BuLi(1.4M シクロヘキサン中、200mL、280mmol)を1時間にわたって滴下した。20分後、THF(150mL)に溶解させたI2(44.6g、175.8mmol)の溶液を30分にわたって滴下した。その後この混合物を−78℃で30分間攪拌した。飽和NH4Clを加え、冷却浴を取り除いた。反応混合物をH2OとEtOAcとの間に分配した。2層を分離し、有機層を10% Na2SO3、H2O、およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM(50mL)に懸濁させ、ヘプタン(300mL)を加えた。残りの懸濁液から白色粉末3(18.1g、39%)をサクションフィルターによって回収し、空気乾燥させた。ろ液を減圧下で濃縮し、残留物をヘプタン(200mL)に懸濁させた。別バッチの3(3.8g、8%)をサクション濾過によって回収し、空気乾燥させた。シリカゲルクロマトグラフィーによるろ液の精製により、追加の生成物を得ることができた。
1H NMR(CDCl3)δ7.30−17.10(m,2H)、6.16(br s,1H)、4.26(q,J=7.1Hz,2H)、1.32(t,J=7.1Hz,3H);19F NMR(CDCl3)δ−56.90(s,3F)、−114.35(d、J=8.5Hz,1F);MS 394(M+1、100%)、374、364、321、267.
【0132】
工程C:(6−フルオロ−3−トリフルオロメトキシ−2−トリメチルシラニルエチニル−フェニル)−カルバミン酸エチルエステル(4)の製造
【化23】
脱気したTHF(180mL)中に溶解した3(18.1g、45.9mmol)、Et3N(12.8mL、91.9mmol)、Pd(PPh)2Cl2(1.6g、5%mol)、CuI(0.7g、8%mol)、およびTMS−アセチレン(19.6mL、137.8mmol)混合物を60℃で一晩加熱した。混合物を室温まで冷却し、その後H2OとEtOAcの間に分配した。混合物をセライトを通して濾過し、不溶性物質を除去した。2層のろ液を分離し、有機層をH2O およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAcを用いてシリカゲル上で精製し、15.6g(93%)の生成物4をベージュの固形物として得た。
1H NMR(CDCl3)δ7.15−7.00(m,2H)、6.41(br s,1H)、4.26(q,J=7.1Hz,2H)、1.31(t,J=7.1Hz,3H)、0.27(s,9H);19F NMR(CDCl3)δ−57.59(s,3F)、−118.15(s,1F);MS 364(M+1、100%).
【0133】
工程D:7−フルオロ−4−トリフルオロメトキシ−1H−インドール(5)の製造
【化24】
脱気したt−BuOH(300mL)に溶解した4(28.9g、79.6mmol)およびKOH(35.7g、636.7mmol)の混合物を70℃で一晩加熱した。LC/MSが、反応が完了したことを示した。その混合物を室温まで冷却し、H2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を、溶離液としてヘプタン/EtOAc(100/0〜60/40)を用いてシリカゲル上で精製し、16g(91%)の5を黄色液体として得た。
1H NMR(CDCl3)δ8.47(br s,1H)、7.35−7.20(m,1H)、6.95−6.80(m,2H)、6.68(d、J=2.5Hz,1H);19F NMR(CDCl3)δ−57.63(s,3F)、−136.10(d、J=8.5Hz,1F);MS 220(M+1、100%)、200.
【0134】
工程E:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール(6)の製造
【化25】
DMSO(150mL)に溶解させた5(16g、72.8mmol)およびKOH粉末(20.4g、364.2mmol)の混合物を室温で10分間攪拌した。2−メトキシエチル臭化物(10.3mL、109.2mmol)を加えた。その混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。この混合物をH2OとEt2Oの間に分配した。2層を分離し、水層をEt2O(2X)で抽出した。合わせた有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、19.3g(95%)の6を黄色液体として得た。
1H NMR(CDCl3)δ7.15(d、J=2.1Hz,1H)、6.90−6.75(m,2H)、6.56(t,J=2.5Hz,1 H)、3.72(t,J=5.2Hz,2H)、3.72(t,J=5.2Hz,2H)、3.31(s,3H);19F NMR(CDCl3)δ−57.54(s,3F)、−137.00(d、J=11.3Hz,1F);MS 278(M+1、100%).
【0135】
工程F:2,2,2−トリフルオロ−1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−エタノン(7)の製造
【化26】
DMF(135mL)に溶解させた6(19.3g、69.7mmol)の混合物にTFAA(26.2mL、188.2mmol)を加えた。この混合物を40℃で一晩加熱した。TLCが、反応が完了したことを示した。混合物を室温まで冷却し、その後H2O
とEt2Oの間に分配した。2層を分離し、有機層を飽和NaHCO3(2X)、H2O およびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(100/0〜50/50)を用いてシリカゲル上で精製し、23.4g(89%)の7をわずかに緑色の固形物として得た。
1H NMR(CDCl3)δ8.03(d、J=1.4Hz,1H)、7.20−6.95(m,2H)、4.54(t,J=4.9Hz,2H)、3.76(t,J=4.8Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−71.10(s,3F)、−134.95(d、J=11.5Hz,1F);MS 374(M+1、100%).
【0136】
工程G:7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボン酸(8)の製造
【化27】
MeOH(100mL)および5M NaOH(100mL)に7(23.4g、62.6mmol)を溶解させた混合物を80℃で一晩加熱した。LC/MSが、反応が完了したことを示した。その反応混合物を室温まで冷却し、その後減圧下で濃縮して大部分のMeOHを除去した。残留物をH2Oに溶解させて、その後Et2Oで一度洗浄した。水層を濃HClを用いてpH2までゆっくりと酸性化させた。酸性化させた懸濁液をEt2Oで抽出し、有機抽出物をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。残留物をDCM/ヘプタン(10/90)に懸濁させた。懸濁液中の白色粉末8(19.4g、96%)をサクションフィルターで回収し、空気乾燥させた。
1H NMR(CDCl3)δ8.02(s,1H)、7.15−7.05(m,1H)、7.00−6.90(m,1H)、4.49(t,J=5.0Hz,2H)、3.75(t,J=4.9Hz,2H)、3.33(s,3H);19F NMR(CDCl3)δ−57.74(s,3F)、−135.65(d、J=11.3Hz,1F);MS 363(M+CH3CN+1)、322(M+1、100%).
【0137】
工程H:2,2,2−トリフルオロ−N−(4−フルオロ−3−{1−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−カルボニル]−ピペリジン−4−イル}−ベンジル)−アセトアミド(9)の製造
【化28】
CH2Cl2に8(19.1g、59.6mmol)、Et3N(24.8mL、177.9mmol)、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピペリジン−4−イル−ベンジル)−アセトアミド塩酸塩(11、26.4g、77.5mmol)(14)、およびEDCI(17.1g、89.3mmol)を溶解させた混合物を室温で一晩攪拌した。TLCとLC/MSの両方が、反応が完了したことを示した。混合物をH2OとCH2Cl2の間に分配した。2層を分離し、有機層をブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮した。粗製品を溶離液としてヘプタン/EtOAc(40/60〜0/100)を用いてシリカゲル上で精製し、9(36g、99%)を白色泡沫として得た。
1H NMR(CDCl3)δ7.37(s,1H)、7.20−7.10(m,2H)、7.10−6.85(m,4H)、4.95(br s,1H)、4.60−4.35(m,4H)、3.90(br s,1 H)、3.73(t,J=5.0Hz,2H)、3.32(s,3H)、3.25−2.70(m,3H)、2.05−1.50(m,4H);19F NMR(CDCl3)δ−57.54(s,3F)、−75.39(s,3F)、−119.31(s,1F)、−134.96(d、J=11.3Hz,1F);MS 608(M+1、100%).
【0138】
工程I:[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン塩酸塩(10)の製造
【化29】
MeOH(400mL)に9(36g、59.3mmol)を溶解した混合物にK2CO3水溶液(65.5g、474mmol、120mL H2Oに溶解)を加えた。この混合物を室温で一晩攪拌した。LC/MSが、反応が完了したことを示した。反応混合物を減圧下で濃縮し、大部分のメタノールを除去した。残留物をH2OとEtOAcの間に分配した。2層を分離し、有機層をH2Oおよびブラインで洗浄し、MgSO4で乾燥させ、濾過して減圧下で濃縮して27.5g(90%)の10を透明な無色の粘着性ゴムとして得た。
1H NMR(CDCl3)δ7.42(s,1H)、7.25−7.10(m,2H)、7.05−6.85(m,3H)、4.92(br s,1H)、4.46(t,J=5.2Hz,2H)、3.86(br s,3 H)、3.74(t,J=5.1Hz,2H)、3.32(s,3H)、3.30−2.75(m,3H)、2.24(br s,2H)、2.05−1.55(m,4H);19F NMR(CDCl3)δ−57.52(s,3F)、−121.64(s,1F)、−136.03(d、J=11.3Hz,1F);MS 512(M+1、100%).
【0139】
Et2O(30mL)に溶解させた上記の物質(2.856g、5.59mmol)の溶液に2N HCl/Et2O(3mL、6mmol)を滴下した。固体沈殿物が形成し、エーテル溶液をデカントした。固体をさらなるEt2Oで洗浄してその後デカントした。残りの淡黄色固形物を温MeOH(10mL)に溶解し、その後Et2O(50mL)をその溶液がわずかににごるまで加えた。約2時間後、固体沈殿物が出現した。さらなるEt2O(5−10mL)を加え、その後懸濁液を冷蔵庫に一晩置いておいた。白色の結晶生成物(2.475g、4.52mmol)を回収し、高減圧下で4時間乾燥させた。
1H NMR(DMSO−d6)δ8.32(br s,2H)、7.71(s,1H)、7.43(d、1H、J=7.2Hz)、7.36(m,1H)、7.26−7.20(m,1H)、7.12−7.08(m,2H)、4.49(t,J=5.1Hz、2H)、4.00(s,2H)、3.71(t,J= 5.1Hz、2H)、3.32(s,3H)、3.21−3.07(m,3H)、2.99(br s,2H)、1.80−1.62(m,4H);19F NMR(DMSO−d6)δ−56.79(s,3F)、−119.34(s,1F)、−134.53(d、J=9.6Hz,1F);MS 512(M+1、100%). CHN:理論値:C 53.06%、H 5.16%、N 7.42%(calc’d as 1.0 H2O). 実測値:C 53.03%、H
4.82%、N 7.22、Cl 6.64%.
【0140】
参考実施例3
式Iの化合物の安息香酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(1320g、2.58mol)を含んでいると思われるトルエン溶液をすでに入れてある20Lのガラスジャケット反応器を攪拌し、61℃まで加熱した。安息香酸(316g、2.58mol)を加え、全ての安息香酸が溶解した後、シクロヘキサン(6.04L)を加えた。上記のバッチからの[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエート(0.100g)で結晶が生じる77℃まで反応物を加熱した。結晶化を77℃で進行させ、15分後、その反応物を−10℃/hのランプで冷却した。反応が61℃まで到達したときに攪拌も冷却も停止させ、反応を室温まで冷却した。一晩放置した後、攪拌を再開し、生成物を濾過により回収した。濾過ケークをトルエン(3L)およびシクロヘキサン(1.5L)より調製した溶媒混合物で洗浄した。サクションにより部分的に乾燥した後、生成物を40℃で乾燥させる乾燥オーブンに移し、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゾエートを無色の固形物として得た。:1408.8g(86%)、mp=156−159℃.
【0141】
参考実施例4
式Iの化合物のベシル酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン ベンゼンスルホン酸塩
アセトニトリル(12mL)にベンゼンスルホン酸一水和物(698mg、3.84mmol)を溶解した溶液を、攪拌したアセトニトリル(5mL)に溶解させた[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(2.0g、3.91mmol)の懸濁液に滴下した。溶解した遊離塩基の最期として混合物からベンゼンスルホン酸塩が結晶化し始めた。2時間後、生成物を濾過により回収し、アセトニトリルで洗浄した。フィルターケークを一晩乾燥させた。固形物を砕き、減圧オーブン中(43〜44℃、6.8−7.3Hg)で7.5時間窒素を流しながら乾燥させ、[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノンベンゼンスルホン酸塩を無色の固体として得た。:2.27g 1(86.7%)、mp=215−218℃.C25H26F5N3O3.C6H6O3Sについての分析算出値:C、55.60;H、4.82;N、6.27. 実測値:C、55.65;H、4.65;N、6.27. Karl Fischer:<0.10.赤外線スペクトルの項目(cm−1):1587、1545、1445、1210、1167、1125、1036、1018.
【0142】
参考実施例5
式Iの化合物のセスキフマル酸塩
[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン セスキフマル酸塩 一水和物
丸底フラスコを、[4−(5−アミノメチル−2−フルオロフェニル)−ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノン(10.4g、20.4mmol)およびフマル酸(4.74g、40.7mmol)で満たした。イソプロパノール(IPA、62mL)を加え、得られた混合物をスチームバスで加熱した。塩の結晶化が起こる前に、ほとんどの物質が溶解した。スチームバスで加熱している間、さらなるIPAを30mLのポーションで加えた。合計152mLのIPAを加えた後、完全な溶液に到達した。得られた溶液を濾過し、そのろ液を室温まで冷却した。ろ液をさらに氷浴で1.5時間冷却し、その後生成物を濾過により回収した。回収した生成物を冷IPA(50mL)で洗浄し、サクションにより部分的に乾燥させ、45℃で乾燥させる乾燥オーブンに移した。一晩乾燥させた後、所望の生成物を無色の固体として得た。:11.8g(84%).IR(cm-1):3122−2700、2920、2824、1698、1584、1512、1443、1397−1368、1293−1217、822、794、639.1H NMR(300MHz、DMSO−d6):δ 10.07(br、3H)、7.71(s,1H)、7.43(dd,J=2.4、7.1、1H)、7.36(ddd、J=2.4、4.9、8.4、1H)、7.19(d、J=8.4、10.7、1H)、7.10(d、J=8.7、11.7、1H)、7.05(ddd、J=1.4、3.3、8.7、1H)、6.50(s,3H)、4.69(br、1H)、4.48(t,J=5.3、2H)、3.97(s,2H)、3.69(t,J=5.4、2H)、3.24(s,3H)、3.08(dddd、J= 3.5、3.5、12.1、12.1、1H)、2.91(br、2H)、1.75(br、2H)、1.63(br、2H).C25H26F5N3O3−1.5C4H4O4についての分析算出値:C、54.31;H、4.70;N、6.13.実測値:C、54.30;H、4.62;N、6.04. MS(ESI)m/z 512.2(M+H).
【0143】
参考実施例6
式Iの化合物のトシル化塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノンp−トルエンスルホン酸
アセトニトリル(3mL)に[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(488mg、0.95mmol)を溶解させた混合物に、p−トルエンスルホン酸一水和物(181mg、0.95mmol)を加えた、この混合物を冷蔵庫に一晩貯蔵した。得られたベージュの結晶をサクションフィルターで回収し、トルエンで洗浄し、減圧下の50℃で一晩乾燥させた。453mg(69%)得られた。
1H NMR(DMSO−d6)δ8.08(bs、3H)、7.70(s,1H)、7.80−6.95(m,9H)、5.00−4.30(m,3H)、4.20−3.90(m,2H)、3.80−3.60(m,3H)、3.23(s,3H)、3.25−2.80(m,3H)、2.28(s,3H)、1.95−1.45(m,4H);19F NMR(DMSO−d6)δ−55.61(s,3F)、−118.98(s,1F)、−134.33(d、J=9.3Hz,1F);LC 2.627 min;MS 512(M+1、100%).Mp 219℃.赤外線スペクトルの項目(cm−1):1583、1548、1511、1501、1250、1200、1169、1123、1115.
【0144】
参考実施例7
式Iの化合物の硫酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(423mg、0.827mmol)を20mLのガラスバイアル中に秤量した。この固体に硫酸(1.0N 試薬、1.5当量、1.30mmol、2.60mL)および1.7mL 水の溶液を加えた。室温で2時間攪拌した後、結晶性生成物が沈殿した。濾過および乾燥後、固体がアモルファスとして発見された。数滴の水で処理すると、そのアモルファス固体は結晶形態に戻った。Mp 62℃.赤外線スペクトルの項目(cm−1):1574、1545、1511、1483、1362、1267、1219、1212、1162、1096、1051.
【0145】
参考実施例8
式Iの化合物のクエン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(265mg、0.52mmol)を20mLのガラスバイアル中に秤量した。この溶液に2:1(v/v)アセトニトリル/水に溶解させたクエン酸溶液(0.158mmol/mlクエン酸3.30mL)を加えた。全ての固体が素早く溶解し、得られた無色の溶液を室温で1時間置いておいた。溶液を窒素ガス流の元でエバポレートし、その後室温の減圧下で乾燥させた。固体を最小限の量の水を加えた温アセトニトリル中で再結晶化させて無色の溶液を得た。冷却するとその溶液に、室温に置いておくとプレート状にに変形するような非常に長い繊維の粒子状の生成物が沈殿した。Mp 112℃.赤外線スペクトルの項目(cm−1):1721、1590、1553、1369、1245、1174、1155、1119。
【0146】
参考実施例9
式Iの化合物のメタンスルホン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(0.250g、0.489mmol)を20mLのガラスバイアル中に秤量した。水に溶解させたメタンスルホン酸(0.50mmol/ml溶液0.98mL)を加え、その混合物を攪拌しながら60℃まで加熱した。全ての固形物が溶解したわけではないが、さらに25mlのメタンスルホン酸溶液を加えて透明な溶液を得た。室温で1時間攪拌した後、溶液を、ロータリーエバポレーターにより減圧下でエバポレートして非常に粘性のオイルを得た。このオイルを、アセトニトリル中で正方形プレートに再結晶化させた。赤外線スペクトルの項目(cm−1):1596、1540、1214、1159、1112、1040、1020.
【0147】
参考実施例10
式Iの化合物の酒石酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(0.250g、0.554mmol)を、20mLのガラスバイアル中に秤量した。5:1(v/v)アセトニトリル/水に溶解させたL−(+)−酒石酸溶液2.66mmol/mlを調製し、0.2084mLのこの溶液を秤量した固体に加え、攪拌しながら60℃まで加熱して透明の溶液を得た。その後この溶液を、ロータリーエバポレーターを用いて減圧下でエバポレートして温酢酸イソプロピル中で再結晶化したガラス状の固形物を分離し、そこに最小限の量のイソプロパノールを加えて透明な溶液を得た。濾過によって単離した結晶生成物を冷却し、室温にて減圧下で乾燥させた。
【0148】
参考実施例11
式Iの化合物のリン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(133.9mg、0.262mmol)に、リン酸溶液(1mmole/mL イソプロパノールに溶解、1.1当量)を加えた。この混合物を、マグネットスターラーを用いて攪拌しながら500μLのイソプロパノールに室温で溶解させた。この物質をエバポレートして室温で乾燥させ、結晶性の物質は単離されなかった。この物質を500μLのアセトン、500μLの酢酸エチル、および1mLのヘプタンに再溶解させた。この物質をオイルとして分離した。この混合物をエバポレートして窒素流下で乾燥させた。一旦乾燥させ、酢酸エチル(500μL)およびトルエン(500μL)を加え、その物質をオイルとして分離した。この混合物をエバポレートして一晩室温で乾燥させた。メチルイソブチルケトン(1mL)およびトルエン(500μL)を加え、その物質を溶解させた。混合物を室温で一晩エバポレートした。結晶が出現し、室温での減圧濾過により、回収した。物質を室温での減圧オーブン(300mbar)中で一晩乾燥させた。
【0149】
参考実施例12
式Iの化合物のグルタミン酸塩
[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−[7−フルオロ−1−(2−メトキシ−エチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]−メタノン(138.8mg、0.271mmol)、グルタミン酸溶液(162.4mg/20mL(水に溶解)、1.1当量)を加えた。メタノール(2mL)を加えて物質を溶解させた。その混合物を室温で一晩エバポレートし、白色のアモルファス物質が沈殿した。その物質にイソプロパノール(600μL)をく加えた。結晶が出現し、それを室温にて減圧濾過により回収した。物質を減圧オーブン(約300mbar)中で一晩乾燥させた。
【0150】
参考実施例13
式Iの化合物の安息香酸塩の結晶形態
サンプル準備:材料を上記の参考実施例3のように準備した。安息香酸塩の懸濁液をナノピュアウオーター中に50mg/mLの遊離塩基当量(1mLの水中に63.6mgの塩)として調製した。サンプルを500rpmで一晩攪拌し、4時間放置したのち遠心分離した(懸濁として計29時間)。13000rpmで8分間遠心分離し、回収した固体を、湿潤サンプルとしてXRPD(x−ray power diffraction)で分析し、顕微鏡で評価した。その後この湿潤固体を周囲室温で一晩空気乾燥し、乾燥サンプルとしてXRPDおよび熱分析により分析した。そのままの薬物物質を初期の物質と比較した。遊離塩基薬物物質のXRPDもまた比較として用いた。XRPDが水量の異なる複数のピークを示しており、出現した安息香酸塩は可変の水和物であると考えられる。
【0151】
装置パラメータ
XRPD法
Cu対陰極を備えるジーメンスモデル D5000
プログラム:1.0 Sec.dql
範囲:2°〜40°.2−θスケール
工程サイズ:0.02°
雰囲気:周囲条件の温度および湿度
標準的なトップロード、低容量のキャビティー試験片マウントを用いた
【0152】
DSC−TGA:
TA Instruments Model Q−600 Simultaneous
DSC−TGA
パージガス:ヘリウム100mL/分
温度プログラム:10℃/分 線形加熱速度
サンプル準備:約3−5mgの粉末を開口アルミニウムパンに移し、TGA中にロードした。空のアルミニウムパンを参照として用いた。
【0153】
結果:
XRPDおよび熱分析を湿潤および乾燥サンプルで実施した。湿潤サンプルのXRPDは、ベースラインのいくらかのシフトと上昇を示した。しかし乾燥(一晩)サンプルでは、XRPDは、初期の物質と比較してピークの向上した分解を示した。乾燥サンプルの熱分析は、初期と同じTGAプロフィールを示した。XRPDおよび熱分析に基づいて、遊離塩基または水和物形態への転換は報告されなかった。遊離塩基のXRPDより、以下を得た:
【0154】
図1は、式Iの化合物の安息香酸塩の結晶形態AについてのXRPD結果を示す。この図は、サンプルについて角度に対する相対強度(%)を示している。ピークは、以下の角度で見られた:7.75、10.13、17.03、17.16、17.99、18.39、20.51、21.33、21.88、23.19、23.43、および27.59.
【0155】
図2は、式Iの化合物の安息香酸塩の結晶形態AについてのDSC結果を示している。この図は、160.29℃で溶解が開始し、162℃でその形態が溶解することを示している。
【0156】
生物活性
本発明の化合物の特性は、1)そのβ−トリプターゼ阻害能力(IC50およびKi値)によって証明される。
【0157】
インビトロ試験手順
背景技術の章に記載されたように、トリプターゼの全ての作用がその触媒活性に依存しているので、その触媒活性を阻害する化合物は、トリプターゼの作用を強力に阻害する。この触媒活性の阻害は、インビトロの酵素アッセイおよび細胞アッセイにより測定される。
【0158】
単離ヒト肺トリプターゼまたは酵母細胞内に発現させた組換えヒトβトリプターゼのいずれかを使用して、トリプターゼの阻害活性を確認する。単離天然酵素または発現酵素を使用して、本質的に同等の結果が得られる。アッセイ手順は、基質としてL−ピログルタミル−L−プロリル−L−アルギニン−パラ−ニトロアニリド(S2366:Quadratech)を使用する、96ウェルのマイクロプレート(Costar3590)を利用する(本質的にはMcEuen et al.Biochem Pharm.1996,52,331−340頁に記載されている)。アッセイは0.5mM基質(2×Km)を用いて50mM Tris(pH8.2)、100nM NaCl、0.05% Tween20、50μg/mlヘパリン中で実施され、そしてマイクロプレートは405nmの波長でマイクロプレートリーダー(Beckman Biomek Plate reader)上で読み取られる。
【0159】
プロトコル(IC50およびKiの測定)
化合物は2連で、以下の最終濃度:0.01、0.03、0.1、0.3、1、3、10μMで添加することを除いて、プロトコルは本質的に上記と同様である(全ての希釈は手作業で行なう)。各アッセイについて、一点測定またはIC50測定のいずれにおいても、標準化合物を使用して、比較のためのIC50を導く。IC50値から、式:Ki=IC50/(1+[基質]/Km)を使用して、Kiを計算し得る。
【0160】
式Iの化合物についてのβ−トリプターゼ阻害能力は、26±5nMのKi値であった。
【0161】
抗原誘導性気道過敏症アッセイのプロトコル
抗原感作およびチャレンジ:雄のHartleyモルモット(225〜250g)をオボアルブミン(1%溶液0.5mL、i.p.およびs.c.)で1日目(8/25/08)に感作した。4日目(8/28/08)に、動物は1%オボアルブミン(0.5mL)のブースタ注射を受けた(i.p.)。21日目(09/16/08)に、抗原チャレンジの24時間前に、動物にビヒクル(0.5%メチルセルロース/0.2%Tween 80)または試験化合物のいずれかを経口投薬した(2mL/kg)。また、抗原チャレンジの30分前に、動物にメピラミン(10mg/kg、i.p.)を注射し、アナフィラキシー性の衰弱(anaphylactic collapse)を予防した。次いで、動物を、DeVilbiss Ultranebネブライザーを使用して、1%オボアルブミンのエアロゾルに20分間暴露させた。ネガティブコントロールの動物はチャレンジしなかった。感作溶液:鶏卵白(Sigma A55031G;lot#087K7004)由来のアルブミン1gを100mLの生理食塩水に加え、溶液中に入れた。
【0162】
気道抵抗測定:チャレンジの18〜25時間後、動物に麻酔(ケタミン(62mg/kg)、キシラジン(30mg/kg)およびプロメース(promace)(1.5mg/kg)を含む反応混液0.5ml用量(i.m.))をかけ、外科的準備をしてその後全身を体積変動記録計に置いた。動物を、気管カニューレを介して50呼吸/分の速度で1mL/100gの1回呼吸量を送達するUgo−Basile人工呼吸器につないだ。ヒスタミンチャレンジのために頚静脈にもカニューレを挿入した。肺圧差を記録できるように、水を満たした食道カニューレを取り付けた。異なる圧力のトランスデューサを使用して、気管と食道カニューレとの間の差異として肺圧差を測定した。容量、気流および肺圧差のシグナルを、肺分析システム(Buxco XAソフトウェア)を使用してモニターし、そしてこれらを使用して肺抵抗(cm H2O/mL/s)および動的コンプライアンス(mL/cm H2O)を計算した。気道抵抗および動的コンプライアンスを呼吸基礎(breath basis)による呼吸数で計算した。ヒスタミンを静脈内に投与し、そして増大する濃度(1〜20μg/kg)についての反応性を評価した。
【0163】
式Iの化合物のフマル酸塩についてのこのアッセイ結果を以下の表に示す。このアッセイは、喘息を潜在的に治療するための化合物の有効性に関する。式Iの化合物のフマル酸塩は、これらの表によって示されるように、アレルゲンチャレンジの24時間前に投与された場合、抗原誘導性気道過敏症の用量関連阻害を示した。
【0164】
【表1】
【0165】
【表2】
【0166】
本発明は本明細書中に記載した特定の実施態様によって範囲は制限されない。実際、本明細書中に記載されたものに加えて、本発明の種々の変更が、上記の説明および添付の図面から当業者に明らかとなる。このような変更は、添付の特許請求の範囲内に属することが意図される。
【0167】
種々の文献を本明細書中で引用し、この開示はその内容が参照によって援用される。
【図面の簡単な説明】
【0168】
【図1】式Iの化合物の安息香酸塩の結晶形態AについてのXRPD結果を示す。
【図2】式Iの化合物の安息香酸塩の結晶形態AについてのDSC結果を示している。
【特許請求の範囲】
【請求項1】
式I:
【化1】
の化合物またはそれらのプロドラッグ、薬学的に受容可能な塩もしくは溶媒和物。
【請求項2】
薬学的に受容可能な塩としての請求項1に記載の化合物。
【請求項3】
薬学的に受容可能な塩が以下:
塩酸塩、フマル酸塩、ベシル酸塩、トシラート、硫酸塩、クエン酸塩、メタンスルホン酸塩、酒石酸塩、リン酸塩、グルタミン酸塩および安息香酸塩から選択される、請求項2に記載の化合物。
【請求項4】
塩が塩酸塩である、請求項3に記載の化合物。
【請求項5】
塩がフマル酸塩である、請求項3に記載の化合物。
【請求項6】
塩がベシル酸塩である、請求項3に記載の化合物。
【請求項7】
塩がトシラートである、請求項3に記載の化合物。
【請求項8】
塩が硫酸塩である、請求項3に記載の化合物。
【請求項9】
塩がクエン酸塩である、請求項3に記載の化合物。
【請求項10】
塩がメタンスルホン酸塩である、請求項3に記載の化合物。
【請求項11】
塩が酒石酸塩である、請求項3に記載の化合物。
【請求項12】
塩がリン酸塩である、請求項3に記載の化合物。
【請求項13】
塩がグルタミン酸塩である、請求項3に記載の化合物。
【請求項14】
塩が安息香酸塩である、請求項3に記載の化合物。
【請求項15】
請求項14に記載の化合物の結晶形態A。
【請求項16】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88のθ角のうち少なくとも2つにおいてXRPDピークを有する、請求項14に記載の化合物の結晶形態。
【請求項17】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも3つにおいてXRPDピークを有する、請求項16に記載の化合物の結晶形態。
【請求項18】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも4つにおいてXRPDピークを有する、請求項17に記載の化合物の結晶形態。
【請求項19】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも5つにおいてXRPDピークを有する、請求項18に記載の化合物の結晶形態。
【請求項20】
結晶形態は162℃で溶解する、請求項14に記載の化合物の結晶形態。
【請求項21】
トリプターゼインヒビターの改善が必要な生理学的症状を罹患しているかまたは罹患しやすい患者を治療する方法であって、治療有効量の請求項1の化合物を該患者に投与することを包含する、方法。
【請求項22】
生理学的症状が、炎症性疾患、関節軟骨崩壊疾患、結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺炎、線維症、慢性閉塞性肺疾患、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫、肥厚性瘢痕、皮膚科的症状、アテローム動脈硬化性プラーク破壊に関連する症状、歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍および合胞体ウイルス感染からなる群から選択される、請求項21に記載の方法。
【請求項23】
生理学的症状が炎症性疾患である、請求項22に記載の方法。
【請求項24】
炎症性疾患が、関節の炎症、炎症性腸疾患、関節炎、関節リウマチ、リウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎、喘息または変形性関節症である、請求項23に記載の方法。
【請求項25】
炎症性疾患が炎症性腸疾患である、請求項22に記載の方法。
【請求項26】
生理学的症状がCOPDである、請求項22に記載の方法。
【請求項27】
生理学的症状が喘息である、請求項24に記載の方法。
【請求項28】
生理学的症状が皮膚科的症状である、請求項22に記載の方法。
【請求項29】
皮膚科的症状が、過敏性皮膚炎、乾癬および湿疹より選択される、請求項28に記載の方法。
【請求項30】
皮膚科的症状が過敏性皮膚炎である、請求項29に記載の方法。
【請求項31】
皮膚科的症状が乾癬である、請求項29に記載の方法。
【請求項32】
喘息を患っている患者を治療する方法であって、治療有効量の請求項1に記載の化合物ならびにβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビター、および抗炎症薬からなる群から選択される第二の化合物との組み合わせを患者に投与することを包含する、方法。
【請求項33】
投与が、請求項1に記載の化合物が血漿と比較して肺組織に優先的に拡散するようなものである、請求項32に記載の方法。
【請求項34】
治療有効量の請求項1に記載の化合物およびその薬学的に受容可能な担体を含有する医薬組成物。
【請求項35】
βアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症薬からなる群より選択される治療有効量の第二の化合物をさらに含む、請求項34に記載の医薬組成物。
【請求項36】
第二の化合物がβアドレナリン作用性アゴニストである、請求項35に記載の医薬組成物。
【請求項37】
βアドレナリン作用性アゴニストが、アルブテロール、テルブタリン、ホルモテロール、フェノテロール、またはプレナリンより選択される、請求項36に記載の医薬組成物。
【請求項38】
第二の化合物が抗コリン作用薬である、請求項35に記載の医薬組成物。
【請求項39】
抗コリン作用薬が臭化イプラトロピウムまたはチオトロピウムである、請求項38に記載の医薬組成物。
【請求項40】
第二の化合物が抗炎症性コルチコステロイドである、請求項35に記載の医薬組成物。
【請求項41】
抗炎症性コルチコステロイドが、ベクロメタゾンジプロピオネート、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンおよびデキサメタゾンより選択される、請求項40に記載の医薬組成物。
【請求項42】
第二の化合物が抗炎症薬である、請求項35に記載の医薬組成物。
【請求項43】
抗炎症薬が、クロモグリク酸ナトリウムまたはネドクロミルナトリウムである、請求項42に記載の医薬組成物。
【請求項44】
ロイコトリエンアンタゴニストがモンテルカストである、請求項35に記載の医薬組成物。
【請求項45】
ホスホジエステラーゼ−4 インヒビターがロフルミラストおよびシフルモラストより選択される、請求項35に記載の医薬組成物。
【請求項46】
以下の式を有する化合物;
【化2】
【請求項47】
以下の式を有する化合物;
【化3】
【請求項48】
以下の式を有する化合物;
【化4】
【請求項49】
以下の式を有する化合物;
【化5】
【請求項50】
以下の式を有する化合物;
【化6】
【請求項51】
以下の式を有する化合物;
【化7】
【請求項52】
以下の式を有する化合物;
【化8】
【請求項53】
以下の式を有する化合物;
【化9】
【請求項54】
以下の式を有する化合物;
【化10】
【請求項55】
以下の式を有する化合物;
【化11】
【請求項56】
以下の式を有する化合物;
【化12】
【請求項57】
鈴木カップリング条件下で3−ブロモ−4−フルオロベンジルアミン塩酸塩とピリジン−4−ボロン酸をカップリングして、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩を得ることを含む、請求項2に記載の2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩の製造方法。
【請求項58】
鈴木カップリング条件は、少なくともi−プロピルアルコールの沸点を持つアルコール性溶媒、極性非プロトン生溶媒、もしくはエーテル溶媒、または前出の溶媒と水もしくはトルエンの任意の混合物から選択される鈴木カップリング溶媒を用いる、請求項4に記載の方法。
【請求項59】
鈴木カップリング溶媒は、少なくともi−プロピルアルコールの沸点を持つアルコール性溶媒である、請求項5に記載の方法。
【請求項1】
式I:
【化1】
の化合物またはそれらのプロドラッグ、薬学的に受容可能な塩もしくは溶媒和物。
【請求項2】
薬学的に受容可能な塩としての請求項1に記載の化合物。
【請求項3】
薬学的に受容可能な塩が以下:
塩酸塩、フマル酸塩、ベシル酸塩、トシラート、硫酸塩、クエン酸塩、メタンスルホン酸塩、酒石酸塩、リン酸塩、グルタミン酸塩および安息香酸塩から選択される、請求項2に記載の化合物。
【請求項4】
塩が塩酸塩である、請求項3に記載の化合物。
【請求項5】
塩がフマル酸塩である、請求項3に記載の化合物。
【請求項6】
塩がベシル酸塩である、請求項3に記載の化合物。
【請求項7】
塩がトシラートである、請求項3に記載の化合物。
【請求項8】
塩が硫酸塩である、請求項3に記載の化合物。
【請求項9】
塩がクエン酸塩である、請求項3に記載の化合物。
【請求項10】
塩がメタンスルホン酸塩である、請求項3に記載の化合物。
【請求項11】
塩が酒石酸塩である、請求項3に記載の化合物。
【請求項12】
塩がリン酸塩である、請求項3に記載の化合物。
【請求項13】
塩がグルタミン酸塩である、請求項3に記載の化合物。
【請求項14】
塩が安息香酸塩である、請求項3に記載の化合物。
【請求項15】
請求項14に記載の化合物の結晶形態A。
【請求項16】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88のθ角のうち少なくとも2つにおいてXRPDピークを有する、請求項14に記載の化合物の結晶形態。
【請求項17】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも3つにおいてXRPDピークを有する、請求項16に記載の化合物の結晶形態。
【請求項18】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも4つにおいてXRPDピークを有する、請求項17に記載の化合物の結晶形態。
【請求項19】
結晶形態が、以下:7.75、10.13、17.03、17.16、18.39、21.33、および21.88の角度のうち少なくとも5つにおいてXRPDピークを有する、請求項18に記載の化合物の結晶形態。
【請求項20】
結晶形態は162℃で溶解する、請求項14に記載の化合物の結晶形態。
【請求項21】
トリプターゼインヒビターの改善が必要な生理学的症状を罹患しているかまたは罹患しやすい患者を治療する方法であって、治療有効量の請求項1の化合物を該患者に投与することを包含する、方法。
【請求項22】
生理学的症状が、炎症性疾患、関節軟骨崩壊疾患、結膜炎、春季結膜炎、炎症性腸疾患、喘息、アレルギー性鼻炎、間質性肺炎、線維症、慢性閉塞性肺疾患、強皮症、肺線維症、肝硬変、心筋線維症、神経線維腫、肥厚性瘢痕、皮膚科的症状、アテローム動脈硬化性プラーク破壊に関連する症状、歯周病、糖尿病性網膜症、腫瘍増殖、過敏症、多発性硬化症、消化性潰瘍および合胞体ウイルス感染からなる群から選択される、請求項21に記載の方法。
【請求項23】
生理学的症状が炎症性疾患である、請求項22に記載の方法。
【請求項24】
炎症性疾患が、関節の炎症、炎症性腸疾患、関節炎、関節リウマチ、リウマチ様脊椎炎、痛風性関節炎、外傷性関節炎、風疹関節炎、乾癬性関節炎、喘息または変形性関節症である、請求項23に記載の方法。
【請求項25】
炎症性疾患が炎症性腸疾患である、請求項22に記載の方法。
【請求項26】
生理学的症状がCOPDである、請求項22に記載の方法。
【請求項27】
生理学的症状が喘息である、請求項24に記載の方法。
【請求項28】
生理学的症状が皮膚科的症状である、請求項22に記載の方法。
【請求項29】
皮膚科的症状が、過敏性皮膚炎、乾癬および湿疹より選択される、請求項28に記載の方法。
【請求項30】
皮膚科的症状が過敏性皮膚炎である、請求項29に記載の方法。
【請求項31】
皮膚科的症状が乾癬である、請求項29に記載の方法。
【請求項32】
喘息を患っている患者を治療する方法であって、治療有効量の請求項1に記載の化合物ならびにβアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビター、および抗炎症薬からなる群から選択される第二の化合物との組み合わせを患者に投与することを包含する、方法。
【請求項33】
投与が、請求項1に記載の化合物が血漿と比較して肺組織に優先的に拡散するようなものである、請求項32に記載の方法。
【請求項34】
治療有効量の請求項1に記載の化合物およびその薬学的に受容可能な担体を含有する医薬組成物。
【請求項35】
βアドレナリン作用性アゴニスト、抗コリン作用薬、抗炎症性コルチコステロイド、ロイコトリエン受容体アンタゴニスト、リポキシゲナーゼインヒビター、ホスホジエステラーゼ−4 インヒビターおよび抗炎症薬からなる群より選択される治療有効量の第二の化合物をさらに含む、請求項34に記載の医薬組成物。
【請求項36】
第二の化合物がβアドレナリン作用性アゴニストである、請求項35に記載の医薬組成物。
【請求項37】
βアドレナリン作用性アゴニストが、アルブテロール、テルブタリン、ホルモテロール、フェノテロール、またはプレナリンより選択される、請求項36に記載の医薬組成物。
【請求項38】
第二の化合物が抗コリン作用薬である、請求項35に記載の医薬組成物。
【請求項39】
抗コリン作用薬が臭化イプラトロピウムまたはチオトロピウムである、請求項38に記載の医薬組成物。
【請求項40】
第二の化合物が抗炎症性コルチコステロイドである、請求項35に記載の医薬組成物。
【請求項41】
抗炎症性コルチコステロイドが、ベクロメタゾンジプロピオネート、トリアムシノロンアセトニド、フルニソリド、プロピオン酸フルチカゾン、フロ酸モメタゾン、メチルプレドニゾン、プレドニゾロンおよびデキサメタゾンより選択される、請求項40に記載の医薬組成物。
【請求項42】
第二の化合物が抗炎症薬である、請求項35に記載の医薬組成物。
【請求項43】
抗炎症薬が、クロモグリク酸ナトリウムまたはネドクロミルナトリウムである、請求項42に記載の医薬組成物。
【請求項44】
ロイコトリエンアンタゴニストがモンテルカストである、請求項35に記載の医薬組成物。
【請求項45】
ホスホジエステラーゼ−4 インヒビターがロフルミラストおよびシフルモラストより選択される、請求項35に記載の医薬組成物。
【請求項46】
以下の式を有する化合物;
【化2】
【請求項47】
以下の式を有する化合物;
【化3】
【請求項48】
以下の式を有する化合物;
【化4】
【請求項49】
以下の式を有する化合物;
【化5】
【請求項50】
以下の式を有する化合物;
【化6】
【請求項51】
以下の式を有する化合物;
【化7】
【請求項52】
以下の式を有する化合物;
【化8】
【請求項53】
以下の式を有する化合物;
【化9】
【請求項54】
以下の式を有する化合物;
【化10】
【請求項55】
以下の式を有する化合物;
【化11】
【請求項56】
以下の式を有する化合物;
【化12】
【請求項57】
鈴木カップリング条件下で3−ブロモ−4−フルオロベンジルアミン塩酸塩とピリジン−4−ボロン酸をカップリングして、2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩を得ることを含む、請求項2に記載の2,2,2−トリフルオロ−N−(4−フルオロ−3−ピリジン−4−イル−ベンジル)−アセトアミド塩酸塩の製造方法。
【請求項58】
鈴木カップリング条件は、少なくともi−プロピルアルコールの沸点を持つアルコール性溶媒、極性非プロトン生溶媒、もしくはエーテル溶媒、または前出の溶媒と水もしくはトルエンの任意の混合物から選択される鈴木カップリング溶媒を用いる、請求項4に記載の方法。
【請求項59】
鈴木カップリング溶媒は、少なくともi−プロピルアルコールの沸点を持つアルコール性溶媒である、請求項5に記載の方法。
【図1】

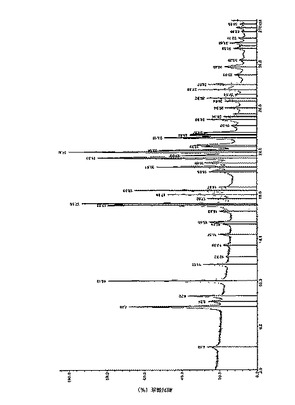
【図2】
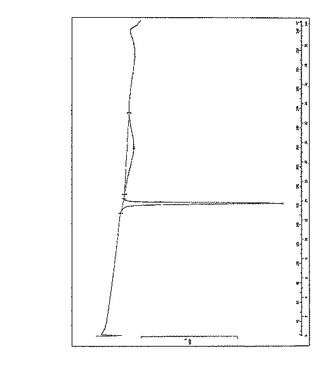
【図2】
【公表番号】特表2012−500794(P2012−500794A)
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願番号】特願2011−523975(P2011−523975)
【出願日】平成21年8月20日(2009.8.20)
【国際出願番号】PCT/US2009/054381
【国際公開番号】WO2010/022196
【国際公開日】平成22年2月25日(2010.2.25)
【出願人】(504456798)サノフイ (433)
【Fターム(参考)】
【公表日】平成24年1月12日(2012.1.12)
【国際特許分類】
【出願日】平成21年8月20日(2009.8.20)
【国際出願番号】PCT/US2009/054381
【国際公開番号】WO2010/022196
【国際公開日】平成22年2月25日(2010.2.25)
【出願人】(504456798)サノフイ (433)
【Fターム(参考)】
[ Back to top ]