
胃腸管保護および治療活性増強のためのレシチン油およびNSAIDSの製剤を用いる方法および組成物
【課題】
低い胃腸(GI)毒性ならびに炎症、疼痛、発熱、血小板凝集、組織の潰瘍形成および/または他の組織の障害を治療するための高い治療活性を有する非ステロイド性抗炎症薬含有製剤の開発。
【解決手段】
非ステロイド性抗炎症薬(NSAID)をレシチン油などのリン脂質含有油またはリン脂質が加えられている生体適合性油に直接加えて、低い胃腸(GI)毒性ならびに炎症、疼痛、発熱、血小板凝集、組織の潰瘍形成および/または他の組織の障害を治療するための高い治療活性を有する、NSAID含有製剤を製造する、新規医薬組成物が提供される。本発明の組成物は、内部、経口、直接または局所投与のための、非水性溶液、ペースト、懸濁液、分散液、コロイド懸濁液の形、または水性乳濁液もしくはマイクロエマルジョンの形である。
低い胃腸(GI)毒性ならびに炎症、疼痛、発熱、血小板凝集、組織の潰瘍形成および/または他の組織の障害を治療するための高い治療活性を有する非ステロイド性抗炎症薬含有製剤の開発。
【解決手段】
非ステロイド性抗炎症薬(NSAID)をレシチン油などのリン脂質含有油またはリン脂質が加えられている生体適合性油に直接加えて、低い胃腸(GI)毒性ならびに炎症、疼痛、発熱、血小板凝集、組織の潰瘍形成および/または他の組織の障害を治療するための高い治療活性を有する、NSAID含有製剤を製造する、新規医薬組成物が提供される。本発明の組成物は、内部、経口、直接または局所投与のための、非水性溶液、ペースト、懸濁液、分散液、コロイド懸濁液の形、または水性乳濁液もしくはマイクロエマルジョンの形である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、生体適合性油および非ステロイド性抗炎症薬(NSAID)を含む独特の組成物であって、油またはその成分がNSAIDのGI(胃腸)毒性を軽減し、かつ炎症、疼痛、発熱および血栓症ならびに卒中、外傷性脳損傷、脊髄損傷、心血管疾患、卵巣癌、大腸癌(colon cancer)、アルツハイマー病、関節炎、ブドウ膜炎、および粘膜炎などの他の疾患を治療する薬物の治療活性を増強する際に有効である組成物に関する。
【0002】
特に、本発明は、内部、経口および/または局所投与することができ、溶液、ペースト、半固体、分散液、懸濁液、コロイドまたはその混合物であってもよい医薬品を形成するために、NSAIDを粉末で、リン脂質を含む生体適合性油に直接混合する製剤に関する。
【背景技術】
【0003】
NSAIDは、発熱、炎症、疼痛、血栓症および発癌を含むいくつかの生体発病過程を阻害する能力を有する化合物群を構成し、その中で最初に発見されたものはアスピリンである1。その高い治療能力のゆえに、NSAIDは世界中の一般大衆の間で、一般大衆薬および処方薬の両方で大量に消費されている。有用性が高いため、慢性関節リウマチおよび骨関節症に苦しむ3000万〜4000万人の米国人、ならびに他の炎症性状態または損傷が原因の炎症および疼痛、月経困難の疼痛;発熱;血栓症および関連する心血管疾患の発症;卵巣癌、大腸癌およびアルツハイマー病を治療/予防するために医薬品を使用している数え切れない他の人々を含む、かなりのパーセンテージの大衆が定期的にNSAIDを消費している1,2。常に増え続けるNSAID使用の傾向に伴う問題、特に高齢者における問題は、これらの薬物が一般に胃腸(GI)副作用を引き起こすということである3~6。
【0004】
胃および小腸で、薬物が消化不良(胃障害、胸やけ、胃内ガス貯留、または悪心)、びらん、胃炎/十二指腸炎および潰瘍を引き起こす者もある。NSAID使用者では胃腸出血も起こることがあり、(様々な重症度の)貧血、または最も重篤な症例では生命を脅かすこともある出血をきたしうる7,8。一つまたは複数のこれらのGI合併症は、定期的NSAID使用者の20〜40%で起こると推定されている。NSAIDの市場が大きいことを考慮すれば、頻度の低いGI合併症でも、年間推定76,000人の米国人を病院送りにし、推定7,600人が死亡している。
【0005】
NSAID作用の理解に役立つ重要な研究の一つは1970年代前半のVaneらの先駆的研究で、NSAIDファミリーの化学的に異なるメンバーが、アラキドン酸からプロスタグランジンG2およびH2への酸化および過酸化の連続的段階による変換を触媒する酵素、シクロオキシゲナーゼ(COX)の活性を阻害する能力を共有することを報告している9~11。プロスタグランジンH2は、特定のプロスタグランジン合成酵素によって触媒される過程により、標的細胞でいくつかのエイコサノイドの一つに変換されることになる。したがって、可逆的または不可逆的にCOX活性を阻害することにより、NSAIDは特定の組織または細胞液からプロスタグランジンを枯渇させると考えられ、これは組織の炎症を促進することが明らかにされている12。これらが判明した直後、Upjohn CompanyのRobertらは特定のプロスタグランジン類がいくつかの潰瘍発生性化合物および/または状態からGI上皮を保護する顕著な性質を共有することを示し、これらの脂質メディエーターの「細胞保護」特性を示した13。これら二つの重要な研究に基づき、NSAIDは粘膜COX活性を阻害し、組織から「細胞保護性」プロスタグランジンを枯渇させることにより、GI上皮の損傷および潰瘍形成を引き起こすと結論づけられた。
【0006】
アラキドン酸代謝についての我々の理解における次の最も最近の発展は1990年代前半に訪れ、当時、何人かの研究者14~18が、最初に記載された酵素(現在ではCOX−1と呼ばれている)に構造および機能的に関連している第二のCOXアイソザイム(現在ではCOX−2と呼ばれている)を同定およびクローニングした。GI粘膜を含むほとんどの組織で構成的に発現されるCOX−1とは対照的に、COX−2は主にサイトカインおよび他の炎症メディエーターによって誘導されることが判明した。これらの知見、およびCOX−2が炎症部位で選択的に発現され、非炎症性GI粘膜では低レベルまたは検出できないレベルでしか発現されないという証拠19~23に基づき、いくつかの製薬会社がCOX−2を選択的に阻害する化合物の開発を開始した。
【0007】
この努力はついに、最初の二つのCOX−2選択的阻害剤、セレブレックス(セレコキシブ)およびビオックス(ロフェコキシブ)の発売に結びついた。これまでに発表されている前臨床および臨床データより、これらの化合物は治療上有効であり、GI粘膜への毒性が低いことが明らかにされている。このニュースは医学界および一般地域社会両方において大きな興奮をもたらし、セレブレックスおよびビオックスが市場に出されて最初の二年間に調合されたこれら薬剤の処方は記録的な数となった24。
【0008】
本発明者およびNSAIDによるGI損傷を研究している他の研究者の幾人かの主な関心は、COX阻害とGI損傷および出血との間のつながりはあまり強くないということである。例えば、Ligumskyらは1980年代前半に、COX阻害を粘膜損傷から切り離したように見えるラットおよびイヌでの一連の論文を発表した25~27。当初、彼らは、アスピリンは組織から「細胞保護性」のプロスタグランジンを枯渇させる一方、その代謝物であるサリチル酸はCOX阻害活性をまったく示さないが、アスピリンとサリチル酸はイヌの胃粘膜への損傷を引き起こす同等の能力を有していることを示した25。続く齧歯類での試験で、アスピリンを皮下または胃内のいずれに投与したかに関わらず、粘膜のCOX活性は>90%阻害されるが、NSAIDを胃内に投与すると、潰瘍はラットの胃でのみ形成されることが判明した26,27。Whittleも、小腸の損傷はインドメタシン投与の48時間後、すなわちCOX活性(活性はインドメタシン投与後<3時間で完全に阻害される)が正常に戻った時点でしか生じ始めなかったため、インドメタシンのCOX阻害を誘導する効果と、小腸の粘膜損傷とは別物であると報告した28。
【0009】
粘膜COXの阻害はNSAIDによる腸疾患の病因に直接関与していないことを示唆する証拠はいくつかの臨床試験によっても支持されていることを指摘しておくべきで、これらはアスピリンをi.v.投与しても、NSAIDの経口投与とは対照的に、ヒト胃粘膜に対して検出できる組織学的損傷を引き起こさないことを報告している29。また、NSAID治療の2〜4週間後、ヒトの胃粘膜は経口アスピリンまたはインドメタシンの有害な作用に対して抵抗性となり、この適応反応はCOX活性の回復とつながっておらず、COX活性は試験期間中完全に阻害されたままであることも報告された30。
【0010】
最後に、NSAIDが主に粘膜COX−1を阻害することによってGI損傷を引き起こすとの仮説から、標的指向遺伝子破壊によりアイソザイムが欠損しているマウスは、自発粘膜潰瘍を発生しやすく、その野生型同腹仔に比べてNSAIDに対する感受性が高いと予測される。Langenbachら31は、COX−1を持たない動物は検出可能なGI疾患を持たず、むしろインドメタシンによる潰瘍発生に対して抵抗性であると報告している。さらに混乱することには、Morhamら32はその後の試験で、COX−2ノックアウトマウスは生存不可能で、腹膜炎ならびに腎疾患で死ぬことが多いことを報告している。COX−2阻害が有害である可能性は、動物を選択的COX−2阻害剤で治療した場合に、近位および遠位の腸の潰瘍治癒が悪化することを示すいくつかの動物試験によっても支持されている33,34。ヒトにおける類似の合併症はこれまでのところ報告されていない。
【発明の概要】
【発明が解決しようとする課題】
【0011】
前述の証拠に基づき、NSAIDがGI粘膜損傷を引き起こす他のメカニズムを研究し、これらの化合物のGI毒性を軽減または予防するための代替戦略開発において、この情報をいかに利用することができるかを調べるための説得力のある主張を行うことができる。NSAIDによる胃腸疾患に対し可能性のある他の標的は、これらの薬物の以下の能力である:粘膜血流量を低下させ、血管壁への白血球の付着を引き起こす能力;酸化的リン酸化を脱共役させる能力;そのプロトノフォア特性により細胞酸性化を引き起こす能力;および粘膜の疎水性、すなわち非湿潤性の特徴を弱め、それにより管腔の酸に対する組織の感受性を増大させる能力35~40。発明者の研究室では過去15〜20年間、この最後の性質に焦点を合わせている。
【0012】
1983年に、発明者の研究室は、イヌの胃粘膜を接触角分析により調べて、これが独特の疎水性表面を有するという初の知見を得た41,42。それ以来、発明者および他の研究室では、胃粘膜のこの非湿潤性表面特性が、齧歯類およびヒトを含む他のいくつかの種で見いだされることを明らかにしてきた40,43,44。さらに、生化学的技法と形態学的技法の両方を用いて、この性質は粘膜ゲル層内および層をコーティングしている界面活性剤様のリン脂質の細胞外ライニングに起因しうることが示された45~47。発明者の研究室でも、NSAIDを含む、胃粘膜に損傷を与える多くの物質が、組織を非湿潤性(疎水性)から湿潤性(親水性)の状態に速やかに転換する能力を有しており、この有害な作用は合成または精製リン脂質の投与によって軽減しうることを見いだした48~51。
【0013】
近年、研究の焦点は、NSAID−リン脂質相互作用のメカニズムにあてられている。これらの試験において、発明者の研究室では、NSAIDが粘膜ゲル層の内部および表面上のホスファチジルコリン(PC)などの双性イオンと化学的に結合することによって粘膜損傷を引き起こすと考えられ、静電結合部位は双性イオンリン脂質であるホスファチジルコリン(PC)の正に荷電したコリンヘッド基と、NSAIDの負に荷電した(カルボキシルまたはスルホニル)基との間であるという、説得力のある証拠を得ている52。この情報に基づき、発明者らのグループはラットで、投与前に合成または精製PCとあらかじめ化学的に結合させたいくつかのNSAIDのGI(胃腸)毒性を評価し、GI損傷形成および出血に関して、これらの新規薬物が非修飾NSAIDに比べてはるかに有害性が低いという証拠を得た。最近行われた予備的二重盲検交差試験において、四日間でヒト被検者に生じる胃損傷を比べると、精製(純度93%)PCを用いてのPC−アスピリンでは非修飾アスピリンよりも胃損傷が有意に少ないことが判明し、このアプローチのヒト疾患への適用性が確認された53。
【0014】
興味深いことに、発明者の研究室では、発熱、炎症/疼痛、血栓症および骨粗鬆症の動物モデルにおいて、PC−NSAIDが非修飾薬物よりもすぐれた治療上の最大効力および用量効力を有することを見いだし、これはその胃への低毒性がバイオアベイラビリティの低下では単純に説明できないことを示している52,54。
【0015】
PC(他の類似のリン脂質)およびNSAIDの組み合わせはNSAID投与の病因効果(pathogeni ceffect, 病原作用、発病作用)を軽減することになるが、これらの組み合わせの経口投与では、NSAID単独に比べて有効用量あたりの必要量が大量となるため、十分とは言えない。したがって、当技術分野において、組成物中の高いNSAID濃度を可能にするNSAIDおよび担体の組成物であって、担体がNSAIDの病因効果を軽減し、経口、内部または局所投与に適した形態である組成物が必要とされている。さらに、当技術分野において、特にアスピリン含有医薬品について、経年変化(self−life)が改善されたNSAID組成物が必要とされている。
【課題を解決するための手段】
【0016】
一般組成物
本発明は、非水性液体担体中に比較的高濃度の非ステロイド性抗炎症薬(NSAID)を含む組成物を提供する。
【0017】
本発明は、非水性液体担体中のNSAID組成物であって、担体が生体適合性油およびリン脂質を含む組成物を提供する。
【0018】
本発明は、非水性液体担体中のNSAID組成物であって、担体がリン脂質に富む生体適合性油を含む組成物を提供する。
【0019】
本発明は、非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、ヒトを含む動物において、担体またはその成分がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高める作用をする組成物も提供する。
【0020】
本発明は、非水性液体担体中に比較的高濃度のNSAID、リン脂質を含む組成物であって、ヒトを含む動物において、リン脂質がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在する組成物も提供する。
【0021】
本発明は、リン脂質および生体適合性油を含む非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、ヒトを含む動物において、リン脂質がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在する組成物も提供する。
【0022】
リン脂質の存在はNSAIDの一般病因性および/または毒性も軽減する。したがって、リン脂質はアセトアミノフェンの投与による肝損傷ならびに/またはイブプロフェンもしくはCOX−2阻害剤などの他のNSAIDの投与による腎および/もしくは心血管副作用を軽減ならびに/または予防する。
【0023】
一般組成物の一般製造法
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0024】
本発明は、リン脂質を含む非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合してリン脂質−NSAID結合複合体を含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0025】
本発明は、ホスファチジルコリン含有生体適合性油を含む非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合してホスファチジルコリン−NSAID結合複合体を含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0026】
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して、担体がリン脂質含有生体適合性油もしくは生体適合性油およびリン脂質またはその混合物を含む、溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0027】
乳化組成物
本発明は、非水性担体を含む組成物の水性乳濁液であって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもある組成物も提供する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、乳化組成物の粒径は、有害作用を引き起こすことなく組成物の経口服用または組織もしくは臓器部位への注射を可能にするのに十分小さい。静脈内または動脈内注射剤形のためには、マイクロエマルジョンが好ましく、平均粒径は0.5から約10μmの間まで下げることができ、好ましくは約1から5μmの間である。
【0028】
本発明は、非水性担体を含む組成物の水性マイクロエマルジョンであって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもあるマイクロエマルジョンも提供する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。
【0029】
乳化組成物の製造法
本発明は、本発明の水性乳濁液の製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌する段階を含み、乳化剤は、存在する場合には、安定な乳濁液を生成するのに十分な量で存在する方法も提供する。
【0030】
本発明は、本発明の水性マイクロエマルジョンの製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌し、乳濁液をマイクロエマルジョン生成(microemulsifying)条件下で剪断してマイクロエマルジョンを生成する段階を含み、乳化剤は、存在する場合には、安定なマイクロエマルジョンを生成するのに十分な量で存在する方法も提供する。
【0031】
乳化剤が存在しても存在しなくてもよい理由は、リン脂質自体がある程度の乳化性を有しているからである。
【0032】
炎症を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、組織の炎症を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシット(illicit)するのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い組成物も提供する。
【0033】
本発明は、組織、臓器および/または切開部の炎症ならびに他の結果を軽減するための術後治療であって、組成物が治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含むか、または組成物がその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含み、組成物がリン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い治療も提供する。当然のことながら、組成物は軟膏、噴霧剤、拭き取り布など(wipe)にコーティングしたもの、生体分解性の基板にコーティングしたものなどであってもよい。
【0034】
血小板凝集を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体、またはその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、血小板凝集を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で血小板凝集を軽減し、粘膜毒性および/または刺激が低い組成物も提供する。
【0035】
発熱状態を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体、またはその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、抗発熱活性のための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で抗発熱活性を有し、粘膜毒性および/または刺激が低い組成物も提供する。
【0036】
潰瘍化組織を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液もしくはマイクロエマルジョン、またはリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む非水性includingを含む潰瘍化組織を治療するための組成物であって、リン脂質は組織の潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、組織の潰瘍化領域の炎症を軽減する組成物も提供する。
【0037】
口の潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む含嗽剤であって、リン脂質は口の潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口の潰瘍化領域の炎症を軽減し、リン脂質の量は口の潰瘍形成を軽減するのに十分であるのみならず、NSAIDによる組織損傷を軽減または提示するのにも十分である組成物も提供する。
【0038】
口、食道およびGI管潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む飲用医薬品であって、リン脂質は口、食道、および/またはGI管潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口、食道、および/またはGI管の潰瘍化領域の炎症を軽減し、リン脂質の量は口、食道、および/またはGI管潰瘍形成を軽減するのに十分であるのみならず、NSAIDが存在する場合にはそれによる組織損傷を軽減するのにも十分である医薬品も提供する。
【0039】
眼の炎症を治療するための組成物
本発明は、水性溶液中にリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む点眼剤であって、リン脂質は眼の炎症および/または潰瘍形成もしくは刺激を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、眼の強膜、ブドウ膜、水晶体または脈絡網膜領域の炎症を軽減し、リン脂質の量は眼の炎症を軽減するのに十分であるのみならず、NSAIDによる組織損傷を軽減または提示するのにも十分である薬剤も提供する。
【0040】
潰瘍化組織の治療法
本発明は、本発明の乳濁液またはマイクロエマルジョンの投与による、口、食道、GI管、および/または眼の炎症および/または潰瘍形成障害の治療法も提供する。
【0041】
中枢および/または末梢神経系の外傷を治療するための組成物
本発明は、リン脂質および治療上有効な量のNSAIDを含む非水性担体またはリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む非水性封入物(including)を含む、脊髄損傷、卒中および/または外傷性脳損傷を経口または内部治療するための組成物であって、リン脂質が血液脳関門を通過しての、または中枢神経系(CNS)もしくは末梢神経系(PNS)内へのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、NSAIDが炎症、血小板凝集、疼痛(侵害受容)感覚、細胞死および/または炎症によるアポトーシスを軽減する組成物も提供する。
【0042】
中枢および/または末梢神経系の外傷の治療法
本発明は、本発明の組成物の経口投与および/または注射による直接投与によって、脊髄損傷、卒中および/または外傷性脳損傷を治療する方法であって、直接投与は静脈内(i.v.投与)、動脈内(i.a.投与)または外傷部位に直接(直接投与)のいずれであってもよく、i.v.およびi.a.投与については、リン脂質が血液脳関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、すべての投与様式において、リン脂質がNSAIDの病因効果を軽減する方法も提供する。
【0043】
本発明は、リン脂質を含む油性担体中に比較的高濃度のNSAIDを含む水性乳濁液またはマイクロエマルジョンである、脊髄損傷(例えば、慢性疼痛症候群)、卒中および/または外傷性脳損傷の症状を改善するための医薬品であって、NSAIDおよびリン脂質が医薬品中で結合複合体を形成し、組成物が外傷組織上で外傷組織の腫脹を軽減するのに十分な濃度のNSAIDおよびNSAIDの病因効果を軽減するのに十分な濃度のリン脂質を含む医薬品も提供する。
【0044】
アルツハイマー病を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、アルツハイマー病に関連する症状を予防、治療または改善するための組成物であって、NSAIDおよびリン脂質がアルツハイマー病の症状の発現を予防するか、またはアルツハイマー病の症状を改善する作用をする組成物も提供する。
【0045】
アルツハイマー病の治療法
本発明は、アルツハイマー病に関連する症状の予防、治療または改善法であって、治療プロトコルに従い経口および/または内部に本発明の組成物を経口または内部投与する段階を含む方法も提供する。
【0046】
本発明は、同様の要素は同じ番号が付されている添付の例示的図面と共に、下記の詳細な説明を参照すればより十分に理解することができる。
【0047】
語句の定義
下記の語句は以下に示す意味を有し、これらの意味はその一般的に受け入れられている意味に対応しても、しなくてもよい。
【0048】
「NSAID」なる語句は、非ステロイド性抗炎症薬として一般に分類される薬物のいかなる変種も意味し、イブプロフェン、ピロキシカム、サリチル酸塩、アスピリン、ナプロキセン、インドメタシン、ジクロフェナク、アセトアミノフェン、COX2阻害剤またはそのいかなる混合物も含まれるが、これらに限定されることはない。
【0049】
「本質的に含まない」なる語句は、所与の成分を生物学的に不活性および/または活性でない量で含む組成物を意味し、好ましくは成分は約0.10重量%未満の所与の成分量で存在し、特に約0.01重量%未満が好ましい。
【0050】
「比較的高濃度」なる語句は、NSAIDの担体に対する重量比が約10:1から約1:10であることを意味する。好ましくは、NSAIDの担体に対する重量比は約5:1から約1:5、特に約2:1から1:2、とりわけ約2:1から1:1である。
【0051】
双性イオンリン脂質なる語句は広範なリン脂質を意味し、ホスファチジルコリン、ホスファチジルセリン、ホスファチジルエタノールアミン、スフィンゴミエリンおよび他のセラミド、ならびに様々な他の双性イオンリン脂質が含まれるが、これらに限定されることはない。
【0052】
「生体適合性油」なる語句は、動物の消費またはFDAによってヒトの消費が承認されている、いかなる油も意味する。
【0053】
「内部投与」または「内部投与した」なる語句は、最初に消化管を通過することなく、組成物を血流、組織部位、臓器などに直接提供する、いかなる技法による投与も意味する。
【0054】
「経口投与」または「経口投与した」なる語句は、口からの投与を意味する。
【0055】
「局所投与」または「局所投与した」なる語句は、皮膚、粘膜ゲル層、眼、組織および/または外科処置中に曝露される臓器、もしくは任意の他の曝露される体組織などの表面上への投与を意味する。
【0056】
「結合複合体」なる語句は、NSAIDおよび双性イオンリン脂質の間の相互作用などの、NSAIDおよびリン脂質の間の非共有結合による化学的および/または物理的相互作用を意味する。
【0057】
「双性イオン(zwitter ion)」なる語句は、生理的pHで分子が正に荷電した官能基および負に荷電した官能基の両方を含むことを意味する。
【0058】
「陰イオン性リン脂質」なる語句は、生理的pHで全体に負の電荷を有するリン脂質を意味する。
【0059】
「中性リン脂質」なる語句は、非荷電脂質を意味する。
【0060】
「乳濁液」なる語句は、一つの不混和相のもう一つの不混和相中の懸濁液であって、第二相中の第一相の小滴の形での懸濁液を意味する。本明細書において用いられる乳濁液なる語句は、速やかに分離するか、または全く分離しない懸濁液を含み、したがって、安定および不安定乳濁液を含む。
【0061】
「安定乳濁液」なる語句は、調整後少なくとも1日は分離せず、好ましくは少なくとも1週間は分離せず、特に少なくとも1ヶ月後まで分離せず、とりわけ無期限に乳濁液のままである、水中油混合物を意味する。
【0062】
「安定マイクロエマルジョン」なる語句は、調整後少なくとも1日は分離せず、好ましくは少なくとも1週間は分離せず、特に少なくとも1ヶ月後まで分離せず、とりわけ無期限に乳濁液のままである、水中油混合物を意味する。
【0063】
「比較的疎水性の関門」なる語句は、疎水性を有し、関門を通過しての親水性試薬の輸送を一般に阻止するか、または低減する、いかなる外部、内部、細胞性または細胞下関門も意味する。そのような関門には、親水性物質よりも疎水性物質をより容易にそれを通して輸送する、ヒトを含む動物の粘膜ゲル層、原形質膜(細胞膜)、血液脳関門、またはいかなる他の関門も含まれるが、これらに限定されることはない。
【発明を実施するための最良の形態】
【0064】
発明者は、リン脂質および場合によりNSAIDを含む非水性液体生体適合性担体を含む独特の医薬製剤を製造して、粘膜組織潰瘍化の修復を改善し、かつ/またはNSAID投与の病因効果を軽減しうることを見いだした。NSAIDが存在する場合には、NSAIDの担体に対する重量比は一般に約10:1から約1:10であり、これはNSAIDとしては低いGI毒性および高い治療活性の予想外の性質を有する、担体中に高度に濃縮されたNSAID混合物となる。好ましくは、NSAIDの担体に対する重量比は約5:1から約1:5、特に約2:1から1:2、とりわけ約2:1から1:1である。
【0065】
NSAIDを含む組成物について、本発明を縮小してNSAIDによる潰瘍疾患、および後足の急性炎症の齧歯類モデルで実施した。製剤は溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物の形であってもよい。
【0066】
NSAIDを含まない組成物について、リン脂質自体が組織の潰瘍形成、特に放射線療法および/または化学療法が原因の組織潰瘍形成の予防および/または軽減において治療上有益な効果を付与することができる。
【0067】
非水性液体生体適合性担体は生体適合性油または生体適合性油もしくは油様物質の混合物を含む。生体適合性油または油混合物はリン脂質を元々含んでいてもよく、またはそれにリン脂質が加えられていてもよい。元々または担体への添加により存在するリン脂質の量は、組織の潰瘍形成を予防、軽減または治療するために十分であるか、あるいは製剤がNSAIDを含む場合には、GI潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管疾患などのNSAIDの病因効果、ならびに/または高血圧、アテローム性動脈硬化症、血栓症、狭心症、卒中および心筋梗塞などの副作用を軽減するのに十分である。
【0068】
発明者は、前述の組成物の水性乳濁液またはマイクロエマルジョンを生成して、様々な型の癌の放射線療法および/または化学療法の結果生じる、またはこれらが原因の口、食道およびGI潰瘍形成を治療しうることも見いだした。乳濁液またはマイクロエマルジョンは、放射線療法および/もしくは化学療法の後、最中、前に投与してもよく、またはその前、最中、および/もしくは後の投与を含む混合プロトコルで投与してもよい。
【0069】
発明者および他の研究者らによる以前の出版物および特許において、リン脂質およびNSAIDを含む組成物は、成分をメタノール、エタノールもしくはクロロホルムなどの有機溶媒にまず溶解し、蒸留もしくは蒸発によって溶媒を除去することにより生成するか、またはNSAIDをリン脂質を加えた水性溶液に溶解した後、凍結乾燥した。これらの工程は二つの成分を化学的に相互作用させて複合体を形成させる。これらの工程は、ジパルミトイルホスファチジルコリン(DPPC)などの合成的に製造されたリン脂質として、または精製もしくは半精製化合物としてホスファチジルコリンを用いることが最も多かった。
【0070】
本発明は、概括的には、リン脂質および場合によりNSAIDを含む非水性液体生体適合性担体を含む医薬製剤または組成物であって、リン脂質は組織の潰瘍形成および/または炎症を予防、軽減または治療するために十分な量であり、NSAIDが存在する場合には、リン脂質はNSAIDの病因効果を軽減することができる量で存在する医薬製剤または組成物に関する。製剤は一般に、粘稠溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物であり、経口投与、直接投与、内部投与または局所投与することができる。
【0071】
本発明は、概括的には、リン脂質およびNSAIDを含む非水性液体生体適合性担体を含む医薬製剤または組成物であって、リン脂質は組織の潰瘍形成を予防、軽減または治療し、NSAIDのGI毒性を軽減するために十分な量である医薬製剤または組成物に関する。非水性液体を使用すると、NSAIDの有効治療量の体積を下げるための高濃度のNSAIDを有する組成物を生成することができる。製剤は一般に、粘稠溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物であり、経口投与、内部投与または局所投与することができる。
【0072】
本発明は、概括的には、NSAIDの病因効果が軽減された高濃縮NSAID組成物を生成するための医薬製剤の製造法であって、固体NSAIDを非水性担体と混合する段階を含み、担体がリン脂質含有生体適合性油もしくは生体適合性油およびリン脂質、またはその混合物を含む方法にも関する。
【0073】
本発明は、概括的には、NSAIDおよびリン脂質を含む非水性液体担体を含む医薬製剤の有効量を投与することによる炎症、疼痛または他のNSAIDで治療可能な病態の治療法であって、リン脂質はNSAIDによる病態を軽減するのに十分な量で存在し、NSAIDは治療上有効な量で存在し、リン脂質−NSAIDの組み合わせによって一用量あたりのNSAID投与量をリン脂質非存在下で同じ治療効果をイリシットするNSAIDの等価の量よりも少なくすることができる方法にも関する。
【0074】
本発明は、概括的には、非水性担体中にリン脂質および場合によりNSAIDを含む医薬製剤の有効量を投与することにより潰瘍化組織を予防、軽減および/もしくは治療する、ならびに/あるいは炎症、疼痛、または組織の炎症および/もしくは潰瘍形成に関連するNSAIDで治療可能な他の病態を軽減する方法であって、担体が生体適合性油またはその混合物である方法にも関する。
【0075】
特に、発明者は、リン脂質を元々含む非精製レシチン油などのリン脂質を含む生体適合性油を含む独特の医薬製剤であって、得られる製剤が低いGI毒性および高い治療活性の予想外の性質を有する、溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液もしくはその混合物または組成物である医薬製剤を見いだした。
【0076】
組成物は生体適合性油とリン脂質および場合によりNSAIDとを混合することによって容易に製造され、ここでNSAIDを粉末で粗製または半粗製レシチン油に直接加えて、経口投与のためにゼラチン軟もしくは硬カプセルまたはカリフォルニア州のVitaHerb Nutraceuticals of Placentiaから入手できるベジキャップ(vegicap)に加えるか、内部投与のために注射するか、あるいは局所投与のために皮膚に塗布することができる、ペースト、半固体、分散液もしくはコロイド懸濁液または同様の組成物を生成する。予想外の知見は、図1、2および15に示すとおり、この単純な製剤は前述の通常の方法で製造されるPC−NSAIDと同様、NSAIDによる潰瘍疾患の齧歯類モデルにおいて著しく低い胃腸(GI)毒性を有し、図3および4に示すとおり、足の炎症の急性モデルにおいて、ならびに図7および8に示すとおり、脊髄損傷の慢性モデルにおいて、炎症/疼痛を治療する高い治療活性も有することであった。
【0077】
一般に、NSAIDのリン脂質含有油に対する重量比は約10:1から約1:10、好ましくは約4:1から約1:4、特に約2:1から約1:2、とりわけ約2:1から約1:1の範囲である。リン脂質を元々含む、本発明の実施において有用な油について、油は一般に約10から約15重量%のリン脂質、好ましくは約10重量%から約20重量%のリン脂質、特に約10重量%から約40重量%のリン脂質を含む。しかし、これよりも多い量および少ない量のリン脂質も同様に用いることができる。しかし、約10重量%よりもはるかに少ない重量%では、有効治療量またはいかなる添加NSAIDとも結合するために十分な量が問題となり、その一方で約40重量%よりも高い重量%では、レシチン油などのリン脂質を元々含む生体適合性油に精製リン脂質を加えなければならないこともある。少量のリン脂質(約10重量%未満)を含む生体適合性油に対しては、油にリン脂質を加える。そのような油−リン脂質の組み合わせは、約90重量%という高いリン脂質濃度で製造することができる。しかし、好ましい組み合わせは約10重量%から約90重量%の間、特に約20重量%から約80重量%の間、特に約20重量%から約60重量%の間、とりわけ約20重量%から約40重量%の間の量のリン脂質を含む。
【0078】
一般に、一般用途のための本発明の組成物を含むNSAIDの用量は、一用量あたり5mgから一用量あたり500mgの範囲である。当然のことながら、これよりも低用量および高用量の製剤を製造することもできる。しかし、この用量範囲は市販のNSAIDで典型的に見られる範囲を含む。好ましくは、NSAIDの用量範囲は一用量あたり約10mgから約325mg、特に一用量あたり約25mgから約200mg、とりわけ一用量あたり約50mgから約100mgである。各NSAIDは錠剤または類似のもの一つあたり異なる用量範囲を有し、これらの範囲は、患者がその語句が本明細書において用いられるとおりのNSAIDを含む製剤を用いる場合に、一般に投与されることになるすべての範囲を含むことを意味することが理解されるべきである。また、本発明の組成物はNSAIDを単に含むのではなく、リン脂質を含む非水性液体担体中にNSAIDを含み、リン脂質の量は、NSAIDが存在する場合には、NSAIDの治療効果を増強する一方で、NSAIDの病因効果を軽減するのに十分であることも理解されるべきである。これらのNSAIDの病因効果は、当然のことながら、NSAIDに特有であるが、潰瘍形成、出血などのGI損傷(全てではないにしてもほとんどのNSAID)、肝損傷(例えばアセトアミノフェン)、腎損傷(例えばイブプロフェン、アセトアミノフェン、COX−2阻害剤)、心損傷(例えばCOX−2阻害剤)などが含まれ、これらに限定されることはない。NSAID−リン脂質結合複合体のため、所与の治療効果または反応(熱の低下、炎症の軽減、血小板凝集の軽減など)をイリシットするのに必要とされるNSAIDの用量(mg)が低下する。低下は用量(mg)の1倍低下から用量(mg)の15分の1への低下の範囲でありうる。好ましくは、範囲はNSAID用量(mg)の約1倍低下から約10分の1への低下である。リン脂質−NSAIDの組み合わせを含む組成物によって増大した生物活性は、NSAIDの毒性の同等の増大をきたすことはないが、驚くべきことに、本明細書に示すデータが証明するとおり、NSAIDの毒性の低下を引き起こす。
【0079】
慢性関節リウマチ、アルツハイマー病、CNSおよびPNS外傷などのより重度の状態、またはNSAIDおよび/またはリン脂質で治療可能な他のより重度の状態に対しては、NSAIDの必要一日用量は一般にはるかに高い。典型的には、一日用量は一日あたり約100mgから約5000mg、好ましくは一日あたり約500mgから約3000mg、特に一日あたり約750mgから約3000mg、とりわけ一日あたり約1000mgから約3000mgの範囲である。さらに、リン脂質−NSAIDの組み合わせの有効性が高まったことにより、NSAIDの病因性または毒性が同時に高まることなく、より高い治療効果をイリシットすることが可能となる。当然のことながら、NSAIDのこの生体活性の増大によって、投与するNSAIDの用量を引き下げることが可能となる。
【0080】
一般的な経験により、NSAID活性を増強するのに十分な量のリン脂質を含む一方で、NSAID毒性を軽減する、非水性担体、生体適合性油にNSAIDが溶解、分散、懸濁または他の方法により混合されている、本発明の製剤でNSAIDを投与する場合、必要投与量は患者、特定の状態および他の要因に応じて、その状態を治療するのに必要とされる推奨用量の5%という低い量からその用量の100%まででありうる。好ましくは、投与量は特定の状態を治療するのに必要とされる推奨用量の約10%から約90%、特に特定の状態を治療するのに必要とされる推奨用量の約10%から約50%である。所与の状態のための所与のNSAIDに対する必要推奨投与量は、Physicians Desk Reference (PDR)、AMAの出版物、FDAの出版物などの出版物中に見いだされ、これらは十分に確立された基準である。
【0081】
本発明の組成物は以下も含むことができる:(1)医薬品として許容される量のビタミンA、ビタミンC、ビタミンEまたはFDAによってヒトおよび動物の消費が承認されている他の抗酸化剤、ならびにその混合物または組み合わせからなる群より選択される抗酸化剤;(2)医薬品として許容される量の銅、亜鉛、金、アルミニウムおよびカルシウムならびにその混合物または組み合わせからなる群より選択される多価陽イオン;(3)医薬品として許容される量のジメチルスルホキシド/DMSO、プロピレングリコール/PPG、および中鎖トリグリセリド/MCTならびにその混合物または組み合わせからなる群より選択される流動性、拡散性、または浸透性促進剤;(4)医薬品として許容される量の食品着色料または非毒性色素;(5)医薬品として許容される量の香味増強剤;(6)賦形剤;および/または(7)補助剤。
【0082】
一般組成物
本発明は、非水性液体担体中に比較的高濃度の非ステロイド性抗炎症薬(NSAID)を含む組成物に関する。好ましくは、担体は生体適合性油およびリン脂質またはリン脂質に富む生体適合性油を含む。担体は元々および/または添加により、ヒトの体を含む動物の体内で、NSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量のリン脂質を含む。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0083】
本発明は、リン脂質および生体適合性油を含む非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、リン脂質がヒトの体を含む動物の体内で、NSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在し、組成物用量がNSAIDおよび/またはリン脂質の治療上有効な量を送達するのに十分であり、NSAIDの量がリン脂質非存在下で同等の治療効果をイリシットするのに必要とされるNSAIDの量の1〜10分の1である組成物に関する。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0084】
本発明の組成物中のリン脂質の存在はNSAIDの一般および特定の病因性および/または毒性も軽減する。したがって、リン脂質はアセトアミノフェンの投与による肝損傷ならびに/またはイブプロフェンもしくはCOX−2阻害剤などの他のNSAIDの投与による腎および/もしくは心損傷を軽減ならびに/または予防する。
【0085】
一般組成物の一般製造法
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して、比較的高濃度のNSAIDを含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法にも関する。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0086】
乳化組成物
本発明は、非水性担体を含む組成物の水性乳濁液であって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもある組成物にも関する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる乳濁液は比較的高濃度のリン脂質−NSAID結合複合体を有する組成物を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0087】
本発明は、非水性担体を含む組成物の水性マイクロエマルジョンであって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもあるマイクロエマルジョンにも関する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。水性マイクロエマルジョンは、組成物を長期間マイクロエマルジョンの状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる乳濁液は比較的高濃度のリン脂質−NSAID結合複合体を有する組成物を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0088】
乳化組成物の製造法
本発明は、本発明の水性乳濁液の製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌する段階を含み、乳化剤は、存在する場合には、安定な乳濁液を生成するのに十分な量で存在する方法にも関する。
【0089】
本発明は、本発明の水性マイクロエマルジョンの製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌し、乳濁液をマイクロエマルジョン生成(microemulsifying)条件下で剪断してマイクロエマルジョンを生成する段階を含み、乳化剤は、存在する場合には、安定なマイクロエマルジョンを生成するのに十分な量で存在する方法にも関する。
【0090】
乳化剤が存在しても存在しなくてもよい理由は、リン脂質自体がある程度の乳化性を有しているからである。
【0091】
炎症を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、組織の炎症を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い組成物にも関する。
【0092】
血小板凝集を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、血小板凝集を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で血小板凝集を軽減し、粘膜毒性および/または刺激が低い組成物にも関する。
【0093】
発熱状態を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、抗発熱活性のための組成物であって、リン脂質非存在下で同等の治療反応をillicit(イリシット)するのに典型的に必要とされる用量よりも低いNSAID用量で抗発熱活性を有し、粘膜毒性および/または刺激が低い組成物にも関する。
【0094】
潰瘍化および/または炎症組織を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む潰瘍化組織を治療するための組成物であって、リン脂質は組織の炎症および/または潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、組織の患部領域の炎症を軽減する組成物にも関する。
【0095】
口の潰瘍形成および/または炎症を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む含嗽剤であって、リン脂質は口潰瘍形成および/または炎症を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口の患部領域の炎症を軽減する組成物にも関する。
【0096】
口、食道およびGI管潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む飲用医薬品であって、リン脂質は口、食道、および/またはGI管の炎症および/または潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口、食道、および/またはGI路の患部領域の炎症を軽減する医薬品にも関する。
【0097】
眼の炎症を治療するための組成物
本発明は、水性溶液中にリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む点眼剤であって、リン脂質は眼の炎症または刺激を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、ブドウ膜炎または関連眼障害に関連する眼の炎症を軽減する薬剤にも関する。
【0098】
潰瘍化および/または炎症組織の治療法
本発明は、本発明の乳濁液またはマイクロエマルジョンの投与による、口、食道、GI管、眼および/または他の炎症および/または潰瘍化組織部位の炎症性および/または潰瘍性障害の治療法にも関する。
【0099】
中枢および/または末梢神経系の外傷を治療するための組成物
本発明は、リン脂質および治療上有効な量のNSAIDを含む非水性担体またはその中にリン脂質および治療上有効な量のNSAIDを含む非水性担体が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、脊髄損傷、卒中および/または外傷性脳損傷を経口または内部治療するための組成物であって、リン脂質が血液脳関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、NSAIDが炎症、血小板凝集、抗発熱活性および/または炎症による細胞死を軽減する組成物にも関する。
【0100】
中枢および/または末梢神経系の外傷の治療法
本発明は、本発明の組成物を静脈内(i.v.投与)、動脈内(i.a.投与)または外傷部位に直接(直接投与)注射することによって、脊髄損傷、卒中および/または外傷性脳損傷を治療する方法であって、i.v.およびi.a.投与については、リン脂質が血液脳関門または他の神経性関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、すべての投与様式において、リン脂質がNSAIDの病因効果を軽減する方法にも関する。
【0101】
本発明は、リン脂質を含む油性または水性担体中に比較的高濃度のNSAIDを含む、脊髄損傷、卒中および/または外傷性脳損傷の症状を改善するための医薬品であって、NSAIDおよびリン脂質が医薬品中で結合複合体を形成し、組成物が外傷組織上で外傷組織の腫脹を軽減するのに十分な濃度のNSAIDおよびNSAIDの病因効果を軽減するのに十分な濃度のリン脂質を含む医薬品にも関する。
【0102】
アルツハイマー病を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、アルツハイマー病に関連する症状を予防、治療または改善するための組成物であって、NSAIDおよびリン脂質がアルツハイマー病の症状の発現を予防するか、またはアルツハイマー病の症状を改善する作用をする組成物にも関する。
【0103】
アルツハイマー病の治療法
本発明は、アルツハイマー病に関連する症状の予防、治療または改善法であって、治療プロトコルに従い経口および/または内部に本発明の組成物を経口または内部投与する段階を含む方法にも関する。
【0104】
切開部を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、切開の結果生じる手術による局所炎症を軽減し、治癒を促進するための、切開部を治療するための組成物であって、NSAIDおよびリン脂質が炎症および関連症状を軽減し、治癒を促進する作用をする組成物にも関する。
【0105】
切開部の治療法
本発明は、切開の結果生じる手術による局所炎症を軽減し、治癒を促進するための、切開部の治療法であって、手術中および手術後であるが、縫合前に、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む組成物を手術部位に適用することを含み、NSAIDおよびリン脂質が炎症および関連症状を軽減し、治癒を促進する作用をする方法にも関する。本発明の好ましい治療製剤には、乳濁液もしくはマイクロエマルジョンまたは本発明の組成物の類似の製剤の噴霧適用が含まれる。
【0106】
放射線療法および/または化学療法が原因の潰瘍形成および/または炎症を改善するための組成物
本発明は、生体適合性油、リン脂質および場合により治療上有効な量のNSAIDを含む、粘膜炎または関連状態などの、特定の癌の放射線療法および/または化学療法によって引き起こされる組織の潰瘍形成を改善するための組成物であって、リン脂質は粘膜炎に関連する潰瘍形成または炎症を予防および/または軽減するのに十分な量で存在し、NSAIDが存在する場合には、リン脂質は潰瘍形成または炎症を予防および/または軽減するのみならず、NSAIDが確実に状態をさらに悪化させないようにするのにも十分な量で存在する組成物にも関する。好ましくは、化学療法に対しては、化学療法剤を適当に製剤した本発明の組成物と共に投与する。したがって、化学療法剤を経口投与する場合、化学療法剤と本発明の組成物の成分との間に有害相互作用が起こらないならば、化学療法剤を適当に製剤した本発明の組成物と混合して、患者に投与してもよい。化学療法剤と本発明の組成物の成分との間に有害相互作用が起こる場合、または化学療法剤を注射によって投与する場合、本発明の組成物を化学療法剤と共に、および十分な時間の後に経口投与し、粘膜炎発現を予防または期間短縮する。
【0107】
放射線療法および/または化学療法が原因の潰瘍形成および/または炎症の改善法
本発明は、放射線療法および/または化学療法などの医学的治療によって引き起こされる粘膜炎または他の潰瘍形成状態を予防および/または治療する方法であって、生体適合性油、リン脂質および場合により治療上有効な量のNSAIDを含み、リン脂質は粘膜炎に関連する潰瘍形成および/または炎症を予防および/または軽減するのに十分な量で存在し、NSAIDが存在する場合には、リン脂質は潰瘍形成を予防および/または軽減するのみならず、NSAIDが確実に状態をさらに悪化させないようにするのにも十分な量で存在する、本発明の組成物の治療上有効な量を、放射線療法または化学療法の前、同時、および/または後に体の患部に投与する段階を含む方法にも関する。好ましくは、組成物は経口投与用に設計され、放射線療法および/または化学療法の前および同時に投与して、粘膜炎発現を予防および/または治療および/または期間短縮する。
【0108】
本発明の組成物の経口投与のために、組成物を好ましくは乳濁液、マイクロエマルジョンなどの形で水性溶液中に小滴として分散する。小滴は、乳化剤、懸濁化剤、および含嗽剤などで一般に見られる他の成分を含んでいてもよい。本発明の組成物は、米国特許第5,407,663号、第5,236,699号、第5,130,146号、第5,085,850号に記載の製剤を含む含嗽剤または口腔衛生製剤と共に用いることもでき、前述の開示は参照として本明細書に組み入れられる。本発明の組成物は、ペースト、ロゼンジ、または経口投与用に一般に用いられるいかなる他の様式で経口投与することもできる。当然のことながら、組成物はカプセル、ゲルカプセルなどに含まれていてもよい。
【0109】
局所投与のために、本発明の組成物は、軟膏、ペースト、油、乳濁液、マイクロエマルジョン、またはその混合物もしくは組み合わせの形であってもよい。さらに、組成物は、軟膏および美容産業で一般に用いられる他の成分と混合することもできる。
【0110】
乳濁液
本発明の組成物は、乳濁液として製造および製剤することができる。乳濁液は典型的には一つの液体のもう一つの液体中の、通常は直径0.1μmを越える液滴の形での不均質系である。(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245; Block in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第2巻, p. 335; Higuchiら, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 301)。乳濁液は、十分に混合し、互いに分散した二つの不混和性の液相からなる二相系であることが多い。一般に、乳濁液は油中水(w/o)または水中油(o/w)のいずれかでありうる。水相が細かく分割され、微小液滴として大量の油相中に分散されている場合、得られる組成物は油中水(w/o)乳濁液と呼ばれる。あるいは、油相が細かく分割され、微小液滴として大量の水相中に分散されている場合、得られる組成物は水中油(o/w)乳濁液と呼ばれる。乳濁液は分散相および水相、油相いずれかの溶液として、またはそれ自体分離相として存在しうる活性薬物に加えて、追加の成分を含んでいてもよい。乳化剤、安定化剤、色素、および抗酸化剤などの医薬品賦形剤も、必要に応じて乳濁液中に存在してもよい。医薬品乳濁液は、例えば油中水中油(o/w/o)および水中油中水(w/o/w)乳濁液の場合などの、三相以上からなる多相乳剤であってもよい。そのような複雑な製剤は、単純な二成分乳濁液にはない特定の利点を提供することが多い。o/w乳濁液の個々の油滴が小さい水滴を封入している多相乳濁液はw/o/w乳濁液を構成する。同様に、油性連続相中に安定化された水滴中に封入された油滴系は、o/w/o乳濁液を提供する。
【0111】
乳濁液は熱力学的安定性が非常に小さいか、またはないことによって特徴付けられる。しばしば、乳濁液の分散または不連続相が外側または連続相に十分に分散し、乳化剤または製剤の粘性によりこの形に維持される。乳濁液型の軟膏基剤およびクリームの場合のように、乳濁液の相のいずれかは半固体または固体であってもよい。乳濁液を安定化する他の手段は、乳濁液のいずれかの相に取り込むことができる乳化剤の使用を必要とする。乳化剤は次の四つの範疇に広く分類される:合成界面活性剤、天然乳化剤、吸収基剤、および細かく分散された固体(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。
【0112】
合成界面活性剤は表面活性剤としても知られ、乳濁液の製剤において広範に適用されており、文献に総説が掲載されている(Rieger, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 285 ; Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), Marcel Dekker, Inc., New York, N. Y., 1988, 第1巻, p. 199)。界面活性剤は典型的には両親媒性で、親水性および疎水性部分を含む。界面活性剤の親水性と疎水性との比は親水性−親油性バランスと呼ばれ、製剤を製造する際に界面活性剤を分類および選択するための有用な道具である。界面活性剤は親油性基の性質に基づいて次の異なるクラスに分類することができる:非イオン性、陰イオン性、陽イオン性および両性(Rieger, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 285)。
【0113】
乳濁液製剤において用いられる天然乳化剤には、ラノリン、蜜蝋、ホスファチド、レシチンおよびアラビアゴムが含まれる。吸収基剤は、無水ラノリンおよび親水性ワセリンなどの、水を吸収してw/o乳濁液を形成するが、その半固体の稠性を保持するような親水性を有する。細かく分割された固体も、特に複数の界面活性剤の組み合わせおよび粘稠製剤において、良好な乳化剤として用いられている。これらには、重金属水酸化物などの極性無機固体、ベントナイト、アタパルジャイト、ヘクトライト、カオリン、モンモリロナイト、コロイド状ケイ酸アルミニウムおよびコロイド状ケイ酸アルミニウムマグネシウムなどの非膨潤性クレー、色素ならびに炭素またはトリステアリン酸グリセリルなどの非極性固体が含まれる。
【0114】
多様な非乳化物質も乳濁液製剤に含まれ、乳濁液の性質に寄与する。これらには、脂肪、油、ワックス、脂肪酸、脂肪アルコール、脂肪酸エステル、湿潤剤、親水性コロイド、保存剤および抗酸化剤が含まれる(Block, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 335 ; Idson, in Pharmaceutical
Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York,
N. Y., 第1巻, p. 199)。
【0115】
親水性コロイドまたはハイドロコロイドには、天然ゴムおよび多糖などの合成ポリマー(例えば、アラビアゴム、寒天、アルギン酸、カラゲナン、グアーゴム、カラヤゴム、およびトラガカント)、セルロース誘導体(例えば、カルボキシメチルセルロースおよびカルボキシプロピルセルロース)、ならびに合成ポリマー(例えば、カルボマー、セルロースエーテル、およびカルボキシビニルポリマー)が含まれる。これらは水中で分散または膨潤して、分散相の液滴周囲に強い界面フィルムを生成すること、および外側の相の粘度を高めることにより乳濁液を安定化するコロイド溶液を生成する。
【0116】
乳濁液は、微生物の増殖を容易に支持しうる、炭水化物、蛋白質、明らかにされているステロールおよびホスファチドなどのいくつかの成分を含むことが多いため、これらの製剤は保存剤を組み入れることが多い。乳濁液製剤に含まれて一般的に用いられる保存剤には、メチルパラベン、プロピルパラベン、四級アンモニウム塩、塩化ベンザルコニウム、p−ヒドロキシ安息香酸のエステルおよびホウ酸が含まれる。製剤の劣化を防ぐために、抗酸化剤も乳濁液製剤に一般に加えられる。用いられる抗酸化剤は、トコフェロール、没食子酸アルキル、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエンなどのフリーラジカル捕捉剤、またはアスコルビン酸およびメタ重亜硫酸ナトリウムなどの還元剤、ならびにクエン酸、酒石酸、およびレシチンなどの抗酸化剤相乗剤であってもよい。
【0117】
乳濁液製剤の経皮、経口および非経口経路による適用、ならびにそれらの製造法は文献に総説が掲載されている(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。経口送達用の乳濁液製剤は、製剤の容易さ、吸収およびバイオアベイラビリティの見地からの有効性といった理由により、非常に広く用いられている。(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245 ; Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。鉱油を基剤とする緩下剤、油溶性ビタミンおよび高脂栄養製剤も、o/w乳濁液として一般に経口投与されている物質に含まれる。
【0118】
マイクロエマルジョン
本発明の一つの態様において、本発明の組成物をマイクロエマルジョンとして製剤する。マイクロエマルジョンは、単一の光学的等方で、熱力学的に安定な液体溶液である、水、油および両親媒性物質の系と定義することができる(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245)。典型的には、マイクロエマルジョンはまず油を水性界面活性剤溶液に分散させ、次いで十分な量の第四の成分、一般には中鎖アルコールを加えて、透明な系を生成することによって調製される系である。したがって、マイクロエマルジョンは、界面活性分子の界面フィルムによって安定化される、二つの不混和性の液体の、熱力学的に安定で、等方性の透明分散液とも記載されている(LeungおよびShah, in: Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., 編, 1989, VCH Publishers, New York, pages 185-215)。マイクロエマルジョンは一般に、油、水、界面活性剤および電解質を含む三つから五つの成分の組み合わせによって調製する。マイクロエマルジョンが油中水(w/o)または水中油(o/w)型のいずれであるかは、用いる油および界面活性剤の性質、ならびに界面活性剤分子の極性頭部および炭化水素尾部の構造および幾何学的充填に依存する(Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 271)。
【0119】
相図を用いる現象学的アプローチは広く研究されており、マイクロエマルジョンをどのようにして製剤するかについての包括的知識を当業者にもたらしている(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245 ; Block, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 335)。通常の乳濁液と比べて、マイクロエマルジョンは、自然に生成する熱力学的に安定な液滴の製剤中に、水不溶性薬物を可溶化する利点を提供する。
【0120】
マイクロエマルジョンの調製に用いられる界面活性剤には、イオン性界面活性剤、非イオン性界面活性剤、Brij 96、ポリオキシエチレンオレイルエーテル、ポリグリセロール脂肪酸エステル、テトラグリセロールモノラウレート(ML310)、テトラグリセロールモノオレエート(MO310)、ヘキサグリセロールモノオレエート(PO310)、ヘキサグリセロールペンタオレエート(PO500)、デカグリセロールモノカプレート(MCA750)、デカグリセロールモノオレエート(MO750)、デカグリセロールセキオレエート(S0750)、デカグリセロールデカオレエート(DAO750)単独またはコサーファクタントとの組み合わせで含まれるが、これらに限定されることはない。コサーファクタントは通常はエタノール、1−プロパノール、および1−ブタノールなどの短鎖アルコールで、界面活性剤フィルム中に浸透し、したがって界面活性剤分子の間に生じた空隙のために無秩序なフィルムを生成することによって、界面の流動性を高める作用をする。しかし、マイクロエマルジョンはコサーファクタントを使用せずに調製することもでき、アルコールを含まない自己乳化マイクロエマルジョン系が当技術分野において知られている。水相は典型的には水、薬物の水溶液、グリセロール、PEG300、PEG400、ポリグリセロール、プロピレングリコール、およびエチレングリコールの誘導体であってもよいが、これらに限定されることはない。油相にはCaptex 300、Captex 355、Capmul MCM、脂肪酸エステル、中鎖(C8〜C12)モノ、ジ、およびトリグリセリド、ポリオキシエチル化グリセリル脂肪酸エステル、脂肪アルコール、ポリグリコール化グリセリド、飽和ポリグリコール化C8〜C10グリセリド、植物油ならびにシリコーン油等の物質が含まれるが、これらに限定されることはない。
【0121】
マイクロエマルジョンは、薬物の可溶化および薬物の吸収促進の見地から特に興味が持たれる。脂質を基剤とするマイクロエマルジョン(o/wおよびw/oの両方)は、ペプチドを含む薬物の経口バイオアベイラビリティを高めることが提唱されている(Constantinidesら, Pharmaceutical Research, 1994,11,1385-1390; Ritschel, Meth. Find. Exp. Clin. Pharmacol., 1993,13,205)。マイクロエマルジョンは改善された薬物の可溶性、酵素加水分解からの薬物の保護、界面活性剤が引き起こす膜の流動性および浸透性変更による薬物吸収促進の可能性、調製の容易さ、固体剤形に比べての経口投与の容易さ、改善された臨床効力、および低毒性なる利点を提供する(Constantinidesら, Pharmaceutical Research, 1994,11, 1385 ; Hoら, J. Pharm. Sci., 1996,85,138-143)。マイクロエマルジョンは、その成分が室温で混合すると、自然に生成することが多い。マイクロエマルジョンは、美容および医薬両方の適用における活性成分の経皮送達でも有効である。本発明のマイクロエマルジョン組成物および製剤は、胃腸管を介しての経口投与では、リン脂質および/またはNSAID−リン脂質の組み合わせからの治療反応の増大を促進し、同様に胃腸管、CNS、PNS、膣、口、食道、頬腔、鼻腔、副鼻洞および他の投与領域内の関門などの疎水性の関門を通過しての、リン脂質および/またはNSAID−リン脂質の組み合わせの局所細胞の治療反応および取り込みの改善を促進すると予想される。
【0122】
本発明のマイクロエマルジョンは、製剤の性質を改善し、本発明の製剤を含むリン脂質および/またはNSAID−リン脂質の組み合わせの吸収を促進するために、モノステアリン酸ソルビタン(Grill 3)、ラブラソル(Labrasol)、および浸透性促進剤などの追加の成分および添加物を含むこともできる。本発明のマイクロエマルジョンにおいて用いられる浸透性促進剤は、次の広く五つに分けた範疇の一つに属するとして分類することができる:界面活性剤、脂肪酸、胆汁酸、キレート剤、および非キレート非界面活性剤(Leeら, Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92)。これらのクラスはそれぞれ前述されている。
【0123】
本発明において用いるのに適したリン脂質には、ジミリストイルホスファチジルコリン、ジステアロイルホスファチジルコリン、ジリノレオイル−ホスファチジルコリン(DLL−PC)、ジパルミトイル−ホスファチジルコリン(DPPC)、ダイズホスファチジルコリン(ダイズ−PCまたはPCS)、および卵ホスファチジルコリン(卵−PCまたはPCE)が含まれるが、これらに限定されることはない。飽和リン脂質であるDPPCにおいて、飽和脂肪族置換基R1およびR2はCH3−(CH2)14であり、R3はCH3であり、XはHである。不飽和リン脂質であるDLL−PCにおいて、R1およびR2はCH3−(CH2)4−CH=CH−CH2−CH=CH−(CH2)7であり、R3はCH3であり、XhaHである。不飽和リン脂質の混合物である卵PCにおいて、R1は主に飽和脂肪族置換基(例えば、パルミチン酸またはステアリン酸)を含み、R2は主に不飽和脂肪族置換基(例えば、オレイン酸またはアラキドン酸)である。ダイズ−PC(Soy-PC)中では、飽和リン脂質(パルミチン酸およびステアリン酸)に加えて不飽和リン脂質の混合物(オレイン酸、リノール酸、およびリノレン酸)である。好ましい双性イオンリン脂質には、ジパルミトイルホスファチジルコリン、ホスファチジルコリン、またはその混合物が含まれるが、これらに限定されることはない。
【0124】
適当なNSAIDには、フェノプロフェンカルシウム(登録商標Nalfon)、フルルビプロフェン(登録商標Ansaid)、スプロフェン。ベノキサプロフェン、イブプロフェン(処方薬、登録商標Motrin)、イブプロフェン(200mg一般薬、登録商標Nuprin、Motrin 1B)、ケトプロフェン(登録商標Orduis、Oruvall)、ナプロキセン(登録商標Naprosyn)、ナプロキセンナトリウム(登録商標Aleve、Anaprox、Aflaxen)、オキサプロジン(登録商標Daypro)などのプロピオン酸薬;ジクロフェナクナトリウム(登録商標Voltaren)、ジクロフェナクカリウム(登録商標Cataflam)、エトドラク(登録商標Lodine)、インドメタシン(登録商標Indocin)、ケトロラクトロメタミン(筋肉内注射用登録商標Acular、Toradol)、ケトロラク(経口用登録商標Toradol)などの酢酸薬;ナブメトン(登録商標Relafen)、スリンダク(登録商標Clinoril)、トルメチンナトリウム(登録商標Tolectin)などのケトン薬;メクロフェナメートナトリウム(登録商標Meclomen)、メフェナム酸(登録商標Ponstel)などのフェナメート薬;ピロキシカム(登録商標Dolibid)などのオキシカム薬;ジフルニサル(登録商標Feldene)、アスピリンなどのサリチル酸薬;オキシフェンブタゾン(登録商標Tandearil)、フェニルブタゾン(登録商標Butazolidin)などのピラゾリン酸薬;アセトアミノフェン(登録商標Tylenol)など;セレブレックス、ビオックスなどのCOX−2阻害剤またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0125】
適当な生体適合性乳化剤には、ヒトまたは動物の消費または体内での使用が承認されている、いかなるイオン性または非イオン性乳化剤または界面活性剤も含まれるが、これらに限定されることはない。典型例には、アセチル化モノグリセリド、脂肪酸のアルミニウム塩、アラビノガラクタン、パン酵母グリカン、炭酸カルシウム、脂肪酸のカルシウム塩、イナゴマメガム(ローカストビーンガム)、カードラン、食用油脂または食用脂生成脂肪酸のモノおよびジグリセチドのジアセチル酒石酸エステル、スルホコハク酸ジオクチルナトリウム、リン酸2ナトリウム(相互参照(X−ref)−1、2および3ナトリウムリン酸塩)、エトキシル化モノおよびジグリセリド、キリンサイEucheuma cottonii抽出物、キリンサイEucheuma spinosum抽出物、脂肪酸、(アルミニウム、カルシウム、マグネシウム、カリウム、およびナトリウム)の塩、ベータ−アミラーゼ処理したn−オクチルコハク酸無水物でエステル化した食用デンプン、フラゾリドン、フルセレラン、フルセレラン、アンモニウム、カルシウム、カリウム、またはナトリウム塩、ガッチゴム、スギノリ抽出物、脂肪酸のグリセリル−ラクトエステル、ヘキシトールオレエート、ヒドロキシル化レシチン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、グリセロールおよびプロピレングリコールのラクチル化脂肪酸エステル、脂肪酸のラクチル(Lactylic)エステル、レシチン、ヒドロキシル化レシチン、メチルエチルセルロース、食用油脂または食用脂生成酸のモノおよびジグリセリド、クエン酸モノイソプロピル、食用油脂または食用脂生成脂肪酸のモノおよびジグリセチドのリン酸1ナトリウム誘導体、Myrj45(8−ステアリン酸ポリオキシエチレン)、ウシ胆汁酸抽出物、ペクチン(修飾ペクチンを含む)、ポリエチレングリコール(400)ジオレエート、脂肪酸のポリグリセロールエステル、ポリオキシエチレングリコール(400)モノおよびジオレエート、ポリソルベート60(モノステアリン酸ポリオキシエチレン(20)ソルビタン)、ポリソルベート65(トリステアリン酸ポリオキシエチレン(20)ソルビタン)、ポリソルベート80(モノオレイン酸ポリオキシエチレン(20)ソルビタン)、脂肪酸のカリウム塩、プロピレングリコールアルギネーyト(アルギン酸のプロピレングリコールエステル)、脂肪および脂肪酸のプロピレングリコールモノおよびジエステル、ナタネ油、完全硬化、過グリセリン化ピロリン酸水素ナトリウム、リン酸ナトリウムアルミニウム、次亜リン酸ナトリウム、メチル硫酸ナトリウム、ペクチニン酸ナトリウム、脂肪酸のナトリウム塩、ステアロイルラクチレートナトリウム、スルホ酢酸ナトリウム誘導体(モノおよびジグリセリド)、ソルビタンモノオレエート、ソルビタンモノステアレート、サクシニル化モノグリセリド、サクシステアリン(コハク酸水素ステアロイルプロピレングリコール)、ショ糖酢酸イソ酪酸エステル(SAIB)、ショ糖脂肪酸エステル、硫酸化オレイン酸ブチル、リン酸3ナトリウム、ザンサンガムなど、またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0126】
適当な中性脂質には、トリグリセリドなどのいかなる中性脂質も含まれるが、これらに限定されることはない。トリグリセリドなどの代表的中性脂質を部分的一覧のために、米国特許第4,950,656号および第5,043,329号を特に参照する。飽和および不飽和両方のトリグリセリドを本発明の組成物において用いることができ、トリパルミチン(飽和)、トリオレインおよびトリリノレイン(不飽和)などのトリグリセリドが含まれる。しかし、これらの特定のトリグリセリドは便宜上本明細書に記載するにすぎず、様々な有用なトリグリセリドの単なる具体例で、包括的であることをさらに意図するものではない。
【0127】
適当な生体適合性、生体分解性ポリマーの非限定例には、ポリ乳酸、ポリグリコール酸、ポリカプロラクトン、ポリ酸無水物、ポリアミド、ポリウレタン、ポリエステルアミド、ポリオルトエステル、ポリジオキサノン、ポリアセタール、ポリケタール、ポリカーボネート、ポリオルトカーボネート、ポリフォスファーゼン、ポリヒドロキシ酪酸、ポリヒドロキシ吉草酸、ポリアルキレンオキザレート、ポリアルキレンサクシネート、ポリ(リンゴ酸)、ポリ(アミノ酸)、ポリ(メチルビニルエーテル)、ポリ(マレイン酸無水物)、キチン、キトサン、およびそのコポリマー、ターポリマー、もしくは高級モノマー重合体(higher poly-monomer polymers)、またはその組み合わせもしくは混合物が含まれる。好ましい生体分解性ポリマーはすべて加水分解によって分解される。
【0128】
典型的には、ポリマーはポリ酸無水物などの表面侵食性ポリマーまたはポリオルトエステルなどの塊状侵食性ポリマーのいずれかとなる。ポリ(l−乳酸)(PILA)、ポリ(dl−乳酸)(PLA)、ポリ(グリコール酸)(PGA)、ポリカプロラクトン、そのコポリマー、ターポリマー、もしくは高級モノマー重合体、またはその組み合わせもしくは混合物が好ましい生体適合性、生体分解性ポリマーである。好ましい生体分解性コポリマーは、乳酸およびグリコール酸のコポリマーで、ポリ(dl−乳酸−グリコール酸)コポリマー(PLG)と呼ばれることもある。ポリ(dl−乳酸−グリコール酸)コポリマーの共重合モノマー(乳酸:グリコール酸)の比は、好ましくは乳酸:グリコール酸が約100:0から約50:50の間である。最も好ましくは、共重合モノマーの比は乳酸:グリコール酸が約85:15から約50:50の間である。PLG:PLAが、好ましくは約85:15から約50:50のPLAとPLGの配合物(blend)を用いて、ポリマー物質を調製することもある。
【0129】
PLA,PILA、PGA、PLGおよびその組み合わせまたは混合物または配合物は、ヒトの臨床使用が承認されている合成ポリマーに含まれる。これらは現在、手術の縫合材料として、および制御放出装置において、ならびに他の医学および薬学上の適用において用いられている。これらは生体適合性で、その分解産物は乳酸およびグリコール酸などの、通常の代謝経路に入る低分子量化合物である。さらに、ポリ(乳酸−グリコール酸)コポリマーは、乳酸とグリコール酸のコポリマー比を単に変動させることにより、分解速度が数日から数年までの広範囲に及ぶという利点を提供する。
【0130】
生物学的適用において用いられるポリマーの生体分解を促進するために、本発明の組成物は組成物において用いられるポリマーの生体分解を助けうる酵素の添加も含むことができる。好ましい酵素または類似の試薬は、エステル加水分解能力を有するプロテアーゼまたは加水分解酵素である。そのような酵素には、プロテイナーゼK、ブロメライン、プロナーゼE、セルラーゼ、デキストラナーゼ、エラスターゼ、プラスミンストレプトキナーゼ、トリプシン、キモトリプシン、パパイン、キモパパイン、コラゲナーゼ、スブチリシン、クロストリドペプチダーゼA、フィシン、カルボキシペプチダーゼA、ペクチナーゼ、ペクチンエステラーゼ、酸化還元酵素、酸化酵素などが含まれるが、これらに限定されることはない。
【0131】
適当な化学および/または放射線療法剤(商品名)には、白金錯体、金(III)錯体、パラジウム錯体、アリトレチノイン(Panretin)、アロプリノール(Zyloprim)、アルトレタミン(Hexalen)、アミフォスチン(Ethyol)、アミフォスチン(Ethyol)、アミフォスチン(Ethyol)、アナストロゾール(Arimidex)、アナストロゾール(Arimidex)、三酸化二ヒ素(Trisenox)、ベキサロテン(Targretin)、ベキサロテン(Targretin)、ブレオマイシン(Blenoxane)、静注用ブスルファン(Busulfex)、経口用ブスルファン(Myleran)、カペシタビン(Xeloda)、カペシタビン(Xeloda)、カペシタビン(Xeloda)、カルボプラチン(Paraplatin)、カルボプラチン(Paraplatin)、植込用カルムスチン+ポリフェプロサン20(Gliadel Wafer)、セレコキシブ(Celebrex)、クロランブシル(Leukeran)、シスプラチン(Platinol)、シスプラチン(Platinol)、シスプラチン(Platinol)、クラドリビン(Leustatin(2−CdA)、シクロホスファミド(Cytoxan)、シタラビンリポソーム(DepoCyt)、ダウノルビシンリポソーム(DanuoXome)、ダウノルビシンダウノマイシン(Daunorubicin)、ダウノルビシン(ダウノマイシン(Cerubidine)、デクスラゾキサン(Zinecard)、ドセタキセル(Taxotere)、ドセタキセル(Taxotere)、ドセタキセル(Taxotere)、ドキソルビシン(Adriamycin PFS注射剤)、ドキソルビシンリポソーム(Doxil)、ドキソルビシンリポソーム(Doxil)、エリオットB溶液(Elliott’s B Solution)、エピルビシン(Ellence)、エストラムスチン(Emcyt)、リン酸エトポシド(Etopophos)、リン酸エトポシド(Etopophos)、リン酸エトポシド(Etopophos)、エトポシド(VP−16 (Vepesid)、エトポシド(VP−16 (Vepesid)、エキセメスタン(Aromasin)、フルダラビン(Fludara)、フルオロウラシル(5−FU (Adrucil)、ゲムシタビン(Gemzar)、ゲムシタビン(Gemzar)、ゲムツズマブ−オゾガミシン(Mylotarg)、酢酸ゴセレリン(Zoladex植込剤)、ヒドロキシ尿素(Hydreaカプセル)、イダルビシン(Idamycin)、イダルビシン(Idamycin)、イフォスファミド(IFEX)、メシル酸イマチニブ(Gleevec)、イリノテカン(Camptosar)、イリノテカン(Camptosar)、イリノテカン(Camptosar)、レトロゾール(Femara)、レトロゾール(Femara)ロイコボリン(Leucovorin)、レバミソール(Ergamisol)、メルファランL−PAM(Alkeran)、メスナ(Mesnex)、メトトレキセート(Methotrexate)、メトキシサレン(Uvadex)、ミトキサントロン(Novantrone)、ミトキサントロン(Novantrone)、パクリタキセル(Paxene)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パラメーター深泥ネート(Aredia)、ペガデマーゼ(Adagen(ウシメガデマーゼ))、ペントスタチン(Nipent)、ペントスタチン(Nipent)、ポルフィマーナトリウム(Photofrin)、ポルフィマーナトリウム(Photofrin)、ポルフィマーナトリウム(Photofrin)、ストレプトゾシン(Zanosar)、タルク(Sclerosol)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、テモゾラミド(Temodar)、テニポシドVM−26(Vumon)、トポテカン(Hycamtin)、トポテカン(Hycamtin)、トレミフェン(Fareston)、トレチノインATRA(Vesanoid)、バルルビシン(Valstar)、ビノレルビン(Navelbine)、またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0132】
本発明は好ましくは未精製レシチン油の使用に関するが、本発明は、リン脂質を含むヒトが消費可能ないかなる油も含むが、これらに限定されることはない、リン脂質を含むいかなる生体適合性油も使用することができる。
【0133】
適当な生体適合性には、植物もしくは動物油またはそれらの誘導体などの天然油あるいは合成油、特にダイズ由来のレシチン油などのリン脂質に富む天然油を含む、FDAによってヒトまたは動物の消費が承認されているいかなる油も含まれるが、これらに限定されることはない。そのような油の典型例には、落花生油、ナタネ油、アボカド油、ベニバナ油、オリーブ油、トウモロコシ油、ダイズ油、ゴマ油、ビタミンA、ビタミンD、ビタミンE、魚油などの精油、植物油、硬化植物油、動物油が含まれる。
【0134】
本発明の製剤または組成物は、抗酸化剤(例えば、ビタミンA、C、D、Eなど)、微量金属および/または多価陽イオン(アルミニウム、金、銅、亜鉛、カルシウムなど)、界面活性剤および/または溶媒(例えば、プロピレングリコール/PPG、ジメチルスルホキシド/DMSO、中鎖トリグリセリド/MCTなど)、非毒性色素、ならびに香味増強剤などの他の化学物質も含むことができ、これらはアンテ遺影、流動性/拡散性、浸透性、有効性および消費者の認容性を改善するために製造されているため、これらを製剤に加えることができる。
【0135】
リン脂質、好ましくはPCおよびNSAIDを含む本発明の製剤は、経口投与用のゼラチン軟カプセルもしくは硬カプセルまたはベジキャップに充填するために用いることができ、あるいは炎症、潰瘍化および/または被刺激組織または皮膚に局所適用するために、そのまま溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液、またはその混合物として用いることもできる。
【0136】
本製剤の一つの好ましい態様はレシチン油を基剤とするPC−NSAID組成物で、これはGI毒性について試験されている。試験された三つの製剤には、アスピリン、インドメタシンおよびイブプロフェンと組み合わせたレシチン油が含まれる。この試験において、アスピリンを35%PCを含むダイズレシチン油、Phosal 35 SBと組み合わせ、アスピリン用量18mg/kg(NSAID:レシチン油の重量比を1:0.5から1:1、1:2まで系統的に変動させた)で絶食ラットに胃内投与した。加えて、他のラット群に等用量のアスピリンをレシチン油非存在下で投与するか、または等しい体積の生理食塩水(以下、「食塩水」とも記載する)を投与した。45分後、すべてのラットを1mlの0.6N HClで胃内攻撃(チャレンジ)し、15分後、ラットを安楽死させ、胃を切開して、確立された方法50-52によって胃損傷を評価した。
【0137】
図1に示すとおり、データはアスピリンを単独投与したラットで観察された多数の胃損傷とは対照的に、三種すべてのアスピリン:レシチン製剤で治療したラットはすべて、胃損傷が有意に少なかったことを示していた。
【0138】
非アスピリンNSAIDであるインドメタシンおよびイブプロフェンの胃毒性を評価するために、もう一つの潰瘍モデルを用いた。すなわち、以前に記載されているとおり52、GI出血が終点であった。このモデルにおいて、NSAIDを絶食ラットに単独またはレシチン油Phosal 35 SBとの組み合わせ(NSAID:レシチンの重量比1:1)のいずれかで胃内投与した。対照ラットには等しい体積の食塩水を投与した。ラットをNSAIDのGI損傷効果に対してより敏感にするために、すべてのラットに酸化窒素(NO)合成酵素阻害剤であるL−NAME(20mg/kg)も、18〜20時間の試験期間中に三回注射し、その後ラットを安楽死させ、GI管の遠位20cmに2mlの食塩水を流し、GI出血の指標としてのヘモグロビン分析のために流出液を回収した。これらの実験の結果を下記の図2および3に示している。
【0139】
図2を参照すると、データはインドメタシンは10mg/kgの用量でGI出血の重度の増大を引き起こし、これは等用量のインドメタシンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは顕著かつ有意に軽減されたことを示していた。
【0140】
図3を参照すると、データはイブプロフェン(齧歯類モデル系において通常のNSAIDの中では最も毒性が低いものの一つと考えられている)は100mg/kgの用量でGI出血の適度の増大を引き起こし、これは等用量のイブプロフェンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは有意に軽減されたことを示していた。
【0141】
次いで、前述の方法を用いて、NSAID−レシチン製剤の抗炎症/鎮痛活性を(NSAID単独と比べて)評価した。これは、完全フロイントアジュバント(CFA)0.1mlをラットの左後足に注射して急性炎症反応を誘導することにより達成した。四日後、終夜絶食させたラットに、NSAID(アスピリン、インドメタシンまたはイブプロフェンのいずれか)単独またはPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)(アスピリンだけは他の比も評価した)を胃内投与した。2時間後、ラットの圧に対する疼痛感度を、ランダル−セリート法(55)を用いて測定した。これは、炎症足または対側の非炎症足に適用した圧を、ラットが疼痛感覚の最初の徴候(発声または試験中の後足の指の伸展)を示すまで漸増させることにより達成し、この値をラットの疼痛閾値として記した。したがって、低い疼痛閾値は炎症足が圧に対して非常に敏感であることを示す一方で、高い疼痛閾値は低い疼痛感度または鎮痛を意味する。結果を図4〜6に示している。
【0142】
図4を参照すると、データはアスピリンは10mg/kgの用量でラットの患側の足の疼痛閾値を高める適度の能力を有するが、等用量のアスピリンをレシチン油との組み合わせで投与した場合には、試験したすべての重量比で鎮痛活性が有意に増強されたことを示していた。
【0143】
図5を参照すると、データはイブプロフェンは25mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のイブプロフェンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示していた。
【0144】
図6を参照すると、データはインドメタシンは4mg/kgの用量でを示していた。データは、ペースト様組成物が非修飾INDOに比べて疼痛処理活性を改善し、対照食塩水に比べて疼痛処理活性を非常に改善することを示している。
【0145】
NSAIDと中枢および末梢神経系外傷および損傷
炎症の過程は、脊髄損傷(SCI)などのCNSへの急性外傷性損傷[A1]ならびにアルツハイマー病(AD)などの遅延型神経変性疾患[A2]両方に関連する進行性病態生理における主要な成分である。炎症の過程は、SCIで一般に観察される運動機能の進行性の劣化および慢性疼痛の発生、ならびにADで観察される記憶および認知機能の損失を直接引き起こすか、またはそれらに寄与すると考えられる。最近、抗炎症薬の使用が外傷性SCIの齧歯類モデルで組織損失および機能欠損の減弱において効果を示している[A3]。
【0146】
さらに重要なことには、いくつかの最近の疫学調査は、非ステロイド性抗炎症薬(NSAID)の長期消費がADのリスクを50%まで低下させる可能性を示唆している[A4]。炎症状態の性質に応じて短期または長期いずれかのNSAID治療戦略を用いうることが予想されるため、NSAIDが低用量で有効であり、かつその耐容性が高く、副作用が最小であることは極めて重要である。一般大衆の約40%が長期NSAID消費に反応して、消化不良から生命を脅かす消化性潰瘍および出血の発現誘導に及ぶ胃腸(GI)症状を発症することはよく確立されている[A5]。
【0147】
1995年に、PIの研究室は、シクロオキシゲナーゼ(COX)活性の阻害に加えて、NSAIDが上部GI管の表面疎水性関門を、おそらくはリン脂質の表面ライニングに化学的に結合することによって、減弱させる能力を有すると報告した[A6]。さらに、発明者らは、実験動物とヒトの両方で、合成種または精製抽出物(例えば、ダイズレシチン由来)のいずれかとして存在する最も有名なリン脂質、ホスファチジルコリン(PC)とNSAIDが化学的に結合している場合には、NSAIDの有害作用を防止しうることを示した[A6、A7]。興味深いことに、PC−NSAIDは、低いGI毒性に加え、おそらくPC−NSAIDの高い膜浸透性とCOX阻害活性に起因する、発熱、炎症および疼痛を阻害する高い治療活性も有していることが明らかにされた[A6、A8、A9]。
【0148】
したがって、本発明の組成物は神経の炎症を減弱し、SCIおよびADを含むいくつかの神経学的状態に関連する病態生理を軽減する。
【0149】
経口投与したPC−イブプロフェンは末梢神経結紮に関連する炎症依存性痛覚過敏の発生を低下させる
坐骨神経周囲を4本のクロミックグット縫合糸で緩く結紮すると、手術の2〜4日後に、同側後足に適用した圧または熱に対する痛覚過敏反応によって示されるとおり、患部神経の重度の末梢神経炎症および神経障害性疼痛の誘導を引き起こすことが報告されている[A10、A11]。ラットにおけるこの誘導法を用いた末梢神経炎症のPC−NSAID治療の効果および痛覚過敏の軽減。この齧歯類モデルを用いて、右または左いずれかの坐骨神経の神経炎症を誘導した。擬似手術を対側に実施した。手術の2日後、ラットを次の実験群に無作為に分類した(ラット12匹/群):食塩水対照;イブプロフェン(15mg/kg);およびPC−イブプロフェン(等用量のNSAID)。次の2日間、ラットに試験NSAID製剤を1日2回投与し、疼痛感覚のいくつかの行動指標を、2日間の投与期間の前後両方の後足で評価した。有効性を評価するために用いた行動解析は次のとおりであった:患部後足の保護行動;熱に対する後足引っ込め潜時;フォイの毛刺激に対する足引っ込め反応;および後足への圧適用に対する疼痛反応[A8]。安楽死にあたり、結紮および対照神経を摘出して、炎症の指標について肉眼検査および組織検査の両方を行った。これらの試験の結果は、後足炎症のモデル(フロイントアジュバントで誘導)においてPC−イブプロフェンの鎮痛活性はイブプロフェン単独よりも著しく高く、またPC−イブプロフェンは坐骨神経結紮による疼痛感覚の緩和においても、フォイの毛刺激および熱の両方に対する足引っ込め反応を測定することにより評価して、より有効であることを示している。
【0150】
経口投与したPC−イブプロフェンは挫傷性SCIのラットモデルにおける組織損失、移動運動機能を低下させ、慢性疼痛症候群の発生を減弱する
最近、ラット成獣において抗炎症薬の単回用量送達が脊髄傷害のサイズを低下させることが明らかにされた[A3]。これらのNSAID治療ラットは治療していないラットに比べてより大きい移動運動活性と、神経障害性疼痛に特徴的な痛覚過敏および機械的異痛症(接触による疼痛)の症状軽減を示した。慢性神経障害性疼痛の発生は、脊髄損傷後に非常に頻繁に起こり、患者の永久的な負担になりうる。慢性中枢疼痛の発生を防止または減弱するための耐容性が高く、有効な治療法の開発が痛切に必要とされている。経口投与したPC−NSAIDはSCIに関連する組織損傷を軽減し、移動運動の転帰を改善し、慢性疼痛症候群を予防する。
【0151】
PC−イブプロフェンはADのトランスジェニックマウスモデルにおけるアルツハイマー様病態生理の発生低減においてイブプロフェンよりも有効である
最近の臨床での証拠は、NSAIDがAD発症のリスクを著しく低下させる可能性を示唆している。主な問題は、ADに対する治療戦略の設計に十分な動物モデルが不足しているということである。最近確立されたヒトβ−アミロイド過剰発現Tg2576マウスは、年齢依存性記憶および認知欠損、ならびにアミロイド斑形成、小膠細胞活性化、星状細胞反応性および異栄養性神経突起を含む組織病理学的欠損を示す、都合のよい齧歯類モデルを提供する[A19〜A21]。イブプロフェンは最近、Tg2576マウスADモデルにおいてアミロイド斑、異栄養性神経突起および活性化小膠細胞の数を減少させることが明らかにされた[A21]。
【0152】
PC−NSAIDの貯蔵寿命最適化
PC−NSAIDの商品化成功には、室温条件下で長期間安定なままである製剤が必要とされる。これはイブプロフェンのようなほとんどのNSAIDにとっては問題ではないが、水に曝露されると速やかに水解されてサリチル酸となるアスピリンにとってはまだ問題である。レシチン油またはリン脂質を含む任意の他の生体適合性油などの非水性担体中に溶解および/または分散されたNSAIDを主薬とする本発明の製剤。そのような環境は疎水性であるため、これらはアスピリンを主薬とする製剤におけるアスピリンの安定性を増強することになりうる。
【0153】
中枢および末梢神経系の外傷および損傷についての実験結果
これらの実験は、PC−イブプロフェンが脊髄損傷(SCI)の有用な治療薬であることを示している。結果は、ラットの脊髄損傷(SCI)後に、体重1kgあたり25mgのNSAIDで1日2回6週間治療した際の効果を、PC−イブプロフェン、イブプロフェンおよび食塩水を比較して証明している。
【0154】
図7Aおよび7Bを参照すると、グラフ化されたデータは、ランダル−セリート法を用い、食塩水で治療したSCIラットの疼痛圧閾値の低下によって証明されるとおり、SCIがラットで痛覚過敏を引き起こしたことを示している。これとは対照的に、SCIによる痛覚過敏は、イブプロフェンまたはPC−イブプロフェンのいずれかで治療したSCIラットでは見られず、PC−イブプロフェンは非修飾イブプロフェンよりも優れているようであった。このデータを二つの様式で示している。図7Aでは、データを記録されたまま直接プロット(正規化せずに)した一方で、図7Bでは、各ラットのデータ値を、それ自体の術前基準値と比較している。このデータのグラフはおそらく、SCI後のNSAID投与の有益な効果について最も説得力のあるものである。
【0155】
図8を参照すると、SCIラットにおけるPC−イブプロフェンの優れた鎮痛活性が、漸増する直径(力に相当する)を有するフォイの毛への後足の刺激に対する後足反応の%を測定する、第二の行動試験でも示されている。ランダル−セリート試験では、高い疼痛圧閾値が鎮痛の指標であるのに対し、この場合、低い数値が鎮痛の指標であることに留意されたい。
【0156】
図9を参照すると、食塩水またはPC−イブプロフェンで治療したラットとは対照的に、イブプロフェンで治療したSCIラットでは、6週間の試験期間中に体重が増加しないという証拠。SCIラットはイブプロフェン単独に対して軽度の中毒反応を有するというこの示唆は、血液尿素窒素(腎毒性の証拠)および乳酸脱水素酵素(LDH、肝毒性の証拠)のわずかな上昇によっても示されている。
【0157】
図10を参照すると、確立されたBBB試験により評価した、SCI後の運動機能の回復は、非修飾イブプロフェンで治療したラットでは低下するが、この運動機能回復の指標において、食塩水とPC−イブプロフェンとの間には差がなかったという証拠。
【0158】
血栓性障害治療用の有効な製剤としてのPC−NSAID
生体適合性油中にホスファチジルコリン(PC)などのリン脂質およびNSAID、特にアスピリンを含む、本発明の製剤は、血栓症、卒中、および心筋梗塞を含む血栓性障害治療用の有効な製剤である。改善されたGI安全性に加えて、PC−アスピリンは通常のアスピリンよりも強力な血小板凝集および血栓形成阻害剤である。アスピリン(ASA)は双性イオンリン脂質と化学的に結合して、天然アスピリンに比べて同等または高い発熱、疼痛および炎症軽減活性を有するが、潰瘍および出血といったアスピリンの重篤な胃腸副作用は持たない結合複合体を形成する。リン脂質複合アスピリンが、動脈血栓症のインビボモデルで血栓形成を防止する際に、アスピリン単独よりも強力であることは興味深い。したがって、本発明のPC−アスピリン製剤は血小板凝集および血栓形成を阻害して、血栓性障害の症状を軽減する。
【0159】
心筋梗塞(MI)および卒中などの血栓性動脈閉塞性疾患は、米国および西洋社会における死亡の第一の原因である。米国心臓学会(American Heart Association)によれば、来る一年間に100万人を越える米国人が急性心筋梗塞を患うことになる。動脈血栓症の発生率を効果的に低下させうる薬物は、臨床上非常に重要である。血栓症は動脈閉塞性疾患の開始および拡大における重大な過程であるため、新規な特定の抗血栓薬を開発する説得力のある理由がある。動脈血栓症は、血球、血管壁および血漿蛋白質の間の一連の細胞および生化学的相互作用を含む複雑な過程である(B1)。血小板はこれらの相互作用において中心的役割を果たす(B2)。血小板は損傷血管壁に接着し、細胞の活性化、分泌および凝集を起こす。活性化された血小板は血液凝固を加速し、その分泌された分子は血管平滑筋の細胞増殖を促進する。血小板が動脈血栓症で果たす中心的役割を考慮して、血小板機能の阻害に基づく抗血栓症薬を開発するため、長年にわたって多大な努力が払われてきた(B3)。しかし、臨床上有用な化合物はほとんどない。事実、アスピリンはその有効性と費用を考慮することにより、いまだに臨床で用いられる抗血小板薬の主な薬剤であり、原型である。アスピリンはMIおよび卒中の一次および二次予防において有効である(B4〜B7)。しかし、アスピリンの最適な治療用量については不確実な点が残り、40%を越える患者が胃腸毒性のためにアスピリンまたは腸溶アスピリンでさえも使用することができない。特に関連があるのは、非常に低用量のアスピリン(10〜80mg)でさえもかなりの数のヒト被検者で胃のびらん性損傷および出血を引き起こしたという最近の報告である。このことは、現在のところGI出血のために入院する最大の患者群は、心血管リスク低下のために低用量アスピリンを長期服用している個人であることの説明となりうる(B8)。
【0160】
アスピリンおよび他の非ステロイド性抗炎症薬(NSAID)の主な作用機序は、それらのプロスタグランジン合成を阻害する能力を通じてであることがかなり前から知られている。シクロオキシゲナーゼの二つのアイソザイム、COX−1およびCOX−2が記載されており、NSAIDは両方のCOXイソ型の活性を阻止する(B9〜B12)。アスピリンはその抗血小板作用を、血小板のCOX−1活性を阻害することによりトロンボキサンA2(TXA2)産生を阻止することで発揮する。しかし、アスピリンは血管内皮細胞でも同じ酵素を阻害し、したがってプロスタサイクリン(PGI2)の産生を防止する(B13、B14)。この内皮COXの阻害は血栓症またはアテローム性動脈硬化症の進行を促進することもあり、「アスピリンジレンマ」と呼ばれ、アスピリンの臨床での有用性における欠点である。このジレンマが低用量アスピリンの使用につながり、TXA2の産生を阻害しながらPGI2を残すことで、血小板凝集を低減しながら血管拡張を維持するための最適な抗血栓状態が得られることを示唆している。したがって、好ましいPGI2とTXA2の比は、不安定狭心症、心筋梗塞、一過性脳虚血発作、および卒中の治療および予防において深い意味を有すると考えられる。適当な投与処方、製剤、および送達速度により、アスピリンはおそらく血管PGI2産生の妨害を最小限に抑えて、血小板TXA2精製を防止することができる(B15〜B17)。
【0161】
血栓症の治療および/または予防におけるNSAIDの使用
アスピリンを含むNSAIDは一般大衆の間で最も大量に消費される薬物であり、発熱、疼痛および炎症の治療においてこの薬物群が高い効力を有するため、その使用は過去10年間に指数関数的速度で増大している(B18)。アスピリンを長期服用している個人では一般母集団に比べて心血管疾患(狭心症、心筋梗塞、血栓症、および卒中)の発生率が低いという最近の証拠により、この薬物を自身で処方する人の数が増え続けることになり、アスピリンの年間総売上高の35〜40%を占めている(B19)。その結果、我々の母集団の約1%が毎日アスピリンを服用していると推定されている。その結果、FDAは卒中、狭心症および心臓発作のリスク低下のためのアスピリンの使用を承認した。しかし、アスピリンの最適な治療用量については不確実な点が残っている。我々の母集団の平均年齢が上がるため増え続けると予想される、NSAIDの使用の指数関数的増加を妨害する一つの局面は、この化合物群が使用者のかなりのパーセンテージで重篤な副作用を引き起こし、最も顕著な副作用は胃腸出血および潰瘍形成であるということである(B20)。特に関連があるのは、非常に低用量のアスピリン(10〜75mg)でさえもヒト被検者で著しい胃のびらん性損傷および出血を引き起こしたという最近の報告である(B8)。
【0162】
アスピリン用量はPCなどの双性イオンリン脂質との結合によってかなり下げることができ、薬物の利益:危険比の著しい改善が得られる。リン脂質複合アスピリンは血管PGI2に対して血小板TXA2産生を選択的に阻害し、リン脂質/アスピリン複合体では、複合していないアスピリンの作用に比べて、血小板および内皮COX活性を区別する作用があることは、その高い抗血栓活性にとって重要であると考えられる。この知見は、動脈血栓症のインビボモデルにおいて、低用量のリン脂質複合アスピリンは実験期間中を通して血栓形成および血管閉塞を防止したが、この閾値よりも低い用量のアスピリン単独では、血栓形成を防止することができず、1時間以内に血管が閉塞したという事実によってさらに裏付けられる。PC−アスピリンの追加の利点は、これにより生じる胃粘膜損傷が通常のアスピリンよりも著しく少ないことで、したがってPC−アスピリン複合体を胃腸への副作用がない有効な抗血栓薬として開発することで、広範な費用効果の高い臨床適用が可能になることも強調すべきである。
【0163】
主な研究者による結果
1995年に、Lichtenbergerら(B21)は、ラットで経口投与したアスピリンおよびいくつかの他のNSAID(ジクロフェナク、インドメタシンナプロキセンを含む)の急性GI有害作用は、薬物を合成PCのジパルミトイルホスファチジルコリン(DPPC)またはダイズレシチンの精製抽出物とあらかじめ結合させれば、著しく軽減しうることを報告した。最近、発明者は、16名の健常志願者に4日間の試験期間中アスピリンまたはPCと複合したアスピリン(1日3回650mg)のいずれかを経口投与し、粘膜損傷をビデオ内視鏡検査によって評価した、試験的二重盲検交叉臨床試験の結果を報告した(B22)。図11に示すとおり、PC−アスピリン複合体は等用量の非修飾アスピリンに比べて、感受性の高い個人において胃のびらんの数を有意に70%減少させた。図12に示すとおり、アスピリンおよびPC−アスピリンはいずれも洞のCOX活性を>85%阻害する同等の能力を有していたため、この胃毒性の低減は薬物のCOX阻害活性の変化には関係していなかった。
【0164】
発明者の研究室では、ラットの発熱、疼痛および炎症を阻害するアスピリンの治療活性は、NSAIDをPCと化学的に結合させて投与した場合に一貫して増強され、その治療効力は非修飾NSAIDの5〜10倍に増大することも報告した(B23)。関節炎の齧歯類モデルを用いた他の試験でも、関節の炎症および疼痛を阻害するNSAIDの効力が、薬物をPCと化学的に結合させて胃内投与した場合に増強される、確証的な証拠が得られた。
【0165】
血小板および血管組織においてそれぞれTXA2およびPGI2を産生する能力に対するリン脂質−アスピリン複合体(等モル濃度の二つの物質を含む)の効果のエクスビボ動物試験を調べた。ラットに非修飾またはDPPC結合アスピリン(20mg/kg用量)のいずれかを胃内投与し、30分後、血液を採取し、血小板に富む血漿を調製し、アラキドン酸により凝集を誘導した。非修飾アスピリンまたはDPPC−アスピリンで個々に治療したラットの血小板では、食塩水対照に比べてTXB2(TXA2の安定代謝物)産生の低下が見られた。図13Aに示すとおり、PC−アスピリンはTXB2の産生をさらに抑制した。
【0166】
次いで、このエクスビボアプローチを用いて、血管(内皮)PGI2を測定した。薬物投与の1時間後に摘出した腹部大動脈を、アラキドン酸(AA、25mM)と共にインキュベートすることにより、6−ケトPGF1α(PGI2の安定代謝物)を産生する能力について評価した。図13Bに示すとおり、アスピリンは対照に比べて6−ケトPGF1αの産生を有意に阻害したが、DPPC単独およびASAと複合したDPPCは効果を示さなかった。したがって、PC−アスピリンの投与によって、血小板シクロオキシゲナーゼの選択的阻害を達成し、血管プロスタサイクリン生合成を保存することが可能である。
【0167】
PCと複合した、または複合していないアスピリンの抗血栓効果を、動脈血栓形成のインビボモデルにおいて評価した。プロトコル(B24)に従い、麻酔したウサギの左頚動脈に、平均頚動脈血流速度が対照値よりも50%高くなる点(これは形成した血栓による血管の50%閉塞に対応する)まで陽極電流をかけた。この点で、電流を止め、その後2〜3時間にわたり血流をモニターしながら試験薬物を静脈内投与した。図14Aから、食塩水またはリン脂質単独で治療した対照ウサギでは、<60分で頚動脈の血流速度はゼロ(血管の完全な血栓閉塞を示す)に低下したことが認められる(閉鎖までの平均時間=40±17)。これに対して、非修飾アスピリンを5〜20mg/kgの用量範囲で投与したウサギは、2〜3時間の実験期間を通して、血管閉塞を全く示さなかった(データは示していない)。興味深いことに、アスピリンの用量を2.5mg/kgに下げると、図14Aに示すとおり、リン脂質と複合したアスピリンはまだ血栓形成の防止において有効である(複合体投与の>180分後)が、アスピリン単独(この閾値よりも低い用量で)では血栓形成を防止することはできず、血管は61±15分胃内に閉塞する(n=4)ことが観察された。さらに、図14Bに示すとおり、アスピリン−リン脂質複合体を2.5mg/kg用量で投与したウサギで形成された血栓の重量は、天然アスピリン、食塩水またはリン脂質単独で治療したウサギに比べて有意に小さかった。
【0168】
6−ケトPGF1α(PGI2の代謝物)およびTXA2濃度も患部頚動脈で測定した。食塩水対照ウサギにおいて、血栓が形成されている患部動脈におけるPGI2のTXA2に対する比は、血栓が精製していない非患部(正常)頚動脈よりも有意に低かった(TXB2産生の増加のため)。このPGI2のTXA2に対する比は、ウサギをアスピリン(2.5mg/kg)またはリン脂質単独で治療した場合には、わずかに改善されたが、血栓形成を阻止するのに十分ではなかった。これに対して、図14Cに示すとおり、同じ用量のPC−アスピリンを投与したウサギのPGI2/TXA2比は、有意に改善され、食塩水で治療したウサギの正常動脈(陽極電流に曝露していない)で求めた比と有意な差はなかった。
【0169】
PC−アセトアミノフェン製剤
図15は、絶食Sprague Dawleyラットにアセトアミノフェン(Tylenol)単独またはTylenol:P35SBの組み合わせで経口攻撃した(経口チャレンジ)24時間後に、Tylenol:P35SB(1:2)がラットに肝酵素アスパラギン酸アミノ基転移酵素(AST)の上昇によって示される肝損傷からの保護を提供することを示すデータを示している。データは、いくつかの統計検定を用いることによって、ASTレベルがTylenolで治療したラットでは食塩水対照値に対して有意に上昇するが、Tylenol:P35SBの組み合わせ(重量比1:2)を投与したラットでは上昇しないと思われることを示している。
【0170】
引用文献
1. Furst DE, Paulus HE. Aspirin and other nonsteroidal anti-inflammatory drugs. In: Arthritis and Allied Conditions (McCarty DJ, Koopman WJ, Eds) Lea & Febiger, Philadelphia, 1993, pg 567-602.
2. Pelletier J-P. Pathological pathways of osteoarthritis. In: Non-steroidal Antiinflammatory Drugs: A Research and Clinical Perspective. Royal Society of Medicine Press, London, 1994, 1-14.
3. Jiang Y, Zhao J, Genant HK, Dequeker J, Geusens P. Bone mineral density and biomechanical properties of spine and femur of ovariectomized rats treated with naproxen. Bone 22: 509-514, 1996.
4. Walt R., Katschinski B, Logan R, Ashley J, Langman M. Rising frequency of ulcer perforation in elderly people in the United Kingdom. Lancet 489-492, 1986.
5. Allison MC, Howatson AG, Torrance CJ, Lee FD, Russel RI: Gastrointestinal damage associated with the use of nonsteroidal anti-inflammatory drugs. N. Engl J. Med. 327: 749-754, 1992.
6. Kurata JH, Abbey DE. The effect of chronic aspirin use on duodenal and gastric ulcer hospitalizations. R Clin. Gastroenterol. 12 (3): 260-266, 1990.
7. Symmons DPM. Mortality in rheumatoid arthritis. Br. J Rheum. 27 (Suppl 1): 44-54, 1988.
8. Henry DA, Johnston A, Dobson A, Duggan J. Fatal peptic ulcer complications and the use of non-steroidal anti-inflammatory drugs, aspirin and corticosteroids. Br. Med. J 295: 1227-1229, 1987.
9. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action of aspirinlike drugs. Nature 231: 232-251, 1971.
10. Ferreira SH Vane JR. New aspects of the mode of action of NSAIDs. Ann Rev Pharmacol 14: 57-. 70, 1974
11. Whittle BJR, Higgs GA, Eakin KE, Moncada S, Vane JR. Selective inhibition of prostaglandin production in inflammatory exudates and gastric mucosa. Nature 284: 271273, 1980.
12. Bergstrom S, Duner H, von Euler US, Pernow B, Sjovall J. Observations on the effects of infusions of prostaglandin E in man. Acta Physiol Scand. 45: 145-152, 1959.
13. Robert A. Nezamis JE, Lancaster C, Hanchar AJ: Cytoprotection by prostaglandins in rats: prevention of gastric necrosis produced by alcohol, HCL, NaOH, hypertonic NaCl and thermal injury. Gastroenterology 70: 359-370, 1979.
14. Mitchell JA, Akarasreenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of NSAIDs as inhibitors of constitutive and inducible cyclo-oxygenase. P. N. A. S. 90: 1169311697, 1993.
15. Masferrer JL, Zioeifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclo-oxygenase-2 in vivo is antiinflammatory and non-ulcerogenic. P. N. A. S 91: 3228-3232, 1994.
16. Xie W, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen responsive gene encoding prostaglandin synthesis is regulated by mRNA splicing. P. N. A. S. 88: 2692-2696, 1991.
17. O'Banion MK, Sardowski HB, Winn V, Young DA. A serum and glucocorticoid regulated 4-kilobase RNA encodes a cyclooxygenase-related protein. J Biol Chem 266: 23261-7, 1991.
18. Meade EA, Smith WL, Dewitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other nonsteroidal antiinflammatory drugs. JBiol Chem 268: 6610-6614, 1993.
19. Masferrer JL, Zioeifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclo-oxygenase-2 in vivo is antiinflammatory and non-ulcerogenic. P. NA. S. 91: 3228-3232, 1994. mRNA encodes a cyclooxygenase-related protein. JBiol Chem 1991; 266: 23261-7.
20. Lipsky PE, Isakson PC. Outcome of specific COX-2 inhibition in rheumatoid arthritis. J Rheumatol24 (Suppl 49): 9-14, 1997.
21. Bjarnason I, Macpherson A, Rotman H, Schupp, Hayllar J. A randomized doubleblind, cross-over study on the gastroduodenal tolerability of a highly specific cyclooxygenase-2 inhibitor, flosulide and naproxen. Scand JGastroenterol 32: 126-130, 1997.
22. Simon LS, Lanza FL, Lipsky PE et. al. Preliminary safety and efficacy of SC-58635, a novel COX-2 inhibitor. Arthritis Rheum 41: 1591-1602, 1998.
23. Laine L, Harper S, Simon T, Bath T, Johnson J, Schwartz H, Stern S, Quan H, Bolognese J. A randomized trial comparing the effect of Rofecoxib, a cyclooxygenase 2specific inhibitor, with that of ibuprofen on the gastroduodenal mucosa of patients with osteoarthritis. Gastroenterology 117: 776-783, 1999.
24. Will super aspirin supersede aspirin Modern DrugDiscovery May/June 54-59, 1999.
25. Ligumsky M, Grossman MI, Kauffman Jr GL. Endogenous gastric mucosal prostaglandins: their role in mucosal integrity. Am. J physio 242: G337-341, 1982..
26. Ligumsky M, Golanska EM, Hansen DG, Kauffman Jr GL. Aspirin can inhibit gastric mucosal cyclo-oxygenase without causing lesions in the rat. Gastroenterology 84; 756-761, 1983.
27. Ligumsky M, Sestieri M, karmeli F, Zimmerman J, Okon E, Rachmilewitz D. Rectal administration of nonsteroidal antiinflammatoruy drugs. Gastroenterology 98: 12451249, 1990.
28. Whittle BJR. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology 80: 94-98, 1981.
29. Ivey KK, Paone DB, krause WJ. Acute effect of systemic aspirin on gastric mucosa in man. Dig. Dis Sci. 25: 97-99, 1980.
30. Konturek JW, Dembinski A, Konturek SJ, Stachura J, Domschke W. Infection of Helicobacter pylori in gastric adaptation to continued aspirin administration in human subjects. Gastroenterology 114: 245-255, 1998.
31. Langerbach R, Morham SG, Tiano HF, Loftin CD et. al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacininduced gastric ulceration. Cell 83: 483-492, 1995.
32. Morham SG, Langenbach R, Loftin CD et. al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83: 473-482, 1995.
33. Mizuno H, Sakamoto C, Matsuda K et. al. Induction of COX-2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology 112: 387-397, 1997.
34. Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest 98: 2076-2085, 1996.
35. Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology 112: 1000-1016, 1997.
36. Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Sm X Physiol. 259: G462-467, 1990.
37. McCafferty D-M, Granger DN, Wallace JL. Indomethacin-induced gastric injury and leukocyte adherence in arthritic vs healthy rats. Gastroenterology 109 ; 1173-1180, 1995.
38. Mahmud T, Rafi, SS, Scott, DL, Wrigglesworth JM, Bjarnason I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum 39: 1998-2003, 1996.
39. McCormack K, Brune K. Classical absorption theory and the development of gastric mucosal damage associated with non-steroidal anti-inflammatory drugs. Arch Toxicol 60: 261-269, 1987.
40. Lichtenberger, LM. The hydrophobic barrier properties of gastrointestinal mucus. Ann. Rev. Physiol. 57: 565-583, 1995.
41. Hills BA, Butler BD, Lichtenberger LM. Gastric Mucosal Barrier: The hydrophobic lining to the lumen of the stomach. Am. J ; Physiol.: Gastrointestinal and Liver Physiology 7: G561-68, 1983.
42. Lichtenberger LM, Graziani LA, Dial EJ, Butler BD, Hills BA. Role of surface-active phospholipids in gastric cytoprotection. Science 219: 1327-29, 1983.
43. Spychal RT, Marrero JM, Saverymuttu SH, Northfield TC. Measurement of the surface hydrophobicity of human gastrointestinal mucosa. Gastroenterology 97: 104-11, 1989.
44. Go MF, Lew GM, Lichtenberger LM, Genta RM, Graham DY. Gastric mucosal hydrophobicity and Helicobacter pylori: response to antimicrobial therapy. Am J Gastroenterol 88: 1362-65, 1993.
45. Butler BD, Lichtenberger LM, Hills BA. Distribution of surfactants in the canine GI tract and their ability to lubricate. Am. J Physiol: Gastointestinal and Liver Physiology 7: G 645-51, 1983..
46. Kao Y-CJ, Lichtenberger LM. A method to preserve extracellular surfactant-like phospholipids on the luminal surface of the rodent gastric mucosa. J Histochem. Cytochem. 38: 427-31, 1990.
47. Kao Y-CJ, Lichtenberger LM. Phospholipid-and neutral-lipid-containing organelles of rat gastroduodenal mucous cells. Gastroenterology 101: 7-21, 1991.
48. Goddard PJ, Lichtenberger LM. Does aspirin damage the canine gastric mucosa by reducing its surface hydrophobicity ? Am. J Physiology: Gastrointestinal and Liver Physiology 15: G421-30, 1987.
49. Goddard PJ, Kao Y-CJ, Lichtenberger LM. Luminal surface hydrophobicity of canine gastric mucosa is dependent on a surface mucous gel. Gastroenterology 98: 361-70, 1990.
50. Dial EJ, Lichtenberger LM. A role for milk phospholipids in protection against gastric acid. Gastroenterology 87: 379-385, 1984.
51. Lichtenberger LM, Romero JJ, Kao Y-C, Dial EJ. Gastric protective activity of mixtures of saturated polar and neutral lipids in rats. Gastroenterology 99 ; 311-326, 1990.
52. Lichtenberger LM, Wang Z-M, Romero JJ, Ulloa C, Perez JC, Giraud M-N, Barreto JC. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nature Medicine 1: 154-158, 1995.
53. Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Phospholipid association reduces the gastric toxicity of aspirin in human subjects. Am J Gastroenterol 94: 1818 1822, 1999.
54. Lichtenberger LM, Ulloa C, Vanous AL, Romero JJ, Dial EJ, Illich PA, Walters ET. Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. JPET 1996; 277: 1221-1227.
55. Randall LO, Selitto JJ. A method for measurement of analgesic activity of inflamed tissue. Arch. Int. Pharmacodyn. 111: 409-411, 1957.0
A1. Faden, A. I., Experimental neurobiology of central nervous system trauma. Crit Rev
Neurobiol, 1993.7 (3-4): p. 175-86.
A2. Rogers, J., et al., Inflammation and Alzheimer's disease pathogenesis. Neurobiol
Aging, 1996.17 (5): p. 681-6.
A3. Hains, B. C., J. A. Yucra, and C. E. Hulsebosch, Reduction of pathological and behavioral deficitsfollowingspinal cord contusion injurywith the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma, 2001.18 (4): p. 409-23.
A4. Stewart, W. F., et al., Risk of Alzheimer's disease and duration oJ7NSAID use. Neurology, 1997.48 (3): p. 626-32.
A5. Gabriel, S. E., L. Jaakkimainen, and C. Bombardier, Riskfor serious gastrointestinal complications related to use ofnonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med, 1991.115 (10): p. 787-96.
A6. Lichtenberger, L. M., et al., Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal ofNSAID-induced gastrointestinal injury. Nat Med, 1995.1 (2): p. 154-8.
A7. Anand, B. S., et al., Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Am J Gastroenterol, 1999.94 (7): p. 1818-22.
A8. Lichtenberger, L. M., et al., Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. J Pharmacol Exp Ther, 1996.277 (3): p. 1221-7.
A9. Lichtenberger, L. M., et al., Phosphatidylcholine association increases the anti inflammatoyy and analgesic activity of ibuprofen in acute and chronic rodent models ofjoint inflammation: relationship to alterations in bioavailability and cyclooxygenase-inhibitory potency. J Pharmacol Exp Ther, 2001.298 (1): p. 279-87.
A10. Clatworthy, A. L., et al., Role of peri-axonal inflammation in the development of thermal hyperalgesia and guarding behavior in a rat model of neuropathic pain. Neurosci Lett, 1995.184 (1): p. 5-8.
A11. Coggeshall, R. E., et al., Is large myelinatedfiber loss associated with hyperalgesia in a model of experimental peripheral neuropathy in the rat? Pain, 1993.52 (2): p. 233-42.
A12. Carlson, S. L., et al., Acute inflammatory response in spinal cordfollowing impact injury. Exp Neurol, 1998.151 (1): p. 77-88.
A13. Hirst, W. D., et al., Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Mol Cell Neurosci, 1999.13 (1): p. 57-68.
A14. Resnick, D. K., et al., Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma, 1998.15 (12): p. 1005-13.
A15. Plunkett, J. A., et al., Effects of interleukin-10 (IL-10) on pain behavior and gene expression following excitotoxic spinal cord injury in the rat. Exp Neurol, 2001.168 (1): p. 144-54.
A16. Basso, D. M., M. S. Beattie, and J. C. Bresnahan, A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma, 1995.12 (1): p. 1-21.
A17. Grill, R., et al., Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth andpartialfunctional recovery after spinal cord injury. J Neurosci, 1997.17 (14): p. 5560-72.
A18. Rabchevsky, A. G., et al., CyclosporinA treatmentfollowingspinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing J Neurotrauma, 2001. 18 (5): p. 513-22.
A19. Hsiao, K., et al., Correlative memory deficits, Abeta elevation, and amyloidplaques in transgenic mice. Science, 1996.274 (5284): p. 99-102.
A20. Hsiao, K., Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol, 1998.33 (7-8): p. 883-9.
A21. Lim, G. P., et al., Ibuprofensuppressesplaquepathologyand inflammation inamouse modelfor Alzheimer's disease. J Neurosci, 2000.20 (15): p. 5709-14.
A22. Morris, R., Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Met, 1984.11 (1): p. 47-60.
A23. Lichtenberger, L. M., R. Darling, and J. J. Romero, Effect of luminal damaging agents on the gastric mucosal barrier and prostaglandin metabolism in cyclooxygenase (COX) knockout mice. Gastroenterology, 2001.120: p. A-143.
B1. Wu KK. Thrombogenesis, Atherogenesis and Hypercoagulability in "Thromboembolic Disorders"edited by Wu KK. PSG Publisher, Littleton, Mass, 1984, pp 5-18.
B2. Schafer AI, Handin RI. The role of platelets in thrombotic and vascular disease. Progr
Cardiovasc Dis 22: 31, 1979.
B3. Fuster V, Chesbro JH. Platelet inhibitor drugs in management of arterial thromboembolic and atherosclerotic disease. Mayo Clinic Proc. 56: 265, 1981.
B4. Fields WS, Lemak NA, Frankowsk RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Stroke 8: 301-314, 1977.
B5. Canadian Cooperative Study Group. A randomized trial of aspirin and sulfide pyrazone in threatened stroke. New Eng J Med 299: 53-59, 1978.
B6. Lewis HD Jr, Davis JW, Arclirbald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in man with unstable anginas. Results of a VA cooperative study. N Eng J Med 313: 396, 1983.
B7. The Steering Committee of the Physicians Health Study Research Group Preliminary Report: Findings from the aspirin component of the ongoing physicians health study. N Eng J Med 318: 362, 1988.
B8. Cryer B, Feldman M. Effects of very low dose daily, long term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury. Gastroenterology 117: 17-25, 1999.
B9. Vane J. Towards a better aspirin. Nature 367: 215-216, 1994.
B10. Smith WL, DeWitt DL. Biochemistry of prostaglandin endoperoxide H synthase-1 and synthase-2 and their differential susceptibility to non-steroidal anti-inflammatory drugs. Seminars in Nephro. 15: 179, 1995.
B11. Rome LH, Lands WEM. Structure requirements for time dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci USA 72: 48634865, 1975.
B12. Laneuville O, Breuer DK, DeWitt DL et. al. Differential inhibition of human prostaglandin endoperoxide H synthase-1 and-2 by non steroidal anti-inflammatory drugs. J Pharm Exp Ther 271: 927-934, 1994.
B13. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action of aspirinlike drugs. Nature 231: 232, 1971.
B14. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets I. Acetylation of a particular fraction protein. J Clin Invest 56: 624-632, 1975.
B15. Hennekens CH, Buring JE. Aspirin and cardiovascular disease. Bull N Y Acad Med 65: 57-68, 1989.
B16. Viinikka L. Acetylsalicylic acid and the balance between prostacyclin and thromboxane. Scand J Clin Lab Invest 50 (supple 201): 103, 1990.
B17. Lekstrom JA, Bell WR. Aspirin in the prevention of thrombosis. Med 70: 161, 1991.
B18. Gabriel SE, Fehring RA. Trends in the utilization of non-steroidal anti-inflammatory drugs in the United States, 1986-1990. J Clin Epidemiol 45: 1041-1044, 1992.
B19. Keifer DM; A century of pain relief. Todays Chemist at Work, December 38-42, 1997.
B20. Gabriel SE, Jaakkimainen R, Bombardier C. Risk for serious gastrointestinal complications related to the use of nonsteroidal anti-inflammatory drugs. Ann Int Med 115: 787-796, 1991.
B21. Lichtenberger LM, Wang ZM, Romero JJ, Ulloa C, Perez J, Giraud M-N, Barreto JC. NSAIDs associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced G. I. injury. Nature Medicine 1: 154-158, 1995.
B22. Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Evidence that phospholipid reduces the gastric toxicity of aspirin in human subjects. Am J Gastroenterol 94: 1818-1822, 1999.
B23. Lichtenberger LM, Ulloa C, Vanous AL, Romero JJ, Dial EJ, Illich PA, Walters ET. Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. J Pharm Exp Therap 277: 1221-1227, 1996.
B24. Benedict CR, Refine CJ, Keyt BA, Pakala R, Paoni NF, Thomas R, Bennett WF. New variant of human tissue plasminigen activator (TPA) with enhanced efficacy and lower incidence of bleeding compared with recombinant human TPA. Circulation 92: 3032-3040, 1995.
B25. Blake PR, Summers MF. NOESY-1-1 Ech spectroscopy with eliminated radiation damping. J Magn Res 86: 622-625, 1990.
B26. Pinon JF. In vivo study of platelet aggregation in rats. J Pharmaco Methods 12: 79, 1984.
B27. Triplett DA, Harms CS, Newhouse P, Clark C. Platelet Function: Laboratory evaluation and clinical application. Edited by Triplett DA. American Society of Clinical Pathologists, Chicago, 1978.
B28. Sanduja SK, Mehta K, Xu X-M, Sanduja R and Wu KK. Differentiation associated expression of prostaglandin H and thromboxane A synthases in monocytoid leukemia cell lines. Blood 78: 3178-3185, 1991.
B29. Sanduja SK, Tsai AL, Aleksic NM, Wu, K. K. Kinetic of Prostacyclin Synthesis in PGHS-1 Overexpressed Endothelial cells. Am. JPhysiol. 267: C1459-1466, 1994.
B30. Gambino MC, Cerletti C, Marchi S, Garattini S, Gaetano GD. How intravenous administration of low dose aspirin inhibits both vascular and platelet cyclooxygenase activity: an experimental study in the rats. Expt Bio Med 182: 287, 1986.
B31. Pierangeli SS, Barker JH, Stikovac D, Ackerman D, Anderson G, Barquinero J, Acland R, Harris EN. Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost 71: 670-674, 1994.
B32. Edwards MH, Pierangeli S, Liu X, Barker JH, Anderson G, Harris EN. Hydroxychloroquine reverses thrombogenic antibodies in mice. Circulation 96: 4380-4384, 1997.
B33. Pierangeli SS, Liu X, Antonov JT, Sparrow JT, Harris EN, Myones BL. Induction of pathogenic anticardiolipin antibodies in a murine model. Arthritis Rheum 41: S135, 1998.
B34. Myones BL, Antonov IV, Fedorova LI, Volgin AY, Liu X, Espinola R, Harris EN, Pierangeli SS. Complexes of protein and saturated cardiolipin are capable of binding antiphospholipid antibodies and inducing thrombogenic antiphospholipid antibodies in a murine model. Arthritis Rheum 42: S369, 1999.
【0171】
結び
本明細書において引用するすべての文献は参照として本明細書に組み入れられる。本発明を十分かつ完全に記載してきたが、添付の特許請求の範囲内で、本発明を具体的に記載している以外の様式で実施しうることを理解すべきである。本発明をその好ましい態様を参照しながら開示してきたが、当業者であれば、本記載を読むことにより、上で記載し、添付の特許請求の範囲で主張する本発明の範囲および精神から逸脱することのない変更および改変を加えうることを理解すると思われる。
【図面の簡単な説明】
【0172】
【図1】図1は、アスピリン(ASA)を単独投与したラットで観察された多数の胃損傷とは対照的に、アスピリン:レシチン(LEC、レシチン油、Phosal 35 SB使用)重量比が約1:0.5、1:1および約1:2である三種のASA:LEC製剤で治療したラットはすべて、胃損傷が有意に少なかったことを示す図である。
【図2】図2は、インドメタシンは10mg/kgの用量でGI出血の重度の増大を引き起こし、これは等用量のインドメタシンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは顕著かつ有意に軽減されたことを示す図である。
【図3】図3は、イブプロフェン(ラットにおいて通常のNSAIDの中では最も毒性が低いものの一つと考えられている)は100mg/kgの用量でGI出血の適度の増大を引き起こし、これは等用量のイブプロフェンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは有意に軽減されたことを示す図である。
【図4】図4は、アスピリンは10mg/kgの用量でラットの患側の足の疼痛閾値を高める適度の能力を有するが、等用量のアスピリンをレシチン油との組み合わせで投与した場合には、試験したすべての重量比で鎮痛活性が有意に増強されたことを示す図である。
【図5】図5は、イブプロフェンは25mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のイブプロフェンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示す図である。
【図6】図6は、インドメタシンは4mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のインドメタシンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示す図である。
【図7A】図7Aは、脊髄損傷(SCI)によって誘導された痛覚過敏がPC−イブプロフェンおよびイブプロフェンによる治療で逆転することに関するデータを示す図である。
【図7B】図7Bは、脊髄損傷(SCI)によって誘導された痛覚過敏がPC−イブプロフェンおよびイブプロフェンによる治療で逆転することに関するデータを示す図である。
【図8】図8は、脊髄損傷後5週間のラットにおけるPC−イブプロフェンおよびイブプロフェンの鎮痛活性に関するデータを示す図である。
【図9】図9は、PC−イブプロフェンおよびイブプロフェンで治療した脊髄損傷ラットにおける6週間の間の体重増加に関するデータを示す図である。
【図10】図10は、PC−イブプロフェンおよびイブプロフェンで治療した脊髄損傷(PCI)後の運動機能回復に関するデータを示す図である。
【図11】図11は、PC−アスピリン複合体は等用量の非修飾アスピリンに比べて、感受性の高い個人において胃のびらんの数を有意に70%減少させ、この胃毒性の低減は薬物のCOX阻害活性の変化には関係していなかったことを示す図である。
【図12】図12は、アスピリンおよびPC−アスピリンはいずれも洞のCOX活性を>85%阻害する同等の能力を有していたことを示す図である。
【図13A】図13AおよびBは、食塩水、DPPC、ASA(20mg/kg)、またはDPPCと複合したASAの経口投与後30分のラット血小板におけるTXB2濃度を示す図である。PRPを調製し、AA(2mM)によって凝集を誘導した。TXB2はRIAによって測定した。結果は平均±SEMで表している(n=3)。*=ASAに対しp<0.050。略語:DPPC=ジパルミトイルホスファチジルコリン;AA=アラキドン酸;pRP=血小板を多く含む血漿;TXB=トロンボキサン。
【図13B】図13Bは、20mg/kgのASA単独またはDPPCとの複合体をラットに胃内投与した場合の、腹部大動脈による6KPGF1a産生に対する効果を示す図である。1時間後、大動脈を摘出し、各大動脈環を25mMのAAを含むトリス−HCl緩衝液中、370Cで10分間インキュベートした。6KPGF1aはRIAによって測定した。*=ASAに対しp<0.050;**=食塩水に対しp<0.001;n=4。
【図14A】図14Aは、食塩水またはPC対照ならびに2.5mg/kgの非修飾アスピリンまたはDPPCと複合したアスピリンを投与したウサギの血栓形成中の血流速度(kHz)の代表的記録を示す図である。
【図14B】図14Bは、ウサギ動脈血栓症モデルにおける2.5mg/kgのアスピリン単独またはアスピリン−DPPCの血栓重量に対する効果を示す図である。
【図14C】図14Cは、ウサギ動脈血栓症モデルにおける2.5mg/kgのアスピリン単独またはアスピリン−DPPCの頚動脈のPGI2対TXA2比に対する効果を示す図である。
【図15】図15は、ラットにおける肝損傷に関するデータを、絶食ラットにアセトアミノフェン(800mg/kg)単独またはP35SBとの重量比1:1および1:2の組み合わせを経口投与した後24時間のアスパラギン酸アミノ基転移酵素(AST)の血漿レベル上昇によって示す図である。
【技術分野】
【0001】
本発明は、生体適合性油および非ステロイド性抗炎症薬(NSAID)を含む独特の組成物であって、油またはその成分がNSAIDのGI(胃腸)毒性を軽減し、かつ炎症、疼痛、発熱および血栓症ならびに卒中、外傷性脳損傷、脊髄損傷、心血管疾患、卵巣癌、大腸癌(colon cancer)、アルツハイマー病、関節炎、ブドウ膜炎、および粘膜炎などの他の疾患を治療する薬物の治療活性を増強する際に有効である組成物に関する。
【0002】
特に、本発明は、内部、経口および/または局所投与することができ、溶液、ペースト、半固体、分散液、懸濁液、コロイドまたはその混合物であってもよい医薬品を形成するために、NSAIDを粉末で、リン脂質を含む生体適合性油に直接混合する製剤に関する。
【背景技術】
【0003】
NSAIDは、発熱、炎症、疼痛、血栓症および発癌を含むいくつかの生体発病過程を阻害する能力を有する化合物群を構成し、その中で最初に発見されたものはアスピリンである1。その高い治療能力のゆえに、NSAIDは世界中の一般大衆の間で、一般大衆薬および処方薬の両方で大量に消費されている。有用性が高いため、慢性関節リウマチおよび骨関節症に苦しむ3000万〜4000万人の米国人、ならびに他の炎症性状態または損傷が原因の炎症および疼痛、月経困難の疼痛;発熱;血栓症および関連する心血管疾患の発症;卵巣癌、大腸癌およびアルツハイマー病を治療/予防するために医薬品を使用している数え切れない他の人々を含む、かなりのパーセンテージの大衆が定期的にNSAIDを消費している1,2。常に増え続けるNSAID使用の傾向に伴う問題、特に高齢者における問題は、これらの薬物が一般に胃腸(GI)副作用を引き起こすということである3~6。
【0004】
胃および小腸で、薬物が消化不良(胃障害、胸やけ、胃内ガス貯留、または悪心)、びらん、胃炎/十二指腸炎および潰瘍を引き起こす者もある。NSAID使用者では胃腸出血も起こることがあり、(様々な重症度の)貧血、または最も重篤な症例では生命を脅かすこともある出血をきたしうる7,8。一つまたは複数のこれらのGI合併症は、定期的NSAID使用者の20〜40%で起こると推定されている。NSAIDの市場が大きいことを考慮すれば、頻度の低いGI合併症でも、年間推定76,000人の米国人を病院送りにし、推定7,600人が死亡している。
【0005】
NSAID作用の理解に役立つ重要な研究の一つは1970年代前半のVaneらの先駆的研究で、NSAIDファミリーの化学的に異なるメンバーが、アラキドン酸からプロスタグランジンG2およびH2への酸化および過酸化の連続的段階による変換を触媒する酵素、シクロオキシゲナーゼ(COX)の活性を阻害する能力を共有することを報告している9~11。プロスタグランジンH2は、特定のプロスタグランジン合成酵素によって触媒される過程により、標的細胞でいくつかのエイコサノイドの一つに変換されることになる。したがって、可逆的または不可逆的にCOX活性を阻害することにより、NSAIDは特定の組織または細胞液からプロスタグランジンを枯渇させると考えられ、これは組織の炎症を促進することが明らかにされている12。これらが判明した直後、Upjohn CompanyのRobertらは特定のプロスタグランジン類がいくつかの潰瘍発生性化合物および/または状態からGI上皮を保護する顕著な性質を共有することを示し、これらの脂質メディエーターの「細胞保護」特性を示した13。これら二つの重要な研究に基づき、NSAIDは粘膜COX活性を阻害し、組織から「細胞保護性」プロスタグランジンを枯渇させることにより、GI上皮の損傷および潰瘍形成を引き起こすと結論づけられた。
【0006】
アラキドン酸代謝についての我々の理解における次の最も最近の発展は1990年代前半に訪れ、当時、何人かの研究者14~18が、最初に記載された酵素(現在ではCOX−1と呼ばれている)に構造および機能的に関連している第二のCOXアイソザイム(現在ではCOX−2と呼ばれている)を同定およびクローニングした。GI粘膜を含むほとんどの組織で構成的に発現されるCOX−1とは対照的に、COX−2は主にサイトカインおよび他の炎症メディエーターによって誘導されることが判明した。これらの知見、およびCOX−2が炎症部位で選択的に発現され、非炎症性GI粘膜では低レベルまたは検出できないレベルでしか発現されないという証拠19~23に基づき、いくつかの製薬会社がCOX−2を選択的に阻害する化合物の開発を開始した。
【0007】
この努力はついに、最初の二つのCOX−2選択的阻害剤、セレブレックス(セレコキシブ)およびビオックス(ロフェコキシブ)の発売に結びついた。これまでに発表されている前臨床および臨床データより、これらの化合物は治療上有効であり、GI粘膜への毒性が低いことが明らかにされている。このニュースは医学界および一般地域社会両方において大きな興奮をもたらし、セレブレックスおよびビオックスが市場に出されて最初の二年間に調合されたこれら薬剤の処方は記録的な数となった24。
【0008】
本発明者およびNSAIDによるGI損傷を研究している他の研究者の幾人かの主な関心は、COX阻害とGI損傷および出血との間のつながりはあまり強くないということである。例えば、Ligumskyらは1980年代前半に、COX阻害を粘膜損傷から切り離したように見えるラットおよびイヌでの一連の論文を発表した25~27。当初、彼らは、アスピリンは組織から「細胞保護性」のプロスタグランジンを枯渇させる一方、その代謝物であるサリチル酸はCOX阻害活性をまったく示さないが、アスピリンとサリチル酸はイヌの胃粘膜への損傷を引き起こす同等の能力を有していることを示した25。続く齧歯類での試験で、アスピリンを皮下または胃内のいずれに投与したかに関わらず、粘膜のCOX活性は>90%阻害されるが、NSAIDを胃内に投与すると、潰瘍はラットの胃でのみ形成されることが判明した26,27。Whittleも、小腸の損傷はインドメタシン投与の48時間後、すなわちCOX活性(活性はインドメタシン投与後<3時間で完全に阻害される)が正常に戻った時点でしか生じ始めなかったため、インドメタシンのCOX阻害を誘導する効果と、小腸の粘膜損傷とは別物であると報告した28。
【0009】
粘膜COXの阻害はNSAIDによる腸疾患の病因に直接関与していないことを示唆する証拠はいくつかの臨床試験によっても支持されていることを指摘しておくべきで、これらはアスピリンをi.v.投与しても、NSAIDの経口投与とは対照的に、ヒト胃粘膜に対して検出できる組織学的損傷を引き起こさないことを報告している29。また、NSAID治療の2〜4週間後、ヒトの胃粘膜は経口アスピリンまたはインドメタシンの有害な作用に対して抵抗性となり、この適応反応はCOX活性の回復とつながっておらず、COX活性は試験期間中完全に阻害されたままであることも報告された30。
【0010】
最後に、NSAIDが主に粘膜COX−1を阻害することによってGI損傷を引き起こすとの仮説から、標的指向遺伝子破壊によりアイソザイムが欠損しているマウスは、自発粘膜潰瘍を発生しやすく、その野生型同腹仔に比べてNSAIDに対する感受性が高いと予測される。Langenbachら31は、COX−1を持たない動物は検出可能なGI疾患を持たず、むしろインドメタシンによる潰瘍発生に対して抵抗性であると報告している。さらに混乱することには、Morhamら32はその後の試験で、COX−2ノックアウトマウスは生存不可能で、腹膜炎ならびに腎疾患で死ぬことが多いことを報告している。COX−2阻害が有害である可能性は、動物を選択的COX−2阻害剤で治療した場合に、近位および遠位の腸の潰瘍治癒が悪化することを示すいくつかの動物試験によっても支持されている33,34。ヒトにおける類似の合併症はこれまでのところ報告されていない。
【発明の概要】
【発明が解決しようとする課題】
【0011】
前述の証拠に基づき、NSAIDがGI粘膜損傷を引き起こす他のメカニズムを研究し、これらの化合物のGI毒性を軽減または予防するための代替戦略開発において、この情報をいかに利用することができるかを調べるための説得力のある主張を行うことができる。NSAIDによる胃腸疾患に対し可能性のある他の標的は、これらの薬物の以下の能力である:粘膜血流量を低下させ、血管壁への白血球の付着を引き起こす能力;酸化的リン酸化を脱共役させる能力;そのプロトノフォア特性により細胞酸性化を引き起こす能力;および粘膜の疎水性、すなわち非湿潤性の特徴を弱め、それにより管腔の酸に対する組織の感受性を増大させる能力35~40。発明者の研究室では過去15〜20年間、この最後の性質に焦点を合わせている。
【0012】
1983年に、発明者の研究室は、イヌの胃粘膜を接触角分析により調べて、これが独特の疎水性表面を有するという初の知見を得た41,42。それ以来、発明者および他の研究室では、胃粘膜のこの非湿潤性表面特性が、齧歯類およびヒトを含む他のいくつかの種で見いだされることを明らかにしてきた40,43,44。さらに、生化学的技法と形態学的技法の両方を用いて、この性質は粘膜ゲル層内および層をコーティングしている界面活性剤様のリン脂質の細胞外ライニングに起因しうることが示された45~47。発明者の研究室でも、NSAIDを含む、胃粘膜に損傷を与える多くの物質が、組織を非湿潤性(疎水性)から湿潤性(親水性)の状態に速やかに転換する能力を有しており、この有害な作用は合成または精製リン脂質の投与によって軽減しうることを見いだした48~51。
【0013】
近年、研究の焦点は、NSAID−リン脂質相互作用のメカニズムにあてられている。これらの試験において、発明者の研究室では、NSAIDが粘膜ゲル層の内部および表面上のホスファチジルコリン(PC)などの双性イオンと化学的に結合することによって粘膜損傷を引き起こすと考えられ、静電結合部位は双性イオンリン脂質であるホスファチジルコリン(PC)の正に荷電したコリンヘッド基と、NSAIDの負に荷電した(カルボキシルまたはスルホニル)基との間であるという、説得力のある証拠を得ている52。この情報に基づき、発明者らのグループはラットで、投与前に合成または精製PCとあらかじめ化学的に結合させたいくつかのNSAIDのGI(胃腸)毒性を評価し、GI損傷形成および出血に関して、これらの新規薬物が非修飾NSAIDに比べてはるかに有害性が低いという証拠を得た。最近行われた予備的二重盲検交差試験において、四日間でヒト被検者に生じる胃損傷を比べると、精製(純度93%)PCを用いてのPC−アスピリンでは非修飾アスピリンよりも胃損傷が有意に少ないことが判明し、このアプローチのヒト疾患への適用性が確認された53。
【0014】
興味深いことに、発明者の研究室では、発熱、炎症/疼痛、血栓症および骨粗鬆症の動物モデルにおいて、PC−NSAIDが非修飾薬物よりもすぐれた治療上の最大効力および用量効力を有することを見いだし、これはその胃への低毒性がバイオアベイラビリティの低下では単純に説明できないことを示している52,54。
【0015】
PC(他の類似のリン脂質)およびNSAIDの組み合わせはNSAID投与の病因効果(pathogeni ceffect, 病原作用、発病作用)を軽減することになるが、これらの組み合わせの経口投与では、NSAID単独に比べて有効用量あたりの必要量が大量となるため、十分とは言えない。したがって、当技術分野において、組成物中の高いNSAID濃度を可能にするNSAIDおよび担体の組成物であって、担体がNSAIDの病因効果を軽減し、経口、内部または局所投与に適した形態である組成物が必要とされている。さらに、当技術分野において、特にアスピリン含有医薬品について、経年変化(self−life)が改善されたNSAID組成物が必要とされている。
【課題を解決するための手段】
【0016】
一般組成物
本発明は、非水性液体担体中に比較的高濃度の非ステロイド性抗炎症薬(NSAID)を含む組成物を提供する。
【0017】
本発明は、非水性液体担体中のNSAID組成物であって、担体が生体適合性油およびリン脂質を含む組成物を提供する。
【0018】
本発明は、非水性液体担体中のNSAID組成物であって、担体がリン脂質に富む生体適合性油を含む組成物を提供する。
【0019】
本発明は、非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、ヒトを含む動物において、担体またはその成分がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高める作用をする組成物も提供する。
【0020】
本発明は、非水性液体担体中に比較的高濃度のNSAID、リン脂質を含む組成物であって、ヒトを含む動物において、リン脂質がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在する組成物も提供する。
【0021】
本発明は、リン脂質および生体適合性油を含む非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、ヒトを含む動物において、リン脂質がNSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在する組成物も提供する。
【0022】
リン脂質の存在はNSAIDの一般病因性および/または毒性も軽減する。したがって、リン脂質はアセトアミノフェンの投与による肝損傷ならびに/またはイブプロフェンもしくはCOX−2阻害剤などの他のNSAIDの投与による腎および/もしくは心血管副作用を軽減ならびに/または予防する。
【0023】
一般組成物の一般製造法
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0024】
本発明は、リン脂質を含む非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合してリン脂質−NSAID結合複合体を含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0025】
本発明は、ホスファチジルコリン含有生体適合性油を含む非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合してホスファチジルコリン−NSAID結合複合体を含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0026】
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して、担体がリン脂質含有生体適合性油もしくは生体適合性油およびリン脂質またはその混合物を含む、溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法も提供する。
【0027】
乳化組成物
本発明は、非水性担体を含む組成物の水性乳濁液であって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもある組成物も提供する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、乳化組成物の粒径は、有害作用を引き起こすことなく組成物の経口服用または組織もしくは臓器部位への注射を可能にするのに十分小さい。静脈内または動脈内注射剤形のためには、マイクロエマルジョンが好ましく、平均粒径は0.5から約10μmの間まで下げることができ、好ましくは約1から5μmの間である。
【0028】
本発明は、非水性担体を含む組成物の水性マイクロエマルジョンであって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもあるマイクロエマルジョンも提供する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。
【0029】
乳化組成物の製造法
本発明は、本発明の水性乳濁液の製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌する段階を含み、乳化剤は、存在する場合には、安定な乳濁液を生成するのに十分な量で存在する方法も提供する。
【0030】
本発明は、本発明の水性マイクロエマルジョンの製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌し、乳濁液をマイクロエマルジョン生成(microemulsifying)条件下で剪断してマイクロエマルジョンを生成する段階を含み、乳化剤は、存在する場合には、安定なマイクロエマルジョンを生成するのに十分な量で存在する方法も提供する。
【0031】
乳化剤が存在しても存在しなくてもよい理由は、リン脂質自体がある程度の乳化性を有しているからである。
【0032】
炎症を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、組織の炎症を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシット(illicit)するのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い組成物も提供する。
【0033】
本発明は、組織、臓器および/または切開部の炎症ならびに他の結果を軽減するための術後治療であって、組成物が治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含むか、または組成物がその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含み、組成物がリン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い治療も提供する。当然のことながら、組成物は軟膏、噴霧剤、拭き取り布など(wipe)にコーティングしたもの、生体分解性の基板にコーティングしたものなどであってもよい。
【0034】
血小板凝集を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体、またはその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、血小板凝集を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で血小板凝集を軽減し、粘膜毒性および/または刺激が低い組成物も提供する。
【0035】
発熱状態を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体、またはその中に非水性担体組成物が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、抗発熱活性のための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で抗発熱活性を有し、粘膜毒性および/または刺激が低い組成物も提供する。
【0036】
潰瘍化組織を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液もしくはマイクロエマルジョン、またはリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む非水性includingを含む潰瘍化組織を治療するための組成物であって、リン脂質は組織の潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、組織の潰瘍化領域の炎症を軽減する組成物も提供する。
【0037】
口の潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む含嗽剤であって、リン脂質は口の潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口の潰瘍化領域の炎症を軽減し、リン脂質の量は口の潰瘍形成を軽減するのに十分であるのみならず、NSAIDによる組織損傷を軽減または提示するのにも十分である組成物も提供する。
【0038】
口、食道およびGI管潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む飲用医薬品であって、リン脂質は口、食道、および/またはGI管潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口、食道、および/またはGI管の潰瘍化領域の炎症を軽減し、リン脂質の量は口、食道、および/またはGI管潰瘍形成を軽減するのに十分であるのみならず、NSAIDが存在する場合にはそれによる組織損傷を軽減するのにも十分である医薬品も提供する。
【0039】
眼の炎症を治療するための組成物
本発明は、水性溶液中にリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む点眼剤であって、リン脂質は眼の炎症および/または潰瘍形成もしくは刺激を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、眼の強膜、ブドウ膜、水晶体または脈絡網膜領域の炎症を軽減し、リン脂質の量は眼の炎症を軽減するのに十分であるのみならず、NSAIDによる組織損傷を軽減または提示するのにも十分である薬剤も提供する。
【0040】
潰瘍化組織の治療法
本発明は、本発明の乳濁液またはマイクロエマルジョンの投与による、口、食道、GI管、および/または眼の炎症および/または潰瘍形成障害の治療法も提供する。
【0041】
中枢および/または末梢神経系の外傷を治療するための組成物
本発明は、リン脂質および治療上有効な量のNSAIDを含む非水性担体またはリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む非水性封入物(including)を含む、脊髄損傷、卒中および/または外傷性脳損傷を経口または内部治療するための組成物であって、リン脂質が血液脳関門を通過しての、または中枢神経系(CNS)もしくは末梢神経系(PNS)内へのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、NSAIDが炎症、血小板凝集、疼痛(侵害受容)感覚、細胞死および/または炎症によるアポトーシスを軽減する組成物も提供する。
【0042】
中枢および/または末梢神経系の外傷の治療法
本発明は、本発明の組成物の経口投与および/または注射による直接投与によって、脊髄損傷、卒中および/または外傷性脳損傷を治療する方法であって、直接投与は静脈内(i.v.投与)、動脈内(i.a.投与)または外傷部位に直接(直接投与)のいずれであってもよく、i.v.およびi.a.投与については、リン脂質が血液脳関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、すべての投与様式において、リン脂質がNSAIDの病因効果を軽減する方法も提供する。
【0043】
本発明は、リン脂質を含む油性担体中に比較的高濃度のNSAIDを含む水性乳濁液またはマイクロエマルジョンである、脊髄損傷(例えば、慢性疼痛症候群)、卒中および/または外傷性脳損傷の症状を改善するための医薬品であって、NSAIDおよびリン脂質が医薬品中で結合複合体を形成し、組成物が外傷組織上で外傷組織の腫脹を軽減するのに十分な濃度のNSAIDおよびNSAIDの病因効果を軽減するのに十分な濃度のリン脂質を含む医薬品も提供する。
【0044】
アルツハイマー病を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、アルツハイマー病に関連する症状を予防、治療または改善するための組成物であって、NSAIDおよびリン脂質がアルツハイマー病の症状の発現を予防するか、またはアルツハイマー病の症状を改善する作用をする組成物も提供する。
【0045】
アルツハイマー病の治療法
本発明は、アルツハイマー病に関連する症状の予防、治療または改善法であって、治療プロトコルに従い経口および/または内部に本発明の組成物を経口または内部投与する段階を含む方法も提供する。
【0046】
本発明は、同様の要素は同じ番号が付されている添付の例示的図面と共に、下記の詳細な説明を参照すればより十分に理解することができる。
【0047】
語句の定義
下記の語句は以下に示す意味を有し、これらの意味はその一般的に受け入れられている意味に対応しても、しなくてもよい。
【0048】
「NSAID」なる語句は、非ステロイド性抗炎症薬として一般に分類される薬物のいかなる変種も意味し、イブプロフェン、ピロキシカム、サリチル酸塩、アスピリン、ナプロキセン、インドメタシン、ジクロフェナク、アセトアミノフェン、COX2阻害剤またはそのいかなる混合物も含まれるが、これらに限定されることはない。
【0049】
「本質的に含まない」なる語句は、所与の成分を生物学的に不活性および/または活性でない量で含む組成物を意味し、好ましくは成分は約0.10重量%未満の所与の成分量で存在し、特に約0.01重量%未満が好ましい。
【0050】
「比較的高濃度」なる語句は、NSAIDの担体に対する重量比が約10:1から約1:10であることを意味する。好ましくは、NSAIDの担体に対する重量比は約5:1から約1:5、特に約2:1から1:2、とりわけ約2:1から1:1である。
【0051】
双性イオンリン脂質なる語句は広範なリン脂質を意味し、ホスファチジルコリン、ホスファチジルセリン、ホスファチジルエタノールアミン、スフィンゴミエリンおよび他のセラミド、ならびに様々な他の双性イオンリン脂質が含まれるが、これらに限定されることはない。
【0052】
「生体適合性油」なる語句は、動物の消費またはFDAによってヒトの消費が承認されている、いかなる油も意味する。
【0053】
「内部投与」または「内部投与した」なる語句は、最初に消化管を通過することなく、組成物を血流、組織部位、臓器などに直接提供する、いかなる技法による投与も意味する。
【0054】
「経口投与」または「経口投与した」なる語句は、口からの投与を意味する。
【0055】
「局所投与」または「局所投与した」なる語句は、皮膚、粘膜ゲル層、眼、組織および/または外科処置中に曝露される臓器、もしくは任意の他の曝露される体組織などの表面上への投与を意味する。
【0056】
「結合複合体」なる語句は、NSAIDおよび双性イオンリン脂質の間の相互作用などの、NSAIDおよびリン脂質の間の非共有結合による化学的および/または物理的相互作用を意味する。
【0057】
「双性イオン(zwitter ion)」なる語句は、生理的pHで分子が正に荷電した官能基および負に荷電した官能基の両方を含むことを意味する。
【0058】
「陰イオン性リン脂質」なる語句は、生理的pHで全体に負の電荷を有するリン脂質を意味する。
【0059】
「中性リン脂質」なる語句は、非荷電脂質を意味する。
【0060】
「乳濁液」なる語句は、一つの不混和相のもう一つの不混和相中の懸濁液であって、第二相中の第一相の小滴の形での懸濁液を意味する。本明細書において用いられる乳濁液なる語句は、速やかに分離するか、または全く分離しない懸濁液を含み、したがって、安定および不安定乳濁液を含む。
【0061】
「安定乳濁液」なる語句は、調整後少なくとも1日は分離せず、好ましくは少なくとも1週間は分離せず、特に少なくとも1ヶ月後まで分離せず、とりわけ無期限に乳濁液のままである、水中油混合物を意味する。
【0062】
「安定マイクロエマルジョン」なる語句は、調整後少なくとも1日は分離せず、好ましくは少なくとも1週間は分離せず、特に少なくとも1ヶ月後まで分離せず、とりわけ無期限に乳濁液のままである、水中油混合物を意味する。
【0063】
「比較的疎水性の関門」なる語句は、疎水性を有し、関門を通過しての親水性試薬の輸送を一般に阻止するか、または低減する、いかなる外部、内部、細胞性または細胞下関門も意味する。そのような関門には、親水性物質よりも疎水性物質をより容易にそれを通して輸送する、ヒトを含む動物の粘膜ゲル層、原形質膜(細胞膜)、血液脳関門、またはいかなる他の関門も含まれるが、これらに限定されることはない。
【発明を実施するための最良の形態】
【0064】
発明者は、リン脂質および場合によりNSAIDを含む非水性液体生体適合性担体を含む独特の医薬製剤を製造して、粘膜組織潰瘍化の修復を改善し、かつ/またはNSAID投与の病因効果を軽減しうることを見いだした。NSAIDが存在する場合には、NSAIDの担体に対する重量比は一般に約10:1から約1:10であり、これはNSAIDとしては低いGI毒性および高い治療活性の予想外の性質を有する、担体中に高度に濃縮されたNSAID混合物となる。好ましくは、NSAIDの担体に対する重量比は約5:1から約1:5、特に約2:1から1:2、とりわけ約2:1から1:1である。
【0065】
NSAIDを含む組成物について、本発明を縮小してNSAIDによる潰瘍疾患、および後足の急性炎症の齧歯類モデルで実施した。製剤は溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物の形であってもよい。
【0066】
NSAIDを含まない組成物について、リン脂質自体が組織の潰瘍形成、特に放射線療法および/または化学療法が原因の組織潰瘍形成の予防および/または軽減において治療上有益な効果を付与することができる。
【0067】
非水性液体生体適合性担体は生体適合性油または生体適合性油もしくは油様物質の混合物を含む。生体適合性油または油混合物はリン脂質を元々含んでいてもよく、またはそれにリン脂質が加えられていてもよい。元々または担体への添加により存在するリン脂質の量は、組織の潰瘍形成を予防、軽減または治療するために十分であるか、あるいは製剤がNSAIDを含む場合には、GI潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管疾患などのNSAIDの病因効果、ならびに/または高血圧、アテローム性動脈硬化症、血栓症、狭心症、卒中および心筋梗塞などの副作用を軽減するのに十分である。
【0068】
発明者は、前述の組成物の水性乳濁液またはマイクロエマルジョンを生成して、様々な型の癌の放射線療法および/または化学療法の結果生じる、またはこれらが原因の口、食道およびGI潰瘍形成を治療しうることも見いだした。乳濁液またはマイクロエマルジョンは、放射線療法および/もしくは化学療法の後、最中、前に投与してもよく、またはその前、最中、および/もしくは後の投与を含む混合プロトコルで投与してもよい。
【0069】
発明者および他の研究者らによる以前の出版物および特許において、リン脂質およびNSAIDを含む組成物は、成分をメタノール、エタノールもしくはクロロホルムなどの有機溶媒にまず溶解し、蒸留もしくは蒸発によって溶媒を除去することにより生成するか、またはNSAIDをリン脂質を加えた水性溶液に溶解した後、凍結乾燥した。これらの工程は二つの成分を化学的に相互作用させて複合体を形成させる。これらの工程は、ジパルミトイルホスファチジルコリン(DPPC)などの合成的に製造されたリン脂質として、または精製もしくは半精製化合物としてホスファチジルコリンを用いることが最も多かった。
【0070】
本発明は、概括的には、リン脂質および場合によりNSAIDを含む非水性液体生体適合性担体を含む医薬製剤または組成物であって、リン脂質は組織の潰瘍形成および/または炎症を予防、軽減または治療するために十分な量であり、NSAIDが存在する場合には、リン脂質はNSAIDの病因効果を軽減することができる量で存在する医薬製剤または組成物に関する。製剤は一般に、粘稠溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物であり、経口投与、直接投与、内部投与または局所投与することができる。
【0071】
本発明は、概括的には、リン脂質およびNSAIDを含む非水性液体生体適合性担体を含む医薬製剤または組成物であって、リン脂質は組織の潰瘍形成を予防、軽減または治療し、NSAIDのGI毒性を軽減するために十分な量である医薬製剤または組成物に関する。非水性液体を使用すると、NSAIDの有効治療量の体積を下げるための高濃度のNSAIDを有する組成物を生成することができる。製剤は一般に、粘稠溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物であり、経口投与、内部投与または局所投与することができる。
【0072】
本発明は、概括的には、NSAIDの病因効果が軽減された高濃縮NSAID組成物を生成するための医薬製剤の製造法であって、固体NSAIDを非水性担体と混合する段階を含み、担体がリン脂質含有生体適合性油もしくは生体適合性油およびリン脂質、またはその混合物を含む方法にも関する。
【0073】
本発明は、概括的には、NSAIDおよびリン脂質を含む非水性液体担体を含む医薬製剤の有効量を投与することによる炎症、疼痛または他のNSAIDで治療可能な病態の治療法であって、リン脂質はNSAIDによる病態を軽減するのに十分な量で存在し、NSAIDは治療上有効な量で存在し、リン脂質−NSAIDの組み合わせによって一用量あたりのNSAID投与量をリン脂質非存在下で同じ治療効果をイリシットするNSAIDの等価の量よりも少なくすることができる方法にも関する。
【0074】
本発明は、概括的には、非水性担体中にリン脂質および場合によりNSAIDを含む医薬製剤の有効量を投与することにより潰瘍化組織を予防、軽減および/もしくは治療する、ならびに/あるいは炎症、疼痛、または組織の炎症および/もしくは潰瘍形成に関連するNSAIDで治療可能な他の病態を軽減する方法であって、担体が生体適合性油またはその混合物である方法にも関する。
【0075】
特に、発明者は、リン脂質を元々含む非精製レシチン油などのリン脂質を含む生体適合性油を含む独特の医薬製剤であって、得られる製剤が低いGI毒性および高い治療活性の予想外の性質を有する、溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液もしくはその混合物または組成物である医薬製剤を見いだした。
【0076】
組成物は生体適合性油とリン脂質および場合によりNSAIDとを混合することによって容易に製造され、ここでNSAIDを粉末で粗製または半粗製レシチン油に直接加えて、経口投与のためにゼラチン軟もしくは硬カプセルまたはカリフォルニア州のVitaHerb Nutraceuticals of Placentiaから入手できるベジキャップ(vegicap)に加えるか、内部投与のために注射するか、あるいは局所投与のために皮膚に塗布することができる、ペースト、半固体、分散液もしくはコロイド懸濁液または同様の組成物を生成する。予想外の知見は、図1、2および15に示すとおり、この単純な製剤は前述の通常の方法で製造されるPC−NSAIDと同様、NSAIDによる潰瘍疾患の齧歯類モデルにおいて著しく低い胃腸(GI)毒性を有し、図3および4に示すとおり、足の炎症の急性モデルにおいて、ならびに図7および8に示すとおり、脊髄損傷の慢性モデルにおいて、炎症/疼痛を治療する高い治療活性も有することであった。
【0077】
一般に、NSAIDのリン脂質含有油に対する重量比は約10:1から約1:10、好ましくは約4:1から約1:4、特に約2:1から約1:2、とりわけ約2:1から約1:1の範囲である。リン脂質を元々含む、本発明の実施において有用な油について、油は一般に約10から約15重量%のリン脂質、好ましくは約10重量%から約20重量%のリン脂質、特に約10重量%から約40重量%のリン脂質を含む。しかし、これよりも多い量および少ない量のリン脂質も同様に用いることができる。しかし、約10重量%よりもはるかに少ない重量%では、有効治療量またはいかなる添加NSAIDとも結合するために十分な量が問題となり、その一方で約40重量%よりも高い重量%では、レシチン油などのリン脂質を元々含む生体適合性油に精製リン脂質を加えなければならないこともある。少量のリン脂質(約10重量%未満)を含む生体適合性油に対しては、油にリン脂質を加える。そのような油−リン脂質の組み合わせは、約90重量%という高いリン脂質濃度で製造することができる。しかし、好ましい組み合わせは約10重量%から約90重量%の間、特に約20重量%から約80重量%の間、特に約20重量%から約60重量%の間、とりわけ約20重量%から約40重量%の間の量のリン脂質を含む。
【0078】
一般に、一般用途のための本発明の組成物を含むNSAIDの用量は、一用量あたり5mgから一用量あたり500mgの範囲である。当然のことながら、これよりも低用量および高用量の製剤を製造することもできる。しかし、この用量範囲は市販のNSAIDで典型的に見られる範囲を含む。好ましくは、NSAIDの用量範囲は一用量あたり約10mgから約325mg、特に一用量あたり約25mgから約200mg、とりわけ一用量あたり約50mgから約100mgである。各NSAIDは錠剤または類似のもの一つあたり異なる用量範囲を有し、これらの範囲は、患者がその語句が本明細書において用いられるとおりのNSAIDを含む製剤を用いる場合に、一般に投与されることになるすべての範囲を含むことを意味することが理解されるべきである。また、本発明の組成物はNSAIDを単に含むのではなく、リン脂質を含む非水性液体担体中にNSAIDを含み、リン脂質の量は、NSAIDが存在する場合には、NSAIDの治療効果を増強する一方で、NSAIDの病因効果を軽減するのに十分であることも理解されるべきである。これらのNSAIDの病因効果は、当然のことながら、NSAIDに特有であるが、潰瘍形成、出血などのGI損傷(全てではないにしてもほとんどのNSAID)、肝損傷(例えばアセトアミノフェン)、腎損傷(例えばイブプロフェン、アセトアミノフェン、COX−2阻害剤)、心損傷(例えばCOX−2阻害剤)などが含まれ、これらに限定されることはない。NSAID−リン脂質結合複合体のため、所与の治療効果または反応(熱の低下、炎症の軽減、血小板凝集の軽減など)をイリシットするのに必要とされるNSAIDの用量(mg)が低下する。低下は用量(mg)の1倍低下から用量(mg)の15分の1への低下の範囲でありうる。好ましくは、範囲はNSAID用量(mg)の約1倍低下から約10分の1への低下である。リン脂質−NSAIDの組み合わせを含む組成物によって増大した生物活性は、NSAIDの毒性の同等の増大をきたすことはないが、驚くべきことに、本明細書に示すデータが証明するとおり、NSAIDの毒性の低下を引き起こす。
【0079】
慢性関節リウマチ、アルツハイマー病、CNSおよびPNS外傷などのより重度の状態、またはNSAIDおよび/またはリン脂質で治療可能な他のより重度の状態に対しては、NSAIDの必要一日用量は一般にはるかに高い。典型的には、一日用量は一日あたり約100mgから約5000mg、好ましくは一日あたり約500mgから約3000mg、特に一日あたり約750mgから約3000mg、とりわけ一日あたり約1000mgから約3000mgの範囲である。さらに、リン脂質−NSAIDの組み合わせの有効性が高まったことにより、NSAIDの病因性または毒性が同時に高まることなく、より高い治療効果をイリシットすることが可能となる。当然のことながら、NSAIDのこの生体活性の増大によって、投与するNSAIDの用量を引き下げることが可能となる。
【0080】
一般的な経験により、NSAID活性を増強するのに十分な量のリン脂質を含む一方で、NSAID毒性を軽減する、非水性担体、生体適合性油にNSAIDが溶解、分散、懸濁または他の方法により混合されている、本発明の製剤でNSAIDを投与する場合、必要投与量は患者、特定の状態および他の要因に応じて、その状態を治療するのに必要とされる推奨用量の5%という低い量からその用量の100%まででありうる。好ましくは、投与量は特定の状態を治療するのに必要とされる推奨用量の約10%から約90%、特に特定の状態を治療するのに必要とされる推奨用量の約10%から約50%である。所与の状態のための所与のNSAIDに対する必要推奨投与量は、Physicians Desk Reference (PDR)、AMAの出版物、FDAの出版物などの出版物中に見いだされ、これらは十分に確立された基準である。
【0081】
本発明の組成物は以下も含むことができる:(1)医薬品として許容される量のビタミンA、ビタミンC、ビタミンEまたはFDAによってヒトおよび動物の消費が承認されている他の抗酸化剤、ならびにその混合物または組み合わせからなる群より選択される抗酸化剤;(2)医薬品として許容される量の銅、亜鉛、金、アルミニウムおよびカルシウムならびにその混合物または組み合わせからなる群より選択される多価陽イオン;(3)医薬品として許容される量のジメチルスルホキシド/DMSO、プロピレングリコール/PPG、および中鎖トリグリセリド/MCTならびにその混合物または組み合わせからなる群より選択される流動性、拡散性、または浸透性促進剤;(4)医薬品として許容される量の食品着色料または非毒性色素;(5)医薬品として許容される量の香味増強剤;(6)賦形剤;および/または(7)補助剤。
【0082】
一般組成物
本発明は、非水性液体担体中に比較的高濃度の非ステロイド性抗炎症薬(NSAID)を含む組成物に関する。好ましくは、担体は生体適合性油およびリン脂質またはリン脂質に富む生体適合性油を含む。担体は元々および/または添加により、ヒトの体を含む動物の体内で、NSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量のリン脂質を含む。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0083】
本発明は、リン脂質および生体適合性油を含む非水性液体担体中に比較的高濃度のNSAIDを含む組成物であって、リン脂質がヒトの体を含む動物の体内で、NSAIDの病因効果を軽減し、NSAIDのバイオアベイラビリティを高め、かつ比較的疎水性の関門を通過してのNSAIDの有効性を高めるのに十分な量で存在し、組成物用量がNSAIDおよび/またはリン脂質の治療上有効な量を送達するのに十分であり、NSAIDの量がリン脂質非存在下で同等の治療効果をイリシットするのに必要とされるNSAIDの量の1〜10分の1である組成物に関する。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0084】
本発明の組成物中のリン脂質の存在はNSAIDの一般および特定の病因性および/または毒性も軽減する。したがって、リン脂質はアセトアミノフェンの投与による肝損傷ならびに/またはイブプロフェンもしくはCOX−2阻害剤などの他のNSAIDの投与による腎および/もしくは心損傷を軽減ならびに/または予防する。
【0085】
一般組成物の一般製造法
本発明は、非水性液体担体中にNSAIDを含む組成物の製造法であって、NSAIDを担体と混合して、比較的高濃度のNSAIDを含む溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液またはその混合物を形成する段階を含む方法にも関する。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる組成物は比較的高濃度のリン脂質−NSAID結合複合体を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0086】
乳化組成物
本発明は、非水性担体を含む組成物の水性乳濁液であって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもある組成物にも関する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる乳濁液は比較的高濃度のリン脂質−NSAID結合複合体を有する組成物を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0087】
本発明は、非水性担体を含む組成物の水性マイクロエマルジョンであって、担体が生体適合性油、治療上有益な効果を生じるのに十分な量のリン脂質、およびゼロから治療上有効な量のNSAIDを含み、NSAIDが存在する場合にはリン脂質の量はNSAIDの病因効果を軽減するのに十分でもあるマイクロエマルジョンにも関する。水性乳濁液は、組成物を長期間乳濁液の状態に保つための生体適合性乳化剤を含むこともできる。水性マイクロエマルジョンは、組成物を長期間マイクロエマルジョンの状態に保つための生体適合性乳化剤を含むこともできる。好ましくは、担体はリン脂質含有生体適合性油または生体適合性油およびリン脂質を含む。好ましくは、得られる乳濁液は比較的高濃度のリン脂質−NSAID結合複合体を有する組成物を含む。特に、得られる組成物は比較的高濃度のホスファチジルコリン−NSAID結合複合体を含む。
【0088】
乳化組成物の製造法
本発明は、本発明の水性乳濁液の製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌する段階を含み、乳化剤は、存在する場合には、安定な乳濁液を生成するのに十分な量で存在する方法にも関する。
【0089】
本発明は、本発明の水性マイクロエマルジョンの製造法であって、所与の量の本発明の望ましい非水性組成物を水性溶液に、乳化剤非存在下または存在下で加え、組成物および溶液を乳濁液を生成するのに十分な時間撹拌し、乳濁液をマイクロエマルジョン生成(microemulsifying)条件下で剪断してマイクロエマルジョンを生成する段階を含み、乳化剤は、存在する場合には、安定なマイクロエマルジョンを生成するのに十分な量で存在する方法にも関する。
【0090】
乳化剤が存在しても存在しなくてもよい理由は、リン脂質自体がある程度の乳化性を有しているからである。
【0091】
炎症を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、組織の炎症を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で組織の炎症を軽減し、粘膜毒性および/または刺激が低い組成物にも関する。
【0092】
血小板凝集を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、血小板凝集を軽減するための組成物であって、リン脂質非存在下で同等の治療反応をイリシットするのに典型的に必要とされる用量よりも低いNSAID用量で血小板凝集を軽減し、粘膜毒性および/または刺激が低い組成物にも関する。
【0093】
発熱状態を治療するための組成物
本発明は、治療上有効な量のNSAIDおよびNSAIDの病因効果を軽減するのに十分な量のリン脂質を含む非水性担体を含む、抗発熱活性のための組成物であって、リン脂質非存在下で同等の治療反応をillicit(イリシット)するのに典型的に必要とされる用量よりも低いNSAID用量で抗発熱活性を有し、粘膜毒性および/または刺激が低い組成物にも関する。
【0094】
潰瘍化および/または炎症組織を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む潰瘍化組織を治療するための組成物であって、リン脂質は組織の炎症および/または潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、組織の患部領域の炎症を軽減する組成物にも関する。
【0095】
口の潰瘍形成および/または炎症を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む含嗽剤であって、リン脂質は口潰瘍形成および/または炎症を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口の患部領域の炎症を軽減する組成物にも関する。
【0096】
口、食道およびGI管潰瘍形成を治療するための組成物
本発明は、リン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む飲用医薬品であって、リン脂質は口、食道、および/またはGI管の炎症および/または潰瘍形成を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、口、食道、および/またはGI路の患部領域の炎症を軽減する医薬品にも関する。
【0097】
眼の炎症を治療するための組成物
本発明は、水性溶液中にリン脂質、生体適合性油およびゼロから治療上有効な量のNSAIDを含む水性乳濁液またはマイクロエマルジョンを含む点眼剤であって、リン脂質は眼の炎症または刺激を軽減するのに十分な量で存在し、NSAIDは、存在する場合には、ブドウ膜炎または関連眼障害に関連する眼の炎症を軽減する薬剤にも関する。
【0098】
潰瘍化および/または炎症組織の治療法
本発明は、本発明の乳濁液またはマイクロエマルジョンの投与による、口、食道、GI管、眼および/または他の炎症および/または潰瘍化組織部位の炎症性および/または潰瘍性障害の治療法にも関する。
【0099】
中枢および/または末梢神経系の外傷を治療するための組成物
本発明は、リン脂質および治療上有効な量のNSAIDを含む非水性担体またはその中にリン脂質および治療上有効な量のNSAIDを含む非水性担体が分散されている水性溶液(例えば、乳濁液またはマイクロエマルジョン)を含む、脊髄損傷、卒中および/または外傷性脳損傷を経口または内部治療するための組成物であって、リン脂質が血液脳関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、NSAIDが炎症、血小板凝集、抗発熱活性および/または炎症による細胞死を軽減する組成物にも関する。
【0100】
中枢および/または末梢神経系の外傷の治療法
本発明は、本発明の組成物を静脈内(i.v.投与)、動脈内(i.a.投与)または外傷部位に直接(直接投与)注射することによって、脊髄損傷、卒中および/または外傷性脳損傷を治療する方法であって、i.v.およびi.a.投与については、リン脂質が血液脳関門または他の神経性関門を通過してのNSAIDの輸送を増大させて、より多くのNSAIDが外傷部位に到達し、炎症を軽減することを可能にし、すべての投与様式において、リン脂質がNSAIDの病因効果を軽減する方法にも関する。
【0101】
本発明は、リン脂質を含む油性または水性担体中に比較的高濃度のNSAIDを含む、脊髄損傷、卒中および/または外傷性脳損傷の症状を改善するための医薬品であって、NSAIDおよびリン脂質が医薬品中で結合複合体を形成し、組成物が外傷組織上で外傷組織の腫脹を軽減するのに十分な濃度のNSAIDおよびNSAIDの病因効果を軽減するのに十分な濃度のリン脂質を含む医薬品にも関する。
【0102】
アルツハイマー病を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、アルツハイマー病に関連する症状を予防、治療または改善するための組成物であって、NSAIDおよびリン脂質がアルツハイマー病の症状の発現を予防するか、またはアルツハイマー病の症状を改善する作用をする組成物にも関する。
【0103】
アルツハイマー病の治療法
本発明は、アルツハイマー病に関連する症状の予防、治療または改善法であって、治療プロトコルに従い経口および/または内部に本発明の組成物を経口または内部投与する段階を含む方法にも関する。
【0104】
切開部を治療するための組成物
本発明は、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む、切開の結果生じる手術による局所炎症を軽減し、治癒を促進するための、切開部を治療するための組成物であって、NSAIDおよびリン脂質が炎症および関連症状を軽減し、治癒を促進する作用をする組成物にも関する。
【0105】
切開部の治療法
本発明は、切開の結果生じる手術による局所炎症を軽減し、治癒を促進するための、切開部の治療法であって、手術中および手術後であるが、縫合前に、生体適合性油、リン脂質および治療上有効な量のNSAIDを含む組成物を手術部位に適用することを含み、NSAIDおよびリン脂質が炎症および関連症状を軽減し、治癒を促進する作用をする方法にも関する。本発明の好ましい治療製剤には、乳濁液もしくはマイクロエマルジョンまたは本発明の組成物の類似の製剤の噴霧適用が含まれる。
【0106】
放射線療法および/または化学療法が原因の潰瘍形成および/または炎症を改善するための組成物
本発明は、生体適合性油、リン脂質および場合により治療上有効な量のNSAIDを含む、粘膜炎または関連状態などの、特定の癌の放射線療法および/または化学療法によって引き起こされる組織の潰瘍形成を改善するための組成物であって、リン脂質は粘膜炎に関連する潰瘍形成または炎症を予防および/または軽減するのに十分な量で存在し、NSAIDが存在する場合には、リン脂質は潰瘍形成または炎症を予防および/または軽減するのみならず、NSAIDが確実に状態をさらに悪化させないようにするのにも十分な量で存在する組成物にも関する。好ましくは、化学療法に対しては、化学療法剤を適当に製剤した本発明の組成物と共に投与する。したがって、化学療法剤を経口投与する場合、化学療法剤と本発明の組成物の成分との間に有害相互作用が起こらないならば、化学療法剤を適当に製剤した本発明の組成物と混合して、患者に投与してもよい。化学療法剤と本発明の組成物の成分との間に有害相互作用が起こる場合、または化学療法剤を注射によって投与する場合、本発明の組成物を化学療法剤と共に、および十分な時間の後に経口投与し、粘膜炎発現を予防または期間短縮する。
【0107】
放射線療法および/または化学療法が原因の潰瘍形成および/または炎症の改善法
本発明は、放射線療法および/または化学療法などの医学的治療によって引き起こされる粘膜炎または他の潰瘍形成状態を予防および/または治療する方法であって、生体適合性油、リン脂質および場合により治療上有効な量のNSAIDを含み、リン脂質は粘膜炎に関連する潰瘍形成および/または炎症を予防および/または軽減するのに十分な量で存在し、NSAIDが存在する場合には、リン脂質は潰瘍形成を予防および/または軽減するのみならず、NSAIDが確実に状態をさらに悪化させないようにするのにも十分な量で存在する、本発明の組成物の治療上有効な量を、放射線療法または化学療法の前、同時、および/または後に体の患部に投与する段階を含む方法にも関する。好ましくは、組成物は経口投与用に設計され、放射線療法および/または化学療法の前および同時に投与して、粘膜炎発現を予防および/または治療および/または期間短縮する。
【0108】
本発明の組成物の経口投与のために、組成物を好ましくは乳濁液、マイクロエマルジョンなどの形で水性溶液中に小滴として分散する。小滴は、乳化剤、懸濁化剤、および含嗽剤などで一般に見られる他の成分を含んでいてもよい。本発明の組成物は、米国特許第5,407,663号、第5,236,699号、第5,130,146号、第5,085,850号に記載の製剤を含む含嗽剤または口腔衛生製剤と共に用いることもでき、前述の開示は参照として本明細書に組み入れられる。本発明の組成物は、ペースト、ロゼンジ、または経口投与用に一般に用いられるいかなる他の様式で経口投与することもできる。当然のことながら、組成物はカプセル、ゲルカプセルなどに含まれていてもよい。
【0109】
局所投与のために、本発明の組成物は、軟膏、ペースト、油、乳濁液、マイクロエマルジョン、またはその混合物もしくは組み合わせの形であってもよい。さらに、組成物は、軟膏および美容産業で一般に用いられる他の成分と混合することもできる。
【0110】
乳濁液
本発明の組成物は、乳濁液として製造および製剤することができる。乳濁液は典型的には一つの液体のもう一つの液体中の、通常は直径0.1μmを越える液滴の形での不均質系である。(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199; Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245; Block in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第2巻, p. 335; Higuchiら, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 301)。乳濁液は、十分に混合し、互いに分散した二つの不混和性の液相からなる二相系であることが多い。一般に、乳濁液は油中水(w/o)または水中油(o/w)のいずれかでありうる。水相が細かく分割され、微小液滴として大量の油相中に分散されている場合、得られる組成物は油中水(w/o)乳濁液と呼ばれる。あるいは、油相が細かく分割され、微小液滴として大量の水相中に分散されている場合、得られる組成物は水中油(o/w)乳濁液と呼ばれる。乳濁液は分散相および水相、油相いずれかの溶液として、またはそれ自体分離相として存在しうる活性薬物に加えて、追加の成分を含んでいてもよい。乳化剤、安定化剤、色素、および抗酸化剤などの医薬品賦形剤も、必要に応じて乳濁液中に存在してもよい。医薬品乳濁液は、例えば油中水中油(o/w/o)および水中油中水(w/o/w)乳濁液の場合などの、三相以上からなる多相乳剤であってもよい。そのような複雑な製剤は、単純な二成分乳濁液にはない特定の利点を提供することが多い。o/w乳濁液の個々の油滴が小さい水滴を封入している多相乳濁液はw/o/w乳濁液を構成する。同様に、油性連続相中に安定化された水滴中に封入された油滴系は、o/w/o乳濁液を提供する。
【0111】
乳濁液は熱力学的安定性が非常に小さいか、またはないことによって特徴付けられる。しばしば、乳濁液の分散または不連続相が外側または連続相に十分に分散し、乳化剤または製剤の粘性によりこの形に維持される。乳濁液型の軟膏基剤およびクリームの場合のように、乳濁液の相のいずれかは半固体または固体であってもよい。乳濁液を安定化する他の手段は、乳濁液のいずれかの相に取り込むことができる乳化剤の使用を必要とする。乳化剤は次の四つの範疇に広く分類される:合成界面活性剤、天然乳化剤、吸収基剤、および細かく分散された固体(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。
【0112】
合成界面活性剤は表面活性剤としても知られ、乳濁液の製剤において広範に適用されており、文献に総説が掲載されている(Rieger, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 285 ; Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), Marcel Dekker, Inc., New York, N. Y., 1988, 第1巻, p. 199)。界面活性剤は典型的には両親媒性で、親水性および疎水性部分を含む。界面活性剤の親水性と疎水性との比は親水性−親油性バランスと呼ばれ、製剤を製造する際に界面活性剤を分類および選択するための有用な道具である。界面活性剤は親油性基の性質に基づいて次の異なるクラスに分類することができる:非イオン性、陰イオン性、陽イオン性および両性(Rieger, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 285)。
【0113】
乳濁液製剤において用いられる天然乳化剤には、ラノリン、蜜蝋、ホスファチド、レシチンおよびアラビアゴムが含まれる。吸収基剤は、無水ラノリンおよび親水性ワセリンなどの、水を吸収してw/o乳濁液を形成するが、その半固体の稠性を保持するような親水性を有する。細かく分割された固体も、特に複数の界面活性剤の組み合わせおよび粘稠製剤において、良好な乳化剤として用いられている。これらには、重金属水酸化物などの極性無機固体、ベントナイト、アタパルジャイト、ヘクトライト、カオリン、モンモリロナイト、コロイド状ケイ酸アルミニウムおよびコロイド状ケイ酸アルミニウムマグネシウムなどの非膨潤性クレー、色素ならびに炭素またはトリステアリン酸グリセリルなどの非極性固体が含まれる。
【0114】
多様な非乳化物質も乳濁液製剤に含まれ、乳濁液の性質に寄与する。これらには、脂肪、油、ワックス、脂肪酸、脂肪アルコール、脂肪酸エステル、湿潤剤、親水性コロイド、保存剤および抗酸化剤が含まれる(Block, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 335 ; Idson, in Pharmaceutical
Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York,
N. Y., 第1巻, p. 199)。
【0115】
親水性コロイドまたはハイドロコロイドには、天然ゴムおよび多糖などの合成ポリマー(例えば、アラビアゴム、寒天、アルギン酸、カラゲナン、グアーゴム、カラヤゴム、およびトラガカント)、セルロース誘導体(例えば、カルボキシメチルセルロースおよびカルボキシプロピルセルロース)、ならびに合成ポリマー(例えば、カルボマー、セルロースエーテル、およびカルボキシビニルポリマー)が含まれる。これらは水中で分散または膨潤して、分散相の液滴周囲に強い界面フィルムを生成すること、および外側の相の粘度を高めることにより乳濁液を安定化するコロイド溶液を生成する。
【0116】
乳濁液は、微生物の増殖を容易に支持しうる、炭水化物、蛋白質、明らかにされているステロールおよびホスファチドなどのいくつかの成分を含むことが多いため、これらの製剤は保存剤を組み入れることが多い。乳濁液製剤に含まれて一般的に用いられる保存剤には、メチルパラベン、プロピルパラベン、四級アンモニウム塩、塩化ベンザルコニウム、p−ヒドロキシ安息香酸のエステルおよびホウ酸が含まれる。製剤の劣化を防ぐために、抗酸化剤も乳濁液製剤に一般に加えられる。用いられる抗酸化剤は、トコフェロール、没食子酸アルキル、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエンなどのフリーラジカル捕捉剤、またはアスコルビン酸およびメタ重亜硫酸ナトリウムなどの還元剤、ならびにクエン酸、酒石酸、およびレシチンなどの抗酸化剤相乗剤であってもよい。
【0117】
乳濁液製剤の経皮、経口および非経口経路による適用、ならびにそれらの製造法は文献に総説が掲載されている(Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。経口送達用の乳濁液製剤は、製剤の容易さ、吸収およびバイオアベイラビリティの見地からの有効性といった理由により、非常に広く用いられている。(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245 ; Idson, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 199)。鉱油を基剤とする緩下剤、油溶性ビタミンおよび高脂栄養製剤も、o/w乳濁液として一般に経口投与されている物質に含まれる。
【0118】
マイクロエマルジョン
本発明の一つの態様において、本発明の組成物をマイクロエマルジョンとして製剤する。マイクロエマルジョンは、単一の光学的等方で、熱力学的に安定な液体溶液である、水、油および両親媒性物質の系と定義することができる(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245)。典型的には、マイクロエマルジョンはまず油を水性界面活性剤溶液に分散させ、次いで十分な量の第四の成分、一般には中鎖アルコールを加えて、透明な系を生成することによって調製される系である。したがって、マイクロエマルジョンは、界面活性分子の界面フィルムによって安定化される、二つの不混和性の液体の、熱力学的に安定で、等方性の透明分散液とも記載されている(LeungおよびShah, in: Controlled Release of Drugs: Polymers and Aggregate Systems, Rosoff, M., 編, 1989, VCH Publishers, New York, pages 185-215)。マイクロエマルジョンは一般に、油、水、界面活性剤および電解質を含む三つから五つの成分の組み合わせによって調製する。マイクロエマルジョンが油中水(w/o)または水中油(o/w)型のいずれであるかは、用いる油および界面活性剤の性質、ならびに界面活性剤分子の極性頭部および炭化水素尾部の構造および幾何学的充填に依存する(Schott, in Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa., 1985, p. 271)。
【0119】
相図を用いる現象学的アプローチは広く研究されており、マイクロエマルジョンをどのようにして製剤するかについての包括的知識を当業者にもたらしている(Rosoff, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 245 ; Block, in Pharmaceutical Dosage Forms, Lieberman, RiegerおよびBanker (編), 1988, Marcel Dekker, Inc., New York, N. Y., 第1巻, p. 335)。通常の乳濁液と比べて、マイクロエマルジョンは、自然に生成する熱力学的に安定な液滴の製剤中に、水不溶性薬物を可溶化する利点を提供する。
【0120】
マイクロエマルジョンの調製に用いられる界面活性剤には、イオン性界面活性剤、非イオン性界面活性剤、Brij 96、ポリオキシエチレンオレイルエーテル、ポリグリセロール脂肪酸エステル、テトラグリセロールモノラウレート(ML310)、テトラグリセロールモノオレエート(MO310)、ヘキサグリセロールモノオレエート(PO310)、ヘキサグリセロールペンタオレエート(PO500)、デカグリセロールモノカプレート(MCA750)、デカグリセロールモノオレエート(MO750)、デカグリセロールセキオレエート(S0750)、デカグリセロールデカオレエート(DAO750)単独またはコサーファクタントとの組み合わせで含まれるが、これらに限定されることはない。コサーファクタントは通常はエタノール、1−プロパノール、および1−ブタノールなどの短鎖アルコールで、界面活性剤フィルム中に浸透し、したがって界面活性剤分子の間に生じた空隙のために無秩序なフィルムを生成することによって、界面の流動性を高める作用をする。しかし、マイクロエマルジョンはコサーファクタントを使用せずに調製することもでき、アルコールを含まない自己乳化マイクロエマルジョン系が当技術分野において知られている。水相は典型的には水、薬物の水溶液、グリセロール、PEG300、PEG400、ポリグリセロール、プロピレングリコール、およびエチレングリコールの誘導体であってもよいが、これらに限定されることはない。油相にはCaptex 300、Captex 355、Capmul MCM、脂肪酸エステル、中鎖(C8〜C12)モノ、ジ、およびトリグリセリド、ポリオキシエチル化グリセリル脂肪酸エステル、脂肪アルコール、ポリグリコール化グリセリド、飽和ポリグリコール化C8〜C10グリセリド、植物油ならびにシリコーン油等の物質が含まれるが、これらに限定されることはない。
【0121】
マイクロエマルジョンは、薬物の可溶化および薬物の吸収促進の見地から特に興味が持たれる。脂質を基剤とするマイクロエマルジョン(o/wおよびw/oの両方)は、ペプチドを含む薬物の経口バイオアベイラビリティを高めることが提唱されている(Constantinidesら, Pharmaceutical Research, 1994,11,1385-1390; Ritschel, Meth. Find. Exp. Clin. Pharmacol., 1993,13,205)。マイクロエマルジョンは改善された薬物の可溶性、酵素加水分解からの薬物の保護、界面活性剤が引き起こす膜の流動性および浸透性変更による薬物吸収促進の可能性、調製の容易さ、固体剤形に比べての経口投与の容易さ、改善された臨床効力、および低毒性なる利点を提供する(Constantinidesら, Pharmaceutical Research, 1994,11, 1385 ; Hoら, J. Pharm. Sci., 1996,85,138-143)。マイクロエマルジョンは、その成分が室温で混合すると、自然に生成することが多い。マイクロエマルジョンは、美容および医薬両方の適用における活性成分の経皮送達でも有効である。本発明のマイクロエマルジョン組成物および製剤は、胃腸管を介しての経口投与では、リン脂質および/またはNSAID−リン脂質の組み合わせからの治療反応の増大を促進し、同様に胃腸管、CNS、PNS、膣、口、食道、頬腔、鼻腔、副鼻洞および他の投与領域内の関門などの疎水性の関門を通過しての、リン脂質および/またはNSAID−リン脂質の組み合わせの局所細胞の治療反応および取り込みの改善を促進すると予想される。
【0122】
本発明のマイクロエマルジョンは、製剤の性質を改善し、本発明の製剤を含むリン脂質および/またはNSAID−リン脂質の組み合わせの吸収を促進するために、モノステアリン酸ソルビタン(Grill 3)、ラブラソル(Labrasol)、および浸透性促進剤などの追加の成分および添加物を含むこともできる。本発明のマイクロエマルジョンにおいて用いられる浸透性促進剤は、次の広く五つに分けた範疇の一つに属するとして分類することができる:界面活性剤、脂肪酸、胆汁酸、キレート剤、および非キレート非界面活性剤(Leeら, Critical Reviews in Therapeutic Drug Carrier Systems, 1991, p. 92)。これらのクラスはそれぞれ前述されている。
【0123】
本発明において用いるのに適したリン脂質には、ジミリストイルホスファチジルコリン、ジステアロイルホスファチジルコリン、ジリノレオイル−ホスファチジルコリン(DLL−PC)、ジパルミトイル−ホスファチジルコリン(DPPC)、ダイズホスファチジルコリン(ダイズ−PCまたはPCS)、および卵ホスファチジルコリン(卵−PCまたはPCE)が含まれるが、これらに限定されることはない。飽和リン脂質であるDPPCにおいて、飽和脂肪族置換基R1およびR2はCH3−(CH2)14であり、R3はCH3であり、XはHである。不飽和リン脂質であるDLL−PCにおいて、R1およびR2はCH3−(CH2)4−CH=CH−CH2−CH=CH−(CH2)7であり、R3はCH3であり、XhaHである。不飽和リン脂質の混合物である卵PCにおいて、R1は主に飽和脂肪族置換基(例えば、パルミチン酸またはステアリン酸)を含み、R2は主に不飽和脂肪族置換基(例えば、オレイン酸またはアラキドン酸)である。ダイズ−PC(Soy-PC)中では、飽和リン脂質(パルミチン酸およびステアリン酸)に加えて不飽和リン脂質の混合物(オレイン酸、リノール酸、およびリノレン酸)である。好ましい双性イオンリン脂質には、ジパルミトイルホスファチジルコリン、ホスファチジルコリン、またはその混合物が含まれるが、これらに限定されることはない。
【0124】
適当なNSAIDには、フェノプロフェンカルシウム(登録商標Nalfon)、フルルビプロフェン(登録商標Ansaid)、スプロフェン。ベノキサプロフェン、イブプロフェン(処方薬、登録商標Motrin)、イブプロフェン(200mg一般薬、登録商標Nuprin、Motrin 1B)、ケトプロフェン(登録商標Orduis、Oruvall)、ナプロキセン(登録商標Naprosyn)、ナプロキセンナトリウム(登録商標Aleve、Anaprox、Aflaxen)、オキサプロジン(登録商標Daypro)などのプロピオン酸薬;ジクロフェナクナトリウム(登録商標Voltaren)、ジクロフェナクカリウム(登録商標Cataflam)、エトドラク(登録商標Lodine)、インドメタシン(登録商標Indocin)、ケトロラクトロメタミン(筋肉内注射用登録商標Acular、Toradol)、ケトロラク(経口用登録商標Toradol)などの酢酸薬;ナブメトン(登録商標Relafen)、スリンダク(登録商標Clinoril)、トルメチンナトリウム(登録商標Tolectin)などのケトン薬;メクロフェナメートナトリウム(登録商標Meclomen)、メフェナム酸(登録商標Ponstel)などのフェナメート薬;ピロキシカム(登録商標Dolibid)などのオキシカム薬;ジフルニサル(登録商標Feldene)、アスピリンなどのサリチル酸薬;オキシフェンブタゾン(登録商標Tandearil)、フェニルブタゾン(登録商標Butazolidin)などのピラゾリン酸薬;アセトアミノフェン(登録商標Tylenol)など;セレブレックス、ビオックスなどのCOX−2阻害剤またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0125】
適当な生体適合性乳化剤には、ヒトまたは動物の消費または体内での使用が承認されている、いかなるイオン性または非イオン性乳化剤または界面活性剤も含まれるが、これらに限定されることはない。典型例には、アセチル化モノグリセリド、脂肪酸のアルミニウム塩、アラビノガラクタン、パン酵母グリカン、炭酸カルシウム、脂肪酸のカルシウム塩、イナゴマメガム(ローカストビーンガム)、カードラン、食用油脂または食用脂生成脂肪酸のモノおよびジグリセチドのジアセチル酒石酸エステル、スルホコハク酸ジオクチルナトリウム、リン酸2ナトリウム(相互参照(X−ref)−1、2および3ナトリウムリン酸塩)、エトキシル化モノおよびジグリセリド、キリンサイEucheuma cottonii抽出物、キリンサイEucheuma spinosum抽出物、脂肪酸、(アルミニウム、カルシウム、マグネシウム、カリウム、およびナトリウム)の塩、ベータ−アミラーゼ処理したn−オクチルコハク酸無水物でエステル化した食用デンプン、フラゾリドン、フルセレラン、フルセレラン、アンモニウム、カルシウム、カリウム、またはナトリウム塩、ガッチゴム、スギノリ抽出物、脂肪酸のグリセリル−ラクトエステル、ヘキシトールオレエート、ヒドロキシル化レシチン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、グリセロールおよびプロピレングリコールのラクチル化脂肪酸エステル、脂肪酸のラクチル(Lactylic)エステル、レシチン、ヒドロキシル化レシチン、メチルエチルセルロース、食用油脂または食用脂生成酸のモノおよびジグリセリド、クエン酸モノイソプロピル、食用油脂または食用脂生成脂肪酸のモノおよびジグリセチドのリン酸1ナトリウム誘導体、Myrj45(8−ステアリン酸ポリオキシエチレン)、ウシ胆汁酸抽出物、ペクチン(修飾ペクチンを含む)、ポリエチレングリコール(400)ジオレエート、脂肪酸のポリグリセロールエステル、ポリオキシエチレングリコール(400)モノおよびジオレエート、ポリソルベート60(モノステアリン酸ポリオキシエチレン(20)ソルビタン)、ポリソルベート65(トリステアリン酸ポリオキシエチレン(20)ソルビタン)、ポリソルベート80(モノオレイン酸ポリオキシエチレン(20)ソルビタン)、脂肪酸のカリウム塩、プロピレングリコールアルギネーyト(アルギン酸のプロピレングリコールエステル)、脂肪および脂肪酸のプロピレングリコールモノおよびジエステル、ナタネ油、完全硬化、過グリセリン化ピロリン酸水素ナトリウム、リン酸ナトリウムアルミニウム、次亜リン酸ナトリウム、メチル硫酸ナトリウム、ペクチニン酸ナトリウム、脂肪酸のナトリウム塩、ステアロイルラクチレートナトリウム、スルホ酢酸ナトリウム誘導体(モノおよびジグリセリド)、ソルビタンモノオレエート、ソルビタンモノステアレート、サクシニル化モノグリセリド、サクシステアリン(コハク酸水素ステアロイルプロピレングリコール)、ショ糖酢酸イソ酪酸エステル(SAIB)、ショ糖脂肪酸エステル、硫酸化オレイン酸ブチル、リン酸3ナトリウム、ザンサンガムなど、またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0126】
適当な中性脂質には、トリグリセリドなどのいかなる中性脂質も含まれるが、これらに限定されることはない。トリグリセリドなどの代表的中性脂質を部分的一覧のために、米国特許第4,950,656号および第5,043,329号を特に参照する。飽和および不飽和両方のトリグリセリドを本発明の組成物において用いることができ、トリパルミチン(飽和)、トリオレインおよびトリリノレイン(不飽和)などのトリグリセリドが含まれる。しかし、これらの特定のトリグリセリドは便宜上本明細書に記載するにすぎず、様々な有用なトリグリセリドの単なる具体例で、包括的であることをさらに意図するものではない。
【0127】
適当な生体適合性、生体分解性ポリマーの非限定例には、ポリ乳酸、ポリグリコール酸、ポリカプロラクトン、ポリ酸無水物、ポリアミド、ポリウレタン、ポリエステルアミド、ポリオルトエステル、ポリジオキサノン、ポリアセタール、ポリケタール、ポリカーボネート、ポリオルトカーボネート、ポリフォスファーゼン、ポリヒドロキシ酪酸、ポリヒドロキシ吉草酸、ポリアルキレンオキザレート、ポリアルキレンサクシネート、ポリ(リンゴ酸)、ポリ(アミノ酸)、ポリ(メチルビニルエーテル)、ポリ(マレイン酸無水物)、キチン、キトサン、およびそのコポリマー、ターポリマー、もしくは高級モノマー重合体(higher poly-monomer polymers)、またはその組み合わせもしくは混合物が含まれる。好ましい生体分解性ポリマーはすべて加水分解によって分解される。
【0128】
典型的には、ポリマーはポリ酸無水物などの表面侵食性ポリマーまたはポリオルトエステルなどの塊状侵食性ポリマーのいずれかとなる。ポリ(l−乳酸)(PILA)、ポリ(dl−乳酸)(PLA)、ポリ(グリコール酸)(PGA)、ポリカプロラクトン、そのコポリマー、ターポリマー、もしくは高級モノマー重合体、またはその組み合わせもしくは混合物が好ましい生体適合性、生体分解性ポリマーである。好ましい生体分解性コポリマーは、乳酸およびグリコール酸のコポリマーで、ポリ(dl−乳酸−グリコール酸)コポリマー(PLG)と呼ばれることもある。ポリ(dl−乳酸−グリコール酸)コポリマーの共重合モノマー(乳酸:グリコール酸)の比は、好ましくは乳酸:グリコール酸が約100:0から約50:50の間である。最も好ましくは、共重合モノマーの比は乳酸:グリコール酸が約85:15から約50:50の間である。PLG:PLAが、好ましくは約85:15から約50:50のPLAとPLGの配合物(blend)を用いて、ポリマー物質を調製することもある。
【0129】
PLA,PILA、PGA、PLGおよびその組み合わせまたは混合物または配合物は、ヒトの臨床使用が承認されている合成ポリマーに含まれる。これらは現在、手術の縫合材料として、および制御放出装置において、ならびに他の医学および薬学上の適用において用いられている。これらは生体適合性で、その分解産物は乳酸およびグリコール酸などの、通常の代謝経路に入る低分子量化合物である。さらに、ポリ(乳酸−グリコール酸)コポリマーは、乳酸とグリコール酸のコポリマー比を単に変動させることにより、分解速度が数日から数年までの広範囲に及ぶという利点を提供する。
【0130】
生物学的適用において用いられるポリマーの生体分解を促進するために、本発明の組成物は組成物において用いられるポリマーの生体分解を助けうる酵素の添加も含むことができる。好ましい酵素または類似の試薬は、エステル加水分解能力を有するプロテアーゼまたは加水分解酵素である。そのような酵素には、プロテイナーゼK、ブロメライン、プロナーゼE、セルラーゼ、デキストラナーゼ、エラスターゼ、プラスミンストレプトキナーゼ、トリプシン、キモトリプシン、パパイン、キモパパイン、コラゲナーゼ、スブチリシン、クロストリドペプチダーゼA、フィシン、カルボキシペプチダーゼA、ペクチナーゼ、ペクチンエステラーゼ、酸化還元酵素、酸化酵素などが含まれるが、これらに限定されることはない。
【0131】
適当な化学および/または放射線療法剤(商品名)には、白金錯体、金(III)錯体、パラジウム錯体、アリトレチノイン(Panretin)、アロプリノール(Zyloprim)、アルトレタミン(Hexalen)、アミフォスチン(Ethyol)、アミフォスチン(Ethyol)、アミフォスチン(Ethyol)、アナストロゾール(Arimidex)、アナストロゾール(Arimidex)、三酸化二ヒ素(Trisenox)、ベキサロテン(Targretin)、ベキサロテン(Targretin)、ブレオマイシン(Blenoxane)、静注用ブスルファン(Busulfex)、経口用ブスルファン(Myleran)、カペシタビン(Xeloda)、カペシタビン(Xeloda)、カペシタビン(Xeloda)、カルボプラチン(Paraplatin)、カルボプラチン(Paraplatin)、植込用カルムスチン+ポリフェプロサン20(Gliadel Wafer)、セレコキシブ(Celebrex)、クロランブシル(Leukeran)、シスプラチン(Platinol)、シスプラチン(Platinol)、シスプラチン(Platinol)、クラドリビン(Leustatin(2−CdA)、シクロホスファミド(Cytoxan)、シタラビンリポソーム(DepoCyt)、ダウノルビシンリポソーム(DanuoXome)、ダウノルビシンダウノマイシン(Daunorubicin)、ダウノルビシン(ダウノマイシン(Cerubidine)、デクスラゾキサン(Zinecard)、ドセタキセル(Taxotere)、ドセタキセル(Taxotere)、ドセタキセル(Taxotere)、ドキソルビシン(Adriamycin PFS注射剤)、ドキソルビシンリポソーム(Doxil)、ドキソルビシンリポソーム(Doxil)、エリオットB溶液(Elliott’s B Solution)、エピルビシン(Ellence)、エストラムスチン(Emcyt)、リン酸エトポシド(Etopophos)、リン酸エトポシド(Etopophos)、リン酸エトポシド(Etopophos)、エトポシド(VP−16 (Vepesid)、エトポシド(VP−16 (Vepesid)、エキセメスタン(Aromasin)、フルダラビン(Fludara)、フルオロウラシル(5−FU (Adrucil)、ゲムシタビン(Gemzar)、ゲムシタビン(Gemzar)、ゲムツズマブ−オゾガミシン(Mylotarg)、酢酸ゴセレリン(Zoladex植込剤)、ヒドロキシ尿素(Hydreaカプセル)、イダルビシン(Idamycin)、イダルビシン(Idamycin)、イフォスファミド(IFEX)、メシル酸イマチニブ(Gleevec)、イリノテカン(Camptosar)、イリノテカン(Camptosar)、イリノテカン(Camptosar)、レトロゾール(Femara)、レトロゾール(Femara)ロイコボリン(Leucovorin)、レバミソール(Ergamisol)、メルファランL−PAM(Alkeran)、メスナ(Mesnex)、メトトレキセート(Methotrexate)、メトキシサレン(Uvadex)、ミトキサントロン(Novantrone)、ミトキサントロン(Novantrone)、パクリタキセル(Paxene)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パクリタキセル(Taxol)、パラメーター深泥ネート(Aredia)、ペガデマーゼ(Adagen(ウシメガデマーゼ))、ペントスタチン(Nipent)、ペントスタチン(Nipent)、ポルフィマーナトリウム(Photofrin)、ポルフィマーナトリウム(Photofrin)、ポルフィマーナトリウム(Photofrin)、ストレプトゾシン(Zanosar)、タルク(Sclerosol)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、タモキシフェン(Nolvadex)、テモゾラミド(Temodar)、テニポシドVM−26(Vumon)、トポテカン(Hycamtin)、トポテカン(Hycamtin)、トレミフェン(Fareston)、トレチノインATRA(Vesanoid)、バルルビシン(Valstar)、ビノレルビン(Navelbine)、またはその混合物もしくは組み合わせが含まれるが、これらに限定されることはない。
【0132】
本発明は好ましくは未精製レシチン油の使用に関するが、本発明は、リン脂質を含むヒトが消費可能ないかなる油も含むが、これらに限定されることはない、リン脂質を含むいかなる生体適合性油も使用することができる。
【0133】
適当な生体適合性には、植物もしくは動物油またはそれらの誘導体などの天然油あるいは合成油、特にダイズ由来のレシチン油などのリン脂質に富む天然油を含む、FDAによってヒトまたは動物の消費が承認されているいかなる油も含まれるが、これらに限定されることはない。そのような油の典型例には、落花生油、ナタネ油、アボカド油、ベニバナ油、オリーブ油、トウモロコシ油、ダイズ油、ゴマ油、ビタミンA、ビタミンD、ビタミンE、魚油などの精油、植物油、硬化植物油、動物油が含まれる。
【0134】
本発明の製剤または組成物は、抗酸化剤(例えば、ビタミンA、C、D、Eなど)、微量金属および/または多価陽イオン(アルミニウム、金、銅、亜鉛、カルシウムなど)、界面活性剤および/または溶媒(例えば、プロピレングリコール/PPG、ジメチルスルホキシド/DMSO、中鎖トリグリセリド/MCTなど)、非毒性色素、ならびに香味増強剤などの他の化学物質も含むことができ、これらはアンテ遺影、流動性/拡散性、浸透性、有効性および消費者の認容性を改善するために製造されているため、これらを製剤に加えることができる。
【0135】
リン脂質、好ましくはPCおよびNSAIDを含む本発明の製剤は、経口投与用のゼラチン軟カプセルもしくは硬カプセルまたはベジキャップに充填するために用いることができ、あるいは炎症、潰瘍化および/または被刺激組織または皮膚に局所適用するために、そのまま溶液、ペースト、半固体、分散液、懸濁液、コロイド懸濁液、またはその混合物として用いることもできる。
【0136】
本製剤の一つの好ましい態様はレシチン油を基剤とするPC−NSAID組成物で、これはGI毒性について試験されている。試験された三つの製剤には、アスピリン、インドメタシンおよびイブプロフェンと組み合わせたレシチン油が含まれる。この試験において、アスピリンを35%PCを含むダイズレシチン油、Phosal 35 SBと組み合わせ、アスピリン用量18mg/kg(NSAID:レシチン油の重量比を1:0.5から1:1、1:2まで系統的に変動させた)で絶食ラットに胃内投与した。加えて、他のラット群に等用量のアスピリンをレシチン油非存在下で投与するか、または等しい体積の生理食塩水(以下、「食塩水」とも記載する)を投与した。45分後、すべてのラットを1mlの0.6N HClで胃内攻撃(チャレンジ)し、15分後、ラットを安楽死させ、胃を切開して、確立された方法50-52によって胃損傷を評価した。
【0137】
図1に示すとおり、データはアスピリンを単独投与したラットで観察された多数の胃損傷とは対照的に、三種すべてのアスピリン:レシチン製剤で治療したラットはすべて、胃損傷が有意に少なかったことを示していた。
【0138】
非アスピリンNSAIDであるインドメタシンおよびイブプロフェンの胃毒性を評価するために、もう一つの潰瘍モデルを用いた。すなわち、以前に記載されているとおり52、GI出血が終点であった。このモデルにおいて、NSAIDを絶食ラットに単独またはレシチン油Phosal 35 SBとの組み合わせ(NSAID:レシチンの重量比1:1)のいずれかで胃内投与した。対照ラットには等しい体積の食塩水を投与した。ラットをNSAIDのGI損傷効果に対してより敏感にするために、すべてのラットに酸化窒素(NO)合成酵素阻害剤であるL−NAME(20mg/kg)も、18〜20時間の試験期間中に三回注射し、その後ラットを安楽死させ、GI管の遠位20cmに2mlの食塩水を流し、GI出血の指標としてのヘモグロビン分析のために流出液を回収した。これらの実験の結果を下記の図2および3に示している。
【0139】
図2を参照すると、データはインドメタシンは10mg/kgの用量でGI出血の重度の増大を引き起こし、これは等用量のインドメタシンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは顕著かつ有意に軽減されたことを示していた。
【0140】
図3を参照すると、データはイブプロフェン(齧歯類モデル系において通常のNSAIDの中では最も毒性が低いものの一つと考えられている)は100mg/kgの用量でGI出血の適度の増大を引き起こし、これは等用量のイブプロフェンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは有意に軽減されたことを示していた。
【0141】
次いで、前述の方法を用いて、NSAID−レシチン製剤の抗炎症/鎮痛活性を(NSAID単独と比べて)評価した。これは、完全フロイントアジュバント(CFA)0.1mlをラットの左後足に注射して急性炎症反応を誘導することにより達成した。四日後、終夜絶食させたラットに、NSAID(アスピリン、インドメタシンまたはイブプロフェンのいずれか)単独またはPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)(アスピリンだけは他の比も評価した)を胃内投与した。2時間後、ラットの圧に対する疼痛感度を、ランダル−セリート法(55)を用いて測定した。これは、炎症足または対側の非炎症足に適用した圧を、ラットが疼痛感覚の最初の徴候(発声または試験中の後足の指の伸展)を示すまで漸増させることにより達成し、この値をラットの疼痛閾値として記した。したがって、低い疼痛閾値は炎症足が圧に対して非常に敏感であることを示す一方で、高い疼痛閾値は低い疼痛感度または鎮痛を意味する。結果を図4〜6に示している。
【0142】
図4を参照すると、データはアスピリンは10mg/kgの用量でラットの患側の足の疼痛閾値を高める適度の能力を有するが、等用量のアスピリンをレシチン油との組み合わせで投与した場合には、試験したすべての重量比で鎮痛活性が有意に増強されたことを示していた。
【0143】
図5を参照すると、データはイブプロフェンは25mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のイブプロフェンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示していた。
【0144】
図6を参照すると、データはインドメタシンは4mg/kgの用量でを示していた。データは、ペースト様組成物が非修飾INDOに比べて疼痛処理活性を改善し、対照食塩水に比べて疼痛処理活性を非常に改善することを示している。
【0145】
NSAIDと中枢および末梢神経系外傷および損傷
炎症の過程は、脊髄損傷(SCI)などのCNSへの急性外傷性損傷[A1]ならびにアルツハイマー病(AD)などの遅延型神経変性疾患[A2]両方に関連する進行性病態生理における主要な成分である。炎症の過程は、SCIで一般に観察される運動機能の進行性の劣化および慢性疼痛の発生、ならびにADで観察される記憶および認知機能の損失を直接引き起こすか、またはそれらに寄与すると考えられる。最近、抗炎症薬の使用が外傷性SCIの齧歯類モデルで組織損失および機能欠損の減弱において効果を示している[A3]。
【0146】
さらに重要なことには、いくつかの最近の疫学調査は、非ステロイド性抗炎症薬(NSAID)の長期消費がADのリスクを50%まで低下させる可能性を示唆している[A4]。炎症状態の性質に応じて短期または長期いずれかのNSAID治療戦略を用いうることが予想されるため、NSAIDが低用量で有効であり、かつその耐容性が高く、副作用が最小であることは極めて重要である。一般大衆の約40%が長期NSAID消費に反応して、消化不良から生命を脅かす消化性潰瘍および出血の発現誘導に及ぶ胃腸(GI)症状を発症することはよく確立されている[A5]。
【0147】
1995年に、PIの研究室は、シクロオキシゲナーゼ(COX)活性の阻害に加えて、NSAIDが上部GI管の表面疎水性関門を、おそらくはリン脂質の表面ライニングに化学的に結合することによって、減弱させる能力を有すると報告した[A6]。さらに、発明者らは、実験動物とヒトの両方で、合成種または精製抽出物(例えば、ダイズレシチン由来)のいずれかとして存在する最も有名なリン脂質、ホスファチジルコリン(PC)とNSAIDが化学的に結合している場合には、NSAIDの有害作用を防止しうることを示した[A6、A7]。興味深いことに、PC−NSAIDは、低いGI毒性に加え、おそらくPC−NSAIDの高い膜浸透性とCOX阻害活性に起因する、発熱、炎症および疼痛を阻害する高い治療活性も有していることが明らかにされた[A6、A8、A9]。
【0148】
したがって、本発明の組成物は神経の炎症を減弱し、SCIおよびADを含むいくつかの神経学的状態に関連する病態生理を軽減する。
【0149】
経口投与したPC−イブプロフェンは末梢神経結紮に関連する炎症依存性痛覚過敏の発生を低下させる
坐骨神経周囲を4本のクロミックグット縫合糸で緩く結紮すると、手術の2〜4日後に、同側後足に適用した圧または熱に対する痛覚過敏反応によって示されるとおり、患部神経の重度の末梢神経炎症および神経障害性疼痛の誘導を引き起こすことが報告されている[A10、A11]。ラットにおけるこの誘導法を用いた末梢神経炎症のPC−NSAID治療の効果および痛覚過敏の軽減。この齧歯類モデルを用いて、右または左いずれかの坐骨神経の神経炎症を誘導した。擬似手術を対側に実施した。手術の2日後、ラットを次の実験群に無作為に分類した(ラット12匹/群):食塩水対照;イブプロフェン(15mg/kg);およびPC−イブプロフェン(等用量のNSAID)。次の2日間、ラットに試験NSAID製剤を1日2回投与し、疼痛感覚のいくつかの行動指標を、2日間の投与期間の前後両方の後足で評価した。有効性を評価するために用いた行動解析は次のとおりであった:患部後足の保護行動;熱に対する後足引っ込め潜時;フォイの毛刺激に対する足引っ込め反応;および後足への圧適用に対する疼痛反応[A8]。安楽死にあたり、結紮および対照神経を摘出して、炎症の指標について肉眼検査および組織検査の両方を行った。これらの試験の結果は、後足炎症のモデル(フロイントアジュバントで誘導)においてPC−イブプロフェンの鎮痛活性はイブプロフェン単独よりも著しく高く、またPC−イブプロフェンは坐骨神経結紮による疼痛感覚の緩和においても、フォイの毛刺激および熱の両方に対する足引っ込め反応を測定することにより評価して、より有効であることを示している。
【0150】
経口投与したPC−イブプロフェンは挫傷性SCIのラットモデルにおける組織損失、移動運動機能を低下させ、慢性疼痛症候群の発生を減弱する
最近、ラット成獣において抗炎症薬の単回用量送達が脊髄傷害のサイズを低下させることが明らかにされた[A3]。これらのNSAID治療ラットは治療していないラットに比べてより大きい移動運動活性と、神経障害性疼痛に特徴的な痛覚過敏および機械的異痛症(接触による疼痛)の症状軽減を示した。慢性神経障害性疼痛の発生は、脊髄損傷後に非常に頻繁に起こり、患者の永久的な負担になりうる。慢性中枢疼痛の発生を防止または減弱するための耐容性が高く、有効な治療法の開発が痛切に必要とされている。経口投与したPC−NSAIDはSCIに関連する組織損傷を軽減し、移動運動の転帰を改善し、慢性疼痛症候群を予防する。
【0151】
PC−イブプロフェンはADのトランスジェニックマウスモデルにおけるアルツハイマー様病態生理の発生低減においてイブプロフェンよりも有効である
最近の臨床での証拠は、NSAIDがAD発症のリスクを著しく低下させる可能性を示唆している。主な問題は、ADに対する治療戦略の設計に十分な動物モデルが不足しているということである。最近確立されたヒトβ−アミロイド過剰発現Tg2576マウスは、年齢依存性記憶および認知欠損、ならびにアミロイド斑形成、小膠細胞活性化、星状細胞反応性および異栄養性神経突起を含む組織病理学的欠損を示す、都合のよい齧歯類モデルを提供する[A19〜A21]。イブプロフェンは最近、Tg2576マウスADモデルにおいてアミロイド斑、異栄養性神経突起および活性化小膠細胞の数を減少させることが明らかにされた[A21]。
【0152】
PC−NSAIDの貯蔵寿命最適化
PC−NSAIDの商品化成功には、室温条件下で長期間安定なままである製剤が必要とされる。これはイブプロフェンのようなほとんどのNSAIDにとっては問題ではないが、水に曝露されると速やかに水解されてサリチル酸となるアスピリンにとってはまだ問題である。レシチン油またはリン脂質を含む任意の他の生体適合性油などの非水性担体中に溶解および/または分散されたNSAIDを主薬とする本発明の製剤。そのような環境は疎水性であるため、これらはアスピリンを主薬とする製剤におけるアスピリンの安定性を増強することになりうる。
【0153】
中枢および末梢神経系の外傷および損傷についての実験結果
これらの実験は、PC−イブプロフェンが脊髄損傷(SCI)の有用な治療薬であることを示している。結果は、ラットの脊髄損傷(SCI)後に、体重1kgあたり25mgのNSAIDで1日2回6週間治療した際の効果を、PC−イブプロフェン、イブプロフェンおよび食塩水を比較して証明している。
【0154】
図7Aおよび7Bを参照すると、グラフ化されたデータは、ランダル−セリート法を用い、食塩水で治療したSCIラットの疼痛圧閾値の低下によって証明されるとおり、SCIがラットで痛覚過敏を引き起こしたことを示している。これとは対照的に、SCIによる痛覚過敏は、イブプロフェンまたはPC−イブプロフェンのいずれかで治療したSCIラットでは見られず、PC−イブプロフェンは非修飾イブプロフェンよりも優れているようであった。このデータを二つの様式で示している。図7Aでは、データを記録されたまま直接プロット(正規化せずに)した一方で、図7Bでは、各ラットのデータ値を、それ自体の術前基準値と比較している。このデータのグラフはおそらく、SCI後のNSAID投与の有益な効果について最も説得力のあるものである。
【0155】
図8を参照すると、SCIラットにおけるPC−イブプロフェンの優れた鎮痛活性が、漸増する直径(力に相当する)を有するフォイの毛への後足の刺激に対する後足反応の%を測定する、第二の行動試験でも示されている。ランダル−セリート試験では、高い疼痛圧閾値が鎮痛の指標であるのに対し、この場合、低い数値が鎮痛の指標であることに留意されたい。
【0156】
図9を参照すると、食塩水またはPC−イブプロフェンで治療したラットとは対照的に、イブプロフェンで治療したSCIラットでは、6週間の試験期間中に体重が増加しないという証拠。SCIラットはイブプロフェン単独に対して軽度の中毒反応を有するというこの示唆は、血液尿素窒素(腎毒性の証拠)および乳酸脱水素酵素(LDH、肝毒性の証拠)のわずかな上昇によっても示されている。
【0157】
図10を参照すると、確立されたBBB試験により評価した、SCI後の運動機能の回復は、非修飾イブプロフェンで治療したラットでは低下するが、この運動機能回復の指標において、食塩水とPC−イブプロフェンとの間には差がなかったという証拠。
【0158】
血栓性障害治療用の有効な製剤としてのPC−NSAID
生体適合性油中にホスファチジルコリン(PC)などのリン脂質およびNSAID、特にアスピリンを含む、本発明の製剤は、血栓症、卒中、および心筋梗塞を含む血栓性障害治療用の有効な製剤である。改善されたGI安全性に加えて、PC−アスピリンは通常のアスピリンよりも強力な血小板凝集および血栓形成阻害剤である。アスピリン(ASA)は双性イオンリン脂質と化学的に結合して、天然アスピリンに比べて同等または高い発熱、疼痛および炎症軽減活性を有するが、潰瘍および出血といったアスピリンの重篤な胃腸副作用は持たない結合複合体を形成する。リン脂質複合アスピリンが、動脈血栓症のインビボモデルで血栓形成を防止する際に、アスピリン単独よりも強力であることは興味深い。したがって、本発明のPC−アスピリン製剤は血小板凝集および血栓形成を阻害して、血栓性障害の症状を軽減する。
【0159】
心筋梗塞(MI)および卒中などの血栓性動脈閉塞性疾患は、米国および西洋社会における死亡の第一の原因である。米国心臓学会(American Heart Association)によれば、来る一年間に100万人を越える米国人が急性心筋梗塞を患うことになる。動脈血栓症の発生率を効果的に低下させうる薬物は、臨床上非常に重要である。血栓症は動脈閉塞性疾患の開始および拡大における重大な過程であるため、新規な特定の抗血栓薬を開発する説得力のある理由がある。動脈血栓症は、血球、血管壁および血漿蛋白質の間の一連の細胞および生化学的相互作用を含む複雑な過程である(B1)。血小板はこれらの相互作用において中心的役割を果たす(B2)。血小板は損傷血管壁に接着し、細胞の活性化、分泌および凝集を起こす。活性化された血小板は血液凝固を加速し、その分泌された分子は血管平滑筋の細胞増殖を促進する。血小板が動脈血栓症で果たす中心的役割を考慮して、血小板機能の阻害に基づく抗血栓症薬を開発するため、長年にわたって多大な努力が払われてきた(B3)。しかし、臨床上有用な化合物はほとんどない。事実、アスピリンはその有効性と費用を考慮することにより、いまだに臨床で用いられる抗血小板薬の主な薬剤であり、原型である。アスピリンはMIおよび卒中の一次および二次予防において有効である(B4〜B7)。しかし、アスピリンの最適な治療用量については不確実な点が残り、40%を越える患者が胃腸毒性のためにアスピリンまたは腸溶アスピリンでさえも使用することができない。特に関連があるのは、非常に低用量のアスピリン(10〜80mg)でさえもかなりの数のヒト被検者で胃のびらん性損傷および出血を引き起こしたという最近の報告である。このことは、現在のところGI出血のために入院する最大の患者群は、心血管リスク低下のために低用量アスピリンを長期服用している個人であることの説明となりうる(B8)。
【0160】
アスピリンおよび他の非ステロイド性抗炎症薬(NSAID)の主な作用機序は、それらのプロスタグランジン合成を阻害する能力を通じてであることがかなり前から知られている。シクロオキシゲナーゼの二つのアイソザイム、COX−1およびCOX−2が記載されており、NSAIDは両方のCOXイソ型の活性を阻止する(B9〜B12)。アスピリンはその抗血小板作用を、血小板のCOX−1活性を阻害することによりトロンボキサンA2(TXA2)産生を阻止することで発揮する。しかし、アスピリンは血管内皮細胞でも同じ酵素を阻害し、したがってプロスタサイクリン(PGI2)の産生を防止する(B13、B14)。この内皮COXの阻害は血栓症またはアテローム性動脈硬化症の進行を促進することもあり、「アスピリンジレンマ」と呼ばれ、アスピリンの臨床での有用性における欠点である。このジレンマが低用量アスピリンの使用につながり、TXA2の産生を阻害しながらPGI2を残すことで、血小板凝集を低減しながら血管拡張を維持するための最適な抗血栓状態が得られることを示唆している。したがって、好ましいPGI2とTXA2の比は、不安定狭心症、心筋梗塞、一過性脳虚血発作、および卒中の治療および予防において深い意味を有すると考えられる。適当な投与処方、製剤、および送達速度により、アスピリンはおそらく血管PGI2産生の妨害を最小限に抑えて、血小板TXA2精製を防止することができる(B15〜B17)。
【0161】
血栓症の治療および/または予防におけるNSAIDの使用
アスピリンを含むNSAIDは一般大衆の間で最も大量に消費される薬物であり、発熱、疼痛および炎症の治療においてこの薬物群が高い効力を有するため、その使用は過去10年間に指数関数的速度で増大している(B18)。アスピリンを長期服用している個人では一般母集団に比べて心血管疾患(狭心症、心筋梗塞、血栓症、および卒中)の発生率が低いという最近の証拠により、この薬物を自身で処方する人の数が増え続けることになり、アスピリンの年間総売上高の35〜40%を占めている(B19)。その結果、我々の母集団の約1%が毎日アスピリンを服用していると推定されている。その結果、FDAは卒中、狭心症および心臓発作のリスク低下のためのアスピリンの使用を承認した。しかし、アスピリンの最適な治療用量については不確実な点が残っている。我々の母集団の平均年齢が上がるため増え続けると予想される、NSAIDの使用の指数関数的増加を妨害する一つの局面は、この化合物群が使用者のかなりのパーセンテージで重篤な副作用を引き起こし、最も顕著な副作用は胃腸出血および潰瘍形成であるということである(B20)。特に関連があるのは、非常に低用量のアスピリン(10〜75mg)でさえもヒト被検者で著しい胃のびらん性損傷および出血を引き起こしたという最近の報告である(B8)。
【0162】
アスピリン用量はPCなどの双性イオンリン脂質との結合によってかなり下げることができ、薬物の利益:危険比の著しい改善が得られる。リン脂質複合アスピリンは血管PGI2に対して血小板TXA2産生を選択的に阻害し、リン脂質/アスピリン複合体では、複合していないアスピリンの作用に比べて、血小板および内皮COX活性を区別する作用があることは、その高い抗血栓活性にとって重要であると考えられる。この知見は、動脈血栓症のインビボモデルにおいて、低用量のリン脂質複合アスピリンは実験期間中を通して血栓形成および血管閉塞を防止したが、この閾値よりも低い用量のアスピリン単独では、血栓形成を防止することができず、1時間以内に血管が閉塞したという事実によってさらに裏付けられる。PC−アスピリンの追加の利点は、これにより生じる胃粘膜損傷が通常のアスピリンよりも著しく少ないことで、したがってPC−アスピリン複合体を胃腸への副作用がない有効な抗血栓薬として開発することで、広範な費用効果の高い臨床適用が可能になることも強調すべきである。
【0163】
主な研究者による結果
1995年に、Lichtenbergerら(B21)は、ラットで経口投与したアスピリンおよびいくつかの他のNSAID(ジクロフェナク、インドメタシンナプロキセンを含む)の急性GI有害作用は、薬物を合成PCのジパルミトイルホスファチジルコリン(DPPC)またはダイズレシチンの精製抽出物とあらかじめ結合させれば、著しく軽減しうることを報告した。最近、発明者は、16名の健常志願者に4日間の試験期間中アスピリンまたはPCと複合したアスピリン(1日3回650mg)のいずれかを経口投与し、粘膜損傷をビデオ内視鏡検査によって評価した、試験的二重盲検交叉臨床試験の結果を報告した(B22)。図11に示すとおり、PC−アスピリン複合体は等用量の非修飾アスピリンに比べて、感受性の高い個人において胃のびらんの数を有意に70%減少させた。図12に示すとおり、アスピリンおよびPC−アスピリンはいずれも洞のCOX活性を>85%阻害する同等の能力を有していたため、この胃毒性の低減は薬物のCOX阻害活性の変化には関係していなかった。
【0164】
発明者の研究室では、ラットの発熱、疼痛および炎症を阻害するアスピリンの治療活性は、NSAIDをPCと化学的に結合させて投与した場合に一貫して増強され、その治療効力は非修飾NSAIDの5〜10倍に増大することも報告した(B23)。関節炎の齧歯類モデルを用いた他の試験でも、関節の炎症および疼痛を阻害するNSAIDの効力が、薬物をPCと化学的に結合させて胃内投与した場合に増強される、確証的な証拠が得られた。
【0165】
血小板および血管組織においてそれぞれTXA2およびPGI2を産生する能力に対するリン脂質−アスピリン複合体(等モル濃度の二つの物質を含む)の効果のエクスビボ動物試験を調べた。ラットに非修飾またはDPPC結合アスピリン(20mg/kg用量)のいずれかを胃内投与し、30分後、血液を採取し、血小板に富む血漿を調製し、アラキドン酸により凝集を誘導した。非修飾アスピリンまたはDPPC−アスピリンで個々に治療したラットの血小板では、食塩水対照に比べてTXB2(TXA2の安定代謝物)産生の低下が見られた。図13Aに示すとおり、PC−アスピリンはTXB2の産生をさらに抑制した。
【0166】
次いで、このエクスビボアプローチを用いて、血管(内皮)PGI2を測定した。薬物投与の1時間後に摘出した腹部大動脈を、アラキドン酸(AA、25mM)と共にインキュベートすることにより、6−ケトPGF1α(PGI2の安定代謝物)を産生する能力について評価した。図13Bに示すとおり、アスピリンは対照に比べて6−ケトPGF1αの産生を有意に阻害したが、DPPC単独およびASAと複合したDPPCは効果を示さなかった。したがって、PC−アスピリンの投与によって、血小板シクロオキシゲナーゼの選択的阻害を達成し、血管プロスタサイクリン生合成を保存することが可能である。
【0167】
PCと複合した、または複合していないアスピリンの抗血栓効果を、動脈血栓形成のインビボモデルにおいて評価した。プロトコル(B24)に従い、麻酔したウサギの左頚動脈に、平均頚動脈血流速度が対照値よりも50%高くなる点(これは形成した血栓による血管の50%閉塞に対応する)まで陽極電流をかけた。この点で、電流を止め、その後2〜3時間にわたり血流をモニターしながら試験薬物を静脈内投与した。図14Aから、食塩水またはリン脂質単独で治療した対照ウサギでは、<60分で頚動脈の血流速度はゼロ(血管の完全な血栓閉塞を示す)に低下したことが認められる(閉鎖までの平均時間=40±17)。これに対して、非修飾アスピリンを5〜20mg/kgの用量範囲で投与したウサギは、2〜3時間の実験期間を通して、血管閉塞を全く示さなかった(データは示していない)。興味深いことに、アスピリンの用量を2.5mg/kgに下げると、図14Aに示すとおり、リン脂質と複合したアスピリンはまだ血栓形成の防止において有効である(複合体投与の>180分後)が、アスピリン単独(この閾値よりも低い用量で)では血栓形成を防止することはできず、血管は61±15分胃内に閉塞する(n=4)ことが観察された。さらに、図14Bに示すとおり、アスピリン−リン脂質複合体を2.5mg/kg用量で投与したウサギで形成された血栓の重量は、天然アスピリン、食塩水またはリン脂質単独で治療したウサギに比べて有意に小さかった。
【0168】
6−ケトPGF1α(PGI2の代謝物)およびTXA2濃度も患部頚動脈で測定した。食塩水対照ウサギにおいて、血栓が形成されている患部動脈におけるPGI2のTXA2に対する比は、血栓が精製していない非患部(正常)頚動脈よりも有意に低かった(TXB2産生の増加のため)。このPGI2のTXA2に対する比は、ウサギをアスピリン(2.5mg/kg)またはリン脂質単独で治療した場合には、わずかに改善されたが、血栓形成を阻止するのに十分ではなかった。これに対して、図14Cに示すとおり、同じ用量のPC−アスピリンを投与したウサギのPGI2/TXA2比は、有意に改善され、食塩水で治療したウサギの正常動脈(陽極電流に曝露していない)で求めた比と有意な差はなかった。
【0169】
PC−アセトアミノフェン製剤
図15は、絶食Sprague Dawleyラットにアセトアミノフェン(Tylenol)単独またはTylenol:P35SBの組み合わせで経口攻撃した(経口チャレンジ)24時間後に、Tylenol:P35SB(1:2)がラットに肝酵素アスパラギン酸アミノ基転移酵素(AST)の上昇によって示される肝損傷からの保護を提供することを示すデータを示している。データは、いくつかの統計検定を用いることによって、ASTレベルがTylenolで治療したラットでは食塩水対照値に対して有意に上昇するが、Tylenol:P35SBの組み合わせ(重量比1:2)を投与したラットでは上昇しないと思われることを示している。
【0170】
引用文献
1. Furst DE, Paulus HE. Aspirin and other nonsteroidal anti-inflammatory drugs. In: Arthritis and Allied Conditions (McCarty DJ, Koopman WJ, Eds) Lea & Febiger, Philadelphia, 1993, pg 567-602.
2. Pelletier J-P. Pathological pathways of osteoarthritis. In: Non-steroidal Antiinflammatory Drugs: A Research and Clinical Perspective. Royal Society of Medicine Press, London, 1994, 1-14.
3. Jiang Y, Zhao J, Genant HK, Dequeker J, Geusens P. Bone mineral density and biomechanical properties of spine and femur of ovariectomized rats treated with naproxen. Bone 22: 509-514, 1996.
4. Walt R., Katschinski B, Logan R, Ashley J, Langman M. Rising frequency of ulcer perforation in elderly people in the United Kingdom. Lancet 489-492, 1986.
5. Allison MC, Howatson AG, Torrance CJ, Lee FD, Russel RI: Gastrointestinal damage associated with the use of nonsteroidal anti-inflammatory drugs. N. Engl J. Med. 327: 749-754, 1992.
6. Kurata JH, Abbey DE. The effect of chronic aspirin use on duodenal and gastric ulcer hospitalizations. R Clin. Gastroenterol. 12 (3): 260-266, 1990.
7. Symmons DPM. Mortality in rheumatoid arthritis. Br. J Rheum. 27 (Suppl 1): 44-54, 1988.
8. Henry DA, Johnston A, Dobson A, Duggan J. Fatal peptic ulcer complications and the use of non-steroidal anti-inflammatory drugs, aspirin and corticosteroids. Br. Med. J 295: 1227-1229, 1987.
9. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action of aspirinlike drugs. Nature 231: 232-251, 1971.
10. Ferreira SH Vane JR. New aspects of the mode of action of NSAIDs. Ann Rev Pharmacol 14: 57-. 70, 1974
11. Whittle BJR, Higgs GA, Eakin KE, Moncada S, Vane JR. Selective inhibition of prostaglandin production in inflammatory exudates and gastric mucosa. Nature 284: 271273, 1980.
12. Bergstrom S, Duner H, von Euler US, Pernow B, Sjovall J. Observations on the effects of infusions of prostaglandin E in man. Acta Physiol Scand. 45: 145-152, 1959.
13. Robert A. Nezamis JE, Lancaster C, Hanchar AJ: Cytoprotection by prostaglandins in rats: prevention of gastric necrosis produced by alcohol, HCL, NaOH, hypertonic NaCl and thermal injury. Gastroenterology 70: 359-370, 1979.
14. Mitchell JA, Akarasreenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of NSAIDs as inhibitors of constitutive and inducible cyclo-oxygenase. P. N. A. S. 90: 1169311697, 1993.
15. Masferrer JL, Zioeifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclo-oxygenase-2 in vivo is antiinflammatory and non-ulcerogenic. P. N. A. S 91: 3228-3232, 1994.
16. Xie W, Chipman JG, Robertson DL, Erikson RL, Simmons DL. Expression of a mitogen responsive gene encoding prostaglandin synthesis is regulated by mRNA splicing. P. N. A. S. 88: 2692-2696, 1991.
17. O'Banion MK, Sardowski HB, Winn V, Young DA. A serum and glucocorticoid regulated 4-kilobase RNA encodes a cyclooxygenase-related protein. J Biol Chem 266: 23261-7, 1991.
18. Meade EA, Smith WL, Dewitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other nonsteroidal antiinflammatory drugs. JBiol Chem 268: 6610-6614, 1993.
19. Masferrer JL, Zioeifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclo-oxygenase-2 in vivo is antiinflammatory and non-ulcerogenic. P. NA. S. 91: 3228-3232, 1994. mRNA encodes a cyclooxygenase-related protein. JBiol Chem 1991; 266: 23261-7.
20. Lipsky PE, Isakson PC. Outcome of specific COX-2 inhibition in rheumatoid arthritis. J Rheumatol24 (Suppl 49): 9-14, 1997.
21. Bjarnason I, Macpherson A, Rotman H, Schupp, Hayllar J. A randomized doubleblind, cross-over study on the gastroduodenal tolerability of a highly specific cyclooxygenase-2 inhibitor, flosulide and naproxen. Scand JGastroenterol 32: 126-130, 1997.
22. Simon LS, Lanza FL, Lipsky PE et. al. Preliminary safety and efficacy of SC-58635, a novel COX-2 inhibitor. Arthritis Rheum 41: 1591-1602, 1998.
23. Laine L, Harper S, Simon T, Bath T, Johnson J, Schwartz H, Stern S, Quan H, Bolognese J. A randomized trial comparing the effect of Rofecoxib, a cyclooxygenase 2specific inhibitor, with that of ibuprofen on the gastroduodenal mucosa of patients with osteoarthritis. Gastroenterology 117: 776-783, 1999.
24. Will super aspirin supersede aspirin Modern DrugDiscovery May/June 54-59, 1999.
25. Ligumsky M, Grossman MI, Kauffman Jr GL. Endogenous gastric mucosal prostaglandins: their role in mucosal integrity. Am. J physio 242: G337-341, 1982..
26. Ligumsky M, Golanska EM, Hansen DG, Kauffman Jr GL. Aspirin can inhibit gastric mucosal cyclo-oxygenase without causing lesions in the rat. Gastroenterology 84; 756-761, 1983.
27. Ligumsky M, Sestieri M, karmeli F, Zimmerman J, Okon E, Rachmilewitz D. Rectal administration of nonsteroidal antiinflammatoruy drugs. Gastroenterology 98: 12451249, 1990.
28. Whittle BJR. Temporal relationship between cyclooxygenase inhibition, as measured by prostacyclin biosynthesis and the gastrointestinal damage induced by indomethacin in the rat. Gastroenterology 80: 94-98, 1981.
29. Ivey KK, Paone DB, krause WJ. Acute effect of systemic aspirin on gastric mucosa in man. Dig. Dis Sci. 25: 97-99, 1980.
30. Konturek JW, Dembinski A, Konturek SJ, Stachura J, Domschke W. Infection of Helicobacter pylori in gastric adaptation to continued aspirin administration in human subjects. Gastroenterology 114: 245-255, 1998.
31. Langerbach R, Morham SG, Tiano HF, Loftin CD et. al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacininduced gastric ulceration. Cell 83: 483-492, 1995.
32. Morham SG, Langenbach R, Loftin CD et. al. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83: 473-482, 1995.
33. Mizuno H, Sakamoto C, Matsuda K et. al. Induction of COX-2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology 112: 387-397, 1997.
34. Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest 98: 2076-2085, 1996.
35. Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology 112: 1000-1016, 1997.
36. Wallace JL, Keenan CM, Granger DN. Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process. Sm X Physiol. 259: G462-467, 1990.
37. McCafferty D-M, Granger DN, Wallace JL. Indomethacin-induced gastric injury and leukocyte adherence in arthritic vs healthy rats. Gastroenterology 109 ; 1173-1180, 1995.
38. Mahmud T, Rafi, SS, Scott, DL, Wrigglesworth JM, Bjarnason I. Nonsteroidal antiinflammatory drugs and uncoupling of mitochondrial oxidative phosphorylation. Arthritis Rheum 39: 1998-2003, 1996.
39. McCormack K, Brune K. Classical absorption theory and the development of gastric mucosal damage associated with non-steroidal anti-inflammatory drugs. Arch Toxicol 60: 261-269, 1987.
40. Lichtenberger, LM. The hydrophobic barrier properties of gastrointestinal mucus. Ann. Rev. Physiol. 57: 565-583, 1995.
41. Hills BA, Butler BD, Lichtenberger LM. Gastric Mucosal Barrier: The hydrophobic lining to the lumen of the stomach. Am. J ; Physiol.: Gastrointestinal and Liver Physiology 7: G561-68, 1983.
42. Lichtenberger LM, Graziani LA, Dial EJ, Butler BD, Hills BA. Role of surface-active phospholipids in gastric cytoprotection. Science 219: 1327-29, 1983.
43. Spychal RT, Marrero JM, Saverymuttu SH, Northfield TC. Measurement of the surface hydrophobicity of human gastrointestinal mucosa. Gastroenterology 97: 104-11, 1989.
44. Go MF, Lew GM, Lichtenberger LM, Genta RM, Graham DY. Gastric mucosal hydrophobicity and Helicobacter pylori: response to antimicrobial therapy. Am J Gastroenterol 88: 1362-65, 1993.
45. Butler BD, Lichtenberger LM, Hills BA. Distribution of surfactants in the canine GI tract and their ability to lubricate. Am. J Physiol: Gastointestinal and Liver Physiology 7: G 645-51, 1983..
46. Kao Y-CJ, Lichtenberger LM. A method to preserve extracellular surfactant-like phospholipids on the luminal surface of the rodent gastric mucosa. J Histochem. Cytochem. 38: 427-31, 1990.
47. Kao Y-CJ, Lichtenberger LM. Phospholipid-and neutral-lipid-containing organelles of rat gastroduodenal mucous cells. Gastroenterology 101: 7-21, 1991.
48. Goddard PJ, Lichtenberger LM. Does aspirin damage the canine gastric mucosa by reducing its surface hydrophobicity ? Am. J Physiology: Gastrointestinal and Liver Physiology 15: G421-30, 1987.
49. Goddard PJ, Kao Y-CJ, Lichtenberger LM. Luminal surface hydrophobicity of canine gastric mucosa is dependent on a surface mucous gel. Gastroenterology 98: 361-70, 1990.
50. Dial EJ, Lichtenberger LM. A role for milk phospholipids in protection against gastric acid. Gastroenterology 87: 379-385, 1984.
51. Lichtenberger LM, Romero JJ, Kao Y-C, Dial EJ. Gastric protective activity of mixtures of saturated polar and neutral lipids in rats. Gastroenterology 99 ; 311-326, 1990.
52. Lichtenberger LM, Wang Z-M, Romero JJ, Ulloa C, Perez JC, Giraud M-N, Barreto JC. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nature Medicine 1: 154-158, 1995.
53. Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Phospholipid association reduces the gastric toxicity of aspirin in human subjects. Am J Gastroenterol 94: 1818 1822, 1999.
54. Lichtenberger LM, Ulloa C, Vanous AL, Romero JJ, Dial EJ, Illich PA, Walters ET. Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. JPET 1996; 277: 1221-1227.
55. Randall LO, Selitto JJ. A method for measurement of analgesic activity of inflamed tissue. Arch. Int. Pharmacodyn. 111: 409-411, 1957.0
A1. Faden, A. I., Experimental neurobiology of central nervous system trauma. Crit Rev
Neurobiol, 1993.7 (3-4): p. 175-86.
A2. Rogers, J., et al., Inflammation and Alzheimer's disease pathogenesis. Neurobiol
Aging, 1996.17 (5): p. 681-6.
A3. Hains, B. C., J. A. Yucra, and C. E. Hulsebosch, Reduction of pathological and behavioral deficitsfollowingspinal cord contusion injurywith the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma, 2001.18 (4): p. 409-23.
A4. Stewart, W. F., et al., Risk of Alzheimer's disease and duration oJ7NSAID use. Neurology, 1997.48 (3): p. 626-32.
A5. Gabriel, S. E., L. Jaakkimainen, and C. Bombardier, Riskfor serious gastrointestinal complications related to use ofnonsteroidal anti-inflammatory drugs. A meta-analysis. Ann Intern Med, 1991.115 (10): p. 787-96.
A6. Lichtenberger, L. M., et al., Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal ofNSAID-induced gastrointestinal injury. Nat Med, 1995.1 (2): p. 154-8.
A7. Anand, B. S., et al., Phospholipid association reduces the gastric mucosal toxicity of aspirin in human subjects. Am J Gastroenterol, 1999.94 (7): p. 1818-22.
A8. Lichtenberger, L. M., et al., Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. J Pharmacol Exp Ther, 1996.277 (3): p. 1221-7.
A9. Lichtenberger, L. M., et al., Phosphatidylcholine association increases the anti inflammatoyy and analgesic activity of ibuprofen in acute and chronic rodent models ofjoint inflammation: relationship to alterations in bioavailability and cyclooxygenase-inhibitory potency. J Pharmacol Exp Ther, 2001.298 (1): p. 279-87.
A10. Clatworthy, A. L., et al., Role of peri-axonal inflammation in the development of thermal hyperalgesia and guarding behavior in a rat model of neuropathic pain. Neurosci Lett, 1995.184 (1): p. 5-8.
A11. Coggeshall, R. E., et al., Is large myelinatedfiber loss associated with hyperalgesia in a model of experimental peripheral neuropathy in the rat? Pain, 1993.52 (2): p. 233-42.
A12. Carlson, S. L., et al., Acute inflammatory response in spinal cordfollowing impact injury. Exp Neurol, 1998.151 (1): p. 77-88.
A13. Hirst, W. D., et al., Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Mol Cell Neurosci, 1999.13 (1): p. 57-68.
A14. Resnick, D. K., et al., Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma, 1998.15 (12): p. 1005-13.
A15. Plunkett, J. A., et al., Effects of interleukin-10 (IL-10) on pain behavior and gene expression following excitotoxic spinal cord injury in the rat. Exp Neurol, 2001.168 (1): p. 144-54.
A16. Basso, D. M., M. S. Beattie, and J. C. Bresnahan, A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma, 1995.12 (1): p. 1-21.
A17. Grill, R., et al., Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth andpartialfunctional recovery after spinal cord injury. J Neurosci, 1997.17 (14): p. 5560-72.
A18. Rabchevsky, A. G., et al., CyclosporinA treatmentfollowingspinal cord injury to the rat: behavioral effects and stereological assessment of tissue sparing J Neurotrauma, 2001. 18 (5): p. 513-22.
A19. Hsiao, K., et al., Correlative memory deficits, Abeta elevation, and amyloidplaques in transgenic mice. Science, 1996.274 (5284): p. 99-102.
A20. Hsiao, K., Transgenic mice expressing Alzheimer amyloid precursor proteins. Exp Gerontol, 1998.33 (7-8): p. 883-9.
A21. Lim, G. P., et al., Ibuprofensuppressesplaquepathologyand inflammation inamouse modelfor Alzheimer's disease. J Neurosci, 2000.20 (15): p. 5709-14.
A22. Morris, R., Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Met, 1984.11 (1): p. 47-60.
A23. Lichtenberger, L. M., R. Darling, and J. J. Romero, Effect of luminal damaging agents on the gastric mucosal barrier and prostaglandin metabolism in cyclooxygenase (COX) knockout mice. Gastroenterology, 2001.120: p. A-143.
B1. Wu KK. Thrombogenesis, Atherogenesis and Hypercoagulability in "Thromboembolic Disorders"edited by Wu KK. PSG Publisher, Littleton, Mass, 1984, pp 5-18.
B2. Schafer AI, Handin RI. The role of platelets in thrombotic and vascular disease. Progr
Cardiovasc Dis 22: 31, 1979.
B3. Fuster V, Chesbro JH. Platelet inhibitor drugs in management of arterial thromboembolic and atherosclerotic disease. Mayo Clinic Proc. 56: 265, 1981.
B4. Fields WS, Lemak NA, Frankowsk RF, Hardy RJ. Controlled trial of aspirin in cerebral ischemia. Stroke 8: 301-314, 1977.
B5. Canadian Cooperative Study Group. A randomized trial of aspirin and sulfide pyrazone in threatened stroke. New Eng J Med 299: 53-59, 1978.
B6. Lewis HD Jr, Davis JW, Arclirbald DG, et al. Protective effects of aspirin against acute myocardial infarction and death in man with unstable anginas. Results of a VA cooperative study. N Eng J Med 313: 396, 1983.
B7. The Steering Committee of the Physicians Health Study Research Group Preliminary Report: Findings from the aspirin component of the ongoing physicians health study. N Eng J Med 318: 362, 1988.
B8. Cryer B, Feldman M. Effects of very low dose daily, long term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury. Gastroenterology 117: 17-25, 1999.
B9. Vane J. Towards a better aspirin. Nature 367: 215-216, 1994.
B10. Smith WL, DeWitt DL. Biochemistry of prostaglandin endoperoxide H synthase-1 and synthase-2 and their differential susceptibility to non-steroidal anti-inflammatory drugs. Seminars in Nephro. 15: 179, 1995.
B11. Rome LH, Lands WEM. Structure requirements for time dependent inhibition of prostaglandin biosynthesis by anti-inflammatory drugs. Proc Natl Acad Sci USA 72: 48634865, 1975.
B12. Laneuville O, Breuer DK, DeWitt DL et. al. Differential inhibition of human prostaglandin endoperoxide H synthase-1 and-2 by non steroidal anti-inflammatory drugs. J Pharm Exp Ther 271: 927-934, 1994.
B13. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action of aspirinlike drugs. Nature 231: 232, 1971.
B14. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets I. Acetylation of a particular fraction protein. J Clin Invest 56: 624-632, 1975.
B15. Hennekens CH, Buring JE. Aspirin and cardiovascular disease. Bull N Y Acad Med 65: 57-68, 1989.
B16. Viinikka L. Acetylsalicylic acid and the balance between prostacyclin and thromboxane. Scand J Clin Lab Invest 50 (supple 201): 103, 1990.
B17. Lekstrom JA, Bell WR. Aspirin in the prevention of thrombosis. Med 70: 161, 1991.
B18. Gabriel SE, Fehring RA. Trends in the utilization of non-steroidal anti-inflammatory drugs in the United States, 1986-1990. J Clin Epidemiol 45: 1041-1044, 1992.
B19. Keifer DM; A century of pain relief. Todays Chemist at Work, December 38-42, 1997.
B20. Gabriel SE, Jaakkimainen R, Bombardier C. Risk for serious gastrointestinal complications related to the use of nonsteroidal anti-inflammatory drugs. Ann Int Med 115: 787-796, 1991.
B21. Lichtenberger LM, Wang ZM, Romero JJ, Ulloa C, Perez J, Giraud M-N, Barreto JC. NSAIDs associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced G. I. injury. Nature Medicine 1: 154-158, 1995.
B22. Anand BS, Romero JJ, Sanduja SK, Lichtenberger LM. Evidence that phospholipid reduces the gastric toxicity of aspirin in human subjects. Am J Gastroenterol 94: 1818-1822, 1999.
B23. Lichtenberger LM, Ulloa C, Vanous AL, Romero JJ, Dial EJ, Illich PA, Walters ET. Zwitterionic phospholipids enhance aspirin's therapeutic activity, as demonstrated in rodent model systems. J Pharm Exp Therap 277: 1221-1227, 1996.
B24. Benedict CR, Refine CJ, Keyt BA, Pakala R, Paoni NF, Thomas R, Bennett WF. New variant of human tissue plasminigen activator (TPA) with enhanced efficacy and lower incidence of bleeding compared with recombinant human TPA. Circulation 92: 3032-3040, 1995.
B25. Blake PR, Summers MF. NOESY-1-1 Ech spectroscopy with eliminated radiation damping. J Magn Res 86: 622-625, 1990.
B26. Pinon JF. In vivo study of platelet aggregation in rats. J Pharmaco Methods 12: 79, 1984.
B27. Triplett DA, Harms CS, Newhouse P, Clark C. Platelet Function: Laboratory evaluation and clinical application. Edited by Triplett DA. American Society of Clinical Pathologists, Chicago, 1978.
B28. Sanduja SK, Mehta K, Xu X-M, Sanduja R and Wu KK. Differentiation associated expression of prostaglandin H and thromboxane A synthases in monocytoid leukemia cell lines. Blood 78: 3178-3185, 1991.
B29. Sanduja SK, Tsai AL, Aleksic NM, Wu, K. K. Kinetic of Prostacyclin Synthesis in PGHS-1 Overexpressed Endothelial cells. Am. JPhysiol. 267: C1459-1466, 1994.
B30. Gambino MC, Cerletti C, Marchi S, Garattini S, Gaetano GD. How intravenous administration of low dose aspirin inhibits both vascular and platelet cyclooxygenase activity: an experimental study in the rats. Expt Bio Med 182: 287, 1986.
B31. Pierangeli SS, Barker JH, Stikovac D, Ackerman D, Anderson G, Barquinero J, Acland R, Harris EN. Effect of human IgG antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost 71: 670-674, 1994.
B32. Edwards MH, Pierangeli S, Liu X, Barker JH, Anderson G, Harris EN. Hydroxychloroquine reverses thrombogenic antibodies in mice. Circulation 96: 4380-4384, 1997.
B33. Pierangeli SS, Liu X, Antonov JT, Sparrow JT, Harris EN, Myones BL. Induction of pathogenic anticardiolipin antibodies in a murine model. Arthritis Rheum 41: S135, 1998.
B34. Myones BL, Antonov IV, Fedorova LI, Volgin AY, Liu X, Espinola R, Harris EN, Pierangeli SS. Complexes of protein and saturated cardiolipin are capable of binding antiphospholipid antibodies and inducing thrombogenic antiphospholipid antibodies in a murine model. Arthritis Rheum 42: S369, 1999.
【0171】
結び
本明細書において引用するすべての文献は参照として本明細書に組み入れられる。本発明を十分かつ完全に記載してきたが、添付の特許請求の範囲内で、本発明を具体的に記載している以外の様式で実施しうることを理解すべきである。本発明をその好ましい態様を参照しながら開示してきたが、当業者であれば、本記載を読むことにより、上で記載し、添付の特許請求の範囲で主張する本発明の範囲および精神から逸脱することのない変更および改変を加えうることを理解すると思われる。
【図面の簡単な説明】
【0172】
【図1】図1は、アスピリン(ASA)を単独投与したラットで観察された多数の胃損傷とは対照的に、アスピリン:レシチン(LEC、レシチン油、Phosal 35 SB使用)重量比が約1:0.5、1:1および約1:2である三種のASA:LEC製剤で治療したラットはすべて、胃損傷が有意に少なかったことを示す図である。
【図2】図2は、インドメタシンは10mg/kgの用量でGI出血の重度の増大を引き起こし、これは等用量のインドメタシンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは顕著かつ有意に軽減されたことを示す図である。
【図3】図3は、イブプロフェン(ラットにおいて通常のNSAIDの中では最も毒性が低いものの一つと考えられている)は100mg/kgの用量でGI出血の適度の増大を引き起こし、これは等用量のイブプロフェンをPhosal 35 SBとの組み合わせ(NSAID:レシチン重量比1:1)で胃内投与したラットでは有意に軽減されたことを示す図である。
【図4】図4は、アスピリンは10mg/kgの用量でラットの患側の足の疼痛閾値を高める適度の能力を有するが、等用量のアスピリンをレシチン油との組み合わせで投与した場合には、試験したすべての重量比で鎮痛活性が有意に増強されたことを示す図である。
【図5】図5は、イブプロフェンは25mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のイブプロフェンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示す図である。
【図6】図6は、インドメタシンは4mg/kgの用量でラットの炎症足の疼痛圧閾値を高める、有意ではないが適度の能力を有するが、等用量のインドメタシンをレシチン油と重量比1:1の組み合わせで投与した場合には、鎮痛活性が有意に増強されたことを示す図である。
【図7A】図7Aは、脊髄損傷(SCI)によって誘導された痛覚過敏がPC−イブプロフェンおよびイブプロフェンによる治療で逆転することに関するデータを示す図である。
【図7B】図7Bは、脊髄損傷(SCI)によって誘導された痛覚過敏がPC−イブプロフェンおよびイブプロフェンによる治療で逆転することに関するデータを示す図である。
【図8】図8は、脊髄損傷後5週間のラットにおけるPC−イブプロフェンおよびイブプロフェンの鎮痛活性に関するデータを示す図である。
【図9】図9は、PC−イブプロフェンおよびイブプロフェンで治療した脊髄損傷ラットにおける6週間の間の体重増加に関するデータを示す図である。
【図10】図10は、PC−イブプロフェンおよびイブプロフェンで治療した脊髄損傷(PCI)後の運動機能回復に関するデータを示す図である。
【図11】図11は、PC−アスピリン複合体は等用量の非修飾アスピリンに比べて、感受性の高い個人において胃のびらんの数を有意に70%減少させ、この胃毒性の低減は薬物のCOX阻害活性の変化には関係していなかったことを示す図である。
【図12】図12は、アスピリンおよびPC−アスピリンはいずれも洞のCOX活性を>85%阻害する同等の能力を有していたことを示す図である。
【図13A】図13AおよびBは、食塩水、DPPC、ASA(20mg/kg)、またはDPPCと複合したASAの経口投与後30分のラット血小板におけるTXB2濃度を示す図である。PRPを調製し、AA(2mM)によって凝集を誘導した。TXB2はRIAによって測定した。結果は平均±SEMで表している(n=3)。*=ASAに対しp<0.050。略語:DPPC=ジパルミトイルホスファチジルコリン;AA=アラキドン酸;pRP=血小板を多く含む血漿;TXB=トロンボキサン。
【図13B】図13Bは、20mg/kgのASA単独またはDPPCとの複合体をラットに胃内投与した場合の、腹部大動脈による6KPGF1a産生に対する効果を示す図である。1時間後、大動脈を摘出し、各大動脈環を25mMのAAを含むトリス−HCl緩衝液中、370Cで10分間インキュベートした。6KPGF1aはRIAによって測定した。*=ASAに対しp<0.050;**=食塩水に対しp<0.001;n=4。
【図14A】図14Aは、食塩水またはPC対照ならびに2.5mg/kgの非修飾アスピリンまたはDPPCと複合したアスピリンを投与したウサギの血栓形成中の血流速度(kHz)の代表的記録を示す図である。
【図14B】図14Bは、ウサギ動脈血栓症モデルにおける2.5mg/kgのアスピリン単独またはアスピリン−DPPCの血栓重量に対する効果を示す図である。
【図14C】図14Cは、ウサギ動脈血栓症モデルにおける2.5mg/kgのアスピリン単独またはアスピリン−DPPCの頚動脈のPGI2対TXA2比に対する効果を示す図である。
【図15】図15は、ラットにおける肝損傷に関するデータを、絶食ラットにアセトアミノフェン(800mg/kg)単独またはP35SBとの重量比1:1および1:2の組み合わせを経口投与した後24時間のアスパラギン酸アミノ基転移酵素(AST)の血漿レベル上昇によって示す図である。
【特許請求の範囲】
【請求項1】
組織の潰瘍形成、組織の炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を予防、軽減および/または治療する、リン脂質を含む非水性担体および治療上有効な量のNSAIDを含む組成物であって、リン脂質が胃腸潰瘍形成、出血、肝損傷、腎損傷、および/または心血管副作用を含むNSAIDの病因効果を軽減するのに十分な量で存在する組成物。
【請求項2】
担体が生体適合性油、リン脂質含有生体適合性油、またはその混合物を含む、請求項1に記載の組成物。
【請求項3】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を含むレシチン油である、請求項1に記載の組成物。
【請求項4】
濃度が約10から約30重量%である、請求項3に記載の組成物。
【請求項5】
濃度が約10から約20重量%である、請求項3に記載の組成物。
【請求項6】
NSAIDの担体に対する重量比が約10:1から約1:10の間である、請求項1に記載の組成物。
【請求項7】
重量比が約2:1から約1:1の間である、請求項6に記載の組成物。
【請求項8】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項1に記載の組成物。
【請求項9】
リン脂質がNSAIDの胃腸毒性を軽減する、請求項1から16のいずれか一項に記載の組成物。
【請求項10】
炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療および/または予防するためのNSAIDの治療活性および/または効力を増大させる、請求項1から16のいずれか一項に記載の組成物。
【請求項11】
NSAID含有製剤の製造法であって、治療上有効な量のNSAIDを十分な量のリン脂質を含む非水性担体に加えて、非水性溶液、ペースト、半固体、懸濁液、分散液、コロイド懸濁液またはその混合物を形成する段階を含み、組成物の胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果が低いか、またはNSAIDの治療活性および/もしくは効力が増強されており、組成物が炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する方法。
【請求項12】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項11に記載の方法。
【請求項13】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項12に記載の方法。
【請求項14】
濃度が約10から約30重量%である、請求項13に記載の方法。
【請求項15】
濃度が約10から約20重量%である、請求項13に記載の方法。
【請求項16】
NSAID:担体の重量比が約10:1から約1:10である、請求項11に記載の方法。
【請求項17】
重量比が約2:1から約1:1である、請求項16に記載の方法。
【請求項18】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項11に記載の方法。
【請求項19】
組織の潰瘍形成、組織の炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を予防、軽減および/または治療する、水相および非水相ならびに場合により懸濁化剤を含む水性乳濁液またはマイクロエマルジョンを含む組成物であって、水相は生体適合性水溶液であり、非水相はリン脂質および場合により治療上有効な量のNSAIDを含む非水性担体を含み、リン脂質は組織の潰瘍形成を防止または軽減するのに十分な量で存在し、NSAIDが存在する場合には、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/または心血管副作用を含むNSAIDの病因効果を軽減するのに十分な量で存在する組成物。
【請求項20】
担体が生体適合性油、リン脂質含有生体適合性油、またはその混合物を含む、請求項19に記載の組成物。
【請求項21】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を含むレシチン油である、請求項19に記載の組成物。
【請求項22】
濃度が約10から約30重量%である、請求項21に記載の組成物。
【請求項23】
濃度が約10から約20重量%である、請求項21に記載の組成物。
【請求項24】
NSAIDの担体に対する重量比が約10:1から約1:10の間である、請求項19に記載の組成物。
【請求項25】
重量比が約2:1から約1:1の間である、請求項24に記載の組成物。
【請求項26】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項19に記載の組成物。
【請求項27】
リン脂質がNSAIDの胃腸毒性を軽減する、請求項19から26のいずれか一項に記載の組成物。
【請求項28】
炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療および/または予防するためのNSAIDの治療活性および/または効力を増大させる、請求項19から26のいずれか一項に記載の組成物。
【請求項29】
粘膜炎による、口腔、歯肉および歯を含む口の潰瘍形成および/または炎症の経口治療のための含嗽剤または洗口剤である、請求項19から28のいずれか一項に記載の組成物。
【請求項30】
ブドウ膜炎による眼の炎症治療のための点眼剤または洗眼剤である、請求項19から28のいずれか一項に記載の組成物。
【請求項31】
粘膜炎による口、食道および/または胃腸管治療のための飲用である、請求項19から28のいずれか一項に記載の組成物。
【請求項32】
NSAID含有製剤の製造法であって:
治療上有効な量のNSAIDを十分な量のリン脂質を含む非水性担体に加えて、溶液、ペースト、半固体、懸濁液、分散液、コロイド懸濁液またはその混合物の形の非水性組成物を形成する段階;および
非水性組成物を生体適合性水溶液中、乳化剤存在下または非存在下で乳化する段階を含み、
組成物の胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果が低く、ならびに/またはNSAIDの治療活性および/もしくは効力が増強されており、組成物が炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する方法。
【請求項33】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項29に記載の方法。
【請求項34】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項30に記載の方法。
【請求項35】
濃度が約10から約30重量%である、請求項31に記載の方法。
【請求項36】
濃度が約10から約20重量%である、請求項31に記載の方法。
【請求項37】
NSAID:担体の重量比が約10:1から約1:10である、請求項29に記載の方法。
【請求項38】
重量比が約2:1から約1:1である、請求項34に記載の方法。
【請求項39】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項29に記載の方法。
【請求項40】
乳化物質を剪断してマイクロエマルジョンを形成する段階をさらに含む、請求項29に記載の方法。
【請求項41】
炎症の治療法であって:
ヒトを含む動物に、治療上有効な量のNSAIDならびに、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果を軽減し、かつ/またはNSAIDの治療活性および/もしくは効力を増強するのに十分な量のリン脂質を含む非水性担体を含み、炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する組成物を投与する段階を含む方法。
【請求項42】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項41に記載の方法。
【請求項43】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項42に記載の方法。
【請求項44】
濃度が約10から約30重量%である、請求項43に記載の方法。
【請求項45】
濃度が約10から約20重量%である、請求項43に記載の方法。
【請求項46】
NSAID:担体の重量比が約10:1から約1:10である、請求項41に記載の方法。
【請求項47】
重量比が約2:1から約1:1である、請求項46に記載の方法。
【請求項48】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項41に記載の方法。
【請求項49】
組織の潰瘍形成および/または炎症の予防または治療法であって:
ヒトを含む動物に、場合により治療上有効な量のNSAIDならびに、化学および/または放射線療法が原因の粘膜炎による潰瘍形成および/または炎症を予防し、NSAIDが存在する場合には、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果を軽減し、かつ/または化学および/もしくは放射線療法に関連する炎症、疼痛、および/もしくは発熱の軽減におけるNSAIDの治療活性および/もしくは効力を増強するのに十分な量のリン脂質を含む非水性担体を含む組成物を投与する段階を含む方法。
【請求項50】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項49に記載の方法。
【請求項51】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項50に記載の方法。
【請求項52】
濃度が約10から約30重量%である、請求項51に記載の方法。
【請求項53】
濃度が約10から約20重量%である、請求項51に記載の方法。
【請求項54】
NSAID:担体の重量比が約10:1から約1:10である、請求項49に記載の方法。
【請求項55】
重量比が約2:1から約1:1である、請求項54に記載の方法。
【請求項56】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項49に記載の方法。
【請求項1】
組織の潰瘍形成、組織の炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を予防、軽減および/または治療する、リン脂質を含む非水性担体および治療上有効な量のNSAIDを含む組成物であって、リン脂質が胃腸潰瘍形成、出血、肝損傷、腎損傷、および/または心血管副作用を含むNSAIDの病因効果を軽減するのに十分な量で存在する組成物。
【請求項2】
担体が生体適合性油、リン脂質含有生体適合性油、またはその混合物を含む、請求項1に記載の組成物。
【請求項3】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を含むレシチン油である、請求項1に記載の組成物。
【請求項4】
濃度が約10から約30重量%である、請求項3に記載の組成物。
【請求項5】
濃度が約10から約20重量%である、請求項3に記載の組成物。
【請求項6】
NSAIDの担体に対する重量比が約10:1から約1:10の間である、請求項1に記載の組成物。
【請求項7】
重量比が約2:1から約1:1の間である、請求項6に記載の組成物。
【請求項8】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項1に記載の組成物。
【請求項9】
リン脂質がNSAIDの胃腸毒性を軽減する、請求項1から16のいずれか一項に記載の組成物。
【請求項10】
炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療および/または予防するためのNSAIDの治療活性および/または効力を増大させる、請求項1から16のいずれか一項に記載の組成物。
【請求項11】
NSAID含有製剤の製造法であって、治療上有効な量のNSAIDを十分な量のリン脂質を含む非水性担体に加えて、非水性溶液、ペースト、半固体、懸濁液、分散液、コロイド懸濁液またはその混合物を形成する段階を含み、組成物の胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果が低いか、またはNSAIDの治療活性および/もしくは効力が増強されており、組成物が炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する方法。
【請求項12】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項11に記載の方法。
【請求項13】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項12に記載の方法。
【請求項14】
濃度が約10から約30重量%である、請求項13に記載の方法。
【請求項15】
濃度が約10から約20重量%である、請求項13に記載の方法。
【請求項16】
NSAID:担体の重量比が約10:1から約1:10である、請求項11に記載の方法。
【請求項17】
重量比が約2:1から約1:1である、請求項16に記載の方法。
【請求項18】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項11に記載の方法。
【請求項19】
組織の潰瘍形成、組織の炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を予防、軽減および/または治療する、水相および非水相ならびに場合により懸濁化剤を含む水性乳濁液またはマイクロエマルジョンを含む組成物であって、水相は生体適合性水溶液であり、非水相はリン脂質および場合により治療上有効な量のNSAIDを含む非水性担体を含み、リン脂質は組織の潰瘍形成を防止または軽減するのに十分な量で存在し、NSAIDが存在する場合には、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/または心血管副作用を含むNSAIDの病因効果を軽減するのに十分な量で存在する組成物。
【請求項20】
担体が生体適合性油、リン脂質含有生体適合性油、またはその混合物を含む、請求項19に記載の組成物。
【請求項21】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を含むレシチン油である、請求項19に記載の組成物。
【請求項22】
濃度が約10から約30重量%である、請求項21に記載の組成物。
【請求項23】
濃度が約10から約20重量%である、請求項21に記載の組成物。
【請求項24】
NSAIDの担体に対する重量比が約10:1から約1:10の間である、請求項19に記載の組成物。
【請求項25】
重量比が約2:1から約1:1の間である、請求項24に記載の組成物。
【請求項26】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項19に記載の組成物。
【請求項27】
リン脂質がNSAIDの胃腸毒性を軽減する、請求項19から26のいずれか一項に記載の組成物。
【請求項28】
炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療および/または予防するためのNSAIDの治療活性および/または効力を増大させる、請求項19から26のいずれか一項に記載の組成物。
【請求項29】
粘膜炎による、口腔、歯肉および歯を含む口の潰瘍形成および/または炎症の経口治療のための含嗽剤または洗口剤である、請求項19から28のいずれか一項に記載の組成物。
【請求項30】
ブドウ膜炎による眼の炎症治療のための点眼剤または洗眼剤である、請求項19から28のいずれか一項に記載の組成物。
【請求項31】
粘膜炎による口、食道および/または胃腸管治療のための飲用である、請求項19から28のいずれか一項に記載の組成物。
【請求項32】
NSAID含有製剤の製造法であって:
治療上有効な量のNSAIDを十分な量のリン脂質を含む非水性担体に加えて、溶液、ペースト、半固体、懸濁液、分散液、コロイド懸濁液またはその混合物の形の非水性組成物を形成する段階;および
非水性組成物を生体適合性水溶液中、乳化剤存在下または非存在下で乳化する段階を含み、
組成物の胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果が低く、ならびに/またはNSAIDの治療活性および/もしくは効力が増強されており、組成物が炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する方法。
【請求項33】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項29に記載の方法。
【請求項34】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項30に記載の方法。
【請求項35】
濃度が約10から約30重量%である、請求項31に記載の方法。
【請求項36】
濃度が約10から約20重量%である、請求項31に記載の方法。
【請求項37】
NSAID:担体の重量比が約10:1から約1:10である、請求項29に記載の方法。
【請求項38】
重量比が約2:1から約1:1である、請求項34に記載の方法。
【請求項39】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項29に記載の方法。
【請求項40】
乳化物質を剪断してマイクロエマルジョンを形成する段階をさらに含む、請求項29に記載の方法。
【請求項41】
炎症の治療法であって:
ヒトを含む動物に、治療上有効な量のNSAIDならびに、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果を軽減し、かつ/またはNSAIDの治療活性および/もしくは効力を増強するのに十分な量のリン脂質を含む非水性担体を含み、炎症、疼痛、発熱、心血管疾患、卵巣癌、大腸癌、および/またはアルツハイマー病を治療、予防または軽減する組成物を投与する段階を含む方法。
【請求項42】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項41に記載の方法。
【請求項43】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項42に記載の方法。
【請求項44】
濃度が約10から約30重量%である、請求項43に記載の方法。
【請求項45】
濃度が約10から約20重量%である、請求項43に記載の方法。
【請求項46】
NSAID:担体の重量比が約10:1から約1:10である、請求項41に記載の方法。
【請求項47】
重量比が約2:1から約1:1である、請求項46に記載の方法。
【請求項48】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項41に記載の方法。
【請求項49】
組織の潰瘍形成および/または炎症の予防または治療法であって:
ヒトを含む動物に、場合により治療上有効な量のNSAIDならびに、化学および/または放射線療法が原因の粘膜炎による潰瘍形成および/または炎症を予防し、NSAIDが存在する場合には、胃腸潰瘍形成、出血、肝損傷、腎損傷、および/もしくは心血管副作用を含むNSAIDの病因効果を軽減し、かつ/または化学および/もしくは放射線療法に関連する炎症、疼痛、および/もしくは発熱の軽減におけるNSAIDの治療活性および/もしくは効力を増強するのに十分な量のリン脂質を含む非水性担体を含む組成物を投与する段階を含む方法。
【請求項50】
担体が生体適合性油およびリン脂質、リン脂質含有生体適合性油、またはその混合物を含む、請求項49に記載の方法。
【請求項51】
リン脂質含有生体適合性油が約10から約40重量%の濃度のリン脂質を有するレシチン油である、請求項50に記載の方法。
【請求項52】
濃度が約10から約30重量%である、請求項51に記載の方法。
【請求項53】
濃度が約10から約20重量%である、請求項51に記載の方法。
【請求項54】
NSAID:担体の重量比が約10:1から約1:10である、請求項49に記載の方法。
【請求項55】
重量比が約2:1から約1:1である、請求項54に記載の方法。
【請求項56】
NSAIDがアスピリン、サリチル酸塩、ナプロキセン、ジクロフェナク、インドメタシン、スリンダク、イブプロフェン、ケトプロフェン、オキサプロゼン、ケトロラク、スリンダク、ナブメトン、メクロフェナミック、ピロキシカム、ジフルニサル、オキシフェンブタゾン、フェニルブタゾン、セレコキシブ、ロフェコキシブ、COX2阻害剤、アセトアミノフェン、またはその混合物および組み合わせからなる群より選択される、請求項49に記載の方法。
【図1】


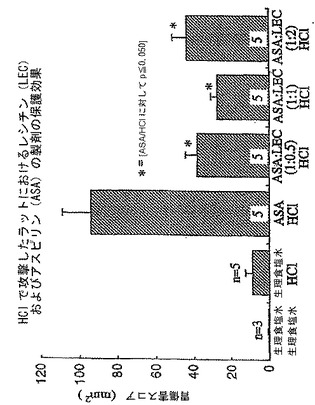
【図2】
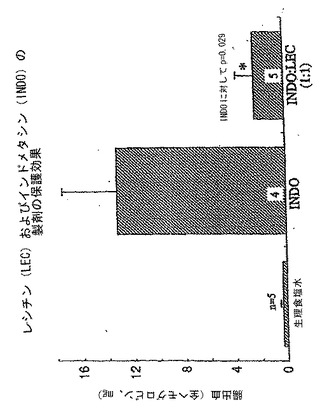
【図3】

【図4】
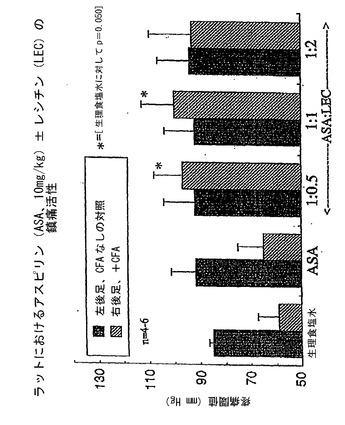
【図5】
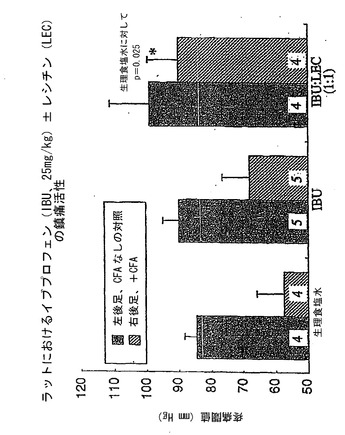
【図6】
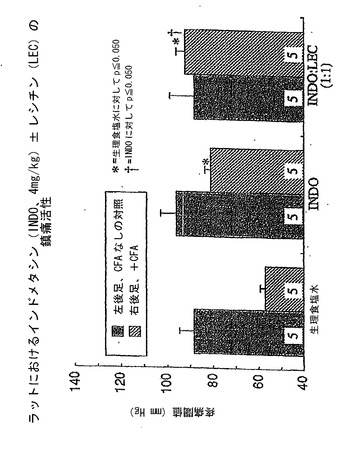
【図7A】
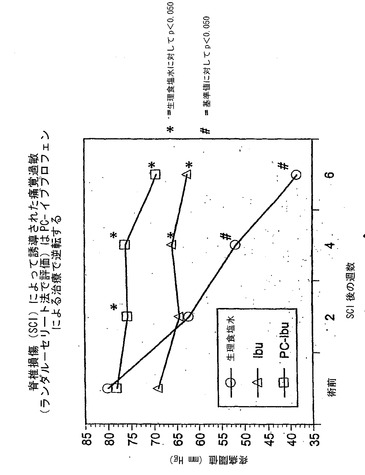
【図7B】
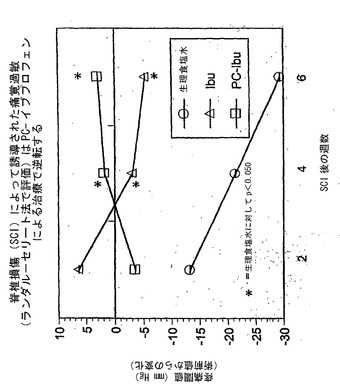
【図8】

【図9】
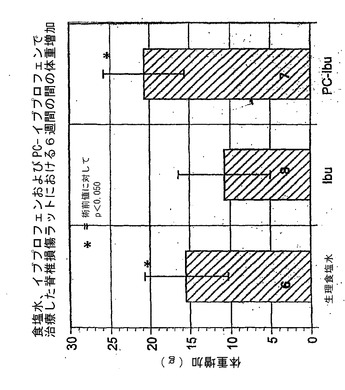
【図10】
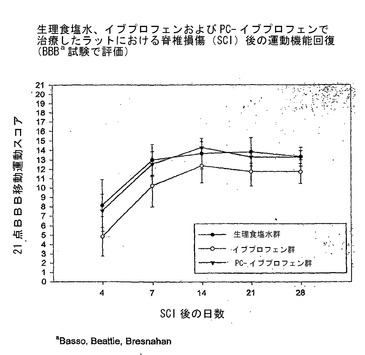
【図11】
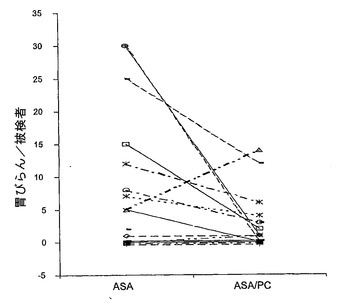
【図12】

【図13A】
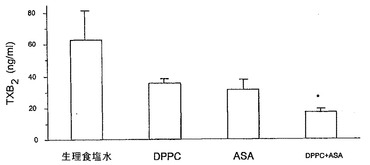
【図13B】
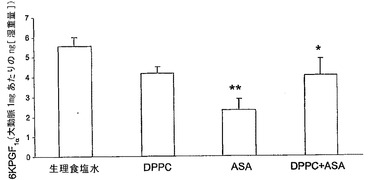
【図14A】

【図14B】
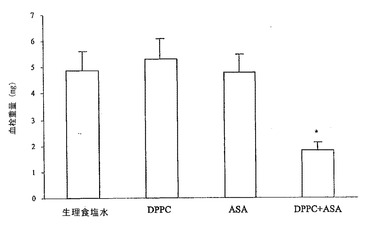
【図14C】

【図15】
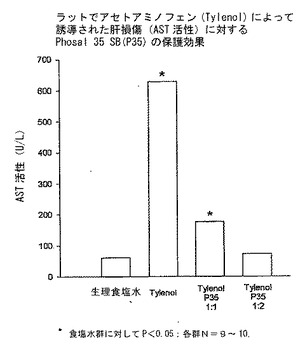
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13A】
【図13B】
【図14A】
【図14B】
【図14C】
【図15】
【公開番号】特開2010−31044(P2010−31044A)
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願番号】特願2009−255329(P2009−255329)
【出願日】平成21年11月6日(2009.11.6)
【分割の表示】特願2002−582987(P2002−582987)の分割
【原出願日】平成13年12月19日(2001.12.19)
【出願人】(501449540)ザ・ボード・オブ・リージェンツ・オブ・ザ・ユニバーシティ・オブ・テキサス・システム (4)
【氏名又は名称原語表記】THE BOARD OF REGENTS OF THE UNIVERSITY OF TEXAS SYSTEM
【Fターム(参考)】
【公開日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願日】平成21年11月6日(2009.11.6)
【分割の表示】特願2002−582987(P2002−582987)の分割
【原出願日】平成13年12月19日(2001.12.19)
【出願人】(501449540)ザ・ボード・オブ・リージェンツ・オブ・ザ・ユニバーシティ・オブ・テキサス・システム (4)
【氏名又は名称原語表記】THE BOARD OF REGENTS OF THE UNIVERSITY OF TEXAS SYSTEM
【Fターム(参考)】
[ Back to top ]