
胃酸分泌の抑制を必要とする病状を治療するための組成物
本発明は、胃酸分泌インヒビターとしての不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI)、胃管腔における壁細胞のアクチベーターとしてのペンタガストリン(PG)またはPG類似体を含む新規経口組成物に関する。好ましい態様において、本発明の組成物は、胃液中でPGの有効性を保存する少なくとも一の薬剤を更に含み、これによりPGは胃において局所的に作用することができる。予期し得ないことに、本発明の組成物は、胃において局所的に抗酸活性を示し、これは食事に依存しておらず、かつ迅速発現および長期の酸分泌阻害を示す。

【発明の詳細な説明】
【発明の分野】
【0001】
本発明は、胃酸分泌に対して迅速発現性長期阻害効果を保持し、かつ食事に依存しない、胃酸分泌阻害のための新規経口組成物に関する。
【発明の背景】
【0002】
多数の病状は、胃酸分泌を抑制する必要性により特徴づけられる。かかる症状には、ゾリンジャー/エリソン症候群(ZES)、胃食道逆流疾患(GERD)、消化性潰瘍疾患、十二指腸潰瘍、食道炎などが含まれるがこれらに限定されない。消化性潰瘍などの症状は、重い合併症を発症することがあり、先進工業国では最も流行性の疾患の幾つかに相当する。
【0003】
GERDおよび消化性潰瘍疾患の治療において現在採用されている主な治療には、たとえばヒスタミンH2−受容体アンタゴニストまたはプロトンポンプインヒビター(PPI’s)を用いることにより胃の酸性度を低下させる薬剤が含まれる。PPI’sは、壁細胞からの酸分泌を担う壁細胞H+/K+ ATPアーゼプロトンポンプを阻害することにより作用する。PPI’s、たとえばオメプラゾール、およびその薬学的に許容可能な塩は、たとえばEP 05129、EP 124495およびUS Patent No. 4,255,431に開示される。
【0004】
PPI薬剤は、腸溶性コーティング顆粒剤で通常投与される酸不安定性プロドラッグである。PPIsは、小腸で吸収された後、弱塩基であるため、壁細胞の酸性環境(milieu)内に優先的に蓄積する。壁細胞の酸性環境(milieu)内の酸性状況(environment)により、当該プロドラッグは、活性なスルフェンアミドに変換され、これは、壁細胞H+/K+ ATPアーゼポンプに結合し阻害する活性薬剤である。
【0005】
PPIsの薬効は文書により充分裏付けられているが、PPIsには注目すべき制限がある。投薬および食事摂取の時期は、これら薬剤の薬物動態並びに胃酸分泌の抑制能力に影響を及ぼし得る(Hatlebakk et al., Aliment Pharmacol Ther. 2000; 14 (10): 1267-72)。具体的には、胃酸分泌の最適な抑制を達成するためには、PPIは食物の摂取前に服用しなければならない。更に、PPIsは、薬理作用の開始が比較的遅く、最大限の酸抑制および症状の緩和を達成するのに数日を要することがあり、オン・デマンド(on-demand)GERD療法にその有用性は限定される(Sachs G, Eur J Gastroenterol Hepatol. 2001; 13 Suppl 1: S35-41)。更に、PPIsは、胃酸の24h抑制を提供することはできず、夜間の酸ブレークスルーは、GERD患者において胸やけの痛みにつながり、PPIsの1日に2回の投薬を用いてもこれは発生する(Tytgat GN, Eur J Gastroenterol Hepatol. 2001; 13 Suppl 1: S29-33)。最終的に、これら薬剤は、薬物動態において患者間の実質的な変動性を示し、他の薬剤と重大な相互作用を示すことがある(Hatlebakk et al., Clin Pharmacokinet. 1996; 31 (5): 386-406)。よって、PPI-介在性活性の改良は、胃腸病学において充分に認識された課題(challenge)である。
【0006】
ペンタガストリン(PG)(β−アラニル−L−トリプトフィル−L−メチオニル−L−アスパルチル−L−フェニル−アラニルアミド;配列番号2)は、ガストリンのカルボキシ末端のテトラペプチドを含有するペンタペプチドである。このカルボキシ末端のテトラペプチドは、ほぼすべての天然のガストリンに見られる活性部分である。動物においてPGは、主に、胃に存在する類エンテロクロマフィン(ECL)細胞からのヒスタミンの放出を誘導することにより、胃酸の分泌を誘導するように作用する。ヒスタミンの放出およびその後の壁細胞に存在するヒスタミン受容体の活性化は、壁細胞の活性化につながり、プロトンイオンを胃管腔に活発に分泌する。PGは壁細胞に直接作用し、その活性化を誘導することもできる。PGは、胃酸分泌機能を評価するための診断用薬剤として当該技術分野で典型的に使用される。
【0007】
酸性状況でのPGの溶解度の低さ、およびPGが胃でペプシン分解されやすいという事実より、本出願人が発見するまで明らかに予測できない経口投与後の胃酸分泌のインデユーサーとしてのPGの用途が提示された。本出願人の発見前には、PGは非経口ルートにより投与された場合のみ、酸分泌の誘導において効果的に活性であると当業者により考えられていた。実際、酸分泌に対する効果は、PGの経口投与を受けた正常な4被検体では検出されなかったが、胃腸の異常を伴う別の3患者では幾らかの効果が検出された(Morrell & Keynes Lancet. 1975; 2 (7937): 712)。実際、この研究は、経口投与したときにPG活性が失われる証拠として薬理学の教科書に引用されている(Martindale Thirty-second edition, pl616, the Chapter:"Supplementary Drugs and Other Substances")。
【0008】
Pisegna et al.のWO01/22985('985公報)は、プロトンポンプインヒビター(PPI)と併用して全身に投与されるPGの使用を開示する。'985公報によれば、PPIと併用してPGを投与することにより、過剰な胃酸分泌を低減する/軽減するPPIの薬効は増大する。'985公報は、PGは好ましくは注射(たとえば皮下注射)により投与されるべきであることを開示し教示する。しかし、'985公報は、PGおよびPPIが静脈内、非経口、または経口的手段により投与され得ることも一般的に開示する。また'985公報は、PPIおよびPIが錠剤で調製され得ることを一般的に開示する。しかし、'985公報は、使用すべき特定の投与量や処方を開示しておらず実施例を提示していない。更に、'985公報は、PGが経口的にデリバーされると効果的でないという従来技術の教示をどのようにして回避するか教示も示唆もしておらず、効果的と思われるPGの経口投与量の例を提案していない。加えて、'985公報は、PGが胃で局所的に活性であることを開示しておらず、これは、本発明者らが驚くべきことに発見したことである。また'985公報は、胃管腔で局所的効果を達成するために、PG保存剤を使用して、胃においてPG活性の生物学的活性を保存することを教示していない。本発明の時点の技術水準に照らして、'985公報の一般的開示は、局所デリバリーのためのPGを含む経口組成物を調製する動機付けを当業者に与えるものではない。
【0009】
De Graef et al., Gastroenterology, 91, 333-337 (1986)(De Graef公報)は、オメプラゾールは、静脈注射によりPGで前処理したイヌに投与した場合、胃酸分泌の阻害において、より効果的であることを開示する。De Graef公報には、PGの経口投与が、胃管腔で局所的に作用することにより効果的であり、オメプラゾールの効果を増強することは記載されていない。
【0010】
Phillipsの米国特許Nos. 6,489, 346; 6,645, 988; および6,699, 885 (まとめて"Phillips特許")は、PPI、少なくとも一の緩衝化剤、および具体的な壁細胞アクチベーターから成る経口組成物を用いた、酸原因の胃腸障害を治療する薬学的組成物および方法を開示する。Phillips特許に開示される壁細胞アクチベーターには、たとえば、チョコレート、重炭酸ナトリウム、カルシウム、ハッカ油、スペアミント油、コーヒー、茶およびコラ、カフェイン、テオフィリン、テオブロミンおよびアミノ酸残基が含まれる。Phillips特許に示されるとおり、提案されるこれら壁細胞アクチベーターはすべて、酸分泌に対して阻害効果および刺激効果の両方を発揮する内在性ガストリンの放出を誘導する。しかし、Phillips特許は、PGの使用を開示も示唆もしておらず、PGは、(CCK-AおよびCCK-Bレセプターの両方を活性化し、阻害と刺激の両効果を含む)Phillips特許に記載される壁細胞アクチベーターとは異なり、刺激活性のみを保持し、CCK-B受容体にのみ結合する。
【0011】
胃酸分泌の阻害が必要な病状を効果的に治療する方法を開発することにより、長期の切実な要求が満たされるでしょう。PPI’sの広範囲の使用にもかかわらず、PPIの薬効を高める必要性がなお存在しており、たとえば迅速で効果的な発現、夜間の酸のブレークスルーを含む長時間の効果、少ない投与量での高い効果、および食事に依存しない投与が挙げられる。
【発明の概要】
【0012】
本発明の目的は、食事に依存せず、かつ胃酸分泌に対して迅速発現性長期阻害効果を示す、胃酸分泌阻害のための経口組成物を提供することである。
【0013】
本発明の別の目的は、不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI)および壁細胞のアクチベーターを含む、胃酸分泌阻害のための経口組成物であって、PPIの抗酸活性が、食事に依存せず、かつ酸分泌に対して迅速発現性長期阻害効果を示す組成物を提供することである。
【0014】
本発明の一つの態様において、経口組成物は、胃酸分泌インヒビターとしての不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI)、壁細胞のアクチベーターとしてのペンタガストリン(PG)および/またはPG類似体、および胃液中でPGの有効性を保存する一以上の薬剤を含み、その結果、PGの生物学的活性は維持され、これによりPGが胃で局所的に作用することが可能である。予測外なことに、本発明の組成物は、食事に依存しない胃における抗酸活性を保持し、かつ酸分泌に対して迅速発現性長期阻害を示す。本発明の組成物は、胃における酸分泌の抑制が必要な慢性または急性障害に罹患している被検体を治療するために使用され得る。
【0015】
本発明によるプロトンポンプインヒビター(PPIs)は、胃壁細胞においてH+/K+-アデノシントリホスファターゼ(ATPアーゼ)プロトンポンプの活性を阻害する化合物である。PPIは、そのプロドラッグ形態において、イオン化していないため、壁細胞の細胞膜を通過することができる。非イオン化PPIは、壁細胞に到達すると、活性化壁細胞の酸分泌部分、分泌細管に移動する。分泌細管にトラップされたPPIは、プロトン化され、プロトンポンプのアルファサブユニットのシステイン残基とジスルフィド共有結合を形成することができる活性なスルフェンアミド形態に変換され、これにより不可逆的にプロトンポンプを阻害する。
【0016】
上述したとおり、本発明は、PGが、経口投与された場合に、好ましくは胃管腔で局所的に作用して壁細胞を活性化することにより、局所的に活性であるという驚くべき発明者らの発見に基くものである。活性な壁細胞は、酸性pHを保持しており、これは、PPIが活性なプロトン化スルフェンアミド形態に変換されるために必要である。したがって、PGが胃管腔に直接作用することにより壁細胞が同時に活性化されると、PPIによるポンプの阻害は最大限に発揮される。
【0017】
本発明の経口組成物は、胃酸分泌の低減を目的とした公知のPPIベースの組成物と比べて以下の利点を示す。本発明の組成物は、胃管腔における局所的効果により、PGの全身投与と関連した副作用を生じることなく、PGによる壁細胞の活性化を可能にする。PGによる壁細胞のプレ活性化は、PPIの活性なスルフェンアミド形態への変換を促進し、これによりPPIの効果が迅速に発現する。更に、本発明の組成物は、食事に依存しない様式で、胃において抗酸活性の迅速な発現を示す。このように、経口組成物において活性薬剤を組合せることにより、酸分泌の迅速な低減が必要な急性症状に対して有効な解決策が提供される。最終的に、本発明の経口組成物は、単回の投薬を用いて、少なくとも24hの間、胃酸分泌の長期抑制を提供する。
【0018】
本発明による経口組成物は、胃管腔における壁細胞の局所的アクチベーターとして、PGまたはPG類似体を含む。アミノ酸配列βAla-Trp-Met-Asp-PheNH2(配列番号2)を含むPGに加えて、本発明は、壁細胞のアクチベーターとして、ガストリンまたはPG類似体またはその誘導体の使用を想定する。かかる変異体には、ガストリンの34-、17-、および14-アミノ酸種、および完全な薬理活性を有することが文献(Tracey and Gregory (1964) Nature (London), 204: 935参照)で報告されているガストリンの活性なC末端テトラペプチドTrp-Met-Asp-PheNH2(配列番号1)を含む他のトランケーション変異体を含むが、これらに限定されない。
【0019】
天然のアミノ酸が保存的置換で置換されているガストリンの変異体および/またはトランケートされたガストリンも含まれる。これら分子の種々の類似体には、PGのN−保護誘導体Boc-βAla-Trp-Met-Asp-PheNH2(Bocはtert-ブチルオキシカルボニル基である)またはF-Moc-βAla-Trp-Met-Asp-PheNH2(Mocはメトキシカルボニルである)が含まれるがこれらに限定されない。
【0020】
非限定的な態様において、本発明による経口組成物は、酸性胃液中でPGの有効性を保存する一以上の薬剤を更に含む。これら薬剤は、好ましくは、胃液中でPGの溶解性を保持しその分解を防ぐことにより胃液中でPGの有効性を保存するのに充分な量で存在し、その結果、胃においてPGの局所的な生物学的活性が保存される。これにより、PGは胃で局所的に作用し壁細胞を活性化することができる。かかる薬剤は、好ましくは、胃液中に溶解されたときに、ペプシンを阻害する値まで胃液のpHを一時的に上昇させることにより、ペプシンによる胃液中のPGの分解を阻害することができる制酸剤またはアルカリ性薬剤である。PGは、アルカリ性状況でのみ可溶であるため、胃液のpHの一時的な上昇は、PGの少なくともかなりの割合が胃液中に可溶なまま残ることを保証する。
【0021】
任意の弱塩基または強塩基(およびその混合物)が、本発明の経口組成物においてアルカリ性薬剤として利用可能であることに注目されたい。アルカリ性薬剤または制酸剤は、酸性胃液においてPGの安定性および溶解性を実質的に保存するのに充分な量で組成物中に存在する。したがって、本発明のアルカリ性薬剤は、胃液に溶解されたときに、PGの十分な有効性を達成し治療作用を発揮するのに充分な値まで胃のpHを上昇させることができる。
【0022】
好ましい態様に従って、組成物中のアルカリ性薬剤は、PGが胃で壁細胞に到達しそれを活性化するのに充分な期間にわたって、4を超える値、より好ましくは5を超える値まで胃液のpHを上昇させるのに充分な量で存在する。より好ましい態様において、アルカリ性薬剤は、5〜60分の期間、好ましくは5〜30分の期間にわたって、5を超える値まで胃液のpHを上昇させることができる。このように、本発明によるアルカリ性薬剤は、PGが壁細胞を活性化するのに充分な期間にわたって、胃液中でPGの溶解性を保存する。更に、胃液中の一時的なアルカリ性状況は、酸性pHにおいてのみ活性であるペプシンによるPGの分解を妨害する。
【0023】
種々の態様に従って、本発明の組成物は、酸性胃液中でPGの有効性を保存する他の薬剤を更に含む。かかる薬剤は、たとえば、胃におけるペプチドの分解を低減するペプシンインヒビター(すなわちペプスタチンおよびその誘導体バシトラシン−環状ドデカペプチド)、または胃粘膜の粘度を低減し、これにより酸分泌を担う細胞に到達するPGの能力を促進する粘液溶解剤である。かかる粘液溶解剤は、還元剤、たとえばN-アセチルシステイン、ジチオトレイトール、クエン酸またはマンニトールである。本発明の組成物は、胃に存在する細菌に対して効果的な抗生物質を更に含んでいてもよい。
【0024】
本発明の有効成分は、単一の経口投与形態、好ましくは固体投与形態で調合され得る。また、懸濁剤などの液体投与形態が使用されてもよい。よって、一つの態様において、PPI、PGおよび胃液中でPGの有効性を保存する薬剤は、多層錠剤、懸濁錠剤、発泡錠剤、粉剤、ペレット、顆粒剤、マルチプルビーズを含む硬質ゼラチンカプセル剤、または脂質ベースの賦形剤を含む軟質ゼラチンカプセル剤として調合され得る。
【0025】
一つの態様に従って、本発明の固体投与形態は、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされたPPI粒子、PGの粒子、および一以上のアルカリ性薬剤の粒子を含有するカプセル剤または多層錠剤である。PGによる胃管腔における壁細胞の活性化が、小腸の近位部におけるPPIの吸収と同調するようにするために、単一の経口投与形態は、胃でのPG放出時間を延長する時間依存性放出ポリマーでコーティングされたPGビーズを含んでいてもよい。このように、胃においてPG放出が延長されると、壁細胞上でPGの活性とPPIの活性とが同調するようになる。
【0026】
本発明の有効成分は、別個の投与形態で調合されてもよい。たとえば、PGおよび胃液中でPGの有効性を保存する薬剤は、経口懸濁剤または固体投与形態、たとえばカプセル剤、錠剤、懸濁錠剤、または発泡錠剤で調合され、PPIは別の固体投与形態、好ましくは腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーを備えたビーズを含有するカプセル剤または錠剤で調合され得る。別個の投与形態は、PGおよび胃液中でPGの有効性を保存する薬剤を一つの投与形態で含有し、PPIを別の投与形態で含有するキットとして提供されてもよい。この場合、それらの生理活性の時間的オーバーラップが少なくとも存在するように、PGはPPIと共に投与される。PPIおよびPGは、同時におよび/または連続して投与することができる。
【0027】
本発明で使用されるPPI粒子は、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされていてもよいし、コーティング層なしであってもよい。胃を通過する間の非コーティングPPIの安定性は、組成物に存在する一以上のアルカリ性薬剤により保存される。小腸の近位部における非腸溶性コーティングPPIの緩衝化懸濁剤の吸収は、腸溶性コーティングPPI顆粒剤の吸収より迅速であることが以前に実証されている(Pilbrantand Cederberg, Scand. J. Gastroenterol 1985: 20 (supp. 108): 113-120)。従って、非コーティングPPI粒子を組成物で使用する場合、胃でのPGの放出を遅延させる必要はない。しかし、コーティングPPI粒子を使用する場合、たとえばポリマーコーティングPG粒子を使用して胃でのPGの放出を遅延させることにより、PPIの放出をPGの放出と同調させることが必要である。
【0028】
別の態様において、本発明は、胃酸分泌の抑制が必要な障害または胃酸分泌の抑制により通常治療される障害に罹患した被検体を治療する方法に関する。本方法は、胃酸分泌インヒビターとしてのPPI、胃管腔における壁細胞のアクチベーターとしてのPGまたはPG類似体、および胃管腔でPGの有効性を保存するのに充分な量の少なくとも一の保存剤を含む薬学的組成物を被検体に投与することを含む。
【0029】
本発明の組成物は、胃酸分泌の阻害が必要な哺乳類の病状を予防または治療するために使用することができる。好ましくは、哺乳類はヒトである。本発明の組成物は、かかる病状を治療する際、および発症前にかかる病状の発症リスクを最小限にする際の両方において効果的である。
【0030】
本発明の薬学的組成物は、胃酸分泌の抑制により治療される多数の病的症状に使用することができる。かかる症状には、ゾリンジャー/エリソン症候群(ZES)、胃食道逆流疾患(GERD)、食道炎、消化性潰瘍疾患、十二指腸潰瘍、胃炎および胃粘膜びらん、消化不良症などが含まれるがこれらに限定されない。
【0031】
また本発明は、経口薬学的キットを含む。キットは、典型的に、薬学的に効果的な量の(i)配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液中の前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む。一つの態様において、有効成分は、別々の投与ユニット形態で調合される。キットは、その有効成分を被検体に投与することにより、胃酸分泌の抑制が必要な被検体の障害を治療または予防するために使用することができる。ペプチドは、典型的には、PPIの投与と同時、その前またはその後に投与される。
【0032】
これらの態様および更なる態様は、後述の詳細な説明および実施例から明らかでしょう。
【発明の詳細な説明】
【0033】
「アルカリ性薬剤」の用語は、PGとともに(たとえば、PGの前、間および/または後に)調合されるかまたはデリバーされたときに、胃内でPGの有効性を実質的に保存する値まで胃管腔のpHを一時的に上昇させるように機能する任意の薬学的に適切な弱塩基または強塩基(およびその混合物)をいう。
【0034】
「胃内でPGの有効性を保存する薬剤」の用語は、胃におけるPGの溶解性および安定性を維持することができる任意の薬剤をいう。具体的には、かかる薬剤は、可溶性形態で少なくともかなりの量のPGを維持することができ、かつ胃液で分解されず、その結果、胃におけるPGの生物学的活性が維持される。
【0035】
「胃におけるPGの生物学的活性」の用語は、胃管腔に位置する壁細胞の活性化をいう。
【0036】
「と併用して(in conjunction with)」の用語は、PPIおよびPGを別個の投与形態で投与するときに、それらの生理活性の時間的オーバーラップが少なくとも存在することを意味する。よって、PPIおよびPGは、同時におよび/または連続して投与することができる。
【0037】
本発明は、PGが経口投与後に活性なまま残り、好ましくは胃で局所的に作用することにより壁細胞を活性化することができるという驚くべき発見に基くものである。重要なこととして、壁細胞の活性化には、PPIプロドラッグが、胃H+/K+-ATPアーゼプロトンポンプの不可逆性インヒビターとして作用する活性な形態に変換されることが必要である。本発明の経口組成物は、胃酸分泌の阻害においてPPIの薬効を増大する活性薬剤の特有の組合せを提供する。
【0038】
本発明の組成物は、胃酸分泌の阻害が必要な哺乳類の病状を予防または治療するために使用することができる。本発明の組成物は、病状を治療する際、およびかかる病状の発症リスクを発症前に最小限にする際の両方に効果的である。かかる病状には、たとえば、逆流性食道炎、胃炎、十二指腸炎、胃潰瘍および十二指腸潰瘍が含まれる。更に、本発明の組成物は、胃酸阻害効果が望まれる他の胃腸疾患を治療または予防するために使用することができ、たとえば、(低用量アスピリンを含む)非ステロイド性抗炎症薬(NSAID)治療中の患者、非潰瘍性消化不良の患者、症候性胃食道逆流疾患(GERD)の患者、およびガストリン産生腫瘍の患者に使用することができる。また、集中治療の状況におかれた患者、急性消化管出血の患者、胃酸の吸引を予防する術前および術後の状況、およびストレス性潰瘍を予防および治療する術前および術後の状況において使用することもできる。更に、ヘリコバクター感染症およびこれに関連した疾患の治療に有用であり得る。治療に適した他の症状には、ゾリンジャー−エリソン症候群(ZES)、ウェルナー症候群、および全身性肥満細胞症が含まれるがこれらに限定されない。
【0039】
本発明による壁細胞アクチベーターは、好ましくは、配列番号2で示されるアミノ酸配列を有するPGである。しかし、ガストリンのC末端テトラペプチドTrp-Met-Asp-PheNH2(配列番号1で示される)を含む任意のPG類似体が、壁細胞アクチベーターとして使用され得る。かかる類似体には、ガストリンの34-、17-、および14-アミノ酸種、および他のトランケーション変異体が含まれるがこれらに限定されない。また、天然のアミノ酸が保存的置換により置換された、ガストリンの変異体および/またはトランケートされたガストリンも含まれる。また、たとえばPGのN−保護誘導体を含むがこれに限定されない、これら分子の種々の類似体も含まれる。PGの適切な保護基には、当該技術分野で公知の標準的ヒドロキシル保護基、たとえばメトキシメチル(MOM)、β−メトキシエトキシメチル(MEM)、トリアルキルシリル、トリフェニルメチル(trityl)、tert−ブトキシカルボニル(t-BOC)、エトキシエチル(EE)、f-MOC(メトキシカルボニル)、およびTROCなどが含まれる。保護基は、当業者に一般に公知の標準的手法を用いることにより除去し、所望のPG誘導体を得ることができる(T. W. Green, Protective Groups in Organic Synthesis, Chapter 2, pages 10-69 (1981))。
【0040】
ガストリン、ペンタガストリン、またはその類似体は、商業的に入手可能である。加えて、合成のプロトコールが周知である。このため、たとえば、PGは周知のペプチド合成方法を用いて化学的に合成することができる(たとえば、Barany and Merrifield Solid-Phase Peptide Synthesis; pp. 3-284 in The Peptides: Analysis, Synthesis, Biology. Vol. 2: Special methods in peptide synthesis, part a.; Merrifield et al. (1963) J. Am. Chem. Soc., 85: 2149-2156; and Stewart et al. (1984) Solid Phase Peptide Synthesis, 2nd ed. Pierce Chem. Co., Rockford, ILL.を参照)。更にPGは、たとえばBoc-Ala残基をテトラペプチドTrp-Met-Asp-PheNH2に結合させることにより化学的に合成することができる。
【0041】
本発明の組成物は、過度な有害な副作用を起こすことなく、壁細胞に対して薬理効果を達成するのに効果的な量でPGまたはその類似体を含む。組成物に存在するPGの標準的な概算量は、好ましくは1-100 mg、より好ましくは2-60 mg、最も好ましくは4-40 mgのPG量(または等価な量のPG類似体)である。
【0042】
本発明の組成物は、胃H+/K+-ATPアーゼプロトンポンプの不可逆性インヒビターとして作用するPPIを更に含む。本発明で使用されるPPIは、H+, K+-ATPアーゼ阻害活性を有する任意の置換ベンズイミダゾール化合物とすることができる。本発明の目的において、「PPI」の用語は、H+, K+-ATPアーゼインヒビターとしての薬理活性を保持する任意の置換ベンズイミダゾールを意味し、オメプラゾール、ランソプラゾール(lansoprazole)、パントプラゾール、ラベプラゾール、ドントプラゾール、ペルプラゾール(s-オメプラゾールマグネシウム)、ハベプラゾール、ランソプラゾール(ransoprazol)、パリプラゾール、およびレミノプラゾールの中性形態または塩の形態、単一のエナンチオマーまたは異性体または他の誘導体またはエナンチオマーのアルカリ性塩を含むが、これらに限定されない。
【0043】
本発明で使用され得る胃H+/K+-ATPアーゼプロトンポンプインヒビターの例は、たとえば、プロトンポンプインヒビターとして効果的な新規チアジアゾール化合物を記載する米国特許第6,093,738号に開示される。プロトンポンプインヒビターとして、欧州特許第322133号および第404322号はキナゾリン誘導体を開示し、欧州特許第259174号はキノリン誘導体を記載し、WO 91/13337および米国特許第5,750,531号はピリミジン誘導体を開示する。また、適切なプロトンポンプインヒビターは、たとえば、EP-A1-174726、EP-A1-166287、GB 2 163 747およびW090/06925、W091/19711、W091/19712、W094/27988およびW095/01977にも開示される。
【0044】
本発明による組成物においてPPI粒子は、コーティングされていてもコーティングされていなくてもよい。オメプラゾールなどのPPIを含む腸溶性コーティング粒子の調製は、たとえば、米国特許第4,786,505号および第4,853,230号に開示される。
【0045】
本発明の組成物は、過度な有害な副作用を起こすことなく、薬理効果または治療的改善を達成するのに効果的な量でPPIを含む。治療的改善には、胃pHの上昇、消化管出血の減少、または症状の改善もしくは除去を含むがこれらに限定されない。好ましい態様に従って、PPIの典型的な一日量は変動し、患者の個々の必要量および治療すべき疾患などの種々の因子に依存する。一般に、PPIの一日量は、1-400 mgの範囲である。組成物中に存在するPPIの好ましい標準的概算量は、典型的には、約20-40 mgのオメプラゾール、約30 mgのランソプラゾール(lansoprazole)、約40 mgのパントプラゾール、約20 mgのラベプラゾール、並びに薬理的に等価な用量の以下のPPIs:ハベプラゾール、パリプラゾール、ドントプラゾール、ランソプラゾール(ransoprazole)、ペルプラゾール(s-オメプラゾールマグネシウム)、およびレミノプラゾールである。
【0046】
好ましい態様において、本発明の組成物は、酸性胃液中でPGの有効性を保存する一以上の薬剤を更に含む。より具体的には、保存剤は、胃液中でPGの安定性または溶解性を維持する。これにより、PGが胃において局所的に作用し壁細胞を活性化することが可能になる。かかる薬剤は、好ましくは、それらを胃液に溶解したときに、胃に存在するペプチダーゼを阻害し、かつ少なくともかなりの割合のPGが胃液中に可溶なまま残るpHまで、胃液のpHを上昇させることができるアルカリ性薬剤または制酸剤である。
【0047】
本発明で使用されるアルカリ性薬剤には、たとえば以下のものが含まれる:重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、重炭酸カルシウム、およびその他のカルシウム塩。重炭酸ナトリウムは水に容易に溶解するが、炭酸カルシウムは水不溶性であり、酸性環境でのみゆっくり溶解できることに注意されたい。したがって炭酸カルシウムは、胃内でアルカリ性薬剤の持続した溶解が望まれるときに有効であり得る。
【0048】
本発明で使用される制酸剤の例には、一以上の以下のものが含まれる:アルミナ、炭酸カルシウム、および重炭酸ナトリウム;アルミナおよびマグネシア;アルミナ、マグネシア、炭酸カルシウム、およびシメチコン;アルミナ、マグネシア、および炭酸マグネシウム;アルミナ、マグネシア、炭酸マグネシウム、およびシメチコン;アルミナ、マグネシア、およびシメチコン;アルミナ、アルギン酸マグネシウム、および炭酸マグネシウム;アルミナおよび炭酸マグネシウム;アルミナ、炭酸マグネシウム、およびシメチコン;アルミナ、炭酸マグネシウム、および重炭酸ナトリウム;アルミナおよび三ケイ酸マグネシウム;アルミナ、三ケイ酸マグネシウム、および重炭酸ナトリウム;アルミナおよびシメチコン;アルミナおよび重炭酸ナトリウム;塩基性炭酸アルミニウム;塩基性炭酸アルミニウムおよびシメチコン;水酸化アルミニウム;炭酸カルシウム;炭酸カルシウムおよびマグネシア;炭酸カルシウム、マグネシア、およびシメチコン;炭酸カルシウムおよびシメチコン;炭酸カルシウムおよびマグネシウム;マガルドレート;マガルドレートおよびシメチコン;炭酸マグネシウムおよび重炭酸ナトリウム;水酸化マグネシウム;酸化マグネシウム。
【0049】
好ましくは、本発明の組成物は、一以上のアルカリ性薬剤または制酸剤を、薬理効果を達成するのに効果的な量で含む。具体的には、組成物中のアルカリ性薬剤または制酸剤は、PGが胃の壁細胞を活性化するのに充分な期間にわたって、胃に存在するプロテアーゼに最適なpHより高いpHまで胃液のpHを上昇させるのに充分な量で存在する。好ましい態様において、アルカリ性薬剤または制酸剤は、5〜60分の期間にわたって、好ましくは5〜30分の期間にわたって、5を超えるpHまで胃液のpHを上昇させるのに充分な量で存在する。本発明の組成物に必要なアルカリ性薬剤の量は、使用されるアルカリ性薬剤のタイプおよび所定のアルカリ性薬剤により提供される塩基に相当するものにより必然的に変動する。実際、胃においてPGの優れた有効性を提供するのに必要な量は、(米国薬局方(USP)ガイドラインに従って調製される)200ミリリットルの人工胃液の溶液に添加したときに、そのHCl溶液のpHを少なくともpH5.0まで上昇させる量である。好ましくは少なくとも100ミリグラム、より好ましくは少なくとも300ミリグラム、最も好ましくは少なくとも500ミリグラムのアルカリ性薬剤が、本発明の薬学的組成物で使用される。
【0050】
別の態様において、本発明の組成物は、酸性胃液中でPGの有効性を保存する他の薬剤を更に含む。たとえば、本発明の組成物は、ペプシンインヒビター、たとえば活性化されたペンタペプチドペプスタチン、および天然または合成由来のその誘導体を含んでいてもよい。これらインヒビターは、ペプシンによるPGの分解を減少させる。更に、本発明の組成物は、胃粘膜の粘度を減少させることによりPGが壁細胞に到達する能力を促進する粘液溶解剤を含んでいてもよい。かかる粘液溶解剤は、たとえば還元剤、たとえばN-アセチルシステイン、ジチオトレイトール、クエン酸またはマンニトールである。あるいは、本発明の組成物は、PGのためのポリマーコーティング、たとえば胃の酸性環境からPGを保護するポリマーの腸溶性コーティングを含んでいてもよい。
【0051】
本発明の有効成分は、好ましくは、すべての有効成分を含有する単一の経口投与形態で調合される。本発明の組成物は、固体または液体形態の何れかで調合され得る。液体製剤と比較して固体製剤の安定性が改良されていることから固体製剤が好ましいことに注意されたい。
【0052】
一つの態様において、PPI粒子、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、単一の固体投与形態、たとえば多層錠剤、懸濁錠剤、発泡錠剤、粉剤、ペレット、顆粒剤、または複数のビーズを含むカプセル剤で調合される。別の態様において、活性薬剤は、単一の液体投与形態、たとえば全ての有効成分を含有する懸濁剤、または使用前に再構成されるドライ懸濁剤で調合されていてもよい。
【0053】
単一の投与形態において、PPI粒子およびPG粒子は、胃におけるPGの局所的生物学的活性と壁細胞に対するPPIの全身効果とを同調させるために、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされていてもよい。たとえば、コーティングPPI粒子を使用して血液での吸収を遅延させる場合、PG粒子もコーティングしてその放出を遅延させることが望ましい。一つの具体的な態様において、PPI粒子が厚い非腸溶性層でコーティングされ、PPIの放出が好ましくは20−80分、より好ましくは25−75分、最も好ましくは30−60分遅延され、PG粒子が薄い非腸溶性ポリマー層でコーティングされ、PGの放出が好ましくは5−60分、より好ましくは8−45分、最も好ましくは10−30分遅延される。これら条件により、薬理学的PPI血漿濃度を達成する前に、PGによる壁細胞のプレ活性化が可能になる。
【0054】
本発明で使用される適切なpH依存性腸溶性ポリマーの非限定的な例は、以下のとおりである:酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、フタル酸ポリ酢酸ビニル、メタクリル酸共重合体、シェラック、コハク酸ヒドロキシプロピルメチルセルロース、トリメリット酸酢酸セルロース、および上記の何れかの混合物。適切な商業的に入手可能な腸溶性材料は、たとえば商標Eudragit L 100-55で販売されている。このコーティングは、基質にスプレーコーティングすることができる。
【0055】
非腸溶性時間依存性放出ポリマーには、たとえば、胃液から水分を吸収して胃で膨張することにより、粒子のサイズを増大させて厚いコーティング層を作成する一以上のポリマーが含まれる。時間依存性放出コーティングは、一般に、外部の水性媒体のpHに依存しない侵食および/または拡散特性を有する。よって、有効成分は、拡散により、または胃内で粒子がゆっくり侵食された後に、粒子からゆっくり放出される。
【0056】
流体と投与形態の表面との相互作用により生じる胃内でのポリマーの侵食特性は、ポリマーの分子量および薬剤/ポリマーの比により主に決定される。PGおよびPPIの放出を確実に約10分〜約60分遅延させるためには、ポリマーの分子量が約105〜約107グラム/molの範囲であることが推奨される。更に、PGまたはPPI/ポリマーの比が約2:3〜約9:1、好ましくは約3:2〜9:1、最も好ましくは約4:1〜9:1であることが推奨される。
【0057】
適切な非腸溶性時間依存性放出コーティングは、たとえば、フィルム形成化合物、たとえばセルロース誘導体、たとえばメチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシエチルセルロース、および/またはアクリルポリマー、たとえばEudragit商標ポリマーの非腸溶性形態である。他のフィルム形成材料を、単独で、または互いに組み合わせて、または上述のものと一緒に使用してもよい。これらの他のフィルム形成材料には、一般に、ポリ(ビニルピロリドン)、ゼイン、ポリ(エチレングリコール)、ポリ(エチレンオキシド)、ポリ(ビニルアルコール)、ポリ(ビニルアセテート)、およびエチルセルロース、並びに他の薬学的に許容可能な親水性および疎水性フィルム形成材料が含まれる。これらフィルム形成材料は、賦形剤または溶媒システムとして水を用いて基質コアに適用してもよい。また、水性アルコールシステムをフィルム形成のための賦形剤として採用してもよい。
【0058】
本発明の時間依存性放出コーティングを作成するのに適した他の材料には、たとえば以下のものが挙げられるがこれらに限定されない:水溶性ポリサッカリドガム、たとえばカラーギンナン、フコイダン(fucoidan)、ガッチゴム、トラガカント、アラビノガラクタン、ペクチン、およびキサンタン;ポリサッカリドガムの水溶性塩、たとえばアルギン酸ナトリウム、トラガカントナトリウム(sodium tragacanthin)、およびガッチゴムナトリウム(sodium gum ghattate);アルキル基が直鎖もしくは分枝の1〜7の炭素の水溶性ヒドロキシアルキルセルロース、たとえばヒドロキシメチルセルロース、ヒドロキシエチルセルロース、およびヒドロキシプロピルセルロース;合成水溶性セルロース系ラミナ形成材(former)、たとえばメチルセルロースおよびそのヒドロキシアルキルメチルセルロース誘導体、たとえばヒドロキシエチルメチルセルロース、ヒドロキシプロピルメチルセルロース、およびヒドロキシブチルメチルセルロースから成る群より選択されるメンバー;他のセルロースポリマー、たとえばカルボキシメチルセルロースナトリウム;および当業者に公知の他の材料。この目的のために使用することができる他のラミナ形成材料には、ポリ(ビニルピロリドン)、ポリビニルアルコール、ポリエチレンオキシド、ゼラチンとポリビニルピロリドンの混合物、ゼラチン、グルコース、サッカリド、ポビドン、コポビドン、ポリ(ビニルピロリドン)−ポリ(酢酸ビニル)共重合体が挙げられる。
【0059】
胃でのPGの放出を遅延させるための別のアプローチは、胃液より低い密度の浮遊粒子の使用により粒子からPGの放出を遅延させることである。一つの好ましい態様において、浮遊粒子は、胃液との接触により、エチルセルロースコーティング重炭酸ナトリウムビーズ内に二酸化炭素が放出することにより得られる。エチルセルロースコーティング重炭酸ナトリウムコアからの二酸化炭素の放出は、粒子の浮力を許容し、粒子からのPGの放出を遅延させる。
【0060】
胃でのPGの放出を遅延させるために、他の遅延性胃内容物排出アプローチを使用してもよい。これらアプローチには、消化しにくいポリマーまたは胃の運動性パターンを食事状態に変更する脂肪酸塩の使用により、胃内容物排出速度を低下させ、薬剤放出の大幅な延長を許容することが含まれる(たとえばSingh and Kim, J. of Controlled Release 63 (2000) 235-259に開示される)。
【0061】
ある状況においては、胃内容物排出に抵抗するサイズまで、胃内で急速に広がる投与形態を用いることにより、胃でのPGの保持時間を延長することが望ましい。かかるシステムは、長期間にわたって完全な状態を保持し、小さなピースへの分解が起こるまで、胃から排出されることはない。Caldwell (Caldwell, L. J., Gardener, C. R., Cargill, R. C. (1988), 米国特許第4,767,627号)は、侵食性ポリマーから作成され、硬質ゼラチンカプセルに折り畳まれ挿入された薬剤を充填した十字デバイスを記載する。経口投与後、ゼラチンシェルは崩壊し、折り畳まれたデバイスは展開する。システムは、最小サイズ1.6 cmおよび最大サイズ5 cmであるため、胃からの通過が可能なほど充分小さくなるポイントまでポリマーが侵食してはじめて、胃から幽門へ移動する。
【0062】
胃でのPGの保持時間を延長する別のアプローチは、ヒトへの投与に都合がよい親水性侵食性ポリマーシステム、たとえばポリ(エチレンオキシド)(Polyox)およびヒドロキシプロピル−メチルセルロース(HPMC)を使用することである。このシステムは、流体を吸収すると、胃内の保持を延長させるサイズまで短期間で膨張し、上部消化管において含有薬剤の吸収部位への持続性デリバリーを可能にする。これらシステムは、侵食性および親水性ポリマーまたはポリマー混合物からつくられるため、相当な期間をかけて容易に侵食され、胃から出ていく。膨張の期間は、これが食道で起こらないような期間であり、もしシステムが部分的に膨張した状態で腸に移動すると、水和ポリマーの侵食性および弾性が、デバイスによる腸閉塞の機会を除去する。
【0063】
一つの具体的な例において、本発明の組成物は、硬質または軟質ゼラチンカプセルに含有される複数のビーズを含む単一投与形態として調合される。カプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズまたは時間依存性放出ポリマーでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよび重炭酸ナトリウムエチルセルロースを含むビーズ、PG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースでコーティングされたビーズ。組成物中のセルロース系ポリマーにより、PGビーズの浮遊が許容され、ビーズからのPGの放出が遅延する。PG放出速度は、ヒドロキシプロピルメチルセルロースの厚みおよび侵食速度により決定される。
【0064】
別の具体的な例において、ゼラチンカプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズまたは時間依存性放出コーティングでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよびPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースでコーティングされたアルギン酸塩を含むビーズ。
【0065】
更に別の具体的な例において、ゼラチンカプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズ、時間依存性放出ポリマーでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよびPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースを含むミニタブ(mini-tab)の形態の粒子。
【0066】
更に別の例において、本発明の組成物は、一つの層に腸溶性コーティングPPIを含み、第二の層にPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースを含む、プレスコート錠剤または二層の錠剤として調合される。
【0067】
更に別の例において、本発明の組成物は、硬質ゼラチンカプセル内に二層の非水系半固体充填物として調合されてもよく、ここでは、PPIが、室温以上で液体であるが冷却すると半固体を形成する脂質ベース(非水系、迅速性放出)に可溶化されているため、硬質ゼラチンカプセルに充填することが可能である。脂溶性アルカリ性薬剤、たとえばアミンまたは重炭酸ナトリウムのファイン懸濁液を含めてもよい。
【0068】
本発明の単一投与形態の組成物は、好ましくは、腸溶性コーティングPPI粒子または時間依存性放出粒子の代わりに、非コーティングPPIを含む。非コーティングPPIの小腸上部における吸収は、コーティングPPIの吸収より速い。したがって、組成物に非コーティングPPIを使用することにより、PGの放出を遅延させる必要もなく、胃におけるPGの生物学的活性とPPIが活性である期間との間を、より正確に同調させることができる。このように種々の好ましい態様に従って、本発明の組成物は、PG、非コーティングPPIおよび一以上のアルカリ性薬剤を含む、二層の錠剤、プレスコート錠剤、発泡錠剤または懸濁錠剤として調合される。
【0069】
本発明の有効成分は、複数の経口投与形態で調合されてもよく、ここではPGおよび胃液中でPGの有効性を保存する一以上の薬剤が、別々の投与形態であるがPPIと結合させて投与される。たとえば、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、経口懸濁剤または固体投与形態、たとえばカプセル剤、錠剤、懸濁錠剤、または発泡錠剤で調合されてもよく、PPIは、別の固体投与形態、好ましくはカプセル剤または錠剤に含有される腸溶性コーティングビーズまたは時間依存性放出ビーズで調合されてもよい。
【0070】
複数の経口投与形態を用いる場合、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、PPIより前、PPIと同時、またはPPIの後に投与することができる。連続的投与では、PPIを投与したとき、またはPPIが活性になったときに、PGが生理的効果を発揮する限り、PGの投与とPPIの投与との間に実質的な遅れ(たとえば、分または数時間)があってもよい。好ましい態様において、投与されるPPIは、腸溶性コーティング形態または時間依存性放出形態である。この態様によれば、PGが胃で活性である間に、小腸の近位部で吸収されたPPIがH+/K+−ATPアーゼポンプを阻害するのに確実に有効であるようにするため、PPIの投与は、PGの投与より先に行われることが好ましい。
【0071】
本発明の有効成分は、不活性な薬学的に許容可能なビーズ内に組み込まれてもよい。この場合、薬剤は、ビーズにコーティングされる前に、更なる成分と混合されてもよい。成分には、結合剤、界面活性剤、充填剤、崩壊剤、アルカリ性添加剤または他の薬学的に許容可能な成分の単一物または混合物が含まれるがこれに限定されない。結合剤には、たとえば、セルロース、たとえばヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびカルボキシメチル−セルロースナトリウム、ポリビニルピロリドン、糖、デンプン、および凝集性を備えた他の薬学的に許容可能な物質が含まれる。適切な界面活性剤には、薬学的に許容可能な非イオン系またはイオン系界面活性剤が含まれる。適切な界面活性剤の例は、ラウリル硫酸ナトリウムである。
【0072】
粒子は、慣用的な技術により、パックされた経口摂取用の塊に形成されてもよい。たとえば、粒子は、公知のカプセル化の手法および材料を用いて、「硬質充填カプセル」としてカプセル化されてもよい。カプセル化材料は、カプセルが摂取された後に粒子が胃で迅速に分散するように、胃液中で可溶性が高くなければならない。
【0073】
別の態様において、本発明の有効成分は、圧縮錠剤にパッケージングされる。「圧縮錠剤」の用語は、一般に、一回の圧縮により、またはプレ圧密タッピングとその後の最終圧縮により調製される、経口摂取用の扁平な非コーティング錠剤をいう。かかる固体形態は、当該技術分野で周知のとおり製造することができる。錠剤形態は、たとえば、一以上のラクトース、マンニトール、コーンスターチ、ポテトスターチ、微結晶性セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、着色剤、希釈剤、緩衝化剤、湿潤剤、保存剤、香味剤、および薬学的に適合可能なキャリアを含むことができる。製造プロセスは、4つの確立された方法の一つまたはその組合せを採用することができる:(1)乾燥混合;(2)直接圧縮;(3)圧延(milling);および(4)非水系顆粒化。Lachman et al., The Theory and Practice of Industrial Pharmacy (1986)。かかる錠剤は、フィルムコーティングを備えていてもよく、これは、好ましくは経口摂取したときまたは希釈剤と接触したときに溶解する。
【0074】
かかる錠剤に使用することができるアルカリ性薬剤の非限定的な例には、重炭酸ナトリウム、アルカリ土類金属塩、たとえば炭酸カルシウム、水酸化カルシウム、乳酸カルシウム、グルセロリン酸カルシウム、酢酸カルシウム、炭酸マグネシウム、水酸化マグネシウム、ケイ酸マグネシウム、アルミン酸マグネシウム、水酸化アルミニウムまたは水酸化マグネシウムアルミニウムが含まれる。制酸錠剤を作成するのに有用な特別なアルカリ土類金属塩は、炭酸カルシウムである。
【0075】
別の代案において、本発明の組成物は、圧縮形態、たとえば懸濁錠剤および発泡錠剤で調合され、水または他の希釈剤と反応したときに、経口投与のための組成物の水性形態がつくられる。これらの形態は、錠剤の飲込みまたは咀嚼よりずっと受け入れられる用法で、子供や老人などに投薬するために特に有用である。本発明の薬学的錠剤または他の固体投与形態は、わずかな振盪または攪拌により、アルカリ性薬剤を崩壊させる。
【0076】
本明細書で使用される「懸濁錠剤」の用語は、水中に置かれた後急速に崩壊する圧縮錠剤をいい、容易に分散可能であり、正確な投与量のPPI、PGおよびアルカリ性薬剤を含有する懸濁液を形成する。一つの非限定的な例において、懸濁錠剤は、20-40 mgオメプラゾール、4 mg PG、およびアルカリ性薬剤として約1-4グラムの重炭酸ナトリウムまたはカルシウムを含み得る。錠剤の急速な崩壊を達成するために、クロスカルメロースナトリウムなどの崩壊剤を製剤に添加してもよい。崩壊剤は、単独で、または圧縮困難な錠剤の材料の圧縮性を高める能力に関して周知である微結晶性セルロースと組合せて、圧縮錠剤に混合され得る。また、微結晶性セルロースは、単独で、または他の成分と共にプロセスされた状態で、圧縮錠剤のための一般的な添加剤であり、圧縮困難な錠剤の材料の圧縮性を高める能力に関して周知である。これは、商標Avicelで商業的に入手可能である。
【0077】
懸濁錠剤組成物は、当業者に明らかなように、上述の成分に加えて、薬学的錠剤でしばしば用いられる他の成分、たとえば香味剤、甘味剤、フロー助剤、潤滑剤、または他の一般的な錠剤アジュバントを含有してもよい。他の崩壊剤、たとえばクロスピビドン(crospividone)およびグリコール酸デンプンナトリウム(sodium starch glycolate)を使用してもよいが、クロスカルメロースナトリウムが好ましい。
【0078】
上記成分に加えて、上述の経口投与形態は、薬学の分野で慣用的な適量の他の材料、たとえば希釈剤、潤滑剤、結合剤、顆粒化助剤、着色剤、香味剤および滑り剤(glidant)を含有してもよい。経口投与形態を調合するために使用され得る薬学的に許容可能なキャリアおよび賦形剤の具体的な例は、Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986)に記載され、これは参照により本明細書に組み込まれる。
【0079】
以下の実施例は、本発明のある態様を更に充分に説明するために提示する。しかし、これらを本発明の広い範囲を限定するものと解釈すべきではない。当業者であれば、本発明の範囲から逸脱することなく、本明細書に開示される原理の多くの変更および改変を容易に考案することができる。
【実施例】
【0080】
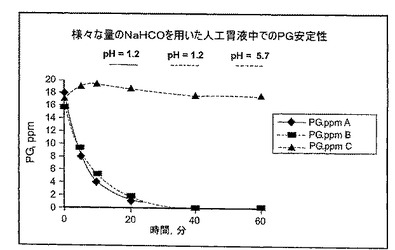
例1:NaHCO3は人工胃液中でPGの安定性を保存する
NaHCO3の存在下で酸性pHにおけるPGの安定性を、人工胃液を用いてインビトロで試験した。人工胃液は、米国薬局方(USP) 2000 Ed., P.235に従って調製した。200 mlの胃液を調製するために、0.4 gのNaClおよび0.64 gのペプシンを、16 ml 1M HClおよび184 mlの水に溶解した。胃液のpHは1.2であった。10または20 mlの8.4%(1M)NaHCO3(それぞれ最終濃度3.72 mg/mlまたは7.12 mg/ml)および16 mlの250 ppm PG 溶液 (0.25 mg/ml)を、当該溶液に添加した。最終溶液におけるPGの濃度は16 ppmであった。表示された場合には、オメプラゾール顆粒を更に添加した(溶液BおよびC)。最終溶液中でのPGの経時的安定性を決定するために、調製後の以下の時点: 0'(調製直後)、5'、10'、20'、40'、60'で採取したサンプルについてHPLC分析を行った。反応を停止させるために、NH4OHを用いてpHを7.5−8.5に調整した。
【0081】
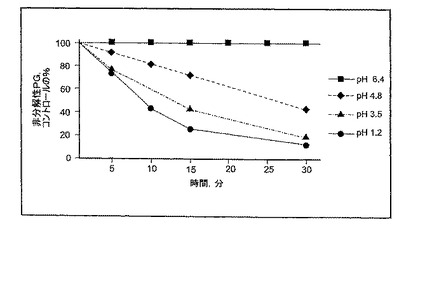
図1に示されるとおり、3.72 mg/mlのNaHCO3(pH 1.2)の存在下にPGを含む溶液AおよびBでは、PGの迅速な分解が観察された。しかし、7.12 mg/mlのNaHCO3(pH 5.7)を含む溶液Cでは、PGは1h安定なままであった。これらの結果から、pHを5.0より大きくするのに充分な濃度でNaHCO3などのアルカリ性薬剤を添加すると、ペプシンによるPGの分解が妨害されることが分かる。図2は、少なくとも80%のPGがpH4.8において少なくとも15分間分解されないまま残ることを更に示す。
【0082】
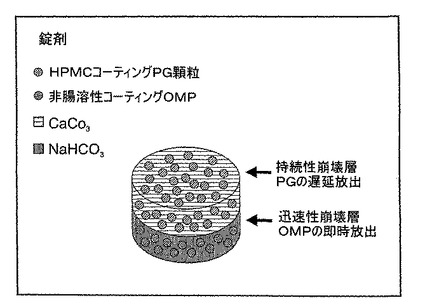
A.製剤の説明−非腸溶性コーティングオメプラゾールを含有する錠剤
例2:PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含むプレスコート錠剤または二層の錠剤
プレスコート錠剤または二層の錠剤を、単一の投与形態として調合し、各錠剤は、以下の成分を含有する:
オメプラゾール (パウダー) 40 mg
PG 4 mg
NaHCO3 500 mg
CaCO3 500 mg
クロスカルメロースナトリウム
ヒドロキシプロピルメチルセルロース (HPMC)
微結晶性セルロース (Avicel)
ステアリン酸マグネシウム
デンプン。
【0083】
プレスコート錠剤または二層の錠剤は、2段階のプロセスで調製される。単一の錠剤のために、4 mg PG、250 mg 炭酸カルシウムおよび微結晶性セルロースを混合して、これを錠剤の第一の層にプレ圧縮する。PGを含有するこの層を、錠剤からのPGの放出を10−15分遅延させるHPMCの薄層で更にコーティングする。第二の層については、40 mgの非腸溶性コーティングオメプラゾールのパウダーを、500 mg NaHC03、250 mg CaCO3および適切な結合剤とともにPG層の上に圧縮し、錠剤の第二の層を形成する。錠剤の第二の層は、消化直後に崩壊し、オメプラゾールはすぐに放出される。PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含む二層の錠剤の概略図を図3に示す。
【0084】
例3:PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含む迅速性崩壊錠剤
迅速性崩壊錠剤を、以下の成分を含有する単一の投与形態として調合する:
オメプラゾール (パウダー) 40 mg
PG 4 mg
NaHCO3 500 mg
CaCO3 500 mg
クロスカルメロースナトリウム
微結晶性セルロース
ステアリン酸マグネシウム
デンプン。
【0085】
非腸溶性コーティングオメプラゾール(40 mg)、PG(4 mg)、NaHCO3、CaCO3、クロスカルメロースナトリウム、微結晶性セルロースおよびステアリン酸マグネシウムを混合し、得られた混合物を、標準的な錠剤プレッシングを用いて錠剤に圧縮し、迅速性崩壊錠剤(intravescent)を得る。
【0086】
例4:PG、腸溶性コーティングオメプラゾール、および重炭酸ナトリウムを含む発泡サック
発泡錠剤を、以下の成分を含有する単一の投与形態として調合する:
オメプラゾール 40 mg
PG 4 mg
NaHCO3 958 mg
クエン酸 832 mg
炭酸カリウム 312 mg
ステアリン酸マグネシウム
デンプン。
【0087】
腸溶性コーティングオメプラゾール(40 mg)およびPG(4 mg)を乳鉢に置き、乳棒で微粉に粉砕する。重炭酸ナトリウム、クエン酸、炭酸カリウムおよび他の全ての賦形剤を混合物に添加し、発泡性粉末の均質な混合物を形成する。得られた粉末を、40 mg腸溶性コーティングオメプラゾールと混合し、単位用量のパケットにパックする。
【0088】
B.製剤の説明−コーティングオメプラゾールを含有するマルチ粒子カプセル剤
例5:エチルセルロース−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
この例は、マルチ粒子硬質ゼラチンカプセルの製造に関与するステップを説明する。硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
エチルセルロースコーティング重炭酸ナトリウムビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0089】
PGを炭酸アンモニウム緩衝液pH 8に溶解することによりPG溶液を調製する。PG溶液を、流動層式装置でエチルセルロースコーティング重炭酸ナトリウムビーズにスプレーする。乾燥後、PG-重炭酸ナトリウムビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0090】
ゼラチンカプセルが胃の胃液中に溶解すると、PG含有ビーズのHPMC層が膨張し、胃酸が重炭酸ナトリウムと反応し、ビーズのコア内部にCO2を生成する。エチルセルロースコーティング重炭酸ナトリウムコアからの二酸化炭素の放出が、粒子の浮揚性を許容し、これにより粒子からのPGおよび炭酸カルシウムの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。CaCO3は、長期間にわたって胃のpHを上昇させ、放出の際のPGを保護する。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0091】
例6:アルギン酸塩−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
アルギン酸塩粒子に充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0092】
アルギン酸塩粒子は、アルギン酸塩溶液を塩化カルシウム溶液に滴下し、その後凍結乾燥してアルギン酸塩粒子を得ることにより作成する。例5のとおり調製されたPG溶液を、流動層式装置でアルギン酸塩粒子にスプレーする。乾燥後、PG-アルギン酸塩ビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0093】
ゼラチンカプセルが胃に溶解すると、PGビーズは、HPMC層と胃液の接触により膨張する。凍結乾燥されたアルギン酸塩粒子が、その低密度により粒子の浮揚性を許容し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0094】
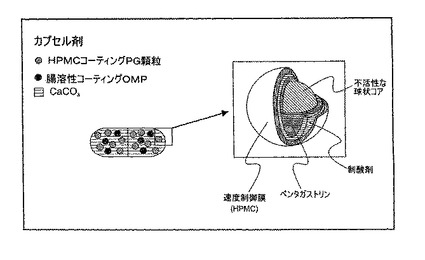
例7:スクロース−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
不活性な糖ビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0095】
PG溶液を、流動層式装置で不活性な糖ペレット(Nu-Pareils, 25/30)にスプレーする。乾燥後、PG-糖ビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。PG顆粒の概略図を図4に示す。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0096】
ゼラチンカプセルが胃に溶解すると、PGビーズは、PG含有ビーズのHPMC層と胃液の接触により膨張し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0097】
例8:HPMC−PGミニタブ(minitab)、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
不活性な糖ビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0098】
PGを、HPMCおよびCaCO3と組合せて顆粒状にし、ミニタブに圧縮する。ミニタブは、胃の胃液と接触したときに迅速に膨張する能力を有していることにより、胃での保持を可能にする。PGの胃への放出は、膨張したミニタブのポリマーマトリクスの侵食速度によりコントロールされる。PGミニタブを、腸溶性コーティングオメプラゾールビーズと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0099】
例9:非腸溶性時間依存性放出コーティングでコーティングされたオメプラゾールおよびPGビーズを含有するマルチ粒子カプセル剤
この例は、マルチ粒子硬質ゼラチンカプセルの製造に関与するステップを説明する。カプセルを、以下の混合集団の粒子を含む単一の投与形態として調合する:時間依存性放出コーティングでコーティングされたPGビーズ、時間依存性放出コーティングでコーティングされたオメプラゾールビーズ、および炭酸カルシウム。カプセルの概略図を図5に示す。各カプセルは、以下の成分を含有する:
・薄いHPMC層でコーティングされた40 mgオメプラゾールビーズ
・糖の球体上に充填され、薄いHPMC層でコーティングされた4 mg PG
・600 mg炭酸カルシウム(CaCO3)。
【0100】
コーティングの組成は、媒体がコアと接触するようになったときに、コアが水性環境に急速に崩壊するようにデザインされる。この目的のために、糖の球体は、制酸剤(NaHCO3またはCaCO3)層でコーティングされる。PGを炭酸アンモニウム緩衝液pH 8に溶解することによりPG溶液を調製する。PG溶液を、流動層式装置で上記制酸剤コーティングビーズにスプレーする。乾燥後、ビーズを、HPMCの薄層で更にコーティングし、約10分の遅延放出のPG粒子を作成する。オメプラゾールは、制酸剤コーティング糖球体上に層状に重ねられ、厚い時間放出性HPMCコーティングで被覆される。崩壊剤を粒子のコアに添加して、HPMCが溶解した後オメプラゾールがすぐに放出されるよう促進してもよい。コーティングオメプラゾールビーズは、胃を通過することを目的とし、HPMCが溶解された後、小腸の上部で吸収され、すぐにオメプラゾールが放出される。最終PG粒子を、オメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。PGおよびOMPの放出速度は、ビーズのHMPC層の厚みおよび侵食速度により決定される。CaCO3は、長期間にわたって胃のpHを上昇させ、放出の際のPGを保護する。
【0101】
C 製剤の説明−腸溶性コーティングオメプラゾールを含有する錠剤
例10:PG、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むプレスコーティング錠剤
プレスコーティング錠剤を、以下の成分を含有する単一の投与形態として調合する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
4 mg PG顆粒
炭酸カルシウム
ヒドロキプロピルメチルセルロース(HPMC)。
【0102】
プレスコーティング錠剤は、2段階の方法で調製される。単一の錠剤については、4 mg PG、900 mg炭酸カルシウムおよびHPMCを混合し、錠剤の中心コアにプレ圧縮する。40 mgの腸溶性コーティングオメプラゾールビーズを、PGコアにプレスコーティングし、錠剤の外層を形成する。最終錠剤は、制御された放出のPGコア層とオメプラゾール腸溶性コーティングビーズの即時放出性外層から構成される。別の例において、有効成分は、二層の錠剤に圧縮され、ここで第一の層は、4 mg PG、900 mg炭酸カルシウムおよびHPMCを含み、第二の層は、40 mgの腸溶性コーティングオメプラゾールビーズを含む。
【0103】
圧縮された錠剤は、以下の賦形剤の一以上を含んでいてもよい:ラクトース、マンニトール、コーンスターチ、ポテトスターチ、微結晶性セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、着色剤、希釈剤、緩衝化剤、湿潤剤、保存剤、香味剤、および薬学的に適合可能なキャリア。
【0104】
例11:PG、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムを含む迅速崩壊性錠剤
迅速崩壊性懸濁錠剤を、以下の成分を含有する単一の投与形態として調合する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
4 mg PG顆粒
900 mg炭酸カルシウム
クロスカルメロースナトリウム
微結晶性セルロース
ステアリン酸マグネシウム
ヒドロキシプロピルメチルセルロース(HPMC)。
【0105】
PG顆粒を、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)でコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび上述の賦形剤と混合し、得られた混合物を標準的な錠剤プレッシングを用いて錠剤に圧縮する。得られた錠剤は、迅速な崩壊時間を有し、胃で迅速に崩壊するために水で飲み込んでもよい。
【0106】
懸濁錠剤の崩壊の際に、PG粒子は、PG含有ビーズのHPMC層と水性環境の接触により膨張し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0107】
D インビボ実験
例12:ラットにおけるPG経口投与後の胃酸分泌の刺激
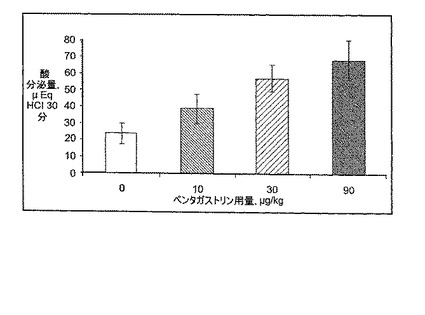
PGおよびPPIの併用による胃酸分泌の阻害は、経口投与されたPGが胃内で局所的に酸分泌を誘発する能力に基く。この問題に取り組むために、麻酔をかけたラットに、増加する量のPGを(経口)投与し、胃酸分泌を幽門結紮の胃でモニターした。増加する量(10、30、および90μg/kg)のPGを、幽門結紮のラットに経口胃管栄養法により投与した。30分の処置後、胃液を胃管腔から回収し、酸濃度をNaOHで滴定することにより決定し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。結果を、各実験グループに由来する7−8の動物の平均±SEMで表現する。図6に示されるとおり、経口投与されたPGは、用量依存的に有意に胃酸分泌を増大させ、このことは、経口投与されたPGが局所的に胃酸分泌を誘導することを示す。
【0108】
例13:オメプラゾールとともに投与されたPGの胃内pHに対する効果
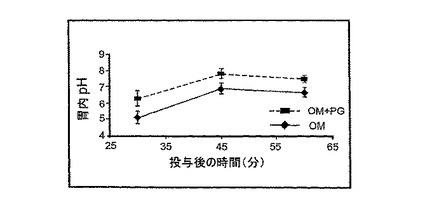
胃酸分泌の抑制に対するPG-PPIの併用効果を試験するために、麻酔をかけたラットに、オメプラゾール(10 mg/kg)を単独でまたはPG(350μg/kg)と併用して胃内に注入した。併用処置したラットは、オメプラゾールの15分前にPGを受けとった。処置から30、45、および60分後に吸引により胃液を回収し、胃酸分泌に対する薬剤の効果を、pHをモニターすることにより検出した。そのデータは、全ての時点において胃内pH値が、オメプラゾール単独で処置したラットよりPGとオメプラゾールを併用して処置したラットにおいて著しく高いことを示した(図7)。これら結果は、PGがラットにおいてPPIの抗分泌活性を増大させることを示す。
【0109】
例14:ランソプラゾール(Lansoprazole)は理性的(conscious)動物で用量依存的に胃酸分泌を阻害する
この実験では、様々なモデルの幽門結紮のラットを使用し、これは、理性的動物において胃酸分泌に対する薬剤効果の分析を許容する。このモデルは、胃酸分泌に対する麻酔の効果を排除する。研究薬剤を単独でまたは併用して経口投与した。1または2時間後、麻酔ガス機を用いて、幽門結紮を行い腹部を閉じるのに充分な短い時間(5分)、動物に麻酔をかけた。その後、回復のために動物をかごに戻した。数時間後、動物を屠殺し、食道のまわりに結紮糸を置き、胃を取り出し、胃内容物を回収した。遠心分離後、胃液サンプルを0.01 N NaOHを用いて終点pH 7に自動滴定し、滴定酸分泌量を計算した。
【0110】
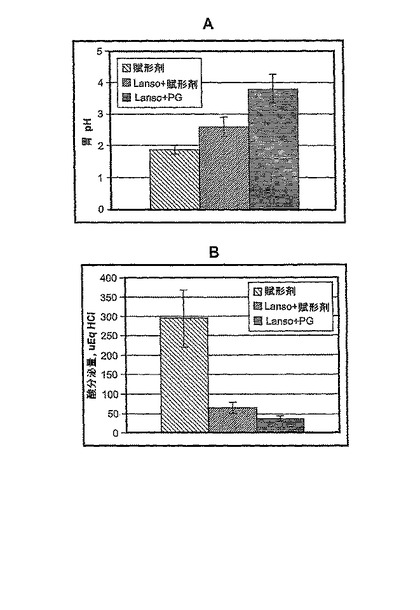
ランソプラゾールを、単純懸濁液(simplified suspension;SLS)として経口胃管栄養法により投与した。SLSは以下のとおり調製した:30 mgカプセル(Zoton)の内容物を8.4%重炭酸ナトリウムに懸濁した。幽門結紮の2時間前に、3つの用量のランソプラゾール(20、5および1.25 mg/kg)でラットを処置した。プラセボとしてコントロールグループに8.4% NaHCO3を投与した。図8は、ランソプラゾールが用量依存的に胃酸分泌を阻害することを示す。
【0111】
例15:理性的な幽門結紮ラットの胃酸分泌に対する、PGと併用して投与したランソプラゾール(Lansoprazole)の効果
この実験では、PG(300μg/kg)の15分前(A)または15分後(B)に、ラットを用量5 mg/kgのSLSで処置した。コントロールのラットは、プラセボとして8.4% NaHCO3およびPG-賦形剤を併用して注入した。すべての薬剤は、幽門結紮の2時間前に、経口胃管栄養法により投与した。胃液は3時間かけて回収した。データは平均±SEMで表す。各実験グループの動物数は8−9である。
【0112】
図9Aから分かるとおり、PGの15分前にSLSを投与すると、ランソプラゾール単独の場合と比較して、酸阻害の程度が高いが、PGで前処置した後SLSで処置したラットの酸分泌量は、ランソプラゾール単独で処置したラットと変わらなかった(図9B)。これらの結果は、PGが、胃酸分泌の遮断におけるランソプラゾールの効果を高めることを示す。更に、二つの化合物のタイミングは、PG/ランソプラゾールの併用処置の有効性を高めるために重要である。
【0113】
別の実験では、ラットを、用量2.5 mg/kgのSLSおよび賦形剤、あるいはSLSおよびPG(300μg/kg)で3日連続して1日に1回処置した。SLSは、PGまたは賦形剤の15分前に投与した。コントロールのラットは、プラセボとして8.4% NaHCO3およびPG-賦形剤を併用して注入した。すべての薬剤は、経口胃管栄養法により投与した。幽門結紮は、3日目に、処置の2時間後に実施した。胃液は3時間かけて回収した。データは平均±SEMで表す。各実験グループの動物数は8である。図10Aおよび10Bに示すとおり、SLSをPGと併用して3日間連続して投与すると、SLS単独の場合と比較して、胃内pHが有意に高くなった。同様に、SLS/PGを併用して3日間連続で処置したラットの胃酸分泌は、SLSを単独で投与したラットの胃酸分泌より少なかった。
【0114】
例16:ラットのPG介在性胃酸分泌に対するCCK-Bアンタゴニストの効果
PGはガストリンホルモン類似体であるため、その局所的効果は、ガストリン経路を介して、すなわちガストリンレセプター(CCKB)の活性化を介して介在されると考えられる。この仮説を試験するために、PG介在性酸分泌に対する特定のCCKBアンタゴニスト(Itriglumide)の効果を調査した。
【0115】
この研究では、ラットにKetamineおよびDomitor混合物で麻酔をかけ、20 mg/kgのItriglumideを十二指腸内に(i.d.)投与した。15分後、胃の幽門を結紮し、300μg/kg PGを胃内に(i.g.)投与した。30分後、胃液を得て、遠心分離し、上清の体積とpHを測定した。酸濃度(滴定酸性度)を、胃液サンプルをNaOHで滴定することにより分析し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。下記表1に示される結果から明らかなように、CCKBアンタゴニスト(ant.)の十二指腸内注入により、ラットの胃酸分泌に対するPGの局所的効果は阻害される。
【表1】
【0116】
例17:麻酔され幽門結紮されたラットの酸分泌に対するPGの十二指腸内注入の効果
麻酔され幽門結紮されたラットの酸分泌に対するPGの十二指腸内注入の効果を調査した。この研究では、300μg/kg PGを、麻酔され幽門結紮されたラットの十二指腸内に投与し、胃酸分泌レベルを30分後に測定した。胃液を得て、遠心分離し、上清の体積とpHを測定した。酸濃度(滴定酸性度)を、胃液サンプルをNaOHで滴定することにより分析し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。コントロールとして、等量のPGを胃内に注入し、胃液分泌に対するPGの効果を測定した。表2に示されるとおり、PGの胃内注入および十二指腸内注入のいずれも、麻酔され幽門結紮されたラットにおいて胃酸分泌を誘導する。
【表2】
【0117】
本発明が、特別に示されたものおよび上述のものに限定されないことは当業者に認識されるでしょう。むしろ、本発明の範囲は特許請求の範囲により規定される。
【図面の簡単な説明】
【0118】
【図1】図1は、NaHCO3が人工胃液中でPGの安定性を保存することを示す。
【図2】図2は、種々のpH値における非分解性PGのパーセンテージを示す。
【図3】図3は、PG、非腸溶性コーティングオメプラゾールおよび緩衝化剤を含む二層の錠剤の概略図である。
【図4】図4は、マルチ粒子カプセル調合物で使用されるPG顆粒の概略図である。
【図5】図5は、時間放出性コーティングビーズを含むカプセル剤の概略図である。
【図6】図6は、PGが、ラットにおいて用量依存的に胃酸分泌を刺激することを示す。
【図7】図7は、PGが、ラットにおいて胃酸分泌に対するPPI介在性効果を増大させることを示す。
【図8】図8は、ランソプラゾール(Lansoprazole)が、理性的動物において用量依存的に胃酸分泌を阻害することを示す。
【図9】図9は、ランソプラゾール(Lansoprazole)をPGの前に投与した場合(A)、PGが、ランソプラゾールの胃酸分泌の遮断効果を増大し、ランソプラゾールをPGの後に投与した場合(B)、PGが、ランソプラゾールの胃酸分泌の遮断効果を増大しないことを示す。
【図10】図10は、ランソプラゾール(Lansoprazole)をPGと併用して3日連続投与すると、ランソプラゾール単独と比較して、胃内pHが有意に高くなり(A)、胃酸分泌が有意に低くなる(B)ことを示す。
【発明の分野】
【0001】
本発明は、胃酸分泌に対して迅速発現性長期阻害効果を保持し、かつ食事に依存しない、胃酸分泌阻害のための新規経口組成物に関する。
【発明の背景】
【0002】
多数の病状は、胃酸分泌を抑制する必要性により特徴づけられる。かかる症状には、ゾリンジャー/エリソン症候群(ZES)、胃食道逆流疾患(GERD)、消化性潰瘍疾患、十二指腸潰瘍、食道炎などが含まれるがこれらに限定されない。消化性潰瘍などの症状は、重い合併症を発症することがあり、先進工業国では最も流行性の疾患の幾つかに相当する。
【0003】
GERDおよび消化性潰瘍疾患の治療において現在採用されている主な治療には、たとえばヒスタミンH2−受容体アンタゴニストまたはプロトンポンプインヒビター(PPI’s)を用いることにより胃の酸性度を低下させる薬剤が含まれる。PPI’sは、壁細胞からの酸分泌を担う壁細胞H+/K+ ATPアーゼプロトンポンプを阻害することにより作用する。PPI’s、たとえばオメプラゾール、およびその薬学的に許容可能な塩は、たとえばEP 05129、EP 124495およびUS Patent No. 4,255,431に開示される。
【0004】
PPI薬剤は、腸溶性コーティング顆粒剤で通常投与される酸不安定性プロドラッグである。PPIsは、小腸で吸収された後、弱塩基であるため、壁細胞の酸性環境(milieu)内に優先的に蓄積する。壁細胞の酸性環境(milieu)内の酸性状況(environment)により、当該プロドラッグは、活性なスルフェンアミドに変換され、これは、壁細胞H+/K+ ATPアーゼポンプに結合し阻害する活性薬剤である。
【0005】
PPIsの薬効は文書により充分裏付けられているが、PPIsには注目すべき制限がある。投薬および食事摂取の時期は、これら薬剤の薬物動態並びに胃酸分泌の抑制能力に影響を及ぼし得る(Hatlebakk et al., Aliment Pharmacol Ther. 2000; 14 (10): 1267-72)。具体的には、胃酸分泌の最適な抑制を達成するためには、PPIは食物の摂取前に服用しなければならない。更に、PPIsは、薬理作用の開始が比較的遅く、最大限の酸抑制および症状の緩和を達成するのに数日を要することがあり、オン・デマンド(on-demand)GERD療法にその有用性は限定される(Sachs G, Eur J Gastroenterol Hepatol. 2001; 13 Suppl 1: S35-41)。更に、PPIsは、胃酸の24h抑制を提供することはできず、夜間の酸ブレークスルーは、GERD患者において胸やけの痛みにつながり、PPIsの1日に2回の投薬を用いてもこれは発生する(Tytgat GN, Eur J Gastroenterol Hepatol. 2001; 13 Suppl 1: S29-33)。最終的に、これら薬剤は、薬物動態において患者間の実質的な変動性を示し、他の薬剤と重大な相互作用を示すことがある(Hatlebakk et al., Clin Pharmacokinet. 1996; 31 (5): 386-406)。よって、PPI-介在性活性の改良は、胃腸病学において充分に認識された課題(challenge)である。
【0006】
ペンタガストリン(PG)(β−アラニル−L−トリプトフィル−L−メチオニル−L−アスパルチル−L−フェニル−アラニルアミド;配列番号2)は、ガストリンのカルボキシ末端のテトラペプチドを含有するペンタペプチドである。このカルボキシ末端のテトラペプチドは、ほぼすべての天然のガストリンに見られる活性部分である。動物においてPGは、主に、胃に存在する類エンテロクロマフィン(ECL)細胞からのヒスタミンの放出を誘導することにより、胃酸の分泌を誘導するように作用する。ヒスタミンの放出およびその後の壁細胞に存在するヒスタミン受容体の活性化は、壁細胞の活性化につながり、プロトンイオンを胃管腔に活発に分泌する。PGは壁細胞に直接作用し、その活性化を誘導することもできる。PGは、胃酸分泌機能を評価するための診断用薬剤として当該技術分野で典型的に使用される。
【0007】
酸性状況でのPGの溶解度の低さ、およびPGが胃でペプシン分解されやすいという事実より、本出願人が発見するまで明らかに予測できない経口投与後の胃酸分泌のインデユーサーとしてのPGの用途が提示された。本出願人の発見前には、PGは非経口ルートにより投与された場合のみ、酸分泌の誘導において効果的に活性であると当業者により考えられていた。実際、酸分泌に対する効果は、PGの経口投与を受けた正常な4被検体では検出されなかったが、胃腸の異常を伴う別の3患者では幾らかの効果が検出された(Morrell & Keynes Lancet. 1975; 2 (7937): 712)。実際、この研究は、経口投与したときにPG活性が失われる証拠として薬理学の教科書に引用されている(Martindale Thirty-second edition, pl616, the Chapter:"Supplementary Drugs and Other Substances")。
【0008】
Pisegna et al.のWO01/22985('985公報)は、プロトンポンプインヒビター(PPI)と併用して全身に投与されるPGの使用を開示する。'985公報によれば、PPIと併用してPGを投与することにより、過剰な胃酸分泌を低減する/軽減するPPIの薬効は増大する。'985公報は、PGは好ましくは注射(たとえば皮下注射)により投与されるべきであることを開示し教示する。しかし、'985公報は、PGおよびPPIが静脈内、非経口、または経口的手段により投与され得ることも一般的に開示する。また'985公報は、PPIおよびPIが錠剤で調製され得ることを一般的に開示する。しかし、'985公報は、使用すべき特定の投与量や処方を開示しておらず実施例を提示していない。更に、'985公報は、PGが経口的にデリバーされると効果的でないという従来技術の教示をどのようにして回避するか教示も示唆もしておらず、効果的と思われるPGの経口投与量の例を提案していない。加えて、'985公報は、PGが胃で局所的に活性であることを開示しておらず、これは、本発明者らが驚くべきことに発見したことである。また'985公報は、胃管腔で局所的効果を達成するために、PG保存剤を使用して、胃においてPG活性の生物学的活性を保存することを教示していない。本発明の時点の技術水準に照らして、'985公報の一般的開示は、局所デリバリーのためのPGを含む経口組成物を調製する動機付けを当業者に与えるものではない。
【0009】
De Graef et al., Gastroenterology, 91, 333-337 (1986)(De Graef公報)は、オメプラゾールは、静脈注射によりPGで前処理したイヌに投与した場合、胃酸分泌の阻害において、より効果的であることを開示する。De Graef公報には、PGの経口投与が、胃管腔で局所的に作用することにより効果的であり、オメプラゾールの効果を増強することは記載されていない。
【0010】
Phillipsの米国特許Nos. 6,489, 346; 6,645, 988; および6,699, 885 (まとめて"Phillips特許")は、PPI、少なくとも一の緩衝化剤、および具体的な壁細胞アクチベーターから成る経口組成物を用いた、酸原因の胃腸障害を治療する薬学的組成物および方法を開示する。Phillips特許に開示される壁細胞アクチベーターには、たとえば、チョコレート、重炭酸ナトリウム、カルシウム、ハッカ油、スペアミント油、コーヒー、茶およびコラ、カフェイン、テオフィリン、テオブロミンおよびアミノ酸残基が含まれる。Phillips特許に示されるとおり、提案されるこれら壁細胞アクチベーターはすべて、酸分泌に対して阻害効果および刺激効果の両方を発揮する内在性ガストリンの放出を誘導する。しかし、Phillips特許は、PGの使用を開示も示唆もしておらず、PGは、(CCK-AおよびCCK-Bレセプターの両方を活性化し、阻害と刺激の両効果を含む)Phillips特許に記載される壁細胞アクチベーターとは異なり、刺激活性のみを保持し、CCK-B受容体にのみ結合する。
【0011】
胃酸分泌の阻害が必要な病状を効果的に治療する方法を開発することにより、長期の切実な要求が満たされるでしょう。PPI’sの広範囲の使用にもかかわらず、PPIの薬効を高める必要性がなお存在しており、たとえば迅速で効果的な発現、夜間の酸のブレークスルーを含む長時間の効果、少ない投与量での高い効果、および食事に依存しない投与が挙げられる。
【発明の概要】
【0012】
本発明の目的は、食事に依存せず、かつ胃酸分泌に対して迅速発現性長期阻害効果を示す、胃酸分泌阻害のための経口組成物を提供することである。
【0013】
本発明の別の目的は、不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI)および壁細胞のアクチベーターを含む、胃酸分泌阻害のための経口組成物であって、PPIの抗酸活性が、食事に依存せず、かつ酸分泌に対して迅速発現性長期阻害効果を示す組成物を提供することである。
【0014】
本発明の一つの態様において、経口組成物は、胃酸分泌インヒビターとしての不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI)、壁細胞のアクチベーターとしてのペンタガストリン(PG)および/またはPG類似体、および胃液中でPGの有効性を保存する一以上の薬剤を含み、その結果、PGの生物学的活性は維持され、これによりPGが胃で局所的に作用することが可能である。予測外なことに、本発明の組成物は、食事に依存しない胃における抗酸活性を保持し、かつ酸分泌に対して迅速発現性長期阻害を示す。本発明の組成物は、胃における酸分泌の抑制が必要な慢性または急性障害に罹患している被検体を治療するために使用され得る。
【0015】
本発明によるプロトンポンプインヒビター(PPIs)は、胃壁細胞においてH+/K+-アデノシントリホスファターゼ(ATPアーゼ)プロトンポンプの活性を阻害する化合物である。PPIは、そのプロドラッグ形態において、イオン化していないため、壁細胞の細胞膜を通過することができる。非イオン化PPIは、壁細胞に到達すると、活性化壁細胞の酸分泌部分、分泌細管に移動する。分泌細管にトラップされたPPIは、プロトン化され、プロトンポンプのアルファサブユニットのシステイン残基とジスルフィド共有結合を形成することができる活性なスルフェンアミド形態に変換され、これにより不可逆的にプロトンポンプを阻害する。
【0016】
上述したとおり、本発明は、PGが、経口投与された場合に、好ましくは胃管腔で局所的に作用して壁細胞を活性化することにより、局所的に活性であるという驚くべき発明者らの発見に基くものである。活性な壁細胞は、酸性pHを保持しており、これは、PPIが活性なプロトン化スルフェンアミド形態に変換されるために必要である。したがって、PGが胃管腔に直接作用することにより壁細胞が同時に活性化されると、PPIによるポンプの阻害は最大限に発揮される。
【0017】
本発明の経口組成物は、胃酸分泌の低減を目的とした公知のPPIベースの組成物と比べて以下の利点を示す。本発明の組成物は、胃管腔における局所的効果により、PGの全身投与と関連した副作用を生じることなく、PGによる壁細胞の活性化を可能にする。PGによる壁細胞のプレ活性化は、PPIの活性なスルフェンアミド形態への変換を促進し、これによりPPIの効果が迅速に発現する。更に、本発明の組成物は、食事に依存しない様式で、胃において抗酸活性の迅速な発現を示す。このように、経口組成物において活性薬剤を組合せることにより、酸分泌の迅速な低減が必要な急性症状に対して有効な解決策が提供される。最終的に、本発明の経口組成物は、単回の投薬を用いて、少なくとも24hの間、胃酸分泌の長期抑制を提供する。
【0018】
本発明による経口組成物は、胃管腔における壁細胞の局所的アクチベーターとして、PGまたはPG類似体を含む。アミノ酸配列βAla-Trp-Met-Asp-PheNH2(配列番号2)を含むPGに加えて、本発明は、壁細胞のアクチベーターとして、ガストリンまたはPG類似体またはその誘導体の使用を想定する。かかる変異体には、ガストリンの34-、17-、および14-アミノ酸種、および完全な薬理活性を有することが文献(Tracey and Gregory (1964) Nature (London), 204: 935参照)で報告されているガストリンの活性なC末端テトラペプチドTrp-Met-Asp-PheNH2(配列番号1)を含む他のトランケーション変異体を含むが、これらに限定されない。
【0019】
天然のアミノ酸が保存的置換で置換されているガストリンの変異体および/またはトランケートされたガストリンも含まれる。これら分子の種々の類似体には、PGのN−保護誘導体Boc-βAla-Trp-Met-Asp-PheNH2(Bocはtert-ブチルオキシカルボニル基である)またはF-Moc-βAla-Trp-Met-Asp-PheNH2(Mocはメトキシカルボニルである)が含まれるがこれらに限定されない。
【0020】
非限定的な態様において、本発明による経口組成物は、酸性胃液中でPGの有効性を保存する一以上の薬剤を更に含む。これら薬剤は、好ましくは、胃液中でPGの溶解性を保持しその分解を防ぐことにより胃液中でPGの有効性を保存するのに充分な量で存在し、その結果、胃においてPGの局所的な生物学的活性が保存される。これにより、PGは胃で局所的に作用し壁細胞を活性化することができる。かかる薬剤は、好ましくは、胃液中に溶解されたときに、ペプシンを阻害する値まで胃液のpHを一時的に上昇させることにより、ペプシンによる胃液中のPGの分解を阻害することができる制酸剤またはアルカリ性薬剤である。PGは、アルカリ性状況でのみ可溶であるため、胃液のpHの一時的な上昇は、PGの少なくともかなりの割合が胃液中に可溶なまま残ることを保証する。
【0021】
任意の弱塩基または強塩基(およびその混合物)が、本発明の経口組成物においてアルカリ性薬剤として利用可能であることに注目されたい。アルカリ性薬剤または制酸剤は、酸性胃液においてPGの安定性および溶解性を実質的に保存するのに充分な量で組成物中に存在する。したがって、本発明のアルカリ性薬剤は、胃液に溶解されたときに、PGの十分な有効性を達成し治療作用を発揮するのに充分な値まで胃のpHを上昇させることができる。
【0022】
好ましい態様に従って、組成物中のアルカリ性薬剤は、PGが胃で壁細胞に到達しそれを活性化するのに充分な期間にわたって、4を超える値、より好ましくは5を超える値まで胃液のpHを上昇させるのに充分な量で存在する。より好ましい態様において、アルカリ性薬剤は、5〜60分の期間、好ましくは5〜30分の期間にわたって、5を超える値まで胃液のpHを上昇させることができる。このように、本発明によるアルカリ性薬剤は、PGが壁細胞を活性化するのに充分な期間にわたって、胃液中でPGの溶解性を保存する。更に、胃液中の一時的なアルカリ性状況は、酸性pHにおいてのみ活性であるペプシンによるPGの分解を妨害する。
【0023】
種々の態様に従って、本発明の組成物は、酸性胃液中でPGの有効性を保存する他の薬剤を更に含む。かかる薬剤は、たとえば、胃におけるペプチドの分解を低減するペプシンインヒビター(すなわちペプスタチンおよびその誘導体バシトラシン−環状ドデカペプチド)、または胃粘膜の粘度を低減し、これにより酸分泌を担う細胞に到達するPGの能力を促進する粘液溶解剤である。かかる粘液溶解剤は、還元剤、たとえばN-アセチルシステイン、ジチオトレイトール、クエン酸またはマンニトールである。本発明の組成物は、胃に存在する細菌に対して効果的な抗生物質を更に含んでいてもよい。
【0024】
本発明の有効成分は、単一の経口投与形態、好ましくは固体投与形態で調合され得る。また、懸濁剤などの液体投与形態が使用されてもよい。よって、一つの態様において、PPI、PGおよび胃液中でPGの有効性を保存する薬剤は、多層錠剤、懸濁錠剤、発泡錠剤、粉剤、ペレット、顆粒剤、マルチプルビーズを含む硬質ゼラチンカプセル剤、または脂質ベースの賦形剤を含む軟質ゼラチンカプセル剤として調合され得る。
【0025】
一つの態様に従って、本発明の固体投与形態は、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされたPPI粒子、PGの粒子、および一以上のアルカリ性薬剤の粒子を含有するカプセル剤または多層錠剤である。PGによる胃管腔における壁細胞の活性化が、小腸の近位部におけるPPIの吸収と同調するようにするために、単一の経口投与形態は、胃でのPG放出時間を延長する時間依存性放出ポリマーでコーティングされたPGビーズを含んでいてもよい。このように、胃においてPG放出が延長されると、壁細胞上でPGの活性とPPIの活性とが同調するようになる。
【0026】
本発明の有効成分は、別個の投与形態で調合されてもよい。たとえば、PGおよび胃液中でPGの有効性を保存する薬剤は、経口懸濁剤または固体投与形態、たとえばカプセル剤、錠剤、懸濁錠剤、または発泡錠剤で調合され、PPIは別の固体投与形態、好ましくは腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーを備えたビーズを含有するカプセル剤または錠剤で調合され得る。別個の投与形態は、PGおよび胃液中でPGの有効性を保存する薬剤を一つの投与形態で含有し、PPIを別の投与形態で含有するキットとして提供されてもよい。この場合、それらの生理活性の時間的オーバーラップが少なくとも存在するように、PGはPPIと共に投与される。PPIおよびPGは、同時におよび/または連続して投与することができる。
【0027】
本発明で使用されるPPI粒子は、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされていてもよいし、コーティング層なしであってもよい。胃を通過する間の非コーティングPPIの安定性は、組成物に存在する一以上のアルカリ性薬剤により保存される。小腸の近位部における非腸溶性コーティングPPIの緩衝化懸濁剤の吸収は、腸溶性コーティングPPI顆粒剤の吸収より迅速であることが以前に実証されている(Pilbrantand Cederberg, Scand. J. Gastroenterol 1985: 20 (supp. 108): 113-120)。従って、非コーティングPPI粒子を組成物で使用する場合、胃でのPGの放出を遅延させる必要はない。しかし、コーティングPPI粒子を使用する場合、たとえばポリマーコーティングPG粒子を使用して胃でのPGの放出を遅延させることにより、PPIの放出をPGの放出と同調させることが必要である。
【0028】
別の態様において、本発明は、胃酸分泌の抑制が必要な障害または胃酸分泌の抑制により通常治療される障害に罹患した被検体を治療する方法に関する。本方法は、胃酸分泌インヒビターとしてのPPI、胃管腔における壁細胞のアクチベーターとしてのPGまたはPG類似体、および胃管腔でPGの有効性を保存するのに充分な量の少なくとも一の保存剤を含む薬学的組成物を被検体に投与することを含む。
【0029】
本発明の組成物は、胃酸分泌の阻害が必要な哺乳類の病状を予防または治療するために使用することができる。好ましくは、哺乳類はヒトである。本発明の組成物は、かかる病状を治療する際、および発症前にかかる病状の発症リスクを最小限にする際の両方において効果的である。
【0030】
本発明の薬学的組成物は、胃酸分泌の抑制により治療される多数の病的症状に使用することができる。かかる症状には、ゾリンジャー/エリソン症候群(ZES)、胃食道逆流疾患(GERD)、食道炎、消化性潰瘍疾患、十二指腸潰瘍、胃炎および胃粘膜びらん、消化不良症などが含まれるがこれらに限定されない。
【0031】
また本発明は、経口薬学的キットを含む。キットは、典型的に、薬学的に効果的な量の(i)配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液中の前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む。一つの態様において、有効成分は、別々の投与ユニット形態で調合される。キットは、その有効成分を被検体に投与することにより、胃酸分泌の抑制が必要な被検体の障害を治療または予防するために使用することができる。ペプチドは、典型的には、PPIの投与と同時、その前またはその後に投与される。
【0032】
これらの態様および更なる態様は、後述の詳細な説明および実施例から明らかでしょう。
【発明の詳細な説明】
【0033】
「アルカリ性薬剤」の用語は、PGとともに(たとえば、PGの前、間および/または後に)調合されるかまたはデリバーされたときに、胃内でPGの有効性を実質的に保存する値まで胃管腔のpHを一時的に上昇させるように機能する任意の薬学的に適切な弱塩基または強塩基(およびその混合物)をいう。
【0034】
「胃内でPGの有効性を保存する薬剤」の用語は、胃におけるPGの溶解性および安定性を維持することができる任意の薬剤をいう。具体的には、かかる薬剤は、可溶性形態で少なくともかなりの量のPGを維持することができ、かつ胃液で分解されず、その結果、胃におけるPGの生物学的活性が維持される。
【0035】
「胃におけるPGの生物学的活性」の用語は、胃管腔に位置する壁細胞の活性化をいう。
【0036】
「と併用して(in conjunction with)」の用語は、PPIおよびPGを別個の投与形態で投与するときに、それらの生理活性の時間的オーバーラップが少なくとも存在することを意味する。よって、PPIおよびPGは、同時におよび/または連続して投与することができる。
【0037】
本発明は、PGが経口投与後に活性なまま残り、好ましくは胃で局所的に作用することにより壁細胞を活性化することができるという驚くべき発見に基くものである。重要なこととして、壁細胞の活性化には、PPIプロドラッグが、胃H+/K+-ATPアーゼプロトンポンプの不可逆性インヒビターとして作用する活性な形態に変換されることが必要である。本発明の経口組成物は、胃酸分泌の阻害においてPPIの薬効を増大する活性薬剤の特有の組合せを提供する。
【0038】
本発明の組成物は、胃酸分泌の阻害が必要な哺乳類の病状を予防または治療するために使用することができる。本発明の組成物は、病状を治療する際、およびかかる病状の発症リスクを発症前に最小限にする際の両方に効果的である。かかる病状には、たとえば、逆流性食道炎、胃炎、十二指腸炎、胃潰瘍および十二指腸潰瘍が含まれる。更に、本発明の組成物は、胃酸阻害効果が望まれる他の胃腸疾患を治療または予防するために使用することができ、たとえば、(低用量アスピリンを含む)非ステロイド性抗炎症薬(NSAID)治療中の患者、非潰瘍性消化不良の患者、症候性胃食道逆流疾患(GERD)の患者、およびガストリン産生腫瘍の患者に使用することができる。また、集中治療の状況におかれた患者、急性消化管出血の患者、胃酸の吸引を予防する術前および術後の状況、およびストレス性潰瘍を予防および治療する術前および術後の状況において使用することもできる。更に、ヘリコバクター感染症およびこれに関連した疾患の治療に有用であり得る。治療に適した他の症状には、ゾリンジャー−エリソン症候群(ZES)、ウェルナー症候群、および全身性肥満細胞症が含まれるがこれらに限定されない。
【0039】
本発明による壁細胞アクチベーターは、好ましくは、配列番号2で示されるアミノ酸配列を有するPGである。しかし、ガストリンのC末端テトラペプチドTrp-Met-Asp-PheNH2(配列番号1で示される)を含む任意のPG類似体が、壁細胞アクチベーターとして使用され得る。かかる類似体には、ガストリンの34-、17-、および14-アミノ酸種、および他のトランケーション変異体が含まれるがこれらに限定されない。また、天然のアミノ酸が保存的置換により置換された、ガストリンの変異体および/またはトランケートされたガストリンも含まれる。また、たとえばPGのN−保護誘導体を含むがこれに限定されない、これら分子の種々の類似体も含まれる。PGの適切な保護基には、当該技術分野で公知の標準的ヒドロキシル保護基、たとえばメトキシメチル(MOM)、β−メトキシエトキシメチル(MEM)、トリアルキルシリル、トリフェニルメチル(trityl)、tert−ブトキシカルボニル(t-BOC)、エトキシエチル(EE)、f-MOC(メトキシカルボニル)、およびTROCなどが含まれる。保護基は、当業者に一般に公知の標準的手法を用いることにより除去し、所望のPG誘導体を得ることができる(T. W. Green, Protective Groups in Organic Synthesis, Chapter 2, pages 10-69 (1981))。
【0040】
ガストリン、ペンタガストリン、またはその類似体は、商業的に入手可能である。加えて、合成のプロトコールが周知である。このため、たとえば、PGは周知のペプチド合成方法を用いて化学的に合成することができる(たとえば、Barany and Merrifield Solid-Phase Peptide Synthesis; pp. 3-284 in The Peptides: Analysis, Synthesis, Biology. Vol. 2: Special methods in peptide synthesis, part a.; Merrifield et al. (1963) J. Am. Chem. Soc., 85: 2149-2156; and Stewart et al. (1984) Solid Phase Peptide Synthesis, 2nd ed. Pierce Chem. Co., Rockford, ILL.を参照)。更にPGは、たとえばBoc-Ala残基をテトラペプチドTrp-Met-Asp-PheNH2に結合させることにより化学的に合成することができる。
【0041】
本発明の組成物は、過度な有害な副作用を起こすことなく、壁細胞に対して薬理効果を達成するのに効果的な量でPGまたはその類似体を含む。組成物に存在するPGの標準的な概算量は、好ましくは1-100 mg、より好ましくは2-60 mg、最も好ましくは4-40 mgのPG量(または等価な量のPG類似体)である。
【0042】
本発明の組成物は、胃H+/K+-ATPアーゼプロトンポンプの不可逆性インヒビターとして作用するPPIを更に含む。本発明で使用されるPPIは、H+, K+-ATPアーゼ阻害活性を有する任意の置換ベンズイミダゾール化合物とすることができる。本発明の目的において、「PPI」の用語は、H+, K+-ATPアーゼインヒビターとしての薬理活性を保持する任意の置換ベンズイミダゾールを意味し、オメプラゾール、ランソプラゾール(lansoprazole)、パントプラゾール、ラベプラゾール、ドントプラゾール、ペルプラゾール(s-オメプラゾールマグネシウム)、ハベプラゾール、ランソプラゾール(ransoprazol)、パリプラゾール、およびレミノプラゾールの中性形態または塩の形態、単一のエナンチオマーまたは異性体または他の誘導体またはエナンチオマーのアルカリ性塩を含むが、これらに限定されない。
【0043】
本発明で使用され得る胃H+/K+-ATPアーゼプロトンポンプインヒビターの例は、たとえば、プロトンポンプインヒビターとして効果的な新規チアジアゾール化合物を記載する米国特許第6,093,738号に開示される。プロトンポンプインヒビターとして、欧州特許第322133号および第404322号はキナゾリン誘導体を開示し、欧州特許第259174号はキノリン誘導体を記載し、WO 91/13337および米国特許第5,750,531号はピリミジン誘導体を開示する。また、適切なプロトンポンプインヒビターは、たとえば、EP-A1-174726、EP-A1-166287、GB 2 163 747およびW090/06925、W091/19711、W091/19712、W094/27988およびW095/01977にも開示される。
【0044】
本発明による組成物においてPPI粒子は、コーティングされていてもコーティングされていなくてもよい。オメプラゾールなどのPPIを含む腸溶性コーティング粒子の調製は、たとえば、米国特許第4,786,505号および第4,853,230号に開示される。
【0045】
本発明の組成物は、過度な有害な副作用を起こすことなく、薬理効果または治療的改善を達成するのに効果的な量でPPIを含む。治療的改善には、胃pHの上昇、消化管出血の減少、または症状の改善もしくは除去を含むがこれらに限定されない。好ましい態様に従って、PPIの典型的な一日量は変動し、患者の個々の必要量および治療すべき疾患などの種々の因子に依存する。一般に、PPIの一日量は、1-400 mgの範囲である。組成物中に存在するPPIの好ましい標準的概算量は、典型的には、約20-40 mgのオメプラゾール、約30 mgのランソプラゾール(lansoprazole)、約40 mgのパントプラゾール、約20 mgのラベプラゾール、並びに薬理的に等価な用量の以下のPPIs:ハベプラゾール、パリプラゾール、ドントプラゾール、ランソプラゾール(ransoprazole)、ペルプラゾール(s-オメプラゾールマグネシウム)、およびレミノプラゾールである。
【0046】
好ましい態様において、本発明の組成物は、酸性胃液中でPGの有効性を保存する一以上の薬剤を更に含む。より具体的には、保存剤は、胃液中でPGの安定性または溶解性を維持する。これにより、PGが胃において局所的に作用し壁細胞を活性化することが可能になる。かかる薬剤は、好ましくは、それらを胃液に溶解したときに、胃に存在するペプチダーゼを阻害し、かつ少なくともかなりの割合のPGが胃液中に可溶なまま残るpHまで、胃液のpHを上昇させることができるアルカリ性薬剤または制酸剤である。
【0047】
本発明で使用されるアルカリ性薬剤には、たとえば以下のものが含まれる:重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、重炭酸カルシウム、およびその他のカルシウム塩。重炭酸ナトリウムは水に容易に溶解するが、炭酸カルシウムは水不溶性であり、酸性環境でのみゆっくり溶解できることに注意されたい。したがって炭酸カルシウムは、胃内でアルカリ性薬剤の持続した溶解が望まれるときに有効であり得る。
【0048】
本発明で使用される制酸剤の例には、一以上の以下のものが含まれる:アルミナ、炭酸カルシウム、および重炭酸ナトリウム;アルミナおよびマグネシア;アルミナ、マグネシア、炭酸カルシウム、およびシメチコン;アルミナ、マグネシア、および炭酸マグネシウム;アルミナ、マグネシア、炭酸マグネシウム、およびシメチコン;アルミナ、マグネシア、およびシメチコン;アルミナ、アルギン酸マグネシウム、および炭酸マグネシウム;アルミナおよび炭酸マグネシウム;アルミナ、炭酸マグネシウム、およびシメチコン;アルミナ、炭酸マグネシウム、および重炭酸ナトリウム;アルミナおよび三ケイ酸マグネシウム;アルミナ、三ケイ酸マグネシウム、および重炭酸ナトリウム;アルミナおよびシメチコン;アルミナおよび重炭酸ナトリウム;塩基性炭酸アルミニウム;塩基性炭酸アルミニウムおよびシメチコン;水酸化アルミニウム;炭酸カルシウム;炭酸カルシウムおよびマグネシア;炭酸カルシウム、マグネシア、およびシメチコン;炭酸カルシウムおよびシメチコン;炭酸カルシウムおよびマグネシウム;マガルドレート;マガルドレートおよびシメチコン;炭酸マグネシウムおよび重炭酸ナトリウム;水酸化マグネシウム;酸化マグネシウム。
【0049】
好ましくは、本発明の組成物は、一以上のアルカリ性薬剤または制酸剤を、薬理効果を達成するのに効果的な量で含む。具体的には、組成物中のアルカリ性薬剤または制酸剤は、PGが胃の壁細胞を活性化するのに充分な期間にわたって、胃に存在するプロテアーゼに最適なpHより高いpHまで胃液のpHを上昇させるのに充分な量で存在する。好ましい態様において、アルカリ性薬剤または制酸剤は、5〜60分の期間にわたって、好ましくは5〜30分の期間にわたって、5を超えるpHまで胃液のpHを上昇させるのに充分な量で存在する。本発明の組成物に必要なアルカリ性薬剤の量は、使用されるアルカリ性薬剤のタイプおよび所定のアルカリ性薬剤により提供される塩基に相当するものにより必然的に変動する。実際、胃においてPGの優れた有効性を提供するのに必要な量は、(米国薬局方(USP)ガイドラインに従って調製される)200ミリリットルの人工胃液の溶液に添加したときに、そのHCl溶液のpHを少なくともpH5.0まで上昇させる量である。好ましくは少なくとも100ミリグラム、より好ましくは少なくとも300ミリグラム、最も好ましくは少なくとも500ミリグラムのアルカリ性薬剤が、本発明の薬学的組成物で使用される。
【0050】
別の態様において、本発明の組成物は、酸性胃液中でPGの有効性を保存する他の薬剤を更に含む。たとえば、本発明の組成物は、ペプシンインヒビター、たとえば活性化されたペンタペプチドペプスタチン、および天然または合成由来のその誘導体を含んでいてもよい。これらインヒビターは、ペプシンによるPGの分解を減少させる。更に、本発明の組成物は、胃粘膜の粘度を減少させることによりPGが壁細胞に到達する能力を促進する粘液溶解剤を含んでいてもよい。かかる粘液溶解剤は、たとえば還元剤、たとえばN-アセチルシステイン、ジチオトレイトール、クエン酸またはマンニトールである。あるいは、本発明の組成物は、PGのためのポリマーコーティング、たとえば胃の酸性環境からPGを保護するポリマーの腸溶性コーティングを含んでいてもよい。
【0051】
本発明の有効成分は、好ましくは、すべての有効成分を含有する単一の経口投与形態で調合される。本発明の組成物は、固体または液体形態の何れかで調合され得る。液体製剤と比較して固体製剤の安定性が改良されていることから固体製剤が好ましいことに注意されたい。
【0052】
一つの態様において、PPI粒子、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、単一の固体投与形態、たとえば多層錠剤、懸濁錠剤、発泡錠剤、粉剤、ペレット、顆粒剤、または複数のビーズを含むカプセル剤で調合される。別の態様において、活性薬剤は、単一の液体投与形態、たとえば全ての有効成分を含有する懸濁剤、または使用前に再構成されるドライ懸濁剤で調合されていてもよい。
【0053】
単一の投与形態において、PPI粒子およびPG粒子は、胃におけるPGの局所的生物学的活性と壁細胞に対するPPIの全身効果とを同調させるために、腸溶性pH依存性放出ポリマーまたは非腸溶性時間依存性放出ポリマーの何れかでコーティングされていてもよい。たとえば、コーティングPPI粒子を使用して血液での吸収を遅延させる場合、PG粒子もコーティングしてその放出を遅延させることが望ましい。一つの具体的な態様において、PPI粒子が厚い非腸溶性層でコーティングされ、PPIの放出が好ましくは20−80分、より好ましくは25−75分、最も好ましくは30−60分遅延され、PG粒子が薄い非腸溶性ポリマー層でコーティングされ、PGの放出が好ましくは5−60分、より好ましくは8−45分、最も好ましくは10−30分遅延される。これら条件により、薬理学的PPI血漿濃度を達成する前に、PGによる壁細胞のプレ活性化が可能になる。
【0054】
本発明で使用される適切なpH依存性腸溶性ポリマーの非限定的な例は、以下のとおりである:酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、フタル酸ポリ酢酸ビニル、メタクリル酸共重合体、シェラック、コハク酸ヒドロキシプロピルメチルセルロース、トリメリット酸酢酸セルロース、および上記の何れかの混合物。適切な商業的に入手可能な腸溶性材料は、たとえば商標Eudragit L 100-55で販売されている。このコーティングは、基質にスプレーコーティングすることができる。
【0055】
非腸溶性時間依存性放出ポリマーには、たとえば、胃液から水分を吸収して胃で膨張することにより、粒子のサイズを増大させて厚いコーティング層を作成する一以上のポリマーが含まれる。時間依存性放出コーティングは、一般に、外部の水性媒体のpHに依存しない侵食および/または拡散特性を有する。よって、有効成分は、拡散により、または胃内で粒子がゆっくり侵食された後に、粒子からゆっくり放出される。
【0056】
流体と投与形態の表面との相互作用により生じる胃内でのポリマーの侵食特性は、ポリマーの分子量および薬剤/ポリマーの比により主に決定される。PGおよびPPIの放出を確実に約10分〜約60分遅延させるためには、ポリマーの分子量が約105〜約107グラム/molの範囲であることが推奨される。更に、PGまたはPPI/ポリマーの比が約2:3〜約9:1、好ましくは約3:2〜9:1、最も好ましくは約4:1〜9:1であることが推奨される。
【0057】
適切な非腸溶性時間依存性放出コーティングは、たとえば、フィルム形成化合物、たとえばセルロース誘導体、たとえばメチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシエチルセルロース、および/またはアクリルポリマー、たとえばEudragit商標ポリマーの非腸溶性形態である。他のフィルム形成材料を、単独で、または互いに組み合わせて、または上述のものと一緒に使用してもよい。これらの他のフィルム形成材料には、一般に、ポリ(ビニルピロリドン)、ゼイン、ポリ(エチレングリコール)、ポリ(エチレンオキシド)、ポリ(ビニルアルコール)、ポリ(ビニルアセテート)、およびエチルセルロース、並びに他の薬学的に許容可能な親水性および疎水性フィルム形成材料が含まれる。これらフィルム形成材料は、賦形剤または溶媒システムとして水を用いて基質コアに適用してもよい。また、水性アルコールシステムをフィルム形成のための賦形剤として採用してもよい。
【0058】
本発明の時間依存性放出コーティングを作成するのに適した他の材料には、たとえば以下のものが挙げられるがこれらに限定されない:水溶性ポリサッカリドガム、たとえばカラーギンナン、フコイダン(fucoidan)、ガッチゴム、トラガカント、アラビノガラクタン、ペクチン、およびキサンタン;ポリサッカリドガムの水溶性塩、たとえばアルギン酸ナトリウム、トラガカントナトリウム(sodium tragacanthin)、およびガッチゴムナトリウム(sodium gum ghattate);アルキル基が直鎖もしくは分枝の1〜7の炭素の水溶性ヒドロキシアルキルセルロース、たとえばヒドロキシメチルセルロース、ヒドロキシエチルセルロース、およびヒドロキシプロピルセルロース;合成水溶性セルロース系ラミナ形成材(former)、たとえばメチルセルロースおよびそのヒドロキシアルキルメチルセルロース誘導体、たとえばヒドロキシエチルメチルセルロース、ヒドロキシプロピルメチルセルロース、およびヒドロキシブチルメチルセルロースから成る群より選択されるメンバー;他のセルロースポリマー、たとえばカルボキシメチルセルロースナトリウム;および当業者に公知の他の材料。この目的のために使用することができる他のラミナ形成材料には、ポリ(ビニルピロリドン)、ポリビニルアルコール、ポリエチレンオキシド、ゼラチンとポリビニルピロリドンの混合物、ゼラチン、グルコース、サッカリド、ポビドン、コポビドン、ポリ(ビニルピロリドン)−ポリ(酢酸ビニル)共重合体が挙げられる。
【0059】
胃でのPGの放出を遅延させるための別のアプローチは、胃液より低い密度の浮遊粒子の使用により粒子からPGの放出を遅延させることである。一つの好ましい態様において、浮遊粒子は、胃液との接触により、エチルセルロースコーティング重炭酸ナトリウムビーズ内に二酸化炭素が放出することにより得られる。エチルセルロースコーティング重炭酸ナトリウムコアからの二酸化炭素の放出は、粒子の浮力を許容し、粒子からのPGの放出を遅延させる。
【0060】
胃でのPGの放出を遅延させるために、他の遅延性胃内容物排出アプローチを使用してもよい。これらアプローチには、消化しにくいポリマーまたは胃の運動性パターンを食事状態に変更する脂肪酸塩の使用により、胃内容物排出速度を低下させ、薬剤放出の大幅な延長を許容することが含まれる(たとえばSingh and Kim, J. of Controlled Release 63 (2000) 235-259に開示される)。
【0061】
ある状況においては、胃内容物排出に抵抗するサイズまで、胃内で急速に広がる投与形態を用いることにより、胃でのPGの保持時間を延長することが望ましい。かかるシステムは、長期間にわたって完全な状態を保持し、小さなピースへの分解が起こるまで、胃から排出されることはない。Caldwell (Caldwell, L. J., Gardener, C. R., Cargill, R. C. (1988), 米国特許第4,767,627号)は、侵食性ポリマーから作成され、硬質ゼラチンカプセルに折り畳まれ挿入された薬剤を充填した十字デバイスを記載する。経口投与後、ゼラチンシェルは崩壊し、折り畳まれたデバイスは展開する。システムは、最小サイズ1.6 cmおよび最大サイズ5 cmであるため、胃からの通過が可能なほど充分小さくなるポイントまでポリマーが侵食してはじめて、胃から幽門へ移動する。
【0062】
胃でのPGの保持時間を延長する別のアプローチは、ヒトへの投与に都合がよい親水性侵食性ポリマーシステム、たとえばポリ(エチレンオキシド)(Polyox)およびヒドロキシプロピル−メチルセルロース(HPMC)を使用することである。このシステムは、流体を吸収すると、胃内の保持を延長させるサイズまで短期間で膨張し、上部消化管において含有薬剤の吸収部位への持続性デリバリーを可能にする。これらシステムは、侵食性および親水性ポリマーまたはポリマー混合物からつくられるため、相当な期間をかけて容易に侵食され、胃から出ていく。膨張の期間は、これが食道で起こらないような期間であり、もしシステムが部分的に膨張した状態で腸に移動すると、水和ポリマーの侵食性および弾性が、デバイスによる腸閉塞の機会を除去する。
【0063】
一つの具体的な例において、本発明の組成物は、硬質または軟質ゼラチンカプセルに含有される複数のビーズを含む単一投与形態として調合される。カプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズまたは時間依存性放出ポリマーでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよび重炭酸ナトリウムエチルセルロースを含むビーズ、PG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースでコーティングされたビーズ。組成物中のセルロース系ポリマーにより、PGビーズの浮遊が許容され、ビーズからのPGの放出が遅延する。PG放出速度は、ヒドロキシプロピルメチルセルロースの厚みおよび侵食速度により決定される。
【0064】
別の具体的な例において、ゼラチンカプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズまたは時間依存性放出コーティングでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよびPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースでコーティングされたアルギン酸塩を含むビーズ。
【0065】
更に別の具体的な例において、ゼラチンカプセルは、以下から選択される混合集団のビーズを含有する:腸溶性コーティングPPIを含むビーズ、時間依存性放出ポリマーでコーティングされたPPIを含むビーズ、炭酸カルシウムを含むビーズおよびPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースを含むミニタブ(mini-tab)の形態の粒子。
【0066】
更に別の例において、本発明の組成物は、一つの層に腸溶性コーティングPPIを含み、第二の層にPG、炭酸カルシウムおよびヒドロキシプロピルメチルセルロースを含む、プレスコート錠剤または二層の錠剤として調合される。
【0067】
更に別の例において、本発明の組成物は、硬質ゼラチンカプセル内に二層の非水系半固体充填物として調合されてもよく、ここでは、PPIが、室温以上で液体であるが冷却すると半固体を形成する脂質ベース(非水系、迅速性放出)に可溶化されているため、硬質ゼラチンカプセルに充填することが可能である。脂溶性アルカリ性薬剤、たとえばアミンまたは重炭酸ナトリウムのファイン懸濁液を含めてもよい。
【0068】
本発明の単一投与形態の組成物は、好ましくは、腸溶性コーティングPPI粒子または時間依存性放出粒子の代わりに、非コーティングPPIを含む。非コーティングPPIの小腸上部における吸収は、コーティングPPIの吸収より速い。したがって、組成物に非コーティングPPIを使用することにより、PGの放出を遅延させる必要もなく、胃におけるPGの生物学的活性とPPIが活性である期間との間を、より正確に同調させることができる。このように種々の好ましい態様に従って、本発明の組成物は、PG、非コーティングPPIおよび一以上のアルカリ性薬剤を含む、二層の錠剤、プレスコート錠剤、発泡錠剤または懸濁錠剤として調合される。
【0069】
本発明の有効成分は、複数の経口投与形態で調合されてもよく、ここではPGおよび胃液中でPGの有効性を保存する一以上の薬剤が、別々の投与形態であるがPPIと結合させて投与される。たとえば、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、経口懸濁剤または固体投与形態、たとえばカプセル剤、錠剤、懸濁錠剤、または発泡錠剤で調合されてもよく、PPIは、別の固体投与形態、好ましくはカプセル剤または錠剤に含有される腸溶性コーティングビーズまたは時間依存性放出ビーズで調合されてもよい。
【0070】
複数の経口投与形態を用いる場合、PGおよび胃液中でPGの有効性を保存する一以上の薬剤は、PPIより前、PPIと同時、またはPPIの後に投与することができる。連続的投与では、PPIを投与したとき、またはPPIが活性になったときに、PGが生理的効果を発揮する限り、PGの投与とPPIの投与との間に実質的な遅れ(たとえば、分または数時間)があってもよい。好ましい態様において、投与されるPPIは、腸溶性コーティング形態または時間依存性放出形態である。この態様によれば、PGが胃で活性である間に、小腸の近位部で吸収されたPPIがH+/K+−ATPアーゼポンプを阻害するのに確実に有効であるようにするため、PPIの投与は、PGの投与より先に行われることが好ましい。
【0071】
本発明の有効成分は、不活性な薬学的に許容可能なビーズ内に組み込まれてもよい。この場合、薬剤は、ビーズにコーティングされる前に、更なる成分と混合されてもよい。成分には、結合剤、界面活性剤、充填剤、崩壊剤、アルカリ性添加剤または他の薬学的に許容可能な成分の単一物または混合物が含まれるがこれに限定されない。結合剤には、たとえば、セルロース、たとえばヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびカルボキシメチル−セルロースナトリウム、ポリビニルピロリドン、糖、デンプン、および凝集性を備えた他の薬学的に許容可能な物質が含まれる。適切な界面活性剤には、薬学的に許容可能な非イオン系またはイオン系界面活性剤が含まれる。適切な界面活性剤の例は、ラウリル硫酸ナトリウムである。
【0072】
粒子は、慣用的な技術により、パックされた経口摂取用の塊に形成されてもよい。たとえば、粒子は、公知のカプセル化の手法および材料を用いて、「硬質充填カプセル」としてカプセル化されてもよい。カプセル化材料は、カプセルが摂取された後に粒子が胃で迅速に分散するように、胃液中で可溶性が高くなければならない。
【0073】
別の態様において、本発明の有効成分は、圧縮錠剤にパッケージングされる。「圧縮錠剤」の用語は、一般に、一回の圧縮により、またはプレ圧密タッピングとその後の最終圧縮により調製される、経口摂取用の扁平な非コーティング錠剤をいう。かかる固体形態は、当該技術分野で周知のとおり製造することができる。錠剤形態は、たとえば、一以上のラクトース、マンニトール、コーンスターチ、ポテトスターチ、微結晶性セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、着色剤、希釈剤、緩衝化剤、湿潤剤、保存剤、香味剤、および薬学的に適合可能なキャリアを含むことができる。製造プロセスは、4つの確立された方法の一つまたはその組合せを採用することができる:(1)乾燥混合;(2)直接圧縮;(3)圧延(milling);および(4)非水系顆粒化。Lachman et al., The Theory and Practice of Industrial Pharmacy (1986)。かかる錠剤は、フィルムコーティングを備えていてもよく、これは、好ましくは経口摂取したときまたは希釈剤と接触したときに溶解する。
【0074】
かかる錠剤に使用することができるアルカリ性薬剤の非限定的な例には、重炭酸ナトリウム、アルカリ土類金属塩、たとえば炭酸カルシウム、水酸化カルシウム、乳酸カルシウム、グルセロリン酸カルシウム、酢酸カルシウム、炭酸マグネシウム、水酸化マグネシウム、ケイ酸マグネシウム、アルミン酸マグネシウム、水酸化アルミニウムまたは水酸化マグネシウムアルミニウムが含まれる。制酸錠剤を作成するのに有用な特別なアルカリ土類金属塩は、炭酸カルシウムである。
【0075】
別の代案において、本発明の組成物は、圧縮形態、たとえば懸濁錠剤および発泡錠剤で調合され、水または他の希釈剤と反応したときに、経口投与のための組成物の水性形態がつくられる。これらの形態は、錠剤の飲込みまたは咀嚼よりずっと受け入れられる用法で、子供や老人などに投薬するために特に有用である。本発明の薬学的錠剤または他の固体投与形態は、わずかな振盪または攪拌により、アルカリ性薬剤を崩壊させる。
【0076】
本明細書で使用される「懸濁錠剤」の用語は、水中に置かれた後急速に崩壊する圧縮錠剤をいい、容易に分散可能であり、正確な投与量のPPI、PGおよびアルカリ性薬剤を含有する懸濁液を形成する。一つの非限定的な例において、懸濁錠剤は、20-40 mgオメプラゾール、4 mg PG、およびアルカリ性薬剤として約1-4グラムの重炭酸ナトリウムまたはカルシウムを含み得る。錠剤の急速な崩壊を達成するために、クロスカルメロースナトリウムなどの崩壊剤を製剤に添加してもよい。崩壊剤は、単独で、または圧縮困難な錠剤の材料の圧縮性を高める能力に関して周知である微結晶性セルロースと組合せて、圧縮錠剤に混合され得る。また、微結晶性セルロースは、単独で、または他の成分と共にプロセスされた状態で、圧縮錠剤のための一般的な添加剤であり、圧縮困難な錠剤の材料の圧縮性を高める能力に関して周知である。これは、商標Avicelで商業的に入手可能である。
【0077】
懸濁錠剤組成物は、当業者に明らかなように、上述の成分に加えて、薬学的錠剤でしばしば用いられる他の成分、たとえば香味剤、甘味剤、フロー助剤、潤滑剤、または他の一般的な錠剤アジュバントを含有してもよい。他の崩壊剤、たとえばクロスピビドン(crospividone)およびグリコール酸デンプンナトリウム(sodium starch glycolate)を使用してもよいが、クロスカルメロースナトリウムが好ましい。
【0078】
上記成分に加えて、上述の経口投与形態は、薬学の分野で慣用的な適量の他の材料、たとえば希釈剤、潤滑剤、結合剤、顆粒化助剤、着色剤、香味剤および滑り剤(glidant)を含有してもよい。経口投与形態を調合するために使用され得る薬学的に許容可能なキャリアおよび賦形剤の具体的な例は、Handbook of Pharmaceutical Excipients, American Pharmaceutical Association (1986)に記載され、これは参照により本明細書に組み込まれる。
【0079】
以下の実施例は、本発明のある態様を更に充分に説明するために提示する。しかし、これらを本発明の広い範囲を限定するものと解釈すべきではない。当業者であれば、本発明の範囲から逸脱することなく、本明細書に開示される原理の多くの変更および改変を容易に考案することができる。
【実施例】
【0080】
例1:NaHCO3は人工胃液中でPGの安定性を保存する
NaHCO3の存在下で酸性pHにおけるPGの安定性を、人工胃液を用いてインビトロで試験した。人工胃液は、米国薬局方(USP) 2000 Ed., P.235に従って調製した。200 mlの胃液を調製するために、0.4 gのNaClおよび0.64 gのペプシンを、16 ml 1M HClおよび184 mlの水に溶解した。胃液のpHは1.2であった。10または20 mlの8.4%(1M)NaHCO3(それぞれ最終濃度3.72 mg/mlまたは7.12 mg/ml)および16 mlの250 ppm PG 溶液 (0.25 mg/ml)を、当該溶液に添加した。最終溶液におけるPGの濃度は16 ppmであった。表示された場合には、オメプラゾール顆粒を更に添加した(溶液BおよびC)。最終溶液中でのPGの経時的安定性を決定するために、調製後の以下の時点: 0'(調製直後)、5'、10'、20'、40'、60'で採取したサンプルについてHPLC分析を行った。反応を停止させるために、NH4OHを用いてpHを7.5−8.5に調整した。
【0081】
図1に示されるとおり、3.72 mg/mlのNaHCO3(pH 1.2)の存在下にPGを含む溶液AおよびBでは、PGの迅速な分解が観察された。しかし、7.12 mg/mlのNaHCO3(pH 5.7)を含む溶液Cでは、PGは1h安定なままであった。これらの結果から、pHを5.0より大きくするのに充分な濃度でNaHCO3などのアルカリ性薬剤を添加すると、ペプシンによるPGの分解が妨害されることが分かる。図2は、少なくとも80%のPGがpH4.8において少なくとも15分間分解されないまま残ることを更に示す。
【0082】
A.製剤の説明−非腸溶性コーティングオメプラゾールを含有する錠剤
例2:PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含むプレスコート錠剤または二層の錠剤
プレスコート錠剤または二層の錠剤を、単一の投与形態として調合し、各錠剤は、以下の成分を含有する:
オメプラゾール (パウダー) 40 mg
PG 4 mg
NaHCO3 500 mg
CaCO3 500 mg
クロスカルメロースナトリウム
ヒドロキシプロピルメチルセルロース (HPMC)
微結晶性セルロース (Avicel)
ステアリン酸マグネシウム
デンプン。
【0083】
プレスコート錠剤または二層の錠剤は、2段階のプロセスで調製される。単一の錠剤のために、4 mg PG、250 mg 炭酸カルシウムおよび微結晶性セルロースを混合して、これを錠剤の第一の層にプレ圧縮する。PGを含有するこの層を、錠剤からのPGの放出を10−15分遅延させるHPMCの薄層で更にコーティングする。第二の層については、40 mgの非腸溶性コーティングオメプラゾールのパウダーを、500 mg NaHC03、250 mg CaCO3および適切な結合剤とともにPG層の上に圧縮し、錠剤の第二の層を形成する。錠剤の第二の層は、消化直後に崩壊し、オメプラゾールはすぐに放出される。PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含む二層の錠剤の概略図を図3に示す。
【0084】
例3:PG、非腸溶性コーティングオメプラゾール、重炭酸ナトリウムおよび炭酸カルシウムを含む迅速性崩壊錠剤
迅速性崩壊錠剤を、以下の成分を含有する単一の投与形態として調合する:
オメプラゾール (パウダー) 40 mg
PG 4 mg
NaHCO3 500 mg
CaCO3 500 mg
クロスカルメロースナトリウム
微結晶性セルロース
ステアリン酸マグネシウム
デンプン。
【0085】
非腸溶性コーティングオメプラゾール(40 mg)、PG(4 mg)、NaHCO3、CaCO3、クロスカルメロースナトリウム、微結晶性セルロースおよびステアリン酸マグネシウムを混合し、得られた混合物を、標準的な錠剤プレッシングを用いて錠剤に圧縮し、迅速性崩壊錠剤(intravescent)を得る。
【0086】
例4:PG、腸溶性コーティングオメプラゾール、および重炭酸ナトリウムを含む発泡サック
発泡錠剤を、以下の成分を含有する単一の投与形態として調合する:
オメプラゾール 40 mg
PG 4 mg
NaHCO3 958 mg
クエン酸 832 mg
炭酸カリウム 312 mg
ステアリン酸マグネシウム
デンプン。
【0087】
腸溶性コーティングオメプラゾール(40 mg)およびPG(4 mg)を乳鉢に置き、乳棒で微粉に粉砕する。重炭酸ナトリウム、クエン酸、炭酸カリウムおよび他の全ての賦形剤を混合物に添加し、発泡性粉末の均質な混合物を形成する。得られた粉末を、40 mg腸溶性コーティングオメプラゾールと混合し、単位用量のパケットにパックする。
【0088】
B.製剤の説明−コーティングオメプラゾールを含有するマルチ粒子カプセル剤
例5:エチルセルロース−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
この例は、マルチ粒子硬質ゼラチンカプセルの製造に関与するステップを説明する。硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
エチルセルロースコーティング重炭酸ナトリウムビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0089】
PGを炭酸アンモニウム緩衝液pH 8に溶解することによりPG溶液を調製する。PG溶液を、流動層式装置でエチルセルロースコーティング重炭酸ナトリウムビーズにスプレーする。乾燥後、PG-重炭酸ナトリウムビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0090】
ゼラチンカプセルが胃の胃液中に溶解すると、PG含有ビーズのHPMC層が膨張し、胃酸が重炭酸ナトリウムと反応し、ビーズのコア内部にCO2を生成する。エチルセルロースコーティング重炭酸ナトリウムコアからの二酸化炭素の放出が、粒子の浮揚性を許容し、これにより粒子からのPGおよび炭酸カルシウムの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。CaCO3は、長期間にわたって胃のpHを上昇させ、放出の際のPGを保護する。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0091】
例6:アルギン酸塩−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
アルギン酸塩粒子に充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0092】
アルギン酸塩粒子は、アルギン酸塩溶液を塩化カルシウム溶液に滴下し、その後凍結乾燥してアルギン酸塩粒子を得ることにより作成する。例5のとおり調製されたPG溶液を、流動層式装置でアルギン酸塩粒子にスプレーする。乾燥後、PG-アルギン酸塩ビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0093】
ゼラチンカプセルが胃に溶解すると、PGビーズは、HPMC層と胃液の接触により膨張する。凍結乾燥されたアルギン酸塩粒子が、その低密度により粒子の浮揚性を許容し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0094】
例7:スクロース−PGビーズ、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングビーズとしての40 mgオメプラゾール
不活性な糖ビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0095】
PG溶液を、流動層式装置で不活性な糖ペレット(Nu-Pareils, 25/30)にスプレーする。乾燥後、PG-糖ビーズを、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)で更にコーティングし、最終PG粒子を形成する。PG顆粒の概略図を図4に示す。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0096】
ゼラチンカプセルが胃に溶解すると、PGビーズは、PG含有ビーズのHPMC層と胃液の接触により膨張し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0097】
例8:HPMC−PGミニタブ(minitab)、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むカプセル剤
硬質ゼラチンカプセルを、混合集団の粒子を含む単一の投与形態として調合する。各カプセルは、以下の成分を含有する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
不活性な糖ビーズに充填した4 mg PG
600 mg炭酸カルシウム(CaCO3)
ヒドロキシプロピルメチルセルロース(HPMC)。
【0098】
PGを、HPMCおよびCaCO3と組合せて顆粒状にし、ミニタブに圧縮する。ミニタブは、胃の胃液と接触したときに迅速に膨張する能力を有していることにより、胃での保持を可能にする。PGの胃への放出は、膨張したミニタブのポリマーマトリクスの侵食速度によりコントロールされる。PGミニタブを、腸溶性コーティングオメプラゾールビーズと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。
【0099】
例9:非腸溶性時間依存性放出コーティングでコーティングされたオメプラゾールおよびPGビーズを含有するマルチ粒子カプセル剤
この例は、マルチ粒子硬質ゼラチンカプセルの製造に関与するステップを説明する。カプセルを、以下の混合集団の粒子を含む単一の投与形態として調合する:時間依存性放出コーティングでコーティングされたPGビーズ、時間依存性放出コーティングでコーティングされたオメプラゾールビーズ、および炭酸カルシウム。カプセルの概略図を図5に示す。各カプセルは、以下の成分を含有する:
・薄いHPMC層でコーティングされた40 mgオメプラゾールビーズ
・糖の球体上に充填され、薄いHPMC層でコーティングされた4 mg PG
・600 mg炭酸カルシウム(CaCO3)。
【0100】
コーティングの組成は、媒体がコアと接触するようになったときに、コアが水性環境に急速に崩壊するようにデザインされる。この目的のために、糖の球体は、制酸剤(NaHCO3またはCaCO3)層でコーティングされる。PGを炭酸アンモニウム緩衝液pH 8に溶解することによりPG溶液を調製する。PG溶液を、流動層式装置で上記制酸剤コーティングビーズにスプレーする。乾燥後、ビーズを、HPMCの薄層で更にコーティングし、約10分の遅延放出のPG粒子を作成する。オメプラゾールは、制酸剤コーティング糖球体上に層状に重ねられ、厚い時間放出性HPMCコーティングで被覆される。崩壊剤を粒子のコアに添加して、HPMCが溶解した後オメプラゾールがすぐに放出されるよう促進してもよい。コーティングオメプラゾールビーズは、胃を通過することを目的とし、HPMCが溶解された後、小腸の上部で吸収され、すぐにオメプラゾールが放出される。最終PG粒子を、オメプラゾールビーズおよび炭酸カルシウムパウダーと共に、カプセルにつき40 mgオメプラゾール、4 mg PGおよび600 mg炭酸カルシウムに相当する量で、サイズ0の硬質ゼラチンカプセルにパックする。PGおよびOMPの放出速度は、ビーズのHMPC層の厚みおよび侵食速度により決定される。CaCO3は、長期間にわたって胃のpHを上昇させ、放出の際のPGを保護する。
【0101】
C 製剤の説明−腸溶性コーティングオメプラゾールを含有する錠剤
例10:PG、腸溶性コーティングオメプラゾールビーズ、および炭酸カルシウムを含むプレスコーティング錠剤
プレスコーティング錠剤を、以下の成分を含有する単一の投与形態として調合する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
4 mg PG顆粒
炭酸カルシウム
ヒドロキプロピルメチルセルロース(HPMC)。
【0102】
プレスコーティング錠剤は、2段階の方法で調製される。単一の錠剤については、4 mg PG、900 mg炭酸カルシウムおよびHPMCを混合し、錠剤の中心コアにプレ圧縮する。40 mgの腸溶性コーティングオメプラゾールビーズを、PGコアにプレスコーティングし、錠剤の外層を形成する。最終錠剤は、制御された放出のPGコア層とオメプラゾール腸溶性コーティングビーズの即時放出性外層から構成される。別の例において、有効成分は、二層の錠剤に圧縮され、ここで第一の層は、4 mg PG、900 mg炭酸カルシウムおよびHPMCを含み、第二の層は、40 mgの腸溶性コーティングオメプラゾールビーズを含む。
【0103】
圧縮された錠剤は、以下の賦形剤の一以上を含んでいてもよい:ラクトース、マンニトール、コーンスターチ、ポテトスターチ、微結晶性セルロース、アカシア、ゼラチン、コロイド状二酸化ケイ素、クロスカルメロースナトリウム、タルク、ステアリン酸マグネシウム、ステアリン酸、および他の賦形剤、着色剤、希釈剤、緩衝化剤、湿潤剤、保存剤、香味剤、および薬学的に適合可能なキャリア。
【0104】
例11:PG、腸溶性コーティングオメプラゾールビーズおよび炭酸カルシウムを含む迅速崩壊性錠剤
迅速崩壊性懸濁錠剤を、以下の成分を含有する単一の投与形態として調合する:
腸溶性コーティングオメプラゾールビーズとしての40 mgオメプラゾール
4 mg PG顆粒
900 mg炭酸カルシウム
クロスカルメロースナトリウム
微結晶性セルロース
ステアリン酸マグネシウム
ヒドロキシプロピルメチルセルロース(HPMC)。
【0105】
PG顆粒を、CaCO3およびヒドロキシプロピルメチルセルロース(HPMC)でコーティングし、最終PG粒子を形成する。最終PG粒子を、腸溶性コーティングオメプラゾールビーズおよび上述の賦形剤と混合し、得られた混合物を標準的な錠剤プレッシングを用いて錠剤に圧縮する。得られた錠剤は、迅速な崩壊時間を有し、胃で迅速に崩壊するために水で飲み込んでもよい。
【0106】
懸濁錠剤の崩壊の際に、PG粒子は、PG含有ビーズのHPMC層と水性環境の接触により膨張し、これにより粒子からのPGの放出が遅延する。PG放出速度は、PGビーズのHMPC層の厚みおよび侵食速度により決定される。腸溶性コーティングオメプラゾールビーズは、胃を通過し、オメプラゾールは、遅延することなく小腸の上部に吸収される。
【0107】
D インビボ実験
例12:ラットにおけるPG経口投与後の胃酸分泌の刺激
PGおよびPPIの併用による胃酸分泌の阻害は、経口投与されたPGが胃内で局所的に酸分泌を誘発する能力に基く。この問題に取り組むために、麻酔をかけたラットに、増加する量のPGを(経口)投与し、胃酸分泌を幽門結紮の胃でモニターした。増加する量(10、30、および90μg/kg)のPGを、幽門結紮のラットに経口胃管栄養法により投与した。30分の処置後、胃液を胃管腔から回収し、酸濃度をNaOHで滴定することにより決定し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。結果を、各実験グループに由来する7−8の動物の平均±SEMで表現する。図6に示されるとおり、経口投与されたPGは、用量依存的に有意に胃酸分泌を増大させ、このことは、経口投与されたPGが局所的に胃酸分泌を誘導することを示す。
【0108】
例13:オメプラゾールとともに投与されたPGの胃内pHに対する効果
胃酸分泌の抑制に対するPG-PPIの併用効果を試験するために、麻酔をかけたラットに、オメプラゾール(10 mg/kg)を単独でまたはPG(350μg/kg)と併用して胃内に注入した。併用処置したラットは、オメプラゾールの15分前にPGを受けとった。処置から30、45、および60分後に吸引により胃液を回収し、胃酸分泌に対する薬剤の効果を、pHをモニターすることにより検出した。そのデータは、全ての時点において胃内pH値が、オメプラゾール単独で処置したラットよりPGとオメプラゾールを併用して処置したラットにおいて著しく高いことを示した(図7)。これら結果は、PGがラットにおいてPPIの抗分泌活性を増大させることを示す。
【0109】
例14:ランソプラゾール(Lansoprazole)は理性的(conscious)動物で用量依存的に胃酸分泌を阻害する
この実験では、様々なモデルの幽門結紮のラットを使用し、これは、理性的動物において胃酸分泌に対する薬剤効果の分析を許容する。このモデルは、胃酸分泌に対する麻酔の効果を排除する。研究薬剤を単独でまたは併用して経口投与した。1または2時間後、麻酔ガス機を用いて、幽門結紮を行い腹部を閉じるのに充分な短い時間(5分)、動物に麻酔をかけた。その後、回復のために動物をかごに戻した。数時間後、動物を屠殺し、食道のまわりに結紮糸を置き、胃を取り出し、胃内容物を回収した。遠心分離後、胃液サンプルを0.01 N NaOHを用いて終点pH 7に自動滴定し、滴定酸分泌量を計算した。
【0110】
ランソプラゾールを、単純懸濁液(simplified suspension;SLS)として経口胃管栄養法により投与した。SLSは以下のとおり調製した:30 mgカプセル(Zoton)の内容物を8.4%重炭酸ナトリウムに懸濁した。幽門結紮の2時間前に、3つの用量のランソプラゾール(20、5および1.25 mg/kg)でラットを処置した。プラセボとしてコントロールグループに8.4% NaHCO3を投与した。図8は、ランソプラゾールが用量依存的に胃酸分泌を阻害することを示す。
【0111】
例15:理性的な幽門結紮ラットの胃酸分泌に対する、PGと併用して投与したランソプラゾール(Lansoprazole)の効果
この実験では、PG(300μg/kg)の15分前(A)または15分後(B)に、ラットを用量5 mg/kgのSLSで処置した。コントロールのラットは、プラセボとして8.4% NaHCO3およびPG-賦形剤を併用して注入した。すべての薬剤は、幽門結紮の2時間前に、経口胃管栄養法により投与した。胃液は3時間かけて回収した。データは平均±SEMで表す。各実験グループの動物数は8−9である。
【0112】
図9Aから分かるとおり、PGの15分前にSLSを投与すると、ランソプラゾール単独の場合と比較して、酸阻害の程度が高いが、PGで前処置した後SLSで処置したラットの酸分泌量は、ランソプラゾール単独で処置したラットと変わらなかった(図9B)。これらの結果は、PGが、胃酸分泌の遮断におけるランソプラゾールの効果を高めることを示す。更に、二つの化合物のタイミングは、PG/ランソプラゾールの併用処置の有効性を高めるために重要である。
【0113】
別の実験では、ラットを、用量2.5 mg/kgのSLSおよび賦形剤、あるいはSLSおよびPG(300μg/kg)で3日連続して1日に1回処置した。SLSは、PGまたは賦形剤の15分前に投与した。コントロールのラットは、プラセボとして8.4% NaHCO3およびPG-賦形剤を併用して注入した。すべての薬剤は、経口胃管栄養法により投与した。幽門結紮は、3日目に、処置の2時間後に実施した。胃液は3時間かけて回収した。データは平均±SEMで表す。各実験グループの動物数は8である。図10Aおよび10Bに示すとおり、SLSをPGと併用して3日間連続して投与すると、SLS単独の場合と比較して、胃内pHが有意に高くなった。同様に、SLS/PGを併用して3日間連続で処置したラットの胃酸分泌は、SLSを単独で投与したラットの胃酸分泌より少なかった。
【0114】
例16:ラットのPG介在性胃酸分泌に対するCCK-Bアンタゴニストの効果
PGはガストリンホルモン類似体であるため、その局所的効果は、ガストリン経路を介して、すなわちガストリンレセプター(CCKB)の活性化を介して介在されると考えられる。この仮説を試験するために、PG介在性酸分泌に対する特定のCCKBアンタゴニスト(Itriglumide)の効果を調査した。
【0115】
この研究では、ラットにKetamineおよびDomitor混合物で麻酔をかけ、20 mg/kgのItriglumideを十二指腸内に(i.d.)投与した。15分後、胃の幽門を結紮し、300μg/kg PGを胃内に(i.g.)投与した。30分後、胃液を得て、遠心分離し、上清の体積とpHを測定した。酸濃度(滴定酸性度)を、胃液サンプルをNaOHで滴定することにより分析し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。下記表1に示される結果から明らかなように、CCKBアンタゴニスト(ant.)の十二指腸内注入により、ラットの胃酸分泌に対するPGの局所的効果は阻害される。
【表1】
【0116】
例17:麻酔され幽門結紮されたラットの酸分泌に対するPGの十二指腸内注入の効果
麻酔され幽門結紮されたラットの酸分泌に対するPGの十二指腸内注入の効果を調査した。この研究では、300μg/kg PGを、麻酔され幽門結紮されたラットの十二指腸内に投与し、胃酸分泌レベルを30分後に測定した。胃液を得て、遠心分離し、上清の体積とpHを測定した。酸濃度(滴定酸性度)を、胃液サンプルをNaOHで滴定することにより分析し、μEq HClで表されるトータル酸分泌量を、サンプル体積と酸濃度を掛け合わせることにより計算した。コントロールとして、等量のPGを胃内に注入し、胃液分泌に対するPGの効果を測定した。表2に示されるとおり、PGの胃内注入および十二指腸内注入のいずれも、麻酔され幽門結紮されたラットにおいて胃酸分泌を誘導する。
【表2】
【0117】
本発明が、特別に示されたものおよび上述のものに限定されないことは当業者に認識されるでしょう。むしろ、本発明の範囲は特許請求の範囲により規定される。
【図面の簡単な説明】
【0118】
【図1】図1は、NaHCO3が人工胃液中でPGの安定性を保存することを示す。
【図2】図2は、種々のpH値における非分解性PGのパーセンテージを示す。
【図3】図3は、PG、非腸溶性コーティングオメプラゾールおよび緩衝化剤を含む二層の錠剤の概略図である。
【図4】図4は、マルチ粒子カプセル調合物で使用されるPG顆粒の概略図である。
【図5】図5は、時間放出性コーティングビーズを含むカプセル剤の概略図である。
【図6】図6は、PGが、ラットにおいて用量依存的に胃酸分泌を刺激することを示す。
【図7】図7は、PGが、ラットにおいて胃酸分泌に対するPPI介在性効果を増大させることを示す。
【図8】図8は、ランソプラゾール(Lansoprazole)が、理性的動物において用量依存的に胃酸分泌を阻害することを示す。
【図9】図9は、ランソプラゾール(Lansoprazole)をPGの前に投与した場合(A)、PGが、ランソプラゾールの胃酸分泌の遮断効果を増大し、ランソプラゾールをPGの後に投与した場合(B)、PGが、ランソプラゾールの胃酸分泌の遮断効果を増大しないことを示す。
【図10】図10は、ランソプラゾール(Lansoprazole)をPGと併用して3日連続投与すると、ランソプラゾール単独と比較して、胃内pHが有意に高くなり(A)、胃酸分泌が有意に低くなる(B)ことを示す。
【特許請求の範囲】
【請求項1】
薬学的に効果的な量の(i)壁細胞を活性化する、配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液中の前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む経口薬学的組成物。
【請求項2】
前記ペプチドが、配列番号2のアミノ酸配列を有するペンタガストリン(PG)またはその合成類似体である、請求項1に記載の経口組成物。
【請求項3】
前記保存薬剤が、一以上のアルカリ性薬剤であり、前記アルカリ性薬剤の量が、PGの生物学的活性を維持するように、胃においてPGの有効性を保存するのに充分である、請求項2に記載の経口組成物。
【請求項4】
前記アルカリ性薬剤が、炭酸カルシウム、重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、および重炭酸カルシウムから成る群より選択される、請求項3に記載の経口組成物。
【請求項5】
前記経口組成物が単一の単位投与形態で調合され、前記アルカリ性薬剤が少なくとも300 mgの量である、請求項3に記載の経口組成物。
【請求項6】
前記ペプチドが、胃管腔に位置する壁細胞を局所的に活性化するのに充分な量である、請求項2に記載の経口組成物。
【請求項7】
前記有効成分が、単一の単位投与形態で調合される、請求項2に記載の経口組成物。
【請求項8】
前記PGの量が2〜60 mgである、請求項7に記載の経口組成物、
【請求項9】
前記単一の単位投与形態が、PPIビーズ、PGビーズおよび少なくとも一のアルカリ性薬剤を含む、二層の錠剤、プレスコート錠剤、マルチ粒子カプセル剤、発泡錠剤、懸濁錠剤、液剤、または懸濁剤である、請求項7に記載の経口組成物。
【請求項10】
前記アルカリ性薬剤の量が、PGの生物学的活性を維持するように、胃においてPGの有効性を保存するのに充分である、請求項9に記載の経口組成物。
【請求項11】
前記PPIビーズおよび前記PGビーズが、腸溶性コーティングまたは時間依存性放出ポリマーでコーティングされ、前記PPIビーズからのPPIの放出が、前記PGビーズからのPGの放出に比べて遅延する、請求項10に記載の経口組成物。
【請求項12】
前記時間依存性放出ポリマーが、水性環境において膨張可能な少なくとも一のポリマーを含む、請求項11に記載の経口組成物。
【請求項13】
少なくとも一のポリマーが、合成ポリマーおよびセルロース系ポリマーから成る群より選択されるか、またはその置換誘導体である、請求項12に記載の経口組成物。
【請求項14】
前記PGビーズが、PGビーズ内にトラップされた二酸化炭素を形成するために胃酸と反応可能な少なくとも一の炭酸塩を更に含み、これにより胃液に対して前記PGビーズの浮力を誘導する、請求項11に記載の経口組成物。
【請求項15】
前記炭酸塩が、重炭酸ナトリウムまたは炭酸カルシウムである、請求項14に記載に記載の経口組成物。
【請求項16】
非コーティングPPIビーズ、PGビーズおよび少なくとも一のアルカリ性薬剤を含み、前記PGビーズからのPGの放出が、前記PPIビーズからのPPIの放出に比べて遅延する、請求項10に記載の経口組成物。
【請求項17】
前記一以上のアルカリ性薬剤が、炭酸カルシウム、重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、および重炭酸カルシウムから成る群より選択される、請求項16に記載の経口組成物。
【請求項18】
前記組成物が、非コーティングPPIビーズ、PGビーズおよび一以上のアルカリ性薬剤を含み、プレスコート錠剤、二層の錠剤、マルチ粒子カプセル剤、発泡錠剤、懸濁錠剤、液剤、または懸濁剤の形態で調合される、請求項16に記載の経口組成物。
【請求項19】
前記PPIが、ラベプラゾール、オメプラゾール、イソメプラゾール、ランソプラゾール(lansoprazole)、パントプラゾール、レミノプラゾール、その単一の鏡像異性体、そのアルカリ塩、およびその混合物から成る群より選択される、請求項1に記載の経口組成物。
【請求項20】
前記組成物が、ペプシンインヒビター、粘液溶解剤、または胃に存在する細菌に対して効果的な抗生物質を更に含む、請求項1に記載の経口組成物。
【請求項21】
前記ペプチドが、メトキシメチル(MOM)、β−メトキシエトキシメチル(MEM)、トリアルキルシリル、トリフェニルメチル(trityl)、TIPSO、tert−ブトキシカルボニル(t-BOC)、エトキシエチル(EE)、F-MOC、およびTROCから成る群より選択されるPGのN−保護誘導体である、請求項2に記載の経口組成物。
【請求項22】
薬学的に効果的な量の(i)配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液における前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む経口薬学的キット。
【請求項23】
前記有効成分が、別々の単位投与形態で調合される、請求項22に記載のキット。
【請求項24】
胃酸分泌の抑制が必要な被検体の障害を治療または予防する方法であって、かかる治療の必要な被検体に、治療上効果的な量の請求項1に記載の組成物を投与することを含む方法。
【請求項25】
前記障害が、逆流性食道炎、胃炎、十二指腸炎、胃潰瘍、十二指腸潰瘍、非ステロイド性抗炎症薬(NSAID)と関連した病状、非潰瘍性消化不良、胃食道逆流疾患、ガストリン産生腫瘍、急性上部消化管出血、ストレス潰瘍、ヘリコバクターピロリ感染症、ゾリンジャー−エリソン症候群(ZES)、ウェルナー症候群、および全身性肥満細胞症から成るより選択される、請求項24に記載の方法。
【請求項26】
前記被検体がヒト被検体である、請求項24に記載の方法。
【請求項27】
胃酸分泌の抑制が必要な被検体の障害を治療または予防する方法であって、かかる治療の必要な被検体に、治療上効果的な量の請求項22に記載の組成物を投与することを含む方法。
【請求項28】
前記ペプチドが、PPIの投与と同時、投与前、または投与後に投与される、請求項27に記載の方法。
【請求項1】
薬学的に効果的な量の(i)壁細胞を活性化する、配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液中の前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む経口薬学的組成物。
【請求項2】
前記ペプチドが、配列番号2のアミノ酸配列を有するペンタガストリン(PG)またはその合成類似体である、請求項1に記載の経口組成物。
【請求項3】
前記保存薬剤が、一以上のアルカリ性薬剤であり、前記アルカリ性薬剤の量が、PGの生物学的活性を維持するように、胃においてPGの有効性を保存するのに充分である、請求項2に記載の経口組成物。
【請求項4】
前記アルカリ性薬剤が、炭酸カルシウム、重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、および重炭酸カルシウムから成る群より選択される、請求項3に記載の経口組成物。
【請求項5】
前記経口組成物が単一の単位投与形態で調合され、前記アルカリ性薬剤が少なくとも300 mgの量である、請求項3に記載の経口組成物。
【請求項6】
前記ペプチドが、胃管腔に位置する壁細胞を局所的に活性化するのに充分な量である、請求項2に記載の経口組成物。
【請求項7】
前記有効成分が、単一の単位投与形態で調合される、請求項2に記載の経口組成物。
【請求項8】
前記PGの量が2〜60 mgである、請求項7に記載の経口組成物、
【請求項9】
前記単一の単位投与形態が、PPIビーズ、PGビーズおよび少なくとも一のアルカリ性薬剤を含む、二層の錠剤、プレスコート錠剤、マルチ粒子カプセル剤、発泡錠剤、懸濁錠剤、液剤、または懸濁剤である、請求項7に記載の経口組成物。
【請求項10】
前記アルカリ性薬剤の量が、PGの生物学的活性を維持するように、胃においてPGの有効性を保存するのに充分である、請求項9に記載の経口組成物。
【請求項11】
前記PPIビーズおよび前記PGビーズが、腸溶性コーティングまたは時間依存性放出ポリマーでコーティングされ、前記PPIビーズからのPPIの放出が、前記PGビーズからのPGの放出に比べて遅延する、請求項10に記載の経口組成物。
【請求項12】
前記時間依存性放出ポリマーが、水性環境において膨張可能な少なくとも一のポリマーを含む、請求項11に記載の経口組成物。
【請求項13】
少なくとも一のポリマーが、合成ポリマーおよびセルロース系ポリマーから成る群より選択されるか、またはその置換誘導体である、請求項12に記載の経口組成物。
【請求項14】
前記PGビーズが、PGビーズ内にトラップされた二酸化炭素を形成するために胃酸と反応可能な少なくとも一の炭酸塩を更に含み、これにより胃液に対して前記PGビーズの浮力を誘導する、請求項11に記載の経口組成物。
【請求項15】
前記炭酸塩が、重炭酸ナトリウムまたは炭酸カルシウムである、請求項14に記載に記載の経口組成物。
【請求項16】
非コーティングPPIビーズ、PGビーズおよび少なくとも一のアルカリ性薬剤を含み、前記PGビーズからのPGの放出が、前記PPIビーズからのPPIの放出に比べて遅延する、請求項10に記載の経口組成物。
【請求項17】
前記一以上のアルカリ性薬剤が、炭酸カルシウム、重炭酸ナトリウムまたはカリウム、酸化、水酸化または炭酸マグネシウム、乳酸マグネシウム、グルコン酸マグネシウム、水酸化アルミニウム、炭酸、リン酸またはクエン酸のアルミニウム、カルシウム、ナトリウムまたはカリウムとの塩、炭酸二ナトリウム、リン酸水素二ナトリウム、グリシン酸アルミニウムとバッファーとの混合物、水酸化カルシウム、乳酸カルシウム、炭酸カルシウム、および重炭酸カルシウムから成る群より選択される、請求項16に記載の経口組成物。
【請求項18】
前記組成物が、非コーティングPPIビーズ、PGビーズおよび一以上のアルカリ性薬剤を含み、プレスコート錠剤、二層の錠剤、マルチ粒子カプセル剤、発泡錠剤、懸濁錠剤、液剤、または懸濁剤の形態で調合される、請求項16に記載の経口組成物。
【請求項19】
前記PPIが、ラベプラゾール、オメプラゾール、イソメプラゾール、ランソプラゾール(lansoprazole)、パントプラゾール、レミノプラゾール、その単一の鏡像異性体、そのアルカリ塩、およびその混合物から成る群より選択される、請求項1に記載の経口組成物。
【請求項20】
前記組成物が、ペプシンインヒビター、粘液溶解剤、または胃に存在する細菌に対して効果的な抗生物質を更に含む、請求項1に記載の経口組成物。
【請求項21】
前記ペプチドが、メトキシメチル(MOM)、β−メトキシエトキシメチル(MEM)、トリアルキルシリル、トリフェニルメチル(trityl)、TIPSO、tert−ブトキシカルボニル(t-BOC)、エトキシエチル(EE)、F-MOC、およびTROCから成る群より選択されるPGのN−保護誘導体である、請求項2に記載の経口組成物。
【請求項22】
薬学的に効果的な量の(i)配列番号1のアミノ酸配列を含むペプチド;(ii)不可逆性の胃H+/K+-ATPアーゼプロトンポンプインヒビター(PPI);および(iii)胃液における前記ペプチドの有効性を保存する少なくとも一の薬剤を、有効成分として含む経口薬学的キット。
【請求項23】
前記有効成分が、別々の単位投与形態で調合される、請求項22に記載のキット。
【請求項24】
胃酸分泌の抑制が必要な被検体の障害を治療または予防する方法であって、かかる治療の必要な被検体に、治療上効果的な量の請求項1に記載の組成物を投与することを含む方法。
【請求項25】
前記障害が、逆流性食道炎、胃炎、十二指腸炎、胃潰瘍、十二指腸潰瘍、非ステロイド性抗炎症薬(NSAID)と関連した病状、非潰瘍性消化不良、胃食道逆流疾患、ガストリン産生腫瘍、急性上部消化管出血、ストレス潰瘍、ヘリコバクターピロリ感染症、ゾリンジャー−エリソン症候群(ZES)、ウェルナー症候群、および全身性肥満細胞症から成るより選択される、請求項24に記載の方法。
【請求項26】
前記被検体がヒト被検体である、請求項24に記載の方法。
【請求項27】
胃酸分泌の抑制が必要な被検体の障害を治療または予防する方法であって、かかる治療の必要な被検体に、治療上効果的な量の請求項22に記載の組成物を投与することを含む方法。
【請求項28】
前記ペプチドが、PPIの投与と同時、投与前、または投与後に投与される、請求項27に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】

【図9】

【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2007−503427(P2007−503427A)
【公表日】平成19年2月22日(2007.2.22)
【国際特許分類】
【出願番号】特願2006−524456(P2006−524456)
【出願日】平成16年8月25日(2004.8.25)
【国際出願番号】PCT/IB2004/002745
【国際公開番号】WO2005/020879
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(506068070)ベクタ・リミテッド (6)
【Fターム(参考)】
【公表日】平成19年2月22日(2007.2.22)
【国際特許分類】
【出願日】平成16年8月25日(2004.8.25)
【国際出願番号】PCT/IB2004/002745
【国際公開番号】WO2005/020879
【国際公開日】平成17年3月10日(2005.3.10)
【出願人】(506068070)ベクタ・リミテッド (6)
【Fターム(参考)】
[ Back to top ]