
脂肪細胞へのsiRNAの導入方法
一般に、核酸配列を脂肪細胞に導入するのは困難だった。Akt1、Akt2およびMyo1cを標的とする配列の導入を含む、siRNAなどの核酸を脂肪細胞に導入する方法を記述する。グルコース輸送に関与する遺伝子の同定およびインスリン応答モジュレータの同定の方法を記述する。治療方法も提供する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
政府の権利
本願発明は、部分的にNIHが交付する政府助成金第DK30648号によって行われた。政府は本願発明の権利を有することができる。
【0002】
関連出願
本願明細書は、2002年12月11日に提出された米国特許仮出願第60/432,427号、表題「Method of Introducing siRNA
into Adipocytes(脂肪細胞へのsiRNAの導入方法)」の利益を主張する。上に引用された特許出願の内容全体は、引用をもって本願明細書に援用するものとする。
【0003】
背景
培養された脂肪細胞は、グルコース輸送へのインスリンの作用を研究するための主要なモデルシステムである。他に、グルコース輸送体GLUT4をこれだけ高濃度に発現し、グルコース輸送を増やすことによってこれだけ強くインスリンに応答する培養細胞種はない。インスリンのGLUT4へのこの作用は、2型糖尿病の中核を成す。なぜなら、脂肪および筋肉中のインスリン耐性がこの疾患を引き起こす主要な異常だからである。インスリンシグナル伝達経路の構成要素が発見されたことは、インスリン耐性を理解し、2型糖尿病の治療法のための薬物標的候補を提供する上で重要である。
【0004】
そのようなインスリンシグナル伝達経路の構成要素の発見は、この経路への関与が疑われるタンパク質をコードする特定の遺伝子の発現を削除することによって、促進される。脂肪細胞中の遺伝子またはタンパク質発現の特異的な削除を容易に行うことができる方法はない。これらの細胞は扱いが難しく、線維芽細胞などその他の細胞で作用する試薬では容易にトランスフェクトできない。
【0005】
グルコースの恒常性は、インスリンによる筋肉および脂肪細胞へのグルコース輸送の調節によって、一部制御されている。PI 3-キナーゼ下流のAktタンパク質キナーゼ(タンパク質キナーゼB (PKB)としても知られる)は、このインスリンシグナル伝達経路に関与していると考えられているが、Akt1-/- およびAkt2-/- マウスで得られた結果ははっきりしない。Aktは、インスリンシグナル伝達と、GLUT4輸送を調節する構成要素との、PI3キナーゼ依存性経路を介した連結に強く関与する下流エフェクタの1つである。このAktへの役割は、培養細胞での研究と遺伝子ノックアウトマウスモデルでの研究に基づいている(Kohn et al., 1996, J. Biol. Chem. 49:31372、Hill et al., 1999, Mol. Cell. Biol. 19:7771、Wang et al., 1999, Mol. Cell. Biol. 19:4008、Ueki et al., 1998, J. Biol. Chem. 273:5315、Cho et al., 2001 (a), Science 292:1728)。しかし、この重要な問題は見解が一致しておらず(Kotani et al.,
1998, Mol. Cell. Biol. 18:6971)、 Akt1イソ型が欠如しているマウスから得られた最近のデータでは、インスリンの感受性に低下は認められなかった(Cho et al.,
2001 (b), J. Biol. Chem. 276:38349)。筋肉と脂肪中に発現する主要なイソ型であるAkt2を欠損するマウスから採取した骨格筋は、低レベルのインスリンに対する感受性がやや低下しただけで、実際には最大量のインスリンには普通に応答する。これらのマウスに認められた糖尿病は、肝グルコース新生へのインスリン作用の劇的な減衰によるところが大きいと考えられている(Cho, 2001 (a),
supra)。
【0006】
要約
本願発明は、siRNAを脂肪細胞に導入する方法の発見に、部分的に基づく。前記方法は、Akt1、Akt2、CISK およびMyo1cを含む、いくつかの異なる配列を標的とするsiRNAを用いて実証される。
【0007】
したがって、本願発明には核酸を脂肪細胞に導入する方法が含まれる。前記方法には、脂肪細胞を得るステップと、前記脂肪細胞を核酸分子に接触させるステップと、混合物を形成するステップと、前記混合物を電気穿孔するステップと、が含まれる。前記核酸は、たとえばAkt1、Akt2、CISKまたはMyo1c核酸配列を標的とするsiRNAなどのsiRNAであってよい。 一般に、前記電気穿孔は、室温で、約0.18 kVおよび約960 μFの静電容量で行う。ある実施態様では、本願発明にはグルコース輸送に影響する遺伝子を同定する方法が含まれる。前記方法には、脂肪細胞を提供するステップと、上述の通り電気穿孔を用いて前記脂肪細胞に前記遺伝子を標的とするsiRNAを導入するステップと、前記標的遺伝子の発現に好適な条件下において前記細胞を培養するステップと、グルコース輸送の減少が前記標的遺伝子がグルコース輸送を示唆するように、前記細胞中のグルコース輸送をアッセイするステップが含まれる。
【0008】
別の実施態様では、本願発明には細胞におけるAkt1またはAkt2の発現もしくは活性を阻害する方法が含まれる。前記方法には、Akt1またはAkt2を発現する細胞を得るステップと、前記細胞にAkt1核酸配列またはAkt2核酸配列を標的とするsiRNAを導入するステップが含まれる(たとえば図3Aに記載の通り)。ある実施態様では、前記細胞は脂肪細胞である。ある局面では、Akt1およびAkt2の両方の発現または活性を前記方法を用いて阻害する。この局面では、Akt1を標的とするsiRNAおよびAkt2を標的とするsiRNAを前記細胞に導入することもできる。代替的には、Akt1およびAkt2の両方を標的とするsiRNAを前記細胞に導入することもできる。
【0009】
前記発明には、インスリン媒介GSK3リン酸化を阻害する方法も含まれる。前記方法には、インスリン選択性細胞を得るステップと、前記細胞をAkt2発現または活性を阻害する作用物質と接触させるステップとが含まれる。ある局面では、前記作用物質はAkt2を標的とするsiRNAである。
【0010】
別の実施態様では、本願発明はヘキソース輸送を阻害する方法に関連する。前記方法には、ヘキソース輸送を行うことができる細胞を得るステップと、前記細胞をAkt1、Akt2またはその両方の発現または活性を特異的に阻害する作用物質と接触させるステップとが含まれる。ある実施態様では、Akt1またはAkt2の発現もしくは活性、およびヘキソース輸送を特異的に阻害する作用物質を部分的に阻害する。前記作用物質は、Akt1、Akt2またはその両方を特異的に標的とするsiRNAであってよい。
【0011】
ある局面では、本願発明はグルコース輸送に影響する遺伝子を同定する方法であって、(a) 脂肪細胞を提供するステップと、本願明細書に提供される方法を用いて前記脂肪細胞に前記遺伝子を標的とするsiRNAを導入するステップと、(b) 前記標的遺伝子の発現に好適な条件下において前記細胞を培養するステップと、(c)前記細胞中のグルコース輸送をアッセイするステップであって、当該ステップにおいてグルコース輸送の減少が前記標的遺伝子がグルコース輸送を示唆し、それによりグルコース輸送に影響する遺伝子を同定するためのステップと、が含まれる方法を提供する。
本願発明はまた、インスリン応答モジュレータを同定する方法であって、(a) グルコース輸送に影響するタンパク質を発現する細胞をテスト化合物と接触させるステップであって、当該ステップにおいて前記タンパク質が本願明細書に提供される方法に従って同定された遺伝子によってコードされるステップと、(b)インスリン応答モジュレータが同定されるように、前記タンパク質の活性を調節するテスト化合物の能力を決定するステップと、が含まれる方法も提供する。
【0012】
関連する局面では、本願発明はインスリン応答モジュレータを同定する方法であって、(a) グルコース輸送に影響するタンパク質を発現する細胞をテスト化合物と接触させるステップであって、当該ステップにおいて前記タンパク質が本願明細書に提供される方法に従って同定された遺伝子によってコードされるステップと、(b)インスリン応答モジュレータが同定されるように、前記タンパク質の発現を調節するテスト化合物の能力を決定するステップと、が含まれる方法も提供する。
【0013】
さまざまな実施態様において、前記モジュレータは陽性モジュレータまたは陰性モジュレータである。本願発明はさらに、本願発明の方法によって同定されるモジュレータを特徴とする。
【0014】
別の局面では、本願発明は、本願発明のインスリン応答モジュレータを対象に投与するステップを含む、前記対象におけるグルコースの恒常性を調節する方法を提供する。別の局面では、本願発明は、本願発明のインスリン応答モジュレータを対象に投与するステップを含む、前記対象における血中グルコース値を調節する方法を提供する。
【0015】
さらに、本願明細書において同定されたモジュレータを含む薬学的組成物を特徴とする。
【0016】
別の局面では、本願発明は、本願発明の薬学的組成物を投与するステップを含む、インスリン応答疾患または障害を治療する方法を提供する。さまざまな実施態様では、前記疾患または障害は2型糖尿病、インスリン耐性、および肥満からなるグループから選択される。
【0017】
標的遺伝子は、siRNA(標的化siRNA)またはsiRNA誘導体によって、RNAi媒介遺伝子ノックダウンを標的とした遺伝子である。siRNAのある部分は、前記標的遺伝子のmRNAの部分に完全に相補的である。
【0018】
対照細胞または対照培養物は、修飾siRNAまたはsiRNA誘導体と接触したことがない細胞または培養物である。前記対照細胞または培養物は、一般に、RNAi媒介ノックダウンを目的とする1つ以上の内因性遺伝子を発現する1つ以上のレポータ遺伝子を含む。本願発明のある実施態様では、前記対照細胞または対照培養物は、対象のレポータ遺伝子または内因性遺伝子を標的としたsiRANを含む。ある場合、前記対照細胞または対照培養物は、siRNAまたは修飾siRNAのアンチセンス鎖に相当するアンチセンス鎖、またはsiRNAまたは修飾siRNAのスクランブル配列を含むアンチセンス鎖などの、導入された対照配列を含む。
【0019】
テスト細胞またはテスト培養物は、RNAi媒介遺伝子ノックダウンの目的で発現された、または1つ以上発現された内因性遺伝子である1つ以上のレポータ遺伝子を含み、また対象のレポータ遺伝子または内因性遺伝子を標的とした修飾siRNAまたはsiRNA誘導体も含む。
【0020】
siRNAを脂肪細胞に導入する方法(たとえばリポフェクタミンTMなどの作用剤を用いたトランスフェクションなど)は、一般に成功したことがない。電気穿孔を用いた本願発明の方法は、トランスフェクション剤を用いない、siRNAの脂肪細胞への導入方法を提供する。したがって、本願発明は、たとえば、グルコース輸送および糖尿病などの障害など、脂肪細胞機能に関連する治療の研究および開発のためにsiRNAを用いる、遺伝子ノックダウン法を用いることができる。
【0021】
他に定義のない限り、本願明細書に用いられたすべての技術および科学用語は、本願発明が属する当業者によって普通に理解されるものと同じ意味を有する。本願明細書に記載のものに類似するまたはそれに相当する方法および材料は、本願発明の実施またはテストに用いることができ、好適な方法および材料を以下に説明する。本願明細書に引用されるすべての出版物、特許出願、特許、およびその他の参考文献は、その全体を引用によりここに援用する。さらに、当該材料、方法、および例は、具体例であって、制限を意図しない
【0022】
本願発明のその他の特徴および利点は、以下の発明の詳細な説明、図面、および特許請求の範囲から明らかになるであろう。
【0023】
発明の詳細な説明
本願発明は、たとえばsiRNAなどの核酸を脂肪細胞に導入する方法を提供する。この新規の方法は、特定の条件下で脂肪細胞の電気穿孔を用いる。前記方法は、テストタンパク質ラミンA/C を用いて開発され、Akt(タンパク質キナーゼB;PKB)の役割を調べるために用いられてきた。この新規の方法を用いて、他のタンパク質を正常に発現させる一方で、標的化されたタンパク質の発現の完全な阻害を実施することができる。
【0024】
この方法は、たとえば、2型糖尿病の治療に用いることができるインスリン増感剤の同定、および肥満を軽減する薬物などの薬物発見などに有用である。
【0025】
遺伝子特異的ノックアウトマウスはシグナル伝達分子の急性作用を研究するツールを提供するが、マウスにおいて認められた表現型は、遺伝子発現および発育制御における無関係の変化に起因する可能性がある。たとえば、本願発明は、たとえば無傷の培養脂肪細胞におけるAktタンパク質キナーゼなどの、脂肪細胞における発現の選択的阻害を、RNA干渉(RNAi)の機序によって可能にする方法を提供する。この強力なアプローチは、Akt1およびAkt2遺伝子を両方とも喪失すると致死的な場合の、マウスの遺伝子ノックアウト(つまりトランスジェニックマウス)において遭遇する問題点を克服する。
【0026】
培養細胞、特に一次または二次細胞中の遺伝子の選択的阻害を可能にする方法は、遺伝子標的を定めるために遺伝子の機能を明らかにする上で有用である。そのような方法は、特定の遺伝子の発現が阻害される場合に期待される効果の一覧を作成するのにも有用である。前記一覧は、化合物をスクリーニングして、効果が前記の特定の遺伝子の発現の調節に限定されている化合物を同定する場合に用いることができる。本願明細書に記載されているようなシステムは、脂肪細胞において特に有用である。それは、脂肪細胞が肥満および糖尿病(たとえば2型糖尿病)など、薬物が探索されている障害に関連しているからである。
【0027】
本願発明をより容易に理解するため、特定の用語を以下のように義する。
【0028】
「ヌクレオシド」という用語は、リボースまたはデオキシリボース糖に共有結合したプリンまたはピリミジンを有する分子を意味する。例示的なヌクレオチドには、アデノシン、グアシン、シチジン、ウリジン、およびチミジンが含まれる。「ヌクレオチド」という用語は、糖部分にエステル結合したリン酸基を1つ以上有するヌクレオシドを意味する。例示的なヌクレオシドには、1リン酸ヌクレオシド、2リン酸ヌクレオシド、および3リン酸ヌクレオシドが含まれる。「ポリヌクレオチド」および「核酸分子」という用語は、本願明細書では交換可能に用いられ、5’および3’炭素原子の間のホスホジエステル結合によって結合したヌクレオシドのポリマーを意味する。
【0029】
「RNA」または「RNA分子」または「リボ核酸分子」という用語は、一般に、リボヌクレオチドのポリマーを意味する。「DNA」または「DNA分子」または「デオキシリボ核酸分子」という用語は、一般にデオキシリボヌクレオチドのポリマーを意味する。DNAおよびRNA分子は自然に合成することができる(たとえば、それぞれDNA複製またはDNAの転写によって)。RNA分子は転写後に修飾することができる。DNAおよびRNA分子は化学的にも合成することができる。DNAおよびRNA分子は1本鎖であっても(すなわちそれぞれssRNAおよびssDNA)、または多重鎖(たとえば2本鎖、すなわちそれぞれdsRNAおよびdsDNA)であってもよい。しかし、本願発明の性質に基づき、「RNA」または「RNA分子」、または「リボ核酸分子」という用語は、主に(つまり80%超、または好ましくは90%超)リボヌクレオチドを含むポリマーも意味するが、任意で例えば少なくとも1つのデオキシリボヌクレオチドおよび/または少なくとも1つのヌクレオチド類似体などの、少なくとも1つの非リボヌクレオチド分子を含むこともできる。
【0030】
本願明細書では「改変ヌクレオチド」または「修飾ヌクレオチド」も意味する「ヌクレオチド類似体」という用語は、天然に存在しないリボヌクレオチドまたはデオキシリボヌクレオチドを含む、標準的ではないヌクレオチドを意味する。好ましいヌクレオチド類似体は、任意の位置を修飾し、ヌクレオチドの特定の化学的特性を変えつつ、核酸類似体の目的の機能を実行する能力を維持できるようにする。
【0031】
「RNA類似体」という用語は、相当する未改変または未修飾RNAと比較して少なくとも1つの改変またな修飾ヌクレオチドを有するが、前記の相当する未改変または未修飾RNAと同一のまたは類似する性質または機能を保持する、ポリヌクレオチド(たとえば化学合成されたポリヌクレオチドなど)を意味する。上述の通り、前記オリゴヌクレオチドは、ホスホジエステル結合を有するRNA分子と比較してRNA類似体の加水分解が低速度になる結合で結合していてよい。例示的なRNA類似体には、糖および/または骨格修飾リボヌクレオチドおよび/またはデオキシリボヌクレオチドが含まれる。そのような改変または修飾にはさらに、RNA末端または内部などへの非ヌクレオチド物質の付加(RNAの1つ以上のヌクレオチド)が含まれる。RNA類似体は、RNA干渉を媒介する能力を有する天然のRNAに十分類似していればい。
【0032】
本願明細書に用いられる「RNA干渉」または「RNAi」という用語は、一般に、標的分子(たとえば標的遺伝子、タンパク質、またはRNA)を下方調節することによる配列特異的または選択的なプロセスを意味する。ある実施態様では、「RNA干渉」または「RNAi」のプロセスは、たとえば細胞内のRNA分子など、RNA分子の分解であって、前記分解がRNA作用物質によって引き起こされる、分解を特徴とする。分解は、酵素的RNA誘導サイレンシング複合体(RISC)によって触媒される。RNAi は、細胞内で外来のRNA(たとえばウイルス性RNA)を除去するため自然にに生じる。天然のRNAiは、分解メカニズムがその他の同様のRNA配列に方向付けられている遊離dsRNAから切断された断片によって進行する。代替的には、RNAiは、たとえば標的遺伝子の発現をサイレンシングさせるなど、人工的に開始させることもできる。
【0033】
本願明細書に記載の「低分子干渉RNA」(「siRNA」)という用語(「短分子干渉RNA」ともいう)は、RNA干渉を方向付けるまたは媒介する能力のある約10乃至50ヌクレオチドを含むRNA(またはRNA類似体)を意味する。RNAiを方向付けるまたは媒介する標的mRNA配列に十分に相補的な配列を有する「siRNA」とは、siRNAがRNAi機構(たとえばRISC複合体)またはプロセスによって標的mRNAの破壊を惹起するのに十分な配列を有することを意味する。
【0034】
本願明細書に記載の「shRNA」または「短分子ヘアピンRNA」は、ステムループ構造を有するRNA作用物質であって、相補配列の第1および第2の領域であって、前記領域の相補性および配向は前記領域の間で塩基対が生じるのに十分な程度であって、前記第1および第2の領域はループ領域で結合しており、前記ループは前記ループ領域内のヌクレオチド(またはヌクレオチド類似体)の間の塩基対の欠損から生じる、第1および第2の領域を含む、RNA作用物質を含む。
【0035】
「mRNA」または「メッセンジャRNA」は、1つ以上のポリペプチド鎖のアミノ酸配列を特定する1本鎖RNAである。この情報は、リボソームが前記mRNAに結合する際にタンパク質合成中に翻訳される。
【0036】
障害に「関与する」遺伝子には、疾患または障害もしくは前記疾患または障害の少なくとも1つの症状に作用するまたはそれを生じる、正常なまたは異常な発現または機能の遺伝子が含まれる。
【0037】
本願発明の様々な方法論には、本願明細書では「適切な対照」とも交換可能な意味である「好適な対照」と、価値、レベル、特色、特徴、性質などの比較に関与するステップを含む。「好適な対照」または「適切な対照」は、比較の目的で有用な、当業者が精通している任意の対照または標準である。ある実施態様では、「好適な対照」または「適切な対照」は、本願明細書に記載の通り、RNAi方法論を実行する前に決定する、価値、レベル、特色、特徴、性質などである。たとえば、転写速度、mRNAレベル、翻訳速度、タンパク質レベル、生物学的活性、細胞の特徴または性質、ゲノタイプ、表現型などは、細胞または生物に本願発明のsiRNAを導入する前に決定することができる。別の実施態様では、「好適な対照」または「適切な対照」は、細胞、またはたとえば正常な形質を示す対象、または正常細胞もしくは生物などの生物において決定される価値、レベル、特色、特徴、性質などである。さらに別の実施態様では、「好適な対照」または「適切な対照」は、予め決定された価値、レベル、特色、特徴、性質などである。
【0038】
他に定義のない限り、本願明細書に用いられたすべての技術および科学用語は、本願発明が属する当業者によって普通に理解されるものと同じ意味を有する。本願明細書に記載のものに類似するまたはそれに相当する方法および材料は、本願発明の実施またはテストに用いることができ、好適な方法および材料を以下に説明する。
【0039】
I. RNAi分子
RNA干渉(RNAi)は、配列特異的、転写後遺伝子サイレンシングであって、抑制される遺伝子に相同の2本鎖RNA(dsRNA)によって開始される。dsRNAは細胞リボヌクレアーゼIIIであるダイサーによって処理され、配列特異的mRNA分解を媒介する3’オーバーハング(低分子干渉RNA、siRNA)を有する約21ntの2本鎖を生じる。哺乳類細胞では、siRNA分子は、非特異的インターフェロン応答経路を誘導しない、特異的なサイレンシング遺伝子を発現することができる。したがって、siRNAは、機能喪失型表現型を分析するための、アンチセンスヌクレオチドおよびリボザイムなどのその他の遺伝子ツールに代わる新規で強力な代替物である。siRNA 2本鎖を用いて特異的遺伝子の発現と干渉させるには、標的へのアクセスのしやすさ、標的細胞へのsiRNAの効果的な輸送、および一部の用途においては長期的なsiRNA発現の知識が必要である。本願発明は、本願明細書に記載のとおり、電気穿孔法を用いて、たとえば脂肪細胞などの細胞に短2本鎖siRNA分子を形質導入することによって、siRNAをたとえば脂肪細胞などの細胞培養物中の標的細胞へ輸送する効果的な戦略を提供する。本願発明の方法はまた、各種のPol III プロモータによって機能型siRNAまたはその前駆体の転写を可能にする発現カセットを、たとえば脂肪細胞などの細胞に導入するのにも有用である。
【0040】
RNAiはたとえばsiRNAおよび/または長分子dsRNAが動物および植物細胞において標的mRNAの配列特異的分解を誘発する非常に有効なプロセスである(Hutvagner and
Zamore, Curr. Opin. Genet. Dev.:12, 225-232 (2002); Sharp, Genes Dev.,
15:485-490 (2001))。哺乳類細胞において、RNAiは、低分子干渉RNA(siRNA)の21ヌクレオチド(nt)2本鎖(Chiu et al., Mol. Cell. 10:549-561 (2002); Elbashir et al., Nature
411:494-498 (2001))、またはマイクロRNA(miRNA)、機能型低分子ヘアピンRNA(shRNA)、またはRNAポリメラーゼIIIプロモータを有するDNAテンプレートを用いてin vivoにおいて発現させることができるその他のdsRNAによって惹起させることができる(Zeng et al., Mol. Cell 9:1327-1333
(2002); Paddison et al., Genes Dev. 16:948-958
(2002); Lee et al., Nature Biotechnol. 20:500-505
(2002); Paul et al., Nature Biotechnol. 20:505-508
(2002); Tuschl, T., Nature Biotechnol. 20:440-448
(2002); Yu et al., Proc. Natl. Acad. Sci. USA
99(9):6047-6052 (2002); McManus et al., RNA 8:842-850 (2002); Sui et
al., Proc. Natl. Acad. Sci. USA 99(6):5515-5520 (2002).)。
【0041】
したがって、本願発明は、たとえばAkt1、Akt2、Myo1cまたはCISKなどのグルコース輸送に影響する遺伝子のmRNA配列を標的とする核酸分子(つまりRNAi分子)を提供する。ある実施態様では、グルコース輸送に影響する遺伝子は、表1に記載のRNAi標的ヌクレオチド配列を含む。
【0042】
本願発明のRNAi分子には、siRNA分子が含まれる。本願発明のsiRNA作製物には、各鎖にたとえば16、17、18、19、20、21、22、23、24、25、26、27、28、29、または30 ヌクレオチドなど約16乃至30ヌクレオチドを含むdsRNAであって、当該分子において片方の鎖がmRNAにおける標的領域に実質的にたとえば少なくとも80%(またはそれを超える、たとえば85%、90%、95%、または100%)実質的に同一であって、もう片方の鎖は第1の鎖と同一であるかまたは実質的に同一である、dsRNAが含まれる。siRNAは典型的に、2乃至3ヌクレオチドの3’オーバーハング末端、5’リン酸末端(細胞から抽出した)および3’ヒドロキシル末端を有する。RNAを合成する方法、およびRNAを修飾する方法は当業に知られている(たとえば Hwang et al., 1999, Proc. Nat. Acad. Sci. USA 96:12997-13002; およびHuq and Rana, 1997, Biochem. 36:12592-12599)。siRNAは市販されている(たとえばダーマコン・リサーチ社(コロラド州レイファイエット)、アンビオン社(テキサス州オースチン))。
【0043】
本願発明のsiRNA分子は、化学的に合成することができ、またはin vitroではDNAテンプレートから、もしくはin vivoではたとえばshRNAから、もしくはin vitro転写dsRNAテンプレートをRNAiを切断して媒介する20もしくは21bpの2本鎖RNAのプールをつくるためにヒトDICER酵素を用いて、転写することができる。 siRNA分子は当業に知られる任意の方法、たとえば以下のプロトコルを用いることによってデザインすることができる。
【0044】
1. AUG開始コドンから始めてAAジヌクレオチド配列を探す。各AAおよび3’側に16ヌクレオチド以上離れたところが、siRNA標的の候補である(図15、16、34、35、36参照)。5’非翻訳領域(UTR)および開始コドンに近い領域(約75塩基ほど)から得たsiRNAは有用性が低いと考えられる。なぜなら、それらは制御性タンパク質結合部位により豊富に存在しており、結合タンパク質および/または翻訳開始複合体がsiRNPまたはRISCエンドヌクレアーゼ複合体の結合を妨げる可能性があるからである。したがって、ある実施態様では、前記核酸分子は、開始コドンの少なくとも50乃至100nt下流から始まるcDNA配列の領域から選択される。さらに、G/C含有量が少ないsiRNA(35乃至55%)は、G/C含有量が55%を超えるものよりも活性であることもある。したがってある実施態様では、前記発明には35乃至55%のG/C含有量を有する核酸分子が含まれる。さらに、siRNAの鎖は、たとえば2ヌクレオチドなど、1乃至4ヌクレオチドの3’オーバーハングを有するように対を形成させることができる。したがって別の実施態様では、前記核酸分子はTTなどの2ヌクレオチドの3’オーバーハングを有することができる。前記のオーバーハングしているヌクレオチドは、RNAまたはDNAのいずれかであってよい。
【0045】
2. 当業に知られる任意の方法を用いて、標的候補と適切なゲノムデータベース(ヒト、マウス、ラットなど)を比較し、他のコード配列に顕著に相同性を有する任意の標的配列を考慮から除外する。そのような配列相同性の探索に使われる1つの方法はBLASTとして知られ、国立衛生研究所の国立バイオテクノロジー情報センターのウェブサイトで入手することができる。
【0046】
3. 評価基準に合う1つ以上の配列を選択する。さらに、siRNAのデザインおよび使用についての一般的な情報は、ロックフェラー大学のThomas Tuschl 博士の研究所のウエブサイトで入手できる「The siRNA User Guide(siRNAユーザーガイド)」の中に見つけることができる。
【0047】
本願発明のある実施態様では、siRNAは比較的低レベルの毒性を示す。たとえば、標的配列の発現がsiRNAで処理されていない細胞中の発現と比較して50%減少する場合、siRNAで処理された培養物中の細胞の少なくとも50%が生存する場合、前記標的配列の発現を阻害するsiRNAの濃度は比較的低い毒性を有している。低い毒性は、たとえば少なくとも60%、75%、85%、90%、95%、または100%など、より高い細胞生存性に関連していると考えられる。細胞の生存性を測定する方法は当業に知られており、トリパンブルー排除法が含まれる。これは、たとえば標的遺伝子のノックアウト(たとえばトランスジェニック動物など)とは対称的である。したがって、本願発明の利点は、遺伝子を操作してその遺伝子の発現を操作する場合、細胞または動物に致死的である可能性がある遺伝子の発現の操作が可能になることである。
【0048】
本願発明のRNAi作用物質には、低分子ヘアピンRNA(shRNA)、およびshRNAを発現するためにつくられた発現作製物が含まれてよい。shRNAの転写は、ポリメラーゼIII(pol III)プロモータから開始し、4-5チミン転写終了部位の2位で終了すると考えられている。発現後、shRNAは3’UUオーバーハングを有するステム-ループ構造に折り畳まれると考えられ、その後、これらのshRNAの末端が処理されて、shRNAが約21ヌクレオチドのsiRNA様分子に転換される。Brummelkamp et
al., Science 296:550-553 (2002); Lee et al, (2002). supra; Miyagishi
and Taira, Nature Biotechnol. 20:497-500 (2002);
Paddison et al. (2002), supra;
Paul (2002), supra; Sui (2002) supra; Yu et al. (2002), supra。shRNAデザインおよび使用についてのさらなる情報は、インターネットの以下のアドレス katahdin.cshl.org:9331/RNAi/docs/BseRI-BamHI_Strategy.pdf および katahdin.cshl.org:9331/RNAi/docs/Web_version_of_PCR_strategy1.pdfで見つけることができる。
【0049】
本願発明の発現作製物には、適切な発現システムに使用するのに好適な任意の作製物が含まれ、当業に知られるレトロウイルスベクタ、直線状発現カセット、プラスミドおよびウイルスまたはウイルス由来ベクタがそれらに限定されずに含まれる。そのような発現作製物には、1つ以上の誘導プロモータ、U6 snRNAプロモータもしくはH1 RNAポリメラーゼIIIプロモータなどのRNA Pol IIIプロモータシステム、または当業に知られるその他のプロモータが含まれてよい。両方の鎖を発現する発現作製物には、両方の鎖を連結するループ構造が含まれてもよく、または各鎖は同一の作製物内の個別のプロモータから別々に転写されてもよい。各鎖は個別の発現作製物から転写されてもよい。Tuschl (2002), supra。
【0050】
細胞内における長期間の標的遺伝子の抑制を可能にするために、細胞内で組換えDNA作製物からsiRNA 2本鎖を発現させる方法は当業に知られており、機能的な2本鎖siRNAを発現することが可能な哺乳類Pol III プロモータシステム(たとえばH1またはU6/snRNAプロモータシステム(Tuschl (2002), supra) )が含まれる(Bagella et al., J. Cell. Physiol.
177:206-213 (1998); Lee et al. (2002),
supra; Miyagishi et al. (2002),
supra; Paul et al. (2002), supra;
Yu et al. (2002), supra;
Sui et al. (2002), supra)。RNA Pol IIIによる転写終了は、一連のDNAテンプレートの連続する4つのT残基で生じ、特定の配列においてsiRNA転写物を終了させるメカニズムを提供する。前記siRNAは5’-3’および3’-5’の方向に向いた前記標的遺伝子の配列に相補的であって、siRNAの2本鎖は同一の作製物または別々の作製物中に発現することができる。H1またはU6 snRNAプロモータによって駆動され、細胞内で発現するヘアピンsiRNAは、標的遺伝子の発現を阻害することができる(Bagella et al. (1998), supra; Lee et
al. (2002), supra; Miyagishi
et al. (2002), supra; Paul et
al. (2002), supra; Yu et al. (2002), supra; Sui et al. (2002) supra)。T7プロモータの制御下にあるsiRNA配列を含有する作製物もまた、T7 RNA ポリメラーゼを発現するベクタを有する細胞に同時トランスフェクトされると、機能性siRNAをつくる。1つの作製物は、同一の遺伝子または複数の遺伝子に方向付けられているたとえばAkt1、Akt2、CISKまたはMyo1cなどをコードする遺伝子の複数の領域など、siRNAをコードする複数の配列を含有することがあり、たとえば、別々のPolIIIプロモータ部位によって駆動することができる。
【0051】
動物細胞はミクロRNA(miRNA)とよばれる約22ヌクレオチドの非コードRNAの領域を発現する。ミクロRNAは動物の成長中に転写後または転写レベルで遺伝子発現を制御することができる。miRNAのある共通の性質は、おそらくダイサー、RNase III型酵素、またはその相同体によって、約70ヌクレオチドの前駆体RNAステム-ループからすべて切除されることである。標的mRNAに相補的な配列を有するmiRNA前駆体のステム配列を置換することによって、新規miRNAを発現するベクター作製物を用いて、哺乳類細胞の中で特異的mRNA標的に対してRNAiを開始するためにsiRNAを産生することができる(Zeng (2002), supra)。ポリメラーゼIIIプロモータを含有するDNAベクタによって発現する場合、ミクロRNAでデザインされたヘアピンは遺伝子発現をサイレンシングすることができる(McManus (2002), supra)。動物モデルにおいて、全胚電気穿孔は、合成siRNAを効率的に移植後マウス胚合成siRNAに送達させることができる。成体マウスにおいて、siRNAの効率的な送達は、「高圧」送達技術によって実現することができる。これは溶液を含有する大量のsiRNAを、尾部静脈を介して動物体内へ迅速に(5秒以内に)注入する技術である。ナノ粒子およびリポソームを用いて、siRNAを動物体内へ送達することもできる。
【0052】
陰性対照siRNAは、選択されたsiRNAと同一のヌクレオチド組成を有するべきだが、適切なゲノムに相補的な重要な配列は有さない。そのような陰性対照は、選択されたsiRNAのヌクレオチド配列をランダムにスクランブルしてデザインしてもよく、相同性探索は、前記陰性対照が適切なゲノムにおける任意のその他の遺伝子に対する相同性を欠損していることを確認するために行うことができる。さらに、陰性対照siRNAは、前記配列に1つ以上の塩基ミスマッチを導入することによってデザインすることができる。
【0053】
本願発明の核酸組成物には、架橋siRNA誘導体など、当業に知られる未修飾siRNAおよび修飾siRNAの両方が含まれる。たとえば体内の半減期を延長するためなど、前記組成物の薬物動態学を変化させるために、架橋を用いることができる。したがって、本願発明には、2本の鎖が架橋されるように、核酸の相補的な2本の鎖を有するsiRNA誘導体を含む、siRNA誘導体が含まれる。たとえば、片方の鎖の3’OH末端を修飾し、2本鎖を架橋させて3’OH末端で修飾することができる。前記siRNA誘導体は1つの架橋(たとえばソラーレン架橋)を含有することができる。siRNA誘導体の修飾は、細胞取り込みが改善するか、もしくは相当するsiRNAと比較して、得られたsiRNA誘導体の細胞の標的活性を促進するか、前記細胞のsiRNA誘導体を追跡するのに有用であるか、または相当するsiRNAと比較したsiRNA誘導体の安定性を向上することもある。
【0054】
本願発明の核酸組成物は、たとえば吸収、有効性、バイオアベイラビリティ、および/または半減期などの薬物動態学的パラメタなどの前記組成物の性質を向上させるナノ粒子などの別の部分と結合させることもでき、または結合させないでもよい。その結合は、たとえば、Lambert et al.,
Drug Deliv. Rev.:47(1), 99-112 (2001) (ポリアルキルシアノアクリル酸(PACA)ナノ粒子に装着された核酸について記載)、Fattal et al.,
J. Control Release 53(1-3):137-43 (1998) (dナノ粒子に結合させた核酸について記載)、 Schwab et al.,
Ann. Oncol. 5 Suppl. 4:55-8 (1994) (挿入剤、疎水基、ポリカチオンPACAナノ粒子に結合させた核酸について記載)、およびGodard et al.,
Eur. J. Biochem. 232(2):404-10
(1995) (ナノ粒子に結合させた核酸について記載)の方法を用いるなど、当業に知られる方法によって実現することができる。
【0055】
本願発明の核酸分子は、当業に知られる任意の方法を用いてラベルすることもでき、たとえば前記核酸分子組成物がたとえばCy3、フルオレセイン、ローダミンなどのフルオロフォアで標識することができる。その標識は、たとえばSILENCERTM
siRNA ラベリングキット(アンビオン)など、キットを用いて行うことができる。さらに、前記siRNAは、たとえば3H、32Pまたはその他の適切な同位体などを用いて放射標識することができる。
【0056】
本願明細書に記載の細胞(たとえば脂肪細胞など)または組織全体に導入された組換えRNA前駆体によって、望ましいsiRNA分子の産生が引き起こされるだろう。そこで、そのようなsiRNA分子は、切断および破壊のための特異的なmRNA配列に結合し、それを標的とするために、RNAi経路の内因性タンパク質要素に関連するだろう。このようにすれば、組換えRNA前駆体から作製されたsiRNAが標的とするmRNAは、前記細胞および組織から大幅に減少し、前記細胞または組織中のmRNAによってコードされるタンパク質の濃度が減少するだろう。RNA前駆体は、典型的にはdsRNAの1本鎖を個別にコードするか、またはRNAヘアピンループ構造のヌクレオチド配列全体をコードする核酸分子である。
【0057】
さらに、RNAiは、少なくとも1つの1本鎖RNA中間体を経る過程であると考えられるため、当業者は、ss-siRNA(たとえばds-siRNAのアンチセンス鎖など、本願明細書に記載のとおりデザイン(たとえば化学合成用に)、作製(たとえば酵素的に作製)または発現(たとえばベクターまたはプラスミドから)することもでき、本願特許請求の方法論にしたがって利用することができるということは理解されよう。さらに、無脊椎動物では、RNAiは長いdsRNA(たとえば長さが約100乃至1000ヌクレオチド、好ましくは約200乃至500であって、たとえば、約250、300、350、400、または450ヌクレオチドのdsRNA)であって、RNAiのエフェクタT細胞として作用するRNAiは長いdsRNAによって効果的に引き起こすことができる。(Brondani et al., Proc Natl Acad Sci U S A. 2001
Dec 4;98(25):14428-33. Epub 2001 Nov 27)。
【0058】
本願発明のsiRNA分子は、前記標的遺伝子の以下の配列を標的とすることができ、たとえば前記siRNA分子が、その鎖の1つとして、前記DNA配列の任意の1つに相当するRNA配列(たとえば前記siRNA2本差のセンス鎖など)、およびその対立遺伝子変異体の相当する配列を含んでよい。表の配列は、標的遺伝子配列(すなわちcDNA配列)を表す。しかし、当業者は、たとえばセンス鎖などのsiRNA鎖が相当するリボヌクレオチドを含んでおり、アンチセンス鎖が相補的リボヌクレオチド配列を含むということを理解するだろう。さらなるデオキシチミジンオーバーハングも、本願明細書に記載されるとおり意図される。
【0059】
【表1】

【0060】
II. 脂肪細胞培養物
本願発明は、たとえばsiRNAなどの核酸を脂肪細胞に導入に関連する。脂肪細胞は当業に知られる方法を用いて培養することができる(たとえば Adipose
Tissue Protocols, Methods in Molecular Biology, v.
155, ed. Gerard Ailhaud. Totowa, NJ,
Humana Press, 2001を参照)。例1(後述)は、3T3-L1細胞を脂肪細胞に分化させるそのような1つの方法を説明する。使用することができるその他の方法には、解剖した(一般に細胞を切除するための処理後の)脂肪組織から得た脂肪細胞を用いた、間葉細胞の脂肪細胞への分化(たとえばステムセル・テクノロジーズ社(カナダ、バンクーバー)などから入手可能な試薬を用いた)、および脂肪細胞を作成する市販の方法(たとえば脂肪細胞分化キット、ストラタジーン社、プロモセル(独、ハイデルベルグ))が含まれる。
【0061】
本願明細書に記載の方法を用いた核酸の導入のための細胞を調製するために、脂肪細胞を培養基質から剥離するか、または標準方法で解離させる。
【0062】
III. 核酸分子の脂肪細胞への導入
一般に、リボ核酸、デオキシ核酸、およびこれらの変異体は、脂肪細胞に導入することができる。前記核酸は、たとえば、脂肪細胞、アンチセンスRNA、siRNA、siRNA前駆体、またはsiRNA前駆体を発現するベクターに発現させる、遺伝子産生物または遺伝子産生物の断片をコードする配列を含有するなどの、核酸ベクターであってよい。前記核酸は、2本鎖または1本鎖であってよい。
【0063】
脂肪細胞に導入される核酸配列は、たとえば長さ約2乃至5,000塩基、2乃至1000塩基、2乃至500塩基、2乃至300塩基、2乃至200塩基、2乃至100塩基、2乃至50塩基、または2乃至25塩基、もしくは1乃至15塩基などの、約2乃至10000塩基である。前記核酸がsiRNAである場合、各鎖は一般に、たとえば長さ16、17、18、19、20、21、22、23、24、25、26、27、28、29、または30ヌクレオチドなどの、約16乃至30ヌクレオチドである。そのような核酸を調節する方法は、当業に知られる。典型的には、本願明細書に記載の方法に用いられる核酸サンプルは、脱塩してから細胞に導入する。
【0064】
一般に、核酸分子は、たとえばリン酸バッファ食塩水(PBS)など好適なバッファ中で洗浄した後に細胞に導入し、それからたとえばPBSなどの好適なバッファ中で再懸濁するその他の好適なバッファには、Krebs/リンガー/HEPESバッファがそれに限定されずに含まれる。再懸濁した細胞は、PBSに再懸濁した核酸か、または電気穿孔の手順に好適なその他のバッファ中に混合する。たとえば、0.1乃至80nmol、典型的には約20nMのsiRNAを用いる。約1乃至1000万個の細胞を前記方法に用いてよく、典型的には各手順に約500万個の細胞を用いる。典型的には、前記細胞は室温で電気穿孔されるが、他の温度であってもよい。その後、前記混合物を、たとえば約0.001乃至20 kV、約0.01乃至2.0 kV、約0.02乃至1.0 kV、約0.05乃至0.5 kV、約0.1乃至0.25kVの設定、および好ましくは0.11、0.12、0.13、0.14、0.15、0.16、0.17、0.18、0.19、0.20、0.21、0.22、0.23、0.24または0.25 kVの設定、ならびに約 10-5000 μF、約 100-3000 μF、約 200-1800 μF、約 350-1550 μF、約 5-1350 μF、約 750-1150 μF、約 850-1050 μF、約 900-1000 μFの設定、および好ましくは約900、910、920、930、940、950、960、970、980、990、または100 μFの静電容量の設定など、前記細胞へsiRNAを効率的に導入できるような設定で、好適にな装置(たとえばバイオラッド・ジーンパルサーIIシステム、バイオラッドラボラトリーズ社(カリフォルニア州ハーキュリーズ))を用いて電気穿孔のパルスに曝露する。好ましい実施態様では、前記混合物を、0.18kVおよび960μFの設定で電気穿孔のパルスに曝露する。典型的には約400乃至600μLの細胞、たとえば450μLの細胞などを、前記手順に用いる。電気穿孔後、前記細胞をただちに新鮮培地と混合し、37℃で10分間インキュベートしてからデオキシグルコース取り込みアッセイ、ウエスタンブロッティング、または免疫蛍光顕微分析法などのその後の用途に好適な多穴プレートに再播種する。再播種後、約2、4、8、12、16、20、24、30、36、42、48、54、60、72、78、94または約120分経過時点など、再播種後好適な時点に、分析を行う。好ましい実施態様では、分析は再播種後24乃至48時間経過時点で行う。
【0065】
本願明細書に記載の細胞または生物全体に導入したsiRNAは、切断および破壊のための特異的なmRNA配列に結合し、標的とするRNAi経路の内因性タンパク質要素に関連するだろう。このようにすれば、siRNAが標的とするmRNAは、前記細胞および生物から激減し、前記細胞または組織中のmRNAによってコードされるタンパク質の濃度が減少する。
【0066】
前記方法は、たとえば、発現(または過剰発現)すると異常なグルコース代謝を引き起こす核酸配列の活性を低減する低分子を発見するために探索する場合には有用である。主要な疑問は、この核酸配列の発現または活性を減少させると細胞に予期せぬ作用、とくに悪性の作用を及ぼすかどうかである。RNAi経路による破壊のために脂肪細胞中の核酸配列をコードするmRNAを標的とするsiRNA誘導体を発現することによって、そのような薬物候補の悪性作用を決定することができる。つまり、本願明細書に記載の方法では、前記核酸配列および薬物標的としてのその産生物の適切性を迅速に評価することができる。
【0067】
RNAiは遺伝子機能を解明するための新規のアプローチを提供する。RNAi媒介遺伝子ノックダウンは、遺伝子機能のゲノムワイドな分析、および治療用に用いられる可能性のある遺伝子の標的バリデーションに有用である。siRNAは哺乳類遺伝子機能を研究する細胞生物学者にとって有用なツールである。たとえば、siRNAは有子分裂、RNA転写物のプロセッシングおよび輸送、細胞接合の形成、および膜輸送などの一般的な細胞生物学的機序の分析に有用である。そのような分析に用いることができる試薬(たとえば、相当する未修飾siRNAと比較した細胞における高安定性を有するsiRNA)は、そのような研究用の商品価値を有する。
【0068】
特定の遺伝子はsiRNAを用いることによってノックダウンすることができ、得られた表現型を観察することができる。しかし、基本的な遺伝子のノックダウンは致死的または有毒である可能性もあり、細胞における多くの経路に影響を与える可能性がある。したがって、ある場合には、前記細胞に、ノックダウン時には有効性が最大ではないsiRNAを提供することが好ましい。過度に有効なノックダウンの悪性作用は、相当するsiRNAと比較して低いRNAi活性を有するsiRNA誘導体を前記細胞と接触させることによって調節することができる。siRNA誘導体は修飾siRNAである。修飾には、架橋または3’末端の阻害が、それらに限定されずに含まれる(Chiu and Rana,
2002, Mol Cell 10:549-561)。この目的に用いられるsiRNA誘導体の好適な濃度には、RNAi活性を最大に阻害することがなく、siRNAの望ましくない作用を軽減する濃度が含まれる。相当するsiRNAよりも低い効率のノックダウンを生じることができるsiRNA誘導体の量は、当業に知られる方法を用いて決定することができる。たとえば、特定の核酸配列を標的とするsiRNAをトランスフェクトした細胞における発現レベルと、前記siRNAの誘導体をトランスフェクトした細胞における特定の核酸配列の発現量を比較する。siRNA誘導体をトランスフェクトした細胞における高発現レベルは、前記siRNAは、発現のノックダウンの効率がよくないということを示唆している。そのような誘導体は、効率の高いノックダウンが前記細胞に致死的であるノックダウン実験に有用である。したがって、ある場合では、有用なsiRNA誘導体はRNAiの阻害が100%未満のsiRNA誘導体である。たとえば、siRNAのRNAi作用を低下させるのに有用なsiRNA誘導体は、たとえば90%、75%、50%、25%、または10%未満など、RNAi活性を阻害することができる。
【0069】
IV 核酸標的
本願明細書に記載のsiRNAの核酸標的は、2型糖尿病、インスリン耐性もしくは肥満に関連する任意の遺伝子、または、たとえばAkt1、Akt2、CISK、またはMyo1cをコードする遺伝子などをそれらに限定せずに含む、グルコース輸送またはインスリン応答に重要な任意の遺伝子であってよい。ある実施態様では、前記標的遺伝子は、表1に記載の標的ヌクレオチド配列を含む。
【0070】
したがって、Akt1のmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号NM_009652 (GI:6753033;Mus musculus) (配列番号20); NM_005163 (GI:4885060; Homo
sapiens) (配列番号22)がそれらに限定されずに含まれるマウスもしくはヒトAkt1に、実質的に同一の配列など、Akt1の任意のオルソログであってよい。Akt2のmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号U22445 (GI:942577; Mus musculus) (配列番号21); NM_001626 (GI:6715585; Homo
sapiens) (配列番号23)がそれらに限定されずに含まれるマウスもしくはヒトAkt2に、実質的に同一の配列など、Akt2の任意のオルソログであってよい。CISKのmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号AF312007 (GI:11321320; Mus musculus) (配列番号24); ジェンバンク受入番号NM_013257.3 (GI:25168264; Homo sapiens, variant 1); ジェンバンク受入番号NM_170709 (GI:25168266; Homo sapiens,
variant 2)がそれらに限定されずに含まれるマウスもしくはヒトCISKに、実質的に同一の配列など、CISKの任意のオルソログであってよい。Myo1cのmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号U22445 NM_008659 (GI:31543277; Mus
musculus) (配列番号25); NM_033375 (GI:24415399; Homo
sapiens) (配列番号26)がそれらに限定されずに含まれるマウスもしくはヒトMyo1cに、実質的に同一の配列など、Myo1cの任意のオルソログであってよい。
【0071】
本願明細書に記載の「オルソログ」という用語は、参照配列に実質的に同一の配列を意味する。本願明細書に記載の「実質的に同一の」という用語は、十分な数のもしくは最小数の同一または等価のアミノ酸残基またはヌクレオチドを含有する第1のアミノ酸またはヌクレオチド配列と、第2のアミノ酸またはヌクレオチド配列であって、前記第1および第2のアミノ酸またはヌクレオチド配列が共通の構造ドメインまたは共通の機能的活性を有する、第1のアミノ酸配列を意味する。たとえば、少なくとも約60%または65% の同一性、おそらく75% の同一性、よりおそらくは85%、90%の同一性を有する共通の構造ドメインを含有するアミノ酸またはヌクレオチド配列である。本願明細書において、91%、92%、93%、94%、95%、96%、97%、98% または99%の同一性は、実質的に同一であると定義する。
【0072】
配列間の相同性または配列同一性(前記用語は本願明細書においては交換可能である)の計算は以下の通り行われる。
【0073】
2つのアミノ酸配列、または2つの核酸配列の同一性の割合を決定するために、その配列を最適な比較の目的のためにアラインメントする(たとえば最適なアラインメントのために第1および第2のアミノ酸または核酸配列の片方または両方にギャップを導入してもよく、非相同成敗列を比較目的のために無視してもよい)。ある実施態様では、比較の目的のためのアラインメントされた参照配列の長さは、前記参照配列の長さの少なくとも50%、少なくとも60%、少なくとも70%、80%、90%、100%である。それから、対応するアミノ酸の位置またはヌクレオチドの位置にあるアミノ酸残基またはヌクレオチドを比較する。第1の配列のある位置が第2の配列の対応する位置として同一のアミノ酸残基またはヌクレオチドによって占められていれば、その分子はその位置において同一である(本願明細書に記載の通り、アミノ酸または核酸の「同一性」はアミノ酸または核酸の「相同性」と等価である)。前記の2つの配列間の同一性の割合は、前記配列によって共有された同一の位置の数の関数であり、2つの配列の最適化アラインメントに導入する必要のあるギャップ数、および各ギャップの長さを考慮する。
【0074】
配列の比較と2つの配列間の同一性の割合の決定は、数学的アルゴリズムを用いて行うことができる。ある実施態様では、2つのアミノ酸配列間の同一性の割合は、ブロッサム62マトリックスまたはPAM250マトリックスのいずれか、GCGソフトウエアパッケージ(Accelrysのオフィシャルウェブサイトで入手可能)のGAPプログラムに導入された、Needleman and
Wunsch (J. Mol. Biol.
48:444-453 (1970))アルゴリズムを用いて、16、14、12、10、8、6、または4のギャップ重みおよび1、2、3、4、5、または6の長さ重みを用いて決定される。さらに別の実施態様では、2つの核酸配列間の同一性の割合は、GCGソフトウエアパッケージ(Accelrysのオフィシャルウェブサイトで入手可能)のGAPプログラムを用いて、NWSgapdna.CMP マトリックス、および40、50、60、70、または80のギャップ重みおよび1、2、3、4、5、または6の長さ重みを用いて決定される。あるセットのパラメタ(および、分子が配列同一性または相同性の本願発明の制限内に入っているかどうかを決定するためにどのようなパラメタを用いてよいのか、実行者がよく知らない場合に用いることができるパラメタ)は、ギャップペナルティ12、ギャップの延長ペナルティ4、およびフレームシフトギャップペナルティ5を有するブロッサム62スコアリングマトリックスである。
【0075】
アミノ酸またはヌクレオチド配列間の同一性の割合は、ALIGNプログラム(バージョン2.0)に組み込まれたMeyers and W.
Miller (CABIOS, 4:11-17 (1989)) のアルゴリズムを用いて、PAM120重み残基表、ギャップ長さペナルティ12、ギャップペナルティ4を用いて、決定することができる。
【0076】
本願明細書に記載の核酸およびタンパク質配列は「クエリ配列」として用いることができ、たとえばファミリメンバまたは関連配列などのその他のオルトログを同定するための好適データベースに対する検索を実行することができる。そのような検索は、Altschul, et al.
J. Mol. Biol. 215:403-10 (1990)のNBLAST およびXBLASTプログラム (version 2.0) を用いて行うことができる。BLASTヌクレオチド探索は、本願発明の既知のTEF核酸分子に相同なヌクレオチド配列を得るために、NBLASTプログラム、スコア= 100、ワード長=12を用いて行うことができる。BLASTタンパク質探索は、既知のTEF核酸分子に相同なアミノ酸配列を得るために、XBLASTプログラム、スコア= 50、ワード長=3を用いて行うことができる。比較目的のためのギャップアラインメントを得るために、Altschul et al., (1997) Nucleic
Acids Research 25(17):3389-3402に記載のようにギャップBLASTを用いることができる。BLASTおよびギャップBLASTプログラムを用いる場合、それぞれのプログラム(たとえばXBLASTおよびNBLAST)のデフォルトパラメータを用いてよい。国立衛生研究所の国立バイオテクノロジインフォメーションセンタのウェブサイトを参照のこと。
オルソログは、たとえばヒトcDNAライブラリなどのcDNAライブラリのスクリーニングなど当業に知られる任意の他のルーチン法を用いて、参照配列に実質的に同一な配列を同定するためにデザインされたプローブを用いても、同定することができる。
【0077】
V. 機能的アッセイ
機能的アッセイは、たとえば脂肪細胞のsiRNA方法論を用いた標的細胞の減少後など、特定の遺伝子のノックダウンの効果を決定するために用いるられる。そのようなアッセイは、糖尿病においてグルコース輸送に関与する、またはエフェクタとして示唆される特定の遺伝子ノックダウンの効果を決定するために、特に有用である。そのようなアッセイは当業に知られており、たとえば例1に記載されるとおり、その一部は本願明細書に記載されている。そのようなアッセイには、デオキシグルコース取り込み、グルコース・トランスロケーションアッセイ用のまたはMYCタグGlut4をアッセイするステップが、それらに限定されずに含まれる。
【0078】
A. 細胞アッセイ
ある実施態様では、グルコース輸送に関与する遺伝子によってコードされるポリペプチド、または生物学的に活性なその部分を発現する能力を有する細胞をテスト化合物と接触させ、テスト化合物の、前記ポリペプチドまたは生物学的に活性なその部分の発現を調節する能力を決定する、細胞アッセイである。 別の実施態様では、アッセイはグルコース輸送に関与する遺伝子によってコードされるポリペプチド(または生物学的に活性なその部分)を発現させる細胞をテスト化合物と接触させ、テスト化合物の、前記ポリペプチド(または生物学的に活性なその部分)の発現を調節する能力を決定する、細胞アッセイである。たとえば、その細胞は哺乳類由来または酵母細胞であってよい。好ましい実施態様では、前記細胞は脂肪細胞である。たとえば、前記ポリペプチドは、たとえば前記細胞などの前記細胞を非相同的に発現するか、または前記細胞に由来してよい。前記テスト化合物の、グルコース輸送(または生物学的に活性なその部分)に関与する遺伝子によってコードされたポリペプチドの活性を調節する能力の決定は、たとえば本願明細書に記載のAkt1またはAkt2の活性など、当業に標準的に知られるようなポリペプチドに起因する活性の任意の1つをアッセイすることによって実施することができる。前記テスト化合物がグルコース輸送に関与する遺伝子によってコードされるポリペプチド(または生物学的に活性なその部分)の活性を調節する能力の決定は、前記ポリペプチドの基質標的分子の活性をアッセイすることによって実施することができる。ある実施態様では、前記テスト化合物がグルコース輸送に関与する遺伝子によってコードされるポリペプチドまたは生物学的に活性なその部分の活性を調節する能力の決定は、前記ポリペプチドまたは生物学的に活性なその部分の基質標的分子に結合する能力をアッセイすることによって実施することができる。ある好ましい実施態様では、前記細胞はグルコース輸送に関与する遺伝子によってコードされるポリペプチドまたは生物学的に活性なその部分を過剰発現する。
【0079】
本願明細書に記載の「生物学的に活性な」断片という用語には、たとえば細胞によるインスリン媒介グルコース取り込みなどを含む、少なくとも1つのグルコース輸送関連活性を示す、または発揮するのに十分な、グルコース輸送に関与するポリペプチドの任意の部分(たとえば近接するアミノ酸の部分)が含まれる。各種の実施態様では、グルコース輸送に関与する遺伝子は、たとえばAkt1、Akt2、CISKまたはMyo1cであってよい。
【0080】
本願発明の細胞アッセイによると、テスト化合物の、グルコース輸送に関与するポリペプチドまたは生物学的に活性なその部分の活性を調節する能力の決定は、本願明細書に記載の、Akt、CISKまたはMyo1cポリペプチドの活性と同時に生じる天然の活性または間接的な活性をアッセイすることによって決定することができる。たとえば、Akt発現細胞のインスリン依存性のグルコース取り込み能力に前記テスト化合物が与える作用は、本願テスト化合物の存在下においてアッセイすることができる。また、好ましい実施態様では、本願発明の細胞アッセイは、インスリン応答のモジュレータとしてテスト化合物を同定する最終ステップを含むことが意図される。
【0081】
B. ハイスループットアッセイ
脂肪細胞中に標的遺伝子ノックアウトを用いたハイスループットアッセイも、標的をテストするのに有用である。たとえば、脂肪細胞中に発現される核酸配列を標的とした各種のsiRNAのプールを、本願明細書に記載の方法を用いて脂肪細胞にトランスフェクトする。それから、前記のトランスフェクトされた細胞を、1つ以上の機能(たとえばグルコース輸送またはGLUT4トランスロケーション)のかく乱についてテストする。そのようなかく乱を証明するsiRNAのプールは、さらに小さいプールに分割され、そのプロセスを、認められたかく乱(作用)に関連する1つ以上の配列が同定されるまで繰り返す。この方法は、2つ以上の種類のsiRNAが細胞によって取り込まれる可能性もあるために、2つ以上の配列のノックダウンを必要とする作用を同定するためにも有用である可能性がある。そのような技術は、たとえば、糖尿病および肥満などの脂肪細胞関連疾患のための薬物発見に関連する研究などに有用である。たとえば、そのような方法は、グルコース輸送および糖尿病などの脂肪細胞に関連する機能および障害の研究に有用な薬物標的のライブラリを作成するために用いることができる。
【0082】
VI. テスト化合物
本願発明のテスト化合物は、当業に知られるコンビナトリアルライブラリの方法における多数のアプローチのいずれかを用いて得ることができる。その方法には、生物学的ライブラリ、空間指定平行固相または液層ライブラリ、畳込みを要する合成ライブラリ法、1ビーズ1化合物ライブラリ法、および親和性クロマトグラフィ選択を用いた合成ライブラリ法などが含まれる。生物学的ライブラリアプローチはペプチドライブラリに限定されるが、一方で、その他の4つのアプローチはペプチド、非ペプチドオリゴマーまたは化合物の低分子ライブラリに適用可能である(Lam, K.S. (1997)
Anticancer Drug Des. 12:145)。
【0083】
分子ライブラリの合成のための方法の例は、たとえばDeWitt et al. (1993) Proc. Natl. Acad. Sci. U.S.A. 90:6909、Erb et al.
(1994)
Proc. Natl. Acad. Sci. USA
91:11422、Zuckermann et al. (1994). J. Med. Chem.
37:2678、Cho et al. (1993)
Science 261:1303、Carrell et al. (1994) Angew. Chem. Int. Ed. Engl. 33:2059、Carell et al.
(1994) Angew. Chem. Int. Ed. Engl. 33:2061、and in Gallop et
al. (1994) J. Med. Chem.
37:1233など、当業に見いだすことができる。
【0084】
化合物のライブラリは、溶液中(e.g.,
Houghten (1992) Biotechniques 13:412-421)、ビーズ上(Lam (1991) Nature
354:82-84)、チップ(Fodor (1993) Nature 364:555-556)、細菌 (Ladner USP
5,223,409)、胞子(Ladner USP '409)、プラスミド(Cull et al.
(1992) Proc Natl Acad Sci USA
89:1865-1869)、またはファージ上(Scott and Smith (1990) Science 249:386-390)、(Devlin (1990) Science
249:404-406)、(Cwirla et al. (1990)
Proc. Natl. Acad. Sci. 87:6378-6382)、(Felici (1991) J.
Mol. Biol. 222:301-310)、(Ladner supra.)に存在することもできる。
【0085】
好ましい実施態様では、前記ライブラリは天然産生物ライブラリである。
【0086】
VII 薬学的組成物
本願発明はさらに、上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータに関連する。上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータは、好適な動物モデルにおいてテストすることができる。たとえば、本願明細書に記載のとおり同定されたインスリン応答モジュレータを動物モデルに用いて、そのようなモジュレータでの処置の有効性、毒性、または副作用を決定することができる。 代替的には、本願明細書に記載の通り同定されたモジュレータを動物モデルに用い、そのような作用薬の作用機序を決定することができる。 さらに、本願発明は、後述のの治療のための上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータの使用に関連する。
【0087】
したがって、本願発明のインスリン応答モジュレータを、投与に好適な薬学的組成物に組み込むことができる。そのような組成物は、典型的には、核酸分子、タンパク質、抗体、または調節性化合物、および薬学的に許容な担体が含まれる。本願明細書に記載の「薬学的に許容な担体」という用語は、薬学的投与に適合する、溶媒、分散媒、コーティング、抗細菌および抗真菌剤、ならびに等張および吸収遅延剤など、いずれかおよびすべてを含むことを意図する。薬学的に活性な物質へのそのような媒質および物質の使用は、当業に公知である。任意の従来の媒質または作用物質が活性化合物と適合性がない場合以外は、前記組成物へのそれらの使用が考慮される。補助的な活性化合物も、当該組成物に組み込むことができる。
【0088】
本願発明の薬学的組成物は、意図する投与経路と適合するように調剤される。投与経路の例には、たとえば静脈内、皮内、皮下、口腔内(たとえば吸入)、経皮(局所)、経粘膜、および直腸投与などの非経口投与が含まれる。非経口、皮内、または皮下投与に使用する溶液または懸濁液には、以下の成分、注射用の水、食塩水、固定油、ポリエチレングリコール、グリセリン、プロピレングリコール、またはその他の合成溶媒などの滅菌希釈剤、ベンジルアルコールまたはメチルパラベンなどの抗菌剤、アスコルビン酸または亜硫酸ナトリウムなどの抗酸化剤、エチレンジアミン四酢酸などおキレート剤、酢酸塩、クエン酸塩、またはリン酸塩などのバッファ、および塩化ナトリウムまたはデキストロースなどの張力調整剤などが含まれていてよい。非経口製剤は、アンプル、ガラスまたはプラスチック製の使い捨て用シリンジ、または複数の投与用バイアルの中に封入されていてよい。
【0089】
注射に使用するために好適な薬学的組成物には、滅菌水溶液(水溶性)、または分散液、および滅菌注射用溶液または分散液の即時調整用滅菌粉末が含まれる。静脈内投与に好適な担体には、生理食塩水、静菌水、クレモホールELTM(BASF、ニュージャージー州パシッパニー)、またはリン酸緩衝食塩水(PBS)が含まれる。すべての場合において、前記組成物は滅菌状態であって、注射容器への充填が容易な程度、流動的でなければならない。前記組成物は、製造および保存の条件下において安定でなければならず、細菌および真菌などの微生物の汚染作用から保護されなければならない。前記担体は、たとえば水、エタノール、ポリオール(たとえばグリセロール、ポリエチレングリコール、および液体状ポリエチレングリコールなど)、それらの好適な混合物、ならびに植物油などを含む溶媒又は分散媒であってよい。適切な流動性は、たとえばレシチンなどのコーティングの使用、分散液の場合は所要の粒子サイズの維持、および表面活性剤の使用などによって維持することができる。微生物の作用は、たとえばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどの各種抗細菌剤および抗真菌剤によって防ぐことができる。多くの場合、たとえば糖、マニトールなどのポリアルコール、ソルビトール、塩化ナトリウムなどの等張剤を前記化合物に含めることが望ましいだろう。注射可能な組成物の吸収を持続させるために、たとえばモノステアリン酸アルミニウムおよびゼラチンなどの吸収遅延剤を組成物に含ませてもよい。
【0090】
滅菌された注射可能な溶液は、適切な溶媒中に入れた必要量の前記活性化合物を、必要に応じて上述の成分1つまたはそれらの組み合わせに組み入れてからろ過滅菌することによって調製することができる。一般に、分散液は、塩基性分散媒および上述の物質のうちの必要なその他の成分を含む滅菌担体に前記活性化合物を組み入れて調製する。滅菌注射用溶液の調製のための滅菌粉末の場合、好ましい調製方法は、前記活性成分の粉末に加えて予め滅菌ろ過されたその溶液から得られた任意の追加の望ましい成分を得られる、真空乾燥および凍結乾燥である。
【0091】
経口組成物には一般に、不活性な希釈剤または食用担体が含まれる。それらは、ゼラチンカプセルに封入されるか、または錠剤に圧縮されてよい。治療用の経口投与の場合、前記活性化合物を賦形剤に組み込み、錠剤、トローチ、またはカプセルなどの形状で用いることができる。経口用組成物はまた、洗口液として用いるための液状担体であって、当該担体において前記液状担体中の前記化合物が口腔内の塗布され、うがいして吐き出すかまたは飲み込まれる、液状担体を用いて調製することもできる。薬学的に適合な結合剤、および/またはアジュバント物質は、前記組成物の一部分として含めることもできる。前記錠剤、丸剤、カプセル、およびトローチなどは、以下の成分、もしくは同様の性質の化合物のいずれかを含むことができる。それは、微結晶性セルロース、ガムトラガカント、もしくはゼラチンなどの結合剤、デンプンもしくはラクトースなどの賦形剤、アルギン酸、プリモゲル、もしくはトウモロコシデンプンなどの崩壊剤、ステアリン酸マグネシウムもしくはステロート(Sterote)などの潤滑剤、コロイド状二酸化ケイ素などの流動促進剤、ショ糖もしくはサッカリンなどの甘味剤、またはペパーミント、サリチル酸メチル、もしくはオレンジ香味料などの香味剤である。
【0092】
吸入による投与の場合、前記化合物は、たとえば二酸化炭素などの好適な高圧ガスを含む加圧容器もしくはディスペンサ、またはネビュライザからのエアロゾルスプレーの形状で送達する。
全身投与はまた、経粘膜または経皮手段でもよい。経粘膜または経皮投与の場合、透過させようとする関門に適切な浸透剤を、前記製剤に用いる。そのような浸透剤は一般に当業に知られており、たとえば経粘膜投与の場合、界面活性剤、胆汁塩、およびフシジン酸誘導体が含まれる。経粘膜投与は、経鼻スプレーまたは坐剤の使用によって実施することができる。経皮投与の場合、前記活性化合物は、一般に当業に知られる軟膏(ointment)、軟膏(salve)、ゲル、またはクリームに調製する。
【0093】
前記化合物はまた、坐剤(たとえばカカオバターおよびその他のグリセリドなどの従来の坐剤ベース)または直腸送達用の停留浣腸の形状に調製してもよい。
【0094】
ある実施態様では、前記活性化合物は、インプラントおよびマイクロカプセル封入送達システムを含む、制御放出製剤などの、体外への急速な排出から前記化合物を保護する担体とともに調製する。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸などの生分解性、生体適合性ポリマーを用いることができる。そのような製剤の調製の方法は当業に明らかであろう。前記物質はまた、アルザ社およびノバファーマシューティカルズ社から市販されており入手することもできる。リポソーム懸濁液(ウイルス性抗原に対するモノクローナル抗体を有する感染細胞を標的とするリポソームを含む)はまた、薬学的に許容な担体として用いることもできる。これらは、米国特許第4,522,811号などに記載の、当業に知られる方法に従って調製することができる。
【0095】
投与の簡便性と製剤の均一性のために、経口または非経口組成物を投与単位剤形に調剤することは、特に有利である。本願明細書に用いられる投与単位剤形は、治療される対象に単位投与量として好適な物理的に分散した単位であって、必要な薬学的担体とともに望ましい治療効果を生じるように計算された、予め決められた量の活性化合物を含む各単位を意味する。本願発明の投与単位剤形の仕様は、前記活性化合物に特有の性質および達成されるべき特定の治療効果、ならびに個体の治療用の当該活性化合物を調合する技術に固有の限界によって支配され、およびそれらに依存する。
【0096】
当該化合物の毒性および治療上の有効性は、たとえばLD50(個体群の50%に致死的な投与量)およびED50(個体群の50%に治療上有効な投与量)など、細胞培養物または実験動物における標準的な薬学的方法によって決定することができる。毒性作用および治療上有効な作用の投与量の比は治療指数であって、LD50/ED50の比で表すことができる。大きい治療指数を示す化合物が好ましい。有毒な副作用を示す化合物を用いる場合、感染していない細胞に与える可能性のある損傷を最小にして副作用を減少するよう、そのような化合物を罹患組織の部位に狙わせる送達システムをデザインするように、注意が払われなければならない。
【0097】
細胞培養アッセイおよび動物研究から得られるデータは、ヒトにおける使用のための用量の範囲を決定する際に用いることができる。そのような化合物の用量は、好ましくは毒性が小さいかまたは皆無のED50を含む循環濃度の範囲内にある。前記用量は、用いられた投与剤形および使用された投与経路によって、この範囲内で変化させてもよい。本願発明の方法に用いられるいずれの化合物の場合も、治療上有効な投与量は、最初は細胞培養アッセイで推定することができる。1回投与量は、動物モデルにおいて、細胞培養物で決定したIC50(すなわち症状の最大半減抑制を達成する前記テスト化合物の濃度)を含む循環血漿濃度の範囲になるように調製してもよい。このような知見は、ヒトにおける有用な投与量をより正確に決定するために用いることができる。血漿濃度は、たとえば高性能液体クロマトグラフィによって測定することもできる。
【0098】
薬学的組成物は、投与法の説明書とともに、容器、パック、またはディスペンサに包含されていてよい。
【0099】
VIII. 治療方法
本願発明はまた、処置方法または治療方法も特徴とする。ある実施態様では、本願発明は、本願発明にしたがって同定された調節性化合物で、望ましい治療効果が実現できるように、対象(たとえばその必要のあるヒト対象)を処置する方法を特徴とする別の実施態様では、前記方法は、本願明細書に記載の方法論したがって同定された調節性化合物を、望ましい治療効果が実現できるように、前記対象から単離した組織または細胞株に投与するステップに関与する。好ましい実施態様では、本願発明は、たとえばインスリンへの低感受性もしくはインスリン耐性、または糖尿病(2型糖尿病)など、インスリン応答障害を有する対象を処置する方法を特徴とする。本願発明はまた、前糖尿病もしくはその症状、高血糖、および/または1型糖尿病を有する対象を処置する治療方法も提供する。好ましい治療効果はグルコース取り込みおよび/または輸送の調節である。望ましい治療効果にはまた、前記対象の治癒または治療、前記対象における疾患もしくは障害、または前記対象における前記疾患もしくは障害の少なくとも1つの症状の緩和、軽減、改変、もしくは改善、または前記対象の健康のその他の改善または作用が、それらに限定されずに含まれる。本願発明の好ましい局面は、グルコース輸送に作用するとして本願明細書に提供される方法によって同定される遺伝子を調節する方法、またはグルコース輸送またはインスリンの応答性に作用する治療目的の既知の遺伝子に関連する。
【0100】
本願明細書に開示された方法によって同定されたモジュレータを対象に用いて、インスリン応答の調節、グルコース輸送の制御、糖新生の制御、グルコース恒常性の制御、および血糖値の制御を行うこともできる。
【0101】
インスリン応答モジュレータでの対象の処置の有効性は、(i)好適なモジュレータで処置する前に、前記対象においてインスリン応答または代替的にはグルコース耐性のレベルを検出するステップと、(ii)前記モジュレータで処置後の前記対象におけるインスリン応答または代替的にはグルコース耐性のレベルを検出するステップと、(iii)投与前および投与後のレベルを比較するステップと、(iv)前記対象への前記モジュレータの投与を適宜変更するステップと、によって達成することができる。たとえば、前記対象が非感受性のインスリン応答を示し続ける場合、前記モジュレータの大量投与が望ましい場合がある。
【0102】
代替的には、インスリン応答モジュレータでの対象の処置の有効性は、(i)好適なモジュレータで処置する前に、前記対象において血糖またはグルコース耐性を検出するステップと、(ii)前記モジュレータで処置後の前記対象における前記血糖値または代替的にはグルコース耐性のレベルを検出するステップと、(iii)投与前および投与後のレベルを比較するステップと、(iv)前記対象への前記モジュレータの投与を適宜変更するステップと、によって実現することができる。たとえば、前記対象が十分に血糖を排除できない場合、前記モジュレータの大量投与または持続的投与が望ましい場合がある。
【0103】
IX. ノックアウトおよび/またはノックダウン細胞または生物
本願発明のsiRNA分子(またはベクターもしくはそれをコードする導入遺伝子)のさらなる好ましい使用は、真核細胞、または真核性非ヒト生物、好ましくは哺乳類細胞または生物、および最も好ましくはヒト細胞において行われる機能性分析である。標的特異的RNA干渉を方向付けるために、標的mRNA配列に十分相補的な、好適なsiRNA分子を投与することによって、脂肪細胞などの細胞培養物、または標的生物などの標的細胞において、特定のノックアウトまたはノックダウン表現型を得ることができる。
【0104】
したがって、本願発明のさらなる対象物質は、たとえば脂肪細胞などの真核細胞、または少なくとも1つの内因性標的遺伝子の完全なまたは少なくとも部分的に不足した発現を含む標的遺伝子特異的ノックアウトまたはノックダウン表現型を示す非ヒト真核性生物であって、前記細胞または生物が前記標的遺伝子の発現を阻害する能力があるsiRNA分子をコードするDNAを含む少なくとも1つのベクタをトランスフェクト(たとえば電気穿孔法によって)された、細胞または生物である。本願発明は、前記siRNAの特異性による数種類の異なる内因性遺伝子の標的特異的なノックアウトまたはノックダウンを可能にするという点は特筆されるべきである。
【0105】
細胞または非ヒト生物、特にヒト細胞または非ヒト哺乳類の遺伝子特異的ノックアウトまたはノックダウン表現型を、たとえば、遺伝子発現プロフィールおよび/またはプロテオームの分析など、複雑な生理学的プロセスの機能性および/または表現型分析など、分析手順に用いてもよい。好ましくは、前記分析は、オリゴヌクレオチドを用いたチップを使用したハイスループット方法によって行われる。
【0106】
RNAiを用いたノックアウトまたはノックダウン技術を使用して、内因性標的遺伝子の発現を、標的細胞または標的生物において阻害することもできる。前記内因性遺伝子は、たとえばアフィニティタグ、特に多重アフィニティタグなどの検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列に、任意で融合することができる、遺伝子またはDNAなど、前記標的タンパク質または標的タンパク質の変異体もしくは突然変異型をコードする外因性標的核酸によって補完されてもよい。
【0107】
前記標的遺伝子の変異体または突然変異型は、前記内因性遺伝子産生物と、1アミノ酸または複数のアミノ酸の置換、挿入、および/または削除によるアミノ酸レベルで異なる遺伝子産生物をコードする点において、前記内因性標的遺伝子と異なる。前記変異体または突然変異型は、前記内因性標的遺伝子と同一の生物学的活性を有する場合もある。その一方で、前記変異体または突然変異型標的遺伝子は、たとえば部分的に欠失した活性、相補的に欠失した活性、高い活性など、内因性標的遺伝子の生物学的活性と異なる生物学的活性も有していてよい。補完は、たとえば前記標的タンパク質、および前記アフィニティタグ、ならびに前記標的細胞中の内因性遺伝子をノックアウトするための2本鎖RNA分子など、前記内因性核酸によってコードされたポリペプチドを含むことによって、達成することもできる。この加圧は、たとえば前記タグ修飾標的タンパク質などの内因性核酸によってコードされたポリペプチド、および前記2本鎖RNA分子の両方を発現する好適な発現ベクタを用いて、または代替的には発現ベクタの組み合わせを用いることによって達成することもできる。前記標的細胞においてde novoで合成されたタンパク質およびタンパク質複合体は、たとえば修飾された融合タンパク質など、内因性遺伝子産生物を含有するだろう。前記siRNAi分子による外因性遺伝子産生物の抑制を避けるために、前記外因性核酸をコードする核酸配列を、前記siRNA分子に相同的な配列の一部分においてDNAレベルで(アミノ酸レベルでの突然変異を生じて、または生じずに)変化させてもよい。代替的には、前記内因性標的遺伝子は、たとえばマウスなどその他の種から得た対応するヌクレオチド配列によって補完させることもできる。
【0108】
使用
本願明細書に記載の方法は、発現が脂肪細胞の発達または代謝に影響を与える遺伝子を同定するために有用である。さらに、特定の遺伝子をノックダウンする方法を用いることによって、細胞上のそのようなノックダウンの作用は、薬物のスクリーニングに有用であると特徴づけることができる。たとえば、脂肪細胞で発現するタンパク質の発現または活性に特異的に影響する薬物の望ましい作用は、前記タンパク質をコードする配列がノックダウンされた脂肪細胞を調べることによって、予測することができる。そのような細胞の表現型は、前記標的遺伝子の発現または活性を阻害するために有用な薬物の特徴を同定するための指針を提供する。
【実施例】
【0109】
例1材料および方法
材料
ヒトインスリンはイーライリリー社から入手した。ヤギポリクローナル抗Akt1抗体(C末端sc-7126近傍の抗原性ヒトAkt1ペプチド)、HRP結合ロバ抗ヤギIgG、マウスモノクローナル抗ラミンA/C (sc7293)、およびモノクローナルGSK3α/βは、サンタクルズバイオテク社(カリフォルニア州サンタクルズ)から入手した。ウサギポリクローナル抗Akt2抗体(ヒトAkt2のC末端にある抗原性ペプチド)は、Morris J. Birnbaum博士(ペンシルバニア大学、Hill et al.,
1999, Mol. Cell. Biol. 19:7771-7781参照)に提供していただいた。Acrp30に対するウサギポリクローナル抗体は、アフィニティ・バイオリエージェント社(コロラド州ゴールデン)から、および非筋肉ミオシンIIBに対する抗体はコバンス社(カリフォルニア州リッチモンド)から入手した。ホスホAktスレオニン308/309、ホスホGSK3α/β(ser21/9)、およびホスホErk1/2 に対するポリクローナル抗体は、セル・シグナリング・テクノロジー社(マサチューセッツ州ベバリー)から入手した。フルオレセインイソチアネート(FITC)結合ヤギ抗マウス抗体はバイオソース・インターナショナル社(カリフォルニア州カマリロ)から入手した。
【0110】
siRNA 2本鎖のデザイン及び合成
RNAオリゴヌクレオチドの21マー・センスおよびアンチセンス鎖を、Elbashir et al.,
2001, Nature 411:94に記載の通りデザインした。RNAオリゴヌクレオチドは、核酸合成器で合成され、ヌクレアーゼ分解を防ぐために2’オルソエステルで保護し、その後、ダーマコン・リサーチ社におけるアニオン交換HPLCで精製した。それから、RNAオリゴヌクレオチドのセンスおよびアンチセンス鎖を、2’脱保護し、アニーリングして、凍結乾燥させ、siRNAを作成した。そのsiRNA 2本鎖を75%エタノールで2回洗浄して脱塩し、Speed-Vacを用いて乾燥させ、そのペレットを無ヌクレアーゼ水に再懸濁させてから培養した脂肪にトランスフェクションした。
【0111】
3T3L1脂肪細胞の細胞培養および電気穿孔
3T3-L1線維芽細胞を、10%ウシ胎仔血清、50μg/mlストレプトマイシン、および50ユニット/mlペニシリンを補充したDMEM培地で成長させた。それは、Harrison et al. (1990,
J. Biol. Chem. 265:20106)に既報の通り、脂肪細胞に分化した。3T3-L1脂肪細胞に、電気穿孔法でsiRNA 2本鎖をトランスフェクトした。簡単に説明すると、前記脂肪細胞の分化5日目に、0.25%トリプシンおよび1mlあたり0.5mgのコラゲナーゼのリン酸バッファ食塩水(PBS)溶液を加えた培養皿から剥離させて、2回洗浄し、PBSに再懸濁させた。その後、約500万個の細胞(p150培養皿1枚の半分の細胞)を、バイオラドジーンパルサIIシステム(バイオラドラボラトリーズ社、カリフォルニア州ハーキュリーズ)で、0.18kV、電気容量960μFの設定で、電気穿孔のパルスによって前記細胞に移したsiRNA 2本鎖と混合した。前記電気穿孔は、細胞450μlを用いて行われた。電気穿孔後、細胞をただちに新鮮培地と混合し、37℃で10分間インキュベートしてからデオキシグルコース取り込みアッセイ、ウエスタンブロッティング、および免疫蛍光顕微分析法のためにデザインされた、多穴プレートに再播種した。分析は一般に、再播種から24または48時間後に行った。
【0112】
免疫蛍光顕微鏡検査
ラミンA/Cを可視化するために、細胞を4%ホルムアルデヒドで固定して、1% FBSおよび0.5%トリトンTMX-100を含有するPBSで透過化した。その後、細胞を1次マウス抗ラットラミンA/C抗体で4℃で一晩インキュベートした。洗浄後、前記細胞をFITC標識ヤギ抗マウスIgGで30分間、室温でインキュベートした。洗浄後、2.5%DABCOを含有する90%グリセロール中でカバースリップをのせた。蛍光顕微鏡検査を、CCDカメラ(ロパー・サイエンティフィック社ニュージャージー州トレントン)およびメタモルフ画像処理ソフトウェア(ユニバーサルイメージング社、ペンシルバニア州ダウニングトン)を装着したIX70倒立顕微鏡(米国オリンパス社、ニューヨーク州メルビル)で行った。
【0113】
ウエスタンブロッティング法
実験的な処理の後、前記細胞を25mM Hepes、pH7.4, 1% NP40、100 mM NaCl、5 mM フッ化ナトリウム、1 mM EDTA、1 mM バナジウム酸ナトリウム、5 mM ピロリン酸ナトリウム、1 mM フッ化フェニルメチルスルホニル(PMSF)、10 μg/ml アプロチニン、および5 μg/ml ロイペプチンに溶解した。Aktスレオニン308/309、GSK3α/βセリン21/9のリン酸化、およびErk1/2のチロシンリン酸化を検出するために、3T3-L1 脂肪細胞ライセートから得た50μgタンパク質を8% SDS-PAGEに溶解し、ニトロセルロース膜に電気移動させた。その後、その膜を抗ホスホ特異的抗体(1:1000希釈)で、4℃で一晩インキュベートしてから、西洋ワサビペルオキシダーゼ(HRP)結合抗ウサギIg抗体(1:10,000希釈)中で、室温で1時間インキュベートした。その膜を、各抗体でインキュベートした後、水バッファ(PBS pH 7.4、0.1% ツイーン 20)で室温で1時間、洗浄した。最後に、セリンリン酸化Aktの量をECLTM キットを用いて検出した。Akt1を、1次ヤギポリクローナル抗体(1:750希釈)および2次HRP結合ロバ抗ヤギ抗体で検出した。Akt2に対する1次ウサギポリクローナル抗体(1:1000希釈)、非筋肉ミオシンIIB (0.1 μg/ml)およびAcrp30 (0.5 μg/ml) を、全細胞ライセートから得たタンパク質25μgを用いてその抗原を検出するために使用した。同じニトロセルロース膜を用いて数種類のタンパク質およびホスホタンパク質を検出するために、前記のブロットをストリッピングバッファ(62.5 mM トリス-HCl、pH 6.7、100 mM 2-メルカプトエタノール、および2% SDS)中で、30乃至45分間、60℃で緩やかに振とうさせてインキュベートし、洗浄用バッファで少なくとも1時間洗浄してから、次の実験用にデザインされた抗体で再ブロッティングした。
【0114】
2-デオキシグルコース取り込みアッセイ
3T3-L1 脂肪細胞におけるインスリン刺激グルコース輸送を、2-デオキシグルコース取り込みを測定することによって推測した。簡単に説明すると、siRNAトランスフェクト細胞を12穴プレート上に再播種し、40時間培養してから、DMEM培地で2回洗浄して、0.5%ウシ血清アルブミン(BSA)を含有するDMEM培地で37℃で4時間インキュベートした。それから、細胞をクレブス-リンガーHepes(KRH)バッファ(130 mM NaCl、5 mM KCl、1.3 mM CaCl2、1.3 mM MgSO4、25 mM Hepes [pH
7.4])で2回洗浄し、さらに、0.5% BSAおよび2 mMピルビン酸ナトリウムを捕捉したKRHバッファ中で1.5時間飢餓状態にした。それから、細胞をインスリンで37℃で30分間刺激した。グルコース取り込みは、[1,2-3H]
2-デオキシ-D-グルコース の添加で開始し、37℃で5分間かけて最終アッセイ濃度100μMにした。アッセイは氷冷KRHバッファで4回洗浄して終了し、前記細胞を0.4mlの1%TritonTM
X-100で可溶化し、トリチウムの取り込み量をシンチレーション計数で測定した。非特異的なデオキシグルコース取り込みを20 μMシトカラシンBの存在下で測定し、それぞれの測定値から差し引いて特異的な取り込みを得た。
【0115】
Myo1cのsiRNA誘導分解
スクランブルしたsiRNA 6 もしくはsiRNA 6 単体のいずれかを20nmol、またはsiRNA2およびsiRNA 6それぞれ10nmolずつを、分化した3T3-L1 脂肪細胞に電気穿孔して挿入した。電気穿孔後、前記細胞を24穴プレートに再播種し、24時間安置した。その後、グルコース取り込みを測定した。スクランブルsiRNAトランスフェクト細胞中の基本取り込み量を1に正規化して対照として用い、増加倍数を計算した。対応のあるt検定を用いて、siRNA 6およびsiRNA2の混合物の存在下に対する、siRNA 6単体の存在下における2-デオキシグルコース取り込みの減少率を求めた。これらの細胞の一部分は、Myo1c、myosin 2bおよびEHD2について、ウェスタンブロット法で分析した。
【0116】
例2:脂肪細胞へのsiRNAの導入
最初の実験を行ったところ、他の細胞種においてsiRNA媒介遺伝子サイレンシングのために開発された条件(Elbashir et al.,
2001, Nature 411:94) は3T3-L1 線維芽細胞では有効に働くが(図1B、右図)、3T3-L1脂肪細胞では有効に働かないことが実証された。 オリゴフェクタミンTMなどのその他の細胞種において有効なトランスフェクションの他の手段では、脂肪細胞に対して相当の毒性を示した。
【0117】
siRNAを用いた技術を脂肪細胞に応用するために、代替的な方法論を開発した。前記技術の開発には、例1に記載の方法を用いて、3T3-L1 脂肪細胞の電気穿孔およびマウスラミンA/Cを標的としたCy3タグsiRNA
2本鎖(図 1A; センスおよびアンチセンス鎖、それぞれ配列番号 1および2、配列番号19のマウスラミンA/Cを標的とする)を用いた。この新規の技術を用いて、Cy3-siRNAを培養した脂肪細胞に実質的に100%の効率で導入し、48時間後までに、スクランブルCy3タグsiRNA種をトランスフェクトした細胞と比較して、ほぼすべての細胞が核ラミンA/Cの喪失を示した(それぞれ配列番号3および4のセンスおよびアンチセンス鎖)(図1B、左図)。これらの結果を定量化したところ、20nmolのsiRNAを5 x 10 6 個の脂肪細胞の懸濁液に加えると、毒性が検出されない約90%の脂肪細胞においてラミンA/Cが喪失する結果となることが示された。これらの知見は、インスリン感受性培養脂肪細胞において毒性が低く信頼性があり有効な遺伝子サイレンシングの方法の基礎を提供する。
【0118】
前記培養物中の実質的にすべての細胞は、siRNAに方向付けられた遺伝子サイレンシングに応答した目的のタンパク質の低発現を示す。これは、脂肪細胞中にプラスミドベクタをトランスフェクトした後のcDNAコードタンパク質の発現とは異なる。この場合、ほとんどの場合において1乃至10%の細胞しか非相同性のタンパク質を発現しない。したがって、この方法を用いたsiRNAに方向付けた遺伝子サイレンシングは、1細胞アッセイが困難な機能について特定の遺伝子産生物の役割を評価するために有用である。ある例はインスリン刺激グルコース輸送である。siRNAなどの核酸を脂肪細胞に導入する新規方法は、たとえばmycタグGLUT-GFPの使用で評価したGLUT4トランスロケーションなどの1細胞アッセイと併用することができる。
【0119】
例3:Akt発現の阻害
核酸を脂肪細胞に導入する新規の方法をさらに調べるために、Aktイソ型であるAkt1およびAkt2のそれぞれに対する2つのsiRNA種を、3T3-L1 脂肪細胞におけるこれらタンパク質キナーゼの発現を阻害する能力についてテストした(図3A乃至B)。Akt1およびAkt2 siRNA 2本鎖は、国立バイオテクノロジー情報センターのデータベースから得たマウスAkt1およびAkt2 mRNA 配列(それぞれ配列番号20および21)にしたがってデザインした。
【0120】
Akt1を標的としたsiRNAは、図2に記載のAkt1の2種類の標的配列“akt1a”および“akt1b”(5’-AACGAUGGCACCUUUAUUGGC; 配列番号 5 および5’-AACCAGGACCACGAGAAGCUG; 配列番号 6)のうちの1つを標的とした。siRNA標的配列“akt1a” のアンチセンス鎖は配列5’-
GCCAAUAAAGGUGCCAUCGdTdT (配列番号 9)を有し、siRNA標的配列“akt1b” のアンチセンス鎖は配列5’- CAGCUUCUCGUGGUCCUGGdTdT (配列番号 10)を有した。Akt2を標的としたsiRNAは、図2に記載のAkt2の2種類の標的配列“akt2a”および“akt2b”(5’-AAACUCCUCGGCAAGGGCACC; 配列番号 7 および5’-AACCAGGACCACGAGCGCCUC; 配列番号 8)のうちの1つを標的とした。siRNA標的配列“akt1a” のアンチセンス鎖は配列5’-
GGUGCCCUUGCCGAGGAGUdTdT (配列番号 11)を有し、siRNA標的配列“akt1b” のアンチセンス鎖は配列5’- GAGGCGCUCGUGGUCCUGGdTdT (配列番号 12)を有した。
【0121】
これらの配列を標的としたsiRNAを、例1に記載の細胞に導入した。Akt1に方向付けられたsiRNA種はそれぞれ、トランスフェクションから24および48時間後の両方の時点でAkt1の発現を阻害した。これらの1つ(akt1b)は、48時間経過時点までに実質的に完全なAkt1の除去を方向付けているが、Akt2の発現には影響はなかった(図3B、4AおよびB)。同様に、Akt2の発現は、siRNA種のakt2bのトランスフェクション後、選択的に約70%低下させることができたが、akt2a siRNAの作用は小さかった(図3B、4Aおよび4B)。最も作用の大きかったakt1bおよびakt2b siRNA種は、Akt1およびAkt2 mRNA 配列の類似の領域を標的とした。これらの配列はAkt1およびAkt2において、それぞれアミノ酸351乃至357 および352乃至358をコードしている(図3A)。それぞれの標的mRNAをサイレンシングするための、akt2b siRNAに対するakt1bの選択性は、21ヌクレオチド中4ヌクレオチドが異なるだけでも明らかである(図1A)。これは、これらのsiRNAをこれらの類似しているが異なる配列を標的用に使用することの特異性を実証している。数種類のその他の無関係なタンパク質(たとえば図3、4A、および6Aに示されているミオシンIIb、および図4Aおよび6Aに示されているAcrp30)の発現には、akt1bまたはakt2b siRNAは影響を与えなかった。このことからも、さらに前記方法の特異性が実証される。
【0122】
Akt1およびAkt2は、活性Tループのスレオニン308または309にあるタンパク質キナーゼPDK1でリン酸化して活性化する。そしてさらなる活性化はセリン473(Akt1)または474(Akt2)におけるリン酸化によって生じる(Alessi et al., 1997, Curr. Biol. 7:776;
Williams et al., 2001, Curr. Biol. 10:439; Brazil et al., 2001, Trends Biochem.
Sci. 26:657)。両方のタンパク質に含有される完全スレオニン308/309 のインスリン刺激リン酸化への、Akt2に対するAkt1の選択的喪失の効果は、抗ホスホThr308抗体を用いたウェスタンブロッティングによって評価した。Akt1タンパク質の完全喪失によって、培養3T3-L1脂肪細胞中のAktタンパク質キナーゼの完全Thr308リン酸化において、10乃至20%しか減少せず、脂肪細胞においてAkt1の量がAkt2よりもはるかに少ないということを示している既報の結果と一致する(Hill et al., 1999, Mol. Cell. Biol.
19:7771; Summers et al., 1999, J. Biol. Chem. 274:23858; Calera et al., 1998,
J. Biol. Chem. 273:7201)。反対に、約70%のAkt2発現の減少により、Aktタンパク質キナーゼのインスリン刺激スレオニンリン酸化が55乃至60%と顕著に減少した。これらをもとに、これらのデータから、インスリン感受性培養脂肪細胞においてAkt1よりもAkt2のほうが優位であることが確認された。
【0123】
Akt1またはAkt2発現の選択的低下がインスリンシグナル伝達の下流標的であるグリコーゲンシンターゼキナーゼ(GSK)3に与える影響(Cross et al.,
1995, Nature 378:785; Cohen et al., 2001, Nat. Rev. Mol. Cell. Biol. 2:769) を評価した。GSK3αは、投与量依存的に前記ホルモンに応じてAktタンパク質キナーゼによってリン酸化されると考えられている。3つの独立した実験において、akt1b siRNAによって方向付けられているAkt1の
95%以上が失われても、インスリン媒介GSK3αリン酸化の有意な低下は生じなかったが(図5Aおよび5B)、これらの研究において10乃至20%の作用は検出されていないかもしれない。反対に、Akt2発現が約70%低下すると、インスリン媒介GSK3αリン酸化の約40%の阻害を生じた(図5A、5B、6Aおよび 6C)。対照研究では、Akt1またはAkt2のいずれかが減少しても、MAPキナーゼErk-1およびErk-2 へのインスリンシグナル伝達の低減は認められず(図5Cおよび5D)、この方法によるAktタンパク質キナーゼのサイレンシング作用の特異性が確認された。これらのデータは、培養脂肪細胞中のGSK3αへのインスリンの作用は特異的にAkt2を要することを示している。したがって、Akt2の発現または活性の阻害を用いて、インスリン媒介GSK3αリン酸化を阻害することができる。
【0124】
この同一のアプローチをヘキソース輸送の調節に応用したところ、Akt1発現を除去すると(図6A)、3T3-L1 脂肪細胞におけるインスリン刺激2-デオキシグルコース取り込みに、小さいが有意な20乃至30%の減少が生じた。Akt2タンパク質を正常値の約70%減少させたところ、インスリンの応答は50乃至58%低下した。これらのデータは、Akt1およびAkt2の両方とも、これらの細胞における完全活性化Aktへの寄与にだいたい比例して培養脂肪細胞におけるヘキソース輸送のインスリンの応答性に寄与することを示している。このような作用を、akt1bおよびakt2b siRNAの組み合わせを用いて3T3-L1脂肪細胞におけるAkt1およびAkt2の両方の減少についてテストした。この併用処置は実質的に完全にAkt1発現を除去し、Akt2発現を65%超低減させる一方で、抗ホスホスレオニン308/309 抗体によって検出された完全Aktのインスリン刺激リン酸化は81%低減した(図6A)。重要なことに、インスリン刺激デオキシグルコース取り込みは、このような条件下において約80%阻害されが、それに対し、Akt2だけを減少させた場合には約58%だった(図6C)。このような条件下において、GLUT4発現には変化がなかった。
【0125】
これらのデータは、Akt2はこの応答において主要なタンパク質キナーゼだが、Akt1も重要な役割を果たすことができることを実証している。したがって、インスリン刺激デオキシグルコース取り込みがAkt2の部分的な減少によって有意に障害される条件下では、残存するインスリンシグナルの半分にはAkt1が必要となる(図6C)。インスリンに応じたGSK3αリン酸化も、Akt2だけが減少した場合に対して両方のタンパク質キナーゼが減少した場合ではより大きく阻害された(図6Aおよび6C)。
【0126】
これらのデータは、たとえばAkt1またはAkt2の発現または活性が欠乏している場合、Akt1またはAktの発現または活性を上げるのはインスリン刺激グルコース取り込みを刺激するのに有用である。これはまた、糖尿病などの脂肪細胞関連疾患の治療の標的を同定するためにsiRNAを脂肪細胞に導入する方法の有用性を実証する。
【0127】
本願明細書に提示した知見は、グルコース輸送およびGSK3αへの正常なインスリンシグナル伝達へのAktタンパク質キナーゼの絶対的な要求を示しており、使用可能なAkt1とAkt2の合計はインスリン応答の程度に正比例していることを示唆している(図6)。この結論は、全Aktの活性化およびグルコース輸送に認められた類似のインスリン用量反応関係性と一致している。Akt1、Akt2または両方の進行的な喪失は、対応するグルコース輸送刺激の進行的な喪失を生じる(図6)。このような考察は、Akt1が部分的にAkt2に代わってグルコース輸送応答性を維持することができることを示している。したがって、Akt1の発現または活性の増大はグルコース輸送応答性の増大に有用である。
【0128】
次に、siRNAによるAkt1およびAkt2の両方の欠乏の組み合わせがインスリン媒介GLUT4に与える影響を、3T3-L1 脂肪細胞で調べた。myc-GLUT4-EGFP プラスミドDNAと、akt1bおよび akt2b siRNAの混合物のコトランスフェクションを行ったところ、48時間後のAkt1およびAkt2タンパク質レベルはそれぞれ約90%および65%の減少が認められた(図7B)。この、Akt1およびAkt2タンパク質の組み合わせノックダウンによって、Myc-GLUT4-GFP作製物をトランスフェクトした脂肪細胞の約70%のインスリン刺激細胞表面Myシグナル(抗Myc Abにより検出)が喪失した(図7AおよびC)。陽性トランスフェクト細胞における全Myc-GLUT4-GFP シグナルに対する細胞表面Myc rim シグナルの割合を定量化したところ、Akt1 およびAkt2の両方の喪失によって前記細胞表面上のインスリン刺激Myc-GLUT4-GFPが60%減少したことが明らかになった。GLUT4応答性の低下は、インスリンの最大濃度および亜最大濃度の両方において認められるが(図7C)、インスリンの欠如下においては、GLUT4転移にはAktの低下の有意な作用は検出されない。
【0129】
要約すると、これらのデータはインスリンのグルコース輸送への作用にはAktが絶対的に要求されていることを実証している。また、たとえばsiRNAなどの核酸を脂肪細胞に導入する方法の有効性を実証している。
【0130】
例4:インスリンシグナル伝達遺伝子のサイレンシング
インスリンシグナル伝達に特に関連する遺伝子が、培養脂肪細胞中のsiRNAでサイレンシングできるかどうかをテストするために、実験を行った。
【0131】
脂肪細胞ライセートのウェスタンブロット分析法を、Akt関連タンパク質キナーゼCISKのsiRNAに方向付けられた遺伝子サイレンシングにしたがって行った。CISKはN末端においてPHドメインではなくPXドメインを含んでいるが、前記タンパク質キナーゼドメインのAktと約50%の配列同一性を示す。CISKはCOS細胞ではIGF-1およびEGFによって活性化され、3T3-L1 脂肪細胞のCISKの活性化はGLUT4トランスロケーションを刺激することができる。したがって、オープンリーディングフレームの配列に対して方向付けられたsiRNAによってCISK発現を低下させる能力をテストした。そのような目的の遺伝子の場合、一般に、数種類の異なるsiRNA種をテスト用に調製する。CISKに対して方向付けられたsiRNA候補を、当業に知られる方法を用いてデザインし、本願明細書に記載の方法を用いてテストした。この実験は、CISKを標的とするそのような2種類のsiRNA種はCISKのサイレンシングに有効であるということを実証した。siRNA標的CISKは、標準技術を用いて合成した(ダーマコン社)。テストした両方のsiRNA CISK種は、トランスフェクションから48時間後のCISK発現の選択的減少において有効だった。24時間経過時点において有意な作用もあったが、48時間経過時点ほど完ぺきではなかった。培養脂肪細胞にラミンに対するsiRNAをトランスフェクトしても、トランスフェクトされなかった細胞と比較して、CISK発現の減少を生じなかった。
【0132】
もう一つの対照として、同様に脂肪細胞で大量に発現される、無関係なタンパク質EHD2のウェスタンブロッティングを行った。EHD2の発現では、CISKに方向付けられたsiRNAトランスフェクションの効果がわずかであるか全くないことが示された。
【0133】
同様に、特殊なミオシンMyolcに対する数種類のsiRNA種をテストした。siRNA(ダーマコン社)を、以下のMyo1c mRNA配列、スクランブル、 5’- CAGUCGCGUUUGCGACUGG (配列番号13)、siRNA 2, 5’-
AAGGCGUUGUACAGCCGGACAUU (配列番号14)、siRNA 6, 5’- AAGCUUCCAGACAGGGAUCCAUG (配列番号15)を標的とするようにデザインした。対応するMyo1c siRNAは、以下の配列、スクランブル、 5’- CCAGUCGCAAACGCGACUGdTdT
(配列番号 16)、siRNA 2, 5’-
UGUCCGGCUGUACAACGCCdTdT (配列番号17)、 および siRNA 6, 5’- UGGAUCCCUGUCUGGAAGCdTdT (配列番号18)を有するアンチセンス鎖を含んだ。
【0134】
トランスフェクションから24時間後、Myo1cタンパク質の量は2種類のsiRNA種の組み合わせをトランスフェクトした細胞において約40%減少し、1種類のsiRNA種をトランスフェクトした細胞では約25%減少した(図8A、B)。2種類のその他の対照タンパク質、ミオシン2bおよびEHD2の発現は、影響を受けなかった(図8A)。siRNA
6またはsiRNA 6およびsiRNA2の両方をトランスフェクトした脂肪細胞の、インスリン刺激2-デオキシグルコース取り込みは、両方とも1nMおよび100nMインスリンの濃度のスクランブルsiRNAをトランスフェクトした細胞と比較して、有意に阻害された。このような結果は、siRNAによるMyo1c発現の減少が、対応するインスリンへのGLUT4応答性の低下を生じることを示唆した。
【0135】
これらのデータは、siRNAに方向付けられた遺伝子サイレンシングを脂肪細胞において、たとえばインスリンシグナル伝達経路におけるタンパク質の機能を評価するための遺伝子発見、および薬物標的の確認のために用いることができるという、さらなる証拠を提供する。
【0136】
その他の実施態様
本願発明は、発明の詳細な説明に関連して説明されているが、上述の説明は具体例を示すことを意図しており本願発明の範囲を制限することは意図しておらず、それは添付の特許請求の範囲によって定義されると理解される。その他の局面、利点、および改善点は、添付の特許請求の範囲内である。
【図面の簡単な説明】
【0137】
【図1A】Cy3標識ラミンA/C およびスクランブルsiRNA 2本鎖の配列を示すチャート。ラミンA/C siRNA は、NCBIデータベースから得たマウスラミンA mRNA配列(受入番号 BC015302) (配列番号 19) にしたがってデザインされており、マウスESTデータベースにしたがってマウスラミンA/C に固有である。ラミンA/C siRNA は配列番号1のセンス鎖、および配列番号2のアンチセンス鎖を含む。マウススクランブルsiRNAは、センス鎖がマウスESTデータベースにおいて得られた任意の遺伝子に相同性を示さない、ランダム配列としてデザインされた。
【図1B】蛍光マイクログラフの一連の複製(1a、1b、2a、2b、3a、3b、4a、および4bとして標識)で、細胞500万個につき80nmolの濃度による電気穿孔によって、Cy3-標識マウスラミンA/CまたはスクランブルsiRNA 2本鎖をトランスフェクトした3T3-L1脂肪細胞(5日目)を示す。48時間再播種した後、ラミンA/Cの核膜局在化を、本願明細書に記載の通り、マウスラミンA/C に対する抗体を用いた免疫蛍光によって可視化した。
【図2】記載の濃度におけるCy3標識スクランブルsiRNAまたはラミンA/C siRNA を分化した脂肪細胞(5日目)に電気穿孔した実験の結果を示す棒グラフ。抗ラミンA/C抗体の免疫染色後、ラミンA/C染色をした細胞を計数した。データは3つの独立した実験の平均±SD を示す。
【図3A】Akt1およびAkt2タンパク質、Akt1およびAkt2 mRNA、ならびにAkt1 (配列番号5および6) およびAkt2 (配列番号 7および8)のsiRNA標的配列の模式図。
【図3B】24または48時間で示された、siRNAの導入後の脂肪細胞ライセートのウェスタンブロットのホホイメージ。ブロットは、Akt1、Akt2およびミオシンIIBについて染色した。Akt1およびAkt2 siRNA 2本鎖は、NCBIのデータベースから得たマウスAkt1およびAkt2 mRNA 配列(それぞれ受入番号NM_009652、配列番号20および受入番号U22445、配列番号21)にしたがってデザインした。Sc: スクランブル、A1a: akt1a siRNA、A1b: akt1b siRNA、A2a: akt2a siRNA、A2b: akt2b siRNA。
【図4A】電気穿孔によってsiRNA(40nmol siRNA 2本鎖/5 x 106細胞)をトランスフェクトし、42時間再播種し、6時間血清を欠乏状態にしてから15分間、37℃でインスリンで処理した3T3-L1 の脂肪細胞の脂肪細胞ライセートのウェスタンブロットのホスホイメージ。タンパク質の完全細胞ライセート50Tg を、Akt1およびホスホ-Thr308/309 Aktの検出に用いた。タンパク質25 Tgを、Akt2、非筋肉ミオシンIIBおよびAcrp30の検出に用いた。
【図4B】Akt1b、またはAkt2b、またはスクランブルsiRNAの導入後の、Akt1およびAkt2タンパク質量の棒グラフ。
【図4C】スクランブル、Akt1b、またはAkt2b siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のAktスレオニン308/309 リン酸化の棒グラフ。データは3つの独立した実験の平均±SD として示される。
【図5A】siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のホスホ-GSK3およびGSK3を検出するウェスタンブロットのホスフォイメージ。
【図5B】siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のホスホ-Erk1/2およびErk1/2 を検出するウェスタンブロットのホスフォイメージ。
【図5C】GSK3I タンパク質バンドの強度に対するホスホGSK3I バンドの強度の割合の測定によって定量化したGSK3I のリン酸化を示す棒グラフ。
【図5D】Erk1/2 タンパク質バンドの強度に対するホスホErk1/2 バンドの強度の割合の測定によって定量化したErk1/2 のリン酸化を示す棒グラフ。データは3つの独立した実験の平均±SD として示される。
【図6A】Aktタンパク質レベル、ホスホスレオニン308/309レベル、およびホスホセリン21-GSK3Iレベルの代表的なウェスタンブロット画像のホスフォイメージ。
【図6B】インスリン刺激デオキシグルコース取り込みの用量依存性を示す実験の棒グラフ。定量的なデータは4つの独立した実験の平均±SD として示される。
【図6C】100nMインスリン誘導グルコース取り込みの阻害、およびノックダウンAkt1およびAkt2タンパク質レベルによるおよびGSK3Iリン酸化を示す実験の棒グラフ。定量的なデータは3つの独立した実験の平均±SD として示される。
【図7A】GFP陽性細胞および顔面外Myc染色の代表的な画像。使用したインスリンの濃度は100nMであった。細胞の辺縁のMycシグナルの輝度が最大の細胞の中心部分に近い光学平面において、Myc染色およびGFPシグナルの画像を撮影した。
【図7B】Myc-Glut4-EGFP およびsiRNAを48時間トランスフェクトした脂肪細胞のAktタンパク質レベルを示すウェスタンブロット。
【図7C】細胞表面上のMyc-GLUT4-GFPを示すトランスフェクト脂肪細胞の割合を示す棒グラフ。データは3つの独立した実験であって、各実験において200個を超える細胞が計数された実験の平均±SD として示される。
【図7D】Myc-GLUT4-GFPを発現する脂肪細胞における合計GFPシグナルに対する細胞表面Mycシグナルの割合を示す棒グラフ。使用したインスリンの濃度は100nMであった。平均±SDとして示されたデータの比較は、対応のないスチューデントt検定を用いることによって行った。*, P < 0.001 はスクランブルsiRNAをトランスフェクトした細胞とakt1b/akt2b siRNAをトランスフェクトした脂肪の差。Scr スクランブルsiRNA、1b akt1b siRNA、2b akt2b siRNA。
【図8A】スクランブルsiRNA (レーン 1)、siRNA 6 (レーン 2)、またはsiRNAs 6 および2(レーン3)の発現後の、分化した3T3-L1 脂肪細胞中のMyo1、ミオシン2bおよびEHD2タンパク質の量を示すウェスタンブロット。
【図8B】スキャンニングデンシトメータを用いた、図8Aに示されるMyo1cタンパク質の定量を示す棒グラフ。
【図8C】スクランブルsiRNA、siRNA 6またはsiRNAs 6 および2のいずれかをトランスフェクトした3T3-L1脂肪細胞中の、インスリン刺激[3H]2デオキシグルコース取り込みを示す棒グラフ。* P < 0.0004、** P < 0.0001。
【配列表】
【図1】
【発明の詳細な説明】
【0001】
政府の権利
本願発明は、部分的にNIHが交付する政府助成金第DK30648号によって行われた。政府は本願発明の権利を有することができる。
【0002】
関連出願
本願明細書は、2002年12月11日に提出された米国特許仮出願第60/432,427号、表題「Method of Introducing siRNA
into Adipocytes(脂肪細胞へのsiRNAの導入方法)」の利益を主張する。上に引用された特許出願の内容全体は、引用をもって本願明細書に援用するものとする。
【0003】
背景
培養された脂肪細胞は、グルコース輸送へのインスリンの作用を研究するための主要なモデルシステムである。他に、グルコース輸送体GLUT4をこれだけ高濃度に発現し、グルコース輸送を増やすことによってこれだけ強くインスリンに応答する培養細胞種はない。インスリンのGLUT4へのこの作用は、2型糖尿病の中核を成す。なぜなら、脂肪および筋肉中のインスリン耐性がこの疾患を引き起こす主要な異常だからである。インスリンシグナル伝達経路の構成要素が発見されたことは、インスリン耐性を理解し、2型糖尿病の治療法のための薬物標的候補を提供する上で重要である。
【0004】
そのようなインスリンシグナル伝達経路の構成要素の発見は、この経路への関与が疑われるタンパク質をコードする特定の遺伝子の発現を削除することによって、促進される。脂肪細胞中の遺伝子またはタンパク質発現の特異的な削除を容易に行うことができる方法はない。これらの細胞は扱いが難しく、線維芽細胞などその他の細胞で作用する試薬では容易にトランスフェクトできない。
【0005】
グルコースの恒常性は、インスリンによる筋肉および脂肪細胞へのグルコース輸送の調節によって、一部制御されている。PI 3-キナーゼ下流のAktタンパク質キナーゼ(タンパク質キナーゼB (PKB)としても知られる)は、このインスリンシグナル伝達経路に関与していると考えられているが、Akt1-/- およびAkt2-/- マウスで得られた結果ははっきりしない。Aktは、インスリンシグナル伝達と、GLUT4輸送を調節する構成要素との、PI3キナーゼ依存性経路を介した連結に強く関与する下流エフェクタの1つである。このAktへの役割は、培養細胞での研究と遺伝子ノックアウトマウスモデルでの研究に基づいている(Kohn et al., 1996, J. Biol. Chem. 49:31372、Hill et al., 1999, Mol. Cell. Biol. 19:7771、Wang et al., 1999, Mol. Cell. Biol. 19:4008、Ueki et al., 1998, J. Biol. Chem. 273:5315、Cho et al., 2001 (a), Science 292:1728)。しかし、この重要な問題は見解が一致しておらず(Kotani et al.,
1998, Mol. Cell. Biol. 18:6971)、 Akt1イソ型が欠如しているマウスから得られた最近のデータでは、インスリンの感受性に低下は認められなかった(Cho et al.,
2001 (b), J. Biol. Chem. 276:38349)。筋肉と脂肪中に発現する主要なイソ型であるAkt2を欠損するマウスから採取した骨格筋は、低レベルのインスリンに対する感受性がやや低下しただけで、実際には最大量のインスリンには普通に応答する。これらのマウスに認められた糖尿病は、肝グルコース新生へのインスリン作用の劇的な減衰によるところが大きいと考えられている(Cho, 2001 (a),
supra)。
【0006】
要約
本願発明は、siRNAを脂肪細胞に導入する方法の発見に、部分的に基づく。前記方法は、Akt1、Akt2、CISK およびMyo1cを含む、いくつかの異なる配列を標的とするsiRNAを用いて実証される。
【0007】
したがって、本願発明には核酸を脂肪細胞に導入する方法が含まれる。前記方法には、脂肪細胞を得るステップと、前記脂肪細胞を核酸分子に接触させるステップと、混合物を形成するステップと、前記混合物を電気穿孔するステップと、が含まれる。前記核酸は、たとえばAkt1、Akt2、CISKまたはMyo1c核酸配列を標的とするsiRNAなどのsiRNAであってよい。 一般に、前記電気穿孔は、室温で、約0.18 kVおよび約960 μFの静電容量で行う。ある実施態様では、本願発明にはグルコース輸送に影響する遺伝子を同定する方法が含まれる。前記方法には、脂肪細胞を提供するステップと、上述の通り電気穿孔を用いて前記脂肪細胞に前記遺伝子を標的とするsiRNAを導入するステップと、前記標的遺伝子の発現に好適な条件下において前記細胞を培養するステップと、グルコース輸送の減少が前記標的遺伝子がグルコース輸送を示唆するように、前記細胞中のグルコース輸送をアッセイするステップが含まれる。
【0008】
別の実施態様では、本願発明には細胞におけるAkt1またはAkt2の発現もしくは活性を阻害する方法が含まれる。前記方法には、Akt1またはAkt2を発現する細胞を得るステップと、前記細胞にAkt1核酸配列またはAkt2核酸配列を標的とするsiRNAを導入するステップが含まれる(たとえば図3Aに記載の通り)。ある実施態様では、前記細胞は脂肪細胞である。ある局面では、Akt1およびAkt2の両方の発現または活性を前記方法を用いて阻害する。この局面では、Akt1を標的とするsiRNAおよびAkt2を標的とするsiRNAを前記細胞に導入することもできる。代替的には、Akt1およびAkt2の両方を標的とするsiRNAを前記細胞に導入することもできる。
【0009】
前記発明には、インスリン媒介GSK3リン酸化を阻害する方法も含まれる。前記方法には、インスリン選択性細胞を得るステップと、前記細胞をAkt2発現または活性を阻害する作用物質と接触させるステップとが含まれる。ある局面では、前記作用物質はAkt2を標的とするsiRNAである。
【0010】
別の実施態様では、本願発明はヘキソース輸送を阻害する方法に関連する。前記方法には、ヘキソース輸送を行うことができる細胞を得るステップと、前記細胞をAkt1、Akt2またはその両方の発現または活性を特異的に阻害する作用物質と接触させるステップとが含まれる。ある実施態様では、Akt1またはAkt2の発現もしくは活性、およびヘキソース輸送を特異的に阻害する作用物質を部分的に阻害する。前記作用物質は、Akt1、Akt2またはその両方を特異的に標的とするsiRNAであってよい。
【0011】
ある局面では、本願発明はグルコース輸送に影響する遺伝子を同定する方法であって、(a) 脂肪細胞を提供するステップと、本願明細書に提供される方法を用いて前記脂肪細胞に前記遺伝子を標的とするsiRNAを導入するステップと、(b) 前記標的遺伝子の発現に好適な条件下において前記細胞を培養するステップと、(c)前記細胞中のグルコース輸送をアッセイするステップであって、当該ステップにおいてグルコース輸送の減少が前記標的遺伝子がグルコース輸送を示唆し、それによりグルコース輸送に影響する遺伝子を同定するためのステップと、が含まれる方法を提供する。
本願発明はまた、インスリン応答モジュレータを同定する方法であって、(a) グルコース輸送に影響するタンパク質を発現する細胞をテスト化合物と接触させるステップであって、当該ステップにおいて前記タンパク質が本願明細書に提供される方法に従って同定された遺伝子によってコードされるステップと、(b)インスリン応答モジュレータが同定されるように、前記タンパク質の活性を調節するテスト化合物の能力を決定するステップと、が含まれる方法も提供する。
【0012】
関連する局面では、本願発明はインスリン応答モジュレータを同定する方法であって、(a) グルコース輸送に影響するタンパク質を発現する細胞をテスト化合物と接触させるステップであって、当該ステップにおいて前記タンパク質が本願明細書に提供される方法に従って同定された遺伝子によってコードされるステップと、(b)インスリン応答モジュレータが同定されるように、前記タンパク質の発現を調節するテスト化合物の能力を決定するステップと、が含まれる方法も提供する。
【0013】
さまざまな実施態様において、前記モジュレータは陽性モジュレータまたは陰性モジュレータである。本願発明はさらに、本願発明の方法によって同定されるモジュレータを特徴とする。
【0014】
別の局面では、本願発明は、本願発明のインスリン応答モジュレータを対象に投与するステップを含む、前記対象におけるグルコースの恒常性を調節する方法を提供する。別の局面では、本願発明は、本願発明のインスリン応答モジュレータを対象に投与するステップを含む、前記対象における血中グルコース値を調節する方法を提供する。
【0015】
さらに、本願明細書において同定されたモジュレータを含む薬学的組成物を特徴とする。
【0016】
別の局面では、本願発明は、本願発明の薬学的組成物を投与するステップを含む、インスリン応答疾患または障害を治療する方法を提供する。さまざまな実施態様では、前記疾患または障害は2型糖尿病、インスリン耐性、および肥満からなるグループから選択される。
【0017】
標的遺伝子は、siRNA(標的化siRNA)またはsiRNA誘導体によって、RNAi媒介遺伝子ノックダウンを標的とした遺伝子である。siRNAのある部分は、前記標的遺伝子のmRNAの部分に完全に相補的である。
【0018】
対照細胞または対照培養物は、修飾siRNAまたはsiRNA誘導体と接触したことがない細胞または培養物である。前記対照細胞または培養物は、一般に、RNAi媒介ノックダウンを目的とする1つ以上の内因性遺伝子を発現する1つ以上のレポータ遺伝子を含む。本願発明のある実施態様では、前記対照細胞または対照培養物は、対象のレポータ遺伝子または内因性遺伝子を標的としたsiRANを含む。ある場合、前記対照細胞または対照培養物は、siRNAまたは修飾siRNAのアンチセンス鎖に相当するアンチセンス鎖、またはsiRNAまたは修飾siRNAのスクランブル配列を含むアンチセンス鎖などの、導入された対照配列を含む。
【0019】
テスト細胞またはテスト培養物は、RNAi媒介遺伝子ノックダウンの目的で発現された、または1つ以上発現された内因性遺伝子である1つ以上のレポータ遺伝子を含み、また対象のレポータ遺伝子または内因性遺伝子を標的とした修飾siRNAまたはsiRNA誘導体も含む。
【0020】
siRNAを脂肪細胞に導入する方法(たとえばリポフェクタミンTMなどの作用剤を用いたトランスフェクションなど)は、一般に成功したことがない。電気穿孔を用いた本願発明の方法は、トランスフェクション剤を用いない、siRNAの脂肪細胞への導入方法を提供する。したがって、本願発明は、たとえば、グルコース輸送および糖尿病などの障害など、脂肪細胞機能に関連する治療の研究および開発のためにsiRNAを用いる、遺伝子ノックダウン法を用いることができる。
【0021】
他に定義のない限り、本願明細書に用いられたすべての技術および科学用語は、本願発明が属する当業者によって普通に理解されるものと同じ意味を有する。本願明細書に記載のものに類似するまたはそれに相当する方法および材料は、本願発明の実施またはテストに用いることができ、好適な方法および材料を以下に説明する。本願明細書に引用されるすべての出版物、特許出願、特許、およびその他の参考文献は、その全体を引用によりここに援用する。さらに、当該材料、方法、および例は、具体例であって、制限を意図しない
【0022】
本願発明のその他の特徴および利点は、以下の発明の詳細な説明、図面、および特許請求の範囲から明らかになるであろう。
【0023】
発明の詳細な説明
本願発明は、たとえばsiRNAなどの核酸を脂肪細胞に導入する方法を提供する。この新規の方法は、特定の条件下で脂肪細胞の電気穿孔を用いる。前記方法は、テストタンパク質ラミンA/C を用いて開発され、Akt(タンパク質キナーゼB;PKB)の役割を調べるために用いられてきた。この新規の方法を用いて、他のタンパク質を正常に発現させる一方で、標的化されたタンパク質の発現の完全な阻害を実施することができる。
【0024】
この方法は、たとえば、2型糖尿病の治療に用いることができるインスリン増感剤の同定、および肥満を軽減する薬物などの薬物発見などに有用である。
【0025】
遺伝子特異的ノックアウトマウスはシグナル伝達分子の急性作用を研究するツールを提供するが、マウスにおいて認められた表現型は、遺伝子発現および発育制御における無関係の変化に起因する可能性がある。たとえば、本願発明は、たとえば無傷の培養脂肪細胞におけるAktタンパク質キナーゼなどの、脂肪細胞における発現の選択的阻害を、RNA干渉(RNAi)の機序によって可能にする方法を提供する。この強力なアプローチは、Akt1およびAkt2遺伝子を両方とも喪失すると致死的な場合の、マウスの遺伝子ノックアウト(つまりトランスジェニックマウス)において遭遇する問題点を克服する。
【0026】
培養細胞、特に一次または二次細胞中の遺伝子の選択的阻害を可能にする方法は、遺伝子標的を定めるために遺伝子の機能を明らかにする上で有用である。そのような方法は、特定の遺伝子の発現が阻害される場合に期待される効果の一覧を作成するのにも有用である。前記一覧は、化合物をスクリーニングして、効果が前記の特定の遺伝子の発現の調節に限定されている化合物を同定する場合に用いることができる。本願明細書に記載されているようなシステムは、脂肪細胞において特に有用である。それは、脂肪細胞が肥満および糖尿病(たとえば2型糖尿病)など、薬物が探索されている障害に関連しているからである。
【0027】
本願発明をより容易に理解するため、特定の用語を以下のように義する。
【0028】
「ヌクレオシド」という用語は、リボースまたはデオキシリボース糖に共有結合したプリンまたはピリミジンを有する分子を意味する。例示的なヌクレオチドには、アデノシン、グアシン、シチジン、ウリジン、およびチミジンが含まれる。「ヌクレオチド」という用語は、糖部分にエステル結合したリン酸基を1つ以上有するヌクレオシドを意味する。例示的なヌクレオシドには、1リン酸ヌクレオシド、2リン酸ヌクレオシド、および3リン酸ヌクレオシドが含まれる。「ポリヌクレオチド」および「核酸分子」という用語は、本願明細書では交換可能に用いられ、5’および3’炭素原子の間のホスホジエステル結合によって結合したヌクレオシドのポリマーを意味する。
【0029】
「RNA」または「RNA分子」または「リボ核酸分子」という用語は、一般に、リボヌクレオチドのポリマーを意味する。「DNA」または「DNA分子」または「デオキシリボ核酸分子」という用語は、一般にデオキシリボヌクレオチドのポリマーを意味する。DNAおよびRNA分子は自然に合成することができる(たとえば、それぞれDNA複製またはDNAの転写によって)。RNA分子は転写後に修飾することができる。DNAおよびRNA分子は化学的にも合成することができる。DNAおよびRNA分子は1本鎖であっても(すなわちそれぞれssRNAおよびssDNA)、または多重鎖(たとえば2本鎖、すなわちそれぞれdsRNAおよびdsDNA)であってもよい。しかし、本願発明の性質に基づき、「RNA」または「RNA分子」、または「リボ核酸分子」という用語は、主に(つまり80%超、または好ましくは90%超)リボヌクレオチドを含むポリマーも意味するが、任意で例えば少なくとも1つのデオキシリボヌクレオチドおよび/または少なくとも1つのヌクレオチド類似体などの、少なくとも1つの非リボヌクレオチド分子を含むこともできる。
【0030】
本願明細書では「改変ヌクレオチド」または「修飾ヌクレオチド」も意味する「ヌクレオチド類似体」という用語は、天然に存在しないリボヌクレオチドまたはデオキシリボヌクレオチドを含む、標準的ではないヌクレオチドを意味する。好ましいヌクレオチド類似体は、任意の位置を修飾し、ヌクレオチドの特定の化学的特性を変えつつ、核酸類似体の目的の機能を実行する能力を維持できるようにする。
【0031】
「RNA類似体」という用語は、相当する未改変または未修飾RNAと比較して少なくとも1つの改変またな修飾ヌクレオチドを有するが、前記の相当する未改変または未修飾RNAと同一のまたは類似する性質または機能を保持する、ポリヌクレオチド(たとえば化学合成されたポリヌクレオチドなど)を意味する。上述の通り、前記オリゴヌクレオチドは、ホスホジエステル結合を有するRNA分子と比較してRNA類似体の加水分解が低速度になる結合で結合していてよい。例示的なRNA類似体には、糖および/または骨格修飾リボヌクレオチドおよび/またはデオキシリボヌクレオチドが含まれる。そのような改変または修飾にはさらに、RNA末端または内部などへの非ヌクレオチド物質の付加(RNAの1つ以上のヌクレオチド)が含まれる。RNA類似体は、RNA干渉を媒介する能力を有する天然のRNAに十分類似していればい。
【0032】
本願明細書に用いられる「RNA干渉」または「RNAi」という用語は、一般に、標的分子(たとえば標的遺伝子、タンパク質、またはRNA)を下方調節することによる配列特異的または選択的なプロセスを意味する。ある実施態様では、「RNA干渉」または「RNAi」のプロセスは、たとえば細胞内のRNA分子など、RNA分子の分解であって、前記分解がRNA作用物質によって引き起こされる、分解を特徴とする。分解は、酵素的RNA誘導サイレンシング複合体(RISC)によって触媒される。RNAi は、細胞内で外来のRNA(たとえばウイルス性RNA)を除去するため自然にに生じる。天然のRNAiは、分解メカニズムがその他の同様のRNA配列に方向付けられている遊離dsRNAから切断された断片によって進行する。代替的には、RNAiは、たとえば標的遺伝子の発現をサイレンシングさせるなど、人工的に開始させることもできる。
【0033】
本願明細書に記載の「低分子干渉RNA」(「siRNA」)という用語(「短分子干渉RNA」ともいう)は、RNA干渉を方向付けるまたは媒介する能力のある約10乃至50ヌクレオチドを含むRNA(またはRNA類似体)を意味する。RNAiを方向付けるまたは媒介する標的mRNA配列に十分に相補的な配列を有する「siRNA」とは、siRNAがRNAi機構(たとえばRISC複合体)またはプロセスによって標的mRNAの破壊を惹起するのに十分な配列を有することを意味する。
【0034】
本願明細書に記載の「shRNA」または「短分子ヘアピンRNA」は、ステムループ構造を有するRNA作用物質であって、相補配列の第1および第2の領域であって、前記領域の相補性および配向は前記領域の間で塩基対が生じるのに十分な程度であって、前記第1および第2の領域はループ領域で結合しており、前記ループは前記ループ領域内のヌクレオチド(またはヌクレオチド類似体)の間の塩基対の欠損から生じる、第1および第2の領域を含む、RNA作用物質を含む。
【0035】
「mRNA」または「メッセンジャRNA」は、1つ以上のポリペプチド鎖のアミノ酸配列を特定する1本鎖RNAである。この情報は、リボソームが前記mRNAに結合する際にタンパク質合成中に翻訳される。
【0036】
障害に「関与する」遺伝子には、疾患または障害もしくは前記疾患または障害の少なくとも1つの症状に作用するまたはそれを生じる、正常なまたは異常な発現または機能の遺伝子が含まれる。
【0037】
本願発明の様々な方法論には、本願明細書では「適切な対照」とも交換可能な意味である「好適な対照」と、価値、レベル、特色、特徴、性質などの比較に関与するステップを含む。「好適な対照」または「適切な対照」は、比較の目的で有用な、当業者が精通している任意の対照または標準である。ある実施態様では、「好適な対照」または「適切な対照」は、本願明細書に記載の通り、RNAi方法論を実行する前に決定する、価値、レベル、特色、特徴、性質などである。たとえば、転写速度、mRNAレベル、翻訳速度、タンパク質レベル、生物学的活性、細胞の特徴または性質、ゲノタイプ、表現型などは、細胞または生物に本願発明のsiRNAを導入する前に決定することができる。別の実施態様では、「好適な対照」または「適切な対照」は、細胞、またはたとえば正常な形質を示す対象、または正常細胞もしくは生物などの生物において決定される価値、レベル、特色、特徴、性質などである。さらに別の実施態様では、「好適な対照」または「適切な対照」は、予め決定された価値、レベル、特色、特徴、性質などである。
【0038】
他に定義のない限り、本願明細書に用いられたすべての技術および科学用語は、本願発明が属する当業者によって普通に理解されるものと同じ意味を有する。本願明細書に記載のものに類似するまたはそれに相当する方法および材料は、本願発明の実施またはテストに用いることができ、好適な方法および材料を以下に説明する。
【0039】
I. RNAi分子
RNA干渉(RNAi)は、配列特異的、転写後遺伝子サイレンシングであって、抑制される遺伝子に相同の2本鎖RNA(dsRNA)によって開始される。dsRNAは細胞リボヌクレアーゼIIIであるダイサーによって処理され、配列特異的mRNA分解を媒介する3’オーバーハング(低分子干渉RNA、siRNA)を有する約21ntの2本鎖を生じる。哺乳類細胞では、siRNA分子は、非特異的インターフェロン応答経路を誘導しない、特異的なサイレンシング遺伝子を発現することができる。したがって、siRNAは、機能喪失型表現型を分析するための、アンチセンスヌクレオチドおよびリボザイムなどのその他の遺伝子ツールに代わる新規で強力な代替物である。siRNA 2本鎖を用いて特異的遺伝子の発現と干渉させるには、標的へのアクセスのしやすさ、標的細胞へのsiRNAの効果的な輸送、および一部の用途においては長期的なsiRNA発現の知識が必要である。本願発明は、本願明細書に記載のとおり、電気穿孔法を用いて、たとえば脂肪細胞などの細胞に短2本鎖siRNA分子を形質導入することによって、siRNAをたとえば脂肪細胞などの細胞培養物中の標的細胞へ輸送する効果的な戦略を提供する。本願発明の方法はまた、各種のPol III プロモータによって機能型siRNAまたはその前駆体の転写を可能にする発現カセットを、たとえば脂肪細胞などの細胞に導入するのにも有用である。
【0040】
RNAiはたとえばsiRNAおよび/または長分子dsRNAが動物および植物細胞において標的mRNAの配列特異的分解を誘発する非常に有効なプロセスである(Hutvagner and
Zamore, Curr. Opin. Genet. Dev.:12, 225-232 (2002); Sharp, Genes Dev.,
15:485-490 (2001))。哺乳類細胞において、RNAiは、低分子干渉RNA(siRNA)の21ヌクレオチド(nt)2本鎖(Chiu et al., Mol. Cell. 10:549-561 (2002); Elbashir et al., Nature
411:494-498 (2001))、またはマイクロRNA(miRNA)、機能型低分子ヘアピンRNA(shRNA)、またはRNAポリメラーゼIIIプロモータを有するDNAテンプレートを用いてin vivoにおいて発現させることができるその他のdsRNAによって惹起させることができる(Zeng et al., Mol. Cell 9:1327-1333
(2002); Paddison et al., Genes Dev. 16:948-958
(2002); Lee et al., Nature Biotechnol. 20:500-505
(2002); Paul et al., Nature Biotechnol. 20:505-508
(2002); Tuschl, T., Nature Biotechnol. 20:440-448
(2002); Yu et al., Proc. Natl. Acad. Sci. USA
99(9):6047-6052 (2002); McManus et al., RNA 8:842-850 (2002); Sui et
al., Proc. Natl. Acad. Sci. USA 99(6):5515-5520 (2002).)。
【0041】
したがって、本願発明は、たとえばAkt1、Akt2、Myo1cまたはCISKなどのグルコース輸送に影響する遺伝子のmRNA配列を標的とする核酸分子(つまりRNAi分子)を提供する。ある実施態様では、グルコース輸送に影響する遺伝子は、表1に記載のRNAi標的ヌクレオチド配列を含む。
【0042】
本願発明のRNAi分子には、siRNA分子が含まれる。本願発明のsiRNA作製物には、各鎖にたとえば16、17、18、19、20、21、22、23、24、25、26、27、28、29、または30 ヌクレオチドなど約16乃至30ヌクレオチドを含むdsRNAであって、当該分子において片方の鎖がmRNAにおける標的領域に実質的にたとえば少なくとも80%(またはそれを超える、たとえば85%、90%、95%、または100%)実質的に同一であって、もう片方の鎖は第1の鎖と同一であるかまたは実質的に同一である、dsRNAが含まれる。siRNAは典型的に、2乃至3ヌクレオチドの3’オーバーハング末端、5’リン酸末端(細胞から抽出した)および3’ヒドロキシル末端を有する。RNAを合成する方法、およびRNAを修飾する方法は当業に知られている(たとえば Hwang et al., 1999, Proc. Nat. Acad. Sci. USA 96:12997-13002; およびHuq and Rana, 1997, Biochem. 36:12592-12599)。siRNAは市販されている(たとえばダーマコン・リサーチ社(コロラド州レイファイエット)、アンビオン社(テキサス州オースチン))。
【0043】
本願発明のsiRNA分子は、化学的に合成することができ、またはin vitroではDNAテンプレートから、もしくはin vivoではたとえばshRNAから、もしくはin vitro転写dsRNAテンプレートをRNAiを切断して媒介する20もしくは21bpの2本鎖RNAのプールをつくるためにヒトDICER酵素を用いて、転写することができる。 siRNA分子は当業に知られる任意の方法、たとえば以下のプロトコルを用いることによってデザインすることができる。
【0044】
1. AUG開始コドンから始めてAAジヌクレオチド配列を探す。各AAおよび3’側に16ヌクレオチド以上離れたところが、siRNA標的の候補である(図15、16、34、35、36参照)。5’非翻訳領域(UTR)および開始コドンに近い領域(約75塩基ほど)から得たsiRNAは有用性が低いと考えられる。なぜなら、それらは制御性タンパク質結合部位により豊富に存在しており、結合タンパク質および/または翻訳開始複合体がsiRNPまたはRISCエンドヌクレアーゼ複合体の結合を妨げる可能性があるからである。したがって、ある実施態様では、前記核酸分子は、開始コドンの少なくとも50乃至100nt下流から始まるcDNA配列の領域から選択される。さらに、G/C含有量が少ないsiRNA(35乃至55%)は、G/C含有量が55%を超えるものよりも活性であることもある。したがってある実施態様では、前記発明には35乃至55%のG/C含有量を有する核酸分子が含まれる。さらに、siRNAの鎖は、たとえば2ヌクレオチドなど、1乃至4ヌクレオチドの3’オーバーハングを有するように対を形成させることができる。したがって別の実施態様では、前記核酸分子はTTなどの2ヌクレオチドの3’オーバーハングを有することができる。前記のオーバーハングしているヌクレオチドは、RNAまたはDNAのいずれかであってよい。
【0045】
2. 当業に知られる任意の方法を用いて、標的候補と適切なゲノムデータベース(ヒト、マウス、ラットなど)を比較し、他のコード配列に顕著に相同性を有する任意の標的配列を考慮から除外する。そのような配列相同性の探索に使われる1つの方法はBLASTとして知られ、国立衛生研究所の国立バイオテクノロジー情報センターのウェブサイトで入手することができる。
【0046】
3. 評価基準に合う1つ以上の配列を選択する。さらに、siRNAのデザインおよび使用についての一般的な情報は、ロックフェラー大学のThomas Tuschl 博士の研究所のウエブサイトで入手できる「The siRNA User Guide(siRNAユーザーガイド)」の中に見つけることができる。
【0047】
本願発明のある実施態様では、siRNAは比較的低レベルの毒性を示す。たとえば、標的配列の発現がsiRNAで処理されていない細胞中の発現と比較して50%減少する場合、siRNAで処理された培養物中の細胞の少なくとも50%が生存する場合、前記標的配列の発現を阻害するsiRNAの濃度は比較的低い毒性を有している。低い毒性は、たとえば少なくとも60%、75%、85%、90%、95%、または100%など、より高い細胞生存性に関連していると考えられる。細胞の生存性を測定する方法は当業に知られており、トリパンブルー排除法が含まれる。これは、たとえば標的遺伝子のノックアウト(たとえばトランスジェニック動物など)とは対称的である。したがって、本願発明の利点は、遺伝子を操作してその遺伝子の発現を操作する場合、細胞または動物に致死的である可能性がある遺伝子の発現の操作が可能になることである。
【0048】
本願発明のRNAi作用物質には、低分子ヘアピンRNA(shRNA)、およびshRNAを発現するためにつくられた発現作製物が含まれてよい。shRNAの転写は、ポリメラーゼIII(pol III)プロモータから開始し、4-5チミン転写終了部位の2位で終了すると考えられている。発現後、shRNAは3’UUオーバーハングを有するステム-ループ構造に折り畳まれると考えられ、その後、これらのshRNAの末端が処理されて、shRNAが約21ヌクレオチドのsiRNA様分子に転換される。Brummelkamp et
al., Science 296:550-553 (2002); Lee et al, (2002). supra; Miyagishi
and Taira, Nature Biotechnol. 20:497-500 (2002);
Paddison et al. (2002), supra;
Paul (2002), supra; Sui (2002) supra; Yu et al. (2002), supra。shRNAデザインおよび使用についてのさらなる情報は、インターネットの以下のアドレス katahdin.cshl.org:9331/RNAi/docs/BseRI-BamHI_Strategy.pdf および katahdin.cshl.org:9331/RNAi/docs/Web_version_of_PCR_strategy1.pdfで見つけることができる。
【0049】
本願発明の発現作製物には、適切な発現システムに使用するのに好適な任意の作製物が含まれ、当業に知られるレトロウイルスベクタ、直線状発現カセット、プラスミドおよびウイルスまたはウイルス由来ベクタがそれらに限定されずに含まれる。そのような発現作製物には、1つ以上の誘導プロモータ、U6 snRNAプロモータもしくはH1 RNAポリメラーゼIIIプロモータなどのRNA Pol IIIプロモータシステム、または当業に知られるその他のプロモータが含まれてよい。両方の鎖を発現する発現作製物には、両方の鎖を連結するループ構造が含まれてもよく、または各鎖は同一の作製物内の個別のプロモータから別々に転写されてもよい。各鎖は個別の発現作製物から転写されてもよい。Tuschl (2002), supra。
【0050】
細胞内における長期間の標的遺伝子の抑制を可能にするために、細胞内で組換えDNA作製物からsiRNA 2本鎖を発現させる方法は当業に知られており、機能的な2本鎖siRNAを発現することが可能な哺乳類Pol III プロモータシステム(たとえばH1またはU6/snRNAプロモータシステム(Tuschl (2002), supra) )が含まれる(Bagella et al., J. Cell. Physiol.
177:206-213 (1998); Lee et al. (2002),
supra; Miyagishi et al. (2002),
supra; Paul et al. (2002), supra;
Yu et al. (2002), supra;
Sui et al. (2002), supra)。RNA Pol IIIによる転写終了は、一連のDNAテンプレートの連続する4つのT残基で生じ、特定の配列においてsiRNA転写物を終了させるメカニズムを提供する。前記siRNAは5’-3’および3’-5’の方向に向いた前記標的遺伝子の配列に相補的であって、siRNAの2本鎖は同一の作製物または別々の作製物中に発現することができる。H1またはU6 snRNAプロモータによって駆動され、細胞内で発現するヘアピンsiRNAは、標的遺伝子の発現を阻害することができる(Bagella et al. (1998), supra; Lee et
al. (2002), supra; Miyagishi
et al. (2002), supra; Paul et
al. (2002), supra; Yu et al. (2002), supra; Sui et al. (2002) supra)。T7プロモータの制御下にあるsiRNA配列を含有する作製物もまた、T7 RNA ポリメラーゼを発現するベクタを有する細胞に同時トランスフェクトされると、機能性siRNAをつくる。1つの作製物は、同一の遺伝子または複数の遺伝子に方向付けられているたとえばAkt1、Akt2、CISKまたはMyo1cなどをコードする遺伝子の複数の領域など、siRNAをコードする複数の配列を含有することがあり、たとえば、別々のPolIIIプロモータ部位によって駆動することができる。
【0051】
動物細胞はミクロRNA(miRNA)とよばれる約22ヌクレオチドの非コードRNAの領域を発現する。ミクロRNAは動物の成長中に転写後または転写レベルで遺伝子発現を制御することができる。miRNAのある共通の性質は、おそらくダイサー、RNase III型酵素、またはその相同体によって、約70ヌクレオチドの前駆体RNAステム-ループからすべて切除されることである。標的mRNAに相補的な配列を有するmiRNA前駆体のステム配列を置換することによって、新規miRNAを発現するベクター作製物を用いて、哺乳類細胞の中で特異的mRNA標的に対してRNAiを開始するためにsiRNAを産生することができる(Zeng (2002), supra)。ポリメラーゼIIIプロモータを含有するDNAベクタによって発現する場合、ミクロRNAでデザインされたヘアピンは遺伝子発現をサイレンシングすることができる(McManus (2002), supra)。動物モデルにおいて、全胚電気穿孔は、合成siRNAを効率的に移植後マウス胚合成siRNAに送達させることができる。成体マウスにおいて、siRNAの効率的な送達は、「高圧」送達技術によって実現することができる。これは溶液を含有する大量のsiRNAを、尾部静脈を介して動物体内へ迅速に(5秒以内に)注入する技術である。ナノ粒子およびリポソームを用いて、siRNAを動物体内へ送達することもできる。
【0052】
陰性対照siRNAは、選択されたsiRNAと同一のヌクレオチド組成を有するべきだが、適切なゲノムに相補的な重要な配列は有さない。そのような陰性対照は、選択されたsiRNAのヌクレオチド配列をランダムにスクランブルしてデザインしてもよく、相同性探索は、前記陰性対照が適切なゲノムにおける任意のその他の遺伝子に対する相同性を欠損していることを確認するために行うことができる。さらに、陰性対照siRNAは、前記配列に1つ以上の塩基ミスマッチを導入することによってデザインすることができる。
【0053】
本願発明の核酸組成物には、架橋siRNA誘導体など、当業に知られる未修飾siRNAおよび修飾siRNAの両方が含まれる。たとえば体内の半減期を延長するためなど、前記組成物の薬物動態学を変化させるために、架橋を用いることができる。したがって、本願発明には、2本の鎖が架橋されるように、核酸の相補的な2本の鎖を有するsiRNA誘導体を含む、siRNA誘導体が含まれる。たとえば、片方の鎖の3’OH末端を修飾し、2本鎖を架橋させて3’OH末端で修飾することができる。前記siRNA誘導体は1つの架橋(たとえばソラーレン架橋)を含有することができる。siRNA誘導体の修飾は、細胞取り込みが改善するか、もしくは相当するsiRNAと比較して、得られたsiRNA誘導体の細胞の標的活性を促進するか、前記細胞のsiRNA誘導体を追跡するのに有用であるか、または相当するsiRNAと比較したsiRNA誘導体の安定性を向上することもある。
【0054】
本願発明の核酸組成物は、たとえば吸収、有効性、バイオアベイラビリティ、および/または半減期などの薬物動態学的パラメタなどの前記組成物の性質を向上させるナノ粒子などの別の部分と結合させることもでき、または結合させないでもよい。その結合は、たとえば、Lambert et al.,
Drug Deliv. Rev.:47(1), 99-112 (2001) (ポリアルキルシアノアクリル酸(PACA)ナノ粒子に装着された核酸について記載)、Fattal et al.,
J. Control Release 53(1-3):137-43 (1998) (dナノ粒子に結合させた核酸について記載)、 Schwab et al.,
Ann. Oncol. 5 Suppl. 4:55-8 (1994) (挿入剤、疎水基、ポリカチオンPACAナノ粒子に結合させた核酸について記載)、およびGodard et al.,
Eur. J. Biochem. 232(2):404-10
(1995) (ナノ粒子に結合させた核酸について記載)の方法を用いるなど、当業に知られる方法によって実現することができる。
【0055】
本願発明の核酸分子は、当業に知られる任意の方法を用いてラベルすることもでき、たとえば前記核酸分子組成物がたとえばCy3、フルオレセイン、ローダミンなどのフルオロフォアで標識することができる。その標識は、たとえばSILENCERTM
siRNA ラベリングキット(アンビオン)など、キットを用いて行うことができる。さらに、前記siRNAは、たとえば3H、32Pまたはその他の適切な同位体などを用いて放射標識することができる。
【0056】
本願明細書に記載の細胞(たとえば脂肪細胞など)または組織全体に導入された組換えRNA前駆体によって、望ましいsiRNA分子の産生が引き起こされるだろう。そこで、そのようなsiRNA分子は、切断および破壊のための特異的なmRNA配列に結合し、それを標的とするために、RNAi経路の内因性タンパク質要素に関連するだろう。このようにすれば、組換えRNA前駆体から作製されたsiRNAが標的とするmRNAは、前記細胞および組織から大幅に減少し、前記細胞または組織中のmRNAによってコードされるタンパク質の濃度が減少するだろう。RNA前駆体は、典型的にはdsRNAの1本鎖を個別にコードするか、またはRNAヘアピンループ構造のヌクレオチド配列全体をコードする核酸分子である。
【0057】
さらに、RNAiは、少なくとも1つの1本鎖RNA中間体を経る過程であると考えられるため、当業者は、ss-siRNA(たとえばds-siRNAのアンチセンス鎖など、本願明細書に記載のとおりデザイン(たとえば化学合成用に)、作製(たとえば酵素的に作製)または発現(たとえばベクターまたはプラスミドから)することもでき、本願特許請求の方法論にしたがって利用することができるということは理解されよう。さらに、無脊椎動物では、RNAiは長いdsRNA(たとえば長さが約100乃至1000ヌクレオチド、好ましくは約200乃至500であって、たとえば、約250、300、350、400、または450ヌクレオチドのdsRNA)であって、RNAiのエフェクタT細胞として作用するRNAiは長いdsRNAによって効果的に引き起こすことができる。(Brondani et al., Proc Natl Acad Sci U S A. 2001
Dec 4;98(25):14428-33. Epub 2001 Nov 27)。
【0058】
本願発明のsiRNA分子は、前記標的遺伝子の以下の配列を標的とすることができ、たとえば前記siRNA分子が、その鎖の1つとして、前記DNA配列の任意の1つに相当するRNA配列(たとえば前記siRNA2本差のセンス鎖など)、およびその対立遺伝子変異体の相当する配列を含んでよい。表の配列は、標的遺伝子配列(すなわちcDNA配列)を表す。しかし、当業者は、たとえばセンス鎖などのsiRNA鎖が相当するリボヌクレオチドを含んでおり、アンチセンス鎖が相補的リボヌクレオチド配列を含むということを理解するだろう。さらなるデオキシチミジンオーバーハングも、本願明細書に記載されるとおり意図される。
【0059】
【表1】
【0060】
II. 脂肪細胞培養物
本願発明は、たとえばsiRNAなどの核酸を脂肪細胞に導入に関連する。脂肪細胞は当業に知られる方法を用いて培養することができる(たとえば Adipose
Tissue Protocols, Methods in Molecular Biology, v.
155, ed. Gerard Ailhaud. Totowa, NJ,
Humana Press, 2001を参照)。例1(後述)は、3T3-L1細胞を脂肪細胞に分化させるそのような1つの方法を説明する。使用することができるその他の方法には、解剖した(一般に細胞を切除するための処理後の)脂肪組織から得た脂肪細胞を用いた、間葉細胞の脂肪細胞への分化(たとえばステムセル・テクノロジーズ社(カナダ、バンクーバー)などから入手可能な試薬を用いた)、および脂肪細胞を作成する市販の方法(たとえば脂肪細胞分化キット、ストラタジーン社、プロモセル(独、ハイデルベルグ))が含まれる。
【0061】
本願明細書に記載の方法を用いた核酸の導入のための細胞を調製するために、脂肪細胞を培養基質から剥離するか、または標準方法で解離させる。
【0062】
III. 核酸分子の脂肪細胞への導入
一般に、リボ核酸、デオキシ核酸、およびこれらの変異体は、脂肪細胞に導入することができる。前記核酸は、たとえば、脂肪細胞、アンチセンスRNA、siRNA、siRNA前駆体、またはsiRNA前駆体を発現するベクターに発現させる、遺伝子産生物または遺伝子産生物の断片をコードする配列を含有するなどの、核酸ベクターであってよい。前記核酸は、2本鎖または1本鎖であってよい。
【0063】
脂肪細胞に導入される核酸配列は、たとえば長さ約2乃至5,000塩基、2乃至1000塩基、2乃至500塩基、2乃至300塩基、2乃至200塩基、2乃至100塩基、2乃至50塩基、または2乃至25塩基、もしくは1乃至15塩基などの、約2乃至10000塩基である。前記核酸がsiRNAである場合、各鎖は一般に、たとえば長さ16、17、18、19、20、21、22、23、24、25、26、27、28、29、または30ヌクレオチドなどの、約16乃至30ヌクレオチドである。そのような核酸を調節する方法は、当業に知られる。典型的には、本願明細書に記載の方法に用いられる核酸サンプルは、脱塩してから細胞に導入する。
【0064】
一般に、核酸分子は、たとえばリン酸バッファ食塩水(PBS)など好適なバッファ中で洗浄した後に細胞に導入し、それからたとえばPBSなどの好適なバッファ中で再懸濁するその他の好適なバッファには、Krebs/リンガー/HEPESバッファがそれに限定されずに含まれる。再懸濁した細胞は、PBSに再懸濁した核酸か、または電気穿孔の手順に好適なその他のバッファ中に混合する。たとえば、0.1乃至80nmol、典型的には約20nMのsiRNAを用いる。約1乃至1000万個の細胞を前記方法に用いてよく、典型的には各手順に約500万個の細胞を用いる。典型的には、前記細胞は室温で電気穿孔されるが、他の温度であってもよい。その後、前記混合物を、たとえば約0.001乃至20 kV、約0.01乃至2.0 kV、約0.02乃至1.0 kV、約0.05乃至0.5 kV、約0.1乃至0.25kVの設定、および好ましくは0.11、0.12、0.13、0.14、0.15、0.16、0.17、0.18、0.19、0.20、0.21、0.22、0.23、0.24または0.25 kVの設定、ならびに約 10-5000 μF、約 100-3000 μF、約 200-1800 μF、約 350-1550 μF、約 5-1350 μF、約 750-1150 μF、約 850-1050 μF、約 900-1000 μFの設定、および好ましくは約900、910、920、930、940、950、960、970、980、990、または100 μFの静電容量の設定など、前記細胞へsiRNAを効率的に導入できるような設定で、好適にな装置(たとえばバイオラッド・ジーンパルサーIIシステム、バイオラッドラボラトリーズ社(カリフォルニア州ハーキュリーズ))を用いて電気穿孔のパルスに曝露する。好ましい実施態様では、前記混合物を、0.18kVおよび960μFの設定で電気穿孔のパルスに曝露する。典型的には約400乃至600μLの細胞、たとえば450μLの細胞などを、前記手順に用いる。電気穿孔後、前記細胞をただちに新鮮培地と混合し、37℃で10分間インキュベートしてからデオキシグルコース取り込みアッセイ、ウエスタンブロッティング、または免疫蛍光顕微分析法などのその後の用途に好適な多穴プレートに再播種する。再播種後、約2、4、8、12、16、20、24、30、36、42、48、54、60、72、78、94または約120分経過時点など、再播種後好適な時点に、分析を行う。好ましい実施態様では、分析は再播種後24乃至48時間経過時点で行う。
【0065】
本願明細書に記載の細胞または生物全体に導入したsiRNAは、切断および破壊のための特異的なmRNA配列に結合し、標的とするRNAi経路の内因性タンパク質要素に関連するだろう。このようにすれば、siRNAが標的とするmRNAは、前記細胞および生物から激減し、前記細胞または組織中のmRNAによってコードされるタンパク質の濃度が減少する。
【0066】
前記方法は、たとえば、発現(または過剰発現)すると異常なグルコース代謝を引き起こす核酸配列の活性を低減する低分子を発見するために探索する場合には有用である。主要な疑問は、この核酸配列の発現または活性を減少させると細胞に予期せぬ作用、とくに悪性の作用を及ぼすかどうかである。RNAi経路による破壊のために脂肪細胞中の核酸配列をコードするmRNAを標的とするsiRNA誘導体を発現することによって、そのような薬物候補の悪性作用を決定することができる。つまり、本願明細書に記載の方法では、前記核酸配列および薬物標的としてのその産生物の適切性を迅速に評価することができる。
【0067】
RNAiは遺伝子機能を解明するための新規のアプローチを提供する。RNAi媒介遺伝子ノックダウンは、遺伝子機能のゲノムワイドな分析、および治療用に用いられる可能性のある遺伝子の標的バリデーションに有用である。siRNAは哺乳類遺伝子機能を研究する細胞生物学者にとって有用なツールである。たとえば、siRNAは有子分裂、RNA転写物のプロセッシングおよび輸送、細胞接合の形成、および膜輸送などの一般的な細胞生物学的機序の分析に有用である。そのような分析に用いることができる試薬(たとえば、相当する未修飾siRNAと比較した細胞における高安定性を有するsiRNA)は、そのような研究用の商品価値を有する。
【0068】
特定の遺伝子はsiRNAを用いることによってノックダウンすることができ、得られた表現型を観察することができる。しかし、基本的な遺伝子のノックダウンは致死的または有毒である可能性もあり、細胞における多くの経路に影響を与える可能性がある。したがって、ある場合には、前記細胞に、ノックダウン時には有効性が最大ではないsiRNAを提供することが好ましい。過度に有効なノックダウンの悪性作用は、相当するsiRNAと比較して低いRNAi活性を有するsiRNA誘導体を前記細胞と接触させることによって調節することができる。siRNA誘導体は修飾siRNAである。修飾には、架橋または3’末端の阻害が、それらに限定されずに含まれる(Chiu and Rana,
2002, Mol Cell 10:549-561)。この目的に用いられるsiRNA誘導体の好適な濃度には、RNAi活性を最大に阻害することがなく、siRNAの望ましくない作用を軽減する濃度が含まれる。相当するsiRNAよりも低い効率のノックダウンを生じることができるsiRNA誘導体の量は、当業に知られる方法を用いて決定することができる。たとえば、特定の核酸配列を標的とするsiRNAをトランスフェクトした細胞における発現レベルと、前記siRNAの誘導体をトランスフェクトした細胞における特定の核酸配列の発現量を比較する。siRNA誘導体をトランスフェクトした細胞における高発現レベルは、前記siRNAは、発現のノックダウンの効率がよくないということを示唆している。そのような誘導体は、効率の高いノックダウンが前記細胞に致死的であるノックダウン実験に有用である。したがって、ある場合では、有用なsiRNA誘導体はRNAiの阻害が100%未満のsiRNA誘導体である。たとえば、siRNAのRNAi作用を低下させるのに有用なsiRNA誘導体は、たとえば90%、75%、50%、25%、または10%未満など、RNAi活性を阻害することができる。
【0069】
IV 核酸標的
本願明細書に記載のsiRNAの核酸標的は、2型糖尿病、インスリン耐性もしくは肥満に関連する任意の遺伝子、または、たとえばAkt1、Akt2、CISK、またはMyo1cをコードする遺伝子などをそれらに限定せずに含む、グルコース輸送またはインスリン応答に重要な任意の遺伝子であってよい。ある実施態様では、前記標的遺伝子は、表1に記載の標的ヌクレオチド配列を含む。
【0070】
したがって、Akt1のmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号NM_009652 (GI:6753033;Mus musculus) (配列番号20); NM_005163 (GI:4885060; Homo
sapiens) (配列番号22)がそれらに限定されずに含まれるマウスもしくはヒトAkt1に、実質的に同一の配列など、Akt1の任意のオルソログであってよい。Akt2のmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号U22445 (GI:942577; Mus musculus) (配列番号21); NM_001626 (GI:6715585; Homo
sapiens) (配列番号23)がそれらに限定されずに含まれるマウスもしくはヒトAkt2に、実質的に同一の配列など、Akt2の任意のオルソログであってよい。CISKのmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号AF312007 (GI:11321320; Mus musculus) (配列番号24); ジェンバンク受入番号NM_013257.3 (GI:25168264; Homo sapiens, variant 1); ジェンバンク受入番号NM_170709 (GI:25168266; Homo sapiens,
variant 2)がそれらに限定されずに含まれるマウスもしくはヒトCISKに、実質的に同一の配列など、CISKの任意のオルソログであってよい。Myo1cのmRNA配列は、S.セレビシエ、C.エレガンス、D.メラノガスター、ジェンバンク受入番号U22445 NM_008659 (GI:31543277; Mus
musculus) (配列番号25); NM_033375 (GI:24415399; Homo
sapiens) (配列番号26)がそれらに限定されずに含まれるマウスもしくはヒトMyo1cに、実質的に同一の配列など、Myo1cの任意のオルソログであってよい。
【0071】
本願明細書に記載の「オルソログ」という用語は、参照配列に実質的に同一の配列を意味する。本願明細書に記載の「実質的に同一の」という用語は、十分な数のもしくは最小数の同一または等価のアミノ酸残基またはヌクレオチドを含有する第1のアミノ酸またはヌクレオチド配列と、第2のアミノ酸またはヌクレオチド配列であって、前記第1および第2のアミノ酸またはヌクレオチド配列が共通の構造ドメインまたは共通の機能的活性を有する、第1のアミノ酸配列を意味する。たとえば、少なくとも約60%または65% の同一性、おそらく75% の同一性、よりおそらくは85%、90%の同一性を有する共通の構造ドメインを含有するアミノ酸またはヌクレオチド配列である。本願明細書において、91%、92%、93%、94%、95%、96%、97%、98% または99%の同一性は、実質的に同一であると定義する。
【0072】
配列間の相同性または配列同一性(前記用語は本願明細書においては交換可能である)の計算は以下の通り行われる。
【0073】
2つのアミノ酸配列、または2つの核酸配列の同一性の割合を決定するために、その配列を最適な比較の目的のためにアラインメントする(たとえば最適なアラインメントのために第1および第2のアミノ酸または核酸配列の片方または両方にギャップを導入してもよく、非相同成敗列を比較目的のために無視してもよい)。ある実施態様では、比較の目的のためのアラインメントされた参照配列の長さは、前記参照配列の長さの少なくとも50%、少なくとも60%、少なくとも70%、80%、90%、100%である。それから、対応するアミノ酸の位置またはヌクレオチドの位置にあるアミノ酸残基またはヌクレオチドを比較する。第1の配列のある位置が第2の配列の対応する位置として同一のアミノ酸残基またはヌクレオチドによって占められていれば、その分子はその位置において同一である(本願明細書に記載の通り、アミノ酸または核酸の「同一性」はアミノ酸または核酸の「相同性」と等価である)。前記の2つの配列間の同一性の割合は、前記配列によって共有された同一の位置の数の関数であり、2つの配列の最適化アラインメントに導入する必要のあるギャップ数、および各ギャップの長さを考慮する。
【0074】
配列の比較と2つの配列間の同一性の割合の決定は、数学的アルゴリズムを用いて行うことができる。ある実施態様では、2つのアミノ酸配列間の同一性の割合は、ブロッサム62マトリックスまたはPAM250マトリックスのいずれか、GCGソフトウエアパッケージ(Accelrysのオフィシャルウェブサイトで入手可能)のGAPプログラムに導入された、Needleman and
Wunsch (J. Mol. Biol.
48:444-453 (1970))アルゴリズムを用いて、16、14、12、10、8、6、または4のギャップ重みおよび1、2、3、4、5、または6の長さ重みを用いて決定される。さらに別の実施態様では、2つの核酸配列間の同一性の割合は、GCGソフトウエアパッケージ(Accelrysのオフィシャルウェブサイトで入手可能)のGAPプログラムを用いて、NWSgapdna.CMP マトリックス、および40、50、60、70、または80のギャップ重みおよび1、2、3、4、5、または6の長さ重みを用いて決定される。あるセットのパラメタ(および、分子が配列同一性または相同性の本願発明の制限内に入っているかどうかを決定するためにどのようなパラメタを用いてよいのか、実行者がよく知らない場合に用いることができるパラメタ)は、ギャップペナルティ12、ギャップの延長ペナルティ4、およびフレームシフトギャップペナルティ5を有するブロッサム62スコアリングマトリックスである。
【0075】
アミノ酸またはヌクレオチド配列間の同一性の割合は、ALIGNプログラム(バージョン2.0)に組み込まれたMeyers and W.
Miller (CABIOS, 4:11-17 (1989)) のアルゴリズムを用いて、PAM120重み残基表、ギャップ長さペナルティ12、ギャップペナルティ4を用いて、決定することができる。
【0076】
本願明細書に記載の核酸およびタンパク質配列は「クエリ配列」として用いることができ、たとえばファミリメンバまたは関連配列などのその他のオルトログを同定するための好適データベースに対する検索を実行することができる。そのような検索は、Altschul, et al.
J. Mol. Biol. 215:403-10 (1990)のNBLAST およびXBLASTプログラム (version 2.0) を用いて行うことができる。BLASTヌクレオチド探索は、本願発明の既知のTEF核酸分子に相同なヌクレオチド配列を得るために、NBLASTプログラム、スコア= 100、ワード長=12を用いて行うことができる。BLASTタンパク質探索は、既知のTEF核酸分子に相同なアミノ酸配列を得るために、XBLASTプログラム、スコア= 50、ワード長=3を用いて行うことができる。比較目的のためのギャップアラインメントを得るために、Altschul et al., (1997) Nucleic
Acids Research 25(17):3389-3402に記載のようにギャップBLASTを用いることができる。BLASTおよびギャップBLASTプログラムを用いる場合、それぞれのプログラム(たとえばXBLASTおよびNBLAST)のデフォルトパラメータを用いてよい。国立衛生研究所の国立バイオテクノロジインフォメーションセンタのウェブサイトを参照のこと。
オルソログは、たとえばヒトcDNAライブラリなどのcDNAライブラリのスクリーニングなど当業に知られる任意の他のルーチン法を用いて、参照配列に実質的に同一な配列を同定するためにデザインされたプローブを用いても、同定することができる。
【0077】
V. 機能的アッセイ
機能的アッセイは、たとえば脂肪細胞のsiRNA方法論を用いた標的細胞の減少後など、特定の遺伝子のノックダウンの効果を決定するために用いるられる。そのようなアッセイは、糖尿病においてグルコース輸送に関与する、またはエフェクタとして示唆される特定の遺伝子ノックダウンの効果を決定するために、特に有用である。そのようなアッセイは当業に知られており、たとえば例1に記載されるとおり、その一部は本願明細書に記載されている。そのようなアッセイには、デオキシグルコース取り込み、グルコース・トランスロケーションアッセイ用のまたはMYCタグGlut4をアッセイするステップが、それらに限定されずに含まれる。
【0078】
A. 細胞アッセイ
ある実施態様では、グルコース輸送に関与する遺伝子によってコードされるポリペプチド、または生物学的に活性なその部分を発現する能力を有する細胞をテスト化合物と接触させ、テスト化合物の、前記ポリペプチドまたは生物学的に活性なその部分の発現を調節する能力を決定する、細胞アッセイである。 別の実施態様では、アッセイはグルコース輸送に関与する遺伝子によってコードされるポリペプチド(または生物学的に活性なその部分)を発現させる細胞をテスト化合物と接触させ、テスト化合物の、前記ポリペプチド(または生物学的に活性なその部分)の発現を調節する能力を決定する、細胞アッセイである。たとえば、その細胞は哺乳類由来または酵母細胞であってよい。好ましい実施態様では、前記細胞は脂肪細胞である。たとえば、前記ポリペプチドは、たとえば前記細胞などの前記細胞を非相同的に発現するか、または前記細胞に由来してよい。前記テスト化合物の、グルコース輸送(または生物学的に活性なその部分)に関与する遺伝子によってコードされたポリペプチドの活性を調節する能力の決定は、たとえば本願明細書に記載のAkt1またはAkt2の活性など、当業に標準的に知られるようなポリペプチドに起因する活性の任意の1つをアッセイすることによって実施することができる。前記テスト化合物がグルコース輸送に関与する遺伝子によってコードされるポリペプチド(または生物学的に活性なその部分)の活性を調節する能力の決定は、前記ポリペプチドの基質標的分子の活性をアッセイすることによって実施することができる。ある実施態様では、前記テスト化合物がグルコース輸送に関与する遺伝子によってコードされるポリペプチドまたは生物学的に活性なその部分の活性を調節する能力の決定は、前記ポリペプチドまたは生物学的に活性なその部分の基質標的分子に結合する能力をアッセイすることによって実施することができる。ある好ましい実施態様では、前記細胞はグルコース輸送に関与する遺伝子によってコードされるポリペプチドまたは生物学的に活性なその部分を過剰発現する。
【0079】
本願明細書に記載の「生物学的に活性な」断片という用語には、たとえば細胞によるインスリン媒介グルコース取り込みなどを含む、少なくとも1つのグルコース輸送関連活性を示す、または発揮するのに十分な、グルコース輸送に関与するポリペプチドの任意の部分(たとえば近接するアミノ酸の部分)が含まれる。各種の実施態様では、グルコース輸送に関与する遺伝子は、たとえばAkt1、Akt2、CISKまたはMyo1cであってよい。
【0080】
本願発明の細胞アッセイによると、テスト化合物の、グルコース輸送に関与するポリペプチドまたは生物学的に活性なその部分の活性を調節する能力の決定は、本願明細書に記載の、Akt、CISKまたはMyo1cポリペプチドの活性と同時に生じる天然の活性または間接的な活性をアッセイすることによって決定することができる。たとえば、Akt発現細胞のインスリン依存性のグルコース取り込み能力に前記テスト化合物が与える作用は、本願テスト化合物の存在下においてアッセイすることができる。また、好ましい実施態様では、本願発明の細胞アッセイは、インスリン応答のモジュレータとしてテスト化合物を同定する最終ステップを含むことが意図される。
【0081】
B. ハイスループットアッセイ
脂肪細胞中に標的遺伝子ノックアウトを用いたハイスループットアッセイも、標的をテストするのに有用である。たとえば、脂肪細胞中に発現される核酸配列を標的とした各種のsiRNAのプールを、本願明細書に記載の方法を用いて脂肪細胞にトランスフェクトする。それから、前記のトランスフェクトされた細胞を、1つ以上の機能(たとえばグルコース輸送またはGLUT4トランスロケーション)のかく乱についてテストする。そのようなかく乱を証明するsiRNAのプールは、さらに小さいプールに分割され、そのプロセスを、認められたかく乱(作用)に関連する1つ以上の配列が同定されるまで繰り返す。この方法は、2つ以上の種類のsiRNAが細胞によって取り込まれる可能性もあるために、2つ以上の配列のノックダウンを必要とする作用を同定するためにも有用である可能性がある。そのような技術は、たとえば、糖尿病および肥満などの脂肪細胞関連疾患のための薬物発見に関連する研究などに有用である。たとえば、そのような方法は、グルコース輸送および糖尿病などの脂肪細胞に関連する機能および障害の研究に有用な薬物標的のライブラリを作成するために用いることができる。
【0082】
VI. テスト化合物
本願発明のテスト化合物は、当業に知られるコンビナトリアルライブラリの方法における多数のアプローチのいずれかを用いて得ることができる。その方法には、生物学的ライブラリ、空間指定平行固相または液層ライブラリ、畳込みを要する合成ライブラリ法、1ビーズ1化合物ライブラリ法、および親和性クロマトグラフィ選択を用いた合成ライブラリ法などが含まれる。生物学的ライブラリアプローチはペプチドライブラリに限定されるが、一方で、その他の4つのアプローチはペプチド、非ペプチドオリゴマーまたは化合物の低分子ライブラリに適用可能である(Lam, K.S. (1997)
Anticancer Drug Des. 12:145)。
【0083】
分子ライブラリの合成のための方法の例は、たとえばDeWitt et al. (1993) Proc. Natl. Acad. Sci. U.S.A. 90:6909、Erb et al.
(1994)
Proc. Natl. Acad. Sci. USA
91:11422、Zuckermann et al. (1994). J. Med. Chem.
37:2678、Cho et al. (1993)
Science 261:1303、Carrell et al. (1994) Angew. Chem. Int. Ed. Engl. 33:2059、Carell et al.
(1994) Angew. Chem. Int. Ed. Engl. 33:2061、and in Gallop et
al. (1994) J. Med. Chem.
37:1233など、当業に見いだすことができる。
【0084】
化合物のライブラリは、溶液中(e.g.,
Houghten (1992) Biotechniques 13:412-421)、ビーズ上(Lam (1991) Nature
354:82-84)、チップ(Fodor (1993) Nature 364:555-556)、細菌 (Ladner USP
5,223,409)、胞子(Ladner USP '409)、プラスミド(Cull et al.
(1992) Proc Natl Acad Sci USA
89:1865-1869)、またはファージ上(Scott and Smith (1990) Science 249:386-390)、(Devlin (1990) Science
249:404-406)、(Cwirla et al. (1990)
Proc. Natl. Acad. Sci. 87:6378-6382)、(Felici (1991) J.
Mol. Biol. 222:301-310)、(Ladner supra.)に存在することもできる。
【0085】
好ましい実施態様では、前記ライブラリは天然産生物ライブラリである。
【0086】
VII 薬学的組成物
本願発明はさらに、上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータに関連する。上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータは、好適な動物モデルにおいてテストすることができる。たとえば、本願明細書に記載のとおり同定されたインスリン応答モジュレータを動物モデルに用いて、そのようなモジュレータでの処置の有効性、毒性、または副作用を決定することができる。 代替的には、本願明細書に記載の通り同定されたモジュレータを動物モデルに用い、そのような作用薬の作用機序を決定することができる。 さらに、本願発明は、後述のの治療のための上述のスクリーニングアッセイによって同定されたインスリン応答モジュレータの使用に関連する。
【0087】
したがって、本願発明のインスリン応答モジュレータを、投与に好適な薬学的組成物に組み込むことができる。そのような組成物は、典型的には、核酸分子、タンパク質、抗体、または調節性化合物、および薬学的に許容な担体が含まれる。本願明細書に記載の「薬学的に許容な担体」という用語は、薬学的投与に適合する、溶媒、分散媒、コーティング、抗細菌および抗真菌剤、ならびに等張および吸収遅延剤など、いずれかおよびすべてを含むことを意図する。薬学的に活性な物質へのそのような媒質および物質の使用は、当業に公知である。任意の従来の媒質または作用物質が活性化合物と適合性がない場合以外は、前記組成物へのそれらの使用が考慮される。補助的な活性化合物も、当該組成物に組み込むことができる。
【0088】
本願発明の薬学的組成物は、意図する投与経路と適合するように調剤される。投与経路の例には、たとえば静脈内、皮内、皮下、口腔内(たとえば吸入)、経皮(局所)、経粘膜、および直腸投与などの非経口投与が含まれる。非経口、皮内、または皮下投与に使用する溶液または懸濁液には、以下の成分、注射用の水、食塩水、固定油、ポリエチレングリコール、グリセリン、プロピレングリコール、またはその他の合成溶媒などの滅菌希釈剤、ベンジルアルコールまたはメチルパラベンなどの抗菌剤、アスコルビン酸または亜硫酸ナトリウムなどの抗酸化剤、エチレンジアミン四酢酸などおキレート剤、酢酸塩、クエン酸塩、またはリン酸塩などのバッファ、および塩化ナトリウムまたはデキストロースなどの張力調整剤などが含まれていてよい。非経口製剤は、アンプル、ガラスまたはプラスチック製の使い捨て用シリンジ、または複数の投与用バイアルの中に封入されていてよい。
【0089】
注射に使用するために好適な薬学的組成物には、滅菌水溶液(水溶性)、または分散液、および滅菌注射用溶液または分散液の即時調整用滅菌粉末が含まれる。静脈内投与に好適な担体には、生理食塩水、静菌水、クレモホールELTM(BASF、ニュージャージー州パシッパニー)、またはリン酸緩衝食塩水(PBS)が含まれる。すべての場合において、前記組成物は滅菌状態であって、注射容器への充填が容易な程度、流動的でなければならない。前記組成物は、製造および保存の条件下において安定でなければならず、細菌および真菌などの微生物の汚染作用から保護されなければならない。前記担体は、たとえば水、エタノール、ポリオール(たとえばグリセロール、ポリエチレングリコール、および液体状ポリエチレングリコールなど)、それらの好適な混合物、ならびに植物油などを含む溶媒又は分散媒であってよい。適切な流動性は、たとえばレシチンなどのコーティングの使用、分散液の場合は所要の粒子サイズの維持、および表面活性剤の使用などによって維持することができる。微生物の作用は、たとえばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどの各種抗細菌剤および抗真菌剤によって防ぐことができる。多くの場合、たとえば糖、マニトールなどのポリアルコール、ソルビトール、塩化ナトリウムなどの等張剤を前記化合物に含めることが望ましいだろう。注射可能な組成物の吸収を持続させるために、たとえばモノステアリン酸アルミニウムおよびゼラチンなどの吸収遅延剤を組成物に含ませてもよい。
【0090】
滅菌された注射可能な溶液は、適切な溶媒中に入れた必要量の前記活性化合物を、必要に応じて上述の成分1つまたはそれらの組み合わせに組み入れてからろ過滅菌することによって調製することができる。一般に、分散液は、塩基性分散媒および上述の物質のうちの必要なその他の成分を含む滅菌担体に前記活性化合物を組み入れて調製する。滅菌注射用溶液の調製のための滅菌粉末の場合、好ましい調製方法は、前記活性成分の粉末に加えて予め滅菌ろ過されたその溶液から得られた任意の追加の望ましい成分を得られる、真空乾燥および凍結乾燥である。
【0091】
経口組成物には一般に、不活性な希釈剤または食用担体が含まれる。それらは、ゼラチンカプセルに封入されるか、または錠剤に圧縮されてよい。治療用の経口投与の場合、前記活性化合物を賦形剤に組み込み、錠剤、トローチ、またはカプセルなどの形状で用いることができる。経口用組成物はまた、洗口液として用いるための液状担体であって、当該担体において前記液状担体中の前記化合物が口腔内の塗布され、うがいして吐き出すかまたは飲み込まれる、液状担体を用いて調製することもできる。薬学的に適合な結合剤、および/またはアジュバント物質は、前記組成物の一部分として含めることもできる。前記錠剤、丸剤、カプセル、およびトローチなどは、以下の成分、もしくは同様の性質の化合物のいずれかを含むことができる。それは、微結晶性セルロース、ガムトラガカント、もしくはゼラチンなどの結合剤、デンプンもしくはラクトースなどの賦形剤、アルギン酸、プリモゲル、もしくはトウモロコシデンプンなどの崩壊剤、ステアリン酸マグネシウムもしくはステロート(Sterote)などの潤滑剤、コロイド状二酸化ケイ素などの流動促進剤、ショ糖もしくはサッカリンなどの甘味剤、またはペパーミント、サリチル酸メチル、もしくはオレンジ香味料などの香味剤である。
【0092】
吸入による投与の場合、前記化合物は、たとえば二酸化炭素などの好適な高圧ガスを含む加圧容器もしくはディスペンサ、またはネビュライザからのエアロゾルスプレーの形状で送達する。
全身投与はまた、経粘膜または経皮手段でもよい。経粘膜または経皮投与の場合、透過させようとする関門に適切な浸透剤を、前記製剤に用いる。そのような浸透剤は一般に当業に知られており、たとえば経粘膜投与の場合、界面活性剤、胆汁塩、およびフシジン酸誘導体が含まれる。経粘膜投与は、経鼻スプレーまたは坐剤の使用によって実施することができる。経皮投与の場合、前記活性化合物は、一般に当業に知られる軟膏(ointment)、軟膏(salve)、ゲル、またはクリームに調製する。
【0093】
前記化合物はまた、坐剤(たとえばカカオバターおよびその他のグリセリドなどの従来の坐剤ベース)または直腸送達用の停留浣腸の形状に調製してもよい。
【0094】
ある実施態様では、前記活性化合物は、インプラントおよびマイクロカプセル封入送達システムを含む、制御放出製剤などの、体外への急速な排出から前記化合物を保護する担体とともに調製する。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸などの生分解性、生体適合性ポリマーを用いることができる。そのような製剤の調製の方法は当業に明らかであろう。前記物質はまた、アルザ社およびノバファーマシューティカルズ社から市販されており入手することもできる。リポソーム懸濁液(ウイルス性抗原に対するモノクローナル抗体を有する感染細胞を標的とするリポソームを含む)はまた、薬学的に許容な担体として用いることもできる。これらは、米国特許第4,522,811号などに記載の、当業に知られる方法に従って調製することができる。
【0095】
投与の簡便性と製剤の均一性のために、経口または非経口組成物を投与単位剤形に調剤することは、特に有利である。本願明細書に用いられる投与単位剤形は、治療される対象に単位投与量として好適な物理的に分散した単位であって、必要な薬学的担体とともに望ましい治療効果を生じるように計算された、予め決められた量の活性化合物を含む各単位を意味する。本願発明の投与単位剤形の仕様は、前記活性化合物に特有の性質および達成されるべき特定の治療効果、ならびに個体の治療用の当該活性化合物を調合する技術に固有の限界によって支配され、およびそれらに依存する。
【0096】
当該化合物の毒性および治療上の有効性は、たとえばLD50(個体群の50%に致死的な投与量)およびED50(個体群の50%に治療上有効な投与量)など、細胞培養物または実験動物における標準的な薬学的方法によって決定することができる。毒性作用および治療上有効な作用の投与量の比は治療指数であって、LD50/ED50の比で表すことができる。大きい治療指数を示す化合物が好ましい。有毒な副作用を示す化合物を用いる場合、感染していない細胞に与える可能性のある損傷を最小にして副作用を減少するよう、そのような化合物を罹患組織の部位に狙わせる送達システムをデザインするように、注意が払われなければならない。
【0097】
細胞培養アッセイおよび動物研究から得られるデータは、ヒトにおける使用のための用量の範囲を決定する際に用いることができる。そのような化合物の用量は、好ましくは毒性が小さいかまたは皆無のED50を含む循環濃度の範囲内にある。前記用量は、用いられた投与剤形および使用された投与経路によって、この範囲内で変化させてもよい。本願発明の方法に用いられるいずれの化合物の場合も、治療上有効な投与量は、最初は細胞培養アッセイで推定することができる。1回投与量は、動物モデルにおいて、細胞培養物で決定したIC50(すなわち症状の最大半減抑制を達成する前記テスト化合物の濃度)を含む循環血漿濃度の範囲になるように調製してもよい。このような知見は、ヒトにおける有用な投与量をより正確に決定するために用いることができる。血漿濃度は、たとえば高性能液体クロマトグラフィによって測定することもできる。
【0098】
薬学的組成物は、投与法の説明書とともに、容器、パック、またはディスペンサに包含されていてよい。
【0099】
VIII. 治療方法
本願発明はまた、処置方法または治療方法も特徴とする。ある実施態様では、本願発明は、本願発明にしたがって同定された調節性化合物で、望ましい治療効果が実現できるように、対象(たとえばその必要のあるヒト対象)を処置する方法を特徴とする別の実施態様では、前記方法は、本願明細書に記載の方法論したがって同定された調節性化合物を、望ましい治療効果が実現できるように、前記対象から単離した組織または細胞株に投与するステップに関与する。好ましい実施態様では、本願発明は、たとえばインスリンへの低感受性もしくはインスリン耐性、または糖尿病(2型糖尿病)など、インスリン応答障害を有する対象を処置する方法を特徴とする。本願発明はまた、前糖尿病もしくはその症状、高血糖、および/または1型糖尿病を有する対象を処置する治療方法も提供する。好ましい治療効果はグルコース取り込みおよび/または輸送の調節である。望ましい治療効果にはまた、前記対象の治癒または治療、前記対象における疾患もしくは障害、または前記対象における前記疾患もしくは障害の少なくとも1つの症状の緩和、軽減、改変、もしくは改善、または前記対象の健康のその他の改善または作用が、それらに限定されずに含まれる。本願発明の好ましい局面は、グルコース輸送に作用するとして本願明細書に提供される方法によって同定される遺伝子を調節する方法、またはグルコース輸送またはインスリンの応答性に作用する治療目的の既知の遺伝子に関連する。
【0100】
本願明細書に開示された方法によって同定されたモジュレータを対象に用いて、インスリン応答の調節、グルコース輸送の制御、糖新生の制御、グルコース恒常性の制御、および血糖値の制御を行うこともできる。
【0101】
インスリン応答モジュレータでの対象の処置の有効性は、(i)好適なモジュレータで処置する前に、前記対象においてインスリン応答または代替的にはグルコース耐性のレベルを検出するステップと、(ii)前記モジュレータで処置後の前記対象におけるインスリン応答または代替的にはグルコース耐性のレベルを検出するステップと、(iii)投与前および投与後のレベルを比較するステップと、(iv)前記対象への前記モジュレータの投与を適宜変更するステップと、によって達成することができる。たとえば、前記対象が非感受性のインスリン応答を示し続ける場合、前記モジュレータの大量投与が望ましい場合がある。
【0102】
代替的には、インスリン応答モジュレータでの対象の処置の有効性は、(i)好適なモジュレータで処置する前に、前記対象において血糖またはグルコース耐性を検出するステップと、(ii)前記モジュレータで処置後の前記対象における前記血糖値または代替的にはグルコース耐性のレベルを検出するステップと、(iii)投与前および投与後のレベルを比較するステップと、(iv)前記対象への前記モジュレータの投与を適宜変更するステップと、によって実現することができる。たとえば、前記対象が十分に血糖を排除できない場合、前記モジュレータの大量投与または持続的投与が望ましい場合がある。
【0103】
IX. ノックアウトおよび/またはノックダウン細胞または生物
本願発明のsiRNA分子(またはベクターもしくはそれをコードする導入遺伝子)のさらなる好ましい使用は、真核細胞、または真核性非ヒト生物、好ましくは哺乳類細胞または生物、および最も好ましくはヒト細胞において行われる機能性分析である。標的特異的RNA干渉を方向付けるために、標的mRNA配列に十分相補的な、好適なsiRNA分子を投与することによって、脂肪細胞などの細胞培養物、または標的生物などの標的細胞において、特定のノックアウトまたはノックダウン表現型を得ることができる。
【0104】
したがって、本願発明のさらなる対象物質は、たとえば脂肪細胞などの真核細胞、または少なくとも1つの内因性標的遺伝子の完全なまたは少なくとも部分的に不足した発現を含む標的遺伝子特異的ノックアウトまたはノックダウン表現型を示す非ヒト真核性生物であって、前記細胞または生物が前記標的遺伝子の発現を阻害する能力があるsiRNA分子をコードするDNAを含む少なくとも1つのベクタをトランスフェクト(たとえば電気穿孔法によって)された、細胞または生物である。本願発明は、前記siRNAの特異性による数種類の異なる内因性遺伝子の標的特異的なノックアウトまたはノックダウンを可能にするという点は特筆されるべきである。
【0105】
細胞または非ヒト生物、特にヒト細胞または非ヒト哺乳類の遺伝子特異的ノックアウトまたはノックダウン表現型を、たとえば、遺伝子発現プロフィールおよび/またはプロテオームの分析など、複雑な生理学的プロセスの機能性および/または表現型分析など、分析手順に用いてもよい。好ましくは、前記分析は、オリゴヌクレオチドを用いたチップを使用したハイスループット方法によって行われる。
【0106】
RNAiを用いたノックアウトまたはノックダウン技術を使用して、内因性標的遺伝子の発現を、標的細胞または標的生物において阻害することもできる。前記内因性遺伝子は、たとえばアフィニティタグ、特に多重アフィニティタグなどの検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列に、任意で融合することができる、遺伝子またはDNAなど、前記標的タンパク質または標的タンパク質の変異体もしくは突然変異型をコードする外因性標的核酸によって補完されてもよい。
【0107】
前記標的遺伝子の変異体または突然変異型は、前記内因性遺伝子産生物と、1アミノ酸または複数のアミノ酸の置換、挿入、および/または削除によるアミノ酸レベルで異なる遺伝子産生物をコードする点において、前記内因性標的遺伝子と異なる。前記変異体または突然変異型は、前記内因性標的遺伝子と同一の生物学的活性を有する場合もある。その一方で、前記変異体または突然変異型標的遺伝子は、たとえば部分的に欠失した活性、相補的に欠失した活性、高い活性など、内因性標的遺伝子の生物学的活性と異なる生物学的活性も有していてよい。補完は、たとえば前記標的タンパク質、および前記アフィニティタグ、ならびに前記標的細胞中の内因性遺伝子をノックアウトするための2本鎖RNA分子など、前記内因性核酸によってコードされたポリペプチドを含むことによって、達成することもできる。この加圧は、たとえば前記タグ修飾標的タンパク質などの内因性核酸によってコードされたポリペプチド、および前記2本鎖RNA分子の両方を発現する好適な発現ベクタを用いて、または代替的には発現ベクタの組み合わせを用いることによって達成することもできる。前記標的細胞においてde novoで合成されたタンパク質およびタンパク質複合体は、たとえば修飾された融合タンパク質など、内因性遺伝子産生物を含有するだろう。前記siRNAi分子による外因性遺伝子産生物の抑制を避けるために、前記外因性核酸をコードする核酸配列を、前記siRNA分子に相同的な配列の一部分においてDNAレベルで(アミノ酸レベルでの突然変異を生じて、または生じずに)変化させてもよい。代替的には、前記内因性標的遺伝子は、たとえばマウスなどその他の種から得た対応するヌクレオチド配列によって補完させることもできる。
【0108】
使用
本願明細書に記載の方法は、発現が脂肪細胞の発達または代謝に影響を与える遺伝子を同定するために有用である。さらに、特定の遺伝子をノックダウンする方法を用いることによって、細胞上のそのようなノックダウンの作用は、薬物のスクリーニングに有用であると特徴づけることができる。たとえば、脂肪細胞で発現するタンパク質の発現または活性に特異的に影響する薬物の望ましい作用は、前記タンパク質をコードする配列がノックダウンされた脂肪細胞を調べることによって、予測することができる。そのような細胞の表現型は、前記標的遺伝子の発現または活性を阻害するために有用な薬物の特徴を同定するための指針を提供する。
【実施例】
【0109】
例1材料および方法
材料
ヒトインスリンはイーライリリー社から入手した。ヤギポリクローナル抗Akt1抗体(C末端sc-7126近傍の抗原性ヒトAkt1ペプチド)、HRP結合ロバ抗ヤギIgG、マウスモノクローナル抗ラミンA/C (sc7293)、およびモノクローナルGSK3α/βは、サンタクルズバイオテク社(カリフォルニア州サンタクルズ)から入手した。ウサギポリクローナル抗Akt2抗体(ヒトAkt2のC末端にある抗原性ペプチド)は、Morris J. Birnbaum博士(ペンシルバニア大学、Hill et al.,
1999, Mol. Cell. Biol. 19:7771-7781参照)に提供していただいた。Acrp30に対するウサギポリクローナル抗体は、アフィニティ・バイオリエージェント社(コロラド州ゴールデン)から、および非筋肉ミオシンIIBに対する抗体はコバンス社(カリフォルニア州リッチモンド)から入手した。ホスホAktスレオニン308/309、ホスホGSK3α/β(ser21/9)、およびホスホErk1/2 に対するポリクローナル抗体は、セル・シグナリング・テクノロジー社(マサチューセッツ州ベバリー)から入手した。フルオレセインイソチアネート(FITC)結合ヤギ抗マウス抗体はバイオソース・インターナショナル社(カリフォルニア州カマリロ)から入手した。
【0110】
siRNA 2本鎖のデザイン及び合成
RNAオリゴヌクレオチドの21マー・センスおよびアンチセンス鎖を、Elbashir et al.,
2001, Nature 411:94に記載の通りデザインした。RNAオリゴヌクレオチドは、核酸合成器で合成され、ヌクレアーゼ分解を防ぐために2’オルソエステルで保護し、その後、ダーマコン・リサーチ社におけるアニオン交換HPLCで精製した。それから、RNAオリゴヌクレオチドのセンスおよびアンチセンス鎖を、2’脱保護し、アニーリングして、凍結乾燥させ、siRNAを作成した。そのsiRNA 2本鎖を75%エタノールで2回洗浄して脱塩し、Speed-Vacを用いて乾燥させ、そのペレットを無ヌクレアーゼ水に再懸濁させてから培養した脂肪にトランスフェクションした。
【0111】
3T3L1脂肪細胞の細胞培養および電気穿孔
3T3-L1線維芽細胞を、10%ウシ胎仔血清、50μg/mlストレプトマイシン、および50ユニット/mlペニシリンを補充したDMEM培地で成長させた。それは、Harrison et al. (1990,
J. Biol. Chem. 265:20106)に既報の通り、脂肪細胞に分化した。3T3-L1脂肪細胞に、電気穿孔法でsiRNA 2本鎖をトランスフェクトした。簡単に説明すると、前記脂肪細胞の分化5日目に、0.25%トリプシンおよび1mlあたり0.5mgのコラゲナーゼのリン酸バッファ食塩水(PBS)溶液を加えた培養皿から剥離させて、2回洗浄し、PBSに再懸濁させた。その後、約500万個の細胞(p150培養皿1枚の半分の細胞)を、バイオラドジーンパルサIIシステム(バイオラドラボラトリーズ社、カリフォルニア州ハーキュリーズ)で、0.18kV、電気容量960μFの設定で、電気穿孔のパルスによって前記細胞に移したsiRNA 2本鎖と混合した。前記電気穿孔は、細胞450μlを用いて行われた。電気穿孔後、細胞をただちに新鮮培地と混合し、37℃で10分間インキュベートしてからデオキシグルコース取り込みアッセイ、ウエスタンブロッティング、および免疫蛍光顕微分析法のためにデザインされた、多穴プレートに再播種した。分析は一般に、再播種から24または48時間後に行った。
【0112】
免疫蛍光顕微鏡検査
ラミンA/Cを可視化するために、細胞を4%ホルムアルデヒドで固定して、1% FBSおよび0.5%トリトンTMX-100を含有するPBSで透過化した。その後、細胞を1次マウス抗ラットラミンA/C抗体で4℃で一晩インキュベートした。洗浄後、前記細胞をFITC標識ヤギ抗マウスIgGで30分間、室温でインキュベートした。洗浄後、2.5%DABCOを含有する90%グリセロール中でカバースリップをのせた。蛍光顕微鏡検査を、CCDカメラ(ロパー・サイエンティフィック社ニュージャージー州トレントン)およびメタモルフ画像処理ソフトウェア(ユニバーサルイメージング社、ペンシルバニア州ダウニングトン)を装着したIX70倒立顕微鏡(米国オリンパス社、ニューヨーク州メルビル)で行った。
【0113】
ウエスタンブロッティング法
実験的な処理の後、前記細胞を25mM Hepes、pH7.4, 1% NP40、100 mM NaCl、5 mM フッ化ナトリウム、1 mM EDTA、1 mM バナジウム酸ナトリウム、5 mM ピロリン酸ナトリウム、1 mM フッ化フェニルメチルスルホニル(PMSF)、10 μg/ml アプロチニン、および5 μg/ml ロイペプチンに溶解した。Aktスレオニン308/309、GSK3α/βセリン21/9のリン酸化、およびErk1/2のチロシンリン酸化を検出するために、3T3-L1 脂肪細胞ライセートから得た50μgタンパク質を8% SDS-PAGEに溶解し、ニトロセルロース膜に電気移動させた。その後、その膜を抗ホスホ特異的抗体(1:1000希釈)で、4℃で一晩インキュベートしてから、西洋ワサビペルオキシダーゼ(HRP)結合抗ウサギIg抗体(1:10,000希釈)中で、室温で1時間インキュベートした。その膜を、各抗体でインキュベートした後、水バッファ(PBS pH 7.4、0.1% ツイーン 20)で室温で1時間、洗浄した。最後に、セリンリン酸化Aktの量をECLTM キットを用いて検出した。Akt1を、1次ヤギポリクローナル抗体(1:750希釈)および2次HRP結合ロバ抗ヤギ抗体で検出した。Akt2に対する1次ウサギポリクローナル抗体(1:1000希釈)、非筋肉ミオシンIIB (0.1 μg/ml)およびAcrp30 (0.5 μg/ml) を、全細胞ライセートから得たタンパク質25μgを用いてその抗原を検出するために使用した。同じニトロセルロース膜を用いて数種類のタンパク質およびホスホタンパク質を検出するために、前記のブロットをストリッピングバッファ(62.5 mM トリス-HCl、pH 6.7、100 mM 2-メルカプトエタノール、および2% SDS)中で、30乃至45分間、60℃で緩やかに振とうさせてインキュベートし、洗浄用バッファで少なくとも1時間洗浄してから、次の実験用にデザインされた抗体で再ブロッティングした。
【0114】
2-デオキシグルコース取り込みアッセイ
3T3-L1 脂肪細胞におけるインスリン刺激グルコース輸送を、2-デオキシグルコース取り込みを測定することによって推測した。簡単に説明すると、siRNAトランスフェクト細胞を12穴プレート上に再播種し、40時間培養してから、DMEM培地で2回洗浄して、0.5%ウシ血清アルブミン(BSA)を含有するDMEM培地で37℃で4時間インキュベートした。それから、細胞をクレブス-リンガーHepes(KRH)バッファ(130 mM NaCl、5 mM KCl、1.3 mM CaCl2、1.3 mM MgSO4、25 mM Hepes [pH
7.4])で2回洗浄し、さらに、0.5% BSAおよび2 mMピルビン酸ナトリウムを捕捉したKRHバッファ中で1.5時間飢餓状態にした。それから、細胞をインスリンで37℃で30分間刺激した。グルコース取り込みは、[1,2-3H]
2-デオキシ-D-グルコース の添加で開始し、37℃で5分間かけて最終アッセイ濃度100μMにした。アッセイは氷冷KRHバッファで4回洗浄して終了し、前記細胞を0.4mlの1%TritonTM
X-100で可溶化し、トリチウムの取り込み量をシンチレーション計数で測定した。非特異的なデオキシグルコース取り込みを20 μMシトカラシンBの存在下で測定し、それぞれの測定値から差し引いて特異的な取り込みを得た。
【0115】
Myo1cのsiRNA誘導分解
スクランブルしたsiRNA 6 もしくはsiRNA 6 単体のいずれかを20nmol、またはsiRNA2およびsiRNA 6それぞれ10nmolずつを、分化した3T3-L1 脂肪細胞に電気穿孔して挿入した。電気穿孔後、前記細胞を24穴プレートに再播種し、24時間安置した。その後、グルコース取り込みを測定した。スクランブルsiRNAトランスフェクト細胞中の基本取り込み量を1に正規化して対照として用い、増加倍数を計算した。対応のあるt検定を用いて、siRNA 6およびsiRNA2の混合物の存在下に対する、siRNA 6単体の存在下における2-デオキシグルコース取り込みの減少率を求めた。これらの細胞の一部分は、Myo1c、myosin 2bおよびEHD2について、ウェスタンブロット法で分析した。
【0116】
例2:脂肪細胞へのsiRNAの導入
最初の実験を行ったところ、他の細胞種においてsiRNA媒介遺伝子サイレンシングのために開発された条件(Elbashir et al.,
2001, Nature 411:94) は3T3-L1 線維芽細胞では有効に働くが(図1B、右図)、3T3-L1脂肪細胞では有効に働かないことが実証された。 オリゴフェクタミンTMなどのその他の細胞種において有効なトランスフェクションの他の手段では、脂肪細胞に対して相当の毒性を示した。
【0117】
siRNAを用いた技術を脂肪細胞に応用するために、代替的な方法論を開発した。前記技術の開発には、例1に記載の方法を用いて、3T3-L1 脂肪細胞の電気穿孔およびマウスラミンA/Cを標的としたCy3タグsiRNA
2本鎖(図 1A; センスおよびアンチセンス鎖、それぞれ配列番号 1および2、配列番号19のマウスラミンA/Cを標的とする)を用いた。この新規の技術を用いて、Cy3-siRNAを培養した脂肪細胞に実質的に100%の効率で導入し、48時間後までに、スクランブルCy3タグsiRNA種をトランスフェクトした細胞と比較して、ほぼすべての細胞が核ラミンA/Cの喪失を示した(それぞれ配列番号3および4のセンスおよびアンチセンス鎖)(図1B、左図)。これらの結果を定量化したところ、20nmolのsiRNAを5 x 10 6 個の脂肪細胞の懸濁液に加えると、毒性が検出されない約90%の脂肪細胞においてラミンA/Cが喪失する結果となることが示された。これらの知見は、インスリン感受性培養脂肪細胞において毒性が低く信頼性があり有効な遺伝子サイレンシングの方法の基礎を提供する。
【0118】
前記培養物中の実質的にすべての細胞は、siRNAに方向付けられた遺伝子サイレンシングに応答した目的のタンパク質の低発現を示す。これは、脂肪細胞中にプラスミドベクタをトランスフェクトした後のcDNAコードタンパク質の発現とは異なる。この場合、ほとんどの場合において1乃至10%の細胞しか非相同性のタンパク質を発現しない。したがって、この方法を用いたsiRNAに方向付けた遺伝子サイレンシングは、1細胞アッセイが困難な機能について特定の遺伝子産生物の役割を評価するために有用である。ある例はインスリン刺激グルコース輸送である。siRNAなどの核酸を脂肪細胞に導入する新規方法は、たとえばmycタグGLUT-GFPの使用で評価したGLUT4トランスロケーションなどの1細胞アッセイと併用することができる。
【0119】
例3:Akt発現の阻害
核酸を脂肪細胞に導入する新規の方法をさらに調べるために、Aktイソ型であるAkt1およびAkt2のそれぞれに対する2つのsiRNA種を、3T3-L1 脂肪細胞におけるこれらタンパク質キナーゼの発現を阻害する能力についてテストした(図3A乃至B)。Akt1およびAkt2 siRNA 2本鎖は、国立バイオテクノロジー情報センターのデータベースから得たマウスAkt1およびAkt2 mRNA 配列(それぞれ配列番号20および21)にしたがってデザインした。
【0120】
Akt1を標的としたsiRNAは、図2に記載のAkt1の2種類の標的配列“akt1a”および“akt1b”(5’-AACGAUGGCACCUUUAUUGGC; 配列番号 5 および5’-AACCAGGACCACGAGAAGCUG; 配列番号 6)のうちの1つを標的とした。siRNA標的配列“akt1a” のアンチセンス鎖は配列5’-
GCCAAUAAAGGUGCCAUCGdTdT (配列番号 9)を有し、siRNA標的配列“akt1b” のアンチセンス鎖は配列5’- CAGCUUCUCGUGGUCCUGGdTdT (配列番号 10)を有した。Akt2を標的としたsiRNAは、図2に記載のAkt2の2種類の標的配列“akt2a”および“akt2b”(5’-AAACUCCUCGGCAAGGGCACC; 配列番号 7 および5’-AACCAGGACCACGAGCGCCUC; 配列番号 8)のうちの1つを標的とした。siRNA標的配列“akt1a” のアンチセンス鎖は配列5’-
GGUGCCCUUGCCGAGGAGUdTdT (配列番号 11)を有し、siRNA標的配列“akt1b” のアンチセンス鎖は配列5’- GAGGCGCUCGUGGUCCUGGdTdT (配列番号 12)を有した。
【0121】
これらの配列を標的としたsiRNAを、例1に記載の細胞に導入した。Akt1に方向付けられたsiRNA種はそれぞれ、トランスフェクションから24および48時間後の両方の時点でAkt1の発現を阻害した。これらの1つ(akt1b)は、48時間経過時点までに実質的に完全なAkt1の除去を方向付けているが、Akt2の発現には影響はなかった(図3B、4AおよびB)。同様に、Akt2の発現は、siRNA種のakt2bのトランスフェクション後、選択的に約70%低下させることができたが、akt2a siRNAの作用は小さかった(図3B、4Aおよび4B)。最も作用の大きかったakt1bおよびakt2b siRNA種は、Akt1およびAkt2 mRNA 配列の類似の領域を標的とした。これらの配列はAkt1およびAkt2において、それぞれアミノ酸351乃至357 および352乃至358をコードしている(図3A)。それぞれの標的mRNAをサイレンシングするための、akt2b siRNAに対するakt1bの選択性は、21ヌクレオチド中4ヌクレオチドが異なるだけでも明らかである(図1A)。これは、これらのsiRNAをこれらの類似しているが異なる配列を標的用に使用することの特異性を実証している。数種類のその他の無関係なタンパク質(たとえば図3、4A、および6Aに示されているミオシンIIb、および図4Aおよび6Aに示されているAcrp30)の発現には、akt1bまたはakt2b siRNAは影響を与えなかった。このことからも、さらに前記方法の特異性が実証される。
【0122】
Akt1およびAkt2は、活性Tループのスレオニン308または309にあるタンパク質キナーゼPDK1でリン酸化して活性化する。そしてさらなる活性化はセリン473(Akt1)または474(Akt2)におけるリン酸化によって生じる(Alessi et al., 1997, Curr. Biol. 7:776;
Williams et al., 2001, Curr. Biol. 10:439; Brazil et al., 2001, Trends Biochem.
Sci. 26:657)。両方のタンパク質に含有される完全スレオニン308/309 のインスリン刺激リン酸化への、Akt2に対するAkt1の選択的喪失の効果は、抗ホスホThr308抗体を用いたウェスタンブロッティングによって評価した。Akt1タンパク質の完全喪失によって、培養3T3-L1脂肪細胞中のAktタンパク質キナーゼの完全Thr308リン酸化において、10乃至20%しか減少せず、脂肪細胞においてAkt1の量がAkt2よりもはるかに少ないということを示している既報の結果と一致する(Hill et al., 1999, Mol. Cell. Biol.
19:7771; Summers et al., 1999, J. Biol. Chem. 274:23858; Calera et al., 1998,
J. Biol. Chem. 273:7201)。反対に、約70%のAkt2発現の減少により、Aktタンパク質キナーゼのインスリン刺激スレオニンリン酸化が55乃至60%と顕著に減少した。これらをもとに、これらのデータから、インスリン感受性培養脂肪細胞においてAkt1よりもAkt2のほうが優位であることが確認された。
【0123】
Akt1またはAkt2発現の選択的低下がインスリンシグナル伝達の下流標的であるグリコーゲンシンターゼキナーゼ(GSK)3に与える影響(Cross et al.,
1995, Nature 378:785; Cohen et al., 2001, Nat. Rev. Mol. Cell. Biol. 2:769) を評価した。GSK3αは、投与量依存的に前記ホルモンに応じてAktタンパク質キナーゼによってリン酸化されると考えられている。3つの独立した実験において、akt1b siRNAによって方向付けられているAkt1の
95%以上が失われても、インスリン媒介GSK3αリン酸化の有意な低下は生じなかったが(図5Aおよび5B)、これらの研究において10乃至20%の作用は検出されていないかもしれない。反対に、Akt2発現が約70%低下すると、インスリン媒介GSK3αリン酸化の約40%の阻害を生じた(図5A、5B、6Aおよび 6C)。対照研究では、Akt1またはAkt2のいずれかが減少しても、MAPキナーゼErk-1およびErk-2 へのインスリンシグナル伝達の低減は認められず(図5Cおよび5D)、この方法によるAktタンパク質キナーゼのサイレンシング作用の特異性が確認された。これらのデータは、培養脂肪細胞中のGSK3αへのインスリンの作用は特異的にAkt2を要することを示している。したがって、Akt2の発現または活性の阻害を用いて、インスリン媒介GSK3αリン酸化を阻害することができる。
【0124】
この同一のアプローチをヘキソース輸送の調節に応用したところ、Akt1発現を除去すると(図6A)、3T3-L1 脂肪細胞におけるインスリン刺激2-デオキシグルコース取り込みに、小さいが有意な20乃至30%の減少が生じた。Akt2タンパク質を正常値の約70%減少させたところ、インスリンの応答は50乃至58%低下した。これらのデータは、Akt1およびAkt2の両方とも、これらの細胞における完全活性化Aktへの寄与にだいたい比例して培養脂肪細胞におけるヘキソース輸送のインスリンの応答性に寄与することを示している。このような作用を、akt1bおよびakt2b siRNAの組み合わせを用いて3T3-L1脂肪細胞におけるAkt1およびAkt2の両方の減少についてテストした。この併用処置は実質的に完全にAkt1発現を除去し、Akt2発現を65%超低減させる一方で、抗ホスホスレオニン308/309 抗体によって検出された完全Aktのインスリン刺激リン酸化は81%低減した(図6A)。重要なことに、インスリン刺激デオキシグルコース取り込みは、このような条件下において約80%阻害されが、それに対し、Akt2だけを減少させた場合には約58%だった(図6C)。このような条件下において、GLUT4発現には変化がなかった。
【0125】
これらのデータは、Akt2はこの応答において主要なタンパク質キナーゼだが、Akt1も重要な役割を果たすことができることを実証している。したがって、インスリン刺激デオキシグルコース取り込みがAkt2の部分的な減少によって有意に障害される条件下では、残存するインスリンシグナルの半分にはAkt1が必要となる(図6C)。インスリンに応じたGSK3αリン酸化も、Akt2だけが減少した場合に対して両方のタンパク質キナーゼが減少した場合ではより大きく阻害された(図6Aおよび6C)。
【0126】
これらのデータは、たとえばAkt1またはAkt2の発現または活性が欠乏している場合、Akt1またはAktの発現または活性を上げるのはインスリン刺激グルコース取り込みを刺激するのに有用である。これはまた、糖尿病などの脂肪細胞関連疾患の治療の標的を同定するためにsiRNAを脂肪細胞に導入する方法の有用性を実証する。
【0127】
本願明細書に提示した知見は、グルコース輸送およびGSK3αへの正常なインスリンシグナル伝達へのAktタンパク質キナーゼの絶対的な要求を示しており、使用可能なAkt1とAkt2の合計はインスリン応答の程度に正比例していることを示唆している(図6)。この結論は、全Aktの活性化およびグルコース輸送に認められた類似のインスリン用量反応関係性と一致している。Akt1、Akt2または両方の進行的な喪失は、対応するグルコース輸送刺激の進行的な喪失を生じる(図6)。このような考察は、Akt1が部分的にAkt2に代わってグルコース輸送応答性を維持することができることを示している。したがって、Akt1の発現または活性の増大はグルコース輸送応答性の増大に有用である。
【0128】
次に、siRNAによるAkt1およびAkt2の両方の欠乏の組み合わせがインスリン媒介GLUT4に与える影響を、3T3-L1 脂肪細胞で調べた。myc-GLUT4-EGFP プラスミドDNAと、akt1bおよび akt2b siRNAの混合物のコトランスフェクションを行ったところ、48時間後のAkt1およびAkt2タンパク質レベルはそれぞれ約90%および65%の減少が認められた(図7B)。この、Akt1およびAkt2タンパク質の組み合わせノックダウンによって、Myc-GLUT4-GFP作製物をトランスフェクトした脂肪細胞の約70%のインスリン刺激細胞表面Myシグナル(抗Myc Abにより検出)が喪失した(図7AおよびC)。陽性トランスフェクト細胞における全Myc-GLUT4-GFP シグナルに対する細胞表面Myc rim シグナルの割合を定量化したところ、Akt1 およびAkt2の両方の喪失によって前記細胞表面上のインスリン刺激Myc-GLUT4-GFPが60%減少したことが明らかになった。GLUT4応答性の低下は、インスリンの最大濃度および亜最大濃度の両方において認められるが(図7C)、インスリンの欠如下においては、GLUT4転移にはAktの低下の有意な作用は検出されない。
【0129】
要約すると、これらのデータはインスリンのグルコース輸送への作用にはAktが絶対的に要求されていることを実証している。また、たとえばsiRNAなどの核酸を脂肪細胞に導入する方法の有効性を実証している。
【0130】
例4:インスリンシグナル伝達遺伝子のサイレンシング
インスリンシグナル伝達に特に関連する遺伝子が、培養脂肪細胞中のsiRNAでサイレンシングできるかどうかをテストするために、実験を行った。
【0131】
脂肪細胞ライセートのウェスタンブロット分析法を、Akt関連タンパク質キナーゼCISKのsiRNAに方向付けられた遺伝子サイレンシングにしたがって行った。CISKはN末端においてPHドメインではなくPXドメインを含んでいるが、前記タンパク質キナーゼドメインのAktと約50%の配列同一性を示す。CISKはCOS細胞ではIGF-1およびEGFによって活性化され、3T3-L1 脂肪細胞のCISKの活性化はGLUT4トランスロケーションを刺激することができる。したがって、オープンリーディングフレームの配列に対して方向付けられたsiRNAによってCISK発現を低下させる能力をテストした。そのような目的の遺伝子の場合、一般に、数種類の異なるsiRNA種をテスト用に調製する。CISKに対して方向付けられたsiRNA候補を、当業に知られる方法を用いてデザインし、本願明細書に記載の方法を用いてテストした。この実験は、CISKを標的とするそのような2種類のsiRNA種はCISKのサイレンシングに有効であるということを実証した。siRNA標的CISKは、標準技術を用いて合成した(ダーマコン社)。テストした両方のsiRNA CISK種は、トランスフェクションから48時間後のCISK発現の選択的減少において有効だった。24時間経過時点において有意な作用もあったが、48時間経過時点ほど完ぺきではなかった。培養脂肪細胞にラミンに対するsiRNAをトランスフェクトしても、トランスフェクトされなかった細胞と比較して、CISK発現の減少を生じなかった。
【0132】
もう一つの対照として、同様に脂肪細胞で大量に発現される、無関係なタンパク質EHD2のウェスタンブロッティングを行った。EHD2の発現では、CISKに方向付けられたsiRNAトランスフェクションの効果がわずかであるか全くないことが示された。
【0133】
同様に、特殊なミオシンMyolcに対する数種類のsiRNA種をテストした。siRNA(ダーマコン社)を、以下のMyo1c mRNA配列、スクランブル、 5’- CAGUCGCGUUUGCGACUGG (配列番号13)、siRNA 2, 5’-
AAGGCGUUGUACAGCCGGACAUU (配列番号14)、siRNA 6, 5’- AAGCUUCCAGACAGGGAUCCAUG (配列番号15)を標的とするようにデザインした。対応するMyo1c siRNAは、以下の配列、スクランブル、 5’- CCAGUCGCAAACGCGACUGdTdT
(配列番号 16)、siRNA 2, 5’-
UGUCCGGCUGUACAACGCCdTdT (配列番号17)、 および siRNA 6, 5’- UGGAUCCCUGUCUGGAAGCdTdT (配列番号18)を有するアンチセンス鎖を含んだ。
【0134】
トランスフェクションから24時間後、Myo1cタンパク質の量は2種類のsiRNA種の組み合わせをトランスフェクトした細胞において約40%減少し、1種類のsiRNA種をトランスフェクトした細胞では約25%減少した(図8A、B)。2種類のその他の対照タンパク質、ミオシン2bおよびEHD2の発現は、影響を受けなかった(図8A)。siRNA
6またはsiRNA 6およびsiRNA2の両方をトランスフェクトした脂肪細胞の、インスリン刺激2-デオキシグルコース取り込みは、両方とも1nMおよび100nMインスリンの濃度のスクランブルsiRNAをトランスフェクトした細胞と比較して、有意に阻害された。このような結果は、siRNAによるMyo1c発現の減少が、対応するインスリンへのGLUT4応答性の低下を生じることを示唆した。
【0135】
これらのデータは、siRNAに方向付けられた遺伝子サイレンシングを脂肪細胞において、たとえばインスリンシグナル伝達経路におけるタンパク質の機能を評価するための遺伝子発見、および薬物標的の確認のために用いることができるという、さらなる証拠を提供する。
【0136】
その他の実施態様
本願発明は、発明の詳細な説明に関連して説明されているが、上述の説明は具体例を示すことを意図しており本願発明の範囲を制限することは意図しておらず、それは添付の特許請求の範囲によって定義されると理解される。その他の局面、利点、および改善点は、添付の特許請求の範囲内である。
【図面の簡単な説明】
【0137】
【図1A】Cy3標識ラミンA/C およびスクランブルsiRNA 2本鎖の配列を示すチャート。ラミンA/C siRNA は、NCBIデータベースから得たマウスラミンA mRNA配列(受入番号 BC015302) (配列番号 19) にしたがってデザインされており、マウスESTデータベースにしたがってマウスラミンA/C に固有である。ラミンA/C siRNA は配列番号1のセンス鎖、および配列番号2のアンチセンス鎖を含む。マウススクランブルsiRNAは、センス鎖がマウスESTデータベースにおいて得られた任意の遺伝子に相同性を示さない、ランダム配列としてデザインされた。
【図1B】蛍光マイクログラフの一連の複製(1a、1b、2a、2b、3a、3b、4a、および4bとして標識)で、細胞500万個につき80nmolの濃度による電気穿孔によって、Cy3-標識マウスラミンA/CまたはスクランブルsiRNA 2本鎖をトランスフェクトした3T3-L1脂肪細胞(5日目)を示す。48時間再播種した後、ラミンA/Cの核膜局在化を、本願明細書に記載の通り、マウスラミンA/C に対する抗体を用いた免疫蛍光によって可視化した。
【図2】記載の濃度におけるCy3標識スクランブルsiRNAまたはラミンA/C siRNA を分化した脂肪細胞(5日目)に電気穿孔した実験の結果を示す棒グラフ。抗ラミンA/C抗体の免疫染色後、ラミンA/C染色をした細胞を計数した。データは3つの独立した実験の平均±SD を示す。
【図3A】Akt1およびAkt2タンパク質、Akt1およびAkt2 mRNA、ならびにAkt1 (配列番号5および6) およびAkt2 (配列番号 7および8)のsiRNA標的配列の模式図。
【図3B】24または48時間で示された、siRNAの導入後の脂肪細胞ライセートのウェスタンブロットのホホイメージ。ブロットは、Akt1、Akt2およびミオシンIIBについて染色した。Akt1およびAkt2 siRNA 2本鎖は、NCBIのデータベースから得たマウスAkt1およびAkt2 mRNA 配列(それぞれ受入番号NM_009652、配列番号20および受入番号U22445、配列番号21)にしたがってデザインした。Sc: スクランブル、A1a: akt1a siRNA、A1b: akt1b siRNA、A2a: akt2a siRNA、A2b: akt2b siRNA。
【図4A】電気穿孔によってsiRNA(40nmol siRNA 2本鎖/5 x 106細胞)をトランスフェクトし、42時間再播種し、6時間血清を欠乏状態にしてから15分間、37℃でインスリンで処理した3T3-L1 の脂肪細胞の脂肪細胞ライセートのウェスタンブロットのホスホイメージ。タンパク質の完全細胞ライセート50Tg を、Akt1およびホスホ-Thr308/309 Aktの検出に用いた。タンパク質25 Tgを、Akt2、非筋肉ミオシンIIBおよびAcrp30の検出に用いた。
【図4B】Akt1b、またはAkt2b、またはスクランブルsiRNAの導入後の、Akt1およびAkt2タンパク質量の棒グラフ。
【図4C】スクランブル、Akt1b、またはAkt2b siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のAktスレオニン308/309 リン酸化の棒グラフ。データは3つの独立した実験の平均±SD として示される。
【図5A】siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のホスホ-GSK3およびGSK3を検出するウェスタンブロットのホスフォイメージ。
【図5B】siRNAの導入後、0、l nM、または100 nMインスリン中でインキュベーションした後のホスホ-Erk1/2およびErk1/2 を検出するウェスタンブロットのホスフォイメージ。
【図5C】GSK3I タンパク質バンドの強度に対するホスホGSK3I バンドの強度の割合の測定によって定量化したGSK3I のリン酸化を示す棒グラフ。
【図5D】Erk1/2 タンパク質バンドの強度に対するホスホErk1/2 バンドの強度の割合の測定によって定量化したErk1/2 のリン酸化を示す棒グラフ。データは3つの独立した実験の平均±SD として示される。
【図6A】Aktタンパク質レベル、ホスホスレオニン308/309レベル、およびホスホセリン21-GSK3Iレベルの代表的なウェスタンブロット画像のホスフォイメージ。
【図6B】インスリン刺激デオキシグルコース取り込みの用量依存性を示す実験の棒グラフ。定量的なデータは4つの独立した実験の平均±SD として示される。
【図6C】100nMインスリン誘導グルコース取り込みの阻害、およびノックダウンAkt1およびAkt2タンパク質レベルによるおよびGSK3Iリン酸化を示す実験の棒グラフ。定量的なデータは3つの独立した実験の平均±SD として示される。
【図7A】GFP陽性細胞および顔面外Myc染色の代表的な画像。使用したインスリンの濃度は100nMであった。細胞の辺縁のMycシグナルの輝度が最大の細胞の中心部分に近い光学平面において、Myc染色およびGFPシグナルの画像を撮影した。
【図7B】Myc-Glut4-EGFP およびsiRNAを48時間トランスフェクトした脂肪細胞のAktタンパク質レベルを示すウェスタンブロット。
【図7C】細胞表面上のMyc-GLUT4-GFPを示すトランスフェクト脂肪細胞の割合を示す棒グラフ。データは3つの独立した実験であって、各実験において200個を超える細胞が計数された実験の平均±SD として示される。
【図7D】Myc-GLUT4-GFPを発現する脂肪細胞における合計GFPシグナルに対する細胞表面Mycシグナルの割合を示す棒グラフ。使用したインスリンの濃度は100nMであった。平均±SDとして示されたデータの比較は、対応のないスチューデントt検定を用いることによって行った。*, P < 0.001 はスクランブルsiRNAをトランスフェクトした細胞とakt1b/akt2b siRNAをトランスフェクトした脂肪の差。Scr スクランブルsiRNA、1b akt1b siRNA、2b akt2b siRNA。
【図8A】スクランブルsiRNA (レーン 1)、siRNA 6 (レーン 2)、またはsiRNAs 6 および2(レーン3)の発現後の、分化した3T3-L1 脂肪細胞中のMyo1、ミオシン2bおよびEHD2タンパク質の量を示すウェスタンブロット。
【図8B】スキャンニングデンシトメータを用いた、図8Aに示されるMyo1cタンパク質の定量を示す棒グラフ。
【図8C】スクランブルsiRNA、siRNA 6またはsiRNAs 6 および2のいずれかをトランスフェクトした3T3-L1脂肪細胞中の、インスリン刺激[3H]2デオキシグルコース取り込みを示す棒グラフ。* P < 0.0004、** P < 0.0001。
【配列表】
【図1】
【特許請求の範囲】
【請求項1】
核酸を脂肪細胞に導入する方法であって、当該方法が
(a) 細胞膜を有する脂肪細胞と核酸分子を接触させて、混合物を形成するステップと、
(b) 前記核酸が前記脂肪細胞に導入されるように、前記細胞膜が透過化するような条件下で、前記混合物を電気穿孔するステップと、
を含む、方法。
【請求項2】
請求項1に記載の方法であって、当該方法において前記核酸分子がsiRNAである、方法。
【請求項3】
請求項1に記載の方法であって、当該方法において前記電気穿孔を、約0.05 kV 乃至約0.5 kV、および電気容量が約750 μF乃至約 1150 μFで行う、方法。
【請求項4】
請求項1に記載の方法であって、当該方法において前記電気穿孔を、約0.1 kV 乃至約0.25 kV、および電気容量が約850 μF乃至約 1050 μFで行う、方法。
【請求項5】
請求項1に記載の方法であって、当該方法において前記電気穿孔を0.18 kVおよび電気容量960 μFで行う、方法。
【請求項6】
請求項1に記載の方法であって、当該方法において前記電気穿孔を室温で行う、方法。
【請求項7】
請求項2に記載の方法であって、当該方法において前記siRNAがAkt1またはAkt2 核酸配列を標的とする、方法。
【請求項8】
請求項2に記載の方法であって、当該方法において前記siRNAがCISK核酸配列を標的とする、方法。
【請求項9】
請求項2に記載の方法であって、当該方法において前記siRNAがMyo1c核酸配列を標的とする、方法。
【請求項10】
細胞におけるAkt1発現または活性を阻害する方法であって、当該方法が
(a) 配列番号5または配列番号6のAkt1配列を標的とするsiRNAを、Akt1を発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のAkt1発現または活性を阻害するステップと、
を含む、方法。
【請求項11】
請求10に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項12】
脂肪細胞におけるAkt1発現または活性を阻害する方法であって、当該方法が
(a) 配列番号5または配列番号6のAkt1配列を標的とするsiRNAをAkt1を発現する細胞に導入するステップであって、当該siRNAが請求項1に記載の方法に従って導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のAkt1発現または活性を阻害するステップと、
を含む、方法。
【請求項13】
細胞へのAkt2発現または活性を阻害する方法であって、当該方法が
(a) 配列番号7または配列番号8のAkt2配列を標的とするsiRNAを、Akt2を発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のAkt2発現または活性を阻害するステップと、
を含む、方法。
【請求項14】
請求13に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項15】
脂肪細胞におけるAkt2発現または活性を阻害する方法であって、当該方法が
(a) 配列番号7または配列番号8のAkt2配列を標的とするsiRNAをAkt2を発現する細胞に導入するステップであって、当該siRNAが請求項1に記載の方法に従って導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のAkt2発現または活性を阻害するステップと、
を含む、方法。
【請求項16】
細胞におけるMyo1c発現または活性を阻害する方法であって、当該方法が
(a) 配列番号14または配列番号15のMyo1c配列を標的とするsiRNAを、Myo1cを発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のMyo1c発現または活性を阻害するステップと、
を含む、方法。
【請求項17】
請求13に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項18】
脂肪細胞におけるMyo1c発現または活性を阻害する方法であって、当該方法が
(a) 配列番号14または配列番号15のMyo1c配列を標的とするsiRNAを、Myo1cを発現する細胞に導入するステップであって、当該ステップにおいて前記siRNAが請求項1に記載の方法によって導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のMyo1c発現または活性を阻害するステップと、
を含む、方法。
【請求項19】
インスリン媒介GSK3αリン酸化を阻害する方法であって、当該方法が、インスリン感受性細胞とAkt2発現または活性を阻害する作用物質を接触させて、インスリン媒介GSK3αリン酸化を阻害するステップを含む、方法。
【請求項20】
請求19に記載の方法であって、当該方法において前記作用物質がAkt2を標的としたsiRNAである、方法。
【請求項21】
請求20に記載の方法であって、当該方法において前記siRNAが配列番号7または配列番号8のAkt2配列を標的とする、方法。
【請求項22】
ヘキソース輸送を阻害する方法であって、当該方法が、ヘキソース輸送を行うことができる細胞とAkt1、Akt2、またはその両方の発現または活性を特異的に阻害する作用物質を接触させて、ヘキソース輸送を阻害するステップを含む、方法。。
【請求項23】
請求項22に記載の方法であって、当該方法において、前記作用物質がAkt1またはAkt2の発現もしくは活性を特異的に阻害し、ヘキソース輸送が部分的に阻害される、方法。
【請求項24】
請求22に記載の方法であって、当該方法において前記作用物質がAkt1、Akt2またはその両方を特異的に標的としたsiRNAである、方法。
【請求項25】
請求24に記載の方法であって、当該方法においてAkt2を標的とするsiRNAが配列番号7または配列番号8の配列を標的とする、方法。
【請求項26】
請求24に記載の方法であって、当該方法において前記Akt1を標的とするsiRNAが配列番号5または配列番号6の配列を標的とする、方法。
【請求項27】
グルコース輸送に影響する遺伝子を同定する方法であって、当該方法が
(a) 請求項1に記載の方法を用いて前記遺伝子を標的としたsiRNAを脂肪細胞に導入するステップと、
(b) 前記標的遺伝子の発現に好適な条件下で前記細胞を培養するステップと、
(c) 前記細胞中のグルコース輸送を評価するステップであって、当該ステップにおいてグルコース輸送の低下が、標的遺伝子がグルコース輸送に影響を与えることによってグルコース輸送に影響を与える遺伝子を同定することを示唆する、ステップと、
を含む、方法。
【請求項28】
インスリン応答モジュレータを同定する方法であって、当該方法が
(a) グルコース輸送に影響を与えるタンパク質を発現する細胞とテスト化合物を接触させるステップであって、当該ステップにおいて前記タンパク質が請求項27に記載の方法にしたがって同定された遺伝子によってコードされている、ステップと、
(b) 前記テスト化合物の、前記タンパク質の活性を調節する能力を決定するステップであって、当該ステップによって前記インスリン応答モジュレータを同定する、ステップと、
を含む、方法。
【請求項29】
インスリン応答モジュレータを同定する方法であって、当該方法が
(a) グルコース輸送に影響を与えるタンパク質を発現する細胞とテスト化合物を接触させるステップであって、当該ステップにおいて前記タンパク質が請求項27に記載の方法にしたがって同定された遺伝子によってコードされている、ステップと、
(b) 前記テスト化合物の、前記タンパク質の発現を調節する能力を決定するステップであって、当該ステップによって前記インスリン応答モジュレータを同定する、ステップと、
を含む、方法。
【請求項30】
請求項28または29に記載の方法であって、当該方法において前記モジュレータは陽性モジュレータである、方法。
【請求項31】
請求項28または29に記載の方法であって、当該方法において前記モジュレータは陰性モジュレータである、方法。
【請求項32】
請求項28または29に記載の方法によって同定されたモジュレータ。
【請求項33】
請求項32に記載のモジュレータを含む、薬学的組成物。
【請求項34】
対象におけるグルコースの恒常性を調節する方法であって、当該方法が請求項32に記載のインスリン応答モジュレータを前記対象に投与するステップを含む、方法。
【請求項35】
対象における血糖値を調節する方法であって、当該方法が請求項32に記載のインスリン応答モジュレータを前記対象に投与するステップを含む、方法。
【請求項36】
インスリン応答疾患または障害を治療する方法であって、当該方法において請求項33に記載の薬学的組成物を投与するステップを含む、方法を提供する。
【請求項37】
請求項36に記載の方法であって、当該方法において前記疾患または障害は2型糖尿病、インスリン耐性、および肥満からなるグループから選択される、方法。
【請求項1】
核酸を脂肪細胞に導入する方法であって、当該方法が
(a) 細胞膜を有する脂肪細胞と核酸分子を接触させて、混合物を形成するステップと、
(b) 前記核酸が前記脂肪細胞に導入されるように、前記細胞膜が透過化するような条件下で、前記混合物を電気穿孔するステップと、
を含む、方法。
【請求項2】
請求項1に記載の方法であって、当該方法において前記核酸分子がsiRNAである、方法。
【請求項3】
請求項1に記載の方法であって、当該方法において前記電気穿孔を、約0.05 kV 乃至約0.5 kV、および電気容量が約750 μF乃至約 1150 μFで行う、方法。
【請求項4】
請求項1に記載の方法であって、当該方法において前記電気穿孔を、約0.1 kV 乃至約0.25 kV、および電気容量が約850 μF乃至約 1050 μFで行う、方法。
【請求項5】
請求項1に記載の方法であって、当該方法において前記電気穿孔を0.18 kVおよび電気容量960 μFで行う、方法。
【請求項6】
請求項1に記載の方法であって、当該方法において前記電気穿孔を室温で行う、方法。
【請求項7】
請求項2に記載の方法であって、当該方法において前記siRNAがAkt1またはAkt2 核酸配列を標的とする、方法。
【請求項8】
請求項2に記載の方法であって、当該方法において前記siRNAがCISK核酸配列を標的とする、方法。
【請求項9】
請求項2に記載の方法であって、当該方法において前記siRNAがMyo1c核酸配列を標的とする、方法。
【請求項10】
細胞におけるAkt1発現または活性を阻害する方法であって、当該方法が
(a) 配列番号5または配列番号6のAkt1配列を標的とするsiRNAを、Akt1を発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のAkt1発現または活性を阻害するステップと、
を含む、方法。
【請求項11】
請求10に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項12】
脂肪細胞におけるAkt1発現または活性を阻害する方法であって、当該方法が
(a) 配列番号5または配列番号6のAkt1配列を標的とするsiRNAをAkt1を発現する細胞に導入するステップであって、当該siRNAが請求項1に記載の方法に従って導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のAkt1発現または活性を阻害するステップと、
を含む、方法。
【請求項13】
細胞へのAkt2発現または活性を阻害する方法であって、当該方法が
(a) 配列番号7または配列番号8のAkt2配列を標的とするsiRNAを、Akt2を発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のAkt2発現または活性を阻害するステップと、
を含む、方法。
【請求項14】
請求13に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項15】
脂肪細胞におけるAkt2発現または活性を阻害する方法であって、当該方法が
(a) 配列番号7または配列番号8のAkt2配列を標的とするsiRNAをAkt2を発現する細胞に導入するステップであって、当該siRNAが請求項1に記載の方法に従って導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のAkt2発現または活性を阻害するステップと、
を含む、方法。
【請求項16】
細胞におけるMyo1c発現または活性を阻害する方法であって、当該方法が
(a) 配列番号14または配列番号15のMyo1c配列を標的とするsiRNAを、Myo1cを発現する細胞に導入するステップと、
(b) siRNAがRNAiを媒介するような条件下で前記細胞を培養して、前記細胞中のMyo1c発現または活性を阻害するステップと、
を含む、方法。
【請求項17】
請求13に記載の方法であって、当該方法において前記細胞が脂肪細胞である、方法。
【請求項18】
脂肪細胞におけるMyo1c発現または活性を阻害する方法であって、当該方法が
(a) 配列番号14または配列番号15のMyo1c配列を標的とするsiRNAを、Myo1cを発現する細胞に導入するステップであって、当該ステップにおいて前記siRNAが請求項1に記載の方法によって導入されるステップと、
(b) siRNAがRNAiを媒介するような条件下で前記脂肪細胞を培養して、前記細胞中のMyo1c発現または活性を阻害するステップと、
を含む、方法。
【請求項19】
インスリン媒介GSK3αリン酸化を阻害する方法であって、当該方法が、インスリン感受性細胞とAkt2発現または活性を阻害する作用物質を接触させて、インスリン媒介GSK3αリン酸化を阻害するステップを含む、方法。
【請求項20】
請求19に記載の方法であって、当該方法において前記作用物質がAkt2を標的としたsiRNAである、方法。
【請求項21】
請求20に記載の方法であって、当該方法において前記siRNAが配列番号7または配列番号8のAkt2配列を標的とする、方法。
【請求項22】
ヘキソース輸送を阻害する方法であって、当該方法が、ヘキソース輸送を行うことができる細胞とAkt1、Akt2、またはその両方の発現または活性を特異的に阻害する作用物質を接触させて、ヘキソース輸送を阻害するステップを含む、方法。。
【請求項23】
請求項22に記載の方法であって、当該方法において、前記作用物質がAkt1またはAkt2の発現もしくは活性を特異的に阻害し、ヘキソース輸送が部分的に阻害される、方法。
【請求項24】
請求22に記載の方法であって、当該方法において前記作用物質がAkt1、Akt2またはその両方を特異的に標的としたsiRNAである、方法。
【請求項25】
請求24に記載の方法であって、当該方法においてAkt2を標的とするsiRNAが配列番号7または配列番号8の配列を標的とする、方法。
【請求項26】
請求24に記載の方法であって、当該方法において前記Akt1を標的とするsiRNAが配列番号5または配列番号6の配列を標的とする、方法。
【請求項27】
グルコース輸送に影響する遺伝子を同定する方法であって、当該方法が
(a) 請求項1に記載の方法を用いて前記遺伝子を標的としたsiRNAを脂肪細胞に導入するステップと、
(b) 前記標的遺伝子の発現に好適な条件下で前記細胞を培養するステップと、
(c) 前記細胞中のグルコース輸送を評価するステップであって、当該ステップにおいてグルコース輸送の低下が、標的遺伝子がグルコース輸送に影響を与えることによってグルコース輸送に影響を与える遺伝子を同定することを示唆する、ステップと、
を含む、方法。
【請求項28】
インスリン応答モジュレータを同定する方法であって、当該方法が
(a) グルコース輸送に影響を与えるタンパク質を発現する細胞とテスト化合物を接触させるステップであって、当該ステップにおいて前記タンパク質が請求項27に記載の方法にしたがって同定された遺伝子によってコードされている、ステップと、
(b) 前記テスト化合物の、前記タンパク質の活性を調節する能力を決定するステップであって、当該ステップによって前記インスリン応答モジュレータを同定する、ステップと、
を含む、方法。
【請求項29】
インスリン応答モジュレータを同定する方法であって、当該方法が
(a) グルコース輸送に影響を与えるタンパク質を発現する細胞とテスト化合物を接触させるステップであって、当該ステップにおいて前記タンパク質が請求項27に記載の方法にしたがって同定された遺伝子によってコードされている、ステップと、
(b) 前記テスト化合物の、前記タンパク質の発現を調節する能力を決定するステップであって、当該ステップによって前記インスリン応答モジュレータを同定する、ステップと、
を含む、方法。
【請求項30】
請求項28または29に記載の方法であって、当該方法において前記モジュレータは陽性モジュレータである、方法。
【請求項31】
請求項28または29に記載の方法であって、当該方法において前記モジュレータは陰性モジュレータである、方法。
【請求項32】
請求項28または29に記載の方法によって同定されたモジュレータ。
【請求項33】
請求項32に記載のモジュレータを含む、薬学的組成物。
【請求項34】
対象におけるグルコースの恒常性を調節する方法であって、当該方法が請求項32に記載のインスリン応答モジュレータを前記対象に投与するステップを含む、方法。
【請求項35】
対象における血糖値を調節する方法であって、当該方法が請求項32に記載のインスリン応答モジュレータを前記対象に投与するステップを含む、方法。
【請求項36】
インスリン応答疾患または障害を治療する方法であって、当該方法において請求項33に記載の薬学的組成物を投与するステップを含む、方法を提供する。
【請求項37】
請求項36に記載の方法であって、当該方法において前記疾患または障害は2型糖尿病、インスリン耐性、および肥満からなるグループから選択される、方法。
【図2】

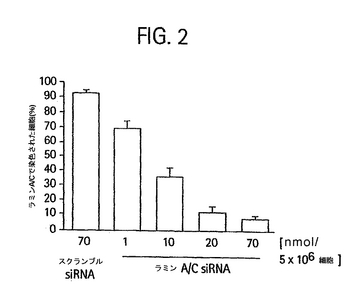
【図3A】
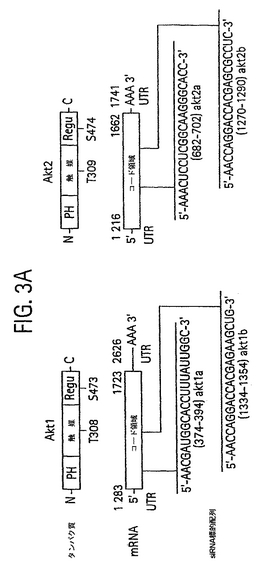
【図3B】
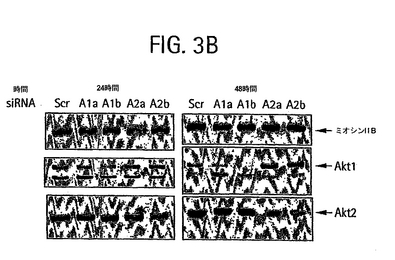
【図4A】
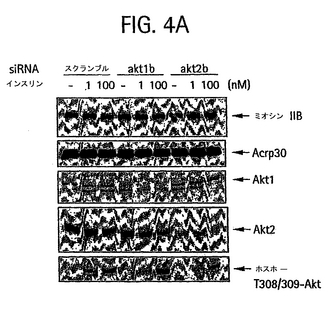
【図4B】
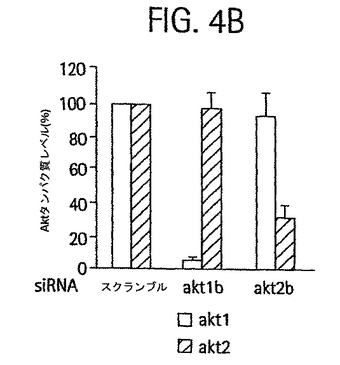
【図4C】
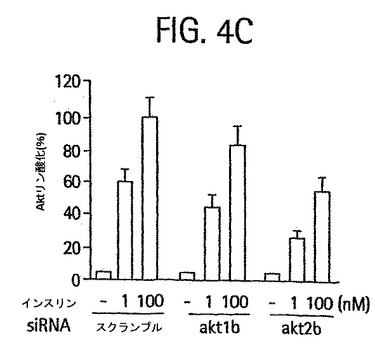
【図5】
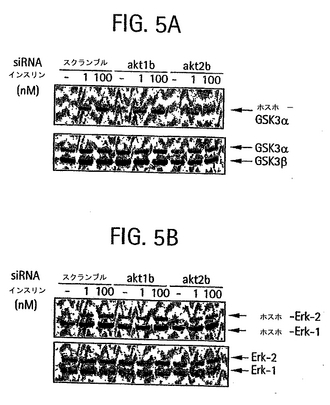
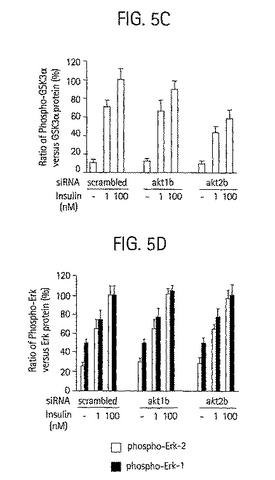
【図6】
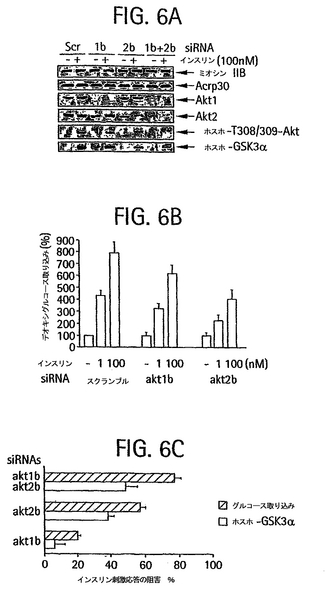
【図7】
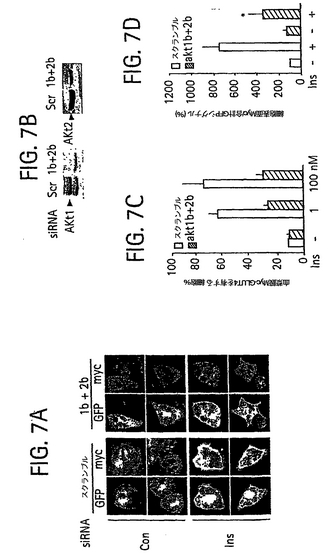
【図8】
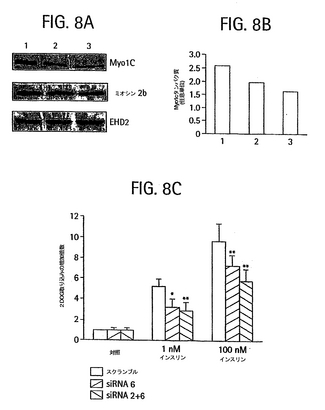
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2006−509504(P2006−509504A)
【公表日】平成18年3月23日(2006.3.23)
【国際特許分類】
【出願番号】特願2004−558215(P2004−558215)
【出願日】平成15年12月11日(2003.12.11)
【国際出願番号】PCT/US2003/039774
【国際公開番号】WO2004/053103
【国際公開日】平成16年6月24日(2004.6.24)
【出願人】(505220170)ユニバーシティー オブ マサチューセッツ (6)
【氏名又は名称原語表記】University of Massachusetts
【住所又は居所原語表記】1 Beacon Street, 26th Floor,Boston, MA 02108 (US).
【Fターム(参考)】
【公表日】平成18年3月23日(2006.3.23)
【国際特許分類】
【出願日】平成15年12月11日(2003.12.11)
【国際出願番号】PCT/US2003/039774
【国際公開番号】WO2004/053103
【国際公開日】平成16年6月24日(2004.6.24)
【出願人】(505220170)ユニバーシティー オブ マサチューセッツ (6)
【氏名又は名称原語表記】University of Massachusetts
【住所又は居所原語表記】1 Beacon Street, 26th Floor,Boston, MA 02108 (US).
【Fターム(参考)】
[ Back to top ]