
脂肪酸不飽和化酵素遺伝子、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物
【課題】 哺乳動物において、n−3系多価不飽和脂肪酸を効率よく合成できる、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を提供する。
【解決手段】 本発明は、脂肪酸不飽和化酵素遺伝子を含有する組換え発現ベクター、およびこの組換え発現ベクターを用いた脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物細胞、および、脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物である。
【解決手段】 本発明は、脂肪酸不飽和化酵素遺伝子を含有する組換え発現ベクター、およびこの組換え発現ベクターを用いた脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物細胞、および、脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、非ヒト動物細胞に脂肪酸不飽和化酵素遺伝子を導入する組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物に関する。
【背景技術】
【0002】
脂肪は、エネルギー源として非常に重要である。脂肪は、同時に、動物にとって正常な発育と機能維持に欠くことができない、不飽和脂肪酸(リノール酸(18:2n−6)、α−リノレン酸(18:3n−3)、およびアラキドン酸(20:4n−6))などの摂取源でもある。脂肪酸の合成では、Δ12脂肪酸不飽和化酵素により、オレイン酸(18:1)からリノール酸(18:2n−6)が合成され、さらに、ω3脂肪酸不飽和化酵素により、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)が合成される。
【0003】
ここで、多価不飽和脂肪酸は、生合成の経路別に、n−3系、n−6系、n−9系などに分類される。ここで、「n−」に続く数字3、6および9は、多価不飽和脂肪酸の炭素鎖のメチル基から数えて最初の二重結合が何番目の炭素にあるかを示す。例えば、「n−3」系は、炭素数が18個の脂肪酸のメチル基の炭素を1位の炭素とし、カルボキシル基側に向かって順次2位、3位・・・としたとき、メチル基から数えて最初の二重結合が3位の炭素にある多価不飽和脂肪酸を示す。「n−3」系は、「ω3」系とも表示される。また、「n−」の前に記載される数字は、一分子内に含まれる二重結合の数を示す。一方、「Δ」は、多価不飽和脂肪酸の炭素鎖のカルボキシル基から数えて最初の二重結合が何番目の炭素にあるかを示す。また、ω3脂肪酸不飽和化酵素、Δ12脂肪酸不飽和化酵素は、それぞれ、脂肪酸の炭素鎖のメチル基から数えて3位の位置に二重結合を作る酵素を、脂肪酸の炭素鎖のカルボキシル基から数えて12位の位置に二重結合を作る酵素を、意味する。
【0004】
しかし、哺乳動物などの高等動物では、上記の酵素を有さない。したがって、動物によれば、オレイン酸(18:1)からリノール酸(18:2n−6)の合成や、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成は行えないものもいる。
【0005】
α−リノレン酸(18:3n−3)からはエイコサペンタエン酸(EPA、20:5n−3)が、EPAからはドコサヘキサエン酸(DHA、22:6n−3)などが合成される。EPAやDHAなどのn−3系多価不飽和脂肪酸は、アトピー性皮膚炎、血栓性疾患、ガン、冠動脈心疾患などに低減効果があることが報告されている(例えば、非特許文献1、2参照)。
【0006】
これらのn−3系多価不飽和脂肪酸は、魚類や海草類などの海産物に多く含まれている。したがって、海産物を経口摂取することが非常に重要である。しかし、牛や豚などの畜肉やパーム油などの植物油に代表される陸生生物由来食品を過剰に摂取し、海産物の摂食が不足している、現在の先進国の食生活では、n−3系多価不飽和脂肪酸は摂取されにくい。
【0007】
このため、高等動物細胞が合成できない多価不飽和脂肪酸を高等動物細胞や高等動物中で合成しようとすることが試みられている。
【0008】
本発明者らは、ブタに、ホウレンソウ(Spinacia olerancea)根部由来のリノール酸(18:2n−6)を生成するΔ12脂肪酸不飽和化酵素をコードする遺伝子である、FAD2(fatty acid desaturation 2)を導入したものを提案した(例えば、特許文献1参照)。また、FAD2が導入されたブタでは、リノール酸(18:2n−6)が新規に合成されることを証明した。
【0009】
リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)を合成するω3脂肪酸不飽和化酵素については、例えば、植物等におけるω3脂肪酸不飽和化酵素の発現量や活性を、遺伝子操作等によって人為的に制御するための技術に使用できるω3脂肪酸不飽和化酵素遺伝子等が提供されている(例えば、特許文献2参照)。
【0010】
また、子嚢菌と脂質生成菌であるモルティエレラ・アルピナのΔ12脂肪酸不飽和酵素と、子嚢菌のω3脂肪酸不飽和化酵素の推定アミノ酸配列を比較し、相同性の高いアミノ酸配列に対応するプライマーを設計し、これから、ω3脂肪酸不飽和化活性を有するポリペプチドを得る技術が開示されている(例えば、特許文献3参照)。この文献中には、設計したプライマーから得られるポリヌクレオチドを含むベクターを動物に導入してよいことが記載されている。
【0011】
また、アラキドン酸産生糸状菌であるモルティエレラ・アルピナのΔ12脂肪酸不飽和化酵素遺伝子をマウスのL細胞に導入することで、哺乳動物細胞内でΔ12脂肪酸不飽和化酵素遺伝子が機能することを証明している。また、リノール酸だけでなく、リノール酸以降のn−6系脂肪酸の増加も確認している。このことから、哺乳動物がもたないΔ12脂肪酸不飽和化酵素遺伝子を哺乳動物細胞に導入でき、機能的に発現することが示されている(例えば、非特許文献3参照)。
【0012】
線虫由来のω3脂肪酸不飽和化酵素遺伝子であるfat−1をマウスの乳腺で発現させ、乳中のn−3系脂肪酸の増加を確認しており、哺乳動物でω3脂肪酸不飽和化酵素遺伝子が機能的に発現することが示されている。また、乳腺をターゲットにすることで乳の脂肪酸組成を改変できることを示し、授乳した仔マウスへの効果も確認されている(例えば、非特許文献4参照)。
【非特許文献1】Connor WE, Importance of n−3 fatty acids in health and disease. Am. J. Clin. Nutr. 71, 171S−175S (2000).
【非特許文献2】Terry PD et al., Intakes of fish and marine fatty acids and the risks of cancers of the breast and prostate and of other hormone−related cancers: A review of the epidemiologic evidence. Am. J. Clin. Nutr. 77,p.532−5543 (2003).
【非特許文献3】Kelder B et al., Expression of fungal desaturase genes in cultured mammalian cells. Molecula and Cellular Biochemistry 219,p.7−11(2001)
【非特許文献4】Kao BT et al., Endogenous production and elevated levels of long−chain n−3 fatty acids in the milk of transgenic mice. J. Dairy Sci. 89, p.3195−3201(2006)
【特許文献1】特開2003−245070号公報
【特許文献2】特開2001−95588
【特許文献3】特開2006−55104
【発明の開示】
【発明が解決しようとする課題】
【0013】
哺乳動物において、n−3系多価不飽和脂肪酸が合成されれば、食肉等からn−3系多価不飽和脂肪酸を効率よく摂取することができる。しかし、いずれの文献においても、哺乳動物において、ω3脂肪酸不飽和化酵素が機能し、n−3系多価不飽和脂肪酸の合成されたことについては、開示されていない。非特許文献4の場合では、遺伝子改変マウスの作出効率が極めて低く、中性脂肪への影響も少なかった。
【0014】
すなわち、本発明は、上記問題に鑑みなされたものであり、その目的は、哺乳動物において、n−3系多価不飽和脂肪酸を効率よく合成できる、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を提供することにある。
【課題を解決するための手段】
【0015】
本発明者らは、上記課題を解決するために鋭意検討をした結果、α−リノレン酸(18:3n−3)の合成に対して酵素活性の高い脂肪酸不飽和化酵素遺伝子を用いて、組換え発現ベクターとすることで、α−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸が多く合成される、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を提供できることを見出し、本発明を完成させた。すなわち、本発明は以下のとおりである。
【0016】
本発明の脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(D)のいずれかの塩基配列からなるポリヌクレオチドである。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【0017】
本発明の組換え発現ベクターは、脂肪酸不飽和化酵素遺伝子を含有する。
【0018】
上記脂肪酸不飽和化酵素遺伝子は、ω3脂肪酸不飽和化酵素遺伝子である。
【0019】
上記脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(C)のいずれかの塩基配列からなる遺伝子であるとよい。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド、
(B)配列番号1に示される塩基配列において、1若しくは数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド、
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
【0020】
上記脂肪酸不飽和化酵素遺伝子は、ヒト化遺伝子であると好ましい。
【0021】
上記組換え発現ベクターは、レポーター遺伝子、IRES配列、および選択マーカー遺伝子を含むと、好ましい。
【0022】
本発明の非ヒト動物細胞は、上記組換え発現ベクターを用いて、脂肪酸不飽和化酵素遺伝子が導入されている。
【0023】
本発明の非ヒト動物は、脂肪酸不飽和化酵素遺伝子が導入されている。
【発明の効果】
【0024】
本発明の組換え発現ベクターを用いて非ヒト動物を形質変換すれば、非ヒト動物内で、非ヒト動物に内在する脂肪酸を不飽和化する反応を触媒することができる。この結果、従来動物ではできなかった、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成を可能とする。
【0025】
また、非ヒト動物内で、α−リノレン酸(18:3n−3)を基質とするn−3系多価不飽和脂肪酸が合成される。この結果、n−3系多価不飽和脂肪酸に富む食肉、乳製品などの畜産品の供給を可能とする。
【発明を実施するための最良の形態】
【0026】
以下に、本発明を詳細に説明する。
【0027】
[脂肪酸不飽和化酵素遺伝子]
本発明の組換え発現ベクターに用いられる脂肪酸不飽和化酵素遺伝子は、α−リノレン酸の生成に関わるω3脂肪酸不飽和酵素をコードする遺伝子(例えば、fatty acid desaturation 3、FAD3など)である。FAD3は、通常、植物、細菌、線虫などの下等動物に多く存在する遺伝子である。FAD3は、シロイヌナズナ、セイヨウアブラナ、大豆などから単離されている。本発明で用いることのできるFAD3は、FAD3を有する植物から取得すればよく、その由来は問わない。好ましくは、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成能力の高い植物のFAD3を用いればよい。例えば、アマ科の植物である。また、植物由来のFAD3以外に、線虫から単離されるfat−1を用いてもよい。
【0028】
また、植物由来の遺伝子は、動物細胞内では発現されにくい場合がある。本発明で用いられる脂肪酸不飽和化酵素遺伝子は、植物由来の脂肪酸不飽和化酵素遺伝子をそのまま用いてもよく、この遺伝子をヒト化処理したものを用いてもよい(Kang JX, et al.,Transgenic mice:fat−1 mice convert n−6 to n−3 fatty acids. Nature,427,p.504(2004)、Lai L, al., Generation of cloned transgenic pigs rich in omega−3 fatty acids. Nature Biotechnology,24,p.1472−1473 (2006))。ヒト化処理を行うと、遺伝子の発現効率を向上させることができる。本明細書中で、ヒト化処理とは、植物由来の遺伝子を、アミノ酸配列を変更することなく、哺乳動物でのコドン使用頻度に準じて、塩基配列の変更を行うことをいう。ヒト化処理は、例えば以下のように行う。以下の例では、アマ由来のFAD3を用いる。
【0029】
アマ由来のFAD3は、以下のようにして得られる。ベニバナアマ(Linum grandiflorum)の幼若種子から抽出したtotal RNAからcDNAを合成する。縮重ポリメレースチェーン反応(degenerate PCR)法と、RACE法とを用いて、FAD3 cDNAの配列を調べる。
【0030】
植物の脂肪酸不飽和化酵素には、葉緑体で機能する型と小胞体膜上で機能する型がある。この植物の脂肪酸不飽和化酵素を動物で機能させるには、小胞体型の脂肪酸不飽和化酵素を用いる必要がある。小胞体型の脂肪酸不飽和化酵素は、細胞の小胞体膜に結合したタンパク質であり、3つのヒスチジンクラスターに鉄(Fe)原子が挟まれた活性中心を有する。得られたcDNAがFAD3遺伝子であるかどうかは、まず得られたcDNAに、小胞体移行シグナルを有していることを確認する。小胞体移行シグナルは、アミノ酸3’末端に「KXKXX」(K:リシン、Xはアミノ酸)配列を有する。FAD3の小胞体移行シグナルは、「KSKSA」(S:セリン、A:アラニン)である。したがって、cDNA中に上記FAD3の小胞体移行シグナルのアミノ酸配列に該当する塩基配列を有していることを確認すればよい。
【0031】
次に、3つのヒスチジンクラスターがあることを確認する。3つのヒスチジンクラスターは、「HCDH」、「HxxxxxHRTHH」、「HHxxxxHVIHH」(H:ヒスチジン、C:システイン、D:アスパラギン酸、R:アルギニン、T:スレオニン、V:バリン、I:イソロイシン、X:アミノ酸)配列である。3つのヒスチジンクラスターの存在は、この配列と配列が存在する場所で特定することができる。FAD3の3つのヒスチジンクラスターは、「HCDH」、「HGWKISHRTHH」、「HHDIGTHVIHH」(G:グリシン、W:トリプトファン)である。したがって、cDNA中に上記FAD3の3つヒスチジンクラスターのアミノ酸配列に該当する塩基配列を有していることを確認すればよい。
【0032】
次に、推定された配列をもとにプライマーを設計し、PCR法により、遺伝子FAD3を得る。得られたアマ由来cDNAの酵素活性を検討するため、公知の方法により酵母発現ベクターを構築し、酵母での酵素活性を確認する(Reed DW et al., Characterization of the Brassica napus extraplastidial linoleate desaturase by expression in Saccharomyces cerevisiae. Plant Physiology 122,p.715−720(2000))。非形質転換酵母と比較して、形質転換酵母においてα―リノレン酸(18:3n−3)の増加を確認して、得られたcDNAをアマ由来のFAD3とする。
【0033】
哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度を、Codon Usage Databese(http://www.kazusa.or.jp/codon/)を用いて比較する。その結果をもとに、FAD3遺伝子の配列を設計する。たとえば、アルギニンは、ヒトではCGCのコドンの使用頻度が高いが、アマ由来FAD3では、CGCのコドンの使用頻度は低く、代わりにAGAの使用頻度が高い。また、アスパラギンは、ヒトではAAUの使用頻度が78%を占めるが、アマ由来FAD3では54%とやや低くなる。ヒト化処理では、これらのコドン使用頻度を比較して、アマ由来FAD3の使用するコドンを、ヒトのコドンに代えて、ヒト化FAD3(hFAD3)遺伝子の配列を設計する。なお、コドンの変更は、必ず、ヒトのコドンの使用頻度に合わせる必要はない。コドンの使用頻度に近づければよい。また、遺伝子発現を効率化するために開始コドン付近にコザック配列を挿入してもよい。
【0034】
配列番号1に示される塩基配列からなるポリヌクレオチドは、アマ由来脂肪酸不飽和化酵素遺伝子FAD3をヒト化処理した(humanized)ポリヌクレオチド(hFAD3)の一例を示す。
(塩基配列1)
【表1】

【0035】
本発明では、前記脂肪酸不飽和化酵素遺伝子としては、以下の(A)〜(D)のいずれかの塩基配列からなる遺伝子を使用することができる。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド、
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド、
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【0036】
上記「1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列」とは、例えば1〜20個、好ましくは1〜15個、より好ましくは1〜10個、さらに好ましくは1〜5個の任意の数の塩基が欠失、置換若しくは付加された塩基配列を意味する。
【0037】
例えば、これら1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなるDNA(変異DNA)は、化学合成、遺伝子工学的手法、突然変異誘発などの当業者に既知の任意の方法により作製することもできる。具体的には、配列番号1に示される塩基配列からなるDNAに対し、変異原となる薬剤と接触作用させる方法、紫外線を照射する方法、遺伝子工学的な手法等を用いて、これらDNAに変異を導入することにより、変異DNAを取得することができる。遺伝子工学的手法の一つである部位特異的変異誘発法は特定の位置に特定の変異を導入できる手法であることから有用であり、Molecular Cloning:A laboratory Mannual,2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor,NY.,1989.以後“モレキュラークローニング第2版”と略す)、Current Protocols in Molecular Biology, Supplement 1〜38,John Wiley & Sons (1987−1997)等に記載の方法に準じて行うことができる。この変異DNAを適切な発現系を用いて発現させることにより、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質を得ることができる。
【0038】
上記「ストリンジェントな条件下でハイブリダイズする塩基配列」とは、DNA又はRNAなどの核酸をプローブとして使用し、コロニー・ハイブリダイゼーション法、プラークハイブリダイゼーション法、あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られる塩基配列を意味し、具体的には、コロニーあるいはプラーク由来のDNAまたは該DNAの断片を固定化したフィルターを用いて、0.7〜1.0MのNaCl存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍程度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウム)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、モレキュラークローニング第2版等に記載されている方法に準じて行うことができる。
【0039】
例えば、ストリンジェントな条件下でハイブリダイズすることができるDNAとしては、プローブとして使用するDNAの塩基配列と一定以上の相同性を有するDNAが挙げることができ、例えば60%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAを好適に例示することができる。
【0040】
3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチドは、上記した3つのヒスチジンクラスターと小胞体膜移行シグナルの確認方法と、真核細胞内でω3脂肪酸不飽和化酵素活性の発現を確認することにより行う。
【0041】
[脂肪酸不飽和化酵素遺伝子の作成]
本発明で用いる脂肪酸不飽和化酵素遺伝子は、例えば以下のようにして作成する。
【0042】
まず、上記で設計したhFAD3から最適化遺伝子の塩基配列を設計する。具体的には、上記配列番号1の塩基配列の5’末端と3’末端には、ベクターに導入する際に用いる、制限酵素認識部位を設ける。例えば、5’末端にHindIII認識部位を設け、3’末端にBamHI認識部位を設けるなどである。また、効率的に翻訳を行うために、開始コドンの近傍に、リボソーム結合部位であるコザック(Kozak)配列を挿入すると好ましい。
【0043】
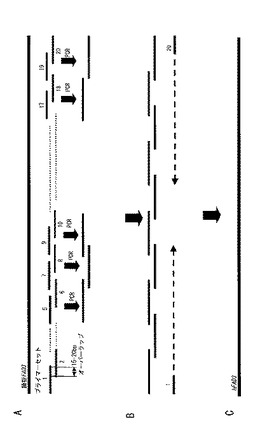
このようにして設計した最適化遺伝子の塩基配列から、Sinclairら(2002)と、Kimら(1997)のPCR法を用いて、hFAD3の再構築遺伝子を作成する。図1は、hFAD3の再構築遺伝子の作成方法を説明する図である。
【0044】
設計した鋳型hFAD3の塩基配列をもとに、70〜90塩基ずつのPCRプライマーを設計する。これらのプライマーは、相互に補足するように12〜20塩基をオーバーラップさせる。表2には、このようにして設計した10組20個のPCRプライマーの例を示す。
【表2】
【0045】
次に、設計したプライマーを組み合わせて、PCR法で、hFAD3遺伝子の合成を行う。上記の例では、No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20とを組み合わせてPCRを行う。各々のプライマーの組み合わせにおいて、事前にアニーリング温度の最適条件を求めておくと良い。
【0046】
上記各々のプライマーセットを用いて得られたPCR産物を合わせて、No1と20のプライマーを組み合わせてPCRを行う。これにより、hFAD3を含むPCR産物が得られる。
【0047】
hFAD3を含むPCR産物から、所望の大きさ(上記の例では、1.2kb)のDNAを抽出する。DNAの抽出は、公知のDNA抽出キットを用いればよい。抽出したDNAは、エタノールを用いて沈殿させて回収する。沈殿したDNAを、トリス塩酸バッファーを加えて溶解し、以下の操作に用いる。
【0048】
得られたPCR産物を、TAクローニングにより、直接ベクターにクローニングする。PCRに用いるTaq DNAポリメラーゼは弱い末端転移酵素活性を持っており、PCR反応によって合成された産物の末端にA(アデニン)を1塩基付加することができる。PCR産物をプラスミドにクローニングする際、開環したプラスミドベクターの末端にT(チミン)を1塩基付加したものを用いると、AとTが水素結合を作ることによって、平滑末端同士のライゲーションよりもクローニング効率が上がる。使用するベクターとしては、プロメガ社のpGEM−T easyが挙げられる。このベクターでは挿入断片を制限酵素EcoRIに切り出すことが可能である。
【0049】
クローニング産物を、選択培養用の大腸菌に入れて形質転換する。選択培地中で生育した大腸菌のコロニーを、液体培地中で増殖させる。増殖した大腸菌培養液を公知のアルカリプレップ法を用いて、大腸菌の細胞壁を分解し、プラスミドを回収する。
【0050】
回収したプラスミドをポリエチレングリコールで沈殿させる。これをPCR法で増幅させる。必要に応じて、PCR産物の一部を公知の方法で、DNA塩基配列の分析をし、hFAD3の配列であることを確認する。
【0051】
[組換え発現ベクター]
本発明の組換え発現ベクターは、上記脂肪酸不飽和化酵素を発現するためのベクターである。本発明の組換え発現ベクターは、非ヒト動物で脂肪酸不飽和化酵素遺伝子を発現できるものであればよい。
【0052】
本発明の発現ベクターとして用いられるベクターには、遺伝子操作技術において広く用いられているものが含まれ、例えば、バクテリオファージ、プラスミド、コスミド、ウイルス、またはレトロウイルスなどが挙げられる。
【0053】
本発明の組換え発現ベクターは、RNAポリメラーゼが転写を開始するためのプロモーター領域を有する。プロモーター領域の下流に、脂肪酸不飽和化酵素遺伝子を導入する。
【0054】
脂肪酸不飽和化酵素遺伝子の発現を調節するプロモーターとしては、例えば、ウイルス(例、サイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルスなど)に由来する遺伝子のプロモーター、哺乳動物(例、ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)に由来する遺伝子(例、アルブミン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、I型およびII型コラーゲン、サイクリックAMP依存蛋白キナーゼβIサブユニット、心房ナトリウム利尿性因子、ドーパミンβ−水酸化酵素、ニューロフィラメント軽鎖、メタロチオネインIおよびIIA、メタロプロティナーゼ1組織インヒビター、平滑筋αアクチン、ポリペプチド鎖伸長因子1α(EF1−α)、βアクチン、α及びβミオシン重鎖、ミオシン軽鎖1及び2、ミエリン塩基性蛋白、血清アミロイドPコンポーネント、レニンなど。また、脂肪細胞特異的に発現するアディポサイトP2(aP2)プロモーターや褐色脂肪細胞特異的に発現する脱共役タンパク質(uncoupling
protein,UCP)プロモーターなど)のプロモーターなどが挙げられ、なかでもニワトリアクチンプロモーターを含むCAGプロモーターなどを用いることができる。CAGプロモーターは、哺乳動物細胞で最も強く発現するので、好ましい。
【0055】
上記ベクターは、遺伝子導入哺乳動物において、目的とするmRNAの転写を終結する配列(ポリA、一般にターミネーターと呼ばれる)を有していることが好ましく、例えば、ウイルス由来、哺乳動物および鳥類由来の各遺伝子の配列を用いて遺伝子発現を操作することが出来る。好ましくは、シミアンウイルスのSV40ターミネーターなどが用いられる。
【0056】
本発明の組換え発現ベクターは、目的とする脂肪酸不飽和化酵素遺伝子の発現を同定するためのレポーター遺伝子を含んでいてもよい。レポーター遺伝子としては、ホタルルシフェラーゼ遺伝子、改良型ホタルルシフェラーゼ遺伝子、ウミシイタケルシフェラーゼ遺伝子、青緑色蛍光タンパク質(GFP)遺伝子、強化青緑色蛍光タンパク質(EGFP)遺伝子、青色蛍光タンパク質(BFP)遺伝子、赤色蛍光タンパク質(CFP)遺伝子などが好ましい。また、遺伝子導入するのではなく、胚の成育段階に応じて胚の表面に生じる表面抗原を利用してもよい。
【0057】
特に、プロモーターとしてCAGプロモーターを用いる場合は、レポーター遺伝子としてEGFPを用いるとよい。EGFPとは、オワンクラゲ由来の緑色蛍光蛋白質(GFP)の遺伝子改変体で、紫外光で蛋白質を励起した際より強い蛍光を発する蛋白質である。この遺伝子はClontech社などから購入することができる。CAGプロモーターとともに導入された細胞でEGFP遺伝子の発現が起こり、蛍光を発する細胞として、同定でき、遺伝子導入を確認できる。
【0058】
ベクターへのレポーター遺伝子の導入は、公知の方法が利用できる。好ましくは、Internal Ribosome Entry Site(IRES)とともに、IRESの下流に導入するとよい。IRESを含むベクターは、同一のプロモーターから複数の遺伝子を共発現させることができる。したがって、本発明のベクターがIRESを含んでいると、脂肪酸不飽和化酵素遺伝子とレポーター遺伝子とを共発現させることができるので、レポーター遺伝子の発現、すなわち発色を確認することで、脂肪酸不飽和化酵素遺伝子の発現を同定することができる。
【0059】
IRESを含むベクターとしては、市販のバイシストロン性ベクターを用いることもできる。
【0060】
また、本発明のベクターには、導入遺伝子が安定に組み込まれたクローンを選択するための選択マーカー遺伝子(例、ネオマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、アンピシリン耐性、ストレプトマイシン耐性遺伝子、カナマイシン耐性遺伝子などの薬剤耐性遺伝子など)をさらに含むことが好ましい。これにより、適当な選択薬剤を使用することにより、目的遺伝子が導入されたベクターを選択的に回収できる。
【0061】
[非ヒト動物]
本発明において脂肪酸不飽和化酵素遺伝子が導入される非ヒト動物としては、魚類、鳥類、哺乳類など食用にできる非ヒト動物であればよい。高等動物は、n−3系脂肪酸を産生していない。したがって、本発明の脂肪酸不飽和化酵素遺伝子を導入することで、n−3系脂肪酸の合成ができる非ヒト動物を得ることができる。
【0062】
使用できる魚類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する魚類であればよい。例えば、養殖の対象とされている魚類、筋肉組織等の可食部分において選択的に不飽和脂肪酸含量が高まる魚類や、子孫にまで安定的に脂肪酸不飽和化酵素遺伝子が発現する魚類等が好ましく、魚の種類としては硬骨魚類が好ましく、硬骨魚類の中でもで、Cyprinidae、Cichlidae、Salmonidae、Claridae、Siluridae、Ictaluridaeに属する魚類が好ましい。具体的には、ブリ、マダイ、ヒラメ、トラフグ、ヒラマサ、クルマエビ、ニジマス、カンパチ、シマアジ、マハタ、クロマグロなどの養殖魚の他、コイ、ゼブラフィッシュ、アフリカナマズ、テラピア、タイセイヨウサケ、メダカなどを好適に例示することができる。
【0063】
使用できる鳥類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する鳥類であればよい。具体的には、ニワトリ、ウズラ、ダチョウ、アイガモなどである。
【0064】
使用できる哺乳類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する非ヒト哺乳類であればよい。例えば、ウシ、スイギュウ、ブタ、ヒツジ、ヤギ、ウマ、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなどが挙げられる。好ましくは、ウシ、スイギュウ、ブタ、ヒツジ、ヤギ、ウマなどのヒトが食用にできる非ヒト哺乳動物であり、さらに好ましくはウシである
【0065】
本発明の組換え発現ベクターが導入される非ヒト動物細胞としては、上記非ヒト動物由来の細胞であれば、初期胚、体細胞、胚性幹細胞であってもよい。好ましくは、体細胞であり、さらに好ましくは筋衛星細胞である。筋衛星細胞は骨格筋内に通常、休止状態で存在する組織特異的幹細胞で、骨格筋の再生や肥大に中心的な役割を担うことが知られている(Seale and Rudnicki,A new look at the origin, function, and “stem cell” status of muscle satellite cells. Developmental Biology 218, 115−124 (2000))。また、適切な細胞外環境を整えることにより、骨、軟骨、心筋、脂肪細胞などの他の細胞群へも分化する性質を有する多能性幹細胞としても位置づけられている(Asakura et al., Muscle satellite cells are multipotential stem cells that exhibit myogenic,osteogenic,and adipogenic differentiation.Differentiation 68,p.245−253(2001))。
【0066】
[非ヒト動物細胞への遺伝子導入]
本発明の組換え発現ベクターを導入する非ヒト動物細胞としては、非ヒト動物の体細胞から樹立した細胞を培養して用いる。体細胞の培養培地、培養条件は、公知の方法を用いる。
【0067】
上記培養した体細胞培養細胞の培地中に、遺伝子導入試薬と組換え発現ベクターとを加え、所定期間培養して、体細胞培養細胞中に遺伝子を導入する。
【0068】
上記遺伝子導入細胞をさらに培地中で培養を続け、細胞数を増加させる。その後、遺伝子導入細胞を、抗生物質などの選択薬剤を添加した培地中で選択培養する。選択培地中で生存した細胞について、顕微鏡下観察し、蛍光が認められたものを、クローニングリングにより単離し、通常の培地中で継代培養する。所定期間培養を続けた後にも、安定して蛍光が観察されたものを、遺伝子導入細胞とする。
【0069】
本発明の非ヒト動物細胞では、この遺伝子導入細胞の一部を脂肪細胞に分化させて、脂肪酸組成を分析することにより、遺伝子導入細胞のα−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能を、事前に評価することができる。
【0070】
[脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物]
(クローン胚の作成)
クローン胚のもとになるレシピエント卵子としては、成熟卵子であれば特に制限されるものではなく、屠場由来の卵巣から採取した卵子や生体の卵巣から経膣的に採取した卵子を体外成熟させたもののほか、プロスタグランジンF2α、クロプロステノール、絨毛ゴナドトロピン等のホルモン投与による過排卵処理により得られる体内成熟卵子であっても使用できる。卵丘細胞が付着している卵子はヒアルロニダーゼ処理を行って、卵丘細胞を除去することが好ましい。
【0071】
上記レシピエント卵子からの除核は、細胞骨格形成阻害剤であるサイトカラシンB処理を施した卵子を使用するのが好ましい。より具体的には、体外成熟卵子等のレシピエント卵子をホールディングピペットで保定し、微細なガラス針によりその透明帯を迅速・的確に切開したのち、サイトカラシンBを含有する培地でそれらのレシピエント卵子を処理し、除核操作用シャーレのサイトカラシン入りドロップに移して、除核用ピペットあるいは除核用ガラス針により第一極体の付近の細胞質を極体ごと吸引又は押し出して行う。なお、それらの極体を含む細胞質を調べることにより除核できていることを確認することが好ましく、また除核卵子からはサイトカラシンBを除去することが好ましい。
【0072】
ドナー細胞としては、上記遺伝子導入細胞を培養したものを用いる。また、ドナー細胞の細胞周期は特に制限されるものではないが、細胞周期G0/G1期に同調させた体細胞が好ましい。なお、細胞周期G0/G1期に同調させた体細胞は、例えば一定期間培養し続けコンフルエントな状態まで培養するあるいは、培養液中の血清濃度を極端に低下させた培養液に交換し血清飢餓状態で培養することにより得ることができる。
【0073】
体細胞クローン胚は、定法に従って、例えば、ドナー細胞を細胞導入用ピペットに吸い込み、このピペットの先端を除核されたレシピエント卵子の囲卵腔に差し込んでドナー細胞を注入したのち、細胞融合装置による電気融合処理、カルシウムショックによる活性化処理を行うことにより作製される。
【0074】
上記クローン胚を、所定の培養培地を用いて、体外培養する。クローン胚の卵割、胚盤胞期胚への発生を観察し、卵割しない胚、胚盤胞期胚へ発生しない胚を除く。次に、これらの胚において、リポーター遺伝子の発生を、蛍光により観察する。蛍光が認められるクローン胚を選択して、仮母の子宮内に移植することで、哺乳動物において、n−3系多価不飽和脂肪酸を効率よく合成できる非ヒト動物を得ることができる。
【0075】
本発明では、ベクター中に、蛍光により、脂肪酸不飽和化酵素遺伝子の発現を確認できるリポーター遺伝子を含む。このため、ベクターを非ヒト動物細胞に導入した状態と、クローン胚を作成した状態において、脂肪酸不飽和化酵素遺伝子の発現を容易に確認できる。
【0076】
また、脂肪酸不飽和化酵素遺伝子の発現を確認した非ヒト動物細胞を脂肪細胞に分化させることができる。これにより、脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物細胞のα−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能をあらかじめ評価することができる。
【0077】
すなわち、本発明の組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を用いれば、α−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能の高い非ヒト動物を容易に得ることができる。
【実施例】
【0078】
以下本発明を詳細に説明するため実施例を挙げて説明するが、本発明は実施例に限定されるものではない。
【0079】
[hFAD3遺伝子]
ベニバナアマ(Linum grandiflorum)の幼若種子から抽出したtotal RNAからcDNAを合成した。縮重ポリメレースチェーン反応(degenerate PCR)法と、RACE法とを用いて、FAD3 cDNAの配列を調べた。推定された配列をもとにプライマーを設計し、PCR法により、全長のcDNAを得た。このアマ由来のcDNAは、既知の植物のFAD3との相同性が高く、FAD3の特徴を有していた。このcDNAの酵素活性を、酵母を用いて検討したところ、α−リノレン酸(18:3n−3)が増加したことから、このcDNAを遺伝子FAD3とした。
【0080】
(ヒト化脂肪酸不飽和化酵素遺伝子の設計)
哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度を、Codon Usage Databese(http://www.kazusa.or.jp/codon/)を用いて比較した。その結果をもとに、FAD3遺伝子の配列を設計した。結果を表3に示す。表3は、アミノ酸:Ala(アラニン)、Arg(アルギニン)、Asn(アスパラギン)、Asp(アスパラギン酸)、Leu(ロイシン)、Gln(グルタミン)、Glu(グルタミン酸)、Gly(グリシン)、His(ヒスチジン)、Ile(イソロイシン)、Lys(リシン)、Pro(プロリン)、Cys(システイン)、Phe(フェニルアラニン)、Ser(セリン)、Thr(スレオニン)、Tyr(チロシン)、Val(バリン)における哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度と、本実施例で設計した遺伝子hFAD3におけるコドン使用頻度を示している。Ala(アラニン)に基づいて説明すると、Alaには、「GCU」、「GCC」、「GCA」、「GCC」という4個のコドンが存在する。ヒトのコドンでは、「GCU」は17%、「GCC」は53%、「GCA」は13%、「GCC」は14%使用されている。一方、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドンでは、「GCU」は28%、「GCC」は48%、「GCA」は17%、「GCC」は7%使用されている。この結果に基づいて、設計する遺伝子hFAD3においては、ヒトのコドンに近づけるように、「GCU」を17%、「GCC」を52%、「GCA」を14%、「GCC」を17%使用することとしたものである。他のアミノ酸に基づく、コドンについても同様にした。なお、表中、hFAD3で空欄になっているものは、使用頻度を変更しなかったものを意味する。
【表3】
【0081】
上記設計されたヒト化脂肪酸不飽和化酵素遺伝子(hFAD3)の塩基配列は、表1に示す。
【0082】
[脂肪酸不飽和化酵素遺伝子の作成]
次に、上記配列番号1の塩基配列の5’末端と3’末端には、ベクターに導入する際に用いる、制限酵素認識部位として、5’末端にHindIII認識部位を設け、3’末端にBamHI認識部位を設けた。また、効率的に翻訳を行うために、開始コドンの近傍に、リボソーム結合部位であるコザック(Kozak)配列を挿入した。
【0083】
このようにして設計した最適化遺伝子の塩基配列から、Sinclairら(Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expression and Purification 26,p.96−105(2002))と、Kimら(Codon optimization for high−level expression of human erythropoietin (EPO) in mammalian cells. Gene 199, p.293−301(1997))のPCR法を用いて、hFAD3の再構築遺伝子を作成した。
【0084】
表1に示す、設計した鋳型hFAD3の塩基配列をもとに、70〜90塩基ずつのPCRプライマーを設計した。これらのプライマーは、相互に補足するように12〜20塩基をオーバーラップさせた。
【0085】
(各プライマーセットにおけるPCR条件の検討)
表2に示す、No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20の組み合わせ10サンプルについて、PCR条件の検討を、以下のように行った。
【0086】
アマ由来FAD3 cDNA400ngを鋳型とし、4μl 10×ExTaq Buffer、3.2μl dNTP mix、0.2μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、0.5pmol/μl 各プライマーセットを合わせて、全量を40μlとした。これを10μlずつ、4本のPCRチューブに分注した。アニーリング温度を、それぞれ55℃、58℃、60℃、63℃に設定して、PCRを行った。PCR条件は、以下のとおりである。まず、94℃で10分間反応させた後、94℃で30秒、アニーリング30秒、72℃で1分を、35サイクル行った。PCR産物を、4%アガロース/TAE(トリス酢酸緩衝液)を用い、1×TAE中で25分間電気泳動を行った。泳動後、エチジウムブロマイド(EtBr)で染色し、UVトランスイルミネーターで、DNAの泳動パターンを観察した。泳導パターンがより明確なバンドを示したサンプルにおける、アニーリング温度を、最適のアニーリング温度とした。各プライマーセットにおける最適のアニーリング温度を表4に示す。
【表4】
【0087】
次に、設計したプライマーを組み合わせて、PCR法で、hFAD3遺伝子の合成を行った。No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20とを組み合わせてPCRを行った。アマ由来FAD3 cDNA100ngを鋳型とし、1μl 10×ExTaq Buffer、0.8μl dNTP mix、0.05μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、0.5pmol/μl 各プライマーセットを合わせて、全量を10μlとした。これらを、94℃で10分間反応させた後、94℃で30秒、最適のアニーリング温度で30秒、72℃で1分を35サイクルの条件でPCRを行った。
【0088】
上記PCR反応後のそれぞれのPCR産物 3.5μlに、5μl 10×ExTaq Buffer、4μl dNTP mix、0.25μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、2.5μlのNo.1とNo.20のプライマーを加え、全量を50μlとした。これを、94℃で10分間反応させた後、94℃で30秒、58℃で30秒、72℃で1分を35サイクルの条件でPCRを行った。これにより、hFAD3を含むPCR産物を得た。
【0089】
(PCR産物のサブクローニング)
上記得られたhFAD3を含むPCR産物から、1.2kbのDNAをDNA抽出キット(GFX(登録商標) PCR DNA and Gel Band Purification Kit、 GEヘルスケアバイオサイエンス株式会社製)を用いて回収した。抽出したDNAは、エタノールを用いて沈殿させて回収した。沈殿したDNAを、トリス塩酸バッファーを加えて溶解し、以下の操作に用いた。
【0090】
得られた1.2kbのDNA断片を、DNA ligation Kit Ver2(タカラバイオ(株)製)を用いて、pGEM−T EASYベクター(プロメガ社製)へ、TAクローニングした。
【0091】
クローニング産物を、大腸菌E.coli JM109 compotent cell(タカラバイオ(株)製)に入れて形質転換した。この形質転換した大腸菌を抗生物質カルベニシリン(和光純薬(株)製)を用いて、LBプレート(ナカライテスク製)で選択培養した。生育したコロニーをLB/Car(カルベニシリン)培地で、37℃、一晩振とう培養した。増殖した大腸菌をアルカリプレップ法を用いて、大腸菌の細胞壁を分解し、プラスミドを回収した。
【0092】
(取得クローンの塩基配列解析)
回収したプラスミドをポリエチレングリコールで沈殿させた後、DNAの濃度測定をした。得られたDNA 200ngに、0.8μlの2.0μM M13 プライマー(終濃度1.6μM)、0.5μl Big dye premix(アプライド バイオシステムズ製)、1.5μl 5×sequencing buffer(アプライド バイオシステムズ製)を加え、滅菌水で10μlにした。この液を、96℃で1分の後、96℃で10秒、50℃で5秒、60℃で4分を25サイクルの条件で、PCR法を行った。反応後、PCR産物をエタノール沈殿し、乾燥させた。乾燥させて得たペレットに、15μl Hi−Di ホルムアミド(アプライド バイオシステムズ製)を加えた。ペレットを溶解させ、攪拌した後、95℃で3分間変性処理をした。その後、氷上で急冷し、3170 DNA アナライザー(アプライド バイオシステムズ製)により、塩基配列を調べた。得られた塩基配列は、設計したhFAD3の塩基配列と同じであることを確認した。
【0093】
[組換え発現ベクターの作成]
ネオマイシン耐性遺伝子を有するpIRES2/EGFPベクター(タカラバイオ(株)製)を、NotI(タカラバイオ(株)製)で消化した。この末端をTakara Blunting Kit(タカラバイオ(株)製)で平滑化し、NotIサイトを除去した。このベクターのマルチクローニングサイトのEcoRIサイトを消化し、脱リン酸化処理を行った。
【0094】
上記pGEM−T Easy ベクター(プロメガ社製)にクローニングされたhFAD3配列をEcoRIで消化して切り出した。この切り出したhFAD3配列を、上記pIRES2/EGFPベクターの脱リン酸化処理を行ったEcoRIサイトに連結し、pIRES2/EGFP−hFAD3とした。
【0095】
次に、pIRES2/EGFP−hFAD3ベクターを、AseI、BglII(タカラバイオ(株)製)で消化し、CMVプロモーター配列を除去した。この部分に、両端にBglIIに結合できる配列を持ち、内側にNotIサイトを配した合成ポリヌクレオチドを挿入した。次に、このベクターを、SacI(タカラバイオ(株)製)で消化した。この部分に、両端にSacI配列結合を持ち、内側にMluI配列を持つ、合成ポリヌクレオチドを挿入した。
【0096】
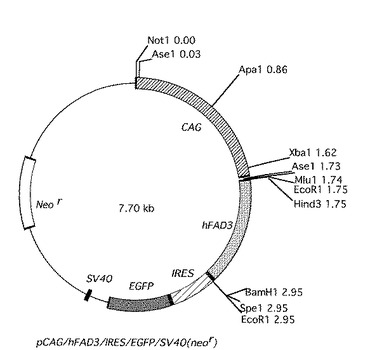
次に、pBluscriptIIKS(Stratagene製)にサブクローニングされた、CAGプロモーターを、NotIおよびMluI(タカラバイオ(株)製)で消化し、切り出した。切り出されたCAGプロモーターを、上記NotIおよびMluIで消化したpIRES2/EGFP−hFAD3ベクターに組み込んだ。これを、pCAG/hFAD3/IRES2/EGFP/SV40(neor)とした。構成したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を図2に示す。
【0097】
上記構築したベクターを大腸菌に形質転換して大量培養した。培養した大腸菌を、アルカリプレップ法により、プラスミドを回収した。回収したプラスミドを、終濃度が5μg/μlとなるように、精製PBS(ギブコ社(製))で溶解した。
【0098】
[ウシ筋衛星細胞への遺伝子導入および導入細胞の単離]
(ウシ筋衛星細胞株の樹立と培養)
ウシ筋衛星細胞は、2週齢雌ウシの胸腺付近の筋組織より樹立した。培地には、5ng/ml FGF(線維芽細胞増殖因子)−2/basic FGF(bFGF、アップステート製)および20%FBS(ウシ胎児血清)を添加したDMEM培地(bFGF−FBS−DMEM)を用いた。まず、ウシより得た筋組織を細切してトリプシン処理した。この後、500gで4分間の遠心分離を3回繰り返し、そのつど上清を回収、事前播種した。2時間後浮遊細胞を回収し、この浮遊細胞を、コラーゲン(高研製)でコートしたディッシュに播種した。播種24時間後に培地を交換し、得られた細胞の培養を行った。これらの細胞は継代培養した後に、以下の実験に用いた。
【0099】
(ウシ筋衛星細胞への遺伝子導入)
上記細胞株を樹立したウシ筋衛星細胞を、培地bFGF−FBS−DMEMを入れた4ウェル ディッシュに播種し、37℃、5% CO2、飽和湿度で24時間培養した。その後、培地を交換した(150μl/ウェル)。次に、1.5mlの試験管に無血清の100μl DMEMと3μlの遺伝子導入試薬(Gene Jammer(登録商標)トランスフェクション試薬)を加え、10分間室温で静置した。そこに、1μg/μlに調整したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を1μl加え、10分間室温で静置した。上記筋衛星細胞を入れた培養液に、この遺伝子導入試薬と導入遺伝子の混合液を100μl加え、37℃、5% CO2、飽和湿度で3時間培養した。その後、400μlのbFGF−FBS−DMEMを加え、37℃、5% CO2、飽和湿度で24時間培養した。
【0100】
(選択培養と遺伝子導入細胞の獲得)
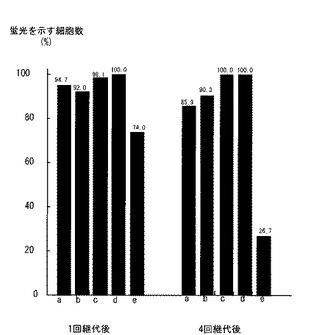
遺伝子を導入したウシ筋衛星細胞を培地bFGF−FBS−DMEM中で細胞数が5倍になるよう継代し、24時間培養した。その後、500μg/ml アミノグリコシド系抗生物質であるG418添加、bFGF−FBS−DMEMを培地として選択培養を行った。選択培養は、9日間行い、培養液は3日ごとに交換した。9日間培養を行うとほとんどの細胞は死滅したが、一部の細胞は生存し、コロニー状に増殖していた。増殖した細胞を蛍光顕微鏡下で観察した。EGFP蛍光が陽性の細胞群をクローニングリングを用いて単離した。単離した遺伝子導入細胞を培地bFGF−FBS−DMEM中で継代培養した(細胞群a〜e)。4代以上継代培養した後、再び蛍光顕微鏡下でEGFP蛍光を観察した。安定してEGFP蛍光が観察された細胞を、遺伝子導入細胞とした(細胞群b〜d)。図3は、選択培養した後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)と、継代培養後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)とを示すグラフである。このグラフから、細胞群b、c、dは、安定してEGFP蛍光が観察される(92.0→90.3、98.1→100.0、100.0→100.0)ことがわかる。細胞群a、eは、EGFP蛍光が安定していない(94.7→85.9、74.0→26.7)ことがわかる。また、細胞群aは他の細胞群と比較して蛍光強度が極端に低かった。以下の実験では、細胞群b、c、dを用いた。
【0101】
(脂肪細胞への分化能力の検討)
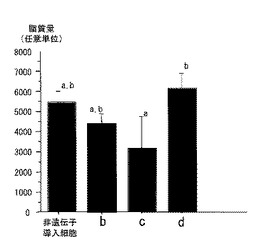
細胞群b、c、dの各細胞における、脂質を蓄積する能力を検討した。まず、1μM デキサメタゾン(シグマ社製)、2mM オクタン酸(シグマ社製)を添加したbFGF−FBS−DMEM(分化誘導培地)を用いて、細胞群b、c、dの細胞を、脂肪細胞へ分化誘導した。脂肪細胞への分化は、具体的には以下のようにして行った。hFAD3導入ウシ筋衛星細胞を、培地bFGF−FBS−DMEM中で70%集密になるまで培養した。その後、この分化誘導培地を用いて9日間培養し、多胞性の脂肪細胞へ分化誘導した。培地は3日おきに交換した。分化誘導した後、Oil Red 0により脂質を染色した。これを、NIHimage(ver。1.62、http://rsb.info.nih.gov/nih−image/、1999)で、染色された面積を測定して、蓄積された脂質量を定量した。結果を図4に示す。図4は、非遺伝子導入細胞と、細胞群b、c、dの細胞における脂質の蓄積を調べた結果を示すグラフである。各棒の上に記載されている「a」「b」は、異なる文字間で有意差(p<0.05)があることを示す。細胞群bおよびdは、非遺伝子導入細胞と脂質量が同様であった(p<0.05)が、細胞群cの脂質蓄積量は、非遺伝子導入細胞と比べ少なかった(p<0.05)。
【0102】
[染色体検査]
70%集密になるまで培養した遺伝子導入細胞である細胞群b、c、dの細胞を、0.02μg/ml デメコルシン(シグマ社製)を加えた培地で3時間培養し、デメコルシン処理を行った。その後、酢酸メタノールで30分間固定し、スライドグラスに滴下した。これを、ギムザ染色し、カバーグラスをかぶせマニキュアで封入して染色体標本を作成した。対照区として遺伝子が導入されていない細胞についても、同様に行った。倒立顕微鏡下1000倍で、50個の細胞について1個あたりの染色体を調べた。結果を表5に示す。図5は、pCAG/hFAD3/IRES2/EGFP/SV40(neor)が導入されたウシ筋衛星細胞において染色体数が正常な細胞の割合を示す表である。
【表5】
【0103】
表5から、細胞株c、dにおける正常細胞(染色体数60)の割合は、対照区の遺伝子導入細胞と同等であった(p<0.01)。一方、細胞群bでは、正常な染色体数を有する細胞の割合は、26%と小さかった(p<0.01)。以下の実験では、細胞群c、dの遺伝子導入細胞を用いて行った。
【0104】
[遺伝子導入細胞におけるhFAD3遺伝子の発現]
hFAD3遺伝子の下流には、IRESとEGFP遺伝子が連結されているので、染色体数が正常な細胞群c、dのhFAD3遺伝子の発現を、EGFP蛍光量として定量した。
【0105】
分化誘導前の遺伝子導入細胞は、10μM リノール酸ナトリウムを添加した、培地bFGF−FBS−DMEM中で、9日間培養し、100倍の倍率で明視野とUV照射下で、それぞれ撮影を行った。培地は、3日おきに交換した。
【0106】
分化誘導後の遺伝子導入細胞は、1μM デキサメタゾン、2mM オクタン酸、および10μM リノール酸ナトリウムを添加した、bFGF−FBS−DMEM(分化誘導培地)中で、9日間培養し、分化誘導していない細胞と同条件で写真の撮影を行った。培地は、3日おきに交換した。
【0107】
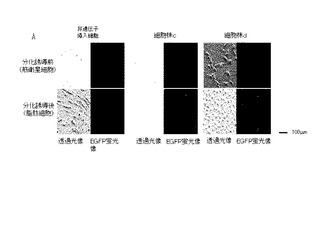
撮影後、画像解析ソフト・アクアコスモス(浜松フォトニクス(株)製)を用いて、取得した画像フィルムの蛍光部分のピクセル数を計測することで、細胞の蛍光強度を測定した。結果を、図5、6に示す。図5は、非遺伝子導入細胞と、細胞群c、dの分化誘導前と分化誘導後の、透過光像と、EGFP蛍光像である。また、図6は、細胞群c、dの分化誘導前と分化誘導後の傾向強度の変化を示すグラフである。
【0108】
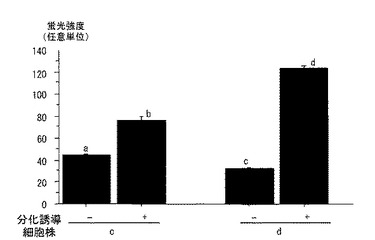
図5から、筋衛星細胞では、分化誘導により脂肪細胞に分化し、脂肪が蓄積されることがわかる。非遺伝子導入細胞からは、分化誘導後の脂肪細胞においても蛍光が発生していないことがわかった。また、遺伝子を導入した細胞群からは、透過光像で認められた分化誘導後の脂肪細胞から、EGFP蛍光が認められた。図6:分化誘導前の細胞であるc「−」、d「−」から、分化誘導前の細胞では、細胞群c、dの細胞とも遺伝子発現量は低いことがわかった。一方、図6:分化誘導後の細胞であるc「+」、d「+」から、分化誘導して、脂肪滴を蓄積した脂肪細胞では、細胞群c、dの細胞とも、EGFP蛍光量は多かった(p<0.05)。また、細胞群dは、細胞群cより、EGFPの発現は多かった(p<0.05)。
【0109】
[遺伝子導入細胞の脂肪酸組成解析]
(分化誘導を行う前の遺伝子導入細胞の脂肪酸解析)
hFAD3遺伝子を導入したウシ筋衛星細胞を10μMリノール酸ナトリウムを添加した、培地bFGF−FBS−DMEM中で9日間培養した。培地は、3日おきに交換した。培養後、細胞をPBS(リン酸緩衝液)で3回洗浄し、培地を除去した。次に、0.5% 2,6−ジ−ターシャルブチル−4−メチル−フェノール(BHT)を添加したメタノール(ナカライテスク製)を10ml加え、セルスクレーパー(旭テクノグラス製)を用いて、細胞ごと掻き採ることで、全脂質を抽出した。細胞の脂質は、プラスチックシャーレ(直径100mm、ベクトン・ディッキンソン アンド カンパニー製)10枚を1サンプルとして、0.5% BHT添加メタノールごとガラス試験管(17ml)に移した。これら脂質の脂肪酸は、カラムDB−23(φ0.25mm×30m、膜厚0.25μm、J&W サイエンテフィック製)を用いてガスクロマトグラフィ(GC−1700、島津製作所(株)製)を用いて分析した(検出器:FID、温度:試料注入口 250℃、検出器:250℃、カラム:50℃で1分間保持し、1分間につき10℃、170℃まで昇温させた。170℃に達した後、1分間につき1.2℃、210℃まで昇温させた。試料導入系:スプリットレス、ガス流量:ヘリウム(キャリヤーガス)1.5ml/分、ヘリウム(メイクアップガス)80kPa、ガス圧力:水素60kPa、空気 50kPa(AOCS official Method 1b−89(2005)に準じた。))。脂質の分析は、遺伝子非導入細胞と、細胞群c、dの細胞について、分化誘導前と分化誘導後の細胞で行った。結果を、表6、7に示す。表6は、分化誘導前の細胞の脂質の分析結果であり、表7は、分化誘導後の細胞の脂質の分析結果である。表中、脂肪酸組成で、「:」の前に記載されている数字は、脂肪酸の炭素数を、「:」の後に記載されている数字は、二重結合の数を、(n−)の後に記載されている数字は、多価不飽和脂肪酸の炭素鎖のメチル基から数えて最初の二重結合が存在する位置を示す。「↑」は、コントロール(遺伝子非導入細胞)と細胞群cまたはdの細胞間で脂肪酸が有意に上昇しているものを、「↓」は、コントロール(遺伝子非導入細胞)と細胞群cまたはdの細胞間で脂肪酸が有意に減少しているものを示した。
【0110】
脂肪酸組成は、遺伝子導入細胞の脂肪細胞への分化実験を3回反復した。各区で抽出した全脂質の脂肪酸組成比を分散分析法及びFisherのPLSDテストによる多重比較検定を行った。有意差水準は、p<0.05とした。
【表6】
【表7】
【0111】
表6から、分化誘導前の筋衛星細胞では、細胞群c、dの細胞ともに、遺伝子非導入細胞と比べて、α−リノレン酸(18:3n−3)をはじめとするn−3系脂肪酸脂肪酸などの脂肪酸組成に変化は認められなかったことがわかる(p<0.05)。
【0112】
表7から、分化誘導後の細胞では、hFAD3が低発現の細胞群cの細胞では、α−リノレン酸(18:3n−3)が0.30%と、対照区の遺伝子非導入細胞0.03%に対し、有意に増加していたことがわかる(p<0.05)。FAD3の基質であるリノール酸(18:2n−6)は9.37%と、対照区の遺伝子非導入細胞12.4%に対し、有意に減少していた(p<0.05)。n−3系の脂肪酸のドコサペンタエン酸(DPA、22:5n−3)は、5.80%と、対照区の遺伝子非導入細胞4.27%に対し、1.5%増加していた(p<0.05)。n−3系の脂肪酸の総計では、11.5%と、対照区の遺伝子非導入細胞9.1%に対し、約2.4%増加していた(p<0.05)。
【0113】
分化誘導後の細胞では、hFAD3が強発現の細胞群dの細胞でも、α−リノレン酸(18:3n−3)が0.27%と、対照区の遺伝子非導入細胞0.03%に対し、有意に増加していた(p<0.05)。FAD3の基質であるリノール酸(18:2n−6)は、9.40%と、対照区の遺伝子非導入細胞12.4%に対し、3%と、大いに減少していた(p<0.05)。n−3系の脂肪酸のドコサペンタエン酸(DPA、22:5n−3)は、5.30%と、対照区の遺伝子非導入細胞4.27%に対し、約1%増加していた(p<0.05)。n−3系の脂肪酸のドコサヘキサエン酸(DHA、22:5n−3)は、6.40%と、対照区の遺伝子非導入細胞3.57%に対し、約2.8%と大いに増加していた(p<0.05)。n−3系の脂肪酸の総計では、12.5%と、対照区の遺伝子非導入細胞9.1%に対し、約2.4%増加していた(p<0.05)。n−6系の脂肪酸の総計では、19.23%と、対照区の遺伝子非導入細胞23.9%に対し、約4.7%も減少していた(p<0.05)。
【0114】
また、(n−6)系の脂肪酸/(n−3)系の脂肪酸の割合は、細胞群cの細胞では、2.10、細胞群dの細胞では、1.57と、対照区の非遺伝子導入細胞の2.63に比べ有意に減少していることがわかった(p<0.05)。
【0115】
以上から、分化誘導した本発明の遺伝子導入細胞では、(n−6)系の脂肪酸から(n−3)系の脂肪酸への変化が進行していることがわかった。
【0116】
[遺伝子導入細胞をドナー細胞に用いたクローン胚の作製]
(ドナー細胞の調整)
遺伝子を導入した細胞群c、dの細胞を集密になるまで培養した。その後、細胞の増殖を停止させて、G0/G1期に細胞周期を同調させた。培養後、細胞c、dの細胞を回収し、培地bFGF−FBS−DMEMで洗浄し、核移植に用いた。また、コントロールとして、遺伝子非導入筋衛星細胞を、同様に培養、回収して用いた。
【0117】
(ウシ卵子の体外熟成)
大阪市食肉衛生検査所において、ウシ屠体より採取されたウシの卵巣を用いた。卵巣から21Gの注射針(テルモ社製)と10mlのシリンジ(テルモ社製)を用いて、ウシ卵子を卵胞液とともに吸引し回収した。卵胞より回収した卵胞液から、実体顕微鏡下で2〜4重層の卵丘細胞が付着した卵丘細胞卵子複合体のみを選別して回収した。その後、洗浄用成熟培養ドロップに、20個ずつ移し、洗浄した。次に、成熟培養ドロップに10個ずつ移動させ、39℃、5%CO2、95%空気、飽和湿度の炭酸ガスインキュベーター内で21時間培養した。
【0118】
(ウシ卵子の裸化)
21時間成熟培養した卵丘細胞卵子複合体を、0.5% FBS(ウシ胎児血清)および2.5ml/mlゲンタマイシン溶液を添加した25mM ヘペス緩衝TCM−199(ギブコ社製)に、0.25% ヒアルロニダーゼを加えた培地(199H+5% NBCS+0.25% ヒアルロニダーゼ)に移し、5分間静置した。静置後、ピペッティングにより、卵丘細胞卵子複合体を完全に取り除いた。
【0119】
(除核)
倒立顕微鏡下において、第1極体を放出し、第二減数分裂中期(MetaphaseII期)に達し、成熟していると思われる正常な卵子を選別した。この卵子の第1極体付近の透明帯を透明帯カット用ニードルで切開した。透明帯を切開した卵子を除核用培地(199H+5% NBCS+5μg/ml サイトカラシンB)に入れ、15分間静置した。静置後、除核用スティックで卵子を上から押さえつけ、第一極体及びその周辺卵細胞質を切開部位から2〜3割ほど押し出した。この卵子を、(199(Ea)+5% NBCS)培地に移し、ピペッティングにより卵子と押し出した卵細胞質に切り離した。切り離した卵細胞質のみを12μg/ml ヘキストNo.33342添加199(Ea)に移し、39℃、5%CO2、95%空気、飽和湿度で、30分間培養し、染色した。倒立顕微鏡下でUV照射により、核の確認を行った。核が確認できた卵細胞質に対応した卵子のみをレシピエント卵子として用いた。
【0120】
(細胞の導入)
除核した卵子を199(H)+5% NBCS培地に、上記ドナー細胞をPVP(ポリビニルピロリドン K−90(ナカライ社製))に移した。細胞導入用ピペットにドナー細胞を5〜10個吸引した後、除核卵子の透明帯切開部位から囲卵腔内にドナー細胞を1個ずつ導入した。細胞導入したウシ卵子を、199(H)+5% NBCS培地に移した。
【0121】
(電気融合)
電気融合には、ECM200(BTX社製)に接続した微小電極(ユニークメディカ イマダ 製)を用いた。除核卵子を、199(H)+5% NBCS培地に移し、ミネラルオイルを洗い流した。この除核卵子を、新たに、199(H)+5% NBCS培地に移し、(199(H)+5% NBCS)培地+細胞融合液混合液(1:1)、細胞融合液の順に移していった。卵子が底に沈んだ後、細胞融合液の楕円形のドロップを作製し、細胞導入した卵子をそこに移した。微小電極でウシ卵子細胞質および導入電極が一直線になるように挟み、交流2.7kV/cm・10μ秒を2回印加し、電気パルスによって融合処理を行った。その後、電気融合処理卵を細胞融合液、細胞融合液+(199(H)+5% NBCS)培地混合液(1:1)、(199(H)+5% NBCS)培地の順に移動させた。最後に、電気融合処理卵を、(199(Ea)+5% NBCS)培地で洗浄し、(199(Ea)+5% NBCS)培地のドロップへ移し、39℃、5%CO2、5%O2、90%N2、飽和湿度で、30分間培養した。培養後、実体顕微鏡下でドナー細胞と卵子細胞質との融合を確認した。
【0122】
(活性化処理)
電気融合処理後、遮光下でカルシウムイオノフォア活性化処理を行った。融合確認ができた再構築胚を、5μM Ca2+イオノマイシン培地に移し、5分間静置した。静置後、シクロへキシミド処理液をCa2+イオノマイシン培地に加え、反応を停止させた。カルシウムイオノフォア処理後の再構築胚を、修正合成卵管液培地(modified Synthetic Ovidautal Fluid (mSOF)medium、合成卵管液培養液(synthetic oviduct fluid medium, SOFM, Tervit et al., 1972)に必須および非必須アミノ酸を添加して修正した培養液(Takahashi and First, 1992)からさらに無機リン酸塩を除いた培養液(modified SOF medium,mSOFM))+10μg/ml シクロへキシミド培地のドロップに移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で6時間培養し、活性化を誘起した。
【0123】
(体外培養)
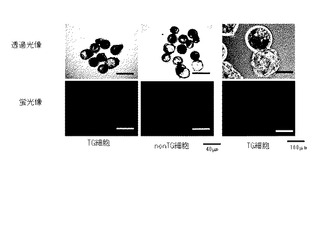
カルシウムイオノフォア−シクロへキシミド複合活性化処理の6時間後に、再構築胚をmSOFm培地に移し、洗浄した。洗浄後、mSOF培地のドロップに、1ドロップに付き20〜40個の再構築胚を移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で120時間培養した。120時間培養後、再構築胚を、20% FBS199+ベータメルカプトエタノール(βME)培地に移し洗浄した。洗浄後、20% FBS199+βME培地のドロップに移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で48時間培養した。48時間培養後、再構築胚の卵割率および胚盤胞期胚への発生率を検討した。また、胚盤胞期胚におけるEGFP蛍光の観察、胚盤胞期胚の回収を行った。表8に、再構築胚の卵割率および胚盤胞期胚への発生率を示す。コントロールとして、遺伝子非導入細胞を用いたクローン胚(表中、「nonTG細胞」と記載)を用いた。表中、「卵割胚数」は、2、4、8細胞期の卵数を、%は培養した胚数に対する卵割した胚数の割合を示す。また、「胚盤胞期胚数」の%は、培養した胚数に対する胚盤胞期胚の割合を示す。
【表8】
【0124】
表8から、細胞群dの細胞を導入したクローン胚(表中、「TG細胞」と記載)の胚盤胞への発生率は21%と、遺伝子非導入細胞を用いたクローン胚の35%と比較してやや低かったが、有意差は認められなかった(p>0.05)。
【0125】
また、図7に示すように、胚盤胞胚期へ発現した遺伝子導入クローン胚の全ての胚で蛍光を確認した。
【0126】
[遺伝子導入細胞クローン胚における導入遺伝子の発現の検討]
活性化処理後168時間で胚盤胞排気まで発生した胚10個を1サンプルとして回収した。このサンプルから、RNAqueous−Micro kit(Ambion社製)を用いて、RNA抽出を常法に従って行った。
【0127】
抽出したRNAを用いて、SperScript III First−Strand Synthesis System for RT−PCR kit(インビトロジェン社製)を使って、cDNAを合成した。
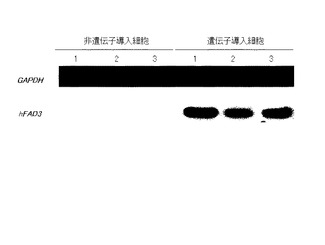
【0128】
得られたcDNAは、TaKaRa Ex Taq polymerase(TaKaRa Ex Taq、タカラバイオ(株)製)を用いて、PCR法により増幅した。PCR反応は、hFADのプライマーを用い、94℃で5分間反応させた後、94℃で1分間、55℃で1分30秒、72℃で1分30秒を35サイクルで行った。また、コントロールとしてハウスキーピング遺伝子GAPDHのプライマーを用いた。
【0129】
反応後、PCR産物を、1.2%寒天/TAEを用い、1×TAE中で25分間電気泳動を行った。泳動後、エチレンブロマイド(EtBr)で染色し、UVトランスイルミネーターでDNAの泳動パターンを観察した。結果を、図8に示す。図中、用いた胚の数は、非遺伝子導入細胞については、1:10個、2:10個、3:9個、遺伝子導入細胞では、1:10個、2:6個、3:10個であった。
【0130】
図8から、遺伝子導入株dを用いて作製したクローン胚では、hFAD3が発現していることがわかる。
【図面の簡単な説明】
【0131】
【図1】図1は、hFAD3の再構築遺伝子の作成方法を説明する図である。
【図2】図2は、構成したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を示す図である。
【図3】図3は、選択培養した後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)と、継代培養後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)とを示すグラフである。
【図4】図4は、非遺伝子導入細胞と、細胞群b、c、dの細胞における脂質の蓄積を調べた結果を示すグラフである。
【図5】図5は、pCAG/hFAD3/IRES2/EGFP/SV40(neor)を導入したウシ筋衛星細胞の分化誘導前と分化誘導後の透過光像および蛍光像である。
【図6】図6は、細胞群c、dの分化誘導前と分化誘導後の傾向強度の変化を示すグラフである。
【図7】図7は、遺伝子導入細胞株dを用いたクローン胚の胚盤胞期胚の透過光像と、EGFP蛍光像を示す図である。
【図8】図8は、胚盤胞に発生したクローン胚を用いた、RT−PCRによる、hFAD3遺伝子の発現を確認した図である。
【技術分野】
【0001】
本発明は、非ヒト動物細胞に脂肪酸不飽和化酵素遺伝子を導入する組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物に関する。
【背景技術】
【0002】
脂肪は、エネルギー源として非常に重要である。脂肪は、同時に、動物にとって正常な発育と機能維持に欠くことができない、不飽和脂肪酸(リノール酸(18:2n−6)、α−リノレン酸(18:3n−3)、およびアラキドン酸(20:4n−6))などの摂取源でもある。脂肪酸の合成では、Δ12脂肪酸不飽和化酵素により、オレイン酸(18:1)からリノール酸(18:2n−6)が合成され、さらに、ω3脂肪酸不飽和化酵素により、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)が合成される。
【0003】
ここで、多価不飽和脂肪酸は、生合成の経路別に、n−3系、n−6系、n−9系などに分類される。ここで、「n−」に続く数字3、6および9は、多価不飽和脂肪酸の炭素鎖のメチル基から数えて最初の二重結合が何番目の炭素にあるかを示す。例えば、「n−3」系は、炭素数が18個の脂肪酸のメチル基の炭素を1位の炭素とし、カルボキシル基側に向かって順次2位、3位・・・としたとき、メチル基から数えて最初の二重結合が3位の炭素にある多価不飽和脂肪酸を示す。「n−3」系は、「ω3」系とも表示される。また、「n−」の前に記載される数字は、一分子内に含まれる二重結合の数を示す。一方、「Δ」は、多価不飽和脂肪酸の炭素鎖のカルボキシル基から数えて最初の二重結合が何番目の炭素にあるかを示す。また、ω3脂肪酸不飽和化酵素、Δ12脂肪酸不飽和化酵素は、それぞれ、脂肪酸の炭素鎖のメチル基から数えて3位の位置に二重結合を作る酵素を、脂肪酸の炭素鎖のカルボキシル基から数えて12位の位置に二重結合を作る酵素を、意味する。
【0004】
しかし、哺乳動物などの高等動物では、上記の酵素を有さない。したがって、動物によれば、オレイン酸(18:1)からリノール酸(18:2n−6)の合成や、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成は行えないものもいる。
【0005】
α−リノレン酸(18:3n−3)からはエイコサペンタエン酸(EPA、20:5n−3)が、EPAからはドコサヘキサエン酸(DHA、22:6n−3)などが合成される。EPAやDHAなどのn−3系多価不飽和脂肪酸は、アトピー性皮膚炎、血栓性疾患、ガン、冠動脈心疾患などに低減効果があることが報告されている(例えば、非特許文献1、2参照)。
【0006】
これらのn−3系多価不飽和脂肪酸は、魚類や海草類などの海産物に多く含まれている。したがって、海産物を経口摂取することが非常に重要である。しかし、牛や豚などの畜肉やパーム油などの植物油に代表される陸生生物由来食品を過剰に摂取し、海産物の摂食が不足している、現在の先進国の食生活では、n−3系多価不飽和脂肪酸は摂取されにくい。
【0007】
このため、高等動物細胞が合成できない多価不飽和脂肪酸を高等動物細胞や高等動物中で合成しようとすることが試みられている。
【0008】
本発明者らは、ブタに、ホウレンソウ(Spinacia olerancea)根部由来のリノール酸(18:2n−6)を生成するΔ12脂肪酸不飽和化酵素をコードする遺伝子である、FAD2(fatty acid desaturation 2)を導入したものを提案した(例えば、特許文献1参照)。また、FAD2が導入されたブタでは、リノール酸(18:2n−6)が新規に合成されることを証明した。
【0009】
リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)を合成するω3脂肪酸不飽和化酵素については、例えば、植物等におけるω3脂肪酸不飽和化酵素の発現量や活性を、遺伝子操作等によって人為的に制御するための技術に使用できるω3脂肪酸不飽和化酵素遺伝子等が提供されている(例えば、特許文献2参照)。
【0010】
また、子嚢菌と脂質生成菌であるモルティエレラ・アルピナのΔ12脂肪酸不飽和酵素と、子嚢菌のω3脂肪酸不飽和化酵素の推定アミノ酸配列を比較し、相同性の高いアミノ酸配列に対応するプライマーを設計し、これから、ω3脂肪酸不飽和化活性を有するポリペプチドを得る技術が開示されている(例えば、特許文献3参照)。この文献中には、設計したプライマーから得られるポリヌクレオチドを含むベクターを動物に導入してよいことが記載されている。
【0011】
また、アラキドン酸産生糸状菌であるモルティエレラ・アルピナのΔ12脂肪酸不飽和化酵素遺伝子をマウスのL細胞に導入することで、哺乳動物細胞内でΔ12脂肪酸不飽和化酵素遺伝子が機能することを証明している。また、リノール酸だけでなく、リノール酸以降のn−6系脂肪酸の増加も確認している。このことから、哺乳動物がもたないΔ12脂肪酸不飽和化酵素遺伝子を哺乳動物細胞に導入でき、機能的に発現することが示されている(例えば、非特許文献3参照)。
【0012】
線虫由来のω3脂肪酸不飽和化酵素遺伝子であるfat−1をマウスの乳腺で発現させ、乳中のn−3系脂肪酸の増加を確認しており、哺乳動物でω3脂肪酸不飽和化酵素遺伝子が機能的に発現することが示されている。また、乳腺をターゲットにすることで乳の脂肪酸組成を改変できることを示し、授乳した仔マウスへの効果も確認されている(例えば、非特許文献4参照)。
【非特許文献1】Connor WE, Importance of n−3 fatty acids in health and disease. Am. J. Clin. Nutr. 71, 171S−175S (2000).
【非特許文献2】Terry PD et al., Intakes of fish and marine fatty acids and the risks of cancers of the breast and prostate and of other hormone−related cancers: A review of the epidemiologic evidence. Am. J. Clin. Nutr. 77,p.532−5543 (2003).
【非特許文献3】Kelder B et al., Expression of fungal desaturase genes in cultured mammalian cells. Molecula and Cellular Biochemistry 219,p.7−11(2001)
【非特許文献4】Kao BT et al., Endogenous production and elevated levels of long−chain n−3 fatty acids in the milk of transgenic mice. J. Dairy Sci. 89, p.3195−3201(2006)
【特許文献1】特開2003−245070号公報
【特許文献2】特開2001−95588
【特許文献3】特開2006−55104
【発明の開示】
【発明が解決しようとする課題】
【0013】
哺乳動物において、n−3系多価不飽和脂肪酸が合成されれば、食肉等からn−3系多価不飽和脂肪酸を効率よく摂取することができる。しかし、いずれの文献においても、哺乳動物において、ω3脂肪酸不飽和化酵素が機能し、n−3系多価不飽和脂肪酸の合成されたことについては、開示されていない。非特許文献4の場合では、遺伝子改変マウスの作出効率が極めて低く、中性脂肪への影響も少なかった。
【0014】
すなわち、本発明は、上記問題に鑑みなされたものであり、その目的は、哺乳動物において、n−3系多価不飽和脂肪酸を効率よく合成できる、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を提供することにある。
【課題を解決するための手段】
【0015】
本発明者らは、上記課題を解決するために鋭意検討をした結果、α−リノレン酸(18:3n−3)の合成に対して酵素活性の高い脂肪酸不飽和化酵素遺伝子を用いて、組換え発現ベクターとすることで、α−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸が多く合成される、組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を提供できることを見出し、本発明を完成させた。すなわち、本発明は以下のとおりである。
【0016】
本発明の脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(D)のいずれかの塩基配列からなるポリヌクレオチドである。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【0017】
本発明の組換え発現ベクターは、脂肪酸不飽和化酵素遺伝子を含有する。
【0018】
上記脂肪酸不飽和化酵素遺伝子は、ω3脂肪酸不飽和化酵素遺伝子である。
【0019】
上記脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(C)のいずれかの塩基配列からなる遺伝子であるとよい。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド、
(B)配列番号1に示される塩基配列において、1若しくは数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド、
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
【0020】
上記脂肪酸不飽和化酵素遺伝子は、ヒト化遺伝子であると好ましい。
【0021】
上記組換え発現ベクターは、レポーター遺伝子、IRES配列、および選択マーカー遺伝子を含むと、好ましい。
【0022】
本発明の非ヒト動物細胞は、上記組換え発現ベクターを用いて、脂肪酸不飽和化酵素遺伝子が導入されている。
【0023】
本発明の非ヒト動物は、脂肪酸不飽和化酵素遺伝子が導入されている。
【発明の効果】
【0024】
本発明の組換え発現ベクターを用いて非ヒト動物を形質変換すれば、非ヒト動物内で、非ヒト動物に内在する脂肪酸を不飽和化する反応を触媒することができる。この結果、従来動物ではできなかった、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成を可能とする。
【0025】
また、非ヒト動物内で、α−リノレン酸(18:3n−3)を基質とするn−3系多価不飽和脂肪酸が合成される。この結果、n−3系多価不飽和脂肪酸に富む食肉、乳製品などの畜産品の供給を可能とする。
【発明を実施するための最良の形態】
【0026】
以下に、本発明を詳細に説明する。
【0027】
[脂肪酸不飽和化酵素遺伝子]
本発明の組換え発現ベクターに用いられる脂肪酸不飽和化酵素遺伝子は、α−リノレン酸の生成に関わるω3脂肪酸不飽和酵素をコードする遺伝子(例えば、fatty acid desaturation 3、FAD3など)である。FAD3は、通常、植物、細菌、線虫などの下等動物に多く存在する遺伝子である。FAD3は、シロイヌナズナ、セイヨウアブラナ、大豆などから単離されている。本発明で用いることのできるFAD3は、FAD3を有する植物から取得すればよく、その由来は問わない。好ましくは、リノール酸(18:2n−6)からα−リノレン酸(18:3n−3)の合成能力の高い植物のFAD3を用いればよい。例えば、アマ科の植物である。また、植物由来のFAD3以外に、線虫から単離されるfat−1を用いてもよい。
【0028】
また、植物由来の遺伝子は、動物細胞内では発現されにくい場合がある。本発明で用いられる脂肪酸不飽和化酵素遺伝子は、植物由来の脂肪酸不飽和化酵素遺伝子をそのまま用いてもよく、この遺伝子をヒト化処理したものを用いてもよい(Kang JX, et al.,Transgenic mice:fat−1 mice convert n−6 to n−3 fatty acids. Nature,427,p.504(2004)、Lai L, al., Generation of cloned transgenic pigs rich in omega−3 fatty acids. Nature Biotechnology,24,p.1472−1473 (2006))。ヒト化処理を行うと、遺伝子の発現効率を向上させることができる。本明細書中で、ヒト化処理とは、植物由来の遺伝子を、アミノ酸配列を変更することなく、哺乳動物でのコドン使用頻度に準じて、塩基配列の変更を行うことをいう。ヒト化処理は、例えば以下のように行う。以下の例では、アマ由来のFAD3を用いる。
【0029】
アマ由来のFAD3は、以下のようにして得られる。ベニバナアマ(Linum grandiflorum)の幼若種子から抽出したtotal RNAからcDNAを合成する。縮重ポリメレースチェーン反応(degenerate PCR)法と、RACE法とを用いて、FAD3 cDNAの配列を調べる。
【0030】
植物の脂肪酸不飽和化酵素には、葉緑体で機能する型と小胞体膜上で機能する型がある。この植物の脂肪酸不飽和化酵素を動物で機能させるには、小胞体型の脂肪酸不飽和化酵素を用いる必要がある。小胞体型の脂肪酸不飽和化酵素は、細胞の小胞体膜に結合したタンパク質であり、3つのヒスチジンクラスターに鉄(Fe)原子が挟まれた活性中心を有する。得られたcDNAがFAD3遺伝子であるかどうかは、まず得られたcDNAに、小胞体移行シグナルを有していることを確認する。小胞体移行シグナルは、アミノ酸3’末端に「KXKXX」(K:リシン、Xはアミノ酸)配列を有する。FAD3の小胞体移行シグナルは、「KSKSA」(S:セリン、A:アラニン)である。したがって、cDNA中に上記FAD3の小胞体移行シグナルのアミノ酸配列に該当する塩基配列を有していることを確認すればよい。
【0031】
次に、3つのヒスチジンクラスターがあることを確認する。3つのヒスチジンクラスターは、「HCDH」、「HxxxxxHRTHH」、「HHxxxxHVIHH」(H:ヒスチジン、C:システイン、D:アスパラギン酸、R:アルギニン、T:スレオニン、V:バリン、I:イソロイシン、X:アミノ酸)配列である。3つのヒスチジンクラスターの存在は、この配列と配列が存在する場所で特定することができる。FAD3の3つのヒスチジンクラスターは、「HCDH」、「HGWKISHRTHH」、「HHDIGTHVIHH」(G:グリシン、W:トリプトファン)である。したがって、cDNA中に上記FAD3の3つヒスチジンクラスターのアミノ酸配列に該当する塩基配列を有していることを確認すればよい。
【0032】
次に、推定された配列をもとにプライマーを設計し、PCR法により、遺伝子FAD3を得る。得られたアマ由来cDNAの酵素活性を検討するため、公知の方法により酵母発現ベクターを構築し、酵母での酵素活性を確認する(Reed DW et al., Characterization of the Brassica napus extraplastidial linoleate desaturase by expression in Saccharomyces cerevisiae. Plant Physiology 122,p.715−720(2000))。非形質転換酵母と比較して、形質転換酵母においてα―リノレン酸(18:3n−3)の増加を確認して、得られたcDNAをアマ由来のFAD3とする。
【0033】
哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度を、Codon Usage Databese(http://www.kazusa.or.jp/codon/)を用いて比較する。その結果をもとに、FAD3遺伝子の配列を設計する。たとえば、アルギニンは、ヒトではCGCのコドンの使用頻度が高いが、アマ由来FAD3では、CGCのコドンの使用頻度は低く、代わりにAGAの使用頻度が高い。また、アスパラギンは、ヒトではAAUの使用頻度が78%を占めるが、アマ由来FAD3では54%とやや低くなる。ヒト化処理では、これらのコドン使用頻度を比較して、アマ由来FAD3の使用するコドンを、ヒトのコドンに代えて、ヒト化FAD3(hFAD3)遺伝子の配列を設計する。なお、コドンの変更は、必ず、ヒトのコドンの使用頻度に合わせる必要はない。コドンの使用頻度に近づければよい。また、遺伝子発現を効率化するために開始コドン付近にコザック配列を挿入してもよい。
【0034】
配列番号1に示される塩基配列からなるポリヌクレオチドは、アマ由来脂肪酸不飽和化酵素遺伝子FAD3をヒト化処理した(humanized)ポリヌクレオチド(hFAD3)の一例を示す。
(塩基配列1)
【表1】
【0035】
本発明では、前記脂肪酸不飽和化酵素遺伝子としては、以下の(A)〜(D)のいずれかの塩基配列からなる遺伝子を使用することができる。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド、
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド、
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【0036】
上記「1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列」とは、例えば1〜20個、好ましくは1〜15個、より好ましくは1〜10個、さらに好ましくは1〜5個の任意の数の塩基が欠失、置換若しくは付加された塩基配列を意味する。
【0037】
例えば、これら1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなるDNA(変異DNA)は、化学合成、遺伝子工学的手法、突然変異誘発などの当業者に既知の任意の方法により作製することもできる。具体的には、配列番号1に示される塩基配列からなるDNAに対し、変異原となる薬剤と接触作用させる方法、紫外線を照射する方法、遺伝子工学的な手法等を用いて、これらDNAに変異を導入することにより、変異DNAを取得することができる。遺伝子工学的手法の一つである部位特異的変異誘発法は特定の位置に特定の変異を導入できる手法であることから有用であり、Molecular Cloning:A laboratory Mannual,2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor,NY.,1989.以後“モレキュラークローニング第2版”と略す)、Current Protocols in Molecular Biology, Supplement 1〜38,John Wiley & Sons (1987−1997)等に記載の方法に準じて行うことができる。この変異DNAを適切な発現系を用いて発現させることにより、1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなるタンパク質を得ることができる。
【0038】
上記「ストリンジェントな条件下でハイブリダイズする塩基配列」とは、DNA又はRNAなどの核酸をプローブとして使用し、コロニー・ハイブリダイゼーション法、プラークハイブリダイゼーション法、あるいはサザンブロットハイブリダイゼーション法等を用いることにより得られる塩基配列を意味し、具体的には、コロニーあるいはプラーク由来のDNAまたは該DNAの断片を固定化したフィルターを用いて、0.7〜1.0MのNaCl存在下、65℃でハイブリダイゼーションを行った後、0.1〜2倍程度のSSC溶液(1倍濃度のSSC溶液の組成は、150mM塩化ナトリウム、15mMクエン酸ナトリウム)を用い、65℃条件下でフィルターを洗浄することにより同定できるDNAをあげることができる。ハイブリダイゼーションは、モレキュラークローニング第2版等に記載されている方法に準じて行うことができる。
【0039】
例えば、ストリンジェントな条件下でハイブリダイズすることができるDNAとしては、プローブとして使用するDNAの塩基配列と一定以上の相同性を有するDNAが挙げることができ、例えば60%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上の相同性を有するDNAを好適に例示することができる。
【0040】
3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチドは、上記した3つのヒスチジンクラスターと小胞体膜移行シグナルの確認方法と、真核細胞内でω3脂肪酸不飽和化酵素活性の発現を確認することにより行う。
【0041】
[脂肪酸不飽和化酵素遺伝子の作成]
本発明で用いる脂肪酸不飽和化酵素遺伝子は、例えば以下のようにして作成する。
【0042】
まず、上記で設計したhFAD3から最適化遺伝子の塩基配列を設計する。具体的には、上記配列番号1の塩基配列の5’末端と3’末端には、ベクターに導入する際に用いる、制限酵素認識部位を設ける。例えば、5’末端にHindIII認識部位を設け、3’末端にBamHI認識部位を設けるなどである。また、効率的に翻訳を行うために、開始コドンの近傍に、リボソーム結合部位であるコザック(Kozak)配列を挿入すると好ましい。
【0043】
このようにして設計した最適化遺伝子の塩基配列から、Sinclairら(2002)と、Kimら(1997)のPCR法を用いて、hFAD3の再構築遺伝子を作成する。図1は、hFAD3の再構築遺伝子の作成方法を説明する図である。
【0044】
設計した鋳型hFAD3の塩基配列をもとに、70〜90塩基ずつのPCRプライマーを設計する。これらのプライマーは、相互に補足するように12〜20塩基をオーバーラップさせる。表2には、このようにして設計した10組20個のPCRプライマーの例を示す。
【表2】
【0045】
次に、設計したプライマーを組み合わせて、PCR法で、hFAD3遺伝子の合成を行う。上記の例では、No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20とを組み合わせてPCRを行う。各々のプライマーの組み合わせにおいて、事前にアニーリング温度の最適条件を求めておくと良い。
【0046】
上記各々のプライマーセットを用いて得られたPCR産物を合わせて、No1と20のプライマーを組み合わせてPCRを行う。これにより、hFAD3を含むPCR産物が得られる。
【0047】
hFAD3を含むPCR産物から、所望の大きさ(上記の例では、1.2kb)のDNAを抽出する。DNAの抽出は、公知のDNA抽出キットを用いればよい。抽出したDNAは、エタノールを用いて沈殿させて回収する。沈殿したDNAを、トリス塩酸バッファーを加えて溶解し、以下の操作に用いる。
【0048】
得られたPCR産物を、TAクローニングにより、直接ベクターにクローニングする。PCRに用いるTaq DNAポリメラーゼは弱い末端転移酵素活性を持っており、PCR反応によって合成された産物の末端にA(アデニン)を1塩基付加することができる。PCR産物をプラスミドにクローニングする際、開環したプラスミドベクターの末端にT(チミン)を1塩基付加したものを用いると、AとTが水素結合を作ることによって、平滑末端同士のライゲーションよりもクローニング効率が上がる。使用するベクターとしては、プロメガ社のpGEM−T easyが挙げられる。このベクターでは挿入断片を制限酵素EcoRIに切り出すことが可能である。
【0049】
クローニング産物を、選択培養用の大腸菌に入れて形質転換する。選択培地中で生育した大腸菌のコロニーを、液体培地中で増殖させる。増殖した大腸菌培養液を公知のアルカリプレップ法を用いて、大腸菌の細胞壁を分解し、プラスミドを回収する。
【0050】
回収したプラスミドをポリエチレングリコールで沈殿させる。これをPCR法で増幅させる。必要に応じて、PCR産物の一部を公知の方法で、DNA塩基配列の分析をし、hFAD3の配列であることを確認する。
【0051】
[組換え発現ベクター]
本発明の組換え発現ベクターは、上記脂肪酸不飽和化酵素を発現するためのベクターである。本発明の組換え発現ベクターは、非ヒト動物で脂肪酸不飽和化酵素遺伝子を発現できるものであればよい。
【0052】
本発明の発現ベクターとして用いられるベクターには、遺伝子操作技術において広く用いられているものが含まれ、例えば、バクテリオファージ、プラスミド、コスミド、ウイルス、またはレトロウイルスなどが挙げられる。
【0053】
本発明の組換え発現ベクターは、RNAポリメラーゼが転写を開始するためのプロモーター領域を有する。プロモーター領域の下流に、脂肪酸不飽和化酵素遺伝子を導入する。
【0054】
脂肪酸不飽和化酵素遺伝子の発現を調節するプロモーターとしては、例えば、ウイルス(例、サイトメガロウイルス、モロニー白血病ウイルス、JCウイルス、乳癌ウイルスなど)に由来する遺伝子のプロモーター、哺乳動物(例、ヒト、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)および鳥類(ニワトリなど)に由来する遺伝子(例、アルブミン、エンドセリン、オステオカルシン、筋クレアチンキナーゼ、I型およびII型コラーゲン、サイクリックAMP依存蛋白キナーゼβIサブユニット、心房ナトリウム利尿性因子、ドーパミンβ−水酸化酵素、ニューロフィラメント軽鎖、メタロチオネインIおよびIIA、メタロプロティナーゼ1組織インヒビター、平滑筋αアクチン、ポリペプチド鎖伸長因子1α(EF1−α)、βアクチン、α及びβミオシン重鎖、ミオシン軽鎖1及び2、ミエリン塩基性蛋白、血清アミロイドPコンポーネント、レニンなど。また、脂肪細胞特異的に発現するアディポサイトP2(aP2)プロモーターや褐色脂肪細胞特異的に発現する脱共役タンパク質(uncoupling
protein,UCP)プロモーターなど)のプロモーターなどが挙げられ、なかでもニワトリアクチンプロモーターを含むCAGプロモーターなどを用いることができる。CAGプロモーターは、哺乳動物細胞で最も強く発現するので、好ましい。
【0055】
上記ベクターは、遺伝子導入哺乳動物において、目的とするmRNAの転写を終結する配列(ポリA、一般にターミネーターと呼ばれる)を有していることが好ましく、例えば、ウイルス由来、哺乳動物および鳥類由来の各遺伝子の配列を用いて遺伝子発現を操作することが出来る。好ましくは、シミアンウイルスのSV40ターミネーターなどが用いられる。
【0056】
本発明の組換え発現ベクターは、目的とする脂肪酸不飽和化酵素遺伝子の発現を同定するためのレポーター遺伝子を含んでいてもよい。レポーター遺伝子としては、ホタルルシフェラーゼ遺伝子、改良型ホタルルシフェラーゼ遺伝子、ウミシイタケルシフェラーゼ遺伝子、青緑色蛍光タンパク質(GFP)遺伝子、強化青緑色蛍光タンパク質(EGFP)遺伝子、青色蛍光タンパク質(BFP)遺伝子、赤色蛍光タンパク質(CFP)遺伝子などが好ましい。また、遺伝子導入するのではなく、胚の成育段階に応じて胚の表面に生じる表面抗原を利用してもよい。
【0057】
特に、プロモーターとしてCAGプロモーターを用いる場合は、レポーター遺伝子としてEGFPを用いるとよい。EGFPとは、オワンクラゲ由来の緑色蛍光蛋白質(GFP)の遺伝子改変体で、紫外光で蛋白質を励起した際より強い蛍光を発する蛋白質である。この遺伝子はClontech社などから購入することができる。CAGプロモーターとともに導入された細胞でEGFP遺伝子の発現が起こり、蛍光を発する細胞として、同定でき、遺伝子導入を確認できる。
【0058】
ベクターへのレポーター遺伝子の導入は、公知の方法が利用できる。好ましくは、Internal Ribosome Entry Site(IRES)とともに、IRESの下流に導入するとよい。IRESを含むベクターは、同一のプロモーターから複数の遺伝子を共発現させることができる。したがって、本発明のベクターがIRESを含んでいると、脂肪酸不飽和化酵素遺伝子とレポーター遺伝子とを共発現させることができるので、レポーター遺伝子の発現、すなわち発色を確認することで、脂肪酸不飽和化酵素遺伝子の発現を同定することができる。
【0059】
IRESを含むベクターとしては、市販のバイシストロン性ベクターを用いることもできる。
【0060】
また、本発明のベクターには、導入遺伝子が安定に組み込まれたクローンを選択するための選択マーカー遺伝子(例、ネオマイシン耐性遺伝子、ハイグロマイシン耐性遺伝子、アンピシリン耐性、ストレプトマイシン耐性遺伝子、カナマイシン耐性遺伝子などの薬剤耐性遺伝子など)をさらに含むことが好ましい。これにより、適当な選択薬剤を使用することにより、目的遺伝子が導入されたベクターを選択的に回収できる。
【0061】
[非ヒト動物]
本発明において脂肪酸不飽和化酵素遺伝子が導入される非ヒト動物としては、魚類、鳥類、哺乳類など食用にできる非ヒト動物であればよい。高等動物は、n−3系脂肪酸を産生していない。したがって、本発明の脂肪酸不飽和化酵素遺伝子を導入することで、n−3系脂肪酸の合成ができる非ヒト動物を得ることができる。
【0062】
使用できる魚類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する魚類であればよい。例えば、養殖の対象とされている魚類、筋肉組織等の可食部分において選択的に不飽和脂肪酸含量が高まる魚類や、子孫にまで安定的に脂肪酸不飽和化酵素遺伝子が発現する魚類等が好ましく、魚の種類としては硬骨魚類が好ましく、硬骨魚類の中でもで、Cyprinidae、Cichlidae、Salmonidae、Claridae、Siluridae、Ictaluridaeに属する魚類が好ましい。具体的には、ブリ、マダイ、ヒラメ、トラフグ、ヒラマサ、クルマエビ、ニジマス、カンパチ、シマアジ、マハタ、クロマグロなどの養殖魚の他、コイ、ゼブラフィッシュ、アフリカナマズ、テラピア、タイセイヨウサケ、メダカなどを好適に例示することができる。
【0063】
使用できる鳥類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する鳥類であればよい。具体的には、ニワトリ、ウズラ、ダチョウ、アイガモなどである。
【0064】
使用できる哺乳類としては、導入される脂肪酸不飽和化酵素遺伝子が発現し、これによりその体内の不飽和脂肪酸含量が増加する非ヒト哺乳類であればよい。例えば、ウシ、スイギュウ、ブタ、ヒツジ、ヤギ、ウマ、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなどが挙げられる。好ましくは、ウシ、スイギュウ、ブタ、ヒツジ、ヤギ、ウマなどのヒトが食用にできる非ヒト哺乳動物であり、さらに好ましくはウシである
【0065】
本発明の組換え発現ベクターが導入される非ヒト動物細胞としては、上記非ヒト動物由来の細胞であれば、初期胚、体細胞、胚性幹細胞であってもよい。好ましくは、体細胞であり、さらに好ましくは筋衛星細胞である。筋衛星細胞は骨格筋内に通常、休止状態で存在する組織特異的幹細胞で、骨格筋の再生や肥大に中心的な役割を担うことが知られている(Seale and Rudnicki,A new look at the origin, function, and “stem cell” status of muscle satellite cells. Developmental Biology 218, 115−124 (2000))。また、適切な細胞外環境を整えることにより、骨、軟骨、心筋、脂肪細胞などの他の細胞群へも分化する性質を有する多能性幹細胞としても位置づけられている(Asakura et al., Muscle satellite cells are multipotential stem cells that exhibit myogenic,osteogenic,and adipogenic differentiation.Differentiation 68,p.245−253(2001))。
【0066】
[非ヒト動物細胞への遺伝子導入]
本発明の組換え発現ベクターを導入する非ヒト動物細胞としては、非ヒト動物の体細胞から樹立した細胞を培養して用いる。体細胞の培養培地、培養条件は、公知の方法を用いる。
【0067】
上記培養した体細胞培養細胞の培地中に、遺伝子導入試薬と組換え発現ベクターとを加え、所定期間培養して、体細胞培養細胞中に遺伝子を導入する。
【0068】
上記遺伝子導入細胞をさらに培地中で培養を続け、細胞数を増加させる。その後、遺伝子導入細胞を、抗生物質などの選択薬剤を添加した培地中で選択培養する。選択培地中で生存した細胞について、顕微鏡下観察し、蛍光が認められたものを、クローニングリングにより単離し、通常の培地中で継代培養する。所定期間培養を続けた後にも、安定して蛍光が観察されたものを、遺伝子導入細胞とする。
【0069】
本発明の非ヒト動物細胞では、この遺伝子導入細胞の一部を脂肪細胞に分化させて、脂肪酸組成を分析することにより、遺伝子導入細胞のα−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能を、事前に評価することができる。
【0070】
[脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物]
(クローン胚の作成)
クローン胚のもとになるレシピエント卵子としては、成熟卵子であれば特に制限されるものではなく、屠場由来の卵巣から採取した卵子や生体の卵巣から経膣的に採取した卵子を体外成熟させたもののほか、プロスタグランジンF2α、クロプロステノール、絨毛ゴナドトロピン等のホルモン投与による過排卵処理により得られる体内成熟卵子であっても使用できる。卵丘細胞が付着している卵子はヒアルロニダーゼ処理を行って、卵丘細胞を除去することが好ましい。
【0071】
上記レシピエント卵子からの除核は、細胞骨格形成阻害剤であるサイトカラシンB処理を施した卵子を使用するのが好ましい。より具体的には、体外成熟卵子等のレシピエント卵子をホールディングピペットで保定し、微細なガラス針によりその透明帯を迅速・的確に切開したのち、サイトカラシンBを含有する培地でそれらのレシピエント卵子を処理し、除核操作用シャーレのサイトカラシン入りドロップに移して、除核用ピペットあるいは除核用ガラス針により第一極体の付近の細胞質を極体ごと吸引又は押し出して行う。なお、それらの極体を含む細胞質を調べることにより除核できていることを確認することが好ましく、また除核卵子からはサイトカラシンBを除去することが好ましい。
【0072】
ドナー細胞としては、上記遺伝子導入細胞を培養したものを用いる。また、ドナー細胞の細胞周期は特に制限されるものではないが、細胞周期G0/G1期に同調させた体細胞が好ましい。なお、細胞周期G0/G1期に同調させた体細胞は、例えば一定期間培養し続けコンフルエントな状態まで培養するあるいは、培養液中の血清濃度を極端に低下させた培養液に交換し血清飢餓状態で培養することにより得ることができる。
【0073】
体細胞クローン胚は、定法に従って、例えば、ドナー細胞を細胞導入用ピペットに吸い込み、このピペットの先端を除核されたレシピエント卵子の囲卵腔に差し込んでドナー細胞を注入したのち、細胞融合装置による電気融合処理、カルシウムショックによる活性化処理を行うことにより作製される。
【0074】
上記クローン胚を、所定の培養培地を用いて、体外培養する。クローン胚の卵割、胚盤胞期胚への発生を観察し、卵割しない胚、胚盤胞期胚へ発生しない胚を除く。次に、これらの胚において、リポーター遺伝子の発生を、蛍光により観察する。蛍光が認められるクローン胚を選択して、仮母の子宮内に移植することで、哺乳動物において、n−3系多価不飽和脂肪酸を効率よく合成できる非ヒト動物を得ることができる。
【0075】
本発明では、ベクター中に、蛍光により、脂肪酸不飽和化酵素遺伝子の発現を確認できるリポーター遺伝子を含む。このため、ベクターを非ヒト動物細胞に導入した状態と、クローン胚を作成した状態において、脂肪酸不飽和化酵素遺伝子の発現を容易に確認できる。
【0076】
また、脂肪酸不飽和化酵素遺伝子の発現を確認した非ヒト動物細胞を脂肪細胞に分化させることができる。これにより、脂肪酸不飽和化酵素遺伝子が導入された非ヒト動物細胞のα−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能をあらかじめ評価することができる。
【0077】
すなわち、本発明の組換え発現ベクター、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞および非ヒト動物を用いれば、α−リノレン酸(18:3n−3)およびn−3系不飽和脂肪酸の合成能の高い非ヒト動物を容易に得ることができる。
【実施例】
【0078】
以下本発明を詳細に説明するため実施例を挙げて説明するが、本発明は実施例に限定されるものではない。
【0079】
[hFAD3遺伝子]
ベニバナアマ(Linum grandiflorum)の幼若種子から抽出したtotal RNAからcDNAを合成した。縮重ポリメレースチェーン反応(degenerate PCR)法と、RACE法とを用いて、FAD3 cDNAの配列を調べた。推定された配列をもとにプライマーを設計し、PCR法により、全長のcDNAを得た。このアマ由来のcDNAは、既知の植物のFAD3との相同性が高く、FAD3の特徴を有していた。このcDNAの酵素活性を、酵母を用いて検討したところ、α−リノレン酸(18:3n−3)が増加したことから、このcDNAを遺伝子FAD3とした。
【0080】
(ヒト化脂肪酸不飽和化酵素遺伝子の設計)
哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度を、Codon Usage Databese(http://www.kazusa.or.jp/codon/)を用いて比較した。その結果をもとに、FAD3遺伝子の配列を設計した。結果を表3に示す。表3は、アミノ酸:Ala(アラニン)、Arg(アルギニン)、Asn(アスパラギン)、Asp(アスパラギン酸)、Leu(ロイシン)、Gln(グルタミン)、Glu(グルタミン酸)、Gly(グリシン)、His(ヒスチジン)、Ile(イソロイシン)、Lys(リシン)、Pro(プロリン)、Cys(システイン)、Phe(フェニルアラニン)、Ser(セリン)、Thr(スレオニン)、Tyr(チロシン)、Val(バリン)における哺乳動物(ヒト)のコドンの使用頻度と、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドン使用頻度と、本実施例で設計した遺伝子hFAD3におけるコドン使用頻度を示している。Ala(アラニン)に基づいて説明すると、Alaには、「GCU」、「GCC」、「GCA」、「GCC」という4個のコドンが存在する。ヒトのコドンでは、「GCU」は17%、「GCC」は53%、「GCA」は13%、「GCC」は14%使用されている。一方、アマ由来脂肪酸不飽和化酵素遺伝子FAD3のコドンでは、「GCU」は28%、「GCC」は48%、「GCA」は17%、「GCC」は7%使用されている。この結果に基づいて、設計する遺伝子hFAD3においては、ヒトのコドンに近づけるように、「GCU」を17%、「GCC」を52%、「GCA」を14%、「GCC」を17%使用することとしたものである。他のアミノ酸に基づく、コドンについても同様にした。なお、表中、hFAD3で空欄になっているものは、使用頻度を変更しなかったものを意味する。
【表3】
【0081】
上記設計されたヒト化脂肪酸不飽和化酵素遺伝子(hFAD3)の塩基配列は、表1に示す。
【0082】
[脂肪酸不飽和化酵素遺伝子の作成]
次に、上記配列番号1の塩基配列の5’末端と3’末端には、ベクターに導入する際に用いる、制限酵素認識部位として、5’末端にHindIII認識部位を設け、3’末端にBamHI認識部位を設けた。また、効率的に翻訳を行うために、開始コドンの近傍に、リボソーム結合部位であるコザック(Kozak)配列を挿入した。
【0083】
このようにして設計した最適化遺伝子の塩基配列から、Sinclairら(Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expression and Purification 26,p.96−105(2002))と、Kimら(Codon optimization for high−level expression of human erythropoietin (EPO) in mammalian cells. Gene 199, p.293−301(1997))のPCR法を用いて、hFAD3の再構築遺伝子を作成した。
【0084】
表1に示す、設計した鋳型hFAD3の塩基配列をもとに、70〜90塩基ずつのPCRプライマーを設計した。これらのプライマーは、相互に補足するように12〜20塩基をオーバーラップさせた。
【0085】
(各プライマーセットにおけるPCR条件の検討)
表2に示す、No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20の組み合わせ10サンプルについて、PCR条件の検討を、以下のように行った。
【0086】
アマ由来FAD3 cDNA400ngを鋳型とし、4μl 10×ExTaq Buffer、3.2μl dNTP mix、0.2μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、0.5pmol/μl 各プライマーセットを合わせて、全量を40μlとした。これを10μlずつ、4本のPCRチューブに分注した。アニーリング温度を、それぞれ55℃、58℃、60℃、63℃に設定して、PCRを行った。PCR条件は、以下のとおりである。まず、94℃で10分間反応させた後、94℃で30秒、アニーリング30秒、72℃で1分を、35サイクル行った。PCR産物を、4%アガロース/TAE(トリス酢酸緩衝液)を用い、1×TAE中で25分間電気泳動を行った。泳動後、エチジウムブロマイド(EtBr)で染色し、UVトランスイルミネーターで、DNAの泳動パターンを観察した。泳導パターンがより明確なバンドを示したサンプルにおける、アニーリング温度を、最適のアニーリング温度とした。各プライマーセットにおける最適のアニーリング温度を表4に示す。
【表4】
【0087】
次に、設計したプライマーを組み合わせて、PCR法で、hFAD3遺伝子の合成を行った。No.1と2、No.3と4、No.5と6、No.7と8、No.9と10、No.11と12、No.13と14、No.15と16、No.17と18、No.19と20とを組み合わせてPCRを行った。アマ由来FAD3 cDNA100ngを鋳型とし、1μl 10×ExTaq Buffer、0.8μl dNTP mix、0.05μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、0.5pmol/μl 各プライマーセットを合わせて、全量を10μlとした。これらを、94℃で10分間反応させた後、94℃で30秒、最適のアニーリング温度で30秒、72℃で1分を35サイクルの条件でPCRを行った。
【0088】
上記PCR反応後のそれぞれのPCR産物 3.5μlに、5μl 10×ExTaq Buffer、4μl dNTP mix、0.25μl Taq polymerase(Takara Ex Taq、タカラバイオ社製)、2.5μlのNo.1とNo.20のプライマーを加え、全量を50μlとした。これを、94℃で10分間反応させた後、94℃で30秒、58℃で30秒、72℃で1分を35サイクルの条件でPCRを行った。これにより、hFAD3を含むPCR産物を得た。
【0089】
(PCR産物のサブクローニング)
上記得られたhFAD3を含むPCR産物から、1.2kbのDNAをDNA抽出キット(GFX(登録商標) PCR DNA and Gel Band Purification Kit、 GEヘルスケアバイオサイエンス株式会社製)を用いて回収した。抽出したDNAは、エタノールを用いて沈殿させて回収した。沈殿したDNAを、トリス塩酸バッファーを加えて溶解し、以下の操作に用いた。
【0090】
得られた1.2kbのDNA断片を、DNA ligation Kit Ver2(タカラバイオ(株)製)を用いて、pGEM−T EASYベクター(プロメガ社製)へ、TAクローニングした。
【0091】
クローニング産物を、大腸菌E.coli JM109 compotent cell(タカラバイオ(株)製)に入れて形質転換した。この形質転換した大腸菌を抗生物質カルベニシリン(和光純薬(株)製)を用いて、LBプレート(ナカライテスク製)で選択培養した。生育したコロニーをLB/Car(カルベニシリン)培地で、37℃、一晩振とう培養した。増殖した大腸菌をアルカリプレップ法を用いて、大腸菌の細胞壁を分解し、プラスミドを回収した。
【0092】
(取得クローンの塩基配列解析)
回収したプラスミドをポリエチレングリコールで沈殿させた後、DNAの濃度測定をした。得られたDNA 200ngに、0.8μlの2.0μM M13 プライマー(終濃度1.6μM)、0.5μl Big dye premix(アプライド バイオシステムズ製)、1.5μl 5×sequencing buffer(アプライド バイオシステムズ製)を加え、滅菌水で10μlにした。この液を、96℃で1分の後、96℃で10秒、50℃で5秒、60℃で4分を25サイクルの条件で、PCR法を行った。反応後、PCR産物をエタノール沈殿し、乾燥させた。乾燥させて得たペレットに、15μl Hi−Di ホルムアミド(アプライド バイオシステムズ製)を加えた。ペレットを溶解させ、攪拌した後、95℃で3分間変性処理をした。その後、氷上で急冷し、3170 DNA アナライザー(アプライド バイオシステムズ製)により、塩基配列を調べた。得られた塩基配列は、設計したhFAD3の塩基配列と同じであることを確認した。
【0093】
[組換え発現ベクターの作成]
ネオマイシン耐性遺伝子を有するpIRES2/EGFPベクター(タカラバイオ(株)製)を、NotI(タカラバイオ(株)製)で消化した。この末端をTakara Blunting Kit(タカラバイオ(株)製)で平滑化し、NotIサイトを除去した。このベクターのマルチクローニングサイトのEcoRIサイトを消化し、脱リン酸化処理を行った。
【0094】
上記pGEM−T Easy ベクター(プロメガ社製)にクローニングされたhFAD3配列をEcoRIで消化して切り出した。この切り出したhFAD3配列を、上記pIRES2/EGFPベクターの脱リン酸化処理を行ったEcoRIサイトに連結し、pIRES2/EGFP−hFAD3とした。
【0095】
次に、pIRES2/EGFP−hFAD3ベクターを、AseI、BglII(タカラバイオ(株)製)で消化し、CMVプロモーター配列を除去した。この部分に、両端にBglIIに結合できる配列を持ち、内側にNotIサイトを配した合成ポリヌクレオチドを挿入した。次に、このベクターを、SacI(タカラバイオ(株)製)で消化した。この部分に、両端にSacI配列結合を持ち、内側にMluI配列を持つ、合成ポリヌクレオチドを挿入した。
【0096】
次に、pBluscriptIIKS(Stratagene製)にサブクローニングされた、CAGプロモーターを、NotIおよびMluI(タカラバイオ(株)製)で消化し、切り出した。切り出されたCAGプロモーターを、上記NotIおよびMluIで消化したpIRES2/EGFP−hFAD3ベクターに組み込んだ。これを、pCAG/hFAD3/IRES2/EGFP/SV40(neor)とした。構成したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を図2に示す。
【0097】
上記構築したベクターを大腸菌に形質転換して大量培養した。培養した大腸菌を、アルカリプレップ法により、プラスミドを回収した。回収したプラスミドを、終濃度が5μg/μlとなるように、精製PBS(ギブコ社(製))で溶解した。
【0098】
[ウシ筋衛星細胞への遺伝子導入および導入細胞の単離]
(ウシ筋衛星細胞株の樹立と培養)
ウシ筋衛星細胞は、2週齢雌ウシの胸腺付近の筋組織より樹立した。培地には、5ng/ml FGF(線維芽細胞増殖因子)−2/basic FGF(bFGF、アップステート製)および20%FBS(ウシ胎児血清)を添加したDMEM培地(bFGF−FBS−DMEM)を用いた。まず、ウシより得た筋組織を細切してトリプシン処理した。この後、500gで4分間の遠心分離を3回繰り返し、そのつど上清を回収、事前播種した。2時間後浮遊細胞を回収し、この浮遊細胞を、コラーゲン(高研製)でコートしたディッシュに播種した。播種24時間後に培地を交換し、得られた細胞の培養を行った。これらの細胞は継代培養した後に、以下の実験に用いた。
【0099】
(ウシ筋衛星細胞への遺伝子導入)
上記細胞株を樹立したウシ筋衛星細胞を、培地bFGF−FBS−DMEMを入れた4ウェル ディッシュに播種し、37℃、5% CO2、飽和湿度で24時間培養した。その後、培地を交換した(150μl/ウェル)。次に、1.5mlの試験管に無血清の100μl DMEMと3μlの遺伝子導入試薬(Gene Jammer(登録商標)トランスフェクション試薬)を加え、10分間室温で静置した。そこに、1μg/μlに調整したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を1μl加え、10分間室温で静置した。上記筋衛星細胞を入れた培養液に、この遺伝子導入試薬と導入遺伝子の混合液を100μl加え、37℃、5% CO2、飽和湿度で3時間培養した。その後、400μlのbFGF−FBS−DMEMを加え、37℃、5% CO2、飽和湿度で24時間培養した。
【0100】
(選択培養と遺伝子導入細胞の獲得)
遺伝子を導入したウシ筋衛星細胞を培地bFGF−FBS−DMEM中で細胞数が5倍になるよう継代し、24時間培養した。その後、500μg/ml アミノグリコシド系抗生物質であるG418添加、bFGF−FBS−DMEMを培地として選択培養を行った。選択培養は、9日間行い、培養液は3日ごとに交換した。9日間培養を行うとほとんどの細胞は死滅したが、一部の細胞は生存し、コロニー状に増殖していた。増殖した細胞を蛍光顕微鏡下で観察した。EGFP蛍光が陽性の細胞群をクローニングリングを用いて単離した。単離した遺伝子導入細胞を培地bFGF−FBS−DMEM中で継代培養した(細胞群a〜e)。4代以上継代培養した後、再び蛍光顕微鏡下でEGFP蛍光を観察した。安定してEGFP蛍光が観察された細胞を、遺伝子導入細胞とした(細胞群b〜d)。図3は、選択培養した後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)と、継代培養後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)とを示すグラフである。このグラフから、細胞群b、c、dは、安定してEGFP蛍光が観察される(92.0→90.3、98.1→100.0、100.0→100.0)ことがわかる。細胞群a、eは、EGFP蛍光が安定していない(94.7→85.9、74.0→26.7)ことがわかる。また、細胞群aは他の細胞群と比較して蛍光強度が極端に低かった。以下の実験では、細胞群b、c、dを用いた。
【0101】
(脂肪細胞への分化能力の検討)
細胞群b、c、dの各細胞における、脂質を蓄積する能力を検討した。まず、1μM デキサメタゾン(シグマ社製)、2mM オクタン酸(シグマ社製)を添加したbFGF−FBS−DMEM(分化誘導培地)を用いて、細胞群b、c、dの細胞を、脂肪細胞へ分化誘導した。脂肪細胞への分化は、具体的には以下のようにして行った。hFAD3導入ウシ筋衛星細胞を、培地bFGF−FBS−DMEM中で70%集密になるまで培養した。その後、この分化誘導培地を用いて9日間培養し、多胞性の脂肪細胞へ分化誘導した。培地は3日おきに交換した。分化誘導した後、Oil Red 0により脂質を染色した。これを、NIHimage(ver。1.62、http://rsb.info.nih.gov/nih−image/、1999)で、染色された面積を測定して、蓄積された脂質量を定量した。結果を図4に示す。図4は、非遺伝子導入細胞と、細胞群b、c、dの細胞における脂質の蓄積を調べた結果を示すグラフである。各棒の上に記載されている「a」「b」は、異なる文字間で有意差(p<0.05)があることを示す。細胞群bおよびdは、非遺伝子導入細胞と脂質量が同様であった(p<0.05)が、細胞群cの脂質蓄積量は、非遺伝子導入細胞と比べ少なかった(p<0.05)。
【0102】
[染色体検査]
70%集密になるまで培養した遺伝子導入細胞である細胞群b、c、dの細胞を、0.02μg/ml デメコルシン(シグマ社製)を加えた培地で3時間培養し、デメコルシン処理を行った。その後、酢酸メタノールで30分間固定し、スライドグラスに滴下した。これを、ギムザ染色し、カバーグラスをかぶせマニキュアで封入して染色体標本を作成した。対照区として遺伝子が導入されていない細胞についても、同様に行った。倒立顕微鏡下1000倍で、50個の細胞について1個あたりの染色体を調べた。結果を表5に示す。図5は、pCAG/hFAD3/IRES2/EGFP/SV40(neor)が導入されたウシ筋衛星細胞において染色体数が正常な細胞の割合を示す表である。
【表5】
【0103】
表5から、細胞株c、dにおける正常細胞(染色体数60)の割合は、対照区の遺伝子導入細胞と同等であった(p<0.01)。一方、細胞群bでは、正常な染色体数を有する細胞の割合は、26%と小さかった(p<0.01)。以下の実験では、細胞群c、dの遺伝子導入細胞を用いて行った。
【0104】
[遺伝子導入細胞におけるhFAD3遺伝子の発現]
hFAD3遺伝子の下流には、IRESとEGFP遺伝子が連結されているので、染色体数が正常な細胞群c、dのhFAD3遺伝子の発現を、EGFP蛍光量として定量した。
【0105】
分化誘導前の遺伝子導入細胞は、10μM リノール酸ナトリウムを添加した、培地bFGF−FBS−DMEM中で、9日間培養し、100倍の倍率で明視野とUV照射下で、それぞれ撮影を行った。培地は、3日おきに交換した。
【0106】
分化誘導後の遺伝子導入細胞は、1μM デキサメタゾン、2mM オクタン酸、および10μM リノール酸ナトリウムを添加した、bFGF−FBS−DMEM(分化誘導培地)中で、9日間培養し、分化誘導していない細胞と同条件で写真の撮影を行った。培地は、3日おきに交換した。
【0107】
撮影後、画像解析ソフト・アクアコスモス(浜松フォトニクス(株)製)を用いて、取得した画像フィルムの蛍光部分のピクセル数を計測することで、細胞の蛍光強度を測定した。結果を、図5、6に示す。図5は、非遺伝子導入細胞と、細胞群c、dの分化誘導前と分化誘導後の、透過光像と、EGFP蛍光像である。また、図6は、細胞群c、dの分化誘導前と分化誘導後の傾向強度の変化を示すグラフである。
【0108】
図5から、筋衛星細胞では、分化誘導により脂肪細胞に分化し、脂肪が蓄積されることがわかる。非遺伝子導入細胞からは、分化誘導後の脂肪細胞においても蛍光が発生していないことがわかった。また、遺伝子を導入した細胞群からは、透過光像で認められた分化誘導後の脂肪細胞から、EGFP蛍光が認められた。図6:分化誘導前の細胞であるc「−」、d「−」から、分化誘導前の細胞では、細胞群c、dの細胞とも遺伝子発現量は低いことがわかった。一方、図6:分化誘導後の細胞であるc「+」、d「+」から、分化誘導して、脂肪滴を蓄積した脂肪細胞では、細胞群c、dの細胞とも、EGFP蛍光量は多かった(p<0.05)。また、細胞群dは、細胞群cより、EGFPの発現は多かった(p<0.05)。
【0109】
[遺伝子導入細胞の脂肪酸組成解析]
(分化誘導を行う前の遺伝子導入細胞の脂肪酸解析)
hFAD3遺伝子を導入したウシ筋衛星細胞を10μMリノール酸ナトリウムを添加した、培地bFGF−FBS−DMEM中で9日間培養した。培地は、3日おきに交換した。培養後、細胞をPBS(リン酸緩衝液)で3回洗浄し、培地を除去した。次に、0.5% 2,6−ジ−ターシャルブチル−4−メチル−フェノール(BHT)を添加したメタノール(ナカライテスク製)を10ml加え、セルスクレーパー(旭テクノグラス製)を用いて、細胞ごと掻き採ることで、全脂質を抽出した。細胞の脂質は、プラスチックシャーレ(直径100mm、ベクトン・ディッキンソン アンド カンパニー製)10枚を1サンプルとして、0.5% BHT添加メタノールごとガラス試験管(17ml)に移した。これら脂質の脂肪酸は、カラムDB−23(φ0.25mm×30m、膜厚0.25μm、J&W サイエンテフィック製)を用いてガスクロマトグラフィ(GC−1700、島津製作所(株)製)を用いて分析した(検出器:FID、温度:試料注入口 250℃、検出器:250℃、カラム:50℃で1分間保持し、1分間につき10℃、170℃まで昇温させた。170℃に達した後、1分間につき1.2℃、210℃まで昇温させた。試料導入系:スプリットレス、ガス流量:ヘリウム(キャリヤーガス)1.5ml/分、ヘリウム(メイクアップガス)80kPa、ガス圧力:水素60kPa、空気 50kPa(AOCS official Method 1b−89(2005)に準じた。))。脂質の分析は、遺伝子非導入細胞と、細胞群c、dの細胞について、分化誘導前と分化誘導後の細胞で行った。結果を、表6、7に示す。表6は、分化誘導前の細胞の脂質の分析結果であり、表7は、分化誘導後の細胞の脂質の分析結果である。表中、脂肪酸組成で、「:」の前に記載されている数字は、脂肪酸の炭素数を、「:」の後に記載されている数字は、二重結合の数を、(n−)の後に記載されている数字は、多価不飽和脂肪酸の炭素鎖のメチル基から数えて最初の二重結合が存在する位置を示す。「↑」は、コントロール(遺伝子非導入細胞)と細胞群cまたはdの細胞間で脂肪酸が有意に上昇しているものを、「↓」は、コントロール(遺伝子非導入細胞)と細胞群cまたはdの細胞間で脂肪酸が有意に減少しているものを示した。
【0110】
脂肪酸組成は、遺伝子導入細胞の脂肪細胞への分化実験を3回反復した。各区で抽出した全脂質の脂肪酸組成比を分散分析法及びFisherのPLSDテストによる多重比較検定を行った。有意差水準は、p<0.05とした。
【表6】
【表7】
【0111】
表6から、分化誘導前の筋衛星細胞では、細胞群c、dの細胞ともに、遺伝子非導入細胞と比べて、α−リノレン酸(18:3n−3)をはじめとするn−3系脂肪酸脂肪酸などの脂肪酸組成に変化は認められなかったことがわかる(p<0.05)。
【0112】
表7から、分化誘導後の細胞では、hFAD3が低発現の細胞群cの細胞では、α−リノレン酸(18:3n−3)が0.30%と、対照区の遺伝子非導入細胞0.03%に対し、有意に増加していたことがわかる(p<0.05)。FAD3の基質であるリノール酸(18:2n−6)は9.37%と、対照区の遺伝子非導入細胞12.4%に対し、有意に減少していた(p<0.05)。n−3系の脂肪酸のドコサペンタエン酸(DPA、22:5n−3)は、5.80%と、対照区の遺伝子非導入細胞4.27%に対し、1.5%増加していた(p<0.05)。n−3系の脂肪酸の総計では、11.5%と、対照区の遺伝子非導入細胞9.1%に対し、約2.4%増加していた(p<0.05)。
【0113】
分化誘導後の細胞では、hFAD3が強発現の細胞群dの細胞でも、α−リノレン酸(18:3n−3)が0.27%と、対照区の遺伝子非導入細胞0.03%に対し、有意に増加していた(p<0.05)。FAD3の基質であるリノール酸(18:2n−6)は、9.40%と、対照区の遺伝子非導入細胞12.4%に対し、3%と、大いに減少していた(p<0.05)。n−3系の脂肪酸のドコサペンタエン酸(DPA、22:5n−3)は、5.30%と、対照区の遺伝子非導入細胞4.27%に対し、約1%増加していた(p<0.05)。n−3系の脂肪酸のドコサヘキサエン酸(DHA、22:5n−3)は、6.40%と、対照区の遺伝子非導入細胞3.57%に対し、約2.8%と大いに増加していた(p<0.05)。n−3系の脂肪酸の総計では、12.5%と、対照区の遺伝子非導入細胞9.1%に対し、約2.4%増加していた(p<0.05)。n−6系の脂肪酸の総計では、19.23%と、対照区の遺伝子非導入細胞23.9%に対し、約4.7%も減少していた(p<0.05)。
【0114】
また、(n−6)系の脂肪酸/(n−3)系の脂肪酸の割合は、細胞群cの細胞では、2.10、細胞群dの細胞では、1.57と、対照区の非遺伝子導入細胞の2.63に比べ有意に減少していることがわかった(p<0.05)。
【0115】
以上から、分化誘導した本発明の遺伝子導入細胞では、(n−6)系の脂肪酸から(n−3)系の脂肪酸への変化が進行していることがわかった。
【0116】
[遺伝子導入細胞をドナー細胞に用いたクローン胚の作製]
(ドナー細胞の調整)
遺伝子を導入した細胞群c、dの細胞を集密になるまで培養した。その後、細胞の増殖を停止させて、G0/G1期に細胞周期を同調させた。培養後、細胞c、dの細胞を回収し、培地bFGF−FBS−DMEMで洗浄し、核移植に用いた。また、コントロールとして、遺伝子非導入筋衛星細胞を、同様に培養、回収して用いた。
【0117】
(ウシ卵子の体外熟成)
大阪市食肉衛生検査所において、ウシ屠体より採取されたウシの卵巣を用いた。卵巣から21Gの注射針(テルモ社製)と10mlのシリンジ(テルモ社製)を用いて、ウシ卵子を卵胞液とともに吸引し回収した。卵胞より回収した卵胞液から、実体顕微鏡下で2〜4重層の卵丘細胞が付着した卵丘細胞卵子複合体のみを選別して回収した。その後、洗浄用成熟培養ドロップに、20個ずつ移し、洗浄した。次に、成熟培養ドロップに10個ずつ移動させ、39℃、5%CO2、95%空気、飽和湿度の炭酸ガスインキュベーター内で21時間培養した。
【0118】
(ウシ卵子の裸化)
21時間成熟培養した卵丘細胞卵子複合体を、0.5% FBS(ウシ胎児血清)および2.5ml/mlゲンタマイシン溶液を添加した25mM ヘペス緩衝TCM−199(ギブコ社製)に、0.25% ヒアルロニダーゼを加えた培地(199H+5% NBCS+0.25% ヒアルロニダーゼ)に移し、5分間静置した。静置後、ピペッティングにより、卵丘細胞卵子複合体を完全に取り除いた。
【0119】
(除核)
倒立顕微鏡下において、第1極体を放出し、第二減数分裂中期(MetaphaseII期)に達し、成熟していると思われる正常な卵子を選別した。この卵子の第1極体付近の透明帯を透明帯カット用ニードルで切開した。透明帯を切開した卵子を除核用培地(199H+5% NBCS+5μg/ml サイトカラシンB)に入れ、15分間静置した。静置後、除核用スティックで卵子を上から押さえつけ、第一極体及びその周辺卵細胞質を切開部位から2〜3割ほど押し出した。この卵子を、(199(Ea)+5% NBCS)培地に移し、ピペッティングにより卵子と押し出した卵細胞質に切り離した。切り離した卵細胞質のみを12μg/ml ヘキストNo.33342添加199(Ea)に移し、39℃、5%CO2、95%空気、飽和湿度で、30分間培養し、染色した。倒立顕微鏡下でUV照射により、核の確認を行った。核が確認できた卵細胞質に対応した卵子のみをレシピエント卵子として用いた。
【0120】
(細胞の導入)
除核した卵子を199(H)+5% NBCS培地に、上記ドナー細胞をPVP(ポリビニルピロリドン K−90(ナカライ社製))に移した。細胞導入用ピペットにドナー細胞を5〜10個吸引した後、除核卵子の透明帯切開部位から囲卵腔内にドナー細胞を1個ずつ導入した。細胞導入したウシ卵子を、199(H)+5% NBCS培地に移した。
【0121】
(電気融合)
電気融合には、ECM200(BTX社製)に接続した微小電極(ユニークメディカ イマダ 製)を用いた。除核卵子を、199(H)+5% NBCS培地に移し、ミネラルオイルを洗い流した。この除核卵子を、新たに、199(H)+5% NBCS培地に移し、(199(H)+5% NBCS)培地+細胞融合液混合液(1:1)、細胞融合液の順に移していった。卵子が底に沈んだ後、細胞融合液の楕円形のドロップを作製し、細胞導入した卵子をそこに移した。微小電極でウシ卵子細胞質および導入電極が一直線になるように挟み、交流2.7kV/cm・10μ秒を2回印加し、電気パルスによって融合処理を行った。その後、電気融合処理卵を細胞融合液、細胞融合液+(199(H)+5% NBCS)培地混合液(1:1)、(199(H)+5% NBCS)培地の順に移動させた。最後に、電気融合処理卵を、(199(Ea)+5% NBCS)培地で洗浄し、(199(Ea)+5% NBCS)培地のドロップへ移し、39℃、5%CO2、5%O2、90%N2、飽和湿度で、30分間培養した。培養後、実体顕微鏡下でドナー細胞と卵子細胞質との融合を確認した。
【0122】
(活性化処理)
電気融合処理後、遮光下でカルシウムイオノフォア活性化処理を行った。融合確認ができた再構築胚を、5μM Ca2+イオノマイシン培地に移し、5分間静置した。静置後、シクロへキシミド処理液をCa2+イオノマイシン培地に加え、反応を停止させた。カルシウムイオノフォア処理後の再構築胚を、修正合成卵管液培地(modified Synthetic Ovidautal Fluid (mSOF)medium、合成卵管液培養液(synthetic oviduct fluid medium, SOFM, Tervit et al., 1972)に必須および非必須アミノ酸を添加して修正した培養液(Takahashi and First, 1992)からさらに無機リン酸塩を除いた培養液(modified SOF medium,mSOFM))+10μg/ml シクロへキシミド培地のドロップに移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で6時間培養し、活性化を誘起した。
【0123】
(体外培養)
カルシウムイオノフォア−シクロへキシミド複合活性化処理の6時間後に、再構築胚をmSOFm培地に移し、洗浄した。洗浄後、mSOF培地のドロップに、1ドロップに付き20〜40個の再構築胚を移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で120時間培養した。120時間培養後、再構築胚を、20% FBS199+ベータメルカプトエタノール(βME)培地に移し洗浄した。洗浄後、20% FBS199+βME培地のドロップに移し、39℃、5%CO2、5%O2、90%N2、飽和湿度のインキュベーター内で48時間培養した。48時間培養後、再構築胚の卵割率および胚盤胞期胚への発生率を検討した。また、胚盤胞期胚におけるEGFP蛍光の観察、胚盤胞期胚の回収を行った。表8に、再構築胚の卵割率および胚盤胞期胚への発生率を示す。コントロールとして、遺伝子非導入細胞を用いたクローン胚(表中、「nonTG細胞」と記載)を用いた。表中、「卵割胚数」は、2、4、8細胞期の卵数を、%は培養した胚数に対する卵割した胚数の割合を示す。また、「胚盤胞期胚数」の%は、培養した胚数に対する胚盤胞期胚の割合を示す。
【表8】
【0124】
表8から、細胞群dの細胞を導入したクローン胚(表中、「TG細胞」と記載)の胚盤胞への発生率は21%と、遺伝子非導入細胞を用いたクローン胚の35%と比較してやや低かったが、有意差は認められなかった(p>0.05)。
【0125】
また、図7に示すように、胚盤胞胚期へ発現した遺伝子導入クローン胚の全ての胚で蛍光を確認した。
【0126】
[遺伝子導入細胞クローン胚における導入遺伝子の発現の検討]
活性化処理後168時間で胚盤胞排気まで発生した胚10個を1サンプルとして回収した。このサンプルから、RNAqueous−Micro kit(Ambion社製)を用いて、RNA抽出を常法に従って行った。
【0127】
抽出したRNAを用いて、SperScript III First−Strand Synthesis System for RT−PCR kit(インビトロジェン社製)を使って、cDNAを合成した。
【0128】
得られたcDNAは、TaKaRa Ex Taq polymerase(TaKaRa Ex Taq、タカラバイオ(株)製)を用いて、PCR法により増幅した。PCR反応は、hFADのプライマーを用い、94℃で5分間反応させた後、94℃で1分間、55℃で1分30秒、72℃で1分30秒を35サイクルで行った。また、コントロールとしてハウスキーピング遺伝子GAPDHのプライマーを用いた。
【0129】
反応後、PCR産物を、1.2%寒天/TAEを用い、1×TAE中で25分間電気泳動を行った。泳動後、エチレンブロマイド(EtBr)で染色し、UVトランスイルミネーターでDNAの泳動パターンを観察した。結果を、図8に示す。図中、用いた胚の数は、非遺伝子導入細胞については、1:10個、2:10個、3:9個、遺伝子導入細胞では、1:10個、2:6個、3:10個であった。
【0130】
図8から、遺伝子導入株dを用いて作製したクローン胚では、hFAD3が発現していることがわかる。
【図面の簡単な説明】
【0131】
【図1】図1は、hFAD3の再構築遺伝子の作成方法を説明する図である。
【図2】図2は、構成したpCAG/hFAD3/IRES2/EGFP/SV40(neor)を示す図である。
【図3】図3は、選択培養した後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)と、継代培養後にEGFP蛍光が陽性の細胞群における蛍光が観察された細胞数の割合(%)とを示すグラフである。
【図4】図4は、非遺伝子導入細胞と、細胞群b、c、dの細胞における脂質の蓄積を調べた結果を示すグラフである。
【図5】図5は、pCAG/hFAD3/IRES2/EGFP/SV40(neor)を導入したウシ筋衛星細胞の分化誘導前と分化誘導後の透過光像および蛍光像である。
【図6】図6は、細胞群c、dの分化誘導前と分化誘導後の傾向強度の変化を示すグラフである。
【図7】図7は、遺伝子導入細胞株dを用いたクローン胚の胚盤胞期胚の透過光像と、EGFP蛍光像を示す図である。
【図8】図8は、胚盤胞に発生したクローン胚を用いた、RT−PCRによる、hFAD3遺伝子の発現を確認した図である。
【特許請求の範囲】
【請求項1】
以下の(A)〜(D)のいずれかの塩基配列からなるポリヌクレオチドである、脂肪酸不飽和化酵素遺伝子。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【請求項2】
脂肪酸不飽和化酵素遺伝子を含有する、組換え発現ベクター。
【請求項3】
前記脂肪酸不飽和化酵素遺伝子は、ω3脂肪酸不飽和化酵素遺伝子である、請求項2に記載の組換え発現ベクター。
【請求項4】
前記脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(C)のいずれかの塩基配列からなる遺伝子である、請求項2に記載の組換え発現ベクター。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【請求項5】
前記脂肪酸不飽和化酵素遺伝子は、ヒト化遺伝子である、請求項2ないし4のいずれかに記載の組換え発現ベクター。
【請求項6】
前記組換え発現ベクターは、レポーター遺伝子、IRES配列、および選択マーカー遺伝子を含む、請求項2〜5のいずれかに記載の組換え発現ベクター
【請求項7】
請求項2〜6のいずれかに記載の組換え発現ベクターを用いて、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞。
【請求項8】
脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物。
【請求項1】
以下の(A)〜(D)のいずれかの塩基配列からなるポリヌクレオチドである、脂肪酸不飽和化酵素遺伝子。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【請求項2】
脂肪酸不飽和化酵素遺伝子を含有する、組換え発現ベクター。
【請求項3】
前記脂肪酸不飽和化酵素遺伝子は、ω3脂肪酸不飽和化酵素遺伝子である、請求項2に記載の組換え発現ベクター。
【請求項4】
前記脂肪酸不飽和化酵素遺伝子は、以下の(A)〜(C)のいずれかの塩基配列からなる遺伝子である、請求項2に記載の組換え発現ベクター。
(A)配列番号1に示される塩基配列からなるポリヌクレオチド
(B)配列番号1に示される塩基配列において、1若しくは複数個の塩基が、欠失、置換、若しくは付加された塩基配列からなり、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(C)配列番号1に示される塩基配列とストリンジェントな条件下でハイブリダイズし、かつω3脂肪酸不飽和化酵素活性を有するタンパク質をコードするポリヌクレオチド
(D)3つのヒスチジンクラスターと小胞体膜移行シグナルを有し、発現させると真核細胞内でω3脂肪酸不飽和化酵素活性を示すポリヌクレオチド
【請求項5】
前記脂肪酸不飽和化酵素遺伝子は、ヒト化遺伝子である、請求項2ないし4のいずれかに記載の組換え発現ベクター。
【請求項6】
前記組換え発現ベクターは、レポーター遺伝子、IRES配列、および選択マーカー遺伝子を含む、請求項2〜5のいずれかに記載の組換え発現ベクター
【請求項7】
請求項2〜6のいずれかに記載の組換え発現ベクターを用いて、脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物細胞。
【請求項8】
脂肪酸不飽和化酵素遺伝子が導入された、非ヒト動物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2009−55876(P2009−55876A)
【公開日】平成21年3月19日(2009.3.19)
【国際特許分類】
【出願番号】特願2007−227818(P2007−227818)
【出願日】平成19年9月3日(2007.9.3)
【出願人】(000125347)学校法人近畿大学 (389)
【Fターム(参考)】
【公開日】平成21年3月19日(2009.3.19)
【国際特許分類】
【出願日】平成19年9月3日(2007.9.3)
【出願人】(000125347)学校法人近畿大学 (389)
【Fターム(参考)】
[ Back to top ]