
脂質修飾された免疫応答調整剤
脂質修飾された免疫応答調整剤化合物と、該化合物を含有する薬学的組成物と、動物のサイトカインの生合成を誘発または阻害するための、そしてウィルス性疾患および腫瘍性疾患を含む疾患の治療における、これらの化合物の免疫調節剤としての使用方法とが開示される。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本出願は、2003年8月14日出願の米国特許出願第10/640904号、ならびに2003年10月30日出願の米国仮特許出願第60/515604号、および2004年2月13日出願の同第60/544561号への優先権を主張し、これらはそれぞれ、参照によってその全体が本明細書に援用される。
【背景技術】
【0002】
1950年代に、1H−イミダゾ[4,5−c]キノリン環系が開発され、抗マラリア剤としての可能性のある用途のために1−(6−メトキシ−8−キノリニル)−2−メチル−1H−イミダゾ[4,5−c]キノリンが合成された。続いて、様々な置換1H−イミダゾ[4,5−c]キノリンの合成が報告された。例えば、可能性のある鎮痙薬および心臓血管薬として、1−[2−(4−ピペリジル)エチル]−1H−イミダゾ[4,5−c]キノリンが合成された。また、いくつかの2−オキソイミダゾ[4,5−c]キノリンが報告されている。
【0003】
特定の1H−イミダゾ[4,5−c]キノリン−4−アミン、ならびにその1−および2−置換誘導体は、抗ウィルス剤、気管支拡張薬および免疫調節剤として有用であることがその後発見された。続いて、特定の置換1H−イミダゾ[4,5−c]ピリジン−4−アミン、キノリン−4−アミン、テトラヒドロキノリン−4−アミン、ナフチリジン−4−アミン、およびテトラヒドロナフチリジン−4−アミン化合物、ならびに特定の類似のチアゾロおよびオキサゾロ化合物が合成され、免疫応答調整剤(IRM)として有用であることが分かり、これらの化合物は様々な疾患の治療において有用であるとされた。
【0004】
サイトカインの生合成の誘発またはその他のメカニズムによって免疫応答を調節することができる化合物への関心および必要性が引き続き存在している。
【発明の開示】
【課題を解決するための手段】
【0005】
サイトカインの生合成を調節するために有用な新しい種類の化合物が今発見された。1つの態様では、本発明は、R1基(R1は、以下で定義されるとおりである)に共有結合したIRM化合物およびその薬学的に許容可能な塩を提供する。1つの実施形態では、本発明は、式I:
【化1】
(式中、RA、RB、R1、およびR”は以下で定義されるとおりである)である前記化合物およびその薬学的に許容可能な塩を提供する。
【0006】
このような化合物の例には、以下の式II、III、IV、V、VI、およびVII:
【化2】
(式中、R、RA、RB、R1、R2、およびnは、以下で定義されるとおりである)を有するものおよびその薬学的に許容可能な塩が含まれる。
【0007】
式Iの化合物を含む、R1基に共有結合したIRM化合物は、動物に投与されると、サイトカインの生合成を誘発または阻害する(例えば、1つまたは複数のサイトカインの生合成または産生を誘発または阻害する)、そして他の方法で免疫応答を調節することができるため、免疫応答調整剤(IRM)として有用である。これにより該化合物は、このような免疫応答の変化に応答するウィルス性疾患、腫瘍性疾患、および自己免疫性疾患などの様々な状態の治療において有用であるとされる。
【0008】
もう1つの態様では、本発明は、免疫応答調整剤化合物を含有する薬学的組成物、ならびに有効量の式Iの1つまたは複数の化合物および/またはその薬学的に許容可能な塩を動物に投与することによって、動物のサイトカインの生合成を誘発または阻害する方法、動物のウィルス性疾患を治療する方法、および動物の腫瘍性疾患を治療する方法を提供する。
【0009】
本明細書における使用では、「a」、「an」、「the」、「少なくとも1つの」、および「1つまたは複数の」は、置き換え可能に使用される。
【0010】
「含む(comprising)」という用語およびその変化形は、説明および特許請求の範囲のこれらの用語が見られるところで、限定的な意味を持たない。
【0011】
本発明の上記の概要は、本発明の開示される各実施形態または全ての実行を説明することは意図されない。以下に続く説明は、実例となる実施形態をより詳細に例示する。また、本明細書中には実施例の一覧による案内も提供され、様々な組み合わせで使用することができる。それぞれの場合において、列挙される一覧は代表的なグループとしての役割を果たすだけであり、排他的な一覧として解釈されてはならない。
【発明を実施するための最良の形態】
【0012】
本発明は、IRM化合物がR1基(R1は、以下で定義されるとおりである)に共有結合した新しい種類の化合物およびその薬学的に許容可能な塩を提供する。より具体的には、本発明は、以下の式I〜式VII:
【化3】
(式中、R、RA、RB、R1、R2、R”、およびnは、以下で定義されるとおりである)を有する化合物およびその薬学的に許容可能な塩を提供する。
【0013】
1つの態様では、本発明は、R1基に共有結合したIRM化合物またはその薬学的に許容可能な塩を提供し、ここでR1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、式中、
アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断(好ましくは、1つの−O−基により中断)され、
Lは、結合または官能性連結基であり、
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むが、ただし、式I:
【化4】
の化合物では、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、式Iの化合物では、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合は、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。
【0014】
1つの実施形態では、本発明は、以下の式I:
【化5】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R”は、水素または非妨害性置換基であり、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)もしくは縮合飽和5〜7員環(任意に、1つのヘテロ原子を含有する)を形成し、ここで該ヘテロ原子はNおよびSからなる群から選択され、上記アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されていて、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0015】
1つの実施形態では、本発明は、以下の式II:
【化6】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意で、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)を形成し、ここでアリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されていて、ここでRは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0016】
もう1つの実施形態では、本発明は、以下の式II:
【化7】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
R3は、水素およびアルキルからなる群から選択される。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0017】
もう1つの実施形態では、本発明は、以下の式III:
【化8】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0〜4であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、nが0の場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0018】
その他の実施形態では、本発明は、以下の式IV、V、VIおよびVII:
【化9】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0または1であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−C(O)−であり、nが0の場合には、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0019】
本発明との関連では、「脂肪族」基という用語は、飽和または不飽和の線状または分枝状炭化水素基を意味する。この用語は、例えば、アルキル、アルケニル、およびアルキニル基を包含するために使用される。
【0020】
本明細書における使用では、用語「アルキル」、「アルケニル」、「アルキニル」および接頭語「alk−」は、直鎖基および分枝鎖基の両方ならびに環状基、すなわちシクロアルキルおよびシクロアルケニルを含めたものである。他に指定されない限りは、これらの基は1〜20個の炭素原子を含有し、アルケニル基は2〜20個の炭素原子を含有し、アルキニル基は2〜20個の炭素原子を含有する。いくつかの実施形態では、これらの基は、全部で10個までの炭素原子、8個までの炭素原子、6個までの炭素原子、または4個までの炭素原子を含有する。環状基は単環式でも多環式でもよく、好ましくは、3〜10個の環炭素原子を有する。典型的な環状基には、シクロプロピル、シクロプロピルメチル、シクロペンチル、シクロヘキシル、アダマンチル、ならびに置換および非置換ボルニル、ノルボルニルおよびノルボルネニルが含まれる。
【0021】
他に指定されない限りは、「アルキレン」、「アルケニレン」および「アルキニレン」は、上記で定義された「アルキル」、「アルケニル」および「アルキニル」基の2価の形である。同様に、「アルキレニル」、「アルケニレニル」および「アルキニレニル」は、上記で定義された「アルキル」、「アルケニル」および「アルキニル」基の2価の形である。例えば、アリールアルキレニル基は、アリール基が付いたアルキレン部分を含む。
【0022】
「ハロアルキル」という用語は、過フッ素化基を含む、1つまたは複数のハロゲン原子で置換された基を含めたものである。これは、接頭語「ハロ−」を含むその他の基にも当てはまる。適切なハロアルキル基の例は、クロロメチル、トリフルオロメチルなどである。
【0023】
「アリール」という用語は、本明細書における使用では、炭素環式芳香環または環系を含む。アリール基の例としては、フェニル、ナフチル、ビフェニル、フルオレニルおよびインデニルがあげられる。
【0024】
「ヘテロアリール」という用語は、少なくとも1つの環へテロ原子(例えば、O、S、N)を含有する芳香環または環系を含む。適切なヘテロアリール基としては、フリル、チエニル、ピリジル、キノリニル、イソキノリニル、インドリル、イソインドリル、トリアゾリル、ピロリル、テトラゾリル、イミダゾリル、ピラゾリル、オキサゾリル、チアゾリル、ベンゾフラニル、ベンゾチオフェニル、カルバゾリル、ベンゾオキサゾリル、ピリミジニル、ベンゾイミダゾリル、キノキサリニル、ベンゾチアゾリル、ナフチリジニル、イソオキサゾリル、イソチアゾリル、プリニル、キナゾリニル、ピラジニル、1−オキシドピリジルなどがある。
【0025】
「ヘテロシクリル」という用語は、少なくとも1つの環へテロ原子(例えば、O、S、N)を含有する非芳香族環または環系を含み、そして上記のヘテロアリール基の完全飽和および部分不飽和誘導体の全てを含む。典型的なヘテロ環状基としては、ピロリジニル、テトラヒドロフラニル、モルホリニル、チオモルホリニル、ピペリジニル、ピペラジニル、チアゾリジニル、イミダゾリジニル、イソチアゾリジニル、テトラヒドロピラニル、キヌクリジニル、ホモピペリジニルなどがある。
【0026】
「アリーレン」、「ヘテロアリーレン」、および「ヘテロシクリレン」という用語は、上記で定義した「アリール」、「ヘテロアリール」、および「ヘテロシクリル」基の2価の形である。同様に、「アリーレニル」、「ヘテロアリーレニル」、および「ヘテロシクリレニル」は、上記で定義した「アリール」、「ヘテロアリール」、および「ヘテロシクリル」基の2価の形である。例えば、アルキルアリーレニル基は、アルキル基が付いたアリーレン部分を含む。
【0027】
本明細書中に記載される任意の式中に基(または置換基または変数)が2つ以上存在する場合、明白に記載されていてもいなくても、各基(または置換基または変数)は独立して選択される。例えば、式−N(R3)2では、各R3基は独立して選択される。もう1つの例では、2つ以上のR基が存在し、各R基が1つまたは複数の−N(R3)2基を含有する場合には、各R基は独立して選択され、各R3基は独立して選択される。
【0028】
本発明は、本明細書中に記載される化合物およびその塩を、異性体(例えば、ジアステレオマーおよびエナンチオマー)、溶媒和物、多形体などを含むその薬学的に許容可能な任意の形で含めたものである。特に、化合物が光学活性であれば、本発明は、特に、化合物のエナンチオマーのそれぞれおよびエナンチオマーのラセミ混合物を含む。
【0029】
いくつかの実施形態では、式I〜VIIの化合物は、1つまたは複数のサイトカインの生合成を誘発する。
【0030】
本明細書中に提示されるどの化合物についても、その実施形態における以下の変数(例えば、R、R”、R1、R2、RA、RB、n、Lなど)のそれぞれは、当業者には理解できるように、その実施形態の他の変数のうちの1つまたは複数と組み合わせられ得る。得られる変数の組み合わせのそれぞれは、本発明の実施形態である。
【0031】
特定の実施形態では、R”は、水素または非妨害性置換基である。本明細書では、「非妨害性」は、化合物または塩が1つまたは複数のサイトカインの生合成を調節(例えば、誘発または阻害)する能力が、非妨害性置換基によって破壊されないことを意味する。実例となる非妨害性R”基は、R2について本明細書中に記載されるものを含む。R”およびR2の好ましい実施形態は、以下に記載される。
【0032】
本発明は、R1基に共有結合したIRM化合物を提供する。本明細書では、R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、式中、アルキレン、アルケニレン、およびアルキニレン基は1つまたは複数の−O−基により任意に中断され、Lは結合または官能性連結基であり、そしてR1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、IRM化合物はイミキモド(imiquimod)ではない。
【0033】
式I〜VIIのいくつかの実施形態では、R1内のアルキレン、アルケニレン、およびアルキニレン基は、線状または分枝状である。特定の実施形態では、R1内のアルキレン、アルケニレン、およびアルキニレン基は線状である。いくつかの実施形態では、アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により中断される。いくつかの実施形態では、アルキレン、アルケニレン、およびアルキニレン基は、1つの−O−基により中断される。
【0034】
本明細書では、R1は、Q−L−R1-1とも称され、式中Qは、アルキレン、アルケニレン、またはアルキニレンであり、1つまたは複数の−O−基によって任意に中断される。いくつかの実施形態では、Qはアルキレンであり、1つの酸素原子(すなわち、−O−基)によって任意に中断される。いくつかの実施形態では、R1は式アルキレン−L−R1-1(すなわち、Q−L−R1-1)を有し、そしてアルキレン(Q)は、1つの酸素原子によって任意に中断される。いくつかの実施形態では、R1は、式C1〜5アルキレン−L−R1-1を有し、そしてC1〜5アルキレンは、1つの−O−基によって任意に中断される。あるいは、記載されるQは、好ましくは、C1〜5アルキレンであり、1つの−O−基によって任意に中断される。好ましいQ基の例としては、−(CH2)2−、−(CH2)3−、−(CH2)4−、−(CH2)5−、および−(CH2)2−O−(CH2)2−があげられる。
【0035】
いくつかの実施形態では、Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基である。
【0036】
いくつかの実施形態では、Lは、結合または−NH−C(O)−、−NH−S(O)2−、および−NH−C(O)−N(R3)−からなる群から選択される官能性連結基である。
【0037】
いくつかの実施形態では、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合、R1-1は、16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、Lが−NH−S(O2)−であり、nが0である場合、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。
【0038】
いくつかの実施形態では、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、Lが−NH−C(O)−であり、nが0である場合、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。
【0039】
いくつかの実施形態では、R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、R1-1は、11〜20個の炭素原子(好ましくは、12〜20個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、R1-1は、11〜20個の炭素原子(好ましくは、12〜20個の炭素原子)を有する線状(すなわち、直鎖)アルキル基である。
【0040】
このようなR1-1置換基は、本発明の化合物に脂質のような特徴を提供するので望ましい。これは、これらの脂質部分が、適用部位におけるIRMの封鎖を助けることができるので有利である。すなわち、脂質部分は、IRMが投与部位から離れて急速に拡散するのを防止するために役立つことができる。この封鎖はIRMのアジュバント性(adjuvancy)の増強もたらすことができ、これは、所望の部位における抗原提示細胞の漸増(recruitment)および活性化の増強によって明らかにされ得る。さらに、この封鎖の結果、IRMの全身への分配が少なくなり、そしてより少ない量のIRMの使用が可能になる。
【0041】
いくつかの実施形態では、RAおよびRBはそれぞれ独立して、水素、ハロゲン、アルキル、アルケニル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。
【0042】
いくつかの実施形態では、RAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)もしくは縮合飽和5〜7員環(任意に、1つのヘテロ原子を含有する)を形成し、ここでヘテロ原子はNおよびSからなる群から選択され、アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されている。好ましくは、置換基は、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。
【0043】
いくつかの実施形態では、RAおよびRBは一緒になって、縮合アリール環、またはNおよびSからなる群から選択される1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで、アリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になってNおよびSからなる群から選択される1つのヘテロ原子を任意に含有する縮合飽和5〜7員環を形成し、1つまたは複数のR基で非置換または置換されている。
【0044】
いくつかの実施形態では、RAおよびRBは一緒になって、NおよびSからなる群から選択される1つのヘテロ原子を任意に含有する縮合飽和5〜7員環を形成し、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される1つまたは複数の置換基で非置換または置換されている。
【0045】
いくつかの実施形態では、RAおよびRBは縮合アリールまたはヘテロアリール環を形成する。
【0046】
いくつかの実施形態では、RAおよびRBは、縮合飽和5〜7員環を形成する。
【0047】
いくつかの実施形態では、RAおよびRBは、非置換である縮合ベンゼン環を形成する。
【0048】
いくつかの実施形態では、RAおよびRBは、非置換である縮合ピリジン環を形成する。
【0049】
いくつかの実施形態では、Rは、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。いくつかの実施形態では、R”およびR2は、水素、アルキル、アルケニル、アリール、ヘテロアリール、ヘテロシクリル、アルキレン−Y−アルキル、アルキレン−Y−アルケニル、アルキレン−Y−アリール、および置換アルキルまたはアルケニルからなる群から選択され、該置換アルキルまたはアルケニルは、−OH、ハロゲン、−N(R4)2、−C(O)−C1〜10アルキル、−C(O)−O−C1〜10アルキル、−N3、アリール、ヘテロアリール、ヘテロシクリル、−C(O)−アリール、および−C(O)−ヘテロアリールからなる群から選択される1つまたは複数の置換基によって置換される。好ましくは、このような実施形態では、Yは−O−または−S(O)0〜2−であり、各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
【0050】
いくつかの実施形態では、R”およびR2は、水素、アルキル、およびアルキレン−O−アルキルからなる群から選択される。
【0051】
いくつかの実施形態では、各R3は独立して、水素およびアルキルからなる群から選択される。
【0052】
いくつかの実施形態では、各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
【0053】
いくつかの実施形態では、Yは、−O−または−S(O)0〜2−である。
【0054】
いくつかの実施形態では、nは0〜4である。いくつかの実施形態では、nは0または1である。いくつかの実施形態では、nは0である。
【0055】
化合物の調製
本発明の化合物は、イミダゾキノリン、テトラヒドロイミダゾキノリン、イミダゾピリジン、イミダゾナフチリジン、およびテトラヒドロイミダゾナフチリジンの調製において有用であることが分かっている合成法を用いて調製することができる。
【0056】
例えば、Lが−NH−C(O)−である本発明の化合物は、米国特許第6,451,810号明細書、同第6,545,016号明細書、同第6,194,425号明細書、同第6,660,747号明細書、および同第6,664,265号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、ステアリン酸、パルミチン酸、およびリノール酸などの通常の脂肪酸から調製することができる。
【0057】
Lが−NH−S(O)2−である本発明の化合物は、米国特許第6,331,539号明細書、同第6,525,064号明細書、同第6,194,425号明細書、同第6,677,347号明細書、同第6,677,349号明細書、および同第6,683,088号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、式R1-1S(O)2Clの塩化スルホニルから調製することができる。
【0058】
Lが−NH−C(O)−N(R3)−または−NH−C(S)−N(R3)−である本発明の化合物は、米国特許第6,541,485号明細書、同第6,573,273号明細書、同第6,656,938号明細書、同第6,660,735号明細書、および同第6,545,017号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、それぞれ式R1-1C=N=OおよびR1-1C=N=Sのイソシアネートまたはチオイソシアネート(thioisocyante)から調製することができる。
【0059】
Lが結合である本発明の化合物は、米国特許第4,689,338号明細書、同第4,929,624号明細書、同第5,268,376号明細書、同第5,389,640号明細書、同第5,352,784号明細書、および同第5,446,153号明細書に記載される合成法を用いて、式R1-1NH2のアミンから調製することができる。
【0060】
Lが−S−または−S(O)2−である本発明の化合物は、米国特許第6,664,264号明細書および同第6,667,312号明細書に記載される合成法を用いて、式R1-1SHのメルカプタンから調製することができる。
【0061】
薬学的組成物および生物活性
本発明の薬学的組成物は、治療的に有効な量の上記の本発明の化合物を、薬学的に許容可能なキャリアと組み合わせて含有する。
【0062】
「治療的に有効な量」または「有効量」という用語は、サイトカインの誘発、サイトカインの阻害、免疫調節、抗腫瘍活性、および/または抗ウィルス活性などの治療または予防効果を誘発するのに十分な化合物の量を意味する。本発明の薬学的組成物で使用される活性化合物の正確な量は、化合物の物理的および化学的性質、キャリアの性質、ならびに意図される投薬計画などの当業者には既知の因子に従って変わり得るが、本発明の組成物は十分な活性成分を含有して、約100ナノグラム/キログラム(ng/kg)〜約50ミリグラム/キログラム(mg/kg)の用量、好ましくは約10マイクログラム/キログラム(μg/kg)〜約5mg/kgの用量の化合物を被験体に提供することが予想される。錠剤、ロゼンジ、カプセル剤、非経口剤形、シロップ剤、クリーム、軟膏、煙霧剤形、経皮パッチ、経粘膜パッチなどの様々な剤形を使用することができる。
【0063】
本発明の化合物は、治療計画において単一の治療薬として投与することができる。あるいは、本発明の化合物は、互いに組み合わせて、または更なる免疫応答調整剤、抗ウィルス剤、抗生物質、抗体、タンパク質、ペプチド、オリゴヌクレオドなどを含む他の活性剤と組み合わせて投与されてもよい。
【0064】
本発明の化合物は、以下に記載される試験セットに従って実行される実験において特定のサイトカインの産生を誘発することが示され、本発明の特定の化合物は、特定のサイトカインの産生を阻害し得る。これらの結果は、化合物が、多数の異なる方法で免疫応答を調節することができる免疫応答調整剤として有用であることを示しており、これにより化合物は、様々な疾患の治療において有用とされる。
【0065】
本発明に従う化合物の投与によってその産生が誘発され得るサイトカインには、一般に、インターフェロン−α(IFN−α)および/または腫瘍壊死因子−α(TNF−α)ならびに特定のインターロイキン(IL)が含まれる。本発明の化合物によってその生合成が誘発され得るサイトカインには、IFN−α、TNF−α、IL−1、IL−6、IL−10およびIL−12、ならびに様々な他のサイトカインが含まれる。いくつかの効果の中でも特に、これらおよびその他のサイトカインは、ウィルスの産生および腫瘍細胞の成長を阻害し、ウィルス性疾患および腫瘍性疾患の治療において化合物を有用にすることができる。従って、本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法を提供する。サイトカインの生合成の誘発のために化合物または組成物が投与される動物は、以下に記載されるような疾患、例えば、ウィルス性疾患または腫瘍性疾患を有し、化合物の投与は治療的な処置を提供することができる。あるいは、化合物の投与が予防的処置を提供するように、動物が疾患にかかる前に化合物が動物に投与されてもよい。
【0066】
サイトカインの産生を誘発できることに加えて、本発明の化合物は、先天性の免疫応答の他の様相にも影響を与え得る。例えば、ナチュラルキラー細胞の活性が刺激され、サイトカイン誘発による効果であり得る。また化合物はマクロファージも活性化することができ、これは次に、一酸化窒素の分泌および更なるサイトカインの産生を刺激する。さらに、化合物は、Bリンパ球の増殖および分化を引き起こすことができる。
【0067】
また本発明の化合物は、後天性の免疫応答にも影響を与える。例えば、化合物が投与されると、Tヘルパー型1(TH1)サイトカインIFN−γの産生が間接的に誘発され、Tヘルパー型2(TH2)サイトカインIL−4、IL−5およびIL−13の産生は阻害され得る。
【0068】
本発明に従う特定の化合物の投与によってその産生が阻害され得るその他のサイトカインには、腫瘍壊死因子−α(TNF−α)が含まれる。いくつかの効果の中でも特に、TNF−αの産生の阻害は、TNFが仲介される動物の疾患の予防的または治療的な処置を提供することができ、例えば、自己免疫性疾患の治療において化合物を有用にする。従って、本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物におけるTNF−αの生合成の阻害方法を提供する。TNF−αの生合成の阻害のために化合物または組成物が投与される動物は、以下に記載されるような疾患、例えば自己免疫性疾患を有し、化合物の投与は治療的な処置を提供することができる。あるいは、化合物の投与が予防的処置を提供するように、動物が疾患にかかる前に化合物が動物に投与されてもよい。
【0069】
疾患の予防的処置でも治療的処置でも、そして先天性または後天性のいずれの免疫をもたらすためでも、化合物または組成物は単独で投与されてもよいし、例えば、ワクチンアジュバントの場合のように1つまたは複数の活性成分と組み合わせて投与されてもよい。他の成分と共に投与される場合、化合物および他の成分は、別々に投与されてもよいし、溶液の場合などのように一緒であるが独立して投与されてもよいし、あるいは、(a)共有結合される、または(b)例えばコロイド懸濁液の場合のように非共有結合で連結される、などのように一緒にそして互いに関連して投与されてもよい。
【0070】
本明細書中で同定されるIRMを治療に使用することができる状態には、
(a)例えば、アデノウィルス、ヘルペスウィルス(例えば、HSV−I、HSV−II、CMV、またはVZV)、ポックスウィルス(例えば、痘瘡または痘疹などのオルトポックスウィルス、または伝染性軟属腫)、ピコルナウィルス(例えば、ライノウィルスまたはエンテロウィルス)、オルトミクソウィルス(例えば、インフルエンザウィルス)、パラミクソウィルス(例えば、パラインフルエンザウィルス、流行性耳下腺炎ウィルス、麻疹ウィルス、および呼吸器合胞体ウィルス(RSV)、コロナウィルス(例えば、SARS)、パポバウィルス(例えば、性器いぼ、尋常性いぼ、または足底いぼを引き起こすものなどのパピローマウィルス)、ヘパドナウィルス(例えば、B型肝炎ウィルス)、フラビウィルス(例えば、C型肝炎ウィルスまたはデング熱ウィルス)、もしくはレトロウィルス(例えば、HIVなどのレンチウィルス)による感染から生じる疾患などのウィルス性疾患と、
(b)例えば、例えばエシェリキア(Escherichia)属、エンテロバクター属(Enterobacter)、サルモネラ属(Salmonella)、ブドウ球菌属(Staphylococci)、赤痢菌属(Shigella)、リステリア属(Listeria)、アエロバクター属(Aerobacter)、ヘリコバクター属(Helicobacter)、クレブシエラ属(Klebsiella)、プロテウス属(Proteus)、シュードモナス属(Pseudomonas)、連鎖球菌属(Streptococcus)、クラミジア属(Chlamydia)、マイコプラスマ属(Mycoplasma)、肺炎球菌属(Pneumococcus)、ナイセリア属(Neisseria)、クロストリジウム属(Clostridium)、バシラス属(Bacillus)、コリネバクテリウム属(Corynebacterium)、ミコバクテリウム属(Mycobacterium)、カンピロバクター属(Campylobacter)、ビブリオ属(Vibrio)、セラチア属(Serratia)、プロビデンシア属(Providencia)、クロモバクテリウム属(Chromobacterium)、ブルセラ属(Brucella)、エルジニア属(Yersinia)、ヘモフィルス属(Haemophilus)、またはボルデテラ属(Bordetella)の細菌による感染から生じる疾患などの細菌性疾患と、
(c)クラミジアなどのその他の感染性の疾患、例えばカンジダ症、アスペルギルス症、ヒストプラスマ症(histoplasmonsis)、クリプトコッカス髄膜炎などの真菌性疾患、または、例えばマラリア、ニューモシスティスカリニ肺炎、リーシュマニア症、クリプトスポリジウム症、トキソプラスマ症、およびトリパノソーマ感染症などの寄生虫病と、
(d)上皮内新形成、子宮頚部形成異常、光線性角化症、基底細胞癌、扁平上皮癌、腎細胞白血病(aenal cell leukemia)、カポージ肉腫(Karposi’s sarcoma)、メラノーマ、胃細胞癌、例えば、骨髄性白血病、慢性リンパ性白血病、および多発性骨髄腫などの白血病、非ホジキンリンパ腫、皮膚T細胞性リンパ腫、B細胞リンパ腫、ヘアリーセル白血病、ならびにその他の癌などの腫瘍性疾患と、
(e)アトピー性皮膚炎または湿疹、好酸球増加症、喘息、アレルギー、アレルギー性鼻炎、全身性エリテマトーデス、本態性血小板血症(essential thrombocythaemia)、多発性硬化症、オーメン症候群(Ommen’s syndrome)、円板状ループス、円形脱毛症、ケロイド形成および他のタイプの瘢痕化の阻害、ならびに慢性の創傷を含む創傷の治癒の増強などの、TH2に仲介されるアトピー性および自己免疫性疾患と、
が含まれるが、これらに限定されない。
【0071】
本明細書中で同定されるIRMは、例えば、BCG、コレラ、疫病(plague)、腸チフス、A型肝炎、B型肝炎、およびC型肝炎、インフルエンザAおよびインフルエンザB、パラインフルエンザ、ポリオ、狂犬病、麻疹、流行性耳下腺炎、風疹、黄熱、破傷風、ジフテリア、ヘモフィルスインフルエンザb、結核、髄膜炎菌および肺炎球菌ワクチン、アデノウィルス、HIV、水痘、サイトメガロウィルス、デング熱、ネコ白血病、家畜の疫病、HSV−1およびHSV−2、豚コレラ、日本脳炎、呼吸器合胞体ウィルス、ロタウィルス、パピローマウィルス、黄熱、およびアルツハイマー病に関連して使用するために、例えば、生ウィルス性、細菌性、または寄生虫性の免疫原と、不活性ウィルス性、腫瘍由来、原生動物性、有機体由来、真菌性、または細菌性の免疫原、トキソイド、トキシンと、自己抗原と、多糖類と、タンパク質と、糖タンパク質と、ペプチドと、細胞ワクチンと、DNAワクチンと、組換えタンパク質と、その他の同様のものなどの、体液および/または細胞に仲介される免疫応答のいずれかを引き起こす物質と共に使用するためのワクチンアジュバントとしても有用であり得る。
【0072】
またIRMは、欠陥がある免疫機能を有する個体において、特に役に立つことができる。例えば、IRM化合物は、例えば、移植患者、癌患者およびHIV患者において細胞性免疫の抑制後に生じ得る日和見感染および腫瘍を治療するためにも使用することができる。
【0073】
従って、上記の疾患または疾患のタイプのうちの1つまたは複数、例えば、ウィルス性疾患または腫瘍性疾患は、治療的に有効な量の式I、II、III、IV、V、VI、VIIの化合物または塩もしくはこれらの組み合わせを動物に投与することによって、それを必要としている(疾患を有する)動物において治療され得る。また有効な量の式I、II、III、IV、V、VI、VIIの化合物または塩もしくはこれらの組み合わせをワクチンアジュバントとして動物に投与することによって、動物はワクチン接種され得る。
【0074】
サイトカインの生合成を誘発するのに有効な化合物の量は、単球、マクロファージ、樹状細胞およびB細胞などの1つまたは複数の細胞タイプに、例えば、IFN−α、TNF−α、IL−1、IL−6、IL−10およびIL−12などの1つまたは複数のサイトカインを、このようなサイトカインの背景レベルよりも増大された量で産生させるのに十分な量である。正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。また本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物のウィルス感染の治療方法および動物の腫瘍性疾患の治療方法も提供する。
【0075】
ウィルス感染を治療または阻害するのに有効な量は、ウィルス性病変、ウィルス量、ウィルス産生速度、および未処置のコントロール動物と比較した死亡率などのウィルス感染の発現のうちの1つまたは複数の減少を引き起こし得る量である。このような治療に有効である正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。腫瘍性の状態を治療するのに有効な化合物の量は、腫瘍の大きさまたは腫瘍病巣の数の減少を引き起こし得る量である。この場合も同様に、正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。
【0076】
特定の実施形態では、本明細書中に記載される有効量の化合物または塩を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法が提供される。もう1つの実施形態では、本明細書中に記載される治療的に有効な量の化合物または塩を動物に投与することを含む、動物におけるウィルス性疾患の治療方法が提供される。もう1つの実施形態では、本明細書中に記載される治療的に有効な量の化合物または塩を動物に投与することを含む、動物における腫瘍性疾患の治療方法が提供される。もう1つの実施形態では、本明細書中に記載される有効量の化合物または塩をワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。
【0077】
本発明の目的および利点は、以下の実施例によってさらに説明されるが、これらの実施例で列挙される特定の物質およびその量、ならびに他の条件および詳細は、本発明を不当に制限すると解釈されてはならない。
【実施例】
【0078】
実施例1
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドの調製
【化10】
パートA
180mLのテトラヒドロフラン(THF)中の2−(2−アミノエトキシ)エタノール(29.0g、0.276mol)の溶液をN2中で0℃に冷却し、140mLの2NのNaOH溶液で処理した。次に、急速に攪拌した溶液に、180mLのTHF中の二炭酸ジ−tert−ブチル(60.2g、0.276mol)溶液を、1時間にわたって1滴ずつ添加した。次に反応混合物を室温まで暖め、さらに18時間攪拌した。次にTHFを減圧下で除去し、残った水性スラリーを、150mLの1MのH2SO4溶液の添加によりpH3にした。次に、これを酢酸エチル(300mL、100mL)で抽出し、合わせた有機層をH2O(2×)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、tert−ブチル2−(2−ヒドロキシエトキシ)エチルカルバメートが無色のオイルとして与えられた(47.1g)。
【0079】
パートB
1Lの無水CH2Cl2中のtert−ブチル2−(2−ヒドロキシエトキシ)エチルカルバメート(47.1g、0.230mol)の急速に攪拌した溶液をN2中で0℃に冷却し、トリエチルアミン(48.0mL、0.345mol)で処理した。次に塩化メタンスルホニル(19.6mL、0.253mol)を30分間かけて1滴ずつ添加した。次に反応混合物を室温まで暖め、さらに22時間攪拌した。500mLの飽和NaHCO3溶液の添加により反応を停止させ、有機層を分離した。次に有機相をH2O(3×500mL)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、2−{2−[(tert−ブトキシカルボニル)アミノ]エトキシ}エチルメタンスルホネートが茶色のオイルとして与えられた(63.5g)。
【0080】
パートC
400mLのN,N−ジメチルホルムアミド(DMF)中の2−{2−[(tert−ブトキシカルボニル)アミノ]エトキシ}エチルメタンスルホネート(63.5g、0.224mol)の攪拌溶液をNaN3(16.1g、0.247mol)で処理し、反応混合物をN2中で90℃に加熱した。5時間後、溶液を室温まで冷却し、500mLの冷H2Oで処理した。次に反応混合物をEt2O(3×300mL)で抽出した。合わせた有機抽出物をH2O(4×100mL)および塩水(2×100mL)で洗浄した。有機部分をMgSO4で乾燥させ、濃縮して、52.0gのtert−ブチル2−(2−アジドエトキシ)エチルカルバメートが薄茶色のオイルとして与えられた。
【0081】
パートD
tert−ブチル2−(2−アジドエトキシ)エチルカルバメート(47.0g、0.204mol)のMeOH溶液を4gの10%Pd炭素(Pd on carbon)で処理し、H2(3Kg/cm2)中で24時間振とうした。次に溶液をセライト(CELITE)パッドによりろ過し、濃縮して、35.3gの粗tert−ブチル2−(2−アミノエトキシ)エチルカルバメートが無色の液体として与えられ、さらに精製することなくこれを使用した。
【0082】
パートE
500mLの無水CH2Cl2中の4−クロロ−3−ニトロキノリン(31.4g、0.151mol)の攪拌溶液を、N2中で、トリエチルアミン(43mL、0.308mol)およびtert−ブチル2−(2−アミノエトキシ)エチルカルバメート(0.151mol)により処理した。一晩攪拌した後、反応混合物をH2O(2×300mL)および塩水(300mL)で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、鮮やかな黄色の固体が与えられた。酢酸エチル/へキサンからの再結晶により、43.6gのtert−ブチル2−{2−[(3−ニトロキノリン−4−イル)アミノ]エトキシ}エチルカルバメートが鮮やかな黄色の結晶として与えられた。
【0083】
パートF
tert−ブチル2−{2−[(3−ニトロキノリン−4−イル)アミノ]エトキシ}エチルカルバメート(7.52g、20.0mmol)のトルエン溶液を1.5gの5%Pt炭素(Pt on carbon)で処理し、H2(3Kg/cm2)中で24時間振とうした。次に溶液をセライトパッドによりろ過し、濃縮して、6.92gの粗tert−ブチル2−{2−[(3−アミノキノリン−4−イル)アミノ]エトキシ}エチルカルバメートが黄色のシロップとして与えられた。
【0084】
パートG
250mLの無水CH2Cl2中のtert−ブチル2−{2−[(3−アミノキノリン−4−イル)アミノ]エトキシ}エチルカルバメート(10.2g、29.5mmol)の溶液を0℃に冷却し、トリエチルアミン(4.18mL、30.0mmol)で処理した。次に塩化メトキシプロピオニル(3.30mL、30.3mmol)を5分間にわたって1滴ずつ添加した。次に反応を室温まで暖め、攪拌を1時間継続した。次に反応混合物を減圧下で濃縮して、オレンジ色の固体が与えられた。これを250mLEtOH中に溶解させ、12.5mLのトリエチルアミンを添加した。混合物を加熱して還流させ、N2中で一晩攪拌した。次に反応を減圧下で濃縮乾燥し、300mLのEt2Oで処理した。次に混合物をろ過し、ろ液を減圧下で濃縮して、茶色の固体が与えられた。固体を200mLの熱いメタノールに溶解させ、活性炭で処理した。熱い溶液をろ過し、濃縮して、11.1gのtert−ブチル2−{2−[2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートが黄色のシロップとして与えられた。
【0085】
パートH
250mLのCHCl3中のtert−ブチル2−{2−[2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.22g、24.7mmol)の溶液を3−クロロ過安息香酸(3−chloroperbenozic acid)(77%、9.12g、40.8mmol)で処理した。30分間攪拌した後、反応混合物を1%のNa2CO3溶液(2×75mL)および塩水で洗浄した。次に有機層をNa2SO4で乾燥させ、濃縮して、10.6gのtert−ブチル2−{2−[2−(2−メトキシエチル)−5−オキシド−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートがオレンジ色のフォームとして与えられ、さらに精製することなくこれを用いた。
【0086】
パートI
100mLの1,2−ジクロロエタン中のtert−ブチル2−{2−[2−(2−メトキシエチル)−5−オキシド−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.6g、24.6mmol)の溶液を60℃に加熱し、10mLの濃NH4OH溶液で処理した。急速に攪拌した溶液に、10分の間にわたって固体の塩化p−トルエンスルホニル(7.05g、37.0mmol)を添加した。反応混合物をさらに1mLの濃NH4OH溶液で処理し、次に加圧容器中に密封し、加熱を2時間継続した。次に反応混合物を冷却し、100mLのCHCl3で処理した。次に反応混合物をH2O、1%のNa2CO3溶液(2×)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、10.6gのtert−ブチル2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートが茶色のフォームとして与えられた。
【0087】
パートJ
tert−ブチル2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.6g、24.6mmol)をエタノール中の75mLの2MのHClで処理し、混合物を攪拌しながら加熱して還流させた。1.5時間後、反応混合物を冷却し、ろ過して、粘着性の固体が与えられた。固体をエタノールおよびEt2Oで洗浄し、真空乾燥させて、塩酸塩が薄茶色の固体として与えられた。塩酸塩を50mLのH2O中に溶解させ、10%のNaOH溶液で処理することによって遊離塩基を作った。次に水性懸濁液を濃縮乾燥させ、残渣をCHCl3で処理した。得られた塩をろ過により除去し、ろ液を濃縮して、3.82gの1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミンが黄褐色の粉体として与えられた。
MS330(M+H)+、
1H NMR(300MHz、DMSO−d6)δ8.10(d、J=8.1Hz、1H)、7.66(d、J=8.2Hz、1H)、7.40(m、1H)、7.25(m、1H)、6.88(brs、2H)、4.78(t、J=5.4Hz、2H)、3.89(t、J=4.8Hz、2H)、3.84(t、J=6.9Hz、2H)、3.54(t、J=5.4Hz、2H)、3.31(s、3H)、3.23(t、J=6.6Hz、2H)、2.88(t、J=5.3Hz、2 H)。
【0088】
パートK
窒素雰囲気下で、ジクロロメタン(3.5mL)およびトリエチルアミン(150μL、1.07mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(140.5mg、0.428mmol)の懸濁液を0℃に冷却した。塩化パルミトイル(130μL、0.428mmol)をゆっくり添加した。反応混合物を0℃で2時間攪拌させ、この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(30mL)で希釈し、飽和重炭酸ナトリウム溶液(2×5mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に減圧下で濃縮した。得られた残渣をカラムクロマトグラフィ(12gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製して、183mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドが白色粉体として提供された。
C33H53N5O3の分析計算値:%C,69.80、%H,9.41、%N,12.33、実測値:%C,69.60、%H,9.28、%N,11.99。
【0089】
実施例2
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドの調製
【化11】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(468μL、3.56mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(442.6mg、1.344mmol)の混合物を0℃に冷却した。塩化ステアロイル(454μL、1.34mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させ、この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に、減圧下で濃縮した。残渣を高真空下で乾燥させ、834mgの粗生成物が提供された。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%メタノールで溶出)により精製し、596mgの生成物が提供された。この物質を酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA(80%クロロホルム/18%メタノール/2%水酸化アンモニウム)300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、およびクロロホルム中6%のCMA500mLで順次溶出)により精製して、23.8mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドが白色のワックス状固体(融点80〜83℃)として提供された。
C35H57N5O3の分析計算値:0.694%のH2O、%C,70.06、%H,9.65、%N,11.67、実測値:%C,70.60、%H,9.91、%N,11.46。
【0090】
実施例3
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドの調製
【化12】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(551μL、4.00mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(527.0mg、1.600mmol)の混合物を0℃に冷却した。塩化ラウロイル(370μL、1.60mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させた。この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に、減圧下で濃縮した。残渣を高真空下で乾燥させ、821mgの粗生成物が提供された。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製し、527mgの生成物が提供された。この物質を酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、クロロホルム中6%のCMA750mL、100%のCMA500mLで順次溶出)により精製し、22.4mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドが白色のワックス状固体(融点80〜83℃)として提供された。
C29H45N5O3の分析計算値:1.66%のH2O、%C,66.94、%H,8.90、%N,13.46、実測値:%C,66.94、%H,9.37、%N,13.28。
【0091】
実施例4
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドの調製
【化13】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(470μL、3.37mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(444.mg、1.349mmol)の混合物を0℃に冷却した。塩化ミリストイル(367μL、1.35mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させた。この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に減圧下で濃縮した。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製した後、酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、およびクロロホルム中6%のCMA600mLで順次溶出)により精製して、9.5mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドが白色のワックス状固体(融点85〜87℃)として提供された。
【0092】
その他の典型的な化合物
特定の他の典型的な化合物は、式(VIII〜X)および以下の置換基を有し、表の各行は、第1列の見出し語に示されるように、式VIII、XIおよび/またはXの特定の化合物を表す。
【化14】
【0093】
【表1】
【表2】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【0094】
実施例5
免疫化
200μlのリン酸緩衝食塩水(PBS)中の複合体(conjugate)(1mgのオボアルブミンおよび200μgのIRM)を用いて、皮下または腹腔内のいずれかで、C57BL/6マウスを免疫化した。コントロールマウスは、200μlのPBS中の1mgのオボアルブミンで免疫化した。一次応答の分析のために、免疫化の5〜7日後にマウスを犠牲にした。二次応答の分析のために、マウスを最初の免疫化の7〜15日後に、追加免疫し、5〜7日後に犠牲にした。他に示されない限りは、皮下で免疫化したマウスから分析のためにリンパ節を収集し、腹腔内で免疫化したマウスから分析のために脾臓細胞を収集した。
【0095】
DMSO中に10mg/mlの濃度で溶解させることによって、N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドのストックIRM溶液を調製した。オボアルブミンをPBS中に50mg/mlの濃度で溶解させた。50μlのストックIRM溶液を150μlのPBSに添加し、次にボルテックスすることにより混合した。50μlのオボアルブミンをストックIRM溶液に添加し、ボルテックスすることにより混合した。IRMおよびオボアルブミンの濁ったコロイド懸濁液が生じた。
【0096】
上記のように(a)オボアルブミン単独、または(b)50μlのオボアルブミンおよびIRMのコロイド懸濁液のいずれかを用いて、0日目にマウスを皮下で免疫化した。6日目に、排出(draining)リンパ節を除去し、均質化し、H−2Kb/SIINFEKLテトラマーで染色して、オボアルブミン特異的T細胞を同定した。図1は、オボアルブミン単独で免疫化したコントロールマウスからのフローサイトメトリーデータを示し、図2および3は、コロイド懸濁液で免疫化した2つの異なるマウスからのデータを示す。
【0097】
試薬
オボアルブミンは、シグマ・ケミカル・カンパニー(Sigma Chemical Company)(ミズーリ州セントルイス(St.Louis,MO))から入手した。優性のオボアルブミンペプチドSIINFEKLに結合したMHCクラスI分子H−2Kbのテトラマーは、ケドル(Kedl)らのJ Exp Med,192:1105−13(2000)に記載されるように生成した。
【0098】
以下に記載される方法を用いて試験した場合に、本発明の化合物はサイトカインの生合成を誘発し、そして特定の化合物はサイトカインの生合成を阻害し得ることがわかった。実施例1〜4の化合物は、以下に記載される「ヒト細胞におけるサイトカインの誘発」アッセイを用いて試験した場合に、インターフェロンおよび腫瘍壊死因子の両方を誘発した。
【0099】
ヒト細胞におけるサイトカインの誘発
インビトロのヒト血液細胞系を用いて、サイトカインの誘発を評価する。テスターマン(Testerman)らにより「免疫調節剤のイミキモドおよびS−27609によるサイトカインの誘発」、Journal of Leukocyte Biology,58,365−372(1995年9月)に記載されるように、活性は、培地に分泌されるインターフェロン−αおよび腫瘍壊死因子−α(それぞれ、IFN−αおよびTNF−α)の測定に基づく。
【0100】
培養のための血液細胞の調製
健康なヒトのドナーからの全血を、静脈穿刺によってEDTAバキュテイナー(vacutainer)管内に採取する。HISTOPAQUE−1077を用いる密度勾配遠心分離法によって、末梢血単核細胞(PBMC)を全血から分離する。ダルベッコのリン酸緩衝食塩水(DPBS)またはハンクス平衡塩溶液(HBSS)により血液を1:1で希釈する。PBMC層を採取し、DPBSまたはHBSSで2回洗浄し、RPMI完全培地中に4×106個の細胞/mLで再懸濁させる。PBMC懸濁液を、試験化合物を含有する同体積のRPMI完全培地を含有する48ウェルの平底無菌組織培養プレート(マサチューセッツ州ケンブリッジのコスター(Costar,Cambridge,MA)またはニュージャージー州リンカーンパークのベクトン・ディッキンソン・ラブウェア(Becton Dickinson Labware,Lincoln Park,NJ))に添加する。
【0101】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、一般に、30〜0.014マイクロモル(μM)の範囲の濃度で試験される。
【0102】
インキュベーション
試験化合物の溶液を、RPMI完全培地を含有する第1のウェルに60μMで添加し、ウェルにおいて連続的な3倍希釈を行なう。次にPBMC懸濁液を同体積でウェルに添加して、試験化合物濃度を所望の範囲(30〜0.014μM)にする。PBMC懸濁液の最終濃度は2×106個の細胞/mLである。プレートを無菌プラスチック蓋で覆い、静かに混ぜてから、5%の二酸化炭素雰囲気中、37℃で18〜24時間インキュベーションを行なう。
【0103】
分離
インキュベーションに続いて、1000rpm(約200×g)において、4℃で10分間プレートを遠心分離する。無菌ポリプロピレンピペットで無細胞の培養の上澄みを取り出し、無菌ポリプロピレン管に移す。分析するまでサンプルを−30〜−70℃に保持する。ELISAによりIFN−αについて、そしてELISAまたはIGENアッセイによりTNF−αについて、サンプルを分析する。
【0104】
ELISAによるIFN−αおよびTNF−αの分析
ニュージャージー州ニューブランズウィックのPBLバイオメディカル・ラボラトリーズ(PBL Biomedical Laboratories,New Brunswick,NJ)からのヒト・マルチスピーシーズ・キットを用いて、ELISAによりIFN−αの濃度を決定する。結果は、pg/mLで表される。
【0105】
カリフォルニア州カマリロのバイオソース・インターナショナル(Biosource International,Camarillo,CA)から入手可能なELISAキットを用いて、TNF−αの濃度を決定する。あるいは、TNF−αの濃度は、オリジン(ORIGEN)M−シリーズイムノアッセイによって決定し、メリーランド州ゲーサーズバーグのアイジェン・インターナショナル(IGEN International,Gaithersburg、MD)からのアイジェン(IGEN)M−8アナライザーにおいて読み取ることができる。イムノアッセイは、カリフォルニア州カマリロのバイオソース・インターナショナルからのヒトのTNF−α捕獲および検出抗体ペアを用いる。結果は、pg/mLで表される。
【0106】
マウス細胞におけるサイトカインの阻害
マウスのマクロファージ細胞株Raw264.7を用いて、リポ多糖(LPS)により刺激されたときに化合物が腫瘍壊死因子−α(TNF−α)の産生を阻害する能力を評価する。
【0107】
単一濃度アッセイ
培養のための血液細胞の調製
生細胞(ATCC)を優しくこすって収集し、カウントする。細胞懸濁液を、10%ウシ胎児血清(FBS)を有するRPMI中で3×105個の細胞/mLにする。細胞懸濁液(100μL)を96ウェルの平底無菌組織培養プレート(ニュージャージー州リンカーンパークのベクトン・ディッキンソン・ラブウェア)に添加する。細胞の最終濃度は、3×104個の細胞/ウェルである。プレートを3時間インキュベートする。試験化合物を添加する前に、培地を、3%FBSを有する無色のRPMI培地に取り替える。
【0108】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、5μMで試験する。LPS(ネズミチフス菌(Salmonella typhimurium)からのリポ多糖、シグマ−アルドリッチ(Sigma−Aldrich))を、用量反応アッセイで測定されるようにEC70濃度に無色のRPMIで希釈する。
【0109】
インキュベーション
試験化合物の溶液(1μl)を各ウェルに添加する。マイクロタイタープレート振とう器においてプレートを1分間混ぜ、次にインキュベータ内に置く。20分後に、LPSの溶液(1μL、EC70濃度約10ng/ml)を添加し、プレートを振とう器で1分間混ぜる。5%の二酸化炭素雰囲気中、37℃でプレートを18〜24時間インキュベートする。
【0110】
TNF−αの分析
インキュベーションに続いて、上澄みをピペットで取り出す。マウスTNF−αキット(カリフォルニア州カマリロのバイオソース・インターナショナルから)を用いて、ELISAによりTNF−α濃度を決定する。結果は、pg/mLで表される。LPS刺激のみにおけるTNF−αの発現は、100%の応答であると考えられる。
【0111】
用量反応アッセイ
培養のための血液細胞の調製
生細胞(ATCC)を優しくこすって収集し、カウントする。細胞懸濁液を、10%FBSを有するRPMI中で4×105個の細胞/mLにする。細胞懸濁液(250μL)を48ウェルの平底無菌組織培養プレート(マサチューセッツ州ケンブリッジのコスター)に添加する。細胞の最終濃度は、1×105個の細胞/ウェルである。プレートを3時間インキュベートする。試験化合物を添加する前に、培地を、3%FBSを有する無色のRPMI培地に取り替える。
【0112】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、0.03、0.1、0.3、1、3、5および10μMで試験した。LPS(ネズミチフス菌からのリポ多糖、シグマ−アルドリッチ)を、用量反応アッセイで測定されるようにEC70濃度に無色のRPMIで希釈する。
【0113】
インキュベーション
試験化合物の溶液(200μl)を各ウェルに添加する。マイクロタイタープレート振とう器においてプレートを1分間混ぜ、次にインキュベータ内に置く。20分後に、LPSの溶液(200μL、EC70濃度約10ng/ml)を添加し、プレートを振とう器で1分間混ぜる。5%の二酸化炭素雰囲気中、37℃でプレートを18〜24時間インキュベートする。
【0114】
TNF−αの分析
インキュベーションに続いて、上澄みをピペットで取り出す。マウスTNF−αキット(カリフォルニア州カマリロのバイオソース・インターナショナルから)を用いて、ELISAによりTNF−α濃度を決定する。結果は、pg/mLで表される。LPS刺激のみにおけるTNF−αの発現は、100%の応答であると考えられる。
【0115】
本明細書中で引用される特許、特許文献、および刊行物の全ての開示は、あたかもそれぞれが個々に援用されたかのように、参照によってその全体が援用される。本発明は、そのいくつかの実施形態を参照して説明された。上記の実例となる実施形態および実施例は、理解を明確にするだけのために提供されたものであり、そこから不要な限定が理解されてはならない。本発明の精神および範囲から逸脱することなく、記載された実施形態に多くの変化が成され得ることは、当業者には明らかであろう。したがって、本発明の範囲は、特許請求の範囲によってのみ限定されることが意図される。
【図面の簡単な説明】
【0116】
【図1】実施例5に記載されるように、オボアルブミンによる免疫化後の抗原特異的CD8+T細胞の拡大を示す。
【図2】実施例5に記載されるように、IRMおよびオボアルブミンのコロイド懸濁液による免疫化後の1つの被験体の抗原特異的CD8+T細胞の拡大を示す。
【図3】実施例5に記載されるように、IRMおよびオボアルブミンのコロイド懸濁液による免疫化後の第2の被験体の抗原特異的CD8+T細胞の拡大を示す。
【技術分野】
【0001】
関連出願の相互参照
本出願は、2003年8月14日出願の米国特許出願第10/640904号、ならびに2003年10月30日出願の米国仮特許出願第60/515604号、および2004年2月13日出願の同第60/544561号への優先権を主張し、これらはそれぞれ、参照によってその全体が本明細書に援用される。
【背景技術】
【0002】
1950年代に、1H−イミダゾ[4,5−c]キノリン環系が開発され、抗マラリア剤としての可能性のある用途のために1−(6−メトキシ−8−キノリニル)−2−メチル−1H−イミダゾ[4,5−c]キノリンが合成された。続いて、様々な置換1H−イミダゾ[4,5−c]キノリンの合成が報告された。例えば、可能性のある鎮痙薬および心臓血管薬として、1−[2−(4−ピペリジル)エチル]−1H−イミダゾ[4,5−c]キノリンが合成された。また、いくつかの2−オキソイミダゾ[4,5−c]キノリンが報告されている。
【0003】
特定の1H−イミダゾ[4,5−c]キノリン−4−アミン、ならびにその1−および2−置換誘導体は、抗ウィルス剤、気管支拡張薬および免疫調節剤として有用であることがその後発見された。続いて、特定の置換1H−イミダゾ[4,5−c]ピリジン−4−アミン、キノリン−4−アミン、テトラヒドロキノリン−4−アミン、ナフチリジン−4−アミン、およびテトラヒドロナフチリジン−4−アミン化合物、ならびに特定の類似のチアゾロおよびオキサゾロ化合物が合成され、免疫応答調整剤(IRM)として有用であることが分かり、これらの化合物は様々な疾患の治療において有用であるとされた。
【0004】
サイトカインの生合成の誘発またはその他のメカニズムによって免疫応答を調節することができる化合物への関心および必要性が引き続き存在している。
【発明の開示】
【課題を解決するための手段】
【0005】
サイトカインの生合成を調節するために有用な新しい種類の化合物が今発見された。1つの態様では、本発明は、R1基(R1は、以下で定義されるとおりである)に共有結合したIRM化合物およびその薬学的に許容可能な塩を提供する。1つの実施形態では、本発明は、式I:
【化1】
(式中、RA、RB、R1、およびR”は以下で定義されるとおりである)である前記化合物およびその薬学的に許容可能な塩を提供する。
【0006】
このような化合物の例には、以下の式II、III、IV、V、VI、およびVII:
【化2】
(式中、R、RA、RB、R1、R2、およびnは、以下で定義されるとおりである)を有するものおよびその薬学的に許容可能な塩が含まれる。
【0007】
式Iの化合物を含む、R1基に共有結合したIRM化合物は、動物に投与されると、サイトカインの生合成を誘発または阻害する(例えば、1つまたは複数のサイトカインの生合成または産生を誘発または阻害する)、そして他の方法で免疫応答を調節することができるため、免疫応答調整剤(IRM)として有用である。これにより該化合物は、このような免疫応答の変化に応答するウィルス性疾患、腫瘍性疾患、および自己免疫性疾患などの様々な状態の治療において有用であるとされる。
【0008】
もう1つの態様では、本発明は、免疫応答調整剤化合物を含有する薬学的組成物、ならびに有効量の式Iの1つまたは複数の化合物および/またはその薬学的に許容可能な塩を動物に投与することによって、動物のサイトカインの生合成を誘発または阻害する方法、動物のウィルス性疾患を治療する方法、および動物の腫瘍性疾患を治療する方法を提供する。
【0009】
本明細書における使用では、「a」、「an」、「the」、「少なくとも1つの」、および「1つまたは複数の」は、置き換え可能に使用される。
【0010】
「含む(comprising)」という用語およびその変化形は、説明および特許請求の範囲のこれらの用語が見られるところで、限定的な意味を持たない。
【0011】
本発明の上記の概要は、本発明の開示される各実施形態または全ての実行を説明することは意図されない。以下に続く説明は、実例となる実施形態をより詳細に例示する。また、本明細書中には実施例の一覧による案内も提供され、様々な組み合わせで使用することができる。それぞれの場合において、列挙される一覧は代表的なグループとしての役割を果たすだけであり、排他的な一覧として解釈されてはならない。
【発明を実施するための最良の形態】
【0012】
本発明は、IRM化合物がR1基(R1は、以下で定義されるとおりである)に共有結合した新しい種類の化合物およびその薬学的に許容可能な塩を提供する。より具体的には、本発明は、以下の式I〜式VII:
【化3】
(式中、R、RA、RB、R1、R2、R”、およびnは、以下で定義されるとおりである)を有する化合物およびその薬学的に許容可能な塩を提供する。
【0013】
1つの態様では、本発明は、R1基に共有結合したIRM化合物またはその薬学的に許容可能な塩を提供し、ここでR1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、式中、
アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断(好ましくは、1つの−O−基により中断)され、
Lは、結合または官能性連結基であり、
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むが、ただし、式I:
【化4】
の化合物では、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、式Iの化合物では、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合は、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。
【0014】
1つの実施形態では、本発明は、以下の式I:
【化5】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R”は、水素または非妨害性置換基であり、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)もしくは縮合飽和5〜7員環(任意に、1つのヘテロ原子を含有する)を形成し、ここで該ヘテロ原子はNおよびSからなる群から選択され、上記アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されていて、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0015】
1つの実施形態では、本発明は、以下の式II:
【化6】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意で、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)を形成し、ここでアリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されていて、ここでRは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0016】
もう1つの実施形態では、本発明は、以下の式II:
【化7】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
R3は、水素およびアルキルからなる群から選択される。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0017】
もう1つの実施形態では、本発明は、以下の式III:
【化8】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0〜4であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、nが0の場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0018】
その他の実施形態では、本発明は、以下の式IV、V、VIおよびVII:
【化9】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基(好ましくは、1つの−O−基)により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0または1であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−C(O)−であり、nが0の場合には、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする。)
の化合物またはその薬学的に許容可能な塩を提供する。
【0019】
本発明との関連では、「脂肪族」基という用語は、飽和または不飽和の線状または分枝状炭化水素基を意味する。この用語は、例えば、アルキル、アルケニル、およびアルキニル基を包含するために使用される。
【0020】
本明細書における使用では、用語「アルキル」、「アルケニル」、「アルキニル」および接頭語「alk−」は、直鎖基および分枝鎖基の両方ならびに環状基、すなわちシクロアルキルおよびシクロアルケニルを含めたものである。他に指定されない限りは、これらの基は1〜20個の炭素原子を含有し、アルケニル基は2〜20個の炭素原子を含有し、アルキニル基は2〜20個の炭素原子を含有する。いくつかの実施形態では、これらの基は、全部で10個までの炭素原子、8個までの炭素原子、6個までの炭素原子、または4個までの炭素原子を含有する。環状基は単環式でも多環式でもよく、好ましくは、3〜10個の環炭素原子を有する。典型的な環状基には、シクロプロピル、シクロプロピルメチル、シクロペンチル、シクロヘキシル、アダマンチル、ならびに置換および非置換ボルニル、ノルボルニルおよびノルボルネニルが含まれる。
【0021】
他に指定されない限りは、「アルキレン」、「アルケニレン」および「アルキニレン」は、上記で定義された「アルキル」、「アルケニル」および「アルキニル」基の2価の形である。同様に、「アルキレニル」、「アルケニレニル」および「アルキニレニル」は、上記で定義された「アルキル」、「アルケニル」および「アルキニル」基の2価の形である。例えば、アリールアルキレニル基は、アリール基が付いたアルキレン部分を含む。
【0022】
「ハロアルキル」という用語は、過フッ素化基を含む、1つまたは複数のハロゲン原子で置換された基を含めたものである。これは、接頭語「ハロ−」を含むその他の基にも当てはまる。適切なハロアルキル基の例は、クロロメチル、トリフルオロメチルなどである。
【0023】
「アリール」という用語は、本明細書における使用では、炭素環式芳香環または環系を含む。アリール基の例としては、フェニル、ナフチル、ビフェニル、フルオレニルおよびインデニルがあげられる。
【0024】
「ヘテロアリール」という用語は、少なくとも1つの環へテロ原子(例えば、O、S、N)を含有する芳香環または環系を含む。適切なヘテロアリール基としては、フリル、チエニル、ピリジル、キノリニル、イソキノリニル、インドリル、イソインドリル、トリアゾリル、ピロリル、テトラゾリル、イミダゾリル、ピラゾリル、オキサゾリル、チアゾリル、ベンゾフラニル、ベンゾチオフェニル、カルバゾリル、ベンゾオキサゾリル、ピリミジニル、ベンゾイミダゾリル、キノキサリニル、ベンゾチアゾリル、ナフチリジニル、イソオキサゾリル、イソチアゾリル、プリニル、キナゾリニル、ピラジニル、1−オキシドピリジルなどがある。
【0025】
「ヘテロシクリル」という用語は、少なくとも1つの環へテロ原子(例えば、O、S、N)を含有する非芳香族環または環系を含み、そして上記のヘテロアリール基の完全飽和および部分不飽和誘導体の全てを含む。典型的なヘテロ環状基としては、ピロリジニル、テトラヒドロフラニル、モルホリニル、チオモルホリニル、ピペリジニル、ピペラジニル、チアゾリジニル、イミダゾリジニル、イソチアゾリジニル、テトラヒドロピラニル、キヌクリジニル、ホモピペリジニルなどがある。
【0026】
「アリーレン」、「ヘテロアリーレン」、および「ヘテロシクリレン」という用語は、上記で定義した「アリール」、「ヘテロアリール」、および「ヘテロシクリル」基の2価の形である。同様に、「アリーレニル」、「ヘテロアリーレニル」、および「ヘテロシクリレニル」は、上記で定義した「アリール」、「ヘテロアリール」、および「ヘテロシクリル」基の2価の形である。例えば、アルキルアリーレニル基は、アルキル基が付いたアリーレン部分を含む。
【0027】
本明細書中に記載される任意の式中に基(または置換基または変数)が2つ以上存在する場合、明白に記載されていてもいなくても、各基(または置換基または変数)は独立して選択される。例えば、式−N(R3)2では、各R3基は独立して選択される。もう1つの例では、2つ以上のR基が存在し、各R基が1つまたは複数の−N(R3)2基を含有する場合には、各R基は独立して選択され、各R3基は独立して選択される。
【0028】
本発明は、本明細書中に記載される化合物およびその塩を、異性体(例えば、ジアステレオマーおよびエナンチオマー)、溶媒和物、多形体などを含むその薬学的に許容可能な任意の形で含めたものである。特に、化合物が光学活性であれば、本発明は、特に、化合物のエナンチオマーのそれぞれおよびエナンチオマーのラセミ混合物を含む。
【0029】
いくつかの実施形態では、式I〜VIIの化合物は、1つまたは複数のサイトカインの生合成を誘発する。
【0030】
本明細書中に提示されるどの化合物についても、その実施形態における以下の変数(例えば、R、R”、R1、R2、RA、RB、n、Lなど)のそれぞれは、当業者には理解できるように、その実施形態の他の変数のうちの1つまたは複数と組み合わせられ得る。得られる変数の組み合わせのそれぞれは、本発明の実施形態である。
【0031】
特定の実施形態では、R”は、水素または非妨害性置換基である。本明細書では、「非妨害性」は、化合物または塩が1つまたは複数のサイトカインの生合成を調節(例えば、誘発または阻害)する能力が、非妨害性置換基によって破壊されないことを意味する。実例となる非妨害性R”基は、R2について本明細書中に記載されるものを含む。R”およびR2の好ましい実施形態は、以下に記載される。
【0032】
本発明は、R1基に共有結合したIRM化合物を提供する。本明細書では、R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、式中、アルキレン、アルケニレン、およびアルキニレン基は1つまたは複数の−O−基により任意に中断され、Lは結合または官能性連結基であり、そしてR1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、IRM化合物はイミキモド(imiquimod)ではない。
【0033】
式I〜VIIのいくつかの実施形態では、R1内のアルキレン、アルケニレン、およびアルキニレン基は、線状または分枝状である。特定の実施形態では、R1内のアルキレン、アルケニレン、およびアルキニレン基は線状である。いくつかの実施形態では、アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により中断される。いくつかの実施形態では、アルキレン、アルケニレン、およびアルキニレン基は、1つの−O−基により中断される。
【0034】
本明細書では、R1は、Q−L−R1-1とも称され、式中Qは、アルキレン、アルケニレン、またはアルキニレンであり、1つまたは複数の−O−基によって任意に中断される。いくつかの実施形態では、Qはアルキレンであり、1つの酸素原子(すなわち、−O−基)によって任意に中断される。いくつかの実施形態では、R1は式アルキレン−L−R1-1(すなわち、Q−L−R1-1)を有し、そしてアルキレン(Q)は、1つの酸素原子によって任意に中断される。いくつかの実施形態では、R1は、式C1〜5アルキレン−L−R1-1を有し、そしてC1〜5アルキレンは、1つの−O−基によって任意に中断される。あるいは、記載されるQは、好ましくは、C1〜5アルキレンであり、1つの−O−基によって任意に中断される。好ましいQ基の例としては、−(CH2)2−、−(CH2)3−、−(CH2)4−、−(CH2)5−、および−(CH2)2−O−(CH2)2−があげられる。
【0035】
いくつかの実施形態では、Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基である。
【0036】
いくつかの実施形態では、Lは、結合または−NH−C(O)−、−NH−S(O)2−、および−NH−C(O)−N(R3)−からなる群から選択される官能性連結基である。
【0037】
いくつかの実施形態では、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合、R1-1は、16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、Lが−NH−S(O2)−であり、nが0である場合、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。
【0038】
いくつかの実施形態では、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、Lが−NH−C(O)−であり、nが0である場合、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。
【0039】
いくつかの実施形態では、R1-1は、少なくとも11個の炭素原子(好ましくは、少なくとも12個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、R1-1は、11〜20個の炭素原子(好ましくは、12〜20個の炭素原子)を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む。いくつかの実施形態では、R1-1は、11〜20個の炭素原子(好ましくは、12〜20個の炭素原子)を有する線状(すなわち、直鎖)アルキル基である。
【0040】
このようなR1-1置換基は、本発明の化合物に脂質のような特徴を提供するので望ましい。これは、これらの脂質部分が、適用部位におけるIRMの封鎖を助けることができるので有利である。すなわち、脂質部分は、IRMが投与部位から離れて急速に拡散するのを防止するために役立つことができる。この封鎖はIRMのアジュバント性(adjuvancy)の増強もたらすことができ、これは、所望の部位における抗原提示細胞の漸増(recruitment)および活性化の増強によって明らかにされ得る。さらに、この封鎖の結果、IRMの全身への分配が少なくなり、そしてより少ない量のIRMの使用が可能になる。
【0041】
いくつかの実施形態では、RAおよびRBはそれぞれ独立して、水素、ハロゲン、アルキル、アルケニル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。
【0042】
いくつかの実施形態では、RAおよびRBは一緒になって、縮合アリール環または縮合ヘテロアリール環(1つのヘテロ原子を含有する)もしくは縮合飽和5〜7員環(任意に、1つのヘテロ原子を含有する)を形成し、ここでヘテロ原子はNおよびSからなる群から選択され、アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されている。好ましくは、置換基は、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。
【0043】
いくつかの実施形態では、RAおよびRBは一緒になって、縮合アリール環、またはNおよびSからなる群から選択される1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで、アリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になってNおよびSからなる群から選択される1つのヘテロ原子を任意に含有する縮合飽和5〜7員環を形成し、1つまたは複数のR基で非置換または置換されている。
【0044】
いくつかの実施形態では、RAおよびRBは一緒になって、NおよびSからなる群から選択される1つのヘテロ原子を任意に含有する縮合飽和5〜7員環を形成し、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される1つまたは複数の置換基で非置換または置換されている。
【0045】
いくつかの実施形態では、RAおよびRBは縮合アリールまたはヘテロアリール環を形成する。
【0046】
いくつかの実施形態では、RAおよびRBは、縮合飽和5〜7員環を形成する。
【0047】
いくつかの実施形態では、RAおよびRBは、非置換である縮合ベンゼン環を形成する。
【0048】
いくつかの実施形態では、RAおよびRBは、非置換である縮合ピリジン環を形成する。
【0049】
いくつかの実施形態では、Rは、ハロゲン、ヒドロキシ、アルキル、アルケニル、ハロアルキル、アルコキシ、アルキルチオ、および−N(R3)2からなる群から選択される。いくつかの実施形態では、R”およびR2は、水素、アルキル、アルケニル、アリール、ヘテロアリール、ヘテロシクリル、アルキレン−Y−アルキル、アルキレン−Y−アルケニル、アルキレン−Y−アリール、および置換アルキルまたはアルケニルからなる群から選択され、該置換アルキルまたはアルケニルは、−OH、ハロゲン、−N(R4)2、−C(O)−C1〜10アルキル、−C(O)−O−C1〜10アルキル、−N3、アリール、ヘテロアリール、ヘテロシクリル、−C(O)−アリール、および−C(O)−ヘテロアリールからなる群から選択される1つまたは複数の置換基によって置換される。好ましくは、このような実施形態では、Yは−O−または−S(O)0〜2−であり、各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
【0050】
いくつかの実施形態では、R”およびR2は、水素、アルキル、およびアルキレン−O−アルキルからなる群から選択される。
【0051】
いくつかの実施形態では、各R3は独立して、水素およびアルキルからなる群から選択される。
【0052】
いくつかの実施形態では、各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される。
【0053】
いくつかの実施形態では、Yは、−O−または−S(O)0〜2−である。
【0054】
いくつかの実施形態では、nは0〜4である。いくつかの実施形態では、nは0または1である。いくつかの実施形態では、nは0である。
【0055】
化合物の調製
本発明の化合物は、イミダゾキノリン、テトラヒドロイミダゾキノリン、イミダゾピリジン、イミダゾナフチリジン、およびテトラヒドロイミダゾナフチリジンの調製において有用であることが分かっている合成法を用いて調製することができる。
【0056】
例えば、Lが−NH−C(O)−である本発明の化合物は、米国特許第6,451,810号明細書、同第6,545,016号明細書、同第6,194,425号明細書、同第6,660,747号明細書、および同第6,664,265号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、ステアリン酸、パルミチン酸、およびリノール酸などの通常の脂肪酸から調製することができる。
【0057】
Lが−NH−S(O)2−である本発明の化合物は、米国特許第6,331,539号明細書、同第6,525,064号明細書、同第6,194,425号明細書、同第6,677,347号明細書、同第6,677,349号明細書、および同第6,683,088号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、式R1-1S(O)2Clの塩化スルホニルから調製することができる。
【0058】
Lが−NH−C(O)−N(R3)−または−NH−C(S)−N(R3)−である本発明の化合物は、米国特許第6,541,485号明細書、同第6,573,273号明細書、同第6,656,938号明細書、同第6,660,735号明細書、および同第6,545,017号明細書、ならびにPCT公開国際公開第03/103584号パンフレットに記載される合成法を用いて、それぞれ式R1-1C=N=OおよびR1-1C=N=Sのイソシアネートまたはチオイソシアネート(thioisocyante)から調製することができる。
【0059】
Lが結合である本発明の化合物は、米国特許第4,689,338号明細書、同第4,929,624号明細書、同第5,268,376号明細書、同第5,389,640号明細書、同第5,352,784号明細書、および同第5,446,153号明細書に記載される合成法を用いて、式R1-1NH2のアミンから調製することができる。
【0060】
Lが−S−または−S(O)2−である本発明の化合物は、米国特許第6,664,264号明細書および同第6,667,312号明細書に記載される合成法を用いて、式R1-1SHのメルカプタンから調製することができる。
【0061】
薬学的組成物および生物活性
本発明の薬学的組成物は、治療的に有効な量の上記の本発明の化合物を、薬学的に許容可能なキャリアと組み合わせて含有する。
【0062】
「治療的に有効な量」または「有効量」という用語は、サイトカインの誘発、サイトカインの阻害、免疫調節、抗腫瘍活性、および/または抗ウィルス活性などの治療または予防効果を誘発するのに十分な化合物の量を意味する。本発明の薬学的組成物で使用される活性化合物の正確な量は、化合物の物理的および化学的性質、キャリアの性質、ならびに意図される投薬計画などの当業者には既知の因子に従って変わり得るが、本発明の組成物は十分な活性成分を含有して、約100ナノグラム/キログラム(ng/kg)〜約50ミリグラム/キログラム(mg/kg)の用量、好ましくは約10マイクログラム/キログラム(μg/kg)〜約5mg/kgの用量の化合物を被験体に提供することが予想される。錠剤、ロゼンジ、カプセル剤、非経口剤形、シロップ剤、クリーム、軟膏、煙霧剤形、経皮パッチ、経粘膜パッチなどの様々な剤形を使用することができる。
【0063】
本発明の化合物は、治療計画において単一の治療薬として投与することができる。あるいは、本発明の化合物は、互いに組み合わせて、または更なる免疫応答調整剤、抗ウィルス剤、抗生物質、抗体、タンパク質、ペプチド、オリゴヌクレオドなどを含む他の活性剤と組み合わせて投与されてもよい。
【0064】
本発明の化合物は、以下に記載される試験セットに従って実行される実験において特定のサイトカインの産生を誘発することが示され、本発明の特定の化合物は、特定のサイトカインの産生を阻害し得る。これらの結果は、化合物が、多数の異なる方法で免疫応答を調節することができる免疫応答調整剤として有用であることを示しており、これにより化合物は、様々な疾患の治療において有用とされる。
【0065】
本発明に従う化合物の投与によってその産生が誘発され得るサイトカインには、一般に、インターフェロン−α(IFN−α)および/または腫瘍壊死因子−α(TNF−α)ならびに特定のインターロイキン(IL)が含まれる。本発明の化合物によってその生合成が誘発され得るサイトカインには、IFN−α、TNF−α、IL−1、IL−6、IL−10およびIL−12、ならびに様々な他のサイトカインが含まれる。いくつかの効果の中でも特に、これらおよびその他のサイトカインは、ウィルスの産生および腫瘍細胞の成長を阻害し、ウィルス性疾患および腫瘍性疾患の治療において化合物を有用にすることができる。従って、本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法を提供する。サイトカインの生合成の誘発のために化合物または組成物が投与される動物は、以下に記載されるような疾患、例えば、ウィルス性疾患または腫瘍性疾患を有し、化合物の投与は治療的な処置を提供することができる。あるいは、化合物の投与が予防的処置を提供するように、動物が疾患にかかる前に化合物が動物に投与されてもよい。
【0066】
サイトカインの産生を誘発できることに加えて、本発明の化合物は、先天性の免疫応答の他の様相にも影響を与え得る。例えば、ナチュラルキラー細胞の活性が刺激され、サイトカイン誘発による効果であり得る。また化合物はマクロファージも活性化することができ、これは次に、一酸化窒素の分泌および更なるサイトカインの産生を刺激する。さらに、化合物は、Bリンパ球の増殖および分化を引き起こすことができる。
【0067】
また本発明の化合物は、後天性の免疫応答にも影響を与える。例えば、化合物が投与されると、Tヘルパー型1(TH1)サイトカインIFN−γの産生が間接的に誘発され、Tヘルパー型2(TH2)サイトカインIL−4、IL−5およびIL−13の産生は阻害され得る。
【0068】
本発明に従う特定の化合物の投与によってその産生が阻害され得るその他のサイトカインには、腫瘍壊死因子−α(TNF−α)が含まれる。いくつかの効果の中でも特に、TNF−αの産生の阻害は、TNFが仲介される動物の疾患の予防的または治療的な処置を提供することができ、例えば、自己免疫性疾患の治療において化合物を有用にする。従って、本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物におけるTNF−αの生合成の阻害方法を提供する。TNF−αの生合成の阻害のために化合物または組成物が投与される動物は、以下に記載されるような疾患、例えば自己免疫性疾患を有し、化合物の投与は治療的な処置を提供することができる。あるいは、化合物の投与が予防的処置を提供するように、動物が疾患にかかる前に化合物が動物に投与されてもよい。
【0069】
疾患の予防的処置でも治療的処置でも、そして先天性または後天性のいずれの免疫をもたらすためでも、化合物または組成物は単独で投与されてもよいし、例えば、ワクチンアジュバントの場合のように1つまたは複数の活性成分と組み合わせて投与されてもよい。他の成分と共に投与される場合、化合物および他の成分は、別々に投与されてもよいし、溶液の場合などのように一緒であるが独立して投与されてもよいし、あるいは、(a)共有結合される、または(b)例えばコロイド懸濁液の場合のように非共有結合で連結される、などのように一緒にそして互いに関連して投与されてもよい。
【0070】
本明細書中で同定されるIRMを治療に使用することができる状態には、
(a)例えば、アデノウィルス、ヘルペスウィルス(例えば、HSV−I、HSV−II、CMV、またはVZV)、ポックスウィルス(例えば、痘瘡または痘疹などのオルトポックスウィルス、または伝染性軟属腫)、ピコルナウィルス(例えば、ライノウィルスまたはエンテロウィルス)、オルトミクソウィルス(例えば、インフルエンザウィルス)、パラミクソウィルス(例えば、パラインフルエンザウィルス、流行性耳下腺炎ウィルス、麻疹ウィルス、および呼吸器合胞体ウィルス(RSV)、コロナウィルス(例えば、SARS)、パポバウィルス(例えば、性器いぼ、尋常性いぼ、または足底いぼを引き起こすものなどのパピローマウィルス)、ヘパドナウィルス(例えば、B型肝炎ウィルス)、フラビウィルス(例えば、C型肝炎ウィルスまたはデング熱ウィルス)、もしくはレトロウィルス(例えば、HIVなどのレンチウィルス)による感染から生じる疾患などのウィルス性疾患と、
(b)例えば、例えばエシェリキア(Escherichia)属、エンテロバクター属(Enterobacter)、サルモネラ属(Salmonella)、ブドウ球菌属(Staphylococci)、赤痢菌属(Shigella)、リステリア属(Listeria)、アエロバクター属(Aerobacter)、ヘリコバクター属(Helicobacter)、クレブシエラ属(Klebsiella)、プロテウス属(Proteus)、シュードモナス属(Pseudomonas)、連鎖球菌属(Streptococcus)、クラミジア属(Chlamydia)、マイコプラスマ属(Mycoplasma)、肺炎球菌属(Pneumococcus)、ナイセリア属(Neisseria)、クロストリジウム属(Clostridium)、バシラス属(Bacillus)、コリネバクテリウム属(Corynebacterium)、ミコバクテリウム属(Mycobacterium)、カンピロバクター属(Campylobacter)、ビブリオ属(Vibrio)、セラチア属(Serratia)、プロビデンシア属(Providencia)、クロモバクテリウム属(Chromobacterium)、ブルセラ属(Brucella)、エルジニア属(Yersinia)、ヘモフィルス属(Haemophilus)、またはボルデテラ属(Bordetella)の細菌による感染から生じる疾患などの細菌性疾患と、
(c)クラミジアなどのその他の感染性の疾患、例えばカンジダ症、アスペルギルス症、ヒストプラスマ症(histoplasmonsis)、クリプトコッカス髄膜炎などの真菌性疾患、または、例えばマラリア、ニューモシスティスカリニ肺炎、リーシュマニア症、クリプトスポリジウム症、トキソプラスマ症、およびトリパノソーマ感染症などの寄生虫病と、
(d)上皮内新形成、子宮頚部形成異常、光線性角化症、基底細胞癌、扁平上皮癌、腎細胞白血病(aenal cell leukemia)、カポージ肉腫(Karposi’s sarcoma)、メラノーマ、胃細胞癌、例えば、骨髄性白血病、慢性リンパ性白血病、および多発性骨髄腫などの白血病、非ホジキンリンパ腫、皮膚T細胞性リンパ腫、B細胞リンパ腫、ヘアリーセル白血病、ならびにその他の癌などの腫瘍性疾患と、
(e)アトピー性皮膚炎または湿疹、好酸球増加症、喘息、アレルギー、アレルギー性鼻炎、全身性エリテマトーデス、本態性血小板血症(essential thrombocythaemia)、多発性硬化症、オーメン症候群(Ommen’s syndrome)、円板状ループス、円形脱毛症、ケロイド形成および他のタイプの瘢痕化の阻害、ならびに慢性の創傷を含む創傷の治癒の増強などの、TH2に仲介されるアトピー性および自己免疫性疾患と、
が含まれるが、これらに限定されない。
【0071】
本明細書中で同定されるIRMは、例えば、BCG、コレラ、疫病(plague)、腸チフス、A型肝炎、B型肝炎、およびC型肝炎、インフルエンザAおよびインフルエンザB、パラインフルエンザ、ポリオ、狂犬病、麻疹、流行性耳下腺炎、風疹、黄熱、破傷風、ジフテリア、ヘモフィルスインフルエンザb、結核、髄膜炎菌および肺炎球菌ワクチン、アデノウィルス、HIV、水痘、サイトメガロウィルス、デング熱、ネコ白血病、家畜の疫病、HSV−1およびHSV−2、豚コレラ、日本脳炎、呼吸器合胞体ウィルス、ロタウィルス、パピローマウィルス、黄熱、およびアルツハイマー病に関連して使用するために、例えば、生ウィルス性、細菌性、または寄生虫性の免疫原と、不活性ウィルス性、腫瘍由来、原生動物性、有機体由来、真菌性、または細菌性の免疫原、トキソイド、トキシンと、自己抗原と、多糖類と、タンパク質と、糖タンパク質と、ペプチドと、細胞ワクチンと、DNAワクチンと、組換えタンパク質と、その他の同様のものなどの、体液および/または細胞に仲介される免疫応答のいずれかを引き起こす物質と共に使用するためのワクチンアジュバントとしても有用であり得る。
【0072】
またIRMは、欠陥がある免疫機能を有する個体において、特に役に立つことができる。例えば、IRM化合物は、例えば、移植患者、癌患者およびHIV患者において細胞性免疫の抑制後に生じ得る日和見感染および腫瘍を治療するためにも使用することができる。
【0073】
従って、上記の疾患または疾患のタイプのうちの1つまたは複数、例えば、ウィルス性疾患または腫瘍性疾患は、治療的に有効な量の式I、II、III、IV、V、VI、VIIの化合物または塩もしくはこれらの組み合わせを動物に投与することによって、それを必要としている(疾患を有する)動物において治療され得る。また有効な量の式I、II、III、IV、V、VI、VIIの化合物または塩もしくはこれらの組み合わせをワクチンアジュバントとして動物に投与することによって、動物はワクチン接種され得る。
【0074】
サイトカインの生合成を誘発するのに有効な化合物の量は、単球、マクロファージ、樹状細胞およびB細胞などの1つまたは複数の細胞タイプに、例えば、IFN−α、TNF−α、IL−1、IL−6、IL−10およびIL−12などの1つまたは複数のサイトカインを、このようなサイトカインの背景レベルよりも増大された量で産生させるのに十分な量である。正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。また本発明は、有効量の本発明の化合物または組成物を動物に投与することを含む、動物のウィルス感染の治療方法および動物の腫瘍性疾患の治療方法も提供する。
【0075】
ウィルス感染を治療または阻害するのに有効な量は、ウィルス性病変、ウィルス量、ウィルス産生速度、および未処置のコントロール動物と比較した死亡率などのウィルス感染の発現のうちの1つまたは複数の減少を引き起こし得る量である。このような治療に有効である正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。腫瘍性の状態を治療するのに有効な化合物の量は、腫瘍の大きさまたは腫瘍病巣の数の減少を引き起こし得る量である。この場合も同様に、正確な量は、当該技術分野において既知の因子に従って変わり得るが、約100ng/kg〜約50mg/kg、好ましくは約10μg/kg〜約5mg/kgの用量であることが予想される。
【0076】
特定の実施形態では、本明細書中に記載される有効量の化合物または塩を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法が提供される。もう1つの実施形態では、本明細書中に記載される治療的に有効な量の化合物または塩を動物に投与することを含む、動物におけるウィルス性疾患の治療方法が提供される。もう1つの実施形態では、本明細書中に記載される治療的に有効な量の化合物または塩を動物に投与することを含む、動物における腫瘍性疾患の治療方法が提供される。もう1つの実施形態では、本明細書中に記載される有効量の化合物または塩をワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。もう1つの実施形態では、有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドをワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法が提供される。
【0077】
本発明の目的および利点は、以下の実施例によってさらに説明されるが、これらの実施例で列挙される特定の物質およびその量、ならびに他の条件および詳細は、本発明を不当に制限すると解釈されてはならない。
【実施例】
【0078】
実施例1
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドの調製
【化10】
パートA
180mLのテトラヒドロフラン(THF)中の2−(2−アミノエトキシ)エタノール(29.0g、0.276mol)の溶液をN2中で0℃に冷却し、140mLの2NのNaOH溶液で処理した。次に、急速に攪拌した溶液に、180mLのTHF中の二炭酸ジ−tert−ブチル(60.2g、0.276mol)溶液を、1時間にわたって1滴ずつ添加した。次に反応混合物を室温まで暖め、さらに18時間攪拌した。次にTHFを減圧下で除去し、残った水性スラリーを、150mLの1MのH2SO4溶液の添加によりpH3にした。次に、これを酢酸エチル(300mL、100mL)で抽出し、合わせた有機層をH2O(2×)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、tert−ブチル2−(2−ヒドロキシエトキシ)エチルカルバメートが無色のオイルとして与えられた(47.1g)。
【0079】
パートB
1Lの無水CH2Cl2中のtert−ブチル2−(2−ヒドロキシエトキシ)エチルカルバメート(47.1g、0.230mol)の急速に攪拌した溶液をN2中で0℃に冷却し、トリエチルアミン(48.0mL、0.345mol)で処理した。次に塩化メタンスルホニル(19.6mL、0.253mol)を30分間かけて1滴ずつ添加した。次に反応混合物を室温まで暖め、さらに22時間攪拌した。500mLの飽和NaHCO3溶液の添加により反応を停止させ、有機層を分離した。次に有機相をH2O(3×500mL)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、2−{2−[(tert−ブトキシカルボニル)アミノ]エトキシ}エチルメタンスルホネートが茶色のオイルとして与えられた(63.5g)。
【0080】
パートC
400mLのN,N−ジメチルホルムアミド(DMF)中の2−{2−[(tert−ブトキシカルボニル)アミノ]エトキシ}エチルメタンスルホネート(63.5g、0.224mol)の攪拌溶液をNaN3(16.1g、0.247mol)で処理し、反応混合物をN2中で90℃に加熱した。5時間後、溶液を室温まで冷却し、500mLの冷H2Oで処理した。次に反応混合物をEt2O(3×300mL)で抽出した。合わせた有機抽出物をH2O(4×100mL)および塩水(2×100mL)で洗浄した。有機部分をMgSO4で乾燥させ、濃縮して、52.0gのtert−ブチル2−(2−アジドエトキシ)エチルカルバメートが薄茶色のオイルとして与えられた。
【0081】
パートD
tert−ブチル2−(2−アジドエトキシ)エチルカルバメート(47.0g、0.204mol)のMeOH溶液を4gの10%Pd炭素(Pd on carbon)で処理し、H2(3Kg/cm2)中で24時間振とうした。次に溶液をセライト(CELITE)パッドによりろ過し、濃縮して、35.3gの粗tert−ブチル2−(2−アミノエトキシ)エチルカルバメートが無色の液体として与えられ、さらに精製することなくこれを使用した。
【0082】
パートE
500mLの無水CH2Cl2中の4−クロロ−3−ニトロキノリン(31.4g、0.151mol)の攪拌溶液を、N2中で、トリエチルアミン(43mL、0.308mol)およびtert−ブチル2−(2−アミノエトキシ)エチルカルバメート(0.151mol)により処理した。一晩攪拌した後、反応混合物をH2O(2×300mL)および塩水(300mL)で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、鮮やかな黄色の固体が与えられた。酢酸エチル/へキサンからの再結晶により、43.6gのtert−ブチル2−{2−[(3−ニトロキノリン−4−イル)アミノ]エトキシ}エチルカルバメートが鮮やかな黄色の結晶として与えられた。
【0083】
パートF
tert−ブチル2−{2−[(3−ニトロキノリン−4−イル)アミノ]エトキシ}エチルカルバメート(7.52g、20.0mmol)のトルエン溶液を1.5gの5%Pt炭素(Pt on carbon)で処理し、H2(3Kg/cm2)中で24時間振とうした。次に溶液をセライトパッドによりろ過し、濃縮して、6.92gの粗tert−ブチル2−{2−[(3−アミノキノリン−4−イル)アミノ]エトキシ}エチルカルバメートが黄色のシロップとして与えられた。
【0084】
パートG
250mLの無水CH2Cl2中のtert−ブチル2−{2−[(3−アミノキノリン−4−イル)アミノ]エトキシ}エチルカルバメート(10.2g、29.5mmol)の溶液を0℃に冷却し、トリエチルアミン(4.18mL、30.0mmol)で処理した。次に塩化メトキシプロピオニル(3.30mL、30.3mmol)を5分間にわたって1滴ずつ添加した。次に反応を室温まで暖め、攪拌を1時間継続した。次に反応混合物を減圧下で濃縮して、オレンジ色の固体が与えられた。これを250mLEtOH中に溶解させ、12.5mLのトリエチルアミンを添加した。混合物を加熱して還流させ、N2中で一晩攪拌した。次に反応を減圧下で濃縮乾燥し、300mLのEt2Oで処理した。次に混合物をろ過し、ろ液を減圧下で濃縮して、茶色の固体が与えられた。固体を200mLの熱いメタノールに溶解させ、活性炭で処理した。熱い溶液をろ過し、濃縮して、11.1gのtert−ブチル2−{2−[2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートが黄色のシロップとして与えられた。
【0085】
パートH
250mLのCHCl3中のtert−ブチル2−{2−[2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.22g、24.7mmol)の溶液を3−クロロ過安息香酸(3−chloroperbenozic acid)(77%、9.12g、40.8mmol)で処理した。30分間攪拌した後、反応混合物を1%のNa2CO3溶液(2×75mL)および塩水で洗浄した。次に有機層をNa2SO4で乾燥させ、濃縮して、10.6gのtert−ブチル2−{2−[2−(2−メトキシエチル)−5−オキシド−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートがオレンジ色のフォームとして与えられ、さらに精製することなくこれを用いた。
【0086】
パートI
100mLの1,2−ジクロロエタン中のtert−ブチル2−{2−[2−(2−メトキシエチル)−5−オキシド−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.6g、24.6mmol)の溶液を60℃に加熱し、10mLの濃NH4OH溶液で処理した。急速に攪拌した溶液に、10分の間にわたって固体の塩化p−トルエンスルホニル(7.05g、37.0mmol)を添加した。反応混合物をさらに1mLの濃NH4OH溶液で処理し、次に加圧容器中に密封し、加熱を2時間継続した。次に反応混合物を冷却し、100mLのCHCl3で処理した。次に反応混合物をH2O、1%のNa2CO3溶液(2×)および塩水で洗浄した。有機部分をNa2SO4で乾燥させ、濃縮して、10.6gのtert−ブチル2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメートが茶色のフォームとして与えられた。
【0087】
パートJ
tert−ブチル2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチルカルバメート(10.6g、24.6mmol)をエタノール中の75mLの2MのHClで処理し、混合物を攪拌しながら加熱して還流させた。1.5時間後、反応混合物を冷却し、ろ過して、粘着性の固体が与えられた。固体をエタノールおよびEt2Oで洗浄し、真空乾燥させて、塩酸塩が薄茶色の固体として与えられた。塩酸塩を50mLのH2O中に溶解させ、10%のNaOH溶液で処理することによって遊離塩基を作った。次に水性懸濁液を濃縮乾燥させ、残渣をCHCl3で処理した。得られた塩をろ過により除去し、ろ液を濃縮して、3.82gの1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミンが黄褐色の粉体として与えられた。
MS330(M+H)+、
1H NMR(300MHz、DMSO−d6)δ8.10(d、J=8.1Hz、1H)、7.66(d、J=8.2Hz、1H)、7.40(m、1H)、7.25(m、1H)、6.88(brs、2H)、4.78(t、J=5.4Hz、2H)、3.89(t、J=4.8Hz、2H)、3.84(t、J=6.9Hz、2H)、3.54(t、J=5.4Hz、2H)、3.31(s、3H)、3.23(t、J=6.6Hz、2H)、2.88(t、J=5.3Hz、2 H)。
【0088】
パートK
窒素雰囲気下で、ジクロロメタン(3.5mL)およびトリエチルアミン(150μL、1.07mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(140.5mg、0.428mmol)の懸濁液を0℃に冷却した。塩化パルミトイル(130μL、0.428mmol)をゆっくり添加した。反応混合物を0℃で2時間攪拌させ、この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(30mL)で希釈し、飽和重炭酸ナトリウム溶液(2×5mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に減圧下で濃縮した。得られた残渣をカラムクロマトグラフィ(12gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製して、183mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドが白色粉体として提供された。
C33H53N5O3の分析計算値:%C,69.80、%H,9.41、%N,12.33、実測値:%C,69.60、%H,9.28、%N,11.99。
【0089】
実施例2
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドの調製
【化11】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(468μL、3.56mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(442.6mg、1.344mmol)の混合物を0℃に冷却した。塩化ステアロイル(454μL、1.34mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させ、この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に、減圧下で濃縮した。残渣を高真空下で乾燥させ、834mgの粗生成物が提供された。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%メタノールで溶出)により精製し、596mgの生成物が提供された。この物質を酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA(80%クロロホルム/18%メタノール/2%水酸化アンモニウム)300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、およびクロロホルム中6%のCMA500mLで順次溶出)により精製して、23.8mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドが白色のワックス状固体(融点80〜83℃)として提供された。
C35H57N5O3の分析計算値:0.694%のH2O、%C,70.06、%H,9.65、%N,11.67、実測値:%C,70.60、%H,9.91、%N,11.46。
【0090】
実施例3
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドの調製
【化12】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(551μL、4.00mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(527.0mg、1.600mmol)の混合物を0℃に冷却した。塩化ラウロイル(370μL、1.60mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させた。この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に、減圧下で濃縮した。残渣を高真空下で乾燥させ、821mgの粗生成物が提供された。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製し、527mgの生成物が提供された。この物質を酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、クロロホルム中6%のCMA750mL、100%のCMA500mLで順次溶出)により精製し、22.4mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドが白色のワックス状固体(融点80〜83℃)として提供された。
C29H45N5O3の分析計算値:1.66%のH2O、%C,66.94、%H,8.90、%N,13.46、実測値:%C,66.94、%H,9.37、%N,13.28。
【0091】
実施例4
N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドの調製
【化13】
窒素雰囲気下で、ジクロロメタン(20.0mL)およびトリエチルアミン(470μL、3.37mmol)の混合物中の1−[2−(2−アミノエトキシ)エチル]−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−4−アミン(444.mg、1.349mmol)の混合物を0℃に冷却した。塩化ミリストイル(367μL、1.35mmol)を10分間にわたってゆっくり添加した。反応混合物を0℃で1時間攪拌させた。この時点で、薄層クロマトグラフィによる分析は、出発物質が残っていないことを示した。反応混合物をジクロロメタン(50mL)で希釈し、飽和重炭酸ナトリウム溶液(2×15mL)で洗浄し、硫酸マグネシウムで乾燥させ、次に減圧下で濃縮した。粗生成物をカラムクロマトグラフィ(20gのシリカゲル、ジクロロメタン中2%のメタノールで溶出)により精製した後、酢酸エチル(1.2mL)から再結晶し、次にさらにカラムクロマトグラフィ(25gのシリカゲル、クロロホルム中1%のCMA300mL、クロロホルム中2%のCMA500mL、クロロホルム中3%のCMA500mL、クロロホルム中4%のCMA500mL、クロロホルム中5%のCMA750mL、およびクロロホルム中6%のCMA600mLで順次溶出)により精製して、9.5mgのN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドが白色のワックス状固体(融点85〜87℃)として提供された。
【0092】
その他の典型的な化合物
特定の他の典型的な化合物は、式(VIII〜X)および以下の置換基を有し、表の各行は、第1列の見出し語に示されるように、式VIII、XIおよび/またはXの特定の化合物を表す。
【化14】
【0093】
【表1】
【表2】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
【表17】
【表18】
【0094】
実施例5
免疫化
200μlのリン酸緩衝食塩水(PBS)中の複合体(conjugate)(1mgのオボアルブミンおよび200μgのIRM)を用いて、皮下または腹腔内のいずれかで、C57BL/6マウスを免疫化した。コントロールマウスは、200μlのPBS中の1mgのオボアルブミンで免疫化した。一次応答の分析のために、免疫化の5〜7日後にマウスを犠牲にした。二次応答の分析のために、マウスを最初の免疫化の7〜15日後に、追加免疫し、5〜7日後に犠牲にした。他に示されない限りは、皮下で免疫化したマウスから分析のためにリンパ節を収集し、腹腔内で免疫化したマウスから分析のために脾臓細胞を収集した。
【0095】
DMSO中に10mg/mlの濃度で溶解させることによって、N−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドのストックIRM溶液を調製した。オボアルブミンをPBS中に50mg/mlの濃度で溶解させた。50μlのストックIRM溶液を150μlのPBSに添加し、次にボルテックスすることにより混合した。50μlのオボアルブミンをストックIRM溶液に添加し、ボルテックスすることにより混合した。IRMおよびオボアルブミンの濁ったコロイド懸濁液が生じた。
【0096】
上記のように(a)オボアルブミン単独、または(b)50μlのオボアルブミンおよびIRMのコロイド懸濁液のいずれかを用いて、0日目にマウスを皮下で免疫化した。6日目に、排出(draining)リンパ節を除去し、均質化し、H−2Kb/SIINFEKLテトラマーで染色して、オボアルブミン特異的T細胞を同定した。図1は、オボアルブミン単独で免疫化したコントロールマウスからのフローサイトメトリーデータを示し、図2および3は、コロイド懸濁液で免疫化した2つの異なるマウスからのデータを示す。
【0097】
試薬
オボアルブミンは、シグマ・ケミカル・カンパニー(Sigma Chemical Company)(ミズーリ州セントルイス(St.Louis,MO))から入手した。優性のオボアルブミンペプチドSIINFEKLに結合したMHCクラスI分子H−2Kbのテトラマーは、ケドル(Kedl)らのJ Exp Med,192:1105−13(2000)に記載されるように生成した。
【0098】
以下に記載される方法を用いて試験した場合に、本発明の化合物はサイトカインの生合成を誘発し、そして特定の化合物はサイトカインの生合成を阻害し得ることがわかった。実施例1〜4の化合物は、以下に記載される「ヒト細胞におけるサイトカインの誘発」アッセイを用いて試験した場合に、インターフェロンおよび腫瘍壊死因子の両方を誘発した。
【0099】
ヒト細胞におけるサイトカインの誘発
インビトロのヒト血液細胞系を用いて、サイトカインの誘発を評価する。テスターマン(Testerman)らにより「免疫調節剤のイミキモドおよびS−27609によるサイトカインの誘発」、Journal of Leukocyte Biology,58,365−372(1995年9月)に記載されるように、活性は、培地に分泌されるインターフェロン−αおよび腫瘍壊死因子−α(それぞれ、IFN−αおよびTNF−α)の測定に基づく。
【0100】
培養のための血液細胞の調製
健康なヒトのドナーからの全血を、静脈穿刺によってEDTAバキュテイナー(vacutainer)管内に採取する。HISTOPAQUE−1077を用いる密度勾配遠心分離法によって、末梢血単核細胞(PBMC)を全血から分離する。ダルベッコのリン酸緩衝食塩水(DPBS)またはハンクス平衡塩溶液(HBSS)により血液を1:1で希釈する。PBMC層を採取し、DPBSまたはHBSSで2回洗浄し、RPMI完全培地中に4×106個の細胞/mLで再懸濁させる。PBMC懸濁液を、試験化合物を含有する同体積のRPMI完全培地を含有する48ウェルの平底無菌組織培養プレート(マサチューセッツ州ケンブリッジのコスター(Costar,Cambridge,MA)またはニュージャージー州リンカーンパークのベクトン・ディッキンソン・ラブウェア(Becton Dickinson Labware,Lincoln Park,NJ))に添加する。
【0101】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、一般に、30〜0.014マイクロモル(μM)の範囲の濃度で試験される。
【0102】
インキュベーション
試験化合物の溶液を、RPMI完全培地を含有する第1のウェルに60μMで添加し、ウェルにおいて連続的な3倍希釈を行なう。次にPBMC懸濁液を同体積でウェルに添加して、試験化合物濃度を所望の範囲(30〜0.014μM)にする。PBMC懸濁液の最終濃度は2×106個の細胞/mLである。プレートを無菌プラスチック蓋で覆い、静かに混ぜてから、5%の二酸化炭素雰囲気中、37℃で18〜24時間インキュベーションを行なう。
【0103】
分離
インキュベーションに続いて、1000rpm(約200×g)において、4℃で10分間プレートを遠心分離する。無菌ポリプロピレンピペットで無細胞の培養の上澄みを取り出し、無菌ポリプロピレン管に移す。分析するまでサンプルを−30〜−70℃に保持する。ELISAによりIFN−αについて、そしてELISAまたはIGENアッセイによりTNF−αについて、サンプルを分析する。
【0104】
ELISAによるIFN−αおよびTNF−αの分析
ニュージャージー州ニューブランズウィックのPBLバイオメディカル・ラボラトリーズ(PBL Biomedical Laboratories,New Brunswick,NJ)からのヒト・マルチスピーシーズ・キットを用いて、ELISAによりIFN−αの濃度を決定する。結果は、pg/mLで表される。
【0105】
カリフォルニア州カマリロのバイオソース・インターナショナル(Biosource International,Camarillo,CA)から入手可能なELISAキットを用いて、TNF−αの濃度を決定する。あるいは、TNF−αの濃度は、オリジン(ORIGEN)M−シリーズイムノアッセイによって決定し、メリーランド州ゲーサーズバーグのアイジェン・インターナショナル(IGEN International,Gaithersburg、MD)からのアイジェン(IGEN)M−8アナライザーにおいて読み取ることができる。イムノアッセイは、カリフォルニア州カマリロのバイオソース・インターナショナルからのヒトのTNF−α捕獲および検出抗体ペアを用いる。結果は、pg/mLで表される。
【0106】
マウス細胞におけるサイトカインの阻害
マウスのマクロファージ細胞株Raw264.7を用いて、リポ多糖(LPS)により刺激されたときに化合物が腫瘍壊死因子−α(TNF−α)の産生を阻害する能力を評価する。
【0107】
単一濃度アッセイ
培養のための血液細胞の調製
生細胞(ATCC)を優しくこすって収集し、カウントする。細胞懸濁液を、10%ウシ胎児血清(FBS)を有するRPMI中で3×105個の細胞/mLにする。細胞懸濁液(100μL)を96ウェルの平底無菌組織培養プレート(ニュージャージー州リンカーンパークのベクトン・ディッキンソン・ラブウェア)に添加する。細胞の最終濃度は、3×104個の細胞/ウェルである。プレートを3時間インキュベートする。試験化合物を添加する前に、培地を、3%FBSを有する無色のRPMI培地に取り替える。
【0108】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、5μMで試験する。LPS(ネズミチフス菌(Salmonella typhimurium)からのリポ多糖、シグマ−アルドリッチ(Sigma−Aldrich))を、用量反応アッセイで測定されるようにEC70濃度に無色のRPMIで希釈する。
【0109】
インキュベーション
試験化合物の溶液(1μl)を各ウェルに添加する。マイクロタイタープレート振とう器においてプレートを1分間混ぜ、次にインキュベータ内に置く。20分後に、LPSの溶液(1μL、EC70濃度約10ng/ml)を添加し、プレートを振とう器で1分間混ぜる。5%の二酸化炭素雰囲気中、37℃でプレートを18〜24時間インキュベートする。
【0110】
TNF−αの分析
インキュベーションに続いて、上澄みをピペットで取り出す。マウスTNF−αキット(カリフォルニア州カマリロのバイオソース・インターナショナルから)を用いて、ELISAによりTNF−α濃度を決定する。結果は、pg/mLで表される。LPS刺激のみにおけるTNF−αの発現は、100%の応答であると考えられる。
【0111】
用量反応アッセイ
培養のための血液細胞の調製
生細胞(ATCC)を優しくこすって収集し、カウントする。細胞懸濁液を、10%FBSを有するRPMI中で4×105個の細胞/mLにする。細胞懸濁液(250μL)を48ウェルの平底無菌組織培養プレート(マサチューセッツ州ケンブリッジのコスター)に添加する。細胞の最終濃度は、1×105個の細胞/ウェルである。プレートを3時間インキュベートする。試験化合物を添加する前に、培地を、3%FBSを有する無色のRPMI培地に取り替える。
【0112】
化合物の調製
化合物をジメチルスルホキシド(DMSO)に可溶性にする。DMSO濃度は、培養ウェルへ添加するための1%の最終濃度を超えてはならない。化合物は、0.03、0.1、0.3、1、3、5および10μMで試験した。LPS(ネズミチフス菌からのリポ多糖、シグマ−アルドリッチ)を、用量反応アッセイで測定されるようにEC70濃度に無色のRPMIで希釈する。
【0113】
インキュベーション
試験化合物の溶液(200μl)を各ウェルに添加する。マイクロタイタープレート振とう器においてプレートを1分間混ぜ、次にインキュベータ内に置く。20分後に、LPSの溶液(200μL、EC70濃度約10ng/ml)を添加し、プレートを振とう器で1分間混ぜる。5%の二酸化炭素雰囲気中、37℃でプレートを18〜24時間インキュベートする。
【0114】
TNF−αの分析
インキュベーションに続いて、上澄みをピペットで取り出す。マウスTNF−αキット(カリフォルニア州カマリロのバイオソース・インターナショナルから)を用いて、ELISAによりTNF−α濃度を決定する。結果は、pg/mLで表される。LPS刺激のみにおけるTNF−αの発現は、100%の応答であると考えられる。
【0115】
本明細書中で引用される特許、特許文献、および刊行物の全ての開示は、あたかもそれぞれが個々に援用されたかのように、参照によってその全体が援用される。本発明は、そのいくつかの実施形態を参照して説明された。上記の実例となる実施形態および実施例は、理解を明確にするだけのために提供されたものであり、そこから不要な限定が理解されてはならない。本発明の精神および範囲から逸脱することなく、記載された実施形態に多くの変化が成され得ることは、当業者には明らかであろう。したがって、本発明の範囲は、特許請求の範囲によってのみ限定されることが意図される。
【図面の簡単な説明】
【0116】
【図1】実施例5に記載されるように、オボアルブミンによる免疫化後の抗原特異的CD8+T細胞の拡大を示す。
【図2】実施例5に記載されるように、IRMおよびオボアルブミンのコロイド懸濁液による免疫化後の1つの被験体の抗原特異的CD8+T細胞の拡大を示す。
【図3】実施例5に記載されるように、IRMおよびオボアルブミンのコロイド懸濁液による免疫化後の第2の被験体の抗原特異的CD8+T細胞の拡大を示す。
【特許請求の範囲】
【請求項1】
以下の式I:
【化1】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断され、
Lは、結合または官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R”は、水素または非妨害性置換基であり、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、あるいはRAおよびRBは一緒になって縮合アリール環または1つのヘテロ原子を含有する縮合ヘテロアリール環もしくは任意に1つのヘテロ原子を含有する縮合飽和5〜7員環を形成し、ここで該ヘテロ原子はNおよびSからなる群から選択され、さらにここで、上記アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されており、そして、
各R3は、独立して水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そしてLが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項2】
RAおよびRBが一緒になって縮合飽和5〜7員環を形成し、NおよびSからなる群から選択される1つのヘテロ原子を任意に含有し、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される1つまたは複数の置換基で非置換または置換されている、請求項1に記載の化合物または塩。
【請求項3】
RAおよびRBが、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される、請求項1に記載の化合物または塩。
【請求項4】
RAおよびRBが、縮合アリールまたは縮合ヘテロアリール環を形成する、請求項1に記載の化合物または塩。
【請求項5】
RAおよびRBが、縮合飽和5〜7員環を形成する、請求項1に記載の化合物または塩。
【請求項6】
RAおよびRBが一緒になって、縮合アリール環、またはNおよびSからなる群から選択される1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで、前記アリールまたはヘテロアリール環が1つまたは複数のR基で非置換または置換されているか、
あるいは、RAおよびRBが一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されており、
各Rが独立して、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される、請求項1に記載の化合物または塩。
【請求項7】
RAおよびRBが、非置換の縮合ベンゼン環を形成する、請求項6に記載の化合物または塩。
【請求項8】
RAおよびRBが、非置換の縮合ピリジン環を形成する、請求項6に記載の化合物または塩。
【請求項9】
R”が、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基によって置換されている前記置換アルキルまたはアルケニル、
からなる群から選択され、ここで、Yが−O−または−S(O)0〜2−であり、各R4が独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される、請求項1〜8のいずれか一項に記載の化合物または塩。
【請求項10】
Lが、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基である、請求項1〜9のいずれか一項に記載の化合物または塩。
【請求項11】
Lが、結合または−NH−C(O)−、−NH−S(O)2−、および−NH−C(O)−N(R3)−からなる群から選択される官能性連結基である、請求項10に記載の化合物または塩。
【請求項12】
R1-1が、11〜20個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む、請求項1〜11のいずれか一項に記載の化合物または塩。
【請求項13】
R1-1が、12〜20個の炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含む、請求項12に記載の化合物または塩。
【請求項14】
R1-1が直鎖C12〜C20アルキルである、請求項13に記載の化合物または塩。
【請求項15】
以下の式II:
【化2】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは、一緒になって縮合アリール環または1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで上記アリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されており、ここでRは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、そして、
R3は、水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項16】
R1が、式アルキレン−L−R1-1を有し、該アルキレンが、1つの−O−基により任意に中断される、請求項15に記載の化合物または塩。
【請求項17】
R1が、式C1〜5アルキレン−L−R1-1を有し、該C1〜5アルキレンが、1つの−O−基により任意に中断される、請求項16に記載の化合物または塩。
【請求項18】
R2が水素、アルキル、およびアルキレン−O−アルキルからなる群から選択される、請求項15に記載の化合物または塩。
【請求項19】
以下の式II:
【化3】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、そして、
R3は、水素およびアルキルからなる群から選択される)
を有する化合物またはその薬学的に許容可能な塩。
【請求項20】
以下の式III:
【化4】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0〜4であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、そして、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、nが0である場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項21】
nが0である、請求項20に記載の化合物または塩。
【請求項22】
以下の式IV、V、VI、およびVII:
【化5】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意で、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0または1であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−C(O)−であり、nが0である場合には、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする)
からなる群から選択される化合物またはその薬学的に許容可能な塩。
【請求項23】
nが0である、請求項22に記載の化合物または塩。
【請求項24】
薬学的に許容可能なキャリアとともに、治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を含む薬学的組成物。
【請求項25】
有効量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法。
【請求項26】
治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物におけるウィルス性疾患の治療方法。
【請求項27】
治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物における腫瘍性疾患の治療方法。
【請求項28】
有効量の請求項1〜23のいずれか一項に記載の化合物または塩をワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法。
【請求項29】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項30】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項31】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項32】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項1】
以下の式I:
【化1】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断され、
Lは、結合または官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R”は、水素または非妨害性置換基であり、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、あるいはRAおよびRBは一緒になって縮合アリール環または1つのヘテロ原子を含有する縮合ヘテロアリール環もしくは任意に1つのヘテロ原子を含有する縮合飽和5〜7員環を形成し、ここで該ヘテロ原子はNおよびSからなる群から選択され、さらにここで、上記アリール、ヘテロアリール、または飽和5〜7員環は、1つまたは複数の非妨害性置換基で非置換または置換されており、そして、
各R3は、独立して水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O)2−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は16個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そしてLが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項2】
RAおよびRBが一緒になって縮合飽和5〜7員環を形成し、NおよびSからなる群から選択される1つのヘテロ原子を任意に含有し、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される1つまたは複数の置換基で非置換または置換されている、請求項1に記載の化合物または塩。
【請求項3】
RAおよびRBが、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される、請求項1に記載の化合物または塩。
【請求項4】
RAおよびRBが、縮合アリールまたは縮合ヘテロアリール環を形成する、請求項1に記載の化合物または塩。
【請求項5】
RAおよびRBが、縮合飽和5〜7員環を形成する、請求項1に記載の化合物または塩。
【請求項6】
RAおよびRBが一緒になって、縮合アリール環、またはNおよびSからなる群から選択される1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで、前記アリールまたはヘテロアリール環が1つまたは複数のR基で非置換または置換されているか、
あるいは、RAおよびRBが一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されており、
各Rが独立して、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択される、請求項1に記載の化合物または塩。
【請求項7】
RAおよびRBが、非置換の縮合ベンゼン環を形成する、請求項6に記載の化合物または塩。
【請求項8】
RAおよびRBが、非置換の縮合ピリジン環を形成する、請求項6に記載の化合物または塩。
【請求項9】
R”が、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基によって置換されている前記置換アルキルまたはアルケニル、
からなる群から選択され、ここで、Yが−O−または−S(O)0〜2−であり、各R4が独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択される、請求項1〜8のいずれか一項に記載の化合物または塩。
【請求項10】
Lが、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基である、請求項1〜9のいずれか一項に記載の化合物または塩。
【請求項11】
Lが、結合または−NH−C(O)−、−NH−S(O)2−、および−NH−C(O)−N(R3)−からなる群から選択される官能性連結基である、請求項10に記載の化合物または塩。
【請求項12】
R1-1が、11〜20個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含む、請求項1〜11のいずれか一項に記載の化合物または塩。
【請求項13】
R1-1が、12〜20個の炭素原子を有する線状または分枝状脂肪族基であり、任意に1つまたは複数の不飽和炭素−炭素結合を含む、請求項12に記載の化合物または塩。
【請求項14】
R1-1が直鎖C12〜C20アルキルである、請求項13に記載の化合物または塩。
【請求項15】
以下の式II:
【化2】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、任意に、1つまたは複数の−O−基により中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択されるか、
あるいはRAおよびRBは、一緒になって縮合アリール環または1つのヘテロ原子を含有する縮合ヘテロアリール環を形成し、ここで上記アリールまたはヘテロアリール環は、1つまたは複数のR基で非置換または置換されているか、あるいは、RAおよびRBは一緒になって縮合飽和5〜7員環を形成し、任意に、NおよびSからなる群から選択される1つのヘテロ原子を含有し、1つまたは複数のR基で非置換または置換されており、ここでRは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、そして、
R3は、水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、RAおよびRBが結合して非置換ベンゼン環を形成する場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とし、そして、Lが−NH−C(O)−であり、RAおよびRBが結合して非置換ピリジン環を形成する場合には、R1-1は、11個よりも多い炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを更なる条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項16】
R1が、式アルキレン−L−R1-1を有し、該アルキレンが、1つの−O−基により任意に中断される、請求項15に記載の化合物または塩。
【請求項17】
R1が、式C1〜5アルキレン−L−R1-1を有し、該C1〜5アルキレンが、1つの−O−基により任意に中断される、請求項16に記載の化合物または塩。
【請求項18】
R2が水素、アルキル、およびアルキレン−O−アルキルからなる群から選択される、請求項15に記載の化合物または塩。
【請求項19】
以下の式II:
【化3】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、ここでYは、−O−または−S(O)0〜2−であり、そして各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
RAおよびRBは、それぞれ独立して、以下の:
水素、
ハロゲン、
アルキル、
アルケニル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、そして、
R3は、水素およびアルキルからなる群から選択される)
を有する化合物またはその薬学的に許容可能な塩。
【請求項20】
以下の式III:
【化4】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0〜4であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、そして、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−S(O2)−であり、nが0である場合には、R1-1は、少なくとも16個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする)
を有する化合物またはその薬学的に許容可能な塩。
【請求項21】
nが0である、請求項20に記載の化合物または塩。
【請求項22】
以下の式IV、V、VI、およびVII:
【化5】
(式中、
R1は、式アルキレン−L−R1-1、アルケニレン−L−R1-1、またはアルキニレン−L−R1-1を有し、ここで、
上記アルキレン、アルケニレン、およびアルキニレン基は、1つまたは複数の−O−基により任意に中断され、
Lは、結合または−NH−S(O)2−、−NH−C(O)−、−NH−C(S)−、−NH−S(O)2−NR3−、−NH−C(O)−NR3−、−NH−C(S)−NR3−、−NH−C(O)−O−、−O−、−S−、および−S(O)2−からなる群から選択される官能性連結基であり、そして
R1-1は、少なくとも11個の炭素原子を有する線状または分枝状脂肪族基であり、任意で、1つまたは複数の不飽和炭素−炭素結合を含み、
Rは、以下の:
ハロゲン、
ヒドロキシ、
アルキル、
アルケニル、
ハロアルキル、
アルコキシ、
アルキルチオ、および
−N(R3)2
からなる群から選択され、
nは0または1であり、
R2は、以下の:
水素、
アルキル、
アルケニル、
アリール、
ヘテロアリール、
ヘテロシクリル、
アルキレン−Y−アルキル、
アルキレン−Y−アルケニル、
アルキレン−Y−アリール、および
置換アルキルまたはアルケニルであって、以下の:
−OH、
ハロゲン、
−N(R4)2、
−C(O)−C1〜10アルキル、
−C(O)−O−C1〜10アルキル、
−N3、
アリール、
ヘテロアリール、
ヘテロシクリル、
−C(O)−アリール、および
−C(O)−ヘテロアリール
からなる群から選択される1つまたは複数の置換基で置換されている前記置換アルキルまたはアルケニル
からなる群から選択され、
Yは、−O−または−S(O)0〜2−であり、
各R4は独立して、水素、C1〜10アルキル、およびC2〜10アルケニルからなる群から選択され、
R3は水素およびアルキルからなる群から選択され、
ただし、Lが−NH−C(O)−であり、nが0である場合には、R1-1は、少なくとも12個の炭素原子を有する線状または分枝状脂肪族基であり、任意に、1つまたは複数の不飽和炭素−炭素結合を含むことを条件とする)
からなる群から選択される化合物またはその薬学的に許容可能な塩。
【請求項23】
nが0である、請求項22に記載の化合物または塩。
【請求項24】
薬学的に許容可能なキャリアとともに、治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を含む薬学的組成物。
【請求項25】
有効量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物におけるサイトカインの生合成の誘発方法。
【請求項26】
治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物におけるウィルス性疾患の治療方法。
【請求項27】
治療的に有効な量の請求項1〜23のいずれか一項に記載の化合物または塩を動物に投与することを含む、動物における腫瘍性疾患の治療方法。
【請求項28】
有効量の請求項1〜23のいずれか一項に記載の化合物または塩をワクチンアジュバントとして動物に投与することを含む、動物へのワクチン接種方法。
【請求項29】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ヘキサデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項30】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)オクタデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項31】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)ドデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【請求項32】
有効量のN−(2−{2−[4−アミノ−2−(2−メトキシエチル)−1H−イミダゾ[4,5−c]キノリン−1−イル]エトキシ}エチル)テトラデカンアミドをワクチンアジュバントとして動物に投与することを含む、該動物へのワクチン接種方法。
【図1】

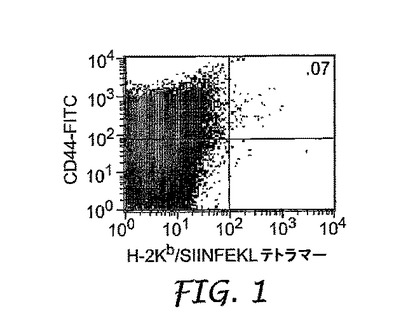
【図2】
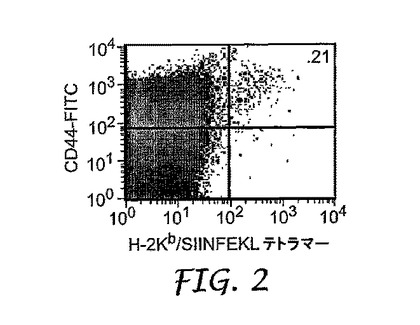
【図3】
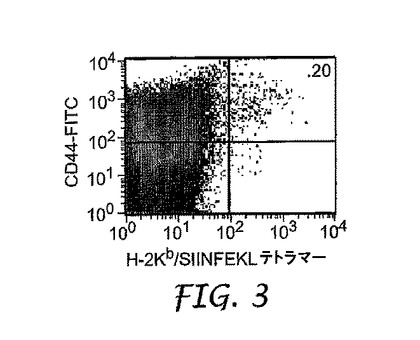
【図2】
【図3】
【公表番号】特表2007−521317(P2007−521317A)
【公表日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願番号】特願2006−523370(P2006−523370)
【出願日】平成16年8月12日(2004.8.12)
【国際出願番号】PCT/US2004/026157
【国際公開番号】WO2005/018555
【国際公開日】平成17年3月3日(2005.3.3)
【出願人】(599056437)スリーエム イノベイティブ プロパティズ カンパニー (1,802)
【Fターム(参考)】
【公表日】平成19年8月2日(2007.8.2)
【国際特許分類】
【出願日】平成16年8月12日(2004.8.12)
【国際出願番号】PCT/US2004/026157
【国際公開番号】WO2005/018555
【国際公開日】平成17年3月3日(2005.3.3)
【出願人】(599056437)スリーエム イノベイティブ プロパティズ カンパニー (1,802)
【Fターム(参考)】
[ Back to top ]