
脳損傷の予後改善薬のスクリーニング方法
【課題】脳の局所的な炎症の遷延による組織損傷を抑止し、予後の改善をはかることのできる化合物の提供。および、そのような化合物をスクリーングする方法の提供。
【解決手段】造血器型プロスタグランジンD合成酵素(H−PGDS)の阻害剤を活性成分として含む医薬組成物の使用。また、ヒト造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを用いて、これらの薬効を有する化合物を容易にスクリーニングできる。
【解決手段】造血器型プロスタグランジンD合成酵素(H−PGDS)の阻害剤を活性成分として含む医薬組成物の使用。また、ヒト造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを用いて、これらの薬効を有する化合物を容易にスクリーニングできる。
【発明の詳細な説明】
【技術分野】
【0001】
この発明は、脳損傷を治療または予防する化合物及びそのスクリーニング方法に関するものである。さらに詳しくは、この発明は、脳血管障害、脳変性疾患、脱髄疾患等の疾患による脳損傷部位のミクログリア細胞やマクロファージにおいて誘導される造血器型プロスタグランジンD合成酵素(以下、「H−PGDS」ということがある)を阻害したり、損傷部位の周辺のアストログリア細胞で発現するプロスタグランジンD受容体(以下、「DP受容体」ということがある)の活性化を阻害することにより、プロスタグランジンD2が関与する脳損傷を治療または予防する化合物と、これらの物質の薬効を、ヒト造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを用いて試験する方法に関するものである。
【背景技術】
【0002】
医療技術の発達により、頭部に大規模な損傷を負っても一命を取り留める人が増えている。交通事故や、労働中の災害、スポーツなどにより頭部外傷を負う人は多く、実に交通事故で死亡する人の半数は頭部外傷が原因である。これらは頭蓋骨に外力がかかった際、直下に生じる脳挫傷、あるいは脳浮腫に起因している。脳浮腫とは、脳血管に存在する脳血液関門の破綻が原因であり、血漿成分が血管外に漏出して脳が腫れてくることである。近年この治療法として、低体温法が注目されているが、これは脳代謝を抑えることで脳浮腫の増悪や頭蓋内圧の上昇を抑制し、二次的な脳損傷を防ごうとする治療方法である。重要な治療方針ではあるが、しかし、低体温による心肺機能や免疫機構の低下による問題が生じる場合もある(非特許文献1を参照)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】N. Engl. J. Med., 2001; 344:556-563
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は脳の局所的な炎症の遷延による組織損傷を抑止し、予後の改善をはかることのできる化合物を提供することを目的とする。
本発明はまたそのような化合物をスクリーングする方法を提供することも目的とする。
【課題を解決するための手段】
【0005】
上記目的を達成するため本発明者は鋭意研究を行ない、次のような知見を得たことに基いて本発明を完成させた。
1)遺伝性であるか外傷性であるかを問わず脳損傷が生じた場合、H−PGDS及びDP受容体の発現が増加する。
2)H−PGDSは脳損傷の局所のミクログリア細胞やマクロファージにおいて、DP受容体は損傷部位の周辺のアストログリア細胞で発現が誘導される。
3)損傷部位ではマクロファージの集積、アストログリア細胞の活性化が顕著である。
4)H−PGDSの阻害剤又はDP受容体の拮抗剤を投与するとDP受容体の発現が減少し、アストログリア細胞の活性化が抑制される。
5)H−PGDS大量発現トランスジェニックマウスでは野性型マウスに比べ脳損傷が増悪される。
6)H−PGDS遺伝子欠損マウスとDP受容体欠損マウスでは野性型マウスに比べ損傷部位での出血やアストログリア細胞の活性化が軽微である。
【0006】
即ち、本発明は、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物を要旨とする。
【0007】
「脳損傷」には交通事故等による外傷性のものに止まらず、脳梗塞や脳出血等の脳血管障害によるもの、アルツハイマー病、多発性硬化症等を含む脳変性疾患、脱髄疾患等も含む。「脳損傷の治療または予防」とは、脳挫傷、脳浮腫、脳梗塞、脳出血、虚血性脳症、アルツハイマー病、多発性硬化症、脱髄疾患の治療または予防を包含する
【0008】
本発明は、プロスタグランジンD受容体の拮抗薬を有効成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0009】
本発明は、有効量の造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤を投与することを含む脳損傷の治療方法をも要旨とする。
【0010】
本発明は、脳損傷の治療用薬剤を製造するための、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤の使用をも要旨とする。
本発明は、有効量のプロスタグランジンD受容体の拮抗薬を投与することを含む脳損傷の治療方法をも要旨とする。
【0011】
本発明は、脳損傷の治療用薬剤を製造するための、プロスタグランジンD受容体の拮抗薬の使用をも要旨とする
【0012】
本発明は、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤およびプロスタグランジンD受容体の拮抗薬を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0013】
本発明は、1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物をトランスジェニックマウスに投与し、
3)該マウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する、
ことを含む脳損傷の治療または予防に用いる化合物のスクリーニング方法をも要旨とする。
【発明の効果】
【0014】
本発明の医薬組成物は、外傷性脳損傷、脳血管障害、脳変性疾患、または脱髄疾患等脳損傷の治療に有効である。また本発明のスクリーニング方法によれば脳損傷の治療または予防に用いることのできる化合物を簡単な方法でスクリーニングできる。
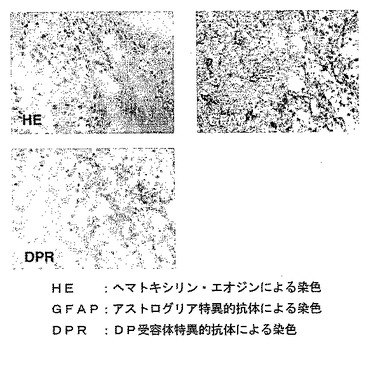
【図面の簡単な説明】
【0015】
【図1】Twitcherマウスの大脳と小脳におけるH−PGDSおよびDP受容体mRNAの発現レベルの変化を示すグラフである。
【図2】免疫組織染色法によるTwitcherマウスのミクログリア細胞やマクロファージ細胞に局在しているH−PGDSを示す模写図である。
【図3】活性化アストログリア細胞に発現したDP受容体を示す模写図である。
【図4】実験的自己免疫性脳脊髄炎マウスにおけるH−PGDSおよびDP受容体mRNAの発現レベルの変化を示すグラフである。
【図5】実験的自己免疫性脳脊髄炎マウスでのH−PGDS発現レベルの変化を示す模写図である。
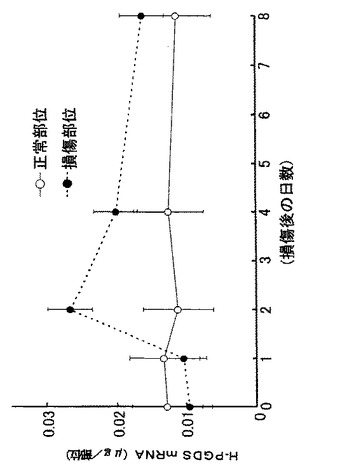
【図6】外傷性脳損傷モデルでのH−PGDSのmRNA発現量の経時変化を示すグラフである。
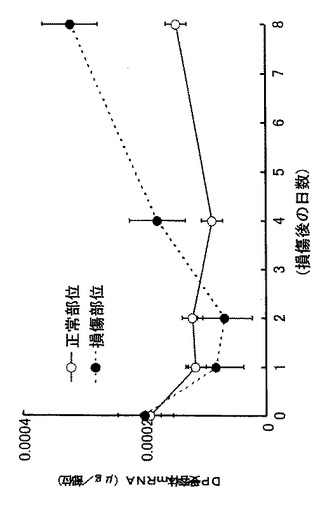
【図7】外傷性脳損傷モデルでのDP受容体のmRNA発現量の経時変化示すグラフである。
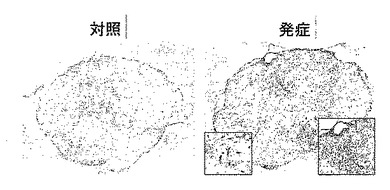
【図8】外傷性脳損傷モデルでの炎症の経時変化を示す模写図である。
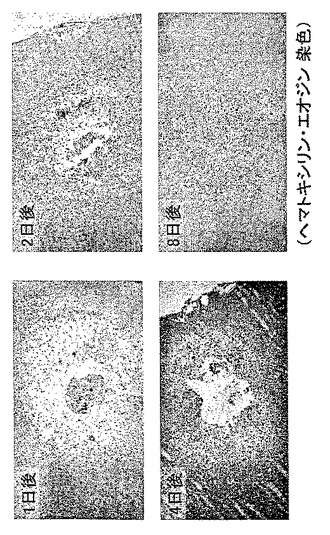
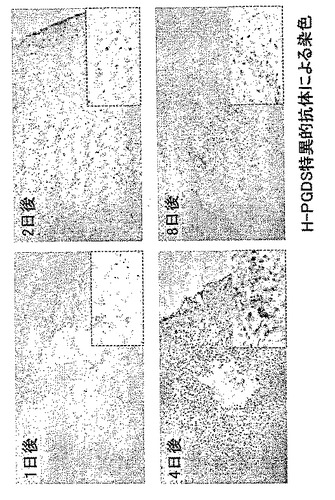
【図9】外傷性脳損傷モデルにおけるH−PGDS発現の経時変化を示す模写図である。
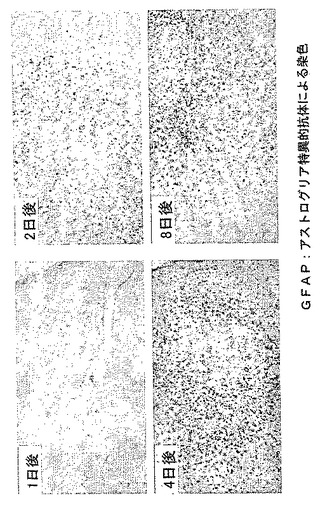
【図10】外傷性脳損傷モデルにおけるアストログリア細胞の活性化の経時変化を示す模写図である。
【図11】外傷性脳損傷周辺の活性化アストログリア細胞でのDP受容体の発現を示す模写図である。
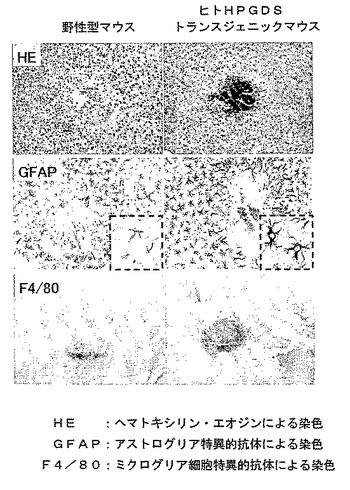
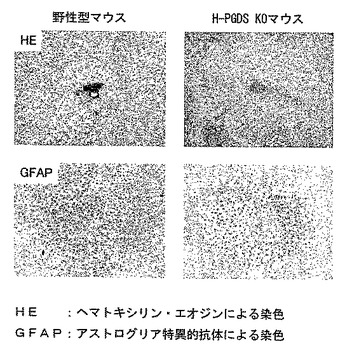
【図12】外傷性脳損傷から4日後の野生型マウスとヒトH−PGDS大量発現トランスジェニックマウスの脳損傷の比較を示す模写図である。
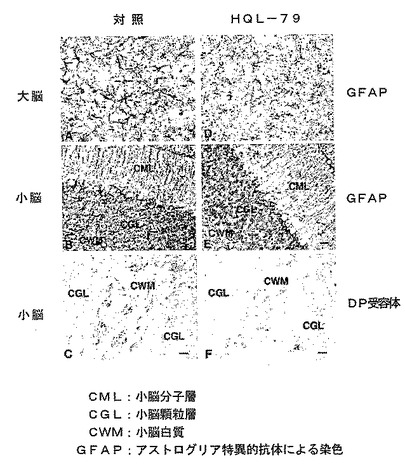
【図13】TwitcherマウスでのHQL−79によるアストログリア細胞の活性化とDP受容体発現の抑制を示す模写図である。
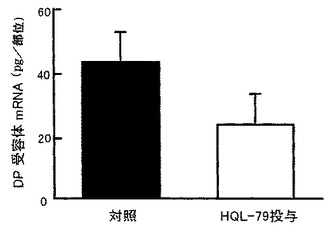
【図14】外傷性脳損傷後のDP受容体発現に対するHQL−79の抑制効果を示すグラフである。
【図15】外傷性脳損傷に対するHQL−79の回復促進効果を示す模写図である。
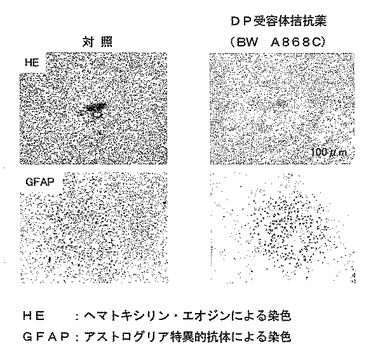
【図16】外傷性脳損傷に対するDP受容体拮抗薬の回復促進効果を示す模写図である。
【図17】外傷性脳損傷から4日後の野生型マウスとH−PGDS 遺伝子欠損マウスの脳損傷の比較を示す模写図である。
【図18】トランスジェニックマウスの作製に用いた導入ベクターの構造を示す図である。
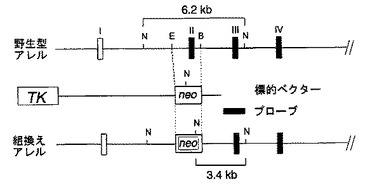
【図19】マウスH−PGDS遺伝子の構造(上段)、ターゲティングベクターにおける変異配列の構造(中段)および相同組換え後のマウスゲノムDNA(下段)を示す図である。
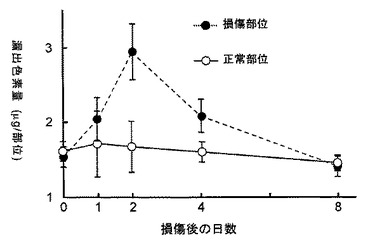
【図20】外傷性脳損傷モデルでの炎症の経時変化を組織へのエバンスブルー色素漏出反応を指標に調べた結果を示すグラフである。
【図21】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示すグラフである(傷害を与える1時間前からHQL−79の投与を開始した)。
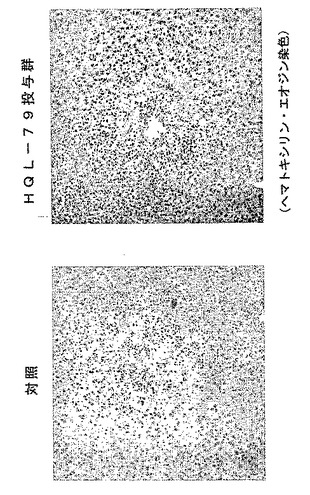
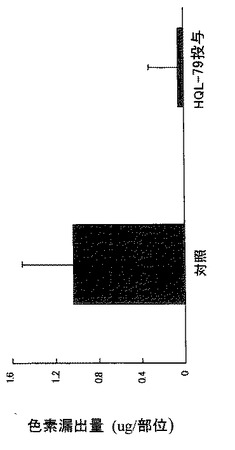
【図22】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示す模写図である(傷害を与えた1日後からHQL−79の投与を開始した)。
【図23】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示すグラフである(傷害を与えた1日後からHQL−79の投与を開始した)。
【図24】外傷性脳損傷に伴うエバンスブルー色素の漏出に対するピナグランジンの抑制効果を示すグラフである。
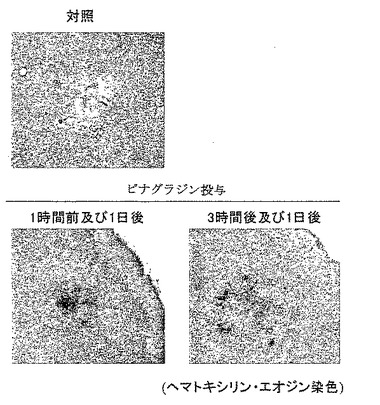
【図25】外傷性脳損傷に対するピナグランジンの進展抑制効果を示す模写図である。
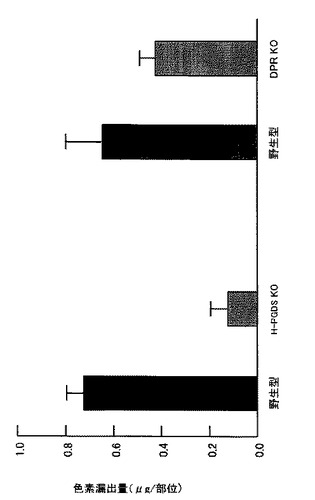
【図26】外傷性脳損傷から2日後の野生型マウスとHPGDS KOあるいはDPR KOマウスの脳損傷を傷害部位への色素漏出を指標に比較したグラフである。
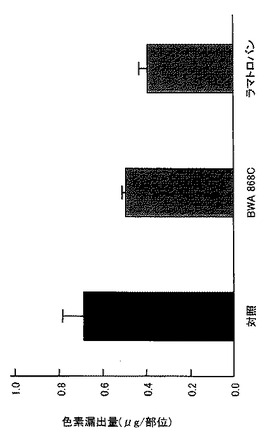
【図27】外傷性脳損傷後の組織傷害(色素漏出)に対するBWA 868Cおよびラマトロバンの抑制効果を示すグラフである。
【発明を実施するための形態】
【0016】
H−PGDS阻害剤の例は、4−ベンズヒドリルオキシ−1−{3−(1H−テトラゾール−5−イル)−プロピル}ピペリジン(HQL−79)、1−アミノ−4−{4−[4−クロロ−6−(2−スルホ−フェニルアミノ)−[1,3,5]トリアジン−2−イルメチル]−3−スルホ−フェニルアミノ}−9,10−ジオキソ−9,10−ジヒドロ−アントラセン−2−スルホン酸(チバクローンブルー)、1−アミノ−4−(4−スルファモイルアニリノ)−アントラキノン−2−スルホン酸 (PGD−042)もしくはその製薬上許容される塩またはそれらの水和物、2−(2’−ベンゾチアゾリル)−5−スチリルー3−(4’−フタルヒドラジディル)テトラゾリウム塩化物(PGD−016)またはそれらの水和物を含む。
【0017】
本明細書中、「製薬上許容される塩」とは、塩基性塩として、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩;N,N−ジベンジルエチレンジアミン等のアラルキルアミン塩;ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等のヘテロ環芳香族アミン塩;テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩、リジン塩等の塩基性アミノ酸塩等が挙げられる。酸性塩としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。これらの塩は、通常行われる方法によって形成させることができる。水和物を形成する時は、任意の数の水分子と配位していてもよい。
【0018】
本発明はまた、プロスタグランジンD受容体の拮抗薬を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0019】
プロスタグランジンD受容体の拮抗薬の例は、(±)−3−ベンジル−5−(6−カルボキシヘキシル)−1−(2−シクロヘキシル−2−ヒドロキシエチルアミノ)−ヒダントイン(BW A868C)、(+)−(3R)−3−(4−フルオロベンゼンスルホンアミド)−1,2,3,4−テトラヒドロカルバゾール−9−プロピオン酸(ラマトロバン)、(Z)−7−[(1R,2R,3S,5S)−2−(5−ヒドロキシベンンゾ[b]チオフェン−3−イルカルボニルアミノ)−10−ノルピナン−3−イル]ヘプタ−5−エン酸、(Z)−7−[(1R,2R,3S,5S)−2−(ベンゾ[b]チオフェン−3−イルカルボニルアミノ)−10−ノルピナン−3−イル]ヘプタ−5−エン酸(ピナグラジン)もしくはその製薬上許容される塩またはそれらの水和物である。
【0020】
更なるプロスタグランジンD受容体の拮抗薬の例は、式(I):
【化1】

(式中、
【化2】
Rは水素、アルキル、アルコキシ、ハロゲン、ヒドロキシ、アシルオキシまたは置換されていてもよいアリールスルホニルオキシを表わし、Xは水素またはアルキルを表わし、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物を含む。
【0021】
好ましい態様では、プロスタグランジンD受容体の拮抗薬は、式(IA):
【化3】
(式中、RおよびXは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物である。
【0022】
更に、好ましくはプロスタグランジンD受容体の拮抗薬は、式(IA−a):
【化4】
(式中、RおよびXは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物である
一般式(I)で表わされる化合物は公知であり以下のようにして製造できる。
【化5】
(式中、Y環、XおよびRは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
【0023】
一般式(I)で示される化合物は、上記反応式で示されるごとく、一般式(II)で示されるアミノ化合物に一般式(III)で示されるカルボン酸またはその反応性誘導体を反応させることにより製造することができる。
【0024】
本反応法における原料化合物(II)中、
【化6】
特公平6−23170号明細書に記載されている。また、
【化7】
特開昭61−49号明細書および特開平2−180862号明細書に記載されている。
【0025】
一般式(III)で示されるカルボン酸とは、4−ブロモベンゾ[b]チオフェン−3−カルボン酸、5−ブロモベンゾ[b]チオフェン−3−カルボン酸、6−ブロモベンゾ[b]チオフェン−3−カルボン酸、7−ブロモベンゾ[b]チオフェン−3−カルボン酸、5−フルオロベンゾ[b]チオフェン−3−カルボン酸、6−フルオロベンゾ[b]チオフェン−3−カルボン酸、4−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、5−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、6−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、7−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、5−アセトキシベンゾ[b]チオフェン−3−カルボン酸、ベンゾ[b]チオフェン−3−カルボン酸、5−ベンゾスルホニルオキシベンゾ[b]チオフェン−3−カルボン酸、5−メチルベンゾ[b]チオフェン−3−カルボン酸、6−メチルベンゾ[b]チオフェン−3−カルボン酸、5−メトキシベンゾ[b]チオフェン−3−カルボン酸、6−メトキシベンゾ[b]チオフェン−3−カルボン酸である。これらのカルボン酸は、それぞれ前記定義の置換基を有することができる。
【0026】
これらのカルボン酸は、日本化学雑誌88巻、7号、758−763(1967)、日本化学雑誌86巻、10号、1067-1072(1965)、J. Chem. Soc (c) 1899-1905(1967)、J.Heterocycle. Chem. 10巻、679-681(1973)、J.Heterocyclic Chem. 19巻、1131-1136(1982)、J. Med. Chem. 29巻、1637-1643(1986)記載の方法に準じて合成できるものである。
【0027】
一般式(III)で示されるカルボン酸の反応性誘導体とは、対応する酸ハロゲン化物(例えば、塩化物、臭化物、沃化物)、酸無水物(例えば、ぎ酸もしくは酢酸との混合酸無水物)、活性エステル(例えば、スクシンイミドエステル)などを意味し、通常アミノ基のアシル化に使用するアシル化剤を包含する。例えば、酸ハロゲン化物とするときは、ハロゲン化チオニル(例えば、塩化チオニル)、ハロゲン化リン(例えば、三塩化リン、五塩化リン)、ハロゲン化オギザリル(例えば、塩化オギザリル)等と公知の方法(例えば、新実験化学講座14巻1787頁(1978);Synthesis 852-854(1986);新実験化学講座22巻115頁(1992))に従って反応させればよい。
反応は通常のアミノ基のアシル化反応の条件に従って行えばよく、例えば、酸ハロゲン化物による縮合反応の場合、溶媒としてエーテル系溶媒(例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン)、ベンゼン系溶媒(例えば、ベンゼン、トルエン、キシレン)、ハロゲン化炭化水素系溶媒(例えば、ジクロロメタン、ジクロロエタン、クロロホルム)、その他、酢酸エチル、ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリルなどを使用し、要すれば塩基(例えば、トリエチルアミン、ピリジン、N、N−ジメチルアミノピリジン、N−メチルモルホリンなどの有機塩基、あるいは水酸化ナトリウム、水酸化カリウム、炭酸カリウムなどの無機塩基)の存在下、冷却下ないし室温あるいは加熱下、好ましくは−20℃ないし氷冷下あるいは室温ないし反応系の加熱還流温度で、数分ないし数10時間、好ましくは0.5時間ないし24時間、より好ましくは1時間ないし12時間実施すればよい。また、カルボン酸を反応性誘導体とはせずに、遊離のまま使用する場合には、アミンとカルボン酸の縮合反応に使用する縮合剤(例えば、ジシクロヘキシルカルボジイミド(DCC)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド、N,N’−カルボニルジイミダゾール)の存在下に反応させる。
本発明の医薬組成物は造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤およびプロスタグランジンD受容体の拮抗薬の両方を活性成分として用いてもよい。
【0028】
本発明で使用する脳損傷の治療または予防に用いる化合物は上記のようにH−PGDS阻害剤又はプロスタグランジンD受容体の拮抗薬から選択することもできるが、次のようにしてスクリーニングすることもできる。
すなわち。
1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物を該トランスジェニックマウスに投与し、
3)該マウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する。
ヒトH−PGDS大量発現トランスジェニックマウスの製造方法は2000年10月5日に出願された国際出願PCT/JP00/06963(WO 01/24627)に開示されている。その全体が本明細書の一部を構成する。
【0029】
製造例1
造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスの作製
WO 01/24627号に開示されている方法に従って造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを作製した。
ヒト細胞のmRNAから調製したcDNAライブラリーから、ラットH−PGDS遺伝子のcDNA(Cell 90:1085-10975,1997; GenBank Accession No. D82071)をプローブしてヒトH−PGDSのcDNA(Eur. J. Biochem. 267:3315-3322,2000; GenBank Accession No.NM-014485)をクローニングした。次にベクターpCAGGS(Gene 108: 193-199 (1991))のクローニング部位(Sal I/Not I)にヒトH−PGDSのcDNAを挿入結合し、導入ベクターを構築した。図18はこの導入ベクターにおける導入遺伝子の構成である。この導入遺伝子はCMVエンハンサーとチキンβ―アクチンプロモーターをH−PGDScDNAの上流に有しており、マウスの染色体に導入されると、これらのエンハンサーおよびプロモーターの作用によりH−PGDSmRNAを大量に発現される。この導入ベクターをマイクロインジェクション法によりFVBマウス(National Institute of Health Animal Genetic Resourceより入手)の受精卵に注入した。遺伝子導入受精卵は定法に従って仮親の卵管に移植し、個体へと発生させ出生させた。得られたマウスの尾部からDNAを抽出し、導入遺伝子の配列に基き合成されたプローブを用いて、サザンブロット法によりトランスジェニックマウスを選別した。
【0030】
製造例2
造血器型プロスタグランジンD合成酵素遺伝子欠損 (H−PGDS KO) マウスの作製
2002年1月28日に出願された特願2002−18666号に教示されている方法に従って造血器型プロスタグランジンD合成酵素遺伝子欠損マウスを作製した。
公知のマウスH−PGDS遺伝子のエクソンII(H−PGDSの蛋白質翻訳開始領域)を含む領域をNeor遺伝子に置換し、さらにH−PGDS遺伝子の約7Kb上流にヘルペスウイルスのサイミジンカイネース遺伝子(HSV−tk遺伝子)を組み込んで変異配列を調製し、この変異配列をベクターに組み込んでターゲティング・ベクターを作製した(図19参照)。
電気穿孔法により、未分化の培養ES細胞(1.2×107個)にターゲティング・ベクターを48μg/mlの割合で導入して遺伝子導入ES細胞を得た。これらの細胞をプレートに播き、2日後にG418およびガンシクロビルを培地に添加して更に7日間培養し、G418およびガンシクロビルに耐性を示すコロニーを得た。これらのコロニーを個別に分離し、さらに培養したのち、DNAを抽出してサザンブロッティングにより相同組換えES細胞を選別した。
次いで、この相同組換えES細胞を、C57BL/6系マウスの胚盤胞へ常法により注入し、仮親マウスへ移植して個体へと発生させた。
その結果、10匹のキメラマウスを得た。得られたキメラマウスのうち、雄の個体と雌の野生型C57BL/6系マウスとを交配させて初代(F1)マウスを得た。これらのF1マウスから、サザンブロット分析により2倍体染色体の一方に変異配列が確認された個体(♂、♀)を選別し、これらを交配させて第2世代(F2)マウスを得た。
最終的に、これらF2マウスから、サザンブロット分析により2倍体染色体の両方に変異配列が確認された個体(ホモ接合体)および片方に変異配列が確認された個体(ヘテロ接合体)を選別しH−PGDS遺伝子欠損マウスを作製した。
【実施例1】
【0031】
実施例1
遺伝性脱髄疾患における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
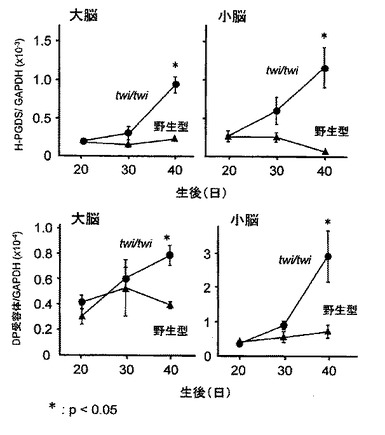
Galactosylceramidase欠損症であるヒトKrabbe病のモデルマウスTwitcher(Kobayashi T, et al., Brain Res., 202:479-483, 1980; Duchen LW,et al., Brain, 103:695-710, 1980; Sakai N, et al., J. Neurochem., 66:1118-1124, 1996; Taniike M. et al., J. Neuropathol. Exp. Neurol., 58:644-653, 1999)を用いて、遺伝性の脱髄による脳損傷に伴うH−PGDSとDP受容体のmRNAの変化を、定量的RT−PCR法により定量した(図1参照)。H−PGDSとDP受容体のmRNAの発現量は、共に、脱髄による脳損傷に伴い増加する。
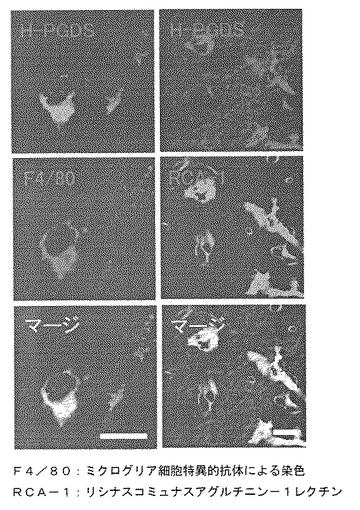
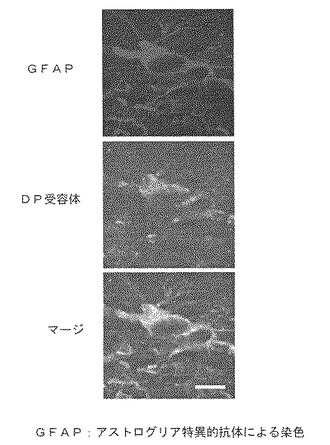
免疫組織染色法により、H−PGDSはミクログリア細胞と脱髄の進んだ組織局所に集積するAmeboid細胞やマクロファージ細胞に発現することを同定した(図2参照)。一方、DP受容体は、脱髄の進んだ組織の周辺に分布する活性化されたアストログリア細胞に発現することを同定した(図3参照)。
【実施例2】
【0032】
実施例2
自己免疫性脱髄疾患における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
ヒト多発性硬化症のモデルである実験的自己免疫性脳脊髄炎マウス(Ichikawa M., et al., Cell Immunol., 191:97-104, 1999; Bernhard Hemmer, et al., Nature Review Neuroscience, 3:291-301, 2002)においても、定量的RT−PCR法により測定したH−PGDSとDP受容体のmRNAの発現量は、共に、脱髄による脳損傷と相関した増加を示す(図4参照)。
免疫組織染色法による観察では、H−PGDSはミクログリア細胞と脱髄の進んだ組織局所に集積するAmeboid細胞やマクロファージ細胞に発現する(図5参照)。
【実施例3】
【0033】
実施例3
外傷性脳損傷における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
外傷性大脳皮質傷害(Stab wound)モデル(Salhia B, et al., Brain Res., 888:87-97, 2000; Asahi M., et al., J. Neurosci., 21:7724-7732, 2001; Garcia de Yebenes E., et al., J. Neurochem., 73:812-820, 1999)を用いて、脳損傷におけるH−PGDSとDP受容体のmRNAの発現を調べた結果、H−PGDSは損傷後2日目に最大値をとり(図6参照)、DP受容体は2日目から8日目にかけて持続的に増加した(図7参照)。
損傷の24時間後から傷害部位周囲に集積するミクログリア細胞とマクロファージでH−PGDSの誘導が起こり(図8と図9参照)、傷害部位周辺のアストログリア細胞ではGFAPとDP受容体の発現が増強し、これらの現象は損傷の8日後まで持続した(図10と図11参照)。
傷害の程度を損傷部位への色素(エバンスブルー)の漏出反応 (Kakimura Y, et al., Nature Medicine, 4:1078-1080, 1998)によって定量化した。損傷後2日目に最大値をとりその後治癒に伴って低下した(図20参照)。
【実施例4】
【0034】
実施例4
造血器型プロスタグランジンD合成酵素阻害剤投与による遺伝性脱髄疾患におけるアストログリア細胞の活性化の抑制
Twitcherマウスに、H−PGDS阻害剤であるHQL−79(4−ベンズヒドリルオキシ−1−{3−(1H−テトラゾール−5−イル)−プロピル}ピペリジン)を30mg/kg/日の用量で、14日間、皮下投与すると、アストログリア細胞の活性化が抑制され、同時に、アストログリア細胞でのDP受容体の発現が低下した(図13参照)。
【実施例5】
【0035】
実施例5
造血器型プロスタグランジンD合成酵素阻害剤投与による外傷性脳損傷におけるDP受容体の誘導の抑制と脳損傷の回復促進
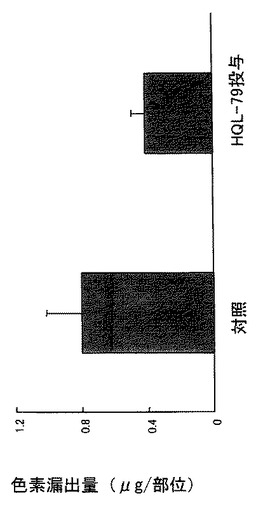
H−PGDS阻害剤であるHQL−79を30mg/kg/日の用量で、傷害を与える1時間前から4日間、マウスに経口投与すると、Stab woundモデルにおける組織損傷領域でのDP受容体mRNA量は低下し(図14参照)、脳損傷の回復促進が認められた(図15参照)。この治療効果は傷害部位への色素漏出反応を指標とした実験によっても確認された(図21参照)。
さらに、傷害を与えた後からH−PGDS阻害剤の投与を開始しても損傷の拡大を抑制することが確認された(図22、23参照)。
【実施例6】
【0036】
実施例6
プロスタグランジンD受容体拮抗薬投与による外傷性脳損傷の軽減
Stab wound モデルマウスに、DP受容体拮抗薬であるBW A868C ((±)−3−ベンジル−5−(6−カルボキシヘキシル)−1−(2−シクロヘキシル−2−ヒドロキシエチルアミノ)ヒダントインを損傷の当日から1 mg/kg/日の用量で4日間、静脈内投与すると脳損傷の回復促進が認められ、組織損傷部位周辺のアストログリアの活性化が抑制された(図16参照)。
Stab woundモデルマウスに対するプロスタグランジンD受容体拮抗薬であるBW A868Cあるいはラマトロバン((+)−(3R)−3−(4−フルオロベンゼンスルホンアミド)−1,2,3,4−テトラヒドロカルバゾール−9−プロピオン酸)の効果を損傷部位への色素漏出量を指標に評価した。すなわち、脳損傷惹起2日後にエバンスブルー色素を静脈内投与し、その後2時間の組織への色素漏出量を測定した。BW A868C(1mg/kg)を脳損傷惹起3時間後および1日後に静脈内投与すると傷害部位への色素漏出を抑制した(図27参照)。
ラマトロバン(30mg/kg)を惹起3時間後および1日後に経口投与すると傷害部位への色素漏出を抑制した(図27参照)。
【実施例7】
【0037】
実施例7
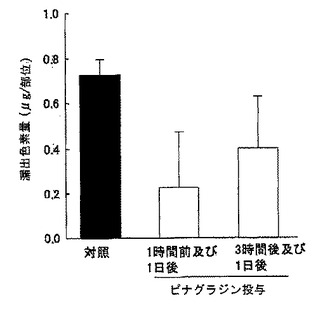
Stab wound モデルに対するDP受容体拮抗薬であるピナグラジンの効果を損傷部位への色素漏出量で評価した。すなわち、脳損傷惹起2日後にエバンスブルー色素を静注し、その後2時間における色素の組織中漏出量を測定した。その結果、ピナグラジン(10mg/kg)を脳損傷惹起1時間前および1日後に経口投与すると、脳損傷に伴って認められるエバンスブルー色素漏出量の増加が抑制された。またピナグラジン(10mg/kg)は惹起3時間および1日後の後処置によっても色素の漏出を抑制した(図24参照)。さらに病理組織学的検討においてピナグラジンはいずれの投与方法によってもStab wound モデルにおける脳損傷の進展を抑制した(図25参照)。
【実施例8】
【0038】
実施例8
ヒト造血器型プロスタグランジンD合成酵素大量発現による外傷性脳損傷の増悪
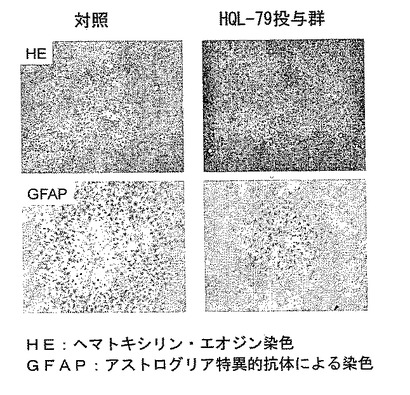
製造例1で作成したヒトH−PGDS大量発現トランスジェニックマウスを用いたStab woundモデルでは、損傷部位でのマクロファージの集積、および、抗GFAP抗体を用いて免疫組織化学的に調べたアストログリア細胞の活性化が、野生型マウスに比べて顕著であり治癒は遅延した(図12参照)。
【実施例9】
【0039】
実施例9
造血器型プロスタグランジンD合成酵素遺伝子欠損による外傷性脳損傷の軽減
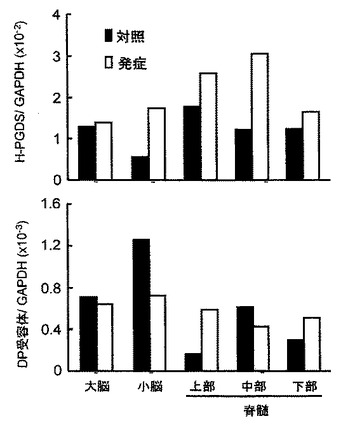
製造例2で作製した造血器型プロスタグランジンD合成酵素遺伝子欠損(HPGDS KO)マウス(ホモ接合体)を用いたStab wound モデルでは、損傷部位での出血、抗 GFAP 抗体を用いて免疫組織化学的に調べたアストログリアの活性化が、さらに傷害部位での色素漏出が野生型マウスに比べて軽微であった(図17および図26参照)。
一方、DP受容体遺伝子欠損(DPR KO)マウス(Matsuoka T, et al., Science, 17;287(5460):2013-2017, 2000)を用いたStab woundモデルでは傷害部位での色素漏出が野生型マウスに比べて軽微であった(図26参照)。
【0040】
本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬を治療に用いるには、通常の経口又は非経口投与用の製剤として製剤化する。本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬を含有する医薬組成物は、経口及び非経口投与のための剤形をとることができる。即ち、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などの経口投与製剤、あるいは、静脈注射、筋肉注射、皮下注射などの注射用溶液又は懸濁液、吸入薬、点眼薬、点鼻薬、坐剤、もしくは軟膏剤、貼布剤、パップ剤などの経皮投与用製剤、経皮吸収剤などの非経口製剤とすることもできる。好ましくは、経口剤または注射用薬剤として用いる。
【0041】
これらの製剤は当業者既知の適当な担体、賦形剤、溶媒、基剤等を用いて製造することができる。例えば、錠剤の場合、活性成分と補助成分を一緒に圧縮又は成型する。補助成分としては、製剤的に許容される賦形剤、例えば結合剤(例、トウモロコシでん粉)、充填剤(例、ラクトース、微結晶性セルロース)、崩壊剤(例、でん粉グリコール酸ナトリウム)又は滑沢剤(例、ステアリン酸マグネシウム)などが用いられる。錠剤は、適宜、コーティングしてもよい。シロップ剤、液剤、懸濁剤などの液体製剤の場合、例えば、懸濁化剤(例、メチルセルロース)、乳化剤(例、レシチン)、保存剤などを用いる。注射用製剤の場合、溶液、懸濁液又は油性もしくは水性乳濁液の形態のいずれでもよく、これらは懸濁安定剤又は分散剤などを含有していてもよい。軟膏剤、貼布剤、パップ剤などの経皮投与用製剤、経皮吸収剤の場合は、水性基剤(水、低級アルコール、ポリオール)または油性基剤(高級脂肪酸エステル類(イソプロピルミリステート)、親油性アルコール)を用いて製剤化する。
【0042】
本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬は、投与形態、患者の症状、年令、体重、性別、あるいは併用される薬物(あるとすれば)などにより異なり、最終的には医師の判断に委ねられるが、経口投与の場合、体重1kgあたり、1日0.01〜100mg、好ましくは0.01〜50mg、より好ましくは0.01〜30mg、非経口投与の場合、体重1kgあたり、1日0.001〜100mg、好ましくは0.001〜5mg、より好ましくは0.001〜3mgを投与する。これを1〜4回に分割して投与すればよい。
【0043】
製剤例
製剤例1
以下の成分を含有する錠剤を製造する:
式(I)で表わされる化合物 10 mg
乳糖 90 mg
微結晶セルロース 30 mg
CMC−Na 15 mg
ステアリン酸マグネシウム 5 mg
150 mg
式(I)で表わされる化合物、乳糖、微結晶セルロース、CMC−Na(カルボキシメチルセルロース ナトリウム塩)を60メッシュのふるいに通し、混合する。混合末にステアリン酸マグネシウム混合し、製錠用混合末を得る。本混合末を直打し、150mgの錠剤を得る。
【0044】
製剤例2
活性成分50mgを含む懸濁剤は次のように製造する:
式(I)で表わされる化合物 50mg
ナトリウムカルボキシメチルセルロース 50mg
シロップ 1.25ml
安息香酸溶液 0.10ml
香料 q.v.
色素 q.v.
精製水を加え合計 5ml
活性成分をNo.45メッシュU.S.のふるいにかけ、ナトリウムカルボキシメチルセルロースおよびシロップと混合して滑らかなペーストにする。安息香酸溶液および香料を水の一部で希釈して加え、攪拌する。ついで水を十分量加えて必要な体積にする。
【0045】
製剤例3
静脈用製剤は次のように製造する:
式(I)で表わされる化合物 100mg
飽和脂肪酸グリセリド 1000ml
上記成分の溶液は通常、1分間に1mlの速度で患者に静脈内投与される。
【0046】
製剤例4
常法により次の組成のゼラチン硬カプセル剤を調製した。
HQL−79 10mg
デンプン 50mg
ステアリン酸マグネシウム 10mg
【0047】
製剤例5
常法により次の組成の錠剤を調製した。
HQL−79 10mg
セルロース、微晶質 500mg
二酸化ケイ素 10mg
ステアリン酸マグネシウム 10mg
【産業上の利用可能性】
【0048】
以上詳述したように、本発明の組成物は脳血管障害、脳変性疾患、脱髄疾患等の疾患の処置に使用できる。
脳損傷局所のミクログリア細胞やマクロファージで誘導されるH−PGDSを阻害したり、損傷部位の周辺のアストログリア細胞で発現するDP受容体の活性化を阻害することにより、プロスタグランジンD2が関与する脳損傷を治療または予防することが可能となる。
プロスタグランジンD2は多発性硬化症患者で生合成が活発であり(Science 294:1731-11735, 2001)、そのモデルである実験的自己免疫性脳脊髄炎マウスの発症過程で増悪因子として働いている可能性が高いので、H−PGDSの特異的な阻害剤や受容体拮抗薬を用いると、現在行われているステロイド療法や免疫抑制剤に代わる、多発性硬化症の治療や予防に有益な療法になりうる。さらにこれらの薬剤は、自己免疫性の細胞障害や神経細胞死に伴う局所炎症反応を抑止することで、アルツハイマー病を含む神経原繊維変化を伴う難治疾患の治療にも応用できる。
【技術分野】
【0001】
この発明は、脳損傷を治療または予防する化合物及びそのスクリーニング方法に関するものである。さらに詳しくは、この発明は、脳血管障害、脳変性疾患、脱髄疾患等の疾患による脳損傷部位のミクログリア細胞やマクロファージにおいて誘導される造血器型プロスタグランジンD合成酵素(以下、「H−PGDS」ということがある)を阻害したり、損傷部位の周辺のアストログリア細胞で発現するプロスタグランジンD受容体(以下、「DP受容体」ということがある)の活性化を阻害することにより、プロスタグランジンD2が関与する脳損傷を治療または予防する化合物と、これらの物質の薬効を、ヒト造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを用いて試験する方法に関するものである。
【背景技術】
【0002】
医療技術の発達により、頭部に大規模な損傷を負っても一命を取り留める人が増えている。交通事故や、労働中の災害、スポーツなどにより頭部外傷を負う人は多く、実に交通事故で死亡する人の半数は頭部外傷が原因である。これらは頭蓋骨に外力がかかった際、直下に生じる脳挫傷、あるいは脳浮腫に起因している。脳浮腫とは、脳血管に存在する脳血液関門の破綻が原因であり、血漿成分が血管外に漏出して脳が腫れてくることである。近年この治療法として、低体温法が注目されているが、これは脳代謝を抑えることで脳浮腫の増悪や頭蓋内圧の上昇を抑制し、二次的な脳損傷を防ごうとする治療方法である。重要な治療方針ではあるが、しかし、低体温による心肺機能や免疫機構の低下による問題が生じる場合もある(非特許文献1を参照)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】N. Engl. J. Med., 2001; 344:556-563
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は脳の局所的な炎症の遷延による組織損傷を抑止し、予後の改善をはかることのできる化合物を提供することを目的とする。
本発明はまたそのような化合物をスクリーングする方法を提供することも目的とする。
【課題を解決するための手段】
【0005】
上記目的を達成するため本発明者は鋭意研究を行ない、次のような知見を得たことに基いて本発明を完成させた。
1)遺伝性であるか外傷性であるかを問わず脳損傷が生じた場合、H−PGDS及びDP受容体の発現が増加する。
2)H−PGDSは脳損傷の局所のミクログリア細胞やマクロファージにおいて、DP受容体は損傷部位の周辺のアストログリア細胞で発現が誘導される。
3)損傷部位ではマクロファージの集積、アストログリア細胞の活性化が顕著である。
4)H−PGDSの阻害剤又はDP受容体の拮抗剤を投与するとDP受容体の発現が減少し、アストログリア細胞の活性化が抑制される。
5)H−PGDS大量発現トランスジェニックマウスでは野性型マウスに比べ脳損傷が増悪される。
6)H−PGDS遺伝子欠損マウスとDP受容体欠損マウスでは野性型マウスに比べ損傷部位での出血やアストログリア細胞の活性化が軽微である。
【0006】
即ち、本発明は、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物を要旨とする。
【0007】
「脳損傷」には交通事故等による外傷性のものに止まらず、脳梗塞や脳出血等の脳血管障害によるもの、アルツハイマー病、多発性硬化症等を含む脳変性疾患、脱髄疾患等も含む。「脳損傷の治療または予防」とは、脳挫傷、脳浮腫、脳梗塞、脳出血、虚血性脳症、アルツハイマー病、多発性硬化症、脱髄疾患の治療または予防を包含する
【0008】
本発明は、プロスタグランジンD受容体の拮抗薬を有効成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0009】
本発明は、有効量の造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤を投与することを含む脳損傷の治療方法をも要旨とする。
【0010】
本発明は、脳損傷の治療用薬剤を製造するための、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤の使用をも要旨とする。
本発明は、有効量のプロスタグランジンD受容体の拮抗薬を投与することを含む脳損傷の治療方法をも要旨とする。
【0011】
本発明は、脳損傷の治療用薬剤を製造するための、プロスタグランジンD受容体の拮抗薬の使用をも要旨とする
【0012】
本発明は、造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤およびプロスタグランジンD受容体の拮抗薬を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0013】
本発明は、1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物をトランスジェニックマウスに投与し、
3)該マウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する、
ことを含む脳損傷の治療または予防に用いる化合物のスクリーニング方法をも要旨とする。
【発明の効果】
【0014】
本発明の医薬組成物は、外傷性脳損傷、脳血管障害、脳変性疾患、または脱髄疾患等脳損傷の治療に有効である。また本発明のスクリーニング方法によれば脳損傷の治療または予防に用いることのできる化合物を簡単な方法でスクリーニングできる。
【図面の簡単な説明】
【0015】
【図1】Twitcherマウスの大脳と小脳におけるH−PGDSおよびDP受容体mRNAの発現レベルの変化を示すグラフである。
【図2】免疫組織染色法によるTwitcherマウスのミクログリア細胞やマクロファージ細胞に局在しているH−PGDSを示す模写図である。
【図3】活性化アストログリア細胞に発現したDP受容体を示す模写図である。
【図4】実験的自己免疫性脳脊髄炎マウスにおけるH−PGDSおよびDP受容体mRNAの発現レベルの変化を示すグラフである。
【図5】実験的自己免疫性脳脊髄炎マウスでのH−PGDS発現レベルの変化を示す模写図である。
【図6】外傷性脳損傷モデルでのH−PGDSのmRNA発現量の経時変化を示すグラフである。
【図7】外傷性脳損傷モデルでのDP受容体のmRNA発現量の経時変化示すグラフである。
【図8】外傷性脳損傷モデルでの炎症の経時変化を示す模写図である。
【図9】外傷性脳損傷モデルにおけるH−PGDS発現の経時変化を示す模写図である。
【図10】外傷性脳損傷モデルにおけるアストログリア細胞の活性化の経時変化を示す模写図である。
【図11】外傷性脳損傷周辺の活性化アストログリア細胞でのDP受容体の発現を示す模写図である。
【図12】外傷性脳損傷から4日後の野生型マウスとヒトH−PGDS大量発現トランスジェニックマウスの脳損傷の比較を示す模写図である。
【図13】TwitcherマウスでのHQL−79によるアストログリア細胞の活性化とDP受容体発現の抑制を示す模写図である。
【図14】外傷性脳損傷後のDP受容体発現に対するHQL−79の抑制効果を示すグラフである。
【図15】外傷性脳損傷に対するHQL−79の回復促進効果を示す模写図である。
【図16】外傷性脳損傷に対するDP受容体拮抗薬の回復促進効果を示す模写図である。
【図17】外傷性脳損傷から4日後の野生型マウスとH−PGDS 遺伝子欠損マウスの脳損傷の比較を示す模写図である。
【図18】トランスジェニックマウスの作製に用いた導入ベクターの構造を示す図である。
【図19】マウスH−PGDS遺伝子の構造(上段)、ターゲティングベクターにおける変異配列の構造(中段)および相同組換え後のマウスゲノムDNA(下段)を示す図である。
【図20】外傷性脳損傷モデルでの炎症の経時変化を組織へのエバンスブルー色素漏出反応を指標に調べた結果を示すグラフである。
【図21】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示すグラフである(傷害を与える1時間前からHQL−79の投与を開始した)。
【図22】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示す模写図である(傷害を与えた1日後からHQL−79の投与を開始した)。
【図23】外傷性脳損傷後の組織傷害(色素漏出)に対するHQL−79の抑制効果を示すグラフである(傷害を与えた1日後からHQL−79の投与を開始した)。
【図24】外傷性脳損傷に伴うエバンスブルー色素の漏出に対するピナグランジンの抑制効果を示すグラフである。
【図25】外傷性脳損傷に対するピナグランジンの進展抑制効果を示す模写図である。
【図26】外傷性脳損傷から2日後の野生型マウスとHPGDS KOあるいはDPR KOマウスの脳損傷を傷害部位への色素漏出を指標に比較したグラフである。
【図27】外傷性脳損傷後の組織傷害(色素漏出)に対するBWA 868Cおよびラマトロバンの抑制効果を示すグラフである。
【発明を実施するための形態】
【0016】
H−PGDS阻害剤の例は、4−ベンズヒドリルオキシ−1−{3−(1H−テトラゾール−5−イル)−プロピル}ピペリジン(HQL−79)、1−アミノ−4−{4−[4−クロロ−6−(2−スルホ−フェニルアミノ)−[1,3,5]トリアジン−2−イルメチル]−3−スルホ−フェニルアミノ}−9,10−ジオキソ−9,10−ジヒドロ−アントラセン−2−スルホン酸(チバクローンブルー)、1−アミノ−4−(4−スルファモイルアニリノ)−アントラキノン−2−スルホン酸 (PGD−042)もしくはその製薬上許容される塩またはそれらの水和物、2−(2’−ベンゾチアゾリル)−5−スチリルー3−(4’−フタルヒドラジディル)テトラゾリウム塩化物(PGD−016)またはそれらの水和物を含む。
【0017】
本明細書中、「製薬上許容される塩」とは、塩基性塩として、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩;N,N−ジベンジルエチレンジアミン等のアラルキルアミン塩;ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等のヘテロ環芳香族アミン塩;テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩、リジン塩等の塩基性アミノ酸塩等が挙げられる。酸性塩としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。これらの塩は、通常行われる方法によって形成させることができる。水和物を形成する時は、任意の数の水分子と配位していてもよい。
【0018】
本発明はまた、プロスタグランジンD受容体の拮抗薬を活性成分として含む、脳損傷の治療または予防に用いる医薬組成物をも要旨とする。
【0019】
プロスタグランジンD受容体の拮抗薬の例は、(±)−3−ベンジル−5−(6−カルボキシヘキシル)−1−(2−シクロヘキシル−2−ヒドロキシエチルアミノ)−ヒダントイン(BW A868C)、(+)−(3R)−3−(4−フルオロベンゼンスルホンアミド)−1,2,3,4−テトラヒドロカルバゾール−9−プロピオン酸(ラマトロバン)、(Z)−7−[(1R,2R,3S,5S)−2−(5−ヒドロキシベンンゾ[b]チオフェン−3−イルカルボニルアミノ)−10−ノルピナン−3−イル]ヘプタ−5−エン酸、(Z)−7−[(1R,2R,3S,5S)−2−(ベンゾ[b]チオフェン−3−イルカルボニルアミノ)−10−ノルピナン−3−イル]ヘプタ−5−エン酸(ピナグラジン)もしくはその製薬上許容される塩またはそれらの水和物である。
【0020】
更なるプロスタグランジンD受容体の拮抗薬の例は、式(I):
【化1】
(式中、
【化2】
Rは水素、アルキル、アルコキシ、ハロゲン、ヒドロキシ、アシルオキシまたは置換されていてもよいアリールスルホニルオキシを表わし、Xは水素またはアルキルを表わし、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物を含む。
【0021】
好ましい態様では、プロスタグランジンD受容体の拮抗薬は、式(IA):
【化3】
(式中、RおよびXは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物である。
【0022】
更に、好ましくはプロスタグランジンD受容体の拮抗薬は、式(IA−a):
【化4】
(式中、RおよびXは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
で示される化合物もしくはその製薬上許容される塩またはそれらの水和物である
一般式(I)で表わされる化合物は公知であり以下のようにして製造できる。
【化5】
(式中、Y環、XおよびRは前記と同意義であり、α鎖の二重結合はE配置またはZ配置を表わす)
【0023】
一般式(I)で示される化合物は、上記反応式で示されるごとく、一般式(II)で示されるアミノ化合物に一般式(III)で示されるカルボン酸またはその反応性誘導体を反応させることにより製造することができる。
【0024】
本反応法における原料化合物(II)中、
【化6】
特公平6−23170号明細書に記載されている。また、
【化7】
特開昭61−49号明細書および特開平2−180862号明細書に記載されている。
【0025】
一般式(III)で示されるカルボン酸とは、4−ブロモベンゾ[b]チオフェン−3−カルボン酸、5−ブロモベンゾ[b]チオフェン−3−カルボン酸、6−ブロモベンゾ[b]チオフェン−3−カルボン酸、7−ブロモベンゾ[b]チオフェン−3−カルボン酸、5−フルオロベンゾ[b]チオフェン−3−カルボン酸、6−フルオロベンゾ[b]チオフェン−3−カルボン酸、4−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、5−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、6−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、7−ヒドロキシベンゾ[b]チオフェン−3−カルボン酸、5−アセトキシベンゾ[b]チオフェン−3−カルボン酸、ベンゾ[b]チオフェン−3−カルボン酸、5−ベンゾスルホニルオキシベンゾ[b]チオフェン−3−カルボン酸、5−メチルベンゾ[b]チオフェン−3−カルボン酸、6−メチルベンゾ[b]チオフェン−3−カルボン酸、5−メトキシベンゾ[b]チオフェン−3−カルボン酸、6−メトキシベンゾ[b]チオフェン−3−カルボン酸である。これらのカルボン酸は、それぞれ前記定義の置換基を有することができる。
【0026】
これらのカルボン酸は、日本化学雑誌88巻、7号、758−763(1967)、日本化学雑誌86巻、10号、1067-1072(1965)、J. Chem. Soc (c) 1899-1905(1967)、J.Heterocycle. Chem. 10巻、679-681(1973)、J.Heterocyclic Chem. 19巻、1131-1136(1982)、J. Med. Chem. 29巻、1637-1643(1986)記載の方法に準じて合成できるものである。
【0027】
一般式(III)で示されるカルボン酸の反応性誘導体とは、対応する酸ハロゲン化物(例えば、塩化物、臭化物、沃化物)、酸無水物(例えば、ぎ酸もしくは酢酸との混合酸無水物)、活性エステル(例えば、スクシンイミドエステル)などを意味し、通常アミノ基のアシル化に使用するアシル化剤を包含する。例えば、酸ハロゲン化物とするときは、ハロゲン化チオニル(例えば、塩化チオニル)、ハロゲン化リン(例えば、三塩化リン、五塩化リン)、ハロゲン化オギザリル(例えば、塩化オギザリル)等と公知の方法(例えば、新実験化学講座14巻1787頁(1978);Synthesis 852-854(1986);新実験化学講座22巻115頁(1992))に従って反応させればよい。
反応は通常のアミノ基のアシル化反応の条件に従って行えばよく、例えば、酸ハロゲン化物による縮合反応の場合、溶媒としてエーテル系溶媒(例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン)、ベンゼン系溶媒(例えば、ベンゼン、トルエン、キシレン)、ハロゲン化炭化水素系溶媒(例えば、ジクロロメタン、ジクロロエタン、クロロホルム)、その他、酢酸エチル、ジメチルホルムアミド、ジメチルスルホキシド、アセトニトリルなどを使用し、要すれば塩基(例えば、トリエチルアミン、ピリジン、N、N−ジメチルアミノピリジン、N−メチルモルホリンなどの有機塩基、あるいは水酸化ナトリウム、水酸化カリウム、炭酸カリウムなどの無機塩基)の存在下、冷却下ないし室温あるいは加熱下、好ましくは−20℃ないし氷冷下あるいは室温ないし反応系の加熱還流温度で、数分ないし数10時間、好ましくは0.5時間ないし24時間、より好ましくは1時間ないし12時間実施すればよい。また、カルボン酸を反応性誘導体とはせずに、遊離のまま使用する場合には、アミンとカルボン酸の縮合反応に使用する縮合剤(例えば、ジシクロヘキシルカルボジイミド(DCC)、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド、N,N’−カルボニルジイミダゾール)の存在下に反応させる。
本発明の医薬組成物は造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤およびプロスタグランジンD受容体の拮抗薬の両方を活性成分として用いてもよい。
【0028】
本発明で使用する脳損傷の治療または予防に用いる化合物は上記のようにH−PGDS阻害剤又はプロスタグランジンD受容体の拮抗薬から選択することもできるが、次のようにしてスクリーニングすることもできる。
すなわち。
1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物を該トランスジェニックマウスに投与し、
3)該マウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する。
ヒトH−PGDS大量発現トランスジェニックマウスの製造方法は2000年10月5日に出願された国際出願PCT/JP00/06963(WO 01/24627)に開示されている。その全体が本明細書の一部を構成する。
【0029】
製造例1
造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスの作製
WO 01/24627号に開示されている方法に従って造血器型プロスタグランジンD合成酵素大量発現トランスジェニックマウスを作製した。
ヒト細胞のmRNAから調製したcDNAライブラリーから、ラットH−PGDS遺伝子のcDNA(Cell 90:1085-10975,1997; GenBank Accession No. D82071)をプローブしてヒトH−PGDSのcDNA(Eur. J. Biochem. 267:3315-3322,2000; GenBank Accession No.NM-014485)をクローニングした。次にベクターpCAGGS(Gene 108: 193-199 (1991))のクローニング部位(Sal I/Not I)にヒトH−PGDSのcDNAを挿入結合し、導入ベクターを構築した。図18はこの導入ベクターにおける導入遺伝子の構成である。この導入遺伝子はCMVエンハンサーとチキンβ―アクチンプロモーターをH−PGDScDNAの上流に有しており、マウスの染色体に導入されると、これらのエンハンサーおよびプロモーターの作用によりH−PGDSmRNAを大量に発現される。この導入ベクターをマイクロインジェクション法によりFVBマウス(National Institute of Health Animal Genetic Resourceより入手)の受精卵に注入した。遺伝子導入受精卵は定法に従って仮親の卵管に移植し、個体へと発生させ出生させた。得られたマウスの尾部からDNAを抽出し、導入遺伝子の配列に基き合成されたプローブを用いて、サザンブロット法によりトランスジェニックマウスを選別した。
【0030】
製造例2
造血器型プロスタグランジンD合成酵素遺伝子欠損 (H−PGDS KO) マウスの作製
2002年1月28日に出願された特願2002−18666号に教示されている方法に従って造血器型プロスタグランジンD合成酵素遺伝子欠損マウスを作製した。
公知のマウスH−PGDS遺伝子のエクソンII(H−PGDSの蛋白質翻訳開始領域)を含む領域をNeor遺伝子に置換し、さらにH−PGDS遺伝子の約7Kb上流にヘルペスウイルスのサイミジンカイネース遺伝子(HSV−tk遺伝子)を組み込んで変異配列を調製し、この変異配列をベクターに組み込んでターゲティング・ベクターを作製した(図19参照)。
電気穿孔法により、未分化の培養ES細胞(1.2×107個)にターゲティング・ベクターを48μg/mlの割合で導入して遺伝子導入ES細胞を得た。これらの細胞をプレートに播き、2日後にG418およびガンシクロビルを培地に添加して更に7日間培養し、G418およびガンシクロビルに耐性を示すコロニーを得た。これらのコロニーを個別に分離し、さらに培養したのち、DNAを抽出してサザンブロッティングにより相同組換えES細胞を選別した。
次いで、この相同組換えES細胞を、C57BL/6系マウスの胚盤胞へ常法により注入し、仮親マウスへ移植して個体へと発生させた。
その結果、10匹のキメラマウスを得た。得られたキメラマウスのうち、雄の個体と雌の野生型C57BL/6系マウスとを交配させて初代(F1)マウスを得た。これらのF1マウスから、サザンブロット分析により2倍体染色体の一方に変異配列が確認された個体(♂、♀)を選別し、これらを交配させて第2世代(F2)マウスを得た。
最終的に、これらF2マウスから、サザンブロット分析により2倍体染色体の両方に変異配列が確認された個体(ホモ接合体)および片方に変異配列が確認された個体(ヘテロ接合体)を選別しH−PGDS遺伝子欠損マウスを作製した。
【実施例1】
【0031】
実施例1
遺伝性脱髄疾患における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
Galactosylceramidase欠損症であるヒトKrabbe病のモデルマウスTwitcher(Kobayashi T, et al., Brain Res., 202:479-483, 1980; Duchen LW,et al., Brain, 103:695-710, 1980; Sakai N, et al., J. Neurochem., 66:1118-1124, 1996; Taniike M. et al., J. Neuropathol. Exp. Neurol., 58:644-653, 1999)を用いて、遺伝性の脱髄による脳損傷に伴うH−PGDSとDP受容体のmRNAの変化を、定量的RT−PCR法により定量した(図1参照)。H−PGDSとDP受容体のmRNAの発現量は、共に、脱髄による脳損傷に伴い増加する。
免疫組織染色法により、H−PGDSはミクログリア細胞と脱髄の進んだ組織局所に集積するAmeboid細胞やマクロファージ細胞に発現することを同定した(図2参照)。一方、DP受容体は、脱髄の進んだ組織の周辺に分布する活性化されたアストログリア細胞に発現することを同定した(図3参照)。
【実施例2】
【0032】
実施例2
自己免疫性脱髄疾患における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
ヒト多発性硬化症のモデルである実験的自己免疫性脳脊髄炎マウス(Ichikawa M., et al., Cell Immunol., 191:97-104, 1999; Bernhard Hemmer, et al., Nature Review Neuroscience, 3:291-301, 2002)においても、定量的RT−PCR法により測定したH−PGDSとDP受容体のmRNAの発現量は、共に、脱髄による脳損傷と相関した増加を示す(図4参照)。
免疫組織染色法による観察では、H−PGDSはミクログリア細胞と脱髄の進んだ組織局所に集積するAmeboid細胞やマクロファージ細胞に発現する(図5参照)。
【実施例3】
【0033】
実施例3
外傷性脳損傷における造血器型プロスタグランジンD合成酵素とDP受容体の誘導
外傷性大脳皮質傷害(Stab wound)モデル(Salhia B, et al., Brain Res., 888:87-97, 2000; Asahi M., et al., J. Neurosci., 21:7724-7732, 2001; Garcia de Yebenes E., et al., J. Neurochem., 73:812-820, 1999)を用いて、脳損傷におけるH−PGDSとDP受容体のmRNAの発現を調べた結果、H−PGDSは損傷後2日目に最大値をとり(図6参照)、DP受容体は2日目から8日目にかけて持続的に増加した(図7参照)。
損傷の24時間後から傷害部位周囲に集積するミクログリア細胞とマクロファージでH−PGDSの誘導が起こり(図8と図9参照)、傷害部位周辺のアストログリア細胞ではGFAPとDP受容体の発現が増強し、これらの現象は損傷の8日後まで持続した(図10と図11参照)。
傷害の程度を損傷部位への色素(エバンスブルー)の漏出反応 (Kakimura Y, et al., Nature Medicine, 4:1078-1080, 1998)によって定量化した。損傷後2日目に最大値をとりその後治癒に伴って低下した(図20参照)。
【実施例4】
【0034】
実施例4
造血器型プロスタグランジンD合成酵素阻害剤投与による遺伝性脱髄疾患におけるアストログリア細胞の活性化の抑制
Twitcherマウスに、H−PGDS阻害剤であるHQL−79(4−ベンズヒドリルオキシ−1−{3−(1H−テトラゾール−5−イル)−プロピル}ピペリジン)を30mg/kg/日の用量で、14日間、皮下投与すると、アストログリア細胞の活性化が抑制され、同時に、アストログリア細胞でのDP受容体の発現が低下した(図13参照)。
【実施例5】
【0035】
実施例5
造血器型プロスタグランジンD合成酵素阻害剤投与による外傷性脳損傷におけるDP受容体の誘導の抑制と脳損傷の回復促進
H−PGDS阻害剤であるHQL−79を30mg/kg/日の用量で、傷害を与える1時間前から4日間、マウスに経口投与すると、Stab woundモデルにおける組織損傷領域でのDP受容体mRNA量は低下し(図14参照)、脳損傷の回復促進が認められた(図15参照)。この治療効果は傷害部位への色素漏出反応を指標とした実験によっても確認された(図21参照)。
さらに、傷害を与えた後からH−PGDS阻害剤の投与を開始しても損傷の拡大を抑制することが確認された(図22、23参照)。
【実施例6】
【0036】
実施例6
プロスタグランジンD受容体拮抗薬投与による外傷性脳損傷の軽減
Stab wound モデルマウスに、DP受容体拮抗薬であるBW A868C ((±)−3−ベンジル−5−(6−カルボキシヘキシル)−1−(2−シクロヘキシル−2−ヒドロキシエチルアミノ)ヒダントインを損傷の当日から1 mg/kg/日の用量で4日間、静脈内投与すると脳損傷の回復促進が認められ、組織損傷部位周辺のアストログリアの活性化が抑制された(図16参照)。
Stab woundモデルマウスに対するプロスタグランジンD受容体拮抗薬であるBW A868Cあるいはラマトロバン((+)−(3R)−3−(4−フルオロベンゼンスルホンアミド)−1,2,3,4−テトラヒドロカルバゾール−9−プロピオン酸)の効果を損傷部位への色素漏出量を指標に評価した。すなわち、脳損傷惹起2日後にエバンスブルー色素を静脈内投与し、その後2時間の組織への色素漏出量を測定した。BW A868C(1mg/kg)を脳損傷惹起3時間後および1日後に静脈内投与すると傷害部位への色素漏出を抑制した(図27参照)。
ラマトロバン(30mg/kg)を惹起3時間後および1日後に経口投与すると傷害部位への色素漏出を抑制した(図27参照)。
【実施例7】
【0037】
実施例7
Stab wound モデルに対するDP受容体拮抗薬であるピナグラジンの効果を損傷部位への色素漏出量で評価した。すなわち、脳損傷惹起2日後にエバンスブルー色素を静注し、その後2時間における色素の組織中漏出量を測定した。その結果、ピナグラジン(10mg/kg)を脳損傷惹起1時間前および1日後に経口投与すると、脳損傷に伴って認められるエバンスブルー色素漏出量の増加が抑制された。またピナグラジン(10mg/kg)は惹起3時間および1日後の後処置によっても色素の漏出を抑制した(図24参照)。さらに病理組織学的検討においてピナグラジンはいずれの投与方法によってもStab wound モデルにおける脳損傷の進展を抑制した(図25参照)。
【実施例8】
【0038】
実施例8
ヒト造血器型プロスタグランジンD合成酵素大量発現による外傷性脳損傷の増悪
製造例1で作成したヒトH−PGDS大量発現トランスジェニックマウスを用いたStab woundモデルでは、損傷部位でのマクロファージの集積、および、抗GFAP抗体を用いて免疫組織化学的に調べたアストログリア細胞の活性化が、野生型マウスに比べて顕著であり治癒は遅延した(図12参照)。
【実施例9】
【0039】
実施例9
造血器型プロスタグランジンD合成酵素遺伝子欠損による外傷性脳損傷の軽減
製造例2で作製した造血器型プロスタグランジンD合成酵素遺伝子欠損(HPGDS KO)マウス(ホモ接合体)を用いたStab wound モデルでは、損傷部位での出血、抗 GFAP 抗体を用いて免疫組織化学的に調べたアストログリアの活性化が、さらに傷害部位での色素漏出が野生型マウスに比べて軽微であった(図17および図26参照)。
一方、DP受容体遺伝子欠損(DPR KO)マウス(Matsuoka T, et al., Science, 17;287(5460):2013-2017, 2000)を用いたStab woundモデルでは傷害部位での色素漏出が野生型マウスに比べて軽微であった(図26参照)。
【0040】
本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬を治療に用いるには、通常の経口又は非経口投与用の製剤として製剤化する。本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬を含有する医薬組成物は、経口及び非経口投与のための剤形をとることができる。即ち、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などの経口投与製剤、あるいは、静脈注射、筋肉注射、皮下注射などの注射用溶液又は懸濁液、吸入薬、点眼薬、点鼻薬、坐剤、もしくは軟膏剤、貼布剤、パップ剤などの経皮投与用製剤、経皮吸収剤などの非経口製剤とすることもできる。好ましくは、経口剤または注射用薬剤として用いる。
【0041】
これらの製剤は当業者既知の適当な担体、賦形剤、溶媒、基剤等を用いて製造することができる。例えば、錠剤の場合、活性成分と補助成分を一緒に圧縮又は成型する。補助成分としては、製剤的に許容される賦形剤、例えば結合剤(例、トウモロコシでん粉)、充填剤(例、ラクトース、微結晶性セルロース)、崩壊剤(例、でん粉グリコール酸ナトリウム)又は滑沢剤(例、ステアリン酸マグネシウム)などが用いられる。錠剤は、適宜、コーティングしてもよい。シロップ剤、液剤、懸濁剤などの液体製剤の場合、例えば、懸濁化剤(例、メチルセルロース)、乳化剤(例、レシチン)、保存剤などを用いる。注射用製剤の場合、溶液、懸濁液又は油性もしくは水性乳濁液の形態のいずれでもよく、これらは懸濁安定剤又は分散剤などを含有していてもよい。軟膏剤、貼布剤、パップ剤などの経皮投与用製剤、経皮吸収剤の場合は、水性基剤(水、低級アルコール、ポリオール)または油性基剤(高級脂肪酸エステル類(イソプロピルミリステート)、親油性アルコール)を用いて製剤化する。
【0042】
本発明に係る造血器型プロスタグランジンD合成酵素(H−PGDS)阻害剤および/またはプロスタグランジンD受容体の拮抗薬は、投与形態、患者の症状、年令、体重、性別、あるいは併用される薬物(あるとすれば)などにより異なり、最終的には医師の判断に委ねられるが、経口投与の場合、体重1kgあたり、1日0.01〜100mg、好ましくは0.01〜50mg、より好ましくは0.01〜30mg、非経口投与の場合、体重1kgあたり、1日0.001〜100mg、好ましくは0.001〜5mg、より好ましくは0.001〜3mgを投与する。これを1〜4回に分割して投与すればよい。
【0043】
製剤例
製剤例1
以下の成分を含有する錠剤を製造する:
式(I)で表わされる化合物 10 mg
乳糖 90 mg
微結晶セルロース 30 mg
CMC−Na 15 mg
ステアリン酸マグネシウム 5 mg
150 mg
式(I)で表わされる化合物、乳糖、微結晶セルロース、CMC−Na(カルボキシメチルセルロース ナトリウム塩)を60メッシュのふるいに通し、混合する。混合末にステアリン酸マグネシウム混合し、製錠用混合末を得る。本混合末を直打し、150mgの錠剤を得る。
【0044】
製剤例2
活性成分50mgを含む懸濁剤は次のように製造する:
式(I)で表わされる化合物 50mg
ナトリウムカルボキシメチルセルロース 50mg
シロップ 1.25ml
安息香酸溶液 0.10ml
香料 q.v.
色素 q.v.
精製水を加え合計 5ml
活性成分をNo.45メッシュU.S.のふるいにかけ、ナトリウムカルボキシメチルセルロースおよびシロップと混合して滑らかなペーストにする。安息香酸溶液および香料を水の一部で希釈して加え、攪拌する。ついで水を十分量加えて必要な体積にする。
【0045】
製剤例3
静脈用製剤は次のように製造する:
式(I)で表わされる化合物 100mg
飽和脂肪酸グリセリド 1000ml
上記成分の溶液は通常、1分間に1mlの速度で患者に静脈内投与される。
【0046】
製剤例4
常法により次の組成のゼラチン硬カプセル剤を調製した。
HQL−79 10mg
デンプン 50mg
ステアリン酸マグネシウム 10mg
【0047】
製剤例5
常法により次の組成の錠剤を調製した。
HQL−79 10mg
セルロース、微晶質 500mg
二酸化ケイ素 10mg
ステアリン酸マグネシウム 10mg
【産業上の利用可能性】
【0048】
以上詳述したように、本発明の組成物は脳血管障害、脳変性疾患、脱髄疾患等の疾患の処置に使用できる。
脳損傷局所のミクログリア細胞やマクロファージで誘導されるH−PGDSを阻害したり、損傷部位の周辺のアストログリア細胞で発現するDP受容体の活性化を阻害することにより、プロスタグランジンD2が関与する脳損傷を治療または予防することが可能となる。
プロスタグランジンD2は多発性硬化症患者で生合成が活発であり(Science 294:1731-11735, 2001)、そのモデルである実験的自己免疫性脳脊髄炎マウスの発症過程で増悪因子として働いている可能性が高いので、H−PGDSの特異的な阻害剤や受容体拮抗薬を用いると、現在行われているステロイド療法や免疫抑制剤に代わる、多発性硬化症の治療や予防に有益な療法になりうる。さらにこれらの薬剤は、自己免疫性の細胞障害や神経細胞死に伴う局所炎症反応を抑止することで、アルツハイマー病を含む神経原繊維変化を伴う難治疾患の治療にも応用できる。
【特許請求の範囲】
【請求項1】
1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物をトランスジェニックマウスに投与し、
3)候補化合物を投与したマウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する
ことを含む脳損傷の治療または予防に用いる化合物のスクリーニング方法。
【請求項2】
さらに、4)候補化合物を投与したマウスにおける外傷が、候補化合物を与えないトランスジェニックマウスにおける外傷よりも軽減されている場合に、該候補化合物を脳損傷の治療または予防に用いる化合物として選択する、ことを含む請求項1記載の方法。
【請求項3】
脳損傷が外傷性脳損傷である請求項1または2に記載の方法。
【請求項1】
1)ヒトH−PGDS大量発現トランスジェニックマウスの脳に外傷を与え、
2)外傷を与える前又は後に候補化合物をトランスジェニックマウスに投与し、
3)候補化合物を投与したマウスにおける外傷の状態を、候補化合物を与えないトランスジェニックマウスにおける状態と比較する
ことを含む脳損傷の治療または予防に用いる化合物のスクリーニング方法。
【請求項2】
さらに、4)候補化合物を投与したマウスにおける外傷が、候補化合物を与えないトランスジェニックマウスにおける外傷よりも軽減されている場合に、該候補化合物を脳損傷の治療または予防に用いる化合物として選択する、ことを含む請求項1記載の方法。
【請求項3】
脳損傷が外傷性脳損傷である請求項1または2に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
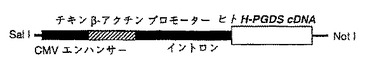
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2010−46062(P2010−46062A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2009−191800(P2009−191800)
【出願日】平成21年8月21日(2009.8.21)
【分割の表示】特願2005−144121(P2005−144121)の分割
【原出願日】平成15年7月14日(2003.7.14)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(390000745)財団法人大阪バイオサイエンス研究所 (32)
【出願人】(000207827)大鵬薬品工業株式会社 (52)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成21年8月21日(2009.8.21)
【分割の表示】特願2005−144121(P2005−144121)の分割
【原出願日】平成15年7月14日(2003.7.14)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(390000745)財団法人大阪バイオサイエンス研究所 (32)
【出願人】(000207827)大鵬薬品工業株式会社 (52)
【Fターム(参考)】
[ Back to top ]