
腫瘍を阻害するための多シストロン性siRNAコンストラクト
【課題】腫瘍に関与する複数の遺伝子を標的とするsiRNA分子を提供する。
【解決手段】本発明は、例えばヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)、ヒトマトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)をコードする配列をコードする複数の遺伝子を標的とする、2つ以上の自己相補的配列を含む多シストロン性低分子干渉RNAコンストラクトを提供する。このコンストラクトを用いて、腫瘍進行が阻害される。
【解決手段】本発明は、例えばヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)、ヒトマトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)をコードする配列をコードする複数の遺伝子を標的とする、2つ以上の自己相補的配列を含む多シストロン性低分子干渉RNAコンストラクトを提供する。このコンストラクトを用いて、腫瘍進行が阻害される。
【発明の詳細な説明】
【背景技術】
【0001】
背景
腫瘍進行は、細胞遊走の間の腫瘍細胞接着のモジュレーションおよび組織浸潤の間の細胞外マトリックス(ECM)の分解を伴う。プロテアーゼの複雑なバランス、それらの活性化因子およびそれらの阻害因子は、腫瘍浸潤の間に、これらの過程の両方を調節する。3つのクラスのECM分解プロテイナーゼは、セリンプロテイナーゼ、メタロプロテイナーゼおよびシステインプロテイナーゼである。ウロキナーゼプラスミノゲン活性化因子(uPA)は、ほとんどのマトリックスおよび基底膜構成要素を分解し、細胞−細胞および細胞−マトリックス相互作用を妨げ得る、プロテアーゼのカスケードを開始させる。細胞表面受容体であるウロキナーゼプラスミノゲン活性化因子受容体(uPAR)に結合するuPAは、プラスミノゲン活性化の数倍の増大によって示されるように、ECM分解に関与する。uPAはまた、ECM構成要素の分解の後に、いくつかの増殖因子を活性化する。uPAの、その受容体であるuPARとの結合は、マイトジェン活性化プロテインキナーゼ(MAPK)ならびに転写(開始)経路のシグナル伝達性転写因子(Stat)経路を含む多数の経路を通じて下流のシグナリング分子を活性化する。
【0002】
最近のRNA干渉(RNAi)の発見によって、癌治療における新しい道が開かれた。RNAiは、標的遺伝子の配列と相同な二本鎖RNA分子を通じて影響を受ける、配列特異的転写後遺伝子サイレンシング機構である。
【0003】
RNA干渉(RNAi)は、配列特異的転写後遺伝子サイレンシング機構であり、二本鎖RNA(dsRNA)によって引き起こされ、dsRNAに相同な配列を有するmRNAの分解を引き起こす。RNAiは、アンチセンス鎖が標的とする遺伝子の転写産物に相補的である、二本鎖RNA(dsRNA)の形成に依存する。配列特異的阻害RNAiはまた、哺乳動物細胞中で誘導され得る。RNAiのある実行において、哺乳動物細胞中の標的mRNAの選択的分解は、二本鎖の低分子干渉RNA(siRNA)でのトランスフェクションによって達成され、標的の迅速で効率的な分解につながった。これらのsiRNAは、文書によって十分立証された、哺乳動物細胞中のより長い二本鎖RNAによって引き起こされる非特異的効果を避けることが示された。
【0004】
前立腺癌は、米国人男性で2番目に多い悪性腫瘍であり、2004年には推定230,110人の新しい症例およびおよそ30,000件の死亡例があった。このように、前立腺癌は、米国および世界中で主要な公衆の健康問題を提起する。現在、転移性前立腺癌は不治であり、最終的には患者の生命を奪う。前立腺癌の相対的な重大さの要因は、転移を引き起こす、構成要素の腫瘍細胞の侵襲性である。腫瘍細胞の侵襲性の性質は、癌転移に特徴的である。腫瘍細胞浸潤および転移は、3つの顕著な段階である、細胞外マトリックスへの悪性(新生物)細胞の接着、マトリックスが分解されて原発腫瘤から細胞が放出されること、および腫瘍細胞が二次標的へ遊走することの、複雑な過程である。
【0005】
多形神経膠芽腫(GBM)は、高度に抗療性の、高度に悪性の中枢神経系原発新生物である。神経膠芽腫を治療に対して抵抗性にしているある特性は、腫瘍細胞が正常な脳組織に侵潤する傾向である。正常な脳組織に影響する療法は、許容されない。したがって、侵襲性は、ヒト神経膠腫の悪性の性質の主要な決定要因である。びまん性単細胞浸潤は、組織学的グレードにかかわらず全ての神経膠腫瘍で起こり、宿主の細胞およびECM障壁を通る、新生物細胞の移動と定義される。悪性神経膠腫は、正常な脳組織と比較してより高レベルのuPA、uPARおよびMMP−9を発現する。
【0006】
MMPは、細胞外マトリックスタンパク質を分解し、運動性を促進するシグナル伝達カスケードを活性化し、GBM運動性に関与するトランスホーミング増殖因子β等の増殖因子を活性化することによって、腫瘍細胞浸潤を促進する。ゼラチナーゼMMP−2およびMMP−9の発現は、神経膠腫を含む種々の癌の侵潤能力および転移能力と相関がある。MMP−9レベルは、神経膠腫悪性腫瘍の組織学的グレードと高度に相関があった。MMP−9は、インビトロで神経膠/内皮共培養物中の内皮細胞形態形成および毛細血管形成に関連性がある。MMPはまた、腫瘍新脈管形成を調節し、腫瘍新生血管形成の間に起こる「新脈管形成スイッチ」に必要かもしれない。
【0007】
システインプロテアーゼであるカテプシンBのタンパク質分解活性は、フィブロネクチン、I型およびIV型コラーゲンおよびラミニンを含む、ECMタンパク質の直接の分解を伴う。カテプシンBはまた、メタロプロテイナーゼならびに可溶性および受容体結合型の両方のウロキナーゼプラスミノゲン活性化因子(uPA)を含む、ECM分解を媒介するタンパク質分解カスケードに関係する他の酵素を、間接的に活性化する。また、カテプシンBは、マトリックスメタロプロテイナーゼの組織阻害因子(TIMP)を不活性化することによって、MMP活性を増大させることが示唆されている。したがって、カテプシンBは、プロ−uPA/プラスミノゲンおよびプロ−MMPの活性化における、重要な上流の調節因子であり得る。カテプシンBはまた、シトクロムc放出ならびにカスパーゼ9および3の活性化(アポトーシスのミトコンドリア経路における重要な現象)を引き起こすことによって、アポトーシスに寄与することが示されている。カテプシンB発現の増大およびその阻害因子レベルの減少は、種々の癌の腫瘍増殖、血管新生、浸潤および転移と関連付けられた。
【0008】
腫瘍に関与する複数の遺伝子を標的とするsiRNA分子が、治療組成物を開発するのに望まれる。
【発明の概要】
【課題を解決するための手段】
【0009】
概要
多シストロン性低分子干渉RNAコンストラクトは、例えばヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)、ヒトマトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)をコードする配列をコードする複数の遺伝子を標的とする、2つ以上の自己相補的配列を含む。コンストラクトを用いて、腫瘍進行が阻害される。
本発明は例えば、以下の項目を提供する:
(項目1)
腫瘍形成の阻害および予め形成された腫瘍の退行に使用される、少なくとも第一および第二の自己相補的配列を含む多シストロン性低分子干渉RNAコンストラクト。
(項目2)
前記第一の自己相補的配列がヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列およびその相補物を含み、前記第二の自己相補的配列がヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列およびその相補物を含む、項目1に記載の多シストロン性コンストラクト。
(項目3)
前記uPAの自己相補的配列が
【化1】

であり、前記uPARの自己相補的配列が
【化2】
である、項目2に記載の多シストロン性コンストラクト。
(項目4)
前記uPARおよびuPAの自己相補的配列が、約22〜35塩基対の長さの介在配列によって分離されている、項目3に記載の多シストロン性コンストラクト。
(項目5)
前記介在配列が
【化3】
である、項目4に記載の多シストロン性コンストラクト。
(項目6)
前記第一の自己相補的配列がウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列およびその相補物を含み、前記第二の自己相補的配列がマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列およびその相補物を含む、項目1に記載の多シストロン性コンストラクト。
(項目7)
前記uPARの自己相補的配列が
【化4】
であり、前記MMP−9の自己相補的配列が
【化5】
である、項目6に記載の多シストロン性コンストラクト。
(項目8)
各ループが9個のヌクレオチドを含む、項目3または7に記載の多シストロン性コンストラクト。
(項目9)
各ループが
【化6】
である、項目8に記載の多シストロン性コンストラクト。
(項目10)
前記自己相補的配列が、長さ約22〜35塩基対の介在配列によって分離されている、項目1に記載の多シストロン性コンストラクト。
(項目11)
前記介在配列が
【化7】
である、項目10に記載の多シストロン性コンストラクト。
(項目12)
前記コンストラクトが環状核酸である、項目1に記載の多シストロン性コンストラクト。
(項目13)
(a)項目1に記載の低分子干渉RNA多シストロン性コンストラクトを腫瘍に投与すること、
(b)該腫瘍において発現されて低分子干渉RNAコンストラクトに標的とされる複数の遺伝子の発現を減少させて、それによって該腫瘍を阻害すること
を包含する、腫瘍形成の阻害または予め形成された腫瘍の退行の方法。
(項目14)
前記低分子干渉RNA多シストロン性コンストラクトが、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とする、項目13に記載の方法。
(項目15)
前記低分子干渉RNA多シストロン性コンストラクトが
【化8】
を含む、項目14に記載の方法。
(項目16)
腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって腫瘍が阻害される、項目1に記載の方法。
(項目17)
前記腫瘍が、前立腺癌、神経膠腫、乳癌、および黒色腫からなる群より選択される、項目1に記載の方法。
(項目18)
前記コンストラクトが直接の送達によって投与される、項目17に記載の方法。
(項目19)
前記低分子干渉RNA多シストロン性コンストラクトが、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とする、項目13に記載の方法。
(項目20)
前記低分子干渉RNA多シストロン性コンストラクトが
【化9】
を含む、項目13に記載の方法。
(項目21)
(a)ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子であって、該低分子干渉
RNA分子が核酸配列
【化10】
を含む、RNA分子、ならびに
(b)ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とするRNA分子であって、該低分子干渉RNA分子が核酸配列
【化11】
を含む、RNA分子
からなる群より選択される、低分子干渉RNA分子。
(項目22)
項目1に記載の多シストロン性コンストラクトで形質転換された、組換え細胞。
(項目23)
項目1に記載の多シストロン性コンストラクトで形質転換された、組換えウイルス。
【0010】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するのに用いられる第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補物はヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列を含む。
【0011】
uPAの自己相補的配列は、
【0012】
【化12】
であり、uPARの自己相補的配列は
【0013】
【化13】
である。ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列ヌクレオチド配列は
【0014】
【化14】
であり、「適した」は、適切に作用することを意味する。uPARおよびuPAの自己相補的配列は、一般に、長さ約22〜35塩基対の介在配列によって分離される。例えば、介在配列は
【0015】
【化15】
である。多シストロン性コンストラクト中のuPARおよびuPAの自己相補的配列は、プロモーター、通常は単一のものに、操作作可能に連結されている。uPAおよびuPARの多シストロン性コンストラクトは、環状核酸または線状核酸である。
【0016】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補的配列はウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0017】
【化16】
であり、MMP−9の自己相補的配列は
【0018】
【化17】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列としては、ヌクレオチド配列
【0019】
【化18】
が挙げられる。uPARおよびMMP−9の自己相補的配列は、一般に、長さ約22〜35塩基対の介在配列によって分離される。介在配列は、
【0020】
【化19】
である。uPAR−MMP−9コンストラクトは、環状または線状核酸である。
【0021】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するのに用いられる、第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補物は、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はカテプシンB(CB)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0022】
【化20】
であり、CBの自己相補的配列は
【0023】
【化21】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列としては、ヌクレオチド配列
【0024】
【化22】
が挙げられる。uPARおよびCBの自己相補的配列は、一般に、長さ約22〜68塩基対の介在配列によって分離される。介在配列は、
【0025】
【化23】
である。uPAR−CBコンストラクトは、環状または線状核酸である。
【0026】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための、第一、第二および第三の自己相補的配列を含む。第一の自己相補物はウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補物はウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列を含み、第三の自己相補物はマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0027】
【化24】
であり、uPAの自己相補的配列は
【0028】
【化25】
であり、MMP−9の自己相補的配列は
【0029】
【化26】
である。ループは、GCの欠乏した約9個のヌクレオチドを含む。ループ配列としては、ヌクレオチド配列
【0030】
【化27】
が挙げられる。uPARおよびMMP−9の自己相補的配列は、一般に、長さ約22〜68塩基対の介在配列によって分離される。介在配列は、
【0031】
【化28】
である。uPA−uPAR領域は、
【0032】
【化29】
のヌクレオチド配列を含む約22〜35塩基によって分離される。uPA−uPAR−MMP−9コンストラクトは、線状または環状核酸である。
【0033】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための、第一および第二の自己相補的配列を含む。第一の自己相補的配列は、マトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含み、第二の自己相補的配列は、カテプシンB(CB)のヌクレオチド配列を含む。MMP−9の自己相補的配列は
【0034】
【化30】
であり、CBの自己相補的配列は
【0035】
【化31】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。ループ配列としては、ヌクレオチド配列
【0036】
【化32】
が挙げられる。MMP−9およびCBの自己相補的配列は、一般に、長さ約22〜37塩基対の介在配列によって分離される。介在配列は、
【0037】
【化33】
である。MMP−9−CBコンストラクトは、環状または線状核酸である。
【0038】
腫瘍を阻害する方法、方法は、
(a)低分子干渉RNA多シストロン性コンストラクトを投与する工程、および
(b)腫瘍中で発現される複数の遺伝子の発現を減少させ、それによって腫瘍の形成または増殖を阻害し、すでに存在する腫瘍を退行させる工程
を含む。
【0039】
例えばウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とし、それによってuPARおよびuPAの発現を減少させ、腫瘍を阻害する、低分子形成または干渉RNA多シストロン性コンストラクトを使用する方法。低分子干渉RNA多シストロン性コンストラクトとしては、ヌクレオチド配列
【0040】
【化34】
が挙げられる。「介在配列」の別の用語は、「スペーサー」である。腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって、腫瘍が阻害される。腫瘍としては、前立腺癌、神経膠腫、乳癌、および黒色腫が挙げられる。コンストラクトは、ウイルスベクターを通じて送達されるか、または直接の送達を通じて、もしくは当業者に公知の任意の適した方法によって、投与される。
【0041】
低分子干渉RNA多シストロン性コンストラクトを使用する方法はまた、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とし、それによってuPARおよびMMP−9の発現を減少させ、腫瘍を阻害する。低分子干渉RNA多シストロン性コンストラクトとしては、ヌクレオチド配列
【0042】
【化35】
が挙げられる。腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって、腫瘍が阻害される。腫瘍としては、前立腺癌、神経膠腫、乳癌、および黒色腫が挙げられる。コンストラクトは、ウイルスベクターを通じて送達されるか、または直接の送達を通じて、もしくは当業者に公知の任意の適した方法によって、投与される。
【0043】
腫瘍を阻害する方法は、
(a)uPA、uPAR、MMP−9およびCBの少なくとも1つを標的とする多シストロン性コンストラクトを投与する工程、ならびに
(b)uPA、uPAR、MMP−9およびCBの少なくとも1つの発現を減少させ、それによって腫瘍を阻害する工程
を含む。
【0044】
低分子干渉RNA分子としては、
(a)核酸配列
【0045】
【化36】
を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子、
(b)核酸配列
【0046】
【化37】
を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とするRNA分子、
(c)
【0047】
【化38】
の核酸配列を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびカテプシンB(CB)を標的とするRNA分子、
(d)
【0048】
【化39】
の核酸配列を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ウロキナーゼ型プラスミノゲン活性化因子(uPA)、およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子、ならびに
(e)
【0049】
【化40】
の核酸配列を含む、マトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)を標的とするRNA分子
が挙げられる。
【0050】
uPA−uPARまたはuPAR−MMP−9またはuPAR−CB、またはuPA−uPAR−MMP−9またはMMP−9−CBの多シストロン性コンストラクトで形質転換した組換え細胞が、本明細書中に開示される。
【0051】
uPA−uPARまたはuPAR−MMP−9またはuPAR−CB、またはuPA−uPAR−MMP−9またはMMP−9−CBの多シストロン性コンストラクトで形質転換した組換えウイルスが、本明細書中に開示される。
略語
uPAはウロキナーゼ型プラスミノゲン活性化因子を意味する。uPARはウロキナーゼ型プラスミノゲン活性化因子受容体を意味する。MMP−9はマトリックスメタロプロテイナーゼ9を意味する。CBはカテプシンBを意味する。CMVはサイトメガロウイルスを意味する。SV40はシミアンウイルス40型を意味する。GFPは緑色蛍光タンパク質を意味する。ECMは細胞外マトリックスを意味する。siRNAは低分子干渉RNAを意味する。shRNAは低分子ヘアピンRNAを意味する。RNAiはRNA干渉を意味する。
【図面の簡単な説明】
【0052】
【図1】図1は、uPAおよびuPARタンパク質発現レベル、ならびにヒト前立腺癌細胞系の浸潤能力と相互関係のある、uPA活性を示す。内在性uPAおよびuPARタンパク質発現を、LNCaP、DU145およびPC3の前立腺癌細胞系から単離された全細胞タンパク質の免疫ブロット解析によって調べた。等しい量の、3つ全ての細胞系の細胞抽出物から単離されたタンパク質を、抗uPA、抗uPARおよび抗GAPDH抗体での免疫ブロットに供した。GAPDHをローディング対照(A)として利用した。前立腺癌細胞系におけるuPA活性を、フィブリンザイモグラフィーによって評価した。等しい量の、血清を含まない培地中の前立腺癌細胞由来のタンパク質を、非還元性条件下で、フィブリノーゲンおよびプラスミノゲンを含有する10%ゲルでのSDS−PAGEによって分離した。Triton X−100洗浄でSDSを交換した後、ゲルをグリシンバッファー(0.1M、pH8.0)中でインキュベートした。アミノブラック染色およびそれに続くメタノール−酢酸での脱染の後の明確な溶解バンドとして、フィブリン溶解活性を検出した(B)。前立腺癌細胞系のインビトロ浸潤能力の比較(C)。マトリゲルで被覆された12μmの孔を有するポリカーボネートフィルターを含む12ウェルトランスウェルチャンバー中で浸潤アッセイを行った。フィルターの下面に通過した細胞を染色し、200×倍率で、顕微鏡下で写真を撮った(C)。200×倍率で、顕微鏡下、3つの無作為な視野で、マトリゲルを通して浸潤している細胞を計数した。各バーは3つの視野の平均値±SDを表し、検出不可能なuPAおよびuPARタンパク質発現を示した低または非転移LNCaP細胞からの有意な差をアスタリスク*で現す(P<0.05)(D)。
【図2】図2は、前立腺癌細胞系PC3における、uPAおよびuPAR発現のRNAiノックダウンを示す。sh−uPAuPARプラスミドコンストラクトの概略図(A)。コンストラクトは、ヒトCMVプロモーターならびにuPAおよびuPARを標的とする相同配列からなる。発現の後、強力なCMVプロモーターが、uPAおよびuPARに特異的な低分子ヘアピン分子の形成を駆動する。ウシ成長ホルモン(BGH)ポリアデニル化配列を、RNAポリメラーゼIIベースのCMVプロモーター終止シグナルとする。Dicer/Droshaは、uPAおよびuPARに特異的なshRNAをプロセシングし、得られるsiRNA分子は、uPAおよびuPARの標的遺伝子と相互作用する。この相互作用によって、uPAおよびuPAR遺伝子発現の同時のノックダウンが生じる。shRNAトランスフェクトPC3細胞から抽出されたRNAの半定量的逆転写PCR(B)。グリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)mRNAを対照として同時増幅した。shRNAトランスフェクトPC3細胞から抽出された全タンパク質溶解物の免疫ブロット(C)。mock、EVおよびSVトランスフェクト細胞中にuPAおよびuPARの両方のバンドが存在する。したがって、各遺伝子特異的shRNAレーンは、適切なバンドの有意な減少を示す。GAPDHが、ローディング対照として含まれた。uPAおよびuPARタンパク質発現レベルを、PC3細胞中での間接免疫蛍光法を用いても検出した。EV、SVおよびmock細胞でトランスフェクトしたPC3細胞は、uPA(FITC)およびuPAR(テキサスレッド)の免疫蛍光検出に陽性に染色した(D)。遺伝子特異的shRNAトランスフェクト細胞は、その後、EV/SVトランスフェクトおよびmock細胞と比較して、uPAおよびuPARの細胞染色プロフィールを変化させた。DAPIで、核対比染色を得た。(結果は、少なくとも3つの別々の実験の代表である。)
【図3】図3は、uPAおよびuPAR発現のRNAiノックダウンがPC3細胞の浸潤能力を阻害することを示す。mock細胞および示されたshRNAプラスミドでトランスフェクトされた細胞の浸潤能力を、マトリゲル浸潤アッセイで調べた(1つの実験の代表的な視野)(A)。本明細書中に記載されるように行った浸潤アッセイ(図1C参照)。マトリゲルを通って浸潤している細胞の代表的な数を、顕微鏡下、3つの無作為な視野で、200×で計数した(B)。各バーは、計数した3つの視野の平均値SDを表す。対照(すなわち、mockまたはスクランブルベクタートランスフェクト細胞)からの有意な差を、アスタリスク*によって示す(P<0.05)。
【図4】図4は、uPAおよびuPAR発現のRNAiノックダウンが細胞増殖を阻害し、PC3細胞におけるアポトーシスを誘導することを示す。遺伝子特異的shRNAプラスミドまたは対照(mockまたはEV/SVトランスフェクト細胞)のいずれかでトランスフェクトしたPC3細胞の生存能を、3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)アッセイによって明らかにした(A)。各バーは、平均値SDの三連の解析を表し、対照からの有意な差をアスタリスク*で表す(P<0.05)。代表的な免疫ブロットは、uPA−uPARノックダウンPC3細胞におけるアポトーシス促進性遺伝子発現の変化を示す(B)。GAPDHを、ローディング対照として用いた。活性化したカスパーゼに結合する細胞透過性カスパーゼ阻害因子であるFAM−VAD−FAKを用いた蛍光標識(下のパネル)で、カスパーゼ活性をインサイチューで検出した(C)。DAPIで核染色を行った(上のパネル)。sh−uPAuPARでトランスフェクトした相当な数の細胞が、緑色蛍光を示した。共焦点顕微鏡下の3つの無作為な視野からのDAPI/FMK−VAD−FAK標識細胞比の定量的データを示す棒の図表(D)。DAPI対FMK−VAD−FAKの比は、sh−uPAuPARでトランスフェクトした細胞で有意に増大した。mockまたはEV/SVトランスフェクト細胞からの有意な差を、アスタリスク*で示す(P<0.05)。uPA−uPAR shRNAでトランスフェクトした細胞およびアクチノマイシンD(ActD、0.2g/ml)で処理した細胞で、DNA標識が観察された(E)。エチジウムブロマイドでアガロースゲルを染色し、UV光下で写真を撮った。キロベースペアの参照として、2.0、1.5、1.0、0.75および0.5kbの標準バンドのDNAマーカーを電気泳動した(レーンM)。
【図5】図5は、uPAおよびuPAR発現のRNAiノックダウンがPC3細胞で下流のシグナリングを阻害することを示す。mockおよびshRNAトランスフェクト細胞における、全体的およびリン酸化形態の細胞外シグナル調節キナーゼ(ERK)、p38およびJNKの免疫ブロット解析(A)。mock、EV、SV、sh−uPA、sh−uPARおよびsh−uPAuPARでトランスフェクトしたPC3細胞を72時間後に溶解し、SDS−PAGEと、それに続く、全体的およびリン酸化形態のERK、p38およびJNK抗体での免疫ブロットに供した。GAPDH抗体を用いて、各レーンに同じ量のタンパク質がロードされたことを確かめた。mockおよびshRNAトランスフェクト細胞におけるStat3タンパク質の免疫ブロット解析(B)。等しい量のタンパク質をロードし、チロシン705に対するリン酸特異的Stat3抗体および非リン酸化形態のStat3に対する抗体を用いて、免疫ブロットを行った。GAPDHが、ローディング対照として含まれた。mockおよびshRNAトランスフェクト細胞の電気泳動移動度シフトアッセイ(C)。タンパク質−DNA複合体を、6%ポリアクリルアミドゲルで分離し、乾燥し、放射能写真を撮った。示されるshRNAトランスフェクト細胞から調製された核抽出物の特異的DNA結合活性を、上に示す。遊離プローブの位置を示す。
【図6】図6は、uPA−uPAR発現のRNAiノックダウンが同所マウス前立腺腫瘍モデルにおいて腫瘍増殖を排除することを示す。同所PC3腫瘍を有するマウスの各処理群からの代表的なインサイチューの写真(A)。原発性前立腺腫瘍を破線の矢印で標識し、一色の矢印は転移の位置を示す。PC3細胞をヌードマウスに前立腺内移植し、PC3前立腺腫瘍を確立し、uPA、uPARおよびuPAuPARに特異的なshRNAで処理した。これらのコンストラクトの4週間の処理後、マウスを屠殺し、原発性前立腺腫瘍増殖および転移を視覚的に評価した。各shRNA処置群からの切開した前立腺腫瘍の比較(B)。各バーは、1つの群あたり6匹の動物の平均腫瘍重量SDを表す。対照群(すなわち、mockまたはEV/SV処理)からの有意な差を、アスタリスク*で表す(P<0.05)。1つの群あたり6匹の動物のPC3前立腺腫瘍から抽出したタンパク質試料を、uPAおよびuPAR発現レベルに関する免疫ブロットを用いて解析した(C)。GAPDHが、ローディング対照として含まれた。
【図7】図7は、uPAおよびuPAR発現のRNAiノックダウンが同所マウス前立腺腫瘍モデルにおいて腫瘍増殖を同時に排除することを示す。同所PC3腫瘍を有するマウスの各処理群からの代表的なインサイチューの写真(A)。原発性前立腺腫瘍を破線の矢印で標識し、一色の矢印は転移の位置を示す。PC3細胞をヌードマウスに前立腺内移植し、PC3前立腺腫瘍を確立し、sh−uPAおよびsh−uPARベクターの両方を同時注射した。これらのコンストラクトの4週間の処理後、マウスを屠殺し、原発性前立腺腫瘍増殖および転移を視覚的に評価した。各shRNA処置群からの切開した前立腺腫瘍の比較(B)。各バーは、1つの群あたり6匹の動物の平均腫瘍重量SDを表す。対照群(すなわち、mockまたはEV/SV処理)からの有意な差を、アスタリスク*で表す(P<0.05)。1つの群あたり6匹の動物のPC3前立腺腫瘍から抽出したタンパク質試料を、uPAおよびuPAR発現レベルに関する免疫ブロットを用いて解析した(C)。GAPDHが、ローディング対照として含まれた。同所PC3前立腺腫瘍の代表的なヘマトキシリンおよびエオシン切片(D)。実験の終わりに、各処置群から原発性前立腺腫瘍を採取した。ホルマリン中で腫瘍を固定し、パラフィンに包埋した。組織切片(5m)を調製し、組織病理学的解析のために、H&Eで染色した。示される処置群からの確立した前立腺腫瘍からの顕微解剖したパラフィン切片のDNA fragEL染色(E)。DNA断片末端標識アッセイを行った。200×倍率である四角を除いて、結果を40×倍率で示す。1つの処置群あたり6つの無作為な視野からのDNA fragEL標識細胞の定量的データを示す棒の図表(F)。対照群からの有意な差を、アスタリスク*で示す(P<0.05)。
【図8】図8は、pUMベクターからのuPARおよびMMP−9のsiRNA発現の概略図を示す。uPARおよびMMP−9に対するCMVプロモーターによって駆動する2つの相補的逆方向反復を有するpcDNA3プラスミドコンストラクトを開発した。CMVプロモーターは、次に二本鎖RNA認識酵素DICERによってプロセシングされて実行可能なsiRNA分子を形成する、二重のヘアピン構造の形成を駆動する。ウシ成長ホルモン(BGH)ポリaシグナル配列によって駆動するポリA尾部を有するmRNA分子によく似ている分子の二次構造のために、二重のヘアピン分子の安定性が確実になる。
【図9】図9は、長いヘアピン(hp)RNAがsiRNAにプロセシングされ、分子がSNB19細胞において対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトされるかどうかを示し、細胞はまた、非GFP細胞においてGFPを標的とする関連のないコンストラクトによってもトランスフェクトして、適切なsiRNA分子のプロセシングを測定した。2%アガロースゲルで分画した低分子RNA分子を、6×SSC存在下で、適切なDIG標識センスオリゴでハイブリダイズさせた。得られたハイブリッド溶液を15%ポリアクリルアミドゲルに流し、ナイロン膜にエレクトロブロットした。製造業者の使用説明書どおりに、膜を処理して、21bp DNA:RNAハイブリッドを可視化した。使用したプローブを数(表1参照)、1−suPAR、2−sMMP−9および3−sGFPとして表す(図9A)。当業者に公知の標準的なプロトコールどおりに、SNB19細胞を、対照/EV(レーンa)、SV(レーンb)、puPAR(レーンc)、pMMP−9(レーンd)およびpUM(レーンe)でトランスフェクトした。72時間後、全RNAを単離し、cDNA合成キット(インビトロジェン)を用いて、ファーストストランドcDNAを合成した(図9B)。ファーストストランドcDNAをuPARおよびMMP−9の鋳型として用いて、PCR反応を構成した。GAPDHのためのPCRもまた構成し、ローディング対照とした(表1参照)。
【図10】図10は、uPARのウエスタンブロット解析およびMMP−9のゼラチンザイモグラフィーを特徴付ける。SNB19細胞を、mock、空の/スクランブルベクターならびにuPARおよびMMP−9の単一または2シストロン性siRNAをコードするベクター(puPAR、pMMP−9、pUM)でトランスフェクトした。(A)EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞からの細胞溶解物中のuPARタンパク質発現のウエスタンブロット解析。uPARに特異的な抗体を用いて、ウエスタンブロット解析を行った。GAPDHを同時に免疫検出して、同じ量の細胞溶解物をロードしたことを確かめた。(B)EV/SV、puPAR、pMMP−9およびpUMからのトランスフェクトした細胞からの等しい量のタンパク質(20μg)を含有する馴化培地を、ラエムリ試料バッファーと混合し、0.1%ゼラチンを含む10%SDS−PAGEゲルに流して、ゼラチンザイモグラフィーによってMMP−9活性を測定した。
【図11】図11は、uPARおよびMMP−9のRNAi媒介性ダウンレギュレーションがSNB19神経膠腫細胞増殖を減少させることを示す。簡潔に述べると、EV/SV、puPAR、pMMP−9およびpCMでトランスフェクトした5×104個のSNB19細胞を、血清を含まない条件下で、VNコートした96ウェルマイクロプレートに播種した。生存能のある細胞の数を、MTTアッセイによって評価した。
【図12】図12は、免疫組織化学的解析および腫瘍誘導性新脈管形成によって示されるように、RNAiがuPARおよびMMP−9レベルを減少させたことを示す。(A)SNB19細胞を、EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトした。対照または非トランスフェクト細胞もまた用いた。トランスフェクションの72時間後、細胞を固定し、処理して、インビボのuPARおよびMMP−9発現を可視化した。DAPIを含む封入剤を用いて細胞をマウントして、核を可視化した。(B)SNB19細胞(2×104)を8ウェルチャンバースライドに播種し、mock EV、puPAR、pMMP−9およびpUMでトランスフェクトした。24時間のインキュベーション後、培地を除去し、細胞を4×104個のヒト微小血管皮膚内皮細胞とともに共培養した。72時間後、内皮細胞を第VIII因子抗原に対する抗体またはH&E染色でプローブし、共焦点走査型レーザー顕微鏡下で調べた。(C)mock EV、puPAR、pMMP−9およびpUMベクターまたはpUMベクターでトランスフェクトした共培養物における新脈管形成の定量化。値は、3つの異なる実験からの平均値±S.D.である。マウス背側ウインドウアッセイによる、pUMベクターにおける腫瘍新脈管形成の阻害(D)。PV、既存の脈管構造、TN、腫瘍が脈管構造を誘導する。
【図13】図13は、uPARおよびMMP−9のsiRNAがSNB19細胞の浸潤を阻害することを示す。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞(1×106)をマトリゲルでコートされたトランスウェルインサート(8μmの孔)を通して24時間遊走させた。マトリゲルでコートしたインサートを通して浸潤した細胞を染色し、計数し、光学顕微鏡下、20×倍率で写真を撮った。(A)浸潤のパーセンテージを定量化した。値は、5つの異なる実験からの平均値±S.D.である(P<0.001)。直径100〜200μmのSNB19細胞スフェロイド(蛍光)を選択し、EV/SV、puPAR、puMMP−9およびpUMでトランスフェクトし、ラット胎仔脳凝集物(緑色蛍光)とともに共培養した。ラット胎仔脳凝集物の進行性の破壊およびSNB19細胞の浸潤を、共焦点レーザー走査型顕微鏡を用いて、72時間にわたって観察した。(C)。24、48および72時間の脳凝集物または腫瘍スフェロイドの残りの体積を、画像解析ソフトウエアを用いて定量化した(D)。
【図14】図14は、予め確立された大脳内の細胞増殖でのRNAi媒介性退行を解析する。SNB19−GFP神経膠芽腫細胞をヌードマウスに大脳内注射した(10μlのリン酸緩衝生理食塩水中2×106個の細胞)。10日後、mock EV、puPAR、pMMP−9およびpUM(Alzetミニポンプを用いて、0.25μl/時間の速度で150μgの各ベクターを脳に注射した(各群中8匹のマウス))。腫瘍切片の顕微鏡写真を、GFP蛍光(A)に関して調べ、それに続いて、ヘマトキシリンおよびエオシンで染色した(B)。これらの細胞の大脳内注射の4〜6週間後のmock EV、puPAR、pMMP−9およびpUMベクター処置群における腫瘍体積の半定量化(C)。示されるデータは、各群からの8匹の動物からの±S.D.値である。他の組の実験において、SNB19 GFP細胞の大脳内注射の10日後に、pUMベクターを3日の間隔で2回腹腔内注射し、4ヵ月後に動物を屠殺した。
【図15】図15は、uPARおよびMMP−9のRNAi媒介性ダウンレギュレーションがERK、MAPKおよびAKTのリン酸化を減少させることを示す。全体的およびリン酸化形態のMAPK、ERKおよびAKTのウエスタンブロット解析。SNB19細胞を、血清を含まない馴化細胞下で、VNコートしたプレート上で、EV/SV、puPAR、MMP−9およびpUMでトランスフェクトし、72時間後に溶解し、SDS−PAGE、ならびに全体的およびリン酸化形態のMAPK、ERKおよびAKT抗体での免疫ブロットに供した。GAPDH抗体を用いて、各レーンに同じ量のタンパク質がロードされたことを確かめた。
【図16】図16は、uPARおよびMMP−9ベクター(A)ならびに細胞表面での細胞の現象(B)の該略図である。プラスミンへのプラスミノゲンの活性化の後、これは次にMMP、ECMの分解後のいくつかの増殖因子のuPA放出を活性化する。該略図は、いくつかのシグナリング経路分子におけるインテグリンの関与を示した。
【図17】図17は、カテプシンBおよびuPARの単一のCMV駆動二重逆方向反復コンストラクトからのhpRNA分子の形成を示す概略図である。CMVウイルスプロモーターは、カテプシンBおよびuPARの自己相補的逆方向反復を有するRNA分子の形成を駆動させる。
【図18】図18は、uPARおよびカテプシンBのウエスタンブロット解析を示す。SNB19細胞を、mock、空のベクターならびにカテプシンBおよびuPARのsiRNAをコードするベクター(pCU)でトランスフェクトした。mock、空のベクターおよびpCUでトランスフェクトしたSNB19細胞からの細胞溶解物中のカテプシンB(A)uPAR(C)タンパク質レベルのウエスタンブロット解析を、カテプシンBおよびuPARに特異的な抗体を用いて行った。ローディング対照のために、β−アクチンを同時に免疫プローブした。カテプシンB(B)およびuPARタンパク質(D)の定量化を、放射能写真を濃度計でスキャンすることによって得た。
【図19】図19は、RNAiが腫瘍細胞誘導性毛細血管網形成を阻害することを示す。SNB19細胞を、mock、空のベクター、pC、pUおよびpCUで24時間トランスフェクトした。次いで、細胞をヒト皮膚内皮細胞とともに48時間共培養した。インキュベーションの後、細胞を固定し、2%ウシ血清アルブミンで1時間ブロックし、内皮細胞を第VIII因子抗原に対する抗体(第VIII因子抗体、DAKOコーポレーション、カリフォルニア州カーピンテリア)またはH&E染色でプローブし、レーザー走査型共焦点顕微鏡下で調べた(A)。mock、空のベクターまたはpCUベクターで感染させた共培養物における新脈管形成の定量化(B)。マウス背側ウインドウアッセイによる、pCUベクターで感染させたSNB19細胞における腫瘍新脈管形成の阻害(C)。PV−既存の脈管構造、TN−腫瘍誘導性脈管構造。
【図20】図20は、RNAiが神経膠腫細胞遊走および浸潤を阻害することを示す。SNB19 GFPスフェロイドを、mock、空のベクター、pC、pUおよびpCUで感染させた。72時間後、単一の神経膠腫スフェロイドを、96ウェルプレート中、ビトロネクチンコートしたウェルの中央に入れ、48時間培養した。遊走アッセイの終わりに、スフェロイドを固定し、写真を撮った(A)。ステージおよび接眼マイクロメータで較正した顕微鏡を用いて、スフェロイドからの細胞の遊走を測定した(B)。示されるデータは、各群からの4つの独立した実験からの結果の平均値±S.D.を示す。SNB19細胞を、mock、空のベクター、pC、pUおよびpcUでのトランスフェクションの72時間後にトリプシン化し、PBSで洗浄し、血清を含まない培地に再懸濁した。マトリゲルでコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット(コスター、マサチューセッツ州ケンブリッジ)中、浸潤アッセイを行った。24時間のインキュベーション期間の後、フィルターからウェル下部へ通過した細胞を染色し、計数し、光学顕微鏡下で写真を撮った(C)。浸潤のパーセンテージを定量化した(D)。値は、5つの異なる実験からの平均値±S.D.である(P<0.001)。SNB19細胞のスフェロイドを、mock、空のベクター、pC、pUおよびpcUでトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。腫瘍スフェロイドによる胎仔脳凝集物の進行性の破壊を観察した(E)。空のベクター、pC、pUまたはpCUベクターで感染させたSNB19スフェロイドによる残りの胎仔脳凝集物の定量化(F)。
【図21】図21は、uPARおよびカテプシンBのRNAi媒介性ダウンレギュレーションがSNB19神経膠腫細胞増殖を減少させることを示す。増殖アッセイ。簡潔に述べると、PBS、EV、SV、pU、pCおよびpCUでトランスフェクトした5×104個のSNB19細胞を、血清を含まない条件下で、VNコートした96ウェルマイクロプレートに播種した。生存能のある細胞の数を、MTTアッセイによって評価した。3つの別々の実験からの平均値(±S.D.)値が示される。
【図22】図22は、uPARおよびカテプシンBのRNAi媒介性ダウンレギュレーションがERKおよびFAKのリン酸化を減少させることを示す。mock、EV、SV、pU、pCおよびpCUコンストラクトでのSNB19細胞のトランスフェクションの後の、特異的抗体を用いた、全体的およびホスホERK、FAK、タンパク質のウエスタンブロット解析。β−アクチンレベルをローディング対照とした。
【図23】図23は、予め確立された腫瘍増殖のRNAi媒介性退行を示す。定位フレームの助けを借りて、ヌードマウスにSNB19 GFP腫瘍細胞を大脳内注射した。1週間後、空のベクター、またはカテプシンBおよびuPARのsiRNAを発現するベクター(pCU)のいずれかを、またはpCもしくはpUによって別々に、Alzetミニポンプを用いて脳に注射した。腫瘍切片の顕微鏡写真を、GFP蛍光で観察し(A)、それに続いてヘマトキシリンおよびエオシンで染色した(B)。5週間後のmock/空のベクター、pU、pCおよびpCUベクター処置群における腫瘍体積の半定量化。示される値は、各群からの6匹の動物の±S.D.値である(*P<0.001)(C)。
【図24】図24は、uPAR、uPAおよびMMP−9の単一のCMV稼動三重逆方向反復コンストラクトからの、hpRNA分子の形成の概略である。強力なCMVウイルスプロモーターが、uPA、uPARおよびMMP−9の自己相補的逆方向反復を有するRNA分子の形成を駆動させる。
【図25】図25は、uPAおよびMMP−9の、ウエスタンブロット、フィブリン/ゼラチンザイモグラフィーおよび免疫組織学的解析である。SNB19細胞を、mockトランスフェクトするか、または空のベクター/スクランブルベクター(EV/SV)およびsiRNA uPAR(puPAR)、uPA(puPA)、MMP−9(pMMP−9)および3つの組み合わせ(pU2M)をコードするベクターでトランスフェクトした。3日間のインキュベーション期間の後、全細胞溶解物を抽出バッファー中で調製し、これらの試料からの50μgのタンパク質を、12%非還元性SDS−PAGEによって分離し、抗uPAR抗体で免疫ブロットした(A)。ローディング対照として、GAPDHを同時に免疫プローブした。これらの試料から馴化培地を回収し(20μg)、ゼラチンおよびフィブリンザイモグラフィーを行って、MMP−9(B)およびuPA活性(C)を検出した。(D)SNB19細胞(1×104)を、Lab−Tek IIチャンバースライド上に播種し、mockトランスフェクトするか、またはEV/SV、ならびにsiRNA puPA、puPARおよびpMMP−9を単独で、または組み合わせて(pU2M)コードするベクターでトランスフェクトした。72時間後、細胞を固定し、ブロッキングバッファーで1時間洗浄し、uPA、uPARおよびMMP−9に対する特異的な抗体を用いて、uPAR、uPAおよびMMP−9発現に関して染色した。
【図26】図26は、siRNAコンストラクトによる、神経膠腫新脈管形成および浸潤の阻害を示す。SNB19細胞(2×104)を、8ウェルチャンバースライド上に播種し、EV/SV、ならびにsiRNA uPAR(puPAR)、uPA(puPA)、MMP−9(pMMP−9)および3つの組み合わせ(pU2M)をコードするベクターでトランスフェクトした。24時間のインキュベーション期間の後、培地を除去し、細胞を、4×104個のヒト内皮細胞とともに共培養するか、または4×104個のヒト内皮細胞のみを馴化培地の存在下で増殖させた。72時間後、内皮細胞を、共培養物中の第VIII因子抗原に関して染色した(緑色蛍光)。保存された馴化培地中で増殖した細胞をH&E染色し、蛍光顕微鏡または明視野顕微鏡下のいずれかで調べた(A)。(B)EV/SV、puPAR、puPA、pMMP−9およびpU2Mベクターに感染させた共培養物における新脈管形成の定量化。値は、4つの実験の平均値±SDである。EV/SV、puPAR、puPA、pMMP−9およびpU2Mでのトランスフェクションの72時間後に、SNB19細胞をトリプシン化し、PBSで洗浄し、血清を含まない培地中に再懸濁した。(C)マトリゲル(0.7mg ml−1)でコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット中で、浸潤アッセイを行った。24時間のインキュベーション期間の後、ウェル下部へフィルターを通過した細胞を染色し、計数し、明視野顕微鏡下で写真を撮った。(D)浸潤のパーセンテージを定量化した。
【図27】図27は、スフェロイドおよび大脳内アッセイによる、siRNAによる侵襲性および腫瘍増殖の阻害および退行を示す。(A)神経膠腫スフェロイドをラット胎仔脳凝集物とともに共培養することによって、神経膠腫スフェロイドの侵襲性を測定した。SNB19細胞のスフェロイドをEV/SV、puPA、puPAR、pMMP−9およびpU2Mでトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。示される時点で、共焦点レーザー走査型顕微鏡を用いて、共培養物の表面から中央までの、連続した厚さ1μmの切片を得た。(B)EV/SV、puPA、puPAR、pMMP−9およびpU2Mでトランスフェクトしたラット脳凝集物の残りの体積を測定した。値は、3つの実験の平均値±SDである。(C、D)予め確立された腫瘍増殖の、RNA媒介性退行。懸濁液中のSNB19GFP細胞(10μlの、血清を含まない培地中、2×106個)を大脳内注射した。1週間後、Alzetミニ浸透圧ポンプを用いて、マウスにEV/SVまたはsiRNA発現ベクター(puPAR、puPA、pMMP−9およびpU2M)のいずれかを注射した(PBS中1.5μg ml−1に希釈したコンストラクトを、各群の6匹のマウスに0.25μg時間−1で注射した)。5週間の追跡期間の後、マウスを屠殺し、それらの脳を除去し、パラフィン包埋および切片化した。蛍光顕微鏡下で、GFP発現細胞に関して切片を観察した。(D)対照、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置群における腫瘍体積の半定量化を、5週間後に評価した。データを、各群からの6匹の動物の平均値±SDで示す。
【図28】図28は、uPAR、uPAおよびMMP−9のRNAi媒介性ダウンレギュレーションがERKのリン酸化を減少させることを示す。EV/SV、puPAR、puPA、pMMP−9およびpU2Mコンストラクトでの神経膠芽腫細胞のトランスフェクション後の全体的およびリン酸化ERK(pERK)タンパク質のウエスタンブロット解析。GAPDHレベルをローディング対照とした。
【図29】図29は、pCMベクターからのカテプシンBおよびMMP−9のsiRNA発現の概略図である。カテプシンBおよびMMP−9に対するCMVプロモーターによって駆動される2つの相補的な逆方向反復を有するpCDNA3プラスミドコンストラクトを開発した。CMVプロモーターは、二重のヘアピン構造の形成を駆動させ、これは次に、二本鎖RNA認識酵素のDICERによってプロセシングされて、実行可能なSiRNA分子を形成する。二重のヘアピン構造の安定性は、ウシ成長ホルモン(BGH)ポリaシグナル配列によって駆動するポリA尾部を有するmRNA分子によく似ている分子の二次構造のために確実になった。
【図30】図30は、RNA干渉がSNB19細胞におけるカテプシンBおよびMMP−9レベルを減少させることを確かめる。全細胞溶解物、および血清を含まない培地を、mock、空のベクターまたはMMP−9(pM)およびカテプシンB(pC)および組み合わせ(pCM)のsiRNAをコードするベクターでトランスフェクトしたSNB19細胞から回収した。それに続いて、これらの試料からの30μgのタンパク質を、非還元性条件下、8%〜12%SDS−PAGEで分離し、ニトロセルロース膜に移した。カテプシンB(A)およびMMP−9(C)に対する抗体ならびに適切な二次抗体(ホースラディッシュペルオキシダーゼ複合体)で膜をプローブし、製造業者(アマシャム、イリノイ州アーリントンハイツ)の使用説明書どおりに展開した。β−アクチンを同時に免疫検出して、同じ量の細胞溶解物がロードしたことを確かめた。血清を含まない培地中で3日間空のベクター、pC、pMまたはpCMベクターを感染させたSNB19細胞のMMP−9活性を、ゼラチンザイモグラフィーによって測定した(B)。
【図31】図31は、RNAiが腫瘍細胞誘導性毛細血管網形成を阻害することを示す。SNB19細胞を、mock、空のベクター、pM、pCおよびpCMで24時間トランスフェクトした。次いで、細胞をヒト皮膚内皮細胞とともに48時間共培養した。インキュベーション後、細胞を固定し、2%ウシ血清アルブミンで1時間ブロックし、第VIII因子抗原に対する抗体(第VIII因子抗体、DAKOコーポレーション、カリフォルニア州カーピンテリア)で内皮細胞をプローブし、適切なFITC複合二次抗体でプローブした後、蛍光顕微鏡下で調べた。SNB19(対照、空のベクター、PM、pC、またはpCMトランスフェクト)馴化培地存在下で増殖した内皮細胞を、HおよびE染色し、写真を撮った(A)。mock、空のベクターまたはpCMベクターに感染させた共培養物における新脈管形成の定量化(B)。マウス皮下脂肪アッセイによる、pCMベクターに感染させたSNB19細胞における腫瘍新脈管形成の阻害(C)。PV−既存の脈管構造、TN−腫瘍誘導性脈管構造。光学顕微鏡を用いて(上のパネル)、およびFITC蛍光に関して(下のパネル)写真を撮り、新しく発生した脈管構造の存在を測定した。
【図32】図32は、RNAiが神経膠腫細胞遊走および浸潤を阻害することを示す。SNB19 GFPスフェロイドを、mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)に感染させた。3日後、単一の神経膠腫スフェロイドを、96ウェルプレート中のビトロネクチンコートしたウェルの中央に入れ、48時間培養した。遊走アッセイの終わりに、スフェロイドを固定し、写真を撮った(A)。mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)でのトランスフェクションの3日後に、SNB19細胞をトリプシン化し、PBSで洗浄し、血清を含まない培地中に再懸濁した。マトリゲルでコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット(コスター、マサチューセッツ州ケンブリッジ)中で、浸潤アッセイを行った。24時間のインキュベーション期間の後、ウェル下部へフィルターを通過した細胞を染色し、計数し、光学顕微鏡下で写真を撮った(B)。SNB19細胞のスフェロイドを、mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)でトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。腫瘍スフェロイドによる胎仔脳凝集物の進行性の破壊を観察した(C)。
【図33】図33は、予め確立された腫瘍増殖のRNAi媒介性退行を示す。定位フレームの助けを借りて、SNB19 GFP腫瘍細胞をヌードマウスに大脳内注射した。1週間後、空のベクター、またはカテプシンBおよびMMP−9(pCM)、カテプシンB(pC)もしくはMMP−9(pM)のsiRNAを発現するベクターのいずれかを、アルゼットミニ浸透圧ポンプを用いて脳に注射した。GFP蛍光に関して腫瘍切片の写真を観察し(A)、それに続いて、ヘマトキシリンおよびエオシンで染色した(B)。5週間後の、mock/空のベクター、pM、pCおよびpCMベクター処置群における腫瘍体積の半定量化を行った。示されるデータは、各群からの6匹の動物からの±S.D.値を示す(*P<0.001)(C)。別の実験において、大脳内注射の10日後に、pCMベクターを2回腹腔内注射し、4ヵ月後に動物を屠殺した(D)。
【図34】図34は、RNAiの誘導のための、RNAポリメラーゼIIプロモーター(CMV)の概略図である。
【図35】図35は、RNAiの誘導のための、RNAポリメラーゼベースのIIIプロモーター(U6)の概略図である。
【図36】図36は、組換えアデノウイルス生成の概略図である。
【図37】図37は、GRP、またはGRP発現を示すmockのRNAiでトランスフェクトした、SNB19 GFP細胞を示す。
【図38】図38は、RT−PCRによって測定した、RNAポリメラーゼII(CMV)またはRNAポリメラーゼIII(U6)によって駆動するRNAiベクターでトランスフェクトしたSNB19細胞におけるOAS1発現を示す。GAPDHのRT−PCRTを対照とした。
【図39】図39は、U6またはCMV駆動プロモーターのいずれかでトランスフェクトした細胞からのSNB19細胞溶解物のウエスタンブロット解析およびフィブリンザイモグラフィーによって測定した、uPARおよびuPAのダウンレギュレーションを示す。スクランブルベクター(SV)のRNAiプラスミド、uPAR(puPAR)、uPA(puPA)およびuPAR−uPA2シストロン性コンストラクト(pU2)のRNAi発現プラスミド。ローディング対照のために、GAPDHをプローブした。
【図40】図40は、細胞性免疫応答の誘導を測定するのに用いたsiRNA発現コンストラクトの概略図である。
【図41】図41は、RT−PCRによる、空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)のsiRNA発現コンストラクトおよびuPARおよびuPAの2シストロン性コンストラクト(pU2)の環状(C)、線状(L)、またはポリAシグナルを欠失した(ΔA)発現カセットでトランスフェクトしたSNB19細胞における2’5’−オリゴアデニル酸シンテターゼ(OAS1)発現の測定を示す。RT−PCRをGAPDHで標準化した。OAS1発現を、示されるように定量化した。
【図42】図42は、実行可能なRNAi誘発因子様構造を示さないEVおよびSV転写産物の予想される二次構造、ならびにポリAありまたはポリAなし(pU2ΔA)の、予想されるpuPAR、puPAおよびpU2転写産物の概略図である。
【図43】図43は、空間二次構造を示す、pU2転写産物の部分的二次構造の概略図である。空間二次構造は、mRNAに対する類似性を有さない。
【図44】図44は、mRNAレベルの、対照(C)、アンチセンスuPAR(uPARとして)、アンチセンスuPA(uPAとして)、ならびにuPAR(puPAR)、uPA(puPA)、uPAR−uPA(pU2)、GFP(pGFP)のRNAiコンストラクト、空のベクター(pEV)、ならびにuPA、uPARおよびOAS1のスクランブルベクター(pSV)から単離された全RNAでのRT−PCRを示す。GAPDHを対照とした。
【図45】図45は、静脈内注射マウスにおけるCMVプロモーターのインサイチューハイブリダイゼーションを示す。生理食塩水(Mock)、空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)、およびuPAR−uPA2シストロン性コンストラクト(pU2)のRNAi発現ベクターを腹腔内注射されたマウスの頭蓋切片を、プローブDNAをアルカリホスファターゼ(AP)で標識することによって、CMVプロモーターを含むDNAの存在に関してプローブした。ウエスタンブルーAP基質(プロメガ、ウィスコンシン州マディソン)によって、AP活性を検出した。
【発明を実施するための形態】
【0053】
詳細な説明
低分子ヘアピンRNA(shRNA)は、低分子干渉RNA(siRNA)とも呼ばれ、uPAおよびuPAR等のヒト遺伝子を標的として、腫瘍成長、腫瘍浸潤、および腫瘍増殖を阻害する。siRNAコンストラクトは、高度に転移性の前立腺癌細胞系PC3において、mRNAおよびタンパク質の両方のレベルで、uPAおよびuPAR発現を有意に阻害した。PC3細胞におけるuPA−uPARノックダウンは、例えばマトリゲル浸潤アッセイによって示されるように、腫瘍細胞浸潤の有意な減少を生じた。uPAおよびuPARの両方のshRNAを発現する単一のプラスミドコンストラクトを用いた、uPAおよびuPARの遺伝子の同時のサイレンシングは、細胞生存能を有意に減少させ、同様に、アポトーシス性細胞死の誘導を生じた。uPAおよびuPARのRNAiはまた、細胞外シグナル制御キナーゼ1/2(ERK1/2)およびシグナル伝達性転写因子3(Stat3)等の下流の標的分子へのuPA−uPARシグナリングを排除した。uPAおよびuPARのshRNAを発現するプラスミドコンストラクトの腫瘍内注射は、同所マウス前立腺癌モデルにおいて、確立された腫瘍増殖および生存を、有意に阻害した。前立腺癌細胞の腫瘍浸潤、腫瘍増殖および生存を活発に進行させるuPA−uPARの下流で機能するシグナリングネットワークの証拠が明らかである。uPAおよびuPARのRNAi誘導性標的化ならびに対応するsiRNAは、前立腺癌を含む癌治療のための、新規の治療剤である。
【0054】
ヒト神経膠腫細胞系における、uPARおよびMMP−9のsiRNA媒介性標的RNA分解は、腫瘍阻害を生じた。uPARおよびMMP−9に対するRNAiは、uPARおよびMMP−9の特異的阻害を達成した。2シストロン性コンストラクト(pUM)は、新脈管形成の、インビトロおよびインビボの両方のモデルにおいて、毛細血管様構造の形成を阻害した。uPARおよびMMP−9の発現をブロックすることは、マトリゲルおよびスフェロイド浸潤アッセイモデルにおいて、神経膠腫腫瘍浸潤の有意な阻害を生じた。uPARおよびMMP−9のRNAiは細胞増殖を阻害し、親および空のベクター/スクランブルベクター(EV/SV)トランスフェクトSNB19細胞と比較して、MAPK、ERKおよびAKTシグナリング経路のリン酸化形態のレベルを減少させた。さらに、2つのプロテアーゼ分子を同時に標的とするためのRNAiを用いること、およびアルゼットミニポンプを用いてこれらのコンストラクトをインビボで大脳内注射することもしくは腹腔内注射は、予め確立された大脳内腫瘍増殖の有意な退行を生じた。uPARおよびMMP−9のヘアピンsiRNA発現ベクターの使用によって、神経膠芽腫を含む癌治療のための効果的な治療手段が提供される。
【0055】
別の実施形態において、RNAiアプローチは、uPARおよびカテプシンB発現をサイレンシングした。RNAiを用いて、腫瘍進行の特徴的な特性である細胞外マトリックス分解に関与するプロテアーゼの発現を阻害した。uPARおよびカテプシンBのRNAiは、インビトロおよびインビボモデルにおいて、神経膠腫細胞浸潤および新脈管形成を減少させた。uPARおよびカテプシンBのhpRNA(siRNA)を発現するプラスミドベクターの腫瘍内注射は、予め確立された大脳内腫瘍の退行を生じた。uPARおよびカテプシンBのRNAiは、細胞増殖を阻害し、対照と比較して、pERKおよびpFAKのレベルを減少させた。RNAiはヒト神経膠腫細胞中で機能し、神経膠芽腫を含む癌遺伝子治療の基礎を提供する。
【0056】
RNAiアプローチは、腫瘍細胞において、uPA、uPARおよびMMP−9発現をサイレンシングした。単一の3シストロン性コンストラクト中のサイトメガロウイルス(CMV)プロモーター駆動DNA鋳型は、ヘアピンRNA(hpRNA)誘発性RNAiを誘導し、単一のコンストラクトでuPA、uPARおよびMMP−9遺伝子発現を阻害した。uPARタンパク質レベルならびにuPAおよびMMP−9の酵素活性は、uPAR、uPAおよびMMP−9のヘアピンsiRNAを発現するプラスミドでトランスフェクトされた細胞を有意に減少させることがわかった。pU2MトランスフェクトSNB19細胞は、免疫組織学的解析によって測定したmockおよびEV/SVトランスフェクト細胞と比較して、uPA、uPARおよびMMP−9発現を有意に減少させた。これらの分子の単一のコンストラクトは、免疫組織化学的解析によって示されるように、それらのそれぞれのタンパク質レベルの特異的な阻害を生じた。uPA、uPARおよびMMP−9のdsRNAを発現するプラスミドベクターでのトランスフェクションの後、マトリゲル浸潤アッセイおよびスフェロイド浸潤アッセイによって示されたmockおよびEV/SV処置群と比較して、神経膠腫細胞浸潤が遅らされた。RNAiを用いたuPA、uPARおよびMMP−9のダウンレギュレーションは、インビトロ(共培養)モデルにおいて、新脈管形成を阻害した。uPA、uPARおよびMMP−9のhpRNAを発現するプラスミドDNAの直接の腫瘍内注射はまた、ヌードマウスにおいて、予め確立された大脳内腫瘍を有意に退行させた。uPAR、uPAおよびMMP−9のRNAiで処理した細胞は、親およびEV/SV処置SNB19細胞と比較して、減少したpERKレベルを示した。uPAR、uPAおよびMMP−9の同時抑制は、癌を治療するための治療手段である。
【0057】
単一のコンストラクトからのサイトメガロウイルス(CMV)プロモーター駆動DNAヘアピンRNA(hpRNA、siRNA)は、MMP−9およびカテプシンB遺伝子発現をブロックした。MMP−9およびカテプシンBのdsRNAを発現するプラスミドベクターのトランスフェクションは、MMP−9およびカテプシンB発現を有意に阻害し、マトリゲルおよびスフェロイド浸潤モデルにおいて神経膠芽腫細胞系である、SNB19の侵襲性の性質を減少させた。SNB19細胞における、RNAiを用いたMMP−9およびカテプシンBのダウンレギュレーションもまた、ヒト微小血管内皮細胞の細胞−細胞相互作用を減少させ、インビトロおよびインビボの両方のモデルにおいて、毛細血管網形成の中断を生じた。MMP−9およびカテプシンBのhpRNAを発現するプラスミドDNAの直接の腫瘍内注射は、インビボの大脳内腫瘍において、確立された神経膠腫腫瘍増殖および浸潤を有意に阻害した。MMP−9およびカテプシンBのhpRNAを発現するプラスミドDNAの腹腔内(ip)注射は、相当な期間にわたって、予め確立された腫瘍を完全に退行させた。MMP−9およびカテプシンBの同時のRNAi媒介性標的化は、ヒト神経膠腫のための、適した治療方法である。
【0058】
uPAR、uPAおよびMMP−9を標的とするプラスミドベースのCMVプロモーター駆動hpRNAは、単独で、または同時に、SNB19ヒト神経膠腫細胞系において、RNAiを誘導する。SNB19ヒト神経膠腫細胞における、uPAR、uPAおよびMMP−9の、同時のRNAi媒介性ダウンレギュレーションは、
(1)インビトロでの浸潤および新脈管形成の阻害。
(2)インビボのヌードマウスにおける、予め確立された大脳内腫瘍の退行。
(3)ERK1および2シグナリング分子のリン酸化の減少。
を引き起こした。
【0059】
環状プラスミド(例えば、uPA、uPAR、MMP−9、およびカテプシンBまたはそれらの任意の組み合わせ)から追い出されるsiRNAまたはshRNAまたはhpRNAが、インビトロのRNA干渉を誘導するのに適している。OAS1誘導によって示されるように、環状プラスミドからのsiRNAが適しており、望ましくない免疫応答を誘導しない。uPA、uPAR、MMP−9、およびカテプシンBまたはそれらの任意の組み合わせの線状コンストラクトもまた、RNAiを誘導するのに適している。
【0060】
本明細書中に開示されるsiRNAまたはshRNAまたはhpRNAは、本質的にuPA、uPAR、MMP−9、カテプシンBまたはそれらの組み合わせの自己相補的配列からなる核酸を含む。ループ領域およびスペーサー(介在領域)は、標的配列、コンストラクト、および治療上の使用に依存して、長さおよび配列の両方が変化し得る。本明細書中に開示される核酸分子を、核酸類似物、誘導体、または任意の適した修飾によって、適切に修飾して、RNAi誘導の安定性または有効性を向上させ得る。本明細書中に開示される核酸分子はまた、他の腫瘍特異的な免疫活性化因子、腫瘍標的化因子、および任意の適した薬学的に許容され得る担体もしくは佐剤と組み合わせて投与され得る。本明細書中に開示される核酸分子はまた、放射線治療または化学療法等の他の癌治療と関連して投与され得る。
【実施例】
【0061】
以下の実施例は例示の目的のためだけであって、開示の範囲を限定するよう解釈されることを意図しない。
【0062】
(実施例1)
内在性uPAおよびuPARタンパク質発現はヒト前立腺癌細胞のインビトロの侵襲性に関連している。
【0063】
PC3細胞は、高度に転移性であるが、DU145およびLNCaP細胞はそれぞれ、中程度の転移性がある、および転移性が乏しい。uPAおよびその受容体uPARは、腫瘍浸潤および転移に関係する。転移能力の異なる3つのヒト前立腺癌細胞系におけるこれらのタンパク質のレベルを比較した。図1Aに示されるように、uPAおよびuPARタンパク質レベルは、望ましくないレベルのこれらのタンパク質を発現した転移性の乏しいLNCaP細胞と比較して、PC3およびDU145細胞において有意に高かった。フィブリンザイモグラフィーによって評価したuPA活性で、同様の傾向が見られた(図1B)。したがって、uPAおよびuPARタンパク質レベルならびにuPA活性は、それらの公知の転移能力と、正に相関があった。前立腺癌が、基底膜タンパク質で構成されるゲル層であるマトリゲルに浸潤する能力を、調べた。このアッセイは、腫瘍浸襲性を評価するためのインビトロモデルにおいて確立している。高度に転移性のある前立腺癌細胞系PC3が、最も高いレベルの侵襲性を示し、DU145およびLNCaP細胞系がそれに続き、順序はそれらの公知の転移能力と一致した(図1C、および1D)。LNCaP細胞よりも、PC3細胞は14倍多く侵襲性があり、DU145は9倍多く侵襲性があった。uPAおよびuPARタンパク質レベルと転移能力の異なるヒト前立腺癌細胞系の浸潤能力との間の強力な相関関係の浸潤能力を実証した。PC3およびDU145細胞系は、LNCaP細胞よりも侵襲性があり、それらの公知の転移能力と一致した。uPAおよびuPARの発現パターンと使用した前立腺癌細胞系の浸潤能力の間に、強力な相関関係が存在する(図1)。前立腺癌細胞系における促進されたuPAおよびuPAR発現は、増大した侵襲性および転移能力と関連がある。
【0064】
(実施例2)
RNAiを用いた、ヒトPC3前立腺癌細胞におけるuPAおよびuPAR遺伝子発現の効率的ノックダウン
低分子ヘアピンRNAを用いて、uPAおよびuPARならびに高い転移能力を発現するヒト前立腺癌細胞系PC3における内在性uPAおよびuPAR遺伝子発現をノックダウンし、前立腺腫瘍進行におけるuPAおよびuPARの生物学的な役割を調査した。uPAまたはuPARのいずれかに対応する19または21nt二本鎖RNAiオリゴヌクレオチドを生じることのできる、低分子ヘアピンコンストラクトを含むpcDNA3−CMVベクターを開発した。また、サイトメガロウイルス(CMV)プロモーターによって駆動してuPAおよびuPARの両方を標的とする二重のヘアピンを生じる、単一の2シストロン性コンストラクトを構築して、2つの内在性遺伝子の発現を同時に阻害する効果を試験した(図2A)。uPA、uPARおよびuPA−uPARの組み合わせのshRNAを発現するベクターをPC3細胞にトランスフェクトした。
【0065】
図2Bに示されるように、半定量的逆転写PCRを通じたuPAおよびuPAR発現のためのshRNAトランスフェクト細胞は、EV/SVトランスフェクト細胞またはmock細胞と比較して、各遺伝子のmRNAレベルの特定の減少を示した。しかしながら、RNAi効果は、uPAおよびuPARを同時に標的とするshRNAベクターで、より大きかった(図2B)。細胞抽出物の免疫ブロット解析を行って、観察されたような減少したmRNA発現が、遺伝子産物の翻訳の減少と相関があるかどうかを決定した。免疫ブロットアッセイによっても、同様の傾向が観察された(図2C)。mRNAレベルならびにタンパク質レベルで、特異性およびローディングの内部対照として使用されたGAPDHの発現に対するRNAiの効果は観察されなかった。また、EV/SVトランスフェクト細胞はまた、RNAi依存性uPAおよびuPARノックダウンが特異的であることを示した(図2Bおよび2C)。
【0066】
また、抗uPAおよび抗uPAR抗体での二重免疫染色を用いて、uPAおよびuPARタンパク質発現に対する遺伝子特異的shRNAの効果が、PC3細胞において検出された。図2Dに示されるように、uPAおよびuPAR染色は、遺伝子特異的shRNAによって、EV/SVトランスフェクト細胞と比較して、劇的に減少した。uPAおよびuPAR二重免疫染色は、sh−uPAuPARでトランスフェクトした細胞において、完全に消失した(図2D)。対照的に、EVおよびSVでトランスフェクトしたPC3細胞は、mock細胞と同様な染色強度およびパターンを示した。これらの免疫蛍光研究によって、RT−PCRおよび免疫ブロットアッセイが確認された。
【0067】
プラスミドベースのsiRNA系を用いた2つの遺伝子の同時の阻害は、有用な手段である。RNAiは、前立腺癌細胞系PC3において、uPAおよびuPAR mRNAならびにタンパク質発現を効果的にダウンレギュレーションした(図2Bおよび2C)。これらの遺伝子特異的RNAiプラスミドは、mockまたはEV/SVトランスフェクト細胞と比較して、uPAおよびuPAR発現を実質的に減少させた。免疫組織化学的染色データから、mock、またはEV/SVでトランスフェクトした細胞がuPAおよびuPARに対する正の染色を現すが、sh−uPAuPARでトランスフェクトした細胞はDAPI核染色を除いてほとんど染色されないことが示され、uPAおよびuPARタンパク質発現のノックダウンが示唆された(図2D)。同様に、uPAおよびuPARに対する免疫染色の強度は、遺伝子特異的RNAiによって減少した。
【0068】
(実施例3)
RNAiによるuPAおよびuPAR発現のノックダウンは、PC3細胞のマトリゲル浸潤を阻害した。
【0069】
uPAおよびuPARの機能の1つは、腫瘍転移に必要な過程である、浸潤の促進である。PC3細胞浸潤に対するuPAおよびuPARノックダウンの影響を、shRNAトランスフェクト細胞を用いたマトリゲル浸潤アッセイによって評価した。mock細胞、またはEV/SVでトランスフェトした細胞と比較して、sh−uPAuPARトランスフェクト細胞は、浸潤能力の実質的な減少を示した(図3A)。PC3細胞の浸潤は、sh−uPAによって対照のものの(すなわち、mockまたはEV/SVトランスフェクト細胞)75%まで減少し、sh−uPAuPARによって90%まで減少した(図3B)。uPAR単独のノックダウンは浸潤の有意な減少を示さなかったが、uPAならびにuPARのノックダウンは、有意な効果を有し(図3Aおよび3B)、マトリゲルへのPC3細胞浸潤が、uPAおよびuPARの共同した機能によって実質的に制御されていることが示唆された。これらの結果から、uPAおよびuPAR発現が前立腺癌浸潤ならびに転移に必要とされることが示される。sh−uPAuPAR効果は細胞がマトリゲル膜を通ってほとんど浸潤できないほど有意であり、RNAiが、タンパク質分解活性および細胞生存能を媒介するuPA−uPAR系に有意に干渉したことが示唆された。
【0070】
(実施例4)
RNAiによるuPAおよびuPAR発現のノックダウンは、細胞増殖を阻害し、アポトーシスを誘導する。
【0071】
細胞増殖および生存に対するRNAi媒介性uPAおよびuPARサイレンシングの効果を、uPAおよびuPARに特異的なshRNAでのトランスフェクションの72時間後に、MTT解析によって調べた(図4A)。uPAを標的とするRNAiは、PC3細胞の増殖能力に対して効果がなかったが、uPARに特異的なRNAiは、低い阻害効果を有した。対照的に、uPAおよびuPARを同時に標的とするRNAiで、PC3細胞の増殖の劇的な減少が観察された(図4A)。生存能のある細胞のパーセンテージはuPARおよびuPA−uPAR RNAi存在下で、対照細胞と比較して、それぞれ平均しておよそ30%および60%減少した。これらの結果から、腫瘍細胞における増大したuPAおよび/またはuPARレベルが細胞に促進された増殖および生存能力を与え得ることが示唆される。このように、uPAおよびuPARレベルを減少させることによって、癌細胞におけるアポトーシスが誘導され得る。
【0072】
この可能性を調べるために、PC3細胞を、sh−uPA、sh−uPARまたはsh−uPAuPARを発現するプラスミドでトランスフェクトした。PC3細胞タンパク質抽出物の分子解析から、sh−uPAuPARトランスフェクションがBax、Bcl−XS/L、カスパーゼ9を含むアポトーシス促進性遺伝子を誘導したことが明らかになった(図4B)。また、FAM−VAD−FMKでのsh−uPAuPARトランスフェクトPC3細胞の蛍光色素染色から、mock細胞またはEV/SVトランスフェクト細胞のいずれかでも検出されなかった、促進されたカスパーゼ活性が明らかになった(図4Cおよび4D)。DNA断片化解析は、アポトーシス誘導のさらなる証拠を提供した。図4Eが示すように、sh−uPAuPARトランスフェクトPC3細胞は、mockまたはEV/SVトランスフェクト細胞のいずれかでは検出されなかった、アガロースゲル電気泳動での、アポトーシスの典型的な特徴であるDNA標識を示した。このDNA標識は、アクチノマイシンD処理によって誘導されるアポトーシスのものと同様であり(図4E)、それによって、観察された細胞死がアポトーシスの結果であることが確認された。また、sh−uPAまたはsh−uPARでトランスフェクトした細胞において、DNA標識は観察されなかった。これらの結果はまた、FAM−VAD−FMKによって測定されるカスパーゼ9誘導および一般的な促進されたカスパーゼ活性と一致している。
【0073】
(実施例5)
RNAiによるuPAおよびuPAR発現のノックダウンは、その下流のシグナリングおよびヌードマウスにおける腫瘍形成を阻害する。
【0074】
uPAおよびuPARサイレンシングによる生物学的結果は、uPA−uPAR媒介性シグナリングおよびそれに続く下流の機能の変化の結果であり得る。uPAおよびuPARの増大した発現はERK1/2シグナリングを活性化するので、uPA−uPARノックダウン細胞におけるマイトジェン活性化タンパク質キナーゼの状態を調べた。免疫ブロット解析から、sh−uPAuPARトランスフェクト細胞においてERK1/2リン酸化が完全に排除されたが対照細胞では排除されなかったことが示される(図5A)。ERKリン酸化は、sh−uPAまたはsh−uPARのいずれかでトランスフェクトした細胞を変化させなかった。Stat3の、リン酸化を含む全体の活性は、sh−uPAuPARでトランスフェクトした細胞において、対照細胞と比較して、実質的に抑制された(図5B)。さらに、sh−uPAuPARでトランスフェクトした細胞からの核抽出物での電気泳動移動度シフトアッセイ(EMSA)から、uPAおよびuPAR発現のノックダウンが、標識されたStat3結合部位への抽出物の結合を阻害したことが示された(図5C)。Stat3活性化は抗アポトーシス経路の刺激に寄与するので、減少したレベルのホスホ−Stat3ならびにDNA結合活性は、sh−uPAuPARトランスフェクトPC3細胞の、アポトーシス性細胞死に対する増大した感受性を説明し得る。あるいは、構成的ERK活性化は、細胞生存に寄与し得る。
【0075】
uPA−uPAR RNAiが、ヌードマウスにおける予め確立されたPC3同所腫瘍の腫瘍形成も抑制するかどうかを調査した。PC3細胞を、前立腺の側葉に前立腺内接種した。移植後7および14日後に、腫瘍に、sh−uPA、sh−uPARまたはsh−uPAuPARを発現するプラスミドコンストラクトを注射した。次いで、対照群で形成された腫瘍の結果として生じた病的状態によって余儀なくされたように、RNAi処置の2回目の投与の14〜15日後にマウスを屠殺した。原発腫瘍および転移の部位の全体の形態を調べた(図6A)。sh−uPA、sh−uPARおよびsh−uPAuPARプラスミドで処置したマウスにおいて二次腫瘍を視覚的に観察したが、EV、SVおよびmockで処置したマウスは、前立腺内に原発腫瘍に加えて二次腫瘍を示した(図6A)。腫瘍を切開し、計量した(図6B)。原発ならびに二次腫瘍の90〜100%の発生率が、mock、EVおよびSV処置群で観察された。sh−uPAおよびsh−uPAR処置は腫瘍増殖を完全には阻害しなかったが、これらの処置群からの形成された腫瘤の重量は、対照処置群からの腫瘍よりも小さかった(図6B)。sh−uPAuPARで処置したマウスにおいて、腫瘍重量の有意な減少が観察された。腫瘍試料中のタンパク質レベルの免疫ブロット解析から、sh−uPAuPARで処置した腫瘍が有意に減少したuPAおよびuPARレベルを有したことが確認された(図6C)。さらなる150μgのsh−uPAuPARは、予め確立された前立腺癌を完全に退行させた。
【0076】
採取したパラフィン包埋腫瘍組織で免疫組織化学的解析を行い、PC3細胞のインビボの性質に対するsh−uPAuPARの効果を評価した。sh−uPAuPAR依存性RNAi発現は、前立腺の全般的な構造を変化させず、H&E染色は大部分で正常な組織構造を示したが、染色は、対照群において腫瘍および宿主細胞の両方を明らかにした。uPAまたはuPARのいずれかのみを標的とするRNAiは、対照群と比較して、腫瘍細胞をわずかに減少させた。おそらく、uPA−uPAR RNAi誘導性アポトーシスからのインビボの救済はない。これを試験するために、クレノウ−FragEL DNA断片化解析を用いて、全ての処置群からのパラフィン包埋腫瘍切片をアポトーシスマーカーで染色した。このアポトーシス細胞の末端標識は、処置群の間の有意な差を示す。mock、EVおよびSV処置群の腫瘍は、FragELに対する、一般化された、低レベルの染色を示し、したがって、腫瘍細胞が健康であることを示した。対照的に、sh−uPAuPAR処置PC3前立腺腫瘍の領域のほとんどは、クレノウ染色に対して陽性であった。
【0077】
sh−uPAおよびsh−uPARの腫瘍内同時注射はまた、予め確立された前立腺腫瘍増殖のほとんど完全な退行を生じたが、mock、EVおよびSVの対照群は、再生性の、優位な腫瘍を示す(図7Aおよび7B)。免疫ブロット解析は、sh−uPAおよびsh−uPARコンストラクトで同時処置した腫瘍におけるuPAおよびuPARタンパク質レベルの選択的ノックダウンを示した(図7C)。この同時処置は、H&E染色によって大部分が正常な組織構造を示したが、mock、EVおよびSV処置群の腫瘍は、前立腺の全般的な構造の観察可能な変化を示し、H&E染色は、腫瘍および宿主細胞の両方を示した(図7D)。同時処置を通じたuPAおよびuPARのノックダウンはまた、正のクレノウ染色によって明らかであるように、アポトーシス性細胞死の有意な誘導を生じた(図7Eおよび7F)。図6A〜Cおよび7A〜Fに示されるデータと組み合わせると、これらの結果は、しかしながら、sh−uPAuPARでの処置が、確立された腫瘍サイズの有意な減少を引き起こすsh−uPAおよびsh−uPARでの同時処置と比較して、能力のあるRNAi効果を有することを示す。
【0078】
インビトロDNA標識解析およびインビボDNA断片末端標識アッセイの結果は、shRNAベースのRNAiによるuPAおよびuPARの同時のノックダウンがアポトーシスを誘導することを示す。uPA−uPAR媒介性下流シグナリングは、ホルモン非依存性前立腺癌の治療のための優れた標的である。uPA−uPAR媒介性下流シグナリングは、前立腺癌細胞系PC3における細胞浸潤、生存および増殖に必要とされるようである。
【0079】
(実施例6)
uPAおよびuPAR機能的シグナリングおよび腫瘍形成におけるそれらの役割
uPAおよびuPARの異常な発現は、前立腺癌の進行した段階における最も頻繁な変化の1つであることがわかった。uPAおよびuPARが前立腺癌の進行した段階でのみ過剰発現されることから、uPAおよびuPARが、増大した増殖および浸潤等の、進行した段階の癌の表現型の決定に関連のある機能経路に影響することが示唆される。細胞外マトリックスを通る浸潤は、腫瘍転移における特徴的な段階である。腫瘍形成を抑制するuPAまたはuPAR発現のいずれかの排除は、いくつかの異なるアプローチを用いて達成されてきた。uPAとuPARのカップリングは、増殖、遊走、浸潤、新脈管形成および転移等のいくつかの異なる型の生物学的応答の独特のネットワークを形成する、いくつかの異なるシグナリング分子を組み合わせる。uPA−uPAR結合に対するこれらの生物学的応答は、細胞型、下流シグナリング分子の性質およびその発現のレベルに高度に特異的であるようである。uPAとuPARの結合は、ERK1および2を活性化し、この誘導されたERK活性は、uPA誘導性MCF−7乳癌細胞遊走に必要である。FAK、SrcおよびShcを含むシグナリングカスケードは、uPA誘導性ERK活性化および細胞遊走の原因である。対照的に、uPA誘導性血管平滑筋細胞(VSMC)遊走および増殖は、Stat経路の活性化を必要とした。ヒト乳癌細胞において、uPA誘導性分裂促進活性は、StatおよびERK経路の両方の活性化を必要とする。アンチセンスuPAは、神経膠芽腫細胞系SNB19において、PI3K/Aktシグナリングを阻害し、細胞をスタウロスポリンによるアポトーシスに対して感作した。uPAとuPARの結合は、細胞遊走、浸潤、増殖および生存を調節するために、シグナリングカスケードを活性化するようである。
【0080】
PC3細胞におけるuPA−uPARに対するRNAiは、浸潤および増殖の顕著な抑制ならびにアポトーシスの誘導を示した(図3〜4)。sh−uPAuPARトランスフェクトPC3細胞において、uPA−uPAR系および下流のシグナリング分子(ERKおよびStat3)の抑制が観察されたが、mockまたはEV/SVトランスフェクト細胞では観察されなかった(図5)。このことから、これらの細胞における観察された表現型変化の全てはuPA−uPAR相互作用およびその下流の分子のリン酸化状態を抑制することによって媒介されたと示唆される。uPA−uPARシグナリングは、StatおよびERK経路の両方を刺激し、癌細胞を死から保護する。いくつかの筋の証拠によって、ERKおよびStat3経路の両方が細胞をアポトーシス性細胞死から保護できることが示されている。sh−uPAまたはsh−uPARのいずれかでのトランスフェクションは、PC3細胞においてアポトーシスを引き起こさなかった(図4)。これは、uPAまたはuPARのいずれか単独のブロッキングは下流のシグナリング分子ERKおよびStat3に十分影響しないかもしれないためかもしれない。
【0081】
uPA−uPARシグナリングはERKおよびStat3発現を調節するので、uPAおよびuPARの同時の阻害は、これらの経路を損なって、増殖阻害およびアポトーシスの誘導につながり得る。uPA−uPAR系は、癌の2つの特徴である細胞増殖および生存を容易にすることによって、細胞生存の正の調節因子として機能するようである。したがって、癌において過剰発現されると、uPAおよびuPARは、癌細胞に、増大した増殖性および/またはアポトーシスに対する増大した耐性を与える。対照的に、uPA−uPAR発現または機能のノックダウンは、癌細胞増殖を阻害し、アポトーシスを誘導するべきである。uPAおよびuPARのノックダウンを通じてuPA−uPAR媒介性シグナリングを遮断することによって、癌細胞増殖が同時に阻害され、アポトーシスが誘導された。注目すべきことに、uPA−uPAR RNAiは、ホルモン耐性前立腺癌細胞系PC3中で機能した。このことから、RNAiによるuPA−uPAR発現のノックダウンは、ホルモン耐性前立腺癌増殖および生存を阻害する戦略であることが示唆される。
【0082】
ヌードマウスにおけるヒト癌細胞の同所移植は、特に転移の発達に関して、ヒトにおけるこれらの細胞の生物学的性質によりよく似ている。このことは、ヌードマウスに異所に移植された場合によりかなり低い効率で原発腫瘍および転移を形成するヒト前立腺癌細胞で特に正しいことがわかっている。shRNAベースのRNAiプラスミド系は、免疫ブロット解析によって測定されたように、同所前立腺腫瘍におけるuPA−uPAR発現を効果的に抑制し得る戦略を代表する(図6C)。さらに、予め確立された同所腫瘍の、sh−uPAuPAR依存性RNAiでのインビボ処置は、腫瘍増殖のほとんど全体的な阻害を示したが、sh−uPAまたはsh−uPAR RNAiでは、部分的な減少しか観察されなかった(図6B)。また、予め確立された同所腫瘍の、sh−uPAおよびsh−uPARでの同時処置もまた、腫瘍増殖をほとんど完全に阻害した(図7)。RNAi処置動物群において、mockまたはEV/SV処置群と比較して、有害な効果は注目されなかった。さらに、このアプローチは、様々な腫瘍型を標的とし、uPA−uPAR依存性悪性表現型をインビトロならびにインビボで阻害することができる。したがって、このRNAi系は、強力な新しい治療手段を提供し、uPA−uPAR下流シグナリング経路を解析することは、同様に、臨床的妥当性を有する、癌介入の治療選択肢を提供する。さらに、RNAiは、uPA−uPAR発現に干渉し、癌生物学におけるそれらのシグナリングの生物学的重要性を研究する、新規の、簡便で選択的な方法を提供する。
【0083】
ヒト癌の分子機構の理解の進歩にもかかわらず、ヒト悪性腫瘍の臨床的治療のための治療アプローチの開発は、主要な課題のままである。uPA−uPAR発現のノックダウンは、PC3細胞の増殖をインビトロならびにインビボで有意に阻害し、最終的には、アポトーシス性細胞死を生じた。異なる標的遺伝子(ERKおよびStat3)が、uPA−uPARシグナルの下流で調節された。PC3細胞は、低いStat3リン酸化およびERKリン酸化を示し、sh−uPAuPARでトランスフェクトした場合、リン酸化は完全に廃された。uPA−uPARのRNAiは、PC3細胞における細胞死をインビトロならびにインビボで誘導した。
【0084】
(実施例7)
uPARおよびMMP−9を標的とするプラスミドベースのCMVプロモーター駆動21bp逆方向反復は、siRNAにプロセシングされる。
【0085】
CMVプロモーター駆動転写産物(uPARおよびMMP−9標的化)がsiRNAに正しくプロセシングされるかどうかを決定するために、SNB19細胞を対照/EV、SV、puPAR、pMMP−9およびpUMでトランスファーした。図8は、コンストラクトの概略図を示す。細胞はまた、非GFP細胞においてGFPを標的とする、関連のないコンストラクトでもトランスフェクトし、RNA転写産物の、siRNAへのプロセシングを決定し、得られた結果が標的遺伝子の分解産物だけではなかったことを確認した。pGFPでトランスフェクトした非GFP SNB19細胞は、RNA転写産物の、siRNAへのプロセシングを生じた(図9A)。同様に、puPAR、pMMP−9およびpUMでトランスフェクトした細胞は、RNA転写産物の、適切なsiRNAへのプロセシングを生じた。EVトランスフェクト細胞は、uPARまたはMMP−9を標的とするいかなるsiRNA様断片も生成せず、見られたsiRNA断片が、コンストラクトに組み込まれた逆方向反復ループからプロセシングされたことを示した。SVトランスフェクト細胞はまた、uPARまたはMMP−9を標的とするいかなるsiRNA様断片も生成しなかった。SVは、いかなる公知の遺伝子にも相同性を有さない、不完全な逆方向反復配列で構成された。SVの21bセンスオリゴでプローブすると、21bpDNA:RNAハイブリッドは見られず、このコンストラクトがsiRNA様断片をプロセシングしなかったことが示された。
【0086】
pUMでトランスフェクトしたSNB19細胞は、uPARおよびMMP−9 mRNAの両方のダウンレギュレーションを引き起こした。uPARおよびMMP−9に相同性のある逆方向21塩基対配列を含むプラスミドコンストラクトがRNAiを誘導するかどうかを決定するために、SNF19細胞を、対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトした。トランスフェクト細胞から全RNAを抽出し、cDNA合成キット(インビトロジェン)を用いてファーストストランドcDNAを合成した。次いで、当業者に公知の標準的なプロトコールに従って、cDNAをPCRに供した。対照/EVおよびSVでトランスフェクトした細胞においてuPAR、MMP−9、およびGAPDHに対する特異的プライマー(表1参照)を用いると、uPARまたはMMP−9のレベルの減少はなかったが、puPARでトランスフェクトした細胞およびuPARのレベルでは、mRNAが有意に減少し、MMP−9 mRNAのレベルは変化しなかった。pMMP−9でトランスフェクトした細胞において、MMP−9 mRNAのレベルが減少したが、uPAR mRNAレベルは変化せず、標的分子に対するベクターの特異性を示した。pUMでトランスフェクトした細胞は、uPARおよびMMP−9 mRNAレベルの両方の減少を示した。GAPDHレベルは変化しなかった(図9B)。
【0087】
(実施例8)
RNA干渉による、MMP活性およびuPARタンパク質レベルの阻害
SNB19細胞を、EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトし、次いで細胞溶解物中のuPARおよびMMP−9レベルをウエスタンブロッティングによって、および馴化培地中のuPARおよびMMP−9レベルをゼラチンザイモグラフィーによって、それぞれ測定した。ウエスタンブロッティングによって、pUMベクターでトランスフェクトしたSNB19細胞は、親およびEV/SV処置細胞と比較して、減少した量のuPARタンパク質を発現した(図10A)。この阻害効果がuPARに対して特異的かどうかを決定するために、同じブロットにおいて、β−アクチンレベルを評価した。β−アクチンレベルは、全てのレーンで同様であり、全てのレーンでの等しいローディングが確認された。pUM感染SNB19細胞からの馴化培地は、mockおよび空の/スクランブルベクタートランスフェクト細胞と比較して、有意に低いレベルのMMP−9活性を示した(図10B)。MMP−2レベルは変化せず、標的タンパク質の特異的な阻害が示された。濃度計によるuPARおよびMMP−9バンドの定量的解析から、親およびEV/SVトランスフェクト細胞と比較して、pUMトランスフェクト細胞において、uPARタンパク質(12〜14倍)およびMMP−9活性(8〜10倍)の有意な減少が明らかになった。puPARおよびpMMP−9ベクターでトランスフェクトした細胞は、2シストロン性コンストラクトとほとんど同じようにuPARおよびMMP−9のレベルを阻害した(図10Aおよび9B)が、標的分子のダウンレギュレーションは、単一のコンストラクトと比較して、2シストロン性コンストラクトで、より明白であった。
【0088】
uPARおよびMMP−9の低分子ヘアピンRNA(shRNA)のベクター媒介性発現は、インビトロおよびインビボで、神経膠腫細胞系において効果的で安定な遺伝子サイレンシングを達成し得る。RNAiベースの遺伝子サイレンシングは、有意な治療能力で、神経膠腫で過剰発現しているタンパク質を標的とするよう適合され得る。合成siRNA分子もまた、内在性MMP−9およびuPARレベルの抑制において、同じ効果を有する。
【0089】
(実施例9)
uPARおよびMMP−9のsiRNAによる細胞増殖の阻害
MTTアッセイを用いて、ビトロネクチンコートされたマイクロプレート上で培養された細胞の増殖に対する、siRNAベクター(EV/SV、puPAR、pMMP−9およびpUM)の効果を評価した。3日間の感染の後、puPAR、puPAR、pMMP−9およびpUMベクター感染SNB19細胞は、親およびEV/SVトランスフェクトSNB19細胞のものと比較して、増殖の減少を示した(図11)。pUMベクター効果は、単一のsiRNAコンストラクト(puPARおよびpMMP−9)と比較して、SNB19増殖がかなり高かった。親およびEV/SVトランスフェクトSNB19細胞の間に、増殖の差はなかった。
【0090】
(実施例10)
RNA干渉は、uPARおよびMMP−9免疫蛍光および腫瘍誘導性新脈管形成を阻害した。
【0091】
puPARおよびpMMP−9を感染させたSNB19細胞は、免疫細胞化学によって測定すると、それぞれuPARおよびMMP−9タンパク質レベルのダウンレギュレーションを引き起こした。pUMでトランスフェクトした細胞は、免疫細胞化学によって測定すると、uPARおよびMMP−9タンパク質レベルの両方のダウンレギュレーションを引き起こした(図12A)。uPARおよびMMP−9の両方のsiRNAを発現する組み合わせたコンストラクトの効果を測定するために、SNB19細胞をpuPAR、pMMP−9およびpUMでトランスフェクトした。細胞はまた、EVおよびSVでトランスフェクトし、対照とした。結果から、puPARのみでトランスフェクトした細胞がuPARタンパク質レベルのダウンレギュレーションを示したことは明らかである。pMMP−9でトランスフェクトした細胞は、MMP−9のみのダウンレギュレーションを示したが、pUMでトランスフェクトした細胞は、uPARおよびMMP−9タンパク質レベルの両方のダウンレギュレーションを引き起こし、二重のコンストラクトが標的タンパク質レベルのダウンレギュレーションで、同じくらい、あるいはそれ以上効率的であったことが示された。uPARおよびMMP−9のsiRNAが腫瘍誘導性毛細血管形成も阻害し得るかどうかを試験するために、トランスフェクトおよび非トランスフェクトSNB19神経膠腫細胞を、ヒト内皮細胞とともに共培養した。第VIII因子抗原を用いて免疫組織化学的解析を行い、インビトロの共培養系における腫瘍誘導性血管形成、ならびにEV/SV、puPARまたはpMMP−9およびpUMでのトランスフェクションの後のこれらの共培養物のH&E染色を評価した。図12Bは、SNB19細胞とともに培養した内皮細胞が、mockおよび空のベクターでトランスフェクトした培養物において24〜48時間以内に明確な毛細血管様網を形成したことを示す。対照的に、pUMトランスフェクトSNB19細胞は、内皮細胞において、毛細血管様網形成を誘導しなかった。分枝点および分枝の数の定量化は、親および空の/スクランブルベクターでトランスフェクトした共培養物と比較して、pUMトランスフェクト共培養において有意に減少した(図12C)。さらに、効果は、毛細血管様構造形成に関して親EV/SV処置群と比較して、puPARおよびpMMP−9ベクターにおいて50%未満であり、puPAおよびpMMPベクターでトランスフェクトした共培養物において50%未満であった。親EVトランスフェクトSNB19細胞を含むチャンバーの移植は、湾曲した細い構造および多くの小さな出血点を有する微小血管発生を生じた。対照的に、pUMベクターでトランスフェクトしたSNB19細胞の移植は、いかなるさらなる微小血管の発生も生じなかった(図12D)。
【0092】
(実施例11)
uPARおよびMMP−9のsiRNAは、SNB19細胞の浸潤を阻害する。
【0093】
siRNA発現はuPARおよびMMP−9を阻害したので、その細胞浸潤を阻害する能力を評価した。EV/SV、puPAR、pMMP−9およびpUMベクターでトランスフェクトしたSNB19細胞を、マトリゲルコートされたフィルターを通して浸潤させた。図13Aは、pUMトランスフェクトSNB19細胞の染色が親およびEV/SVトランスフェクト細胞のものよりも有意に低かったことを示す。細胞の定量的解析から、親およびEV/SVトランスフェクト細胞と比較して、8%のpUMトランスフェクトのみが浸潤したことが示された(図13B)。さらに、puPARおよびpMMP−9ベクターでトランスフェクトしたSNB19細胞の浸潤の定量的解析は、親およびEV/SVトランスフェクトSNB19細胞と比較して、25%および50%浸潤した(図13Aおよび13B)。RNAiはまた、三次元スフェロイド浸潤モデルにおいてSNB19細胞の浸潤を阻害した。図13Cは、mockおよび空の/スクランブルベクターでトランスフェクトした神経膠腫スフェロイドがラット脳凝集物に結合し、集合物に漸進的に浸潤したことを示す。しかしながら、pUMトランスフェクト神経膠腫スフェロイドとの共培養物はラット脳凝集物に結合できず、浸潤しなかった。定量的解析は、親およびEV/SVトランスフェクトスフェロイドでは2〜4%のラット胎仔脳凝集物のみが残ったが、pUMトランスフェクトスフェロイドでは90〜95%のラット胎仔脳凝集物が残ったことを示した(図13D)。72時間目に、puPARおよびpMMP−9トランスフェクト共培養物において、ラット脳凝集物はおよそ25%および45%の浸潤を現した。まとめて考えると、これらの発見から、uPARおよびMMP−9のRNAi媒介性サイレンシングが両方のインビトロモデルにおいて、uPARおよびMMP−9の単一のsiRNAコンストラクトと比較して、神経膠腫細胞浸潤を大いに阻害するという強力な証拠が提供される。これらの結果から、uPARの単一のsiRNAコンストラクトが、MMP−9の単一のsiRNAコンストラクトよりも効果的であったことが示された。
【0094】
(実施例12)
uPARおよびMMP−9のsiRNAの治療効果
腫瘍進行におけるuPARおよびMMP−9遺伝子発現のRNAi媒介性干渉の効果を評価するために、pUMベクターを、定位ポンプを用いて、腫瘍を有するマウスに注射した。侵襲性腫瘍細胞の検出を容易にするために、ヒト神経膠芽腫細胞(SNB19)を、緑色蛍光タンパク質のcDNAで評価した(SNB19−GFP)。脳切片の顕微鏡検査から、PBSまたは空のベクター(EV)のみを施した対照動物は、GFP蛍光および同様の切片のH&E染色で可視化すると、5週間の追跡期間の後、相当な腫瘍増殖を発生したことが明らかになった。対照的に、腫瘍増殖またはGFP蛍光もしくはH&E染色は、同じ条件下でpUMベクターを施した動物では検出されなかった(図14Aおよび14B)。処置に関して盲検化された神経病理学者によるヘマトキシリンおよびエオシン染色した脳切片またはGFP切片の定量化から、対照と空のベクター処置群との間に腫瘍サイズの差がないことが明らかになった。しかしながら、腫瘍の全体的退行が、pUMベクター処置群で現れた(図14C)。uPARおよびMMP−9の単一のsiRNA処置コンストラクトの場合、予め確立された大脳内腫瘍増殖は、それぞれ70%および40%阻害された。pUMベクターの腹腔内注射は、6ヵ月もの長い間、予め確立された大脳内腫瘍増殖の完全な退行を生じた。これらの結果から、uPARおよびMMP−9のRNAi媒介性抑制が、予め確立された大脳内腫瘍増殖を劇的に阻害したことが示された。
【0095】
uPARおよびMMP−9のRNAi媒介性阻害は、いくつかの独立した様式で腫瘍増殖を阻害し得る。DNA断片化によって測定したアポトーシスは、アンチセンスuPAR安定性クローンを注射した動物の脳において、親細胞系と比較して、より高かった。大脳内腫瘍モデルにおいて観察された抗腫瘍効果は、腫瘍細胞死の誘導のためかもしれなかった。
【0096】
腫瘍細胞は、生存および増殖するのに新脈管形成に依存する。uPARおよびMMP−9のRNAi媒介性阻害は、インビトロ共培養系において、腫瘍誘導性を有意に阻害した。pUMベクターの抗新脈管形成効果は、腫瘍細胞が生存に必要な血管を補充する能力を抑制し、抗新脈管形成効果を腫瘍細胞自体に向けた。uPARのsiRNAが腫瘍進行をブロックする能力はまた、uPAの抗アポトーシスおよび新脈管形成効果のブロッキングを含む。一般に、腫瘍が100mmに達した後に腫瘍負荷を減少させることのできる、前臨床モデルにおける単独療法として用いられる単一の抗新脈管形成剤(アンジオスタチンおよびエンドスタチンを含む)はない。PIg、uPA、またはtPAがないことによって、実験的脈絡膜新生血管の発生が、野生型またはuPAR欠損マウスと比較して、有意に減少したことが報告されている。この効果は、部分的にマトリックスメタロプロテイナーゼ活性の調節によると示唆された。研究から合成MMP阻害因子の抗新脈管形成効果が示されたが、これらの阻害因子の実質的に全ては、単一のMMPに対する特異性を欠いている。例えば、MMP−1、−3、および−9を阻害するKB−R7785で処置したマウスの原発腫瘍および転移において、減少した血管密度および増大した腫瘍細胞アポトーシスが観察された。MMPIは、進行した疾患を有する患者において単独療法として用いた場合、わずかな臨床的利益しか示さなかった。したがって、MMP阻害と他のモダリティーとを組み合わせた使用もまた、癌治療の戦略である。
【0097】
(実施例13)
uPARおよびMMP−9に対するsiRNAは、リン酸化ERK、MAPKおよびAKTのレベルを阻害する。
【0098】
ERK、MAPKおよびAKT経路は、細胞増殖および生存において主要な役割を果たす。EV/SV、puPAR、pMMP−9およびpUMでのSNB19細胞のトランスフェクションの後に、これらの分子に特異的な特異的抗体を用いることによって、ウエスタンブロッティングを用いて、全体的およびリン酸化形態のERK、MAPKおよびAKTのレベルを比較した。EV/SV、puPAR、pMMP−9およびpUMコンストラクトによって、全体的MAPK、ERKおよびAKTの量に有意な差はなかった(図15)。しかしながら、リン酸化形態のMAPK、ERKおよびAKTのレベルは、EV/SV、puPARおよびpuPAトランスフェクトSNB19細胞と比較して、pUMによって有意に減少した(図15)。
【0099】
MCF−7細胞における、uPAの、uPARへの結合は、細胞移動性に必要な、ERK1およびERK2を活性化する。前立腺癌細胞系(PC3MLN4)において、MAPK、ERKおよびp38キナーゼシグナリング経路を通じて媒介され得るuPARの発現をアップレギュレーションすることによって、低酸素によって腫瘍細胞浸潤が減少した。さらに、低酸素におけるBcl2によるuPAR発現のアップレギュレーションは、ERKシグナリング経路を通じて、SP1 DNA結合活性によって媒介された。EGFRなしでは、代替的な経路がuPARをERKに連結する。しかしながら、この経路はEGFR発現によってサイレンシングされ、そのため、細胞移動性におけるuPARの関与を示す。PTEN(ホスファターゼおよびテンシン類似物)の安定したトランスフェクションによって、焦点接着キナーゼおよびERK1/ERK2シグナリングのヒアルロン酸誘導性リン酸化によって引き起こされるMMP−9分泌が減少した。EGFR VIII増幅を有する神経膠芽腫は、最も高いレベルのMMP−9を示した。mt ERKおよびmt JNKでのSNB19細胞の一時的トランスフェクションによってMMP−9プロモーターが抑制され、いずれかの経路に干渉することでMMP−9発現の阻害が生じることが示唆された。種々の刺激による、または異なる細胞条件でのMMP−9活性化の調節は、異なるシグナル伝達経路を伴い得る。MEK特異的阻害因子によるERKの阻害は、乳癌細胞においてMMP−9発現をブロックし、MMP−9生成を減少させ、頭部および頸部扁平上皮癌細胞におけるインビボの侵襲性を和らげた。mt−ERK安定性トランスフェクト細胞は、侵襲性が低く、MMP−9レベルが有意に減少していた。これらの2つのシグナリング経路(MAPKおよびERK1/2)は、uPAがuPAR pUMコンストラクトに結合してこれらのシグナリング経路分子のリン酸化形態を阻害すると、活性化されることが報告されている(図15〜16)。
【0100】
(実施例14)
RNAi媒介性癌治療およびsiRNAの送達
例えばuPARおよびMMP−9過剰発現等の、遺伝子のsiRNA阻害は、ヒト癌の治療のための利用可能な治療モダリティーのリストを拡張させる。アンチセンスオリゴヌクレオチドおよびリボザイム技術を含むアンチセンスアプローチが利用可能であるが、それらの効率は十分ではない。uPARおよびMMP−9のRNAi媒介性阻害は、ヌードマウスにおいて、予め確立された神経膠腫腫瘍増殖を完全に抑制した。したがって、RNAiは、標的遺伝子発現を減少させることにおいて、アンチセンスオリゴヌクレオチドおよびリボザイム技術等の他の遺伝学的手段の強力な代替手段である。RNAiまたはRNAi様効果は、標的遺伝子発現を減少させることにおいて、アンチセンス効果よりも能力があり、RNAiの潜在的な適用性が示唆された。環状構造の腫瘍誘導アルギニン−グリシン−アスパラギン酸モチーフ、DNA結合オリゴリシンおよびヒスチジル残基を含むペプチドベクターを用いて、細胞質への送達を容易にした。ペプチドベクターは、siRNAの担体として機能することができる。RNAiベースの遺伝子療法は、神経膠腫ならびに前立腺癌、乳癌、および黒色腫を含む他の転移性腫瘍の治療のための、新規のアプローチである。
【0101】
(実施例15)
全細胞抽出物中のカテプシンBおよびuPARタンパク質レベルに対するpCUベクターの効果
タンパク質分解性分解を標的とするRNAiは、癌細胞浸潤を妨げるための介入である(図17)。カテプシンBおよびuPARは、ECM分解において重要な役割を果たすことが示されている。カテプシンBおよびuPARのsiRNAを発現するベクター(pCU)でのSNB19細胞のトランスフェクションは、mockおよび空のベクター(EV)対照と比較して、両方のタンパク質の発現を強力に阻害した(図18AおよびC)。β−アクチンのレベルから、等しい量のタンパク質がゲルにロードされたことが決定された(図18)。濃度計によるカテプシンBおよびuPARバンドの定量的解析から、mockおよび空のベクターでトランスフェクトした細胞と比較して、カテプシンB(14〜16倍)およびuPARタンパク質(10〜12倍)およびpCUトランスフェクト細胞における有意な(P<0.001)減少が明らかになった(図18BおよびD)。pUおよびpCベクターでトランスフェクトした細胞は、それぞれuPARおよびカテプシンBのレベルを阻害した(図18AおよびC)。
【0102】
(実施例16)
pCUベクターによる、腫瘍細胞誘導性毛細血管網形成の阻害
新生の腫瘍は、腫瘍増殖にエネルギーを供給する新しい血管の形成に依存する。カテプシンBおよびuPARは新脈管形成を調節すると報告されているので、腫瘍細胞誘導性新脈管形成に対するpCUの効果を評価した。インビトロ共培養系における腫瘍誘導性血管形成を評価するための第VIII因子抗原、およびmock、空のベクター、pC、pUまたはpCUでのトランスフェクション後のSNB19細胞の馴化培地の存在下で増殖した内皮細胞のH&E染色を用いて、免疫組織化学的解析を行った。結果から、mockおよび空のベクターでトランスフェクトした細胞の存在下で内皮細胞が48時間以内に毛細血管様構造を形成するが、pCUベクターは腫瘍細胞誘導性毛細血管様網形成を有意に阻害したことが示された(図19A)。分枝点および分枝の数の定量化は、mockおよび空のベクターと比較して、pCUトランスフェクト共培養物において検出できなかった(図19B)。さらに、効果は、毛細血管様構造に関して、pCUベクターと比較して、pCまたはpU処置した共培養物では50%未満であった。インビトロ共培養実験を確かめるために、背側チャンバーモデルによって評価して、pCUベクターが腫瘍新脈管形成を阻害できるかどうかをインビボで調べた。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞を含有する移植したチャンバーは、湾曲した細い構造および多くの小さな出血点を有する微小血管(矢印で示す)の発生を生じた。対照的に、pCUでトランスフェクトしたSNB19細胞の、移植したチャンバーは、いかなるさらなる微小血管の発生も生じなかった(図19C)。
【0103】
(実施例17)
siRNAによる、SNB19スフェロイドの遊走の阻害
カテプシンBおよびuPAR siRNA発現が腫瘍細胞遊走および増殖に影響を及ぼすことができるかどうかを決定するために、SNB19スフェロイドをpCUベクターでトランスフェクトした。図20Aに示すように、mockおよび空のベクター(EV)でトランスフェクトしたスフェロイドからの、かなり高い細胞遊走があり、単一のコンストラクトでトランスフェクトしたスフェロイドでは、遊走の50%までの阻害が観察された。しかしながら、腫瘍スフェロイドからの細胞遊走は、pCUベクターでトランスフェクトしたスフェロイドにおいて、完全に阻害された。mockおよび空のベクターでトランスフェクトしたスフェロイドの遊走は、スフェロイド外に遊走した細胞の数によって定量化すると、pC、pUおよびpCUトランスフェクトスフェロイドと比較して、相当高かった(P<0.001)(図20B)。スフェロイドからの細胞の遊走は、これらの分子の単一のRNAiコンストラクトと比較して、2シストロン性コンストラクトで阻害された。mockおよび空のベクター対照のものと比較して、少ない細胞がpCUトランスフェクトSNB19スフェロイドから遊走したので、それによって、細胞遊走におけるカテプシンBおよびuPARの役割が示された。
【0104】
(実施例18)
カテプシンBおよびuPARに対するsiRNAは、腫瘍細胞浸潤を阻害する。
【0105】
神経膠腫侵襲性に対するカテプシンBおよびuPARのsiRNA媒介性阻害の影響を評価するために、2モデルの系を使用した。mockおよび空のベクターでトランスフェクトしたSNB19細胞は、細胞の強い染色によって観察すると、マトリゲルコートしたトランスウェルインサートに広範に浸潤した。対照的に、pC、pUおよびpCUトランスフェクト培養物は、mockおよび空のベクターでトランスフェクトした細胞と比較して、再構成された基底膜を通る、より低い侵襲性を有した(図20C)。浸潤の定量的測定によって、pC、pUおよびpCUベクターでトランスフェクトしたSNB19細胞が、mockおよび空のベクターでトランスフェクトした対照と比較して、それぞれ55%、40%および6%しか浸潤しなかったことが確認された(図20D)。マトリゲル浸潤アッセイによって測定したこれらの細胞の侵襲性の性質の阻害は、単一のコンストラクトと比較して、2シストロン性コンストラクトトランスフェクト細胞においてかなり高かった。
【0106】
スフェロイド浸潤アッセイにおけるpCUの効果の程度を試験した。スフェロイド共培養アッセイにおいて、対照神経膠腫スフェロイド、および空のベクターでトランスフェクトしたスフェロイドは、ラット胎仔脳凝集物に漸進的に浸潤し、pCUでトランスフェクトしたスフェロイドの、部分的〜ほとんど完全な阻害を生じた(図20E)。ラット胎仔脳凝集物の定量化から、神経膠腫スフェロイドが、24時間以内に25%、48時間以内に70%未満、および72時間以内に90%未満、ラット胎仔脳凝集物を浸潤したことが明らかになった。対照的に、pCUベクターでトランスフェクトした腫瘍スフェロイドは、ラット胎仔脳凝集物を浸潤しなかった。72時間目に、ラット脳凝集物は、mock空のベクター、pC、およびpUトランスフェクト共培養物においておよそ90%、85%、55%および35%の浸潤を現したが、pCUトランスフェクト共培養物では、2%〜3%の浸潤しか現さなかった((図20F)。まとめて考えると、これらの発見から、カテプシンBおよびuPARのRNAi媒介性サイレンシングが神経膠腫細胞浸潤を両方のインビトロモデルで強力に阻害すること、および単一のコンストラクトと比較して、2シストロン性コンストラクトで効果がかなり高かったことの、強力な証拠が提供される。
【0107】
腫瘍細胞浸襲性の獲得は、腫瘍進行の側面の1つである。カテプシンBおよびuPARの発現が浸潤過程の必須の構成要素であることを示す、いくつかの報告がある。pCUベクターでのトランスフェクションによって、マトリゲル浸潤およびスフェロイド共培養アッセイにおいて、SNB19細胞およびスフェロイドの侵襲性が阻害された。カテプシンBのマトリゲル浸潤の要件は、プロテアーゼのネットワークとのその相互作用によるかもしれない。カテプシンBは、プロ−ストロメライシン等の、プロ−uPAおよびメタロプロテイナーゼ等の、セリンプロテイナーゼの前駆体をその活性形態に活性化することが示されている。形質転換ヒト乳房上皮細胞系の、マトリゲルを通る侵襲性は、カテプシンB発現と関連があり、システインプロテイナーゼ阻害因子によって阻害された。卵巣癌細胞において、細胞表面カテプシンBの阻害はプロ−uPAの活性化を防ぎ、それに続いて、マトリゲルを通る癌腫細胞の浸潤を防いだ。ヒト結腸癌におけるカテプシンB活性は、癌細胞、内皮細胞および炎症細胞の侵襲性ならびにアポトーシス性および壊死性細胞死に関連がある。uPAおよびuPARは、乳癌、卵巣癌、および胃癌を含む種々の悪性腫瘍において過剰発現されることが公知であり、侵襲性および転移性表現型の維持に必須であることが示されている。
【0108】
(実施例19)
カテプシンBおよびuPARのsiRNA媒介性ダウンレギュレーションは、SNB19細胞の増殖を減少させる。
【0109】
標準的MTTアッセイを用いて、ビトロネクチンコートされたマイクロプレート上で培養した細胞の増殖に関して、siRNAベクター(対照、EV、SV、pC、pUおよびpCU)の効果を評価した(図21)。感染の72時間後に、pC、pUおよびpCUベクター感染SNB19細胞は、SNB19およびベクター対照のものと比較して、増殖の減少を示した。pCUトランスフェクト細胞は、7日間のトランスフェクションの後でも、測定できるいかなる増殖も示さなかった。7日間のアッセイの後でも、いずれのトランスフェクト細胞でも浮遊細胞または細胞残屑は見られず、アポトーシスがないことが示された。
【0110】
(実施例20)
uPARおよびカテプシンBのsiRNA媒介性ダウンレギュレーションが、ERK1/2およびFAKリン酸化を阻害する。
【0111】
シグナリング経路分子に対するuPARおよびカテプシンBのダウンレギュレーションの効果を測定するために、ともに腫瘍細胞生存、遊走および増殖に直接関係するERKおよびFAKのリン酸化を、ウエスタンブロッティングによってアッセイした。図22は、uPARおよびカテプシンBのRNAi媒介性の同時のダウンレギュレーションによってERK1/2およびFAKのリン酸化が遅れ、単一のコンストラクトでは効果がかなり少なかったことを示す。
【0112】
結果から、uPARおよびカテプシンBのダウンレギュレーションによって、細胞生存および増殖の直接の原因であるERK1/2およびFAKリン酸化のダウンレギュレーションが誘導されたことが示された。ERK−FAKカスケードにおけるuPARの関与は、ヒト癌腫細胞HEp3において以前に報告されているが、カテプシンBの役割は未だ不明である。uPARおよびカテプシンBの組み合わせのダウンレギュレーションは、ERK1/2およびFAKのリン酸化の阻害に、より効果的である。
【0113】
(実施例21)
カテプシンBおよびuPAR RNAiは、大脳内腫瘍増殖を抑制する。
【0114】
大脳内腫瘍モデルを用いて、予め確立されたインビボの腫瘍増殖に対する、RNAi媒介性阻害の潜在的効果を評価した。未処置(mock)およびEV処置対照群の脳切片は、5週間の追跡期間の後のH&E染色および高いGFP蛍光による、大きな広がった腫瘍増殖によって特徴付けられた(図23AおよびB)。しかしながら、pCUベクターで処置したマウスの脳切片で、GFP蛍光は検出されなかった(図23AおよびB)。処置に関して盲検化された神経病理学者によって点数付けされたH&E染色脳切片のさらなる定量化から、mockおよび空のベクター処置群の間の腫瘍サイズの差は明らかにならず、pCおよびpU処置群で、対照と比較して55%および65%の腫瘍増殖の有意な退行が明らかになった。しかしながら、pCU処置群においては、予め確立された腫瘍の全体的退行が明らかになった(図23)。これらの結果から、カテプシンBおよびuPARのRNAi媒介性抑制によって大脳内腫瘍増殖が有意に阻害されたことが示された。
【0115】
ミニ浸透圧ポンプを用いたpCUの局所的大脳内送達によって、ヒト悪性神経膠腫増殖が効果的に阻害された。ミニ浸透圧ポンプは、相当な時間にわたって明確で一貫したパターンの薬物曝露を維持し、脳への作用物質の送達にうまく用いられ得る。カテプシンBおよびuPARのダウンレギュレーションは、腫瘍誘導性新脈管形成の阻害を生じる。共培養アッセイをインビトロで用いて、新脈管形成に対するpCUの効果を試験した。結果から、カテプシンBおよびuPARが新脈管形成の刺激において関連性のある役割を果たすことが示され、大脳内腫瘍モデルにおけるpCUのインビボ抗腫瘍活性の作用の、可能性のある機構が示唆された。新生血管の内皮細胞にカテプシンBの強い染色が存在するが、前立腺の既存の微小血管系には存在しない。同様に、ラット脳微小血管内皮細胞において、それらがインビトロで毛細血管を形成すると、カテプシンBの強い免疫染色が観察された。カテプシンBはTIMPの阻害因子であることが示されており、TIMPは新脈管形成の阻害因子なので、カテプシンBはまた、腫瘍の広がりにおいて関連性のある役割を有する新脈管形成を刺激し得る。おそらく新脈管形成および侵襲性が阻害されたために、カテプシンBおよびuPARのRNAi媒介性標的化によって、予め確立された大脳内腫瘍増殖が抑制された。これらの結果はまた、標的遺伝子発現のsiRNA媒介性ダウンレギュレーションが脳微環境内で十分安定していることを支持する。
【0116】
(実施例22)
SNB19神経膠芽腫細胞における、uPARタンパク質、ならびにuPAおよびMMP−9酵素活性に対するsiRNAコンストラクトの効果
3つの内在性遺伝子をヘアピンsiRNAで同時に阻害するために、CMVプロモーター制御下でuPAR(RNAでのヒトuPARの77〜98塩基)、uPA(RNAでのヒトuPAの346〜367塩基)およびMMP−9(RNAでのヒトMMP−9の360〜381塩基)のsiRNAを発現するベクター(pU2M)を構築した(図24)。塩基は、全長コード配列中の位置を示す。ウエスタンブロット解析を行って、SNB19細胞中のuPARタンパク質濃度に対する、空のベクター/スクランブルベクター(EV/SV)、puPAR、puPA、MMP−9およびpU2Mトランスフェクションの効果を調べた。EV/SV、puPAおよびpMMP−9でトランスフェクトしたSNB19細胞にuPARタンパク質バンドが存在したが、これはpuPARおよびpU2M処置細胞では有意に減少した(図25A)。3シストロン性コンストラクト(pU2M)の効果は、puPARよりも大きかった(図25A)。GAPDHのレベルから、等しい量のタンパク質がゲルにロードされたことが決定された(図25A)。フィブリンザイモグラフィーを行って、uPA酵素活性に対する、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置SNB19細胞の効果を調べた。ゼラチンザイモグラフィーを行って、SNB19細胞中のMMP−9のレベルに対するこれらのコンストラクトの効果を測定した。MMP−9レベルは、親、EV/SVおよびpuPA処置細胞と比較して、puPAR、pMMP−9およびpU2Mで処置したSNB19細胞で有意に減少した(図25B)。興味深いことに、pU2M処置細胞において、MMP−2レベルもまたダウンレギュレーションされた。等しい量のタンパク質がロードされるように、注意が払われた。(図25B)。uPA酵素活性(MR 55 000)は、親、EV/SV、puPARおよびpMMP−9処置群と比較して、puPAおよびpU2M処置細胞で有意に減少した(図25C)。
【0117】
3シストロン性コンストラクトの効果は、これらの分子の単一のsiRNAコンストラクトのものよりも明白であった。免疫組織化学的解析によって測定すると、puPAR、puPA、pMMP−9およびpU2Mトランスフェクションによって、SNB19細胞中のuPAR、uPAおよびMMP−9濃度が減少した。図25Dは、uPAR、uPAおよびMMP−9に対する特異的抗体を用いた、親、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置細胞中のuPAR、uPAおよびMMP−9のタンパク質レベルを示す。uPAR、uPAおよびMMP−9のそれぞれの強度は親細胞中およびEV/SVでトランスフェクトした細胞で高かった。対照的に、uPAR強度は、puPARおよびpU2MでトランスフェクトしたSNB19細胞で減少した。puPAおよびpU2Mトランスフェクションによって、親、EV/SV、puPARおよびpMMP−9トランスフェクト細胞と比較して、uPAタンパク質の強度が有意に減少した。さらに、MMP−9タンパク質濃度は、親、EV/SV、puPAおよびpuPARトランスフェクト細胞と比較して、pMMP−9およびpU2Mトランスフェクト細胞で有意に減少した。これらの結果から、単一のコンストラクトの効果が分子特異的であること、および3シストロン性コンストラクトの効果が単一のコンストラクト単独のものよりかなり明白であることが示された。
【0118】
(実施例23)
puPAR、puPA、pMMP−9およびpU2Mは、腫瘍誘導性毛細血管網形成を阻害する。
【0119】
神経膠腫腫瘍の増殖は、腫瘤の発生を支持するのに必要な、新しい毛細血管の誘導に依存する。神経膠腫細胞からの馴化培地によって微小血管内皮細胞が誘導されて毛細血管様構造を形成する共培養系を用いて、uPAR、uPAおよびMMP−9のRNAi媒介性抑制の効果を調べた。インビトロ共培養系における腫瘍誘導性血管形成を評価するための第VIII因子抗原を用いた免疫組織化学的解析、および行ったH&E染色。内皮細胞は、SNB19親およびEV/SVトランスフェクト細胞からの馴化培地の存在下で、毛細血管様構造を形成する(図26A)。対照的に、uPA、uPARおよびMMP−9のsiRNAを発現するベクターでのSNB19細胞のトランスフェクションによって、個々に、または組み合わせてのいずれかで、腫瘍誘導性微小血管形成が部分的または完全に阻害された(図26A)。EV/SV処置細胞と比較して、pU2M処置細胞では、新しい分枝点および/または分枝の数の増大は検出されなかった(図26A)。さらに、EV/SV処置細胞と比較して、毛細血管様構造の形成が、puPAR処置培養物において約55%、puPA処置培養物において約36%、およびpMMP−9処置細胞において約60%阻害された(図26B)。
【0120】
pU2Mでトランスフェクトした神経膠芽腫細胞系からの馴化培地は、mockまたは空のベクターと比較して、毛細血管様構造を阻害した(図26A〜B)。このことから、新脈管形成の誘導に必要な新脈管形成シグナルはpU2M処置細胞中に存在しなかったことが示される。pU2MトランスフェクトSNB19神経膠腫細胞に新脈管形成誘導がないことによって示されるように、hpRNAによるuPA、uPARおよびMMP−9のダウンレギュレーションは、新脈管形成因子のダウンレギュレーションを引き起こした。uPAまたは組織型プラスミノゲン活性化因子(tPA)がないことによって、野生型(WT)またはuPAR欠損マウス(uPA−/−)と比較して、実験的脈絡膜新生血管の発生が有意に減少した。uPA−/−およびプラスミノゲン活性化因子阻害因子−1欠損(PAI−1−/−)マウスにおいて、WTマウスと比較して有意に減少した原発腫瘍増殖が起こったことが報告されており、uPA−/−およびPAI−/−マウスの腫瘍は、WTマウスと比較して、より低い増殖性およびより高いアポトーシス指数を示し、異なる新血管形態も示した。バクテリオファージディスプレイによってuPA結合を阻害することが示されているいくつかのペプチドは、同系のマウスにおいて新脈管形成および原発腫瘍増殖を阻害する。
【0121】
(実施例24)
puPAR、puPA、pMMP−9およびpU2Mは、SNB19細胞における浸潤を阻害する。
【0122】
ECM構成要素のタンパク質分解性分解は、腫瘍細胞浸潤に関連性がある。神経膠腫侵襲性に対するuPAR、uPAおよびMMP−9のsiRNA媒介性阻害の影響を評価するために、2つのモデルを利用した。第一のモデルにおいて、puPAR、puPA、pMMP−9およびpU2Mを感染させたSNB19細胞の浸潤能力を、EV/SVベクターを感染させたものと比較した。EV/SVでトランスフェクトしたSNB19細胞および親細胞は、細胞の強い染色によって示されるように、マトリゲルコートされたトランスウェルインサートを通る、広範な浸潤を示した。対照的に、puPAR、puPA、pMMP−9およびpU2Mトランスフェクト培養物は、対照と比較した染色強度によって示されるように、再構成された基底膜を通しての侵襲性が低かった(図26C)。定量化によって、puPAR、puPA、pMMP−9およびpU2Mベクターでのトランスフェクションによって、SNB19細胞による浸潤が、親およびEV/SVトランスフェクト対照と比較して、それぞれ9%、40%、15%および2%に減少したことが確かめられた(図26D)。浸潤の阻害は、単一のコンストラクトのみと比較して、3シストロン性コンストラクトでトランスフェクトした細胞でより高かった。
【0123】
スフェロイド浸潤アッセイを用いて、puPAR、puPA、pMMP−9およびpU2Mベクターの効果を調べた。神経膠腫スフェロイドを用いることの相当な潜在的利益は、三次元培養で増殖した腫瘍細胞がインビボの腫瘍のものとより密接に類似した特性を示すことである。スフェロイド共培養系において、対照スフェロイドおよびEV/SVベクターでトランスフェクトしたスフェロイドは、ラット胎仔脳凝集物に漸進的に浸潤したが、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトしたスフェロイドは、部分的〜ほとんど完全な浸潤の阻害を示した(図27A)。定量化によって、神経膠腫スフェロイドがラット胎仔脳凝集物に、1日以内に30%、2日以内に55%、および3日以内に95%浸潤し、このとき腫瘍スフェロイドおよび脳凝集物は組み合わされて単一の実体になったことが明らかになった(図27B)。EV/SVベクターでトランスフェクトした神経膠腫スフェロイドで、同様の傾向が観察された。対照的に、pU2Mベクターでトランスフェクトした腫瘍スフェロイドは、ラット胎仔脳凝集物に浸潤しなかった。3日目までに、ラット脳凝集物は、親、EV/SV、puPA、pMMP−9およびpuPARトランスフェクト共培養物において、およそ96%、95%、45%、25%および15%浸潤され、pU2Mトランスフェクト共培養物では1%浸潤された(図27B)。これらの結果から、pU2Mが両方のインビトロモデルで神経膠腫浸潤を強力に阻害することの強力な証拠が提供される。
【0124】
(実施例25)
pU2Mは、大脳内腫瘍増殖を完全に退行させる。
【0125】
単一または3シストロン性コンストラクトを用いて、ヌードマウスにおいて予め確立された大脳内腫瘍増殖の退行を引き起こすuPAR、uPAおよびMMP−9レベルのダウンレギュレーションを調べた。対照およびEV/SV処置群における全ての動物は、強いGFP蛍光によって特徴付けられる、インタクトな脳腫瘍を有した(図27C)が、puPAR、puPAおよびpMMP−9で処置したマウスの脳切片は、最小のGFP蛍光によって示される小さな腫瘍を有した。注目すべきことに、pU2Mで処置したマウスの脳切片において、GFP蛍光は検出されなかった(図27C)。これらの切片のさらなる定量化(処置条件に対して盲検化された神経病理学者によって点数付けされた)から、親およびEV/SV処置群の間の腫瘍サイズの差、ならびに、対照群と比較した、puPAR、puPAおよびpMMP−9で処置した群における予め確立された腫瘍増殖の有意な退行は明らかにならなかった(それぞれ80%、55%、および68%)(図27D)。しかしながら、pU2M処置群において、予め確立された大脳内腫瘍増殖の完全な退行が明らかになった。これらの結果から、3シストロン性コンストラクトを用いたuPAR、uPAおよびMMP−9のRNAi媒介性抑制が、ヌードマウスにおいて悪性神経膠腫増殖を完全に根絶したことが示される。
【0126】
(実施例26)
ERK1/2リン酸化の阻害
シグナリング経路に対するuPAR、uPAおよびMMP−9のsiRNA媒介性ダウンレギュレーションの効果をよりよく理解するために、腫瘍細胞生存、遊走および増殖に直接関係する、ERK1/2の全体的およびリン酸化レベルをアッセイした。ウエスタンブロットから、puPAR、puPA、pMMP−9およびpU2Mトランスフェクト細胞と比較して、対照およびEV/SVトランスフェクト細胞における全ERK1/2濃度に有意な差がなかったことが示された(図28)。しかしながら、ホスホ−ERK1/2の濃度は、対照、EV/SV、puPAR、puPAおよびpMMP−9トランスフェクトSNB19細胞と比較して、pU2MベクターでトランスフェクトしたSNB19細胞で有意に減少した。注目すべきことに、単一のコンストラクトのいずれかでトランスフェクトしたSNB19細胞において、ホスホ−ERKのレベルに対する効果はなかった。GAPDHレベルから、等しい量のタンパク質がゲルにロードされたことが示された(図28)。
【0127】
(実施例27)
遺伝子特異的siRNAは、神経膠腫細胞系におけるMMP−9およびカテプシンBタンパク質の発現を低下させる。
【0128】
2つの内在性遺伝子をヘアピンsiRNA発現ベクターで同時に阻害することの効果を試験するために、カテプシンB(ヒトカテプシンB mRNAの732〜753塩基)およびMMP−9(ヒトMMP−9 mRNAの360〜381塩基)のsiRNAを発現するベクターを、ヒトサイトメガロウイルス(CMV)プロモーターの制御下に構築した(pCM)(図29)。塩基は、全長コード配列中の位置を示す。図30Aは、pCおよびpCMベクターでのトランスフェクションが、mock、空の、およびpMベクター対照と比較して、カテプシンBレベルを特異的に阻害することを示す。同じブロットで評価したβ−アクチンレベルは、カテプシンBの阻害が特異的であることを示し、等しい量の試料ローディングが確認された。トランスフェクト細胞の馴化培地中で、MMP−2およびMMP−9レベルを測定した。mockおよび空のベクター(EV)トランスフェクト細胞から放出されたMMP−9の量は、同じであった。pMおよびpCMベクターでトランスフェクトした細胞は、mock、EV、およびpC対照と比較して、低いレベルのMMP−9を発現した。MMP−2の発現の変化はなく、pMおよびpCMベクターの配列特異的阻害が示された(図30B)。MMP−9活性の減少がタンパク質発現の減少のためであることを確かめるために、MMP−9特異的抗体での免疫ブロットを用いて、馴化培地を解析した。pMおよびpCMベクターでトランスフェクトした細胞からの馴化培地の免疫ブロットによって、MMP−9タンパク質バンドが劇的に減少したが、バンドは有意に、空のベクターまたはpCベクターを感染させた細胞からの馴化培地においてかなりより強かった(図30C)。
【0129】
(実施例28)
PCMベクターによる、腫瘍細胞誘導性毛細血管網形成の阻害
神経膠腫腫瘍の増殖は、発生する腫瘤を支持するのに必要なので、新しい毛細血管の誘導に依存する。カテプシンBおよびMMP−9のRNAi媒介性抑制を調べるために、微小血管内皮細胞が神経膠腫細胞によって誘導されて毛細血管様構造を形成する、共培養系を用いた。SNB19細胞は、72時間以内に、内皮細胞を誘導して、毛細血管様構造に分化した。対照的に、カテプシンBおよびMMP−9のsiRNAを発現するベクターでのSNB19細胞のトランスフェクションによって、腫瘍細胞誘導性微小血管形態形成が完全に阻害された(図31A)。分枝点および分枝の数のさらなる定量化は、mockおよび空のベクターと比較して、pCMトランスフェクト共培養物中で検出できなかった(図31B)。さらに、効果は、毛細血管様構造に関して、pCMベクターと比較して、pCまたはpM処置共培養物でわずか50%であった。インビトロ共培養実験を確かめるために、背側ウインドウモデルによって評価して、pCMベクターが腫瘍新脈管形成を阻害できるかどうかをインビボで調べた。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞を含有するチャンバーの移植は、湾曲した細い構造および多くの小さな出血点を有する微小血管(矢印で示す)の発生を生じた。対照的に、pCMベクターでトランスフェクトしたSNB19細胞の移植は、いかなるさらなる微小血管の発生も生じなかった(図31C)。
【0130】
悪性腫瘍の増殖維持は、腫瘍に栄養分を供給する血管網の発生と密接に関連している。pCMベクターで処置した細胞における、閉じた多角形および複雑なメッシュ様構造によって特徴付けられる血管ネットワークの形成は、観察されなかった。このネットワークは、典型的には、神経膠腫細胞が内皮細胞とともに共培養された場合に観察される。細胞外マトリックス構成要素のタンパク質分解によって、内皮細胞が遊走できるようになり、貯蔵された新脈管形成シグナリング分子が細胞外マトリックスから放出される。免疫組織化学的解析から、正常な脳組織と比較して、悪性未分化星状細胞腫および神経膠芽腫においてカテプシンBが強く発現されたことが示された。
【0131】
これらの結果から、MMPが新脈管形成を促進し得ること、およびMMPが完全にないと新しい血管形成が予防され得ることが示される。本開示における組み合わせの療法によって達成される腫瘍退行は、カテプシンBおよびMMP−9の相補的作用による。腫瘍細胞中のカテプシンBおよびMMP−9の発現を標的とすることは、新脈管形成および腫瘍増殖を制御する効果的なアプローチである。
【0132】
(実施例29)
神経膠腫遊走および浸潤に対するpCMベクターの抑制効果
細胞遊走は、細胞−細胞結合、細胞−マトリックス結合およびマトリックスリモデリングの共同した調節を必要とする。細胞がスフェロイド遊走アッセイにおいてビトロネクチン上で遊走する能力に対する、カテプシンBおよびMMP−9を抑制することの効果を、研究した。アガロースでコートした6ウェルプレート中で、SNB19−GFP細胞から多細胞神経膠腫スフェロイドを増殖させた。形態およびトリパンブルー排除を用いて生存能をチェックした後、同様の直径(100〜200μm)のスフェロイドを、mock、空のベクター(EV)、またはカテプシンBおよびMMP−9のsiRNAを発現するpCMベクターでトランスフェクトした。3日後、単一のスフェロイドをビトロネクチンコートされたプレートに入れて遊走させた。図32Aは、対照スフェロイドおよび空のベクターを感染させたスフェロイドからの細胞が、pCMベクター感染細胞と比較して、細胞が遊走する有意に高い能力を示したことを示す。細胞外マトリックス構成要素のタンパク質分解性分解は、腫瘍細胞浸潤に関連性がある。カテプシンBおよびMMP−9のsiRNAの発現が神経膠腫侵襲性において役割を果たすかどうかを調査するために、mockおよび空のベクターを感染させた細胞に対する、pCMベクターでトランスフェクトしたSNB19細胞の浸潤能力を比較した。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞は、マトリゲルを通って浸透したpCMベクタートランスフェクト細胞と比較してより広範に、マトリゲルを通って浸潤した(図32B)。
【0133】
スフェロイド浸潤アッセイにおけるsiRNAの抑制効果の程度を調べた。神経膠腫スフェロイドを使用することの潜在的利点は、三次元培養で増殖した腫瘍細胞がインビボの腫瘍のものとより密接に類似した特性を示すことであると示されている。mockおよび空のベクターでトランスフェクトしたスフェロイドは1日以内に正常な脳凝集物の25%、2日以内で50%が浸潤し、3日目までに、腫瘍スフェロイドの95%が浸潤し、脳凝集物は組み合わされて単一の実体になった(図32C)。対照的に、カテプシンBおよびMMP−9のsiRNAを発現するpCMベクターでトランスフェクトした神経膠腫スフェロイドは、正常な脳凝集物から分離したままであった。
【0134】
本開示は、カテプシンBおよびMMP−9に対するsiRNA(pCM)のCMVプロモーター駆動発現が、ウエスタンブロッティングおよびゼラチンザイモグラフィーによって解析すると、SNB19神経膠腫細胞系において、カテプシンBおよびMMP−9発現をうまくサイレンシングし得ることを示す。結果から、pCMベクターで処置した神経膠腫細胞の浸潤能力が有意に阻害されることも示された。癌細胞は、遊走および浸潤するには近隣の細胞および細胞外マトリックス構成要素から分離しなければならない。マトリックスタンパク質分解は、接着部位の除去または結合部位の露出のいずれかによって細胞−マトリックス接着を直接調節し得、これが次に細胞遊走に影響し得る。カテプシンBおよびMMP−9のRNAi媒介性阻害によって、スフェロイド遊走アッセイにおいて示されるように、SNB19神経膠腫細胞の遊走が有意にブロックされた。
【0135】
(実施例30)
RNAiは、ヌードマウスにおいて神経膠芽腫腫瘍の完全な退行を誘導する。
【0136】
MMP−9およびカテプシンBのsiRNAが大脳内SNB19腫瘍の退行を阻害する能力を、ヌードマウスにおいて試験した。予め確立された神経膠腫増殖を有するマウスに、PBS(mock)、空のベクター(EV)、pC、pMおよびpCMベクターを定位注射した。mockおよびEVで処置したマウスの脳切片は、急速な腫瘍増殖を示したが、予め確立された腫瘍にミニ浸透圧ポンプを用いてpCMベクターを注射したマウスは、5週間にわたって腫瘍増殖の完全な阻害を生じた(図32Aおよび32B)。腫瘍サイズの定量化は、mockまたは空のベクターと比較した、pCMベクター処置群における腫瘍の全体的退行を示した(図33C)。pCまたはpMベクター処置群で処置したマウスの脳切片は、対照群と比較して、約50%の腫瘍退行を生じた。ベクターの腹腔内注射もまた、予め確立された大脳内腫瘍増殖の完全な退行を生じ、数ヵ月の長い期間にわたって腫瘍細胞を示さなかった(図33D)。したがって、RNAiは、このヌードマウスモデルにおいて、悪性神経膠腫腫瘍増殖を完全に根絶することができた。神経膠腫増殖の持続的抑制は、siRNA増幅のためであるかもしれない。カテプシンBおよびMMP−9に対するsiRNAは、神経膠腫増殖を、MMP−9およびカテプシンBのアンチセンスオリゴデオシヌクレオチドよりも、より効率的に抑制した。したがって、カテプシンBおよびMMP−9発現の両方の制御は、腫瘍進行の調節において、相当な重要性を有する。
【0137】
カテプシンBおよびMMP−9のRNAi媒介性阻害の抗癌効能が示された。哺乳動物細胞における、アンチセンスオリゴヌクレオチドおよび同じ標的に対するsiRNAの抑制効果の比較から、siRNAのIC50値が、アンチセンスオリゴヌクレオチドのものよりも約100倍低いことが明らかになった。siRNAが配列特異的標的遺伝子をサイレンシングする能力、および遺伝子発現を阻害するのに必要なより低い濃度によって、RNAiは遺伝子治療の強力な手段になる。
【0138】
(実施例31)
環状プラスミドからのsiRNAに対する減少した免疫原性応答
uPAおよびuPARのヘアピンsiRNA分子を生成することのできるベクターを開発するために、哺乳動物発現プラスミドベクターpCDNA3を用いた。図34〜35は、種々の形態のU6およびCMV駆動RNAiコンストラクトの概略図を示す。uPAを(346〜367)およびuPAR(77〜89)標的とする、9塩基のGCの欠乏した領域によって間隔が空けられた自己相補的逆方向反復配列を合成した。uPAのオリゴはHindIII部位で終わり、uPARのオリゴはBamHIで終わり、100℃に5分間加熱することによって自己アニーリングし、6×SSC中室温まで冷却し、それぞれの粘着制限酵素認識部位末端を有する二本鎖DNA分子の形成を生じた。これらのdsDNA分子を、pCDNA3プラスミドベクターのBamHIおよびHindIII部位にライゲートし、CMVプロモーターの下流のuPAおよびuPARの逆方向反復を含み、BGHターミネーターによって終了するプラスミドの形成を生じた。得られたプラスミドはpU2と名づけ、哺乳動物細胞にトランスフェクトすると、uPAおよびuPARの両方を標的とする二重のヘアピンsiRNA分子の生成を生じ、これはdsRNA認識酵素(DICER)によってさらにプロセシングされて、RNAiを誘導する個々のsiRNA分子を生成した。スクランブルベクターの構築において、GFPに相同性のある配列を用いた。完全なヘアピン構造を形成しない不完全な配列を用いて、スクランブルベクターを開発した。2つの自己相補的オリゴを合成およびアニーリングして、HindIII部位を有するdsDNA分子を生じた。このdsDNA分子をpCDNA3プラスミドのHindIII部位でライゲートさせた。得られたプラスミドをpSVと呼んだ。得られたCMV駆動転写産物は、ヘアピン様構造を有さず、いかなる天然の遺伝子にも相同性を有さなかった。
【0139】
uPAおよびuPARのsiRNAを発現する発現カセットを、RNAポリメラーゼIIまたはRNAポリメラーゼIIIプロモーターのいずれかで駆動するΔE1領域で、Ad5シャトルベクターにサブクローニングした。得られたプラスミドを、293個の複製許容性細胞にAd5ゲノムプラスミド(PJM17等)でコトランスフェクトして、uPAおよびuPARのsiRNA発現カセットを含む、組換え複製欠損Ad5ウイルス粒子を生じた(図36)。GFP RNAiベクターを構築して、RNAiを用いた標的化の特異性を決定した。GFPを発現する安定なSNB19細胞を対照として用い、GFPに対するRNAiでトランスフェクトした。GFP RNAiでトランスフェクトした細胞はGFP発現を失った(図37)が、RT−PCR反応に見られるように、GAPDH mRNAの発現には変化がなかった(図44)。
【0140】
U6(RNAポリメラーゼIII)またはCMV(RNAポリメラーゼII)プロモーターのいずれかを有する環状プラスミドが細胞レベル免疫応答を誘導するかどうかを決定するために、U6またはCMVプロモーターのいずれかを有する5つのコンストラクトを用いた。SNB19ヒト神経膠腫細胞を、U6またはCMVプロモーターのいずれかを含む環状プラスミドでトランスフェクトし、以下のものを駆動した。空のベクター(EV)と呼ばれる、インサートなし、スクランブルベクター(SV)と呼ばれる、完全なヘアピン構造を形成しないGFP RNAiインサート、uPARのRNAiヘアピンエクスプレッサー(puPAR)、uPAのRNAiヘアピンエクスプレッサー(puPA)、ならびにuPARおよびuPAの両方の二重のRNAiヘアピン発現(pU2)。RT−PCRを行って、OAS1発現レベルを測定した。48時間のトランスフェクション後にトランスフェクト細胞のそれぞれから全RNAを単離し、RT−PCRを行って、全RNA 50ngあたりのOAS1発現のレベルを測定した(RT−PCRは、製造業者の使用説明書(インビトロジェン)どおりに行った)。U6またはCMVプロモーターのいずれかを含む環状プラスミドでトランスフェクトしたSNB19細胞において、OAS1 mRNAのレベルまたはGAPDH mRNAレベルに変化はなかった(図38)。
【0141】
(実施例32)
RNAiの開始のためのプロモーターとしての、RNAポリメラーゼII(CMV)およびRNAポリメラーゼIII(U6)の比較
RNAポリメラーゼIIおよびRNAポリメラーゼIIIの活性および効果を測定するために、pcDNA3の場合のように、スクランブルベクター、uPAR、uPAおよびuPAR−uPAの組み合わせのために、pSilencerプラスミド(アンビオン、テキサス州オースティン)においてRNAiベクターを構築した。pSilencerコンストラクトを、製造業者の使用説明書どおりに、テトラTで終了させた。SNB19細胞を2組でトランスフェクトし、一方の組はRNAポリメラーゼIIプロモーターCMVを含み、第二の組はRNAポリメラーゼIIIプロモーターU6を含んだ(C、SV、puPAR、puPAおよびpU2)。トランスフェクションの48時間後、標準的なプロトコールどおりに細胞からタンパク質を抽出し、12%ポリアクリルアミドSDSゲルにロードした(10μg/レーン)。標準的なプロトコールどおりにウエスタンブロッティングおよびフィブリンザイモグラフィーを行い、uPARおよびuPAに関してプローブし、GAPDHに関するプロービングによってローディング対照を測定した。図39から、RNAポリメラーゼIIIプロモーターコンストラクトと比較して、標的分子のダウンレギュレーションにおいてRNAポリメラーゼIIプロモーターコンストラクトがより効率的であることが明らかであった。
【0142】
(実施例33)
インターフェロン応答遺伝子OAS1の決定
空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)、ならびにuPARおよびuPAの2シストロン性コンストラクト(pU2)のプラスミドコンストラクトを用いて、SNB19ヒト神経膠腫細胞系におけるインターフェロン誘導のレベルを測定した。OAS1遺伝子発現を、インターフェロン誘導の指標として用いた。環状プラスミド(C)、線状発現カセット(L)、およびBGHポリAシグナル配列欠失線状発現カセット(ΔA)を用いた。SNB19細胞を等しい量の上記のプラスミドまたは発現カセット(C、L、ΔA概略図)でトランスフェクトし、標準的なプロトコールを用いて、48時間のトランスフェクションの後に全RNAを単離した。上記の試料に関してRT−PCRを行い、アガロースゲル上でOAS1単位複製配列のレベルを測定した。OAS1増幅に使用したプライマーは、
【0143】
【化41】
および
【0144】
【化42】
であった。上記のプラスミド(EV、SV、puPAR、puPAおよびpU2)由来の発現カセットを増幅するのに使用したプライマーは、フォワードプライマー
【0145】
【化43】
およびリバースプライマー
【0146】
【化44】
であった(図40)。
【0147】
OAS1(2’5’−オリゴアデニル酸シンテターゼ)mRNA誘導のRT−PCRを行って、ポリAシグナル配列の関連性を決定した。環状、線状(発現カセット単独)、およびポリAシグナル配列を欠失した発現カセットを用いた(それぞれC、LおよびΔA)。EVおよびSVの場合、OAS1 mRNAの誘導は検出されなかった(図41)。EVの場合、図に見られるように、ポリA尾部ありまたはなしで、転写産物の全体の長さは1kbよりも大きいとは期待されず、転写産物の予想される構造は、免疫応答を誘導する有意なdsRNA構造を有さなかった(SVも同様)(図44)。対照的に、puPAR、puPAおよびpU2では、予想される二次構造はdsRNA構造を有したが、ポリA尾部ありでは、依然として免疫応答の誘導は検出されなかった(OAS1発現)。ポリAシグナル配列が存在するがトランスフェクトコンストラクトが線状である発現カセット単独の場合、これは免疫応答を誘導した。このことから、環状分子の存在が実行可能なポリA尾部を生成したことが示された。また、線状コンストラクトはポリAシグナル配列の直後で終了したので、生存能のあるポリA尾部の開始は始められなかったか、または不完全だった。ポリAシグナル配列が欠失した線状コンストラクトの場合、免疫応答が開始し、免疫応答の予防および転写RNA分子の安定性にポリA尾部の存在が必要であり得ることが示された(図41〜42)。
【0148】
2シストロン性コンストラクトからの予想されるmRNAは、miRNAに類似性を有さず、uPARおよびuPA配列の両方の、完璧なヘアピンループ構造を有した。24塩基の部分的dsRNAを形成する48塩基の配列を、uPARとuPA配列との間に導入し、両方のsiRNA分子の効率的な転写を可能にした。2シストロン性配列は、BGHポリ−ΔAシグナル配列によってコードされるポリA配列で終わった(図43)。
【0149】
典型的な抗ウイルス応答遺伝子であるOAS1遺伝子のRT−PCRから、トランスフェクトした対照細胞およびEV/SVのように、免疫応答がなかったことが示された。アンチセンスおよびRNAiトランスフェクト細胞におけるuPAおよびuPAR転写産物にもRT−PCRを行った。RT−PCRによって測定すると、アンチセンストランスフェクト細胞ではuPARまたはuPA mRNA転写産物の変化は見られなかったが、RNAiトランスフェクト細胞ではuPARまたはuPAのmRNAレベルが減少し、それぞれのmRNAの破壊が示された(24時間)。RNAiの機構は、標的mRNA分子の破壊を伴う。OAS1発現は、対照群(pGFP、pEVおよびpSV)と同様であり、細胞レベルの免疫応答がないことが示された(図44)。
【0150】
(実施例34)
RNAi発現ベクターのインサイチュー局在化
ヌクレアーゼを含まない条件下で、標準的なプロトコールどおりに、パラフィン包埋切片を脱パラフィンおよび再水和した。これらの切片をプロテイナーゼKで処理して、タンパク質に結合しているDNAを明らかにした。標準的なプロトコールどおりにDNAを変性した。pcDNA3プラスミドを採取し、CMVプロモーターを含む発現カセット(NruI HindIII消化)を熱安定性アルカリホスファターゼ(アマシャム・バイオサイエンス、ニュージャージー州ピスカタウェイ)で標識し、処置切片にハイブリダイズした。製造業者の使用説明書どおりに、ハイブリダイゼーションを行った。mock注射マウスは、アルカリホスファターゼのいかなる活性も示さなかったが、EV、SV、puPAR、puPAまたはpU2の腹腔内注射で処置したマウスはアルカリホスファターゼの活性を示し、CMVプロモーターの存在が示された。アルカリホスファターゼの活性は、ほとんどの場合、血管の周辺に局在し、脈管構造の四方に広がるパターンを示し、CMVを有するプラスミドベクターが血液脳関門を横切ったことを示した(図45)。
【0151】
(実施例35)
uPAR−カテプシンB環状プラスミドのインターフェロン応答遺伝子OAS1の決定
空のベクター(EV)、スクランブルベクター(SV)、uPAR(pU)、カテプシンB(pC)、ならびにuPARおよびカテプシンBの2シストロン性コンストラクト(pCU)のプラスミドコンストラクトを用いて、SNB19ヒト神経膠腫細胞系におけるインターフェロン誘導のレベルを測定した。OAS1遺伝子発現を、インターフェロン誘導の指標として用いた。環状プラスミド(C)、線状発現カセット(L)、およびBGHポリAシグナル配列欠失発現カセット(−A)を用いた。等しい量の上記のプラスミドまたは発現カセットでSNB19細胞をトランスフェクトし、48時間のトランスフェクションの後に、標準的なプロトコールを用いて全RNAを単離した。上記の試料に関してRT PCRを行い、アガロースゲルでOAS1単位複製配列のレベルを測定した。OAS1増幅に使用したプライマーは、
【0152】
【化45】
および
【0153】
【化46】
であった。上記のプラスミド(EV、SV、pU、pCおよびpCU)由来の発現カセットを増幅するのに用いたプライマーは、フォワードプライマー
【0154】
【化47】
およびリバースプライマー
【0155】
【化48】
であった。
【0156】
OAS1(2’5’−オリゴアデニル酸シンテターゼ)mRNA誘導のRT−PCRを行って、ポリAシグナル配列の関連性を決定した。環状、線状(発現カセット単独)、およびポリAシグナル配列を欠失した発現カセットを用いた(それぞれC、L、および−A)。EVおよびSVの場合、OAS1 mRNAの過剰な誘導は検出されなかった。EVの場合、図に見られるように、ポリA尾部ありまたはなしで、転写産物の全体の長さは1kbよりも大きいとは期待されず、転写産物の予想される構造は、免疫応答を誘導する有意なdsRNA構造を有さなかった(SVも同様)。対照的に、pU、pCおよびpCUでは、予想される二次構造はdsRNA構造を有したが、ポリA尾部ありでは、依然として免疫応答の誘導は検出されなかった(OAS1発現)。ポリAシグナル配列が存在するがトランスフェクトコンストラクトが線状である発現カセット単独の場合、これは免疫応答を誘導した。このことから、環状分子の存在が実行可能なポリA尾部を生成したことが示された。また、線状コンストラクトはポリAシグナル配列の直後で終了したので、実行可能なポリA尾部の開始は始められなかったか、または不完全であった。ポリAシグナル配列が欠失した線状コンストラクトの場合、免疫応答が開始し、免疫応答の予防および転写されたRNA分子の安定性にポリA尾部の存在が必要であり得ることが示された。
【0157】
EV、SV、pU、pCおよびpCUで処置した大脳内異種移植腫瘍において、OAS1の誘導を調べた。インターフェロンα(3μg/マウス)で大脳内処置した正常なマウスが含まれ、5時間後に屠殺した。正常な対照組織として脾臓および肝臓を用いて、OAS1発現の存在が通常の条件下で存在する場合の抗体の特異性を実証した。免疫組織化学に加えて、センス
【0158】
【化49】
オリゴを用いてこれらの組織においてインサイチューハイブリダイゼーションを行い、OAS1 mRNAレベルを測定した。大脳内腫瘍を有するマウス脳またはpU、pCおよびpCUで処置したマウスの脳では、OAS1 mRNAおよびタンパク質の極めて少ない発現のみ。注目すべきことに、pCU処置群ではOAS1の誘導がなかった。
【0159】
材料および方法
低分子ヘアピンRNA発現プラスミドの構築
uPA−uPAR:346〜367塩基のuPA
【0160】
【化50】
および77〜98塩基のuPAR
【0161】
【化51】
に特異的な低分子干渉オリゴヌクレオチドを合成し、アニーリングした。アニーリングしたDNAオリゴヌクレオチドの対を、uPAに特異的なものをHindIII部位で、およびuPARに特異的なものをBamHI部位で、pcDNA3ベクターに段階的に挿入することによって、ヒトCMVプロモーター制御下でuPAおよびuPARの両方のshRNAを発現するuPA−uPAR RNAiプラスミドベクターを構築した(sh−uPAuPAR)。また、uPA(sh−uPA)およびuPAR(sh−uPAR)のshRNA発現ベクターを、別々に構築した。完全なヘアピン構造を形成しない、不完全な配列を有するpcDNA3スクランブルベクターを用いて、対照として用いるためのスクランブルベクターを開発した。空のベクター(EV)およびスクランブルベクター(SV)対照を複数の細胞系において試験したが、トランスフェクション後のMTTアッセイで示されるように、細胞に対するいかなる毒性も示さず、ハウスキーピング遺伝子であるGAPDHおよびy−アクチンの発現に対する効果を有さなかった。
【0162】
uPARおよびMMP−9。サイトメガロウイルス(CMV)プロモーターの下流でuPARおよびMMP−9の両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた(概略1)。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域およびBamHI部位を有する長さ21塩基のuPAR配列を、末端に組み込んだ
【0163】
【化52】
。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にEcoRI部位が組み込まれた+360〜+381のMMP−9相補配列
【0164】
【化53】
を、uPARのsiRNA配列を含むベクターのEcoRI部位にライゲートした。これは、最終的に、35bpの分離を有する、uPARおよびMMP−9のsiRNA発現プラスミドを生じた。オリゴは自己相補的で左右対称性を有するので、いずれのインサートの配向も重要ではなかった。SV40ターミネーターを、RNA合成の停止シグナルとした。
【0165】
カテプシンBおよびuPA:サイトメガロウイルス(CMV)プロモーターの下流でカテプシンBおよびuPARの両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた(図17)。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域を有し、BamHI部位が末端に組み込まれた長さ21塩基のuPAR配列
【0166】
【化54】
を用いた。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA−3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にXhoI部位が組み込まれた+732〜+753のカテプシンB相補配列
【0167】
【化55】
を、uPARのsiRNA配列を含むベクターのXhoI部位にライゲートした。これは、最終的に、pCUと呼ばれる、カテプシンBおよびuPARのsiRNA発現プラスミドを生じた。uPAR(pU)およびカテプシンB(pC)の単一のsiRNA発現ベクターもまた構築した。オリゴは自己相補的で左右対称性を有したので、単一または2シストロン中のいずれのインサートの配向も重要ではなかった。BGHポリAターミネーターを、3つ全てのコンストラクトのRNA合成の停止シグナルとした。
【0168】
uPAR、uPAおよびMMP−9:サイトメガロウイルス(CMV)プロモーターの下流でuPAR、uPAおよびMMP−9の両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域を有し、BamHI部位が末端に組み込まれた長さ21塩基のuPAR配列
【0169】
【化56】
を用いた。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にHindIII部位が組み込まれた+346〜+367のuPA相補配列
【0170】
【化57】
をHindIII部位にライゲートし、+360〜+381のMMP−9
【0171】
【化58】
を、uPARおよびuPAのsiRNA配列を含むベクターのEcoRI部位にライゲートした。これは、最終的に、pU2Mと呼ばれる、uPAR、uPAおよびMMP−9のsiRNA発現プラスミドを生じた。uPAR(puPAR)、uPA(puPA)およびMMP−9(pMMP−9)の単一のsiRNA発現ベクターもまた構築した。オリゴは自己相補的で左右対称性を有したので、単一または3シストロン性コンストラクト中のいずれのインサートの配向も要因ではなかった。BGHポリAターミネーターを、4つ全てのコンストラクトのRNA合成の停止シグナルとした。
【0172】
カテプシンBおよびMMP−9:カテプシンB(732〜753)およびMMP−9(360〜381)を標的とする、GCの欠乏した9塩基の領域によって間隔を空けられた自己相補的逆方向反復配列を合成した。カテプシンBのオリゴはXhoI部位で終わり、MMP−9のオリゴはEcoRIで終わり、5分間100℃に加熱することによって自己アニーリングさせ、6×SSC中で室温まで冷却して、それぞれの粘着制限酵素認識部位末端を有する、二本鎖DNA分子の形成を生じた。これらのdsDNA分子をpCDNAプラスミドベクターのXhoIおよびEcoRI部位にライゲートして、CMVプロモーターの下流にカテプシンBおよびMMP−9の逆方向反復を含み、SV40ターミネーターによって終了するプラスミドの構築を生じた。哺乳動物細胞にトランスフェクトした、pCMと呼ばれる得られたプラスミドは、カテプシンBおよびMMP−9の両方を標的とする二重ヘアピンsiRNA分子の生成を生じ、これはdsRNA認識酵素(DICER)によってさらにプロセシングされて、RNAiを誘導する個々のsiRNA分子を生成する(概略1)。
【0173】
細胞培養およびトランスフェクション条件:
前立腺癌細胞:ヒト前立腺癌細胞系LNCaP、DU145およびPC3を、アメリカン・タイプ・カルチャー・コレクション(バージニア州マナッサス)から得た。LNCaP細胞を、2mM L−グルタミン、1.5g/L重炭酸ナトリウム、4.5g/Lグルコース、10mM HEPES、および1.0mMピルビン酸ナトリウムを補充したRPMI培地(インビトロジェン、カリフォルニア州カールスバッド)中で増殖させた。PC3およびDU145細胞を最小必須培地中で増殖させた。両方の培地は、10%ウシ血清アルブミン(ギブコBRL、テキサス州ルイスビル)および5%ペニシリン/ストレプトマイシンを含み、5%CO2の加湿雰囲気中で、370Cのインキュベーター中で維持した。製造業者の使用説明書どおりにリポフェクタミン(商標)2000試薬(ライフ・テクノロジー、メリーランド州ロックビル)を用いて、トランスフェクションを行った。72時間のトランスフェクションの後、細胞増殖アッセイ、免疫ブロット解析、RT−PCR解析、マトリゲル浸潤アッセイ、DNA断片化アッセイ、EMSAアッセイおよびカスパーゼ活性アッセイに細胞を用いた。DAPIおよび二重免疫染色のために、Lab−Tek IIチャンバースライド(ナルジェ・ヌンク・インターナショナル、イリノイ州ネーパービル)中でトランスフェクションを行った。
【0174】
神経膠芽腫細胞:ヒト神経膠芽腫細胞系SNB19を、10%FCS、100−μg/mlストレプトマイシンおよび100−単位/mlペニシリン(インビトロジェン、カリフォルニア州カールスバッド)を補充したDMEM F−12(シグマ・ケミカル・カンパニー、ミズーリ州セントルイス)中、37℃、加湿した5%CO2雰囲気下で維持した。製造業者の使用説明書どおりにリポフェクタミン試薬(インビトロジェン、ニューヨーク州グランドアイランド)を用いて、siRNAを発現するpC pUまたはpCUプラスミドで細胞をトランスフェクトした。トランスフェクション後、血清を含まない培地中で48時間細胞をインキュベートした。
【0175】
フィブリンザイモグラフィー:電気泳動で分離された形態のuPAの酵素活性および分子量を、SDS−PAGEによって、前立腺癌細胞系LNCaP、DU145およびPC3の馴化培地中で測定した。簡潔に述べると、SDS−PAGEゲルは、重合前に、精製されたプラスミノゲンおよびフィブリノーゲンが基質である、アクリルアミドを含有する。重合後、試料中の等しい量のタンパク質を電気泳動し、ゲルを洗浄および染色した。EV/SV puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように行った。
【0176】
ゼラチンザイモグラフィー:EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトした細胞から、馴化培地を回収し、遠心分離して細胞残屑を除去した。ゼラチン(0.5mg/ml)を含有する10%ドデシル硫酸ナトリウム−ポリアクリルアミドゲルを用いて、得られた試料の20μgを、ゼラチナーゼ活性に関してアッセイした。アミドブラック(シグマ・アルドリッチ、ミズーリ州セントルイス)でゲルを染色し、ゲル中の明確なバンドの面積として、ゼラチナーゼ活性を可視化した。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように行った。
【0177】
逆転写−PCR解析:
uPA−uPAR:キアジェンRNeasyキットを用いて細胞RNAを単離し、1gのRNAをDNase処理(10単位/RNA 1g、1時間)し、逆転写反応(RT、20l)の鋳型として用いた。RT反応ミックス(インビトロジェン)は、1l(10pm)のプライマーを含んだ。次いで、得られたcDNAをPCR反応において用いて、ゲル電気泳動によって解析した。以下のプライマーを用いた。
uPA−センス:
【0178】
【化59】
uPA−アンチセンス:
【0179】
【化60】
uPAR−センス:
【0180】
【化61】
uPAR−アンチセンス:
【0181】
【化62】
GAPDH−センス:
【0182】
【化63】
GAPDH−アンチセンス:
【0183】
【化64】
【0184】
【表1】
本明細書中に記載されるように、対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞のRT−PCR解析を行った。
【0185】
PCR条件は以下の通りであった。95℃で5分間、その後95℃で1分間、55℃で1分間、および72℃で1分間を35サイクル。最後の伸長は72℃で5分間であった。アニーリング温度は、種々のコンストラクトの配列に依存して変化し、標準的な手順に従って行った。
【0186】
PC3免疫蛍光検出:種々のshRNAプラスミドでトランスフェクトしたPC3細胞を、4%パラホルムアルデヒドで固定し、抗uPA(1:500、バイオメダ、カリフォルニア州フォスターシティ)および/または抗uPAR(1:500、アメリカン・ダイアグノスティクス・インク、コネチカット州グリニッジ)とともにインキュベートした。洗浄後、蛍光二次抗体(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)を1:500希釈で加えた。再度細胞をPBSで3回洗浄し、DAPIで対比染色した。顕微鏡(オリンパス、ニューヨーク州メルヴィル)に接続された電荷結合素子RTスライダースポットカメラ(ダイアグノスティック・インストゥルメンツ・インク、ミシガン州バローズ・スターリング・ハイツ)を用いて、蛍光像を得、スポットRTソフトウエアバージョン3.5(ダイアグノスティック・インストゥルメンツ・インク、ミシガン州バローズ・スターリング・ハイツ)を備えたコンピュータによって管理した。
【0187】
PC3細胞マトリゲル浸潤アッセイ:トランスフェクション後、細胞を分離し、PBS中で2回洗浄した。5×105個の細胞を、マトリゲル(0.7mg/ml)(コラボラティブ・リサーチ・インク、マサチューセッツ州ボストン)でコートしたトランスウェルインサート(12μM孔)の上の方のチャンバーに播種した。下の方のチャンバーに400lのRPMI培地を充填した。24時間のインキュベーション期間の後、上の方のチャンバーの遊走しなかった細胞を穏やかにこすり落とし、インサートの下面に存在する接着細胞をHema−3で染色し、写真を撮った。
【0188】
製造業者の使用説明書どおりにインサイチューカスパーゼ活性アッセイ:ポリカスパーゼ検出キット(イムノケミストリー・テクノロジース、イリノイ州ブルーミントン)を用いて、カスパーゼ活性を検出した。このアッセイにおいて、細胞透過性で非細胞障害性の、カスパーゼの蛍光色素インヒビター(FLICA)が活性のあるカスパーゼヘテロ二量体の大きいサブユニット上の反応性システイン残基に共有結合し、それによってさらなる酵素活性を阻害する。このキットは、ほとんどのカスパーゼの検出のための一般的なプローブであって、緑色蛍光を発光する、多くのカスパーゼ(カスパーゼ1、−3、−4、−5、−6、−7、−8および−9、FAM−VAD−FMK)のカルボキシフルオレセイン標識フルオロメチルケトンペプチド阻害因子を使用する。緑色蛍光シグナルは、試薬を加えた時点での細胞中の活性のあるカスパーゼの量の直接の尺度である。72時間のトランスフェクションの後、細胞をFAM−VAD−FMK色素(インサイチューマーカー)で染色することによって、カスパーゼ活性を検出した。結合したマーカーを、共焦点顕微鏡で観察される蛍光検出によって局在化した。核染色にDAPIを用いた。
【0189】
DNA標識アッセイ:トランスフェクションの後、細胞を採取し、PBS中で2回洗浄した。細胞ペレットを、0.1mg/mlプロテイナーゼK(インビトロジェン)を含有する溶解バッファー(10mM Tris−HCl、400mM NaCl、1mM EDTAおよび1% TritonX−100)に再懸濁し、次いで37℃で2時間インキュベートした。遠心分離によって溶解物からDNAを取り除き、次いで等しい量のフェノール/クロロホルムを用いて抽出し、−80℃で2時間無水エタノールおよび0.3M酢酸ナトリウム(pH5.2)を加えることによって沈殿させた。DNAをTris−EDTAバッファー(10mM Tris−HCl、pH7.5、1mM EDTA)に再懸濁し、37℃で1時間RNaseAで処理し、次いで、エチジウムブロマイド(0.5g/ml)で染色した1.5%アガロースゲルで分離させた。
【0190】
電気泳動移動度シフトアッセイ(EMSA):トランスフェクションの後、製造業者の使用説明書どおりにタンパク質抽出キット(アンビオン、テキサス州オースティン)を用いて、核タンパク質を抽出した。ビシンコニン酸手順(ピアス・バイオケミカル・カンパニー、イリノイ州ロックフォード)を用いて、希釈した試料で核タンパク質の濃度を測定した。パノミクス(カリフォルニア州レッドウッドシティ)の電気泳動移動度シフトアッセイ(EMSA)キットを用いて、タンパク質抽出物中のStat3とDNAプローブとの間の相互作用を調査した。
【0191】
DNA断片末端標識アッセイ:shRNA処置または対照前立腺腫瘍組織切片(厚さ5M)を脱パラフィンおよび再水和した。次に、標本全体をプロテイナーゼK溶液(10mM Tris中20g/mlプロテイナーゼK、pH8)で覆うことによって、組織切片を透過化し、室温で20分間インキュベートした。次いで、組織切片をTris緩衝生理食塩水(1×TBS、20mM Tris pH7.6、140mM NaCl)で洗浄した。組織切片を、メタノール中に希釈した3%過酸化水素に室温で5分間浸漬することによって、内在性ペルオキシダーゼの不活性化を達成した。次いでスライドガラスをクレノウ平衡バッファー(50mM Tris pH8、50mM NaCl、10mM MgCl2)に30分間入れた。次いで、製造業者の使用説明書(クレノウ−FragEL
DNA断片化検出キット、オンコジーン・リサーチ・プロダクツ、マサチューセッツ州ケンブリッジ)に従って、37℃で90分間、加湿チャンバー中で、組織切片を、クレノウでのDNA断片末端標識に最適な比で標識および非標識デオキシヌクレオチドの混合物を含有する60lの溶液とともにインキュベートした。EDTA(0.5M、pH8)との室温で5分間のインキュベーションによって、酵素反応を停止した。次いでスライドをTBSで洗浄し、ブロッキングバッファーに10分間浸漬し(PBS中4%のBSA)、その後ペルオキシダーゼストレプトアビジンを含有する100lの溶液とともに30分間、加湿チャンバー中室温でインキュベートした。次いで、組織切片をTBS中で洗浄し、3,3’ジアミノベンジジン(DAB、0.7mg/ml)、過酸化水素および尿素(0.6mg/ml)を含有する溶液で覆った。次に、スライドを蒸留水で洗浄し、メチルグリーン(0.3%)で30秒間対比染色し、オリンパス蛍光顕微鏡下で調べた。スポットRTソフトウエアバージョン3.5(ダイアグノスティック・インストゥルメンツ、ミシガン州)を用いて、陽性のDNA断片末端標識染色を、6つの無作為に捕捉した像/試料から点数付けした。
【0192】
同所マウス前立腺処置モデル:無胸腺症雄ヌードマウス(nu/nu、6〜8週齢)を、ハーラン・スプラグ−ドーレイ(インディアナ州インディアナポリス)から得た。動物取り扱いおよび実験手順は、イリノイ医科大学動物実験委員会によって承認された。これまでに記載されているように、同所移植を行った。簡潔に述べると、ケタミン(50mg/kg)およびキシラジン(10mg/kg)での全身麻酔の後、下腹部で下正中切開を行った。30μlのPBS中のPC3細胞の懸濁液(1×106)を前立腺の側葉に注射し、外科用金属クリップで傷を閉じた。この細胞濃度は、7日間の移植の間に一貫した局所的腫瘍増殖を達成するのに必要であった。マウスを、1つの処置群あたり6匹のマウスの、5つの処置群に分割した。移植後7および14日目に、下正中切開を行い、腫瘍に、sh−uPA、sh−uPAR、sh−uPA−uPARまたはEV/SV対照(それぞれ75μg/150μg)を発現するプラスミドコンストラクトを注射した。別の組の実験において、7および14日目に、同所に移植したマウスに、sh−uPAおよびsh−uPARプラスミドを腫瘍内に同時注射した(それぞれ150μg)。最後のshRNAプラスミド注射の14〜15日後にマウスを屠殺し、目視検査によって原発腫瘍増殖および転移の部位を決定し、写真を撮った。次いで原発腫瘍を切除し、測定および計量した。H&E染色のために、ホルマリン中で標本を固定し、パラフィンに包埋した。また、組織の一部を免疫ブロットのために直ちに瞬間凍結した。
【0193】
ウエスタンブロッティング。SNB19細胞をmock、空のベクター、pC、pUまたはpCUでトランスフェクトし、48時間インキュベートした。インキュベートの終わりに、細胞を採取し、冷PBSで2回洗浄し、プロテアーゼ阻害因子を含有するバッファー(150mM NaCl、50mM Tris−Hcl、2mM EDTA、1% NP−40、PH7.4)中で溶解した。上清または細胞由来の等しい量のタンパク質(30μg/レーン)を非還元性条件下、10%アクリルアミドゲルで電気泳動した。SDS−PAGEの後、タンパク質をポリ二フッ化ビニリデン膜(バイオ−ラッド)に移した。非特異的結合をブロックするために、5%無脂肪スキムミルクを含む、0.1% Tween−20を有するPBS[T−PBS]中で膜を2時間インキュベートした。その後、それぞれ、T−PBS+5%無脂肪乳中のカテプシンB、uPAR、ERK、pERK、FAKまたはpFAKに対する抗体とともに、膜を2時間インキュベートした。T−PBS中での洗浄の後、製造業者の使用説明書どおりにペルオキシダーゼ標識抗ウサギ抗体を有するECL(商標)検出キット(アマシャム・ファルマシア・バイオテック、英国アマシャム)を用いて、膜上のタンパク質を可視化した。ローディング対照のために、膜を細長い形に切り、標準的なプロトコールどおりに、β−アクチンに対するモノクローナル抗体でプローブした。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞の免疫ブロット解析もまた、本明細書中に記載されるように行った。uPA−uPAR免疫ブロット解析に、以下の抗体を用いた。抗uPA(バイオメダ、カリフォルニア州フォスターシティ)、抗uPAR(アメリカン・ダイアグノスティクス・インク、コネチカット州グリニッジ)、抗Bax(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)、抗Bcl−XS/L(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)、抗カスパーゼ9(セル・シグナリング・テクノロジー・インク、マサチューセッツ州ベヴァリー)、および抗GAPDH(アブカム、マサチューセッツ州ケンブリッジ)。全体的およびリン酸形態のERK、JNK、p38およびStat3に対する抗体を、サンタクルス・バイオテクノロジー(カリフォルニア州サンタクルス)から得た。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞のウエスタンブロッティングもまた、本明細書中に記載されるように行った。
【0194】
免疫組織化学的解析。SNB19細胞(1×104)を、ビトロネクチンコートした8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、pMMP−9およびpU2Mでトランスフェクトした。さらに72時間後、細胞を3.7%ホルムアルデヒドで固定し、PBS中の1%ウシ血清アルブミンとともに室温で1時間、ブロッキングのためにインキュベートした。スライドをPBSで洗浄した後、IgG抗uPAR(ウサギ)またはIgG抗MMP−9(マウス)のいずれかを、1:200の濃度で加えた。スライドを4℃で一晩インキュベートし、PBSで3回洗浄して、過剰な一次抗体を除去した。次いで細胞を抗マウスFITC複合体または抗FITC複合体IgG(1:500希釈)とともに、室温で1時間インキュベートした。次いで、スライドを3回洗浄し、ガラスカバースリップで覆い、蛍光顕微鏡写真を得た。合成合併像を得て、対照/EV、SV、puPAR、pMMP−9およびpUMトランスフェクト細胞におけるuPARおよびMMP−9の発現を可視化した。
【0195】
SNB19細胞増殖アッセイ。MTSアッセイによって細胞増殖を評価した。インビトロでのSNB19細胞の増殖に対するこれらのコンストラクトの効果を検出するために、セルタイター(商標)比色アッセイを用いて、生存能のある細胞質量を測定した。5×103個の神経膠芽腫細胞を三連で96または24ウェルプレートに播種し、24時間増殖させた後、培地のみ(mock)、EV、SV、pC、pUおよびpCUベクターで48時間トランスフェクトした。次いで、これらの細胞、血清含有培地に換え、種々の時間間隔を割り当てた。各時点の前に、MTS試薬を加えて、さらに2時間インキュベーションを継続して発色させた。ELISAプレートリーダーを用いて、各ウェルでA490を測定した。短期間対長期間の細胞培養の吸光度読み取りを比較し、これらのコンストラクトの効果を、対応する未処置/処置群の増殖に関して解釈した。siRNAコンストラクトによる増殖のパーセント阻害を、これらのコンストラクトを除いた同じ培地中での同じ細胞の増殖速度に関して計算した。
【0196】
PC3細胞増殖アッセイ:トランスフェクションの72時間後の細胞の生存能を、MTTアッセイを用いて評価した。MTT[3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド](シグマ)を、500g/mlの濃度で各ウェル中の培地に加え、プレートを37℃で4時間インキュベートした。酸−イソプロパノール(0.04N HCl/イソプロパノール)を全てのウェルに直ちに加え、暗青色の結晶が効果的に溶解するように、勢いよく混合した。570nmで吸光度を測定した(ベンチマーク、バイオラッド、カリフォルニア州ハーキュリーズ)。
【0197】
SNB19インビトロ新脈管形成アッセイ。SNB19細胞(2×104)を8ウェルチャンバースライドに播種し、標準的なプロトコールどおりに、mock、EV、pU、pCおよびpCUでトランスフェクトした。24時間のインキュベーション期間の後、培地を除去し、4×104個のヒト皮膚内皮細胞を播種し、72時間共培養させた。3.7%ホルムアルデヒド中での固定の後、第VIII因子抗原に関して内皮細胞を免疫プローブした。第VIII因子抗体は、DAKOコーポレーション(カリフォルニア州カーピンテリア)から購入した。細胞をPBSで洗浄し、FITC複合二次抗体とともに1時間インキュベートし、次いで洗浄し、蛍光顕微鏡下で調べた。SNB19 mock、EV、pU、pCまたはpCUトランスフェクト細胞からの馴化培地存在下で増殖した内皮細胞の同様のスライドをH&Eで染色して、ネットワーク形成を可視化した。新脈管形成の定量化にイメージプロソフトウエアを使用し、以下の方法によって新脈管形成の程度を測定した。分枝点の数および点あたりの分枝の総数を無作為に数え(10視野あたり)、新脈管形成の程度を示す生成物を対照と比較した。8ウェルチャンバースライドに播種してmock/EV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞(2×104)のインビトロ新脈管形成アッセイを、本明細書中に記載されるように行った。8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞(1×104ウェル−1)の新脈管形成アッセイを、本明細書中に記載されるように行った。
【0198】
背側皮下脂肪チャンバーモデル:無胸腺症ヌードマウス(nu/nu、18匹の雄/雌、28〜32g)を、特定の病原体、微生物を含まない環境内で飼育および維持した。背側皮下脂肪チャンバーモデルの移植技術。無菌の小動物外科技術は以下の通りであった。マウスをケタミン(50mg/kg)ザイラジン(10mg/kg)の腹腔内注射によって麻酔した。一旦動物を完全に麻酔すると、10mlの空気を注射することによって、マウスに背側空気嚢を作製した。密閉剤で「O」リング(フィッシャー)の縁の両側に0.45ミクロンのミリポア膜(フィッシャー)を一直線に並べることによって、拡散チャンバー(フィッシャー)を用意した。一旦チャンバーを乾燥させる(2〜3分)、20分間のUV照射によってそれらを滅菌した。20μlのPBSを用いて、膜を濡らした。100〜150μlの無菌PBSに懸濁した2×106個のSNB19細胞(mock、空のベクターまたはpCUトランスフェクト)を、「O」リングの開口部を通してチャンバーに注射した。少量の骨ろうで開口部を密閉した。背側空気嚢の縁に沿って水平に11/2〜2cmの表面切除を行い、空気嚢を開いた。鉗子の助けを借りて、チャンバーを皮膚の下に入れ、注意深く縫合した。10日後に動物をケタミン/キシラジンで麻酔し、生理食塩水(10ml)と、ついで10mlの10%ホルマリン/0.1Mリン酸溶液およびそれに続くPBS中0.001%FITC溶液の心臓内還流によって屠殺した。動物の、移植したチャンバーの周りの皮を注意深くはぎ、移植したチャンバーを皮下空気筋膜から除去した。可視光下で、およびFITC蛍光に関して、チャンバーを覆っている皮下脂肪の写真を撮った。空気嚢筋膜の領域のチャンバー中の血管の数を計数し、それらの長さを測定した。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように、背側皮下脂肪チャンバーモデルに使用した。
【0199】
SNB19細胞遊走アッセイ:SNB19細胞のGFP発現バリアントの、ダルベッコ改変イーグル培地中の2×106個の細胞の懸濁液を、超低接着100mm組織培養プレート上に播種し、スフェロイド凝集物が形成されるまで培養した。直径が約150μmのスフェロイド(約4×104個の細胞/スフェロイド)を選択し、mock、空のベクター、pC、pUおよびpCUでトランスフェクトして、48時間培養した。トランスフェクションの72時間後、単一の神経膠腫スフェロイドを、ビトロネクチンコートされた(50μg/mL)96ウェルマイクロプレートの各ウェルに入れ、200μlの、血清を含まない培地とともに培養した。スフェロイドを37℃で24時間インキュベートし、その後スフェロイドを固定し、Hema−3で染色し、写真を撮った。ステージおよび接眼マイクロメータで較正した顕微鏡を用いて、スフェロイドから単層への細胞の遊走を測定し、細胞遊走の指数とした。神経膠芽腫細胞を三連で96または24ウェルプレートに播種し、24時間増殖させた後、本明細書中に記載されるように、培地のみ(mock)、EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトした。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞の細胞遊走アッセイを、本明細書中に記載されるように行った。SNB19細胞(1×104ウェル−1)のアッセイを8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0200】
SNB19細胞ボイデンチャンバー浸潤アッセイ:改変ボイデンチャンバーアッセイを用いて、カテプシンBおよびuPARのsiRNAを発現するベクター存在下でのSNB19細胞のインビトロ侵襲性を評価した。SNB19細胞を、カテプシンBおよびuPARのsiRNAを単独で、またはともに発現するmock、EV、pU、pCまたはpCUベクターで48時間トランスフェクトした。1×106個の細胞を、0.2%BSAを補充した600μlの、血清を含まない培地中に懸濁し、マトリゲル(0.7mg/ml)でコートされたトランスウェルチャンバー(コーニング・コスター・フィッシャー・サイエンティフック カタログ番号07−200−158、ペンシルバニア州ピッツバーグ)の上の区画に入れた。チャンバーの下の区画を、200μlの、血清を含まない培地で充填し、細胞を24時間遊走させた。インキュベーションの後、細胞を固定し、Hema−3で染色し、写真を撮った。浸潤アッセイの定量化を行った。SNB19細胞(1×104ウェル−1)のアッセイを、8ウェルチャンバーに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0201】
SNB19スフェロイドアッセイ:SNB19神経膠芽腫細胞(3×106)を、DMEM中で調製した0.75%アガーで予めコートした100mm組織培養プレート(コーニング、ニューヨーク州コーニング)に播種し、スフェロイド凝集物が形成されるまで培養した。直径100〜200μmのスフェロイドを選択し、mock、空のベクター、pC、pUおよびpCUで48時間トランスフェクトした。感染の3日後、SNB19スフェロイドを蛍光色素DiIで染色し、DiOで染色したラット脳凝集物と接触させて置いた。これまでに記載されたように、共焦点レーザー走査型顕微鏡を用いてラット脳凝集物の進行性の破壊およびSNB19細胞の浸潤を観察し、写真を撮った。共培養の間の脳凝集物または腫瘍スフェロイドの残りの体積を、これまでに記載されたように測定した。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞のスフェロイドアッセイを、本明細書中に記載されるように行った。SNB19細胞(1×104ウェル−1)のアッセイを8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0202】
神経膠腫解析のマウス実験。定位フレームを用いて、SNB19 GFP細胞(2×106)をヌードマウスの脳に注射した。8〜10日後、マウスをmock、空のベクターEV/SV、pC、pUおよびpCUで処理した。0.25μl/時間の速度で、アルゼット(ダイレクトコーポレーション、カリフォルニア州キュパージョン)ミニ浸透圧ポンプを用いて、ベクターのインビボ大脳内送達を行い、150μgベクターDNAのmock(PBS)、150μg pC、150μg pU、150μg pCUまたはpuPARベクター(150μg)、pMMP−9ベクター(150μg)およびpUMベクター(150μg)を脳に注射した(マウス1匹あたり100uL)。全ての実験は、ピオリアのイリノイ医科大学での実験を承認するインスティチューショナル・アニマル・コントロール・ユーザーズ・コミッティーによって設定された、機関のガイドラインを遵守して行った。5週間後、または対照マウスが症状を示し始めたときに、ホルムアルデヒドの心臓還流によってマウスを麻酔した。次いで、標準的なプロトコールどおりに、脳を除去し、パラフィン包埋した。切片を調製し、GFP発現に関して観察するか、またはH&Eで染色した。切片は、各例における腫瘍サイズに関して半定量的に、盲検化して検討および点数付けされた。1切片あたりの平均腫瘍面積を用いて腫瘍体積を計算し、対照と処置群との間で比較した。SNB19細胞(1×104ウェル−1)の実験を8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0203】
統計的解析:グラフパッド・プリズムソフトウエア(カリフォルニア州サンディエゴ)を用い、異なる値の間の有意性の解析のためのANOVAを用いて、統計的比較を行った。値を、少なくとも3つの別々の実験からの平均値SDとして表し、P値が0.05未満で、差を有意であると見なした。
【0204】
大脳内腫瘍増殖阻害。大脳内腫瘍モデルのために、2×106個のSNB19細胞をヌードマウスに大脳内注射した。腫瘍を10日間増殖させた。このとき、動物を7つの群に無作為化し、EV/SV、puPAR、puPA、pMMP−9およびpU2M(0.25μl時間−1の速度でアルゼットミニポンプを用いて、150μgの各コンストラクトを脳に注射した(各群あたり6匹のマウス)。腫瘍接種の5週間後、PBS中3.5%ホルムアルデヒドの心臓還流によって、各群からの6匹のマウスを屠殺した。それらの脳を除去し、24時間4%パラホルムアルデヒドに入れ、パラフィン包埋および切片化した。蛍光顕微鏡下でGFP蛍光に関して切片をスクリーニングし、腫瘍増殖を調べた。切片を盲検化して検討し、腫瘍サイズに関して半定量的に点数付けした。各切片中の平均腫瘍面積を用いて腫瘍体積を計算し、対照と処置群との間で比較した。
【0205】
マトリゲル浸潤アッセイ。空のベクター(EV)、またはカテプシンBおよびMMP−9のsiRNAを発現するベクター(pCM)のいずれかでのトランスフェクションの後、ボイデンチャンバー浸潤アッセイを用いて、トランスフェクトSNB19細胞の侵襲性をインビトロで試験した。簡潔に述べると、8μmの孔を有するトランスウェルインサートをマトリゲル(0.7mg/ml)(コラボラティブ・リサーチ,インク、マサチューセッツ州ボストン)でコートした。SNB19細胞をトリプシン化し、500μlの細胞懸濁液(1×106個の細胞/ml)を三連でウェルに加えた。37℃で24時間のインキュベーションの後、ウェル下部へフィルターを通過した細胞を定量化し、ウェル上部および下部の細胞の合計のパーセンテージとして表した。膜の下側の細胞を固定し、Hema−3で染色し、写真を撮った。記載される他のコンストラクトのアッセイもまた、本明細書中に記載されている手順に従って行った。
【0206】
核酸の送達:例えば親油性薬剤、アデノ、アデノ関連またはレンチを含むウイルスまたは修飾されたウイルス等の任意の型の方法を用いて、核酸の送達を達成する。二本鎖RNA、一本鎖RNA、または一方の鎖がDNAで他方がRNAであるか、またはリボースもしくはデオキシリボースバックボーンを同じ鎖上に有するDNA−RNAハイブリッドとしての、平滑または粘着末端を有する、環状または線状であるむき出しのプラスミドコンストラクトもまた、使用する。タンパク質もしくは炭化水素またはそれらの組み合わせでコートされた、またはホルモンもしくはホルモン誘導体と関連して使用される、DNA、RNAまたはDNA−RNAハイブリッド。化学的に修飾されたDNAまたはRNAまたはDNA−RNAハイブリッドもまた、uPAR、uPA、MMP−9およびカテプシンBを標的とするRNAiを誘導する治療分子として、任意の組み合わせで使用され得る。意図される使用は、真核生物または細胞系、好ましくはヒトおよびヒト細胞系である。
【0207】
siRNAまたはshRNAの送達のための当業者に公知の方法および治療組成物は、本開示の範囲内である。例えば、本明細書中に開示されるsiRNA、shRNAおよび他の核酸は、ウイルスベクター送達、リポフェクション、電気化学的および生化学的方法等の技術を通じて、哺乳動物細胞に送達され得る。ウイルスベースの送達は、目的の核酸を含む組換えアデノウイルス、レンチウイルス、レトロウイルスの、または任意の適したベクターを作製すること、および当業者に公知の技術を用いてウイルスベクターを送達することを含む。リポソームベースの送達系としては、リポフェクションおよびカルジオリピンベースの組成物が挙げられる。むき出しの核酸の直接の、および化学的または生化学的佐剤と組み合わせた送達は、本開示の範囲内である。例えばuPA、uPAR、MMP−9、およびカテプシンB等の標的配列に対する、例えばsiRNAを含む環状プラスミドは、直接注射もしくは腫瘍内送達されるか、または腹腔内注射され得る。同様に、合成または化学的に調製された核酸はまた、腫瘍内または腹腔内で送達され得る。また、siRNA、またはsiRNAを発現する核酸の選択的または特異的送達は、ペプチド核酸(PNA)もしくは抗体または任意の適した標的化因子等の別の作用物質との適切なカップリングを通じても達成され得る。上述の技術および方法は、細胞間、細胞内、インビトロ細胞培養物、インビボ、器官内部で、血液脳関門を横切っての、前立腺癌、神経膠腫、乳癌、および結腸癌への核酸配列の送達に適しており、適用可能である。
【0208】
配列:種々のsiRNAコンストラクトの配列(部分的配列)を本明細書中に開示する。下線を引いた部分は、自己相補的逆方向反復を示す。太字は介在ループ配列を示す。
【0209】
UPAR−uPA
【0210】
【化65】
UPAR−MMP−9
【0211】
【化66】
UPAR CB
【0212】
【化67】
MMP9−CB
【0213】
【化68】
UPAR、uPAおよびカテプシンB
【0214】
【化69】
引用した文書
これらの文書は、本開示における材料および方法に関連する範囲にのみ含められる。
【0215】
【化70】
【0216】
【化71】
【0217】
【化72】
【0218】
【化73】
【背景技術】
【0001】
背景
腫瘍進行は、細胞遊走の間の腫瘍細胞接着のモジュレーションおよび組織浸潤の間の細胞外マトリックス(ECM)の分解を伴う。プロテアーゼの複雑なバランス、それらの活性化因子およびそれらの阻害因子は、腫瘍浸潤の間に、これらの過程の両方を調節する。3つのクラスのECM分解プロテイナーゼは、セリンプロテイナーゼ、メタロプロテイナーゼおよびシステインプロテイナーゼである。ウロキナーゼプラスミノゲン活性化因子(uPA)は、ほとんどのマトリックスおよび基底膜構成要素を分解し、細胞−細胞および細胞−マトリックス相互作用を妨げ得る、プロテアーゼのカスケードを開始させる。細胞表面受容体であるウロキナーゼプラスミノゲン活性化因子受容体(uPAR)に結合するuPAは、プラスミノゲン活性化の数倍の増大によって示されるように、ECM分解に関与する。uPAはまた、ECM構成要素の分解の後に、いくつかの増殖因子を活性化する。uPAの、その受容体であるuPARとの結合は、マイトジェン活性化プロテインキナーゼ(MAPK)ならびに転写(開始)経路のシグナル伝達性転写因子(Stat)経路を含む多数の経路を通じて下流のシグナリング分子を活性化する。
【0002】
最近のRNA干渉(RNAi)の発見によって、癌治療における新しい道が開かれた。RNAiは、標的遺伝子の配列と相同な二本鎖RNA分子を通じて影響を受ける、配列特異的転写後遺伝子サイレンシング機構である。
【0003】
RNA干渉(RNAi)は、配列特異的転写後遺伝子サイレンシング機構であり、二本鎖RNA(dsRNA)によって引き起こされ、dsRNAに相同な配列を有するmRNAの分解を引き起こす。RNAiは、アンチセンス鎖が標的とする遺伝子の転写産物に相補的である、二本鎖RNA(dsRNA)の形成に依存する。配列特異的阻害RNAiはまた、哺乳動物細胞中で誘導され得る。RNAiのある実行において、哺乳動物細胞中の標的mRNAの選択的分解は、二本鎖の低分子干渉RNA(siRNA)でのトランスフェクションによって達成され、標的の迅速で効率的な分解につながった。これらのsiRNAは、文書によって十分立証された、哺乳動物細胞中のより長い二本鎖RNAによって引き起こされる非特異的効果を避けることが示された。
【0004】
前立腺癌は、米国人男性で2番目に多い悪性腫瘍であり、2004年には推定230,110人の新しい症例およびおよそ30,000件の死亡例があった。このように、前立腺癌は、米国および世界中で主要な公衆の健康問題を提起する。現在、転移性前立腺癌は不治であり、最終的には患者の生命を奪う。前立腺癌の相対的な重大さの要因は、転移を引き起こす、構成要素の腫瘍細胞の侵襲性である。腫瘍細胞の侵襲性の性質は、癌転移に特徴的である。腫瘍細胞浸潤および転移は、3つの顕著な段階である、細胞外マトリックスへの悪性(新生物)細胞の接着、マトリックスが分解されて原発腫瘤から細胞が放出されること、および腫瘍細胞が二次標的へ遊走することの、複雑な過程である。
【0005】
多形神経膠芽腫(GBM)は、高度に抗療性の、高度に悪性の中枢神経系原発新生物である。神経膠芽腫を治療に対して抵抗性にしているある特性は、腫瘍細胞が正常な脳組織に侵潤する傾向である。正常な脳組織に影響する療法は、許容されない。したがって、侵襲性は、ヒト神経膠腫の悪性の性質の主要な決定要因である。びまん性単細胞浸潤は、組織学的グレードにかかわらず全ての神経膠腫瘍で起こり、宿主の細胞およびECM障壁を通る、新生物細胞の移動と定義される。悪性神経膠腫は、正常な脳組織と比較してより高レベルのuPA、uPARおよびMMP−9を発現する。
【0006】
MMPは、細胞外マトリックスタンパク質を分解し、運動性を促進するシグナル伝達カスケードを活性化し、GBM運動性に関与するトランスホーミング増殖因子β等の増殖因子を活性化することによって、腫瘍細胞浸潤を促進する。ゼラチナーゼMMP−2およびMMP−9の発現は、神経膠腫を含む種々の癌の侵潤能力および転移能力と相関がある。MMP−9レベルは、神経膠腫悪性腫瘍の組織学的グレードと高度に相関があった。MMP−9は、インビトロで神経膠/内皮共培養物中の内皮細胞形態形成および毛細血管形成に関連性がある。MMPはまた、腫瘍新脈管形成を調節し、腫瘍新生血管形成の間に起こる「新脈管形成スイッチ」に必要かもしれない。
【0007】
システインプロテアーゼであるカテプシンBのタンパク質分解活性は、フィブロネクチン、I型およびIV型コラーゲンおよびラミニンを含む、ECMタンパク質の直接の分解を伴う。カテプシンBはまた、メタロプロテイナーゼならびに可溶性および受容体結合型の両方のウロキナーゼプラスミノゲン活性化因子(uPA)を含む、ECM分解を媒介するタンパク質分解カスケードに関係する他の酵素を、間接的に活性化する。また、カテプシンBは、マトリックスメタロプロテイナーゼの組織阻害因子(TIMP)を不活性化することによって、MMP活性を増大させることが示唆されている。したがって、カテプシンBは、プロ−uPA/プラスミノゲンおよびプロ−MMPの活性化における、重要な上流の調節因子であり得る。カテプシンBはまた、シトクロムc放出ならびにカスパーゼ9および3の活性化(アポトーシスのミトコンドリア経路における重要な現象)を引き起こすことによって、アポトーシスに寄与することが示されている。カテプシンB発現の増大およびその阻害因子レベルの減少は、種々の癌の腫瘍増殖、血管新生、浸潤および転移と関連付けられた。
【0008】
腫瘍に関与する複数の遺伝子を標的とするsiRNA分子が、治療組成物を開発するのに望まれる。
【発明の概要】
【課題を解決するための手段】
【0009】
概要
多シストロン性低分子干渉RNAコンストラクトは、例えばヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)、ヒトマトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)をコードする配列をコードする複数の遺伝子を標的とする、2つ以上の自己相補的配列を含む。コンストラクトを用いて、腫瘍進行が阻害される。
本発明は例えば、以下の項目を提供する:
(項目1)
腫瘍形成の阻害および予め形成された腫瘍の退行に使用される、少なくとも第一および第二の自己相補的配列を含む多シストロン性低分子干渉RNAコンストラクト。
(項目2)
前記第一の自己相補的配列がヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列およびその相補物を含み、前記第二の自己相補的配列がヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列およびその相補物を含む、項目1に記載の多シストロン性コンストラクト。
(項目3)
前記uPAの自己相補的配列が
【化1】
であり、前記uPARの自己相補的配列が
【化2】
である、項目2に記載の多シストロン性コンストラクト。
(項目4)
前記uPARおよびuPAの自己相補的配列が、約22〜35塩基対の長さの介在配列によって分離されている、項目3に記載の多シストロン性コンストラクト。
(項目5)
前記介在配列が
【化3】
である、項目4に記載の多シストロン性コンストラクト。
(項目6)
前記第一の自己相補的配列がウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列およびその相補物を含み、前記第二の自己相補的配列がマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列およびその相補物を含む、項目1に記載の多シストロン性コンストラクト。
(項目7)
前記uPARの自己相補的配列が
【化4】
であり、前記MMP−9の自己相補的配列が
【化5】
である、項目6に記載の多シストロン性コンストラクト。
(項目8)
各ループが9個のヌクレオチドを含む、項目3または7に記載の多シストロン性コンストラクト。
(項目9)
各ループが
【化6】
である、項目8に記載の多シストロン性コンストラクト。
(項目10)
前記自己相補的配列が、長さ約22〜35塩基対の介在配列によって分離されている、項目1に記載の多シストロン性コンストラクト。
(項目11)
前記介在配列が
【化7】
である、項目10に記載の多シストロン性コンストラクト。
(項目12)
前記コンストラクトが環状核酸である、項目1に記載の多シストロン性コンストラクト。
(項目13)
(a)項目1に記載の低分子干渉RNA多シストロン性コンストラクトを腫瘍に投与すること、
(b)該腫瘍において発現されて低分子干渉RNAコンストラクトに標的とされる複数の遺伝子の発現を減少させて、それによって該腫瘍を阻害すること
を包含する、腫瘍形成の阻害または予め形成された腫瘍の退行の方法。
(項目14)
前記低分子干渉RNA多シストロン性コンストラクトが、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とする、項目13に記載の方法。
(項目15)
前記低分子干渉RNA多シストロン性コンストラクトが
【化8】
を含む、項目14に記載の方法。
(項目16)
腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって腫瘍が阻害される、項目1に記載の方法。
(項目17)
前記腫瘍が、前立腺癌、神経膠腫、乳癌、および黒色腫からなる群より選択される、項目1に記載の方法。
(項目18)
前記コンストラクトが直接の送達によって投与される、項目17に記載の方法。
(項目19)
前記低分子干渉RNA多シストロン性コンストラクトが、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とする、項目13に記載の方法。
(項目20)
前記低分子干渉RNA多シストロン性コンストラクトが
【化9】
を含む、項目13に記載の方法。
(項目21)
(a)ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子であって、該低分子干渉
RNA分子が核酸配列
【化10】
を含む、RNA分子、ならびに
(b)ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とするRNA分子であって、該低分子干渉RNA分子が核酸配列
【化11】
を含む、RNA分子
からなる群より選択される、低分子干渉RNA分子。
(項目22)
項目1に記載の多シストロン性コンストラクトで形質転換された、組換え細胞。
(項目23)
項目1に記載の多シストロン性コンストラクトで形質転換された、組換えウイルス。
【0010】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するのに用いられる第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補物はヒトウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はヒトウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列を含む。
【0011】
uPAの自己相補的配列は、
【0012】
【化12】
であり、uPARの自己相補的配列は
【0013】
【化13】
である。ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列ヌクレオチド配列は
【0014】
【化14】
であり、「適した」は、適切に作用することを意味する。uPARおよびuPAの自己相補的配列は、一般に、長さ約22〜35塩基対の介在配列によって分離される。例えば、介在配列は
【0015】
【化15】
である。多シストロン性コンストラクト中のuPARおよびuPAの自己相補的配列は、プロモーター、通常は単一のものに、操作作可能に連結されている。uPAおよびuPARの多シストロン性コンストラクトは、環状核酸または線状核酸である。
【0016】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補的配列はウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0017】
【化16】
であり、MMP−9の自己相補的配列は
【0018】
【化17】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列としては、ヌクレオチド配列
【0019】
【化18】
が挙げられる。uPARおよびMMP−9の自己相補的配列は、一般に、長さ約22〜35塩基対の介在配列によって分離される。介在配列は、
【0020】
【化19】
である。uPAR−MMP−9コンストラクトは、環状または線状核酸である。
【0021】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するのに用いられる、第一および第二の自己相補的配列を含む。ある実施形態において、第一の自己相補物は、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補的配列はカテプシンB(CB)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0022】
【化20】
であり、CBの自己相補的配列は
【0023】
【化21】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。適したループ配列としては、ヌクレオチド配列
【0024】
【化22】
が挙げられる。uPARおよびCBの自己相補的配列は、一般に、長さ約22〜68塩基対の介在配列によって分離される。介在配列は、
【0025】
【化23】
である。uPAR−CBコンストラクトは、環状または線状核酸である。
【0026】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための、第一、第二および第三の自己相補的配列を含む。第一の自己相補物はウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)のヌクレオチド配列を含み、第二の自己相補物はウロキナーゼ型プラスミノゲン活性化因子(uPA)のヌクレオチド配列を含み、第三の自己相補物はマトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含む。uPARの自己相補的配列は
【0027】
【化24】
であり、uPAの自己相補的配列は
【0028】
【化25】
であり、MMP−9の自己相補的配列は
【0029】
【化26】
である。ループは、GCの欠乏した約9個のヌクレオチドを含む。ループ配列としては、ヌクレオチド配列
【0030】
【化27】
が挙げられる。uPARおよびMMP−9の自己相補的配列は、一般に、長さ約22〜68塩基対の介在配列によって分離される。介在配列は、
【0031】
【化28】
である。uPA−uPAR領域は、
【0032】
【化29】
のヌクレオチド配列を含む約22〜35塩基によって分離される。uPA−uPAR−MMP−9コンストラクトは、線状または環状核酸である。
【0033】
多シストロン性低分子干渉RNAコンストラクトは、少なくとも、腫瘍を阻害するための、第一および第二の自己相補的配列を含む。第一の自己相補的配列は、マトリックスメタロプロテイナーゼ9(MMP−9)のヌクレオチド配列を含み、第二の自己相補的配列は、カテプシンB(CB)のヌクレオチド配列を含む。MMP−9の自己相補的配列は
【0034】
【化30】
であり、CBの自己相補的配列は
【0035】
【化31】
であり、ループは、GCの欠乏した約9個のヌクレオチドを含む。ループ配列としては、ヌクレオチド配列
【0036】
【化32】
が挙げられる。MMP−9およびCBの自己相補的配列は、一般に、長さ約22〜37塩基対の介在配列によって分離される。介在配列は、
【0037】
【化33】
である。MMP−9−CBコンストラクトは、環状または線状核酸である。
【0038】
腫瘍を阻害する方法、方法は、
(a)低分子干渉RNA多シストロン性コンストラクトを投与する工程、および
(b)腫瘍中で発現される複数の遺伝子の発現を減少させ、それによって腫瘍の形成または増殖を阻害し、すでに存在する腫瘍を退行させる工程
を含む。
【0039】
例えばウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とし、それによってuPARおよびuPAの発現を減少させ、腫瘍を阻害する、低分子形成または干渉RNA多シストロン性コンストラクトを使用する方法。低分子干渉RNA多シストロン性コンストラクトとしては、ヌクレオチド配列
【0040】
【化34】
が挙げられる。「介在配列」の別の用語は、「スペーサー」である。腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって、腫瘍が阻害される。腫瘍としては、前立腺癌、神経膠腫、乳癌、および黒色腫が挙げられる。コンストラクトは、ウイルスベクターを通じて送達されるか、または直接の送達を通じて、もしくは当業者に公知の任意の適した方法によって、投与される。
【0041】
低分子干渉RNA多シストロン性コンストラクトを使用する方法はまた、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とし、それによってuPARおよびMMP−9の発現を減少させ、腫瘍を阻害する。低分子干渉RNA多シストロン性コンストラクトとしては、ヌクレオチド配列
【0042】
【化35】
が挙げられる。腫瘍細胞増殖、腫瘍細胞浸潤、腫瘍細胞遊走および新脈管形成の少なくとも1つを減少させることによって、腫瘍が阻害される。腫瘍としては、前立腺癌、神経膠腫、乳癌、および黒色腫が挙げられる。コンストラクトは、ウイルスベクターを通じて送達されるか、または直接の送達を通じて、もしくは当業者に公知の任意の適した方法によって、投与される。
【0043】
腫瘍を阻害する方法は、
(a)uPA、uPAR、MMP−9およびCBの少なくとも1つを標的とする多シストロン性コンストラクトを投与する工程、ならびに
(b)uPA、uPAR、MMP−9およびCBの少なくとも1つの発現を減少させ、それによって腫瘍を阻害する工程
を含む。
【0044】
低分子干渉RNA分子としては、
(a)核酸配列
【0045】
【化36】
を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子、
(b)核酸配列
【0046】
【化37】
を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびウロキナーゼ型プラスミノゲン活性化因子(uPA)を標的とするRNA分子、
(c)
【0047】
【化38】
の核酸配列を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)およびカテプシンB(CB)を標的とするRNA分子、
(d)
【0048】
【化39】
の核酸配列を含む、ウロキナーゼ型プラスミノゲン活性化因子受容体(uPAR)、ウロキナーゼ型プラスミノゲン活性化因子(uPA)、およびマトリックスメタロプロテイナーゼ9(MMP−9)を標的とするRNA分子、ならびに
(e)
【0049】
【化40】
の核酸配列を含む、マトリックスメタロプロテイナーゼ9(MMP−9)およびカテプシンB(CB)を標的とするRNA分子
が挙げられる。
【0050】
uPA−uPARまたはuPAR−MMP−9またはuPAR−CB、またはuPA−uPAR−MMP−9またはMMP−9−CBの多シストロン性コンストラクトで形質転換した組換え細胞が、本明細書中に開示される。
【0051】
uPA−uPARまたはuPAR−MMP−9またはuPAR−CB、またはuPA−uPAR−MMP−9またはMMP−9−CBの多シストロン性コンストラクトで形質転換した組換えウイルスが、本明細書中に開示される。
略語
uPAはウロキナーゼ型プラスミノゲン活性化因子を意味する。uPARはウロキナーゼ型プラスミノゲン活性化因子受容体を意味する。MMP−9はマトリックスメタロプロテイナーゼ9を意味する。CBはカテプシンBを意味する。CMVはサイトメガロウイルスを意味する。SV40はシミアンウイルス40型を意味する。GFPは緑色蛍光タンパク質を意味する。ECMは細胞外マトリックスを意味する。siRNAは低分子干渉RNAを意味する。shRNAは低分子ヘアピンRNAを意味する。RNAiはRNA干渉を意味する。
【図面の簡単な説明】
【0052】
【図1】図1は、uPAおよびuPARタンパク質発現レベル、ならびにヒト前立腺癌細胞系の浸潤能力と相互関係のある、uPA活性を示す。内在性uPAおよびuPARタンパク質発現を、LNCaP、DU145およびPC3の前立腺癌細胞系から単離された全細胞タンパク質の免疫ブロット解析によって調べた。等しい量の、3つ全ての細胞系の細胞抽出物から単離されたタンパク質を、抗uPA、抗uPARおよび抗GAPDH抗体での免疫ブロットに供した。GAPDHをローディング対照(A)として利用した。前立腺癌細胞系におけるuPA活性を、フィブリンザイモグラフィーによって評価した。等しい量の、血清を含まない培地中の前立腺癌細胞由来のタンパク質を、非還元性条件下で、フィブリノーゲンおよびプラスミノゲンを含有する10%ゲルでのSDS−PAGEによって分離した。Triton X−100洗浄でSDSを交換した後、ゲルをグリシンバッファー(0.1M、pH8.0)中でインキュベートした。アミノブラック染色およびそれに続くメタノール−酢酸での脱染の後の明確な溶解バンドとして、フィブリン溶解活性を検出した(B)。前立腺癌細胞系のインビトロ浸潤能力の比較(C)。マトリゲルで被覆された12μmの孔を有するポリカーボネートフィルターを含む12ウェルトランスウェルチャンバー中で浸潤アッセイを行った。フィルターの下面に通過した細胞を染色し、200×倍率で、顕微鏡下で写真を撮った(C)。200×倍率で、顕微鏡下、3つの無作為な視野で、マトリゲルを通して浸潤している細胞を計数した。各バーは3つの視野の平均値±SDを表し、検出不可能なuPAおよびuPARタンパク質発現を示した低または非転移LNCaP細胞からの有意な差をアスタリスク*で現す(P<0.05)(D)。
【図2】図2は、前立腺癌細胞系PC3における、uPAおよびuPAR発現のRNAiノックダウンを示す。sh−uPAuPARプラスミドコンストラクトの概略図(A)。コンストラクトは、ヒトCMVプロモーターならびにuPAおよびuPARを標的とする相同配列からなる。発現の後、強力なCMVプロモーターが、uPAおよびuPARに特異的な低分子ヘアピン分子の形成を駆動する。ウシ成長ホルモン(BGH)ポリアデニル化配列を、RNAポリメラーゼIIベースのCMVプロモーター終止シグナルとする。Dicer/Droshaは、uPAおよびuPARに特異的なshRNAをプロセシングし、得られるsiRNA分子は、uPAおよびuPARの標的遺伝子と相互作用する。この相互作用によって、uPAおよびuPAR遺伝子発現の同時のノックダウンが生じる。shRNAトランスフェクトPC3細胞から抽出されたRNAの半定量的逆転写PCR(B)。グリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)mRNAを対照として同時増幅した。shRNAトランスフェクトPC3細胞から抽出された全タンパク質溶解物の免疫ブロット(C)。mock、EVおよびSVトランスフェクト細胞中にuPAおよびuPARの両方のバンドが存在する。したがって、各遺伝子特異的shRNAレーンは、適切なバンドの有意な減少を示す。GAPDHが、ローディング対照として含まれた。uPAおよびuPARタンパク質発現レベルを、PC3細胞中での間接免疫蛍光法を用いても検出した。EV、SVおよびmock細胞でトランスフェクトしたPC3細胞は、uPA(FITC)およびuPAR(テキサスレッド)の免疫蛍光検出に陽性に染色した(D)。遺伝子特異的shRNAトランスフェクト細胞は、その後、EV/SVトランスフェクトおよびmock細胞と比較して、uPAおよびuPARの細胞染色プロフィールを変化させた。DAPIで、核対比染色を得た。(結果は、少なくとも3つの別々の実験の代表である。)
【図3】図3は、uPAおよびuPAR発現のRNAiノックダウンがPC3細胞の浸潤能力を阻害することを示す。mock細胞および示されたshRNAプラスミドでトランスフェクトされた細胞の浸潤能力を、マトリゲル浸潤アッセイで調べた(1つの実験の代表的な視野)(A)。本明細書中に記載されるように行った浸潤アッセイ(図1C参照)。マトリゲルを通って浸潤している細胞の代表的な数を、顕微鏡下、3つの無作為な視野で、200×で計数した(B)。各バーは、計数した3つの視野の平均値SDを表す。対照(すなわち、mockまたはスクランブルベクタートランスフェクト細胞)からの有意な差を、アスタリスク*によって示す(P<0.05)。
【図4】図4は、uPAおよびuPAR発現のRNAiノックダウンが細胞増殖を阻害し、PC3細胞におけるアポトーシスを誘導することを示す。遺伝子特異的shRNAプラスミドまたは対照(mockまたはEV/SVトランスフェクト細胞)のいずれかでトランスフェクトしたPC3細胞の生存能を、3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド(MTT)アッセイによって明らかにした(A)。各バーは、平均値SDの三連の解析を表し、対照からの有意な差をアスタリスク*で表す(P<0.05)。代表的な免疫ブロットは、uPA−uPARノックダウンPC3細胞におけるアポトーシス促進性遺伝子発現の変化を示す(B)。GAPDHを、ローディング対照として用いた。活性化したカスパーゼに結合する細胞透過性カスパーゼ阻害因子であるFAM−VAD−FAKを用いた蛍光標識(下のパネル)で、カスパーゼ活性をインサイチューで検出した(C)。DAPIで核染色を行った(上のパネル)。sh−uPAuPARでトランスフェクトした相当な数の細胞が、緑色蛍光を示した。共焦点顕微鏡下の3つの無作為な視野からのDAPI/FMK−VAD−FAK標識細胞比の定量的データを示す棒の図表(D)。DAPI対FMK−VAD−FAKの比は、sh−uPAuPARでトランスフェクトした細胞で有意に増大した。mockまたはEV/SVトランスフェクト細胞からの有意な差を、アスタリスク*で示す(P<0.05)。uPA−uPAR shRNAでトランスフェクトした細胞およびアクチノマイシンD(ActD、0.2g/ml)で処理した細胞で、DNA標識が観察された(E)。エチジウムブロマイドでアガロースゲルを染色し、UV光下で写真を撮った。キロベースペアの参照として、2.0、1.5、1.0、0.75および0.5kbの標準バンドのDNAマーカーを電気泳動した(レーンM)。
【図5】図5は、uPAおよびuPAR発現のRNAiノックダウンがPC3細胞で下流のシグナリングを阻害することを示す。mockおよびshRNAトランスフェクト細胞における、全体的およびリン酸化形態の細胞外シグナル調節キナーゼ(ERK)、p38およびJNKの免疫ブロット解析(A)。mock、EV、SV、sh−uPA、sh−uPARおよびsh−uPAuPARでトランスフェクトしたPC3細胞を72時間後に溶解し、SDS−PAGEと、それに続く、全体的およびリン酸化形態のERK、p38およびJNK抗体での免疫ブロットに供した。GAPDH抗体を用いて、各レーンに同じ量のタンパク質がロードされたことを確かめた。mockおよびshRNAトランスフェクト細胞におけるStat3タンパク質の免疫ブロット解析(B)。等しい量のタンパク質をロードし、チロシン705に対するリン酸特異的Stat3抗体および非リン酸化形態のStat3に対する抗体を用いて、免疫ブロットを行った。GAPDHが、ローディング対照として含まれた。mockおよびshRNAトランスフェクト細胞の電気泳動移動度シフトアッセイ(C)。タンパク質−DNA複合体を、6%ポリアクリルアミドゲルで分離し、乾燥し、放射能写真を撮った。示されるshRNAトランスフェクト細胞から調製された核抽出物の特異的DNA結合活性を、上に示す。遊離プローブの位置を示す。
【図6】図6は、uPA−uPAR発現のRNAiノックダウンが同所マウス前立腺腫瘍モデルにおいて腫瘍増殖を排除することを示す。同所PC3腫瘍を有するマウスの各処理群からの代表的なインサイチューの写真(A)。原発性前立腺腫瘍を破線の矢印で標識し、一色の矢印は転移の位置を示す。PC3細胞をヌードマウスに前立腺内移植し、PC3前立腺腫瘍を確立し、uPA、uPARおよびuPAuPARに特異的なshRNAで処理した。これらのコンストラクトの4週間の処理後、マウスを屠殺し、原発性前立腺腫瘍増殖および転移を視覚的に評価した。各shRNA処置群からの切開した前立腺腫瘍の比較(B)。各バーは、1つの群あたり6匹の動物の平均腫瘍重量SDを表す。対照群(すなわち、mockまたはEV/SV処理)からの有意な差を、アスタリスク*で表す(P<0.05)。1つの群あたり6匹の動物のPC3前立腺腫瘍から抽出したタンパク質試料を、uPAおよびuPAR発現レベルに関する免疫ブロットを用いて解析した(C)。GAPDHが、ローディング対照として含まれた。
【図7】図7は、uPAおよびuPAR発現のRNAiノックダウンが同所マウス前立腺腫瘍モデルにおいて腫瘍増殖を同時に排除することを示す。同所PC3腫瘍を有するマウスの各処理群からの代表的なインサイチューの写真(A)。原発性前立腺腫瘍を破線の矢印で標識し、一色の矢印は転移の位置を示す。PC3細胞をヌードマウスに前立腺内移植し、PC3前立腺腫瘍を確立し、sh−uPAおよびsh−uPARベクターの両方を同時注射した。これらのコンストラクトの4週間の処理後、マウスを屠殺し、原発性前立腺腫瘍増殖および転移を視覚的に評価した。各shRNA処置群からの切開した前立腺腫瘍の比較(B)。各バーは、1つの群あたり6匹の動物の平均腫瘍重量SDを表す。対照群(すなわち、mockまたはEV/SV処理)からの有意な差を、アスタリスク*で表す(P<0.05)。1つの群あたり6匹の動物のPC3前立腺腫瘍から抽出したタンパク質試料を、uPAおよびuPAR発現レベルに関する免疫ブロットを用いて解析した(C)。GAPDHが、ローディング対照として含まれた。同所PC3前立腺腫瘍の代表的なヘマトキシリンおよびエオシン切片(D)。実験の終わりに、各処置群から原発性前立腺腫瘍を採取した。ホルマリン中で腫瘍を固定し、パラフィンに包埋した。組織切片(5m)を調製し、組織病理学的解析のために、H&Eで染色した。示される処置群からの確立した前立腺腫瘍からの顕微解剖したパラフィン切片のDNA fragEL染色(E)。DNA断片末端標識アッセイを行った。200×倍率である四角を除いて、結果を40×倍率で示す。1つの処置群あたり6つの無作為な視野からのDNA fragEL標識細胞の定量的データを示す棒の図表(F)。対照群からの有意な差を、アスタリスク*で示す(P<0.05)。
【図8】図8は、pUMベクターからのuPARおよびMMP−9のsiRNA発現の概略図を示す。uPARおよびMMP−9に対するCMVプロモーターによって駆動する2つの相補的逆方向反復を有するpcDNA3プラスミドコンストラクトを開発した。CMVプロモーターは、次に二本鎖RNA認識酵素DICERによってプロセシングされて実行可能なsiRNA分子を形成する、二重のヘアピン構造の形成を駆動する。ウシ成長ホルモン(BGH)ポリaシグナル配列によって駆動するポリA尾部を有するmRNA分子によく似ている分子の二次構造のために、二重のヘアピン分子の安定性が確実になる。
【図9】図9は、長いヘアピン(hp)RNAがsiRNAにプロセシングされ、分子がSNB19細胞において対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトされるかどうかを示し、細胞はまた、非GFP細胞においてGFPを標的とする関連のないコンストラクトによってもトランスフェクトして、適切なsiRNA分子のプロセシングを測定した。2%アガロースゲルで分画した低分子RNA分子を、6×SSC存在下で、適切なDIG標識センスオリゴでハイブリダイズさせた。得られたハイブリッド溶液を15%ポリアクリルアミドゲルに流し、ナイロン膜にエレクトロブロットした。製造業者の使用説明書どおりに、膜を処理して、21bp DNA:RNAハイブリッドを可視化した。使用したプローブを数(表1参照)、1−suPAR、2−sMMP−9および3−sGFPとして表す(図9A)。当業者に公知の標準的なプロトコールどおりに、SNB19細胞を、対照/EV(レーンa)、SV(レーンb)、puPAR(レーンc)、pMMP−9(レーンd)およびpUM(レーンe)でトランスフェクトした。72時間後、全RNAを単離し、cDNA合成キット(インビトロジェン)を用いて、ファーストストランドcDNAを合成した(図9B)。ファーストストランドcDNAをuPARおよびMMP−9の鋳型として用いて、PCR反応を構成した。GAPDHのためのPCRもまた構成し、ローディング対照とした(表1参照)。
【図10】図10は、uPARのウエスタンブロット解析およびMMP−9のゼラチンザイモグラフィーを特徴付ける。SNB19細胞を、mock、空の/スクランブルベクターならびにuPARおよびMMP−9の単一または2シストロン性siRNAをコードするベクター(puPAR、pMMP−9、pUM)でトランスフェクトした。(A)EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞からの細胞溶解物中のuPARタンパク質発現のウエスタンブロット解析。uPARに特異的な抗体を用いて、ウエスタンブロット解析を行った。GAPDHを同時に免疫検出して、同じ量の細胞溶解物をロードしたことを確かめた。(B)EV/SV、puPAR、pMMP−9およびpUMからのトランスフェクトした細胞からの等しい量のタンパク質(20μg)を含有する馴化培地を、ラエムリ試料バッファーと混合し、0.1%ゼラチンを含む10%SDS−PAGEゲルに流して、ゼラチンザイモグラフィーによってMMP−9活性を測定した。
【図11】図11は、uPARおよびMMP−9のRNAi媒介性ダウンレギュレーションがSNB19神経膠腫細胞増殖を減少させることを示す。簡潔に述べると、EV/SV、puPAR、pMMP−9およびpCMでトランスフェクトした5×104個のSNB19細胞を、血清を含まない条件下で、VNコートした96ウェルマイクロプレートに播種した。生存能のある細胞の数を、MTTアッセイによって評価した。
【図12】図12は、免疫組織化学的解析および腫瘍誘導性新脈管形成によって示されるように、RNAiがuPARおよびMMP−9レベルを減少させたことを示す。(A)SNB19細胞を、EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトした。対照または非トランスフェクト細胞もまた用いた。トランスフェクションの72時間後、細胞を固定し、処理して、インビボのuPARおよびMMP−9発現を可視化した。DAPIを含む封入剤を用いて細胞をマウントして、核を可視化した。(B)SNB19細胞(2×104)を8ウェルチャンバースライドに播種し、mock EV、puPAR、pMMP−9およびpUMでトランスフェクトした。24時間のインキュベーション後、培地を除去し、細胞を4×104個のヒト微小血管皮膚内皮細胞とともに共培養した。72時間後、内皮細胞を第VIII因子抗原に対する抗体またはH&E染色でプローブし、共焦点走査型レーザー顕微鏡下で調べた。(C)mock EV、puPAR、pMMP−9およびpUMベクターまたはpUMベクターでトランスフェクトした共培養物における新脈管形成の定量化。値は、3つの異なる実験からの平均値±S.D.である。マウス背側ウインドウアッセイによる、pUMベクターにおける腫瘍新脈管形成の阻害(D)。PV、既存の脈管構造、TN、腫瘍が脈管構造を誘導する。
【図13】図13は、uPARおよびMMP−9のsiRNAがSNB19細胞の浸潤を阻害することを示す。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞(1×106)をマトリゲルでコートされたトランスウェルインサート(8μmの孔)を通して24時間遊走させた。マトリゲルでコートしたインサートを通して浸潤した細胞を染色し、計数し、光学顕微鏡下、20×倍率で写真を撮った。(A)浸潤のパーセンテージを定量化した。値は、5つの異なる実験からの平均値±S.D.である(P<0.001)。直径100〜200μmのSNB19細胞スフェロイド(蛍光)を選択し、EV/SV、puPAR、puMMP−9およびpUMでトランスフェクトし、ラット胎仔脳凝集物(緑色蛍光)とともに共培養した。ラット胎仔脳凝集物の進行性の破壊およびSNB19細胞の浸潤を、共焦点レーザー走査型顕微鏡を用いて、72時間にわたって観察した。(C)。24、48および72時間の脳凝集物または腫瘍スフェロイドの残りの体積を、画像解析ソフトウエアを用いて定量化した(D)。
【図14】図14は、予め確立された大脳内の細胞増殖でのRNAi媒介性退行を解析する。SNB19−GFP神経膠芽腫細胞をヌードマウスに大脳内注射した(10μlのリン酸緩衝生理食塩水中2×106個の細胞)。10日後、mock EV、puPAR、pMMP−9およびpUM(Alzetミニポンプを用いて、0.25μl/時間の速度で150μgの各ベクターを脳に注射した(各群中8匹のマウス))。腫瘍切片の顕微鏡写真を、GFP蛍光(A)に関して調べ、それに続いて、ヘマトキシリンおよびエオシンで染色した(B)。これらの細胞の大脳内注射の4〜6週間後のmock EV、puPAR、pMMP−9およびpUMベクター処置群における腫瘍体積の半定量化(C)。示されるデータは、各群からの8匹の動物からの±S.D.値である。他の組の実験において、SNB19 GFP細胞の大脳内注射の10日後に、pUMベクターを3日の間隔で2回腹腔内注射し、4ヵ月後に動物を屠殺した。
【図15】図15は、uPARおよびMMP−9のRNAi媒介性ダウンレギュレーションがERK、MAPKおよびAKTのリン酸化を減少させることを示す。全体的およびリン酸化形態のMAPK、ERKおよびAKTのウエスタンブロット解析。SNB19細胞を、血清を含まない馴化細胞下で、VNコートしたプレート上で、EV/SV、puPAR、MMP−9およびpUMでトランスフェクトし、72時間後に溶解し、SDS−PAGE、ならびに全体的およびリン酸化形態のMAPK、ERKおよびAKT抗体での免疫ブロットに供した。GAPDH抗体を用いて、各レーンに同じ量のタンパク質がロードされたことを確かめた。
【図16】図16は、uPARおよびMMP−9ベクター(A)ならびに細胞表面での細胞の現象(B)の該略図である。プラスミンへのプラスミノゲンの活性化の後、これは次にMMP、ECMの分解後のいくつかの増殖因子のuPA放出を活性化する。該略図は、いくつかのシグナリング経路分子におけるインテグリンの関与を示した。
【図17】図17は、カテプシンBおよびuPARの単一のCMV駆動二重逆方向反復コンストラクトからのhpRNA分子の形成を示す概略図である。CMVウイルスプロモーターは、カテプシンBおよびuPARの自己相補的逆方向反復を有するRNA分子の形成を駆動させる。
【図18】図18は、uPARおよびカテプシンBのウエスタンブロット解析を示す。SNB19細胞を、mock、空のベクターならびにカテプシンBおよびuPARのsiRNAをコードするベクター(pCU)でトランスフェクトした。mock、空のベクターおよびpCUでトランスフェクトしたSNB19細胞からの細胞溶解物中のカテプシンB(A)uPAR(C)タンパク質レベルのウエスタンブロット解析を、カテプシンBおよびuPARに特異的な抗体を用いて行った。ローディング対照のために、β−アクチンを同時に免疫プローブした。カテプシンB(B)およびuPARタンパク質(D)の定量化を、放射能写真を濃度計でスキャンすることによって得た。
【図19】図19は、RNAiが腫瘍細胞誘導性毛細血管網形成を阻害することを示す。SNB19細胞を、mock、空のベクター、pC、pUおよびpCUで24時間トランスフェクトした。次いで、細胞をヒト皮膚内皮細胞とともに48時間共培養した。インキュベーションの後、細胞を固定し、2%ウシ血清アルブミンで1時間ブロックし、内皮細胞を第VIII因子抗原に対する抗体(第VIII因子抗体、DAKOコーポレーション、カリフォルニア州カーピンテリア)またはH&E染色でプローブし、レーザー走査型共焦点顕微鏡下で調べた(A)。mock、空のベクターまたはpCUベクターで感染させた共培養物における新脈管形成の定量化(B)。マウス背側ウインドウアッセイによる、pCUベクターで感染させたSNB19細胞における腫瘍新脈管形成の阻害(C)。PV−既存の脈管構造、TN−腫瘍誘導性脈管構造。
【図20】図20は、RNAiが神経膠腫細胞遊走および浸潤を阻害することを示す。SNB19 GFPスフェロイドを、mock、空のベクター、pC、pUおよびpCUで感染させた。72時間後、単一の神経膠腫スフェロイドを、96ウェルプレート中、ビトロネクチンコートしたウェルの中央に入れ、48時間培養した。遊走アッセイの終わりに、スフェロイドを固定し、写真を撮った(A)。ステージおよび接眼マイクロメータで較正した顕微鏡を用いて、スフェロイドからの細胞の遊走を測定した(B)。示されるデータは、各群からの4つの独立した実験からの結果の平均値±S.D.を示す。SNB19細胞を、mock、空のベクター、pC、pUおよびpcUでのトランスフェクションの72時間後にトリプシン化し、PBSで洗浄し、血清を含まない培地に再懸濁した。マトリゲルでコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット(コスター、マサチューセッツ州ケンブリッジ)中、浸潤アッセイを行った。24時間のインキュベーション期間の後、フィルターからウェル下部へ通過した細胞を染色し、計数し、光学顕微鏡下で写真を撮った(C)。浸潤のパーセンテージを定量化した(D)。値は、5つの異なる実験からの平均値±S.D.である(P<0.001)。SNB19細胞のスフェロイドを、mock、空のベクター、pC、pUおよびpcUでトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。腫瘍スフェロイドによる胎仔脳凝集物の進行性の破壊を観察した(E)。空のベクター、pC、pUまたはpCUベクターで感染させたSNB19スフェロイドによる残りの胎仔脳凝集物の定量化(F)。
【図21】図21は、uPARおよびカテプシンBのRNAi媒介性ダウンレギュレーションがSNB19神経膠腫細胞増殖を減少させることを示す。増殖アッセイ。簡潔に述べると、PBS、EV、SV、pU、pCおよびpCUでトランスフェクトした5×104個のSNB19細胞を、血清を含まない条件下で、VNコートした96ウェルマイクロプレートに播種した。生存能のある細胞の数を、MTTアッセイによって評価した。3つの別々の実験からの平均値(±S.D.)値が示される。
【図22】図22は、uPARおよびカテプシンBのRNAi媒介性ダウンレギュレーションがERKおよびFAKのリン酸化を減少させることを示す。mock、EV、SV、pU、pCおよびpCUコンストラクトでのSNB19細胞のトランスフェクションの後の、特異的抗体を用いた、全体的およびホスホERK、FAK、タンパク質のウエスタンブロット解析。β−アクチンレベルをローディング対照とした。
【図23】図23は、予め確立された腫瘍増殖のRNAi媒介性退行を示す。定位フレームの助けを借りて、ヌードマウスにSNB19 GFP腫瘍細胞を大脳内注射した。1週間後、空のベクター、またはカテプシンBおよびuPARのsiRNAを発現するベクター(pCU)のいずれかを、またはpCもしくはpUによって別々に、Alzetミニポンプを用いて脳に注射した。腫瘍切片の顕微鏡写真を、GFP蛍光で観察し(A)、それに続いてヘマトキシリンおよびエオシンで染色した(B)。5週間後のmock/空のベクター、pU、pCおよびpCUベクター処置群における腫瘍体積の半定量化。示される値は、各群からの6匹の動物の±S.D.値である(*P<0.001)(C)。
【図24】図24は、uPAR、uPAおよびMMP−9の単一のCMV稼動三重逆方向反復コンストラクトからの、hpRNA分子の形成の概略である。強力なCMVウイルスプロモーターが、uPA、uPARおよびMMP−9の自己相補的逆方向反復を有するRNA分子の形成を駆動させる。
【図25】図25は、uPAおよびMMP−9の、ウエスタンブロット、フィブリン/ゼラチンザイモグラフィーおよび免疫組織学的解析である。SNB19細胞を、mockトランスフェクトするか、または空のベクター/スクランブルベクター(EV/SV)およびsiRNA uPAR(puPAR)、uPA(puPA)、MMP−9(pMMP−9)および3つの組み合わせ(pU2M)をコードするベクターでトランスフェクトした。3日間のインキュベーション期間の後、全細胞溶解物を抽出バッファー中で調製し、これらの試料からの50μgのタンパク質を、12%非還元性SDS−PAGEによって分離し、抗uPAR抗体で免疫ブロットした(A)。ローディング対照として、GAPDHを同時に免疫プローブした。これらの試料から馴化培地を回収し(20μg)、ゼラチンおよびフィブリンザイモグラフィーを行って、MMP−9(B)およびuPA活性(C)を検出した。(D)SNB19細胞(1×104)を、Lab−Tek IIチャンバースライド上に播種し、mockトランスフェクトするか、またはEV/SV、ならびにsiRNA puPA、puPARおよびpMMP−9を単独で、または組み合わせて(pU2M)コードするベクターでトランスフェクトした。72時間後、細胞を固定し、ブロッキングバッファーで1時間洗浄し、uPA、uPARおよびMMP−9に対する特異的な抗体を用いて、uPAR、uPAおよびMMP−9発現に関して染色した。
【図26】図26は、siRNAコンストラクトによる、神経膠腫新脈管形成および浸潤の阻害を示す。SNB19細胞(2×104)を、8ウェルチャンバースライド上に播種し、EV/SV、ならびにsiRNA uPAR(puPAR)、uPA(puPA)、MMP−9(pMMP−9)および3つの組み合わせ(pU2M)をコードするベクターでトランスフェクトした。24時間のインキュベーション期間の後、培地を除去し、細胞を、4×104個のヒト内皮細胞とともに共培養するか、または4×104個のヒト内皮細胞のみを馴化培地の存在下で増殖させた。72時間後、内皮細胞を、共培養物中の第VIII因子抗原に関して染色した(緑色蛍光)。保存された馴化培地中で増殖した細胞をH&E染色し、蛍光顕微鏡または明視野顕微鏡下のいずれかで調べた(A)。(B)EV/SV、puPAR、puPA、pMMP−9およびpU2Mベクターに感染させた共培養物における新脈管形成の定量化。値は、4つの実験の平均値±SDである。EV/SV、puPAR、puPA、pMMP−9およびpU2Mでのトランスフェクションの72時間後に、SNB19細胞をトリプシン化し、PBSで洗浄し、血清を含まない培地中に再懸濁した。(C)マトリゲル(0.7mg ml−1)でコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット中で、浸潤アッセイを行った。24時間のインキュベーション期間の後、ウェル下部へフィルターを通過した細胞を染色し、計数し、明視野顕微鏡下で写真を撮った。(D)浸潤のパーセンテージを定量化した。
【図27】図27は、スフェロイドおよび大脳内アッセイによる、siRNAによる侵襲性および腫瘍増殖の阻害および退行を示す。(A)神経膠腫スフェロイドをラット胎仔脳凝集物とともに共培養することによって、神経膠腫スフェロイドの侵襲性を測定した。SNB19細胞のスフェロイドをEV/SV、puPA、puPAR、pMMP−9およびpU2Mでトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。示される時点で、共焦点レーザー走査型顕微鏡を用いて、共培養物の表面から中央までの、連続した厚さ1μmの切片を得た。(B)EV/SV、puPA、puPAR、pMMP−9およびpU2Mでトランスフェクトしたラット脳凝集物の残りの体積を測定した。値は、3つの実験の平均値±SDである。(C、D)予め確立された腫瘍増殖の、RNA媒介性退行。懸濁液中のSNB19GFP細胞(10μlの、血清を含まない培地中、2×106個)を大脳内注射した。1週間後、Alzetミニ浸透圧ポンプを用いて、マウスにEV/SVまたはsiRNA発現ベクター(puPAR、puPA、pMMP−9およびpU2M)のいずれかを注射した(PBS中1.5μg ml−1に希釈したコンストラクトを、各群の6匹のマウスに0.25μg時間−1で注射した)。5週間の追跡期間の後、マウスを屠殺し、それらの脳を除去し、パラフィン包埋および切片化した。蛍光顕微鏡下で、GFP発現細胞に関して切片を観察した。(D)対照、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置群における腫瘍体積の半定量化を、5週間後に評価した。データを、各群からの6匹の動物の平均値±SDで示す。
【図28】図28は、uPAR、uPAおよびMMP−9のRNAi媒介性ダウンレギュレーションがERKのリン酸化を減少させることを示す。EV/SV、puPAR、puPA、pMMP−9およびpU2Mコンストラクトでの神経膠芽腫細胞のトランスフェクション後の全体的およびリン酸化ERK(pERK)タンパク質のウエスタンブロット解析。GAPDHレベルをローディング対照とした。
【図29】図29は、pCMベクターからのカテプシンBおよびMMP−9のsiRNA発現の概略図である。カテプシンBおよびMMP−9に対するCMVプロモーターによって駆動される2つの相補的な逆方向反復を有するpCDNA3プラスミドコンストラクトを開発した。CMVプロモーターは、二重のヘアピン構造の形成を駆動させ、これは次に、二本鎖RNA認識酵素のDICERによってプロセシングされて、実行可能なSiRNA分子を形成する。二重のヘアピン構造の安定性は、ウシ成長ホルモン(BGH)ポリaシグナル配列によって駆動するポリA尾部を有するmRNA分子によく似ている分子の二次構造のために確実になった。
【図30】図30は、RNA干渉がSNB19細胞におけるカテプシンBおよびMMP−9レベルを減少させることを確かめる。全細胞溶解物、および血清を含まない培地を、mock、空のベクターまたはMMP−9(pM)およびカテプシンB(pC)および組み合わせ(pCM)のsiRNAをコードするベクターでトランスフェクトしたSNB19細胞から回収した。それに続いて、これらの試料からの30μgのタンパク質を、非還元性条件下、8%〜12%SDS−PAGEで分離し、ニトロセルロース膜に移した。カテプシンB(A)およびMMP−9(C)に対する抗体ならびに適切な二次抗体(ホースラディッシュペルオキシダーゼ複合体)で膜をプローブし、製造業者(アマシャム、イリノイ州アーリントンハイツ)の使用説明書どおりに展開した。β−アクチンを同時に免疫検出して、同じ量の細胞溶解物がロードしたことを確かめた。血清を含まない培地中で3日間空のベクター、pC、pMまたはpCMベクターを感染させたSNB19細胞のMMP−9活性を、ゼラチンザイモグラフィーによって測定した(B)。
【図31】図31は、RNAiが腫瘍細胞誘導性毛細血管網形成を阻害することを示す。SNB19細胞を、mock、空のベクター、pM、pCおよびpCMで24時間トランスフェクトした。次いで、細胞をヒト皮膚内皮細胞とともに48時間共培養した。インキュベーション後、細胞を固定し、2%ウシ血清アルブミンで1時間ブロックし、第VIII因子抗原に対する抗体(第VIII因子抗体、DAKOコーポレーション、カリフォルニア州カーピンテリア)で内皮細胞をプローブし、適切なFITC複合二次抗体でプローブした後、蛍光顕微鏡下で調べた。SNB19(対照、空のベクター、PM、pC、またはpCMトランスフェクト)馴化培地存在下で増殖した内皮細胞を、HおよびE染色し、写真を撮った(A)。mock、空のベクターまたはpCMベクターに感染させた共培養物における新脈管形成の定量化(B)。マウス皮下脂肪アッセイによる、pCMベクターに感染させたSNB19細胞における腫瘍新脈管形成の阻害(C)。PV−既存の脈管構造、TN−腫瘍誘導性脈管構造。光学顕微鏡を用いて(上のパネル)、およびFITC蛍光に関して(下のパネル)写真を撮り、新しく発生した脈管構造の存在を測定した。
【図32】図32は、RNAiが神経膠腫細胞遊走および浸潤を阻害することを示す。SNB19 GFPスフェロイドを、mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)に感染させた。3日後、単一の神経膠腫スフェロイドを、96ウェルプレート中のビトロネクチンコートしたウェルの中央に入れ、48時間培養した。遊走アッセイの終わりに、スフェロイドを固定し、写真を撮った(A)。mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)でのトランスフェクションの3日後に、SNB19細胞をトリプシン化し、PBSで洗浄し、血清を含まない培地中に再懸濁した。マトリゲルでコートされた8μmの孔を有するポリカーボネートフィルター上の12ウェルトランスウェルユニット(コスター、マサチューセッツ州ケンブリッジ)中で、浸潤アッセイを行った。24時間のインキュベーション期間の後、ウェル下部へフィルターを通過した細胞を染色し、計数し、光学顕微鏡下で写真を撮った(B)。SNB19細胞のスフェロイドを、mock、空のベクター、ならびにカテプシンBおよびMMP−9のsiRNAをコードするベクター(pCM)でトランスフェクトし、DiIで染色し、DiO染色したラット胎仔脳凝集物とともに共培養した。腫瘍スフェロイドによる胎仔脳凝集物の進行性の破壊を観察した(C)。
【図33】図33は、予め確立された腫瘍増殖のRNAi媒介性退行を示す。定位フレームの助けを借りて、SNB19 GFP腫瘍細胞をヌードマウスに大脳内注射した。1週間後、空のベクター、またはカテプシンBおよびMMP−9(pCM)、カテプシンB(pC)もしくはMMP−9(pM)のsiRNAを発現するベクターのいずれかを、アルゼットミニ浸透圧ポンプを用いて脳に注射した。GFP蛍光に関して腫瘍切片の写真を観察し(A)、それに続いて、ヘマトキシリンおよびエオシンで染色した(B)。5週間後の、mock/空のベクター、pM、pCおよびpCMベクター処置群における腫瘍体積の半定量化を行った。示されるデータは、各群からの6匹の動物からの±S.D.値を示す(*P<0.001)(C)。別の実験において、大脳内注射の10日後に、pCMベクターを2回腹腔内注射し、4ヵ月後に動物を屠殺した(D)。
【図34】図34は、RNAiの誘導のための、RNAポリメラーゼIIプロモーター(CMV)の概略図である。
【図35】図35は、RNAiの誘導のための、RNAポリメラーゼベースのIIIプロモーター(U6)の概略図である。
【図36】図36は、組換えアデノウイルス生成の概略図である。
【図37】図37は、GRP、またはGRP発現を示すmockのRNAiでトランスフェクトした、SNB19 GFP細胞を示す。
【図38】図38は、RT−PCRによって測定した、RNAポリメラーゼII(CMV)またはRNAポリメラーゼIII(U6)によって駆動するRNAiベクターでトランスフェクトしたSNB19細胞におけるOAS1発現を示す。GAPDHのRT−PCRTを対照とした。
【図39】図39は、U6またはCMV駆動プロモーターのいずれかでトランスフェクトした細胞からのSNB19細胞溶解物のウエスタンブロット解析およびフィブリンザイモグラフィーによって測定した、uPARおよびuPAのダウンレギュレーションを示す。スクランブルベクター(SV)のRNAiプラスミド、uPAR(puPAR)、uPA(puPA)およびuPAR−uPA2シストロン性コンストラクト(pU2)のRNAi発現プラスミド。ローディング対照のために、GAPDHをプローブした。
【図40】図40は、細胞性免疫応答の誘導を測定するのに用いたsiRNA発現コンストラクトの概略図である。
【図41】図41は、RT−PCRによる、空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)のsiRNA発現コンストラクトおよびuPARおよびuPAの2シストロン性コンストラクト(pU2)の環状(C)、線状(L)、またはポリAシグナルを欠失した(ΔA)発現カセットでトランスフェクトしたSNB19細胞における2’5’−オリゴアデニル酸シンテターゼ(OAS1)発現の測定を示す。RT−PCRをGAPDHで標準化した。OAS1発現を、示されるように定量化した。
【図42】図42は、実行可能なRNAi誘発因子様構造を示さないEVおよびSV転写産物の予想される二次構造、ならびにポリAありまたはポリAなし(pU2ΔA)の、予想されるpuPAR、puPAおよびpU2転写産物の概略図である。
【図43】図43は、空間二次構造を示す、pU2転写産物の部分的二次構造の概略図である。空間二次構造は、mRNAに対する類似性を有さない。
【図44】図44は、mRNAレベルの、対照(C)、アンチセンスuPAR(uPARとして)、アンチセンスuPA(uPAとして)、ならびにuPAR(puPAR)、uPA(puPA)、uPAR−uPA(pU2)、GFP(pGFP)のRNAiコンストラクト、空のベクター(pEV)、ならびにuPA、uPARおよびOAS1のスクランブルベクター(pSV)から単離された全RNAでのRT−PCRを示す。GAPDHを対照とした。
【図45】図45は、静脈内注射マウスにおけるCMVプロモーターのインサイチューハイブリダイゼーションを示す。生理食塩水(Mock)、空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)、およびuPAR−uPA2シストロン性コンストラクト(pU2)のRNAi発現ベクターを腹腔内注射されたマウスの頭蓋切片を、プローブDNAをアルカリホスファターゼ(AP)で標識することによって、CMVプロモーターを含むDNAの存在に関してプローブした。ウエスタンブルーAP基質(プロメガ、ウィスコンシン州マディソン)によって、AP活性を検出した。
【発明を実施するための形態】
【0053】
詳細な説明
低分子ヘアピンRNA(shRNA)は、低分子干渉RNA(siRNA)とも呼ばれ、uPAおよびuPAR等のヒト遺伝子を標的として、腫瘍成長、腫瘍浸潤、および腫瘍増殖を阻害する。siRNAコンストラクトは、高度に転移性の前立腺癌細胞系PC3において、mRNAおよびタンパク質の両方のレベルで、uPAおよびuPAR発現を有意に阻害した。PC3細胞におけるuPA−uPARノックダウンは、例えばマトリゲル浸潤アッセイによって示されるように、腫瘍細胞浸潤の有意な減少を生じた。uPAおよびuPARの両方のshRNAを発現する単一のプラスミドコンストラクトを用いた、uPAおよびuPARの遺伝子の同時のサイレンシングは、細胞生存能を有意に減少させ、同様に、アポトーシス性細胞死の誘導を生じた。uPAおよびuPARのRNAiはまた、細胞外シグナル制御キナーゼ1/2(ERK1/2)およびシグナル伝達性転写因子3(Stat3)等の下流の標的分子へのuPA−uPARシグナリングを排除した。uPAおよびuPARのshRNAを発現するプラスミドコンストラクトの腫瘍内注射は、同所マウス前立腺癌モデルにおいて、確立された腫瘍増殖および生存を、有意に阻害した。前立腺癌細胞の腫瘍浸潤、腫瘍増殖および生存を活発に進行させるuPA−uPARの下流で機能するシグナリングネットワークの証拠が明らかである。uPAおよびuPARのRNAi誘導性標的化ならびに対応するsiRNAは、前立腺癌を含む癌治療のための、新規の治療剤である。
【0054】
ヒト神経膠腫細胞系における、uPARおよびMMP−9のsiRNA媒介性標的RNA分解は、腫瘍阻害を生じた。uPARおよびMMP−9に対するRNAiは、uPARおよびMMP−9の特異的阻害を達成した。2シストロン性コンストラクト(pUM)は、新脈管形成の、インビトロおよびインビボの両方のモデルにおいて、毛細血管様構造の形成を阻害した。uPARおよびMMP−9の発現をブロックすることは、マトリゲルおよびスフェロイド浸潤アッセイモデルにおいて、神経膠腫腫瘍浸潤の有意な阻害を生じた。uPARおよびMMP−9のRNAiは細胞増殖を阻害し、親および空のベクター/スクランブルベクター(EV/SV)トランスフェクトSNB19細胞と比較して、MAPK、ERKおよびAKTシグナリング経路のリン酸化形態のレベルを減少させた。さらに、2つのプロテアーゼ分子を同時に標的とするためのRNAiを用いること、およびアルゼットミニポンプを用いてこれらのコンストラクトをインビボで大脳内注射することもしくは腹腔内注射は、予め確立された大脳内腫瘍増殖の有意な退行を生じた。uPARおよびMMP−9のヘアピンsiRNA発現ベクターの使用によって、神経膠芽腫を含む癌治療のための効果的な治療手段が提供される。
【0055】
別の実施形態において、RNAiアプローチは、uPARおよびカテプシンB発現をサイレンシングした。RNAiを用いて、腫瘍進行の特徴的な特性である細胞外マトリックス分解に関与するプロテアーゼの発現を阻害した。uPARおよびカテプシンBのRNAiは、インビトロおよびインビボモデルにおいて、神経膠腫細胞浸潤および新脈管形成を減少させた。uPARおよびカテプシンBのhpRNA(siRNA)を発現するプラスミドベクターの腫瘍内注射は、予め確立された大脳内腫瘍の退行を生じた。uPARおよびカテプシンBのRNAiは、細胞増殖を阻害し、対照と比較して、pERKおよびpFAKのレベルを減少させた。RNAiはヒト神経膠腫細胞中で機能し、神経膠芽腫を含む癌遺伝子治療の基礎を提供する。
【0056】
RNAiアプローチは、腫瘍細胞において、uPA、uPARおよびMMP−9発現をサイレンシングした。単一の3シストロン性コンストラクト中のサイトメガロウイルス(CMV)プロモーター駆動DNA鋳型は、ヘアピンRNA(hpRNA)誘発性RNAiを誘導し、単一のコンストラクトでuPA、uPARおよびMMP−9遺伝子発現を阻害した。uPARタンパク質レベルならびにuPAおよびMMP−9の酵素活性は、uPAR、uPAおよびMMP−9のヘアピンsiRNAを発現するプラスミドでトランスフェクトされた細胞を有意に減少させることがわかった。pU2MトランスフェクトSNB19細胞は、免疫組織学的解析によって測定したmockおよびEV/SVトランスフェクト細胞と比較して、uPA、uPARおよびMMP−9発現を有意に減少させた。これらの分子の単一のコンストラクトは、免疫組織化学的解析によって示されるように、それらのそれぞれのタンパク質レベルの特異的な阻害を生じた。uPA、uPARおよびMMP−9のdsRNAを発現するプラスミドベクターでのトランスフェクションの後、マトリゲル浸潤アッセイおよびスフェロイド浸潤アッセイによって示されたmockおよびEV/SV処置群と比較して、神経膠腫細胞浸潤が遅らされた。RNAiを用いたuPA、uPARおよびMMP−9のダウンレギュレーションは、インビトロ(共培養)モデルにおいて、新脈管形成を阻害した。uPA、uPARおよびMMP−9のhpRNAを発現するプラスミドDNAの直接の腫瘍内注射はまた、ヌードマウスにおいて、予め確立された大脳内腫瘍を有意に退行させた。uPAR、uPAおよびMMP−9のRNAiで処理した細胞は、親およびEV/SV処置SNB19細胞と比較して、減少したpERKレベルを示した。uPAR、uPAおよびMMP−9の同時抑制は、癌を治療するための治療手段である。
【0057】
単一のコンストラクトからのサイトメガロウイルス(CMV)プロモーター駆動DNAヘアピンRNA(hpRNA、siRNA)は、MMP−9およびカテプシンB遺伝子発現をブロックした。MMP−9およびカテプシンBのdsRNAを発現するプラスミドベクターのトランスフェクションは、MMP−9およびカテプシンB発現を有意に阻害し、マトリゲルおよびスフェロイド浸潤モデルにおいて神経膠芽腫細胞系である、SNB19の侵襲性の性質を減少させた。SNB19細胞における、RNAiを用いたMMP−9およびカテプシンBのダウンレギュレーションもまた、ヒト微小血管内皮細胞の細胞−細胞相互作用を減少させ、インビトロおよびインビボの両方のモデルにおいて、毛細血管網形成の中断を生じた。MMP−9およびカテプシンBのhpRNAを発現するプラスミドDNAの直接の腫瘍内注射は、インビボの大脳内腫瘍において、確立された神経膠腫腫瘍増殖および浸潤を有意に阻害した。MMP−9およびカテプシンBのhpRNAを発現するプラスミドDNAの腹腔内(ip)注射は、相当な期間にわたって、予め確立された腫瘍を完全に退行させた。MMP−9およびカテプシンBの同時のRNAi媒介性標的化は、ヒト神経膠腫のための、適した治療方法である。
【0058】
uPAR、uPAおよびMMP−9を標的とするプラスミドベースのCMVプロモーター駆動hpRNAは、単独で、または同時に、SNB19ヒト神経膠腫細胞系において、RNAiを誘導する。SNB19ヒト神経膠腫細胞における、uPAR、uPAおよびMMP−9の、同時のRNAi媒介性ダウンレギュレーションは、
(1)インビトロでの浸潤および新脈管形成の阻害。
(2)インビボのヌードマウスにおける、予め確立された大脳内腫瘍の退行。
(3)ERK1および2シグナリング分子のリン酸化の減少。
を引き起こした。
【0059】
環状プラスミド(例えば、uPA、uPAR、MMP−9、およびカテプシンBまたはそれらの任意の組み合わせ)から追い出されるsiRNAまたはshRNAまたはhpRNAが、インビトロのRNA干渉を誘導するのに適している。OAS1誘導によって示されるように、環状プラスミドからのsiRNAが適しており、望ましくない免疫応答を誘導しない。uPA、uPAR、MMP−9、およびカテプシンBまたはそれらの任意の組み合わせの線状コンストラクトもまた、RNAiを誘導するのに適している。
【0060】
本明細書中に開示されるsiRNAまたはshRNAまたはhpRNAは、本質的にuPA、uPAR、MMP−9、カテプシンBまたはそれらの組み合わせの自己相補的配列からなる核酸を含む。ループ領域およびスペーサー(介在領域)は、標的配列、コンストラクト、および治療上の使用に依存して、長さおよび配列の両方が変化し得る。本明細書中に開示される核酸分子を、核酸類似物、誘導体、または任意の適した修飾によって、適切に修飾して、RNAi誘導の安定性または有効性を向上させ得る。本明細書中に開示される核酸分子はまた、他の腫瘍特異的な免疫活性化因子、腫瘍標的化因子、および任意の適した薬学的に許容され得る担体もしくは佐剤と組み合わせて投与され得る。本明細書中に開示される核酸分子はまた、放射線治療または化学療法等の他の癌治療と関連して投与され得る。
【実施例】
【0061】
以下の実施例は例示の目的のためだけであって、開示の範囲を限定するよう解釈されることを意図しない。
【0062】
(実施例1)
内在性uPAおよびuPARタンパク質発現はヒト前立腺癌細胞のインビトロの侵襲性に関連している。
【0063】
PC3細胞は、高度に転移性であるが、DU145およびLNCaP細胞はそれぞれ、中程度の転移性がある、および転移性が乏しい。uPAおよびその受容体uPARは、腫瘍浸潤および転移に関係する。転移能力の異なる3つのヒト前立腺癌細胞系におけるこれらのタンパク質のレベルを比較した。図1Aに示されるように、uPAおよびuPARタンパク質レベルは、望ましくないレベルのこれらのタンパク質を発現した転移性の乏しいLNCaP細胞と比較して、PC3およびDU145細胞において有意に高かった。フィブリンザイモグラフィーによって評価したuPA活性で、同様の傾向が見られた(図1B)。したがって、uPAおよびuPARタンパク質レベルならびにuPA活性は、それらの公知の転移能力と、正に相関があった。前立腺癌が、基底膜タンパク質で構成されるゲル層であるマトリゲルに浸潤する能力を、調べた。このアッセイは、腫瘍浸襲性を評価するためのインビトロモデルにおいて確立している。高度に転移性のある前立腺癌細胞系PC3が、最も高いレベルの侵襲性を示し、DU145およびLNCaP細胞系がそれに続き、順序はそれらの公知の転移能力と一致した(図1C、および1D)。LNCaP細胞よりも、PC3細胞は14倍多く侵襲性があり、DU145は9倍多く侵襲性があった。uPAおよびuPARタンパク質レベルと転移能力の異なるヒト前立腺癌細胞系の浸潤能力との間の強力な相関関係の浸潤能力を実証した。PC3およびDU145細胞系は、LNCaP細胞よりも侵襲性があり、それらの公知の転移能力と一致した。uPAおよびuPARの発現パターンと使用した前立腺癌細胞系の浸潤能力の間に、強力な相関関係が存在する(図1)。前立腺癌細胞系における促進されたuPAおよびuPAR発現は、増大した侵襲性および転移能力と関連がある。
【0064】
(実施例2)
RNAiを用いた、ヒトPC3前立腺癌細胞におけるuPAおよびuPAR遺伝子発現の効率的ノックダウン
低分子ヘアピンRNAを用いて、uPAおよびuPARならびに高い転移能力を発現するヒト前立腺癌細胞系PC3における内在性uPAおよびuPAR遺伝子発現をノックダウンし、前立腺腫瘍進行におけるuPAおよびuPARの生物学的な役割を調査した。uPAまたはuPARのいずれかに対応する19または21nt二本鎖RNAiオリゴヌクレオチドを生じることのできる、低分子ヘアピンコンストラクトを含むpcDNA3−CMVベクターを開発した。また、サイトメガロウイルス(CMV)プロモーターによって駆動してuPAおよびuPARの両方を標的とする二重のヘアピンを生じる、単一の2シストロン性コンストラクトを構築して、2つの内在性遺伝子の発現を同時に阻害する効果を試験した(図2A)。uPA、uPARおよびuPA−uPARの組み合わせのshRNAを発現するベクターをPC3細胞にトランスフェクトした。
【0065】
図2Bに示されるように、半定量的逆転写PCRを通じたuPAおよびuPAR発現のためのshRNAトランスフェクト細胞は、EV/SVトランスフェクト細胞またはmock細胞と比較して、各遺伝子のmRNAレベルの特定の減少を示した。しかしながら、RNAi効果は、uPAおよびuPARを同時に標的とするshRNAベクターで、より大きかった(図2B)。細胞抽出物の免疫ブロット解析を行って、観察されたような減少したmRNA発現が、遺伝子産物の翻訳の減少と相関があるかどうかを決定した。免疫ブロットアッセイによっても、同様の傾向が観察された(図2C)。mRNAレベルならびにタンパク質レベルで、特異性およびローディングの内部対照として使用されたGAPDHの発現に対するRNAiの効果は観察されなかった。また、EV/SVトランスフェクト細胞はまた、RNAi依存性uPAおよびuPARノックダウンが特異的であることを示した(図2Bおよび2C)。
【0066】
また、抗uPAおよび抗uPAR抗体での二重免疫染色を用いて、uPAおよびuPARタンパク質発現に対する遺伝子特異的shRNAの効果が、PC3細胞において検出された。図2Dに示されるように、uPAおよびuPAR染色は、遺伝子特異的shRNAによって、EV/SVトランスフェクト細胞と比較して、劇的に減少した。uPAおよびuPAR二重免疫染色は、sh−uPAuPARでトランスフェクトした細胞において、完全に消失した(図2D)。対照的に、EVおよびSVでトランスフェクトしたPC3細胞は、mock細胞と同様な染色強度およびパターンを示した。これらの免疫蛍光研究によって、RT−PCRおよび免疫ブロットアッセイが確認された。
【0067】
プラスミドベースのsiRNA系を用いた2つの遺伝子の同時の阻害は、有用な手段である。RNAiは、前立腺癌細胞系PC3において、uPAおよびuPAR mRNAならびにタンパク質発現を効果的にダウンレギュレーションした(図2Bおよび2C)。これらの遺伝子特異的RNAiプラスミドは、mockまたはEV/SVトランスフェクト細胞と比較して、uPAおよびuPAR発現を実質的に減少させた。免疫組織化学的染色データから、mock、またはEV/SVでトランスフェクトした細胞がuPAおよびuPARに対する正の染色を現すが、sh−uPAuPARでトランスフェクトした細胞はDAPI核染色を除いてほとんど染色されないことが示され、uPAおよびuPARタンパク質発現のノックダウンが示唆された(図2D)。同様に、uPAおよびuPARに対する免疫染色の強度は、遺伝子特異的RNAiによって減少した。
【0068】
(実施例3)
RNAiによるuPAおよびuPAR発現のノックダウンは、PC3細胞のマトリゲル浸潤を阻害した。
【0069】
uPAおよびuPARの機能の1つは、腫瘍転移に必要な過程である、浸潤の促進である。PC3細胞浸潤に対するuPAおよびuPARノックダウンの影響を、shRNAトランスフェクト細胞を用いたマトリゲル浸潤アッセイによって評価した。mock細胞、またはEV/SVでトランスフェトした細胞と比較して、sh−uPAuPARトランスフェクト細胞は、浸潤能力の実質的な減少を示した(図3A)。PC3細胞の浸潤は、sh−uPAによって対照のものの(すなわち、mockまたはEV/SVトランスフェクト細胞)75%まで減少し、sh−uPAuPARによって90%まで減少した(図3B)。uPAR単独のノックダウンは浸潤の有意な減少を示さなかったが、uPAならびにuPARのノックダウンは、有意な効果を有し(図3Aおよび3B)、マトリゲルへのPC3細胞浸潤が、uPAおよびuPARの共同した機能によって実質的に制御されていることが示唆された。これらの結果から、uPAおよびuPAR発現が前立腺癌浸潤ならびに転移に必要とされることが示される。sh−uPAuPAR効果は細胞がマトリゲル膜を通ってほとんど浸潤できないほど有意であり、RNAiが、タンパク質分解活性および細胞生存能を媒介するuPA−uPAR系に有意に干渉したことが示唆された。
【0070】
(実施例4)
RNAiによるuPAおよびuPAR発現のノックダウンは、細胞増殖を阻害し、アポトーシスを誘導する。
【0071】
細胞増殖および生存に対するRNAi媒介性uPAおよびuPARサイレンシングの効果を、uPAおよびuPARに特異的なshRNAでのトランスフェクションの72時間後に、MTT解析によって調べた(図4A)。uPAを標的とするRNAiは、PC3細胞の増殖能力に対して効果がなかったが、uPARに特異的なRNAiは、低い阻害効果を有した。対照的に、uPAおよびuPARを同時に標的とするRNAiで、PC3細胞の増殖の劇的な減少が観察された(図4A)。生存能のある細胞のパーセンテージはuPARおよびuPA−uPAR RNAi存在下で、対照細胞と比較して、それぞれ平均しておよそ30%および60%減少した。これらの結果から、腫瘍細胞における増大したuPAおよび/またはuPARレベルが細胞に促進された増殖および生存能力を与え得ることが示唆される。このように、uPAおよびuPARレベルを減少させることによって、癌細胞におけるアポトーシスが誘導され得る。
【0072】
この可能性を調べるために、PC3細胞を、sh−uPA、sh−uPARまたはsh−uPAuPARを発現するプラスミドでトランスフェクトした。PC3細胞タンパク質抽出物の分子解析から、sh−uPAuPARトランスフェクションがBax、Bcl−XS/L、カスパーゼ9を含むアポトーシス促進性遺伝子を誘導したことが明らかになった(図4B)。また、FAM−VAD−FMKでのsh−uPAuPARトランスフェクトPC3細胞の蛍光色素染色から、mock細胞またはEV/SVトランスフェクト細胞のいずれかでも検出されなかった、促進されたカスパーゼ活性が明らかになった(図4Cおよび4D)。DNA断片化解析は、アポトーシス誘導のさらなる証拠を提供した。図4Eが示すように、sh−uPAuPARトランスフェクトPC3細胞は、mockまたはEV/SVトランスフェクト細胞のいずれかでは検出されなかった、アガロースゲル電気泳動での、アポトーシスの典型的な特徴であるDNA標識を示した。このDNA標識は、アクチノマイシンD処理によって誘導されるアポトーシスのものと同様であり(図4E)、それによって、観察された細胞死がアポトーシスの結果であることが確認された。また、sh−uPAまたはsh−uPARでトランスフェクトした細胞において、DNA標識は観察されなかった。これらの結果はまた、FAM−VAD−FMKによって測定されるカスパーゼ9誘導および一般的な促進されたカスパーゼ活性と一致している。
【0073】
(実施例5)
RNAiによるuPAおよびuPAR発現のノックダウンは、その下流のシグナリングおよびヌードマウスにおける腫瘍形成を阻害する。
【0074】
uPAおよびuPARサイレンシングによる生物学的結果は、uPA−uPAR媒介性シグナリングおよびそれに続く下流の機能の変化の結果であり得る。uPAおよびuPARの増大した発現はERK1/2シグナリングを活性化するので、uPA−uPARノックダウン細胞におけるマイトジェン活性化タンパク質キナーゼの状態を調べた。免疫ブロット解析から、sh−uPAuPARトランスフェクト細胞においてERK1/2リン酸化が完全に排除されたが対照細胞では排除されなかったことが示される(図5A)。ERKリン酸化は、sh−uPAまたはsh−uPARのいずれかでトランスフェクトした細胞を変化させなかった。Stat3の、リン酸化を含む全体の活性は、sh−uPAuPARでトランスフェクトした細胞において、対照細胞と比較して、実質的に抑制された(図5B)。さらに、sh−uPAuPARでトランスフェクトした細胞からの核抽出物での電気泳動移動度シフトアッセイ(EMSA)から、uPAおよびuPAR発現のノックダウンが、標識されたStat3結合部位への抽出物の結合を阻害したことが示された(図5C)。Stat3活性化は抗アポトーシス経路の刺激に寄与するので、減少したレベルのホスホ−Stat3ならびにDNA結合活性は、sh−uPAuPARトランスフェクトPC3細胞の、アポトーシス性細胞死に対する増大した感受性を説明し得る。あるいは、構成的ERK活性化は、細胞生存に寄与し得る。
【0075】
uPA−uPAR RNAiが、ヌードマウスにおける予め確立されたPC3同所腫瘍の腫瘍形成も抑制するかどうかを調査した。PC3細胞を、前立腺の側葉に前立腺内接種した。移植後7および14日後に、腫瘍に、sh−uPA、sh−uPARまたはsh−uPAuPARを発現するプラスミドコンストラクトを注射した。次いで、対照群で形成された腫瘍の結果として生じた病的状態によって余儀なくされたように、RNAi処置の2回目の投与の14〜15日後にマウスを屠殺した。原発腫瘍および転移の部位の全体の形態を調べた(図6A)。sh−uPA、sh−uPARおよびsh−uPAuPARプラスミドで処置したマウスにおいて二次腫瘍を視覚的に観察したが、EV、SVおよびmockで処置したマウスは、前立腺内に原発腫瘍に加えて二次腫瘍を示した(図6A)。腫瘍を切開し、計量した(図6B)。原発ならびに二次腫瘍の90〜100%の発生率が、mock、EVおよびSV処置群で観察された。sh−uPAおよびsh−uPAR処置は腫瘍増殖を完全には阻害しなかったが、これらの処置群からの形成された腫瘤の重量は、対照処置群からの腫瘍よりも小さかった(図6B)。sh−uPAuPARで処置したマウスにおいて、腫瘍重量の有意な減少が観察された。腫瘍試料中のタンパク質レベルの免疫ブロット解析から、sh−uPAuPARで処置した腫瘍が有意に減少したuPAおよびuPARレベルを有したことが確認された(図6C)。さらなる150μgのsh−uPAuPARは、予め確立された前立腺癌を完全に退行させた。
【0076】
採取したパラフィン包埋腫瘍組織で免疫組織化学的解析を行い、PC3細胞のインビボの性質に対するsh−uPAuPARの効果を評価した。sh−uPAuPAR依存性RNAi発現は、前立腺の全般的な構造を変化させず、H&E染色は大部分で正常な組織構造を示したが、染色は、対照群において腫瘍および宿主細胞の両方を明らかにした。uPAまたはuPARのいずれかのみを標的とするRNAiは、対照群と比較して、腫瘍細胞をわずかに減少させた。おそらく、uPA−uPAR RNAi誘導性アポトーシスからのインビボの救済はない。これを試験するために、クレノウ−FragEL DNA断片化解析を用いて、全ての処置群からのパラフィン包埋腫瘍切片をアポトーシスマーカーで染色した。このアポトーシス細胞の末端標識は、処置群の間の有意な差を示す。mock、EVおよびSV処置群の腫瘍は、FragELに対する、一般化された、低レベルの染色を示し、したがって、腫瘍細胞が健康であることを示した。対照的に、sh−uPAuPAR処置PC3前立腺腫瘍の領域のほとんどは、クレノウ染色に対して陽性であった。
【0077】
sh−uPAおよびsh−uPARの腫瘍内同時注射はまた、予め確立された前立腺腫瘍増殖のほとんど完全な退行を生じたが、mock、EVおよびSVの対照群は、再生性の、優位な腫瘍を示す(図7Aおよび7B)。免疫ブロット解析は、sh−uPAおよびsh−uPARコンストラクトで同時処置した腫瘍におけるuPAおよびuPARタンパク質レベルの選択的ノックダウンを示した(図7C)。この同時処置は、H&E染色によって大部分が正常な組織構造を示したが、mock、EVおよびSV処置群の腫瘍は、前立腺の全般的な構造の観察可能な変化を示し、H&E染色は、腫瘍および宿主細胞の両方を示した(図7D)。同時処置を通じたuPAおよびuPARのノックダウンはまた、正のクレノウ染色によって明らかであるように、アポトーシス性細胞死の有意な誘導を生じた(図7Eおよび7F)。図6A〜Cおよび7A〜Fに示されるデータと組み合わせると、これらの結果は、しかしながら、sh−uPAuPARでの処置が、確立された腫瘍サイズの有意な減少を引き起こすsh−uPAおよびsh−uPARでの同時処置と比較して、能力のあるRNAi効果を有することを示す。
【0078】
インビトロDNA標識解析およびインビボDNA断片末端標識アッセイの結果は、shRNAベースのRNAiによるuPAおよびuPARの同時のノックダウンがアポトーシスを誘導することを示す。uPA−uPAR媒介性下流シグナリングは、ホルモン非依存性前立腺癌の治療のための優れた標的である。uPA−uPAR媒介性下流シグナリングは、前立腺癌細胞系PC3における細胞浸潤、生存および増殖に必要とされるようである。
【0079】
(実施例6)
uPAおよびuPAR機能的シグナリングおよび腫瘍形成におけるそれらの役割
uPAおよびuPARの異常な発現は、前立腺癌の進行した段階における最も頻繁な変化の1つであることがわかった。uPAおよびuPARが前立腺癌の進行した段階でのみ過剰発現されることから、uPAおよびuPARが、増大した増殖および浸潤等の、進行した段階の癌の表現型の決定に関連のある機能経路に影響することが示唆される。細胞外マトリックスを通る浸潤は、腫瘍転移における特徴的な段階である。腫瘍形成を抑制するuPAまたはuPAR発現のいずれかの排除は、いくつかの異なるアプローチを用いて達成されてきた。uPAとuPARのカップリングは、増殖、遊走、浸潤、新脈管形成および転移等のいくつかの異なる型の生物学的応答の独特のネットワークを形成する、いくつかの異なるシグナリング分子を組み合わせる。uPA−uPAR結合に対するこれらの生物学的応答は、細胞型、下流シグナリング分子の性質およびその発現のレベルに高度に特異的であるようである。uPAとuPARの結合は、ERK1および2を活性化し、この誘導されたERK活性は、uPA誘導性MCF−7乳癌細胞遊走に必要である。FAK、SrcおよびShcを含むシグナリングカスケードは、uPA誘導性ERK活性化および細胞遊走の原因である。対照的に、uPA誘導性血管平滑筋細胞(VSMC)遊走および増殖は、Stat経路の活性化を必要とした。ヒト乳癌細胞において、uPA誘導性分裂促進活性は、StatおよびERK経路の両方の活性化を必要とする。アンチセンスuPAは、神経膠芽腫細胞系SNB19において、PI3K/Aktシグナリングを阻害し、細胞をスタウロスポリンによるアポトーシスに対して感作した。uPAとuPARの結合は、細胞遊走、浸潤、増殖および生存を調節するために、シグナリングカスケードを活性化するようである。
【0080】
PC3細胞におけるuPA−uPARに対するRNAiは、浸潤および増殖の顕著な抑制ならびにアポトーシスの誘導を示した(図3〜4)。sh−uPAuPARトランスフェクトPC3細胞において、uPA−uPAR系および下流のシグナリング分子(ERKおよびStat3)の抑制が観察されたが、mockまたはEV/SVトランスフェクト細胞では観察されなかった(図5)。このことから、これらの細胞における観察された表現型変化の全てはuPA−uPAR相互作用およびその下流の分子のリン酸化状態を抑制することによって媒介されたと示唆される。uPA−uPARシグナリングは、StatおよびERK経路の両方を刺激し、癌細胞を死から保護する。いくつかの筋の証拠によって、ERKおよびStat3経路の両方が細胞をアポトーシス性細胞死から保護できることが示されている。sh−uPAまたはsh−uPARのいずれかでのトランスフェクションは、PC3細胞においてアポトーシスを引き起こさなかった(図4)。これは、uPAまたはuPARのいずれか単独のブロッキングは下流のシグナリング分子ERKおよびStat3に十分影響しないかもしれないためかもしれない。
【0081】
uPA−uPARシグナリングはERKおよびStat3発現を調節するので、uPAおよびuPARの同時の阻害は、これらの経路を損なって、増殖阻害およびアポトーシスの誘導につながり得る。uPA−uPAR系は、癌の2つの特徴である細胞増殖および生存を容易にすることによって、細胞生存の正の調節因子として機能するようである。したがって、癌において過剰発現されると、uPAおよびuPARは、癌細胞に、増大した増殖性および/またはアポトーシスに対する増大した耐性を与える。対照的に、uPA−uPAR発現または機能のノックダウンは、癌細胞増殖を阻害し、アポトーシスを誘導するべきである。uPAおよびuPARのノックダウンを通じてuPA−uPAR媒介性シグナリングを遮断することによって、癌細胞増殖が同時に阻害され、アポトーシスが誘導された。注目すべきことに、uPA−uPAR RNAiは、ホルモン耐性前立腺癌細胞系PC3中で機能した。このことから、RNAiによるuPA−uPAR発現のノックダウンは、ホルモン耐性前立腺癌増殖および生存を阻害する戦略であることが示唆される。
【0082】
ヌードマウスにおけるヒト癌細胞の同所移植は、特に転移の発達に関して、ヒトにおけるこれらの細胞の生物学的性質によりよく似ている。このことは、ヌードマウスに異所に移植された場合によりかなり低い効率で原発腫瘍および転移を形成するヒト前立腺癌細胞で特に正しいことがわかっている。shRNAベースのRNAiプラスミド系は、免疫ブロット解析によって測定されたように、同所前立腺腫瘍におけるuPA−uPAR発現を効果的に抑制し得る戦略を代表する(図6C)。さらに、予め確立された同所腫瘍の、sh−uPAuPAR依存性RNAiでのインビボ処置は、腫瘍増殖のほとんど全体的な阻害を示したが、sh−uPAまたはsh−uPAR RNAiでは、部分的な減少しか観察されなかった(図6B)。また、予め確立された同所腫瘍の、sh−uPAおよびsh−uPARでの同時処置もまた、腫瘍増殖をほとんど完全に阻害した(図7)。RNAi処置動物群において、mockまたはEV/SV処置群と比較して、有害な効果は注目されなかった。さらに、このアプローチは、様々な腫瘍型を標的とし、uPA−uPAR依存性悪性表現型をインビトロならびにインビボで阻害することができる。したがって、このRNAi系は、強力な新しい治療手段を提供し、uPA−uPAR下流シグナリング経路を解析することは、同様に、臨床的妥当性を有する、癌介入の治療選択肢を提供する。さらに、RNAiは、uPA−uPAR発現に干渉し、癌生物学におけるそれらのシグナリングの生物学的重要性を研究する、新規の、簡便で選択的な方法を提供する。
【0083】
ヒト癌の分子機構の理解の進歩にもかかわらず、ヒト悪性腫瘍の臨床的治療のための治療アプローチの開発は、主要な課題のままである。uPA−uPAR発現のノックダウンは、PC3細胞の増殖をインビトロならびにインビボで有意に阻害し、最終的には、アポトーシス性細胞死を生じた。異なる標的遺伝子(ERKおよびStat3)が、uPA−uPARシグナルの下流で調節された。PC3細胞は、低いStat3リン酸化およびERKリン酸化を示し、sh−uPAuPARでトランスフェクトした場合、リン酸化は完全に廃された。uPA−uPARのRNAiは、PC3細胞における細胞死をインビトロならびにインビボで誘導した。
【0084】
(実施例7)
uPARおよびMMP−9を標的とするプラスミドベースのCMVプロモーター駆動21bp逆方向反復は、siRNAにプロセシングされる。
【0085】
CMVプロモーター駆動転写産物(uPARおよびMMP−9標的化)がsiRNAに正しくプロセシングされるかどうかを決定するために、SNB19細胞を対照/EV、SV、puPAR、pMMP−9およびpUMでトランスファーした。図8は、コンストラクトの概略図を示す。細胞はまた、非GFP細胞においてGFPを標的とする、関連のないコンストラクトでもトランスフェクトし、RNA転写産物の、siRNAへのプロセシングを決定し、得られた結果が標的遺伝子の分解産物だけではなかったことを確認した。pGFPでトランスフェクトした非GFP SNB19細胞は、RNA転写産物の、siRNAへのプロセシングを生じた(図9A)。同様に、puPAR、pMMP−9およびpUMでトランスフェクトした細胞は、RNA転写産物の、適切なsiRNAへのプロセシングを生じた。EVトランスフェクト細胞は、uPARまたはMMP−9を標的とするいかなるsiRNA様断片も生成せず、見られたsiRNA断片が、コンストラクトに組み込まれた逆方向反復ループからプロセシングされたことを示した。SVトランスフェクト細胞はまた、uPARまたはMMP−9を標的とするいかなるsiRNA様断片も生成しなかった。SVは、いかなる公知の遺伝子にも相同性を有さない、不完全な逆方向反復配列で構成された。SVの21bセンスオリゴでプローブすると、21bpDNA:RNAハイブリッドは見られず、このコンストラクトがsiRNA様断片をプロセシングしなかったことが示された。
【0086】
pUMでトランスフェクトしたSNB19細胞は、uPARおよびMMP−9 mRNAの両方のダウンレギュレーションを引き起こした。uPARおよびMMP−9に相同性のある逆方向21塩基対配列を含むプラスミドコンストラクトがRNAiを誘導するかどうかを決定するために、SNF19細胞を、対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトした。トランスフェクト細胞から全RNAを抽出し、cDNA合成キット(インビトロジェン)を用いてファーストストランドcDNAを合成した。次いで、当業者に公知の標準的なプロトコールに従って、cDNAをPCRに供した。対照/EVおよびSVでトランスフェクトした細胞においてuPAR、MMP−9、およびGAPDHに対する特異的プライマー(表1参照)を用いると、uPARまたはMMP−9のレベルの減少はなかったが、puPARでトランスフェクトした細胞およびuPARのレベルでは、mRNAが有意に減少し、MMP−9 mRNAのレベルは変化しなかった。pMMP−9でトランスフェクトした細胞において、MMP−9 mRNAのレベルが減少したが、uPAR mRNAレベルは変化せず、標的分子に対するベクターの特異性を示した。pUMでトランスフェクトした細胞は、uPARおよびMMP−9 mRNAレベルの両方の減少を示した。GAPDHレベルは変化しなかった(図9B)。
【0087】
(実施例8)
RNA干渉による、MMP活性およびuPARタンパク質レベルの阻害
SNB19細胞を、EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトし、次いで細胞溶解物中のuPARおよびMMP−9レベルをウエスタンブロッティングによって、および馴化培地中のuPARおよびMMP−9レベルをゼラチンザイモグラフィーによって、それぞれ測定した。ウエスタンブロッティングによって、pUMベクターでトランスフェクトしたSNB19細胞は、親およびEV/SV処置細胞と比較して、減少した量のuPARタンパク質を発現した(図10A)。この阻害効果がuPARに対して特異的かどうかを決定するために、同じブロットにおいて、β−アクチンレベルを評価した。β−アクチンレベルは、全てのレーンで同様であり、全てのレーンでの等しいローディングが確認された。pUM感染SNB19細胞からの馴化培地は、mockおよび空の/スクランブルベクタートランスフェクト細胞と比較して、有意に低いレベルのMMP−9活性を示した(図10B)。MMP−2レベルは変化せず、標的タンパク質の特異的な阻害が示された。濃度計によるuPARおよびMMP−9バンドの定量的解析から、親およびEV/SVトランスフェクト細胞と比較して、pUMトランスフェクト細胞において、uPARタンパク質(12〜14倍)およびMMP−9活性(8〜10倍)の有意な減少が明らかになった。puPARおよびpMMP−9ベクターでトランスフェクトした細胞は、2シストロン性コンストラクトとほとんど同じようにuPARおよびMMP−9のレベルを阻害した(図10Aおよび9B)が、標的分子のダウンレギュレーションは、単一のコンストラクトと比較して、2シストロン性コンストラクトで、より明白であった。
【0088】
uPARおよびMMP−9の低分子ヘアピンRNA(shRNA)のベクター媒介性発現は、インビトロおよびインビボで、神経膠腫細胞系において効果的で安定な遺伝子サイレンシングを達成し得る。RNAiベースの遺伝子サイレンシングは、有意な治療能力で、神経膠腫で過剰発現しているタンパク質を標的とするよう適合され得る。合成siRNA分子もまた、内在性MMP−9およびuPARレベルの抑制において、同じ効果を有する。
【0089】
(実施例9)
uPARおよびMMP−9のsiRNAによる細胞増殖の阻害
MTTアッセイを用いて、ビトロネクチンコートされたマイクロプレート上で培養された細胞の増殖に対する、siRNAベクター(EV/SV、puPAR、pMMP−9およびpUM)の効果を評価した。3日間の感染の後、puPAR、puPAR、pMMP−9およびpUMベクター感染SNB19細胞は、親およびEV/SVトランスフェクトSNB19細胞のものと比較して、増殖の減少を示した(図11)。pUMベクター効果は、単一のsiRNAコンストラクト(puPARおよびpMMP−9)と比較して、SNB19増殖がかなり高かった。親およびEV/SVトランスフェクトSNB19細胞の間に、増殖の差はなかった。
【0090】
(実施例10)
RNA干渉は、uPARおよびMMP−9免疫蛍光および腫瘍誘導性新脈管形成を阻害した。
【0091】
puPARおよびpMMP−9を感染させたSNB19細胞は、免疫細胞化学によって測定すると、それぞれuPARおよびMMP−9タンパク質レベルのダウンレギュレーションを引き起こした。pUMでトランスフェクトした細胞は、免疫細胞化学によって測定すると、uPARおよびMMP−9タンパク質レベルの両方のダウンレギュレーションを引き起こした(図12A)。uPARおよびMMP−9の両方のsiRNAを発現する組み合わせたコンストラクトの効果を測定するために、SNB19細胞をpuPAR、pMMP−9およびpUMでトランスフェクトした。細胞はまた、EVおよびSVでトランスフェクトし、対照とした。結果から、puPARのみでトランスフェクトした細胞がuPARタンパク質レベルのダウンレギュレーションを示したことは明らかである。pMMP−9でトランスフェクトした細胞は、MMP−9のみのダウンレギュレーションを示したが、pUMでトランスフェクトした細胞は、uPARおよびMMP−9タンパク質レベルの両方のダウンレギュレーションを引き起こし、二重のコンストラクトが標的タンパク質レベルのダウンレギュレーションで、同じくらい、あるいはそれ以上効率的であったことが示された。uPARおよびMMP−9のsiRNAが腫瘍誘導性毛細血管形成も阻害し得るかどうかを試験するために、トランスフェクトおよび非トランスフェクトSNB19神経膠腫細胞を、ヒト内皮細胞とともに共培養した。第VIII因子抗原を用いて免疫組織化学的解析を行い、インビトロの共培養系における腫瘍誘導性血管形成、ならびにEV/SV、puPARまたはpMMP−9およびpUMでのトランスフェクションの後のこれらの共培養物のH&E染色を評価した。図12Bは、SNB19細胞とともに培養した内皮細胞が、mockおよび空のベクターでトランスフェクトした培養物において24〜48時間以内に明確な毛細血管様網を形成したことを示す。対照的に、pUMトランスフェクトSNB19細胞は、内皮細胞において、毛細血管様網形成を誘導しなかった。分枝点および分枝の数の定量化は、親および空の/スクランブルベクターでトランスフェクトした共培養物と比較して、pUMトランスフェクト共培養において有意に減少した(図12C)。さらに、効果は、毛細血管様構造形成に関して親EV/SV処置群と比較して、puPARおよびpMMP−9ベクターにおいて50%未満であり、puPAおよびpMMPベクターでトランスフェクトした共培養物において50%未満であった。親EVトランスフェクトSNB19細胞を含むチャンバーの移植は、湾曲した細い構造および多くの小さな出血点を有する微小血管発生を生じた。対照的に、pUMベクターでトランスフェクトしたSNB19細胞の移植は、いかなるさらなる微小血管の発生も生じなかった(図12D)。
【0092】
(実施例11)
uPARおよびMMP−9のsiRNAは、SNB19細胞の浸潤を阻害する。
【0093】
siRNA発現はuPARおよびMMP−9を阻害したので、その細胞浸潤を阻害する能力を評価した。EV/SV、puPAR、pMMP−9およびpUMベクターでトランスフェクトしたSNB19細胞を、マトリゲルコートされたフィルターを通して浸潤させた。図13Aは、pUMトランスフェクトSNB19細胞の染色が親およびEV/SVトランスフェクト細胞のものよりも有意に低かったことを示す。細胞の定量的解析から、親およびEV/SVトランスフェクト細胞と比較して、8%のpUMトランスフェクトのみが浸潤したことが示された(図13B)。さらに、puPARおよびpMMP−9ベクターでトランスフェクトしたSNB19細胞の浸潤の定量的解析は、親およびEV/SVトランスフェクトSNB19細胞と比較して、25%および50%浸潤した(図13Aおよび13B)。RNAiはまた、三次元スフェロイド浸潤モデルにおいてSNB19細胞の浸潤を阻害した。図13Cは、mockおよび空の/スクランブルベクターでトランスフェクトした神経膠腫スフェロイドがラット脳凝集物に結合し、集合物に漸進的に浸潤したことを示す。しかしながら、pUMトランスフェクト神経膠腫スフェロイドとの共培養物はラット脳凝集物に結合できず、浸潤しなかった。定量的解析は、親およびEV/SVトランスフェクトスフェロイドでは2〜4%のラット胎仔脳凝集物のみが残ったが、pUMトランスフェクトスフェロイドでは90〜95%のラット胎仔脳凝集物が残ったことを示した(図13D)。72時間目に、puPARおよびpMMP−9トランスフェクト共培養物において、ラット脳凝集物はおよそ25%および45%の浸潤を現した。まとめて考えると、これらの発見から、uPARおよびMMP−9のRNAi媒介性サイレンシングが両方のインビトロモデルにおいて、uPARおよびMMP−9の単一のsiRNAコンストラクトと比較して、神経膠腫細胞浸潤を大いに阻害するという強力な証拠が提供される。これらの結果から、uPARの単一のsiRNAコンストラクトが、MMP−9の単一のsiRNAコンストラクトよりも効果的であったことが示された。
【0094】
(実施例12)
uPARおよびMMP−9のsiRNAの治療効果
腫瘍進行におけるuPARおよびMMP−9遺伝子発現のRNAi媒介性干渉の効果を評価するために、pUMベクターを、定位ポンプを用いて、腫瘍を有するマウスに注射した。侵襲性腫瘍細胞の検出を容易にするために、ヒト神経膠芽腫細胞(SNB19)を、緑色蛍光タンパク質のcDNAで評価した(SNB19−GFP)。脳切片の顕微鏡検査から、PBSまたは空のベクター(EV)のみを施した対照動物は、GFP蛍光および同様の切片のH&E染色で可視化すると、5週間の追跡期間の後、相当な腫瘍増殖を発生したことが明らかになった。対照的に、腫瘍増殖またはGFP蛍光もしくはH&E染色は、同じ条件下でpUMベクターを施した動物では検出されなかった(図14Aおよび14B)。処置に関して盲検化された神経病理学者によるヘマトキシリンおよびエオシン染色した脳切片またはGFP切片の定量化から、対照と空のベクター処置群との間に腫瘍サイズの差がないことが明らかになった。しかしながら、腫瘍の全体的退行が、pUMベクター処置群で現れた(図14C)。uPARおよびMMP−9の単一のsiRNA処置コンストラクトの場合、予め確立された大脳内腫瘍増殖は、それぞれ70%および40%阻害された。pUMベクターの腹腔内注射は、6ヵ月もの長い間、予め確立された大脳内腫瘍増殖の完全な退行を生じた。これらの結果から、uPARおよびMMP−9のRNAi媒介性抑制が、予め確立された大脳内腫瘍増殖を劇的に阻害したことが示された。
【0095】
uPARおよびMMP−9のRNAi媒介性阻害は、いくつかの独立した様式で腫瘍増殖を阻害し得る。DNA断片化によって測定したアポトーシスは、アンチセンスuPAR安定性クローンを注射した動物の脳において、親細胞系と比較して、より高かった。大脳内腫瘍モデルにおいて観察された抗腫瘍効果は、腫瘍細胞死の誘導のためかもしれなかった。
【0096】
腫瘍細胞は、生存および増殖するのに新脈管形成に依存する。uPARおよびMMP−9のRNAi媒介性阻害は、インビトロ共培養系において、腫瘍誘導性を有意に阻害した。pUMベクターの抗新脈管形成効果は、腫瘍細胞が生存に必要な血管を補充する能力を抑制し、抗新脈管形成効果を腫瘍細胞自体に向けた。uPARのsiRNAが腫瘍進行をブロックする能力はまた、uPAの抗アポトーシスおよび新脈管形成効果のブロッキングを含む。一般に、腫瘍が100mmに達した後に腫瘍負荷を減少させることのできる、前臨床モデルにおける単独療法として用いられる単一の抗新脈管形成剤(アンジオスタチンおよびエンドスタチンを含む)はない。PIg、uPA、またはtPAがないことによって、実験的脈絡膜新生血管の発生が、野生型またはuPAR欠損マウスと比較して、有意に減少したことが報告されている。この効果は、部分的にマトリックスメタロプロテイナーゼ活性の調節によると示唆された。研究から合成MMP阻害因子の抗新脈管形成効果が示されたが、これらの阻害因子の実質的に全ては、単一のMMPに対する特異性を欠いている。例えば、MMP−1、−3、および−9を阻害するKB−R7785で処置したマウスの原発腫瘍および転移において、減少した血管密度および増大した腫瘍細胞アポトーシスが観察された。MMPIは、進行した疾患を有する患者において単独療法として用いた場合、わずかな臨床的利益しか示さなかった。したがって、MMP阻害と他のモダリティーとを組み合わせた使用もまた、癌治療の戦略である。
【0097】
(実施例13)
uPARおよびMMP−9に対するsiRNAは、リン酸化ERK、MAPKおよびAKTのレベルを阻害する。
【0098】
ERK、MAPKおよびAKT経路は、細胞増殖および生存において主要な役割を果たす。EV/SV、puPAR、pMMP−9およびpUMでのSNB19細胞のトランスフェクションの後に、これらの分子に特異的な特異的抗体を用いることによって、ウエスタンブロッティングを用いて、全体的およびリン酸化形態のERK、MAPKおよびAKTのレベルを比較した。EV/SV、puPAR、pMMP−9およびpUMコンストラクトによって、全体的MAPK、ERKおよびAKTの量に有意な差はなかった(図15)。しかしながら、リン酸化形態のMAPK、ERKおよびAKTのレベルは、EV/SV、puPARおよびpuPAトランスフェクトSNB19細胞と比較して、pUMによって有意に減少した(図15)。
【0099】
MCF−7細胞における、uPAの、uPARへの結合は、細胞移動性に必要な、ERK1およびERK2を活性化する。前立腺癌細胞系(PC3MLN4)において、MAPK、ERKおよびp38キナーゼシグナリング経路を通じて媒介され得るuPARの発現をアップレギュレーションすることによって、低酸素によって腫瘍細胞浸潤が減少した。さらに、低酸素におけるBcl2によるuPAR発現のアップレギュレーションは、ERKシグナリング経路を通じて、SP1 DNA結合活性によって媒介された。EGFRなしでは、代替的な経路がuPARをERKに連結する。しかしながら、この経路はEGFR発現によってサイレンシングされ、そのため、細胞移動性におけるuPARの関与を示す。PTEN(ホスファターゼおよびテンシン類似物)の安定したトランスフェクションによって、焦点接着キナーゼおよびERK1/ERK2シグナリングのヒアルロン酸誘導性リン酸化によって引き起こされるMMP−9分泌が減少した。EGFR VIII増幅を有する神経膠芽腫は、最も高いレベルのMMP−9を示した。mt ERKおよびmt JNKでのSNB19細胞の一時的トランスフェクションによってMMP−9プロモーターが抑制され、いずれかの経路に干渉することでMMP−9発現の阻害が生じることが示唆された。種々の刺激による、または異なる細胞条件でのMMP−9活性化の調節は、異なるシグナル伝達経路を伴い得る。MEK特異的阻害因子によるERKの阻害は、乳癌細胞においてMMP−9発現をブロックし、MMP−9生成を減少させ、頭部および頸部扁平上皮癌細胞におけるインビボの侵襲性を和らげた。mt−ERK安定性トランスフェクト細胞は、侵襲性が低く、MMP−9レベルが有意に減少していた。これらの2つのシグナリング経路(MAPKおよびERK1/2)は、uPAがuPAR pUMコンストラクトに結合してこれらのシグナリング経路分子のリン酸化形態を阻害すると、活性化されることが報告されている(図15〜16)。
【0100】
(実施例14)
RNAi媒介性癌治療およびsiRNAの送達
例えばuPARおよびMMP−9過剰発現等の、遺伝子のsiRNA阻害は、ヒト癌の治療のための利用可能な治療モダリティーのリストを拡張させる。アンチセンスオリゴヌクレオチドおよびリボザイム技術を含むアンチセンスアプローチが利用可能であるが、それらの効率は十分ではない。uPARおよびMMP−9のRNAi媒介性阻害は、ヌードマウスにおいて、予め確立された神経膠腫腫瘍増殖を完全に抑制した。したがって、RNAiは、標的遺伝子発現を減少させることにおいて、アンチセンスオリゴヌクレオチドおよびリボザイム技術等の他の遺伝学的手段の強力な代替手段である。RNAiまたはRNAi様効果は、標的遺伝子発現を減少させることにおいて、アンチセンス効果よりも能力があり、RNAiの潜在的な適用性が示唆された。環状構造の腫瘍誘導アルギニン−グリシン−アスパラギン酸モチーフ、DNA結合オリゴリシンおよびヒスチジル残基を含むペプチドベクターを用いて、細胞質への送達を容易にした。ペプチドベクターは、siRNAの担体として機能することができる。RNAiベースの遺伝子療法は、神経膠腫ならびに前立腺癌、乳癌、および黒色腫を含む他の転移性腫瘍の治療のための、新規のアプローチである。
【0101】
(実施例15)
全細胞抽出物中のカテプシンBおよびuPARタンパク質レベルに対するpCUベクターの効果
タンパク質分解性分解を標的とするRNAiは、癌細胞浸潤を妨げるための介入である(図17)。カテプシンBおよびuPARは、ECM分解において重要な役割を果たすことが示されている。カテプシンBおよびuPARのsiRNAを発現するベクター(pCU)でのSNB19細胞のトランスフェクションは、mockおよび空のベクター(EV)対照と比較して、両方のタンパク質の発現を強力に阻害した(図18AおよびC)。β−アクチンのレベルから、等しい量のタンパク質がゲルにロードされたことが決定された(図18)。濃度計によるカテプシンBおよびuPARバンドの定量的解析から、mockおよび空のベクターでトランスフェクトした細胞と比較して、カテプシンB(14〜16倍)およびuPARタンパク質(10〜12倍)およびpCUトランスフェクト細胞における有意な(P<0.001)減少が明らかになった(図18BおよびD)。pUおよびpCベクターでトランスフェクトした細胞は、それぞれuPARおよびカテプシンBのレベルを阻害した(図18AおよびC)。
【0102】
(実施例16)
pCUベクターによる、腫瘍細胞誘導性毛細血管網形成の阻害
新生の腫瘍は、腫瘍増殖にエネルギーを供給する新しい血管の形成に依存する。カテプシンBおよびuPARは新脈管形成を調節すると報告されているので、腫瘍細胞誘導性新脈管形成に対するpCUの効果を評価した。インビトロ共培養系における腫瘍誘導性血管形成を評価するための第VIII因子抗原、およびmock、空のベクター、pC、pUまたはpCUでのトランスフェクション後のSNB19細胞の馴化培地の存在下で増殖した内皮細胞のH&E染色を用いて、免疫組織化学的解析を行った。結果から、mockおよび空のベクターでトランスフェクトした細胞の存在下で内皮細胞が48時間以内に毛細血管様構造を形成するが、pCUベクターは腫瘍細胞誘導性毛細血管様網形成を有意に阻害したことが示された(図19A)。分枝点および分枝の数の定量化は、mockおよび空のベクターと比較して、pCUトランスフェクト共培養物において検出できなかった(図19B)。さらに、効果は、毛細血管様構造に関して、pCUベクターと比較して、pCまたはpU処置した共培養物では50%未満であった。インビトロ共培養実験を確かめるために、背側チャンバーモデルによって評価して、pCUベクターが腫瘍新脈管形成を阻害できるかどうかをインビボで調べた。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞を含有する移植したチャンバーは、湾曲した細い構造および多くの小さな出血点を有する微小血管(矢印で示す)の発生を生じた。対照的に、pCUでトランスフェクトしたSNB19細胞の、移植したチャンバーは、いかなるさらなる微小血管の発生も生じなかった(図19C)。
【0103】
(実施例17)
siRNAによる、SNB19スフェロイドの遊走の阻害
カテプシンBおよびuPAR siRNA発現が腫瘍細胞遊走および増殖に影響を及ぼすことができるかどうかを決定するために、SNB19スフェロイドをpCUベクターでトランスフェクトした。図20Aに示すように、mockおよび空のベクター(EV)でトランスフェクトしたスフェロイドからの、かなり高い細胞遊走があり、単一のコンストラクトでトランスフェクトしたスフェロイドでは、遊走の50%までの阻害が観察された。しかしながら、腫瘍スフェロイドからの細胞遊走は、pCUベクターでトランスフェクトしたスフェロイドにおいて、完全に阻害された。mockおよび空のベクターでトランスフェクトしたスフェロイドの遊走は、スフェロイド外に遊走した細胞の数によって定量化すると、pC、pUおよびpCUトランスフェクトスフェロイドと比較して、相当高かった(P<0.001)(図20B)。スフェロイドからの細胞の遊走は、これらの分子の単一のRNAiコンストラクトと比較して、2シストロン性コンストラクトで阻害された。mockおよび空のベクター対照のものと比較して、少ない細胞がpCUトランスフェクトSNB19スフェロイドから遊走したので、それによって、細胞遊走におけるカテプシンBおよびuPARの役割が示された。
【0104】
(実施例18)
カテプシンBおよびuPARに対するsiRNAは、腫瘍細胞浸潤を阻害する。
【0105】
神経膠腫侵襲性に対するカテプシンBおよびuPARのsiRNA媒介性阻害の影響を評価するために、2モデルの系を使用した。mockおよび空のベクターでトランスフェクトしたSNB19細胞は、細胞の強い染色によって観察すると、マトリゲルコートしたトランスウェルインサートに広範に浸潤した。対照的に、pC、pUおよびpCUトランスフェクト培養物は、mockおよび空のベクターでトランスフェクトした細胞と比較して、再構成された基底膜を通る、より低い侵襲性を有した(図20C)。浸潤の定量的測定によって、pC、pUおよびpCUベクターでトランスフェクトしたSNB19細胞が、mockおよび空のベクターでトランスフェクトした対照と比較して、それぞれ55%、40%および6%しか浸潤しなかったことが確認された(図20D)。マトリゲル浸潤アッセイによって測定したこれらの細胞の侵襲性の性質の阻害は、単一のコンストラクトと比較して、2シストロン性コンストラクトトランスフェクト細胞においてかなり高かった。
【0106】
スフェロイド浸潤アッセイにおけるpCUの効果の程度を試験した。スフェロイド共培養アッセイにおいて、対照神経膠腫スフェロイド、および空のベクターでトランスフェクトしたスフェロイドは、ラット胎仔脳凝集物に漸進的に浸潤し、pCUでトランスフェクトしたスフェロイドの、部分的〜ほとんど完全な阻害を生じた(図20E)。ラット胎仔脳凝集物の定量化から、神経膠腫スフェロイドが、24時間以内に25%、48時間以内に70%未満、および72時間以内に90%未満、ラット胎仔脳凝集物を浸潤したことが明らかになった。対照的に、pCUベクターでトランスフェクトした腫瘍スフェロイドは、ラット胎仔脳凝集物を浸潤しなかった。72時間目に、ラット脳凝集物は、mock空のベクター、pC、およびpUトランスフェクト共培養物においておよそ90%、85%、55%および35%の浸潤を現したが、pCUトランスフェクト共培養物では、2%〜3%の浸潤しか現さなかった((図20F)。まとめて考えると、これらの発見から、カテプシンBおよびuPARのRNAi媒介性サイレンシングが神経膠腫細胞浸潤を両方のインビトロモデルで強力に阻害すること、および単一のコンストラクトと比較して、2シストロン性コンストラクトで効果がかなり高かったことの、強力な証拠が提供される。
【0107】
腫瘍細胞浸襲性の獲得は、腫瘍進行の側面の1つである。カテプシンBおよびuPARの発現が浸潤過程の必須の構成要素であることを示す、いくつかの報告がある。pCUベクターでのトランスフェクションによって、マトリゲル浸潤およびスフェロイド共培養アッセイにおいて、SNB19細胞およびスフェロイドの侵襲性が阻害された。カテプシンBのマトリゲル浸潤の要件は、プロテアーゼのネットワークとのその相互作用によるかもしれない。カテプシンBは、プロ−ストロメライシン等の、プロ−uPAおよびメタロプロテイナーゼ等の、セリンプロテイナーゼの前駆体をその活性形態に活性化することが示されている。形質転換ヒト乳房上皮細胞系の、マトリゲルを通る侵襲性は、カテプシンB発現と関連があり、システインプロテイナーゼ阻害因子によって阻害された。卵巣癌細胞において、細胞表面カテプシンBの阻害はプロ−uPAの活性化を防ぎ、それに続いて、マトリゲルを通る癌腫細胞の浸潤を防いだ。ヒト結腸癌におけるカテプシンB活性は、癌細胞、内皮細胞および炎症細胞の侵襲性ならびにアポトーシス性および壊死性細胞死に関連がある。uPAおよびuPARは、乳癌、卵巣癌、および胃癌を含む種々の悪性腫瘍において過剰発現されることが公知であり、侵襲性および転移性表現型の維持に必須であることが示されている。
【0108】
(実施例19)
カテプシンBおよびuPARのsiRNA媒介性ダウンレギュレーションは、SNB19細胞の増殖を減少させる。
【0109】
標準的MTTアッセイを用いて、ビトロネクチンコートされたマイクロプレート上で培養した細胞の増殖に関して、siRNAベクター(対照、EV、SV、pC、pUおよびpCU)の効果を評価した(図21)。感染の72時間後に、pC、pUおよびpCUベクター感染SNB19細胞は、SNB19およびベクター対照のものと比較して、増殖の減少を示した。pCUトランスフェクト細胞は、7日間のトランスフェクションの後でも、測定できるいかなる増殖も示さなかった。7日間のアッセイの後でも、いずれのトランスフェクト細胞でも浮遊細胞または細胞残屑は見られず、アポトーシスがないことが示された。
【0110】
(実施例20)
uPARおよびカテプシンBのsiRNA媒介性ダウンレギュレーションが、ERK1/2およびFAKリン酸化を阻害する。
【0111】
シグナリング経路分子に対するuPARおよびカテプシンBのダウンレギュレーションの効果を測定するために、ともに腫瘍細胞生存、遊走および増殖に直接関係するERKおよびFAKのリン酸化を、ウエスタンブロッティングによってアッセイした。図22は、uPARおよびカテプシンBのRNAi媒介性の同時のダウンレギュレーションによってERK1/2およびFAKのリン酸化が遅れ、単一のコンストラクトでは効果がかなり少なかったことを示す。
【0112】
結果から、uPARおよびカテプシンBのダウンレギュレーションによって、細胞生存および増殖の直接の原因であるERK1/2およびFAKリン酸化のダウンレギュレーションが誘導されたことが示された。ERK−FAKカスケードにおけるuPARの関与は、ヒト癌腫細胞HEp3において以前に報告されているが、カテプシンBの役割は未だ不明である。uPARおよびカテプシンBの組み合わせのダウンレギュレーションは、ERK1/2およびFAKのリン酸化の阻害に、より効果的である。
【0113】
(実施例21)
カテプシンBおよびuPAR RNAiは、大脳内腫瘍増殖を抑制する。
【0114】
大脳内腫瘍モデルを用いて、予め確立されたインビボの腫瘍増殖に対する、RNAi媒介性阻害の潜在的効果を評価した。未処置(mock)およびEV処置対照群の脳切片は、5週間の追跡期間の後のH&E染色および高いGFP蛍光による、大きな広がった腫瘍増殖によって特徴付けられた(図23AおよびB)。しかしながら、pCUベクターで処置したマウスの脳切片で、GFP蛍光は検出されなかった(図23AおよびB)。処置に関して盲検化された神経病理学者によって点数付けされたH&E染色脳切片のさらなる定量化から、mockおよび空のベクター処置群の間の腫瘍サイズの差は明らかにならず、pCおよびpU処置群で、対照と比較して55%および65%の腫瘍増殖の有意な退行が明らかになった。しかしながら、pCU処置群においては、予め確立された腫瘍の全体的退行が明らかになった(図23)。これらの結果から、カテプシンBおよびuPARのRNAi媒介性抑制によって大脳内腫瘍増殖が有意に阻害されたことが示された。
【0115】
ミニ浸透圧ポンプを用いたpCUの局所的大脳内送達によって、ヒト悪性神経膠腫増殖が効果的に阻害された。ミニ浸透圧ポンプは、相当な時間にわたって明確で一貫したパターンの薬物曝露を維持し、脳への作用物質の送達にうまく用いられ得る。カテプシンBおよびuPARのダウンレギュレーションは、腫瘍誘導性新脈管形成の阻害を生じる。共培養アッセイをインビトロで用いて、新脈管形成に対するpCUの効果を試験した。結果から、カテプシンBおよびuPARが新脈管形成の刺激において関連性のある役割を果たすことが示され、大脳内腫瘍モデルにおけるpCUのインビボ抗腫瘍活性の作用の、可能性のある機構が示唆された。新生血管の内皮細胞にカテプシンBの強い染色が存在するが、前立腺の既存の微小血管系には存在しない。同様に、ラット脳微小血管内皮細胞において、それらがインビトロで毛細血管を形成すると、カテプシンBの強い免疫染色が観察された。カテプシンBはTIMPの阻害因子であることが示されており、TIMPは新脈管形成の阻害因子なので、カテプシンBはまた、腫瘍の広がりにおいて関連性のある役割を有する新脈管形成を刺激し得る。おそらく新脈管形成および侵襲性が阻害されたために、カテプシンBおよびuPARのRNAi媒介性標的化によって、予め確立された大脳内腫瘍増殖が抑制された。これらの結果はまた、標的遺伝子発現のsiRNA媒介性ダウンレギュレーションが脳微環境内で十分安定していることを支持する。
【0116】
(実施例22)
SNB19神経膠芽腫細胞における、uPARタンパク質、ならびにuPAおよびMMP−9酵素活性に対するsiRNAコンストラクトの効果
3つの内在性遺伝子をヘアピンsiRNAで同時に阻害するために、CMVプロモーター制御下でuPAR(RNAでのヒトuPARの77〜98塩基)、uPA(RNAでのヒトuPAの346〜367塩基)およびMMP−9(RNAでのヒトMMP−9の360〜381塩基)のsiRNAを発現するベクター(pU2M)を構築した(図24)。塩基は、全長コード配列中の位置を示す。ウエスタンブロット解析を行って、SNB19細胞中のuPARタンパク質濃度に対する、空のベクター/スクランブルベクター(EV/SV)、puPAR、puPA、MMP−9およびpU2Mトランスフェクションの効果を調べた。EV/SV、puPAおよびpMMP−9でトランスフェクトしたSNB19細胞にuPARタンパク質バンドが存在したが、これはpuPARおよびpU2M処置細胞では有意に減少した(図25A)。3シストロン性コンストラクト(pU2M)の効果は、puPARよりも大きかった(図25A)。GAPDHのレベルから、等しい量のタンパク質がゲルにロードされたことが決定された(図25A)。フィブリンザイモグラフィーを行って、uPA酵素活性に対する、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置SNB19細胞の効果を調べた。ゼラチンザイモグラフィーを行って、SNB19細胞中のMMP−9のレベルに対するこれらのコンストラクトの効果を測定した。MMP−9レベルは、親、EV/SVおよびpuPA処置細胞と比較して、puPAR、pMMP−9およびpU2Mで処置したSNB19細胞で有意に減少した(図25B)。興味深いことに、pU2M処置細胞において、MMP−2レベルもまたダウンレギュレーションされた。等しい量のタンパク質がロードされるように、注意が払われた。(図25B)。uPA酵素活性(MR 55 000)は、親、EV/SV、puPARおよびpMMP−9処置群と比較して、puPAおよびpU2M処置細胞で有意に減少した(図25C)。
【0117】
3シストロン性コンストラクトの効果は、これらの分子の単一のsiRNAコンストラクトのものよりも明白であった。免疫組織化学的解析によって測定すると、puPAR、puPA、pMMP−9およびpU2Mトランスフェクションによって、SNB19細胞中のuPAR、uPAおよびMMP−9濃度が減少した。図25Dは、uPAR、uPAおよびMMP−9に対する特異的抗体を用いた、親、EV/SV、puPAR、puPA、pMMP−9およびpU2M処置細胞中のuPAR、uPAおよびMMP−9のタンパク質レベルを示す。uPAR、uPAおよびMMP−9のそれぞれの強度は親細胞中およびEV/SVでトランスフェクトした細胞で高かった。対照的に、uPAR強度は、puPARおよびpU2MでトランスフェクトしたSNB19細胞で減少した。puPAおよびpU2Mトランスフェクションによって、親、EV/SV、puPARおよびpMMP−9トランスフェクト細胞と比較して、uPAタンパク質の強度が有意に減少した。さらに、MMP−9タンパク質濃度は、親、EV/SV、puPAおよびpuPARトランスフェクト細胞と比較して、pMMP−9およびpU2Mトランスフェクト細胞で有意に減少した。これらの結果から、単一のコンストラクトの効果が分子特異的であること、および3シストロン性コンストラクトの効果が単一のコンストラクト単独のものよりかなり明白であることが示された。
【0118】
(実施例23)
puPAR、puPA、pMMP−9およびpU2Mは、腫瘍誘導性毛細血管網形成を阻害する。
【0119】
神経膠腫腫瘍の増殖は、腫瘤の発生を支持するのに必要な、新しい毛細血管の誘導に依存する。神経膠腫細胞からの馴化培地によって微小血管内皮細胞が誘導されて毛細血管様構造を形成する共培養系を用いて、uPAR、uPAおよびMMP−9のRNAi媒介性抑制の効果を調べた。インビトロ共培養系における腫瘍誘導性血管形成を評価するための第VIII因子抗原を用いた免疫組織化学的解析、および行ったH&E染色。内皮細胞は、SNB19親およびEV/SVトランスフェクト細胞からの馴化培地の存在下で、毛細血管様構造を形成する(図26A)。対照的に、uPA、uPARおよびMMP−9のsiRNAを発現するベクターでのSNB19細胞のトランスフェクションによって、個々に、または組み合わせてのいずれかで、腫瘍誘導性微小血管形成が部分的または完全に阻害された(図26A)。EV/SV処置細胞と比較して、pU2M処置細胞では、新しい分枝点および/または分枝の数の増大は検出されなかった(図26A)。さらに、EV/SV処置細胞と比較して、毛細血管様構造の形成が、puPAR処置培養物において約55%、puPA処置培養物において約36%、およびpMMP−9処置細胞において約60%阻害された(図26B)。
【0120】
pU2Mでトランスフェクトした神経膠芽腫細胞系からの馴化培地は、mockまたは空のベクターと比較して、毛細血管様構造を阻害した(図26A〜B)。このことから、新脈管形成の誘導に必要な新脈管形成シグナルはpU2M処置細胞中に存在しなかったことが示される。pU2MトランスフェクトSNB19神経膠腫細胞に新脈管形成誘導がないことによって示されるように、hpRNAによるuPA、uPARおよびMMP−9のダウンレギュレーションは、新脈管形成因子のダウンレギュレーションを引き起こした。uPAまたは組織型プラスミノゲン活性化因子(tPA)がないことによって、野生型(WT)またはuPAR欠損マウス(uPA−/−)と比較して、実験的脈絡膜新生血管の発生が有意に減少した。uPA−/−およびプラスミノゲン活性化因子阻害因子−1欠損(PAI−1−/−)マウスにおいて、WTマウスと比較して有意に減少した原発腫瘍増殖が起こったことが報告されており、uPA−/−およびPAI−/−マウスの腫瘍は、WTマウスと比較して、より低い増殖性およびより高いアポトーシス指数を示し、異なる新血管形態も示した。バクテリオファージディスプレイによってuPA結合を阻害することが示されているいくつかのペプチドは、同系のマウスにおいて新脈管形成および原発腫瘍増殖を阻害する。
【0121】
(実施例24)
puPAR、puPA、pMMP−9およびpU2Mは、SNB19細胞における浸潤を阻害する。
【0122】
ECM構成要素のタンパク質分解性分解は、腫瘍細胞浸潤に関連性がある。神経膠腫侵襲性に対するuPAR、uPAおよびMMP−9のsiRNA媒介性阻害の影響を評価するために、2つのモデルを利用した。第一のモデルにおいて、puPAR、puPA、pMMP−9およびpU2Mを感染させたSNB19細胞の浸潤能力を、EV/SVベクターを感染させたものと比較した。EV/SVでトランスフェクトしたSNB19細胞および親細胞は、細胞の強い染色によって示されるように、マトリゲルコートされたトランスウェルインサートを通る、広範な浸潤を示した。対照的に、puPAR、puPA、pMMP−9およびpU2Mトランスフェクト培養物は、対照と比較した染色強度によって示されるように、再構成された基底膜を通しての侵襲性が低かった(図26C)。定量化によって、puPAR、puPA、pMMP−9およびpU2Mベクターでのトランスフェクションによって、SNB19細胞による浸潤が、親およびEV/SVトランスフェクト対照と比較して、それぞれ9%、40%、15%および2%に減少したことが確かめられた(図26D)。浸潤の阻害は、単一のコンストラクトのみと比較して、3シストロン性コンストラクトでトランスフェクトした細胞でより高かった。
【0123】
スフェロイド浸潤アッセイを用いて、puPAR、puPA、pMMP−9およびpU2Mベクターの効果を調べた。神経膠腫スフェロイドを用いることの相当な潜在的利益は、三次元培養で増殖した腫瘍細胞がインビボの腫瘍のものとより密接に類似した特性を示すことである。スフェロイド共培養系において、対照スフェロイドおよびEV/SVベクターでトランスフェクトしたスフェロイドは、ラット胎仔脳凝集物に漸進的に浸潤したが、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトしたスフェロイドは、部分的〜ほとんど完全な浸潤の阻害を示した(図27A)。定量化によって、神経膠腫スフェロイドがラット胎仔脳凝集物に、1日以内に30%、2日以内に55%、および3日以内に95%浸潤し、このとき腫瘍スフェロイドおよび脳凝集物は組み合わされて単一の実体になったことが明らかになった(図27B)。EV/SVベクターでトランスフェクトした神経膠腫スフェロイドで、同様の傾向が観察された。対照的に、pU2Mベクターでトランスフェクトした腫瘍スフェロイドは、ラット胎仔脳凝集物に浸潤しなかった。3日目までに、ラット脳凝集物は、親、EV/SV、puPA、pMMP−9およびpuPARトランスフェクト共培養物において、およそ96%、95%、45%、25%および15%浸潤され、pU2Mトランスフェクト共培養物では1%浸潤された(図27B)。これらの結果から、pU2Mが両方のインビトロモデルで神経膠腫浸潤を強力に阻害することの強力な証拠が提供される。
【0124】
(実施例25)
pU2Mは、大脳内腫瘍増殖を完全に退行させる。
【0125】
単一または3シストロン性コンストラクトを用いて、ヌードマウスにおいて予め確立された大脳内腫瘍増殖の退行を引き起こすuPAR、uPAおよびMMP−9レベルのダウンレギュレーションを調べた。対照およびEV/SV処置群における全ての動物は、強いGFP蛍光によって特徴付けられる、インタクトな脳腫瘍を有した(図27C)が、puPAR、puPAおよびpMMP−9で処置したマウスの脳切片は、最小のGFP蛍光によって示される小さな腫瘍を有した。注目すべきことに、pU2Mで処置したマウスの脳切片において、GFP蛍光は検出されなかった(図27C)。これらの切片のさらなる定量化(処置条件に対して盲検化された神経病理学者によって点数付けされた)から、親およびEV/SV処置群の間の腫瘍サイズの差、ならびに、対照群と比較した、puPAR、puPAおよびpMMP−9で処置した群における予め確立された腫瘍増殖の有意な退行は明らかにならなかった(それぞれ80%、55%、および68%)(図27D)。しかしながら、pU2M処置群において、予め確立された大脳内腫瘍増殖の完全な退行が明らかになった。これらの結果から、3シストロン性コンストラクトを用いたuPAR、uPAおよびMMP−9のRNAi媒介性抑制が、ヌードマウスにおいて悪性神経膠腫増殖を完全に根絶したことが示される。
【0126】
(実施例26)
ERK1/2リン酸化の阻害
シグナリング経路に対するuPAR、uPAおよびMMP−9のsiRNA媒介性ダウンレギュレーションの効果をよりよく理解するために、腫瘍細胞生存、遊走および増殖に直接関係する、ERK1/2の全体的およびリン酸化レベルをアッセイした。ウエスタンブロットから、puPAR、puPA、pMMP−9およびpU2Mトランスフェクト細胞と比較して、対照およびEV/SVトランスフェクト細胞における全ERK1/2濃度に有意な差がなかったことが示された(図28)。しかしながら、ホスホ−ERK1/2の濃度は、対照、EV/SV、puPAR、puPAおよびpMMP−9トランスフェクトSNB19細胞と比較して、pU2MベクターでトランスフェクトしたSNB19細胞で有意に減少した。注目すべきことに、単一のコンストラクトのいずれかでトランスフェクトしたSNB19細胞において、ホスホ−ERKのレベルに対する効果はなかった。GAPDHレベルから、等しい量のタンパク質がゲルにロードされたことが示された(図28)。
【0127】
(実施例27)
遺伝子特異的siRNAは、神経膠腫細胞系におけるMMP−9およびカテプシンBタンパク質の発現を低下させる。
【0128】
2つの内在性遺伝子をヘアピンsiRNA発現ベクターで同時に阻害することの効果を試験するために、カテプシンB(ヒトカテプシンB mRNAの732〜753塩基)およびMMP−9(ヒトMMP−9 mRNAの360〜381塩基)のsiRNAを発現するベクターを、ヒトサイトメガロウイルス(CMV)プロモーターの制御下に構築した(pCM)(図29)。塩基は、全長コード配列中の位置を示す。図30Aは、pCおよびpCMベクターでのトランスフェクションが、mock、空の、およびpMベクター対照と比較して、カテプシンBレベルを特異的に阻害することを示す。同じブロットで評価したβ−アクチンレベルは、カテプシンBの阻害が特異的であることを示し、等しい量の試料ローディングが確認された。トランスフェクト細胞の馴化培地中で、MMP−2およびMMP−9レベルを測定した。mockおよび空のベクター(EV)トランスフェクト細胞から放出されたMMP−9の量は、同じであった。pMおよびpCMベクターでトランスフェクトした細胞は、mock、EV、およびpC対照と比較して、低いレベルのMMP−9を発現した。MMP−2の発現の変化はなく、pMおよびpCMベクターの配列特異的阻害が示された(図30B)。MMP−9活性の減少がタンパク質発現の減少のためであることを確かめるために、MMP−9特異的抗体での免疫ブロットを用いて、馴化培地を解析した。pMおよびpCMベクターでトランスフェクトした細胞からの馴化培地の免疫ブロットによって、MMP−9タンパク質バンドが劇的に減少したが、バンドは有意に、空のベクターまたはpCベクターを感染させた細胞からの馴化培地においてかなりより強かった(図30C)。
【0129】
(実施例28)
PCMベクターによる、腫瘍細胞誘導性毛細血管網形成の阻害
神経膠腫腫瘍の増殖は、発生する腫瘤を支持するのに必要なので、新しい毛細血管の誘導に依存する。カテプシンBおよびMMP−9のRNAi媒介性抑制を調べるために、微小血管内皮細胞が神経膠腫細胞によって誘導されて毛細血管様構造を形成する、共培養系を用いた。SNB19細胞は、72時間以内に、内皮細胞を誘導して、毛細血管様構造に分化した。対照的に、カテプシンBおよびMMP−9のsiRNAを発現するベクターでのSNB19細胞のトランスフェクションによって、腫瘍細胞誘導性微小血管形態形成が完全に阻害された(図31A)。分枝点および分枝の数のさらなる定量化は、mockおよび空のベクターと比較して、pCMトランスフェクト共培養物中で検出できなかった(図31B)。さらに、効果は、毛細血管様構造に関して、pCMベクターと比較して、pCまたはpM処置共培養物でわずか50%であった。インビトロ共培養実験を確かめるために、背側ウインドウモデルによって評価して、pCMベクターが腫瘍新脈管形成を阻害できるかどうかをインビボで調べた。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞を含有するチャンバーの移植は、湾曲した細い構造および多くの小さな出血点を有する微小血管(矢印で示す)の発生を生じた。対照的に、pCMベクターでトランスフェクトしたSNB19細胞の移植は、いかなるさらなる微小血管の発生も生じなかった(図31C)。
【0130】
悪性腫瘍の増殖維持は、腫瘍に栄養分を供給する血管網の発生と密接に関連している。pCMベクターで処置した細胞における、閉じた多角形および複雑なメッシュ様構造によって特徴付けられる血管ネットワークの形成は、観察されなかった。このネットワークは、典型的には、神経膠腫細胞が内皮細胞とともに共培養された場合に観察される。細胞外マトリックス構成要素のタンパク質分解によって、内皮細胞が遊走できるようになり、貯蔵された新脈管形成シグナリング分子が細胞外マトリックスから放出される。免疫組織化学的解析から、正常な脳組織と比較して、悪性未分化星状細胞腫および神経膠芽腫においてカテプシンBが強く発現されたことが示された。
【0131】
これらの結果から、MMPが新脈管形成を促進し得ること、およびMMPが完全にないと新しい血管形成が予防され得ることが示される。本開示における組み合わせの療法によって達成される腫瘍退行は、カテプシンBおよびMMP−9の相補的作用による。腫瘍細胞中のカテプシンBおよびMMP−9の発現を標的とすることは、新脈管形成および腫瘍増殖を制御する効果的なアプローチである。
【0132】
(実施例29)
神経膠腫遊走および浸潤に対するpCMベクターの抑制効果
細胞遊走は、細胞−細胞結合、細胞−マトリックス結合およびマトリックスリモデリングの共同した調節を必要とする。細胞がスフェロイド遊走アッセイにおいてビトロネクチン上で遊走する能力に対する、カテプシンBおよびMMP−9を抑制することの効果を、研究した。アガロースでコートした6ウェルプレート中で、SNB19−GFP細胞から多細胞神経膠腫スフェロイドを増殖させた。形態およびトリパンブルー排除を用いて生存能をチェックした後、同様の直径(100〜200μm)のスフェロイドを、mock、空のベクター(EV)、またはカテプシンBおよびMMP−9のsiRNAを発現するpCMベクターでトランスフェクトした。3日後、単一のスフェロイドをビトロネクチンコートされたプレートに入れて遊走させた。図32Aは、対照スフェロイドおよび空のベクターを感染させたスフェロイドからの細胞が、pCMベクター感染細胞と比較して、細胞が遊走する有意に高い能力を示したことを示す。細胞外マトリックス構成要素のタンパク質分解性分解は、腫瘍細胞浸潤に関連性がある。カテプシンBおよびMMP−9のsiRNAの発現が神経膠腫侵襲性において役割を果たすかどうかを調査するために、mockおよび空のベクターを感染させた細胞に対する、pCMベクターでトランスフェクトしたSNB19細胞の浸潤能力を比較した。mockおよび空のベクター(EV)でトランスフェクトしたSNB19細胞は、マトリゲルを通って浸透したpCMベクタートランスフェクト細胞と比較してより広範に、マトリゲルを通って浸潤した(図32B)。
【0133】
スフェロイド浸潤アッセイにおけるsiRNAの抑制効果の程度を調べた。神経膠腫スフェロイドを使用することの潜在的利点は、三次元培養で増殖した腫瘍細胞がインビボの腫瘍のものとより密接に類似した特性を示すことであると示されている。mockおよび空のベクターでトランスフェクトしたスフェロイドは1日以内に正常な脳凝集物の25%、2日以内で50%が浸潤し、3日目までに、腫瘍スフェロイドの95%が浸潤し、脳凝集物は組み合わされて単一の実体になった(図32C)。対照的に、カテプシンBおよびMMP−9のsiRNAを発現するpCMベクターでトランスフェクトした神経膠腫スフェロイドは、正常な脳凝集物から分離したままであった。
【0134】
本開示は、カテプシンBおよびMMP−9に対するsiRNA(pCM)のCMVプロモーター駆動発現が、ウエスタンブロッティングおよびゼラチンザイモグラフィーによって解析すると、SNB19神経膠腫細胞系において、カテプシンBおよびMMP−9発現をうまくサイレンシングし得ることを示す。結果から、pCMベクターで処置した神経膠腫細胞の浸潤能力が有意に阻害されることも示された。癌細胞は、遊走および浸潤するには近隣の細胞および細胞外マトリックス構成要素から分離しなければならない。マトリックスタンパク質分解は、接着部位の除去または結合部位の露出のいずれかによって細胞−マトリックス接着を直接調節し得、これが次に細胞遊走に影響し得る。カテプシンBおよびMMP−9のRNAi媒介性阻害によって、スフェロイド遊走アッセイにおいて示されるように、SNB19神経膠腫細胞の遊走が有意にブロックされた。
【0135】
(実施例30)
RNAiは、ヌードマウスにおいて神経膠芽腫腫瘍の完全な退行を誘導する。
【0136】
MMP−9およびカテプシンBのsiRNAが大脳内SNB19腫瘍の退行を阻害する能力を、ヌードマウスにおいて試験した。予め確立された神経膠腫増殖を有するマウスに、PBS(mock)、空のベクター(EV)、pC、pMおよびpCMベクターを定位注射した。mockおよびEVで処置したマウスの脳切片は、急速な腫瘍増殖を示したが、予め確立された腫瘍にミニ浸透圧ポンプを用いてpCMベクターを注射したマウスは、5週間にわたって腫瘍増殖の完全な阻害を生じた(図32Aおよび32B)。腫瘍サイズの定量化は、mockまたは空のベクターと比較した、pCMベクター処置群における腫瘍の全体的退行を示した(図33C)。pCまたはpMベクター処置群で処置したマウスの脳切片は、対照群と比較して、約50%の腫瘍退行を生じた。ベクターの腹腔内注射もまた、予め確立された大脳内腫瘍増殖の完全な退行を生じ、数ヵ月の長い期間にわたって腫瘍細胞を示さなかった(図33D)。したがって、RNAiは、このヌードマウスモデルにおいて、悪性神経膠腫腫瘍増殖を完全に根絶することができた。神経膠腫増殖の持続的抑制は、siRNA増幅のためであるかもしれない。カテプシンBおよびMMP−9に対するsiRNAは、神経膠腫増殖を、MMP−9およびカテプシンBのアンチセンスオリゴデオシヌクレオチドよりも、より効率的に抑制した。したがって、カテプシンBおよびMMP−9発現の両方の制御は、腫瘍進行の調節において、相当な重要性を有する。
【0137】
カテプシンBおよびMMP−9のRNAi媒介性阻害の抗癌効能が示された。哺乳動物細胞における、アンチセンスオリゴヌクレオチドおよび同じ標的に対するsiRNAの抑制効果の比較から、siRNAのIC50値が、アンチセンスオリゴヌクレオチドのものよりも約100倍低いことが明らかになった。siRNAが配列特異的標的遺伝子をサイレンシングする能力、および遺伝子発現を阻害するのに必要なより低い濃度によって、RNAiは遺伝子治療の強力な手段になる。
【0138】
(実施例31)
環状プラスミドからのsiRNAに対する減少した免疫原性応答
uPAおよびuPARのヘアピンsiRNA分子を生成することのできるベクターを開発するために、哺乳動物発現プラスミドベクターpCDNA3を用いた。図34〜35は、種々の形態のU6およびCMV駆動RNAiコンストラクトの概略図を示す。uPAを(346〜367)およびuPAR(77〜89)標的とする、9塩基のGCの欠乏した領域によって間隔が空けられた自己相補的逆方向反復配列を合成した。uPAのオリゴはHindIII部位で終わり、uPARのオリゴはBamHIで終わり、100℃に5分間加熱することによって自己アニーリングし、6×SSC中室温まで冷却し、それぞれの粘着制限酵素認識部位末端を有する二本鎖DNA分子の形成を生じた。これらのdsDNA分子を、pCDNA3プラスミドベクターのBamHIおよびHindIII部位にライゲートし、CMVプロモーターの下流のuPAおよびuPARの逆方向反復を含み、BGHターミネーターによって終了するプラスミドの形成を生じた。得られたプラスミドはpU2と名づけ、哺乳動物細胞にトランスフェクトすると、uPAおよびuPARの両方を標的とする二重のヘアピンsiRNA分子の生成を生じ、これはdsRNA認識酵素(DICER)によってさらにプロセシングされて、RNAiを誘導する個々のsiRNA分子を生成した。スクランブルベクターの構築において、GFPに相同性のある配列を用いた。完全なヘアピン構造を形成しない不完全な配列を用いて、スクランブルベクターを開発した。2つの自己相補的オリゴを合成およびアニーリングして、HindIII部位を有するdsDNA分子を生じた。このdsDNA分子をpCDNA3プラスミドのHindIII部位でライゲートさせた。得られたプラスミドをpSVと呼んだ。得られたCMV駆動転写産物は、ヘアピン様構造を有さず、いかなる天然の遺伝子にも相同性を有さなかった。
【0139】
uPAおよびuPARのsiRNAを発現する発現カセットを、RNAポリメラーゼIIまたはRNAポリメラーゼIIIプロモーターのいずれかで駆動するΔE1領域で、Ad5シャトルベクターにサブクローニングした。得られたプラスミドを、293個の複製許容性細胞にAd5ゲノムプラスミド(PJM17等)でコトランスフェクトして、uPAおよびuPARのsiRNA発現カセットを含む、組換え複製欠損Ad5ウイルス粒子を生じた(図36)。GFP RNAiベクターを構築して、RNAiを用いた標的化の特異性を決定した。GFPを発現する安定なSNB19細胞を対照として用い、GFPに対するRNAiでトランスフェクトした。GFP RNAiでトランスフェクトした細胞はGFP発現を失った(図37)が、RT−PCR反応に見られるように、GAPDH mRNAの発現には変化がなかった(図44)。
【0140】
U6(RNAポリメラーゼIII)またはCMV(RNAポリメラーゼII)プロモーターのいずれかを有する環状プラスミドが細胞レベル免疫応答を誘導するかどうかを決定するために、U6またはCMVプロモーターのいずれかを有する5つのコンストラクトを用いた。SNB19ヒト神経膠腫細胞を、U6またはCMVプロモーターのいずれかを含む環状プラスミドでトランスフェクトし、以下のものを駆動した。空のベクター(EV)と呼ばれる、インサートなし、スクランブルベクター(SV)と呼ばれる、完全なヘアピン構造を形成しないGFP RNAiインサート、uPARのRNAiヘアピンエクスプレッサー(puPAR)、uPAのRNAiヘアピンエクスプレッサー(puPA)、ならびにuPARおよびuPAの両方の二重のRNAiヘアピン発現(pU2)。RT−PCRを行って、OAS1発現レベルを測定した。48時間のトランスフェクション後にトランスフェクト細胞のそれぞれから全RNAを単離し、RT−PCRを行って、全RNA 50ngあたりのOAS1発現のレベルを測定した(RT−PCRは、製造業者の使用説明書(インビトロジェン)どおりに行った)。U6またはCMVプロモーターのいずれかを含む環状プラスミドでトランスフェクトしたSNB19細胞において、OAS1 mRNAのレベルまたはGAPDH mRNAレベルに変化はなかった(図38)。
【0141】
(実施例32)
RNAiの開始のためのプロモーターとしての、RNAポリメラーゼII(CMV)およびRNAポリメラーゼIII(U6)の比較
RNAポリメラーゼIIおよびRNAポリメラーゼIIIの活性および効果を測定するために、pcDNA3の場合のように、スクランブルベクター、uPAR、uPAおよびuPAR−uPAの組み合わせのために、pSilencerプラスミド(アンビオン、テキサス州オースティン)においてRNAiベクターを構築した。pSilencerコンストラクトを、製造業者の使用説明書どおりに、テトラTで終了させた。SNB19細胞を2組でトランスフェクトし、一方の組はRNAポリメラーゼIIプロモーターCMVを含み、第二の組はRNAポリメラーゼIIIプロモーターU6を含んだ(C、SV、puPAR、puPAおよびpU2)。トランスフェクションの48時間後、標準的なプロトコールどおりに細胞からタンパク質を抽出し、12%ポリアクリルアミドSDSゲルにロードした(10μg/レーン)。標準的なプロトコールどおりにウエスタンブロッティングおよびフィブリンザイモグラフィーを行い、uPARおよびuPAに関してプローブし、GAPDHに関するプロービングによってローディング対照を測定した。図39から、RNAポリメラーゼIIIプロモーターコンストラクトと比較して、標的分子のダウンレギュレーションにおいてRNAポリメラーゼIIプロモーターコンストラクトがより効率的であることが明らかであった。
【0142】
(実施例33)
インターフェロン応答遺伝子OAS1の決定
空のベクター(EV)、スクランブルベクター(SV)、uPAR(puPAR)、uPA(puPA)、ならびにuPARおよびuPAの2シストロン性コンストラクト(pU2)のプラスミドコンストラクトを用いて、SNB19ヒト神経膠腫細胞系におけるインターフェロン誘導のレベルを測定した。OAS1遺伝子発現を、インターフェロン誘導の指標として用いた。環状プラスミド(C)、線状発現カセット(L)、およびBGHポリAシグナル配列欠失線状発現カセット(ΔA)を用いた。SNB19細胞を等しい量の上記のプラスミドまたは発現カセット(C、L、ΔA概略図)でトランスフェクトし、標準的なプロトコールを用いて、48時間のトランスフェクションの後に全RNAを単離した。上記の試料に関してRT−PCRを行い、アガロースゲル上でOAS1単位複製配列のレベルを測定した。OAS1増幅に使用したプライマーは、
【0143】
【化41】
および
【0144】
【化42】
であった。上記のプラスミド(EV、SV、puPAR、puPAおよびpU2)由来の発現カセットを増幅するのに使用したプライマーは、フォワードプライマー
【0145】
【化43】
およびリバースプライマー
【0146】
【化44】
であった(図40)。
【0147】
OAS1(2’5’−オリゴアデニル酸シンテターゼ)mRNA誘導のRT−PCRを行って、ポリAシグナル配列の関連性を決定した。環状、線状(発現カセット単独)、およびポリAシグナル配列を欠失した発現カセットを用いた(それぞれC、LおよびΔA)。EVおよびSVの場合、OAS1 mRNAの誘導は検出されなかった(図41)。EVの場合、図に見られるように、ポリA尾部ありまたはなしで、転写産物の全体の長さは1kbよりも大きいとは期待されず、転写産物の予想される構造は、免疫応答を誘導する有意なdsRNA構造を有さなかった(SVも同様)(図44)。対照的に、puPAR、puPAおよびpU2では、予想される二次構造はdsRNA構造を有したが、ポリA尾部ありでは、依然として免疫応答の誘導は検出されなかった(OAS1発現)。ポリAシグナル配列が存在するがトランスフェクトコンストラクトが線状である発現カセット単独の場合、これは免疫応答を誘導した。このことから、環状分子の存在が実行可能なポリA尾部を生成したことが示された。また、線状コンストラクトはポリAシグナル配列の直後で終了したので、生存能のあるポリA尾部の開始は始められなかったか、または不完全だった。ポリAシグナル配列が欠失した線状コンストラクトの場合、免疫応答が開始し、免疫応答の予防および転写RNA分子の安定性にポリA尾部の存在が必要であり得ることが示された(図41〜42)。
【0148】
2シストロン性コンストラクトからの予想されるmRNAは、miRNAに類似性を有さず、uPARおよびuPA配列の両方の、完璧なヘアピンループ構造を有した。24塩基の部分的dsRNAを形成する48塩基の配列を、uPARとuPA配列との間に導入し、両方のsiRNA分子の効率的な転写を可能にした。2シストロン性配列は、BGHポリ−ΔAシグナル配列によってコードされるポリA配列で終わった(図43)。
【0149】
典型的な抗ウイルス応答遺伝子であるOAS1遺伝子のRT−PCRから、トランスフェクトした対照細胞およびEV/SVのように、免疫応答がなかったことが示された。アンチセンスおよびRNAiトランスフェクト細胞におけるuPAおよびuPAR転写産物にもRT−PCRを行った。RT−PCRによって測定すると、アンチセンストランスフェクト細胞ではuPARまたはuPA mRNA転写産物の変化は見られなかったが、RNAiトランスフェクト細胞ではuPARまたはuPAのmRNAレベルが減少し、それぞれのmRNAの破壊が示された(24時間)。RNAiの機構は、標的mRNA分子の破壊を伴う。OAS1発現は、対照群(pGFP、pEVおよびpSV)と同様であり、細胞レベルの免疫応答がないことが示された(図44)。
【0150】
(実施例34)
RNAi発現ベクターのインサイチュー局在化
ヌクレアーゼを含まない条件下で、標準的なプロトコールどおりに、パラフィン包埋切片を脱パラフィンおよび再水和した。これらの切片をプロテイナーゼKで処理して、タンパク質に結合しているDNAを明らかにした。標準的なプロトコールどおりにDNAを変性した。pcDNA3プラスミドを採取し、CMVプロモーターを含む発現カセット(NruI HindIII消化)を熱安定性アルカリホスファターゼ(アマシャム・バイオサイエンス、ニュージャージー州ピスカタウェイ)で標識し、処置切片にハイブリダイズした。製造業者の使用説明書どおりに、ハイブリダイゼーションを行った。mock注射マウスは、アルカリホスファターゼのいかなる活性も示さなかったが、EV、SV、puPAR、puPAまたはpU2の腹腔内注射で処置したマウスはアルカリホスファターゼの活性を示し、CMVプロモーターの存在が示された。アルカリホスファターゼの活性は、ほとんどの場合、血管の周辺に局在し、脈管構造の四方に広がるパターンを示し、CMVを有するプラスミドベクターが血液脳関門を横切ったことを示した(図45)。
【0151】
(実施例35)
uPAR−カテプシンB環状プラスミドのインターフェロン応答遺伝子OAS1の決定
空のベクター(EV)、スクランブルベクター(SV)、uPAR(pU)、カテプシンB(pC)、ならびにuPARおよびカテプシンBの2シストロン性コンストラクト(pCU)のプラスミドコンストラクトを用いて、SNB19ヒト神経膠腫細胞系におけるインターフェロン誘導のレベルを測定した。OAS1遺伝子発現を、インターフェロン誘導の指標として用いた。環状プラスミド(C)、線状発現カセット(L)、およびBGHポリAシグナル配列欠失発現カセット(−A)を用いた。等しい量の上記のプラスミドまたは発現カセットでSNB19細胞をトランスフェクトし、48時間のトランスフェクションの後に、標準的なプロトコールを用いて全RNAを単離した。上記の試料に関してRT PCRを行い、アガロースゲルでOAS1単位複製配列のレベルを測定した。OAS1増幅に使用したプライマーは、
【0152】
【化45】
および
【0153】
【化46】
であった。上記のプラスミド(EV、SV、pU、pCおよびpCU)由来の発現カセットを増幅するのに用いたプライマーは、フォワードプライマー
【0154】
【化47】
およびリバースプライマー
【0155】
【化48】
であった。
【0156】
OAS1(2’5’−オリゴアデニル酸シンテターゼ)mRNA誘導のRT−PCRを行って、ポリAシグナル配列の関連性を決定した。環状、線状(発現カセット単独)、およびポリAシグナル配列を欠失した発現カセットを用いた(それぞれC、L、および−A)。EVおよびSVの場合、OAS1 mRNAの過剰な誘導は検出されなかった。EVの場合、図に見られるように、ポリA尾部ありまたはなしで、転写産物の全体の長さは1kbよりも大きいとは期待されず、転写産物の予想される構造は、免疫応答を誘導する有意なdsRNA構造を有さなかった(SVも同様)。対照的に、pU、pCおよびpCUでは、予想される二次構造はdsRNA構造を有したが、ポリA尾部ありでは、依然として免疫応答の誘導は検出されなかった(OAS1発現)。ポリAシグナル配列が存在するがトランスフェクトコンストラクトが線状である発現カセット単独の場合、これは免疫応答を誘導した。このことから、環状分子の存在が実行可能なポリA尾部を生成したことが示された。また、線状コンストラクトはポリAシグナル配列の直後で終了したので、実行可能なポリA尾部の開始は始められなかったか、または不完全であった。ポリAシグナル配列が欠失した線状コンストラクトの場合、免疫応答が開始し、免疫応答の予防および転写されたRNA分子の安定性にポリA尾部の存在が必要であり得ることが示された。
【0157】
EV、SV、pU、pCおよびpCUで処置した大脳内異種移植腫瘍において、OAS1の誘導を調べた。インターフェロンα(3μg/マウス)で大脳内処置した正常なマウスが含まれ、5時間後に屠殺した。正常な対照組織として脾臓および肝臓を用いて、OAS1発現の存在が通常の条件下で存在する場合の抗体の特異性を実証した。免疫組織化学に加えて、センス
【0158】
【化49】
オリゴを用いてこれらの組織においてインサイチューハイブリダイゼーションを行い、OAS1 mRNAレベルを測定した。大脳内腫瘍を有するマウス脳またはpU、pCおよびpCUで処置したマウスの脳では、OAS1 mRNAおよびタンパク質の極めて少ない発現のみ。注目すべきことに、pCU処置群ではOAS1の誘導がなかった。
【0159】
材料および方法
低分子ヘアピンRNA発現プラスミドの構築
uPA−uPAR:346〜367塩基のuPA
【0160】
【化50】
および77〜98塩基のuPAR
【0161】
【化51】
に特異的な低分子干渉オリゴヌクレオチドを合成し、アニーリングした。アニーリングしたDNAオリゴヌクレオチドの対を、uPAに特異的なものをHindIII部位で、およびuPARに特異的なものをBamHI部位で、pcDNA3ベクターに段階的に挿入することによって、ヒトCMVプロモーター制御下でuPAおよびuPARの両方のshRNAを発現するuPA−uPAR RNAiプラスミドベクターを構築した(sh−uPAuPAR)。また、uPA(sh−uPA)およびuPAR(sh−uPAR)のshRNA発現ベクターを、別々に構築した。完全なヘアピン構造を形成しない、不完全な配列を有するpcDNA3スクランブルベクターを用いて、対照として用いるためのスクランブルベクターを開発した。空のベクター(EV)およびスクランブルベクター(SV)対照を複数の細胞系において試験したが、トランスフェクション後のMTTアッセイで示されるように、細胞に対するいかなる毒性も示さず、ハウスキーピング遺伝子であるGAPDHおよびy−アクチンの発現に対する効果を有さなかった。
【0162】
uPARおよびMMP−9。サイトメガロウイルス(CMV)プロモーターの下流でuPARおよびMMP−9の両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた(概略1)。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域およびBamHI部位を有する長さ21塩基のuPAR配列を、末端に組み込んだ
【0163】
【化52】
。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にEcoRI部位が組み込まれた+360〜+381のMMP−9相補配列
【0164】
【化53】
を、uPARのsiRNA配列を含むベクターのEcoRI部位にライゲートした。これは、最終的に、35bpの分離を有する、uPARおよびMMP−9のsiRNA発現プラスミドを生じた。オリゴは自己相補的で左右対称性を有するので、いずれのインサートの配向も重要ではなかった。SV40ターミネーターを、RNA合成の停止シグナルとした。
【0165】
カテプシンBおよびuPA:サイトメガロウイルス(CMV)プロモーターの下流でカテプシンBおよびuPARの両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた(図17)。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域を有し、BamHI部位が末端に組み込まれた長さ21塩基のuPAR配列
【0166】
【化54】
を用いた。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA−3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にXhoI部位が組み込まれた+732〜+753のカテプシンB相補配列
【0167】
【化55】
を、uPARのsiRNA配列を含むベクターのXhoI部位にライゲートした。これは、最終的に、pCUと呼ばれる、カテプシンBおよびuPARのsiRNA発現プラスミドを生じた。uPAR(pU)およびカテプシンB(pC)の単一のsiRNA発現ベクターもまた構築した。オリゴは自己相補的で左右対称性を有したので、単一または2シストロン中のいずれのインサートの配向も重要ではなかった。BGHポリAターミネーターを、3つ全てのコンストラクトのRNA合成の停止シグナルとした。
【0168】
uPAR、uPAおよびMMP−9:サイトメガロウイルス(CMV)プロモーターの下流でuPAR、uPAおよびMMP−9の両方のsiRNAを発現するベクターの構築のために、pcDNA3を用いた。+77〜+98のuPAR配列を標的配列として用い、便宜上、自己相補的オリゴを用いた。9塩基のループ領域を有し、BamHI部位が末端に組み込まれた長さ21塩基のuPAR配列
【0169】
【化56】
を用いた。標準的なプロトコールを用いて6×SSC中でオリゴを自己アニーリングさせ、pcDNA3ベクタープラスミド中のBamHI部位にライゲートした。同様に、末端にHindIII部位が組み込まれた+346〜+367のuPA相補配列
【0170】
【化57】
をHindIII部位にライゲートし、+360〜+381のMMP−9
【0171】
【化58】
を、uPARおよびuPAのsiRNA配列を含むベクターのEcoRI部位にライゲートした。これは、最終的に、pU2Mと呼ばれる、uPAR、uPAおよびMMP−9のsiRNA発現プラスミドを生じた。uPAR(puPAR)、uPA(puPA)およびMMP−9(pMMP−9)の単一のsiRNA発現ベクターもまた構築した。オリゴは自己相補的で左右対称性を有したので、単一または3シストロン性コンストラクト中のいずれのインサートの配向も要因ではなかった。BGHポリAターミネーターを、4つ全てのコンストラクトのRNA合成の停止シグナルとした。
【0172】
カテプシンBおよびMMP−9:カテプシンB(732〜753)およびMMP−9(360〜381)を標的とする、GCの欠乏した9塩基の領域によって間隔を空けられた自己相補的逆方向反復配列を合成した。カテプシンBのオリゴはXhoI部位で終わり、MMP−9のオリゴはEcoRIで終わり、5分間100℃に加熱することによって自己アニーリングさせ、6×SSC中で室温まで冷却して、それぞれの粘着制限酵素認識部位末端を有する、二本鎖DNA分子の形成を生じた。これらのdsDNA分子をpCDNAプラスミドベクターのXhoIおよびEcoRI部位にライゲートして、CMVプロモーターの下流にカテプシンBおよびMMP−9の逆方向反復を含み、SV40ターミネーターによって終了するプラスミドの構築を生じた。哺乳動物細胞にトランスフェクトした、pCMと呼ばれる得られたプラスミドは、カテプシンBおよびMMP−9の両方を標的とする二重ヘアピンsiRNA分子の生成を生じ、これはdsRNA認識酵素(DICER)によってさらにプロセシングされて、RNAiを誘導する個々のsiRNA分子を生成する(概略1)。
【0173】
細胞培養およびトランスフェクション条件:
前立腺癌細胞:ヒト前立腺癌細胞系LNCaP、DU145およびPC3を、アメリカン・タイプ・カルチャー・コレクション(バージニア州マナッサス)から得た。LNCaP細胞を、2mM L−グルタミン、1.5g/L重炭酸ナトリウム、4.5g/Lグルコース、10mM HEPES、および1.0mMピルビン酸ナトリウムを補充したRPMI培地(インビトロジェン、カリフォルニア州カールスバッド)中で増殖させた。PC3およびDU145細胞を最小必須培地中で増殖させた。両方の培地は、10%ウシ血清アルブミン(ギブコBRL、テキサス州ルイスビル)および5%ペニシリン/ストレプトマイシンを含み、5%CO2の加湿雰囲気中で、370Cのインキュベーター中で維持した。製造業者の使用説明書どおりにリポフェクタミン(商標)2000試薬(ライフ・テクノロジー、メリーランド州ロックビル)を用いて、トランスフェクションを行った。72時間のトランスフェクションの後、細胞増殖アッセイ、免疫ブロット解析、RT−PCR解析、マトリゲル浸潤アッセイ、DNA断片化アッセイ、EMSAアッセイおよびカスパーゼ活性アッセイに細胞を用いた。DAPIおよび二重免疫染色のために、Lab−Tek IIチャンバースライド(ナルジェ・ヌンク・インターナショナル、イリノイ州ネーパービル)中でトランスフェクションを行った。
【0174】
神経膠芽腫細胞:ヒト神経膠芽腫細胞系SNB19を、10%FCS、100−μg/mlストレプトマイシンおよび100−単位/mlペニシリン(インビトロジェン、カリフォルニア州カールスバッド)を補充したDMEM F−12(シグマ・ケミカル・カンパニー、ミズーリ州セントルイス)中、37℃、加湿した5%CO2雰囲気下で維持した。製造業者の使用説明書どおりにリポフェクタミン試薬(インビトロジェン、ニューヨーク州グランドアイランド)を用いて、siRNAを発現するpC pUまたはpCUプラスミドで細胞をトランスフェクトした。トランスフェクション後、血清を含まない培地中で48時間細胞をインキュベートした。
【0175】
フィブリンザイモグラフィー:電気泳動で分離された形態のuPAの酵素活性および分子量を、SDS−PAGEによって、前立腺癌細胞系LNCaP、DU145およびPC3の馴化培地中で測定した。簡潔に述べると、SDS−PAGEゲルは、重合前に、精製されたプラスミノゲンおよびフィブリノーゲンが基質である、アクリルアミドを含有する。重合後、試料中の等しい量のタンパク質を電気泳動し、ゲルを洗浄および染色した。EV/SV puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように行った。
【0176】
ゼラチンザイモグラフィー:EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトした細胞から、馴化培地を回収し、遠心分離して細胞残屑を除去した。ゼラチン(0.5mg/ml)を含有する10%ドデシル硫酸ナトリウム−ポリアクリルアミドゲルを用いて、得られた試料の20μgを、ゼラチナーゼ活性に関してアッセイした。アミドブラック(シグマ・アルドリッチ、ミズーリ州セントルイス)でゲルを染色し、ゲル中の明確なバンドの面積として、ゼラチナーゼ活性を可視化した。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように行った。
【0177】
逆転写−PCR解析:
uPA−uPAR:キアジェンRNeasyキットを用いて細胞RNAを単離し、1gのRNAをDNase処理(10単位/RNA 1g、1時間)し、逆転写反応(RT、20l)の鋳型として用いた。RT反応ミックス(インビトロジェン)は、1l(10pm)のプライマーを含んだ。次いで、得られたcDNAをPCR反応において用いて、ゲル電気泳動によって解析した。以下のプライマーを用いた。
uPA−センス:
【0178】
【化59】
uPA−アンチセンス:
【0179】
【化60】
uPAR−センス:
【0180】
【化61】
uPAR−アンチセンス:
【0181】
【化62】
GAPDH−センス:
【0182】
【化63】
GAPDH−アンチセンス:
【0183】
【化64】
【0184】
【表1】
本明細書中に記載されるように、対照/EV、SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞のRT−PCR解析を行った。
【0185】
PCR条件は以下の通りであった。95℃で5分間、その後95℃で1分間、55℃で1分間、および72℃で1分間を35サイクル。最後の伸長は72℃で5分間であった。アニーリング温度は、種々のコンストラクトの配列に依存して変化し、標準的な手順に従って行った。
【0186】
PC3免疫蛍光検出:種々のshRNAプラスミドでトランスフェクトしたPC3細胞を、4%パラホルムアルデヒドで固定し、抗uPA(1:500、バイオメダ、カリフォルニア州フォスターシティ)および/または抗uPAR(1:500、アメリカン・ダイアグノスティクス・インク、コネチカット州グリニッジ)とともにインキュベートした。洗浄後、蛍光二次抗体(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)を1:500希釈で加えた。再度細胞をPBSで3回洗浄し、DAPIで対比染色した。顕微鏡(オリンパス、ニューヨーク州メルヴィル)に接続された電荷結合素子RTスライダースポットカメラ(ダイアグノスティック・インストゥルメンツ・インク、ミシガン州バローズ・スターリング・ハイツ)を用いて、蛍光像を得、スポットRTソフトウエアバージョン3.5(ダイアグノスティック・インストゥルメンツ・インク、ミシガン州バローズ・スターリング・ハイツ)を備えたコンピュータによって管理した。
【0187】
PC3細胞マトリゲル浸潤アッセイ:トランスフェクション後、細胞を分離し、PBS中で2回洗浄した。5×105個の細胞を、マトリゲル(0.7mg/ml)(コラボラティブ・リサーチ・インク、マサチューセッツ州ボストン)でコートしたトランスウェルインサート(12μM孔)の上の方のチャンバーに播種した。下の方のチャンバーに400lのRPMI培地を充填した。24時間のインキュベーション期間の後、上の方のチャンバーの遊走しなかった細胞を穏やかにこすり落とし、インサートの下面に存在する接着細胞をHema−3で染色し、写真を撮った。
【0188】
製造業者の使用説明書どおりにインサイチューカスパーゼ活性アッセイ:ポリカスパーゼ検出キット(イムノケミストリー・テクノロジース、イリノイ州ブルーミントン)を用いて、カスパーゼ活性を検出した。このアッセイにおいて、細胞透過性で非細胞障害性の、カスパーゼの蛍光色素インヒビター(FLICA)が活性のあるカスパーゼヘテロ二量体の大きいサブユニット上の反応性システイン残基に共有結合し、それによってさらなる酵素活性を阻害する。このキットは、ほとんどのカスパーゼの検出のための一般的なプローブであって、緑色蛍光を発光する、多くのカスパーゼ(カスパーゼ1、−3、−4、−5、−6、−7、−8および−9、FAM−VAD−FMK)のカルボキシフルオレセイン標識フルオロメチルケトンペプチド阻害因子を使用する。緑色蛍光シグナルは、試薬を加えた時点での細胞中の活性のあるカスパーゼの量の直接の尺度である。72時間のトランスフェクションの後、細胞をFAM−VAD−FMK色素(インサイチューマーカー)で染色することによって、カスパーゼ活性を検出した。結合したマーカーを、共焦点顕微鏡で観察される蛍光検出によって局在化した。核染色にDAPIを用いた。
【0189】
DNA標識アッセイ:トランスフェクションの後、細胞を採取し、PBS中で2回洗浄した。細胞ペレットを、0.1mg/mlプロテイナーゼK(インビトロジェン)を含有する溶解バッファー(10mM Tris−HCl、400mM NaCl、1mM EDTAおよび1% TritonX−100)に再懸濁し、次いで37℃で2時間インキュベートした。遠心分離によって溶解物からDNAを取り除き、次いで等しい量のフェノール/クロロホルムを用いて抽出し、−80℃で2時間無水エタノールおよび0.3M酢酸ナトリウム(pH5.2)を加えることによって沈殿させた。DNAをTris−EDTAバッファー(10mM Tris−HCl、pH7.5、1mM EDTA)に再懸濁し、37℃で1時間RNaseAで処理し、次いで、エチジウムブロマイド(0.5g/ml)で染色した1.5%アガロースゲルで分離させた。
【0190】
電気泳動移動度シフトアッセイ(EMSA):トランスフェクションの後、製造業者の使用説明書どおりにタンパク質抽出キット(アンビオン、テキサス州オースティン)を用いて、核タンパク質を抽出した。ビシンコニン酸手順(ピアス・バイオケミカル・カンパニー、イリノイ州ロックフォード)を用いて、希釈した試料で核タンパク質の濃度を測定した。パノミクス(カリフォルニア州レッドウッドシティ)の電気泳動移動度シフトアッセイ(EMSA)キットを用いて、タンパク質抽出物中のStat3とDNAプローブとの間の相互作用を調査した。
【0191】
DNA断片末端標識アッセイ:shRNA処置または対照前立腺腫瘍組織切片(厚さ5M)を脱パラフィンおよび再水和した。次に、標本全体をプロテイナーゼK溶液(10mM Tris中20g/mlプロテイナーゼK、pH8)で覆うことによって、組織切片を透過化し、室温で20分間インキュベートした。次いで、組織切片をTris緩衝生理食塩水(1×TBS、20mM Tris pH7.6、140mM NaCl)で洗浄した。組織切片を、メタノール中に希釈した3%過酸化水素に室温で5分間浸漬することによって、内在性ペルオキシダーゼの不活性化を達成した。次いでスライドガラスをクレノウ平衡バッファー(50mM Tris pH8、50mM NaCl、10mM MgCl2)に30分間入れた。次いで、製造業者の使用説明書(クレノウ−FragEL
DNA断片化検出キット、オンコジーン・リサーチ・プロダクツ、マサチューセッツ州ケンブリッジ)に従って、37℃で90分間、加湿チャンバー中で、組織切片を、クレノウでのDNA断片末端標識に最適な比で標識および非標識デオキシヌクレオチドの混合物を含有する60lの溶液とともにインキュベートした。EDTA(0.5M、pH8)との室温で5分間のインキュベーションによって、酵素反応を停止した。次いでスライドをTBSで洗浄し、ブロッキングバッファーに10分間浸漬し(PBS中4%のBSA)、その後ペルオキシダーゼストレプトアビジンを含有する100lの溶液とともに30分間、加湿チャンバー中室温でインキュベートした。次いで、組織切片をTBS中で洗浄し、3,3’ジアミノベンジジン(DAB、0.7mg/ml)、過酸化水素および尿素(0.6mg/ml)を含有する溶液で覆った。次に、スライドを蒸留水で洗浄し、メチルグリーン(0.3%)で30秒間対比染色し、オリンパス蛍光顕微鏡下で調べた。スポットRTソフトウエアバージョン3.5(ダイアグノスティック・インストゥルメンツ、ミシガン州)を用いて、陽性のDNA断片末端標識染色を、6つの無作為に捕捉した像/試料から点数付けした。
【0192】
同所マウス前立腺処置モデル:無胸腺症雄ヌードマウス(nu/nu、6〜8週齢)を、ハーラン・スプラグ−ドーレイ(インディアナ州インディアナポリス)から得た。動物取り扱いおよび実験手順は、イリノイ医科大学動物実験委員会によって承認された。これまでに記載されているように、同所移植を行った。簡潔に述べると、ケタミン(50mg/kg)およびキシラジン(10mg/kg)での全身麻酔の後、下腹部で下正中切開を行った。30μlのPBS中のPC3細胞の懸濁液(1×106)を前立腺の側葉に注射し、外科用金属クリップで傷を閉じた。この細胞濃度は、7日間の移植の間に一貫した局所的腫瘍増殖を達成するのに必要であった。マウスを、1つの処置群あたり6匹のマウスの、5つの処置群に分割した。移植後7および14日目に、下正中切開を行い、腫瘍に、sh−uPA、sh−uPAR、sh−uPA−uPARまたはEV/SV対照(それぞれ75μg/150μg)を発現するプラスミドコンストラクトを注射した。別の組の実験において、7および14日目に、同所に移植したマウスに、sh−uPAおよびsh−uPARプラスミドを腫瘍内に同時注射した(それぞれ150μg)。最後のshRNAプラスミド注射の14〜15日後にマウスを屠殺し、目視検査によって原発腫瘍増殖および転移の部位を決定し、写真を撮った。次いで原発腫瘍を切除し、測定および計量した。H&E染色のために、ホルマリン中で標本を固定し、パラフィンに包埋した。また、組織の一部を免疫ブロットのために直ちに瞬間凍結した。
【0193】
ウエスタンブロッティング。SNB19細胞をmock、空のベクター、pC、pUまたはpCUでトランスフェクトし、48時間インキュベートした。インキュベートの終わりに、細胞を採取し、冷PBSで2回洗浄し、プロテアーゼ阻害因子を含有するバッファー(150mM NaCl、50mM Tris−Hcl、2mM EDTA、1% NP−40、PH7.4)中で溶解した。上清または細胞由来の等しい量のタンパク質(30μg/レーン)を非還元性条件下、10%アクリルアミドゲルで電気泳動した。SDS−PAGEの後、タンパク質をポリ二フッ化ビニリデン膜(バイオ−ラッド)に移した。非特異的結合をブロックするために、5%無脂肪スキムミルクを含む、0.1% Tween−20を有するPBS[T−PBS]中で膜を2時間インキュベートした。その後、それぞれ、T−PBS+5%無脂肪乳中のカテプシンB、uPAR、ERK、pERK、FAKまたはpFAKに対する抗体とともに、膜を2時間インキュベートした。T−PBS中での洗浄の後、製造業者の使用説明書どおりにペルオキシダーゼ標識抗ウサギ抗体を有するECL(商標)検出キット(アマシャム・ファルマシア・バイオテック、英国アマシャム)を用いて、膜上のタンパク質を可視化した。ローディング対照のために、膜を細長い形に切り、標準的なプロトコールどおりに、β−アクチンに対するモノクローナル抗体でプローブした。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞の免疫ブロット解析もまた、本明細書中に記載されるように行った。uPA−uPAR免疫ブロット解析に、以下の抗体を用いた。抗uPA(バイオメダ、カリフォルニア州フォスターシティ)、抗uPAR(アメリカン・ダイアグノスティクス・インク、コネチカット州グリニッジ)、抗Bax(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)、抗Bcl−XS/L(サンタクルス・バイオテクノロジー、カリフォルニア州サンタクルス)、抗カスパーゼ9(セル・シグナリング・テクノロジー・インク、マサチューセッツ州ベヴァリー)、および抗GAPDH(アブカム、マサチューセッツ州ケンブリッジ)。全体的およびリン酸形態のERK、JNK、p38およびStat3に対する抗体を、サンタクルス・バイオテクノロジー(カリフォルニア州サンタクルス)から得た。EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞のウエスタンブロッティングもまた、本明細書中に記載されるように行った。
【0194】
免疫組織化学的解析。SNB19細胞(1×104)を、ビトロネクチンコートした8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、pMMP−9およびpU2Mでトランスフェクトした。さらに72時間後、細胞を3.7%ホルムアルデヒドで固定し、PBS中の1%ウシ血清アルブミンとともに室温で1時間、ブロッキングのためにインキュベートした。スライドをPBSで洗浄した後、IgG抗uPAR(ウサギ)またはIgG抗MMP−9(マウス)のいずれかを、1:200の濃度で加えた。スライドを4℃で一晩インキュベートし、PBSで3回洗浄して、過剰な一次抗体を除去した。次いで細胞を抗マウスFITC複合体または抗FITC複合体IgG(1:500希釈)とともに、室温で1時間インキュベートした。次いで、スライドを3回洗浄し、ガラスカバースリップで覆い、蛍光顕微鏡写真を得た。合成合併像を得て、対照/EV、SV、puPAR、pMMP−9およびpUMトランスフェクト細胞におけるuPARおよびMMP−9の発現を可視化した。
【0195】
SNB19細胞増殖アッセイ。MTSアッセイによって細胞増殖を評価した。インビトロでのSNB19細胞の増殖に対するこれらのコンストラクトの効果を検出するために、セルタイター(商標)比色アッセイを用いて、生存能のある細胞質量を測定した。5×103個の神経膠芽腫細胞を三連で96または24ウェルプレートに播種し、24時間増殖させた後、培地のみ(mock)、EV、SV、pC、pUおよびpCUベクターで48時間トランスフェクトした。次いで、これらの細胞、血清含有培地に換え、種々の時間間隔を割り当てた。各時点の前に、MTS試薬を加えて、さらに2時間インキュベーションを継続して発色させた。ELISAプレートリーダーを用いて、各ウェルでA490を測定した。短期間対長期間の細胞培養の吸光度読み取りを比較し、これらのコンストラクトの効果を、対応する未処置/処置群の増殖に関して解釈した。siRNAコンストラクトによる増殖のパーセント阻害を、これらのコンストラクトを除いた同じ培地中での同じ細胞の増殖速度に関して計算した。
【0196】
PC3細胞増殖アッセイ:トランスフェクションの72時間後の細胞の生存能を、MTTアッセイを用いて評価した。MTT[3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロマイド](シグマ)を、500g/mlの濃度で各ウェル中の培地に加え、プレートを37℃で4時間インキュベートした。酸−イソプロパノール(0.04N HCl/イソプロパノール)を全てのウェルに直ちに加え、暗青色の結晶が効果的に溶解するように、勢いよく混合した。570nmで吸光度を測定した(ベンチマーク、バイオラッド、カリフォルニア州ハーキュリーズ)。
【0197】
SNB19インビトロ新脈管形成アッセイ。SNB19細胞(2×104)を8ウェルチャンバースライドに播種し、標準的なプロトコールどおりに、mock、EV、pU、pCおよびpCUでトランスフェクトした。24時間のインキュベーション期間の後、培地を除去し、4×104個のヒト皮膚内皮細胞を播種し、72時間共培養させた。3.7%ホルムアルデヒド中での固定の後、第VIII因子抗原に関して内皮細胞を免疫プローブした。第VIII因子抗体は、DAKOコーポレーション(カリフォルニア州カーピンテリア)から購入した。細胞をPBSで洗浄し、FITC複合二次抗体とともに1時間インキュベートし、次いで洗浄し、蛍光顕微鏡下で調べた。SNB19 mock、EV、pU、pCまたはpCUトランスフェクト細胞からの馴化培地存在下で増殖した内皮細胞の同様のスライドをH&Eで染色して、ネットワーク形成を可視化した。新脈管形成の定量化にイメージプロソフトウエアを使用し、以下の方法によって新脈管形成の程度を測定した。分枝点の数および点あたりの分枝の総数を無作為に数え(10視野あたり)、新脈管形成の程度を示す生成物を対照と比較した。8ウェルチャンバースライドに播種してmock/EV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞(2×104)のインビトロ新脈管形成アッセイを、本明細書中に記載されるように行った。8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2MでトランスフェクトしたSNB19細胞(1×104ウェル−1)の新脈管形成アッセイを、本明細書中に記載されるように行った。
【0198】
背側皮下脂肪チャンバーモデル:無胸腺症ヌードマウス(nu/nu、18匹の雄/雌、28〜32g)を、特定の病原体、微生物を含まない環境内で飼育および維持した。背側皮下脂肪チャンバーモデルの移植技術。無菌の小動物外科技術は以下の通りであった。マウスをケタミン(50mg/kg)ザイラジン(10mg/kg)の腹腔内注射によって麻酔した。一旦動物を完全に麻酔すると、10mlの空気を注射することによって、マウスに背側空気嚢を作製した。密閉剤で「O」リング(フィッシャー)の縁の両側に0.45ミクロンのミリポア膜(フィッシャー)を一直線に並べることによって、拡散チャンバー(フィッシャー)を用意した。一旦チャンバーを乾燥させる(2〜3分)、20分間のUV照射によってそれらを滅菌した。20μlのPBSを用いて、膜を濡らした。100〜150μlの無菌PBSに懸濁した2×106個のSNB19細胞(mock、空のベクターまたはpCUトランスフェクト)を、「O」リングの開口部を通してチャンバーに注射した。少量の骨ろうで開口部を密閉した。背側空気嚢の縁に沿って水平に11/2〜2cmの表面切除を行い、空気嚢を開いた。鉗子の助けを借りて、チャンバーを皮膚の下に入れ、注意深く縫合した。10日後に動物をケタミン/キシラジンで麻酔し、生理食塩水(10ml)と、ついで10mlの10%ホルマリン/0.1Mリン酸溶液およびそれに続くPBS中0.001%FITC溶液の心臓内還流によって屠殺した。動物の、移植したチャンバーの周りの皮を注意深くはぎ、移植したチャンバーを皮下空気筋膜から除去した。可視光下で、およびFITC蛍光に関して、チャンバーを覆っている皮下脂肪の写真を撮った。空気嚢筋膜の領域のチャンバー中の血管の数を計数し、それらの長さを測定した。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞もまた、本明細書中に記載されるように、背側皮下脂肪チャンバーモデルに使用した。
【0199】
SNB19細胞遊走アッセイ:SNB19細胞のGFP発現バリアントの、ダルベッコ改変イーグル培地中の2×106個の細胞の懸濁液を、超低接着100mm組織培養プレート上に播種し、スフェロイド凝集物が形成されるまで培養した。直径が約150μmのスフェロイド(約4×104個の細胞/スフェロイド)を選択し、mock、空のベクター、pC、pUおよびpCUでトランスフェクトして、48時間培養した。トランスフェクションの72時間後、単一の神経膠腫スフェロイドを、ビトロネクチンコートされた(50μg/mL)96ウェルマイクロプレートの各ウェルに入れ、200μlの、血清を含まない培地とともに培養した。スフェロイドを37℃で24時間インキュベートし、その後スフェロイドを固定し、Hema−3で染色し、写真を撮った。ステージおよび接眼マイクロメータで較正した顕微鏡を用いて、スフェロイドから単層への細胞の遊走を測定し、細胞遊走の指数とした。神経膠芽腫細胞を三連で96または24ウェルプレートに播種し、24時間増殖させた後、本明細書中に記載されるように、培地のみ(mock)、EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトした。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞の細胞遊走アッセイを、本明細書中に記載されるように行った。SNB19細胞(1×104ウェル−1)のアッセイを8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0200】
SNB19細胞ボイデンチャンバー浸潤アッセイ:改変ボイデンチャンバーアッセイを用いて、カテプシンBおよびuPARのsiRNAを発現するベクター存在下でのSNB19細胞のインビトロ侵襲性を評価した。SNB19細胞を、カテプシンBおよびuPARのsiRNAを単独で、またはともに発現するmock、EV、pU、pCまたはpCUベクターで48時間トランスフェクトした。1×106個の細胞を、0.2%BSAを補充した600μlの、血清を含まない培地中に懸濁し、マトリゲル(0.7mg/ml)でコートされたトランスウェルチャンバー(コーニング・コスター・フィッシャー・サイエンティフック カタログ番号07−200−158、ペンシルバニア州ピッツバーグ)の上の区画に入れた。チャンバーの下の区画を、200μlの、血清を含まない培地で充填し、細胞を24時間遊走させた。インキュベーションの後、細胞を固定し、Hema−3で染色し、写真を撮った。浸潤アッセイの定量化を行った。SNB19細胞(1×104ウェル−1)のアッセイを、8ウェルチャンバーに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0201】
SNB19スフェロイドアッセイ:SNB19神経膠芽腫細胞(3×106)を、DMEM中で調製した0.75%アガーで予めコートした100mm組織培養プレート(コーニング、ニューヨーク州コーニング)に播種し、スフェロイド凝集物が形成されるまで培養した。直径100〜200μmのスフェロイドを選択し、mock、空のベクター、pC、pUおよびpCUで48時間トランスフェクトした。感染の3日後、SNB19スフェロイドを蛍光色素DiIで染色し、DiOで染色したラット脳凝集物と接触させて置いた。これまでに記載されたように、共焦点レーザー走査型顕微鏡を用いてラット脳凝集物の進行性の破壊およびSNB19細胞の浸潤を観察し、写真を撮った。共培養の間の脳凝集物または腫瘍スフェロイドの残りの体積を、これまでに記載されたように測定した。EV/SV、puPAR、pMMP−9およびpUMでトランスフェクトしたSNB19細胞のスフェロイドアッセイを、本明細書中に記載されるように行った。SNB19細胞(1×104ウェル−1)のアッセイを8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0202】
神経膠腫解析のマウス実験。定位フレームを用いて、SNB19 GFP細胞(2×106)をヌードマウスの脳に注射した。8〜10日後、マウスをmock、空のベクターEV/SV、pC、pUおよびpCUで処理した。0.25μl/時間の速度で、アルゼット(ダイレクトコーポレーション、カリフォルニア州キュパージョン)ミニ浸透圧ポンプを用いて、ベクターのインビボ大脳内送達を行い、150μgベクターDNAのmock(PBS)、150μg pC、150μg pU、150μg pCUまたはpuPARベクター(150μg)、pMMP−9ベクター(150μg)およびpUMベクター(150μg)を脳に注射した(マウス1匹あたり100uL)。全ての実験は、ピオリアのイリノイ医科大学での実験を承認するインスティチューショナル・アニマル・コントロール・ユーザーズ・コミッティーによって設定された、機関のガイドラインを遵守して行った。5週間後、または対照マウスが症状を示し始めたときに、ホルムアルデヒドの心臓還流によってマウスを麻酔した。次いで、標準的なプロトコールどおりに、脳を除去し、パラフィン包埋した。切片を調製し、GFP発現に関して観察するか、またはH&Eで染色した。切片は、各例における腫瘍サイズに関して半定量的に、盲検化して検討および点数付けされた。1切片あたりの平均腫瘍面積を用いて腫瘍体積を計算し、対照と処置群との間で比較した。SNB19細胞(1×104ウェル−1)の実験を8ウェルチャンバースライドに播種し、24時間インキュベートし、EV/SV、puPAR、puPA、pMMP−9およびpU2Mでトランスフェクトし、本明細書中に記載されるように行った。
【0203】
統計的解析:グラフパッド・プリズムソフトウエア(カリフォルニア州サンディエゴ)を用い、異なる値の間の有意性の解析のためのANOVAを用いて、統計的比較を行った。値を、少なくとも3つの別々の実験からの平均値SDとして表し、P値が0.05未満で、差を有意であると見なした。
【0204】
大脳内腫瘍増殖阻害。大脳内腫瘍モデルのために、2×106個のSNB19細胞をヌードマウスに大脳内注射した。腫瘍を10日間増殖させた。このとき、動物を7つの群に無作為化し、EV/SV、puPAR、puPA、pMMP−9およびpU2M(0.25μl時間−1の速度でアルゼットミニポンプを用いて、150μgの各コンストラクトを脳に注射した(各群あたり6匹のマウス)。腫瘍接種の5週間後、PBS中3.5%ホルムアルデヒドの心臓還流によって、各群からの6匹のマウスを屠殺した。それらの脳を除去し、24時間4%パラホルムアルデヒドに入れ、パラフィン包埋および切片化した。蛍光顕微鏡下でGFP蛍光に関して切片をスクリーニングし、腫瘍増殖を調べた。切片を盲検化して検討し、腫瘍サイズに関して半定量的に点数付けした。各切片中の平均腫瘍面積を用いて腫瘍体積を計算し、対照と処置群との間で比較した。
【0205】
マトリゲル浸潤アッセイ。空のベクター(EV)、またはカテプシンBおよびMMP−9のsiRNAを発現するベクター(pCM)のいずれかでのトランスフェクションの後、ボイデンチャンバー浸潤アッセイを用いて、トランスフェクトSNB19細胞の侵襲性をインビトロで試験した。簡潔に述べると、8μmの孔を有するトランスウェルインサートをマトリゲル(0.7mg/ml)(コラボラティブ・リサーチ,インク、マサチューセッツ州ボストン)でコートした。SNB19細胞をトリプシン化し、500μlの細胞懸濁液(1×106個の細胞/ml)を三連でウェルに加えた。37℃で24時間のインキュベーションの後、ウェル下部へフィルターを通過した細胞を定量化し、ウェル上部および下部の細胞の合計のパーセンテージとして表した。膜の下側の細胞を固定し、Hema−3で染色し、写真を撮った。記載される他のコンストラクトのアッセイもまた、本明細書中に記載されている手順に従って行った。
【0206】
核酸の送達:例えば親油性薬剤、アデノ、アデノ関連またはレンチを含むウイルスまたは修飾されたウイルス等の任意の型の方法を用いて、核酸の送達を達成する。二本鎖RNA、一本鎖RNA、または一方の鎖がDNAで他方がRNAであるか、またはリボースもしくはデオキシリボースバックボーンを同じ鎖上に有するDNA−RNAハイブリッドとしての、平滑または粘着末端を有する、環状または線状であるむき出しのプラスミドコンストラクトもまた、使用する。タンパク質もしくは炭化水素またはそれらの組み合わせでコートされた、またはホルモンもしくはホルモン誘導体と関連して使用される、DNA、RNAまたはDNA−RNAハイブリッド。化学的に修飾されたDNAまたはRNAまたはDNA−RNAハイブリッドもまた、uPAR、uPA、MMP−9およびカテプシンBを標的とするRNAiを誘導する治療分子として、任意の組み合わせで使用され得る。意図される使用は、真核生物または細胞系、好ましくはヒトおよびヒト細胞系である。
【0207】
siRNAまたはshRNAの送達のための当業者に公知の方法および治療組成物は、本開示の範囲内である。例えば、本明細書中に開示されるsiRNA、shRNAおよび他の核酸は、ウイルスベクター送達、リポフェクション、電気化学的および生化学的方法等の技術を通じて、哺乳動物細胞に送達され得る。ウイルスベースの送達は、目的の核酸を含む組換えアデノウイルス、レンチウイルス、レトロウイルスの、または任意の適したベクターを作製すること、および当業者に公知の技術を用いてウイルスベクターを送達することを含む。リポソームベースの送達系としては、リポフェクションおよびカルジオリピンベースの組成物が挙げられる。むき出しの核酸の直接の、および化学的または生化学的佐剤と組み合わせた送達は、本開示の範囲内である。例えばuPA、uPAR、MMP−9、およびカテプシンB等の標的配列に対する、例えばsiRNAを含む環状プラスミドは、直接注射もしくは腫瘍内送達されるか、または腹腔内注射され得る。同様に、合成または化学的に調製された核酸はまた、腫瘍内または腹腔内で送達され得る。また、siRNA、またはsiRNAを発現する核酸の選択的または特異的送達は、ペプチド核酸(PNA)もしくは抗体または任意の適した標的化因子等の別の作用物質との適切なカップリングを通じても達成され得る。上述の技術および方法は、細胞間、細胞内、インビトロ細胞培養物、インビボ、器官内部で、血液脳関門を横切っての、前立腺癌、神経膠腫、乳癌、および結腸癌への核酸配列の送達に適しており、適用可能である。
【0208】
配列:種々のsiRNAコンストラクトの配列(部分的配列)を本明細書中に開示する。下線を引いた部分は、自己相補的逆方向反復を示す。太字は介在ループ配列を示す。
【0209】
UPAR−uPA
【0210】
【化65】
UPAR−MMP−9
【0211】
【化66】
UPAR CB
【0212】
【化67】
MMP9−CB
【0213】
【化68】
UPAR、uPAおよびカテプシンB
【0214】
【化69】
引用した文書
これらの文書は、本開示における材料および方法に関連する範囲にのみ含められる。
【0215】
【化70】
【0216】
【化71】
【0217】
【化72】
【0218】
【化73】
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

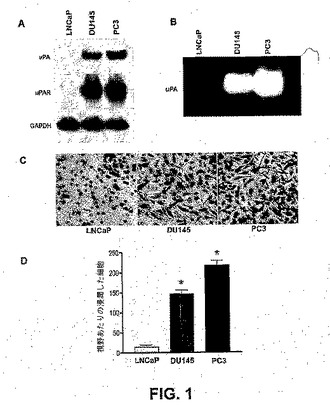
【図2】

【図3】
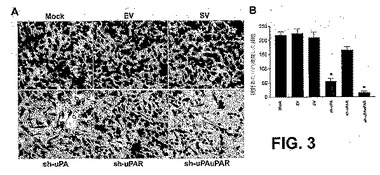
【図4】
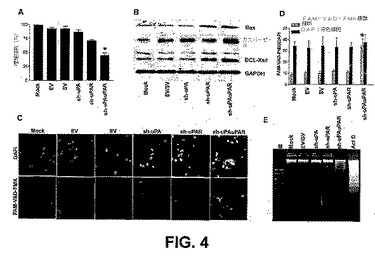
【図5】
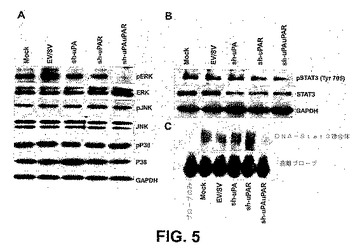
【図6】
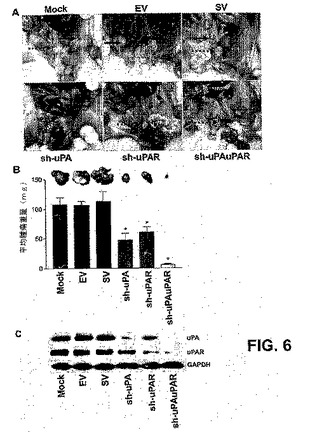
【図7】
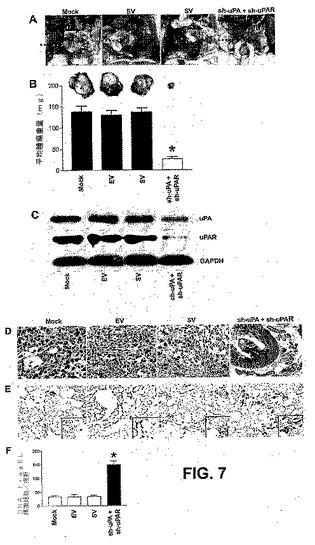
【図8】
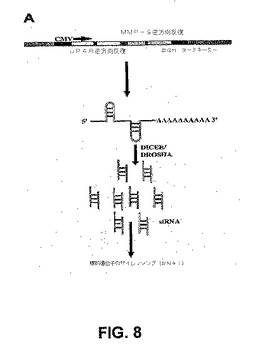
【図9】
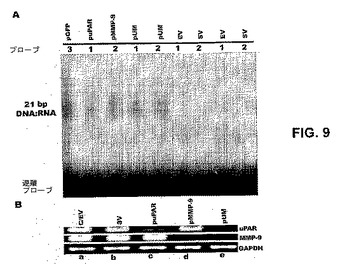
【図10】
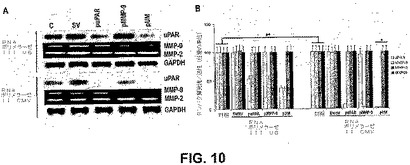
【図11】
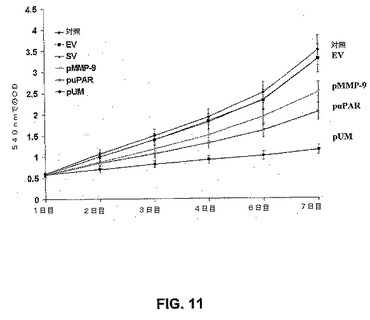
【図12】
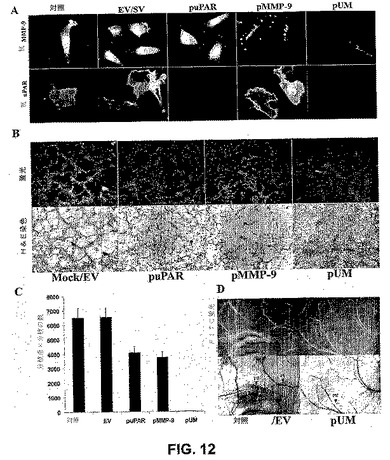
【図13】
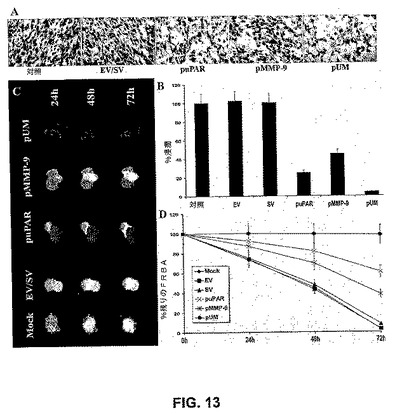
【図14】
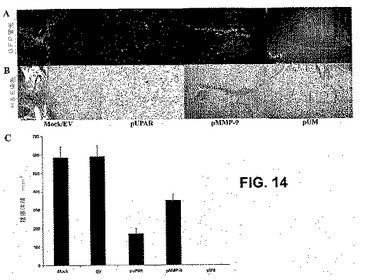
【図15】
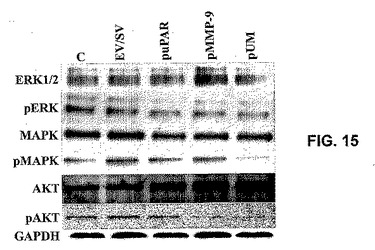
【図16】
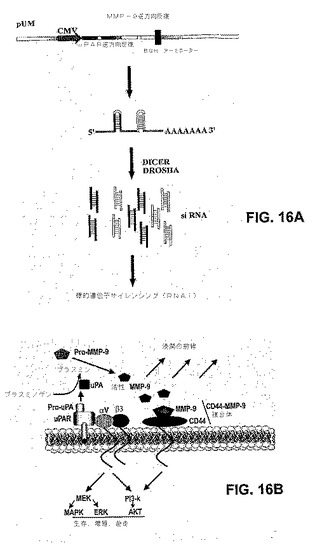
【図17】
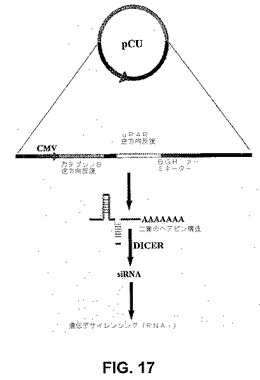
【図18】
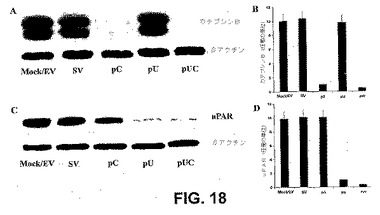
【図19】
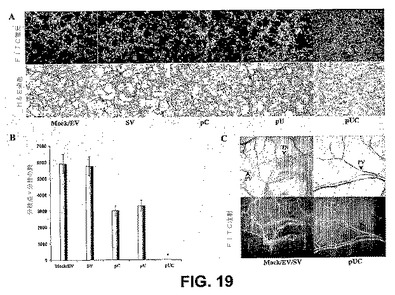
【図20】
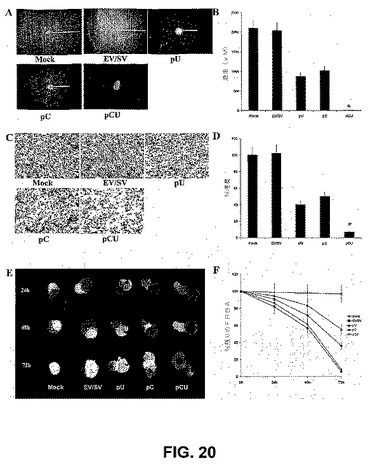
【図21】
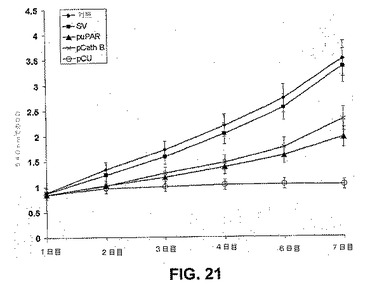
【図22】
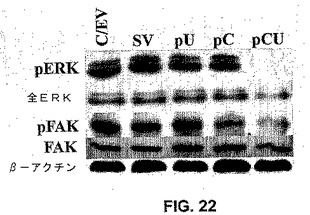
【図23】
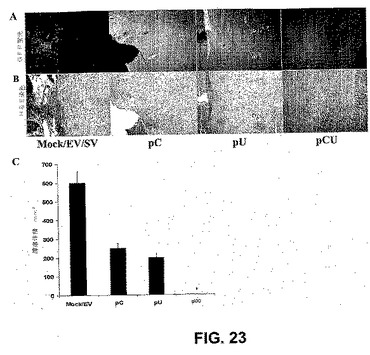
【図24】
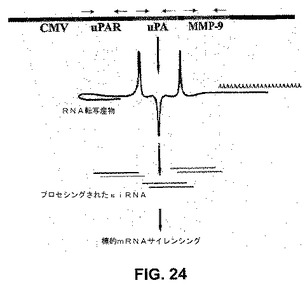
【図25】
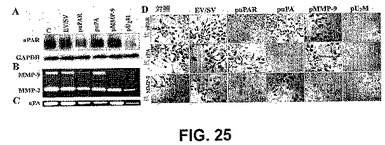
【図26】
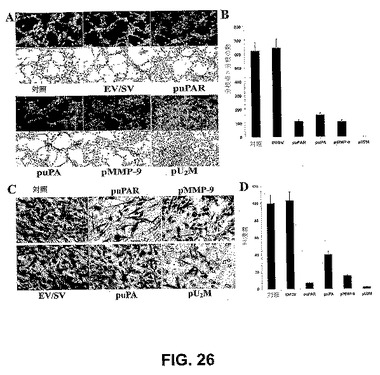
【図27】
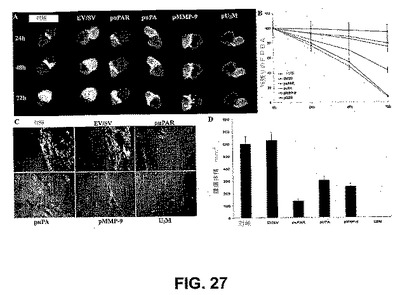
【図28】

【図29】
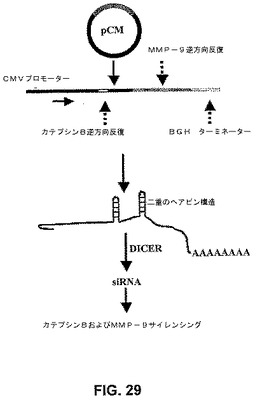
【図30】
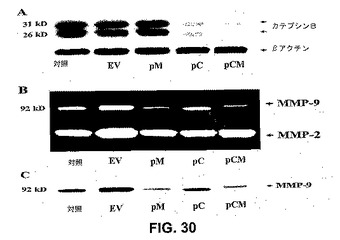
【図31】
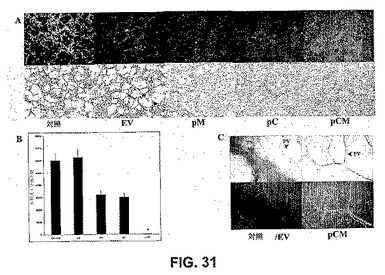
【図32】
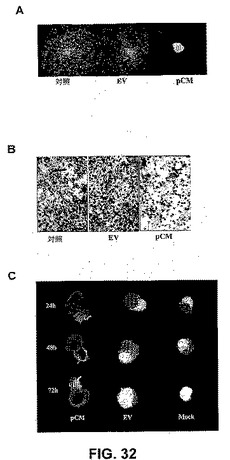
【図33】
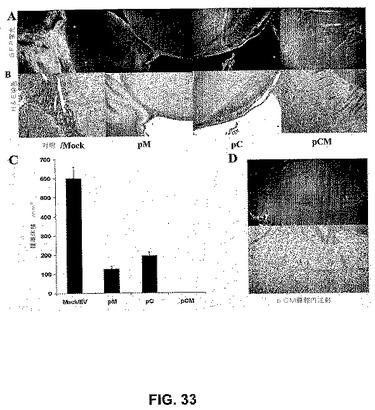
【図34】
【図35】
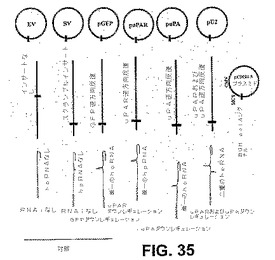
【図36】
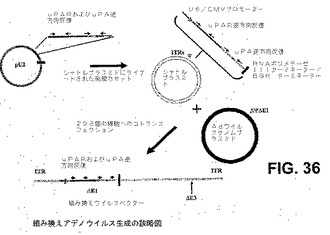
【図37】
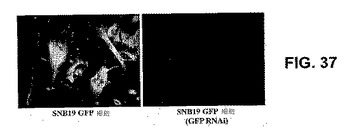
【図38】
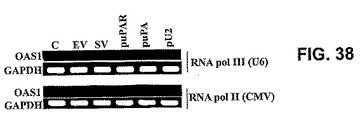
【図39】
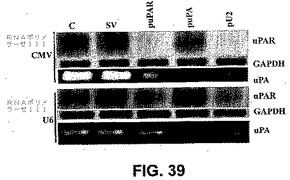
【図40】
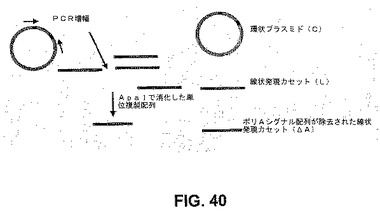
【図41】
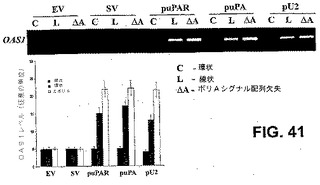
【図42】
【図43】
【図44】
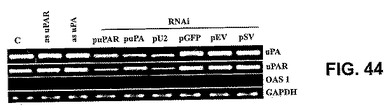
【図45】
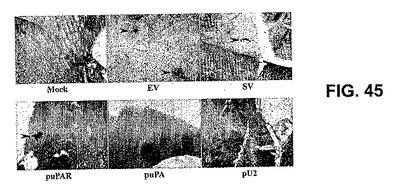
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【公開番号】特開2013−27405(P2013−27405A)
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−237627(P2012−237627)
【出願日】平成24年10月29日(2012.10.29)
【分割の表示】特願2007−543245(P2007−543245)の分割
【原出願日】平成17年11月17日(2005.11.17)
【出願人】(503060525)ザ ボード オブ トラスティーズ オブ ザ ユニバーシティ オブ イリノイ (25)
【Fターム(参考)】
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願番号】特願2012−237627(P2012−237627)
【出願日】平成24年10月29日(2012.10.29)
【分割の表示】特願2007−543245(P2007−543245)の分割
【原出願日】平成17年11月17日(2005.11.17)
【出願人】(503060525)ザ ボード オブ トラスティーズ オブ ザ ユニバーシティ オブ イリノイ (25)
【Fターム(参考)】
[ Back to top ]