
腫瘍壊死因子α阻害ペプチドおよびその使用方法
本発明は、腫瘍壊死因子α(TNFアルファもしくはTNF−α)阻害ペプチドとその調製方法に係わる。本発明は、本発明のTNF−α阻害ペプチドを含有する医薬組成物、およびTNF−α媒介炎症性疾患の治療への該組成物の使用にも係わる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は生物活性ペプチドとその調製方法に係わる。本発明はさらに、腫瘍壊死因子α(TNF−αもしくはTNFアルファ)阻害ペプチドとその調製方法に係わる。本発明は、上記のようなペプチド分子を含有する医薬組成物、並びに慢性関節リウマチ、乾癬性関節炎、クローン病および敗血症などの腫瘍壊死因子α(TNF−αもしくはTNFアルファ)媒介炎症性疾患の治療への該組成物の使用にも係わる。
【背景技術】
【0002】
サイトカインは、活性化された免疫細胞、すなわちB細胞、T細胞並びに単球およびマクロファージによって産生される一群のシグナルタンパク質である。サイトカインにはインターロイキンファミリー(IL−1〜23)、インターフェロン(α、βおよびγ)、並びにTNF−αおよびβが含まれる(非特許文献1および2)。IL−1およびTNF−αは敗血症、RAなどの自己免疫性状態、皮膚疾患、炎症性腸疾患といった疾病における炎症応答の主要メディエーターとして重要な役割を果たすことが示唆されている(非特許文献3〜7)。またサイトカイン調節研究により、TNF−αが炎症性病態を惹起する最も重要なサイトカインであることも示唆されている(非特許文献8)。
【0003】
TNF−αは元来、マウスの腫瘍に出血性壊死を引き起こす分子として発見された(非特許文献9)。その後、「カケクチン」として知られ、悪液質状態の誘因と考えられた血清タンパク質についての一連の研究から、カケクチンはTNF−αと同じものであることが最終的に判明した(非特許文献10)。TNF−αは今や、広範な活性を有し、様々な臨床状態に関与する炎症メディエーターとして確立されている。
【0004】
TNF−αは、数種の細胞、特に活性化されたマクロファージによって産生される分子量17kDaのタンパク質である。TNF−αは始め、安定な三量体に構成された膜貫通タンパク質として合成される。次いで、メタロプロテアーゼTNF−α変換酵素(TACE)により切断されてホモ三量体の可溶性TNF(sTNF)となり、遍在発現したそのコグネイト受容体(TNF RI、p55、およびTNF RII、p75)に結合する。細胞特異的なエフェクターを伴ったTNF受容体の遍在発現は、TNF−αに媒介される細胞応答がきわめて様々であることを説明するものであり、そのような応答の幾つかは有害で、命に関わる。単核細胞から離脱したTNF受容体は掃討作用によって、また天然の阻害剤として作用することによってTNF−α濃度を低下させる。
【0005】
TNF−αはきわめて様々な細胞応答を誘発し、その多くは有害な結果をもたらす。例えば、TNF−αは、食欲不振に起因する不十分な摂食に伴う脂肪の減少および全身のタンパク質の枯渇から帰結する状態である悪液質を誘発する。悪液質は癌患者に普通に認められ、後天性免疫不全症候群(AIDS)の患者でも観察されている。加えて、動物にTNF−αを大量に注射すると、敗血症性ショック症状の殆どが発現する。多発性硬化症および慢性関節リウマチなどの自己免疫疾患、乾癬、乾癬性関節炎、過敏症、免疫複合体病および移植片対宿主病、並びに移植拒絶反応においてTNF−αが役割を有することも示されている。TNF−αはマラリアおよび肺線維症とすら関連付けられている。従って、疾病状態におけるTNF−α産生の阻止を目差すことには多大の関心が寄せられており、その達成は治療上有益である。
【0006】
TNF関連疾患の治療
TNF−αの副作用を打ち消す方法としては、抗TNF抗体および可溶性RNF−Rの使用が注目されている。動物モデルでは、TNF−α関連炎症性疾患の治療にTNF−α特異的な抗体が有効であった(非特許文献11〜13)。抗TNF抗体のキメラ形態がヒトでの臨床試験用に構築された(非特許文献14〜16)。さらに、可溶性TNF受容体融合タンパク質がTNF拮抗薬としてヒトの患者に導入された(非特許文献17〜19)。
【0007】
特許文献1は、TNF受容体スーパーファミリーに属する分子の結合ループから設計される環状ペプチドおよびペプチド類似体に係わり、それらのペプチドおよびペプチド類似体はTNF−αとTNF受容体との相互結合作用に干渉してin vitro、並びにin vivoで阻害活性を示し、in vivoでのTNFの望ましくない生物活性に拮抗する。この文献に記載された発明では環化ペプチドが好ましく、なぜならタンパク質−タンパク質相互作用ではループおよびターンが機能上重要な役割を果たすからである。具体的な実施形態ではTNF−R p55の三つの結合ループから環状ペプチドが設計され、それらのペプチドはTNF−αと結合して、TNF−αがその細胞受容体に結合するのを阻害する。最も好ましい実施形態において、ペプチドは少なくとも7個のアミノ酸を有し、より小形の直鎖状ペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0008】
特許文献2は、TNF−αとTNF受容体との結合およびTNF−α機能を有効量の阻害ペプチドの投与によって阻害する方法に係わる。この特許は、TNF受容体と結合し、それによってTNF−αの、細胞上のTNF−α受容体に結合してこれを活性化する能力を妨げるペプチドに係わる。特許された発明は特に、7個、および12個のアミノ酸残基を有するペプチドをTNF−αとTNF受容体との結合およびTNF機能の阻害に使用する方法に係わる。特許文献2に記載された発明は、TNF受容体に結合し得る小形ペプチドをスクリーニングすることを目的とする。同定された分子で最も小形のものは7アミノ酸長の配列を有する。より小形のペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0009】
特許文献3は尿由来のTNF阻害ペプチドに係わる。この特許は、TNF−αに対して活性である精製形態のTNF阻害剤にも係わる。また、TNFに対して活性を示す医薬製剤として有用である精製形態のTNF阻害剤にも係わる。この特許はさらに、その精製形態において得られた30kDaタンパク質および40kDaタンパク質に係わる。開示されたこれらのタンパク質のアミノ酸配列は15アミノ酸以上の長さを有し、より小形のペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0010】
特許文献4は、グリシン、アラニンおよびセリンから成る群から選択された少なくとも1種のアミノ酸またはその生理学的に許容される塩を、腫瘍壊死因子(TNF)濃度が生理的恒常性および局所的炎症を媒介する濃度を上回る患者のTNF濃度を低下させる医薬または栄養製剤の調製に使用する方法に係わる。この特許は、TNF濃度の低下がマクロファージ型細胞によるTNF産生、マクロファージ型細胞によるTNF放出、またはTNF受容体によるTNFの結合を阻害もしくは低減することにより可能であることを開示している。この特許は、TNF濃度を低下させる医薬または栄養製剤の調製にグリシン、アラニンおよびセリンのみを使用する方法に係わるものである。この特許は複数のアミノ酸の組み合わせやそのような組み合わせを含むペプチドを使用する方法は一切開示していない。
【0011】
特許文献5は、ヒトTNF−αの複数の分子表面から成る群から選択された少なくとも一つのヒトTNF−α分子表面に実質的に類似する分子表面を有するTNF−α拮抗化合物に係わる。この文献に記載された発明の化合物は、両端に結合した連結部分と、スペーサー部分とを有する。発明化合物によって、25個のアミノ酸並びに連結部分およびスペーサー部分を有するTNF−α阻害ペプチドが開示されている。特許文献5に記載されたTNF−α拮抗化合物はTNF p55受容体および/またはTNF p75受容体に結合し、TNF−αに媒介される細胞毒性を阻害する。特許文献5はスペーサー部分を有しない、より小形のペプチドを示唆してはいない。
【0012】
当該技術分野で現在使用可能な治療薬は、可溶性TNF受容体または抗TNFモノクローナル抗体の使用によってTNF−αを中和するべく設計されている(非特許文献20および21)。可溶性TNF受容体や抗TNFモノクローナル抗体は循環するTNF−αと結合し、それによって、TNF−αが細胞表面のTNF−Rへアクセスし、その後炎症経路を活性化するのを制限する。TNF−α濃度の抑制に使用可能な治療薬は、(1)インフリキシマブ(レミケード): マウス−ヒトキメラ型抗ヒトTNF−αモノクローナル抗体、(2)ヒュミラ: 完全ヒト型抗TNF−αモノクローナル抗体、(3)エタネルセプト: ヒトIgGのFc領域と融合した可溶性p75 sTNF−RIIの二量体融合タンパク質である。これらの分子は様々な自己免疫疾患で効力を示すものの、幾つかの制約、すなわち低いバイオアベイラビリティーおよび安定性、重篤な免疫反応の誘発、並びに高いコストを伴う。TNF−α活性の阻害には別の手段を講じることが妥当であろう。
【0013】
抗TNF抗体やTNF受容体の配列を利用して生物活性ペプチドフラグメントが合成されている(非特許文献22〜24)が、それらは大形で、難溶性である。このことは、恐らくは小形分子の形態を取る改良されたTNF−α阻害化合物の必要性を強調している。
【0014】
1997年に為された上記のような合成の一研究においてTakasaki等は、TNF−RIの三つの結合ループから設計されるペプチド類似体について検討した(非特許文献25)。結合ループを構成するアミノ酸配列に基づいて複数のペプチドが作製された。それらのペプチド配列の中で、TNF−RIのドメイン3のループ1(残基107〜114)に基づく環状WP9(配列WSENL)がTNF−α阻害活性に関して最も有望であることが判明した。Takasaki等はWP9の配列WSENLを鋳型として用いて、環状ペプチド模倣体WP9Q、WP9ELY、WP9YおよびWP9QYを設計した。ペプチド模倣体WP9QYは治療上の有効性を示し、マウスにおいて実験的自己免疫性脳脊髄炎(EAE)および慢性関節リウマチ(RA)の重篤度を低下させた。しかしこの物質は、生理緩衝液に比較的溶解しにくいため、潜在的治療薬としてのヒトへの使用は制限される(非特許文献25)。Takasaki等の1997年の研究では、ペプチドの環化および芳香族化によって安定性およびバイオアベイラビリティーは向上したが、溶解性の向上または活性の向上への効果はほとんど、または全く認められなかった。Takasakiは、WP9QYは今日までに開発された最小のペプチド模倣体であり、次世代の非ペプチド性阻害剤のためのリード化合物として用いられるかもしれないと述べている。このように、この文献は、当時公知の最小ペプチドに基づき非ペプチド性TNF阻害剤の開発を試みることを当業者に示唆している。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】米国特許第6,265,535号
【特許文献2】米国特許第6,344,443号
【特許文献3】米国特許第6,143,866号
【特許文献4】米国特許第6,048,543号
【特許文献5】米国特許第6,107,273号
【非特許文献】
【0016】
【非特許文献1】Janeway, C.A. et al., Immunobiology, 4th Ed., New York, Garland, 1999
【非特許文献2】Roitt, I. et al., Immunology, 5th ed., London, Mosby, 2002
【非特許文献3】Locksley, R.M. et al., Cell 104(4): 487−501, 2001
【非特許文献4】Feldmann, M. et al., Ann. Rev. Immunol. 14: 397−440, 1996
【非特許文献5】Buchan, G. et al., Clin. Exp. Immuol. 73: 449−455, 1988
【非特許文献6】Canto, E. et al., Clin. Immunol. 119(2): 156−165, 2006
【非特許文献7】Fantuzzi, F. et al., Expert Opi. Ther. Targets 12(9): 1085−1096, 2008
【非特許文献8】Feldmann et al., Ann. Rheum. Dis. 58: (Suppl. 1)127−131, 1999
【非特許文献9】Carswell et al., Proc. Natl. Acad. Sci. U.S.A. 72: 3666, 1975
【非特許文献10】Beutler et al., Annu. Rev. Immunol. 7: 625, 1989
【非特許文献11】Williams et al., Proc. Natl. Acad. Sci. U.S.A. 89: 9784−9788, 1992
【非特許文献12】Baker et al., Eur. J. Immunol. 24: 2040, 1994
【非特許文献13】Suitters et al., J. Exp. Med. 179: 849, 1994
【非特許文献14】Lorenz et al., J. Immunol. 156: 1646, 1996
【非特許文献15】Walker et al., J. Infect. Dis. 174: 63, 1996
【非特許文献16】Tak et al., Arthritis Rheumat. 39: 1077, 1996
【非特許文献17】Peppel et al., J. Exp. Med. 174: 1483, 1991
【非特許文献18】Williams et al., Immunol. 84: 433, 1995
【非特許文献19】Baumgartner et al., Arthritis Rheumat. 39 (Suppl.): S74, 1996
【非特許文献20】Piguet et al., Immunology 77: 510−514, 1992
【非特許文献21】Elliot et al., Arthritis Rheum. 36: 1681−1690, 1993
【非特許文献22】Weisong, Q. et al., Mol. Immunol. 43: 660−666, 2006
【非特許文献23】Zhang, J. et al., Biochem. Biophy. Res. Comm. 7: 1181−1187, 2003
【非特許文献24】Aoki et al., J. Clin. Invest. 116(6): 1525−1534, 2006
【非特許文献25】Takasaki et al., Nat. Biotechnol. Nov. 15(12): 1266−1270, 1997
【発明の概要】
【発明が解決しようとする課題】
【0017】
上記背景技術の考察から、生理緩衝液に溶解しやすく、安定性およびバイオアベイラビリティーが高く、僅かな副作用しか有せず、かつ低コストであるペプチドの設計を目差して改良された次世代TNF−α阻害剤を開発する必要性は明らかである。
【課題を解決するための手段】
【0018】
本発明によれば、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体が提供され、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0019】
本発明の別の態様によれば、式:
X1−X2−X3
を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体が提供され、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0020】
本発明は、本発明の生物活性ペプチドをTNF−α関連疾病状態の治療に使用する方法にも係わる。本発明はまた、本発明の生物活性ペプチドを含有する医薬組成物にも係わる。
【図面の簡単な説明】
【0021】
【図1】図1は、ペプチドの抗TNF−α活性をL929バイオアッセイを用いて示したグラフである。X軸上の数字1〜10は配列番号1〜10のペプチドを意味する。Y軸はTNF−α媒介細胞毒性の阻害パーセンテージを、各々二重に行なわれた3回の独立した実験の平均値±SEとして表わす。X軸上の「+ve」は陽性対照、すなわち公知のTNF−α阻害ペプチドである環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)を意味する。
【図2】図2は、配列番号2および6の直鎖状ペプチドと、配列番号9および10の環状ペプチドと、陽性対照すなわち環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)ペプチドとをTNF−αに誘発される細胞毒性の阻害について比較したグラフである。結果はTNF−α媒介細胞毒性の阻害パーセンテージの、各々三重に行なわれた3回の独立した実験から得られた平均値±SEとして示されている。
【図3】図3は、配列番号6のペプチドおよびエタネルセプト(Et)によるTNF−α媒介細胞毒性の阻害を比較したグラフである。結果はTNF−α媒介細胞毒性の阻害パーセンテージの、各々三重に行なわれた2回の独立した実験から得られた平均値±SEとして示されている。
【図4】図4は、ペプチドとTNF−αとの結合のフローサイトメトリーによる定量を示すグラフである。配列番号2および6のペプチドの存在下でのTNF−αと細胞受容体との結合の蛍光活性化セルソーター(FACS)分析結果を図4aに示す。Y軸はTNF−RI発現に関して陽性である細胞のパーセンテージを、3回の独立した実験から得られた平均値±SEとして表わす。棒グラフ(図4a)のX軸上、およびフローサイトメトリーヒストグラム(図4b)中の数字1〜5の意味は次のとおりである。 1=二次抗体としての抗マウスIgG FITCで染色したU937細胞。 2=マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 3=組み換えTNF−αで処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 4=組み換えTNF−αと配列番号2との複合体で処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 5=組み換えTNF−αと配列番号6のペプチドとの複合体で処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。
【図5】図5は、配列番号6のペプチドとU937細胞上に発現したTNF−R1との結合を示すグラフである。配列番号6のペプチドと、U937細胞上に発現したTNF−RIとの結合の蛍光活性化セルソーター(FACS)分析結果を図5に示す。Y軸はTNF−RI発現に関して陽性である細胞のパーセンテージを、2回の独立した実験から得られた平均値±SEとして表わす。棒グラフのX軸上、および重畳表示されたフローサイトメトリーヒストグラム(図5)中の数字は以下の試料を意味する。 1: 未処理のU937細胞。 2: U937細胞+TNF−α。 3: U937細胞+配列番号6のペプチド。
【図6】図6は、媒体(CFA)マウス、対照(PBS)マウス、およびコラーゲン免疫マウスの血清からの抗コラーゲンIgG濃度の推定を示すグラフである。
【図7】図7は、コラーゲン誘発関節炎のマウスモデルにおいて、異なる用量および投与スケジュールで用いられる配列番号6のペプチドでの処理の前(Pre Tx)および後(Post Tx)に得られた右足根骨部位での足の太さ(paw thickness)の平均値を示すグラフである。動物群はそれぞれ4匹または5匹の動物から成っていた。Y軸上の値は、群毎に得られる足の太さの平均値±SEである。X軸上のアルファベットは動物群を意味する。 A: 1.25mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 B: 2.5mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 C: 5mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 D: 7.5mg/kgの配列番号6のペプチドを、毎週1回の投与を4週間行なうスケジュールで用いて処理した関節炎マウス群。 E: 7.5mg/kgの配列番号6のペプチドを、1週目に1回投与し、3週間後に2回目の投与を行なうスケジュールで用いて処理した関節炎マウス群。 F: 対照としてのPBSで処理した関節炎マウス群。 G: 対照動物群、すなわち健康な雄のC57BL/6マウスの群。 処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図8a】図8aは、配列番号6のペプチド、配列番号2のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた左足根骨および右足根骨部位での足の太さの平均値±SEを示すグラフである。PBS処理動物は未処理動物と見なされ、また図中「対照」とは健康な雄のC57BL/6マウスのことである。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図8b】図8bは、配列番号6のペプチド、配列番号2のペプチド、およびエタネルセプト(Et)で処理した動物における処理後の抗コラーゲンIgG1/IgG2a比を比較したグラフである。PBS処理動物は未処理動物と見なされ、また図中「対照」とは健康な雄のC57BL/6マウスのことである。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図9a】図9aは、配列番号6のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた右足根骨および右関節部位での足の太さの平均値を示すグラフである。PBS処理動物は未処理動物と見なされる。動物群はそれぞれ3匹の動物から成っていた。
【図9b】図9bは、アジュバント誘発関節炎のラットモデルにおいて、配列番号6のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた臨床スコアを比較したグラフである。PBS処理動物は未処理動物と見なされる。動物群はそれぞれ3匹の動物から成っていた。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図10】図10は、配列番号6のペプチド、エタネルセプト(Et)、および陰性対照としてのPBSでの処理の前後に関節炎ラットにおいて撮影した足および関節の腫脹の代表的な写真を示す説明図である。
【発明を実施するための形態】
【0022】
本発明は、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTryを含む群から選択され得る。
【0023】
本発明は、式:
X1−X2−X3
を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0024】
本発明はまた、TNF−α阻害剤として作用する生物活性ペプチドに係わる。本発明による生物活性ペプチドは、例えば下記のような機構により作用し得るが、これらに限定はされない。
a) 本発明のペプチドはTNF−αと結合して複合体を形成し、それによってTNF−αと受容体TNF−R1との結合を防止することができる。
b) 本発明のペプチドは直接受容体TNF−R1に結合することができ、TNF−αが受容体TNF−R1に結合するのを防止することができる。
【0025】
本発明は、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を含有する医薬組成物にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0026】
本発明はさらに、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0027】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0028】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0029】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を含有する医薬組成物に係わる。
【0030】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法に係わる。
【0031】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTrp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0032】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTrp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0033】
本発明は、式:
X1−X2−X3
〔式中X1、X2およびX3はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0034】
本発明は、式:
X1−X2−X3
〔式中X1、X2およびX3はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0035】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0036】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0037】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物に係わる。
【0038】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法に係わる。
【0039】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0040】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0041】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0042】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0043】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0044】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0045】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0046】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0047】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0048】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0049】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0050】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0051】
本明細書中に用いた「ペプチド」という語は、ペプチド結合で連結された複数の天然アミノ酸サブユニットから成る重合体を意味する。アミノ酸という語は、天然ペプチドに類する部分を有するが、非天然部分も有する成分を意味する場合も有る。すなわち、ペプチドは改変されたアミノ酸や結合を有し得る。生物活性ペプチドという語は、哺乳動物への投与時所与の種類/量の薬理学的または生物学的効果を発揮するペプチドを意味する。
【0052】
本明細書中に用いた「アミノ酸/アミノ酸残基」という語は、遺伝的にコードされるL−アミノ酸、遺伝的にコードされない天然アミノ酸、合成L−アミノ酸、もしくは前記いずれかのD−鏡像異性体、またはその製薬学的に許容される塩/誘導体を意味し得る。本明細書中、遺伝的にコードされる20種のL−アミノ酸、およびコードされない一般的なアミノ酸の表記には次のような通常の略号が適用されている。
【0053】
【表1】
【0054】
【表2】
【0055】
本発明の範囲内に含まれるペプチドは、指定されたクラスのアミノ酸残基に関して部分的に規定される。アミノ酸は、主にアミノ酸側鎖の特徴に依存して三つの主要なクラス、すなわち親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸に分類される。これらの主要クラスはさらにサブクラスに分割され得る。親水性アミノ酸には酸性、塩基性または極性側鎖を有するアミノ酸が含まれ、疎水性アミノ酸には芳香族側鎖または非極性側鎖を有するアミノ酸が含まれる。さらに、非極性アミノ酸には特に脂肪族アミノ酸が含まれる。
【0056】
「疎水性アミノ酸」とは、その側鎖が生理的pHにおいて非荷電であり、かつ水溶液によって反発されるアミノ酸のことである。遺伝的にコードされる疎水性アミノ酸の例にはIle、Leu、およびValが含まれる。遺伝的にコードされない疎水性アミノ酸の例にはt−BuAが含まれる。
【0057】
「芳香族アミノ酸」とは、共役π電子系を有する環(芳香族基)を少なくとも1個含む側鎖を有する疎水性アミノ酸のことである。芳香族基は、アルキル、アルケニル、アルキニル、ヒドロキシ、スルファニル、ニトロおよびアミノ基などの置換基でさらに置換されていてもよい。遺伝的にコードされる芳香族アミノ酸の例にはフェニルアラニン、チロシン、およびトリプトファンが含まれる。遺伝的にコードされない芳香族アミノ酸で一般的なものとしては、フェニルグリシン、2−ナフチルアラニン、β−2−チエニルアラニン、1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸、4−クロロフェニルアラニン、2−フルオロフェニルアラニン、3−フルオロフェニルアラニン、および4−フルオロフェニルアラニンなどが挙げられる。
【0058】
「非極性アミノ酸」とは、生理的pHにおいて通常非荷電であり、かつ極性を有しない側鎖を有する疎水性アミノ酸のことである。遺伝的にコードされる非極性アミノ酸の例にはグリシン、プロリン、およびメチオニンが含まれる。コードされない非極性アミノ酸の例にはChaが含まれる。
【0059】
「脂肪族アミノ酸」とは、飽和または不飽和の直鎖状、分枝鎖状もしくは環状炭化水素側鎖を有する非極性アミノ酸のことである。遺伝的にコードされる脂肪族アミノ酸の例にはAla、Leu、Val、およびIleが含まれる。コードされない脂肪族アミノ酸の例にはNleが含まれる。
【0060】
「親水性アミノ酸」とは、その側鎖が水溶液によって誘引されるアミノ酸のことである。遺伝的にコードされる親水性アミノ酸の例にはSerおよびLysが含まれる。コードされない親水性アミノ酸の例にはCitおよびhCysが含まれる。
【0061】
「酸性アミノ酸」とは側鎖pΚ値が7未満の親水性アミノ酸のことである。酸性アミノ酸は典型的には、生理的pHにおいて水素イオンが失われることに起因して負に荷電される側鎖を有する。遺伝的にコードされる酸性アミノ酸の例にはアスパラギン酸(アスパルタート)およびグルタミン酸(グルタマート)が含まれる。
【0062】
「塩基性アミノ酸」とは、側鎖pΚ値が7より大きい親水性アミノ酸のことである。塩基性アミノ酸は典型的には、生理的pHにおいてヒドロニウムイオンとの会合に起因して正に荷電される側鎖を有する。遺伝的にコードされる塩基性アミノ酸の例にはアルギニン、リジン、およびヒスチジンが含まれる。遺伝的にコードされない塩基性アミノ酸の例には非環状アミノ酸のオルニチン、2,3−ジアミノプロピオン酸、2,4−ジアミノ酪酸、およびホモアルギニンが含まれる。
【0063】
「極性アミノ酸」とは、生理的pHにおいて非荷電であるが、2個の原子に共有された電子対が一方の原子により近接して保持されている結合を含む側鎖を有する親水性アミノ酸のことである。遺伝的にコードされる極性アミノ酸の例にはアスパラギンおよびグルタミンが含まれる。遺伝的にコードされない極性アミノ酸の例にはシトルリン、N−アセチルリジン、およびメチオニンスルホキシドが含まれる。
【0064】
「システイン様アミノ酸」とは、その側鎖が別のアミノ酸残基の側鎖とジスルフィド結合などの共有結合を形成し得るアミノ酸のことである。システイン様アミノ酸は典型的には、少なくとも1個のチオール(SH)基を含む側鎖を有する。遺伝的にコードされるシステイン様アミノ酸の例にはシステインが含まれる。遺伝的にコードされないシステイン様アミノ酸の例にはホモシステインおよびペニシラミンが含まれる。
【0065】
当業者には理解されようが、上述の分類は絶対的なものではない。アミノ酸によっては上記特徴のうちの二つ以上を具え、従って二つ以上のカテゴリーに包含され得る。例えば、チロシンは芳香環および極性ヒドロキシ基を両方とも有する。すなわち、チロシンは二重の特性を有しており、芳香族アミノ酸と極性アミノ酸との両カテゴリーに含まれ得る。同様に、システインはジスルフィド結合を形成し得るみのでなく、非極性でもある。厳密には疎水性もしくは非極性アミノ酸には分類されないものの、システインは多くの場合ペプチドに疎水性を付与するべく用いられ得る。
【0066】
本発明のペプチドおよびペプチド類似体を構成し得る一般的なアミノ酸で遺伝的にコードされないものには、β−アラニン(b−Ala)および他のω−アミノ酸、例えば3−アミノプロピオン酸(Dap)、2,3−ジアミノプロピオン酸(Dpr)、4−アミノ酪酸など; α−アミノイソ酪酸(Aib); ε−アミノヘキサン酸(Aha); δ−アミノ吉草酸(Ava); N−メチルグリシンもしくはサルコシン(MeGly); オルニチン(Orn); シトルリン(Cit); t−ブチルアラニン(t−BuA); t−ブチルグリシン(t−BuG); N−メチルイソロイシン(MeIle); フェニルグリシン(Phg); シクロヘキシルアラニン(Cha); ノルロイシン(Nle); 2−ナフチルアラニン(2−Nal); 4−クロロフェニルアラニン(Phe(4−Cl)); 2−フルオロフェニルアラニン(Phe(2−F)); 3−フルオロフェニルアラニン(Phe(3−F)); 4−フルオロフェニルアラニン(Phe(4−F)); ペニシラミン(Pen); 1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Tic); β−2−チエニルアラニン(Thi); メチオニンスルホキシド(MSO); ホモアルギニン(hArg); N−アセチルリジン(AcLys); 2,3−ジアミノ酪酸(Dab); 2,4−ジアミノ酪酸(Dbu); p−アミノフェニルアラニン(Phe(pNH2)); N−メチルバリン(MeVal); ホモシステイン(hCys)、およびホモセリン(hSer)が非限定的に含まれる。これらのアミノ酸も、先に定義したカテゴリーに適宜分類される。
【0067】
上記遺伝的にコードされる、およびコードされないアミノ酸の分類を次の表2にまとめる。表2は単なる説明用であり、本明細書に開示されたペプチドおよびペプチド類似体を構成し得るアミノ酸残基の網羅的なリストではないと理解されるべきである。本明細書に開示されたペプチドおよびペプチド類似体の作製に有用である他のアミノ酸残基は、例えばFasman, CRC Practical Handbook of Biochemistry and Molecular Biology, CRC Press, Inc., 1989とその引用文献中に見出され得る。本明細書で具体的に言及されていないアミノ酸は、公知の挙動に基づき、および/またはその化学的および/または物理的諸特性を具体的に同定されたアミノ酸と比較することにより上述のカテゴリーに適宜分類することができる。
【0068】
【表3】
【0069】
好ましくは、本発明の生物活性ペプチドには以下の配列が非限定的に含まれる。
配列番号1: Trp−Ser−Gln(WSQ)
配列番号2: Trp−Ser−Leu(WSL)
配列番号3: Trp−Gln−Tyr(WQY)
配列番号4: Ser−Gln−Tyr(SQY)
配列番号5: Ser−Gln−Leu(SQL)
配列番号6: Ser−Asn−Tyr(SNY)
配列番号7: Gln−Tyr−Leu(QYL)
配列番号8: Gln−Asn−Tyr(QNY)
配列番号9: 環状Trp−Ser−Leu CY(cy WSL)
配列番号10: 環状Ser−Asn−Tyr CY(cy SNY)
配列番号11: Trp−Gln−Gln(WQQ)
配列番号12: Trp−Asn−Gln(WNQ)
配列番号13: Trp−Tyr−Gln(WYQ)
配列番号14: Trp−Gln−Leu(WQL)
配列番号15: Trp−Asn−Leu(WNL)
配列番号16: Trp−Tyr−Leu(WYL)
配列番号17: Trp−Ser−Tyr(WSY)
配列番号18: Trp−Asn−Tyr(WNY)
配列番号19: Trp−Tyr−Tyr(WYY)
配列番号20: Ser−Ser−Gln(SSQ)
配列番号21: Ser−Gln−Gln(SQQ)
配列番号22: Ser−Asn−Gln(SNQ)
配列番号23: Ser−Ser−Leu(SSL)
配列番号24: Ser−Asn−Leu(SNL)
配列番号25: Ser−Tyr−Leu(SYL)
配列番号26: Ser−Ser−Tyr(SSY)
配列番号27: Ser−Tyr−Gln(SYQ)
配列番号28: Ser−Tyr−Tyr(SYY)
配列番号29: Gln−Ser−Gln(QSQ)
配列番号30: Gln−Gln−Gln(QQQ)
配列番号31: Gln−Asn−Gln(QNQ)
配列番号32: Gln−Tyr−Gln(QYQ)
配列番号33: Gln−Ser−Leu(QSL)
配列番号34: Gln−Gln−Leu(QQL)
配列番号35: Gln−Asn−Leu(QNL)
配列番号36: Gln−Ser−Tyr(QSY)
配列番号37: Gln−Gln−Tyr(QQY)
配列番号38: Gln−Tyr−Tyr(QYY)
【0070】
より好ましくは、本発明の生物活性ペプチドにはSer−Asn−Tyr(SNY)すなわち配列番号6、およびTrp−Ser−Leu(WSL)すなわち配列番号2が非限定的に含まれる。
【0071】
本発明のペプチドは直鎖状であっても環状であってもよいが、好ましくは直鎖状である。
【0072】
公知のTNF−α阻害活性を有するペプチド配列である環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu CY)を、in vitro研究でのTNF−α拮抗活性評価に陽性対照として用いた。
【0073】
本発明によるペプチドの表記において、アミノ酸残基Xn間の記号「−」は通常、骨格を成すアミノ酸同士の結合を表わす。従って、記号「−」は普通アミド結合(−C(O)−NH)を表わす。しかし、具体的な実施形態として本明細書に開示されたペプチドのいずれにおいても、場合によっては1個以上のアミド結合を他の結合、好ましくは置換アミド結合、またはアミド結合の同配体に置き換えてもよいと理解されるべきである。
【0074】
(本明細書中に用いた)「TNFアルファ」という語と「TNF−α」という語とは同じもの、すなわちTNFアルファもしくは腫瘍壊死因子αを意味する。
【0075】
本発明のペプチドは、当該技術分野で公知のいずれの方法で合成してもよい。好ましい一法として、本発明の新規なペプチドを、1.00 mmolスケールにおいて自動ペプチド合成機(Applied Biosystems社製433A型ペプチドシンセサイザ)上でFmoc法を用いる固相法により合成した。ペプチドの構築はC末端からN末端へと行なった。Wang樹脂を用いてペプチドを合成した。合成に用いた樹脂は、Novabiochem社から調達したWang樹脂(100〜200メッシュ)であった(置換率1.2mmol/g樹脂)。
【0076】
合成の際、化学成分を用いてペプチドの反応性側鎖を保護した。N−末端アミノ基を9−フルオレニルメトキシカルボニル(Fmoc)基で保護した。ロイシンは側鎖を保護せずに用いた。トリプトファンの側鎖はtert−ブトキシカルボニル(Boc)で保護した。アスパラギンおよびグルタミンの側鎖はトリチル(trt)で保護した。セリンおよびチロシンは、側鎖をt−ブチル(tBu)で保護して用いた。システインはS−アセトアミドメチル(Acm)で保護した。最初のアミノ酸をWang樹脂上に、4−ジメチルアミノピリジン(DMAP)およびジイソプロピルカルボジイミド(DIC)を用いて導入し、その後無水酢酸を用いてキャップ化を行なった。アミノ酸と樹脂とのカップリングに用いられる活性化試薬には、2−(1H−ベンゾトリアゾル−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスファート/1−ヒドロキシベンゾトリアゾール(HBRU/HOBt)、およびジイソプロピルエチルアミン(DIEA)が含まれる。カップリング反応はNMP中で生起させた。ペプチド鎖の構築完了後、ペプチド−樹脂をメタノールで洗浄し、乾燥した。トリフルオロ酢酸、結晶性フェノール、チオアニソール、エタンジチオールおよび脱イオン水から成る切断用混合物での処理を室温で2〜3時間行なうことにより、ペプチドを樹脂から切り離した。冷乾燥エーテルで沈澱させて粗なペプチドを得た。次に、沈澱物を濾別して水に溶解させ、Vertis凍結乾燥機で凍結乾燥した。このようにして得られた粗なペプチドを、Phenomenex C18(250×22.1)逆相カラムを用い、かつ所与の濃度勾配の0.1%TFA含有アセトニトリル/水を用いる分取HPLCによって精製した。溶出画分を、Phenomenex C18(250×4.6)逆相カラムを用いるHPLC分析システム(株式会社島津製作所(日本))で再分析した。アセトニトリルを蒸発させ、画分を凍結乾燥して精製ペプチドを得た。各ペプチドを質量スペクトルによって同定した。
【0077】
ジメチルホルムアミド(DMF)中のヨウ素を用いて、樹脂上のペプチドを環化した。樹脂を自動振盪機で穏やかに振盪しながらDMF中の6倍モル過剰のヨウ素で処理した。反応の進行をHPLCで監視した。反応完了後、樹脂をDMF中の0.4Mアスコルビン酸でクエンチした。次に、樹脂をDMFおよびメタノールで洗浄し、数分間真空乾燥した。最後に環化ペプチドを樹脂から切り離し、RP−HPLCで分析した。粗なペプチドを分取HPLCによって精製し、エレクトロスプレー質量分析法で特性解析を行なった。本発明のペプチドを、高速液体クロマトグラフィー(HPLC)、イオン交換クロマトグラフィー、ゲル電気泳動、アフィニティークロマトグラフィーといった、当該技術分野で公知の方法で精製したが、好ましいのはクロマトグラフィーによる方法であった。より好ましくは、本発明のペプチドは、RP C−18カラムを用いる半分取型Shimatzu HPLCシステムで精製される。実際に用いられる諸条件は、正味荷電、疎水性、および親水性などの要因に依存する。
【0078】
本発明のペプチドは、質量分析法、SDS−PAGE、等電集束法、2D−電気泳動、クロマトグラフィー、ゲル濾過(大きさに基づく分離)、イオン交換(電荷に基づく分離)、配列決定、プロテアーゼ特異性測定、HPLC、X線結晶解析法といった、当該技術分野で公知の方法で分析され得る。好ましい一法として、本発明のペプチドを質量分析法で分析した。
【0079】
ペプチドの純度は、当該技術分野で公知であるいずれの方法でも測定可能である。ペプチドの純度を逆相HPLCで測定した。本発明のペプチドを純度について分析した。本発明のペプチドは比較的高い純度を有する。
【0080】
本発明のペプチドは水や、酢酸緩衝液、リン酸緩衝液、DMSO含有PBSといった生理的緩衝液に可溶である。
【0081】
本発明によるTNF−α阻害剤は、正常で健康な被検者における濃度を上回る濃度で存在するTNF−αに関連する病態もしくは状態の治療に有用である。そのような病態には、慢性関節リウマチ、全身性エリテマトーデス、および乾癬など、急性および慢性の免疫性および自己免疫性病態; 悪液質を含めた敗血症症候群; 急性または慢性細菌感染が原因の循環虚脱及びショック; 細菌感染、ウイルス感染、および真菌感染を含めた急性および慢性の寄生または感染プロセス; クローン病、およびTNF−α分泌腫瘍を伴う悪性病態が非限定的に含まれる。
【0082】
製剤化および投与経路
本発明の化合物は被検者に対してそのまま投与しても、医薬組成物の形態で投与してもよい。本発明のペプチドを含有する医薬組成物は、通常の混合法、溶解法、顆粒化法、糖剤形成法、湿式粉砕法、乳化法、カプセル封入法、捕捉法、または凍結乾燥法によって製造され得る。医薬組成物は、活性なペプチドまたはペプチド類似体を医薬として用い得る製剤へと加工することを容易にする、生理学的に許容されるキャリヤー、稀釈剤、添加剤または補助剤を1種以上用いて通常のように製剤化され得る。適正な製剤化は選択された投与経路に依存する。
【0083】
適当な製薬用キャリヤーは、当該技術分野での標準的な参考文献であるA.Osol編Remington’s Pharmaceutical Sciencesの最新版に記載されている。
【0084】
本発明の医薬組成物は、活性物質が哺乳動物の体内においてその物質の作用部位に到達することを可能にするいずれの手段で投与されてもよい。本発明のペプチドは、当該技術分野で公知であるいずれの投与経路からも投与され得る。様々な経路からの投与には、局所投与、非経口投与、経粘膜投与、経口投与、口腔内投与、直腸内投与、吸入投与、鼻腔内投与、膣内投与、および舌下投与が非限定的に含まれる。
【0085】
局所投与用としては、本発明のペプチドは当該技術分野で良く知られている溶液剤、ゲル剤、軟膏、クリーム剤、懸濁剤などに製剤化され得るが、これらに限定はされない。
【0086】
全身用製剤は、注射、例えば皮下注射、静脈内注射、筋肉内注射、髄腔内注射または腹腔内注射による投与用に設計された製剤、および経皮投与、経粘膜投与、経口投与または肺内投与用に設計された製剤を包含する。
【0087】
注射用としては、本発明のペプチドは水溶液中、好ましくはハンクス液、リンゲル液、または生理食塩水緩衝液といった生理学的に適合する緩衝液中に製剤化され得る。溶液は懸濁化剤、安定剤および/または分散剤などの剤形維持剤(formulatory agents)を含有してもよい。
【0088】
あるいは他の場合には、ペプチドを用時適当な賦形剤、例えば滅菌発熱性物質除去蒸留水で構成される散剤の形態としてもよい。
【0089】
経粘膜投与用製剤には、透過するべき関門に適した浸透剤が用いられる。そのような浸透剤は通常、当該技術分野で公知である。
【0090】
経口投与用製剤は、活性ペプチドを製薬学的に許容される、当該技術分野で公知のキャリヤーと配合することにより容易に製造できる。キャリヤーは本発明のペプチドを、治療される患者の経口摂取用に錠剤、丸剤、糖剤、カプセル剤、液剤、ゲル剤、シロップ剤、スラリー剤、懸濁剤等に製剤化することを可能にする。
所望であれば、固形製剤を標準的な方法で被覆してもよい。
【0091】
例えば懸濁剤、エリキシル剤および溶液剤などの経口液剤に好適なキャリヤー、添加剤または稀釈剤には、水、グリコール、油、アルコール等が含まれる。着香料、防腐剤、着色料などを添加することも可能である。
【0092】
口腔内投与用には、ペプチドは通常のように製剤化されて錠剤、トローチ剤等の形態を取り得る。
【0093】
吸入投与用に本発明により用いられるペプチドは、適当なプロペラント、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素または他の適当な気体を用いた加圧パッケージまたはネブライザーから噴霧エアゾール剤の形態で好適に送出される。加圧エアゾール剤の場合、単位用量は、バルブを設けて所定量を送出することにより決定され得る。化合物と、乳糖やデンプンといった適当な粉末状基剤とを混合した粉末混合物を収容した、例えばゼラチン製のカプセルやカートリッジを吸入器またはインサフレーター用に調製してもよい。
【0094】
ペプチドを、例えばカカオ脂や他のグリセリドなど、通常の坐剤用基剤を含有する坐剤や停留浣腸剤などの直腸内または膣内投与用製剤とすることも可能である。
【0095】
上述の諸製剤に加え、ペプチドをデポー剤に製剤化することも可能である。この持続性製剤は埋め込み(例えば皮下または筋肉内埋め込み)によってか、または筋肉内注射によって投与され得る。従ってペプチドは、例えば適当なポリマーまたは疎水性材料を用いて(例えば許容される油を分散媒とした乳剤に)製剤化しても、イオン交換樹脂を用いて製剤化しても、あるいはまたやや難溶の誘導体、例えばやや難溶の塩として製剤化してもよい。
【0096】
上記以外の医薬送達系を採用することも可能である。本発明のペプチドおよびペプチド類似体の送達に用い得る送達媒体の良く知られた例に、リポソームおよび乳剤が有る。ジメチルスルホキシドなど、幾つかの有機溶媒も用い得るが、通常比較的高い毒性という不都合を伴う。ペプチドの送達には持続放出システムも用い得る。様々な持続放出性材料が確立されており、それらは当業者に良く知られている。治療試薬の化学的性質および生物学的安定性次第では、タンパク質安定化のための付加的な方策を用いてもよい。
【0097】
本発明によれば、製薬学的に許容される塩および誘導体とは、遊離塩基の生物活性を実質的に保有する塩および誘導体のことである。製薬学的に許容される塩および誘導体には、当業者によって製造され得る塩および誘導体が含まれる。
【0098】
本発明のペプチドは通常、所期の目的の達成に有効な量で用いられる。TNF関連疾患の治療または予防のための使用では、本発明のペプチドまたはその医薬組成物は治療有効量で投与または適用される。治療有効量とは、症状の改善もしくは予防、または治療される患者の延命に有効な量のことである。治療有効量は、特に本明細書の詳細な開示に照らせば当業者には容易に決定できる。
【0099】
投与薬剤の用量は、当然ながら、特定薬剤の薬力学特性並びにその投与モードおよび投与経路; 被投与者の年齢、健康状態および体重; 症状の種類および程度、併用治療の種類、治療頻度、並びに所望の効果など、周知の要因に依存して変動する。
【0100】
初期量は、当該技術分野で良く知られた方法を用いてin vivoデータ、例えば動物モデルから推定することもできる。当業者であれば、動物データに基づいてヒトへの投与を最適化することは容易に可能である。
【0101】
投与量および投与間隔は、ペプチドの血漿中濃度を治療効果の維持に十分なものとするべく個別に調整され得る。注射による投与の場合、患者に対する用量は普通約0.1〜50mg/kg/日、好ましくは約0.5〜10mg/kg/日である。典型的には、1〜10mg/kg/日の量を1日につき複数回(6回)に分けて投与するか、1日1回隔日投与するか、または持続放出形態で投与することが所望の結果を得る上で有効である。病的状態の改善に有効な血漿中濃度を達成するべく投与量および投与間隔を個別に調整してもよい。治療上有効な血清中濃度は、投与を毎日複数回に分けて行なうことによって達成され得る。
【0102】
局所投与または選択的摂取の場合、有効な局所的ペプチド濃度は血漿中濃度と関わりが無いかもしれない。当業者であれば、甚だしい実験を行なわなくとも治療上有効な局所投与量を最適化することができる。
【0103】
当然ながら、投与されるペプチドの量は、治療される被検者、被検者の体重、疾病の重篤度、投与方法、および処方医師の判断に依存する。
【0104】
治療は症状が検出される間、さらには症状が検出されない場合にも断続的に反復され得る。治療は単独で行なっても、他の薬物を併用して行なってもよい。
【0105】
本出願明細書中各所で様々な刊行物および特許が引用され、特許はその番号、他の刊行物は著者名および発行年で特定されている。完全な引用文献リストを後段に示す。その結果、本発明の属する分野の技術水準をより十全に説明するべく、引用された刊行物および特許の開示はその全体が援用により本明細書に含まれる。
【0106】
本発明をここに詳述したが、用いられた用語は説明のためのものであり、限定のためのものではないと理解されるべきである。
【0107】
本発明の様々な改良および変形が上記教示に照らして可能であることは明らかである。従って、添付の請求項の範囲内であれば、本発明は具体的に記載された以外の形態でも実施され得ると理解されるべきである。
【0108】
方法概略および結果
【実施例1】
【0109】
配列番号6のペプチドの調製方法
実施例1(a)―配列番号6のペプチドの合成: ペプチド固相合成法を用い、自動または半自動ペプチド合成装置でFmoc化学に則っり配列番号6のペプチドを合成した。Wang樹脂を用いて合成を行なった。樹脂の置換率は0.6mmol/gから1.2mmol/gまでであった。チロシンおよびセリンの側鎖をt−ブチル基で保護し、アスパラギン側鎖をトリチル基で保護した。DMF中のDIC/HOBt(3〜5eq)およびDMAP(1〜2eq)を室温で約5〜8時間用いて、最初のアミノ酸チロシンをWang樹脂上に導入した。DMF中の20%ピペリジンを20〜30分間用い、N末端を保護するFmoc基を除去して脱保護を行なった。1mmolの反応に15〜20mlのDMFを用いた。脱保護したチロシンにFmoc−Asp(tBu)−OH(3〜5eq)を、DMF中のDIC/HOBt(3〜5eq)を室温で約3〜4時間用いることによりカップリングさせた。反応の完了をKaiser試験で監視した。Kaiser試験では、青色の消失によってカップリング反応の完了が示される。反応完了後、反応混合物をDMFで3回洗浄した。Fmoc基を上記のように除去して脱保護を行ない、その後反応混合物をDMFで3回洗浄した。上記と同様の操作で、Fmoc−Ser(tBu)−OHをアスパラギンにカップリングさせ、脱保護した。固相上に構築されたペプチドの平均収率は90〜95%であった。
【0110】
樹脂からのペプチド切断:
TFA:フェノール:TIS:DIT:水を82.5:5.0:2.5:5.0:5.0の比率で含有する試薬混合物(150ml)を用いてペプチドを樹脂から切り離した。不断の攪拌下、配列番号6のペプチドを担持した樹脂を切断試薬中に氷冷環境下で15分間維持し、次いでやはり不断の攪拌下に室温で2時間維持した。反応完了後、混合物を焼結漏斗で濾過し、濾液に冷ジエチルエーテルを添加することによってペプチドを沈澱させた。
沈澱したペプチドを焼結漏斗で濾過し、乾燥し、水に溶解させ、最後に凍結乾燥して粗なペプチドを得た。粗ペプチド収率は85〜90%であった。
【0111】
ペプチドの精製:
粗なペプチドを、溶離剤としてアセトニトリルおよび水を用いる分析用HPLCで分析した。C−18分取カラム(250×50mm、10μ)を用いるHPLC LC−8A装置(島津製作所)において粗BRC605−1の精製を、流速80〜120ml/分、検出波長210nmで行なうアセトニトリル(0.1%TFA)−水(0.1%TFA)混合物での定組成溶離によって実施した。精製画分を分析用HPLCで分析して所望の画分をプールし、凍結乾燥し、特性解析を行なった。この方法による総収率は70%を越えることが判明した。
【0112】
ペプチドの特性解析:
MS分析―配列番号6のペプチドの全バッチについて、質量スペクトル分析による特性解析を行なった。各バッチの分子質量は383.5となる(陽性モード)。
ペプチド配列決定―配列番号6のペプチドの全バッチについて、ペプチド配列決定による特性解析を行なった。各バッチのペプチド配列は実際の配列を与える。
【実施例2】
【0113】
L929細胞のTNF−α媒介細胞毒性のペプチド配列による阻害
マウス線維芽細胞系L929を用いて、本発明のペプチドをTNF−α媒介細胞毒性の阻害について分析した。L929細胞(ATCC)にTNF−αを添加すると細胞毒性が誘発され、そのことは、生細胞をMTT(3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロミド、テトラゾール系)やクリスタルバイオレットといった生体用色素で染色し、その後色素をメタノールで抽出することによって推定できる。抽出した色素の吸光度は595nmで測定し得る(Hansen et al., Journal of Immunological Methods, 119: 203−210, 1989)。アッセイは下記のように行なった。
【0114】
L929細胞(10%FCSを補充したダルベッコ変法イーグル培地中に維持)をマイクロタイタープレートに2×105細胞/mlの密度で播種し、濃度1μg/mlのアクチノマイシンD(ACT−D)の存在下、37℃および5%CO2において2時間インキュベートした。75μMのペプチド溶液と共に37℃でプレインキュベートしたTNF−α(100pg/ml)をL929細胞に添加し、37℃および5%CO2において一晩インキュベートした。
【0115】
環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)ペプチドを、TNF関連細胞毒性の阻害を推定するための陽性対照として用いた。陽性対照ペプチド溶液(75μM)と共に37℃でプレインキュベートしたTNF−α(100pg/ml)を別のウェル内のL929細胞に添加し、37℃および5%CO2において一晩インキュベートした。
【0116】
試験プレートおよび陽性対照プレートの生細胞を100μl/ウェルの0.05%クリスタルバイオレットで染色し、室温で15分間インキュベートし、PBSで洗浄し、かつ室温で一晩乾燥した。クリスタルバイオレットを100μl/ウェルのメタノールで細胞から抽出し、抽出したクリスタルバイオレット色素の吸光度を595nmで測定した。細胞毒性の阻害パーセンテージを式:
[(ODtest−ODTNF)/(ODctrl−ODTNF)]×100
によって算出した。
TNF−α誘発細胞毒性阻害の評価は、本発明のペプチドを75μMの濃度で用いて行なった。
【0117】
実施例2(a):
配列番号1〜10のペプチドをTNF−α誘発細胞毒性の阻害について分析した。配列番号1、2、6および8のペプチドはTNF−α誘発細胞毒性を陽性対照よりも強く阻害した。配列番号1、2、6および8のペプチドのTNF−α誘発細胞毒性阻害率(各々二重に行なわれた複数回の実験に基づき算出された阻害率(%)の平均値±SE)はそれぞれ65±9.2%、48±6%、62±6.2%、および51±2.3%であった。
【0118】
実施例2(b)―配列番号2および6のペプチドの直鎖状および環状形態の、TNF−α誘発細胞毒性を阻害する能力の比較:
配列番号2および6の直鎖状ペプチド並びに配列番号9および10の環状ペプチドを分析して、TNF−α誘発細胞毒性を阻害する能力を比較した(分析方法は上述したとおり)。配列番号9および10の環状ペプチドより配列番号2および6の直鎖状ペプチドの方が能力の高いTNF−α誘発細胞毒性抑制物質であることが判明した。図2に示したように、配列番号9および10の環状ペプチドによる阻害はごく低率であった。
【0119】
陽性対照ペプチドすなわち環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)についてもその直鎖状および環状形態を分析し、TNF−α誘発細胞毒性を阻害する能力を比較した。興味深いことに逆のパターンが観察され、すなわち、陽性対照ペプチドの環状形態は比較的高いTNF−α誘発細胞毒性阻害率(60±10%)を示したが、陽性対照ペプチドの直鎖状形態はTNF−α誘発細胞毒性の阻害を有効に惹起しなかった(図2)。
【0120】
配列番号2および6の直鎖状ペプチドは、公知のTNF−α阻害ペプチドである陽性対照の環状形態と同等の高い阻害能力を有することが判明した。
【0121】
実施例2(c)―配列番号6のペプチドおよびエタネルセプト(「エンブレル」として市販されている公知のTNF−α阻害剤)の、TNF−α誘発細胞毒性を阻害する能力の比較:
配列番号6のペプチドおよびエタネルセプトを分析して、TNF−α誘発細胞毒性を阻害する能力を比較した(分析方法は実施例2において上述したとおり)。配列番号6のペプチドはエタネルセプトに匹敵するTNF−α誘発細胞毒性阻害能力を有することが判明した(図3)。
【0122】
ペプチドの結合に関する研究
ペプチドとTNF−α、またはU937細胞(TNF受容体発現ヒト白血病細胞系)上のTNF−R1との結合を、フローサイトメトリーアッセイ法を用いて調べた。ペプチドとTNF−αとの結合はTNF−αとTNF−R1との相互作用を妨げ、このことはフローサイトメトリーによって分析することができる。ペプチドが直接TNF−R1に結合することによってTNF−R1陽性細胞のパーセンテージが低下することもフローサイトメトリーにより分析可能である。
【実施例3】
【0123】
ペプチドによるTNF−αと細胞上の受容体TNF−R1との結合の阻害
TNF−αはU937細胞(ATCC)上のコグネイト受容体TNF−R1に結合し、その結果TNF−R1に関して陽性である細胞のパーセンテージは低下する。TNF−α結合ペプチドはTNF−αに結合し、TNF−αとTNF−RIとの相互作用を妨害する。TNF−R1に関して陽性である細胞のパーセンテージは、蛍光色素結合抗TNF−RI抗体での染色によって定量できる。
【0124】
配列番号2および6のペプチドを、TNF−αとU937細胞上の受容体TNF−R1との結合の阻害について分析した。配列番号2または6のペプチドによるTNF−αとU937細胞上のTNF−RIとの結合の阻害を蛍光活性化セルソーター(Becton Dickinson(米国)のFACS、FACSCalibur)を用いて推定した。
【0125】
0.5%BSA(ウシ血清アルブミン)および0.05%NaN3(結合緩衝液)を含有するPBS中にU937細胞(10%FCSを補充したRPMI−1640培地(Sigma−Aldrich(米国))中に維持)を、緩衝液100μl当たり1×105細胞の密度で懸濁させた。上記ペプチド(配列番号2および6)をそれぞれPBSに加えた2種のペプチド溶液(50μl)をTNF−α(5ng)と共に別々の試験管に入れ、37℃で1時間プレインキュベートした。次いで、ペプチドとTNF−αとの複合体をU937細胞に添加し、4℃で1時間インキュベートした。別の試験管にU937細胞(1×105個)をTNF−α(5ng)と共に入れ、4℃で1時間インキュベートした(並列実験)。その後、細胞を結合緩衝液で洗浄し、この細胞に5μl(1mg/ml)のヒト抗マウスTNF受容体抗体(クローン番号HTR−9; Novus Biologicals)を4℃で1時間添加した。次に、細胞を結合緩衝液で洗浄し、暗中4℃で30分間、10μl(10μg/ml)のフルオレセイン結合ヤギ抗マウスIgG二次抗体(GIBCO BRL, Gaithersburg, MD)で染色した。結合緩衝液で2回洗浄後、フローサイトメーターFACSCalibur(Becton Dickinson)を用いて細胞を分析した。ゲートを生細胞集団に設定し、ペプチドによるTNF−α/細胞結合の阻害の程度をTNF−RI発現陽性細胞のパーセンテージに基づき推定した。
【0126】
未処理のU937細胞は約78±1%がTNF−R1発現に関して陽性であることが判明した。U937細胞をTNF−αと共にプレインキュベートすると、TNF−R1発現陽性細胞は32±10%に減少した(図4aおよび4b)。U937細胞を配列番号2のペプチドとTNF−αとの複合体と共にプレインキュベートすると、TNF−R1発現陽性細胞の比率は37±8.5%となった。U937細胞を配列番号6のペプチドとTNF−αとの複合体と共にプレインキュベートすると、TNF−R1発現陽性細胞の比率は57±8%となった(図4aおよび4b)。
【0127】
上記の結果から、配列番号2および6のペプチドがTNF−αと結合してTNF−αとTNF−R1との結合を防止し、受容体TNF−R1に関して陽性である細胞の減少をTNF−αが未処理である場合と比較して抑制することは明らかである。
【実施例4】
【0128】
フローサイトメトリーによるペプチドとU937細胞上のTNF−R1との結合の評価
U937細胞を用いてペプチドとTNF−R1との結合を定量した。U937細胞に添加されたTNF−αはU937細胞上のコグネイト受容体TNF−R1に結合し、その結果TNF−R1陽性細胞のパーセンテージは低下する。本発明によるTNF−α阻害ペプチドはTNF−R1に結合し、U937細胞を該ペプチドと共にインキュベートするとTNF−R1陽性細胞が減少する。
【0129】
配列番号6のペプチドをU937細胞上のTNF−R1への結合について、フローサイトメトリーを用いて分析した。U937細胞を配列番号6のペプチドと共にインキュベートして、配列番号6のペプチドが直接受容体TNF−R1に結合することを実証した。配列番号6のペプチド(250μM)およびTNF−α(10ng)それぞれと共にインキュベートしたU937細胞を2回洗浄し、これを4℃で1時間、5μl(1mg/ml)のヒト抗マウスTNF受容体抗体(クローン番号HTR−9; Novus Biologicals)で染色した。次に、細胞を結合緩衝液で洗浄し、暗中4℃で30分間、10μl(10μg/ml)のフルオレセイン結合ヤギ抗マウスIgG二次抗体(GIBCO BRL, Gaithersburg, MD)で染色した。結合緩衝液で2回洗浄後、フローサイトメーターFACSCalibur(Becton Dickinson)を用いて細胞を分析した。ゲートを生細胞集団に設定し、TNF−R1への結合の程度をTNF−RI発現陽性細胞のパーセンテージに基づき推定した。
【0130】
83%のU937細胞上にTNF−R1が発現していることが判明した。U937細胞を組み換えTNF−α(10ng)と共にインキュベートすると、TNF−R1陽性細胞のパーセンテージは31.6%に低下した。U937細胞を配列番号6のペプチドと共にインキュベートすると、TNF−R1陽性細胞のパーセンテージは32%に低下した(図5)。TNF−R1発現に関して陽性である細胞のパーセンテージの低下が配列番号6と共にインキュベートしたあととTNF−αと共にインキュベートしたあととで同等であることが判明したが、このことはペプチドがTNF−R1に結合することを明らかに示している。
【実施例5】
【0131】
慢性関節リウマチのマウスモデルにおけるペプチドのin vivo効力
実施例5(a)―関節炎マウスモデルの開発: Lalruに所在するパナセア バイオテックの動物飼育施設から入手した雄のC57BL/6マウスを用いて、慢性関節リウマチのマウスモデルを開発した。完全フロイントアジュバント(CFA)(Sigma)中に乳濁させた150μgのトリタイプIIコラーゲン(Sigma)の皮内注射によってマウスを免疫した。一次免疫後第17日に、不完全フロイントアジュバントに加えた100μgのトリタイプIIコラーゲンでの追加免疫を動物に対して行なった(Ethan M.Shevach, Curr. Prot. Immunol. 2002: 15.0.1−15.0.6; Inglis, J. et al., Nature Protocols 4: 612−618, 2008)。対照(健康な雄のC57BL/6マウス)と比較しての足の太さ(paw thickness)の増加および抗コラーゲンIgG抗体の存在を、関節炎の発症を評価するパラメーターとした。動物の足の太さはデジタルノギスで測定し、抗コラーゲン抗体は免疫後第35日にマウス血清中で測定した。
【0132】
実施例5(b)―マウス血清中抗コラーゲンIgG濃度:
トリタイプIIコラーゲン(Sigma)をPBSに1μg/mlの量で加えたものをELISAプレートに塗布し、4℃で一晩インキュベートした。PBS−0.05%Tween 20(PBS−T)で3回洗浄後、コラーゲン、媒体(CFA)、およびPBS(対照)を注射したマウスから得た血清を別々のウェルに稀釈比1:100で添加した。プレートを室温で2時間インキュベートした。PBS−Tで5回洗浄後、ウサギ抗マウスIgG HRP(Bethyl Laboratories)をウェルに稀釈比1:10000で添加し、室温で45分間インキュベートした。プレートをPBS−Tで洗浄後、基質のOPD(オルトフェニルジアミン)を添加し、発色を観察した。発色を2N HClで停止させた。490nmでの吸光度を記録した(Ethan M.Shevach, Curr. Prot. Immunol. 2002: 15.0.1−15.0.6)。コラーゲン免疫マウスが示した抗コラーゲンIgGの血清中濃度は媒体(CFA)群および対照群(PBS注射)と比較して高かった(図6)。
【0133】
実施例5(c)―マウスモデルにおける用量および投与スケジュールの最適化:
コラーゲン誘発関節炎のマウスモデル(上述の方法で用意)において、配列番号6のペプチドの最適な用量および投与スケジュールを決定した。関節炎マウスを足の太さに従ってランダム化し、各々4匹または5匹から成る複数の群に分けて、配列番号6のペプチドの静脈内注射を行なった。各群に適用した用量および投与スケジュールを表3に示す。
【0134】
【表4】
【0135】
用量7.5mg/kgで4週間毎週1回投与した配列番号6のペプチドは足の太さを、対照動物で観察されたものと比較して僅かに減少させた。また、5mg/kgの用量、および1週間に3回、その後3週間毎週3回投与するスケジュールで投与した配列番号6のペプチドが足の太さを対照動物のものに比べて有意に減少させることが観察された。従って、用量5mg/kgで1週間に3回、その後3週間毎週3回投与する投与計画が最適なものとして選択された(図7)。
【0136】
実施例5(d)―マウスモデルにおける配列番号6および2のペプチド並びにエタネルセプトの効力の比較:
コラーゲン誘発関節炎のマウスモデル(上述の方法で用意)を用いて、配列番号6および2のペプチドの効力をエタネルセプト(商品名「エンブレル」の下に市販されている認可されたTNF−α阻害剤)のものと比較した。関節炎マウスに配列番号6および2のペプチド並びにエタネルセプトを用量5mg/kgで第1週に3回、その後3週間毎週1回静脈内投与した。
【0137】
配列番号6のペプチドおよびエタネルセプトの投与は足の太さを、正常な対照動物(対照: 健康な雄のC57BL/6マウス)と同等の太さにまで有意に減少させることが観察された(p<0.01)(図8a)。配列番号2のペプチドの投与も足の太さを有意に減少させた(p<0.05)(図8a)。これらの結果は、配列番号2および6のペプチドがエタネルセプトに匹敵する効力を有することを明らかに示している。
【0138】
実施例5(e)―配列番号2および6のペプチド並びにエタネルセプトで処理した動物において処理後のIgG1/IgG2a比を測定する比較研究:
コラーゲン誘発関節炎の発症には、Th1応答もしくは炎症誘発応答の増大が伴う。IgG1/IgG2a濃度比を測定して、本発明のペプチドおよびエタネルセプトでの処理がマウスモデルにおいて臨床疾患をTh1応答の低下により改善したかどうかを判断した。小さいIgG1/IgG2a比ほどTh1応答もしくは炎症誘発応答のダウンレギュレーションを示唆することになろう。
【0139】
関節炎マウスに配列番号2および6のペプチド、エタネルセプト、並びにPBSを用量5mg/kgで第1週に3回、その後3週間毎週1回静脈内投与した。配列番号6のペプチドおよびエタネルセプトを投与した場合、処理後のIgG1/IgG2a比は未処理の関節炎動物(PBS処理動物を未処理動物と見なす)と比較して小さくなった(図8b)。未処理の関節炎動物(PBS処理動物を未処理動物と見なす)のIgG1/IgG2a比は進行中の炎症に起因して大きかった。このことは、配列番号6のペプチドでの処理が関節炎マウスにおいてTh1応答を低下させ、その結果IgG1/IgG2a比を対照動物(対照: 健康な雄のC57BL/6マウス)のIgG1/IgG2a比に匹敵するほど小さくして症状の寛解をもたらすことを示唆している。
【実施例6】
【0140】
アジュバント誘発関節炎のラットモデルにおける配列番号6のペプチドのin vivo効力
配列番号6のペプチドのin vivo効力を「アジュバント」誘発関節炎のラットモデルにおいて評価した。このモデルは抗炎症性薬物の開発及び試験で広く用いられている。ラットにおけるアジュバント関節炎(AA)はヒトの炎症性関節炎の諸特徴を模倣する(Pearson et al., Proc. Soc. Exp. Biol. Med. 112: 95−110, 1956)。150μgの完全フロイントアジュバントで皮内免疫を行ない、その後第7日に不完全フロイントアジュバントで追加免疫を行なうことによって雄のWistarラットにAAを誘発した。動物の関節炎を臨床的スコアリングと、デジタルノギスを用いた足(paw)および関節の太さの測定とにより評価した。臨床スコアは以下の基準に従って付与した。
0=紅斑も腫脹も無し
1=後肢または前肢の1本の指に僅かな紅斑または腫脹
2=後肢または前肢の2本以上の指に紅斑および腫脹
3=足関節または手関節に紅斑および腫脹
4=後肢または前肢の指および足関節または手関節の全体に紅斑および腫脹が認められ、かつ足関節または手関節が屈曲不能
【0141】
関節炎ラットに配列番号6のペプチドを2.5mg/kgの用量で、第1週には6日間毎日1回投与し、第2週には1日おきに3回投与した。別の関節炎動物群にはエタネルセプト(Et)を同様にして投与した。配列番号6のペプチドを投与した動物においても、エタネルセプトを投与した動物においても、未処理の関節炎ラット(PBSで処理したラットを未処理の関節炎ラットと見なす)に比べて足の太さが減少した(図9a)。配列番号6のペプチドまたはエタネルセプトで処理した場合には臨床スコアの有意な低下も認められた(p<0.05)(図9bおよび10)。総体的に、得られた結果は、配列番号6のペプチドが関節炎ラットにおける足の太さの減少や臨床スコアの低下に関してエタネルセプトに匹敵する効力を有することを示唆している(図9aおよび9b)。
【0142】
本明細書および添付の請求項中、「a」、「an」、および「the」を付した単数形は、文脈上明らかに別様に解されないかぎり複数形の意味を包含することに留意されたい。
【技術分野】
【0001】
本発明は生物活性ペプチドとその調製方法に係わる。本発明はさらに、腫瘍壊死因子α(TNF−αもしくはTNFアルファ)阻害ペプチドとその調製方法に係わる。本発明は、上記のようなペプチド分子を含有する医薬組成物、並びに慢性関節リウマチ、乾癬性関節炎、クローン病および敗血症などの腫瘍壊死因子α(TNF−αもしくはTNFアルファ)媒介炎症性疾患の治療への該組成物の使用にも係わる。
【背景技術】
【0002】
サイトカインは、活性化された免疫細胞、すなわちB細胞、T細胞並びに単球およびマクロファージによって産生される一群のシグナルタンパク質である。サイトカインにはインターロイキンファミリー(IL−1〜23)、インターフェロン(α、βおよびγ)、並びにTNF−αおよびβが含まれる(非特許文献1および2)。IL−1およびTNF−αは敗血症、RAなどの自己免疫性状態、皮膚疾患、炎症性腸疾患といった疾病における炎症応答の主要メディエーターとして重要な役割を果たすことが示唆されている(非特許文献3〜7)。またサイトカイン調節研究により、TNF−αが炎症性病態を惹起する最も重要なサイトカインであることも示唆されている(非特許文献8)。
【0003】
TNF−αは元来、マウスの腫瘍に出血性壊死を引き起こす分子として発見された(非特許文献9)。その後、「カケクチン」として知られ、悪液質状態の誘因と考えられた血清タンパク質についての一連の研究から、カケクチンはTNF−αと同じものであることが最終的に判明した(非特許文献10)。TNF−αは今や、広範な活性を有し、様々な臨床状態に関与する炎症メディエーターとして確立されている。
【0004】
TNF−αは、数種の細胞、特に活性化されたマクロファージによって産生される分子量17kDaのタンパク質である。TNF−αは始め、安定な三量体に構成された膜貫通タンパク質として合成される。次いで、メタロプロテアーゼTNF−α変換酵素(TACE)により切断されてホモ三量体の可溶性TNF(sTNF)となり、遍在発現したそのコグネイト受容体(TNF RI、p55、およびTNF RII、p75)に結合する。細胞特異的なエフェクターを伴ったTNF受容体の遍在発現は、TNF−αに媒介される細胞応答がきわめて様々であることを説明するものであり、そのような応答の幾つかは有害で、命に関わる。単核細胞から離脱したTNF受容体は掃討作用によって、また天然の阻害剤として作用することによってTNF−α濃度を低下させる。
【0005】
TNF−αはきわめて様々な細胞応答を誘発し、その多くは有害な結果をもたらす。例えば、TNF−αは、食欲不振に起因する不十分な摂食に伴う脂肪の減少および全身のタンパク質の枯渇から帰結する状態である悪液質を誘発する。悪液質は癌患者に普通に認められ、後天性免疫不全症候群(AIDS)の患者でも観察されている。加えて、動物にTNF−αを大量に注射すると、敗血症性ショック症状の殆どが発現する。多発性硬化症および慢性関節リウマチなどの自己免疫疾患、乾癬、乾癬性関節炎、過敏症、免疫複合体病および移植片対宿主病、並びに移植拒絶反応においてTNF−αが役割を有することも示されている。TNF−αはマラリアおよび肺線維症とすら関連付けられている。従って、疾病状態におけるTNF−α産生の阻止を目差すことには多大の関心が寄せられており、その達成は治療上有益である。
【0006】
TNF関連疾患の治療
TNF−αの副作用を打ち消す方法としては、抗TNF抗体および可溶性RNF−Rの使用が注目されている。動物モデルでは、TNF−α関連炎症性疾患の治療にTNF−α特異的な抗体が有効であった(非特許文献11〜13)。抗TNF抗体のキメラ形態がヒトでの臨床試験用に構築された(非特許文献14〜16)。さらに、可溶性TNF受容体融合タンパク質がTNF拮抗薬としてヒトの患者に導入された(非特許文献17〜19)。
【0007】
特許文献1は、TNF受容体スーパーファミリーに属する分子の結合ループから設計される環状ペプチドおよびペプチド類似体に係わり、それらのペプチドおよびペプチド類似体はTNF−αとTNF受容体との相互結合作用に干渉してin vitro、並びにin vivoで阻害活性を示し、in vivoでのTNFの望ましくない生物活性に拮抗する。この文献に記載された発明では環化ペプチドが好ましく、なぜならタンパク質−タンパク質相互作用ではループおよびターンが機能上重要な役割を果たすからである。具体的な実施形態ではTNF−R p55の三つの結合ループから環状ペプチドが設計され、それらのペプチドはTNF−αと結合して、TNF−αがその細胞受容体に結合するのを阻害する。最も好ましい実施形態において、ペプチドは少なくとも7個のアミノ酸を有し、より小形の直鎖状ペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0008】
特許文献2は、TNF−αとTNF受容体との結合およびTNF−α機能を有効量の阻害ペプチドの投与によって阻害する方法に係わる。この特許は、TNF受容体と結合し、それによってTNF−αの、細胞上のTNF−α受容体に結合してこれを活性化する能力を妨げるペプチドに係わる。特許された発明は特に、7個、および12個のアミノ酸残基を有するペプチドをTNF−αとTNF受容体との結合およびTNF機能の阻害に使用する方法に係わる。特許文献2に記載された発明は、TNF受容体に結合し得る小形ペプチドをスクリーニングすることを目的とする。同定された分子で最も小形のものは7アミノ酸長の配列を有する。より小形のペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0009】
特許文献3は尿由来のTNF阻害ペプチドに係わる。この特許は、TNF−αに対して活性である精製形態のTNF阻害剤にも係わる。また、TNFに対して活性を示す医薬製剤として有用である精製形態のTNF阻害剤にも係わる。この特許はさらに、その精製形態において得られた30kDaタンパク質および40kDaタンパク質に係わる。開示されたこれらのタンパク質のアミノ酸配列は15アミノ酸以上の長さを有し、より小形のペプチドがTNF−α阻害剤として有用であり得るとの示唆は無い。
【0010】
特許文献4は、グリシン、アラニンおよびセリンから成る群から選択された少なくとも1種のアミノ酸またはその生理学的に許容される塩を、腫瘍壊死因子(TNF)濃度が生理的恒常性および局所的炎症を媒介する濃度を上回る患者のTNF濃度を低下させる医薬または栄養製剤の調製に使用する方法に係わる。この特許は、TNF濃度の低下がマクロファージ型細胞によるTNF産生、マクロファージ型細胞によるTNF放出、またはTNF受容体によるTNFの結合を阻害もしくは低減することにより可能であることを開示している。この特許は、TNF濃度を低下させる医薬または栄養製剤の調製にグリシン、アラニンおよびセリンのみを使用する方法に係わるものである。この特許は複数のアミノ酸の組み合わせやそのような組み合わせを含むペプチドを使用する方法は一切開示していない。
【0011】
特許文献5は、ヒトTNF−αの複数の分子表面から成る群から選択された少なくとも一つのヒトTNF−α分子表面に実質的に類似する分子表面を有するTNF−α拮抗化合物に係わる。この文献に記載された発明の化合物は、両端に結合した連結部分と、スペーサー部分とを有する。発明化合物によって、25個のアミノ酸並びに連結部分およびスペーサー部分を有するTNF−α阻害ペプチドが開示されている。特許文献5に記載されたTNF−α拮抗化合物はTNF p55受容体および/またはTNF p75受容体に結合し、TNF−αに媒介される細胞毒性を阻害する。特許文献5はスペーサー部分を有しない、より小形のペプチドを示唆してはいない。
【0012】
当該技術分野で現在使用可能な治療薬は、可溶性TNF受容体または抗TNFモノクローナル抗体の使用によってTNF−αを中和するべく設計されている(非特許文献20および21)。可溶性TNF受容体や抗TNFモノクローナル抗体は循環するTNF−αと結合し、それによって、TNF−αが細胞表面のTNF−Rへアクセスし、その後炎症経路を活性化するのを制限する。TNF−α濃度の抑制に使用可能な治療薬は、(1)インフリキシマブ(レミケード): マウス−ヒトキメラ型抗ヒトTNF−αモノクローナル抗体、(2)ヒュミラ: 完全ヒト型抗TNF−αモノクローナル抗体、(3)エタネルセプト: ヒトIgGのFc領域と融合した可溶性p75 sTNF−RIIの二量体融合タンパク質である。これらの分子は様々な自己免疫疾患で効力を示すものの、幾つかの制約、すなわち低いバイオアベイラビリティーおよび安定性、重篤な免疫反応の誘発、並びに高いコストを伴う。TNF−α活性の阻害には別の手段を講じることが妥当であろう。
【0013】
抗TNF抗体やTNF受容体の配列を利用して生物活性ペプチドフラグメントが合成されている(非特許文献22〜24)が、それらは大形で、難溶性である。このことは、恐らくは小形分子の形態を取る改良されたTNF−α阻害化合物の必要性を強調している。
【0014】
1997年に為された上記のような合成の一研究においてTakasaki等は、TNF−RIの三つの結合ループから設計されるペプチド類似体について検討した(非特許文献25)。結合ループを構成するアミノ酸配列に基づいて複数のペプチドが作製された。それらのペプチド配列の中で、TNF−RIのドメイン3のループ1(残基107〜114)に基づく環状WP9(配列WSENL)がTNF−α阻害活性に関して最も有望であることが判明した。Takasaki等はWP9の配列WSENLを鋳型として用いて、環状ペプチド模倣体WP9Q、WP9ELY、WP9YおよびWP9QYを設計した。ペプチド模倣体WP9QYは治療上の有効性を示し、マウスにおいて実験的自己免疫性脳脊髄炎(EAE)および慢性関節リウマチ(RA)の重篤度を低下させた。しかしこの物質は、生理緩衝液に比較的溶解しにくいため、潜在的治療薬としてのヒトへの使用は制限される(非特許文献25)。Takasaki等の1997年の研究では、ペプチドの環化および芳香族化によって安定性およびバイオアベイラビリティーは向上したが、溶解性の向上または活性の向上への効果はほとんど、または全く認められなかった。Takasakiは、WP9QYは今日までに開発された最小のペプチド模倣体であり、次世代の非ペプチド性阻害剤のためのリード化合物として用いられるかもしれないと述べている。このように、この文献は、当時公知の最小ペプチドに基づき非ペプチド性TNF阻害剤の開発を試みることを当業者に示唆している。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】米国特許第6,265,535号
【特許文献2】米国特許第6,344,443号
【特許文献3】米国特許第6,143,866号
【特許文献4】米国特許第6,048,543号
【特許文献5】米国特許第6,107,273号
【非特許文献】
【0016】
【非特許文献1】Janeway, C.A. et al., Immunobiology, 4th Ed., New York, Garland, 1999
【非特許文献2】Roitt, I. et al., Immunology, 5th ed., London, Mosby, 2002
【非特許文献3】Locksley, R.M. et al., Cell 104(4): 487−501, 2001
【非特許文献4】Feldmann, M. et al., Ann. Rev. Immunol. 14: 397−440, 1996
【非特許文献5】Buchan, G. et al., Clin. Exp. Immuol. 73: 449−455, 1988
【非特許文献6】Canto, E. et al., Clin. Immunol. 119(2): 156−165, 2006
【非特許文献7】Fantuzzi, F. et al., Expert Opi. Ther. Targets 12(9): 1085−1096, 2008
【非特許文献8】Feldmann et al., Ann. Rheum. Dis. 58: (Suppl. 1)127−131, 1999
【非特許文献9】Carswell et al., Proc. Natl. Acad. Sci. U.S.A. 72: 3666, 1975
【非特許文献10】Beutler et al., Annu. Rev. Immunol. 7: 625, 1989
【非特許文献11】Williams et al., Proc. Natl. Acad. Sci. U.S.A. 89: 9784−9788, 1992
【非特許文献12】Baker et al., Eur. J. Immunol. 24: 2040, 1994
【非特許文献13】Suitters et al., J. Exp. Med. 179: 849, 1994
【非特許文献14】Lorenz et al., J. Immunol. 156: 1646, 1996
【非特許文献15】Walker et al., J. Infect. Dis. 174: 63, 1996
【非特許文献16】Tak et al., Arthritis Rheumat. 39: 1077, 1996
【非特許文献17】Peppel et al., J. Exp. Med. 174: 1483, 1991
【非特許文献18】Williams et al., Immunol. 84: 433, 1995
【非特許文献19】Baumgartner et al., Arthritis Rheumat. 39 (Suppl.): S74, 1996
【非特許文献20】Piguet et al., Immunology 77: 510−514, 1992
【非特許文献21】Elliot et al., Arthritis Rheum. 36: 1681−1690, 1993
【非特許文献22】Weisong, Q. et al., Mol. Immunol. 43: 660−666, 2006
【非特許文献23】Zhang, J. et al., Biochem. Biophy. Res. Comm. 7: 1181−1187, 2003
【非特許文献24】Aoki et al., J. Clin. Invest. 116(6): 1525−1534, 2006
【非特許文献25】Takasaki et al., Nat. Biotechnol. Nov. 15(12): 1266−1270, 1997
【発明の概要】
【発明が解決しようとする課題】
【0017】
上記背景技術の考察から、生理緩衝液に溶解しやすく、安定性およびバイオアベイラビリティーが高く、僅かな副作用しか有せず、かつ低コストであるペプチドの設計を目差して改良された次世代TNF−α阻害剤を開発する必要性は明らかである。
【課題を解決するための手段】
【0018】
本発明によれば、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体が提供され、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0019】
本発明の別の態様によれば、式:
X1−X2−X3
を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体が提供され、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0020】
本発明は、本発明の生物活性ペプチドをTNF−α関連疾病状態の治療に使用する方法にも係わる。本発明はまた、本発明の生物活性ペプチドを含有する医薬組成物にも係わる。
【図面の簡単な説明】
【0021】
【図1】図1は、ペプチドの抗TNF−α活性をL929バイオアッセイを用いて示したグラフである。X軸上の数字1〜10は配列番号1〜10のペプチドを意味する。Y軸はTNF−α媒介細胞毒性の阻害パーセンテージを、各々二重に行なわれた3回の独立した実験の平均値±SEとして表わす。X軸上の「+ve」は陽性対照、すなわち公知のTNF−α阻害ペプチドである環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)を意味する。
【図2】図2は、配列番号2および6の直鎖状ペプチドと、配列番号9および10の環状ペプチドと、陽性対照すなわち環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)ペプチドとをTNF−αに誘発される細胞毒性の阻害について比較したグラフである。結果はTNF−α媒介細胞毒性の阻害パーセンテージの、各々三重に行なわれた3回の独立した実験から得られた平均値±SEとして示されている。
【図3】図3は、配列番号6のペプチドおよびエタネルセプト(Et)によるTNF−α媒介細胞毒性の阻害を比較したグラフである。結果はTNF−α媒介細胞毒性の阻害パーセンテージの、各々三重に行なわれた2回の独立した実験から得られた平均値±SEとして示されている。
【図4】図4は、ペプチドとTNF−αとの結合のフローサイトメトリーによる定量を示すグラフである。配列番号2および6のペプチドの存在下でのTNF−αと細胞受容体との結合の蛍光活性化セルソーター(FACS)分析結果を図4aに示す。Y軸はTNF−RI発現に関して陽性である細胞のパーセンテージを、3回の独立した実験から得られた平均値±SEとして表わす。棒グラフ(図4a)のX軸上、およびフローサイトメトリーヒストグラム(図4b)中の数字1〜5の意味は次のとおりである。 1=二次抗体としての抗マウスIgG FITCで染色したU937細胞。 2=マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 3=組み換えTNF−αで処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 4=組み換えTNF−αと配列番号2との複合体で処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。 5=組み換えTNF−αと配列番号6のペプチドとの複合体で処理し、マウス抗ヒトTNF受容体抗体および抗マウスIgG FITCで染色したU937細胞。
【図5】図5は、配列番号6のペプチドとU937細胞上に発現したTNF−R1との結合を示すグラフである。配列番号6のペプチドと、U937細胞上に発現したTNF−RIとの結合の蛍光活性化セルソーター(FACS)分析結果を図5に示す。Y軸はTNF−RI発現に関して陽性である細胞のパーセンテージを、2回の独立した実験から得られた平均値±SEとして表わす。棒グラフのX軸上、および重畳表示されたフローサイトメトリーヒストグラム(図5)中の数字は以下の試料を意味する。 1: 未処理のU937細胞。 2: U937細胞+TNF−α。 3: U937細胞+配列番号6のペプチド。
【図6】図6は、媒体(CFA)マウス、対照(PBS)マウス、およびコラーゲン免疫マウスの血清からの抗コラーゲンIgG濃度の推定を示すグラフである。
【図7】図7は、コラーゲン誘発関節炎のマウスモデルにおいて、異なる用量および投与スケジュールで用いられる配列番号6のペプチドでの処理の前(Pre Tx)および後(Post Tx)に得られた右足根骨部位での足の太さ(paw thickness)の平均値を示すグラフである。動物群はそれぞれ4匹または5匹の動物から成っていた。Y軸上の値は、群毎に得られる足の太さの平均値±SEである。X軸上のアルファベットは動物群を意味する。 A: 1.25mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 B: 2.5mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 C: 5mg/kgの配列番号6のペプチドを、1週間に3回投与し、その後毎週1回の投与を3週間行なうスケジュールで用いて処理した関節炎マウス群。 D: 7.5mg/kgの配列番号6のペプチドを、毎週1回の投与を4週間行なうスケジュールで用いて処理した関節炎マウス群。 E: 7.5mg/kgの配列番号6のペプチドを、1週目に1回投与し、3週間後に2回目の投与を行なうスケジュールで用いて処理した関節炎マウス群。 F: 対照としてのPBSで処理した関節炎マウス群。 G: 対照動物群、すなわち健康な雄のC57BL/6マウスの群。 処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図8a】図8aは、配列番号6のペプチド、配列番号2のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた左足根骨および右足根骨部位での足の太さの平均値±SEを示すグラフである。PBS処理動物は未処理動物と見なされ、また図中「対照」とは健康な雄のC57BL/6マウスのことである。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図8b】図8bは、配列番号6のペプチド、配列番号2のペプチド、およびエタネルセプト(Et)で処理した動物における処理後の抗コラーゲンIgG1/IgG2a比を比較したグラフである。PBS処理動物は未処理動物と見なされ、また図中「対照」とは健康な雄のC57BL/6マウスのことである。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図9a】図9aは、配列番号6のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた右足根骨および右関節部位での足の太さの平均値を示すグラフである。PBS処理動物は未処理動物と見なされる。動物群はそれぞれ3匹の動物から成っていた。
【図9b】図9bは、アジュバント誘発関節炎のラットモデルにおいて、配列番号6のペプチド、およびエタネルセプト(Et)での処理の前(Pre Tx)および後(Post Tx)に得られた臨床スコアを比較したグラフである。PBS処理動物は未処理動物と見なされる。動物群はそれぞれ3匹の動物から成っていた。処理の前後にp値を算出した。図中「**」はp値が0.01未満であることを示し、「*」はp<0.05を示す。
【図10】図10は、配列番号6のペプチド、エタネルセプト(Et)、および陰性対照としてのPBSでの処理の前後に関節炎ラットにおいて撮影した足および関節の腫脹の代表的な写真を示す説明図である。
【発明を実施するための形態】
【0022】
本発明は、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTryを含む群から選択され得る。
【0023】
本発明は、式:
X1−X2−X3
を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0024】
本発明はまた、TNF−α阻害剤として作用する生物活性ペプチドに係わる。本発明による生物活性ペプチドは、例えば下記のような機構により作用し得るが、これらに限定はされない。
a) 本発明のペプチドはTNF−αと結合して複合体を形成し、それによってTNF−αと受容体TNF−R1との結合を防止することができる。
b) 本発明のペプチドは直接受容体TNF−R1に結合することができ、TNF−αが受容体TNF−R1に結合するのを防止することができる。
【0025】
本発明は、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を含有する医薬組成物にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0026】
本発明はさらに、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わり、その際X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得る。
【0027】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0028】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0029】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を含有する医薬組成物に係わる。
【0030】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸は親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸を含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法に係わる。
【0031】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTrp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0032】
本発明は、式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された0〜2個のアミノ酸であり、X3は単一のアミノ酸残基であり、アミノ酸はTrp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択され得、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0033】
本発明は、式:
X1−X2−X3
〔式中X1、X2およびX3はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0034】
本発明は、式:
X1−X2−X3
〔式中X1、X2およびX3はそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0035】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0036】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0037】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物に係わる。
【0038】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択された0〜2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された0〜2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法に係わる。
【0039】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0040】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0041】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0042】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0043】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0044】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0045】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0046】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0047】
本発明はさらに、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体に係わる。
【0048】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体にも係わる。
【0049】
本発明は、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体と、製薬学的に許容されるキャリヤーとを含有する医薬組成物にも係わる。
【0050】
本発明は、TNF−α関連疾病状態を治療する方法であって、式:
X1−X2−X3
〔式中X1はTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである〕を有する生物活性ペプチドまたはその製薬学的に許容される塩および誘導体を投与することを含む方法にも係わる。
【0051】
本明細書中に用いた「ペプチド」という語は、ペプチド結合で連結された複数の天然アミノ酸サブユニットから成る重合体を意味する。アミノ酸という語は、天然ペプチドに類する部分を有するが、非天然部分も有する成分を意味する場合も有る。すなわち、ペプチドは改変されたアミノ酸や結合を有し得る。生物活性ペプチドという語は、哺乳動物への投与時所与の種類/量の薬理学的または生物学的効果を発揮するペプチドを意味する。
【0052】
本明細書中に用いた「アミノ酸/アミノ酸残基」という語は、遺伝的にコードされるL−アミノ酸、遺伝的にコードされない天然アミノ酸、合成L−アミノ酸、もしくは前記いずれかのD−鏡像異性体、またはその製薬学的に許容される塩/誘導体を意味し得る。本明細書中、遺伝的にコードされる20種のL−アミノ酸、およびコードされない一般的なアミノ酸の表記には次のような通常の略号が適用されている。
【0053】
【表1】
【0054】
【表2】
【0055】
本発明の範囲内に含まれるペプチドは、指定されたクラスのアミノ酸残基に関して部分的に規定される。アミノ酸は、主にアミノ酸側鎖の特徴に依存して三つの主要なクラス、すなわち親水性アミノ酸、疎水性アミノ酸、およびシステイン様アミノ酸に分類される。これらの主要クラスはさらにサブクラスに分割され得る。親水性アミノ酸には酸性、塩基性または極性側鎖を有するアミノ酸が含まれ、疎水性アミノ酸には芳香族側鎖または非極性側鎖を有するアミノ酸が含まれる。さらに、非極性アミノ酸には特に脂肪族アミノ酸が含まれる。
【0056】
「疎水性アミノ酸」とは、その側鎖が生理的pHにおいて非荷電であり、かつ水溶液によって反発されるアミノ酸のことである。遺伝的にコードされる疎水性アミノ酸の例にはIle、Leu、およびValが含まれる。遺伝的にコードされない疎水性アミノ酸の例にはt−BuAが含まれる。
【0057】
「芳香族アミノ酸」とは、共役π電子系を有する環(芳香族基)を少なくとも1個含む側鎖を有する疎水性アミノ酸のことである。芳香族基は、アルキル、アルケニル、アルキニル、ヒドロキシ、スルファニル、ニトロおよびアミノ基などの置換基でさらに置換されていてもよい。遺伝的にコードされる芳香族アミノ酸の例にはフェニルアラニン、チロシン、およびトリプトファンが含まれる。遺伝的にコードされない芳香族アミノ酸で一般的なものとしては、フェニルグリシン、2−ナフチルアラニン、β−2−チエニルアラニン、1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸、4−クロロフェニルアラニン、2−フルオロフェニルアラニン、3−フルオロフェニルアラニン、および4−フルオロフェニルアラニンなどが挙げられる。
【0058】
「非極性アミノ酸」とは、生理的pHにおいて通常非荷電であり、かつ極性を有しない側鎖を有する疎水性アミノ酸のことである。遺伝的にコードされる非極性アミノ酸の例にはグリシン、プロリン、およびメチオニンが含まれる。コードされない非極性アミノ酸の例にはChaが含まれる。
【0059】
「脂肪族アミノ酸」とは、飽和または不飽和の直鎖状、分枝鎖状もしくは環状炭化水素側鎖を有する非極性アミノ酸のことである。遺伝的にコードされる脂肪族アミノ酸の例にはAla、Leu、Val、およびIleが含まれる。コードされない脂肪族アミノ酸の例にはNleが含まれる。
【0060】
「親水性アミノ酸」とは、その側鎖が水溶液によって誘引されるアミノ酸のことである。遺伝的にコードされる親水性アミノ酸の例にはSerおよびLysが含まれる。コードされない親水性アミノ酸の例にはCitおよびhCysが含まれる。
【0061】
「酸性アミノ酸」とは側鎖pΚ値が7未満の親水性アミノ酸のことである。酸性アミノ酸は典型的には、生理的pHにおいて水素イオンが失われることに起因して負に荷電される側鎖を有する。遺伝的にコードされる酸性アミノ酸の例にはアスパラギン酸(アスパルタート)およびグルタミン酸(グルタマート)が含まれる。
【0062】
「塩基性アミノ酸」とは、側鎖pΚ値が7より大きい親水性アミノ酸のことである。塩基性アミノ酸は典型的には、生理的pHにおいてヒドロニウムイオンとの会合に起因して正に荷電される側鎖を有する。遺伝的にコードされる塩基性アミノ酸の例にはアルギニン、リジン、およびヒスチジンが含まれる。遺伝的にコードされない塩基性アミノ酸の例には非環状アミノ酸のオルニチン、2,3−ジアミノプロピオン酸、2,4−ジアミノ酪酸、およびホモアルギニンが含まれる。
【0063】
「極性アミノ酸」とは、生理的pHにおいて非荷電であるが、2個の原子に共有された電子対が一方の原子により近接して保持されている結合を含む側鎖を有する親水性アミノ酸のことである。遺伝的にコードされる極性アミノ酸の例にはアスパラギンおよびグルタミンが含まれる。遺伝的にコードされない極性アミノ酸の例にはシトルリン、N−アセチルリジン、およびメチオニンスルホキシドが含まれる。
【0064】
「システイン様アミノ酸」とは、その側鎖が別のアミノ酸残基の側鎖とジスルフィド結合などの共有結合を形成し得るアミノ酸のことである。システイン様アミノ酸は典型的には、少なくとも1個のチオール(SH)基を含む側鎖を有する。遺伝的にコードされるシステイン様アミノ酸の例にはシステインが含まれる。遺伝的にコードされないシステイン様アミノ酸の例にはホモシステインおよびペニシラミンが含まれる。
【0065】
当業者には理解されようが、上述の分類は絶対的なものではない。アミノ酸によっては上記特徴のうちの二つ以上を具え、従って二つ以上のカテゴリーに包含され得る。例えば、チロシンは芳香環および極性ヒドロキシ基を両方とも有する。すなわち、チロシンは二重の特性を有しており、芳香族アミノ酸と極性アミノ酸との両カテゴリーに含まれ得る。同様に、システインはジスルフィド結合を形成し得るみのでなく、非極性でもある。厳密には疎水性もしくは非極性アミノ酸には分類されないものの、システインは多くの場合ペプチドに疎水性を付与するべく用いられ得る。
【0066】
本発明のペプチドおよびペプチド類似体を構成し得る一般的なアミノ酸で遺伝的にコードされないものには、β−アラニン(b−Ala)および他のω−アミノ酸、例えば3−アミノプロピオン酸(Dap)、2,3−ジアミノプロピオン酸(Dpr)、4−アミノ酪酸など; α−アミノイソ酪酸(Aib); ε−アミノヘキサン酸(Aha); δ−アミノ吉草酸(Ava); N−メチルグリシンもしくはサルコシン(MeGly); オルニチン(Orn); シトルリン(Cit); t−ブチルアラニン(t−BuA); t−ブチルグリシン(t−BuG); N−メチルイソロイシン(MeIle); フェニルグリシン(Phg); シクロヘキシルアラニン(Cha); ノルロイシン(Nle); 2−ナフチルアラニン(2−Nal); 4−クロロフェニルアラニン(Phe(4−Cl)); 2−フルオロフェニルアラニン(Phe(2−F)); 3−フルオロフェニルアラニン(Phe(3−F)); 4−フルオロフェニルアラニン(Phe(4−F)); ペニシラミン(Pen); 1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Tic); β−2−チエニルアラニン(Thi); メチオニンスルホキシド(MSO); ホモアルギニン(hArg); N−アセチルリジン(AcLys); 2,3−ジアミノ酪酸(Dab); 2,4−ジアミノ酪酸(Dbu); p−アミノフェニルアラニン(Phe(pNH2)); N−メチルバリン(MeVal); ホモシステイン(hCys)、およびホモセリン(hSer)が非限定的に含まれる。これらのアミノ酸も、先に定義したカテゴリーに適宜分類される。
【0067】
上記遺伝的にコードされる、およびコードされないアミノ酸の分類を次の表2にまとめる。表2は単なる説明用であり、本明細書に開示されたペプチドおよびペプチド類似体を構成し得るアミノ酸残基の網羅的なリストではないと理解されるべきである。本明細書に開示されたペプチドおよびペプチド類似体の作製に有用である他のアミノ酸残基は、例えばFasman, CRC Practical Handbook of Biochemistry and Molecular Biology, CRC Press, Inc., 1989とその引用文献中に見出され得る。本明細書で具体的に言及されていないアミノ酸は、公知の挙動に基づき、および/またはその化学的および/または物理的諸特性を具体的に同定されたアミノ酸と比較することにより上述のカテゴリーに適宜分類することができる。
【0068】
【表3】
【0069】
好ましくは、本発明の生物活性ペプチドには以下の配列が非限定的に含まれる。
配列番号1: Trp−Ser−Gln(WSQ)
配列番号2: Trp−Ser−Leu(WSL)
配列番号3: Trp−Gln−Tyr(WQY)
配列番号4: Ser−Gln−Tyr(SQY)
配列番号5: Ser−Gln−Leu(SQL)
配列番号6: Ser−Asn−Tyr(SNY)
配列番号7: Gln−Tyr−Leu(QYL)
配列番号8: Gln−Asn−Tyr(QNY)
配列番号9: 環状Trp−Ser−Leu CY(cy WSL)
配列番号10: 環状Ser−Asn−Tyr CY(cy SNY)
配列番号11: Trp−Gln−Gln(WQQ)
配列番号12: Trp−Asn−Gln(WNQ)
配列番号13: Trp−Tyr−Gln(WYQ)
配列番号14: Trp−Gln−Leu(WQL)
配列番号15: Trp−Asn−Leu(WNL)
配列番号16: Trp−Tyr−Leu(WYL)
配列番号17: Trp−Ser−Tyr(WSY)
配列番号18: Trp−Asn−Tyr(WNY)
配列番号19: Trp−Tyr−Tyr(WYY)
配列番号20: Ser−Ser−Gln(SSQ)
配列番号21: Ser−Gln−Gln(SQQ)
配列番号22: Ser−Asn−Gln(SNQ)
配列番号23: Ser−Ser−Leu(SSL)
配列番号24: Ser−Asn−Leu(SNL)
配列番号25: Ser−Tyr−Leu(SYL)
配列番号26: Ser−Ser−Tyr(SSY)
配列番号27: Ser−Tyr−Gln(SYQ)
配列番号28: Ser−Tyr−Tyr(SYY)
配列番号29: Gln−Ser−Gln(QSQ)
配列番号30: Gln−Gln−Gln(QQQ)
配列番号31: Gln−Asn−Gln(QNQ)
配列番号32: Gln−Tyr−Gln(QYQ)
配列番号33: Gln−Ser−Leu(QSL)
配列番号34: Gln−Gln−Leu(QQL)
配列番号35: Gln−Asn−Leu(QNL)
配列番号36: Gln−Ser−Tyr(QSY)
配列番号37: Gln−Gln−Tyr(QQY)
配列番号38: Gln−Tyr−Tyr(QYY)
【0070】
より好ましくは、本発明の生物活性ペプチドにはSer−Asn−Tyr(SNY)すなわち配列番号6、およびTrp−Ser−Leu(WSL)すなわち配列番号2が非限定的に含まれる。
【0071】
本発明のペプチドは直鎖状であっても環状であってもよいが、好ましくは直鎖状である。
【0072】
公知のTNF−α阻害活性を有するペプチド配列である環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu CY)を、in vitro研究でのTNF−α拮抗活性評価に陽性対照として用いた。
【0073】
本発明によるペプチドの表記において、アミノ酸残基Xn間の記号「−」は通常、骨格を成すアミノ酸同士の結合を表わす。従って、記号「−」は普通アミド結合(−C(O)−NH)を表わす。しかし、具体的な実施形態として本明細書に開示されたペプチドのいずれにおいても、場合によっては1個以上のアミド結合を他の結合、好ましくは置換アミド結合、またはアミド結合の同配体に置き換えてもよいと理解されるべきである。
【0074】
(本明細書中に用いた)「TNFアルファ」という語と「TNF−α」という語とは同じもの、すなわちTNFアルファもしくは腫瘍壊死因子αを意味する。
【0075】
本発明のペプチドは、当該技術分野で公知のいずれの方法で合成してもよい。好ましい一法として、本発明の新規なペプチドを、1.00 mmolスケールにおいて自動ペプチド合成機(Applied Biosystems社製433A型ペプチドシンセサイザ)上でFmoc法を用いる固相法により合成した。ペプチドの構築はC末端からN末端へと行なった。Wang樹脂を用いてペプチドを合成した。合成に用いた樹脂は、Novabiochem社から調達したWang樹脂(100〜200メッシュ)であった(置換率1.2mmol/g樹脂)。
【0076】
合成の際、化学成分を用いてペプチドの反応性側鎖を保護した。N−末端アミノ基を9−フルオレニルメトキシカルボニル(Fmoc)基で保護した。ロイシンは側鎖を保護せずに用いた。トリプトファンの側鎖はtert−ブトキシカルボニル(Boc)で保護した。アスパラギンおよびグルタミンの側鎖はトリチル(trt)で保護した。セリンおよびチロシンは、側鎖をt−ブチル(tBu)で保護して用いた。システインはS−アセトアミドメチル(Acm)で保護した。最初のアミノ酸をWang樹脂上に、4−ジメチルアミノピリジン(DMAP)およびジイソプロピルカルボジイミド(DIC)を用いて導入し、その後無水酢酸を用いてキャップ化を行なった。アミノ酸と樹脂とのカップリングに用いられる活性化試薬には、2−(1H−ベンゾトリアゾル−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスファート/1−ヒドロキシベンゾトリアゾール(HBRU/HOBt)、およびジイソプロピルエチルアミン(DIEA)が含まれる。カップリング反応はNMP中で生起させた。ペプチド鎖の構築完了後、ペプチド−樹脂をメタノールで洗浄し、乾燥した。トリフルオロ酢酸、結晶性フェノール、チオアニソール、エタンジチオールおよび脱イオン水から成る切断用混合物での処理を室温で2〜3時間行なうことにより、ペプチドを樹脂から切り離した。冷乾燥エーテルで沈澱させて粗なペプチドを得た。次に、沈澱物を濾別して水に溶解させ、Vertis凍結乾燥機で凍結乾燥した。このようにして得られた粗なペプチドを、Phenomenex C18(250×22.1)逆相カラムを用い、かつ所与の濃度勾配の0.1%TFA含有アセトニトリル/水を用いる分取HPLCによって精製した。溶出画分を、Phenomenex C18(250×4.6)逆相カラムを用いるHPLC分析システム(株式会社島津製作所(日本))で再分析した。アセトニトリルを蒸発させ、画分を凍結乾燥して精製ペプチドを得た。各ペプチドを質量スペクトルによって同定した。
【0077】
ジメチルホルムアミド(DMF)中のヨウ素を用いて、樹脂上のペプチドを環化した。樹脂を自動振盪機で穏やかに振盪しながらDMF中の6倍モル過剰のヨウ素で処理した。反応の進行をHPLCで監視した。反応完了後、樹脂をDMF中の0.4Mアスコルビン酸でクエンチした。次に、樹脂をDMFおよびメタノールで洗浄し、数分間真空乾燥した。最後に環化ペプチドを樹脂から切り離し、RP−HPLCで分析した。粗なペプチドを分取HPLCによって精製し、エレクトロスプレー質量分析法で特性解析を行なった。本発明のペプチドを、高速液体クロマトグラフィー(HPLC)、イオン交換クロマトグラフィー、ゲル電気泳動、アフィニティークロマトグラフィーといった、当該技術分野で公知の方法で精製したが、好ましいのはクロマトグラフィーによる方法であった。より好ましくは、本発明のペプチドは、RP C−18カラムを用いる半分取型Shimatzu HPLCシステムで精製される。実際に用いられる諸条件は、正味荷電、疎水性、および親水性などの要因に依存する。
【0078】
本発明のペプチドは、質量分析法、SDS−PAGE、等電集束法、2D−電気泳動、クロマトグラフィー、ゲル濾過(大きさに基づく分離)、イオン交換(電荷に基づく分離)、配列決定、プロテアーゼ特異性測定、HPLC、X線結晶解析法といった、当該技術分野で公知の方法で分析され得る。好ましい一法として、本発明のペプチドを質量分析法で分析した。
【0079】
ペプチドの純度は、当該技術分野で公知であるいずれの方法でも測定可能である。ペプチドの純度を逆相HPLCで測定した。本発明のペプチドを純度について分析した。本発明のペプチドは比較的高い純度を有する。
【0080】
本発明のペプチドは水や、酢酸緩衝液、リン酸緩衝液、DMSO含有PBSといった生理的緩衝液に可溶である。
【0081】
本発明によるTNF−α阻害剤は、正常で健康な被検者における濃度を上回る濃度で存在するTNF−αに関連する病態もしくは状態の治療に有用である。そのような病態には、慢性関節リウマチ、全身性エリテマトーデス、および乾癬など、急性および慢性の免疫性および自己免疫性病態; 悪液質を含めた敗血症症候群; 急性または慢性細菌感染が原因の循環虚脱及びショック; 細菌感染、ウイルス感染、および真菌感染を含めた急性および慢性の寄生または感染プロセス; クローン病、およびTNF−α分泌腫瘍を伴う悪性病態が非限定的に含まれる。
【0082】
製剤化および投与経路
本発明の化合物は被検者に対してそのまま投与しても、医薬組成物の形態で投与してもよい。本発明のペプチドを含有する医薬組成物は、通常の混合法、溶解法、顆粒化法、糖剤形成法、湿式粉砕法、乳化法、カプセル封入法、捕捉法、または凍結乾燥法によって製造され得る。医薬組成物は、活性なペプチドまたはペプチド類似体を医薬として用い得る製剤へと加工することを容易にする、生理学的に許容されるキャリヤー、稀釈剤、添加剤または補助剤を1種以上用いて通常のように製剤化され得る。適正な製剤化は選択された投与経路に依存する。
【0083】
適当な製薬用キャリヤーは、当該技術分野での標準的な参考文献であるA.Osol編Remington’s Pharmaceutical Sciencesの最新版に記載されている。
【0084】
本発明の医薬組成物は、活性物質が哺乳動物の体内においてその物質の作用部位に到達することを可能にするいずれの手段で投与されてもよい。本発明のペプチドは、当該技術分野で公知であるいずれの投与経路からも投与され得る。様々な経路からの投与には、局所投与、非経口投与、経粘膜投与、経口投与、口腔内投与、直腸内投与、吸入投与、鼻腔内投与、膣内投与、および舌下投与が非限定的に含まれる。
【0085】
局所投与用としては、本発明のペプチドは当該技術分野で良く知られている溶液剤、ゲル剤、軟膏、クリーム剤、懸濁剤などに製剤化され得るが、これらに限定はされない。
【0086】
全身用製剤は、注射、例えば皮下注射、静脈内注射、筋肉内注射、髄腔内注射または腹腔内注射による投与用に設計された製剤、および経皮投与、経粘膜投与、経口投与または肺内投与用に設計された製剤を包含する。
【0087】
注射用としては、本発明のペプチドは水溶液中、好ましくはハンクス液、リンゲル液、または生理食塩水緩衝液といった生理学的に適合する緩衝液中に製剤化され得る。溶液は懸濁化剤、安定剤および/または分散剤などの剤形維持剤(formulatory agents)を含有してもよい。
【0088】
あるいは他の場合には、ペプチドを用時適当な賦形剤、例えば滅菌発熱性物質除去蒸留水で構成される散剤の形態としてもよい。
【0089】
経粘膜投与用製剤には、透過するべき関門に適した浸透剤が用いられる。そのような浸透剤は通常、当該技術分野で公知である。
【0090】
経口投与用製剤は、活性ペプチドを製薬学的に許容される、当該技術分野で公知のキャリヤーと配合することにより容易に製造できる。キャリヤーは本発明のペプチドを、治療される患者の経口摂取用に錠剤、丸剤、糖剤、カプセル剤、液剤、ゲル剤、シロップ剤、スラリー剤、懸濁剤等に製剤化することを可能にする。
所望であれば、固形製剤を標準的な方法で被覆してもよい。
【0091】
例えば懸濁剤、エリキシル剤および溶液剤などの経口液剤に好適なキャリヤー、添加剤または稀釈剤には、水、グリコール、油、アルコール等が含まれる。着香料、防腐剤、着色料などを添加することも可能である。
【0092】
口腔内投与用には、ペプチドは通常のように製剤化されて錠剤、トローチ剤等の形態を取り得る。
【0093】
吸入投与用に本発明により用いられるペプチドは、適当なプロペラント、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素または他の適当な気体を用いた加圧パッケージまたはネブライザーから噴霧エアゾール剤の形態で好適に送出される。加圧エアゾール剤の場合、単位用量は、バルブを設けて所定量を送出することにより決定され得る。化合物と、乳糖やデンプンといった適当な粉末状基剤とを混合した粉末混合物を収容した、例えばゼラチン製のカプセルやカートリッジを吸入器またはインサフレーター用に調製してもよい。
【0094】
ペプチドを、例えばカカオ脂や他のグリセリドなど、通常の坐剤用基剤を含有する坐剤や停留浣腸剤などの直腸内または膣内投与用製剤とすることも可能である。
【0095】
上述の諸製剤に加え、ペプチドをデポー剤に製剤化することも可能である。この持続性製剤は埋め込み(例えば皮下または筋肉内埋め込み)によってか、または筋肉内注射によって投与され得る。従ってペプチドは、例えば適当なポリマーまたは疎水性材料を用いて(例えば許容される油を分散媒とした乳剤に)製剤化しても、イオン交換樹脂を用いて製剤化しても、あるいはまたやや難溶の誘導体、例えばやや難溶の塩として製剤化してもよい。
【0096】
上記以外の医薬送達系を採用することも可能である。本発明のペプチドおよびペプチド類似体の送達に用い得る送達媒体の良く知られた例に、リポソームおよび乳剤が有る。ジメチルスルホキシドなど、幾つかの有機溶媒も用い得るが、通常比較的高い毒性という不都合を伴う。ペプチドの送達には持続放出システムも用い得る。様々な持続放出性材料が確立されており、それらは当業者に良く知られている。治療試薬の化学的性質および生物学的安定性次第では、タンパク質安定化のための付加的な方策を用いてもよい。
【0097】
本発明によれば、製薬学的に許容される塩および誘導体とは、遊離塩基の生物活性を実質的に保有する塩および誘導体のことである。製薬学的に許容される塩および誘導体には、当業者によって製造され得る塩および誘導体が含まれる。
【0098】
本発明のペプチドは通常、所期の目的の達成に有効な量で用いられる。TNF関連疾患の治療または予防のための使用では、本発明のペプチドまたはその医薬組成物は治療有効量で投与または適用される。治療有効量とは、症状の改善もしくは予防、または治療される患者の延命に有効な量のことである。治療有効量は、特に本明細書の詳細な開示に照らせば当業者には容易に決定できる。
【0099】
投与薬剤の用量は、当然ながら、特定薬剤の薬力学特性並びにその投与モードおよび投与経路; 被投与者の年齢、健康状態および体重; 症状の種類および程度、併用治療の種類、治療頻度、並びに所望の効果など、周知の要因に依存して変動する。
【0100】
初期量は、当該技術分野で良く知られた方法を用いてin vivoデータ、例えば動物モデルから推定することもできる。当業者であれば、動物データに基づいてヒトへの投与を最適化することは容易に可能である。
【0101】
投与量および投与間隔は、ペプチドの血漿中濃度を治療効果の維持に十分なものとするべく個別に調整され得る。注射による投与の場合、患者に対する用量は普通約0.1〜50mg/kg/日、好ましくは約0.5〜10mg/kg/日である。典型的には、1〜10mg/kg/日の量を1日につき複数回(6回)に分けて投与するか、1日1回隔日投与するか、または持続放出形態で投与することが所望の結果を得る上で有効である。病的状態の改善に有効な血漿中濃度を達成するべく投与量および投与間隔を個別に調整してもよい。治療上有効な血清中濃度は、投与を毎日複数回に分けて行なうことによって達成され得る。
【0102】
局所投与または選択的摂取の場合、有効な局所的ペプチド濃度は血漿中濃度と関わりが無いかもしれない。当業者であれば、甚だしい実験を行なわなくとも治療上有効な局所投与量を最適化することができる。
【0103】
当然ながら、投与されるペプチドの量は、治療される被検者、被検者の体重、疾病の重篤度、投与方法、および処方医師の判断に依存する。
【0104】
治療は症状が検出される間、さらには症状が検出されない場合にも断続的に反復され得る。治療は単独で行なっても、他の薬物を併用して行なってもよい。
【0105】
本出願明細書中各所で様々な刊行物および特許が引用され、特許はその番号、他の刊行物は著者名および発行年で特定されている。完全な引用文献リストを後段に示す。その結果、本発明の属する分野の技術水準をより十全に説明するべく、引用された刊行物および特許の開示はその全体が援用により本明細書に含まれる。
【0106】
本発明をここに詳述したが、用いられた用語は説明のためのものであり、限定のためのものではないと理解されるべきである。
【0107】
本発明の様々な改良および変形が上記教示に照らして可能であることは明らかである。従って、添付の請求項の範囲内であれば、本発明は具体的に記載された以外の形態でも実施され得ると理解されるべきである。
【0108】
方法概略および結果
【実施例1】
【0109】
配列番号6のペプチドの調製方法
実施例1(a)―配列番号6のペプチドの合成: ペプチド固相合成法を用い、自動または半自動ペプチド合成装置でFmoc化学に則っり配列番号6のペプチドを合成した。Wang樹脂を用いて合成を行なった。樹脂の置換率は0.6mmol/gから1.2mmol/gまでであった。チロシンおよびセリンの側鎖をt−ブチル基で保護し、アスパラギン側鎖をトリチル基で保護した。DMF中のDIC/HOBt(3〜5eq)およびDMAP(1〜2eq)を室温で約5〜8時間用いて、最初のアミノ酸チロシンをWang樹脂上に導入した。DMF中の20%ピペリジンを20〜30分間用い、N末端を保護するFmoc基を除去して脱保護を行なった。1mmolの反応に15〜20mlのDMFを用いた。脱保護したチロシンにFmoc−Asp(tBu)−OH(3〜5eq)を、DMF中のDIC/HOBt(3〜5eq)を室温で約3〜4時間用いることによりカップリングさせた。反応の完了をKaiser試験で監視した。Kaiser試験では、青色の消失によってカップリング反応の完了が示される。反応完了後、反応混合物をDMFで3回洗浄した。Fmoc基を上記のように除去して脱保護を行ない、その後反応混合物をDMFで3回洗浄した。上記と同様の操作で、Fmoc−Ser(tBu)−OHをアスパラギンにカップリングさせ、脱保護した。固相上に構築されたペプチドの平均収率は90〜95%であった。
【0110】
樹脂からのペプチド切断:
TFA:フェノール:TIS:DIT:水を82.5:5.0:2.5:5.0:5.0の比率で含有する試薬混合物(150ml)を用いてペプチドを樹脂から切り離した。不断の攪拌下、配列番号6のペプチドを担持した樹脂を切断試薬中に氷冷環境下で15分間維持し、次いでやはり不断の攪拌下に室温で2時間維持した。反応完了後、混合物を焼結漏斗で濾過し、濾液に冷ジエチルエーテルを添加することによってペプチドを沈澱させた。
沈澱したペプチドを焼結漏斗で濾過し、乾燥し、水に溶解させ、最後に凍結乾燥して粗なペプチドを得た。粗ペプチド収率は85〜90%であった。
【0111】
ペプチドの精製:
粗なペプチドを、溶離剤としてアセトニトリルおよび水を用いる分析用HPLCで分析した。C−18分取カラム(250×50mm、10μ)を用いるHPLC LC−8A装置(島津製作所)において粗BRC605−1の精製を、流速80〜120ml/分、検出波長210nmで行なうアセトニトリル(0.1%TFA)−水(0.1%TFA)混合物での定組成溶離によって実施した。精製画分を分析用HPLCで分析して所望の画分をプールし、凍結乾燥し、特性解析を行なった。この方法による総収率は70%を越えることが判明した。
【0112】
ペプチドの特性解析:
MS分析―配列番号6のペプチドの全バッチについて、質量スペクトル分析による特性解析を行なった。各バッチの分子質量は383.5となる(陽性モード)。
ペプチド配列決定―配列番号6のペプチドの全バッチについて、ペプチド配列決定による特性解析を行なった。各バッチのペプチド配列は実際の配列を与える。
【実施例2】
【0113】
L929細胞のTNF−α媒介細胞毒性のペプチド配列による阻害
マウス線維芽細胞系L929を用いて、本発明のペプチドをTNF−α媒介細胞毒性の阻害について分析した。L929細胞(ATCC)にTNF−αを添加すると細胞毒性が誘発され、そのことは、生細胞をMTT(3−(4,5−ジメチルチアゾル−2−イル)−2,5−ジフェニルテトラゾリウムブロミド、テトラゾール系)やクリスタルバイオレットといった生体用色素で染色し、その後色素をメタノールで抽出することによって推定できる。抽出した色素の吸光度は595nmで測定し得る(Hansen et al., Journal of Immunological Methods, 119: 203−210, 1989)。アッセイは下記のように行なった。
【0114】
L929細胞(10%FCSを補充したダルベッコ変法イーグル培地中に維持)をマイクロタイタープレートに2×105細胞/mlの密度で播種し、濃度1μg/mlのアクチノマイシンD(ACT−D)の存在下、37℃および5%CO2において2時間インキュベートした。75μMのペプチド溶液と共に37℃でプレインキュベートしたTNF−α(100pg/ml)をL929細胞に添加し、37℃および5%CO2において一晩インキュベートした。
【0115】
環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)ペプチドを、TNF関連細胞毒性の阻害を推定するための陽性対照として用いた。陽性対照ペプチド溶液(75μM)と共に37℃でプレインキュベートしたTNF−α(100pg/ml)を別のウェル内のL929細胞に添加し、37℃および5%CO2において一晩インキュベートした。
【0116】
試験プレートおよび陽性対照プレートの生細胞を100μl/ウェルの0.05%クリスタルバイオレットで染色し、室温で15分間インキュベートし、PBSで洗浄し、かつ室温で一晩乾燥した。クリスタルバイオレットを100μl/ウェルのメタノールで細胞から抽出し、抽出したクリスタルバイオレット色素の吸光度を595nmで測定した。細胞毒性の阻害パーセンテージを式:
[(ODtest−ODTNF)/(ODctrl−ODTNF)]×100
によって算出した。
TNF−α誘発細胞毒性阻害の評価は、本発明のペプチドを75μMの濃度で用いて行なった。
【0117】
実施例2(a):
配列番号1〜10のペプチドをTNF−α誘発細胞毒性の阻害について分析した。配列番号1、2、6および8のペプチドはTNF−α誘発細胞毒性を陽性対照よりも強く阻害した。配列番号1、2、6および8のペプチドのTNF−α誘発細胞毒性阻害率(各々二重に行なわれた複数回の実験に基づき算出された阻害率(%)の平均値±SE)はそれぞれ65±9.2%、48±6%、62±6.2%、および51±2.3%であった。
【0118】
実施例2(b)―配列番号2および6のペプチドの直鎖状および環状形態の、TNF−α誘発細胞毒性を阻害する能力の比較:
配列番号2および6の直鎖状ペプチド並びに配列番号9および10の環状ペプチドを分析して、TNF−α誘発細胞毒性を阻害する能力を比較した(分析方法は上述したとおり)。配列番号9および10の環状ペプチドより配列番号2および6の直鎖状ペプチドの方が能力の高いTNF−α誘発細胞毒性抑制物質であることが判明した。図2に示したように、配列番号9および10の環状ペプチドによる阻害はごく低率であった。
【0119】
陽性対照ペプチドすなわち環状WSQYL(cy Trp−Ser−Gln−Tyr−Leu)についてもその直鎖状および環状形態を分析し、TNF−α誘発細胞毒性を阻害する能力を比較した。興味深いことに逆のパターンが観察され、すなわち、陽性対照ペプチドの環状形態は比較的高いTNF−α誘発細胞毒性阻害率(60±10%)を示したが、陽性対照ペプチドの直鎖状形態はTNF−α誘発細胞毒性の阻害を有効に惹起しなかった(図2)。
【0120】
配列番号2および6の直鎖状ペプチドは、公知のTNF−α阻害ペプチドである陽性対照の環状形態と同等の高い阻害能力を有することが判明した。
【0121】
実施例2(c)―配列番号6のペプチドおよびエタネルセプト(「エンブレル」として市販されている公知のTNF−α阻害剤)の、TNF−α誘発細胞毒性を阻害する能力の比較:
配列番号6のペプチドおよびエタネルセプトを分析して、TNF−α誘発細胞毒性を阻害する能力を比較した(分析方法は実施例2において上述したとおり)。配列番号6のペプチドはエタネルセプトに匹敵するTNF−α誘発細胞毒性阻害能力を有することが判明した(図3)。
【0122】
ペプチドの結合に関する研究
ペプチドとTNF−α、またはU937細胞(TNF受容体発現ヒト白血病細胞系)上のTNF−R1との結合を、フローサイトメトリーアッセイ法を用いて調べた。ペプチドとTNF−αとの結合はTNF−αとTNF−R1との相互作用を妨げ、このことはフローサイトメトリーによって分析することができる。ペプチドが直接TNF−R1に結合することによってTNF−R1陽性細胞のパーセンテージが低下することもフローサイトメトリーにより分析可能である。
【実施例3】
【0123】
ペプチドによるTNF−αと細胞上の受容体TNF−R1との結合の阻害
TNF−αはU937細胞(ATCC)上のコグネイト受容体TNF−R1に結合し、その結果TNF−R1に関して陽性である細胞のパーセンテージは低下する。TNF−α結合ペプチドはTNF−αに結合し、TNF−αとTNF−RIとの相互作用を妨害する。TNF−R1に関して陽性である細胞のパーセンテージは、蛍光色素結合抗TNF−RI抗体での染色によって定量できる。
【0124】
配列番号2および6のペプチドを、TNF−αとU937細胞上の受容体TNF−R1との結合の阻害について分析した。配列番号2または6のペプチドによるTNF−αとU937細胞上のTNF−RIとの結合の阻害を蛍光活性化セルソーター(Becton Dickinson(米国)のFACS、FACSCalibur)を用いて推定した。
【0125】
0.5%BSA(ウシ血清アルブミン)および0.05%NaN3(結合緩衝液)を含有するPBS中にU937細胞(10%FCSを補充したRPMI−1640培地(Sigma−Aldrich(米国))中に維持)を、緩衝液100μl当たり1×105細胞の密度で懸濁させた。上記ペプチド(配列番号2および6)をそれぞれPBSに加えた2種のペプチド溶液(50μl)をTNF−α(5ng)と共に別々の試験管に入れ、37℃で1時間プレインキュベートした。次いで、ペプチドとTNF−αとの複合体をU937細胞に添加し、4℃で1時間インキュベートした。別の試験管にU937細胞(1×105個)をTNF−α(5ng)と共に入れ、4℃で1時間インキュベートした(並列実験)。その後、細胞を結合緩衝液で洗浄し、この細胞に5μl(1mg/ml)のヒト抗マウスTNF受容体抗体(クローン番号HTR−9; Novus Biologicals)を4℃で1時間添加した。次に、細胞を結合緩衝液で洗浄し、暗中4℃で30分間、10μl(10μg/ml)のフルオレセイン結合ヤギ抗マウスIgG二次抗体(GIBCO BRL, Gaithersburg, MD)で染色した。結合緩衝液で2回洗浄後、フローサイトメーターFACSCalibur(Becton Dickinson)を用いて細胞を分析した。ゲートを生細胞集団に設定し、ペプチドによるTNF−α/細胞結合の阻害の程度をTNF−RI発現陽性細胞のパーセンテージに基づき推定した。
【0126】
未処理のU937細胞は約78±1%がTNF−R1発現に関して陽性であることが判明した。U937細胞をTNF−αと共にプレインキュベートすると、TNF−R1発現陽性細胞は32±10%に減少した(図4aおよび4b)。U937細胞を配列番号2のペプチドとTNF−αとの複合体と共にプレインキュベートすると、TNF−R1発現陽性細胞の比率は37±8.5%となった。U937細胞を配列番号6のペプチドとTNF−αとの複合体と共にプレインキュベートすると、TNF−R1発現陽性細胞の比率は57±8%となった(図4aおよび4b)。
【0127】
上記の結果から、配列番号2および6のペプチドがTNF−αと結合してTNF−αとTNF−R1との結合を防止し、受容体TNF−R1に関して陽性である細胞の減少をTNF−αが未処理である場合と比較して抑制することは明らかである。
【実施例4】
【0128】
フローサイトメトリーによるペプチドとU937細胞上のTNF−R1との結合の評価
U937細胞を用いてペプチドとTNF−R1との結合を定量した。U937細胞に添加されたTNF−αはU937細胞上のコグネイト受容体TNF−R1に結合し、その結果TNF−R1陽性細胞のパーセンテージは低下する。本発明によるTNF−α阻害ペプチドはTNF−R1に結合し、U937細胞を該ペプチドと共にインキュベートするとTNF−R1陽性細胞が減少する。
【0129】
配列番号6のペプチドをU937細胞上のTNF−R1への結合について、フローサイトメトリーを用いて分析した。U937細胞を配列番号6のペプチドと共にインキュベートして、配列番号6のペプチドが直接受容体TNF−R1に結合することを実証した。配列番号6のペプチド(250μM)およびTNF−α(10ng)それぞれと共にインキュベートしたU937細胞を2回洗浄し、これを4℃で1時間、5μl(1mg/ml)のヒト抗マウスTNF受容体抗体(クローン番号HTR−9; Novus Biologicals)で染色した。次に、細胞を結合緩衝液で洗浄し、暗中4℃で30分間、10μl(10μg/ml)のフルオレセイン結合ヤギ抗マウスIgG二次抗体(GIBCO BRL, Gaithersburg, MD)で染色した。結合緩衝液で2回洗浄後、フローサイトメーターFACSCalibur(Becton Dickinson)を用いて細胞を分析した。ゲートを生細胞集団に設定し、TNF−R1への結合の程度をTNF−RI発現陽性細胞のパーセンテージに基づき推定した。
【0130】
83%のU937細胞上にTNF−R1が発現していることが判明した。U937細胞を組み換えTNF−α(10ng)と共にインキュベートすると、TNF−R1陽性細胞のパーセンテージは31.6%に低下した。U937細胞を配列番号6のペプチドと共にインキュベートすると、TNF−R1陽性細胞のパーセンテージは32%に低下した(図5)。TNF−R1発現に関して陽性である細胞のパーセンテージの低下が配列番号6と共にインキュベートしたあととTNF−αと共にインキュベートしたあととで同等であることが判明したが、このことはペプチドがTNF−R1に結合することを明らかに示している。
【実施例5】
【0131】
慢性関節リウマチのマウスモデルにおけるペプチドのin vivo効力
実施例5(a)―関節炎マウスモデルの開発: Lalruに所在するパナセア バイオテックの動物飼育施設から入手した雄のC57BL/6マウスを用いて、慢性関節リウマチのマウスモデルを開発した。完全フロイントアジュバント(CFA)(Sigma)中に乳濁させた150μgのトリタイプIIコラーゲン(Sigma)の皮内注射によってマウスを免疫した。一次免疫後第17日に、不完全フロイントアジュバントに加えた100μgのトリタイプIIコラーゲンでの追加免疫を動物に対して行なった(Ethan M.Shevach, Curr. Prot. Immunol. 2002: 15.0.1−15.0.6; Inglis, J. et al., Nature Protocols 4: 612−618, 2008)。対照(健康な雄のC57BL/6マウス)と比較しての足の太さ(paw thickness)の増加および抗コラーゲンIgG抗体の存在を、関節炎の発症を評価するパラメーターとした。動物の足の太さはデジタルノギスで測定し、抗コラーゲン抗体は免疫後第35日にマウス血清中で測定した。
【0132】
実施例5(b)―マウス血清中抗コラーゲンIgG濃度:
トリタイプIIコラーゲン(Sigma)をPBSに1μg/mlの量で加えたものをELISAプレートに塗布し、4℃で一晩インキュベートした。PBS−0.05%Tween 20(PBS−T)で3回洗浄後、コラーゲン、媒体(CFA)、およびPBS(対照)を注射したマウスから得た血清を別々のウェルに稀釈比1:100で添加した。プレートを室温で2時間インキュベートした。PBS−Tで5回洗浄後、ウサギ抗マウスIgG HRP(Bethyl Laboratories)をウェルに稀釈比1:10000で添加し、室温で45分間インキュベートした。プレートをPBS−Tで洗浄後、基質のOPD(オルトフェニルジアミン)を添加し、発色を観察した。発色を2N HClで停止させた。490nmでの吸光度を記録した(Ethan M.Shevach, Curr. Prot. Immunol. 2002: 15.0.1−15.0.6)。コラーゲン免疫マウスが示した抗コラーゲンIgGの血清中濃度は媒体(CFA)群および対照群(PBS注射)と比較して高かった(図6)。
【0133】
実施例5(c)―マウスモデルにおける用量および投与スケジュールの最適化:
コラーゲン誘発関節炎のマウスモデル(上述の方法で用意)において、配列番号6のペプチドの最適な用量および投与スケジュールを決定した。関節炎マウスを足の太さに従ってランダム化し、各々4匹または5匹から成る複数の群に分けて、配列番号6のペプチドの静脈内注射を行なった。各群に適用した用量および投与スケジュールを表3に示す。
【0134】
【表4】
【0135】
用量7.5mg/kgで4週間毎週1回投与した配列番号6のペプチドは足の太さを、対照動物で観察されたものと比較して僅かに減少させた。また、5mg/kgの用量、および1週間に3回、その後3週間毎週3回投与するスケジュールで投与した配列番号6のペプチドが足の太さを対照動物のものに比べて有意に減少させることが観察された。従って、用量5mg/kgで1週間に3回、その後3週間毎週3回投与する投与計画が最適なものとして選択された(図7)。
【0136】
実施例5(d)―マウスモデルにおける配列番号6および2のペプチド並びにエタネルセプトの効力の比較:
コラーゲン誘発関節炎のマウスモデル(上述の方法で用意)を用いて、配列番号6および2のペプチドの効力をエタネルセプト(商品名「エンブレル」の下に市販されている認可されたTNF−α阻害剤)のものと比較した。関節炎マウスに配列番号6および2のペプチド並びにエタネルセプトを用量5mg/kgで第1週に3回、その後3週間毎週1回静脈内投与した。
【0137】
配列番号6のペプチドおよびエタネルセプトの投与は足の太さを、正常な対照動物(対照: 健康な雄のC57BL/6マウス)と同等の太さにまで有意に減少させることが観察された(p<0.01)(図8a)。配列番号2のペプチドの投与も足の太さを有意に減少させた(p<0.05)(図8a)。これらの結果は、配列番号2および6のペプチドがエタネルセプトに匹敵する効力を有することを明らかに示している。
【0138】
実施例5(e)―配列番号2および6のペプチド並びにエタネルセプトで処理した動物において処理後のIgG1/IgG2a比を測定する比較研究:
コラーゲン誘発関節炎の発症には、Th1応答もしくは炎症誘発応答の増大が伴う。IgG1/IgG2a濃度比を測定して、本発明のペプチドおよびエタネルセプトでの処理がマウスモデルにおいて臨床疾患をTh1応答の低下により改善したかどうかを判断した。小さいIgG1/IgG2a比ほどTh1応答もしくは炎症誘発応答のダウンレギュレーションを示唆することになろう。
【0139】
関節炎マウスに配列番号2および6のペプチド、エタネルセプト、並びにPBSを用量5mg/kgで第1週に3回、その後3週間毎週1回静脈内投与した。配列番号6のペプチドおよびエタネルセプトを投与した場合、処理後のIgG1/IgG2a比は未処理の関節炎動物(PBS処理動物を未処理動物と見なす)と比較して小さくなった(図8b)。未処理の関節炎動物(PBS処理動物を未処理動物と見なす)のIgG1/IgG2a比は進行中の炎症に起因して大きかった。このことは、配列番号6のペプチドでの処理が関節炎マウスにおいてTh1応答を低下させ、その結果IgG1/IgG2a比を対照動物(対照: 健康な雄のC57BL/6マウス)のIgG1/IgG2a比に匹敵するほど小さくして症状の寛解をもたらすことを示唆している。
【実施例6】
【0140】
アジュバント誘発関節炎のラットモデルにおける配列番号6のペプチドのin vivo効力
配列番号6のペプチドのin vivo効力を「アジュバント」誘発関節炎のラットモデルにおいて評価した。このモデルは抗炎症性薬物の開発及び試験で広く用いられている。ラットにおけるアジュバント関節炎(AA)はヒトの炎症性関節炎の諸特徴を模倣する(Pearson et al., Proc. Soc. Exp. Biol. Med. 112: 95−110, 1956)。150μgの完全フロイントアジュバントで皮内免疫を行ない、その後第7日に不完全フロイントアジュバントで追加免疫を行なうことによって雄のWistarラットにAAを誘発した。動物の関節炎を臨床的スコアリングと、デジタルノギスを用いた足(paw)および関節の太さの測定とにより評価した。臨床スコアは以下の基準に従って付与した。
0=紅斑も腫脹も無し
1=後肢または前肢の1本の指に僅かな紅斑または腫脹
2=後肢または前肢の2本以上の指に紅斑および腫脹
3=足関節または手関節に紅斑および腫脹
4=後肢または前肢の指および足関節または手関節の全体に紅斑および腫脹が認められ、かつ足関節または手関節が屈曲不能
【0141】
関節炎ラットに配列番号6のペプチドを2.5mg/kgの用量で、第1週には6日間毎日1回投与し、第2週には1日おきに3回投与した。別の関節炎動物群にはエタネルセプト(Et)を同様にして投与した。配列番号6のペプチドを投与した動物においても、エタネルセプトを投与した動物においても、未処理の関節炎ラット(PBSで処理したラットを未処理の関節炎ラットと見なす)に比べて足の太さが減少した(図9a)。配列番号6のペプチドまたはエタネルセプトで処理した場合には臨床スコアの有意な低下も認められた(p<0.05)(図9bおよび10)。総体的に、得られた結果は、配列番号6のペプチドが関節炎ラットにおける足の太さの減少や臨床スコアの低下に関してエタネルセプトに匹敵する効力を有することを示唆している(図9aおよび9b)。
【0142】
本明細書および添付の請求項中、「a」、「an」、および「the」を付した単数形は、文脈上明らかに別様に解されないかぎり複数形の意味を包含することに留意されたい。
【特許請求の範囲】
【請求項1】
式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸であり、それらのアミノ酸はTrp、Ser、Gln、Leu、TyrおよびAsnから成る群から選択され、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体。
【請求項2】
X1およびX2がそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuから成る群から選択された1個または2個のアミノ酸であり、X3は単一のアミノ酸残基で、Trp、Ser、Gln、Asn、TyrおよびLeuから成る群から選択される、請求項1に記載のTNF−α阻害ペプチド。
【請求項3】
X1がTrp、SerおよびGlnを含む群から選択された1個または2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された1個または2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基である、請求項1に記載のTNF−α阻害ペプチド。
【請求項4】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである、請求項1に記載のTNF−α阻害ペプチド。
【請求項5】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである、請求項1に記載のTNF−α阻害ペプチド。
【請求項6】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである、請求項1に記載のTNF−α阻害ペプチド。
【請求項7】
X1、X2およびX3がそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である、請求項1に記載のTNF−α阻害ペプチド。
【請求項8】
配列番号1〜38から成る群から選択される、請求項1に記載のTNF−α阻害ペプチド。
【請求項9】
直鎖状ペプチドまたは環状ペプチドである、請求項1に記載のTNF−α阻害ペプチド。
【請求項10】
請求項1に記載のTNF−α阻害ペプチドを調製する方法であって、前記ペプチドを固相合成法で調製する方法。
【請求項11】
請求項1または8に記載のTNF−α阻害ペプチド、および製薬学的に許容されるキャリヤーを含有する医薬組成物。
【請求項12】
TNF−α関連疾病状態を治療する方法であって、請求項1または8に記載のTNF−α阻害ペプチドを投与することを含む方法。
【請求項13】
局所投与、非経口投与、経粘膜投与、経口投与、口腔内投与、直腸内投与、吸入投与、鼻腔内投与、直腸内投与、膣内投与、または舌下投与可能である、請求項11に記載の医薬組成物。
【請求項1】
式:
X1−X2−X3
〔式中X1およびX2はそれぞれ独立に0〜2個のアミノ酸であり、X3は単一のアミノ酸であり、それらのアミノ酸はTrp、Ser、Gln、Leu、TyrおよびAsnから成る群から選択され、X1、X2およびX3のアミノ酸は合計で2個以上である〕を有するTNF−α阻害ペプチドまたはその製薬学的に許容される塩および誘導体。
【請求項2】
X1およびX2がそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuから成る群から選択された1個または2個のアミノ酸であり、X3は単一のアミノ酸残基で、Trp、Ser、Gln、Asn、TyrおよびLeuから成る群から選択される、請求項1に記載のTNF−α阻害ペプチド。
【請求項3】
X1がTrp、SerおよびGlnを含む群から選択された1個または2個のアミノ酸であり、X2はSer、Gln、AsnおよびTyrを含む群から選択された1個または2個のアミノ酸であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基である、請求項1に記載のTNF−α阻害ペプチド。
【請求項4】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がTrpである場合X2はSerまたはGlnである、請求項1に記載のTNF−α阻害ペプチド。
【請求項5】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がSerである場合X2はAsnまたはGlnである、請求項1に記載のTNF−α阻害ペプチド。
【請求項6】
X1がTrp、SerおよびGlnを含む群から選択されたアミノ酸残基であり、X2はSer、Gln、AsnおよびTyrを含む群から選択されたアミノ酸残基であり、X3はGln、LeuおよびTyrを含む群から選択されたアミノ酸残基であり、ただしX1がGlnである場合X2はAsnまたはTyrである、請求項1に記載のTNF−α阻害ペプチド。
【請求項7】
X1、X2およびX3がそれぞれ独立に、Trp、Ser、Gln、Asn、TyrおよびLeuを含む群から選択された単一のアミノ酸残基である、請求項1に記載のTNF−α阻害ペプチド。
【請求項8】
配列番号1〜38から成る群から選択される、請求項1に記載のTNF−α阻害ペプチド。
【請求項9】
直鎖状ペプチドまたは環状ペプチドである、請求項1に記載のTNF−α阻害ペプチド。
【請求項10】
請求項1に記載のTNF−α阻害ペプチドを調製する方法であって、前記ペプチドを固相合成法で調製する方法。
【請求項11】
請求項1または8に記載のTNF−α阻害ペプチド、および製薬学的に許容されるキャリヤーを含有する医薬組成物。
【請求項12】
TNF−α関連疾病状態を治療する方法であって、請求項1または8に記載のTNF−α阻害ペプチドを投与することを含む方法。
【請求項13】
局所投与、非経口投与、経粘膜投与、経口投与、口腔内投与、直腸内投与、吸入投与、鼻腔内投与、直腸内投与、膣内投与、または舌下投与可能である、請求項11に記載の医薬組成物。
【図1】

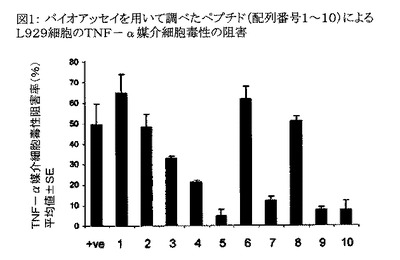
【図2】
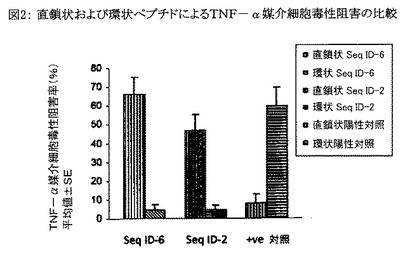
【図3】
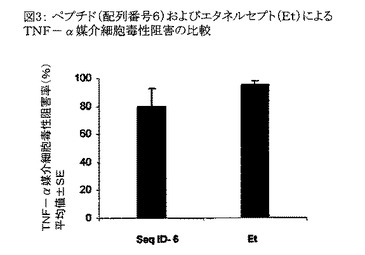
【図4】
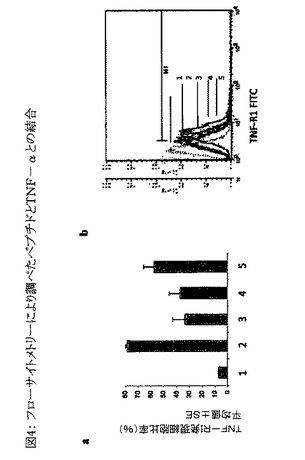
【図5】
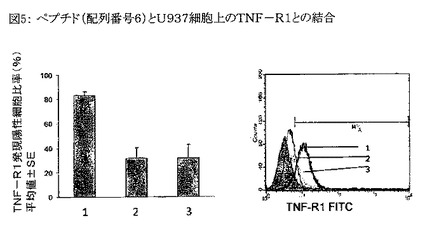
【図6】
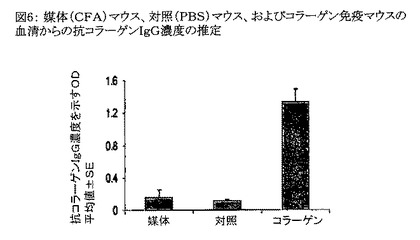
【図8a】
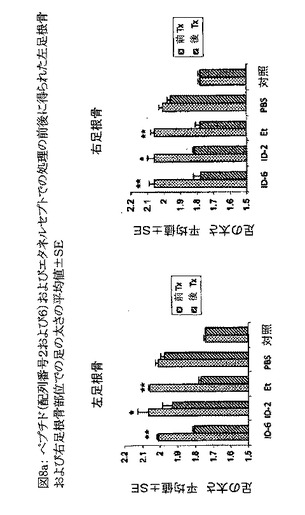
【図8b】
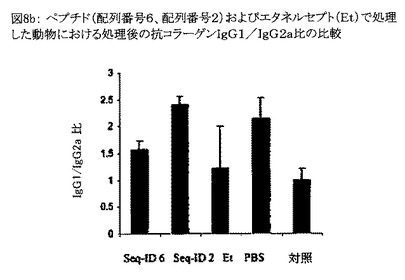
【図9a】
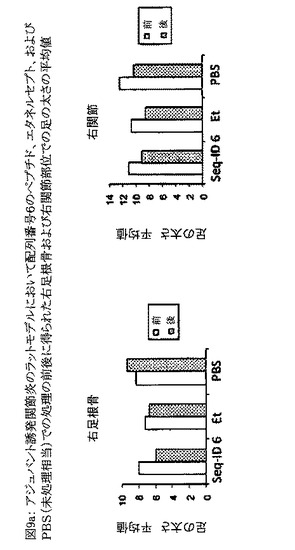
【図9b】
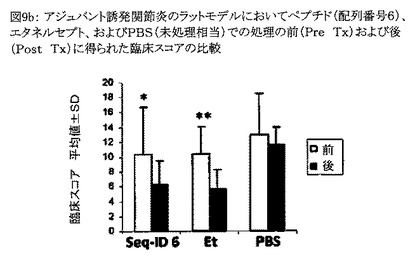
【図7】
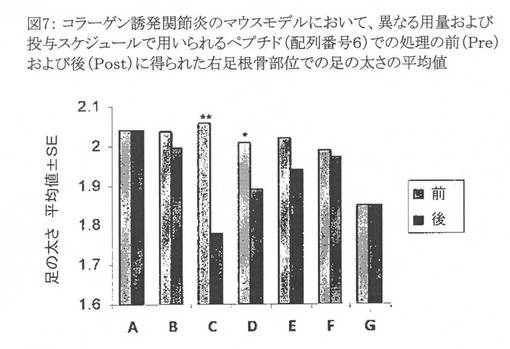
【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図8a】
【図8b】
【図9a】
【図9b】
【図7】
【図10】
【公表番号】特表2012−509312(P2012−509312A)
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願番号】特願2011−537012(P2011−537012)
【出願日】平成21年11月5日(2009.11.5)
【国際出願番号】PCT/IN2009/000626
【国際公開番号】WO2010/058419
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(500445631)パナセア バイオテック リミテッド (29)
【Fターム(参考)】
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願日】平成21年11月5日(2009.11.5)
【国際出願番号】PCT/IN2009/000626
【国際公開番号】WO2010/058419
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(500445631)パナセア バイオテック リミテッド (29)
【Fターム(参考)】
[ Back to top ]