
腫瘍壊死因子アルファ(TNF−α)合成および分泌を調節するための、T細胞におけるKv1.1電位依存性カリウムチャンネルのモデュレーション、およびTNF−αの有害な高または低レベルを介しての人の疾患または傷害の治療
T細胞の電位依存性カリウムチャンネルKv1.1の遮断により、強健で、唯一的にTNF−αの産生が引き起こされ、それ故、T細胞の電位依存性カリウムチャンネルKv1.1の遮断を癌の根絶、感染病原体のより優れた根絶、特定の分子または細胞に対する血管および血液脳関門の透過性の増加、ならびに神経細胞の特色、再生の機能および発達の改善のために使用できる。T細胞の電位依存性カリウムチャンネルKv1.1の遮断は、強健で、唯一的なTNF−αの産生を引き起こす。同様に、遮断されたKv1.1チャンネルの脱遮断またはKv1.1チャンネルの開口は、T細胞のTNF−αの過剰量の産生および分泌を阻止し、従って関節リウマチのような病気およびKv1.1チャンネルの損傷した機能および/または病理学上の遮断に伴う神経疾患の治療に有用であろう。該神経疾患にはKv1抗体と関連したPNH;Kv1抗体に伴う脳炎;および突発性運動失調1型(EA−1)が挙げられ、それらの全てにおいて、Kv1.1チャンネルを遮断されたT細胞は、過剰のTNF−αを分泌し、それにより病態の一因になっている可能性がある。Kv1.1チャンネルの遮断は、インビボあるいはエクスビボにて、デンドロトキシン−KまたはKv1.1チャンネルに対する選択的なモノクローン抗体のような選択的Kv1.1チャンネル遮断分子と接触させることによって達成させてもよい。Kv1.1遮断の阻止は、Kv1.1開口剤、またはKv1.1チャンネルの閉鎖を阻止するような分子によって達成するであろう。

【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
30年前に分離された腫瘍壊死因子アルファ(TNF−α)は、アポトーシスおよび細胞生存、ならびに炎症および免疫において主要な役割を果たす多機能なサイトカインである。その抗腫瘍性から名付けられたが、TNFは広範囲の他の疾患に関わるとされる。現在癌におけるTNFの使用は、局所的に進行した軟組織肉腫、転移メラノーマおよび組織学的な他の切除不可能な腫瘍の局所的な治療において患肢の切断を避けるためのものである。TNF−αは、分離患肢潅流の設定において、細胞増殖抑制剤と相乗的に作用することが示された。TNF−αとTNFの受容体1および2(TNFR−1、TNFR−2)の相互作用は、いくつかのシグナル伝達経路を活性化させ、TNF−αの多様な機能を発揮させる。TNFR1のシグナル伝達分子は、非常に良く解明されているが、シグナル伝達の制御については不明である。これらの分子についての洞察のほかに、過去10年間の研究室における実験は、腫瘍治療期間中のTNF−αの作用に光を当てた。出血性壊死を引き起こす、赤血球およびリンパ球の血管外遊出の他に、TNF−αは、血管内壁の透過性増大および破壊を誘導することによって、腫瘍関連脈管構造(TAV)を標的とする。この結果、腫瘍内での細胞増殖抑制剤の選択的蓄積の即効性、および腫瘍の脈管構造の破壊の遅発性効果が起こる。最近公表された総説(van Horssen等(2006))は、TNF−αについて分子から臨床までを記述し、TNFR−1シグナル伝達の分子的洞察およびTNF−αの抗腫瘍活性の細胞機構から始まり、臨床反応で終わる、癌におけるTNF−αの使用の概観を提供している。更に加えて、TNF−αの活性をモデュレーションしている可能性のある因子についても議論している。
【0002】
TNF−αが、1975年バクテリアエンドトキシンで処理済のマウスの血清から「Coleyの毒素」の活性成分として分離され、マウスの腫瘍に出血性壊死を引き起こすことが示された。ニューヨークの外科医であったWilliam Coleyが、彼の調製した細菌濾液(「Coleyの混合毒素」)でいく人かの癌患者を治療した際に高熱と腫瘍の壊死を観察したのは、殆ど1世紀前であった。その分離の10年後、TNF−αは又「カケクチン」およびT細胞分化因子と位置づけられた。1984年ヒトTNF−α遺伝子がクローン化され、一連の臨床実験が設定され、分離潅流の設定における患肢を脅かすSTSの治療のための、欧州医薬品審査庁からの認可を得た(上記に関してはvan Horssen等(2006)、およびそこに引用した論文を参照)。
【0003】
赤血球を除いて、実際ほとんどすべての細胞型に発現している2つの高親和性受容体、TNFR1(p55)およびTNFR2(p75)を活性化することにより、TNF−αは、後天的免疫性および自然免疫性、ウィルスおよび細菌のような外来の侵入物と闘うために必要な炎症の誘導、腫瘍の根絶、オリゴデンドロサイトの再生と増殖の促進、神経の再ミエリン化その他多くのものを含め、めまぐるしく変化する病気において主要な役割を果たしている。
【0004】
T細胞(Tリンパ球とも呼ばれる)は、TNF−αの主要な源である。T細胞からTNF−αの分泌を引き起こすことが知られている主要な刺激は、T細胞受容体(TCR)の関与であって、自己主要組織適合性複合体分子の構成において、抗原提示細胞によってT細胞に提示される抗原と、T細胞の出会いの時に起こるのと類似している。該「古典的な」TCR活性化により、T細胞は強健なしかし非選択的なサイトカイン分泌(すなわちこれらの細胞によって分泌され得る全てのサイトカインの同時分泌)を引き起こす。
【0005】
TNF−αは、ヒトの生理および病理において二重の役割を果たす:一方ではTNF−αは、疾患と闘い治療するために決定的に必要とされる;他方過剰のTNF−αは、疾患および組織破壊を引き起こすため、有害である。
【0006】
一般的に、先天的免疫反応の活性化におけるTNF−αの主要機能は、種々の型の外来侵入物から体を保護するために、および癌細胞を根絶するために、有益で重要であると一般的に考えられている。TNF−αは又、種々の分子に対する血管および血液脳関門(BBB)の透過性を大幅に増加させ、および組織への移動と侵入に必要とされる、白血球のローリングと付着を増大させる。したがって、TNF−αの欠如または不足は、広範囲で病的な結果を招くことは明らかであり、その結果の中には、免疫能および、感染性生物と対抗し、癌等を根絶する能力の低下が含まれる。
【0007】
TNF−αが癌細胞を殺す能力に基づき、TNF−αは、分離患肢潅流(ILP)の現場において、軟組織肉腫(STS)、種々の組織学的切除不可能な腫瘍、および患肢に限定された移行転移メラノーマに対する癌治療に現在用いられている(van Horssen等(2006)およびそこに引用した論文)。
【0008】
van Horssen等(2006)の表1に、高悪性度(Wallach等、1999;Mayan、2002)STSの治療のため、TNF−αのILPでの適用に対し、1998年のEMEAによりTNF−αが承認されることになった、欧州での多施設臨床試験の概要が示されている。これらの多施設臨床試験においては、76%の全奏功率および82%の患肢救済中央値を観察した。さらにこの表には、多施設における経験を確認する、STSの最大の単一施設での研究を載せている。衝撃的に一貫した、中央値76%(レンジ、58%〜91%)主要な奏功率が、中央値84%(レンジ、58%〜97%)の患肢救済率を伴って観察された。欧州においては、患肢救済のための国内照会形の35の癌センターにおいて、TNF−αに基づくILPが現在行われている。移行転移メラノーマに対するメルファラン単独ILPは、文献報告によれば、約50%の完全奏功(CR)率および80%の全奏功率を引き起こすと報告されている(van Horssen等(2006))およびそれに引用している論文を参照)。この設定におけるTNF−αの導入は、CR率を70%−90%に、全奏功率を95%〜100%に増加させると報告された。これらの結果は、van Horssen等(2006)における表2に要約してある。
【0009】
一方、ヒトにおける不適切なTNF−αまたはTNF−αの過剰産生は、制御されない有害な炎症、組織破壊および器官の損傷を引き起こすこともまた明らかであり、多くの研究において報告されている。該TNF−αの過剰産生は、リウマチ性関節炎、多発性硬化症のような種々の自己免疫疾患、炎症性腸疾患のような炎症性疾患ならびに他の病的状態、そのうち例えば、強直性脊椎炎および脳卒中、において起こる。
【0010】
中枢神経系(CNS)に於いて、TNF−αは又二重の役割を果たす:一方では、TNF−αは有益であって、オリゴデンドロサイトの増殖と再生、神経細胞保護、神経再ミエリン化および種々の他の重要なプロセスに必要とされる。他方、過剰の内在TNF−αは、神経細胞傷害および神経変性に寄与する。中枢神経系(CNS)機能のモデュレーション時に炎症および免疫分子の役割により多くの注目が寄せられている。腫瘍壊死因子−α(TNF−α)は、炎症促進性のサイトカインであり、その受容体はCNS全体の神経細胞およびグリア細胞上で発現している。その二つの受容体の作用を通じて、TNF−αは、神経保護的または神経毒性の広範囲な作用を神経細胞に及ぼす。それは直接的および間接的に、アストロサイト上のグリアのグルタミン酸輸送体を阻害することにより、グルタミン酸の興奮毒性を促進する役割を果たす。さらに、TNF−αは、例えばシナプス上のAMPA受容体の発現を増加させ、グルタミン酸伝達に直接作用する。TNF−αは又、p38MAPキナーゼに依存するプロセスである、長期増強(LTP)を阻害することによりシナプス可塑性の役割を果たす。
【0011】
TNF−αの欠如または不足、すなわち免疫能または他のTNF−αを介した有益な効果の欠如または不足、を特徴とする特定の病態において、他のいかなるサイトカインをも増大させることなくTNF−αの産生、発現および分泌だけを増加させる新規な方法を発見することは、治療価値を有することは、上記から明らかである。
【0012】
以下に、TNF−αの増強および標的細胞に及ぼすその後の作用が治療戦略と成りうる、病的状態の例を示す:
a.腫瘍を殺すまたは除去することができないため、死に至る癌(van Horssen等(2006)、TNF−αの抗腫瘍効果および臨床的有用性についての最近の総説のすべての内容は参照により本明細書に援用される);
b.死に導く可能性のある、ウィルスおよび細菌のような外来侵入物と闘うことができなくなる慢性または急性の免疫不全;および
c.重要な神経機能がついには欠損し、生命を危険にする可能性のある、疾患または傷害に伴う神経細胞の再生の欠如または不足。
【0013】
過剰な又有害なTNF−αを特徴とする特定の病態に於いて、TNF−αの産生、発現および分泌を停止する新規な方法を発見することは、非常に治療価値を有することは、先の序論からも明らかである。
【0014】
以下は、TNF−αを停止または減少させおよびその後の標的細胞に及ぼすその影響が、以下の治療戦略になり得る病的状態の例である;リウマチ性関節炎、乾癬性関節炎、1型糖尿病、クローン病、乾癬、潰瘍性大腸炎、強直性脊椎炎、サルコイドーソスおよび頭部傷害後。
【0015】
発明の要約
本特許出願の根拠としての機能を果たす研究において、本発明の発明者らは、T細胞中のKv1.1電位依存性カリウムチャンネルの選択的閉鎖は、TNF−αの劇的な又独占的な合成および分泌を引き起こすために、単独で十分であって(すなわち任意の他の分子の存在無しに)、試験した他のサイトカインのいずれにも、すなわちインターフェロンガンマ、インターロイキン−4およびインターロイキン−10にも影響を与えないことを発見した。
【0016】
これらの発見に基づいて、T細胞内のKv1.1電位依存性カリウムチャンネルの閉鎖または遮断は、TNF−αを介する効果が治療的価値を有するような状況において、T細胞によるTNF−αの急速、強健および選択的な合成および分泌を達成するであろうことが分かった。
【0017】
T細胞におけるKv1.1電位依存性カリウムチャンネル閉鎖は、デンドロトキシン−K(DTX−K)のような高度に選択的なKv1.1チャンネル遮断剤、または特異的な抗−Kv1.1抗体、または下流経路の遮断によって、細胞外的にもしくは細胞内的に、T細胞のKv1.1チャンネルの選択的遮断を導く任意の他の方法によって本明細書中で達成される。
【0018】
従って、本発明の一つの目的は、T細胞に大量のTNF−αを産生および分泌させ、疾患や傷害と闘うために、TNF−αの欠乏と関連している病態を患っているヒトまたは動物被験体のT細胞内のKv1.1チャンネルを遮断する方法を提供することである。
【0019】
本発明の更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、選択的なKv1.1イオンチャンネル遮断剤である方法を提供することである。
【0020】
本発明の更なる目的は、T細胞に大量のTNF−αを産生および分泌させ、疾患や傷害と闘うために、T細胞によるTNF−αの増大した唯一の分泌により恩恵を受けることができるヒトまたは動物被験体のT細胞内のKv1.1チャンネルを遮断する方法を提供することである。
【0021】
本発明の別の目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、特異的な抗Kv1.1抗体であるような方法を提供することである。
【0022】
本発明の更なる別の目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、T細胞内のKv1.1チャンネルの下流経路を遮断できる分子であるような方法を提供することである。
【0023】
本発明の又更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子を、エクスビボにてT細胞に投与後にヒトまたは動物患者の体内へ接種して戻すような方法を提供することである。
【0024】
本発明の更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、体内に投与される、すなわちヒトまたは動物患者の体内に、静脈内、皮下、腹腔内、腫瘍内、くも膜下腔内または頭蓋内注入により、もしくは経口投与または経皮的に軟膏により投与されるような方法を提供することである。
【0025】
本発明の又更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、ヒトまたは動物被験体の体内に植え込み式の薬剤送達ポンプを用いて投与されるような方法を提供することである。
【0026】
本発明の別の目的は、ヒトまたは動物被験体が癌に罹患しており、それら自身のT細胞を、体外または体内で、T細胞のKv1.1チャンネルを遮断する分子に曝すことにより、TNF−αの強健な合成または分泌を誘発し、TNF−αによって癌を直接、死滅させる方法を提供することである。
【0027】
好ましい一実施形態では、ヒトまたは動物被験体が癌に罹患しており、それら自身のT細胞を、体外または体内で、T細胞のKv1.1チャンネルを遮断する分子に曝すことで、TNF−αの強健な合成または分泌を誘発し、腫瘍を有する器官へのT細胞の侵入を増加させ、腫瘍破壊細胞の動員を向上させるための有益な炎症環境をつくることにより、癌のT細胞免疫療法を増強する。
【0028】
好ましい別の実施形態では、ヒトまたは動物被験体は免疫不全に罹患している。
【0029】
更に好ましい別の実施形態では、ヒトまたは動物患者は、神経細胞傷害または神経疾患の後、神経細胞再生の不全に罹患している。
【0030】
本発明の好ましい別の実施形態では、TNF−αの強健な合成および分泌を得るための手法は、静脈内、皮下、腹腔内、腫瘍内、くも膜下腔内または頭蓋内注入により、もしくは経口投与または経皮的に軟膏により、TNF−αによる増強を動物患者の体の中に達成するのに役立つ。
【0031】
疾患の第二の部類では、TNF−αの有害な過剰は、本発明に従って、仮想の内在疾患関連Kv1.1遮断剤によって誘発されるKv1.1チャンネルを閉鎖/遮断させない分子を用いて、T細胞からのTNF−αの産生と分泌を阻止することにより克服される。もし後者(つまり仮想の内在疾患関連Kv1.1遮断剤)がT細胞における慢性のTNF−α産生および分泌の原因であるならば、例えばKv1.1チャンネル開口剤によってKv1.1チャンネルの遮断をさせないようにすれば、強健なTNF−αの増加の誘発をもまた阻害するであろう。チャンネルを開口し、従ってTNF−αの産生を阻止する分子は、細胞外作用剤であっても良く、またはチャンネルの遮断のシグナルを送る細胞内電位感覚要素を損なう分子であってもよい。該細胞外作用剤の例としては、従来の方法で、Kv1.1分子に対して作製し、作用剤活性に対してスクリーニングを行ったモノクローン抗体が挙げられる。
【0032】
好適な実施態様の詳細な説明
本発明は、正常ヒトT細胞におけるKv1.1電位依存性カリウムチャンネルの選択的遮断により、それのみで強健で、唯一TNF−α蛋白の分泌およびmRNAが増加されるという発見に基づいている。劇的なTNF−αの分泌が、高度に選択的なKv1.1遮断剤、デンドロトキシン−Kによって(しかし9つの他のK+、Na+、Ca2+およびCl−遮断剤によっては誘導されない);市販の抗−Kv1.1抗体によって;および、重要なことには、Kv1.1抗体を保持している末梢神経興奮性亢進症の患者からの血清によって誘導された。Kv1.1遮断は、TNF−α分泌のみを誘発させ、一方「古典的」なTCR−活性化は、TNF−α、IFN−γ、IL−4およびIL−10の分泌を、同時に、非唯一に増加させた。Kv1.1遮断は、またNF−κBの核転移を誘導する。
【0033】
ヒトT細胞におけるKv1.1タンパクおよびmRNAの発現は、本明細書ではRT−PCRおよびフローサイトメトリーによって示した。デンドロトキシン−Kでエクスビボにて前処理をし、従ってTNF−αを過剰分泌しているヒトT細胞をSCIDマウスに注射すると、肝臓への宿主細胞のインビボ動員を引き起こすが、これは脳へTNF−αを注入後に報告されたことに類似している。今まで、これらの発見を予想できることは全く知られていなかった。Kv1.1遮断によって誘発されたTNF−αの増加は、それ故、ある疾患(例えば、癌および免疫不全)ではTNF−αを高め、他(例えばリウマチ性関節炎、クローン病および他の自己免疫/炎症性疾患)では抑止して、治療的に用いることができる可能性がある。
【0034】
本発明により、ヒトT細胞における一種類のイオンチャンネルの選択的遮断が、一種類のサイトカイン、この場合はTNF−α、の唯一の産生と分泌を誘発できることが立証される。該機構は、全てのT細胞サイトカインの同時産生を促進することを特徴とする、種々の分裂促進因子(例えばホルボールエステル)による、「古典的」なTCR活性および非特異的なT細胞刺激の両者とは際立って対照的である。一つのサイトカインを唯一かつ強力に増加させ、一方では他のThサイトカインのレベルを増加させない機構の発見は、特にサイトカインがTNF−αであることから(Dinarelo ら, 2002; Wallach ら, 1999)、重要な臨床的意味を有する。なぜならば、このサイトカインが多くの疾患の病理において重要であり、そのレベルが過剰であるかまたは不十分であるからである。
【0035】
実際、一方ではTNF−αは、健康に基本的で有益な役割を果たす。それは自然免疫反応を活性化し、環境の攻撃から生体を保護する必須な炎症反応を引き起こす(Dinarelo ら, 2002)。更に加えて、TNF−αは、腫瘍の壊死を起こさせ、白血球のローリングと接着の増強し、種々の分子に対する血管と血液脳関門(BBB)の透過性を増加させ(Mayan 2002, van Horssen ら 2006)、オリゴデンドロサイトの増殖と再生を誘導し、神経の再ミエリン形成を促進し(Arnett ら, 2001; Finsen ら, 2002)、細胞を特定の組織へ動員し、ならびに多くの他の重要な機能を誘発する(Dinarelo ら, 2002; Wallach ら, 1999)。
【0036】
他方、TNF−αの過剰産生または無秩序な産生は、通常非常に有害であり、組織破壊に導く重篤な損傷を与える炎症を起こすことがあり、その後に種々の重篤な自己免疫疾患の原因となるが、その中にはリウマチ性関節炎および多発性硬化症、炎症性腸炎を含む炎症性疾患、ならびに外傷、強直性脊椎炎、脳卒中および過眠症のような多様な病理状態の種々なタイプが含まれる。興味あることには、ひとたびTNF−αが過剰になった場合には、抗−TNF−αモノクロナール抗体および可溶性TNF−α受容体のような、TNF−αのシグナル伝達を遮断する治療法に対する適応症のリストはどんどん大きくなっている一方、TNF−αの増加を阻止または遮断(すなわち過剰の有害レベルに達する前に)は、未だ達成されていない。更に、ヒトの疾患におけるTNF−αの慢性産生の原因となる機構については未だ不明なところが多く、他のサイトカインに影響せずに、選択的にTNF−αの増加を促進する機構も未だ解明されていない。
【0037】
T細胞を含む種々の細胞タイプにおいて、特定のイオンチャンネルが主要な細胞反応に影響を与え、指示するという既知の特性(DeCoursey ら, 1984; Deutsch ら, 1986, Lewis ら, 1995; Lewis ら, 1988; Rader ら, 1996; Freedman ら, 1995; Koo ら , 1997; Lin ら, 1993; Ishida ら, 1993; Jansen ら, 1999; Kotturi ら, 2003; Phipps ら, 1996; Verheugen ら, 1997)、およびKv1.3電位依存性カリウムチャンネルのゲーティングは、それ自身で主要T細胞機能を誘発できるという本発明者の研究室における以前の実証(Levite ら, 2000)によって促され、本発明は、正常ヒトT細胞における特定のカリウム(K+)、ナトリウム(Na+)、カルシウム(Ca2+)または塩素イオン(Cl−)チャンネルの単なる遮断が、選択的なサイトカイン分泌、特にTNF−αの分泌を誘発することができるかどうかの研究に基づいている。本結果は、正常のヒトT細胞内の電位依存性K+チャンネルを含むKv1.1サブユニットの選択的な遮断が、強健的で、唯一かつ長期的なTNF−α分泌の増加に対する新規な機構を明示している。
【0038】
本発見は、新規な予想外の機構を明らかにしたが、それは正常な末梢ヒトT細胞内の電位依存性K+チャンネルを含むKv1.1サブユニットの選択的な遮断が、TNF−α分泌およびmRNAレベルを著しく、唯一かつ長期的に増加させたことである。我々の知る限り、一般的に電位依存性K+チャンネル、特にKv1.1についてまたはTNF−αについて、Kv1.1とTNF−αの直接の機能的な関連に関する本発見を予想するようなことは全く知られていない。更に、本発見は、他のThサイトカインを増加させることなく、TNF−αの単独増加に対する新規の機構を明らかにしている。
【0039】
次の段落において、考察がなされる:a)本発明の更なる詳細;b)主としてT細胞中のKv1.1電位依存性K+チャンネルおよびKvファミリーの他のメンバー、ならびにDTX−Kの特殊な特徴;c)PNH,Kv1遮断に伴うヒトの神経疾患;およびd)様々なヒトの疾患においてTNF−αの促進する、または抑止する新規の臨床標的として、T細胞Kv1.1の使用。
【0040】
本発明の更なる詳細
非常に選択的なKv1.1遮断剤DTX−K(Harvey, 2001)および特異的、市販抗−Kv1.1抗体の両者は、T細胞TNF−αを劇的に増加させることを発見した。対照的に、T細胞中で発現しているK+、Na+、Ca2+、Cl−イオンチャンネルまたはグルタミン酸/AMPAイオンチャンネル受容体(Ganor ら, 2003)に対する9つの他の遮断剤にT細胞を曝しても該効果は得られなかった。Kv1.1には特異的ではないが、電位依存性K+チャンネルを含むKv1.1サブユニットにいくらか影響することができる二つのKv1.1遮断剤、KTXおよびMgTXもまたヒトT細胞からTNF−αを誘発したが、程度はずっと低く、Kv1.1チャンネルサブユニットに対するそれらの低い親和性と特異性に見合ったものであった。
【0041】
KTXおよびMgTXは、Kv1.1チャンネルに対してDTX−Kほど特異的ではないが、本発明に従って使用するには十分特異的である。本発明は、PMAによって代表されるような、ホルボールエステルのような非特異的なT細胞活性化剤をすべて考慮に入れているわけではない。しかしながら、本明細書で用いる「選択的」という用語は、PMAのようなホルボールエステルの場合のように、T細胞サイトカインが無差別に産生するのではない限り、他のKvチャンネルにいくらかの活性を有する可能性のあるKv1.1遮断剤を含むことを意図している。Kv1.1チャンネルに対する選択性が大きければ大きいほど、該分子に対する好ましさは大きい。従って、DTX−Kは、現時点において最も好ましい該分子である。しかしながら、DTX−KほどTNF−αを産生しないかも知れない、又DTX−KほどKv1.1に対する特異的選択性がないかも知れないが、KTXおよびMgTXもまた、本発明に従って用いることができる。従って、以下の十分に選択性を有する、および選択性がより低いDv1.1電位依存性イオンチャンネル遮断剤は、Kv1.1チャンネルを遮断し、および少なくともKvチャンネルに対して実質的に選択的である、いかなる他の分子とも同じように、本発明に従って使用できる:
A)デンドロトキシン類
1)トキシンK、デンドロトキシン−K(DTX−K)とも呼ばれる、は、ブラックマンバ、Dendroaspis polylepsisから由来し、Kv1.1チャンネルを優先的に遮断し、ピコモル濃度で活性を有する。
2)アルファーデンドロトキシンはグリーンマンバDendroaspis angusticepsから由来し、クローンされたKv1.1、Kv1.2およびKv1.6チャンネルを、低ナノモル範囲で遮断する。
3)トキシンI、デンドロトキシン−I(DTX−I)とも呼ばれる、は、ブラックマンバ、Dendroaspis polylepsisから由来し、クローンされたKv1.1、Kv1.2およびKv1.6チャンネルを低ナノモル範囲で遮断する。
B)コノトキシン類:イモガイ、Conus virgoの毒素から、ViTx(virgo-toxin)と呼ばれるペプチドが分離され、Kv1.1およびKv1.3のK+−チャンネルを遮断するが、Kv1.2タイプは遮断しない(Kauferestein ら, 2003)。
C)マウロトキシン類:天然および合成のマウロトキシンは両方とも、Xenopus卵母細胞で発現されたKv1.1、Kv1.2およびKv1.3チャンネルを、殆ど同じ40、0.8および150nMの半分有効量(IC50)で遮断する(Rochat ら, 1998)。
D)アギトキシン類
1)rAgitoxin-2 Leiurus q. hebraeusはKv1.1、Kv1.3、Kv1.6を50pM〜10nMで遮断する。(Alomone Labs web site; www.alomone.com/p_products)。
2)rAgitoxin-3 Leiurus q. hebraeusはKv1.1、Kv1.3、Kv1.6を50pM〜10nMで遮断する。(Alomone Labs web site; www.alomone.com/p_products)。
E)r-Hongotoxin-1 Centruroides limbatusはKv1.1、Kv1.2、Kv1.3を0.1〜0.2pMで遮断する。
F)Pi4はさそりPandinus imperatorの毒液由来の38アミノ酸長ペプチドであり、Shaker B K+チャンネル(Kv1.1サブファミリータイプのチャンネル)を、100nM濃度で完全にかつ可逆的に遮断する(Olamendi-Portugal 1998)。
G)Kaliotoxin(KTX):KTXはいくつかのCa2+活性化および電位依存性K+チャンネルを遮断する。Xenopus laevis卵母細胞へのKTXのバス適用(bath application)は、組み換えKv1.3およびKv1.1チャンネルを強力に、又Kv1.2チャンネルをより低い強度で遮断し、それぞれのK(d)値は、0.1、1.5および2.5nMであった(Mourre ら, 1999)。
H)BTK−2:インドのさそりButhus tamulusから精製されたKv1.1カリウムチャンネルの新阻害剤(Dhawan ら, 2003)。
I)新Kv1.1カリウムチャンネル遮断トキシン:Palamneus gravimanus (インド黒さそり)の毒液由来の新Kv1.1カリウムチャンネル遮断トキシン(More ら, 2005)。
J)rMargatoxin:MgTX K+チャンネルは、主としてKv1.3電位依存性K+チャンネルを遮断するが、Kv1.1にも作用することができる。
K)Stichodactyla helianthusペプチド(ShK)は、S. helianthusの毒液から分離された、Kv1.1およびKv1.3チャンネルの既知の高親和性遮断剤である。
【0042】
本実験で用いたKv1.1に対する抗体は、市販されておりAlamone Labs Ltd., Jerusalem, Israel, product no. APC-009から得られる。Alomone.com/System/UpLoadFiles/DGallery/Docs/apc- 009_AN-05.pdfで得られるデータシートを参照。この抗体は、配列HRETE GEEQA QLLHV SSPNL ASDSD LSRRS SSTIS KSEYM EIEED MNNSI AHYRQ ANIRT GNCTT ADQNC VNKSK LLTDV(配列番号7)を有するGST融合タンパクに対して産生させたウサギ抗体であって、マウスKv1.1の残基416−495に相当する(GeneBank Accession P16388)。このエピトープの80残基の内76残基は、対応するヒトタンパクに保存されている。Kv1.1のエピトープ、好ましくはヒトKv1.1に対して産生された他のいかなる抗体も、ポリクローンあるいはモノクローンであれ、それらがKv1.1チャンネルを遮断する能力を有する限り、本発明の目的のために用いることができる。本明細書および請求項で用いる「抗体」という用語はポリクローンまたはモノクローン、ならびにヒト化抗体、単鎖抗体のような遺伝子改変の抗体、およびCDRs、Fab、F(ab)2等のような抗原結合性断片を包含することを意図している。
【0043】
Kv1.1チャンネルに対するモノクローン抗体を作製する際、それらはチャンネルを遮断する(拮抗する)または開口する(作用する)能力に関してスクリーニングをしてもよい。拮抗剤である抗体は、TNF−αの産生の増加が望まれる病気を治療するために用いることができ、作用剤である抗体は、TNF−αの過剰な産生を阻止する必要がある疾患を治療するために用いることができる。
【0044】
TNF−α転写におけるCa2+の既知の役割(Lobo ら, 1999)と合致して、Kv1.1遮断によるTNF−αの誘導にはCa2+流入を必要とすることが発見されたが、それはT細胞内におけるCa2+チャンネルの高度の選択的遮断剤であるR-(+)-Bay K 8644(Kotturi ら, 2003)がこの効果を阻止したからである。驚くべきことには、Kv1.1チャンネルの遮断は、TNF−αの急速な増加を引き起こす一方、IFN−γ、IL−4およびIL−10を増加させず、そのことは、これら全てのThサイトカインは強健だが、非選択的に、又同時に結局増加することになる「古典的な」TCR−活性化から著しく異なっていた。
【0045】
TNF−αを過剰発現するDTX−Kで処理した正常ヒトT細胞を、SCIDマウスに注射すると、TNF−αの脳内への注射後起こることが最近研究で示された結果(Campbell ら, 2005)に類似して、専ら肝臓に存在するマウス常在細胞数が倍増したことから、Kv1.1チャンネルの選択的遮断によってヒトT細胞内に誘導されたTNF−αの増加は、インビボにて機能的な影響をもたらした。
【0046】
Kv1.1電位依存性K+チャンネルおよびそのファミリーの他のメンバー、ならびにDTV−Kの特異な特徴:Kv1.1チャンネルは電位依存性K+チャンネルのファミリーに属しており、そのいくつかのメンバーはT細胞で発現しており、重要な機能的役割を果たしている(DeCourseyら, 1984; Deutsch ら, 1986; Lewis ら, 1995; Lewis ら , 1998; Freedman ら, 1995; Koo ら , 1997; Lin ら, 1993; Levite ら, 2000; Liuら, 2002; Beeton ら, 2001; Leonard ら, 1992; Levite 2001)。これらのチャンネルはC2+活性化K+チャンネルとは異なっており、これらチャンネルはT細胞により発現しており、T細胞機能として重要である(Rader ら, 1996; Jensen ら, 1999; Vreheugen ら, 1997)。様々な種類の電位依存性K+チャンネルのグループ分けおよび独特な特性は、いくつかの研究で考察されている(Grissmer ら, 1994; Mathisら, 1998を参照)。一般的に、電位依存性K+チャンネルの阻害は、脱分極を起こし(Leonard ら, 1992)(すなわち陰性度が低い膜電位へのシフト)、それが、種々の主要な細胞特性および細胞機能に影響を及ぼし、発揮させる(Levite ら, 2000; Leonard ら, 1992; Levite, 2001)。神経細胞においては、電位依存性チャンネルの一つの大きな役割は、膜の興奮性の制御である(Ruteckie, 1992; Smart ら, 1998)。これには活動電位および神経細胞の反復発火の期間を制限し、ならびに膜電位を安定化させることが含まれる。
【0047】
T細胞内の特定の電位依存性K+チャンネルは、β1インテグリン類を活性化し、その後フィブロネクチンと同様にECM成分へのT細胞接着を誘発し(Levite ら, 2000)、膜電位(Levite ら, 2000)、カルシウム流入(Grinstein ら, 1990; Lin et al, 1993; Verheugen ら, 1997)、増殖(Freedman ら, 1992; Leonard ら, 1992)、IL−2産生(Liu ら, 2002, Freedman ら, 1992)、アポトーシス(Bortner ら 1999; maeno ら , 2000; Vu ら, 2001)、および細胞容積(Grinstein ら, 1990)を調節することが、先の研究で示された。
【0048】
従って、全体として、Kvチャンネルは、主要な生理的T細胞機能を調節し、それらの不適切な機能は、TCR−活性化を阻害し、重要なT細胞を介しての免疫反応を阻止する。例えば、Liu ら(2002)は、電気生理学、薬理学およびRT−PCRの方法を組み合わせて、未処理のマウスCD4+T細胞は、Kv1.1、Kv1.2、Kv1.3およびKv1.6電流を発していること、ならびにこれらのKvチャンネルが、該未処理の細胞によるTCR−誘導(抗−CD3および抗−CD28により)IL−2分泌を最大にするために必要となることを示した(Liu ら, 2002)。
【0049】
対照的に、マウスTCR−活性化エフェクターT細胞は、Kv1.3電流(未処理細胞よりは±6倍高い)のみを発するが、K1.3も、他のいかなるKvも、これらの活性化されたT細胞のIL−2、IL−4、またはIPN−γの産生、カルシウムシグナル伝達または膜電位に対して有意に寄与はしていない(Liu ら, 2002)。この後者の研究で行った観察により、末梢マウスCD4+細胞において、分化状態、TCR活性化の強さ、共刺激因子の存在あるいは非存在の表れとして、Kv電流が、質的にも又量的にも変化することを示された(Liu ら, 2002)。自然の電位依存性K+チャンネルはヘテロ多量体であって、種々のKvサブタイプの組み合わせから構成されており、ヘテロ多量体チャンネルが異なれば、親チャンネルと比較して、劇的に異なったチャンネル機能を有することになる。ヘテロダイマーを含む異なったKv1.1もまた異なった遮断剤に対して異なった感受性を有している。例えば、ヘテロダイマーを含むKv1.1中にKv1.2が存在すると、TEAに対する全アレイの感受性が低下する。先の研究では、DTX−Kが、Kv1.1サブユニットとの強く高度に特異的な相互作用という点で、またそれが、これらのチャンネルとそのN−末端(K3、K6)およびβ−ターン(K25、K26)における残基を介して相互作用をするという点で、独特なものであることを示された(Wang ら)。DTX−Kの非常に独特な作用機構により、DTX−KによるTNF−αの増加を(いまだ未調査の機構で)説明できる可能性がある一方、特異性がより低いKv1遮断剤の作用機構により、たとえあったとしても効果はあまり目立たなくなる。
【0050】
ヒトおよびマウスT細胞におけるKv1.1電流試験である別の研究で、異なった結論に達した(Freedman ら, 1995; Cahalan ら, 1985; Liu ら, 2002)。本結果は、特異的なKv1.1RT−PCRおよびフローサイトメトリーによって、正常末梢ヒトT細胞のかなりの割合は、Kv1.1mRNAおよび膜タンパクを明瞭に発現していることを示し、このタンパクの新しい面を明らかにした。しかし、我々の発見は、正常静止状態の末梢ヒトT細胞において機能的な(すなわち電気生理学的に活性を有する)Kv1.1電流の存在を証明するものでは勿論ない。電位依存性K+チャンネルファミリーの他のメンバー、主としてKv1.3、がT細胞の活性化と機能において重要な役割を果たしている証拠が、多くの最近の研究によって示唆された(DeCoursy ら, 1984; Deutsch ら, 1986; Lewis ら, 1995; Lewis ら, 1988; Freedman ら, 1995; Koo ら, 1997; Lin ら, 1993; Leite ら, 2000; Liu ら, 2002; Beeton ら, 2001; Leonard ら, 1992; Rus ら, 2005)。更に、本発明者の研究室は、正常ヒトT細胞の電位依存性K+チャンネルの一つのメンバー、Kv1.3、のゲート開閉は、それ自身のみでβ1インテグリン活性化とT細胞接着を誘発でき、Kv1.3チャンネルは、これらの細胞中で、実際β1インテグリンと物理的に又機能的にカップリングしていることを以前示した(Levite ら, 2000; Levite 2001)。注目すべき点は興味ある相違点である:我々が以前研究したKv1.3チャンネルの場合には、T細胞機能を誘導するのはKv1.3チャンネルを開口した時であり(すなわちT細胞接着を引き起こす)(Leviteら, 2000; Levite 2001)、一方本研究においては、そうする(すなわちTNF−αを増加させる)のはKv1.1チャンネルの遮断である。
【0051】
Kv1.1遮断に伴ういくつかのヒト神経疾患:一般的に神経の興奮性を増加させるKv1.1チャンネルの遮断は少なくとも3つのヒト神経疾患を直接伴っている:
1)Kv1抗体と関連するPNH
2)Kv1抗体と関連する脳炎
3)ヒトKv1.1チャンネルをコードする遺伝子の様々な突然変異に伴う、突発性運動失調1型(EA−1)(Vincent et al, 2004; Adelman et al 1995; Browne et al, 1994)。
【0052】
PNH:Kv1遮断に伴うヒトの神経疾患:マウスにおけるKv1.1チャンネルの欠損は、癲癇、温度感受性振盪および痛覚過敏症を起こすが、これらは神経細胞の過剰興奮性と一致する特徴である(Smart ら, 1998; Zhou et al , 1998)。臨床医は、全般的なPNHの筋肉運動の症状を記述するのに、神経性筋強直症、アイザックス症候群、ミオキミアおよび線維束性痙攣症候群を含む多くの用語を用いる(Hart et al, 2002)。PNHが通常自己免疫を介する(Newsom-Davis ら, 1993)という考えは、最近60人のPNH患者の35%は、100pmol/lを越える血清Kv1抗体タイター有していたという知見によって確証された(Hart et al, 2002)。これらの抗体は病原性があり、Kv1チャンネルを遮断し、従って末梢およびお中枢神経系の両方に神経の過剰興奮性と広範囲の臨床症状を引き起こす。ここで、いく人かのKv1抗体陽性PNH患者の血清は(方法参照)、ヒトT細胞による相当な量のTNF−α分泌を刺激したが、一方IFN−γを増加させなかった(DTX−Kの結果と類似している)。この発見は、T細胞における抗体を介したサイトカイン誘導が、神経の過剰興奮性の病態およびPNHの臨床的特徴ならびに多分自己免疫性脳炎の一因となる可能性を示唆している。本データは又疲労および筋肉痛のようなPNHに伴う、より非特異的な症状は、少なくとも部分的にはKv1抗体を介した血液循環中のTNF−αの増加によって引き起こされた可能性を示唆している。従って、本発明の他の特徴は、1)Kv1抗体に伴うPNH,2)Kv1抗体に伴う脳炎、3)突発性−運動失調1型(EA−1)を循環系におけるTNF−αの力価を減少させる任意の方法で治療する方法であって、TBP−1およびTBP−2のようなTNF−α結合タンパク、抗-TNF−α抗体、Kv1.1チャンネル非遮断剤等のような方法である。
【0053】
T細胞のKv1.1機能不全は、TNF−α過剰に伴うヒトの疾患に寄与する可能性があるので、T細胞におけるKv1.1は新規の治療標的である。ヒトTNF−αは生物活性を有する、233アミノ酸、26kDaの膜結合性タンパクとして合成される。この膜結合性タンパクは、TNF−α変換酵素(TACE)、アダマリシンによって157アミノ酸、17kDa可溶性タンパクに酵素的に切断され、容易にホモ三量体を形成する。ホモ三量体TNF−α(細胞結合型または可溶性のいずれか)のその受容体(TNF−RIおよびTNF−RII)への結合は、静止状態の細胞においてシグナル伝達に必要な受容体のオリゴマー形成を誘導する。
【0054】
TNF−αの不適切な産生または過剰産生は、その発病原因において炎症が関与する広範囲の病気において起こるが、その病気には、炎症性腸疾患、リウマチ性関節炎、多発性硬化症、強直性脊椎炎および脳卒中が含まれる(Baugh ら, 2001)。本発見により、インビボでのKv1.1抗体またはKv1.1遮断剤を介してのT細胞Kv1.1遮断は、少なくとも部分的に、これらの障害での過剰のTNF−αに原因があり、それ故新規の治療標的である、という結論におそらく導かれるであろう。
【0055】
他方、TNF−αは、他の多くの目的のために非常に必要とされることから、TNF−αを長期間過剰発現する自己DTX−K処理T細胞のヒトにおける受動的な輸送は、非常に特定の状況においては、有益であろう。これらには、T細胞による癌細胞根絶の強化、ウイルスおよびバクテリアのT細胞攻撃を高めること(特に免疫不全の種々のタイプの場合)および必要に応じて宿主細胞の肝臓への動員の増加が挙げられる。
【0056】
実施例1:
材料と方法
イオンチャンネル遮断剤:表1も参照。本明細書に用いる各イオンチャンネル遮断剤について特定されるものは、正式名、カッコ内に短縮名と製造者、有効濃度(それぞれの製造者のデータシートおよび/または文献から得られた)である。更なる詳細情報は、International Union of Pharmacology website (Gutman et al, 2003を参照)に見出すことができる。遮断剤としては以下のものが挙げられる:4−アミノピリジン(4−AP,Sigma,St. Louis, MO)、10μM〜1mM;テトラエチルアンモニウム(TEA,Sigma)、100μM〜10mM;キニーネ(Sigma)、1〜10μM;クロトリマゾール(CLT,Agis Industries, Bnei Brak, Israel)、10nM〜10μM;rカリブドトキシン (CTX,Alomone labs, Jerusalem, Israel)、10〜100nM;カリオトキシン(KTX,Alomone)、1〜100nM;rマルガトキシン (MgTX,Alomone)、50pM〜50nM;デンドロトキシン−K(DTX−K,Alomone)、10〜100nM;テトロドトキシン(TTX,Alomone)、100nM〜1μM;NPPB(Torcris Cookson, Avonmouth, UK)、100〜200μM;R-(+)Bay K8644(+Bay K, Tocris)、10nM〜1μM;CNQX(Tocris)、100nM−50μM。
【0057】
【表1】
【0058】
ヒトT細胞:正常ヒトT細胞は、(Levite ら, 2000; Ganor ら, 2003; Levite ら, 1998)に記載されているように健常提供者の末梢血から精製し、得た90%以上のT細胞を含む細胞群を、10%FCS、1%Lグルタミンおよび抗生物質を含むRPMI培地(Biological Industries, Bet Haemek, Israel)に懸濁し、2x106細胞/ml(37℃/5%CO2)で維持した。
【0059】
ELISAによるTNF−α、IFN−γ、IL−10およびIL−4の定量:新たに精製した正常静止ヒトT細胞(2x106/ml)を24穴プレート(Costar, Corning, NY)中でインキュベーションし、種々のチャンネル遮断剤を加えた。重要なことは、他の刺激分子(例えばホルボールエステル、抗原、抗−CD3・CD28mAb)を加えないことであった。サイトカインレベルは、TNF−αとIFN−γについては24時間後、IL−10およびIL−4については72時間後、定量的サンドイッチELISA法(R&D, Minneapolis, MN)で、製造者の指示に従い、上清を測定した。
【0060】
TNF−αおよびKv1.1のRT−PCR:全RNAは、Tri−Reagentプロトコル(MRC, Cincinnati, OH)に従って調製した。第一鎖cDNAは、Reverse Transcription System(Promega, Madison, WI)を用いて合成した。RT−PCRは、50ngのcDNA、5μlのx10 Optibuffer (Bioline, London, UK)、3μlの50mM MgCl2、2.5μlの10mM dNTP混合液(Promega)、4UのBio−X−actDNAポリメラーゼ(Bioline)を含む50μlの反応混合液中で行った。TNF−α/S14同時増幅については、2μlの各TNF−αプライマー(10pmol/μl)および0.25μlの各S14プライマー(10pmol/μl)を用いた。Kv1.1/S14同時増幅については、5μlの各Kv1.1プライマー(100pmol/μl)および2.5μlの各S14プライマー(10pmol/μl)を用いた。プライマー配列(5’−3’)および産物の長さは以下の通りである:
TNF−αプライマーペア−上流プライマー
【表2】
下流プライマー
【表3】
494bp;Kv1.1プライマーペア−上流プライマー
【表4】
下流プライマー
【表5】
709bp;S14プライマーペア−上流プライマー
【表6】
下流プライマー
【表7】
166bp。TNF−αのPCR条件は:94℃1分、63℃40秒および72℃40秒(29サイクル)。Kv1.1のPCR条件は、94℃3分(1サイクル);94℃1分、60/57/54/51/48℃2分および72℃3分(それぞれ3サイクル);94℃1分、50℃2分および72℃3分(25サイクル)。TNF−α/S14RT−PCR産物の濃度分析は、Adobe Photoshop 7.0 MEによって行った。TNF−αバンドの相対強度は、S14バンドの相対強度に対して規格化した。
【0061】
核NF−κBのEMSA:正常ヒトT細胞から新たに分離した核画分は、Nuclear Extract Kit(Active Motif, Carlsbad, CA)用いて製造者の指示に従って採取した。核画分中のNF−κBレベルは、TransAM NF-kB Family Kit(Active Motif)を用いて製造者の指示に従って測定した。
【0062】
NF−κBの免疫蛍光組織化学:DTX−K(100nM 7、15、30および60分間)で前処理または未処理の新たに分離した正常ヒトT細胞を沈殿させ(1200g、10分、40℃)、PBSに1x106細胞/mlで懸濁し、遠心し(1200g、10分)、ガラススライド上にサイトスピン(250μl/スライド、1500rpm、5分;Shandon Elliot, London, UK)で固定し、透過処理を行い(100mlの95%メタノールおよび3ml氷酢酸を含む溶液200μl/スライド)、10%正常ヤギ血清、2%BSA、1%グリシンおよび0.5%のTriton X-100を含むブロッキング培地中で(200μl/スライド)インキュベーションした。それから細胞をウサギ抗−ヒトNF−κB p50ポリクローン抗体(1:50希釈した80μlと30分間室温;Santa Cruz Biotechnology, Santa Cruz, CA)で染色した。この第一回の染色後、細胞をFITC−結合ヤギ抗−ウサギIgG(1:50希釈で30分間室温;Jackson ImmunoResearch, West Grove, PA)に曝し、PBS中で2回洗浄し、それから細胞核に取り込ませるHoechst 33342に曝した(1:2000希釈で5分間室温;Molecular Probes, Eugene, OR)。染色した細胞を、蛍光顕微鏡(Nikon Eclipse E600)で可視化し、ACT−1ソフトウエアープログラムを用いて、x100倍で写真撮影を行った。
【0063】
チャンネルを含有するKv1.1サブユニットの検出のための免疫染色およびフローサイトメトリー:新たに精製した正常ヒトT細胞を、透過処理(70%エタノール、1時間、20℃で)した後、一次抗体としてウサギ抗−Kv1.1ポリクローン抗体(Alomone)を用いて、6μg/ml/1x106細胞/100μl、30分間氷上で、免疫蛍光染色を行った。アイソタイプのコントロールとして、細胞を同じ濃度の以下のように調製した正常ウサギIgGで染色した:健常コントロールの白色ニュージーランド雄のウサギから得た血清に等量の飽和硫酸アンモニウムを滴下する;混合液を1時間4℃で攪拌し、3000g15分、4℃で遠心した。沈殿物をリン酸緩衝食塩水(PBS)に再懸濁し、硫酸アンモニウムのいかなる痕跡をも除くために、同じ溶液に対して一夜3回透析した。IgG濃度の推定はO.D280で定量した(1.45O.D280は1mgIgG/mlとした)。それから細胞を、FITC結合ヤギ抗−ウサギIgGを二次抗体として(1:100希釈した100μl、Jackson)染色した。二次抗体のみで染色した細胞を追加の陰性コントロールとした。蛍光像をFACSortに記録した。
【0064】
PNH患者および健常者個人コントロール:血清は12人のPNH患者から採取したが、すべての患者は筋電図検査によって、全身性後天性疾患を確認し、又多発神経障害の臨床的または電気生理的証拠は無かった。どの患者も、造影CT縦隔上に胸腺肥厚または胸腺腫は有しなかった。血清採取の少なくとも1年前までは、どの患者も免疫療法は受けていなかった。年齢、性および地理的に一致させた4人の健常者個人コントロールから血清の供与を受けた。全ての患者から書面で、この研究に対するインフォームドコンセントを得、この研究は、Sefton Research倫理委員会、リバプール、UKによって許可された。血清Kv1抗体力価は、標準(125)I−アルファーデンドロトキシン免疫沈降アッセイ(Hart et al, 1997)によって検出した。この方法はKv1.1、1.2および1.6に対する抗体を検出する。陽性力価は、100pM以上に設定してある。50pM未満の力価は陰性と見なす。ここで試験した12人のPNH患者のうち7人はKv1抗体に関しては陰性であった。PNH患者の関連した臨床特徴を下に要約する。
PNH Kv1抗体陽性(PNH+)
PNH+(1):女性、40歳、PNH(36歳で発症)および甲状腺機能低下症、Kv1 239pM。
PNH+(2):女性、49歳、PNHのみ(47歳で発症)、Kv1 335pM。
PNH+(3):男性、52歳、PNH(40歳で発症)およびDMタイプ1、Kv1 235pM。
PNH+(4):男性、52歳、PNHのみ(43歳で発症)、Kv1 382pM。
PNH+(5):男性、51歳、PNH(51歳で発症)および軽度の乾癬、Kv1 346pM。
PNH+(6):男性、44歳、PNHのみ(43歳で発症)、Kv1 165pM。
PNH+(4):男性、23歳、PNHのみ(22歳で発症)、Kv1 255pM。
PNH Kv1抗体陰性(PNH−)
PNH−(7):女性、48歳、PNHのみ(41歳で発症)。
PNH−(8):女性、45歳、PNH(37歳で発症)およびレイノー病。
PNH−(10):男性、53歳、PNHのみ(50歳で発症)。
PNH−(11):男性、49歳、PNHのみ(44歳で発症)。
PNH−(12):女性、41歳、PNHのみ(15歳で発症)および湿疹。
【0065】
市販Kv1.1抗体またはPNH患者の血清の、Kv1抗体の存在あるいは非存在下での、正常ヒトT細胞によるTNF−α分泌の誘発能力の試験:新たに精製した正常ヒトT細胞(2x106細胞/ml)を24穴プレート中でインキュベーションし、ウサギ抗−Kv1.1ポリクローン抗体(1:1000希釈;alomone)に24時間曝した。培地に分泌されたTNF−αのレベルは、上記のようにELISA法によって検査した。
【0066】
代わりに、T細胞(2x106細胞/ml)をKv1抗体−陽性PNH患者、Kv1抗体−陰性患者または健常者の血清(1:100希釈)に曝した。T細胞を含まない培養を陰性コントロールとした。24時間インキュベーションした後、上清中のTNF−αおよびIFN−γのレベルを上記のように検査した。培地に分泌されたTNF−αのレベルは、抗−Kv1.1抗体陰性PNH患者の血清に曝されたT細胞によって分泌される平均TNF−αレベルの±2*SDを越えるならば、増加したとした。
【0067】
SCIDマウスへの、DTX−K処理ヒトT細胞のインビボ注射:NOD/SCID雌マウス腹腔内へ(ip)、シクロホスファミド(Sigma、20mg/ml PBS溶液)200μlを細胞注射する24時間前に注射した。新たに精製した正常ヒトT細胞(1x108細胞)をDTX-K(100nM、24時間)またはPBSで前処理し、洗浄し、2’,7’−ビス−(2−カルボキシエチル)−5−(および−6)−カルボキシフルオレッシンアセトキシメチル(BCECF AM、Molecular Probes, Eugene, Oregon)で蛍光標識した。それからヒトT細胞をPBSで洗浄し、懸濁し、標識細胞の200μl(1.5x107細胞に相当)をそれぞれの動物のipに注射した(各群n=5)。24時間後、各動物から血液を採取し、肝臓、脾臓、腎臓、骨髄および脳を取り出した。脳を除く全ての臓器から単細胞懸濁液を調製した。それぞれの臓器における総細胞数ならびに常在SCID細胞(非蛍光)のみまたは注入したヒトT細胞(蛍光)のみの数をFACSortによって計測した。
【0068】
脳の免疫組織化学:脳を取り出し、ブアン固定液に4℃で一夜浸し、PBSで洗浄し、パラフィン包埋を行うまで70%エタノールに貯蔵した。連続7μ厚の冠状切片を切り、脱パラフィン、脱水し、家庭用Samsung電子レンジを用いてマイクロウエーブ処理を行った。これは、1mM EDTAバッファー(pH=8.0)に浸し、沸騰するまで最高出力で加熱し、更に10分間20%効率で加熱し、同じバッファー中で、室温で30分間冷却することにより行った。
【0069】
免疫組織化学については、切片をPBS/20%ウマ血清(HS)(Vector Laboratories, Burlingame, CA)と4℃で一夜前インキュベーションを行った。それから切片を、ラット抗−ヒトCD3モノクローン抗体(PBS/2%HSで1:50希釈;Serotec, Oxford, UK)と4℃で96時間インキュベーションした。PBSで洗浄後、切片をビオチン化抗−ラットIgG(PBS/2%HSで1:10希釈;Vector)と室温で1.5時間インキュベーションし、再び洗浄後Cy3結合したストレプトアビジン(PBSで1:200希釈;Jackson)とインキュベーションした。切片を蛍光顕微鏡(Nikon Eclipse E600)で観察し、ACT−1ソフトウエアープログラムを用いてx40倍率で写真撮影を行った。
【0070】
統計解析:統計的有意差をStudent's t-testによって解析した。患者の異なった群間の定量的な変動を比較するために、非パラメータKruskal-Wallis テストを用い;次いで対での比較は非パラメータMann-Whitney U-testによって行った。
【0071】
実施例2:正常ヒトT細胞のチャンネルを含むKv1.1サブユニットの選択的遮断は、著しい又単独のTNF−α分泌を誘発する。
新たに精製した正常ヒトT細胞(「静止」正常ヒト末梢T細胞)を、他の刺激分子が全く存在しない状態で12の異なったチャンネル遮断剤(表1参照)に曝した。これらの12の遮断剤は、T細胞に発現されるK+、Na+、Ca2+およびCl−チャンネルの殆どのタイプを遮断するそれら報告された能力に基づいて選択された(Deutsch ら, 1986; Lewis ら, 1995; Lewis ら, 1988; Rader ら, 1996; Freedman ら, 1995; Koo ら, 1997; Lin ら, 1993; Ishida ら, 1993; Jensen ら, 1999; Kotturi ら, 2003; Phipps ら, 1996; Verheugen ら, 1997; Levite ら, 2000; Chandyら, 1984)。培地へ分泌されるTNFαのレベルは、ELISA法により24時間後に試験した。陽性コントロールとして、T細胞を非特異的な様式で活性化することが知られている、強力なホルボールエステル、PMAに曝した。結果は図1Aに示してあり、2〜3回の独立した実験において試験したTNF−α分泌の倍増率±SDを表す。各イオンチャンネル遮断剤は、少なくとも2回の独立した実験において、その有効適用範囲のいくつかの濃度で試験した(材料と方法を参照)。各遮断剤の隣の図の数字は用いた最高のモル濃度を表す。統計解析:MgTX、KTX、DTX−KおよびPMAのそれぞれについて、未処理に対して*P=0.0001、0.0005、0.0011および0.0005(Student's t-test)。
【0072】
図1Aは、電位依存性K+チャンネルを含むKv1.1サブユニットの、高度に選択的なKv1.1遮断剤であるデンドロトキシン−K(DTX−K)(Harvey et al , 2001)(又該物として多くの研究に使用され、その内のいくつかは(Harvey et al , 2001)に引用されている)による遮断が、それだけで、TNF−αの分泌を引き起こすことを示している。推奨される至適有効濃度(すなわち多くの他の研究において通常用いられるような、100nM)で使用したDTX−Kは、試験したヒトT細胞によるTNF−α分泌の33.4倍増を誘導したが、これは本明細書で陽性コントロールとして用いた、強力な非特異的活性化剤、ホルボールエステル(PMA、41.3倍増)によって得られるものと同規模であった。TNF−α分泌の有意な増加は、カリオトキシン(KTX)およびrマルガトキシン(MgTX)によってもまた起きた(それぞれ15.1および6.5倍増、図1A):これらは、電位依存性K+チャンネルの遮断剤であって、主としてKv1.3チャンネルに影響するが、Kv1.1チャンネルにも又影響する可能性がある。DTX−Kは、KTXおよびMgTXと比較すれば、Kv1.1に対するより高い親和性と選択性を有している(例えば、Kv1.1に対するKiは、DTX−Kは16pMでありKTXの20nMよりもはるかに高い(Gutman et al , 2003))。
【0073】
DTX−K、KTXおよびMgTXとは対照的に、正常ヒトT細胞を9つの他のイオンチャンネル遮断剤のいくつかの有効濃度に曝した後も、どんな有意なTNF−αの上方調節も観察されなかった(表1)が、その中には更なるK+チャンネルの遮断剤(Kv1.1に対しては選択性がより少ない):4−アミノピリジン(4−AP)、クロトリマゾ−ル(CLT),テトレエチルアンモニウム(TEA)、rカリブドトキシン(CTX)およびキニーネ;Na+チャンネル遮断剤:テトロドトキシン(TTX);Cl−チャンネル遮断剤:NPPB;Ca2+チャンネル遮断剤:R-(+)-Bay K8644 (+Bay K);およびイオンチャンネル(イオンチャンネル内蔵型)グルタミン酸/AMPA受容体(ヒトT細胞中に高レベルで存在することが最近同定された)(Ganor et al, 2003):CNQX。
【0074】
DTX−Kの誘発効果は、図1Bに示したように用量依存性である。10−9M〜10−6MのDTX−Kに対して24時間曝露した後のTNF−α分泌の倍増±SDを示す。行われた三つの実験のうち代表的なものを示す。未処理に対して*P=0.0001である(Student's t-test)。TNF−α分泌は、100nM DTX−Kに対して8.7倍で、1μMDTX−Kに対しては39.5倍であった(注:図1Aおよび1Bに示す結果は、別の二人のヒトから精製したT細胞に関するものである、下を参照のこと)。
【0075】
続く実験で、我々は更なる10人の個体から新たに分離したT細胞を試験し、DTX−Kの効果は、再現性があることが分かった。図1C〜Hは、これらの6人のヒト個体からのT細胞のDTX−Kに対する被曝後(24時間、100nM)培地に分泌されたTNF−αの実際の平均濃度をpg/ml±SDで示したものである(aおよびbに描かれた倍増率ではなく)。統計解析:1、3、5〜6の個体それぞれについて、未処理に対して*P=0.0054、0.0052、0.0063および0.0197(Student's t-test)。本明細書ではpg/mlで示す、個体4および2のT細胞から分泌されたTNF−αは又、倍増率にて上に示している(それぞれ図AおよびB)。図1C〜Hは、T細胞由来のTNF−αのバックグラウンドレベルが異なっているにもかかわらず、DTX−K(24時間、100nM)は、全ての6人の個体のT細胞による著しいTNF−αの分泌を誘発し、それぞれ22.3、8.7、49.0、34.4、2.4および2.0の倍増率の結果になったことを示す。DTX−K誘導性TNF−α増加の最高値は、38pg/mlから1308pg/mlであって、34.4倍増を示している(図1F,個体4)。
【0076】
注:DTX−KはこれらのヒトT細胞の細胞生存または増殖に、有意な影響は持たなかった(データは示さず)。
【0077】
実施例3:チャンネルを含むKv1.1サブユニットのDTX−Kによる遮断は、TNF−αの唯一的な分泌を誘起するが、一方「古典的な」TCRの活性化は、TNF−α、INF−γ、IL−10およびIL−4の強健であるが、非唯一的な分泌を誘導する。
図2A〜Hにおいて、正常ヒトT細胞を、100nM DTX−K(図2A〜D)または抗−CD3および抗−CD28mAbs(図2E〜H)を用いた「古典的な」TCR活性化に曝した。その後24〜72時間内に培地に分泌されるTNF−α、INF−γ、IL−10およびIL−4のレベルを、ELISAによって試験した。少なくとも3回の独立して行った実験のうちの代表的な実験の一つからの、TNF−α(図2Aおよび2E、24時間)、INF−γ(図2Bおよび2F、24時間)、IL−10(図2Cおよび2G、72時間)およびIL−4(図2Dおよび2H,72時間)の分泌の倍増率±SDを示す。統計解析:未処理に対して*P=0.0056(A)、0.0050(E)、0.0051(F)、0.0050(G)および0.0053(H)(Student's t-test)。
【0078】
興味あることには、DTX−KによるKv1.1の遮断はTNF−αのみを増加させたが、INF−γ、IL−4およびIL−10のレベルには影響しないことが分かった(図2A〜D,結果を、全4サイトカインの分泌の倍増率で表す)。これとは著しく対照的に、抗−CD3および抗−D28抗体によって達成したTCR活性化は、全4つのThサイトカインの強健であるが、非選択的な分泌を誘起した(図2E〜2H)。予想したように、TCR活性化後のTNF−α分泌の規模は、DTX−K処理後に得られるものに比べて、ずっと大きかった(図2Eに対する図2A)。TCR活性化の間または後に、DTX−KによってT細胞のチャンネルを含むKv1.1サブユニットを遮断しても、TCR活性化のみによって達成された以上には、TNF−αレベルの更なる増加は起こらなかった(データは示されない)。
【0079】
実施例4:T細胞のチャンネルを含むKv1.1サブユニットの遮断により、長期間のTNF−αの増加が生じる
T細胞のTNF−α分泌は、TCR活性化後、通常は24時間でピークに達し、その後徐々に低下する。それ故、T細胞内のチャンネルを含むKv1.1サブユニットの遮断によって起きるTNF−α分泌を調査し、この時期にピークに達するかどうか、および分泌が一時的かあるいは長期なものかを検討した。図2Iおよび2Jにおいて、個体4(図2I)および個体5(図2J)(既に図1F、2Gに示した同じ二人の個体)からの正常T細胞を100nM、DTX−Kに曝し、その後異なった時点でELISA法により、培地をTNF−αについて検査した。それぞれの個体について、それぞれの時点でのTNF−α分泌の倍増率±SDを示す。個体4のそれぞれ16、24、48および72時間において、未処理に対して*P=0.0053、0.0052、0.0050および0.0053であり;個体5のそれぞれ16、24、48および72時間において、未処理に対して*P=0.0057、0.0063、0.0072および0.00316であった(Student's t-test)。
【0080】
この実験により、DTX−Kに誘導されたTNF−α分泌の動態が(図2I,2J)、それらのバックグラウンドおよびDTX−Kに誘導された、T細胞由来のTNFαレベル(図1F、2G)と比較していく分異なっていることを明らかにする。これらの相違にもかかわらず以下の二つの特徴は共通している:1)有意に増加したTNF−αの分泌は、16時間後において既に明らかである;2)TNF−αの分泌は、DTX−K処理の開始から72時間後であっても有意に高い状態を維持した。
【0081】
実施例5:DTX−K誘導性TNF−α分泌にはT細胞膜を通してCa2+の流入が必要とされる。
Ca2+は、TNF−αの転写に関与していることが知られている(Lobo et al, 1999)。これに基づいて、T細胞のチャンネルを含むKv1.1サブユニットを遮断することによって生じる著しいTNF−αの分泌には、「連続的な」Ca2+の流入が必要となるかどうかを検討した。T細胞内に発現されたL型Ca2+チャンネルの強力で非常に選択的な遮断剤を用いた:(R) - (+) - Bay K 8644であって、DTX−K(100nM)の添加の5分前に正常ヒトT細胞に加えた。選択的なCa2+チャンネル遮断剤は、DTX−KのTNF−αの分泌を引き起こす能力を完全に阻止したが、対照として加えたCl−チャンネル遮断剤、NPPB(表1)は、該阻止能力を有しなかった(図2K)。この実験において、Kv1.1遮断(100nM DTX−K、24時間)に対する反応におけるTNFαの分泌は、T細胞を100μM +Bay-K(高度の選択的な、T細胞中で発現されたCa2+チャンネル遮断剤で、DTX−Kの5分前に加えられた)で前処理した場合は、完全に阻止されたが、関係ないCl−チャンネル遮断剤、NPPBの100μMで前処理を行った場合は阻止されなかった。結果は、実施した3回の実験の一つからのTNF−αの分泌の平均(pg/ml)±SDを表している。統計解析:未処理に対して*P<0.0001、DTX−Kのみに対して**P<0.0001(Student's t-test)。
【0082】
実施例6:T細胞内のチャンネルを含むKv1.1サブユニットの遮断により、またTNF−α mRNAレベルも増加する。
次に、T細胞のチャンネルを含むKv1.1サブユニットの遮断により、TNF−αタンパクだけではなく、TNF−αのmRNAのレベルも増加するかについての問題を研究した。この問題を研究するために、DTX−Kをヒト正常T細胞に24時間加え、RNAを調製し、cDNAに転写し、TNF−α特異的なプライマーを用いて、特異的なTNF−αRT−PCRを行った。逆転写の効率のコントロールとして、内部標準(リボソームS14タンパク)を用いた更なるRT−PCR反応を同時に行った(すなわち同じPCRチューブにおいて)。さらに具体的に言うと、正常末梢ヒトT細胞を100nMDTX−Kに24時間曝した。それからcDNAを調製し、TNFαおよび内部コントロール、すなわちリボソームS14タンパク、に対する特異的なプライマーを用いて、同じPCRチューブ内で同時に二つのRT−PCR反応によって増幅した。結果を図3Aに示す。上段のバンド:TNF−α(予想される産物のサイズ:494bp);下段バンド:S14(予想される産物のサイズ:166bp)。下の棒グラフは、TNF−α/S14の対応するRT−PCRバンドの濃度解析および正規化の後に計算したTNF−αmRNAの倍増率を表す。NC=陰性コントロール(cDNA無し)。
【0083】
図3Aは、チャンネルを含むKv1.1サブユニットのDTX−Kによる遮断によって、実際、TNF−α mRNAレベルが増加することを示している。
【0084】
実施例7:チャンネルを含むKv1.1サブユニットの遮断は、正常ヒトT細胞のNF−
κBの活性化を導く。
TNF−αは(IL−1のような他の炎症促進性のサイトカインのように)NF−κBを活性化させるだけではなく、NF−κBによってもまた誘導される(Siebenlist et al, 1990)。TNF−α遺伝子の場合は、TNFα遺伝子の発現に対して観察されたNF−κBの重要性は、細胞タイプと解析された、エンハンサー/プロモータの区域に依存するのかも知れない(Siebenlist et al, 1990)。これらに基づいて、正常ヒトT細胞(ここでもまた、いかなる他の刺激の完全に存在しない状態で)におけるKv1.1チャンネルの遮断が、NF−κBに影響を及ぼすことができるかを我々は検討した。これを二つの技術によって研究した:1)未処理およびDTX−K処理正常ヒトT細胞から核抽出物を調製し、市販の電気泳動移動度シフトアッセイ(EMSA)によって核NF−κB結合配列に結合したNF−κBの量を測定する;2)抗−ヒトNF−κB p50特異的抗体、FITC結合抗−ヒトIgGおよび細胞核を特異的に染色するHoechst 33342を用いて、免疫組織化学染色法によって、推定されるNF−κBの核への移行を可視化する。
【0085】
核抽出物を数時間加えたが、そのうち7分のみを示す。OD±SDで示した結果は、市販のEMSAによって定量された、核NF−κB結合配列に結合したNF−κBの量を反映している。別個に行った二つの実験の一つを図3Bに示す。統計解析:*P=0.0092(Student's t-test)。
【0086】
OD値が核NF−κB結合配列に結合しているNF−κBの量を表す、図3Bから見られるように、DTX−K添加後7分で核抽出物に存在するNF−κBのレベルは有意に増加した。予想されたように、この増加は一時的なものであって、しばらく経つと試験した点はもはや検出されなかった(データは示していない)。EMSAによって検出したこの結果は、図3Cに示す二重免疫蛍光写真と一致していた。図3Cに結果が示してある実験において、正常ヒトT細胞は、処理しないかまたはDTX−Kで7、15、30および60分処理した(それぞれのカラムの上のDTX−K処理時間を参照)。免疫蛍光組織化学染色は、抗−ヒトNF−κB p50抗体、次いでFITC結合抗−ヒトIgGを用いて行うか、あるいは細胞核に取り込まれるHoechst 33342を用いて行った。拡大率x100の代表的な写真は、NF−κB p50緑色蛍光(上段のパネル)、核の青色蛍光(中段パネル)両者を重ねたもの(下段パネル)を示す。研究したT細胞の外形サイズ(明視野)および核(Hoechst)は、左のカラムに示す。従って、図3Cは、DTX−K添加後7分(左から3番目のカラム)で細胞核(Hoechst青色蛍光)は明瞭にNF−κB p50陽性(緑色蛍光)であるが、未処理の細胞においてはそうではなく、又試験した後の三つの時点:15,30および60分では(図3C,それぞれ三つの右のカラム)NF−κB p50の蛍光は見られなかった。注目すべき点は、DTX−K処理後7分において、細胞質(Hoechst 陰性)は、NF−κB p50に対して陽性に染色されたが(図3C,左から3番目のカラム)、未だ明瞭でない理由により細胞質NF−κB p50は、未処理細胞においては観察されない。唯一の論理的なしかし推測でしかない、考えられる説明は、本明細書で用いる抗−p50抗体は立体構造にいくらか敏感であり、もしそうなら活性化およびNF−κB阻害剤IκB(Siebenlist et al , 1990)の放出後、細胞質p50は、この抗体によって検出しやすくなる可能性がある。
【0087】
実施例8:正常ヒトT細胞は電位依存性Kv1.1タンパク質およびmRNAを発現する。
先の研究は、機能的な(すなわち電気生理的に活性を有する)チャンネルを含むKv1.1サブユニットは、主として脳、心臓、網膜、骨格筋および膵島で発現されていることを示した(Beckh et al , 1990; Klumpp et al , 1991; Matsubara et al, 1991; Roberds et al, 1991; Tsaur et al, 1992)。対照的にT細胞中のKv1.1電流に関しては、先の研究から現れる像は不明瞭である。いくつかの研究では、パッチ−クランプ技法によってヒトT細胞がKv1.1の特徴を発現するかを試験したが、該電流を同定することができなかった(例えば(Cahalan et al, 1985))。しかしこれと対照的に、Freedman et alはマウスCD4−CD8−胸腺細胞によるKv1.1電流/発現を示し(Freedman et al, 1995)、Liu et alは、いくつかの方法の使用に基づいて、未処理のマウスCD4+リンパ球に発現している電流は、Kv1.1、Kv1.1、Kv1.3およびKv1.6と一致すると報告した(Liu et al, 2002)。本明細書においては、Kv1.1の全く新規な面を研究して、正常末梢ヒトT細胞においてKv1.1タンパクおよび、mRNAの発現を確認することを欲した(本研究の範囲を逸脱するKv1.1電流について再び試験することなしに)。本方法は、Kv1.1特異的なプライマーを用いたKv1.1特異的RT−PCR、ならびにKv1.1特異的な抗体を用いたフローサイトメトリー(この目的のためには今まで使用されていなかった方法)を含む。
【0088】
図4Aは、cDNAを正常末梢ヒトT細胞から調製し、Kv1.1特異的プライマーを用いてKv1.1特異的RT−PCRを行った結果を示す(上段バンド;予想産物サイズ:709bp)。コントロールPCRは、同時に同じPCRチューブで、リボソームS14プライマーを用いて行った(下段バンド;予想産物サイズ:166bp)。図4Aは、Kv1.1特異的mRNAが実際に正常ヒトT細胞のcDNAから増幅されることを示す。更に加えて、Kv1.1特異的な免疫蛍光染色を行った。T細胞は、ウサギ抗−Kv1.1ポリクローン抗体(図4B、実太線)あるいはアイソタイプコントロール非特異的染色として、正常ウサギIgG(図4B、破線)で染色し、それからFITC結合抗ウサギIgGで染色した。Kv1.1発現は、FACSortによって評価した。五つの独立した実験(異なった個体のT細胞を用いた)の一つを示す。ここでヒトT細胞の49.0%は、Kv1.1陽性であった。行われた他の四つの実験においては、Kv1.1陽性T細胞の百分率は40−80%の間で変異した。従って、図4Bは、フローサイトメトリー解析によって、Kv1.1タンパク質が正常末梢ヒトT細胞のかなり実質的な割合で細胞表面に明瞭に発現していることを示す。
【0089】
実施例9:市販の抗−Kv1.1チャンネル抗体は顕著なTNF−αの分泌を誘発し、それにより選択的なKv1.1遮断剤の効果を模倣する。
この実験において、抗−Kv1.1特異的抗体が、Kv1.1チャンネル遮断剤、DTX−Kのように作用し、TNF−αの分泌を増加させるかを、我々は検討した。3人の更なる個体(各個体7−9)からの正常ヒトT細胞を、市販のウサギ抗−Kv1.1ポリクローン抗体とインキュベーションし(1:1000倍希釈、24時間)、培地中に分泌されたTNF−αのレベルをELISA法によって試験した。結果を図4C、4Dおよび4Eに示し、各個体のT細胞によって分泌されたTNF−αの平均±SDを提示する。それぞれ未処理の個体7〜9に対して、*P=0.0003、0.0057、および0.0021であった(Student's t-test)。
【0090】
図4C〜4Eは、3人の異なった該被験体に由来する正常ヒトT細胞と24時間インキュベーションした、市販の抗−Kv1.1抗体は、これらの細胞によって分泌されるTNF−αの量を、それぞれ7.2、14.8および23.2倍増を誘導したことを示す。ここで注意すべきことは、ここで用いた抗−Kv1.1抗体はKv1.1チャンネルの細胞内エピトープに対して指向性を有していることである。この抗体は、T細胞内に侵入した後TNF−αの分泌を誘導すると考えられているが、これは抗体の生体細胞内に侵入する能力を明瞭に示した先の報告に基づいている(Alarcon-Segovia et al , 1996; Ruiz-Arguelles et al, 2003)。事実、種々の自己免疫疾患において、病原となる抗体は細胞内エピトープに対して指向性を有しており(例えば、全身性エリテマトーデスにおける抗−DNAおよび抗−リボヌクレタンパク抗体)(Alarcon-Segovia et al , 1996; Ruiz-Arguelles et al, 2003)、細胞内に侵入後にのみそれらの有害な効果を発揮する。
【0091】
実施例10:Kv1抗体陽性末梢神経過剰興奮症(PNH)血清は、正常ヒトT細胞
によるTNF−αの分泌を著しく促進する。
この実験は、PNH患者の血清に存在する病原性のKv1抗体(すなわち、Kv1.1、Kv1.2およびKv1.6抗体)(Hart et al, 2002)が、市販Kv1.1抗体およびDTX−Kの作用を再現するか、又ヒトT細胞によるTNF−α分泌を増加させるかを検討する。PNHの通常提示される臨床症状は、筋肉のれん縮、硬直、こわばりおよび脱力感であって、多汗症、偽性ミオトニー現象、偽性テタニーおよび感覚性の症状である(Newsom- Davis et al, 1993)。加えて、PNH患者は、CNS症状を有することもある(Vincent et al, 2004)。PNHの殆どの症例は自己免疫症であって、Kv1.1、Kv1.2およびKv1.6抗体の混合物である循環性のKv1抗体(Hart et al, 002)によって起こる。
【0092】
この仮説を試験するために、正常末梢ヒトT細胞を健常コントロール被験体(A群)Kv1抗体陽性PHN患者(B群)またはKv1抗体陰性患者(C群)の任意の血清に(1:100倍希釈)に曝した。全ての群は年齢、性および地理的に適合させた(方法を参照)。続く24時間に培地に分泌されたTNF−α(図4F)またはINF−γ(図4G)のレベルは、ELISA法によって試験した。結果は、行われた三つの独立した実験の平均である。図4Fにおいて、水平バーおよびそれぞれの数値は中央値を表す。ここでは下カットオフ(カットオフA)である破線は、コントロール群の血清に曝されたT細胞によって分泌されたTNF−αの平均+2*SDを表し;ここでは上カットオフ(カットオフC)は、Kv1−陰性PNH血清に曝されたT細胞によって分泌されたTNF−αの平均+2*SDを表す。グラフに示されたそれぞれの数値は、分泌されたTNF−αの実際の平均量(pg/ml)を表す。3つのKv1陽性PNH患者(つまり、PNH+5,2および6)の血清は、明瞭にカットオフCの上のTNF−αの分泌の増加を誘導した(しかしINF−γは誘導されない)。統計解析:KW統計=8.0003、P=0.0183,Kruskal-Wallis 検定。対比較はMann-Whitney U検定でおこなった:A群対B群、P=0.0117、A群対C群、P=0.0087、C群対B群、P=0.6282。
【0093】
図4は、いく人かのKv1抗体陽性PNH患者の血清は、健常コントロール被験体または検出可能なKv1抗体を有しないPNH患者からの血清によって誘導されるよりは有意に高い、著しいTNFαの分泌を誘導することを示している。更に、TNF−α誘発効果は、抗体力価と正の関係にあった(材料および方法参照)。PNH患者からの血清の同体積および濃度を含むがT細胞を含まない、陰性コントロール実験培養には、TNF−αレベルに影響は無かった。重要なことには、PNH患者のKv1抗体陽性血清は、TNF−αの増加を誘導したが、DTX−Kで観察された(図2)と同様に、INF−γの増加は誘導せず(図4G)、この効果の選択性を確認している。総合的に、この一連の実験は、増加したKv1抗体力価を含むいくつかのPNH患者の血清は、正常ヒトT細胞による著しいTNFα分泌を誘導し、それによって市販抗−Kv1.1抗体および選択的Kv1.1遮断剤、DTX−Kの効果を模倣することを示した。
【0094】
実施例11:前処理Kv1.1遮断により上昇したTNF−αレベルを分泌する正常ヒトT細胞を、SCIDマウスへ注射することで、肝臓へ宿主細胞を選択的に動員させる。
Kv1.1の選択的遮断に反応しているヒトT細胞によって分泌するTNF−αが、インビボにて機能的であるかを試験するために、我々は、最近の研究を想起したが、それは、脳内へTNF−αを投与に続いて一連の現象が起こり、その中には、特異的に肝臓に蓄積する宿主のED1−陽性細胞(すなわち常在および活性化クッパー細胞/動員された血中単球)の急速な2倍までに増加する、というものであった(Campbell et al, 2005)。我々は、DTX−K処理済のヒトT細胞によって分泌された、増加したTNF−αも該効果を誘導するかを検討した。このことを試験するために、新たに精製した正常ヒトT細胞を、PBSまたは100nM DTX−Kに24時間曝し、洗浄し、蛍光標識を付け、それから後者の細胞が増加したTNF−αを分泌していることを確認した後(データは示していない)、SCIDマウス(すなわち自身のリンパ細胞を欠いている)に注射した(各群n=5)。肝臓、骨髄、脾臓、および腎臓の単細胞懸濁ならびに血液細胞に存在する、細胞の総数、ならびに常在SCID細胞のみ(非蛍光)および注射されたヒトT細胞のみの数を、24時間後FACSortによって計測した。肝臓に関しては、結果は非蛍光SCID宿主細胞の平均±SEM数(図5A)および蛍光標識ヒトT細胞の平均±SEM数(図5B)で表される。他の臓器に関しては、非蛍光SCID宿主細胞の数のみを示す:骨髄(図5C)、血液(図5D)、脾臓(図5E)および腎臓(図5F)、未処理に対して*P<0.0001(Student's t test)。図5Gは、注入されたヒトT細胞の検出のために、エクスビボにてDTX−Kと前インキュベーションしたヒトT細胞を注射したSCIDマウスの脳切片の免疫組織化学像を示す。SCIDマウスにDTX−K処理ヒトT細胞を注射後24時間、CD3陽性細胞が脳皮質内に検出されることを示すx40の拡大率の代表的な写真である。
【0095】
図5AはTNF−αを分泌しているDTX−K処理ヒトT細胞の注入は、肝臓における細胞数を2倍にしたことを示す。我々が依拠した関連研究において(Campbell et al, 2005)、この効果は肝臓に限定されていて、骨髄、血液、脾臓、および腎臓における細胞総数は、有意には変化しなかった(それぞれ図5C〜5F)。更に、予想されるように、肝臓における細胞数の増加はマウス(SCID)宿主細胞の動員に由来するもので(図5A)、受動的に移したヒトT細胞がこの臓器に入ったことに由来するのではなかった(図5B)。技術的な制限によって、動員したマウス細胞の正確な同定は、行わなかった。最後に、脳の切片の免疫組織化学像は、注射されたSCIDマウスの皮質内に、注射されたDTX−K処理済のヒトT細胞の存在を明らかにした(図5G)これは、原則として、肝臓への宿主細胞の生体内動員は、脳内で作動しているTNF−α(すなわちDTX−K処理ヒトT細胞によって脳へ送達されたTNF−α)によって介される可能性を示唆しており、TNFαを脳内に注射後、宿主細胞が肝臓に動員された最近の報告に類似している(Campbell et al, 2005)。実際、TNFαのBBBを混乱させる能力に基づけば(Mayhan 2002)、DTX−K処理細胞が脳に侵入することができることは、驚くには当たらない。脳に到達した未処理およびDTX−K処理ヒト細胞の数の間の統計的に正当な比較は不可能である。最後の注意点として、肝臓に動員されたマウスSCID細胞が、ヒトTNF−α(DTX−K処理ヒトT細胞によって分泌される)に反応できたことは、我々にとって驚くべきことではない。なぜならマウスTNF−α受容体1型は組み換えマウスおよびヒトTNF−αの両方に同様な親和性を有しているからである(Lewis et al, 1991)。更にヒトTNF−αを発現しているトランスジェニックマウスは、リウマチ性関節炎を有するヒトの患者の特徴である多くの徴候を発現し(Keffer et al, 1991; Li et al , 2003)、マウス起源の細胞がヒトTNF−αに応答することができることを示唆している。
【0096】
具体的な実施形態の先の記述は、本発明の一般的な性質を十分に明らかにしているので、他者は、現在の知識を応用することにより該具体的な実施形態を、必要以上の実験を行うこと無く、一般概念から逸脱すること無く、容易に改変しおよび/または種々の応用に適応することができ、それ故該適応および改変は、開示した実施形態と等価の意味と範囲内に包含することを意図すべきであり、また意図している。本明細書で用いられた言葉使いや用語は、記述するためであって、限定するもので無いことを理解されたい。種々の開示した機能を実施するための手段、材料および段階は、本発明から逸脱すること無く種々の代替型を取ってもよい。従って、上記の明細書および/または請求項に存在する可能性のある、「・・・に対する手段」および「・・・のための手段」あるいは機能的な記載に続くいかなる方法段階の用語は、あらゆる構造、物理的、化学的または電気的要素または構造、もしくは現在または将来存在する可能性のある、引用した機能を実施するいかなる方法段階をも包含することを意図している、すなわち同じ機能を実施するための他の手段または段階を用いることができ;該表現はその最も広義に解釈されることを意図している。
【0097】
【表8】
【図面の簡単な説明】
【0098】
【図1】図1Aは、それぞれの試験されたイオンチャンネル遮断剤についての、TNF−αの倍増率を示すグラフである。 図2Bは、用量の関数としてのTNF−αの倍増率を示すグラフである。 図1C〜Hは、6人の異なった該被験体についての、それぞれのDTX−K誘導TNF−α分泌の量を示すグラフである。
【図2】図2A〜Hは、DTX−K(図2A〜D)または抗−CD3および抗−CD28mAb(図2E〜H)に曝された正常ヒトT細胞によって分泌された種々のサイトカインのレベルを示す。TNF−α分泌の倍増率を図2Aおよび2Eに、IFNγを図2BおよびFに、IL−10を図2CおよびGに、ならびにIL−4を図2DおよびHに示す。 図2IおよびJは、二人の異なった該被験体においてそれぞれのTNF−α分泌の倍増率を、時間の関数として示したグラフである。 図2Kは、未処理、DTX−Kのみの処理、またはBay−KまたはNPPSのいずれかで最初に前処理済のT細胞についてのTNF−α分泌を示すグラフである。
【図3】図3Aにおいては、正常末梢ヒトT細胞をDTV−Kに曝した後、上部のバンドはTNF−α(予想産物サイズ:494bp)および下部のバンドはS14(予想産物サイズ:166bp)を示す。下の棒グラフは、濃度測定分析および対応するTNF−α/S14のRT−PCRバンドの規格化の後計算したTNF−αmRNAの倍増率を表す。NC=陰性対照(cDNA存在せず)。 図3Bは、100nMDTX−Kに先に曝された正常T細胞から調製した核抽出物における、核NF−κB−結合配列に結合したNF−κBの量を示す。 図3Cは、正常ヒトT細胞が未処理であるか、または100nM DTX−Kと7、15、30および60分処理済の(上記DTX−K処理時間についてはそれぞれのカラムを参照)、二重免疫蛍光写真を示す。拡大率x100の代表的な写真は、NF−κB p50 緑蛍光(上段パネル)、核 青色蛍光(中段パネル)および両者を重ねたもの(下段パネル)を示す。試験したT細胞(明視野)および核(Hoechst)の外形サイズを左のカラムに示す。
【図4】図4Aは、Kv1.1特異的なプライマーを用いて行ったKv1.1特異的なRT−PCRの結果を示す(上段バンド;予想産物サイズ;709bp)。コントロールRT−PCRは、リボソームS14プライマーを用いて同じPCRチューブの中で同時に行った(下段バンド;予想産物サイズ;166bp)。 図4Bは、T細胞を、ウサギ抗−Kv1.1ポリクローン抗体で(図4B、太実線)、または、アイソタイプコントロール非特異的染色として、正常ウサギIgG(図4B、破線)で染色し、それからFITC結合抗−ウサギIgGで染色した後の、FACSortの結果を示す。 図4C〜Eは、3人の異なった該被験体のそれぞれ個人のT細胞によるTNF−α分泌の平均値の結果を示したもので、それらのT細胞を市販ウサギ抗−Kv1.1ポリクローン抗体とインキュベーションした。 図4FおよびGは、正常末梢ヒトT細胞を、健常コントロールの個人(A群)、Kv1抗体−陽性PHN患者(B群)またはKv1抗体−陰性PHN患者(C群)の血清に曝し、培地中に分泌されたTNF−α(図4F)またはIFN−γのレベルを示す。
【図5】図5A〜Fは、新たに調製した正常ヒトT細胞を、PBSまたは100nM DTX−Kに24時間曝し、洗浄し、蛍光標識を付け、それからSCIDマウスに注射した後の結果を示す。グラフは、肝臓、骨髄、脾臓および腎臓の単細胞懸濁液ならびに血液に存在する細胞の総数、ならびに常在のSCID細胞(非蛍光)のみの数および注射されたT細胞(蛍光)のみの数を示す。図5Aは、肝臓における非蛍光SCID宿主細胞の平均±SEM数を示し、又図5Bは、肝臓における蛍光標識されたヒトT細胞の平均±SEMを示す。他の器官については、非蛍光SCID宿主細胞の数だけを示す:骨髄(図5C)、血液(図5D)、脾臓(図5E)および腎臓(図5F)。 図5Gは、注入されたヒトT細胞の検出のために、DTX−Kとエクスビボにて前インキュベーションしたヒトT細胞を注射したSCIDマウスからの脳切片の免疫組織化学像を示す。SCIDマウスにDTX−K処理したヒトT細胞を注射後24時間、ヒトCD3陽性細胞が皮質内に検出されることを示す、拡大率x40の代表的な写真を提示する。
【技術分野】
【0001】
発明の背景
30年前に分離された腫瘍壊死因子アルファ(TNF−α)は、アポトーシスおよび細胞生存、ならびに炎症および免疫において主要な役割を果たす多機能なサイトカインである。その抗腫瘍性から名付けられたが、TNFは広範囲の他の疾患に関わるとされる。現在癌におけるTNFの使用は、局所的に進行した軟組織肉腫、転移メラノーマおよび組織学的な他の切除不可能な腫瘍の局所的な治療において患肢の切断を避けるためのものである。TNF−αは、分離患肢潅流の設定において、細胞増殖抑制剤と相乗的に作用することが示された。TNF−αとTNFの受容体1および2(TNFR−1、TNFR−2)の相互作用は、いくつかのシグナル伝達経路を活性化させ、TNF−αの多様な機能を発揮させる。TNFR1のシグナル伝達分子は、非常に良く解明されているが、シグナル伝達の制御については不明である。これらの分子についての洞察のほかに、過去10年間の研究室における実験は、腫瘍治療期間中のTNF−αの作用に光を当てた。出血性壊死を引き起こす、赤血球およびリンパ球の血管外遊出の他に、TNF−αは、血管内壁の透過性増大および破壊を誘導することによって、腫瘍関連脈管構造(TAV)を標的とする。この結果、腫瘍内での細胞増殖抑制剤の選択的蓄積の即効性、および腫瘍の脈管構造の破壊の遅発性効果が起こる。最近公表された総説(van Horssen等(2006))は、TNF−αについて分子から臨床までを記述し、TNFR−1シグナル伝達の分子的洞察およびTNF−αの抗腫瘍活性の細胞機構から始まり、臨床反応で終わる、癌におけるTNF−αの使用の概観を提供している。更に加えて、TNF−αの活性をモデュレーションしている可能性のある因子についても議論している。
【0002】
TNF−αが、1975年バクテリアエンドトキシンで処理済のマウスの血清から「Coleyの毒素」の活性成分として分離され、マウスの腫瘍に出血性壊死を引き起こすことが示された。ニューヨークの外科医であったWilliam Coleyが、彼の調製した細菌濾液(「Coleyの混合毒素」)でいく人かの癌患者を治療した際に高熱と腫瘍の壊死を観察したのは、殆ど1世紀前であった。その分離の10年後、TNF−αは又「カケクチン」およびT細胞分化因子と位置づけられた。1984年ヒトTNF−α遺伝子がクローン化され、一連の臨床実験が設定され、分離潅流の設定における患肢を脅かすSTSの治療のための、欧州医薬品審査庁からの認可を得た(上記に関してはvan Horssen等(2006)、およびそこに引用した論文を参照)。
【0003】
赤血球を除いて、実際ほとんどすべての細胞型に発現している2つの高親和性受容体、TNFR1(p55)およびTNFR2(p75)を活性化することにより、TNF−αは、後天的免疫性および自然免疫性、ウィルスおよび細菌のような外来の侵入物と闘うために必要な炎症の誘導、腫瘍の根絶、オリゴデンドロサイトの再生と増殖の促進、神経の再ミエリン化その他多くのものを含め、めまぐるしく変化する病気において主要な役割を果たしている。
【0004】
T細胞(Tリンパ球とも呼ばれる)は、TNF−αの主要な源である。T細胞からTNF−αの分泌を引き起こすことが知られている主要な刺激は、T細胞受容体(TCR)の関与であって、自己主要組織適合性複合体分子の構成において、抗原提示細胞によってT細胞に提示される抗原と、T細胞の出会いの時に起こるのと類似している。該「古典的な」TCR活性化により、T細胞は強健なしかし非選択的なサイトカイン分泌(すなわちこれらの細胞によって分泌され得る全てのサイトカインの同時分泌)を引き起こす。
【0005】
TNF−αは、ヒトの生理および病理において二重の役割を果たす:一方ではTNF−αは、疾患と闘い治療するために決定的に必要とされる;他方過剰のTNF−αは、疾患および組織破壊を引き起こすため、有害である。
【0006】
一般的に、先天的免疫反応の活性化におけるTNF−αの主要機能は、種々の型の外来侵入物から体を保護するために、および癌細胞を根絶するために、有益で重要であると一般的に考えられている。TNF−αは又、種々の分子に対する血管および血液脳関門(BBB)の透過性を大幅に増加させ、および組織への移動と侵入に必要とされる、白血球のローリングと付着を増大させる。したがって、TNF−αの欠如または不足は、広範囲で病的な結果を招くことは明らかであり、その結果の中には、免疫能および、感染性生物と対抗し、癌等を根絶する能力の低下が含まれる。
【0007】
TNF−αが癌細胞を殺す能力に基づき、TNF−αは、分離患肢潅流(ILP)の現場において、軟組織肉腫(STS)、種々の組織学的切除不可能な腫瘍、および患肢に限定された移行転移メラノーマに対する癌治療に現在用いられている(van Horssen等(2006)およびそこに引用した論文)。
【0008】
van Horssen等(2006)の表1に、高悪性度(Wallach等、1999;Mayan、2002)STSの治療のため、TNF−αのILPでの適用に対し、1998年のEMEAによりTNF−αが承認されることになった、欧州での多施設臨床試験の概要が示されている。これらの多施設臨床試験においては、76%の全奏功率および82%の患肢救済中央値を観察した。さらにこの表には、多施設における経験を確認する、STSの最大の単一施設での研究を載せている。衝撃的に一貫した、中央値76%(レンジ、58%〜91%)主要な奏功率が、中央値84%(レンジ、58%〜97%)の患肢救済率を伴って観察された。欧州においては、患肢救済のための国内照会形の35の癌センターにおいて、TNF−αに基づくILPが現在行われている。移行転移メラノーマに対するメルファラン単独ILPは、文献報告によれば、約50%の完全奏功(CR)率および80%の全奏功率を引き起こすと報告されている(van Horssen等(2006))およびそれに引用している論文を参照)。この設定におけるTNF−αの導入は、CR率を70%−90%に、全奏功率を95%〜100%に増加させると報告された。これらの結果は、van Horssen等(2006)における表2に要約してある。
【0009】
一方、ヒトにおける不適切なTNF−αまたはTNF−αの過剰産生は、制御されない有害な炎症、組織破壊および器官の損傷を引き起こすこともまた明らかであり、多くの研究において報告されている。該TNF−αの過剰産生は、リウマチ性関節炎、多発性硬化症のような種々の自己免疫疾患、炎症性腸疾患のような炎症性疾患ならびに他の病的状態、そのうち例えば、強直性脊椎炎および脳卒中、において起こる。
【0010】
中枢神経系(CNS)に於いて、TNF−αは又二重の役割を果たす:一方では、TNF−αは有益であって、オリゴデンドロサイトの増殖と再生、神経細胞保護、神経再ミエリン化および種々の他の重要なプロセスに必要とされる。他方、過剰の内在TNF−αは、神経細胞傷害および神経変性に寄与する。中枢神経系(CNS)機能のモデュレーション時に炎症および免疫分子の役割により多くの注目が寄せられている。腫瘍壊死因子−α(TNF−α)は、炎症促進性のサイトカインであり、その受容体はCNS全体の神経細胞およびグリア細胞上で発現している。その二つの受容体の作用を通じて、TNF−αは、神経保護的または神経毒性の広範囲な作用を神経細胞に及ぼす。それは直接的および間接的に、アストロサイト上のグリアのグルタミン酸輸送体を阻害することにより、グルタミン酸の興奮毒性を促進する役割を果たす。さらに、TNF−αは、例えばシナプス上のAMPA受容体の発現を増加させ、グルタミン酸伝達に直接作用する。TNF−αは又、p38MAPキナーゼに依存するプロセスである、長期増強(LTP)を阻害することによりシナプス可塑性の役割を果たす。
【0011】
TNF−αの欠如または不足、すなわち免疫能または他のTNF−αを介した有益な効果の欠如または不足、を特徴とする特定の病態において、他のいかなるサイトカインをも増大させることなくTNF−αの産生、発現および分泌だけを増加させる新規な方法を発見することは、治療価値を有することは、上記から明らかである。
【0012】
以下に、TNF−αの増強および標的細胞に及ぼすその後の作用が治療戦略と成りうる、病的状態の例を示す:
a.腫瘍を殺すまたは除去することができないため、死に至る癌(van Horssen等(2006)、TNF−αの抗腫瘍効果および臨床的有用性についての最近の総説のすべての内容は参照により本明細書に援用される);
b.死に導く可能性のある、ウィルスおよび細菌のような外来侵入物と闘うことができなくなる慢性または急性の免疫不全;および
c.重要な神経機能がついには欠損し、生命を危険にする可能性のある、疾患または傷害に伴う神経細胞の再生の欠如または不足。
【0013】
過剰な又有害なTNF−αを特徴とする特定の病態に於いて、TNF−αの産生、発現および分泌を停止する新規な方法を発見することは、非常に治療価値を有することは、先の序論からも明らかである。
【0014】
以下は、TNF−αを停止または減少させおよびその後の標的細胞に及ぼすその影響が、以下の治療戦略になり得る病的状態の例である;リウマチ性関節炎、乾癬性関節炎、1型糖尿病、クローン病、乾癬、潰瘍性大腸炎、強直性脊椎炎、サルコイドーソスおよび頭部傷害後。
【0015】
発明の要約
本特許出願の根拠としての機能を果たす研究において、本発明の発明者らは、T細胞中のKv1.1電位依存性カリウムチャンネルの選択的閉鎖は、TNF−αの劇的な又独占的な合成および分泌を引き起こすために、単独で十分であって(すなわち任意の他の分子の存在無しに)、試験した他のサイトカインのいずれにも、すなわちインターフェロンガンマ、インターロイキン−4およびインターロイキン−10にも影響を与えないことを発見した。
【0016】
これらの発見に基づいて、T細胞内のKv1.1電位依存性カリウムチャンネルの閉鎖または遮断は、TNF−αを介する効果が治療的価値を有するような状況において、T細胞によるTNF−αの急速、強健および選択的な合成および分泌を達成するであろうことが分かった。
【0017】
T細胞におけるKv1.1電位依存性カリウムチャンネル閉鎖は、デンドロトキシン−K(DTX−K)のような高度に選択的なKv1.1チャンネル遮断剤、または特異的な抗−Kv1.1抗体、または下流経路の遮断によって、細胞外的にもしくは細胞内的に、T細胞のKv1.1チャンネルの選択的遮断を導く任意の他の方法によって本明細書中で達成される。
【0018】
従って、本発明の一つの目的は、T細胞に大量のTNF−αを産生および分泌させ、疾患や傷害と闘うために、TNF−αの欠乏と関連している病態を患っているヒトまたは動物被験体のT細胞内のKv1.1チャンネルを遮断する方法を提供することである。
【0019】
本発明の更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、選択的なKv1.1イオンチャンネル遮断剤である方法を提供することである。
【0020】
本発明の更なる目的は、T細胞に大量のTNF−αを産生および分泌させ、疾患や傷害と闘うために、T細胞によるTNF−αの増大した唯一の分泌により恩恵を受けることができるヒトまたは動物被験体のT細胞内のKv1.1チャンネルを遮断する方法を提供することである。
【0021】
本発明の別の目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、特異的な抗Kv1.1抗体であるような方法を提供することである。
【0022】
本発明の更なる別の目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、T細胞内のKv1.1チャンネルの下流経路を遮断できる分子であるような方法を提供することである。
【0023】
本発明の又更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子を、エクスビボにてT細胞に投与後にヒトまたは動物患者の体内へ接種して戻すような方法を提供することである。
【0024】
本発明の更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、体内に投与される、すなわちヒトまたは動物患者の体内に、静脈内、皮下、腹腔内、腫瘍内、くも膜下腔内または頭蓋内注入により、もしくは経口投与または経皮的に軟膏により投与されるような方法を提供することである。
【0025】
本発明の又更なる目的は、T細胞内の電位依存性カリウムチャンネルKv1.1を遮断するために用いる分子が、ヒトまたは動物被験体の体内に植え込み式の薬剤送達ポンプを用いて投与されるような方法を提供することである。
【0026】
本発明の別の目的は、ヒトまたは動物被験体が癌に罹患しており、それら自身のT細胞を、体外または体内で、T細胞のKv1.1チャンネルを遮断する分子に曝すことにより、TNF−αの強健な合成または分泌を誘発し、TNF−αによって癌を直接、死滅させる方法を提供することである。
【0027】
好ましい一実施形態では、ヒトまたは動物被験体が癌に罹患しており、それら自身のT細胞を、体外または体内で、T細胞のKv1.1チャンネルを遮断する分子に曝すことで、TNF−αの強健な合成または分泌を誘発し、腫瘍を有する器官へのT細胞の侵入を増加させ、腫瘍破壊細胞の動員を向上させるための有益な炎症環境をつくることにより、癌のT細胞免疫療法を増強する。
【0028】
好ましい別の実施形態では、ヒトまたは動物被験体は免疫不全に罹患している。
【0029】
更に好ましい別の実施形態では、ヒトまたは動物患者は、神経細胞傷害または神経疾患の後、神経細胞再生の不全に罹患している。
【0030】
本発明の好ましい別の実施形態では、TNF−αの強健な合成および分泌を得るための手法は、静脈内、皮下、腹腔内、腫瘍内、くも膜下腔内または頭蓋内注入により、もしくは経口投与または経皮的に軟膏により、TNF−αによる増強を動物患者の体の中に達成するのに役立つ。
【0031】
疾患の第二の部類では、TNF−αの有害な過剰は、本発明に従って、仮想の内在疾患関連Kv1.1遮断剤によって誘発されるKv1.1チャンネルを閉鎖/遮断させない分子を用いて、T細胞からのTNF−αの産生と分泌を阻止することにより克服される。もし後者(つまり仮想の内在疾患関連Kv1.1遮断剤)がT細胞における慢性のTNF−α産生および分泌の原因であるならば、例えばKv1.1チャンネル開口剤によってKv1.1チャンネルの遮断をさせないようにすれば、強健なTNF−αの増加の誘発をもまた阻害するであろう。チャンネルを開口し、従ってTNF−αの産生を阻止する分子は、細胞外作用剤であっても良く、またはチャンネルの遮断のシグナルを送る細胞内電位感覚要素を損なう分子であってもよい。該細胞外作用剤の例としては、従来の方法で、Kv1.1分子に対して作製し、作用剤活性に対してスクリーニングを行ったモノクローン抗体が挙げられる。
【0032】
好適な実施態様の詳細な説明
本発明は、正常ヒトT細胞におけるKv1.1電位依存性カリウムチャンネルの選択的遮断により、それのみで強健で、唯一TNF−α蛋白の分泌およびmRNAが増加されるという発見に基づいている。劇的なTNF−αの分泌が、高度に選択的なKv1.1遮断剤、デンドロトキシン−Kによって(しかし9つの他のK+、Na+、Ca2+およびCl−遮断剤によっては誘導されない);市販の抗−Kv1.1抗体によって;および、重要なことには、Kv1.1抗体を保持している末梢神経興奮性亢進症の患者からの血清によって誘導された。Kv1.1遮断は、TNF−α分泌のみを誘発させ、一方「古典的」なTCR−活性化は、TNF−α、IFN−γ、IL−4およびIL−10の分泌を、同時に、非唯一に増加させた。Kv1.1遮断は、またNF−κBの核転移を誘導する。
【0033】
ヒトT細胞におけるKv1.1タンパクおよびmRNAの発現は、本明細書ではRT−PCRおよびフローサイトメトリーによって示した。デンドロトキシン−Kでエクスビボにて前処理をし、従ってTNF−αを過剰分泌しているヒトT細胞をSCIDマウスに注射すると、肝臓への宿主細胞のインビボ動員を引き起こすが、これは脳へTNF−αを注入後に報告されたことに類似している。今まで、これらの発見を予想できることは全く知られていなかった。Kv1.1遮断によって誘発されたTNF−αの増加は、それ故、ある疾患(例えば、癌および免疫不全)ではTNF−αを高め、他(例えばリウマチ性関節炎、クローン病および他の自己免疫/炎症性疾患)では抑止して、治療的に用いることができる可能性がある。
【0034】
本発明により、ヒトT細胞における一種類のイオンチャンネルの選択的遮断が、一種類のサイトカイン、この場合はTNF−α、の唯一の産生と分泌を誘発できることが立証される。該機構は、全てのT細胞サイトカインの同時産生を促進することを特徴とする、種々の分裂促進因子(例えばホルボールエステル)による、「古典的」なTCR活性および非特異的なT細胞刺激の両者とは際立って対照的である。一つのサイトカインを唯一かつ強力に増加させ、一方では他のThサイトカインのレベルを増加させない機構の発見は、特にサイトカインがTNF−αであることから(Dinarelo ら, 2002; Wallach ら, 1999)、重要な臨床的意味を有する。なぜならば、このサイトカインが多くの疾患の病理において重要であり、そのレベルが過剰であるかまたは不十分であるからである。
【0035】
実際、一方ではTNF−αは、健康に基本的で有益な役割を果たす。それは自然免疫反応を活性化し、環境の攻撃から生体を保護する必須な炎症反応を引き起こす(Dinarelo ら, 2002)。更に加えて、TNF−αは、腫瘍の壊死を起こさせ、白血球のローリングと接着の増強し、種々の分子に対する血管と血液脳関門(BBB)の透過性を増加させ(Mayan 2002, van Horssen ら 2006)、オリゴデンドロサイトの増殖と再生を誘導し、神経の再ミエリン形成を促進し(Arnett ら, 2001; Finsen ら, 2002)、細胞を特定の組織へ動員し、ならびに多くの他の重要な機能を誘発する(Dinarelo ら, 2002; Wallach ら, 1999)。
【0036】
他方、TNF−αの過剰産生または無秩序な産生は、通常非常に有害であり、組織破壊に導く重篤な損傷を与える炎症を起こすことがあり、その後に種々の重篤な自己免疫疾患の原因となるが、その中にはリウマチ性関節炎および多発性硬化症、炎症性腸炎を含む炎症性疾患、ならびに外傷、強直性脊椎炎、脳卒中および過眠症のような多様な病理状態の種々なタイプが含まれる。興味あることには、ひとたびTNF−αが過剰になった場合には、抗−TNF−αモノクロナール抗体および可溶性TNF−α受容体のような、TNF−αのシグナル伝達を遮断する治療法に対する適応症のリストはどんどん大きくなっている一方、TNF−αの増加を阻止または遮断(すなわち過剰の有害レベルに達する前に)は、未だ達成されていない。更に、ヒトの疾患におけるTNF−αの慢性産生の原因となる機構については未だ不明なところが多く、他のサイトカインに影響せずに、選択的にTNF−αの増加を促進する機構も未だ解明されていない。
【0037】
T細胞を含む種々の細胞タイプにおいて、特定のイオンチャンネルが主要な細胞反応に影響を与え、指示するという既知の特性(DeCoursey ら, 1984; Deutsch ら, 1986, Lewis ら, 1995; Lewis ら, 1988; Rader ら, 1996; Freedman ら, 1995; Koo ら , 1997; Lin ら, 1993; Ishida ら, 1993; Jansen ら, 1999; Kotturi ら, 2003; Phipps ら, 1996; Verheugen ら, 1997)、およびKv1.3電位依存性カリウムチャンネルのゲーティングは、それ自身で主要T細胞機能を誘発できるという本発明者の研究室における以前の実証(Levite ら, 2000)によって促され、本発明は、正常ヒトT細胞における特定のカリウム(K+)、ナトリウム(Na+)、カルシウム(Ca2+)または塩素イオン(Cl−)チャンネルの単なる遮断が、選択的なサイトカイン分泌、特にTNF−αの分泌を誘発することができるかどうかの研究に基づいている。本結果は、正常のヒトT細胞内の電位依存性K+チャンネルを含むKv1.1サブユニットの選択的な遮断が、強健的で、唯一かつ長期的なTNF−α分泌の増加に対する新規な機構を明示している。
【0038】
本発見は、新規な予想外の機構を明らかにしたが、それは正常な末梢ヒトT細胞内の電位依存性K+チャンネルを含むKv1.1サブユニットの選択的な遮断が、TNF−α分泌およびmRNAレベルを著しく、唯一かつ長期的に増加させたことである。我々の知る限り、一般的に電位依存性K+チャンネル、特にKv1.1についてまたはTNF−αについて、Kv1.1とTNF−αの直接の機能的な関連に関する本発見を予想するようなことは全く知られていない。更に、本発見は、他のThサイトカインを増加させることなく、TNF−αの単独増加に対する新規の機構を明らかにしている。
【0039】
次の段落において、考察がなされる:a)本発明の更なる詳細;b)主としてT細胞中のKv1.1電位依存性K+チャンネルおよびKvファミリーの他のメンバー、ならびにDTX−Kの特殊な特徴;c)PNH,Kv1遮断に伴うヒトの神経疾患;およびd)様々なヒトの疾患においてTNF−αの促進する、または抑止する新規の臨床標的として、T細胞Kv1.1の使用。
【0040】
本発明の更なる詳細
非常に選択的なKv1.1遮断剤DTX−K(Harvey, 2001)および特異的、市販抗−Kv1.1抗体の両者は、T細胞TNF−αを劇的に増加させることを発見した。対照的に、T細胞中で発現しているK+、Na+、Ca2+、Cl−イオンチャンネルまたはグルタミン酸/AMPAイオンチャンネル受容体(Ganor ら, 2003)に対する9つの他の遮断剤にT細胞を曝しても該効果は得られなかった。Kv1.1には特異的ではないが、電位依存性K+チャンネルを含むKv1.1サブユニットにいくらか影響することができる二つのKv1.1遮断剤、KTXおよびMgTXもまたヒトT細胞からTNF−αを誘発したが、程度はずっと低く、Kv1.1チャンネルサブユニットに対するそれらの低い親和性と特異性に見合ったものであった。
【0041】
KTXおよびMgTXは、Kv1.1チャンネルに対してDTX−Kほど特異的ではないが、本発明に従って使用するには十分特異的である。本発明は、PMAによって代表されるような、ホルボールエステルのような非特異的なT細胞活性化剤をすべて考慮に入れているわけではない。しかしながら、本明細書で用いる「選択的」という用語は、PMAのようなホルボールエステルの場合のように、T細胞サイトカインが無差別に産生するのではない限り、他のKvチャンネルにいくらかの活性を有する可能性のあるKv1.1遮断剤を含むことを意図している。Kv1.1チャンネルに対する選択性が大きければ大きいほど、該分子に対する好ましさは大きい。従って、DTX−Kは、現時点において最も好ましい該分子である。しかしながら、DTX−KほどTNF−αを産生しないかも知れない、又DTX−KほどKv1.1に対する特異的選択性がないかも知れないが、KTXおよびMgTXもまた、本発明に従って用いることができる。従って、以下の十分に選択性を有する、および選択性がより低いDv1.1電位依存性イオンチャンネル遮断剤は、Kv1.1チャンネルを遮断し、および少なくともKvチャンネルに対して実質的に選択的である、いかなる他の分子とも同じように、本発明に従って使用できる:
A)デンドロトキシン類
1)トキシンK、デンドロトキシン−K(DTX−K)とも呼ばれる、は、ブラックマンバ、Dendroaspis polylepsisから由来し、Kv1.1チャンネルを優先的に遮断し、ピコモル濃度で活性を有する。
2)アルファーデンドロトキシンはグリーンマンバDendroaspis angusticepsから由来し、クローンされたKv1.1、Kv1.2およびKv1.6チャンネルを、低ナノモル範囲で遮断する。
3)トキシンI、デンドロトキシン−I(DTX−I)とも呼ばれる、は、ブラックマンバ、Dendroaspis polylepsisから由来し、クローンされたKv1.1、Kv1.2およびKv1.6チャンネルを低ナノモル範囲で遮断する。
B)コノトキシン類:イモガイ、Conus virgoの毒素から、ViTx(virgo-toxin)と呼ばれるペプチドが分離され、Kv1.1およびKv1.3のK+−チャンネルを遮断するが、Kv1.2タイプは遮断しない(Kauferestein ら, 2003)。
C)マウロトキシン類:天然および合成のマウロトキシンは両方とも、Xenopus卵母細胞で発現されたKv1.1、Kv1.2およびKv1.3チャンネルを、殆ど同じ40、0.8および150nMの半分有効量(IC50)で遮断する(Rochat ら, 1998)。
D)アギトキシン類
1)rAgitoxin-2 Leiurus q. hebraeusはKv1.1、Kv1.3、Kv1.6を50pM〜10nMで遮断する。(Alomone Labs web site; www.alomone.com/p_products)。
2)rAgitoxin-3 Leiurus q. hebraeusはKv1.1、Kv1.3、Kv1.6を50pM〜10nMで遮断する。(Alomone Labs web site; www.alomone.com/p_products)。
E)r-Hongotoxin-1 Centruroides limbatusはKv1.1、Kv1.2、Kv1.3を0.1〜0.2pMで遮断する。
F)Pi4はさそりPandinus imperatorの毒液由来の38アミノ酸長ペプチドであり、Shaker B K+チャンネル(Kv1.1サブファミリータイプのチャンネル)を、100nM濃度で完全にかつ可逆的に遮断する(Olamendi-Portugal 1998)。
G)Kaliotoxin(KTX):KTXはいくつかのCa2+活性化および電位依存性K+チャンネルを遮断する。Xenopus laevis卵母細胞へのKTXのバス適用(bath application)は、組み換えKv1.3およびKv1.1チャンネルを強力に、又Kv1.2チャンネルをより低い強度で遮断し、それぞれのK(d)値は、0.1、1.5および2.5nMであった(Mourre ら, 1999)。
H)BTK−2:インドのさそりButhus tamulusから精製されたKv1.1カリウムチャンネルの新阻害剤(Dhawan ら, 2003)。
I)新Kv1.1カリウムチャンネル遮断トキシン:Palamneus gravimanus (インド黒さそり)の毒液由来の新Kv1.1カリウムチャンネル遮断トキシン(More ら, 2005)。
J)rMargatoxin:MgTX K+チャンネルは、主としてKv1.3電位依存性K+チャンネルを遮断するが、Kv1.1にも作用することができる。
K)Stichodactyla helianthusペプチド(ShK)は、S. helianthusの毒液から分離された、Kv1.1およびKv1.3チャンネルの既知の高親和性遮断剤である。
【0042】
本実験で用いたKv1.1に対する抗体は、市販されておりAlamone Labs Ltd., Jerusalem, Israel, product no. APC-009から得られる。Alomone.com/System/UpLoadFiles/DGallery/Docs/apc- 009_AN-05.pdfで得られるデータシートを参照。この抗体は、配列HRETE GEEQA QLLHV SSPNL ASDSD LSRRS SSTIS KSEYM EIEED MNNSI AHYRQ ANIRT GNCTT ADQNC VNKSK LLTDV(配列番号7)を有するGST融合タンパクに対して産生させたウサギ抗体であって、マウスKv1.1の残基416−495に相当する(GeneBank Accession P16388)。このエピトープの80残基の内76残基は、対応するヒトタンパクに保存されている。Kv1.1のエピトープ、好ましくはヒトKv1.1に対して産生された他のいかなる抗体も、ポリクローンあるいはモノクローンであれ、それらがKv1.1チャンネルを遮断する能力を有する限り、本発明の目的のために用いることができる。本明細書および請求項で用いる「抗体」という用語はポリクローンまたはモノクローン、ならびにヒト化抗体、単鎖抗体のような遺伝子改変の抗体、およびCDRs、Fab、F(ab)2等のような抗原結合性断片を包含することを意図している。
【0043】
Kv1.1チャンネルに対するモノクローン抗体を作製する際、それらはチャンネルを遮断する(拮抗する)または開口する(作用する)能力に関してスクリーニングをしてもよい。拮抗剤である抗体は、TNF−αの産生の増加が望まれる病気を治療するために用いることができ、作用剤である抗体は、TNF−αの過剰な産生を阻止する必要がある疾患を治療するために用いることができる。
【0044】
TNF−α転写におけるCa2+の既知の役割(Lobo ら, 1999)と合致して、Kv1.1遮断によるTNF−αの誘導にはCa2+流入を必要とすることが発見されたが、それはT細胞内におけるCa2+チャンネルの高度の選択的遮断剤であるR-(+)-Bay K 8644(Kotturi ら, 2003)がこの効果を阻止したからである。驚くべきことには、Kv1.1チャンネルの遮断は、TNF−αの急速な増加を引き起こす一方、IFN−γ、IL−4およびIL−10を増加させず、そのことは、これら全てのThサイトカインは強健だが、非選択的に、又同時に結局増加することになる「古典的な」TCR−活性化から著しく異なっていた。
【0045】
TNF−αを過剰発現するDTX−Kで処理した正常ヒトT細胞を、SCIDマウスに注射すると、TNF−αの脳内への注射後起こることが最近研究で示された結果(Campbell ら, 2005)に類似して、専ら肝臓に存在するマウス常在細胞数が倍増したことから、Kv1.1チャンネルの選択的遮断によってヒトT細胞内に誘導されたTNF−αの増加は、インビボにて機能的な影響をもたらした。
【0046】
Kv1.1電位依存性K+チャンネルおよびそのファミリーの他のメンバー、ならびにDTV−Kの特異な特徴:Kv1.1チャンネルは電位依存性K+チャンネルのファミリーに属しており、そのいくつかのメンバーはT細胞で発現しており、重要な機能的役割を果たしている(DeCourseyら, 1984; Deutsch ら, 1986; Lewis ら, 1995; Lewis ら , 1998; Freedman ら, 1995; Koo ら , 1997; Lin ら, 1993; Levite ら, 2000; Liuら, 2002; Beeton ら, 2001; Leonard ら, 1992; Levite 2001)。これらのチャンネルはC2+活性化K+チャンネルとは異なっており、これらチャンネルはT細胞により発現しており、T細胞機能として重要である(Rader ら, 1996; Jensen ら, 1999; Vreheugen ら, 1997)。様々な種類の電位依存性K+チャンネルのグループ分けおよび独特な特性は、いくつかの研究で考察されている(Grissmer ら, 1994; Mathisら, 1998を参照)。一般的に、電位依存性K+チャンネルの阻害は、脱分極を起こし(Leonard ら, 1992)(すなわち陰性度が低い膜電位へのシフト)、それが、種々の主要な細胞特性および細胞機能に影響を及ぼし、発揮させる(Levite ら, 2000; Leonard ら, 1992; Levite, 2001)。神経細胞においては、電位依存性チャンネルの一つの大きな役割は、膜の興奮性の制御である(Ruteckie, 1992; Smart ら, 1998)。これには活動電位および神経細胞の反復発火の期間を制限し、ならびに膜電位を安定化させることが含まれる。
【0047】
T細胞内の特定の電位依存性K+チャンネルは、β1インテグリン類を活性化し、その後フィブロネクチンと同様にECM成分へのT細胞接着を誘発し(Levite ら, 2000)、膜電位(Levite ら, 2000)、カルシウム流入(Grinstein ら, 1990; Lin et al, 1993; Verheugen ら, 1997)、増殖(Freedman ら, 1992; Leonard ら, 1992)、IL−2産生(Liu ら, 2002, Freedman ら, 1992)、アポトーシス(Bortner ら 1999; maeno ら , 2000; Vu ら, 2001)、および細胞容積(Grinstein ら, 1990)を調節することが、先の研究で示された。
【0048】
従って、全体として、Kvチャンネルは、主要な生理的T細胞機能を調節し、それらの不適切な機能は、TCR−活性化を阻害し、重要なT細胞を介しての免疫反応を阻止する。例えば、Liu ら(2002)は、電気生理学、薬理学およびRT−PCRの方法を組み合わせて、未処理のマウスCD4+T細胞は、Kv1.1、Kv1.2、Kv1.3およびKv1.6電流を発していること、ならびにこれらのKvチャンネルが、該未処理の細胞によるTCR−誘導(抗−CD3および抗−CD28により)IL−2分泌を最大にするために必要となることを示した(Liu ら, 2002)。
【0049】
対照的に、マウスTCR−活性化エフェクターT細胞は、Kv1.3電流(未処理細胞よりは±6倍高い)のみを発するが、K1.3も、他のいかなるKvも、これらの活性化されたT細胞のIL−2、IL−4、またはIPN−γの産生、カルシウムシグナル伝達または膜電位に対して有意に寄与はしていない(Liu ら, 2002)。この後者の研究で行った観察により、末梢マウスCD4+細胞において、分化状態、TCR活性化の強さ、共刺激因子の存在あるいは非存在の表れとして、Kv電流が、質的にも又量的にも変化することを示された(Liu ら, 2002)。自然の電位依存性K+チャンネルはヘテロ多量体であって、種々のKvサブタイプの組み合わせから構成されており、ヘテロ多量体チャンネルが異なれば、親チャンネルと比較して、劇的に異なったチャンネル機能を有することになる。ヘテロダイマーを含む異なったKv1.1もまた異なった遮断剤に対して異なった感受性を有している。例えば、ヘテロダイマーを含むKv1.1中にKv1.2が存在すると、TEAに対する全アレイの感受性が低下する。先の研究では、DTX−Kが、Kv1.1サブユニットとの強く高度に特異的な相互作用という点で、またそれが、これらのチャンネルとそのN−末端(K3、K6)およびβ−ターン(K25、K26)における残基を介して相互作用をするという点で、独特なものであることを示された(Wang ら)。DTX−Kの非常に独特な作用機構により、DTX−KによるTNF−αの増加を(いまだ未調査の機構で)説明できる可能性がある一方、特異性がより低いKv1遮断剤の作用機構により、たとえあったとしても効果はあまり目立たなくなる。
【0050】
ヒトおよびマウスT細胞におけるKv1.1電流試験である別の研究で、異なった結論に達した(Freedman ら, 1995; Cahalan ら, 1985; Liu ら, 2002)。本結果は、特異的なKv1.1RT−PCRおよびフローサイトメトリーによって、正常末梢ヒトT細胞のかなりの割合は、Kv1.1mRNAおよび膜タンパクを明瞭に発現していることを示し、このタンパクの新しい面を明らかにした。しかし、我々の発見は、正常静止状態の末梢ヒトT細胞において機能的な(すなわち電気生理学的に活性を有する)Kv1.1電流の存在を証明するものでは勿論ない。電位依存性K+チャンネルファミリーの他のメンバー、主としてKv1.3、がT細胞の活性化と機能において重要な役割を果たしている証拠が、多くの最近の研究によって示唆された(DeCoursy ら, 1984; Deutsch ら, 1986; Lewis ら, 1995; Lewis ら, 1988; Freedman ら, 1995; Koo ら, 1997; Lin ら, 1993; Leite ら, 2000; Liu ら, 2002; Beeton ら, 2001; Leonard ら, 1992; Rus ら, 2005)。更に、本発明者の研究室は、正常ヒトT細胞の電位依存性K+チャンネルの一つのメンバー、Kv1.3、のゲート開閉は、それ自身のみでβ1インテグリン活性化とT細胞接着を誘発でき、Kv1.3チャンネルは、これらの細胞中で、実際β1インテグリンと物理的に又機能的にカップリングしていることを以前示した(Levite ら, 2000; Levite 2001)。注目すべき点は興味ある相違点である:我々が以前研究したKv1.3チャンネルの場合には、T細胞機能を誘導するのはKv1.3チャンネルを開口した時であり(すなわちT細胞接着を引き起こす)(Leviteら, 2000; Levite 2001)、一方本研究においては、そうする(すなわちTNF−αを増加させる)のはKv1.1チャンネルの遮断である。
【0051】
Kv1.1遮断に伴ういくつかのヒト神経疾患:一般的に神経の興奮性を増加させるKv1.1チャンネルの遮断は少なくとも3つのヒト神経疾患を直接伴っている:
1)Kv1抗体と関連するPNH
2)Kv1抗体と関連する脳炎
3)ヒトKv1.1チャンネルをコードする遺伝子の様々な突然変異に伴う、突発性運動失調1型(EA−1)(Vincent et al, 2004; Adelman et al 1995; Browne et al, 1994)。
【0052】
PNH:Kv1遮断に伴うヒトの神経疾患:マウスにおけるKv1.1チャンネルの欠損は、癲癇、温度感受性振盪および痛覚過敏症を起こすが、これらは神経細胞の過剰興奮性と一致する特徴である(Smart ら, 1998; Zhou et al , 1998)。臨床医は、全般的なPNHの筋肉運動の症状を記述するのに、神経性筋強直症、アイザックス症候群、ミオキミアおよび線維束性痙攣症候群を含む多くの用語を用いる(Hart et al, 2002)。PNHが通常自己免疫を介する(Newsom-Davis ら, 1993)という考えは、最近60人のPNH患者の35%は、100pmol/lを越える血清Kv1抗体タイター有していたという知見によって確証された(Hart et al, 2002)。これらの抗体は病原性があり、Kv1チャンネルを遮断し、従って末梢およびお中枢神経系の両方に神経の過剰興奮性と広範囲の臨床症状を引き起こす。ここで、いく人かのKv1抗体陽性PNH患者の血清は(方法参照)、ヒトT細胞による相当な量のTNF−α分泌を刺激したが、一方IFN−γを増加させなかった(DTX−Kの結果と類似している)。この発見は、T細胞における抗体を介したサイトカイン誘導が、神経の過剰興奮性の病態およびPNHの臨床的特徴ならびに多分自己免疫性脳炎の一因となる可能性を示唆している。本データは又疲労および筋肉痛のようなPNHに伴う、より非特異的な症状は、少なくとも部分的にはKv1抗体を介した血液循環中のTNF−αの増加によって引き起こされた可能性を示唆している。従って、本発明の他の特徴は、1)Kv1抗体に伴うPNH,2)Kv1抗体に伴う脳炎、3)突発性−運動失調1型(EA−1)を循環系におけるTNF−αの力価を減少させる任意の方法で治療する方法であって、TBP−1およびTBP−2のようなTNF−α結合タンパク、抗-TNF−α抗体、Kv1.1チャンネル非遮断剤等のような方法である。
【0053】
T細胞のKv1.1機能不全は、TNF−α過剰に伴うヒトの疾患に寄与する可能性があるので、T細胞におけるKv1.1は新規の治療標的である。ヒトTNF−αは生物活性を有する、233アミノ酸、26kDaの膜結合性タンパクとして合成される。この膜結合性タンパクは、TNF−α変換酵素(TACE)、アダマリシンによって157アミノ酸、17kDa可溶性タンパクに酵素的に切断され、容易にホモ三量体を形成する。ホモ三量体TNF−α(細胞結合型または可溶性のいずれか)のその受容体(TNF−RIおよびTNF−RII)への結合は、静止状態の細胞においてシグナル伝達に必要な受容体のオリゴマー形成を誘導する。
【0054】
TNF−αの不適切な産生または過剰産生は、その発病原因において炎症が関与する広範囲の病気において起こるが、その病気には、炎症性腸疾患、リウマチ性関節炎、多発性硬化症、強直性脊椎炎および脳卒中が含まれる(Baugh ら, 2001)。本発見により、インビボでのKv1.1抗体またはKv1.1遮断剤を介してのT細胞Kv1.1遮断は、少なくとも部分的に、これらの障害での過剰のTNF−αに原因があり、それ故新規の治療標的である、という結論におそらく導かれるであろう。
【0055】
他方、TNF−αは、他の多くの目的のために非常に必要とされることから、TNF−αを長期間過剰発現する自己DTX−K処理T細胞のヒトにおける受動的な輸送は、非常に特定の状況においては、有益であろう。これらには、T細胞による癌細胞根絶の強化、ウイルスおよびバクテリアのT細胞攻撃を高めること(特に免疫不全の種々のタイプの場合)および必要に応じて宿主細胞の肝臓への動員の増加が挙げられる。
【0056】
実施例1:
材料と方法
イオンチャンネル遮断剤:表1も参照。本明細書に用いる各イオンチャンネル遮断剤について特定されるものは、正式名、カッコ内に短縮名と製造者、有効濃度(それぞれの製造者のデータシートおよび/または文献から得られた)である。更なる詳細情報は、International Union of Pharmacology website (Gutman et al, 2003を参照)に見出すことができる。遮断剤としては以下のものが挙げられる:4−アミノピリジン(4−AP,Sigma,St. Louis, MO)、10μM〜1mM;テトラエチルアンモニウム(TEA,Sigma)、100μM〜10mM;キニーネ(Sigma)、1〜10μM;クロトリマゾール(CLT,Agis Industries, Bnei Brak, Israel)、10nM〜10μM;rカリブドトキシン (CTX,Alomone labs, Jerusalem, Israel)、10〜100nM;カリオトキシン(KTX,Alomone)、1〜100nM;rマルガトキシン (MgTX,Alomone)、50pM〜50nM;デンドロトキシン−K(DTX−K,Alomone)、10〜100nM;テトロドトキシン(TTX,Alomone)、100nM〜1μM;NPPB(Torcris Cookson, Avonmouth, UK)、100〜200μM;R-(+)Bay K8644(+Bay K, Tocris)、10nM〜1μM;CNQX(Tocris)、100nM−50μM。
【0057】
【表1】
【0058】
ヒトT細胞:正常ヒトT細胞は、(Levite ら, 2000; Ganor ら, 2003; Levite ら, 1998)に記載されているように健常提供者の末梢血から精製し、得た90%以上のT細胞を含む細胞群を、10%FCS、1%Lグルタミンおよび抗生物質を含むRPMI培地(Biological Industries, Bet Haemek, Israel)に懸濁し、2x106細胞/ml(37℃/5%CO2)で維持した。
【0059】
ELISAによるTNF−α、IFN−γ、IL−10およびIL−4の定量:新たに精製した正常静止ヒトT細胞(2x106/ml)を24穴プレート(Costar, Corning, NY)中でインキュベーションし、種々のチャンネル遮断剤を加えた。重要なことは、他の刺激分子(例えばホルボールエステル、抗原、抗−CD3・CD28mAb)を加えないことであった。サイトカインレベルは、TNF−αとIFN−γについては24時間後、IL−10およびIL−4については72時間後、定量的サンドイッチELISA法(R&D, Minneapolis, MN)で、製造者の指示に従い、上清を測定した。
【0060】
TNF−αおよびKv1.1のRT−PCR:全RNAは、Tri−Reagentプロトコル(MRC, Cincinnati, OH)に従って調製した。第一鎖cDNAは、Reverse Transcription System(Promega, Madison, WI)を用いて合成した。RT−PCRは、50ngのcDNA、5μlのx10 Optibuffer (Bioline, London, UK)、3μlの50mM MgCl2、2.5μlの10mM dNTP混合液(Promega)、4UのBio−X−actDNAポリメラーゼ(Bioline)を含む50μlの反応混合液中で行った。TNF−α/S14同時増幅については、2μlの各TNF−αプライマー(10pmol/μl)および0.25μlの各S14プライマー(10pmol/μl)を用いた。Kv1.1/S14同時増幅については、5μlの各Kv1.1プライマー(100pmol/μl)および2.5μlの各S14プライマー(10pmol/μl)を用いた。プライマー配列(5’−3’)および産物の長さは以下の通りである:
TNF−αプライマーペア−上流プライマー
【表2】
下流プライマー
【表3】
494bp;Kv1.1プライマーペア−上流プライマー
【表4】
下流プライマー
【表5】
709bp;S14プライマーペア−上流プライマー
【表6】
下流プライマー
【表7】
166bp。TNF−αのPCR条件は:94℃1分、63℃40秒および72℃40秒(29サイクル)。Kv1.1のPCR条件は、94℃3分(1サイクル);94℃1分、60/57/54/51/48℃2分および72℃3分(それぞれ3サイクル);94℃1分、50℃2分および72℃3分(25サイクル)。TNF−α/S14RT−PCR産物の濃度分析は、Adobe Photoshop 7.0 MEによって行った。TNF−αバンドの相対強度は、S14バンドの相対強度に対して規格化した。
【0061】
核NF−κBのEMSA:正常ヒトT細胞から新たに分離した核画分は、Nuclear Extract Kit(Active Motif, Carlsbad, CA)用いて製造者の指示に従って採取した。核画分中のNF−κBレベルは、TransAM NF-kB Family Kit(Active Motif)を用いて製造者の指示に従って測定した。
【0062】
NF−κBの免疫蛍光組織化学:DTX−K(100nM 7、15、30および60分間)で前処理または未処理の新たに分離した正常ヒトT細胞を沈殿させ(1200g、10分、40℃)、PBSに1x106細胞/mlで懸濁し、遠心し(1200g、10分)、ガラススライド上にサイトスピン(250μl/スライド、1500rpm、5分;Shandon Elliot, London, UK)で固定し、透過処理を行い(100mlの95%メタノールおよび3ml氷酢酸を含む溶液200μl/スライド)、10%正常ヤギ血清、2%BSA、1%グリシンおよび0.5%のTriton X-100を含むブロッキング培地中で(200μl/スライド)インキュベーションした。それから細胞をウサギ抗−ヒトNF−κB p50ポリクローン抗体(1:50希釈した80μlと30分間室温;Santa Cruz Biotechnology, Santa Cruz, CA)で染色した。この第一回の染色後、細胞をFITC−結合ヤギ抗−ウサギIgG(1:50希釈で30分間室温;Jackson ImmunoResearch, West Grove, PA)に曝し、PBS中で2回洗浄し、それから細胞核に取り込ませるHoechst 33342に曝した(1:2000希釈で5分間室温;Molecular Probes, Eugene, OR)。染色した細胞を、蛍光顕微鏡(Nikon Eclipse E600)で可視化し、ACT−1ソフトウエアープログラムを用いて、x100倍で写真撮影を行った。
【0063】
チャンネルを含有するKv1.1サブユニットの検出のための免疫染色およびフローサイトメトリー:新たに精製した正常ヒトT細胞を、透過処理(70%エタノール、1時間、20℃で)した後、一次抗体としてウサギ抗−Kv1.1ポリクローン抗体(Alomone)を用いて、6μg/ml/1x106細胞/100μl、30分間氷上で、免疫蛍光染色を行った。アイソタイプのコントロールとして、細胞を同じ濃度の以下のように調製した正常ウサギIgGで染色した:健常コントロールの白色ニュージーランド雄のウサギから得た血清に等量の飽和硫酸アンモニウムを滴下する;混合液を1時間4℃で攪拌し、3000g15分、4℃で遠心した。沈殿物をリン酸緩衝食塩水(PBS)に再懸濁し、硫酸アンモニウムのいかなる痕跡をも除くために、同じ溶液に対して一夜3回透析した。IgG濃度の推定はO.D280で定量した(1.45O.D280は1mgIgG/mlとした)。それから細胞を、FITC結合ヤギ抗−ウサギIgGを二次抗体として(1:100希釈した100μl、Jackson)染色した。二次抗体のみで染色した細胞を追加の陰性コントロールとした。蛍光像をFACSortに記録した。
【0064】
PNH患者および健常者個人コントロール:血清は12人のPNH患者から採取したが、すべての患者は筋電図検査によって、全身性後天性疾患を確認し、又多発神経障害の臨床的または電気生理的証拠は無かった。どの患者も、造影CT縦隔上に胸腺肥厚または胸腺腫は有しなかった。血清採取の少なくとも1年前までは、どの患者も免疫療法は受けていなかった。年齢、性および地理的に一致させた4人の健常者個人コントロールから血清の供与を受けた。全ての患者から書面で、この研究に対するインフォームドコンセントを得、この研究は、Sefton Research倫理委員会、リバプール、UKによって許可された。血清Kv1抗体力価は、標準(125)I−アルファーデンドロトキシン免疫沈降アッセイ(Hart et al, 1997)によって検出した。この方法はKv1.1、1.2および1.6に対する抗体を検出する。陽性力価は、100pM以上に設定してある。50pM未満の力価は陰性と見なす。ここで試験した12人のPNH患者のうち7人はKv1抗体に関しては陰性であった。PNH患者の関連した臨床特徴を下に要約する。
PNH Kv1抗体陽性(PNH+)
PNH+(1):女性、40歳、PNH(36歳で発症)および甲状腺機能低下症、Kv1 239pM。
PNH+(2):女性、49歳、PNHのみ(47歳で発症)、Kv1 335pM。
PNH+(3):男性、52歳、PNH(40歳で発症)およびDMタイプ1、Kv1 235pM。
PNH+(4):男性、52歳、PNHのみ(43歳で発症)、Kv1 382pM。
PNH+(5):男性、51歳、PNH(51歳で発症)および軽度の乾癬、Kv1 346pM。
PNH+(6):男性、44歳、PNHのみ(43歳で発症)、Kv1 165pM。
PNH+(4):男性、23歳、PNHのみ(22歳で発症)、Kv1 255pM。
PNH Kv1抗体陰性(PNH−)
PNH−(7):女性、48歳、PNHのみ(41歳で発症)。
PNH−(8):女性、45歳、PNH(37歳で発症)およびレイノー病。
PNH−(10):男性、53歳、PNHのみ(50歳で発症)。
PNH−(11):男性、49歳、PNHのみ(44歳で発症)。
PNH−(12):女性、41歳、PNHのみ(15歳で発症)および湿疹。
【0065】
市販Kv1.1抗体またはPNH患者の血清の、Kv1抗体の存在あるいは非存在下での、正常ヒトT細胞によるTNF−α分泌の誘発能力の試験:新たに精製した正常ヒトT細胞(2x106細胞/ml)を24穴プレート中でインキュベーションし、ウサギ抗−Kv1.1ポリクローン抗体(1:1000希釈;alomone)に24時間曝した。培地に分泌されたTNF−αのレベルは、上記のようにELISA法によって検査した。
【0066】
代わりに、T細胞(2x106細胞/ml)をKv1抗体−陽性PNH患者、Kv1抗体−陰性患者または健常者の血清(1:100希釈)に曝した。T細胞を含まない培養を陰性コントロールとした。24時間インキュベーションした後、上清中のTNF−αおよびIFN−γのレベルを上記のように検査した。培地に分泌されたTNF−αのレベルは、抗−Kv1.1抗体陰性PNH患者の血清に曝されたT細胞によって分泌される平均TNF−αレベルの±2*SDを越えるならば、増加したとした。
【0067】
SCIDマウスへの、DTX−K処理ヒトT細胞のインビボ注射:NOD/SCID雌マウス腹腔内へ(ip)、シクロホスファミド(Sigma、20mg/ml PBS溶液)200μlを細胞注射する24時間前に注射した。新たに精製した正常ヒトT細胞(1x108細胞)をDTX-K(100nM、24時間)またはPBSで前処理し、洗浄し、2’,7’−ビス−(2−カルボキシエチル)−5−(および−6)−カルボキシフルオレッシンアセトキシメチル(BCECF AM、Molecular Probes, Eugene, Oregon)で蛍光標識した。それからヒトT細胞をPBSで洗浄し、懸濁し、標識細胞の200μl(1.5x107細胞に相当)をそれぞれの動物のipに注射した(各群n=5)。24時間後、各動物から血液を採取し、肝臓、脾臓、腎臓、骨髄および脳を取り出した。脳を除く全ての臓器から単細胞懸濁液を調製した。それぞれの臓器における総細胞数ならびに常在SCID細胞(非蛍光)のみまたは注入したヒトT細胞(蛍光)のみの数をFACSortによって計測した。
【0068】
脳の免疫組織化学:脳を取り出し、ブアン固定液に4℃で一夜浸し、PBSで洗浄し、パラフィン包埋を行うまで70%エタノールに貯蔵した。連続7μ厚の冠状切片を切り、脱パラフィン、脱水し、家庭用Samsung電子レンジを用いてマイクロウエーブ処理を行った。これは、1mM EDTAバッファー(pH=8.0)に浸し、沸騰するまで最高出力で加熱し、更に10分間20%効率で加熱し、同じバッファー中で、室温で30分間冷却することにより行った。
【0069】
免疫組織化学については、切片をPBS/20%ウマ血清(HS)(Vector Laboratories, Burlingame, CA)と4℃で一夜前インキュベーションを行った。それから切片を、ラット抗−ヒトCD3モノクローン抗体(PBS/2%HSで1:50希釈;Serotec, Oxford, UK)と4℃で96時間インキュベーションした。PBSで洗浄後、切片をビオチン化抗−ラットIgG(PBS/2%HSで1:10希釈;Vector)と室温で1.5時間インキュベーションし、再び洗浄後Cy3結合したストレプトアビジン(PBSで1:200希釈;Jackson)とインキュベーションした。切片を蛍光顕微鏡(Nikon Eclipse E600)で観察し、ACT−1ソフトウエアープログラムを用いてx40倍率で写真撮影を行った。
【0070】
統計解析:統計的有意差をStudent's t-testによって解析した。患者の異なった群間の定量的な変動を比較するために、非パラメータKruskal-Wallis テストを用い;次いで対での比較は非パラメータMann-Whitney U-testによって行った。
【0071】
実施例2:正常ヒトT細胞のチャンネルを含むKv1.1サブユニットの選択的遮断は、著しい又単独のTNF−α分泌を誘発する。
新たに精製した正常ヒトT細胞(「静止」正常ヒト末梢T細胞)を、他の刺激分子が全く存在しない状態で12の異なったチャンネル遮断剤(表1参照)に曝した。これらの12の遮断剤は、T細胞に発現されるK+、Na+、Ca2+およびCl−チャンネルの殆どのタイプを遮断するそれら報告された能力に基づいて選択された(Deutsch ら, 1986; Lewis ら, 1995; Lewis ら, 1988; Rader ら, 1996; Freedman ら, 1995; Koo ら, 1997; Lin ら, 1993; Ishida ら, 1993; Jensen ら, 1999; Kotturi ら, 2003; Phipps ら, 1996; Verheugen ら, 1997; Levite ら, 2000; Chandyら, 1984)。培地へ分泌されるTNFαのレベルは、ELISA法により24時間後に試験した。陽性コントロールとして、T細胞を非特異的な様式で活性化することが知られている、強力なホルボールエステル、PMAに曝した。結果は図1Aに示してあり、2〜3回の独立した実験において試験したTNF−α分泌の倍増率±SDを表す。各イオンチャンネル遮断剤は、少なくとも2回の独立した実験において、その有効適用範囲のいくつかの濃度で試験した(材料と方法を参照)。各遮断剤の隣の図の数字は用いた最高のモル濃度を表す。統計解析:MgTX、KTX、DTX−KおよびPMAのそれぞれについて、未処理に対して*P=0.0001、0.0005、0.0011および0.0005(Student's t-test)。
【0072】
図1Aは、電位依存性K+チャンネルを含むKv1.1サブユニットの、高度に選択的なKv1.1遮断剤であるデンドロトキシン−K(DTX−K)(Harvey et al , 2001)(又該物として多くの研究に使用され、その内のいくつかは(Harvey et al , 2001)に引用されている)による遮断が、それだけで、TNF−αの分泌を引き起こすことを示している。推奨される至適有効濃度(すなわち多くの他の研究において通常用いられるような、100nM)で使用したDTX−Kは、試験したヒトT細胞によるTNF−α分泌の33.4倍増を誘導したが、これは本明細書で陽性コントロールとして用いた、強力な非特異的活性化剤、ホルボールエステル(PMA、41.3倍増)によって得られるものと同規模であった。TNF−α分泌の有意な増加は、カリオトキシン(KTX)およびrマルガトキシン(MgTX)によってもまた起きた(それぞれ15.1および6.5倍増、図1A):これらは、電位依存性K+チャンネルの遮断剤であって、主としてKv1.3チャンネルに影響するが、Kv1.1チャンネルにも又影響する可能性がある。DTX−Kは、KTXおよびMgTXと比較すれば、Kv1.1に対するより高い親和性と選択性を有している(例えば、Kv1.1に対するKiは、DTX−Kは16pMでありKTXの20nMよりもはるかに高い(Gutman et al , 2003))。
【0073】
DTX−K、KTXおよびMgTXとは対照的に、正常ヒトT細胞を9つの他のイオンチャンネル遮断剤のいくつかの有効濃度に曝した後も、どんな有意なTNF−αの上方調節も観察されなかった(表1)が、その中には更なるK+チャンネルの遮断剤(Kv1.1に対しては選択性がより少ない):4−アミノピリジン(4−AP)、クロトリマゾ−ル(CLT),テトレエチルアンモニウム(TEA)、rカリブドトキシン(CTX)およびキニーネ;Na+チャンネル遮断剤:テトロドトキシン(TTX);Cl−チャンネル遮断剤:NPPB;Ca2+チャンネル遮断剤:R-(+)-Bay K8644 (+Bay K);およびイオンチャンネル(イオンチャンネル内蔵型)グルタミン酸/AMPA受容体(ヒトT細胞中に高レベルで存在することが最近同定された)(Ganor et al, 2003):CNQX。
【0074】
DTX−Kの誘発効果は、図1Bに示したように用量依存性である。10−9M〜10−6MのDTX−Kに対して24時間曝露した後のTNF−α分泌の倍増±SDを示す。行われた三つの実験のうち代表的なものを示す。未処理に対して*P=0.0001である(Student's t-test)。TNF−α分泌は、100nM DTX−Kに対して8.7倍で、1μMDTX−Kに対しては39.5倍であった(注:図1Aおよび1Bに示す結果は、別の二人のヒトから精製したT細胞に関するものである、下を参照のこと)。
【0075】
続く実験で、我々は更なる10人の個体から新たに分離したT細胞を試験し、DTX−Kの効果は、再現性があることが分かった。図1C〜Hは、これらの6人のヒト個体からのT細胞のDTX−Kに対する被曝後(24時間、100nM)培地に分泌されたTNF−αの実際の平均濃度をpg/ml±SDで示したものである(aおよびbに描かれた倍増率ではなく)。統計解析:1、3、5〜6の個体それぞれについて、未処理に対して*P=0.0054、0.0052、0.0063および0.0197(Student's t-test)。本明細書ではpg/mlで示す、個体4および2のT細胞から分泌されたTNF−αは又、倍増率にて上に示している(それぞれ図AおよびB)。図1C〜Hは、T細胞由来のTNF−αのバックグラウンドレベルが異なっているにもかかわらず、DTX−K(24時間、100nM)は、全ての6人の個体のT細胞による著しいTNF−αの分泌を誘発し、それぞれ22.3、8.7、49.0、34.4、2.4および2.0の倍増率の結果になったことを示す。DTX−K誘導性TNF−α増加の最高値は、38pg/mlから1308pg/mlであって、34.4倍増を示している(図1F,個体4)。
【0076】
注:DTX−KはこれらのヒトT細胞の細胞生存または増殖に、有意な影響は持たなかった(データは示さず)。
【0077】
実施例3:チャンネルを含むKv1.1サブユニットのDTX−Kによる遮断は、TNF−αの唯一的な分泌を誘起するが、一方「古典的な」TCRの活性化は、TNF−α、INF−γ、IL−10およびIL−4の強健であるが、非唯一的な分泌を誘導する。
図2A〜Hにおいて、正常ヒトT細胞を、100nM DTX−K(図2A〜D)または抗−CD3および抗−CD28mAbs(図2E〜H)を用いた「古典的な」TCR活性化に曝した。その後24〜72時間内に培地に分泌されるTNF−α、INF−γ、IL−10およびIL−4のレベルを、ELISAによって試験した。少なくとも3回の独立して行った実験のうちの代表的な実験の一つからの、TNF−α(図2Aおよび2E、24時間)、INF−γ(図2Bおよび2F、24時間)、IL−10(図2Cおよび2G、72時間)およびIL−4(図2Dおよび2H,72時間)の分泌の倍増率±SDを示す。統計解析:未処理に対して*P=0.0056(A)、0.0050(E)、0.0051(F)、0.0050(G)および0.0053(H)(Student's t-test)。
【0078】
興味あることには、DTX−KによるKv1.1の遮断はTNF−αのみを増加させたが、INF−γ、IL−4およびIL−10のレベルには影響しないことが分かった(図2A〜D,結果を、全4サイトカインの分泌の倍増率で表す)。これとは著しく対照的に、抗−CD3および抗−D28抗体によって達成したTCR活性化は、全4つのThサイトカインの強健であるが、非選択的な分泌を誘起した(図2E〜2H)。予想したように、TCR活性化後のTNF−α分泌の規模は、DTX−K処理後に得られるものに比べて、ずっと大きかった(図2Eに対する図2A)。TCR活性化の間または後に、DTX−KによってT細胞のチャンネルを含むKv1.1サブユニットを遮断しても、TCR活性化のみによって達成された以上には、TNF−αレベルの更なる増加は起こらなかった(データは示されない)。
【0079】
実施例4:T細胞のチャンネルを含むKv1.1サブユニットの遮断により、長期間のTNF−αの増加が生じる
T細胞のTNF−α分泌は、TCR活性化後、通常は24時間でピークに達し、その後徐々に低下する。それ故、T細胞内のチャンネルを含むKv1.1サブユニットの遮断によって起きるTNF−α分泌を調査し、この時期にピークに達するかどうか、および分泌が一時的かあるいは長期なものかを検討した。図2Iおよび2Jにおいて、個体4(図2I)および個体5(図2J)(既に図1F、2Gに示した同じ二人の個体)からの正常T細胞を100nM、DTX−Kに曝し、その後異なった時点でELISA法により、培地をTNF−αについて検査した。それぞれの個体について、それぞれの時点でのTNF−α分泌の倍増率±SDを示す。個体4のそれぞれ16、24、48および72時間において、未処理に対して*P=0.0053、0.0052、0.0050および0.0053であり;個体5のそれぞれ16、24、48および72時間において、未処理に対して*P=0.0057、0.0063、0.0072および0.00316であった(Student's t-test)。
【0080】
この実験により、DTX−Kに誘導されたTNF−α分泌の動態が(図2I,2J)、それらのバックグラウンドおよびDTX−Kに誘導された、T細胞由来のTNFαレベル(図1F、2G)と比較していく分異なっていることを明らかにする。これらの相違にもかかわらず以下の二つの特徴は共通している:1)有意に増加したTNF−αの分泌は、16時間後において既に明らかである;2)TNF−αの分泌は、DTX−K処理の開始から72時間後であっても有意に高い状態を維持した。
【0081】
実施例5:DTX−K誘導性TNF−α分泌にはT細胞膜を通してCa2+の流入が必要とされる。
Ca2+は、TNF−αの転写に関与していることが知られている(Lobo et al, 1999)。これに基づいて、T細胞のチャンネルを含むKv1.1サブユニットを遮断することによって生じる著しいTNF−αの分泌には、「連続的な」Ca2+の流入が必要となるかどうかを検討した。T細胞内に発現されたL型Ca2+チャンネルの強力で非常に選択的な遮断剤を用いた:(R) - (+) - Bay K 8644であって、DTX−K(100nM)の添加の5分前に正常ヒトT細胞に加えた。選択的なCa2+チャンネル遮断剤は、DTX−KのTNF−αの分泌を引き起こす能力を完全に阻止したが、対照として加えたCl−チャンネル遮断剤、NPPB(表1)は、該阻止能力を有しなかった(図2K)。この実験において、Kv1.1遮断(100nM DTX−K、24時間)に対する反応におけるTNFαの分泌は、T細胞を100μM +Bay-K(高度の選択的な、T細胞中で発現されたCa2+チャンネル遮断剤で、DTX−Kの5分前に加えられた)で前処理した場合は、完全に阻止されたが、関係ないCl−チャンネル遮断剤、NPPBの100μMで前処理を行った場合は阻止されなかった。結果は、実施した3回の実験の一つからのTNF−αの分泌の平均(pg/ml)±SDを表している。統計解析:未処理に対して*P<0.0001、DTX−Kのみに対して**P<0.0001(Student's t-test)。
【0082】
実施例6:T細胞内のチャンネルを含むKv1.1サブユニットの遮断により、またTNF−α mRNAレベルも増加する。
次に、T細胞のチャンネルを含むKv1.1サブユニットの遮断により、TNF−αタンパクだけではなく、TNF−αのmRNAのレベルも増加するかについての問題を研究した。この問題を研究するために、DTX−Kをヒト正常T細胞に24時間加え、RNAを調製し、cDNAに転写し、TNF−α特異的なプライマーを用いて、特異的なTNF−αRT−PCRを行った。逆転写の効率のコントロールとして、内部標準(リボソームS14タンパク)を用いた更なるRT−PCR反応を同時に行った(すなわち同じPCRチューブにおいて)。さらに具体的に言うと、正常末梢ヒトT細胞を100nMDTX−Kに24時間曝した。それからcDNAを調製し、TNFαおよび内部コントロール、すなわちリボソームS14タンパク、に対する特異的なプライマーを用いて、同じPCRチューブ内で同時に二つのRT−PCR反応によって増幅した。結果を図3Aに示す。上段のバンド:TNF−α(予想される産物のサイズ:494bp);下段バンド:S14(予想される産物のサイズ:166bp)。下の棒グラフは、TNF−α/S14の対応するRT−PCRバンドの濃度解析および正規化の後に計算したTNF−αmRNAの倍増率を表す。NC=陰性コントロール(cDNA無し)。
【0083】
図3Aは、チャンネルを含むKv1.1サブユニットのDTX−Kによる遮断によって、実際、TNF−α mRNAレベルが増加することを示している。
【0084】
実施例7:チャンネルを含むKv1.1サブユニットの遮断は、正常ヒトT細胞のNF−
κBの活性化を導く。
TNF−αは(IL−1のような他の炎症促進性のサイトカインのように)NF−κBを活性化させるだけではなく、NF−κBによってもまた誘導される(Siebenlist et al, 1990)。TNF−α遺伝子の場合は、TNFα遺伝子の発現に対して観察されたNF−κBの重要性は、細胞タイプと解析された、エンハンサー/プロモータの区域に依存するのかも知れない(Siebenlist et al, 1990)。これらに基づいて、正常ヒトT細胞(ここでもまた、いかなる他の刺激の完全に存在しない状態で)におけるKv1.1チャンネルの遮断が、NF−κBに影響を及ぼすことができるかを我々は検討した。これを二つの技術によって研究した:1)未処理およびDTX−K処理正常ヒトT細胞から核抽出物を調製し、市販の電気泳動移動度シフトアッセイ(EMSA)によって核NF−κB結合配列に結合したNF−κBの量を測定する;2)抗−ヒトNF−κB p50特異的抗体、FITC結合抗−ヒトIgGおよび細胞核を特異的に染色するHoechst 33342を用いて、免疫組織化学染色法によって、推定されるNF−κBの核への移行を可視化する。
【0085】
核抽出物を数時間加えたが、そのうち7分のみを示す。OD±SDで示した結果は、市販のEMSAによって定量された、核NF−κB結合配列に結合したNF−κBの量を反映している。別個に行った二つの実験の一つを図3Bに示す。統計解析:*P=0.0092(Student's t-test)。
【0086】
OD値が核NF−κB結合配列に結合しているNF−κBの量を表す、図3Bから見られるように、DTX−K添加後7分で核抽出物に存在するNF−κBのレベルは有意に増加した。予想されたように、この増加は一時的なものであって、しばらく経つと試験した点はもはや検出されなかった(データは示していない)。EMSAによって検出したこの結果は、図3Cに示す二重免疫蛍光写真と一致していた。図3Cに結果が示してある実験において、正常ヒトT細胞は、処理しないかまたはDTX−Kで7、15、30および60分処理した(それぞれのカラムの上のDTX−K処理時間を参照)。免疫蛍光組織化学染色は、抗−ヒトNF−κB p50抗体、次いでFITC結合抗−ヒトIgGを用いて行うか、あるいは細胞核に取り込まれるHoechst 33342を用いて行った。拡大率x100の代表的な写真は、NF−κB p50緑色蛍光(上段のパネル)、核の青色蛍光(中段パネル)両者を重ねたもの(下段パネル)を示す。研究したT細胞の外形サイズ(明視野)および核(Hoechst)は、左のカラムに示す。従って、図3Cは、DTX−K添加後7分(左から3番目のカラム)で細胞核(Hoechst青色蛍光)は明瞭にNF−κB p50陽性(緑色蛍光)であるが、未処理の細胞においてはそうではなく、又試験した後の三つの時点:15,30および60分では(図3C,それぞれ三つの右のカラム)NF−κB p50の蛍光は見られなかった。注目すべき点は、DTX−K処理後7分において、細胞質(Hoechst 陰性)は、NF−κB p50に対して陽性に染色されたが(図3C,左から3番目のカラム)、未だ明瞭でない理由により細胞質NF−κB p50は、未処理細胞においては観察されない。唯一の論理的なしかし推測でしかない、考えられる説明は、本明細書で用いる抗−p50抗体は立体構造にいくらか敏感であり、もしそうなら活性化およびNF−κB阻害剤IκB(Siebenlist et al , 1990)の放出後、細胞質p50は、この抗体によって検出しやすくなる可能性がある。
【0087】
実施例8:正常ヒトT細胞は電位依存性Kv1.1タンパク質およびmRNAを発現する。
先の研究は、機能的な(すなわち電気生理的に活性を有する)チャンネルを含むKv1.1サブユニットは、主として脳、心臓、網膜、骨格筋および膵島で発現されていることを示した(Beckh et al , 1990; Klumpp et al , 1991; Matsubara et al, 1991; Roberds et al, 1991; Tsaur et al, 1992)。対照的にT細胞中のKv1.1電流に関しては、先の研究から現れる像は不明瞭である。いくつかの研究では、パッチ−クランプ技法によってヒトT細胞がKv1.1の特徴を発現するかを試験したが、該電流を同定することができなかった(例えば(Cahalan et al, 1985))。しかしこれと対照的に、Freedman et alはマウスCD4−CD8−胸腺細胞によるKv1.1電流/発現を示し(Freedman et al, 1995)、Liu et alは、いくつかの方法の使用に基づいて、未処理のマウスCD4+リンパ球に発現している電流は、Kv1.1、Kv1.1、Kv1.3およびKv1.6と一致すると報告した(Liu et al, 2002)。本明細書においては、Kv1.1の全く新規な面を研究して、正常末梢ヒトT細胞においてKv1.1タンパクおよび、mRNAの発現を確認することを欲した(本研究の範囲を逸脱するKv1.1電流について再び試験することなしに)。本方法は、Kv1.1特異的なプライマーを用いたKv1.1特異的RT−PCR、ならびにKv1.1特異的な抗体を用いたフローサイトメトリー(この目的のためには今まで使用されていなかった方法)を含む。
【0088】
図4Aは、cDNAを正常末梢ヒトT細胞から調製し、Kv1.1特異的プライマーを用いてKv1.1特異的RT−PCRを行った結果を示す(上段バンド;予想産物サイズ:709bp)。コントロールPCRは、同時に同じPCRチューブで、リボソームS14プライマーを用いて行った(下段バンド;予想産物サイズ:166bp)。図4Aは、Kv1.1特異的mRNAが実際に正常ヒトT細胞のcDNAから増幅されることを示す。更に加えて、Kv1.1特異的な免疫蛍光染色を行った。T細胞は、ウサギ抗−Kv1.1ポリクローン抗体(図4B、実太線)あるいはアイソタイプコントロール非特異的染色として、正常ウサギIgG(図4B、破線)で染色し、それからFITC結合抗ウサギIgGで染色した。Kv1.1発現は、FACSortによって評価した。五つの独立した実験(異なった個体のT細胞を用いた)の一つを示す。ここでヒトT細胞の49.0%は、Kv1.1陽性であった。行われた他の四つの実験においては、Kv1.1陽性T細胞の百分率は40−80%の間で変異した。従って、図4Bは、フローサイトメトリー解析によって、Kv1.1タンパク質が正常末梢ヒトT細胞のかなり実質的な割合で細胞表面に明瞭に発現していることを示す。
【0089】
実施例9:市販の抗−Kv1.1チャンネル抗体は顕著なTNF−αの分泌を誘発し、それにより選択的なKv1.1遮断剤の効果を模倣する。
この実験において、抗−Kv1.1特異的抗体が、Kv1.1チャンネル遮断剤、DTX−Kのように作用し、TNF−αの分泌を増加させるかを、我々は検討した。3人の更なる個体(各個体7−9)からの正常ヒトT細胞を、市販のウサギ抗−Kv1.1ポリクローン抗体とインキュベーションし(1:1000倍希釈、24時間)、培地中に分泌されたTNF−αのレベルをELISA法によって試験した。結果を図4C、4Dおよび4Eに示し、各個体のT細胞によって分泌されたTNF−αの平均±SDを提示する。それぞれ未処理の個体7〜9に対して、*P=0.0003、0.0057、および0.0021であった(Student's t-test)。
【0090】
図4C〜4Eは、3人の異なった該被験体に由来する正常ヒトT細胞と24時間インキュベーションした、市販の抗−Kv1.1抗体は、これらの細胞によって分泌されるTNF−αの量を、それぞれ7.2、14.8および23.2倍増を誘導したことを示す。ここで注意すべきことは、ここで用いた抗−Kv1.1抗体はKv1.1チャンネルの細胞内エピトープに対して指向性を有していることである。この抗体は、T細胞内に侵入した後TNF−αの分泌を誘導すると考えられているが、これは抗体の生体細胞内に侵入する能力を明瞭に示した先の報告に基づいている(Alarcon-Segovia et al , 1996; Ruiz-Arguelles et al, 2003)。事実、種々の自己免疫疾患において、病原となる抗体は細胞内エピトープに対して指向性を有しており(例えば、全身性エリテマトーデスにおける抗−DNAおよび抗−リボヌクレタンパク抗体)(Alarcon-Segovia et al , 1996; Ruiz-Arguelles et al, 2003)、細胞内に侵入後にのみそれらの有害な効果を発揮する。
【0091】
実施例10:Kv1抗体陽性末梢神経過剰興奮症(PNH)血清は、正常ヒトT細胞
によるTNF−αの分泌を著しく促進する。
この実験は、PNH患者の血清に存在する病原性のKv1抗体(すなわち、Kv1.1、Kv1.2およびKv1.6抗体)(Hart et al, 2002)が、市販Kv1.1抗体およびDTX−Kの作用を再現するか、又ヒトT細胞によるTNF−α分泌を増加させるかを検討する。PNHの通常提示される臨床症状は、筋肉のれん縮、硬直、こわばりおよび脱力感であって、多汗症、偽性ミオトニー現象、偽性テタニーおよび感覚性の症状である(Newsom- Davis et al, 1993)。加えて、PNH患者は、CNS症状を有することもある(Vincent et al, 2004)。PNHの殆どの症例は自己免疫症であって、Kv1.1、Kv1.2およびKv1.6抗体の混合物である循環性のKv1抗体(Hart et al, 002)によって起こる。
【0092】
この仮説を試験するために、正常末梢ヒトT細胞を健常コントロール被験体(A群)Kv1抗体陽性PHN患者(B群)またはKv1抗体陰性患者(C群)の任意の血清に(1:100倍希釈)に曝した。全ての群は年齢、性および地理的に適合させた(方法を参照)。続く24時間に培地に分泌されたTNF−α(図4F)またはINF−γ(図4G)のレベルは、ELISA法によって試験した。結果は、行われた三つの独立した実験の平均である。図4Fにおいて、水平バーおよびそれぞれの数値は中央値を表す。ここでは下カットオフ(カットオフA)である破線は、コントロール群の血清に曝されたT細胞によって分泌されたTNF−αの平均+2*SDを表し;ここでは上カットオフ(カットオフC)は、Kv1−陰性PNH血清に曝されたT細胞によって分泌されたTNF−αの平均+2*SDを表す。グラフに示されたそれぞれの数値は、分泌されたTNF−αの実際の平均量(pg/ml)を表す。3つのKv1陽性PNH患者(つまり、PNH+5,2および6)の血清は、明瞭にカットオフCの上のTNF−αの分泌の増加を誘導した(しかしINF−γは誘導されない)。統計解析:KW統計=8.0003、P=0.0183,Kruskal-Wallis 検定。対比較はMann-Whitney U検定でおこなった:A群対B群、P=0.0117、A群対C群、P=0.0087、C群対B群、P=0.6282。
【0093】
図4は、いく人かのKv1抗体陽性PNH患者の血清は、健常コントロール被験体または検出可能なKv1抗体を有しないPNH患者からの血清によって誘導されるよりは有意に高い、著しいTNFαの分泌を誘導することを示している。更に、TNF−α誘発効果は、抗体力価と正の関係にあった(材料および方法参照)。PNH患者からの血清の同体積および濃度を含むがT細胞を含まない、陰性コントロール実験培養には、TNF−αレベルに影響は無かった。重要なことには、PNH患者のKv1抗体陽性血清は、TNF−αの増加を誘導したが、DTX−Kで観察された(図2)と同様に、INF−γの増加は誘導せず(図4G)、この効果の選択性を確認している。総合的に、この一連の実験は、増加したKv1抗体力価を含むいくつかのPNH患者の血清は、正常ヒトT細胞による著しいTNFα分泌を誘導し、それによって市販抗−Kv1.1抗体および選択的Kv1.1遮断剤、DTX−Kの効果を模倣することを示した。
【0094】
実施例11:前処理Kv1.1遮断により上昇したTNF−αレベルを分泌する正常ヒトT細胞を、SCIDマウスへ注射することで、肝臓へ宿主細胞を選択的に動員させる。
Kv1.1の選択的遮断に反応しているヒトT細胞によって分泌するTNF−αが、インビボにて機能的であるかを試験するために、我々は、最近の研究を想起したが、それは、脳内へTNF−αを投与に続いて一連の現象が起こり、その中には、特異的に肝臓に蓄積する宿主のED1−陽性細胞(すなわち常在および活性化クッパー細胞/動員された血中単球)の急速な2倍までに増加する、というものであった(Campbell et al, 2005)。我々は、DTX−K処理済のヒトT細胞によって分泌された、増加したTNF−αも該効果を誘導するかを検討した。このことを試験するために、新たに精製した正常ヒトT細胞を、PBSまたは100nM DTX−Kに24時間曝し、洗浄し、蛍光標識を付け、それから後者の細胞が増加したTNF−αを分泌していることを確認した後(データは示していない)、SCIDマウス(すなわち自身のリンパ細胞を欠いている)に注射した(各群n=5)。肝臓、骨髄、脾臓、および腎臓の単細胞懸濁ならびに血液細胞に存在する、細胞の総数、ならびに常在SCID細胞のみ(非蛍光)および注射されたヒトT細胞のみの数を、24時間後FACSortによって計測した。肝臓に関しては、結果は非蛍光SCID宿主細胞の平均±SEM数(図5A)および蛍光標識ヒトT細胞の平均±SEM数(図5B)で表される。他の臓器に関しては、非蛍光SCID宿主細胞の数のみを示す:骨髄(図5C)、血液(図5D)、脾臓(図5E)および腎臓(図5F)、未処理に対して*P<0.0001(Student's t test)。図5Gは、注入されたヒトT細胞の検出のために、エクスビボにてDTX−Kと前インキュベーションしたヒトT細胞を注射したSCIDマウスの脳切片の免疫組織化学像を示す。SCIDマウスにDTX−K処理ヒトT細胞を注射後24時間、CD3陽性細胞が脳皮質内に検出されることを示すx40の拡大率の代表的な写真である。
【0095】
図5AはTNF−αを分泌しているDTX−K処理ヒトT細胞の注入は、肝臓における細胞数を2倍にしたことを示す。我々が依拠した関連研究において(Campbell et al, 2005)、この効果は肝臓に限定されていて、骨髄、血液、脾臓、および腎臓における細胞総数は、有意には変化しなかった(それぞれ図5C〜5F)。更に、予想されるように、肝臓における細胞数の増加はマウス(SCID)宿主細胞の動員に由来するもので(図5A)、受動的に移したヒトT細胞がこの臓器に入ったことに由来するのではなかった(図5B)。技術的な制限によって、動員したマウス細胞の正確な同定は、行わなかった。最後に、脳の切片の免疫組織化学像は、注射されたSCIDマウスの皮質内に、注射されたDTX−K処理済のヒトT細胞の存在を明らかにした(図5G)これは、原則として、肝臓への宿主細胞の生体内動員は、脳内で作動しているTNF−α(すなわちDTX−K処理ヒトT細胞によって脳へ送達されたTNF−α)によって介される可能性を示唆しており、TNFαを脳内に注射後、宿主細胞が肝臓に動員された最近の報告に類似している(Campbell et al, 2005)。実際、TNFαのBBBを混乱させる能力に基づけば(Mayhan 2002)、DTX−K処理細胞が脳に侵入することができることは、驚くには当たらない。脳に到達した未処理およびDTX−K処理ヒト細胞の数の間の統計的に正当な比較は不可能である。最後の注意点として、肝臓に動員されたマウスSCID細胞が、ヒトTNF−α(DTX−K処理ヒトT細胞によって分泌される)に反応できたことは、我々にとって驚くべきことではない。なぜならマウスTNF−α受容体1型は組み換えマウスおよびヒトTNF−αの両方に同様な親和性を有しているからである(Lewis et al, 1991)。更にヒトTNF−αを発現しているトランスジェニックマウスは、リウマチ性関節炎を有するヒトの患者の特徴である多くの徴候を発現し(Keffer et al, 1991; Li et al , 2003)、マウス起源の細胞がヒトTNF−αに応答することができることを示唆している。
【0096】
具体的な実施形態の先の記述は、本発明の一般的な性質を十分に明らかにしているので、他者は、現在の知識を応用することにより該具体的な実施形態を、必要以上の実験を行うこと無く、一般概念から逸脱すること無く、容易に改変しおよび/または種々の応用に適応することができ、それ故該適応および改変は、開示した実施形態と等価の意味と範囲内に包含することを意図すべきであり、また意図している。本明細書で用いられた言葉使いや用語は、記述するためであって、限定するもので無いことを理解されたい。種々の開示した機能を実施するための手段、材料および段階は、本発明から逸脱すること無く種々の代替型を取ってもよい。従って、上記の明細書および/または請求項に存在する可能性のある、「・・・に対する手段」および「・・・のための手段」あるいは機能的な記載に続くいかなる方法段階の用語は、あらゆる構造、物理的、化学的または電気的要素または構造、もしくは現在または将来存在する可能性のある、引用した機能を実施するいかなる方法段階をも包含することを意図している、すなわち同じ機能を実施するための他の手段または段階を用いることができ;該表現はその最も広義に解釈されることを意図している。
【0097】
【表8】
【図面の簡単な説明】
【0098】
【図1】図1Aは、それぞれの試験されたイオンチャンネル遮断剤についての、TNF−αの倍増率を示すグラフである。 図2Bは、用量の関数としてのTNF−αの倍増率を示すグラフである。 図1C〜Hは、6人の異なった該被験体についての、それぞれのDTX−K誘導TNF−α分泌の量を示すグラフである。
【図2】図2A〜Hは、DTX−K(図2A〜D)または抗−CD3および抗−CD28mAb(図2E〜H)に曝された正常ヒトT細胞によって分泌された種々のサイトカインのレベルを示す。TNF−α分泌の倍増率を図2Aおよび2Eに、IFNγを図2BおよびFに、IL−10を図2CおよびGに、ならびにIL−4を図2DおよびHに示す。 図2IおよびJは、二人の異なった該被験体においてそれぞれのTNF−α分泌の倍増率を、時間の関数として示したグラフである。 図2Kは、未処理、DTX−Kのみの処理、またはBay−KまたはNPPSのいずれかで最初に前処理済のT細胞についてのTNF−α分泌を示すグラフである。
【図3】図3Aにおいては、正常末梢ヒトT細胞をDTV−Kに曝した後、上部のバンドはTNF−α(予想産物サイズ:494bp)および下部のバンドはS14(予想産物サイズ:166bp)を示す。下の棒グラフは、濃度測定分析および対応するTNF−α/S14のRT−PCRバンドの規格化の後計算したTNF−αmRNAの倍増率を表す。NC=陰性対照(cDNA存在せず)。 図3Bは、100nMDTX−Kに先に曝された正常T細胞から調製した核抽出物における、核NF−κB−結合配列に結合したNF−κBの量を示す。 図3Cは、正常ヒトT細胞が未処理であるか、または100nM DTX−Kと7、15、30および60分処理済の(上記DTX−K処理時間についてはそれぞれのカラムを参照)、二重免疫蛍光写真を示す。拡大率x100の代表的な写真は、NF−κB p50 緑蛍光(上段パネル)、核 青色蛍光(中段パネル)および両者を重ねたもの(下段パネル)を示す。試験したT細胞(明視野)および核(Hoechst)の外形サイズを左のカラムに示す。
【図4】図4Aは、Kv1.1特異的なプライマーを用いて行ったKv1.1特異的なRT−PCRの結果を示す(上段バンド;予想産物サイズ;709bp)。コントロールRT−PCRは、リボソームS14プライマーを用いて同じPCRチューブの中で同時に行った(下段バンド;予想産物サイズ;166bp)。 図4Bは、T細胞を、ウサギ抗−Kv1.1ポリクローン抗体で(図4B、太実線)、または、アイソタイプコントロール非特異的染色として、正常ウサギIgG(図4B、破線)で染色し、それからFITC結合抗−ウサギIgGで染色した後の、FACSortの結果を示す。 図4C〜Eは、3人の異なった該被験体のそれぞれ個人のT細胞によるTNF−α分泌の平均値の結果を示したもので、それらのT細胞を市販ウサギ抗−Kv1.1ポリクローン抗体とインキュベーションした。 図4FおよびGは、正常末梢ヒトT細胞を、健常コントロールの個人(A群)、Kv1抗体−陽性PHN患者(B群)またはKv1抗体−陰性PHN患者(C群)の血清に曝し、培地中に分泌されたTNF−α(図4F)またはIFN−γのレベルを示す。
【図5】図5A〜Fは、新たに調製した正常ヒトT細胞を、PBSまたは100nM DTX−Kに24時間曝し、洗浄し、蛍光標識を付け、それからSCIDマウスに注射した後の結果を示す。グラフは、肝臓、骨髄、脾臓および腎臓の単細胞懸濁液ならびに血液に存在する細胞の総数、ならびに常在のSCID細胞(非蛍光)のみの数および注射されたT細胞(蛍光)のみの数を示す。図5Aは、肝臓における非蛍光SCID宿主細胞の平均±SEM数を示し、又図5Bは、肝臓における蛍光標識されたヒトT細胞の平均±SEMを示す。他の器官については、非蛍光SCID宿主細胞の数だけを示す:骨髄(図5C)、血液(図5D)、脾臓(図5E)および腎臓(図5F)。 図5Gは、注入されたヒトT細胞の検出のために、DTX−Kとエクスビボにて前インキュベーションしたヒトT細胞を注射したSCIDマウスからの脳切片の免疫組織化学像を示す。SCIDマウスにDTX−K処理したヒトT細胞を注射後24時間、ヒトCD3陽性細胞が皮質内に検出されることを示す、拡大率x40の代表的な写真を提示する。
【特許請求の範囲】
【請求項1】
T細胞での電位依存性カリウムチャンネルKv1.1の選択的遮断の原因となることを含む、T細胞によるTNF−αの合成および分泌を起こさせる方法。
【請求項2】
前記原因となるステップが、該T細胞をKv1.1チャンネルの選択的遮断の原因となる分子に接触させることを含む、請求項1に記載の方法。
【請求項3】
ヒトまたは他の動物被験体において、TNF−αのインビボ分泌を増加させることにより治療可能な疾患を治療する方法であって、電位依存性カリウムチャンネルKv1.1の選択的遮断を有するT細胞を、該被験体内に存在させることを含む方法。
【請求項4】
該被験体が、癌、免疫不全もしくは神経細胞傷害後または神経疾患後の神経細胞の再生欠損に罹患している、請求項3に記載の方法。
【請求項5】
該被験体が、局所的に進行した軟組織肉腫、転移メラノーマまたは他の切除不可能な組織学的腫瘍に罹患しており、患肢の切断を回避すべきである、請求項4に記載の方法。
【請求項6】
前記原因となるステップが、Kv1.1チャンネルの選択的遮断の原因となる分子を、該被験体に投与することを含む、請求項3〜5のいずれか一項に記載の方法。
【請求項7】
前記原因となるステップが、自己T細胞を該被験体から取り出し、自己T細胞をKv1.1チャンネルの選択的遮断の原因となる分子とエクスビボで接触させ、該処理済の自己T細胞を該被験体の体内へ投与して戻すことを含む、請求項3〜5のいずれか一項に記載の方法。
【請求項8】
該被験体が、局所的に進行した軟組織肉腫、転移メラノーマまたは他の切除不可能な組織学的腫瘍に罹患しており、患肢の切断を回避すべきであり、前記処理済の自己T細胞を分離患肢潅流設定によって投与する、請求項7に記載の方法。
【請求項9】
該被験体が、肝癌に罹患しており、種々の免疫細胞の肝臓への動員が増強され、T細胞攻撃による肝癌の直接攻撃が起こり、該処理済のT細胞によるTNFαの分泌により肝癌の直接根絶が起こる、請求項7に記載の方法。
【請求項10】
それを必要とするヒトまたは他の動物被験体内の血液脳関門および末梢内皮の透過性を増大させる方法であって、電位依存性カリウムチャンネルKv1.1の選択的遮断を有するT細胞が、該被験体内に存在する原因となることを含む方法。
【請求項11】
前記原因ステップが、Kv1.1チャンネルの選択的遮断の原因となる分子を、該被験体に投与することを含む、請求項10に記載の方法。
【請求項12】
前記原因ステップが、自己T細胞を該被験体から取り出し、自己T細胞をKv1.1チャンネルの選択的遮断の原因となる分子とエクスビボで接触させ、処理済の自己T細胞を該被験体の体内へ投与して戻すことを含む、請求項10に記載の方法。
【請求項13】
Kv1.1チャンネルの選択的遮断の原因となる前記分子がデンドロトキシン−Kである、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項14】
Kv1.1チャンネルの選択的遮断の原因となる前記分子が特異的抗−Kv1.1抗体である、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項15】
Kv1.1チャンネルの選択的遮断の原因となる前記分子がKv1.1チャンネルの下流経路を選択的に遮断できる分子である、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項16】
Kv1.1チャンネルを開くか、またはKv1.1チャンネルの遮断を克服することを含む、T細胞によるTNF−αの合成および分泌を阻害する方法。
【請求項17】
ヒトまたは他の動物被験体において、TNF−αのインビボ分泌を減少させることによって治療可能な疾患または病気を治療する方法であって、該被験体のT細胞上のKv1.1チャンネルを開くか、または該被験体のT細胞上のKv1.1チャンネルの遮断を克服することを含む方法。
【請求項18】
該被験体がリウマチ性関節炎または外傷後病気に罹患している、請求項17に記載の方法。
【請求項19】
該疾患または病気が、Kv1抗体に関連するPNHであり;Kv1抗体に関連する脳炎であり;およびヒトKv1.1チャンネルをコードする遺伝子の様々な突然変異を伴う1型突発性運動失調(EA−1)である、請求項17に記載の方法。
【請求項1】
T細胞での電位依存性カリウムチャンネルKv1.1の選択的遮断の原因となることを含む、T細胞によるTNF−αの合成および分泌を起こさせる方法。
【請求項2】
前記原因となるステップが、該T細胞をKv1.1チャンネルの選択的遮断の原因となる分子に接触させることを含む、請求項1に記載の方法。
【請求項3】
ヒトまたは他の動物被験体において、TNF−αのインビボ分泌を増加させることにより治療可能な疾患を治療する方法であって、電位依存性カリウムチャンネルKv1.1の選択的遮断を有するT細胞を、該被験体内に存在させることを含む方法。
【請求項4】
該被験体が、癌、免疫不全もしくは神経細胞傷害後または神経疾患後の神経細胞の再生欠損に罹患している、請求項3に記載の方法。
【請求項5】
該被験体が、局所的に進行した軟組織肉腫、転移メラノーマまたは他の切除不可能な組織学的腫瘍に罹患しており、患肢の切断を回避すべきである、請求項4に記載の方法。
【請求項6】
前記原因となるステップが、Kv1.1チャンネルの選択的遮断の原因となる分子を、該被験体に投与することを含む、請求項3〜5のいずれか一項に記載の方法。
【請求項7】
前記原因となるステップが、自己T細胞を該被験体から取り出し、自己T細胞をKv1.1チャンネルの選択的遮断の原因となる分子とエクスビボで接触させ、該処理済の自己T細胞を該被験体の体内へ投与して戻すことを含む、請求項3〜5のいずれか一項に記載の方法。
【請求項8】
該被験体が、局所的に進行した軟組織肉腫、転移メラノーマまたは他の切除不可能な組織学的腫瘍に罹患しており、患肢の切断を回避すべきであり、前記処理済の自己T細胞を分離患肢潅流設定によって投与する、請求項7に記載の方法。
【請求項9】
該被験体が、肝癌に罹患しており、種々の免疫細胞の肝臓への動員が増強され、T細胞攻撃による肝癌の直接攻撃が起こり、該処理済のT細胞によるTNFαの分泌により肝癌の直接根絶が起こる、請求項7に記載の方法。
【請求項10】
それを必要とするヒトまたは他の動物被験体内の血液脳関門および末梢内皮の透過性を増大させる方法であって、電位依存性カリウムチャンネルKv1.1の選択的遮断を有するT細胞が、該被験体内に存在する原因となることを含む方法。
【請求項11】
前記原因ステップが、Kv1.1チャンネルの選択的遮断の原因となる分子を、該被験体に投与することを含む、請求項10に記載の方法。
【請求項12】
前記原因ステップが、自己T細胞を該被験体から取り出し、自己T細胞をKv1.1チャンネルの選択的遮断の原因となる分子とエクスビボで接触させ、処理済の自己T細胞を該被験体の体内へ投与して戻すことを含む、請求項10に記載の方法。
【請求項13】
Kv1.1チャンネルの選択的遮断の原因となる前記分子がデンドロトキシン−Kである、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項14】
Kv1.1チャンネルの選択的遮断の原因となる前記分子が特異的抗−Kv1.1抗体である、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項15】
Kv1.1チャンネルの選択的遮断の原因となる前記分子がKv1.1チャンネルの下流経路を選択的に遮断できる分子である、請求項2、6〜9、11および12のいずれか一項に記載の方法。
【請求項16】
Kv1.1チャンネルを開くか、またはKv1.1チャンネルの遮断を克服することを含む、T細胞によるTNF−αの合成および分泌を阻害する方法。
【請求項17】
ヒトまたは他の動物被験体において、TNF−αのインビボ分泌を減少させることによって治療可能な疾患または病気を治療する方法であって、該被験体のT細胞上のKv1.1チャンネルを開くか、または該被験体のT細胞上のKv1.1チャンネルの遮断を克服することを含む方法。
【請求項18】
該被験体がリウマチ性関節炎または外傷後病気に罹患している、請求項17に記載の方法。
【請求項19】
該疾患または病気が、Kv1抗体に関連するPNHであり;Kv1抗体に関連する脳炎であり;およびヒトKv1.1チャンネルをコードする遺伝子の様々な突然変異を伴う1型突発性運動失調(EA−1)である、請求項17に記載の方法。
【図1】


【図2】
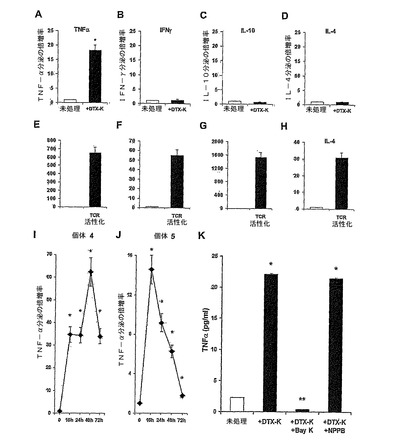
【図3】
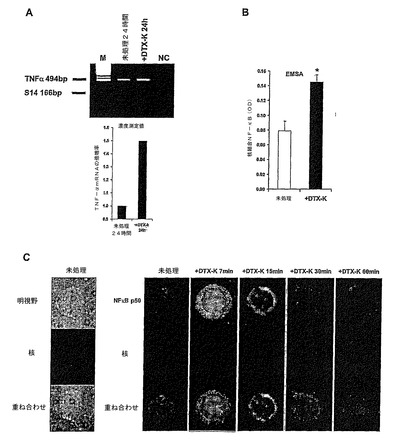
【図4】
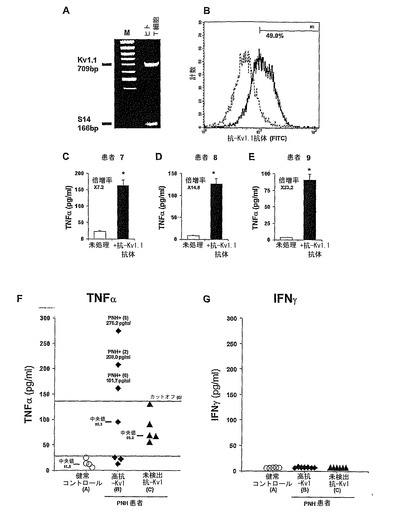
【図5】
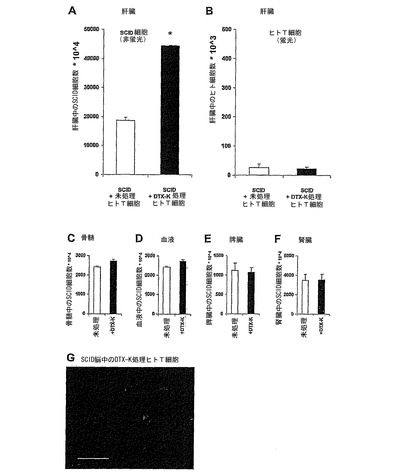
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2009−502206(P2009−502206A)
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願番号】特願2008−525208(P2008−525208)
【出願日】平成18年8月3日(2006.8.3)
【国際出願番号】PCT/US2006/030361
【国際公開番号】WO2007/019267
【国際公開日】平成19年2月15日(2007.2.15)
【出願人】(508036569)ミニュー・セラピューティクス・リミテッド (1)
【氏名又は名称原語表記】MiNEUET THERAPEUTICS LTD.
【Fターム(参考)】
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願日】平成18年8月3日(2006.8.3)
【国際出願番号】PCT/US2006/030361
【国際公開番号】WO2007/019267
【国際公開日】平成19年2月15日(2007.2.15)
【出願人】(508036569)ミニュー・セラピューティクス・リミテッド (1)
【氏名又は名称原語表記】MiNEUET THERAPEUTICS LTD.
【Fターム(参考)】
[ Back to top ]