
腫瘍画像化のためのアミノ酸類似体の立体選択的合成
放射性標識した非天然アミノ酸1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)およびその類似体は、腫瘍細胞に対するその選択的親和性のため、陽電子射出断層撮影法および単光子射出コンピュータ断層撮影法に有用な腫瘍画像化剤の候補である。本発明は、シン−ACBC類似体の立体選択的な合成方法を提供する。開示した合成ストラテジーは、信頼性が高く、かつ効率的であり、グラム量のACBC類似体の様々なシン−異性体、特に、シン−[18F]−1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)およびシン−[123I]−1−アミノ−3−ヨードシクロブタン−1−カルボン酸(IACBC)類似体を合成するのに用いることができる。

【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2005年6月23日出願の米国仮出願番号第60/693,385号に関する優先権を主張するものである。それを、本出願に相反しない範囲で参照により本明細書に組み込む。
【0002】
(連邦研究支援の承認)
本発明は、国立衛生研究所(National Institutes of Health)より付与された認可番号5−R21−CA−098891のもとでの政府支援によって行われた。政府は本発明において一定の権利を有するものである。
【0003】
(発明の背景)
本発明は、シン−アミノ酸類似体を合成する方法、およびその方法によって合成される化合物、具体的にはシン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体に関する。本発明のアミノ酸類似体は、生体系において特異的な結合を有し、陽電子射出断層撮影法(PET)および単光子放出(SPECT)画像検査法で使用することができる。
【背景技術】
【0004】
陽電子射出断層撮影法(PET)および単光子射出コンピュータ断層撮影法(SPECT)を用いる、腫瘍を画像化するための代謝トレーサーとして用いる放射性標識アミノ酸の開発はこれまでかなりの期間行われている。放射性標識アミノ酸は様々な腫瘍タイプに適用されているが、頭蓋内腫瘍へのその適用は、他の画像診断法より優れた潜在的利点のため、相当な注目を集めてきた。脳腫瘍の外科的切除および/または放射線治療の後、CTやMRIなどの従来の画像検査法では、残留腫瘍または再発腫瘍と、治療介入による組織傷害とを信頼性高く識別することはできず、処置の有効性の監視や腫瘍再発の検出に最適ではない[非特許文献1;非特許文献2]。
【0005】
新生物の診断および画像化のための主要なPET用薬剤である2−[18F]フルオロデオキシグルコース(FDG)では、脳腫瘍の画像化に限界がある。放射線治療または外科的治療の後発生する可能性がある炎症組織と同じように、正常な大脳皮質組織は高い[18F]FDGの取り込みを示す;これらの因子は[18F]FDGで得られる画像の解釈を複雑にする可能性がある[非特許文献3;Conti, PS (1995)]。
【0006】
多くの報告は、放射性標識アミノ酸で画像化するPETおよびSPECTは、CT法またはMRI法より良好に正常脳内の腫瘍境界を規定し、それによって、より良好な治療計画が可能になることを示している[非特許文献4;非特許文献5]。さらに、いくつかの研究は、アミノ酸の取り込みの度合が腫瘍の異型度と相関しており、それは重要な徴候情報を提供する可能性がある[非特許文献5]。
【0007】
アミノ酸は腫瘍細胞の増殖に必須の栄養素である。陽電子放出アイソトープの炭素11およびフッ素18を含む様々なアミノ酸が調製されている。これらは脳腫瘍および全身性腫瘍を有する患者の腫瘍を画像化するための臨床腫瘍学的分野で使用できる潜在性があると評価されており、ある種の癌において2−[18F]FDGに対して優れた特徴を有している。これらのアミノ酸候補はさらに2つの主要カテゴリーに分けることができる。第1のカテゴリーは、[11C]バリン、L−[11C]ロイシン、L−[11C]メチオニン(MET)およびL−[1−11C]チロシン、ならびに2−[18F]フルオロ−L−チロシンおよび4−[18F]フルオロ−L−フェニルアラニンなどの構造的に似ている類似体などの放射性標識した天然由来のアミノ酸によって代表される。腫瘍細胞膜を通るこれらのアミノ酸の移動は主に、ナトリウム非依存性ロイシン型「L」アミノ酸輸送系による担体媒介輸送によって起こる。正常組織と比較した、これら天然由来の放射性標識アミノ酸の腫瘍内への取り込みの増大と保持の延長は、タンパク質中への大幅かつ迅速な局部的取り込みに一部起因している。これら放射性標識アミノ酸のうち、[11C]METが臨床的に腫瘍を検出するのに最も広く用いられている。[11C]METは脳腫瘍および全身性腫瘍を検出するのに有用であることが分かっているが、複数の経路を通してのインビボでの代謝の影響を受けやすく、多くの放射性標識代謝産物をもたらす。したがって、腫瘍代謝活性の信頼性のある測定に必要な精度を有する画像分析は不可能である。ヒトにおける[11C]METの腫瘍取り込みの動態解析の研究は、アミノ酸輸送が、タンパク質合成より感度の高い腫瘍細胞増殖の測定を提供できることを強く示唆している。
【0008】
[11C]METに伴う欠点は第2のカテゴリーのアミノ酸を用いて克服することができる。これらは1−アミノシクロブタン−1−[11C]カルボン酸([11C]ACBC)などの非天然性アミノ酸である。[11C]METと比較した[11C]ACBCの利点は、それが代謝されないということである。炭素11アミノ酸を臨床用途に適用するための大きな限界は炭素11の半減期が20分間と短いことである。半減期が20分間であるということは、炭素11アミノ酸を生成するのに現地での粒子加速器を必要とする。さらに、炭素11アミノ酸の各バッチ生成により、単一線量だけまたは比較的少ない線量だけしか発生させることができない。したがって、炭素11アミノ酸は、広範な臨床用途での局部的分配には不適切な候補品である。
【0009】
炭素11の半減期の物理的限界を克服するために、我々は最近、いくつかの新規なフッ素18で標識した非天然性アミノ酸の開発に焦点をあてた。そのいくつかは特許文献1および特許文献2に開示されている。その開示を参照により本明細書に組み込む。これらには、アンチ−1−アミノ−3−[18F]フルオロシクロブチル−1−カルボン酸(アンチ−[18F]FACBC)、シン−1−アミノ−3−[18F]フルオロシクロブチル−1−カルボン酸(シン−[18F]FACBC)、シン−およびアンチ−1−アミノ−3−[18F]フルオロメチル−シクロブタン−1−カルボン酸(シン−およびアンチ−[18F]FMACBC)が含まれる。これらのフッ素−18アミノ酸は、画像技術の陽電子射出断層撮影法(PET)を用いたアミノ酸輸送をもとにして、インビボで脳腫瘍および全身性腫瘍を画像化するのに用いることができる。我々の開発では、主として「L」型の大型中性アミノ酸輸送系を用い、より少ない程度に「A」型アミノ酸輸送系を用いる担体媒介輸送によって、腫瘍毛細管を通って移動するフッ素18標識シクロブチルアミノ酸を用いる。我々の予備的評価では、「L」型輸送系のための主要基質である陽電子放射体で標識したシクロブチルアミノ酸は、脳腫瘍および全身性腫瘍を有する患者の腫瘍を画像化するのに臨床腫瘍学的に優れた潜在性を示した。11C(t1/2=20分間)ではなく、シクロブチル/分鎖アミノ酸の18F−標識を提案する主な理由は、臨床用途において、11C−標識放射性医薬品ではなく18Fを用いて得られる大きな運搬上および経済上の利益である。忙しい核医学部門で、18F標識放射性医薬品を用いる腫瘍画像化の利点は、主に18Fのより長い半減期(t1/2=110分間)によるものである。18Fのより長い半減期により、放射性トレーサーの単一生産ロットからの現地外での分配と複数線量が可能になる。さらに、これらの非代謝アミノ酸は、2−[18F]FDGPETでは十分に画像化されないある種の全身性固形癌に対する画像化剤としても、より広い用途を有している。特許文献3(これを参照により本明細書に組み込む)はさらに、18Fを用いた標識およびPET画像化での使用に適した2−アミノ−3−フルオロ−2−メチルプロパン酸(FAMP)や3−フルオロ−2−メチル−2−(メチルアミノ)プロパン酸(N−MeFAMP)などのα−アミノイソ酪酸(AIB)のフッ素化類似体の例を開示している。AIBは、主としてA型アミノ酸輸送系を介して細胞中へ活発に輸送される非代謝性のα,α−ジアルキルアミノ酸である。システムAのアミノ酸輸送は、細胞増殖および細胞分裂の際に増大し、腫瘍細胞において上方調節されることも示されている[非特許文献6;非特許文献7]。動物において実験的に誘発される腫瘍、およびヒトにおいて自発的に発生する腫瘍の研究は、正常組織に対して、放射性標識AIBの取り込みが増加していることが示されている[非特許文献8;非特許文献9]。AIBのN−メチル類似体、N−MeAIBは、AIBよりA型アミノ酸輸送系に対して、さらにより高い選択性を示す[非特許文献10]。N−MeAIBは炭素11で放射性標識され、ヒトにおいて代謝的に安定である[非特許文献11]。
【特許文献1】米国特許第5,808,146号明細書
【特許文献2】米国特許第5,817,776号明細書
【特許文献3】国際公開第03/093412号パンフレット
【非特許文献1】Buonocore, E(1992年)、Clinical Positron Emission Tomography. Mosby−Year Book, Inc. St. Louis, MO、17〜22頁
【非特許文献2】Langleben, DDら(2000年)、J. Nucl. Med. 41:1861〜1867頁
【非特許文献3】Griffeth, LKら(1993年)、Radiology. 186:37〜44頁
【非特許文献4】Ogawa, Tら(1993年)、Radiology. 186: 45〜53頁
【非特許文献5】Jager, PLら(2001年)、Nucl. Med., 42:432〜445頁
【非特許文献6】Palacin, Mら(1998年)、Physiol. Rev. 78:969〜1054頁
【非特許文献7】Bussolati, Oら(1996年)、FASEB J. 10:920〜926頁
【非特許文献8】Conti, PSら(1986年)、Eur. J. Nucl. Med. 12:353〜356頁
【非特許文献9】Uehara, Hら(1997年)、J. Cereb. Blood Flow Metab. 17:1239〜1253頁
【非特許文献10】Shotwell, MAら(1983年)、Biochim. Biophys. Acta. 737:267〜84頁
【非特許文献11】Nagren, Kら(2000年)、J. Labelled Cpd. Radiopharm. 43:1013〜1021頁
【発明の開示】
【発明が解決しようとする課題】
【0010】
脳腫瘍および全身性腫瘍を有する患者での腫瘍の画像化のための陽電子放出アイソトープを含むアミノ酸類似体の利点は当業界でよく認識されているが、これらの化合物の立体特異的異性体を大量に提供できる、信頼性が高く効率的な合成方法は依然として必要とされている。候補化合物を、細胞および動物モデルにおける有効性の確認研究からヒトでの適用に移行する際、使用する合成技術は、その化合物が定常的にかつ信頼性高く生産されるように適合するものでなければならない。そのために、本発明者等はここに、シン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体を作製するための立体選択的な合成ストラテジーを開発した。この立体選択的な合成ストラテジーは、様々なアミノ酸類似体、特に、PETおよびSPECTを用いた腫瘍画像化用の放射性トレーサーを含むものを合成するのに適用することができることは以下の説明で明らかであろう。
【課題を解決するための手段】
【0011】
(発明の要旨)
本発明は、シン異性体のアミノ酸類似体を合成するための主要前駆体の特定の立体異性体を生成する合成ストラテジーを提供する。このストラテジーはシン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体を合成するのに特に有用である。合成における主要工程には、トランス−アルコールへの前駆体シントンの還元が含まれ、このトランス−アルコールはシン−異性体の最終生成物に転換される。本明細書で開示する合成ストラテジーは信頼性が高く、かつ効率的であり、シン−ACBC類似体の放射線合成のための主要前駆体をグラム単位の規模で調製することが可能になる。さらに、本明細書で開示の合成ストラテジーは、最後の工程として、適切なアイソトープを組み込んでアイソトープの有用な寿命を最大化する。
【0012】
本発明は以下の式を有するトランス−アルコールを提供する:
式1
【0013】
【化4】
[式中、YおよびZ=独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)であり、
R1〜R3=独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルであり、
R4およびR5=独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニル、ハロアルキニルであり、但し、ハロは非放射性のF、Cl、BrおよびIである]
本発明はまた、式1の一般構造を有するトランス−アルコールの合成方法も提供する。上式のトランス−アルコールの合成の主要工程は、ポリマー結合還元剤(例えば、Aldrich32、864−2水素化ほう素ポリマー担持アンバーライトIRA400;Aldrich52、630−4シアノ水素化ほう素ポリマー担持;Aldrich35,994−7水素化ほう素ポリマー担持アンバーライトA−26;Aldrich59、603−5水素化ほう素亜鉛ポリマー(結合))を用いる金属水素化物による直接還元である。本明細書でのスキーム3では、リチウムトリイソブチルボランおよびZnCl2を用いたこの反応を例示する。
【0014】
開示する合成ストラテジーは、脳および身体の腫瘍の検出および評価での使用ならびに他の使用のための様々なアミノ酸化合物のシン−異性体を調製するのに用いることができる。これらの化合物は、1−アミノ−シクロアルキル−1−カルボン酸の有利な特性、すなわちその迅速な取り込みおよび長期的保持と、フッ素18、ヨウ素−123、ヨウ素−125、ヨウ素−131、臭素−75、臭素−76、臭素−77、臭素−82、アスタチン−210、アスタチン−211および他のアスタチンアイソトープなどの特定の有用なハロゲンアイソトープを含むハロゲン置換基の特性とを組み合わせる。さらにこれらの化合物は、キレート化錯体を用いてテクネチウムおよびレニウムアイソトープで標識することができる。詳細な説明についてはWO03/093412および米国特許第5,817,776号を参照されたい。
【0015】
トランス−アルコールを含む本発明の合成ストラテジーを用いて作製することができるシン−アミノ酸類似体には、これらに限定されないが、以下の式を有する化合物が含まれる:
式II
【0016】
【化5】
[式中、YおよびZ=独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)であり、
R1〜R3=独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニル、ハロアルキニルであり、
R4〜R5=独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルケニル、アルキニル、ハロアルケニル、ハロアルキニルであり、但し、ハロは非放射性のF、Cl、BrおよびIであり、
R7=ハロゲン、ハロアルキル、ハロアルケニル、ハロアルキニル、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリール、ハロヘテロアリールであり、但し、ハロ=F−18、I−123、I−124、Tc−99mおよびReキレートなどの標識化合物を含むF、Cl、Br、I、Atである]
本明細書で開示する本発明の方法を用いて作製できる具体的な放射性標識アミノ酸類似体には、これらに限定されないが、上記構造を有するフルオロ−、ブロモ−もしくはヨード−置換のシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシルアミノ酸、またはヘテロ原子、すなわちN、OおよびSならびにSeを含む脂環式化合物が含まれる。
【0017】
本発明により作製されるアミノ酸化合物は、インビボで対象に投与した場合、腫瘍組織に対して高い特異性を有する。したがって、本発明は、本発明の方法により作製されるシン−アミノ酸類似体を含む医薬組成物および診断用組成物も提供する。好ましいアミノ酸化合物は少なくとも2:1の標的と非標的の比を示し、インビボで安定であり、投与後1時間以内で標的にほぼ局在化する。好ましいアミノ酸化合物の例には、シン−[18F]−1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)、シン−[123I]−1−アミノ−3−ヨードシクロブタン−1−カルボン酸(IACBC)およびシン−[18F]−1−アミノ−3−フルオロアルキル−シクロブタン−1−カルボン酸、例えばシン−[18F]−1−アミノ−3−フルオロメチル−シクロブタン−1−カルボン酸(FMACBC)が含まれる。
【0018】
本発明のアミノ酸類似体は対象の腫瘍を検出および/または監視するための画像化剤として有用である。アミノ酸類似の画像化剤はインビボで投与され、その標識に適した手段を用いて監視される。アミノ酸類似の画像化剤をインビボで検出および/または監視するのに好ましい方法には、陽電子断層撮影法(PET)および単光子射出コンピュータ断層撮影法(SPECT)が含まれる。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
本発明は、とりわけ腫瘍画像化に有用なシン−アミノ酸類似体を合成するための新規な方法に関する。本発明者等はここに、シン−ACBC類似体を合成するための、トランス異性体の主要前駆体の立体選択的な合成を可能にする合成ストラテジーを開発した。本発明の合成ストラテジーにより作製されるACBC類似体は実質的に純粋なシン−異性体である。本明細書で用いる「実質的に純粋な」という用語は、生成物が、少なくとも60%のその異性体純度、好ましくは70%の純度、好ましくは90%超のシン−異性体純度であることを意味する。すべての中間体は60%〜100%の値であり、それらが個別にリストに挙げられているかいないかにかかわらず、その範囲にある中間体はすべて包含されるものとする。
【0020】
本明細書で用いる用語および語句は概ね、当業界で周知の標準的な教科書、雑誌文献および文脈を参照すると見出せる当業界で認識されている意味を有する。以下の定義は、本発明に関連してその具体的な使用を明らかにするために提供されるものである。
【0021】
本明細書で用いる「薬学的に受容可能な塩」という用語は、過度の毒性、炎症、アレルギー反応等がなく患者の組織と接触させて用いるのに適しており、妥当な利益/リスク比に相応するものであり、かつ目的の用途に有効な本発明の化合物のカルボン酸塩または酸付加塩、ならびに、可能な場合、本発明の化合物の双性イオンの形態を指す。「薬学的に受容可能な塩」という用語は、本発明の化合物の比較的非毒性の無機および有機酸付加塩を指す。脂肪族モノおよびジカルボン酸、例えば酢酸、フェニル置換アルカン酸、ヒドロキシアルカン酸およびアルカンジオン酸、芳香族酸ならびに脂肪族および芳香族スルホン酸などの非毒性の有機酸から誘導される塩も含まれる。これらの塩は、化合物の最終的な単離や精製の際にその場で調製するか、あるいは、精製されたそのフリーベースの化合物を適切な有機酸または無機酸と別個に反応させ、その生成した塩を単離して調製することができる。他の代表的な塩には、臭化水素酸塩、塩酸塩、硫酸塩、重硫酸塩、硝酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチレート、メシラート、グルコヘプトネート、ラクチオビオネート、ラウリルスルホン酸塩、プロピオン酸塩、ピバル酸塩、サイクラミン酸塩、イセチオン酸塩等が含まれる。これらは、ナトリウム、リチウム、カリウム、カルシウム、マグネシウム等のアルカリ金属およびアルカリ土類金属をもとにしたカチオン、ならびに、これらに限定されないが、アンモニウム、テトラメチルアンモニウム、テトラエチルアンモニウム、メチルアミン、ジメチルアミン、トリメチルアミン、トリエチルアミン、エチルアミン等を含む非毒性アンモニウム、四級アンモニウムおよびアミンカチオンを含むことができる。例えば、Berge S. M,ら、Pharmaceutical Salts, J. Pharm. Sci. 66:1〜19頁(1977年)を参照されたい。これを参照により本明細書に組み込む。
【0022】
同様に、本明細書で用いる「薬学的に受容可能な担体」という用語は、医薬組成物または診断用組成物における本発明の活性成分の担体/安定剤/希釈剤として作用する有機または無機組成物である。いくつかの場合、薬学的に受容可能な担体は塩である。薬学的に受容可能な担体の他の例には、これらに限定されないが、水、リン酸緩衝生理食塩水、生理食塩水、pH調製剤(例えば酸、塩基、緩衝剤)、アスコルビン酸などの安定剤、等張剤(例えば、塩化ナトリウム)、水性溶媒、ポリソルベートまたはTWEEN80などの洗剤(イオン性および非イオン性)が含まれる。
【0023】
それ自体または他の基の一部として、本明細書で用いる「アルキル」という用語は、メチル、エチル、プロピル、イソプロピル、ブチル、t−ブチルおよびイソブチルなどの最大で10個の炭素、好ましくは6個の炭素、より好ましくは4個の炭素の直鎖、分鎖もしくは環状の飽和炭化水素を指す。本発明のアルキル基は、主鎖中の1個もしくは複数の炭素原子がヘテロ原子で置換されていることができ、1個もしくは複数の水素原子がハロゲンまたは−OHで置換されていることができる任意選択で置換されたものも含む。それ自体または他の基の一部として、本明細書で用いる「アリール」という用語は、フェニル、ナフチルまたはテトラヒドロナフチルなどの環部分に5〜12個の炭素、環部分に好ましくは6〜10個の炭素を単環式または二環式の芳香族基を指す。アリール基の1つまたは複数の環は縮合環を含むことができる。アリール基は直鎖、分鎖または環状であってよい1個または複数のアルキル基で置換されていてよい。アリール基は、環位置で、それが存在する化合物または化合物の部分の機能にそれほど決定的に影響を及ぼさない置換基で置換されていてもよい。置換アリール基は、環の中の1個または複数の炭素が、1個または複数のヘテロ原子(例えばN、OまたはSであり、任意選択で、適切な原子価のための水素または置換基を有する)で置き換わっている複素芳香族環を有するものも含む。
【0024】
本明細書で用いる「アルコキシ」という用語は、鎖長がそれに限定されていない限り、酸素原子と結合している上記定義の通りの直鎖または分鎖アルキル基、これらに限定されないが、例えばメトキシ、エトキシ、n−プロポキシ、イソプロポキシなどを意味する。アルコキシ鎖は1〜6個の炭素原子の長さ、より好ましくは1〜4個の炭素原子の長さであることが好ましい。
【0025】
「アシル」基は−CO−基を含む基である。
【0026】
それ自体または他の基の一部として、本明細書で用いる「モノアルキルアミン」という用語は、上記定義の1個のアルキル基で置換されているアミノ基を指す。
【0027】
それ自体または他の基の一部として、本明細書で用いる「ジアルキルアミン」という用語は、上記定義の2個のアルキル基で置換されているアミノ基を指す。
【0028】
それ自体または他の基の一部として、本明細書で用いる「ハロ」という用語は塩素、臭素、フッ素またはヨウ素を指す。
【0029】
本明細書で用いる「複素環」または「複素環式環」という用語は、言及のない限り、炭素原子とN、OおよびSからなる群から選択される1〜3個のヘテロ原子からなる、飽和または不飽和の安定した5〜7員のモノ複素環式環系であって、その窒素およびイオウヘテロ原子が任意選択で酸化されていてよいモノ複素環式環系を表わす。特に有用なものは、1個の酸素もしくはイオウと結合した1個の窒素ヘテロ原子、または2個の窒素ヘテロ原子を含む環である。そうした複素環式基の例には、ピペリジニル、ピロリル、ピロリジニル、イミダゾリル、イミダジニル、イミダゾリジニル、ピリジル、ピラジニル、ピリミジニル、オキサゾリル、オキサゾジニル、イソキサゾリル、イソオキサゾジニル、チアゾリル、チアゾリジニル、イソチアゾリル、ホモピペリジニル、ホモピペラジニル、ピリダジニル、ピラゾイルおよびピラゾリジニルが含まれる。チアモルホリニル、ピペラジニル、およびモルホリニルが最も好ましい。
【0030】
本明細書で用いる「ヘテロ原子」という用語は、酸素原子(「O」)、イオウ原子(「S」)または窒素原子(「N」)を意味する。ヘテロ原子が窒素である場合、RaおよびRbが互いに独立に水素もしくはC1〜4アルキル、C2〜4アミノアルキル、C1〜4ハロアルキル、ハロベンジルであるか、またはRaおよびRbが一緒になってその環内にO、SもしくはNRc(Rcは水素またはC1〜4アルキルである)を任意選択で有する5〜7員の複素環式環を形成するNRaRb部分を形成することを理解されよう。
【0031】
本発明の化合物は腫瘍結合剤およびNMDA受容体結合リガンドとして有用であり、放射性アイソトープはPETおよびSPECTによる画像化を含む腫瘍画像化技術のためのトレーサー化合物として特に有用である。画像化剤として特に有用なものはF−18で標識した化合物である。その理由は、F−18が110分の半減期を有しており、それによって、放射能標識トレーサー中に取り込み、精製し、かつヒトまたは動物対象に投与するために十分な時間が与えられるからである。さらに、サイクロトロンから最大で半径約200マイル以上離れた設備でF−18標識した化合物を用いることができる。
【0032】
SPECT画像化には高エネルギー光子を放出するアイソトープトレーサー(γ−エミッター)を用いる。有用なアイソトープの範囲はPET用より大きいが、SPECTはより低い三次元分解能を提供する。それでも、SPECTは、類似体結合、局在化およびクリアランスの速度についての臨床的に重要な情報を得るために広く用いられる。SPECT画像化に有用なアイソトープは13.3時間の半減期を有する[123I]、γ−エミッターである。[123I]で標識した化合物は、製造場所から最大で約1000マイルまで送出するか、またはアイソトープ自体を現地での合成のために輸送することができる。アイソトープの放出の85%は159KeV光子である。これは現在用いられているSPECT装置で容易に測定される。
【0033】
したがって、PET画像化の代替法としてSPECT分析で用いるために、本発明の化合物は、[123I]で迅速かつ効率的に標識することができる。さらに、同一化合物を、どのアイソトープを用いても標識することができるので、同じトレーサーを用いたPETとSPECTにより得られた結果を比較することができる。
【0034】
他のハロゲンアイソトープを、PETもしくはSPECTの画像化に、または従来のトレーサー標識に役立てることができる。これらには、使用に適した半減期と放射特性を有する75Br、76Br、77Brおよび82Brが含まれる。一般に、上記したアイソトープのためにいずれかのハロゲン部分を置換するのには化学的手段がある。したがって、当業者は、安定したアイソトープハロゲン同族体を含む、本発明の化合物の任意のハロゲン化同族体の生化学的または生理学的活性を利用することができる。アスタチンは他のハロゲンアイソトープを代替することができ、[210At]は8.3時間の半減期を有するα粒子を放出する。したがって、At置換化合物は、結合が十分に腫瘍特異的である腫瘍の治療に有用である。
【0035】
本発明は、PETおよびSPECTを用いて腫瘍を画像化するための方法を提供する。その方法は、適切にアイソトープで標識された画像発生量の本発明の化合物を対象(実験および/または診断の目的のためのヒトまたは動物であってよい)に投与する工程と、次いで、[18F]または他の陽電子エミッターを用いた場合にはPETによって、あるいは[123I]または他のγエミッターを用いた場合にはSPECTによって化合物の分布を測定する工程とを含む。画像発生量は、スキャナーの検出感度やノイズレベル、アイソトープの寿命、対象の身体の大きさおよび投与経路を考慮して、PETまたはSPECTのスキャナーに画像を少なくとも提供することができる量である。上記のすべての変数は周知の例であり、必要以上の実験を用いることなく、当業者に知られている計算および測定によって明らかにされる。
【0036】
本発明の化合物は、構造中の任意の原子または原子の組合せのアイソトープで標識できることを理解されよう。本明細書では[18F]、[123I]および[125I]がPET、SPECTおよびトレーサー分析に特に有用であると強調してきたが、安定したアイソトープ同族体の生理学的または薬理学的特性によりもたらされるものを含む他の使用が考えられ、それらは当業者に明らかであろう。
【0037】
本発明の化合物は、Tc付加体を介してテクネチウム(Tc)標識することもできる。Tc、特にTc99mのアイソトープは腫瘍の画像化に用いられてきた。本発明は、腫瘍画像化に有用である本発明の化合物のTc−錯化付加体を提供する。その付加体は、飽和しているかまたは二重結合もしくは三重結合を有する4〜6個の炭素鎖によって環状アミノ酸と結合しているTc配位錯体である。二重結合が存在する場合、E(トランス)かまたはZ(シス)異性体のどちらかを合成することができ、どちらの異性体も用いることができる。Tcで標識した本発明の化合物は、アイソトープの有用期間を最大にするために、最後の段階で99mTcアイソトープを取り込んで合成される。
【0038】
米国特許第5,817,776号は、(アンチ−[18F]−1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC))を合成するための10工程の一連の反応を開示している。その一連の反応は、工程4に続いて、主要中間体であるシス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸とトランス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸のそれぞれ75:25の混合を、大きな労働力を要するセミ分取高圧液体クロマトグラフィーで分離する工程を含む。次いで、精製された主成分異性体のシス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸を、6工程の一連反応でトリフレート前駆体に転換させる。
【0039】
本発明者等は合成方法の改良を目指して、放射性標識するための前駆体であるシス1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステル(8)とトランス1−アミノ−3−フルオロシクロブタン−1−カルボン酸(アンチ−FACBC)(10)の両方を大規模合成するトランス−(アンチ−)1−アミノ−3−[18F]フルオロシクロブタン−1−カルボン酸(アンチ−[18F]FACBC)の立体選択的な合成法を開発した。スキーム1および2はアンチ−FACBCの合成工程を示す。この合成工程を用いて、7工程の一連反応によりトリフレート前駆体(8)を調製することができる。この合成における重要な工程はシントン3−ベンジルオキシ−シクロブタノン(2)の調製である。シクロブタノン3の調製は、1−ブロモ−2−ベンジルオキシ−3−ブロモプロパン(1)をメチルエチル−スルホキシドとn−ブチルリチウムで処理することによる環化反応を含む。ケトン2を、Bucherer Strecker条件のもとで直接ヒダントイン3および4に転換させた。シス:トランスヒダントインの80:20混合物をフラッシュクロマトグラフィーにより容易に精製して、所望のシスヒダントイン4を得た。トリフレート前駆体、シス1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステル(8)への4の転換を、米国特許第5,817,776号に記載の一連の反応により実施した。この方法を用いて、グラム単位の量の化合物9を調製することができた[McConathyら(2003年)Jour. of Applied Radiation and Isotopes, 58:657〜666頁]。
【0040】
【化6】
トランス−1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステルを大規模に生産するために、スキーム3〜5示すように腫瘍画像化用の、シン−異性体のアミノ酸類似体、具体的にはシス−(シン−)1−アミノ−3−フルオロシクロブタン−1−カルボン酸(シン−FACBC)を十分な量で得るための、新規な包括的合成アプローチを開発した。その合成における主要工程は、シントンである1−トリフルオロアセトアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11a)、1−フタルアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11b)、1−t−ブチルカルバメート−シクロブタン−3−オン−1−カルボン酸メチルエステル(11c)および1−ベンズアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11d)の還元を含むものである。ケトンである11a〜dは、リチウムトリイソブチルボランとZnCl2で処理して、63〜80%の収率で直接トランス−(アンチ−)アルコールに転換された。この方法により、それぞれ95:5、97:3、70:30および90:10のトランス:シスアルコールの混合物の12a、12b、12cおよび12dが得られた。12a〜12dをフラッシュクロマトグラフィーにより容易に精製して、所望のトランスアルコール12a〜dを得た。12a〜dのトリフレート前駆体への転換は、米国特許第5,817,776号に記載されている一連の反応で実施することができる。これらの合成アプローチの開発は、シン−およびアンチ−FACBCが、癌の診断および癌治療の管理に有用な画像化剤として有効であることを確認する将来の多施設臨床試験のために、前駆体をPETセンターへ分配するために容易に利用できる供給法を確立するのに必須である。
【0041】
【化7】
上記反応は以下の仕方で実施した。THF(無水)中ケトン(11a、b、c、またはd)の溶液に、アルゴン雰囲気下、室温(rt)で2当量のZnCl2(無水物、THF中に)を加えた。溶液を室温で30分間攪拌し、続いて−78℃で1.5当量のLiBR’3Hを加えた。混合物を−78℃で2時間攪拌し、次いで室温で終夜攪拌した。NH4Cl(1N水溶液、3当量)を加え、混合物を室温で30分間攪拌した。反応物をブラインで洗浄し、水相を酢酸エチルで再抽出した。一緒にした有機相を硫酸ナトリウムで脱水し、濃縮して乾燥させた。生成物を、溶離液として1:1のヘキサンおよび酢酸エチルを用いてシリカゲルで精製した。収率は約63〜80%であった。
【0042】
スキーム3に示す反応工程は、4つのシントン(11a〜11d)を4つのトランス−アルコール、12a〜12dに還元する例を具体的に示すものであるが、この立体選択的な合成工程は、腫瘍画像化に有用なシン−アミノ酸類似体の合成のための様々なトランス−アルコールの合成に適用することができる。以下のスキーム4は本発明のこの態様を示す。
【0043】
【化8】
スキーム5はシン−FACBCの合成のための工程を例示する。
【0044】
【化9】
スキーム6では、本明細書で開示する立体選択的な合成方法を用いて合成できるアミノ酸類似体、[18F]−1−アミノ−4−フルオロ−シクロヘキサン−1−カルボン酸(FACHC)の合成を例示する。
【0045】
【化10】
スキーム7は、本明細書で開示する立体選択的な合成方法で用いる主要シントンであるシン/アンチ−1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸20の合成を示す。
【0046】
【化11】
スキーム8は、本明細書で開示する立体選択的な合成方法で用いる主要シクロヘキサノン中間体である1−[N−(t−ブトキシカルボニル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(24)、1−アミノ−4−シクロヘキサノン−1−カルボン酸メチルエステル(25)の合成を示す。
【0047】
【化12】
スキーム9は、本明細書で開示する立体選択的な合成方法で調製されたシン/アンチ−1−[N−置換−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル27a〜dの合成を示す。
【0048】
【化13】
【実施例】
【0049】
以下の説明によって、本発明の好ましい実施形態の例としての合成法を提供する。しかし、当業者は本発明の実施にあたって、出発原料、試薬、溶媒、温度、固体基体、合成方法、精製方法、分析方法、および具体的に例示したもの以外の他の反応条件を、必要以上の実験をすることなく用いることができることを理解されよう。そうした任意の原料や方法の当業界で周知の機能的均等物のすべては、本発明に含まれるものとする。使用される用語および表現は説明のための用語として用いるものであって限定的なものではなく、そうした用語および表現の使用において、示しかつ説明した特徴またはその一部のどんな均等物も排除しようとするものではなく、特許請求した本発明の範囲内で様々な変更が可能であることを理解されたい。したがって、好ましい実施形態および任意選択の特徴によって本発明を具体的に開示してきたが、当業者は、本明細書で開示した考え方の改変および変更を行うことができ、そうした改変形態および変更形態は、添付の特許請求の範囲により定義される本発明の範囲内であることを理解すべきである。
【0050】
(実施例1)
シン−およびアンチ−[18F]1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)の合成(スキーム1、2および5)
本明細書で報告される手順において以下の方法を用いた。[18F]−フルオリドは、11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、Seimens社製サイクロトロンで作製した。すべての溶媒および化学薬品は、分析グレード品をさらに精製することなく使用した。化合物の融点はBuchiSP装置を用いて毛細管で測定した。薄層クロマトグラフィー分析(TLC)は、アルミニウム上にコーティングされたシリカゲルGPF−254(Analtech,Inc.Newark,DEから入手)の250mm厚の層を用いて実施した。カラムクロマトグラフィーは、60〜200メッシュシリカゲル(Sigma−Aldrich,St.Louis,MO)を用いて実施した。赤外スペクトル(IR)はNaClプレートを備えたBeckman18A分光光度計で測定した。プロトン核磁気共鳴スペクトル(1H NMR)はNicolet高分解能装置を用いて300MHzで得た。
【0051】
1−ブロモ−2−ベンジルオキシ−3−ブロモプロパン1の合成:
凝縮器を備えたフラスコ中で、臭化ベンジル(83mL、0.70モル)、エピブロモヒドリン(60mL、0.70モル)および塩化水銀(I)(120mg、0.25ミリモル)からなる混合物を150℃で終夜攪拌した。生成物を、30cmのVigreux凝縮器を通した真空蒸留(110〜115℃、0.5mmHg)により単離して、生成物1(152g、70%)を無色液体として得た。1H NMR (CDCl3) δ3.45 (4H, d, J=5.2), 3.66−3.71 (1H, m) 4.55 (2H, s) 7.19−7.27 (5H, m)。
【0052】
3−ベンジルオキシシクロブタノン2の合成:
シクロブタノン2の調製は、Oguraら(1984年)Bull. Chem. Soc. Jpn. 57;1637〜42頁の報告による手順をもとにした。2.4当量部のn−ブチルリチウム(ヘキサン中に1.6M、243mL)を、400mLのテトラヒドロフラン中に2.4当量のメチルメチルスルフィニルメチルスルフィド(41mL、0.39ミリモル)を含む溶液に−10℃で滴下した。次いで反応混合物を−10℃で2時間攪拌し、続いて−70℃に冷却した。黄色の反応混合物を−70℃に保持し、85mLのテトラヒドロフラン中の1当量のジブロモ種1(50g、0.16ミリモル)を滴下した。反応混合物を終夜かけて室温に加温した。反応混合物をブラインに加え、酢酸エチルで2回抽出した。一緒にした有機層に通常の処理を施して約60mLの暗赤褐色液体を得た。このシン−およびアンチ−ジチオケタールS−オキシド中間体の混合物を、シリカゲルカラムクロマトグラフィー(90gシリカ)により3つに分けて精製した。より極性の小さい不純物をまず3:7酢酸エチル:ヘキサンで溶出させ、続いて、生成物を高純度の酢酸エチルで溶出させた。この方法で合計23.8gの中間体を得た。同じ条件を用いて次に2を合成して24.6gを得た。
【0053】
シン−およびアンチ−ジチオケタールS−オキシド中間体(48.4g、0.18モル)を1200mLのジエチルエーテルに溶解し、68mLの35%過塩素酸で処理した。終夜攪拌した後、反応混合物を重炭酸ナトリウムで中和し、続いて通常の後処理を行った。シリカゲルカラムクロマトグラフィー(15:85酢酸エチル:ヘキサン)で精製してケトン2(23.6g、1からは41%)を橙黄色液体として得た。1H NMR δ3.11−3.29 (4H, m) 4.35−4.42 (1H, m) 4.53 (2H, s) 7.30−7.40 (5H, m)。
【0054】
シス/トランス5−(3−ベンジルオキシシクロブタン)ヒダントイン3の合成:
900mLの水の中の10当量の炭酸アンモニウム(125g、1.3モル)および4当量の塩化アンモニウム(27.8g、0.52モル)の溶液に、900mLのエタノール中の1当量のシクロブタノン2(23.6g、0.13モル)を加えた。室温で30分間攪拌した後、4.5当量部のシアン化カリウム(38g、0.58モル)を加え、反応混合物を60℃で終夜加熱した。溶媒を減圧下で除去し、粗製の黄色固体を約1リットルの水で完全に濯いで、塩を除去した。白色の結晶生成物(16.4g、51%)をシン:アンチ異性体の5:1混合物として得た。単離された主成分異性体を、シリカゲルカラムクロマトグラフィー(2:98メタノール:ジクロロメタン)により得た。この手順を用いて、95gのシリカゲルで1.0gの混合物を精製して500〜600mgの純粋な3を1回の試行で得た。シン−5−(3−ベンジルオキシシクロブタン)ヒダントイン(3):1H NMR (CDCl3) δ2.30−2.35 (2H, m) 2.87−2.92 (2H, m) 4.18−4.25 (1H, m) 4.46 (2H, s) 5.66 (1 H, 広幅s) 7.28−7.38 (5H, m) 7.55 (1H, 広幅s)。 アンチ−5−(3−ベンジルオキシシクロブタン)ヒダントイン(4):1H NMR (CDCl3) δ2.44−2.50 (2H, m) 2.77−2.83 (2H, m) 4.21−4.27 (1H, m) 4.46 (2H, s) 5.82 (1H, 広幅s) 7.29−7.38 (6H, m)。
【0055】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ベンジルオキシシクロブタン−1−カルボン酸5の合成:
密封したステンレス製容器の中で、30mLの3N水酸化ナトリウム中の化合物3(1.35g、5.5ミリモル)の懸濁液を180℃で終夜加熱した。冷却した後、反応混合物を濃塩酸でpH6〜7まで中和した。減圧下で水を除去した後、得られた固体を4×30mLの熱エタノールで抽出した。一緒にしたエタノール抽出物を濃縮し、残留物を50mLの9:1メタノール:トリエチルアミンに溶解した。その溶液に、1.3当量部のジ−tert−ブチルジカーボネート(1.56g)を加え、溶液を室温で終夜攪拌した。溶媒を減圧下で除去し、粗生成物を、80mLの氷冷酢酸エチルと80mLの氷冷0.2N塩酸の混合液中で5分間攪拌した。有機層は保持し、水相を2×80mLの氷冷酢酸エチルで抽出した。一緒にした有機層を3×60mLの水で洗浄し、通常の後処理を行った。N−Boc酸5(1.27g、72%)を、さらに精製することなく次の工程で用いるのに適した白色の固体として得た。1H NMR (CDCl3) δ1.44 (9H, s) 2.21−2.26 (2H, m) 3.02−3.08 (2H, 広幅m) 4.12−4.19 (1H, m) 4.44 (2H, s) 5.18 (1H, 広幅s) 7.27−7.37 (5H, m)。
【0056】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ベンジルオキシシクロブタン−1−カルボン酸メチルエステル6の合成:
ヘキサン(1.4mL)中の1.5当量部の2.0Mトリメチルシリルジアゾメタンを、10mLの1:1メタノール:テトラヒドロフラン中のN−Boc酸5(600mg、1.87ミリモル)の溶液に滴下した。添加による発熱の際、相当なガスが発生した。20分間攪拌した後、反応混合物を減圧下で濃縮し、粗生成物をシリカゲルカラムクロマトグラフィー(2:8酢酸エチル:ヘキサン)で精製した。N−Bocメチルエステル6(0.45g、72%)を白色の結晶性固体として得た。1H NMR (CDCl3) δ1.42 (9H, s) 2.24−2.36 (2H, 広幅m) 2.88− 2.96 (2H, m) 3.75 (3H, s) 4.16−4.23 (1H, m) 4.44 (2H, s) 5.13 (1H, s) 7.27−7.36 (5H, m)。
【0057】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ヒドロキシシクロブタン−1−カルボン酸メチルエステル7の合成:
10mLのCH3OH中の6(450mg、1.34ミリモル)の溶液に、アルゴン雰囲気下で200mgの10%Pd/Cを加えた。反応混合物を水素雰囲気下、室温で終夜攪拌した。次いで懸濁液をセライト(Celite)(登録商標)でろ過し、減圧下で濃縮した。シリカゲルカラムクロマトグラフィー(6:4酢酸エチル:ヘキサン)で精製してアルコール7(200mg、61%)を白色の結晶性固体:134〜135℃として得た(128〜130℃、Shoup and Goodman, J Labelled Compd Radiopharm、1999年;42:215〜225頁による報告。1H NMR (CDCl3) δ1.45 (9H, s) 2.54− 2.61 (2H, 広幅m) 2.98−3.04 (2H, m) 3.79 (3H, s) 4.26−4.34 (1H, 広幅m) 5.63 (1H, 広幅s)。 (C11H19NO5) の元素分析: 計算値: C: 53.87 H: 7.81 N: 5.71, 実測値: C: 53.93 H: 8.00 N: 5.71。
【0058】
化合物1−[N−(tert−ブトキシカルボニル)アミノ]−シクロブタン−3−オン−1−カルボン酸メチルエステル11cの合成。
【0059】
4mLのジクロロメタン中の1.1当量部の塩化オキサリル(1.05mL、ジクロロメタン中に2M溶液)に、アルゴン雰囲気下、−50〜−60℃で、1mLのジクロロメタン中の2.2当量のジメチルスルホキシド(290μL)を滴下しながら加えた。この溶液を3分間攪拌し、続いて、2mLジクロロメタンおよび0.8mLのジメチルスルホキシド中に溶解した異性体的に不純な7(458mg、1.9ミリモル)を滴下した。反応混合物を−50〜−60℃で20分間攪拌し、次いで5当量のトリエチルアミン(1.3mL)を加えた。反応混合物を5分間攪拌し、冷却浴を取り外し、溶液をさらに15分間攪拌した。粗生成物をシリカゲルカラムクロマトグラフィー(1:4酢酸エチル:ヘキサン)で精製して11c(456mg、100%の収率)を白色固体:118〜119℃(酢酸エチル/ヘキサン)として得た。1H NMR (CDCl3) δ1.46 (9H, s) 3.49−3.66 (4H, m) 3.83 (3H, s) 5.47 (1H, 広幅s)。 (C11H17NO5) の元素分析: 計算値: C: 54.31 H: 7.04 N: 5.76, 実測値: C: 54.50 H: 6.96 N: 5.61。
【0060】
アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ヒドロキシシクロブタン−1−カルボン酸メチルエステル12cの合成。
【0061】
1mlのTHF(無水)中のケトン(11c、16.4mg、0.067ミリモル)の溶液に、Ar雰囲気下、室温でZnCl2(18mg、THF中に0.134ミリモル)を加えた。溶液を室温で30分間攪拌し、続いてL−セレクトリド(L−Selectride)(19mg、THF中に0.10ミリモルF)を−78℃で加えた。混合物を−78℃で2時間攪拌し、次いで室温で終夜攪拌した。NH4Cl(1N水溶液、3当量)を加え、混合物を室温で30分間攪拌した。反応物をブラインで洗浄し、水相を酢酸エチルで再抽出した。一緒にした有機相を硫酸ナトリウムで脱水し、濃縮して乾燥させた。溶離液として1:1のヘキサンおよび酢酸エチルを用いて、生成物をシリカゲルで精製した。生成物(12c、16mg、100%)を白色固体として得た。1H NMR (CDCl3) δ1.44 (9H, s) 2.53− 2.63 (4H, 広幅m) 3.77 (3H, s) 4.43−4.50 (1H, 広幅m) 5.02 (1H, 広幅s)。
【0062】
アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−トリフルオロメチルスルホンオキシシクロ−ブタン−1−カルボン酸メチルエステル13の合成。
【0063】
アルゴン雰囲気下で、アルコール9(10mg、0.04ミリモル)を2mLのジクロロメタンに溶解した。氷浴で冷却しながら100μL部のピリジンを加え、続いて4.5当量部のトリフルオロメタンスルホン酸無水物(30μL)を加えた。15分間攪拌した後、溶媒を減圧下室温で除去した。粗生成物をシリカゲルカラムクロマトグラフィー(3:7酢酸エチル:ヘキサン)で精製して標識前駆体13を得た。
【0064】
シン−[18F]1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)15の合成:
11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、[18F]−フルオリドを生成させた。水を蒸発させ、アセトニトリルを蒸発させてフルオリドを乾燥させた後、保護されたアミノ酸トリフレート13(20mg)をアセトニトリル溶液(1mL)に導入した。密封した容器中で、非担体担持での(NCA)フッ素化反応を、炭酸カリウムとKryptofix(Trademark Aldrich Chemical Co.,Milwaukee,WI)の存在下、85℃で5分間かけて実施した。反応混合物を塩化メチレンで希釈して未反応18Fを除去し、続いてシリカゲルSeppakを通過させて18F標識生成物14を得た。1mLの6N HClで115℃で15分間処理して14を脱保護させ、次いでシン−[18F]FACBC15を含む水溶液を、イオン遅滞樹脂(AG11A8 50〜100メッシュ)に通した。
【0065】
アンチ−[18F]FACBC10の合成:
11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、[18F]−フルオリドを生成させた。水を蒸発させ、アセトニトリルを蒸発させてフルオリドを乾燥させた後、保護されたアミノ酸トリフレートシン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−トリフルオロメタンスルホンオキシシクロブタン−1−カルボン酸メチルエステル(20mg)をアセトニトリル溶液(1mL)に導入した。密封した容器中で、非担体担持での(NCA)フッ素化反応を、炭酸カリウムとKryptofix(Trademark Aldrich Chemical Co.,Milwaukee,WI)の存在下、85℃で5分間かけて実施した。反応混合物を塩化メチレンで希釈して未反応18Fを除去し、続いてシリカゲルSeppakを通過させて、18F標識生成物14を得た。18F標識生成物、シン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−[18F]フルオロシクロブタン−1−カルボン酸メチルエステルを42%E.O.B.の収率で得た。1mLの4N HClで115℃で15分間処理してシン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−[18F]フルオロシクロブタン−1−カルボン酸メチルエステルを脱保護させ、次いで18FACBC13を含む水溶液を、イオン遅滞樹脂(AG11A8 50〜100メッシュ)に通した。合成はE.O.B.の後60分間で、12%(17.5%E.O.B.)の総括放射化学的収率で完了した。詳細については上記のMcConathyら(2003年)を参照されたい。
【0066】
(実施例2)
シン−およびアンチ−1−アミノ−4−ヒドロキシシクロヘキサン−1−カルボン酸エステル(スキーム7−9)の合成
4−エチレンアセタールシクロヘキサノール(16)
0℃の冷却した50mlメタノール中の1,4−シクロヘキサンジオンモノエチレンアセタール(3.41g、21.8ミリモル)の溶液に、水素化ほう素ナトリウム(0.826g、21.8ミリモル)を分割して加えた。反応物をさらに1.5時間攪拌し、続いて1N HClを加えてpH7にした。混合物を酢酸エチルとブラインに分配させた。沈殿物が形成され始めるまで水層を濃縮し、この水層を酢酸エチルで2回抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。この粗アルコール(3.28g、95.2%)をさらに精製することなく用いた。1H NMR (CDCl3) δ: 1.54−1.87 (8H, m, 4×−CH2−), 3.77 (1H, m, −CH−), 3.91 (4H, t, 2×O−CH2−)。
【0067】
1−エチレンアセタール−4−ベンジルオキシ−シクロヘキサン(17)
0℃の15mlのTHF中の水素化ナトリウム(410mg、17.1ミリモル)の懸濁液に、5mlのTHF中の4−エチレンアセタールシクロヘキサノール(1)(1.36g、8.61ミリモル)を加えた。反応物を0℃で1.5時間攪拌し、臭化ベンジル(1.75g、10.2ミリモル)を加えた。反応物を室温で終夜攪拌した。反応物を塩化アンモニウム(飽和)でクエンチした。生成物を酢酸エチルで抽出し、有機相をブラインで洗浄し、硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物をシリカゲルクロマトグラフィー(ヘキサン中の20%酢酸エチル)で精製して2.17g(100%)のベンジルエーテルを得た。1H NMR (CDCl3) δ: 1.51−1.88 (8H, m, 4×−CH2−), 3.51 (1H, m, −CH−), 3.91 (4H, t, 2×O−CH2−), 4.52 (2H, s, Ph−CH2−), 7.25−7.34 (5H, m, Ph−H)。
【0068】
4−ベンジルオキシ−シクロヘキサノン(18)
50mlのTHF中の1−エチレンアセタール−4−ベンジルオキシ−シクロヘキサン(17)(3.13g、12.6ミリモル)の溶液に、塩酸水溶液(1N、30ml)を室温で加えた。反応物を終夜攪拌し、重炭酸ナトリウム(飽和)で中和した。生成物を酢酸エチルで抽出し、有機相をブラインで洗浄し、硫酸ナトリウムで脱水し、ろ過し、濃縮させた。シリカゲルクロマトグラフィー(ヘキサン中の20%酢酸エチル)で精製して2.45g(95.2%)の標記のケトンを得た。1H NMR (CDCl3) δ: 1.95−2.62 (8H, m, 4×−CH2−), 3.82 (1H, m, −CH−), 4.59 (2H, s, Ph−CH2−), 7.28−7.36 (5H, m, Ph−H)。
【0069】
シン/アンチ−6−(4−ベンジルオキシシクロヘキサン)ヒダントイン(19)
100mlのエタノール中の4−ベンジルオキシ−シクロヘキサノン(18)(2.45g、12ミリモル)の溶液に、100mlの水の中の炭酸アンモニウム(4.6g、48ミリモル)と塩化アンモニウム(1.28g、24ミリモル)の溶液を加えた。混合物を室温で15分間攪拌し、次いでシアン化カリウム(940mg、14.4ミリモル)を加えた。反応物を室温で終夜攪拌した。溶媒を減圧下で除去した。得られた固体を水で繰り返し洗浄して、ろ取した。このヒダントイン(hytantoin)のシン/アンチ粗混合物(3.02g、91.8%)を、さらに精製することなく使用した。1H NMR (CD3OD) δ: 1.58−2.15 (8H, m, 4×−CH2−), 3.48, 3.66 (1H, m, −CH−), 4.52, 4.56 (2H, s, Ph−CH2−), 7.25−7.33 (5H, m, Ph−H)。
【0070】
シン/アンチ−1−アミノ−4−ベンジルオキシシクロヘキサン−1−カルボン酸(20)
シン/アンチヒダントイン(19)(2.72g、9.93ミリモル)を30mlの3N NaOH中に懸濁させ、鋼製円筒容器中に密封し、これを120℃で1日加熱した。室温に冷却した後、濃塩酸溶液を加えて反応液をpH7にした。減圧下で濃縮し、乾燥させてシン/アンチアミノ酸の粗生成物を得た。この生成物を、さらに精製することなく使用した。
【0071】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸(21)
50mlの9:1MeOH/トリエチルアミン中の上記調製によるシン/アンチ−1−アミノ−4−ベンジルオキシシクロヘキサン−1−カルボン酸(20)の懸濁液にジ−t−ブチルジカーボネート(3.25g、14.9ミリモル)を加えた。反応混合物を室温で24時間攪拌した。溶媒を減圧下で除去した。得られた残留物を50mlの氷冷した1:1水/酢酸エチルに溶解した。3N HClで溶液のpHを2〜3に調節した。有機層は保持し、水層を塩化ナトリウムで飽和させて、酢酸エチル(3×25ml)で抽出した。一緒にした有機層を硫酸マグネシウムで脱水し、溶媒を減圧下で除去した。この生成物(3.46g、100%)を、さらに精製することなく使用した。1H NMR (CD3OD) δ: 1.41 [9H, s, −C(CH3)3 ], 1.57−2.25 (8H, m, 4×−CH2−), 3.40, 3.58 (1H, m, −CH−), 4.49, 4.54 (2H, s, Ph−CH2−), 4.84 (1H, br, NH), 7.25−7.33 (5H, m, Ph−H)。
【0072】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸メチルエステル(22)
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸(21)(1.14g、3.26ミリモル)を、40mlベンゼンおよび10mlメタノール中に溶解し、トリメチルシリルジアゾメタン(558mg、4.88ミリモル、2.5mlのヘキサン中、2M溶液)を室温で加えた。反応物を室温で30分間攪拌し、次いで溶媒を減圧下で除去した。ヘキサン中の20%酢酸エチルを用いてフラッシュクロマトグラフィーで精製して1.03g(87.2%)の純粋な生成物を油状物として得た。1H NMR (CD3OD) δ: 1.408, 1.413 [9H, s, −C(CH3)3 ], 1.5−2.3 (8H, m, 4×−CH2−), 3.40, 3.58 (1H, m, −CH−), 3.69, 3.71 (3H, s, COCH3), 4.49, 4.54 (2H, s, Ph−CH2−), 4.77, 4.79 (1H, br, NH), 7.25−7.33 (5H, m, Ph−H)。
【0073】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(23)
50mlのエタノール中のベンジルエーテル(22)(947mg、2.6ミリモル)と活性炭担持の10%パラジウム(142mg)の懸濁液を水素雰囲気下で終夜攪拌した。反応混合物をセライト(登録商標)でろ過し、ろ液を減圧下で濃縮した。シリカゲルカラムクロマトグラフィー(ヘキサン中に50%酢酸エチル)で精製して(23)(701mg、98.4%)を得た。アンチ−対シン−の比は34:66であった。1H NMR (CD3OD) δ: 1.411, 1.416 [9H, s, −C(CH3)3 ], 1.53−2.25 (8H, m, 4×−CH2−), 3.65, 3.91 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.77 (1H, br, NH)。
【0074】
1−[N−(t−ブトキシカルボニル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(24)
アルゴン雰囲気下で、テトラプロピルアンモニウムペルルテナート(26mg、0.075ミリモル)を一括して、ジクロロメタン中の15mlの10%アセトニトリル中のアルコール(23)(410mg、1.5ミリモル)、N−メチル−モルホリンN−オキシド(264mg、2.25ミリモル)および750mg4Aモレキュラーシーブの攪拌混合物に加えた。反応物を室温で1時間攪拌し、次いで溶媒を減圧下で除去した。得られた残留物をジクロロメタン中にとり、シリカゲルカラムクロマトグラフィー(ヘキサン中に30%酢酸エチル)で精製した。ケトン(24)、372mg(91.6%)を白色固体として得た。1H NMR (CD3OD) δ: 1.43 [9H, s, −C(CH3)3 ], 2.32−2.42 (8H, m, 4×−CH2−), 3.74 (3H, s, COCH3), 5.04 (1H, br, NH)。
【0075】
1−アミノ−4−シクロヘキサノン−1−カルボン酸メチルエステル(25)
5mlジクロロメタン中のケトン(24)(325mg、1.2ミリモル)の溶液に、トリフルオロ酢酸(1.37g、12ミリモル)を加えた。反応物を室温で終夜攪拌した。溶媒と試薬を減圧下で除去した。得られた白色の固体を、さらに精製することなく使用した。
【0076】
1−[N−(フタロイル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26b)
10mlトルエン中のアミン(25)(80mg、0.47ミリモル)およびトリエチルアミン(476mg、4.7ミリモル)の懸濁液に、無水フタル酸(77mg、0.52ミリモル)を加えた。混合物を120℃で5時間還流させた。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:4酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26b)(37.6mg、26.6%、2段階で)を白色固体として得た。1H NMR (CD3OD) δ: 2.54−3.14 (8H, m, 4×−CH2−), 3.77 (3H, s, COCH3), 7.73−7.85 (4H, m, Ph−H)。
【0077】
1−[N−(トリフルオロアセチル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26c)
−10℃に冷却した1mlジクロロメタン中のアミン(25)(14mg、0.082ミリモル)およびトリエチルアミン(166mg、1.64ミリモル)の懸濁液に、トリフルオロ酢酸無水物(86mg、0.41ミリモル)を加えた。混合物を室温に加温し、終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:2酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26c)(17.5mg、79.9%)を透明油状物として得た。1H NMR (CD3OD) δ: 2.44−2.56 (8H, m, 4×−CH2−), 3.79 (3H, s, COCH3), 6.86 (1H, br, NH)。
【0078】
1−[N−(ベンゾイル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26d)
0℃に冷却した3mlジクロロメタン中のアミン(25)(50mg、0.29ミリモル)およびピリジン(934mg、11.8ミリモル)の懸濁液に、塩化ベンゾイル(62mg、0.44ミリモル)を加えた。混合物を室温に加温し、終夜攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:2酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26d)(22mg、27.6%)を白色固体として得た。1H NMR (CD3OD) δ: 2.46−2.58 (8H, m, 4×−CH2−), 3.81 (3H, s, COCH3), 6.82 (1H, br, NH), 7.48−8.13 (5H, m, Ph−H)。
【0079】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27a)
1mlのTHF中のケトン(26a)(18mg、0.066ミリモル)の溶液に、室温で塩化亜鉛(18mg、0.13ミリモル、264μlのTHF中0.5M溶液)を加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(19mg、0.10ミリモル、100μlのTHF中の1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27a)(12.7mg、70.5%)を透明油状物として得た。アンチ−とシン−の比は67:33であった。1H NMR (CD3OD) δ: 1.411, 1.415 [9H, s, −C(CH3)3 ], 1.55−2.26 (8H, m, 4×−CH2−), 3.65, 3.92 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.70 (1H, br, NH)。
【0080】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27a)(塩化亜鉛は存在せず)
−78℃に冷却した1mlのTHF中のケトン(24)(21.7mg、0.08ミリモル)の溶液にL−セレクトリド(22.8mg、0.12ミリモル、120μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27a)(3mg、13.7%)を透明油状物として得た。アンチ−とシン−の比は11:89であった。1H NMR (CD3OD) δ: 1.415, 1.420 [9H, s, −C(CH3)3 ], 1.53−2.25 (8H, m, 4×−CH2−), 3.65 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.70 (1H, br, NH)。
【0081】
シン/アンチ−1−[N−(フタロイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27b)
1mlのTHF中のケトン(26b)(20mg、0.066ミリモル)の溶液に塩化亜鉛(18mg、0.13ミリモル、260μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(19mg、0.10ミリモル、100μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27b)(13.2mg、66%)を透明油状物として得た。アンチ−とシン−の比は52:48であった。1H NMR (CD3OD) δ: 1.60−2.01 (8H, m, 4×−CH2−), 3.70, 3.75 (3H, s, COCH3), 3.86 (1H, m, −CH−), 7.69−7.82 (4H, m, Ph−H)。
【0082】
シン/アンチ−1−[N−(フタロイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27b)(塩化亜鉛は存在せず)
−78℃に冷却した1mlのTHF中のケトン(26b)(18mg、0.059ミリモル)の溶液に、L−セレクトリド(17mg、0.09ミリモル、90μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27b)(13.2mg、66%)を透明油状物として得た。アンチ−とシン−の比は52:48であった。
【0083】
シン/アンチ−1−[N−(トリフルオロアセチル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27c)
1mlのTHF中のケトン(26c)(17mg、0.064ミリモル)の溶液に、塩化亜鉛(17mg、0.13ミリモル、256μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(18mg、0.096ミリモル、96μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27c)(13.5mg、78.4%)を透明油状物として得た。アンチ−とシン−の比は66:34であった。1H NMR (CD3OD) δ: 1.67−2.37 (8H, m, 4×−CH2−), 3.72, 3.75 (3H, s, COCH3), 3.97 (1H, m, −CH−), 6.43 (1H, br, NH)。
【0084】
シン/アンチ−1−[N−(ベンゾイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27d)
1mlのTHF中のケトン(26d)(22mg、0.08ミリモル)の溶液に、塩化亜鉛(22mg、0.16ミリモル、320μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(23mg、0.12ミリモル、120μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた(生成物を検出することはできなかった)。
【0085】
(実施例3)
インビトロおよびインビボでのアミノ酸取り込みのアッセイ
最初に腫瘍細胞を、加湿インキュベーター(37℃、5%CO2/95%空気)の中で、ダルベッコ変法イーグル培地(DMEM)を入れたT型フラスコ中で単層として増殖させた。増殖培地を10%ウシ胎仔血清および抗生物質(10,000単位/mlペニシリンおよび10mg/mlストレプトマイシン)で補充した。増殖培地は週に3回取り換え、細胞が1週間で培養密度に達するように細胞を継代させた。
【0086】
単層のコンフルエントに達したら、細胞を以下の方法で実験用に調製した。増殖培地をT型フラスコから取り出し、単層細胞を1×トリプシン:EDTAに約1分間曝して、細胞とフラスコとの間のタンパク質の結合を弱めた。次いで、フラスコをたたいて、細胞を離脱させた。補充した培地を加えてトリプシンのタンパク分解作用を阻害させ、細胞を18Ga針で吸引し、次いでそれらを単分散させた。血球計算板を用いて顕微鏡下で細胞の試料をカウントし、トリパンブルー染色により生存/死亡割合を推定した(>98%の生存率)。細胞の残りを遠心分離管に入れ、75×gで5分間遠心分離し、上澄みを除去した。次いで細胞をアミノ酸/無血清DMEM塩中に再懸濁した。
【0087】
この試験では、約4.55×105細胞を、12×75mmガラスバイアル中、インキュベーター条件下で、3mlのアミノ酸の存在しない培地±輸送体阻害剤(10mM)中の[18F]10(アンチ−FACBC)かまたは[18F]15(シン−FACBC、5μCi)に30分間曝した。各アッセイ条件は2通り実施した。インキュベーションした後、細胞を遠心分離に2回かけ(75×gで5分間)、氷冷したアミノ酸/無血清DMEM塩で濯いで上澄みの残留活性を除去した。バイアルをPackard Cobra II自動γ線計測器中に置き、生のカウント数を減衰補正し、細胞数当たりの活性を測定した。これらの試験によるデータ(対照に対する取り込み%で表わした)を、Excelを用いてグラフ化し、ワンウェイANOVA(GraphPad Prismソフトウェアパッケージ)を用いて分析してグループ間の統計的比較を行った。
【0088】
[18F]10と[18F]15が支配的にL−型アミノ酸輸送系を介して細胞中に入るという仮定を吟味するために、アミノ酸輸送の十分説明した2つの阻害剤の存在下および非存在下で、培養した9L神経膠肉腫と種々のヒト癌細胞株を用いてアミノ酸取り込みアッセイを実施した。N−MeAIBはA型アミノ酸輸送系の選択的、競合的阻害剤であり、他方、2−アミノ−ビシクロ[2.2.1]ヘプラン−2−カルボン酸(BCH)はナトリウム非依存性L型輸送系のための阻害剤として通常用いられるが、この化合物も、ナトリウム依存性B0,+およびB0輸送系によって、アミノ酸取り込みを競合的に阻害する。A型およびL型アミノ酸輸送系は、腫瘍画像化のために用いられる放射性標識アミノ酸のインビボでの取り込みにかかわっていた。
【0089】
阻害剤が存在しないと、[18F]10と[18F]15はどちらも、9L神経膠肉腫細胞および種々のヒト癌細胞株中で同じようなレベルの取り込みを示し、インキュベーション後30分間で、それぞれ、細胞百万個当たり初期線量の0.43%および0.50%の細胞内蓄積であった。阻害剤の効果を比較しやすくするために、データを、表1に示すように、対照条件(阻害剤なし)に対する取り込みパーセントとして表わした。[18F]10および[18F]15の場合、BCHは、対照に対して活性取り込みの>50%を遮断した。対照と比較したBCHによる[18F]10および[18F]15の取り込みの減少は、統計的に有意であった(ワンウェイANOVAにより、それぞれp<0.05、p<0.01)。これらの阻害試験は、[18F]10および[18F]15が、BCHの存在下での両化合物の取り込みの阻害をもとにして試験して、癌細胞におけるL型アミノ酸輸送系のための基質であることを示している。
【0090】
【表1】
腫瘍誘発および動物の処置:
すべての動物実験は人道的条件下で実施されており、Emory Universityの動物管理使用委員会(Institutional Animal Use and Care Committee(IUCAC))による承認を得たものである。ラット9L神経膠肉腫細胞を、雄性フィッシャーラットの脳の中に埋め込んだ。簡単に述べると、頭部定位ホルダーに固定した麻酔したラットに、4×104ラット9L神経膠肉腫細胞(1×107/mL)の懸濁液を、正中線の3mm右側、プレグマの1mm前方に、頭蓋外板から5mmの深度の位置で注射した。注射は2分間にわたって実施し、腫瘍細胞の逆流を最小限に抑えるように針は1分間にわたって引き抜いた。潜り穴と頭皮切り口を閉じ、この処置から回復させた後、動物を元のコロニーに戻した。頭蓋内腫瘍は発達し、腫瘍担持ラットにおける体重減少、感情鈍麻および猫背の姿勢が認められ、この動物を埋め込み後17〜19日間で使用した。解剖すると、腫瘍細胞を埋め込まれた30匹の動物のうち、25匹について裸眼で視認できる腫瘍が進行しており、これらを試験に用いた。図1〜3はその試験結果を示す。
【0091】
齧歯類生体内分布試験:
16匹の健常な雄性フィッシャー344ラット(200〜250g)に、0.3mLの滅菌水中の約85μCiの[18F]10かまたは[18F]15を静脈注射した後、放射能の組織分布を測定した。実験前に、動物に餌と水を適宜与えた。頭蓋内の腫瘤(mass)の存在下での麻酔状態に伴う死亡を回避するために、RTV−190齧歯類拘束装置(Braintree Scientific)を用いて、尾静脈注射を覚醒動物で実施した。線量を注射後、5分間、30分間、60分間および120分間で4匹からなるラットのグループを殺した。動物を解剖し、選択した組織の重量を量り、Packard Cobra II自動γ線計測器で、線量標準と合わせてカウントした。生のカウント数を減衰補正し、カウント数を、グラム組織当たりの合計注射線量(ID/g%)割合として正規化した。切除し、各時間点で比較のために用いた、腫瘍組織と腫瘍の反対側の対応する脳の領域における活性の取り込みの比較を、ワンウェイANOVA(GraphPad Prismソフトウェアパッケージ)を用いて分析した。以下の図1〜3のこれらの試験結果を示す。
【0092】
図1〜3に示すように、頭蓋内に9L神経膠肉腫細胞を埋め込まれたラットでは、腫瘍組織中での放射能の保持は、[18F]10および[18F]15の静脈注射後60分で高く、腫瘍の反対側の脳組織での放射能の取り込みは低く留まった(<0.3%線量/g)。[18F]10についての腫瘍取り込みと正常な脳の取り込みの比は60分および120分間で6.5:1であり、他方、[18F]15については、同一時間点でその比は5.3:1であった。これらの結果は、アンチ−[18F]FACBC、[18F]10と同様にシン−[18F]FACBC[18F]15が、脳腫瘍を画像化するための優れた候補品であることを実証している。
【0093】
本発明の方法により作製された化合物は、溶媒和、特に水和したものであってもよい。水和は、化合物、または化合物を含む組成物の製造の際に起こるか、あるいは水和は化合物の吸湿により経時的に起こる。さらに、本発明の化合物は、水、エタノール等などの薬学的に受容可能な溶媒と、溶媒和していない形態でも溶媒和した形態でも存在することができる。一般に、本発明のためには、溶媒和した形態は溶媒和していない形態と同等であると考えられる。
【0094】
本発明の化合物を画像化剤として用いる場合、それらを123I、131I、18F、76Br、および77Brなどの適切な放射性ハロゲンアイソトープで標識しなければならない。本発明の放射性ハロゲン化化合物は、腫瘍を画像化させるのに必要な材料を備えたキットでも容易に提供することができる。例えば、最終生成物を、使用する場所またはその時間に作製することができるように、キットは、画像化するのに使用できるようにした適切なアイソトープ(例えば、18F)で標識された最終生成物または中間体化合物と、試薬(例えば、溶媒、脱保護剤)と一緒にした標識(例えば、K[18F]F)を含むことができる。
【0095】
腫瘍画像化方法の第1の工程では、標識した本発明の化合物を、検出可能な量で組織または患者に導入する。化合物は一般に医薬組成物の一部であり、当業者に周知の方法により組織または患者に投与される。例えば、化合物は、経口、経直腸、非経口(静脈内、筋肉内または皮下)、嚢内、膣内、腹腔内、膀胱内、局所(粉剤、軟膏またはドロップ)、または頬側もしくは経鼻噴霧のいずれかで投与することができる。
【0096】
本発明の画像検査法では、標識化合物を、検出可能な量で患者に導入し、化合物が腫瘍の組織または細胞と会合するのに十分な時間を経た後、その標識化合物を患者の内部で非侵襲的に検出する。本発明の他の実施形態では、標識した化合物を患者に導入し、化合物が腫瘍組織と会合するのに十分な時間を与え、次いで患者からの組織試料を取り出し、患者からは別個に、その組織中の標識化合物を検出する。あるいは、組織試料を患者から取り出し、標識した本発明の化合物を組織試料中に導入する。化合物が腫瘍組織と結合するのに十分な時間を経た後、化合物を検出する。「組織」という用語は、患者の身体の一部を意味する。組織の例には、脳、心臓、肝臓、血管および動脈が含まれる。検出可能な量は、選択した検出方法で検出するのに必要な標識化合物の量である。検出するために患者の中に導入すべき標識化合物の量は、当業者によって容易に決定される。例えば、選択した検出方法で化合物が検出されるまで、標識化合物の量を増加させて患者に与えることができる。化合物が検出されるように、化合物中に標識を導入する。
【0097】
標識化合物の患者への投与は全身的投与経路でも局部的投与経路でもよい。例えば、標識化合物は、身体全体に送達されるように患者に投与することができる。あるいは、標識化合物を、対象の特定の器官または組織に投与することができる。
【0098】
化合物が腫瘍と会合する十分な時間量を決定することは、当業者に周知である。必要な時間量は、検出可能量の標識した本発明の化合物を患者に導入し、次いで、投与後異なった時間で標識化合物を検出することによって容易に決定することができる。
【0099】
標識化合物を検出するための様々な方法は、当業者に周知である。例えば、放射性標識化合物を検出するために、磁気共鳴映像法(MRI)、陽電子射出断層撮影法(PET)または単光子射出コンピュータ断層撮影法(SPECT)を用いることができる。本発明の化合物を腫瘍画像化剤として用いる場合に、PETおよびSPECTが好ましい。化合物に導入される標識は所望の検出方法に依存することになる。例えば、検出方法としてPETを選択した場合、化合物は、11Cまたは18Fなどの陽電子放出原子を有していなければならない。
【0100】
放射性診断薬は、信頼性のある診断を確実にする十分な放射能と放射能濃度を有していなければならない。例えば、放射性金属がテクネチウム−99mである場合、通常投与の際に約0.5〜5.0ml中に0.1〜50mCiの量を含むことができる。式の化合物の量は、放射性金属と安定したキレート化合物を形成するのに十分な量にすることができる。
【0101】
放射性診断薬としての本発明の化合物は十分に安定であり、したがって、そのまますぐに投与してもよく、また使用時まで貯蔵することもできる。望むなら、放射性診断薬は、pH調製剤(例えば、酸、塩基、緩衝剤)、安定剤(例えば、アスコルビン酸)または等張剤(例えば、塩化ナトリウム)などの任意の添加剤を含むことができる。腫瘍の画像化は、本明細書の化合物を用いて定量的に実施し、それによって所与の腫瘍のためのその治療薬の効能を評価することもできる。
【0102】
画像化に好ましい化合物には、123I、124I、125I、131I、18F、76Br、77Brまたは11Cなどの放射性アイソトープが含まれる。
【0103】
本明細書で説明する合成スキームは、本発明の好ましい実施形態の例としての合成法を示す。しかし、当業者は本発明の実施にあたって、出発原料、試薬、溶媒、温度、固体基体、合成方法、精製方法、分析方法、および具体的に例示したもの以外の他の反応条件を、必要以上の実験をすることなく、用いることができることを理解されよう。そうした任意の原料や方法の当業界で周知の機能的均等物のすべては、本発明に含まれるものとする。使用される用語および表現は説明のための用語として用いるものであって限定的なものではなく、そうした用語および表現の使用において、示しかつ説明した特徴またはその一部のどんな均等物も排除しようとするものではなく、特許請求した本発明の範囲内で様々な変更が可能であることを理解されたい。したがって、好ましい実施形態および任意選択の特徴によって本発明を具体的に開示してきたが、当業者は、本明細書で開示した考え方の改変および変更を行うことができ、そうした改変形態および変更形態は、添付の特許請求の範囲により定義される本発明の範囲内であることを理解すべきである。
【0104】
本明細書で、ある置換基の群が開示される場合、群メンバーの任意の異性体および鏡像異性体を含む、その群およびすべての下位群のすべての個別メンバーが別々に開示されることを理解されたい。マーカシュ群または他のグルーピングを本明細書で用いる場合、その群のすべての個別のメンバーおよびその群に可能なすべての組合せおよび下位組合せは、個別にその開示に含まれるものとする。例えば式や化学名において、化合物が、本明細書で化合物の具体的な異性体または鏡像異性体が指定されずに記述される場合、その記述は、個別にまたは任意の組合せで、記述された化合物の各異性体および鏡像異性体を含むものとする。さらに、別段の指定のない限り、本発明で開示の化合物のすべての同位変異体は本開示に包含されるものとする。例えば、開示した分子中の任意の1個または複数の水素を、重水素または三重水素で置き換えることができることを理解されよう。分子の同位変異体は、分子のアッセイ、および分子またはその使用に関連する化学的または生物学的研究における標準品として一般に有用である。同一化合物を異なって称することができることが当業者に周知である場合、化合物の具体的な名称は例示的なものであるものとする。
【0105】
本明細書で開示の分子の多くは1つまたは複数のイオン性基[陽子をそこから取り外すことができる基(例えば、−COOH)かまたはそれに加えることができる基(例えば、アミン)、あるいは四級化できる基(例えば、アミン)]を含む。そうした分子およびその塩の可能なすべてのイオン形態は、本明細書の開示に個別に包含されるものとする。本明細書の化合物の塩に関しては、当業者は所与の用途のための本発明の塩の調製に適した使用可能な広範囲の対イオンの中から選択することができる。
【0106】
別段の指定のない限り、本明細書で記述するかまたは例示した成分のすべての処方または組合せを、本発明を実施するために用いることができる。
【0107】
明細書において、ある範囲、例えば温度の範囲、時間の範囲、純度の範囲または組成物もしくは濃度の範囲が与えられている場合、すべての中間範囲および下位範囲、ならびに所与の範囲に含まれるすべての個別の値は本開示に包含されるものとする。
【0108】
本明細書で言及されるすべての特許および出版物は、本発明が関係する当業界の技術者の技術レベルを示すものである。出願日時点での最新技術を示すために、本明細書で引用した文献のその全体を参照により本明細書に組み込み、また、必要なら、従来技術の範囲内である特定の実施形態を排除するために、この情報を本明細書で用いることができるものとする。例えば、化合物が特許請求される場合、本明細書で引用する文献に使用可能な開示が提供されている化合物を含む出願者の発明に先行する技術において既知であり使用可能な化合物は、本明細書の特許請求の範囲の組成物には含まれないものと理解されたい。
【0109】
本明細書で用いる、「含む(comprising)」は、「含む(including)」、「含む(containing)」または「ことを特徴とする(characterized by)」と同義的なものであり、かつ包含的である、すなわち幅広く解釈できる(open−ended)ものであり、他の言及されていない要素または方法の工程を排除するものではない。本明細書で用いる「からなる(consisting of)」は、特許請求の範囲の要素で指定されていないどの要素、工程または成分も排除する。本明細書で用いる「本質的にからなる(consisting essentially of)」は、特許請求の範囲の基本的特徴および新規な特徴に実質的に影響を及ぼさない材料または工程を排除しない。本明細書のそれぞれ場合において、「含む(comprising)」、「本質的にからなる」および「からなる」という用語のいずれも、他の2つのうちのいずれかで置き換えることができる。本明細書で例示的に説明した本発明は、本明細書で具体的に開示されていない要素や限定がなくても適切に実施することができる。
【0110】
本明細書で引用した文献を、本出願の開示との不整合がない範囲で参照により本明細書に組み込む。特に、本発明を用いて作製できるアミノ酸類似体の例およびその詳細な合成方法を提供するために、米国特許第5,808,146号、同第5,817,776号およびWO03/093412が本明細書で引用されている。それらを参照により本明細書に組み込む。出発原料、追加の出発原料、追加の試薬、追加の合成方法、追加の分析方法および追加の本発明の使用の供給源に関する詳細を提供するために、本明細書で提供したいくつかの文献を参照により本明細書に組み込む。
【図面の簡単な説明】
【0111】
【図1】図1は、9L腫瘍におけるインビボでの化合物の取り込みを示す。結果を、注射後60分での対照に対する取り込み%で表わす。詳細は実施例2を参照されたい。
【図2】図2は、注射後60分の対側正常脳におけるインビボでの取り込みを示す。
【図3】図3は、注射後60分間のインビボでの腫瘍における化合物の取り込み対正常細胞における化合物の取り込みの比を示す。その比は図1および図2のパーセント値から得た。
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2005年6月23日出願の米国仮出願番号第60/693,385号に関する優先権を主張するものである。それを、本出願に相反しない範囲で参照により本明細書に組み込む。
【0002】
(連邦研究支援の承認)
本発明は、国立衛生研究所(National Institutes of Health)より付与された認可番号5−R21−CA−098891のもとでの政府支援によって行われた。政府は本発明において一定の権利を有するものである。
【0003】
(発明の背景)
本発明は、シン−アミノ酸類似体を合成する方法、およびその方法によって合成される化合物、具体的にはシン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体に関する。本発明のアミノ酸類似体は、生体系において特異的な結合を有し、陽電子射出断層撮影法(PET)および単光子放出(SPECT)画像検査法で使用することができる。
【背景技術】
【0004】
陽電子射出断層撮影法(PET)および単光子射出コンピュータ断層撮影法(SPECT)を用いる、腫瘍を画像化するための代謝トレーサーとして用いる放射性標識アミノ酸の開発はこれまでかなりの期間行われている。放射性標識アミノ酸は様々な腫瘍タイプに適用されているが、頭蓋内腫瘍へのその適用は、他の画像診断法より優れた潜在的利点のため、相当な注目を集めてきた。脳腫瘍の外科的切除および/または放射線治療の後、CTやMRIなどの従来の画像検査法では、残留腫瘍または再発腫瘍と、治療介入による組織傷害とを信頼性高く識別することはできず、処置の有効性の監視や腫瘍再発の検出に最適ではない[非特許文献1;非特許文献2]。
【0005】
新生物の診断および画像化のための主要なPET用薬剤である2−[18F]フルオロデオキシグルコース(FDG)では、脳腫瘍の画像化に限界がある。放射線治療または外科的治療の後発生する可能性がある炎症組織と同じように、正常な大脳皮質組織は高い[18F]FDGの取り込みを示す;これらの因子は[18F]FDGで得られる画像の解釈を複雑にする可能性がある[非特許文献3;Conti, PS (1995)]。
【0006】
多くの報告は、放射性標識アミノ酸で画像化するPETおよびSPECTは、CT法またはMRI法より良好に正常脳内の腫瘍境界を規定し、それによって、より良好な治療計画が可能になることを示している[非特許文献4;非特許文献5]。さらに、いくつかの研究は、アミノ酸の取り込みの度合が腫瘍の異型度と相関しており、それは重要な徴候情報を提供する可能性がある[非特許文献5]。
【0007】
アミノ酸は腫瘍細胞の増殖に必須の栄養素である。陽電子放出アイソトープの炭素11およびフッ素18を含む様々なアミノ酸が調製されている。これらは脳腫瘍および全身性腫瘍を有する患者の腫瘍を画像化するための臨床腫瘍学的分野で使用できる潜在性があると評価されており、ある種の癌において2−[18F]FDGに対して優れた特徴を有している。これらのアミノ酸候補はさらに2つの主要カテゴリーに分けることができる。第1のカテゴリーは、[11C]バリン、L−[11C]ロイシン、L−[11C]メチオニン(MET)およびL−[1−11C]チロシン、ならびに2−[18F]フルオロ−L−チロシンおよび4−[18F]フルオロ−L−フェニルアラニンなどの構造的に似ている類似体などの放射性標識した天然由来のアミノ酸によって代表される。腫瘍細胞膜を通るこれらのアミノ酸の移動は主に、ナトリウム非依存性ロイシン型「L」アミノ酸輸送系による担体媒介輸送によって起こる。正常組織と比較した、これら天然由来の放射性標識アミノ酸の腫瘍内への取り込みの増大と保持の延長は、タンパク質中への大幅かつ迅速な局部的取り込みに一部起因している。これら放射性標識アミノ酸のうち、[11C]METが臨床的に腫瘍を検出するのに最も広く用いられている。[11C]METは脳腫瘍および全身性腫瘍を検出するのに有用であることが分かっているが、複数の経路を通してのインビボでの代謝の影響を受けやすく、多くの放射性標識代謝産物をもたらす。したがって、腫瘍代謝活性の信頼性のある測定に必要な精度を有する画像分析は不可能である。ヒトにおける[11C]METの腫瘍取り込みの動態解析の研究は、アミノ酸輸送が、タンパク質合成より感度の高い腫瘍細胞増殖の測定を提供できることを強く示唆している。
【0008】
[11C]METに伴う欠点は第2のカテゴリーのアミノ酸を用いて克服することができる。これらは1−アミノシクロブタン−1−[11C]カルボン酸([11C]ACBC)などの非天然性アミノ酸である。[11C]METと比較した[11C]ACBCの利点は、それが代謝されないということである。炭素11アミノ酸を臨床用途に適用するための大きな限界は炭素11の半減期が20分間と短いことである。半減期が20分間であるということは、炭素11アミノ酸を生成するのに現地での粒子加速器を必要とする。さらに、炭素11アミノ酸の各バッチ生成により、単一線量だけまたは比較的少ない線量だけしか発生させることができない。したがって、炭素11アミノ酸は、広範な臨床用途での局部的分配には不適切な候補品である。
【0009】
炭素11の半減期の物理的限界を克服するために、我々は最近、いくつかの新規なフッ素18で標識した非天然性アミノ酸の開発に焦点をあてた。そのいくつかは特許文献1および特許文献2に開示されている。その開示を参照により本明細書に組み込む。これらには、アンチ−1−アミノ−3−[18F]フルオロシクロブチル−1−カルボン酸(アンチ−[18F]FACBC)、シン−1−アミノ−3−[18F]フルオロシクロブチル−1−カルボン酸(シン−[18F]FACBC)、シン−およびアンチ−1−アミノ−3−[18F]フルオロメチル−シクロブタン−1−カルボン酸(シン−およびアンチ−[18F]FMACBC)が含まれる。これらのフッ素−18アミノ酸は、画像技術の陽電子射出断層撮影法(PET)を用いたアミノ酸輸送をもとにして、インビボで脳腫瘍および全身性腫瘍を画像化するのに用いることができる。我々の開発では、主として「L」型の大型中性アミノ酸輸送系を用い、より少ない程度に「A」型アミノ酸輸送系を用いる担体媒介輸送によって、腫瘍毛細管を通って移動するフッ素18標識シクロブチルアミノ酸を用いる。我々の予備的評価では、「L」型輸送系のための主要基質である陽電子放射体で標識したシクロブチルアミノ酸は、脳腫瘍および全身性腫瘍を有する患者の腫瘍を画像化するのに臨床腫瘍学的に優れた潜在性を示した。11C(t1/2=20分間)ではなく、シクロブチル/分鎖アミノ酸の18F−標識を提案する主な理由は、臨床用途において、11C−標識放射性医薬品ではなく18Fを用いて得られる大きな運搬上および経済上の利益である。忙しい核医学部門で、18F標識放射性医薬品を用いる腫瘍画像化の利点は、主に18Fのより長い半減期(t1/2=110分間)によるものである。18Fのより長い半減期により、放射性トレーサーの単一生産ロットからの現地外での分配と複数線量が可能になる。さらに、これらの非代謝アミノ酸は、2−[18F]FDGPETでは十分に画像化されないある種の全身性固形癌に対する画像化剤としても、より広い用途を有している。特許文献3(これを参照により本明細書に組み込む)はさらに、18Fを用いた標識およびPET画像化での使用に適した2−アミノ−3−フルオロ−2−メチルプロパン酸(FAMP)や3−フルオロ−2−メチル−2−(メチルアミノ)プロパン酸(N−MeFAMP)などのα−アミノイソ酪酸(AIB)のフッ素化類似体の例を開示している。AIBは、主としてA型アミノ酸輸送系を介して細胞中へ活発に輸送される非代謝性のα,α−ジアルキルアミノ酸である。システムAのアミノ酸輸送は、細胞増殖および細胞分裂の際に増大し、腫瘍細胞において上方調節されることも示されている[非特許文献6;非特許文献7]。動物において実験的に誘発される腫瘍、およびヒトにおいて自発的に発生する腫瘍の研究は、正常組織に対して、放射性標識AIBの取り込みが増加していることが示されている[非特許文献8;非特許文献9]。AIBのN−メチル類似体、N−MeAIBは、AIBよりA型アミノ酸輸送系に対して、さらにより高い選択性を示す[非特許文献10]。N−MeAIBは炭素11で放射性標識され、ヒトにおいて代謝的に安定である[非特許文献11]。
【特許文献1】米国特許第5,808,146号明細書
【特許文献2】米国特許第5,817,776号明細書
【特許文献3】国際公開第03/093412号パンフレット
【非特許文献1】Buonocore, E(1992年)、Clinical Positron Emission Tomography. Mosby−Year Book, Inc. St. Louis, MO、17〜22頁
【非特許文献2】Langleben, DDら(2000年)、J. Nucl. Med. 41:1861〜1867頁
【非特許文献3】Griffeth, LKら(1993年)、Radiology. 186:37〜44頁
【非特許文献4】Ogawa, Tら(1993年)、Radiology. 186: 45〜53頁
【非特許文献5】Jager, PLら(2001年)、Nucl. Med., 42:432〜445頁
【非特許文献6】Palacin, Mら(1998年)、Physiol. Rev. 78:969〜1054頁
【非特許文献7】Bussolati, Oら(1996年)、FASEB J. 10:920〜926頁
【非特許文献8】Conti, PSら(1986年)、Eur. J. Nucl. Med. 12:353〜356頁
【非特許文献9】Uehara, Hら(1997年)、J. Cereb. Blood Flow Metab. 17:1239〜1253頁
【非特許文献10】Shotwell, MAら(1983年)、Biochim. Biophys. Acta. 737:267〜84頁
【非特許文献11】Nagren, Kら(2000年)、J. Labelled Cpd. Radiopharm. 43:1013〜1021頁
【発明の開示】
【発明が解決しようとする課題】
【0010】
脳腫瘍および全身性腫瘍を有する患者での腫瘍の画像化のための陽電子放出アイソトープを含むアミノ酸類似体の利点は当業界でよく認識されているが、これらの化合物の立体特異的異性体を大量に提供できる、信頼性が高く効率的な合成方法は依然として必要とされている。候補化合物を、細胞および動物モデルにおける有効性の確認研究からヒトでの適用に移行する際、使用する合成技術は、その化合物が定常的にかつ信頼性高く生産されるように適合するものでなければならない。そのために、本発明者等はここに、シン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体を作製するための立体選択的な合成ストラテジーを開発した。この立体選択的な合成ストラテジーは、様々なアミノ酸類似体、特に、PETおよびSPECTを用いた腫瘍画像化用の放射性トレーサーを含むものを合成するのに適用することができることは以下の説明で明らかであろう。
【課題を解決するための手段】
【0011】
(発明の要旨)
本発明は、シン異性体のアミノ酸類似体を合成するための主要前駆体の特定の立体異性体を生成する合成ストラテジーを提供する。このストラテジーはシン−1−アミノ−3−シクロブタン−1−カルボン酸(ACBC)類似体を合成するのに特に有用である。合成における主要工程には、トランス−アルコールへの前駆体シントンの還元が含まれ、このトランス−アルコールはシン−異性体の最終生成物に転換される。本明細書で開示する合成ストラテジーは信頼性が高く、かつ効率的であり、シン−ACBC類似体の放射線合成のための主要前駆体をグラム単位の規模で調製することが可能になる。さらに、本明細書で開示の合成ストラテジーは、最後の工程として、適切なアイソトープを組み込んでアイソトープの有用な寿命を最大化する。
【0012】
本発明は以下の式を有するトランス−アルコールを提供する:
式1
【0013】
【化4】
[式中、YおよびZ=独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)であり、
R1〜R3=独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルであり、
R4およびR5=独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニル、ハロアルキニルであり、但し、ハロは非放射性のF、Cl、BrおよびIである]
本発明はまた、式1の一般構造を有するトランス−アルコールの合成方法も提供する。上式のトランス−アルコールの合成の主要工程は、ポリマー結合還元剤(例えば、Aldrich32、864−2水素化ほう素ポリマー担持アンバーライトIRA400;Aldrich52、630−4シアノ水素化ほう素ポリマー担持;Aldrich35,994−7水素化ほう素ポリマー担持アンバーライトA−26;Aldrich59、603−5水素化ほう素亜鉛ポリマー(結合))を用いる金属水素化物による直接還元である。本明細書でのスキーム3では、リチウムトリイソブチルボランおよびZnCl2を用いたこの反応を例示する。
【0014】
開示する合成ストラテジーは、脳および身体の腫瘍の検出および評価での使用ならびに他の使用のための様々なアミノ酸化合物のシン−異性体を調製するのに用いることができる。これらの化合物は、1−アミノ−シクロアルキル−1−カルボン酸の有利な特性、すなわちその迅速な取り込みおよび長期的保持と、フッ素18、ヨウ素−123、ヨウ素−125、ヨウ素−131、臭素−75、臭素−76、臭素−77、臭素−82、アスタチン−210、アスタチン−211および他のアスタチンアイソトープなどの特定の有用なハロゲンアイソトープを含むハロゲン置換基の特性とを組み合わせる。さらにこれらの化合物は、キレート化錯体を用いてテクネチウムおよびレニウムアイソトープで標識することができる。詳細な説明についてはWO03/093412および米国特許第5,817,776号を参照されたい。
【0015】
トランス−アルコールを含む本発明の合成ストラテジーを用いて作製することができるシン−アミノ酸類似体には、これらに限定されないが、以下の式を有する化合物が含まれる:
式II
【0016】
【化5】
[式中、YおよびZ=独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)であり、
R1〜R3=独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニル、ハロアルキニルであり、
R4〜R5=独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルケニル、アルキニル、ハロアルケニル、ハロアルキニルであり、但し、ハロは非放射性のF、Cl、BrおよびIであり、
R7=ハロゲン、ハロアルキル、ハロアルケニル、ハロアルキニル、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリール、ハロヘテロアリールであり、但し、ハロ=F−18、I−123、I−124、Tc−99mおよびReキレートなどの標識化合物を含むF、Cl、Br、I、Atである]
本明細書で開示する本発明の方法を用いて作製できる具体的な放射性標識アミノ酸類似体には、これらに限定されないが、上記構造を有するフルオロ−、ブロモ−もしくはヨード−置換のシクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシルアミノ酸、またはヘテロ原子、すなわちN、OおよびSならびにSeを含む脂環式化合物が含まれる。
【0017】
本発明により作製されるアミノ酸化合物は、インビボで対象に投与した場合、腫瘍組織に対して高い特異性を有する。したがって、本発明は、本発明の方法により作製されるシン−アミノ酸類似体を含む医薬組成物および診断用組成物も提供する。好ましいアミノ酸化合物は少なくとも2:1の標的と非標的の比を示し、インビボで安定であり、投与後1時間以内で標的にほぼ局在化する。好ましいアミノ酸化合物の例には、シン−[18F]−1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)、シン−[123I]−1−アミノ−3−ヨードシクロブタン−1−カルボン酸(IACBC)およびシン−[18F]−1−アミノ−3−フルオロアルキル−シクロブタン−1−カルボン酸、例えばシン−[18F]−1−アミノ−3−フルオロメチル−シクロブタン−1−カルボン酸(FMACBC)が含まれる。
【0018】
本発明のアミノ酸類似体は対象の腫瘍を検出および/または監視するための画像化剤として有用である。アミノ酸類似の画像化剤はインビボで投与され、その標識に適した手段を用いて監視される。アミノ酸類似の画像化剤をインビボで検出および/または監視するのに好ましい方法には、陽電子断層撮影法(PET)および単光子射出コンピュータ断層撮影法(SPECT)が含まれる。
【発明を実施するための最良の形態】
【0019】
(発明の詳細な説明)
本発明は、とりわけ腫瘍画像化に有用なシン−アミノ酸類似体を合成するための新規な方法に関する。本発明者等はここに、シン−ACBC類似体を合成するための、トランス異性体の主要前駆体の立体選択的な合成を可能にする合成ストラテジーを開発した。本発明の合成ストラテジーにより作製されるACBC類似体は実質的に純粋なシン−異性体である。本明細書で用いる「実質的に純粋な」という用語は、生成物が、少なくとも60%のその異性体純度、好ましくは70%の純度、好ましくは90%超のシン−異性体純度であることを意味する。すべての中間体は60%〜100%の値であり、それらが個別にリストに挙げられているかいないかにかかわらず、その範囲にある中間体はすべて包含されるものとする。
【0020】
本明細書で用いる用語および語句は概ね、当業界で周知の標準的な教科書、雑誌文献および文脈を参照すると見出せる当業界で認識されている意味を有する。以下の定義は、本発明に関連してその具体的な使用を明らかにするために提供されるものである。
【0021】
本明細書で用いる「薬学的に受容可能な塩」という用語は、過度の毒性、炎症、アレルギー反応等がなく患者の組織と接触させて用いるのに適しており、妥当な利益/リスク比に相応するものであり、かつ目的の用途に有効な本発明の化合物のカルボン酸塩または酸付加塩、ならびに、可能な場合、本発明の化合物の双性イオンの形態を指す。「薬学的に受容可能な塩」という用語は、本発明の化合物の比較的非毒性の無機および有機酸付加塩を指す。脂肪族モノおよびジカルボン酸、例えば酢酸、フェニル置換アルカン酸、ヒドロキシアルカン酸およびアルカンジオン酸、芳香族酸ならびに脂肪族および芳香族スルホン酸などの非毒性の有機酸から誘導される塩も含まれる。これらの塩は、化合物の最終的な単離や精製の際にその場で調製するか、あるいは、精製されたそのフリーベースの化合物を適切な有機酸または無機酸と別個に反応させ、その生成した塩を単離して調製することができる。他の代表的な塩には、臭化水素酸塩、塩酸塩、硫酸塩、重硫酸塩、硝酸塩、酢酸塩、シュウ酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、ホウ酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチレート、メシラート、グルコヘプトネート、ラクチオビオネート、ラウリルスルホン酸塩、プロピオン酸塩、ピバル酸塩、サイクラミン酸塩、イセチオン酸塩等が含まれる。これらは、ナトリウム、リチウム、カリウム、カルシウム、マグネシウム等のアルカリ金属およびアルカリ土類金属をもとにしたカチオン、ならびに、これらに限定されないが、アンモニウム、テトラメチルアンモニウム、テトラエチルアンモニウム、メチルアミン、ジメチルアミン、トリメチルアミン、トリエチルアミン、エチルアミン等を含む非毒性アンモニウム、四級アンモニウムおよびアミンカチオンを含むことができる。例えば、Berge S. M,ら、Pharmaceutical Salts, J. Pharm. Sci. 66:1〜19頁(1977年)を参照されたい。これを参照により本明細書に組み込む。
【0022】
同様に、本明細書で用いる「薬学的に受容可能な担体」という用語は、医薬組成物または診断用組成物における本発明の活性成分の担体/安定剤/希釈剤として作用する有機または無機組成物である。いくつかの場合、薬学的に受容可能な担体は塩である。薬学的に受容可能な担体の他の例には、これらに限定されないが、水、リン酸緩衝生理食塩水、生理食塩水、pH調製剤(例えば酸、塩基、緩衝剤)、アスコルビン酸などの安定剤、等張剤(例えば、塩化ナトリウム)、水性溶媒、ポリソルベートまたはTWEEN80などの洗剤(イオン性および非イオン性)が含まれる。
【0023】
それ自体または他の基の一部として、本明細書で用いる「アルキル」という用語は、メチル、エチル、プロピル、イソプロピル、ブチル、t−ブチルおよびイソブチルなどの最大で10個の炭素、好ましくは6個の炭素、より好ましくは4個の炭素の直鎖、分鎖もしくは環状の飽和炭化水素を指す。本発明のアルキル基は、主鎖中の1個もしくは複数の炭素原子がヘテロ原子で置換されていることができ、1個もしくは複数の水素原子がハロゲンまたは−OHで置換されていることができる任意選択で置換されたものも含む。それ自体または他の基の一部として、本明細書で用いる「アリール」という用語は、フェニル、ナフチルまたはテトラヒドロナフチルなどの環部分に5〜12個の炭素、環部分に好ましくは6〜10個の炭素を単環式または二環式の芳香族基を指す。アリール基の1つまたは複数の環は縮合環を含むことができる。アリール基は直鎖、分鎖または環状であってよい1個または複数のアルキル基で置換されていてよい。アリール基は、環位置で、それが存在する化合物または化合物の部分の機能にそれほど決定的に影響を及ぼさない置換基で置換されていてもよい。置換アリール基は、環の中の1個または複数の炭素が、1個または複数のヘテロ原子(例えばN、OまたはSであり、任意選択で、適切な原子価のための水素または置換基を有する)で置き換わっている複素芳香族環を有するものも含む。
【0024】
本明細書で用いる「アルコキシ」という用語は、鎖長がそれに限定されていない限り、酸素原子と結合している上記定義の通りの直鎖または分鎖アルキル基、これらに限定されないが、例えばメトキシ、エトキシ、n−プロポキシ、イソプロポキシなどを意味する。アルコキシ鎖は1〜6個の炭素原子の長さ、より好ましくは1〜4個の炭素原子の長さであることが好ましい。
【0025】
「アシル」基は−CO−基を含む基である。
【0026】
それ自体または他の基の一部として、本明細書で用いる「モノアルキルアミン」という用語は、上記定義の1個のアルキル基で置換されているアミノ基を指す。
【0027】
それ自体または他の基の一部として、本明細書で用いる「ジアルキルアミン」という用語は、上記定義の2個のアルキル基で置換されているアミノ基を指す。
【0028】
それ自体または他の基の一部として、本明細書で用いる「ハロ」という用語は塩素、臭素、フッ素またはヨウ素を指す。
【0029】
本明細書で用いる「複素環」または「複素環式環」という用語は、言及のない限り、炭素原子とN、OおよびSからなる群から選択される1〜3個のヘテロ原子からなる、飽和または不飽和の安定した5〜7員のモノ複素環式環系であって、その窒素およびイオウヘテロ原子が任意選択で酸化されていてよいモノ複素環式環系を表わす。特に有用なものは、1個の酸素もしくはイオウと結合した1個の窒素ヘテロ原子、または2個の窒素ヘテロ原子を含む環である。そうした複素環式基の例には、ピペリジニル、ピロリル、ピロリジニル、イミダゾリル、イミダジニル、イミダゾリジニル、ピリジル、ピラジニル、ピリミジニル、オキサゾリル、オキサゾジニル、イソキサゾリル、イソオキサゾジニル、チアゾリル、チアゾリジニル、イソチアゾリル、ホモピペリジニル、ホモピペラジニル、ピリダジニル、ピラゾイルおよびピラゾリジニルが含まれる。チアモルホリニル、ピペラジニル、およびモルホリニルが最も好ましい。
【0030】
本明細書で用いる「ヘテロ原子」という用語は、酸素原子(「O」)、イオウ原子(「S」)または窒素原子(「N」)を意味する。ヘテロ原子が窒素である場合、RaおよびRbが互いに独立に水素もしくはC1〜4アルキル、C2〜4アミノアルキル、C1〜4ハロアルキル、ハロベンジルであるか、またはRaおよびRbが一緒になってその環内にO、SもしくはNRc(Rcは水素またはC1〜4アルキルである)を任意選択で有する5〜7員の複素環式環を形成するNRaRb部分を形成することを理解されよう。
【0031】
本発明の化合物は腫瘍結合剤およびNMDA受容体結合リガンドとして有用であり、放射性アイソトープはPETおよびSPECTによる画像化を含む腫瘍画像化技術のためのトレーサー化合物として特に有用である。画像化剤として特に有用なものはF−18で標識した化合物である。その理由は、F−18が110分の半減期を有しており、それによって、放射能標識トレーサー中に取り込み、精製し、かつヒトまたは動物対象に投与するために十分な時間が与えられるからである。さらに、サイクロトロンから最大で半径約200マイル以上離れた設備でF−18標識した化合物を用いることができる。
【0032】
SPECT画像化には高エネルギー光子を放出するアイソトープトレーサー(γ−エミッター)を用いる。有用なアイソトープの範囲はPET用より大きいが、SPECTはより低い三次元分解能を提供する。それでも、SPECTは、類似体結合、局在化およびクリアランスの速度についての臨床的に重要な情報を得るために広く用いられる。SPECT画像化に有用なアイソトープは13.3時間の半減期を有する[123I]、γ−エミッターである。[123I]で標識した化合物は、製造場所から最大で約1000マイルまで送出するか、またはアイソトープ自体を現地での合成のために輸送することができる。アイソトープの放出の85%は159KeV光子である。これは現在用いられているSPECT装置で容易に測定される。
【0033】
したがって、PET画像化の代替法としてSPECT分析で用いるために、本発明の化合物は、[123I]で迅速かつ効率的に標識することができる。さらに、同一化合物を、どのアイソトープを用いても標識することができるので、同じトレーサーを用いたPETとSPECTにより得られた結果を比較することができる。
【0034】
他のハロゲンアイソトープを、PETもしくはSPECTの画像化に、または従来のトレーサー標識に役立てることができる。これらには、使用に適した半減期と放射特性を有する75Br、76Br、77Brおよび82Brが含まれる。一般に、上記したアイソトープのためにいずれかのハロゲン部分を置換するのには化学的手段がある。したがって、当業者は、安定したアイソトープハロゲン同族体を含む、本発明の化合物の任意のハロゲン化同族体の生化学的または生理学的活性を利用することができる。アスタチンは他のハロゲンアイソトープを代替することができ、[210At]は8.3時間の半減期を有するα粒子を放出する。したがって、At置換化合物は、結合が十分に腫瘍特異的である腫瘍の治療に有用である。
【0035】
本発明は、PETおよびSPECTを用いて腫瘍を画像化するための方法を提供する。その方法は、適切にアイソトープで標識された画像発生量の本発明の化合物を対象(実験および/または診断の目的のためのヒトまたは動物であってよい)に投与する工程と、次いで、[18F]または他の陽電子エミッターを用いた場合にはPETによって、あるいは[123I]または他のγエミッターを用いた場合にはSPECTによって化合物の分布を測定する工程とを含む。画像発生量は、スキャナーの検出感度やノイズレベル、アイソトープの寿命、対象の身体の大きさおよび投与経路を考慮して、PETまたはSPECTのスキャナーに画像を少なくとも提供することができる量である。上記のすべての変数は周知の例であり、必要以上の実験を用いることなく、当業者に知られている計算および測定によって明らかにされる。
【0036】
本発明の化合物は、構造中の任意の原子または原子の組合せのアイソトープで標識できることを理解されよう。本明細書では[18F]、[123I]および[125I]がPET、SPECTおよびトレーサー分析に特に有用であると強調してきたが、安定したアイソトープ同族体の生理学的または薬理学的特性によりもたらされるものを含む他の使用が考えられ、それらは当業者に明らかであろう。
【0037】
本発明の化合物は、Tc付加体を介してテクネチウム(Tc)標識することもできる。Tc、特にTc99mのアイソトープは腫瘍の画像化に用いられてきた。本発明は、腫瘍画像化に有用である本発明の化合物のTc−錯化付加体を提供する。その付加体は、飽和しているかまたは二重結合もしくは三重結合を有する4〜6個の炭素鎖によって環状アミノ酸と結合しているTc配位錯体である。二重結合が存在する場合、E(トランス)かまたはZ(シス)異性体のどちらかを合成することができ、どちらの異性体も用いることができる。Tcで標識した本発明の化合物は、アイソトープの有用期間を最大にするために、最後の段階で99mTcアイソトープを取り込んで合成される。
【0038】
米国特許第5,817,776号は、(アンチ−[18F]−1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC))を合成するための10工程の一連の反応を開示している。その一連の反応は、工程4に続いて、主要中間体であるシス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸とトランス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸のそれぞれ75:25の混合を、大きな労働力を要するセミ分取高圧液体クロマトグラフィーで分離する工程を含む。次いで、精製された主成分異性体のシス1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸を、6工程の一連反応でトリフレート前駆体に転換させる。
【0039】
本発明者等は合成方法の改良を目指して、放射性標識するための前駆体であるシス1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステル(8)とトランス1−アミノ−3−フルオロシクロブタン−1−カルボン酸(アンチ−FACBC)(10)の両方を大規模合成するトランス−(アンチ−)1−アミノ−3−[18F]フルオロシクロブタン−1−カルボン酸(アンチ−[18F]FACBC)の立体選択的な合成法を開発した。スキーム1および2はアンチ−FACBCの合成工程を示す。この合成工程を用いて、7工程の一連反応によりトリフレート前駆体(8)を調製することができる。この合成における重要な工程はシントン3−ベンジルオキシ−シクロブタノン(2)の調製である。シクロブタノン3の調製は、1−ブロモ−2−ベンジルオキシ−3−ブロモプロパン(1)をメチルエチル−スルホキシドとn−ブチルリチウムで処理することによる環化反応を含む。ケトン2を、Bucherer Strecker条件のもとで直接ヒダントイン3および4に転換させた。シス:トランスヒダントインの80:20混合物をフラッシュクロマトグラフィーにより容易に精製して、所望のシスヒダントイン4を得た。トリフレート前駆体、シス1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステル(8)への4の転換を、米国特許第5,817,776号に記載の一連の反応により実施した。この方法を用いて、グラム単位の量の化合物9を調製することができた[McConathyら(2003年)Jour. of Applied Radiation and Isotopes, 58:657〜666頁]。
【0040】
【化6】
トランス−1−t−ブチルカルバメート−3−トリフルオロメタンスルホンオキシ−1−シクロブタン−1−カルボン酸メチルエステルを大規模に生産するために、スキーム3〜5示すように腫瘍画像化用の、シン−異性体のアミノ酸類似体、具体的にはシス−(シン−)1−アミノ−3−フルオロシクロブタン−1−カルボン酸(シン−FACBC)を十分な量で得るための、新規な包括的合成アプローチを開発した。その合成における主要工程は、シントンである1−トリフルオロアセトアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11a)、1−フタルアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11b)、1−t−ブチルカルバメート−シクロブタン−3−オン−1−カルボン酸メチルエステル(11c)および1−ベンズアミド−シクロブタン−3−オン−1−カルボン酸メチルエステル(11d)の還元を含むものである。ケトンである11a〜dは、リチウムトリイソブチルボランとZnCl2で処理して、63〜80%の収率で直接トランス−(アンチ−)アルコールに転換された。この方法により、それぞれ95:5、97:3、70:30および90:10のトランス:シスアルコールの混合物の12a、12b、12cおよび12dが得られた。12a〜12dをフラッシュクロマトグラフィーにより容易に精製して、所望のトランスアルコール12a〜dを得た。12a〜dのトリフレート前駆体への転換は、米国特許第5,817,776号に記載されている一連の反応で実施することができる。これらの合成アプローチの開発は、シン−およびアンチ−FACBCが、癌の診断および癌治療の管理に有用な画像化剤として有効であることを確認する将来の多施設臨床試験のために、前駆体をPETセンターへ分配するために容易に利用できる供給法を確立するのに必須である。
【0041】
【化7】
上記反応は以下の仕方で実施した。THF(無水)中ケトン(11a、b、c、またはd)の溶液に、アルゴン雰囲気下、室温(rt)で2当量のZnCl2(無水物、THF中に)を加えた。溶液を室温で30分間攪拌し、続いて−78℃で1.5当量のLiBR’3Hを加えた。混合物を−78℃で2時間攪拌し、次いで室温で終夜攪拌した。NH4Cl(1N水溶液、3当量)を加え、混合物を室温で30分間攪拌した。反応物をブラインで洗浄し、水相を酢酸エチルで再抽出した。一緒にした有機相を硫酸ナトリウムで脱水し、濃縮して乾燥させた。生成物を、溶離液として1:1のヘキサンおよび酢酸エチルを用いてシリカゲルで精製した。収率は約63〜80%であった。
【0042】
スキーム3に示す反応工程は、4つのシントン(11a〜11d)を4つのトランス−アルコール、12a〜12dに還元する例を具体的に示すものであるが、この立体選択的な合成工程は、腫瘍画像化に有用なシン−アミノ酸類似体の合成のための様々なトランス−アルコールの合成に適用することができる。以下のスキーム4は本発明のこの態様を示す。
【0043】
【化8】
スキーム5はシン−FACBCの合成のための工程を例示する。
【0044】
【化9】
スキーム6では、本明細書で開示する立体選択的な合成方法を用いて合成できるアミノ酸類似体、[18F]−1−アミノ−4−フルオロ−シクロヘキサン−1−カルボン酸(FACHC)の合成を例示する。
【0045】
【化10】
スキーム7は、本明細書で開示する立体選択的な合成方法で用いる主要シントンであるシン/アンチ−1−アミノ−3−ベンジルオキシシクロブタン−1−カルボン酸20の合成を示す。
【0046】
【化11】
スキーム8は、本明細書で開示する立体選択的な合成方法で用いる主要シクロヘキサノン中間体である1−[N−(t−ブトキシカルボニル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(24)、1−アミノ−4−シクロヘキサノン−1−カルボン酸メチルエステル(25)の合成を示す。
【0047】
【化12】
スキーム9は、本明細書で開示する立体選択的な合成方法で調製されたシン/アンチ−1−[N−置換−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル27a〜dの合成を示す。
【0048】
【化13】
【実施例】
【0049】
以下の説明によって、本発明の好ましい実施形態の例としての合成法を提供する。しかし、当業者は本発明の実施にあたって、出発原料、試薬、溶媒、温度、固体基体、合成方法、精製方法、分析方法、および具体的に例示したもの以外の他の反応条件を、必要以上の実験をすることなく用いることができることを理解されよう。そうした任意の原料や方法の当業界で周知の機能的均等物のすべては、本発明に含まれるものとする。使用される用語および表現は説明のための用語として用いるものであって限定的なものではなく、そうした用語および表現の使用において、示しかつ説明した特徴またはその一部のどんな均等物も排除しようとするものではなく、特許請求した本発明の範囲内で様々な変更が可能であることを理解されたい。したがって、好ましい実施形態および任意選択の特徴によって本発明を具体的に開示してきたが、当業者は、本明細書で開示した考え方の改変および変更を行うことができ、そうした改変形態および変更形態は、添付の特許請求の範囲により定義される本発明の範囲内であることを理解すべきである。
【0050】
(実施例1)
シン−およびアンチ−[18F]1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)の合成(スキーム1、2および5)
本明細書で報告される手順において以下の方法を用いた。[18F]−フルオリドは、11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、Seimens社製サイクロトロンで作製した。すべての溶媒および化学薬品は、分析グレード品をさらに精製することなく使用した。化合物の融点はBuchiSP装置を用いて毛細管で測定した。薄層クロマトグラフィー分析(TLC)は、アルミニウム上にコーティングされたシリカゲルGPF−254(Analtech,Inc.Newark,DEから入手)の250mm厚の層を用いて実施した。カラムクロマトグラフィーは、60〜200メッシュシリカゲル(Sigma−Aldrich,St.Louis,MO)を用いて実施した。赤外スペクトル(IR)はNaClプレートを備えたBeckman18A分光光度計で測定した。プロトン核磁気共鳴スペクトル(1H NMR)はNicolet高分解能装置を用いて300MHzで得た。
【0051】
1−ブロモ−2−ベンジルオキシ−3−ブロモプロパン1の合成:
凝縮器を備えたフラスコ中で、臭化ベンジル(83mL、0.70モル)、エピブロモヒドリン(60mL、0.70モル)および塩化水銀(I)(120mg、0.25ミリモル)からなる混合物を150℃で終夜攪拌した。生成物を、30cmのVigreux凝縮器を通した真空蒸留(110〜115℃、0.5mmHg)により単離して、生成物1(152g、70%)を無色液体として得た。1H NMR (CDCl3) δ3.45 (4H, d, J=5.2), 3.66−3.71 (1H, m) 4.55 (2H, s) 7.19−7.27 (5H, m)。
【0052】
3−ベンジルオキシシクロブタノン2の合成:
シクロブタノン2の調製は、Oguraら(1984年)Bull. Chem. Soc. Jpn. 57;1637〜42頁の報告による手順をもとにした。2.4当量部のn−ブチルリチウム(ヘキサン中に1.6M、243mL)を、400mLのテトラヒドロフラン中に2.4当量のメチルメチルスルフィニルメチルスルフィド(41mL、0.39ミリモル)を含む溶液に−10℃で滴下した。次いで反応混合物を−10℃で2時間攪拌し、続いて−70℃に冷却した。黄色の反応混合物を−70℃に保持し、85mLのテトラヒドロフラン中の1当量のジブロモ種1(50g、0.16ミリモル)を滴下した。反応混合物を終夜かけて室温に加温した。反応混合物をブラインに加え、酢酸エチルで2回抽出した。一緒にした有機層に通常の処理を施して約60mLの暗赤褐色液体を得た。このシン−およびアンチ−ジチオケタールS−オキシド中間体の混合物を、シリカゲルカラムクロマトグラフィー(90gシリカ)により3つに分けて精製した。より極性の小さい不純物をまず3:7酢酸エチル:ヘキサンで溶出させ、続いて、生成物を高純度の酢酸エチルで溶出させた。この方法で合計23.8gの中間体を得た。同じ条件を用いて次に2を合成して24.6gを得た。
【0053】
シン−およびアンチ−ジチオケタールS−オキシド中間体(48.4g、0.18モル)を1200mLのジエチルエーテルに溶解し、68mLの35%過塩素酸で処理した。終夜攪拌した後、反応混合物を重炭酸ナトリウムで中和し、続いて通常の後処理を行った。シリカゲルカラムクロマトグラフィー(15:85酢酸エチル:ヘキサン)で精製してケトン2(23.6g、1からは41%)を橙黄色液体として得た。1H NMR δ3.11−3.29 (4H, m) 4.35−4.42 (1H, m) 4.53 (2H, s) 7.30−7.40 (5H, m)。
【0054】
シス/トランス5−(3−ベンジルオキシシクロブタン)ヒダントイン3の合成:
900mLの水の中の10当量の炭酸アンモニウム(125g、1.3モル)および4当量の塩化アンモニウム(27.8g、0.52モル)の溶液に、900mLのエタノール中の1当量のシクロブタノン2(23.6g、0.13モル)を加えた。室温で30分間攪拌した後、4.5当量部のシアン化カリウム(38g、0.58モル)を加え、反応混合物を60℃で終夜加熱した。溶媒を減圧下で除去し、粗製の黄色固体を約1リットルの水で完全に濯いで、塩を除去した。白色の結晶生成物(16.4g、51%)をシン:アンチ異性体の5:1混合物として得た。単離された主成分異性体を、シリカゲルカラムクロマトグラフィー(2:98メタノール:ジクロロメタン)により得た。この手順を用いて、95gのシリカゲルで1.0gの混合物を精製して500〜600mgの純粋な3を1回の試行で得た。シン−5−(3−ベンジルオキシシクロブタン)ヒダントイン(3):1H NMR (CDCl3) δ2.30−2.35 (2H, m) 2.87−2.92 (2H, m) 4.18−4.25 (1H, m) 4.46 (2H, s) 5.66 (1 H, 広幅s) 7.28−7.38 (5H, m) 7.55 (1H, 広幅s)。 アンチ−5−(3−ベンジルオキシシクロブタン)ヒダントイン(4):1H NMR (CDCl3) δ2.44−2.50 (2H, m) 2.77−2.83 (2H, m) 4.21−4.27 (1H, m) 4.46 (2H, s) 5.82 (1H, 広幅s) 7.29−7.38 (6H, m)。
【0055】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ベンジルオキシシクロブタン−1−カルボン酸5の合成:
密封したステンレス製容器の中で、30mLの3N水酸化ナトリウム中の化合物3(1.35g、5.5ミリモル)の懸濁液を180℃で終夜加熱した。冷却した後、反応混合物を濃塩酸でpH6〜7まで中和した。減圧下で水を除去した後、得られた固体を4×30mLの熱エタノールで抽出した。一緒にしたエタノール抽出物を濃縮し、残留物を50mLの9:1メタノール:トリエチルアミンに溶解した。その溶液に、1.3当量部のジ−tert−ブチルジカーボネート(1.56g)を加え、溶液を室温で終夜攪拌した。溶媒を減圧下で除去し、粗生成物を、80mLの氷冷酢酸エチルと80mLの氷冷0.2N塩酸の混合液中で5分間攪拌した。有機層は保持し、水相を2×80mLの氷冷酢酸エチルで抽出した。一緒にした有機層を3×60mLの水で洗浄し、通常の後処理を行った。N−Boc酸5(1.27g、72%)を、さらに精製することなく次の工程で用いるのに適した白色の固体として得た。1H NMR (CDCl3) δ1.44 (9H, s) 2.21−2.26 (2H, m) 3.02−3.08 (2H, 広幅m) 4.12−4.19 (1H, m) 4.44 (2H, s) 5.18 (1H, 広幅s) 7.27−7.37 (5H, m)。
【0056】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ベンジルオキシシクロブタン−1−カルボン酸メチルエステル6の合成:
ヘキサン(1.4mL)中の1.5当量部の2.0Mトリメチルシリルジアゾメタンを、10mLの1:1メタノール:テトラヒドロフラン中のN−Boc酸5(600mg、1.87ミリモル)の溶液に滴下した。添加による発熱の際、相当なガスが発生した。20分間攪拌した後、反応混合物を減圧下で濃縮し、粗生成物をシリカゲルカラムクロマトグラフィー(2:8酢酸エチル:ヘキサン)で精製した。N−Bocメチルエステル6(0.45g、72%)を白色の結晶性固体として得た。1H NMR (CDCl3) δ1.42 (9H, s) 2.24−2.36 (2H, 広幅m) 2.88− 2.96 (2H, m) 3.75 (3H, s) 4.16−4.23 (1H, m) 4.44 (2H, s) 5.13 (1H, s) 7.27−7.36 (5H, m)。
【0057】
シン/アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ヒドロキシシクロブタン−1−カルボン酸メチルエステル7の合成:
10mLのCH3OH中の6(450mg、1.34ミリモル)の溶液に、アルゴン雰囲気下で200mgの10%Pd/Cを加えた。反応混合物を水素雰囲気下、室温で終夜攪拌した。次いで懸濁液をセライト(Celite)(登録商標)でろ過し、減圧下で濃縮した。シリカゲルカラムクロマトグラフィー(6:4酢酸エチル:ヘキサン)で精製してアルコール7(200mg、61%)を白色の結晶性固体:134〜135℃として得た(128〜130℃、Shoup and Goodman, J Labelled Compd Radiopharm、1999年;42:215〜225頁による報告。1H NMR (CDCl3) δ1.45 (9H, s) 2.54− 2.61 (2H, 広幅m) 2.98−3.04 (2H, m) 3.79 (3H, s) 4.26−4.34 (1H, 広幅m) 5.63 (1H, 広幅s)。 (C11H19NO5) の元素分析: 計算値: C: 53.87 H: 7.81 N: 5.71, 実測値: C: 53.93 H: 8.00 N: 5.71。
【0058】
化合物1−[N−(tert−ブトキシカルボニル)アミノ]−シクロブタン−3−オン−1−カルボン酸メチルエステル11cの合成。
【0059】
4mLのジクロロメタン中の1.1当量部の塩化オキサリル(1.05mL、ジクロロメタン中に2M溶液)に、アルゴン雰囲気下、−50〜−60℃で、1mLのジクロロメタン中の2.2当量のジメチルスルホキシド(290μL)を滴下しながら加えた。この溶液を3分間攪拌し、続いて、2mLジクロロメタンおよび0.8mLのジメチルスルホキシド中に溶解した異性体的に不純な7(458mg、1.9ミリモル)を滴下した。反応混合物を−50〜−60℃で20分間攪拌し、次いで5当量のトリエチルアミン(1.3mL)を加えた。反応混合物を5分間攪拌し、冷却浴を取り外し、溶液をさらに15分間攪拌した。粗生成物をシリカゲルカラムクロマトグラフィー(1:4酢酸エチル:ヘキサン)で精製して11c(456mg、100%の収率)を白色固体:118〜119℃(酢酸エチル/ヘキサン)として得た。1H NMR (CDCl3) δ1.46 (9H, s) 3.49−3.66 (4H, m) 3.83 (3H, s) 5.47 (1H, 広幅s)。 (C11H17NO5) の元素分析: 計算値: C: 54.31 H: 7.04 N: 5.76, 実測値: C: 54.50 H: 6.96 N: 5.61。
【0060】
アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−ヒドロキシシクロブタン−1−カルボン酸メチルエステル12cの合成。
【0061】
1mlのTHF(無水)中のケトン(11c、16.4mg、0.067ミリモル)の溶液に、Ar雰囲気下、室温でZnCl2(18mg、THF中に0.134ミリモル)を加えた。溶液を室温で30分間攪拌し、続いてL−セレクトリド(L−Selectride)(19mg、THF中に0.10ミリモルF)を−78℃で加えた。混合物を−78℃で2時間攪拌し、次いで室温で終夜攪拌した。NH4Cl(1N水溶液、3当量)を加え、混合物を室温で30分間攪拌した。反応物をブラインで洗浄し、水相を酢酸エチルで再抽出した。一緒にした有機相を硫酸ナトリウムで脱水し、濃縮して乾燥させた。溶離液として1:1のヘキサンおよび酢酸エチルを用いて、生成物をシリカゲルで精製した。生成物(12c、16mg、100%)を白色固体として得た。1H NMR (CDCl3) δ1.44 (9H, s) 2.53− 2.63 (4H, 広幅m) 3.77 (3H, s) 4.43−4.50 (1H, 広幅m) 5.02 (1H, 広幅s)。
【0062】
アンチ−1−(N−(tert−ブトキシカルボニル)アミノ)−3−トリフルオロメチルスルホンオキシシクロ−ブタン−1−カルボン酸メチルエステル13の合成。
【0063】
アルゴン雰囲気下で、アルコール9(10mg、0.04ミリモル)を2mLのジクロロメタンに溶解した。氷浴で冷却しながら100μL部のピリジンを加え、続いて4.5当量部のトリフルオロメタンスルホン酸無水物(30μL)を加えた。15分間攪拌した後、溶媒を減圧下室温で除去した。粗生成物をシリカゲルカラムクロマトグラフィー(3:7酢酸エチル:ヘキサン)で精製して標識前駆体13を得た。
【0064】
シン−[18F]1−アミノ−3−フルオロシクロブタン−1−カルボン酸(FACBC)15の合成:
11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、[18F]−フルオリドを生成させた。水を蒸発させ、アセトニトリルを蒸発させてフルオリドを乾燥させた後、保護されたアミノ酸トリフレート13(20mg)をアセトニトリル溶液(1mL)に導入した。密封した容器中で、非担体担持での(NCA)フッ素化反応を、炭酸カリウムとKryptofix(Trademark Aldrich Chemical Co.,Milwaukee,WI)の存在下、85℃で5分間かけて実施した。反応混合物を塩化メチレンで希釈して未反応18Fを除去し、続いてシリカゲルSeppakを通過させて18F標識生成物14を得た。1mLの6N HClで115℃で15分間処理して14を脱保護させ、次いでシン−[18F]FACBC15を含む水溶液を、イオン遅滞樹脂(AG11A8 50〜100メッシュ)に通した。
【0065】
アンチ−[18F]FACBC10の合成:
11MeV陽子を用いた95%濃縮[18O]水での18O(p,n)18F反応により、[18F]−フルオリドを生成させた。水を蒸発させ、アセトニトリルを蒸発させてフルオリドを乾燥させた後、保護されたアミノ酸トリフレートシン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−トリフルオロメタンスルホンオキシシクロブタン−1−カルボン酸メチルエステル(20mg)をアセトニトリル溶液(1mL)に導入した。密封した容器中で、非担体担持での(NCA)フッ素化反応を、炭酸カリウムとKryptofix(Trademark Aldrich Chemical Co.,Milwaukee,WI)の存在下、85℃で5分間かけて実施した。反応混合物を塩化メチレンで希釈して未反応18Fを除去し、続いてシリカゲルSeppakを通過させて、18F標識生成物14を得た。18F標識生成物、シン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−[18F]フルオロシクロブタン−1−カルボン酸メチルエステルを42%E.O.B.の収率で得た。1mLの4N HClで115℃で15分間処理してシン−1−(N−(tert−ブトキシカルボニル)アミノ)−3−[18F]フルオロシクロブタン−1−カルボン酸メチルエステルを脱保護させ、次いで18FACBC13を含む水溶液を、イオン遅滞樹脂(AG11A8 50〜100メッシュ)に通した。合成はE.O.B.の後60分間で、12%(17.5%E.O.B.)の総括放射化学的収率で完了した。詳細については上記のMcConathyら(2003年)を参照されたい。
【0066】
(実施例2)
シン−およびアンチ−1−アミノ−4−ヒドロキシシクロヘキサン−1−カルボン酸エステル(スキーム7−9)の合成
4−エチレンアセタールシクロヘキサノール(16)
0℃の冷却した50mlメタノール中の1,4−シクロヘキサンジオンモノエチレンアセタール(3.41g、21.8ミリモル)の溶液に、水素化ほう素ナトリウム(0.826g、21.8ミリモル)を分割して加えた。反応物をさらに1.5時間攪拌し、続いて1N HClを加えてpH7にした。混合物を酢酸エチルとブラインに分配させた。沈殿物が形成され始めるまで水層を濃縮し、この水層を酢酸エチルで2回抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。この粗アルコール(3.28g、95.2%)をさらに精製することなく用いた。1H NMR (CDCl3) δ: 1.54−1.87 (8H, m, 4×−CH2−), 3.77 (1H, m, −CH−), 3.91 (4H, t, 2×O−CH2−)。
【0067】
1−エチレンアセタール−4−ベンジルオキシ−シクロヘキサン(17)
0℃の15mlのTHF中の水素化ナトリウム(410mg、17.1ミリモル)の懸濁液に、5mlのTHF中の4−エチレンアセタールシクロヘキサノール(1)(1.36g、8.61ミリモル)を加えた。反応物を0℃で1.5時間攪拌し、臭化ベンジル(1.75g、10.2ミリモル)を加えた。反応物を室温で終夜攪拌した。反応物を塩化アンモニウム(飽和)でクエンチした。生成物を酢酸エチルで抽出し、有機相をブラインで洗浄し、硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物をシリカゲルクロマトグラフィー(ヘキサン中の20%酢酸エチル)で精製して2.17g(100%)のベンジルエーテルを得た。1H NMR (CDCl3) δ: 1.51−1.88 (8H, m, 4×−CH2−), 3.51 (1H, m, −CH−), 3.91 (4H, t, 2×O−CH2−), 4.52 (2H, s, Ph−CH2−), 7.25−7.34 (5H, m, Ph−H)。
【0068】
4−ベンジルオキシ−シクロヘキサノン(18)
50mlのTHF中の1−エチレンアセタール−4−ベンジルオキシ−シクロヘキサン(17)(3.13g、12.6ミリモル)の溶液に、塩酸水溶液(1N、30ml)を室温で加えた。反応物を終夜攪拌し、重炭酸ナトリウム(飽和)で中和した。生成物を酢酸エチルで抽出し、有機相をブラインで洗浄し、硫酸ナトリウムで脱水し、ろ過し、濃縮させた。シリカゲルクロマトグラフィー(ヘキサン中の20%酢酸エチル)で精製して2.45g(95.2%)の標記のケトンを得た。1H NMR (CDCl3) δ: 1.95−2.62 (8H, m, 4×−CH2−), 3.82 (1H, m, −CH−), 4.59 (2H, s, Ph−CH2−), 7.28−7.36 (5H, m, Ph−H)。
【0069】
シン/アンチ−6−(4−ベンジルオキシシクロヘキサン)ヒダントイン(19)
100mlのエタノール中の4−ベンジルオキシ−シクロヘキサノン(18)(2.45g、12ミリモル)の溶液に、100mlの水の中の炭酸アンモニウム(4.6g、48ミリモル)と塩化アンモニウム(1.28g、24ミリモル)の溶液を加えた。混合物を室温で15分間攪拌し、次いでシアン化カリウム(940mg、14.4ミリモル)を加えた。反応物を室温で終夜攪拌した。溶媒を減圧下で除去した。得られた固体を水で繰り返し洗浄して、ろ取した。このヒダントイン(hytantoin)のシン/アンチ粗混合物(3.02g、91.8%)を、さらに精製することなく使用した。1H NMR (CD3OD) δ: 1.58−2.15 (8H, m, 4×−CH2−), 3.48, 3.66 (1H, m, −CH−), 4.52, 4.56 (2H, s, Ph−CH2−), 7.25−7.33 (5H, m, Ph−H)。
【0070】
シン/アンチ−1−アミノ−4−ベンジルオキシシクロヘキサン−1−カルボン酸(20)
シン/アンチヒダントイン(19)(2.72g、9.93ミリモル)を30mlの3N NaOH中に懸濁させ、鋼製円筒容器中に密封し、これを120℃で1日加熱した。室温に冷却した後、濃塩酸溶液を加えて反応液をpH7にした。減圧下で濃縮し、乾燥させてシン/アンチアミノ酸の粗生成物を得た。この生成物を、さらに精製することなく使用した。
【0071】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸(21)
50mlの9:1MeOH/トリエチルアミン中の上記調製によるシン/アンチ−1−アミノ−4−ベンジルオキシシクロヘキサン−1−カルボン酸(20)の懸濁液にジ−t−ブチルジカーボネート(3.25g、14.9ミリモル)を加えた。反応混合物を室温で24時間攪拌した。溶媒を減圧下で除去した。得られた残留物を50mlの氷冷した1:1水/酢酸エチルに溶解した。3N HClで溶液のpHを2〜3に調節した。有機層は保持し、水層を塩化ナトリウムで飽和させて、酢酸エチル(3×25ml)で抽出した。一緒にした有機層を硫酸マグネシウムで脱水し、溶媒を減圧下で除去した。この生成物(3.46g、100%)を、さらに精製することなく使用した。1H NMR (CD3OD) δ: 1.41 [9H, s, −C(CH3)3 ], 1.57−2.25 (8H, m, 4×−CH2−), 3.40, 3.58 (1H, m, −CH−), 4.49, 4.54 (2H, s, Ph−CH2−), 4.84 (1H, br, NH), 7.25−7.33 (5H, m, Ph−H)。
【0072】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸メチルエステル(22)
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ベンジルオキシシクロヘキサン−1−カルボン酸(21)(1.14g、3.26ミリモル)を、40mlベンゼンおよび10mlメタノール中に溶解し、トリメチルシリルジアゾメタン(558mg、4.88ミリモル、2.5mlのヘキサン中、2M溶液)を室温で加えた。反応物を室温で30分間攪拌し、次いで溶媒を減圧下で除去した。ヘキサン中の20%酢酸エチルを用いてフラッシュクロマトグラフィーで精製して1.03g(87.2%)の純粋な生成物を油状物として得た。1H NMR (CD3OD) δ: 1.408, 1.413 [9H, s, −C(CH3)3 ], 1.5−2.3 (8H, m, 4×−CH2−), 3.40, 3.58 (1H, m, −CH−), 3.69, 3.71 (3H, s, COCH3), 4.49, 4.54 (2H, s, Ph−CH2−), 4.77, 4.79 (1H, br, NH), 7.25−7.33 (5H, m, Ph−H)。
【0073】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(23)
50mlのエタノール中のベンジルエーテル(22)(947mg、2.6ミリモル)と活性炭担持の10%パラジウム(142mg)の懸濁液を水素雰囲気下で終夜攪拌した。反応混合物をセライト(登録商標)でろ過し、ろ液を減圧下で濃縮した。シリカゲルカラムクロマトグラフィー(ヘキサン中に50%酢酸エチル)で精製して(23)(701mg、98.4%)を得た。アンチ−対シン−の比は34:66であった。1H NMR (CD3OD) δ: 1.411, 1.416 [9H, s, −C(CH3)3 ], 1.53−2.25 (8H, m, 4×−CH2−), 3.65, 3.91 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.77 (1H, br, NH)。
【0074】
1−[N−(t−ブトキシカルボニル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(24)
アルゴン雰囲気下で、テトラプロピルアンモニウムペルルテナート(26mg、0.075ミリモル)を一括して、ジクロロメタン中の15mlの10%アセトニトリル中のアルコール(23)(410mg、1.5ミリモル)、N−メチル−モルホリンN−オキシド(264mg、2.25ミリモル)および750mg4Aモレキュラーシーブの攪拌混合物に加えた。反応物を室温で1時間攪拌し、次いで溶媒を減圧下で除去した。得られた残留物をジクロロメタン中にとり、シリカゲルカラムクロマトグラフィー(ヘキサン中に30%酢酸エチル)で精製した。ケトン(24)、372mg(91.6%)を白色固体として得た。1H NMR (CD3OD) δ: 1.43 [9H, s, −C(CH3)3 ], 2.32−2.42 (8H, m, 4×−CH2−), 3.74 (3H, s, COCH3), 5.04 (1H, br, NH)。
【0075】
1−アミノ−4−シクロヘキサノン−1−カルボン酸メチルエステル(25)
5mlジクロロメタン中のケトン(24)(325mg、1.2ミリモル)の溶液に、トリフルオロ酢酸(1.37g、12ミリモル)を加えた。反応物を室温で終夜攪拌した。溶媒と試薬を減圧下で除去した。得られた白色の固体を、さらに精製することなく使用した。
【0076】
1−[N−(フタロイル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26b)
10mlトルエン中のアミン(25)(80mg、0.47ミリモル)およびトリエチルアミン(476mg、4.7ミリモル)の懸濁液に、無水フタル酸(77mg、0.52ミリモル)を加えた。混合物を120℃で5時間還流させた。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:4酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26b)(37.6mg、26.6%、2段階で)を白色固体として得た。1H NMR (CD3OD) δ: 2.54−3.14 (8H, m, 4×−CH2−), 3.77 (3H, s, COCH3), 7.73−7.85 (4H, m, Ph−H)。
【0077】
1−[N−(トリフルオロアセチル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26c)
−10℃に冷却した1mlジクロロメタン中のアミン(25)(14mg、0.082ミリモル)およびトリエチルアミン(166mg、1.64ミリモル)の懸濁液に、トリフルオロ酢酸無水物(86mg、0.41ミリモル)を加えた。混合物を室温に加温し、終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:2酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26c)(17.5mg、79.9%)を透明油状物として得た。1H NMR (CD3OD) δ: 2.44−2.56 (8H, m, 4×−CH2−), 3.79 (3H, s, COCH3), 6.86 (1H, br, NH)。
【0078】
1−[N−(ベンゾイル)アミノ]−4−シクロヘキサノン−1−カルボン酸メチルエステル(26d)
0℃に冷却した3mlジクロロメタン中のアミン(25)(50mg、0.29ミリモル)およびピリジン(934mg、11.8ミリモル)の懸濁液に、塩化ベンゾイル(62mg、0.44ミリモル)を加えた。混合物を室温に加温し、終夜攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:2酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してケトン(26d)(22mg、27.6%)を白色固体として得た。1H NMR (CD3OD) δ: 2.46−2.58 (8H, m, 4×−CH2−), 3.81 (3H, s, COCH3), 6.82 (1H, br, NH), 7.48−8.13 (5H, m, Ph−H)。
【0079】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27a)
1mlのTHF中のケトン(26a)(18mg、0.066ミリモル)の溶液に、室温で塩化亜鉛(18mg、0.13ミリモル、264μlのTHF中0.5M溶液)を加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(19mg、0.10ミリモル、100μlのTHF中の1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27a)(12.7mg、70.5%)を透明油状物として得た。アンチ−とシン−の比は67:33であった。1H NMR (CD3OD) δ: 1.411, 1.415 [9H, s, −C(CH3)3 ], 1.55−2.26 (8H, m, 4×−CH2−), 3.65, 3.92 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.70 (1H, br, NH)。
【0080】
シン/アンチ−1−[N−(t−ブトキシカルボニル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27a)(塩化亜鉛は存在せず)
−78℃に冷却した1mlのTHF中のケトン(24)(21.7mg、0.08ミリモル)の溶液にL−セレクトリド(22.8mg、0.12ミリモル、120μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27a)(3mg、13.7%)を透明油状物として得た。アンチ−とシン−の比は11:89であった。1H NMR (CD3OD) δ: 1.415, 1.420 [9H, s, −C(CH3)3 ], 1.53−2.25 (8H, m, 4×−CH2−), 3.65 (1H, m, −CH−), 3.70, 3.71 (3H, s, COCH3), 4.70 (1H, br, NH)。
【0081】
シン/アンチ−1−[N−(フタロイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27b)
1mlのTHF中のケトン(26b)(20mg、0.066ミリモル)の溶液に塩化亜鉛(18mg、0.13ミリモル、260μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(19mg、0.10ミリモル、100μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27b)(13.2mg、66%)を透明油状物として得た。アンチ−とシン−の比は52:48であった。1H NMR (CD3OD) δ: 1.60−2.01 (8H, m, 4×−CH2−), 3.70, 3.75 (3H, s, COCH3), 3.86 (1H, m, −CH−), 7.69−7.82 (4H, m, Ph−H)。
【0082】
シン/アンチ−1−[N−(フタロイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27b)(塩化亜鉛は存在せず)
−78℃に冷却した1mlのTHF中のケトン(26b)(18mg、0.059ミリモル)の溶液に、L−セレクトリド(17mg、0.09ミリモル、90μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27b)(13.2mg、66%)を透明油状物として得た。アンチ−とシン−の比は52:48であった。
【0083】
シン/アンチ−1−[N−(トリフルオロアセチル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27c)
1mlのTHF中のケトン(26c)(17mg、0.064ミリモル)の溶液に、塩化亜鉛(17mg、0.13ミリモル、256μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(18mg、0.096ミリモル、96μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた。粗生成物を、1:1酢酸エチルおよびヘキサンを用いてフラッシュクロマトグラフィーで精製してアルコール(27c)(13.5mg、78.4%)を透明油状物として得た。アンチ−とシン−の比は66:34であった。1H NMR (CD3OD) δ: 1.67−2.37 (8H, m, 4×−CH2−), 3.72, 3.75 (3H, s, COCH3), 3.97 (1H, m, −CH−), 6.43 (1H, br, NH)。
【0084】
シン/アンチ−1−[N−(ベンゾイル)アミノ]−4−ヒドロキシシクロヘキサン−1−カルボン酸メチルエステル(27d)
1mlのTHF中のケトン(26d)(22mg、0.08ミリモル)の溶液に、塩化亜鉛(22mg、0.16ミリモル、320μlのTHF中0.5M溶液)を室温で加え、混合物を30分間攪拌した。反応物を−78℃に冷却し、L−セレクトリド(23mg、0.12ミリモル、120μlのTHF中1M溶液)を加えた。混合物を−78℃で2時間攪拌し、室温で終夜攪拌した。数滴の1N塩化アンモニウムを加え、30分間攪拌した。反応物をブラインで洗浄し、水層を酢酸エチルで抽出した。一緒にした有機層を硫酸ナトリウムで脱水し、ろ過し、濃縮させた(生成物を検出することはできなかった)。
【0085】
(実施例3)
インビトロおよびインビボでのアミノ酸取り込みのアッセイ
最初に腫瘍細胞を、加湿インキュベーター(37℃、5%CO2/95%空気)の中で、ダルベッコ変法イーグル培地(DMEM)を入れたT型フラスコ中で単層として増殖させた。増殖培地を10%ウシ胎仔血清および抗生物質(10,000単位/mlペニシリンおよび10mg/mlストレプトマイシン)で補充した。増殖培地は週に3回取り換え、細胞が1週間で培養密度に達するように細胞を継代させた。
【0086】
単層のコンフルエントに達したら、細胞を以下の方法で実験用に調製した。増殖培地をT型フラスコから取り出し、単層細胞を1×トリプシン:EDTAに約1分間曝して、細胞とフラスコとの間のタンパク質の結合を弱めた。次いで、フラスコをたたいて、細胞を離脱させた。補充した培地を加えてトリプシンのタンパク分解作用を阻害させ、細胞を18Ga針で吸引し、次いでそれらを単分散させた。血球計算板を用いて顕微鏡下で細胞の試料をカウントし、トリパンブルー染色により生存/死亡割合を推定した(>98%の生存率)。細胞の残りを遠心分離管に入れ、75×gで5分間遠心分離し、上澄みを除去した。次いで細胞をアミノ酸/無血清DMEM塩中に再懸濁した。
【0087】
この試験では、約4.55×105細胞を、12×75mmガラスバイアル中、インキュベーター条件下で、3mlのアミノ酸の存在しない培地±輸送体阻害剤(10mM)中の[18F]10(アンチ−FACBC)かまたは[18F]15(シン−FACBC、5μCi)に30分間曝した。各アッセイ条件は2通り実施した。インキュベーションした後、細胞を遠心分離に2回かけ(75×gで5分間)、氷冷したアミノ酸/無血清DMEM塩で濯いで上澄みの残留活性を除去した。バイアルをPackard Cobra II自動γ線計測器中に置き、生のカウント数を減衰補正し、細胞数当たりの活性を測定した。これらの試験によるデータ(対照に対する取り込み%で表わした)を、Excelを用いてグラフ化し、ワンウェイANOVA(GraphPad Prismソフトウェアパッケージ)を用いて分析してグループ間の統計的比較を行った。
【0088】
[18F]10と[18F]15が支配的にL−型アミノ酸輸送系を介して細胞中に入るという仮定を吟味するために、アミノ酸輸送の十分説明した2つの阻害剤の存在下および非存在下で、培養した9L神経膠肉腫と種々のヒト癌細胞株を用いてアミノ酸取り込みアッセイを実施した。N−MeAIBはA型アミノ酸輸送系の選択的、競合的阻害剤であり、他方、2−アミノ−ビシクロ[2.2.1]ヘプラン−2−カルボン酸(BCH)はナトリウム非依存性L型輸送系のための阻害剤として通常用いられるが、この化合物も、ナトリウム依存性B0,+およびB0輸送系によって、アミノ酸取り込みを競合的に阻害する。A型およびL型アミノ酸輸送系は、腫瘍画像化のために用いられる放射性標識アミノ酸のインビボでの取り込みにかかわっていた。
【0089】
阻害剤が存在しないと、[18F]10と[18F]15はどちらも、9L神経膠肉腫細胞および種々のヒト癌細胞株中で同じようなレベルの取り込みを示し、インキュベーション後30分間で、それぞれ、細胞百万個当たり初期線量の0.43%および0.50%の細胞内蓄積であった。阻害剤の効果を比較しやすくするために、データを、表1に示すように、対照条件(阻害剤なし)に対する取り込みパーセントとして表わした。[18F]10および[18F]15の場合、BCHは、対照に対して活性取り込みの>50%を遮断した。対照と比較したBCHによる[18F]10および[18F]15の取り込みの減少は、統計的に有意であった(ワンウェイANOVAにより、それぞれp<0.05、p<0.01)。これらの阻害試験は、[18F]10および[18F]15が、BCHの存在下での両化合物の取り込みの阻害をもとにして試験して、癌細胞におけるL型アミノ酸輸送系のための基質であることを示している。
【0090】
【表1】
腫瘍誘発および動物の処置:
すべての動物実験は人道的条件下で実施されており、Emory Universityの動物管理使用委員会(Institutional Animal Use and Care Committee(IUCAC))による承認を得たものである。ラット9L神経膠肉腫細胞を、雄性フィッシャーラットの脳の中に埋め込んだ。簡単に述べると、頭部定位ホルダーに固定した麻酔したラットに、4×104ラット9L神経膠肉腫細胞(1×107/mL)の懸濁液を、正中線の3mm右側、プレグマの1mm前方に、頭蓋外板から5mmの深度の位置で注射した。注射は2分間にわたって実施し、腫瘍細胞の逆流を最小限に抑えるように針は1分間にわたって引き抜いた。潜り穴と頭皮切り口を閉じ、この処置から回復させた後、動物を元のコロニーに戻した。頭蓋内腫瘍は発達し、腫瘍担持ラットにおける体重減少、感情鈍麻および猫背の姿勢が認められ、この動物を埋め込み後17〜19日間で使用した。解剖すると、腫瘍細胞を埋め込まれた30匹の動物のうち、25匹について裸眼で視認できる腫瘍が進行しており、これらを試験に用いた。図1〜3はその試験結果を示す。
【0091】
齧歯類生体内分布試験:
16匹の健常な雄性フィッシャー344ラット(200〜250g)に、0.3mLの滅菌水中の約85μCiの[18F]10かまたは[18F]15を静脈注射した後、放射能の組織分布を測定した。実験前に、動物に餌と水を適宜与えた。頭蓋内の腫瘤(mass)の存在下での麻酔状態に伴う死亡を回避するために、RTV−190齧歯類拘束装置(Braintree Scientific)を用いて、尾静脈注射を覚醒動物で実施した。線量を注射後、5分間、30分間、60分間および120分間で4匹からなるラットのグループを殺した。動物を解剖し、選択した組織の重量を量り、Packard Cobra II自動γ線計測器で、線量標準と合わせてカウントした。生のカウント数を減衰補正し、カウント数を、グラム組織当たりの合計注射線量(ID/g%)割合として正規化した。切除し、各時間点で比較のために用いた、腫瘍組織と腫瘍の反対側の対応する脳の領域における活性の取り込みの比較を、ワンウェイANOVA(GraphPad Prismソフトウェアパッケージ)を用いて分析した。以下の図1〜3のこれらの試験結果を示す。
【0092】
図1〜3に示すように、頭蓋内に9L神経膠肉腫細胞を埋め込まれたラットでは、腫瘍組織中での放射能の保持は、[18F]10および[18F]15の静脈注射後60分で高く、腫瘍の反対側の脳組織での放射能の取り込みは低く留まった(<0.3%線量/g)。[18F]10についての腫瘍取り込みと正常な脳の取り込みの比は60分および120分間で6.5:1であり、他方、[18F]15については、同一時間点でその比は5.3:1であった。これらの結果は、アンチ−[18F]FACBC、[18F]10と同様にシン−[18F]FACBC[18F]15が、脳腫瘍を画像化するための優れた候補品であることを実証している。
【0093】
本発明の方法により作製された化合物は、溶媒和、特に水和したものであってもよい。水和は、化合物、または化合物を含む組成物の製造の際に起こるか、あるいは水和は化合物の吸湿により経時的に起こる。さらに、本発明の化合物は、水、エタノール等などの薬学的に受容可能な溶媒と、溶媒和していない形態でも溶媒和した形態でも存在することができる。一般に、本発明のためには、溶媒和した形態は溶媒和していない形態と同等であると考えられる。
【0094】
本発明の化合物を画像化剤として用いる場合、それらを123I、131I、18F、76Br、および77Brなどの適切な放射性ハロゲンアイソトープで標識しなければならない。本発明の放射性ハロゲン化化合物は、腫瘍を画像化させるのに必要な材料を備えたキットでも容易に提供することができる。例えば、最終生成物を、使用する場所またはその時間に作製することができるように、キットは、画像化するのに使用できるようにした適切なアイソトープ(例えば、18F)で標識された最終生成物または中間体化合物と、試薬(例えば、溶媒、脱保護剤)と一緒にした標識(例えば、K[18F]F)を含むことができる。
【0095】
腫瘍画像化方法の第1の工程では、標識した本発明の化合物を、検出可能な量で組織または患者に導入する。化合物は一般に医薬組成物の一部であり、当業者に周知の方法により組織または患者に投与される。例えば、化合物は、経口、経直腸、非経口(静脈内、筋肉内または皮下)、嚢内、膣内、腹腔内、膀胱内、局所(粉剤、軟膏またはドロップ)、または頬側もしくは経鼻噴霧のいずれかで投与することができる。
【0096】
本発明の画像検査法では、標識化合物を、検出可能な量で患者に導入し、化合物が腫瘍の組織または細胞と会合するのに十分な時間を経た後、その標識化合物を患者の内部で非侵襲的に検出する。本発明の他の実施形態では、標識した化合物を患者に導入し、化合物が腫瘍組織と会合するのに十分な時間を与え、次いで患者からの組織試料を取り出し、患者からは別個に、その組織中の標識化合物を検出する。あるいは、組織試料を患者から取り出し、標識した本発明の化合物を組織試料中に導入する。化合物が腫瘍組織と結合するのに十分な時間を経た後、化合物を検出する。「組織」という用語は、患者の身体の一部を意味する。組織の例には、脳、心臓、肝臓、血管および動脈が含まれる。検出可能な量は、選択した検出方法で検出するのに必要な標識化合物の量である。検出するために患者の中に導入すべき標識化合物の量は、当業者によって容易に決定される。例えば、選択した検出方法で化合物が検出されるまで、標識化合物の量を増加させて患者に与えることができる。化合物が検出されるように、化合物中に標識を導入する。
【0097】
標識化合物の患者への投与は全身的投与経路でも局部的投与経路でもよい。例えば、標識化合物は、身体全体に送達されるように患者に投与することができる。あるいは、標識化合物を、対象の特定の器官または組織に投与することができる。
【0098】
化合物が腫瘍と会合する十分な時間量を決定することは、当業者に周知である。必要な時間量は、検出可能量の標識した本発明の化合物を患者に導入し、次いで、投与後異なった時間で標識化合物を検出することによって容易に決定することができる。
【0099】
標識化合物を検出するための様々な方法は、当業者に周知である。例えば、放射性標識化合物を検出するために、磁気共鳴映像法(MRI)、陽電子射出断層撮影法(PET)または単光子射出コンピュータ断層撮影法(SPECT)を用いることができる。本発明の化合物を腫瘍画像化剤として用いる場合に、PETおよびSPECTが好ましい。化合物に導入される標識は所望の検出方法に依存することになる。例えば、検出方法としてPETを選択した場合、化合物は、11Cまたは18Fなどの陽電子放出原子を有していなければならない。
【0100】
放射性診断薬は、信頼性のある診断を確実にする十分な放射能と放射能濃度を有していなければならない。例えば、放射性金属がテクネチウム−99mである場合、通常投与の際に約0.5〜5.0ml中に0.1〜50mCiの量を含むことができる。式の化合物の量は、放射性金属と安定したキレート化合物を形成するのに十分な量にすることができる。
【0101】
放射性診断薬としての本発明の化合物は十分に安定であり、したがって、そのまますぐに投与してもよく、また使用時まで貯蔵することもできる。望むなら、放射性診断薬は、pH調製剤(例えば、酸、塩基、緩衝剤)、安定剤(例えば、アスコルビン酸)または等張剤(例えば、塩化ナトリウム)などの任意の添加剤を含むことができる。腫瘍の画像化は、本明細書の化合物を用いて定量的に実施し、それによって所与の腫瘍のためのその治療薬の効能を評価することもできる。
【0102】
画像化に好ましい化合物には、123I、124I、125I、131I、18F、76Br、77Brまたは11Cなどの放射性アイソトープが含まれる。
【0103】
本明細書で説明する合成スキームは、本発明の好ましい実施形態の例としての合成法を示す。しかし、当業者は本発明の実施にあたって、出発原料、試薬、溶媒、温度、固体基体、合成方法、精製方法、分析方法、および具体的に例示したもの以外の他の反応条件を、必要以上の実験をすることなく、用いることができることを理解されよう。そうした任意の原料や方法の当業界で周知の機能的均等物のすべては、本発明に含まれるものとする。使用される用語および表現は説明のための用語として用いるものであって限定的なものではなく、そうした用語および表現の使用において、示しかつ説明した特徴またはその一部のどんな均等物も排除しようとするものではなく、特許請求した本発明の範囲内で様々な変更が可能であることを理解されたい。したがって、好ましい実施形態および任意選択の特徴によって本発明を具体的に開示してきたが、当業者は、本明細書で開示した考え方の改変および変更を行うことができ、そうした改変形態および変更形態は、添付の特許請求の範囲により定義される本発明の範囲内であることを理解すべきである。
【0104】
本明細書で、ある置換基の群が開示される場合、群メンバーの任意の異性体および鏡像異性体を含む、その群およびすべての下位群のすべての個別メンバーが別々に開示されることを理解されたい。マーカシュ群または他のグルーピングを本明細書で用いる場合、その群のすべての個別のメンバーおよびその群に可能なすべての組合せおよび下位組合せは、個別にその開示に含まれるものとする。例えば式や化学名において、化合物が、本明細書で化合物の具体的な異性体または鏡像異性体が指定されずに記述される場合、その記述は、個別にまたは任意の組合せで、記述された化合物の各異性体および鏡像異性体を含むものとする。さらに、別段の指定のない限り、本発明で開示の化合物のすべての同位変異体は本開示に包含されるものとする。例えば、開示した分子中の任意の1個または複数の水素を、重水素または三重水素で置き換えることができることを理解されよう。分子の同位変異体は、分子のアッセイ、および分子またはその使用に関連する化学的または生物学的研究における標準品として一般に有用である。同一化合物を異なって称することができることが当業者に周知である場合、化合物の具体的な名称は例示的なものであるものとする。
【0105】
本明細書で開示の分子の多くは1つまたは複数のイオン性基[陽子をそこから取り外すことができる基(例えば、−COOH)かまたはそれに加えることができる基(例えば、アミン)、あるいは四級化できる基(例えば、アミン)]を含む。そうした分子およびその塩の可能なすべてのイオン形態は、本明細書の開示に個別に包含されるものとする。本明細書の化合物の塩に関しては、当業者は所与の用途のための本発明の塩の調製に適した使用可能な広範囲の対イオンの中から選択することができる。
【0106】
別段の指定のない限り、本明細書で記述するかまたは例示した成分のすべての処方または組合せを、本発明を実施するために用いることができる。
【0107】
明細書において、ある範囲、例えば温度の範囲、時間の範囲、純度の範囲または組成物もしくは濃度の範囲が与えられている場合、すべての中間範囲および下位範囲、ならびに所与の範囲に含まれるすべての個別の値は本開示に包含されるものとする。
【0108】
本明細書で言及されるすべての特許および出版物は、本発明が関係する当業界の技術者の技術レベルを示すものである。出願日時点での最新技術を示すために、本明細書で引用した文献のその全体を参照により本明細書に組み込み、また、必要なら、従来技術の範囲内である特定の実施形態を排除するために、この情報を本明細書で用いることができるものとする。例えば、化合物が特許請求される場合、本明細書で引用する文献に使用可能な開示が提供されている化合物を含む出願者の発明に先行する技術において既知であり使用可能な化合物は、本明細書の特許請求の範囲の組成物には含まれないものと理解されたい。
【0109】
本明細書で用いる、「含む(comprising)」は、「含む(including)」、「含む(containing)」または「ことを特徴とする(characterized by)」と同義的なものであり、かつ包含的である、すなわち幅広く解釈できる(open−ended)ものであり、他の言及されていない要素または方法の工程を排除するものではない。本明細書で用いる「からなる(consisting of)」は、特許請求の範囲の要素で指定されていないどの要素、工程または成分も排除する。本明細書で用いる「本質的にからなる(consisting essentially of)」は、特許請求の範囲の基本的特徴および新規な特徴に実質的に影響を及ぼさない材料または工程を排除しない。本明細書のそれぞれ場合において、「含む(comprising)」、「本質的にからなる」および「からなる」という用語のいずれも、他の2つのうちのいずれかで置き換えることができる。本明細書で例示的に説明した本発明は、本明細書で具体的に開示されていない要素や限定がなくても適切に実施することができる。
【0110】
本明細書で引用した文献を、本出願の開示との不整合がない範囲で参照により本明細書に組み込む。特に、本発明を用いて作製できるアミノ酸類似体の例およびその詳細な合成方法を提供するために、米国特許第5,808,146号、同第5,817,776号およびWO03/093412が本明細書で引用されている。それらを参照により本明細書に組み込む。出発原料、追加の出発原料、追加の試薬、追加の合成方法、追加の分析方法および追加の本発明の使用の供給源に関する詳細を提供するために、本明細書で提供したいくつかの文献を参照により本明細書に組み込む。
【図面の簡単な説明】
【0111】
【図1】図1は、9L腫瘍におけるインビボでの化合物の取り込みを示す。結果を、注射後60分での対照に対する取り込み%で表わす。詳細は実施例2を参照されたい。
【図2】図2は、注射後60分の対側正常脳におけるインビボでの取り込みを示す。
【図3】図3は、注射後60分間のインビボでの腫瘍における化合物の取り込み対正常細胞における化合物の取り込みの比を示す。その比は図1および図2のパーセント値から得た。
【特許請求の範囲】
【請求項1】
実質的に純粋な式IIのシン−アミノ酸類似体または薬学的に受容可能なその塩の合成方法であって、式IIは、
【化1】
であり、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4〜R5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルケニル、アルキニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択され;R7はハロゲン、ハロアルキル、ハロアルケニル、ハロアルキニル、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリールおよびハロヘテロアリール、そのTc−99mおよびReキレートからなる群から選択され、ここで、ハロまたはハロゲンはF、Cl、Br、I、At、F−18、I−123、I−124およびBr−76からなる群から選択され、
該方法は、ケトンを式Iのトランス−アルコールへと転換させる工程および該トランス−アルコールを式IIのシン−アミノ酸類似体へと転換させる工程を包含し、ここで式Iは、
【化2】
であり、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4およびR5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択される、方法。
【請求項2】
R4およびR5が独立に、H、アルキル、シクロアルキル、アシル、アリール、ヘテロアリール、アルキニルおよびアルケニルからなる群から選択され;R7がハロゲン、ハロアルキルC1〜C6、ハロアルケニルC1〜C6、ハロアルキニルC1〜C6、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリールおよびハロヘテロアリールからなる群から選択され、ここで、R7中のハロまたはハロゲンは18Fまたは123Iのいずれかである、請求項1に記載の方法。
【請求項3】
R1、R2およびR3が独立に、水素、アルキルC1〜C6、ハロアルキルC1〜C6、アルケニルC1〜C6、ハロアルケニルC1〜C6、アルキニルC1〜C6およびハロアルキニルC1〜C6からなる群から選択される、請求項2に記載の方法。
【請求項4】
前記アミノ酸類似体中のYおよびZがCH2である、請求項3に記載の方法。
【請求項5】
R1、R2およびR3が水素またはアルキルC1〜C4である、請求項4に記載の方法。
【請求項6】
R7が18F、18F−アルキルC1〜C4、123Iおよび123I−アルキルC1〜C4からなる群から選択される、請求項1または5に記載の方法。
【請求項7】
前記アミノ酸類似体がシン−3−[18F]FACBCである、請求項6に記載の方法。
【請求項8】
前記アミノ酸類似体がシン−3−[123I]IACBCである、請求項6に記載の方法。
【請求項9】
前記アミノ酸類似体がシン−3−[18F]FMACBCである、請求項6に記載の方法。
【請求項10】
前記アミノ酸類似体がシン−[18F]FACHCである、請求項6に記載の方法。
【請求項11】
実質的に純粋な次式の化合物
【化3】
であって、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4〜R5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択される、化合物。
【請求項12】
R1、R2およびR3が独立に、H、アルキルC1〜C6、ハロアルキルC1〜C6、アルケニルC1〜C6、アルキニルC1〜C6、ハロアルケニルC1〜C6およびハロアルキニルC1〜C6からなる群から選択され、R4およびR5が独立に、水素、アルキルC1〜C6、アリール、ヘテロアリール、アルキニルC1〜C6およびアルケニルC1〜C6からなる群から選択される、請求項11に記載の化合物。
【請求項13】
R1、R2およびR3が水素であり、YおよびZがCH2である、請求項12に記載の化合物。
【請求項14】
R1、R2およびR3が水素であり、YおよびZがC2H4である、請求項12に記載の方法。
【請求項15】
請求項1に記載の方法によって作製される実質的に純粋なシン−アミノ酸類似体。
【請求項16】
前記類似体がシン−3−[18F]FACBCである、請求項15に記載のアミノ酸類似体。
【請求項17】
請求項15に記載のシン−アミノ酸類似体と生理学的に受容可能な担体とを含む、腫瘍を画像化するための薬学的組成物。
【請求項18】
前記アミノ酸類似体がシン−3−[18F]FACBCである、請求項17に記載の組成物。
【請求項19】
陽電子射出断層撮影法または単光子射出コンピュータ断層撮影法による腫瘍の画像化方法であって、
a)腫瘍を有している疑いのある対象に画像生成量の請求項1に記載の標識化合物を投与する工程;
b)該標識化合物が該腫瘍と会合するのに十分な時間を与える工程;および
c)PETまたはSPECTによって、該対象における該標識化合物の分布を測定する工程
を包含する、方法。
【請求項20】
前記標識化合物がシン−3−[18F]FACBCである、請求項19に記載の方法。
【請求項21】
請求項11に記載の化合物と、該化合物をシン−3−[18F]FACBCに転換させるのに必要な試薬とを含む、実質的に純粋なシン−3−[18F]FACBCを合成するためのキット。
【請求項1】
実質的に純粋な式IIのシン−アミノ酸類似体または薬学的に受容可能なその塩の合成方法であって、式IIは、
【化1】
であり、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4〜R5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルケニル、アルキニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択され;R7はハロゲン、ハロアルキル、ハロアルケニル、ハロアルキニル、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリールおよびハロヘテロアリール、そのTc−99mおよびReキレートからなる群から選択され、ここで、ハロまたはハロゲンはF、Cl、Br、I、At、F−18、I−123、I−124およびBr−76からなる群から選択され、
該方法は、ケトンを式Iのトランス−アルコールへと転換させる工程および該トランス−アルコールを式IIのシン−アミノ酸類似体へと転換させる工程を包含し、ここで式Iは、
【化2】
であり、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4およびR5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択される、方法。
【請求項2】
R4およびR5が独立に、H、アルキル、シクロアルキル、アシル、アリール、ヘテロアリール、アルキニルおよびアルケニルからなる群から選択され;R7がハロゲン、ハロアルキルC1〜C6、ハロアルケニルC1〜C6、ハロアルキニルC1〜C6、ハロヘテロアルキル、ハロヘテロアルケニル、ハロヘテロアルキニル、ハロアリールおよびハロヘテロアリールからなる群から選択され、ここで、R7中のハロまたはハロゲンは18Fまたは123Iのいずれかである、請求項1に記載の方法。
【請求項3】
R1、R2およびR3が独立に、水素、アルキルC1〜C6、ハロアルキルC1〜C6、アルケニルC1〜C6、ハロアルケニルC1〜C6、アルキニルC1〜C6およびハロアルキニルC1〜C6からなる群から選択される、請求項2に記載の方法。
【請求項4】
前記アミノ酸類似体中のYおよびZがCH2である、請求項3に記載の方法。
【請求項5】
R1、R2およびR3が水素またはアルキルC1〜C4である、請求項4に記載の方法。
【請求項6】
R7が18F、18F−アルキルC1〜C4、123Iおよび123I−アルキルC1〜C4からなる群から選択される、請求項1または5に記載の方法。
【請求項7】
前記アミノ酸類似体がシン−3−[18F]FACBCである、請求項6に記載の方法。
【請求項8】
前記アミノ酸類似体がシン−3−[123I]IACBCである、請求項6に記載の方法。
【請求項9】
前記アミノ酸類似体がシン−3−[18F]FMACBCである、請求項6に記載の方法。
【請求項10】
前記アミノ酸類似体がシン−[18F]FACHCである、請求項6に記載の方法。
【請求項11】
実質的に純粋な次式の化合物
【化3】
であって、ここで、YおよびZは独立に、CH2、N、O、S、Seおよび(CR4,R5)n(n=1〜4)からなる群から選択され;R1〜R3は独立に、H、アルキル、シクロアルキル、アシル、アリール、アルケニル、アルキニル、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、ハロアルケニルおよびハロアルキニルからなる群から選択され;R4〜R5は独立に、H、アルキル、シクロアルキル、アシル、アリール、ハロ、ハロアルキル、ハロアシル、ヘテロアリール、ハロアリール、ハロヘテロアリール、アルキニル、アルケニル、ハロアルケニルおよびハロアルキニルからなる群から選択され、ここで、ハロは非放射性のF、Cl、BrおよびIからなる群から選択される、化合物。
【請求項12】
R1、R2およびR3が独立に、H、アルキルC1〜C6、ハロアルキルC1〜C6、アルケニルC1〜C6、アルキニルC1〜C6、ハロアルケニルC1〜C6およびハロアルキニルC1〜C6からなる群から選択され、R4およびR5が独立に、水素、アルキルC1〜C6、アリール、ヘテロアリール、アルキニルC1〜C6およびアルケニルC1〜C6からなる群から選択される、請求項11に記載の化合物。
【請求項13】
R1、R2およびR3が水素であり、YおよびZがCH2である、請求項12に記載の化合物。
【請求項14】
R1、R2およびR3が水素であり、YおよびZがC2H4である、請求項12に記載の方法。
【請求項15】
請求項1に記載の方法によって作製される実質的に純粋なシン−アミノ酸類似体。
【請求項16】
前記類似体がシン−3−[18F]FACBCである、請求項15に記載のアミノ酸類似体。
【請求項17】
請求項15に記載のシン−アミノ酸類似体と生理学的に受容可能な担体とを含む、腫瘍を画像化するための薬学的組成物。
【請求項18】
前記アミノ酸類似体がシン−3−[18F]FACBCである、請求項17に記載の組成物。
【請求項19】
陽電子射出断層撮影法または単光子射出コンピュータ断層撮影法による腫瘍の画像化方法であって、
a)腫瘍を有している疑いのある対象に画像生成量の請求項1に記載の標識化合物を投与する工程;
b)該標識化合物が該腫瘍と会合するのに十分な時間を与える工程;および
c)PETまたはSPECTによって、該対象における該標識化合物の分布を測定する工程
を包含する、方法。
【請求項20】
前記標識化合物がシン−3−[18F]FACBCである、請求項19に記載の方法。
【請求項21】
請求項11に記載の化合物と、該化合物をシン−3−[18F]FACBCに転換させるのに必要な試薬とを含む、実質的に純粋なシン−3−[18F]FACBCを合成するためのキット。
【図1】

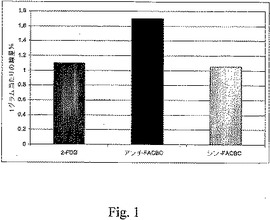
【図2】
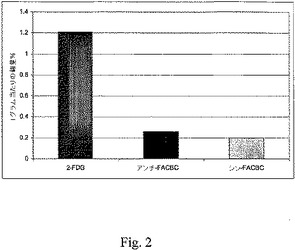
【図3】
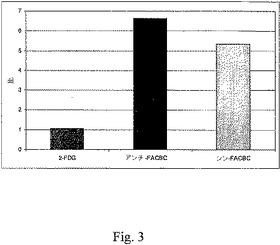
【図2】
【図3】
【公表番号】特表2008−546783(P2008−546783A)
【公表日】平成20年12月25日(2008.12.25)
【国際特許分類】
【出願番号】特願2008−518271(P2008−518271)
【出願日】平成18年6月19日(2006.6.19)
【国際出願番号】PCT/US2006/023740
【国際公開番号】WO2007/001958
【国際公開日】平成19年1月4日(2007.1.4)
【出願人】(598038968)エモリー・ユニバーシティ (19)
【Fターム(参考)】
【公表日】平成20年12月25日(2008.12.25)
【国際特許分類】
【出願日】平成18年6月19日(2006.6.19)
【国際出願番号】PCT/US2006/023740
【国際公開番号】WO2007/001958
【国際公開日】平成19年1月4日(2007.1.4)
【出願人】(598038968)エモリー・ユニバーシティ (19)
【Fターム(参考)】
[ Back to top ]