
腫瘍関連標的抗原を低度から中程度のレベルで発現している腫瘍細胞の三機能二重特異性抗体による破壊
【課題】腫瘍性疾患の予防および治療用の薬学的組成物を製造するための三機能二重特異性抗体の提供。
【解決手段】(a)T細胞に結合する特性;(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;を有する、三機能二重特異性抗体。
【解決手段】(a)T細胞に結合する特性;(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;を有する、三機能二重特異性抗体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、腫瘍性疾患(tumor deseases)の予防および治療用の薬学的組成物を製造するための三機能二重特異性抗体の使用に関する。
【背景技術】
【0002】
免疫療法を行う中で単特異性モノクローナル抗体による腫瘍の治療における現在未解決の問題は、低度から中程度の発現レベルの腫瘍標的抗原を有する腫瘍細胞の破壊の効率が不十分なことである。一つの具体例は、標的抗原Her2/neuを低度から中程度のレベルで発現している乳ガンの治療であり、他の具体例は、CD20腫瘍抗原を低度のレベルで発現している非ホジキンリンパ腫および慢性リンパ性白血病の治療である。
【0003】
転移性乳ガンは、ほとんど常に致死性の疾患である。転移が最初に明らかになってからの生存時間のメジアンは、17から20ヶ月の範囲である。内分泌性、細胞傷害性および生物学上の多数の薬剤が一時的な効果を証明しているが、総意としての療法の基準はなく、治療には重大な副作用を生じることが多い。
【0004】
上皮増殖因子(EGF)ファミリーメンバーであるHer2/neuは、約25〜30%の乳ガン患者の腫瘍試料において過剰発現し、より急速な腫瘍の成長およびより悪い予後の原因とされる。
【0005】
EGF受容体のファミリーは、4つのメンバーEGFR(ERBB1)、Her2/neu(ERBB2)、ERBB3、ERBB4を含む。EGFRおよびHer2/neuは、治療標的として鋭意研究されている。細胞内受容体シグナルを阻害する小分子化合物だけでなく、EGFRおよびHer2/neuの細胞外ドメインを標的とする抗体も、既に臨床で使用され、臨床上の効果が証明されている。しかし、それらの抗腫瘍効果は、前臨床試験から予測されるほど強力でないことが多く、化学的療法と組み合わせることが好ましい。
【0006】
Herceptin(登録商標)は、乳ガンの治療に広く使用される、よく受け入れられたモノクローナル抗体である。しかし、Herceptin(登録商標)は、標的抗原であるHer2/neuの腫瘍細胞上での発現レベルが少なくとも2+/FISH+(いわゆるHercepTest(登録商標)およびFISH法解析陽性で証明される)、または3+である患者に対してしか使用は成功しない。いくつかの研究により、Her2/neuの追加遺伝子増幅(FISH法で示される)とともに相対的に高い標的抗原Her2/neuの発現密度が、統計的に有意な生存を得るために必要だということが示されている。前臨床試験での生体実験によっても、Herceptin(登録商標)による腫瘍細胞の効果的な破壊が見られるのは、腫瘍細胞が標的抗原であるHer2/neuを高レベルに発現している場合だけであることが示されている。
【0007】
これらの制限は、腫瘍上にHer2/neuを高レベルに発現する(前記Dako社のHercepTestでスコア2+/FISH+、または3+とされる)、約25〜30%の転移性乳ガン患者しか、Herceptin(登録商標)を用いて効果的に治療することができないことの原因である。
【0008】
Perezらの研究では、免疫組織化学的検査(IHC)での評価によると、35%の乳ガン患者がHer2のスコア1+、14%が2+、13%が3+とされた(非特許文献1)。
【0009】
Kiewe, P.らは、Her2/neuおよびCD3を標的とする完全な二重特異性モノクローナル抗体の使用について記載している(非特許文献2)。この抗体は、転移性乳ガンの治療にフェーズI試験で使用された。この抗Her2×抗CD3抗体によって生じる副作用の研究を行い、必要な人数である17人の患者の募集(recruiting)を早めるために、前記HercepTest(登録商標)でのスコアが1+(25%)、2+(12.5%)および3+(62.5%)の患者が試験された。この予備的な結果解析では、抗腫瘍応答は、評価可能な15人の患者のうち4人で見られた。しかし、前記HercepTest(登録商標)のスコアが1+の範疇の患者で抗腫瘍応答を示した者はいなかった。これらの応答した者のHer2/neuの発現状態(status)に関しては、全く情報が開示されていない。さらに、このフェーズI試験を計画する時点では、ertumaxomabが、高レベル(スコア+3)のHer2/neuを発現する細胞系統(例えば、Sk−Br−3)を除去するという実験データのみが入手可能であった。
【0010】
全ての典型的なフェーズI試験と同様に、Kieweらが行ったフェーズI試験は、三機能抗体であるertumaxomabの安全性および許容性を調査するために計画された。この試験での第二の変数(secondary variables)は、抗腫瘍活性および種々の免疫学的変数の測定であった。この研究への患者の募集を早めるために、患者の腫瘍細胞上のHer2/neuの発現に関する制限を設けなかった。これが可能であったのは、この研究の第一の目的は、安全性と許容性であり、腫瘍細胞上のHer2/neuの発現レベルは、全く重要では無かったからである。
【0011】
三機能抗体である抗Her2×抗CD3のertumaxomabが、スコア2+および3+に従いHer2/neuを過剰発現する患者においてのみ有効であるという知見は、全く驚きは無かった。なぜなら、Herceptin(登録商標)等の抗体を用いて実施された他の調査でも、Her2/neuを中(2+/FISH+)から高レベル(3+)に発現する患者に対してのみに効果が示されたからである。
【0012】
例えば、Gatzemeierらは、Herceptin等の抗体が、実際に臨床上の利益を提供できるのは、Her2/neuの発現が3+または2+/FISH+の患者に対してだけであるという証拠を提供した(非特許文献3)。
【0013】
同様に、Micromet社は、2006年10月2日に、乳ガンおよび前立腺ガンにおける上皮細胞接着分子EpCAMに対する抗体であるadecatumumabの二つのフェーズII試験についてのデータを公表した。その腫瘍組織において高レベルにEpCAMを発現する患者においてのみ、腫瘍増殖停止時間の有意な延長を示し、EpCAMの発現が低い患者においては有意な利益を示さなかった。adecatumumabには、EpCAMを高度に過剰発現する乳ガンの患者に治療の選択肢を与える可能性があるということが強調された。EpCAMの過剰発現が低い患者には全く有用でないことが見出された。
【0014】
非ホジキンリンパ腫(NHL)および慢性リンパ性白血病(CLL)は、最も広く認められる悪性腫瘍である。NHLは、アメリカ合衆国において5番目に多い悪性疾患(malignant disease)であり、年間発症率は10万人あたり18〜20である。NHLの発症件数は、1990年代中期まで年率5〜10%で増加した(非特許文献4)が、それ以来一定している(非特許文献5)。CLLは、西洋で最も多い白血病であり、年間発症率は10万人あたり3〜4である。
【0015】
抗CD20モノクローナル抗体であるRituximabを用いるB細胞リンパ腫の治療における(特に、化学療法と組み合わせにおける)結果が有望であるにも関わらず、患者は結局再発した(非特許文献6、7)。したがって、さらなる治療の選択肢が必要である。
【0016】
Rituximabおよびalemtuzumab等のモノクローナル抗体の認可以後、治療における大きな改善が達成されたという事実があるにも関わらず、現在、緩徐進行性NHLおよびCLLは、両方とも治癒不能な疾患である(非特許文献8〜11)。抗体だけによる療法は高度には成功していないが、特にCLLにおけるモノクローナル抗体と標準的な化学療法処方との組み合わせは、患者の結果を有意に改善した(非特許文献12、13)。しかし、患者は結局再発し(非特許文献6)、新しい治療法の戦略が必死に求められている。完全ヒト化抗CD20抗体または他の抗原(例えばCD19、CD22、CD23、CD80もしくはHLA−DR)を標的とする抗体等の多くの新薬候補が、現在鋭意検討されている(非特許文献14、15)。
【0017】
新しい標的の探索に加え、抗体療法を改善する別のアプローチとしては、二重特異性抗体の開発があげられる。多くの二重特異性の方式(format)が開発され、試験されている。例えば、クアドローマ細胞で産生させた完全長抗体、化学的にクロスリンクさせたF(ab)2断片、一本鎖抗体(非特許文献16)、二重特異性抗体(非特許文献17)である。これらの抗体の中には、ヒトリンパ腫の治療に試験されたものもある。しかしながら、in vitroにおける効能が有望であるにも関わらず、これらの分子は、限定的な臨床上の利益しか示さなかった(非特許文献18〜20)。
【0018】
CD19またはCD20等のB細胞特異的抗原を標的とする二重特異性抗体は、この10〜15年の間における鋭意研究の対象であるが、穏やかな応答しか示していない限定的な臨床データが、今のところ入手可能な程度である(非特許文献18、21、22)。さらに、製造工程が複雑で効率的でないため、臨床用途のための充分な量の抗体を産生することが難しい。
【0019】
二重特異性抗体(bsAb)は、例えば、悪性細胞等の腫瘍細胞に対してエフェクター細胞を誘導することによって、免疫学的に治療するための道具(tool)である。しかしながら、bsAbは通常エフェクター細胞のうち一つのクラス、すなわちT細胞、NK細胞、FcγRI+またはFcαRI+細胞のいずれかだけしか誘導、活性化しないので、その効能は限定的である。これらの先行技術抗体の全てが、腫瘍関連抗原が中から高レベルで腫瘍細胞表面に発現している場合にのみ、効果的に腫瘍を治療することが記載されている。
【0020】
抗体の免疫学的エフェクター機能を促進することは、抗体に基づくガン治療の効能を改善する一つのアプローチを示す。改善されたエフェクター機能を有する三機能二重特異性抗体は、例えば、Zeidlerら(非特許文献23)、Riesenbergら(非特許文献24)、Ruf&Lindhofer(非特許文献25)またはHeissら(非特許文献26)の出版物において、本発明者らによって、腫瘍細胞およびTリンパ球を二つの結合アームで認識するだけではなく、Fc領域を通じてFcγ受容体陽性修飾細胞にも結合するものとして記載されている。しかしながら、これらの抗体が、Her2/neuまたはCD20のような腫瘍関連抗原を低レベルで発現している腫瘍細胞を治療するのに有用であるということは現在まで知られていなかった。
【0021】
上記で説明したHer2/neu、EpCAMおよびCD20に関する制限は、例えば、G250、GD3,GD2、プロテオグリカン、MHC II、EGF−RおよびCEA等の他の腫瘍関連抗原に関しても当てはまる。
【0022】
要約すると、本発明の優先日までに入手できる全ての実験データは、腫瘍抗原を中程度から高度に過剰発現する患者に対する有意な利益の証拠しか提供していなかった。
【先行技術文献】
【非特許文献】
【0023】
【非特許文献1】Perez EA et al.: Her2 testing in patients with breast cancer: poor correlation between weak positivity by immunohistochemistry and gene amplification by fluorescence in situ hybridization. Mayo Clin Proc 2002; 77: 148−154
【非特許文献2】Kiewe, P. et al.: “Phase I trial of the trifunctional anti−Her2 x anti−CD3 antibody ertumaxomab in metastatic breast cancer”; Journal of Clinical Oncology, vol 23, no. 16S, 1 June 2005 (ASCO Abstract 2205−06−01)
【非特許文献3】Gatzemeier et al., Anals of Oncology 15:19−27, 2004
【非特許文献4】Greiner TC, Medeiros LJ, Jaffe ES. Non−Hodgkin’s lymphoma. Cancer. 1995;75:370−380.
【非特許文献5】Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review, 1975−2003. http://seercancergov. 2006.
【非特許文献6】Smith MR. Rituximab (monoclonal anti−CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359−7368.
【非特許文献7】Ghielmini M. Multimodality therapies and optimal schedule of antibodies: rituximab in lymphoma as an example. Hematology (Am Soc Hematol Educ Program). 2005:321−328.
【非特許文献8】Lin TS, Lucas MS, Byrd JC. Rituximab in B−cell chronic lymphocytic leukemia. Semin Oncol. 2003;30:483−492.
【非特許文献9】Dillman RO. Treatment of low−grade B−cell lymphoma with the monoclonal antibody rituximab. Semin Oncol. 2003;30:434−447.
【非特許文献10】Blum KA, Bartlett NL. Antibodies for the treatment of diffuse large cell lymphoma. Semin Oncol. 2003;30:448−456.
【非特許文献11】Moreton P, Hillmen P. Alemtuzumab therapy in B−cell lymphoproliferative disorders. Semin Oncol. 2003;30:493−501.
【非特許文献12】Hiddemann W, Dreyling M, Unterhalt M. Rituximab plus chemotherapy in follicular and mantle cell lymphomas. Semin Oncol. 2003;30:16−20.
【非特許文献13】Montserrat E. Rituximab in chronic lymphocytic leukemia. Semin Oncol. 2003;30:34−39.
【非特許文献14】Mavromatis BH, Cheson BD. Novel therapies for chronic lymphocytic leukemia. Blood Rev. 2004;18:137−148.
【非特許文献15】Nagy ZA, Hubner B, Lohning C, et al. Fully human, HLA−DR−specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8:801−807.
【非特許文献16】Baeuerle PA, Kufer P, Lutterbuse R. Bispecific antibodies for polyclonal T−cell engagement. Curr Opin Mol Ther. 2003;5:413−419.
【非特許文献17】Peipp M, Valerius T. Bispecific antibodies targeting cancer cells. Biochem Soc Trans. 2002;30:507−511.
【非特許文献18】Manzke O, Tesch H, Borchmann P, et al. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. I. Clinical phase I evaluation. Int J Cancer. 2001;91:508−515.
【非特許文献19】Manzke O, Tesch H, Lorenzen J, Diehl V, Bohlen H. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. II. Assessment of cellular immune responses. Int J Cancer. 2001;91:516−522.
【非特許文献20】Haagen IA. Performance of CD3xCD19 bispecific monoclonal antibodies in B cell malignancy. Leuk Lymphoma. 1995;19:381−393.
【非特許文献21】Weiner GJ, De Gast GC. Bispecific monoclonal antibody therapy of B−cell malignancy. Leuk Lymphoma. 1995;16:199−207.
【非特許文献22】De Gast GC, Van Houten AA, Haagen IA, et al. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother. 1995;4:433−437.
【非特許文献23】Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246−1252.
【非特許文献24】Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem. 2001;49:911−917.
【非特許文献25】Ruf P, Lindhofer H. Induction of a long−lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526−2534.
【非特許文献26】Heiss MM, Strohlein MA, Jager M, et al. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435−443.
【発明の概要】
【発明が解決しようとする課題】
【0024】
したがって、例えば、Her2/neuを低度に発現している腫瘍細胞(前記Dako社のHercepTest(登録商標)において、例えば、1+のスコアである)、腫瘍細胞あたり500,000以下の腫瘍関連抗原という中程度のレベルでHer2/neuを発現し、前記Dako社のHercepTest(登録商標)において、2+のスコアであるが、FISH法陰性のもの、もしくはCD20を低度に発現している腫瘍細胞、または少なくとも1つの上記した特異的な腫瘍関連抗原、特にCD20およびEpCAMを、腫瘍細胞あたり約1,000〜350,000、特に、5,000〜150,000のレベルおよび範囲で発現している腫瘍細胞をも死滅させることが可能な抗体の抗腫瘍活性の改善が必要である。
【課題を解決するための手段】
【0025】
上記した問題は、以下に述べる特性を有する三機能二重特異性抗体から選択され、種々のクラスの免疫細胞の協調を促進する抗体の、腫瘍性疾患の予防および治療用の薬学的組成物を製造するための使用によって解決されることを見出した。
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
前記腫瘍関連抗原は、下記のとおり前記腫瘍細胞上に発現されている。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、または、
・EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【図面の簡単な説明】
【0026】
【図1】図1A−Fは、異なる抗Her2抗体を用いてフローサイトメトリーにより測定された細胞系統Lox、HCT−8およびSKBR3のHer2/neu発現特性を示す。
【図2a】図2aは、Her2+3 SKBR−3細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2b】図2bは、Her2+1 HCT−8細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2c】図2cは、Her2ネガティブ Lox細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2d】図2dは、Her2+1 HCT−8細胞の死滅:MDSワークフロー(workflow)によるドナー2のドットプロット結果の一例を示す。 *印の番号は、表3に対応する。
【図3】図3は、サイトカインビーズアレイシステム(CBA)を用いて測定された24時間後のクローン原性アッセイ上清中のサイトカイン分泌を示す。
【図4】図4は、推定される三細胞複合体(tri−cell−complex)を示す。三機能抗体は、異なる免疫学的メカニズムにより腫瘍細胞の認識と破壊を促進することができる。IFN−γを除く図中のサイトカインに関するリストをまとめなければならない(Bei der Auflistung der Zytokine im Bild sollte man noch IFN−γ aufnehmen)。注:ADCC、抗体依存性細胞傷害;DC、樹状細胞;NK、ナチュラルキラー細胞;DC−CK1、樹状細胞サイトカイン1;IL、インターロイキン;LFA、リンパ球機能関連抗原;TNF−α、腫瘍壊死因子α;CD、表面抗原分類(出典 Ruf&Lindhofer,2001)
【図5A】図5は、Bi20の精製を示す。(図5A)陽イオン交換クロマトグラフィー(CIEX)を示す。親マウス抗体(ピーク1)を、HR10/10 high performance SP−Sepharose column上で、三機能抗体(ピーク2)から分離した。
【図5B】(図5B)Bi20三機能抗体精製の等電点電気泳動(IEF)を示す。親ラット抗体(26II6)を、プロテインAアフィニティークロマトグラフィー(AC Bi20)後に除去し、親マウス抗体(TPA10)を、CIEX(CIEX P1 Bi20)により三機能抗体から分離した。精製されたBi20は、前記陽イオン交換クロマトグラフィーのピーク2(CIEX P2 Bi20)に見出された。
【図6A】図6は、Bi20が特異的に結合するエフェクター細胞を示す。190ngのBi20を、5×105個の標的細胞とともにインキュベートし、その結合をFACS解析により測定した。Ramos細胞(図6A)を、CD20アームの標的細胞として用いた。
【図6B】THP−1細胞(図6B)を、Fc領域用に用いた。拮抗実験のために、細胞を予めRituximab(Rit)とともに所定の過剰量の抗体で30分間インキュベートした。
【図7A】図7は、エフェクター細胞の存在下におけるBi20が媒介する効果的なB細胞の減少を示す。(図7A)1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Mec1)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともにインキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)(neg)前記アッセイにおけるB細胞の絶対数を、100%に設定した。
【図7B】(図7B)1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。前記B細胞の絶対数を、トリパンブルー色素排除集計およびFACS解析により、1日目、2日目、3日目において測定し、B細胞の減少割合を算出した。
【図8A】図8は、Bi20が媒介するT細胞の活性化を示す。(図8A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。CD4+およびCD8+T細胞上におけるCD25の発現のMFIを、FACS解析により測定した。
【図8B】(図8B)PBMCおよびRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。CD4+T細胞上におけるCD25の発現のMFIを、1日目、2日目および3日目に測定した。
【図9】図9は、Bi20が誘導するT細胞の増殖を示す。1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、50ng/mlの濃度のBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。T細胞数を、トリパンブルー色素排除集計並びに抗CD5/4抗体および抗CD5/8抗体を用いたFACS解析により測定した。
【図10A】図10は、Bi20により媒介される単球の活性化を示す。(図10A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)またはrituximabとともに1日間インキュベートした。CD14+単球によるCD25の発現のMFIを、FACS解析により測定した。
【図10B】(図10B)1×106個/mlのPBMCおよび1×105個/mlのTHP−1細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab(Rit)、TPA10、または26II6とともに1日間インキュベートした。CD33+THP−1細胞の活性化を、CD25またはCD40の発現によって測定した。
【図11A】図11は、健康なドナーのエフェクター細胞存在下におけるBi20が誘導するCLL B細胞の減少を示す。(図11A)1×106個/mlのPBMCおよび2×105個/mlのCLL腫瘍細胞を、5、50、もしくは250ng/mlのBi20、250ng/mlのcatumaxomab(Catum)、または250ng/mlのRituximab(Rit)とともに3日間インキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)前記アッセイにおけるB細胞の絶対数を、100%に設定した。MVは、CLL−1からCLL−9の平均値を示す。
【図11B】(図11B)CLL患者検体(1〜14)におけるCD20の発現の概要、細胞系統(Raji、Mec−1)および1体の健康なドナー(PBMC)。MFI=蛍光強度の平均値を示す。
【発明を実施するための形態】
【0027】
本発明の一実施形態において、全ての引用した腫瘍抗原が、腫瘍細胞あたり約5,000〜約150,000の量で腫瘍細胞上に発現している。
【0028】
本発明は、ヒトまたは動物の腫瘍細胞を含む身体、もしくは前記腫瘍細胞を含むこれらの身体の部分を、下記の特性を有する三機能二重特異性抗体の薬学的に有効な量に接触させることにより、腫瘍性疾患を予防および治療するためのヒトまたは動物の治療方法も提供する。
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
前記腫瘍関連抗原は、下記のとおり前記腫瘍細胞上に発現されている。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、または、
・EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【0029】
患者の腫瘍細胞を、前記三機能二重特異性抗体に接触させることは、前記三機能二重特異性抗体を受容可能な服用形式、経口または非経口いずれかで、以下に記述するようにヒトまたは動物の体内に投与することによっても実施される。
【0030】
白血病またはリンパ腫等の免疫システムの細胞が関与する腫瘍性疾患を治療するのであれば、前記接触は、ex vivoで行われてもよい。例えば、米国特許第7,018,632号公報または米国特許第5,985,276号公報が引用され、それらは本明細書に完全に取り込まれている。
【0031】
自己の腫瘍細胞を含む材料は、本明細書に記載の三機能二重特異性抗体とともに、10分間〜10時間、好ましくは5時間以下の短時間インキュベートされ、または、一定時間の長時間インキュベートされることにより、自己の腫瘍細胞は、抗体に襲撃される(charged)。続いて、患者の血液細胞、好ましくは末梢血単核球(PBMC)を加え、この混合物を1〜14日間の長時間インキュベートする。またはそれに代えて、自己の腫瘍細胞を、前記三機能二重特異性抗体および患者の血液細胞、好ましくは末梢血単核球に直接接触させる。このようにして、腫瘍に対する無数の免疫細胞の“プライミング(priming)”が、ex vivoで達成される。その後、これらの細胞は、患者に再注入される。長時間インキュベートすることによっても、抗体の内部移行(internalization)および分解が起こる。前述のとおり、上記で引用した米国特許には、これらの方法が完全に記載されている。しかしながら、上記で示した腫瘍関連抗原について、腫瘍細胞あたり約5,000〜約150,000の腫瘍関連抗原が発現している腫瘍細胞が関与する腫瘍性疾患を治療することも可能なのかという、専門家に向けた成功のための合理的な予測は全く提供されていない。前記方法では、健康なドナーのPBMCを加えずに、同種間または自己設定(autologous setting)で実施してもよい。珍しいことに、腫瘍細胞、特にB細胞の減少は、エフェクター細胞のサイトカイン(例えば、IL−2、GM−SCF)による前活性化(pre−activation)または共活性化(coactivation)には全く依存しない。このことは、従来技術に記載されている他の二重特異性抗体または単特異性抗体に対する典型的な不利な点である。
【0032】
本発明の好ましい実施形態は、従属クレーム並びに以下の記載およびそれに伴う実験データ、図、表に示されている。
【0033】
本発明者らは、驚いたことに、従来技術に記載されている三機能二重特異性抗体が、低度から中程度のレベルで腫瘍細胞上に発現されている、前記腫瘍細胞上の選択された特定の腫瘍関連標的抗原を標的とするのに効果的に使用可能であることを示すことができた。標的抗原として特に好ましいものは、Her2/neu、EpCAMおよびCD20腫瘍関連抗原である。
【0034】
前記腫瘍関連抗原は、前記腫瘍細胞上に発現される前記腫瘍関連抗原のタイプ、およびHer2/neuの場合は、前記腫瘍抗原が増幅されているかどうかに依存して種々のレベルで前記腫瘍細胞上に発現されている。
【0035】
Her2/neuの場合、患者の腫瘍がHer2/neuを腫瘍細胞上に腫瘍細胞あたり約5,000〜約150,000の量で腫瘍関連抗原が発現していれば、本明細書中の記載のように前記患者は三機能二重特異性抗体で治療が成功可能であることが見出されている。これらの患者は、前記DAKO社のHercepTest(登録商標)に従えば、1+のスコアとされることになる。1+の上限を、約100,000または110,000に分類することも正しいと考えられる。最も重要なことは、前記スコアが、“中程度の発現”ではなく、“低度の発現”と分類されなければならないことである。
【0036】
腫瘍細胞あたり500,000分子以下のHer2/neu腫瘍関連抗原を発現し、前記DAKO社のHercepTest(登録商標)に従って2+に分類される患者は、FISH法陰性と分類された場合であっても、治療が成功可能なことも見出されている。従来は、前記DAKO社のHercepTest(登録商標)に従ってHer2/neu値が2+のスコアとされ、腫瘍細胞あたり500,000以下のHer2/neu腫瘍抗原を発現し、FISH法陽性の患者しか治療が成功可能でなかった。Her2/neu値が2+であっても、FISH法陰性の患者は、治療が成功可能でなかった。
【0037】
CD20を発現する腫瘍を有する患者では、驚いたことに、腫瘍細胞あたり約1000〜約350,000の量で腫瘍関連抗原としてCD20を発現している場合でさえも、患者の治療が成功可能であることが見出された。さらに、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAを腫瘍細胞あたり約1000〜約350,000の量で腫瘍関連抗原として発現している患者は、本明細書に記載の前記三機能二重特異性抗体により治療が成功可能である。
【0038】
前記腫瘍関連抗原は、Her2/neuの場合、前記腫瘍細胞上に、好ましくは腫瘍細胞あたり少なくとも約10,000、約20,000、約50,000または約80,000の量で発現しており、好ましくは腫瘍細胞あたり最大で約110,000、約120,000または約130,000の量で発現している。
【0039】
前記DAKO社のHercepTest(登録商標)でHer2/neu値がスコア2+(中程度の発現)とされた患者においては、腫瘍細胞上の腫瘍関連抗原の好ましい発現の量は、500,000以下または450,000、400,000、350,000もしくは300,000以下であり、下限は約100,000か、約110,000より多いか、約150,000、約200,000または約250,000より多い。
【0040】
CD20腫瘍抗原を発現する腫瘍細胞では、腫瘍細胞あたりのCD20分子の数は、約1000〜約350,000の範囲で、B細胞悪性腫瘍の型および体内部位に依存し、好ましくは、約310,00または300,000以下で変化する。腫瘍細胞あたりのCD20分子の数は、特に、約5000、約10,000または約50,000から約100,000、150,000、200,000、250,000または300,000以下で変化することが可能である。前記状況は、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEA等の請求項1で示した他の全ての腫瘍抗原においても同様である。
【0041】
腫瘍関連抗原の発現レベルは、単一の限定された腫瘍の実体から生じた種々の腫瘍細胞上に、より低度の発現値から高度の発現値の間に分布し得ることを申し添えておく。これらの数値は全て、悪性腫瘍のタイプ、患者のタイプ、腫瘍の進行等に依存した平均値であることを理解されなければならない。
【0042】
本発明者らによって選択された腫瘍関連標的抗原が低度から中程度の発現レベルでのみ腫瘍細胞上に永続的かつ安定に発現していることが、極めて重要であることを申し添えておく。ヒートショックタンパク質、MIC分子であるMIC AおよびMIC B等の他のタイプの誘導可能な抗原は、両方ともヒートショックプロモーター要素の制御下にあり、誘導後一生の間増加するレベルで発現されるが、Her2/neuおよびCD20等の腫瘍関連抗原の発現割合は、一般的に一定かつ安定である。
【0043】
本発明者らによって選択された腫瘍関連標的抗原は、特定の腫瘍に関連するか、または特異的である。EpCAMは、典型的には腺ガンに関連し、Her2/neuは、乳ガンだけでなく大腸ガン、肺ガン、胃ガン、膵臓ガンおよび卵巣ガンに関連し、CD20は、非ホジキンリンパ腫または慢性リンパ性白血病等のB細胞リンパ腫に関連し、G250は、腎臓ガンに関連し、プロテオグリカン、GD3およびGD2は、メラノーマに関連し、MHC IIはB細胞リンパ腫に関連し、並びにEGF−RおよびCEAは、上皮性腫瘍に関連する。
【0044】
本発明で用いる三機能二重特異性抗体(略してtrAb)は、それ自体は公知である。例えば、米国特許第6,551,592号公報が引用され、その内容は本明細書に完全に取り込まれている。同様のことがドイツ公開特許第19649223A号公報およびドイツ公開特許第19710495A号公報についても言え、それらに対応する米国特許公報とともに引用され、それらもまた本明細書中に包含されている。
【0045】
本発明で用いる抗体は、カプセル、タブレット、水性の懸濁液、溶液等の許容できるいかなる服用形式で経口投与されてもよい。抗体およびその誘導物は、非経口的に投与されてもよい。それは以下の投与経路を介する。すなわち、皮下、静脈内、腹腔内、筋内、関節内、滑液内、胸骨内、鼻腔内、局部、莢膜内、肝内、腫瘍内、病巣内および頭蓋内への注射または注入技術である。一般的には、抗体は、静脈内への注射または注入により供給される。
【0046】
本発明の抗体は、単独で投与されてもよいし、または、許容できるアジュバント(adjuvant)、賦形剤および添加剤を含む薬学的に許容できるキャリアーとともに投与されてよい。これらは、全て当業者にとって周知である。
【0047】
有効な服用量は、多数の因子に依存する。体重、治療の目的、最大許容服用量、使用された具体的な製剤、投与経路、患者の応答等の多数のパラメーターに従って任意の患者に対して服用量を調整することは、熟練した内科医が優に為し得る範囲である。
【0048】
本発明に従って使用されるtrAbは、一回の注入あたり、好ましくは約5〜約1000μg、さらに好ましくは約10〜約300μg、約10〜約100μg、または約10〜約50μgの量である。最適な量は、実験により、熟練者によって決定され得る。前記三機能抗体は、さらに好ましくは約0.05〜約15μg/kg体重、さらに好ましくは約0.5〜5μg/kg体重、および約0.5〜2μg/kg体重の量で使用される。投与の回数は、患者の必要性、特に疾患の重さ、患者の応答、適用した服用量およびその技術分野で知られる他の多数の因子に従って、医師が選択できる。
【0049】
好ましくは、前記三機能抗体は、抗CD3×抗腫瘍関連抗原抗体および/または抗CD4×抗腫瘍関連抗原抗体および/または抗CD5×抗腫瘍関連抗原抗体および/または抗CD6×抗腫瘍関連抗原抗体および/または抗CD8抗腫瘍関連抗原抗体および/または抗CD2×抗腫瘍関連抗原抗体および/または抗CD28×抗腫瘍関連抗原抗体および/または抗CD44×抗腫瘍関連抗原抗体が選択されるが、特に好ましくは、抗CD3×抗腫瘍関連抗原抗体が使用される。腫瘍関連抗原として特に好ましくは、Her2/neu、EpCAMまたはCD20抗原であり、さらに好ましくは、三機能抗体の抗CD3結合アームと組み合わせる。
【0050】
本発明の前記三機能抗体を使用することで、前記trAbの前記Fc領域が前記Fc受容体陽性細胞に結合することによって前記Fc受容体陽性細胞が活性化される。それにより、サイトカインおよび/または共刺激抗原の発現が開始または増加される。そこで、少なくとも、T細胞の生理的活性化に必要な第二の活性化シグナルが、前記共刺激抗原および/またはサイトカインにより前記T細胞に伝達される。この活性化は、活性化マーカーの上方制御(up−regulation)、腫瘍細胞の殺傷およびT細胞の増殖によって示される。
【0051】
trAbによる前記Fc受容体陽性細胞の活性化は、前記サブクラス、または抗体の重鎖断片の前記サブクラスの組み合わせにそれぞれ依存する。in vitroの実験で証明されたように、例えば、前記マウスIgG2a/ラットIgG2bのサブクラスの組み合わせのtrAbは、Fc受容体陽性細胞に結合し、同時に活性化することが可能で、これらの細胞の表面におけるCD40、CD80もしくはCD86等の共刺激抗原の上方制御または新しい形成(発現)をそれぞれ導く。一方、前記マウスIgG1/ラットIgG2bのサブクラスの組み合わせの二重特異性抗体は、Fc受容体陽性細胞に結合可能だが((1)Haagenら、 J. Immunology, 1995, 154: 1852−1860)、明らかにこれらの細胞を同程度には活性化できない((2)Gastら、 Cancer Immunol. Immunother., 1995, 40: 390)。したがって、trAbのFc領域におけるマウスIgG2a/ラットIgG2bのアイソタイプの組み合わせが特に好ましい。しかしながら、本明細書に記載のとおり、これは他の全てのアイソタイプの組み合わせについても言えることであり、それらは本発明の他の好ましい実施形態において使用することができる。
【0052】
前記trAbが、一つの結合アーム(例えば、抗CD3)でT細胞に同時に結合し、活性化する間、前記trAbの前記Fc領域に結合した前記Fc受容体陽性細胞からの共刺激シグナルが前記T細胞に伝達される可能性がある。すなわち、前記trAbの一つの結合アームを介したT細胞活性化、および前記Fc受容体陽性細胞から前記T細胞への共刺激シグナルの同時期の伝達の組み合わせだけが、効果的なT細胞の活性化につながる。
【0053】
抗腫瘍免疫の誘導における他の重要な側面は、前記trAbによって標的とされる修飾細胞(単球、マクロファージ、NK細胞または樹状細胞)による腫瘍構成物の潜在的な食作用、分解および提示である。この古典的な抗原提示機構により、腫瘍特異的なCD4陽性細胞およびCD8陽性細胞が生じてもよい。さらに、腫瘍特異的CD4細胞は、T/B細胞の協調という状況で体液性免疫応答の誘導に重要な役割を果たす。
【0054】
三機能二重特異性抗体は、第1の結合アームでT細胞のT細胞受容体複合体、第二の結合アームで前記腫瘍細胞上の前記腫瘍関連抗原に結合可能である。それによって、それらは、サイトカインの放出、またはアポトーシスを起こす機構により、腫瘍細胞を破壊するT細胞を活性化する。さらに、三機能抗体による活性化ということでは、これらの受容体を通じて腫瘍特異的抗原を認識するT細胞は再び活性化する可能性があり、そうして長く持続する抗腫瘍免疫へとつながる可能性があるように思われる。この点で特に重要なのは、単球/マクロファージおよび樹状細胞等の修飾細胞への結合を媒介し、それら自身の細胞傷害性を発達させ、および/または、付随して前記T細胞に重要な共刺激シグナルを伝達させる、前記三機能二重特異性抗体の完全なFc領域である。明らかに、この方法により、現在まで未知の腫瘍特異的ペプチドに対して、T細胞の応答が誘導され得る。
【0055】
潜在的にアネルギー化した腫瘍特異的T細胞をtrAbにより腫瘍細胞に誘導し、前記trAbのFc領域に結合した修飾細胞によってそのようなT細胞を同時に共刺激することにより、腫瘍特異的細胞傷害性T細胞(CTL)のアネルギー状態は撤廃可能であった。すなわち、患者中に存在する腫瘍に対して既に存在するT細胞寛容性が、trAbにより撤廃される可能性がある。そして、このことにより、腫瘍細胞の直接の破壊に加えて、長く持続する抗腫瘍免疫が誘導される可能性がある。
【0056】
好ましくは、本発明に従って使用される抗体は、アネルギー状態にある腫瘍特異的T細胞を再活性化し得る。さらに、それらは腫瘍に反応する補体結合抗体を誘導可能であり、したがって体液性免疫応答を誘導する。
【0057】
前記T細胞への結合は、好ましくはCD3、CD2、CD4、CD5、CD6、CD8、CD28および/またはCD44を通じて起こる。前記Fc受容体陽性細胞は少なくとも一つのFcγ受容体IまたはIIIを有する。
本発明に従って使用され得る抗体は、Fcγ受容体Iおよび/またはIII陽性細胞である、単球、マクロファージ、樹状細胞、“ナチュラルキラー”細胞(NK細胞)および/または活性化好中球に結合し得る。
【0058】
本発明に従って使用され得る抗体は、共刺激抗原であるCD40、CD80、CD86、ICAM−1および/もしくはLFA−3の発現の開始もしくは増加、または/並びに、前記Fc受容体陽性細胞によるサイトカイン分泌を導く。好ましくは、前記サイトカインはIL−1、IL−2、IL−4、IL−6、IL−8、IL−12、IFN−γおよび/またはTNF−αである。
好ましくは、前記T細胞への結合は、前記T細胞のT細胞受容体複合体を通じて起こる。
【0059】
好ましくは、前記三機能二重特異性抗体は、異種性の完全なラット/マウス二重特異性抗体である。
【0060】
本発明に従って使用され得るtrAbによって、T細胞は活性化され、腫瘍細胞に対して誘導される。好ましい有用な異種性の三機能二重特異性抗体は、以下のアイソタイプの組み合わせの少なくとも一つから選択される。
ラット−IgG2b/マウス−IgG2a、
ラット−IgG2b/マウス−IgG2b、
ラット−IgG2b/マウス−IgG3;
ラット−IgG2b/ヒト−IgG1、
ラット−IgG2b/ヒト−IgG2、
ラット−IgG2b/ヒト−IgG3[東洋人のアロタイプG3m(st)=プロテインAに結合]、
ラット−IgG2b/ヒト−IgG4、
ラット−IgG2b/ラット−IgG2c;
マウス−IgG2a/ヒト−IgG3[白色人種のアロタイプG3m(b+g)=プロテインAに結合しない、以後*で示す]
マウス−IgG2a/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2a/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2a/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒトIgG4/ラット−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域−CH2−CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG2−[ヒンジ領域−CH2−CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG3−[ヒンジ領域−CH2−CH3、東洋人のアロタイプ]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域−CH2−CH3]
ヒト−IgG1/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG2[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG2[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG2/ヒト−[VH−CH1、VL−CL]−ヒト−IgG2−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
マウス−IgG2b/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2b/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒト−IgG4/ラット−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG1/マウス−[VH−CH1,VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
【0061】
好ましくは、本発明に基づく有用な抗体は、モノクローナル、キメラ、組み換え、合成、半合成であるか、または、例えば、Fv、Fab、scFvもしくはF(ab)2断片を有する化学的に修飾された完全な抗体である。
【0062】
好ましくは、ヒト起源の抗体、またはその誘導体もしくは断片が使用されるか、または、ヒトでの使用に適するように修飾された抗体(いわゆる、“ヒト化抗体”)が使用される(例えば、Shalabyら、 J. Exp. Med. 175 (1992), 217; Mocikatら、 Transplantation 57 (1994), 405を参照)。
【0063】
上述した種々のタイプの抗体および抗体断片の調製は、熟練者にとって周知である。モノクローナル抗体、好ましくは、例えば、ヒト、ラット、マウス、ウサギまたはヤギのような哺乳類起源のモノクローナル抗体の調製は、例えば、KoehlerおよびMilstein(Nature 256 (1975), 495)、HarlowおよびLane(Antibodies, A Laboratory Manual (1988), Cold Spring Harbor)、またはGalfre(Meth. Enzymol. 73 (1981), 3)、またはドイツ公開特許第19531346号公報に記載されている従来法を用いて行うことが可能である。
【0064】
さらに、組み換えDNA技術を用いて、熟練者に知られた技術に従って(Kuruczら、 J. Immunol. 154 (1995), 4576; Hollingerら、 Proc. Natl. Acad. Sci. USA 90 (1993), 6444を参照)、記載された抗体を調製することが可能である。
【0065】
二つの異なる特異性を有する抗体、いわゆる二重特異性抗体の調製は、一方では、組み換えDNA技術を用いて実施することが可能であるが、他方では、いわゆるハイブリッドハイブリドーマ融合技術(例えば、Milsteinら、 Nature 305 (1983), 537を参照)によっても実施することが可能である。この技術は、所望の特異性の一つを有する抗体をそれぞれ産生するハイブリドーマ細胞系統の融合、および、両方の特異性を有する抗体を産生する組み換え細胞系統の同定、単離を含む。
【0066】
本発明の基礎をなす課題は、請求項1で定義した三機能二重特異性抗体を用いて解決される。続いて、二つの特異性を示す抗体の調製をより詳細に記載する。本発明に含まれる三機能二重特異性抗体は先行技術に属し、そのような調製方法を記載した文献は、本明細書中にそれら全体が取り込まれている。
【0067】
三機能二重特異性抗体は、それぞれ特異性を示し、かつ、通常の抗体と同様に、周知のエフェクター機能を発揮するFc領域を有する、二つの抗体セミ分子(それぞれが一つのHおよび一つのL免疫グロブリン鎖を有する)から構成される。それらは、クアドローマ技術を用いて調製することが好ましい。この調製方法は、ドイツ公開特許第4419399A号公報において例証されている。完全な開示のために、この公報は、二重特異性抗体の定義にも関連する引用によりその全体が取り込まれている。他の調製方法も、本発明において要求される上述の定義に基づく三機能二重特異性抗体に導かれる限り、有用であることは理解されるべきである。
【0068】
前記trAbのFcγ−RIへの結合は、最適な抗腫瘍効果という観点から二つの不可欠な利点を示す。
(1)Fcγ−RI陽性細胞は、ADCCによって腫瘍細胞を除去する能力を有する。そのため、前記trAbによって腫瘍細胞に誘導された細胞傷害性T細胞の抗腫瘍効果に相乗的に貢献することができる。
(2)FcγRI陽性細胞(単球/マクロファージ/樹状細胞等)は、T細胞に対して、抗原提示に似た重要な共刺激シグナルを提供し得る。それにより、T細胞アネルギーを阻害する。さらに、腫瘍特異的ペプチド(腫瘍細胞上でMHC抗原を通じて提示される)を認識するT細胞受容体を有するT細胞でさえ、T細胞の修飾細胞と腫瘍細胞とのtrAbを通じた相互作用による望ましい副産物として、刺激可能である。この配置において、T細胞の適切な活性化に必要な共刺激は、修飾細胞(単球等)から提供される。このため、T細胞受容体と独立で、trAbが媒介する直接的な腫瘍の破壊とは別に、本発明の抗体は、前記trAbの分解後も患者内でパトロールを続ける腫瘍特異的T細胞を活性化し、産生することも可能なはずである。これは、遺伝子治療的なアプローチ(例えば、腫瘍細胞中にB−7等の共刺激抗原を取り込むことによる)と似て、三機能二重特異性抗体によって患者の腫瘍寛容性が撤廃され得ることを意味する。
【0069】
後述する実験データは、本明細書中に記載したtrAbが、前記標的抗原、特に好ましくはHer2/neuおよびCD20標的抗原を低度から中程度の発現レベルでしか有さない腫瘍細胞の破壊に、驚くほど高度に効果的に使用可能であることを示す。高度な効果の理由は、前述した前記三機能抗体の作用様式に見出すことができる。前記作用様式は、単特異性抗体とは大きく異なる。本発明において使用される三機能抗体は、抗T細胞結合アームおよびFc領域により、種々のタイプの免疫細胞を誘導し、同時に活性化できる。これらの免疫細胞の典型例は、Tリンパ球並びにFcγ受容体I型(CD64)およびIII型(CD16)を有する修飾細胞である。第一の結合アームは、Her2/neuまたはCD20等の腫瘍細胞上の標的抗原に結合し、第二の結合アームは、例えば、Tリンパ球上のCD3に結合する(図4)。
【0070】
腫瘍抗原の発現レベルを定量化する方法は、その技術分野において公知である。例えば、免疫細胞化学法であり、例えば、ELISA試験、定量的フローサイトメトリー、組織染色法およびサイトスピン解析等の方法である。腫瘍細胞上の腫瘍関連標的抗原の発現を定量的に決定する方法の典型例は、腫瘍組織上のHer2/neuの発現レベルの定量的決定を可能にした、DAKO社(Glostrup、デンマーク)によって開発されたHercepTest(登録商標)等の免疫組織化学法である。HercepTest(登録商標)に関する参考文献が、DAKO Cytomation Autostainerコード番号K5207第1版として、DAKO社により記載されており、出版番号111 781−001およびK5207/EFG/AOS/13.10.04によって同定できる。この文献は引用により本明細書中に完全に取り込まれている。腫瘍細胞の表面に発現する標的抗原のおよその数を表す4つの段階0、1+、2+、3+を含む、Her2/neuによる評価システムが提供されている。このシステムに従って、標的細胞の表面に20,000未満のHer2/neu分子が発現している細胞は、陰性と分類され、約20,000より多く、約100,000〜110,000以下(最大で150,000)の分子を発現する細胞は、1+(低度の発現)と分類され、約500,000以下の分子を発現する細胞は、2+(中程度の発現)、約2,000,000から約10,000,000分子を発現する細胞は、3+(高度の発現)に分類される。本発明によれば、Her2/neuに対する三機能二重特異性抗体は、HercepTest(登録商標)に従ってHer2/neu 1+および2+(後者においては、さらにFISH法陰性の場合)と分類された腫瘍細胞を有する患者の治療に使用可能であることが見出されている。
【0071】
いわゆるFISH法は、当該技術分野において周知である。“FISH”とは「蛍光In Situ Hybridization」を意味し、細胞遺伝学的技術であり、染色体上の特異的DNA配列の有無を検出し、場所を特定するために使用可能である。前記技術自体は確立されており当該技術分野において公知である。引用するとすれば、例えば、Laudadio J、Quigley DI、Tubbs R、Wolff DJ、HER2試験があげられる。検出方法およびそれらの臨床上の実績の総覧は、引用により本明細書中に完全に取り込まれている。
【0072】
FISH法の試験結果において陰性とは、Her2/neu遺伝子は増幅されていないこと、すなわちHer2/neu遺伝子の数が通常の生理学的範囲内にあるということを意味する。しかし、全ての乳ガンのうち15から20%では、Her2/neu遺伝子は増幅されている。この増幅の検出は確立されており、乳ガンの前記療法にとって重要である。Her2/neu遺伝子が増幅されている場合、FISH解析は陽性であり、その場合、例えばHerceptinによる療法が有効である可能性がある。一方、Her2/neuを過剰発現するが、FISH法において陰性の患者は、Herceptin等の抗体では効果的に治療できない。それゆえ、500,000以下のHer2/neuの発現を有するがFISH法において陰性の患者もまた、本明細書に記載されているように前記三機能二重特異性抗体で治療が成功可能であることは、乳ガンの療法における重要な進歩であると考えられなければならない。多数のグループが、B細胞悪性腫瘍でのCD20の発現も調査している。Ginaldiら(J.Clin.Pathol., 51:364, 1998)は、Quantum Simply Cellular microbeadsキット(Sigma社、セントルイス、ミズーリ州、米国)を用いて、種々のB細胞悪性腫瘍のCD20の発現を評価した。彼等は、例えばB−CLLでは白血球細胞あたり平均65,000のCD20分子、マントル細胞リンパ腫では細胞あたり123,000のCD20分子を見出した。この調査により、有毛細胞白血病の場合、平均値は細胞あたり312,000のCD20分子と決定された。
【0073】
Huhら(Am J Clin Pathol 116:437, 2001)は、さらに、例えば、末梢血および骨髄のような種々の部位でのCD20の種々の発現を、B細胞悪性腫瘍で比較可能な方法で調査した。ここにおいて、骨髄試料では、B−CLLにおいて細胞あたりのCD20分子の数は最低であり(平均4,067;範囲222〜46,395)、濾胞性リンパ腫(平均22,240;範囲3,689〜39,643)、マントル細胞リンパ腫(平均29,770;範囲2,785〜97,727)ではより高く、有毛細胞白血病(平均31,563;範囲1,607〜72,540)では最高であった。対照的に、末梢血では、細胞あたりのCD20分子の数は一般的により高く、例えばB−CLLでは平均数は9,051で、563から31,507の範囲であった。
【0074】
まとめると、B細胞悪性腫瘍の場合、細胞あたりのCD20分子数は、約1000から300,000まで、B細胞悪性腫瘍のタイプおよび身体での部位に依存して変化し、最大約350,000であった。
【0075】
次の実施例において、添付の図および表を参照することによって、本発明を、さらに詳細に記載する。第一の実施例は、本発明を、商標Rexomun(登録商標)(国際一般名 ertumaxomab)で入手可能な三機能抗体、および、Rexomun(登録商標)の標的となる前記腫瘍関連抗原Her2/neuの観点から例証する。第二の実施例は、CD3を通じてT細胞に結合し、腫瘍抗原としてCD20を認識するBi20(FBTA05としても知られる)という三機能二重特異性抗体に向けられている。これらの実施例は、請求項1および明細書全体で特定した他の全ての腫瘍関連抗原に関しても、本発明が使用可能であるという説得力のある証拠を提供する。これらの腫瘍関連抗原全ては、従来使用されていた抗体に基づく免疫療法ではほとんど治療不可能なほど少数しか腫瘍細胞の表面上に存在していなくてもよい。
【0076】
本発明を説明するための図および表は、実施例1および2に関連し、次のとおりに説明される。
【0077】
図1:A−F 異なる抗Her2抗体を用いてフローサイトメトリーにより測定された細胞系統Lox、HCT−8およびSKBR3のHer2/neu発現特性
【0078】
図2a:Her2+3 SKBR−3細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0079】
図2b:Her2+1 HCT−8細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0080】
図2c:Her2ネガティブ Lox細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0081】
図2d:Her2+1 HCT−8細胞の死滅:MDSワークフロー(workflow)によるドナー2のドットプロット結果の一例; *印の番号は、表3に対応する。
【0082】
図3:サイトカインビーズアレイシステム(CBA)を用いて測定された24時間後のクローン原性アッセイ上清中のサイトカイン分泌
【0083】
図4:推定される三細胞複合体(tri−cell−complex)。三機能抗体は、異なる免疫学的メカニズムにより腫瘍細胞の認識と破壊を促進することができる。IFN−γを除く図中のサイトカインに関するリストをまとめなければならない(Bei der Auflistung der Zytokine im Bild sollte man noch IFN−γ aufnehmen)。
注:ADCC、抗体依存性細胞傷害;DC、樹状細胞;NK、ナチュラルキラー細胞;DC−CK1、樹状細胞サイトカイン1;IL、インターロイキン;LFA、リンパ球機能関連抗原;TNF−α、腫瘍壊死因子α;CD、表面抗原分類(出典 Ruf&Lindhofer,2001)
【0084】
図5.Bi20の精製。(A)陽イオン交換クロマトグラフィー(CIEX)。親マウス抗体(ピーク1)を、HR10/10 high performance SP−Sepharose column上で、三機能抗体(ピーク2)から分離した。(B)Bi20三機能抗体精製の等電点電気泳動(IEF)。親ラット抗体(26II6)を、プロテインAアフィニティークロマトグラフィー(AC Bi20)後に除去し、親マウス抗体(TPA10)を、CIEX(CIEX P1 Bi20)により三機能抗体から分離した。精製されたBi20は、前記陽イオン交換クロマトグラフィーのピーク2(CIEX P2 Bi20)に見出された。
【0085】
図6.Bi20が特異的に結合するエフェクター細胞。190ngのBi20を、5×105個の標的細胞とともにインキュベートし、その結合をFACS解析により測定した。Ramos細胞(A)を、CD20アームの標的細胞として用いた。THP−1細胞(B)を、Fc領域用に用いた。拮抗実験のために、細胞を予めRituximab(Rit)とともに所定の過剰量の抗体で30分間インキュベートした。
【0086】
図7.エフェクター細胞の存在下におけるBi20が媒介する効果的なB細胞の減少。(A)1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Mec1)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともにインキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)(neg)前記アッセイにおけるB細胞の絶対数を、100%に設定した。(B)1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。前記B細胞の絶対数を、トリパンブルー色素排除集計およびFACS解析により、1日目、2日目、3日目において測定し、B細胞の減少割合を算出した。
【0087】
図8.Bi20が媒介するT細胞の活性化。(A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。CD4+およびCD8+T細胞上におけるCD25の発現のMFIを、FACS解析により測定した。(B)PBMCおよびRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。CD4+T細胞上におけるCD25の発現のMFIを、1日目、2日目および3日目に測定した。
【0088】
図9.Bi20が誘導するT細胞の増殖。1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、50ng/mlの濃度のBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。T細胞数を、トリパンブルー色素排除集計並びに抗CD5/4抗体および抗CD5/8抗体を用いたFACS解析により測定した。
【0089】
図10.Bi20により媒介される単球の活性化。(A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)またはrituximabとともに1日間インキュベートした。CD14+単球によるCD25の発現のMFIを、FACS解析により測定した。(B)1×106個/mlのPBMCおよび1×105個/mlのTHP−1細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab(Rit)、TPA10、または26II6とともに1日間インキュベートした。CD33+THP−1細胞の活性化を、CD25またはCD40の発現によって測定した。
【0090】
図11.健康なドナーのエフェクター細胞存在下におけるBi20が誘導するCLL B細胞の減少。(A)1×106個/mlのPBMCおよび2×105個/mlのCLL腫瘍細胞を、5、50、もしくは250ng/mlのBi20、250ng/mlのcatumaxomab(Catum)、または250ng/mlのRituximab(Rit)とともに3日間インキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)前記アッセイにおけるB細胞の絶対数を、100%に設定した。MV:CLL−1からCLL−9の平均値。(B)CLL患者検体(1〜14)におけるCD20の発現の概要、細胞系統(Raji、Mec−1)および1体の健康なドナー(PBMC)。MFI=蛍光強度の平均値。
【0091】
表1:用いた抗体およびそれらに対応する検出抗体の特性
* TRION Pharma社、ミュンヘン、$ Roche社、# Micromet社、ミュンヘン
【0092】
表2:用いた細胞系統およびそれらのHer2/neuスコア
*:Ross et al., Molecular and Cellular Proteomics 3:379−398, 2004に準拠
【0093】
表3:クローン原性アッセイサンプル
Ab=抗体、μg=マイクログラム、ng=ナノグラム
【0094】
表4.Bi20が媒介するサイトカイン分泌。1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Ramos、Mec1、Granta、DOHH−2)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに、3日間インキュベートした。上清を収集し、サイトカイン分泌を、CBA解析およびFACS解析を用いて測定した。
【0095】
表5.Bi20が媒介する自己設定におけるin vitroでのCLL B細胞の除去。B細胞およびT細胞の割合(%)、B細胞上におけるCD20の発現(MFI)、およびE:T比を、CLL PBMCの調製後直ちにインキュベート前に測定した。CLL細胞を所定量のBi20またはrituximabとともに、0、3、6、および9日目にインキュベートした。B細胞の除去を、患者検体に応じて3日目から10日目の間に測定した。B細胞の除去を、減少したB細胞の割合(%)で示す。T細胞活性化:CD4+およびCD8+T細胞上におけるCD25の発現は、少なくとも5倍(++)上方制御された。エフェクター/ターゲット比(E:T)を、単球数、NK細胞数およびT細胞数に対するB細胞数として定義した。10%以下のB細胞の除去は、ネガティブとみなした。
【実施例】
【0096】
実施例1
実施例1は、Her2/neuに結合する三機能二重特異性抗体をHercepTest(登録商標)におけるスコア1+の患者の治療に用いた場合の性能に関するキャラクタリゼーションについて記載する。
【0097】
方法
[細胞系統および抗体]
Rexomun(登録商標)(Trion Pharma社製、ミュンヘン)は、一方の結合アームにより腫瘍関連抗原Her2/neuを標的にし、他方のアームによりT細胞上のCD3分子に結合する三機能抗体である。さらに、Rexomun(登録商標)は、マウスIgG2aおよびラットIgG2bアイソタイプからなる強力なFc領域を通じてFcγRI陽性およびFcγRIII陽性細胞(例えば、マクロファージ、NK細胞、樹状細胞)に結合する。モノクローナル抗体2502A(Trion Pharma社、ミュンヘン)は、Her2/neuに特異的であり、Rexomun(登録商標)に含まれる親抗体の一つである。26II6(Trion Pharma社、ミュンヘン)は、CD3に特異的であり、Rexomun(登録商標)に含まれる親抗体の他の一つである。520C9(ATCC HB−8696)は、Her2/neu受容体を認識する他のモノクローナル抗体である。Trastuzumabは、マウスの抗Her2/neu抗体であるMab4D5(58)から開発されたヒト化抗Her2モノクローナル抗体である。表1に、用いた抗体およびそれらに対応する検出抗体の特性を示す。
【0098】
SKBR3(ATCC H TB−30)は、IHCにより検出されたHer/neuを強力に過剰発現する(HercepTest(登録商標)スコア+3)乳ガン細胞系統であり(58)、HCT−8(ATCC CCL−244)は、大腸ガン細胞系統であり、Loxは、メラノーマ細胞系統である(表2)。
【0099】
[Her2/neuスコアリング]
HCT−8細胞におけるHer2/neuの状態(status)を、HercepTest(登録商標)キット(DAKO社、グロストラップ、デンマーク)を用いてIHCにより評価し、製造者のマニュアルに従って、ミュンヘン大学(LMU ミュンヘン)の病理学研究所(Institute of Pathology)の力を借りて実施した。
【0100】
[FACS解析]
FACS解析を、5×105個の標的細胞(SKBR3、HCT−8またはLox)を用いて実施した。細胞を、100ngのモノクローナル抗体または100ngの三機能抗体Rexomun(登録商標)とともに、45分間4℃でインキュベートした。それから、検体をMg2+およびCa2+を含まないリン酸緩衝生理食塩水(PBS)(PAN Biotecs社、アイデンバッハ)を用いて洗浄し、対応する検出抗体(Dianova社、ハンブルク)とともに45分間4℃でインキュベートした(表1)。PBSを用いてもう一度洗浄工程を行った後、細胞を0.01μg/mlの臭化エチジウムを含むPBS(Sigma社、ミュンヘン)中に再懸濁した。結合した抗体の量を、オーバーレイヒストグラム(overlay histogram)における蛍光強度の平均値により解析した。抗ヒト検出抗体のみを用いた検体、または26II6(抗CD3)およびそれに対応する検出抗体(アイソタイプコントロール)とともにインキュベートした検体を、ネガティブコントロールとした。すべてのFACS解析は、FACSCalibur(Becton Dickinson社、ハイデルベルク)により実行し、Cell Quest ProまたはWinMDI2.8を用いて解析した。
【0101】
[クローン原性アッセイ]
抗体が媒介する腫瘍の除去を、刺激されていないエフェクター細胞を用いた長期クローン原性アッセイを用いて検討した。そのために、106個のPBMCを、5%の標的細胞(SKBR3、HCT−8またはLox)とともにスパイクし(spike)、24穴プレート(Greiner社)に播種した。抗体を、表3に示す異なる濃度および組み合わせで添加した。標的細胞のみの検体およびいずれの抗体も添加していない標的細胞とエフェクター細胞との混合物からなる検体を、コントロールとした。検体を、37℃/5%の二酸化炭素でインキュベートし、RPMI1640、L−グルタミンおよび10%のウシ胎児血清(FCS)を3日に一度ずつ供給した。8日間のインキュベート後、細胞を前記24穴プレートから収集し、Mg2+/Ca2+を含まないPBSを用いて洗浄し、Neubauer Cell Chamberにおいて、トリパンブルーによる死細胞の排除により計数した。それから、生細胞をスライドあたり2.5×105個の細胞濃度で、サイトスピンスライド(Menzel社)上で遠心沈殿した。遠心沈降後の検体を一晩乾燥させ、続いて免疫細胞化学法にかけた。異なる3体のドナー由来のPBMCを用いて、実験を3回繰り返した。
【0102】
[免疫細胞化学法]
サイトスピンスライド上での非特異的結合特性を、10%のヒト血清を用いて飽和させた。SKBR3およびHCT−8細胞を、スライドあたり1.5μgの抗サイトケラチン8、18、19抗体(A45B−B3、マウスIgG1、Micromet社、ミュンヘン)を用いて染色し、Lox細胞を、スライドあたり1.5μgの抗EGFR抗体(ミュンヘンのDr.Htunに快く提供していただいたマウスIgG2a)を用いて染色した。サイトケラチン染色用の抗マウスIgG1 Alexa Fluor 477(FITC)抗体(Molecular probes社、ユージーン)、またはEGFR染色用の抗マウスIgG2a Alexa Fluor 477(FITC)抗体(Molecular probes社、ユージーン)を、検出抗体として、スライドあたり1.5μgで用いた。サイトスピンスライドを、FITC標識された細胞を計数するコンピュータ化された画像解析を用いたMicrometastasis detection system(MDS、Applied Imaging社)により解析した。
【0103】
[+1のHer2/neu発現を示す大腸ガン細胞系統であるHCT−8]
Her2/neuの+1発現を有する細胞系統を見出すために、Her2/neu発現のレベルを、異なる細胞系統において検討した。
【0104】
そのために、前記モノクローナルHer2/neu抗体である2502A、520C9、Trastuzumabおよび前記三機能抗体Rexomun(登録商標)を用いたFACS解析を実施した。前記モノクローナル抗CD3抗体である26II6を、ニセのコントロールとした。図1A−Fに、SKBR−3、HCT−8またはLox細胞に用いた抗体の結合特性を示す。異なる検出抗体のみを用いたフローサイトメトリー検体を、コントロールとし(図1A)、それに対応する検出抗体を用いた26II6(抗CD3)を、アイソタイプコントロールとした(図1B)。
【0105】
メラノーマ細胞系統であるLoxは、予想どおりHer2/neu受容体の発現を示さなかった(図1C−F)。一方、SKER3細胞は、モノクローナル抗体520C9(図1C)、2502A(図1D)およびTrastuzumab(図1E)を用いたHer2/neuに対して重度に染色された。Rexomun(登録商標)は、SKBR3細胞を低度に染色した。おそらく、その一方のアームの結合特性のためであろう(図1F)。SKBR3細胞と比較して、HCT−8細胞は、すべての用いたHer2抗体に対するHer2/neu受容体の発現が明らかに弱い(図1C−F)。このことは、これらの細胞のさらなる評価を正当化する。加えて、HCT−8細胞のHer2/neu発現を、DAKO社のHercepTest(登録商標)を用いて解析した。前記テストを、ミュンヘン工科大学(TU、ミュンヘン)の病理学研究所の力を借りて実施した。前記大腸ガン細胞系統であるHCT−8は、実施したDAKOテストにおいて、FACS解析の結果を確認することで、Her2/neuの+1発現を示した。表2に、用いた細胞系統、それらのHer2/neuスコアおよびHer2/neu発現の測定に適用された方法の概要を示す。
【0106】
[Rexomun(登録商標)は、in vitroにおいて、Her2/neu+3細胞系統およびHer2/neu+1細胞系統を死滅させることが可能である]
腫瘍部位に対して異なるタイプの免疫細胞を補充すると同時に活性化するRexomun(登録商標)の能力は、Rexomun(登録商標)が、Her2/neuを低度(+1)に発現している腫瘍細胞を除去することも可能であるかもしれないという仮説を導いた。
【0107】
そのために、長期細胞障害性解析(クローン原性アッセイ)を確立した。3体の健康なドナーのPBMCを、Her2/neu+3発現する細胞系統であるSKBR−3、Her2/neuネガティブのLox細胞系統またはHer2/neu+1発現するHCT−8細胞系統とともに、8日間インキュベートした。異なる抗体濃度のTrastuzumab(100μg/ml〜1μg/ml)、またはRexomun(登録商標)(100ng/ml〜1ng/ml)を、表3に示すように添加した。腫瘍細胞の死滅を、8日後に前記細胞を収集し、それらをサイトスピンスライドに移し、前記細胞を抗CK8、18、19抗体(SKBR−3およびHCT−8)、または抗EGFR抗体(Lox)と、これらに対応するAlexa−Fluor FITC抗体とを用いて染色することで評価した。それから、前記染色したサイトスピンスライドを、Micrometastasis Detection System(MDS、Applied Imaging社、英国)上のSLIDE SCANプログラムを用いるコンピュータ化された画像解析により評価した。
【0108】
この研究において、前記三機能抗体であるRexomun(登録商標)(抗Her2/neu×抗CD3)を、低度にHer2/neuを発現する腫瘍細胞系統であるHCT−8(HercepTestスコア:+1)の腫瘍細胞増殖を阻害する能力において、ヒト化抗Her2/neuモノクローナル抗体であるTrastuzumab(登録商標)と比較した。
【0109】
前記細胞系統SKBR3(+3 Hercepスコア)、HCT−8(+1 Hercepスコア)およびLox(Her2/neuネガティブ)におけるHer2/neu発現を、FACS解析により測定した。一方、HCT−8細胞を、HercepTest(登録商標)によってもテストした。Her2/neuに対する種々の抗体を用いたFACS解析は、SKBR3細胞においては強力なHer2発現を示し、HCT−8細胞においてはより弱い発現を示し、Lox細胞においてはHer2/neuの発現を示さなかった。これらの結果は、HCT−8細胞の+1HercepTest(登録商標)スコアと一致する。三機能抗体であるRexomun(登録商標)の蛍光の平均値は、モノクローナル抗体の前記値より、わずかに低い。モノクローナル抗体が二価の結合であるのと比較して、Rexomun(登録商標)が一価の結合であるためである(図1:A−F)。
【0110】
前記低度にHer2/neuを発現する細胞系統であるHCT−8の増殖を阻害するRexomun(登録商標)およびTrastuzumab(登録商標)の能力をテストするために、クローン原性アッセイを実施した。8日後の培養細胞を、培養皿から収集し、サイトスピンスライド上で遠心沈降し、腫瘍細胞を染色した。抗体を添加していないすべてのコントロール検体(24穴プレートあたり12,500個の腫瘍細胞)(図2a〜c、検体A1、B1、C1)は、サイトスピン染色後のMDSにおいて、細胞系統SKBR3、HCT−8およびLoxに関して腫瘍細胞の増殖を示した。スパイクした腫瘍細胞、または抗体を添加していない250000個のPBMCを含むスライドでは、予想どおり全てにおいて腫瘍細胞が見られなかった(図2a〜c、検体A2、B2、C2)。PBMCを5%のSKBR3、HCT−8またはLox細胞とともに、抗体を添加せずに混合することで、腫瘍の増殖を、同種間のコントロールにおいて明確に可視化した(図2a〜c、検体A3、B3、C3)。
【0111】
Her2+3細胞系統であるSKBR−3の腫瘍細胞の増殖は、予想どおり100μg、50μg、10μgおよび1μgのTrastuzumab(登録商標)の添加により阻害された(図2a、検体A4〜A8)。さらに、Rexomun(登録商標)も、わずか100ng、50ng、10ngまたは1ng添加しただけで、腫瘍の増殖を阻害することが可能であった。検体A9〜A12に示すように(図2a)、腫瘍細胞の増殖は、Trastuzumab(登録商標)よりも効果的に阻害された。
【0112】
Her2/neuネガティブ細胞系統であるLoxは、Her2/neu受容体が存在することにより駆り立てられる(driven)腫瘍細胞除去の特異性を示すTrastuzumab(登録商標)、またはRexomun(登録商標)を種々の量で添加しても、腫瘍細胞の増殖は阻害されなかった(図2c、検体C4〜C12)。
【0113】
大腸ガン細胞系統であるHCT−8(Her2/neuスコア+1)の増殖は、種々の量(100μg〜1μg)のTrastuzumab(登録商標)によって、際立っては阻害されなかった(図2b、検体B4〜B8)。このことは、Her2/neu+1の乳ガン細胞系統であるMCF−7(5)に関する従来の知見と一致する。MCF−7細胞(ATCC HTB−22)は、TNF−αによる増殖阻害を示すため、三機能抗体であるremovab(登録商標)(抗EpCAM×抗CD3)に類似するrexomun(登録商標)は、その活性化特性によりTNF−α分泌の引き金を引いてしまうとして、これらの細胞をクローン原性アッセイに用いることができなかった(59)。しかしながら、Rexomun(登録商標)は、1穴あたり100ng、50ng、10ngという低濃度で、+1のHer2/neuの細胞系統であるHCT−8を破壊することが可能であった(図2b、検体B9〜B11)。1ngのRexomun(登録商標)では、HCT−8細胞の腫瘍細胞増殖の阻害に成功しなかった(図2b、検体B12)。図2dに、標的としてHCT−8細胞を用いるドナー2のクローン原性アッセイ関する元のMDSデータプロットを示す。
【0114】
結論として、Rexomun(登録商標)は、+3または+2の過剰発現/遺伝子増幅を示す乳ガン患者のためだけでなく、低度のHer2/neu発現レベルの患者のための治療の選択肢となる。
【0115】
実施例2
ここでは、ヒトCD3およびヒトCD20に対する新規な三機能二重特異性抗体であるBi20の産生と性能に関するキャラクタリゼーションについて記載する。特に、腫瘍抗原であるCD20を低レベルでしか発現していない患者の治療に用いられるその能力について記載する。
【0116】
CD20は、35kDaの糖化されていないリン酸化タンパク質であり、B細胞分化において役割を果たすカルシウムチャネルとして働くと言われている24。しかしながら、CD20欠損マウスが正常なB細胞発生を示し、表現型的にも正常であることから25、その正確な機能は未だに不明である。CD20は、いくつかの理由により免疫療法のための標的として選択される:(i)CD20は、正常なB細胞および悪性のB細胞においては独占的に発現されるが、血液学的な前駆体細胞または他のヒトの組織の細胞においては発現されず26、(ii)CD20は、ほとんどのB細胞リンパ腫細胞において発現され27、(iii)CD20は、抗体との結合のために噴出され、または分泌されない28、29。さらに、腫瘍治療用の標的としてのCD20の適合性は、Rituximabの使用、特に従来の化学療法との組み合わせによる使用の成功により臨床的に確認されている。
【0117】
本発明の研究においては、このtrAbが、効果的にヒトB細胞腫瘍細胞および、低レベルでしかCD20を発現していない慢性リンパ性白病(CLL)患者由来の細胞を死滅させることを実証するために、前記Bi20のin vitroにおける特性について詳細に解析を行った。腫瘍細胞の除去のために、エフェクター細胞を予め活性化させる必要がなく、前記腫瘍細胞の除去は、典型的なTh1サイトカインパターン、並びにT細胞および単球の強力な活性化に伴って起こった。臨床応用のための重要な必要条件としては、Bi20が、容易に精製され、充分な量を産生し得ることである。従って、このtrAbは、治癒不能な非ホジキン悪性リンパ腫(NHL)患者、およびCLL患者のための他の新規な治療の選択肢を提案し得る。
【0118】
材料および方法
[抗体および試薬]
CD20特異的モノクローナルIgG2a抗体を産生するマウスの細胞系統であるTPA10を、26II6を用いてCD3特異的IgG2b抗体を分泌するラットの細胞系統と融合し、結果として、Bi20(FBTA05)を産生するクアドローマ細胞系統であるTPBs05を得た。Catumaxomab(Removab(登録商標))を、26II6由来のCD3特異的アームおよび抗EpCAM特異性からなる三機能性のコントロール抗体として用いた。キメラの抗CD20抗体であるRituximab(Mabthera(登録商標))は、Roche社(バーゼル、スイス)から入手した。前記抗CD20抗体および対応するアイソタイプは、Beckman Coulter Immunotech社(クレフェルト、ドイツ)から入手した。FACS解析に用いた他のすべての抗体は、BD Biosciences社(ハイデルベルク、ドイツ)から購入した。ヨウ化プロピジウムは、Sigma Chemicals社(ダイゼンホッフェン、ドイツ)から入手した。
【0119】
[細胞系統およびPBMCの調製]
CLL細胞系統であるMec1を、Dr. Michael Hallek(GSF、ミュンヘン、ドイツ)に快く提供していただいた。バーキットリンパ腫(Burkitt’s lymphoma(BL))細胞系統であるRamosは、ATCC(米国)から入手した。前記BL細胞系統であるRaji、前記NHL細胞系統であるDOHH−2およびGranta、並びにAML細胞系統であるTHP−1は、DSMZ(ブラウンシュヴァイク、ドイツ)から購入した。細胞を、10%の熱失活したFCS、非必須アミノ酸、ピルビン酸ナトリウムおよびL−グルタミン酸塩(PAN−Biotech社)を添加したRPMI−1640培地(PAN−Biotech社、アイデンバッハ、ドイツ)中で生育した。ヒト抹消血単核球細胞(PBMC)は、PANCOLL(PAN−Biotech社)を用いて密度勾配遠心により、健康なドナーのヘパリン処理した血液から単離した。CLL患者由来の抹消血検体は、インフォームドコンセント(informed consent)の後に入手した。CLLの診断は、標準的な臨床的診断基準および研究室的診断基準(laboratory criteria)に従った。CLL細胞は、精製後直ちに用い、または後の使用のために凍結保存した(cryo−conserved)。CLL細胞は、10%のFCSを添加したIMDM(PAN−Biotech社)中で培養した。自己設定におけるアッセイには、新鮮に調製したCLL細胞のみを用いた。
【0120】
[Bi20の産生および精製]
Bi20を、クアドローマ技術を用いて産生した30。クアドローマ細胞の上清を、標的細胞に結合するtrAb用に、FACS解析によりテストした。trAb産生細胞を、抗体産生の安定性を向上させるために数回にわたってサブクローニングし、マスター細胞バンクを確立した。trAbを、クアドローマ細胞の培養上清から、説明されているように31、AKTA purifier 100(Amersham Biosciences社、ウプサラ、スウェーデン)を用いて、プロテインAアフィニティクロマトグラフィーおよびイオン交換クロマトグラフィーにより精製した。
【0121】
[等電点電気泳動]
抗体検体を、pH7〜11のIsoGel−Agarose−IEF(Cambrex Bio Science社、ロックランド、米国)に、スポットし(spotted)、75分間、1500V、50mA、および25Wで分離した。抗体のアイソフォームを、クーマシーブリリアントブルー(Sigma−Aldrich社、ミズーリ州、米国)を用いて染色することにより可視化した。ゲルを、解析のためにスキャンした。
【0122】
[FACS解析]
標的細胞への抗体の結合、エフェクター細胞の活性化、関連するCD20の発現、および細胞のサブタイプ組成を、488nmのアルゴンレーザーを備えたFACScalibur(BD Biosciences社、ハイデルベルク、ドイツ)を用いて、フローサイトメトリーにより解析した。データを、Cellquest Software(BD Biosciences社)を用いて解析した。
抗体の結合を解析するために、5×105個の細胞(Ramos、JurkatまたはTHP−1)を、所定の濃度のBi20とともに、30分間4℃でインキュベートし、2%の熱失活したFCSを添加したPBSを用いて洗浄し、およびFITC−標識されたマウスの抗ラットIgG検出抗体(Ramos)、またはFITC標識されたラットの抗マウス抗体(Jurkat、THP−1)とともに、30分間4℃でインキュベートした。
Bi20の活性化特性を決定するために、1×106個/mlのPBMCおよび2×105個/mlのヒトの腫瘍細胞を、前記インキュベートした濃度の抗体とともに、1、2、および3日間37℃で、RPMI培地においてインキュベートした。細胞を収集、洗浄し、FITCまたはPE標識されたCD25およびCD69に対する検出抗体とともに、30分間4℃でインキュベートした。
細胞のサブタイプ組成を、下記の抗CD抗体混合物(FITC/PE/APC)を用いて、3×106個のPBMCを染色することにより決定した:14/19/5、4/8/5、4/8/25、45/3/5および16/56/5。
【0123】
[B細胞減少解析]
Bi20が媒介するB細胞減少を、バイオアッセイを用いて測定した。健康なドナーから入手したPBMC(1×106個/ml)に、2×105個/mlの所定の腫瘍細胞(Raji、Ramos、Mec1、DOHH−2もしくはGranta)、またはCLL患者の細胞(エフェクター対標的比 5:1)を添加し、所定の濃度における異なる抗体の存在下において総容積1mlでインキュベートした。1日目、2日目および3日目における細胞を、収集、洗浄して、生きている(viable)B細胞を、PE結合抗CD19検出抗体を用いてFACS解析により解析した。生きている細胞の総数を、トリパンブルー色素排除集計により測定した。
自己CLL患者検体中の抗体が媒介するB細胞減少を検討するために、CLL患者から単離したPBMCを、2×106個/mlの密度で蒔いた。抗体を、所定の濃度で0日目、3日目および6日目に添加した。FACS測定およびトリパンブルー色素排除集計を、所定の時点で実施した。
【0124】
[サイトカインの測定]
Bi20に誘導されたサイトカインの放出を、B細胞減少実験に用いた条件と同じ条件下において測定した。腫瘍B細胞系統および健康なドナーからのPBMCを、50ng/mlの所定の抗体とともにインキュベートした。3日後、上清を収集した。サイトカインを、ヒトTh1/Th2細胞数測定ビーズアレイ(CBA、BD Biosciencess社、ハイデルベルク、ドイツ)を用いて測定した。前記CBAは、INF−γ、TNF−α、IL−2、IL−4、IL−6およびIL−10の解析を同時に行うことが可能である。データの取得および解析を、製造者のプロトコールに従って実施した。
【0125】
結果
[Bi20の産生および精製]
前記trAbであるBi20を、クアドローマ細胞において産生し、プロテインAアフィニティクロマトグラフィーにより獲得した。強力なラットの抗体は、プロテインAに結合しないため、溶出液が不要である。それから、親マウス抗体等の不純物を、陽イオン交換クロマトグラフィー(CIEX)により除去した(図5A)。マウス抗体を、148mMのNaClで溶出した(図5A、ピーク1)。一方、trAbを、320mMのNaClで溶出した。精製されたtrAbの等電点電気泳動は、pIが8.65〜8.15の範囲において明確なバンドパターンを示した。trAbは、CIEX分離のピーク2においてのみ検出された。親抗体の混入は検出されなかった。
【0126】
[Bi20の結合特性]
Bi20の三機能の性質を実証するために、その特異的な標的細胞への結合を検討した。CD20結合アームの機能性を確認するために、CD20+バーキットリンパ腫細胞系統であるRamosを、CD3結合アームのためにCD3+T細胞系統であるJurkatを、FcγRIに結合することを確認するために単球細胞系統であるTHP−1を用いた(図6)。CD20へのBi20の結合を確認するために、Rituximabを用いた競合的結合アッセイを実施した。Ramos細胞上でのBi20の結合を、予め前記細胞を21倍の過剰なRituximabとともにインキュベートすることにより、完全に阻止することができた(図6A)。同様に、THP−1細胞へのBi20の結合を、Rituximabにより、競合することができた(図6B)。Jurkat細胞上のCD3へのBi20の結合は、Ramos細胞上でのCD20へのBi20の結合に匹敵した(データを示さず)。前記Bi20のCD3結合アームは、特性がはっきりしているtrAbであるcatumaxomabおよびertumaxomabにおいて対応するアームと同一であるため、前記CD3結合アームの特異性のさらなる解析を、実施しなかった。要約すれば、これらのデータにより、Bi20の三価の結合機序および結合特性が確認された。
【0127】
[Bi20が媒介するB細胞の除去]
B細胞の死滅についてのBi20の効果を研究するために、FACSに基づくバイオアッセイを開発した。事前の、または同時の刺激のいずれもしていない新鮮に調製したPBMCを、腫瘍B細胞と5:1の比率で混合した。さらに、50ng/mlの所定の抗体を混合し、3日間インキュベートした。細胞を収集し、分集団の組成をFACS解析により決定した。トリパンブルー色素排除集計を、細胞の絶対数を得るために実施した。
標的細胞として、前記CD20+バーキットリンパ腫細胞系統であるRajiを用い、50ng/mlのBi20を用いたことで、効果的なB細胞の死滅が観察された(図7)。コントロールとして、前記trAbである、B細胞と結合しない(データを示さず)catumaxomab(抗EpCAM×抗CD3)、並びに前記親抗体26II6(抗CD3)およびTPA10(抗CD20)を用いた。Catumaxomab(Catum)および親抗体の組み合わせは、いくらかのB細胞減少を誘導した。おそらく、INF−γおよびTNF−α等のサイトカインの放出によるものであろう(表4)。このことは、Bi20の抗腫瘍活性が、実際にはその三機能デザイン(design)により引き起こされることを示している。これらの実験的な条件(低濃度の抗体およびE:T比)下では、Rituximabは、軽微なB細胞数の減少しか媒介しなかった。CLL細胞系統であるMec1を用いた比較可能なデータを入手した。そのデータでは、CD20の発現がRaji細胞より2.5倍低かった(MFI 179対428、データを示さず)。
【0128】
これらの結果は、Bi20が媒介したB細胞の減少をより詳細に解析するのに勢いをつけた。時間−動態解析(Time−kinetic analyses)は、わずか24時間後にRaji細胞の減少を示した(図7B)。少なくとも35%のB細胞が、0.5ng/mlという低濃度の抗体において除去された。250ng/mlおよび50ng/mlの濃度のBi20においては、2日目および3日目それぞれにおいて、完全なB細胞の減少が観察された。Mec1細胞においても匹敵する結果が見出された(データを示さず)。
さらに、B細胞系統における代表的な他のリンパ腫の実体を、効果的に死滅させることができるかどうか検討した。DOHH−2(濾胞性リンパ腫)、Granta(マントル細胞リンパ腫)、またはRamos(バーキットリンパ腫)を標的細胞として用いた場合、効果的なB細胞減少を検出した(データを示さず)。
【0129】
[同時発生的なB細胞結合に依存しないBi20が媒介するT細胞の活性化]
前述した、例えば、抗CD28抗体を用いて予め刺激した後のみにB細胞の細胞傷害性を示す二重特異性抗リンパ腫抗体32とは対照的に、Bi20により誘導された効果的なB細胞の溶解には、T細胞または単球の前活性化が必要ではなかった。そのために、エフェクター細胞が、Bi20が結合することにより活性化され得るかどうかを問うた。PBMCおよびRaji細胞を、前記所定の抗体とともにインキュベートした。CD4+およびCD8+T細胞上の活性化マーカーであるCD25の上方制御を、フローサイトメトリーにより測定した(図8A)。Bi20およびcatumaxomabの両方が、CD4+およびCD8+T細胞上のCD25の明確な上方制御を誘導した。このことは、trAbがCD3およびCD20に同時に結合することが、T細胞の活性化には必要ないことを示している。CD25の上方制御は、前記親抗体26II6(抗CD3)とTPA10(抗CD20)との組み合わせによっても観察された。予想どおり、Rituximabは、いずれのT細胞の活性化も誘導しなかった(図8A)。
【0130】
図8Bに示すように、T細胞の活性化は、インキュベートする期間と抗体の濃度とに依存する。最大のCD25発現は、2日目の最も高い濃度(250ng/ml)のBi20において観察された。CD69等の他の活性化マーカーも上方制御されたが、より低度でより短期間なものであった(データを示さず)。
【0131】
[Bi20が誘導するT細胞の増殖]
T細胞の活性化に加えて、前記異なる抗体により誘導されるCD4+およびCD8+T細胞数の増加を測定した(図9)。Bi20を用いたインキュベーションの結果として、CD4+サブセットと比較して、CD8+サブセットのT細胞が優先的な増殖を示した(145%対85%のT細胞発現)。CD4+:CD8+比は、1.8:1から1.3:1に変化した。Catumaxomabおよび親抗体はともに、T細胞増殖を誘導したが、Bi20より低度であった。予想どおり、Rituximabは、T細胞増殖を刺激しなかった。特に、前記抗体以外の刺激因子は、増殖を誘導するのに必要ではなかった。
【0132】
[単球の活性化は、同時発生的なT細胞結合には依存するが、同時のB細胞結合には依存しない]
つぎに、単球/マクロファージ等のFcγRI/III+細胞も、Bi20により活性化され得るかどうかという問いに取り組んだ。PBMCおよびRaji細胞を、異なる抗体の存在下においてインキュベートし、CD14+細胞の活性化を測定した。図6Aに示すように、Bi20およびcatumaxomabの両trAbは、CD14+単球/マクロファージ上でのCD25の上方制御を誘導した。このことは、同時のB細胞結合は、単球の活性化に必要ではないことを示している。対照的に、B細胞および単球/マクロファージに結合し、そのFc領域を通じてT細胞とは結合しないRituximabは、CD14+修飾細胞の活性化を誘導しなかった。これらの結果から、T細胞の関与が、修飾細胞の活性化には不可欠であると結論付けられる。
【0133】
この結果を、悪性のB細胞が存在しない場合におけるわずかに異なるシステムを用いて確認した。PBMCおよびTHP−1細胞を、異なる抗体の存在下においてインキュベートした。CD33+であるTHP−1細胞の活性化を、CD25およびCD40の発現によりモニターした(図10B)。THP−1およびT細胞に同時に結合するすべての抗体(Bi20、TPBs01、26II6)は、THP−1細胞を活性化した。対照的に、THP−1およびB細胞には結合するが、T細胞には結合しない抗体(TPA10、Rituximab)を用いた場合は、活性化は観察されなかった。PBMCが存在しない場合におけるTHP−1細胞は、いずれの抗体によっても活性化されなかった(データを示さず)。このことは、同時発生的にT細胞および単球と結合することは、本実験条件において単球を活性化するための絶対的な必須条件であることを示している。
【0134】
[Bi20が誘導するTh1類似のサイトカイン特性]
サイトカインの放出は、モノクローナル抗体を治療的に使用した後に発生する免疫反応のひとつであるので33、Bi20により誘導されるサイトカインの特性を研究した。PBMCを、異なる腫瘍細胞(Raji、Ramos、DOHH−2、GrantaまたはMec1)および50ng/mlの抗体とともにインキュベートした。3日後、上清を収集し、IFN−γ、TNF−α、IL−2、IL−4、IL−6およびIL−10の量を解析した(表4)。
【0135】
Bi20は、IL−6を高レベルに誘導した。前記IL−6は、前記他の抗体とともにインキュベートした後の場合には、明らかに低いレベルで検出された。TNF−αは、Bi20またはcatumaxomabによる処理を行った後に検出されたが、Rituximabまたは前記親抗体とともにインキュベートした後には検出されなかった。IL−2の発現における強力な増加が、Bi20とともにインキュベートした後にだけ検出された。これにより、3つの結合パートナー(腫瘍細胞、T細胞および修飾細胞)が、すべて存在した場合にのみIL−2が放出されることを示すtrAbを用いた従来の研究が確認された18。Th2系統のマーカーであるIL−4は、わずかにだけ分泌された。一方、Th1に特異的なサイトカインであるIFN−γは、強力に産生された。興味深いことに、抑制性サイトカイン(inhibitory cytokine)として振る舞い得るIL−10も放出されている。このことは、免疫学的なカウンター反応(counter−reaction)を示唆している。同様の結果は、すべての細胞系統(Raji、Ramos、Mec1、DOHH−2、Granta;表4)においても得られた。
【0136】
[Bi20が媒介するCLL患者由来のB細胞の除去]
つぎに、Bi20が媒介するB細胞の減少が、CLL患者由来の細胞を用いることにより確認できるかを検討した。図11に示すように、9体の異なるドナー由来の細胞を、健康なドナー由来のPBMCおよびBi20(5、50または250ng/ml)、またはコントロール抗体とともに培養した。Bi20の濃度を向上させることにより、CLL B細胞の除去が向上した(最大99%)。Rituximabも、明らかなB細胞減少を誘導したが、250ng/mlの濃度でもB細胞の半分近くが生き残った。このことは、少なくとも本実験条件の範囲内においては、前記trAbであるBi20は、CLL患者由来の原発性腫瘍細胞の除去に関して、Rituximabよりもいっそう強力であることを示している。この場合において、catumaxomabにより媒介された非特異的なB細胞減少は、B細胞系統を用いた従来の実験と比較して、実質的に低下していた(図7および11)。
【0137】
意外なことに、非常に低いレベルでCD20を発現しているCLL B細胞をテストしたときでさえ(CLL2、3および4;図11AおよびB)、ほとんどすべてのB細胞が、Bi20により除去された(それぞれ、99%、93%および98%)。対照的に、Rituximabは、同じ患者グループ由来の細胞を用いた場合、その効果が劣っていたか、またはその効果を示さなかった(それぞれ、76%、79%および1%)。
【0138】
つぎに、Bi20が、健康なドナー由来のエフェクター細胞の添加なしに、自己設定においてCLL B細胞の死滅を誘導できるのかどうかという重大な疑問に取り組んだ。そのために、CLL患者由来のPBMCを、Bi20およびコントロール抗体とともにインキュベートした。好ましくないE:T比および公知のCLLにおけるT細胞アネルギーのために、細胞を、少なくとも6日間(CLL10患者を除く)インキュベートし、抗体を3日おきに添加した。B細胞の減少およびT細胞の活性化の解析を、CLL10患者の場合(3日目)を除いて、6日目から10日目の間に実施した。
【0139】
表2を要約すると、効果的なB細胞の減少(>85%)を、7つの患者検体のうち3つにおいて検出した。1つの検体(CLL13の患者)において、65%の腫瘍B細胞の減少が観察された。興味深いことに、B細胞の減少が、非常に低いレベルでCD20を発現している検体(CLL11、12および13の患者、図11B)でさえ観察された。このことは、同種間の設定(allogeneic setting)において前記結果が得られることにより確認された(図11AおよびB)。同じ濃度でRituximabを用いたコントロール実験では、CLL B細胞が減少しない、または減少が非常に低度であることが示された。
【0140】
効果的なB細胞の減少は、エフェクター対標的比(E:T)に依存すると思われる。最も良い反応を示した患者検体(CLL10、11および12)は、エフェクター細胞の割合も最も高かった。特に、最も好ましいE:T比を有する検体(3.5:1;CLL10の患者由来)は、3日後にだけ効果的なB細胞の除去を示した。
【0141】
CLLのT細胞は、アネルギー性であることが知られており、特にCD4サブセットであることが知られている34。驚いたことに、インキュベーションの終盤におけるCD25の上方制御によるCD4+およびCD8+T細胞の活性化を解析した場合、6つの患者検体のうち5つの検体で前記CD4+およびCD8+サブセットの両方の活性化が効果的に発生していることが見出された(表2)。検体をRituximabとともに処理した場合には、活性化が見られなかった(データを示さず)。これらの結果より、少なくともin vitroにおいてBi20は、腫瘍細胞の存在下でさえCLL検体におけるT細胞アネルギーを克服し得る。
【0142】
前記抗CD20モノクローナル抗体であるRituximabを用いるB細胞リンパ腫の治療における(特に、化学療法との組み合わせにおける)有望な結果にもかかわらず、最終的には、患者は再発してしまう9、35。従って、さらなる治療の選択肢が必要である。二重特異性抗体を使用することが、改善方法の一つであろう。それは、腫瘍細胞に対するT細胞等のエフェクター細胞に向けられる。
【0143】
CD19またはCD20等のB細胞に特異的な抗原に対する二重特異性抗体は、この10〜15年間で集中的に研究がなされてきた。しかし、これまでは中程度の反応を示すわずかな臨床データしか入手できなかった14、36、37。さらに、製造工程が複雑で効果的でなかったため、臨床用途のための充分な量の抗体を産生することが難しかった。ここで、CD20およびCD3に対する新規な三機能二重特異性抗体であるBi20の産生と生産について記載する。特に、マウスIgG2aとラットIgG2bアイソタイプとの組み合わせの使用のみならず、本明細書に記載した他の三機能二重特異性抗体の使用は、種制限的な(species−restricted)重鎖/軽鎖の組み合わせによって、正確に組み合わせられた二重特異性抗体の高収率生産を可能にする30。これらのtrAbは、腫瘍細胞、T細胞およびFcγRI/III+修飾細胞と同時に結合し、腫瘍細胞の根絶を増強することを特徴とする17、18。
【0144】
Bi20は、何らの同時刺激を必要とせずにin vitroにおけるB細胞の除去において非常に効果的であることが示された。B細胞の減少アッセイは、抗体濃度が0.5ng/mlと低度であっても、24時間後に70%近くのRaji腫瘍B細胞を除去することが可能であり、3日後には、腫瘍細胞の完全な根絶が可能であったことが実証された。B細胞系統における代表的な他のNHLの実体(Mec1、GrantaまたはDOHH−2)を用いても、同様の結果が得られた。このことは、腫瘍細胞の除去が、この種の悪性疾患(malignant disease)の特定のサブグループに限定されないことを示している。
【0145】
さらに、前記CLL患者由来のB細胞もまた、アポトーシス耐性が亢進され、CD20の発現レベルが低いという特性を有するCLL B細胞であるにもかかわらず27、38、39、効果的に死滅した(図11AおよびB、表5)。この場合も先と同様に、効果的なCLL B細胞の除去に、T細胞の前活性化または共活性化を必要とせず、前述の他の二重特異性抗体とは対照的であった。例えば、前記二重特異性マウス抗体であるBis20(マウスIgG1 CD20×マウスIgG2b CD3)は、実験に用いたE:T比は同じであるが、10倍の抗体濃度、予め活性化されたT細胞および短いインキュベーション時間において、60%近くのB細胞の減少を誘導した40。抗CD20×抗CD3二重特異性抗体を用いる研究において、腫瘍細胞(Raji)の溶解は、末梢血リンパ球(PBLs)を、予めIL−2を用いて刺激した場合においてのみ達成された。1000ng/mlの濃度の二重特異性抗体、および20:1のE:T比であっても、約35%の細胞しか溶解されなかった41。さらに、Cochloviusらは、1〜2.5μg/mlの抗体濃度における効果的なB細胞の溶解に、CD28による予め活性化されたPBLおよびT細胞の同時刺激が必要である抗CD3×抗CD19二重特異性抗体を示した32、42。
【0146】
Bi20は、健康なドナーのPBMCを添加しない自己設定においてさえ、7つの患者検体のうち4つの検体において効果的な腫瘍細胞の死滅を誘導する。Rituximabとは際立って対照的に、Bi20が媒介するB細胞の減少は、CLL腫瘍細胞による非常に低レベルのCD20発現下においてさえも効果的である。そしてそのB細胞の減少は、自己設定および同種間設定(allogenetic setting)について言える(表5および図11)。この知見は、おそらく、CLL腫瘍細胞によるCD20の発現が低いために、Rituximabが単独療法として限定的な効果しか示さないというCLL研究の臨床データに基づいている3、43。
【0147】
いくつかの他の二重特異性抗体44、45とは対照的に、Bi20は、抗CD28抗体またはIL−2等の任意の同時刺激分子が存在しない場合においても、CD4+およびCD8+T細胞サブセットのいずれも効果的に活性化した。結果的に、高いレベルのINF−γを検出した。前記INF−γは、刺激されたT細胞により放出されることが知られており、Th1型反応の指標(indicator)である46。T細胞の活性化も、T細胞サブセットの両方の増殖により伴って起こる。
【0148】
T細胞の活性化は、健康なドナーのT細胞だけでなく、B−CLL患者由来のT細胞を用いた場合にも観察された(表5)。これらのT細胞は、多くの場合アネルギーの特徴を有する。おそらく、CD28およびT細胞受容体の発現が低いためであろう47、48。本実験においては、実質的なCLL B細胞の減少が観察されない検体においてさえも、CD4+およびCD8+T細胞サブセットの活性化を検出した。いくつかの検体(CLL−2、−4、−14)におけるB細胞の減少が限定的なのは、インキュベート期間が短いためかもしれない。前記インキュベーション期間は、in vitroにおけるB−CLL細胞の生存率がわずかであるために制限されてしまう。活性補助因子(coactivator)依存性T細胞の活性化に基づく比較可能なB細胞の減少が、単鎖CD19×CD3二重特異性抗体に関して近年述べられており、さらに、前記二重特異性抗体の方式(format)の効力を確認している49、50。従って、本発明者らの成果は、Bi20が、in vitroだけでなくin vivoにおいても、CLLのT細胞のアネルギーを克服する可能性を有しているかもしれないことを示す。
【0149】
Bi20は、T細胞に加えて、FcγRI+細胞に結合し、FcγRI+細胞を活性化する。腫瘍標的細胞およびFcγRI+細胞の結合が、活性化にとって充分ではないため、活性化は、親抗体であるTPA10において示されるように、Bi20のT細胞に同時に結合することに依存する(図10AおよびB)。trAbのFc領域の重要性は、免疫担当性のマウスB細胞リンパ腫モデルにおける近年の本発明者等による知見により裏づけられ、長期間持続する抗腫瘍免疫の誘導は、前記trAbのFc領域の機能性に依存した。これらの実験において、trAbのF(ab)2断片の使用は、これらのマウスの生存期間を減少させた20。この結果は、in vitroおよびマウスモデルにおいて、単一の二重特異性抗体と比較して高い抗腫瘍活性を示す抗CD19×抗FcγRIIIと抗CD19×抗CD3との二重特異性抗体の組み合わせを示す近年の出版物と一致する51。しかしながら、両ケースにおいては、抗CD28抗体によるT細胞の付加的な同時刺激が必要であった。これらの結果は、Bi20により媒介されるものとして、同時に起こるT細胞およびFcγR+エフェクター細胞の結合および活性化が、腫瘍細胞とT細胞単独に結合する場合より、さらに効果的に腫瘍細胞を死滅させることが可能であることを示している。
【0150】
総合すれば、本発明者らの成果は、腫瘍細胞によるCD20の発現が非常に低い場合であっても、trAbであるBi20が、強力な腫瘍B細胞減少の誘導因子(inducer)であり、前活性化または共活性化のいずれにも依存しないことを示す。同時に発生するT細胞および修飾免疫細胞の活性化、並びにそれらの腫瘍細胞の減少は、in vitroにおいてだけでなく、in vivoにおいても、前述のCLLのT細胞アネルギーを克服する。従って、本明細書中に記載の前記三機能二重特異性抗体は、かねてから治癒不能とされてきた非ホジキンリンパ腫および、本明細書に記載したCD20の発現が低い患者におけるCLLを治療するための新しい治療の選択肢を提案する。
【0151】
参照文献
1. Greiner TC, Medeiros LJ, Jaffe ES. Non−Hodgkin’s lymphoma. Cancer. 1995;75:370−380.
2. Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review, 1975−2003. http://seercancergov. 2006.
3. Lin TS, Lucas MS, Byrd JC. Rituximab in B−cell chronic lymphocytic leukemia. Semin Oncol. 2003;30:483−492.
4. Dillman RO. Treatment of low−grade B−cell lymphoma with the monoclonal antibody rituximab. Semin Oncol. 2003;30:434−447.
5. Blum KA, Bartlett NL. Antibodies for the treatment of diffuse large cell lymphoma. Semin Oncol. 2003;30:448−456.
6. Moreton P, Hillmen P. Alemtuzumab therapy in B−cell lymphoproliferative disorders. Semin Oncol. 2003;30:493−501.
7. Hiddemann W, Dreyling M, Unterhalt M. Rituximab plus chemotherapy in follicular and mantle cell lymphomas. Semin Oncol. 2003;30:16−20.
8. Montserrat E. Rituximab in chronic lymphocytic leukemia. Semin Oncol. 2003;30:34−39.
9. Smith MR. Rituximab (monoclonal anti−CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359−7368.
10. Mavromatis BH, Cheson BD. Novel therapies for chronic lymphocytic leukemia. Blood Rev. 2004;18:137−148.
11. Nagy ZA, Hubner B, Lohning C, et al. Fully human, HLA−DR−specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8:801−807.
12. Baeuerle PA, Kufer P, Lutterbuse R. Bispecific antibodies for polyclonal T−cell engagement. Curr Opin Mol Ther. 2003;5:413−419.
13. Peipp M, Valerius T. Bispecific antibodies targeting cancer cells. Biochem Soc Trans. 2002;30:507−511.
14. Manzke O, Tesch H, Borchmann P, et al. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. I. Clinical phase I evaluation. Int J Cancer. 2001;91:508−515.
15. Manzke O, Tesch H, Lorenzen J, Diehl V, Bohlen H. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. II. Assessment of cellular immune responses. Int J Cancer. 2001;91:516−522.
16. Haagen IA. Performance of CD3xCD19 bispecific monoclonal antibodies in B cell malignancy. Leuk Lymphoma. 1995;19:381−393.
17. Zeidler R, Mysliwietz J, Csanady M, et al. The Fc−region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261−266.
18. Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246−1252.
19. Lindhofer H, Menzel H, Gunther W, Hultner L, Thierfelder S. Bispecific antibodies target operationally tumor−specific antigens in two leukemia relapse models. Blood. 1996;88:4651−4658.
20. Ruf P, Lindhofer H. Induction of a long−lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526−2534.
21. Heiss MM, Strohlein MA, Jager M, et al. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435−443.
22. Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem. 2001;49:911−917.
23. Kiewe P, Hasmuller S, Kahlert S, et al. Phase I trial of the trifunctional anti−HER2 x anti−CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res. 2006;12:3085−3091.
24. Tedder TF, Engel P. CD20: a regulator of cell−cycle progression of B lymphocytes. Immunol Today. 1994;15:450−454.
25. O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Mice carrying a CD20 gene disruption. Immunogenetics. 1998;48:125−132.
26. Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. 1981;67:134−140.
27. Delgado J, Matutes E, Morilla AM, et al. Diagnostic significance of CD20 and FMC7 expression in B−cell disorders. Am J Clin Pathol. 2003;120:754−759.
28. Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell−associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63:1424−1433.
29. Press OW, Howell−Clark J, Anderson S, Bernstein I. Retention of B−cell−specific monoclonal antibodies by human lymphoma cells. Blood. 1994;83:1390−1397.
30. Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species−restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single−step purification of bispecific antibodies. J Immunol. 1995;155:219−225.
31. Ruf P, Jager M, Ellwart J, Wosch S, Kusterer E, Lindhofer H. Two new trifunctional antibodies for the therapy of human malignant melanoma. Int J Cancer. 2004;108:725−732.
32. Cochlovius B, Kipriyanov SM, Stassar MJ, et al. Treatment of human B cell lymphoma xenografts with a CD3 x CD19 diabody and T cells. J Immunol. 2000;165:888−895.
33. Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine−release syndrome in patients with B−cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti−CD20 monoclonal antibody (rituximab, IDEC−C2B8). Blood. 1999;94:2217−2224.
34. Bartik MM, Welker D, Kay NE. Impairments in immune cell function in B cell chronic lymphocytic leukemia. Semin Oncol. 1998;25:27−33.
35. Ghielmini M. Multimodality therapies and optimal schedule of antibodies: rituximab in lymphoma as an example. Hematology (Am Soc Hematol Educ Program). 2005:321−328.
36. Weiner GJ, De Gast GC. Bispecific monoclonal antibody therapy of B−cell malignancy. Leuk Lymphoma. 1995;16:199−207.
37. De Gast GC, Van Houten AA, Haagen IA, et al. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother. 1995;4:433−437.
38. Huh YO, Keating MJ, Saffer HL, Jilani I, Lerner S, Albitar M. Higher levels of surface CD20 expression on circulating lymphocytes compared with bone marrow and lymph nodes in B−cell chronic lymphocytic leukemia. Am J Clin Pathol. 2001;116:437−443.
39. Reed JC, Kitada S, Kim Y, Byrd J. Modulating apoptosis pathways in low−grade B−cell malignancies using biological response modifiers. Semin Oncol. 2002;29:10−24.
40. Withoff S, Bijman MN, Stel AJ, et al. Characterization of BIS20x3, a bi−specific antibody activating and retargeting T−cells to CD20−positive B−cells. Br J Cancer. 2001;84:1115−1121.
41. Xiong D, Xu Y, Liu H, et al. Efficient inhibition of human B−cell lymphoma xenografts with an anti−CD20 x anti−CD3 bispecific diabody. Cancer Lett. 2002;177:29−39.
42. Kipriyanov SM, Moldenhauer G, Strauss G, Little M. Bispecific CD3 x CD19 diabody for T cell−mediated lysis of malignant human B cells. Int J Cancer. 1998;77:763−772.
43. Nguyen DT, Amess JA, Doughty H, Hendry L, Diamond LW. IDEC−C2B8 anti−CD20 (rituximab) immunotherapy in patients with low−grade non−Hodgkin’s lymphoma and lymphoproliferative disorders: evaluation of response on 48 patients. Eur J Haematol. 1999;62:76−82.
44. Manzke O, Berthold F, Huebel K, Tesch H, Diehl V, Bohlen H. CD3xCD19 bispecific antibodies and CD28 bivalent antibodies enhance T−cell reactivity against autologous leukemic cells in pediatric B−ALL bone marrow. Int J Cancer. 1999;80:715−722.
45. Manzke O, Titzer S, Tesch H, Diehl V, Bohlen H. CD3 x CD19 bispecific antibodies and CD28 costimulation for locoregional treatment of low−malignancy non−Hodgkin’s lymphoma. Cancer Immunol Immunother. 1997;45:198−202.
46. Berenson LS, Ota N, Murphy KM. Issues in T−helper 1 development−−resolved and unresolved. Immunol Rev. 2004;202:157−174.
47. Mellstedt H, Choudhury A. T and B cells in B−chronic lymphocytic leukaemia: Faust, Mephistopheles and the pact with the Devil. Cancer Immunol Immunother. 2006;55:210−220.
48. Rossi E, Matutes E, Morilla R, Owusu−Ankomah K, Heffernan AM, Catovsky D. Zeta chain and CD28 are poorly expressed on T lymphocytes from chronic lymphocytic leukemia. Leukemia. 1996;10:494−497.
49. Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and costimulation−independent cytotoxic T−cell response against lymphoma cells catalyzed by a single−chain bispecific antibody. Int J Cancer. 2002;100:690−697.
50. Loffler A, Gruen M, Wuchter C, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti−CD19/anti−CD3 single−chain antibody construct. Leukemia. 2003;17:900−909.
51. Kipriyanov SM, Cochlovius B, Schafer HJ, et al. Synergistic antitumor effect of bispecific CD19 x CD3 and CD19 x CD16 diabodies in a preclinical model of non−Hodgkin’s lymphoma. J Immunol. 2002;169:137−144.
52. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER−2/neu oncogene. Science 1987;235(4785):177−82.
53. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE et al. Studies of the HER−2/neu proto−oncogene in human breast and ovarian cancer. Science 1989;244(4905):707−12.
54. Hynes NE, Stern DF. The biology of erbB−2/neu/HER−2 and its role in cancer. Biochim.Biophys.Acta 1994;1198(2−3):165−84.
55. Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C et al. Differential responses of human tumor cell lines to anti−p185HER2 monoclonal antibodies. Cancer Immunol.Immunother. 1993;37(4):255−63.
56. Sarup JC, Johnson RM, King KL, Fendly BM, Lipari MT, Napier MA et al. Characterization of an anti−p185HER2 monoclonal antibody that stimulates receptor function and inhibits tumor cell growth. Growth Regul. 1991;1(2):72−82.
57. Paik S, Bryant J, Tan−Chiu E, Romond E, Hiller W, Park K et al. Real−world performance of HER2 testing−−ational Surgical Adjuvant Breast and Bowel Project experience. J.Natl.Cancer Inst. 2002;94(11):852−4.
58. Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol.Cell Biol. 1989;9(3):1165−72.
59. Zeidler R, Mayer A, Gires O, Schmitt B, Mack B, Lindhofer H et al. TNFalpha contributes to the antitumor activity of a bispecific, trifunctional antibody. Anticancer Res. 2001;21(5):3499−503.
【0152】
【表1】

【0153】
【表2】
【0154】
【表3】
【0155】
【表4】
【0156】
【表5】
【技術分野】
【0001】
本発明は、腫瘍性疾患(tumor deseases)の予防および治療用の薬学的組成物を製造するための三機能二重特異性抗体の使用に関する。
【背景技術】
【0002】
免疫療法を行う中で単特異性モノクローナル抗体による腫瘍の治療における現在未解決の問題は、低度から中程度の発現レベルの腫瘍標的抗原を有する腫瘍細胞の破壊の効率が不十分なことである。一つの具体例は、標的抗原Her2/neuを低度から中程度のレベルで発現している乳ガンの治療であり、他の具体例は、CD20腫瘍抗原を低度のレベルで発現している非ホジキンリンパ腫および慢性リンパ性白血病の治療である。
【0003】
転移性乳ガンは、ほとんど常に致死性の疾患である。転移が最初に明らかになってからの生存時間のメジアンは、17から20ヶ月の範囲である。内分泌性、細胞傷害性および生物学上の多数の薬剤が一時的な効果を証明しているが、総意としての療法の基準はなく、治療には重大な副作用を生じることが多い。
【0004】
上皮増殖因子(EGF)ファミリーメンバーであるHer2/neuは、約25〜30%の乳ガン患者の腫瘍試料において過剰発現し、より急速な腫瘍の成長およびより悪い予後の原因とされる。
【0005】
EGF受容体のファミリーは、4つのメンバーEGFR(ERBB1)、Her2/neu(ERBB2)、ERBB3、ERBB4を含む。EGFRおよびHer2/neuは、治療標的として鋭意研究されている。細胞内受容体シグナルを阻害する小分子化合物だけでなく、EGFRおよびHer2/neuの細胞外ドメインを標的とする抗体も、既に臨床で使用され、臨床上の効果が証明されている。しかし、それらの抗腫瘍効果は、前臨床試験から予測されるほど強力でないことが多く、化学的療法と組み合わせることが好ましい。
【0006】
Herceptin(登録商標)は、乳ガンの治療に広く使用される、よく受け入れられたモノクローナル抗体である。しかし、Herceptin(登録商標)は、標的抗原であるHer2/neuの腫瘍細胞上での発現レベルが少なくとも2+/FISH+(いわゆるHercepTest(登録商標)およびFISH法解析陽性で証明される)、または3+である患者に対してしか使用は成功しない。いくつかの研究により、Her2/neuの追加遺伝子増幅(FISH法で示される)とともに相対的に高い標的抗原Her2/neuの発現密度が、統計的に有意な生存を得るために必要だということが示されている。前臨床試験での生体実験によっても、Herceptin(登録商標)による腫瘍細胞の効果的な破壊が見られるのは、腫瘍細胞が標的抗原であるHer2/neuを高レベルに発現している場合だけであることが示されている。
【0007】
これらの制限は、腫瘍上にHer2/neuを高レベルに発現する(前記Dako社のHercepTestでスコア2+/FISH+、または3+とされる)、約25〜30%の転移性乳ガン患者しか、Herceptin(登録商標)を用いて効果的に治療することができないことの原因である。
【0008】
Perezらの研究では、免疫組織化学的検査(IHC)での評価によると、35%の乳ガン患者がHer2のスコア1+、14%が2+、13%が3+とされた(非特許文献1)。
【0009】
Kiewe, P.らは、Her2/neuおよびCD3を標的とする完全な二重特異性モノクローナル抗体の使用について記載している(非特許文献2)。この抗体は、転移性乳ガンの治療にフェーズI試験で使用された。この抗Her2×抗CD3抗体によって生じる副作用の研究を行い、必要な人数である17人の患者の募集(recruiting)を早めるために、前記HercepTest(登録商標)でのスコアが1+(25%)、2+(12.5%)および3+(62.5%)の患者が試験された。この予備的な結果解析では、抗腫瘍応答は、評価可能な15人の患者のうち4人で見られた。しかし、前記HercepTest(登録商標)のスコアが1+の範疇の患者で抗腫瘍応答を示した者はいなかった。これらの応答した者のHer2/neuの発現状態(status)に関しては、全く情報が開示されていない。さらに、このフェーズI試験を計画する時点では、ertumaxomabが、高レベル(スコア+3)のHer2/neuを発現する細胞系統(例えば、Sk−Br−3)を除去するという実験データのみが入手可能であった。
【0010】
全ての典型的なフェーズI試験と同様に、Kieweらが行ったフェーズI試験は、三機能抗体であるertumaxomabの安全性および許容性を調査するために計画された。この試験での第二の変数(secondary variables)は、抗腫瘍活性および種々の免疫学的変数の測定であった。この研究への患者の募集を早めるために、患者の腫瘍細胞上のHer2/neuの発現に関する制限を設けなかった。これが可能であったのは、この研究の第一の目的は、安全性と許容性であり、腫瘍細胞上のHer2/neuの発現レベルは、全く重要では無かったからである。
【0011】
三機能抗体である抗Her2×抗CD3のertumaxomabが、スコア2+および3+に従いHer2/neuを過剰発現する患者においてのみ有効であるという知見は、全く驚きは無かった。なぜなら、Herceptin(登録商標)等の抗体を用いて実施された他の調査でも、Her2/neuを中(2+/FISH+)から高レベル(3+)に発現する患者に対してのみに効果が示されたからである。
【0012】
例えば、Gatzemeierらは、Herceptin等の抗体が、実際に臨床上の利益を提供できるのは、Her2/neuの発現が3+または2+/FISH+の患者に対してだけであるという証拠を提供した(非特許文献3)。
【0013】
同様に、Micromet社は、2006年10月2日に、乳ガンおよび前立腺ガンにおける上皮細胞接着分子EpCAMに対する抗体であるadecatumumabの二つのフェーズII試験についてのデータを公表した。その腫瘍組織において高レベルにEpCAMを発現する患者においてのみ、腫瘍増殖停止時間の有意な延長を示し、EpCAMの発現が低い患者においては有意な利益を示さなかった。adecatumumabには、EpCAMを高度に過剰発現する乳ガンの患者に治療の選択肢を与える可能性があるということが強調された。EpCAMの過剰発現が低い患者には全く有用でないことが見出された。
【0014】
非ホジキンリンパ腫(NHL)および慢性リンパ性白血病(CLL)は、最も広く認められる悪性腫瘍である。NHLは、アメリカ合衆国において5番目に多い悪性疾患(malignant disease)であり、年間発症率は10万人あたり18〜20である。NHLの発症件数は、1990年代中期まで年率5〜10%で増加した(非特許文献4)が、それ以来一定している(非特許文献5)。CLLは、西洋で最も多い白血病であり、年間発症率は10万人あたり3〜4である。
【0015】
抗CD20モノクローナル抗体であるRituximabを用いるB細胞リンパ腫の治療における(特に、化学療法と組み合わせにおける)結果が有望であるにも関わらず、患者は結局再発した(非特許文献6、7)。したがって、さらなる治療の選択肢が必要である。
【0016】
Rituximabおよびalemtuzumab等のモノクローナル抗体の認可以後、治療における大きな改善が達成されたという事実があるにも関わらず、現在、緩徐進行性NHLおよびCLLは、両方とも治癒不能な疾患である(非特許文献8〜11)。抗体だけによる療法は高度には成功していないが、特にCLLにおけるモノクローナル抗体と標準的な化学療法処方との組み合わせは、患者の結果を有意に改善した(非特許文献12、13)。しかし、患者は結局再発し(非特許文献6)、新しい治療法の戦略が必死に求められている。完全ヒト化抗CD20抗体または他の抗原(例えばCD19、CD22、CD23、CD80もしくはHLA−DR)を標的とする抗体等の多くの新薬候補が、現在鋭意検討されている(非特許文献14、15)。
【0017】
新しい標的の探索に加え、抗体療法を改善する別のアプローチとしては、二重特異性抗体の開発があげられる。多くの二重特異性の方式(format)が開発され、試験されている。例えば、クアドローマ細胞で産生させた完全長抗体、化学的にクロスリンクさせたF(ab)2断片、一本鎖抗体(非特許文献16)、二重特異性抗体(非特許文献17)である。これらの抗体の中には、ヒトリンパ腫の治療に試験されたものもある。しかしながら、in vitroにおける効能が有望であるにも関わらず、これらの分子は、限定的な臨床上の利益しか示さなかった(非特許文献18〜20)。
【0018】
CD19またはCD20等のB細胞特異的抗原を標的とする二重特異性抗体は、この10〜15年の間における鋭意研究の対象であるが、穏やかな応答しか示していない限定的な臨床データが、今のところ入手可能な程度である(非特許文献18、21、22)。さらに、製造工程が複雑で効率的でないため、臨床用途のための充分な量の抗体を産生することが難しい。
【0019】
二重特異性抗体(bsAb)は、例えば、悪性細胞等の腫瘍細胞に対してエフェクター細胞を誘導することによって、免疫学的に治療するための道具(tool)である。しかしながら、bsAbは通常エフェクター細胞のうち一つのクラス、すなわちT細胞、NK細胞、FcγRI+またはFcαRI+細胞のいずれかだけしか誘導、活性化しないので、その効能は限定的である。これらの先行技術抗体の全てが、腫瘍関連抗原が中から高レベルで腫瘍細胞表面に発現している場合にのみ、効果的に腫瘍を治療することが記載されている。
【0020】
抗体の免疫学的エフェクター機能を促進することは、抗体に基づくガン治療の効能を改善する一つのアプローチを示す。改善されたエフェクター機能を有する三機能二重特異性抗体は、例えば、Zeidlerら(非特許文献23)、Riesenbergら(非特許文献24)、Ruf&Lindhofer(非特許文献25)またはHeissら(非特許文献26)の出版物において、本発明者らによって、腫瘍細胞およびTリンパ球を二つの結合アームで認識するだけではなく、Fc領域を通じてFcγ受容体陽性修飾細胞にも結合するものとして記載されている。しかしながら、これらの抗体が、Her2/neuまたはCD20のような腫瘍関連抗原を低レベルで発現している腫瘍細胞を治療するのに有用であるということは現在まで知られていなかった。
【0021】
上記で説明したHer2/neu、EpCAMおよびCD20に関する制限は、例えば、G250、GD3,GD2、プロテオグリカン、MHC II、EGF−RおよびCEA等の他の腫瘍関連抗原に関しても当てはまる。
【0022】
要約すると、本発明の優先日までに入手できる全ての実験データは、腫瘍抗原を中程度から高度に過剰発現する患者に対する有意な利益の証拠しか提供していなかった。
【先行技術文献】
【非特許文献】
【0023】
【非特許文献1】Perez EA et al.: Her2 testing in patients with breast cancer: poor correlation between weak positivity by immunohistochemistry and gene amplification by fluorescence in situ hybridization. Mayo Clin Proc 2002; 77: 148−154
【非特許文献2】Kiewe, P. et al.: “Phase I trial of the trifunctional anti−Her2 x anti−CD3 antibody ertumaxomab in metastatic breast cancer”; Journal of Clinical Oncology, vol 23, no. 16S, 1 June 2005 (ASCO Abstract 2205−06−01)
【非特許文献3】Gatzemeier et al., Anals of Oncology 15:19−27, 2004
【非特許文献4】Greiner TC, Medeiros LJ, Jaffe ES. Non−Hodgkin’s lymphoma. Cancer. 1995;75:370−380.
【非特許文献5】Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review, 1975−2003. http://seercancergov. 2006.
【非特許文献6】Smith MR. Rituximab (monoclonal anti−CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359−7368.
【非特許文献7】Ghielmini M. Multimodality therapies and optimal schedule of antibodies: rituximab in lymphoma as an example. Hematology (Am Soc Hematol Educ Program). 2005:321−328.
【非特許文献8】Lin TS, Lucas MS, Byrd JC. Rituximab in B−cell chronic lymphocytic leukemia. Semin Oncol. 2003;30:483−492.
【非特許文献9】Dillman RO. Treatment of low−grade B−cell lymphoma with the monoclonal antibody rituximab. Semin Oncol. 2003;30:434−447.
【非特許文献10】Blum KA, Bartlett NL. Antibodies for the treatment of diffuse large cell lymphoma. Semin Oncol. 2003;30:448−456.
【非特許文献11】Moreton P, Hillmen P. Alemtuzumab therapy in B−cell lymphoproliferative disorders. Semin Oncol. 2003;30:493−501.
【非特許文献12】Hiddemann W, Dreyling M, Unterhalt M. Rituximab plus chemotherapy in follicular and mantle cell lymphomas. Semin Oncol. 2003;30:16−20.
【非特許文献13】Montserrat E. Rituximab in chronic lymphocytic leukemia. Semin Oncol. 2003;30:34−39.
【非特許文献14】Mavromatis BH, Cheson BD. Novel therapies for chronic lymphocytic leukemia. Blood Rev. 2004;18:137−148.
【非特許文献15】Nagy ZA, Hubner B, Lohning C, et al. Fully human, HLA−DR−specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8:801−807.
【非特許文献16】Baeuerle PA, Kufer P, Lutterbuse R. Bispecific antibodies for polyclonal T−cell engagement. Curr Opin Mol Ther. 2003;5:413−419.
【非特許文献17】Peipp M, Valerius T. Bispecific antibodies targeting cancer cells. Biochem Soc Trans. 2002;30:507−511.
【非特許文献18】Manzke O, Tesch H, Borchmann P, et al. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. I. Clinical phase I evaluation. Int J Cancer. 2001;91:508−515.
【非特許文献19】Manzke O, Tesch H, Lorenzen J, Diehl V, Bohlen H. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. II. Assessment of cellular immune responses. Int J Cancer. 2001;91:516−522.
【非特許文献20】Haagen IA. Performance of CD3xCD19 bispecific monoclonal antibodies in B cell malignancy. Leuk Lymphoma. 1995;19:381−393.
【非特許文献21】Weiner GJ, De Gast GC. Bispecific monoclonal antibody therapy of B−cell malignancy. Leuk Lymphoma. 1995;16:199−207.
【非特許文献22】De Gast GC, Van Houten AA, Haagen IA, et al. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother. 1995;4:433−437.
【非特許文献23】Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246−1252.
【非特許文献24】Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem. 2001;49:911−917.
【非特許文献25】Ruf P, Lindhofer H. Induction of a long−lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526−2534.
【非特許文献26】Heiss MM, Strohlein MA, Jager M, et al. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435−443.
【発明の概要】
【発明が解決しようとする課題】
【0024】
したがって、例えば、Her2/neuを低度に発現している腫瘍細胞(前記Dako社のHercepTest(登録商標)において、例えば、1+のスコアである)、腫瘍細胞あたり500,000以下の腫瘍関連抗原という中程度のレベルでHer2/neuを発現し、前記Dako社のHercepTest(登録商標)において、2+のスコアであるが、FISH法陰性のもの、もしくはCD20を低度に発現している腫瘍細胞、または少なくとも1つの上記した特異的な腫瘍関連抗原、特にCD20およびEpCAMを、腫瘍細胞あたり約1,000〜350,000、特に、5,000〜150,000のレベルおよび範囲で発現している腫瘍細胞をも死滅させることが可能な抗体の抗腫瘍活性の改善が必要である。
【課題を解決するための手段】
【0025】
上記した問題は、以下に述べる特性を有する三機能二重特異性抗体から選択され、種々のクラスの免疫細胞の協調を促進する抗体の、腫瘍性疾患の予防および治療用の薬学的組成物を製造するための使用によって解決されることを見出した。
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
前記腫瘍関連抗原は、下記のとおり前記腫瘍細胞上に発現されている。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、または、
・EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【図面の簡単な説明】
【0026】
【図1】図1A−Fは、異なる抗Her2抗体を用いてフローサイトメトリーにより測定された細胞系統Lox、HCT−8およびSKBR3のHer2/neu発現特性を示す。
【図2a】図2aは、Her2+3 SKBR−3細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2b】図2bは、Her2+1 HCT−8細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2c】図2cは、Her2ネガティブ Lox細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析を示す。 *印の番号は、表3に対応する。
【図2d】図2dは、Her2+1 HCT−8細胞の死滅:MDSワークフロー(workflow)によるドナー2のドットプロット結果の一例を示す。 *印の番号は、表3に対応する。
【図3】図3は、サイトカインビーズアレイシステム(CBA)を用いて測定された24時間後のクローン原性アッセイ上清中のサイトカイン分泌を示す。
【図4】図4は、推定される三細胞複合体(tri−cell−complex)を示す。三機能抗体は、異なる免疫学的メカニズムにより腫瘍細胞の認識と破壊を促進することができる。IFN−γを除く図中のサイトカインに関するリストをまとめなければならない(Bei der Auflistung der Zytokine im Bild sollte man noch IFN−γ aufnehmen)。注:ADCC、抗体依存性細胞傷害;DC、樹状細胞;NK、ナチュラルキラー細胞;DC−CK1、樹状細胞サイトカイン1;IL、インターロイキン;LFA、リンパ球機能関連抗原;TNF−α、腫瘍壊死因子α;CD、表面抗原分類(出典 Ruf&Lindhofer,2001)
【図5A】図5は、Bi20の精製を示す。(図5A)陽イオン交換クロマトグラフィー(CIEX)を示す。親マウス抗体(ピーク1)を、HR10/10 high performance SP−Sepharose column上で、三機能抗体(ピーク2)から分離した。
【図5B】(図5B)Bi20三機能抗体精製の等電点電気泳動(IEF)を示す。親ラット抗体(26II6)を、プロテインAアフィニティークロマトグラフィー(AC Bi20)後に除去し、親マウス抗体(TPA10)を、CIEX(CIEX P1 Bi20)により三機能抗体から分離した。精製されたBi20は、前記陽イオン交換クロマトグラフィーのピーク2(CIEX P2 Bi20)に見出された。
【図6A】図6は、Bi20が特異的に結合するエフェクター細胞を示す。190ngのBi20を、5×105個の標的細胞とともにインキュベートし、その結合をFACS解析により測定した。Ramos細胞(図6A)を、CD20アームの標的細胞として用いた。
【図6B】THP−1細胞(図6B)を、Fc領域用に用いた。拮抗実験のために、細胞を予めRituximab(Rit)とともに所定の過剰量の抗体で30分間インキュベートした。
【図7A】図7は、エフェクター細胞の存在下におけるBi20が媒介する効果的なB細胞の減少を示す。(図7A)1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Mec1)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともにインキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)(neg)前記アッセイにおけるB細胞の絶対数を、100%に設定した。
【図7B】(図7B)1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。前記B細胞の絶対数を、トリパンブルー色素排除集計およびFACS解析により、1日目、2日目、3日目において測定し、B細胞の減少割合を算出した。
【図8A】図8は、Bi20が媒介するT細胞の活性化を示す。(図8A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。CD4+およびCD8+T細胞上におけるCD25の発現のMFIを、FACS解析により測定した。
【図8B】(図8B)PBMCおよびRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。CD4+T細胞上におけるCD25の発現のMFIを、1日目、2日目および3日目に測定した。
【図9】図9は、Bi20が誘導するT細胞の増殖を示す。1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、50ng/mlの濃度のBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。T細胞数を、トリパンブルー色素排除集計並びに抗CD5/4抗体および抗CD5/8抗体を用いたFACS解析により測定した。
【図10A】図10は、Bi20により媒介される単球の活性化を示す。(図10A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)またはrituximabとともに1日間インキュベートした。CD14+単球によるCD25の発現のMFIを、FACS解析により測定した。
【図10B】(図10B)1×106個/mlのPBMCおよび1×105個/mlのTHP−1細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab(Rit)、TPA10、または26II6とともに1日間インキュベートした。CD33+THP−1細胞の活性化を、CD25またはCD40の発現によって測定した。
【図11A】図11は、健康なドナーのエフェクター細胞存在下におけるBi20が誘導するCLL B細胞の減少を示す。(図11A)1×106個/mlのPBMCおよび2×105個/mlのCLL腫瘍細胞を、5、50、もしくは250ng/mlのBi20、250ng/mlのcatumaxomab(Catum)、または250ng/mlのRituximab(Rit)とともに3日間インキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)前記アッセイにおけるB細胞の絶対数を、100%に設定した。MVは、CLL−1からCLL−9の平均値を示す。
【図11B】(図11B)CLL患者検体(1〜14)におけるCD20の発現の概要、細胞系統(Raji、Mec−1)および1体の健康なドナー(PBMC)。MFI=蛍光強度の平均値を示す。
【発明を実施するための形態】
【0027】
本発明の一実施形態において、全ての引用した腫瘍抗原が、腫瘍細胞あたり約5,000〜約150,000の量で腫瘍細胞上に発現している。
【0028】
本発明は、ヒトまたは動物の腫瘍細胞を含む身体、もしくは前記腫瘍細胞を含むこれらの身体の部分を、下記の特性を有する三機能二重特異性抗体の薬学的に有効な量に接触させることにより、腫瘍性疾患を予防および治療するためのヒトまたは動物の治療方法も提供する。
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、GD2、GD3、プロテオグリカン、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
前記腫瘍関連抗原は、下記のとおり前記腫瘍細胞上に発現されている。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、または、
・EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【0029】
患者の腫瘍細胞を、前記三機能二重特異性抗体に接触させることは、前記三機能二重特異性抗体を受容可能な服用形式、経口または非経口いずれかで、以下に記述するようにヒトまたは動物の体内に投与することによっても実施される。
【0030】
白血病またはリンパ腫等の免疫システムの細胞が関与する腫瘍性疾患を治療するのであれば、前記接触は、ex vivoで行われてもよい。例えば、米国特許第7,018,632号公報または米国特許第5,985,276号公報が引用され、それらは本明細書に完全に取り込まれている。
【0031】
自己の腫瘍細胞を含む材料は、本明細書に記載の三機能二重特異性抗体とともに、10分間〜10時間、好ましくは5時間以下の短時間インキュベートされ、または、一定時間の長時間インキュベートされることにより、自己の腫瘍細胞は、抗体に襲撃される(charged)。続いて、患者の血液細胞、好ましくは末梢血単核球(PBMC)を加え、この混合物を1〜14日間の長時間インキュベートする。またはそれに代えて、自己の腫瘍細胞を、前記三機能二重特異性抗体および患者の血液細胞、好ましくは末梢血単核球に直接接触させる。このようにして、腫瘍に対する無数の免疫細胞の“プライミング(priming)”が、ex vivoで達成される。その後、これらの細胞は、患者に再注入される。長時間インキュベートすることによっても、抗体の内部移行(internalization)および分解が起こる。前述のとおり、上記で引用した米国特許には、これらの方法が完全に記載されている。しかしながら、上記で示した腫瘍関連抗原について、腫瘍細胞あたり約5,000〜約150,000の腫瘍関連抗原が発現している腫瘍細胞が関与する腫瘍性疾患を治療することも可能なのかという、専門家に向けた成功のための合理的な予測は全く提供されていない。前記方法では、健康なドナーのPBMCを加えずに、同種間または自己設定(autologous setting)で実施してもよい。珍しいことに、腫瘍細胞、特にB細胞の減少は、エフェクター細胞のサイトカイン(例えば、IL−2、GM−SCF)による前活性化(pre−activation)または共活性化(coactivation)には全く依存しない。このことは、従来技術に記載されている他の二重特異性抗体または単特異性抗体に対する典型的な不利な点である。
【0032】
本発明の好ましい実施形態は、従属クレーム並びに以下の記載およびそれに伴う実験データ、図、表に示されている。
【0033】
本発明者らは、驚いたことに、従来技術に記載されている三機能二重特異性抗体が、低度から中程度のレベルで腫瘍細胞上に発現されている、前記腫瘍細胞上の選択された特定の腫瘍関連標的抗原を標的とするのに効果的に使用可能であることを示すことができた。標的抗原として特に好ましいものは、Her2/neu、EpCAMおよびCD20腫瘍関連抗原である。
【0034】
前記腫瘍関連抗原は、前記腫瘍細胞上に発現される前記腫瘍関連抗原のタイプ、およびHer2/neuの場合は、前記腫瘍抗原が増幅されているかどうかに依存して種々のレベルで前記腫瘍細胞上に発現されている。
【0035】
Her2/neuの場合、患者の腫瘍がHer2/neuを腫瘍細胞上に腫瘍細胞あたり約5,000〜約150,000の量で腫瘍関連抗原が発現していれば、本明細書中の記載のように前記患者は三機能二重特異性抗体で治療が成功可能であることが見出されている。これらの患者は、前記DAKO社のHercepTest(登録商標)に従えば、1+のスコアとされることになる。1+の上限を、約100,000または110,000に分類することも正しいと考えられる。最も重要なことは、前記スコアが、“中程度の発現”ではなく、“低度の発現”と分類されなければならないことである。
【0036】
腫瘍細胞あたり500,000分子以下のHer2/neu腫瘍関連抗原を発現し、前記DAKO社のHercepTest(登録商標)に従って2+に分類される患者は、FISH法陰性と分類された場合であっても、治療が成功可能なことも見出されている。従来は、前記DAKO社のHercepTest(登録商標)に従ってHer2/neu値が2+のスコアとされ、腫瘍細胞あたり500,000以下のHer2/neu腫瘍抗原を発現し、FISH法陽性の患者しか治療が成功可能でなかった。Her2/neu値が2+であっても、FISH法陰性の患者は、治療が成功可能でなかった。
【0037】
CD20を発現する腫瘍を有する患者では、驚いたことに、腫瘍細胞あたり約1000〜約350,000の量で腫瘍関連抗原としてCD20を発現している場合でさえも、患者の治療が成功可能であることが見出された。さらに、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAを腫瘍細胞あたり約1000〜約350,000の量で腫瘍関連抗原として発現している患者は、本明細書に記載の前記三機能二重特異性抗体により治療が成功可能である。
【0038】
前記腫瘍関連抗原は、Her2/neuの場合、前記腫瘍細胞上に、好ましくは腫瘍細胞あたり少なくとも約10,000、約20,000、約50,000または約80,000の量で発現しており、好ましくは腫瘍細胞あたり最大で約110,000、約120,000または約130,000の量で発現している。
【0039】
前記DAKO社のHercepTest(登録商標)でHer2/neu値がスコア2+(中程度の発現)とされた患者においては、腫瘍細胞上の腫瘍関連抗原の好ましい発現の量は、500,000以下または450,000、400,000、350,000もしくは300,000以下であり、下限は約100,000か、約110,000より多いか、約150,000、約200,000または約250,000より多い。
【0040】
CD20腫瘍抗原を発現する腫瘍細胞では、腫瘍細胞あたりのCD20分子の数は、約1000〜約350,000の範囲で、B細胞悪性腫瘍の型および体内部位に依存し、好ましくは、約310,00または300,000以下で変化する。腫瘍細胞あたりのCD20分子の数は、特に、約5000、約10,000または約50,000から約100,000、150,000、200,000、250,000または300,000以下で変化することが可能である。前記状況は、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEA等の請求項1で示した他の全ての腫瘍抗原においても同様である。
【0041】
腫瘍関連抗原の発現レベルは、単一の限定された腫瘍の実体から生じた種々の腫瘍細胞上に、より低度の発現値から高度の発現値の間に分布し得ることを申し添えておく。これらの数値は全て、悪性腫瘍のタイプ、患者のタイプ、腫瘍の進行等に依存した平均値であることを理解されなければならない。
【0042】
本発明者らによって選択された腫瘍関連標的抗原が低度から中程度の発現レベルでのみ腫瘍細胞上に永続的かつ安定に発現していることが、極めて重要であることを申し添えておく。ヒートショックタンパク質、MIC分子であるMIC AおよびMIC B等の他のタイプの誘導可能な抗原は、両方ともヒートショックプロモーター要素の制御下にあり、誘導後一生の間増加するレベルで発現されるが、Her2/neuおよびCD20等の腫瘍関連抗原の発現割合は、一般的に一定かつ安定である。
【0043】
本発明者らによって選択された腫瘍関連標的抗原は、特定の腫瘍に関連するか、または特異的である。EpCAMは、典型的には腺ガンに関連し、Her2/neuは、乳ガンだけでなく大腸ガン、肺ガン、胃ガン、膵臓ガンおよび卵巣ガンに関連し、CD20は、非ホジキンリンパ腫または慢性リンパ性白血病等のB細胞リンパ腫に関連し、G250は、腎臓ガンに関連し、プロテオグリカン、GD3およびGD2は、メラノーマに関連し、MHC IIはB細胞リンパ腫に関連し、並びにEGF−RおよびCEAは、上皮性腫瘍に関連する。
【0044】
本発明で用いる三機能二重特異性抗体(略してtrAb)は、それ自体は公知である。例えば、米国特許第6,551,592号公報が引用され、その内容は本明細書に完全に取り込まれている。同様のことがドイツ公開特許第19649223A号公報およびドイツ公開特許第19710495A号公報についても言え、それらに対応する米国特許公報とともに引用され、それらもまた本明細書中に包含されている。
【0045】
本発明で用いる抗体は、カプセル、タブレット、水性の懸濁液、溶液等の許容できるいかなる服用形式で経口投与されてもよい。抗体およびその誘導物は、非経口的に投与されてもよい。それは以下の投与経路を介する。すなわち、皮下、静脈内、腹腔内、筋内、関節内、滑液内、胸骨内、鼻腔内、局部、莢膜内、肝内、腫瘍内、病巣内および頭蓋内への注射または注入技術である。一般的には、抗体は、静脈内への注射または注入により供給される。
【0046】
本発明の抗体は、単独で投与されてもよいし、または、許容できるアジュバント(adjuvant)、賦形剤および添加剤を含む薬学的に許容できるキャリアーとともに投与されてよい。これらは、全て当業者にとって周知である。
【0047】
有効な服用量は、多数の因子に依存する。体重、治療の目的、最大許容服用量、使用された具体的な製剤、投与経路、患者の応答等の多数のパラメーターに従って任意の患者に対して服用量を調整することは、熟練した内科医が優に為し得る範囲である。
【0048】
本発明に従って使用されるtrAbは、一回の注入あたり、好ましくは約5〜約1000μg、さらに好ましくは約10〜約300μg、約10〜約100μg、または約10〜約50μgの量である。最適な量は、実験により、熟練者によって決定され得る。前記三機能抗体は、さらに好ましくは約0.05〜約15μg/kg体重、さらに好ましくは約0.5〜5μg/kg体重、および約0.5〜2μg/kg体重の量で使用される。投与の回数は、患者の必要性、特に疾患の重さ、患者の応答、適用した服用量およびその技術分野で知られる他の多数の因子に従って、医師が選択できる。
【0049】
好ましくは、前記三機能抗体は、抗CD3×抗腫瘍関連抗原抗体および/または抗CD4×抗腫瘍関連抗原抗体および/または抗CD5×抗腫瘍関連抗原抗体および/または抗CD6×抗腫瘍関連抗原抗体および/または抗CD8抗腫瘍関連抗原抗体および/または抗CD2×抗腫瘍関連抗原抗体および/または抗CD28×抗腫瘍関連抗原抗体および/または抗CD44×抗腫瘍関連抗原抗体が選択されるが、特に好ましくは、抗CD3×抗腫瘍関連抗原抗体が使用される。腫瘍関連抗原として特に好ましくは、Her2/neu、EpCAMまたはCD20抗原であり、さらに好ましくは、三機能抗体の抗CD3結合アームと組み合わせる。
【0050】
本発明の前記三機能抗体を使用することで、前記trAbの前記Fc領域が前記Fc受容体陽性細胞に結合することによって前記Fc受容体陽性細胞が活性化される。それにより、サイトカインおよび/または共刺激抗原の発現が開始または増加される。そこで、少なくとも、T細胞の生理的活性化に必要な第二の活性化シグナルが、前記共刺激抗原および/またはサイトカインにより前記T細胞に伝達される。この活性化は、活性化マーカーの上方制御(up−regulation)、腫瘍細胞の殺傷およびT細胞の増殖によって示される。
【0051】
trAbによる前記Fc受容体陽性細胞の活性化は、前記サブクラス、または抗体の重鎖断片の前記サブクラスの組み合わせにそれぞれ依存する。in vitroの実験で証明されたように、例えば、前記マウスIgG2a/ラットIgG2bのサブクラスの組み合わせのtrAbは、Fc受容体陽性細胞に結合し、同時に活性化することが可能で、これらの細胞の表面におけるCD40、CD80もしくはCD86等の共刺激抗原の上方制御または新しい形成(発現)をそれぞれ導く。一方、前記マウスIgG1/ラットIgG2bのサブクラスの組み合わせの二重特異性抗体は、Fc受容体陽性細胞に結合可能だが((1)Haagenら、 J. Immunology, 1995, 154: 1852−1860)、明らかにこれらの細胞を同程度には活性化できない((2)Gastら、 Cancer Immunol. Immunother., 1995, 40: 390)。したがって、trAbのFc領域におけるマウスIgG2a/ラットIgG2bのアイソタイプの組み合わせが特に好ましい。しかしながら、本明細書に記載のとおり、これは他の全てのアイソタイプの組み合わせについても言えることであり、それらは本発明の他の好ましい実施形態において使用することができる。
【0052】
前記trAbが、一つの結合アーム(例えば、抗CD3)でT細胞に同時に結合し、活性化する間、前記trAbの前記Fc領域に結合した前記Fc受容体陽性細胞からの共刺激シグナルが前記T細胞に伝達される可能性がある。すなわち、前記trAbの一つの結合アームを介したT細胞活性化、および前記Fc受容体陽性細胞から前記T細胞への共刺激シグナルの同時期の伝達の組み合わせだけが、効果的なT細胞の活性化につながる。
【0053】
抗腫瘍免疫の誘導における他の重要な側面は、前記trAbによって標的とされる修飾細胞(単球、マクロファージ、NK細胞または樹状細胞)による腫瘍構成物の潜在的な食作用、分解および提示である。この古典的な抗原提示機構により、腫瘍特異的なCD4陽性細胞およびCD8陽性細胞が生じてもよい。さらに、腫瘍特異的CD4細胞は、T/B細胞の協調という状況で体液性免疫応答の誘導に重要な役割を果たす。
【0054】
三機能二重特異性抗体は、第1の結合アームでT細胞のT細胞受容体複合体、第二の結合アームで前記腫瘍細胞上の前記腫瘍関連抗原に結合可能である。それによって、それらは、サイトカインの放出、またはアポトーシスを起こす機構により、腫瘍細胞を破壊するT細胞を活性化する。さらに、三機能抗体による活性化ということでは、これらの受容体を通じて腫瘍特異的抗原を認識するT細胞は再び活性化する可能性があり、そうして長く持続する抗腫瘍免疫へとつながる可能性があるように思われる。この点で特に重要なのは、単球/マクロファージおよび樹状細胞等の修飾細胞への結合を媒介し、それら自身の細胞傷害性を発達させ、および/または、付随して前記T細胞に重要な共刺激シグナルを伝達させる、前記三機能二重特異性抗体の完全なFc領域である。明らかに、この方法により、現在まで未知の腫瘍特異的ペプチドに対して、T細胞の応答が誘導され得る。
【0055】
潜在的にアネルギー化した腫瘍特異的T細胞をtrAbにより腫瘍細胞に誘導し、前記trAbのFc領域に結合した修飾細胞によってそのようなT細胞を同時に共刺激することにより、腫瘍特異的細胞傷害性T細胞(CTL)のアネルギー状態は撤廃可能であった。すなわち、患者中に存在する腫瘍に対して既に存在するT細胞寛容性が、trAbにより撤廃される可能性がある。そして、このことにより、腫瘍細胞の直接の破壊に加えて、長く持続する抗腫瘍免疫が誘導される可能性がある。
【0056】
好ましくは、本発明に従って使用される抗体は、アネルギー状態にある腫瘍特異的T細胞を再活性化し得る。さらに、それらは腫瘍に反応する補体結合抗体を誘導可能であり、したがって体液性免疫応答を誘導する。
【0057】
前記T細胞への結合は、好ましくはCD3、CD2、CD4、CD5、CD6、CD8、CD28および/またはCD44を通じて起こる。前記Fc受容体陽性細胞は少なくとも一つのFcγ受容体IまたはIIIを有する。
本発明に従って使用され得る抗体は、Fcγ受容体Iおよび/またはIII陽性細胞である、単球、マクロファージ、樹状細胞、“ナチュラルキラー”細胞(NK細胞)および/または活性化好中球に結合し得る。
【0058】
本発明に従って使用され得る抗体は、共刺激抗原であるCD40、CD80、CD86、ICAM−1および/もしくはLFA−3の発現の開始もしくは増加、または/並びに、前記Fc受容体陽性細胞によるサイトカイン分泌を導く。好ましくは、前記サイトカインはIL−1、IL−2、IL−4、IL−6、IL−8、IL−12、IFN−γおよび/またはTNF−αである。
好ましくは、前記T細胞への結合は、前記T細胞のT細胞受容体複合体を通じて起こる。
【0059】
好ましくは、前記三機能二重特異性抗体は、異種性の完全なラット/マウス二重特異性抗体である。
【0060】
本発明に従って使用され得るtrAbによって、T細胞は活性化され、腫瘍細胞に対して誘導される。好ましい有用な異種性の三機能二重特異性抗体は、以下のアイソタイプの組み合わせの少なくとも一つから選択される。
ラット−IgG2b/マウス−IgG2a、
ラット−IgG2b/マウス−IgG2b、
ラット−IgG2b/マウス−IgG3;
ラット−IgG2b/ヒト−IgG1、
ラット−IgG2b/ヒト−IgG2、
ラット−IgG2b/ヒト−IgG3[東洋人のアロタイプG3m(st)=プロテインAに結合]、
ラット−IgG2b/ヒト−IgG4、
ラット−IgG2b/ラット−IgG2c;
マウス−IgG2a/ヒト−IgG3[白色人種のアロタイプG3m(b+g)=プロテインAに結合しない、以後*で示す]
マウス−IgG2a/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2a/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2a/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒトIgG4/ラット−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域−CH2−CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG2−[ヒンジ領域−CH2−CH3]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG3−[ヒンジ領域−CH2−CH3、東洋人のアロタイプ]
ラット−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域−CH2−CH3]
ヒト−IgG1/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG2[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG2[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG1/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG2/ヒト−[VH−CH1、VL−CL]−ヒト−IgG2−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4[CH2のN−末端領域]−ヒト−IgG3*[CH2のC−末端領域:>アミノ酸番号251]−ヒト−IgG3*[CH3]
マウス−IgG2b/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2b/ヒト−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−IgG2b/マウス−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG3*−[CH2−CH3]
マウス−[VH−CH1、VL−CL]−ヒト−IgG4/ラット−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG1/ラット−[VH−CH1、VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG1/マウス−[VH−CH1,VL−CL]−ヒト−IgG1−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
ヒト−IgG4/ヒト−[VH−CH1、VL−CL]−ヒト−IgG4−[ヒンジ領域]−ヒト−IgG4−[CH2]−ヒト−IgG3*−[CH3]
【0061】
好ましくは、本発明に基づく有用な抗体は、モノクローナル、キメラ、組み換え、合成、半合成であるか、または、例えば、Fv、Fab、scFvもしくはF(ab)2断片を有する化学的に修飾された完全な抗体である。
【0062】
好ましくは、ヒト起源の抗体、またはその誘導体もしくは断片が使用されるか、または、ヒトでの使用に適するように修飾された抗体(いわゆる、“ヒト化抗体”)が使用される(例えば、Shalabyら、 J. Exp. Med. 175 (1992), 217; Mocikatら、 Transplantation 57 (1994), 405を参照)。
【0063】
上述した種々のタイプの抗体および抗体断片の調製は、熟練者にとって周知である。モノクローナル抗体、好ましくは、例えば、ヒト、ラット、マウス、ウサギまたはヤギのような哺乳類起源のモノクローナル抗体の調製は、例えば、KoehlerおよびMilstein(Nature 256 (1975), 495)、HarlowおよびLane(Antibodies, A Laboratory Manual (1988), Cold Spring Harbor)、またはGalfre(Meth. Enzymol. 73 (1981), 3)、またはドイツ公開特許第19531346号公報に記載されている従来法を用いて行うことが可能である。
【0064】
さらに、組み換えDNA技術を用いて、熟練者に知られた技術に従って(Kuruczら、 J. Immunol. 154 (1995), 4576; Hollingerら、 Proc. Natl. Acad. Sci. USA 90 (1993), 6444を参照)、記載された抗体を調製することが可能である。
【0065】
二つの異なる特異性を有する抗体、いわゆる二重特異性抗体の調製は、一方では、組み換えDNA技術を用いて実施することが可能であるが、他方では、いわゆるハイブリッドハイブリドーマ融合技術(例えば、Milsteinら、 Nature 305 (1983), 537を参照)によっても実施することが可能である。この技術は、所望の特異性の一つを有する抗体をそれぞれ産生するハイブリドーマ細胞系統の融合、および、両方の特異性を有する抗体を産生する組み換え細胞系統の同定、単離を含む。
【0066】
本発明の基礎をなす課題は、請求項1で定義した三機能二重特異性抗体を用いて解決される。続いて、二つの特異性を示す抗体の調製をより詳細に記載する。本発明に含まれる三機能二重特異性抗体は先行技術に属し、そのような調製方法を記載した文献は、本明細書中にそれら全体が取り込まれている。
【0067】
三機能二重特異性抗体は、それぞれ特異性を示し、かつ、通常の抗体と同様に、周知のエフェクター機能を発揮するFc領域を有する、二つの抗体セミ分子(それぞれが一つのHおよび一つのL免疫グロブリン鎖を有する)から構成される。それらは、クアドローマ技術を用いて調製することが好ましい。この調製方法は、ドイツ公開特許第4419399A号公報において例証されている。完全な開示のために、この公報は、二重特異性抗体の定義にも関連する引用によりその全体が取り込まれている。他の調製方法も、本発明において要求される上述の定義に基づく三機能二重特異性抗体に導かれる限り、有用であることは理解されるべきである。
【0068】
前記trAbのFcγ−RIへの結合は、最適な抗腫瘍効果という観点から二つの不可欠な利点を示す。
(1)Fcγ−RI陽性細胞は、ADCCによって腫瘍細胞を除去する能力を有する。そのため、前記trAbによって腫瘍細胞に誘導された細胞傷害性T細胞の抗腫瘍効果に相乗的に貢献することができる。
(2)FcγRI陽性細胞(単球/マクロファージ/樹状細胞等)は、T細胞に対して、抗原提示に似た重要な共刺激シグナルを提供し得る。それにより、T細胞アネルギーを阻害する。さらに、腫瘍特異的ペプチド(腫瘍細胞上でMHC抗原を通じて提示される)を認識するT細胞受容体を有するT細胞でさえ、T細胞の修飾細胞と腫瘍細胞とのtrAbを通じた相互作用による望ましい副産物として、刺激可能である。この配置において、T細胞の適切な活性化に必要な共刺激は、修飾細胞(単球等)から提供される。このため、T細胞受容体と独立で、trAbが媒介する直接的な腫瘍の破壊とは別に、本発明の抗体は、前記trAbの分解後も患者内でパトロールを続ける腫瘍特異的T細胞を活性化し、産生することも可能なはずである。これは、遺伝子治療的なアプローチ(例えば、腫瘍細胞中にB−7等の共刺激抗原を取り込むことによる)と似て、三機能二重特異性抗体によって患者の腫瘍寛容性が撤廃され得ることを意味する。
【0069】
後述する実験データは、本明細書中に記載したtrAbが、前記標的抗原、特に好ましくはHer2/neuおよびCD20標的抗原を低度から中程度の発現レベルでしか有さない腫瘍細胞の破壊に、驚くほど高度に効果的に使用可能であることを示す。高度な効果の理由は、前述した前記三機能抗体の作用様式に見出すことができる。前記作用様式は、単特異性抗体とは大きく異なる。本発明において使用される三機能抗体は、抗T細胞結合アームおよびFc領域により、種々のタイプの免疫細胞を誘導し、同時に活性化できる。これらの免疫細胞の典型例は、Tリンパ球並びにFcγ受容体I型(CD64)およびIII型(CD16)を有する修飾細胞である。第一の結合アームは、Her2/neuまたはCD20等の腫瘍細胞上の標的抗原に結合し、第二の結合アームは、例えば、Tリンパ球上のCD3に結合する(図4)。
【0070】
腫瘍抗原の発現レベルを定量化する方法は、その技術分野において公知である。例えば、免疫細胞化学法であり、例えば、ELISA試験、定量的フローサイトメトリー、組織染色法およびサイトスピン解析等の方法である。腫瘍細胞上の腫瘍関連標的抗原の発現を定量的に決定する方法の典型例は、腫瘍組織上のHer2/neuの発現レベルの定量的決定を可能にした、DAKO社(Glostrup、デンマーク)によって開発されたHercepTest(登録商標)等の免疫組織化学法である。HercepTest(登録商標)に関する参考文献が、DAKO Cytomation Autostainerコード番号K5207第1版として、DAKO社により記載されており、出版番号111 781−001およびK5207/EFG/AOS/13.10.04によって同定できる。この文献は引用により本明細書中に完全に取り込まれている。腫瘍細胞の表面に発現する標的抗原のおよその数を表す4つの段階0、1+、2+、3+を含む、Her2/neuによる評価システムが提供されている。このシステムに従って、標的細胞の表面に20,000未満のHer2/neu分子が発現している細胞は、陰性と分類され、約20,000より多く、約100,000〜110,000以下(最大で150,000)の分子を発現する細胞は、1+(低度の発現)と分類され、約500,000以下の分子を発現する細胞は、2+(中程度の発現)、約2,000,000から約10,000,000分子を発現する細胞は、3+(高度の発現)に分類される。本発明によれば、Her2/neuに対する三機能二重特異性抗体は、HercepTest(登録商標)に従ってHer2/neu 1+および2+(後者においては、さらにFISH法陰性の場合)と分類された腫瘍細胞を有する患者の治療に使用可能であることが見出されている。
【0071】
いわゆるFISH法は、当該技術分野において周知である。“FISH”とは「蛍光In Situ Hybridization」を意味し、細胞遺伝学的技術であり、染色体上の特異的DNA配列の有無を検出し、場所を特定するために使用可能である。前記技術自体は確立されており当該技術分野において公知である。引用するとすれば、例えば、Laudadio J、Quigley DI、Tubbs R、Wolff DJ、HER2試験があげられる。検出方法およびそれらの臨床上の実績の総覧は、引用により本明細書中に完全に取り込まれている。
【0072】
FISH法の試験結果において陰性とは、Her2/neu遺伝子は増幅されていないこと、すなわちHer2/neu遺伝子の数が通常の生理学的範囲内にあるということを意味する。しかし、全ての乳ガンのうち15から20%では、Her2/neu遺伝子は増幅されている。この増幅の検出は確立されており、乳ガンの前記療法にとって重要である。Her2/neu遺伝子が増幅されている場合、FISH解析は陽性であり、その場合、例えばHerceptinによる療法が有効である可能性がある。一方、Her2/neuを過剰発現するが、FISH法において陰性の患者は、Herceptin等の抗体では効果的に治療できない。それゆえ、500,000以下のHer2/neuの発現を有するがFISH法において陰性の患者もまた、本明細書に記載されているように前記三機能二重特異性抗体で治療が成功可能であることは、乳ガンの療法における重要な進歩であると考えられなければならない。多数のグループが、B細胞悪性腫瘍でのCD20の発現も調査している。Ginaldiら(J.Clin.Pathol., 51:364, 1998)は、Quantum Simply Cellular microbeadsキット(Sigma社、セントルイス、ミズーリ州、米国)を用いて、種々のB細胞悪性腫瘍のCD20の発現を評価した。彼等は、例えばB−CLLでは白血球細胞あたり平均65,000のCD20分子、マントル細胞リンパ腫では細胞あたり123,000のCD20分子を見出した。この調査により、有毛細胞白血病の場合、平均値は細胞あたり312,000のCD20分子と決定された。
【0073】
Huhら(Am J Clin Pathol 116:437, 2001)は、さらに、例えば、末梢血および骨髄のような種々の部位でのCD20の種々の発現を、B細胞悪性腫瘍で比較可能な方法で調査した。ここにおいて、骨髄試料では、B−CLLにおいて細胞あたりのCD20分子の数は最低であり(平均4,067;範囲222〜46,395)、濾胞性リンパ腫(平均22,240;範囲3,689〜39,643)、マントル細胞リンパ腫(平均29,770;範囲2,785〜97,727)ではより高く、有毛細胞白血病(平均31,563;範囲1,607〜72,540)では最高であった。対照的に、末梢血では、細胞あたりのCD20分子の数は一般的により高く、例えばB−CLLでは平均数は9,051で、563から31,507の範囲であった。
【0074】
まとめると、B細胞悪性腫瘍の場合、細胞あたりのCD20分子数は、約1000から300,000まで、B細胞悪性腫瘍のタイプおよび身体での部位に依存して変化し、最大約350,000であった。
【0075】
次の実施例において、添付の図および表を参照することによって、本発明を、さらに詳細に記載する。第一の実施例は、本発明を、商標Rexomun(登録商標)(国際一般名 ertumaxomab)で入手可能な三機能抗体、および、Rexomun(登録商標)の標的となる前記腫瘍関連抗原Her2/neuの観点から例証する。第二の実施例は、CD3を通じてT細胞に結合し、腫瘍抗原としてCD20を認識するBi20(FBTA05としても知られる)という三機能二重特異性抗体に向けられている。これらの実施例は、請求項1および明細書全体で特定した他の全ての腫瘍関連抗原に関しても、本発明が使用可能であるという説得力のある証拠を提供する。これらの腫瘍関連抗原全ては、従来使用されていた抗体に基づく免疫療法ではほとんど治療不可能なほど少数しか腫瘍細胞の表面上に存在していなくてもよい。
【0076】
本発明を説明するための図および表は、実施例1および2に関連し、次のとおりに説明される。
【0077】
図1:A−F 異なる抗Her2抗体を用いてフローサイトメトリーにより測定された細胞系統Lox、HCT−8およびSKBR3のHer2/neu発現特性
【0078】
図2a:Her2+3 SKBR−3細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0079】
図2b:Her2+1 HCT−8細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0080】
図2c:Her2ネガティブ Lox細胞の死滅:Micrometastasis Detection Systemを用いたドットプロット解析; *印の番号は、表3に対応する。
【0081】
図2d:Her2+1 HCT−8細胞の死滅:MDSワークフロー(workflow)によるドナー2のドットプロット結果の一例; *印の番号は、表3に対応する。
【0082】
図3:サイトカインビーズアレイシステム(CBA)を用いて測定された24時間後のクローン原性アッセイ上清中のサイトカイン分泌
【0083】
図4:推定される三細胞複合体(tri−cell−complex)。三機能抗体は、異なる免疫学的メカニズムにより腫瘍細胞の認識と破壊を促進することができる。IFN−γを除く図中のサイトカインに関するリストをまとめなければならない(Bei der Auflistung der Zytokine im Bild sollte man noch IFN−γ aufnehmen)。
注:ADCC、抗体依存性細胞傷害;DC、樹状細胞;NK、ナチュラルキラー細胞;DC−CK1、樹状細胞サイトカイン1;IL、インターロイキン;LFA、リンパ球機能関連抗原;TNF−α、腫瘍壊死因子α;CD、表面抗原分類(出典 Ruf&Lindhofer,2001)
【0084】
図5.Bi20の精製。(A)陽イオン交換クロマトグラフィー(CIEX)。親マウス抗体(ピーク1)を、HR10/10 high performance SP−Sepharose column上で、三機能抗体(ピーク2)から分離した。(B)Bi20三機能抗体精製の等電点電気泳動(IEF)。親ラット抗体(26II6)を、プロテインAアフィニティークロマトグラフィー(AC Bi20)後に除去し、親マウス抗体(TPA10)を、CIEX(CIEX P1 Bi20)により三機能抗体から分離した。精製されたBi20は、前記陽イオン交換クロマトグラフィーのピーク2(CIEX P2 Bi20)に見出された。
【0085】
図6.Bi20が特異的に結合するエフェクター細胞。190ngのBi20を、5×105個の標的細胞とともにインキュベートし、その結合をFACS解析により測定した。Ramos細胞(A)を、CD20アームの標的細胞として用いた。THP−1細胞(B)を、Fc領域用に用いた。拮抗実験のために、細胞を予めRituximab(Rit)とともに所定の過剰量の抗体で30分間インキュベートした。
【0086】
図7.エフェクター細胞の存在下におけるBi20が媒介する効果的なB細胞の減少。(A)1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Mec1)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともにインキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)(neg)前記アッセイにおけるB細胞の絶対数を、100%に設定した。(B)1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。前記B細胞の絶対数を、トリパンブルー色素排除集計およびFACS解析により、1日目、2日目、3日目において測定し、B細胞の減少割合を算出した。
【0087】
図8.Bi20が媒介するT細胞の活性化。(A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。CD4+およびCD8+T細胞上におけるCD25の発現のMFIを、FACS解析により測定した。(B)PBMCおよびRaji細胞を、Bi20とともに、Bi20の量を増加させて(0.5〜250ng/ml)インキュベートした。CD4+T細胞上におけるCD25の発現のMFIを、1日目、2日目および3日目に測定した。
【0088】
図9.Bi20が誘導するT細胞の増殖。1×106個/mlのPBMCおよび2×105個/mlのRaji細胞を、50ng/mlの濃度のBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに3日間インキュベートした。T細胞数を、トリパンブルー色素排除集計並びに抗CD5/4抗体および抗CD5/8抗体を用いたFACS解析により測定した。
【0089】
図10.Bi20により媒介される単球の活性化。(A)1×106個/mlのPBMCおよび2×105個/mlのRaji腫瘍細胞を、50ng/mlのBi20、catumaxomab(Catum)またはrituximabとともに1日間インキュベートした。CD14+単球によるCD25の発現のMFIを、FACS解析により測定した。(B)1×106個/mlのPBMCおよび1×105個/mlのTHP−1細胞を、50ng/mlのBi20、catumaxomab(Catum)、rituximab(Rit)、TPA10、または26II6とともに1日間インキュベートした。CD33+THP−1細胞の活性化を、CD25またはCD40の発現によって測定した。
【0090】
図11.健康なドナーのエフェクター細胞存在下におけるBi20が誘導するCLL B細胞の減少。(A)1×106個/mlのPBMCおよび2×105個/mlのCLL腫瘍細胞を、5、50、もしくは250ng/mlのBi20、250ng/mlのcatumaxomab(Catum)、または250ng/mlのRituximab(Rit)とともに3日間インキュベートした。B細胞の除去を、単球、B細胞およびT細胞のトリパンブルー色素排除集計並びにFACS解析により、3日目に測定した。抗体を用いない(ネガティブ)前記アッセイにおけるB細胞の絶対数を、100%に設定した。MV:CLL−1からCLL−9の平均値。(B)CLL患者検体(1〜14)におけるCD20の発現の概要、細胞系統(Raji、Mec−1)および1体の健康なドナー(PBMC)。MFI=蛍光強度の平均値。
【0091】
表1:用いた抗体およびそれらに対応する検出抗体の特性
* TRION Pharma社、ミュンヘン、$ Roche社、# Micromet社、ミュンヘン
【0092】
表2:用いた細胞系統およびそれらのHer2/neuスコア
*:Ross et al., Molecular and Cellular Proteomics 3:379−398, 2004に準拠
【0093】
表3:クローン原性アッセイサンプル
Ab=抗体、μg=マイクログラム、ng=ナノグラム
【0094】
表4.Bi20が媒介するサイトカイン分泌。1×106個/mlのPBMCおよび2×105個/mlの腫瘍細胞(Raji、Ramos、Mec1、Granta、DOHH−2)を、50ng/mlのBi20、catumaxomab(Catum)、rituximab、または親抗体であるTPA10および26II6とともに、3日間インキュベートした。上清を収集し、サイトカイン分泌を、CBA解析およびFACS解析を用いて測定した。
【0095】
表5.Bi20が媒介する自己設定におけるin vitroでのCLL B細胞の除去。B細胞およびT細胞の割合(%)、B細胞上におけるCD20の発現(MFI)、およびE:T比を、CLL PBMCの調製後直ちにインキュベート前に測定した。CLL細胞を所定量のBi20またはrituximabとともに、0、3、6、および9日目にインキュベートした。B細胞の除去を、患者検体に応じて3日目から10日目の間に測定した。B細胞の除去を、減少したB細胞の割合(%)で示す。T細胞活性化:CD4+およびCD8+T細胞上におけるCD25の発現は、少なくとも5倍(++)上方制御された。エフェクター/ターゲット比(E:T)を、単球数、NK細胞数およびT細胞数に対するB細胞数として定義した。10%以下のB細胞の除去は、ネガティブとみなした。
【実施例】
【0096】
実施例1
実施例1は、Her2/neuに結合する三機能二重特異性抗体をHercepTest(登録商標)におけるスコア1+の患者の治療に用いた場合の性能に関するキャラクタリゼーションについて記載する。
【0097】
方法
[細胞系統および抗体]
Rexomun(登録商標)(Trion Pharma社製、ミュンヘン)は、一方の結合アームにより腫瘍関連抗原Her2/neuを標的にし、他方のアームによりT細胞上のCD3分子に結合する三機能抗体である。さらに、Rexomun(登録商標)は、マウスIgG2aおよびラットIgG2bアイソタイプからなる強力なFc領域を通じてFcγRI陽性およびFcγRIII陽性細胞(例えば、マクロファージ、NK細胞、樹状細胞)に結合する。モノクローナル抗体2502A(Trion Pharma社、ミュンヘン)は、Her2/neuに特異的であり、Rexomun(登録商標)に含まれる親抗体の一つである。26II6(Trion Pharma社、ミュンヘン)は、CD3に特異的であり、Rexomun(登録商標)に含まれる親抗体の他の一つである。520C9(ATCC HB−8696)は、Her2/neu受容体を認識する他のモノクローナル抗体である。Trastuzumabは、マウスの抗Her2/neu抗体であるMab4D5(58)から開発されたヒト化抗Her2モノクローナル抗体である。表1に、用いた抗体およびそれらに対応する検出抗体の特性を示す。
【0098】
SKBR3(ATCC H TB−30)は、IHCにより検出されたHer/neuを強力に過剰発現する(HercepTest(登録商標)スコア+3)乳ガン細胞系統であり(58)、HCT−8(ATCC CCL−244)は、大腸ガン細胞系統であり、Loxは、メラノーマ細胞系統である(表2)。
【0099】
[Her2/neuスコアリング]
HCT−8細胞におけるHer2/neuの状態(status)を、HercepTest(登録商標)キット(DAKO社、グロストラップ、デンマーク)を用いてIHCにより評価し、製造者のマニュアルに従って、ミュンヘン大学(LMU ミュンヘン)の病理学研究所(Institute of Pathology)の力を借りて実施した。
【0100】
[FACS解析]
FACS解析を、5×105個の標的細胞(SKBR3、HCT−8またはLox)を用いて実施した。細胞を、100ngのモノクローナル抗体または100ngの三機能抗体Rexomun(登録商標)とともに、45分間4℃でインキュベートした。それから、検体をMg2+およびCa2+を含まないリン酸緩衝生理食塩水(PBS)(PAN Biotecs社、アイデンバッハ)を用いて洗浄し、対応する検出抗体(Dianova社、ハンブルク)とともに45分間4℃でインキュベートした(表1)。PBSを用いてもう一度洗浄工程を行った後、細胞を0.01μg/mlの臭化エチジウムを含むPBS(Sigma社、ミュンヘン)中に再懸濁した。結合した抗体の量を、オーバーレイヒストグラム(overlay histogram)における蛍光強度の平均値により解析した。抗ヒト検出抗体のみを用いた検体、または26II6(抗CD3)およびそれに対応する検出抗体(アイソタイプコントロール)とともにインキュベートした検体を、ネガティブコントロールとした。すべてのFACS解析は、FACSCalibur(Becton Dickinson社、ハイデルベルク)により実行し、Cell Quest ProまたはWinMDI2.8を用いて解析した。
【0101】
[クローン原性アッセイ]
抗体が媒介する腫瘍の除去を、刺激されていないエフェクター細胞を用いた長期クローン原性アッセイを用いて検討した。そのために、106個のPBMCを、5%の標的細胞(SKBR3、HCT−8またはLox)とともにスパイクし(spike)、24穴プレート(Greiner社)に播種した。抗体を、表3に示す異なる濃度および組み合わせで添加した。標的細胞のみの検体およびいずれの抗体も添加していない標的細胞とエフェクター細胞との混合物からなる検体を、コントロールとした。検体を、37℃/5%の二酸化炭素でインキュベートし、RPMI1640、L−グルタミンおよび10%のウシ胎児血清(FCS)を3日に一度ずつ供給した。8日間のインキュベート後、細胞を前記24穴プレートから収集し、Mg2+/Ca2+を含まないPBSを用いて洗浄し、Neubauer Cell Chamberにおいて、トリパンブルーによる死細胞の排除により計数した。それから、生細胞をスライドあたり2.5×105個の細胞濃度で、サイトスピンスライド(Menzel社)上で遠心沈殿した。遠心沈降後の検体を一晩乾燥させ、続いて免疫細胞化学法にかけた。異なる3体のドナー由来のPBMCを用いて、実験を3回繰り返した。
【0102】
[免疫細胞化学法]
サイトスピンスライド上での非特異的結合特性を、10%のヒト血清を用いて飽和させた。SKBR3およびHCT−8細胞を、スライドあたり1.5μgの抗サイトケラチン8、18、19抗体(A45B−B3、マウスIgG1、Micromet社、ミュンヘン)を用いて染色し、Lox細胞を、スライドあたり1.5μgの抗EGFR抗体(ミュンヘンのDr.Htunに快く提供していただいたマウスIgG2a)を用いて染色した。サイトケラチン染色用の抗マウスIgG1 Alexa Fluor 477(FITC)抗体(Molecular probes社、ユージーン)、またはEGFR染色用の抗マウスIgG2a Alexa Fluor 477(FITC)抗体(Molecular probes社、ユージーン)を、検出抗体として、スライドあたり1.5μgで用いた。サイトスピンスライドを、FITC標識された細胞を計数するコンピュータ化された画像解析を用いたMicrometastasis detection system(MDS、Applied Imaging社)により解析した。
【0103】
[+1のHer2/neu発現を示す大腸ガン細胞系統であるHCT−8]
Her2/neuの+1発現を有する細胞系統を見出すために、Her2/neu発現のレベルを、異なる細胞系統において検討した。
【0104】
そのために、前記モノクローナルHer2/neu抗体である2502A、520C9、Trastuzumabおよび前記三機能抗体Rexomun(登録商標)を用いたFACS解析を実施した。前記モノクローナル抗CD3抗体である26II6を、ニセのコントロールとした。図1A−Fに、SKBR−3、HCT−8またはLox細胞に用いた抗体の結合特性を示す。異なる検出抗体のみを用いたフローサイトメトリー検体を、コントロールとし(図1A)、それに対応する検出抗体を用いた26II6(抗CD3)を、アイソタイプコントロールとした(図1B)。
【0105】
メラノーマ細胞系統であるLoxは、予想どおりHer2/neu受容体の発現を示さなかった(図1C−F)。一方、SKER3細胞は、モノクローナル抗体520C9(図1C)、2502A(図1D)およびTrastuzumab(図1E)を用いたHer2/neuに対して重度に染色された。Rexomun(登録商標)は、SKBR3細胞を低度に染色した。おそらく、その一方のアームの結合特性のためであろう(図1F)。SKBR3細胞と比較して、HCT−8細胞は、すべての用いたHer2抗体に対するHer2/neu受容体の発現が明らかに弱い(図1C−F)。このことは、これらの細胞のさらなる評価を正当化する。加えて、HCT−8細胞のHer2/neu発現を、DAKO社のHercepTest(登録商標)を用いて解析した。前記テストを、ミュンヘン工科大学(TU、ミュンヘン)の病理学研究所の力を借りて実施した。前記大腸ガン細胞系統であるHCT−8は、実施したDAKOテストにおいて、FACS解析の結果を確認することで、Her2/neuの+1発現を示した。表2に、用いた細胞系統、それらのHer2/neuスコアおよびHer2/neu発現の測定に適用された方法の概要を示す。
【0106】
[Rexomun(登録商標)は、in vitroにおいて、Her2/neu+3細胞系統およびHer2/neu+1細胞系統を死滅させることが可能である]
腫瘍部位に対して異なるタイプの免疫細胞を補充すると同時に活性化するRexomun(登録商標)の能力は、Rexomun(登録商標)が、Her2/neuを低度(+1)に発現している腫瘍細胞を除去することも可能であるかもしれないという仮説を導いた。
【0107】
そのために、長期細胞障害性解析(クローン原性アッセイ)を確立した。3体の健康なドナーのPBMCを、Her2/neu+3発現する細胞系統であるSKBR−3、Her2/neuネガティブのLox細胞系統またはHer2/neu+1発現するHCT−8細胞系統とともに、8日間インキュベートした。異なる抗体濃度のTrastuzumab(100μg/ml〜1μg/ml)、またはRexomun(登録商標)(100ng/ml〜1ng/ml)を、表3に示すように添加した。腫瘍細胞の死滅を、8日後に前記細胞を収集し、それらをサイトスピンスライドに移し、前記細胞を抗CK8、18、19抗体(SKBR−3およびHCT−8)、または抗EGFR抗体(Lox)と、これらに対応するAlexa−Fluor FITC抗体とを用いて染色することで評価した。それから、前記染色したサイトスピンスライドを、Micrometastasis Detection System(MDS、Applied Imaging社、英国)上のSLIDE SCANプログラムを用いるコンピュータ化された画像解析により評価した。
【0108】
この研究において、前記三機能抗体であるRexomun(登録商標)(抗Her2/neu×抗CD3)を、低度にHer2/neuを発現する腫瘍細胞系統であるHCT−8(HercepTestスコア:+1)の腫瘍細胞増殖を阻害する能力において、ヒト化抗Her2/neuモノクローナル抗体であるTrastuzumab(登録商標)と比較した。
【0109】
前記細胞系統SKBR3(+3 Hercepスコア)、HCT−8(+1 Hercepスコア)およびLox(Her2/neuネガティブ)におけるHer2/neu発現を、FACS解析により測定した。一方、HCT−8細胞を、HercepTest(登録商標)によってもテストした。Her2/neuに対する種々の抗体を用いたFACS解析は、SKBR3細胞においては強力なHer2発現を示し、HCT−8細胞においてはより弱い発現を示し、Lox細胞においてはHer2/neuの発現を示さなかった。これらの結果は、HCT−8細胞の+1HercepTest(登録商標)スコアと一致する。三機能抗体であるRexomun(登録商標)の蛍光の平均値は、モノクローナル抗体の前記値より、わずかに低い。モノクローナル抗体が二価の結合であるのと比較して、Rexomun(登録商標)が一価の結合であるためである(図1:A−F)。
【0110】
前記低度にHer2/neuを発現する細胞系統であるHCT−8の増殖を阻害するRexomun(登録商標)およびTrastuzumab(登録商標)の能力をテストするために、クローン原性アッセイを実施した。8日後の培養細胞を、培養皿から収集し、サイトスピンスライド上で遠心沈降し、腫瘍細胞を染色した。抗体を添加していないすべてのコントロール検体(24穴プレートあたり12,500個の腫瘍細胞)(図2a〜c、検体A1、B1、C1)は、サイトスピン染色後のMDSにおいて、細胞系統SKBR3、HCT−8およびLoxに関して腫瘍細胞の増殖を示した。スパイクした腫瘍細胞、または抗体を添加していない250000個のPBMCを含むスライドでは、予想どおり全てにおいて腫瘍細胞が見られなかった(図2a〜c、検体A2、B2、C2)。PBMCを5%のSKBR3、HCT−8またはLox細胞とともに、抗体を添加せずに混合することで、腫瘍の増殖を、同種間のコントロールにおいて明確に可視化した(図2a〜c、検体A3、B3、C3)。
【0111】
Her2+3細胞系統であるSKBR−3の腫瘍細胞の増殖は、予想どおり100μg、50μg、10μgおよび1μgのTrastuzumab(登録商標)の添加により阻害された(図2a、検体A4〜A8)。さらに、Rexomun(登録商標)も、わずか100ng、50ng、10ngまたは1ng添加しただけで、腫瘍の増殖を阻害することが可能であった。検体A9〜A12に示すように(図2a)、腫瘍細胞の増殖は、Trastuzumab(登録商標)よりも効果的に阻害された。
【0112】
Her2/neuネガティブ細胞系統であるLoxは、Her2/neu受容体が存在することにより駆り立てられる(driven)腫瘍細胞除去の特異性を示すTrastuzumab(登録商標)、またはRexomun(登録商標)を種々の量で添加しても、腫瘍細胞の増殖は阻害されなかった(図2c、検体C4〜C12)。
【0113】
大腸ガン細胞系統であるHCT−8(Her2/neuスコア+1)の増殖は、種々の量(100μg〜1μg)のTrastuzumab(登録商標)によって、際立っては阻害されなかった(図2b、検体B4〜B8)。このことは、Her2/neu+1の乳ガン細胞系統であるMCF−7(5)に関する従来の知見と一致する。MCF−7細胞(ATCC HTB−22)は、TNF−αによる増殖阻害を示すため、三機能抗体であるremovab(登録商標)(抗EpCAM×抗CD3)に類似するrexomun(登録商標)は、その活性化特性によりTNF−α分泌の引き金を引いてしまうとして、これらの細胞をクローン原性アッセイに用いることができなかった(59)。しかしながら、Rexomun(登録商標)は、1穴あたり100ng、50ng、10ngという低濃度で、+1のHer2/neuの細胞系統であるHCT−8を破壊することが可能であった(図2b、検体B9〜B11)。1ngのRexomun(登録商標)では、HCT−8細胞の腫瘍細胞増殖の阻害に成功しなかった(図2b、検体B12)。図2dに、標的としてHCT−8細胞を用いるドナー2のクローン原性アッセイ関する元のMDSデータプロットを示す。
【0114】
結論として、Rexomun(登録商標)は、+3または+2の過剰発現/遺伝子増幅を示す乳ガン患者のためだけでなく、低度のHer2/neu発現レベルの患者のための治療の選択肢となる。
【0115】
実施例2
ここでは、ヒトCD3およびヒトCD20に対する新規な三機能二重特異性抗体であるBi20の産生と性能に関するキャラクタリゼーションについて記載する。特に、腫瘍抗原であるCD20を低レベルでしか発現していない患者の治療に用いられるその能力について記載する。
【0116】
CD20は、35kDaの糖化されていないリン酸化タンパク質であり、B細胞分化において役割を果たすカルシウムチャネルとして働くと言われている24。しかしながら、CD20欠損マウスが正常なB細胞発生を示し、表現型的にも正常であることから25、その正確な機能は未だに不明である。CD20は、いくつかの理由により免疫療法のための標的として選択される:(i)CD20は、正常なB細胞および悪性のB細胞においては独占的に発現されるが、血液学的な前駆体細胞または他のヒトの組織の細胞においては発現されず26、(ii)CD20は、ほとんどのB細胞リンパ腫細胞において発現され27、(iii)CD20は、抗体との結合のために噴出され、または分泌されない28、29。さらに、腫瘍治療用の標的としてのCD20の適合性は、Rituximabの使用、特に従来の化学療法との組み合わせによる使用の成功により臨床的に確認されている。
【0117】
本発明の研究においては、このtrAbが、効果的にヒトB細胞腫瘍細胞および、低レベルでしかCD20を発現していない慢性リンパ性白病(CLL)患者由来の細胞を死滅させることを実証するために、前記Bi20のin vitroにおける特性について詳細に解析を行った。腫瘍細胞の除去のために、エフェクター細胞を予め活性化させる必要がなく、前記腫瘍細胞の除去は、典型的なTh1サイトカインパターン、並びにT細胞および単球の強力な活性化に伴って起こった。臨床応用のための重要な必要条件としては、Bi20が、容易に精製され、充分な量を産生し得ることである。従って、このtrAbは、治癒不能な非ホジキン悪性リンパ腫(NHL)患者、およびCLL患者のための他の新規な治療の選択肢を提案し得る。
【0118】
材料および方法
[抗体および試薬]
CD20特異的モノクローナルIgG2a抗体を産生するマウスの細胞系統であるTPA10を、26II6を用いてCD3特異的IgG2b抗体を分泌するラットの細胞系統と融合し、結果として、Bi20(FBTA05)を産生するクアドローマ細胞系統であるTPBs05を得た。Catumaxomab(Removab(登録商標))を、26II6由来のCD3特異的アームおよび抗EpCAM特異性からなる三機能性のコントロール抗体として用いた。キメラの抗CD20抗体であるRituximab(Mabthera(登録商標))は、Roche社(バーゼル、スイス)から入手した。前記抗CD20抗体および対応するアイソタイプは、Beckman Coulter Immunotech社(クレフェルト、ドイツ)から入手した。FACS解析に用いた他のすべての抗体は、BD Biosciences社(ハイデルベルク、ドイツ)から購入した。ヨウ化プロピジウムは、Sigma Chemicals社(ダイゼンホッフェン、ドイツ)から入手した。
【0119】
[細胞系統およびPBMCの調製]
CLL細胞系統であるMec1を、Dr. Michael Hallek(GSF、ミュンヘン、ドイツ)に快く提供していただいた。バーキットリンパ腫(Burkitt’s lymphoma(BL))細胞系統であるRamosは、ATCC(米国)から入手した。前記BL細胞系統であるRaji、前記NHL細胞系統であるDOHH−2およびGranta、並びにAML細胞系統であるTHP−1は、DSMZ(ブラウンシュヴァイク、ドイツ)から購入した。細胞を、10%の熱失活したFCS、非必須アミノ酸、ピルビン酸ナトリウムおよびL−グルタミン酸塩(PAN−Biotech社)を添加したRPMI−1640培地(PAN−Biotech社、アイデンバッハ、ドイツ)中で生育した。ヒト抹消血単核球細胞(PBMC)は、PANCOLL(PAN−Biotech社)を用いて密度勾配遠心により、健康なドナーのヘパリン処理した血液から単離した。CLL患者由来の抹消血検体は、インフォームドコンセント(informed consent)の後に入手した。CLLの診断は、標準的な臨床的診断基準および研究室的診断基準(laboratory criteria)に従った。CLL細胞は、精製後直ちに用い、または後の使用のために凍結保存した(cryo−conserved)。CLL細胞は、10%のFCSを添加したIMDM(PAN−Biotech社)中で培養した。自己設定におけるアッセイには、新鮮に調製したCLL細胞のみを用いた。
【0120】
[Bi20の産生および精製]
Bi20を、クアドローマ技術を用いて産生した30。クアドローマ細胞の上清を、標的細胞に結合するtrAb用に、FACS解析によりテストした。trAb産生細胞を、抗体産生の安定性を向上させるために数回にわたってサブクローニングし、マスター細胞バンクを確立した。trAbを、クアドローマ細胞の培養上清から、説明されているように31、AKTA purifier 100(Amersham Biosciences社、ウプサラ、スウェーデン)を用いて、プロテインAアフィニティクロマトグラフィーおよびイオン交換クロマトグラフィーにより精製した。
【0121】
[等電点電気泳動]
抗体検体を、pH7〜11のIsoGel−Agarose−IEF(Cambrex Bio Science社、ロックランド、米国)に、スポットし(spotted)、75分間、1500V、50mA、および25Wで分離した。抗体のアイソフォームを、クーマシーブリリアントブルー(Sigma−Aldrich社、ミズーリ州、米国)を用いて染色することにより可視化した。ゲルを、解析のためにスキャンした。
【0122】
[FACS解析]
標的細胞への抗体の結合、エフェクター細胞の活性化、関連するCD20の発現、および細胞のサブタイプ組成を、488nmのアルゴンレーザーを備えたFACScalibur(BD Biosciences社、ハイデルベルク、ドイツ)を用いて、フローサイトメトリーにより解析した。データを、Cellquest Software(BD Biosciences社)を用いて解析した。
抗体の結合を解析するために、5×105個の細胞(Ramos、JurkatまたはTHP−1)を、所定の濃度のBi20とともに、30分間4℃でインキュベートし、2%の熱失活したFCSを添加したPBSを用いて洗浄し、およびFITC−標識されたマウスの抗ラットIgG検出抗体(Ramos)、またはFITC標識されたラットの抗マウス抗体(Jurkat、THP−1)とともに、30分間4℃でインキュベートした。
Bi20の活性化特性を決定するために、1×106個/mlのPBMCおよび2×105個/mlのヒトの腫瘍細胞を、前記インキュベートした濃度の抗体とともに、1、2、および3日間37℃で、RPMI培地においてインキュベートした。細胞を収集、洗浄し、FITCまたはPE標識されたCD25およびCD69に対する検出抗体とともに、30分間4℃でインキュベートした。
細胞のサブタイプ組成を、下記の抗CD抗体混合物(FITC/PE/APC)を用いて、3×106個のPBMCを染色することにより決定した:14/19/5、4/8/5、4/8/25、45/3/5および16/56/5。
【0123】
[B細胞減少解析]
Bi20が媒介するB細胞減少を、バイオアッセイを用いて測定した。健康なドナーから入手したPBMC(1×106個/ml)に、2×105個/mlの所定の腫瘍細胞(Raji、Ramos、Mec1、DOHH−2もしくはGranta)、またはCLL患者の細胞(エフェクター対標的比 5:1)を添加し、所定の濃度における異なる抗体の存在下において総容積1mlでインキュベートした。1日目、2日目および3日目における細胞を、収集、洗浄して、生きている(viable)B細胞を、PE結合抗CD19検出抗体を用いてFACS解析により解析した。生きている細胞の総数を、トリパンブルー色素排除集計により測定した。
自己CLL患者検体中の抗体が媒介するB細胞減少を検討するために、CLL患者から単離したPBMCを、2×106個/mlの密度で蒔いた。抗体を、所定の濃度で0日目、3日目および6日目に添加した。FACS測定およびトリパンブルー色素排除集計を、所定の時点で実施した。
【0124】
[サイトカインの測定]
Bi20に誘導されたサイトカインの放出を、B細胞減少実験に用いた条件と同じ条件下において測定した。腫瘍B細胞系統および健康なドナーからのPBMCを、50ng/mlの所定の抗体とともにインキュベートした。3日後、上清を収集した。サイトカインを、ヒトTh1/Th2細胞数測定ビーズアレイ(CBA、BD Biosciencess社、ハイデルベルク、ドイツ)を用いて測定した。前記CBAは、INF−γ、TNF−α、IL−2、IL−4、IL−6およびIL−10の解析を同時に行うことが可能である。データの取得および解析を、製造者のプロトコールに従って実施した。
【0125】
結果
[Bi20の産生および精製]
前記trAbであるBi20を、クアドローマ細胞において産生し、プロテインAアフィニティクロマトグラフィーにより獲得した。強力なラットの抗体は、プロテインAに結合しないため、溶出液が不要である。それから、親マウス抗体等の不純物を、陽イオン交換クロマトグラフィー(CIEX)により除去した(図5A)。マウス抗体を、148mMのNaClで溶出した(図5A、ピーク1)。一方、trAbを、320mMのNaClで溶出した。精製されたtrAbの等電点電気泳動は、pIが8.65〜8.15の範囲において明確なバンドパターンを示した。trAbは、CIEX分離のピーク2においてのみ検出された。親抗体の混入は検出されなかった。
【0126】
[Bi20の結合特性]
Bi20の三機能の性質を実証するために、その特異的な標的細胞への結合を検討した。CD20結合アームの機能性を確認するために、CD20+バーキットリンパ腫細胞系統であるRamosを、CD3結合アームのためにCD3+T細胞系統であるJurkatを、FcγRIに結合することを確認するために単球細胞系統であるTHP−1を用いた(図6)。CD20へのBi20の結合を確認するために、Rituximabを用いた競合的結合アッセイを実施した。Ramos細胞上でのBi20の結合を、予め前記細胞を21倍の過剰なRituximabとともにインキュベートすることにより、完全に阻止することができた(図6A)。同様に、THP−1細胞へのBi20の結合を、Rituximabにより、競合することができた(図6B)。Jurkat細胞上のCD3へのBi20の結合は、Ramos細胞上でのCD20へのBi20の結合に匹敵した(データを示さず)。前記Bi20のCD3結合アームは、特性がはっきりしているtrAbであるcatumaxomabおよびertumaxomabにおいて対応するアームと同一であるため、前記CD3結合アームの特異性のさらなる解析を、実施しなかった。要約すれば、これらのデータにより、Bi20の三価の結合機序および結合特性が確認された。
【0127】
[Bi20が媒介するB細胞の除去]
B細胞の死滅についてのBi20の効果を研究するために、FACSに基づくバイオアッセイを開発した。事前の、または同時の刺激のいずれもしていない新鮮に調製したPBMCを、腫瘍B細胞と5:1の比率で混合した。さらに、50ng/mlの所定の抗体を混合し、3日間インキュベートした。細胞を収集し、分集団の組成をFACS解析により決定した。トリパンブルー色素排除集計を、細胞の絶対数を得るために実施した。
標的細胞として、前記CD20+バーキットリンパ腫細胞系統であるRajiを用い、50ng/mlのBi20を用いたことで、効果的なB細胞の死滅が観察された(図7)。コントロールとして、前記trAbである、B細胞と結合しない(データを示さず)catumaxomab(抗EpCAM×抗CD3)、並びに前記親抗体26II6(抗CD3)およびTPA10(抗CD20)を用いた。Catumaxomab(Catum)および親抗体の組み合わせは、いくらかのB細胞減少を誘導した。おそらく、INF−γおよびTNF−α等のサイトカインの放出によるものであろう(表4)。このことは、Bi20の抗腫瘍活性が、実際にはその三機能デザイン(design)により引き起こされることを示している。これらの実験的な条件(低濃度の抗体およびE:T比)下では、Rituximabは、軽微なB細胞数の減少しか媒介しなかった。CLL細胞系統であるMec1を用いた比較可能なデータを入手した。そのデータでは、CD20の発現がRaji細胞より2.5倍低かった(MFI 179対428、データを示さず)。
【0128】
これらの結果は、Bi20が媒介したB細胞の減少をより詳細に解析するのに勢いをつけた。時間−動態解析(Time−kinetic analyses)は、わずか24時間後にRaji細胞の減少を示した(図7B)。少なくとも35%のB細胞が、0.5ng/mlという低濃度の抗体において除去された。250ng/mlおよび50ng/mlの濃度のBi20においては、2日目および3日目それぞれにおいて、完全なB細胞の減少が観察された。Mec1細胞においても匹敵する結果が見出された(データを示さず)。
さらに、B細胞系統における代表的な他のリンパ腫の実体を、効果的に死滅させることができるかどうか検討した。DOHH−2(濾胞性リンパ腫)、Granta(マントル細胞リンパ腫)、またはRamos(バーキットリンパ腫)を標的細胞として用いた場合、効果的なB細胞減少を検出した(データを示さず)。
【0129】
[同時発生的なB細胞結合に依存しないBi20が媒介するT細胞の活性化]
前述した、例えば、抗CD28抗体を用いて予め刺激した後のみにB細胞の細胞傷害性を示す二重特異性抗リンパ腫抗体32とは対照的に、Bi20により誘導された効果的なB細胞の溶解には、T細胞または単球の前活性化が必要ではなかった。そのために、エフェクター細胞が、Bi20が結合することにより活性化され得るかどうかを問うた。PBMCおよびRaji細胞を、前記所定の抗体とともにインキュベートした。CD4+およびCD8+T細胞上の活性化マーカーであるCD25の上方制御を、フローサイトメトリーにより測定した(図8A)。Bi20およびcatumaxomabの両方が、CD4+およびCD8+T細胞上のCD25の明確な上方制御を誘導した。このことは、trAbがCD3およびCD20に同時に結合することが、T細胞の活性化には必要ないことを示している。CD25の上方制御は、前記親抗体26II6(抗CD3)とTPA10(抗CD20)との組み合わせによっても観察された。予想どおり、Rituximabは、いずれのT細胞の活性化も誘導しなかった(図8A)。
【0130】
図8Bに示すように、T細胞の活性化は、インキュベートする期間と抗体の濃度とに依存する。最大のCD25発現は、2日目の最も高い濃度(250ng/ml)のBi20において観察された。CD69等の他の活性化マーカーも上方制御されたが、より低度でより短期間なものであった(データを示さず)。
【0131】
[Bi20が誘導するT細胞の増殖]
T細胞の活性化に加えて、前記異なる抗体により誘導されるCD4+およびCD8+T細胞数の増加を測定した(図9)。Bi20を用いたインキュベーションの結果として、CD4+サブセットと比較して、CD8+サブセットのT細胞が優先的な増殖を示した(145%対85%のT細胞発現)。CD4+:CD8+比は、1.8:1から1.3:1に変化した。Catumaxomabおよび親抗体はともに、T細胞増殖を誘導したが、Bi20より低度であった。予想どおり、Rituximabは、T細胞増殖を刺激しなかった。特に、前記抗体以外の刺激因子は、増殖を誘導するのに必要ではなかった。
【0132】
[単球の活性化は、同時発生的なT細胞結合には依存するが、同時のB細胞結合には依存しない]
つぎに、単球/マクロファージ等のFcγRI/III+細胞も、Bi20により活性化され得るかどうかという問いに取り組んだ。PBMCおよびRaji細胞を、異なる抗体の存在下においてインキュベートし、CD14+細胞の活性化を測定した。図6Aに示すように、Bi20およびcatumaxomabの両trAbは、CD14+単球/マクロファージ上でのCD25の上方制御を誘導した。このことは、同時のB細胞結合は、単球の活性化に必要ではないことを示している。対照的に、B細胞および単球/マクロファージに結合し、そのFc領域を通じてT細胞とは結合しないRituximabは、CD14+修飾細胞の活性化を誘導しなかった。これらの結果から、T細胞の関与が、修飾細胞の活性化には不可欠であると結論付けられる。
【0133】
この結果を、悪性のB細胞が存在しない場合におけるわずかに異なるシステムを用いて確認した。PBMCおよびTHP−1細胞を、異なる抗体の存在下においてインキュベートした。CD33+であるTHP−1細胞の活性化を、CD25およびCD40の発現によりモニターした(図10B)。THP−1およびT細胞に同時に結合するすべての抗体(Bi20、TPBs01、26II6)は、THP−1細胞を活性化した。対照的に、THP−1およびB細胞には結合するが、T細胞には結合しない抗体(TPA10、Rituximab)を用いた場合は、活性化は観察されなかった。PBMCが存在しない場合におけるTHP−1細胞は、いずれの抗体によっても活性化されなかった(データを示さず)。このことは、同時発生的にT細胞および単球と結合することは、本実験条件において単球を活性化するための絶対的な必須条件であることを示している。
【0134】
[Bi20が誘導するTh1類似のサイトカイン特性]
サイトカインの放出は、モノクローナル抗体を治療的に使用した後に発生する免疫反応のひとつであるので33、Bi20により誘導されるサイトカインの特性を研究した。PBMCを、異なる腫瘍細胞(Raji、Ramos、DOHH−2、GrantaまたはMec1)および50ng/mlの抗体とともにインキュベートした。3日後、上清を収集し、IFN−γ、TNF−α、IL−2、IL−4、IL−6およびIL−10の量を解析した(表4)。
【0135】
Bi20は、IL−6を高レベルに誘導した。前記IL−6は、前記他の抗体とともにインキュベートした後の場合には、明らかに低いレベルで検出された。TNF−αは、Bi20またはcatumaxomabによる処理を行った後に検出されたが、Rituximabまたは前記親抗体とともにインキュベートした後には検出されなかった。IL−2の発現における強力な増加が、Bi20とともにインキュベートした後にだけ検出された。これにより、3つの結合パートナー(腫瘍細胞、T細胞および修飾細胞)が、すべて存在した場合にのみIL−2が放出されることを示すtrAbを用いた従来の研究が確認された18。Th2系統のマーカーであるIL−4は、わずかにだけ分泌された。一方、Th1に特異的なサイトカインであるIFN−γは、強力に産生された。興味深いことに、抑制性サイトカイン(inhibitory cytokine)として振る舞い得るIL−10も放出されている。このことは、免疫学的なカウンター反応(counter−reaction)を示唆している。同様の結果は、すべての細胞系統(Raji、Ramos、Mec1、DOHH−2、Granta;表4)においても得られた。
【0136】
[Bi20が媒介するCLL患者由来のB細胞の除去]
つぎに、Bi20が媒介するB細胞の減少が、CLL患者由来の細胞を用いることにより確認できるかを検討した。図11に示すように、9体の異なるドナー由来の細胞を、健康なドナー由来のPBMCおよびBi20(5、50または250ng/ml)、またはコントロール抗体とともに培養した。Bi20の濃度を向上させることにより、CLL B細胞の除去が向上した(最大99%)。Rituximabも、明らかなB細胞減少を誘導したが、250ng/mlの濃度でもB細胞の半分近くが生き残った。このことは、少なくとも本実験条件の範囲内においては、前記trAbであるBi20は、CLL患者由来の原発性腫瘍細胞の除去に関して、Rituximabよりもいっそう強力であることを示している。この場合において、catumaxomabにより媒介された非特異的なB細胞減少は、B細胞系統を用いた従来の実験と比較して、実質的に低下していた(図7および11)。
【0137】
意外なことに、非常に低いレベルでCD20を発現しているCLL B細胞をテストしたときでさえ(CLL2、3および4;図11AおよびB)、ほとんどすべてのB細胞が、Bi20により除去された(それぞれ、99%、93%および98%)。対照的に、Rituximabは、同じ患者グループ由来の細胞を用いた場合、その効果が劣っていたか、またはその効果を示さなかった(それぞれ、76%、79%および1%)。
【0138】
つぎに、Bi20が、健康なドナー由来のエフェクター細胞の添加なしに、自己設定においてCLL B細胞の死滅を誘導できるのかどうかという重大な疑問に取り組んだ。そのために、CLL患者由来のPBMCを、Bi20およびコントロール抗体とともにインキュベートした。好ましくないE:T比および公知のCLLにおけるT細胞アネルギーのために、細胞を、少なくとも6日間(CLL10患者を除く)インキュベートし、抗体を3日おきに添加した。B細胞の減少およびT細胞の活性化の解析を、CLL10患者の場合(3日目)を除いて、6日目から10日目の間に実施した。
【0139】
表2を要約すると、効果的なB細胞の減少(>85%)を、7つの患者検体のうち3つにおいて検出した。1つの検体(CLL13の患者)において、65%の腫瘍B細胞の減少が観察された。興味深いことに、B細胞の減少が、非常に低いレベルでCD20を発現している検体(CLL11、12および13の患者、図11B)でさえ観察された。このことは、同種間の設定(allogeneic setting)において前記結果が得られることにより確認された(図11AおよびB)。同じ濃度でRituximabを用いたコントロール実験では、CLL B細胞が減少しない、または減少が非常に低度であることが示された。
【0140】
効果的なB細胞の減少は、エフェクター対標的比(E:T)に依存すると思われる。最も良い反応を示した患者検体(CLL10、11および12)は、エフェクター細胞の割合も最も高かった。特に、最も好ましいE:T比を有する検体(3.5:1;CLL10の患者由来)は、3日後にだけ効果的なB細胞の除去を示した。
【0141】
CLLのT細胞は、アネルギー性であることが知られており、特にCD4サブセットであることが知られている34。驚いたことに、インキュベーションの終盤におけるCD25の上方制御によるCD4+およびCD8+T細胞の活性化を解析した場合、6つの患者検体のうち5つの検体で前記CD4+およびCD8+サブセットの両方の活性化が効果的に発生していることが見出された(表2)。検体をRituximabとともに処理した場合には、活性化が見られなかった(データを示さず)。これらの結果より、少なくともin vitroにおいてBi20は、腫瘍細胞の存在下でさえCLL検体におけるT細胞アネルギーを克服し得る。
【0142】
前記抗CD20モノクローナル抗体であるRituximabを用いるB細胞リンパ腫の治療における(特に、化学療法との組み合わせにおける)有望な結果にもかかわらず、最終的には、患者は再発してしまう9、35。従って、さらなる治療の選択肢が必要である。二重特異性抗体を使用することが、改善方法の一つであろう。それは、腫瘍細胞に対するT細胞等のエフェクター細胞に向けられる。
【0143】
CD19またはCD20等のB細胞に特異的な抗原に対する二重特異性抗体は、この10〜15年間で集中的に研究がなされてきた。しかし、これまでは中程度の反応を示すわずかな臨床データしか入手できなかった14、36、37。さらに、製造工程が複雑で効果的でなかったため、臨床用途のための充分な量の抗体を産生することが難しかった。ここで、CD20およびCD3に対する新規な三機能二重特異性抗体であるBi20の産生と生産について記載する。特に、マウスIgG2aとラットIgG2bアイソタイプとの組み合わせの使用のみならず、本明細書に記載した他の三機能二重特異性抗体の使用は、種制限的な(species−restricted)重鎖/軽鎖の組み合わせによって、正確に組み合わせられた二重特異性抗体の高収率生産を可能にする30。これらのtrAbは、腫瘍細胞、T細胞およびFcγRI/III+修飾細胞と同時に結合し、腫瘍細胞の根絶を増強することを特徴とする17、18。
【0144】
Bi20は、何らの同時刺激を必要とせずにin vitroにおけるB細胞の除去において非常に効果的であることが示された。B細胞の減少アッセイは、抗体濃度が0.5ng/mlと低度であっても、24時間後に70%近くのRaji腫瘍B細胞を除去することが可能であり、3日後には、腫瘍細胞の完全な根絶が可能であったことが実証された。B細胞系統における代表的な他のNHLの実体(Mec1、GrantaまたはDOHH−2)を用いても、同様の結果が得られた。このことは、腫瘍細胞の除去が、この種の悪性疾患(malignant disease)の特定のサブグループに限定されないことを示している。
【0145】
さらに、前記CLL患者由来のB細胞もまた、アポトーシス耐性が亢進され、CD20の発現レベルが低いという特性を有するCLL B細胞であるにもかかわらず27、38、39、効果的に死滅した(図11AおよびB、表5)。この場合も先と同様に、効果的なCLL B細胞の除去に、T細胞の前活性化または共活性化を必要とせず、前述の他の二重特異性抗体とは対照的であった。例えば、前記二重特異性マウス抗体であるBis20(マウスIgG1 CD20×マウスIgG2b CD3)は、実験に用いたE:T比は同じであるが、10倍の抗体濃度、予め活性化されたT細胞および短いインキュベーション時間において、60%近くのB細胞の減少を誘導した40。抗CD20×抗CD3二重特異性抗体を用いる研究において、腫瘍細胞(Raji)の溶解は、末梢血リンパ球(PBLs)を、予めIL−2を用いて刺激した場合においてのみ達成された。1000ng/mlの濃度の二重特異性抗体、および20:1のE:T比であっても、約35%の細胞しか溶解されなかった41。さらに、Cochloviusらは、1〜2.5μg/mlの抗体濃度における効果的なB細胞の溶解に、CD28による予め活性化されたPBLおよびT細胞の同時刺激が必要である抗CD3×抗CD19二重特異性抗体を示した32、42。
【0146】
Bi20は、健康なドナーのPBMCを添加しない自己設定においてさえ、7つの患者検体のうち4つの検体において効果的な腫瘍細胞の死滅を誘導する。Rituximabとは際立って対照的に、Bi20が媒介するB細胞の減少は、CLL腫瘍細胞による非常に低レベルのCD20発現下においてさえも効果的である。そしてそのB細胞の減少は、自己設定および同種間設定(allogenetic setting)について言える(表5および図11)。この知見は、おそらく、CLL腫瘍細胞によるCD20の発現が低いために、Rituximabが単独療法として限定的な効果しか示さないというCLL研究の臨床データに基づいている3、43。
【0147】
いくつかの他の二重特異性抗体44、45とは対照的に、Bi20は、抗CD28抗体またはIL−2等の任意の同時刺激分子が存在しない場合においても、CD4+およびCD8+T細胞サブセットのいずれも効果的に活性化した。結果的に、高いレベルのINF−γを検出した。前記INF−γは、刺激されたT細胞により放出されることが知られており、Th1型反応の指標(indicator)である46。T細胞の活性化も、T細胞サブセットの両方の増殖により伴って起こる。
【0148】
T細胞の活性化は、健康なドナーのT細胞だけでなく、B−CLL患者由来のT細胞を用いた場合にも観察された(表5)。これらのT細胞は、多くの場合アネルギーの特徴を有する。おそらく、CD28およびT細胞受容体の発現が低いためであろう47、48。本実験においては、実質的なCLL B細胞の減少が観察されない検体においてさえも、CD4+およびCD8+T細胞サブセットの活性化を検出した。いくつかの検体(CLL−2、−4、−14)におけるB細胞の減少が限定的なのは、インキュベート期間が短いためかもしれない。前記インキュベーション期間は、in vitroにおけるB−CLL細胞の生存率がわずかであるために制限されてしまう。活性補助因子(coactivator)依存性T細胞の活性化に基づく比較可能なB細胞の減少が、単鎖CD19×CD3二重特異性抗体に関して近年述べられており、さらに、前記二重特異性抗体の方式(format)の効力を確認している49、50。従って、本発明者らの成果は、Bi20が、in vitroだけでなくin vivoにおいても、CLLのT細胞のアネルギーを克服する可能性を有しているかもしれないことを示す。
【0149】
Bi20は、T細胞に加えて、FcγRI+細胞に結合し、FcγRI+細胞を活性化する。腫瘍標的細胞およびFcγRI+細胞の結合が、活性化にとって充分ではないため、活性化は、親抗体であるTPA10において示されるように、Bi20のT細胞に同時に結合することに依存する(図10AおよびB)。trAbのFc領域の重要性は、免疫担当性のマウスB細胞リンパ腫モデルにおける近年の本発明者等による知見により裏づけられ、長期間持続する抗腫瘍免疫の誘導は、前記trAbのFc領域の機能性に依存した。これらの実験において、trAbのF(ab)2断片の使用は、これらのマウスの生存期間を減少させた20。この結果は、in vitroおよびマウスモデルにおいて、単一の二重特異性抗体と比較して高い抗腫瘍活性を示す抗CD19×抗FcγRIIIと抗CD19×抗CD3との二重特異性抗体の組み合わせを示す近年の出版物と一致する51。しかしながら、両ケースにおいては、抗CD28抗体によるT細胞の付加的な同時刺激が必要であった。これらの結果は、Bi20により媒介されるものとして、同時に起こるT細胞およびFcγR+エフェクター細胞の結合および活性化が、腫瘍細胞とT細胞単独に結合する場合より、さらに効果的に腫瘍細胞を死滅させることが可能であることを示している。
【0150】
総合すれば、本発明者らの成果は、腫瘍細胞によるCD20の発現が非常に低い場合であっても、trAbであるBi20が、強力な腫瘍B細胞減少の誘導因子(inducer)であり、前活性化または共活性化のいずれにも依存しないことを示す。同時に発生するT細胞および修飾免疫細胞の活性化、並びにそれらの腫瘍細胞の減少は、in vitroにおいてだけでなく、in vivoにおいても、前述のCLLのT細胞アネルギーを克服する。従って、本明細書中に記載の前記三機能二重特異性抗体は、かねてから治癒不能とされてきた非ホジキンリンパ腫および、本明細書に記載したCD20の発現が低い患者におけるCLLを治療するための新しい治療の選択肢を提案する。
【0151】
参照文献
1. Greiner TC, Medeiros LJ, Jaffe ES. Non−Hodgkin’s lymphoma. Cancer. 1995;75:370−380.
2. Ries LAG, Harkins D, Krapcho M, et al. SEER Cancer Statistics Review, 1975−2003. http://seercancergov. 2006.
3. Lin TS, Lucas MS, Byrd JC. Rituximab in B−cell chronic lymphocytic leukemia. Semin Oncol. 2003;30:483−492.
4. Dillman RO. Treatment of low−grade B−cell lymphoma with the monoclonal antibody rituximab. Semin Oncol. 2003;30:434−447.
5. Blum KA, Bartlett NL. Antibodies for the treatment of diffuse large cell lymphoma. Semin Oncol. 2003;30:448−456.
6. Moreton P, Hillmen P. Alemtuzumab therapy in B−cell lymphoproliferative disorders. Semin Oncol. 2003;30:493−501.
7. Hiddemann W, Dreyling M, Unterhalt M. Rituximab plus chemotherapy in follicular and mantle cell lymphomas. Semin Oncol. 2003;30:16−20.
8. Montserrat E. Rituximab in chronic lymphocytic leukemia. Semin Oncol. 2003;30:34−39.
9. Smith MR. Rituximab (monoclonal anti−CD20 antibody): mechanisms of action and resistance. Oncogene. 2003;22:7359−7368.
10. Mavromatis BH, Cheson BD. Novel therapies for chronic lymphocytic leukemia. Blood Rev. 2004;18:137−148.
11. Nagy ZA, Hubner B, Lohning C, et al. Fully human, HLA−DR−specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat Med. 2002;8:801−807.
12. Baeuerle PA, Kufer P, Lutterbuse R. Bispecific antibodies for polyclonal T−cell engagement. Curr Opin Mol Ther. 2003;5:413−419.
13. Peipp M, Valerius T. Bispecific antibodies targeting cancer cells. Biochem Soc Trans. 2002;30:507−511.
14. Manzke O, Tesch H, Borchmann P, et al. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. I. Clinical phase I evaluation. Int J Cancer. 2001;91:508−515.
15. Manzke O, Tesch H, Lorenzen J, Diehl V, Bohlen H. Locoregional treatment of low−grade B−cell lymphoma with CD3xCD19 bispecific antibodies and CD28 costimulation. II. Assessment of cellular immune responses. Int J Cancer. 2001;91:516−522.
16. Haagen IA. Performance of CD3xCD19 bispecific monoclonal antibodies in B cell malignancy. Leuk Lymphoma. 1995;19:381−393.
17. Zeidler R, Mysliwietz J, Csanady M, et al. The Fc−region of a new class of intact bispecific antibody mediates activation of accessory cells and NK cells and induces direct phagocytosis of tumour cells. Br J Cancer. 2000;83:261−266.
18. Zeidler R, Reisbach G, Wollenberg B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246−1252.
19. Lindhofer H, Menzel H, Gunther W, Hultner L, Thierfelder S. Bispecific antibodies target operationally tumor−specific antigens in two leukemia relapse models. Blood. 1996;88:4651−4658.
20. Ruf P, Lindhofer H. Induction of a long−lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526−2534.
21. Heiss MM, Strohlein MA, Jager M, et al. Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 2005;117:435−443.
22. Riesenberg R, Buchner A, Pohla H, Lindhofer H. Lysis of prostate carcinoma cells by trifunctional bispecific antibodies (alpha EpCAM x alpha CD3). J Histochem Cytochem. 2001;49:911−917.
23. Kiewe P, Hasmuller S, Kahlert S, et al. Phase I trial of the trifunctional anti−HER2 x anti−CD3 antibody ertumaxomab in metastatic breast cancer. Clin Cancer Res. 2006;12:3085−3091.
24. Tedder TF, Engel P. CD20: a regulator of cell−cycle progression of B lymphocytes. Immunol Today. 1994;15:450−454.
25. O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Mice carrying a CD20 gene disruption. Immunogenetics. 1998;48:125−132.
26. Nadler LM, Ritz J, Hardy R, Pesando JM, Schlossman SF, Stashenko P. A unique cell surface antigen identifying lymphoid malignancies of B cell origin. J Clin Invest. 1981;67:134−140.
27. Delgado J, Matutes E, Morilla AM, et al. Diagnostic significance of CD20 and FMC7 expression in B−cell disorders. Am J Clin Pathol. 2003;120:754−759.
28. Anderson KC, Bates MP, Slaughenhoupt BL, Pinkus GS, Schlossman SF, Nadler LM. Expression of human B cell−associated antigens on leukemias and lymphomas: a model of human B cell differentiation. Blood. 1984;63:1424−1433.
29. Press OW, Howell−Clark J, Anderson S, Bernstein I. Retention of B−cell−specific monoclonal antibodies by human lymphoma cells. Blood. 1994;83:1390−1397.
30. Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species−restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single−step purification of bispecific antibodies. J Immunol. 1995;155:219−225.
31. Ruf P, Jager M, Ellwart J, Wosch S, Kusterer E, Lindhofer H. Two new trifunctional antibodies for the therapy of human malignant melanoma. Int J Cancer. 2004;108:725−732.
32. Cochlovius B, Kipriyanov SM, Stassar MJ, et al. Treatment of human B cell lymphoma xenografts with a CD3 x CD19 diabody and T cells. J Immunol. 2000;165:888−895.
33. Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine−release syndrome in patients with B−cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti−CD20 monoclonal antibody (rituximab, IDEC−C2B8). Blood. 1999;94:2217−2224.
34. Bartik MM, Welker D, Kay NE. Impairments in immune cell function in B cell chronic lymphocytic leukemia. Semin Oncol. 1998;25:27−33.
35. Ghielmini M. Multimodality therapies and optimal schedule of antibodies: rituximab in lymphoma as an example. Hematology (Am Soc Hematol Educ Program). 2005:321−328.
36. Weiner GJ, De Gast GC. Bispecific monoclonal antibody therapy of B−cell malignancy. Leuk Lymphoma. 1995;16:199−207.
37. De Gast GC, Van Houten AA, Haagen IA, et al. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother. 1995;4:433−437.
38. Huh YO, Keating MJ, Saffer HL, Jilani I, Lerner S, Albitar M. Higher levels of surface CD20 expression on circulating lymphocytes compared with bone marrow and lymph nodes in B−cell chronic lymphocytic leukemia. Am J Clin Pathol. 2001;116:437−443.
39. Reed JC, Kitada S, Kim Y, Byrd J. Modulating apoptosis pathways in low−grade B−cell malignancies using biological response modifiers. Semin Oncol. 2002;29:10−24.
40. Withoff S, Bijman MN, Stel AJ, et al. Characterization of BIS20x3, a bi−specific antibody activating and retargeting T−cells to CD20−positive B−cells. Br J Cancer. 2001;84:1115−1121.
41. Xiong D, Xu Y, Liu H, et al. Efficient inhibition of human B−cell lymphoma xenografts with an anti−CD20 x anti−CD3 bispecific diabody. Cancer Lett. 2002;177:29−39.
42. Kipriyanov SM, Moldenhauer G, Strauss G, Little M. Bispecific CD3 x CD19 diabody for T cell−mediated lysis of malignant human B cells. Int J Cancer. 1998;77:763−772.
43. Nguyen DT, Amess JA, Doughty H, Hendry L, Diamond LW. IDEC−C2B8 anti−CD20 (rituximab) immunotherapy in patients with low−grade non−Hodgkin’s lymphoma and lymphoproliferative disorders: evaluation of response on 48 patients. Eur J Haematol. 1999;62:76−82.
44. Manzke O, Berthold F, Huebel K, Tesch H, Diehl V, Bohlen H. CD3xCD19 bispecific antibodies and CD28 bivalent antibodies enhance T−cell reactivity against autologous leukemic cells in pediatric B−ALL bone marrow. Int J Cancer. 1999;80:715−722.
45. Manzke O, Titzer S, Tesch H, Diehl V, Bohlen H. CD3 x CD19 bispecific antibodies and CD28 costimulation for locoregional treatment of low−malignancy non−Hodgkin’s lymphoma. Cancer Immunol Immunother. 1997;45:198−202.
46. Berenson LS, Ota N, Murphy KM. Issues in T−helper 1 development−−resolved and unresolved. Immunol Rev. 2004;202:157−174.
47. Mellstedt H, Choudhury A. T and B cells in B−chronic lymphocytic leukaemia: Faust, Mephistopheles and the pact with the Devil. Cancer Immunol Immunother. 2006;55:210−220.
48. Rossi E, Matutes E, Morilla R, Owusu−Ankomah K, Heffernan AM, Catovsky D. Zeta chain and CD28 are poorly expressed on T lymphocytes from chronic lymphocytic leukemia. Leukemia. 1996;10:494−497.
49. Dreier T, Lorenczewski G, Brandl C, et al. Extremely potent, rapid and costimulation−independent cytotoxic T−cell response against lymphoma cells catalyzed by a single−chain bispecific antibody. Int J Cancer. 2002;100:690−697.
50. Loffler A, Gruen M, Wuchter C, et al. Efficient elimination of chronic lymphocytic leukaemia B cells by autologous T cells with a bispecific anti−CD19/anti−CD3 single−chain antibody construct. Leukemia. 2003;17:900−909.
51. Kipriyanov SM, Cochlovius B, Schafer HJ, et al. Synergistic antitumor effect of bispecific CD19 x CD3 and CD19 x CD16 diabodies in a preclinical model of non−Hodgkin’s lymphoma. J Immunol. 2002;169:137−144.
52. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER−2/neu oncogene. Science 1987;235(4785):177−82.
53. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE et al. Studies of the HER−2/neu proto−oncogene in human breast and ovarian cancer. Science 1989;244(4905):707−12.
54. Hynes NE, Stern DF. The biology of erbB−2/neu/HER−2 and its role in cancer. Biochim.Biophys.Acta 1994;1198(2−3):165−84.
55. Lewis GD, Figari I, Fendly B, Wong WL, Carter P, Gorman C et al. Differential responses of human tumor cell lines to anti−p185HER2 monoclonal antibodies. Cancer Immunol.Immunother. 1993;37(4):255−63.
56. Sarup JC, Johnson RM, King KL, Fendly BM, Lipari MT, Napier MA et al. Characterization of an anti−p185HER2 monoclonal antibody that stimulates receptor function and inhibits tumor cell growth. Growth Regul. 1991;1(2):72−82.
57. Paik S, Bryant J, Tan−Chiu E, Romond E, Hiller W, Park K et al. Real−world performance of HER2 testing−−ational Surgical Adjuvant Breast and Bowel Project experience. J.Natl.Cancer Inst. 2002;94(11):852−4.
58. Hudziak RM, Lewis GD, Winget M, Fendly BM, Shepard HM, Ullrich A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol.Cell Biol. 1989;9(3):1165−72.
59. Zeidler R, Mayer A, Gires O, Schmitt B, Mack B, Lindhofer H et al. TNFalpha contributes to the antitumor activity of a bispecific, trifunctional antibody. Anticancer Res. 2001;21(5):3499−503.
【0152】
【表1】
【0153】
【表2】
【0154】
【表3】
【0155】
【表4】
【0156】
【表5】
【特許請求の範囲】
【請求項1】
下記特性を有する三機能二重特異性抗体の使用であって、
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
腫瘍性疾患(tumor deseases)の予防および治療用の薬学的組成物を製造するための使用であり、前記腫瘍関連抗原が、下記のとおり前記腫瘍細胞上に発現されている使用。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性(negative)の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約310,00以下、もしくは約300,00以下の量で、または、
・EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【請求項2】
前記腫瘍関連抗原が、Her2/neuまたはCD20である請求項1記載の使用。
【請求項3】
前記腫瘍抗原が、前記腫瘍細胞上に、腫瘍細胞あたり、少なくとも約5,000、20,000、50,000または80,000の量で発現され、腫瘍細胞あたり、最大で約150,000、110,000または100,000の量で発現されている請求項1または2記載の使用。
【請求項4】
前記三機能抗体が、抗CD3×抗腫瘍関連抗原抗体、および/または抗CD4×抗腫瘍関連抗原抗体、および/または抗CD5×抗腫瘍関連抗原抗体、および/または抗CD6×抗腫瘍関連抗原抗体、および/または抗CD8抗腫瘍関連抗原抗体、および/または抗CD2×抗腫瘍関連抗原抗体、および/または抗CD28×抗腫瘍関連抗原抗体、および/または抗CD44×抗腫瘍関連抗原抗体から選択される請求項1から3のいずれか一項に記載の使用。
【請求項5】
前記三機能抗体が、異種性の二重特異性抗体から選択され、好ましくは、異種性のラット/マウス二重特異性抗体である請求項1から4のいずれか一項に記載の使用。
【請求項6】
前記三機能抗体が、そのFc領域におけるアイソタイプの組み合わせにおいて、下記グループの少なくとも一つのメンバーから選択される請求項1から5のいずれか一項に記載の使用。
ラット−IgG2b/マウス−IgG2a
ラット−IgG2b/マウス−IgG2b
ラット−IgG2b/ヒト−IgG1
ラット−IgG2b/ヒト−IgG2
【請求項7】
前記三機能抗体が、0.05〜15μg/kg体重の量で投与される請求項1から6のいずれか一項に記載の使用。
【請求項8】
前記三機能二重特異性抗体が、Fcγ受容体I型/III型に結合する抗Her2/neu×抗CD3抗体であり、好ましくは、前記ラット−IgG2b/マウス−IgG2aのアイソタイプの組み合わせを有する請求項1から7のいずれか一項に記載の使用。
【請求項9】
前記Her2/neu抗原が、約5,000〜150,000の量で、好ましくは、約20,000〜100,000の量で、乳腺腫瘍細胞上に発現されている請求項1から8のいずれか一項に記載の使用。
【請求項10】
前記腫瘍性疾患が、Dako社のHercep Test(登録商標)に従ってHer2/neu値1+に分類され、または前記Dako社のHercep Test(登録商標)に従ってHer2/neu値2+の場合においてFISH法陰性に分類されるガンであり、前記ガンが、特に乳ガンである請求項1から9のいずれか一項に記載の使用。
【請求項11】
前記三機能二重特異性抗体が、Fcγ受容体I型/III型に結合する抗CD20×抗CD3抗体であり、好ましくは、前記ラット−IgG2b/マウス−IgG2aのアイソタイプの組み合わせを有する請求項1から10のいずれか一項に記載の使用。
【請求項1】
下記特性を有する三機能二重特異性抗体の使用であって、
(a)T細胞に結合する特性;
(b)腫瘍細胞上の少なくとも一つの腫瘍関連抗原に結合し、前記抗原が、Her2/neu、CD20、EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAからなる群から選択される特性;
(c)Fcγ受容体I型および/またはIII型陽性細胞に、Fc領域を通じて結合する特性;
腫瘍性疾患(tumor deseases)の予防および治療用の薬学的組成物を製造するための使用であり、前記腫瘍関連抗原が、下記のとおり前記腫瘍細胞上に発現されている使用。
・Her2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約5,000〜約150,000の量で、または
・FISH法陰性(negative)の腫瘍細胞におけるHer2/neuに関する腫瘍関連抗原が、腫瘍細胞あたり約500,000以下の量で、または、
・CD20に関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約310,00以下、もしくは約300,00以下の量で、または、
・EpCAM、G250、プロテオグリカン、GD2、GD3、MHC II、EGF−RおよびCEAに関する腫瘍関連抗原が、腫瘍細胞あたり約1,000〜約350,000の量で、好ましくは、約300,000以下の量で。
【請求項2】
前記腫瘍関連抗原が、Her2/neuまたはCD20である請求項1記載の使用。
【請求項3】
前記腫瘍抗原が、前記腫瘍細胞上に、腫瘍細胞あたり、少なくとも約5,000、20,000、50,000または80,000の量で発現され、腫瘍細胞あたり、最大で約150,000、110,000または100,000の量で発現されている請求項1または2記載の使用。
【請求項4】
前記三機能抗体が、抗CD3×抗腫瘍関連抗原抗体、および/または抗CD4×抗腫瘍関連抗原抗体、および/または抗CD5×抗腫瘍関連抗原抗体、および/または抗CD6×抗腫瘍関連抗原抗体、および/または抗CD8抗腫瘍関連抗原抗体、および/または抗CD2×抗腫瘍関連抗原抗体、および/または抗CD28×抗腫瘍関連抗原抗体、および/または抗CD44×抗腫瘍関連抗原抗体から選択される請求項1から3のいずれか一項に記載の使用。
【請求項5】
前記三機能抗体が、異種性の二重特異性抗体から選択され、好ましくは、異種性のラット/マウス二重特異性抗体である請求項1から4のいずれか一項に記載の使用。
【請求項6】
前記三機能抗体が、そのFc領域におけるアイソタイプの組み合わせにおいて、下記グループの少なくとも一つのメンバーから選択される請求項1から5のいずれか一項に記載の使用。
ラット−IgG2b/マウス−IgG2a
ラット−IgG2b/マウス−IgG2b
ラット−IgG2b/ヒト−IgG1
ラット−IgG2b/ヒト−IgG2
【請求項7】
前記三機能抗体が、0.05〜15μg/kg体重の量で投与される請求項1から6のいずれか一項に記載の使用。
【請求項8】
前記三機能二重特異性抗体が、Fcγ受容体I型/III型に結合する抗Her2/neu×抗CD3抗体であり、好ましくは、前記ラット−IgG2b/マウス−IgG2aのアイソタイプの組み合わせを有する請求項1から7のいずれか一項に記載の使用。
【請求項9】
前記Her2/neu抗原が、約5,000〜150,000の量で、好ましくは、約20,000〜100,000の量で、乳腺腫瘍細胞上に発現されている請求項1から8のいずれか一項に記載の使用。
【請求項10】
前記腫瘍性疾患が、Dako社のHercep Test(登録商標)に従ってHer2/neu値1+に分類され、または前記Dako社のHercep Test(登録商標)に従ってHer2/neu値2+の場合においてFISH法陰性に分類されるガンであり、前記ガンが、特に乳ガンである請求項1から9のいずれか一項に記載の使用。
【請求項11】
前記三機能二重特異性抗体が、Fcγ受容体I型/III型に結合する抗CD20×抗CD3抗体であり、好ましくは、前記ラット−IgG2b/マウス−IgG2aのアイソタイプの組み合わせを有する請求項1から10のいずれか一項に記載の使用。
【図1】

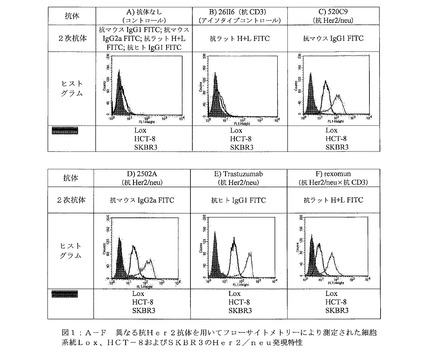
【図2a】
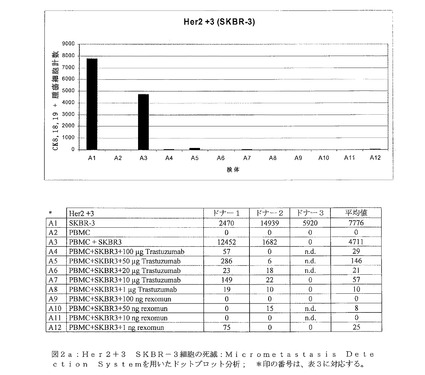
【図2b】
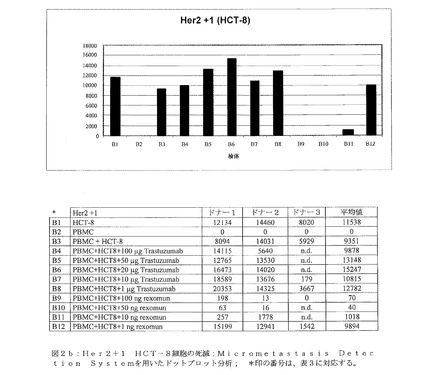
【図2c】
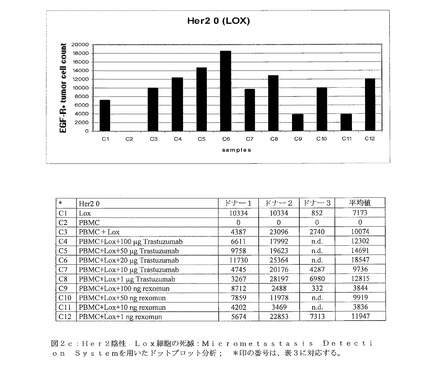
【図2d】
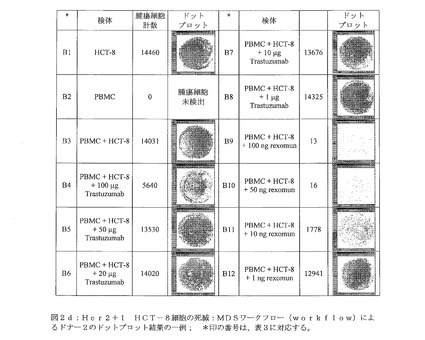
【図3】
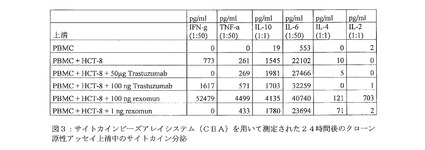
【図4】
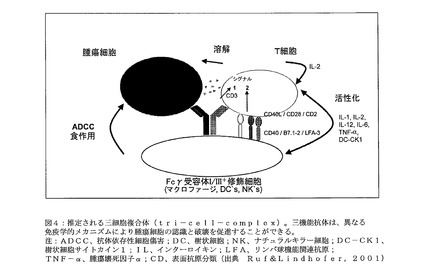
【図5A】
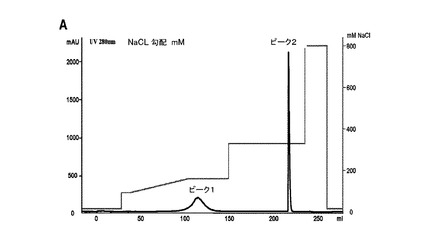
【図6A】
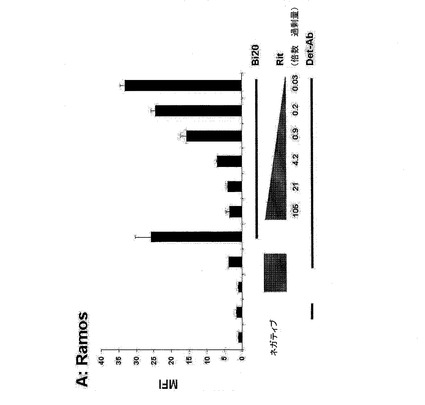
【図6B】
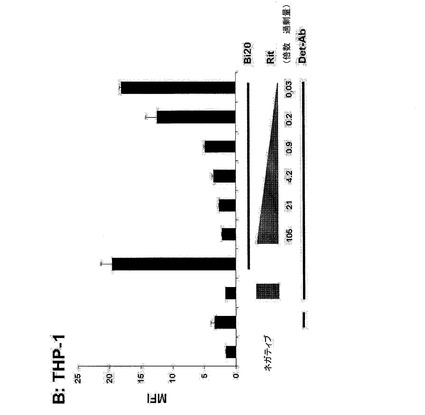
【図7A】
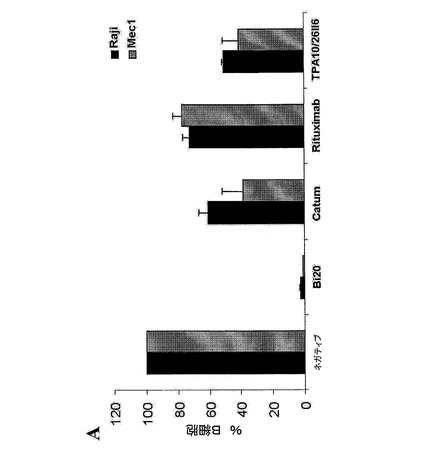
【図7B】
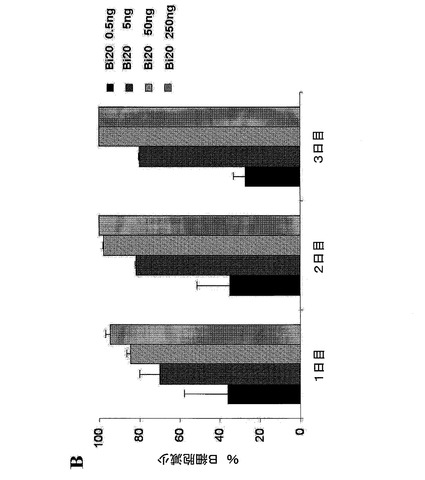
【図8A】
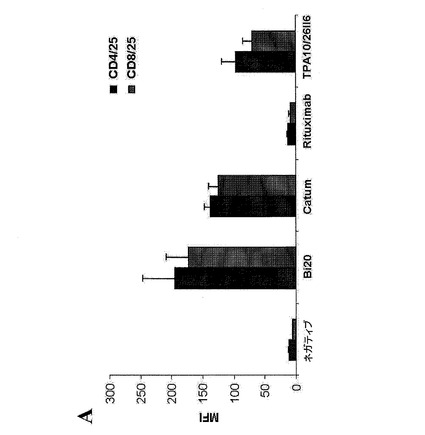
【図8B】
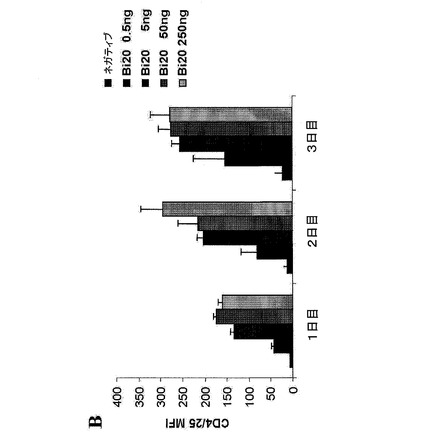
【図9】
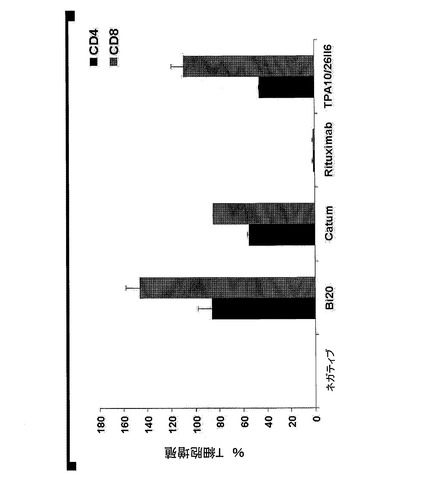
【図10A】
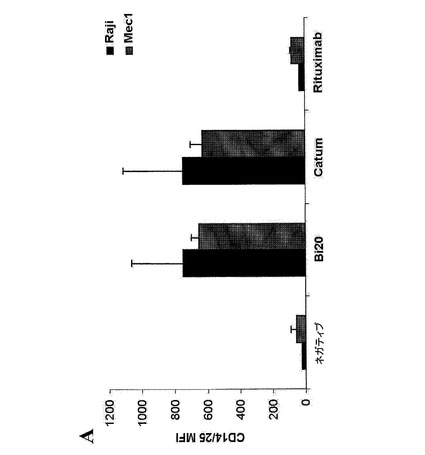
【図10B】
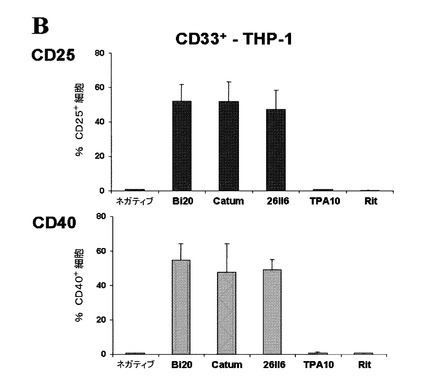
【図11A】
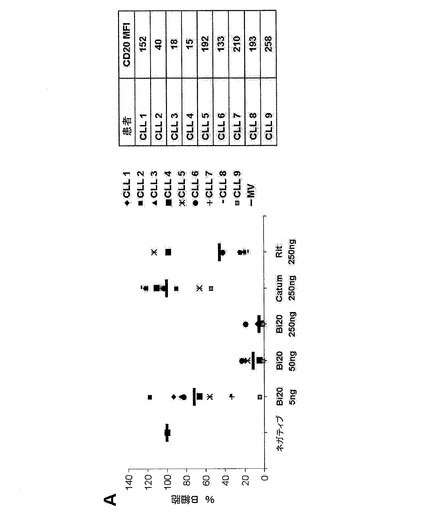
【図11B】
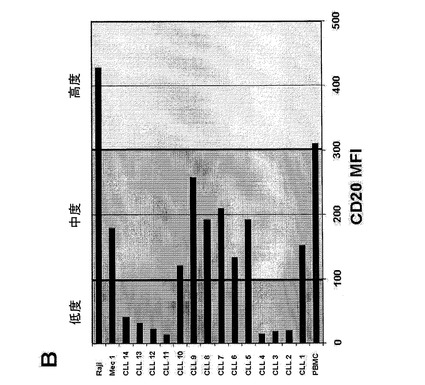
【図5B】
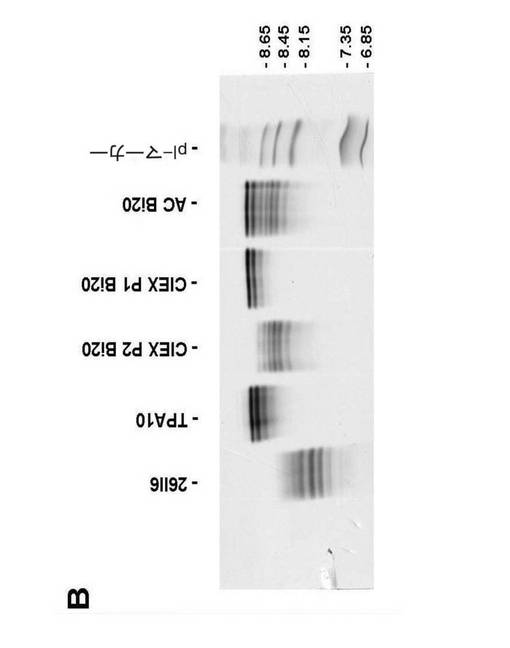
【図2a】
【図2b】
【図2c】
【図2d】
【図3】
【図4】
【図5A】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9】
【図10A】
【図10B】
【図11A】
【図11B】
【図5B】
【公開番号】特開2013−79274(P2013−79274A)
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願番号】特願2012−286212(P2012−286212)
【出願日】平成24年12月27日(2012.12.27)
【分割の表示】特願2008−554780(P2008−554780)の分割
【原出願日】平成19年2月15日(2007.2.15)
【出願人】(508246799)トリオン・ファルマ・ゲーエムベーハー (2)
【Fターム(参考)】
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願日】平成24年12月27日(2012.12.27)
【分割の表示】特願2008−554780(P2008−554780)の分割
【原出願日】平成19年2月15日(2007.2.15)
【出願人】(508246799)トリオン・ファルマ・ゲーエムベーハー (2)
【Fターム(参考)】
[ Back to top ]