
腸管幹/前駆細胞の取得方法
【課題】 腸管上皮細胞の幹/前駆細胞の取得及び培養方法の提供、ならびに腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法の提供。
【解決手段】 腸管細胞の凝集体を培養すること、好ましくは該凝集体をEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得及び培養方法、ならびに該細胞の培養液中からEGFを除去することを含む、腸管上皮細胞の幹/前駆細胞から、腸上皮組織を構築する細胞への分化を誘導する方法。
【解決手段】 腸管細胞の凝集体を培養すること、好ましくは該凝集体をEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得及び培養方法、ならびに該細胞の培養液中からEGFを除去することを含む、腸管上皮細胞の幹/前駆細胞から、腸上皮組織を構築する細胞への分化を誘導する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は腸管上皮細胞の幹/前駆細胞を取得し効率的に培養する方法、該方法によって得られる幹/前駆細胞に関する。さらに本発明は腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法に関する。
【背景技術】
【0002】
薬剤の吸収試験やその他の薬学研究では腸上皮細胞の初代培養細胞を使用することが多い。これまでの培養系では、腸上皮細胞は数日の間に淘汰されてしまい、常に新しい細胞を調製し使用する必要があった。そのため、腫瘍組織由来の細胞株が用いられているが、信頼性の高いデータを得るためには正常細胞を使用する必要があり、正常な腸上皮細胞を簡便に使用できることが望まれている。この場合、培養下で長期間細胞を維持するためには、成熟した腸上皮細胞に加え、それらを常に産生する幹/前駆細胞を培養下で維持する必要があると言える。
【0003】
食物の消化吸収を担う臓器である腸管には、機能的役割の異なる腸上皮細胞群の根幹に位置する幹細胞(腸管幹細胞)が存在し、幹細胞の分裂と維持により腸上皮細胞が継続して供給される。このように、腸管幹細胞は旺盛な増殖能を有するが、その能力を人為的に培養下で保持させ続けることは極めて難しく、これまでに腸管幹細胞の培養に成功した例はない。腸管幹細胞の増殖や分化を培養下で制御できれば、腸管の幹細胞システムに対する基礎研究用ツールとして利用できるだけでなく、薬剤吸収・代謝試験など創薬過程におけるスクリーニング用ツールとして利用できる。また、組織工学的に移植可能な腸上皮組織を供給することができる。
【0004】
一方、初期胚に由来する、多能性の幹細胞株である胚性幹細胞(ES細胞)は、多能性を有するが故に、通常の培養方法では細胞が徐々に分化していってしまう。そのため、ES細胞を効率よく増やすためにはES細胞を未分化状態のままで増殖させる必要がある。マウスのES細胞の場合には白血病阻害因子(LIF)を培地に添加することにより未分化状態を維持できることが知られている(非特許文献1)が、組織幹細胞である腸管幹細胞ではLIFの代替となるような添加因子は知られていない。またES細胞では分化誘導の際に胚様体(embryoid body)と呼ばれる細胞凝集体を作らせることが一般的に知られているが、腸管幹細胞の分化を制御する方法は未だ確立されていない。
【非特許文献1】Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988 Dec 15;336(6200):688-90.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は腸管上皮細胞の幹/前駆細胞の取得を目的とし、さらに該幹/前駆細胞を効率的に培養することを目的とする。さらに本発明の別の目的は、腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法を提供することにある。
【課題を解決するための手段】
【0006】
本発明者らは、上記課題に鑑み、鋭意検討を行った結果、腸管由来の細胞(腸管細胞)を浮遊培養することによって細胞凝集体を得て、当該凝集体を、上皮増殖因子(EGF)を含有する培養液中で培養することによって、腸管の幹/前駆細胞を選択的に取得、増殖させることができることを見出した。さらに、得られた幹/前駆細胞を適当な分化誘導条件下で腸上皮組織を構築する細胞へと分化させることに成功して本発明を完成するに至った。すなわち本発明は以下の通りである。
【0007】
[1]腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[2]腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[3]凝集体が腸管細胞を浮遊培養することによって得られるものである、上記[1]又は[2]記載の方法。
[4]以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を浮遊培養することによって細胞塊(凝集体)を調製する工程
(3)腸管細胞の凝集体を、EGFを含有する培養液中で培養する工程
(4)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
[5]腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、上記[4]記載の方法。
[6]腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む腸管上皮細胞の幹/前駆細胞の取得方法。
[7]腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[8]以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を、EGFを含有する培養液中、高密度で培養する工程
(3)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
[9]腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、上記[8]記載の方法。
[10]上記[1]〜[9]のいずれか1項に記載の方法によって得られた腸管上皮細胞の幹/前駆細胞。
[11]腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[12]腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[13]凝集体が腸管細胞を浮遊培養することによって得られるものである、上記[11]又は[12]記載の方法。
[14]腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[15]腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[16]腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法であって、該幹/前駆細胞の培養液からEGFを除去することを特徴とする方法。
[17]腸上皮組織を構築する細胞が、腸内分泌細胞又は粘液分泌細胞である、上記[16]記載の方法。
[18]血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する、腸管上皮細胞の幹/前駆細胞培養用培地。
【発明の効果】
【0008】
本発明によって得られる腸管上皮細胞の幹/前駆細胞及び該細胞から腸上皮組織を構築する細胞への分化を誘導する方法は、腸組織の発生・分化を理解するための基礎研究用ツールとして有用であり、又、組織工学的に移植可能な腸上皮組織の供給を可能とする。さらに該幹/前駆細胞に由来する腸上皮細胞を用いて薬物の腸管における吸収実験などを効率的に行うことが期待できる。
【発明を実施するための最良の形態】
【0009】
文中で特に断らない限り、本明細書で用いるすべての技術用語及び科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様又は同等の任意の方法及び材料は、本発明の実施又は試験において使用することができるが、好ましい方法及び材料を以下に記載する。本明細書で言及したすべての刊行物及び特許は、例えば、記載された発明に関連して使用されうる刊行物に記載されている、構築物及び方法論を記載及び開示する目的で、参照として本明細書に組み入れられる。
【0010】
本発明の取得方法及び培養方法の対象となる腸管上皮細胞の幹/前駆細胞は、任意の哺乳動物の腸管細胞に由来する。ここで「哺乳動物」とは、具体的にはヒトをはじめウシ、ウマ、イヌ、モルモット、マウス、ラット等が挙げられる。好ましくはヒトであるが、薬物の吸収実験等の研究用ツールとしての利用を考えた場合、マウスやラット等の実験動物としてよく用いられる動物も又、好ましい。
【0011】
腸は、大きく小腸と大腸の2つに分けることができる。小腸は更に口側から、十二指腸、空腸、回腸に分けられ、大腸は盲腸、結腸(上行結腸、横行結腸、下行結腸、S状結腸)、直腸に分けられる。成体の腸は、広範にわたって腺窩と絨毛に折り畳まれている上皮層及び支持間葉系組織によって取り囲まれているルーメンから構成されている。腸の上皮細胞は常に新生され、古くなったものは剥離してゆく。腸上皮細胞の新生は、腸上皮組織中に、幹細胞による細胞再生系が存在するからである。幹細胞は、小腸の絨毛の根元が窪んだ陰窩(crypt)の底のほうに存在する。また、胎児においては、内胚葉細胞層が腸上皮となるよう運命付けられ、重層細胞層が分化した偏極上皮単層へと転換する。腸上皮の形態及び恒常性は細胞増殖、移動、分化及びアポトーシス間での高度に制御された均衡の結果である。本発明の取得方法及び培養方法の対象となる「腸管上皮細胞の幹/前駆細胞」とは、腸上皮細胞へと分化し得る、及び/又は分化することが運命づけられている細胞である。「幹細胞」は自己複製能と多分化能をあわせもつ細胞であって、分化運命が限定されている「前駆細胞」と完全に一致するものではないが、腸上皮細胞へと分化する細胞という点では同一であり、従って、本願発明では特に区別することなく「幹/前駆細胞」と称する。また、「腸管上皮細胞の幹/前駆細胞」を、便宜上「腸管幹/前駆細胞」と称することもある。
【0012】
本明細書中、「腸管細胞」とは、成体あるいは胎児から採取される腸由来の細胞であって、分裂能力を有する腸上皮細胞あるいは腸上皮細胞となり得る細胞の総称である。具体的には、腸管、好ましくは小腸部分を取り出し細かく切り刻んで、振動等の物理的な手段によって、EGTAあるいはEDTA等を用いた化学的な処理によって、あるいはコラゲナーゼ、トリプシン、キモトリプシン、ペプシン等のプロテアーゼ(好ましくはコラゲナーゼ)を用いた酵素的な処理によって細胞を分散することによって得ることができる。上記処理は単独で行っても、複数の処理を組み合わせて行っても良い。胎児の腸管を用いる場合には、円筒状の腸構造から、腸上皮組織中に凸凹が生じ始める時期のものを用いることが好ましい。例えばマウスでは、胎生12〜17日目、好ましくは14.5日の胎児から採取する。また、本発明において用いられる「腸管細胞」は、上記の如く腸管から採取されるものであってもよいが、分裂能力を有する腸上皮細胞あるいは腸上皮細胞となり得る細胞を含有する市販の腸管由来の細胞であってもよい。かかる市販の腸管由来の細胞としては、ACBRI(Applied Cell Biology Research Institute)により分離され(ACBRI 519)、日本では大日本住友製薬株式会社から入手可能なヒト小腸上皮細胞(human intestinal epithelial cells, cat No. CS-ABI-519)が挙げられる。
【0013】
本発明の第一の特徴は、腸管細胞に凝集体をつくらせ、それを培養することにある。凝集体を形成させることによって、より生体内に近い環境に細胞をおくことができる。ここで、「凝集体」とは、細胞が多数集合して形成された三次元構造(球状やぶどうの房状)を有する細胞の塊である。例えば、後述するが、20〜40万細胞/ウェルの濃度で低接着性の96ウェル丸底ディッシュに播種した場合、10〜20個程度の直径200〜500μm程度の凝集体が形成される。凝集体の最外部に細胞のシート状構造が見られる。より生体内に近い環境を提供することが可能な範囲で、凝集体を構成する細胞の数及び大きさは、細胞の状態や培養条件によって変わり得る。
【0014】
腸管細胞の凝集体は、具体的には以下のようにして調製することができる。まず、腸管細胞を適当な媒体中、低接着性の培養容器上で浮遊培養する。例えば低接着性の培養容器として96ウェルの丸底ディッシュを用いた場合には、1ウェルあたり20〜40万細胞を播種し、形成した凝集体を低接着性の6cmディッシュ等に移したりしながら、37℃で、2〜10日間、好ましくは7日間、適当な媒体中で培養する。低接着性の培養容器としては、通常、当分野で用いられている培養容器に低接着性となるような処理を施したものが例示される。培養容器としては、培養ディッシュ、培養フラスコ、回転培養用器具(スピナーフラスコ等)等が挙げられる。低接着性となるような処理としては、ハイドロゲルを共有結合させること(コーティング)によって蛋白質や細胞の接着を抑制するような処理が用いられる。又、市販されているものを用いることもできる。
【0015】
成体由来の腸管細胞は凝集体を形成しにくいが、該腸管細胞を高密度で培養することによって、同様の効果(生体内に近い環境を作り出すという効果)を得ることができる。例えば直径6cm(媒体の量:3.5mL)のペトリディッシュ(接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ;以下、単に普通のペトリディッシュともいう)で培養する場合には、1〜5×106細胞を播種し、37℃で、1〜5日間、好ましくは3日間、適当な媒体中で培養することによって高密度培養を達成することができる。
【0016】
上記した、凝集体の培養及び高密度培養において使用可能な媒体としては、腸管細胞が生存し得て、且つ分化やアポトーシスの誘導が抑制されている状態を維持できるものであれば特に限定されず、当分野で通常用いられる細胞培養用培地(ダルベッコ改変イーグル培地、MEM、RPMI1640培地、ハムF−12培地等)をベースとし、必要に応じて各種の成分を補充したものが用いられる。特に、血清、抗生物質、増殖因子等の成分を補充することが好ましい。
【0017】
媒体中に含められる好適な成分としては、5〜20%、好ましくは10%の血清、0.5〜2μg/mL、好ましくは1μg/mLのインスリン、1×10−8〜1×10−6mol/L、好ましくは1×10−7mol/Lのデキサメサゾン、5〜15mmol/L、好ましくは10mmol/Lのニコチンアミド、1〜5mmol/L、好ましくは2mmol/LのL−グルタミン、10〜100μmol/L、好ましくは50μmol/Lのβ−メルカプトエタノール、ペニシリン/ストレプトマイシン等が挙げられる。いずれも商業的に入手可能である。血清としては、ウマ、ウシ由来の血清等が使用され、好ましくは胎児血清である。インスリンとしては、ウシ、ブタの膵臓から生産されたインスリンや、遺伝子組み換え技術により生産されたインスリンのいずれも使用できるが、安定な品質と供給という点から組換え型インスリンを用いることが好ましい。
【0018】
本発明の第2の特徴は、選択的に腸管幹/前駆細胞を増殖、取得又は培養するために、腸管細胞の培養を、EGF(上皮増殖因子)存在下で行うことである。
【0019】
EGFは、Cohenらによってマウス胎児の早期眼開裂作用を持つ物質としてマウス顎下腺より単離同定され、ついで尿よりヒトEGFが発見された(S. Cohen and G. Carpenteret al., Proc. Natl. Acad. Sci. USA., 72, 1317, 1975; H. Gregory, Nature, 257, 325, 1975)。遺伝子構造も詳細に解明されている(G. I. Bell et al., Nucleic Acids Res., 14, 8427, 1986)。EGFは、53アミノ酸残基及び3つの分子内ジスルフィド結合からなる6045Daの蛋白質であり、細胞表面に存在するEGFR(上皮増殖因子受容体)にリガンドとして結合し、細胞の成長と増殖の調節に重要な役割を果たす。本発明で用いるEGFは選択的に腸管幹/前駆細胞を増殖、取得又は培養することを可能にする限り、その由来は特に限定されず、天然由来のものであっても公知の遺伝子配列やアミノ酸配列等の情報に基づいて合成又は半合成して製造されるものであってもよい。商業的に入手可能なものであってもよい。マウスの幹/前駆細胞を対象とする場合にはマウスの、ヒトの幹/前駆細胞を対象とする場合にはヒトのEGFをそれぞれ用いることが好ましいが、必ずしも動物種を一致させる必要はない。腸管幹/前駆細胞を選択的に増殖、取得又は培養することが可能な範囲で、EGFは改変されたものであってもよい。このような改変には、1個又は複数個のヌクレオチドあるいはアミノ酸の変異、挿入、欠失及び置換が含まれる。改変されたEGFは、当分野で通常実施される手段によって調製することができる。例えば、エキソヌクレアーゼを用いる欠失変異株(deletion mutant)作製法、カセット変異法等の部位特異的変異(site−directed mutagenesis)によってEGF DNAを人為的に改変させ、該改変EGF DNAを用いて、所望蛋白質を調製することができる。
【0020】
EGFは、腸管幹/前駆細胞を取得するために腸管細胞を培養する過程を通じて持続的に媒体中に含めておくことが好ましいが、特に凝集体を形成し、その後長期間培養し増殖させる間は必須的に媒体中に含める。EGFの媒体中の濃度は、20〜40ng/mL、好ましくは20ng/mL程度である。
【0021】
EGFを含有する培養液中で培養することによって、市販の腸管細胞、例えば上述のヒト小腸上皮細胞(Cat No. CS-ABI-519)からもまた、腸管幹/前駆細胞を得ることができる。市販されているヒト小腸上皮細胞を用いた場合には、凝集体の形成や高密度での培養を必要とすることなく、EGFを含有する培養液、特に20〜40ng/mL、好ましくは20ng/mLのEGFに加えて、5〜20%、好ましくは10%の血清、0.5〜2μg/mL、好ましくは1μg/mLのインスリン、1×10−8〜1×10−6mol/L、好ましくは1×10−7mol/Lのデキサメサゾン、5〜15mmol/L、好ましくは10mmol/Lのニコチンアミド、1〜5mmol/L、好ましくは2mmol/LのL−グルタミン、10〜100μmol/L、好ましくは50μmol/Lのβ−メルカプトエタノールを含む培養液を用いて継代培養することによって腸管幹/前駆細胞を取得することができる。
【0022】
10%ウシ胎児血清、インスリン(1μg/mL)、デキサメサゾン(1×10−7mol/L)、ニコチンアミド(10mmol/L)、L−グルタミン(2mmol/L)、β−メルカプトエタノール(50μmol/L)、ペニシリン(100U/mL)+ストレプトマイシン(100μg/mL)及びEGF(20ng/mL)を補充したダルベッコ改変培地−ハムF12の混合培地(1:1)は、腸管細胞を培養する為の媒体(培養液)の好適な一例である。
【0023】
上記で得られた、細胞凝集体を普通のペトリディッシュに再播種し、EGFを含有する媒体(培養液)中で37℃で、2〜6ヶ月、好ましくは3ヶ月程度培養する。成体の腸管由来の細胞は凝集体を形成しにくいので、高密度培養によって細胞を凝集しているのに等しい状態にする。成体の腸管に由来する細胞は、EGFを含有する培養液中で37℃で、6〜12ヶ月、好ましくは8ヶ月程度高密度培養する。
【0024】
長期にわたる凝集体の培養あるいは高密度培養を続けると、線維芽細胞様の細胞に対し上皮細胞様の細胞が多く見られるようになる。もし線維芽細胞様の細胞が残存している場合には、1〜10μg/mL、好ましくは4μg/mL程度のモノヨード酢酸で3〜5時間程度処理することによって線維芽細胞様の細胞のみを死滅させることができる。上皮細胞様の細胞の増殖が観察されれば、その後は、細胞培養用の培養容器にて継代し、解析に用いたり、使用時まで凍結保存したりする。
【0025】
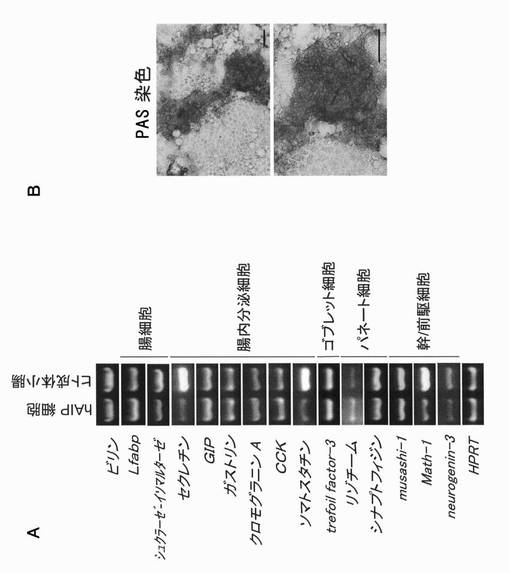
長期にわたる凝集体の培養あるいは高密度培養を続けることによって得られる細胞、あるいはCat No. CS-ABI-519等の市販のヒト小腸上皮細胞をEGFを含有する培養液で継代培養することによって得られる細胞が腸管上皮細胞あるいは腸管幹/前駆細胞であるかの判断は、それらの細胞におけるマーカー蛋白質の発現の有無を調べることによって行うことができる。本明細書中、「マーカー」とは特にことわりのない限り、腸管上皮細胞あるいは腸管幹/前駆細胞に特有な発現様式を示す一連の蛋白質又はそれをコードする遺伝子から構成される群の各々を意味する。かかるマーカー蛋白質は哺乳動物の種類等によってそのアミノ酸配列が異なる場合があるが、本発明においてはその対象となる細胞における発現様式が同じである限り、そのような蛋白質もマーカー蛋白質として使用することができる。上皮細胞のマーカー蛋白質としては、E−カドヘリン(E−cadherin)やZo−1(Laprise P, et al., J Biol Chem. 2004 Mar 12; 279(11):10157-66. Epub 2003 Dec 29.;Escaffit F, et al., Exp Cell Res. 2005 Jan 15;302(2):206-20.)、腸上皮細胞のマーカー蛋白質としては、ビリン(Villin)(Maunoury, R. et al., EMBO J. 7, 3321-3329 (1988).)が挙げられる。腸管幹/前駆細胞のマーカー蛋白質としては、Musashi−1(Potten CS, et al., Differentiation. 2003 Jan;71(1):28-41.)、Math−1(Yang Q, et al., Science 294:2155-2158)、Neurogenin3(Ngn3、Jenny M, et al., EMBO J. 2002 Dec 2;21(23):6338-47.)が挙げられる。
【0026】
E−カドヘリンは、上皮系の細胞上に発現しているカルシウム依存性の細胞間接着分子であり、それ同士が結合するホモフィリックな接着により上皮組織の形成、維持に重要な働きをする。Zo−1は、上皮細胞や内皮細胞に必須である細胞間接着装置タイトジャンクション(TJ)の裏打ち蛋白質として知られており、接着装置の形成やそこを介して細胞増殖の調節に関与していると考えられている蛋白質である。ビリンは、小腸上皮細胞の微絨毛の構成成分であり、フィブリンとともにアクチンを束ねる働きを有している。ビリンは微絨毛にのみ見出される。Musashi−1は、神経系前駆細胞によって発現されるRNA結合蛋白質であり、腸管の幹/前駆細胞にも発現しているという報告がある。Math−1は、bHLH型の転写因子であり、マウス腸管における分泌細胞系への分化に必要であることが知られ、腸管幹/前駆細胞に発現している。同様にbHLH型転写因子であるNgn3は膵内分泌細胞、胃腸管組織の発生に重要であり、腸管幹/前駆細胞に発現している。
【0027】
本発明において、腸管上皮細胞又は腸管幹/前駆細胞であることの確認は、これらのマーカー蛋白質及び/又はそれをコードする遺伝子の発現を、各マーカー蛋白質又はそれをコードする遺伝子に特異的親和性を有する物質を用いて解析する工程を含むことによって行うことができる。本明細書中、「用いて」という用語について、その方法は特に限定されず、具体的には、例えばマーカー蛋白質に特異的親和性を有する物質を用いる場合であれば該マーカー蛋白質の抗体との抗原抗体反応を利用する方法が挙げられ、又、マーカー蛋白質をコードする遺伝子に特異的親和性を有する物質を用いる場合であればハイブリダイゼーション反応を利用する方法が挙げられる。
【0028】
マーカー蛋白質に特異的親和性を有する物質としては例えば当該蛋白質に特異的親和性を有する抗体又はその断片が挙げられ、その特異的親和性とは抗原・抗体反応により該蛋白質を特異的に認識し、結合する能力のことである。該抗体又はその断片は、当該蛋白質と特異的に結合可能なものであれば特に限定されず、ポリクローナル抗体、モノクローナル抗体及びそれらの機能的断片のいずれであってもよい。これらの抗体あるいはその機能的断片は、通常当分野で行なわれている方法によって製せられる。例えばポリクローナル抗体を用いる場合であれば、該蛋白質をマウスやウサギといった動物の背部皮下あるいは腹腔内あるいは静脈等に注射して免疫し、抗体価が上昇するのを待った後に抗血清を採取する方法が挙げられ、またモノクローナル抗体を用いる場合であれば、常法に従いハイブリドーマを作製して、その分泌液を採取する方法が挙げられる。抗体断片を製造する方法としてはクローニングした抗体遺伝子断片を微生物等に発現させる方法がよく用いられている。当該抗体、抗体断片等の純度は、当該蛋白質に対する特異的親和性を保持している限り、特に限定されない。これらの抗体又はその断片は、蛍光物質、酵素やラジオアイソトープ等で標識されていてもよい。
さらに、これらは市販されているものを用いても良い。
【0029】
マーカー蛋白質をコードする遺伝子に特異的親和性を有する物質としては、例えば当該遺伝子に特異的親和性を有するオリゴ又はポリヌクレオチドプローブ(以下、便宜上単にプローブともいう)、ならびにオリゴ又はポリヌクレオチドプライマー対(以下、便宜上単にプライマー対ともいう)が挙げられ、その特異的親和性とは、目的の遺伝子にのみハイブリダイズする性質を意味し、従って当該遺伝子の全部もしくは一部に完全相補的なものか、もしくは上記性質を満たす範囲で1乃至数個のミスマッチを含んでいても良い。該プローブ、プライマー対は、当該遺伝子に特異的親和性を有するものであれば特に限定されないが、例えば当該遺伝子の塩基配列の全部もしくは一部、ならびにそれらの相補配列を含むオリゴ又はポリヌクレオチド等が挙げられ、検出すべき遺伝子の形態に応じて適宜選択する。当該オリゴ又はポリヌクレオチドは当該遺伝子に対する特異的親和性を有している限りはその由来は特に限定されず、合成されたものであっても、当該遺伝子から必要な部分を切り出し、通常行なわれる方法によって精製されたものであってもよい。これらのオリゴ又はポリヌクレオチドは、蛍光物質、酵素やラジオアイソトープ等で標識されていてもよい。
【0030】
又、腸管幹/前駆細胞ではβ−カテニンの転写活性がみられる(He XC, et al., Nat Genet. 2004 Oct;36(10):1117-21.)ので、この活性を指標に幹/前駆細胞であることを確認することもできる。例えば、TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出する方法が利用できる。
【0031】
本発明では、腸管細胞の凝集体(あるいは高密度培養した細胞)をEGFを含有する培養液中で培養することによって、腸管幹/前駆細胞を選択的に増殖させることができ、すなわち該幹/前駆細胞を効率よく取得することができる。さらに該幹/前駆細胞の自己複製能及び多分化能は長期間安定に維持される。本発明の方法により得られた腸管幹/前駆細胞が自己複製能を維持していることは、細胞数の計測、光学顕微鏡下での観察等、細胞増殖を測定し得る種々の公知技術を用いて、培養容器中での腸管幹/前駆細胞の増殖を観察することによって容易に判断することができる。腸管幹/前駆細胞が多分化能を維持していることは、該幹/前駆細胞を分化誘導した場合に腸上皮組織を構築する複数種の細胞が得られるかどうかで判断することができる。腸上皮組織を構築する細胞としては、腸内分泌細胞(ガストリン分泌細胞、セロトニン分泌細胞、クロモグラニンA分泌細胞、セクレチン分泌細胞、ソマトスタチン分泌細胞、GIP(gastric inhibitory polypeptide)分泌細胞、CCK(cholecystokinin)分泌細胞等)及び粘液分泌細胞(ゴブレット細胞、パネート細胞等)、栄養素の吸収作用を担う腸細胞が挙げられる。各細胞に特異的に発現している成分を該成分に特異的な親和性を有する物質を用いて検出することによって、あるいは分泌成分を直接解析することによって目的の細胞であるか否かを判定することができる。「特異的な親和性を有する物質」としては、各分泌成分に特異的親和性を有する抗体又はその断片や、各分泌成分をコードする遺伝子に特異的親和性を有するオリゴ又はポリヌクレオチドプローブが挙げられる。「抗体又はその断片」及び「オリゴ又はポリヌクレオチドプローブ」の詳細は上記に準じる。
ゴブレット細胞に関しては、PAS染色やアルシアンブルー染色等の染色法によっても判定することができる。
【0032】
腸管幹/前駆細胞の、腸管上皮組織を構築する細胞への分化誘導は、培養液中からEGFを除去することによって簡便に実施することができる。具体的にはEGFを含有する培養液をEGFを含有しない培養液と培地交換することによって行うことができる。培地交換しておよそ1週間程度で細胞の分化誘導を確認することができるが、分化誘導に要する期間は、細胞の状態や、分化する細胞のタイプ等によって変動し得る。
【実施例】
【0033】
以下、実施例にそって本発明をさらに詳細に説明するが、これら実施例は本発明の範囲を何ら限定するものではない。また、本発明において使用する試薬や装置、材料は特に言及されない限り、商業的に入手可能である。
【0034】
実施例1:胎児腸管由来の腸管幹/前駆細胞の取得
胎生14.5日のマウス胎児から腸管を取り出し、メスで細かく切り刻み腸組織片とした。得られた腸組織片を、1mM EGTAの入ったHank’s溶液に入れ、37℃のウォーターバスで10分間振とうした後、遠心分離によってPBSで1回洗浄した。遠心分離した後、腸組織片を、1mg/mlのコラゲナーゼ溶液(5mM CaCl2の入ったHank’s溶液に溶解したもの)を加え、37℃のウォーターバスで15〜20分間振とうした。軽くピペッティングした後にメッシュを通して未消化組織を除いて細胞分散液を得た(マウス胎児由来の腸管細胞)。
調製したマウス胎児由来の腸管細胞を2〜4×105細胞/ウェルの濃度で低接着性の96ウェル丸底ディッシュ(Low Cell Binding Plates,cat no:145399;Nunc)に播種し、ダルベッコ改変イーグル培地とF−12培地とを1:1の割合で含有する培養液〔10%ウシ胎仔血清、1μg/mL インスリン、1×10−7mol/L デキサメサゾン、10mmol/L ニコチンアミド、2mmol/L L−グルタミン、50μmmol/L β−メルカプトエタノール、ペニシリン/ストレプトマイシン(100U/mL(ペニシリン)+100μg/mL(ストレプトマイシン))、20ng/mL EGF(Sigma,E9644)を含む〕中で、37℃、5%CO2存在下、インキュベーター内で培養した。以後、EGFを含有するこの培地をEGF(+)培地、あるいは腸管幹/前駆細胞培養用培地とも称する。
2日後、96ウェル丸底ディッシュに播種したマウス胎児腸管細胞が集合して細胞塊を形成した(図1a)。その後、形成した細胞塊を低接着性の6cmディッシュ等に移したりしながら、さらに5日間程度培養後、それら細胞塊をペトリディッシュ(BD,35−1007)に移し、上記EGF(+)培地中、37℃、5%CO2存在下、インキュベーター内で培養を続けた(図1b)。ここで用いたペトリディッシュは、接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュである。一方低接着性ディッシュを用いず、細胞分散液をすぐに接着性のペトリディッシュに播種し、上記EGF(+)培地中で培養した場合には線維芽細胞様の細胞のみディッシュに残った(図1c)。
凝集体を普通のペトリディッシュに移して3ヶ月程度培養を続けると、線維芽細胞様の細胞に対し、上皮細胞様の細胞が多く見られるようになった。もし線維芽細胞様の細胞が残存している場合は、モノヨード酢酸(4μg/mL)で3〜5時間程度処理することで線維芽細胞様の細胞を死滅させることができる。
得られた上皮細胞様の細胞は、細胞培養用ディッシュにて継代し(図2)、解析(後述する)や凍結保存に用いた。
又、後述の実施例からも明らかなように、この上皮細胞様の細胞は、自己複製能、多分化能を有する幹/前駆細胞を含んでいることから、便宜上、FIP(Fetal Intestine progenitor)細胞とも、以後、称する。
【0035】
実施例2:成体腸管由来の腸管幹/前駆細胞の取得
成体マウスから取り出した腸管の内部を、23Gの針を使ってPBSで洗浄し、その後、腸管にハサミを入れて管を開き、シート状にし、さらにハサミで5mm四方程度に切り分けた(腸組織片)。得られた腸組織片を用いて実施例1と同様にして、EGTA、コラゲナーゼ及びピペッティング処理によって細胞分散液を得た(成体マウス腸管細胞)。
成体由来の腸管細胞は、細胞塊を形成しにくいので、単層で凝集させて培養した。すなわち、直径6cmのペトリディッシュ(接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ)に1〜5×106細胞を播種して、高密度培養を行った。実施例1と同組成の培養液を用い37℃、5%CO2存在下、インキュベーター内で培養した。
高密度培養を8ヶ月程度続けると、線維芽細胞様の細胞に対し、上皮細胞様の細胞が多く見られるようになった。もし線維芽細胞様の細胞が残存している場合は、モノヨード酢酸(4μg/mL)で3〜5時間程度処理することで線維芽細胞様の細胞を死滅させることができる。
得られた上皮細胞様の細胞は、細胞培養用ディッシュにて継代し、解析や凍結保存に用いた。
【0036】
実施例3:増殖能
FIP細胞の増殖能について調べた。FIP細胞を、種々の濃度(0ng/mL〜80ng/mL)でEGFを含有する培養液中で培養した。培養液は、EGFの濃度が異なること以外は、実施例1で用いた培養液と同じ組成である。EGFを含まない培養液中で2日間培養した細胞(図3a)、EGFを20ng/mLの濃度で含む培養液中で2日間培養した細胞(図3b)を示す。又、12ウェルの培養プレートに播種し、各種濃度でEGFを含む培養液中で1日間培養した細胞をトリプシン−EDTAではがしてその数を血球計算板により計測した(図3c)。
腸管細胞由来の上皮細胞様の細胞の増殖がEGF依存性であることが示された。
【0037】
実施例4:増殖とアポトーシス
FIP細胞をEGF(+)培地(EGF濃度20ng/mL)中、細胞培養用ディッシュ上で2日間培養した(図4a)。同じ状態のものを2セット用意した。EGF存在下で2日間培養した後、一方は、同じEGF(+)培地で培地交換してさらに1日間培養し(図4b)、もう一方はEGFを含まない培養液に培地交換してさらに1日間培養した(図4c)。EGF存在下では細胞は増殖を続けたが、EGF非存在下では細胞の増殖は抑えられ、死ぬ細胞が確認された。一連の過程における細胞の増殖能の変化を、BrdU免疫染色法により確認した。核染色剤であるDAPIを用いて二重染色した(図5a〜f)。
EGF存在下で2日間培養した後、一方は、EGFを含まない培養液に培地交換してさらに1日間培養し、もう一方は同じEGF(+)培地で培地交換してさらに1日間培養した。その後、BrdU(10μM)(Sigma,B9285)を培地に添加し、1.5時間経過後にBrdUを取り込んだ増殖細胞の割合をフローサイトメトリーおよび免疫染色にて解析した。フローサイトメトリー解析は、BrdU Flow Kit (BD,552598)のマニュアルに沿って細胞を標識後、FACS Aria(BD)を用いて行った。この際、ゲートの設定はネガティブコントロールを指標にして行った。BrdUの免疫染色は、BrdU Flow Kitの試薬と抗BrdU抗体(×50)(BD,347580)、Cy3をコンジュゲートしたロバ抗マウスIgG抗体(×200)(Jackson ImmunoResearch Laboratories,cat no.715−165−150)、およびDAPI(1μg/mL)(Sigma,D9542)を用いて行い、蛍光顕微鏡で観察した。EGF非存在下で培養した細胞についてはBrdUの取り込みが顕著に減少しており、増殖能が低下したことが明らかとなった(図5a〜f)。
また、EGFを培養液から除くと、死ぬ細胞が確認されたことから、EGF非存在下でのアポトーシス誘導について調べた(図5g〜h)。
BrdUの解析時と同様に細胞を培養した。細胞をディッシュから回収した後、冷PBSにて細胞を1回洗い、アネキシンV binding buffer(1×)(BD,556454)にて細胞懸濁液を調製した。そこへ7−AAD(5μL;BD,559925)とAPC−conjugated アネキシンV(5μL;BD,550474)を添加し、室温にて15分間反応させた(遮光)。染色した細胞はFACS Aria(BD)を用いて解析した。ゲートの設定はネガティブコントロールを指標にして行った。アネキシンVはリン脂質結合性タンパク質である。アポトーシスを起こした細胞は細胞膜表面にホスファチジルセリンを露出するようになり、そこにアネキシンVが結合する。従ってアネキシンVはアポトーシス細胞に強い親和性を有する。EGF非存在下ではアネキシンVが結合する細胞が顕著に増加しており、アポトーシス誘導が起こっていることが示された(図5g〜h)。
FIP細胞は、EGFの存在下、活発に分裂増殖したが、EGFを含まない培養条件では増殖活性が著しく減少し、アポトーシスを起こす細胞が増加した。
【0038】
実施例5:マーカー蛋白質の発現
FIP細胞における種々のマーカー蛋白質の発現について調べた。各マーカー蛋白質の発現は、各蛋白質の抗体をそれぞれ用いて免疫染色法により調べた。尚、細胞の存在を示す為に、核染色剤であるDAPIで二重染色した。
細胞をPBSで洗浄し、4%パラホルムアルデヒド(PFA)(PBS中)で5分間、室温で固定した。続いて、25%アセトン(メタノール中)で1分間、室温で処理した。固定後、細胞を0.05%ポリオキシエチレン(20)ソルビタンモノラウレート(Tween 20)(Wako)を含有するPBS(PBS−Tween)で洗浄し、続いて、0.2%Triton X−100(Sigma)で1時間、室温で処理した。非特異的な結合は、10%のロバ血清(Sigma,D9663)で1時間、室温で処理することによりブロッキングした。固定した細胞を1次抗体と湿室内で16時間、4℃でインキュベートした。細胞を、PBS−Tween20で洗浄し、ブロッキングした後、さらに2次抗体と4時間、4℃でインキュベートした。室温でDAPIと5分間インキュベートし、洗浄後、細胞を蛍光顕微鏡で観察した。
用いた抗体は以下の通り。
(1次抗体)
抗E−カドヘリン抗体:BD,610181(2次抗体、マウス用);×100
抗Zo−1抗体:Zymed,61−7300(2次抗体、ウサギ用);×100
抗ビリン抗体:Santa Cruz,sc−7672(2次抗体,ヤギ用);×10
抗Musashi−1抗体: Chemicon,AB5977(2次抗体、ウサギ用);×100
(2次抗体)
マウス用:Alexa 488が結合したロバ抗マウスIgG(Molecular Probes,A21202);×200
ウサギ用:Alexa 488が結合したロバ抗ウサギIgG(Molecular Probes,A21206);×200
ヤギ用:Alexa 488が結合したロバ抗ヤギIgG(Molecular Probes,A11055);×200
FIP細胞は、全細胞において上皮細胞のマーカーであるE−カドヘリンやZo−1、及び、腸上皮細胞のマーカーであるビリンを発現していた。また、一部の細胞は腸管幹/前駆細胞マーカーと考えられているMusashi−1を発現していた(図6)。
【0039】
実施例6:βカテニン転写活性化
TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)をリポフェクトアミン2000(Invitrogen,11668)を用いて細胞に導入し、X−gal染色により陽性細胞を検出した。具体的には以下の手順で行った。
(1) 細胞をPBSで洗浄(×2)
(2) 固定液(2%PFA/0.2%グルタルアルデヒド(GA)/0.05%NP−40/PBS)を添加(6ウェルプレートの1ウェルあたり1.5〜2mL)
(3) 1分間室温で放置
(4) PBSで洗浄(×2)
(5) X−gal溶液を添加(6ウェルプレートの1ウェルあたり1.5〜2mL)
(6) 37℃のインキュベーター中で放置(少なくとも3時間〜一晩)
(7) PBSで洗浄(×2)することで反応を停止させる
(8) 観察(写真撮影)、PBS中4℃で保存
(固定液の調製)
以下の割合で各成分を混合して固定液を調製した。
20% PFA/PBS 1mL(−20℃保存)
25% GA 80μL(−20℃保存)
5% NP−40 80μL(室温保存)
PBS 9mL(室温保存)
(X−gal溶液の調製)
以下の割合で各成分を混合してX−gal溶液を調製した。
500mM K3Fe(CN)6 50μl(室温保存、遮光保存)
500mM K4Fe(CN)6 50μl(室温保存、遮光保存)
1M MgCl2 10μl(室温保存)
20mg/ml X−gal 250μl(−20℃保存、遮光保存)
PBS 4.7ml(室温保存)
X−gal陽性細胞が検出され、βカテニンの転写活性化が確認された(図7)。
【0040】
実施例7:腸上皮組織を構築する細胞への分化誘導
FIP細胞をコンフルエントな状態になるまで培養した。コンフルエントになった状態からEGFを除くと、アポトーシスを起こす細胞の中で生き残る細胞が存在した。これら生き残った細胞をEGF非存在下で培養し続けると分化した腸上皮様細胞の特徴を示す細胞が出現してきた(図8)。
この状態の細胞をPAS染色すると、EGF非存在下で培養し続けた細胞において赤紫色に染色される細胞が多数出現した(図9)。PAS染色は、糖質に含まれる1,2グリコール基群を過ヨウ素酸で酸化し、生じた2分子のアルデヒド基がシッフ試薬1分子と結合して、赤紫色の化合物を形成する2つの反応を利用したものであり、グリコーゲン、中性粘液多糖類、糖蛋白質、粘液蛋白質、糖脂質等の検出に用いられる。腸上皮組織においては、粘液分泌細胞の1種であるゴブレット細胞を染め分けることが知られている。具体的には以下の手順で行った。尚、各試薬については当分野で通常PAS染色を行う際に用いられている試薬を使用することができる。
(1) 細胞をPBSで洗浄(×1)
(2) 0.5%過ヨウ素酸で室温7分間処理
(3) ミリQ水で洗浄(×5)
(4) シッフ試薬で室温7〜10分間処理
(5) 亜硫酸水で処理(室温1分間処理を3回繰り返す)
(6) ミリQ水で洗浄(×5)
(7) ヘマトキシレンで処理(室温で1分間)
(8) ミリQ水で洗浄(×5)
又、各腸上皮組織において分泌、発現している蛋白質に対する抗体を用いて免疫染色を行い、該蛋白質の発現状況を解析した。用いた各蛋白質の抗体は以下の通り。尚、細胞の存在を示す為に、核染色剤であるDAPIで二重染色した。
(1次抗体)
抗セロトニン抗体:Immunostar,20080(2次抗体、ウサギ用);×10
抗クロモグラニンA抗体:Santa Cruz,sc−1488(2次抗体、ヤギ用);×10
抗ガストリン抗体:Santa Cruz,sc−7783(2次抗体、ヤギ用);×10
抗セクレチン抗体:Chemicon,AB981(2次抗体、ウサギ用);×100
抗ソマトスタチン抗体:AFFINITI,SA1268(2次抗体、ウサギ用);×10
抗GIP抗体:Chemicon,AB953(2次抗体、ウサギ用);×10
抗リゾチーム抗体:Dako,A0099(2次抗体、ウサギ用);×100
抗シナプトフィジン抗体:Dako,A0010(2次抗体,ウサギ用);×10
(2次抗体)
ウサギ用:Alexa 488が結合したロバ抗ウサギIgG(Molecular Probes,A21206);×200
ヤギ用:Alexa 488が結合したロバ抗ヤギIgG(Molecular Probes,A11055);×200
結果、EGF非存在下で培養すると、腸内分泌細胞に特徴的なセロトニン、クロモグラニンA、ガストリン、セクレチン、ソマトスタチン、GIPを発現している細胞が検出され、粘液分泌細胞の1種であるパネート細胞に特徴的なリゾチーム及びシナプトフィジンを発現している細胞が検出された。これらの結果より、FIP細胞の培養液からEGFを除去することにより腸内分泌細胞への分化が誘導されたことがわかる。
【0041】
実施例8:RT−PCRによる解析
上記した実施例より、FIP細胞には、腸管幹/前駆細胞が含まれていることが示され、さらにEGF存在下で、該幹/前駆細胞は増殖を続け、EGFを培養液中から除去することにより、細胞はアポトーシスを引き起こし、又、種々の腸上皮組織を構築する細胞へと分化することがわかった(模式図を図10に示す)。
FIP細胞、及び各発生段階(胎生14.5日目、新生児、成体)にあるマウスの腸組織を用いて、腸管の細胞分化に関与すると考えられる遺伝子の発現プロファイルをRT−PCRによって解析した。
FIP細胞をEGF(+)培地で培養後、コンフルエントになった時点でEGFを含まない培地に培地交換し、その後1週間培養した。その細胞からRNAを抽出(RNeasy Mini Kit;QIAGEN)し、逆転写酵素反応(SuperScript III)によりcDNAを得た。腸組織からのRNA抽出では、ホモジナイザー(ポリトロン)を用いて組織を破砕後に同様の方法で抽出と逆転写酵素反応を行った。続いて、それらのcDNAを用いてPCRを行い、各種分化マーカーの発現を解析した。
目的とするマーカーならびにそのプライマーを表1に示す。
【0042】
【表1】

【0043】
PCRサイクルは、95℃(4分間)→[94℃(1分間)→56℃(1分間)→72℃(1分間)]×45サイクル→72℃(10分間)である。PCRにより産生された核酸は、2%アガロースゲルにて展開し、解析した。結果を図11に示す。
【0044】
実施例9:単一細胞レベルでの解析
上記した実施例3〜7での実験はheterogenousな細胞集団での解析であったので、腸管幹/前駆細胞の解析を厳密に行うためには、単一細胞レベルでの解析が必要であると考えた。そこで、フローサイトメトリーを用いてFIP細胞をクローンソーティングし、96ウェルディッシュの各ウェルにて単一細胞培養を試みた。増殖してきたクローンのひとつを用いて同様にして、増殖能、マーカー蛋白質の発現及び分化誘導について調べた。
その結果、単一コロニーから増殖させた細胞についても、E−カドヘリンやZo−1、ビリンを全細胞で発現し、また一部のコロニーについてはMusashi−1を発現していた。さらにEGF依存的な増殖活性や細胞死抑制を示し、さらに、EGF非存在下で複数の腸上皮細胞マーカーを発現するようになった。このように、単一細胞レベルの解析においても増殖能や多分化能が示されたことで、FIP細胞には腸管幹/前駆細胞が含まれることが確認された。
以上の結果より、FIP細胞集団には、自己複製能及び多分化能を有する幹/前駆細胞が含まれ、長期間安定に維持され得ることが示された。本願発明の方法を用いることによって腸管上皮細胞の幹/前駆細胞を選択的に増殖させることが可能となった。かかる選択的増殖によって、効率的に腸管幹/前駆細胞を取得・培養することができる。
【0045】
実施例10:ヒト小腸上皮細胞からの腸管幹/前駆細胞の取得
本実施例では、ACBRIにより分離され、日本では大日本住友製薬株式会社から供給されているヒト小腸上皮細胞(Cat No. CS-ABI-519)を用いた。この細胞を添付のデータシートに記載された方法に従って培養ディッシュに播種し培養した(図12a)。培養ディッシュは、タイプIVコラーゲンコートされたディッシュを用いた。培養液として、データシートに記載され、添付されている培地、すなわちCS−2.0基礎培地に添加剤を加えたもの(以下、CS-2.0 + supplement)を用いた。当該処理は、20ng/mLのEGF存在下で行なった。
CS-2.0 + supplementを用いて培養したところ継代を2回行なった時点で細胞は培養ディッシュに接着せず、全て死滅してしまった(図12c,e)。
そこで、実施例1で調製した、マウス胎児由来の腸管細胞を培養するのに用いたEGFを含有する培地(腸管幹/前駆細胞培養用培地)を用いて同様にして継代培養したところ、継代を重ねても培養ディッシュに接着し、増殖維持することができる細胞が得られた(図12b,d)。
これらの細胞は、購入したヒト上皮細胞集団中に含まれる腸管幹/前駆細胞であると考えられたので、便宜上、hAIP(human adult intestine progenitor)細胞とも、以後、称する。
【0046】
実施例11:マーカータンパク質の発現とβカテニン転写活性化
hAIP細胞について、実施例5と同様にしてマーカータンパク質の発現について調べた。
【0047】
hAIP細胞は、全細胞において上皮細胞のマーカーであるE−カドヘリンやZo−1、及び、腸上皮細胞のマーカーであるビリンを発現していた。また、一部の細胞は腸管幹/前駆細胞マーカーと考えられているMusashi−1を発現していた(図13、上段)。
また、hAIP細胞について、実施例6と同様にして、βカテニンの転写活性化の有無を確認した。X−gal陽性細胞が検出され、βカテニンの転写活性化が確認された(図13、下段)。
【0048】
実施例12:増殖能
実施例3と同様にしてhAIP細胞の増殖能について調べた。実施例1で用いたEGF(+)培地、及びEGFを含まない以外は同じ組成のEGF(−)培地でそれぞれ4日間培養した。EGFの存在下ではhAIP細胞は活発に分裂増殖するが、EGFを含まない培養条件では増殖活性が低下し、アポトーシスを起こす細胞が増加した(図14)。
この結果から、EGFはhAIP細胞の自己複製を促進し、細胞死を抑制することで、培養下における維持を可能にしていることがわかる。
【0049】
実施例13:増殖とアポトーシス
hAIP細胞をEGF(+)培地(EGF濃度20ng/mL)中、4日間培養した。同じ状態のものを2セット用意した。EGF存在下で4日間培養した後、一方は、同じEGF(+)培地で培地交換してさらに1日間培養し(図15A,左)、もう一方はEGFを含まない培養液に培地交換してさらに1日間培養した(図15A,右)。EGF存在下では細胞は増殖を続けたが、EGF非存在下では細胞の増殖は抑えられ、死ぬ細胞が確認された。一連の過程における細胞の増殖能の変化を、BrdU免疫染色法により確認した。核染色剤であるDAPIを用いて二重染色した(図15A)。
EGF存在下で4日間培養した後、一方は、EGFを含まない培養液に培地交換してさらに1日間培養し、もう一方は同じEGF(+)培地で培地交換してさらに1日間培養した。その後、BrdU(10μM)(Sigma,B9285)を培地に添加し、1.5時間経過後にBrdUを取り込んだ増殖細胞の割合をフローサイトメトリーおよび免疫染色にて解析した。フローサイトメトリー解析は、実施例4で記載した方法に従って行なった(図15B,左)。BrdUの解析時と同様に細胞を培養し、アネキシンVとの結合を実施例4で記載した方法に従って解析した。アネキシンVが結合する細胞をアポトーシスを起こしている細胞とみなし、その割合を調べた(図15B,右)。EGFを培養液中から除くことによって、BrdUの取り込みが減少し、すなわち増殖能が低下することがわかる。さらに、EGFを培養液中から除くことによって、アポトーシスを起こす細胞が増加した。
hAIP細胞の増殖あるいはアポトーシスに及ぼすEGFの影響を確認するために、EGFを含有する培養液中に、EGFシグナル伝達系を阻害するAG1478、PD98059、あるいはLY294002を添加した培養液を用いて同様にBrdUの取り込みとアポトーシス誘導について調べた。
PD98059(2’−アミノ−3’−メトキシフラボン)は、MEK1(MAPキナーゼ・キナーゼ)に対する選択的で細胞透過性の阻害剤であり、MEK1の活性化を阻害する結果、続いて起こるMAPK(ERK1/2)のリン酸化−活性化を阻害する。使用濃度:100μM。
LY294002([2−(4−モルホリニル)−8−フェニル−4H−1−ベンゾピラン−4−オン]はホスファチジルイノシトール3−キナーゼ(PI3−キナーゼ)に対する強力で特異的な細胞透過性阻害剤である。使用濃度:100μM
【0050】
実施例14:腸上皮組織を構築する細胞への分化誘導
hAIP細胞は上記した培養条件下で自然に分化誘導され、腸上皮様細胞の特徴を示す細胞が出現してくる。この状態の細胞を実施例7に記載する方法に従ってPAS染色すると、赤紫色に染色される細胞が多数出現したことから、腸上皮組織であるゴブレット細胞の存在を確認することができた(図16B)。
さらに、実施例8と同様な手法で、腸上皮組織を構築する細胞への分化誘導をRT−PCR解析により調べた。誘導される腸上皮組織の種類は、図10に示されている。
hAIP細胞、及びヒト成体小腸からの組織を用いて、腸管の細胞分化に関与すると考えられる遺伝子の発現プロファイルをRT−PCRによって解析した。目的とするマーカー及びそのためのプライマーは、下記表2に示すヒト用のものを用いた。
【0051】
【表2】
【0052】
結果を図16Aに示す。
hAIP細胞培養中には、腸上皮組織を構築する複数の腸内分泌細胞や粘液分泌細胞の特徴を示す細胞が観察された。従ってhAIP細胞は、培養下で増殖し続けるのと同時に、腸を構成する様々な細胞に分化する多分化能を有することが判明した。
【配列表フリーテキスト】
【0053】
配列番号1:ビリン(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号2:ビリン(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号3:Lfabp(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号4:Lfabp(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号5:クロモグラニンA(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号6:クロモグラニンA(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号7:Trefoil factor−3(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号8:Trefoil factor−3(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号9:リゾチーム(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号10:リゾチーム(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号11:シナプトフィジン(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号12:シナプトフィジン(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号13:Musashi−1(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号14:Musashi−1(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号15:Math−1(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号16:Math−1(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号17:ビリン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号18:ビリン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号19:Lfabp(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号20:Lfabp(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号21:シュクラーゼ−イソマルターゼ(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号22:シュクラーゼ−イソマルターゼ(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号23:セクレチン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号24:セクレチン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号25:GIP(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号26:GIP(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号27:ガストリン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号28:ガストリン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号29:クロモグラニンA(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号30:クロモグラニンA(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号31:CCK(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号32:CCK(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号33:ソマトスタチン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号34:ソマトスタチン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号35:Trefoil factor−3(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号36:Trefoil factor−3(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号37:リゾチーム(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号38:リゾチーム(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号39:シナプトフィジン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号40:シナプトフィジン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号41:Musashi−1(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号42:Musashi−1(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号43:Math−1(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号44:Math−1(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号45:Ngn3(Neurogenin−3)(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号46:Ngn3(Neurogenin−3)(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号47:HPRT(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号48:HPRT(ヒト)を検出する為のPCR用プライマー(リバース)である。
【図面の簡単な説明】
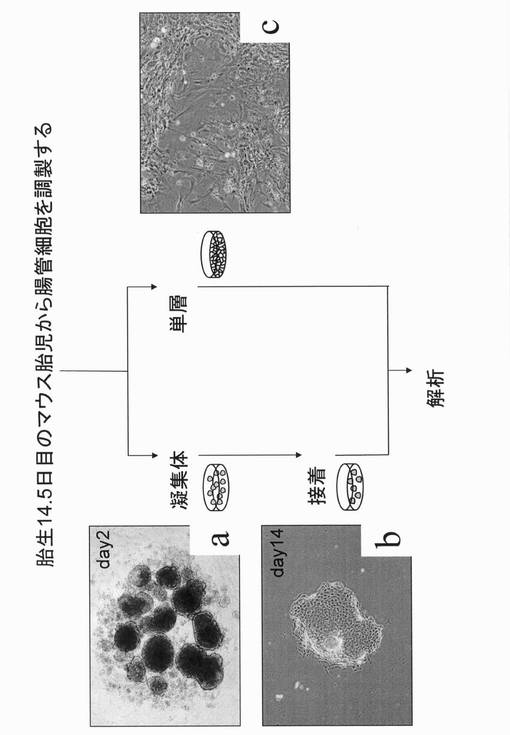
【0054】
【図1】腸管細胞培養の簡単な手順を示す図である。aは、非接着性培養容器上で形成させた凝集体を示す図(位相差像)である。bは、形成させた凝集体を接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ上で培養した状態を示す図(位相差像)である。cは、凝集体を形成させず、腸管細胞を接着性培養容器上で培養した状態を示す図(位相差像)である。
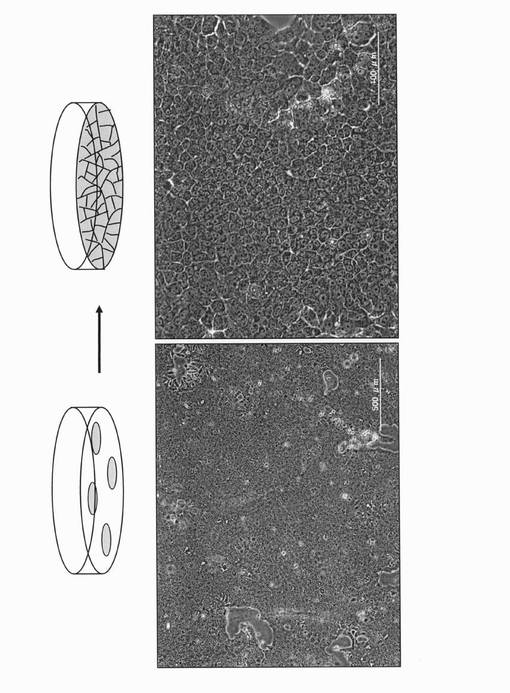
【図2】一連の操作によって得られた腸管細胞由来の、上皮細胞様の細胞を継代培養して増殖させた状態を示す図(位相差像)である。
【図3】FIP細胞のEGF依存性の増殖能を示した図(位相差像)である。aはEGF非存在下で、bはEGF存在下でそれぞれ2日間培養した状態を示す。cは細胞をトリプシン−EDTAではがして血球計算板により計測した結果を示す。横軸は添加したEGFの濃度を、縦軸は細胞数を示す。
【図4】EGF存在下で培養していたFIP細胞をEGF非存在下又は引き続きEGF存在下で培養した場合の細胞の状態を示す図(位相差像)である。aはEGF存在下で2日間FIP細胞を培養した状態を示す。EGF存在下での2日間の培養後、さらにEGFを含む培養液中で1日間(b)、あるいはEGFを含まない培養液中で1日間(c)、それぞれ培養した。
【図5】EGF存在下で培養していたFIP細胞を引き続きEGF存在下で培養した場合又はEGF非存在下で培養した場合の細胞の状態を示す図である。a及びbは、それぞれの細胞のBrdU取り込みの様子をBrdU免疫染色法により調べた結果を表す図(蛍光顕微鏡像)である。c及びdは、それぞれの細胞をBrdU/DAPIの二重染色により染色した結果を表す図である(蛍光顕微鏡像)。e及びfは、EGF存在下、あるいは非存在下でのBrdUの取り込みをFACS解析した結果を表すグラフである。g及びhは、EGF存在下、あるいは非存在下でのアネキシンVの結合をFACS解析した結果を表すグラフである。
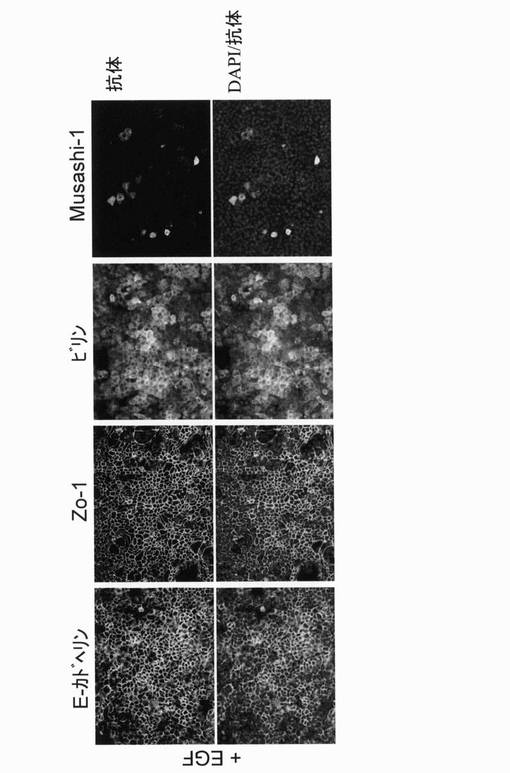
【図6】EGF存在下で培養を続けたFIP細胞における各種マーカー蛋白質(E−カドヘリン、Zo−1、ビリン及びMusashi−1)の発現を調べた結果を示す図である(蛍光顕微鏡像)。
【図7】TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出した結果を示す図である。
【図8】EGF存在下で培養していたFIP細胞(a,c)からEGFを除去し、さらにEGF非存在下で7日間培養した細胞(b,d)の状態を示す図(位相差像)である。
【図9】FIP細胞をEGF非存在下で培養し続けた細胞(腸上皮細胞様の細胞)をPAS染色した像である。aはコンフルエントな状態になったFIP細胞を示し、bはEGF非存在下で7日間培養した細胞を示す。
【図10】FIP細胞とEGFとの関係を模式的に示した図である。
【図11】FIP細胞、胎生14.5日のマウス腸組織、新生児腸組織、成体腸組織における種々の遺伝子発現プロファイルをRT−PCR法により調べた結果を示す図である。
【図12】ヒト小腸上皮細胞(Cat No. CS-ABI-519)をCS-2.0+supplement培地あるいは腸管幹/前駆細胞培養用の培地で継代培養した場合の細胞の様子を示す図である。aは、CS-2.0+supplement培地中に播種した継代1回目の細胞の様子を、b及びdは、腸管幹/前駆細胞培養用の培地で2回継代培養を行なった細胞の様子を示している。播種後1日目(b)と5日目(d)の様子である。c及びeは、CS-2.0+supplement培地で2回継代培養を行なった細胞の様子を示している。播種後1日目(c)と5日目(e)の様子である。位相差像である。
【図13】上段は、EGF存在下で培養を続けたhAIP細胞における各種マーカー蛋白質(E−カドヘリン、Zo−1、ビリン及びMusashi−1)の発現を調べた結果を示す図(蛍光顕微鏡像)である。下段は、TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出した結果を示す図である。
【図14】hAIP細胞のEGF依存性の増殖能を示した図(位相差像)である。a及びcはEGF存在下で、b及びdはEGF非存在下でそれぞれ4日間培養した状態を示す。c及びdは、それぞれa及びbの高倍率像である。
【図15】hAIP細胞の増殖とアポトーシスに及ぼすEGFの影響を解析した結果を示す図である。図15AはEGF存在下で培養していたhAIP細胞を引き続きEGF存在下で培養した場合又はEGF非存在下で培養した場合の細胞の状態を示す図である。それぞれの細胞をBrdU/DAPIの二重染色により染色した結果を表す(蛍光顕微鏡像)。図15Bは、EGF存在下、あるいは非存在下、あるいは種々の阻害剤を用いた場合の、BrdUの取り込みをFACS解析した結果、あるいはアポトーシスを起こした細胞をFACS解析した結果を表すグラフである。
【図16】図16Aは、hAIP細胞、ヒト成体腸組織における種々の遺伝子発現プロファイルをRT−PCR法により調べた結果を示す図である。図16BはhAIP細胞をPAS染色した像である。
【技術分野】
【0001】
本発明は腸管上皮細胞の幹/前駆細胞を取得し効率的に培養する方法、該方法によって得られる幹/前駆細胞に関する。さらに本発明は腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法に関する。
【背景技術】
【0002】
薬剤の吸収試験やその他の薬学研究では腸上皮細胞の初代培養細胞を使用することが多い。これまでの培養系では、腸上皮細胞は数日の間に淘汰されてしまい、常に新しい細胞を調製し使用する必要があった。そのため、腫瘍組織由来の細胞株が用いられているが、信頼性の高いデータを得るためには正常細胞を使用する必要があり、正常な腸上皮細胞を簡便に使用できることが望まれている。この場合、培養下で長期間細胞を維持するためには、成熟した腸上皮細胞に加え、それらを常に産生する幹/前駆細胞を培養下で維持する必要があると言える。
【0003】
食物の消化吸収を担う臓器である腸管には、機能的役割の異なる腸上皮細胞群の根幹に位置する幹細胞(腸管幹細胞)が存在し、幹細胞の分裂と維持により腸上皮細胞が継続して供給される。このように、腸管幹細胞は旺盛な増殖能を有するが、その能力を人為的に培養下で保持させ続けることは極めて難しく、これまでに腸管幹細胞の培養に成功した例はない。腸管幹細胞の増殖や分化を培養下で制御できれば、腸管の幹細胞システムに対する基礎研究用ツールとして利用できるだけでなく、薬剤吸収・代謝試験など創薬過程におけるスクリーニング用ツールとして利用できる。また、組織工学的に移植可能な腸上皮組織を供給することができる。
【0004】
一方、初期胚に由来する、多能性の幹細胞株である胚性幹細胞(ES細胞)は、多能性を有するが故に、通常の培養方法では細胞が徐々に分化していってしまう。そのため、ES細胞を効率よく増やすためにはES細胞を未分化状態のままで増殖させる必要がある。マウスのES細胞の場合には白血病阻害因子(LIF)を培地に添加することにより未分化状態を維持できることが知られている(非特許文献1)が、組織幹細胞である腸管幹細胞ではLIFの代替となるような添加因子は知られていない。またES細胞では分化誘導の際に胚様体(embryoid body)と呼ばれる細胞凝集体を作らせることが一般的に知られているが、腸管幹細胞の分化を制御する方法は未だ確立されていない。
【非特許文献1】Smith AG, Heath JK, Donaldson DD, Wong GG, Moreau J, Stahl M, Rogers D. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988 Dec 15;336(6200):688-90.
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は腸管上皮細胞の幹/前駆細胞の取得を目的とし、さらに該幹/前駆細胞を効率的に培養することを目的とする。さらに本発明の別の目的は、腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法を提供することにある。
【課題を解決するための手段】
【0006】
本発明者らは、上記課題に鑑み、鋭意検討を行った結果、腸管由来の細胞(腸管細胞)を浮遊培養することによって細胞凝集体を得て、当該凝集体を、上皮増殖因子(EGF)を含有する培養液中で培養することによって、腸管の幹/前駆細胞を選択的に取得、増殖させることができることを見出した。さらに、得られた幹/前駆細胞を適当な分化誘導条件下で腸上皮組織を構築する細胞へと分化させることに成功して本発明を完成するに至った。すなわち本発明は以下の通りである。
【0007】
[1]腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[2]腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[3]凝集体が腸管細胞を浮遊培養することによって得られるものである、上記[1]又は[2]記載の方法。
[4]以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を浮遊培養することによって細胞塊(凝集体)を調製する工程
(3)腸管細胞の凝集体を、EGFを含有する培養液中で培養する工程
(4)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
[5]腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、上記[4]記載の方法。
[6]腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む腸管上皮細胞の幹/前駆細胞の取得方法。
[7]腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
[8]以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を、EGFを含有する培養液中、高密度で培養する工程
(3)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
[9]腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、上記[8]記載の方法。
[10]上記[1]〜[9]のいずれか1項に記載の方法によって得られた腸管上皮細胞の幹/前駆細胞。
[11]腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[12]腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[13]凝集体が腸管細胞を浮遊培養することによって得られるものである、上記[11]又は[12]記載の方法。
[14]腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[15]腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
[16]腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法であって、該幹/前駆細胞の培養液からEGFを除去することを特徴とする方法。
[17]腸上皮組織を構築する細胞が、腸内分泌細胞又は粘液分泌細胞である、上記[16]記載の方法。
[18]血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する、腸管上皮細胞の幹/前駆細胞培養用培地。
【発明の効果】
【0008】
本発明によって得られる腸管上皮細胞の幹/前駆細胞及び該細胞から腸上皮組織を構築する細胞への分化を誘導する方法は、腸組織の発生・分化を理解するための基礎研究用ツールとして有用であり、又、組織工学的に移植可能な腸上皮組織の供給を可能とする。さらに該幹/前駆細胞に由来する腸上皮細胞を用いて薬物の腸管における吸収実験などを効率的に行うことが期待できる。
【発明を実施するための最良の形態】
【0009】
文中で特に断らない限り、本明細書で用いるすべての技術用語及び科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様又は同等の任意の方法及び材料は、本発明の実施又は試験において使用することができるが、好ましい方法及び材料を以下に記載する。本明細書で言及したすべての刊行物及び特許は、例えば、記載された発明に関連して使用されうる刊行物に記載されている、構築物及び方法論を記載及び開示する目的で、参照として本明細書に組み入れられる。
【0010】
本発明の取得方法及び培養方法の対象となる腸管上皮細胞の幹/前駆細胞は、任意の哺乳動物の腸管細胞に由来する。ここで「哺乳動物」とは、具体的にはヒトをはじめウシ、ウマ、イヌ、モルモット、マウス、ラット等が挙げられる。好ましくはヒトであるが、薬物の吸収実験等の研究用ツールとしての利用を考えた場合、マウスやラット等の実験動物としてよく用いられる動物も又、好ましい。
【0011】
腸は、大きく小腸と大腸の2つに分けることができる。小腸は更に口側から、十二指腸、空腸、回腸に分けられ、大腸は盲腸、結腸(上行結腸、横行結腸、下行結腸、S状結腸)、直腸に分けられる。成体の腸は、広範にわたって腺窩と絨毛に折り畳まれている上皮層及び支持間葉系組織によって取り囲まれているルーメンから構成されている。腸の上皮細胞は常に新生され、古くなったものは剥離してゆく。腸上皮細胞の新生は、腸上皮組織中に、幹細胞による細胞再生系が存在するからである。幹細胞は、小腸の絨毛の根元が窪んだ陰窩(crypt)の底のほうに存在する。また、胎児においては、内胚葉細胞層が腸上皮となるよう運命付けられ、重層細胞層が分化した偏極上皮単層へと転換する。腸上皮の形態及び恒常性は細胞増殖、移動、分化及びアポトーシス間での高度に制御された均衡の結果である。本発明の取得方法及び培養方法の対象となる「腸管上皮細胞の幹/前駆細胞」とは、腸上皮細胞へと分化し得る、及び/又は分化することが運命づけられている細胞である。「幹細胞」は自己複製能と多分化能をあわせもつ細胞であって、分化運命が限定されている「前駆細胞」と完全に一致するものではないが、腸上皮細胞へと分化する細胞という点では同一であり、従って、本願発明では特に区別することなく「幹/前駆細胞」と称する。また、「腸管上皮細胞の幹/前駆細胞」を、便宜上「腸管幹/前駆細胞」と称することもある。
【0012】
本明細書中、「腸管細胞」とは、成体あるいは胎児から採取される腸由来の細胞であって、分裂能力を有する腸上皮細胞あるいは腸上皮細胞となり得る細胞の総称である。具体的には、腸管、好ましくは小腸部分を取り出し細かく切り刻んで、振動等の物理的な手段によって、EGTAあるいはEDTA等を用いた化学的な処理によって、あるいはコラゲナーゼ、トリプシン、キモトリプシン、ペプシン等のプロテアーゼ(好ましくはコラゲナーゼ)を用いた酵素的な処理によって細胞を分散することによって得ることができる。上記処理は単独で行っても、複数の処理を組み合わせて行っても良い。胎児の腸管を用いる場合には、円筒状の腸構造から、腸上皮組織中に凸凹が生じ始める時期のものを用いることが好ましい。例えばマウスでは、胎生12〜17日目、好ましくは14.5日の胎児から採取する。また、本発明において用いられる「腸管細胞」は、上記の如く腸管から採取されるものであってもよいが、分裂能力を有する腸上皮細胞あるいは腸上皮細胞となり得る細胞を含有する市販の腸管由来の細胞であってもよい。かかる市販の腸管由来の細胞としては、ACBRI(Applied Cell Biology Research Institute)により分離され(ACBRI 519)、日本では大日本住友製薬株式会社から入手可能なヒト小腸上皮細胞(human intestinal epithelial cells, cat No. CS-ABI-519)が挙げられる。
【0013】
本発明の第一の特徴は、腸管細胞に凝集体をつくらせ、それを培養することにある。凝集体を形成させることによって、より生体内に近い環境に細胞をおくことができる。ここで、「凝集体」とは、細胞が多数集合して形成された三次元構造(球状やぶどうの房状)を有する細胞の塊である。例えば、後述するが、20〜40万細胞/ウェルの濃度で低接着性の96ウェル丸底ディッシュに播種した場合、10〜20個程度の直径200〜500μm程度の凝集体が形成される。凝集体の最外部に細胞のシート状構造が見られる。より生体内に近い環境を提供することが可能な範囲で、凝集体を構成する細胞の数及び大きさは、細胞の状態や培養条件によって変わり得る。
【0014】
腸管細胞の凝集体は、具体的には以下のようにして調製することができる。まず、腸管細胞を適当な媒体中、低接着性の培養容器上で浮遊培養する。例えば低接着性の培養容器として96ウェルの丸底ディッシュを用いた場合には、1ウェルあたり20〜40万細胞を播種し、形成した凝集体を低接着性の6cmディッシュ等に移したりしながら、37℃で、2〜10日間、好ましくは7日間、適当な媒体中で培養する。低接着性の培養容器としては、通常、当分野で用いられている培養容器に低接着性となるような処理を施したものが例示される。培養容器としては、培養ディッシュ、培養フラスコ、回転培養用器具(スピナーフラスコ等)等が挙げられる。低接着性となるような処理としては、ハイドロゲルを共有結合させること(コーティング)によって蛋白質や細胞の接着を抑制するような処理が用いられる。又、市販されているものを用いることもできる。
【0015】
成体由来の腸管細胞は凝集体を形成しにくいが、該腸管細胞を高密度で培養することによって、同様の効果(生体内に近い環境を作り出すという効果)を得ることができる。例えば直径6cm(媒体の量:3.5mL)のペトリディッシュ(接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ;以下、単に普通のペトリディッシュともいう)で培養する場合には、1〜5×106細胞を播種し、37℃で、1〜5日間、好ましくは3日間、適当な媒体中で培養することによって高密度培養を達成することができる。
【0016】
上記した、凝集体の培養及び高密度培養において使用可能な媒体としては、腸管細胞が生存し得て、且つ分化やアポトーシスの誘導が抑制されている状態を維持できるものであれば特に限定されず、当分野で通常用いられる細胞培養用培地(ダルベッコ改変イーグル培地、MEM、RPMI1640培地、ハムF−12培地等)をベースとし、必要に応じて各種の成分を補充したものが用いられる。特に、血清、抗生物質、増殖因子等の成分を補充することが好ましい。
【0017】
媒体中に含められる好適な成分としては、5〜20%、好ましくは10%の血清、0.5〜2μg/mL、好ましくは1μg/mLのインスリン、1×10−8〜1×10−6mol/L、好ましくは1×10−7mol/Lのデキサメサゾン、5〜15mmol/L、好ましくは10mmol/Lのニコチンアミド、1〜5mmol/L、好ましくは2mmol/LのL−グルタミン、10〜100μmol/L、好ましくは50μmol/Lのβ−メルカプトエタノール、ペニシリン/ストレプトマイシン等が挙げられる。いずれも商業的に入手可能である。血清としては、ウマ、ウシ由来の血清等が使用され、好ましくは胎児血清である。インスリンとしては、ウシ、ブタの膵臓から生産されたインスリンや、遺伝子組み換え技術により生産されたインスリンのいずれも使用できるが、安定な品質と供給という点から組換え型インスリンを用いることが好ましい。
【0018】
本発明の第2の特徴は、選択的に腸管幹/前駆細胞を増殖、取得又は培養するために、腸管細胞の培養を、EGF(上皮増殖因子)存在下で行うことである。
【0019】
EGFは、Cohenらによってマウス胎児の早期眼開裂作用を持つ物質としてマウス顎下腺より単離同定され、ついで尿よりヒトEGFが発見された(S. Cohen and G. Carpenteret al., Proc. Natl. Acad. Sci. USA., 72, 1317, 1975; H. Gregory, Nature, 257, 325, 1975)。遺伝子構造も詳細に解明されている(G. I. Bell et al., Nucleic Acids Res., 14, 8427, 1986)。EGFは、53アミノ酸残基及び3つの分子内ジスルフィド結合からなる6045Daの蛋白質であり、細胞表面に存在するEGFR(上皮増殖因子受容体)にリガンドとして結合し、細胞の成長と増殖の調節に重要な役割を果たす。本発明で用いるEGFは選択的に腸管幹/前駆細胞を増殖、取得又は培養することを可能にする限り、その由来は特に限定されず、天然由来のものであっても公知の遺伝子配列やアミノ酸配列等の情報に基づいて合成又は半合成して製造されるものであってもよい。商業的に入手可能なものであってもよい。マウスの幹/前駆細胞を対象とする場合にはマウスの、ヒトの幹/前駆細胞を対象とする場合にはヒトのEGFをそれぞれ用いることが好ましいが、必ずしも動物種を一致させる必要はない。腸管幹/前駆細胞を選択的に増殖、取得又は培養することが可能な範囲で、EGFは改変されたものであってもよい。このような改変には、1個又は複数個のヌクレオチドあるいはアミノ酸の変異、挿入、欠失及び置換が含まれる。改変されたEGFは、当分野で通常実施される手段によって調製することができる。例えば、エキソヌクレアーゼを用いる欠失変異株(deletion mutant)作製法、カセット変異法等の部位特異的変異(site−directed mutagenesis)によってEGF DNAを人為的に改変させ、該改変EGF DNAを用いて、所望蛋白質を調製することができる。
【0020】
EGFは、腸管幹/前駆細胞を取得するために腸管細胞を培養する過程を通じて持続的に媒体中に含めておくことが好ましいが、特に凝集体を形成し、その後長期間培養し増殖させる間は必須的に媒体中に含める。EGFの媒体中の濃度は、20〜40ng/mL、好ましくは20ng/mL程度である。
【0021】
EGFを含有する培養液中で培養することによって、市販の腸管細胞、例えば上述のヒト小腸上皮細胞(Cat No. CS-ABI-519)からもまた、腸管幹/前駆細胞を得ることができる。市販されているヒト小腸上皮細胞を用いた場合には、凝集体の形成や高密度での培養を必要とすることなく、EGFを含有する培養液、特に20〜40ng/mL、好ましくは20ng/mLのEGFに加えて、5〜20%、好ましくは10%の血清、0.5〜2μg/mL、好ましくは1μg/mLのインスリン、1×10−8〜1×10−6mol/L、好ましくは1×10−7mol/Lのデキサメサゾン、5〜15mmol/L、好ましくは10mmol/Lのニコチンアミド、1〜5mmol/L、好ましくは2mmol/LのL−グルタミン、10〜100μmol/L、好ましくは50μmol/Lのβ−メルカプトエタノールを含む培養液を用いて継代培養することによって腸管幹/前駆細胞を取得することができる。
【0022】
10%ウシ胎児血清、インスリン(1μg/mL)、デキサメサゾン(1×10−7mol/L)、ニコチンアミド(10mmol/L)、L−グルタミン(2mmol/L)、β−メルカプトエタノール(50μmol/L)、ペニシリン(100U/mL)+ストレプトマイシン(100μg/mL)及びEGF(20ng/mL)を補充したダルベッコ改変培地−ハムF12の混合培地(1:1)は、腸管細胞を培養する為の媒体(培養液)の好適な一例である。
【0023】
上記で得られた、細胞凝集体を普通のペトリディッシュに再播種し、EGFを含有する媒体(培養液)中で37℃で、2〜6ヶ月、好ましくは3ヶ月程度培養する。成体の腸管由来の細胞は凝集体を形成しにくいので、高密度培養によって細胞を凝集しているのに等しい状態にする。成体の腸管に由来する細胞は、EGFを含有する培養液中で37℃で、6〜12ヶ月、好ましくは8ヶ月程度高密度培養する。
【0024】
長期にわたる凝集体の培養あるいは高密度培養を続けると、線維芽細胞様の細胞に対し上皮細胞様の細胞が多く見られるようになる。もし線維芽細胞様の細胞が残存している場合には、1〜10μg/mL、好ましくは4μg/mL程度のモノヨード酢酸で3〜5時間程度処理することによって線維芽細胞様の細胞のみを死滅させることができる。上皮細胞様の細胞の増殖が観察されれば、その後は、細胞培養用の培養容器にて継代し、解析に用いたり、使用時まで凍結保存したりする。
【0025】
長期にわたる凝集体の培養あるいは高密度培養を続けることによって得られる細胞、あるいはCat No. CS-ABI-519等の市販のヒト小腸上皮細胞をEGFを含有する培養液で継代培養することによって得られる細胞が腸管上皮細胞あるいは腸管幹/前駆細胞であるかの判断は、それらの細胞におけるマーカー蛋白質の発現の有無を調べることによって行うことができる。本明細書中、「マーカー」とは特にことわりのない限り、腸管上皮細胞あるいは腸管幹/前駆細胞に特有な発現様式を示す一連の蛋白質又はそれをコードする遺伝子から構成される群の各々を意味する。かかるマーカー蛋白質は哺乳動物の種類等によってそのアミノ酸配列が異なる場合があるが、本発明においてはその対象となる細胞における発現様式が同じである限り、そのような蛋白質もマーカー蛋白質として使用することができる。上皮細胞のマーカー蛋白質としては、E−カドヘリン(E−cadherin)やZo−1(Laprise P, et al., J Biol Chem. 2004 Mar 12; 279(11):10157-66. Epub 2003 Dec 29.;Escaffit F, et al., Exp Cell Res. 2005 Jan 15;302(2):206-20.)、腸上皮細胞のマーカー蛋白質としては、ビリン(Villin)(Maunoury, R. et al., EMBO J. 7, 3321-3329 (1988).)が挙げられる。腸管幹/前駆細胞のマーカー蛋白質としては、Musashi−1(Potten CS, et al., Differentiation. 2003 Jan;71(1):28-41.)、Math−1(Yang Q, et al., Science 294:2155-2158)、Neurogenin3(Ngn3、Jenny M, et al., EMBO J. 2002 Dec 2;21(23):6338-47.)が挙げられる。
【0026】
E−カドヘリンは、上皮系の細胞上に発現しているカルシウム依存性の細胞間接着分子であり、それ同士が結合するホモフィリックな接着により上皮組織の形成、維持に重要な働きをする。Zo−1は、上皮細胞や内皮細胞に必須である細胞間接着装置タイトジャンクション(TJ)の裏打ち蛋白質として知られており、接着装置の形成やそこを介して細胞増殖の調節に関与していると考えられている蛋白質である。ビリンは、小腸上皮細胞の微絨毛の構成成分であり、フィブリンとともにアクチンを束ねる働きを有している。ビリンは微絨毛にのみ見出される。Musashi−1は、神経系前駆細胞によって発現されるRNA結合蛋白質であり、腸管の幹/前駆細胞にも発現しているという報告がある。Math−1は、bHLH型の転写因子であり、マウス腸管における分泌細胞系への分化に必要であることが知られ、腸管幹/前駆細胞に発現している。同様にbHLH型転写因子であるNgn3は膵内分泌細胞、胃腸管組織の発生に重要であり、腸管幹/前駆細胞に発現している。
【0027】
本発明において、腸管上皮細胞又は腸管幹/前駆細胞であることの確認は、これらのマーカー蛋白質及び/又はそれをコードする遺伝子の発現を、各マーカー蛋白質又はそれをコードする遺伝子に特異的親和性を有する物質を用いて解析する工程を含むことによって行うことができる。本明細書中、「用いて」という用語について、その方法は特に限定されず、具体的には、例えばマーカー蛋白質に特異的親和性を有する物質を用いる場合であれば該マーカー蛋白質の抗体との抗原抗体反応を利用する方法が挙げられ、又、マーカー蛋白質をコードする遺伝子に特異的親和性を有する物質を用いる場合であればハイブリダイゼーション反応を利用する方法が挙げられる。
【0028】
マーカー蛋白質に特異的親和性を有する物質としては例えば当該蛋白質に特異的親和性を有する抗体又はその断片が挙げられ、その特異的親和性とは抗原・抗体反応により該蛋白質を特異的に認識し、結合する能力のことである。該抗体又はその断片は、当該蛋白質と特異的に結合可能なものであれば特に限定されず、ポリクローナル抗体、モノクローナル抗体及びそれらの機能的断片のいずれであってもよい。これらの抗体あるいはその機能的断片は、通常当分野で行なわれている方法によって製せられる。例えばポリクローナル抗体を用いる場合であれば、該蛋白質をマウスやウサギといった動物の背部皮下あるいは腹腔内あるいは静脈等に注射して免疫し、抗体価が上昇するのを待った後に抗血清を採取する方法が挙げられ、またモノクローナル抗体を用いる場合であれば、常法に従いハイブリドーマを作製して、その分泌液を採取する方法が挙げられる。抗体断片を製造する方法としてはクローニングした抗体遺伝子断片を微生物等に発現させる方法がよく用いられている。当該抗体、抗体断片等の純度は、当該蛋白質に対する特異的親和性を保持している限り、特に限定されない。これらの抗体又はその断片は、蛍光物質、酵素やラジオアイソトープ等で標識されていてもよい。
さらに、これらは市販されているものを用いても良い。
【0029】
マーカー蛋白質をコードする遺伝子に特異的親和性を有する物質としては、例えば当該遺伝子に特異的親和性を有するオリゴ又はポリヌクレオチドプローブ(以下、便宜上単にプローブともいう)、ならびにオリゴ又はポリヌクレオチドプライマー対(以下、便宜上単にプライマー対ともいう)が挙げられ、その特異的親和性とは、目的の遺伝子にのみハイブリダイズする性質を意味し、従って当該遺伝子の全部もしくは一部に完全相補的なものか、もしくは上記性質を満たす範囲で1乃至数個のミスマッチを含んでいても良い。該プローブ、プライマー対は、当該遺伝子に特異的親和性を有するものであれば特に限定されないが、例えば当該遺伝子の塩基配列の全部もしくは一部、ならびにそれらの相補配列を含むオリゴ又はポリヌクレオチド等が挙げられ、検出すべき遺伝子の形態に応じて適宜選択する。当該オリゴ又はポリヌクレオチドは当該遺伝子に対する特異的親和性を有している限りはその由来は特に限定されず、合成されたものであっても、当該遺伝子から必要な部分を切り出し、通常行なわれる方法によって精製されたものであってもよい。これらのオリゴ又はポリヌクレオチドは、蛍光物質、酵素やラジオアイソトープ等で標識されていてもよい。
【0030】
又、腸管幹/前駆細胞ではβ−カテニンの転写活性がみられる(He XC, et al., Nat Genet. 2004 Oct;36(10):1117-21.)ので、この活性を指標に幹/前駆細胞であることを確認することもできる。例えば、TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出する方法が利用できる。
【0031】
本発明では、腸管細胞の凝集体(あるいは高密度培養した細胞)をEGFを含有する培養液中で培養することによって、腸管幹/前駆細胞を選択的に増殖させることができ、すなわち該幹/前駆細胞を効率よく取得することができる。さらに該幹/前駆細胞の自己複製能及び多分化能は長期間安定に維持される。本発明の方法により得られた腸管幹/前駆細胞が自己複製能を維持していることは、細胞数の計測、光学顕微鏡下での観察等、細胞増殖を測定し得る種々の公知技術を用いて、培養容器中での腸管幹/前駆細胞の増殖を観察することによって容易に判断することができる。腸管幹/前駆細胞が多分化能を維持していることは、該幹/前駆細胞を分化誘導した場合に腸上皮組織を構築する複数種の細胞が得られるかどうかで判断することができる。腸上皮組織を構築する細胞としては、腸内分泌細胞(ガストリン分泌細胞、セロトニン分泌細胞、クロモグラニンA分泌細胞、セクレチン分泌細胞、ソマトスタチン分泌細胞、GIP(gastric inhibitory polypeptide)分泌細胞、CCK(cholecystokinin)分泌細胞等)及び粘液分泌細胞(ゴブレット細胞、パネート細胞等)、栄養素の吸収作用を担う腸細胞が挙げられる。各細胞に特異的に発現している成分を該成分に特異的な親和性を有する物質を用いて検出することによって、あるいは分泌成分を直接解析することによって目的の細胞であるか否かを判定することができる。「特異的な親和性を有する物質」としては、各分泌成分に特異的親和性を有する抗体又はその断片や、各分泌成分をコードする遺伝子に特異的親和性を有するオリゴ又はポリヌクレオチドプローブが挙げられる。「抗体又はその断片」及び「オリゴ又はポリヌクレオチドプローブ」の詳細は上記に準じる。
ゴブレット細胞に関しては、PAS染色やアルシアンブルー染色等の染色法によっても判定することができる。
【0032】
腸管幹/前駆細胞の、腸管上皮組織を構築する細胞への分化誘導は、培養液中からEGFを除去することによって簡便に実施することができる。具体的にはEGFを含有する培養液をEGFを含有しない培養液と培地交換することによって行うことができる。培地交換しておよそ1週間程度で細胞の分化誘導を確認することができるが、分化誘導に要する期間は、細胞の状態や、分化する細胞のタイプ等によって変動し得る。
【実施例】
【0033】
以下、実施例にそって本発明をさらに詳細に説明するが、これら実施例は本発明の範囲を何ら限定するものではない。また、本発明において使用する試薬や装置、材料は特に言及されない限り、商業的に入手可能である。
【0034】
実施例1:胎児腸管由来の腸管幹/前駆細胞の取得
胎生14.5日のマウス胎児から腸管を取り出し、メスで細かく切り刻み腸組織片とした。得られた腸組織片を、1mM EGTAの入ったHank’s溶液に入れ、37℃のウォーターバスで10分間振とうした後、遠心分離によってPBSで1回洗浄した。遠心分離した後、腸組織片を、1mg/mlのコラゲナーゼ溶液(5mM CaCl2の入ったHank’s溶液に溶解したもの)を加え、37℃のウォーターバスで15〜20分間振とうした。軽くピペッティングした後にメッシュを通して未消化組織を除いて細胞分散液を得た(マウス胎児由来の腸管細胞)。
調製したマウス胎児由来の腸管細胞を2〜4×105細胞/ウェルの濃度で低接着性の96ウェル丸底ディッシュ(Low Cell Binding Plates,cat no:145399;Nunc)に播種し、ダルベッコ改変イーグル培地とF−12培地とを1:1の割合で含有する培養液〔10%ウシ胎仔血清、1μg/mL インスリン、1×10−7mol/L デキサメサゾン、10mmol/L ニコチンアミド、2mmol/L L−グルタミン、50μmmol/L β−メルカプトエタノール、ペニシリン/ストレプトマイシン(100U/mL(ペニシリン)+100μg/mL(ストレプトマイシン))、20ng/mL EGF(Sigma,E9644)を含む〕中で、37℃、5%CO2存在下、インキュベーター内で培養した。以後、EGFを含有するこの培地をEGF(+)培地、あるいは腸管幹/前駆細胞培養用培地とも称する。
2日後、96ウェル丸底ディッシュに播種したマウス胎児腸管細胞が集合して細胞塊を形成した(図1a)。その後、形成した細胞塊を低接着性の6cmディッシュ等に移したりしながら、さらに5日間程度培養後、それら細胞塊をペトリディッシュ(BD,35−1007)に移し、上記EGF(+)培地中、37℃、5%CO2存在下、インキュベーター内で培養を続けた(図1b)。ここで用いたペトリディッシュは、接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュである。一方低接着性ディッシュを用いず、細胞分散液をすぐに接着性のペトリディッシュに播種し、上記EGF(+)培地中で培養した場合には線維芽細胞様の細胞のみディッシュに残った(図1c)。
凝集体を普通のペトリディッシュに移して3ヶ月程度培養を続けると、線維芽細胞様の細胞に対し、上皮細胞様の細胞が多く見られるようになった。もし線維芽細胞様の細胞が残存している場合は、モノヨード酢酸(4μg/mL)で3〜5時間程度処理することで線維芽細胞様の細胞を死滅させることができる。
得られた上皮細胞様の細胞は、細胞培養用ディッシュにて継代し(図2)、解析(後述する)や凍結保存に用いた。
又、後述の実施例からも明らかなように、この上皮細胞様の細胞は、自己複製能、多分化能を有する幹/前駆細胞を含んでいることから、便宜上、FIP(Fetal Intestine progenitor)細胞とも、以後、称する。
【0035】
実施例2:成体腸管由来の腸管幹/前駆細胞の取得
成体マウスから取り出した腸管の内部を、23Gの針を使ってPBSで洗浄し、その後、腸管にハサミを入れて管を開き、シート状にし、さらにハサミで5mm四方程度に切り分けた(腸組織片)。得られた腸組織片を用いて実施例1と同様にして、EGTA、コラゲナーゼ及びピペッティング処理によって細胞分散液を得た(成体マウス腸管細胞)。
成体由来の腸管細胞は、細胞塊を形成しにくいので、単層で凝集させて培養した。すなわち、直径6cmのペトリディッシュ(接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ)に1〜5×106細胞を播種して、高密度培養を行った。実施例1と同組成の培養液を用い37℃、5%CO2存在下、インキュベーター内で培養した。
高密度培養を8ヶ月程度続けると、線維芽細胞様の細胞に対し、上皮細胞様の細胞が多く見られるようになった。もし線維芽細胞様の細胞が残存している場合は、モノヨード酢酸(4μg/mL)で3〜5時間程度処理することで線維芽細胞様の細胞を死滅させることができる。
得られた上皮細胞様の細胞は、細胞培養用ディッシュにて継代し、解析や凍結保存に用いた。
【0036】
実施例3:増殖能
FIP細胞の増殖能について調べた。FIP細胞を、種々の濃度(0ng/mL〜80ng/mL)でEGFを含有する培養液中で培養した。培養液は、EGFの濃度が異なること以外は、実施例1で用いた培養液と同じ組成である。EGFを含まない培養液中で2日間培養した細胞(図3a)、EGFを20ng/mLの濃度で含む培養液中で2日間培養した細胞(図3b)を示す。又、12ウェルの培養プレートに播種し、各種濃度でEGFを含む培養液中で1日間培養した細胞をトリプシン−EDTAではがしてその数を血球計算板により計測した(図3c)。
腸管細胞由来の上皮細胞様の細胞の増殖がEGF依存性であることが示された。
【0037】
実施例4:増殖とアポトーシス
FIP細胞をEGF(+)培地(EGF濃度20ng/mL)中、細胞培養用ディッシュ上で2日間培養した(図4a)。同じ状態のものを2セット用意した。EGF存在下で2日間培養した後、一方は、同じEGF(+)培地で培地交換してさらに1日間培養し(図4b)、もう一方はEGFを含まない培養液に培地交換してさらに1日間培養した(図4c)。EGF存在下では細胞は増殖を続けたが、EGF非存在下では細胞の増殖は抑えられ、死ぬ細胞が確認された。一連の過程における細胞の増殖能の変化を、BrdU免疫染色法により確認した。核染色剤であるDAPIを用いて二重染色した(図5a〜f)。
EGF存在下で2日間培養した後、一方は、EGFを含まない培養液に培地交換してさらに1日間培養し、もう一方は同じEGF(+)培地で培地交換してさらに1日間培養した。その後、BrdU(10μM)(Sigma,B9285)を培地に添加し、1.5時間経過後にBrdUを取り込んだ増殖細胞の割合をフローサイトメトリーおよび免疫染色にて解析した。フローサイトメトリー解析は、BrdU Flow Kit (BD,552598)のマニュアルに沿って細胞を標識後、FACS Aria(BD)を用いて行った。この際、ゲートの設定はネガティブコントロールを指標にして行った。BrdUの免疫染色は、BrdU Flow Kitの試薬と抗BrdU抗体(×50)(BD,347580)、Cy3をコンジュゲートしたロバ抗マウスIgG抗体(×200)(Jackson ImmunoResearch Laboratories,cat no.715−165−150)、およびDAPI(1μg/mL)(Sigma,D9542)を用いて行い、蛍光顕微鏡で観察した。EGF非存在下で培養した細胞についてはBrdUの取り込みが顕著に減少しており、増殖能が低下したことが明らかとなった(図5a〜f)。
また、EGFを培養液から除くと、死ぬ細胞が確認されたことから、EGF非存在下でのアポトーシス誘導について調べた(図5g〜h)。
BrdUの解析時と同様に細胞を培養した。細胞をディッシュから回収した後、冷PBSにて細胞を1回洗い、アネキシンV binding buffer(1×)(BD,556454)にて細胞懸濁液を調製した。そこへ7−AAD(5μL;BD,559925)とAPC−conjugated アネキシンV(5μL;BD,550474)を添加し、室温にて15分間反応させた(遮光)。染色した細胞はFACS Aria(BD)を用いて解析した。ゲートの設定はネガティブコントロールを指標にして行った。アネキシンVはリン脂質結合性タンパク質である。アポトーシスを起こした細胞は細胞膜表面にホスファチジルセリンを露出するようになり、そこにアネキシンVが結合する。従ってアネキシンVはアポトーシス細胞に強い親和性を有する。EGF非存在下ではアネキシンVが結合する細胞が顕著に増加しており、アポトーシス誘導が起こっていることが示された(図5g〜h)。
FIP細胞は、EGFの存在下、活発に分裂増殖したが、EGFを含まない培養条件では増殖活性が著しく減少し、アポトーシスを起こす細胞が増加した。
【0038】
実施例5:マーカー蛋白質の発現
FIP細胞における種々のマーカー蛋白質の発現について調べた。各マーカー蛋白質の発現は、各蛋白質の抗体をそれぞれ用いて免疫染色法により調べた。尚、細胞の存在を示す為に、核染色剤であるDAPIで二重染色した。
細胞をPBSで洗浄し、4%パラホルムアルデヒド(PFA)(PBS中)で5分間、室温で固定した。続いて、25%アセトン(メタノール中)で1分間、室温で処理した。固定後、細胞を0.05%ポリオキシエチレン(20)ソルビタンモノラウレート(Tween 20)(Wako)を含有するPBS(PBS−Tween)で洗浄し、続いて、0.2%Triton X−100(Sigma)で1時間、室温で処理した。非特異的な結合は、10%のロバ血清(Sigma,D9663)で1時間、室温で処理することによりブロッキングした。固定した細胞を1次抗体と湿室内で16時間、4℃でインキュベートした。細胞を、PBS−Tween20で洗浄し、ブロッキングした後、さらに2次抗体と4時間、4℃でインキュベートした。室温でDAPIと5分間インキュベートし、洗浄後、細胞を蛍光顕微鏡で観察した。
用いた抗体は以下の通り。
(1次抗体)
抗E−カドヘリン抗体:BD,610181(2次抗体、マウス用);×100
抗Zo−1抗体:Zymed,61−7300(2次抗体、ウサギ用);×100
抗ビリン抗体:Santa Cruz,sc−7672(2次抗体,ヤギ用);×10
抗Musashi−1抗体: Chemicon,AB5977(2次抗体、ウサギ用);×100
(2次抗体)
マウス用:Alexa 488が結合したロバ抗マウスIgG(Molecular Probes,A21202);×200
ウサギ用:Alexa 488が結合したロバ抗ウサギIgG(Molecular Probes,A21206);×200
ヤギ用:Alexa 488が結合したロバ抗ヤギIgG(Molecular Probes,A11055);×200
FIP細胞は、全細胞において上皮細胞のマーカーであるE−カドヘリンやZo−1、及び、腸上皮細胞のマーカーであるビリンを発現していた。また、一部の細胞は腸管幹/前駆細胞マーカーと考えられているMusashi−1を発現していた(図6)。
【0039】
実施例6:βカテニン転写活性化
TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)をリポフェクトアミン2000(Invitrogen,11668)を用いて細胞に導入し、X−gal染色により陽性細胞を検出した。具体的には以下の手順で行った。
(1) 細胞をPBSで洗浄(×2)
(2) 固定液(2%PFA/0.2%グルタルアルデヒド(GA)/0.05%NP−40/PBS)を添加(6ウェルプレートの1ウェルあたり1.5〜2mL)
(3) 1分間室温で放置
(4) PBSで洗浄(×2)
(5) X−gal溶液を添加(6ウェルプレートの1ウェルあたり1.5〜2mL)
(6) 37℃のインキュベーター中で放置(少なくとも3時間〜一晩)
(7) PBSで洗浄(×2)することで反応を停止させる
(8) 観察(写真撮影)、PBS中4℃で保存
(固定液の調製)
以下の割合で各成分を混合して固定液を調製した。
20% PFA/PBS 1mL(−20℃保存)
25% GA 80μL(−20℃保存)
5% NP−40 80μL(室温保存)
PBS 9mL(室温保存)
(X−gal溶液の調製)
以下の割合で各成分を混合してX−gal溶液を調製した。
500mM K3Fe(CN)6 50μl(室温保存、遮光保存)
500mM K4Fe(CN)6 50μl(室温保存、遮光保存)
1M MgCl2 10μl(室温保存)
20mg/ml X−gal 250μl(−20℃保存、遮光保存)
PBS 4.7ml(室温保存)
X−gal陽性細胞が検出され、βカテニンの転写活性化が確認された(図7)。
【0040】
実施例7:腸上皮組織を構築する細胞への分化誘導
FIP細胞をコンフルエントな状態になるまで培養した。コンフルエントになった状態からEGFを除くと、アポトーシスを起こす細胞の中で生き残る細胞が存在した。これら生き残った細胞をEGF非存在下で培養し続けると分化した腸上皮様細胞の特徴を示す細胞が出現してきた(図8)。
この状態の細胞をPAS染色すると、EGF非存在下で培養し続けた細胞において赤紫色に染色される細胞が多数出現した(図9)。PAS染色は、糖質に含まれる1,2グリコール基群を過ヨウ素酸で酸化し、生じた2分子のアルデヒド基がシッフ試薬1分子と結合して、赤紫色の化合物を形成する2つの反応を利用したものであり、グリコーゲン、中性粘液多糖類、糖蛋白質、粘液蛋白質、糖脂質等の検出に用いられる。腸上皮組織においては、粘液分泌細胞の1種であるゴブレット細胞を染め分けることが知られている。具体的には以下の手順で行った。尚、各試薬については当分野で通常PAS染色を行う際に用いられている試薬を使用することができる。
(1) 細胞をPBSで洗浄(×1)
(2) 0.5%過ヨウ素酸で室温7分間処理
(3) ミリQ水で洗浄(×5)
(4) シッフ試薬で室温7〜10分間処理
(5) 亜硫酸水で処理(室温1分間処理を3回繰り返す)
(6) ミリQ水で洗浄(×5)
(7) ヘマトキシレンで処理(室温で1分間)
(8) ミリQ水で洗浄(×5)
又、各腸上皮組織において分泌、発現している蛋白質に対する抗体を用いて免疫染色を行い、該蛋白質の発現状況を解析した。用いた各蛋白質の抗体は以下の通り。尚、細胞の存在を示す為に、核染色剤であるDAPIで二重染色した。
(1次抗体)
抗セロトニン抗体:Immunostar,20080(2次抗体、ウサギ用);×10
抗クロモグラニンA抗体:Santa Cruz,sc−1488(2次抗体、ヤギ用);×10
抗ガストリン抗体:Santa Cruz,sc−7783(2次抗体、ヤギ用);×10
抗セクレチン抗体:Chemicon,AB981(2次抗体、ウサギ用);×100
抗ソマトスタチン抗体:AFFINITI,SA1268(2次抗体、ウサギ用);×10
抗GIP抗体:Chemicon,AB953(2次抗体、ウサギ用);×10
抗リゾチーム抗体:Dako,A0099(2次抗体、ウサギ用);×100
抗シナプトフィジン抗体:Dako,A0010(2次抗体,ウサギ用);×10
(2次抗体)
ウサギ用:Alexa 488が結合したロバ抗ウサギIgG(Molecular Probes,A21206);×200
ヤギ用:Alexa 488が結合したロバ抗ヤギIgG(Molecular Probes,A11055);×200
結果、EGF非存在下で培養すると、腸内分泌細胞に特徴的なセロトニン、クロモグラニンA、ガストリン、セクレチン、ソマトスタチン、GIPを発現している細胞が検出され、粘液分泌細胞の1種であるパネート細胞に特徴的なリゾチーム及びシナプトフィジンを発現している細胞が検出された。これらの結果より、FIP細胞の培養液からEGFを除去することにより腸内分泌細胞への分化が誘導されたことがわかる。
【0041】
実施例8:RT−PCRによる解析
上記した実施例より、FIP細胞には、腸管幹/前駆細胞が含まれていることが示され、さらにEGF存在下で、該幹/前駆細胞は増殖を続け、EGFを培養液中から除去することにより、細胞はアポトーシスを引き起こし、又、種々の腸上皮組織を構築する細胞へと分化することがわかった(模式図を図10に示す)。
FIP細胞、及び各発生段階(胎生14.5日目、新生児、成体)にあるマウスの腸組織を用いて、腸管の細胞分化に関与すると考えられる遺伝子の発現プロファイルをRT−PCRによって解析した。
FIP細胞をEGF(+)培地で培養後、コンフルエントになった時点でEGFを含まない培地に培地交換し、その後1週間培養した。その細胞からRNAを抽出(RNeasy Mini Kit;QIAGEN)し、逆転写酵素反応(SuperScript III)によりcDNAを得た。腸組織からのRNA抽出では、ホモジナイザー(ポリトロン)を用いて組織を破砕後に同様の方法で抽出と逆転写酵素反応を行った。続いて、それらのcDNAを用いてPCRを行い、各種分化マーカーの発現を解析した。
目的とするマーカーならびにそのプライマーを表1に示す。
【0042】
【表1】
【0043】
PCRサイクルは、95℃(4分間)→[94℃(1分間)→56℃(1分間)→72℃(1分間)]×45サイクル→72℃(10分間)である。PCRにより産生された核酸は、2%アガロースゲルにて展開し、解析した。結果を図11に示す。
【0044】
実施例9:単一細胞レベルでの解析
上記した実施例3〜7での実験はheterogenousな細胞集団での解析であったので、腸管幹/前駆細胞の解析を厳密に行うためには、単一細胞レベルでの解析が必要であると考えた。そこで、フローサイトメトリーを用いてFIP細胞をクローンソーティングし、96ウェルディッシュの各ウェルにて単一細胞培養を試みた。増殖してきたクローンのひとつを用いて同様にして、増殖能、マーカー蛋白質の発現及び分化誘導について調べた。
その結果、単一コロニーから増殖させた細胞についても、E−カドヘリンやZo−1、ビリンを全細胞で発現し、また一部のコロニーについてはMusashi−1を発現していた。さらにEGF依存的な増殖活性や細胞死抑制を示し、さらに、EGF非存在下で複数の腸上皮細胞マーカーを発現するようになった。このように、単一細胞レベルの解析においても増殖能や多分化能が示されたことで、FIP細胞には腸管幹/前駆細胞が含まれることが確認された。
以上の結果より、FIP細胞集団には、自己複製能及び多分化能を有する幹/前駆細胞が含まれ、長期間安定に維持され得ることが示された。本願発明の方法を用いることによって腸管上皮細胞の幹/前駆細胞を選択的に増殖させることが可能となった。かかる選択的増殖によって、効率的に腸管幹/前駆細胞を取得・培養することができる。
【0045】
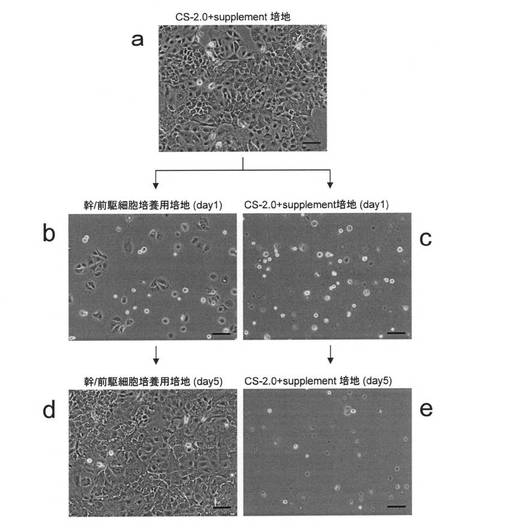
実施例10:ヒト小腸上皮細胞からの腸管幹/前駆細胞の取得
本実施例では、ACBRIにより分離され、日本では大日本住友製薬株式会社から供給されているヒト小腸上皮細胞(Cat No. CS-ABI-519)を用いた。この細胞を添付のデータシートに記載された方法に従って培養ディッシュに播種し培養した(図12a)。培養ディッシュは、タイプIVコラーゲンコートされたディッシュを用いた。培養液として、データシートに記載され、添付されている培地、すなわちCS−2.0基礎培地に添加剤を加えたもの(以下、CS-2.0 + supplement)を用いた。当該処理は、20ng/mLのEGF存在下で行なった。
CS-2.0 + supplementを用いて培養したところ継代を2回行なった時点で細胞は培養ディッシュに接着せず、全て死滅してしまった(図12c,e)。
そこで、実施例1で調製した、マウス胎児由来の腸管細胞を培養するのに用いたEGFを含有する培地(腸管幹/前駆細胞培養用培地)を用いて同様にして継代培養したところ、継代を重ねても培養ディッシュに接着し、増殖維持することができる細胞が得られた(図12b,d)。
これらの細胞は、購入したヒト上皮細胞集団中に含まれる腸管幹/前駆細胞であると考えられたので、便宜上、hAIP(human adult intestine progenitor)細胞とも、以後、称する。
【0046】
実施例11:マーカータンパク質の発現とβカテニン転写活性化
hAIP細胞について、実施例5と同様にしてマーカータンパク質の発現について調べた。
【0047】
hAIP細胞は、全細胞において上皮細胞のマーカーであるE−カドヘリンやZo−1、及び、腸上皮細胞のマーカーであるビリンを発現していた。また、一部の細胞は腸管幹/前駆細胞マーカーと考えられているMusashi−1を発現していた(図13、上段)。
また、hAIP細胞について、実施例6と同様にして、βカテニンの転写活性化の有無を確認した。X−gal陽性細胞が検出され、βカテニンの転写活性化が確認された(図13、下段)。
【0048】
実施例12:増殖能
実施例3と同様にしてhAIP細胞の増殖能について調べた。実施例1で用いたEGF(+)培地、及びEGFを含まない以外は同じ組成のEGF(−)培地でそれぞれ4日間培養した。EGFの存在下ではhAIP細胞は活発に分裂増殖するが、EGFを含まない培養条件では増殖活性が低下し、アポトーシスを起こす細胞が増加した(図14)。
この結果から、EGFはhAIP細胞の自己複製を促進し、細胞死を抑制することで、培養下における維持を可能にしていることがわかる。
【0049】
実施例13:増殖とアポトーシス
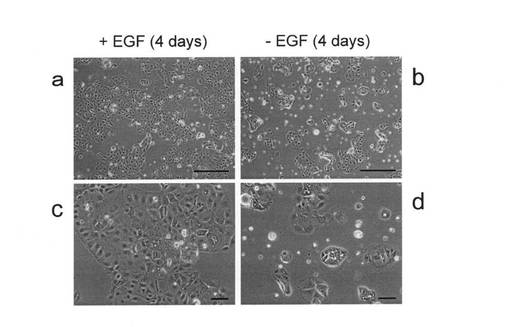
hAIP細胞をEGF(+)培地(EGF濃度20ng/mL)中、4日間培養した。同じ状態のものを2セット用意した。EGF存在下で4日間培養した後、一方は、同じEGF(+)培地で培地交換してさらに1日間培養し(図15A,左)、もう一方はEGFを含まない培養液に培地交換してさらに1日間培養した(図15A,右)。EGF存在下では細胞は増殖を続けたが、EGF非存在下では細胞の増殖は抑えられ、死ぬ細胞が確認された。一連の過程における細胞の増殖能の変化を、BrdU免疫染色法により確認した。核染色剤であるDAPIを用いて二重染色した(図15A)。
EGF存在下で4日間培養した後、一方は、EGFを含まない培養液に培地交換してさらに1日間培養し、もう一方は同じEGF(+)培地で培地交換してさらに1日間培養した。その後、BrdU(10μM)(Sigma,B9285)を培地に添加し、1.5時間経過後にBrdUを取り込んだ増殖細胞の割合をフローサイトメトリーおよび免疫染色にて解析した。フローサイトメトリー解析は、実施例4で記載した方法に従って行なった(図15B,左)。BrdUの解析時と同様に細胞を培養し、アネキシンVとの結合を実施例4で記載した方法に従って解析した。アネキシンVが結合する細胞をアポトーシスを起こしている細胞とみなし、その割合を調べた(図15B,右)。EGFを培養液中から除くことによって、BrdUの取り込みが減少し、すなわち増殖能が低下することがわかる。さらに、EGFを培養液中から除くことによって、アポトーシスを起こす細胞が増加した。
hAIP細胞の増殖あるいはアポトーシスに及ぼすEGFの影響を確認するために、EGFを含有する培養液中に、EGFシグナル伝達系を阻害するAG1478、PD98059、あるいはLY294002を添加した培養液を用いて同様にBrdUの取り込みとアポトーシス誘導について調べた。
PD98059(2’−アミノ−3’−メトキシフラボン)は、MEK1(MAPキナーゼ・キナーゼ)に対する選択的で細胞透過性の阻害剤であり、MEK1の活性化を阻害する結果、続いて起こるMAPK(ERK1/2)のリン酸化−活性化を阻害する。使用濃度:100μM。
LY294002([2−(4−モルホリニル)−8−フェニル−4H−1−ベンゾピラン−4−オン]はホスファチジルイノシトール3−キナーゼ(PI3−キナーゼ)に対する強力で特異的な細胞透過性阻害剤である。使用濃度:100μM
【0050】
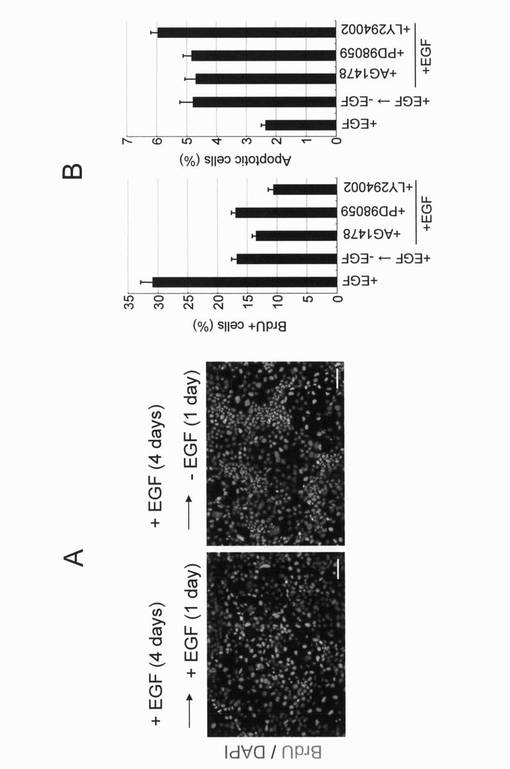
実施例14:腸上皮組織を構築する細胞への分化誘導
hAIP細胞は上記した培養条件下で自然に分化誘導され、腸上皮様細胞の特徴を示す細胞が出現してくる。この状態の細胞を実施例7に記載する方法に従ってPAS染色すると、赤紫色に染色される細胞が多数出現したことから、腸上皮組織であるゴブレット細胞の存在を確認することができた(図16B)。
さらに、実施例8と同様な手法で、腸上皮組織を構築する細胞への分化誘導をRT−PCR解析により調べた。誘導される腸上皮組織の種類は、図10に示されている。
hAIP細胞、及びヒト成体小腸からの組織を用いて、腸管の細胞分化に関与すると考えられる遺伝子の発現プロファイルをRT−PCRによって解析した。目的とするマーカー及びそのためのプライマーは、下記表2に示すヒト用のものを用いた。
【0051】
【表2】
【0052】
結果を図16Aに示す。
hAIP細胞培養中には、腸上皮組織を構築する複数の腸内分泌細胞や粘液分泌細胞の特徴を示す細胞が観察された。従ってhAIP細胞は、培養下で増殖し続けるのと同時に、腸を構成する様々な細胞に分化する多分化能を有することが判明した。
【配列表フリーテキスト】
【0053】
配列番号1:ビリン(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号2:ビリン(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号3:Lfabp(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号4:Lfabp(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号5:クロモグラニンA(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号6:クロモグラニンA(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号7:Trefoil factor−3(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号8:Trefoil factor−3(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号9:リゾチーム(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号10:リゾチーム(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号11:シナプトフィジン(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号12:シナプトフィジン(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号13:Musashi−1(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号14:Musashi−1(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号15:Math−1(マウス)を検出する為のPCR用プライマー(フォワード)である。
配列番号16:Math−1(マウス)を検出する為のPCR用プライマー(リバース)である。
配列番号17:ビリン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号18:ビリン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号19:Lfabp(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号20:Lfabp(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号21:シュクラーゼ−イソマルターゼ(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号22:シュクラーゼ−イソマルターゼ(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号23:セクレチン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号24:セクレチン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号25:GIP(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号26:GIP(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号27:ガストリン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号28:ガストリン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号29:クロモグラニンA(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号30:クロモグラニンA(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号31:CCK(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号32:CCK(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号33:ソマトスタチン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号34:ソマトスタチン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号35:Trefoil factor−3(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号36:Trefoil factor−3(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号37:リゾチーム(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号38:リゾチーム(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号39:シナプトフィジン(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号40:シナプトフィジン(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号41:Musashi−1(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号42:Musashi−1(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号43:Math−1(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号44:Math−1(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号45:Ngn3(Neurogenin−3)(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号46:Ngn3(Neurogenin−3)(ヒト)を検出する為のPCR用プライマー(リバース)である。
配列番号47:HPRT(ヒト)を検出する為のPCR用プライマー(フォワード)である。
配列番号48:HPRT(ヒト)を検出する為のPCR用プライマー(リバース)である。
【図面の簡単な説明】
【0054】
【図1】腸管細胞培養の簡単な手順を示す図である。aは、非接着性培養容器上で形成させた凝集体を示す図(位相差像)である。bは、形成させた凝集体を接着細胞用の処理を行っていない(しかし低接着性処理も行っていない)普通のペトリディッシュ上で培養した状態を示す図(位相差像)である。cは、凝集体を形成させず、腸管細胞を接着性培養容器上で培養した状態を示す図(位相差像)である。
【図2】一連の操作によって得られた腸管細胞由来の、上皮細胞様の細胞を継代培養して増殖させた状態を示す図(位相差像)である。
【図3】FIP細胞のEGF依存性の増殖能を示した図(位相差像)である。aはEGF非存在下で、bはEGF存在下でそれぞれ2日間培養した状態を示す。cは細胞をトリプシン−EDTAではがして血球計算板により計測した結果を示す。横軸は添加したEGFの濃度を、縦軸は細胞数を示す。
【図4】EGF存在下で培養していたFIP細胞をEGF非存在下又は引き続きEGF存在下で培養した場合の細胞の状態を示す図(位相差像)である。aはEGF存在下で2日間FIP細胞を培養した状態を示す。EGF存在下での2日間の培養後、さらにEGFを含む培養液中で1日間(b)、あるいはEGFを含まない培養液中で1日間(c)、それぞれ培養した。
【図5】EGF存在下で培養していたFIP細胞を引き続きEGF存在下で培養した場合又はEGF非存在下で培養した場合の細胞の状態を示す図である。a及びbは、それぞれの細胞のBrdU取り込みの様子をBrdU免疫染色法により調べた結果を表す図(蛍光顕微鏡像)である。c及びdは、それぞれの細胞をBrdU/DAPIの二重染色により染色した結果を表す図である(蛍光顕微鏡像)。e及びfは、EGF存在下、あるいは非存在下でのBrdUの取り込みをFACS解析した結果を表すグラフである。g及びhは、EGF存在下、あるいは非存在下でのアネキシンVの結合をFACS解析した結果を表すグラフである。
【図6】EGF存在下で培養を続けたFIP細胞における各種マーカー蛋白質(E−カドヘリン、Zo−1、ビリン及びMusashi−1)の発現を調べた結果を示す図である(蛍光顕微鏡像)。
【図7】TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出した結果を示す図である。
【図8】EGF存在下で培養していたFIP細胞(a,c)からEGFを除去し、さらにEGF非存在下で7日間培養した細胞(b,d)の状態を示す図(位相差像)である。
【図9】FIP細胞をEGF非存在下で培養し続けた細胞(腸上皮細胞様の細胞)をPAS染色した像である。aはコンフルエントな状態になったFIP細胞を示し、bはEGF非存在下で7日間培養した細胞を示す。
【図10】FIP細胞とEGFとの関係を模式的に示した図である。
【図11】FIP細胞、胎生14.5日のマウス腸組織、新生児腸組織、成体腸組織における種々の遺伝子発現プロファイルをRT−PCR法により調べた結果を示す図である。
【図12】ヒト小腸上皮細胞(Cat No. CS-ABI-519)をCS-2.0+supplement培地あるいは腸管幹/前駆細胞培養用の培地で継代培養した場合の細胞の様子を示す図である。aは、CS-2.0+supplement培地中に播種した継代1回目の細胞の様子を、b及びdは、腸管幹/前駆細胞培養用の培地で2回継代培養を行なった細胞の様子を示している。播種後1日目(b)と5日目(d)の様子である。c及びeは、CS-2.0+supplement培地で2回継代培養を行なった細胞の様子を示している。播種後1日目(c)と5日目(e)の様子である。位相差像である。
【図13】上段は、EGF存在下で培養を続けたhAIP細胞における各種マーカー蛋白質(E−カドヘリン、Zo−1、ビリン及びMusashi−1)の発現を調べた結果を示す図(蛍光顕微鏡像)である。下段は、TCF/b−catenin binding siteによってβガラクトシダーゼ遺伝子がドライブされるコンストラクト(TOPGAL)を細胞に導入し、X−gal染色により陽性細胞を検出した結果を示す図である。
【図14】hAIP細胞のEGF依存性の増殖能を示した図(位相差像)である。a及びcはEGF存在下で、b及びdはEGF非存在下でそれぞれ4日間培養した状態を示す。c及びdは、それぞれa及びbの高倍率像である。
【図15】hAIP細胞の増殖とアポトーシスに及ぼすEGFの影響を解析した結果を示す図である。図15AはEGF存在下で培養していたhAIP細胞を引き続きEGF存在下で培養した場合又はEGF非存在下で培養した場合の細胞の状態を示す図である。それぞれの細胞をBrdU/DAPIの二重染色により染色した結果を表す(蛍光顕微鏡像)。図15Bは、EGF存在下、あるいは非存在下、あるいは種々の阻害剤を用いた場合の、BrdUの取り込みをFACS解析した結果、あるいはアポトーシスを起こした細胞をFACS解析した結果を表すグラフである。
【図16】図16Aは、hAIP細胞、ヒト成体腸組織における種々の遺伝子発現プロファイルをRT−PCR法により調べた結果を示す図である。図16BはhAIP細胞をPAS染色した像である。
【特許請求の範囲】
【請求項1】
腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項2】
腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項3】
凝集体が腸管細胞を浮遊培養することによって得られるものである、請求項1又は2記載の方法。
【請求項4】
以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を浮遊培養することによって細胞塊(凝集体)を調製する工程
(3)腸管細胞の凝集体を、EGFを含有する培養液中で培養する工程
(4)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
【請求項5】
腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、請求項4記載の方法。
【請求項6】
腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項7】
腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項8】
以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を、EGFを含有する培養液中、高密度で培養する工程
(3)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
【請求項9】
腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、請求項8記載の方法。
【請求項10】
請求項1〜9のいずれか1項に記載の方法によって得られた腸管上皮細胞の幹/前駆細胞。
【請求項11】
腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項12】
腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項13】
凝集体が腸管細胞を浮遊培養することによって得られるものである、請求項11又は12記載の方法。
【請求項14】
腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項15】
腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項16】
腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法であって、該幹/前駆細胞の培養液からEGFを除去することを特徴とする方法。
【請求項17】
腸上皮組織を構築する細胞が、腸内分泌細胞又は粘液分泌細胞である、請求項16記載の方法。
【請求項18】
血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する、腸管上皮細胞の幹/前駆細胞培養用培地。
【請求項1】
腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項2】
腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項3】
凝集体が腸管細胞を浮遊培養することによって得られるものである、請求項1又は2記載の方法。
【請求項4】
以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を浮遊培養することによって細胞塊(凝集体)を調製する工程
(3)腸管細胞の凝集体を、EGFを含有する培養液中で培養する工程
(4)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
【請求項5】
腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、請求項4記載の方法。
【請求項6】
腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項7】
腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の取得方法。
【請求項8】
以下の工程を含む、腸管上皮細胞の幹/前駆細胞の取得方法。
(1)腸管から腸管細胞を採取する工程
(2)腸管細胞を、EGFを含有する培養液中、高密度で培養する工程
(3)腸管上皮細胞の幹/前駆細胞マーカーを発現している細胞を腸管上皮細胞の幹/前駆細胞として回収する工程
【請求項9】
腸管上皮細胞の幹/前駆細胞マーカーがMusashi−1である、請求項8記載の方法。
【請求項10】
請求項1〜9のいずれか1項に記載の方法によって得られた腸管上皮細胞の幹/前駆細胞。
【請求項11】
腸管細胞の凝集体を培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項12】
腸管細胞の凝集体を、EGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項13】
凝集体が腸管細胞を浮遊培養することによって得られるものである、請求項11又は12記載の方法。
【請求項14】
腸管細胞を、血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する培養液中で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項15】
腸管細胞を、EGFを含有する培養液中、高密度で培養することを含む、腸管上皮細胞の幹/前駆細胞の培養方法。
【請求項16】
腸管上皮細胞の幹/前駆細胞から腸上皮組織を構築する細胞への分化を誘導する方法であって、該幹/前駆細胞の培養液からEGFを除去することを特徴とする方法。
【請求項17】
腸上皮組織を構築する細胞が、腸内分泌細胞又は粘液分泌細胞である、請求項16記載の方法。
【請求項18】
血清、インスリン、デキサメサゾン、ニコチンアミド、L−グルタミン、β−メルカプトエタノール及びEGFを含有する、腸管上皮細胞の幹/前駆細胞培養用培地。
【図10】


【図1】
【図2】
【図3】

【図4】

【図5】

【図6】
【図7】

【図8】

【図9】

【図11】

【図12】
【図13】

【図14】
【図15】
【図16】
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2008−206510(P2008−206510A)
【公開日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願番号】特願2007−241536(P2007−241536)
【出願日】平成19年9月18日(2007.9.18)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成18年度、文部科学省、科学技術試験研究「再生医療実現化プロジェクト」産業活性化再生特別措置法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
【公開日】平成20年9月11日(2008.9.11)
【国際特許分類】
【出願日】平成19年9月18日(2007.9.18)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成18年度、文部科学省、科学技術試験研究「再生医療実現化プロジェクト」産業活性化再生特別措置法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
[ Back to top ]