
膨潤性の球状核
【課題】物性バラツキがなく容易に製造可能な膨潤性を有する球状核、及び精度が高く容易に薬物溶出制御が可能な膜破裂型の時限放出製剤の提供。
【解決手段】結晶セルロースとクロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上の膨潤剤との質量比が90:10〜30:70からなり、吸水による体積膨潤度が100%〜700%である真球度が0.7以上1以下の球状核、及び球状核の表面が薬物で被覆された球形素顆粒、球形素顆粒の表面が水不溶性物質で被覆された球形顆粒。
【解決手段】結晶セルロースとクロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上の膨潤剤との質量比が90:10〜30:70からなり、吸水による体積膨潤度が100%〜700%である真球度が0.7以上1以下の球状核、及び球状核の表面が薬物で被覆された球形素顆粒、球形素顆粒の表面が水不溶性物質で被覆された球形顆粒。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬品分野の時間薬物療法に用いられる膨潤性を有する球状核、該球状核を用いて製造される球状素顆粒および球状顆粒に関する。また、本発明は球状核の製造方法に関する。
【背景技術】
【0002】
多くの慢性疾病患において病態の周期的変化が起こることが知られている。これはヒトの生理機能の多くが体内時計によって支配され、周期的に変動することに基づくものである。このような背景から、症状が現れる時間帯、あるいは治療上必要な時間帯にのみ薬物の血中濃度を高めて薬効を発現させる、時間薬物療法が検討されてきた。なかでも、服用後所定の時間が経過した後に薬物を放出することにより、薬効の発現を必要な時間遅らせる機能を有する時限放出製剤は、時間薬物療法を通常の投薬法によって実現することが可能となる。放出開始時間を調整することで目的に応じた時限放出製剤を設計できる。
例えば、放出開始時間を数時間単位と調整すれば、小腸や大腸に薬物を作用させることが可能となり、放出開始時間を数分単位と調整すれば、胃などに薬物を作用させることが可能となり、さらに不快な味を有する薬物のマスキングとして用いることもできる。
この時限放出製剤の一形態である膜破裂型の時限放出製剤は、薬物と膨潤性物質を配合した錠剤や顆粒剤に水不溶性の皮膜を被覆したものである。この製剤は、生体に投与した際に水不溶性の皮膜を介して浸透してくる水分によって製剤中の膨潤性物質が膨張し、その膨張力によって皮膜が破れることにより初めて薬物が溶出する仕組みとなっている。
【0003】
特許文献1には、核粒子の周囲に膨潤剤と共に薬物を付着させ、それを水不溶性の被覆物質と添加剤の混合物で被覆した膜破裂型の持続性製剤が開示されている。この製剤の製造方法として、膨潤剤を粉噴霧して核粒子表面に付着させている。
特許文献2には、芯を含有する薬剤に崩壊剤を付着させ、さらに透過性の水不溶性ポリマーとグライダントの混合物で被覆した膜破裂型の遅延放出製剤が開示されている。
特許文献3には、薬物と崩壊剤を湿潤状態で球状化して得られた核に、水溶性の重合体と不溶性の重合体の混合物で被覆した膜破裂型の遅延放出製剤が開示されている。
特許文献4には、薬物と水膨潤性物質を湿潤状態で球状化して得られた核に、水不溶性高分子を含む皮膜で被覆した膜破裂型の時限放出製剤が開示されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開昭62−30709号公報
【特許文献2】特表2003−510346号公報
【特許文献3】特表2004−510812号公報
【特許文献4】特開2009−191034号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
特許文献1では、核粒子に膨潤剤を粉噴霧する方法が記載されているが、核粒子表面に粉末状の膨潤剤が不均一に付着する。そのため、外側の被覆膜が均一な厚みで被覆されず、その結果、得られる製剤の膜破裂制御が非常に難しいという問題があった。
特許文献2では、崩壊剤を有機溶剤中に懸濁させた液を用いて芯を含有する薬剤上にスプレイ(噴霧)しているが、有機溶媒は環境・コスト・残留などの様々な問題がある。加えて、一般的に、有機溶媒系から水溶液系あるいは水性懸濁液系へと転換が図られているが、崩壊剤を水溶液中に懸濁させると、吸水により体積膨張して系の粘度が高くなりスプレイすることが困難となる。また低粘度とするために系を希釈すると、作業効率が非常に悪くなってしまうという問題点が生じる。
さらに特許文献1及び2のように、核(芯)に対して薬物や膨潤剤等を付着させる方法では、最終的に得られる顆粒サイズが大きくなる問題があった。一般的に核として用いられるノンパレル103(粒径:355〜850μm/フロイント産業(株)製)では、得られる顆粒の粒径は最小でも500〜1000μm程度であり、それよりも小さい顆粒を得ることはできない。
【0006】
特許文献3及び4の核は薬物を含有するため、薬物の物性(吸湿性、凝集性、分解性など)の影響で、真球度が高く、かつ薬物含有量や粒径サイズの均一な核を安定に製造することが困難になるという問題があった。また各薬物の種類ごとに核の最適な製造条件が異なるため、薬物の種類を変える度に製造条件の最適化が必要となるという煩わしさがあった。
さらに特許文献3及び4のように、押し出し造粒を用いて顆粒を製造する場合は、真球度が高く、かつ小さい粒径の顆粒を得るのは難しいのが一般的である。特に、押し出しされにくい薬物や膨潤剤などを含む場合は、その配合量や種類が限定される問題があった。特許文献3では顆粒の粒径は500〜4000μmとの記載があり、特許文献4では180〜500μmとの記載がある。しかし実際に特許文献4の実施例には、小さくても420〜500μmの顆粒しか得られていない。
本発明の課題は、精度の高い時限放出製剤を提供すること、その時限放出製剤の核となる膨潤性を有する球状核を提供すること、精度の高い時限放出製剤が容易に得られることである。
【課題を解決するための手段】
【0007】
本発明者らは上記課題に対し鋭意検討した結果、膨潤剤と結晶セルロースがランダムに混在した状態で存在する球状核に、薬物を含有する層と、さらに水不溶性物質を含有する層を被覆することで、上記課題を解決できることを見出し、本発明を完成した。
すなわち、本発明は以下の通りである。
[1]結晶セルロースと膨潤剤を含む混合物からなる球状核。
[2]結晶セルロースと膨潤剤の質量比が90:10〜30:70であることを特徴とする上記[1]に記載の球状核。
[3]結晶セルロースと膨潤剤の質量比が60:40〜40:60であることを特徴とする上記[1]又は[2]に記載の球状核。
[4]吸水による体積膨潤度が100%〜700%であることを特徴とする上記[1]から[3]のいずれかに記載の球状核。
[5]真球度が0.7以上1以下であることを特徴とする上記[1]から[4]のいずれかに記載の球状核。
【0008】
[6]密度が0.8g/ml以上1g/ml以下であることを特徴とする上記[1]から[5]のいずれかに記載の球状核。
[7]粒径が200〜500μmであることを特徴とする上記[1]から[6]のいずれかに記載の球状核。
[8]膨潤剤が、クロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上であることを特徴とする上記[1]から[7]のいずれかに記載の球状核。
[9]上記[1]から[8]のいずれかに記載の球状核の表面が薬物で被覆されていることを特徴とする球状素顆粒。
[10]上記[9]に記載の球状素顆粒の表面が水不溶性物質で被覆されていることを特徴とする球状顆粒。
[11]薬物の放出開始時間が15分から30分である上記[10]に記載の球形顆粒。
[12]結晶セルロースと膨潤剤を湿潤状態で球状化することを特徴とする上記[1]から[8]のいずれかに記載の球状核を製造する方法。
【発明の効果】
【0009】
本発明は、結晶セルロースと膨潤剤がランダムに混在した状態で存在する球状核である。核の表面に膨潤剤を被覆させる方法と比較して、その分の手間が不要となる利点があり、さらに表面が滑らかで真球度の高い核が得られやすい。さらに、膜破裂制御に重要な役割を有する膨潤剤が核中に均一に分布しているので、核の膨張程度が制御しやすくなる。
さらに本発明の球状核は薬物を含有しないため、予め薬物を含有する核と比較して、薬物の物性に影響されず、一定の品質を維持した真球度の高い球状核を製造できる。加えて、膨潤剤が薬物の影響を受けずに水を吸水して膨張するため、精度の高い膜破裂型時限放出製剤を容易に得ることが可能となる。
【図面の簡単な説明】
【0010】
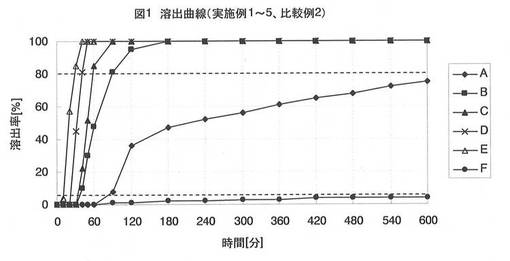
【図1】図1は、実施例1〜5、比較例1で得られた球形顆粒A〜Fの薬物溶出曲線である。
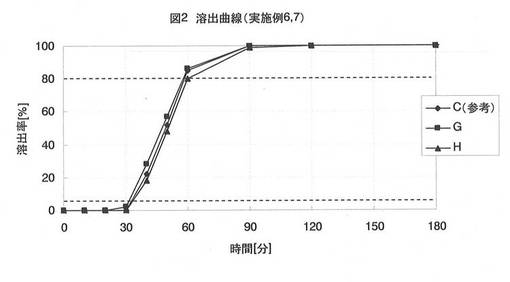
【図2】図2は、実施例3、6、7で得られた球形顆粒C、G、Hの薬物溶出曲線である。
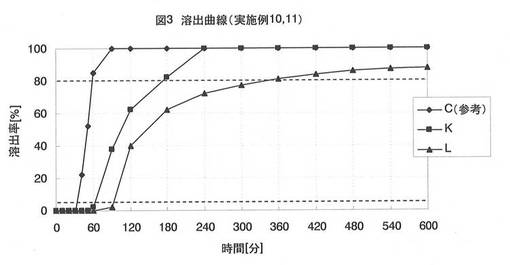
【図3】図3は、実施例10で得られた球形顆粒K、Lの薬物溶出曲線である。
【発明を実施するための形態】
【0011】
以下、本発明について、詳細に説明する。なお、本発明は以下の説明に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
本発明の球状核は、結晶セルロースと膨潤剤を含有する組成物からなり、膨潤剤が結晶セルロースからなる核の周りに被覆されたものではなく、結晶セルロースと膨潤剤が核中にランダムに混在する。核の周りに膨潤剤が被覆された球状核は、水に浸すと表面に付着した膨潤剤が崩壊して核から剥がれてしまうのに対して、本発明の球状核は膨潤剤が核から剥がれずに核そのものが大きく膨張する点に特徴がある。
ここで、本発明において、「核」とは、時限放出製剤として用いる際に、水不溶性成分からなる層や薬物からなる層を外殻に形成するためのものである。この球状核の中には薬物が含まれないため、薬物の影響を受けずに核中の膨潤剤が水を吸水して膨張しやすくなる。この大きな膨張効果が、精度の高い時限放出製剤を得るのに重要な要因となる。
【0012】
本発明において薬物とは、薬学的に活性な薬効成分のことである。
結晶セルロースとは、リンター、パルプ、再生繊維等のセルロース質を酸加水分解あるいはアルカリ酸化分解あるいは両者を組合わせる、あるいは上記の化学的処理のあとに粉砕などの機械的処理を施こすなどして得られるものである。ここでいう結晶セルロースとは、X線回折法により求められる結晶化度が10%以上であるセルロースをいう。好ましくは40%以上である。また、平均重合度は60〜375が好ましい。平均重合度が60以下ではセルロース分子の絡み合いが少なくなるため球状核の磨損度が大きくなり、また375以上では繊維性が現われるため球状になりにくく好ましくない。より好ましくは60〜300である。吸水量は、1.0〜2.8ml/g、200メッシュ留分は80質量%以下であることが好ましい。吸水量は、JIS K5101に記載の吸油量の測定法に準じ、油の代わりに蒸留水を用いる。終点は全体が一つの魂状となった後、離水し始める点とする。また、結晶セルロースは粉体のものを用いることができる。
【0013】
膨潤剤とは、水分を吸収して膨張するような物質である。ここで、本発明において、膨潤剤はセルロースを含まない。膨潤剤とは、具体的には、後で説明するところの沈降体積が2〜30ml/gとなる物質であり、製剤の技術分野において通常用いられる崩壊剤を好適に用いることができる。ここで、崩壊剤とは、例えば医薬品添加物事典2007の用途別索引397頁に、崩壊剤として列記されている物質のことを言う。崩壊剤の具体例としては、例えば、クロスカルメロースナトリウム、カルメロース、カルメロースカルシウム、カルメロースナトリウム、低置換度ヒドロキシプロピルセルロース等のセルロース類、カルボキシメチルスターチナトリウム、ヒドロキシプロピルスターチ、コメデンプン、コムギデンプン、トウモロコシデンプン、バレイショデンプン、部分アルファー化デンプン等のデンプン類、クロスポビドン、クロスポビドンコポリマー等の合成高分子等を挙げることができる。上記から選ばれる2種以上を併用してもよい。膨潤剤は粉体のものを用いることができる。
【0014】
特に好ましくは、次のものが挙げられる(化合物名(商品名/メーカー)と記載する)。クロスカルメロースナトリウム(キッコレート/旭化成ケミカルズ(株)、Primellose/DMV、Ac−Di−Sol/カネダなど)、アルファー化デンプン(SWELSTAR PD−1/旭化成ケミカルズ(株)、LYCATAB PGS/ロケットジャパン、アミコール/日澱化学など)、カルボキシメチルスターチナトリウム(GLYCOLYS/ロケットジャパン、Primojel/DMV、エキスプロタブ/木村産業など)、低置換度ヒドロキシプロピルセルロース(L−HPC/信越化学工業(株)など)、クロスポビドン(クロスポビドン/五協産業、コリドン/BASFジャパン、ポリプラスドン/アイエスピー・ジャパンなど)である。さらに好ましくは、クロスカルメロースナトリウムである。
【0015】
本発明の球状核の好ましい製造方法について記載するが、下記方法に限定されるものではない。
結晶セルロースと膨潤剤を含有する粉体を高速攪拌造粒機で混合しながら、蒸留水を加え練合・造粒する。蒸留水の代わりに、ヒドロキシプロピルセルロース、でんぷん糊、ポリビニルピロリドンなどの水溶液、又は有機溶媒を結合液として用いても良いが、ヒドロキシプロピルセルロース、でんぷん糊、ポリビニルピロリドンなどの水溶液の使用が好ましい。なお、造粒方法として特許文献3及び4に記載されている押し出し造粒は、混合物に結合剤を添加して練合した湿潤物を、一定の大きさのスクリーンから押し出して、乾燥、分級して顆粒を得る造粒方法である。そのため、一般的に、真球度が高く、かつ小さい粒径の顆粒を得るのが難しい。加えて、押し出しされにくい薬物や膨潤剤などを含む場合は、その配合量や種類が限定されるので好ましくない。それに対して、攪拌造粒は、混合物を攪拌翼で攪拌しながら、結合剤を散布し造粒する造粒方法であり、真球度が高く、かつ小さい粒径の顆粒を得やすい。
その後、転動流動層造粒機(転動型流動層コーティング装置)へ移し、蒸留水を噴霧しながら、球形化を行った後、乾燥し、必要により篩分し、球状核を得る。
【0016】
球状核表面に薬物層や水不溶性物質の皮膜層を均一に付着させるためには、粒径サイズが均一で、真球度の高い核であることが好ましい。またコーティング時の破損を防ぐために、密度の高い硬い核であることが好ましい。加えて、球状核の粒径は、小さすぎるとコーティング時に凝集しやすく、大きすぎると口中でザラツクなど触感がよくないため、球状核の粒径は200〜500μmの範囲内であることが好ましい。より好ましくは、300〜500μmである。
上記のような物性を満たす球状核を製造するためには、製造時の適切な加水量や攪拌速度が重要なファクターとなる。結晶セルロースと膨潤剤が同じ質量比率であれば、製造時の加水量を多くすると、得られる球状核の密度と真球度がともに高くなり、粒径が大きくなる傾向を示す。一方、結晶セルロースと膨潤剤が同じ質量比率で攪拌速度を速くすると、得られる球状核の密度と真球度がともに低くなり、粒径が小さくなる傾向を示す。また膨潤剤の比率が多い処方では、より加水量が多く必要となり、固形物の体積が膨張するので装置への仕込み量が少なくなる。このように適した物性を有する球状核を得るために、加水量や攪拌条件を調整しながら製造することが重要である。
【0017】
例えば高速攪拌造粒機を用いて球状核を製造する場合において、好ましい加水量は膨潤剤に対して質量比で6〜20倍であり、好ましい攪拌速度はメインブレードが500m/分以下、クロススクリューが800m/分以下である。一方、特許文献3及び4の「薬物を含有する核」は、膨潤剤に対して、質量比で5倍量以下の水あるいは10%エタノール水溶液を配合して製造されることが記載されている(実施例)。装置の違いだけでなく、核中の薬物の有無が製造時の最適加水量やその他製造条件に大きな影響を与えていると考えられる。
本発明の球状核を製造する際に、薬学的に不活性であれば、結晶セルロース以外の賦形剤(乳糖、白糖、D−マンニトールなどの糖類、コーンスターチ、ポテトスターチなどのでんぷん類、あるいは第二りん酸カルシウム、ケイ酸アルミニウムなどの無機物類など)を、本発明の効果に支障のない程度配合してもよい。
【0018】
本発明の球状核は吸水により体積が膨潤する。この膨潤効果で確実に水不溶性の皮膜を破裂させることが可能となる。後に測定方法を説明する体積膨潤度とは、膨潤前の球状核に対する膨潤後の球状核の体積の増加率である。例えば、体積膨純度が100%の場合、膨潤前の体積に比べて膨潤後の体積が100%増加すること、つまり2倍になることを意味している。本発明においては、膨潤前の球状核の体積に対して100〜700%であることが好ましく、より好ましくは200%〜600%である。
膨潤剤の配合比率が多くなるほど体積膨潤度は大きくなるが、膨潤剤の比率が多くなりすぎると得られる球状核の表面が粗くなり、真球度や密度が低くなる。また吸水により膨潤する際に、球状核が崩壊し形状を維持できなくなる問題が生じる。そのため、球状核は結晶セルロースと膨潤剤を90:10〜30:70の質量比で含有することが好ましい。膨潤剤が10質量%未満になると、球状核が吸水により十分に膨張できずに、膜を破裂させにくくなる。膨潤剤が70質量%より多いと、球状核表面が粗くなり、薬物層や水不溶性物質の皮膜層を均一に付着させることが難しくなる。結晶セルロースと膨潤剤の質量比は、より好ましくは、60:40〜40:60である。
【0019】
本発明の球状核中に薬物を含有しないことが、大きな膨潤効果を得るための重要なポイントとなる。球状核中に薬物が存在すると、膨潤剤が十分に水を吸って膨張するのが阻害される。吸湿性の高い薬物であれば、核内部に進入してきた水を薬物が吸水するため、その分、膨潤剤が十分に吸水・膨張しにくくなる。また疎水性の薬物であれば、核中に疎水性物質が点在することで、水が核内部に進入しにくくなり膨潤剤が十分に吸水・膨張しにくくなる。特許文献3に薬物を含有する核について記載があるが、水和した際の体積が20〜100%しか膨張しないことが記載されている。
本発明の球状核は真球度が0.7以上1以下であることが好ましい。真球度が低いと、球状核表面に薬物層や水不溶性物質の皮膜層を均一に付着させることが困難となり、望み通りの膜破裂制御ができにくくなる。真球度は高ければ高いほど好ましく、0.8以上であるとより好ましい。
また、密度は0.8g/ml以上1g/ml以下であることが好ましい。密度が低いと、コーティング時に球状核の流動が悪くなり、均一なコーティングが困難となる。さらに球状核の凝集や破損も多くなり、高品質な時限放出製剤を得ることが難しくなる。目的とする真球度や密度を得るためには、前述したように球状核製造時の適切な加水量や攪拌速度が重要なファクターとなる。
【0020】
本発明の、球状核の周りに薬物を含有する層を有する球形素顆粒は、例えば以下の方法により製造される。
球状核を遠心流動型コーティング装置中で転動させながら、結合剤含有水溶液を連続的に噴霧し、同時に薬物と必要ならば賦形剤とから成る粉体を供給し、球状核に粉体を被覆し、乾燥して球形素顆粒とする。あるいは球状核を流動層コーティング装置中で流動させながら、結合剤水溶液中に薬物を溶解あるいは懸濁させた液を噴霧し、球状核に薬物を含む粉体を被覆し、乾燥して球形素顆粒とする。
薬物の積層量は薬物の投与量によって決まるが、球状核に対して0.1〜300質量%である。本発明の球形素顆粒は球状核の表面に薬物が付着しているため、特許文献3及び4のように、予め薬物を含有する核と比較して、薬物の配合量が制限されないというメリットがある。
【0021】
結合剤としては、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルセルロース、でんぷん糊、アルファー化でんぷん、ポリビニルピロリドン、アラビアガム、糖シロップ、カルボキシメチルセルロースナトリウム、プルランなどが挙げられるが、薬物が水溶性の場合、薬物水溶液自体が結合剤水溶液として作用することもある。
賦形剤としては、乳糖、コーンスターチ、結晶セルロース、白糖、D−マンニトール、アルファー化でんぷん、部分アルファー化でんぷんなどが挙げられる。
また薬物を付着させた後に、通常用いられる方法に従って球形素顆粒にシールコートを施してもよい。
【0022】
本発明の、球形素顆粒の周りに水不溶性物質を含有する皮膜層を有する球形顆粒は、例えば以下の方法により製造される。
球形素顆粒を流動させながら水不溶性物質を含有する水性懸濁液を噴霧してコーティングし、乾燥させて皮膜層を形成させ、球形顆粒とする。この時、水不溶性物質を懸濁させる溶媒として有機溶媒は好ましくない。
使用するコーティング装置としては、遠心流動型コーティング装置、流動層コーティング装置、転動型流動層コーティング装置、パン型コーティング装置など通常のコーティング機などが挙げられる。
被覆に用いる水不溶性物質の量は、薬物の放出開始時間や球状核の大きさなどによって異なるが、球状核に対して5〜200質量%であり、好ましくは10〜100質量%、さらに好ましくは10〜50質量%である。
【0023】
水不溶性物質としては、エチルセルロース、アクリル酸エチル・メタクリル酸メチルコポリマー、ヒドロキシプロピルメチルセルロースフタレート、セルロースアセテートフタレート、カルボキシメチルエチルセルロース、セルロースアセテート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、セラック、などが挙げられる。上記から選ばれる2種以上を併用してもよい。好ましくは、エチルセルロースである。
水不溶性物質を含有する皮膜層には、水不溶性物質に加えて可塑剤、ワックス、着色剤、香料、滑沢剤、凝集防止剤などの目的で添加剤を配合することができる。
【0024】
本発明の球形顆粒が経口投与されと、皮膜層を介して消化管の水分を吸収することで、中心にある球状核中の膨潤剤が徐々に膨潤する。そして一定時間後に、その球状核中の膨潤剤の膨潤による体積増加により、皮膜の破壊が起き、薬物が瞬時に全量放出される仕組みとなっている。薬物の放出が開始されるまでの時間(放出開始時間)、及び薬物の放出が開始されてから大部分の薬物が放出されるまでの時間(放出時間)は、水不溶性物質の種類や被覆量、及び中心にある球状核中の膨潤剤の種類や配合比率によって自由に調整できる。膨潤剤の配合比率が多く膨潤効果が強い球状核であるほど、また水不溶性物質の被覆量が少なく水分が浸透しやすい皮膜であるほど、放出開始時間、及び放出時間が短くなる傾向を示す。放出開始時間は30分以内が好ましく、より好ましくは15〜30分以内である。また放出時間は90分以内が好ましく、より好ましくは60分以内、さらに好ましくは30分以内である。
なお、特許文献4では、放出開始時間及び放出時間を制御する技術に関する記載があるが、核の製造や皮膜の形成に用いるアルコール濃度を調整することで、その目的を達成している。しかし有機溶媒の使用は、環境・コスト・残留など様々な問題があり、出来れば使用したくないのが一般的である。本発明では、一切有機溶媒を用いずに放出開始時間、及び放出時間を制御することが可能となる。
【実施例】
【0025】
本発明を実施例に基づいて説明する。
本発明で用いられる物性の測定方法及び条件は以下のとおりである。
まず、物性の測定方法を以下にまとめて記す。
<球状核の体積膨潤度[%]>
試料にスポイトで水を垂らして膨潤させ、5分後に水を拭き取る。膨潤前後の顕微鏡写真をとり、短径、長径を計測し、これらの平均値を直径として、膨潤前後の粒子の体積を算出する。粒子20個について実施し、その平均値を求め、以下の式より体積膨潤度とする。
体積膨潤度[%]=(膨潤後の体積−膨潤前の体積)×100/膨潤前の体積
【0026】
<球状核の真球度>
試料の顕微鏡写真をとり、短径/長径比を計測する。粒子150個について実施し、その平均値を真球度とする。
<球状核の密度[g/ml]>
スコットボリュームメーター(筒井理化学器械(株))を用いて測定する。試料を定量フィーダーにて30〜60秒かけて25mlの測定容器内にあふれるまで流下させる。次いで容器の上部に堆積した過剰量の試料をすり落とし、その後、容器に疎充填された重量を量る。容器に疎充填された重量を測定容器の容積で除した値を密度とする。3回測定し、その平均値をとる。
【0027】
<薬物の溶出試験>
溶出試験器DT−610(日本分光(株))及び分光光度計V−530(日本分光(株))を用いて測定する。一般試験法「溶出試験法」に準じて実施し、装置は「装置2」(パドル法)、パドル回転数は100rpm、試験液は日本薬局方「溶出試験第1液」を使用する。3回測定し、その平均値をとる。
<薬物の放出開始時間、放出時間>
薬物の溶出率が5%未満のときは薬物の放出が抑制されているとして、溶出率が5%未満である時間を放出開始時間とする。また放出開始時間から薬物の溶出率が80%以上となるまでの時間を放出時間とする。
【0028】
[実施例1]
結晶セルロース(吸水量2.1ml/g、200メッシュ留分30%、平均重合度220、結晶化度65%)1440gとクロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))160gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水2720g(膨潤剤に対して17.0倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=90:10)を得た。
【0029】
次に、得られた球状造粒物を転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に水分込みで1000g仕込み、温度50℃、速度301m/分(半径8cm、600rpm)で転動しながら、30分かけて蒸留水300gを少しずつ加えた後に、温度25℃、速度301m/分で30分間転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Aを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は52質量%であった。得られた球状核Aを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核A600gを転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に仕込み、薬物水分散液(3.9質量%リボフラビン、0.8質量%ヒドロキシプロピルセルロース、95.3質量%蒸留水)で噴霧・被覆し、球状核に対してリボフラビンを2.0質量%含有する球形素顆粒Aを得た。
【0030】
次いで得られた球形素顆粒A550gを転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に仕込み、水不溶性物質として13質量%エチルセルロース(セリオスコートEC−30A/旭化成ケミカルズ(株))、可塑剤として2質量%クエン酸トリエチル、85質量%蒸留水を含有する水性懸濁液を、球状核に対して10質量%(固形分)まで噴霧・被覆し、乾燥させた。その後105℃のオーブンで1時間加熱することで皮膜層を形成させた。篩で500μm以上の粒子を除去し、エチルセルロースとクエン酸トリエチルを固形分で10質量%被覆させた球形顆粒Aを得た。
【0031】
[実施例2]
結晶セルロース600gとクロスカルメロースナトリウム400gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水3950g(膨潤剤に対して9.9倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=60:40)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Bを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は45質量%であった。得られた球状核Bを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Bを用いて、実施例1と同様にして球形素顆粒Bを得た。
次いで得られた球形素顆粒Bを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Bを得た。
【0032】
[実施例3]
結晶セルロース400gとクロスカルメロースナトリウム400gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水3760g(膨潤剤に対して9.4倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=50:50)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Cを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は32質量%であった。得られた球状核Cを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Cを用いて、実施例1と同様にして球形素顆粒Cを得た。
次いで得られた球形素顆粒Cを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Cを得た。
【0033】
[実施例4]
結晶セルロース320gとクロスカルメロースナトリウム480gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水4400g(膨潤剤に対して9.2倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=40:60)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Dを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は25質量%であった。得られた球状核Dを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Dを用いて、実施例1と同様にして球形素顆粒Dを得た。
次いで得られた球形素顆粒Dを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Dを得た。
【0034】
[実施例5]
結晶セルロース210gとクロスカルメロースナトリウム490gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水4340g(膨潤剤に対して8.9倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=30:70)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Eを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は13質量%であった。得られた球状核Eを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸が見られた。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Eを用いて、実施例1と同様にして球形素顆粒Eを得た。
次いで得られた球形素顆粒Eを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Eを得た。
【0035】
[比較例1]
結晶セルロース(吸水量2.1ml/g、200メッシュ留分30%、平均重合度220、結晶化度65%)のみ1600gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水1520gを少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に水分込みで1000g仕込み、温度50℃、速度301m/分(半径8cm、600rpm)で転動しながら、30分かけて蒸留水300gを少しずつ加えた後に、温度25℃、速度301m/分で30分間転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Fを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は65質量%であった。得られた球状核Fを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Fを用いて、実施例1と同様にして球形素顆粒Fを得た。
次いで得られた球形素顆粒Fを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で10質量%被覆させた球形顆粒Fを得た。
【0036】
[比較例2]
クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))のみ600gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水4500g(膨潤剤に対して7.5倍の質量比)を少しずつ加えた後、60分間攪拌したが、餅状の固まりになり球状化できなかった。
【0037】
[試験例1]
前述した方法に従って、球形顆粒A〜Fにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒A〜Fに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図1に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表1に記載した。
【0038】
【表1】

【0039】
実施例1〜5、比較例1、2では、膨潤剤の配合比率について評価している。
実施例1〜5(表1及び図1)から、結晶セルロースと膨潤剤(カルボキシメチルセルロースナトリウム)の配合比率(90:10〜30:70)によって、物性の異なる球状核が得られることを確認した。また、膨潤剤の配合比率が多いほど放出開始時間、及び放出時間を短縮でき、薬物溶出が制御可能であることを確認した。
結晶セルロースと膨潤剤が90:10の比率である球形顆粒A(実施例1)は、核の膨潤度が小さいために一定の放出開始時間の後に徐放性の溶出パターンを示した。
結晶セルロースと膨潤剤が30:70の比率である球形顆粒E(実施例5)は、10分間の放出開始時間を示したが、その間に、徐々に薬物の溶出が発生し、4%近く溶出した。
【0040】
それに対して、結晶セルロースと膨潤剤が60:40〜40:60の比率である球形顆粒B〜D(実施例2〜4)は、一定の放出開始時間後にそれぞれ1時間以内で80%以上の薬物放出が観察され、非常に精度が高く調整しやすい球形顆粒が得られた。
また、球状核Eの表面に凹凸があり、他の球状核と比較するとやや形状が歪であった。それに対して、球状核A〜Dは表面が滑らかなために、薬物や水不溶性物質の皮膜を均一に形成することができる。
比較例1より、膨潤剤を含まずに結晶セルロースのみで得られる球形顆粒Fは、10時間経っても殆ど薬物が放出されず、膜破裂型時限放出製剤としては不適切であることが分かった。
比較例2より、膨潤剤のみでは球状核が得られないことが分かった。
【0041】
[実施例6]
まずは本発明で好ましい膨潤剤として用いられる、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))、アルファー化デンプン(SWELSTAR PD−1/旭化成ケミカルズ(株))、カルボキシメチルスターチナトリウム(Primojel/DMV)、低置換度ヒドロキシプロピルセルロース(L−HPC LH−21/信越化学工業(株))、クロスポビドン(ポリプラスドンXL−10/ISP)の沈降体積を以下の手順で測定した。
蒸留水75mlをビーカーに入れ、スターラーで攪拌しながら、膨潤剤1.0gを少しずつ加えた。
膨潤剤を全て投入後、3分間撹拌した。
懸濁液を100mlメスシリンダーに移し、100mlにメスアップ後、沈降管に入れた。
懸濁液を16時間静置し、沈降体積を読み取った。
その結果、沈降体積の大きい順に、カルボキシメチルスターチナトリウム:28.8ml/g、低置換度ヒドロキシプロピルセルロース:21.0ml/g、クロスカルメロースナトリウム:16.5ml/g、アルファー化デンプン:12.0ml/g、クロスポビドン:6.1ml/gであった。ここで沈降体積はその膨潤剤の膨潤性に相関すると考えられ、つまり沈降体積が大きいほど膨潤性が高いと推測される。
【0042】
本発明の球状核における体積膨潤度は、膨潤剤の膨潤性に影響されると考えられるので、最も大きな沈降体積を示したカルボキシメチルスターチナトリウム、及び最も小さな沈降体積を示したクロスポビドンの2種類で球状核を製造し、評価した。
実施例3において、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))の代わりに、カルボキシメチルスターチナトリウム(Primojel/DMV)を用いること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Gを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は30質量%であった。得られた球状核Gを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表2に記載した。
球状核Gを用いて、実施例1と同様にして球形素顆粒Gを得た。
次いで得られた球形素顆粒Gを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Gを得た。
【0043】
[実施例7]
実施例3において、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))の代わりに、クロスポビドン(ポリプラスドンXL−10/ISP)を用いること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Hを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は39質量%であった。得られた球状核Hを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表2に記載した。
球状核Hを用いて、実施例1と同様にして球形素顆粒Hを得た。
次いで得られた球形素顆粒Hを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Hを得た。
【0044】
[試験例2]
前述した方法に従って、球形顆粒G、Hにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒G、Hに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図2に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表2に記載した。
【0045】
【表2】
【0046】
実施例3、6、7では、膨潤剤の種類について評価している。
実施例3、6、7(表1、表2及び図2)から、膨潤剤の配合比率が同じであれば、その種類を変えてもほぼ同じ物性の球状核が得られることを確認した。実施例6で列挙した膨潤剤のうち、最も大きな膨潤性を示したカルボキシメチルスターチナトリウム、中程度の膨潤性を示したクロスカルメロースナトリウム、最も小さな膨潤性を示したクロスポビドンの3種類で確認できたことより、他の膨潤剤を用いても同様の結果が得られると予想される。また、球形顆粒G、H(実施例6、7)は、球形顆粒C(実施例3)と同じく、30分の放出開始時間、及び30分の放出時間を示し、同等の効果が確認できた。
【0047】
[実施例8]
実施例3において、高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)で攪拌しながら加える蒸留水の量を2000g(膨潤剤に対して5.0倍の質量比)にすること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Iを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は11質量%であった。得られた球状核Iを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸があった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表3に記載した。
【0048】
[実施例9]
実施例3において、高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)の攪拌条件を、メインブレード879m/分(半径14cm、900rpm)、クロススクリュー754m/分(半径4cm、3000rpm)にすること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Jを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は13質量%であった。得られた球状核Jを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸があった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表3に記載した。
【0049】
【表3】
【0050】
実施例8、9では、球状核の製造条件について評価している。
球状核C(実施例3)と比較すると、球状核Iは形状が歪(真球度が低い)で表面が粗く体積膨潤度も低かった。これは加水量が少ないために膨潤剤が十分に膨潤しきれず、結晶セルロースと膨潤剤が不均一に混合されて球状核が形成されたためと思われ、好ましい球状核を得るためには、最適な加水量があることを確認した。
球状核C(実施例3)と比較すると、球状核Jは体積膨潤度は同等だが形状が歪(真球度が低い)で表面が粗かった。これは攪拌速度が速いために適切に球状化できなかったためと思われ、好ましい球状核を得るためには、最適な攪拌条件があることを確認した。
球状核Cは、形状がほぼ真球であり、良好な製剤が得られる可能性が高い。
【0051】
[実施例10]
実施例3で得られた球形素顆粒Cを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で40質量%被覆させた球形顆粒K、及び60質量%被覆させた球形顆粒Lを得た。
【0052】
[試験例3]
前述した方法に従って、球形顆粒K、Lにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒K、Lに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図3に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表4に示す。
【0053】
【表4】
【0054】
実施例10では、水不溶性物質の被覆量について評価している。
同じ球状核を用いて30質量%被覆させた球形顆粒C(実施例3)の溶出パターンと比較すると、40質量%、60質量%まで被覆させた球形顆粒K、Lは、ともに放出開始時間、及び放出時間が長くなり、水不溶性物質の被覆量によって薬物溶出が制御可能であることを確認した。水不溶性物質の被覆量が多くなると水分が浸透しにくくなるためと考えられる。
【産業上の利用可能性】
【0055】
本発明は、医薬品分野の特に時間薬物療法において好適に利用できる。精度が高く、容易に薬物溶出制御が可能な膜破裂型の時限放出製剤として利用できる。
【技術分野】
【0001】
本発明は、医薬品分野の時間薬物療法に用いられる膨潤性を有する球状核、該球状核を用いて製造される球状素顆粒および球状顆粒に関する。また、本発明は球状核の製造方法に関する。
【背景技術】
【0002】
多くの慢性疾病患において病態の周期的変化が起こることが知られている。これはヒトの生理機能の多くが体内時計によって支配され、周期的に変動することに基づくものである。このような背景から、症状が現れる時間帯、あるいは治療上必要な時間帯にのみ薬物の血中濃度を高めて薬効を発現させる、時間薬物療法が検討されてきた。なかでも、服用後所定の時間が経過した後に薬物を放出することにより、薬効の発現を必要な時間遅らせる機能を有する時限放出製剤は、時間薬物療法を通常の投薬法によって実現することが可能となる。放出開始時間を調整することで目的に応じた時限放出製剤を設計できる。
例えば、放出開始時間を数時間単位と調整すれば、小腸や大腸に薬物を作用させることが可能となり、放出開始時間を数分単位と調整すれば、胃などに薬物を作用させることが可能となり、さらに不快な味を有する薬物のマスキングとして用いることもできる。
この時限放出製剤の一形態である膜破裂型の時限放出製剤は、薬物と膨潤性物質を配合した錠剤や顆粒剤に水不溶性の皮膜を被覆したものである。この製剤は、生体に投与した際に水不溶性の皮膜を介して浸透してくる水分によって製剤中の膨潤性物質が膨張し、その膨張力によって皮膜が破れることにより初めて薬物が溶出する仕組みとなっている。
【0003】
特許文献1には、核粒子の周囲に膨潤剤と共に薬物を付着させ、それを水不溶性の被覆物質と添加剤の混合物で被覆した膜破裂型の持続性製剤が開示されている。この製剤の製造方法として、膨潤剤を粉噴霧して核粒子表面に付着させている。
特許文献2には、芯を含有する薬剤に崩壊剤を付着させ、さらに透過性の水不溶性ポリマーとグライダントの混合物で被覆した膜破裂型の遅延放出製剤が開示されている。
特許文献3には、薬物と崩壊剤を湿潤状態で球状化して得られた核に、水溶性の重合体と不溶性の重合体の混合物で被覆した膜破裂型の遅延放出製剤が開示されている。
特許文献4には、薬物と水膨潤性物質を湿潤状態で球状化して得られた核に、水不溶性高分子を含む皮膜で被覆した膜破裂型の時限放出製剤が開示されている。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開昭62−30709号公報
【特許文献2】特表2003−510346号公報
【特許文献3】特表2004−510812号公報
【特許文献4】特開2009−191034号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
特許文献1では、核粒子に膨潤剤を粉噴霧する方法が記載されているが、核粒子表面に粉末状の膨潤剤が不均一に付着する。そのため、外側の被覆膜が均一な厚みで被覆されず、その結果、得られる製剤の膜破裂制御が非常に難しいという問題があった。
特許文献2では、崩壊剤を有機溶剤中に懸濁させた液を用いて芯を含有する薬剤上にスプレイ(噴霧)しているが、有機溶媒は環境・コスト・残留などの様々な問題がある。加えて、一般的に、有機溶媒系から水溶液系あるいは水性懸濁液系へと転換が図られているが、崩壊剤を水溶液中に懸濁させると、吸水により体積膨張して系の粘度が高くなりスプレイすることが困難となる。また低粘度とするために系を希釈すると、作業効率が非常に悪くなってしまうという問題点が生じる。
さらに特許文献1及び2のように、核(芯)に対して薬物や膨潤剤等を付着させる方法では、最終的に得られる顆粒サイズが大きくなる問題があった。一般的に核として用いられるノンパレル103(粒径:355〜850μm/フロイント産業(株)製)では、得られる顆粒の粒径は最小でも500〜1000μm程度であり、それよりも小さい顆粒を得ることはできない。
【0006】
特許文献3及び4の核は薬物を含有するため、薬物の物性(吸湿性、凝集性、分解性など)の影響で、真球度が高く、かつ薬物含有量や粒径サイズの均一な核を安定に製造することが困難になるという問題があった。また各薬物の種類ごとに核の最適な製造条件が異なるため、薬物の種類を変える度に製造条件の最適化が必要となるという煩わしさがあった。
さらに特許文献3及び4のように、押し出し造粒を用いて顆粒を製造する場合は、真球度が高く、かつ小さい粒径の顆粒を得るのは難しいのが一般的である。特に、押し出しされにくい薬物や膨潤剤などを含む場合は、その配合量や種類が限定される問題があった。特許文献3では顆粒の粒径は500〜4000μmとの記載があり、特許文献4では180〜500μmとの記載がある。しかし実際に特許文献4の実施例には、小さくても420〜500μmの顆粒しか得られていない。
本発明の課題は、精度の高い時限放出製剤を提供すること、その時限放出製剤の核となる膨潤性を有する球状核を提供すること、精度の高い時限放出製剤が容易に得られることである。
【課題を解決するための手段】
【0007】
本発明者らは上記課題に対し鋭意検討した結果、膨潤剤と結晶セルロースがランダムに混在した状態で存在する球状核に、薬物を含有する層と、さらに水不溶性物質を含有する層を被覆することで、上記課題を解決できることを見出し、本発明を完成した。
すなわち、本発明は以下の通りである。
[1]結晶セルロースと膨潤剤を含む混合物からなる球状核。
[2]結晶セルロースと膨潤剤の質量比が90:10〜30:70であることを特徴とする上記[1]に記載の球状核。
[3]結晶セルロースと膨潤剤の質量比が60:40〜40:60であることを特徴とする上記[1]又は[2]に記載の球状核。
[4]吸水による体積膨潤度が100%〜700%であることを特徴とする上記[1]から[3]のいずれかに記載の球状核。
[5]真球度が0.7以上1以下であることを特徴とする上記[1]から[4]のいずれかに記載の球状核。
【0008】
[6]密度が0.8g/ml以上1g/ml以下であることを特徴とする上記[1]から[5]のいずれかに記載の球状核。
[7]粒径が200〜500μmであることを特徴とする上記[1]から[6]のいずれかに記載の球状核。
[8]膨潤剤が、クロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上であることを特徴とする上記[1]から[7]のいずれかに記載の球状核。
[9]上記[1]から[8]のいずれかに記載の球状核の表面が薬物で被覆されていることを特徴とする球状素顆粒。
[10]上記[9]に記載の球状素顆粒の表面が水不溶性物質で被覆されていることを特徴とする球状顆粒。
[11]薬物の放出開始時間が15分から30分である上記[10]に記載の球形顆粒。
[12]結晶セルロースと膨潤剤を湿潤状態で球状化することを特徴とする上記[1]から[8]のいずれかに記載の球状核を製造する方法。
【発明の効果】
【0009】
本発明は、結晶セルロースと膨潤剤がランダムに混在した状態で存在する球状核である。核の表面に膨潤剤を被覆させる方法と比較して、その分の手間が不要となる利点があり、さらに表面が滑らかで真球度の高い核が得られやすい。さらに、膜破裂制御に重要な役割を有する膨潤剤が核中に均一に分布しているので、核の膨張程度が制御しやすくなる。
さらに本発明の球状核は薬物を含有しないため、予め薬物を含有する核と比較して、薬物の物性に影響されず、一定の品質を維持した真球度の高い球状核を製造できる。加えて、膨潤剤が薬物の影響を受けずに水を吸水して膨張するため、精度の高い膜破裂型時限放出製剤を容易に得ることが可能となる。
【図面の簡単な説明】
【0010】
【図1】図1は、実施例1〜5、比較例1で得られた球形顆粒A〜Fの薬物溶出曲線である。
【図2】図2は、実施例3、6、7で得られた球形顆粒C、G、Hの薬物溶出曲線である。
【図3】図3は、実施例10で得られた球形顆粒K、Lの薬物溶出曲線である。
【発明を実施するための形態】
【0011】
以下、本発明について、詳細に説明する。なお、本発明は以下の説明に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
本発明の球状核は、結晶セルロースと膨潤剤を含有する組成物からなり、膨潤剤が結晶セルロースからなる核の周りに被覆されたものではなく、結晶セルロースと膨潤剤が核中にランダムに混在する。核の周りに膨潤剤が被覆された球状核は、水に浸すと表面に付着した膨潤剤が崩壊して核から剥がれてしまうのに対して、本発明の球状核は膨潤剤が核から剥がれずに核そのものが大きく膨張する点に特徴がある。
ここで、本発明において、「核」とは、時限放出製剤として用いる際に、水不溶性成分からなる層や薬物からなる層を外殻に形成するためのものである。この球状核の中には薬物が含まれないため、薬物の影響を受けずに核中の膨潤剤が水を吸水して膨張しやすくなる。この大きな膨張効果が、精度の高い時限放出製剤を得るのに重要な要因となる。
【0012】
本発明において薬物とは、薬学的に活性な薬効成分のことである。
結晶セルロースとは、リンター、パルプ、再生繊維等のセルロース質を酸加水分解あるいはアルカリ酸化分解あるいは両者を組合わせる、あるいは上記の化学的処理のあとに粉砕などの機械的処理を施こすなどして得られるものである。ここでいう結晶セルロースとは、X線回折法により求められる結晶化度が10%以上であるセルロースをいう。好ましくは40%以上である。また、平均重合度は60〜375が好ましい。平均重合度が60以下ではセルロース分子の絡み合いが少なくなるため球状核の磨損度が大きくなり、また375以上では繊維性が現われるため球状になりにくく好ましくない。より好ましくは60〜300である。吸水量は、1.0〜2.8ml/g、200メッシュ留分は80質量%以下であることが好ましい。吸水量は、JIS K5101に記載の吸油量の測定法に準じ、油の代わりに蒸留水を用いる。終点は全体が一つの魂状となった後、離水し始める点とする。また、結晶セルロースは粉体のものを用いることができる。
【0013】
膨潤剤とは、水分を吸収して膨張するような物質である。ここで、本発明において、膨潤剤はセルロースを含まない。膨潤剤とは、具体的には、後で説明するところの沈降体積が2〜30ml/gとなる物質であり、製剤の技術分野において通常用いられる崩壊剤を好適に用いることができる。ここで、崩壊剤とは、例えば医薬品添加物事典2007の用途別索引397頁に、崩壊剤として列記されている物質のことを言う。崩壊剤の具体例としては、例えば、クロスカルメロースナトリウム、カルメロース、カルメロースカルシウム、カルメロースナトリウム、低置換度ヒドロキシプロピルセルロース等のセルロース類、カルボキシメチルスターチナトリウム、ヒドロキシプロピルスターチ、コメデンプン、コムギデンプン、トウモロコシデンプン、バレイショデンプン、部分アルファー化デンプン等のデンプン類、クロスポビドン、クロスポビドンコポリマー等の合成高分子等を挙げることができる。上記から選ばれる2種以上を併用してもよい。膨潤剤は粉体のものを用いることができる。
【0014】
特に好ましくは、次のものが挙げられる(化合物名(商品名/メーカー)と記載する)。クロスカルメロースナトリウム(キッコレート/旭化成ケミカルズ(株)、Primellose/DMV、Ac−Di−Sol/カネダなど)、アルファー化デンプン(SWELSTAR PD−1/旭化成ケミカルズ(株)、LYCATAB PGS/ロケットジャパン、アミコール/日澱化学など)、カルボキシメチルスターチナトリウム(GLYCOLYS/ロケットジャパン、Primojel/DMV、エキスプロタブ/木村産業など)、低置換度ヒドロキシプロピルセルロース(L−HPC/信越化学工業(株)など)、クロスポビドン(クロスポビドン/五協産業、コリドン/BASFジャパン、ポリプラスドン/アイエスピー・ジャパンなど)である。さらに好ましくは、クロスカルメロースナトリウムである。
【0015】
本発明の球状核の好ましい製造方法について記載するが、下記方法に限定されるものではない。
結晶セルロースと膨潤剤を含有する粉体を高速攪拌造粒機で混合しながら、蒸留水を加え練合・造粒する。蒸留水の代わりに、ヒドロキシプロピルセルロース、でんぷん糊、ポリビニルピロリドンなどの水溶液、又は有機溶媒を結合液として用いても良いが、ヒドロキシプロピルセルロース、でんぷん糊、ポリビニルピロリドンなどの水溶液の使用が好ましい。なお、造粒方法として特許文献3及び4に記載されている押し出し造粒は、混合物に結合剤を添加して練合した湿潤物を、一定の大きさのスクリーンから押し出して、乾燥、分級して顆粒を得る造粒方法である。そのため、一般的に、真球度が高く、かつ小さい粒径の顆粒を得るのが難しい。加えて、押し出しされにくい薬物や膨潤剤などを含む場合は、その配合量や種類が限定されるので好ましくない。それに対して、攪拌造粒は、混合物を攪拌翼で攪拌しながら、結合剤を散布し造粒する造粒方法であり、真球度が高く、かつ小さい粒径の顆粒を得やすい。
その後、転動流動層造粒機(転動型流動層コーティング装置)へ移し、蒸留水を噴霧しながら、球形化を行った後、乾燥し、必要により篩分し、球状核を得る。
【0016】
球状核表面に薬物層や水不溶性物質の皮膜層を均一に付着させるためには、粒径サイズが均一で、真球度の高い核であることが好ましい。またコーティング時の破損を防ぐために、密度の高い硬い核であることが好ましい。加えて、球状核の粒径は、小さすぎるとコーティング時に凝集しやすく、大きすぎると口中でザラツクなど触感がよくないため、球状核の粒径は200〜500μmの範囲内であることが好ましい。より好ましくは、300〜500μmである。
上記のような物性を満たす球状核を製造するためには、製造時の適切な加水量や攪拌速度が重要なファクターとなる。結晶セルロースと膨潤剤が同じ質量比率であれば、製造時の加水量を多くすると、得られる球状核の密度と真球度がともに高くなり、粒径が大きくなる傾向を示す。一方、結晶セルロースと膨潤剤が同じ質量比率で攪拌速度を速くすると、得られる球状核の密度と真球度がともに低くなり、粒径が小さくなる傾向を示す。また膨潤剤の比率が多い処方では、より加水量が多く必要となり、固形物の体積が膨張するので装置への仕込み量が少なくなる。このように適した物性を有する球状核を得るために、加水量や攪拌条件を調整しながら製造することが重要である。
【0017】
例えば高速攪拌造粒機を用いて球状核を製造する場合において、好ましい加水量は膨潤剤に対して質量比で6〜20倍であり、好ましい攪拌速度はメインブレードが500m/分以下、クロススクリューが800m/分以下である。一方、特許文献3及び4の「薬物を含有する核」は、膨潤剤に対して、質量比で5倍量以下の水あるいは10%エタノール水溶液を配合して製造されることが記載されている(実施例)。装置の違いだけでなく、核中の薬物の有無が製造時の最適加水量やその他製造条件に大きな影響を与えていると考えられる。
本発明の球状核を製造する際に、薬学的に不活性であれば、結晶セルロース以外の賦形剤(乳糖、白糖、D−マンニトールなどの糖類、コーンスターチ、ポテトスターチなどのでんぷん類、あるいは第二りん酸カルシウム、ケイ酸アルミニウムなどの無機物類など)を、本発明の効果に支障のない程度配合してもよい。
【0018】
本発明の球状核は吸水により体積が膨潤する。この膨潤効果で確実に水不溶性の皮膜を破裂させることが可能となる。後に測定方法を説明する体積膨潤度とは、膨潤前の球状核に対する膨潤後の球状核の体積の増加率である。例えば、体積膨純度が100%の場合、膨潤前の体積に比べて膨潤後の体積が100%増加すること、つまり2倍になることを意味している。本発明においては、膨潤前の球状核の体積に対して100〜700%であることが好ましく、より好ましくは200%〜600%である。
膨潤剤の配合比率が多くなるほど体積膨潤度は大きくなるが、膨潤剤の比率が多くなりすぎると得られる球状核の表面が粗くなり、真球度や密度が低くなる。また吸水により膨潤する際に、球状核が崩壊し形状を維持できなくなる問題が生じる。そのため、球状核は結晶セルロースと膨潤剤を90:10〜30:70の質量比で含有することが好ましい。膨潤剤が10質量%未満になると、球状核が吸水により十分に膨張できずに、膜を破裂させにくくなる。膨潤剤が70質量%より多いと、球状核表面が粗くなり、薬物層や水不溶性物質の皮膜層を均一に付着させることが難しくなる。結晶セルロースと膨潤剤の質量比は、より好ましくは、60:40〜40:60である。
【0019】
本発明の球状核中に薬物を含有しないことが、大きな膨潤効果を得るための重要なポイントとなる。球状核中に薬物が存在すると、膨潤剤が十分に水を吸って膨張するのが阻害される。吸湿性の高い薬物であれば、核内部に進入してきた水を薬物が吸水するため、その分、膨潤剤が十分に吸水・膨張しにくくなる。また疎水性の薬物であれば、核中に疎水性物質が点在することで、水が核内部に進入しにくくなり膨潤剤が十分に吸水・膨張しにくくなる。特許文献3に薬物を含有する核について記載があるが、水和した際の体積が20〜100%しか膨張しないことが記載されている。
本発明の球状核は真球度が0.7以上1以下であることが好ましい。真球度が低いと、球状核表面に薬物層や水不溶性物質の皮膜層を均一に付着させることが困難となり、望み通りの膜破裂制御ができにくくなる。真球度は高ければ高いほど好ましく、0.8以上であるとより好ましい。
また、密度は0.8g/ml以上1g/ml以下であることが好ましい。密度が低いと、コーティング時に球状核の流動が悪くなり、均一なコーティングが困難となる。さらに球状核の凝集や破損も多くなり、高品質な時限放出製剤を得ることが難しくなる。目的とする真球度や密度を得るためには、前述したように球状核製造時の適切な加水量や攪拌速度が重要なファクターとなる。
【0020】
本発明の、球状核の周りに薬物を含有する層を有する球形素顆粒は、例えば以下の方法により製造される。
球状核を遠心流動型コーティング装置中で転動させながら、結合剤含有水溶液を連続的に噴霧し、同時に薬物と必要ならば賦形剤とから成る粉体を供給し、球状核に粉体を被覆し、乾燥して球形素顆粒とする。あるいは球状核を流動層コーティング装置中で流動させながら、結合剤水溶液中に薬物を溶解あるいは懸濁させた液を噴霧し、球状核に薬物を含む粉体を被覆し、乾燥して球形素顆粒とする。
薬物の積層量は薬物の投与量によって決まるが、球状核に対して0.1〜300質量%である。本発明の球形素顆粒は球状核の表面に薬物が付着しているため、特許文献3及び4のように、予め薬物を含有する核と比較して、薬物の配合量が制限されないというメリットがある。
【0021】
結合剤としては、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、メチルセルロース、でんぷん糊、アルファー化でんぷん、ポリビニルピロリドン、アラビアガム、糖シロップ、カルボキシメチルセルロースナトリウム、プルランなどが挙げられるが、薬物が水溶性の場合、薬物水溶液自体が結合剤水溶液として作用することもある。
賦形剤としては、乳糖、コーンスターチ、結晶セルロース、白糖、D−マンニトール、アルファー化でんぷん、部分アルファー化でんぷんなどが挙げられる。
また薬物を付着させた後に、通常用いられる方法に従って球形素顆粒にシールコートを施してもよい。
【0022】
本発明の、球形素顆粒の周りに水不溶性物質を含有する皮膜層を有する球形顆粒は、例えば以下の方法により製造される。
球形素顆粒を流動させながら水不溶性物質を含有する水性懸濁液を噴霧してコーティングし、乾燥させて皮膜層を形成させ、球形顆粒とする。この時、水不溶性物質を懸濁させる溶媒として有機溶媒は好ましくない。
使用するコーティング装置としては、遠心流動型コーティング装置、流動層コーティング装置、転動型流動層コーティング装置、パン型コーティング装置など通常のコーティング機などが挙げられる。
被覆に用いる水不溶性物質の量は、薬物の放出開始時間や球状核の大きさなどによって異なるが、球状核に対して5〜200質量%であり、好ましくは10〜100質量%、さらに好ましくは10〜50質量%である。
【0023】
水不溶性物質としては、エチルセルロース、アクリル酸エチル・メタクリル酸メチルコポリマー、ヒドロキシプロピルメチルセルロースフタレート、セルロースアセテートフタレート、カルボキシメチルエチルセルロース、セルロースアセテート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、セラック、などが挙げられる。上記から選ばれる2種以上を併用してもよい。好ましくは、エチルセルロースである。
水不溶性物質を含有する皮膜層には、水不溶性物質に加えて可塑剤、ワックス、着色剤、香料、滑沢剤、凝集防止剤などの目的で添加剤を配合することができる。
【0024】
本発明の球形顆粒が経口投与されと、皮膜層を介して消化管の水分を吸収することで、中心にある球状核中の膨潤剤が徐々に膨潤する。そして一定時間後に、その球状核中の膨潤剤の膨潤による体積増加により、皮膜の破壊が起き、薬物が瞬時に全量放出される仕組みとなっている。薬物の放出が開始されるまでの時間(放出開始時間)、及び薬物の放出が開始されてから大部分の薬物が放出されるまでの時間(放出時間)は、水不溶性物質の種類や被覆量、及び中心にある球状核中の膨潤剤の種類や配合比率によって自由に調整できる。膨潤剤の配合比率が多く膨潤効果が強い球状核であるほど、また水不溶性物質の被覆量が少なく水分が浸透しやすい皮膜であるほど、放出開始時間、及び放出時間が短くなる傾向を示す。放出開始時間は30分以内が好ましく、より好ましくは15〜30分以内である。また放出時間は90分以内が好ましく、より好ましくは60分以内、さらに好ましくは30分以内である。
なお、特許文献4では、放出開始時間及び放出時間を制御する技術に関する記載があるが、核の製造や皮膜の形成に用いるアルコール濃度を調整することで、その目的を達成している。しかし有機溶媒の使用は、環境・コスト・残留など様々な問題があり、出来れば使用したくないのが一般的である。本発明では、一切有機溶媒を用いずに放出開始時間、及び放出時間を制御することが可能となる。
【実施例】
【0025】
本発明を実施例に基づいて説明する。
本発明で用いられる物性の測定方法及び条件は以下のとおりである。
まず、物性の測定方法を以下にまとめて記す。
<球状核の体積膨潤度[%]>
試料にスポイトで水を垂らして膨潤させ、5分後に水を拭き取る。膨潤前後の顕微鏡写真をとり、短径、長径を計測し、これらの平均値を直径として、膨潤前後の粒子の体積を算出する。粒子20個について実施し、その平均値を求め、以下の式より体積膨潤度とする。
体積膨潤度[%]=(膨潤後の体積−膨潤前の体積)×100/膨潤前の体積
【0026】
<球状核の真球度>
試料の顕微鏡写真をとり、短径/長径比を計測する。粒子150個について実施し、その平均値を真球度とする。
<球状核の密度[g/ml]>
スコットボリュームメーター(筒井理化学器械(株))を用いて測定する。試料を定量フィーダーにて30〜60秒かけて25mlの測定容器内にあふれるまで流下させる。次いで容器の上部に堆積した過剰量の試料をすり落とし、その後、容器に疎充填された重量を量る。容器に疎充填された重量を測定容器の容積で除した値を密度とする。3回測定し、その平均値をとる。
【0027】
<薬物の溶出試験>
溶出試験器DT−610(日本分光(株))及び分光光度計V−530(日本分光(株))を用いて測定する。一般試験法「溶出試験法」に準じて実施し、装置は「装置2」(パドル法)、パドル回転数は100rpm、試験液は日本薬局方「溶出試験第1液」を使用する。3回測定し、その平均値をとる。
<薬物の放出開始時間、放出時間>
薬物の溶出率が5%未満のときは薬物の放出が抑制されているとして、溶出率が5%未満である時間を放出開始時間とする。また放出開始時間から薬物の溶出率が80%以上となるまでの時間を放出時間とする。
【0028】
[実施例1]
結晶セルロース(吸水量2.1ml/g、200メッシュ留分30%、平均重合度220、結晶化度65%)1440gとクロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))160gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水2720g(膨潤剤に対して17.0倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=90:10)を得た。
【0029】
次に、得られた球状造粒物を転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に水分込みで1000g仕込み、温度50℃、速度301m/分(半径8cm、600rpm)で転動しながら、30分かけて蒸留水300gを少しずつ加えた後に、温度25℃、速度301m/分で30分間転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Aを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は52質量%であった。得られた球状核Aを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核A600gを転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に仕込み、薬物水分散液(3.9質量%リボフラビン、0.8質量%ヒドロキシプロピルセルロース、95.3質量%蒸留水)で噴霧・被覆し、球状核に対してリボフラビンを2.0質量%含有する球形素顆粒Aを得た。
【0030】
次いで得られた球形素顆粒A550gを転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に仕込み、水不溶性物質として13質量%エチルセルロース(セリオスコートEC−30A/旭化成ケミカルズ(株))、可塑剤として2質量%クエン酸トリエチル、85質量%蒸留水を含有する水性懸濁液を、球状核に対して10質量%(固形分)まで噴霧・被覆し、乾燥させた。その後105℃のオーブンで1時間加熱することで皮膜層を形成させた。篩で500μm以上の粒子を除去し、エチルセルロースとクエン酸トリエチルを固形分で10質量%被覆させた球形顆粒Aを得た。
【0031】
[実施例2]
結晶セルロース600gとクロスカルメロースナトリウム400gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水3950g(膨潤剤に対して9.9倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=60:40)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Bを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は45質量%であった。得られた球状核Bを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Bを用いて、実施例1と同様にして球形素顆粒Bを得た。
次いで得られた球形素顆粒Bを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Bを得た。
【0032】
[実施例3]
結晶セルロース400gとクロスカルメロースナトリウム400gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水3760g(膨潤剤に対して9.4倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=50:50)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Cを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は32質量%であった。得られた球状核Cを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Cを用いて、実施例1と同様にして球形素顆粒Cを得た。
次いで得られた球形素顆粒Cを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Cを得た。
【0033】
[実施例4]
結晶セルロース320gとクロスカルメロースナトリウム480gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水4400g(膨潤剤に対して9.2倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=40:60)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Dを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は25質量%であった。得られた球状核Dを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Dを用いて、実施例1と同様にして球形素顆粒Dを得た。
次いで得られた球形素顆粒Dを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Dを得た。
【0034】
[実施例5]
結晶セルロース210gとクロスカルメロースナトリウム490gを高速攪拌造粒機に仕込み、実施例1と同じ条件で攪拌しながら蒸留水4340g(膨潤剤に対して8.9倍の質量比)を少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物(結晶セルロース:膨潤剤=30:70)を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置に水分込みで1000g仕込み、実施例1と同じ条件で転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Eを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は13質量%であった。得られた球状核Eを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸が見られた。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Eを用いて、実施例1と同様にして球形素顆粒Eを得た。
次いで得られた球形素顆粒Eを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Eを得た。
【0035】
[比較例1]
結晶セルロース(吸水量2.1ml/g、200メッシュ留分30%、平均重合度220、結晶化度65%)のみ1600gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水1520gを少しずつ加えた後、60分間攪拌・造粒し、湿った状態の球状造粒物を得た。
次に、得られた球状造粒物を転動型流動層コーティング装置(マルチプレックスMP−01/(株)パウレック)に水分込みで1000g仕込み、温度50℃、速度301m/分(半径8cm、600rpm)で転動しながら、30分かけて蒸留水300gを少しずつ加えた後に、温度25℃、速度301m/分で30分間転動し、表面円滑化・球形化を行った。次いで、温度80℃、速度75m/分(半径8cm、150rpm)で転動しながら乾燥し、球状核を得た。
篩で300〜500μmの粒径を有する球状核Fを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は65質量%であった。得られた球状核Fを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表1に記載した。
球状核Fを用いて、実施例1と同様にして球形素顆粒Fを得た。
次いで得られた球形素顆粒Fを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で10質量%被覆させた球形顆粒Fを得た。
【0036】
[比較例2]
クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))のみ600gを高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)に仕込み、温度45℃、メインブレード264m/分(半径14cm、300rpm)、クロススクリュー251m/分(半径4cm、1000rpm)で攪拌しながら蒸留水4500g(膨潤剤に対して7.5倍の質量比)を少しずつ加えた後、60分間攪拌したが、餅状の固まりになり球状化できなかった。
【0037】
[試験例1]
前述した方法に従って、球形顆粒A〜Fにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒A〜Fに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図1に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表1に記載した。
【0038】
【表1】
【0039】
実施例1〜5、比較例1、2では、膨潤剤の配合比率について評価している。
実施例1〜5(表1及び図1)から、結晶セルロースと膨潤剤(カルボキシメチルセルロースナトリウム)の配合比率(90:10〜30:70)によって、物性の異なる球状核が得られることを確認した。また、膨潤剤の配合比率が多いほど放出開始時間、及び放出時間を短縮でき、薬物溶出が制御可能であることを確認した。
結晶セルロースと膨潤剤が90:10の比率である球形顆粒A(実施例1)は、核の膨潤度が小さいために一定の放出開始時間の後に徐放性の溶出パターンを示した。
結晶セルロースと膨潤剤が30:70の比率である球形顆粒E(実施例5)は、10分間の放出開始時間を示したが、その間に、徐々に薬物の溶出が発生し、4%近く溶出した。
【0040】
それに対して、結晶セルロースと膨潤剤が60:40〜40:60の比率である球形顆粒B〜D(実施例2〜4)は、一定の放出開始時間後にそれぞれ1時間以内で80%以上の薬物放出が観察され、非常に精度が高く調整しやすい球形顆粒が得られた。
また、球状核Eの表面に凹凸があり、他の球状核と比較するとやや形状が歪であった。それに対して、球状核A〜Dは表面が滑らかなために、薬物や水不溶性物質の皮膜を均一に形成することができる。
比較例1より、膨潤剤を含まずに結晶セルロースのみで得られる球形顆粒Fは、10時間経っても殆ど薬物が放出されず、膜破裂型時限放出製剤としては不適切であることが分かった。
比較例2より、膨潤剤のみでは球状核が得られないことが分かった。
【0041】
[実施例6]
まずは本発明で好ましい膨潤剤として用いられる、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))、アルファー化デンプン(SWELSTAR PD−1/旭化成ケミカルズ(株))、カルボキシメチルスターチナトリウム(Primojel/DMV)、低置換度ヒドロキシプロピルセルロース(L−HPC LH−21/信越化学工業(株))、クロスポビドン(ポリプラスドンXL−10/ISP)の沈降体積を以下の手順で測定した。
蒸留水75mlをビーカーに入れ、スターラーで攪拌しながら、膨潤剤1.0gを少しずつ加えた。
膨潤剤を全て投入後、3分間撹拌した。
懸濁液を100mlメスシリンダーに移し、100mlにメスアップ後、沈降管に入れた。
懸濁液を16時間静置し、沈降体積を読み取った。
その結果、沈降体積の大きい順に、カルボキシメチルスターチナトリウム:28.8ml/g、低置換度ヒドロキシプロピルセルロース:21.0ml/g、クロスカルメロースナトリウム:16.5ml/g、アルファー化デンプン:12.0ml/g、クロスポビドン:6.1ml/gであった。ここで沈降体積はその膨潤剤の膨潤性に相関すると考えられ、つまり沈降体積が大きいほど膨潤性が高いと推測される。
【0042】
本発明の球状核における体積膨潤度は、膨潤剤の膨潤性に影響されると考えられるので、最も大きな沈降体積を示したカルボキシメチルスターチナトリウム、及び最も小さな沈降体積を示したクロスポビドンの2種類で球状核を製造し、評価した。
実施例3において、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))の代わりに、カルボキシメチルスターチナトリウム(Primojel/DMV)を用いること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Gを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は30質量%であった。得られた球状核Gを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表2に記載した。
球状核Gを用いて、実施例1と同様にして球形素顆粒Gを得た。
次いで得られた球形素顆粒Gを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Gを得た。
【0043】
[実施例7]
実施例3において、クロスカルメロースナトリウム(キッコレートND−2HS/旭化成ケミカルズ(株))の代わりに、クロスポビドン(ポリプラスドンXL−10/ISP)を用いること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Hを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は39質量%であった。得られた球状核Hを高分解能走査型電子顕微鏡(SEM)で観察したところ、円滑な表面であった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表2に記載した。
球状核Hを用いて、実施例1と同様にして球形素顆粒Hを得た。
次いで得られた球形素顆粒Hを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で30質量%被覆させた球形顆粒Hを得た。
【0044】
[試験例2]
前述した方法に従って、球形顆粒G、Hにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒G、Hに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図2に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表2に記載した。
【0045】
【表2】
【0046】
実施例3、6、7では、膨潤剤の種類について評価している。
実施例3、6、7(表1、表2及び図2)から、膨潤剤の配合比率が同じであれば、その種類を変えてもほぼ同じ物性の球状核が得られることを確認した。実施例6で列挙した膨潤剤のうち、最も大きな膨潤性を示したカルボキシメチルスターチナトリウム、中程度の膨潤性を示したクロスカルメロースナトリウム、最も小さな膨潤性を示したクロスポビドンの3種類で確認できたことより、他の膨潤剤を用いても同様の結果が得られると予想される。また、球形顆粒G、H(実施例6、7)は、球形顆粒C(実施例3)と同じく、30分の放出開始時間、及び30分の放出時間を示し、同等の効果が確認できた。
【0047】
[実施例8]
実施例3において、高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)で攪拌しながら加える蒸留水の量を2000g(膨潤剤に対して5.0倍の質量比)にすること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Iを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は11質量%であった。得られた球状核Iを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸があった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表3に記載した。
【0048】
[実施例9]
実施例3において、高速攪拌造粒機(バーチカルグラニュレーターFM−VG−10P/(株)パウレック)の攪拌条件を、メインブレード879m/分(半径14cm、900rpm)、クロススクリュー754m/分(半径4cm、3000rpm)にすること以外は全く同様にして球状核を得た。
篩で300〜500μmの粒径を有する球状核Jを得た。篩分する前の全球状核に対して、300〜500μm粒径の割合は13質量%であった。得られた球状核Jを高分解能走査型電子顕微鏡(SEM)で観察したところ、表面に凹凸があった。さらに体積膨潤度、真球度、密度を前述の方法に従って求め、それらの値を表3に記載した。
【0049】
【表3】
【0050】
実施例8、9では、球状核の製造条件について評価している。
球状核C(実施例3)と比較すると、球状核Iは形状が歪(真球度が低い)で表面が粗く体積膨潤度も低かった。これは加水量が少ないために膨潤剤が十分に膨潤しきれず、結晶セルロースと膨潤剤が不均一に混合されて球状核が形成されたためと思われ、好ましい球状核を得るためには、最適な加水量があることを確認した。
球状核C(実施例3)と比較すると、球状核Jは体積膨潤度は同等だが形状が歪(真球度が低い)で表面が粗かった。これは攪拌速度が速いために適切に球状化できなかったためと思われ、好ましい球状核を得るためには、最適な攪拌条件があることを確認した。
球状核Cは、形状がほぼ真球であり、良好な製剤が得られる可能性が高い。
【0051】
[実施例10]
実施例3で得られた球形素顆粒Cを用いて、実施例1と同様にして、エチルセルロースとクエン酸トリエチルを固形分で40質量%被覆させた球形顆粒K、及び60質量%被覆させた球形顆粒Lを得た。
【0052】
[試験例3]
前述した方法に従って、球形顆粒K、Lにおける薬物の溶出試験を実施した。リボフラビンの溶出量をUV法(波長:445nm)で測定し、球形顆粒K、Lに含まれるリボフラビン量に対する溶出量の百分率で評価した。得られた溶出曲線を図3に示し、それぞれの放出開始時間(薬物の溶出率が5%未満の時間)、及び放出時間(放出開始時間から薬物の溶出率が80%以上となるまでの時間)を表4に示す。
【0053】
【表4】
【0054】
実施例10では、水不溶性物質の被覆量について評価している。
同じ球状核を用いて30質量%被覆させた球形顆粒C(実施例3)の溶出パターンと比較すると、40質量%、60質量%まで被覆させた球形顆粒K、Lは、ともに放出開始時間、及び放出時間が長くなり、水不溶性物質の被覆量によって薬物溶出が制御可能であることを確認した。水不溶性物質の被覆量が多くなると水分が浸透しにくくなるためと考えられる。
【産業上の利用可能性】
【0055】
本発明は、医薬品分野の特に時間薬物療法において好適に利用できる。精度が高く、容易に薬物溶出制御が可能な膜破裂型の時限放出製剤として利用できる。
【特許請求の範囲】
【請求項1】
結晶セルロースと膨潤剤を含む混合物からなる球状核。
【請求項2】
結晶セルロースと膨潤剤の質量比が90:10〜30:70であることを特徴とする請求項1に記載の球状核。
【請求項3】
結晶セルロースと膨潤剤の質量比が60:40〜40:60であることを特徴とする請求項1又は2に記載の球状核。
【請求項4】
吸水による体積膨潤度が100%〜700%であることを特徴とする請求項1から3のいずれかに記載の球状核。
【請求項5】
真球度が0.7以上1以下であることを特徴とする請求項1から4のいずれかに記載の球状核。
【請求項6】
密度が0.8g/ml以上1g/ml以下であることを特徴とする請求項1から5のいずれかに記載の球状核。
【請求項7】
粒径が200〜500μmであることを特徴とする請求項1から6のいずれかに記載の球状核。
【請求項8】
膨潤剤が、クロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上であることを特徴とする請求項1から7のいずれかに記載の球状核。
【請求項9】
請求項1から8のいずれかに記載の球状核の表面が薬物で被覆されていることを特徴とする球状素顆粒。
【請求項10】
請求項9に記載の球状素顆粒の表面が水不溶性物質で被覆されていることを特徴とする球状顆粒。
【請求項11】
薬物の放出開始時間が15分から30分である請求項10に記載の球形顆粒。
【請求項12】
結晶セルロースと膨潤剤を湿潤状態で球状化することを特徴とする請求項1から8のいずれかに記載の球状核を製造する方法。
【請求項1】
結晶セルロースと膨潤剤を含む混合物からなる球状核。
【請求項2】
結晶セルロースと膨潤剤の質量比が90:10〜30:70であることを特徴とする請求項1に記載の球状核。
【請求項3】
結晶セルロースと膨潤剤の質量比が60:40〜40:60であることを特徴とする請求項1又は2に記載の球状核。
【請求項4】
吸水による体積膨潤度が100%〜700%であることを特徴とする請求項1から3のいずれかに記載の球状核。
【請求項5】
真球度が0.7以上1以下であることを特徴とする請求項1から4のいずれかに記載の球状核。
【請求項6】
密度が0.8g/ml以上1g/ml以下であることを特徴とする請求項1から5のいずれかに記載の球状核。
【請求項7】
粒径が200〜500μmであることを特徴とする請求項1から6のいずれかに記載の球状核。
【請求項8】
膨潤剤が、クロスカルメロースナトリウム、アルファー化デンプン、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、クロスポビドンから選ばれる1種以上であることを特徴とする請求項1から7のいずれかに記載の球状核。
【請求項9】
請求項1から8のいずれかに記載の球状核の表面が薬物で被覆されていることを特徴とする球状素顆粒。
【請求項10】
請求項9に記載の球状素顆粒の表面が水不溶性物質で被覆されていることを特徴とする球状顆粒。
【請求項11】
薬物の放出開始時間が15分から30分である請求項10に記載の球形顆粒。
【請求項12】
結晶セルロースと膨潤剤を湿潤状態で球状化することを特徴とする請求項1から8のいずれかに記載の球状核を製造する方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2011−207851(P2011−207851A)
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願番号】特願2010−79685(P2010−79685)
【出願日】平成22年3月30日(2010.3.30)
【出願人】(303046314)旭化成ケミカルズ株式会社 (2,513)
【Fターム(参考)】
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願日】平成22年3月30日(2010.3.30)
【出願人】(303046314)旭化成ケミカルズ株式会社 (2,513)
【Fターム(参考)】
[ Back to top ]