
自己免疫障害を処置するためのグルカゴン様ペプチド−1受容体(GLP−1R)アゴニスト
【課題】 多発性硬化症を含む中枢神経系を冒す自己免疫疾患における中枢神経系組織の白血球浸潤を減少させる方法を提供することを目的とする。
【解決手段】 グルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置を必要とする哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法。
【解決手段】 グルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置を必要とする哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法。
【発明の詳細な説明】
【技術分野】
【0001】
本願は、2009年8月27日に出願された米国仮出願第61/237654号の利益を主張するものであり、この内容は全体が参照により本明細書に組み込まれる。
【0002】
(配列表に関する参照)
本願は電子出願されており、そしてテキスト形式で電子的に提出された配列表を含む。テキストファイルは、2010年8月9日に作成された「PC33892ASeqList.txt」と題する、そして30KBのサイズを有する配列表を含む。本テキストファイルに含まれる配列表は本明細書の一部であり、そしてその全体が参照により本明細書に組み込まれる。
【0003】
本発明は、多発性硬化症を含む中枢神経系を冒す自己免疫疾患の処置に関する。本発明は、中枢神経系組織の白血球浸潤を減少させるためのグルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを提供する。
【背景技術】
【0004】
多発性硬化症(MS)は、炎症、脱髄、及び軸索傷害によって特徴付けられる中枢神経系(CNS)の脱髄性自己免疫疾患である。本疾患は世界で百万人以上の人々を冒し、男性よりも女性で2倍罹患しやすい。MSの症状は、通常20歳〜40歳の間に発現する。
【0005】
MSの特徴は研究されているが、本疾患の病因は不明である。当該特徴としては、CNS組織の損傷、ミクログリアの活性化、炎症性サイトカイン産生、T細胞移動及びクローン型拡大の停止、及びマクロファージエフェクター機能変化、MHCの産生、アップレギュレーション、及びT細胞浸潤によるCNSへの直接攻撃を含む。
【0006】
実験的自己免疫性脳脊髄炎(EAE)は、MSの標準モデルと考えられる。マウス疾患モデルは、MSの病因を研究し、その処置のための薬剤を評価するために使用されている(非特許文献1)。EAEの臨床的特徴は、多数の浸潤リンパ球及びマクロファージによるCNSの炎症及び脱髄を含む。ミエリン塩基性タンパク質(MBP)、プロテオリピドタンパク質(PLP)、及びミエリンオリゴデンドロサイト糖タンパク質(MOG)を含む、ミエリンの幾つかの異なるタンパク質成分によるマウスの能動免疫は、自己免疫抗体の産生及び上行性麻痺の臨床症状を誘発する。疾患は、マウス株及び免疫に使用されるミエリンタンパク質によって急性又は慢性となり得る。EAEは、MSの病因を研究し、その処置のための薬剤を評価するのに使用されている(非特許文献1)。
【0007】
MSは大部分、神経組織に豊富に存在する脂質及び幾つかのタンパク質を含む、ミエリンに対するT細胞反応に起因する。脱髄及び臨床的麻痺は、ミエリン抗原に対して特異性を有するTh1及びTh17表現型のT細胞によるCNS組織浸潤に起因する。Th1細胞は、TNF−α及びIFN−γを含む炎症性サイトカインを産生する。IL−17発現は、MS患者、並びに関節リウマチ及び過敏性腸疾患患者で増加している(非特許文献2、3)。このことは、自己免疫疾患におけるTh17細胞の病原性役割を示唆する。CNSの損傷は、自己免疫抗体の産生及び補体活性化を含む他の免疫反応によっても引き起こされると考えられる。B細胞は、疾患の全期間にわたって認められるイソタイプのMBP特異的及びMOG特異的抗体を有するMS及びEAEの早期及び晩期の期間に関わる。脳及び脊髄組織から調製した切片は、白血球浸潤(特にリンパ球及びマクロファージ)及び神経系の基になる組織の破壊を呈する。
【0008】
グルカゴン様ペプチド(7−36)アミド(GLP−1)は、ラット及びヒトの両方におけるグルコインクレチンである(非特許文献4、5)。インスリン分泌の増加及びグルカゴン分泌の減少に加えて、30アミノ酸のGLP−1ペプチドはプロインスリン遺伝子転写を促進し、胃内容排出時間を減速し、そして食物摂取を減少させる。GLP−1は、グルカゴン様ペプチド1受容体(GLP−1R)に結合することによってその生理作用を示す。GLP−1Rは、多くの重要な生理的及び病態生理的過程を調節する、Gタンパク質結合受容体(GLCR)スーパーファミリーのクラスB受容体サブクラスに属する。GLP−1Rは、Gタンパク質活性化、cAMP産生増加及びPKAの活性化に連結する7回膜貫通受容体である。また、GLP−1Rを通して開始されるPKA非依存性反応である。GLP−1の作用に対する他の反応は、例えば、膵β細胞増殖及びβ細胞アポトーシスの減少と同時に増殖を含む。加えて、GLP−1活性は、膵臓細胞におけるグルコース輸送体−2(GLUT2)及びグルコキナーゼ遺伝子の発現増加をもたらすことができる。
【0009】
GLP−1は、また、孤束核(NTS)の尾部領域のニューロンによっても合成されるニューロペプチドである(非特許文献6)。GLP−1免疫反応性線維及びGLP−1Rは、脳全体に広く発現しているように見える(非特許文献6、7、8)。GLP−1R欠損マウスは、海馬体細胞におけるGlp1r遺伝子導入後の表現型修正を伴う、カイニン酸投与後の発作強度及びニューロン損傷を増強している (非特許文献9)。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Aharoni, R. et al., 2005, J. Neurosci. 25:8217-28
【非特許文献2】Komiyama, Y. et al., 2006, J. Immunol. 177, 566-573
【非特許文献3】Matusevicius, D. et al., 1999, Mult. Scler. 5, 101-104
【非特許文献4】Dupre and Ebert and Creutzfeld, 1987, Diabetes Metab. Rev. 3
【非特許文献5】Mojsov et al., 1986, J. Biol. Chem. 261:11880
【非特許文献6】Jin, S. L. et al., 1988, J Comp Neurol. 271:519-532
【非特許文献7】Larsen, P. J. et al., 1997, Neuroscience 77:257-270
【非特許文献8】Merchenthaler, I. et al., 1999, J Comp Neurol 403:261-280
【非特許文献9】During, M. J. et al., 2003, Nat Med. 9(9):1173-9
【発明の概要】
【発明が解決しようとする課題】
【0011】
1つの態様では、本発明は、GLP−1Rアゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置が必要な哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法を提供する。本発明は、哺乳動物の中枢神経系組織の白血球浸潤を減少させるために使用される薬剤の製造における、GLP−1Rアゴニストの使用を更に提供する。本発明は、哺乳動物のTh1及びTh17細胞数を減少させるために使用される薬剤の製造における、GLP−1Rアゴニストの使用を更に提供する。
【課題を解決するための手段】
【0012】
幾つかの実施態様では、哺乳動物は自己免疫障害を有する。1つの実施態様では、自己免疫障害は実験的自己免疫性脳脊髄炎である。別の実施態様では、自己免疫障害は多発性硬化症である。他の実施態様では、自己免疫障害は、免疫拒絶、移植片対宿主病、ブドウ膜炎、視神経症、視神経炎、横断性脊髄炎、炎症性腸疾患、関節リウマチ、強直性脊椎炎、全身性エリテマトーデス、重症筋無力症、又はグレーブス病と関連している。好ましい実施態様では、哺乳動物はヒトである。
【0013】
幾つかの実施態様では、GLP−1Rアゴニストを含む組成物の投与は、哺乳動物のリ
ンパ節においてTh1及び/又はTh17細胞数を減少させる。幾つかの実施態様では、GLP−1Rアゴニストを含む組成物の投与は、哺乳動物のリンパ節においてTer119+赤血球系細胞を減少させる。
【0014】
好ましい実施態様では、浸潤白血球はT細胞及びマクロファージを含む。特定の実施態様では、浸潤白血球はCD3発現及びCD68発現白血球を含む。好ましい実施態様では、中枢神経系組織は脳組織又は脊髄組織である。
【0015】
幾つかの実施態様では、GLP−1RアゴニストはOAP−189である。他の実施態様では、GLP−1RアゴニストはDPP−4阻害剤である。他の実施態様では、GLP−1Rアゴニストは抗GLP−1Rアゴニスト抗体である。
【0016】
幾つかの実施態様では、GLP−1Rアゴニストは天然GLP−1Rアゴニストである。幾つかの実施態様では、天然GLP−1Rアゴニストはエキセンジン−4である。関連実施態様では、天然GLP−1Rアゴニストはエキセンジン−4のフラグメント又は誘導体を含む。幾つかの実施態様では、エキセンジン−4のフラグメント又は誘導体は、GLP−1Rに結合してそれを活性化する。
【0017】
幾つかの実施態様では、GLP−1Rアゴニストは、GLP−1Rアゴニストペプチド及び抗体を含むGLP−1Rアゴニスト抗体コンジュゲート(「GAC」)を含む。関連する実施態様では、GACは以下の構造:
【化1】

式中、
ペプチドは以下の式:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14
E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40 (配列番号3)]−R2
式中、
R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;R2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であり、そしてX2はAib、A、S、T、V、L、I、D−Alaなどの遮断基であり;X10はV、L、I、又はAであり;X12はS又はKであり;X13はQ又はYであり;X14はG、C、F、Y、W、M、又はLであり;X16はK、D、E、又はGであり;X17はE又はQ;X19はL、I、V、又はAであり;X20はオルニチン、又はK(SH)Rなどの誘導体化リジン基、又はKであり;X21はL又はEであり;X23はI又はLであり;X24はA又はEであり;X25はW又はFであり;X26はL又はIであり;X27はI、K、又はVであり;X28はR、オルニチン、N、又はKであり;X29はAib又はGであり;X30は任意のアミノ酸、好ましくはG又はRであり;X31はP又は不存在であり;X32はS又は不存在であり;X33はS又は不存在であり;X34はG又は不存在であり;X35はA又は不存在であり;X36はP又は不存在であり;X37はP又は不存在であり;X38はP又は不存在であり;X39はS又は不存在であり;そしてX40は連結残基又は不存在であり;そしてここで、X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39、又はX40の1つは、リンカーを介して抗体の結合部位に共有結合した求核性側鎖を含む連結残基で置換され、ここで連結残基はK、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン、及びホモセリンから成る群から選択される;
を有するものであってよい。
【0018】
幾つかの実施態様では、GACは以下の構造:
【化2】
を含むことができる。
【0019】
幾つかの実施態様では、GACは以下の構造:
【化3】
を含むことができる。
【0020】
幾つかの実施態様では、抗体は、完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、二重特異性抗体及びミニ抗体から成る群から選択される。幾つかの実施態様では、抗体は、IgG1、IgG2、IgG3、及びIgG4から成る群から選択される定常ドメインを含む。幾つかの実施態様では、抗体は触媒抗体である。幾つかの実施態様では、抗体はアルドラーゼ抗体である。幾つかの実施態様では、アルドラーゼ抗体は、配列番号11に記載のアミノ酸配列を含む軽鎖、及び配列番号12に記載のアミノ酸配列を含む重鎖アミノ酸配列を含む。
【0021】
他の実施態様では、本発明は、GLP−1Rを活性化するのに有効量のGLP−1Rアゴニスト及び使用説明書を含む、中枢神経系組織の白血球浸潤を低減するための部品キットを提供する。関する実施態様では、本発明は、GLP−1Rを活性化するのに有効量のGLP−1Rアゴニスト及び使用説明書を含む、中枢神経系を冒す自己免疫障害を処置するするための部品キットを提供する。
【0022】
本発明のこれらの及び他の態様は、以下の記述及び実施例から明らかになるであろう。
【図面の簡単な説明】
【0023】
【図1】MOG免疫後のEAE動物中の浸潤細胞体、T細胞、単球及びミクログリアを示す組織化学を図示する。EAE動物(上側パネル)及び健常動物(下側パネル)からの脊髄切片を、浸潤細胞体(左)、浸潤T細胞を検出する抗CD3抗体(中間)、又は単球及びミクログリアを検出する抗CD68抗体(右)を染色するために、クレシルバイオレットで染色した。
【図2】MOG免疫後に1mg/kgのGLP−1Rアゴニストで処置した動物における、EAEの臨床的悪性度を示すグラフを図示する。動物に、1mg/kgエキセンジン−4(EXE4)、2mg/kgニューロトロフィン−4(NT4)、又はPBS(対照)のいずれかを、MOG免疫後に日0から日9まで毎日与えた。マウスを以下のスコアリング・システムによりEAEの臨床徴候を毎日評価した:0=正常;1=尾跛行;2=やや後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。
【図3】MOG免疫後に1mg/kgのGLP−1Rアゴニストで処置したEAE動物における、病的状態を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日、又はGLP−1アゴニストGAC−1(1mg/kg)で週1回処置した。臨床スコアを毎日モニターし、上記のように評価した。
【図4】MOG免疫後に種々の用量のGLP−1Rアゴニスト(エキセンジン−4又はGAC−1)で処置した、EAE動物における病的状態を示すグラフを図示する。エキセンジン−4(EXE4)処置した動物を、MOG免疫後日0から日2まで毎日3mg/kgエキセンジン−4で処置し、そして日3から日8まで毎日1mg/kgエキセンジン−4で処置した。GAC−1処置動物を、日0に3mg/kgGAC−1で処置し、続いて免疫後日7に始めて1mg/kgGAC−1を週1回処置した。対照動物は、対照PBSで毎日処置した。臨床スコアを毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図5】A〜Cは、(A)PBS対照処置及び(B)エキセンジン−4処置及び(C)GAC−1処置をした、EAE動物からの脊髄切片の組織化学染色の結果を図示する。切片のミエリンをルクソールファーストブルーで染色した。黒矢印は、ルクソールファーストブルー染色低下を示す白質の脱髄を示す。
【図6】A〜Fは、PBS対照(A、D)及びエキセンジン−4処置(B、E)及びGAC−1処置(C、F)とした、EAE動物からの脊髄切片の免疫組織化学染色の結果を図示する。切片は、CD3(A〜C)又はCD68(D〜F)に特異的な抗体で染色された。呈示切片は脊髄断面切片の前角である。
【図7】MHCクラスIIに特異的な抗体で染色された脳、及び脊髄からのCD11b+CD45hi細胞(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo細胞(静止ミクログリア)集団の、フローサイトメトリー分析からのデータを示すグラフを図示する。各グラフにおいて、エキセンジン−4(EXE4)処置をしたEAE動物からの細胞のMHCクラスII発現は黒実線(□)で表わされ、そして対照処置をしたEAE動物からの細胞のMHCクラスII発現は点線
【化4】
で表わされる。上側パネル:疾患発症時の動物から採取した細胞。下側パネル:疾患発症時の動物から採取した細胞。
【図8】EAEの養子免疫伝達モデルで脾細胞の病原性を示すグラフを図示する。左のグラフ:動物にPLPp(139−151)免疫後、日0から日4まで1mg/kgエキセンジン−4(EXE4)又はPBS(対照)を毎日与えた。動物を免疫後日5に犠牲にした。右のグラフ:エキセンジン−4処置ドナー(▽)及びPBS処置ドナー(■)からの脾細胞をナイーブSJL/J動物に伝達した。レシピエントマウスの臨床スコア及び体重変化を毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図9】A〜Cは、GLP−1Rアゴニスト処置後のリンパ節重量及び脾臓重量及びサイズを図示する。動物は、MOG免疫後日0から日5まで、1mg/kgエキセンジン−4(EAE−EXE4)、PBS(EAE−ビヒクル)又は4mg/kgデキサメタゾン対照(EAE−Dex)で毎日処置した。ナイーブ動物はMOGで免疫されなかった。(A)鼠径リンパ節重量。(B)脾臓重量。(C)ナイーブ動物、ビヒクルで処置したEAE動物及びエキセンジン−4で処置したEAE動物からの脾臓。
【図10】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ球(A−C)及び単球(D−F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸は、リンパ球(A−C)又は単球(D−F)である細胞の割合を示す。
【図11】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。CD4+細胞(A〜C)及び細胞CD8+(D〜F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸には、表示されたマーカーに対する陽性染色細胞の割合が示される。
【図12】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。CD19+細胞(A〜C)及びTer119+細胞(D〜F)をリンパ球(A〜C)及び単球(D〜F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸には、表示されたマーカーに対する陽性染色細胞の割合が示される。
【図13】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるTer119+細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。脾臓細胞は、赤血球系統マーカーTer119で表面染色された。Ter119に陽性の染色細胞の割合をy−軸に示す。
【図14】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における活性化T細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ節からの活性化T細胞は、高レベルのCD44(CD44hi)を有するCD4+細胞と同定された。y−軸は、処置群及びナイーブ群のそれぞれにおける、高レベルのCD44を有するCD4+細胞の割合を表わす。
【図15】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における増殖細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ節細胞をMOG刺激により培養し、そして細胞増殖を測定するためにBrdUで処置した。y−軸は、処置群及びナイーブ群のそれぞれにおける、BrdU陽性のCD4+の割合を表わす。
【図16】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるIL−17+細胞集団変化を示すグラフを図示する。動物を、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5(左図)及び日7(右図)に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。鼠径リンパ節細胞は、CD4、IL−17及びIFN−γで染色された。IL−17に対して陽性染色のCD4+細胞の割合をy−軸に示す。
【図17】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるインターフェロン−γ(IFN−γ)細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。鼠径リンパ節細胞は、CD4、IL−17及びIFN−γで染色された。IFN−γに対して陽性染色のCD4+細胞の割合をy−軸に示す。
【図18】MOG免疫後にGLP−1Rアゴニスト(エキセンジン−4)又はDPP−4阻害剤(シタグリプチン)で処置した、EAE動物における病的状態を示すグラフを図示する。MOG免疫動物に、1mg/kgシタグリプチン、10mg/kgシタグリプチン、エキセンジン−4、又はメチルセルロースを投与した。臨床スコアを毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図19】GLP−1Rアゴニスト(エキセンジン−4)又はビヒクルで処置した、EAE動物の病的状態を示すグラフを図示する。免疫後日29に始めて、PLP免疫SJL/Jマウスに、1mg/kgエキセンジン−4又はビヒクルを毎日投与した。臨床スコアを毎日モニターした。EAEの臨床徴候を以下のように評価した:0、麻痺なし;1、尾色調欠失;2、後肢弱;3、後肢麻痺;4、後肢及び前脚麻痺;5、瀕死又は死亡。
【図20】エキセンジン−4又はPBSで処置した迷走神経切離又は迷走神経非切離EAE動物における、病的状態を示すグラフを図示する。マウスを、以下のスコアリング・システムに従い、EAEの臨床徴候について毎日評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。
【発明を実施するための形態】
【0024】
本発明は、多発性硬化症及び他の自己免疫障害の処置のためのGLP−1Rアゴニストを使用する方法に関する。
【0025】
一般的方法
本発明は、分子生物学、細胞生物学及び免疫学の分野で使用される従来技術を採用する。当該技術は、「分子クローニング:実験室マニュアル、第2版」(Molecular Cloning: A Laboratory Manual, second edition) (Sambrook, et al., 1989) Cold Spring Harbor Press;「オリゴヌクレオチド合成 」(Oligonucleotide Synthesis) (M.J. Gait, ed., 1984);「分子生物学の方法」(Methods in Molecular Biology), Humana Press;「細胞生物学:実験室ノート」(Cell Biology: A Laboratory Notebook)(J.E. Cellis, ed., 1998) Academic Press;「動物細胞培養」(Animal Cell Culture)(R.I. Freshney, ed., 1987);「細胞及び組織培養入門」(Introduction to Cell and Tissue Culture) (J.P. Mather and P.E. Roberts, 1998) Plenum Press; 「細胞及び組織培養:実験室操作」(Cell and Tissue Culture: Laboratory Procedures) (A. Doyle, J.B. Griffiths, and D.G. Newell, eds., 1993-8) J. Wiley and Sons;「酵素学における方法」(Methods in Enzymology) (Academic Press, Inc.);「実験免疫学便覧」(Handbook of Experimental Immunology) (D.M. Weir and C.C. Blackwell, eds.);「哺乳動物細胞での遺伝子移入ベクター」(Gene Transfer Vectors for Mammalian Cells) (J.M. Miller and M.P. Calos, eds., 1987);「分子生物学の最近のプロトコール」(Current Protocols in Molecular Biology) (F.M. Ausubel, et al., eds., 1987);「PCR:ポリメラーゼ連鎖反応」(PCR: The Polymerase Chain Reaction), (Mullis, et al., eds., 1994);「免疫学における最近のプロトコール」(Current Protocols in Immunology) (J.E. Coligan et al., eds., 1991);「分子生物学における短プロトコール」(Short Protocols in Molecular Biology) (Wiley and Sons, 1999); Immunobiology (C.A. Janeway and P. Travers, 1997);「抗体」(Antibodies) (P. Finch, 1997);「抗体:実践的アプローチ」(Antibodies: a practical approach) (D. Catty., ed., IRL Press, 1988-1989);「モノクローナル抗体:実践的アプローチ」(Monoclonal antibodies : a practical approach) (P. Shepherd and C. Dean, eds., Oxford University Press, 2000);「抗体の使用法:実験室マニュアル」(Using antibodies: a laboratory manual) (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999);「抗体」(The Antibodies) (M. Zanetti and J.D. Capra, eds., Harwood Academic Publishers, 1995)などの参考文献中に記載されている。
【0026】
定義
以下の略語、用語及び語句は、下記に定義されているように本明細書で使用される。特に明記しない限り、本明細書に記載の本発明に関連して使用される科学及び技術用語は、当業者により一般的に理解される意味を有する。更に、文脈のよって規定されない限り、単数用語は複数を含み、そして複数用語は単数を含むものとする。
【0027】
【表1】
【0028】
例えば、D−Ala又はN−Me−D−Ileのような「D」接頭辞により特に指示がない限り、本明細書に記載のペプチド中のアミノ酸及びアミノアシル残基のα炭素の立体化学は、「L」立体配置である。 Cahn-Ingold-Prelog の「R」及び「S」記号は、ペプチドのN−末端で特定のアシル置換基におけるキラル中心の立体化学を明示するために使用される。記号「R、S」は2つのエナンチオマー型のラセミ混合物を示すことを意味する。この命名法は、R. S. Cahn, et al., Angew. Chem. Int. Ed. Engl., 5:385-415 (1966)に記載のものに従う。
【0029】
D−HはDヒスチジンを指す。
本明細書で使用される2−アミノイソ酪酸は、以下の構造:
【化5】
を有する。
【0030】
「ポリペプチド」、「ペプチド」、及び「タンパク質」は、アミノ酸残基の重合体を表すために互換性を持って使用される。本明細書で使用されるこれらの用語は、1つ又はそれ以上のアミノ酸残基が対応する天然アミノ酸の人工化学的アナログである、アミノ酸重合体に適用してもよい。これらの用語は、天然アミノ酸重合体に適用してもよい。ペプチドの結合機能が維持される限り、アミノ酸はL又はD型であってよい。ペプチドは環状であってよく、ペプチド内の2つの非隣接アミノ酸間で分子内結合、例えば、骨格から骨格、側鎖から骨格及び側鎖から側鎖への環化を有してもよい。環状ペプチドは、当技術分野で周知の方法によって製造することができる。例えば、米国特許第6013625号;S. Cheng et al., J Med. Chem. 37:1-8 (1994)を参照されたい。
【0031】
すべてのペプチド配列は一般に認められている慣例に従って書かれるが、それによれば、α−N末端アミノ酸残基は左側であり、そしてα−C末端アミノ酸残基は右側である。本明細書で使用される用語「N末端」は、ペプチド中のアミノ酸の遊離α−アミノ基を指し、そして用語「C末端」はペプチド中のアミノ酸の遊離カルボン酸末端を指す。1つの基を有するN末端であるペプチドは、N末端アミノ酸残基のα−アミノ窒素上に1つの基を保持するペプチドを指す。1つの基を有するN末端であるるアミノ酸は、α−アミノ窒素上に1つの基を保持するアミノ酸を指す。
【0032】
「抗体」は、免疫グロブリン分子の可変領域に位置する少なくとも1つの抗原認識部位を通して、糖質、ポリヌクレオチド、脂質、ポリペプチドなどの標的への特異的結合が可能な免疫グロブリン分子である。本明細書で使用される用語は、インタクトポリクローナル又はモノクローナル抗体だけでなく、そのフラグメント(Fab、Fab’、F(ab’)2、Fv)、一本鎖(scFv)及びドメイン抗体(例えば、サメ及びラクダ科抗体を含む)、及び抗体部分を含む融合タンパク質、更に抗原認識部位を含む免疫グロブリン分子のその他の修飾立体配置のいずれをも包含する。抗体はIgG、IgA、又はIgM(又はそのサブクラス)などいずれのクラスの抗体をも含み、そして抗体はいずれかの特定のクラスである必要はない。その重鎖の定常ドメインの抗体アミノ酸配列に応じて、免疫グロブリンは異なるクラスに帰属することができる。免疫グロブリンには5つの主要クラス:IgA、IgD、IgE、IgG、及びIgMがあり、そしてこれらの幾つかはサブクラス(イソタイプ)、例えば、IgG1、IgG2、IgG3、IgG4、IgA1及びIgA2に更に分けられる。異なるクラスの免疫グロブリンに対応する重鎖定常ドメインは、それぞれα、δ、ε、γ、及びμと呼ばれる。異なるクラスの免疫グロブリンのサブユニット構造及び3次元立体配置は周知である。
【0033】
本明細書で使用される「浸潤白血球」は、自己免疫疾患、優先してCNSを冒す自己免疫疾患の結果として、脳及び脊髄組織を含む中枢神経系(CNS)組織へ侵入、浸潤、又は移動する白血球である。浸潤白血球は、主としてT細胞及び単球であるが、他の白血球が存在してもよい。
【0034】
本明細書で使用される「白血球浸潤減少」は、脳及び脊髄組織を含むCNS組織へ白血球の移動(即ち、侵入又は浸潤)を減少させることを指す。白血球浸潤減少は、特にCNS組織を構成する神経細胞及び/又は他の支持細胞について、白血球浸潤によって媒介される細胞毒性を減少させることを指す。白血球浸潤は、T細胞及び単球によって媒介される侵入を含む。白血球浸潤減少は、自己免疫攻撃からCNS組織を守ることを含む。白血球浸潤によって破壊されるCNSの細胞は、ミエリン発現細胞及び隣接非ミエリン発現細胞を含む。細胞破壊は、アポトーシス、壊死、又はその組み合わせであってよい。白血球浸潤減少は、疾患進行減速、病的状態の発症または重症度の遅延、生存期間の延長、クオリティオブライフの改善、認知、運動又は行動症状の減少又は安定化などの臨床的適応によって特徴付けられる。白血球浸潤減少は、中枢神経系(CNS)組織への白血球の移動リスクを抑制し又は減少させることも含む。白血球浸潤減少は、部分的又は完全であってもよく、例えば、その減少は、本明細書に記載のように相対的又は実質的減少で、約10%、約20%、約30%、約40%、約50%、約60%、約70%、約80%、約90%、又は更に約95%であってよい。
【0035】
本明細書で使用される「GLP−1R」は、GLP−1Rの活性の少なくとも一部を保持する、いずれかの型のGLP−1R及びその変異体を指す。ヒトGLP−1Rに対する特定の言及によるような、特に別の指示がない限り、GLP−1Rは、天然配列GLP−1Rの全哺乳動物種、例えば、ヒト、イヌ、ネコ、ウマ、及びウシを含む。
【0036】
本明細書で使用される「GLP−1受容体アゴニスト」又は「GLP−1Rアゴニスト」は、GLP−1Rの活性化量を増加させる分子であり、天然アゴニストGLP−1及びエキセンジン−4によって産生するものと類似の効果をもたらす。GLP−1Rアゴニストは、例えば、GLP−1Rに結合して活性化させることにより、GLP−1Rに立体配座変化を引き起こすことにより、GLP−1Rに結合したGタンパク質の活性化を引き起こすことにより、GLP−1Rを長期間(無期限を含む)活性化(例えば、活性立体配座において)状態にさせることにより、天然アゴニストの結合を調節することにより、GLP−1のインヒビターをブロック、又はその反対にGLP−1R活性化を調節し若しくはGLP−1R活性化に特徴的な細胞内イベントのカスケードを開始させることにより、GLP−1Rの活性化を増加させる。GLP−1Rアゴニストの好ましい特性は、本明細書に記載されている。本発明のGLP−1Rアゴニストは、少なくとも5%、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%、少なくとも100%、少なくとも200%、又はそれ以上GLP−1Rの活性化を増加することができる。
【0037】
本明細書で使用される「天然GLP−1Rアゴニスト」は、自然に存在しGLP−1Rの活性剤として機能する分子である。既知のGLP−1Rの天然アゴニストは、ペプチドホルモンGLP−1及びエキセンジン−4である。天然GLP−1Rアゴニストは、突然変異したGLP−1R対立遺伝子を有する動物に発現したポリペプチドなどの天然変異体分子を含む。
【0038】
「生物活性」は、本発明のGLP−1Rアゴニストと併せて使用する場合、一般に、GLP−1Rを結合及び活性化する能力及び/又はGLP−1Rシグナル伝達機能によって媒介される下流経路を有することを指す。本明細書で使用される「生物活性」は、GLP−1R発現細胞上で、GLP−1Rの天然リガンドとして、GLP−1の作用によって誘導されるものに共通した1つ又はそれ以上のエフェクター機能を包含する。生物活性は、以下:GLP−1Rを結合し活性化する能力;GLP−1Rに結合し活性化すること、GLP−1Rの立体配座変化を引き起こす能力、GLP−1Rに結合したGタンパク質の活性化を引き起こす能力、GLP−1Rを長期間(無期限を含む)活性化(例えば、活性立体配座において)状態にさせる能力、細胞内cAMPを増加させる能力、インスリン放出を促進する能力、及びGLP−1R活性化に特徴的な細胞内イベントのカスケードを開始させる能力、のいずれか1つ又はそれ以上を含むが、これらに限定されない。
【0039】
本明細書で使用される「完全アゴニスト」は、有効濃度でGLP−1Rの測定可能な効果を、本質的に完全に誘導するアゴニストである。例えば、GLP−1Rの測定可能な効果は、cAMPレベル増加であってよい。部分アゴニストとは、測定可能な効果を部分的に誘導することができるが、しかし最大濃度でも完全アゴニストではないアゴニストを意味する。実質的に完全とは、少なくとも約80%、好ましくは少なくとも約90%、より好ましくは少なくとも約95%、及び最も好ましくは少なくとも98%の測定可能な効果が誘導されることを意味する。関連する「測定可能な効果」は、本明細書に記載されている。
【0040】
本明細書で使用される「フラグメント」ポリペプチドは、少なくとも幾つかの生物特性を保持する大型ポリペプチド、即ち、GLP−1Rを活性化する能力などを有する大型ポリペプチドの一部分である。好ましいフラグメントは、大型ポリペプチドの生物特性をもたらすアミノ酸残基及び/又は構造又は大型ポリペプチドを含む。ポリペプチドフラグメントはペプチドとも呼ばれるが、本明細書ではポリペプチドとペプチド間で区別はされない。典型的フラグメントは本明細書に記載されている。フラグメントは、本明細書に記載のように誘導体化されてもよい。
【0041】
本明細書で使用される「誘導体」ポリペプチドは、官能基又は官能部分の付加又は除去などの、1つ又はそれ以上の共有若しくは非共有修飾を有する。誘導体の例は、本明細書に提供される。
【0042】
本明細書で使用される特定のポリペプチド配列の「変異体」は、デフォルト・パラメータで設定されたClustal V又はBLAST、例えば、「BLAST 2 Sequence」ツール、バージョン2.0.9(1999年5月7日)などのアルゴリズムを用いて、特定長さの1つのポリペプチド配列を超える特定のポリペプチド配列と、少なくとも40%配列同一性を有するポリペプチド配列と定義される。当該の1対のポリペプチドは、1つのポリペプチドのある規定の長さにわたって、例えば、少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも91%、少なくとも92%、少なくとも93%、少なくとも94%、少なくとも95%、少なくとも96%、少なくとも97%、少なくとも98%、又は少なくとも99%以上の配列同一性を示してもよい。
【0043】
ポリペプチド配列に適用され本明細書で使用される「配列同一性」は、Clustal V、MEGALIGN、又はBLASTなどの標準化アルゴリズムを用いて整列された、少なくとも2つのポリペプチド配列間の残基整合の割合を指す。ポリペプチド配列アラインメントの方法は周知である。幾つかのアラインメント法は、保存アミノ酸配列を考慮に入れる。本明細書に記載の当該保存配列は、一般的に、置換部位での電荷と疎水性を保存し、その結果ポリペプチドの構造(及び機能)を保存する。
【0044】
用語「ポリペプチド」、「オリゴペプチド」、「ペプチド」及び「タンパク質」は、いずれの長さの、好ましくは相対的に短い(例えば、10〜100アミノ酸)アミノ酸鎖をも指すように、本明細書では互換性を持って使用される。アミノ酸鎖は直鎖状又は分枝鎖状であってよく、修飾アミノ酸を含んでもよく、及び/又は非アミノ酸によって遮断されてもよい。この用語は、天然修飾又は遮断;例えば、ジスルフィド結合形成、グリコシル化、脂質化、アセチル化、リン酸化、又は標識成分との結合などその他の操作又は修飾によるアミノ酸鎖を包含する。また、例えば、アミノ酸の1つ又はそれ以上のアナログ(例えば、非天然アミノ酸などを含む)、並びに当技術分野で公知の他の修飾を含むポリペプチドも、本定義の範囲内である。当然のことながら、ポリペプチドは、一本鎖又は会合鎖として存在し得る。
【0045】
当技術分野で公知の本明細書において互換性を持って使用される「ポリヌクレオチド」又は「核酸」は、いずれかの長さのヌクレオチド鎖を指し、そしてDNA及びRNAを含む。ヌクレオチドは、デオキシリボヌクレオチド、リボヌクレオチド、修飾ヌクレオチド又は塩基、及び/又はそれらのアナログ、又はDNA若しくはRNAポリメラーゼにより鎖中に取り込むことができるいずれの基質であってもよい。ポリヌクレオチドは、メチル化ヌクレオチド及びそれらのアナログなどの修飾ヌクレオチドを含んでもよい。存在するならば、ヌクレオチド構造への修飾は鎖の会合前又は後になされてもよい。ヌクレオチド配列は、非ヌクレオチド成分により遮断される。ポリヌクレオチドは、標識成分と併せることによるなど、重合後に更に修飾してもよい。他のタイプの修飾は、例えば、「caps」、アナログによる1つ又はそれ以上の天然ヌクレオチドの置換、例えば、非荷電結合(例えば、ホスホン酸メチル、ホスホトリエステル、ホスホアミダート、カルバメートなど)による及び荷電結合(ホスホロチオアート、ホスホロジチオアートなど)によるもの、例えば、タンパク質(例えば、ヌクレアーゼ、毒素、抗体、シグナルペプチド、ポリ−L−リジンなど)などのペンダント部分を含むもの、挿入剤(例えば、アクリジン、プソラレンなど)によるもの、キレート化剤(例えば、金属、放射性金属、硼素、酸化性金属など)を含むもの、アルキル化剤を含むもの、修飾結合(例えば、αアノマー核酸など)によるものなどのヌクレオチド間修飾、並びに1つ又は複数のポリヌクレオチド非修飾型を含む。更に、糖類に通常存在するいずれかのヒドロキシル基も、例えば、ホスホネート基、ホスフェイト基により置換され、標準保護基により保護され、若しくは更なるヌクレオチドへの更なる結合の調製のために活性化されてもよく、又は固体支持体に結合されてもよい。5’及び3’末端OHは、リン酸化され又はアミン若しくは1〜20炭素原子の有機キャッピング基部分で置換することができる。他のヒドロキシル基も、標準的な保護基へ誘導体化してもよい。ポリヌクレオチドは、例えば、2’−O−メチル−、2’−O−アリル、2’−フルオロ−又は2’−アジド−リボース、炭素環糖類アナログ、α又はβアノマー糖類、アラビノース、キシロース又はリキソースなどのエピマー糖類、ピラノース糖類、フラノース糖類、セドヘプツロース、非環式糖アナログ及びメチルリボシドなどの脱塩基ヌクレオチドアナログを含む、一般的に当技術分野で公知のリボース又はデオキシリボース糖類の類似形も含むことができる。1つ又はそれ以上のホスホジエステル結合は、代わりの結合基によって置換してもよい。これらの代わりの結合基は、ホスフェイトがP(O)S(「チオアート」)、P(S)S(「ジチオアート」)、(O)NR2(「アミダート」)、P(O)R、P(O)OR’、CO又はCH2(「ホルムアセタール」)によって置換され、ここで、各R又はR’は、独立に、H、又は場合によりエーテル(−O−)結合、アリール、アルケニル、シクロアルキル、シクロアルケニル又はアラルキルを含む、置換若しくは非置換アルキル(1〜20℃)である実施態様を含むが、これらに限定されない。ポリヌクレオチド中の全結合が同一である必要はない。前記の説明は、RNA及びDNAを含み、本明細書で言及する全ポリヌクレオチドに適用される。
【0046】
本明細書で使用される「実質的に純粋」は、少なくとも50%純粋(即ち、不純物無し)、より好ましくは少なくとも90%純粋、より好ましくは少なくとも95%純粋、尚より好ましくは少なくとも98%純粋、そして最も好ましくは少なくとも99%純粋である物質を指す。
【0047】
「宿主細胞」は、ポリヌクレオチド挿入部分の取り込みのために1つ又は複数のベクターのレシピエントになり得るか又はなっている個々の細胞又は細胞培養液を含む。宿主細胞は単一宿主細胞の子孫を含み、そして子孫は、自然の、偶発的、又は意図的な突然変異の故に元の親細胞と必ずしも完全に同一(形態において又はゲノムDNA相補性において)である必要はない。宿主細胞は、本発明の1つ又は複数のポリヌクレオチドにより、インビボでトランスフェクトされる細胞を含む。
【0048】
本明細書で使用される「処置」は、有益な又は所望の臨床結果を得るためのアプローチである。本発明の目的として、有益な又は所望の臨床結果は、以下:運動改善、脚弱減少、CNSのリンパ球浸潤減少、及びMSを含む自己免疫疾患に起因する脱髄減少の1つ又はそれ以上を含むが、これらに限定されない。
【0049】
「発症減少」は、この病態に一般的に使用される他の薬剤(例えば、それへの露出)の量及び/又は治療の必要性の減少を含んでもよい、いずれかの重症度の減少を意味する。当業者には当然のことながら、個体は処置へのそれらの反応性の点で変動してもよく、そのようなものとして、例えば、「発症を減少させる方法」は、当該投与がその特定の個体にそのような発症減少をもたらす可能性があるという合理的な予測に基づいて、GLP−1Rアゴニストを投与することを反映する。
【0050】
「改善」は、GLP−1Rアゴニストを投与しないことに比べて、1つ又はそれ以上の症状の低減又は改善を意味する。「改善」は、症状の期間の短縮又は減少をも含む。
【0051】
本明細書で使用される「GLP−1Rを活性化するのに有効な量」、又はGLP−1Rアゴニストについての同様の表現は、GLP−1Rアゴニストの投与前のベースラインレベルと比べて、GLP−1R活性化(本明細書に記載され当技術分野で公知)の増加に十分な量を指す。活性の増加は、少なくとも5%、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%、少なくとも10%、少なくとも200%、又はそれ以上であってよい。この量は、投与経路、体内でのGLP−1Rアゴニストの半減期、溶解性、バイオアベイラビリティ、クリアランス率、並びにGLP−1Rアゴニストの他の薬物動力学的特性、動物又は患者などの体重及び代謝のような考慮すべき事項を勘案するものである。
【0052】
本明細書で使用される、薬剤、化合物、又は医薬組成物の「有効投薬量」又は「有効量」は、いずれか1つ又はそれ以上の有益な又は所望の結果をもたらすのに十分な量である。予防的使用では、有益な又は所望の結果は、疾患の生化学的、組織学的及び/又は行動症状、その合併症及び疾患の進展時に現われる中間の病的表現型を含む、疾患のリスクの除去又は減少、重症度の軽減、又は発症遅延を含む。治療的使用では、有益な又は所望の結果は、例えば、CMT疾患の1つ又はそれ以上の症状の減少、疾患を処置するのに必要とされる他の薬剤の用量減少、別の薬剤の効果の強化、及び/又は患者の疾患進行遅延などの臨床結果を含む。有効投薬量は、1つ又はそれ以上の投与方法で投与することができる。本発明の目的のために、薬剤、化合物、又は医薬組成物の有効投薬量は、予防的又は治療的処置を直接的にせよ間接的にせよ達成するのに十分な量である。臨床状況において理解されているように、薬剤、化合物、又は医薬組成物の有効投薬量は、別の薬剤、化合物、又は医薬組成物と併せて達成しても又は達成しなくてもよい。従って、「有効投薬量」は、1つ又はそれ以上の治療薬を投与する状況で考慮してもよく、そして1つ又はそれ以上の他の薬剤と併せて所望の結果が得られる可能性があるか又は得られる場合、単剤は有効量で与えられるものと考えてよい。
【0053】
本明細書で使用される「処置が必要な動物」又は類似の表現は、CNSに関わる自己免疫疾患を発症するリスクを有するか又はその危険性がある動物、好ましくはヒトを含む哺乳動物を意味する。CNSを冒す自己免疫疾患(又は障害、無差別に)の例は、マウスの実験的自己免疫性脳脊髄炎(EAE)、ヒトの多発性硬化症(MS)、及び他の動物に見られる類似の自己免疫疾患である。CNSの自己免疫攻撃は、例えば、免疫拒絶、視神経症、炎症性腸疾患、及びパーキンソン病にも見られる。
【0054】
「個体」又は「対象」は、哺乳動物、より好ましくはヒトである。哺乳動物は、家畜、スポーツ動物、ペット、霊長類、ウマ、イヌ、ネコ、マウス及びラットを含むが、これらに限定されない。
【0055】
本明細書で使用される「ベクター」は、宿主細胞中の関係する1つ又はそれ以上の1つ又は複数の遺伝子若しくは1つ又は複数の配列をデリバリーし、そして好ましくは発現することができる構成物を意味する。ベクターの例は、ウイルスベクター、裸のDNA又はRNA発現ベクター、プラスミド、コスミド又はファージベクター、カチオン性縮合剤に関連するDNA又はRNA発現ベクター、リポソームにカプセル化されたDNA又はRNA発現ベクター、及び生成細胞などの特定の真核細胞を含むが、これらに限定されない。
【0056】
本明細書で使用される「発現調節配列」は、核酸の転写を方向付ける核酸配列を意味する。発現調節配列は、構成的若しくは誘導型プロモーター、又はエンハンサーなどのプロモーターであってよい。発現調節配列は転写する核酸配列に機能的に結合される。
【0057】
本明細書で使用される「薬学的に許容される担体」又は「薬学的に許容される医薬品添加剤」は、有効成分と組み合わせた場合、成分が生物活性を保持することを可能とし、そして対象の免疫系と非反応性であるいずれの物質をも含む。例としては、リン酸緩衝食塩溶液などのいずれかの標準的薬剤担体、水、油/水乳濁液などの乳剤、及び種々のタイプの湿潤剤が挙げられるが、それらに限定されない。エアロゾル又は非経口的投与のための好ましい賦形剤は、リン酸緩衝生理食塩水(PBS)又は生理食塩水(0.9%)である。当該担体を含む組成物は、周知の従来法(例えば、「Remingtonの薬学、第18版」(Remington's Pharmaceutical Sciences, 18th edition), A. Gennaro, ed., Mack Publishing Co., Easton, PA, 1990; 及び「Remington、薬剤学の科学及び実践」(Remington, The Science and Practice of Pharmacy 20th Ed.) Mack Publishing, 2000を参照)によって製剤化される。
【0058】
本明細書で使用される「末梢投与」又は「末梢血管から投与される」は、中枢神経系(CNS)又は血液脳関門(BBB)の外側で対象に薬剤を導入することを指す。末梢投与は、脊椎又は脳への直接投与以外のいずれの投与経路をも包含する。末梢投与は局所的又は全身的であってよい。
【0059】
中枢神経系(CNS)を冒す自己免疫障害の処置のための、GLP−1Rアゴニスト
本発明は、多発性硬化症及び中枢神経系(CNS)を冒す他の自己免疫障害の処置のための、GLP−1Rアゴニストの使用方法を提供する。本発明の1つの特徴として、GLP−1Rアゴニストは、CNSの組織への白血球浸潤を減少し及びCNSの構成的神経組織の破壊を減少し、その結果として脚麻痺などこの疾患の臨床症状を改善するのに有効である。特に、GLP−1Rアゴニストは、ミエリン抗原を提示し、そしてそれらを産生する細胞に対する細胞毒性作用に寄与するのに活性を有する、単球及びT細胞の移動を減少させる。
【0060】
本発明に至る観察は、十分認められているMSの動物モデル、実験的自己免疫性脳脊髄炎(EAE)マウスを用いて行なわれた。EAEは、ヒトのMSと多くの臨床的及び病態的特徴を共有する実験的病状である。多くのFDA承認のMS治療法が先ず発見され、そしてマウス及びラットにおけるEAEモデルを基準に開発された(Steinman and Zamvil,
2006により検討された)。ミエリン塩基性タンパク質(MBP)、プロテオリピドタンパク質(PLP)、及びミエリンオリゴデンドロサイト糖タンパク質(MOG)を含む、ミエリンの幾つかの異なるタンパク質成分によるEAEマウスの能動的免疫は、自己抗体の産生及び上行性麻痺の臨床症状を誘発する。例えば、MOGペプチドアミノ酸35−55(Aharoni, R., et al.)又はPLPペプチドアミノ酸139−151(PLPp(139−151))で免疫後に、EAEをC57BL/6マウスに誘発することができる。MOG又はPLPによる免疫は、MSに類似した脱髄及び病的状態を引き起こすミエリン特異的自己免疫反応を誘導する。EAEの臨床的特徴は、多数の浸潤リンパ球、単球及びマクロファージによるCNSの炎症及び脱髄を含む(図1)。
【0061】
GLP−1Rアゴニストを用いる動物実験
本発明の裏付けとして行なわれた実験では、GLP−1Rアゴニストの直接投与は、CNS特異的自己免疫疾患動物における病的状態及びCNS組織障害を減少させることが示された(図2〜4)。エキセンジン−4(EXE4)は、MOG免疫した後に続いてEAE動物に投与された。エキセンジン−4で処置した動物は、対照動物と比べて病的状態の有意な減少を示した。この結果は、GLP−1Rアゴニストの投与が、慢性EAEの進行を緩徐化させるのに有効なことを証明した。実験は、症状の発症に関してエキセンジン−4によって得られた保護が、trkBアゴニストNT4(PCT国際公開第2008/078179号を参照)によって得られた保護と少なくとも同じ(もし、より良くない場合は)であること、そしてエキセンジン−4による処置は、処置が有効になるために症状発症時点まで継続する必要はないことを示した(図2)。
【0062】
動物実験では、GLP−1Rアゴニスト化合物GAC−1の週1回の投与が、対照と比べてEAE罹病率に対して有意な保護をもたらすことを示した。GAC−1は週1回ベースで投与した場合に有効であった。実験は、GAC−1による週1回の処置が、症状発症の時点まで少なくとも1日1回のエキセンジン−4処置と同様に保護することを示した。
【0063】
組織学的分析は、対照EAE動物からの切片と比べて、GLP−1Rアゴニストによって処置した動物から調製した切片においては、脱髄及び白血球浸潤減少を示した(図5A〜C及び6A〜F)。GLP−1Rアゴニスト処置動物は、CNSCD3+細胞浸潤の証拠を事実上示さなかった。浸潤細胞の特定はCD3発現T細胞及びCD68発現マクロファージの染色によって行なわれた。GLP−1Rアゴニスト処置動物からの脊髄組織は、対照動物と比べた場合、実質的にT細胞及び単球浸潤減少を示した(図6A〜F)。CNS組織切片の組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSのリンパ球及び単球浸潤を減少させることを証明した。
【0064】
EAEマウスでの活性化マーカーMHCクラスII発現の解析は、エキセンジン−4による処置が、脳及び脊髄の両方において、CD11b+CD45hi(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo(静止ミクログリア)細胞集団の両方で、MHCクラスIIの発現レベルを減少させることを示した(図7)。エキセンジン−4を与えたEAEマウスからの静止ミクログリア、活性化ミクログリア及び浸潤マクロファージは、症状発症及び疾患ピークの両方で、ビヒクル処置対照動物からの細胞と比べてMHCクラスII発現の低下を示した。
【0065】
動物実験では、エキセンジン−4処置マウスの脾細胞は、EAEの養子免疫伝達モデルにおいて低病原性であることを示した(図8)。エキセンジン−4処置PLP誘導マウスから分離した脾細胞を受けたマウスは、対照(PBS処置)PLP誘導マウスから分離した脾細胞を受けたマウスよりも実質的に低重度の疾患を発症した。
【0066】
GLP−1Rアゴニスト処置EAEマウスの鼠径リンパ節重量は、ビヒクル処置対照EAE動物のリンパ節よりも小さかった(図9A)。対照処置EAE動物のリンパ節は実質的に重かった。GLP−1Rアゴニスト処置EAEマウスの脾臓はナイーブ動物の脾臓の重量及びサイズと同程度であり、ビヒクル処置対照EAE動物の脾臓と対照的に、実質的に重くそして大きかった(図9B〜C)。
【0067】
EAE動物のGLP−1Rアゴニスト処置に由来する免疫学的変化を解析するために動物実験が行なわれた(図10A〜F、11A〜F及び12A〜F)。GLP−1Rアゴニスト処置は、ナイーブ動物のTer119+細胞数の近くまで、MOG誘発動物でTer119+細胞数の大幅減少をもたらした(図12D〜F及び13)。対照的に、PBS処置対照EAE動物は、Ter119+細胞数を増加した。加えて、GLP−1Rアゴニスト処置は、ナイーブ動物と比べてIL−17+及びIFN−γ+細胞数を増加したPBS処置対照EAE動物と比べて、EAE動物でIL−17+及びIFN−γ+細胞数を減少させた(図16及び17)。免疫学的解析の結果、GLP−1Rアゴニスト処置は、EAE動物でのヘルパーT細胞レベルを減少させることを示唆する。
【0068】
上記の結果は、GLP−1RアゴニストがCNSの脱髄及び白血球浸潤を減少させ、そして広く認められているMSの動物モデルであるEAEの進行を緩徐化させるのに有効であることを証明した。天然のGLP−1Rアゴニストエキセンジン−4及びGLP−1RアゴニストGAC−1は、いずれも疾患進行減速に有効であった。組織化学実験では、GLP−1Rアゴニストが、T細胞及び単球によるCNS組織の浸潤を減少させることを示した。
【0069】
CNS自己免疫障害に対するGLP−1Rアゴニスト
理論に限定されることなく、GLP−1Rアゴニストは、主として白血球の移動を調節し、及び/又はTh1及びTh17細胞レベルを減少させることにより機能すると考えられる。本方法は、更なる治療効果をもたらすために最近の免疫抑制療法と併用してもよい。
【0070】
本発明のGLP−1Rアゴニストは、多数の自己免疫又は関連疾患において、CNS組織の白血球浸潤を減少させるために使用することができる。本発明のGLP−1Rアゴニストは、多数の自己免疫又は関連疾患において、Th1及びTh17細胞レベルを減少させるためにも使用することができる。MSのモデルであることに加えて、EAEマウスは、視神経炎を研究するために使用することもできる。GLP−1Rアゴニストは、とりわけ白血球浸潤及び/又はTh1及びTh17細胞によって媒介されるすべてのこれらの疾患及び他の自己免疫障害におけるCNS免疫浸潤を軽減することが期待される。用語の疾患及び障害は、区別せずに使用することを銘記されたい。
【0071】
本発明の特徴は、自己免疫疾患を患っている動物に対するGLP−1Rアゴニストの直接投与である。本発明の好ましい実施態様はポリペプチドについて記載されているが、本発明は体内においてコード化されたGLP−1Rアゴニストの発現を方向付ける、当該GLP−1Rアゴニストポリペプチドをコード化するポリペプチドの投与を包含する。直接DNA注入及び遺伝子療法デリバリーの方法は当技術分野で公知である。GLP−1Rアゴニストポリペプチド、又はそれらをコード化するポリヌクレオチドは、薬剤の投与により誘導するのと対照的に、動物に直接投与される。本発明は、天然GLP−1Rアゴニスト及び/又はアゴニスト抗体に合致する方法でGLP−1Rを結合し活性化する、ペプチド模倣薬分子をも包含する。
【0072】
本明細書に記載の方法に従って使用するための特定のGLP−1Rアゴニストは、以下に更に詳細に記載される。更なるGLP−1Rアゴニストは、本発明の範囲から逸脱することなく当業者には明らかなものである。
【0073】
天然のGLP−1Rアゴニスト及びそれらの誘導体
GLP−1Rアゴニストは、GLP−1、エキセンジン−4、プロエキセンジン(例えば、米国特許第6723530号を参照)、エキセンジン−3(例えば、米国特許第5424286号を参照)、OAP−189(例えば、米国特許出願公開第20090181885号を参照、その全文は、参照することにより本明細書に組み入れられている)、及びオキシントモジュリンを含む、天然アゴニストポリペプチド、そのフラグメント、変異体、及び誘導体を含むが、これらに限定されない。本明細書で使用されるGLP−1及び/又はエキセンジン−4ポリペプチド配列は、結果として生じるポリペプチドがGLP−1Rに結合してアゴニストとして機能するという条件で、対応するGLP−1Rと同じ種から又は異なる種から生じたものであってよい。
【0074】
GLP−1Rアゴニストは、天然及び変異GLP−1を含む。GLP−1ポリペプチドは、多数の哺乳動物で同定されている。HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号1)は、2位置のアラニンにおいて、ジペプチジルペプチダーゼIV(DPP−4)によるGLP−1の切断によって生成された30アミノ酸GLP−1(7〜36)ペプチドである。D.J.Drucker(2001)「内分泌学」(Endocrinology)142:521-527。GLP−1(7〜36)はGLP−1Rアゴニストとして機能し、グルコース依存性インスリン分泌増加をもたらす。しかしながら、GLP−1(7〜36)の半減期はわずか数分である。ピロテアーゼ切断部位は、GLP−1ポリペプチドの半減期を延長するために除去されても、又はそれらの活性の調節を可能にするために加えられてもよい。GLP−1Rアゴニストは、PEGなどの半減期拡張部分、IgGFc領域、アルブミン、又はMyc、HA(ヘマグルチニン)、His−6、又はFLAGなどのペプチド若しくはエピトープに、結合又は融合してもよい。他の典型的GLP−1ポリペプチドは、GLP−1(7〜37)HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号42)及びGLP−1(1〜45)を含む(例えば、米国特許出願公開第2003/0232754号を参照)。
【0075】
GLP−1Rアゴニストは、更に天然及び変異エキセンジン−4を含む。エキセンジン−4はアメリカドクトカゲの唾液中に同定されている。エキセンジン−4は、GLP−1と約53%相同性の39アミノ酸ペプチドである。アメリカドクトカゲエキセンジン−4の配列は、HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS(配列番号2)である。GLP−1(7〜36)のように、エキセンジン−4はGLP−1Rアゴニストとして機能し、グルコース依存性インスリン分泌を促進する。しかしながら、GLP−1(7〜36)と異なり、エキセンジン−4はDPP−4による切断に対する抵抗性の増加を示す。GLP−1(7〜36)のN末端領域及びエキセンジン−4はほとんど同一であるが、有意な差異は第2アミノ酸残基である。この残基はGLP−1(7〜36)ではアラニンであり、しかしエキセンジン−4ではグリシンである。N末端領域におけるこの単一アミノ酸は、DPP−4消化に対するエキセンジン−4の抵抗性の増加に関与している。エキセンジン−4とGLP−1(7〜36)との間の別な有意な差異は、Trpケージを形成するエキセンジン−4のC末端における9つの更なるアミノ酸残基の存在である。
【0076】
本発明の天然及び変異GLP−1及びエキセンジン−4ポリペプチドは、キメラ、変異体、フラグメント(ペプチドを含む)、及び/又はその誘導体を含む。好ましいフラグメントは、天然ポリペプチドのGLP−1R結合部分、又はキメラの、コンセンサス、又は突然変異した等価結合部分を含む。フラグメントは合成ペプチドを含む。変異体は、保存又は非保存アミノ酸置換を有する天然アミノ酸配列変異体を含む。典型的変異体としては、GLP−1−Tf(米国特許出願公開第20060205037号)、OAP−189、エキセンジン−4−Tf(国際公開公報第2008012629号)、GLP−1、及び米国特許出願公開第20040242853号に開示のエキセンジン−4ペプチドアナログが含まれるが、これらに限定されない。OAP−189の配列は、HAQGTFTSDYSKYLEQELVKYFIQWLKNAGPSKNNIA(配列番号43)である。
【0077】
保存置換は、類似のサイズ、電荷、又は疎水性のアミノ酸残基を含む。例えば、AlaはVal、Leu、又はIleによって置換してもよい。ArgはLys、Gln、又はAsnによって置換してもよい。AsnはGln、His、Lys、又はArgによって置換してもよい。AspはGluによって置換してもよい。CysはSerによって置換してもよい。GlnはAsnによって置換してもよい。GluはAspによって置換してもよい。GlyはProによって置換してもよい。HisはAsn、Gln、Lys、又はArgによって置換してもよい。IleはLeu、Val、Met、Ala、Phe、又はノルロイシンによって置換してもよい。Leuはノルロイシン、Ile、Val、Met、Ala、又はPheによって置換してもよい。LysはArg、Gln、又はAsnによって置換してもよい。MetはLeu、Phe、又はIleによって置換してもよい。PheはLeu、Val、Ile、又はAlaによって置換してもよい。ProはGlyによって置換してもよい。SerはThrによって置換してもよい。ThrはSerによって置換してもよい。TrpはTyrによって置換してもよい。TyrはTrp、Phe、Thr、又はSerによって置換してもよい。ValはIle、Leu、Met、Phe、Ala、又はノルロイシンによって置換してもよい。
【0078】
機能の実質的な修飾は、(a)例えば、シート又はヘリカル立体配座として置換の部位におけるポリペプチドの骨格の構造、(b)標的部位での分子の電荷又は疎水性、又は(c)側鎖の大きさの維持に対するそれらの効果において著しく異なる置換を選択することによって達成されると考えられる。天然残基は共通の側鎖特性(これらのあるものは、幾つかの官能基に分類することができる)に基づいてグループ分けされる:
(1)疎水性:Met、Ala、Val、Leu、Ile、ノルロイシン;
(2)中性親水性:Cys、Ser、Thr;
(3)酸性:Asp、Glu;
(4)塩基性:Asn、Gln、His、Lys、Arg;
(5)芳香族:Trp、Tyr、Phe;及び
(6)たわみを誘導する残基:Gly及びPro。
【0079】
非保存置換は、1つのクラスのメンバーを別のクラスのメンバーと交換し、又は前項で保存性と特定されなかった置換に関わる。
【0080】
アゴニストの適正な立体配座を維持するのに関係しないいずれのシステイン残基も、分子の酸化安定性を改善し又は異常架橋を防止するために一般的にセリンと置換してよい。反対に、1つ又は複数のシステイン結合がその安定性を改善するためにアゴニストに付加されてもよい。
【0081】
アミノ酸修飾は、可変領域など1領域の1つ又はそれ以上のアミノ酸を変化させ又は修飾することから、完全な再設計までの範囲で変動することができる。可変領域の変化は、結合親和性及び/又は特異性を変えることができる。
【0082】
変異体は、結果として得られるポリペプチド又は誘導体がGLP−1Rに結合してアゴニストとして機能するという条件で、天然アミノ酸配列変異体及び改変変異体を含む。GLP−1R活性化を測定するための分析については、本明細書及び引用文献に記載されている。
【0083】
誘導体は、共有結合又は非共有結合修飾ペプチド及びポリペプチド(例えば、アシル化、ペグ化、ファルネシル化、グリコシル化、又はリン酸化ポリペプチド)である。ポリペプチドは、結合及び/又は活性を調節し、生体内画像化を可能にし、半減期を調節し、血液脳関門を通る輸送を調節し、又は特定の細胞型又は組織へのポリペプチドのターゲッティングを補助するために、更なる官能基を含んでもよい。ポリペプチドは、置換がGLP−1Rへのポリペプチドの結合し、又はアゴニスト活性に実質的に影響を与えないことを条件として、修飾(例えば、ペグ化、グリコシル化、又は他の部位の追加)を促進させるためのアミノ酸置換を含んでもよい。
【0084】
一般的な修飾は、生物活性の最小ロスで全身クリアランスを減少させるペグ化である。ポリエチレングリコール重合体(PEG)は、当技術分野で公知の方法(例えば、Roberts et al. (2002),「最新ドラッグデリバリー概説」(Advanced Drug Delivery Reviews)54:459-476; Sakane et al. (1997) Pharm. Res. 14:1085-91を参照)を用いて、GLP−1及びエキセンジン−4ポリペプチド(並びにGLP−1Rアゴニスト抗体)の種々の官能基へ結合してもよい。PEGは、例えば、アミノ基、カルボキシル基、修飾又は天然N末端、アミン基、及びチオール基に結合してもよい。幾つかの実施態様では、1つ又はそれ以上の表面アミノ酸残基はPEG分子で修飾される。PEG分子には様々なサイズ(例えば、約2〜40kDaの範囲)がある。GLP−1、エキセンジン−4、又は他のポリペプチドに結合するPEG分子は、約2,000、10,000、15,000、20,000、25,000、30,000、35,000、40,000Daのいずれかの分子量を有することができる。PEG分子は、単一又は分枝鎖状であってよい。GLP−1RアゴニストポリペプチドへPEGを結合させるために、1つ又は両方の末端に官能基を有するPEGの誘導体を使用してもよい。官能基は、ポリペプチド上の利用可能な反応性基のタイプに基づいて選択される。誘導体をポリペプチドに結合する方法は当技術分野で公知である。PEGのN末端特異的結合の1つの例は、ペグ化を促進するために1の位置の残基をセリン又はトレオニンに突然変異させることである。
【0085】
ポリペプチド又はそれらの誘導体は、直接又は合成リンカーを経て他の分子に結合してもよい。好ましいGLP−1Rアゴニストポリペプチド、そのフラグメント、又は誘導体は、その価値が報告されている天然のGLP−1Rアゴニストと比べて、類似の又はより優れた親和性、選択性、及び活性化を示す。アゴニストポリペプチドの小部分は「ペプチド」と呼ばれるが、この専門用語は、これに限定されると解釈すべきではない。
【0086】
好ましいGLP−1Rアゴニストは、EAE動物モデルを含む本明細書に記載の数々の実験及び分析において、エキセンジン−4及びGAC−1と比べて類似の生物学的特性を示す。
【0087】
GLP−1Rアゴニスト−抗体コンジュゲート
誘導体は、半減期を延長し及び/又は薬効を強化するための、GLP−1Rアゴニストペプチドの抗体への共有結合を更に含む。例えば、抗体の結合部位に直接か又は介在リンカーを介して、結合したGLP−1Rアゴニストペプチド剤を含むGLP−1Rアゴニスト化合物は、米国特許出願公開第20090098130号及びPCT国際公開第2008/081418号に記載されており、いずれも、それらの全文が参照することにより本明細書に組み入れられている。当該GLP−1Rアゴニスト化合物は、本明細書では「GLP−1Rアゴニスト−抗体コンジュゲート」又は「GAC」と呼ばれる。GACは、直接か又はリンカーを経て抗体に結合した、例えば、GLP−1又はエキセンジン−4フラグメント又は誘導体などのGLP−1Rアゴニストペプチドを含む。リンカーが使用される幾つかの実施態様では、リンカーは、GLP−1RアゴニストペプチドとGLP−1R間の抗体制限結合を避けるために、抗体からGLP−1Rアゴニストペプチドを遠ざけるように作用すると考えられる。
【0088】
本明細書に記載のGACは、GLP−1Rアゴニストペプチド、即ち、GLP−1Rの活性化量を増加させるペプチドを含み、これは天然アゴニストGLP−1及びエキセンジン−4によって生じるものと類似の効果をもたらす。幾つかの実施態様では、GLP−1Rペプチドは、1つ又はそれ以上のR基によって隣接してもよい。例えば、幾つかの実施態様では、GACのGLP−1Rアゴニストペプチドは、以下の構造を有してよい:R1−HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号1)−R2、ここで、R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;そしてR2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質である。他の実施態様では、GLP−1Rアゴニストペプチドは、以下の構造を有してよい:R1−HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS(配列番号2)−R2(配列番号4);ここで、R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;そしてR2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質である。GLP−1Rアゴニストペプチドは、配列番号1及び2のアナログを更に含む。当該アナログは、例えば、バイオアベイラビリティ増加、安定性増加、EAE処置プロファイル改善、Th17細胞数減少プロファイル改善、食欲抑制改善、体重管理改善、及び/又は宿主免疫認識減少などの更なる有利な特性を有してもよい。本明細書で使用される配列番号1又は配列番号2のペプチドアナログは、本質的に配列番号1又は配列番号2の配列を有するペプチドであるが、しかし1つ又はそれ以上のアミノ酸の置換、挿入若しくは欠失、又はそれらの組み合わせを有する。
【0089】
特定の実施態様では、本明細書で提供されるGLP−1Rアゴニストペプチドは、配列番号1又は配列番号2を含む。好適な典型的配列番号1及び配列番号2アナログは、例えば、2008年1月4日に出願された米国特許出願第11/969850号(米国特許出願公開第20090098130号として公開)の表IIに記載され、その全文は、参照することにより本明細書に組み入れられている。
【0090】
GLP−1Rアゴニストペプチドは、当技術分野で周知の技術を用いて調製することができる。例えば、GLP−1Rアゴニスト抗体コンジュゲート化合物を製造する方法は、米国特許出願公開第20090098130号に記載されている。一般的には、GLP−1Rアゴニストペプチドの合成が第1工程であり、そして米国特許出願公開第20090098130号に記載のように行なわれる。次いで、GLP−1Rアゴニストペプチドは、結合成分(リンカー)への連結のために誘導体化され、次いで抗体へ結合される。当業者には当然のことながら、使用される特異的合成工程は、3成分の正確な性質によって決まる。このように、本明細書に記載のGLP−1Rアゴニストペプチド−リンカーコンジュゲート及びGACは、容易に合成することができる。
【0091】
本明細書で使用される「抗体」は、B細胞及びその変異体の産物である免疫グロブリン、並びにT細胞及びその変異体の産物であるT細胞受容体(TcR)を含む。結合部位は抗原結合に関与する抗体分子の一部を指す。
【0092】
本明細書に記載のGLP−1Rアゴニストペプチドは、直接か又はリンカーを経て抗体中の結合部位に共有結合してもよい。適切なリンカーは、標的薬剤と抗体間で十分な距離を与えるように選択することができる。他のリンカーの検討としては、得られるGAC及びGLP−1Rアゴニストペプチド−リンカーの物理的又は薬物動力学的特性に対する影響、溶解性、親油性、親水性、疎水性、安定性(概ね安定並びに計画的分解)、剛性、柔軟性、免疫原性、抗体結合の調節、ミセル又はリポソームへの取り込まれ能力などが含まれる。ペプチド組成、リンカー組成、結合残基位置の更に微小な変化は、得られる分子の種々の特性(例えば、安定性、溶解性、半減期、バイオアベイラビリティ、標的結合能力、アゴニスト又はアンタゴニスト活性、薬物動力学など)に対して予測できない及び驚くべき効果を有することができる。
【0093】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式I:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14
E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40(配列番号3)]−R2
式中、
X2は、Aib、A、S、T、V、L、I、D−Alaなどの保護基であり;X10は、V、L、I又はAであり;X12は、S又はKであり;X13は、Q又はYであり;X14は、G、C、F、Y、W、M又はLであり;X16は、K、D、E又はGであり;X17は、E又はQであり;X19は、L、I、V又はAであり;X20は、オルニチン又はK(SH)R若しくはKなどの誘導体化リジン基であり;X21は、L又はEであり;X23は、I又はLであり;X24は、A又はEであり;X25は、W又はFであり;X26は、L又はIであり;X27は、I、K又はVであり;X28は、R、オルニチン、N又はKであり;X29は、Aib
又はGであり;X30は、いずれかのアミノ酸、好ましくはG又はRであり;X31は、P又は不存在であり;X32は、S又は不存在であり;X33は、S又は不存在であり;X34は、G又は不存在であり;X35は、A又は不存在であり;X36は、P又は不存在であり;X37は、P又は不存在であり;X38は、P又は不存在であり;X39は、S又は不存在であり;そしてX40は、連結残基又は不存在であり;かつ、X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39又はX40の1つは、直接又は中間リンカーを経由して抗体の結合部位に共有結合できる求核性側鎖を含む連結残基(−[LR]−)で置換され、そして連結残基はK、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてよい;
のものであってよい。
【0094】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式II:
R1−[H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−V10−S11−S12−Y13−L14−E15−G16−Q17−A18−A19−K20−E21−F22−I23−A24−W25−L26−V27−K28−G29−R30(配列番号4)]−R2
の配列を含む。幾つかの実施態様では、残基の1つは結合残基と置換されてもよい。幾つかの実施態様では、G16又はA24の1つは、直接又は中間リンカーを介して抗体の結合部位に共有結合できる求核性側鎖を含む結合残基で置換され、そして結合残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてもよい。他の実施態様では、リンカーは、R30基のC末端、又は31位置での更なる残基のC末端に共有結合してもよい。
【0095】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式III:
R1−[H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−L10−S11−K12−Q13−M14−E15−E16−E17−A18−V19−R20−L21−F22−I23−E24−W25−L26−K27−N28−G29−G30−P31−S32−S33−G34−A35−P36−P37−P38−S39−X40(配列番号5)]−R2
式中、
X40は、結合残基又は不存在であり、L10、S11、K12、Q13、M14、E16、E17、A19、R20、L21、E24、L26、K27、N28、G32、S33、G34、A35、P36、P37、P38、S39又はX40の1つは、直接又は中間リンカーを介して抗体の結合部位に共有結合できる求核性側鎖を含む結合残基(−[LR]−)で置換され、かつ、結合残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてもよい;
の配列を含んでもよい。
【0096】
式I、II及びIIIでは、X2はAibなどの保護残基であってよく、又はアラニン、(例えば、GLP1に見られるような)、又はグリシン(例えば、エキセンジン−4に見られるような)、又は別の残基であってもよい。
【0097】
式I、II及びIIIでは、R1は、不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3又はC(O)CH(CH3)CH3であってよく;そしてR2は、不存在、OH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であってよい。
【0098】
式I、II及びIIIでは;R1は不存在であってよい。式I、II及びIIIでは;R2はNH2であってよい。
【0099】
式I、II及びIIIでは、結合残基はリジン(K)であってよい。結合残基はCであってよい。式I、II及びIIIでは、結合残基はK(SH):
【化6】
であってよい。
【0100】
結合残基がK、K(SH)又はCの場合、結合残基は下式:
【化7】
のリンカーに更に共有結合してもよい。
【0101】
このリンカーは、下式:
【化8】
のようにβラクタム基を通して抗体に共有結合してもよい。
【0102】
幾つかの実施態様では、リンカーのβラクタム基は、下式:
【化9】
のように、抗体の2つの結合部位の少なくとも1つに共有結合してもよい。
【0103】
幾つかの実施態様では、結合残基及びリンカーは下式:
【化10】
の構造のものであってよい。
【0104】
幾つかの実施態様では、リンカー及び結合残基は、下式:
【化11】
のように、式I、II及びIIIによって例示されたもののようなGLP−1Rアゴニストペプチドに共有結合してもよい。
【0105】
幾つかの実施態様では、ペプチド−リンカー複合体は下式:
【化12】
のように、抗体に共有結合してもよい。
【0106】
幾つかの実施態様では、ペプチド−リンカー複合体は下式:
【化13】
のように、抗体の結合部位の少なくとも1つに結合する。
【0107】
特定の実施態様では、GLP−1Rアゴニストペプチドは、配列:
H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−L10−S11−K12−Q13−M14−E15−E16−E17−A18−V19−R20−L21−F22−I23−E24−W25−L26−K27−N28−G29−G30−P31−S32−S33−G34−A35−P36−P37−P38−S39−X40(配列番号6)]
式中、
M14又はX40の1つは結合残基で置換される;
を含んでもよく、幾つかの実施態様では、GLP−1Rアゴニストペプチド−リンカーは、下式:
【化14】
のようにK40(結合残基)を経由してリンカーAに結合したペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−M−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S−K(配列番号7)]−NH2
を含む。
【0108】
化合物GAC−1では、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−M−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S−K(配列番号7)]−NH2
は、下式:
【化15】
のようにK40(結合残基)を経由してリンカーAに結合し、抗体の結合部位に結合している。
【0109】
従って、GAC−1は下式:
【化16】
の構造を有する。
【0110】
別の実施態様では、GLP−1Rアゴニストペプチド−リンカーは、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−K−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S(配列番号8)]−NH2
を含み、このペプチドは、下式:
【化17】
のようにK14(結合残基)を経由してリンカーAに結合している。
【0111】
化合物GAC−2では、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−K−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S (配列番号8)]−NH2
は、下式:
【化18】
のようにK14(結合残基)を経由してリンカーAに結合し、抗体の結合部位に結合している。
【0112】
従って、GAC−2は以下の構造:
【化19】
を有する。
【0113】
典型的なGACは、下記の表1に与えられるいずれかのペプチド配列を有するGACを含むが、これらに限定されない。幾つかの実施態様では、GACGLP−1Rアゴニスト化合物は、表1からのいずれかのGACである。表1に示されるデータは、米国特許出願公報第20090098130号に記載の方法を用いて得られた。表1に与えられるEC50値は、例えば、米国特許出願公報第20090098130号の実施例30に記載の、グルコース刺激性インスリン分泌(GSIS)分析を用いて測定された。表1において、EC50欄の(P)は、ペプチドを抗体に連結する場合のEC50を示す。
【0114】
【表2】
【0115】
【表3】
【0116】
【表4】
【0117】
上記のように、本発明で使用するための化合物は、抗体に共有結合してもよい。米国特許出願公開第20090098130号(全文が参照することにより本明細書に組み入れられている)は、とりわけ、抗体、有用なフラグメント及び変異体及びその修飾、結合部位及びCDR、抗体調製、発現、ヒト化、アミノ酸修飾、糖鎖付加、ADCC、CDC、抗体の血清半減期増加、発現ベクター、哺乳動物宿主系及び折畳み、そして抗体技術の他の要素を記載している。
【0118】
幾つかの実施態様では、本発明の化合物と併せて使用することができる幾つかの抗体は、抗体結合部位に反応性側鎖を有してもよい。反応性側鎖は天然に存在してもよく、又は突然変異により抗体中に入れられてもよい。抗体結合部位の反応性残基は、残基が抗体を作製するために最初に同定されたリンパ系細胞に存在する核酸によりコード化される場合など、抗体と関連してもよい。代わりに、アミノ酸残基は、特定の残基をコード化するために、DNAを意図的に突然変異させることにより生じてもよい(例えば、Meares et al.による国際公開公報第01/22922号を参照)。反応性残基は、例えば、本明細書に記載の固有のコドン、tRNA、及びアミノアシル−tRNAを用いた生合成取り込みによる非天然残基であってよい。別のアプローチでは、アミノ酸残基又はその反応性官能基(例えば、求核性アミノ基又はスルフヒドリル基)が、抗体結合部位中のアミノ酸残基に結合してもよい。従って、本明細書で使用される「抗体の結合部位中のアミノ酸残基を通して」生じる抗体との共有結合は、結合が抗体結合部位のアミノ酸残基へ直接であっても、又は抗体結合部位におけるアミノ酸残基の側鎖へ結合する化学部分を通してであってもよい。幾つかの実施態様では、アミノ酸はシステインであり、そして側鎖の反応基はスルフヒドリル基である。他の実施態様では、アミノ酸残基はリジンであり、そして側鎖の反応基はε−アミノ基である。
【0119】
触媒抗体は、1つ又はそれ以上の反応性アミノ酸側鎖を含む、好適な結合部位を有する抗体の1つの供給源である。当該抗体は、アルドラーゼ抗体、βラクタマーゼ抗体、エステラーゼ抗体、アミダーゼ抗体などを含む。
【0120】
1つの実施態様は、マウスモノクローナル抗体mAb33F12及びmAb38C2、並びに当該抗体の好適なキメラ及びヒト化バージョン(例えば、h38C2、配列番号11及び12)などのアルドラーゼ抗体を含む。
【0121】
h38C2軽鎖配列(219アミノ酸):
【化20】
【0122】
h38C2h鎖配列(448アミノ酸):
【化21】
【0123】
マウスmAb38C2(及びh38C2)は、HCDR3に近接してしかし外側に反応性リジンを有し、そして反応性免疫及び機構的模倣天然アルドラーゼ酵素によって生成した、新しいクラスの触媒抗体のプロトタイプである。C.F. Barbas 3rd et al., Science 278:2085-2092, 1997を参照。使用してもよい他のアルドラーゼ触媒抗体は、ATCC受入番号PTA−1015を有するハイブリドーマ85A2;ATCC受入番号PTA−1014を有するハイブリドーマ85C7;ATCC受入番号PTA−1017を有するハイブリドーマ92F9;ATCC受入番号PTA−823を有するハイブリドーマ93F3;ATCC受入番号PTA−824を有するハイブリドーマ84G3;ATCC受入番号PTA−1018を有するハイブリドーマ84G11;ATCC受入番号PTA−1019を有するハイブリドーマ84H9;ATCC受入番号PTA−825を有するハイブリドーマ85H6;ATCC 受入番号PTA−1016を有するハイブリドーマ90G8を含む。反応性リジンを通して、これらの抗体は、天然アルドラーゼのエナミンメカニズムを用いて、アルドール及びレトロアルドール反応を触媒する。例えば、J. Wagner et al., Science 270:1797-1800, 1995; C.F. Barbas 3rd et al., Science 278:2085-2092, 1997; G. Zhong et al., Angew. Chem. Int. Ed. Engl. 38:3738-3741, 1999; A. Karlstrom et al., Proc. Natl. Acad. Sci. U.S.A., 97:3878-3883, 2000を参照されたい。アルドラーゼ抗体、及びアルドラーゼ抗体を生成する方法は、米国特許出願公開第6210938号、第6368839号、第6326176号、第6589766号、第5985626号、及び第573375号に記載され、これらは、参照することにより本明細書に組み入れられている。
【0124】
本発明の化合物は、チオエステラーゼ及びエステラーゼ触媒抗体の結合部位に見られるもののように、反応性システインに、本発明の化合物を結合させることによって形成してもよい。好適なチオエステラーゼ触媒抗体は、K.D. Janda et al., Proc. Natl. Acad. Sci. U.S.A. 91:2532-2536 (1994)によって記載されている。 好適なエステラーゼ触媒抗体は、P. Wirsching et al., Science 270:1775-1782 (1995)によって記載されている。反応性アミノ酸含有抗体は、反応性アミノ酸をコードするために抗体結合部位を突然変異させること、又は反応基を含むリンカーで抗体結合部位のアミノ酸側鎖を誘導化することを含み、当技術分野で周知の手段により調製してもよい。
【0125】
幾つかの実施態様では、抗体はヒト化抗体であってよい。好ましくは、ヒト化抗体は、βラクタム基又は結合部位に共有結合を形成することができる他の化学基に対して、高結合親和性を保持する。種々の形態のヒト化マウスアルドラーゼ抗体が意図される。幾つかの実施態様では、抗体は、ヒト定常ドメインCκ及びCγ11を有するh38c2IgG1及びh38c2Fabなどの、ヒト化アルドラーゼ触媒抗体である。C. Rader et al.,
2003, J. Mol. Bio. 332:889-899は、h38c2Fab及びh38c2IgG1を産生するために使用することができる遺伝子配列及びベクターを開示している。ヒト生殖細胞系Vk遺伝子DPK−9及びヒトJk遺伝子JK4を、m38c2のκ軽鎖可変ドメインのヒト化に対するフレームワークとして使用し、そしてヒト生殖細胞系遺伝子DP−47及びJH遺伝子JH4をm38c2の重鎖可変ドメインのヒト化に対するフレームワークとして使用した。G1m(f)アロタイプを有する抗体h38c2IgG1を含む本発明の化合物の特定の実施態様では、リンカーのβラクタム基は、重鎖の99の位置でリジン残基の側鎖に結合してもよい。他の実施態様は、h38c2の可変ドメイン(VL及びVH)及びIgG1、IgG2、IgG3、又はIgG4からの定常ドメインを含むキメラ抗体を使用してもよい。抗体は、h38c2からのVH及びVLドメインを含む完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、VL、二重特異性抗体、又はミニ抗体であってよい。抗体は、h38c2からのVH及びVLドメイン及びIgG1、IgG2、IgG3、又はIgG4から成る群から選択される定常ドメインを含む抗体であってもよい。幾つかの実施態様では、抗体はh38C2IgG1であってもよい。幾つかの実施態様では、抗体は、ヒトIgG、IgA、IgM、IgD、又はIgE抗体からの定常領域を含むマウスアルドラーゼ抗体のヒト化バージョンであってもよい。他の実施態様では、抗体は、マウスアルドラーゼ抗体からの可変領域、及びヒトIgG、IgA、IgM、IgD、又はIgE抗体からの定常領域を含むキメラ抗体であってよい。更なる実施態様では、抗体は、天然又はナイーブヒトIgG、IgA、IgM、IgD、又はIgE抗体からのポリペプチド配列を含む、マウスアルドラーゼ抗体の完全ヒト化バージョンであってよい。
【0126】
ヒト化アルドラーゼ抗体フラグメントの種々の形態も意図される。1つの実施態様では、h38c2F(ab’)2を使用する。h38c2F(ab’)2はh38c2IgG1のタンパク分解によって産生してもよい。別の実施態様は、介在リンカー(Gly4Ser)3により選択的に結合されるh38c2からのVL及びVHドメインを含む、h38c2scFvを使用する。ヒト化に代わる方法として、ヒト抗体を生成することができる。
【0127】
他のGLP−1Rアゴニスト
GLP−1Rアゴニストは、例えば、抗GLP−1Rアゴニスト抗体の、エキセナチド(BYETTA(登録商標))、アルビグルチド、R1583、リラグルチド、AVE−0010、S4P及びBoc5(Chen et al. (2007), Proc. Natl. Acad. Sci. USA.104:943-948を参照)を含むが、これらに限定されない。
【0128】
GLP−1Rアゴニストは、例えば、シタグリプチン、ビルダグリプチン、サキサグリプチン、リナグリプチン、ドゥトグリプチン(dutogliptin)、ゲミグリプチン(gemigliptin)、アログリプチン及びベルベリンなどのDPP−4阻害剤をも含むが、これらに限定されない。
【0129】
幾つかの実施態様では、GLP−1Rアゴニストは、モノクローナル抗GLP−1Rアゴニスト抗体である。モノクローナル抗GLP−1R抗体は当業者により容易に生産することができる。ハイブリドーマによりモノクローナル抗体を作製する一般的手法は、現在当技術分野で周知である。例えば、M. Schreier et al., 「ハイブリドーマ技術」(Hybridoma Techniques) (Cold Spring Harbor Laboratory 1980); Hammerling et al.,「モノクローナル抗体及びT細胞ハイブリドーマ」(Monoclonal Antibodies and T-Cell Hybridomas) (Elsevier Biomedical Press 1981); Kennett et al.,「モノクローナル抗体」(Monoclonal Antibodies) (Plenum Press 1980)を参照されたい。不死化抗体分泌細胞系は、発癌DNA又はEBVによるBリンパ球の直接形質転換など、融合以外の技術によっても産生することができる。必要に応じて、後に不死化細胞系に転換する正常Bリンパ球集団にチャレンジするために、幾つかの抗原供給源を使用することができる。
【0130】
ポリヌクレオチド
幾つかの実施態様では、本発明は、本明細書に記載のいずれかのポリヌクレオチドをコード化するポリヌクレオチドを提供する。ポリヌクレオチドは、当技術分野で公知の手順によって作製し発現することができる。
【0131】
幾つかの実施態様では、本発明は、本発明のいずれかのポリヌクレオチドを含む組成物(医薬組成物など)を提供する。幾つかの実施態様では、組成物は、本明細書に記載のポリペプチドをコード化するポリヌクレオチドを含む発現ベクターを含む。他の実施態様では、組成物は、本明細書に記載のいずれかのポリペプチドをコード化するポリヌクレオチドを含む発現ベクターを含む。発現ベクター及びポリヌクレオチド組成物の投与は、本明細書に更に記載されている。
【0132】
幾つかの実施態様では、本発明は、本明細書に記載のいずれかのポリヌクレオチドを作製する方法を提供する。
【0133】
いずれの当該配列に補完的なポリヌクレオチドも、本発明に包括される。ポリヌクレオチドは、一本鎖(コーディング又はアンチセンス)又は二本鎖であってよく、そしてDNA(ゲノム、又は合成)又はRNA分子であってよい。RNA分子は、イントロンを含み1対1様式でDNA分子に対応するHnRNA分子、及びイントロンを含まないmRNA分子を含む。更なるコード又は非コード配列は、本発明のポリヌクレオチド中に存在してもよいが、しなくてもよく、そしてポリヌクレオチドは、他の分子及び/又は支持物質に結合してもよいが、しなくてもよい。
【0134】
ポリヌクレオチドは、天然配列(即ち、抗体又はその部分をコード化する内在性配列)を含み、又は当該配列の変異体を含んでもよい。ポリヌクレオチド変異体は、コード化ポリペプチドの免疫反応性が天然の免疫反応性分子に比べて低下しないように、1つ又はそれ以上の置換、付加、欠失及び又は挿入を含む。コード化ポリペプチドの免疫反応性に及ぼす影響は、一般的に本明細書に記載のように評価することができる。変異体は、好ましくは、天然抗体又はその部分をコード化するポリヌクレオチド配列と、少なくとも約70%同一性、より好ましくは少なくとも約80%同一性、尚より好ましくは少なくとも約90%同一性、そして最も好ましくは少なくとも約95%同一性を示す。
【0135】
2つのポリヌクレオチド又はポリペプチド配列は、下記に示す最大対応に整列した場合に、2つの配列中のヌクレオチド又はアミノ酸の配列が同じならば「同一」であるといわれる。2つの配列間の比較は、一般的に配列類似性の局所領域を同定し比較する比較窓を通して配列を比較することによって行なわれる。本明細書で使用される「比較窓」は、少なくとも約20の隣接位置、通常は30から約75まで、又は40から約50までのセグメントを指し、その場合、配列は、2つの配列を最適に整列した後で、同数の隣接位置の参照配列と比較してもよい。
【0136】
比較のための配列の最適アラインメントは、初期設定パラメータを用いたバイオインファマティクス・ソフトウェア(DNASTAR, Inc., Madison, WI)のLasergene一式中のMegalignプログラムを用いて実施することができる。このプログラムは、以下の参考文献に記載された幾つかのアラインメント・スキームを具体化する: Dayhoff, M.O., 1978,「タンパク質進化の変化モデル−距離関係を検出するマトリックス」(A model of evolutionary change in proteins - Matrices for detecting distant relationships). In Dayhoff, M.O. (ed.) 「タンパク質の配列と構造の地図」(Atlas of Protein Sequence and Structure), National Biomedical Research Foundation, Washington DC Vol. 5, Suppl. 3, pp. 345-358; Hein J., 1990,「アラインメント及び系統発生論の統一手法」(Unified Approach to Alignment and Phylogenes) pp. 626-645「酵素学の方法」(Methods in Enzymology) vol. 183, Academic Press, Inc., San Diego, CA; Higgins, D.G. and Sharp, P.M., 1989, CABIOS 5:151-153; Myers, E.W. and Muller W., 1988, CABIOS 4:11-17; Robinson, E.D., 1971, Comb. Theor. 11:105; Santou, N., Nes, M., 1987, Mol. Biol. Evol. 4:406-425; Sneath, P.H.A. and Sokal, R.R., 1973,「数量分類学、数量分類学の原理と実際」(Numerical Taxonomy the Principles and Practice of Numerical Taxonomy), Freeman Press, San Francisco, CA; Wilbur, W.J. and Lipman, D.J., 1983, Proc. Natl. Acad. Sci. USA 80:726-730。
【0137】
好ましくは、「配列同一性の割合」は、少なくとも20位置の比較窓を通して2つの最適整列配列を比較することによって決定されるが、ここで、比較窓におけるポリヌクレオチド又はポリペプチド配列の部分は、2つの配列の最適アラインメントに対する参照配列(付加又は欠失を含まない)と比べて、20%以下、通常は5から15%、又は10から12%の付加又は欠失(即ち、ギャップ)を含んでもよい。この割合は、同一の核酸塩基又はアミノ酸残基が整合位置の数を与えるために両配列に生じる位置数を決定すること、参照配列(即ち窓サイズ)における位置の総数によって整合位置数を割ること、そして配列同一性の割合を与えるために結果に100を掛けることによって算出される。
【0138】
又はその代わりに、変異体は、実質的に天然遺伝子又はその部分若しくは相補体に実質的に相同性であってもよい。当該ポリヌクレオチド変異体は、天然ポリペプチド(又は相補配列)をコード化するDNA配列へ中程度のストリンジェントな条件下において、ハイブリダイズすることができる。
【0139】
好適な「中程度ストリンジェントな条件」は、5×SSC、0.5%SDS、1.0mMEDTA(pH8.0)の溶液中で予洗すること;50℃〜65℃、5×SSCで、一夜ハイブリダイズすること;続いて0.1%SDSを含む2×、0.5×及び0.2×SSCのそれぞれで65℃にて20分間2回洗浄することを含む。
【0140】
本明細書で使用される「高度ストリンジェントな条件」又は「高ストリンジェンシー条件」は、(1)洗浄に低イオン強度及び高温、例えば、0.015M塩化ナトリウム/0.0015Mクエン酸ナトリウム/0.1%ドデシル硫酸ナトリウムを50℃で用いる;(2)ハイブリダイゼーション時にホルムアミドなどの変性剤、例えば、50%(v/v)ホルムアミド+0.1%ウシ血清アルブミン/0.1%フィコール/0.1%ポリビニルピロリドン/50mMリン酸緩衝液を、750mM食塩、75mMクエン酸ナトリウムと共にpH6.5で42℃にて用いる;又は(3)50%ホルムアミド、5×SSC(0.75MNaCl、0.075Mクエン酸ナトリウム)、50mMリン酸ナトリウム(pH6.8)、0.1%ピロリン酸ナトリウム、5×デンハルト溶液、超音波分解サケ精子DNA(50μg/ml)、0.1%SDS、及び10%デキストラン硫酸を42℃にて、0.2×SSC(塩化ナトリウム/クエン酸ナトリウム)中42℃にて、そして50%ホルムアミドを55℃にて洗浄液で、続いてEDTAを含む0.1×SSCから成る高ストリンジェンシー洗浄液を55℃にて用いるような条件である。熟練技術者、温度、イオン強度などを調整し、必要に応じてプローブ長さなどの因子を適合させる方法を認識するであろう。
【0141】
当業者には当然のことながら、遺伝コードの縮重の結果として、本明細書に記載のポリペプチドをコード化する多くのヌクレオチド配列がある。これらのポリヌクレオチドの一部は、いずれもの天然遺伝子のヌクレオチド配列に対しても最小の相同性を保有する。それにもかかわらず、コドン使用頻度の差に因って変動するポリヌクレオチドが、本発明によって特に意図される。更に、本明細書で提供されるポリヌクレオチド配列を含む遺伝子の対立遺伝子は、本発明の範囲内である。対立遺伝子は、ヌクレオチドの欠失、付加及び/又は置換などの1つ又はそれ以上の突然変異の結果として変化する、内在性遺伝子である。得られるmRNA及びタンパク質は構造又は機能の変化を有していてもよいが、しかし有していなくてもよい。対立遺伝子は標準技術(ハイブリダイゼーション、増幅及び/又はデータベース配列比較)を用いて同定することができる。
【0142】
本発明のポリヌクレオチドは、化学合成、組み換え法 又はPCRを用いて得ることができる。化学的ポリヌクレオチド合成法は、当技術分野において周知されており、本明細書において詳細に説明する必要はない。当業者は、本明細書で提供される配列及び所望のDNA配列を生産するために、市販のDNA合成装置を使用することができる。
【0143】
組み換え法を用いてポリヌクレオチドを調製するために、所望の配列を含むポリヌクレオチドを好適なベクターに挿入し、そして更に本明細書に記載のようにベクターを複製及び増幅するために、好適な宿主細胞中へ順々に導入することができる。ポリヌクレオチドは、当技術分野で公知のいずれの手段によっても宿主細胞中に挿入し得る。細胞は、直接取り込み、エンドサイトーシス、トランスフェクション、F−交配又はエレクトロポレーションにより外因性ポリヌクレオチドを導入することによって形質転換される。いったん導入されると、外因性ポリヌクレオチドは、細胞内で非組込み型ベクター(プラスミドなど)として保持され、又は宿主細胞ゲノムに組込むことができる。このように増幅したポリヌクレオチドは、当技術分野で周知の方法により宿主細胞から単離することができる。例えば、上記Sambrook et al., 1989を参照されたい。
【0144】
代わりに、PCRはDNAの再生産を可能にする。PCR技術は当技術分野で周知であり、米国特許第4683195号、第4800159号、第4754065号及び第4683202号、並びに「PCR:ポリメラーゼ連鎖反応」(PCR:The Polymerase Chain Reaction), Mullis et al. eds., Birkauswer Press, Boston (1994)に記載されている。
【0145】
RNAは、適切なベクターに分離DNAを用いること、それを好適な宿主細胞へ挿入することにより得ることができる。細胞が複製され、DNAがRNAへ転写される場合、RNAは、例えば、上記のSambrook et al., 1989に記載のように、当業者に周知の方法を用いて分離することができる。
【0146】
好適なクローニングベクターは、標準技術に従って構成してもよく、又は当技術分野で利用可能な多数のクローニングベクターから選択してもよい。選択したクローニングベクターは、使用する目的の宿主細胞によって変化する可能性があると同時に、有用なクローニングベクターは、一般的に自己複製能を有し、特定の制限エンドヌクレアーゼに対して単一標的を有する可能性があり、及び/又はベクターを含むクローンを選択するのに使用することが可能なマーカー遺伝子を保有することができる。好適な例は、プラスミド及び細菌ウイルス、例えば、pUC18、pUC19、Bluescript(例えば、pBSSK+)及びその誘導体、mp18、mp19、pBR322、pMB9、ColE1、pCR1、RP4、ファージDNA、並びにpSA3及びpAT28などのシャトルベクターを含む。これら及び他の多くのクローニングベクターは、BioRad, Strategene、及び Invitrogenなどの販売業者から入手可能である。
【0147】
発現ベクターは、一般的に本発明のポリヌクレオチドを含む複製可能なポリヌクレオチド構成物である。発現ベクターは、宿主細胞内で染色体DNAのエピソームとしてか又は内在性部分として複製可能でなければならないことが示唆される。好適な発現ベクターとしては、例えば、PCT国際公開第87/04462号に開示のように、プラスミド、アデノウイルス、アデノ随伴ウイルス、レトロウイルスなどのウイルスベクター、コスミド、及び1つ又は複数の発現ベクターを含むが、それらに限定されない。ベクター成分は、一般的に1つ又はそれ以上の以下のもの:シグナル配列;複製の起源;1つ又はそれ以上のマーカー遺伝子;好適な転写調節因子(プロモーター、エンハンサー、及びターミネーターなど)を含むが、それらに限定されない。発現(即ち、翻訳)には、リボソーム結合部位、翻訳開始部位、及び終止コドンなどの1つ又はそれ以上の翻訳調節因子が有用である。
【0148】
関係するポリヌクレオチドを含むベクターは、エレクトロポレーション、塩化カルシウム、塩化ルビジウム、リン酸カルシウム、DEAE−デキストラン、又は他の物質を用いるトランスフェクション;マイクロプロジェクタイルボンバードメント法(パーティクルガン法);リポフェクション;及び感染(例えば、ベクターはワクシニアウイルスなどの感染体である)を含む多くの適切な手段のいずれかにより、細胞宿主に導入することができる。ベクター又はポリヌクレオチドを導入する選択は、しばしば宿主細胞の特性によって決まる。
【0149】
本発明は、本明細書に記載のいずれかのポリヌクレオチドを含む宿主細胞をも提供する。ヘテロ接合性DNAを過剰発現することができるいずれの宿主細胞も、関係するポリペプチド又はタンパク質をコード化する遺伝子を分離する目的で使用することができる。哺乳動物宿主細胞の例としては、COS、HeLa、及びCHO細胞が含まれるが、これらに限定されない。PCT国際公開第87/04462号をも参照されたい。好適な非哺乳動物宿主細胞としては、原核細胞(大腸菌又は枯草菌など)及び酵母(S.セレビシアエ、S.ポンベ;又はK.ラクチスなど)が含まれる。好ましくは、宿主細胞は、宿主細胞中に存在する場合、関係する対応の内在性抗体又はタンパク質よりも約5倍高い、より好ましくは10倍高い、尚より好ましくは20倍高いレベルでcDNAを発現する。GLP−1R又はGLP−1Rドメインに特異的に結合する宿主細胞のスクリーニングは、免疫測定又はFACSによって達成される。関係するタンパク質を過剰発現する細胞は同定することができる。
【0150】
GLP−1Rアゴニスト組成物
GLP−1Rアゴニストは、多発性硬化症など中枢神経系を冒す自己免疫疾患の処置のための薬剤製造に使用することができる。このように、GLP−1Rアゴニストを含む組成物は、本明細書に記載のように哺乳動物(ヒト患者を含む)における疾患を処置するのに使用することができる。
【0151】
本発明の方法で使用される組成物は、有効量のGLP−1Rアゴニストを含む。当然のことながら、組成物は1つより多いGLP−1Rアゴニストを含むことができる。
【0152】
GLP−1Rアゴニスト組成物は、更に好適な薬学的に許容される医薬品添加剤を含んでもよい。好適な担体、賦形剤及び医薬品添加剤は当技術分野で周知されており、糖質、ろう、水溶性及び/又は膨潤性重合体、親水性又は疎水性物質、ゼラチン、脂肪油、溶媒、水などの物質を含む。使用される特定の担体、賦形剤及び医薬品添加剤は、本発明の化合物が適用される手段及び目的によって決まるものである。一般的に、安全な溶媒は、水などの非毒性水性溶媒及び水に溶解又は混合可能な他の非毒性溶媒である。好適な水性溶媒としては、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、及びその混合物である。製剤も、薬剤(即ち、本発明の化合物又はその医薬組成物)の洗練された提示、又は医薬品(即ち、薬剤)の製造における補助をもたらすために、1つ又はそれ以上の緩衝剤、安定化剤、界面活性剤、湿潤剤、滑沢剤、乳化剤、懸濁化剤、保存剤、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味剤、芳香剤、着香剤及び他の既知添加剤を含む。幾つかの製剤は、リポソームなどの単体を含む。リポソーム製剤はサイトフェクチン、多重層ベシクル及び単層ベシクルを含むが、それらに限定されない。非経口及び経口ドラッグデリバリーのための医薬品添加剤及び製剤は、Remington, 「薬剤学の科学と実践」(The Science and Practice of Pharmacy) (2000)に記載されている。
【0153】
一般的に、GLP−1Rアゴニスト組成物は、注射(例えば、腹腔内、静脈内、皮下、筋肉内など)による投与のために製剤化されるが、他の投与形態を使用することができる。GLP−1Rアゴニストは、植物性又は他の類似の油脂、合成脂肪酸グリセリド、高級脂肪酸エステル又はプロピレングリコールなどの水性又は非水溶媒にそれらを溶解、懸濁化及び乳化することにより;そして必要に応じて、溶解剤、等張剤、懸濁化剤、乳化剤、安定化剤及び保存剤などの既存の添加剤を用いて、注射製剤に製剤化することができる。
【0154】
投与及び投薬
本明細書に例示した投与計画及び治療投薬量は、動物体重、血中のGLP−1Rアゴニストの半減期、及びGLP−1Rアゴニストの親和性などの当該因子によって決められた。好ましい投与計画及び投薬量は、投与間で体内のGLP−1Rアゴニストの治療量を維持する。エキセンジン−4及びGAC−1の有効投薬量範囲は、本明細書に、Davies et al., 1993に、及び他の参考文献中に例示されている。
【0155】
本発明の裏付けとして行なわれた実験で示されるように、疾患経過中の初期でのエキセンジン−4による「パルス処置」は、動物の病的状態を減少させるのに十分であると考えられる。GLP−1Rアゴニストの継続投与は、処置の有効性に必要ではないと考えられる。GACによる処置などの他の実施態様では、週1回未満の頻度の処置は、処置の有効性に十分と考えられる。
【0156】
特定の薬剤投与計画、即ち、用量、投与時期及び反復は、処置されるヒト及び動物の年齢、病態、及び体重によって決まるといえる。初期薬剤投与計画は、動物実験から外挿してもよい。GLP−1Rアゴニスト投与の時期/頻度は、特定のGLP−1Rアゴニストの循環半減期(又は神経組織中の半減期)、血液脳関門を通過するアゴニストの量、細胞中のアゴニストの半減期、毒性、及びいわゆる副作用を基準にすべきである。
【0157】
本研究では、GLP−1Rアゴニストの投与は、腹腔内 (i.p.) 注射によって行なわれる;しかしながら他の投与経路も有効と予想される(例えば、静脈内、皮下、筋肉内)。頭蓋内又は脊髄内/髄腔内投与(又はCNSの他の組織への投与)は有効と考えられる。他の投与経路は、特定のGLP−1Rアゴニスト、そのコーティング、コンジュゲーション、又は特定の生物学的特性(例えば、経口、舌下、滑液嚢内、粘膜、経皮、関節内、腟内、肛門、尿道内、経鼻、耳内、経吸入、吸入法、経カテーテル、ボーラス投与、ステント又は他の埋め込み型装置、塞栓性組成物、点滴、貼付又は可溶性フィルムなど)に応じて好適であり得る。
【0158】
GLP−1Rアゴニストは、好適な末梢経路を経由して好ましく投与される。それにもかかわらず、当然のことながら、わずかな割合のアゴニストが血液脳関門を通過し、中枢神経系の細胞にデリバリーされると考えられる。幾つかの例では、CNSへの経路をたどる末梢投与GLP−1Rアゴニストの量は小さい(せいぜい1%未満)。
【0159】
本発明の特徴は、自己免疫疾患を患う哺乳動物対象に対するGLP−1Rアゴニストの直接投与である。「直接」は、GLP−1Rアゴニストポリペプチド、又は当該ポリペプチドの発現を方向付けすることができるポリヌクレオチドが、イノキュレーションの標準経路により動物にデリバリーされることを意味する。GLP−1Rアゴニストは、グルココルチコイド、細胞静止薬(例えば、アルキル化剤、代謝拮抗物質、メトトレキサート、アザチオプリン及びメルカプトプリン)などの免疫抑制薬、細胞傷害抗体(例えば、T細胞受容体及びIL−2受容体特異的抗体)、B細胞欠失療法(例えば、抗CD20抗体/RITUXAN(登録商標)、抗BLyS抗体)、T細胞移動障害薬(例えば、抗インテグリンα4/β1抗体/TYSABRI(登録商標)、イムノフィリンに作用する薬剤(例えば、シクロスポリン、タクロリムス、シロリムス、ラパマイシン)、インターフェロン(例えば、IFN−β)、グラチラマー/COPAXONE(登録商標)、オピオイド、TNF結合タンパク質、ミコフェノラート、及び外来抗体又は治療用抗原に対する動物の免疫反応を抑制するために使用される他の生物剤を含み、他の薬剤との組み合わせで動物にデリバリーしてもよい。
【0160】
自己免疫疾患を患うヒトを含む哺乳動物対象の処置において、GLP−1Rアゴニスト又は薬学的に許容される誘導体の治療的有効量が投与される。例えば、GLP−1Rアゴニストは、約0.1mg/kg体重から約15mg/kg体重までの毎日の静脈内点滴で投与することができる。従って、1つの実施態様は、約0.5mg/kg体重の用量を与える。別の実施態様は、約0.75mg/kg体重の用量を与える。別の実施態様は、約1.0mg/kg体重の用量を与える。別の実施態様は、約2.5mg/kg体重の用量を与える。別の実施態様は、約5mg/kg体重の用量を与える。別の実施態様は、約10.0mg/kg体重の用量を与える。別の実施態様は、約15.0mg/kg体重の用量を与える。GLP−1Rアゴニスト又はその薬学的に許容される誘導体の用量を、1日当り約1回から週当たり2回、又は代わりに毎週約1回から月当たり1回の間隔で投与する必要がある。1つの実施態様では、本発明のGLP−1Rアゴニスト又はその薬学的に許容される誘導体のピーク血漿中濃度を達成するために、1用量は約0.002mg/mlから30mg/mlまで投与される。これは、適切な製剤(化学の当業者に公知のいずれの好適な製剤も使用することができる)において投与成分溶液の無菌注射剤によって達成し得る。望ましい血中レベルは、妥当な分析方法により測定した血漿中レベルを確認しながら、本発明のGLP−1Rアゴニストの持続点滴によって維持してもよい。
【0161】
個体に対してGACを投与する1つの方法は、GLP−1Rアゴニストペプチド−リンカーコンジュゲートを個体に投与し、それがインビボで適切な抗体の結合部位と共有化合物を形成することを可能にすることを含む。インビボで形成するGACの抗体部分は、GLP−1Rアゴニストペプチド−リンカーコンジュゲートの投与の前、同時、又は後に個体に投与してもよい。GLP−1Rペプチドは、リンカー/反応性部分を含んでもよく、又は抗体結合部位は標的剤に共有結合するように好適に修飾してもよい。代わりに又は加えて、抗体は、適切な免疫原により免疫後に個体の循環中に存在し得る。例えば、触媒抗体は、担体タンパク質に結合された基質の反応性中間体で免疫することにより作成してもよい。R.A. Lerner and C.F. Barbas 3rd, 1996, Acta Chem. Scand. 50:672-678を参照されたい。特に、アルドラーゼ触媒抗体は、P. Wirsching et al., 1995, Science 270:1775-1782 (commenting on J. Wagner et al., 1995, Science 270:1797-1800)によって記載されているように、ジケトン部分に結合したキーホールリンペットヘモシアニンと投与することにより作成してもよい。
【0162】
天然GLP−1Rアゴニストより優れたGACの利点は、GACが循環タンパク質リガンドと比べて、相対的に長い循環半減期を有する傾向を示すことである。例えば、天然アゴニストが連日投与を必要とする場合、GACは週1回投与するだけでよい。
【0163】
GLP−1Rアゴニストによる処置は、多発性硬化症及び関連疾患に対し、既存の処置法と組み合わせることができる。多発性硬化症の処置及び管理のための既存薬は、以下:ABC(即ち、Avonex-Betaseron/Betaferon-Copaxone) 処置 (例えば、インターフェロン1a(AVONEX(登録商標), REBIF(登録商標))、インターフェロン1b(BETASERON(登録商標), BETAFERON(登録商標))、酢酸グラチラマー(COPAXONE(登録商標));化学療法薬(例えば、ミトキサントロン (NOVANTRONE(登録商標))、アザチオプリン(IMURAN(登録商標))、シクロホスファミド(CYTOXAN(登録商標), NEOSAR(登録商標))、サイクロセリン(SANDIMMUNE(登録商標))、メトトレキサート、及びクラドリビン(LEUSTATIN(登録商標)); コルチコステロイド及び副腎皮質刺激ホルモン(ACTH)(例えば、メチルプレドニゾロン(DEPO-MEDROL(登録商標), SOLU-MEDROL(登録商標))、プレドニゾン(DELTASONE(登録商標))、プレドニゾロン(DELTA-CORTEF(登録商標))、デキサメタゾン(MEDROL(登録商標), DECADRON(登録商標))、副腎皮質刺激ホルモン(ACTH(登録商標))、及びコルチコトロフィン(ACTHAR(登録商標)); 疼痛介在(感覚異常) (例えば、カルバマゼピン(TEGRETOL(登録商標), EPITOL(登録商標), ATRETOL(登録商標), CARBATROL(登録商標))、ガバペンチン(NEURONTIN(登録商標))、トピラマート(TOPAMAX(登録商標))、ゾニサミド(ZONEGRAN(登録商標))、フェニトイン(DILANTIN(登録商標))、デシプラミン(NORPRAMIN(登録商標))、アミトリプチリン(ELAVIL(登録商標))、イミプラミン(TOFRANIL(登録商標), IMAVATE(商標), JANIMINE(登録商標))、ドキセピン(SINEQUAN(登録商標), ADAPIN(登録商標), TRIADAPIN(登録商標), ZONALON(登録商標))、プロトリプチリン(VIVACTIL(登録商標))、大麻及び合成カンナビノイド(MARINOL(登録商標))、ペントキシフィリン(TRENTAL(登録商標))、イブプロフェン(NEUROFEN(登録商標))、アスピリン、アセトアミノフェン、及びヒドロキシジン(ATARAX(登録商標)); 及び他の処置法(例えば、ナタリズマブ(ANTEGREN(登録商標))、アレムツズマブ(CAMPATH-1H(登録商標))、4−アミノピリジン(FAMPRIDINE(登録商標))、3,4−ジアミノピリジン、エリプロジル、IV免疫グロブリン(GAMMAGARD(登録商標), GAMMAR-IV(登録商標), GAMIMUNE N(登録商標), IVEEGAM(登録商標), PANGLOBULIN(登録商標), SANDOGLOBULIN(登録商標), VENOGLOBULIN(登録商標))を含むが、それらに限定されない。
【0164】
キット
本発明は、本発明の方法を実施するための部品のキット(キット)も提供する。本発明のキットは、本明細書に記載のGLP−1Rアゴニストを含む1つ又はそれ以上の容器、及び本明細書に記載の本発明のいずれかの方法に沿った使用説明書を含む。一般的に、これらの使用説明書は、GLP−1Rアゴニストの投与方法についての解説を含む。キットは、処置が必要な動物を特定し、そして処置の有効性をモニター又は測定するための使用説明書を更に含む。使用説明書は、一般的に、投薬量、投与計画(投与の頻度)、及び投与経路に関する情報を含む。キットで提供される使用説明書は、データファイル又は表計算ソフトの形式で書かれ、又は機械/コンピュータ可読であってよい。容器は単位用量、大量包装(例えば、マルチドーズ包装)又はサブユニット用量であってよい。本発明のキットで提供される使用説明書は、ラベル又は添付文書(例えば、キットに含まれるペーパーシート)に関する通常書面での使用説明書であるが、機械可読使用説明書(例えば、磁気又は光学保存ディスク媒体の使用説明書)も認められる。
【0165】
キットは、シリンジ、針、カテーテル、吸入器、ポンプ、アルコール交換、ゲージ、CNS生検器具、組織学的抗体及び染色液などを含む、GLP−1Rアゴニストを投与するための器具をも含む。キットの構成部品は必要に応じて滅菌される。キットは、酢酸グラチラマー(GA)及びデキサメタゾンなどの免疫抑制薬を含む更なる薬剤を提供してもよいが、それらに限定されない。キットは、日付印、不正開封防止包装、及び高周波識別(RFID)タグ又は他の在庫管理機能を含んでもよい。
【0166】
本発明のキットは、好適に包装されている。好適な包装は、バイアル、ボトル、ジャー、フレキシブル包装(例えば、密封Mylar又はプラスチックバッグ)などを含むが、これらに限定されない。吸入器などの特殊器具、経鼻投与器具(例えば、噴霧器)又はミニポンプなどの輸液用器具と組み合わせて使用するための包装も意図される。キットは、滅菌点検口(例えば、容器は皮下注射針によって穿通できる栓を有する静脈注射液バッグ又はバイアルであってよい)を有してよい。容器は、滅菌点検口(例えば、容器は皮下注射針によって穿通できる栓を有する静脈注射液バッグ又はバイアルであってよい)を有してもよい。組成物中の少なくとも1つの活性剤は、GLP−1Rアゴニストである。容器は、第2の薬学低活性剤を含んでもよい。
【0167】
場合により、キットは緩衝剤などの更なる成分及び解説的情報を提供してもよい。通常、キットは、容器及び容器について又はそれに関連するラベル又は1つ又は複数の添付文書を含む。
【実施例】
【0168】
以下の実施例は説明の目的のためだけに提示され、決して本発明の範囲を限定することを意図するものではない。実際、本明細書に示されそして記載されるものに加えられる本発明の種々の改変は、前述の説明から当業者には明らかとなり、そして添付した特許請求の範囲内に属するものである。
【0169】
〔実施例1〕
GLP−1Rアゴニストの同定
GLP−1Rアゴニストは、1つ又はそれ以上の以下の方法を含み、当該技術分野において認められている方法を用いて同定することができる。例えば、細胞内cAMPの濃度増加を検出するアッセイを使用してもよい。このアッセイは、例えばcAMP応答配列の調節下にあるレポーター遺伝子の発現を測定することによる、cAMPの定性的又は定量的測定、並びに選択したGPCRの潜在的アゴニスト又はアンタゴニストの同定及びキャラクタリゼーションに好適である。レポーターを発現するレポーター遺伝子を誘導することができる化合物は、GLP−1Rアゴニストと同定される。1つのこのような分析は、候補化合物へのその反応を検出するために、GLP−1R遺伝子の発現ベクター及びcAMP応答配列の調節下にあるルシフェラーゼレポーター遺伝子の発現ベクターにより、安定的にトランスフェクトされる、ヒト胎児腎細胞を用いる (Thorens, 1992,「細胞生物学」(Cell Biology) 89: 8641-8645; Drucker et al., 1987, Proc. Natl. Acad. Sci. U.S.A. Vol.84: 3434-3438)。
【0170】
アッセイの第1段階は、GPCR、本ケースではGLP−1R、の立体配座変化を含み、ここでこの受容体はcAMP応答配列の調節下にあるレポーター遺伝子を含む真核細胞の細胞膜に存在する。受容体は、内在性受容体又は受容体をコード化する核酸であってよく、又は受容体構築物は、細胞に形質転換されてもよい。典型的には、第1の固相(例えば、第1のアッセイプレートのウェル)は、このような細胞(通常は哺乳動物細胞系)が固相に付着するように細胞の実質的に均質集団でコーティングされる。しばしば、細胞は付着性で、それにより自然に第1固相に付着する。次いで、候補アゴニストなどの被検体は、受容体(例えば、GLP−1R)が被検体に暴露(又はそれと接触)するように、付着細胞を有するウェルに加えられる。このアッセイは、関心あるGPCR(例えば、GLP−1R)のアゴニストリガンドの同定を可能にする。被検体への暴露に続いて、付着細胞はレポーター遺伝子の発現を可能にする所定時間(例えば、約6時間)の間、適した条件下にインキュベートされる。
【0171】
次いで、細胞はアッセイの検出相にかけられる状態になる。レポーター遺伝子の発現は、例えばルシフェラーゼ活性の検出により測定される。ルシフェラーゼ活性の検出は、当技術分野で公知の任意の様々な適した方法によって行なうことができる。
【0172】
最初の同定に続いて、候補(例えば、抗GLP−1Rモノクローナル抗体)のアゴニスト活性は、細胞内cAMPの濃度を測定することにより更に確認し、リファインすることができる。典型的には、第1の固相(例えば、第1のアッセイプレートのウェル)は、このような細胞(通常は、例えばHEK293細胞などの哺乳動物細胞系)が固相に付着するように細胞の実質的に均質集団でコーティングされる。候補は細胞を含むアッセイプレートに接種され、そして例えば約1時間適した条件下にインキュベートされる。細胞内のcAMP濃度は、当業者に公知であって任意の種々の適した方法で検出することができる。例えば、細胞内cAMP濃度は、cAMP−Screen Direct(登録商標) システムキット(Applied Biosystems)の仕様書に従って検出することができる。HitHunter(登録商標)cAMP XS + Assay(DiscoveRX(登録商標))、インビトロベースの競合免疫測定法などの他のcAMPアッセイキットが商業的に入手可能である。簡潔に言えば、細胞溶解物からの遊離cAMPは、β−ガラクトシダーゼ(β−gal)の小分子ペプチドフラグメント、標識ED−cAMPコンジュゲートに対して抗体結合を競合する。遊離cAMPの不存在下では、ED−cAMPコンジュゲートは抗体によって捕捉されて、相補性に利用できないことから、低シグナルの結果となる。遊離cAMPの存在下では、抗体部位は占有され、ED−cAMPコンジュゲートをフリーのままにしてEAを補完できるようにし、基質加水分解に活性なβ−galEFC酵素を形成させて化学発光シグナルを産生する。陽性シグナルは、抗体によって結合された遊離cAMPの量に正比例して発生する。
【0173】
候補(例えば、GLP−1Rモノクローナル抗体)のアゴニスト活性は、標的生物活性を試験するために知られているバイオアッセイにより、更に確認し、リファインすることができる。例えば、候補がGLP−1Rをアゴナイズする能力は、インスリン分泌を測定するアッセイで試験することができる。候補がインスリン分泌を促進させる能力は、RIN−38ラットインスリノーマ細胞系などの培養動物細胞に候補を与え、そして培地への免疫反応性インスリン(IRI)の放出をモニターすることにより測定することができる。代わりに、候補を動物に注射し、そして免疫反応性インスリン(IRI)の血漿レベルをモニターすることができる。
【0174】
IRIの存在は、例えばインスリンを特異的に検出することができる、ラジオイムノアッセイの使用を通して検出することができる。IRIの存在を検出することが可能な任意のラジオイムノアッセイを使用することができる;1つのこのようなアッセイはAlbano, J. D. M., et al., 1972, Acta Endocrinol., 70:487-509の方法の変法である。この変法では、pH7.4のリン酸/アルブミン緩衝液が用いられる。インキュベーションは、500μlのリン酸緩衝液、50μlの灌流液サンプル又は灌流液中のラットインスリン標準、100μlの抗インスリン抗血清(Wellcome Laboratories; 1:40,000希釈)、及び100μlの125Iインスリンの連続添加で調製し、10×75mm使い捨てガラス管中で750μlの総量とした。4℃で2〜3日間インキュベーション後、遊離インスリンは活性炭分離により抗体結合インスリンから分離される。アッセイ感度は1〜2uU/mLである。組織培養で増殖した細胞の細胞培養培地へのIRI放出を測定するために、好ましくは放射標識をプロインスリンに組み込む。ポリペプチドを標識することが可能な任意の放射標識を使用することができるが、標識プロインスリンを得るためには、3Hロイシンを使用することが好ましい。
【0175】
候補GLP−1Rアゴニストがインスリン分泌促進特性を有するかどうかを測定するために、膵注入によって測定することもできる。In situ単離ラット膵灌流アッセイは、Penhos, J. C., et al., 1969, 「糖尿病」(Diabetes), 18:733-738の方法の変法である。350〜600g質量の絶食雄Charles River系アルビノラットをアミタールナトリウム (Eli Lilly and Co.:160ng/kg)の腹腔内注射で麻酔した。腎臓、副腎、胃、及び下部結腸血管を結紮した。全腸を、約4cmの十二指腸及び下行結腸及び直腸を除いて切除した。従って腸の一小部分だけを灌流して、グルカゴン様免疫反応性を有する腸内物質による干渉の可能性を最小化した。灌流液は、4%デキストランT70及び0.2%ウシ血清アルブミン画分Vを備えた改変クレブスリンガー(Krebs-Ringer)重炭酸緩衝液であり、そして95%O2及び5%CO2でバブルさせた。非拍動流、4チャンネルローラーベアリングポンプ(Buchler polystatic, Buchler Instruments Division, Nuclear-Chicago Corp.)を使用し、そして1つの灌流液源から他へのスイッチは、3方コックで切替えることにより達成した。灌流を行なって、モニターし、そして解析する方法は、Weir, G. C., et al., 1974, J. Olin. Inestigat. 54:1403-1412の方法に従うが、これは参照により本明細書に組み込まれる。
【0176】
〔実施例2〕
多数のリンパ球及びマクロファージは自己免疫疾患マウスのCNSに浸潤する
本実施例は、炎症細胞でCNSの浸潤を説明する。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に再度、400ngの百日咳毒素をi.v.注射した。MOG免疫後日18に動物(EAE及びナイーブ)を犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。CD3(Chemicon)及びCD68(Serotec)に対する一次抗体で免疫組織化学を行なった。Alexa 488(Invitrogen)に結合したヤギ抗ラット二次抗体を検出に使用した。クレシルバイオレット及びルクソールファーストブルー染色を使用して、浸潤細胞体及びミエリンを染色した。
図1の染色結果は、EAEマウス(上側)のCNSが、健常マウス(下側)と比べて多数の浸潤T細胞、単球及びミクログリアを有することを示す。
【0177】
〔実施例3〕
GLP−1Rアゴニストの直接投与は自己免疫疾患における罹病率(morbidity)を減少させる
本発明の裏付けとして行なった実験では、GLP−1Rアゴニストの直接投与は、CNS特異的自己免疫疾患の動物における罹病率及びCNS組織損傷を減少させることが示された(図2)。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちにそして48時間後に、再度400ngの百日咳毒素をi.v.注射した。
マウスをMOG免疫後日0から日9まで、1mg/kgエキセンジン−4(n=10)、2mg/kgNT4(陽性対照、n=10)又はビヒクル(PBS、陰性対照、n=10)で毎日処置した。動物の罹病率を以下のようにスコア化した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。エキセンジン−4で処置した動物は、対照動物と比べて有意な罹病率低下を示した(図2)。この結果は、GLP−1Rアゴニストが慢性EAEの進行を緩徐化させるのに有効なことを示した。
【0178】
〔実施例4〕
GLP−1Rアゴニスト化合物は自己免疫疾患の罹病率を減少させるのに有効である
動物実験は、GLP−1Rアゴニスト抗体コンジュゲート化合物GAC−1が、対照免疫グロブリンと比べてEAE罹病率に対して有意な保護をもたらすことを示した(図3及び4)。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスをMOG免疫後日0から日6まで、1mg/kgエキセンジン−4(n=10)か又はビヒクル(n=10)で毎日処置した。別の群のマウスを、1mg/kgGAC−1(n=10)により週1回ベースで処置した。マウスを以下の尺度に従ってEAEの臨床徴候で毎日評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。GAC−1は、MOG免疫後週1回投与した場合、罹病率を減少させるのに有効であった(図3)。
別の実験では、EAEを前記のように誘発した。第1群の動物をMOG免疫後、日0から日2まで3mg/kgエキセンジン−4(n=10)で、そして日3から日8まで1mg/kgエキセンジン−4で処置した。第2群の動物をMOG免疫後日0に3mg/kgGAC−1(n=10)で処置し、その後免疫後日7に開始して1mg/kgGAC−1で週1回処置した。第3群の動物はPBS(対照、n=10)で毎日処置した。臨床スコアを前記の基準に従って毎日モニターした。GAC−1は、3mg/kgの初回用量で投与し、その後1mg/kgの週1回用量で投与した場合にも、尚罹病率を減少させるのにより有効と考えられた(図4)。結果は、3mg/kgGAC−1の初回用量が、1mg/kgだけの用量よりも罹病率に対して一層の保護(即ち、より低い罹病率)をもたらすことを示した。
【0179】
〔実施例5〕
GLP−1RアゴニストはCNSの炎症細胞浸潤をブロックし、自己免疫疾患の脱髄を減少させる
GLP−1Rアゴニストが動物での保護を媒介するメカニズムを更に解明するために、組織学的分析を行なった。脊髄切片を対照、エキセンジン−4処置、及びGAC−1処置EAE誘発動物から調製し、次いでミエリン又は浸潤細胞を同定する白血球マーカーに特異的な抗体を染色するために、ルクソールファーストブルーで染色した。対照動物では、染色はミエリン発現神経細胞のバックグラウンドに対して、白血球浸潤及び細胞破壊の領域を示した(図5A、6A及び6D)。白血球浸潤減少がGLP−1Rアゴニスト処置動物から調製した切片で認められた(図6B、6C、6E及び6F)。かなりのGLP−1Rアゴニスト処置動物は、CNS白血球浸潤の証拠を実質的に示さなかった。
【0180】
脊髄切片を、脱髄を測定するルクソールファーストブルーで染色した(図5A−C)。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を1mg/kgエキセンジン−4又はPBS対照で毎日、又は1mg/kgGAC−1で週1回処置した。動物をMOG免疫後日18に犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。ミエリン染色は、ルクソールファーストブルーを用いて行なった。
【0181】
図5A〜Cは、PBS処置対照EAE動物(図5A)、エキセンジン−4処置EAE動物(図5B)及びGAC−1処置EAE動物(図5C)からの脊髄の代表的な画像を示している。対照マウスの脳は、重度脱髄の領域(即ち、図5Aの黒矢によって示されるように、ルクソールファーストブルー染色の不存在)を示した。GLP−1Rアゴニスト処置動物の切片には、重度脱髄及び/又はニューロン死の領域は認められなかった(図5B及びC)。CNS組織切片の組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSの脱髄を減少させることを実証した。
【0182】
浸潤細胞の同定は、2つの白血球マーカー、T細胞に存在するCD3及びマクロファージに存在するCD68に特異的な抗体を用いて行なわれた。EAEを前記のように雌性C57BL/6マウスに誘発した。動物を1mg/kgエキセンジン−4又はPBS対照で毎日、又は1mg/kgGAC−1で週1回処置した。動物をMOG免疫後日12に犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。免疫組織化学はCD3(Chemicon)及びCD68(Serotec)に対する一次抗体を用いて行なわれた。Alexa488(Invitrogen)に接合したヤギ抗ラット二次抗体が検出に使用された。
【0183】
図6A〜Cは、CD3抗体で染色した結果を示す。多数の明染色断続領域が脱髄領域に対応して対照動物の脊髄に現われた(図6A)。対照的に、エキセンジン−4又はGAC−1で処置したマウスから得た脊髄切片は、CD3に対していずれも事実上染色を示さなかったことから(それぞれ、図6B及び6C)、T細胞浸潤はGLP−1Rアゴニスト処置動物で実質的に減少することが示された。当該観察は、GLP−1Rアゴニスト処置動物からの脊髄組織が、対照動物と比べた場合、T細胞浸潤減少を実質的に示すことを指示した。
【0184】
図6D〜Fは、CD68抗体による染色の結果を示す。対照マウス(図6D)から調製した切片は、エキセンジン−4又はGAC−1処置マウスからの切片(それぞれ、図6E及び6F)よりも強く染色した。当該観察は、GLP−1Rアゴニスト処置動物からの脊髄組織が、対照動物と比べた場合実質的に単球浸潤減少を示すことを指示した。総合すれば、CNS組織切片の免疫組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSのリンパ球及び単球浸潤を減少させることを実証した。
【0185】
〔実施例6〕
GLP−1Rアゴニストは脳及び脊髄のミクログリア及び浸潤マクロファージ中の活性化マーカーを減少させる
本実施例は、疾患発症時(MOG誘発後日15)及び疾患ピーク時(MOG誘発後日28)の両方における、脳及び脊髄のミクログリア及び浸潤マクロファージ中の活性化マーカー、MHC−II発現のGLP−1Rアゴニスト媒介による減少を例証する。
CNS中の抗原提示細胞(APC)に対するエキセンジン−4の作用を評価するために、日0にマウスにMOGを投与した。MOG誘発動物に、日0から日9まで(各処置群n=9)エキセンジン−4(1mg/kg)又はビヒクルを投与した。3匹のエキセンジン−4処置EAEマウス及び3匹のビヒクル処置EAE対照マウスを、日15(疾患発症)及び日28(疾患ピーク)に犠牲にした。ミクログリアを犠牲動物から分離した。簡潔に言えば、マウスをイソフルランで麻酔し、排出液がクリアになるまで左室を通して冷滅菌PBSで灌流した。脊髄及び脳は冷ハンクスの平衡塩類溶液(HBSS)に集めた。脊髄及び脳を、HBSSを用いてナイロン細胞ストレーナー(100μm)を通して押し出した。集めて漉した物質を、遠心分離によりペレット化した。ペレットは、脊髄又は脳当り1mlのHBSSに、コード(cord)当り300U/mlのIV型クロストリジウムコラゲナーゼを用いて再懸濁した。37℃インキュベータ中で60分間のインキュベーションを行った後、脊髄又は脳ホモジネートを30%パーコール(Percoll)に入れ、70%パーコールを下に入れた。そのグラジエントを遠心分離し、そして単球細胞を30%/70%パーコール界面から集めた。残存パーコールを除去するために、350gで5分間、cDMEM中にて2回界面から細胞を洗浄した。
【0186】
精製α−FcgR(2.4G2)でブロック後、単細胞懸濁液を多色フローサイトメトリーで染色した。MHCクラスII(10−3.6)、B220(CD45R)、CD45及びCD11bに特異的なモノクローナル抗体はBD Biosciencesから購入した。死細胞を排除するために、サンプルをフローサイトメトリー解析前に、Hoescht 33342(HO、Calbiochem)と共に氷上で5分間インキュベートした。成熟及び/又は固定アーチファクトを回避するために、細胞懸濁液を染色後直ちに解析した。死細胞(HOhi)及びB220hi細胞は解析中に排除された。ミクログリア及びCNS−マクロファージ解析では、サンプルをLSRIIで解析し、そしてflowjoソフトウェア(FlowJo, Ashland, Oregon)で解析した。
【0187】
ミクログリアサブセットを、CD45及びCD11bの発現に基づいて異なるサブセットに分けた。2つの異なる細胞集団サブセット、CD11b+CD45hi(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo(静止ミクログリア)について、ミクログリア/単球活性化マーカーMHCクラスIIの発現プロファイルを更に解析した。
【0188】
フローサイトメトリー解析の結果は、エキセンジン−4による処置が、症状発症(即ち、誘発後日15)及び疾患ピーク(即ち、誘発後日28)の両時点で、脳及び脊髄の両方において、CD11b+CD45hi及びCD11b+CD45lo細胞集団の両方でMHCクラスIIの発現レベルを減少させることを示す(図7)。図7に図示した各グラフは、示された蛍光強度で染色された全細胞の割合を示す。蛍光強度は、細胞表面でのMHCクラスII発現量を表わす(MHCクラスII発現は蛍光強度と正に相関した)。
【0189】
解析の結果は、ビヒクル処置対照EAE動物の細胞との比較により、GLP−1Rアゴニスト処置が、疾患発症及び疾患ピーク両時点において脳及び脊髄ミクログリア及び浸潤マクロファージでのAPC活性化マーカーMHCクラスIIの発現を抑制することを実証した。
【0190】
〔実施例7〕
エキセンジン−4処置マウスからの脾細胞はEAEの養子免疫伝達モデルにおいて低病原性である
本実施例は、養子免疫伝達モデルにおけるエキセンジン−4処置EAEマウスの脾細胞の病原性減少を例証する。
EAEを、CFA(Difco)に乳化した500μgのPLPp(139−151)の注射により8から12週齢のSJL/J雌マウス(The Jackson Laboratory, Bar Harbor, ME) に誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加した。免疫動物を1mg/kgエキセンジン−4(n=7)か又はPBS(n=7)で毎日処置した。処置動物を免疫後日5に犠牲にし、脾臓及び排出リンパ節を集めた。細胞を20μg/mlPLPp(139−151)と共にインビトロで培養した。48時間後、培養細胞を集め、ナイーブSJL/J動物に尾静脈注射を通して注入した。2千万の細胞を各動物に注射した。動物に、エキセンジン−4処置ドナー(n=7)か又は対照PBS処置ドナー(n=7)からの細胞を投与した。
レシピエントマウスは、臨床スコア及び体重変化を毎日モニターした。EAEの臨床徴候を以下の尺度により評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。図8に示すように、エキセンジン−4処置動物からのドナー細胞を投与したマウスは、PBS処置対照動物からのドナー細胞を投与したマウスよりも有意に低い疾患重症度を有した。この結果は、GLP−1Rアゴニスト処置が、EAEの養子免疫伝達モデルにおける脾細胞の病原性を減少させることを実証した。
【0191】
〔実施例8〕
エキセンジン−4はEAE誘発マウスにおいて二次リンパ器官のサイズを縮小させた
本実施例は、EAEマウスにおける二次リンパ器官のサイズに対するエキセンジン−4の効果を例証する。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。MOG誘発動物を1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も含まれた。疾患誘発後日5に動物を安楽死させた。脾臓及び鼠径リンパ節を解剖して秤量した。リンパ節質量及び脾臓質量及びサイズは、PBSで処置したEAE動物で顕著に増加した(図9A〜9C)。しかしながら、エキセンジン−4処置EAE動物のリンパ節(図9A)及び脾臓(図9B)は、対照動物よりも有意に少ない質量であった。同様に、免疫抑制薬デキサメタゾンもEAE動物のリンパ節及び脾臓質量を減少させた。エキセンジン−4処置EAE動物の脾臓はナイーブ動物の脾臓と同程度のサイズであった(図9C)。対照的に、対照EAE動物の脾臓は有意に肥大した(図9C)。これらの結果は、GLP−1Rアゴニスト処置がEAEの二次リンパ器官のサイズを減少させるのに有効なことを実証した。
【0192】
〔実施例9〕
自己免疫疾患におけるエキセンジン−4治療後の免疫学的変化
本実施例は、エキセンジン−4処置後の免疫学的変化を例証する。
最新のMS治療薬(コルチコステロイド及びCD20抗体RITUXANTMを含む)は、免疫抑制、T細胞枯渇、B細胞枯渇及び/又は免疫反応に関する他の作用を示す免疫調節薬である。GLP−1RアゴニストがEAEにおいてT細胞及びB細胞集団に影響を与えるかどうかを判定するために、EAE動物の免疫学的変化を、エキセンジン−4処置後に評価した。比較のために、動物を陰性対照としてのPBSで処置した。
【0193】
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を免疫後日0から日6まで、1mg/kgエキセンジン−4(n=3)か又はPBS(n=3)で毎日処置した。処置動物を、免疫しなかったナイーブマウス(n=3)と共に免疫後日6に犠牲にした。脾臓、リンパ節及び末梢血を集めた。赤血球細胞の細胞分離及び溶解後に、細胞を発蛍光団結合CD4、CD8、CD19(B細胞マーカー)及びTer119抗体(BD Biosciences)で染色した。細胞を、FACSAriaTMセルソーター(BD Biosciences)による蛍光活性化セルソーティング(FACS)を用いて解析した。
【0194】
脾臓、リンパ節及び血液中のリンパ球レベルは、PBS処置EAE動物と比べてエキセンジン−4処置EAE動物では不変と思われた(図10A〜C)。脾臓中の単球レベルは、脾臓エキセンジン−4処置EAE動物でやや減少したが(図10D)、リンパ節及び血液中では有意に変化しなかった(図10E及び10F)。エキセンジン−4処置はCD4+又はCD8+細胞集団を有意に変化させなかった(図11A〜F)。エキセンジン−4マーカーに由来するCD19+集団の変化は、正に統計的有意水準に達した(図12A〜C)。これらの所見は、CD4+、CD8+及びCD19+細胞を直接殺細胞するコルチコステロイド及びRITUXANTMなどの免疫抑制薬と、GLP−1Rアゴニストの作用メカニズムは異なることを示唆する。
【0195】
EAE動物のエキセンジン−4処置により、脾臓においてTer119+細胞集団がナイーブレベルに戻る大幅な減少が認められた(図12D)。Ter119+細胞はリンパ節には存在せず(図12E)、そして血液中で有意に変化しなかった(図12F)。この結果は、エキセンジン−処置が、EAEマウス脾臓中のTer119+細胞集団を減少させるのに非常に有効なことを実証した。
【0196】
別の実験では、EAEを前記のように雌マウスに誘発させた。動物を1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も実験に含まれた。疾患誘発後日5に動物を安楽死させた。脾臓及び鼠径リンパ節を解剖し、そして細胞を分離し、赤血球分化マーカーTer119及びCD4の表面染色を行ない、FACSで解析した。Ter119+細胞の割合を測定した(図13)。罹患動物はナイーブ動物に比べて高率のTer119+細胞を有する一方で、エキセンジン−4処置EAE動物は、PBS処置EAE動物と比べて低率のTer119+細胞を有した。免疫抑制薬デキサメタゾンによる処置も、EAE動物においてTer119+細胞の20〜25%減少をもたらしたが、エキセンジン−4処置(50〜55%)ほどではなかった。この結果は、エキセンジン−4処置がMOG免疫動物の脾臓中のTer119+細胞の割合を減少させるのに有効なことを実証した。
【0197】
〔実施例10〕
GLP−1Rアゴニスト処置は自己免疫疾患におけるT細胞活性化を減少させた
本実施例は、EAEマウスのエキセンジン−4処置後のT細胞活性化レベルの変化を例証する。
EAEを、前記のようにMOG(35−55)により雌C57BL/6マウスに誘発した。免疫動物は、PBSビヒクル(n=7)、1mg/kgエキセンジン−4(n=7)、又は4mg/kgデキサメタゾン(n=3)で毎日処置した。動物は、免疫しなかったナイーブ群の3匹のマウスと共に、免疫後日5に犠牲にした。リンパ節を集めた。細胞分離及び赤血球細胞の溶解後に、細胞を発蛍光団結合CD4及びCD44抗体(BDbiosciences)で染色した。細胞はFACSariaTMセルソーター (BD Biosciences)を用いて解析した。解析では、FACS像はCD4+細胞でゲートされた。高レベルのCD44(CD44hi)を有する細胞及びCD4に対する陽性染色は、活性化CD4細胞と同定した。CD4+CD44hi細胞の割合を各処置群で解析した。
図14に示すように、約35%のCD4細胞がナイーブ動物で活性化(CD4+CD44hi)された。EAE動物からの約60%のCD4細胞が活性化された(図14)。エキセンジン−4及びデキサメタゾンによる両処置は、活性化T細胞の割合を減少させ、ナイーブレベルに戻した(図14)。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット(Dunnett)事後検定を行なった。2アスタリスク(**)は、P値が、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で0.01より小さいことを示す。
この結果は、エキセンジン−4処置が、 EAE誘発後のT細胞活性化を抑制するのに有効なことを実証した。
【0198】
〔実施例11〕
GLP−1Rアゴニスト処置は自己免疫疾患においてCD4T細胞増殖を阻害した
本実施例は、EAEマウスのエキセンジン−4処置後のT細胞増殖の変化を例証する。
EAEを、前記のようにMOG(35−55)により雌C57BL/6マウスに誘発した。免疫動物は、PBSビヒクル(n=7)、1mg/kgエキセンジン−4(n=7)、又は4mg/kgデキサメタゾン(n=3)で毎日処置した。動物は、免疫しなかったナイーブ群の3匹のマウスと共に、免疫後日5に犠牲にした。リンパ節を集め、48時間MOG刺激で培養し、次いで増殖細胞を同定するためにBrdUで処置した。BrdUの添加の18時間後細胞を採取した。細胞増殖を測定するために、BrdU染色キット(BD Biosciences)をパシフィックブルー結合CD4抗体と一緒に使用した。細胞は、FACSariaTMセルソーター(BD Biosciences)を用いて解析した。CD4+/BrdU+細胞は増殖CD4T細胞と同定した。
図15に示すように、ナイーブ動物からのCD4細胞はMOG刺激に応答してほとんど増殖せず;ナイーブ動物からのわずか約3%のCD4T細胞がBrdUに対して陽性であった。対照的に、EAE動物からの細胞は、MOG刺激に応答してナイーブ動物からの細胞よりもはるかに大幅な程度増殖していた(図15)。エキセンジン−4及びデキサメタゾンによる処置は、CD4細胞増殖を有意に阻害した(図15)。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット事後検定を行なった。3アスタリスク(***)は、P値が、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で0.01より小さいことを示す。
この結果は、エキセンジン−4処置がEAE誘発後にCD4+細胞の増殖を減少させるに有効なことを実証した。
【0199】
〔実施例12〕
GLP−1Rアゴニスト処置は自己免疫疾患における脾細胞サイトカインレベルを変化させた
本実施例は、EAEマウスのエキセンジン−4処置後の脾細胞サイトカインレベル変化を例証する。
GLP−1RアゴニストがEAEのサイトカインレベルに影響を与えるかどうかを判定するために、EAE動物における脾細胞サイトカインレベルの変化を、エキセンジン−4処置後に評価した。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。誘発動物を、対照ビヒクル(PBS)又はエキセンジン−4(1mg/kg)で毎日処置した。脾細胞(表2の第1列の「SPL」)及びリンパ節(表2の第1列の「LN」)細胞をEAE誘発後日7に処置動物から単離し、そしてナイーブ動物(n=7)からも単離した。細胞を20μg/mlのMOGを含む刺激培地中で48時間培養した。培養培地中のサイトカインレベルをProcartaR Cytokine アッセイキット(Panomics(登録商標))を用いて測定した。使用したキットには、32の別な種類のサイトカイン及びケモカイン測定用ビーズ結合抗体が含まれた。
【0200】
PBS処置群(対照、表2の第3列)及びエキセンジン−4処置群(対照、表2の第4列)間で有意なレベル変化を示したサイトカインが、表2にリストアップされている。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット事後検定を行なった。1アスタリスク(*)は、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で、P値が0.05より小さいことを示し、そして2アスタリスク(**)は、P値が0.01より小さいことを示す。
【0201】
【表5】
【0202】
結果は、エキセンジン−4処置が、IL−17、IFN−γ、TNF−αなどの炎症性サイトカインのレベルを低下させ、疾患進行の減弱をもたらすことを示唆する。表2のデータは、エキセンジン−4処置EAE動物からの細胞におけるサイトカイン発現のFACS解析と一致する(例えば、実施例13を参照)。
【0203】
〔実施例13〕
エキセンジン−4処置はEAE誘発後にリンパ節中のIL−17+及びIFN−γ細胞を減少させた
本実施例は、EAEマウスのエキセンジン−4処置後のIL−17+及びIFN−γ細胞集団の変化を例証する。
GLP−1RアゴニストがEAE中のTh17及びTh1細胞集団に影響を与えるかどうかを判定するために、EAE動物中のIL−17+及びIFN−γ細胞集団の変化を、エキセンジン−4処置後に評価した。IL−17はTh17細胞に存在し、IFN−γはTh1細胞に存在する。比較のために、動物を、陰性対照としてPBSで、陽性対照としてデキサメタゾンで処置した。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を、1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も含まれた。疾患誘発後日5又は日7に動物を安楽死させた。鼠径リンパ節を解剖してFACSにより解析した。細胞をCD4について表面染色し、IL−17及びIFN−γについて細胞内染色した。CD4及びIL−17、又はCD4及びIFN−γについて二重陽性細胞を、FACSにより解析した。
【0204】
罹患動物がナイーブ動物と比べて高率のIL−17+細胞を有していた一方で、 エキセンジン−4−処置EAE動物は、MOG免疫後日5及び日7の両日において、PBS処置EAE動物と比べて低率のIL−17+細胞を有していた(図16)。免疫抑制薬デキサメタゾンによる処置も、EAE動物におけるIL−17+細胞の50%減少をもたらしたが、エキセンジン−4処置(80〜90%)ほどではなかった。この結果は、エキセンジン−4処置が、リンパ節におけるTer119+細胞の割合を減少させるのに有効なことを実証した。
【0205】
リンパ節中のIFN−γ+細胞集団の大幅な減少が、EAE動物のエキセンジン−4処置で認められた(図17)。予想通り、免疫抑制薬デキサメタゾンも、EAE動物においてIFN−γ+細胞の減少をもたらした。この結果は、エキセンジン−4処置が、EAEマウスのリンパ節中のIFN−γ+細胞集団を減少させるのに有効なことを実証した。これらの結果は、MOG免疫動物におけるTh17及びTh1の両細胞を減少させるGLP−1Rアゴニストの役割を裏付けるものである。
【0206】
両方の天然GLP−1Rアゴニスト、及びGLP−1RアゴニストCovX体は、EAEの進行を緩徐化させるのに有効である。天然アゴニストエキセンジン−4及びGAC−1は、いずれも疾患進行を緩徐化させるのに有効であった。エキセンジン−4は、EAE症状の発症前に投与した場合に有効であった。GAC−1は、EAE症状の発症前及び発症後週1回ベースで投与した場合に有効であった。GLP−1Rアゴニストの初回量の増加は罹病率減少と関連し、用量依存性を示唆した。組織化学実験は、GLP−1Rアゴニストが、T細胞及び単球によるCNS組織の浸潤を減少させることを示した。ミエリン染色は、GLP−1Rアゴニスト処置動物の神経細胞に対する損傷の減少を示した。GLP−1Rアゴニスト処置は、EAEの発症及びピーク時で、脳及び脊髄ミクログリア、及び浸潤するマクロファージのAPC活性化マーカーMHCクラスIIの発現を抑制した。
【0207】
エキセンジン−4を用いて、GLP−1RアゴニストがEAE進行に影響を与えるメカニズムを更に検討した。CD4+、CD8+及びCD19+細胞を直接殺細胞する、コルチコステロイド及びRITUXANTMなどの免疫抑制薬とGLP−1Rアゴニストの作用メカニズムは異なっているようである。これは、GLP−1Rアゴニストが、EAE誘発動物におけるCD4+又はCD8+細胞集団を有意に変化させなかったという観察における証拠である。しかしながら、GLP−1Rアゴニストは、MOG免疫動物のリンパ節におけるIL−17+及びIFN−g+細胞集団の両方を減少させた。これらの結果は、GLP−1Rアゴニスト処置が、リンパ節におけるTh17及びTh1集団を減少させることを実証した。
【0208】
〔実施例14〕
ジペプチジルペプチダーゼ−4(DPP−4)阻害剤は自己免疫疾患の罹病率を減少させるのに有効である
本実施例は、DPP−4阻害剤の処置によるGLP−1レベル上昇が、EAE進行を減弱させることができることを例証する。動物実験は、DPP−4阻害剤シタグリプチンが、メチルセルローズ陰性対照と比べて、EAE罹病率に対して有意な保護をもたらすことを示した(図18)。
GLP−1は、ジペプチジルペプチダーゼ−4(DPP−4)により生体内で分解される。DPP−4阻害剤は、例えばシタグリプチン、ビルダグリプチン、サクサグリプチン、リナグリプチン、ドュトグリプチン(dutogliptin)、ゲミグリプチン(gemigliptin)、アログリプチン及びベルベリンを含むが、これらに限定されない。DPP−4阻害剤シタグリプチンが、グルコースレベル増加及びGLP−1レベル上昇を抑制することができるかどうかを試験するために、4群の12月齢雄C57Bl/6マウス(n=4〜5)を一夜絶食させ、次の朝に経口ブドウ糖負荷試験にかけた。経口グルコース負荷の1時間前に、試験動物に1mg/kgか又は10mg/kgシタグリプチンを経口投与した。陰性対照群の動物にはメチルセルロースを投与し、陽性対照群にはエキセンジン−4(1mg/kgIP)を投与した。経口グルコース負荷後、グルコースレベルを20、40、60及び120分で試験した。グルコース及びGLP−1レベルをチェックするため、血液サンプルもグルコース負荷の20分後に採取した。エキセンジン−4処置は、ほとんど完全にグルコース上昇をブロックした。いずれのシタグリプチン濃度による処置も、メチルセルロース対照と比べて低いグルコースレベルの傾向を示したが、二元配置ANOVAによる統計的有意水準に至らなかった。いずれの用量でのシタグリプチンも、メチルセルロース対照と比べて経口グルコース負荷の20分後に血中GLP−1レベルを有意に増加させた。
【0209】
自己免疫疾患におけるDPP−4阻害剤の効果を試験するために、EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスに免疫後日0から日28まで、1mg/kgエキセンジン−4、10mg/kgシタグリプチン、1mg/kgシタグリプチン又はメチルセルロース(陰性対照)を毎日1回投与した。免疫後日5に血液サンプルをGLP1レベルの測定のために採取した。10mg/kgシタグリプチン処置動物は、メチルセルロース処置動物と比べて有意に高いGLP1レベルを有した。疾患進行をモニターするために、動物の罹病率を毎日以下のようにスコア化した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。エキセンジン−4又はシタグリプチンで処置した動物は、対照動物と比べて有意な罹病率減少を示した(図18)。図18のグラフに示されるように、エキセンジン−4による処置は日25を通してEAE進行を大きく減弱させ、1mg/kgか又は10mg/kgシタグリプチンによる処置は、病徴を減弱させる傾向を示した。10mg/kgシタグリプチン処置群での疾患スコアは、メチルセルロース処置群と比べて日18から日20まで有意に低かった(図18)。本結果は、エキセンジン−4及びシタグリプチンがいずれも慢性EAEの進行を緩徐化させるのに有効なことを実証した。
【0210】
〔実施例15〕
末梢免疫細胞はエキセンジン−4の標的ではない
本実施例は、末梢免疫細胞がエキセンジン−4の標的ではないことを例証する。
GLP−1Rアゴニスト−抗体コンジュゲートGAC−2(前記)が、エキセンジン−4/GLP−1受容体の発現をチェックするための染色試薬として使用された。GAC−2は、発蛍光団アレクサ488で標識した。本実施例では、GLP−1R発現CHO細胞を陽性対照として使用した。GLP−1R発現CHO細胞又はナイーブCHO細胞を1%FBSでブロックし、次いでGAC−2−アレクサ488で染色して、蛍光活性化セルソーティング(FACS)によって解析した。ナイーブCHO細胞中の全細胞がGAC−2染色に対して陰性なのに、34%のGLP−1R発現CHO細胞はGAC−2について陽性であったことから、GAC−2−アレクサ488は生細胞でのGLP−1R発現を認識することができることが示された。
ナイーブ動物及び及びEAE動物由来の脾臓リンパ節及び血液からの白血球について、GLP−1R又は他の可能なエキセンジン−4受容体の発現を調べた。リンパ球及び単球マクロファージコンパートメントの両方について、エキセンジン−4/GLP−1受容体発現をGAC−2を用いて解析した。いずれのコンパートメントにも陽性シグナルが認められなかったことから、エキセンジン−4/GLP−1受容体は末梢リンパ器官に発現しないか、又はそれらの発現レベルが非常に低く検出閾値以下であることが示された。
エキセンジン−4が末梢免疫細胞に直接作用するかどうかを試験するために、リンパ球細胞をエキセンジン−4(10μg/ml)又はビヒクル(陰性対照)の存在下MOGでインビトロ処理した。MOGの代わりに、PBSを活性化の陰性対照として使用した。MOG処理リンパ球細胞の活性化は、CD44発現(T−細胞活性化)及びMHCII発現(APC活性化)を解析することにより測定した。MOG処理細胞にエキセンジン−4を与えた場合、活性化阻害は認められなかった。これらの結果は、末梢免疫細胞はエキセンジン−4の標的ではないことを示す。
【0211】
〔実施例16〕
進行中のEAEマウスの罹病率を減少させるエキセンジン−4処置
本実施例は、進行中のEAEマウスの処置におけるエキセンジン−4の有効性を例証する。
本発明の裏付けのために行なわれた実験では、GLP−1Rアゴニストの直接投与は、進行中のEAEマウスの罹病率を減少させることを示した(図19)。EAEを、4mg/mlの結核加熱死菌H37Ra(Difco Laboratories)を含む完全フロイントアジュバント(CFA)に溶解した200μgのPLP(p139−151)による免疫で、SJL/Jマウス(8から12週齢、the Jackson Laboratory)に誘発した(Youssef et al., Nature (2002) 420:78-84を参照)。免疫マウスの体重測定及びEAEの臨床徴候を毎日調べてスコア化した。急性EAEをSJL/Jに樹立後、マウス(n=28)を臨床スコア及び体重に基づいて2群に無作為割付けした。
エキセンジン−4処置は、再発の初めに開始した(免疫後日29)。エキセンジン−4(1mg/kg)か又はビヒクル(PBS;対照用)を毎日1回投与した(i.p.)。マウスを無作為割付けした後、日29から日63まで毎日処置した。動物の罹病率を以下のようにスコア化した:0、麻痺なし;1、尾色調消失;2、後肢弱;3、後肢麻痺;4、後肢及び前脚麻痺;5、瀕死又は死亡。エキセンジン−4で処置した動物は、対照動物と比べて有意な罹病率減少を示した。この結果は、GLP−1Rアゴニストが進行中のEAEマウスの罹病率を減少させるのに有効なことを実証した。
【0212】
〔実施例17〕
インビボで細胞のエキセンジン−4処置は抗原特異的CD4+T細胞増殖をブロックする
本実施例は、エキセンジン−4でインビボ処置後の細胞の抗原特異的CD4+T細胞増殖の阻害を例証する。
本発明の裏付けのために行なわれた実験では、GLP−1Rアゴニストでの細胞のインビボ処置は、抗原特異的CD4+T細胞増殖を阻害することが示された(図19)。SJL/JEAE動物及びC57BL/6EAE動物の両方からの抗原特異的CD4+T細胞の増殖はブロックされた。SJL/J雌マウス(8から12週齢、the Jackson Laboratory)において、EAEを、4mg/mlの結核加熱死菌H37Ra(Difco Laboratories)を含む完全フロイントアジュバント(CFA)に溶解した200μgのPLP(p139−151)による免疫で誘発した(Youssef et al., Nature (2002) 420:78-84を参照)。雌C57BL/6マウスにおいて、EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスをエキセンジン−4(1mg/kg)か又はビヒクルのみ(PBS;対照として)で毎日処置した。
【0213】
免疫後日5に、排出リンパ節及び脾臓を処置動物から単離した。リンパ節及び脾臓からの細胞を洗浄し、1×106細胞/mlの最終濃度で予熱したPBS/0.1%BSAに再懸濁した。細胞増殖をモニターするために、細胞をCFSEで標識し、次いでPLP又はMOGペプチドの存在下で増殖させた。CFSE溶液(Invitrogen, catalog #C34554)を1μMの最終濃度まで加えた。細胞を37℃で10分間色素と共にインキュベートし、次いで5容の氷冷培養培地の添加及び5分の氷上インキュベーションによってクエンチした。細胞を遠心分離によりペレット化し、新鮮培地にて全部を3洗液で洗浄した。細胞を計数し、そしてPLPp139−151(SJL/Jマウスからの細胞)か又はMOGp35−55(C57BL/6マウスからの細胞)ペプチドのいずれかと共に、0、10、及び50μg/mlで72時間インキュベートした。細胞はフローサイトメトリーで分析した。CFSA染色を欠く細胞を増殖T細胞と同定した。そのデータを表3に要約する。
【0214】
【表6】
【0215】
表3中のデータは、SJL/J−EAE及びC57BL/6EAEマウスのエキセンジン−4処置が、対照動物と比べて増殖抗原特異的CD4+T細胞の減少をもたらすことを示す。この結果は、細胞をインビボにてGLP−1Rアゴニストで処置した場合に、EAE動物からの抗原特異的CD4+T細胞の増殖がブロックされることを実証した。
【0216】
前記のインビボエキセンジン−4処置の結果と対照的に、エキセンジン−4は、インビトロで脾細胞に投与した場合、CD4+T細胞の増殖に影響を与えなかった。簡潔に言えば、CD4+T細胞は、ナイーブC57BL/6J脾細胞から単離した。細胞増殖をモニターするために、細胞を前記のようにCFSEで標識し、次いでプレート結合抗CD3(1μg/ml)及び抗CD28(0.5μg/ml)で活性化した。エキセンジン−4(0ng/ml、10ng/ml、300ng/ml及び10μg/mlで)又はビヒクル(PBS)を細胞培養物に加え、そして細胞を72時間処置した。処置細胞をフローサイトメトリーで分析した。CFSA染色を欠く細胞を増殖性T細胞と同定した。インビトロにおいてエキセンジン−4で処置したCD4+T細胞は、対照細胞と同等に増殖した。この結果は、抗原特異的CD4+T細胞の増殖が、細胞をインビボでGLP−1Rアゴニストで処置した場合には影響されないことを実証した。
【0217】
〔実施例18〕
表面GLP−1Rは末梢免疫細胞で検出されない
本実施例は、末梢免疫細胞におけるエキセンジン−4染色の不存在を例証する。
GLP−1R発現CHO細胞及び親CHO細胞を、固定又は透過処理(permeabilization)なしでFITC標識GAC−2細胞と共にインキュベートした。細胞を一次抗体(トータル50μl中で10μg/ml)と共に室温で30分間インキュベートした。細胞をフローサイトメトリーを用いて分析した。FITCシグナルはGLP−1R発現CHO細胞で検出されたが、親CHO細胞では検出されなかった。このように、GAC−2は、細胞表面でGLP−1Rを検出することができる。
脾臓、リンパ節及び血液からの末梢免疫細胞は、MOGp35−55による免疫の5日後にナイーブ動物及びEAE動物から単離された。単離細胞をFITC標識GAC−2で染色し、フローサイトメトリーで分析しリンパ球及び単球にゲーティングした。GLP−1R染色は、これらの細胞集団のいずれでも検出されなかった。この結果は、これらの末梢リンパ器官細胞がそれらの表面でGLP−1Rを発現しないこと、又は表面GLP−1R発現が検出レベル以下であることを示唆する。
【0218】
〔実施例19〕
エキセンジン−4は2D2マウスモデルにおいてインビトロで免疫細胞のMOG活性化に影響を与えない
本実施例は、エキセンジン−4が免疫細胞のインビトロでMOG活性化をブロックしないことを例証する。
脾臓及びリンパ節細胞を7週齢の2D2マウスから単離し、そしてMOG刺激の有り又は無しで48時間培養した。エキセンジン−4(10μg/ml)又はビヒクル(PBS)をMOG刺激2D2細胞培養溶液に加えた。培養において48時間のMOG刺激後、細胞を集め、抗CD44抗体及び抗MHCクラスII抗体で染色し、そしてフローサイトメトリーで分析した。抗CD44抗体をT細胞活性化のマーカーとして使用した。抗MHCクラスII抗体をAPC活性化のマーカーとして使用した。
MOGで刺激した脾臓及びリンパ節細胞は、活性化T細胞の存在について陽性として染色した。エキセンジン−4処置はT細胞活性化をブロックしなかった。MOG刺激の不存在下に培養された細胞は、活性化T細胞について有意な染色を欠いていた。この結果は、2D2脾細胞及びリンパ節T細胞がMOG刺激によってエキソビボで活性化し得ること、及びエキセンジン−4処置がエキソビボで活性化を直接ブロックしないことを示す。
MOGで刺激した脾細胞及びリンパ節細胞は、活性化APC細胞の存在について陽性として染色した。エキセンジン−4処置はAPC細胞活性化をブロックしなかった。MOG刺激の不存在下に培養された細胞は、活性化APC細胞についての有意な染色を欠いていた。この結果は、2D2脾細胞及びリンパ節抗原提示細胞が、MOG刺激によってエキソビボで活性化し得ることを示唆するが、エキセンジン−4はエキソビボで活性化を直接ブロックしないようである。
【0219】
〔実施例20〕
エキセンジン−4処置の効果は炎症性サイトカイン産生を阻害する
本実施例は、炎症性サイトカイン産生に対するGLP−1Rの効果を例証する。
SJL/JEAEマウスモデルを用いて、炎症性サイトカイン産生に対するインビボでのエキセンジン−4処置の効果を検討した。再発寛解EAEを、PLP(p139−151)免疫により8週齢SJL/J動物に誘発した。1群の動物を1mg/kgエキセンジン−4で毎日処置し、そして第2群の動物をPBS(対照)で処置した。免疫後日5に、脾細胞を両群の動物から単離し、MOG刺激によりインビトロで培養した。MOG刺激による培養の48時間後に培養培地を集め、IFN−γ及びIL−17のLUMINEX(登録商標)サイトカインアッセイを用いてサイトカイン産生レベルを解析した。
【0220】
エキセンジン−4処置EAE動物からの脾細胞は、対照EAE動物からの脾細胞と比べて、有意に少ない量のIFN−γ及びIL−17を産生した。このことは、エキセンジン−4がPLP(p139〜151)免疫後にTh1及びTh17細胞を正常化するのに有効なことを示す。
【0221】
2D2脾細胞培養を用いて、エキソビボ系において炎症性サイトカイン産生に対するエキセンジン−4の効果を解析した。7週齢2D2動物からの脾細胞を単離し、MOG刺激の有り又は無しで培養した。MOG刺激による脾細胞培養において、エキセンジン−4がエキソビボでTh1又はTh17細胞を減少させるかどうかを試験するために、種々の濃度のエキセンジン−4(10ng/ml、300ng/ml及び10μg/ml)を加えた。PBSを陰性対照として用いた。エキセンジン−4又はPBS添加の48時間後に、培養培地を集め、サイトカインレベルを測定した。MOGで刺激した細胞からの培地は、高レベルのIFN−γ及びIL−17を有した。対照的に、MOGで刺激しなかった細胞からの培地は、最小レベルのIFN−γ及びIL−17を含んでいた。培養液中のエキセンジン−4の存在は、IFN−γの産生に対して効果がなかった。更に、10ng/mlエキセンジン−4で処置した細胞により産生したIL−17のレベルは、エキセンジン−4で処置しなかった細胞により産生したレベルと差がなかった。しかしながら、10ng/mlか又は300ng/mlで処置した細胞からの培地はやや低いIL−17のレベルを有するようであるが、その差は統計的に有意ではなかった。
【0222】
これらの結果は、エキセンジン−4がインビボのEAEマウスモデルにおいて、炎症性サイトカイン産生を減少させることを実証した。しかしながら、対照的に、エキセンジン−4は2D2エキソビボモデルにおける炎症性サイトカイン産生を減少させなかった。これらの結果は、エキセンジン−4が、脾臓又はリンパ節を直接標的化しないようであることを実証した。
【0223】
〔実施例21〕
エキセンジン−4処置はEAE動物の胸腺中のCD4、CD8二重陽性細胞を激減させる
本実施例は、EAE動物の胸腺中のCD4、CD8二重陽性細胞の減少を例証する。
EAEを誘発するために、8週齢のC57Bl/6マウスを皮下によりMOG/CFAで免疫し、免疫後直ちに、そして48時間後に百日咳毒素をi.p.注射した。1群の動物をエキセンジン−4(1mg/kg;n=3)で毎日処置し、そして別の群を対照PBS(n=4)で処置した。免疫後日4に、エキセンジン−4処置及び対照動物の胸腺を採取した。4ナイーブ動物の胸腺を同時に採取した。胸腺を測定し、そして胸腺細胞を単離し、計数し、染色し、次いでフローサイトメトリーで分析した。そのデータを下の表4に要約する。
【0224】
【表7】
【0225】
表4に示されるように、対照EAE動物の胸腺はナイーブ動物の胸腺と比べて有意に小さかった(p=0.007)。エキセンジン−4処置EAE動物の胸腺は、対照EAE動物の胸腺よりも更に小さかった(p=0.002)。対照EAE動物、エキセンジン−4処置EAE動物、及びナイーブ動物の胸腺からの胸腺細胞を単離した。前記の胸腺計測と一致して、対照EAE動物からの総胸腺細胞数は、ナイーブ動物よりも有意に少なかった。エキセンジン−4処置EAE動物からの総胸腺細胞数は、対照動物よりも有意に少なかった。
【0226】
胸腺細胞を抗CD4及び抗CD8抗体で染色し、そしてフローサイトメトリーで分析した。ナイーブ動物からの総胸腺細胞集団の84.6±0.3%はCD4、CD8二重陽性であった。対照EAE動物の総胸腺細胞集団の30.3±11.9%はCD4、CD8二重陽性であった。このデータは、MOG刺激に応答して胸腺中の二重陽性T細胞の分化及び成熟を反映している。エキセンジン−4で処置したEAE動物では、CD4、CD8二重陽性T細胞集団は、ほとんど完全に消失した−エキセンジン−4で処置したEAE動物からの総胸腺細胞集団のわずか2.3±0.3%が、CD4、CD8二重陽性であった。更なる研究は、エキセンジン−4で処置した動物において脾臓及びリンパ節細胞数の増加はないことを実証した。これらの結果は、エキセンジン−4が細胞死によるCD4、CD8二重陽性細胞の減少を引き起こす可能性を示唆する。
【0227】
別の研究では、T細胞成熟ナイーブ動物、及びEAE動物に対するエキセンジン−4の効果を検討した。EAEを、MOG/CFAの皮下注射し、続いて百日咳毒素をi.p.注射することにより8週齢C57BL/6マウスに誘発した。ナイーブ及びEAE動物を3群(n=3)に分けて、それぞれPBS(対照)、エキセンジン−4(1mg/kg)又はデキサメタゾン(4mg/kg)で処置した。デキサメタゾンは、T細胞死を引き起こすことが知られている。免疫後日6に胸腺を採取した。胸腺細胞を単離し、抗CD4及び抗CD8抗体で染色し、そしてフローサイトメトリーで分析した。データを下の表5に要約する。
【0228】
【表8】
【0229】
EAE対照動物の胸腺細胞では、CD4、CD8二重陽性細胞集団(21.7±6.3%)は、ナイーブコントロール動物(86.3±1.1%)と比べて減少した。エキセンジン−4処置及びデキサメタゾン処置の両方は、EAE動物のCD4、CD8二重陽性細胞集団(それぞれ、5.6±2.6%及び1.5±0.5%)を急激に減少させた。
【0230】
ナイーブ動物からの総胸腺細胞集団の86.3±1.1%はCD4、CD8二重陽性であった。デキサメタゾン処置は、CD4、CD8二重陽性細胞集団の有意な減少を引き起こした−デキサメタゾンで処置したナイーブ動物からの総胸腺細胞集団のわずか12.4±3.3%がCD4、CD8二重陽性であった。しかしながら、エキセンジン−4処置は、ナイーブ動物の胸腺のCD4、CD8二重陽性細胞集団に影響を与えなかった。エキセンジン−4で処置したナイーブ動物からの総胸腺細胞集団の81.5±2.0%は、CD4、CD8二重陽性であった。
【0231】
これらの結果は、エキセンジン−4処置はEAE動物の胸腺においてCD4、CD8二重陽性細胞の減少をもたらすが、ナイーブ動物では減少をもたらさないことを実証した。
【0232】
〔実施例22〕
迷走神経切離術は体重減少に対するエキセンジン−4の効果を消失させるが、EAE進行に対するエキセンジン−4の効果に影響を与えない
本実施例は、エキセンジン−4処置に対する迷走神経切離術の効果を例証する。
EAE進行に対するエキセンジン−4の効果が迷走神経を通して媒介されるかどうかを検討するために、EAEを、迷走神経切離した8週齢C57Bl/6動物(The Jackson Laboratory)に誘発した。EAEを、MOG/CFAの皮下免疫により、続いてその直後、及び48時間後に、百日咳毒素をi.p.注射することにより、迷走神経切離及び迷走神経非切離マウスに誘発した。免疫動物を2群(n=7〜8)に分けた。1群はエキセンジン−4(1mg/kg)で毎日処置し、そして他の群はPBS(対照)で処置した。体重及びEAE臨床スコアを毎日モニターした。日20の実験終了時に、動物を安楽死させた。胃計測の結果は、迷走神経切離動物の迷走神経が適正に切断されたことを確認した。
【0233】
MOG免疫の最初の4日以内に、エキセンジン−4処置迷走神経非切離EAEマウスは、対照の迷走神経非切離EAEマウスと比べてそれらの体重の10%が減量した。迷走神経切離マウスでは、、エキセンジン−4処置EAEマウスと対照EAEマウス間で体重の差は認められなかった。このように、迷走神経切離術は、体重減少に対するエキセンジン−4の効果を消失させた。この結果は、体重変化に対するエキセンジン−4の効果が、インタクト迷走神経に依存していることを示唆する。
【0234】
EAE疾患進行は、迷走神経切離術によって影響を受けなかった。迷走神経切離及び迷走神経非切離マウスの両方では、エキセンジン−4処置はPBS処置動物と比べてEAE進行を顕著に遅延させた(図20)。このように、迷走神経切離術は、EAE疾患進行のエキセンジン−4媒介遅延に影響を与えた。この結果は、EAEに対するエキセンジン−4の効果が、迷走神経によって媒介されないことを示唆する。
【0235】
開示された教示は、種々の応用、方法、キット及び組成物に関連して記載されているが、当然のことながら、種々の変更及び改変は、本明細書に記載の教示及び以下の特許請求の範囲から逸脱しない範囲で行なうことができる。前述の実施例は、開示された教示をより的確に例証するために提供されるものであって、本明細書に記載の教示の範囲を限定することを意図するものではない。本教示はこれらの典型的な実施態様について記載されているが、熟練技術者には当然のことながら、これらの典型的な実施態様の変形及び改変は、過度の実験なしに可能である。そのようなすべての変形及び改変は、本教示の範囲内である。
【0236】
本明細書で使用されるセクションの見出しは、系統立てる目的のためだけであり、決して記載された主題を限定するものと解釈すべきではない。当然のことながら、本教示に論じられる温度、濃度、時間などの前に「約」が存在し、その結果、僅かなそして実体のない偏差は、本明細書中の本教示の範囲内であることを暗示する。本願では、特に明記しない限り、単数の使用は複数を含んでいる。また、「含む(comprise)」、「含む(comprises)」、「含んでいる」(comprising)、「含有する(contain)」、「含有する(contains)」、「含有している(containing)」、「包む(include)」、「含む(includes)」及び「含んでいる(including)」の使用は限定することを意図するものではない。当然のことながら、前述の概要及びその後の詳細な説明は、単に例示しそして説明するためであって、本発明を制限するものではない。
【0237】
特許、特許出願、学術論文、教科書など、及びそれらがすでに存在しない程度までそれらに引用されている参考文献を含む、本明細書で引用されたすべての参考文献は、参照によりそれらの全体が本明細書に組み込まれている。1つ又はそれ以上の組み込まれた文献及び類似の資料が、定義された用語、用語の用法、記載技術など(これらに限定されない)を含め本出願と異なり又は相反する事象では、本願が規制する。
【0238】
前述の記載及び実施例は、本発明の一定の格別な実施態様を詳述し、発明者によって意図されたベストモードを記載している。しかし、当然のことながら、いかに前述の記載がテキスト中に詳述されていても、本発明は様々な方法で実施することができ、そして本発明は、添付の特許請求の範囲及びその任意の均等物のいずれかに従って解釈すべきものである。
【技術分野】
【0001】
本願は、2009年8月27日に出願された米国仮出願第61/237654号の利益を主張するものであり、この内容は全体が参照により本明細書に組み込まれる。
【0002】
(配列表に関する参照)
本願は電子出願されており、そしてテキスト形式で電子的に提出された配列表を含む。テキストファイルは、2010年8月9日に作成された「PC33892ASeqList.txt」と題する、そして30KBのサイズを有する配列表を含む。本テキストファイルに含まれる配列表は本明細書の一部であり、そしてその全体が参照により本明細書に組み込まれる。
【0003】
本発明は、多発性硬化症を含む中枢神経系を冒す自己免疫疾患の処置に関する。本発明は、中枢神経系組織の白血球浸潤を減少させるためのグルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを提供する。
【背景技術】
【0004】
多発性硬化症(MS)は、炎症、脱髄、及び軸索傷害によって特徴付けられる中枢神経系(CNS)の脱髄性自己免疫疾患である。本疾患は世界で百万人以上の人々を冒し、男性よりも女性で2倍罹患しやすい。MSの症状は、通常20歳〜40歳の間に発現する。
【0005】
MSの特徴は研究されているが、本疾患の病因は不明である。当該特徴としては、CNS組織の損傷、ミクログリアの活性化、炎症性サイトカイン産生、T細胞移動及びクローン型拡大の停止、及びマクロファージエフェクター機能変化、MHCの産生、アップレギュレーション、及びT細胞浸潤によるCNSへの直接攻撃を含む。
【0006】
実験的自己免疫性脳脊髄炎(EAE)は、MSの標準モデルと考えられる。マウス疾患モデルは、MSの病因を研究し、その処置のための薬剤を評価するために使用されている(非特許文献1)。EAEの臨床的特徴は、多数の浸潤リンパ球及びマクロファージによるCNSの炎症及び脱髄を含む。ミエリン塩基性タンパク質(MBP)、プロテオリピドタンパク質(PLP)、及びミエリンオリゴデンドロサイト糖タンパク質(MOG)を含む、ミエリンの幾つかの異なるタンパク質成分によるマウスの能動免疫は、自己免疫抗体の産生及び上行性麻痺の臨床症状を誘発する。疾患は、マウス株及び免疫に使用されるミエリンタンパク質によって急性又は慢性となり得る。EAEは、MSの病因を研究し、その処置のための薬剤を評価するのに使用されている(非特許文献1)。
【0007】
MSは大部分、神経組織に豊富に存在する脂質及び幾つかのタンパク質を含む、ミエリンに対するT細胞反応に起因する。脱髄及び臨床的麻痺は、ミエリン抗原に対して特異性を有するTh1及びTh17表現型のT細胞によるCNS組織浸潤に起因する。Th1細胞は、TNF−α及びIFN−γを含む炎症性サイトカインを産生する。IL−17発現は、MS患者、並びに関節リウマチ及び過敏性腸疾患患者で増加している(非特許文献2、3)。このことは、自己免疫疾患におけるTh17細胞の病原性役割を示唆する。CNSの損傷は、自己免疫抗体の産生及び補体活性化を含む他の免疫反応によっても引き起こされると考えられる。B細胞は、疾患の全期間にわたって認められるイソタイプのMBP特異的及びMOG特異的抗体を有するMS及びEAEの早期及び晩期の期間に関わる。脳及び脊髄組織から調製した切片は、白血球浸潤(特にリンパ球及びマクロファージ)及び神経系の基になる組織の破壊を呈する。
【0008】
グルカゴン様ペプチド(7−36)アミド(GLP−1)は、ラット及びヒトの両方におけるグルコインクレチンである(非特許文献4、5)。インスリン分泌の増加及びグルカゴン分泌の減少に加えて、30アミノ酸のGLP−1ペプチドはプロインスリン遺伝子転写を促進し、胃内容排出時間を減速し、そして食物摂取を減少させる。GLP−1は、グルカゴン様ペプチド1受容体(GLP−1R)に結合することによってその生理作用を示す。GLP−1Rは、多くの重要な生理的及び病態生理的過程を調節する、Gタンパク質結合受容体(GLCR)スーパーファミリーのクラスB受容体サブクラスに属する。GLP−1Rは、Gタンパク質活性化、cAMP産生増加及びPKAの活性化に連結する7回膜貫通受容体である。また、GLP−1Rを通して開始されるPKA非依存性反応である。GLP−1の作用に対する他の反応は、例えば、膵β細胞増殖及びβ細胞アポトーシスの減少と同時に増殖を含む。加えて、GLP−1活性は、膵臓細胞におけるグルコース輸送体−2(GLUT2)及びグルコキナーゼ遺伝子の発現増加をもたらすことができる。
【0009】
GLP−1は、また、孤束核(NTS)の尾部領域のニューロンによっても合成されるニューロペプチドである(非特許文献6)。GLP−1免疫反応性線維及びGLP−1Rは、脳全体に広く発現しているように見える(非特許文献6、7、8)。GLP−1R欠損マウスは、海馬体細胞におけるGlp1r遺伝子導入後の表現型修正を伴う、カイニン酸投与後の発作強度及びニューロン損傷を増強している (非特許文献9)。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Aharoni, R. et al., 2005, J. Neurosci. 25:8217-28
【非特許文献2】Komiyama, Y. et al., 2006, J. Immunol. 177, 566-573
【非特許文献3】Matusevicius, D. et al., 1999, Mult. Scler. 5, 101-104
【非特許文献4】Dupre and Ebert and Creutzfeld, 1987, Diabetes Metab. Rev. 3
【非特許文献5】Mojsov et al., 1986, J. Biol. Chem. 261:11880
【非特許文献6】Jin, S. L. et al., 1988, J Comp Neurol. 271:519-532
【非特許文献7】Larsen, P. J. et al., 1997, Neuroscience 77:257-270
【非特許文献8】Merchenthaler, I. et al., 1999, J Comp Neurol 403:261-280
【非特許文献9】During, M. J. et al., 2003, Nat Med. 9(9):1173-9
【発明の概要】
【発明が解決しようとする課題】
【0011】
1つの態様では、本発明は、GLP−1Rアゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置が必要な哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法を提供する。本発明は、哺乳動物の中枢神経系組織の白血球浸潤を減少させるために使用される薬剤の製造における、GLP−1Rアゴニストの使用を更に提供する。本発明は、哺乳動物のTh1及びTh17細胞数を減少させるために使用される薬剤の製造における、GLP−1Rアゴニストの使用を更に提供する。
【課題を解決するための手段】
【0012】
幾つかの実施態様では、哺乳動物は自己免疫障害を有する。1つの実施態様では、自己免疫障害は実験的自己免疫性脳脊髄炎である。別の実施態様では、自己免疫障害は多発性硬化症である。他の実施態様では、自己免疫障害は、免疫拒絶、移植片対宿主病、ブドウ膜炎、視神経症、視神経炎、横断性脊髄炎、炎症性腸疾患、関節リウマチ、強直性脊椎炎、全身性エリテマトーデス、重症筋無力症、又はグレーブス病と関連している。好ましい実施態様では、哺乳動物はヒトである。
【0013】
幾つかの実施態様では、GLP−1Rアゴニストを含む組成物の投与は、哺乳動物のリ
ンパ節においてTh1及び/又はTh17細胞数を減少させる。幾つかの実施態様では、GLP−1Rアゴニストを含む組成物の投与は、哺乳動物のリンパ節においてTer119+赤血球系細胞を減少させる。
【0014】
好ましい実施態様では、浸潤白血球はT細胞及びマクロファージを含む。特定の実施態様では、浸潤白血球はCD3発現及びCD68発現白血球を含む。好ましい実施態様では、中枢神経系組織は脳組織又は脊髄組織である。
【0015】
幾つかの実施態様では、GLP−1RアゴニストはOAP−189である。他の実施態様では、GLP−1RアゴニストはDPP−4阻害剤である。他の実施態様では、GLP−1Rアゴニストは抗GLP−1Rアゴニスト抗体である。
【0016】
幾つかの実施態様では、GLP−1Rアゴニストは天然GLP−1Rアゴニストである。幾つかの実施態様では、天然GLP−1Rアゴニストはエキセンジン−4である。関連実施態様では、天然GLP−1Rアゴニストはエキセンジン−4のフラグメント又は誘導体を含む。幾つかの実施態様では、エキセンジン−4のフラグメント又は誘導体は、GLP−1Rに結合してそれを活性化する。
【0017】
幾つかの実施態様では、GLP−1Rアゴニストは、GLP−1Rアゴニストペプチド及び抗体を含むGLP−1Rアゴニスト抗体コンジュゲート(「GAC」)を含む。関連する実施態様では、GACは以下の構造:
【化1】
式中、
ペプチドは以下の式:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14
E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40 (配列番号3)]−R2
式中、
R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;R2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であり、そしてX2はAib、A、S、T、V、L、I、D−Alaなどの遮断基であり;X10はV、L、I、又はAであり;X12はS又はKであり;X13はQ又はYであり;X14はG、C、F、Y、W、M、又はLであり;X16はK、D、E、又はGであり;X17はE又はQ;X19はL、I、V、又はAであり;X20はオルニチン、又はK(SH)Rなどの誘導体化リジン基、又はKであり;X21はL又はEであり;X23はI又はLであり;X24はA又はEであり;X25はW又はFであり;X26はL又はIであり;X27はI、K、又はVであり;X28はR、オルニチン、N、又はKであり;X29はAib又はGであり;X30は任意のアミノ酸、好ましくはG又はRであり;X31はP又は不存在であり;X32はS又は不存在であり;X33はS又は不存在であり;X34はG又は不存在であり;X35はA又は不存在であり;X36はP又は不存在であり;X37はP又は不存在であり;X38はP又は不存在であり;X39はS又は不存在であり;そしてX40は連結残基又は不存在であり;そしてここで、X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39、又はX40の1つは、リンカーを介して抗体の結合部位に共有結合した求核性側鎖を含む連結残基で置換され、ここで連結残基はK、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン、及びホモセリンから成る群から選択される;
を有するものであってよい。
【0018】
幾つかの実施態様では、GACは以下の構造:
【化2】
を含むことができる。
【0019】
幾つかの実施態様では、GACは以下の構造:
【化3】
を含むことができる。
【0020】
幾つかの実施態様では、抗体は、完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、二重特異性抗体及びミニ抗体から成る群から選択される。幾つかの実施態様では、抗体は、IgG1、IgG2、IgG3、及びIgG4から成る群から選択される定常ドメインを含む。幾つかの実施態様では、抗体は触媒抗体である。幾つかの実施態様では、抗体はアルドラーゼ抗体である。幾つかの実施態様では、アルドラーゼ抗体は、配列番号11に記載のアミノ酸配列を含む軽鎖、及び配列番号12に記載のアミノ酸配列を含む重鎖アミノ酸配列を含む。
【0021】
他の実施態様では、本発明は、GLP−1Rを活性化するのに有効量のGLP−1Rアゴニスト及び使用説明書を含む、中枢神経系組織の白血球浸潤を低減するための部品キットを提供する。関する実施態様では、本発明は、GLP−1Rを活性化するのに有効量のGLP−1Rアゴニスト及び使用説明書を含む、中枢神経系を冒す自己免疫障害を処置するするための部品キットを提供する。
【0022】
本発明のこれらの及び他の態様は、以下の記述及び実施例から明らかになるであろう。
【図面の簡単な説明】
【0023】
【図1】MOG免疫後のEAE動物中の浸潤細胞体、T細胞、単球及びミクログリアを示す組織化学を図示する。EAE動物(上側パネル)及び健常動物(下側パネル)からの脊髄切片を、浸潤細胞体(左)、浸潤T細胞を検出する抗CD3抗体(中間)、又は単球及びミクログリアを検出する抗CD68抗体(右)を染色するために、クレシルバイオレットで染色した。
【図2】MOG免疫後に1mg/kgのGLP−1Rアゴニストで処置した動物における、EAEの臨床的悪性度を示すグラフを図示する。動物に、1mg/kgエキセンジン−4(EXE4)、2mg/kgニューロトロフィン−4(NT4)、又はPBS(対照)のいずれかを、MOG免疫後に日0から日9まで毎日与えた。マウスを以下のスコアリング・システムによりEAEの臨床徴候を毎日評価した:0=正常;1=尾跛行;2=やや後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。
【図3】MOG免疫後に1mg/kgのGLP−1Rアゴニストで処置したEAE動物における、病的状態を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日、又はGLP−1アゴニストGAC−1(1mg/kg)で週1回処置した。臨床スコアを毎日モニターし、上記のように評価した。
【図4】MOG免疫後に種々の用量のGLP−1Rアゴニスト(エキセンジン−4又はGAC−1)で処置した、EAE動物における病的状態を示すグラフを図示する。エキセンジン−4(EXE4)処置した動物を、MOG免疫後日0から日2まで毎日3mg/kgエキセンジン−4で処置し、そして日3から日8まで毎日1mg/kgエキセンジン−4で処置した。GAC−1処置動物を、日0に3mg/kgGAC−1で処置し、続いて免疫後日7に始めて1mg/kgGAC−1を週1回処置した。対照動物は、対照PBSで毎日処置した。臨床スコアを毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図5】A〜Cは、(A)PBS対照処置及び(B)エキセンジン−4処置及び(C)GAC−1処置をした、EAE動物からの脊髄切片の組織化学染色の結果を図示する。切片のミエリンをルクソールファーストブルーで染色した。黒矢印は、ルクソールファーストブルー染色低下を示す白質の脱髄を示す。
【図6】A〜Fは、PBS対照(A、D)及びエキセンジン−4処置(B、E)及びGAC−1処置(C、F)とした、EAE動物からの脊髄切片の免疫組織化学染色の結果を図示する。切片は、CD3(A〜C)又はCD68(D〜F)に特異的な抗体で染色された。呈示切片は脊髄断面切片の前角である。
【図7】MHCクラスIIに特異的な抗体で染色された脳、及び脊髄からのCD11b+CD45hi細胞(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo細胞(静止ミクログリア)集団の、フローサイトメトリー分析からのデータを示すグラフを図示する。各グラフにおいて、エキセンジン−4(EXE4)処置をしたEAE動物からの細胞のMHCクラスII発現は黒実線(□)で表わされ、そして対照処置をしたEAE動物からの細胞のMHCクラスII発現は点線
【化4】
で表わされる。上側パネル:疾患発症時の動物から採取した細胞。下側パネル:疾患発症時の動物から採取した細胞。
【図8】EAEの養子免疫伝達モデルで脾細胞の病原性を示すグラフを図示する。左のグラフ:動物にPLPp(139−151)免疫後、日0から日4まで1mg/kgエキセンジン−4(EXE4)又はPBS(対照)を毎日与えた。動物を免疫後日5に犠牲にした。右のグラフ:エキセンジン−4処置ドナー(▽)及びPBS処置ドナー(■)からの脾細胞をナイーブSJL/J動物に伝達した。レシピエントマウスの臨床スコア及び体重変化を毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図9】A〜Cは、GLP−1Rアゴニスト処置後のリンパ節重量及び脾臓重量及びサイズを図示する。動物は、MOG免疫後日0から日5まで、1mg/kgエキセンジン−4(EAE−EXE4)、PBS(EAE−ビヒクル)又は4mg/kgデキサメタゾン対照(EAE−Dex)で毎日処置した。ナイーブ動物はMOGで免疫されなかった。(A)鼠径リンパ節重量。(B)脾臓重量。(C)ナイーブ動物、ビヒクルで処置したEAE動物及びエキセンジン−4で処置したEAE動物からの脾臓。
【図10】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ球(A−C)及び単球(D−F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸は、リンパ球(A−C)又は単球(D−F)である細胞の割合を示す。
【図11】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。CD4+細胞(A〜C)及び細胞CD8+(D〜F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸には、表示されたマーカーに対する陽性染色細胞の割合が示される。
【図12】A〜Fは、MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における免疫学的変化を示すグラフを図示する。動物を、MOG免疫後日0から日6まで、1mg/kgエキセンジン−4(EXE4)又はPBSで毎日処置した。動物を、免疫後日6に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。CD19+細胞(A〜C)及びTer119+細胞(D〜F)をリンパ球(A〜C)及び単球(D〜F)を、脾臓(A、D)、リンパ節(B、E)及び末梢血(C、F)から測定した。y−軸には、表示されたマーカーに対する陽性染色細胞の割合が示される。
【図13】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるTer119+細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。脾臓細胞は、赤血球系統マーカーTer119で表面染色された。Ter119に陽性の染色細胞の割合をy−軸に示す。
【図14】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における活性化T細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ節からの活性化T細胞は、高レベルのCD44(CD44hi)を有するCD4+細胞と同定された。y−軸は、処置群及びナイーブ群のそれぞれにおける、高レベルのCD44を有するCD4+細胞の割合を表わす。
【図15】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物における増殖細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。リンパ節細胞をMOG刺激により培養し、そして細胞増殖を測定するためにBrdUで処置した。y−軸は、処置群及びナイーブ群のそれぞれにおける、BrdU陽性のCD4+の割合を表わす。
【図16】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるIL−17+細胞集団変化を示すグラフを図示する。動物を、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5(左図)及び日7(右図)に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。鼠径リンパ節細胞は、CD4、IL−17及びIFN−γで染色された。IL−17に対して陽性染色のCD4+細胞の割合をy−軸に示す。
【図17】MOG免疫に続いてGLP−1Rアゴニストで処置した後の、EAE動物におけるインターフェロン−γ(IFN−γ)細胞集団変化を示すグラフを図示する。動物を、日0から日5まで、PBS(EAE−PBS)、1mg/kgエキセンジン−4(EAE−EXE4)又は4mg/kgデキサメタゾン(EAE−dex)で毎日処置した。動物を、免疫後日5に、免疫されなかった(ナイーブ)マウスと共に犠牲にした。鼠径リンパ節細胞は、CD4、IL−17及びIFN−γで染色された。IFN−γに対して陽性染色のCD4+細胞の割合をy−軸に示す。
【図18】MOG免疫後にGLP−1Rアゴニスト(エキセンジン−4)又はDPP−4阻害剤(シタグリプチン)で処置した、EAE動物における病的状態を示すグラフを図示する。MOG免疫動物に、1mg/kgシタグリプチン、10mg/kgシタグリプチン、エキセンジン−4、又はメチルセルロースを投与した。臨床スコアを毎日モニターした。EAEの臨床徴候を上記のように評価した。
【図19】GLP−1Rアゴニスト(エキセンジン−4)又はビヒクルで処置した、EAE動物の病的状態を示すグラフを図示する。免疫後日29に始めて、PLP免疫SJL/Jマウスに、1mg/kgエキセンジン−4又はビヒクルを毎日投与した。臨床スコアを毎日モニターした。EAEの臨床徴候を以下のように評価した:0、麻痺なし;1、尾色調欠失;2、後肢弱;3、後肢麻痺;4、後肢及び前脚麻痺;5、瀕死又は死亡。
【図20】エキセンジン−4又はPBSで処置した迷走神経切離又は迷走神経非切離EAE動物における、病的状態を示すグラフを図示する。マウスを、以下のスコアリング・システムに従い、EAEの臨床徴候について毎日評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。
【発明を実施するための形態】
【0024】
本発明は、多発性硬化症及び他の自己免疫障害の処置のためのGLP−1Rアゴニストを使用する方法に関する。
【0025】
一般的方法
本発明は、分子生物学、細胞生物学及び免疫学の分野で使用される従来技術を採用する。当該技術は、「分子クローニング:実験室マニュアル、第2版」(Molecular Cloning: A Laboratory Manual, second edition) (Sambrook, et al., 1989) Cold Spring Harbor Press;「オリゴヌクレオチド合成 」(Oligonucleotide Synthesis) (M.J. Gait, ed., 1984);「分子生物学の方法」(Methods in Molecular Biology), Humana Press;「細胞生物学:実験室ノート」(Cell Biology: A Laboratory Notebook)(J.E. Cellis, ed., 1998) Academic Press;「動物細胞培養」(Animal Cell Culture)(R.I. Freshney, ed., 1987);「細胞及び組織培養入門」(Introduction to Cell and Tissue Culture) (J.P. Mather and P.E. Roberts, 1998) Plenum Press; 「細胞及び組織培養:実験室操作」(Cell and Tissue Culture: Laboratory Procedures) (A. Doyle, J.B. Griffiths, and D.G. Newell, eds., 1993-8) J. Wiley and Sons;「酵素学における方法」(Methods in Enzymology) (Academic Press, Inc.);「実験免疫学便覧」(Handbook of Experimental Immunology) (D.M. Weir and C.C. Blackwell, eds.);「哺乳動物細胞での遺伝子移入ベクター」(Gene Transfer Vectors for Mammalian Cells) (J.M. Miller and M.P. Calos, eds., 1987);「分子生物学の最近のプロトコール」(Current Protocols in Molecular Biology) (F.M. Ausubel, et al., eds., 1987);「PCR:ポリメラーゼ連鎖反応」(PCR: The Polymerase Chain Reaction), (Mullis, et al., eds., 1994);「免疫学における最近のプロトコール」(Current Protocols in Immunology) (J.E. Coligan et al., eds., 1991);「分子生物学における短プロトコール」(Short Protocols in Molecular Biology) (Wiley and Sons, 1999); Immunobiology (C.A. Janeway and P. Travers, 1997);「抗体」(Antibodies) (P. Finch, 1997);「抗体:実践的アプローチ」(Antibodies: a practical approach) (D. Catty., ed., IRL Press, 1988-1989);「モノクローナル抗体:実践的アプローチ」(Monoclonal antibodies : a practical approach) (P. Shepherd and C. Dean, eds., Oxford University Press, 2000);「抗体の使用法:実験室マニュアル」(Using antibodies: a laboratory manual) (E. Harlow and D. Lane (Cold Spring Harbor Laboratory Press, 1999);「抗体」(The Antibodies) (M. Zanetti and J.D. Capra, eds., Harwood Academic Publishers, 1995)などの参考文献中に記載されている。
【0026】
定義
以下の略語、用語及び語句は、下記に定義されているように本明細書で使用される。特に明記しない限り、本明細書に記載の本発明に関連して使用される科学及び技術用語は、当業者により一般的に理解される意味を有する。更に、文脈のよって規定されない限り、単数用語は複数を含み、そして複数用語は単数を含むものとする。
【0027】
【表1】
【0028】
例えば、D−Ala又はN−Me−D−Ileのような「D」接頭辞により特に指示がない限り、本明細書に記載のペプチド中のアミノ酸及びアミノアシル残基のα炭素の立体化学は、「L」立体配置である。 Cahn-Ingold-Prelog の「R」及び「S」記号は、ペプチドのN−末端で特定のアシル置換基におけるキラル中心の立体化学を明示するために使用される。記号「R、S」は2つのエナンチオマー型のラセミ混合物を示すことを意味する。この命名法は、R. S. Cahn, et al., Angew. Chem. Int. Ed. Engl., 5:385-415 (1966)に記載のものに従う。
【0029】
D−HはDヒスチジンを指す。
本明細書で使用される2−アミノイソ酪酸は、以下の構造:
【化5】
を有する。
【0030】
「ポリペプチド」、「ペプチド」、及び「タンパク質」は、アミノ酸残基の重合体を表すために互換性を持って使用される。本明細書で使用されるこれらの用語は、1つ又はそれ以上のアミノ酸残基が対応する天然アミノ酸の人工化学的アナログである、アミノ酸重合体に適用してもよい。これらの用語は、天然アミノ酸重合体に適用してもよい。ペプチドの結合機能が維持される限り、アミノ酸はL又はD型であってよい。ペプチドは環状であってよく、ペプチド内の2つの非隣接アミノ酸間で分子内結合、例えば、骨格から骨格、側鎖から骨格及び側鎖から側鎖への環化を有してもよい。環状ペプチドは、当技術分野で周知の方法によって製造することができる。例えば、米国特許第6013625号;S. Cheng et al., J Med. Chem. 37:1-8 (1994)を参照されたい。
【0031】
すべてのペプチド配列は一般に認められている慣例に従って書かれるが、それによれば、α−N末端アミノ酸残基は左側であり、そしてα−C末端アミノ酸残基は右側である。本明細書で使用される用語「N末端」は、ペプチド中のアミノ酸の遊離α−アミノ基を指し、そして用語「C末端」はペプチド中のアミノ酸の遊離カルボン酸末端を指す。1つの基を有するN末端であるペプチドは、N末端アミノ酸残基のα−アミノ窒素上に1つの基を保持するペプチドを指す。1つの基を有するN末端であるるアミノ酸は、α−アミノ窒素上に1つの基を保持するアミノ酸を指す。
【0032】
「抗体」は、免疫グロブリン分子の可変領域に位置する少なくとも1つの抗原認識部位を通して、糖質、ポリヌクレオチド、脂質、ポリペプチドなどの標的への特異的結合が可能な免疫グロブリン分子である。本明細書で使用される用語は、インタクトポリクローナル又はモノクローナル抗体だけでなく、そのフラグメント(Fab、Fab’、F(ab’)2、Fv)、一本鎖(scFv)及びドメイン抗体(例えば、サメ及びラクダ科抗体を含む)、及び抗体部分を含む融合タンパク質、更に抗原認識部位を含む免疫グロブリン分子のその他の修飾立体配置のいずれをも包含する。抗体はIgG、IgA、又はIgM(又はそのサブクラス)などいずれのクラスの抗体をも含み、そして抗体はいずれかの特定のクラスである必要はない。その重鎖の定常ドメインの抗体アミノ酸配列に応じて、免疫グロブリンは異なるクラスに帰属することができる。免疫グロブリンには5つの主要クラス:IgA、IgD、IgE、IgG、及びIgMがあり、そしてこれらの幾つかはサブクラス(イソタイプ)、例えば、IgG1、IgG2、IgG3、IgG4、IgA1及びIgA2に更に分けられる。異なるクラスの免疫グロブリンに対応する重鎖定常ドメインは、それぞれα、δ、ε、γ、及びμと呼ばれる。異なるクラスの免疫グロブリンのサブユニット構造及び3次元立体配置は周知である。
【0033】
本明細書で使用される「浸潤白血球」は、自己免疫疾患、優先してCNSを冒す自己免疫疾患の結果として、脳及び脊髄組織を含む中枢神経系(CNS)組織へ侵入、浸潤、又は移動する白血球である。浸潤白血球は、主としてT細胞及び単球であるが、他の白血球が存在してもよい。
【0034】
本明細書で使用される「白血球浸潤減少」は、脳及び脊髄組織を含むCNS組織へ白血球の移動(即ち、侵入又は浸潤)を減少させることを指す。白血球浸潤減少は、特にCNS組織を構成する神経細胞及び/又は他の支持細胞について、白血球浸潤によって媒介される細胞毒性を減少させることを指す。白血球浸潤は、T細胞及び単球によって媒介される侵入を含む。白血球浸潤減少は、自己免疫攻撃からCNS組織を守ることを含む。白血球浸潤によって破壊されるCNSの細胞は、ミエリン発現細胞及び隣接非ミエリン発現細胞を含む。細胞破壊は、アポトーシス、壊死、又はその組み合わせであってよい。白血球浸潤減少は、疾患進行減速、病的状態の発症または重症度の遅延、生存期間の延長、クオリティオブライフの改善、認知、運動又は行動症状の減少又は安定化などの臨床的適応によって特徴付けられる。白血球浸潤減少は、中枢神経系(CNS)組織への白血球の移動リスクを抑制し又は減少させることも含む。白血球浸潤減少は、部分的又は完全であってもよく、例えば、その減少は、本明細書に記載のように相対的又は実質的減少で、約10%、約20%、約30%、約40%、約50%、約60%、約70%、約80%、約90%、又は更に約95%であってよい。
【0035】
本明細書で使用される「GLP−1R」は、GLP−1Rの活性の少なくとも一部を保持する、いずれかの型のGLP−1R及びその変異体を指す。ヒトGLP−1Rに対する特定の言及によるような、特に別の指示がない限り、GLP−1Rは、天然配列GLP−1Rの全哺乳動物種、例えば、ヒト、イヌ、ネコ、ウマ、及びウシを含む。
【0036】
本明細書で使用される「GLP−1受容体アゴニスト」又は「GLP−1Rアゴニスト」は、GLP−1Rの活性化量を増加させる分子であり、天然アゴニストGLP−1及びエキセンジン−4によって産生するものと類似の効果をもたらす。GLP−1Rアゴニストは、例えば、GLP−1Rに結合して活性化させることにより、GLP−1Rに立体配座変化を引き起こすことにより、GLP−1Rに結合したGタンパク質の活性化を引き起こすことにより、GLP−1Rを長期間(無期限を含む)活性化(例えば、活性立体配座において)状態にさせることにより、天然アゴニストの結合を調節することにより、GLP−1のインヒビターをブロック、又はその反対にGLP−1R活性化を調節し若しくはGLP−1R活性化に特徴的な細胞内イベントのカスケードを開始させることにより、GLP−1Rの活性化を増加させる。GLP−1Rアゴニストの好ましい特性は、本明細書に記載されている。本発明のGLP−1Rアゴニストは、少なくとも5%、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%、少なくとも100%、少なくとも200%、又はそれ以上GLP−1Rの活性化を増加することができる。
【0037】
本明細書で使用される「天然GLP−1Rアゴニスト」は、自然に存在しGLP−1Rの活性剤として機能する分子である。既知のGLP−1Rの天然アゴニストは、ペプチドホルモンGLP−1及びエキセンジン−4である。天然GLP−1Rアゴニストは、突然変異したGLP−1R対立遺伝子を有する動物に発現したポリペプチドなどの天然変異体分子を含む。
【0038】
「生物活性」は、本発明のGLP−1Rアゴニストと併せて使用する場合、一般に、GLP−1Rを結合及び活性化する能力及び/又はGLP−1Rシグナル伝達機能によって媒介される下流経路を有することを指す。本明細書で使用される「生物活性」は、GLP−1R発現細胞上で、GLP−1Rの天然リガンドとして、GLP−1の作用によって誘導されるものに共通した1つ又はそれ以上のエフェクター機能を包含する。生物活性は、以下:GLP−1Rを結合し活性化する能力;GLP−1Rに結合し活性化すること、GLP−1Rの立体配座変化を引き起こす能力、GLP−1Rに結合したGタンパク質の活性化を引き起こす能力、GLP−1Rを長期間(無期限を含む)活性化(例えば、活性立体配座において)状態にさせる能力、細胞内cAMPを増加させる能力、インスリン放出を促進する能力、及びGLP−1R活性化に特徴的な細胞内イベントのカスケードを開始させる能力、のいずれか1つ又はそれ以上を含むが、これらに限定されない。
【0039】
本明細書で使用される「完全アゴニスト」は、有効濃度でGLP−1Rの測定可能な効果を、本質的に完全に誘導するアゴニストである。例えば、GLP−1Rの測定可能な効果は、cAMPレベル増加であってよい。部分アゴニストとは、測定可能な効果を部分的に誘導することができるが、しかし最大濃度でも完全アゴニストではないアゴニストを意味する。実質的に完全とは、少なくとも約80%、好ましくは少なくとも約90%、より好ましくは少なくとも約95%、及び最も好ましくは少なくとも98%の測定可能な効果が誘導されることを意味する。関連する「測定可能な効果」は、本明細書に記載されている。
【0040】
本明細書で使用される「フラグメント」ポリペプチドは、少なくとも幾つかの生物特性を保持する大型ポリペプチド、即ち、GLP−1Rを活性化する能力などを有する大型ポリペプチドの一部分である。好ましいフラグメントは、大型ポリペプチドの生物特性をもたらすアミノ酸残基及び/又は構造又は大型ポリペプチドを含む。ポリペプチドフラグメントはペプチドとも呼ばれるが、本明細書ではポリペプチドとペプチド間で区別はされない。典型的フラグメントは本明細書に記載されている。フラグメントは、本明細書に記載のように誘導体化されてもよい。
【0041】
本明細書で使用される「誘導体」ポリペプチドは、官能基又は官能部分の付加又は除去などの、1つ又はそれ以上の共有若しくは非共有修飾を有する。誘導体の例は、本明細書に提供される。
【0042】
本明細書で使用される特定のポリペプチド配列の「変異体」は、デフォルト・パラメータで設定されたClustal V又はBLAST、例えば、「BLAST 2 Sequence」ツール、バージョン2.0.9(1999年5月7日)などのアルゴリズムを用いて、特定長さの1つのポリペプチド配列を超える特定のポリペプチド配列と、少なくとも40%配列同一性を有するポリペプチド配列と定義される。当該の1対のポリペプチドは、1つのポリペプチドのある規定の長さにわたって、例えば、少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも91%、少なくとも92%、少なくとも93%、少なくとも94%、少なくとも95%、少なくとも96%、少なくとも97%、少なくとも98%、又は少なくとも99%以上の配列同一性を示してもよい。
【0043】
ポリペプチド配列に適用され本明細書で使用される「配列同一性」は、Clustal V、MEGALIGN、又はBLASTなどの標準化アルゴリズムを用いて整列された、少なくとも2つのポリペプチド配列間の残基整合の割合を指す。ポリペプチド配列アラインメントの方法は周知である。幾つかのアラインメント法は、保存アミノ酸配列を考慮に入れる。本明細書に記載の当該保存配列は、一般的に、置換部位での電荷と疎水性を保存し、その結果ポリペプチドの構造(及び機能)を保存する。
【0044】
用語「ポリペプチド」、「オリゴペプチド」、「ペプチド」及び「タンパク質」は、いずれの長さの、好ましくは相対的に短い(例えば、10〜100アミノ酸)アミノ酸鎖をも指すように、本明細書では互換性を持って使用される。アミノ酸鎖は直鎖状又は分枝鎖状であってよく、修飾アミノ酸を含んでもよく、及び/又は非アミノ酸によって遮断されてもよい。この用語は、天然修飾又は遮断;例えば、ジスルフィド結合形成、グリコシル化、脂質化、アセチル化、リン酸化、又は標識成分との結合などその他の操作又は修飾によるアミノ酸鎖を包含する。また、例えば、アミノ酸の1つ又はそれ以上のアナログ(例えば、非天然アミノ酸などを含む)、並びに当技術分野で公知の他の修飾を含むポリペプチドも、本定義の範囲内である。当然のことながら、ポリペプチドは、一本鎖又は会合鎖として存在し得る。
【0045】
当技術分野で公知の本明細書において互換性を持って使用される「ポリヌクレオチド」又は「核酸」は、いずれかの長さのヌクレオチド鎖を指し、そしてDNA及びRNAを含む。ヌクレオチドは、デオキシリボヌクレオチド、リボヌクレオチド、修飾ヌクレオチド又は塩基、及び/又はそれらのアナログ、又はDNA若しくはRNAポリメラーゼにより鎖中に取り込むことができるいずれの基質であってもよい。ポリヌクレオチドは、メチル化ヌクレオチド及びそれらのアナログなどの修飾ヌクレオチドを含んでもよい。存在するならば、ヌクレオチド構造への修飾は鎖の会合前又は後になされてもよい。ヌクレオチド配列は、非ヌクレオチド成分により遮断される。ポリヌクレオチドは、標識成分と併せることによるなど、重合後に更に修飾してもよい。他のタイプの修飾は、例えば、「caps」、アナログによる1つ又はそれ以上の天然ヌクレオチドの置換、例えば、非荷電結合(例えば、ホスホン酸メチル、ホスホトリエステル、ホスホアミダート、カルバメートなど)による及び荷電結合(ホスホロチオアート、ホスホロジチオアートなど)によるもの、例えば、タンパク質(例えば、ヌクレアーゼ、毒素、抗体、シグナルペプチド、ポリ−L−リジンなど)などのペンダント部分を含むもの、挿入剤(例えば、アクリジン、プソラレンなど)によるもの、キレート化剤(例えば、金属、放射性金属、硼素、酸化性金属など)を含むもの、アルキル化剤を含むもの、修飾結合(例えば、αアノマー核酸など)によるものなどのヌクレオチド間修飾、並びに1つ又は複数のポリヌクレオチド非修飾型を含む。更に、糖類に通常存在するいずれかのヒドロキシル基も、例えば、ホスホネート基、ホスフェイト基により置換され、標準保護基により保護され、若しくは更なるヌクレオチドへの更なる結合の調製のために活性化されてもよく、又は固体支持体に結合されてもよい。5’及び3’末端OHは、リン酸化され又はアミン若しくは1〜20炭素原子の有機キャッピング基部分で置換することができる。他のヒドロキシル基も、標準的な保護基へ誘導体化してもよい。ポリヌクレオチドは、例えば、2’−O−メチル−、2’−O−アリル、2’−フルオロ−又は2’−アジド−リボース、炭素環糖類アナログ、α又はβアノマー糖類、アラビノース、キシロース又はリキソースなどのエピマー糖類、ピラノース糖類、フラノース糖類、セドヘプツロース、非環式糖アナログ及びメチルリボシドなどの脱塩基ヌクレオチドアナログを含む、一般的に当技術分野で公知のリボース又はデオキシリボース糖類の類似形も含むことができる。1つ又はそれ以上のホスホジエステル結合は、代わりの結合基によって置換してもよい。これらの代わりの結合基は、ホスフェイトがP(O)S(「チオアート」)、P(S)S(「ジチオアート」)、(O)NR2(「アミダート」)、P(O)R、P(O)OR’、CO又はCH2(「ホルムアセタール」)によって置換され、ここで、各R又はR’は、独立に、H、又は場合によりエーテル(−O−)結合、アリール、アルケニル、シクロアルキル、シクロアルケニル又はアラルキルを含む、置換若しくは非置換アルキル(1〜20℃)である実施態様を含むが、これらに限定されない。ポリヌクレオチド中の全結合が同一である必要はない。前記の説明は、RNA及びDNAを含み、本明細書で言及する全ポリヌクレオチドに適用される。
【0046】
本明細書で使用される「実質的に純粋」は、少なくとも50%純粋(即ち、不純物無し)、より好ましくは少なくとも90%純粋、より好ましくは少なくとも95%純粋、尚より好ましくは少なくとも98%純粋、そして最も好ましくは少なくとも99%純粋である物質を指す。
【0047】
「宿主細胞」は、ポリヌクレオチド挿入部分の取り込みのために1つ又は複数のベクターのレシピエントになり得るか又はなっている個々の細胞又は細胞培養液を含む。宿主細胞は単一宿主細胞の子孫を含み、そして子孫は、自然の、偶発的、又は意図的な突然変異の故に元の親細胞と必ずしも完全に同一(形態において又はゲノムDNA相補性において)である必要はない。宿主細胞は、本発明の1つ又は複数のポリヌクレオチドにより、インビボでトランスフェクトされる細胞を含む。
【0048】
本明細書で使用される「処置」は、有益な又は所望の臨床結果を得るためのアプローチである。本発明の目的として、有益な又は所望の臨床結果は、以下:運動改善、脚弱減少、CNSのリンパ球浸潤減少、及びMSを含む自己免疫疾患に起因する脱髄減少の1つ又はそれ以上を含むが、これらに限定されない。
【0049】
「発症減少」は、この病態に一般的に使用される他の薬剤(例えば、それへの露出)の量及び/又は治療の必要性の減少を含んでもよい、いずれかの重症度の減少を意味する。当業者には当然のことながら、個体は処置へのそれらの反応性の点で変動してもよく、そのようなものとして、例えば、「発症を減少させる方法」は、当該投与がその特定の個体にそのような発症減少をもたらす可能性があるという合理的な予測に基づいて、GLP−1Rアゴニストを投与することを反映する。
【0050】
「改善」は、GLP−1Rアゴニストを投与しないことに比べて、1つ又はそれ以上の症状の低減又は改善を意味する。「改善」は、症状の期間の短縮又は減少をも含む。
【0051】
本明細書で使用される「GLP−1Rを活性化するのに有効な量」、又はGLP−1Rアゴニストについての同様の表現は、GLP−1Rアゴニストの投与前のベースラインレベルと比べて、GLP−1R活性化(本明細書に記載され当技術分野で公知)の増加に十分な量を指す。活性の増加は、少なくとも5%、少なくとも10%、少なくとも20%、少なくとも30%、少なくとも50%、少なくとも10%、少なくとも200%、又はそれ以上であってよい。この量は、投与経路、体内でのGLP−1Rアゴニストの半減期、溶解性、バイオアベイラビリティ、クリアランス率、並びにGLP−1Rアゴニストの他の薬物動力学的特性、動物又は患者などの体重及び代謝のような考慮すべき事項を勘案するものである。
【0052】
本明細書で使用される、薬剤、化合物、又は医薬組成物の「有効投薬量」又は「有効量」は、いずれか1つ又はそれ以上の有益な又は所望の結果をもたらすのに十分な量である。予防的使用では、有益な又は所望の結果は、疾患の生化学的、組織学的及び/又は行動症状、その合併症及び疾患の進展時に現われる中間の病的表現型を含む、疾患のリスクの除去又は減少、重症度の軽減、又は発症遅延を含む。治療的使用では、有益な又は所望の結果は、例えば、CMT疾患の1つ又はそれ以上の症状の減少、疾患を処置するのに必要とされる他の薬剤の用量減少、別の薬剤の効果の強化、及び/又は患者の疾患進行遅延などの臨床結果を含む。有効投薬量は、1つ又はそれ以上の投与方法で投与することができる。本発明の目的のために、薬剤、化合物、又は医薬組成物の有効投薬量は、予防的又は治療的処置を直接的にせよ間接的にせよ達成するのに十分な量である。臨床状況において理解されているように、薬剤、化合物、又は医薬組成物の有効投薬量は、別の薬剤、化合物、又は医薬組成物と併せて達成しても又は達成しなくてもよい。従って、「有効投薬量」は、1つ又はそれ以上の治療薬を投与する状況で考慮してもよく、そして1つ又はそれ以上の他の薬剤と併せて所望の結果が得られる可能性があるか又は得られる場合、単剤は有効量で与えられるものと考えてよい。
【0053】
本明細書で使用される「処置が必要な動物」又は類似の表現は、CNSに関わる自己免疫疾患を発症するリスクを有するか又はその危険性がある動物、好ましくはヒトを含む哺乳動物を意味する。CNSを冒す自己免疫疾患(又は障害、無差別に)の例は、マウスの実験的自己免疫性脳脊髄炎(EAE)、ヒトの多発性硬化症(MS)、及び他の動物に見られる類似の自己免疫疾患である。CNSの自己免疫攻撃は、例えば、免疫拒絶、視神経症、炎症性腸疾患、及びパーキンソン病にも見られる。
【0054】
「個体」又は「対象」は、哺乳動物、より好ましくはヒトである。哺乳動物は、家畜、スポーツ動物、ペット、霊長類、ウマ、イヌ、ネコ、マウス及びラットを含むが、これらに限定されない。
【0055】
本明細書で使用される「ベクター」は、宿主細胞中の関係する1つ又はそれ以上の1つ又は複数の遺伝子若しくは1つ又は複数の配列をデリバリーし、そして好ましくは発現することができる構成物を意味する。ベクターの例は、ウイルスベクター、裸のDNA又はRNA発現ベクター、プラスミド、コスミド又はファージベクター、カチオン性縮合剤に関連するDNA又はRNA発現ベクター、リポソームにカプセル化されたDNA又はRNA発現ベクター、及び生成細胞などの特定の真核細胞を含むが、これらに限定されない。
【0056】
本明細書で使用される「発現調節配列」は、核酸の転写を方向付ける核酸配列を意味する。発現調節配列は、構成的若しくは誘導型プロモーター、又はエンハンサーなどのプロモーターであってよい。発現調節配列は転写する核酸配列に機能的に結合される。
【0057】
本明細書で使用される「薬学的に許容される担体」又は「薬学的に許容される医薬品添加剤」は、有効成分と組み合わせた場合、成分が生物活性を保持することを可能とし、そして対象の免疫系と非反応性であるいずれの物質をも含む。例としては、リン酸緩衝食塩溶液などのいずれかの標準的薬剤担体、水、油/水乳濁液などの乳剤、及び種々のタイプの湿潤剤が挙げられるが、それらに限定されない。エアロゾル又は非経口的投与のための好ましい賦形剤は、リン酸緩衝生理食塩水(PBS)又は生理食塩水(0.9%)である。当該担体を含む組成物は、周知の従来法(例えば、「Remingtonの薬学、第18版」(Remington's Pharmaceutical Sciences, 18th edition), A. Gennaro, ed., Mack Publishing Co., Easton, PA, 1990; 及び「Remington、薬剤学の科学及び実践」(Remington, The Science and Practice of Pharmacy 20th Ed.) Mack Publishing, 2000を参照)によって製剤化される。
【0058】
本明細書で使用される「末梢投与」又は「末梢血管から投与される」は、中枢神経系(CNS)又は血液脳関門(BBB)の外側で対象に薬剤を導入することを指す。末梢投与は、脊椎又は脳への直接投与以外のいずれの投与経路をも包含する。末梢投与は局所的又は全身的であってよい。
【0059】
中枢神経系(CNS)を冒す自己免疫障害の処置のための、GLP−1Rアゴニスト
本発明は、多発性硬化症及び中枢神経系(CNS)を冒す他の自己免疫障害の処置のための、GLP−1Rアゴニストの使用方法を提供する。本発明の1つの特徴として、GLP−1Rアゴニストは、CNSの組織への白血球浸潤を減少し及びCNSの構成的神経組織の破壊を減少し、その結果として脚麻痺などこの疾患の臨床症状を改善するのに有効である。特に、GLP−1Rアゴニストは、ミエリン抗原を提示し、そしてそれらを産生する細胞に対する細胞毒性作用に寄与するのに活性を有する、単球及びT細胞の移動を減少させる。
【0060】
本発明に至る観察は、十分認められているMSの動物モデル、実験的自己免疫性脳脊髄炎(EAE)マウスを用いて行なわれた。EAEは、ヒトのMSと多くの臨床的及び病態的特徴を共有する実験的病状である。多くのFDA承認のMS治療法が先ず発見され、そしてマウス及びラットにおけるEAEモデルを基準に開発された(Steinman and Zamvil,
2006により検討された)。ミエリン塩基性タンパク質(MBP)、プロテオリピドタンパク質(PLP)、及びミエリンオリゴデンドロサイト糖タンパク質(MOG)を含む、ミエリンの幾つかの異なるタンパク質成分によるEAEマウスの能動的免疫は、自己抗体の産生及び上行性麻痺の臨床症状を誘発する。例えば、MOGペプチドアミノ酸35−55(Aharoni, R., et al.)又はPLPペプチドアミノ酸139−151(PLPp(139−151))で免疫後に、EAEをC57BL/6マウスに誘発することができる。MOG又はPLPによる免疫は、MSに類似した脱髄及び病的状態を引き起こすミエリン特異的自己免疫反応を誘導する。EAEの臨床的特徴は、多数の浸潤リンパ球、単球及びマクロファージによるCNSの炎症及び脱髄を含む(図1)。
【0061】
GLP−1Rアゴニストを用いる動物実験
本発明の裏付けとして行なわれた実験では、GLP−1Rアゴニストの直接投与は、CNS特異的自己免疫疾患動物における病的状態及びCNS組織障害を減少させることが示された(図2〜4)。エキセンジン−4(EXE4)は、MOG免疫した後に続いてEAE動物に投与された。エキセンジン−4で処置した動物は、対照動物と比べて病的状態の有意な減少を示した。この結果は、GLP−1Rアゴニストの投与が、慢性EAEの進行を緩徐化させるのに有効なことを証明した。実験は、症状の発症に関してエキセンジン−4によって得られた保護が、trkBアゴニストNT4(PCT国際公開第2008/078179号を参照)によって得られた保護と少なくとも同じ(もし、より良くない場合は)であること、そしてエキセンジン−4による処置は、処置が有効になるために症状発症時点まで継続する必要はないことを示した(図2)。
【0062】
動物実験では、GLP−1Rアゴニスト化合物GAC−1の週1回の投与が、対照と比べてEAE罹病率に対して有意な保護をもたらすことを示した。GAC−1は週1回ベースで投与した場合に有効であった。実験は、GAC−1による週1回の処置が、症状発症の時点まで少なくとも1日1回のエキセンジン−4処置と同様に保護することを示した。
【0063】
組織学的分析は、対照EAE動物からの切片と比べて、GLP−1Rアゴニストによって処置した動物から調製した切片においては、脱髄及び白血球浸潤減少を示した(図5A〜C及び6A〜F)。GLP−1Rアゴニスト処置動物は、CNSCD3+細胞浸潤の証拠を事実上示さなかった。浸潤細胞の特定はCD3発現T細胞及びCD68発現マクロファージの染色によって行なわれた。GLP−1Rアゴニスト処置動物からの脊髄組織は、対照動物と比べた場合、実質的にT細胞及び単球浸潤減少を示した(図6A〜F)。CNS組織切片の組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSのリンパ球及び単球浸潤を減少させることを証明した。
【0064】
EAEマウスでの活性化マーカーMHCクラスII発現の解析は、エキセンジン−4による処置が、脳及び脊髄の両方において、CD11b+CD45hi(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo(静止ミクログリア)細胞集団の両方で、MHCクラスIIの発現レベルを減少させることを示した(図7)。エキセンジン−4を与えたEAEマウスからの静止ミクログリア、活性化ミクログリア及び浸潤マクロファージは、症状発症及び疾患ピークの両方で、ビヒクル処置対照動物からの細胞と比べてMHCクラスII発現の低下を示した。
【0065】
動物実験では、エキセンジン−4処置マウスの脾細胞は、EAEの養子免疫伝達モデルにおいて低病原性であることを示した(図8)。エキセンジン−4処置PLP誘導マウスから分離した脾細胞を受けたマウスは、対照(PBS処置)PLP誘導マウスから分離した脾細胞を受けたマウスよりも実質的に低重度の疾患を発症した。
【0066】
GLP−1Rアゴニスト処置EAEマウスの鼠径リンパ節重量は、ビヒクル処置対照EAE動物のリンパ節よりも小さかった(図9A)。対照処置EAE動物のリンパ節は実質的に重かった。GLP−1Rアゴニスト処置EAEマウスの脾臓はナイーブ動物の脾臓の重量及びサイズと同程度であり、ビヒクル処置対照EAE動物の脾臓と対照的に、実質的に重くそして大きかった(図9B〜C)。
【0067】
EAE動物のGLP−1Rアゴニスト処置に由来する免疫学的変化を解析するために動物実験が行なわれた(図10A〜F、11A〜F及び12A〜F)。GLP−1Rアゴニスト処置は、ナイーブ動物のTer119+細胞数の近くまで、MOG誘発動物でTer119+細胞数の大幅減少をもたらした(図12D〜F及び13)。対照的に、PBS処置対照EAE動物は、Ter119+細胞数を増加した。加えて、GLP−1Rアゴニスト処置は、ナイーブ動物と比べてIL−17+及びIFN−γ+細胞数を増加したPBS処置対照EAE動物と比べて、EAE動物でIL−17+及びIFN−γ+細胞数を減少させた(図16及び17)。免疫学的解析の結果、GLP−1Rアゴニスト処置は、EAE動物でのヘルパーT細胞レベルを減少させることを示唆する。
【0068】
上記の結果は、GLP−1RアゴニストがCNSの脱髄及び白血球浸潤を減少させ、そして広く認められているMSの動物モデルであるEAEの進行を緩徐化させるのに有効であることを証明した。天然のGLP−1Rアゴニストエキセンジン−4及びGLP−1RアゴニストGAC−1は、いずれも疾患進行減速に有効であった。組織化学実験では、GLP−1Rアゴニストが、T細胞及び単球によるCNS組織の浸潤を減少させることを示した。
【0069】
CNS自己免疫障害に対するGLP−1Rアゴニスト
理論に限定されることなく、GLP−1Rアゴニストは、主として白血球の移動を調節し、及び/又はTh1及びTh17細胞レベルを減少させることにより機能すると考えられる。本方法は、更なる治療効果をもたらすために最近の免疫抑制療法と併用してもよい。
【0070】
本発明のGLP−1Rアゴニストは、多数の自己免疫又は関連疾患において、CNS組織の白血球浸潤を減少させるために使用することができる。本発明のGLP−1Rアゴニストは、多数の自己免疫又は関連疾患において、Th1及びTh17細胞レベルを減少させるためにも使用することができる。MSのモデルであることに加えて、EAEマウスは、視神経炎を研究するために使用することもできる。GLP−1Rアゴニストは、とりわけ白血球浸潤及び/又はTh1及びTh17細胞によって媒介されるすべてのこれらの疾患及び他の自己免疫障害におけるCNS免疫浸潤を軽減することが期待される。用語の疾患及び障害は、区別せずに使用することを銘記されたい。
【0071】
本発明の特徴は、自己免疫疾患を患っている動物に対するGLP−1Rアゴニストの直接投与である。本発明の好ましい実施態様はポリペプチドについて記載されているが、本発明は体内においてコード化されたGLP−1Rアゴニストの発現を方向付ける、当該GLP−1Rアゴニストポリペプチドをコード化するポリペプチドの投与を包含する。直接DNA注入及び遺伝子療法デリバリーの方法は当技術分野で公知である。GLP−1Rアゴニストポリペプチド、又はそれらをコード化するポリヌクレオチドは、薬剤の投与により誘導するのと対照的に、動物に直接投与される。本発明は、天然GLP−1Rアゴニスト及び/又はアゴニスト抗体に合致する方法でGLP−1Rを結合し活性化する、ペプチド模倣薬分子をも包含する。
【0072】
本明細書に記載の方法に従って使用するための特定のGLP−1Rアゴニストは、以下に更に詳細に記載される。更なるGLP−1Rアゴニストは、本発明の範囲から逸脱することなく当業者には明らかなものである。
【0073】
天然のGLP−1Rアゴニスト及びそれらの誘導体
GLP−1Rアゴニストは、GLP−1、エキセンジン−4、プロエキセンジン(例えば、米国特許第6723530号を参照)、エキセンジン−3(例えば、米国特許第5424286号を参照)、OAP−189(例えば、米国特許出願公開第20090181885号を参照、その全文は、参照することにより本明細書に組み入れられている)、及びオキシントモジュリンを含む、天然アゴニストポリペプチド、そのフラグメント、変異体、及び誘導体を含むが、これらに限定されない。本明細書で使用されるGLP−1及び/又はエキセンジン−4ポリペプチド配列は、結果として生じるポリペプチドがGLP−1Rに結合してアゴニストとして機能するという条件で、対応するGLP−1Rと同じ種から又は異なる種から生じたものであってよい。
【0074】
GLP−1Rアゴニストは、天然及び変異GLP−1を含む。GLP−1ポリペプチドは、多数の哺乳動物で同定されている。HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号1)は、2位置のアラニンにおいて、ジペプチジルペプチダーゼIV(DPP−4)によるGLP−1の切断によって生成された30アミノ酸GLP−1(7〜36)ペプチドである。D.J.Drucker(2001)「内分泌学」(Endocrinology)142:521-527。GLP−1(7〜36)はGLP−1Rアゴニストとして機能し、グルコース依存性インスリン分泌増加をもたらす。しかしながら、GLP−1(7〜36)の半減期はわずか数分である。ピロテアーゼ切断部位は、GLP−1ポリペプチドの半減期を延長するために除去されても、又はそれらの活性の調節を可能にするために加えられてもよい。GLP−1Rアゴニストは、PEGなどの半減期拡張部分、IgGFc領域、アルブミン、又はMyc、HA(ヘマグルチニン)、His−6、又はFLAGなどのペプチド若しくはエピトープに、結合又は融合してもよい。他の典型的GLP−1ポリペプチドは、GLP−1(7〜37)HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号42)及びGLP−1(1〜45)を含む(例えば、米国特許出願公開第2003/0232754号を参照)。
【0075】
GLP−1Rアゴニストは、更に天然及び変異エキセンジン−4を含む。エキセンジン−4はアメリカドクトカゲの唾液中に同定されている。エキセンジン−4は、GLP−1と約53%相同性の39アミノ酸ペプチドである。アメリカドクトカゲエキセンジン−4の配列は、HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS(配列番号2)である。GLP−1(7〜36)のように、エキセンジン−4はGLP−1Rアゴニストとして機能し、グルコース依存性インスリン分泌を促進する。しかしながら、GLP−1(7〜36)と異なり、エキセンジン−4はDPP−4による切断に対する抵抗性の増加を示す。GLP−1(7〜36)のN末端領域及びエキセンジン−4はほとんど同一であるが、有意な差異は第2アミノ酸残基である。この残基はGLP−1(7〜36)ではアラニンであり、しかしエキセンジン−4ではグリシンである。N末端領域におけるこの単一アミノ酸は、DPP−4消化に対するエキセンジン−4の抵抗性の増加に関与している。エキセンジン−4とGLP−1(7〜36)との間の別な有意な差異は、Trpケージを形成するエキセンジン−4のC末端における9つの更なるアミノ酸残基の存在である。
【0076】
本発明の天然及び変異GLP−1及びエキセンジン−4ポリペプチドは、キメラ、変異体、フラグメント(ペプチドを含む)、及び/又はその誘導体を含む。好ましいフラグメントは、天然ポリペプチドのGLP−1R結合部分、又はキメラの、コンセンサス、又は突然変異した等価結合部分を含む。フラグメントは合成ペプチドを含む。変異体は、保存又は非保存アミノ酸置換を有する天然アミノ酸配列変異体を含む。典型的変異体としては、GLP−1−Tf(米国特許出願公開第20060205037号)、OAP−189、エキセンジン−4−Tf(国際公開公報第2008012629号)、GLP−1、及び米国特許出願公開第20040242853号に開示のエキセンジン−4ペプチドアナログが含まれるが、これらに限定されない。OAP−189の配列は、HAQGTFTSDYSKYLEQELVKYFIQWLKNAGPSKNNIA(配列番号43)である。
【0077】
保存置換は、類似のサイズ、電荷、又は疎水性のアミノ酸残基を含む。例えば、AlaはVal、Leu、又はIleによって置換してもよい。ArgはLys、Gln、又はAsnによって置換してもよい。AsnはGln、His、Lys、又はArgによって置換してもよい。AspはGluによって置換してもよい。CysはSerによって置換してもよい。GlnはAsnによって置換してもよい。GluはAspによって置換してもよい。GlyはProによって置換してもよい。HisはAsn、Gln、Lys、又はArgによって置換してもよい。IleはLeu、Val、Met、Ala、Phe、又はノルロイシンによって置換してもよい。Leuはノルロイシン、Ile、Val、Met、Ala、又はPheによって置換してもよい。LysはArg、Gln、又はAsnによって置換してもよい。MetはLeu、Phe、又はIleによって置換してもよい。PheはLeu、Val、Ile、又はAlaによって置換してもよい。ProはGlyによって置換してもよい。SerはThrによって置換してもよい。ThrはSerによって置換してもよい。TrpはTyrによって置換してもよい。TyrはTrp、Phe、Thr、又はSerによって置換してもよい。ValはIle、Leu、Met、Phe、Ala、又はノルロイシンによって置換してもよい。
【0078】
機能の実質的な修飾は、(a)例えば、シート又はヘリカル立体配座として置換の部位におけるポリペプチドの骨格の構造、(b)標的部位での分子の電荷又は疎水性、又は(c)側鎖の大きさの維持に対するそれらの効果において著しく異なる置換を選択することによって達成されると考えられる。天然残基は共通の側鎖特性(これらのあるものは、幾つかの官能基に分類することができる)に基づいてグループ分けされる:
(1)疎水性:Met、Ala、Val、Leu、Ile、ノルロイシン;
(2)中性親水性:Cys、Ser、Thr;
(3)酸性:Asp、Glu;
(4)塩基性:Asn、Gln、His、Lys、Arg;
(5)芳香族:Trp、Tyr、Phe;及び
(6)たわみを誘導する残基:Gly及びPro。
【0079】
非保存置換は、1つのクラスのメンバーを別のクラスのメンバーと交換し、又は前項で保存性と特定されなかった置換に関わる。
【0080】
アゴニストの適正な立体配座を維持するのに関係しないいずれのシステイン残基も、分子の酸化安定性を改善し又は異常架橋を防止するために一般的にセリンと置換してよい。反対に、1つ又は複数のシステイン結合がその安定性を改善するためにアゴニストに付加されてもよい。
【0081】
アミノ酸修飾は、可変領域など1領域の1つ又はそれ以上のアミノ酸を変化させ又は修飾することから、完全な再設計までの範囲で変動することができる。可変領域の変化は、結合親和性及び/又は特異性を変えることができる。
【0082】
変異体は、結果として得られるポリペプチド又は誘導体がGLP−1Rに結合してアゴニストとして機能するという条件で、天然アミノ酸配列変異体及び改変変異体を含む。GLP−1R活性化を測定するための分析については、本明細書及び引用文献に記載されている。
【0083】
誘導体は、共有結合又は非共有結合修飾ペプチド及びポリペプチド(例えば、アシル化、ペグ化、ファルネシル化、グリコシル化、又はリン酸化ポリペプチド)である。ポリペプチドは、結合及び/又は活性を調節し、生体内画像化を可能にし、半減期を調節し、血液脳関門を通る輸送を調節し、又は特定の細胞型又は組織へのポリペプチドのターゲッティングを補助するために、更なる官能基を含んでもよい。ポリペプチドは、置換がGLP−1Rへのポリペプチドの結合し、又はアゴニスト活性に実質的に影響を与えないことを条件として、修飾(例えば、ペグ化、グリコシル化、又は他の部位の追加)を促進させるためのアミノ酸置換を含んでもよい。
【0084】
一般的な修飾は、生物活性の最小ロスで全身クリアランスを減少させるペグ化である。ポリエチレングリコール重合体(PEG)は、当技術分野で公知の方法(例えば、Roberts et al. (2002),「最新ドラッグデリバリー概説」(Advanced Drug Delivery Reviews)54:459-476; Sakane et al. (1997) Pharm. Res. 14:1085-91を参照)を用いて、GLP−1及びエキセンジン−4ポリペプチド(並びにGLP−1Rアゴニスト抗体)の種々の官能基へ結合してもよい。PEGは、例えば、アミノ基、カルボキシル基、修飾又は天然N末端、アミン基、及びチオール基に結合してもよい。幾つかの実施態様では、1つ又はそれ以上の表面アミノ酸残基はPEG分子で修飾される。PEG分子には様々なサイズ(例えば、約2〜40kDaの範囲)がある。GLP−1、エキセンジン−4、又は他のポリペプチドに結合するPEG分子は、約2,000、10,000、15,000、20,000、25,000、30,000、35,000、40,000Daのいずれかの分子量を有することができる。PEG分子は、単一又は分枝鎖状であってよい。GLP−1RアゴニストポリペプチドへPEGを結合させるために、1つ又は両方の末端に官能基を有するPEGの誘導体を使用してもよい。官能基は、ポリペプチド上の利用可能な反応性基のタイプに基づいて選択される。誘導体をポリペプチドに結合する方法は当技術分野で公知である。PEGのN末端特異的結合の1つの例は、ペグ化を促進するために1の位置の残基をセリン又はトレオニンに突然変異させることである。
【0085】
ポリペプチド又はそれらの誘導体は、直接又は合成リンカーを経て他の分子に結合してもよい。好ましいGLP−1Rアゴニストポリペプチド、そのフラグメント、又は誘導体は、その価値が報告されている天然のGLP−1Rアゴニストと比べて、類似の又はより優れた親和性、選択性、及び活性化を示す。アゴニストポリペプチドの小部分は「ペプチド」と呼ばれるが、この専門用語は、これに限定されると解釈すべきではない。
【0086】
好ましいGLP−1Rアゴニストは、EAE動物モデルを含む本明細書に記載の数々の実験及び分析において、エキセンジン−4及びGAC−1と比べて類似の生物学的特性を示す。
【0087】
GLP−1Rアゴニスト−抗体コンジュゲート
誘導体は、半減期を延長し及び/又は薬効を強化するための、GLP−1Rアゴニストペプチドの抗体への共有結合を更に含む。例えば、抗体の結合部位に直接か又は介在リンカーを介して、結合したGLP−1Rアゴニストペプチド剤を含むGLP−1Rアゴニスト化合物は、米国特許出願公開第20090098130号及びPCT国際公開第2008/081418号に記載されており、いずれも、それらの全文が参照することにより本明細書に組み入れられている。当該GLP−1Rアゴニスト化合物は、本明細書では「GLP−1Rアゴニスト−抗体コンジュゲート」又は「GAC」と呼ばれる。GACは、直接か又はリンカーを経て抗体に結合した、例えば、GLP−1又はエキセンジン−4フラグメント又は誘導体などのGLP−1Rアゴニストペプチドを含む。リンカーが使用される幾つかの実施態様では、リンカーは、GLP−1RアゴニストペプチドとGLP−1R間の抗体制限結合を避けるために、抗体からGLP−1Rアゴニストペプチドを遠ざけるように作用すると考えられる。
【0088】
本明細書に記載のGACは、GLP−1Rアゴニストペプチド、即ち、GLP−1Rの活性化量を増加させるペプチドを含み、これは天然アゴニストGLP−1及びエキセンジン−4によって生じるものと類似の効果をもたらす。幾つかの実施態様では、GLP−1Rペプチドは、1つ又はそれ以上のR基によって隣接してもよい。例えば、幾つかの実施態様では、GACのGLP−1Rアゴニストペプチドは、以下の構造を有してよい:R1−HAEGTFTSDVSSYLEGQAAKEFIAWLVKGR(配列番号1)−R2、ここで、R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;そしてR2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質である。他の実施態様では、GLP−1Rアゴニストペプチドは、以下の構造を有してよい:R1−HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPPS(配列番号2)−R2(配列番号4);ここで、R1は不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;そしてR2はOH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質である。GLP−1Rアゴニストペプチドは、配列番号1及び2のアナログを更に含む。当該アナログは、例えば、バイオアベイラビリティ増加、安定性増加、EAE処置プロファイル改善、Th17細胞数減少プロファイル改善、食欲抑制改善、体重管理改善、及び/又は宿主免疫認識減少などの更なる有利な特性を有してもよい。本明細書で使用される配列番号1又は配列番号2のペプチドアナログは、本質的に配列番号1又は配列番号2の配列を有するペプチドであるが、しかし1つ又はそれ以上のアミノ酸の置換、挿入若しくは欠失、又はそれらの組み合わせを有する。
【0089】
特定の実施態様では、本明細書で提供されるGLP−1Rアゴニストペプチドは、配列番号1又は配列番号2を含む。好適な典型的配列番号1及び配列番号2アナログは、例えば、2008年1月4日に出願された米国特許出願第11/969850号(米国特許出願公開第20090098130号として公開)の表IIに記載され、その全文は、参照することにより本明細書に組み入れられている。
【0090】
GLP−1Rアゴニストペプチドは、当技術分野で周知の技術を用いて調製することができる。例えば、GLP−1Rアゴニスト抗体コンジュゲート化合物を製造する方法は、米国特許出願公開第20090098130号に記載されている。一般的には、GLP−1Rアゴニストペプチドの合成が第1工程であり、そして米国特許出願公開第20090098130号に記載のように行なわれる。次いで、GLP−1Rアゴニストペプチドは、結合成分(リンカー)への連結のために誘導体化され、次いで抗体へ結合される。当業者には当然のことながら、使用される特異的合成工程は、3成分の正確な性質によって決まる。このように、本明細書に記載のGLP−1Rアゴニストペプチド−リンカーコンジュゲート及びGACは、容易に合成することができる。
【0091】
本明細書で使用される「抗体」は、B細胞及びその変異体の産物である免疫グロブリン、並びにT細胞及びその変異体の産物であるT細胞受容体(TcR)を含む。結合部位は抗原結合に関与する抗体分子の一部を指す。
【0092】
本明細書に記載のGLP−1Rアゴニストペプチドは、直接か又はリンカーを経て抗体中の結合部位に共有結合してもよい。適切なリンカーは、標的薬剤と抗体間で十分な距離を与えるように選択することができる。他のリンカーの検討としては、得られるGAC及びGLP−1Rアゴニストペプチド−リンカーの物理的又は薬物動力学的特性に対する影響、溶解性、親油性、親水性、疎水性、安定性(概ね安定並びに計画的分解)、剛性、柔軟性、免疫原性、抗体結合の調節、ミセル又はリポソームへの取り込まれ能力などが含まれる。ペプチド組成、リンカー組成、結合残基位置の更に微小な変化は、得られる分子の種々の特性(例えば、安定性、溶解性、半減期、バイオアベイラビリティ、標的結合能力、アゴニスト又はアンタゴニスト活性、薬物動力学など)に対して予測できない及び驚くべき効果を有することができる。
【0093】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式I:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14
E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40(配列番号3)]−R2
式中、
X2は、Aib、A、S、T、V、L、I、D−Alaなどの保護基であり;X10は、V、L、I又はAであり;X12は、S又はKであり;X13は、Q又はYであり;X14は、G、C、F、Y、W、M又はLであり;X16は、K、D、E又はGであり;X17は、E又はQであり;X19は、L、I、V又はAであり;X20は、オルニチン又はK(SH)R若しくはKなどの誘導体化リジン基であり;X21は、L又はEであり;X23は、I又はLであり;X24は、A又はEであり;X25は、W又はFであり;X26は、L又はIであり;X27は、I、K又はVであり;X28は、R、オルニチン、N又はKであり;X29は、Aib
又はGであり;X30は、いずれかのアミノ酸、好ましくはG又はRであり;X31は、P又は不存在であり;X32は、S又は不存在であり;X33は、S又は不存在であり;X34は、G又は不存在であり;X35は、A又は不存在であり;X36は、P又は不存在であり;X37は、P又は不存在であり;X38は、P又は不存在であり;X39は、S又は不存在であり;そしてX40は、連結残基又は不存在であり;かつ、X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39又はX40の1つは、直接又は中間リンカーを経由して抗体の結合部位に共有結合できる求核性側鎖を含む連結残基(−[LR]−)で置換され、そして連結残基はK、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてよい;
のものであってよい。
【0094】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式II:
R1−[H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−V10−S11−S12−Y13−L14−E15−G16−Q17−A18−A19−K20−E21−F22−I23−A24−W25−L26−V27−K28−G29−R30(配列番号4)]−R2
の配列を含む。幾つかの実施態様では、残基の1つは結合残基と置換されてもよい。幾つかの実施態様では、G16又はA24の1つは、直接又は中間リンカーを介して抗体の結合部位に共有結合できる求核性側鎖を含む結合残基で置換され、そして結合残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてもよい。他の実施態様では、リンカーは、R30基のC末端、又は31位置での更なる残基のC末端に共有結合してもよい。
【0095】
幾つかの実施態様では、本発明を用いて使用するためのペプチドは、式III:
R1−[H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−L10−S11−K12−Q13−M14−E15−E16−E17−A18−V19−R20−L21−F22−I23−E24−W25−L26−K27−N28−G29−G30−P31−S32−S33−G34−A35−P36−P37−P38−S39−X40(配列番号5)]−R2
式中、
X40は、結合残基又は不存在であり、L10、S11、K12、Q13、M14、E16、E17、A19、R20、L21、E24、L26、K27、N28、G32、S33、G34、A35、P36、P37、P38、S39又はX40の1つは、直接又は中間リンカーを介して抗体の結合部位に共有結合できる求核性側鎖を含む結合残基(−[LR]−)で置換され、かつ、結合残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン及びホモセリンから成る群から選択されてもよい;
の配列を含んでもよい。
【0096】
式I、II及びIIIでは、X2はAibなどの保護残基であってよく、又はアラニン、(例えば、GLP1に見られるような)、又はグリシン(例えば、エキセンジン−4に見られるような)、又は別の残基であってもよい。
【0097】
式I、II及びIIIでは、R1は、不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3又はC(O)CH(CH3)CH3であってよく;そしてR2は、不存在、OH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であってよい。
【0098】
式I、II及びIIIでは;R1は不存在であってよい。式I、II及びIIIでは;R2はNH2であってよい。
【0099】
式I、II及びIIIでは、結合残基はリジン(K)であってよい。結合残基はCであってよい。式I、II及びIIIでは、結合残基はK(SH):
【化6】
であってよい。
【0100】
結合残基がK、K(SH)又はCの場合、結合残基は下式:
【化7】
のリンカーに更に共有結合してもよい。
【0101】
このリンカーは、下式:
【化8】
のようにβラクタム基を通して抗体に共有結合してもよい。
【0102】
幾つかの実施態様では、リンカーのβラクタム基は、下式:
【化9】
のように、抗体の2つの結合部位の少なくとも1つに共有結合してもよい。
【0103】
幾つかの実施態様では、結合残基及びリンカーは下式:
【化10】
の構造のものであってよい。
【0104】
幾つかの実施態様では、リンカー及び結合残基は、下式:
【化11】
のように、式I、II及びIIIによって例示されたもののようなGLP−1Rアゴニストペプチドに共有結合してもよい。
【0105】
幾つかの実施態様では、ペプチド−リンカー複合体は下式:
【化12】
のように、抗体に共有結合してもよい。
【0106】
幾つかの実施態様では、ペプチド−リンカー複合体は下式:
【化13】
のように、抗体の結合部位の少なくとも1つに結合する。
【0107】
特定の実施態様では、GLP−1Rアゴニストペプチドは、配列:
H1−(Aib)2−E3−G4−T5−F6−T7−S8−D9−L10−S11−K12−Q13−M14−E15−E16−E17−A18−V19−R20−L21−F22−I23−E24−W25−L26−K27−N28−G29−G30−P31−S32−S33−G34−A35−P36−P37−P38−S39−X40(配列番号6)]
式中、
M14又はX40の1つは結合残基で置換される;
を含んでもよく、幾つかの実施態様では、GLP−1Rアゴニストペプチド−リンカーは、下式:
【化14】
のようにK40(結合残基)を経由してリンカーAに結合したペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−M−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S−K(配列番号7)]−NH2
を含む。
【0108】
化合物GAC−1では、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−M−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S−K(配列番号7)]−NH2
は、下式:
【化15】
のようにK40(結合残基)を経由してリンカーAに結合し、抗体の結合部位に結合している。
【0109】
従って、GAC−1は下式:
【化16】
の構造を有する。
【0110】
別の実施態様では、GLP−1Rアゴニストペプチド−リンカーは、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−K−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S(配列番号8)]−NH2
を含み、このペプチドは、下式:
【化17】
のようにK14(結合残基)を経由してリンカーAに結合している。
【0111】
化合物GAC−2では、ペプチド:
NH2−[H−(Aib)−E−G−T−F−T−S−D−L−S−K−Q−K−E−E−E−A−V−R−L−F−I−E−W−L−K−N−G−G−P−S−S−G−A−P−P−P−S (配列番号8)]−NH2
は、下式:
【化18】
のようにK14(結合残基)を経由してリンカーAに結合し、抗体の結合部位に結合している。
【0112】
従って、GAC−2は以下の構造:
【化19】
を有する。
【0113】
典型的なGACは、下記の表1に与えられるいずれかのペプチド配列を有するGACを含むが、これらに限定されない。幾つかの実施態様では、GACGLP−1Rアゴニスト化合物は、表1からのいずれかのGACである。表1に示されるデータは、米国特許出願公報第20090098130号に記載の方法を用いて得られた。表1に与えられるEC50値は、例えば、米国特許出願公報第20090098130号の実施例30に記載の、グルコース刺激性インスリン分泌(GSIS)分析を用いて測定された。表1において、EC50欄の(P)は、ペプチドを抗体に連結する場合のEC50を示す。
【0114】
【表2】
【0115】
【表3】
【0116】
【表4】
【0117】
上記のように、本発明で使用するための化合物は、抗体に共有結合してもよい。米国特許出願公開第20090098130号(全文が参照することにより本明細書に組み入れられている)は、とりわけ、抗体、有用なフラグメント及び変異体及びその修飾、結合部位及びCDR、抗体調製、発現、ヒト化、アミノ酸修飾、糖鎖付加、ADCC、CDC、抗体の血清半減期増加、発現ベクター、哺乳動物宿主系及び折畳み、そして抗体技術の他の要素を記載している。
【0118】
幾つかの実施態様では、本発明の化合物と併せて使用することができる幾つかの抗体は、抗体結合部位に反応性側鎖を有してもよい。反応性側鎖は天然に存在してもよく、又は突然変異により抗体中に入れられてもよい。抗体結合部位の反応性残基は、残基が抗体を作製するために最初に同定されたリンパ系細胞に存在する核酸によりコード化される場合など、抗体と関連してもよい。代わりに、アミノ酸残基は、特定の残基をコード化するために、DNAを意図的に突然変異させることにより生じてもよい(例えば、Meares et al.による国際公開公報第01/22922号を参照)。反応性残基は、例えば、本明細書に記載の固有のコドン、tRNA、及びアミノアシル−tRNAを用いた生合成取り込みによる非天然残基であってよい。別のアプローチでは、アミノ酸残基又はその反応性官能基(例えば、求核性アミノ基又はスルフヒドリル基)が、抗体結合部位中のアミノ酸残基に結合してもよい。従って、本明細書で使用される「抗体の結合部位中のアミノ酸残基を通して」生じる抗体との共有結合は、結合が抗体結合部位のアミノ酸残基へ直接であっても、又は抗体結合部位におけるアミノ酸残基の側鎖へ結合する化学部分を通してであってもよい。幾つかの実施態様では、アミノ酸はシステインであり、そして側鎖の反応基はスルフヒドリル基である。他の実施態様では、アミノ酸残基はリジンであり、そして側鎖の反応基はε−アミノ基である。
【0119】
触媒抗体は、1つ又はそれ以上の反応性アミノ酸側鎖を含む、好適な結合部位を有する抗体の1つの供給源である。当該抗体は、アルドラーゼ抗体、βラクタマーゼ抗体、エステラーゼ抗体、アミダーゼ抗体などを含む。
【0120】
1つの実施態様は、マウスモノクローナル抗体mAb33F12及びmAb38C2、並びに当該抗体の好適なキメラ及びヒト化バージョン(例えば、h38C2、配列番号11及び12)などのアルドラーゼ抗体を含む。
【0121】
h38C2軽鎖配列(219アミノ酸):
【化20】
【0122】
h38C2h鎖配列(448アミノ酸):
【化21】
【0123】
マウスmAb38C2(及びh38C2)は、HCDR3に近接してしかし外側に反応性リジンを有し、そして反応性免疫及び機構的模倣天然アルドラーゼ酵素によって生成した、新しいクラスの触媒抗体のプロトタイプである。C.F. Barbas 3rd et al., Science 278:2085-2092, 1997を参照。使用してもよい他のアルドラーゼ触媒抗体は、ATCC受入番号PTA−1015を有するハイブリドーマ85A2;ATCC受入番号PTA−1014を有するハイブリドーマ85C7;ATCC受入番号PTA−1017を有するハイブリドーマ92F9;ATCC受入番号PTA−823を有するハイブリドーマ93F3;ATCC受入番号PTA−824を有するハイブリドーマ84G3;ATCC受入番号PTA−1018を有するハイブリドーマ84G11;ATCC受入番号PTA−1019を有するハイブリドーマ84H9;ATCC受入番号PTA−825を有するハイブリドーマ85H6;ATCC 受入番号PTA−1016を有するハイブリドーマ90G8を含む。反応性リジンを通して、これらの抗体は、天然アルドラーゼのエナミンメカニズムを用いて、アルドール及びレトロアルドール反応を触媒する。例えば、J. Wagner et al., Science 270:1797-1800, 1995; C.F. Barbas 3rd et al., Science 278:2085-2092, 1997; G. Zhong et al., Angew. Chem. Int. Ed. Engl. 38:3738-3741, 1999; A. Karlstrom et al., Proc. Natl. Acad. Sci. U.S.A., 97:3878-3883, 2000を参照されたい。アルドラーゼ抗体、及びアルドラーゼ抗体を生成する方法は、米国特許出願公開第6210938号、第6368839号、第6326176号、第6589766号、第5985626号、及び第573375号に記載され、これらは、参照することにより本明細書に組み入れられている。
【0124】
本発明の化合物は、チオエステラーゼ及びエステラーゼ触媒抗体の結合部位に見られるもののように、反応性システインに、本発明の化合物を結合させることによって形成してもよい。好適なチオエステラーゼ触媒抗体は、K.D. Janda et al., Proc. Natl. Acad. Sci. U.S.A. 91:2532-2536 (1994)によって記載されている。 好適なエステラーゼ触媒抗体は、P. Wirsching et al., Science 270:1775-1782 (1995)によって記載されている。反応性アミノ酸含有抗体は、反応性アミノ酸をコードするために抗体結合部位を突然変異させること、又は反応基を含むリンカーで抗体結合部位のアミノ酸側鎖を誘導化することを含み、当技術分野で周知の手段により調製してもよい。
【0125】
幾つかの実施態様では、抗体はヒト化抗体であってよい。好ましくは、ヒト化抗体は、βラクタム基又は結合部位に共有結合を形成することができる他の化学基に対して、高結合親和性を保持する。種々の形態のヒト化マウスアルドラーゼ抗体が意図される。幾つかの実施態様では、抗体は、ヒト定常ドメインCκ及びCγ11を有するh38c2IgG1及びh38c2Fabなどの、ヒト化アルドラーゼ触媒抗体である。C. Rader et al.,
2003, J. Mol. Bio. 332:889-899は、h38c2Fab及びh38c2IgG1を産生するために使用することができる遺伝子配列及びベクターを開示している。ヒト生殖細胞系Vk遺伝子DPK−9及びヒトJk遺伝子JK4を、m38c2のκ軽鎖可変ドメインのヒト化に対するフレームワークとして使用し、そしてヒト生殖細胞系遺伝子DP−47及びJH遺伝子JH4をm38c2の重鎖可変ドメインのヒト化に対するフレームワークとして使用した。G1m(f)アロタイプを有する抗体h38c2IgG1を含む本発明の化合物の特定の実施態様では、リンカーのβラクタム基は、重鎖の99の位置でリジン残基の側鎖に結合してもよい。他の実施態様は、h38c2の可変ドメイン(VL及びVH)及びIgG1、IgG2、IgG3、又はIgG4からの定常ドメインを含むキメラ抗体を使用してもよい。抗体は、h38c2からのVH及びVLドメインを含む完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、VL、二重特異性抗体、又はミニ抗体であってよい。抗体は、h38c2からのVH及びVLドメイン及びIgG1、IgG2、IgG3、又はIgG4から成る群から選択される定常ドメインを含む抗体であってもよい。幾つかの実施態様では、抗体はh38C2IgG1であってもよい。幾つかの実施態様では、抗体は、ヒトIgG、IgA、IgM、IgD、又はIgE抗体からの定常領域を含むマウスアルドラーゼ抗体のヒト化バージョンであってもよい。他の実施態様では、抗体は、マウスアルドラーゼ抗体からの可変領域、及びヒトIgG、IgA、IgM、IgD、又はIgE抗体からの定常領域を含むキメラ抗体であってよい。更なる実施態様では、抗体は、天然又はナイーブヒトIgG、IgA、IgM、IgD、又はIgE抗体からのポリペプチド配列を含む、マウスアルドラーゼ抗体の完全ヒト化バージョンであってよい。
【0126】
ヒト化アルドラーゼ抗体フラグメントの種々の形態も意図される。1つの実施態様では、h38c2F(ab’)2を使用する。h38c2F(ab’)2はh38c2IgG1のタンパク分解によって産生してもよい。別の実施態様は、介在リンカー(Gly4Ser)3により選択的に結合されるh38c2からのVL及びVHドメインを含む、h38c2scFvを使用する。ヒト化に代わる方法として、ヒト抗体を生成することができる。
【0127】
他のGLP−1Rアゴニスト
GLP−1Rアゴニストは、例えば、抗GLP−1Rアゴニスト抗体の、エキセナチド(BYETTA(登録商標))、アルビグルチド、R1583、リラグルチド、AVE−0010、S4P及びBoc5(Chen et al. (2007), Proc. Natl. Acad. Sci. USA.104:943-948を参照)を含むが、これらに限定されない。
【0128】
GLP−1Rアゴニストは、例えば、シタグリプチン、ビルダグリプチン、サキサグリプチン、リナグリプチン、ドゥトグリプチン(dutogliptin)、ゲミグリプチン(gemigliptin)、アログリプチン及びベルベリンなどのDPP−4阻害剤をも含むが、これらに限定されない。
【0129】
幾つかの実施態様では、GLP−1Rアゴニストは、モノクローナル抗GLP−1Rアゴニスト抗体である。モノクローナル抗GLP−1R抗体は当業者により容易に生産することができる。ハイブリドーマによりモノクローナル抗体を作製する一般的手法は、現在当技術分野で周知である。例えば、M. Schreier et al., 「ハイブリドーマ技術」(Hybridoma Techniques) (Cold Spring Harbor Laboratory 1980); Hammerling et al.,「モノクローナル抗体及びT細胞ハイブリドーマ」(Monoclonal Antibodies and T-Cell Hybridomas) (Elsevier Biomedical Press 1981); Kennett et al.,「モノクローナル抗体」(Monoclonal Antibodies) (Plenum Press 1980)を参照されたい。不死化抗体分泌細胞系は、発癌DNA又はEBVによるBリンパ球の直接形質転換など、融合以外の技術によっても産生することができる。必要に応じて、後に不死化細胞系に転換する正常Bリンパ球集団にチャレンジするために、幾つかの抗原供給源を使用することができる。
【0130】
ポリヌクレオチド
幾つかの実施態様では、本発明は、本明細書に記載のいずれかのポリヌクレオチドをコード化するポリヌクレオチドを提供する。ポリヌクレオチドは、当技術分野で公知の手順によって作製し発現することができる。
【0131】
幾つかの実施態様では、本発明は、本発明のいずれかのポリヌクレオチドを含む組成物(医薬組成物など)を提供する。幾つかの実施態様では、組成物は、本明細書に記載のポリペプチドをコード化するポリヌクレオチドを含む発現ベクターを含む。他の実施態様では、組成物は、本明細書に記載のいずれかのポリペプチドをコード化するポリヌクレオチドを含む発現ベクターを含む。発現ベクター及びポリヌクレオチド組成物の投与は、本明細書に更に記載されている。
【0132】
幾つかの実施態様では、本発明は、本明細書に記載のいずれかのポリヌクレオチドを作製する方法を提供する。
【0133】
いずれの当該配列に補完的なポリヌクレオチドも、本発明に包括される。ポリヌクレオチドは、一本鎖(コーディング又はアンチセンス)又は二本鎖であってよく、そしてDNA(ゲノム、又は合成)又はRNA分子であってよい。RNA分子は、イントロンを含み1対1様式でDNA分子に対応するHnRNA分子、及びイントロンを含まないmRNA分子を含む。更なるコード又は非コード配列は、本発明のポリヌクレオチド中に存在してもよいが、しなくてもよく、そしてポリヌクレオチドは、他の分子及び/又は支持物質に結合してもよいが、しなくてもよい。
【0134】
ポリヌクレオチドは、天然配列(即ち、抗体又はその部分をコード化する内在性配列)を含み、又は当該配列の変異体を含んでもよい。ポリヌクレオチド変異体は、コード化ポリペプチドの免疫反応性が天然の免疫反応性分子に比べて低下しないように、1つ又はそれ以上の置換、付加、欠失及び又は挿入を含む。コード化ポリペプチドの免疫反応性に及ぼす影響は、一般的に本明細書に記載のように評価することができる。変異体は、好ましくは、天然抗体又はその部分をコード化するポリヌクレオチド配列と、少なくとも約70%同一性、より好ましくは少なくとも約80%同一性、尚より好ましくは少なくとも約90%同一性、そして最も好ましくは少なくとも約95%同一性を示す。
【0135】
2つのポリヌクレオチド又はポリペプチド配列は、下記に示す最大対応に整列した場合に、2つの配列中のヌクレオチド又はアミノ酸の配列が同じならば「同一」であるといわれる。2つの配列間の比較は、一般的に配列類似性の局所領域を同定し比較する比較窓を通して配列を比較することによって行なわれる。本明細書で使用される「比較窓」は、少なくとも約20の隣接位置、通常は30から約75まで、又は40から約50までのセグメントを指し、その場合、配列は、2つの配列を最適に整列した後で、同数の隣接位置の参照配列と比較してもよい。
【0136】
比較のための配列の最適アラインメントは、初期設定パラメータを用いたバイオインファマティクス・ソフトウェア(DNASTAR, Inc., Madison, WI)のLasergene一式中のMegalignプログラムを用いて実施することができる。このプログラムは、以下の参考文献に記載された幾つかのアラインメント・スキームを具体化する: Dayhoff, M.O., 1978,「タンパク質進化の変化モデル−距離関係を検出するマトリックス」(A model of evolutionary change in proteins - Matrices for detecting distant relationships). In Dayhoff, M.O. (ed.) 「タンパク質の配列と構造の地図」(Atlas of Protein Sequence and Structure), National Biomedical Research Foundation, Washington DC Vol. 5, Suppl. 3, pp. 345-358; Hein J., 1990,「アラインメント及び系統発生論の統一手法」(Unified Approach to Alignment and Phylogenes) pp. 626-645「酵素学の方法」(Methods in Enzymology) vol. 183, Academic Press, Inc., San Diego, CA; Higgins, D.G. and Sharp, P.M., 1989, CABIOS 5:151-153; Myers, E.W. and Muller W., 1988, CABIOS 4:11-17; Robinson, E.D., 1971, Comb. Theor. 11:105; Santou, N., Nes, M., 1987, Mol. Biol. Evol. 4:406-425; Sneath, P.H.A. and Sokal, R.R., 1973,「数量分類学、数量分類学の原理と実際」(Numerical Taxonomy the Principles and Practice of Numerical Taxonomy), Freeman Press, San Francisco, CA; Wilbur, W.J. and Lipman, D.J., 1983, Proc. Natl. Acad. Sci. USA 80:726-730。
【0137】
好ましくは、「配列同一性の割合」は、少なくとも20位置の比較窓を通して2つの最適整列配列を比較することによって決定されるが、ここで、比較窓におけるポリヌクレオチド又はポリペプチド配列の部分は、2つの配列の最適アラインメントに対する参照配列(付加又は欠失を含まない)と比べて、20%以下、通常は5から15%、又は10から12%の付加又は欠失(即ち、ギャップ)を含んでもよい。この割合は、同一の核酸塩基又はアミノ酸残基が整合位置の数を与えるために両配列に生じる位置数を決定すること、参照配列(即ち窓サイズ)における位置の総数によって整合位置数を割ること、そして配列同一性の割合を与えるために結果に100を掛けることによって算出される。
【0138】
又はその代わりに、変異体は、実質的に天然遺伝子又はその部分若しくは相補体に実質的に相同性であってもよい。当該ポリヌクレオチド変異体は、天然ポリペプチド(又は相補配列)をコード化するDNA配列へ中程度のストリンジェントな条件下において、ハイブリダイズすることができる。
【0139】
好適な「中程度ストリンジェントな条件」は、5×SSC、0.5%SDS、1.0mMEDTA(pH8.0)の溶液中で予洗すること;50℃〜65℃、5×SSCで、一夜ハイブリダイズすること;続いて0.1%SDSを含む2×、0.5×及び0.2×SSCのそれぞれで65℃にて20分間2回洗浄することを含む。
【0140】
本明細書で使用される「高度ストリンジェントな条件」又は「高ストリンジェンシー条件」は、(1)洗浄に低イオン強度及び高温、例えば、0.015M塩化ナトリウム/0.0015Mクエン酸ナトリウム/0.1%ドデシル硫酸ナトリウムを50℃で用いる;(2)ハイブリダイゼーション時にホルムアミドなどの変性剤、例えば、50%(v/v)ホルムアミド+0.1%ウシ血清アルブミン/0.1%フィコール/0.1%ポリビニルピロリドン/50mMリン酸緩衝液を、750mM食塩、75mMクエン酸ナトリウムと共にpH6.5で42℃にて用いる;又は(3)50%ホルムアミド、5×SSC(0.75MNaCl、0.075Mクエン酸ナトリウム)、50mMリン酸ナトリウム(pH6.8)、0.1%ピロリン酸ナトリウム、5×デンハルト溶液、超音波分解サケ精子DNA(50μg/ml)、0.1%SDS、及び10%デキストラン硫酸を42℃にて、0.2×SSC(塩化ナトリウム/クエン酸ナトリウム)中42℃にて、そして50%ホルムアミドを55℃にて洗浄液で、続いてEDTAを含む0.1×SSCから成る高ストリンジェンシー洗浄液を55℃にて用いるような条件である。熟練技術者、温度、イオン強度などを調整し、必要に応じてプローブ長さなどの因子を適合させる方法を認識するであろう。
【0141】
当業者には当然のことながら、遺伝コードの縮重の結果として、本明細書に記載のポリペプチドをコード化する多くのヌクレオチド配列がある。これらのポリヌクレオチドの一部は、いずれもの天然遺伝子のヌクレオチド配列に対しても最小の相同性を保有する。それにもかかわらず、コドン使用頻度の差に因って変動するポリヌクレオチドが、本発明によって特に意図される。更に、本明細書で提供されるポリヌクレオチド配列を含む遺伝子の対立遺伝子は、本発明の範囲内である。対立遺伝子は、ヌクレオチドの欠失、付加及び/又は置換などの1つ又はそれ以上の突然変異の結果として変化する、内在性遺伝子である。得られるmRNA及びタンパク質は構造又は機能の変化を有していてもよいが、しかし有していなくてもよい。対立遺伝子は標準技術(ハイブリダイゼーション、増幅及び/又はデータベース配列比較)を用いて同定することができる。
【0142】
本発明のポリヌクレオチドは、化学合成、組み換え法 又はPCRを用いて得ることができる。化学的ポリヌクレオチド合成法は、当技術分野において周知されており、本明細書において詳細に説明する必要はない。当業者は、本明細書で提供される配列及び所望のDNA配列を生産するために、市販のDNA合成装置を使用することができる。
【0143】
組み換え法を用いてポリヌクレオチドを調製するために、所望の配列を含むポリヌクレオチドを好適なベクターに挿入し、そして更に本明細書に記載のようにベクターを複製及び増幅するために、好適な宿主細胞中へ順々に導入することができる。ポリヌクレオチドは、当技術分野で公知のいずれの手段によっても宿主細胞中に挿入し得る。細胞は、直接取り込み、エンドサイトーシス、トランスフェクション、F−交配又はエレクトロポレーションにより外因性ポリヌクレオチドを導入することによって形質転換される。いったん導入されると、外因性ポリヌクレオチドは、細胞内で非組込み型ベクター(プラスミドなど)として保持され、又は宿主細胞ゲノムに組込むことができる。このように増幅したポリヌクレオチドは、当技術分野で周知の方法により宿主細胞から単離することができる。例えば、上記Sambrook et al., 1989を参照されたい。
【0144】
代わりに、PCRはDNAの再生産を可能にする。PCR技術は当技術分野で周知であり、米国特許第4683195号、第4800159号、第4754065号及び第4683202号、並びに「PCR:ポリメラーゼ連鎖反応」(PCR:The Polymerase Chain Reaction), Mullis et al. eds., Birkauswer Press, Boston (1994)に記載されている。
【0145】
RNAは、適切なベクターに分離DNAを用いること、それを好適な宿主細胞へ挿入することにより得ることができる。細胞が複製され、DNAがRNAへ転写される場合、RNAは、例えば、上記のSambrook et al., 1989に記載のように、当業者に周知の方法を用いて分離することができる。
【0146】
好適なクローニングベクターは、標準技術に従って構成してもよく、又は当技術分野で利用可能な多数のクローニングベクターから選択してもよい。選択したクローニングベクターは、使用する目的の宿主細胞によって変化する可能性があると同時に、有用なクローニングベクターは、一般的に自己複製能を有し、特定の制限エンドヌクレアーゼに対して単一標的を有する可能性があり、及び/又はベクターを含むクローンを選択するのに使用することが可能なマーカー遺伝子を保有することができる。好適な例は、プラスミド及び細菌ウイルス、例えば、pUC18、pUC19、Bluescript(例えば、pBSSK+)及びその誘導体、mp18、mp19、pBR322、pMB9、ColE1、pCR1、RP4、ファージDNA、並びにpSA3及びpAT28などのシャトルベクターを含む。これら及び他の多くのクローニングベクターは、BioRad, Strategene、及び Invitrogenなどの販売業者から入手可能である。
【0147】
発現ベクターは、一般的に本発明のポリヌクレオチドを含む複製可能なポリヌクレオチド構成物である。発現ベクターは、宿主細胞内で染色体DNAのエピソームとしてか又は内在性部分として複製可能でなければならないことが示唆される。好適な発現ベクターとしては、例えば、PCT国際公開第87/04462号に開示のように、プラスミド、アデノウイルス、アデノ随伴ウイルス、レトロウイルスなどのウイルスベクター、コスミド、及び1つ又は複数の発現ベクターを含むが、それらに限定されない。ベクター成分は、一般的に1つ又はそれ以上の以下のもの:シグナル配列;複製の起源;1つ又はそれ以上のマーカー遺伝子;好適な転写調節因子(プロモーター、エンハンサー、及びターミネーターなど)を含むが、それらに限定されない。発現(即ち、翻訳)には、リボソーム結合部位、翻訳開始部位、及び終止コドンなどの1つ又はそれ以上の翻訳調節因子が有用である。
【0148】
関係するポリヌクレオチドを含むベクターは、エレクトロポレーション、塩化カルシウム、塩化ルビジウム、リン酸カルシウム、DEAE−デキストラン、又は他の物質を用いるトランスフェクション;マイクロプロジェクタイルボンバードメント法(パーティクルガン法);リポフェクション;及び感染(例えば、ベクターはワクシニアウイルスなどの感染体である)を含む多くの適切な手段のいずれかにより、細胞宿主に導入することができる。ベクター又はポリヌクレオチドを導入する選択は、しばしば宿主細胞の特性によって決まる。
【0149】
本発明は、本明細書に記載のいずれかのポリヌクレオチドを含む宿主細胞をも提供する。ヘテロ接合性DNAを過剰発現することができるいずれの宿主細胞も、関係するポリペプチド又はタンパク質をコード化する遺伝子を分離する目的で使用することができる。哺乳動物宿主細胞の例としては、COS、HeLa、及びCHO細胞が含まれるが、これらに限定されない。PCT国際公開第87/04462号をも参照されたい。好適な非哺乳動物宿主細胞としては、原核細胞(大腸菌又は枯草菌など)及び酵母(S.セレビシアエ、S.ポンベ;又はK.ラクチスなど)が含まれる。好ましくは、宿主細胞は、宿主細胞中に存在する場合、関係する対応の内在性抗体又はタンパク質よりも約5倍高い、より好ましくは10倍高い、尚より好ましくは20倍高いレベルでcDNAを発現する。GLP−1R又はGLP−1Rドメインに特異的に結合する宿主細胞のスクリーニングは、免疫測定又はFACSによって達成される。関係するタンパク質を過剰発現する細胞は同定することができる。
【0150】
GLP−1Rアゴニスト組成物
GLP−1Rアゴニストは、多発性硬化症など中枢神経系を冒す自己免疫疾患の処置のための薬剤製造に使用することができる。このように、GLP−1Rアゴニストを含む組成物は、本明細書に記載のように哺乳動物(ヒト患者を含む)における疾患を処置するのに使用することができる。
【0151】
本発明の方法で使用される組成物は、有効量のGLP−1Rアゴニストを含む。当然のことながら、組成物は1つより多いGLP−1Rアゴニストを含むことができる。
【0152】
GLP−1Rアゴニスト組成物は、更に好適な薬学的に許容される医薬品添加剤を含んでもよい。好適な担体、賦形剤及び医薬品添加剤は当技術分野で周知されており、糖質、ろう、水溶性及び/又は膨潤性重合体、親水性又は疎水性物質、ゼラチン、脂肪油、溶媒、水などの物質を含む。使用される特定の担体、賦形剤及び医薬品添加剤は、本発明の化合物が適用される手段及び目的によって決まるものである。一般的に、安全な溶媒は、水などの非毒性水性溶媒及び水に溶解又は混合可能な他の非毒性溶媒である。好適な水性溶媒としては、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば、PEG400、PEG300)など、及びその混合物である。製剤も、薬剤(即ち、本発明の化合物又はその医薬組成物)の洗練された提示、又は医薬品(即ち、薬剤)の製造における補助をもたらすために、1つ又はそれ以上の緩衝剤、安定化剤、界面活性剤、湿潤剤、滑沢剤、乳化剤、懸濁化剤、保存剤、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色剤、甘味剤、芳香剤、着香剤及び他の既知添加剤を含む。幾つかの製剤は、リポソームなどの単体を含む。リポソーム製剤はサイトフェクチン、多重層ベシクル及び単層ベシクルを含むが、それらに限定されない。非経口及び経口ドラッグデリバリーのための医薬品添加剤及び製剤は、Remington, 「薬剤学の科学と実践」(The Science and Practice of Pharmacy) (2000)に記載されている。
【0153】
一般的に、GLP−1Rアゴニスト組成物は、注射(例えば、腹腔内、静脈内、皮下、筋肉内など)による投与のために製剤化されるが、他の投与形態を使用することができる。GLP−1Rアゴニストは、植物性又は他の類似の油脂、合成脂肪酸グリセリド、高級脂肪酸エステル又はプロピレングリコールなどの水性又は非水溶媒にそれらを溶解、懸濁化及び乳化することにより;そして必要に応じて、溶解剤、等張剤、懸濁化剤、乳化剤、安定化剤及び保存剤などの既存の添加剤を用いて、注射製剤に製剤化することができる。
【0154】
投与及び投薬
本明細書に例示した投与計画及び治療投薬量は、動物体重、血中のGLP−1Rアゴニストの半減期、及びGLP−1Rアゴニストの親和性などの当該因子によって決められた。好ましい投与計画及び投薬量は、投与間で体内のGLP−1Rアゴニストの治療量を維持する。エキセンジン−4及びGAC−1の有効投薬量範囲は、本明細書に、Davies et al., 1993に、及び他の参考文献中に例示されている。
【0155】
本発明の裏付けとして行なわれた実験で示されるように、疾患経過中の初期でのエキセンジン−4による「パルス処置」は、動物の病的状態を減少させるのに十分であると考えられる。GLP−1Rアゴニストの継続投与は、処置の有効性に必要ではないと考えられる。GACによる処置などの他の実施態様では、週1回未満の頻度の処置は、処置の有効性に十分と考えられる。
【0156】
特定の薬剤投与計画、即ち、用量、投与時期及び反復は、処置されるヒト及び動物の年齢、病態、及び体重によって決まるといえる。初期薬剤投与計画は、動物実験から外挿してもよい。GLP−1Rアゴニスト投与の時期/頻度は、特定のGLP−1Rアゴニストの循環半減期(又は神経組織中の半減期)、血液脳関門を通過するアゴニストの量、細胞中のアゴニストの半減期、毒性、及びいわゆる副作用を基準にすべきである。
【0157】
本研究では、GLP−1Rアゴニストの投与は、腹腔内 (i.p.) 注射によって行なわれる;しかしながら他の投与経路も有効と予想される(例えば、静脈内、皮下、筋肉内)。頭蓋内又は脊髄内/髄腔内投与(又はCNSの他の組織への投与)は有効と考えられる。他の投与経路は、特定のGLP−1Rアゴニスト、そのコーティング、コンジュゲーション、又は特定の生物学的特性(例えば、経口、舌下、滑液嚢内、粘膜、経皮、関節内、腟内、肛門、尿道内、経鼻、耳内、経吸入、吸入法、経カテーテル、ボーラス投与、ステント又は他の埋め込み型装置、塞栓性組成物、点滴、貼付又は可溶性フィルムなど)に応じて好適であり得る。
【0158】
GLP−1Rアゴニストは、好適な末梢経路を経由して好ましく投与される。それにもかかわらず、当然のことながら、わずかな割合のアゴニストが血液脳関門を通過し、中枢神経系の細胞にデリバリーされると考えられる。幾つかの例では、CNSへの経路をたどる末梢投与GLP−1Rアゴニストの量は小さい(せいぜい1%未満)。
【0159】
本発明の特徴は、自己免疫疾患を患う哺乳動物対象に対するGLP−1Rアゴニストの直接投与である。「直接」は、GLP−1Rアゴニストポリペプチド、又は当該ポリペプチドの発現を方向付けすることができるポリヌクレオチドが、イノキュレーションの標準経路により動物にデリバリーされることを意味する。GLP−1Rアゴニストは、グルココルチコイド、細胞静止薬(例えば、アルキル化剤、代謝拮抗物質、メトトレキサート、アザチオプリン及びメルカプトプリン)などの免疫抑制薬、細胞傷害抗体(例えば、T細胞受容体及びIL−2受容体特異的抗体)、B細胞欠失療法(例えば、抗CD20抗体/RITUXAN(登録商標)、抗BLyS抗体)、T細胞移動障害薬(例えば、抗インテグリンα4/β1抗体/TYSABRI(登録商標)、イムノフィリンに作用する薬剤(例えば、シクロスポリン、タクロリムス、シロリムス、ラパマイシン)、インターフェロン(例えば、IFN−β)、グラチラマー/COPAXONE(登録商標)、オピオイド、TNF結合タンパク質、ミコフェノラート、及び外来抗体又は治療用抗原に対する動物の免疫反応を抑制するために使用される他の生物剤を含み、他の薬剤との組み合わせで動物にデリバリーしてもよい。
【0160】
自己免疫疾患を患うヒトを含む哺乳動物対象の処置において、GLP−1Rアゴニスト又は薬学的に許容される誘導体の治療的有効量が投与される。例えば、GLP−1Rアゴニストは、約0.1mg/kg体重から約15mg/kg体重までの毎日の静脈内点滴で投与することができる。従って、1つの実施態様は、約0.5mg/kg体重の用量を与える。別の実施態様は、約0.75mg/kg体重の用量を与える。別の実施態様は、約1.0mg/kg体重の用量を与える。別の実施態様は、約2.5mg/kg体重の用量を与える。別の実施態様は、約5mg/kg体重の用量を与える。別の実施態様は、約10.0mg/kg体重の用量を与える。別の実施態様は、約15.0mg/kg体重の用量を与える。GLP−1Rアゴニスト又はその薬学的に許容される誘導体の用量を、1日当り約1回から週当たり2回、又は代わりに毎週約1回から月当たり1回の間隔で投与する必要がある。1つの実施態様では、本発明のGLP−1Rアゴニスト又はその薬学的に許容される誘導体のピーク血漿中濃度を達成するために、1用量は約0.002mg/mlから30mg/mlまで投与される。これは、適切な製剤(化学の当業者に公知のいずれの好適な製剤も使用することができる)において投与成分溶液の無菌注射剤によって達成し得る。望ましい血中レベルは、妥当な分析方法により測定した血漿中レベルを確認しながら、本発明のGLP−1Rアゴニストの持続点滴によって維持してもよい。
【0161】
個体に対してGACを投与する1つの方法は、GLP−1Rアゴニストペプチド−リンカーコンジュゲートを個体に投与し、それがインビボで適切な抗体の結合部位と共有化合物を形成することを可能にすることを含む。インビボで形成するGACの抗体部分は、GLP−1Rアゴニストペプチド−リンカーコンジュゲートの投与の前、同時、又は後に個体に投与してもよい。GLP−1Rペプチドは、リンカー/反応性部分を含んでもよく、又は抗体結合部位は標的剤に共有結合するように好適に修飾してもよい。代わりに又は加えて、抗体は、適切な免疫原により免疫後に個体の循環中に存在し得る。例えば、触媒抗体は、担体タンパク質に結合された基質の反応性中間体で免疫することにより作成してもよい。R.A. Lerner and C.F. Barbas 3rd, 1996, Acta Chem. Scand. 50:672-678を参照されたい。特に、アルドラーゼ触媒抗体は、P. Wirsching et al., 1995, Science 270:1775-1782 (commenting on J. Wagner et al., 1995, Science 270:1797-1800)によって記載されているように、ジケトン部分に結合したキーホールリンペットヘモシアニンと投与することにより作成してもよい。
【0162】
天然GLP−1Rアゴニストより優れたGACの利点は、GACが循環タンパク質リガンドと比べて、相対的に長い循環半減期を有する傾向を示すことである。例えば、天然アゴニストが連日投与を必要とする場合、GACは週1回投与するだけでよい。
【0163】
GLP−1Rアゴニストによる処置は、多発性硬化症及び関連疾患に対し、既存の処置法と組み合わせることができる。多発性硬化症の処置及び管理のための既存薬は、以下:ABC(即ち、Avonex-Betaseron/Betaferon-Copaxone) 処置 (例えば、インターフェロン1a(AVONEX(登録商標), REBIF(登録商標))、インターフェロン1b(BETASERON(登録商標), BETAFERON(登録商標))、酢酸グラチラマー(COPAXONE(登録商標));化学療法薬(例えば、ミトキサントロン (NOVANTRONE(登録商標))、アザチオプリン(IMURAN(登録商標))、シクロホスファミド(CYTOXAN(登録商標), NEOSAR(登録商標))、サイクロセリン(SANDIMMUNE(登録商標))、メトトレキサート、及びクラドリビン(LEUSTATIN(登録商標)); コルチコステロイド及び副腎皮質刺激ホルモン(ACTH)(例えば、メチルプレドニゾロン(DEPO-MEDROL(登録商標), SOLU-MEDROL(登録商標))、プレドニゾン(DELTASONE(登録商標))、プレドニゾロン(DELTA-CORTEF(登録商標))、デキサメタゾン(MEDROL(登録商標), DECADRON(登録商標))、副腎皮質刺激ホルモン(ACTH(登録商標))、及びコルチコトロフィン(ACTHAR(登録商標)); 疼痛介在(感覚異常) (例えば、カルバマゼピン(TEGRETOL(登録商標), EPITOL(登録商標), ATRETOL(登録商標), CARBATROL(登録商標))、ガバペンチン(NEURONTIN(登録商標))、トピラマート(TOPAMAX(登録商標))、ゾニサミド(ZONEGRAN(登録商標))、フェニトイン(DILANTIN(登録商標))、デシプラミン(NORPRAMIN(登録商標))、アミトリプチリン(ELAVIL(登録商標))、イミプラミン(TOFRANIL(登録商標), IMAVATE(商標), JANIMINE(登録商標))、ドキセピン(SINEQUAN(登録商標), ADAPIN(登録商標), TRIADAPIN(登録商標), ZONALON(登録商標))、プロトリプチリン(VIVACTIL(登録商標))、大麻及び合成カンナビノイド(MARINOL(登録商標))、ペントキシフィリン(TRENTAL(登録商標))、イブプロフェン(NEUROFEN(登録商標))、アスピリン、アセトアミノフェン、及びヒドロキシジン(ATARAX(登録商標)); 及び他の処置法(例えば、ナタリズマブ(ANTEGREN(登録商標))、アレムツズマブ(CAMPATH-1H(登録商標))、4−アミノピリジン(FAMPRIDINE(登録商標))、3,4−ジアミノピリジン、エリプロジル、IV免疫グロブリン(GAMMAGARD(登録商標), GAMMAR-IV(登録商標), GAMIMUNE N(登録商標), IVEEGAM(登録商標), PANGLOBULIN(登録商標), SANDOGLOBULIN(登録商標), VENOGLOBULIN(登録商標))を含むが、それらに限定されない。
【0164】
キット
本発明は、本発明の方法を実施するための部品のキット(キット)も提供する。本発明のキットは、本明細書に記載のGLP−1Rアゴニストを含む1つ又はそれ以上の容器、及び本明細書に記載の本発明のいずれかの方法に沿った使用説明書を含む。一般的に、これらの使用説明書は、GLP−1Rアゴニストの投与方法についての解説を含む。キットは、処置が必要な動物を特定し、そして処置の有効性をモニター又は測定するための使用説明書を更に含む。使用説明書は、一般的に、投薬量、投与計画(投与の頻度)、及び投与経路に関する情報を含む。キットで提供される使用説明書は、データファイル又は表計算ソフトの形式で書かれ、又は機械/コンピュータ可読であってよい。容器は単位用量、大量包装(例えば、マルチドーズ包装)又はサブユニット用量であってよい。本発明のキットで提供される使用説明書は、ラベル又は添付文書(例えば、キットに含まれるペーパーシート)に関する通常書面での使用説明書であるが、機械可読使用説明書(例えば、磁気又は光学保存ディスク媒体の使用説明書)も認められる。
【0165】
キットは、シリンジ、針、カテーテル、吸入器、ポンプ、アルコール交換、ゲージ、CNS生検器具、組織学的抗体及び染色液などを含む、GLP−1Rアゴニストを投与するための器具をも含む。キットの構成部品は必要に応じて滅菌される。キットは、酢酸グラチラマー(GA)及びデキサメタゾンなどの免疫抑制薬を含む更なる薬剤を提供してもよいが、それらに限定されない。キットは、日付印、不正開封防止包装、及び高周波識別(RFID)タグ又は他の在庫管理機能を含んでもよい。
【0166】
本発明のキットは、好適に包装されている。好適な包装は、バイアル、ボトル、ジャー、フレキシブル包装(例えば、密封Mylar又はプラスチックバッグ)などを含むが、これらに限定されない。吸入器などの特殊器具、経鼻投与器具(例えば、噴霧器)又はミニポンプなどの輸液用器具と組み合わせて使用するための包装も意図される。キットは、滅菌点検口(例えば、容器は皮下注射針によって穿通できる栓を有する静脈注射液バッグ又はバイアルであってよい)を有してよい。容器は、滅菌点検口(例えば、容器は皮下注射針によって穿通できる栓を有する静脈注射液バッグ又はバイアルであってよい)を有してもよい。組成物中の少なくとも1つの活性剤は、GLP−1Rアゴニストである。容器は、第2の薬学低活性剤を含んでもよい。
【0167】
場合により、キットは緩衝剤などの更なる成分及び解説的情報を提供してもよい。通常、キットは、容器及び容器について又はそれに関連するラベル又は1つ又は複数の添付文書を含む。
【実施例】
【0168】
以下の実施例は説明の目的のためだけに提示され、決して本発明の範囲を限定することを意図するものではない。実際、本明細書に示されそして記載されるものに加えられる本発明の種々の改変は、前述の説明から当業者には明らかとなり、そして添付した特許請求の範囲内に属するものである。
【0169】
〔実施例1〕
GLP−1Rアゴニストの同定
GLP−1Rアゴニストは、1つ又はそれ以上の以下の方法を含み、当該技術分野において認められている方法を用いて同定することができる。例えば、細胞内cAMPの濃度増加を検出するアッセイを使用してもよい。このアッセイは、例えばcAMP応答配列の調節下にあるレポーター遺伝子の発現を測定することによる、cAMPの定性的又は定量的測定、並びに選択したGPCRの潜在的アゴニスト又はアンタゴニストの同定及びキャラクタリゼーションに好適である。レポーターを発現するレポーター遺伝子を誘導することができる化合物は、GLP−1Rアゴニストと同定される。1つのこのような分析は、候補化合物へのその反応を検出するために、GLP−1R遺伝子の発現ベクター及びcAMP応答配列の調節下にあるルシフェラーゼレポーター遺伝子の発現ベクターにより、安定的にトランスフェクトされる、ヒト胎児腎細胞を用いる (Thorens, 1992,「細胞生物学」(Cell Biology) 89: 8641-8645; Drucker et al., 1987, Proc. Natl. Acad. Sci. U.S.A. Vol.84: 3434-3438)。
【0170】
アッセイの第1段階は、GPCR、本ケースではGLP−1R、の立体配座変化を含み、ここでこの受容体はcAMP応答配列の調節下にあるレポーター遺伝子を含む真核細胞の細胞膜に存在する。受容体は、内在性受容体又は受容体をコード化する核酸であってよく、又は受容体構築物は、細胞に形質転換されてもよい。典型的には、第1の固相(例えば、第1のアッセイプレートのウェル)は、このような細胞(通常は哺乳動物細胞系)が固相に付着するように細胞の実質的に均質集団でコーティングされる。しばしば、細胞は付着性で、それにより自然に第1固相に付着する。次いで、候補アゴニストなどの被検体は、受容体(例えば、GLP−1R)が被検体に暴露(又はそれと接触)するように、付着細胞を有するウェルに加えられる。このアッセイは、関心あるGPCR(例えば、GLP−1R)のアゴニストリガンドの同定を可能にする。被検体への暴露に続いて、付着細胞はレポーター遺伝子の発現を可能にする所定時間(例えば、約6時間)の間、適した条件下にインキュベートされる。
【0171】
次いで、細胞はアッセイの検出相にかけられる状態になる。レポーター遺伝子の発現は、例えばルシフェラーゼ活性の検出により測定される。ルシフェラーゼ活性の検出は、当技術分野で公知の任意の様々な適した方法によって行なうことができる。
【0172】
最初の同定に続いて、候補(例えば、抗GLP−1Rモノクローナル抗体)のアゴニスト活性は、細胞内cAMPの濃度を測定することにより更に確認し、リファインすることができる。典型的には、第1の固相(例えば、第1のアッセイプレートのウェル)は、このような細胞(通常は、例えばHEK293細胞などの哺乳動物細胞系)が固相に付着するように細胞の実質的に均質集団でコーティングされる。候補は細胞を含むアッセイプレートに接種され、そして例えば約1時間適した条件下にインキュベートされる。細胞内のcAMP濃度は、当業者に公知であって任意の種々の適した方法で検出することができる。例えば、細胞内cAMP濃度は、cAMP−Screen Direct(登録商標) システムキット(Applied Biosystems)の仕様書に従って検出することができる。HitHunter(登録商標)cAMP XS + Assay(DiscoveRX(登録商標))、インビトロベースの競合免疫測定法などの他のcAMPアッセイキットが商業的に入手可能である。簡潔に言えば、細胞溶解物からの遊離cAMPは、β−ガラクトシダーゼ(β−gal)の小分子ペプチドフラグメント、標識ED−cAMPコンジュゲートに対して抗体結合を競合する。遊離cAMPの不存在下では、ED−cAMPコンジュゲートは抗体によって捕捉されて、相補性に利用できないことから、低シグナルの結果となる。遊離cAMPの存在下では、抗体部位は占有され、ED−cAMPコンジュゲートをフリーのままにしてEAを補完できるようにし、基質加水分解に活性なβ−galEFC酵素を形成させて化学発光シグナルを産生する。陽性シグナルは、抗体によって結合された遊離cAMPの量に正比例して発生する。
【0173】
候補(例えば、GLP−1Rモノクローナル抗体)のアゴニスト活性は、標的生物活性を試験するために知られているバイオアッセイにより、更に確認し、リファインすることができる。例えば、候補がGLP−1Rをアゴナイズする能力は、インスリン分泌を測定するアッセイで試験することができる。候補がインスリン分泌を促進させる能力は、RIN−38ラットインスリノーマ細胞系などの培養動物細胞に候補を与え、そして培地への免疫反応性インスリン(IRI)の放出をモニターすることにより測定することができる。代わりに、候補を動物に注射し、そして免疫反応性インスリン(IRI)の血漿レベルをモニターすることができる。
【0174】
IRIの存在は、例えばインスリンを特異的に検出することができる、ラジオイムノアッセイの使用を通して検出することができる。IRIの存在を検出することが可能な任意のラジオイムノアッセイを使用することができる;1つのこのようなアッセイはAlbano, J. D. M., et al., 1972, Acta Endocrinol., 70:487-509の方法の変法である。この変法では、pH7.4のリン酸/アルブミン緩衝液が用いられる。インキュベーションは、500μlのリン酸緩衝液、50μlの灌流液サンプル又は灌流液中のラットインスリン標準、100μlの抗インスリン抗血清(Wellcome Laboratories; 1:40,000希釈)、及び100μlの125Iインスリンの連続添加で調製し、10×75mm使い捨てガラス管中で750μlの総量とした。4℃で2〜3日間インキュベーション後、遊離インスリンは活性炭分離により抗体結合インスリンから分離される。アッセイ感度は1〜2uU/mLである。組織培養で増殖した細胞の細胞培養培地へのIRI放出を測定するために、好ましくは放射標識をプロインスリンに組み込む。ポリペプチドを標識することが可能な任意の放射標識を使用することができるが、標識プロインスリンを得るためには、3Hロイシンを使用することが好ましい。
【0175】
候補GLP−1Rアゴニストがインスリン分泌促進特性を有するかどうかを測定するために、膵注入によって測定することもできる。In situ単離ラット膵灌流アッセイは、Penhos, J. C., et al., 1969, 「糖尿病」(Diabetes), 18:733-738の方法の変法である。350〜600g質量の絶食雄Charles River系アルビノラットをアミタールナトリウム (Eli Lilly and Co.:160ng/kg)の腹腔内注射で麻酔した。腎臓、副腎、胃、及び下部結腸血管を結紮した。全腸を、約4cmの十二指腸及び下行結腸及び直腸を除いて切除した。従って腸の一小部分だけを灌流して、グルカゴン様免疫反応性を有する腸内物質による干渉の可能性を最小化した。灌流液は、4%デキストランT70及び0.2%ウシ血清アルブミン画分Vを備えた改変クレブスリンガー(Krebs-Ringer)重炭酸緩衝液であり、そして95%O2及び5%CO2でバブルさせた。非拍動流、4チャンネルローラーベアリングポンプ(Buchler polystatic, Buchler Instruments Division, Nuclear-Chicago Corp.)を使用し、そして1つの灌流液源から他へのスイッチは、3方コックで切替えることにより達成した。灌流を行なって、モニターし、そして解析する方法は、Weir, G. C., et al., 1974, J. Olin. Inestigat. 54:1403-1412の方法に従うが、これは参照により本明細書に組み込まれる。
【0176】
〔実施例2〕
多数のリンパ球及びマクロファージは自己免疫疾患マウスのCNSに浸潤する
本実施例は、炎症細胞でCNSの浸潤を説明する。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に再度、400ngの百日咳毒素をi.v.注射した。MOG免疫後日18に動物(EAE及びナイーブ)を犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。CD3(Chemicon)及びCD68(Serotec)に対する一次抗体で免疫組織化学を行なった。Alexa 488(Invitrogen)に結合したヤギ抗ラット二次抗体を検出に使用した。クレシルバイオレット及びルクソールファーストブルー染色を使用して、浸潤細胞体及びミエリンを染色した。
図1の染色結果は、EAEマウス(上側)のCNSが、健常マウス(下側)と比べて多数の浸潤T細胞、単球及びミクログリアを有することを示す。
【0177】
〔実施例3〕
GLP−1Rアゴニストの直接投与は自己免疫疾患における罹病率(morbidity)を減少させる
本発明の裏付けとして行なった実験では、GLP−1Rアゴニストの直接投与は、CNS特異的自己免疫疾患の動物における罹病率及びCNS組織損傷を減少させることが示された(図2)。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちにそして48時間後に、再度400ngの百日咳毒素をi.v.注射した。
マウスをMOG免疫後日0から日9まで、1mg/kgエキセンジン−4(n=10)、2mg/kgNT4(陽性対照、n=10)又はビヒクル(PBS、陰性対照、n=10)で毎日処置した。動物の罹病率を以下のようにスコア化した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。エキセンジン−4で処置した動物は、対照動物と比べて有意な罹病率低下を示した(図2)。この結果は、GLP−1Rアゴニストが慢性EAEの進行を緩徐化させるのに有効なことを示した。
【0178】
〔実施例4〕
GLP−1Rアゴニスト化合物は自己免疫疾患の罹病率を減少させるのに有効である
動物実験は、GLP−1Rアゴニスト抗体コンジュゲート化合物GAC−1が、対照免疫グロブリンと比べてEAE罹病率に対して有意な保護をもたらすことを示した(図3及び4)。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスをMOG免疫後日0から日6まで、1mg/kgエキセンジン−4(n=10)か又はビヒクル(n=10)で毎日処置した。別の群のマウスを、1mg/kgGAC−1(n=10)により週1回ベースで処置した。マウスを以下の尺度に従ってEAEの臨床徴候で毎日評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。GAC−1は、MOG免疫後週1回投与した場合、罹病率を減少させるのに有効であった(図3)。
別の実験では、EAEを前記のように誘発した。第1群の動物をMOG免疫後、日0から日2まで3mg/kgエキセンジン−4(n=10)で、そして日3から日8まで1mg/kgエキセンジン−4で処置した。第2群の動物をMOG免疫後日0に3mg/kgGAC−1(n=10)で処置し、その後免疫後日7に開始して1mg/kgGAC−1で週1回処置した。第3群の動物はPBS(対照、n=10)で毎日処置した。臨床スコアを前記の基準に従って毎日モニターした。GAC−1は、3mg/kgの初回用量で投与し、その後1mg/kgの週1回用量で投与した場合にも、尚罹病率を減少させるのにより有効と考えられた(図4)。結果は、3mg/kgGAC−1の初回用量が、1mg/kgだけの用量よりも罹病率に対して一層の保護(即ち、より低い罹病率)をもたらすことを示した。
【0179】
〔実施例5〕
GLP−1RアゴニストはCNSの炎症細胞浸潤をブロックし、自己免疫疾患の脱髄を減少させる
GLP−1Rアゴニストが動物での保護を媒介するメカニズムを更に解明するために、組織学的分析を行なった。脊髄切片を対照、エキセンジン−4処置、及びGAC−1処置EAE誘発動物から調製し、次いでミエリン又は浸潤細胞を同定する白血球マーカーに特異的な抗体を染色するために、ルクソールファーストブルーで染色した。対照動物では、染色はミエリン発現神経細胞のバックグラウンドに対して、白血球浸潤及び細胞破壊の領域を示した(図5A、6A及び6D)。白血球浸潤減少がGLP−1Rアゴニスト処置動物から調製した切片で認められた(図6B、6C、6E及び6F)。かなりのGLP−1Rアゴニスト処置動物は、CNS白血球浸潤の証拠を実質的に示さなかった。
【0180】
脊髄切片を、脱髄を測定するルクソールファーストブルーで染色した(図5A−C)。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を1mg/kgエキセンジン−4又はPBS対照で毎日、又は1mg/kgGAC−1で週1回処置した。動物をMOG免疫後日18に犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。ミエリン染色は、ルクソールファーストブルーを用いて行なった。
【0181】
図5A〜Cは、PBS処置対照EAE動物(図5A)、エキセンジン−4処置EAE動物(図5B)及びGAC−1処置EAE動物(図5C)からの脊髄の代表的な画像を示している。対照マウスの脳は、重度脱髄の領域(即ち、図5Aの黒矢によって示されるように、ルクソールファーストブルー染色の不存在)を示した。GLP−1Rアゴニスト処置動物の切片には、重度脱髄及び/又はニューロン死の領域は認められなかった(図5B及びC)。CNS組織切片の組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSの脱髄を減少させることを実証した。
【0182】
浸潤細胞の同定は、2つの白血球マーカー、T細胞に存在するCD3及びマクロファージに存在するCD68に特異的な抗体を用いて行なわれた。EAEを前記のように雌性C57BL/6マウスに誘発した。動物を1mg/kgエキセンジン−4又はPBS対照で毎日、又は1mg/kgGAC−1で週1回処置した。動物をMOG免疫後日12に犠牲にした。脊髄を採取し、4%パラホルムアルデヒドで固定し、そして20μMで凍結切片化した。免疫組織化学はCD3(Chemicon)及びCD68(Serotec)に対する一次抗体を用いて行なわれた。Alexa488(Invitrogen)に接合したヤギ抗ラット二次抗体が検出に使用された。
【0183】
図6A〜Cは、CD3抗体で染色した結果を示す。多数の明染色断続領域が脱髄領域に対応して対照動物の脊髄に現われた(図6A)。対照的に、エキセンジン−4又はGAC−1で処置したマウスから得た脊髄切片は、CD3に対していずれも事実上染色を示さなかったことから(それぞれ、図6B及び6C)、T細胞浸潤はGLP−1Rアゴニスト処置動物で実質的に減少することが示された。当該観察は、GLP−1Rアゴニスト処置動物からの脊髄組織が、対照動物と比べた場合、T細胞浸潤減少を実質的に示すことを指示した。
【0184】
図6D〜Fは、CD68抗体による染色の結果を示す。対照マウス(図6D)から調製した切片は、エキセンジン−4又はGAC−1処置マウスからの切片(それぞれ、図6E及び6F)よりも強く染色した。当該観察は、GLP−1Rアゴニスト処置動物からの脊髄組織が、対照動物と比べた場合実質的に単球浸潤減少を示すことを指示した。総合すれば、CNS組織切片の免疫組織化学染色の結果は、GLP−1Rアゴニスト処置がCNSのリンパ球及び単球浸潤を減少させることを実証した。
【0185】
〔実施例6〕
GLP−1Rアゴニストは脳及び脊髄のミクログリア及び浸潤マクロファージ中の活性化マーカーを減少させる
本実施例は、疾患発症時(MOG誘発後日15)及び疾患ピーク時(MOG誘発後日28)の両方における、脳及び脊髄のミクログリア及び浸潤マクロファージ中の活性化マーカー、MHC−II発現のGLP−1Rアゴニスト媒介による減少を例証する。
CNS中の抗原提示細胞(APC)に対するエキセンジン−4の作用を評価するために、日0にマウスにMOGを投与した。MOG誘発動物に、日0から日9まで(各処置群n=9)エキセンジン−4(1mg/kg)又はビヒクルを投与した。3匹のエキセンジン−4処置EAEマウス及び3匹のビヒクル処置EAE対照マウスを、日15(疾患発症)及び日28(疾患ピーク)に犠牲にした。ミクログリアを犠牲動物から分離した。簡潔に言えば、マウスをイソフルランで麻酔し、排出液がクリアになるまで左室を通して冷滅菌PBSで灌流した。脊髄及び脳は冷ハンクスの平衡塩類溶液(HBSS)に集めた。脊髄及び脳を、HBSSを用いてナイロン細胞ストレーナー(100μm)を通して押し出した。集めて漉した物質を、遠心分離によりペレット化した。ペレットは、脊髄又は脳当り1mlのHBSSに、コード(cord)当り300U/mlのIV型クロストリジウムコラゲナーゼを用いて再懸濁した。37℃インキュベータ中で60分間のインキュベーションを行った後、脊髄又は脳ホモジネートを30%パーコール(Percoll)に入れ、70%パーコールを下に入れた。そのグラジエントを遠心分離し、そして単球細胞を30%/70%パーコール界面から集めた。残存パーコールを除去するために、350gで5分間、cDMEM中にて2回界面から細胞を洗浄した。
【0186】
精製α−FcgR(2.4G2)でブロック後、単細胞懸濁液を多色フローサイトメトリーで染色した。MHCクラスII(10−3.6)、B220(CD45R)、CD45及びCD11bに特異的なモノクローナル抗体はBD Biosciencesから購入した。死細胞を排除するために、サンプルをフローサイトメトリー解析前に、Hoescht 33342(HO、Calbiochem)と共に氷上で5分間インキュベートした。成熟及び/又は固定アーチファクトを回避するために、細胞懸濁液を染色後直ちに解析した。死細胞(HOhi)及びB220hi細胞は解析中に排除された。ミクログリア及びCNS−マクロファージ解析では、サンプルをLSRIIで解析し、そしてflowjoソフトウェア(FlowJo, Ashland, Oregon)で解析した。
【0187】
ミクログリアサブセットを、CD45及びCD11bの発現に基づいて異なるサブセットに分けた。2つの異なる細胞集団サブセット、CD11b+CD45hi(活性化ミクログリア及び浸潤マクロファージ)及びCD11b+CD45lo(静止ミクログリア)について、ミクログリア/単球活性化マーカーMHCクラスIIの発現プロファイルを更に解析した。
【0188】
フローサイトメトリー解析の結果は、エキセンジン−4による処置が、症状発症(即ち、誘発後日15)及び疾患ピーク(即ち、誘発後日28)の両時点で、脳及び脊髄の両方において、CD11b+CD45hi及びCD11b+CD45lo細胞集団の両方でMHCクラスIIの発現レベルを減少させることを示す(図7)。図7に図示した各グラフは、示された蛍光強度で染色された全細胞の割合を示す。蛍光強度は、細胞表面でのMHCクラスII発現量を表わす(MHCクラスII発現は蛍光強度と正に相関した)。
【0189】
解析の結果は、ビヒクル処置対照EAE動物の細胞との比較により、GLP−1Rアゴニスト処置が、疾患発症及び疾患ピーク両時点において脳及び脊髄ミクログリア及び浸潤マクロファージでのAPC活性化マーカーMHCクラスIIの発現を抑制することを実証した。
【0190】
〔実施例7〕
エキセンジン−4処置マウスからの脾細胞はEAEの養子免疫伝達モデルにおいて低病原性である
本実施例は、養子免疫伝達モデルにおけるエキセンジン−4処置EAEマウスの脾細胞の病原性減少を例証する。
EAEを、CFA(Difco)に乳化した500μgのPLPp(139−151)の注射により8から12週齢のSJL/J雌マウス(The Jackson Laboratory, Bar Harbor, ME) に誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加した。免疫動物を1mg/kgエキセンジン−4(n=7)か又はPBS(n=7)で毎日処置した。処置動物を免疫後日5に犠牲にし、脾臓及び排出リンパ節を集めた。細胞を20μg/mlPLPp(139−151)と共にインビトロで培養した。48時間後、培養細胞を集め、ナイーブSJL/J動物に尾静脈注射を通して注入した。2千万の細胞を各動物に注射した。動物に、エキセンジン−4処置ドナー(n=7)か又は対照PBS処置ドナー(n=7)からの細胞を投与した。
レシピエントマウスは、臨床スコア及び体重変化を毎日モニターした。EAEの臨床徴候を以下の尺度により評価した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。図8に示すように、エキセンジン−4処置動物からのドナー細胞を投与したマウスは、PBS処置対照動物からのドナー細胞を投与したマウスよりも有意に低い疾患重症度を有した。この結果は、GLP−1Rアゴニスト処置が、EAEの養子免疫伝達モデルにおける脾細胞の病原性を減少させることを実証した。
【0191】
〔実施例8〕
エキセンジン−4はEAE誘発マウスにおいて二次リンパ器官のサイズを縮小させた
本実施例は、EAEマウスにおける二次リンパ器官のサイズに対するエキセンジン−4の効果を例証する。
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。MOG誘発動物を1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も含まれた。疾患誘発後日5に動物を安楽死させた。脾臓及び鼠径リンパ節を解剖して秤量した。リンパ節質量及び脾臓質量及びサイズは、PBSで処置したEAE動物で顕著に増加した(図9A〜9C)。しかしながら、エキセンジン−4処置EAE動物のリンパ節(図9A)及び脾臓(図9B)は、対照動物よりも有意に少ない質量であった。同様に、免疫抑制薬デキサメタゾンもEAE動物のリンパ節及び脾臓質量を減少させた。エキセンジン−4処置EAE動物の脾臓はナイーブ動物の脾臓と同程度のサイズであった(図9C)。対照的に、対照EAE動物の脾臓は有意に肥大した(図9C)。これらの結果は、GLP−1Rアゴニスト処置がEAEの二次リンパ器官のサイズを減少させるのに有効なことを実証した。
【0192】
〔実施例9〕
自己免疫疾患におけるエキセンジン−4治療後の免疫学的変化
本実施例は、エキセンジン−4処置後の免疫学的変化を例証する。
最新のMS治療薬(コルチコステロイド及びCD20抗体RITUXANTMを含む)は、免疫抑制、T細胞枯渇、B細胞枯渇及び/又は免疫反応に関する他の作用を示す免疫調節薬である。GLP−1RアゴニストがEAEにおいてT細胞及びB細胞集団に影響を与えるかどうかを判定するために、EAE動物の免疫学的変化を、エキセンジン−4処置後に評価した。比較のために、動物を陰性対照としてのPBSで処置した。
【0193】
EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を免疫後日0から日6まで、1mg/kgエキセンジン−4(n=3)か又はPBS(n=3)で毎日処置した。処置動物を、免疫しなかったナイーブマウス(n=3)と共に免疫後日6に犠牲にした。脾臓、リンパ節及び末梢血を集めた。赤血球細胞の細胞分離及び溶解後に、細胞を発蛍光団結合CD4、CD8、CD19(B細胞マーカー)及びTer119抗体(BD Biosciences)で染色した。細胞を、FACSAriaTMセルソーター(BD Biosciences)による蛍光活性化セルソーティング(FACS)を用いて解析した。
【0194】
脾臓、リンパ節及び血液中のリンパ球レベルは、PBS処置EAE動物と比べてエキセンジン−4処置EAE動物では不変と思われた(図10A〜C)。脾臓中の単球レベルは、脾臓エキセンジン−4処置EAE動物でやや減少したが(図10D)、リンパ節及び血液中では有意に変化しなかった(図10E及び10F)。エキセンジン−4処置はCD4+又はCD8+細胞集団を有意に変化させなかった(図11A〜F)。エキセンジン−4マーカーに由来するCD19+集団の変化は、正に統計的有意水準に達した(図12A〜C)。これらの所見は、CD4+、CD8+及びCD19+細胞を直接殺細胞するコルチコステロイド及びRITUXANTMなどの免疫抑制薬と、GLP−1Rアゴニストの作用メカニズムは異なることを示唆する。
【0195】
EAE動物のエキセンジン−4処置により、脾臓においてTer119+細胞集団がナイーブレベルに戻る大幅な減少が認められた(図12D)。Ter119+細胞はリンパ節には存在せず(図12E)、そして血液中で有意に変化しなかった(図12F)。この結果は、エキセンジン−処置が、EAEマウス脾臓中のTer119+細胞集団を減少させるのに非常に有効なことを実証した。
【0196】
別の実験では、EAEを前記のように雌マウスに誘発させた。動物を1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も実験に含まれた。疾患誘発後日5に動物を安楽死させた。脾臓及び鼠径リンパ節を解剖し、そして細胞を分離し、赤血球分化マーカーTer119及びCD4の表面染色を行ない、FACSで解析した。Ter119+細胞の割合を測定した(図13)。罹患動物はナイーブ動物に比べて高率のTer119+細胞を有する一方で、エキセンジン−4処置EAE動物は、PBS処置EAE動物と比べて低率のTer119+細胞を有した。免疫抑制薬デキサメタゾンによる処置も、EAE動物においてTer119+細胞の20〜25%減少をもたらしたが、エキセンジン−4処置(50〜55%)ほどではなかった。この結果は、エキセンジン−4処置がMOG免疫動物の脾臓中のTer119+細胞の割合を減少させるのに有効なことを実証した。
【0197】
〔実施例10〕
GLP−1Rアゴニスト処置は自己免疫疾患におけるT細胞活性化を減少させた
本実施例は、EAEマウスのエキセンジン−4処置後のT細胞活性化レベルの変化を例証する。
EAEを、前記のようにMOG(35−55)により雌C57BL/6マウスに誘発した。免疫動物は、PBSビヒクル(n=7)、1mg/kgエキセンジン−4(n=7)、又は4mg/kgデキサメタゾン(n=3)で毎日処置した。動物は、免疫しなかったナイーブ群の3匹のマウスと共に、免疫後日5に犠牲にした。リンパ節を集めた。細胞分離及び赤血球細胞の溶解後に、細胞を発蛍光団結合CD4及びCD44抗体(BDbiosciences)で染色した。細胞はFACSariaTMセルソーター (BD Biosciences)を用いて解析した。解析では、FACS像はCD4+細胞でゲートされた。高レベルのCD44(CD44hi)を有する細胞及びCD4に対する陽性染色は、活性化CD4細胞と同定した。CD4+CD44hi細胞の割合を各処置群で解析した。
図14に示すように、約35%のCD4細胞がナイーブ動物で活性化(CD4+CD44hi)された。EAE動物からの約60%のCD4細胞が活性化された(図14)。エキセンジン−4及びデキサメタゾンによる両処置は、活性化T細胞の割合を減少させ、ナイーブレベルに戻した(図14)。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット(Dunnett)事後検定を行なった。2アスタリスク(**)は、P値が、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で0.01より小さいことを示す。
この結果は、エキセンジン−4処置が、 EAE誘発後のT細胞活性化を抑制するのに有効なことを実証した。
【0198】
〔実施例11〕
GLP−1Rアゴニスト処置は自己免疫疾患においてCD4T細胞増殖を阻害した
本実施例は、EAEマウスのエキセンジン−4処置後のT細胞増殖の変化を例証する。
EAEを、前記のようにMOG(35−55)により雌C57BL/6マウスに誘発した。免疫動物は、PBSビヒクル(n=7)、1mg/kgエキセンジン−4(n=7)、又は4mg/kgデキサメタゾン(n=3)で毎日処置した。動物は、免疫しなかったナイーブ群の3匹のマウスと共に、免疫後日5に犠牲にした。リンパ節を集め、48時間MOG刺激で培養し、次いで増殖細胞を同定するためにBrdUで処置した。BrdUの添加の18時間後細胞を採取した。細胞増殖を測定するために、BrdU染色キット(BD Biosciences)をパシフィックブルー結合CD4抗体と一緒に使用した。細胞は、FACSariaTMセルソーター(BD Biosciences)を用いて解析した。CD4+/BrdU+細胞は増殖CD4T細胞と同定した。
図15に示すように、ナイーブ動物からのCD4細胞はMOG刺激に応答してほとんど増殖せず;ナイーブ動物からのわずか約3%のCD4T細胞がBrdUに対して陽性であった。対照的に、EAE動物からの細胞は、MOG刺激に応答してナイーブ動物からの細胞よりもはるかに大幅な程度増殖していた(図15)。エキセンジン−4及びデキサメタゾンによる処置は、CD4細胞増殖を有意に阻害した(図15)。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット事後検定を行なった。3アスタリスク(***)は、P値が、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で0.01より小さいことを示す。
この結果は、エキセンジン−4処置がEAE誘発後にCD4+細胞の増殖を減少させるに有効なことを実証した。
【0199】
〔実施例12〕
GLP−1Rアゴニスト処置は自己免疫疾患における脾細胞サイトカインレベルを変化させた
本実施例は、EAEマウスのエキセンジン−4処置後の脾細胞サイトカインレベル変化を例証する。
GLP−1RアゴニストがEAEのサイトカインレベルに影響を与えるかどうかを判定するために、EAE動物における脾細胞サイトカインレベルの変化を、エキセンジン−4処置後に評価した。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。誘発動物を、対照ビヒクル(PBS)又はエキセンジン−4(1mg/kg)で毎日処置した。脾細胞(表2の第1列の「SPL」)及びリンパ節(表2の第1列の「LN」)細胞をEAE誘発後日7に処置動物から単離し、そしてナイーブ動物(n=7)からも単離した。細胞を20μg/mlのMOGを含む刺激培地中で48時間培養した。培養培地中のサイトカインレベルをProcartaR Cytokine アッセイキット(Panomics(登録商標))を用いて測定した。使用したキットには、32の別な種類のサイトカイン及びケモカイン測定用ビーズ結合抗体が含まれた。
【0200】
PBS処置群(対照、表2の第3列)及びエキセンジン−4処置群(対照、表2の第4列)間で有意なレベル変化を示したサイトカインが、表2にリストアップされている。データを一元配置ANOVAによって解析し、続いてペアワイズ比較のためのダネット事後検定を行なった。1アスタリスク(*)は、PBS処置EAEとエキセンジン−4処置EAE群間で、ペアワイズ比較で、P値が0.05より小さいことを示し、そして2アスタリスク(**)は、P値が0.01より小さいことを示す。
【0201】
【表5】
【0202】
結果は、エキセンジン−4処置が、IL−17、IFN−γ、TNF−αなどの炎症性サイトカインのレベルを低下させ、疾患進行の減弱をもたらすことを示唆する。表2のデータは、エキセンジン−4処置EAE動物からの細胞におけるサイトカイン発現のFACS解析と一致する(例えば、実施例13を参照)。
【0203】
〔実施例13〕
エキセンジン−4処置はEAE誘発後にリンパ節中のIL−17+及びIFN−γ細胞を減少させた
本実施例は、EAEマウスのエキセンジン−4処置後のIL−17+及びIFN−γ細胞集団の変化を例証する。
GLP−1RアゴニストがEAE中のTh17及びTh1細胞集団に影響を与えるかどうかを判定するために、EAE動物中のIL−17+及びIFN−γ細胞集団の変化を、エキセンジン−4処置後に評価した。IL−17はTh17細胞に存在し、IFN−γはTh1細胞に存在する。比較のために、動物を、陰性対照としてPBSで、陽性対照としてデキサメタゾンで処置した。EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。動物を、1mg/kgエキセンジン−4(n=7)、PBS(n=7)又は4mg/kgデキサメタゾン(n=3)対照で毎日処置した。1群のナイーブ動物(n=3)も含まれた。疾患誘発後日5又は日7に動物を安楽死させた。鼠径リンパ節を解剖してFACSにより解析した。細胞をCD4について表面染色し、IL−17及びIFN−γについて細胞内染色した。CD4及びIL−17、又はCD4及びIFN−γについて二重陽性細胞を、FACSにより解析した。
【0204】
罹患動物がナイーブ動物と比べて高率のIL−17+細胞を有していた一方で、 エキセンジン−4−処置EAE動物は、MOG免疫後日5及び日7の両日において、PBS処置EAE動物と比べて低率のIL−17+細胞を有していた(図16)。免疫抑制薬デキサメタゾンによる処置も、EAE動物におけるIL−17+細胞の50%減少をもたらしたが、エキセンジン−4処置(80〜90%)ほどではなかった。この結果は、エキセンジン−4処置が、リンパ節におけるTer119+細胞の割合を減少させるのに有効なことを実証した。
【0205】
リンパ節中のIFN−γ+細胞集団の大幅な減少が、EAE動物のエキセンジン−4処置で認められた(図17)。予想通り、免疫抑制薬デキサメタゾンも、EAE動物においてIFN−γ+細胞の減少をもたらした。この結果は、エキセンジン−4処置が、EAEマウスのリンパ節中のIFN−γ+細胞集団を減少させるのに有効なことを実証した。これらの結果は、MOG免疫動物におけるTh17及びTh1の両細胞を減少させるGLP−1Rアゴニストの役割を裏付けるものである。
【0206】
両方の天然GLP−1Rアゴニスト、及びGLP−1RアゴニストCovX体は、EAEの進行を緩徐化させるのに有効である。天然アゴニストエキセンジン−4及びGAC−1は、いずれも疾患進行を緩徐化させるのに有効であった。エキセンジン−4は、EAE症状の発症前に投与した場合に有効であった。GAC−1は、EAE症状の発症前及び発症後週1回ベースで投与した場合に有効であった。GLP−1Rアゴニストの初回量の増加は罹病率減少と関連し、用量依存性を示唆した。組織化学実験は、GLP−1Rアゴニストが、T細胞及び単球によるCNS組織の浸潤を減少させることを示した。ミエリン染色は、GLP−1Rアゴニスト処置動物の神経細胞に対する損傷の減少を示した。GLP−1Rアゴニスト処置は、EAEの発症及びピーク時で、脳及び脊髄ミクログリア、及び浸潤するマクロファージのAPC活性化マーカーMHCクラスIIの発現を抑制した。
【0207】
エキセンジン−4を用いて、GLP−1RアゴニストがEAE進行に影響を与えるメカニズムを更に検討した。CD4+、CD8+及びCD19+細胞を直接殺細胞する、コルチコステロイド及びRITUXANTMなどの免疫抑制薬とGLP−1Rアゴニストの作用メカニズムは異なっているようである。これは、GLP−1Rアゴニストが、EAE誘発動物におけるCD4+又はCD8+細胞集団を有意に変化させなかったという観察における証拠である。しかしながら、GLP−1Rアゴニストは、MOG免疫動物のリンパ節におけるIL−17+及びIFN−g+細胞集団の両方を減少させた。これらの結果は、GLP−1Rアゴニスト処置が、リンパ節におけるTh17及びTh1集団を減少させることを実証した。
【0208】
〔実施例14〕
ジペプチジルペプチダーゼ−4(DPP−4)阻害剤は自己免疫疾患の罹病率を減少させるのに有効である
本実施例は、DPP−4阻害剤の処置によるGLP−1レベル上昇が、EAE進行を減弱させることができることを例証する。動物実験は、DPP−4阻害剤シタグリプチンが、メチルセルローズ陰性対照と比べて、EAE罹病率に対して有意な保護をもたらすことを示した(図18)。
GLP−1は、ジペプチジルペプチダーゼ−4(DPP−4)により生体内で分解される。DPP−4阻害剤は、例えばシタグリプチン、ビルダグリプチン、サクサグリプチン、リナグリプチン、ドュトグリプチン(dutogliptin)、ゲミグリプチン(gemigliptin)、アログリプチン及びベルベリンを含むが、これらに限定されない。DPP−4阻害剤シタグリプチンが、グルコースレベル増加及びGLP−1レベル上昇を抑制することができるかどうかを試験するために、4群の12月齢雄C57Bl/6マウス(n=4〜5)を一夜絶食させ、次の朝に経口ブドウ糖負荷試験にかけた。経口グルコース負荷の1時間前に、試験動物に1mg/kgか又は10mg/kgシタグリプチンを経口投与した。陰性対照群の動物にはメチルセルロースを投与し、陽性対照群にはエキセンジン−4(1mg/kgIP)を投与した。経口グルコース負荷後、グルコースレベルを20、40、60及び120分で試験した。グルコース及びGLP−1レベルをチェックするため、血液サンプルもグルコース負荷の20分後に採取した。エキセンジン−4処置は、ほとんど完全にグルコース上昇をブロックした。いずれのシタグリプチン濃度による処置も、メチルセルロース対照と比べて低いグルコースレベルの傾向を示したが、二元配置ANOVAによる統計的有意水準に至らなかった。いずれの用量でのシタグリプチンも、メチルセルロース対照と比べて経口グルコース負荷の20分後に血中GLP−1レベルを有意に増加させた。
【0209】
自己免疫疾患におけるDPP−4阻害剤の効果を試験するために、EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により雌C57BL/6マウスに誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスに免疫後日0から日28まで、1mg/kgエキセンジン−4、10mg/kgシタグリプチン、1mg/kgシタグリプチン又はメチルセルロース(陰性対照)を毎日1回投与した。免疫後日5に血液サンプルをGLP1レベルの測定のために採取した。10mg/kgシタグリプチン処置動物は、メチルセルロース処置動物と比べて有意に高いGLP1レベルを有した。疾患進行をモニターするために、動物の罹病率を毎日以下のようにスコア化した:0=正常;1=尾跛行;2=中等度後肢弱;3=準じて重度後肢弱(動物は困難を伴なうがまだ歩行可能);4=重度後肢弱(動物はまだ後肢を動かせるが歩行不能);5=完全後肢麻痺;及び6=死亡。エキセンジン−4又はシタグリプチンで処置した動物は、対照動物と比べて有意な罹病率減少を示した(図18)。図18のグラフに示されるように、エキセンジン−4による処置は日25を通してEAE進行を大きく減弱させ、1mg/kgか又は10mg/kgシタグリプチンによる処置は、病徴を減弱させる傾向を示した。10mg/kgシタグリプチン処置群での疾患スコアは、メチルセルロース処置群と比べて日18から日20まで有意に低かった(図18)。本結果は、エキセンジン−4及びシタグリプチンがいずれも慢性EAEの進行を緩徐化させるのに有効なことを実証した。
【0210】
〔実施例15〕
末梢免疫細胞はエキセンジン−4の標的ではない
本実施例は、末梢免疫細胞がエキセンジン−4の標的ではないことを例証する。
GLP−1Rアゴニスト−抗体コンジュゲートGAC−2(前記)が、エキセンジン−4/GLP−1受容体の発現をチェックするための染色試薬として使用された。GAC−2は、発蛍光団アレクサ488で標識した。本実施例では、GLP−1R発現CHO細胞を陽性対照として使用した。GLP−1R発現CHO細胞又はナイーブCHO細胞を1%FBSでブロックし、次いでGAC−2−アレクサ488で染色して、蛍光活性化セルソーティング(FACS)によって解析した。ナイーブCHO細胞中の全細胞がGAC−2染色に対して陰性なのに、34%のGLP−1R発現CHO細胞はGAC−2について陽性であったことから、GAC−2−アレクサ488は生細胞でのGLP−1R発現を認識することができることが示された。
ナイーブ動物及び及びEAE動物由来の脾臓リンパ節及び血液からの白血球について、GLP−1R又は他の可能なエキセンジン−4受容体の発現を調べた。リンパ球及び単球マクロファージコンパートメントの両方について、エキセンジン−4/GLP−1受容体発現をGAC−2を用いて解析した。いずれのコンパートメントにも陽性シグナルが認められなかったことから、エキセンジン−4/GLP−1受容体は末梢リンパ器官に発現しないか、又はそれらの発現レベルが非常に低く検出閾値以下であることが示された。
エキセンジン−4が末梢免疫細胞に直接作用するかどうかを試験するために、リンパ球細胞をエキセンジン−4(10μg/ml)又はビヒクル(陰性対照)の存在下MOGでインビトロ処理した。MOGの代わりに、PBSを活性化の陰性対照として使用した。MOG処理リンパ球細胞の活性化は、CD44発現(T−細胞活性化)及びMHCII発現(APC活性化)を解析することにより測定した。MOG処理細胞にエキセンジン−4を与えた場合、活性化阻害は認められなかった。これらの結果は、末梢免疫細胞はエキセンジン−4の標的ではないことを示す。
【0211】
〔実施例16〕
進行中のEAEマウスの罹病率を減少させるエキセンジン−4処置
本実施例は、進行中のEAEマウスの処置におけるエキセンジン−4の有効性を例証する。
本発明の裏付けのために行なわれた実験では、GLP−1Rアゴニストの直接投与は、進行中のEAEマウスの罹病率を減少させることを示した(図19)。EAEを、4mg/mlの結核加熱死菌H37Ra(Difco Laboratories)を含む完全フロイントアジュバント(CFA)に溶解した200μgのPLP(p139−151)による免疫で、SJL/Jマウス(8から12週齢、the Jackson Laboratory)に誘発した(Youssef et al., Nature (2002) 420:78-84を参照)。免疫マウスの体重測定及びEAEの臨床徴候を毎日調べてスコア化した。急性EAEをSJL/Jに樹立後、マウス(n=28)を臨床スコア及び体重に基づいて2群に無作為割付けした。
エキセンジン−4処置は、再発の初めに開始した(免疫後日29)。エキセンジン−4(1mg/kg)か又はビヒクル(PBS;対照用)を毎日1回投与した(i.p.)。マウスを無作為割付けした後、日29から日63まで毎日処置した。動物の罹病率を以下のようにスコア化した:0、麻痺なし;1、尾色調消失;2、後肢弱;3、後肢麻痺;4、後肢及び前脚麻痺;5、瀕死又は死亡。エキセンジン−4で処置した動物は、対照動物と比べて有意な罹病率減少を示した。この結果は、GLP−1Rアゴニストが進行中のEAEマウスの罹病率を減少させるのに有効なことを実証した。
【0212】
〔実施例17〕
インビボで細胞のエキセンジン−4処置は抗原特異的CD4+T細胞増殖をブロックする
本実施例は、エキセンジン−4でインビボ処置後の細胞の抗原特異的CD4+T細胞増殖の阻害を例証する。
本発明の裏付けのために行なわれた実験では、GLP−1Rアゴニストでの細胞のインビボ処置は、抗原特異的CD4+T細胞増殖を阻害することが示された(図19)。SJL/JEAE動物及びC57BL/6EAE動物の両方からの抗原特異的CD4+T細胞の増殖はブロックされた。SJL/J雌マウス(8から12週齢、the Jackson Laboratory)において、EAEを、4mg/mlの結核加熱死菌H37Ra(Difco Laboratories)を含む完全フロイントアジュバント(CFA)に溶解した200μgのPLP(p139−151)による免疫で誘発した(Youssef et al., Nature (2002) 420:78-84を参照)。雌C57BL/6マウスにおいて、EAEを、CFA(Difco)に乳化した200μgの脳炎誘発性ペプチドMOG(35−55)のs.c.注射により誘発し、そして4mg/mlの結核菌 (Difco)を実験日0に追加し、その後直ちに、そして48時間後に、再度400ngの百日咳毒素をi.v.注射した。マウスをエキセンジン−4(1mg/kg)か又はビヒクルのみ(PBS;対照として)で毎日処置した。
【0213】
免疫後日5に、排出リンパ節及び脾臓を処置動物から単離した。リンパ節及び脾臓からの細胞を洗浄し、1×106細胞/mlの最終濃度で予熱したPBS/0.1%BSAに再懸濁した。細胞増殖をモニターするために、細胞をCFSEで標識し、次いでPLP又はMOGペプチドの存在下で増殖させた。CFSE溶液(Invitrogen, catalog #C34554)を1μMの最終濃度まで加えた。細胞を37℃で10分間色素と共にインキュベートし、次いで5容の氷冷培養培地の添加及び5分の氷上インキュベーションによってクエンチした。細胞を遠心分離によりペレット化し、新鮮培地にて全部を3洗液で洗浄した。細胞を計数し、そしてPLPp139−151(SJL/Jマウスからの細胞)か又はMOGp35−55(C57BL/6マウスからの細胞)ペプチドのいずれかと共に、0、10、及び50μg/mlで72時間インキュベートした。細胞はフローサイトメトリーで分析した。CFSA染色を欠く細胞を増殖T細胞と同定した。そのデータを表3に要約する。
【0214】
【表6】
【0215】
表3中のデータは、SJL/J−EAE及びC57BL/6EAEマウスのエキセンジン−4処置が、対照動物と比べて増殖抗原特異的CD4+T細胞の減少をもたらすことを示す。この結果は、細胞をインビボにてGLP−1Rアゴニストで処置した場合に、EAE動物からの抗原特異的CD4+T細胞の増殖がブロックされることを実証した。
【0216】
前記のインビボエキセンジン−4処置の結果と対照的に、エキセンジン−4は、インビトロで脾細胞に投与した場合、CD4+T細胞の増殖に影響を与えなかった。簡潔に言えば、CD4+T細胞は、ナイーブC57BL/6J脾細胞から単離した。細胞増殖をモニターするために、細胞を前記のようにCFSEで標識し、次いでプレート結合抗CD3(1μg/ml)及び抗CD28(0.5μg/ml)で活性化した。エキセンジン−4(0ng/ml、10ng/ml、300ng/ml及び10μg/mlで)又はビヒクル(PBS)を細胞培養物に加え、そして細胞を72時間処置した。処置細胞をフローサイトメトリーで分析した。CFSA染色を欠く細胞を増殖性T細胞と同定した。インビトロにおいてエキセンジン−4で処置したCD4+T細胞は、対照細胞と同等に増殖した。この結果は、抗原特異的CD4+T細胞の増殖が、細胞をインビボでGLP−1Rアゴニストで処置した場合には影響されないことを実証した。
【0217】
〔実施例18〕
表面GLP−1Rは末梢免疫細胞で検出されない
本実施例は、末梢免疫細胞におけるエキセンジン−4染色の不存在を例証する。
GLP−1R発現CHO細胞及び親CHO細胞を、固定又は透過処理(permeabilization)なしでFITC標識GAC−2細胞と共にインキュベートした。細胞を一次抗体(トータル50μl中で10μg/ml)と共に室温で30分間インキュベートした。細胞をフローサイトメトリーを用いて分析した。FITCシグナルはGLP−1R発現CHO細胞で検出されたが、親CHO細胞では検出されなかった。このように、GAC−2は、細胞表面でGLP−1Rを検出することができる。
脾臓、リンパ節及び血液からの末梢免疫細胞は、MOGp35−55による免疫の5日後にナイーブ動物及びEAE動物から単離された。単離細胞をFITC標識GAC−2で染色し、フローサイトメトリーで分析しリンパ球及び単球にゲーティングした。GLP−1R染色は、これらの細胞集団のいずれでも検出されなかった。この結果は、これらの末梢リンパ器官細胞がそれらの表面でGLP−1Rを発現しないこと、又は表面GLP−1R発現が検出レベル以下であることを示唆する。
【0218】
〔実施例19〕
エキセンジン−4は2D2マウスモデルにおいてインビトロで免疫細胞のMOG活性化に影響を与えない
本実施例は、エキセンジン−4が免疫細胞のインビトロでMOG活性化をブロックしないことを例証する。
脾臓及びリンパ節細胞を7週齢の2D2マウスから単離し、そしてMOG刺激の有り又は無しで48時間培養した。エキセンジン−4(10μg/ml)又はビヒクル(PBS)をMOG刺激2D2細胞培養溶液に加えた。培養において48時間のMOG刺激後、細胞を集め、抗CD44抗体及び抗MHCクラスII抗体で染色し、そしてフローサイトメトリーで分析した。抗CD44抗体をT細胞活性化のマーカーとして使用した。抗MHCクラスII抗体をAPC活性化のマーカーとして使用した。
MOGで刺激した脾臓及びリンパ節細胞は、活性化T細胞の存在について陽性として染色した。エキセンジン−4処置はT細胞活性化をブロックしなかった。MOG刺激の不存在下に培養された細胞は、活性化T細胞について有意な染色を欠いていた。この結果は、2D2脾細胞及びリンパ節T細胞がMOG刺激によってエキソビボで活性化し得ること、及びエキセンジン−4処置がエキソビボで活性化を直接ブロックしないことを示す。
MOGで刺激した脾細胞及びリンパ節細胞は、活性化APC細胞の存在について陽性として染色した。エキセンジン−4処置はAPC細胞活性化をブロックしなかった。MOG刺激の不存在下に培養された細胞は、活性化APC細胞についての有意な染色を欠いていた。この結果は、2D2脾細胞及びリンパ節抗原提示細胞が、MOG刺激によってエキソビボで活性化し得ることを示唆するが、エキセンジン−4はエキソビボで活性化を直接ブロックしないようである。
【0219】
〔実施例20〕
エキセンジン−4処置の効果は炎症性サイトカイン産生を阻害する
本実施例は、炎症性サイトカイン産生に対するGLP−1Rの効果を例証する。
SJL/JEAEマウスモデルを用いて、炎症性サイトカイン産生に対するインビボでのエキセンジン−4処置の効果を検討した。再発寛解EAEを、PLP(p139−151)免疫により8週齢SJL/J動物に誘発した。1群の動物を1mg/kgエキセンジン−4で毎日処置し、そして第2群の動物をPBS(対照)で処置した。免疫後日5に、脾細胞を両群の動物から単離し、MOG刺激によりインビトロで培養した。MOG刺激による培養の48時間後に培養培地を集め、IFN−γ及びIL−17のLUMINEX(登録商標)サイトカインアッセイを用いてサイトカイン産生レベルを解析した。
【0220】
エキセンジン−4処置EAE動物からの脾細胞は、対照EAE動物からの脾細胞と比べて、有意に少ない量のIFN−γ及びIL−17を産生した。このことは、エキセンジン−4がPLP(p139〜151)免疫後にTh1及びTh17細胞を正常化するのに有効なことを示す。
【0221】
2D2脾細胞培養を用いて、エキソビボ系において炎症性サイトカイン産生に対するエキセンジン−4の効果を解析した。7週齢2D2動物からの脾細胞を単離し、MOG刺激の有り又は無しで培養した。MOG刺激による脾細胞培養において、エキセンジン−4がエキソビボでTh1又はTh17細胞を減少させるかどうかを試験するために、種々の濃度のエキセンジン−4(10ng/ml、300ng/ml及び10μg/ml)を加えた。PBSを陰性対照として用いた。エキセンジン−4又はPBS添加の48時間後に、培養培地を集め、サイトカインレベルを測定した。MOGで刺激した細胞からの培地は、高レベルのIFN−γ及びIL−17を有した。対照的に、MOGで刺激しなかった細胞からの培地は、最小レベルのIFN−γ及びIL−17を含んでいた。培養液中のエキセンジン−4の存在は、IFN−γの産生に対して効果がなかった。更に、10ng/mlエキセンジン−4で処置した細胞により産生したIL−17のレベルは、エキセンジン−4で処置しなかった細胞により産生したレベルと差がなかった。しかしながら、10ng/mlか又は300ng/mlで処置した細胞からの培地はやや低いIL−17のレベルを有するようであるが、その差は統計的に有意ではなかった。
【0222】
これらの結果は、エキセンジン−4がインビボのEAEマウスモデルにおいて、炎症性サイトカイン産生を減少させることを実証した。しかしながら、対照的に、エキセンジン−4は2D2エキソビボモデルにおける炎症性サイトカイン産生を減少させなかった。これらの結果は、エキセンジン−4が、脾臓又はリンパ節を直接標的化しないようであることを実証した。
【0223】
〔実施例21〕
エキセンジン−4処置はEAE動物の胸腺中のCD4、CD8二重陽性細胞を激減させる
本実施例は、EAE動物の胸腺中のCD4、CD8二重陽性細胞の減少を例証する。
EAEを誘発するために、8週齢のC57Bl/6マウスを皮下によりMOG/CFAで免疫し、免疫後直ちに、そして48時間後に百日咳毒素をi.p.注射した。1群の動物をエキセンジン−4(1mg/kg;n=3)で毎日処置し、そして別の群を対照PBS(n=4)で処置した。免疫後日4に、エキセンジン−4処置及び対照動物の胸腺を採取した。4ナイーブ動物の胸腺を同時に採取した。胸腺を測定し、そして胸腺細胞を単離し、計数し、染色し、次いでフローサイトメトリーで分析した。そのデータを下の表4に要約する。
【0224】
【表7】
【0225】
表4に示されるように、対照EAE動物の胸腺はナイーブ動物の胸腺と比べて有意に小さかった(p=0.007)。エキセンジン−4処置EAE動物の胸腺は、対照EAE動物の胸腺よりも更に小さかった(p=0.002)。対照EAE動物、エキセンジン−4処置EAE動物、及びナイーブ動物の胸腺からの胸腺細胞を単離した。前記の胸腺計測と一致して、対照EAE動物からの総胸腺細胞数は、ナイーブ動物よりも有意に少なかった。エキセンジン−4処置EAE動物からの総胸腺細胞数は、対照動物よりも有意に少なかった。
【0226】
胸腺細胞を抗CD4及び抗CD8抗体で染色し、そしてフローサイトメトリーで分析した。ナイーブ動物からの総胸腺細胞集団の84.6±0.3%はCD4、CD8二重陽性であった。対照EAE動物の総胸腺細胞集団の30.3±11.9%はCD4、CD8二重陽性であった。このデータは、MOG刺激に応答して胸腺中の二重陽性T細胞の分化及び成熟を反映している。エキセンジン−4で処置したEAE動物では、CD4、CD8二重陽性T細胞集団は、ほとんど完全に消失した−エキセンジン−4で処置したEAE動物からの総胸腺細胞集団のわずか2.3±0.3%が、CD4、CD8二重陽性であった。更なる研究は、エキセンジン−4で処置した動物において脾臓及びリンパ節細胞数の増加はないことを実証した。これらの結果は、エキセンジン−4が細胞死によるCD4、CD8二重陽性細胞の減少を引き起こす可能性を示唆する。
【0227】
別の研究では、T細胞成熟ナイーブ動物、及びEAE動物に対するエキセンジン−4の効果を検討した。EAEを、MOG/CFAの皮下注射し、続いて百日咳毒素をi.p.注射することにより8週齢C57BL/6マウスに誘発した。ナイーブ及びEAE動物を3群(n=3)に分けて、それぞれPBS(対照)、エキセンジン−4(1mg/kg)又はデキサメタゾン(4mg/kg)で処置した。デキサメタゾンは、T細胞死を引き起こすことが知られている。免疫後日6に胸腺を採取した。胸腺細胞を単離し、抗CD4及び抗CD8抗体で染色し、そしてフローサイトメトリーで分析した。データを下の表5に要約する。
【0228】
【表8】
【0229】
EAE対照動物の胸腺細胞では、CD4、CD8二重陽性細胞集団(21.7±6.3%)は、ナイーブコントロール動物(86.3±1.1%)と比べて減少した。エキセンジン−4処置及びデキサメタゾン処置の両方は、EAE動物のCD4、CD8二重陽性細胞集団(それぞれ、5.6±2.6%及び1.5±0.5%)を急激に減少させた。
【0230】
ナイーブ動物からの総胸腺細胞集団の86.3±1.1%はCD4、CD8二重陽性であった。デキサメタゾン処置は、CD4、CD8二重陽性細胞集団の有意な減少を引き起こした−デキサメタゾンで処置したナイーブ動物からの総胸腺細胞集団のわずか12.4±3.3%がCD4、CD8二重陽性であった。しかしながら、エキセンジン−4処置は、ナイーブ動物の胸腺のCD4、CD8二重陽性細胞集団に影響を与えなかった。エキセンジン−4で処置したナイーブ動物からの総胸腺細胞集団の81.5±2.0%は、CD4、CD8二重陽性であった。
【0231】
これらの結果は、エキセンジン−4処置はEAE動物の胸腺においてCD4、CD8二重陽性細胞の減少をもたらすが、ナイーブ動物では減少をもたらさないことを実証した。
【0232】
〔実施例22〕
迷走神経切離術は体重減少に対するエキセンジン−4の効果を消失させるが、EAE進行に対するエキセンジン−4の効果に影響を与えない
本実施例は、エキセンジン−4処置に対する迷走神経切離術の効果を例証する。
EAE進行に対するエキセンジン−4の効果が迷走神経を通して媒介されるかどうかを検討するために、EAEを、迷走神経切離した8週齢C57Bl/6動物(The Jackson Laboratory)に誘発した。EAEを、MOG/CFAの皮下免疫により、続いてその直後、及び48時間後に、百日咳毒素をi.p.注射することにより、迷走神経切離及び迷走神経非切離マウスに誘発した。免疫動物を2群(n=7〜8)に分けた。1群はエキセンジン−4(1mg/kg)で毎日処置し、そして他の群はPBS(対照)で処置した。体重及びEAE臨床スコアを毎日モニターした。日20の実験終了時に、動物を安楽死させた。胃計測の結果は、迷走神経切離動物の迷走神経が適正に切断されたことを確認した。
【0233】
MOG免疫の最初の4日以内に、エキセンジン−4処置迷走神経非切離EAEマウスは、対照の迷走神経非切離EAEマウスと比べてそれらの体重の10%が減量した。迷走神経切離マウスでは、、エキセンジン−4処置EAEマウスと対照EAEマウス間で体重の差は認められなかった。このように、迷走神経切離術は、体重減少に対するエキセンジン−4の効果を消失させた。この結果は、体重変化に対するエキセンジン−4の効果が、インタクト迷走神経に依存していることを示唆する。
【0234】
EAE疾患進行は、迷走神経切離術によって影響を受けなかった。迷走神経切離及び迷走神経非切離マウスの両方では、エキセンジン−4処置はPBS処置動物と比べてEAE進行を顕著に遅延させた(図20)。このように、迷走神経切離術は、EAE疾患進行のエキセンジン−4媒介遅延に影響を与えた。この結果は、EAEに対するエキセンジン−4の効果が、迷走神経によって媒介されないことを示唆する。
【0235】
開示された教示は、種々の応用、方法、キット及び組成物に関連して記載されているが、当然のことながら、種々の変更及び改変は、本明細書に記載の教示及び以下の特許請求の範囲から逸脱しない範囲で行なうことができる。前述の実施例は、開示された教示をより的確に例証するために提供されるものであって、本明細書に記載の教示の範囲を限定することを意図するものではない。本教示はこれらの典型的な実施態様について記載されているが、熟練技術者には当然のことながら、これらの典型的な実施態様の変形及び改変は、過度の実験なしに可能である。そのようなすべての変形及び改変は、本教示の範囲内である。
【0236】
本明細書で使用されるセクションの見出しは、系統立てる目的のためだけであり、決して記載された主題を限定するものと解釈すべきではない。当然のことながら、本教示に論じられる温度、濃度、時間などの前に「約」が存在し、その結果、僅かなそして実体のない偏差は、本明細書中の本教示の範囲内であることを暗示する。本願では、特に明記しない限り、単数の使用は複数を含んでいる。また、「含む(comprise)」、「含む(comprises)」、「含んでいる」(comprising)、「含有する(contain)」、「含有する(contains)」、「含有している(containing)」、「包む(include)」、「含む(includes)」及び「含んでいる(including)」の使用は限定することを意図するものではない。当然のことながら、前述の概要及びその後の詳細な説明は、単に例示しそして説明するためであって、本発明を制限するものではない。
【0237】
特許、特許出願、学術論文、教科書など、及びそれらがすでに存在しない程度までそれらに引用されている参考文献を含む、本明細書で引用されたすべての参考文献は、参照によりそれらの全体が本明細書に組み込まれている。1つ又はそれ以上の組み込まれた文献及び類似の資料が、定義された用語、用語の用法、記載技術など(これらに限定されない)を含め本出願と異なり又は相反する事象では、本願が規制する。
【0238】
前述の記載及び実施例は、本発明の一定の格別な実施態様を詳述し、発明者によって意図されたベストモードを記載している。しかし、当然のことながら、いかに前述の記載がテキスト中に詳述されていても、本発明は様々な方法で実施することができ、そして本発明は、添付の特許請求の範囲及びその任意の均等物のいずれかに従って解釈すべきものである。
【特許請求の範囲】
【請求項1】
グルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置を必要とする哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法。
【請求項2】
哺乳動物が自己免疫障害を有する、請求項1に記載の方法。
【請求項3】
自己免疫障害が多発性硬化症である、請求項2に記載の方法。
【請求項4】
請求項2に記載の方法であって、自己免疫障害が、免疫拒絶、移植片対宿主病、ブドウ膜炎、視神経症、視神経炎、横断性脊髄炎、炎症性腸疾患、関節リウマチ、強直性脊椎炎、全身性エリテマトーデス、重症筋無力症、又はグレーブス病と関連している、上記方法。
【請求項5】
哺乳動物がヒトである、請求項1に記載の方法。
【請求項6】
GLP−1RアゴニストがOAP−189である、請求項1に記載の方法。
【請求項7】
GLP−1RアゴニストがDPP−4阻害剤である、請求項1に記載の方法。
【請求項8】
GLP−1Rアゴニストが抗GLP−1Rアゴニスト抗体である、請求項1に記載の方法。
【請求項9】
請求項1に記載の方法であって、GLP−1Rアゴニストがエキセンジン−4のフラグメント又は誘導体を含み、ここでエキセンジン−4のフラグメント又は誘導体がGLP−1Rに結合しそれを活性化する、上記方法。
【請求項10】
GLPアゴニストが、GLP−1Rアゴニストペプチド及び抗体を含むGLP−1Rアゴニスト−抗体コンジュゲート(GAC)である、請求項1に記載の方法。
【請求項11】
請求項10に記載の方法であって、GACが以下の構造:
【化1】
〔式中、
ペプチドは以下の式:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14 E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40 (配列番号3)]−R2
[式中、
R1は、不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;
R2は、OH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6
H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であり;そして
X2は、Aib、A、S、T、V、L、I、D−Alaなどの遮断基であり;
X10はV、L、I、又はAであり;
X12は、S又はKであり;
X13は、Q又はYであり;
X14は、G、C、F、Y、W、M、又はLであり;
X16は、K、D、E、又はGであり;
X17は、E又はQであり;
X19は、L、I、V、又はAであり;
X20は、オルニチン、又はK(SH)Rなどの誘導体化リジン基、又はKであり;
X21は、L又はEであり;
X23は、I又はLであり;
X24は、A又はEであり;
X25は、W又はFであり;
X26は、L又はIであり;
X27は、I、K、又はVであり;
X28は、R、オルニチン、N、又はKであり;
X29は、Aib又はGであり;
X30は、任意のアミノ酸、好ましくはG又はRであり;
X31は、P又は不存在であり;
X32は、S又は不存在であり;
X33は、S又は不存在であり;
X34は、G又は不存在であり;
X35は、A又は不存在であり;
X36は、P又は不存在であり;
X37は、P又は不存在であり;
X38は、P又は不存在であり;
X39は、S又は不存在であり;そして
X40は、連結残基又は不存在であり;
そして、ここで、
X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39、又はX40の1つは、リンカーを介して抗体の結合部位に共有結合した求核性側鎖を含む連結残基で置換され、ここで連結残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン、及びホモセリンから成る群から選択される]のものである〕
のものである、上記方法。
【請求項12】
GACが以下の構造:
【化3】
を含む、請求項11に記載の方法。
【請求項13】
GACが以下の構造:
【化4】
を含む、請求項11に記載の方法。
【請求項14】
抗体が完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、二重特異性抗体及びミニ抗体から成る群から選択される、請求項10〜13のいずれか1項に記載の方法。
【請求項15】
抗体が、IgG1、IgG2、IgG3、及びIgG4から成る群から選択される定常ドメインを含む、請求項10〜13のいずれか1項に記載の方法。
【請求項1】
グルカゴン様ペプチド−1受容体(GLP−1R)アゴニストを含む組成物を、GLP−1Rを活性化する有効量で当該処置を必要とする哺乳動物に投与し、それによって中枢神経系組織の白血球浸潤を減少させることを含む、中枢神経系組織の白血球浸潤を減少させる方法。
【請求項2】
哺乳動物が自己免疫障害を有する、請求項1に記載の方法。
【請求項3】
自己免疫障害が多発性硬化症である、請求項2に記載の方法。
【請求項4】
請求項2に記載の方法であって、自己免疫障害が、免疫拒絶、移植片対宿主病、ブドウ膜炎、視神経症、視神経炎、横断性脊髄炎、炎症性腸疾患、関節リウマチ、強直性脊椎炎、全身性エリテマトーデス、重症筋無力症、又はグレーブス病と関連している、上記方法。
【請求項5】
哺乳動物がヒトである、請求項1に記載の方法。
【請求項6】
GLP−1RアゴニストがOAP−189である、請求項1に記載の方法。
【請求項7】
GLP−1RアゴニストがDPP−4阻害剤である、請求項1に記載の方法。
【請求項8】
GLP−1Rアゴニストが抗GLP−1Rアゴニスト抗体である、請求項1に記載の方法。
【請求項9】
請求項1に記載の方法であって、GLP−1Rアゴニストがエキセンジン−4のフラグメント又は誘導体を含み、ここでエキセンジン−4のフラグメント又は誘導体がGLP−1Rに結合しそれを活性化する、上記方法。
【請求項10】
GLPアゴニストが、GLP−1Rアゴニストペプチド及び抗体を含むGLP−1Rアゴニスト−抗体コンジュゲート(GAC)である、請求項1に記載の方法。
【請求項11】
請求項10に記載の方法であって、GACが以下の構造:
【化1】
〔式中、
ペプチドは以下の式:
R1−[H1 X2 E3 G4 T5 F6 T7 S8 D9 X10 S11 X12 X13 X14 E15 X16 X17 A18 X19 X20 X21 F22 X23 X24 X25 X26 X27 X28 X29 X30 X31 X32 X33 X34 X35 X36 X37 X38 X39 X40 (配列番号3)]−R2
[式中、
R1は、不存在、CH3、C(O)CH3、C(O)CH2CH3、C(O)CH2CH2CH3、又はC(O)CH(CH3)CH3であり;
R2は、OH、NH2、NH(CH3)、NHCH2CH3、NHCH2CH2CH3、NHCH(CH3)CH3、NHCH2CH2CH2CH3、NHCH(CH3)CH2CH3、NHC6
H5、NHCH2CH2OCH3、NHOCH3、NHOCH2CH3、カルボキシ保護基、脂質脂肪酸基又は糖質であり;そして
X2は、Aib、A、S、T、V、L、I、D−Alaなどの遮断基であり;
X10はV、L、I、又はAであり;
X12は、S又はKであり;
X13は、Q又はYであり;
X14は、G、C、F、Y、W、M、又はLであり;
X16は、K、D、E、又はGであり;
X17は、E又はQであり;
X19は、L、I、V、又はAであり;
X20は、オルニチン、又はK(SH)Rなどの誘導体化リジン基、又はKであり;
X21は、L又はEであり;
X23は、I又はLであり;
X24は、A又はEであり;
X25は、W又はFであり;
X26は、L又はIであり;
X27は、I、K、又はVであり;
X28は、R、オルニチン、N、又はKであり;
X29は、Aib又はGであり;
X30は、任意のアミノ酸、好ましくはG又はRであり;
X31は、P又は不存在であり;
X32は、S又は不存在であり;
X33は、S又は不存在であり;
X34は、G又は不存在であり;
X35は、A又は不存在であり;
X36は、P又は不存在であり;
X37は、P又は不存在であり;
X38は、P又は不存在であり;
X39は、S又は不存在であり;そして
X40は、連結残基又は不存在であり;
そして、ここで、
X10、S11、X12、X13、X14、X16、X17、X19、X20、X21、X24、X26、X27、X28、X32、X33、X34、X35、X36、X37、X38、X39、又はX40の1つは、リンカーを介して抗体の結合部位に共有結合した求核性側鎖を含む連結残基で置換され、ここで連結残基は、K、R、Y、C、T、S、リジンのホモログ(K(SH)を含む)、ホモシステイン、及びホモセリンから成る群から選択される]のものである〕
のものである、上記方法。
【請求項12】
GACが以下の構造:
【化3】
を含む、請求項11に記載の方法。
【請求項13】
GACが以下の構造:
【化4】
を含む、請求項11に記載の方法。
【請求項14】
抗体が完全長抗体、Fab、Fab’、F(ab’)2、Fv、dsFv、scFv、VH、二重特異性抗体及びミニ抗体から成る群から選択される、請求項10〜13のいずれか1項に記載の方法。
【請求項15】
抗体が、IgG1、IgG2、IgG3、及びIgG4から成る群から選択される定常ドメインを含む、請求項10〜13のいずれか1項に記載の方法。
【図1】

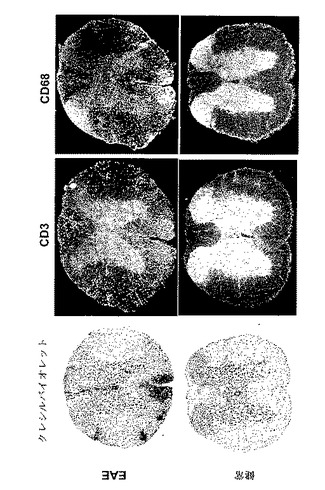
【図2】
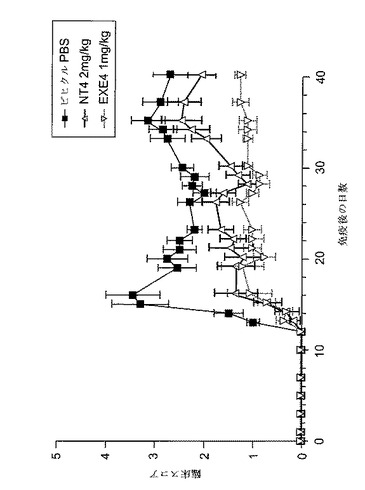
【図3】
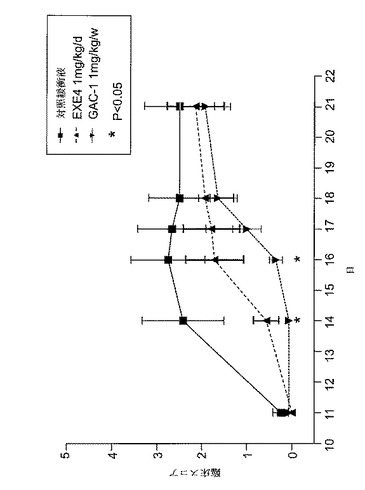
【図4】
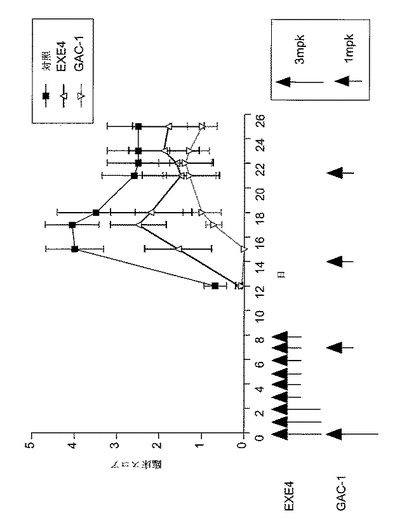
【図5】
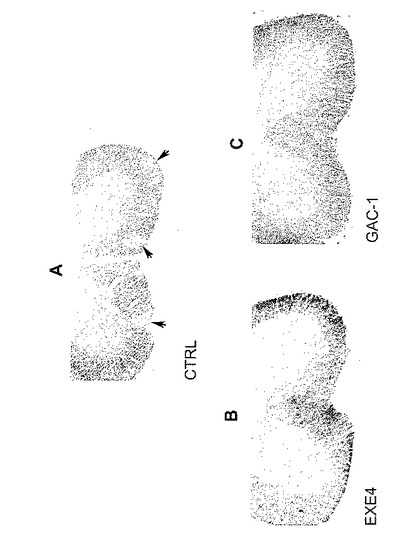
【図6】
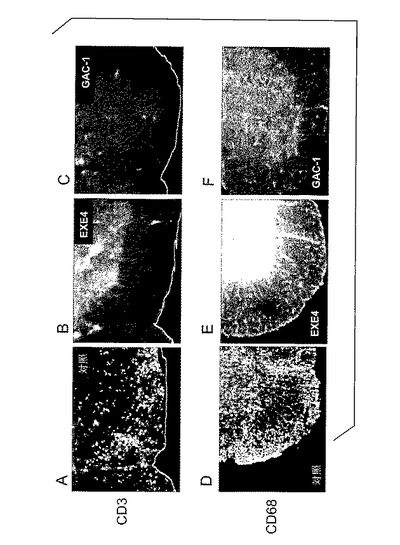
【図7】
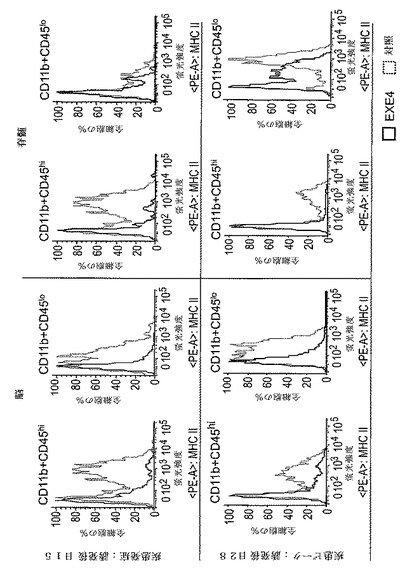
【図8】
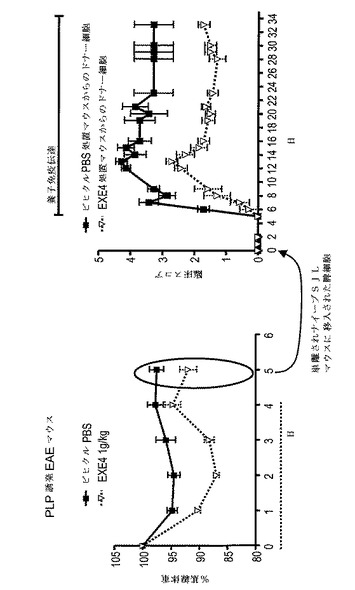
【図9】

【図10】
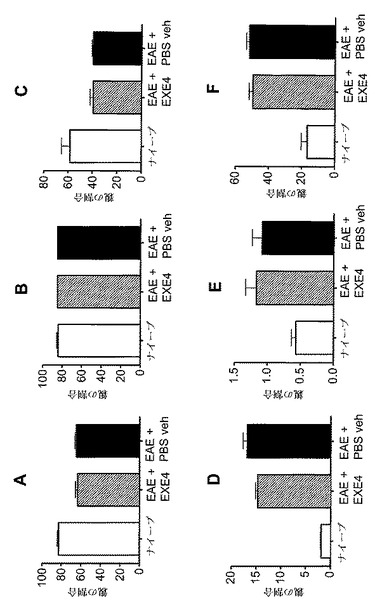
【図11】
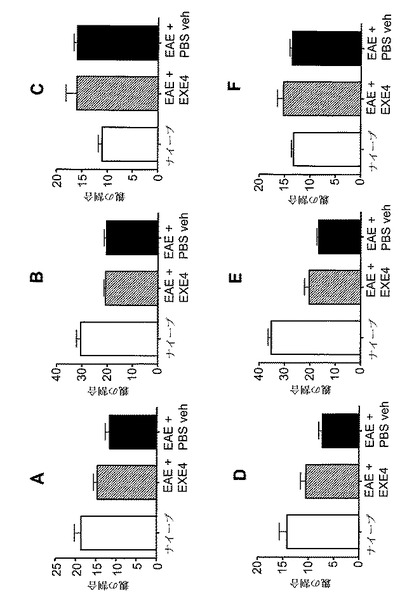
【図12】
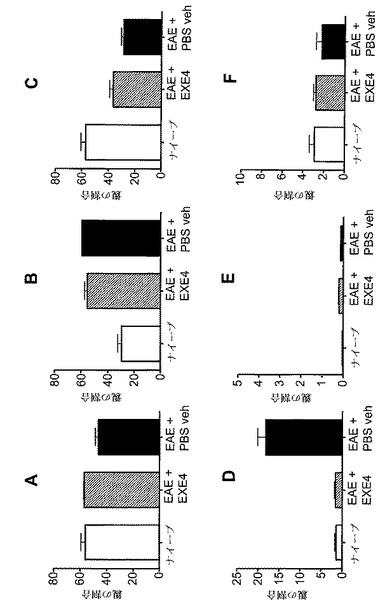
【図13】
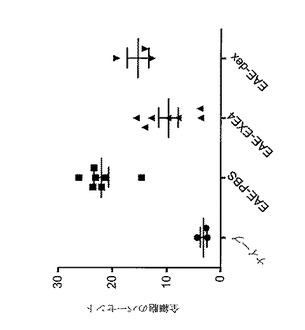
【図14】
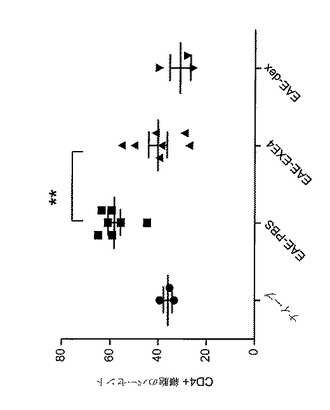
【図15】
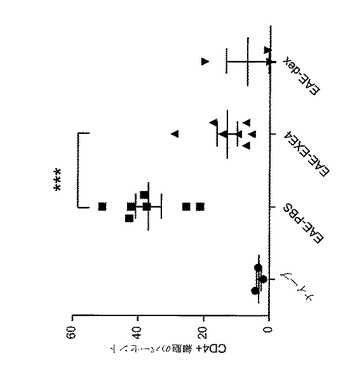
【図16】
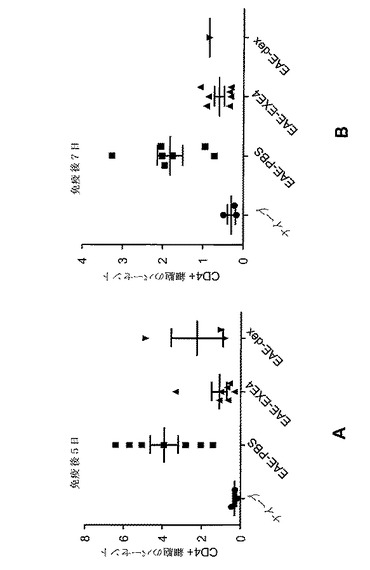
【図17】
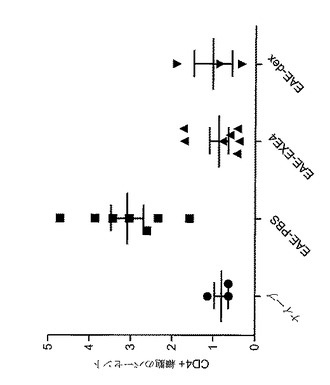
【図18】
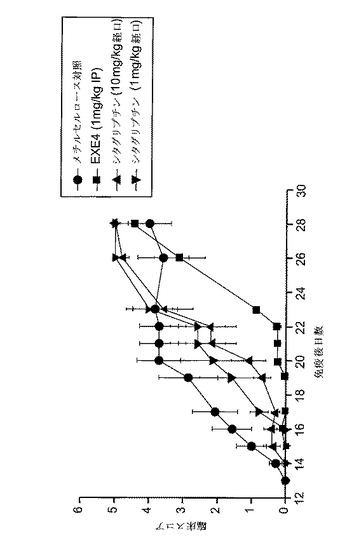
【図19】
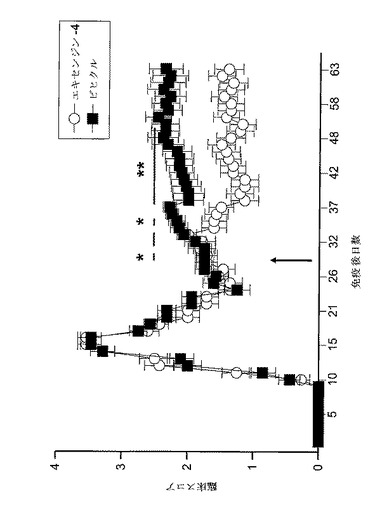
【図20】
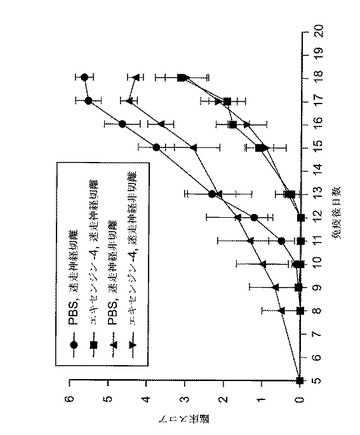
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【公開番号】特開2011−57670(P2011−57670A)
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−189054(P2010−189054)
【出願日】平成22年8月26日(2010.8.26)
【出願人】(505129415)ライナット ニューロサイエンス コーポレイション (33)
【Fターム(参考)】
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願番号】特願2010−189054(P2010−189054)
【出願日】平成22年8月26日(2010.8.26)
【出願人】(505129415)ライナット ニューロサイエンス コーポレイション (33)
【Fターム(参考)】
[ Back to top ]