
薬剤化合物の溶解性を増加させるPEG−脂質抱合体
ジアシルリピッド−ポリマー抱合体が水溶液中において親油性薬物の溶解性を促進するために使用される。この抱合体は骨格鎖と、2つの親油性アシル基と、親水性ポリマーとを含んでいる。
【発明の詳細な説明】
【技術分野】
【0001】
優先権主張
本願は2009年1月23日に出願された米国仮特許願61/205840「薬剤化合物の溶解性を促進させるPEG−脂質抱合体」の優先権を主張する。
【0002】
発明の分野
本発明は体内送達を増進するために薬剤の溶解性を促進させる組成物およびその方法に関する。さらに特定するならば、本発明はそのような増加した溶解性と送達とを提供するジアシルリピッド(脂質)−ポリマー(重合体)抱合体(結合体、複合体)に関する。
【背景技術】
【0003】
発明の背景
薬剤科学においては疎水性薬剤化合物の作用部位への送達は現在においても研究対象の問題である。利用されている薬物送達ビヒクルの種類には、シクロデキストラン、薬物−脂質複合物、リポソームおよびクレモフォール(Cremophor)(登録商標)のごとき溶解剤が含まれる。様々なPEG−脂質抱合体がこれらビヒクルに利用されてきた。しかしながら、多くの有用な薬物は、その溶解性、薬物動態特性および生体内利用性によって未だに限定を受けている。よって、本発明の1目的は、親油性剤を調製および送達する新規な組成物並びに方法を提供することである。
【発明の概要】
【課題を解決するための手段】
【0004】
発明の概略
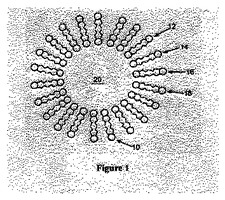
ジアシルリピッド−ポリマー抱合体は水溶液内で親油性薬物の溶解性を促進するために利用される。この抱合体は骨格鎖(主鎖)と、2個の親油性アシル基と、親水性ポリマーとを含む。
【図面の簡単な説明】
【0005】
添付図面は本明細書の一部であり、本発明の実施例を図示しており、以下の詳細な説明と共に本発明の原理および実施形態を説明するものである。
【0006】
以下は図面の要約である。
【0007】
【図1】図1は脂質分子で構成されたリポソームの断面を図示する概略図である。
【図2】図2は脂質分子で構成されたミセルの断面を図示する概略図である。
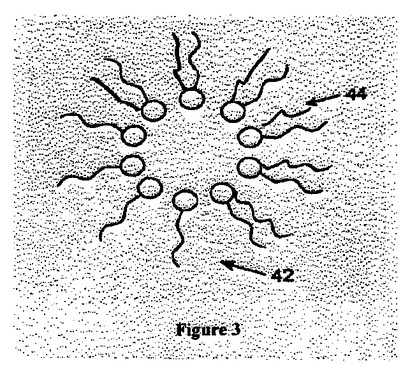
【図3】図3は頭部(ヘッド基)と較べて大きな尾部を有した脂質分子で構成された構造物の断面を図示する概略図である。
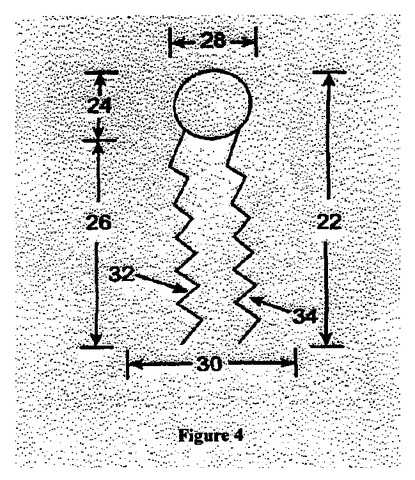
【図4】図4は極性頭部と非極性炭化水素鎖を有した脂質分子の空間詰概略図である。
【図5】図5はゲフィチニブ調合物の薬物動態特性を示す。
【発明を実施するための形態】
【0008】
詳細な説明
以下では薬剤の溶解性を促進し、薬剤の送達を増進するためのジアシルリピッド−ポリマー抱合体の形態による本発明の実施態様が説明されている。添付図面で図示されているごとき本発明の実施態様を詳細に説明する。図面および以下の詳細な説明を通して同一参照符号(番号)は同一要素または類似要素を表す。
【0009】
簡略性を高めるため、本発明の実施態様で現れる全ての型通りの特徴を一々図示および解説してはいない。もちろん、そのような実際の利用形態の開発において開発者の特定目標、例えば利用形態およびビジネス関連に動機付けられた目標を達成するためには多数の実施形態特有の決定が必要であることは理解されよう。また、これらの特有目標は実施形態によって異なり、開発者によっても異なることも理解されるであろう。さらに、このような開発努力は複雑で複合的なものであり、開発には長時間を要するが、いずれにせよ本明細書の開示内容を活用することで当該技術分野の当業者によって自身の通常の研究活動に利用できるであろう。
【0010】
当該分野の当業者であれば本発明の以下の解説が単に本発明の説明を目的としており、本発明を限定するものではないことを理解するであろう。本発明の他の実施態様も、ここでの開示の恩恵を受ける当業者には容易に理解できよう。
【0011】
本発明は薬剤組成物と、その薬剤組成物を調合して使用する方法とを含む。この薬剤組成物は親油性薬物並びに1以上のポリマー−脂質抱合体を含んで成る。この抱合体は骨格鎖、2個のアシル基およびポリマー(重合体)鎖を有している。好適には、それらはヒトの体温よりも低い溶解温度を有し、さらに好適には室温では液体である。薬物を含んだ抱合体の水溶液はリポソームを含まないが、その代わりに様々な微小構造物を含んでいる。
【0012】
定義:ここで使用される“親油性”とは、リピッド(脂質)内で溶解する性能及び/又は生体膜に浸透し、生体膜と相互作用し、及び/又はトラバースすることができる性能のことである。
【0013】
米国特許6610322およびその関連特許文献は、ジアシルリピッド−ポリエチレングリコール(PEG)抱合体を採用することで自然発生性リポソームの形成を教示する。この特許は、その抱合体を水溶液に加えるだけでリポソームを形成するこのようなPEG−脂質抱合体をいかに選択するかを解説する。他の類似したポリマー−脂質分子はリポソームを形成せずに疎水性薬物を溶解させるのに有用であることが発見されている。
【0014】
米国特許6610322が解説するように、所定の条件が満たされるとPEG−脂質抱合体は水溶液と混合されて自然発生的にリポソームを形成する。ジアシルグリセロール−PEG(DAG−PEG)においては、1つの条件は、全脂質(単一脂質または複数の脂質の組み合わせ)のパッキング(充填)パラメータがリポソームを形成させる範囲内に存在しければならないことである。簡単に述べれば、脂質は一般的に円錐形状であって、自動的にリポソームが構成されるようにテーパー状でなければならない。もし頭部(PEGを含む)が疎水性部分または疎水性分子に比して大き過ぎたり小さ過ぎたりすればリポソームは形成されない。
【化1】

化学構造1
【0015】
化学構造1は本発明のポリマー−脂質抱合体の一般形状である。AG1とAG2はアシル基である。すなわち分子の疎水性部分である。これらアシル基は同じであっても異なっていてもよい。Pは親水性ポリマーであり、典型的にはPEGである。Rは親水性ポリマーの先端のモイエティ(部分)である。Bは骨格鎖である。L1は骨格鎖を親水性ポリマーに接続するリンカーである。L2とL3は骨格鎖をアシル基に接続するリンカーである。Bと、L1、L2およびL3を含んだ分子部分は延伸骨格鎖とも考えられる。延伸骨格鎖並びにPおよびRは分子の親水性頭部または親水性頭部基を含む。アシル鎖は親油性尾部を含む。
【0016】
脂質−ポリマー抱合体の両親媒性は水溶液中での親油性薬物の溶解性を促進する性能を与える。アシル基はその薬物と協調し、頭部は水と自由に作用する。骨格鎖とリンカーは一定種類の薬物を溶解させ、及び/又は送達する利点を提供するように選択できるが、骨格鎖およびリンカーは一般的に親水性ポリマーやアシル基よりも機能的には重要性が低い。よって抱合体の単純化バージョンが化学構造2で説明できる。このEBは延伸骨格鎖である。
【化2】
化学構造2
【0017】
リポソームを形成するため、脂質頭部と炭化水素鎖は自動的に、自身を折畳ませるような曲率半径を備えた二重層にならなければならない(図1参照)。もし炭化水素鎖(アシル基)が頭部に較べて小さ過ぎれば、曲率半径は小さ過ぎてミセルが発生するであろう(図2参照)。もし炭化水素鎖が頭部に較べて大き過ぎれば、曲率半径は正負が逆になり、リポソームは形成できない(図3参照)
【0018】
図1は脂質分子で構成されたリポソームの概略断面図である。リポソーム10は水性空間20を囲む脂質分子(例:12、14、16、18)で成る脂質二重層を含む。
【0019】
図4は極性頭部と非極性炭化水素鎖とを有した脂質分子の空間充填図である。脂質分子22は親水性基24と疎水性尾部26とを含んでいる。疎水性尾部26は2つの炭化水素鎖32、34(AG1とAG2)を含んでいる。その化学結合は脂質分子をフレキシブルにしているが、頭部は直径28の領域を満たしており、尾部は直径30の領域を満たしている。脂質分子はリポソームを形成するために二重層内で構成されなければならないため、リポソームは、頭部径と尾部径との比が大き過ぎても小さ過ぎても形成されない。
【0020】
図2は脂質分子で構成されたミセルの概略断面図である。ミセル36は脂質分子(例:38、40)を含む。脂質分子の尾部は頭部に較べて小さな径であるため、脂質分子は小曲率半径を備え、二重層は形成されない。
【0021】
図3は頭部に較べて大きな尾部を備えた脂質分子で成る構造物の概略断面図である。図3で示すように、頭部に較べて大きな尾部を備えた脂質(例:44)が水溶液と混合されると構造物42が形成される。頭部と尾部とサイズ比は二重層の形成を不可能にしている。
【0022】
図1、図2および図3は1種の脂質分子を使用して充填パラメータの基本原理を図示したが、同一原理が脂質の混合物に適用できることは理解されよう。例えば、小さ過ぎて単種としてのリポソームを形成できない炭化水素鎖を有した脂質はコレステロールと混合することができ、適した充填パラメータを有した組成物が得られる。別例では、自身が適した充填パラメータを有している脂質は、自身は適した充填パラメータを有していない限定量の他の脂質を取り込んだリポソームを形成できる。単一脂質および複数脂質の混合物の両方は公知方法で計算できる充填パラメータを有する。一般的にリポソームを形成させるリポソーム組成物は、約0.84から0.88の間の充填パラメータ測定値Paと、約0.88と0.93の間の充填パラメータ測定値Pvを有する。
【0023】
Paは表面に関する充填パラメータであり、Pvは体積に関する充填パラメータである(DDラシック著、リポソーム:「物理学から利用まで」エルセビエル誌、51ページ、1993年)。これらパラメータは式HCa/Ta≡PaおよびHCv/Tv≡Pvから導かれる。このHCaは炭化水素鎖面積であり、Taは全分子面積であり、HCvは炭化水素鎖の体積であり、Tvは全分子の体積である。
【0024】
脂質混合物のための充填パラメータが計算できる。なぜなら脂質の理想的な混合は個々の特性の計算平均となるからである。例えば、理想混合の場合には二元混合のHCa/Ta≡Paは次の式になる。
<Pa>=X1P1+X2P2、X1+X2=1
【0025】
さらに一般化すれば、所定の混合物を含んだi脂質の場合には次の式で表される。
<Pa>=ΣiXiPi及びΣiXi=1
このXiは混合物内の脂質のモル分率であり、Piはその脂質の表面に関する充填パラメータである。
【0026】
2つの一般的な要因が脂質分子の頭部サイズに影響を及ぼす。その1つは頭部の実際の物理的サイズである。例えば、さらに長いPEG鎖の採用は頭部をさらに大きくする。もう1つは頭部に関わる電荷である。例えば、PEG鎖がリン酸ジエステル結合により骨格鎖に抱合するなら、リン酸塩は頭部に電荷を分与し、そのサイズを効果的に増加させるであろうが、そのようなことは起こらない。本発明では、非リン脂質が好適であり、頭部サイズを変える一般的手段はポリマー長を変えること、並びに骨格鎖とリンカーを変えることである。
【0027】
脂質の尾部のサイズは炭化水素鎖の長さと、脂質鎖の飽和度によって影響を受ける。2個の鎖を備えたリピッドを含んだリポソームには組み込めるが、単鎖脂質は一般的にリポソームを形成しない。同様に1個の長鎖と1個の短鎖とを備えた脂質は相対的に小さな尾部サイズを有するであろう。
【0028】
ステロール及び/又は薬剤化合物の存在は予想されるようにリポソームの形成に影響を及ぼすであろう。これら全ての要因は、単一ポリマー−脂質抱合体がリポソームを形成するか否か、またはポリマー−脂質抱合体を含んだ化学的組み合わせが水溶液内でリポソームを形成するか否かを計算して予測する際に考察しなければならない。
【0029】
場合によっては、リポソームは好適な薬物送達ビヒクルであるが、多くの親油性薬物はリポソーム縣濁剤に調合することが困難であることが知られている。問題とは、保存、生体内安定性、薬物と脂質の比率その他である。本発明のポリマー−脂質抱合体を含んだ非リポソーム調合物は多くの場合に有用であり、特定の場合には非常に有効である。
【0030】
米国特許6610322で解説されている充填パラメータに基づいてリポソームを形成するために、PEG−脂質抱合体を選択することが可能であるのと同様に、ポリマー−脂質抱合体の純水溶液またはリポソームを形成しないポリマー−脂質抱合体を含んだ混合物を選択することは可能である。このような選択はポリマー−脂質抱合体の充填パラメータを計算することで数学的に実施できる。この選択は、様々なポリマー−脂質抱合体を試験することで経験的に実行できる。組み合わせる取り組みも利用できる。ここでは充填パラメータに影響を及ぼす要因の知識が活用され、リポソーム形成のための試験を実施する前に充填パラメータを厳格に計算することなく、どのような抱合体が利用可能であるのかを予測する。
【0031】
表1は本発明での利用性を確認するために単一脂質として試験された複数の脂質を示す。この表は米国特許6610322の欄6の表と同じである。脂質は水溶液中にて2重量%の状態で試験された。GDLとはグリセロールジラウリン酸であり、GDOとはグリセロールジオレイン酸であり、GDMとはグリセロールジミリスチン酸であり、GDPとはグリセロールジパルミチン酸であり、GDSとはグリセロールジステアリン酸である。各脂質に対して「PEG」の後の数字はPEG鎖内のC2H4Oサブユニットの数を示す。不飽和ジオレイン酸脂質は飽和ジミリスチン酸脂質と類似した充填パラメータを有する。
【表1】
【0032】
表1はいくつかのポリマー−脂質抱合体のリポソーム形成特性を示す。この表を理解するには、自然リポソーム形成には、脂質または脂質混合物がリポソーム形成温度において流体でなければならないことを知っている必要がある。例えば、PEG−12GDMは試験した全ての温度でリポソームを自然形成する。なぜなら、そのような温度ではリピッドは流体であり、特定の範囲内で充填パラメータを有することに加え、PEGを含むからである。同様に、PEG−12GDO(PEG−12GDMとほぼ同一特性)は試験された全温度でリポソームを自然形成する。高融点を備えたPEG−23GDSは39.8℃の融点以上でリポソームを形成するだけである。
【0033】
好適には本発明のポリマー−脂質抱合体は、人体温度、すなわち約37℃未満の温度で液体となる溶融温度を有する。特に好適には、この抱合体は室温未満、すなわち約25℃未満の溶融温度を有する。充填パラメータの場合と同様に溶融温度は脂質の混合物のために決定できる。
【0034】
調製の容易性を考慮すればさらに低い溶融温度の抱合体が好適である。しかしながら、さらに高い温度で調合物を調製することで、さらに高い溶融温度の抱合体でも利用できる。
【0035】
GDMシリーズの脂質は充填パラメータの重要性を解説する。これら脂質は全てPEGを含んでおり、60℃で液体であり、好適な充填パラメータのおかげでPEG−12GDMだけがリポソームを形成する。一連のDGS脂質は60℃で同一ポイントを示す。
【0036】
当該技術の専門家であれば、リポソームを形成しないポリマー−脂質抱合体の組成物を予測して造りだすために充填パラメータの知識を活用して本発明を実施することができる。例えば、PEG鎖のサイズを変え、または脂質組成物内のPEG含有脂質の濃度を変えると、予測可能な状態で充填パラメータが変わる。同様に骨格鎖及び/又はリンカーの変更も充填パラメータの予測可能な変化をもたらす。ポリマー鎖の長さを変え、及び/又はアシル鎖の長さと飽和状態を変えることで溶融温度も変えることが可能である。
【0037】
いくつかの基本的な条件が満たされている限りは、多様なポリマー−脂質抱合体が本発明の実施に有効である。まず抱合体は疎水性ポリマーを含まなければならない。次に、抱合体は親油性薬物と関連するために2つのアシル鎖を含まねばならない。さらに、リポソームが水溶液内で優勢とならないように調剤の充填パラメータを選択しなければならない。アシル鎖と親水性ポリマーは、溶融温度と充填パラメータに関し、抱合体の化学特性に対して支配的となる傾向はあるものの、骨格鎖も重要である。
【0038】
水溶液を使用して本発明を実施するとき、調合物によっては一部がリポソーム形態であることはあり得る。しかし、リポソームはせいぜい調合物のほんの一部を構成するだけである。多くの場合に、このリポソームは非常に少量である。一般的に本発明の非リポソーム調合物は、希釈水溶液内で10%未満のリポソーム形態の脂質−ポリマー抱合体を有している。すなわちリポソームは抱合体の支配的な構造形態ではないということができる。
【0039】
有用な抱合体は次の2つの範疇に分類できる。すなわち尾部と比較して頭部が大き過ぎるのでリポソームを形成しないものと、尾部と比較して頭部が小さ過ぎるのでリポソームを形成しないものとである。本発明の目的は水溶液中にて親油性化合物の溶解性を促進することであるため、第1の範疇が一般的には好まれる。なぜならこの化合物は水溶要素の割合が高いからである。しかしながら、さらに大きな親油性尾部を有した抱合体は、さらに小さな尾部を有した抱合体よりもさらに多量に薬物を運搬できる。よって、好適なアシル基は少なくとも8個の炭素原子を有する。
【0040】
有用なアシル基(AG1およびAG2)は表2と表3に掲載されたものを含む。
【表2】
【表3】
【0041】
グリセロールは好適な骨格鎖であるが、他の有用な骨格鎖(B)には、3−アミノ−1,2−プロパンジオール、1−アミノ−2,3−プロパンジオール、2−アミノ−1,3−プロパンジオール、3−メルカプト−1,2−プロパンジオール、1−メルカプト−2,3−プロパンジオール、2−メルカプト−1,3−プロパンジオール、ジヒドロキシブチル酸およびグリセリン酸等の3−炭素鎖が含まれる。あるいは、糖類が骨格鎖として利用できる。可能な糖類骨格鎖には単糖類および二糖類が含まれる。骨格鎖は4−炭素鎖または2−炭素鎖も含むことができる。骨格鎖はN(1個の窒素原子)またはCHで、AG1、AF2およびPがそれらCまたはNの骨格鎖に結合したような単純なものでよい(この場合にはリンカーは共有電子対であると考えられる)。さらにアミノ酸が骨格鎖として利用できる。可能なアミノ酸骨格鎖には、システイン、セリン、トレオニンおよびアスパラギンが含まれる。環式プロパン等の環式アルカン、およびアゾールのごとき複素環式化合物も骨格鎖として利用できる。
【0042】
芳香族化合物の特性は特別な配慮を要するが、ベンゼン等の芳香族化合物も骨格鎖として利用できる。まず芳香環は大きくて頑丈であり、頭部の相対サイズを増加させるであろう。次に、芳香環は、典型的には抱合体の親水性部分で親油性である。続いて、芳香環間のスタッキングは予測不能に微小構造の形成に影響を及ぼすであろう。しかし、これらユニークな特性は状況によっては有利に作用する。
【0043】
本発明のポリマー−脂質抱合体を形成する際に有用な親水性ポリマーには、ポリエチレングリコール(PEG)および他のポリアルケン酸化物ポリマー、ポリオキシエチレンアルキルエーテル、ポリビニルピロリドン、ポリ(アリルアミン)、ポリ(1−グリセロールメタクリレート)、ポリ(2−エチル−2−オキサゾリン)、ポリ(2−ヒドロキシエチルメタクリレート/メタクリル酸)/ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(2−ビニルピリジン)、ポリ(アクリルアミド/アクリル酸)、ポリ(アクリル酸)、ポリ(ブタジエン/マレイン酸)、ポリ(エチルアクリレート/アクリル酸)、ポリ(エチレン酸化物−b−プロピレン酸化物)、ポリ(エチレン/アクリル酸)、ポリ(メタクリル酸)、ポリ(マレイン酸)、ポリ(N−イソ−プロピルアクリルアミド)、ポリ(N−ビニルピロリドン/ビニルアセテート)、ポリ(スチレンスルフォン酸)、ポリ(スチレンスルフォン酸/マレイン酸)、ポリ(ビニルアセテート)、ポリ(ビニルリン酸)、ポリ(ビニルアミン)、ポリアクリルアミド、ポリアクリル酸、ポリアニリン、ポリエチレンイミン、プルラン、ポリメタクリルアミドが含まれる。上記のリストに基づいた共重合体とブロック共重合体も利用できる。遊離ポリマーは室温で水溶解性であり、無毒でもある。それらは哺乳動物内に目立った免疫遺伝レスポンスを導き出さない。狭い分子量分布の親水性ポリマーが好適である。製薬ビジネスで既に利用が認められているため、PEGは好適な親水性ポリマーである。
【0044】
様々なR基をポリマーの先端に含むことができる。有用なR基にはアルキル基が含まれ、例えばメチル、アルコキシモイエティ、アミノ酸、および、1、2、3、4、あるいはそれ以上の単糖ユニットをそれぞれ含んだ単糖類、二糖類、三糖類、および多糖類等の糖類が含まれる。さらに抗体部分のごとき標的モイエティ(部分)とビタミン類がR基として利用できる。一般的にR基は非常に水溶解性である。R基の分子量は好適には約200未満であり、さらに好適には約50未満である。大抵の利用においては、血流内で非特定結合や短縮循環時間を回避するためにR基は好適には陽イオン性ではない。しかし、陽イオン性R基は、局部用ゲルおよび口および喉を標的とした口内溶液のごとき投与形態では好適に利用できる。
【0045】
有用なリンカー(L1、L2、L3)にはオキシ、エステルおよびカルボキシル並びに表4で示すものが含まれる。
【表4】
【0046】
リンカーは特定の利用目的で本発明をカスタマイズするように選択できる。例えば、低pHで変動性が高いリンカーは適用形態によっては好適である。他の場合には、溶解した薬剤化合物の一部と水素結合する能力を有するリンカーが有利であろう。しかしながら、大抵の場合には、オキシ、チオール、およびカルボキシ等の単純なリンカーが適している。
【0047】
本発明では広範囲な脂質−ポリマー抱合体が有用である。前述したようにPEGを親水性ポリマーとして有することが望まれる。他の好適実施態様には好適な分子量範囲の抱合体を利用することが含まれる。相対的に大きな頭部の抱合体を使用するときには、延伸骨格鎖のMWが約105から250の範囲であり、P+RのMWがアシル基の組み合わせMWよりも大きいのが好適である。本発明の抱合体は約8から約13の親水性−親油性バランス率(レーティング)を有する。
【0048】
ラブラソール(登録商標)とラブラフィル(登録商標)のごとき現存する溶解性促進剤はPEGを杏ピット油のごとき天然グリセリド混合物と抱合させることで製造される。これでかなり広範囲な構成成分との溶解性混合物となる。一方、本発明の溶解性促進剤は別個の工程で合成され、純粋で明確な調合物が得られる。このように制御することで本発明を特定の親油性薬物を溶解させるべく使用するときに技術者に個別の調整をさせる。場合によっては特定の親油性薬剤のための溶解性促進剤は、本発明の単一で化学的に純粋な脂質−ポリマー抱合体でよい。
【0049】
本発明の抱合体は他の溶解性促進賦形剤と共に使用できる。複数の溶解性促進賦形剤の調製で、本発明の脂質−ポリマー抱合体は好適には全溶解性促進賦形剤の少なくとも約25重量%含まれ、さらに好適には少なくとも約50重量%含まれる。親油性薬物で調製されるときには、本発明の脂質−ポリマー抱合体は好適には全溶解性促進剤と薬物の組み合わせの少なくとも約20重量%含まれ、さらに好適には少なくとも約40重量%含まれる。よって、低い溶解性促進剤と脂質との比率で、本発明の脂質−ポリマー抱合体は全溶解促進剤のさらに高い割合で含まれる。一部の代表的調合物を表5で示す。バッファ剤、水および溶解性を促進しない賦形剤(例:風味剤)はこれらの計算には含まれない。
【表5】
【0050】
ここで「脂質−ポリマー抱合体」とは本発明の抱合体を含むものだけを意味している。現存する溶解性促進剤は脂質とポリマーを含むものもあろうが、それらは本明細書中の本発明の目的に沿った脂質−ポリマー抱合体の概念には含まれない。
【0051】
リン脂質のごとき自然発生脂質とは異なり、本発明の抱合体は臨界的なミセル濃度(CMC)を有しない。ミセルは界面活性剤の濃度がCMCよりも大きく、システムの温度が臨界ミセル温度よりも高い場合のみ形成される。本発明のポリマー−脂質抱合体は任意の濃度で自然に塊物を形成する。
【0052】
水溶液中でのポリマー−脂質抱合体との薬物の調製は粒子のサイジング処理によってさらに増進できる。粒子サイズ減少のためにマイクロナイジング、ウェットミリングまたはスプレードライ等の従来技術が利用できる。好適な粒子サイズは0.05から25ミクロンであり、さらに好適には0.05から10ミクロンである。最も好適には粒子サイズは0.1から5ミクロンである。
【0053】
本発明は様々な状況で有用であり、いくつかの異なる点で従来技術に勝る。親油性薬剤の調製、溶解性および送達に関わる問題は本発明の採用により低減できる。
【0054】
脂質−ポリマー抱合体を使用した薬剤調合物は様々な方法で利用できる。水溶液内での高濃度(約20%w/w以上)にて調合物はゲル状で局部投与に適している。ゲルは経口投与または直腸投与用にカプセル化することもできる。さらに希釈した溶液の調合物は経口投与用、静脈注射用、局部塗布用および皮下注射用として有用である。
【0055】
本発明はその投与形態にかかわらず、いかなる形態の親油性薬剤であってもその溶解性を促進させるのに使用できる。本明細書においては特定の親油性薬物に言及するが、本発明は全ての親油性薬物においても有効に使用される。この脂質−ポリマー抱合体はゲル形態または希釈溶液形態で化粧品、栄養剤および栄養薬剤の製造にも使用できる。
【0056】
本発明の1形態では、本発明は親油性薬物の溶解性を促進する組成物である。この組成物は少なくとも25重量%のジアシルリピッド−ポリマー抱合体を含む。このジアシルリピッド−ポリマー抱合体は次の化学式で表される。
【化3】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含み、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0057】
本発明の別形態では、本発明は親油性活性剤と、ジアシルリピッド−ポリマー抱合体を含んだ1種以上の溶解性増加剤を含んだ薬剤組成物である。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化4】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0058】
本発明の別形態では、本発明は親油性薬物の溶解性を促進させる方法であり、この方法は、水溶液内の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化5】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0059】
本発明の別形態では、本発明は親油性薬物で哺乳対象を治療する方法であり、この方法は、水溶液中の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化6】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0060】
本発明の別形態では、本発明は親油性薬物の溶解性を促進する組成物である。この組成物は少なくとも25重量%の1以上のジアシルリピッド−ポリマー抱合体を含む。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化7】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は割合的に小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0061】
本発明の別形態では、本発明は活性剤と1種以上のジアシルリピッド−ポリマー抱合体を含んだ薬剤組成物である。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化8】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は組成物の少なくとも約20%の量を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施形態では頭部は割合的に大き過ぎてリポソームを形成させない。別実施形態では頭部は小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。組成物はさらに水溶液を含むことができる。
【0062】
本発明の別形態では、本発明は親油性薬物の溶解性を促進させる方法であり、この方法は、水溶液中の薬物をジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化9】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は割合的に小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0063】
本発明の別形態では、本発明は親油性薬物で哺乳対象を治療する方法であり、この方法は、水溶液中の薬物をジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでいる。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化10】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0064】
本発明の実施例と適用形態を解説したが、専門家であれば、本明細書で説明した本発明の範囲から逸脱せずにこれらを改良することは可能であろう。よって本発明はそれらによって限定されず、「請求の範囲」によってのみ限定される。
[実施例1]
【0065】
実施例1:リポソーム形成には大き過ぎる頭部を有したPEG−脂質抱合体による薬剤化合物の溶解性促進(予測例)
【0066】
表6に記載の化合物の存在下で増加された水溶解性を確認するために様々な疎水性薬剤化合物がテストされる。表の抱合体は化学構造2で表されるものである。
【表6】
【0067】
表6の化合物1から3は表1にもPEG−23GDM、PEG−23GDOおよびPEG−23GDLとしてそれぞれ現れる。その他の化合物のアシル基と親水性ポリマーは記載されたどのタイプのリンカーによっても骨格鎖に連結される。骨格鎖のタイプに関係なくAG1とAG2を相互に接近させておくことが望ましい。
【0068】
抱合体は室温で液体である。あるいは各薬剤化合物との予備混合前に溶解状態である。例えば、40mgの選択薬物がまず1:5のw/w比のポリマー−脂質抱合体と予備混合される。20mMのリン酸塩バッファ(pH6)の水溶液が加えられてさらに混合され、全部で2mLの溶液が得られる。分割量の溶液の薬物濃度が測定される。すべての場合において抱合体の存在により溶液内の薬物濃度は抱合体のない場合の溶液の薬物濃度よりも高くなる。
【0069】
試験される化合物はアンホテリシンB、イトラコナゾール、ポサコナゾール、シクロスポリンおよびインドメタシンである。
【0070】
この薬剤化合物の存在下あるいは非存在下の両方でポリマー−脂質抱合体の水溶液の物理形態が検査される。10%未満の抱合体または抱合体/薬物複合体がリポソームの形態である。
[実施例2]
【0071】
実施例2:リポソーム形成には小さ過ぎる頭部を有したPEG−脂質抱合体による薬剤化合物の溶解性促進(予測例)
【0072】
表7に掲載された化合物の存在下で増加された水溶解性を確認するために様々な疎水性薬剤化合物がテストされる。表の抱合体は化学構造2で表されるものである。
【表7】
【0073】
アシル基と親水性ポリマーは掲載されている任意のリンカーを使用して骨格鎖に連結できる。しかし、これら実施例では頭部のサイズを規定することが必須であるため、リンカーは延伸骨格鎖の全分子量を約250未満に保つことが重要である。骨格鎖のタイプに関係なくAG1とAG2を互いに接近させておくことが望ましい。
【0074】
抱合体は室温で液体である。あるいは各薬剤化合物との予備混合前に溶解状態である。例えば、40mgの選択薬物がまず1:8のw/w比のポリマー−脂質抱合体と予備混合される。20mMのリン酸塩バッファ(pH6)の水溶液が加えられてさらに混合され、全部で2mLの溶液が得られる。分割量の溶液の薬物濃度が測定される。すべての場合に抱合体の存在により溶液内の薬物濃度は抱合体が存在しない溶液の薬物濃度よりも高くなる。
【0075】
試験される化合物はアンホテリシンB、イトラコナゾール、ポサコナゾール、シクロスポリンおよびインドメタシンである。
【0076】
薬剤化合物の存在下あるいは非存在下の両方においてポリマー−脂質抱合体の水溶液の物理形態が検査される。10%未満の抱合体または抱合体/薬物複合体がリポソームの形態である。
[実施例3]
【0077】
実施例3:抗癌薬物経口溶液
【0078】
ゲフィチニブの経口送達のために適したポリマー−脂質抱合体が調製された。ポリマー−脂質がミキサープロペラを備えた容器に加えられた。薬剤物質は常時混合しながら加えられた。混合は薬物が脂質内に視覚的に分散するまで継続された。水中の予備溶解された賦形剤が容器にゆっくりと加えられ、適切に混合された。混合は完全に均質な溶液となるまで継続された。サンプル調剤は表8に記載されている。
【表8】
【0079】
PEG−脂質抱合体はDDGBであるか、または表6あるいは表7に掲載されている任意の抱合体でよい。水酸化ナトリウムが純水中の10%w/wの溶液の調製に使用される。目標のpHは4.0から7.0である。必要であればNaOH溶液がpHの調整に使用される。薬物と脂質(すなわち溶解性促進剤)の比は好適には約1:20よりも大きく、さらに好適には約1:5よりも大きい。有機酸は酪酸またはピルビン酸あるいはグリコール酸でよい。なかでも酪酸が最も好適である。有機酸の濃度は好適には1%から10%であり、さらに好適には約2%から5%である。
[実施例4]
【0080】
実施例4:抗癌薬物IV注射溶液
【0081】
IV溶液が実施例3と同様に調製された。しかし人工風味料や保存剤は加えられなかった。目標のpH範囲は6.5から7.5であった。サンプル調剤は表9に掲載されている。
【表9】
【0082】
PEG−脂質抱合体はDDGBであっても、または表6あるいは表7の任意の抱合体であってもよい。アシル基はカルボキシ結合によりグリセロール骨格鎖に取り付けられる。水酸化ナトリウムが純水中で10%w/wの溶液の調製に使用される。目標のpHは4.0から7.0の範囲である。必要であればNaOH溶液がpHの調整に使用される。薬物と脂質の比は約1:20よりも大きく、さらに好適には約1:5よりも大きい。有機酸は酪酸またはピルビン酸あるいはグリコール酸でよい。なかでも酪酸が最も好適である。有機酸の濃度は好適には1%から5%であり、さらに好適には約1%から3%である。
[実施例5]
【0083】
実施例5:抗菌局部クリーム
【0084】
PEG−脂質抱合体がプロペラタイプの混合ブレードを備えたステンレス鋼容器に加えられた。常時混合しながら薬剤物質が加えられた。混合は60℃から65℃の温度で薬物が脂質内で視覚的に分散するまで継続された。有機酸、コレステロールおよびグリセリンが混合しながら加えられた。エタノールとエチオキシジグリコールが混合しながら加えられた。最後に、カルボポルETD2020、純水およびトリエチルアミンが混合しながら加えられた。混合は完全に均質なクリームとなるまで継続された。調剤は表9に記されている。
【表10】
【0085】
薬物はポサコナゾール、エクアコナゾール、イトラコナゾール、テルビナフィンおよびメトロニダゾールを含む抗カビ剤でも殺菌剤でもよい。抱合体は表6または表7に掲載されたどの抱合体であってもよい。有機酸は酪酸、ピルビン酸あるいはグリコール酸でよい。必要であれば水酸化ナトリウムがpHの調整に使用できる。目標のpH範囲は3.5から7.0である。
[実施例7]
【0086】
実施例7:抗菌局部溶液
【0087】
活性剤がまず有機酸とエタノールに溶解されたこと以外は局部用溶液が実施例6と同様に調製された。サンプル調剤は表11で解説されている。
【表11】
【0088】
抱合体は表6または表7に記載のどの抱合体であってもよい。有機酸は酪酸、ピルビン酸あるいはグリコール酸でよい。必要であれば水酸化ナトリウムがpHの調整に使用できる。目標のpH範囲は3.5から7.0である。
[実施例8]
【0089】
実施例8:合成3,4−ジ(オレオイロキシ)−デカエチレングリコール−安息香酸(DDGB)
【0090】
250mLのベンゼン中の0.10モルの3,4−ジヒドロキシ安息香酸と0.1モルの無水ピリジンの溶液が、250mLのベンゼン中の0.22モルの新鮮蒸留塩化オレオイルの氷冷溶液に無水条件下で撹拌しながらゆっくりと加えられ、混合物は無水条件下で20時間にわたって40℃で保存された。冷却後、450mLのクロロホルムが混合物に加えられ、その後に濾過された。溶媒は真空内で蒸発され、白色の未精製固体が得られた。この固体はさらに次のように純化できる。未精製界面活性剤はシリカゲル(G60、グレード60、メッシュ240〜400)カラムでクロマトグラフィにより純化される。このカラムは1:1(v/v)のジエチルエーテル/アセトン混合物で溶離される。溶離された第1部分は非反応試薬であり、廃棄される。この部分は254nmのピーク(UVモニター)で回収された。真空での溶媒の乾燥によって約70%の3,4−ジ(オレオイロキシ)安息香酸が得られた。
【0091】
0.2モルの3,4−ジ(オレイロキシ)安息香酸と、0.2モルのデカエチレングリコールと、0.21モルのp−塩化トルエンスルホニルと、0.11モルの4−ジメチルアミノピリジンと、0.5モルのテトラブチルアンモニウム硫酸水素と、0.14モルの炭酸カリウムの混合物が600mLのテトラヒドロフラン内で縣濁された。この反応混合物は50℃で22時間にわたって撹拌された。反応混合物は室温に冷却され、1500mLの水内に注がれ、300mLの塩化メチレンが加えられた。溶液は希塩酸で酸化され、pH1に調整された。有機層は分離され、350mLの水で5回洗浄され、硫化マグネシウムで乾燥された。溶媒は真空下で除去され、残留物は200mLのエタノールで再溶解された。無色の溶液は4℃で12時間にわたって放置され、または白色沈降物が形成されるまで放置された。真空下で沈降物の濾過により約80%の最終製品が得られた(化学構造3)。
【化11】
3,4−ジ(オレオイロキシ)デカエチレン(PEG−12)安息香酸グリコール
化学構造3
[実施例9]
【0092】
実施例9:ゲフィチニブ製剤の薬剤動態特性および生体内利用性
【0093】
静脈注射と経口投与の後のゲフィチニブ製剤の血液内量を決定するために実験が実施された。比較のためにゲフィチニブ製剤も試験された。3匹の雄マウス(B6D2F1)の群が研究に使用された。ボーラスIV(静脈)注射後、典型的には0時間、0.5時間、1時間、2時間、4時間、7時間、16時間、および24時間後に得られたヘパリン添加されたマウス血清サンプルに対してHPLC−MS分析が実行された。経口給餌後、分析は0時間、0.5時間、1時間、2時間、4時間、7時間、16時間、および24時間後に行われた。各薬物の溶解量を決定するため、薬物はまずサンプル前処理した血清から単離された。アセトニトリルが使用され、サンプル内のタンパク質が除去された。無勾配JPLC−MS/M法が利用され、薬物が可能な妨害物から分離された。複数の反応モニタリング(MRM)モードで薬物量はMS検出により測定された。ウィンノンリンプログラム(5.2版、Pharsight)分析区分モデルの分析を活用してPKデータが分析された。その結果、本発明の化合物の調剤がクレモフォールEL(登録商標)よりも優れたPK特性を有することが証明された。
【0094】
図5は、(1)10mMのリン酸ナトリウムバッファ(pH6.0)中の6%のPEG−12GDOと、(2)10mMのリン酸ナトリウムバッファ(pH6.0)中の6%のDDGBと、(3)10mMのリン酸ナトリウムバッファ(PpH6.0)中の15%のクレモフォールEL(登録商標)のゲフィチニブマウスの経口PK特性を示す。この薬物は経口投与され、投薬濃度は25mg/kgであった。薬物は静脈注射でも投与され、投薬濃度は15%のクレモフォールEL(登録商標)および10mMのリン酸ナトリウムバッファ(pH6.0)中で25mg/kgであった。静脈内投与から得られたAUC(31.3)は相対生物利用性(BA)の計算に使用された。生物利用性はGDO−12,DDGBおよびクレモフォールEL(登録商標)に対してそれぞれ69.4%、56.0%および28.5%であった。
[実施例10]
【0095】
実施例10:固体投与調合物
【0096】
本発明の抱合体は非水溶性薬物の固体経口投与調合物の調製に有効である。この点で抱合体または抱合体の混合物は薬剤物質と混合される。その後、さらに高い融点を有した賦形剤が導入される。賦形剤は適した溶媒中で溶解する。最終混合物は冷却され、溶媒が除去されて混合物はカプセル形態、錠剤形態、等々に形成される。
【0097】
固体投与調合物を調製するために抱合体(全量の約42%)が混合ブレードを備えたステンレス鋼容器に加えられる。薬剤物質(全量の約15%)が混合しながら加えられる。加熱下での混合は薬物が事実上完全に分散するまで継続される。別の容器にはd−α−トコフェリルプロピレングリコール−1000琥珀酸塩(TPGS−VE)または類似化合物(全量の約42%)がエタノール(全量の約1%)内に溶解され。その後に前記のステンレス鋼容器に加えられる。混合は均質な溶液が得られるまで継続される。真空下でエタノールは除去される。
【技術分野】
【0001】
優先権主張
本願は2009年1月23日に出願された米国仮特許願61/205840「薬剤化合物の溶解性を促進させるPEG−脂質抱合体」の優先権を主張する。
【0002】
発明の分野
本発明は体内送達を増進するために薬剤の溶解性を促進させる組成物およびその方法に関する。さらに特定するならば、本発明はそのような増加した溶解性と送達とを提供するジアシルリピッド(脂質)−ポリマー(重合体)抱合体(結合体、複合体)に関する。
【背景技術】
【0003】
発明の背景
薬剤科学においては疎水性薬剤化合物の作用部位への送達は現在においても研究対象の問題である。利用されている薬物送達ビヒクルの種類には、シクロデキストラン、薬物−脂質複合物、リポソームおよびクレモフォール(Cremophor)(登録商標)のごとき溶解剤が含まれる。様々なPEG−脂質抱合体がこれらビヒクルに利用されてきた。しかしながら、多くの有用な薬物は、その溶解性、薬物動態特性および生体内利用性によって未だに限定を受けている。よって、本発明の1目的は、親油性剤を調製および送達する新規な組成物並びに方法を提供することである。
【発明の概要】
【課題を解決するための手段】
【0004】
発明の概略
ジアシルリピッド−ポリマー抱合体は水溶液内で親油性薬物の溶解性を促進するために利用される。この抱合体は骨格鎖(主鎖)と、2個の親油性アシル基と、親水性ポリマーとを含む。
【図面の簡単な説明】
【0005】
添付図面は本明細書の一部であり、本発明の実施例を図示しており、以下の詳細な説明と共に本発明の原理および実施形態を説明するものである。
【0006】
以下は図面の要約である。
【0007】
【図1】図1は脂質分子で構成されたリポソームの断面を図示する概略図である。
【図2】図2は脂質分子で構成されたミセルの断面を図示する概略図である。
【図3】図3は頭部(ヘッド基)と較べて大きな尾部を有した脂質分子で構成された構造物の断面を図示する概略図である。
【図4】図4は極性頭部と非極性炭化水素鎖を有した脂質分子の空間詰概略図である。
【図5】図5はゲフィチニブ調合物の薬物動態特性を示す。
【発明を実施するための形態】
【0008】
詳細な説明
以下では薬剤の溶解性を促進し、薬剤の送達を増進するためのジアシルリピッド−ポリマー抱合体の形態による本発明の実施態様が説明されている。添付図面で図示されているごとき本発明の実施態様を詳細に説明する。図面および以下の詳細な説明を通して同一参照符号(番号)は同一要素または類似要素を表す。
【0009】
簡略性を高めるため、本発明の実施態様で現れる全ての型通りの特徴を一々図示および解説してはいない。もちろん、そのような実際の利用形態の開発において開発者の特定目標、例えば利用形態およびビジネス関連に動機付けられた目標を達成するためには多数の実施形態特有の決定が必要であることは理解されよう。また、これらの特有目標は実施形態によって異なり、開発者によっても異なることも理解されるであろう。さらに、このような開発努力は複雑で複合的なものであり、開発には長時間を要するが、いずれにせよ本明細書の開示内容を活用することで当該技術分野の当業者によって自身の通常の研究活動に利用できるであろう。
【0010】
当該分野の当業者であれば本発明の以下の解説が単に本発明の説明を目的としており、本発明を限定するものではないことを理解するであろう。本発明の他の実施態様も、ここでの開示の恩恵を受ける当業者には容易に理解できよう。
【0011】
本発明は薬剤組成物と、その薬剤組成物を調合して使用する方法とを含む。この薬剤組成物は親油性薬物並びに1以上のポリマー−脂質抱合体を含んで成る。この抱合体は骨格鎖、2個のアシル基およびポリマー(重合体)鎖を有している。好適には、それらはヒトの体温よりも低い溶解温度を有し、さらに好適には室温では液体である。薬物を含んだ抱合体の水溶液はリポソームを含まないが、その代わりに様々な微小構造物を含んでいる。
【0012】
定義:ここで使用される“親油性”とは、リピッド(脂質)内で溶解する性能及び/又は生体膜に浸透し、生体膜と相互作用し、及び/又はトラバースすることができる性能のことである。
【0013】
米国特許6610322およびその関連特許文献は、ジアシルリピッド−ポリエチレングリコール(PEG)抱合体を採用することで自然発生性リポソームの形成を教示する。この特許は、その抱合体を水溶液に加えるだけでリポソームを形成するこのようなPEG−脂質抱合体をいかに選択するかを解説する。他の類似したポリマー−脂質分子はリポソームを形成せずに疎水性薬物を溶解させるのに有用であることが発見されている。
【0014】
米国特許6610322が解説するように、所定の条件が満たされるとPEG−脂質抱合体は水溶液と混合されて自然発生的にリポソームを形成する。ジアシルグリセロール−PEG(DAG−PEG)においては、1つの条件は、全脂質(単一脂質または複数の脂質の組み合わせ)のパッキング(充填)パラメータがリポソームを形成させる範囲内に存在しければならないことである。簡単に述べれば、脂質は一般的に円錐形状であって、自動的にリポソームが構成されるようにテーパー状でなければならない。もし頭部(PEGを含む)が疎水性部分または疎水性分子に比して大き過ぎたり小さ過ぎたりすればリポソームは形成されない。
【化1】
化学構造1
【0015】
化学構造1は本発明のポリマー−脂質抱合体の一般形状である。AG1とAG2はアシル基である。すなわち分子の疎水性部分である。これらアシル基は同じであっても異なっていてもよい。Pは親水性ポリマーであり、典型的にはPEGである。Rは親水性ポリマーの先端のモイエティ(部分)である。Bは骨格鎖である。L1は骨格鎖を親水性ポリマーに接続するリンカーである。L2とL3は骨格鎖をアシル基に接続するリンカーである。Bと、L1、L2およびL3を含んだ分子部分は延伸骨格鎖とも考えられる。延伸骨格鎖並びにPおよびRは分子の親水性頭部または親水性頭部基を含む。アシル鎖は親油性尾部を含む。
【0016】
脂質−ポリマー抱合体の両親媒性は水溶液中での親油性薬物の溶解性を促進する性能を与える。アシル基はその薬物と協調し、頭部は水と自由に作用する。骨格鎖とリンカーは一定種類の薬物を溶解させ、及び/又は送達する利点を提供するように選択できるが、骨格鎖およびリンカーは一般的に親水性ポリマーやアシル基よりも機能的には重要性が低い。よって抱合体の単純化バージョンが化学構造2で説明できる。このEBは延伸骨格鎖である。
【化2】
化学構造2
【0017】
リポソームを形成するため、脂質頭部と炭化水素鎖は自動的に、自身を折畳ませるような曲率半径を備えた二重層にならなければならない(図1参照)。もし炭化水素鎖(アシル基)が頭部に較べて小さ過ぎれば、曲率半径は小さ過ぎてミセルが発生するであろう(図2参照)。もし炭化水素鎖が頭部に較べて大き過ぎれば、曲率半径は正負が逆になり、リポソームは形成できない(図3参照)
【0018】
図1は脂質分子で構成されたリポソームの概略断面図である。リポソーム10は水性空間20を囲む脂質分子(例:12、14、16、18)で成る脂質二重層を含む。
【0019】
図4は極性頭部と非極性炭化水素鎖とを有した脂質分子の空間充填図である。脂質分子22は親水性基24と疎水性尾部26とを含んでいる。疎水性尾部26は2つの炭化水素鎖32、34(AG1とAG2)を含んでいる。その化学結合は脂質分子をフレキシブルにしているが、頭部は直径28の領域を満たしており、尾部は直径30の領域を満たしている。脂質分子はリポソームを形成するために二重層内で構成されなければならないため、リポソームは、頭部径と尾部径との比が大き過ぎても小さ過ぎても形成されない。
【0020】
図2は脂質分子で構成されたミセルの概略断面図である。ミセル36は脂質分子(例:38、40)を含む。脂質分子の尾部は頭部に較べて小さな径であるため、脂質分子は小曲率半径を備え、二重層は形成されない。
【0021】
図3は頭部に較べて大きな尾部を備えた脂質分子で成る構造物の概略断面図である。図3で示すように、頭部に較べて大きな尾部を備えた脂質(例:44)が水溶液と混合されると構造物42が形成される。頭部と尾部とサイズ比は二重層の形成を不可能にしている。
【0022】
図1、図2および図3は1種の脂質分子を使用して充填パラメータの基本原理を図示したが、同一原理が脂質の混合物に適用できることは理解されよう。例えば、小さ過ぎて単種としてのリポソームを形成できない炭化水素鎖を有した脂質はコレステロールと混合することができ、適した充填パラメータを有した組成物が得られる。別例では、自身が適した充填パラメータを有している脂質は、自身は適した充填パラメータを有していない限定量の他の脂質を取り込んだリポソームを形成できる。単一脂質および複数脂質の混合物の両方は公知方法で計算できる充填パラメータを有する。一般的にリポソームを形成させるリポソーム組成物は、約0.84から0.88の間の充填パラメータ測定値Paと、約0.88と0.93の間の充填パラメータ測定値Pvを有する。
【0023】
Paは表面に関する充填パラメータであり、Pvは体積に関する充填パラメータである(DDラシック著、リポソーム:「物理学から利用まで」エルセビエル誌、51ページ、1993年)。これらパラメータは式HCa/Ta≡PaおよびHCv/Tv≡Pvから導かれる。このHCaは炭化水素鎖面積であり、Taは全分子面積であり、HCvは炭化水素鎖の体積であり、Tvは全分子の体積である。
【0024】
脂質混合物のための充填パラメータが計算できる。なぜなら脂質の理想的な混合は個々の特性の計算平均となるからである。例えば、理想混合の場合には二元混合のHCa/Ta≡Paは次の式になる。
<Pa>=X1P1+X2P2、X1+X2=1
【0025】
さらに一般化すれば、所定の混合物を含んだi脂質の場合には次の式で表される。
<Pa>=ΣiXiPi及びΣiXi=1
このXiは混合物内の脂質のモル分率であり、Piはその脂質の表面に関する充填パラメータである。
【0026】
2つの一般的な要因が脂質分子の頭部サイズに影響を及ぼす。その1つは頭部の実際の物理的サイズである。例えば、さらに長いPEG鎖の採用は頭部をさらに大きくする。もう1つは頭部に関わる電荷である。例えば、PEG鎖がリン酸ジエステル結合により骨格鎖に抱合するなら、リン酸塩は頭部に電荷を分与し、そのサイズを効果的に増加させるであろうが、そのようなことは起こらない。本発明では、非リン脂質が好適であり、頭部サイズを変える一般的手段はポリマー長を変えること、並びに骨格鎖とリンカーを変えることである。
【0027】
脂質の尾部のサイズは炭化水素鎖の長さと、脂質鎖の飽和度によって影響を受ける。2個の鎖を備えたリピッドを含んだリポソームには組み込めるが、単鎖脂質は一般的にリポソームを形成しない。同様に1個の長鎖と1個の短鎖とを備えた脂質は相対的に小さな尾部サイズを有するであろう。
【0028】
ステロール及び/又は薬剤化合物の存在は予想されるようにリポソームの形成に影響を及ぼすであろう。これら全ての要因は、単一ポリマー−脂質抱合体がリポソームを形成するか否か、またはポリマー−脂質抱合体を含んだ化学的組み合わせが水溶液内でリポソームを形成するか否かを計算して予測する際に考察しなければならない。
【0029】
場合によっては、リポソームは好適な薬物送達ビヒクルであるが、多くの親油性薬物はリポソーム縣濁剤に調合することが困難であることが知られている。問題とは、保存、生体内安定性、薬物と脂質の比率その他である。本発明のポリマー−脂質抱合体を含んだ非リポソーム調合物は多くの場合に有用であり、特定の場合には非常に有効である。
【0030】
米国特許6610322で解説されている充填パラメータに基づいてリポソームを形成するために、PEG−脂質抱合体を選択することが可能であるのと同様に、ポリマー−脂質抱合体の純水溶液またはリポソームを形成しないポリマー−脂質抱合体を含んだ混合物を選択することは可能である。このような選択はポリマー−脂質抱合体の充填パラメータを計算することで数学的に実施できる。この選択は、様々なポリマー−脂質抱合体を試験することで経験的に実行できる。組み合わせる取り組みも利用できる。ここでは充填パラメータに影響を及ぼす要因の知識が活用され、リポソーム形成のための試験を実施する前に充填パラメータを厳格に計算することなく、どのような抱合体が利用可能であるのかを予測する。
【0031】
表1は本発明での利用性を確認するために単一脂質として試験された複数の脂質を示す。この表は米国特許6610322の欄6の表と同じである。脂質は水溶液中にて2重量%の状態で試験された。GDLとはグリセロールジラウリン酸であり、GDOとはグリセロールジオレイン酸であり、GDMとはグリセロールジミリスチン酸であり、GDPとはグリセロールジパルミチン酸であり、GDSとはグリセロールジステアリン酸である。各脂質に対して「PEG」の後の数字はPEG鎖内のC2H4Oサブユニットの数を示す。不飽和ジオレイン酸脂質は飽和ジミリスチン酸脂質と類似した充填パラメータを有する。
【表1】
【0032】
表1はいくつかのポリマー−脂質抱合体のリポソーム形成特性を示す。この表を理解するには、自然リポソーム形成には、脂質または脂質混合物がリポソーム形成温度において流体でなければならないことを知っている必要がある。例えば、PEG−12GDMは試験した全ての温度でリポソームを自然形成する。なぜなら、そのような温度ではリピッドは流体であり、特定の範囲内で充填パラメータを有することに加え、PEGを含むからである。同様に、PEG−12GDO(PEG−12GDMとほぼ同一特性)は試験された全温度でリポソームを自然形成する。高融点を備えたPEG−23GDSは39.8℃の融点以上でリポソームを形成するだけである。
【0033】
好適には本発明のポリマー−脂質抱合体は、人体温度、すなわち約37℃未満の温度で液体となる溶融温度を有する。特に好適には、この抱合体は室温未満、すなわち約25℃未満の溶融温度を有する。充填パラメータの場合と同様に溶融温度は脂質の混合物のために決定できる。
【0034】
調製の容易性を考慮すればさらに低い溶融温度の抱合体が好適である。しかしながら、さらに高い温度で調合物を調製することで、さらに高い溶融温度の抱合体でも利用できる。
【0035】
GDMシリーズの脂質は充填パラメータの重要性を解説する。これら脂質は全てPEGを含んでおり、60℃で液体であり、好適な充填パラメータのおかげでPEG−12GDMだけがリポソームを形成する。一連のDGS脂質は60℃で同一ポイントを示す。
【0036】
当該技術の専門家であれば、リポソームを形成しないポリマー−脂質抱合体の組成物を予測して造りだすために充填パラメータの知識を活用して本発明を実施することができる。例えば、PEG鎖のサイズを変え、または脂質組成物内のPEG含有脂質の濃度を変えると、予測可能な状態で充填パラメータが変わる。同様に骨格鎖及び/又はリンカーの変更も充填パラメータの予測可能な変化をもたらす。ポリマー鎖の長さを変え、及び/又はアシル鎖の長さと飽和状態を変えることで溶融温度も変えることが可能である。
【0037】
いくつかの基本的な条件が満たされている限りは、多様なポリマー−脂質抱合体が本発明の実施に有効である。まず抱合体は疎水性ポリマーを含まなければならない。次に、抱合体は親油性薬物と関連するために2つのアシル鎖を含まねばならない。さらに、リポソームが水溶液内で優勢とならないように調剤の充填パラメータを選択しなければならない。アシル鎖と親水性ポリマーは、溶融温度と充填パラメータに関し、抱合体の化学特性に対して支配的となる傾向はあるものの、骨格鎖も重要である。
【0038】
水溶液を使用して本発明を実施するとき、調合物によっては一部がリポソーム形態であることはあり得る。しかし、リポソームはせいぜい調合物のほんの一部を構成するだけである。多くの場合に、このリポソームは非常に少量である。一般的に本発明の非リポソーム調合物は、希釈水溶液内で10%未満のリポソーム形態の脂質−ポリマー抱合体を有している。すなわちリポソームは抱合体の支配的な構造形態ではないということができる。
【0039】
有用な抱合体は次の2つの範疇に分類できる。すなわち尾部と比較して頭部が大き過ぎるのでリポソームを形成しないものと、尾部と比較して頭部が小さ過ぎるのでリポソームを形成しないものとである。本発明の目的は水溶液中にて親油性化合物の溶解性を促進することであるため、第1の範疇が一般的には好まれる。なぜならこの化合物は水溶要素の割合が高いからである。しかしながら、さらに大きな親油性尾部を有した抱合体は、さらに小さな尾部を有した抱合体よりもさらに多量に薬物を運搬できる。よって、好適なアシル基は少なくとも8個の炭素原子を有する。
【0040】
有用なアシル基(AG1およびAG2)は表2と表3に掲載されたものを含む。
【表2】
【表3】
【0041】
グリセロールは好適な骨格鎖であるが、他の有用な骨格鎖(B)には、3−アミノ−1,2−プロパンジオール、1−アミノ−2,3−プロパンジオール、2−アミノ−1,3−プロパンジオール、3−メルカプト−1,2−プロパンジオール、1−メルカプト−2,3−プロパンジオール、2−メルカプト−1,3−プロパンジオール、ジヒドロキシブチル酸およびグリセリン酸等の3−炭素鎖が含まれる。あるいは、糖類が骨格鎖として利用できる。可能な糖類骨格鎖には単糖類および二糖類が含まれる。骨格鎖は4−炭素鎖または2−炭素鎖も含むことができる。骨格鎖はN(1個の窒素原子)またはCHで、AG1、AF2およびPがそれらCまたはNの骨格鎖に結合したような単純なものでよい(この場合にはリンカーは共有電子対であると考えられる)。さらにアミノ酸が骨格鎖として利用できる。可能なアミノ酸骨格鎖には、システイン、セリン、トレオニンおよびアスパラギンが含まれる。環式プロパン等の環式アルカン、およびアゾールのごとき複素環式化合物も骨格鎖として利用できる。
【0042】
芳香族化合物の特性は特別な配慮を要するが、ベンゼン等の芳香族化合物も骨格鎖として利用できる。まず芳香環は大きくて頑丈であり、頭部の相対サイズを増加させるであろう。次に、芳香環は、典型的には抱合体の親水性部分で親油性である。続いて、芳香環間のスタッキングは予測不能に微小構造の形成に影響を及ぼすであろう。しかし、これらユニークな特性は状況によっては有利に作用する。
【0043】
本発明のポリマー−脂質抱合体を形成する際に有用な親水性ポリマーには、ポリエチレングリコール(PEG)および他のポリアルケン酸化物ポリマー、ポリオキシエチレンアルキルエーテル、ポリビニルピロリドン、ポリ(アリルアミン)、ポリ(1−グリセロールメタクリレート)、ポリ(2−エチル−2−オキサゾリン)、ポリ(2−ヒドロキシエチルメタクリレート/メタクリル酸)/ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(2−ビニルピリジン)、ポリ(アクリルアミド/アクリル酸)、ポリ(アクリル酸)、ポリ(ブタジエン/マレイン酸)、ポリ(エチルアクリレート/アクリル酸)、ポリ(エチレン酸化物−b−プロピレン酸化物)、ポリ(エチレン/アクリル酸)、ポリ(メタクリル酸)、ポリ(マレイン酸)、ポリ(N−イソ−プロピルアクリルアミド)、ポリ(N−ビニルピロリドン/ビニルアセテート)、ポリ(スチレンスルフォン酸)、ポリ(スチレンスルフォン酸/マレイン酸)、ポリ(ビニルアセテート)、ポリ(ビニルリン酸)、ポリ(ビニルアミン)、ポリアクリルアミド、ポリアクリル酸、ポリアニリン、ポリエチレンイミン、プルラン、ポリメタクリルアミドが含まれる。上記のリストに基づいた共重合体とブロック共重合体も利用できる。遊離ポリマーは室温で水溶解性であり、無毒でもある。それらは哺乳動物内に目立った免疫遺伝レスポンスを導き出さない。狭い分子量分布の親水性ポリマーが好適である。製薬ビジネスで既に利用が認められているため、PEGは好適な親水性ポリマーである。
【0044】
様々なR基をポリマーの先端に含むことができる。有用なR基にはアルキル基が含まれ、例えばメチル、アルコキシモイエティ、アミノ酸、および、1、2、3、4、あるいはそれ以上の単糖ユニットをそれぞれ含んだ単糖類、二糖類、三糖類、および多糖類等の糖類が含まれる。さらに抗体部分のごとき標的モイエティ(部分)とビタミン類がR基として利用できる。一般的にR基は非常に水溶解性である。R基の分子量は好適には約200未満であり、さらに好適には約50未満である。大抵の利用においては、血流内で非特定結合や短縮循環時間を回避するためにR基は好適には陽イオン性ではない。しかし、陽イオン性R基は、局部用ゲルおよび口および喉を標的とした口内溶液のごとき投与形態では好適に利用できる。
【0045】
有用なリンカー(L1、L2、L3)にはオキシ、エステルおよびカルボキシル並びに表4で示すものが含まれる。
【表4】
【0046】
リンカーは特定の利用目的で本発明をカスタマイズするように選択できる。例えば、低pHで変動性が高いリンカーは適用形態によっては好適である。他の場合には、溶解した薬剤化合物の一部と水素結合する能力を有するリンカーが有利であろう。しかしながら、大抵の場合には、オキシ、チオール、およびカルボキシ等の単純なリンカーが適している。
【0047】
本発明では広範囲な脂質−ポリマー抱合体が有用である。前述したようにPEGを親水性ポリマーとして有することが望まれる。他の好適実施態様には好適な分子量範囲の抱合体を利用することが含まれる。相対的に大きな頭部の抱合体を使用するときには、延伸骨格鎖のMWが約105から250の範囲であり、P+RのMWがアシル基の組み合わせMWよりも大きいのが好適である。本発明の抱合体は約8から約13の親水性−親油性バランス率(レーティング)を有する。
【0048】
ラブラソール(登録商標)とラブラフィル(登録商標)のごとき現存する溶解性促進剤はPEGを杏ピット油のごとき天然グリセリド混合物と抱合させることで製造される。これでかなり広範囲な構成成分との溶解性混合物となる。一方、本発明の溶解性促進剤は別個の工程で合成され、純粋で明確な調合物が得られる。このように制御することで本発明を特定の親油性薬物を溶解させるべく使用するときに技術者に個別の調整をさせる。場合によっては特定の親油性薬剤のための溶解性促進剤は、本発明の単一で化学的に純粋な脂質−ポリマー抱合体でよい。
【0049】
本発明の抱合体は他の溶解性促進賦形剤と共に使用できる。複数の溶解性促進賦形剤の調製で、本発明の脂質−ポリマー抱合体は好適には全溶解性促進賦形剤の少なくとも約25重量%含まれ、さらに好適には少なくとも約50重量%含まれる。親油性薬物で調製されるときには、本発明の脂質−ポリマー抱合体は好適には全溶解性促進剤と薬物の組み合わせの少なくとも約20重量%含まれ、さらに好適には少なくとも約40重量%含まれる。よって、低い溶解性促進剤と脂質との比率で、本発明の脂質−ポリマー抱合体は全溶解促進剤のさらに高い割合で含まれる。一部の代表的調合物を表5で示す。バッファ剤、水および溶解性を促進しない賦形剤(例:風味剤)はこれらの計算には含まれない。
【表5】
【0050】
ここで「脂質−ポリマー抱合体」とは本発明の抱合体を含むものだけを意味している。現存する溶解性促進剤は脂質とポリマーを含むものもあろうが、それらは本明細書中の本発明の目的に沿った脂質−ポリマー抱合体の概念には含まれない。
【0051】
リン脂質のごとき自然発生脂質とは異なり、本発明の抱合体は臨界的なミセル濃度(CMC)を有しない。ミセルは界面活性剤の濃度がCMCよりも大きく、システムの温度が臨界ミセル温度よりも高い場合のみ形成される。本発明のポリマー−脂質抱合体は任意の濃度で自然に塊物を形成する。
【0052】
水溶液中でのポリマー−脂質抱合体との薬物の調製は粒子のサイジング処理によってさらに増進できる。粒子サイズ減少のためにマイクロナイジング、ウェットミリングまたはスプレードライ等の従来技術が利用できる。好適な粒子サイズは0.05から25ミクロンであり、さらに好適には0.05から10ミクロンである。最も好適には粒子サイズは0.1から5ミクロンである。
【0053】
本発明は様々な状況で有用であり、いくつかの異なる点で従来技術に勝る。親油性薬剤の調製、溶解性および送達に関わる問題は本発明の採用により低減できる。
【0054】
脂質−ポリマー抱合体を使用した薬剤調合物は様々な方法で利用できる。水溶液内での高濃度(約20%w/w以上)にて調合物はゲル状で局部投与に適している。ゲルは経口投与または直腸投与用にカプセル化することもできる。さらに希釈した溶液の調合物は経口投与用、静脈注射用、局部塗布用および皮下注射用として有用である。
【0055】
本発明はその投与形態にかかわらず、いかなる形態の親油性薬剤であってもその溶解性を促進させるのに使用できる。本明細書においては特定の親油性薬物に言及するが、本発明は全ての親油性薬物においても有効に使用される。この脂質−ポリマー抱合体はゲル形態または希釈溶液形態で化粧品、栄養剤および栄養薬剤の製造にも使用できる。
【0056】
本発明の1形態では、本発明は親油性薬物の溶解性を促進する組成物である。この組成物は少なくとも25重量%のジアシルリピッド−ポリマー抱合体を含む。このジアシルリピッド−ポリマー抱合体は次の化学式で表される。
【化3】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含み、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0057】
本発明の別形態では、本発明は親油性活性剤と、ジアシルリピッド−ポリマー抱合体を含んだ1種以上の溶解性増加剤を含んだ薬剤組成物である。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化4】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0058】
本発明の別形態では、本発明は親油性薬物の溶解性を促進させる方法であり、この方法は、水溶液内の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化5】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0059】
本発明の別形態では、本発明は親油性薬物で哺乳対象を治療する方法であり、この方法は、水溶液中の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化6】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pはポリエチレングリコールであり、Rは陽イオンではなく、AG1とAG2は共に抱合体の親油性尾部を含んでおり、B、L1、L2、L3、PおよびRは共に抱合体の親水性頭部を含んでおり、この頭部は尾部に較べて大き過ぎ、抱合体の純粋な水溶液に支配的にリポソームを形成させない。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。骨格鎖は好適にはグリセロールまたは糖類である。
【0060】
本発明の別形態では、本発明は親油性薬物の溶解性を促進する組成物である。この組成物は少なくとも25重量%の1以上のジアシルリピッド−ポリマー抱合体を含む。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化7】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は割合的に小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0061】
本発明の別形態では、本発明は活性剤と1種以上のジアシルリピッド−ポリマー抱合体を含んだ薬剤組成物である。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化8】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は組成物の少なくとも約20%の量を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施形態では頭部は割合的に大き過ぎてリポソームを形成させない。別実施形態では頭部は小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。組成物はさらに水溶液を含むことができる。
【0062】
本発明の別形態では、本発明は親油性薬物の溶解性を促進させる方法であり、この方法は、水溶液中の薬物をジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化9】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は割合的に小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0063】
本発明の別形態では、本発明は親油性薬物で哺乳対象を治療する方法であり、この方法は、水溶液中の薬物をジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでいる。このジアシルリピッド−ポリマー抱合体は次の式で表される。
【化10】
AG1とAG2はアシル基であり、L1、L2およびL3はリンカーであり、Bは骨格鎖であり、Pは親水性ポリマーであり、Rは約200未満の分子量であり、ジアシルリピッド−ポリマー抱合体はリポソームを形成させない充填パラメータを有している。抱合体は薬物と全溶解性促進剤の組み合わせ量の少なくとも約20%を含む。抱合体の溶融温度は好適には約36℃以下であり、さらに好適には約25℃以下である。1実施態様では頭部は割合的に大き過ぎてリポソームを形成させない。別実施態様では頭部は小さ過ぎてリポソームを形成させない。骨格鎖は好適にはグリセロールまたは糖類である。ポリマーは好適にはPEGである。
【0064】
本発明の実施例と適用形態を解説したが、専門家であれば、本明細書で説明した本発明の範囲から逸脱せずにこれらを改良することは可能であろう。よって本発明はそれらによって限定されず、「請求の範囲」によってのみ限定される。
[実施例1]
【0065】
実施例1:リポソーム形成には大き過ぎる頭部を有したPEG−脂質抱合体による薬剤化合物の溶解性促進(予測例)
【0066】
表6に記載の化合物の存在下で増加された水溶解性を確認するために様々な疎水性薬剤化合物がテストされる。表の抱合体は化学構造2で表されるものである。
【表6】
【0067】
表6の化合物1から3は表1にもPEG−23GDM、PEG−23GDOおよびPEG−23GDLとしてそれぞれ現れる。その他の化合物のアシル基と親水性ポリマーは記載されたどのタイプのリンカーによっても骨格鎖に連結される。骨格鎖のタイプに関係なくAG1とAG2を相互に接近させておくことが望ましい。
【0068】
抱合体は室温で液体である。あるいは各薬剤化合物との予備混合前に溶解状態である。例えば、40mgの選択薬物がまず1:5のw/w比のポリマー−脂質抱合体と予備混合される。20mMのリン酸塩バッファ(pH6)の水溶液が加えられてさらに混合され、全部で2mLの溶液が得られる。分割量の溶液の薬物濃度が測定される。すべての場合において抱合体の存在により溶液内の薬物濃度は抱合体のない場合の溶液の薬物濃度よりも高くなる。
【0069】
試験される化合物はアンホテリシンB、イトラコナゾール、ポサコナゾール、シクロスポリンおよびインドメタシンである。
【0070】
この薬剤化合物の存在下あるいは非存在下の両方でポリマー−脂質抱合体の水溶液の物理形態が検査される。10%未満の抱合体または抱合体/薬物複合体がリポソームの形態である。
[実施例2]
【0071】
実施例2:リポソーム形成には小さ過ぎる頭部を有したPEG−脂質抱合体による薬剤化合物の溶解性促進(予測例)
【0072】
表7に掲載された化合物の存在下で増加された水溶解性を確認するために様々な疎水性薬剤化合物がテストされる。表の抱合体は化学構造2で表されるものである。
【表7】
【0073】
アシル基と親水性ポリマーは掲載されている任意のリンカーを使用して骨格鎖に連結できる。しかし、これら実施例では頭部のサイズを規定することが必須であるため、リンカーは延伸骨格鎖の全分子量を約250未満に保つことが重要である。骨格鎖のタイプに関係なくAG1とAG2を互いに接近させておくことが望ましい。
【0074】
抱合体は室温で液体である。あるいは各薬剤化合物との予備混合前に溶解状態である。例えば、40mgの選択薬物がまず1:8のw/w比のポリマー−脂質抱合体と予備混合される。20mMのリン酸塩バッファ(pH6)の水溶液が加えられてさらに混合され、全部で2mLの溶液が得られる。分割量の溶液の薬物濃度が測定される。すべての場合に抱合体の存在により溶液内の薬物濃度は抱合体が存在しない溶液の薬物濃度よりも高くなる。
【0075】
試験される化合物はアンホテリシンB、イトラコナゾール、ポサコナゾール、シクロスポリンおよびインドメタシンである。
【0076】
薬剤化合物の存在下あるいは非存在下の両方においてポリマー−脂質抱合体の水溶液の物理形態が検査される。10%未満の抱合体または抱合体/薬物複合体がリポソームの形態である。
[実施例3]
【0077】
実施例3:抗癌薬物経口溶液
【0078】
ゲフィチニブの経口送達のために適したポリマー−脂質抱合体が調製された。ポリマー−脂質がミキサープロペラを備えた容器に加えられた。薬剤物質は常時混合しながら加えられた。混合は薬物が脂質内に視覚的に分散するまで継続された。水中の予備溶解された賦形剤が容器にゆっくりと加えられ、適切に混合された。混合は完全に均質な溶液となるまで継続された。サンプル調剤は表8に記載されている。
【表8】
【0079】
PEG−脂質抱合体はDDGBであるか、または表6あるいは表7に掲載されている任意の抱合体でよい。水酸化ナトリウムが純水中の10%w/wの溶液の調製に使用される。目標のpHは4.0から7.0である。必要であればNaOH溶液がpHの調整に使用される。薬物と脂質(すなわち溶解性促進剤)の比は好適には約1:20よりも大きく、さらに好適には約1:5よりも大きい。有機酸は酪酸またはピルビン酸あるいはグリコール酸でよい。なかでも酪酸が最も好適である。有機酸の濃度は好適には1%から10%であり、さらに好適には約2%から5%である。
[実施例4]
【0080】
実施例4:抗癌薬物IV注射溶液
【0081】
IV溶液が実施例3と同様に調製された。しかし人工風味料や保存剤は加えられなかった。目標のpH範囲は6.5から7.5であった。サンプル調剤は表9に掲載されている。
【表9】
【0082】
PEG−脂質抱合体はDDGBであっても、または表6あるいは表7の任意の抱合体であってもよい。アシル基はカルボキシ結合によりグリセロール骨格鎖に取り付けられる。水酸化ナトリウムが純水中で10%w/wの溶液の調製に使用される。目標のpHは4.0から7.0の範囲である。必要であればNaOH溶液がpHの調整に使用される。薬物と脂質の比は約1:20よりも大きく、さらに好適には約1:5よりも大きい。有機酸は酪酸またはピルビン酸あるいはグリコール酸でよい。なかでも酪酸が最も好適である。有機酸の濃度は好適には1%から5%であり、さらに好適には約1%から3%である。
[実施例5]
【0083】
実施例5:抗菌局部クリーム
【0084】
PEG−脂質抱合体がプロペラタイプの混合ブレードを備えたステンレス鋼容器に加えられた。常時混合しながら薬剤物質が加えられた。混合は60℃から65℃の温度で薬物が脂質内で視覚的に分散するまで継続された。有機酸、コレステロールおよびグリセリンが混合しながら加えられた。エタノールとエチオキシジグリコールが混合しながら加えられた。最後に、カルボポルETD2020、純水およびトリエチルアミンが混合しながら加えられた。混合は完全に均質なクリームとなるまで継続された。調剤は表9に記されている。
【表10】
【0085】
薬物はポサコナゾール、エクアコナゾール、イトラコナゾール、テルビナフィンおよびメトロニダゾールを含む抗カビ剤でも殺菌剤でもよい。抱合体は表6または表7に掲載されたどの抱合体であってもよい。有機酸は酪酸、ピルビン酸あるいはグリコール酸でよい。必要であれば水酸化ナトリウムがpHの調整に使用できる。目標のpH範囲は3.5から7.0である。
[実施例7]
【0086】
実施例7:抗菌局部溶液
【0087】
活性剤がまず有機酸とエタノールに溶解されたこと以外は局部用溶液が実施例6と同様に調製された。サンプル調剤は表11で解説されている。
【表11】
【0088】
抱合体は表6または表7に記載のどの抱合体であってもよい。有機酸は酪酸、ピルビン酸あるいはグリコール酸でよい。必要であれば水酸化ナトリウムがpHの調整に使用できる。目標のpH範囲は3.5から7.0である。
[実施例8]
【0089】
実施例8:合成3,4−ジ(オレオイロキシ)−デカエチレングリコール−安息香酸(DDGB)
【0090】
250mLのベンゼン中の0.10モルの3,4−ジヒドロキシ安息香酸と0.1モルの無水ピリジンの溶液が、250mLのベンゼン中の0.22モルの新鮮蒸留塩化オレオイルの氷冷溶液に無水条件下で撹拌しながらゆっくりと加えられ、混合物は無水条件下で20時間にわたって40℃で保存された。冷却後、450mLのクロロホルムが混合物に加えられ、その後に濾過された。溶媒は真空内で蒸発され、白色の未精製固体が得られた。この固体はさらに次のように純化できる。未精製界面活性剤はシリカゲル(G60、グレード60、メッシュ240〜400)カラムでクロマトグラフィにより純化される。このカラムは1:1(v/v)のジエチルエーテル/アセトン混合物で溶離される。溶離された第1部分は非反応試薬であり、廃棄される。この部分は254nmのピーク(UVモニター)で回収された。真空での溶媒の乾燥によって約70%の3,4−ジ(オレオイロキシ)安息香酸が得られた。
【0091】
0.2モルの3,4−ジ(オレイロキシ)安息香酸と、0.2モルのデカエチレングリコールと、0.21モルのp−塩化トルエンスルホニルと、0.11モルの4−ジメチルアミノピリジンと、0.5モルのテトラブチルアンモニウム硫酸水素と、0.14モルの炭酸カリウムの混合物が600mLのテトラヒドロフラン内で縣濁された。この反応混合物は50℃で22時間にわたって撹拌された。反応混合物は室温に冷却され、1500mLの水内に注がれ、300mLの塩化メチレンが加えられた。溶液は希塩酸で酸化され、pH1に調整された。有機層は分離され、350mLの水で5回洗浄され、硫化マグネシウムで乾燥された。溶媒は真空下で除去され、残留物は200mLのエタノールで再溶解された。無色の溶液は4℃で12時間にわたって放置され、または白色沈降物が形成されるまで放置された。真空下で沈降物の濾過により約80%の最終製品が得られた(化学構造3)。
【化11】
3,4−ジ(オレオイロキシ)デカエチレン(PEG−12)安息香酸グリコール
化学構造3
[実施例9]
【0092】
実施例9:ゲフィチニブ製剤の薬剤動態特性および生体内利用性
【0093】
静脈注射と経口投与の後のゲフィチニブ製剤の血液内量を決定するために実験が実施された。比較のためにゲフィチニブ製剤も試験された。3匹の雄マウス(B6D2F1)の群が研究に使用された。ボーラスIV(静脈)注射後、典型的には0時間、0.5時間、1時間、2時間、4時間、7時間、16時間、および24時間後に得られたヘパリン添加されたマウス血清サンプルに対してHPLC−MS分析が実行された。経口給餌後、分析は0時間、0.5時間、1時間、2時間、4時間、7時間、16時間、および24時間後に行われた。各薬物の溶解量を決定するため、薬物はまずサンプル前処理した血清から単離された。アセトニトリルが使用され、サンプル内のタンパク質が除去された。無勾配JPLC−MS/M法が利用され、薬物が可能な妨害物から分離された。複数の反応モニタリング(MRM)モードで薬物量はMS検出により測定された。ウィンノンリンプログラム(5.2版、Pharsight)分析区分モデルの分析を活用してPKデータが分析された。その結果、本発明の化合物の調剤がクレモフォールEL(登録商標)よりも優れたPK特性を有することが証明された。
【0094】
図5は、(1)10mMのリン酸ナトリウムバッファ(pH6.0)中の6%のPEG−12GDOと、(2)10mMのリン酸ナトリウムバッファ(pH6.0)中の6%のDDGBと、(3)10mMのリン酸ナトリウムバッファ(PpH6.0)中の15%のクレモフォールEL(登録商標)のゲフィチニブマウスの経口PK特性を示す。この薬物は経口投与され、投薬濃度は25mg/kgであった。薬物は静脈注射でも投与され、投薬濃度は15%のクレモフォールEL(登録商標)および10mMのリン酸ナトリウムバッファ(pH6.0)中で25mg/kgであった。静脈内投与から得られたAUC(31.3)は相対生物利用性(BA)の計算に使用された。生物利用性はGDO−12,DDGBおよびクレモフォールEL(登録商標)に対してそれぞれ69.4%、56.0%および28.5%であった。
[実施例10]
【0095】
実施例10:固体投与調合物
【0096】
本発明の抱合体は非水溶性薬物の固体経口投与調合物の調製に有効である。この点で抱合体または抱合体の混合物は薬剤物質と混合される。その後、さらに高い融点を有した賦形剤が導入される。賦形剤は適した溶媒中で溶解する。最終混合物は冷却され、溶媒が除去されて混合物はカプセル形態、錠剤形態、等々に形成される。
【0097】
固体投与調合物を調製するために抱合体(全量の約42%)が混合ブレードを備えたステンレス鋼容器に加えられる。薬剤物質(全量の約15%)が混合しながら加えられる。加熱下での混合は薬物が事実上完全に分散するまで継続される。別の容器にはd−α−トコフェリルプロピレングリコール−1000琥珀酸塩(TPGS−VE)または類似化合物(全量の約42%)がエタノール(全量の約1%)内に溶解され。その後に前記のステンレス鋼容器に加えられる。混合は均質な溶液が得られるまで継続される。真空下でエタノールは除去される。
【特許請求の範囲】
【請求項1】
親油性薬物の溶解性を促進する組成物であって、少なくとも25重量%のジアシルリピッド−ポリマー抱合体を含有しており、該ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化1】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させないことを特徴とする組成物。
【請求項2】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項1記載の組成物。
【請求項3】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項1記載の組成物。
【請求項4】
前記骨格鎖はグリセロールであることを特徴とする請求項1記載の組成物。
【請求項5】
前記骨格鎖は糖類であることを特徴とする請求項1記載の組成物。
【請求項6】
親油性活性剤と、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤とを含有した薬剤組成物であって、該ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化2】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでいることを特徴とする薬剤組成物。
【請求項7】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項6記載の薬剤組成物。
【請求項8】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項6記載の薬剤組成物。
【請求項9】
前記骨格鎖はグリセロールであることを特徴とする請求項6記載の薬剤組成物。
【請求項10】
前記骨格鎖は糖類であることを特徴とする請求項6記載の薬剤組成物。
【請求項11】
親油性薬物の溶解性を促進する方法であって、水溶液中の親油性薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、前記ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化3】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでいることを特徴とする方法。
【請求項12】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項11記載の方法。
【請求項13】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項11記載の方法。
【請求項14】
前記骨格鎖はグリセロールであることを特徴とする請求項11記載の方法。
【請求項15】
前記骨格鎖は糖類であることを特徴とする請求項11記載の方法。
【請求項16】
親油性薬物で哺乳動物を治療する方法であって、水溶液中の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、前記ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化4】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでおり、本方法はさらに、
哺乳動物に対して前記水溶液を投与するステップを含んでいることを特徴とする方法。
【請求項17】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項16記載の方法。
【請求項18】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項16記載の方法。
【請求項19】
前記骨格鎖はグリセロールであることを特徴とする請求項16記載の方法。
【請求項20】
前記骨格鎖は糖類であることを特徴とする請求項16記載の方法。
【請求項1】
親油性薬物の溶解性を促進する組成物であって、少なくとも25重量%のジアシルリピッド−ポリマー抱合体を含有しており、該ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化1】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させないことを特徴とする組成物。
【請求項2】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項1記載の組成物。
【請求項3】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項1記載の組成物。
【請求項4】
前記骨格鎖はグリセロールであることを特徴とする請求項1記載の組成物。
【請求項5】
前記骨格鎖は糖類であることを特徴とする請求項1記載の組成物。
【請求項6】
親油性活性剤と、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤とを含有した薬剤組成物であって、該ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化2】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでいることを特徴とする薬剤組成物。
【請求項7】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項6記載の薬剤組成物。
【請求項8】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項6記載の薬剤組成物。
【請求項9】
前記骨格鎖はグリセロールであることを特徴とする請求項6記載の薬剤組成物。
【請求項10】
前記骨格鎖は糖類であることを特徴とする請求項6記載の薬剤組成物。
【請求項11】
親油性薬物の溶解性を促進する方法であって、水溶液中の親油性薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、前記ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化3】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでいることを特徴とする方法。
【請求項12】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項11記載の方法。
【請求項13】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項11記載の方法。
【請求項14】
前記骨格鎖はグリセロールであることを特徴とする請求項11記載の方法。
【請求項15】
前記骨格鎖は糖類であることを特徴とする請求項11記載の方法。
【請求項16】
親油性薬物で哺乳動物を治療する方法であって、水溶液中の薬物を、ジアシルリピッド−ポリマー抱合体を含んだ1以上の溶解性促進剤と組み合わせるステップを含んでおり、前記ジアシルリピッド−ポリマー抱合体は以下化学式の構造のものであり、
【化4】
ここでAG1とAG2はアシル基であり、
L1、L2およびL3はリンカーであり、
Bは骨格鎖であり、
Pはポリエチレングリコールであり、
Rは陽イオンではなく、
AG1とAG2は共に前記抱合体の親油性尾部を含んでおり、
B、L1、L2、L3、PおよびRは共に前記抱合体の親水性頭部を含んでおり、
前記頭部は前記尾部に比して大き過ぎ、前記抱合体の純粋な水溶液にリポソームを支配的に形成させず、
前記抱合体は少なくとも20%の薬物と全溶解性促進剤との組み合わせた量を含んでおり、本方法はさらに、
哺乳動物に対して前記水溶液を投与するステップを含んでいることを特徴とする方法。
【請求項17】
前記抱合体の溶融温度は36℃以下であることを特徴とする請求項16記載の方法。
【請求項18】
前記抱合体の溶融温度は25℃以下であることを特徴とする請求項16記載の方法。
【請求項19】
前記骨格鎖はグリセロールであることを特徴とする請求項16記載の方法。
【請求項20】
前記骨格鎖は糖類であることを特徴とする請求項16記載の方法。
【図1】

【図2】

【図3】
【図4】
【図5】

【図2】
【図3】
【図4】
【図5】
【公表番号】特表2012−515774(P2012−515774A)
【公表日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願番号】特願2011−547960(P2011−547960)
【出願日】平成22年1月22日(2010.1.22)
【国際出願番号】PCT/US2010/000165
【国際公開番号】WO2010/085347
【国際公開日】平成22年7月29日(2010.7.29)
【出願人】(510184601)
【出願人】(510184612)
【Fターム(参考)】
【公表日】平成24年7月12日(2012.7.12)
【国際特許分類】
【出願日】平成22年1月22日(2010.1.22)
【国際出願番号】PCT/US2010/000165
【国際公開番号】WO2010/085347
【国際公開日】平成22年7月29日(2010.7.29)
【出願人】(510184601)
【出願人】(510184612)
【Fターム(参考)】
[ Back to top ]