
薬物のキャリアとして有用なIgGFc断片およびその製造方法
【課題】結合する薬物の生体内持続性を向上させ、生体内活性の減少を最小化すること。
【解決手段】薬物のキャリアとして有用なIgG Fc断片、これを発現するための組み換えベクター、前記組み換えベクターによって形質転換された形質転換体、および前記形質転換体を培養してIgG Fc断片を製造する方法を提供する。
【解決手段】薬物のキャリアとして有用なIgG Fc断片、これを発現するための組み換えベクター、前記組み換えベクターによって形質転換された形質転換体、および前記形質転換体を培養してIgG Fc断片を製造する方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬物のキャリアとして有用なIgG Fc断片に関するものであり、より具体的には、IgG2 Fc並びにIgG4 Fc断片、この組み合わせ(combination)およびこのハイブリッド(hybrid)に関するものである。また、本発明は、前記IgG Fc断片を発現させるための発現ベクター、前記発現ベクターによって形質転換された形質転換体および前記形質転換体を培養して免疫グロブリンFc断片を製造する方法に関するものである。
【背景技術】
【0002】
過去、数多くの薬学者および化学者は、天然的に存在する生理活性分子の生体活性を化学的に変化および/または修飾しようと努力した。このような努力の大部分は、生理活性物質の特定の生体活性を増加させたり、生体活性を持続させたり、毒性を減少させたり、副作用を除去または減少させたり、特定の生理活性を修飾することであった。生理活性物質が化学的に修飾される場合、大部分は生理活性が除去されるかあるいは相当減少するが、場合によっては生理活性が増加または変化することもある。したがって、目的とするところに応じて生理活性を変化させることが可能な化学修飾についての研究が多く行われており、大部分の研究は、生理活性物質(薬物)を生理学的に許容可能なキャリアと共有結合させることであった。
【0003】
国際特許公開公報第WO01/93911号には、多数の酸性残基を持つ重合体が薬物のキャリアとして用いられた例が開示されており、国際特許公開第WO03/00778号には、陰イオン基を含有する両親媒性ブロック共重合体をキャリアとして用いて陽イオン性薬物の安定性を増加させた例が開示されており、ヨーロッパ特許第0681481号には、シクロデキストリンと酸をキャリアとして塩基性薬物の特性を改善させる技術が開示されている。一方、疎水性薬物の場合、生体内での安定性の低下は、主に疎水性薬物の低い水溶解特性に起因する。このような疎水性薬物の低い水溶解特性を改善させるために、脂質(Lipid)をキャリアとして用いた技術が国際特許公開第WO04/064731号に開示されている。ところが、今まで免疫グロブリンFc断片を薬物のキャリアとして用いた例は、公知となっていない。
【0004】
一方、一般的に、ポリペプチドは、安定性が低くて変性し易く、血液内蛋白質加水分解酵素によって分解されて腎臓または肝臓を介して容易に除去されるため、薬理成分としてポリペプチドを含む蛋白質医薬品の血中濃度および力価を保つためには、蛋白質薬物を患者に頻繁に投与する必要がある。ところが、大部分注射剤の形態で患者に投与される蛋白質医薬品の場合、活性ポリペプチドの血中濃度を保つために頻繁に注射を打つことは、患者に夥しい苦痛を引き起こす。このような問題点を解決するために、蛋白質薬物の血中安定性を増加させ、血中薬物濃度を長期間高く持続させて薬効を最大化しようという努力が続けられてきた。このような蛋白質薬物の持続性製剤は、蛋白質薬物の安定性を高めると同時に薬物自体の力価が十分高く維持されなければならず、患者に免疫反応を誘発してはならない。
【0005】
蛋白質を安定化させ、蛋白質加水分解酵素との接触および腎臓による消失を抑制するための方法として、従来では、ポリエチレングリコール(polyethylene glycol:以下「PEG」という)のように溶解度の高い高分子を蛋白質薬物の表面に化学的に付加させる方法が使用された。PEGは、目的蛋白質の特定の部位または様々な部位に非特異的に結合して溶解度を高めることにより、蛋白質を安定化させ且つ蛋白質の加水分解を防止するのに効果があり、しかも、深刻な副作用を起こさないものとして知られている(Sada et al., J. Fermentation Bioengineering 71: 137-139, 1991)。ところが、PEGの結合により、蛋白質の安定性は増加できるが、生理活性蛋白質の力価が著しく低くなるという問題を生じる。さらに、PEGの分子量が増加するほど蛋白質との反応性が低くなり、その結果、収率が減少するという問題を生じる。
【0006】
最近は、PEGの両末端に同一の蛋白質薬物を結合させた二重体を作って活性の増加を図ったり(米国特許第5,738,846号)、あるいはお互い異なる種類の蛋白質薬物をPEGの両末端に結合させて同時に2種の活性を示す蛋白質(国際出願公開第WO92/16221号)も開発されたが、これらは蛋白質薬物の活性を持続させる面では明確な効果を示していない。
【0007】
一方、Kinstler等は、顆粒球コロニー刺激因子(G−CSF)とヒトアルブミンとをPEGに結合させた融合蛋白質が、増加した安定性を示すと報告した(Kinstler et al., Pharmaceutical Research 12(12): 1883-1888, 1995)。ところが、G−CSF−PEG−アルブミン構造を有する前記文献の修飾された薬物は、体内残留時間が天然型薬物を単独で投与した場合に比べて約4倍増加するに止まり、血中半減期の増加が微々であって、蛋白質薬物の効果的な持続性製剤として実用化されていない。
【0008】
生理活性蛋白質の生体内安定性を高める別の方法として、遺伝子組み換えにより血中安定性の高い蛋白質遺伝子と生理活性蛋白質遺伝子を連結した後、前記組み換え遺伝子で形質転換された動物細胞などを培養して融合蛋白質を生産する方法が開発されている。例えば、現在まで蛋白質の安定性の増加に最も効果が高いものと知られているアルブミンまたはその断片を、遺伝子組み換えによって目的の生理活性蛋白質に結合させて生産した融合蛋白質が報告されている(国際出願公開第WO93/15199号および第WO93/15200号、ヨーロッパ特許公開第EP413,622号)。
【0009】
また、ヒューマンゲノムサイエンス(Human Genome Science)社が酵母で生産したインターフェロンアルファとアルブミンの融合蛋白質(製品名:AlbuferonTM)は、猿でインターフェロンの半減期を5時間から93時間に増加させたが、修飾していないインターフェロンに比べて生体活性度が5%未満に著しく減少するという問題がある(Osborn et al., J. Phar. Exp. Ther. 303(2): 540-548, 2002)。すなわち、今まで蛋白質薬物の生体内持続性と安定性を増加させながら生体内活性を同時に維持することが可能な技術は、未だ開示されたことがない。
【0010】
一方、免疫グロブリンおよびこの断片を用いて蛋白質薬物の安定性を高めようとする試みも多く行われた。例えば、米国特許第5,045,312号では、架橋剤を用いてヒトの成長ホルモンに血清アルブミンまたはラットの免疫グロブリンを結合させることにより、修飾していない成長ホルモンに比べて成長ホルモンの活性を増加させることができることを開示している。
【0011】
また、蛋白質薬物と免疫グロブリンFcを融合させようとする試みも行われた。例えば、インターフェロン(大韓民国特許公開第2003−9464号)、およびインターロイキン−4受容体、インターロイキン−7受容体または赤血球生成因子受容体(大韓民国特許登録第249572号)を免疫グロブリンのFc断片と融合している形態で哺乳動物で発現させた。国際特許公開第WO01/03737号では、サイトカインまたは成長因子をペプチド結合によって免疫グロブリンのFc断片に結合させた融合蛋白質を製造した。また、米国特許第5,116,964号では、免疫グロブリンFc断片のアミノ末端またはカルボキシル末端に遺伝子組み換え方法によって融合させた蛋白質を開示しており、米国特許第5,349,053号では、IL−2を免疫グロブリンFc断片にペプチド結合によって融合させた融合蛋白質を開示している。
【0012】
その他にも、遺伝子組み換えによって製造されたFc融合蛋白質の例として、インターフェロン−βまたはその誘導体と免疫グロブリンFc断片の融合蛋白質(国際特許公開第WO00/23472号)、IL−5受容体と免疫グロブリンFc断片の融合蛋白質(米国特許第5,712,121号)が開示されている。ところが、免疫グロブリンFc断片を用いて生理活性ポリペプチド薬物の持続性を改善させようとする技術は、大部分、免疫グロブリンFc断片を融合パートナーとして用いた技術に焦点が合わせられており、免疫グロブリンFc断片をキャリアとして用いる技術は未だ報告されていない。
【0013】
一方、免疫グロブリンFc断片のアミノ酸を修飾する技術も公知になっているが、米国特許第5,605,690号では、免疫グロブリンFc断片で特に補体結合部位または受容体結合部位のアミノ酸を改変させたFcを用いて遺伝子組み換え方法によって製造されたTNFR−IgG1 Fc融合蛋白質を開示している。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】国際特許公開WO01/93911号
【特許文献2】国際特許公開WO03/00778号
【特許文献3】ヨーロッパ特許0681481号
【特許文献4】国際特許公開WO04/064731号
【特許文献5】米国特許第5,738,846号
【特許文献6】国際出願公開WO92/16221号
【特許文献7】国際出願公開WO93/15199号
【特許文献8】国際出願公開WO93/15200号、
【特許文献9】ヨーロッパ特許公開EP413,622号
【特許文献10】米国特許第5,045,312号
【特許文献11】大韓民国特許公開2003−9464号
【特許文献12】大韓民国特許登録第249572号
【特許文献13】国際特許公開WO01/03737号
【特許文献14】米国特許第5,116,964号
【特許文献15】米国特許第5,349,053号
【特許文献16】国際特許公開WO00/23472号
【特許文献17】米国特許第5,712,121号
【特許文献18】米国特許第5,605,690号
【非特許文献】
【0015】
【非特許文献1】Sada et al., J. Fermentation Bioengineering 71: 137-139, 1991
【非特許文献2】Kinstler et al., Pharmaceutical Research 12(12): 1883-1888, 1995
【非特許文献3】Osborn et al., J. Phar. Exp. Ther. 303(2): 540-548, 2002
【発明の概要】
【発明が解決しようとする課題】
【0016】
ところが、このような遺伝子組み換え方法によって生産されたFc融合蛋白質は、免疫グロブリンFc領域の特定の部位、すなわちアミノ末端またはカルボキシル末端でのみ蛋白質融合が可能であり、同種二量体の形態でのみ発現され、単量体の形態では生産が不可能であり、糖鎖化蛋白質間の融合または非糖鎖化蛋白質間の融合のみが可能であって、糖鎖化蛋白質と非糖鎖化蛋白質との融合は不可能であるという欠点がある。また、融合によって新たに発生したアミノ酸配列により免疫反応が誘発されるおそれがあるだけでなく、リンカー部位に対する蛋白質加水分解酵素の敏感性が増加するおそれがあるという問題点を持っている。
【0017】
このような問題を解決するために、本発明者は、前記融合技術の欠点を克服しながら薬物の生体内持続性を向上させ且つ生体内活性の減少を最小化するための方法を研究し、IgG Fc断片、より具体的にはIgG2またはIgG4 Fc断片が薬物と結合する場合、薬物の生体内持続性を向上させかつ生体内活性の減少を最小化することができることを見出し、本発明を完成した。
【課題を解決するための手段】
【0018】
本発明の一目的は、薬物のキャリアとして有用な免疫グロブリンG断片を提供することにある。
【0019】
本発明の他の目的は、免疫グロブリンG断片を製造するための組み換えベクターを提供することにある。
【0020】
本発明の別の目的は、免疫グロブリンG断片を製造するための組み換えベクターで形質転換された形質転換体を提供することにある。
【0021】
本発明の別の目的は、免疫グロブリンG断片を製造するための組み換えベクターで形質転換された形質転換体を培養して免疫グロブリンG断片を製造する方法を提供することにある。
【0022】
本発明の別の目的は、免疫グロブリンG断片を含む薬剤学的組成物を提供することにある。
【図面の簡単な説明】
【0023】
【図1】図1は、免疫グロブリンFcを大腸菌で発現させた後、非還元条件でウエスタンブロットによって分析した結果である。
【図2】図2は、免疫グロブリンFcを15%クライテリオンゲル(Criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させた結果である。
【図3】図3は、免疫グロブリンFcを15%クライテリオンゲル(Criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させた結果である。
【図4】図4は、免疫グロブリンをパパインで切断して得た免疫グロブリンFc断片をクロマトグラフィで精製した結果である。
【図5】図5は、精製された免疫グロブリンFc断片をSDS−PAGEで分析した結果である。
【図6】図6は、カップリング反応後に生成されたIFNα−PEG−Fc(A)、17Ser−G−CSF−PEG−Fc(B)およびEPO−PEG−Fc(C)結合体をSDS−PAGEで分析した結果である。
【図7】図7は、カップリング反応後に精製されたIFNα−PEG−Fc結合体をサイズ排除クロマトグラフィで分析した結果である。
【図8】図8は、EPO−PEG−Fc結合体をMALDI−TOF質量分析器で分析した結果である。
【図9a】天然型免疫グロブリンFcと脱糖鎖化免疫グロブリンFc(deglycosylated Fc:DG Fc)のMALDI−TOF質量分析器で分析した結果である。
【図9b】天然型免疫グロブリンFcと脱糖鎖化免疫グロブリンFc(deglycosylated Fc:DG Fc)のSDS−PAGEで分析した結果である。
【図10】図10は、IFNα−PEG−Fc結合体およびIFNα−PEG−DG Fc結合体をMALDI−TOF質量分析器で分析した結果である。
【図11】図11は、IFNα−PEG−Fc結合体、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体結合体を逆相HPLCで分析した結果である。
【図12】図12は、天然型IFNα、IFNα−40K PEG、IFNα−PEG−アルブミン結合体およびIFNα−PEG−Fc結合体の薬物動態解析グラフである。
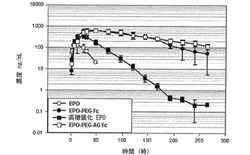
【図13】図13は、天然型EPO、高糖鎖化EPO、EPO−PEG−FcおよびEPO−PEG−AG Fc結合体の薬物動態を分析したグラフである。
【図14】IFNα−PEG−Fc結合体、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体結合体の薬物動態を分析したグラフである。
【図15】図15は、Fab’、Fab’−S−40K PEG連結体、Fab’−N−PEG−N−Fc結合体およびFab’−S−PEG−N−Fc結合体の薬物動態を分析したグラフである。
【図16】図16は、Fab’、Fab’−S−40K PEG連結体、Fab’−N−PEG−N−Fc結合体およびFab’−S−PEG−N−Fc結合体の細胞内活性を分析したグラフである。
【図17】図17は、ヒトIgGサブクラスにおける補体Clqの結合活性を比較分析したグラフである。
【図18】図18は、糖鎖化Fc、酵素を用いて糖を除去したDG Fc、および大腸菌で生産したAG Fcをキャリアとして用いたインターフェロン−PEG−キャリア結合体における補体Clqの結合活性を比較分析したグラフである。
【発明を実施するための形態】
【0024】
一つの様態として、本発明は、薬物のキャリアとして有用な免疫グロブリンG Fc断片に関し、より好ましくはIgG2 FcおよびIgG4 Fc断片に関するものである。
本発明において、「キャリア」とは、薬物と共に結合する物質を意味し、一般に薬物に結合して薬物の生理活性を増減または除去する。ところが、本発明におけるキャリアは、本発明の目的上、結合する薬物の生理活性減少を最小化すると同時に薬物の生体内安定性は増加させるためのものである。
【0025】
このような薬物のキャリアとして、従来から脂質(Lipid)や重合体(polymer)など多くの物質が研究されてきたが、免疫グロブリンFc断片を用いた技術は未だ公知となっていない。すなわち、本発明は、結合する薬物の生体内持続性の増進と生体内活性減少の最小化のためのキャリアとして、使用可能な物質の中でも特にIgG Fc断片を提供することを特徴とし、より好ましくはIgG2 FcおよびIgG4 Fc断片を提供することを特徴とする。
【0026】
本発明において、「免疫グロブリンG(以下、「IgG」と混用する)とは、抗原に選択的に作用して生体免疫に関与する蛋白質の総称であって、ヒトおよび動物から起源することができる。このような免疫グロブリンの一般構造を考察すると、免疫グロブリンは重鎖と軽鎖からなっており、重鎖不変領域はガンマ(γ)、ミュー(μ)、アルファ(α)、デルタ(δ)、エプシロン(ε)タイプを有し、サブクラスとしてガンマ1(γ1)、ガンマ2(γ2)、ガンマ3(γ3)、ガンマ4(γ4)、アルファ1(α1)およびアルファ2(α2)を有する。軽鎖の不変領域はカッパ(κ)およびラムダ(λ)タイプを有する(Coleman et al., Fundamental Immunology, 2nd Ed., 1989, 55-73)。これらにより、免疫グロブリンは、IgG、IgA、IgD、IgEおよびIgMの亜型(isotype)に分けられる。IgGはさらにIgG1、IgG2、IgG3およびIgG4に分けられる。
【0027】
また、免疫グロブリンは構造的にいろいろの断片が知られているが、その例としてFab、F(ab’)、F(ab’)2、Fv、scFv、Fd、Fcなどがある。この中でも、免疫グロブリン断片Fabは、軽鎖および重鎖の可変領域、軽鎖の不変領域および重鎖の第1不変領域(CH1)を持つ構造であって、1つの抗原結合部位を持つ。免疫グロブリン断片Fab’は、重鎖CH1ドメインのC末端に一つ以上のシステイン残基を含むヒンジ領域(hinge region)を持つという点でFabとは差異がある。断片F(ab’)2は、Fab’のヒンジ領域のシステイン残基がジスルフィド結合を成しながら生成される。Fvは、重鎖可変部位および軽鎖可変部位のみを持っている最小の抗体断片であり、scFvは、重鎖可変部位および軽鎖可変部位のみがペプチドリンカーで連結された単一ポリペプチド鎖である。また、Fd断片は、重鎖可変領域およびCH1ドメインのみから構成された断片である。
【0028】
すなわち、前記のような公知の様々な免疫グロブリンのタイプおよびその機能的、構造的断片の中でも、本発明は、薬物のキャリアとして有用なIgG Fc断片を提供し、特にIgG2 FcおよびIgG4 Fc断片を提供することを特徴とする。
【0029】
本発明の免疫グロブリンG Fc断片(以下、「IgG Fc断片」または「本発明のFc断片」と混用する)は、免疫グロブリンGの重鎖と軽鎖可変領域、重鎖不変領域1(CH1)と軽鎖不変領域1(CL1)を除いた、重鎖不変領域2(CH2)および重鎖不変領域3(CH3)部分を意味し、重鎖不変領域にヒンジ(hinge)部分を含むこともある。また、本発明のIgG Fc領域は、天然型と実質的に同等あるいはより向上した効果を持つ限り、IgGの重鎖と軽鎖可変領域のみを除き、一部または全体重鎖不変領域1(CH1)および/または軽鎖不変領域1(CL1)を含んだ、拡張されたFc断片であり、あるいはCH2および/またはCH3に該当する相当長い一部のアミノ酸配列が除去された断片である。
【0030】
このような本発明のFc断片は、天然型アミノ酸配列だけでなく、この配列誘導体(mutant)も含む。アミノ酸配列誘導体とは、天然アミノ酸配列中の一つ以上のアミノ酸残基が欠失、挿入、非保全的または保全的置換、またはこれらの組み合わせによって相異なる配列を持つものを意味する。例えば、IgG Fcの場合、結合に重要であると知られている214〜238、297〜299、318〜322または327〜331番のアミノ酸残基が、修飾のための適当な部位として利用できる。また、ジスルフィド結合を形成することが可能な部位が除去されるか、あるいは天然型FcからN末端の幾つかのアミノ酸が除去されるか、あるいは天然型FcのN末端にメチオニン残基が付加されることもあるなど様々な種類の誘導体が可能である。また、エフェクター機能を無くすために、補体結合部位、例えばClq結合部位またはADCC部位が除去されることがある。このような免疫グロブリンFc領域の配列誘導体を製造する技術は、国際特許公開第WO97/34631号、国際特許公開第WO96/32478号などに開示されている。
【0031】
分子の活性を全体的に変更させない蛋白質およびペプチドにおけるアミノ酸交換は、当該分野に公知となっている(H. Neurath, R. L. Hill, The Proteins, Academic Press, New York, 1979)。最も通常的に起こる交換は、アミノ酸残基Ala/Ser、Val/Ile、Asp/Glu、Thr/Ser、Ala/Gly、Ala/Thr、Ser/Asn、Ala/Val、Ser/Gly、Thy/Phe、Ala/Pro、Lys/Arg、Asp/Asn、Leu/Ile、Leu/Val、Ala/Glu、Asp/Gly間の交換である。
【0032】
場合によっては、リン酸化(phosphorylation)、硫化(sulfation)、アクリル化(acrylation)、糖化(glycosylation)、メチル化(methylation)、ファルネシル化(farnesylation)、アセチル化(acetylation)、アミド化(amidation)などで修飾(modification)されることもできる。
【0033】
前述したFc誘導体は、本発明のFc断片と同一の生物学的活性を示すが、熱やpHなどに対するFc断片の構造的安定性を増大させた誘導体である。
【0034】
また、このようなFc断片は、ヒト、および牛や山羊、豚、マウス、ウサギ、ハムスター、ラット、モルモットなどの動物の生体内から分離して得られた天然型であり、あるいは形質転換された動物細胞または微生物から得られた組み換え型またはその誘導体である。ここで、天然型から得る方法では、全体免疫グロブリンをヒトまたは動物の生体から分離した後、蛋白質分解酵素を処理して得ることができる。パパインを処理する場合にはFabおよびFcに切断され、ペプシンを処理する場合にはpF’cおよびF(ab’)2に切断される。これらの断片からサイズ排除クロマトグラフィ(size-exclusion chromatography)などを用いてFcまたはpF’cを分離することができる。好ましくは、ヒト由来のFc断片は微生物から収得した組み換え型IgG Fc断片である。すなわち、本発明は、微生物から収得したヒト由来の組み換え型IgG2 FcおよびIgG4 Fc断片が好ましい。
【0035】
また、本発明のFc断片は、天然型糖鎖、天然型に比べて増加した糖鎖、天然型に比べて減少した糖鎖、または糖鎖が除去された形態である。このようなFc断片の糖鎖の増減または除去には、化学的方法、酵素学的方法および微生物を用いた遺伝工学的方法などの当該分野における通常の方法が利用できる。ここで、Fcから糖鎖が除去されたFc断片は、補体(Clq)への結合力が著しく低下し、抗体依存性細胞毒性または補体依存性細胞毒性が減少または除去されるので、生体内で不要な免疫反応を誘発しない。このような点において、薬物のキャリアとしての本来の目的にさらに符合する形態は、脱糖鎖化免疫グロブリンFc断片、または非糖鎖化免疫グロブリンFc断片である。
【0036】
図18からも明らかなように、糖鎖化Fcは、非糖鎖化FcよりCDCの活性が高くて免疫反応を誘発する危険が高いと言えるので、本発明の目的上、非糖鎖化Fc断片あるいは脱糖鎖化Fc断片が好ましく、非糖鎖化IgG2 FcおよびIgG4 Fc断片、この組み合わせおよびこのハイブリッドがより好ましい。
【0037】
本発明において、「脱糖鎖化(Deglycosylation)」はFc断片から酵素によって糖を除去することを意味し、「非糖鎖化(Aglycosylation)」はFc断片を糖鎖化されていない形態で原核細胞、好ましくは大腸菌で生産することを意味する。
【0038】
一方、本発明において、「組み合わせ(combination)」とは、二量体または多量体を形成するとき、同一起源の単鎖免疫グロブリンFc断片を暗号化するポリペプチドが相異なる起源の単鎖ポリペプチドと結合を形成することを意味する。すなわち、IgG1 Fc、IgG2 Fc、IgG3 FcおよびIgG4 Fc断片よりなる群から選択された2つ以上の断片から二量体または多量体の製造が可能である。
【0039】
本発明において、「ハイブリッド(hybrid)」とは、単鎖の免疫グロブリンFc断片内に、相異なる起源の2つ以上の免疫グロブリンFc断片に該当する配列が存在することを意味する用語である。本発明の場合、様々な形態のハイブリッドが可能である。すなわち、IgG1 Fc、IgG2 Fc、IgG3 FcおよびIgG4 FcのCH1、CH2、CH3およびCH4よりなる群から選択された1〜4つのドメインからなるドメインのハイブリッドが可能であり、ヒンジ領域を含むことができる。
【0040】
一方、本発明の図17および図18から分かるように、IgGのいろいろなサブクラスのうち、特にIgG4は、補体(Clq)に対する結合力が最も低い。このように補体に対する結合力が低下すると、抗体依存性細胞毒性(ADCC、Antibody-dependent cell cytotoxicity)および補体依存性細胞毒性(CDC、Complement-dependent cytotoxicity)が減少または除去されるので、生体内で不要な免疫反応を誘発しなくなる。このようなClqに対する結合力は、IgG2およびIgG4 Fc断片がIgG1より低く、この中でもIgG4 Fc断片が最も低いことを確認することができる。したがって、薬物のキャリアとして用いるためには、結合するFc断片の抗体依存性細胞毒性および補体依存性細胞毒性のようなエフェクター(effector)機能がより低いものを使用することが好ましいので、本発明の目的上、IgG2 FcおよびIgG4 Fc断片が好ましく、より好ましくはIgG4 Fc断片であり、最も好ましくは配列番号8、10および23のアミノ酸配列を持つFc断片である。
【0041】
本発明の別の様態として、本発明は、IgG Fc断片、好ましくはIgG2 FcおよびIgG4 Fc断片をコードする遺伝子に関するもので、より好ましくは配列番号8、10および23のアミノ酸をコードする遺伝子に関するものである。このような本発明のFcをコードする遺伝子は、天然型核酸配列だけでなく、この配列誘導体も含む。核酸配列誘導体とは、天然核酸配列中の一つ以上の核酸残基が欠失、挿入、非保全的または保全的置換、またはこれらの組み合わせによって相異なる配列を持つものを意味する。
【0042】
本発明の配列番号8のアミノ酸配列をもつFc断片をコードする遺伝子は、好ましくは配列番号4の核酸配列を持つ遺伝子であり、配列番号10のアミノ酸配列を持つFc断片をコードする遺伝子は、好ましくは配列番号9の核酸配列を持つ遺伝子であり、配列番号23のアミノ酸配列を持つFc断片をコードする遺伝子は、好ましくは配列番号22の核酸配列を持つ遺伝子である。本発明のFc断片をコードする核酸配列は、一つ以上の塩基が置換、欠失、挿入またはこれらの組み合わせによって変異できる。前記核酸配列は、天然から分離するか、あるいは人為的に合成または遺伝子組み換え方法によって製造することができる。
【0043】
本発明のFc断片をコードする核酸配列は、これを発現するベクターによって提供される。
【0044】
別の様態として、本発明は、IgG Fc断片を含む組み換えベクターを提供する。
【0045】
本発明において、「ベクター」は、宿主細胞にDNAを導入して抗体または抗体断片を発現させるための手段であって、プラスミドベクター、コスミドベクター、バクテリオファージベクター、およびアデノウイルスベクター、レトロウイルスベクター、アデノ随伴ウイルスベクターのようなウイルスベクターなどがあり、プラスミドベクターが好ましい。本発明の目的上、発現ベクターは、プロモータ、開始コドン、終結コドン、ポリアデニル化シグナルおよびエンハンサーなどの発現調節エレメントの他にも膜標的化または分泌のためのシグナル配列を含むことができる。
【0046】
本発明において、「シグナル配列(signal sequence)」は、蛋白質の細胞質の外への輸送および分泌を可能とする特定のアミノ酸配列を意味する。このようなシグナル配列には、様々な形態が当該分野において公知となっているが、本発明では、好ましくは大腸菌を宿主細胞として用いるため、本発明のシグナル配列は大腸菌で分泌される蛋白質が持つ大腸菌由来のシグナル配列であることが好ましい。一方、大腸菌由来のシグナル配列にはアルカリホスファターゼ、ペニシリナーゼ、Ipp、熱安定性エンテロトキシンII、LamB、PhoE、PelB、OmpAまたはマルトース結合蛋白質などがあるが、最も好ましくは熱安定性エンテロトキシンIIである。
【0047】
一方、開始コドンおよび終結コドンは、一般に免疫原性標的蛋白質をコードするヌクレオチド配列の一部と見なされ、遺伝子作製物が投与されたとき、個体において必ず作用を示さなければならず、コーディング配列とインフレーム(in frame)になければならない。
【0048】
一般なプロモータは、構成的または誘導性である。原核細胞にはlac、tac、T3およびT7プロモータがあるが、これに制限されない。真核細胞には、サルウイルス40(SV40)プロモータ、マウス乳房腫瘍ウイルス(MMTV)プロモータ、ヒト免疫不全ウイルス(HIV)プロモータ(例えば、HIVの長い末端反復部(LTR)プロモータ)、モロニーウイルスプロモータ、サイトメガロウイルス(CMV)プロモータ、エプスタインバーウイルス(EBV)プロモータ、ラウス肉腫ウイルス(RSV)プロモータだけでなく、ヒトβ−アクチン、ヒトヘモグロビン、ヒト筋肉クレアチン、ヒトメタロチオネインなどヒト遺伝子由来のプロモータがあるが、これに制限されない。
【0049】
また、発現ベクターは、ベクターを含有する宿主細胞を選択するための選択性マーカーを含み、複製可能な発現ベクターの場合、複製起点を含む。抗生剤または薬物耐性を与える生成物をコードする遺伝子が普遍的な選択性マーカーとして用いられるが、原核細胞の場合、β−ラクタマーゼ遺伝子(アンピシリン耐性)およびTet遺伝子(テトラサイクリン耐性)が使用でき、真核細胞の場合、ネオマイシン(G418またはジェネティシン)、gpt(ミコフェノール酸)、アンピシリン、およびハイグロマイシン耐性遺伝子が使用できる。ジヒドロ葉酸還元酵素マーカー遺伝子は、様々な宿主でメトトレキサートによって選択できる。宿主の栄養要求性マーカーの遺伝子生成物をコードする遺伝子、例えばLEU2、URA3およびHIS3は酵母において選択性マーカーとして度々使用される。そして、ウイルス(例えば、バキュロウイルス)またはファージベクター、およびレトロウイルスベクターのような宿主細胞のゲノム内に挿入できるベクターも使用可能である。
【0050】
本発明の目的に符合するIgG Fc断片を製作するために、配列番号8、10および23のアミノ酸をコードする遺伝子が挿入されたベクターを用いる。本発明では、pT14S1SH−4T20V22Q(大韓民国特許第38061号)を出発ベクターとして用いて、配列番号23のアミノ酸をコードする配列番号22の遺伝子が挿入されたpSTIIdCG2Fcを製作し、配列番号8のアミノ酸をコードする配列番号4の遺伝子が挿入されたpSTIIdCG4Fcを製作し、前記pSTIIdCG4Fcプラスミドを鋳型としてPCRを行うことにより、前記PCRによって増幅された遺伝子のうち、二量体の形成に必要なヒンジ領域が除去された配列番号10のアミノ酸配列をコードする配列番号9の遺伝子が挿入されたpSTIIG4Moを製作した。
【0051】
別の様態として、本発明は、前記組み換えベクターで形質転換された形質転換体に関するものである。
【0052】
宿主細胞に応じて蛋白質の発現量と修飾などが異なるので、目的に最も適した宿主細胞を選択して使用すればよい。宿主細胞としては、大腸菌(Escherichia coli)、枯草菌(Bacillus subtilis)、ストレプトミセス (Streptomyces)、シュードモナス(Pseudomonas)、プロテウスミラビリス(Proteus mirabilis)またはスタヒロコッカス(Staphylococcus)のような原核宿主細胞があるが、これに制限されない。また、真菌(例えば、アスペルギルス(Aspergillus))、酵母(例えば、ピキアパストリス(Pichia pastoris)、サッカロミセスセレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス(Schizosaccharomyces), アカパンカビ(Neurospora crassa)などの下等真核細胞、昆虫細胞、植物細胞、哺乳動物などを含む高等真核生物由来の細胞を宿主細胞として使用することができる。ところが、本発明の目的上、免疫グロブリンFc断片が糖鎖化されないことが有利なので、原核細胞を使用することが好ましく、特に大腸菌を使用することが最も好ましい。
【0053】
本発明において、宿主細胞への「形質転換」および/または「トランスフェクション(transfection)」は、核酸を有機体、細胞、組織または器官に導入するいずれの方法も含まれるもので、当該分野で公知となっているように、宿主細胞に応じて適切な標準技術を選択して行うことができる。このような方法には、エレクトロポレーション(electroporation)、原形質融合、リン酸カルシウム(CaPO4)沈殿、塩化カルシウム(CaCl2)沈殿、炭化珪素繊維を用いた攪拌、アグロバクテリアを介した形質転換、PEG、硫酸デキストラン、リポフェクトアミンおよび乾燥/抑制を介した形質転換方法などが含まれるが、これに制限されない。
【0054】
例えば、塩化カルシウムを用いるカルシウム処理またはエレクトロポレーションは、一般に原核細胞に対して使用される((Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual (New York: Cold Spring Harbor Laboratory Press))。アグロバクテリウムツメファシエンス(Agrobacterium tumefaciens)を用いたトランスフェクションは、特定の植物細胞の形質転換に使用される(Shaw et al., 1983, Gene, 23:315);国際公開特許WO89/05859)。細胞壁がない哺乳動物細胞の場合、リン酸カルシウム沈殿法を用いることができる(Graham et al, 1978, Virology, 52:456-457)。哺乳動物宿主細胞への形質転換の一般的な方法および特徴は、米国特許第4,399,216号に記載されている。酵母内への形質転換は、一般にVan Solingenなどの文献(J. Bact., 1977, 130:946)およびHsiaoなどの文献(Proc. Natl. Acad. Sci. (USA), 1979, 76:3829)の方法によって行われる。
【0055】
前記本発明に係る発現ベクターが宿主細胞に形質転換された本発明の形質転換体は、HM10932(pSTIIdCG4Fc導入形質転換体)、HM10933(pSTIIG4Mo導入形質転換体)およびHM10936(pSTIIdCG2Fc導入形質転換体)である。
【0056】
別の様態としては、本発明は、IgG Fc断片、好ましくはIgG2 Fc、IgG4 Fc断片、この組み合わせおよびこのハイブリッドを発現することが可能なベクターで形質転換された形質転換体を適切な条件で培養して本発明の免疫グロブリン断片を製造する方法を提供する。
【0057】
前記免疫グロブリン断片の製造において、形質転換体の培養は、当該分野に知られている適当な培地と培養条件に応じて行われる。このような培養過程は、当業者であれば、選択される菌株に応じて容易に調整して使用することができる。
【0058】
形質転換体を培養して得た本発明の免疫グロブリン断片は、精製していない状態で使用でき、さらに様々な通常の方法、例えば透析、塩沈殿およびクロマトグラフィなどを用いて高純度に精製して使用できる。その中でもクロマトグラフィを用いる方法が最も多用されている。カラムの種類と順序の選択にはいずれの場合にも適用できる法則はなく、目的蛋白質である抗体の特性、培養方法などに応じてイオン交換クロマトグラフィ、サイズ排除クロマトグラフィ、親和性クロマトグラフィ、プロテインA親和カラムなどから選択することができる。本発明では、プロテインA親和カラム(protein-A affinity column)、SPセファロースFFカラムなどを用いて精製した。
【0059】
こうして得られるFc断片が遊離体の場合には、自体公知の方法またはこれに準ずる方法によって塩に変換することができ、逆にFc断片が塩の場合には、自体公知の方法またはこれに準ずる方法によって遊離体または他の塩に変換することができる。そして、組み換え体が生産するFc断片に、精製前または精製後に適当な蛋白質修飾酵素を作用させることにより、任意に修飾を加えるか、あるいはポリペプチドを部分的に除去することができる。蛋白質修飾酵素としては、例えばトリプシン、キモトリプシン、アルギニルエンドペプチダーゼ、プロテインキナーゼ、グリコシダーゼなどが使用される。
【0060】
このように製造された本発明のFc断片は、薬物のキャリアとして作用して薬物と結合体を形成する。
【0061】
本発明において、「薬物結合体」または「結合体」は、一つ以上の薬物が一つ以上の免疫グロブリンFcとお互い連結されているものを意味する。
本発明において、「薬物」は、ヒトまたは動物に投与される場合、治療的活性を示す物質を意味し、ポリペプチド、化合物、抽出物、核酸などを含むが、これに制限されない。薬物は、好ましくはポリペプチド薬物である。
【0062】
本発明において、「生理活性ポリペプチド薬物」、「ポリペプチド薬物」および「蛋白質薬物」は同一の意味で使われており、生体内で様々な生理的現象に生理作用を示す生理活性型であることを特徴とする。
【0063】
このようなポリペプチド薬物は、変性し易い、あるいは生体内に存在する蛋白質分解酵素によってよく分解されるといった理由によって、長時間にわたって生理学的活性を持続することができないという欠点がある。しかし、本発明の免疫グロブリンFc断片とポリペプチドを結合させた結合体の場合、薬物の構造的安定性が増加し、分解半減期が増加する。そのうえ、Fc領域の結合によるポリペプチドの生理学的活性の減少は、その他に公知となっている他のポリペプチド薬物製剤に比べて非常に軽微であるといえる。したがって、既存のポリペプチド薬物の生体内利用率に比べて、本発明のポリペプチドおよび本発明のFc断片の結合体は、生体内利用率が著しく増加したことを特徴とする。これは、下記本発明の実施例によっても明白に開示されているところであるが、本発明のFc断片が結合したIFNα、G−CSF、hGHなどが、PEGのみが結合しあるいはPEGとアルブミンが結合した既存の製剤に比べて約2〜6倍程度増加したことを確認することができる(表8、表9および表10)。
【0064】
一方、本発明のFc断片と蛋白質の結合は、従来の組み換え的な方法による融合ではないことを特徴とする。免疫グロブリンFc領域と薬物として用いられる活性ポリペプチドが組み換え的な方法によって融合している形態は、Fc領域のN末端またはC末端にポリペプチドがペプチド結合によって連結された形態であって、これをコードする核酸配列において一つのポリペプチドとして発現されて折りたたまれる。
【0065】
蛋白質の生理学的機能体としての活性が構造によって決定されるという点に基づくとき、これは融合蛋白質活性の急激な減少をもたらす。したがって、ポリペプチド薬物がFcと組み換え的な方法によって融合する場合、構造的安定性が増加したとしても、生体内利用率の面で効果がない。また、このような融合蛋白質は、誤って折りたたまれ(misfolding)て凝集体の形態で発現される傾向があるので、蛋白質の製造、分離の収率面で不経済である。また、活性型ポリペプチドが糖鎖化された形態の場合、真核細胞で発現させなければならず、このような場合、Fcも糖鎖化されるので、生体内で不適切な免疫反応を引き起こすおそれもある。
【0066】
すなわち、本発明によってこそ始めて、糖鎖化された活性型ポリペプチドを非糖鎖化された免疫グロブリンFc領域に連結させた結合体を製造することが可能となり、最上のシステムでそれぞれが製造、分離されるので、蛋白質獲得の収率を高めることができるなど、前記の問題点を全て克服することができる。
【0067】
本発明の免疫グロブリンFc断片に連結可能な蛋白質薬物の例としては、ヒト成長ホルモン、成長ホルモン放出ホルモン、成長ホルモン放出ペプチド、インターフェロン類とインターフェロン受容体類(例えば、インターフェロン−α、−βおよび−γ、水溶性タイプIインターフェロン受容体など)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、グルカゴン様ペプチド類(例えば、GLP−1など)、G蛋白質共役型受容体(G-protein-coupled receptor)、インターロイキン類(例えば、IL−1受容体、IL−4受容体など)、酵素類(例えば、グルコセレブロシダーゼ(glucocerebrosidase)、イズロネート−2−スルファターゼ(iduronate-2-sulfatase)、α−ガラクトシダーゼ−A(alpha-galactosidase-A)、アガルシダーゼα(agalsidase alpha)、アガルシダーゼβ、α−L−イズロニダーゼ(alpha-L-iduronidase)、ブチリルコリンエステラーゼ(butyrylcholinesterase)、キチナーゼ(chitinase)、グルタミン酸デカルボキシラーゼ(glutamate decarboxylase)、イミグルセラーゼ(imiglucerase)、リパーゼ(lipase)、 ウリカーゼ(uricase)、血小板−活性因子アセチルハイドロラーゼ(platelet-activating factor acetylhydrolase)、中性エンドペプチダーゼ(neutral endopeptidase)、ミエロペルオキシダーゼ(myeloperoxidase)など)、インターロイキンおよびサイトカイン結合蛋白質類(例えば、IL−18bp、TNF−結合蛋白質など)、マクロファージ活性因子、マクロファージペプチド、B細胞因子、T細胞因子、プロテインA、アレルギー抑制因子、細胞壊死糖蛋白質、免疫毒素、リンホトキシン、腫瘍壊死因子、腫瘍抑制因子、転移成長因子、アルファ−1アンチトリプシン、アルブミン、α−ラクトアルブミン(alpha-lactalbumin)、アポリポ蛋白質−E、赤血球生成因子、高糖鎖化赤血球生成促進因子、アンジオポエチン類(angiopoietin)、ヘモグロビン、トロンビン(thrombin)、トロンビン受容体活性ペプチド、トロンボモジュリン(thrombomodulin)、血液因子VII、血液因子VIIa、血液因子VIII、血液因子IX、血液因子XIII、プラスミノゲン活性因子、フィブリン−結合ペプチド、ウロキナーゼ、ストレプトキナーゼ、ヒルジン(hirudin)、プロテインC、C−反応性蛋白質、レニン抑制剤、コラゲナーゼ抑制剤、スーパーオキシドジスムターゼ、レプチン、血小板由来成長因子、上皮細胞成長因子、表皮細胞成長因子、アンギオスタチン(angiostatin)、アンギオテンシン(angiotensin)、骨形成成長因子、骨形成促進蛋白質、カルシトニン、インスリン、アトリオペプチン、軟骨誘導因子、エルカトニン(elcatonin)、結合組織活性因子、組織因子経路抑制剤(tissue factor pathway inhibitor)、卵胞刺激ホルモン、黄体形成ホルモン、黄体形成ホルモン放出ホルモン、神経成長因子類(例えば、神経成長因子、毛様体神経栄養因子(cilliary neurotrophic factor)、アキソジェネシス因子−1(axogenesis factor-1)、脳−ナトリウム利尿ペプチド(brain-natriuretic peptide)、神経膠由来神経栄養因子(glial derived neurotrophic factor)、ネトリン(netrin)、好中球抑制因子(neurophil inhibitor factor)、神経栄養因子、ニューチュリン(neuturin)など)、 副甲状腺ホルモン、レラキシン、セクレチン、ソマトメジン、インスリン様成長因子、副腎皮質ホルモン、グルカゴン、コレシストキニン、膵臓ポリペプチド、ガストリン放出ペプチド、コルチコトロピン放出因子、甲状腺刺激ホルモン、オートタキシン(autotaxin)、ラクトフェリン(lactoferrin)、ミオスタチン(myostatin)、受容体類(例えば、TNFR(P75)、TNFR(P55)、IL−1受容体、VEGF受容体、B細胞活性因子受容体など)、受容体拮抗物質(例えば、IL1−Raなど)、細胞表面抗原(例えば、CD2、3、4、5、7、11a、11b、18、19、20、23、25、33、40、45、69など)、モノクローナル抗体、ポリクローナル抗体、抗体断片類(例えば、scFv、Fab、Fab’、F(ab’)2およびFd)、ウイルス由来ワクチン抗原など様々な種類を含み、前記例示された種類に限定されない。抗体断片は、特定の抗原に結合することが可能な能力をもったFab、Fab’、F(ab’)2、FdまたはscFvであり、好ましくはFab’である。
【0068】
特に好ましい生理活性ポリペプチドは、疾病の治療または予防の目的で人体に投与されるとき、投与頻度の高いヒト成長ホルモン、インターフェロン類(インターフェロン−α、−β、−γなど)、顆粒球コロニー刺激因子、赤血球生成因子(EPO)および抗体断片類などである。また、前記生理活性ポリペプチドの天然型と実質的に同等或いはより増加した機能、構造、活性または安定性を有する限り、任意の誘導体または誘導体も本発明の生理活性ポリペプチドの範囲に含まれる。本発明の最も好適なポリペプチドの薬物はインターフェロン−アルファである。
【0069】
本発明では、ポリペプチド薬物以外に他の薬物も使用可能であり、テトラサイクリン、ミノサイクリン、ドキシサイクリン、オフロキサシン、レボフロキサシン、シプロフロキサシン、クラリスロマイシン、エリスロマイシン、セファクロール、セフォタキシム、イミペネム、ペニシリン、ゲンタマイシン、ストレプトマイシン、バンコマイシンなどの誘導体および混合物から選択される抗生剤;メトトレキサート、カルボプラチン、タクソール、シスプラチン、5−フルオロウラシル、ドキソルビシン、エトポシド、パクリタキセル、カンプトセシン、シトシンアラビノシドなどの誘導体および混合物から選択される抗癌剤;インドメタシン、イブプロフェン、ケトプロフェン、ピロキシカム、フルルビプロフェン、ジクロフェナクなどの誘導体および混合物から選択される抗炎症剤;アシクロビル、ロバビンなどの誘導体よび混合物から選択される抗ウイルス剤;およびケトコナゾール、イトラコナゾール、フルコナゾール、アンホテリシン−B、グリセオフルビンなどの誘導体および混合物から選択される抗菌剤を例示することができるが、これに限定されない。
【0070】
一方、本発明のFc断片は、リンカーを媒介として薬物に連結された結合体を形成することができる。
【0071】
このようなリンカーは、ペプチド性リンカーまたは非ペプチド性リンカーを含むが、好ましくは非ペプチド性リンカーであり、より好ましくは非ペプチド性重合体である。ここで、ペプチド性リンカーとは、アミノ酸、好ましくはペプチド結合で連結された1〜20個のアミノ酸を意味し、糖鎖化された形態であってもよい。このようなペプチド性リンカーは、Gly、Ser反復単位を持つペプチドであって、T細胞に対して免疫学的に不活性のペプチドを使用することが好ましい。非ペプチド性重合体の例としては、ポリ(エチレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールの共重合体、ポリオキシエチル化ポリオール、ポリビニルアルコール、多糖類、デキストラン、ポリビニルエーテル、PLA(poly(lactic acid))およびPLGA(poly(lactic-glycolic acid))のような生分解性高分子、脂質重合体、キチン類、ヒアルロン酸などを挙げることができ、最も好ましくはポリ(エチレングリコール)(PEG)などがある。
【0072】
本発明のFc断片−薬物または本発明のFc断片−リンカー−薬物は、様々なモル比の結合が可能である。すなわち、一つのポリペプチド薬物に結合するFc断片および/またはリンカーの数は制限されない。ところが、好ましくは本発明の薬物結合体において薬物とFc断片は、1:1〜10:1、好ましくは1:1〜2:1のモル比で結合できる。
【0073】
また、本発明のFc断片、任意のリンカーおよび薬物の結合は、遺伝子組み換え方法によってFc断片とポリペプチド薬物が融合蛋白質の形態で発現される場合のペプチド結合(peptide bond)を除いた全ての共有結合と、水素結合、イオン結合、ファンデルワールス力、疎水性相互作用のような全ての種類の非共有結合を含む。しかし、薬物の生理活性の面で共有結合であることが好ましい。
【0074】
また、本発明のFc断片、任意のリンカーおよび薬物は、薬物の任意の部位(site)との結合が可能である。一方、本発明のFc断片およびポリペプチド薬物の場合は、N末端、C末端の結合も可能であるが、遊離基と結合することがより好ましく、特にこれらのアミノ末端、リジンのアミノ残基、ヒスチジンのアミノ残基または遊離システイン残基で共有結合がよく形成される。
【0075】
一方、本発明のFc断片、任意のリンカーおよび薬物の結合は、任意の方向に結合できる。すなわち、リンカーと免疫グロブリンFc領域のN末端、C末端または遊離基との結合が可能であり、リンカーと蛋白質薬物のN末端、C末端または遊離基との結合が可能である。リンカーがペプチドリンカーの場合、任意の結合部位における結合が可能である。
また、本発明の結合体は、いろいろの架橋剤(coupling agent)で結合可能である。このような架橋剤の種類は、当該分野に公知となっており、例えば1,1−ビス(ジアゾアセチル)−2−フェニルエタン、グルタルアルデヒド、N−ヒドロキシスクシンイミドエステル、例えば4−アジドサリチル酸とのエステル、イミドエステル、例えばジスクシンイミジルエステル、例えば3,3’−ジチオビス(スクシンイミジルプロピオネート)および二官能性(bifunctional)マレイミド、例えばビス−N−マレイミド−1,8−オクタンを含むが、これに制限されない。
【0076】
一方、本発明に係る新規のFc断片および薬物の結合体は、様々な薬剤学的組成物を製造することができる。
【0077】
本発明において、「投与」はある適切な方法で患者に所定の物質を導入することを意味し、前記結合体は薬物が目的の組織に到達することができる限り、いずれの一般的な経路を介しても投与できる。例えば、腹腔内投与、静脈内投与、筋肉内投与、皮下投与、皮内投与、経口投与、局所投与、鼻内投与、肺内投与、直腸内投与が可能であるが、これに制限されない。しかしながら、経口投与の際、ペプチドは消化されるため、経口用組成物は活性薬剤をコートするか、或いは胃における分解から保護するために剤形化することが好ましい。好ましくは、注射剤の形態で投与できる。また、製薬組成物は、活性物質が標的細胞に移動することが可能な任意の装置によって投与できる。
【0078】
本発明の結合体を含んだ薬剤学的組成物は、薬剤学的に許容可能な担体を含むことができる。薬剤学的に許容される担体は、経口投与の場合には結合剤、滑沢剤、崩壊剤、賦形剤、可溶化剤、分散剤、安定化剤、懸濁化剤、色素、香料などを使用することができ、注射剤の場合には緩衝剤、保存剤、無痛化剤、可溶化剤、等張剤、安定化剤などを混合して使用することができ、局所投与の場合には基剤、賦形剤、潤滑剤、保存剤などを使用することができる。本発明の薬剤学的組成物の剤形は、上述したような薬剤学的に許容される担体と混合して様々に製造できる。例えば、経口投与の場合には錠剤、トローチ剤、カプセル、エリキシル剤(elixir)、サスペンション、シロップ、ウェーハなどの形態で製造することができ、注射剤の場合には、単位投薬アンプルまたは多数回投薬の形態で製造することができる。その他、溶液、懸濁液、錠剤、カプセル、徐方形製剤などに剤形化することができる。
【0079】
一方、製剤化に適した担体、賦形剤および希釈剤の例としては、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、キシリトール、エリスリトール、マルチトール、澱粉、アカシアゴム、アルギン酸塩、ゼラチン、リン酸カルシウム、珪酸カルシウム、セルロース、メチルセルロース、微結晶性セルロース、ポリビニルピロリドン、水、メチルヒドロキシ安息香酸、タルク、ステアリン酸マグネシウムまたは鉱物油などが使用できる。また、充填剤、抗凝集剤、潤滑剤、湿潤剤、香料、乳化剤、防腐剤などをさらに含むことができる。
【0080】
本発明において、Fc断片がキャリアとして用いられた薬物の実際投与量は、治療する疾患、投与経路、患者の年齢、性別並びに体重、および疾患の重症度などのいろいろの関連因子とともに、活性成分である薬物の種類によって決定される。本発明の薬剤学的組成物は、生体内持続性に非常に優れるので、本発明の薬剤学的製剤の投与回数および頻度を著しく減少させることができる。
【0081】
以下、下記の実施例によって本発明をより詳細に説明する。但し、下記の実施例は、本発明を例示するためのものに過ぎず、本発明の範囲を制限するものではない。
【実施例】
【0082】
免疫グロブリンFc断片の製造実施例
(実施例1)ヒト免疫グロブリンIgG4 Fc発現ベクターの製作
(1−1)二量体IgG4 Fc発現ベクターの製作
ヒト免疫グロブリンIgG4のFc遺伝子をクローニングするために、ヒトの血液から収得した血球細胞のRNAを鋳型として次のようにRT−PCRを行った。まず、Qiamp RNA血液キット(Qiagen社)を用いて約6mLの血液から全体RNAを分離した後、このRNAを鋳型とし、One−Step RT−PCRキット(Qiagen社)を用いて遺伝子を増幅した。この際、前記遺伝子配列を収得するために配列番号1と配列番号2で表わされるプライマー対を使用した。配列番号1は、IgG4ヒンジ領域の12個(Glu Ser Lys Tyr Gly Pro Pro Cys Pro Ser Cys Pro、配列番号:3)のアミノ酸配列のうち10番目のアミノ酸配列であるセリンから始まる配列であり、配列番号2は、終結コドンを含むBamHI制限酵素認識部位を挿入したものである。前記プライマー対で増幅された遺伝子は、配列番号4で表わされる塩基配列を持つことが確認され、全体IgG4 Fc中のヒンジ領域のセリン−システイン−プロリンで始まるアミノ末端とCH2およびCH3ドメインから構成される。
【0083】
前記増幅されたIgG4 Fc遺伝子配列を、大腸菌シグナル配列を含む発現ベクターにクローニングするために、本発明者によって開発された発現ベクターpT14S1SH−4T20V22Q(大韓民国特許第38061号)を出発ベクターとして用いた。前記発現ベクターは、配列番号5で表わされる塩基配列を持つ大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む。クローニングを容易にするため、pT14S1SH−4T20V22Qプラスミドの大腸菌熱安定性エンテロトキシンシグナル配列誘導体に配列番号6および7のプライマー対を使用した特定部位の置換突然変異法(site-directed mutagenesis)を行い、シグナル配列の最後のアミノ酸をコードする遺伝子配列を突然変異誘発(mutagenesis)してStuI制限酵素認識部位を挿入した。塩基配列分析法によって、正確にStuI制限酵素認識部位が生成されたことを確認した。pT14S1SH−4T20V22QプラスミドにStuI制限酵素認識部位が生成されたプラスミドをpmSTIIと命名した。前記のように製作されたプラスミドpmSTIIをStuIおよびBamHI制限酵素で処理した後、アガロースゲル電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記増幅されたIgG4 Fc遺伝子断片をBamHI制限酵素で処理した後、前記で回収された発現ベクターの断片に挿入してプラスミドpSTIIdCG4Fcを製作した。このベクターによって発現される蛋白質は、配列番号8のアミノ酸配列を持ち、ヒンジ領域のシステイン残基によるジスルフィド結合によって二量体の形態で存在する。前記製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG4Fc(HM10932)を製造し、これらを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10597)。
【0084】
(1−2)単量体IgG4 Fc発現ベクターの製作
単量体の形態で発現されるIgG4 Fc断片をクローニングするために、配列番号9および2を用いて、前記実施例(1−1)で製作したpSTIIdCG4Fcプラスミドを鋳型としてPCRを行った。前記のPCRによって増幅された遺伝子は、IgG4 Fc配列で二量体の形成に必要なヒンジ領域が除去されたまま、IgG4 CH2−3遺伝子のみが増幅されて単量体の形態で存在する。前記(1−1)と同一の発現ベクターpmSTIIに同一の過程でクローニングしてプラスミドpSTIIG4Moを製作した。前記発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIG4Mo(HM10933)を製造し、これを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10598)。前記発現ベクターによって発現される蛋白質は、配列番号10のアミノ酸配列を持ち、ヒンジ領域がないため、CH2ドメインから発現されて単量体の形態で存在する。
【0085】
(実施例2)ヒト免疫グロブリンIgG1 Fc発現ベクターの製作
(2−1)二量体IgG1 Fc発現ベクターの製作
IgG1のFc遺伝子をクローニングするために、前記実施例(1−1)で行った方法と同様に、ヒトの血液から収得した血球細胞のRNAを鋳型としてOne−Step RT−PCRキット(Qiagen社)を用いてRT−PCRを行った。この際、前記遺伝子配列を収得するために、配列番号11と配列番号12で表わされるプライマー対を使用した。
【0086】
配列番号11は、15個のアミノ酸配列(Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro;配列番号:13)からなるヒンジ領域蛋白質配列のうち13番目のアミノ酸配列であるプロリンから始まる配列であり、配列番号11および12のプライマー対で増幅された遺伝子は、全体IgG1 Fc遺伝子配列中のヒンジ領域のプロリン−システイン−プロリンで始まるアミノ末端とCH2、CH3ドメインからなり、配列番号14の遺伝子配列を持つ。
【0087】
前記増幅されたIgG1 Fc遺伝子配列を大腸菌シグナル配列を含む発現ベクターにクローニングするために、前述したpmSTIIベクターを使用した。前記実施例(1−1)で行ったクローニング過程と類似の方法で、プラスミドpmSTIIをStuI/BamHI制限酵素で処理した後、アガロースゲルに電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記で増幅されたIgG1 Fc遺伝子をBamHI制限酵素で処理した後、前記で回収された発現ベクターの断片に挿入してpSTIIdCG1Fcを製作した。このように製作された発現ベクターを介して発現される産物は、宿主細胞で発現するときに配列番号15のアミノ酸配列を持ち、ヒンジ領域のシステイン残基間のジスルフィド結合によって二量体を形成することができる。製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG1Fc(HM10927)を製造し、これらを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10588)。
【0088】
(2−2)単量体IgG1 Fc発現ベクターの製作
単量体の形態で発現されるIgG1 Fcを製造するために、配列番号16および12のプライマー対を用いて、前記実施例(2−1)で製作したプラスミドpSTIIdCG4Fcを鋳型として遺伝子を増幅した。増幅された遺伝子の断片を実施例(2−1)と同様の方法で発現ベクターpmSTIIに挿入し、配列番号17で表わされる塩基配列を含有するPSTIIG1Moを製作した。前記発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIG1Mo(HM10930)を製造し、これを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10595)。前記発現ベクターによって発現される蛋白質は、二量体の形成を可能とするシステイン残基を含有するヒンジ領域が除去されたため、CH2ドメインから発現されて単量体の形態で存在し、配列番号18で表わされるアミノ酸配列を持つ。
【0089】
(実施例3)ヒト免疫グロブリンIgG2 Fc発現ベクターの製作
ヒトIgG2のFc遺伝子をクローニングするために、前記実施例(1−1)で行った方法と同様に、ヒトの血液から収得した血球細胞のRNAを鋳型としてOne−Step RT−PCRキット(Qiagen社)を用いてRT−PCRを行った。この際、前記遺伝子配列を収得するために、配列番号19と配列番号20で表わされるプライマー対を使用した。配列番号19は12個のアミノ酸配列(Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro;配列番号:21)からなるヒンジ領域蛋白質配列のうち10番目のアミノ酸配列であるプロリンから始まる配列であり、配列番号19および20のプライマー対で増幅された遺伝子は、全体IgG2 Fc遺伝子配列中のヒンジ領域のプロリン−システイン−プロリンで始まるアミノ末端とCH2、CH3ドメインからなり、配列番号22の遺伝子配列を持つ。前記増幅されたIgG2 Fc遺伝子配列を大腸菌シグナル配列を含む発現ベクターにクローニングするために、前述したpmSTIIベクターを使用した。前記実施例(1−1)で行ったクローニング過程と類似の方法で、プラスミドpmSTIIをStuI/BamHI制限酵素で処理した後、アガロースゲル電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記増幅されたIgG2 Fc遺伝子をBamHI制限酵素で処理した後、前記回収された発現ベクターの断片に挿入してpSTIIdCG2Fcを製作した。このように製作された発現ベクターによって発現される産物は、宿主細胞で発現するときに配列番号23のアミノ酸配列を持ち、ヒンジ領域のシステイン残基間のジスルフィド結合によって二量体を形成することができる。それぞれ製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG2Fc(HM10936)を製造した。
【0090】
(実施例4)免疫グロブリンFcの発現および精製
(4−1)免疫グロブリンFcの発現の確認
前記実施例1、2および3で収得した微生物形質転換体を発酵器(Marubishi社)に接種して発酵させた後、免疫グロブリンFc断片の発現の有無を確認した。
まず、LB培地100mLに前記形質転換体をそれぞれ一晩中振盪培養した後、発酵器に接種して本培養を行った。発酵器の温度は、35℃あるいは30℃を維持し、嫌気性状態になることを防止するために、空気を20vvmで投入しながら500rpmで攪拌した。発酵の間、微生物の成長のために足りないエネルギー源を補うために、葡萄糖(glucose)と酵母抽出液(yeast extract)とを微生物の発酵状態に応じて投与した。吸光度600nmでOD値が80〜110となる時期に誘導物質(inducer)IPTGを20μM〜4mMとなるように投与して発現を誘導した。これを、さらに40〜45時間高濃度で培養して吸光度600nmでのOD値が100〜120となるようにした。
【0091】
前記大腸菌形質転換体から免疫グロブリンFcの発現の有無、発現場所、水溶性および二量体形成の有無を確認するために、下記のような試験を行った。発現産物が前記発現ベクターに融合しているシグナル配列によって発酵液またはペリプラスム空間に発現されるかを確認するために、発酵液を遠心分離して、細胞を除去した発酵液溶液を収得し、且つ細胞を回収した。前記細胞を除去した発酵液溶液と、前記回収した細胞に浸透圧ショック(osmotic shock)をかけることによって得られたペリプラスム空間溶液とをウエスタンブロットによって解析し、免疫グロブリンFcの発現の有無を確認したが、その量は極めて少量であった。前記細胞をウルトラ−ソニケーション(Misonix社)を用いて破砕して細胞溶解液(cell lysate)を得、その細胞溶解液を遠心分離して水溶性物質と不溶性物質を分離した後、水溶性物質を次のような方法でウエスタンブロットした。前記水溶性物質をDTTまたはβ−メルカプトエタノールのような還元剤を含まない蛋白質サンプルバッファと混ぜた後、15%SDS−PAGE(Criterion Gel、Bio−Rad社)に電気泳動した。電気泳動済みのゲルをニトロセルロース(nitro-cellulose)膜に転写した後、HRPが融合している抗ヒトFc抗体(Sigma)を用いて検出(detect)を行った。図1に示すように、免疫グロブリンFcは、大腸菌の細胞質内に水溶性の状態で過量に発現されることが分かるとともに、ヒンジ領域の一部が挿入された形質転換体から発現された産物は二量体を形成することが分かる。図1のレーン1、2、3はそれぞれ形質転換体HM10927、HM10932、HM10936で発現された産物であり、レーン4は動物細胞で生産された免疫グロブリンをパパイン処理して生成されたFcであって、糖が存在するため、大腸菌で生成されたものよりやや大きく展開されることが分かる。
【0092】
(4−2)N末端配列の確認
前記実施例において大腸菌の細胞質内で水溶性二量体の形態で発現されるFcは、シグナル配列が融合しているために分泌されず、細胞質内に存在するとき、シグナル配列のプラセシングなしで融合したままで存在するかを確認するために、N末端アミノ酸配列の分析を基礎科学支援研究所のソウル分所に依頼した。分析する試料は下記のように準備した。
【0093】
まず、PVDF膜(Bio−Rad社)をメタノールに約2〜3秒間浸漬して活性化させた後、ブロッキングバッファ(170mMグリシン、25mM Tris−HCl(pH8)、20%メタノール)に十分濡らした。実施例(4−1)で展開した非還元条件のSDS−PAGEゲルを、前記のように準備されたPVDF膜に、ブロッティングキット(Hoefer Semi−Dry Transfer unit、Amersham)を用いて約1時間の転写を行った。PVDF膜に転写された蛋白質を蛋白質染色薬であるCoomassie Blue R−250(Amnesco社)を用いてしばらく(3〜4秒間)染色した後、脱色溶液(水:酢酸:メタノールが5:1:4)で洗浄した。洗浄済みの膜から、蛋白質が含まれている部位をハサミで切断してN末端配列の分析を依頼した。
【0094】
IgG1 Fc蛋白質のN末端配列はPro−Cys−Pro−Ala−Pro−Glu−Leu−Leu−Gly−Glyであり、IgG4 Fc蛋白質のN末端配列はSer−Cysl−Pro−Ala−Pro−Glu−Phe−Leu−Gly−Glyである。また、IgG2 Fc蛋白質のN末端配列はPro−Cys−Pro−Ala−Pro−Pro−Val−Ala−Gly−Proと確認された。以上のようなアミノ酸配列の分析結果から明らかなように、本発明の大腸菌形質転換体から発現されるFc断片は、正確なN末端配列を持つものと確認された。また、シグナル配列を融合させて発現させる際に、細胞外膜またはペリプラスムに分泌されず、細胞質内に過発現されてもシグナル配列が正確にプロセシングされて水溶性の形態で存在するという新しい事実が確認された。
【0095】
(4−3)免疫グロブリンFcの精製
免疫グロブリンに強い親和性があると知られているプロテインA親和性カラム(protein-A affinity column)を用いて下記のような方法で精製した。
【0096】
遠心分離によって発酵液から回収された大腸菌細胞をMicrofluizer(Microfludics社)を用いて破砕して細胞溶解液(cell lysate)を得、細胞質内に存在する組み換え免疫グロブリンFc断片を2段階カラムクロマトグラフィによって精製した。プロテインA親和性カラム(Pharmacia社)5mLをPBSで平衡化させた後、細胞溶解液を5mL/分の流速で負荷した。結合していない分画はPBSで洗浄した後、100mMのクエン酸溶液(pH3.0)で溶出させた。溶出した分画を脱塩カラム(desalting column Hiprep 26/10、Pharmacia社)を用いて10mM Tris緩衝液(pH8.0)で交換した。Q HP 26/10カラム(Pharmacia社)50mLを用いて2次陰イオン交換カラムクロマトグラフィを行った。1次精製された組み換え免疫グロブリンFc分画を前記カラムに結合させた後、10mM Tris緩衝液(pH8.0)条件下で直線濃度勾配(塩化ナトリウムの濃度0M→0.2M)法によって溶出し、高純度の分画を得ることができた。各産物をプロテインAカラムを用いて部分精製した後、組み換え免疫グロブリンFc断片の発現量が下記に示したように決定された。
【0097】
【化1】

【0098】
前記のように収得された免疫グロブリンFcは、重鎖の二量体あるいは単量体の形態なので、還元条件のSDS−PAGEと非還元条件のSDS−PAGEにおいて蛋白質の移動様相が異なる。発現された産物を精製した後、純度を確認するために行ったSDS−PAGE結果を、図2および図3に示した。
【0099】
図2および図3は、このように精製された二量体の形態と単量体の形態の免疫グロブリンFc蛋白質を15%クライテリオンゲル(criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させてその移動様相を比較したものである。図2のAは非還元条件でSDS−PAGEゲルに展開したもので、Bは還元条件でSDS−PAGEゲルに展開したものである。レーンMは予め染色された低範囲標準蛋白質マーカー(prestained low-range standard marker、Bio−Rad社)であり、レーン1〜4はそれぞれ形質転換体HM10927、HM10928(韓国微生物保存センターに2004年9月15日付で寄託した。寄託番号:KCCM−10589)、HM10929(韓国微生物保存センターに2004年9月15日付で寄託した。寄託番号:KCCM−10594)およびHM10932によって生産された免疫グロブリンFc試料である。図2に示すように、還元条件のSDS−PAGEでは、免疫グロブリンFc断片は、ヒンジ領域のシステイン残基によるジスルフィド結合が還元されて、それぞれ単量体の形態で存在するので、単量体の大きさだけ移動した。一方、非還元条件のSDS−PAGEでは、免疫グロブリンFc断片は、それぞれの単量体がジスルフィド結合で連結された二量体の形態で存在し、その結果、約42kDaの移動距離を持つことを確認した。
【0100】
図3のAは、非還元条件で、蛋白質をSDS−PAGEゲルに展開したものであり、Bは還元条件で、蛋白質をSDS−PAGEゲルに展開したものである。レーンMは、前記標準蛋白質マーカーであり、レーン1および2は、形質転換体HM10930およびHM10933によって生産された免疫グロブリンFc試料である。図3に示すように、還元条件と非還元条件とにおける移動距離はあまり差異がなく、分子内ジスルフィド結合の還元有無によって移動距離に若干の差異があることを確認した。
【0101】
免疫グロブリンFcおよび薬物の結合体製造
(実施例5)インターフェロンアルファ(IFNα)−PEG−免疫グロブリンFc領域(Fc)結合体の製造I
(段階1)免疫グロブリンを用いた免疫グロブリンFc領域の製造
免疫グロブリンFc領域を製造するために、10mMのリン酸塩緩衝液に溶解された分子量150kDaの免疫グロブリンG(Immunoglobulin G;IgG、緑十字)200mgを、蛋白質加水分解酵素パパイン(Papain、Sigma社)2mgを用い、37℃で2時間徐々に攪拌しながら処理した。酵素反応後、生成された免疫グロブリンFc領域を精製するために、Superdexカラム、プロテインAカラムおよび陽イオン交換樹脂カラムクロマトグラフィを順次行った。具体的には、反応液を、10mMのリン酸ナトリウム緩衝液(PBS、pH7.3)によって平衡化させたSuperdex200カラム(Pharmacia社)に滴下し、同一の緩衝液を用いて1mL/分の流速で溶出させた。免疫グロブリンFc領域より分子量が相対的に大きい未反応免疫グロブリン(IgG)やF(ab’)2などは、早く溶出するので、これを先に除去した。免疫グロブリンFc領域と類似の分子量を持つFabは、次のように、プロテインAカラムクロマトグラフィを行って除去した(図4)。20mMのリン酸塩緩衝液(pH7.0)で平衡化させたプロテインAカラム(Pharmacia社)にSuperdex200カラムから溶出した免疫グロブリンFc領域含有分画を5mL/分の流速で負荷した後、カラムに結合していない蛋白質を除去するために同一の緩衝液で十分洗浄した。次に、100mMのクエン酸ナトリウム(Na citrate、pH3.0)緩衝液を流して高純度の免疫グロブリンFc領域を溶出させた。最後に、プロテインAカラムで精製されたFc分画を陽イオン交換樹脂カラム(polyCAT、PolyLC社)を用いて精製した。プロテインAカラムで精製されたFc分画を滴下した前記陽イオン交換樹脂カラムは、10mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法によって溶出し、高純度のFc分画を得た。前記高純度のFc分画を12%SDS−PAGEによって解析した(図5のレーン2)。
【0102】
(段階2)IFNα−PEG連結体の製造
両末端にアルデヒド反応基を持つ分子量3.4kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を、ヒトインターフェロンアルファ−2b(hIFNα−2b、分子量20kDa)が5mg/mLの濃度で溶解された100mMのリン酸塩緩衝液に、IFNα:PEGのモル比が1:1、1:2.5、1:5、1:10および1:20となるように添加した。ここに還元剤のナトリウムシアノボロハイドライド(NaCNBH3、Sigma社)を最終濃度が20mMとなるように添加し、4℃で徐々に攪拌しながら3時間反応させた。インターフェロンアルファのアミノ末端部位に選択的にPEGを連結され、PEGとインターフェロンアルファが1:1で結合した連結体を得るために、前記反応混合物をSuperdexRカラム(Pharmacia社)を用いたサイズ排除(size exclusion)クロマトグラフィにかけた。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてIFNα−PEG連結体を精製し、PEGと結合していないインターフェロンアルファ、未反応PEGおよび2つのインターフェロンアルファがPEGと連結された二量体副産物を除去した。精製されたIFNα−PEG連結体を5mg/mLの濃度に濃縮した。これより、反応性が最も良くて二量体などの副産物が少ないIFNα:PEGの最適反応モル比は1:2.5〜1:5であることを確認した。
【0103】
(段階3)IFNα−PEG−Fc結合体の形成
前記段階2で精製されたIFNα−PEG連結体に免疫グロブリンFc領域のN末端に結合させるために、段階1で準備された免疫グロブリンFc領域(約53kDa)を10mMのリン酸塩緩衝液に溶解させた後、IFNα−PEG連結体:Fcのモル比がそれぞれ1:1、1:2、1:4および1:8となるように、IFNα−PEG連結体と混合して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加し、4℃で20時間徐々に攪拌しながら反応させた。これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:Fcの最適反応モル比は1:2であることを確認した。
【0104】
(段階4)IFNα−PEG−Fc結合体の分離および精製
前記段階3の結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−Fc蛋白質結合体を精製するためにSuperdexサイズ排除クロマトグラフィを行った。反応混合物を濃縮した後、10mMのリン酸塩緩衝液(pH7.3)を用いて流速2.5mL/分でカラムを通過させ、結合していないFcおよび未反応物質を除去し、IFNα−PEG−Fc蛋白質結合体分画を得た。得られたIFNα−PEG−Fc蛋白質結合体分画には不純物として少量の未反応Fcおよびインターフェロンアルファ二量体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−Fc蛋白質結合体分画を10mMの酢酸ナトリウム(pH4.5)で平衡化させたPolyCAT LPカラム(PolyLC社)に仕込み、1Mの塩化ナトリウム(NaCl)を含む10mMの酢酸ナトリウム(pH4.5)緩衝液を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.5M)法で溶出してさらに精製した。最後に、陰イオン交換カラムを用いてIFNα−PEG−Fc蛋白質結合体を精製した。PolyWAX LPカラム(PolyLC社)を10mMのTris−HCl(pH7.5)緩衝液で平衡化させた後、精製されたIFNα−PEG−Fc蛋白質結合体分画を負荷し、1Mの塩化ナトリウムを含む10mMのTris−HCl(pH7.5)緩衝液を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.3M)法で溶出して純粋なIFNα−PEG−Fc蛋白質結合体を精製した。
【0105】
(実施例6)IFNα−PEG−Fc結合体の製造II
(段階1)Fc−PEG連結体の製造
両末端にアルデヒド反応基を持つ分子量3.4kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を、前記実施例5の段階1で準備された免疫グロブリンFc領域が15mg/mLの濃度で溶解された100mMのリン酸塩緩衝液に、免疫グロブリンFc:PEGのモル比がそれぞれ1:1、1:2.5、1:5、1:10および1:20となるように添加した。還元剤のNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しながら3時間反応させた。免疫グロブリンFc領域のアミノ末端部位に選択的にPEG:Fcが1:1で結合した連結体を得るために、前記反応混合物を持ってSuperdex(SuperdexR、Pharmacia社)サイズ排除クロマトグラフィを行った。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてFc−PEG連結体を精製し、PEGと結合していない免疫グロブリンFc領域、未反応PEGおよび2つの免疫グロブリンFc領域がPEGに連結された二量体副産物を除去した。精製されたFc−PEG連結体を約15mg/mLの濃度で濃縮した。これより、反応性が最も良くて二量体などの副産物が少ないFc:PEGの最適反応モル比は1:3〜1:10であることを確認した。
【0106】
(段階2)Fc−PEG連結体とインターフェロンアルファの結合体の形成および精製
前記段階1で精製されたFc−PEG連結体をIFNαのN末端に結合させるために、10mMのリン酸塩緩衝液に溶解されたインターフェロンアルファを使用し、Fc−PEG連結体:IFNαのモル比がそれぞれ1:1、1:1.5、1:3および1:6となるように添加して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。反応後、実施例5の段階4と同一の方法で精製して未反応物質および副産物を除去し、これから生成されたFc−PEG−IFNα蛋白質結合体を純粋に分離した。
【0107】
(実施例7)ヒト成長ホルモン(hGH)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト成長ホルモン(hGH、分子量22kDa)を使用し、hGH:PEGのモル比を1:5とする以外は、実施例5と同一の方法でhGH−PEG−Fc蛋白質結合体を製造および精製した。
【0108】
(実施例8)ヒト顆粒球コロニー刺激因子(G−CSF)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト顆粒球コロニー刺激因子(G−CSF)を使用し、G−CSF:PEGのモル比を1:5とする以外は、実施例5と同一の方法でG−CSF−PEG−Fc蛋白質結合体を製造および精製した。
【0109】
一方、天然型G−CSFの17番目のアミノ酸がセリンで置換された誘導体(17S−G−CSF)を用いて前記と同一の方法で17S−G−CSF−PEG−Fc蛋白質結合体を製造および精製した。
【0110】
(実施例9)ヒト赤血球生成因子(EPO)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト赤血球生成因子(Erythropoietin;EPO)を使用し、EPO:PEGのモル比を1:5とする以外は、実施例5と同一の方法でEPO−PEG−Fc蛋白質結合体を製造および精製した。
【0111】
(実施例10)反応基の種類を異にするPEGを用いた蛋白質結合体の製造
両末端の反応基が全てスクシンイミジルプロピオネート(Succinimidyl propionate;SPA)であるPEGを用いてIFNα−PEG−Fc蛋白質結合体を次のように製造した。インターフェロンアルファ10mgが溶解された100mMのリン酸塩緩衝液に、両末端にSPA反応基を持つ分子量3.4kDaのポリエチレングリコールSPA−PEG−SPA(Shearwater社)をIFNα:PEGのモル比がそれぞれ1:1、1:2.5、1:5、1:10および1:20となるように添加し、常温で徐々に攪拌しながら2時間反応させた。インターフェロンアルファのリジン残基のアミノ基部位に選択的にPEGが1:1で結合したPEG−IFNα連結体を得るために、反応混合物をもってSuperdexサイズ排除クロマトグラフィを行った。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてIFNα−PEG連結体を精製し、PEGと結合していないインターフェロンアルファ、未反応PEG、およびPEGの両末端に2つのインターフェロンアルファが連結された二量体副産物を除去した。IFNα−PEG連結体を免疫グロブリンFcのリジン残基のアミノ基部位に結合させるために精製されたIFNα−PEG連結体を約5mg/mLの濃度で濃縮した後、実施例5の段階3および4と同一の方法でIFNα−PEG−Fc結合体を製造および精製した。これより、反応性が最も良くて二量体などの副産物が少ないインターフェロンアルファ:PEGの最適反応モル比は1:2.5〜1:5であることを確認した。
【0112】
一方、前記と同一の方法を行うが、両末端の反応基が全てN−ヒドロキシスクシンイミジル(N-hydroxysuccinimidyl;NHS)であるPEG(NHS−PEG−NHS;Shearwater社)、または両末端の反応基が全てブチルアルデヒド(buthyl aldehyde)であるPEG(BUA−PEG−BUA;Shearwater社)を用いてIFNα−PEG−Fc結合体の生成を確認した。
【0113】
(実施例11)分子量の異なるPEGを用いた蛋白質結合体の製造
両末端にアルデヒド反応基を持つ分子量10kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を用いて実施例5の段階2と同一の方法でIFNα−10K PEG連結体を製造および精製した。この際、反応性が最も良くて二量体などの副産物が少ないインターフェロンアルファ:10K PEGの最適モル比は1:2.5〜1:5であることが確認された。精製されたIFNα−PEG連結体を約5mg/mLとなるように濃縮した後、これを用いて実施例5の段階3および4と同一の方法によってIFNα−10K PEG−Fc蛋白質結合体を製造および精製した。
【0114】
(実施例12)Fab’−S−PEG−N−Fc結合体の製造(−SH基)
(段階1)Fab’の発現および精製
抗腫瘍壊死因子−アルファFab’を発現する大腸菌形質転換体BL21/poDLHF(寄託番号:KCCM−10511)をLB培地100mLに接種して一晩中振盪培養した後、5Lの発酵器(Marubishi)に接種して温度30℃、空気投入量20vvm、攪拌速度500rpmの条件下で培養した。発酵が進むにつれて、微生物の成長のために足りないエネルギー源を補うために、葡萄糖(glucose)と酵母抽出液(yeast extract)とを微生物の発酵状態に応じて投与し、吸光度600nmでのOD値が80となる時期にIPTGを投与して蛋白質の発現を誘導した。これを40〜45時間高濃度で培養して吸光度600nmでのOD値が120〜140となるようにした。得られた発酵液を遠心分離(20,000g、30分)して沈殿物は捨て、上澄み液のみを取った。
【0115】
得られた上澄み液から次の3段階のカラムクロマトグラフィを経て抗腫瘍壊死因子−アルファFab’を純粋に精製した。20mMのリン酸塩緩衝液(pH7.0)で平衡化させたHiTrap蛋白質Gカラム(5mL、Pharmacia社)に前記上澄み液を滴下した後、100mMのグリシン(Glycine、pH3.0)緩衝液を用いて溶出させた。溶出したFab’分画を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200, Pharmacia社)に滴下し、同一の緩衝液で溶出させた。溶出したFab’分画をpolyCAT 21×250カラム(PolyLC社)を用いて最終精製したが、10mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法で溶出して純粋な抗腫瘍壊死因子−アルファFab’分画を得た。
【0116】
(段階2)Fc−PEG連結体の製造および精製
免疫グロブリンFcのN末端のアミノ基にリンカーPEGを結合させるために、前記実施例5の段階1と同一の方法で製造された免疫グロブリンFcを100mMのリン酸ナトリウム緩衝液(pH6.0)に5mg/mLの濃度で溶解させ、ここにNHS−PEG−MAL(3.4kDa、Shearwater社)をFc:PEGのモル比が1:10となるように添加した後、4℃で徐々に攪拌しながら12時間反応させた。
【0117】
反応終了後、反応緩衝液を20mMのリン酸ナトリウム緩衝液(pH6.0)で交換しながら、反応していないNHS−PEG−MALを除去した。緩衝液の交換後、反応物をpolyCATカラム(PolyLC社)に負荷し、20mMのリン酸ナトリウム緩衝液(pH6.0)を直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)方法で流して免疫グロブリンFc−PEG連結体を先に溶出させて得た後、反応していない免疫グロブリンFcを溶出させて除去した。
【0118】
(段階3)Fab’−S−PEG−N−Fc結合体(−SH基)の製造および精製
Fab’の遊離システイン基に免疫グロブリンFc−PEG連結体を結合させるために、段階1で精製されたFab’を100mMのリン酸ナトリウム緩衝液(pH7.3)に2mg/mLの濃度で溶解させた後、同一の緩衝液に段階2で準備された免疫グロブリンFc−PEG連結体をFab’:連結体のモル比が1:5となるように入れた。最終蛋白質の濃度が50mg/mLとなるように濃縮し、4℃で徐々に攪拌しながら24時間反応させた。
【0119】
カップリング反応の終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。カップリングされたFab’−S−PEG−N−Fc結合体は、分子量が大きいので先に溶出し、反応していない免疫グロブリンFc−PEG連結体およびFab’は後で溶出するため、除去することができた。残存する未反応免疫グロブリンFcを完全に除去するために、溶出したFab’−S−PEG−N−Fc結合体分画をさらにpolyCAT 21×250カラム(PolyLC社)に滴下し、20mMのリン酸ナトリウム緩衝液(pH6.0)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、Fc−PEG連結体がFab’のC末端付近の−SH基に連結されたFab’−S−PEG−N−Fc結合体を純粋に得た。
【0120】
(実施例13)Fab’−N−PEG−N−Fc結合体の製造(N末端)
(段階1)Fab’−PEG連結体(N末端)の製造および精製
前記実施例12の段階1で得た、精製されたFab’40mgを100mMのリン酸ナトリウム緩衝液(pH6.0)に5mg/mLの濃度で溶解させた後、ブチルALD−PEG−ブチルALD(3.4kDa、Nektar社)をFab’:PEGのモル比が1:5となるように添加した。還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しがら2時間反応させた。
【0121】
反応終了後、反応緩衝液を20mMのリン酸ナトリウム緩衝液(pH6.0)で交換した。緩衝液の交換後、反応物をpolyCATカラム(PolyLC社)に負荷し、20mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法で溶出し、Fab’のN末端にリンカーPEGが結合したFab’−PEG連結体分画を先に溶出させて得た後、反応していないFab’を溶出させて除去した。
【0122】
(段階2)Fab’−N−PEG−N−Fc結合体の製造および精製
段階1で精製されたFab’−PEG連結体を免疫グロブリンFcのN末端に結合させるために、100mMのリン酸ナトリウム緩衝液(pH6.0)に10mg/mLの濃度で溶解させた後、同一の緩衝液に溶解された免疫グロブリンFcをFab’−PEG連結体:Fcのモル比が1:5となるように入れた。最終蛋白質の濃度が50mg/mLとなるように濃縮し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しながら24時間反応させた。
【0123】
カップリング反応の終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。カップリングされたFab’−N−PEG−N−Fc結合体は、分子量が大きいので先に溶出し、反応していない免疫グロブリンFcおよびFab’−PEG連結体は後で溶出するため除去することができた。残存する未反応免疫グロブリンFcを完全に除去するために、溶出したFab’−N−PEG−N−Fc結合体分画をさらにpolyCAT 21×250カラム(PolyLC社)に負荷し、20mMのリン酸ナトリウム緩衝液(pH6.0)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、免疫グロブリンFc−PEG連結体がFab’のN末端に連結されたFab’−N−PEG−N−Fc結合体を純粋に得た。
【0124】
(実施例14)脱糖鎖化免疫グロブリンFcの製造および精製
前記実施例5と同一の方法で製造した免疫グロブリンFc200mgを、100mMのリン酸塩緩衝液(pH7.5)に2mg/mLの濃度となるように溶解した後、脱糖鎖化酵素PNGase F(NEB社)を300U/mgとなるように添加した。反応混合物を37℃で24時間徐々に攪拌しながら反応させた。反応終了後、脱糖鎖化免疫グロブリンFcを精製するために、反応物をSPセファロースFFカラム(Pharmacia社)に負荷し、10mMの酢酸塩緩衝液(pH4.5)条件で1MのNaClを使用した直線濃度勾配(0.1M→0.6M)法で溶出させ、天然型免疫グロブリンFc分画を先に溶出させた後、脱糖鎖化免疫グロブリンFc(deglycosylated FC:DG Fc)を溶出させて得た。
【0125】
(実施例15)IFNα−PEG−DG Fc結合体の製造
前記実施例5の段階2で精製されたIFNα−PEG連結体に、前記実施例14で製造された脱糖鎖化免疫グロブリンFcを結合させるために、IFNα−PEG連結体を10mMのリン酸塩緩衝液に溶解されたDG FcにIFNα−PEG連結体:DG Fcのモル比がそれぞれ1:1、1:2、1:4および1:8となるように添加して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。
【0126】
これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:DG Fcの最適反応モル比は、1:2であることを確認した。
【0127】
結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−DG Fc蛋白質結合体を精製するために、反応混合物を用いてSuperdex(SuperdexR、Pharmacia社)サイズ排除クロマトグラフィを行った。リン酸塩緩衝液(pH7.3)を用いて流速2.5mL/分でカラムに通過させ、結合していないDG Fcおよび未反応物質を除去し、IFNα−PEG−DG Fc蛋白質結合体分画を精製した。
【0128】
得られたIFNα−PEG−DG Fc蛋白質結合体分画には、不純物として少量の未反応DG FcおよびIFNα−PEG連結体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−DG Fc蛋白質結合体分画を10mMの酢酸ナトリウム(pH4.5)で平衡化させたPolyCAT LPカラム(PolyLC社)に仕込み、1MのNaClを含んだ10mMの酢酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.6M)法で溶出してさらに精製し、最後に陰イオン交換カラムを用いてIFNα−PEG−DG Fc蛋白質結合体を純粋に獲得した。PolyWAX LPカラム(PolyLC社)を10mMのTris−HCl緩衝液(pH7.5)で平衡化させた後、精製されたIFNα−PEG−DG Fc蛋白質結合体分画を負荷し、1Mの塩化ナトリウムを含んだ10mMのTris−HCl緩衝液(pH7.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.3M)法で溶出して純粋なIFNα−PEG−DG Fc蛋白質結合体を精製した。
【0129】
(実施例16)IFNα−PEG連結体と組み換えAG Fc誘導体の結合体の製造
前記実施例5および15と同一の方法により、実施例1で製造されたAG Fc誘導体であるIgG4 delta−CysのN末端にIFNα−PEG連結体を結合させた。実施例1で製造されたAG Fc誘導体であるIgG4 delta−CysをIFNα−PEG連結体と結合させた。結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−AG Fc蛋白質結合体(I)を精製するために、Q HP 26/10カラム(Pharmacia社)50mLを用いて1次精製した後、高圧カラムのpolyCAT 21.5×250カラム(PolyLC社)で高純度の結合体を精製した。カップリング反応液を脱塩カラムHiPrep 26/10(Pharmacia社)を用いて10mMのTris緩衝液(pH8.0)で交換した後、Q HP26/10 50mLカラムに8mL/分の流速で負荷して結合させた後、直線濃度勾配(塩化ナトリウムの濃度0M→0.2M)法によって所望の分画を得た。溶出した分画を10mMの酢酸塩緩衝液(pH5.2)で平衡化されたpolyCAT21.5×250カラムに15mL/分の流速でさらに結合させた後、直線濃度勾配(塩化ナトリウムの濃度0.1M→0.3M)法で溶出させて高純度の分画を得ることができた。同一の方法により、実施例12で製造された別のAG Fc誘導体であるIgG4単量体を用いてIFNα−PEG−AG Fc蛋白質結合体(II)を製造した。
【0130】
(実施例17)ヒト赤血球生成因子(EPO)−PEG−組み換えAG Fc誘導体の結合体の製造
前記実施例16と同一の方法により、EPO−PEG連結体とAG Fc誘導体であるIgG4 delta−Cysとが連結された結合体を製造した。
【0131】
(比較例1)IFNα−40K PEG連結体の製造
100mMのリン酸カリウム緩衝液(pH6.0)にインターフェロンアルファ5mgを溶解して最終体積が5mLとなるように調製した後、PEGの分子量40kDaの活性化メトキシ−PEG−アルデヒド(Shearwater社)をインターフェロンアルファ:40K PEGのモル比が1:4となるように前記溶液に添加した。前記反応溶液に還元剤NaCNBH3を最終濃度が20mMとなるように添加した後、4℃で18時間徐々に攪拌させながら反応させた。インターフェロンアルファに反応していないPEGを不活性化させるために、エタノールアミンを最終濃度が50mMとなるように添加した。
【0132】
未反応PEGの分離および緩衝液の交替のためにSephadexG−25カラム(Pharmacia社)を用いた。まず、2カラム体積(column volume;CV)の10mM Tris−HCl緩衝液(pH7.5)を用いてカラムを平衡化させた後、反応混合物を滴下した。溶出画分は、紫外部吸光計により波長260nmの吸収を測定することによって検出した。前記カラムを同一の緩衝液で溶出させると、大きさが更に大きいPEGによって(N末端に:挿入KYM)修飾されたインターフェロンアルファが先に溶出し、未反応PEGは時差をおいて後で溶出するため、IFNα−40K PEGのみを分離することができる。
【0133】
前記で得た溶出液からIFNα−40K PEG連結体をさらに純粋に分離、精製するために、次のようにクロマトグラフィを行った。3mLのPolyWAX LPカラム(Polywax社)を10mMのTris−HCl緩衝液(pH7.5)で平衡化させた。PEG−IFNα連結体を含有する溶出液を1mL/分の流速でカラムに負荷した後、15mLの平衡緩衝液で洗浄した。30mLの1M NaCl緩衝液を用いた0〜100%の塩濃度勾配法を用いてトリ−、ジ−およびモノ−PEGが結合したインターフェロンアルファを順序とおりに溶出させた。
【0134】
さらに純粋なモノ−PEG結合インターフェロンアルファを分離するために、前記で収得したモノ−PEGとインターフェロンアルファとの連結体溶出分画をサイズ排除クロマトグラフィにかけた。10mMのリン酸ナトリウム緩衝液(pH7.0)で平衡化させたSuperdex200(Superdex 200、Pharmacia社)に前記溶出液を濃縮して滴下した後、同一の緩衝液で溶出させた。この際、1mL/分の流速で緩衝液を流した。トリ−およびジ−PEGが結合したインターフェロンアルファは、モノ−PEGが結合したインターフェロンアルファより溶出時間が相対的に早いので、これを除去して、モノ−PEGが結合したインターフェロンアルファのみを純粋に分離した。
同一の方法により、ヒト成長ホルモン、顆粒球コロニー刺激因子およびその誘導体のアミノ末端に40K PEGが結合したhGH−40K PEG、G−CSF−40K PEGおよび40K PEG−17S−G−CSF誘導体の連結体を製造した。
【0135】
(比較例2)IFNα−PEG−アルブミン結合体の製造
実施例1の段階2で精製されたIFNαF−PEG連結体をアルブミンのアミノ末端に結合させるために、10mMのリン酸塩緩衝液に溶解されたヒトアルブミン(human serum albumin、HSA、約67kDa、緑十字)をIFNα−PEG連結体:アルブミンのモル比が1:1、1:2、1:4および1:8となるように添加した後、反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:アルブミンの最適反応モル比は、1:2であることを確認した。
【0136】
結合反応の後、未反応物質および副産物を除去し、生成されたIFNα−PEG−アルブミン蛋白質結合体を精製するために、Superdexサイズ排除クロマトグラフィを行った。各反応混合物を濃縮した後、10mMの酢酸ナトリウム緩衝液を2.5mL/分の流速でカラムに通過させ、結合していないアルブミンおよび未反応物質を除去し、IFNα−PEG−アルブミン蛋白質結合体のみを精製した。得られたIFNα−PEG−アルブミン蛋白質結合体分画には、不純物として少量の未反応アルブミンおよびインターフェロンアルファ二量体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−アルブミン蛋白質結合体分画を10mMの酢酸ナトリウム溶液(pH4.5)で平衡化させたカラム(SP5PW、Waters社)に負荷し、1Mの塩化ナトリウム(NaCl)を含んだ10mMの酢酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.5M)法で溶出してさらに精製し、IFNα−PEG−アルブミン蛋白質結合体を純粋に獲得した。
【0137】
同一の方法により、ヒト成長ホルモン、G−CSFおよびその誘導体にそれぞれアルブミンが結合したhGH−PEG−アルブミン、G−CSF−PEG−アルブミンおよび17S−G−CSF−PEG−アルブミン結合体を製造した。
【0138】
(比較例3)Fab’−S−40K PEG連結体の製造
実施例8の段階1で精製されたFab’に存在する遊離システイン基を活性化させるために、活性化緩衝液(20mMの酢酸ナトリウム(pH4.0)、0.2mMのDTT)に1時間放置した。PEG修飾緩衝液である50mMのリン酸カリウム(pH6.5)で緩衝液を交換した後、マレイミド−PEG(分子量40kDa、Shearwater社)をFab’:40K PEGのモル比が1:10となるように添加した後、4℃で徐々に攪拌しながら24時間反応させた。
【0139】
反応終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。40K PEGが修飾されたFab’−40K PEG連結体は、分子量が大きいので、反応していないFab’よりも早く溶出し、Fab’は後で溶出するため、除去することができた。完全に除去されていないFab’を除去するために、溶出したFab’−40K PEG連結体分画をpolyCAT 21×250カラム(PolyLC社)を用いて最終精製した。20mMのリン酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、Fab’の−SH基に40K PEGが連結されたFab’−S−40K PEG連結体を純粋に得た。
【0140】
(実験例1)蛋白質結合体の確認および定量
(1−1)蛋白質結合体の確認
前記実施例で製造した蛋白質結合体は、4〜20%の濃度勾配ゲルおよび12%のゲルを用いた非還元性SDS−PAGEおよびELISA(R&D system社)法で確認した。
【0141】
図6に示すように、SDS−PAGEの結果から、生理活性ポリペプチド、非ペプチド性重合体であるPEGおよび免疫グロブリンFc領域のカップリング反応によって、IFNα−PEG−Fc結合体(A)、17Ser−G−CSF−PEG−Fc結合体(B)およびEPO−PEG−Fc結合体(C)が生成されたことを確認した。
【0142】
また、前記実施例10で製造したDG Fcを確認するために非還元性12%SDS−PAGEを行った結果、図9bに示すように、天然型Fcに比べて除去された糖鎖分子量だけ減少した位置でDG Fcバンドが検出された。
【0143】
(1−2)蛋白質結合体の定量
前記実施例で製造したそれぞれの蛋白質結合体の量は、Superdexカラム(HiLoad 26/60 Superdex 75、Pharmacia社)と10mMのリン酸カリウム緩衝液(pH6.0)を溶出液として用いるサイズ排除クロマトグラフィでのピーク面積を対照と比較して換算する方法によって計算した。既に定量されているIFNα、hGH、G−CSF、17S−G−CSF、EPOおよびFcを用いてそれぞれサイズ排除クロマトグラフィを行った後、濃度とピーク面積間の換算係数を測定した。各蛋白質結合体の一定量を使用して同一のサイズ排除クロマトグラフィを行い、ここから得られたピーク面積から免疫グロブリンFc領域に該当するピーク面積を差し引いた値を、各蛋白質結合体に存在する生理活性蛋白質の定量値として決定した。図7は精製されたIFNα−PEG−Fc結合体のサイズ排除クロマトグラフィカラムの分析結果を示すもので、二量体以上の多量体不純物がない単一ピークを確認した。
【0144】
Fcが生理活性ポリペプチドに結合すると、生理活性ポリペプチドの抗体でその量を定量するとき、抗体と前記ポリペプチドとの結合が阻害され、クロマトグラフィによって計算される実際値より少なく定量される。ELISA分析の結果から、IFNα−PEG−Fcの場合には、ELISAによって測定された値が大略実際値の約30%程度であると確認された。
【0145】
(1−3)蛋白質結合体の純度および質量確認
それぞれの実施例で得た蛋白質結合体に対してサイズ排除クロマトグラフィを行い、280nmの吸光を測定した。その結果、IFNα−PEG−Fc、hGH−PEG−Fc、G−CSF−PEG−Fcおよび17Ser−G−CSF−PEG−Fcは、分子量70〜80kDaの物質の滞留時間帯で単一ピークを示した。
【0146】
一方、実施例5、15および16で獲得した蛋白質結合体IFNα−PEG−Fc、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体試料の純度を分析するために、逆相HPLCを行った。逆相カラム(Vydac社、259 VHP54カラム)を用いて分析し、0.5%TFAの存在下のアセトニトリル溶媒を用いた濃度勾配(40〜100%)法によって溶出した。純度は、280nmの吸収を測定することによって解析された。その結果、図11から分かるように、結合していないインターフェロンまたは免疫グロブリンFcは存在しておらず、IFNα−PEG−Fc結合体(A)、IFNα−PEG−DG Fc結合体(B)およびIFNα−PEG−組み換えAG Fc誘導体結合体(C)のいずれも96%以上の純度で純粋に精製された。
【0147】
各精製された試料の正確な分子量を確認するために、各試料の質量をMALDI−TOF(Voyager DE−STR、Applied Biosystems社)超高速質量分析器を用いて分析した。マトリックスとしてはシナピン酸(sinapinic acid)を使用し、それぞれの試験用試料0.5μLを試料スライドに塗布して自然乾燥させた後、同量のマトリックス溶液を添加し、その後さらに自然乾燥させてイオン源(ion source)に導入した。検出はポジティブ方式でリニアモードTOF方式の装置を用いて行った。イオンは、遅延したイオン抽出(DE)を用いる分割抽出供給源で、遅延した抽出時間を750nsec/1500nsecとして、約2.5kVの全体電位差によって加速化した。
【0148】
表1は前記実施例で得たそれぞれのFc蛋白質結合体のMLADI−TOF質量分析結果を数値化して示したものであり、図8はEPO−PEG−Fc結合体、図10はIFNα−PEG−FcおよびIFNα−PEG−DG Fc結合体のMALDI−TOF質量分析結果を示したものである。その結果、収得されたEPO−PEG−Fc蛋白質結合体の純度は95%以上であり、理論値と非常に近い分子量を示した。また、免疫グロブリンFc領域に赤血球生成因子(EPO)が1:1で結合した形態であると確認された。
【0149】
【表1】
【0150】
また、MALDI−TOF質量分析法により、実施例14で製造されたFcおよびDG Fcの分子量を測定した結果、DG Fcの分子量は天然型Fcより3kDa程度小さい50kDaと確認された(図9a)。減少した3kDaは理論的糖鎖サイズに該当する分子量であって、糖鎖が完全に除去されたことを確認することができた。
【0151】
下記表2は、前記実施例11で製造されたIFNα−PEG−DG Fc結合体と、実施例16で製造されたIFNα−PEG−組み換えAG Fc誘導体結合体(IおよびII)のMALDI−TOF質量分析結果を示したものである。IFNα−PEG−Fc結合体の分子量(75.9kDa)と比較したとき、IFNα−PEG−DG Fc結合体は約3kDa程度小さい分子量を示し、IFNα−PEG−組み換えAG Fc誘導体結合体(I)は約3〜4kDa程度小さい分子量を示した。Fc単量体に連結されたIFNα−PEG−組み換えAG Fc誘導体結合体(II)は、Fc単量体分子量に該当する24.5kDaが減少した分子量を示した。
【0152】
【表2】
【0153】
(実験例2)薬物動態学的な調査I
各群当たり5匹のSDラットにおいて、天然型生理活性蛋白質(対照群)と前記実施例および比較例で製造した−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、−PEG−DG Fc結合体、および−PEG−組み換えAG Fc誘導体結合体の血液内安定性および薬物動態学的係数(pharmacokinetic parameters)を比較した。対照群および−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、−PEG−DG Fc結合体、および−PEG−組み換えAG Fc誘導体結合体(試験群)を各100μg/kgずつ皮下注射した後、対照群は注射してから0.5.1、2、4、6、12、24、30、48、72および96時間後に採血し、試験群は注射してから1、6、12、24、30、48、72、96、120、240および288時間後に採血した。ヘパリンを含有するチューブに血液試料を集めて凝固を防止し、エッペンドルフ高速マイクロ遠心分離機で5分間遠心分離して細胞を除去した。血漿内の蛋白質量は、各生理活性蛋白質に対する抗体を用いてELISA方法によって測定した。
IFNα、hGH、G−CSFまたはEPOの天然型蛋白質、これらの−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、および−PEG−DG Fc結合体の薬物動態の分析結果を下記表3〜表7に示した。下記表において、Tmaxは最高薬物濃度に到達する時間を、T1/2は薬物の血中半減期を、MRT(mean residence time)は薬物分子の平均的な体内滞留時間をそれぞれ意味する。
【0154】
【表3】
【0155】
【表4】
【0156】
【表5】
【0157】
【表6】
【0158】
【表7】
【0159】
前記表3および図12の薬物動態グラフから分かるように、IFNα−PEG−Fc蛋白質結合体の場合、血中半減期は90.4時間であって天然型に比べて約50倍増加した。これは比較例1で製造したIFNα−40K PEGの半減期である35.8時間より約2.5倍増加したものである。また、IFNα−PEG−アルブミンの半減期である17.1時間に比べても、本発明のIFNα−PEG−Fc蛋白質結合体が著しく優れた血中半減期を示すことを確認した。
【0160】
一方、表3および図14に示すように、IFNα−PEG−DG Fc結合体の場合には、血中半減期が71.0時間であって、IFNα−PEG−Fc結合体とほぼ同等なので、糖鎖がなくても生体内安定性には大きい影響がなく、組み換え方法で生産された組み換えAG Fc誘導体を用いた結合体も天然型由来のDG Fcと同一の効果を示すことを確認した。ところが、Fc単量体を使用した結合体の場合には、正常的なFc二量体の結合体と比較したとき、約半分に減少した血中半減期を示した。
【0161】
ヒト成長ホルモンの場合も、表4に示すように、本発明の蛋白質結合体が示す血中半減期の増加効果を確認することができる。すなわち、天然型(1.1時間)に比べてhGH−40K PEG連結体およびhGH−PEG−アルブミン結合体の半減期が7.7時間および5.9時間と多少増加した反面、本発明のhGH−PEG−Fc蛋白質結合体の場合には血中半減期が11.8時間と画期的に増加した。
【0162】
表5および表6における顆粒球コロニー刺激因子とその誘導体に対する薬物動態の分析結果においても、天然型、−40K PEG連結体および−PEG−アルブミン結合体に比べて本発明のG−CSF−PEG−Fcおよび17S−G−CSF−PEG−Fc結合体が一層長い血中半減期を示した。蛋白質の血中持続性を増加させる免疫グロブリンFc領域の効果は、天然型生理活性蛋白質だけでなく、一部のアミノ酸を変形させた誘導体においても天然型とほぼ同一の程度であることを確認することができ、このような結果により、本発明の方法が他の蛋白質の誘導体においても類似の効果を示すことを容易に予想することができる。
【0163】
表7および図13に示すように、糖鎖化されている天然型蛋白質としての赤血球生成因子に対しても本発明の蛋白質結合体の血中半減期増加効果が確認された。すなわち、天然型赤血球生成因子の血中半減期が9.4時間であり、EPOを高糖鎖化させて血中安定性を高めたダーベポエチン−α(Darbepoetin-α、Aranesp、Amgen社)の場合は半減期が18.4時間と増加することが分かる。赤血球生成因子に免疫グロブリンFc領域を結合させた本発明のEPO−PEG−Fc結合体の場合には、血中半減期がおおよそ61.5時間と画期的に増加し、糖鎖が除去された大腸菌由来の組み換えAG Fc誘導体を使用した結合体の場合には87.9時間まで半減期の増加を示し、糖鎖が除去されても、血中安定性には影響を及ぼすことなく、抗体機能がない結合体を製造することができることを確認した。
【0164】
前記結果から分かるように、本発明によって免疫グロブリンFc領域および非ペプチド性重合体と共有結合した蛋白質結合体は、天然型蛋白質に比べて数倍〜数十倍以上増加した血中半減期を示した。また、免疫グロブリンFcを遺伝子組み換え法によって大腸菌で生産し、あるいは酵素処理によって糖鎖を除去しても、これを用いて製造された蛋白質結合体における血中半減期の増加効果はほぼ同様に維持された。
【0165】
特に、既存の蛋白質血中持続性を増加させるためのPEG剤形のうち持続性が最も高い40kDa PEGを修飾した蛋白質との比較においても、免疫グロブリンFc蛋白質結合体が著しく高い血中安定性を示した。また、免疫グロブリンFcの代わりにアルブミンを結合させた蛋白質結合体との比較試験においても、本発明の蛋白質結合体が、卓越した血中安定性を示すことから、本発明の蛋白質結合体が持続型蛋白質薬物製剤の開発に効果的であることを確認することができた。点突然変異(point mutation)によるコロニー刺激因子誘導体まで広範囲な範囲の蛋白質において従来のPEG結合蛋白質またはアルブミン蛋白質結合体より卓越した血中安定性と平均滞留時間(MRT)を示す結果からみて、本発明の蛋白質結合体による安定性および持続性増加効果は他の生理活性ポリペプチドにも適用可能であることが分かる。
【0166】
一方、非ペプチド性重合体として10kDa PEGを使用したIFNα−10K PEG−Fc蛋白質結合体(実施例11)を用いて前記と同一の方法で血中半減期を測定したとき、血中半減期は48.8時間であって、分子量3.4kDaのPEGを使用した蛋白質結合体の血中半減期である79.7時間より多少減少した。
【0167】
また、非ペプチド性重合体PEGの分子量の増加に伴って血中半減期はむしろ多少減少したが、このような結果から、蛋白質結合体の血中安定性および持続性の増加の主な要因が非ペプチド性重合体の分子量よりは結合した免疫グロブリンFc領域による効果であることが確認できる。
【0168】
PEGの反応基をアルデヒド反応基以外の反応基に変化させた場合でも、見掛け分子量と血中半減期が、アルデヒド反応基を持つPEGを使用した場合と類似の様相を示した。
【0169】
(実験例3)薬物動態学的な調査II
実施例12および13で製造されたFab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体および比較例3で製造されたFab’−S−40K PEG連結体の血中半減期を測定するために、Fab’を対照群として前記結合体または連結体を用いて実験例2と同一の方法で薬物動態学的な調査を行い、その結果を図15に示した。
図15より、Fab’−S−PEG−N−FcおよびFab’−N−PEG−N−Fc結合体が、Fab’またはFab’−S−40K PEG連結体に比べて2〜3倍延長された血中半減期を示すことが分かる。
【0170】
(実験例4)細胞内活性の測定
(4−1)インターフェロンアルファ蛋白質結合体の細胞内活性比較
インターフェロンアルファ蛋白質結合体の細胞内活性比較のために、IFNα−PEG−Fc(実施例5)、IFNα−PEG−DG Fc(実施例15)、IFNα−PEG−組み換えAG Fc誘導体(実施例16)、IFNα−40K PEG(比較例1)およびIFNα−PEG−アルブミン(比較例2)の抗ウイルス活性を水泡性口内炎ウイルスで飽和させたMDBK(Madin Darby Bovine Kidney、ATCC CCL−22)を使用する細胞培養生検で測定した。この際、PEGが結合していないインターフェロンアルファ−2b(NIBSC国際標準品)を標準物質として使用した。
【0171】
MDBK細胞を、MEM(minimum essential medium:JBI社)に10%FBSおよび1%ペニシリン/ストレプトマイシンが添加された培地で37℃、5%CO2の条件で培養した。測定しようとする試料および標準物質を一定の濃度で細胞培養培地に希釈して96ウェルプレートの各ウェルに100μLずつ分注した。前記培養された細胞を回収し、試料が分注されているプレートに100μLずつ加えた後、37℃、5%CO2の条件で約1時間程度培養した。1時間後、ウイルスの濃度が5〜7×103PFUとなるように調節されたVSV(Vesilculer stomatitis virus)を50μLずつプレートに添加し、37℃、5%CO2の条件で約16〜20時間さらに培養した。試料および標準物質を入れないで細胞とウイルスのみを入れたウェルを陰性対照群として使用し、ウイルス希釈溶液を入れないで細胞のみを入れたウェルを陽性対照群として使用した。
【0172】
培養液を除去し、生きている細胞を染色するために、ニュートラルレッド(neutral red)溶液を100μLずつ添加した後、37℃、5%CO2の条件で2時間培養した。上澄み液を除去した後、100%のエタノールと1%の酢酸とを1:1の比率で混ぜた溶液100μLを各ウェルに入れた。染色された細胞をよく振って溶解させた後、540nmで吸光度を測定した。陰性対照群をブランク(blank)とし、陽性対照群を細胞成長100%と見なして50%細胞成長時の濃度(ED50)を計算した。
【0173】
【表8】
【0174】
その結果、図8に示すように、IFNα−40K PEGは、その活性度が天然型の4.8%水準に低下することが確認できる。特に、修飾されたPEGの大きさが大きいほど、血中安定性は増加し、相対的に活性度は徐々に減少したが、インターフェロンアルファの場合、12kDa PEGが修飾された場合には25%、40kDa PEGが修飾された場合には約7%程度の試験管内活性を持つと報告されたことがある(P. Bailon et al., Bioconjugate Chem. 12: 195-202, 2001)。すなわち、PEGの分子量が増加すると、血中半減期は長くなるが、相対的に活性は急激に減少するので、血中半減期が長いながら活性も優れた蛋白質結合体の開発が要求されている。また、IFNα−PEG−アルブミン結合体も天然型に比べて約5.2%程度と低い活性を示したが、本発明のIFNα−PEG−Fc結合体およびIFNα−PEG−DG Fc結合体は、天然型に比べて相対的活性度が28.1%および25.7%と画期的に高くなることを確認することができ、組み換えAG Fc誘導体を使用した結合体においても類似の活性増加効果を示した。このような結果から、血中半減期の画期的増加に伴ってインターフェロンアルファと免疫グロブリンFc領域の結合体の生体内薬効が非常に優れるものと期待される。
【0175】
(4−2)ヒト成長ホルモン蛋白質結合体の細胞内活性比較
ヒト成長ホルモン蛋白質結合体の細胞内活性を比較するために、hGH−PEG−Fc、hGH−40K PEGおよびhGH−PEG−アルブミンの細胞内活性を比較試験した。
【0176】
ヒト成長ホルモン依存的に分裂を行う細胞としてのラット結節リンパ腫(rat node lymphoma)細胞株であるNb2細胞(European Collection of Cell Cultures (ECACC) #97041101)を用いて細胞内活性度を試験管内の分析によって確認した。
【0177】
Nb2細胞を、培養液(Fisher’s medium)に10%の牛胎児血清(FBS、fetal bovine serum)、0.075%のNaCO3、0.05mMの2−メルカプトエタノールおよび2mMのグルタミンを添加した培地で培養した後、10%の牛胎児血清を除いた同一の培地で24時間さらに培養した。培養液で培養された細胞の数を数えて約2×104個の細胞を96ウェルプレートの各ウェルに入れた後、hGH−PEG−Fc、hGH−40K PEG、hGH−PEG−アルブミン、対照群である国際標準品(National Institute for Biological Standards and Controls、NIBSC)および天然型ヒト成長ホルモン(HM−hGH)をそれぞれ希釈して濃度別に添加した。その後、37℃、CO2の培養器で48時間培養した。その後、細胞の成長程度(各ウェルの細胞数)を測定するために、細胞染色薬(Cell Titer 96 Aqueous One Solution、Promega社)を各ウェルに25μLずつ入れた後、4時間培養した。その後、490nmの吸光度を測定して各試料の力価を計算し、その結果を下記表9に示した。
【0178】
【表9】
【0179】
表9から分かるように、ヒト成長ホルモンの場合も、hGH−40K PEGの活性度は天然型の約7.6%と低下し、hGH−PEG−アルブミン結合体の試験管内活性は天然型に比べて約5.2%程度と低い活性を示した。ところが、本発明のhGH−PEG−Fc結合体は、相対的活性度が天然型に比べて28%以上画期的に増加した。このような結果より、血中半減期の画期的増加に伴ってヒト成長ホルモンと免疫グロブリンFc領域の蛋白質結合体の生体内薬効が非常に優れるものと期待することができる。また、本発明の免疫グロブリンFc蛋白質結合体の活性増加は、免疫グロブリンFcによる血中安定性の増加および受容体との結合力保存、または非ペプチド性重合体によって形成された空間的余裕によるものと把握される。このような作用は、他の生理活性蛋白質の免疫グロブリンFc蛋白質結合体においても類似であるものと期待される。
【0180】
(4−3)顆粒球コロニー刺激因子蛋白質結合体の細胞内活性比較
顆粒球コロニー刺激因子誘導体の蛋白質結合体の細胞内活性を比較するために、天然型G−CSF(Filgrastim、第一薬品(株))、17Ser−G−CSF誘導体、20K PEG−G−CSF(Neulasta社)、40K PEG−17S−G−CSF、17Ser−G−CSF−PEG−アルブミンおよび17S−G−CSF−PEG−Fcの細胞内活性を測定した。
【0181】
まず、ヒト骨髄由来の細胞株であるHL−60(ATCC CCL−240、Promyelocytic leukemia patient/36 yr old Caucasian female)を10%の牛胎児血清を含むRPMI1640倍地で培養してから、細胞の数を約2.2×105細胞/mLとなるように調整した後、DMSO(dimethylsulfoxide、culture grade、Sigma社)を最終1.25%(v/v)となるように添加した。前記の細胞株を96ウェルプレート(Corning/low evaporation 96 well plate)に90μLずつ入れてウェル当たり細胞が約2×104個となるようにした後、5%のCO2が供給される37℃の培養器で約72時間培養した。
【0182】
G−CSF ELISAキット(R&D systems社)を用いて、濃度が決定された各試料を同一の濃度となるようにRPMI1640で希釈し、最終濃度が10μg/mLとなるようにし、これをさらにRPMI1640で1/2に希釈することを19回繰り返し行った。こうして作られた試料を培養中のHL−60細胞株の各ウェルに10μLずつ加え、最終濃度が1μg/mLから連続的に半減するようにした。前記で製造した蛋白質試料を処理した細胞株は、37℃の培養器で72時間、さらに培養した。
【0183】
培養後、細胞株の増加程度を調べるために、CellTiter96TM(Cat.No.G4100、Promega社)を用いて分析し、増加した細胞株の数は、670nm波長における吸光度を測定することによって決定した。
【0184】
【表10】
【0185】
その結果、表10から分かるように、G−CSFのアミノ酸を置換させた17Ser−G−CSF誘導体の免疫グロブリンFc蛋白質結合体も天然型蛋白質の蛋白質結合体と類似の効果を示した。17Ser−G−CSF−PEGの場合には、PEGで修飾されていないものに比べて、血中半減期は増加するが活性度は低下すると既に報告されたことがある(大韓民国特許公開第2004−83268号)。特に、修飾されたPEGの大きさが大きいほど、血中安定性は増加するが、相対的に活性度は徐々に低下することが分かり、17Ser−G−CSF−40K PEGは天然型に比べて約10%以下の非常に低い活性度を示した。すなわち、PEGの分子量が増加すると、血中半減期は長くなるが相対的に活性は急激に減少するので、血中半減期が長くて活性も優れた蛋白質結合体の開発が要求されている。17Ser−G−CSF−PEG−アルブミンも天然型に比べて約23%程度と低い活性を示したが、17Ser−G−CSF−PEG−Fcは天然型に比べて相対的活性度が51%以上と高くなることを確認することができた。これにより、血中半減期の画期的増加に伴って免疫グロブリンFc領域と17Ser−G−CSF誘導体の結合体の生体内薬効が非常に優れるものと期待することができる。
【0186】
(4−4)Fab’結合体の細胞毒性中和試験
実施例8および9で製造されたFab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体、および比較例3で製造されたFab’−S−40K PEG連結体の試験管内活性実験を行った。マウス繊維亜細胞株L929(ATCC CRL−2148)を用いてTNFαの細胞毒性を測定する実験を基本骨格として、Fab’がTNFαの細胞壊死活性をどれくらい中和させるかを測定した。
【0187】
Fab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体およびFab’−S−40K PEG連結体をそれぞれ2倍ずつ順次希釈して96ウェルプレートの各ウェルに100μLずつ分注した後、rhTNF−α(R&D systems社)およびRNA合成の阻害剤として用いられるアクチノマイシンD(actinomycin D、Sigma社)をそれぞれ最終濃度が10ng/mLおよび1μg/mLとなるように添加し、37℃、5%CO2で30分間反応させた後、分析用マイクロプレートに移した。プレートの各ウェルにL929細胞株を5×104個/50μL培地となるように添加し、37℃、5%CO2の培養器で24時間培養した。マイクロプレート内の培養液を除去した後、PBSに5mg/mLの濃度で溶けているMTT(Sigma社)を50μLずつ入れた。約4時間37℃、5%CO2の培養器で培養した。150μLのDMSOを添加した後、540nmの吸光度を測定して細胞毒性中和の度合いを確認した。対照群としては、実施例8の段階1で精製したFab’を使用した。
【0188】
その結果、図16に示すように、実験に使用した全ての蛋白質結合体は、Fab’と類似の力価を示しており、このような結果から、Fab’のN末端またはC末端近くの遊離システイン残基にPEGを介して免疫グロブリンFcを結合させた蛋白質結合体は、Fab’の血中半減期を画期的に増加させることは勿論のこと、生体内活性度も高く維持させることが分かる。
【0189】
(4−5)CDC活性の測定
前記実施例で製造した誘導体と大腸菌形質転換体から発現されて精製された免疫グロブリン不変領域蛋白質とがヒトClqに結合するか否かを確認するために、下記のように固相酵素免疫検定法(ELISA)を行った。実験群として、形質転換体HM10932およびHM10927から生産された免疫グロブリン不変領域試料と前記実施例で製造した誘導体とを使用し、比較群として、糖が結合している免疫グロブリン(IVIG−グロブリンS、緑十字PBM)を始めとして、商業化されて治療用抗体として使われているいろいろの抗体を使用した。前記実験群と比較群の試料を10mMのカーボネート緩衝液(pH9.6)に1μg/mLの濃度で調製した。準備された試料を96ウェルプレート(Nunc)にウェル当たり200ngの量で分注した後、4℃で一晩中コートし、その後ウェルプレートをPBS−T溶液(137mMのNaCl、2mMのKCl、10mMのNa2HPO4、2mMのKH2PO4、0.05%のツイン20)で3回洗浄した。牛血清アルブミンを1%の濃度でPBS−T溶液に溶解させて準備したブロッキングバッファ250μLを各ウェルに添加した後、常温で1時間放置し、同一のPBS−T溶液で3回洗浄した。標準液と試料を適切な濃度でPBS−T溶液によって希釈した後、抗体がコートされたウェルに加えて常温で1時間放置させて反応させ、その後さらにPBS−T溶液で3回洗浄した。ブロッキング反応が完了したプレートに2μg/mLのClq(R&D systems社、米国)を添加した後、2時間常温で反応させ、反応済みのプレートを前記PBS−T溶液で6回洗浄した。ヒトの抗ヒトClq抗体とペルオキシダーゼとの結合体(Biogenesis社、米国)をブロッキングバッファに1000:1に希釈して各ウェルに200μLずつ加えた後、1時間常温で反応させた。反応が完了した後、各ウェルをPBS−T溶液で3回洗浄した後、発色溶液AとB(カラーA−安定化ペルオキシダーゼ[Color A-Stabilized peroxide]溶液およびカラーB−安定化クロモゲン[Color B-stabilized chromogen]溶液、DY999、R&D systems社)を同量で混合し、当該混合液を各ウェルに200μLずつ添加し、30分間放置した。その後、反応停止溶液である2Mの硫酸を50μLずつ添加して反応を停止させた。反応済みのウェルプレートは、マイクロプレートリーダー(Molecular Device社)を用い、標準液と検液の450nmの波長の吸光度を測定し、その結果を図17、図18にそれぞれ示した。
【0190】
免疫グロブリンのFc領域における補体活性をサブクラスによって比較した結果、ヒト免疫グロブリンIgG1(Fitzgerald)、IgG2(Fitzgerald)、IgG4(Fitzgerald)の順にClqに対する高い結合力を持っていることが確認でき、これにより補体活性もサブクラス間に差異があることが分かった。前記実験に使用したIVIGの場合、IgGサブクラスの集合体であるが、IgG1がほぼ大部分を占めているので、分離精製されたIgG1とほぼ類似の親和度を示した。このような対照群と比較するとき、非糖鎖化によるClqの親和度の変化は、補体活性が最も強いIgG1 Fcで著しく減少することが分かり、既に補体反応がないものと知られているIgG4 FcではClqに対する結合力が殆どないので、補体活性のない優れた組み換えキャリアであることが分かった(図17)。
【0191】
Clq親和度が除去されたキャリアの特性が生理活性ペプチドと結合体を作った後にも依然として維持されるか否かを調べるために、IFNαをモデルとして、糖鎖化Fc、酵素を用いた脱糖鎖Fcおよび非糖鎖化組み換えFcをキャリアとして用いて各結合体を作った後、Clqに対する結合力を検討した。糖鎖化FcとIFNα結合体(IFNα−PEG−Fc:Glycosylated IgG1Fc)は、Clqに対して依然として高い親和度を維持するが、ここにPNGaseFなどを用いて脱糖鎖化させたインターフェロン結合体(IFNα−PEG−DGFc:Deglycosylated IgG1Fc)は、結合力が著しく低くなることを確認し、その程度は大腸菌由来の非糖鎖化Fc結合体と同様の水準のClqに対する結合力を示した。それだけでなく、IgG1の非糖鎖化Fcを用いたインターフェロン結合体(IFNα−PEG−AGFcG1:Aglycosylated IgG1Fc)をIgG1からIgG4に変えたインターフェロン結合体(IFNα−PEG−FcG4誘導体1:Aglycosylated IgG4Fc)の場合、Clqに対する親和度が完全に除去されることを確認することができ、結合体を非糖鎖化IgG4 Fc単量体に変えた結合体(IFNα−PEG−FcG4誘導体2:Aglycosylated IgG4Fc)の場合にも、やはりClqに対する結合能力は完全に除去されることからみて、抗体断片のエフェクター(effector)機能がない良いキャリアであることを確認することができた(図18)。
【0192】
本発明は、以下の態様でも提供され得る:
[1]非ペプチド性リンカーを使用して薬物との結合体を形成するための薬物キャリアとして、薬物とは独立的に組換え的な方法によって製造されたものである、IgG Fc断片。
[2]前記IgGがIgG2またはIgG4である1に記載のFc断片。
[3]前記IgGがIgG4である2に記載のFc断片。
[4]非糖鎖化された1に記載のFc断片。
[5]非糖鎖化されたIgG4 Fc断片である4に記載のFc断片。
[6]ヒト由来の非糖鎖化されたIgG4Fc断片である5に記載のFc断片。
[7]配列番号8、10または23のアミノ酸配列を持つ1に記載のFc断片。
[8]1のFc断片をコードする遺伝子。
[9]配列番号4、9または22である8に記載の遺伝子。
[10]9の遺伝子の中から選択される配列を持つ組み換えベクター。
[11]10の組み換えベクターで形質転換された形質転換体。
[12]11の形質転換された組み換え微生物を培養してFc蛋白質を収得する方法。
[13]1のFc断片をキャリアとして含む薬剤学的組成物。
【産業上の利用可能性】
【0193】
本発明のIgG Fc断片は、キャリアとして用いられる場合、薬物の血中半減期を増加させ、薬物の生体内活性を維持させる。特にポリペプチド薬物の場合、ポリペプチド薬物の血中半減期の増加が、従来報告されていたいずれの修飾蛋白質よりも高く、既存の持続型製剤(long-acting formulation)の最も大きい欠点である力価の減少を克服することにより、従来最も効果が良いものと知られているアルブミンを用いた場合よりも著しい血中持続性と生体内活性を有する。しかも免疫反応誘発の危険が殆どないため、蛋白質薬物の持続型製剤の開発に有用に利用できる。また、本発明に係る蛋白質薬物の持続型製剤は、頻繁な注射による患者の苦痛を減少させることができ、活性ポリペプチドの血中濃度を持続的に維持して薬効を安定的に示すことができる。
【技術分野】
【0001】
本発明は、薬物のキャリアとして有用なIgG Fc断片に関するものであり、より具体的には、IgG2 Fc並びにIgG4 Fc断片、この組み合わせ(combination)およびこのハイブリッド(hybrid)に関するものである。また、本発明は、前記IgG Fc断片を発現させるための発現ベクター、前記発現ベクターによって形質転換された形質転換体および前記形質転換体を培養して免疫グロブリンFc断片を製造する方法に関するものである。
【背景技術】
【0002】
過去、数多くの薬学者および化学者は、天然的に存在する生理活性分子の生体活性を化学的に変化および/または修飾しようと努力した。このような努力の大部分は、生理活性物質の特定の生体活性を増加させたり、生体活性を持続させたり、毒性を減少させたり、副作用を除去または減少させたり、特定の生理活性を修飾することであった。生理活性物質が化学的に修飾される場合、大部分は生理活性が除去されるかあるいは相当減少するが、場合によっては生理活性が増加または変化することもある。したがって、目的とするところに応じて生理活性を変化させることが可能な化学修飾についての研究が多く行われており、大部分の研究は、生理活性物質(薬物)を生理学的に許容可能なキャリアと共有結合させることであった。
【0003】
国際特許公開公報第WO01/93911号には、多数の酸性残基を持つ重合体が薬物のキャリアとして用いられた例が開示されており、国際特許公開第WO03/00778号には、陰イオン基を含有する両親媒性ブロック共重合体をキャリアとして用いて陽イオン性薬物の安定性を増加させた例が開示されており、ヨーロッパ特許第0681481号には、シクロデキストリンと酸をキャリアとして塩基性薬物の特性を改善させる技術が開示されている。一方、疎水性薬物の場合、生体内での安定性の低下は、主に疎水性薬物の低い水溶解特性に起因する。このような疎水性薬物の低い水溶解特性を改善させるために、脂質(Lipid)をキャリアとして用いた技術が国際特許公開第WO04/064731号に開示されている。ところが、今まで免疫グロブリンFc断片を薬物のキャリアとして用いた例は、公知となっていない。
【0004】
一方、一般的に、ポリペプチドは、安定性が低くて変性し易く、血液内蛋白質加水分解酵素によって分解されて腎臓または肝臓を介して容易に除去されるため、薬理成分としてポリペプチドを含む蛋白質医薬品の血中濃度および力価を保つためには、蛋白質薬物を患者に頻繁に投与する必要がある。ところが、大部分注射剤の形態で患者に投与される蛋白質医薬品の場合、活性ポリペプチドの血中濃度を保つために頻繁に注射を打つことは、患者に夥しい苦痛を引き起こす。このような問題点を解決するために、蛋白質薬物の血中安定性を増加させ、血中薬物濃度を長期間高く持続させて薬効を最大化しようという努力が続けられてきた。このような蛋白質薬物の持続性製剤は、蛋白質薬物の安定性を高めると同時に薬物自体の力価が十分高く維持されなければならず、患者に免疫反応を誘発してはならない。
【0005】
蛋白質を安定化させ、蛋白質加水分解酵素との接触および腎臓による消失を抑制するための方法として、従来では、ポリエチレングリコール(polyethylene glycol:以下「PEG」という)のように溶解度の高い高分子を蛋白質薬物の表面に化学的に付加させる方法が使用された。PEGは、目的蛋白質の特定の部位または様々な部位に非特異的に結合して溶解度を高めることにより、蛋白質を安定化させ且つ蛋白質の加水分解を防止するのに効果があり、しかも、深刻な副作用を起こさないものとして知られている(Sada et al., J. Fermentation Bioengineering 71: 137-139, 1991)。ところが、PEGの結合により、蛋白質の安定性は増加できるが、生理活性蛋白質の力価が著しく低くなるという問題を生じる。さらに、PEGの分子量が増加するほど蛋白質との反応性が低くなり、その結果、収率が減少するという問題を生じる。
【0006】
最近は、PEGの両末端に同一の蛋白質薬物を結合させた二重体を作って活性の増加を図ったり(米国特許第5,738,846号)、あるいはお互い異なる種類の蛋白質薬物をPEGの両末端に結合させて同時に2種の活性を示す蛋白質(国際出願公開第WO92/16221号)も開発されたが、これらは蛋白質薬物の活性を持続させる面では明確な効果を示していない。
【0007】
一方、Kinstler等は、顆粒球コロニー刺激因子(G−CSF)とヒトアルブミンとをPEGに結合させた融合蛋白質が、増加した安定性を示すと報告した(Kinstler et al., Pharmaceutical Research 12(12): 1883-1888, 1995)。ところが、G−CSF−PEG−アルブミン構造を有する前記文献の修飾された薬物は、体内残留時間が天然型薬物を単独で投与した場合に比べて約4倍増加するに止まり、血中半減期の増加が微々であって、蛋白質薬物の効果的な持続性製剤として実用化されていない。
【0008】
生理活性蛋白質の生体内安定性を高める別の方法として、遺伝子組み換えにより血中安定性の高い蛋白質遺伝子と生理活性蛋白質遺伝子を連結した後、前記組み換え遺伝子で形質転換された動物細胞などを培養して融合蛋白質を生産する方法が開発されている。例えば、現在まで蛋白質の安定性の増加に最も効果が高いものと知られているアルブミンまたはその断片を、遺伝子組み換えによって目的の生理活性蛋白質に結合させて生産した融合蛋白質が報告されている(国際出願公開第WO93/15199号および第WO93/15200号、ヨーロッパ特許公開第EP413,622号)。
【0009】
また、ヒューマンゲノムサイエンス(Human Genome Science)社が酵母で生産したインターフェロンアルファとアルブミンの融合蛋白質(製品名:AlbuferonTM)は、猿でインターフェロンの半減期を5時間から93時間に増加させたが、修飾していないインターフェロンに比べて生体活性度が5%未満に著しく減少するという問題がある(Osborn et al., J. Phar. Exp. Ther. 303(2): 540-548, 2002)。すなわち、今まで蛋白質薬物の生体内持続性と安定性を増加させながら生体内活性を同時に維持することが可能な技術は、未だ開示されたことがない。
【0010】
一方、免疫グロブリンおよびこの断片を用いて蛋白質薬物の安定性を高めようとする試みも多く行われた。例えば、米国特許第5,045,312号では、架橋剤を用いてヒトの成長ホルモンに血清アルブミンまたはラットの免疫グロブリンを結合させることにより、修飾していない成長ホルモンに比べて成長ホルモンの活性を増加させることができることを開示している。
【0011】
また、蛋白質薬物と免疫グロブリンFcを融合させようとする試みも行われた。例えば、インターフェロン(大韓民国特許公開第2003−9464号)、およびインターロイキン−4受容体、インターロイキン−7受容体または赤血球生成因子受容体(大韓民国特許登録第249572号)を免疫グロブリンのFc断片と融合している形態で哺乳動物で発現させた。国際特許公開第WO01/03737号では、サイトカインまたは成長因子をペプチド結合によって免疫グロブリンのFc断片に結合させた融合蛋白質を製造した。また、米国特許第5,116,964号では、免疫グロブリンFc断片のアミノ末端またはカルボキシル末端に遺伝子組み換え方法によって融合させた蛋白質を開示しており、米国特許第5,349,053号では、IL−2を免疫グロブリンFc断片にペプチド結合によって融合させた融合蛋白質を開示している。
【0012】
その他にも、遺伝子組み換えによって製造されたFc融合蛋白質の例として、インターフェロン−βまたはその誘導体と免疫グロブリンFc断片の融合蛋白質(国際特許公開第WO00/23472号)、IL−5受容体と免疫グロブリンFc断片の融合蛋白質(米国特許第5,712,121号)が開示されている。ところが、免疫グロブリンFc断片を用いて生理活性ポリペプチド薬物の持続性を改善させようとする技術は、大部分、免疫グロブリンFc断片を融合パートナーとして用いた技術に焦点が合わせられており、免疫グロブリンFc断片をキャリアとして用いる技術は未だ報告されていない。
【0013】
一方、免疫グロブリンFc断片のアミノ酸を修飾する技術も公知になっているが、米国特許第5,605,690号では、免疫グロブリンFc断片で特に補体結合部位または受容体結合部位のアミノ酸を改変させたFcを用いて遺伝子組み換え方法によって製造されたTNFR−IgG1 Fc融合蛋白質を開示している。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】国際特許公開WO01/93911号
【特許文献2】国際特許公開WO03/00778号
【特許文献3】ヨーロッパ特許0681481号
【特許文献4】国際特許公開WO04/064731号
【特許文献5】米国特許第5,738,846号
【特許文献6】国際出願公開WO92/16221号
【特許文献7】国際出願公開WO93/15199号
【特許文献8】国際出願公開WO93/15200号、
【特許文献9】ヨーロッパ特許公開EP413,622号
【特許文献10】米国特許第5,045,312号
【特許文献11】大韓民国特許公開2003−9464号
【特許文献12】大韓民国特許登録第249572号
【特許文献13】国際特許公開WO01/03737号
【特許文献14】米国特許第5,116,964号
【特許文献15】米国特許第5,349,053号
【特許文献16】国際特許公開WO00/23472号
【特許文献17】米国特許第5,712,121号
【特許文献18】米国特許第5,605,690号
【非特許文献】
【0015】
【非特許文献1】Sada et al., J. Fermentation Bioengineering 71: 137-139, 1991
【非特許文献2】Kinstler et al., Pharmaceutical Research 12(12): 1883-1888, 1995
【非特許文献3】Osborn et al., J. Phar. Exp. Ther. 303(2): 540-548, 2002
【発明の概要】
【発明が解決しようとする課題】
【0016】
ところが、このような遺伝子組み換え方法によって生産されたFc融合蛋白質は、免疫グロブリンFc領域の特定の部位、すなわちアミノ末端またはカルボキシル末端でのみ蛋白質融合が可能であり、同種二量体の形態でのみ発現され、単量体の形態では生産が不可能であり、糖鎖化蛋白質間の融合または非糖鎖化蛋白質間の融合のみが可能であって、糖鎖化蛋白質と非糖鎖化蛋白質との融合は不可能であるという欠点がある。また、融合によって新たに発生したアミノ酸配列により免疫反応が誘発されるおそれがあるだけでなく、リンカー部位に対する蛋白質加水分解酵素の敏感性が増加するおそれがあるという問題点を持っている。
【0017】
このような問題を解決するために、本発明者は、前記融合技術の欠点を克服しながら薬物の生体内持続性を向上させ且つ生体内活性の減少を最小化するための方法を研究し、IgG Fc断片、より具体的にはIgG2またはIgG4 Fc断片が薬物と結合する場合、薬物の生体内持続性を向上させかつ生体内活性の減少を最小化することができることを見出し、本発明を完成した。
【課題を解決するための手段】
【0018】
本発明の一目的は、薬物のキャリアとして有用な免疫グロブリンG断片を提供することにある。
【0019】
本発明の他の目的は、免疫グロブリンG断片を製造するための組み換えベクターを提供することにある。
【0020】
本発明の別の目的は、免疫グロブリンG断片を製造するための組み換えベクターで形質転換された形質転換体を提供することにある。
【0021】
本発明の別の目的は、免疫グロブリンG断片を製造するための組み換えベクターで形質転換された形質転換体を培養して免疫グロブリンG断片を製造する方法を提供することにある。
【0022】
本発明の別の目的は、免疫グロブリンG断片を含む薬剤学的組成物を提供することにある。
【図面の簡単な説明】
【0023】
【図1】図1は、免疫グロブリンFcを大腸菌で発現させた後、非還元条件でウエスタンブロットによって分析した結果である。
【図2】図2は、免疫グロブリンFcを15%クライテリオンゲル(Criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させた結果である。
【図3】図3は、免疫グロブリンFcを15%クライテリオンゲル(Criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させた結果である。
【図4】図4は、免疫グロブリンをパパインで切断して得た免疫グロブリンFc断片をクロマトグラフィで精製した結果である。
【図5】図5は、精製された免疫グロブリンFc断片をSDS−PAGEで分析した結果である。
【図6】図6は、カップリング反応後に生成されたIFNα−PEG−Fc(A)、17Ser−G−CSF−PEG−Fc(B)およびEPO−PEG−Fc(C)結合体をSDS−PAGEで分析した結果である。
【図7】図7は、カップリング反応後に精製されたIFNα−PEG−Fc結合体をサイズ排除クロマトグラフィで分析した結果である。
【図8】図8は、EPO−PEG−Fc結合体をMALDI−TOF質量分析器で分析した結果である。
【図9a】天然型免疫グロブリンFcと脱糖鎖化免疫グロブリンFc(deglycosylated Fc:DG Fc)のMALDI−TOF質量分析器で分析した結果である。
【図9b】天然型免疫グロブリンFcと脱糖鎖化免疫グロブリンFc(deglycosylated Fc:DG Fc)のSDS−PAGEで分析した結果である。
【図10】図10は、IFNα−PEG−Fc結合体およびIFNα−PEG−DG Fc結合体をMALDI−TOF質量分析器で分析した結果である。
【図11】図11は、IFNα−PEG−Fc結合体、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体結合体を逆相HPLCで分析した結果である。
【図12】図12は、天然型IFNα、IFNα−40K PEG、IFNα−PEG−アルブミン結合体およびIFNα−PEG−Fc結合体の薬物動態解析グラフである。
【図13】図13は、天然型EPO、高糖鎖化EPO、EPO−PEG−FcおよびEPO−PEG−AG Fc結合体の薬物動態を分析したグラフである。
【図14】IFNα−PEG−Fc結合体、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体結合体の薬物動態を分析したグラフである。
【図15】図15は、Fab’、Fab’−S−40K PEG連結体、Fab’−N−PEG−N−Fc結合体およびFab’−S−PEG−N−Fc結合体の薬物動態を分析したグラフである。
【図16】図16は、Fab’、Fab’−S−40K PEG連結体、Fab’−N−PEG−N−Fc結合体およびFab’−S−PEG−N−Fc結合体の細胞内活性を分析したグラフである。
【図17】図17は、ヒトIgGサブクラスにおける補体Clqの結合活性を比較分析したグラフである。
【図18】図18は、糖鎖化Fc、酵素を用いて糖を除去したDG Fc、および大腸菌で生産したAG Fcをキャリアとして用いたインターフェロン−PEG−キャリア結合体における補体Clqの結合活性を比較分析したグラフである。
【発明を実施するための形態】
【0024】
一つの様態として、本発明は、薬物のキャリアとして有用な免疫グロブリンG Fc断片に関し、より好ましくはIgG2 FcおよびIgG4 Fc断片に関するものである。
本発明において、「キャリア」とは、薬物と共に結合する物質を意味し、一般に薬物に結合して薬物の生理活性を増減または除去する。ところが、本発明におけるキャリアは、本発明の目的上、結合する薬物の生理活性減少を最小化すると同時に薬物の生体内安定性は増加させるためのものである。
【0025】
このような薬物のキャリアとして、従来から脂質(Lipid)や重合体(polymer)など多くの物質が研究されてきたが、免疫グロブリンFc断片を用いた技術は未だ公知となっていない。すなわち、本発明は、結合する薬物の生体内持続性の増進と生体内活性減少の最小化のためのキャリアとして、使用可能な物質の中でも特にIgG Fc断片を提供することを特徴とし、より好ましくはIgG2 FcおよびIgG4 Fc断片を提供することを特徴とする。
【0026】
本発明において、「免疫グロブリンG(以下、「IgG」と混用する)とは、抗原に選択的に作用して生体免疫に関与する蛋白質の総称であって、ヒトおよび動物から起源することができる。このような免疫グロブリンの一般構造を考察すると、免疫グロブリンは重鎖と軽鎖からなっており、重鎖不変領域はガンマ(γ)、ミュー(μ)、アルファ(α)、デルタ(δ)、エプシロン(ε)タイプを有し、サブクラスとしてガンマ1(γ1)、ガンマ2(γ2)、ガンマ3(γ3)、ガンマ4(γ4)、アルファ1(α1)およびアルファ2(α2)を有する。軽鎖の不変領域はカッパ(κ)およびラムダ(λ)タイプを有する(Coleman et al., Fundamental Immunology, 2nd Ed., 1989, 55-73)。これらにより、免疫グロブリンは、IgG、IgA、IgD、IgEおよびIgMの亜型(isotype)に分けられる。IgGはさらにIgG1、IgG2、IgG3およびIgG4に分けられる。
【0027】
また、免疫グロブリンは構造的にいろいろの断片が知られているが、その例としてFab、F(ab’)、F(ab’)2、Fv、scFv、Fd、Fcなどがある。この中でも、免疫グロブリン断片Fabは、軽鎖および重鎖の可変領域、軽鎖の不変領域および重鎖の第1不変領域(CH1)を持つ構造であって、1つの抗原結合部位を持つ。免疫グロブリン断片Fab’は、重鎖CH1ドメインのC末端に一つ以上のシステイン残基を含むヒンジ領域(hinge region)を持つという点でFabとは差異がある。断片F(ab’)2は、Fab’のヒンジ領域のシステイン残基がジスルフィド結合を成しながら生成される。Fvは、重鎖可変部位および軽鎖可変部位のみを持っている最小の抗体断片であり、scFvは、重鎖可変部位および軽鎖可変部位のみがペプチドリンカーで連結された単一ポリペプチド鎖である。また、Fd断片は、重鎖可変領域およびCH1ドメインのみから構成された断片である。
【0028】
すなわち、前記のような公知の様々な免疫グロブリンのタイプおよびその機能的、構造的断片の中でも、本発明は、薬物のキャリアとして有用なIgG Fc断片を提供し、特にIgG2 FcおよびIgG4 Fc断片を提供することを特徴とする。
【0029】
本発明の免疫グロブリンG Fc断片(以下、「IgG Fc断片」または「本発明のFc断片」と混用する)は、免疫グロブリンGの重鎖と軽鎖可変領域、重鎖不変領域1(CH1)と軽鎖不変領域1(CL1)を除いた、重鎖不変領域2(CH2)および重鎖不変領域3(CH3)部分を意味し、重鎖不変領域にヒンジ(hinge)部分を含むこともある。また、本発明のIgG Fc領域は、天然型と実質的に同等あるいはより向上した効果を持つ限り、IgGの重鎖と軽鎖可変領域のみを除き、一部または全体重鎖不変領域1(CH1)および/または軽鎖不変領域1(CL1)を含んだ、拡張されたFc断片であり、あるいはCH2および/またはCH3に該当する相当長い一部のアミノ酸配列が除去された断片である。
【0030】
このような本発明のFc断片は、天然型アミノ酸配列だけでなく、この配列誘導体(mutant)も含む。アミノ酸配列誘導体とは、天然アミノ酸配列中の一つ以上のアミノ酸残基が欠失、挿入、非保全的または保全的置換、またはこれらの組み合わせによって相異なる配列を持つものを意味する。例えば、IgG Fcの場合、結合に重要であると知られている214〜238、297〜299、318〜322または327〜331番のアミノ酸残基が、修飾のための適当な部位として利用できる。また、ジスルフィド結合を形成することが可能な部位が除去されるか、あるいは天然型FcからN末端の幾つかのアミノ酸が除去されるか、あるいは天然型FcのN末端にメチオニン残基が付加されることもあるなど様々な種類の誘導体が可能である。また、エフェクター機能を無くすために、補体結合部位、例えばClq結合部位またはADCC部位が除去されることがある。このような免疫グロブリンFc領域の配列誘導体を製造する技術は、国際特許公開第WO97/34631号、国際特許公開第WO96/32478号などに開示されている。
【0031】
分子の活性を全体的に変更させない蛋白質およびペプチドにおけるアミノ酸交換は、当該分野に公知となっている(H. Neurath, R. L. Hill, The Proteins, Academic Press, New York, 1979)。最も通常的に起こる交換は、アミノ酸残基Ala/Ser、Val/Ile、Asp/Glu、Thr/Ser、Ala/Gly、Ala/Thr、Ser/Asn、Ala/Val、Ser/Gly、Thy/Phe、Ala/Pro、Lys/Arg、Asp/Asn、Leu/Ile、Leu/Val、Ala/Glu、Asp/Gly間の交換である。
【0032】
場合によっては、リン酸化(phosphorylation)、硫化(sulfation)、アクリル化(acrylation)、糖化(glycosylation)、メチル化(methylation)、ファルネシル化(farnesylation)、アセチル化(acetylation)、アミド化(amidation)などで修飾(modification)されることもできる。
【0033】
前述したFc誘導体は、本発明のFc断片と同一の生物学的活性を示すが、熱やpHなどに対するFc断片の構造的安定性を増大させた誘導体である。
【0034】
また、このようなFc断片は、ヒト、および牛や山羊、豚、マウス、ウサギ、ハムスター、ラット、モルモットなどの動物の生体内から分離して得られた天然型であり、あるいは形質転換された動物細胞または微生物から得られた組み換え型またはその誘導体である。ここで、天然型から得る方法では、全体免疫グロブリンをヒトまたは動物の生体から分離した後、蛋白質分解酵素を処理して得ることができる。パパインを処理する場合にはFabおよびFcに切断され、ペプシンを処理する場合にはpF’cおよびF(ab’)2に切断される。これらの断片からサイズ排除クロマトグラフィ(size-exclusion chromatography)などを用いてFcまたはpF’cを分離することができる。好ましくは、ヒト由来のFc断片は微生物から収得した組み換え型IgG Fc断片である。すなわち、本発明は、微生物から収得したヒト由来の組み換え型IgG2 FcおよびIgG4 Fc断片が好ましい。
【0035】
また、本発明のFc断片は、天然型糖鎖、天然型に比べて増加した糖鎖、天然型に比べて減少した糖鎖、または糖鎖が除去された形態である。このようなFc断片の糖鎖の増減または除去には、化学的方法、酵素学的方法および微生物を用いた遺伝工学的方法などの当該分野における通常の方法が利用できる。ここで、Fcから糖鎖が除去されたFc断片は、補体(Clq)への結合力が著しく低下し、抗体依存性細胞毒性または補体依存性細胞毒性が減少または除去されるので、生体内で不要な免疫反応を誘発しない。このような点において、薬物のキャリアとしての本来の目的にさらに符合する形態は、脱糖鎖化免疫グロブリンFc断片、または非糖鎖化免疫グロブリンFc断片である。
【0036】
図18からも明らかなように、糖鎖化Fcは、非糖鎖化FcよりCDCの活性が高くて免疫反応を誘発する危険が高いと言えるので、本発明の目的上、非糖鎖化Fc断片あるいは脱糖鎖化Fc断片が好ましく、非糖鎖化IgG2 FcおよびIgG4 Fc断片、この組み合わせおよびこのハイブリッドがより好ましい。
【0037】
本発明において、「脱糖鎖化(Deglycosylation)」はFc断片から酵素によって糖を除去することを意味し、「非糖鎖化(Aglycosylation)」はFc断片を糖鎖化されていない形態で原核細胞、好ましくは大腸菌で生産することを意味する。
【0038】
一方、本発明において、「組み合わせ(combination)」とは、二量体または多量体を形成するとき、同一起源の単鎖免疫グロブリンFc断片を暗号化するポリペプチドが相異なる起源の単鎖ポリペプチドと結合を形成することを意味する。すなわち、IgG1 Fc、IgG2 Fc、IgG3 FcおよびIgG4 Fc断片よりなる群から選択された2つ以上の断片から二量体または多量体の製造が可能である。
【0039】
本発明において、「ハイブリッド(hybrid)」とは、単鎖の免疫グロブリンFc断片内に、相異なる起源の2つ以上の免疫グロブリンFc断片に該当する配列が存在することを意味する用語である。本発明の場合、様々な形態のハイブリッドが可能である。すなわち、IgG1 Fc、IgG2 Fc、IgG3 FcおよびIgG4 FcのCH1、CH2、CH3およびCH4よりなる群から選択された1〜4つのドメインからなるドメインのハイブリッドが可能であり、ヒンジ領域を含むことができる。
【0040】
一方、本発明の図17および図18から分かるように、IgGのいろいろなサブクラスのうち、特にIgG4は、補体(Clq)に対する結合力が最も低い。このように補体に対する結合力が低下すると、抗体依存性細胞毒性(ADCC、Antibody-dependent cell cytotoxicity)および補体依存性細胞毒性(CDC、Complement-dependent cytotoxicity)が減少または除去されるので、生体内で不要な免疫反応を誘発しなくなる。このようなClqに対する結合力は、IgG2およびIgG4 Fc断片がIgG1より低く、この中でもIgG4 Fc断片が最も低いことを確認することができる。したがって、薬物のキャリアとして用いるためには、結合するFc断片の抗体依存性細胞毒性および補体依存性細胞毒性のようなエフェクター(effector)機能がより低いものを使用することが好ましいので、本発明の目的上、IgG2 FcおよびIgG4 Fc断片が好ましく、より好ましくはIgG4 Fc断片であり、最も好ましくは配列番号8、10および23のアミノ酸配列を持つFc断片である。
【0041】
本発明の別の様態として、本発明は、IgG Fc断片、好ましくはIgG2 FcおよびIgG4 Fc断片をコードする遺伝子に関するもので、より好ましくは配列番号8、10および23のアミノ酸をコードする遺伝子に関するものである。このような本発明のFcをコードする遺伝子は、天然型核酸配列だけでなく、この配列誘導体も含む。核酸配列誘導体とは、天然核酸配列中の一つ以上の核酸残基が欠失、挿入、非保全的または保全的置換、またはこれらの組み合わせによって相異なる配列を持つものを意味する。
【0042】
本発明の配列番号8のアミノ酸配列をもつFc断片をコードする遺伝子は、好ましくは配列番号4の核酸配列を持つ遺伝子であり、配列番号10のアミノ酸配列を持つFc断片をコードする遺伝子は、好ましくは配列番号9の核酸配列を持つ遺伝子であり、配列番号23のアミノ酸配列を持つFc断片をコードする遺伝子は、好ましくは配列番号22の核酸配列を持つ遺伝子である。本発明のFc断片をコードする核酸配列は、一つ以上の塩基が置換、欠失、挿入またはこれらの組み合わせによって変異できる。前記核酸配列は、天然から分離するか、あるいは人為的に合成または遺伝子組み換え方法によって製造することができる。
【0043】
本発明のFc断片をコードする核酸配列は、これを発現するベクターによって提供される。
【0044】
別の様態として、本発明は、IgG Fc断片を含む組み換えベクターを提供する。
【0045】
本発明において、「ベクター」は、宿主細胞にDNAを導入して抗体または抗体断片を発現させるための手段であって、プラスミドベクター、コスミドベクター、バクテリオファージベクター、およびアデノウイルスベクター、レトロウイルスベクター、アデノ随伴ウイルスベクターのようなウイルスベクターなどがあり、プラスミドベクターが好ましい。本発明の目的上、発現ベクターは、プロモータ、開始コドン、終結コドン、ポリアデニル化シグナルおよびエンハンサーなどの発現調節エレメントの他にも膜標的化または分泌のためのシグナル配列を含むことができる。
【0046】
本発明において、「シグナル配列(signal sequence)」は、蛋白質の細胞質の外への輸送および分泌を可能とする特定のアミノ酸配列を意味する。このようなシグナル配列には、様々な形態が当該分野において公知となっているが、本発明では、好ましくは大腸菌を宿主細胞として用いるため、本発明のシグナル配列は大腸菌で分泌される蛋白質が持つ大腸菌由来のシグナル配列であることが好ましい。一方、大腸菌由来のシグナル配列にはアルカリホスファターゼ、ペニシリナーゼ、Ipp、熱安定性エンテロトキシンII、LamB、PhoE、PelB、OmpAまたはマルトース結合蛋白質などがあるが、最も好ましくは熱安定性エンテロトキシンIIである。
【0047】
一方、開始コドンおよび終結コドンは、一般に免疫原性標的蛋白質をコードするヌクレオチド配列の一部と見なされ、遺伝子作製物が投与されたとき、個体において必ず作用を示さなければならず、コーディング配列とインフレーム(in frame)になければならない。
【0048】
一般なプロモータは、構成的または誘導性である。原核細胞にはlac、tac、T3およびT7プロモータがあるが、これに制限されない。真核細胞には、サルウイルス40(SV40)プロモータ、マウス乳房腫瘍ウイルス(MMTV)プロモータ、ヒト免疫不全ウイルス(HIV)プロモータ(例えば、HIVの長い末端反復部(LTR)プロモータ)、モロニーウイルスプロモータ、サイトメガロウイルス(CMV)プロモータ、エプスタインバーウイルス(EBV)プロモータ、ラウス肉腫ウイルス(RSV)プロモータだけでなく、ヒトβ−アクチン、ヒトヘモグロビン、ヒト筋肉クレアチン、ヒトメタロチオネインなどヒト遺伝子由来のプロモータがあるが、これに制限されない。
【0049】
また、発現ベクターは、ベクターを含有する宿主細胞を選択するための選択性マーカーを含み、複製可能な発現ベクターの場合、複製起点を含む。抗生剤または薬物耐性を与える生成物をコードする遺伝子が普遍的な選択性マーカーとして用いられるが、原核細胞の場合、β−ラクタマーゼ遺伝子(アンピシリン耐性)およびTet遺伝子(テトラサイクリン耐性)が使用でき、真核細胞の場合、ネオマイシン(G418またはジェネティシン)、gpt(ミコフェノール酸)、アンピシリン、およびハイグロマイシン耐性遺伝子が使用できる。ジヒドロ葉酸還元酵素マーカー遺伝子は、様々な宿主でメトトレキサートによって選択できる。宿主の栄養要求性マーカーの遺伝子生成物をコードする遺伝子、例えばLEU2、URA3およびHIS3は酵母において選択性マーカーとして度々使用される。そして、ウイルス(例えば、バキュロウイルス)またはファージベクター、およびレトロウイルスベクターのような宿主細胞のゲノム内に挿入できるベクターも使用可能である。
【0050】
本発明の目的に符合するIgG Fc断片を製作するために、配列番号8、10および23のアミノ酸をコードする遺伝子が挿入されたベクターを用いる。本発明では、pT14S1SH−4T20V22Q(大韓民国特許第38061号)を出発ベクターとして用いて、配列番号23のアミノ酸をコードする配列番号22の遺伝子が挿入されたpSTIIdCG2Fcを製作し、配列番号8のアミノ酸をコードする配列番号4の遺伝子が挿入されたpSTIIdCG4Fcを製作し、前記pSTIIdCG4Fcプラスミドを鋳型としてPCRを行うことにより、前記PCRによって増幅された遺伝子のうち、二量体の形成に必要なヒンジ領域が除去された配列番号10のアミノ酸配列をコードする配列番号9の遺伝子が挿入されたpSTIIG4Moを製作した。
【0051】
別の様態として、本発明は、前記組み換えベクターで形質転換された形質転換体に関するものである。
【0052】
宿主細胞に応じて蛋白質の発現量と修飾などが異なるので、目的に最も適した宿主細胞を選択して使用すればよい。宿主細胞としては、大腸菌(Escherichia coli)、枯草菌(Bacillus subtilis)、ストレプトミセス (Streptomyces)、シュードモナス(Pseudomonas)、プロテウスミラビリス(Proteus mirabilis)またはスタヒロコッカス(Staphylococcus)のような原核宿主細胞があるが、これに制限されない。また、真菌(例えば、アスペルギルス(Aspergillus))、酵母(例えば、ピキアパストリス(Pichia pastoris)、サッカロミセスセレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス(Schizosaccharomyces), アカパンカビ(Neurospora crassa)などの下等真核細胞、昆虫細胞、植物細胞、哺乳動物などを含む高等真核生物由来の細胞を宿主細胞として使用することができる。ところが、本発明の目的上、免疫グロブリンFc断片が糖鎖化されないことが有利なので、原核細胞を使用することが好ましく、特に大腸菌を使用することが最も好ましい。
【0053】
本発明において、宿主細胞への「形質転換」および/または「トランスフェクション(transfection)」は、核酸を有機体、細胞、組織または器官に導入するいずれの方法も含まれるもので、当該分野で公知となっているように、宿主細胞に応じて適切な標準技術を選択して行うことができる。このような方法には、エレクトロポレーション(electroporation)、原形質融合、リン酸カルシウム(CaPO4)沈殿、塩化カルシウム(CaCl2)沈殿、炭化珪素繊維を用いた攪拌、アグロバクテリアを介した形質転換、PEG、硫酸デキストラン、リポフェクトアミンおよび乾燥/抑制を介した形質転換方法などが含まれるが、これに制限されない。
【0054】
例えば、塩化カルシウムを用いるカルシウム処理またはエレクトロポレーションは、一般に原核細胞に対して使用される((Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual (New York: Cold Spring Harbor Laboratory Press))。アグロバクテリウムツメファシエンス(Agrobacterium tumefaciens)を用いたトランスフェクションは、特定の植物細胞の形質転換に使用される(Shaw et al., 1983, Gene, 23:315);国際公開特許WO89/05859)。細胞壁がない哺乳動物細胞の場合、リン酸カルシウム沈殿法を用いることができる(Graham et al, 1978, Virology, 52:456-457)。哺乳動物宿主細胞への形質転換の一般的な方法および特徴は、米国特許第4,399,216号に記載されている。酵母内への形質転換は、一般にVan Solingenなどの文献(J. Bact., 1977, 130:946)およびHsiaoなどの文献(Proc. Natl. Acad. Sci. (USA), 1979, 76:3829)の方法によって行われる。
【0055】
前記本発明に係る発現ベクターが宿主細胞に形質転換された本発明の形質転換体は、HM10932(pSTIIdCG4Fc導入形質転換体)、HM10933(pSTIIG4Mo導入形質転換体)およびHM10936(pSTIIdCG2Fc導入形質転換体)である。
【0056】
別の様態としては、本発明は、IgG Fc断片、好ましくはIgG2 Fc、IgG4 Fc断片、この組み合わせおよびこのハイブリッドを発現することが可能なベクターで形質転換された形質転換体を適切な条件で培養して本発明の免疫グロブリン断片を製造する方法を提供する。
【0057】
前記免疫グロブリン断片の製造において、形質転換体の培養は、当該分野に知られている適当な培地と培養条件に応じて行われる。このような培養過程は、当業者であれば、選択される菌株に応じて容易に調整して使用することができる。
【0058】
形質転換体を培養して得た本発明の免疫グロブリン断片は、精製していない状態で使用でき、さらに様々な通常の方法、例えば透析、塩沈殿およびクロマトグラフィなどを用いて高純度に精製して使用できる。その中でもクロマトグラフィを用いる方法が最も多用されている。カラムの種類と順序の選択にはいずれの場合にも適用できる法則はなく、目的蛋白質である抗体の特性、培養方法などに応じてイオン交換クロマトグラフィ、サイズ排除クロマトグラフィ、親和性クロマトグラフィ、プロテインA親和カラムなどから選択することができる。本発明では、プロテインA親和カラム(protein-A affinity column)、SPセファロースFFカラムなどを用いて精製した。
【0059】
こうして得られるFc断片が遊離体の場合には、自体公知の方法またはこれに準ずる方法によって塩に変換することができ、逆にFc断片が塩の場合には、自体公知の方法またはこれに準ずる方法によって遊離体または他の塩に変換することができる。そして、組み換え体が生産するFc断片に、精製前または精製後に適当な蛋白質修飾酵素を作用させることにより、任意に修飾を加えるか、あるいはポリペプチドを部分的に除去することができる。蛋白質修飾酵素としては、例えばトリプシン、キモトリプシン、アルギニルエンドペプチダーゼ、プロテインキナーゼ、グリコシダーゼなどが使用される。
【0060】
このように製造された本発明のFc断片は、薬物のキャリアとして作用して薬物と結合体を形成する。
【0061】
本発明において、「薬物結合体」または「結合体」は、一つ以上の薬物が一つ以上の免疫グロブリンFcとお互い連結されているものを意味する。
本発明において、「薬物」は、ヒトまたは動物に投与される場合、治療的活性を示す物質を意味し、ポリペプチド、化合物、抽出物、核酸などを含むが、これに制限されない。薬物は、好ましくはポリペプチド薬物である。
【0062】
本発明において、「生理活性ポリペプチド薬物」、「ポリペプチド薬物」および「蛋白質薬物」は同一の意味で使われており、生体内で様々な生理的現象に生理作用を示す生理活性型であることを特徴とする。
【0063】
このようなポリペプチド薬物は、変性し易い、あるいは生体内に存在する蛋白質分解酵素によってよく分解されるといった理由によって、長時間にわたって生理学的活性を持続することができないという欠点がある。しかし、本発明の免疫グロブリンFc断片とポリペプチドを結合させた結合体の場合、薬物の構造的安定性が増加し、分解半減期が増加する。そのうえ、Fc領域の結合によるポリペプチドの生理学的活性の減少は、その他に公知となっている他のポリペプチド薬物製剤に比べて非常に軽微であるといえる。したがって、既存のポリペプチド薬物の生体内利用率に比べて、本発明のポリペプチドおよび本発明のFc断片の結合体は、生体内利用率が著しく増加したことを特徴とする。これは、下記本発明の実施例によっても明白に開示されているところであるが、本発明のFc断片が結合したIFNα、G−CSF、hGHなどが、PEGのみが結合しあるいはPEGとアルブミンが結合した既存の製剤に比べて約2〜6倍程度増加したことを確認することができる(表8、表9および表10)。
【0064】
一方、本発明のFc断片と蛋白質の結合は、従来の組み換え的な方法による融合ではないことを特徴とする。免疫グロブリンFc領域と薬物として用いられる活性ポリペプチドが組み換え的な方法によって融合している形態は、Fc領域のN末端またはC末端にポリペプチドがペプチド結合によって連結された形態であって、これをコードする核酸配列において一つのポリペプチドとして発現されて折りたたまれる。
【0065】
蛋白質の生理学的機能体としての活性が構造によって決定されるという点に基づくとき、これは融合蛋白質活性の急激な減少をもたらす。したがって、ポリペプチド薬物がFcと組み換え的な方法によって融合する場合、構造的安定性が増加したとしても、生体内利用率の面で効果がない。また、このような融合蛋白質は、誤って折りたたまれ(misfolding)て凝集体の形態で発現される傾向があるので、蛋白質の製造、分離の収率面で不経済である。また、活性型ポリペプチドが糖鎖化された形態の場合、真核細胞で発現させなければならず、このような場合、Fcも糖鎖化されるので、生体内で不適切な免疫反応を引き起こすおそれもある。
【0066】
すなわち、本発明によってこそ始めて、糖鎖化された活性型ポリペプチドを非糖鎖化された免疫グロブリンFc領域に連結させた結合体を製造することが可能となり、最上のシステムでそれぞれが製造、分離されるので、蛋白質獲得の収率を高めることができるなど、前記の問題点を全て克服することができる。
【0067】
本発明の免疫グロブリンFc断片に連結可能な蛋白質薬物の例としては、ヒト成長ホルモン、成長ホルモン放出ホルモン、成長ホルモン放出ペプチド、インターフェロン類とインターフェロン受容体類(例えば、インターフェロン−α、−βおよび−γ、水溶性タイプIインターフェロン受容体など)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、グルカゴン様ペプチド類(例えば、GLP−1など)、G蛋白質共役型受容体(G-protein-coupled receptor)、インターロイキン類(例えば、IL−1受容体、IL−4受容体など)、酵素類(例えば、グルコセレブロシダーゼ(glucocerebrosidase)、イズロネート−2−スルファターゼ(iduronate-2-sulfatase)、α−ガラクトシダーゼ−A(alpha-galactosidase-A)、アガルシダーゼα(agalsidase alpha)、アガルシダーゼβ、α−L−イズロニダーゼ(alpha-L-iduronidase)、ブチリルコリンエステラーゼ(butyrylcholinesterase)、キチナーゼ(chitinase)、グルタミン酸デカルボキシラーゼ(glutamate decarboxylase)、イミグルセラーゼ(imiglucerase)、リパーゼ(lipase)、 ウリカーゼ(uricase)、血小板−活性因子アセチルハイドロラーゼ(platelet-activating factor acetylhydrolase)、中性エンドペプチダーゼ(neutral endopeptidase)、ミエロペルオキシダーゼ(myeloperoxidase)など)、インターロイキンおよびサイトカイン結合蛋白質類(例えば、IL−18bp、TNF−結合蛋白質など)、マクロファージ活性因子、マクロファージペプチド、B細胞因子、T細胞因子、プロテインA、アレルギー抑制因子、細胞壊死糖蛋白質、免疫毒素、リンホトキシン、腫瘍壊死因子、腫瘍抑制因子、転移成長因子、アルファ−1アンチトリプシン、アルブミン、α−ラクトアルブミン(alpha-lactalbumin)、アポリポ蛋白質−E、赤血球生成因子、高糖鎖化赤血球生成促進因子、アンジオポエチン類(angiopoietin)、ヘモグロビン、トロンビン(thrombin)、トロンビン受容体活性ペプチド、トロンボモジュリン(thrombomodulin)、血液因子VII、血液因子VIIa、血液因子VIII、血液因子IX、血液因子XIII、プラスミノゲン活性因子、フィブリン−結合ペプチド、ウロキナーゼ、ストレプトキナーゼ、ヒルジン(hirudin)、プロテインC、C−反応性蛋白質、レニン抑制剤、コラゲナーゼ抑制剤、スーパーオキシドジスムターゼ、レプチン、血小板由来成長因子、上皮細胞成長因子、表皮細胞成長因子、アンギオスタチン(angiostatin)、アンギオテンシン(angiotensin)、骨形成成長因子、骨形成促進蛋白質、カルシトニン、インスリン、アトリオペプチン、軟骨誘導因子、エルカトニン(elcatonin)、結合組織活性因子、組織因子経路抑制剤(tissue factor pathway inhibitor)、卵胞刺激ホルモン、黄体形成ホルモン、黄体形成ホルモン放出ホルモン、神経成長因子類(例えば、神経成長因子、毛様体神経栄養因子(cilliary neurotrophic factor)、アキソジェネシス因子−1(axogenesis factor-1)、脳−ナトリウム利尿ペプチド(brain-natriuretic peptide)、神経膠由来神経栄養因子(glial derived neurotrophic factor)、ネトリン(netrin)、好中球抑制因子(neurophil inhibitor factor)、神経栄養因子、ニューチュリン(neuturin)など)、 副甲状腺ホルモン、レラキシン、セクレチン、ソマトメジン、インスリン様成長因子、副腎皮質ホルモン、グルカゴン、コレシストキニン、膵臓ポリペプチド、ガストリン放出ペプチド、コルチコトロピン放出因子、甲状腺刺激ホルモン、オートタキシン(autotaxin)、ラクトフェリン(lactoferrin)、ミオスタチン(myostatin)、受容体類(例えば、TNFR(P75)、TNFR(P55)、IL−1受容体、VEGF受容体、B細胞活性因子受容体など)、受容体拮抗物質(例えば、IL1−Raなど)、細胞表面抗原(例えば、CD2、3、4、5、7、11a、11b、18、19、20、23、25、33、40、45、69など)、モノクローナル抗体、ポリクローナル抗体、抗体断片類(例えば、scFv、Fab、Fab’、F(ab’)2およびFd)、ウイルス由来ワクチン抗原など様々な種類を含み、前記例示された種類に限定されない。抗体断片は、特定の抗原に結合することが可能な能力をもったFab、Fab’、F(ab’)2、FdまたはscFvであり、好ましくはFab’である。
【0068】
特に好ましい生理活性ポリペプチドは、疾病の治療または予防の目的で人体に投与されるとき、投与頻度の高いヒト成長ホルモン、インターフェロン類(インターフェロン−α、−β、−γなど)、顆粒球コロニー刺激因子、赤血球生成因子(EPO)および抗体断片類などである。また、前記生理活性ポリペプチドの天然型と実質的に同等或いはより増加した機能、構造、活性または安定性を有する限り、任意の誘導体または誘導体も本発明の生理活性ポリペプチドの範囲に含まれる。本発明の最も好適なポリペプチドの薬物はインターフェロン−アルファである。
【0069】
本発明では、ポリペプチド薬物以外に他の薬物も使用可能であり、テトラサイクリン、ミノサイクリン、ドキシサイクリン、オフロキサシン、レボフロキサシン、シプロフロキサシン、クラリスロマイシン、エリスロマイシン、セファクロール、セフォタキシム、イミペネム、ペニシリン、ゲンタマイシン、ストレプトマイシン、バンコマイシンなどの誘導体および混合物から選択される抗生剤;メトトレキサート、カルボプラチン、タクソール、シスプラチン、5−フルオロウラシル、ドキソルビシン、エトポシド、パクリタキセル、カンプトセシン、シトシンアラビノシドなどの誘導体および混合物から選択される抗癌剤;インドメタシン、イブプロフェン、ケトプロフェン、ピロキシカム、フルルビプロフェン、ジクロフェナクなどの誘導体および混合物から選択される抗炎症剤;アシクロビル、ロバビンなどの誘導体よび混合物から選択される抗ウイルス剤;およびケトコナゾール、イトラコナゾール、フルコナゾール、アンホテリシン−B、グリセオフルビンなどの誘導体および混合物から選択される抗菌剤を例示することができるが、これに限定されない。
【0070】
一方、本発明のFc断片は、リンカーを媒介として薬物に連結された結合体を形成することができる。
【0071】
このようなリンカーは、ペプチド性リンカーまたは非ペプチド性リンカーを含むが、好ましくは非ペプチド性リンカーであり、より好ましくは非ペプチド性重合体である。ここで、ペプチド性リンカーとは、アミノ酸、好ましくはペプチド結合で連結された1〜20個のアミノ酸を意味し、糖鎖化された形態であってもよい。このようなペプチド性リンカーは、Gly、Ser反復単位を持つペプチドであって、T細胞に対して免疫学的に不活性のペプチドを使用することが好ましい。非ペプチド性重合体の例としては、ポリ(エチレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールの共重合体、ポリオキシエチル化ポリオール、ポリビニルアルコール、多糖類、デキストラン、ポリビニルエーテル、PLA(poly(lactic acid))およびPLGA(poly(lactic-glycolic acid))のような生分解性高分子、脂質重合体、キチン類、ヒアルロン酸などを挙げることができ、最も好ましくはポリ(エチレングリコール)(PEG)などがある。
【0072】
本発明のFc断片−薬物または本発明のFc断片−リンカー−薬物は、様々なモル比の結合が可能である。すなわち、一つのポリペプチド薬物に結合するFc断片および/またはリンカーの数は制限されない。ところが、好ましくは本発明の薬物結合体において薬物とFc断片は、1:1〜10:1、好ましくは1:1〜2:1のモル比で結合できる。
【0073】
また、本発明のFc断片、任意のリンカーおよび薬物の結合は、遺伝子組み換え方法によってFc断片とポリペプチド薬物が融合蛋白質の形態で発現される場合のペプチド結合(peptide bond)を除いた全ての共有結合と、水素結合、イオン結合、ファンデルワールス力、疎水性相互作用のような全ての種類の非共有結合を含む。しかし、薬物の生理活性の面で共有結合であることが好ましい。
【0074】
また、本発明のFc断片、任意のリンカーおよび薬物は、薬物の任意の部位(site)との結合が可能である。一方、本発明のFc断片およびポリペプチド薬物の場合は、N末端、C末端の結合も可能であるが、遊離基と結合することがより好ましく、特にこれらのアミノ末端、リジンのアミノ残基、ヒスチジンのアミノ残基または遊離システイン残基で共有結合がよく形成される。
【0075】
一方、本発明のFc断片、任意のリンカーおよび薬物の結合は、任意の方向に結合できる。すなわち、リンカーと免疫グロブリンFc領域のN末端、C末端または遊離基との結合が可能であり、リンカーと蛋白質薬物のN末端、C末端または遊離基との結合が可能である。リンカーがペプチドリンカーの場合、任意の結合部位における結合が可能である。
また、本発明の結合体は、いろいろの架橋剤(coupling agent)で結合可能である。このような架橋剤の種類は、当該分野に公知となっており、例えば1,1−ビス(ジアゾアセチル)−2−フェニルエタン、グルタルアルデヒド、N−ヒドロキシスクシンイミドエステル、例えば4−アジドサリチル酸とのエステル、イミドエステル、例えばジスクシンイミジルエステル、例えば3,3’−ジチオビス(スクシンイミジルプロピオネート)および二官能性(bifunctional)マレイミド、例えばビス−N−マレイミド−1,8−オクタンを含むが、これに制限されない。
【0076】
一方、本発明に係る新規のFc断片および薬物の結合体は、様々な薬剤学的組成物を製造することができる。
【0077】
本発明において、「投与」はある適切な方法で患者に所定の物質を導入することを意味し、前記結合体は薬物が目的の組織に到達することができる限り、いずれの一般的な経路を介しても投与できる。例えば、腹腔内投与、静脈内投与、筋肉内投与、皮下投与、皮内投与、経口投与、局所投与、鼻内投与、肺内投与、直腸内投与が可能であるが、これに制限されない。しかしながら、経口投与の際、ペプチドは消化されるため、経口用組成物は活性薬剤をコートするか、或いは胃における分解から保護するために剤形化することが好ましい。好ましくは、注射剤の形態で投与できる。また、製薬組成物は、活性物質が標的細胞に移動することが可能な任意の装置によって投与できる。
【0078】
本発明の結合体を含んだ薬剤学的組成物は、薬剤学的に許容可能な担体を含むことができる。薬剤学的に許容される担体は、経口投与の場合には結合剤、滑沢剤、崩壊剤、賦形剤、可溶化剤、分散剤、安定化剤、懸濁化剤、色素、香料などを使用することができ、注射剤の場合には緩衝剤、保存剤、無痛化剤、可溶化剤、等張剤、安定化剤などを混合して使用することができ、局所投与の場合には基剤、賦形剤、潤滑剤、保存剤などを使用することができる。本発明の薬剤学的組成物の剤形は、上述したような薬剤学的に許容される担体と混合して様々に製造できる。例えば、経口投与の場合には錠剤、トローチ剤、カプセル、エリキシル剤(elixir)、サスペンション、シロップ、ウェーハなどの形態で製造することができ、注射剤の場合には、単位投薬アンプルまたは多数回投薬の形態で製造することができる。その他、溶液、懸濁液、錠剤、カプセル、徐方形製剤などに剤形化することができる。
【0079】
一方、製剤化に適した担体、賦形剤および希釈剤の例としては、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、キシリトール、エリスリトール、マルチトール、澱粉、アカシアゴム、アルギン酸塩、ゼラチン、リン酸カルシウム、珪酸カルシウム、セルロース、メチルセルロース、微結晶性セルロース、ポリビニルピロリドン、水、メチルヒドロキシ安息香酸、タルク、ステアリン酸マグネシウムまたは鉱物油などが使用できる。また、充填剤、抗凝集剤、潤滑剤、湿潤剤、香料、乳化剤、防腐剤などをさらに含むことができる。
【0080】
本発明において、Fc断片がキャリアとして用いられた薬物の実際投与量は、治療する疾患、投与経路、患者の年齢、性別並びに体重、および疾患の重症度などのいろいろの関連因子とともに、活性成分である薬物の種類によって決定される。本発明の薬剤学的組成物は、生体内持続性に非常に優れるので、本発明の薬剤学的製剤の投与回数および頻度を著しく減少させることができる。
【0081】
以下、下記の実施例によって本発明をより詳細に説明する。但し、下記の実施例は、本発明を例示するためのものに過ぎず、本発明の範囲を制限するものではない。
【実施例】
【0082】
免疫グロブリンFc断片の製造実施例
(実施例1)ヒト免疫グロブリンIgG4 Fc発現ベクターの製作
(1−1)二量体IgG4 Fc発現ベクターの製作
ヒト免疫グロブリンIgG4のFc遺伝子をクローニングするために、ヒトの血液から収得した血球細胞のRNAを鋳型として次のようにRT−PCRを行った。まず、Qiamp RNA血液キット(Qiagen社)を用いて約6mLの血液から全体RNAを分離した後、このRNAを鋳型とし、One−Step RT−PCRキット(Qiagen社)を用いて遺伝子を増幅した。この際、前記遺伝子配列を収得するために配列番号1と配列番号2で表わされるプライマー対を使用した。配列番号1は、IgG4ヒンジ領域の12個(Glu Ser Lys Tyr Gly Pro Pro Cys Pro Ser Cys Pro、配列番号:3)のアミノ酸配列のうち10番目のアミノ酸配列であるセリンから始まる配列であり、配列番号2は、終結コドンを含むBamHI制限酵素認識部位を挿入したものである。前記プライマー対で増幅された遺伝子は、配列番号4で表わされる塩基配列を持つことが確認され、全体IgG4 Fc中のヒンジ領域のセリン−システイン−プロリンで始まるアミノ末端とCH2およびCH3ドメインから構成される。
【0083】
前記増幅されたIgG4 Fc遺伝子配列を、大腸菌シグナル配列を含む発現ベクターにクローニングするために、本発明者によって開発された発現ベクターpT14S1SH−4T20V22Q(大韓民国特許第38061号)を出発ベクターとして用いた。前記発現ベクターは、配列番号5で表わされる塩基配列を持つ大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む。クローニングを容易にするため、pT14S1SH−4T20V22Qプラスミドの大腸菌熱安定性エンテロトキシンシグナル配列誘導体に配列番号6および7のプライマー対を使用した特定部位の置換突然変異法(site-directed mutagenesis)を行い、シグナル配列の最後のアミノ酸をコードする遺伝子配列を突然変異誘発(mutagenesis)してStuI制限酵素認識部位を挿入した。塩基配列分析法によって、正確にStuI制限酵素認識部位が生成されたことを確認した。pT14S1SH−4T20V22QプラスミドにStuI制限酵素認識部位が生成されたプラスミドをpmSTIIと命名した。前記のように製作されたプラスミドpmSTIIをStuIおよびBamHI制限酵素で処理した後、アガロースゲル電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記増幅されたIgG4 Fc遺伝子断片をBamHI制限酵素で処理した後、前記で回収された発現ベクターの断片に挿入してプラスミドpSTIIdCG4Fcを製作した。このベクターによって発現される蛋白質は、配列番号8のアミノ酸配列を持ち、ヒンジ領域のシステイン残基によるジスルフィド結合によって二量体の形態で存在する。前記製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG4Fc(HM10932)を製造し、これらを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10597)。
【0084】
(1−2)単量体IgG4 Fc発現ベクターの製作
単量体の形態で発現されるIgG4 Fc断片をクローニングするために、配列番号9および2を用いて、前記実施例(1−1)で製作したpSTIIdCG4Fcプラスミドを鋳型としてPCRを行った。前記のPCRによって増幅された遺伝子は、IgG4 Fc配列で二量体の形成に必要なヒンジ領域が除去されたまま、IgG4 CH2−3遺伝子のみが増幅されて単量体の形態で存在する。前記(1−1)と同一の発現ベクターpmSTIIに同一の過程でクローニングしてプラスミドpSTIIG4Moを製作した。前記発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIG4Mo(HM10933)を製造し、これを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10598)。前記発現ベクターによって発現される蛋白質は、配列番号10のアミノ酸配列を持ち、ヒンジ領域がないため、CH2ドメインから発現されて単量体の形態で存在する。
【0085】
(実施例2)ヒト免疫グロブリンIgG1 Fc発現ベクターの製作
(2−1)二量体IgG1 Fc発現ベクターの製作
IgG1のFc遺伝子をクローニングするために、前記実施例(1−1)で行った方法と同様に、ヒトの血液から収得した血球細胞のRNAを鋳型としてOne−Step RT−PCRキット(Qiagen社)を用いてRT−PCRを行った。この際、前記遺伝子配列を収得するために、配列番号11と配列番号12で表わされるプライマー対を使用した。
【0086】
配列番号11は、15個のアミノ酸配列(Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro;配列番号:13)からなるヒンジ領域蛋白質配列のうち13番目のアミノ酸配列であるプロリンから始まる配列であり、配列番号11および12のプライマー対で増幅された遺伝子は、全体IgG1 Fc遺伝子配列中のヒンジ領域のプロリン−システイン−プロリンで始まるアミノ末端とCH2、CH3ドメインからなり、配列番号14の遺伝子配列を持つ。
【0087】
前記増幅されたIgG1 Fc遺伝子配列を大腸菌シグナル配列を含む発現ベクターにクローニングするために、前述したpmSTIIベクターを使用した。前記実施例(1−1)で行ったクローニング過程と類似の方法で、プラスミドpmSTIIをStuI/BamHI制限酵素で処理した後、アガロースゲルに電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記で増幅されたIgG1 Fc遺伝子をBamHI制限酵素で処理した後、前記で回収された発現ベクターの断片に挿入してpSTIIdCG1Fcを製作した。このように製作された発現ベクターを介して発現される産物は、宿主細胞で発現するときに配列番号15のアミノ酸配列を持ち、ヒンジ領域のシステイン残基間のジスルフィド結合によって二量体を形成することができる。製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG1Fc(HM10927)を製造し、これらを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10588)。
【0088】
(2−2)単量体IgG1 Fc発現ベクターの製作
単量体の形態で発現されるIgG1 Fcを製造するために、配列番号16および12のプライマー対を用いて、前記実施例(2−1)で製作したプラスミドpSTIIdCG4Fcを鋳型として遺伝子を増幅した。増幅された遺伝子の断片を実施例(2−1)と同様の方法で発現ベクターpmSTIIに挿入し、配列番号17で表わされる塩基配列を含有するPSTIIG1Moを製作した。前記発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIG1Mo(HM10930)を製造し、これを韓国微生物保存センター(KCCM)に2004年9月15日付で寄託した(寄託番号:KCCM−10595)。前記発現ベクターによって発現される蛋白質は、二量体の形成を可能とするシステイン残基を含有するヒンジ領域が除去されたため、CH2ドメインから発現されて単量体の形態で存在し、配列番号18で表わされるアミノ酸配列を持つ。
【0089】
(実施例3)ヒト免疫グロブリンIgG2 Fc発現ベクターの製作
ヒトIgG2のFc遺伝子をクローニングするために、前記実施例(1−1)で行った方法と同様に、ヒトの血液から収得した血球細胞のRNAを鋳型としてOne−Step RT−PCRキット(Qiagen社)を用いてRT−PCRを行った。この際、前記遺伝子配列を収得するために、配列番号19と配列番号20で表わされるプライマー対を使用した。配列番号19は12個のアミノ酸配列(Glu Arg Lys Cys Cys Val Glu Cys Pro Pro Cys Pro;配列番号:21)からなるヒンジ領域蛋白質配列のうち10番目のアミノ酸配列であるプロリンから始まる配列であり、配列番号19および20のプライマー対で増幅された遺伝子は、全体IgG2 Fc遺伝子配列中のヒンジ領域のプロリン−システイン−プロリンで始まるアミノ末端とCH2、CH3ドメインからなり、配列番号22の遺伝子配列を持つ。前記増幅されたIgG2 Fc遺伝子配列を大腸菌シグナル配列を含む発現ベクターにクローニングするために、前述したpmSTIIベクターを使用した。前記実施例(1−1)で行ったクローニング過程と類似の方法で、プラスミドpmSTIIをStuI/BamHI制限酵素で処理した後、アガロースゲル電気泳動を行い、大腸菌熱安定性エンテロトキシンシグナル配列誘導体を含む大きい断片(4.7kb)を回収した。前記増幅されたIgG2 Fc遺伝子をBamHI制限酵素で処理した後、前記回収された発現ベクターの断片に挿入してpSTIIdCG2Fcを製作した。このように製作された発現ベクターによって発現される産物は、宿主細胞で発現するときに配列番号23のアミノ酸配列を持ち、ヒンジ領域のシステイン残基間のジスルフィド結合によって二量体を形成することができる。それぞれ製作された発現ベクターを大腸菌BL21(DE3)に形質転換させて大腸菌形質転換体BL21/pSTIIdCG2Fc(HM10936)を製造した。
【0090】
(実施例4)免疫グロブリンFcの発現および精製
(4−1)免疫グロブリンFcの発現の確認
前記実施例1、2および3で収得した微生物形質転換体を発酵器(Marubishi社)に接種して発酵させた後、免疫グロブリンFc断片の発現の有無を確認した。
まず、LB培地100mLに前記形質転換体をそれぞれ一晩中振盪培養した後、発酵器に接種して本培養を行った。発酵器の温度は、35℃あるいは30℃を維持し、嫌気性状態になることを防止するために、空気を20vvmで投入しながら500rpmで攪拌した。発酵の間、微生物の成長のために足りないエネルギー源を補うために、葡萄糖(glucose)と酵母抽出液(yeast extract)とを微生物の発酵状態に応じて投与した。吸光度600nmでOD値が80〜110となる時期に誘導物質(inducer)IPTGを20μM〜4mMとなるように投与して発現を誘導した。これを、さらに40〜45時間高濃度で培養して吸光度600nmでのOD値が100〜120となるようにした。
【0091】
前記大腸菌形質転換体から免疫グロブリンFcの発現の有無、発現場所、水溶性および二量体形成の有無を確認するために、下記のような試験を行った。発現産物が前記発現ベクターに融合しているシグナル配列によって発酵液またはペリプラスム空間に発現されるかを確認するために、発酵液を遠心分離して、細胞を除去した発酵液溶液を収得し、且つ細胞を回収した。前記細胞を除去した発酵液溶液と、前記回収した細胞に浸透圧ショック(osmotic shock)をかけることによって得られたペリプラスム空間溶液とをウエスタンブロットによって解析し、免疫グロブリンFcの発現の有無を確認したが、その量は極めて少量であった。前記細胞をウルトラ−ソニケーション(Misonix社)を用いて破砕して細胞溶解液(cell lysate)を得、その細胞溶解液を遠心分離して水溶性物質と不溶性物質を分離した後、水溶性物質を次のような方法でウエスタンブロットした。前記水溶性物質をDTTまたはβ−メルカプトエタノールのような還元剤を含まない蛋白質サンプルバッファと混ぜた後、15%SDS−PAGE(Criterion Gel、Bio−Rad社)に電気泳動した。電気泳動済みのゲルをニトロセルロース(nitro-cellulose)膜に転写した後、HRPが融合している抗ヒトFc抗体(Sigma)を用いて検出(detect)を行った。図1に示すように、免疫グロブリンFcは、大腸菌の細胞質内に水溶性の状態で過量に発現されることが分かるとともに、ヒンジ領域の一部が挿入された形質転換体から発現された産物は二量体を形成することが分かる。図1のレーン1、2、3はそれぞれ形質転換体HM10927、HM10932、HM10936で発現された産物であり、レーン4は動物細胞で生産された免疫グロブリンをパパイン処理して生成されたFcであって、糖が存在するため、大腸菌で生成されたものよりやや大きく展開されることが分かる。
【0092】
(4−2)N末端配列の確認
前記実施例において大腸菌の細胞質内で水溶性二量体の形態で発現されるFcは、シグナル配列が融合しているために分泌されず、細胞質内に存在するとき、シグナル配列のプラセシングなしで融合したままで存在するかを確認するために、N末端アミノ酸配列の分析を基礎科学支援研究所のソウル分所に依頼した。分析する試料は下記のように準備した。
【0093】
まず、PVDF膜(Bio−Rad社)をメタノールに約2〜3秒間浸漬して活性化させた後、ブロッキングバッファ(170mMグリシン、25mM Tris−HCl(pH8)、20%メタノール)に十分濡らした。実施例(4−1)で展開した非還元条件のSDS−PAGEゲルを、前記のように準備されたPVDF膜に、ブロッティングキット(Hoefer Semi−Dry Transfer unit、Amersham)を用いて約1時間の転写を行った。PVDF膜に転写された蛋白質を蛋白質染色薬であるCoomassie Blue R−250(Amnesco社)を用いてしばらく(3〜4秒間)染色した後、脱色溶液(水:酢酸:メタノールが5:1:4)で洗浄した。洗浄済みの膜から、蛋白質が含まれている部位をハサミで切断してN末端配列の分析を依頼した。
【0094】
IgG1 Fc蛋白質のN末端配列はPro−Cys−Pro−Ala−Pro−Glu−Leu−Leu−Gly−Glyであり、IgG4 Fc蛋白質のN末端配列はSer−Cysl−Pro−Ala−Pro−Glu−Phe−Leu−Gly−Glyである。また、IgG2 Fc蛋白質のN末端配列はPro−Cys−Pro−Ala−Pro−Pro−Val−Ala−Gly−Proと確認された。以上のようなアミノ酸配列の分析結果から明らかなように、本発明の大腸菌形質転換体から発現されるFc断片は、正確なN末端配列を持つものと確認された。また、シグナル配列を融合させて発現させる際に、細胞外膜またはペリプラスムに分泌されず、細胞質内に過発現されてもシグナル配列が正確にプロセシングされて水溶性の形態で存在するという新しい事実が確認された。
【0095】
(4−3)免疫グロブリンFcの精製
免疫グロブリンに強い親和性があると知られているプロテインA親和性カラム(protein-A affinity column)を用いて下記のような方法で精製した。
【0096】
遠心分離によって発酵液から回収された大腸菌細胞をMicrofluizer(Microfludics社)を用いて破砕して細胞溶解液(cell lysate)を得、細胞質内に存在する組み換え免疫グロブリンFc断片を2段階カラムクロマトグラフィによって精製した。プロテインA親和性カラム(Pharmacia社)5mLをPBSで平衡化させた後、細胞溶解液を5mL/分の流速で負荷した。結合していない分画はPBSで洗浄した後、100mMのクエン酸溶液(pH3.0)で溶出させた。溶出した分画を脱塩カラム(desalting column Hiprep 26/10、Pharmacia社)を用いて10mM Tris緩衝液(pH8.0)で交換した。Q HP 26/10カラム(Pharmacia社)50mLを用いて2次陰イオン交換カラムクロマトグラフィを行った。1次精製された組み換え免疫グロブリンFc分画を前記カラムに結合させた後、10mM Tris緩衝液(pH8.0)条件下で直線濃度勾配(塩化ナトリウムの濃度0M→0.2M)法によって溶出し、高純度の分画を得ることができた。各産物をプロテインAカラムを用いて部分精製した後、組み換え免疫グロブリンFc断片の発現量が下記に示したように決定された。
【0097】
【化1】
【0098】
前記のように収得された免疫グロブリンFcは、重鎖の二量体あるいは単量体の形態なので、還元条件のSDS−PAGEと非還元条件のSDS−PAGEにおいて蛋白質の移動様相が異なる。発現された産物を精製した後、純度を確認するために行ったSDS−PAGE結果を、図2および図3に示した。
【0099】
図2および図3は、このように精製された二量体の形態と単量体の形態の免疫グロブリンFc蛋白質を15%クライテリオンゲル(criterion gel、Bio−Rad社)を用いて非還元条件のSDS−PAGEゲルと還元条件のSDS−PAGEゲルに展開させてその移動様相を比較したものである。図2のAは非還元条件でSDS−PAGEゲルに展開したもので、Bは還元条件でSDS−PAGEゲルに展開したものである。レーンMは予め染色された低範囲標準蛋白質マーカー(prestained low-range standard marker、Bio−Rad社)であり、レーン1〜4はそれぞれ形質転換体HM10927、HM10928(韓国微生物保存センターに2004年9月15日付で寄託した。寄託番号:KCCM−10589)、HM10929(韓国微生物保存センターに2004年9月15日付で寄託した。寄託番号:KCCM−10594)およびHM10932によって生産された免疫グロブリンFc試料である。図2に示すように、還元条件のSDS−PAGEでは、免疫グロブリンFc断片は、ヒンジ領域のシステイン残基によるジスルフィド結合が還元されて、それぞれ単量体の形態で存在するので、単量体の大きさだけ移動した。一方、非還元条件のSDS−PAGEでは、免疫グロブリンFc断片は、それぞれの単量体がジスルフィド結合で連結された二量体の形態で存在し、その結果、約42kDaの移動距離を持つことを確認した。
【0100】
図3のAは、非還元条件で、蛋白質をSDS−PAGEゲルに展開したものであり、Bは還元条件で、蛋白質をSDS−PAGEゲルに展開したものである。レーンMは、前記標準蛋白質マーカーであり、レーン1および2は、形質転換体HM10930およびHM10933によって生産された免疫グロブリンFc試料である。図3に示すように、還元条件と非還元条件とにおける移動距離はあまり差異がなく、分子内ジスルフィド結合の還元有無によって移動距離に若干の差異があることを確認した。
【0101】
免疫グロブリンFcおよび薬物の結合体製造
(実施例5)インターフェロンアルファ(IFNα)−PEG−免疫グロブリンFc領域(Fc)結合体の製造I
(段階1)免疫グロブリンを用いた免疫グロブリンFc領域の製造
免疫グロブリンFc領域を製造するために、10mMのリン酸塩緩衝液に溶解された分子量150kDaの免疫グロブリンG(Immunoglobulin G;IgG、緑十字)200mgを、蛋白質加水分解酵素パパイン(Papain、Sigma社)2mgを用い、37℃で2時間徐々に攪拌しながら処理した。酵素反応後、生成された免疫グロブリンFc領域を精製するために、Superdexカラム、プロテインAカラムおよび陽イオン交換樹脂カラムクロマトグラフィを順次行った。具体的には、反応液を、10mMのリン酸ナトリウム緩衝液(PBS、pH7.3)によって平衡化させたSuperdex200カラム(Pharmacia社)に滴下し、同一の緩衝液を用いて1mL/分の流速で溶出させた。免疫グロブリンFc領域より分子量が相対的に大きい未反応免疫グロブリン(IgG)やF(ab’)2などは、早く溶出するので、これを先に除去した。免疫グロブリンFc領域と類似の分子量を持つFabは、次のように、プロテインAカラムクロマトグラフィを行って除去した(図4)。20mMのリン酸塩緩衝液(pH7.0)で平衡化させたプロテインAカラム(Pharmacia社)にSuperdex200カラムから溶出した免疫グロブリンFc領域含有分画を5mL/分の流速で負荷した後、カラムに結合していない蛋白質を除去するために同一の緩衝液で十分洗浄した。次に、100mMのクエン酸ナトリウム(Na citrate、pH3.0)緩衝液を流して高純度の免疫グロブリンFc領域を溶出させた。最後に、プロテインAカラムで精製されたFc分画を陽イオン交換樹脂カラム(polyCAT、PolyLC社)を用いて精製した。プロテインAカラムで精製されたFc分画を滴下した前記陽イオン交換樹脂カラムは、10mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法によって溶出し、高純度のFc分画を得た。前記高純度のFc分画を12%SDS−PAGEによって解析した(図5のレーン2)。
【0102】
(段階2)IFNα−PEG連結体の製造
両末端にアルデヒド反応基を持つ分子量3.4kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を、ヒトインターフェロンアルファ−2b(hIFNα−2b、分子量20kDa)が5mg/mLの濃度で溶解された100mMのリン酸塩緩衝液に、IFNα:PEGのモル比が1:1、1:2.5、1:5、1:10および1:20となるように添加した。ここに還元剤のナトリウムシアノボロハイドライド(NaCNBH3、Sigma社)を最終濃度が20mMとなるように添加し、4℃で徐々に攪拌しながら3時間反応させた。インターフェロンアルファのアミノ末端部位に選択的にPEGを連結され、PEGとインターフェロンアルファが1:1で結合した連結体を得るために、前記反応混合物をSuperdexRカラム(Pharmacia社)を用いたサイズ排除(size exclusion)クロマトグラフィにかけた。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてIFNα−PEG連結体を精製し、PEGと結合していないインターフェロンアルファ、未反応PEGおよび2つのインターフェロンアルファがPEGと連結された二量体副産物を除去した。精製されたIFNα−PEG連結体を5mg/mLの濃度に濃縮した。これより、反応性が最も良くて二量体などの副産物が少ないIFNα:PEGの最適反応モル比は1:2.5〜1:5であることを確認した。
【0103】
(段階3)IFNα−PEG−Fc結合体の形成
前記段階2で精製されたIFNα−PEG連結体に免疫グロブリンFc領域のN末端に結合させるために、段階1で準備された免疫グロブリンFc領域(約53kDa)を10mMのリン酸塩緩衝液に溶解させた後、IFNα−PEG連結体:Fcのモル比がそれぞれ1:1、1:2、1:4および1:8となるように、IFNα−PEG連結体と混合して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加し、4℃で20時間徐々に攪拌しながら反応させた。これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:Fcの最適反応モル比は1:2であることを確認した。
【0104】
(段階4)IFNα−PEG−Fc結合体の分離および精製
前記段階3の結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−Fc蛋白質結合体を精製するためにSuperdexサイズ排除クロマトグラフィを行った。反応混合物を濃縮した後、10mMのリン酸塩緩衝液(pH7.3)を用いて流速2.5mL/分でカラムを通過させ、結合していないFcおよび未反応物質を除去し、IFNα−PEG−Fc蛋白質結合体分画を得た。得られたIFNα−PEG−Fc蛋白質結合体分画には不純物として少量の未反応Fcおよびインターフェロンアルファ二量体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−Fc蛋白質結合体分画を10mMの酢酸ナトリウム(pH4.5)で平衡化させたPolyCAT LPカラム(PolyLC社)に仕込み、1Mの塩化ナトリウム(NaCl)を含む10mMの酢酸ナトリウム(pH4.5)緩衝液を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.5M)法で溶出してさらに精製した。最後に、陰イオン交換カラムを用いてIFNα−PEG−Fc蛋白質結合体を精製した。PolyWAX LPカラム(PolyLC社)を10mMのTris−HCl(pH7.5)緩衝液で平衡化させた後、精製されたIFNα−PEG−Fc蛋白質結合体分画を負荷し、1Mの塩化ナトリウムを含む10mMのTris−HCl(pH7.5)緩衝液を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.3M)法で溶出して純粋なIFNα−PEG−Fc蛋白質結合体を精製した。
【0105】
(実施例6)IFNα−PEG−Fc結合体の製造II
(段階1)Fc−PEG連結体の製造
両末端にアルデヒド反応基を持つ分子量3.4kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を、前記実施例5の段階1で準備された免疫グロブリンFc領域が15mg/mLの濃度で溶解された100mMのリン酸塩緩衝液に、免疫グロブリンFc:PEGのモル比がそれぞれ1:1、1:2.5、1:5、1:10および1:20となるように添加した。還元剤のNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しながら3時間反応させた。免疫グロブリンFc領域のアミノ末端部位に選択的にPEG:Fcが1:1で結合した連結体を得るために、前記反応混合物を持ってSuperdex(SuperdexR、Pharmacia社)サイズ排除クロマトグラフィを行った。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてFc−PEG連結体を精製し、PEGと結合していない免疫グロブリンFc領域、未反応PEGおよび2つの免疫グロブリンFc領域がPEGに連結された二量体副産物を除去した。精製されたFc−PEG連結体を約15mg/mLの濃度で濃縮した。これより、反応性が最も良くて二量体などの副産物が少ないFc:PEGの最適反応モル比は1:3〜1:10であることを確認した。
【0106】
(段階2)Fc−PEG連結体とインターフェロンアルファの結合体の形成および精製
前記段階1で精製されたFc−PEG連結体をIFNαのN末端に結合させるために、10mMのリン酸塩緩衝液に溶解されたインターフェロンアルファを使用し、Fc−PEG連結体:IFNαのモル比がそれぞれ1:1、1:1.5、1:3および1:6となるように添加して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。反応後、実施例5の段階4と同一の方法で精製して未反応物質および副産物を除去し、これから生成されたFc−PEG−IFNα蛋白質結合体を純粋に分離した。
【0107】
(実施例7)ヒト成長ホルモン(hGH)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト成長ホルモン(hGH、分子量22kDa)を使用し、hGH:PEGのモル比を1:5とする以外は、実施例5と同一の方法でhGH−PEG−Fc蛋白質結合体を製造および精製した。
【0108】
(実施例8)ヒト顆粒球コロニー刺激因子(G−CSF)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト顆粒球コロニー刺激因子(G−CSF)を使用し、G−CSF:PEGのモル比を1:5とする以外は、実施例5と同一の方法でG−CSF−PEG−Fc蛋白質結合体を製造および精製した。
【0109】
一方、天然型G−CSFの17番目のアミノ酸がセリンで置換された誘導体(17S−G−CSF)を用いて前記と同一の方法で17S−G−CSF−PEG−Fc蛋白質結合体を製造および精製した。
【0110】
(実施例9)ヒト赤血球生成因子(EPO)−PEG−Fc結合体の製造
インターフェロンアルファの代わりにヒト赤血球生成因子(Erythropoietin;EPO)を使用し、EPO:PEGのモル比を1:5とする以外は、実施例5と同一の方法でEPO−PEG−Fc蛋白質結合体を製造および精製した。
【0111】
(実施例10)反応基の種類を異にするPEGを用いた蛋白質結合体の製造
両末端の反応基が全てスクシンイミジルプロピオネート(Succinimidyl propionate;SPA)であるPEGを用いてIFNα−PEG−Fc蛋白質結合体を次のように製造した。インターフェロンアルファ10mgが溶解された100mMのリン酸塩緩衝液に、両末端にSPA反応基を持つ分子量3.4kDaのポリエチレングリコールSPA−PEG−SPA(Shearwater社)をIFNα:PEGのモル比がそれぞれ1:1、1:2.5、1:5、1:10および1:20となるように添加し、常温で徐々に攪拌しながら2時間反応させた。インターフェロンアルファのリジン残基のアミノ基部位に選択的にPEGが1:1で結合したPEG−IFNα連結体を得るために、反応混合物をもってSuperdexサイズ排除クロマトグラフィを行った。溶出液として10mMのリン酸カリウム緩衝液(pH6.0)を用いてIFNα−PEG連結体を精製し、PEGと結合していないインターフェロンアルファ、未反応PEG、およびPEGの両末端に2つのインターフェロンアルファが連結された二量体副産物を除去した。IFNα−PEG連結体を免疫グロブリンFcのリジン残基のアミノ基部位に結合させるために精製されたIFNα−PEG連結体を約5mg/mLの濃度で濃縮した後、実施例5の段階3および4と同一の方法でIFNα−PEG−Fc結合体を製造および精製した。これより、反応性が最も良くて二量体などの副産物が少ないインターフェロンアルファ:PEGの最適反応モル比は1:2.5〜1:5であることを確認した。
【0112】
一方、前記と同一の方法を行うが、両末端の反応基が全てN−ヒドロキシスクシンイミジル(N-hydroxysuccinimidyl;NHS)であるPEG(NHS−PEG−NHS;Shearwater社)、または両末端の反応基が全てブチルアルデヒド(buthyl aldehyde)であるPEG(BUA−PEG−BUA;Shearwater社)を用いてIFNα−PEG−Fc結合体の生成を確認した。
【0113】
(実施例11)分子量の異なるPEGを用いた蛋白質結合体の製造
両末端にアルデヒド反応基を持つ分子量10kDaのポリエチレングリコールALD−PEG−ALD(Shearwater社)を用いて実施例5の段階2と同一の方法でIFNα−10K PEG連結体を製造および精製した。この際、反応性が最も良くて二量体などの副産物が少ないインターフェロンアルファ:10K PEGの最適モル比は1:2.5〜1:5であることが確認された。精製されたIFNα−PEG連結体を約5mg/mLとなるように濃縮した後、これを用いて実施例5の段階3および4と同一の方法によってIFNα−10K PEG−Fc蛋白質結合体を製造および精製した。
【0114】
(実施例12)Fab’−S−PEG−N−Fc結合体の製造(−SH基)
(段階1)Fab’の発現および精製
抗腫瘍壊死因子−アルファFab’を発現する大腸菌形質転換体BL21/poDLHF(寄託番号:KCCM−10511)をLB培地100mLに接種して一晩中振盪培養した後、5Lの発酵器(Marubishi)に接種して温度30℃、空気投入量20vvm、攪拌速度500rpmの条件下で培養した。発酵が進むにつれて、微生物の成長のために足りないエネルギー源を補うために、葡萄糖(glucose)と酵母抽出液(yeast extract)とを微生物の発酵状態に応じて投与し、吸光度600nmでのOD値が80となる時期にIPTGを投与して蛋白質の発現を誘導した。これを40〜45時間高濃度で培養して吸光度600nmでのOD値が120〜140となるようにした。得られた発酵液を遠心分離(20,000g、30分)して沈殿物は捨て、上澄み液のみを取った。
【0115】
得られた上澄み液から次の3段階のカラムクロマトグラフィを経て抗腫瘍壊死因子−アルファFab’を純粋に精製した。20mMのリン酸塩緩衝液(pH7.0)で平衡化させたHiTrap蛋白質Gカラム(5mL、Pharmacia社)に前記上澄み液を滴下した後、100mMのグリシン(Glycine、pH3.0)緩衝液を用いて溶出させた。溶出したFab’分画を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200, Pharmacia社)に滴下し、同一の緩衝液で溶出させた。溶出したFab’分画をpolyCAT 21×250カラム(PolyLC社)を用いて最終精製したが、10mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法で溶出して純粋な抗腫瘍壊死因子−アルファFab’分画を得た。
【0116】
(段階2)Fc−PEG連結体の製造および精製
免疫グロブリンFcのN末端のアミノ基にリンカーPEGを結合させるために、前記実施例5の段階1と同一の方法で製造された免疫グロブリンFcを100mMのリン酸ナトリウム緩衝液(pH6.0)に5mg/mLの濃度で溶解させ、ここにNHS−PEG−MAL(3.4kDa、Shearwater社)をFc:PEGのモル比が1:10となるように添加した後、4℃で徐々に攪拌しながら12時間反応させた。
【0117】
反応終了後、反応緩衝液を20mMのリン酸ナトリウム緩衝液(pH6.0)で交換しながら、反応していないNHS−PEG−MALを除去した。緩衝液の交換後、反応物をpolyCATカラム(PolyLC社)に負荷し、20mMのリン酸ナトリウム緩衝液(pH6.0)を直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)方法で流して免疫グロブリンFc−PEG連結体を先に溶出させて得た後、反応していない免疫グロブリンFcを溶出させて除去した。
【0118】
(段階3)Fab’−S−PEG−N−Fc結合体(−SH基)の製造および精製
Fab’の遊離システイン基に免疫グロブリンFc−PEG連結体を結合させるために、段階1で精製されたFab’を100mMのリン酸ナトリウム緩衝液(pH7.3)に2mg/mLの濃度で溶解させた後、同一の緩衝液に段階2で準備された免疫グロブリンFc−PEG連結体をFab’:連結体のモル比が1:5となるように入れた。最終蛋白質の濃度が50mg/mLとなるように濃縮し、4℃で徐々に攪拌しながら24時間反応させた。
【0119】
カップリング反応の終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。カップリングされたFab’−S−PEG−N−Fc結合体は、分子量が大きいので先に溶出し、反応していない免疫グロブリンFc−PEG連結体およびFab’は後で溶出するため、除去することができた。残存する未反応免疫グロブリンFcを完全に除去するために、溶出したFab’−S−PEG−N−Fc結合体分画をさらにpolyCAT 21×250カラム(PolyLC社)に滴下し、20mMのリン酸ナトリウム緩衝液(pH6.0)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、Fc−PEG連結体がFab’のC末端付近の−SH基に連結されたFab’−S−PEG−N−Fc結合体を純粋に得た。
【0120】
(実施例13)Fab’−N−PEG−N−Fc結合体の製造(N末端)
(段階1)Fab’−PEG連結体(N末端)の製造および精製
前記実施例12の段階1で得た、精製されたFab’40mgを100mMのリン酸ナトリウム緩衝液(pH6.0)に5mg/mLの濃度で溶解させた後、ブチルALD−PEG−ブチルALD(3.4kDa、Nektar社)をFab’:PEGのモル比が1:5となるように添加した。還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しがら2時間反応させた。
【0121】
反応終了後、反応緩衝液を20mMのリン酸ナトリウム緩衝液(pH6.0)で交換した。緩衝液の交換後、反応物をpolyCATカラム(PolyLC社)に負荷し、20mMの酢酸塩緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.4M)法で溶出し、Fab’のN末端にリンカーPEGが結合したFab’−PEG連結体分画を先に溶出させて得た後、反応していないFab’を溶出させて除去した。
【0122】
(段階2)Fab’−N−PEG−N−Fc結合体の製造および精製
段階1で精製されたFab’−PEG連結体を免疫グロブリンFcのN末端に結合させるために、100mMのリン酸ナトリウム緩衝液(pH6.0)に10mg/mLの濃度で溶解させた後、同一の緩衝液に溶解された免疫グロブリンFcをFab’−PEG連結体:Fcのモル比が1:5となるように入れた。最終蛋白質の濃度が50mg/mLとなるように濃縮し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で徐々に攪拌しながら24時間反応させた。
【0123】
カップリング反応の終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。カップリングされたFab’−N−PEG−N−Fc結合体は、分子量が大きいので先に溶出し、反応していない免疫グロブリンFcおよびFab’−PEG連結体は後で溶出するため除去することができた。残存する未反応免疫グロブリンFcを完全に除去するために、溶出したFab’−N−PEG−N−Fc結合体分画をさらにpolyCAT 21×250カラム(PolyLC社)に負荷し、20mMのリン酸ナトリウム緩衝液(pH6.0)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、免疫グロブリンFc−PEG連結体がFab’のN末端に連結されたFab’−N−PEG−N−Fc結合体を純粋に得た。
【0124】
(実施例14)脱糖鎖化免疫グロブリンFcの製造および精製
前記実施例5と同一の方法で製造した免疫グロブリンFc200mgを、100mMのリン酸塩緩衝液(pH7.5)に2mg/mLの濃度となるように溶解した後、脱糖鎖化酵素PNGase F(NEB社)を300U/mgとなるように添加した。反応混合物を37℃で24時間徐々に攪拌しながら反応させた。反応終了後、脱糖鎖化免疫グロブリンFcを精製するために、反応物をSPセファロースFFカラム(Pharmacia社)に負荷し、10mMの酢酸塩緩衝液(pH4.5)条件で1MのNaClを使用した直線濃度勾配(0.1M→0.6M)法で溶出させ、天然型免疫グロブリンFc分画を先に溶出させた後、脱糖鎖化免疫グロブリンFc(deglycosylated FC:DG Fc)を溶出させて得た。
【0125】
(実施例15)IFNα−PEG−DG Fc結合体の製造
前記実施例5の段階2で精製されたIFNα−PEG連結体に、前記実施例14で製造された脱糖鎖化免疫グロブリンFcを結合させるために、IFNα−PEG連結体を10mMのリン酸塩緩衝液に溶解されたDG FcにIFNα−PEG連結体:DG Fcのモル比がそれぞれ1:1、1:2、1:4および1:8となるように添加して反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。
【0126】
これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:DG Fcの最適反応モル比は、1:2であることを確認した。
【0127】
結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−DG Fc蛋白質結合体を精製するために、反応混合物を用いてSuperdex(SuperdexR、Pharmacia社)サイズ排除クロマトグラフィを行った。リン酸塩緩衝液(pH7.3)を用いて流速2.5mL/分でカラムに通過させ、結合していないDG Fcおよび未反応物質を除去し、IFNα−PEG−DG Fc蛋白質結合体分画を精製した。
【0128】
得られたIFNα−PEG−DG Fc蛋白質結合体分画には、不純物として少量の未反応DG FcおよびIFNα−PEG連結体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−DG Fc蛋白質結合体分画を10mMの酢酸ナトリウム(pH4.5)で平衡化させたPolyCAT LPカラム(PolyLC社)に仕込み、1MのNaClを含んだ10mMの酢酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.6M)法で溶出してさらに精製し、最後に陰イオン交換カラムを用いてIFNα−PEG−DG Fc蛋白質結合体を純粋に獲得した。PolyWAX LPカラム(PolyLC社)を10mMのTris−HCl緩衝液(pH7.5)で平衡化させた後、精製されたIFNα−PEG−DG Fc蛋白質結合体分画を負荷し、1Mの塩化ナトリウムを含んだ10mMのTris−HCl緩衝液(pH7.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.3M)法で溶出して純粋なIFNα−PEG−DG Fc蛋白質結合体を精製した。
【0129】
(実施例16)IFNα−PEG連結体と組み換えAG Fc誘導体の結合体の製造
前記実施例5および15と同一の方法により、実施例1で製造されたAG Fc誘導体であるIgG4 delta−CysのN末端にIFNα−PEG連結体を結合させた。実施例1で製造されたAG Fc誘導体であるIgG4 delta−CysをIFNα−PEG連結体と結合させた。結合反応後、未反応物質および副産物を除去し、生成されたIFNα−PEG−AG Fc蛋白質結合体(I)を精製するために、Q HP 26/10カラム(Pharmacia社)50mLを用いて1次精製した後、高圧カラムのpolyCAT 21.5×250カラム(PolyLC社)で高純度の結合体を精製した。カップリング反応液を脱塩カラムHiPrep 26/10(Pharmacia社)を用いて10mMのTris緩衝液(pH8.0)で交換した後、Q HP26/10 50mLカラムに8mL/分の流速で負荷して結合させた後、直線濃度勾配(塩化ナトリウムの濃度0M→0.2M)法によって所望の分画を得た。溶出した分画を10mMの酢酸塩緩衝液(pH5.2)で平衡化されたpolyCAT21.5×250カラムに15mL/分の流速でさらに結合させた後、直線濃度勾配(塩化ナトリウムの濃度0.1M→0.3M)法で溶出させて高純度の分画を得ることができた。同一の方法により、実施例12で製造された別のAG Fc誘導体であるIgG4単量体を用いてIFNα−PEG−AG Fc蛋白質結合体(II)を製造した。
【0130】
(実施例17)ヒト赤血球生成因子(EPO)−PEG−組み換えAG Fc誘導体の結合体の製造
前記実施例16と同一の方法により、EPO−PEG連結体とAG Fc誘導体であるIgG4 delta−Cysとが連結された結合体を製造した。
【0131】
(比較例1)IFNα−40K PEG連結体の製造
100mMのリン酸カリウム緩衝液(pH6.0)にインターフェロンアルファ5mgを溶解して最終体積が5mLとなるように調製した後、PEGの分子量40kDaの活性化メトキシ−PEG−アルデヒド(Shearwater社)をインターフェロンアルファ:40K PEGのモル比が1:4となるように前記溶液に添加した。前記反応溶液に還元剤NaCNBH3を最終濃度が20mMとなるように添加した後、4℃で18時間徐々に攪拌させながら反応させた。インターフェロンアルファに反応していないPEGを不活性化させるために、エタノールアミンを最終濃度が50mMとなるように添加した。
【0132】
未反応PEGの分離および緩衝液の交替のためにSephadexG−25カラム(Pharmacia社)を用いた。まず、2カラム体積(column volume;CV)の10mM Tris−HCl緩衝液(pH7.5)を用いてカラムを平衡化させた後、反応混合物を滴下した。溶出画分は、紫外部吸光計により波長260nmの吸収を測定することによって検出した。前記カラムを同一の緩衝液で溶出させると、大きさが更に大きいPEGによって(N末端に:挿入KYM)修飾されたインターフェロンアルファが先に溶出し、未反応PEGは時差をおいて後で溶出するため、IFNα−40K PEGのみを分離することができる。
【0133】
前記で得た溶出液からIFNα−40K PEG連結体をさらに純粋に分離、精製するために、次のようにクロマトグラフィを行った。3mLのPolyWAX LPカラム(Polywax社)を10mMのTris−HCl緩衝液(pH7.5)で平衡化させた。PEG−IFNα連結体を含有する溶出液を1mL/分の流速でカラムに負荷した後、15mLの平衡緩衝液で洗浄した。30mLの1M NaCl緩衝液を用いた0〜100%の塩濃度勾配法を用いてトリ−、ジ−およびモノ−PEGが結合したインターフェロンアルファを順序とおりに溶出させた。
【0134】
さらに純粋なモノ−PEG結合インターフェロンアルファを分離するために、前記で収得したモノ−PEGとインターフェロンアルファとの連結体溶出分画をサイズ排除クロマトグラフィにかけた。10mMのリン酸ナトリウム緩衝液(pH7.0)で平衡化させたSuperdex200(Superdex 200、Pharmacia社)に前記溶出液を濃縮して滴下した後、同一の緩衝液で溶出させた。この際、1mL/分の流速で緩衝液を流した。トリ−およびジ−PEGが結合したインターフェロンアルファは、モノ−PEGが結合したインターフェロンアルファより溶出時間が相対的に早いので、これを除去して、モノ−PEGが結合したインターフェロンアルファのみを純粋に分離した。
同一の方法により、ヒト成長ホルモン、顆粒球コロニー刺激因子およびその誘導体のアミノ末端に40K PEGが結合したhGH−40K PEG、G−CSF−40K PEGおよび40K PEG−17S−G−CSF誘導体の連結体を製造した。
【0135】
(比較例2)IFNα−PEG−アルブミン結合体の製造
実施例1の段階2で精製されたIFNαF−PEG連結体をアルブミンのアミノ末端に結合させるために、10mMのリン酸塩緩衝液に溶解されたヒトアルブミン(human serum albumin、HSA、約67kDa、緑十字)をIFNα−PEG連結体:アルブミンのモル比が1:1、1:2、1:4および1:8となるように添加した後、反応させた。反応液を100mMのリン酸塩緩衝液に調製し、還元剤としてNaCNBH3を最終濃度が20mMとなるように添加した後、4℃で20時間徐々に攪拌しながら反応させた。これにより、反応性が最も良くて二量体などの副産物が少ないIFNα−PEG連結体:アルブミンの最適反応モル比は、1:2であることを確認した。
【0136】
結合反応の後、未反応物質および副産物を除去し、生成されたIFNα−PEG−アルブミン蛋白質結合体を精製するために、Superdexサイズ排除クロマトグラフィを行った。各反応混合物を濃縮した後、10mMの酢酸ナトリウム緩衝液を2.5mL/分の流速でカラムに通過させ、結合していないアルブミンおよび未反応物質を除去し、IFNα−PEG−アルブミン蛋白質結合体のみを精製した。得られたIFNα−PEG−アルブミン蛋白質結合体分画には、不純物として少量の未反応アルブミンおよびインターフェロンアルファ二量体が混在しているので、これを除去するためにさらに陽イオン交換樹脂クロマトグラフィを行った。IFNα−PEG−アルブミン蛋白質結合体分画を10mMの酢酸ナトリウム溶液(pH4.5)で平衡化させたカラム(SP5PW、Waters社)に負荷し、1Mの塩化ナトリウム(NaCl)を含んだ10mMの酢酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0M→0.5M)法で溶出してさらに精製し、IFNα−PEG−アルブミン蛋白質結合体を純粋に獲得した。
【0137】
同一の方法により、ヒト成長ホルモン、G−CSFおよびその誘導体にそれぞれアルブミンが結合したhGH−PEG−アルブミン、G−CSF−PEG−アルブミンおよび17S−G−CSF−PEG−アルブミン結合体を製造した。
【0138】
(比較例3)Fab’−S−40K PEG連結体の製造
実施例8の段階1で精製されたFab’に存在する遊離システイン基を活性化させるために、活性化緩衝液(20mMの酢酸ナトリウム(pH4.0)、0.2mMのDTT)に1時間放置した。PEG修飾緩衝液である50mMのリン酸カリウム(pH6.5)で緩衝液を交換した後、マレイミド−PEG(分子量40kDa、Shearwater社)をFab’:40K PEGのモル比が1:10となるように添加した後、4℃で徐々に攪拌しながら24時間反応させた。
【0139】
反応終了後、反応液を10mMのリン酸ナトリウム緩衝液(pH7.3)で平衡化させたSuperdex200カラム(Superdex 200、Pharmacia社)に滴下し、同一の緩衝液を1mL/分の流速で流して溶出させた。40K PEGが修飾されたFab’−40K PEG連結体は、分子量が大きいので、反応していないFab’よりも早く溶出し、Fab’は後で溶出するため、除去することができた。完全に除去されていないFab’を除去するために、溶出したFab’−40K PEG連結体分画をpolyCAT 21×250カラム(PolyLC社)を用いて最終精製した。20mMのリン酸ナトリウム緩衝液(pH4.5)を用いた直線濃度勾配(塩化ナトリウムの濃度0.15M→0.5M)法で溶出し、Fab’の−SH基に40K PEGが連結されたFab’−S−40K PEG連結体を純粋に得た。
【0140】
(実験例1)蛋白質結合体の確認および定量
(1−1)蛋白質結合体の確認
前記実施例で製造した蛋白質結合体は、4〜20%の濃度勾配ゲルおよび12%のゲルを用いた非還元性SDS−PAGEおよびELISA(R&D system社)法で確認した。
【0141】
図6に示すように、SDS−PAGEの結果から、生理活性ポリペプチド、非ペプチド性重合体であるPEGおよび免疫グロブリンFc領域のカップリング反応によって、IFNα−PEG−Fc結合体(A)、17Ser−G−CSF−PEG−Fc結合体(B)およびEPO−PEG−Fc結合体(C)が生成されたことを確認した。
【0142】
また、前記実施例10で製造したDG Fcを確認するために非還元性12%SDS−PAGEを行った結果、図9bに示すように、天然型Fcに比べて除去された糖鎖分子量だけ減少した位置でDG Fcバンドが検出された。
【0143】
(1−2)蛋白質結合体の定量
前記実施例で製造したそれぞれの蛋白質結合体の量は、Superdexカラム(HiLoad 26/60 Superdex 75、Pharmacia社)と10mMのリン酸カリウム緩衝液(pH6.0)を溶出液として用いるサイズ排除クロマトグラフィでのピーク面積を対照と比較して換算する方法によって計算した。既に定量されているIFNα、hGH、G−CSF、17S−G−CSF、EPOおよびFcを用いてそれぞれサイズ排除クロマトグラフィを行った後、濃度とピーク面積間の換算係数を測定した。各蛋白質結合体の一定量を使用して同一のサイズ排除クロマトグラフィを行い、ここから得られたピーク面積から免疫グロブリンFc領域に該当するピーク面積を差し引いた値を、各蛋白質結合体に存在する生理活性蛋白質の定量値として決定した。図7は精製されたIFNα−PEG−Fc結合体のサイズ排除クロマトグラフィカラムの分析結果を示すもので、二量体以上の多量体不純物がない単一ピークを確認した。
【0144】
Fcが生理活性ポリペプチドに結合すると、生理活性ポリペプチドの抗体でその量を定量するとき、抗体と前記ポリペプチドとの結合が阻害され、クロマトグラフィによって計算される実際値より少なく定量される。ELISA分析の結果から、IFNα−PEG−Fcの場合には、ELISAによって測定された値が大略実際値の約30%程度であると確認された。
【0145】
(1−3)蛋白質結合体の純度および質量確認
それぞれの実施例で得た蛋白質結合体に対してサイズ排除クロマトグラフィを行い、280nmの吸光を測定した。その結果、IFNα−PEG−Fc、hGH−PEG−Fc、G−CSF−PEG−Fcおよび17Ser−G−CSF−PEG−Fcは、分子量70〜80kDaの物質の滞留時間帯で単一ピークを示した。
【0146】
一方、実施例5、15および16で獲得した蛋白質結合体IFNα−PEG−Fc、IFNα−PEG−DG FcおよびIFNα−PEG−組み換えAG Fc誘導体試料の純度を分析するために、逆相HPLCを行った。逆相カラム(Vydac社、259 VHP54カラム)を用いて分析し、0.5%TFAの存在下のアセトニトリル溶媒を用いた濃度勾配(40〜100%)法によって溶出した。純度は、280nmの吸収を測定することによって解析された。その結果、図11から分かるように、結合していないインターフェロンまたは免疫グロブリンFcは存在しておらず、IFNα−PEG−Fc結合体(A)、IFNα−PEG−DG Fc結合体(B)およびIFNα−PEG−組み換えAG Fc誘導体結合体(C)のいずれも96%以上の純度で純粋に精製された。
【0147】
各精製された試料の正確な分子量を確認するために、各試料の質量をMALDI−TOF(Voyager DE−STR、Applied Biosystems社)超高速質量分析器を用いて分析した。マトリックスとしてはシナピン酸(sinapinic acid)を使用し、それぞれの試験用試料0.5μLを試料スライドに塗布して自然乾燥させた後、同量のマトリックス溶液を添加し、その後さらに自然乾燥させてイオン源(ion source)に導入した。検出はポジティブ方式でリニアモードTOF方式の装置を用いて行った。イオンは、遅延したイオン抽出(DE)を用いる分割抽出供給源で、遅延した抽出時間を750nsec/1500nsecとして、約2.5kVの全体電位差によって加速化した。
【0148】
表1は前記実施例で得たそれぞれのFc蛋白質結合体のMLADI−TOF質量分析結果を数値化して示したものであり、図8はEPO−PEG−Fc結合体、図10はIFNα−PEG−FcおよびIFNα−PEG−DG Fc結合体のMALDI−TOF質量分析結果を示したものである。その結果、収得されたEPO−PEG−Fc蛋白質結合体の純度は95%以上であり、理論値と非常に近い分子量を示した。また、免疫グロブリンFc領域に赤血球生成因子(EPO)が1:1で結合した形態であると確認された。
【0149】
【表1】
【0150】
また、MALDI−TOF質量分析法により、実施例14で製造されたFcおよびDG Fcの分子量を測定した結果、DG Fcの分子量は天然型Fcより3kDa程度小さい50kDaと確認された(図9a)。減少した3kDaは理論的糖鎖サイズに該当する分子量であって、糖鎖が完全に除去されたことを確認することができた。
【0151】
下記表2は、前記実施例11で製造されたIFNα−PEG−DG Fc結合体と、実施例16で製造されたIFNα−PEG−組み換えAG Fc誘導体結合体(IおよびII)のMALDI−TOF質量分析結果を示したものである。IFNα−PEG−Fc結合体の分子量(75.9kDa)と比較したとき、IFNα−PEG−DG Fc結合体は約3kDa程度小さい分子量を示し、IFNα−PEG−組み換えAG Fc誘導体結合体(I)は約3〜4kDa程度小さい分子量を示した。Fc単量体に連結されたIFNα−PEG−組み換えAG Fc誘導体結合体(II)は、Fc単量体分子量に該当する24.5kDaが減少した分子量を示した。
【0152】
【表2】
【0153】
(実験例2)薬物動態学的な調査I
各群当たり5匹のSDラットにおいて、天然型生理活性蛋白質(対照群)と前記実施例および比較例で製造した−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、−PEG−DG Fc結合体、および−PEG−組み換えAG Fc誘導体結合体の血液内安定性および薬物動態学的係数(pharmacokinetic parameters)を比較した。対照群および−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、−PEG−DG Fc結合体、および−PEG−組み換えAG Fc誘導体結合体(試験群)を各100μg/kgずつ皮下注射した後、対照群は注射してから0.5.1、2、4、6、12、24、30、48、72および96時間後に採血し、試験群は注射してから1、6、12、24、30、48、72、96、120、240および288時間後に採血した。ヘパリンを含有するチューブに血液試料を集めて凝固を防止し、エッペンドルフ高速マイクロ遠心分離機で5分間遠心分離して細胞を除去した。血漿内の蛋白質量は、各生理活性蛋白質に対する抗体を用いてELISA方法によって測定した。
IFNα、hGH、G−CSFまたはEPOの天然型蛋白質、これらの−40K PEG連結体、−PEG−アルブミン結合体、−PEG−Fc結合体、および−PEG−DG Fc結合体の薬物動態の分析結果を下記表3〜表7に示した。下記表において、Tmaxは最高薬物濃度に到達する時間を、T1/2は薬物の血中半減期を、MRT(mean residence time)は薬物分子の平均的な体内滞留時間をそれぞれ意味する。
【0154】
【表3】
【0155】
【表4】
【0156】
【表5】
【0157】
【表6】
【0158】
【表7】
【0159】
前記表3および図12の薬物動態グラフから分かるように、IFNα−PEG−Fc蛋白質結合体の場合、血中半減期は90.4時間であって天然型に比べて約50倍増加した。これは比較例1で製造したIFNα−40K PEGの半減期である35.8時間より約2.5倍増加したものである。また、IFNα−PEG−アルブミンの半減期である17.1時間に比べても、本発明のIFNα−PEG−Fc蛋白質結合体が著しく優れた血中半減期を示すことを確認した。
【0160】
一方、表3および図14に示すように、IFNα−PEG−DG Fc結合体の場合には、血中半減期が71.0時間であって、IFNα−PEG−Fc結合体とほぼ同等なので、糖鎖がなくても生体内安定性には大きい影響がなく、組み換え方法で生産された組み換えAG Fc誘導体を用いた結合体も天然型由来のDG Fcと同一の効果を示すことを確認した。ところが、Fc単量体を使用した結合体の場合には、正常的なFc二量体の結合体と比較したとき、約半分に減少した血中半減期を示した。
【0161】
ヒト成長ホルモンの場合も、表4に示すように、本発明の蛋白質結合体が示す血中半減期の増加効果を確認することができる。すなわち、天然型(1.1時間)に比べてhGH−40K PEG連結体およびhGH−PEG−アルブミン結合体の半減期が7.7時間および5.9時間と多少増加した反面、本発明のhGH−PEG−Fc蛋白質結合体の場合には血中半減期が11.8時間と画期的に増加した。
【0162】
表5および表6における顆粒球コロニー刺激因子とその誘導体に対する薬物動態の分析結果においても、天然型、−40K PEG連結体および−PEG−アルブミン結合体に比べて本発明のG−CSF−PEG−Fcおよび17S−G−CSF−PEG−Fc結合体が一層長い血中半減期を示した。蛋白質の血中持続性を増加させる免疫グロブリンFc領域の効果は、天然型生理活性蛋白質だけでなく、一部のアミノ酸を変形させた誘導体においても天然型とほぼ同一の程度であることを確認することができ、このような結果により、本発明の方法が他の蛋白質の誘導体においても類似の効果を示すことを容易に予想することができる。
【0163】
表7および図13に示すように、糖鎖化されている天然型蛋白質としての赤血球生成因子に対しても本発明の蛋白質結合体の血中半減期増加効果が確認された。すなわち、天然型赤血球生成因子の血中半減期が9.4時間であり、EPOを高糖鎖化させて血中安定性を高めたダーベポエチン−α(Darbepoetin-α、Aranesp、Amgen社)の場合は半減期が18.4時間と増加することが分かる。赤血球生成因子に免疫グロブリンFc領域を結合させた本発明のEPO−PEG−Fc結合体の場合には、血中半減期がおおよそ61.5時間と画期的に増加し、糖鎖が除去された大腸菌由来の組み換えAG Fc誘導体を使用した結合体の場合には87.9時間まで半減期の増加を示し、糖鎖が除去されても、血中安定性には影響を及ぼすことなく、抗体機能がない結合体を製造することができることを確認した。
【0164】
前記結果から分かるように、本発明によって免疫グロブリンFc領域および非ペプチド性重合体と共有結合した蛋白質結合体は、天然型蛋白質に比べて数倍〜数十倍以上増加した血中半減期を示した。また、免疫グロブリンFcを遺伝子組み換え法によって大腸菌で生産し、あるいは酵素処理によって糖鎖を除去しても、これを用いて製造された蛋白質結合体における血中半減期の増加効果はほぼ同様に維持された。
【0165】
特に、既存の蛋白質血中持続性を増加させるためのPEG剤形のうち持続性が最も高い40kDa PEGを修飾した蛋白質との比較においても、免疫グロブリンFc蛋白質結合体が著しく高い血中安定性を示した。また、免疫グロブリンFcの代わりにアルブミンを結合させた蛋白質結合体との比較試験においても、本発明の蛋白質結合体が、卓越した血中安定性を示すことから、本発明の蛋白質結合体が持続型蛋白質薬物製剤の開発に効果的であることを確認することができた。点突然変異(point mutation)によるコロニー刺激因子誘導体まで広範囲な範囲の蛋白質において従来のPEG結合蛋白質またはアルブミン蛋白質結合体より卓越した血中安定性と平均滞留時間(MRT)を示す結果からみて、本発明の蛋白質結合体による安定性および持続性増加効果は他の生理活性ポリペプチドにも適用可能であることが分かる。
【0166】
一方、非ペプチド性重合体として10kDa PEGを使用したIFNα−10K PEG−Fc蛋白質結合体(実施例11)を用いて前記と同一の方法で血中半減期を測定したとき、血中半減期は48.8時間であって、分子量3.4kDaのPEGを使用した蛋白質結合体の血中半減期である79.7時間より多少減少した。
【0167】
また、非ペプチド性重合体PEGの分子量の増加に伴って血中半減期はむしろ多少減少したが、このような結果から、蛋白質結合体の血中安定性および持続性の増加の主な要因が非ペプチド性重合体の分子量よりは結合した免疫グロブリンFc領域による効果であることが確認できる。
【0168】
PEGの反応基をアルデヒド反応基以外の反応基に変化させた場合でも、見掛け分子量と血中半減期が、アルデヒド反応基を持つPEGを使用した場合と類似の様相を示した。
【0169】
(実験例3)薬物動態学的な調査II
実施例12および13で製造されたFab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体および比較例3で製造されたFab’−S−40K PEG連結体の血中半減期を測定するために、Fab’を対照群として前記結合体または連結体を用いて実験例2と同一の方法で薬物動態学的な調査を行い、その結果を図15に示した。
図15より、Fab’−S−PEG−N−FcおよびFab’−N−PEG−N−Fc結合体が、Fab’またはFab’−S−40K PEG連結体に比べて2〜3倍延長された血中半減期を示すことが分かる。
【0170】
(実験例4)細胞内活性の測定
(4−1)インターフェロンアルファ蛋白質結合体の細胞内活性比較
インターフェロンアルファ蛋白質結合体の細胞内活性比較のために、IFNα−PEG−Fc(実施例5)、IFNα−PEG−DG Fc(実施例15)、IFNα−PEG−組み換えAG Fc誘導体(実施例16)、IFNα−40K PEG(比較例1)およびIFNα−PEG−アルブミン(比較例2)の抗ウイルス活性を水泡性口内炎ウイルスで飽和させたMDBK(Madin Darby Bovine Kidney、ATCC CCL−22)を使用する細胞培養生検で測定した。この際、PEGが結合していないインターフェロンアルファ−2b(NIBSC国際標準品)を標準物質として使用した。
【0171】
MDBK細胞を、MEM(minimum essential medium:JBI社)に10%FBSおよび1%ペニシリン/ストレプトマイシンが添加された培地で37℃、5%CO2の条件で培養した。測定しようとする試料および標準物質を一定の濃度で細胞培養培地に希釈して96ウェルプレートの各ウェルに100μLずつ分注した。前記培養された細胞を回収し、試料が分注されているプレートに100μLずつ加えた後、37℃、5%CO2の条件で約1時間程度培養した。1時間後、ウイルスの濃度が5〜7×103PFUとなるように調節されたVSV(Vesilculer stomatitis virus)を50μLずつプレートに添加し、37℃、5%CO2の条件で約16〜20時間さらに培養した。試料および標準物質を入れないで細胞とウイルスのみを入れたウェルを陰性対照群として使用し、ウイルス希釈溶液を入れないで細胞のみを入れたウェルを陽性対照群として使用した。
【0172】
培養液を除去し、生きている細胞を染色するために、ニュートラルレッド(neutral red)溶液を100μLずつ添加した後、37℃、5%CO2の条件で2時間培養した。上澄み液を除去した後、100%のエタノールと1%の酢酸とを1:1の比率で混ぜた溶液100μLを各ウェルに入れた。染色された細胞をよく振って溶解させた後、540nmで吸光度を測定した。陰性対照群をブランク(blank)とし、陽性対照群を細胞成長100%と見なして50%細胞成長時の濃度(ED50)を計算した。
【0173】
【表8】
【0174】
その結果、図8に示すように、IFNα−40K PEGは、その活性度が天然型の4.8%水準に低下することが確認できる。特に、修飾されたPEGの大きさが大きいほど、血中安定性は増加し、相対的に活性度は徐々に減少したが、インターフェロンアルファの場合、12kDa PEGが修飾された場合には25%、40kDa PEGが修飾された場合には約7%程度の試験管内活性を持つと報告されたことがある(P. Bailon et al., Bioconjugate Chem. 12: 195-202, 2001)。すなわち、PEGの分子量が増加すると、血中半減期は長くなるが、相対的に活性は急激に減少するので、血中半減期が長いながら活性も優れた蛋白質結合体の開発が要求されている。また、IFNα−PEG−アルブミン結合体も天然型に比べて約5.2%程度と低い活性を示したが、本発明のIFNα−PEG−Fc結合体およびIFNα−PEG−DG Fc結合体は、天然型に比べて相対的活性度が28.1%および25.7%と画期的に高くなることを確認することができ、組み換えAG Fc誘導体を使用した結合体においても類似の活性増加効果を示した。このような結果から、血中半減期の画期的増加に伴ってインターフェロンアルファと免疫グロブリンFc領域の結合体の生体内薬効が非常に優れるものと期待される。
【0175】
(4−2)ヒト成長ホルモン蛋白質結合体の細胞内活性比較
ヒト成長ホルモン蛋白質結合体の細胞内活性を比較するために、hGH−PEG−Fc、hGH−40K PEGおよびhGH−PEG−アルブミンの細胞内活性を比較試験した。
【0176】
ヒト成長ホルモン依存的に分裂を行う細胞としてのラット結節リンパ腫(rat node lymphoma)細胞株であるNb2細胞(European Collection of Cell Cultures (ECACC) #97041101)を用いて細胞内活性度を試験管内の分析によって確認した。
【0177】
Nb2細胞を、培養液(Fisher’s medium)に10%の牛胎児血清(FBS、fetal bovine serum)、0.075%のNaCO3、0.05mMの2−メルカプトエタノールおよび2mMのグルタミンを添加した培地で培養した後、10%の牛胎児血清を除いた同一の培地で24時間さらに培養した。培養液で培養された細胞の数を数えて約2×104個の細胞を96ウェルプレートの各ウェルに入れた後、hGH−PEG−Fc、hGH−40K PEG、hGH−PEG−アルブミン、対照群である国際標準品(National Institute for Biological Standards and Controls、NIBSC)および天然型ヒト成長ホルモン(HM−hGH)をそれぞれ希釈して濃度別に添加した。その後、37℃、CO2の培養器で48時間培養した。その後、細胞の成長程度(各ウェルの細胞数)を測定するために、細胞染色薬(Cell Titer 96 Aqueous One Solution、Promega社)を各ウェルに25μLずつ入れた後、4時間培養した。その後、490nmの吸光度を測定して各試料の力価を計算し、その結果を下記表9に示した。
【0178】
【表9】
【0179】
表9から分かるように、ヒト成長ホルモンの場合も、hGH−40K PEGの活性度は天然型の約7.6%と低下し、hGH−PEG−アルブミン結合体の試験管内活性は天然型に比べて約5.2%程度と低い活性を示した。ところが、本発明のhGH−PEG−Fc結合体は、相対的活性度が天然型に比べて28%以上画期的に増加した。このような結果より、血中半減期の画期的増加に伴ってヒト成長ホルモンと免疫グロブリンFc領域の蛋白質結合体の生体内薬効が非常に優れるものと期待することができる。また、本発明の免疫グロブリンFc蛋白質結合体の活性増加は、免疫グロブリンFcによる血中安定性の増加および受容体との結合力保存、または非ペプチド性重合体によって形成された空間的余裕によるものと把握される。このような作用は、他の生理活性蛋白質の免疫グロブリンFc蛋白質結合体においても類似であるものと期待される。
【0180】
(4−3)顆粒球コロニー刺激因子蛋白質結合体の細胞内活性比較
顆粒球コロニー刺激因子誘導体の蛋白質結合体の細胞内活性を比較するために、天然型G−CSF(Filgrastim、第一薬品(株))、17Ser−G−CSF誘導体、20K PEG−G−CSF(Neulasta社)、40K PEG−17S−G−CSF、17Ser−G−CSF−PEG−アルブミンおよび17S−G−CSF−PEG−Fcの細胞内活性を測定した。
【0181】
まず、ヒト骨髄由来の細胞株であるHL−60(ATCC CCL−240、Promyelocytic leukemia patient/36 yr old Caucasian female)を10%の牛胎児血清を含むRPMI1640倍地で培養してから、細胞の数を約2.2×105細胞/mLとなるように調整した後、DMSO(dimethylsulfoxide、culture grade、Sigma社)を最終1.25%(v/v)となるように添加した。前記の細胞株を96ウェルプレート(Corning/low evaporation 96 well plate)に90μLずつ入れてウェル当たり細胞が約2×104個となるようにした後、5%のCO2が供給される37℃の培養器で約72時間培養した。
【0182】
G−CSF ELISAキット(R&D systems社)を用いて、濃度が決定された各試料を同一の濃度となるようにRPMI1640で希釈し、最終濃度が10μg/mLとなるようにし、これをさらにRPMI1640で1/2に希釈することを19回繰り返し行った。こうして作られた試料を培養中のHL−60細胞株の各ウェルに10μLずつ加え、最終濃度が1μg/mLから連続的に半減するようにした。前記で製造した蛋白質試料を処理した細胞株は、37℃の培養器で72時間、さらに培養した。
【0183】
培養後、細胞株の増加程度を調べるために、CellTiter96TM(Cat.No.G4100、Promega社)を用いて分析し、増加した細胞株の数は、670nm波長における吸光度を測定することによって決定した。
【0184】
【表10】
【0185】
その結果、表10から分かるように、G−CSFのアミノ酸を置換させた17Ser−G−CSF誘導体の免疫グロブリンFc蛋白質結合体も天然型蛋白質の蛋白質結合体と類似の効果を示した。17Ser−G−CSF−PEGの場合には、PEGで修飾されていないものに比べて、血中半減期は増加するが活性度は低下すると既に報告されたことがある(大韓民国特許公開第2004−83268号)。特に、修飾されたPEGの大きさが大きいほど、血中安定性は増加するが、相対的に活性度は徐々に低下することが分かり、17Ser−G−CSF−40K PEGは天然型に比べて約10%以下の非常に低い活性度を示した。すなわち、PEGの分子量が増加すると、血中半減期は長くなるが相対的に活性は急激に減少するので、血中半減期が長くて活性も優れた蛋白質結合体の開発が要求されている。17Ser−G−CSF−PEG−アルブミンも天然型に比べて約23%程度と低い活性を示したが、17Ser−G−CSF−PEG−Fcは天然型に比べて相対的活性度が51%以上と高くなることを確認することができた。これにより、血中半減期の画期的増加に伴って免疫グロブリンFc領域と17Ser−G−CSF誘導体の結合体の生体内薬効が非常に優れるものと期待することができる。
【0186】
(4−4)Fab’結合体の細胞毒性中和試験
実施例8および9で製造されたFab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体、および比較例3で製造されたFab’−S−40K PEG連結体の試験管内活性実験を行った。マウス繊維亜細胞株L929(ATCC CRL−2148)を用いてTNFαの細胞毒性を測定する実験を基本骨格として、Fab’がTNFαの細胞壊死活性をどれくらい中和させるかを測定した。
【0187】
Fab’−S−PEG−N−Fc、Fab’−N−PEG−N−Fc結合体およびFab’−S−40K PEG連結体をそれぞれ2倍ずつ順次希釈して96ウェルプレートの各ウェルに100μLずつ分注した後、rhTNF−α(R&D systems社)およびRNA合成の阻害剤として用いられるアクチノマイシンD(actinomycin D、Sigma社)をそれぞれ最終濃度が10ng/mLおよび1μg/mLとなるように添加し、37℃、5%CO2で30分間反応させた後、分析用マイクロプレートに移した。プレートの各ウェルにL929細胞株を5×104個/50μL培地となるように添加し、37℃、5%CO2の培養器で24時間培養した。マイクロプレート内の培養液を除去した後、PBSに5mg/mLの濃度で溶けているMTT(Sigma社)を50μLずつ入れた。約4時間37℃、5%CO2の培養器で培養した。150μLのDMSOを添加した後、540nmの吸光度を測定して細胞毒性中和の度合いを確認した。対照群としては、実施例8の段階1で精製したFab’を使用した。
【0188】
その結果、図16に示すように、実験に使用した全ての蛋白質結合体は、Fab’と類似の力価を示しており、このような結果から、Fab’のN末端またはC末端近くの遊離システイン残基にPEGを介して免疫グロブリンFcを結合させた蛋白質結合体は、Fab’の血中半減期を画期的に増加させることは勿論のこと、生体内活性度も高く維持させることが分かる。
【0189】
(4−5)CDC活性の測定
前記実施例で製造した誘導体と大腸菌形質転換体から発現されて精製された免疫グロブリン不変領域蛋白質とがヒトClqに結合するか否かを確認するために、下記のように固相酵素免疫検定法(ELISA)を行った。実験群として、形質転換体HM10932およびHM10927から生産された免疫グロブリン不変領域試料と前記実施例で製造した誘導体とを使用し、比較群として、糖が結合している免疫グロブリン(IVIG−グロブリンS、緑十字PBM)を始めとして、商業化されて治療用抗体として使われているいろいろの抗体を使用した。前記実験群と比較群の試料を10mMのカーボネート緩衝液(pH9.6)に1μg/mLの濃度で調製した。準備された試料を96ウェルプレート(Nunc)にウェル当たり200ngの量で分注した後、4℃で一晩中コートし、その後ウェルプレートをPBS−T溶液(137mMのNaCl、2mMのKCl、10mMのNa2HPO4、2mMのKH2PO4、0.05%のツイン20)で3回洗浄した。牛血清アルブミンを1%の濃度でPBS−T溶液に溶解させて準備したブロッキングバッファ250μLを各ウェルに添加した後、常温で1時間放置し、同一のPBS−T溶液で3回洗浄した。標準液と試料を適切な濃度でPBS−T溶液によって希釈した後、抗体がコートされたウェルに加えて常温で1時間放置させて反応させ、その後さらにPBS−T溶液で3回洗浄した。ブロッキング反応が完了したプレートに2μg/mLのClq(R&D systems社、米国)を添加した後、2時間常温で反応させ、反応済みのプレートを前記PBS−T溶液で6回洗浄した。ヒトの抗ヒトClq抗体とペルオキシダーゼとの結合体(Biogenesis社、米国)をブロッキングバッファに1000:1に希釈して各ウェルに200μLずつ加えた後、1時間常温で反応させた。反応が完了した後、各ウェルをPBS−T溶液で3回洗浄した後、発色溶液AとB(カラーA−安定化ペルオキシダーゼ[Color A-Stabilized peroxide]溶液およびカラーB−安定化クロモゲン[Color B-stabilized chromogen]溶液、DY999、R&D systems社)を同量で混合し、当該混合液を各ウェルに200μLずつ添加し、30分間放置した。その後、反応停止溶液である2Mの硫酸を50μLずつ添加して反応を停止させた。反応済みのウェルプレートは、マイクロプレートリーダー(Molecular Device社)を用い、標準液と検液の450nmの波長の吸光度を測定し、その結果を図17、図18にそれぞれ示した。
【0190】
免疫グロブリンのFc領域における補体活性をサブクラスによって比較した結果、ヒト免疫グロブリンIgG1(Fitzgerald)、IgG2(Fitzgerald)、IgG4(Fitzgerald)の順にClqに対する高い結合力を持っていることが確認でき、これにより補体活性もサブクラス間に差異があることが分かった。前記実験に使用したIVIGの場合、IgGサブクラスの集合体であるが、IgG1がほぼ大部分を占めているので、分離精製されたIgG1とほぼ類似の親和度を示した。このような対照群と比較するとき、非糖鎖化によるClqの親和度の変化は、補体活性が最も強いIgG1 Fcで著しく減少することが分かり、既に補体反応がないものと知られているIgG4 FcではClqに対する結合力が殆どないので、補体活性のない優れた組み換えキャリアであることが分かった(図17)。
【0191】
Clq親和度が除去されたキャリアの特性が生理活性ペプチドと結合体を作った後にも依然として維持されるか否かを調べるために、IFNαをモデルとして、糖鎖化Fc、酵素を用いた脱糖鎖Fcおよび非糖鎖化組み換えFcをキャリアとして用いて各結合体を作った後、Clqに対する結合力を検討した。糖鎖化FcとIFNα結合体(IFNα−PEG−Fc:Glycosylated IgG1Fc)は、Clqに対して依然として高い親和度を維持するが、ここにPNGaseFなどを用いて脱糖鎖化させたインターフェロン結合体(IFNα−PEG−DGFc:Deglycosylated IgG1Fc)は、結合力が著しく低くなることを確認し、その程度は大腸菌由来の非糖鎖化Fc結合体と同様の水準のClqに対する結合力を示した。それだけでなく、IgG1の非糖鎖化Fcを用いたインターフェロン結合体(IFNα−PEG−AGFcG1:Aglycosylated IgG1Fc)をIgG1からIgG4に変えたインターフェロン結合体(IFNα−PEG−FcG4誘導体1:Aglycosylated IgG4Fc)の場合、Clqに対する親和度が完全に除去されることを確認することができ、結合体を非糖鎖化IgG4 Fc単量体に変えた結合体(IFNα−PEG−FcG4誘導体2:Aglycosylated IgG4Fc)の場合にも、やはりClqに対する結合能力は完全に除去されることからみて、抗体断片のエフェクター(effector)機能がない良いキャリアであることを確認することができた(図18)。
【0192】
本発明は、以下の態様でも提供され得る:
[1]非ペプチド性リンカーを使用して薬物との結合体を形成するための薬物キャリアとして、薬物とは独立的に組換え的な方法によって製造されたものである、IgG Fc断片。
[2]前記IgGがIgG2またはIgG4である1に記載のFc断片。
[3]前記IgGがIgG4である2に記載のFc断片。
[4]非糖鎖化された1に記載のFc断片。
[5]非糖鎖化されたIgG4 Fc断片である4に記載のFc断片。
[6]ヒト由来の非糖鎖化されたIgG4Fc断片である5に記載のFc断片。
[7]配列番号8、10または23のアミノ酸配列を持つ1に記載のFc断片。
[8]1のFc断片をコードする遺伝子。
[9]配列番号4、9または22である8に記載の遺伝子。
[10]9の遺伝子の中から選択される配列を持つ組み換えベクター。
[11]10の組み換えベクターで形質転換された形質転換体。
[12]11の形質転換された組み換え微生物を培養してFc蛋白質を収得する方法。
[13]1のFc断片をキャリアとして含む薬剤学的組成物。
【産業上の利用可能性】
【0193】
本発明のIgG Fc断片は、キャリアとして用いられる場合、薬物の血中半減期を増加させ、薬物の生体内活性を維持させる。特にポリペプチド薬物の場合、ポリペプチド薬物の血中半減期の増加が、従来報告されていたいずれの修飾蛋白質よりも高く、既存の持続型製剤(long-acting formulation)の最も大きい欠点である力価の減少を克服することにより、従来最も効果が良いものと知られているアルブミンを用いた場合よりも著しい血中持続性と生体内活性を有する。しかも免疫反応誘発の危険が殆どないため、蛋白質薬物の持続型製剤の開発に有用に利用できる。また、本発明に係る蛋白質薬物の持続型製剤は、頻繁な注射による患者の苦痛を減少させることができ、活性ポリペプチドの血中濃度を持続的に維持して薬効を安定的に示すことができる。
【特許請求の範囲】
【請求項1】
非ペプチド性リンカーによってポリペプチド薬物との結合体を製造するためのキャリアとしての、IgG Fc断片の使用(たたし、該結合体は、組換え的な方法によって製造された融合蛋白質でない。)。
【請求項2】
前記IgG Fc断片が非糖鎖化されたものである請求項1に記載の使用。
【請求項3】
前記IgG Fc断片がCH1、CH2、CH3及びCH4ドメインよりなる群から選択される1〜4個のドメインから構成される請求項1に記載の使用。
【請求項4】
前記IgG Fc断片がヒンジ領域をさらに含む請求項3に記載の使用。
【請求項5】
前記IgG Fc断片がIgG1、IgG2、IgG3、IgG4、これらの組み合わせ及びこれらのハイブリッドのFc断片よりなる群から選択される請求項1に記載の使用。
【請求項6】
前記IgG Fc断片がIgG4 Fc断片である請求項5に記載の使用。
【請求項7】
前記IgG Fc断片がヒト非糖鎖化IgG4 Fc断片である請求項6に記載の使用。
【請求項8】
前記IgG Fc断片が配列番号8、10または23のアミノ酸配列を有するFc断片である請求項1に記載の使用。
【請求項9】
前記IgG Fc断片が配列番号4、9または22の塩基配列によってコードされる請求項1に記載の使用。
【請求項10】
前記IgG Fc断片が原核細胞宿主を使用して製造した組換え型IgG Fc断片であり、前記ポリペプチド薬物が真核細胞宿主を使用して製造した組換え型ポリペプチド薬物である請求項1に記載の使用。
【請求項11】
前記IgG Fc断片が非糖鎖化されたものであり、前記ポリペプチド薬物が糖鎖化されたものである請求項1に記載の使用。
【請求項12】
前記IgG Fc断片がポリペプチド薬物に対して1:1〜1:10のモル比で使用される請求項1に記載の使用。
【請求項13】
前記非ペプチド性リンカーがポリ(エチレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールの共重合体、ポリオキシエチル化ポリオール、ポリビニルアルコール、多糖類、デキストラン、ポリビニルエーテル、生分解性高分子、脂質重合体、キチン及びヒアルロン酸よりなる群から選択される請求項1に記載の使用。
【請求項1】
非ペプチド性リンカーによってポリペプチド薬物との結合体を製造するためのキャリアとしての、IgG Fc断片の使用(たたし、該結合体は、組換え的な方法によって製造された融合蛋白質でない。)。
【請求項2】
前記IgG Fc断片が非糖鎖化されたものである請求項1に記載の使用。
【請求項3】
前記IgG Fc断片がCH1、CH2、CH3及びCH4ドメインよりなる群から選択される1〜4個のドメインから構成される請求項1に記載の使用。
【請求項4】
前記IgG Fc断片がヒンジ領域をさらに含む請求項3に記載の使用。
【請求項5】
前記IgG Fc断片がIgG1、IgG2、IgG3、IgG4、これらの組み合わせ及びこれらのハイブリッドのFc断片よりなる群から選択される請求項1に記載の使用。
【請求項6】
前記IgG Fc断片がIgG4 Fc断片である請求項5に記載の使用。
【請求項7】
前記IgG Fc断片がヒト非糖鎖化IgG4 Fc断片である請求項6に記載の使用。
【請求項8】
前記IgG Fc断片が配列番号8、10または23のアミノ酸配列を有するFc断片である請求項1に記載の使用。
【請求項9】
前記IgG Fc断片が配列番号4、9または22の塩基配列によってコードされる請求項1に記載の使用。
【請求項10】
前記IgG Fc断片が原核細胞宿主を使用して製造した組換え型IgG Fc断片であり、前記ポリペプチド薬物が真核細胞宿主を使用して製造した組換え型ポリペプチド薬物である請求項1に記載の使用。
【請求項11】
前記IgG Fc断片が非糖鎖化されたものであり、前記ポリペプチド薬物が糖鎖化されたものである請求項1に記載の使用。
【請求項12】
前記IgG Fc断片がポリペプチド薬物に対して1:1〜1:10のモル比で使用される請求項1に記載の使用。
【請求項13】
前記非ペプチド性リンカーがポリ(エチレングリコール)、ポリ(プロピレングリコール)、エチレングリコールとプロピレングリコールの共重合体、ポリオキシエチル化ポリオール、ポリビニルアルコール、多糖類、デキストラン、ポリビニルエーテル、生分解性高分子、脂質重合体、キチン及びヒアルロン酸よりなる群から選択される請求項1に記載の使用。
【図1】

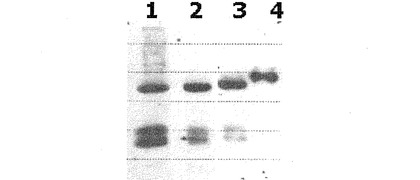
【図2】
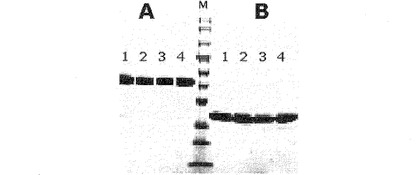
【図3】
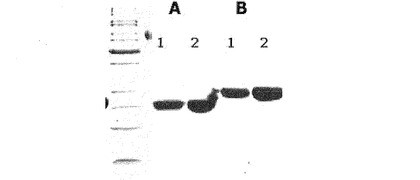
【図4】
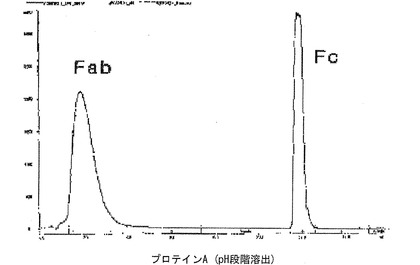
【図5】
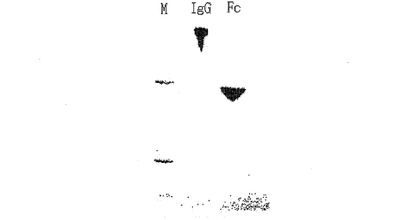
【図6】
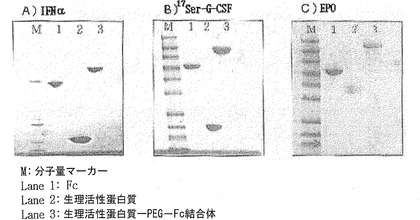
【図7】
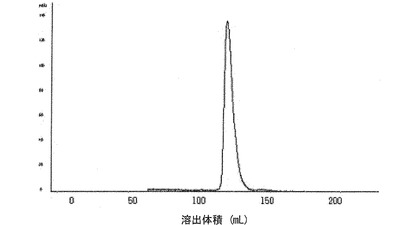
【図8】
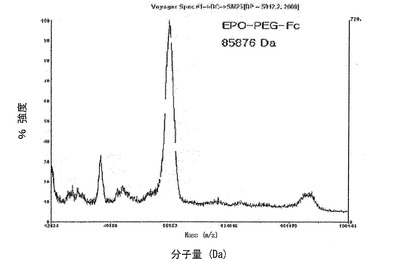
【図9a】
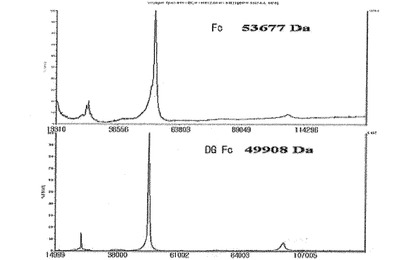
【図9b】
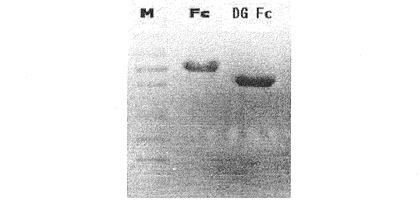
【図10】
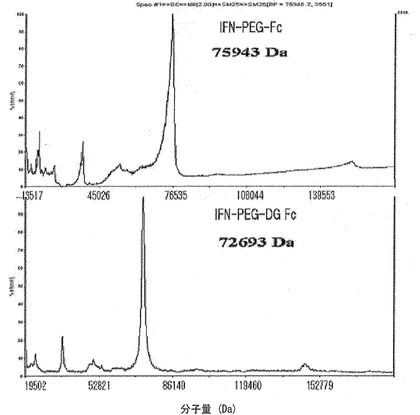
【図11】
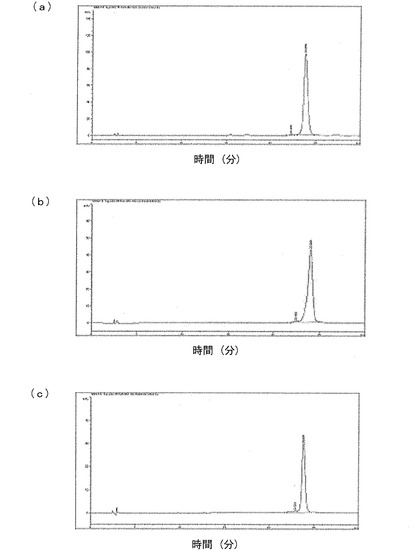
【図12】
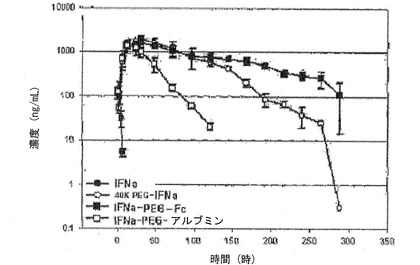
【図13】
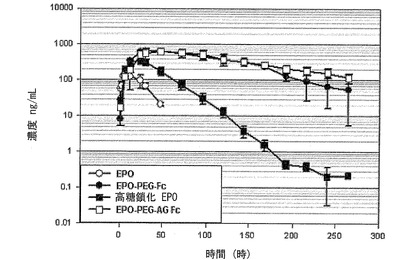
【図14】
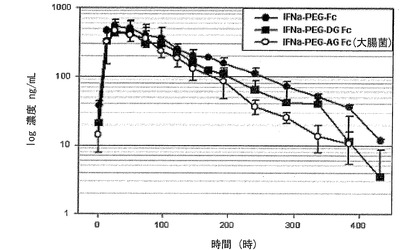
【図15】
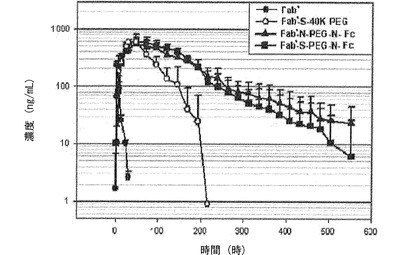
【図16】
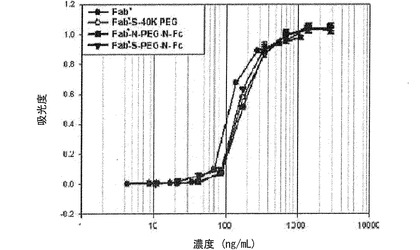
【図17】
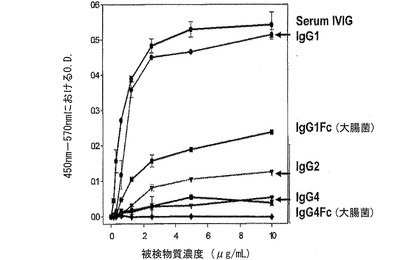
【図18】
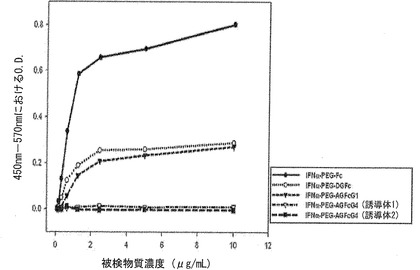
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9a】
【図9b】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2012−1560(P2012−1560A)
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願番号】特願2011−202199(P2011−202199)
【出願日】平成23年9月15日(2011.9.15)
【分割の表示】特願2006−539396(P2006−539396)の分割
【原出願日】平成16年11月13日(2004.11.13)
【出願人】(507293549)ハンミ ホールディングス カンパニー リミテッド (8)
【Fターム(参考)】
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願日】平成23年9月15日(2011.9.15)
【分割の表示】特願2006−539396(P2006−539396)の分割
【原出願日】平成16年11月13日(2004.11.13)
【出願人】(507293549)ハンミ ホールディングス カンパニー リミテッド (8)
【Fターム(参考)】
[ Back to top ]