
薬物投与のための組成物
本発明は、対象における治療薬の生物学的利用能を高めるための組成物および方法、ならびに片頭痛緩和をもたらすための組成物および方法を提供する。本組成物は、少なくとも1つのアルキルグリコシド、および5-HT受容体アゴニストなどの少なくとも1つの治療薬を含み、ここでアルキルグリコシドのアルキル鎖の長さは炭素原子約10〜約16個である。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は全体的には、強化された生物学的利用能をもたらす非刺激性で無毒性の組成物に関し、より具体的には、片頭痛緩和をもたらすための方法および組成物を含む、対象に対する5-HT受容体アゴニストなどの治療薬の送達のためのアルキルグリコシド組成物またはサッカリドアルキルエステル組成物に関する。
【背景技術】
【0002】
背景情報
治療薬は多くの場合、さまざまな表面活性物質(surfactant)と組み合わされる。しかしながら、表面活性物質は往々にして、皮膚、および鼻、口、眼、膣、直腸、食道、腸管などに認められるもののような粘膜を含む他の組織に対して刺激性がある。また、多くの表面活性物質はタンパク質を変性させ、それ故にそれらの生物活性も消失させる。そのような薬剤の開発および使用の上でのもう1つの重大な制約は、それらを作用部位に対して安全に、非侵襲性に、効率的におよび安定して送達させる能力である。したがって、理想的な強化性(enhancing)表面活性物質は、治療薬を安定化し、無毒性で皮膚または粘膜の表面に対して非刺激性であり、抗菌活性を有し、さまざまな膜障壁を通しての治療薬の通過または吸収を膜の構造的完全性および生物学的機能を損なうことなく強化し、かつ、その薬剤の生物学的利用能を高めると考えられる。
【0003】
迅速崩壊性またはいわゆる「急速分散性」の剤形を製造するための製剤化アプローチはこれまでに数多く記載されている。口腔内で崩壊すると薬物物質は嚥下され、その結果、前胃吸収および最終的には胃吸収が起こる。「前胃吸収」という用語は、胃の前の消化管の部分への有効成分の吸収を指すために一般的に用いられ、これには頬側吸収、舌下吸収、口咽頭吸収および食道吸収が含まれる。「胃吸収」という用語は、胃および腸における有効成分の吸収のことを指して一般的に用いられる。薬物が消化管の前胃部分を通過する際に、さまざまな量の薬物が吸収される可能性がある。しかし、薬物の大部分は胃内に移行し、錠剤、カプセル剤または液剤などの腸溶性剤形が吸収される通常の胃吸収様式で吸収される。薬物が腸から吸収されると、薬物は肝臓内に直接運ばれ、そこで、その固有の化学構造に応じて、肝細胞における通常の解毒プロセスを遂行する酵素によって代謝されるかまたは排出される。この排出は、肝臓における「初回通過」代謝または「初回通過」効果と呼ばれる。ほとんどの場合は元の薬物と比較して実質的または完全に不活性である、その結果生じた代謝産物は、多くの場合は血流中を循環する状態で認められ、その後に尿中および/または便中に排出される。迅速崩壊性または迅速分散性の剤形を製造するための製剤化アプローチは、米国特許出願第2006/0134194号(特許文献1)に提示されており、これは参照により本明細書に組み入れられる。
【0004】
以前に記載された急速分散性剤形は、有効成分の前胃吸収または胃吸収を促進させるために、口内に置かれた場合に崩壊または溶解する剤形を提供しているが、本発明の急速分散性剤形は、薬物作用の開始を早めること、および初回通過効果薬物代謝を減少させることといった改善された特性を提供する。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願第2006/0134194号
【発明の概要】
【0006】
本発明は、一部には、薬物の吸収および生物学的利用能を高め、それと同時に薬物のさまざまな有害毒性作用を避けるために有用な薬物強化剤(drug enhancing agent)を含む治療用組成物の開発に基づく。特に、本発明の薬物強化剤は、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルからなる無毒性の表面活性物質を含む。本発明の治療用組成物の1つの利点は、それらが高い生物学的利用能での治療薬の投与および送達を、強化剤のいわゆる「最大無毒性レベル」(それらのNOAEL)よりも著しく低い濃度で、可能にすることである。したがって、本発明は、アルキルグリコシドおよび/またはサッカリドアルキルエステルならびに治療薬(例えば、小分子有機薬物分子、Exenatide、GLP-1などの低分子量ペプチド、タンパク質、ならびに低分子量ヘパリンおよび阻害性RNAなどの非ペプチド性治療用ポリマー)を含む組成物、例えば経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)または脳脊髄液(CSF)送達経路などを介した組成物の投与および使用の方法、ならびにそのような組成物の投与によって対象における疾病状態を改善する方法を提供する。
【0007】
1つの局面において、本発明は、少なくとも1つのアルキルグリコシドおよび/または少なくとも1つのサッカリドアルキルエステルを有する表面活性物質組成物であって、治療薬、薬物または生物活性化合物と混和、混合または配合された場合に、表面活性物質がその薬物の生物活性を安定化し、かつその生物学的利用能を高めるような表面活性物質組成物に関する。
【0008】
したがって、1つの局面において、本発明は、少なくとも1つの生物活性化合物および少なくとも1つの表面活性物質を有する治療用組成物であって、表面活性物質がさらに、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルもしくはスクロースエステルからなり、治療用組成物が生物活性化合物を少なくとも約6カ月またはそれ以上にわたり、約4℃から約25℃までの範囲で安定化する治療用組成物を提供する。
【0009】
本発明はまた、少なくとも1つの治療薬または薬物または生物活性化合物と混和、混合または配合されて、対象に投与または送達される、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む表面活性物質を有する治療用組成物を投与する方法であって、アルキルが約10〜24個、10〜20個、10〜16個または10〜14個の炭素原子を有し、表面活性物質が治療薬の安定性および生物学的利用能を高めるような方法も提供する。
【0010】
さらにもう1つの態様において、本発明は、対象の循環器系への低分子量化合物の吸収を、適した表面活性物質の吸収増加量(absorption increasing amount)と化合物を混和、混合または配合した場合に経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法であって、表面活性物質が親水性サッカリドとの結合によって連結された無毒性および非イオン性の疎水性アルキルであるような方法を提供する。そのような低分子量化合物には、ニコチン、インターフェロン、PYY、GLP-1、合成エキセンディン-4、副甲状腺ホルモン、ヒト成長ホルモンまたは小有機分子が非限定的に含まれる。そのほかの低分子量化合物には、アンチセンスオリゴヌクレオチドまたは干渉性RNA分子(例えば、siRNAまたはRNAi)が含まれる。
【0011】
本発明はまた、糖尿病を治療する方法であって、それを必要とする対象に対して、治療用組成物、例えばインクレチン模倣薬またはその機能的等価物の血糖低下量、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入もしくは肺、または口腔(舌下または頬粘膜細胞)を介して投与し、それにより、対象においてインクレチン模倣薬またはインスリンの吸収を増加させ、血糖のレベルを低下させて、糖尿病を治療する段階を含む方法も提供する。
【0012】
本発明はまた、対象におけるうっ血性心不全を治療する方法であって、それを必要とする対象に対して、GLP-1ペプチドまたはその機能的等価物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の治療的有効量を、経口、眼、鼻、鼻涙管または吸入送達経路を介して投与し、それにより、対象を治療する段階を含む方法も提供する。
【0013】
もう1つの局面において、本発明は、対象における肥満または肥満に伴う糖尿病を治療する方法であって、それを必要とする対象に対して、PYYペプチドまたはその機能的等価物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の治療的有効量を、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して投与し、それにより、対象を治療する方法を提供する。
【0014】
もう1つの局面において、本発明は、対象の循環器系への低分子量治療用化合物の吸収を、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して投与することによって増加させる方法であって、化合物が約1〜30kDであり、ただし投与の経路が経口、眼、鼻または鼻涙管である場合にはその化合物がインスリン、カルシトニンまたはグルカゴンでないことを条件とする方法を提供する。
【0015】
本発明はまた、対象の循環器系への低分子量治療用化合物の吸収を、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法であって、化合物が約1〜30キロダルトン(kD)であり、ただし送達が経口、眼、鼻または鼻涙管経路を介する場合には対象が糖尿病を有しないことを条件とする方法も提供する。
【0016】
本発明の1つの局面においては、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、Exenatide(エキセンディン-4)の治療的有効量との組み合わせで有する薬学的組成物が提供される。
【0017】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、GLP-1の治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0018】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、ニコチンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0019】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、インターフェロンの治療的有効量との組み合わせで含む薬学的組成物を提供する。
【0020】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、PYYの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0021】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、副甲状腺ホルモンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0022】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、約1〜75kDの分子量を有するペプチドの治療的有効量との組み合わせで有する薬学的組成物であって、ただし、ペプチドがインスリン、カルシトニンおよびグルカゴンでないことを条件とする薬学的組成物を提供する。
【0023】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、エリスロポエチンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0024】
1つの局面において、本発明は、オリゴヌクレオチドの治療的有効量を、アルキルグリコシドの吸収増加量との組み合わせで有する薬学的組成物を提供する。オリゴヌクレオチドは、アンチセンスオリゴヌクレオチド、またはsiRNAもしくはRNAiなどの干渉性RNA分子でありうる。オリゴヌクレオチドは典型的には、約1〜20kDの分子量を有し、約1〜100、1〜50、1〜30、1〜25または15〜25ヌクレオチド長である。もう1つの局面において、オリゴヌクレオチドは約5〜10kDの分子量を有する。1つの局面において、アルキルグリコシドはテトラデシル-β-D-マルトシドである。
【0025】
さらにもう1つの態様において、本発明は、対象における低分子量オリゴヌクレオチドの生物学的利用能を、その化合物をアルキルグリコシドの吸収増加量とともに投与し、それにより、対象における化合物の生物学的利用能を高めることによって高める方法を提供する。1つの局面において、アルキルグリコシドはテトラデシルβ-D-マルトシドである。
【0026】
1つの局面において、本発明は、化合物および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量が鼻腔内に投与された対象のCSF中への化合物の吸収を増加させる方法を提供する。
【0027】
さらにもう1つの態様において、本発明は、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、以下のものから選択される粘膜送達強化剤との組み合わせで有する薬学的組成物を提供する:
(a)凝集阻害剤;
(b)電荷修飾剤;
(c)pH制御剤;
(d)分解酵素阻害剤;
(e)粘液溶解剤または粘液除去剤;
(f)繊毛静止剤;
(g)以下から選択される膜透過強化剤:
(i)表面活性物質;(ii)胆汁酸塩;(ii)リン脂質添加物、混合ミセル、リポソームまたは担体;(iii)アルコール;(iv)エナミン;(v)NOドナー化合物;(vi)長鎖両親媒性分子;(vii)小型の疎水性透過強化物質;(viii)ナトリウムまたはサリチル酸誘導体;(ix)アセト酢酸のグリセロールエステル;(x)シクロデキストリンまたはβ-シクロデキストリン誘導体;(xi)中鎖脂肪酸;(xii)キレート剤;(xiii)アミノ酸またはその塩;(xiv)N-アセチルアミノ酸またはその塩;(xv)選択された膜成分に対する分解作用のある酵素;(ix)脂肪酸合成の阻害薬;(x)コレステロール合成の阻害薬;および(xi)(i)〜(x)に列挙された膜透過強化剤の任意の組み合わせ;
(h)上皮結合生理機能の調節剤;
(i)血管拡張薬;
(j)選択的輸送強化剤;ならびに
(k)化合物と有効に組み合わされ、会合し、含有され、封入され、または結合し、その結果、鼻粘膜送達の強化のための化合物の安定化をもたらす、安定化性の送達媒体、担体、粘膜付着剤(mucoadhesive)、支持体または複合体形成種であって、化合物と鼻腔内送達強化剤との製剤化が対象の血漿における化合物の生物学的利用能の増大をもたらすもの。
【0028】
1つの局面において、本発明は、対象の循環器系への低分子量化合物の吸収を、(a)化合物;(b)親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量;および(c)粘膜送達強化剤を、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法を提供する。
【0029】
1つの局面において、本発明は、エキセンディン-4または関連GLP-1ペプチドの治療的有効量を、Intravailアルキルサッカリドの有効量とともに有する組成物を投与することによって、カロリー摂取を制御する方法を提供する。
【0030】
もう1つの局面において、本発明は、エキセンディン-4または関連GLP-1ペプチドの治療的有効量を、Intravailアルキルサッカリドの有効量とともに含む組成物を対象に投与することによって、対象における血糖レベルを制御する方法を提供する。
【0031】
さらに、もう1つの局面において、本発明は、以下のものを含む、制御放出投薬組成物を提供する:
(a)以下のものを含むコア:
(i)少なくとも1つの治療薬または薬物;
(ii)少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル;ならびに
(b)コアを取り囲む少なくとも1つの膜コーティングであって、コーティングが非透過性、透過性、半透過性または多孔性であり、胃腸管の内容物との持続的接触によってより透過性になる膜コーティング。
【0032】
もう1つの態様において、本発明は、アルキル鎖の長さが炭素原子約12〜約14個である少なくとも1つのアルキルグリコシド(alkyglycoside)、抗菌活性を有する少なくとも1つのサッカリド、および少なくとも1つの治療薬、の治療的有効量を投与することにより、アルキルグリコシド組成物を投与する方法を提供する。
【0033】
さらにもう1つの態様において、本発明は、インスリン、PYY、エキセンディン-4または他のGLP-1関連ペプチド、ヒト成長ホルモン、カルシトニン、副甲状腺ホルモン、PTH 1-34などの短縮型副甲状腺ホルモンペプチド、EPO、インターフェロンα、インターフェロンβ、インターフェロンγおよびGCSFからなる群より選択される少なくとも1つの薬物、ならびに抗菌活性を有する少なくとも1つのアルキルサッカリドを有する組成物を提供する。
【0034】
1つの局面において、本発明は、n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドまたはn-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む抗菌性アルキルサッカリド組成物を提供する。
【0035】
さらに、もう1つの局面において、本発明は、少なくとも1つの薬物および少なくとも1つの抗菌薬を約0.05%〜約0.5%の濃度で有する、経粘膜的または経皮的投与のための水性薬物組成物を提供する。
【0036】
もう1つの局面において、本発明は、マトリックス材料およびアルキルサッカリドを含む急速分散性薬物製剤を提供する。本製剤は、アルキルサッカリドを含まない同等の製剤について観察されるものよりも実質的に短いTmaxおよび実質的に小さい初回通過効果を有しうる。1つの態様において、本製剤は約0.1%〜10%のアルキルサッカリドを含んでよく、6時間よりも短いTmaxおよび40%よりも小さい初回通過効果を示す。アルキルグリコシドは任意の適したアルキルグリコシドであってよく、1つの好ましい局面において、ドデシルマルトシド、テトラデシルマルトシド、ドデカン酸スクロース、またはモノ-およびジ-ステアリン酸スクロースである。本製剤は、メラトニン、ラロキシフェン、オランザピン(olanzapene)およびジフェンヒドラミンなどの、ただしそれらには限定されない多種多様な治療薬を含みうる。
【0037】
もう1つの局面において、本発明は、胃送達と頬側送達とのバランスをとるために、薬物製剤中のアルキルサッカリド濃度を薄めることにより、長期間にわたる吸収曲線を提供するための方法を提供する。例えば、これは、マトリックス材料、ならびにアルキルサッカリドを含まない同等の製剤について観察されるものよりも実質的に短いTmaxおよび実質的に小さい初回通過効果を有するアルキルサッカリドを含む薬物製剤を提供することによって行われる。
【0038】
1つの局面において、本発明は、ビスホスホネート類似体またはトリプタン類似体の治療的有効量を、アルキルグリコシドの吸収増加量との組み合わせで有する薬学的組成物を提供する。さまざまな態様において、ビスホスホネート類似体は、エチドロネート、クロドロネート、チルドロネート、パミドロネート、ネリドロネート、オルパドロネート、アレンドロネート、イバンドロネート、リセドロネート、ゾレドロネート、および/またはそれらの薬学的に許容される類似体であってよい。1つの例示的な態様において、ビスホスホネート類似体はアレンドロネート、またはその薬学的に許容される類似体である。さまざまな態様において、トリプタン類似体は、スマトリプタン、リザトリプタン、ナラトリプタン、ゾルミトリプタン、エレトリプタン、アルモトリプタン、フロバトリプタン、および/またはそれらの薬学的に許容される類似体であってよい。1つの例示的な態様において、トリプタン類似体はスマトリプタンまたはその薬学的に許容される類似体である。さまざまな態様において、アルキルグリコシドはテトラデシル-β-D-マルトシドである。
【0039】
さらにもう1つの態様において、本発明は、対象におけるビスホスホネート類似体またはトリプタン類似体の生物学的利用能を、その化合物をアルキルグリコシドの吸収増加量とともに投与し、それにより、対象における化合物の生物学的利用能を高めることによって高める方法を提供する。
【0040】
さらにもう1つの態様において、本発明は、5-HT受容体アゴニストおよびアルキルサッカリドの治療的有効量を含む組成物を提供する。さまざまな態様において、5-HTアゴニストは、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである。
【0041】
さらにもう1つの態様において、本発明は、対象に対して、5-HTアゴニストの減量されているが治療的には有効な量を提供する方法を提供する。本方法は、5-HTアゴニストの治療的有効量;およびアルキルサッカリドを含む組成物である鼻腔内組成物を投与する段階を含み、ここでそのAUCは、アルキルサッカリドの非存在下で投与された5-HTアゴニストの増量された治療的有効量によってもたらされるAUCと比較してほぼ等しい。
【0042】
さらにもう1つの態様において、本発明は、対象における片頭痛緩和の迅速開始を提供する方法であって、5-HTアゴニストおよびアルキルサッカリドの治療的有効量を含む組成物であって対象において約30分またはそれ未満のTmaxを呈する組成物を投与し、片頭痛緩和の迅速開始を提供する段階を含む方法を提供する。
【0043】
さらにもう1つの態様において、本発明は、対象における片頭痛再発の発生率の低下を提供する方法であって、5-HTアゴニストおよびアルキルサッカリドの治療的有効量を含み、約20分未満のTmaxを提供する組成物である組成物を投与し、それにより、対象における片頭痛再発の発生率の低下を提供する段階を含む方法を提供する。
【図面の簡単な説明】
【0044】
【図1】アルキルグリコシドの存在下および非存在下でのMIACALCIN(登録商標)(サケカルシトニン)に関する、静脈内注射と比較した鼻腔内生物学的利用率および対象間変動係数を示しているグラフである。
【図2】血糖レベルを低下させる上での、インスリン/0.25% TDMの鼻腔内投与(●)およびインスリンのみの鼻腔内投与(○)の効果を示しているグラフである。
【図3】グルコースの腹腔内(IP)注射後の血糖レベルを低下させる上での(すなわち、いわゆる「ブドウ糖負荷試験」における)、エキセンディン-4/0.25% TDMの鼻腔内注射(▲)および腹腔内注射(IP)(●)投与、ならびにTDMを含まない食塩水のみのIP注射(○)の効果を示しているグラフである。
【図4】胃管による投与後の雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の1mgのマウスp-Leu-4]OB3の取り込みを示しているグラフである。
【図5】経口投与および直腸投与の両方についての、イヌによる、0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中のスマトリプタンの取り込みを示しているグラフである。
【図6】スマトリプタンが経鼻投与された患者の平均血漿レベルのグラフである。
【図7】スマトリプタンが経鼻投与された患者の平均血漿レベルのグラフである。
【発明を実施するための形態】
【0045】
発明の詳細な説明
本発明は、具体的な態様およびそれらの中に含まれる実施例の詳細な説明の参照によって、より容易に理解されるであろう。
【0046】
本発明は、少なくとも1つの薬物および少なくとも1つの表面活性物質を含む治療用組成物であって、表面活性物質が、薬物の生物学的利用能を高め、かつ対象に投与された場合に観察可能な有害作用を有しない、少なくとも1つのアルキルグリコシドおよび/または少なくとも1つのサッカリドアルキルエステルである、治療用組成物の発見に基づく。
【0047】
「治療用組成物」は、有機性または無機性の担体または添加剤との混合物からなることができ、例えば、錠剤、ペレット剤、カプセル剤、坐薬、液剤、乳剤、懸濁剤、または使用のために適した他の形態のための通常の無毒性の薬学的に許容される担体とともに調合することができる。担体には、以上に開示したものに加えて、グルコース、ラクトース、マンノース、アラビアゴム、ゼラチン、マンニトール、デンプン糊、三ケイ酸マグネシウム、タルク、コーンスターチ、ケラチン、コロイドシリカ、ジャガイモデンプン、尿素、中鎖長トリグリセリド、デキストラン、および固体、半固体または液体の形態にある調合物の製造に用いるために適した他の担体が含まれうる。加えて、補助的な安定剤、増粘剤または着色剤、例えばトリウロースなどの乾燥安定剤を用いることもできる。
【0048】
「薬物」とは、核酸、小分子、タンパク質、ポリペプチドまたはペプチドなどを非限定的に含む、任意の治療用化合物、または分子、または治療薬、または生物活性化合物のことである。
【0049】
「核酸」または「オリゴヌクレオチド」という用語はまた、翻訳領域および非翻訳領域をコードする、またはペプチドもしくはタンパク質をコードする構造遺伝子の翻訳領域もしくは非翻訳領域または調節領域を阻害する、DNA、cDNA、RNA、siRNA、RNAi、dsRNAなども表す。例えば、本発明の核酸は、5'および3'非翻訳調節ヌクレオチド配列、ならびに構造遺伝子に付随する翻訳配列を含みうる。本明細書で用いる場合、「核酸」または「オリゴヌクレオチド」または文法的等価物は、共有結合して一つになった少なくとも2つのヌクレオチドのことを指す。
【0050】
さらに、「オリゴヌクレオチド」という用語は、修飾された糖モイエティー、修飾された塩基モイエティーまたは修飾された糖連結モイエティーなどの修飾された部分を含む構造のことも指す。これらの修飾された部分は、天然塩基、天然糖および天然ホスホジエステル結合と類似した様式で機能する。したがって、オリゴヌクレオチドは、改変された塩基モイエティー、改変された糖モイエティーまたは改変された糖間(inter-sugar)結合を有してもよい。修飾された結合は、例えば、ホスホルアミド、ホスホロチエオート、ホスホロジチオエート、メチルホスホネート、ホスホトリエステル、ホスホルアミデート、O-メチルホスホロアミダイト結合、またはペプチド核酸骨格および結合であってよい。他の類似体には、正の骨格(positive backbone)、非イオン性骨格および非リボース骨格を有するオリゴヌクレオチドが含まれうる。核酸はゲノム性およびcDNAの両方のDNA、RNAまたはハイブリッドであってよく、ここで核酸は、デオキシリボヌクレオチドおよびリボヌクレオチドの任意の組み合わせ、ならびにウラシル、アデニン、チミン、シトシン、グアニン、イノシン、キサンチン、ヒポキサンチン、イソシトシン、イソグアニン、ハロゲン化塩基(halogentated base)などを含む天然の塩基または修飾された塩基の任意の組み合わせを含む。他の修飾には、例えば、天然のプリンおよびピリミジン塩基の代わりに用いられるデアザまたはアザプリンおよびピリミジン;5位もしくは6位に置換基を有するピリミジン塩基、2位、6位もしくは8位に改変された置換基もしくは置き換え置換基を有するプリン塩基、またはそれらの2'位に置換基を有する糖、糖の水素原子の1つもしくは複数の置換、または炭素環式糖もしくは非環式糖が含まれうる。
【0051】
「アンチセンス」という用語は、本明細書で用いる場合、特定の核酸配列に対して相補的な任意の組成物のことを指す。「アンチセンス鎖」という用語は、「センス」鎖に対して相補的な核酸鎖を指して用いられる。アンチセンス分子は、合成または転写を含む任意の方法によって作製しうる。細胞内にひとたび導入されると、相補的ヌクレオチドは細胞によって産生された天然配列と組み合わされて、二重鎖を形成し、転写または翻訳のいずれかを阻止する。
【0052】
アンチセンス分子には、標的受容体またはリガンドmRNA(センス)もしくはDNA(アンチセンス)配列と結合することのできる一本鎖核酸配列(RNAまたはDNAのいずれか)を含むオリゴヌクレオチドが含まれる。アンチセンスまたはセンスオリゴヌクレオチドを導き出す能力は、所与のタンパク質をコードするcDNA配列に基づく。アンチセンスまたはセンスオリゴヌクレオチドにはさらに、修飾された糖-ホスホジエステル骨格を有するオリゴヌクレオチドが含まれ、そのような糖結合は内因性ヌクレアーゼに対して抵抗性である。抵抗性の糖結合を有するそのようなオリゴヌクレオチドはインビボで安定である(すなわち、酵素分解に抵抗することができる)が、標的ヌクレオチド配列と結合することのできる配列特異性は保っている。
【0053】
RNAiは、多様な範囲の生物および細胞種へのdsRNAの導入が相補的mRNAの分解を引き起こす現象である。細胞内で、長いdsRNAはリボヌクレアーゼによって短い(例えば、21〜25ヌクレオチド)の短鎖干渉性RNA(siRNA)へと切断される。siRNAはその後、タンパク質成分と集合してRNA誘導サイレンシング複合体(RISC)となり、そのプロセスにおいて巻き戻し(unwinding)を行う。活性化されたRISCは続いて、siRNAアンチセンス鎖とmRNAとの間の塩基対合相互作用によって相補的転写物と結合する。結合したmRNAは続いて切断され、mRNAの配列特異的分解の結果、遺伝子サイレンシングが起こる。本明細書で用いる場合、「サイレンシング」とは、細胞が染色体DNAの大きな区域を停止させ、その結果、特定の遺伝子の発現を抑制する機構のことを指す。RNAi機構は、ゲノムを内因性転移因子およびウイルス感染から防御するために進化したように思われる。このため、RNAiは、分解させようとする標的mRNAに対して相補的な核酸分子を導入することによって誘導することができる。
【0054】
センスまたはアンチセンスオリゴヌクレオチドの他の例には、ポリ-(L-リジン)のような、標的核酸配列に対するオリゴヌクレオチドの親和性を高める有機モイエティーおよび他のモイエティーと共有結合したオリゴヌクレオチドが含まれる。さらになお、標的ヌクレオチド配列に対するアンチセンスまたはセンスオリゴヌクレオチドの結合特異性を修飾するために、エリプチシンなどの挿入剤、およびアルキル化剤または金属複合体を、センスまたはアンチセンスオリゴヌクレオチドと結びつけることもできる。
【0055】
本発明のペプチドは、小型ないし中程度のサイズ(すなわち、例えば最大で約15kD、30kD、40kD、50kD、60kD、70kD、80kD、90kD、100kD)の、任意の医学的または診断的に有用なペプチドまたはタンパク質であってよい。ポリペプチド吸収の改善の機序は、米国特許第5,661,130号に記載されており、これはその全体が参照により本明細書に組み入れられる。本発明の組成物はそのようなあらゆるペプチドと混合することができるが、ペプチドの利点が改善される度合いは、ペプチドの分子量ならびに物理的および化学的性状、ならびに用いられる特定の表面活性物質に応じて異なると考えられる。ポリペプチドの例には、バソプレシン、バソプレシンポリペプチド類似体、デスモプレシン、グルカゴン、コルチコトロピン(ACTH)、ゴナドトロピン、カルシトニン、インスリンのC-ペプチド、副甲状腺ホルモン(PTH)、成長ホルモン(HG)、ヒト成長ホルモン(hGH)、成長ホルモン放出ホルモン(GHRH)、オキシトシン、コルチコトロピン放出ホルモン(CRH)、ソマトスタチンまたはソマトスタチンポリペプチド類似体、ゴナドトロピンアゴニストまたはゴナドトロフィン(gonadotrophin)アゴニストポリペプチド類似体、ヒト心房性ナトリウム利尿ペプチド(ANP)、ヒトチロキシン放出ホルモン(TRH)、濾胞刺激ホルモン(FSH)、プロラクチン、インスリン、インスリン様増殖因子-I(IGF-I) ソマトメジン-C(SM-C)、カルシトニン、レプチンおよびレプチン由来短ペプチドOB-3、メラトニン、GLP-1またはグルカゴン様ペプチド-1、GiP、ニューロペプチド 下垂体アデニル酸シクラーゼ、GM-1ガングリオシド、神経成長因子(NGF)、ナファレリン、D-tryp6)-LHRH、FGF、VEGFアンタゴニスト、ロイプロリド、インターフェロン(例えば、α、β、γ)低分子量ヘパリン、PYY、LHRHアンタゴニスト、ケラチノサイト増殖因子(KGF)、グリア由来神経栄養因子(GDNF)、グレリンおよびグレリンアンタゴニストが含まれる。さらに、いくつかの局面において、ペプチドまたはタンパク質は、増殖因子、インターロイキン、ポリペプチドワクチン、酵素、エンドルフィン、糖タンパク質、リポタンパク質、または血液凝固カスケードに関与するポリペプチドから選択される。
【0056】
他の薬物または治療用化合物、分子および/または作用物質には、中枢神経系に影響を及ぼす神経伝達物質または神経イオンチャンネルの化合物または分子(すなわち、抗うつ薬(ビュープロピオン))、選択的セロトニン2c受容体アゴニスト、抗けいれん薬(トピラマート、ゾニサミド)、いくつかのドーパミンアンタゴニスト、およびカンナビノイド-1受容体アンタゴニスト(リモバナン));レプチン/インスリン/中枢神経系経路作用薬(すなわち、レプチン類似体、レプチン輸送体および/またはレプチン受容体プロモーター、毛様体神経栄養因子(Axokine)、ニューロペプチドYおよびagouti関連ペプチドアンタゴニスト、プロオピオメラノコルチン、コカイン・アンフェタミン調節転写産物プロモーター、α-メラノサイト刺激ホルモン類似体、メラノコルチン-4受容体アゴニスト、タンパク質チロシンホスファターゼ-1B阻害薬、ペルオキシソーム増殖因子活性化受容体-γ受容体アンタゴニスト、短時間作用型ブロモクリプチン(エルゴセット(ergoset))、ソマトスタチンアゴニスト(オクトレオチド)およびアディポネクチン);消化管-神経経路作用薬(すなわち、グルカゴン様ペプチド-1活性を上昇させる作用物質(エクステンディン-4(extendin-4)、リラグルチド、ジペプチジルペプチダーゼIV阻害薬)、タンパク質YY3-36、グレリン、グレリンアンタゴニスト、アミリン類似体(プラムリンチド));ならびに、安静時代謝率を高める可能性のある化合物または分子 「選択的」β-3刺激物質/アゴニスト、メラニン凝集ホルモンアンタゴニスト、フィトスタノール(phytostanol)類似体、機能性油、P57、アミラーゼ阻害薬、成長ホルモン断片、硫酸デヒドロエピアンドロステロンの合成類似体、脂肪細胞11B-ヒドロキシステロイドデヒドロゲナーゼ1型活性のアンタゴニスト、コルチコトロピン-放出ホルモンアゴニスト、脂肪酸合成の阻害薬、カルボキシペプチダーゼ阻害薬、消化管リパーゼ阻害薬(ATL962)、メラトニン、ラロキシフェン、オランザピンおよびジフェンヒドラミンが含まれる。
【0057】
他の薬物または治療用化合物には、ビスホスホネート類似体などの骨粗鬆症薬が含まれる。ビスホスホネート類似体は、ジホスホネートとしても知られ、骨粗鬆症、変形性骨炎(骨パジェット病)、骨転移(高カルシウム血症を伴うかまたは伴わない)、多発性骨髄腫、骨形成不全症、および骨脆弱性を特徴とする他の病状などの病状の治療のために臨床的に用いられている。このクラスの薬物は、破骨細胞の活動および骨再吸収を阻害する。本明細書に記載された組成物中に用いるためにアルキルサッカリドと混和されるビスホスホネートの例には、N非含有性およびN含有性の両方のビスホスホネート類似体が含まれる。N非含有性ビスホスホネートの例には、エチドロネート(Didronel(商標))、クロドロネート(Bonefos(商標)、Loron(商標))、チルドロネート(Skelid(商標))、およびそれらの薬学的に許容される類似体が含まれる。N含有性ビスホスホネートの例には、パミドロネート(Aredia(商標))、ネリドロネート、オルパドロネート、アレンドロネート(Fosamax(商標)またはFosamax+D(商標))、イバンドロネート(Boniva(商標))、リセドロネート(Actonel(商標))およびゾレドロネート(Zometa(商標)またはReclast(商標))、およびそれらの薬学的に許容される類似体が含まれる。
【0058】
他の薬物または治療用化合物には、トリプタン類似体などの薬物が含まれる。トリプタン類似体は一般に、片頭痛および頭痛の治療のために用いられる、トリプタミンをベースとする薬物のファミリーである。それらの作用は、神経終末および頭蓋内血管におけるセロトニン受容体に対するそれらの結合(それらの収縮を引き起こす)、ならびにそれに引き続く炎症誘発性ニューロペプチド放出の阻害に起因する。本明細書に記載の組成物中に用いるためにアルキルサッカリドと混和されるトリプタンの例には、スマトリプタン(Imitrex(商標)およびImigran(商標))、リザトリプタン(Maxalt(商標))、ナラトリプタン(Amerge(商標)およびNaramig(商標))、ゾルミトリプタン(Zomig(商標))、エレトリプタン(Relpax(商標))、アルモトリプタン(Axert(商標)およびAlmogran(商標))、フロバトリプタン(Frova(商標)およびMigard(商標))、およびそれらの薬学的に許容される塩が含まれる。薬学的に許容される塩の例には、ナラトリプタン-HCl;硫酸スマトリプタンおよび安息香酸リザトリプタンにおけるような、塩酸塩、硫酸塩または安息香酸塩が含まれる。トリプタンの塩形態は「遊離塩基」または非荷電形態と比較してより高い水溶性を示し、このため、本明細書中のさまざまなトリプタンの水性製剤の記載においては、トリプタンの可溶性塩形態を添加すること、またはトリプタンの遊離塩基形態に対する対応する酸(塩酸、硫酸、安息香酸など)の添加によってインサイチューで調製することを意図していることが理解されるべきである。
【0059】
本発明の治療用組成物は、薬物および薬物吸収強化剤、例えば表面活性物質を含む。「表面活性物質」という用語は、水の界面張力を変更させる任意の表面活性剤(surface active agent)のことである。典型的には、表面活性物質は、分子内に1つの親油基および1つの親水基を有する。大まかに言って、この群には、石鹸、界面活性剤(detergent)、乳化剤、分散剤および湿潤剤、ならびに消毒薬のいくつかの群が含まれる。より具体的には、表面活性物質には、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウムおよびモノステアリン酸グリセリン;ならびにポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロースおよびヒドロキシプロピルセルロースなどの親水性ポリマーが含まれる。
【0060】
好ましくは、本発明の表面活性物質は、少なくとも1つの適したアルキルグリコシドからなる。本明細書で用いる場合、「アルキルグリコシド」とは、当技術分野において公知であるように、任意の疎水性アルキルとの結合によって連結された任意の糖のことを指す。任意の「適した」アルキルグリコシドとは、本発明を限定する特性、すなわち、アルキルグリコシドが無毒性および非イオン性であること、ならびにそれが化合物の吸収を、それが化合物とともに眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して投与された場合に増加させることを満たす。適した化合物は、本明細書に記述する方法を用いて決定することができる。
【0061】
本発明のアルキルグリコシドは、公知の手順により、例えば、Rosevear et al., Biochemistry 19:4108-4115 (1980)もしくはKoeltzow and Urfer, J Am. Oil Chem. Soc., 61:1651-1655 (1984)、米国特許第3,219,656号および米国特許第3,839,318号に記載されたように化学的に、または例えば、Li et al., J. Biol. Chem., 266:10723-10726 (1991)もしくはGopalan et al., J. Biol. Chem. 267:9629-9638 (1992)に記載されたように酵素的に合成することができる。
【0062】
本発明のアルキルグリコシドには、以下のものが非限定的に含まれうる:アルキルグリコシド、例えばオクチル-、ノニル-、デシル-、ウンデシル-、ドデシル-、トリデシル-、テトラデシル-、ペンタデシル-、ヘキサデシル-、ヘプタデシル-およびオクタデシル-α-またはβ-D-マルトシド、-グルコシドまたは-スクロシドなど(Koeltzow and Urferに従って合成;Anatrace Inc., Maumee, Ohio;Calbiochem, San Diego, Calif.; Fluka Chemie, Switzerland);アルキルチオマルトシド、例えばヘプチル、オクチル、ドデシル-、トリデシル-およびテトラデシル-β-D-チオマルトシドなど(Defaye, J. and Pederson, C., "Hydrogen Fluoride, Solvent and Reagent for Carbohydrate Conversion Technology" in Carbohydrates as Organic Raw Materials, 247-265 (F. W. Lichtenthaler, ed.) YCH Publishers, New York (1991);Ferenci, T., J. Bacteriol, 144:7-11 (1980)に従って合成);アルキルチオグルコシド、例えばヘプチル-またはオクチル1-チオαまたはβ-D-グルコピラノシド(Anatrace, Inc., Maumee, Ohio;Saito, S. and Tsuchiya, T. Chem. Pharm. Bull. 33:503-508 (1985)を参照);アルキルチオスクロース(例えば、Binder, T. P. and Robyt, J. F., Carbohydr. Res. 140:9-20 (1985)に従って合成);アルキルマルトトリオシド(Koeltzow and Urferに従って合成);スクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸アミド;(オーストラリア特許第382,381号(1987);Chem. Abstr., 108:114719 (1988)およびGruber and Greber pp. 95-116に従って合成);アミド結合によってアルキル鎖と連結されたパラチノースおよびイソマルタミンの誘導体(Kunz, M., "Sucrose-based Hydrophilic Building Blocks as Intermediates for the Synthesis of Surfactants and Polymers" in Carbohydrates as Organic Raw Materials, 127-153に従って合成);尿素によってアルキル鎖と連結されたイソマルタミンの誘導体(Kunzに従って合成);スクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸ウレイド(Gruber and Greber, pp. 95-116に従って合成);ならびにスクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸アミド(オーストラリア特許第382,381号(1987)、Chem. Abstr., 108:114719 (1988)およびGruber and Greber, pp. 95-116に従って合成)。
【0063】
アルキルグリコシドおよび/またはスクロースエステルからなる本発明の表面活性物質は、特徴的な親水性-親油性バランス(HLB)数値を有し、それは経験的に計算または決定することができる(Schick, M. J. Nonionic Surfactants, p. 607 (New York: Marcel Dekker, Inc. (1967))。HLB数値は表面活性物質の親水性特性を直接反映したものであり、すなわち、HLB数値が大きいほど化合物の親水性は高い。HLB数値は、式:(親水性成分のMWの20倍)/(疎水性成分のMW+親水性成分のMW)によって計算することができ、式中、MW=分子量である(Rosen, M. J., Surfactants and Interfacial Phenomena, pp. 242-245, John Wiley, New York (1978))。HLB数値は、表面活性物質の親水性特性を直接表現したものであり、すなわち、HLB数値が大きいほど化合物の親水性は高い。好ましい表面活性物質は約10〜20のHLB数値を有し、さらにより好ましい範囲は約11〜15である。
【0064】
上記の通り、疎水性アルキルは、サッカリドモイエティーの所望の疎水性および親水性に応じて、任意の所望のサイズのものを選択することができる。例えば、アルキル鎖の1つの好ましい範囲は炭素原子が約9〜約24個である。さらにより好ましい範囲は炭素原子が約9〜約16個または約14個である。同様に、いくつかの好ましいグリコシドは、グリコシド結合によって炭素原子が9、10、12、13、14、16、18、20、22または24個のアルキル鎖と連結されたマルトース、スクロースおよびグルコース、例えば、ノニル-、デシル-、ドデシル-およびテトラデシルスクロシド、グルコシドおよびマルトシドなどである。これらの組成物は、アルコールおよびオリゴサッカリドに分解され、両親媒性であることから無毒性である。
【0065】
また、本発明の表面活性物質にはサッカリドも含まれる。本明細書で用いる場合、「サッカリド」は、モノサッカリド、直鎖形態または環状形態にあるオリゴサッカリドもしくはポリサッカリド、またはサッカリド鎖を形成するそれらの組み合わせを含む。オリゴサッカリドは、2つまたはそれ以上のモノサッカリド残基を有するサッカリドのことである。サッカリドは、例えば、現在市販されている任意のサッカリド種から選択することもでき、または合成することもできる。用いうる可能性のある多くのサッカリドのいくつかの例には、グルコース、マルトース、マルトトリオース、マルトテトラオース、スクロースおよびトレハロースが含まれる。好ましいサッカリドには、マルトース、スクロースおよびグルコースが含まれる。
【0066】
本発明の表面活性物質は、同様にスクロースエステルからなることもできる。本明細書で用いる場合、「スクロースエステル」とは脂肪酸のスクロースエステルのことであり、これはスクロースおよび脂肪酸の複合体である。スクロースエステルは、スクロース中に反応に利用しうるヒドロキシル基が8つあり、酢酸基からより大きくより嵩の高い脂肪酸までに至る多くの脂肪酸基がスクロースと反応しうるため、多くの形態をとることができる。この融通性は、用いる脂肪酸モイエティーに基づき、多くの生成物および官能性を適応させることができることを意味する。スクロースエステルは、食品用および非食品用の用途を有し、特に表面活性物質および乳化剤としての用途を有し、医薬品、化粧品、表面活性物質および食品添加物における適用が増えている。それらは生分解性であり、皮膚に対して無毒性であるとともに刺激性が少ない。
【0067】
本発明の表面活性物質は、親水性サッカリドと連結した疎水性アルキル基を有する。疎水性アルキル基と親水性サッカリドとの間の結合には、他にも可能性はあるものの、グリコシド、チオグリコシド(Horton)、アミド(Carbohydrates as Organic Raw Materials, F. W. Lichtenthaler ed., VCH Publishers, New York, 1991)、ウレイド(オーストラリア特許第386,414号(1988);Chem. Abstr. 11O:137536p (1989);Gruber, H. and Greber, G., "Reactive Sucrose Derivatives" in Carbohydrates as Organic Raw Materials, pp. 95-116を参照)またはエステル結合(Sugar Esters: Preparation and Application, J. C. Colbert ed., (Noyes Data Corp., New Jersey), (1974))が含まれうる。さらに、好ましいグリコシドには、グリコシド結合によって炭素原子約9〜16個のアルキル鎖と連結されたマルトース、スクロースおよびグルコース、例えば、ノニル-、デシル-、ドデシル-およびテトラデシルスクロシド、グルコシドおよびマルトシドが含まれうる。この場合も、これらの組成物は両親媒性であり、それらはアルコールおよびオリゴサッカリドへと分解されることから無毒性である。
【0068】
上記の例は、本明細書において特許請求を行う方法に用いられるグリコシドの種類の例示であるが、このリストは網羅的ではない。特許請求の範囲の基準に当てはまる上記の化合物の誘導体も、グリコシドを選択する場合に考慮されるべきである。化合物はすべて、本明細書および実施例に教示された方法に従って有効性に関してスクリーニングすることができる。
【0069】
本発明の組成物は、錠剤、カプセル剤、坐薬、滴剤、噴霧剤、エアロゾル、および持続放出形式または遅延バースト形式からなる群より選択される形式で投与することができる。噴霧剤およびエアロゾルは、適切なディスペンサーの使用によって実現することができる。持続放出形式は、眼インサート(ocular insert)、侵食性(erodible)微粒子、膨張性粘膜付着剤粒子、pH感受性微粒子、ナノ粒子/ラテックス系、イオン交換樹脂ならびに他のポリマー性ゲルおよびインプラントでありうる(Ocusert, Alza Corp., California;Joshi, A., S. Ping and K. J. Himmelstein, 特許出願第WO 91/19481号)。これらの系は、吸収性表面との長期にわたる薬物接触を維持し、流出および非生産的な薬物喪失を防止する。長期にわたる薬物接触は皮膚および粘膜の表面に対して無毒性である。
【0070】
本発明の表面活性物質組成物は安定である。例えば、Baudysらは、米国特許第5,726,154号において、SDS(ドデシル硫酸ナトリウム、表面活性物質)および有機酸を含む水性液体組成物中のカルシトニンが、少なくとも6カ月にわたって安定であることを示している。同様に、本発明の表面活性物質組成物は、薬物と混和された場合に、改善された安定化特性を有する。これらの製剤中に有機酸は必要でない。例えば、本発明の組成物は、約4℃〜25℃に維持された場合に、タンパク質およびペプチド治療薬の安定性を約6カ月またはそれ以上にわたって維持する。
【0071】
表面活性物質組成物の安定性は、一部には、それらの高い最大無毒性レベル(NOAEL)に起因する。米国環境保護庁(Environmental Protection Agency)(EPA)は、最大無毒性レベル(NOAEL)を、曝露された集団とその適切な対照との間で有害作用の頻度または重症度に関して統計学的にも生物学的にも有意な増加がみられない曝露レベルと定義している。それ故に、「最大無毒性レベル」(またはNOAEL)という用語は、規定された条件下で標的生物の形態、機能的能力、成長、発達または寿命の検出可能で有害な変化を引き起こさない、実験または観察によって見いだされる、物質の最大の濃度または量のことである。
【0072】
世界保健機構(World Health Organization)(WHO)の国連食糧農業機関(Food and Agriculture Organization of the United Nations)(FAO)は、いくつかのアルキルグリコシドが非常に高いNOAELを有し、いかなる有害作用も伴わずにこれらのアルキルグリコシドの摂取増加が可能であることを示している。この報告は、ワールドワイドウェブ上のinchem.org/documents/jecfa/jecmono/v1Oje11.htmで見ることができる。例えば、食品中に用いられるスクロースエステルの1つであるドデカン酸スクロースのNOAELは、約20〜30グラム/キログラム/日であり、例えば、70キログラムの人(約154ポンド)は、1日当たり約1400〜2100グラム(または約3〜4.6ポンド)のドデカン酸スクロースを、いかなる観察可能な有害作用も伴わずに摂取することができる。典型的には、ヒトの1日許容摂取量はNOAELの約1%であり、これは無期限に1日当たり約14〜21グラムまたは1400万マイクログラム〜2100万マイクログラムに換算される。NOAELの定義および他の関連した定義は、ワールドワイドウェブ上のepa.gov/OCEPAtermsで見ることができる。このため、本発明において予期されるアルキルグリコシドのレベルで、ある程度の影響は生じる可能性はあるが、それらのレベルは有害であるとも有害作用の前兆であるとも考えられない。
【0073】
したがって、少なくとも1つのアルキルグリコシド、例えば、テトラデシルマルトシド(TDM;またはIntravail A)を、重量比で約0.125%の濃度のアルキルグリコシドを有する本発明の表面活性物質組成物により、治療レジメンに応じて1日2回または1日3回またはそれ以上治療される対象は、1日当たり合計で約200〜300マイクログラムのTDMを摂取する。そのため、TDMの有効量はNOAELの少なくとも1000分の1(すなわち、1/1000)であり、1日許容摂取量であるNOAELの1%よりもはるかに少ない;またはこの場合には1日許容摂取量の約1/50,000である。別の言い方をすれば、本発明のアルキルグリコシドは、本発明において用いられるアルキルグリコシドの量または濃度が有害作用を引き起こさず、いかなる有害作用も伴わずに安全に摂取しうるような高いNOAELを有する。
【0074】
本発明の表面活性物質組成物はまた、それらが生理的に無毒性および非刺激性であるため、安定でもある。「無毒性」という用語は、アルキルグリコシド分子が、ヒトへの投与および摂取のために適する十分に低い毒性を有することを意味する。好ましいアルキルグリコシドは、それらが適用される組織に対して非刺激性である。用いられる任意のアルキルグリコシドは、それが細胞に障害を引き起こさないように非常にわずかな毒性しか有しないかまたは全く毒性を有しないべきである。しかしながら、任意の所与のアルキルグリコシドの毒性は、用いられるアルキルグリコシドの濃度に応じて異なりうる。選択されたアルキルグリコシドが身体によって代謝または排出され、この代謝または排出が有害な毒性をもたらさない様式で行われることも有益である。「非刺激性」という用語は、その作用物質が、皮膚表面または粘膜との即時的、長期的または反復的な接触後に炎症を引き起こさないことを意味する。
【0075】
その上、表面活性物質組成物の、特にスクロースエステルの1つの態様は、抗菌作用薬として役立つ。ある作用物質は、その作用物質またはその等価物が細菌を破壊するかまたは細菌の増殖もしくは繁殖を抑制するならば、「抗菌」薬または物質である。スクロースエステルおよびそれらの脂肪酸の抗菌活性は報告されている。Tetsuaki et al. (1997) "Lysis of Bacillus subtilis cells by glycerol and sucrose esters of fatty acids," Applied and Environmental Microbiology, 53(3):505-508。Watanabeら(2000)は、ラウリン酸ガラクトースおよびラウリン酸フルクトース(galactose and fructose laureate)が特に有効な糖質モノエステルであることを記載している。Watanabe et al., (2000) "Antibacterial carbohydrate monoesters suppressing cell growth of Streptococcus mutan in the presence of sucrose," Curr Microbiol 41(3): 210-213。それ故に、本発明は、本明細書に記載されたスクロースエステルには限定されず、細菌の増殖および繁殖を抑制する、ガラクトースエステルおよびフルクトースエステルを含む他の糖質エステルも範囲に含む。Sutton and Porter (2002), "Development of the antimicrobial effectiveness test as USP Chapter <51>," 56(6): 300-311を参照、これはその全体が参照により本明細書に組み入れられる。例えば、塩化ベンザルコニウムのように一般的に用いられる抗菌薬は、鼻腔内製剤で一般的に用いられるよりもはるかに低いベンザルコニウム濃度で粘膜繊毛表面の破壊が観察されている電子顕微鏡検査によって示されているように、毒性が高度である。例えば、Sebahattin Cureoglu, Murat Akkus, Ustun Osma, Mehmet Yaldiz, Faruk Oktay, Belgin Can, Cengiz Guven, Muhanunet Tekin, and Faruk Menc (2002), "The effect of benzalkonium chloride an electron microscopy study," Eur Arch Otorhinolaryngol 259:362-364を参照。
【0076】
本発明の表面活性物質組成物は、典型的には、重量比で約0.01%〜20%のレベルで存在する。より好ましい組み込みレベルは、重量比で約0.01%〜5%、重量比で約0.01%〜2%、約0.01%〜1%、最も好ましくは重量比で約0.01%〜0.125%である。表面活性物質は好ましくは、組成物中に存在する他の成分と適合するように製剤化される。液体またはゲルまたはカプセルまたは注射用または噴霧薬の組成物において、表面活性物質は最も好ましくは、それがこれらの組成物中の任意のタンパク質または酵素の安定性を促進するか、または少なくともそれを劣化させないように製剤化される。さらに、本発明は、依然として所望の効果を維持しながら吸収強化因子の濃度をできるだけ低く保つことにより、濃度を最適化する。
【0077】
本発明の組成物は、対象に投与された場合、生物活性化合物または薬物の粘膜送達の強化をもたらし、対象への等価濃度の化合物の筋肉内注射後の組織(例えば、CNS)または体液または血漿における化合物のCmaxと比較して、対象の組織または体液または血漿中における化合物のピーク濃度(またはCmax)が約15%、20%または50%またはそれ以上である。
【0078】
薬物または化合物が設定期間、例えば、24時間で血流にどの程度達するかの指標を、24時間またはより長い期間の中のさまざまな時間で薬物血中濃度をプロットし、続いて0〜24時間の間の曲線下面積(AUC)を測定することによって算出することもできる。同様に、薬物有効性の指標を、約0.1〜1.0時間の間の、対象の組織(例えば、CNS)または体液または血漿における生物活性化合物の最大濃度到達時間(Tmax)から決定することもできる。本発明の治療用組成物は、薬物作用の開始の速さを約1.5倍〜2倍に高める(すなわち、Tmaxを減少させる)。
【0079】
また、本発明の治療用組成物または製剤を、必要とする対象に全身的または局所的に投与または送達することもできる。適した経路には、例えば、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)、経粘膜投与、膣、直腸のほか、筋肉内、皮下、静脈内、腹腔内またはCSF送達を含む避腸的送達が含まれる。その上、送達の様式、例えば、液体、ゲル、錠剤、噴霧剤などは、対象への送達の方法にも依存すると考えられる。
【0080】
さらに、本発明の治療用組成物は、薬学的に許容される担体からなることもできる。「薬学的に許容される担体」とは、水性または非水性の作用物質、例えばアルコール性または油性のもの、またはそれらの混合物であり、表面活性物質、皮膚軟化剤、潤滑剤、安定剤、色素、香料、保存料、pHの調整のための酸もしくは塩基、溶媒、乳化剤、ゲル化剤、保湿剤(moisturizer)、安定剤、湿潤剤、持続放出剤、保湿剤(humectant)、または特定の薬学的組成物の形態に一般的に含まれる他の成分が含まれうる。薬学的に許容される担体は当技術分野において周知であり、これには例えば、水もしくは生理的緩衝食塩水などの水性溶液、またはグリコール、グリセロールなどの他の溶媒または媒体、およびオリーブ油または注射可能な有機エステルなどの油が含まれる。薬学的に許容される担体には、例えば、特定の阻害薬の吸収を安定化または増加させるように作用する生理的に許容される化合物、例えば、グルコース、スクロースもしくはデキストランなどの糖質、アスコルビン酸もしくはグルタチオンなどの酸化防止剤、キレート剤、低分子量タンパク質、または他の安定剤もしくは添加剤が含まれうる。また、薬学的に許容される担体を、蒸留水、ベンジルアルコール、ラクトース、デンプン、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、アルギン酸、コロイドシリカ、二酸化チタンおよび香味剤などの物質から選択することもできる。
【0081】
さらに、薬物の加水分解に対するアルキルサッカリドまたはサッカリドアルキルエステルの感受性を低下させるために、薬物内部のさまざまな酸素原子をイオウで置換することもできる(Defaye, J. and Gelas, J. in Studies in Natural Product Chemistry (Atta-ur-Rahman, ed.) Vol. 8, pp. 315-357, Elsevier, Amsterdam, 1991)。例えば、糖環のヘテロ原子は酸素もしくはイオウのいずれであってもよく、またはオリゴサッカリド中のモノサッカリド間の結合が酸素もしくはイオウであってもよい(Horton, D. and Wander, J. D., "Thio Sugars and Derivatives," The Carbohydrates: Chemistry and Biochemistry, 2d. Ed. Vol. IB, (W. Reyman and D. Horton eds.), pp. 799-842, (Academic Press, New York), (1972))。オリゴサッカリドはα(アルファ)またはβ(ベータ)アノマー立体配置のいずれであってもよい(Pacsu, E., et al. in Methods in Carbohydrate Chemistry (R. L. Whistler, et al., eds.) Vol. 2, pp. 376-385, Academic Press, New York 1963)を参照。
【0082】
本発明の組成物は、治療薬または薬物、ならびに本発明による1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル、ならびに適切な薬学的担体または添加剤、例えばマンニトール、コーンスターチ、ポリビニルピロリドンなどを混合し、その混合物を顆粒化し、最後にそれをコーンスターチ、ステアリン酸マグネシウムなどの薬学的担体の存在下で圧縮することにより、錠剤形態に調製することができる。必要に応じて、そのようにして調製された製剤が、糖コーティングもしくは腸溶性コーティングを含んでもよく、または例えば適切なpH媒質中で活性成分が徐々に放出されるような様式で覆われていてもよい。
【0083】
「腸溶性コーティング」という用語は、治療用組成物またはコアを包み込む、取り囲む、またはその周りに層もしくは膜を形成するポリマーのことである。また、腸溶性コーティングが、コーティングに対して適合性または不適合性の薬物を含んでもよい。1つの錠剤組成物は、比較的高いpHレベル(例えば、4.0を上回るpH、4.5を上回るpH、5.0を上回るpH、またはそれ以上)では溶解するかもしくは薬物を放出するが低いpHレベル(例えば、pH 4またはそれ未満)ではそうでない適合性薬物;またはその反対である適合性薬物を有する腸溶性コーティングポリマーを含みうる。
【0084】
1つの好ましい態様において、本発明の用量依存的放出形態は、以下のものを含む錠剤である:
(a)以下のものを含むコア:
(i)治療薬または薬物;
(ii)少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む表面活性物質;ならびに
(b)コアを取り囲む少なくとも1つの膜コーティング、ここでこのコーティングは非透過性、透過性、半透過性または多孔性コーティングであり、規定されたpHの水性環境と接触すると、より透過性または多孔性になる。
【0085】
「膜」という用語は、「コーティング」またはその等価物と同義である。これらの用語は、医薬品、例えば錠剤の、水性溶液もしくは体液に対して、および/またはその中に封入された治療薬もしくは薬物に対して非透過性、透過性、半透過性または多孔性である領域を特定するために用いられる。膜が薬物に対して透過性、半透過性または多孔性であるならば、薬物は溶液中またはインビボで、膜の開口部または細孔を通って放出されうる。多孔性膜は、機械的に製造することもでき(例えば、膜層の中にレーザーを用いて微細な穴または細孔を開ける)、またはそれをコーティングポリマーの物理化学的性状によって付与することもできる。本発明の膜またはコーティングポリマーは当技術分野において周知であり、これにはセルロースエステル、セルロースジエステル、セルローストリエステル、セルロースエーテル、セルロースエステルエーテル、セルロースアシレート、セルロースジアシレート(cellulose diacylate)、セルローストリアシレート(cellulose triacylate)、酢酸セルロース、二酢酸セルロース、三酢酸セルロース、セルロースアセテートプロピオネートおよびセルロースアセテートブチレートが含まれる。
【0086】
その他の適したポリマーは、参照により本明細書に組み入れられる米国特許第3,845,770号、第3,916,899号、第4,008,719号、第4,036,228号および第4,11210号に記載されている。
【0087】
さらに、本発明による腸溶性コーティングは、可塑剤、および、溶液中またはインビボで懸濁液のpHを達成または調整するのに十分な量の水酸化ナトリウム(NaOH)を含みうる。可塑剤の例には、クエン酸トリエチル、トリアセチン、セバシン酸トリブチル(tributyl sebecate)またはポリエチレングリコールが含まれる。また、水酸化カリウム、炭酸カルシウム、カルボキシメチルセルロースナトリウム、酸化マグネシウムおよび水酸化マグネシウムを含む他のアルカリ化剤を、溶液中またはインビボで懸濁液のpHを達成または調整するために用いることもできる。
【0088】
したがって、1つの態様において、腸溶性コーティングは、ある特定のpHまたはpH範囲を有するある特定の媒質中で、1つまたは複数の薬物のある特定のパーセンテージを放出するように設計することができる。例えば、本発明の治療用組成物は、酸性環境(例えば、胃)において化学的に不安定な少なくとも1つの薬物を包み込むかまたは保護する少なくとも1つの腸溶性コーティングを含みうる。腸溶性コーティングは、その薬物を酸性環境(例えば、pH<3)から保護するとともに、より酸性度の低い場所、例えば、pHが3または4または5またはそれ以上である小腸および大腸の領域では、薬物を放出する。この性質を持つ医薬品は、胃腸管の1つの領域から別の領域に運ばれると考えられ、例えば、薬物が胃から小腸(十二指腸、空腸および回腸)に移動するまでに約2〜約4時間かかる。この通過または移行の間に、pHは約3(例えば、胃)、4または5から、約6または7またはそれ以上のpHに変化する。このため、腸溶性コーティングは、薬物を含むコアが実質的に元のままに保たれるのを可能にするとともに、時期尚早な薬物放出、または酸が浸透して薬物を不安定化することを防止する。
【0089】
適した腸溶性ポリマーの例には、酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸フタル酸ポリビニル、メタクリル酸コポリマー、セラック、酢酸トリメット酸セルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸フタル酸セルロース、酢酸コハク酸セルロース、酢酸フマル酸セルロース、安息香酸フタル酸セルロース、プロピオン酸フタル酸セルロース、フタル酸メチルセルロース、カルボキシメチルエチルセルロース、フタル酸エチルヒドロキシエチルセルロース、セラック、スチレン-アクリル酸コポリマー、アクリル酸メチル-アクリル酸コポリマー、アクリル酸メチル-メタクリル酸コポリマー、アクリル酸ブチル-スチレン-アクリル酸コポリマー、メタクリル酸-メタクリル酸メチルコポリマー、メタクリル酸-アクリル酸エチルコポリマー、アクリル酸メチル-メタクリル酸-アクリル酸オクチルコポリマー、酢酸ビニル-無水マレイン酸コポリマー、スチレン-無水マレイン酸コポリマー、スチレン-マレイン酸モノエステルコポリマー、ビニルメチルエーテル-無水マレイン酸コポリマー、エチレン-無水マレイン酸コポリマー、ビニルブチルエーテル-無水マレイン酸コポリマー、アクリロニトリル-アクリル酸メチル-無水マレイン酸コポリマー、アクリル酸ブチル-スチレン-無水マレイン酸コポリマー、ポリビニルアルコールフタレート、ポリビニルアセタールフタレート、ポリビニルブチレートフタレートおよびポリビニルアセトアセタールフタレート、またはそれらの組み合わせが非限定的に含まれる。当業者は、本発明によるコーティングの全体または一部として、他の親水性、疎水性および腸溶性コーティングポリマーを単独または任意の組み合わせで容易に用いうることを認識しているであろう。
【0090】
錠剤の形態にある本発明の治療用組成物は、複数のコーティング、例えば、親水性コーティング(例えば、ヒドロキシプロピルメチルセルロース)、および/または疎水性コーティング(例えば、アルキルセルロース)、および/または腸溶性コーティングを有することができる。例えば、錠剤コアを、親水性、疎水性または腸溶性コーティングから選択される、同じ種類の複数のコーティングまたは複数の異なる種類のコーティングによって包み込むことができる。それ故に、錠剤は、標的組織、または1つもしくは複数の薬物の目的に応じて、少なくとも1つの層を有するが、同じまたは異なるコーティングからなる複数の層も有しうる設計することができると期待される。例えば、錠剤コア層が、第1のコーティング層(例えば、親水性、疎水性または腸溶性コーティング)によって囲まれた第1の組成物、および第2のコーティング層に囲まれた同じまたは異なる用量を有する第2の同じまたは異なる組成物または薬物、などを有してもよい。さまざまなコーティングによるこの多層化は、同じまたは異なる薬物を含む組成物の、第1、第2、第3またはそれ以上の段階的または用量依存的な放出を提供する。
【0091】
1つの好ましい態様においては、本発明の第1の組成物の第1の用量を錠剤コアの中に腸溶性コーティングとともに含め、その結果、腸溶性コーティングがその中に含まれる組成物を崩壊または胃内部への放出から保護し、それらを防止するようにする。もう1つの例では、治療用組成物の第1の負荷用量は第1の層内に含められ、製剤または錠剤の中に含まれる全組成物の総量の約10%〜約40%を占める。第2の負荷用量では、組成物の総用量の別のパーセンテージが放出される。本発明は、治療レジメンにおいて必要とされるだけの多数回の放出用量を想定している。したがって、ある局面において、単一のコーティングまたは複数のコーティング層は、コーティングされた単位剤形の重量比で約2%〜6%、好ましくは約2%〜約5%、さらにより好ましくは重量比で約2%〜約3%の範囲にわたる量にある。
【0092】
したがって、本発明の組成物調合物は、特定の領域のために適したpH溶解性ポリマーを選択することにより、硬カプセル剤または錠剤の内容物を、胃腸管(例えば、小腸および大腸)のより遠隔部にある所望の部位で選択的に放出させることを可能にする。また、組成物調合物の機械的圧出を、水を吸収すると半透過性硬カプセルの内部で膨張し、それ故に硬カプセル剤の開口部を通して組成物を排出させる吸水性ポリマーを含めることによって達成することもできる。
【0093】
用量依存的持続放出に特に適する薬物には、インスリン様増殖因子-I(IGF-I)、ソマトメジン-C(SM-C;糖尿病、神経機能、腎機能)、インスリン(糖尿病)、カルシトニン(骨粗鬆症)、レプチン(肥満;不妊症)、レプチン由来短ペプチド(OB-3)、hGH(エイズ性消耗、低身長症)、ヒト副甲状腺ホルモン(PTH)(骨粗鬆症)、メラトニン(睡眠)、GLP-1またはグルカゴン様ペプチド-1(糖尿病)、GiP(糖尿病)、下垂体アデニル酸シクラーゼ活性化ポリペプチド(PACAP)および膵島機能(糖尿病)、GM-1ガングリオシド(アルツハイマー病)、神経成長因子(NGF)(アルツハイマー病)、ナファレリン(子宮内膜症)、Synarel(登録商標)(酢酸ナファレリン点鼻液),(D-tryp6)-LHRH(妊孕性)、FGF(十二指腸潰瘍、黄斑変性症、熱傷、創傷、脊髄損傷、骨および軟骨損傷の修復)、VEGFアンタゴニスト(受容体を遮断するため)、VEGF(アゴニスト)新生児呼吸窮迫症候群;ALS)、ロイプロリド(前立腺癌および乳癌)、インターフェロン-α(慢性C型肝炎)、低分子量ヘパリン(血液凝固、深部静脈血栓症)、PYY(肥満)、LHRHアンタゴニスト(妊孕性)、LH(黄体形成ホルモン)、グレリンアンタゴニスト(肥満)、KGF(パーキンソン病)、GDNF(パーキンソン病)、G-CSF(癌における赤血球形成)、イミトレックス(Imitrex)(片頭痛)、インテグレリン(Integrelin)(抗凝固)、Natrecor(登録商標)(うっ血性心不全)、ヒトB型ナトリウム利尿ペプチド(hBNP)、SYNAREL(登録商標)(Searl;酢酸ナファレリン点鼻液)、サンドスタチン(Sandostatin)(成長ホルモン補充)、フォルテオ(Forteo)(骨粗鬆症)、DDAVP(登録商標)鼻噴霧薬(酢酸デスモプレシン)、Cetrotide(登録商標)(注射用酢酸セトロレリクス)、Antagon(商標)(酢酸ガニレリクス)、アンジオマックス(Angiomax)(ビバリルジン;トロンビン阻害薬)、Accolate(登録商標)(ザフィルルカスト;注射用)、エキセンディン-4(Exanatide;糖尿病)、SYMLIN(登録商標)(酢酸プラムリンチド;合成アミリン;糖尿病)、デスモプレシン、グルカゴン、ACTH(コルチコトロピン)、インスリンCペプチド、GHRHおよび類似体(GnRHa)、成長ホルモン放出ホルモン、オキシトシン、コルチコトロピン放出ホルモン(CRH)、心房性ナトリウム利尿ペプチド(ANP)、チロキシン放出ホルモン(TRHrh)、濾胞刺激ホルモン(FSH)、プロラクチン、眼用トブラマイシン(角膜感染)、バソプレシン、デスモプレシン、フューゼオン(Fuzeon)(Roche;HIV融合阻害薬 MW 4492)およびエプチフィバチド(Eptifibatide)が非限定的に含まれる。
【0094】
さらに、治療を必要とする任意の特定の対象に対する具体的な用量レベルおよび投薬の頻度はさまざまであってよく、それは用いる具体的な化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全般的健康状態、性別、食事、投与の様式および回数、排泄速度、薬物併用、特定の病状の重症度、ならびに治療法を受ける宿主を含む種々の要因に依存することが、当業者には理解されるであろう。
【0095】
アルキルグリコシド、特にアルキルマルトシド、より具体的にはドデシルマルトシド(DDM)およびテトラデシルマルトシド(TDM)は、溶液中のインスリンを安定化し、ペプチドの凝集を防止することが示されている。Hovgaard et al., "Insulin Stabilization and GI absorption," J. Control. Rel, 19 (1992) 458-463、これはHovgaard et al., "Stabilization of insulin by alkylmaltosides: A spectroscopic evaluation," Int. J. Pharmaceutics 132 (1996) 107-113中に引用(本明細書では以後「Hovgaard-1」)。さらに、Hovgaard-1は、57日後でさえもDDM-インスリン複合体は安定に保たれ、ほぼ完全な生物活性を有していた。この複合体の安定性はアルキル基の長さ(炭素原子の数)に起因していて、DDMとインスリンとの比が高いほどより優れるという仮説が立てられた(例えば、4:1および16:1;Hovgaard 1における図1参照)。しかし、Hovgaard-1によれば、DDM-インスリン複合体は安定であったものの、他のマルトシドについては同じ安定性は示されなかった。なおかつ、1件の関連した研究において、Hovgaardら(1996)は、DDM-インスリンを動物にインビボで経口投与した場合には、この複合体の生物学的利用能は低い(例えば、0.5%〜1%という生物学的利用能)ことを示している。Hovgaard et al., "Stabilization of insulin by alkylmaltoside. B. Oral absorption in vivo in rats," Int. J. Pharmaceutics 132 (1996) 115-121(Hovgaard-2)。それ故に、本発明の改良された1つの局面は、表面活性物質が標的組織、臓器、系などに対する薬物の生物学的利用能を高めること、さらには薬物安定性も高めることである。
【0096】
したがって、本発明の1つの局面は、少なくとも1つの薬物および1つの表面活性物質を有する治療用組成物であって、表面活性物質がさらに、その薬物の生物学的利用能を強化する少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル製剤からなる、治療用組成物を提供することである。薬物製剤の生物学的利用能の決定については本明細書に記載している。本明細書で用いる場合、「生物学的利用能」とは、活性物質またはモイエティーが、元のままの薬物として体循環に到達する比率および程度のことである。あらゆる薬物の生物学的利用能は、いかに良く吸着するか、およびそのうちどれだけ多くが肝臓からの除去を免れるかに依存すると考えられる。
【0097】
絶対的生物学的利用能を決定するためには、被験薬物および投与様式の測定を、静脈内参照用量と対比して行う。静脈内用量の生物学的利用能は定義上100%である。例えば、動物または志願者に対して薬物の静脈内注射および対応する経口用量を投与する。尿試料または血漿試料をある期間にわたって採取し、その期間にわたる薬物のレベルを決定する。
【0098】
血漿中薬物濃度-時間曲線の曲線下面積(AUC)を、静脈内用量および経口用量の両方についてプロットし、両方の製剤の生物学的利用能の計算を単比例によって行う。例えば、同一の静脈内用量および経口用量を投与して、経口AUCが静脈内AUCの50%であるならば、その経口製剤の生物学的利用能は50%である。任意の薬物の生物学的利用能は、不完全な吸収、初回通過クリアランスまたはこれらの組み合わせを含む、多くの要因に依存する(以下でより詳細に考察する)。さらに、血漿中薬物濃度のピーク濃度(またはCmax)も測定し、その薬物の等価濃度の筋肉内(IM)注射後の血漿中薬物濃度のピーク濃度(Cmax)と対比する。その上、血漿中薬物の最大濃度到達時間(またはtmax)は約0.1〜1.0時間である。
【0099】
薬物の複数の製剤(例えば、アルキルグリコシドまたはサッカリドアルキルエステルの薬物製剤)の相対的生物学的利用能を決定するためには、一方または両方の薬物が初回通過クリアランス(以下でより詳細に考察)を受け、それ故に検出されない可能性があるため、その製剤の生物学的利用能を互いと対比して評価する。例えば、第1の経口製剤を第2の経口製剤と対比して評価する。第2の製剤は、第1のものの生物学的利用能を評価するための参照として用いる。この種の検討は、薬物が吸収されることについての2つの製剤の相対的成績の指標を提供する。
【0100】
薬物の生物学的利用能は一致せず、薬物毎に大きく異なる。例えば、女性の閉経後骨粗鬆症の治療のための処方医薬品であるMIACALCIN(登録商標)(Novartis社のサケカルシトニン)鼻噴霧薬の生物学的利用能は、約3%という平均生物学的利用能を有する(0.3%〜30.6%の範囲;図1参照)。MIACALCIN(登録商標)の製品情報シートは、ワールドワイドウェブ上のmiacalcin.com/info/howWorks/index.jspおよびdrugs.com/PDR/Miacalcin_Nasal_Spray.htmlに見ることができる。異なる方法および被験者を用いてさまざまな研究者によって得られたMIACALCIN(登録商標)に関するデータは、薬物の生物学的利用能の大きなばらつきを示しており、例えば、健常志願者では経鼻投与した用量のわずか3%ほどしか生物学的に利用されず、これは筋肉内注射によって投与した同じ用量(MIACALCIN(登録商標)製品挿入物)とは対照的であった。このことは、2桁のばらつきがあり、消費者にとって望ましくないことを表している。
【0101】
薬物の生物学的利用能の低さは、筋肉内ビタミンB12療法後の寛解期にある患者の治療および維持のために用いられる、NASCOBAL(登録商標)(Nastech)またはシアノコバラミンでも観察される。このゲル製剤を鼻腔内に投与し、B12の生物学的利用能を筋肉内B12注射と比較した。B12のピーク濃度(またはTmax)は鼻腔内投与から12時間後に到達し、筋肉内注射と対比した相対的な、B12鼻腔用ゲルの生物学的利用能は約8.9%であった(90%信頼区間、7.1%〜11.2%)。
【0102】
本発明のアルキルグリコシドまたはスクロースエステルには、現在知られている、または将来発見される、あらゆる化合物が含まれる。本発明のアルキルグリコシドおよび/またはサッカリドアルキルエステルとの混和に特によく適している薬物には、他の方法によって投与するのが難しいもの、例えば、胃腸(GI)管の中で分解される薬物、またはGI管から吸収されないもの、または注射などの伝統的な方法の代わりに眼、鼻、鼻涙管、吸入もしくはCSF送達経路を介して自己投与することが可能な薬物がある。いくつかの具体的な例には、ペプチド、ポリペプチド、タンパク質、核酸および他の高分子、例えば、インスリンおよびカルシトニン、エンケファリン、グルカゴンなどのペプチドホルモン、ならびにトルブタミドおよびグリブリドなどの血糖降下薬、ならびに経腸経路によってはほとんど吸収されない作用物質、例えば抗真菌薬の1つであるグリセオフルビンなどが含まれる。他の化合物には、例えば、ニコチン、インターフェロン(例えば、α、β、γ)、PYY、GLP-1、合成エキセンディン-4(Exenatide)、副甲状腺ホルモンおよびヒト成長ホルモン、または他の低分子量ペプチドおよびタンパク質が含まれる。
【0103】
または、薬物の生物学的利用能を、肝臓による薬物の初回通過クリアランスのレベルを測定することによって決定することもできる。鼻腔内に、または口腔(舌下細胞もしくは頬粘膜細胞)を介して投与された、本発明のアルキルグリコシドおよび/またはサッカリドアルキルエステル組成物は、肝門脈血系には入らず、その結果、肝臓による初回通過クリアランスを回避する。肝臓によるこれらの製剤の初回通過クリアランスの回避については本明細書で記載している。「初回通過肝クリアランス」という用語は、薬物が、門脈血から肝臓を通って体循環へと至るその初回通過の間に肝臓によって除去される程度のことである。これはまた、初回通過代謝または初回通過抽出とも呼ばれる。
【0104】
身体からの薬物排出の経路には、薬物が変化することのない、腎臓による排泄;および、薬物が代謝される、肝臓による排出という主に2つがある。これらの2つの経路間のバランスは、この2つの過程の相対的効率に依存する。本発明は本明細書において肝臓または肝クリアランスによる排出について述べる。初回通過肝クリアランスは、Birkett et al. (1990 and 1991)によって記載されており、これはその全体が参照により組み入れられる。Birkett et al., Aust Prescr, 13(1990):88-9;およびBirkett et al., Austra Prescr 14:14-16 (1991)。
【0105】
体循環からの薬物を運ぶ血液は門脈を介して肝臓に入り、続いて肝臓が、その薬物のある特定のパーセンテージまたは比(すなわち、0.5または50%)を抽出する。残った残留物(すなわち、0.2または20%)は肝静脈を介して再び体循環に入る。薬物のこのクリアランスの比率は肝抽出比と呼ばれる。これは肝臓を通っての血液の初回通過の間に非可逆的に除去(または抽出)される、血液中の薬物の割合である。薬物が全く抽出されないならば肝抽出比はゼロである。その反対に、薬物が肝臓を通る初回通過において高度に抽出されるならば、肝抽出比は100%または1.0という高さになることもある。一般に、肝臓による薬物のクリアランスは、その場合、その薬物の肝臓への送達(または肝血流)の速度およびその薬物の除去の効率(または抽出比)に依存する。
【0106】
したがって、肝クリアランスを決定するために用いられる最終的な式は、以下である:
(肝クリアランス−血流)=(非結合率*固有クリアランス)/血流+(非結合率*固有クリアランス)(1)
【0107】
薬物の「非結合率」は、薬物が血液中のタンパク質および細胞とどの程度強固に結合するかに依存する。一般に、血液から肝細胞内への拡散に供されうるのは、この非結合(または遊離)薬物のみである。肝血流およびタンパク質結合がない場合、「固有クリアランス」とは、肝臓がその薬物を除去(または代謝)する能力のことである。生化学の用語では、これは特定の薬物基質に対する肝酵素活性の指標である。ところが、固有クリアランスが高くても、薬物は肝臓内に送られたものよりも迅速には取り除かれない可能性がある。簡単に言えば、2つの状況がある:肝酵素活性が非常に高い場合または非常に低い場合(すなわち、高い抽出比または低い抽出比)。
【0108】
肝酵素活性が低い場合、この式は以下のものに単純化される:
肝クリアランス=非結合率*固有クリアランス(2)
【0109】
クリアランスはこの場合、血流には依存せず、その代わり、血液中でのタンパク質結合の度合いおよびその薬物に向かう薬物代謝酵素の活性に直接依存する。
【0110】
対照的に、肝酵素活性が高い場合、この式は以下になる:
肝クリアランス=肝血流(3)
【0111】
このケースでは、酵素の活性が非常に高いため、肝臓はそこに送られた薬物の大部分を除去し、抽出比が高くなる。したがって、実際の肝クリアランスを決定する唯一の要因は、肝臓への薬物の供給(または肝血流)の速度である。
【0112】
初回通過肝クリアランスは、薬物の抽出の小さな変化でさえも生物学的利用能の大きな変化を引き起こす可能性があるため、重要である。例えば、薬物Aの、それが体循環に到達する時点までの経口投与による生物学的利用能が20%であり、静脈内投与による同じ薬物Aのそれが100%であり、他の複雑化要因が存在しなければ、故に、同程度の血漿中濃度を達成するためには経口用量は静脈内用量の5倍でなければならないと考えられる。
【0113】
第2に、肝酵素活性が非常に高いいくつかの場合には、体循環へと直接に薬物通過が行われ、初回通過肝クリアランスがすべて回避されるように薬物製剤を設計すべきである。例えば、鼻腔内、舌下、頬側、直腸、膣などに投与された薬物は、体循環に直接入り、肝臓によって部分的または完全に抽出されることになる肝門脈血循環には入らない。または、薬物を上記の手段によって投与することができない場合には、胃の中(すなわち、高度の酸性環境)での薬物の放出を防止する少なくとも1つの腸溶性コーティング層を有する錠剤を提供する。したがって、本発明の1つの目的は、薬物をこれらの代替的経路を用いて投与することである。
【0114】
さらに、初回通過肝クリアランスは、多くの患者が複数の薬物レジメンを受けており、このことが肝酵素活性を上昇または低下させる薬物相互作用を引き起こし;その結果、関心対象の薬物の代謝を増加または減少させる(肝抽出比を増加または減少させる)可能性があることからも、重要な要因である。
【0115】
それ故に、本発明の治療用組成物は、体循環系に直接投与し、初回通過肝クリアランスを回避させることができる。初回通過クリアランスを回避することで、より多く薬物が系に供されることが保証される。言い方を変えれば、初回通過肝クリアランスを回避することにより、薬物の生物学的利用能は高くなる。
【0116】
本発明はまた、対象の循環器系への低分子化合物の吸収を増加させる方法であって、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を投与する段階を含む方法にも関する。
【0117】
組成物製剤は、経口投与(経口調合物)、外部投与(例えば、軟膏)、注射(注射用調合物)および粘膜投与(例えば、頬側および坐薬)などといった投与経路に従って適切に選択される。例えば、添加剤(例えば、デンプン、ラクトース、結晶セルロース、乳酸カルシウム、メタケイ酸アルミン酸マグネシウムおよび無水ケイ酸塩)、崩壊剤(例えば、カルボキシメチルセルロースおよびカルボキシメチルセルロースカルシウム)、潤滑剤(例えば、ステアリン酸マグネシウムおよびタルク)、コーティング剤(例えば、ヒドロキシエチルセルロース)および香味剤を、経口製剤および粘膜用製剤に対して用いることができる;一方、水性注射剤を形成させることのできる溶解補助剤および補助的溶解補助剤(例えば、注射用蒸留水、生理的食塩水およびプロピレングリコール)、懸濁剤(例えば、ポリソルベート80などの表面活性物質)、pH調節剤(例えば、有機酸およびその金属塩)ならびに安定剤が注射剤に対して用いられる;ならびに、水性または油性の溶解補助剤および補助的溶解補助剤(例えば、アルコールおよび脂肪酸エステル)、粘着防止剤(例えば、カルボキシビニルポリマーおよびポリサッカリド)ならびに乳化剤(例えば、表面活性物質)が、外部用薬剤に対して用いられる。薬物およびアルキルグリコシドは、投与の前に、上記の添加剤、崩壊剤、コーティング用ポリマー、溶解補助剤、懸濁剤などと混和、混合もしくは配合することもでき、またはそれらを任意の順序で逐次的に投与することもできる。それらを投与前に混合することが好ましい。
【0118】
「粘膜送達強化剤」という用語には、化合物(例えば、生物活性化合物)の放出または溶解性(例えば、製剤送達媒体からの)、拡散速度、透過の能力およびタイミング、取り込み、滞留時間、安定性、有効半減期、ピーク濃度または持続濃度レベル、クリアランスならびに他の所望の粘膜送達特性(例えば、血流または中枢神経系といった、送達部位、または選択された活性標的部位で測定されるもの)を強化する作用物質が含まれる。粘膜送達の強化は、例えば、化合物の拡散、輸送、存続性もしくは安定性を高めること、膜流動性を高めること、細胞内または傍細胞透過を調節するカルシウムおよび他のイオンの利用能または作用をモジュレートすること、粘膜成分(例えば、脂質)を可溶化すること、粘膜組織における非タンパク質およびタンパク質のスルフヒドリルレベルを変化させること、粘膜表面を越える水の流れを増加させること、上皮結合生理機能をモジュレートすること、粘膜上皮を覆う粘液の粘度を低下させること、粘液線毛クリアランス速度を低下させること、ならびに他の機序によるものを含む、種々の機構の任意のものによって生じうる。
【0119】
例示的な粘膜送達強化剤には、以下の作用物質およびそれらの任意の組み合わせが含まれる:
(a)凝集阻害剤;
(b)電荷修飾剤;
(c)pH制御剤;
(d)分解酵素阻害剤;
(e)粘液溶解剤または粘液除去剤;
(f)繊毛静止剤;
(g)以下から選択される膜透過強化剤:
(i)表面活性物質;(ii)胆汁酸塩;(ii)リン脂質添加物、混合ミセル、リポソームまたは担体;(iii)アルコール;(iv)エナミン;(v)NOドナー化合物;(vi)長鎖両親媒性分子;(vii)小型の疎水性透過強化物質;(viii)ナトリウムまたはサリチル酸誘導体;(ix)アセト酢酸のグリセロールエステル;(x)シクロデキストリンまたはβ-シクロデキストリン誘導体;(xi)中鎖脂肪酸;(xii)キレート剤;(xiii)アミノ酸またはその塩;(xiv)N-アセチルアミノ酸またはその塩;(xv)選択された膜成分に対する分解作用のある酵素;(ix)脂肪酸合成の阻害薬;(x)コレステロール合成の阻害薬;および(xi)(i)〜(x)に列挙された膜透過強化剤の任意の組み合わせ;
(h)上皮結合生理機能の調節剤;
(i)血管拡張薬;
(j)選択的輸送強化剤;ならびに
(k)化合物と有効に組み合わされ、会合し、含有され、封入され、または結合し、その結果、鼻粘膜送達の強化のための化合物の安定化をもたらす、安定化性の送達媒体、担体、粘膜付着剤、支持体または複合体形成種であって、化合物と鼻腔内送達強化剤との製剤化が対象の血漿における化合物の生物学的利用能の増大をもたらすもの。
【0120】
そのほかの粘膜送達強化剤には、例えば、クエン酸、クエン酸ナトリウム、プロピレングリコール、グリセリン、アスコルビン酸(例えば、L-アスコルビン酸)、二亜硫酸ナトリウム、エチレンジアミン四酢酸(EDTA)二ナトリウム、塩化ベンザルコニウム、水酸化ナトリウム、およびそれらの混合物が含まれる。例えば、EDTAまたはその塩(例えば、ナトリウム塩またはカリウム塩)を、アルキルサッカリド保存料を含む組成物の重量比で約0.01%〜2%の範囲にわたる量で用いる。
【0121】
本発明の治療薬または薬物は、医学的または診断的に有用で、サイズが小さいかもしくは中程度である、例えば、最大で約15kD、30kD、50kD、75kDなどであるペプチドもしくはタンパク質、または約1〜300個もしくはそれ以上のアミノ酸を有するタンパク質でありうる。本発明の方法はまた、小分子、例えば、3kD未満または1.5kD未満の分子量を有する有機化合物の使用も予期している。
【0122】
本発明による薬物吸収の改善の機序は一般に該当性があり、そのようなすべてのペプチドまたはタンパク質に当てはまるべきであるが、それらの吸収が改善される度合いは、ペプチドまたはタンパク質の分子量(MW)および物理化学的性状、ならびに用いる特定の強化物質によって異なりうる。ペプチドまたはタンパク質の例には、バソプレシン、バソプレシンポリペプチド類似体、デスモプレシン、グルカゴン、コルチコトロピン(ACTH)、ゴナドトロピン、カルシトニン、インスリンのC-ペプチド、副甲状腺ホルモン(PTH)、成長ホルモン(HG)、ヒト成長ホルモン(hGH)、成長ホルモン放出ホルモン(GHRH)、オキシトシン、コルチコトロピン放出ホルモン(CRH)、ソマトスタチンまたはソマトスタチンポリペプチド類似体、ゴナドトロピンアゴニストまたはゴナドトロフィンアゴニストポリペプチド類似体、ヒト心房性ナトリウム利尿ペプチド(ANP)、ヒトチロキシン放出ホルモン(TRH)、濾胞刺激ホルモン(FSH)およびプロラクチンが含まれる。
【0123】
本発明の1つの好ましい組成物は、Exenatide(またはエキセンディン-4)およびアルキルグリコシドであるペプチド薬物である。Exenatideは合成型のエキセンディン-4であり、Amylin(商標)Pharmaceuticalsによる臨床試験に用いられている。エキセンディン-4は、インクレチン模倣薬またはホルモンとして知られる新たなクラスの治療用医薬品の最初のものである低分子量ペプチドである。インクレチンホルモンとは、インスリン分泌の強力な刺激物質として、例えば、胃抑制ポリペプチド(GIP)、グルカゴン様ペプチド-1(GLP-1)、またはExenatideもしくはエキセンディン-4、またはそれらの等価物などとして作用する、さまざまな胃腸(GI)ホルモンおよび因子の任意のもののことである。
【0124】
エキセンディン-4は、アメリカドクトカゲ(Gila Monster Lizard)の唾液分泌物から単離された、天然に存在する39アミノ酸のペプチドである。Eng et al., "Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas," J. Biol. Chem. 267(15):7402-7405(1992)。Exenatideは、グルカゴン様ペプチドまたはGLP-1と類似したグルコース低下作用を示す。Exenatideは、2型糖尿病を有する多くの人々の、未だに満たされていない重要な医学的需要に対処する能力に関して試験が実施中である。臨床試験により、Exenatide治療が血糖を標的レベルに向けて低下させること、および体重減少を伴うことが示唆されている。Exenatide治療によって観察された血糖コントロールに対する効果は、天然に存在するインクレチンホルモンGLP-1のものに類似したいくつかの作用に起因する可能性が高い(実施例7参照)。これらの作用には、血糖レベルの上昇に応答してインスリンを産生する身体の能力を刺激すること、食後のグルカゴン放出を阻害すること、および栄養分が血流中に吸収される速度を遅らせることが含まれる。動物試験において、Exenatide投与は、2型糖尿病が進行するにつれて機能しなくなる膵臓内のインスリン産生細胞であるβ細胞の保持および新たなβ細胞の形成をもたらした。
【0125】
Exenatide、インクレチン模倣薬またはそれらの等価物は、不安定型糖尿病、化学的糖尿病もしくは耐糖能障害、妊娠性糖尿病、尿崩症、中枢性尿崩症、腎性尿崩症、下垂体性尿崩症、潜在性糖尿病、脂肪萎縮性糖尿病、若年発症成人型糖尿病(MODY)、真性糖尿病(DM)、成人発症型真性糖尿病(2型DM)、インスリン依存性真性糖尿病(IDDMまたは1型DM)、インスリン非依存性真性糖尿病(NIDDM)、若年性もしくは若年発症型真性糖尿病、ケトーシスを起こしやすい真性糖尿病、ケトーシス抵抗性真性糖尿病、栄養不良関連真性糖尿病(MRDM)、熱帯性もしくは熱帯性膵性真性糖尿病、真性糖尿病、発症前糖尿病、またはさまざまな薬物によって誘発される糖尿病、例えばサイアザイド糖尿病、ステロイド糖尿病など、またはアロキサン糖尿病および穿刺性糖尿病を非限定的に含むさまざまな糖尿病動物モデルを非限定的に含む、さまざまな型の糖尿病を治療するために用いることができる。
【0126】
もう1つの局面において、本発明の治療用組成物は、肥満を治療するために用いられる。肥満は、成人および青少年の両方においてよくみられる問題である。例えば、PYY3-36(またはAC162352)は、食欲を低下させる上で決定的な役割を果たすホルモンである。消化管ホルモン断片ペプチドPYY3-36(PYY)は、正常体重の対象に注入されると食欲および食物摂取を低下させる。脂肪細胞ホルモンであるレプチンと同様に、PYYは視床下部における食欲回路をモジュレートすることによって食物摂取を低下させる。しかし、肥満患者では、レプチンの作用に対する抵抗性があり、その結果、レプチンの治療有効性が制限される。さらに他の研究も、PYYが食物摂取を低下させることを示している。PYYの注射は、彼らが食事するのを通常よりも平均30%減らし、その結果として体重減少が起こることが明らかになっている。それ故に、PYY 3-36は、肥満に対する治療薬として可能性がある。Amylin(商標)Pharmaceuticalsは、2003年にPYY 3-36の新薬臨床試験開始申請を提出している。
【0127】
本発明の方法によって吸収を増加させることのできる化合物には、他の方法によって投与することが困難な、特定の薬物または治療用の化合物、分子もしくは作用物質の中の、現在知られている、または将来発見される、任意の化合物が含まれる。例えば、胃腸(GI)管の中で分解される薬物、またはGI管からはあまり吸収されないもの、または対象が、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して、注射などの伝統的な自己投与方法よりも容易に自分で投与しうると考えられる薬物が含まれる。いくつかの具体的な例には、ペプチド、ポリペプチド、タンパク質および他の高分子、例えば、インスリンおよびカルシトニン、エンケファリン、グルカゴンなどのペプチドホルモン、ならびにトルブタミドおよびグリブリドなどの血糖降下薬、ならびに経腸経路によってはほとんど吸収されない作用物質、例えば抗真菌薬の1つであるグリセオフルビンなどが含まれる。他の化合物には、例えば、ニコチン、インターフェロン(例えば、α、β、γ)、PYY、GLP-1、合成エキセンディン-4(Exenatide)、副甲状腺ホルモン(PTH)、およびヒト成長ホルモン、または他の低分子量ペプチドおよびタンパク質が含まれる。
【0128】
本明細書において考察した通り、薬物が消化管の頬側、舌下、口咽頭および食道前胃部分を通過する際に、さまざまな量の薬物が吸収される可能性がある。薬物の大部分は胃内に移行し、錠剤、カプセル剤または液剤などの腸溶性剤形が吸収される通常の胃吸収様式で吸収される。薬物が腸から吸収されると、薬物は肝臓内に直接運ばれ、そこで、その固有の化学構造に応じて、肝細胞における通常の解毒プロセスを遂行する酵素によって代謝されるかまたは排出される。以前に考察したように、この排出は、肝臓における「初回通過」代謝または「初回通過」効果と呼ばれる。ほとんどの場合は元の薬物と比較して実質的または完全に不活性である、その結果生じた代謝産物は、多くの場合は血流中を循環している状態で認められ、その後に尿中および/または便中に排出される。
【0129】
本発明の諸局面は、急速分散性剤形に含められた場合に、ある種のアルキルサッカリドの添加が、初回通過効果を受ける薬物の割合をモジュレートし、それ故に、一定量の薬物がより大きな臨床的有益性を発揮することを可能にする、またはより少量の薬物が、他の点では同様なより高い用量と比較して同程度の臨床的有益性を達成することを可能にするという発見に基づく。
【0130】
本発明の別の諸局面は、急速分散性剤形に含められた特定のアルキルサッカリドの量を増加させるかまたは減少させることが、薬物の吸収部位を変更またはモジュレートさせて、頬側組織を通して吸収される薬物の割合を、消化管の他の部位と比較してそれぞれ増加させるかまたは減少させるという発見に基づく。薬物作用の開始を早めながらも、標準的な経口錠剤でみられる通常の比較的長いTmaxは保持することが望ましい場合には、薬物の一部分は頬側で直ちに吸収されて迅速開始をもたらすが、その残りはより緩徐な胃吸収過程で吸収されるように頬側吸収を弱めるために、アルキルサッカリドの含有量を減らすことができる。このようにして、最大または最大に近い頬側吸収を生じることが実験によって見いだされているアルキルサッカリドの濃度よりも低い、例えば20%低いアルキルサッカリド濃度を選択することによって、「全身薬物レベル」-時間グラフにおいて、臨床的に望ましいと判断される、全体的にみてより幅広い吸収ピークを達成することができることが見いだされた。
【0131】
以下の実施例においてさらに考察するように、急速分散性錠剤に対する、特定のアルキル鎖長を有するある種のアルキルサッカリドの添加は、前胃薬物吸収の薬物動態を有益な様式で変化させる。具体的には、約0.2%〜0.3%、0.3%〜0.4%、0.4%〜0.5%、0.5%〜1.0%、1.0%〜2.0%、2.0%〜3.0%、3.0%〜4.0%、4.0%〜5.0%、5.0%〜6.0%、6.0%〜7.0%、7.0%〜8.0%、9.0%〜10.0%、および10%を上回るアルキルグリコシドの組み込みは、前胃薬物吸収の薬物動態を有益な様式で変化させる。例示的な態様において、アルキルサッカリドはドデシルマルトシド、テトラデシルマルトシドおよび/またはドデカン酸スクロースであり、これらは急速分散性錠剤形式中に組み込まれると、体循環に入る薬物を増加させて、肝臓における「初回通過」効果によって排出される薬物を減少させる。さらに、最大薬物レベル到達時間は、典型的には1〜6時間からおよそ15〜45分へと著しく短縮される。精神病エピソードを来している攻撃的患者の治療に用いる場合、薬物のこのより迅速な吸収はより迅速な作用開始をもたらし、それは非常に有益であると考えられる。
【0132】
さらに、本発明の他の諸局面は、ある特定の種類の急速溶解性または急速分散性の錠剤を頬と歯肉との間、または口腔内に頬側組織に近接させて置くと、薬物のさらに多くの割合が体循環に直接吸収され、より少ない量がその後に肝臓において初回通過排出を受けるという発見に基づく。最後に、この効果に関して特に好都合な口腔内の位置は、鼻のすぐ下の、上唇の中央部の内側の、唇と歯肉との間であることが発見された。例示的な局面において、これらの種類の急速溶解性投薬製剤は、凍結乾燥または真空乾燥によって調製される。1つの例示的な局面において、投薬製剤は、実質的に多孔性である投薬製剤がもたらされる様式で調製される。
【0133】
「急速分散性剤形」という用語は、口腔内ですべてまたは一部が溶解しうる、あらゆる種類の剤形を範囲に含むものとする。しかし、例示的な局面において、急速分散性剤形は、有効成分の固体の急速分散性ネットワークであり、有効成分および添加剤に対して不活性である水溶性または水分散性の担体マトリックスである。さまざまな態様において、このネットワークは、固体状態にある組成物から溶媒を凍結乾燥または昇華させることによって得ることができ、この組成物は有効成分、アルキルサッカリド、および溶媒中にある担体の溶液を含む。種々の溶媒がこの用途に適することが当技術分野において公知であるが、本発明とともに用いるのに特に適した1つの溶媒は水である。また、水-アルコール混合物を、この混合溶媒中で薬物溶解性が強化される場合には用いることもできる。水難溶性の薬物の場合は、小さな薬物粒子の分散体を、凍結乾燥または昇華の過程において実質的に不溶性の薬の均一な分布を維持させる水性ゲル中に懸濁させることができる。
【0134】
1つの態様において、水性ゲルは、モノ-およびジ-ステアリン酸スクロースならびに/またはテトラデシル-マルトシドなどの選択されたアルキルサッカリドを用いて形成される、参照により本明細書に組み入れられる米国特許出願第60/957,960号に記載された自己集合性ヒドロゲルであってよい。さまざまな局面において、本発明の急速溶解性組成物は、口腔内に置かれてから20秒以内に、好ましくは10秒未満で崩壊する。
【0135】
本発明の急速溶解性製剤に用いるのに適したマトリックス形成性物質は、本出願の全体を通じて記載される。そのような作用物質には、ゼラチン、コラーゲン、デキストリンならびにダイズ、コムギおよびアメリカオオバコの種子タンパク質などの動物性または植物性タンパク質に由来する材料;アラビアゴム、グアーガム、寒天およびキサンタンなどのゴム類;ポリサッカリド;アルギナート;カラゲナン;デキストラン;カルボキシメチルセルロース;ペクチン;ポリビニルピロリドンなどの合成ポリマー;ならびにゼラチン-アラビアゴム複合体などのポリペプチド/タンパク質またはポリサッカリド複合体が含まれる。例示的な局面においては、ゼラチン、特に魚肉ゼラチンまたはブタゼラチンが用いられる。
【0136】
本明細書に記載された急速溶解性投薬製剤には事実上あらゆる薬物を組み込むことができると考えているが、特によく適した薬物には、メラトニン、ラロキシフェン、オランザピンおよびジフェンヒドラミンが含まれる。
【0137】
さらに、本発明の治療用組成物は、非ペプチド性の薬物または治療薬も想定している。例えば、米国特許第5,552,534号には、種々のペプチドの化学的および/または生物的活性を模倣または阻害する非ペプチド性化合物が開示されている。そのような化合物は、テトラヒドロピラニル環などのある特定の種に対して、化合物をペプチドに対して少なくとも部分的に交差反応性にする化学官能基を付属させることにより、生成させることができる。認識されているであろうが、ペプチドを模倣または阻害する化合物は、それに対してさまざまな度合いの交差反応性を有する。ペプチド模倣物を調製するための他の手法は、米国特許第5,550,251号および第5,288,707号に開示されている。上記の米国特許は、それらの全体が参照により組み入れられる。
【0138】
本発明の方法はまた、アルキルグリコシドおよびタンパク質またはペプチドとともに、タンパク質またはペプチドが体内のその活性部位に活性状態で到達するのを助けるために(すなわち、タンパク質が依然として適正に機能しうる程度に分解が最小限になるように)、プロテアーゼ阻害薬またはペプチダーゼ阻害薬、アプロチニン、ベスタチン、α1プロテイナーゼ阻害薬、ダイズトリプシン阻害薬、組換え分泌性白血球プロテアーゼ阻害薬、カプトプリルおよび他のアンジオテンシン変換酵素(ACE)阻害薬、ならびにチオルファンの投与も含むことができる。プロテアーゼ阻害薬またはペプチダーゼ阻害薬をアルキルグリコシドおよび薬物と混合し、続いて投与することもでき、またはそれをグリコシドもしくは薬物の投与の前もしくは後のいずれかに別個に投与することもできる。
【0139】
本発明はまた、対象における血糖レベルを低下させる方法であって、インスリン、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の血糖低下量を投与し、それにより、インスリンの吸収を増加させ、かつ血糖レベルを低下させる段階を含む方法も提供する。そのような組成物の「血糖低下量」とは、本明細書において教示されるように、血糖レベルを低下させる効果を生じさせることのできる量のことである。血糖を正常血糖範囲または正常血糖に近い範囲に減少させる量が好ましい。血糖レベルの持続的低下を引き起こす量も好ましい。さらにより好ましいのは、血糖レベルを低下させることによって真性糖尿病(DM)を含む糖尿病を治療するのに十分な量である。したがって、本方法は真性糖尿病を治療するために用いることができる。好ましいアルキルグリコシドは、上記のものおよび実施例において例示しているものと同じである。
【0140】
グルカゴン、ならびに少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む血糖上昇量を投与することにより、対象における血糖レベルを上昇させる方法も同じく提供される。組成物がインスリンを含む場合には、血流中でインスリンの公知の効果を引き起こすため、すなわち、対象における血糖レベルを低下させるためにそれを用いることができる。そのような投与は、真性糖尿病または関連疾患を治療するために用いることができる。そのような組成物におけるグルカゴンの「血糖上昇量」とは、血糖レベルを上昇させる効果を生じさせることのできる量のことである。ある好ましい量は、血糖を正常血糖範囲または正常血糖に近い範囲に増加させるものである。もう1つの好ましい量は、血糖レベルの持続的上昇を引き起こすものである。さらにより好ましいのは、血糖レベルを上昇させることによって低血糖症を治療するのに十分な量である。したがって、この方法は低血糖症を治療するために用いることができる。好ましいアルキルグリコシドは、上記のものおよび実施例において例示しているものと同じである。
【0141】
同様に、この組成物がグルカゴンを含む場合には、血流中でグルカゴンの公知の効果を引き起こすため、すなわち、対象における血糖レベルを上昇させるためにそれを用いることができる。したがって、そのような投与は、低血糖クリーゼを含む低血糖症を治療するために用いることができる。
【0142】
本発明はまた、脳脊髄液(CSF)に治療薬を投与する段階を含む、神経障害を改善するための方法も提供する。「神経障害」という用語は、適切な治療薬に反応する、脳、脊柱、および髄膜などの関連組織における任意の障害を表す。神経障害を改善する本発明の治療薬の驚くべき能力は、治療薬の提示を脳室腔内に存続させることに起因する。治療薬を神経障害の領域に存続させることを可能にする本発明の方法の能力は、そのような障害を治療するための特に有効な手段を提供する。
【0143】
しかし、治療を必要とする任意の特定の対象に対する具体的な用量レベルおよび投薬頻度はさまざまであってよく、用いる具体的な化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全般的健康状態、性別、食事、投与の様式および回数、排泄速度、薬物併用、特定の病状の重症度、ならびに治療法を受ける宿主を含む種々の要因に依存することは理解されるであろう。しかし、一般に、投薬は、その具体的な化合物の公知の投与方法にとって典型的であるものに近似すると考えられる。例えば、インスリンの鼻腔内投与に関して、近似的な投与量は約0.5単位/kgのブタ・レギュラーインスリンであると考えられる(Moses et al.)。血糖レベルに影響を及ぼす化合物の投与量は、最適には、適正なグルコースレベル、例えば約5〜6.7mMの正常範囲を達成するために必要なものであると考えられる。さらに、適切な量が、本明細書における教示を考慮して、定型的なものに留まる検査のみを用いて、当業者によって決定されてもよい(実施例を参照)。
【0144】
さらに、本発明の組成物を、滴剤、噴霧剤、エアロゾルおよび持続放出形式からなる群より選択される形式で投与することもできる。噴霧剤およびエアロゾルは、適切なディスペンサーの使用によって実現することができる。持続放出形式は、眼インサート、侵食性微粒子、膨張性粘膜付着剤粒子、pH感受性微粒子、ナノ粒子/ラテックス系、イオン交換樹脂ならびに他のポリマー性ゲルおよびインプラントでありうる(Ocusert, Alza Corp., California;Joshi, A., S. Ping and K. J. Himmelstein, 特許出願第WO 91/19481号)。これらの系は、吸収性表面との長期にわたる薬物接触を維持し、流出および非生産的な薬物喪失を防止する。
【0145】
さまざまな局面において、5-HT受容体アゴニストを含む本組成物の特性をIMITREX(登録商標)溶液と比較する。本明細書で用いる場合、溶液は以下の通りである。IMITREX(登録商標)(コハク酸スマトリプタン)注射剤は、選択的5-ヒドロキシトリプタミン1受容体サブタイプアゴニストである。コハク酸スマトリプタンは、化学的にはコハク酸3-[2-(ジメチルアミノ)エチル]-N-メチル-インドール-5-メタンスルホンアミド(1:1)と称される。IMITREX(登録商標)注射剤は、透明で、無色ないし淡黄色で、無菌性で、発熱物質を含まない皮下注射用の溶液である。IMITREX(登録商標)注射剤の8mg/mL溶液の各0.5mLは、コハク酸塩としての4mgのスマトリプタン(基剤)および3.8mgのUSP塩化ナトリウムを、USP注射用蒸留水の中に含む。IMITREX(登録商標)注射剤の12mg/mL溶液の各0.5mLは、コハク酸塩としての6mgのスマトリプタン(基剤)および3.5mgのUSP塩化ナトリウムを、USP注射用蒸留水の中に含む。どちらの溶液のpH範囲もおよそ4.2〜5.3である。どちらの注射剤のオスモル濃度も291mOsmolである。IMITREX(登録商標)(コハク酸スマトリプタン)鼻噴霧薬は、5mgまたは20mgのスマトリプタンを、NFリン酸二水素カリウム、USP無水リン酸二ナトリウム、NF硫酸、NF水酸化ナトリウムおよびUSP精製水を含む100μL単位用量の水性緩衝溶液中に含む。溶液のpHはおよそ5.5である。溶液のオスモル濃度は、5mgおよび20mgのIMITREX(登録商標)鼻噴霧薬でそれぞれ372または742mOsmolである。
【0146】
本明細書で用いる場合、5-HT受容体には、5-HT1から5-HT7までの受容体ファミリーの任意の受容体が含まれる。そのような受容体には、5-HT1A、5-HT1B、5-HT1D、5-HT1E、5-HT1F、5-HT2A、5-HT2B、5-HT2C、5-HT3、5-HT4、5-HT5A、5-HT6および5-HT7が含まれる。
【0147】
本発明を以下の実施例においてより詳細に説明するが、当業者にはさまざまな変更物および変形物が明らかであると考えられるため、これらは例示に過ぎないことを意図している。以下の実施例は、本発明を限定するのではなく、それを例示することを意図している。
【実施例】
【0148】
実施例1
アルキルグリコシドおよび/またはスクロースエステル製剤は粘膜の刺激も破壊も引き起こさない
鼻粘膜は高度に血管が発達しており、それ故に高度の薬物透過のために最適である。その上、鼻粘膜を通しての薬物の吸収は、中枢神経系(CNS)にとって利用可能である。薬物の局所適用は望ましいものの、この投与方法の課題は粘膜刺激性である。
【0149】
市販の一般用医薬品(OTC)鼻腔用食塩水中のアルキルグリコシド(0.125% TDM)からなる製剤を、インビボでヒト鼻上皮に対して1カ月を上回る期間にわたって投与した。この0.125% TDM製剤を、対照、すなわち同じ市販(OTC)の鼻腔用食塩水と同じ期間にわたって比較した。その結果は、33日間にわたる毎日のTDM投与(すなわち、試験の継続期間)の間および後に、鼻粘膜に対して観察可能な刺激はみられなかったことを示している(非提示データ)。したがって、本発明の組成物は無毒性および非刺激性であり、慢性および進行中の疾患を有する患者にとって有益である反復的および長期的な鼻腔内投与を実現させる。
【0150】
同様の試験を、スクロースエステルの1つであるドデカン酸スクロースを用いて行った。ドデカン酸スクロースをインビボでヒト鼻上皮に対して投与したところ、47日間(すなわち、試験の継続期間)の間および後に、観察可能な刺激は検出されなかった(非提示データ)。したがって、これらの結果は、本発明のアルキルグリコシドおよびスクロースエステルが無毒性であり、長期間にわたって毎日投与した場合に粘膜刺激を引き起こさないことを示している。
【0151】
実施例2
アルキルグリコシドおよび/またはスクロースエステル組成物は、薬物の生物学的利用能を高め、薬物の生物学的利用能のばらつきを減少させることにより、薬物を安定化する
アルキルグリコシドの安定性は、一部には、炭素原子の数、またはアルキル鎖および他の長いアルキル鎖の長さに依存し、テトラデシルマルトシド(TDM)が最大の効果を有する;しかし、DDMを含む他の高度に分枝したアルキル鎖も安定化効果を有する。高いアルキルグリコシド-薬物比の好ましさを記載しているHovgaard-1とは対照的に、本発明はこの比がはるかに低いことを示している。例えば、重量比で約0.01%〜約6%の範囲にあるアルキルグリコシドは、薬物の優れた安定化をもたらす;一方、Hovgaard-1は、はるかに高いアルキルグリコシド-薬物比(10:1および16:1)でのみ安定化が達成されたことを示している。さらにより興味深いことに、約0.01%〜約6%の範囲にある本発明のアルキルグリコシドでは、生物学的利用能が高まっていた(図1参照)。これは、高いアルキルグリコシド比(10:1および16:1)で相対的に低い生物学的利用能(0.5〜1%)を示したHovgaard-2とは極めて対照的である。
【0152】
図1は、アルキルグリコシド(TDM)の存在下および非存在下における薬物MIACALCIN(登録商標)(Novartis社のサケカルシトニン)の生物学的利用能を比較しているグラフである。MIACALCIN(登録商標)は鼻噴霧薬であり、鼻上皮または鼻粘膜に対して直接投与される。図1は、アルキルグリコシドを伴うMIACALCIN(登録商標)をラットに投与した場合と比較して、アルキルグリコシドを伴わないMIACALCIN(登録商標)がヒトにおいて極めて低い生物学的利用能レベルを有する(MIACALCIN(登録商標)製品仕様書挿入物)ことを示している。より具体的には、0.125%および0.250%のアルキルグリコシド(TDM)を伴うMIACALCIN(登録商標)の鼻腔内送達は、それぞれ約43%〜約90%の生物学的利用能をもたらした。アルキルグリコシドを伴わないMIACALCIN(登録商標)の鼻腔内投与の生物学的利用能は、ヒトでは約3%に過ぎず、ラットでは検出不能であり、このことはラットがヒトにおける鼻腔内薬物吸収を評価するための厳格なモデルであることを示唆している。このように、本発明のアルキルグリコシドは薬物の吸収を強化し、その生物学的利用能を高める。
【0153】
さらに、薬物の生物学的利用能を高めることに加えて、本発明のアルキルグリコシド組成物は、薬物の生物学的利用能のばらつきを効果的に低下させる。図1は、アルキルグリコシド(0.125%または0.25%)を伴うMIACALCIN(登録商標)の鼻腔内への投与は+/-8%という生物学的利用能のばらつきを有し、一方、アルキルグリコシドを伴わない場合の生物学的利用能のばらつきは0.3%〜30%すなわち2桁の変化であることを示している。生物学的利用能の増大および生物学的利用能のばらつきの低下は、患者間変動も同じく減少することを確かなものにする。図1に示されている結果は鼻腔内への投与であるが、同様の結果は、経口、頬側、膣、直腸送達などについても、さらには異なるアルキルグリコシド濃度でも期待される。
【0154】
このように、当技術分野とは対照的に、本発明のアルキルグリコシド組成物は、約0.01%〜約6%の範囲で、生物学的利用能の増大および生物学的利用能のばらつきの低下をもたらす。このことは他には報告されていない。
【0155】
実施例3
アルキルサッカリド+インスリンの眼投与はインビボで血糖低下効果を生じさせる
正常ラットに、それらの血糖レベルを上昇させるためにキシラジン/ケタミンの混合物による麻酔を施した。麻酔に反応して起こるD-グルコースレベルの上昇は、薬物投与、例えばインスリンを含む点眼薬の全身血糖低下作用を測定するための最適な系を与える。この動物モデルは、糖尿病性の動物およびヒトで認められる高血糖状態によく似ている。実験動物群では、麻酔ラットにインスリンを含む点眼薬を投与する。実験群による血糖レベルを、インスリンを伴わない点眼薬を投与した麻酔ラットと比較する。血糖レベルの変化および全身反応の違いは、投与経路、例えば眼経路を介して吸収されたインスリンの降下を反映する。
【0156】
成体雄性Sprague-Dawleyラット(250〜350g)に飼料を自由に摂取させ、10:00a.m.〜3:00p.m.の間に実験を実施した。腹腔内(IP)に投与したキシラジン(7.5mg/kg)およびケタミン(50mg/kg)混合物によってラットに麻酔を施し、50〜90分間かけて安定させた後に点眼薬を投与した。キシラジン/ケタミンによる正常ラットの麻酔は血糖値の上昇を生じさせ、それはインスリンを含む点眼薬の全身血糖低下作用を判定するための最適な状態をもたらす。血中D-グルコース値は、実験全体を通じて尾静脈から5〜10分間隔で血液小滴を採取し、その血液を、グルコメーター試験紙(Chemstrip bG)に、装置(Accu-Chek II, Boehringer Mannheim Diagnostics;Indianapolis, Ind.)に用意された指示に従って適用することによって測定した。麻酔された非糖尿病性ラットにおける血中D-グルコース値は200〜400mg/dlの範囲にわたった。
【0157】
50〜90分間の安定化期間の後に、時間0の時点で、0.2%ブタ・レギュラーインスリンを含むかまたは含まず、試験しようとする0.125%〜0.5%の吸収強化性アルキルグリコシド(例えば、TDM)を含むリン酸緩衝食塩水(PBS)で構成される20μlの点眼薬をラットに投与した。点眼薬は、時間0の時点で、眼を開けたままにしてプラスチック製使い捨てピペットチップを用いて滴下し、プロトコールの全体を通じてラットを加温パッド(37℃)上で水平位に保った。ラットが覚醒の徴候を示した場合には追加の麻酔を施した。ラットの各眼に、各個体につき合計2Uの0.2%(50U/ml)ブタ・レギュラーインスリン(Squibb-Novo, Inc.)を含む(実験)かまたは含まない(対照)リン酸緩衝食塩水、pH 7.4中にある0.125〜0.5%吸収強化物質20μlを投与した。オクチル-β-D-マルトシド、デシル-β-D-マルトシド、ドデシル-μ-D-マルトシド、トリデシル-β-D-マルトシドおよびテトラデシル-β-D-マルトシドは、Anatrace, Inc.(Maumee, Ohio)から入手した。ヘキシルグリコピラノシド、ヘプチルグルコピラノシド、ノニルグルコピラノシド、デシルスクロースおよびドデシルスクロースはCalbiochem, Inc.(San Diego, Calif.)から入手した;サポニン、BL-9およびBrij 78はSigma Chemical Co.(St. Louis, Mo)から入手した。
【0158】
1)食塩水のみ;2)食塩水中の0.2%ブタ・レギュラーインスリンのみ;または3)吸収強化物質のみを含む点眼薬をラットに投与したところ、血中D-グルコースレベルは上昇したまま保たれた。しかし、0.2%ブタ・レギュラーインスリンおよびいくつかのアルキルマルトシド化合物またはアルキルスクロース化合物を含む点眼薬を投与した場合には、血中D-グルコース値の顕著な低下が生じ、これは最長2時間にわたって維持された。0.5%ドデシル-β-D-マルトシド(表I参照)または0.5%デシル-β-Dマルトシド(表III参照)とともに眼投与したインスリンは、血糖レベルの即時的かつ持続な降下をもたらし、これは実験の2時間の継続期間にわたって正常血糖(80〜120mg/dl)または正常血糖に近い(120〜160mg/dl)範囲に維持される。それ故に、少なくとも2種のアルキルマルトシドは、眼経路を介して送達されたインスリンの十分な吸収を達成させて、実験的高血糖動物における血糖レベルの即時的かつ持続的な降下を生じさせるのに有効である。したがって、本発明の表面活性物質組成物は、点眼薬の形態で眼経路を介して送達されたインスリンならびに他のペプチド/タンパク質、例えばグルカゴンおよび高分子薬物およびヘパリンなどの全身吸収を達成するために有用である。
【0159】
他のいくつかのアルキルマルトシドも、0.5%トリデシルマルトシド(表III参照)ならびに0.125%(表II)および0.5%のテトラデシルマルトシドを含むインスリンの眼投与に対する吸収強化物質として有効である。これらの試験は、より長いアルキル鎖(または炭素原子数)を有するアルキルマルトシド、例えば、ドデシル-、トリデシル-およびテトラデシル-β-D-マルトシドがより有効なことを示している。炭素原子数の増加も、より大きな疎水性/親水性構造バランスおよび吸収強化効果に寄与する。アルキル鎖がより短い(炭素原子がより少ない)もの、例えばデシルマルトシド、または全く有しないもの(or no)、例えばオクチルマルトシドは、より弱い吸収強化活性を生じさせる。最も有効なアルキルマルトシドはサポニンなどの他の吸収強化物質で認められるものと同等かまたはそれを上回る効果を生じさせる上、全身吸収後に無毒性生成物に代謝されるというさらなる利点があることが注目される。
【0160】
吸収強化物質としてのアルキルマルトシドが、インスリンと併用された場合に血糖低下効果を生じさせる効果は、0.125〜0.5%の範囲にわたる異なる濃度の効果を検討することによって認められるように用量依存的である。一方、0.5%および0.375%のドデシルマルトシドはインスリンの全身吸収および血糖レベルの低下を達成する上で等しく有効であり、0.25%はより小さくかつより一過性の効果を有し、0.125%は無効であるように思われる(表I)。同様に、トリデシルマルトシドもインスリンと併用された場合に血糖濃度を低下させる上で用量依存的効果を示すが、0.25%の吸収強化物質によって達成された効果でさえも実験の2時間の時間経過にわたって持続する。このように、アルキルマルトシドの用量依存的効果は、それらが眼経路を介したタンパク質吸収の強化を、その作用物質の濃度に比例した段階的な様式で達成することを示唆する。
【0161】
(表I)インスリン+さまざまな濃度のドデシルマルトシドを含む点眼薬の、ラットにおける血糖値(mg/dl)に対する効果

【0162】
アルキルサッカリドの吸収強化効果は、ドデシルスクロース(0.125%、0.25%、0.375%)もインスリンの眼吸収および血糖レベルの低下を生じさせる上で用量依存的効果を示すことから、アルキルマルトシドのみには限定されない。この効果は0.125%アルキルサッカリドでさえも観察される(0分時点の335mg/dl.+-.26mg/dlから、120分時点では150mg/dl+-.44mg/dlに)。0.5%デシルスクロースも血糖レベルを低下させる上では有効であったが、アルキルマルトシドに関して示されているように、分子のアルキル鎖の長さおよびそれ故に疎水性の減少はアルキルスクロース化合物の効力を低下させるように思われる。しかし、0.5%デシルスクロースにより、有意かつ持続的な血糖レベルの低下が達成されている(0分時点の313mg/dl.+-.15mg/dlから、120分時点では164mg/dl+-.51mg/dlに)。2つの異なる二糖モイエティーを有するアルキルサッカリドの吸収強化能力は、それがそれらの活性にとって決定的な化合物の物理化学的性状であること、ならびに他のアルキルサッカリド、例えばドデシルラクトースが、アルキルサッカリド強化剤の代謝性状および無毒性性状を保ちながら吸収強化物質として等しくまたはより有効であるという正しい性状バランスを有することを示唆する。これらのアルキルサッカリドは本発明によって予期されている。
【0163】
アルキルグルコシドを用いた試験も実施した;0.5%ヘキシルグルコシドおよび0.5%ヘプチルグルコシドは眼からのインスリン吸収を促進させるのに無効であったが、0.5%ノニルグルコシドはインスリン吸収および血糖レベル低下(297mg/dlから150mg/dlに)を有効に刺激した。この結果は、アルキル鎖の長さならびに糖質モイエティーがインスリン吸収を有効に強化する上で決定的な役割を果たすことをさらに裏づける。
【0164】
これらの試験に用いたアルキルマルトシドまたはアルキルスクロース作用物質のいずれにおいても、眼表面に対する損傷作用が観察されなかったこと(すなわち、非刺激性)に注目すべきである。さらに、インスリンとの併用下でこれらの作用物質によって生じた即時的かつ持続な血糖低下効果は、これらの吸収強化物質が、それらの非変性的で穏和な表面活性物質性状に一致して、そのホルモンの生物活性に有害な影響を及ぼさないことを示唆する。
【0165】
このように、少なくとも1つのアルキルグリコシドおよび薬物からなる本発明の治療用組成物は安定であり、アルキルグリコシドは薬物の吸収を強化する。
【0166】
実施例4
TDM+グルカゴンの眼投与および鼻腔内投与はインビボで血糖低下効果を生じさせる
以前の実施例により、インスリンなどの薬物を伴った吸収強化物質の点眼薬を介した投与が鼻涙管排液系を介した薬物の有意な吸収をもたらすことが示されたことから、ここでは、鼻腔内投与によるインスリンのアルキルマルトシド、アルキルスクロースおよび同様の作用物質との治療的に有効な投与に関して検討した。
【0167】
インスリンと併用したテトラデシルマルトシド(TDM)は、眼経路による点眼薬を介した場合と同じく、点鼻薬の形態で鼻腔内に投与した場合にも血中D-グルコースレベルの低下を生じさせた。0.2%ブタ・レギュラーインスリンを0.125%テトラデシルマルトシドとともに含む点眼薬を、以前に記載した通りにラットに投与する。この組成物の投与は、血糖レベルの即時的かつ顕著な低下を生じさせる。血糖レベルの低下は、同じ濃度のインスリンを0.5%テトラデシルマルトシドとともに含む点鼻薬の投与によってより一層大きくなる(表II)。このように、アルキルサッカリドと薬物との鼻腔内送達および投与は血糖レベルの低下をもたらす。
【0168】
(表II)0.125%テトラデシルマルトシドを含むインスリン点眼薬および0.5%テトラデシルマルトシドを含む点鼻薬の、ラットにおける血糖値に対する効果
【0169】
実施例5
アルキルサッカリド+インスリンの眼投与はインビボで血糖低下効果を生じさせる
以前の研究により、眼からのインスリン吸収がサポニン、BL-9およびBrij-78によって刺激されることが示されている。BL-9およびBrij-78は眼からのグルカゴンの吸収を刺激するのには無効であり、一方、サポニンは有効である。さまざまな表面活性物質+グルカゴン(30μg)(Eli Lilly, Indianapolis, Indiana)を含む点眼薬を投与したラットにおいて、血中D-グルコースレベルの上昇をモニターすることにより、眼からのグルカゴン吸収を測定した。これらの実験では、キシラジン/ケタミンではなくペントバルビタールナトリウムによってラットに麻酔を施した。この手順の変更は、正常血糖範囲にある基礎血糖レベルをもたらし、眼から吸収されたグルカゴンの血糖上昇作用を容易にモニターすることを可能にした。
【0170】
表面活性物質のみまたはグルカゴンのみを含む点眼薬を投与した対のラットを、表面活性物質+グルカゴンを含む点眼薬を投与したラットと比較した。0.5%サポニン+グルカゴンを含む点眼薬をラットに投与すると血中のD-グルコースのレベルは有意に上昇するが、0.5% BL-9または0.5% Brij-78+グルカゴンを含む点眼薬ではそのような効果は観察されない。興味深いことに、ドデシルスクロース、デシルマルトースまたはトリデシルマルトース+グルカゴンを含む点眼薬を、これらの表面活性物質作用物質+インスリンを含む点眼薬による処置を前もって行ったラットに投与すると、グルカゴンが吸収され、血中D-グルコース値が有意に上昇する(表III)。この結果は、ある種のアルキルサッカリドの眼投与が、グルカゴンおよびインスリンを含む薬物の吸収を強化することを裏づけている。その上、本発明の少なくとも1つのアルキルサッカリドを有する製剤を用いて、低血糖クリーゼを治療することも今や可能である。
【0171】
(表III)インスリンまたはグルカゴン、および0.5%デシルマルトシド、0.5%ドデシルスクロースまたは0.5%トリデシルマルトシドを含む点眼薬の、ラットにおける血糖値に対する効果
【0172】
実施例6
0.25% TDM+インスリンの鼻腔内投与はインビボで血糖レベルを低下させる
薬物または作用物質の鼻腔内投与は、例えばマウスおよびラットなどの動物モデルにおいて、鼻の開口は極めて狭いが、可能である。ここに記載した実験および結果では、麻酔誘発性高血糖モデルを用いた(上記の実施例に記載)。キシラジン-ケタミンを含む腹腔内(IP)注射によって高血糖動物を誘発し、血糖レベルをある期間にわたってモニターした。キシラジン-ケタミン注射の直後に、図2に示されているように血糖レベルの上昇がみられ(●)、血糖レベルは約450mg/dlとなった。血糖レベルの上昇は膵インスリン分泌の阻害が原因で起こる。血糖レベルは、キシラジン-ケタミン注射から30分後に約482mg/dlのピークとなった(図2)。続いて、キシラジン-ケタミン注射のおよそ33分後に、0.25%テトラデシルマルトシド(TDM;またはIntravail A)中の6μLのインスリン(Humalog)を、細長いマイクロピペットチップを用いて鼻腔内に投与し、血糖レベルを約15分間隔でモニターした。0.25% TDM/インスリン組成物の投与後に血糖レベルの急速な低下がみられ、約60分の時点、またはインスリン投与から約30分後には約80mg/dlという低さに達した(図2)。約75分の時点には、血糖レベルは正常血糖マウスにおけるベースラインレベル、または約80〜100mg/dlに段階的に復帰した。
【0173】
以上の結果を、0.25% TDMを含まないインスリンのみ(同じ投与量)によって処置した動物と比較した(図2、○)。インスリンのみによる処置は、約120分のタイムマーク、またはインスリン投与の約110分後までは低下を始めない血糖レベルを示した。さらに、インスリンのみによって処置した動物において観察された血糖レベルは、インスリン+0.25% TDMを投与した動物において観察されたような正常血糖レベルには復帰することはなかった(図2)。
【0174】
このように、これらの結果はやはり、ある種のアルキルグリコシドまたはアルキルサッカリド+インスリンなどの薬物からなる本発明の組成物が血糖レベルを有効に低下させること、およびこれらの効果が薬物投与のすぐ後に測定可能であることを示している。
【0175】
実施例7
0.25% TDM(Intravail A)+エキセンディン-4の鼻腔内投与はインビボで血糖レベルを低下させる
ここに記載した試験には、ob/obマウスモデルを利用した。Friedman, J. M., Nature 404, 632-634 (2000)。すべてのマウスに、耐糖能を判定する目的で2g/kgグルコースのボーラスを腹腔内(IP)注射した。時間0の時点で、実験動物に、約100マイクログラム/kgのエキセンディン-4/0.25% TDM(American Peptide社のエキセンディン-4)を10μlの点鼻薬(図3;▲)として、もしくはIP注射(図3;●)により、または食塩水のみのIP注射(薬物を含まずTDMも含まない;図3;○)による投与を行った。対照動物については事前に実施し、薬物は投与しなかった。この試験の結果は図3に示されている。
【0176】
図3は、マウスにグルコースボーラスを投与した時間0の時点での血糖レベルがさまざまであることから、これらの動物の耐糖能が異なることを示している。時間0時点での耐糖能レベルにかかわらず、グルコースボーラスの注射直後に、3匹のマウスすべてで血糖レベルが上昇した。食塩水のみのIP注射を受けた動物の血糖レベルは、薬物を投与された実験動物ほど迅速には低下しなかった。その上、食塩水のみのIP注射を受けた動物は正常血糖レベルに到達することはなかった(図3、○)。対照的に、実験動物は、エキセンディン-4/TDMの点鼻薬の投与またはエキセンディン-4/TDMのIP注射の後に、血糖レベルの迅速かつ即座の低下を示した。
【0177】
グルコースボーラスの約15〜30分前に投与されたエキセンディン-4(図3中の時間0の前;非提示データ)も、さらにより顕著な血糖低下効果を生じさせたが、これはこのホルモンが吸収されて活性化するまでにはある程度の時間がかかるためである。このように、ヒトの臨床試験が現在行われているエキセンディン-4(またはExenatide)は、本発明のアルキルグリコシドと併用した場合に、高血糖対象の血糖レベルを低下させることにより、高血糖状態を有効に治療する。
【0178】
実施例8
アルキルグリコシド(alkyglycoside)は細菌対数増殖を低下させることによって抗菌活性を有する
カンジダ・アルビカンス(Candida albicans)(ATCC No. 10231)、黒色アスペルギルス(Aspergillus niger)(ATCC No. 16404)、大腸菌(Escherichia coli)(ATCC No. 8739)、緑膿菌(Pseudomonas aeruginosa)(ATCC No. 9027)および黄色ブドウ球菌(Staphylococcus aureus)(ATCC No. 6538)の培養物を、American Type Culture Collection, 10801 University Boulevard, Manassas, VA 20110-2209から入手した。本発明に用いた生菌は、元のATCC培養物から取り出して5回を上回る継代を行っていないものとした。本明細書において記載する場合、1回の継代は樹立培養物からの微生物の新たな培地への移行と定義し、すべての移行を算入した。
【0179】
ATCCから受け取った培養物を、ATTCによって用意された指示に従って蘇生させた。ブロス中で増殖させた細胞を遠心によってペレット化し、1/20倍容積の新たな維持用ブロス中に再懸濁させ、同じ容積の20%(v/v、水中)滅菌グリセロールと混ぜ合わせた。寒天培地上で増殖させた細胞を表面から掻き取り、同じく10%グリセロールブロスを含む維持用ブロス中に入れた。懸濁液の少量アリコートを滅菌バイアルに小分けし、バイアルを液体窒素中または温度が約-50℃を上回らない冷凍庫内で貯蔵した。新たな播種用ストックバイアルが必要になった時点で、それを取り出して、一連の作業用ストック培養物の接種に用いた。続いて、これらの作業用ストック培養物を定期的に用いて(細菌および酵母の場合には毎日)、培養物の接種を開始した。
【0180】
本明細書に記載した培地はすべて、被験微生物として上記に指し示した微生物を用いて増殖促進に関して検査すべきである。
【0181】
本発明のアルキルサッカリドが増殖を阻害するか、すなわち抗菌活性を有するか否かを判定するために、適した体積の固形寒天培地の表面に、指定された微生物のそれぞれの新たに復活させたストック培養物からの接種を行った。接種物培養のための培養条件は、表IVに記載されたものと実質的に同じである。例えば、適した培地には、ダイズ-カゼイン消化物またはサブローデキストロース寒天培地が非限定的に含まれうる。細菌およびC.アルビカンスの培養物は、TS滅菌食塩水を用いて表面増殖物を洗浄し、それを適した容器内に集めて、1mL当たり約1×108コロニー形成単位(cfu)の微生物数を得るために十分なTS滅菌食塩水を添加することによって収集した。黒色アスペルギルスの細胞を収集するためには、0.05%のポリソルベート80を含むTS滅菌食塩水を用い、続いて1mL当たり約1×1O8cfuの数を得るために十分なTS滅菌食塩水を添加した。
【0182】
または、ストック培養生物を任意の適した液体培地(例えば、ダイズ-カゼイン消化ブロスまたはサブローデキストロースブロス)中で増殖させて、細胞を遠心によって収集して洗浄し、TS滅菌食塩水中に希釈して1mL当たり約1×108cfuの微生物を得ることもできる。接種物濃度の推定値は、チャレンジ微生物の濁度測定によって決定した。懸濁液は、それを2時間以内に用いない場合には冷蔵すべきである。1mL当たりの初期cfuの推定値を確かめるためには、各懸濁液における1mL当たりのcfu数を、表IVに列記された培地および微生物回収のインキュベーション時間の条件(例えば、約3〜約7日)を用いて決定した。この値は、試験に用いる接種物のサイズを較正するために役立つ。細菌および酵母の懸濁液は収集から24時間以内に用いた;一方、真菌調製物は冷蔵下で最長7日間貯蔵することができる。
【0183】
(表IV)接種物調製のための培養条件
【0184】
製剤をpH 7のリン酸緩衝食塩水(PBS)中にて調製し、どのアルキルグリコシド製剤が抗菌活性を有するかを判定した。栄養分の供給源として、1.5mg/mLのウシ血清アルブミン(BSA;表VおよびVIを参照)または1mg/mLのPYYを培地に添加した(表VIIを参照)。BSA(CAS Number:9048-46-8)はSigma-Aldrich, St. Louis, MO, USAから入手し、n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドおよびn-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドはAnatrace Inc., Maumee, OH, USAから入手し、PYYはBachem California Inc., Torrance, CA, USAから入手した。
【0185】
アルキルグリコシドの抗菌活性を、十分な容積のアルキルグリコシド溶液を移し入れた適したサイズの4つの滅菌蓋付き細菌用容器において調べた。調製して標準化した接種物を各容器に1つずつ接種し、混合した。懸濁液接種物の容積は、アルキルグリコシド溶液の容積の約0.5%〜約1.0%であった。アルキルグリコシド溶液に添加する被験微生物の濃度は、接種後の被験調製物の濃度がアルキルグリコシド溶液1mL当たり約1×105および1×106cfuとなるようにした。増殖阻害または増殖の低下のレベルを対数尺度に基づいて決定するために、各被験調製物中の生菌の初期濃度を、プレート計数法によって決定される、標準化された接種物のそれぞれにおける微生物の濃度に基づいて推定した。接種した容器を、続いて約22.5℃±2.5でインキュベートした。各培養物/容器における微生物の増殖または非増殖を、第14日および第28日に再び判定した。各計数物中に存在するcfu数を、当技術分野において標準的なプレート計数手順によって適用可能な間隔で決定した。続いて、細菌および/または真菌の桁数の変化を、適用可能な試験間隔(例えば、第14日および第28日;表V、VIおよびVIIを参照)での各微生物に関する1mL当たりのcfu濃度のlog10値から、出発時または開始時(例えば、第0日)に存在した1mL当たりのcfu濃度の第1の算出log10値を差し引くことによって求めた。
【0186】
(表V)0.125% n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物における微生物の対数減少値
【0187】
(表VI)0.2% n-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物における対数減少値
【0188】
(表VII)0.25% n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物の対数減少値
【0189】
他のアルキルグリコシドの抗菌活性の決定は、実質的に本明細書に記載された通りに行われると考えられる。
【0190】
実施例9
霊長動物に対するアルキルグリコシド(alkyglycoside)とアンチセンスオリゴヌクレオチドとの投与
修飾された骨格を有するおよそ7,000ダルトンのアンチセンスオリゴヌクレオチド(ASO)(米国特許第7,132,530号に記載されたようなホスホロチエオートオリゴヌクレオチド)をアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標))と混合したものを、空腸内にカニューレを挿入した6匹のカニクイザルに用量10mg/kgで投与した。これらの動物は投与前に絶食させた。被験作用物質をPBS緩衝液中に溶解させ、表VIIIに記載したように、カニューレを通して各動物の空腸内に1.5mL容積を注入するか、または皮下投与(s.c.)した。
【0191】
(表VIII)テトラデシル-β-D-マルトシドとともに投与したアンチセンス薬物の生物学的利用能
【0192】
このプロトコールは、各動物に対して、表VIII中の最初の3つの被験作用物質を3つの異なる日付に投与するという3者間クロスオーバーを伴った。投薬日の間には1週間のウォッシュアウト期間を置いた。これらの動物のうち2匹にはその後、吸収強化物質として5%カプリン酸ナトリウムを含む第4の被験作用物質を投与した。血中レベルの分析は、カチオン性ポリスチレンナノ粒子を用いた固相抽出を伴う定量分析を用いて実施した。
【0193】
血液試料の固相抽出をまず行った。既知量のオリゴヌクレオチドを各試料のアリコート(200〜400μl)に添加し、脱イオン水中の50mM Tris-HCl(pH 9)800μlで希釈したものを用いて、ナノ粒子-オリゴヌクレオチド結合物を形成させた。この混合物を短時間ボルテックス処理した後に、正の表面電荷を誘導するために水溶性カチオン性開始剤を用いて、表面活性物質を含まないエマルション重合によって調製した、200μlのポリスチレンナノ粒子懸濁液を添加した(固形分:およそ10mg/ml)。この混合物をその後に再びボルテックス処理した。5〜10分間のインキュベーション後に懸濁液を遠心し、上清を除去した。粒子を脱イオン水/エタノール(1:1)中の0.5M酢酸の1ml溶液中に再懸濁させ、遠心によって洗浄溶液から分離した。上清を除去した後に、粒子を1mlの脱イオン水中に再懸濁させ、別の遠心段階によって分離した。アンモニア水(25%)/アセトニトリル(60/40)中にある150μM SDS溶液200μlをナノ粒子-オリゴヌクレオチド結合物に添加し、遊離したオリゴヌクレオチドを遠心によって担体から分離した。試料への残留粒子の混入をなくすために、上清を別の1.5mlチューブに入れて再び遠心した。その後に、試料を回転蒸発または凍結乾燥によって乾燥させ、分析時まで-20℃で貯蔵した。
【0194】
抽出した試料のキャピラリーゲル電気泳動によって定量分析を行った。キャピラリーゲル電気泳動(CGE)は、キャピラリー電気泳動システムを用いて行った。ポリビニルアルコール(PVA)でコーティングしたキャピラリー、ポリマー溶液Bおよびオリゴヌクレオチド緩衝液を含むオリゴヌクレオチド分析キットを入手した。PVAでコーティングしたキャピラリーを用い、製造元のプロトコールを用いて分析を行った。
【0195】
CGE分析から得られたデータを用いて、ホスホロチエオートオリゴヌクレオチドの定量を行った。試料中のオリゴヌクレオチドの量(nON)は、以下の式を用いて算出した:
nON=nStd(εStd/εON)((A0N/TON)/(AStd/TStd))、
式中、nStdは試料に添加された標準的なオリゴヌクレオチドの量であり、εStdおよびεONはモル吸光係数であり、AStd/TStdおよびAON/TONはそれぞれ、標準物質および調べようとする化合物の補正ピーク面積(ピーク面積および移動時間の商)である。分析物および標準物質の補正ピーク面積の商は正規化面積と呼ばれる。
【0196】
AUCは、240分間の期間にわたる濃度-時間曲線(cure)から算出した。相対的生物学的利用能は、各AUCを静脈内投与した薬物のAUCによって除算した比として求めた。Intravail(商標)(テトラデシル-β-D-マルトシド)添加剤は最大18%の生物学的利用能をもたらした。表面活性物質添加剤を伴わない対照は検出可能な吸収を示さなかった。カプリン酸ナトリウム製剤は9%という平均生物学的利用能を示した。
【0197】
実施例10
オランザピンの急速分散性剤形の調製
オランザピンの急速分散性剤形を以下の通りに調製した。オランザピン、CAS# 132539-06-1は、SynFine(Ontario, Canada)から入手する。10mM酢酸ナトリウム緩衝液、pH 5.0およびpH 6.5を、以下の通りに調製する。容積測定用の目印の付いた適切なサイズの清浄容器に、495mLの滅菌注射用蒸留水を入れる。0.286mLの酢酸を添加する。1N NaOHを添加してpHを5.00(またはpH 6.5)にする。適正なpHが得られたところでさらに水を添加して総容積を500mLにし、pHを再び確かめる。
【0198】
過程の全体を通じて撹拌を行いながら、魚肉ゼラチンまたは豚皮ゼラチンを酢酸緩衝液にゆっくりと添加し、溶解するのに十分な時間を置くことにより、以下の表IXに含まれた組成物を有する液体製剤を作る。魚肉ゼラチンまたは豚皮ゼラチンが完全に溶解したところで、マンニトールを添加して溶解させる。続いて、甘味料を添加する。これが十分に分散したところで、本発明の化合物のための例の1つである有効成分であるオランザピンを添加し、最終的な溶液を作製する。保存料、酸化防止剤、表面活性物質、粘性強化物質、着色剤、香味剤、甘味料または味遮蔽剤などの二次的成分を組成物に組み込んでもよい。適した着色剤には、赤色、黒色および黄色の酸化鉄、ならびにEllis & Everardから入手しうるFD & C blue No. 2およびFD & C red No. 40などのFD & C色素が含まれうる。適した香味剤には、ミント、ラズベリー、カンゾウ、オレンジ、レモン、グレープフルーツ、キャラメル、バニラ、チェリーおよびグレープの着香料ならびにこれらの組み合わせが含まれうる。適した甘味料には、アスパルテーム、アセスルファムKおよびタウマチンが含まれる。適した味遮蔽剤には炭酸水素ナトリウムが含まれる。シクロデキストリンは、これらの添加剤の有効性を低下させる包接化合物をアルキルサッカリドと形成するため、避けるべきである。
【0199】
上記の薬物溶液の1mLずつのアリコートを、24ウェル使い捨てマイクロウェルプラスチックプレートのウェルに入れる。液体アリコートを含むマイクロウェルプレートを凍結プレートにて-70゜で凍結させ、LabConco Freezone Model 4.5卓上凍結乾燥器に取り付けたガラス製凍結乾燥フラスコに入れて、真空下で凍結乾燥させる。凍結乾燥の後に、迅速分散性錠剤を試験時までマイクロウェルプレート中にて乾燥環境で貯蔵する。モノ-およびジ-ステアリン酸スクロース混合物はCroda Inc.によって寄贈され、これはCRODESTA F-110と命名されている。ドデシルマルトシド、テトラデシルマルトシドおよびスクロースモノデカノエートはAnatrace Inc., Maumee, OHから入手する。
【0200】
(表IX)オランザピン(olanzopine)製剤
1 SynFine, Ontario, Canada
2 Croda Colloids Ltd(加水分解されていない、噴霧乾燥させた魚肉ゼラチン)
4 ドデカン酸スクロース(モノエステル)-Anatrace Inc.
5 Sigma Aldrich(ゼラチンA型、豚皮‐G6144)
Qs=下記量にするために十分な量
【0201】
薬物オランザピンは、Zyprexaとも呼ばれ、「丸飲み(whole-swallowed)」錠剤として投与された場合によく吸収され、経口投与からおよそ6時間後にピーク濃度に達することが知られている。これは初回通過代謝によって高度に排出され、用量のおよそ40%が体循環に達する前に代謝される。薬物動態試験により、口腔内に置いた場合に約3秒〜10秒で崩壊する「丸飲み」オランザピン錠剤、およびこの実施例に上述した様式で凍結乾燥によって調製された迅速分散性オランザピン錠剤は生物学的に同等であり、投与から約6時間後にピーク濃度を示すことが示されている。同様に、肝臓における初回通過効果により、用量のおよそ40%が体循環に達する前に排出される。
【0202】
本実施例において、10mgのオランザピンを含む急速分散性錠剤は、この実施例において上述した通りに凍結乾燥によって調製される。頬側組織と接触させることによって急速分散性オランザピン錠剤を投与することにより、急速分散性オランザピン錠剤に対する特定のアルキル鎖長を有するある種のアルキルサッカリドの添加は、代謝されていない活性薬物と比較した体循環中のオランザピン代謝産物の相対的割合の低下によって認められるように、オランザピンの初回通過効果代謝の実質的な低下をもたらすことが発見されている。血清中または血漿中のオランザピンおよびオランザピン代謝産物の相対的割合は、HPLC Chromatograph, Perkin Elmer 200を、恒温セルを装着したRefractive Index Detectorとともに用いて決定することができる。Lichro sorb RP-18(Merck, Darmstadt, Germany)250mmなどの適した固相吸着剤を、アセトニトリル:水勾配からなる移動相とともに用いることができる。Perkin Elmer 200 auto-samplerを用いた20μLの注射容積および0.8mL/分の流速がこの目的には好適である。具体的には、0.2%から最大10%までのドデシルマルトシドまたはテトラデシルマルトシドまたはドデカン酸スクロースを急速分散性錠剤形式中に組み入れることで、体循環に入る薬物が増加し、肝臓における「初回通過」効果によって排出される薬物が減少する。さらに、最大薬物レベル到達時間は、典型的には1〜6時間からおよそ15〜45分へと著しく短縮される。精神病エピソードを来している攻撃的患者の治療に用いる場合、薬物のこのより迅速な吸収はより迅速な作用開始をもたらし、それは非常に有益であると考えられる。
【0203】
実施例11
メラトニンの急速分散性剤形の調製
メラトニンまたは5-メトキシ-N-アセチルトリプタミンは、睡眠障害の患者における睡眠覚醒サイクルを調節するために用いられる神経ホルモンである。内因性メラトニンは、概日または概年リズムを示すすべての動物で松果体によって分泌される。メラトニンは睡眠覚醒リズムの維持に役割を果たすことが実証されており、その補充は、時差ぼけ、交代制勤務、うつ病およびさまざまな神経学的機能異常に伴って起こる睡眠障害を調節する助けになる可能性がある。
【0204】
市販されているメラトニン製剤には、経口および舌下錠剤、カプセル剤、茶剤、ロゼンジ剤および経口噴霧剤送達系が含まれる。経口メラトニン投与は、内因性ホルモンのものとは異なる薬物動態プロファイルをたどる。経口投与後に、メラトニンはかなりの初回通過肝代謝を受けて6-スルファトキシメラトニン(6-sulfaoxy melatonin)となることから、30〜50%と推定されるメラトニンの生物学的利用能がもたらされる。DeMuroら(2000)は、健常志願者において試験された経口メラトニン錠剤の絶対的生物学的利用能はやや低く、およそ15%であると報告している。メラトニンの平均排出半減期は概ね45分である。
【0205】
1mg、5mg、10mgおよび20mgを含む急速分散性メラトニン錠剤は、上記の実施例10に記載された方法に従い、実施例10に記載されたような1%〜2%のアルキルサッカリドを伴うかまたは伴わずに調製される。投薬が容易になるように、麻酔をかけるニュージーランドホワイトウサギを制限ボックスに入れ、耳介静脈への注射によって投与されるアセプロマジン/ケタミン(0.7mg/0.O3mg、0.1mL中)の単回投与を用いて麻酔を施す。これによって約10分間の麻酔が得られ、その間にウサギに被験物質を投与する。その後にウサギは意識を回復する。2時間の期間にわたる個々の時点で、1mLの血液試料を中心耳動脈から収集する。収集後に、リチウム/ヘパリンを抗凝固薬として用いて、各血液試料から血漿を直ちに調製する。試料はすべて、メラトニンに関してアッセイするまで-70℃で貯蔵する。メラトニンは、GenWay Biotech Inc., San Diego, CAによって製造されている市販のELISAキットを用いて測定する。急速崩壊性錠剤を口腔上部の頬側組織と接触させることによって投与した場合、メラトニンは、アルキルサッカリドの存在下における曲線下面積による測定では少なくとも75%の生物学的利用能で、アルキルサッカリドの非存在下では50%未満で吸収される。メラトニンは、GenWay Biotech Inc., San Diego, CAによって製造されている市販のELISA kit(No. 40-371-25005)を用いて測定する。加えて、アルキルサッカリドを含む錠剤の場合には、メラトニンの最大濃度に、アルキルサッカリドを含まない錠剤の場合に要する時間のおよそ半分で到達する。
【0206】
実施例12
ラロキシフェンの急速分散性剤形の調製
ラロキシフェンは、Evista(登録商標)とも呼ばれ、閉経後女性における骨粗鬆症の治療および予防、骨粗鬆症を有する閉経後女性における浸潤性乳癌のリスク低減、ならびに浸潤性乳癌のリスクが高い閉経後女性における浸潤性乳癌のリスク低減のために用いられている。推奨投与量は60mg錠剤の1日1回投与である。ラロキシフェンの経口用量のおよそ60%は経口投与後に迅速に吸収されるが、全身循環以前の(presystemic)グルクロニド抱合が高度であり、その結果、ラロキシフェンの絶対的生物学的利用能は2%に過ぎない。米国特許第5,576,014号または第6,696,085 B2号または第6,024,981号に記載された通りに調製された急速分散性60mgラロキシフェン錠剤は、およそ2%の絶対的生物学的利用能という極めて類似した薬物動態を有することが見いだされている。しかし、噴霧乾燥分散(Bend Research Inc., Bend Oregon、またはAzoPharma, Miramar, FL)によって、またはより一般的に用いられる標準的な薬学的粉砕もしくは製粉過程によって調製される、10mgまたはそれ未満の微粉化ラロキシフェン、および0.5%〜5%のドデシルマルトシドを含む急速分散性錠剤は、頬側投与された場合、60mg経口錠剤によって達成されるものに類似した全身薬物レベルを達成し、同時に循環中の不活性ラロキシフェングルクロニドがより少なくなる。
【0207】
臨床的有益性は主として非抱合型薬物に起因するが、副作用は、活性薬物、およびグルクロニド抱合を受けた実質的に不活性な薬物のいずれかまたは両方によって媒介される可能性がある。このため、この場合には活性薬物の30倍もの高さの濃度である不活性薬物抱合体に対する曝露を減らすことで、副作用の恐れを低下させる有意な臨床的有益性が得られる可能性がある。ラロキシフェンの水溶解度はおよそ0.25mg/Lである。その結果として、実施例10に記載したような急速分散性製剤を調製するための凍結乾燥のための準備においてラロキシフェンを水中に溶解させることは不可能である。
【0208】
この場合には、1%〜30% w/wのCRODESTA F-110を適した緩衝液に添加し、それをボルテックス処理して、45℃に1時間加熱することによって自己集合性ヒドロゲルを形成させることができる。続いて、微粒子または微粉の形態にあるラロキシフェンをこの温かい液体に添加して、懸濁下で60mg/mLの濃度を達成させ、固体が均一に懸濁化および分散されるまで、それを再びボルテックス処理によって混合する。室温に冷却すると、小分けすることができ、それでも均一な懸濁を維持している安定なチキソトロープヒドロゲルが形成される。pH 2〜pH 7のpH範囲にある酢酸緩衝液は、この目的に特によく適することが見いだされている。ラロキシフェンのゲル懸濁液の1mLアリコートを24ウェル使い捨てマイクロウェルプラスチックプレートのプレートに入れ、実施例1に記載した通りに凍結乾燥する。
【0209】
頬側組織に対する提示によるこの急速分散性製剤の投与は、絶対的生物学的利用能(absolute bioavailabilty)の少なくとも4%への増大(倍加)、および、それに対応した、循環中のラロキシフェングルクロニド抱合体濃度と非抱合型ラロキシフェンとの比の測定可能な低下をもたらす。
【0210】
実施例13
ジフェンヒドラミンの急速分散性剤形の調製
ジフェンヒドラミンは、顕著な中枢鎮静性を有する鎮静性抗ヒスタミン薬であり、不眠症の短期的管理、蕁麻疹および血管性浮腫、鼻炎および結膜炎、掻痒性皮膚障害、悪心および嘔吐を含むアレルギー性病状の症状緩和、乗り物酔い、めまい、ある種の精神科薬物の副作用に起因する不随意運動の予防および治療、ならびにその抗ムスカリン性状によるパーキンソニズムの管理において、睡眠薬として用いられている。ジフェンヒドラミンの特に望ましい特徴は、それが依存症を生み出すという証拠が見たところ存在しないことである。その優れた安全性プロフィールのため、これは一般用医薬品として販売されており、Ambien(登録商標)およびLunesta(登録商標)といったより新たな睡眠薬のいくつかが、夢遊症および睡眠中の食事-むちゃ食いなどの奇妙な挙動を、時として起こる重度のアレルギー反応および顔面腫脹とともに引き起こす恐れがあり、これらのより新たな処方医薬品のこれらの副作用についてFDAが警告表示を求める原因となったこととは異なる。
【0211】
塩酸ジフェンヒドラミンは、25〜50mgの常用量で1日3回または4回投与される。成人および小児における最大用量は1日約300mgである。20〜50mgの用量は、成人および12歳以上の小児において睡眠薬として用いることができる。この薬物は胃腸管からよく吸収される;しかし、これは全身薬物レベルに影響を及ぼすように思われる高度の初回通過代謝を受ける。ピーク血漿中濃度は経口投与から約1〜4時間後に達成される。ジフェンヒドラミンはCNSを含む全身に広く分布し、肝臓における高度の代謝のために、この薬物は主として尿中に代謝産物として排泄され、少量の未変化薬物が存在することが認められる。
【0212】
ジフェンヒドラミンは不眠症および他の障害の治療のために安全かつ有効であると考えられるが、ピーク血漿中濃度の達成までの1〜4時間の遅れに起因する比較的遅い作用開始は不都合であり、安全かつ有効なこの薬物の実用性を減じさせている。静脈内に投与されたジフェンヒドラミンは迅速な作用開始を及ぼす;しかし、静脈内投与は、外来患者の使用にとっても、重篤でない医学的適応症にとっても現実的ではない。ジフェンヒドラミンの迅速作用開始製剤の必要性は明らかである。不眠症の場合には、患者は、長時間の落ち着かない不眠の恐れを最小限に抑えるために、この薬物がその所望の薬理効果を発揮するのに十分な全身薬物レベルを達成するのを待ちながら、この薬物の現行の経口剤形を就眠よりもはるかに前に服用する必要がある。ジフェンヒドラミンの制吐的適用の場合にも、悪心および嘔吐をできるだけ早く緩和するために、迅速作用開始が非常に望ましい。これは乗り物酔いおよびめまいの治療の場合も、これらの症状は予期せずに起こることがあるという理由から同様であり、経口投与した薬物がその有益な効果を達成するのに十分な全身薬物レベルを達成するまでに1〜4時間待たねばならないことは不都合であり、かつ望ましくない。
【0213】
ジフェンヒドラミンの水溶解度はおよそ3.06mg/mLである。このため、実施例12に記載した方法を用いて、50mgの薬物および1%〜2%のアルキルサッカリドを含む急速分散性ジフェンヒドラミン錠剤を調製することができる。ジフェンヒドラミンはやや苦いため、味を改善するために、薬学的に許容される着香料および甘味料の味遮蔽量を添加してもよい。このようにして調製された急速分散性錠剤は、「丸飲み」錠剤シロップ、チュアブル錠、ロゼンジ剤または可食フィルムストリップと比較して作用開始がより迅速であり、より少ない初回通過代謝も示す。
【0214】
実施例14
マウスに対するアルキルグリコシドと抗肥満ペプチド・マウス[D-Leu4]OB3との投与
この実施例は、雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の抗肥満ペプチド、マウス[D-Leu-4]OB3の取り込みを示す。0.3%アルキルグリコシドテトラデシルβ-D-マルトシド(Intravail(商標)A3)と混合した合成レプチンアゴニスト[D-Leu-4]OB3を、6週齢雄性Swiss Websterマウスに対して、胃管により1mgの用量で投与した。
【0215】
マウス[D-Leu-4]OB3(1mg/200ulの濃度)を、PBS(pH 7.2)、またはPBS(pH 7.2)中に再構成した0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)のいずれかに溶解させて、各時点につき4匹ずつのマウスのそれぞれに、麻酔を行わずに胃管によって投与した。10、30、50、70、90または120分後の時点で、イソフルラン(5%)の吸入によってマウスを安楽死させ、尾部大静脈の穿刺によって全瀉血を行った。ペプチドを投与されていない4匹のマウスからも血液を収集した(前採血)。この期間内の4匹のマウスのそれぞれからの血液をプールし、血清試料を調製した。プールした試料のマウス[D-Leu-4]OB3含有量は競合的ELISAによって測定した。
【0216】
これらの実験を2回繰り返した。1回の実験から収集されたデータを表Xおよび図4に提示している。このデータは高度に再現性があると判定された。取り込み曲線をMicrosoft(商標) Excelを用いてプロットし、画像用プログラムSigmaPlot 8.0(商標)(SPSS Science, Chicage, IL)の機能を用いてAUCを算出した。得られた最小のAUC値を1.0に任意に設定した。他のすべてのAUC値を1.0と比較することにより、相対的生物学的利用能を求めた。
【0217】
(表X)胃管による投与後の雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の1mgのマウスp-Leu-4]OB3の取り込み
【0218】
表Xおよび図4から明らかなように、0.3%のアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)の添加は、PBSのみの中のペプチドと比較して、OB-3ペプチドの相対的吸収を4倍に増加させた。
【0219】
実施例15
イヌに対するアルキルグリコシドとスマトリプタンとの投与
この実施例は、イヌによる、0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中のスマトリプタンの取り込みを示す。0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)と混合したスマトリプタンを、イヌに対して、25mgの用量で、経口投与および直腸投与の両方によって投与した。
【0220】
図5から明らかなように、0.5%のアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)の添加は、現在利用可能な25mg経口錠剤と比較して、経口投与および直腸投与の両方で、スマトリプタンのCmaxを増加させた。現在利用可能な錠剤のCmaxは、図5中の横向きの点線によって表されているように、イヌでは104ng/mlと決定された。
【0221】
実施例16
アルキルグリコシドとトリプタンとの投与は生物学的利用能を高める
硫酸スマトリプタン、ナラトリプタン-HCl;または安息香酸リザトリプタンを、0%、0.02%、0.05%、0.1%、0.2%または1.0%のアルキルサッカリドを含む20mM酢酸ナトリウム緩衝液、pH 5.5中に溶解させる。薬物溶液の各セットを、各ラットの1つの鼻孔への2OuLの点滴注入により、ラット8匹ずつからなる6つの群に投与する。3時間の期間にわたり、0、5、10、15、30、60、120および180分の時点で、200μlの血液試料を眼窩出血によって採取する。最後の血液試料を収集した後に、各ラットをCO2によって安楽死させる。採血後に、リチウム/ヘパリンを抗凝固薬として用いて、各血液試料から血漿を直ちに調製する。血漿試料は分析するまで-70℃で貯蔵する。血漿中薬物レベルを、Boultonによって記載された方法を用いるHPLC、または類似のHPLC法によって決定する。濃度-時間データをプロットして、各アルキルサッカリド濃度でのCmaxを求め、アルキルサッカリドが存在する場合と存在しない場合のCmaxの比を算出し、表XIに示されているように記録する。各プロットからのTmax観測値を、濃度-時間プロットの検査によって求め、表XIIに示されているように記録する。この表に明示されている用量は、いずれの場合もトリプタン遊離塩基の量を反映している。
【0222】
(表XI)トリプタン類似物投与のCmax比
【0223】
(表XII)トリプタン類似物投与のTmax
【0224】
実施例17
スマトリプタンとアルキルグリコシドとの鼻投与は生物学的利用能およびCMAXを高め、全身吸収の開始を早める
硫酸スマトリプタンを、0.18%ドデシルマルトシド(「製剤A」)を含むかまたはドデシルマルトシド添加剤を含まない(「製剤B」)、水1L中に0.2gのリン酸二ナトリウムおよび10.0gのリン酸一カリウムを溶解させることによって調製し、pH 5.5に調整したリン酸緩衝液中に溶解させ、100マイクロリットルの噴霧薬当たり20mgという最終的なスマトリプタン濃度にする。最終的なpH調整は、硫酸または水酸化ナトリウム溶液を用いて行う。第3の製剤である、GlaxoSmithKlineによって製造されている100マイクロリットル当たり20mgのImitrex(登録商標)スマトリプタン鼻噴霧薬を「参照」とする。18人の患者に対して、各薬物溶液を、Ing. Erich Pfeiffer GmbH, Radolfzell, Germany、Valois Pharma, Le Neubourg, FranceまたはBecton Dickinson, New Jersey, USAによって製造されているもののような標準的な定量鼻噴霧薬装置を用いた100マイクロリットルの定量鼻噴霧薬として、投薬間に少なくとも3日間のウォッシュアウト期間を置いた3者間クロスオーバー試験の形で投与する。K2 EDTA(EDTA二カリウム)を抗凝固薬として用いる各血液試料からの血漿の調製のために、各患者から、血液試料を、時間指定した間隔で、例えば、図6および7に示されているように0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間の時点で収集する。スマトリプタンの血漿レベルを、高速液体クロマトグラフィー(Ge, Tessier et al. 2004)によって決定する。
【0225】
図6は、Imitrex(登録商標)鼻噴霧薬参照、およびアルキルサッカリド添加剤を含まない製剤Bに関する、図示したさまざまな時点での全患者の平均血漿レベルの比較を標準偏差とともに示している。参照および製剤Bは、Cmaxが1mL当たりおよそ15ng、およびTmaxが1〜2時間というほぼ等しい成績を示した。
【0226】
図7は、Imitrex(登録商標)鼻噴霧薬参照、および0.18%ドデシルマルトシド添加剤を含む製剤Aに関する、図示したさまざまな時点での全患者の平均血漿レベルの比較を標準偏差とともに示している。製剤AのCmaxは1mL当たりおよそ60ng、またはImitrex鼻噴霧薬参照で観察されたCmaxのおよそ4倍である。T-maxは、参照での1〜2時間から、アルキルサッカリドを含む製剤Aではおよそ8〜10分に短縮しており、参照製剤によって1〜2時間で達成される15ng/mLという治療的に意味があると推定されるCmax値に、製剤Aの場合にはおよそ2分で到達した。
【0227】
得られたデータから以下のパラメーターを算出する:
AUC0-t=投与時間から最後の測定可能な濃度の時間までの薬物血漿中濃度-時間曲線下面積。
AUC0-∞=投与時間から無限時までの薬物血漿中濃度-時間曲線下面積。
Cmax=最大薬物血漿中濃度。
Tmax=最大薬物血漿中濃度到達時間。
半減期=投与後に薬物血漿中濃度がその最大濃度の半分になるまでの時間
Kel=排出速度定数。
薬物動態成績の平均を以下に表としてまとめている:
【0228】
(表XIII)
【0229】
すべての対象に関する、0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間といった個々の時点での平均Cmaxデータを、図7にプロットしている。
【0230】
実施例18
リザトリプタン(安息香酸塩)と重量比で最大2%までのアルキルグリコシドとの舌下/頬側投与は、生物学的利用能を有意には高めず、全身吸収の開始も早めない
鼻腔内の粘膜を越えるスマトリプタンの生物学的利用能はドデシルマルトシドの存在下で著しく高まるため、他のトリプタンについても、アルキルサッカリドの存在下で、類似の構成を有する口腔の舌下または頬側の膜を越える同等の生物学的利用能が同様に得られる可能性が高いと考えられる。
【0231】
(表XIV)頬側/舌下送達用のリザトリプタンの製剤
【0232】
製造手順:すべての添加剤を形状的に混合し、適した丸形の浅い凹状工具を用いて圧縮して錠剤にする。または、リザトリプタンおよびアルキルグリコシドを水中または水性アルコール混合物中に溶解させ、その溶液を用いて添加剤を顆粒化し、続いて顆粒を乾燥させ、製粉し、圧縮して錠剤にする。Maxalt-MLT(安息香酸塩リザトリプタン)経口崩壊性錠剤10mgを「参照」とする。製剤Aはアルキルグリコシドを含まない。製剤Bは0.5%アルキルグリコシドを含む。製剤Cは2%アルキルグリコシドを含む。参照、ならびにそれぞれ0%、0.5%および2%のドデシルマルトシドを含む、上記の通りに調製した崩壊性錠剤の3種の製剤を、20人の患者に対して、投薬間に少なくとも3日間のウォッシュアウト期間を置いた4者間4期間クロスオーバー試験の形で投与する。錠剤を舌下または頬と歯肉との間(頬側)のいずれかとして口腔内に置き、唾液との接触によって崩壊させる。血液試料を、K2 EDTA(EDTA二カリウム)を抗凝固薬として用いる各血液試料からの血漿の調製のために、各患者から、血液試料を、時間指定した間隔で、例えば0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間の時点で収集する。リザトリプタンの血漿レベルを、適切なリザトリプタン標準物質を含める(Ge, Tessier et al. 2004)の方法の変法により、高速液体クロマトグラフィーによって決定する。
【0233】
驚いたことに、以下の表XVに見られるように、アルキルサッカリドの添加による舌下または頬側生物学的利用能の増大はほとんどみられないか、または検出不能である。同様に、CmaxおよびTmax値もわずかな変化しか示さない。
【0234】
(表XV)
【0235】
実施例19
ナラトリプタンと重量比で最大0.2%までのアルキルグリコシドとの舌下噴霧投与は、生物学的利用能を有意には高めず、全身吸収の開始も早めない
【0236】
(表XVI)頬側/舌下送達用のナラトリプタン舌下噴霧薬の製剤
【0237】
製造過程:ナラトリプタン塩酸塩を水中に溶解させて、必要な数量のポリエチレングリコールまたはエタノールを添加する。次に、アルキルグリコシド、着香料、アスパルテームをこの溶液に溶解させる。容積の最終的な調整は水で行う。ドデシルマルトシドアルキルサッカリドを伴うかまたは伴わない舌下製剤を、20人の患者に対して、定量噴霧ポンプを用いて2者間2期間クロスオーバー試験の形で投与する。驚いたことに、実施例18の場合と同様に、0.18〜0.2%アルキルサッカリドの存在下で、舌下粘膜を越えるナラトリプタン生物学的利用能の増大はほとんど観察されない。
【0238】
本出願の全体を通じて、さまざまな刊行物を参照している。これらの刊行物の開示内容は、本発明の属する最先端の技術水準をより十分に説明するために、それらの全体が参照により本出願に組み入れられる。
【0239】
【0240】
上記の実施例において、本過程をその特定の態様の具体的な詳細を参照しながら説明してきたが、その変更物および変形物が本発明の趣旨および範囲に含まれることは理解されるであろう。したがって、本発明は以下の特許請求の範囲のみによって限定される。
【技術分野】
【0001】
発明の分野
本発明は全体的には、強化された生物学的利用能をもたらす非刺激性で無毒性の組成物に関し、より具体的には、片頭痛緩和をもたらすための方法および組成物を含む、対象に対する5-HT受容体アゴニストなどの治療薬の送達のためのアルキルグリコシド組成物またはサッカリドアルキルエステル組成物に関する。
【背景技術】
【0002】
背景情報
治療薬は多くの場合、さまざまな表面活性物質(surfactant)と組み合わされる。しかしながら、表面活性物質は往々にして、皮膚、および鼻、口、眼、膣、直腸、食道、腸管などに認められるもののような粘膜を含む他の組織に対して刺激性がある。また、多くの表面活性物質はタンパク質を変性させ、それ故にそれらの生物活性も消失させる。そのような薬剤の開発および使用の上でのもう1つの重大な制約は、それらを作用部位に対して安全に、非侵襲性に、効率的におよび安定して送達させる能力である。したがって、理想的な強化性(enhancing)表面活性物質は、治療薬を安定化し、無毒性で皮膚または粘膜の表面に対して非刺激性であり、抗菌活性を有し、さまざまな膜障壁を通しての治療薬の通過または吸収を膜の構造的完全性および生物学的機能を損なうことなく強化し、かつ、その薬剤の生物学的利用能を高めると考えられる。
【0003】
迅速崩壊性またはいわゆる「急速分散性」の剤形を製造するための製剤化アプローチはこれまでに数多く記載されている。口腔内で崩壊すると薬物物質は嚥下され、その結果、前胃吸収および最終的には胃吸収が起こる。「前胃吸収」という用語は、胃の前の消化管の部分への有効成分の吸収を指すために一般的に用いられ、これには頬側吸収、舌下吸収、口咽頭吸収および食道吸収が含まれる。「胃吸収」という用語は、胃および腸における有効成分の吸収のことを指して一般的に用いられる。薬物が消化管の前胃部分を通過する際に、さまざまな量の薬物が吸収される可能性がある。しかし、薬物の大部分は胃内に移行し、錠剤、カプセル剤または液剤などの腸溶性剤形が吸収される通常の胃吸収様式で吸収される。薬物が腸から吸収されると、薬物は肝臓内に直接運ばれ、そこで、その固有の化学構造に応じて、肝細胞における通常の解毒プロセスを遂行する酵素によって代謝されるかまたは排出される。この排出は、肝臓における「初回通過」代謝または「初回通過」効果と呼ばれる。ほとんどの場合は元の薬物と比較して実質的または完全に不活性である、その結果生じた代謝産物は、多くの場合は血流中を循環する状態で認められ、その後に尿中および/または便中に排出される。迅速崩壊性または迅速分散性の剤形を製造するための製剤化アプローチは、米国特許出願第2006/0134194号(特許文献1)に提示されており、これは参照により本明細書に組み入れられる。
【0004】
以前に記載された急速分散性剤形は、有効成分の前胃吸収または胃吸収を促進させるために、口内に置かれた場合に崩壊または溶解する剤形を提供しているが、本発明の急速分散性剤形は、薬物作用の開始を早めること、および初回通過効果薬物代謝を減少させることといった改善された特性を提供する。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】米国特許出願第2006/0134194号
【発明の概要】
【0006】
本発明は、一部には、薬物の吸収および生物学的利用能を高め、それと同時に薬物のさまざまな有害毒性作用を避けるために有用な薬物強化剤(drug enhancing agent)を含む治療用組成物の開発に基づく。特に、本発明の薬物強化剤は、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルからなる無毒性の表面活性物質を含む。本発明の治療用組成物の1つの利点は、それらが高い生物学的利用能での治療薬の投与および送達を、強化剤のいわゆる「最大無毒性レベル」(それらのNOAEL)よりも著しく低い濃度で、可能にすることである。したがって、本発明は、アルキルグリコシドおよび/またはサッカリドアルキルエステルならびに治療薬(例えば、小分子有機薬物分子、Exenatide、GLP-1などの低分子量ペプチド、タンパク質、ならびに低分子量ヘパリンおよび阻害性RNAなどの非ペプチド性治療用ポリマー)を含む組成物、例えば経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)または脳脊髄液(CSF)送達経路などを介した組成物の投与および使用の方法、ならびにそのような組成物の投与によって対象における疾病状態を改善する方法を提供する。
【0007】
1つの局面において、本発明は、少なくとも1つのアルキルグリコシドおよび/または少なくとも1つのサッカリドアルキルエステルを有する表面活性物質組成物であって、治療薬、薬物または生物活性化合物と混和、混合または配合された場合に、表面活性物質がその薬物の生物活性を安定化し、かつその生物学的利用能を高めるような表面活性物質組成物に関する。
【0008】
したがって、1つの局面において、本発明は、少なくとも1つの生物活性化合物および少なくとも1つの表面活性物質を有する治療用組成物であって、表面活性物質がさらに、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルもしくはスクロースエステルからなり、治療用組成物が生物活性化合物を少なくとも約6カ月またはそれ以上にわたり、約4℃から約25℃までの範囲で安定化する治療用組成物を提供する。
【0009】
本発明はまた、少なくとも1つの治療薬または薬物または生物活性化合物と混和、混合または配合されて、対象に投与または送達される、少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む表面活性物質を有する治療用組成物を投与する方法であって、アルキルが約10〜24個、10〜20個、10〜16個または10〜14個の炭素原子を有し、表面活性物質が治療薬の安定性および生物学的利用能を高めるような方法も提供する。
【0010】
さらにもう1つの態様において、本発明は、対象の循環器系への低分子量化合物の吸収を、適した表面活性物質の吸収増加量(absorption increasing amount)と化合物を混和、混合または配合した場合に経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法であって、表面活性物質が親水性サッカリドとの結合によって連結された無毒性および非イオン性の疎水性アルキルであるような方法を提供する。そのような低分子量化合物には、ニコチン、インターフェロン、PYY、GLP-1、合成エキセンディン-4、副甲状腺ホルモン、ヒト成長ホルモンまたは小有機分子が非限定的に含まれる。そのほかの低分子量化合物には、アンチセンスオリゴヌクレオチドまたは干渉性RNA分子(例えば、siRNAまたはRNAi)が含まれる。
【0011】
本発明はまた、糖尿病を治療する方法であって、それを必要とする対象に対して、治療用組成物、例えばインクレチン模倣薬またはその機能的等価物の血糖低下量、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入もしくは肺、または口腔(舌下または頬粘膜細胞)を介して投与し、それにより、対象においてインクレチン模倣薬またはインスリンの吸収を増加させ、血糖のレベルを低下させて、糖尿病を治療する段階を含む方法も提供する。
【0012】
本発明はまた、対象におけるうっ血性心不全を治療する方法であって、それを必要とする対象に対して、GLP-1ペプチドまたはその機能的等価物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の治療的有効量を、経口、眼、鼻、鼻涙管または吸入送達経路を介して投与し、それにより、対象を治療する段階を含む方法も提供する。
【0013】
もう1つの局面において、本発明は、対象における肥満または肥満に伴う糖尿病を治療する方法であって、それを必要とする対象に対して、PYYペプチドまたはその機能的等価物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の治療的有効量を、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して投与し、それにより、対象を治療する方法を提供する。
【0014】
もう1つの局面において、本発明は、対象の循環器系への低分子量治療用化合物の吸収を、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して投与することによって増加させる方法であって、化合物が約1〜30kDであり、ただし投与の経路が経口、眼、鼻または鼻涙管である場合にはその化合物がインスリン、カルシトニンまたはグルカゴンでないことを条件とする方法を提供する。
【0015】
本発明はまた、対象の循環器系への低分子量治療用化合物の吸収を、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法であって、化合物が約1〜30キロダルトン(kD)であり、ただし送達が経口、眼、鼻または鼻涙管経路を介する場合には対象が糖尿病を有しないことを条件とする方法も提供する。
【0016】
本発明の1つの局面においては、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、Exenatide(エキセンディン-4)の治療的有効量との組み合わせで有する薬学的組成物が提供される。
【0017】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、GLP-1の治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0018】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、ニコチンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0019】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、インターフェロンの治療的有効量との組み合わせで含む薬学的組成物を提供する。
【0020】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、PYYの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0021】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、副甲状腺ホルモンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0022】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、約1〜75kDの分子量を有するペプチドの治療的有効量との組み合わせで有する薬学的組成物であって、ただし、ペプチドがインスリン、カルシトニンおよびグルカゴンでないことを条件とする薬学的組成物を提供する。
【0023】
1つの局面において、本発明は、薬学的に許容される担体中に、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、エリスロポエチンの治療的有効量との組み合わせで有する薬学的組成物を提供する。
【0024】
1つの局面において、本発明は、オリゴヌクレオチドの治療的有効量を、アルキルグリコシドの吸収増加量との組み合わせで有する薬学的組成物を提供する。オリゴヌクレオチドは、アンチセンスオリゴヌクレオチド、またはsiRNAもしくはRNAiなどの干渉性RNA分子でありうる。オリゴヌクレオチドは典型的には、約1〜20kDの分子量を有し、約1〜100、1〜50、1〜30、1〜25または15〜25ヌクレオチド長である。もう1つの局面において、オリゴヌクレオチドは約5〜10kDの分子量を有する。1つの局面において、アルキルグリコシドはテトラデシル-β-D-マルトシドである。
【0025】
さらにもう1つの態様において、本発明は、対象における低分子量オリゴヌクレオチドの生物学的利用能を、その化合物をアルキルグリコシドの吸収増加量とともに投与し、それにより、対象における化合物の生物学的利用能を高めることによって高める方法を提供する。1つの局面において、アルキルグリコシドはテトラデシルβ-D-マルトシドである。
【0026】
1つの局面において、本発明は、化合物および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量が鼻腔内に投与された対象のCSF中への化合物の吸収を増加させる方法を提供する。
【0027】
さらにもう1つの態様において、本発明は、親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドを、以下のものから選択される粘膜送達強化剤との組み合わせで有する薬学的組成物を提供する:
(a)凝集阻害剤;
(b)電荷修飾剤;
(c)pH制御剤;
(d)分解酵素阻害剤;
(e)粘液溶解剤または粘液除去剤;
(f)繊毛静止剤;
(g)以下から選択される膜透過強化剤:
(i)表面活性物質;(ii)胆汁酸塩;(ii)リン脂質添加物、混合ミセル、リポソームまたは担体;(iii)アルコール;(iv)エナミン;(v)NOドナー化合物;(vi)長鎖両親媒性分子;(vii)小型の疎水性透過強化物質;(viii)ナトリウムまたはサリチル酸誘導体;(ix)アセト酢酸のグリセロールエステル;(x)シクロデキストリンまたはβ-シクロデキストリン誘導体;(xi)中鎖脂肪酸;(xii)キレート剤;(xiii)アミノ酸またはその塩;(xiv)N-アセチルアミノ酸またはその塩;(xv)選択された膜成分に対する分解作用のある酵素;(ix)脂肪酸合成の阻害薬;(x)コレステロール合成の阻害薬;および(xi)(i)〜(x)に列挙された膜透過強化剤の任意の組み合わせ;
(h)上皮結合生理機能の調節剤;
(i)血管拡張薬;
(j)選択的輸送強化剤;ならびに
(k)化合物と有効に組み合わされ、会合し、含有され、封入され、または結合し、その結果、鼻粘膜送達の強化のための化合物の安定化をもたらす、安定化性の送達媒体、担体、粘膜付着剤(mucoadhesive)、支持体または複合体形成種であって、化合物と鼻腔内送達強化剤との製剤化が対象の血漿における化合物の生物学的利用能の増大をもたらすもの。
【0028】
1つの局面において、本発明は、対象の循環器系への低分子量化合物の吸収を、(a)化合物;(b)親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量;および(c)粘膜送達強化剤を、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して投与することによって増加させる方法を提供する。
【0029】
1つの局面において、本発明は、エキセンディン-4または関連GLP-1ペプチドの治療的有効量を、Intravailアルキルサッカリドの有効量とともに有する組成物を投与することによって、カロリー摂取を制御する方法を提供する。
【0030】
もう1つの局面において、本発明は、エキセンディン-4または関連GLP-1ペプチドの治療的有効量を、Intravailアルキルサッカリドの有効量とともに含む組成物を対象に投与することによって、対象における血糖レベルを制御する方法を提供する。
【0031】
さらに、もう1つの局面において、本発明は、以下のものを含む、制御放出投薬組成物を提供する:
(a)以下のものを含むコア:
(i)少なくとも1つの治療薬または薬物;
(ii)少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル;ならびに
(b)コアを取り囲む少なくとも1つの膜コーティングであって、コーティングが非透過性、透過性、半透過性または多孔性であり、胃腸管の内容物との持続的接触によってより透過性になる膜コーティング。
【0032】
もう1つの態様において、本発明は、アルキル鎖の長さが炭素原子約12〜約14個である少なくとも1つのアルキルグリコシド(alkyglycoside)、抗菌活性を有する少なくとも1つのサッカリド、および少なくとも1つの治療薬、の治療的有効量を投与することにより、アルキルグリコシド組成物を投与する方法を提供する。
【0033】
さらにもう1つの態様において、本発明は、インスリン、PYY、エキセンディン-4または他のGLP-1関連ペプチド、ヒト成長ホルモン、カルシトニン、副甲状腺ホルモン、PTH 1-34などの短縮型副甲状腺ホルモンペプチド、EPO、インターフェロンα、インターフェロンβ、インターフェロンγおよびGCSFからなる群より選択される少なくとも1つの薬物、ならびに抗菌活性を有する少なくとも1つのアルキルサッカリドを有する組成物を提供する。
【0034】
1つの局面において、本発明は、n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドまたはn-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む抗菌性アルキルサッカリド組成物を提供する。
【0035】
さらに、もう1つの局面において、本発明は、少なくとも1つの薬物および少なくとも1つの抗菌薬を約0.05%〜約0.5%の濃度で有する、経粘膜的または経皮的投与のための水性薬物組成物を提供する。
【0036】
もう1つの局面において、本発明は、マトリックス材料およびアルキルサッカリドを含む急速分散性薬物製剤を提供する。本製剤は、アルキルサッカリドを含まない同等の製剤について観察されるものよりも実質的に短いTmaxおよび実質的に小さい初回通過効果を有しうる。1つの態様において、本製剤は約0.1%〜10%のアルキルサッカリドを含んでよく、6時間よりも短いTmaxおよび40%よりも小さい初回通過効果を示す。アルキルグリコシドは任意の適したアルキルグリコシドであってよく、1つの好ましい局面において、ドデシルマルトシド、テトラデシルマルトシド、ドデカン酸スクロース、またはモノ-およびジ-ステアリン酸スクロースである。本製剤は、メラトニン、ラロキシフェン、オランザピン(olanzapene)およびジフェンヒドラミンなどの、ただしそれらには限定されない多種多様な治療薬を含みうる。
【0037】
もう1つの局面において、本発明は、胃送達と頬側送達とのバランスをとるために、薬物製剤中のアルキルサッカリド濃度を薄めることにより、長期間にわたる吸収曲線を提供するための方法を提供する。例えば、これは、マトリックス材料、ならびにアルキルサッカリドを含まない同等の製剤について観察されるものよりも実質的に短いTmaxおよび実質的に小さい初回通過効果を有するアルキルサッカリドを含む薬物製剤を提供することによって行われる。
【0038】
1つの局面において、本発明は、ビスホスホネート類似体またはトリプタン類似体の治療的有効量を、アルキルグリコシドの吸収増加量との組み合わせで有する薬学的組成物を提供する。さまざまな態様において、ビスホスホネート類似体は、エチドロネート、クロドロネート、チルドロネート、パミドロネート、ネリドロネート、オルパドロネート、アレンドロネート、イバンドロネート、リセドロネート、ゾレドロネート、および/またはそれらの薬学的に許容される類似体であってよい。1つの例示的な態様において、ビスホスホネート類似体はアレンドロネート、またはその薬学的に許容される類似体である。さまざまな態様において、トリプタン類似体は、スマトリプタン、リザトリプタン、ナラトリプタン、ゾルミトリプタン、エレトリプタン、アルモトリプタン、フロバトリプタン、および/またはそれらの薬学的に許容される類似体であってよい。1つの例示的な態様において、トリプタン類似体はスマトリプタンまたはその薬学的に許容される類似体である。さまざまな態様において、アルキルグリコシドはテトラデシル-β-D-マルトシドである。
【0039】
さらにもう1つの態様において、本発明は、対象におけるビスホスホネート類似体またはトリプタン類似体の生物学的利用能を、その化合物をアルキルグリコシドの吸収増加量とともに投与し、それにより、対象における化合物の生物学的利用能を高めることによって高める方法を提供する。
【0040】
さらにもう1つの態様において、本発明は、5-HT受容体アゴニストおよびアルキルサッカリドの治療的有効量を含む組成物を提供する。さまざまな態様において、5-HTアゴニストは、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである。
【0041】
さらにもう1つの態様において、本発明は、対象に対して、5-HTアゴニストの減量されているが治療的には有効な量を提供する方法を提供する。本方法は、5-HTアゴニストの治療的有効量;およびアルキルサッカリドを含む組成物である鼻腔内組成物を投与する段階を含み、ここでそのAUCは、アルキルサッカリドの非存在下で投与された5-HTアゴニストの増量された治療的有効量によってもたらされるAUCと比較してほぼ等しい。
【0042】
さらにもう1つの態様において、本発明は、対象における片頭痛緩和の迅速開始を提供する方法であって、5-HTアゴニストおよびアルキルサッカリドの治療的有効量を含む組成物であって対象において約30分またはそれ未満のTmaxを呈する組成物を投与し、片頭痛緩和の迅速開始を提供する段階を含む方法を提供する。
【0043】
さらにもう1つの態様において、本発明は、対象における片頭痛再発の発生率の低下を提供する方法であって、5-HTアゴニストおよびアルキルサッカリドの治療的有効量を含み、約20分未満のTmaxを提供する組成物である組成物を投与し、それにより、対象における片頭痛再発の発生率の低下を提供する段階を含む方法を提供する。
【図面の簡単な説明】
【0044】
【図1】アルキルグリコシドの存在下および非存在下でのMIACALCIN(登録商標)(サケカルシトニン)に関する、静脈内注射と比較した鼻腔内生物学的利用率および対象間変動係数を示しているグラフである。
【図2】血糖レベルを低下させる上での、インスリン/0.25% TDMの鼻腔内投与(●)およびインスリンのみの鼻腔内投与(○)の効果を示しているグラフである。
【図3】グルコースの腹腔内(IP)注射後の血糖レベルを低下させる上での(すなわち、いわゆる「ブドウ糖負荷試験」における)、エキセンディン-4/0.25% TDMの鼻腔内注射(▲)および腹腔内注射(IP)(●)投与、ならびにTDMを含まない食塩水のみのIP注射(○)の効果を示しているグラフである。
【図4】胃管による投与後の雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の1mgのマウスp-Leu-4]OB3の取り込みを示しているグラフである。
【図5】経口投与および直腸投与の両方についての、イヌによる、0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中のスマトリプタンの取り込みを示しているグラフである。
【図6】スマトリプタンが経鼻投与された患者の平均血漿レベルのグラフである。
【図7】スマトリプタンが経鼻投与された患者の平均血漿レベルのグラフである。
【発明を実施するための形態】
【0045】
発明の詳細な説明
本発明は、具体的な態様およびそれらの中に含まれる実施例の詳細な説明の参照によって、より容易に理解されるであろう。
【0046】
本発明は、少なくとも1つの薬物および少なくとも1つの表面活性物質を含む治療用組成物であって、表面活性物質が、薬物の生物学的利用能を高め、かつ対象に投与された場合に観察可能な有害作用を有しない、少なくとも1つのアルキルグリコシドおよび/または少なくとも1つのサッカリドアルキルエステルである、治療用組成物の発見に基づく。
【0047】
「治療用組成物」は、有機性または無機性の担体または添加剤との混合物からなることができ、例えば、錠剤、ペレット剤、カプセル剤、坐薬、液剤、乳剤、懸濁剤、または使用のために適した他の形態のための通常の無毒性の薬学的に許容される担体とともに調合することができる。担体には、以上に開示したものに加えて、グルコース、ラクトース、マンノース、アラビアゴム、ゼラチン、マンニトール、デンプン糊、三ケイ酸マグネシウム、タルク、コーンスターチ、ケラチン、コロイドシリカ、ジャガイモデンプン、尿素、中鎖長トリグリセリド、デキストラン、および固体、半固体または液体の形態にある調合物の製造に用いるために適した他の担体が含まれうる。加えて、補助的な安定剤、増粘剤または着色剤、例えばトリウロースなどの乾燥安定剤を用いることもできる。
【0048】
「薬物」とは、核酸、小分子、タンパク質、ポリペプチドまたはペプチドなどを非限定的に含む、任意の治療用化合物、または分子、または治療薬、または生物活性化合物のことである。
【0049】
「核酸」または「オリゴヌクレオチド」という用語はまた、翻訳領域および非翻訳領域をコードする、またはペプチドもしくはタンパク質をコードする構造遺伝子の翻訳領域もしくは非翻訳領域または調節領域を阻害する、DNA、cDNA、RNA、siRNA、RNAi、dsRNAなども表す。例えば、本発明の核酸は、5'および3'非翻訳調節ヌクレオチド配列、ならびに構造遺伝子に付随する翻訳配列を含みうる。本明細書で用いる場合、「核酸」または「オリゴヌクレオチド」または文法的等価物は、共有結合して一つになった少なくとも2つのヌクレオチドのことを指す。
【0050】
さらに、「オリゴヌクレオチド」という用語は、修飾された糖モイエティー、修飾された塩基モイエティーまたは修飾された糖連結モイエティーなどの修飾された部分を含む構造のことも指す。これらの修飾された部分は、天然塩基、天然糖および天然ホスホジエステル結合と類似した様式で機能する。したがって、オリゴヌクレオチドは、改変された塩基モイエティー、改変された糖モイエティーまたは改変された糖間(inter-sugar)結合を有してもよい。修飾された結合は、例えば、ホスホルアミド、ホスホロチエオート、ホスホロジチオエート、メチルホスホネート、ホスホトリエステル、ホスホルアミデート、O-メチルホスホロアミダイト結合、またはペプチド核酸骨格および結合であってよい。他の類似体には、正の骨格(positive backbone)、非イオン性骨格および非リボース骨格を有するオリゴヌクレオチドが含まれうる。核酸はゲノム性およびcDNAの両方のDNA、RNAまたはハイブリッドであってよく、ここで核酸は、デオキシリボヌクレオチドおよびリボヌクレオチドの任意の組み合わせ、ならびにウラシル、アデニン、チミン、シトシン、グアニン、イノシン、キサンチン、ヒポキサンチン、イソシトシン、イソグアニン、ハロゲン化塩基(halogentated base)などを含む天然の塩基または修飾された塩基の任意の組み合わせを含む。他の修飾には、例えば、天然のプリンおよびピリミジン塩基の代わりに用いられるデアザまたはアザプリンおよびピリミジン;5位もしくは6位に置換基を有するピリミジン塩基、2位、6位もしくは8位に改変された置換基もしくは置き換え置換基を有するプリン塩基、またはそれらの2'位に置換基を有する糖、糖の水素原子の1つもしくは複数の置換、または炭素環式糖もしくは非環式糖が含まれうる。
【0051】
「アンチセンス」という用語は、本明細書で用いる場合、特定の核酸配列に対して相補的な任意の組成物のことを指す。「アンチセンス鎖」という用語は、「センス」鎖に対して相補的な核酸鎖を指して用いられる。アンチセンス分子は、合成または転写を含む任意の方法によって作製しうる。細胞内にひとたび導入されると、相補的ヌクレオチドは細胞によって産生された天然配列と組み合わされて、二重鎖を形成し、転写または翻訳のいずれかを阻止する。
【0052】
アンチセンス分子には、標的受容体またはリガンドmRNA(センス)もしくはDNA(アンチセンス)配列と結合することのできる一本鎖核酸配列(RNAまたはDNAのいずれか)を含むオリゴヌクレオチドが含まれる。アンチセンスまたはセンスオリゴヌクレオチドを導き出す能力は、所与のタンパク質をコードするcDNA配列に基づく。アンチセンスまたはセンスオリゴヌクレオチドにはさらに、修飾された糖-ホスホジエステル骨格を有するオリゴヌクレオチドが含まれ、そのような糖結合は内因性ヌクレアーゼに対して抵抗性である。抵抗性の糖結合を有するそのようなオリゴヌクレオチドはインビボで安定である(すなわち、酵素分解に抵抗することができる)が、標的ヌクレオチド配列と結合することのできる配列特異性は保っている。
【0053】
RNAiは、多様な範囲の生物および細胞種へのdsRNAの導入が相補的mRNAの分解を引き起こす現象である。細胞内で、長いdsRNAはリボヌクレアーゼによって短い(例えば、21〜25ヌクレオチド)の短鎖干渉性RNA(siRNA)へと切断される。siRNAはその後、タンパク質成分と集合してRNA誘導サイレンシング複合体(RISC)となり、そのプロセスにおいて巻き戻し(unwinding)を行う。活性化されたRISCは続いて、siRNAアンチセンス鎖とmRNAとの間の塩基対合相互作用によって相補的転写物と結合する。結合したmRNAは続いて切断され、mRNAの配列特異的分解の結果、遺伝子サイレンシングが起こる。本明細書で用いる場合、「サイレンシング」とは、細胞が染色体DNAの大きな区域を停止させ、その結果、特定の遺伝子の発現を抑制する機構のことを指す。RNAi機構は、ゲノムを内因性転移因子およびウイルス感染から防御するために進化したように思われる。このため、RNAiは、分解させようとする標的mRNAに対して相補的な核酸分子を導入することによって誘導することができる。
【0054】
センスまたはアンチセンスオリゴヌクレオチドの他の例には、ポリ-(L-リジン)のような、標的核酸配列に対するオリゴヌクレオチドの親和性を高める有機モイエティーおよび他のモイエティーと共有結合したオリゴヌクレオチドが含まれる。さらになお、標的ヌクレオチド配列に対するアンチセンスまたはセンスオリゴヌクレオチドの結合特異性を修飾するために、エリプチシンなどの挿入剤、およびアルキル化剤または金属複合体を、センスまたはアンチセンスオリゴヌクレオチドと結びつけることもできる。
【0055】
本発明のペプチドは、小型ないし中程度のサイズ(すなわち、例えば最大で約15kD、30kD、40kD、50kD、60kD、70kD、80kD、90kD、100kD)の、任意の医学的または診断的に有用なペプチドまたはタンパク質であってよい。ポリペプチド吸収の改善の機序は、米国特許第5,661,130号に記載されており、これはその全体が参照により本明細書に組み入れられる。本発明の組成物はそのようなあらゆるペプチドと混合することができるが、ペプチドの利点が改善される度合いは、ペプチドの分子量ならびに物理的および化学的性状、ならびに用いられる特定の表面活性物質に応じて異なると考えられる。ポリペプチドの例には、バソプレシン、バソプレシンポリペプチド類似体、デスモプレシン、グルカゴン、コルチコトロピン(ACTH)、ゴナドトロピン、カルシトニン、インスリンのC-ペプチド、副甲状腺ホルモン(PTH)、成長ホルモン(HG)、ヒト成長ホルモン(hGH)、成長ホルモン放出ホルモン(GHRH)、オキシトシン、コルチコトロピン放出ホルモン(CRH)、ソマトスタチンまたはソマトスタチンポリペプチド類似体、ゴナドトロピンアゴニストまたはゴナドトロフィン(gonadotrophin)アゴニストポリペプチド類似体、ヒト心房性ナトリウム利尿ペプチド(ANP)、ヒトチロキシン放出ホルモン(TRH)、濾胞刺激ホルモン(FSH)、プロラクチン、インスリン、インスリン様増殖因子-I(IGF-I) ソマトメジン-C(SM-C)、カルシトニン、レプチンおよびレプチン由来短ペプチドOB-3、メラトニン、GLP-1またはグルカゴン様ペプチド-1、GiP、ニューロペプチド 下垂体アデニル酸シクラーゼ、GM-1ガングリオシド、神経成長因子(NGF)、ナファレリン、D-tryp6)-LHRH、FGF、VEGFアンタゴニスト、ロイプロリド、インターフェロン(例えば、α、β、γ)低分子量ヘパリン、PYY、LHRHアンタゴニスト、ケラチノサイト増殖因子(KGF)、グリア由来神経栄養因子(GDNF)、グレリンおよびグレリンアンタゴニストが含まれる。さらに、いくつかの局面において、ペプチドまたはタンパク質は、増殖因子、インターロイキン、ポリペプチドワクチン、酵素、エンドルフィン、糖タンパク質、リポタンパク質、または血液凝固カスケードに関与するポリペプチドから選択される。
【0056】
他の薬物または治療用化合物、分子および/または作用物質には、中枢神経系に影響を及ぼす神経伝達物質または神経イオンチャンネルの化合物または分子(すなわち、抗うつ薬(ビュープロピオン))、選択的セロトニン2c受容体アゴニスト、抗けいれん薬(トピラマート、ゾニサミド)、いくつかのドーパミンアンタゴニスト、およびカンナビノイド-1受容体アンタゴニスト(リモバナン));レプチン/インスリン/中枢神経系経路作用薬(すなわち、レプチン類似体、レプチン輸送体および/またはレプチン受容体プロモーター、毛様体神経栄養因子(Axokine)、ニューロペプチドYおよびagouti関連ペプチドアンタゴニスト、プロオピオメラノコルチン、コカイン・アンフェタミン調節転写産物プロモーター、α-メラノサイト刺激ホルモン類似体、メラノコルチン-4受容体アゴニスト、タンパク質チロシンホスファターゼ-1B阻害薬、ペルオキシソーム増殖因子活性化受容体-γ受容体アンタゴニスト、短時間作用型ブロモクリプチン(エルゴセット(ergoset))、ソマトスタチンアゴニスト(オクトレオチド)およびアディポネクチン);消化管-神経経路作用薬(すなわち、グルカゴン様ペプチド-1活性を上昇させる作用物質(エクステンディン-4(extendin-4)、リラグルチド、ジペプチジルペプチダーゼIV阻害薬)、タンパク質YY3-36、グレリン、グレリンアンタゴニスト、アミリン類似体(プラムリンチド));ならびに、安静時代謝率を高める可能性のある化合物または分子 「選択的」β-3刺激物質/アゴニスト、メラニン凝集ホルモンアンタゴニスト、フィトスタノール(phytostanol)類似体、機能性油、P57、アミラーゼ阻害薬、成長ホルモン断片、硫酸デヒドロエピアンドロステロンの合成類似体、脂肪細胞11B-ヒドロキシステロイドデヒドロゲナーゼ1型活性のアンタゴニスト、コルチコトロピン-放出ホルモンアゴニスト、脂肪酸合成の阻害薬、カルボキシペプチダーゼ阻害薬、消化管リパーゼ阻害薬(ATL962)、メラトニン、ラロキシフェン、オランザピンおよびジフェンヒドラミンが含まれる。
【0057】
他の薬物または治療用化合物には、ビスホスホネート類似体などの骨粗鬆症薬が含まれる。ビスホスホネート類似体は、ジホスホネートとしても知られ、骨粗鬆症、変形性骨炎(骨パジェット病)、骨転移(高カルシウム血症を伴うかまたは伴わない)、多発性骨髄腫、骨形成不全症、および骨脆弱性を特徴とする他の病状などの病状の治療のために臨床的に用いられている。このクラスの薬物は、破骨細胞の活動および骨再吸収を阻害する。本明細書に記載された組成物中に用いるためにアルキルサッカリドと混和されるビスホスホネートの例には、N非含有性およびN含有性の両方のビスホスホネート類似体が含まれる。N非含有性ビスホスホネートの例には、エチドロネート(Didronel(商標))、クロドロネート(Bonefos(商標)、Loron(商標))、チルドロネート(Skelid(商標))、およびそれらの薬学的に許容される類似体が含まれる。N含有性ビスホスホネートの例には、パミドロネート(Aredia(商標))、ネリドロネート、オルパドロネート、アレンドロネート(Fosamax(商標)またはFosamax+D(商標))、イバンドロネート(Boniva(商標))、リセドロネート(Actonel(商標))およびゾレドロネート(Zometa(商標)またはReclast(商標))、およびそれらの薬学的に許容される類似体が含まれる。
【0058】
他の薬物または治療用化合物には、トリプタン類似体などの薬物が含まれる。トリプタン類似体は一般に、片頭痛および頭痛の治療のために用いられる、トリプタミンをベースとする薬物のファミリーである。それらの作用は、神経終末および頭蓋内血管におけるセロトニン受容体に対するそれらの結合(それらの収縮を引き起こす)、ならびにそれに引き続く炎症誘発性ニューロペプチド放出の阻害に起因する。本明細書に記載の組成物中に用いるためにアルキルサッカリドと混和されるトリプタンの例には、スマトリプタン(Imitrex(商標)およびImigran(商標))、リザトリプタン(Maxalt(商標))、ナラトリプタン(Amerge(商標)およびNaramig(商標))、ゾルミトリプタン(Zomig(商標))、エレトリプタン(Relpax(商標))、アルモトリプタン(Axert(商標)およびAlmogran(商標))、フロバトリプタン(Frova(商標)およびMigard(商標))、およびそれらの薬学的に許容される塩が含まれる。薬学的に許容される塩の例には、ナラトリプタン-HCl;硫酸スマトリプタンおよび安息香酸リザトリプタンにおけるような、塩酸塩、硫酸塩または安息香酸塩が含まれる。トリプタンの塩形態は「遊離塩基」または非荷電形態と比較してより高い水溶性を示し、このため、本明細書中のさまざまなトリプタンの水性製剤の記載においては、トリプタンの可溶性塩形態を添加すること、またはトリプタンの遊離塩基形態に対する対応する酸(塩酸、硫酸、安息香酸など)の添加によってインサイチューで調製することを意図していることが理解されるべきである。
【0059】
本発明の治療用組成物は、薬物および薬物吸収強化剤、例えば表面活性物質を含む。「表面活性物質」という用語は、水の界面張力を変更させる任意の表面活性剤(surface active agent)のことである。典型的には、表面活性物質は、分子内に1つの親油基および1つの親水基を有する。大まかに言って、この群には、石鹸、界面活性剤(detergent)、乳化剤、分散剤および湿潤剤、ならびに消毒薬のいくつかの群が含まれる。より具体的には、表面活性物質には、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウムおよびモノステアリン酸グリセリン;ならびにポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロースおよびヒドロキシプロピルセルロースなどの親水性ポリマーが含まれる。
【0060】
好ましくは、本発明の表面活性物質は、少なくとも1つの適したアルキルグリコシドからなる。本明細書で用いる場合、「アルキルグリコシド」とは、当技術分野において公知であるように、任意の疎水性アルキルとの結合によって連結された任意の糖のことを指す。任意の「適した」アルキルグリコシドとは、本発明を限定する特性、すなわち、アルキルグリコシドが無毒性および非イオン性であること、ならびにそれが化合物の吸収を、それが化合物とともに眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して投与された場合に増加させることを満たす。適した化合物は、本明細書に記述する方法を用いて決定することができる。
【0061】
本発明のアルキルグリコシドは、公知の手順により、例えば、Rosevear et al., Biochemistry 19:4108-4115 (1980)もしくはKoeltzow and Urfer, J Am. Oil Chem. Soc., 61:1651-1655 (1984)、米国特許第3,219,656号および米国特許第3,839,318号に記載されたように化学的に、または例えば、Li et al., J. Biol. Chem., 266:10723-10726 (1991)もしくはGopalan et al., J. Biol. Chem. 267:9629-9638 (1992)に記載されたように酵素的に合成することができる。
【0062】
本発明のアルキルグリコシドには、以下のものが非限定的に含まれうる:アルキルグリコシド、例えばオクチル-、ノニル-、デシル-、ウンデシル-、ドデシル-、トリデシル-、テトラデシル-、ペンタデシル-、ヘキサデシル-、ヘプタデシル-およびオクタデシル-α-またはβ-D-マルトシド、-グルコシドまたは-スクロシドなど(Koeltzow and Urferに従って合成;Anatrace Inc., Maumee, Ohio;Calbiochem, San Diego, Calif.; Fluka Chemie, Switzerland);アルキルチオマルトシド、例えばヘプチル、オクチル、ドデシル-、トリデシル-およびテトラデシル-β-D-チオマルトシドなど(Defaye, J. and Pederson, C., "Hydrogen Fluoride, Solvent and Reagent for Carbohydrate Conversion Technology" in Carbohydrates as Organic Raw Materials, 247-265 (F. W. Lichtenthaler, ed.) YCH Publishers, New York (1991);Ferenci, T., J. Bacteriol, 144:7-11 (1980)に従って合成);アルキルチオグルコシド、例えばヘプチル-またはオクチル1-チオαまたはβ-D-グルコピラノシド(Anatrace, Inc., Maumee, Ohio;Saito, S. and Tsuchiya, T. Chem. Pharm. Bull. 33:503-508 (1985)を参照);アルキルチオスクロース(例えば、Binder, T. P. and Robyt, J. F., Carbohydr. Res. 140:9-20 (1985)に従って合成);アルキルマルトトリオシド(Koeltzow and Urferに従って合成);スクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸アミド;(オーストラリア特許第382,381号(1987);Chem. Abstr., 108:114719 (1988)およびGruber and Greber pp. 95-116に従って合成);アミド結合によってアルキル鎖と連結されたパラチノースおよびイソマルタミンの誘導体(Kunz, M., "Sucrose-based Hydrophilic Building Blocks as Intermediates for the Synthesis of Surfactants and Polymers" in Carbohydrates as Organic Raw Materials, 127-153に従って合成);尿素によってアルキル鎖と連結されたイソマルタミンの誘導体(Kunzに従って合成);スクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸ウレイド(Gruber and Greber, pp. 95-116に従って合成);ならびにスクロースβ-アミノ-アルキルエーテルの長鎖脂肪族カルボン酸アミド(オーストラリア特許第382,381号(1987)、Chem. Abstr., 108:114719 (1988)およびGruber and Greber, pp. 95-116に従って合成)。
【0063】
アルキルグリコシドおよび/またはスクロースエステルからなる本発明の表面活性物質は、特徴的な親水性-親油性バランス(HLB)数値を有し、それは経験的に計算または決定することができる(Schick, M. J. Nonionic Surfactants, p. 607 (New York: Marcel Dekker, Inc. (1967))。HLB数値は表面活性物質の親水性特性を直接反映したものであり、すなわち、HLB数値が大きいほど化合物の親水性は高い。HLB数値は、式:(親水性成分のMWの20倍)/(疎水性成分のMW+親水性成分のMW)によって計算することができ、式中、MW=分子量である(Rosen, M. J., Surfactants and Interfacial Phenomena, pp. 242-245, John Wiley, New York (1978))。HLB数値は、表面活性物質の親水性特性を直接表現したものであり、すなわち、HLB数値が大きいほど化合物の親水性は高い。好ましい表面活性物質は約10〜20のHLB数値を有し、さらにより好ましい範囲は約11〜15である。
【0064】
上記の通り、疎水性アルキルは、サッカリドモイエティーの所望の疎水性および親水性に応じて、任意の所望のサイズのものを選択することができる。例えば、アルキル鎖の1つの好ましい範囲は炭素原子が約9〜約24個である。さらにより好ましい範囲は炭素原子が約9〜約16個または約14個である。同様に、いくつかの好ましいグリコシドは、グリコシド結合によって炭素原子が9、10、12、13、14、16、18、20、22または24個のアルキル鎖と連結されたマルトース、スクロースおよびグルコース、例えば、ノニル-、デシル-、ドデシル-およびテトラデシルスクロシド、グルコシドおよびマルトシドなどである。これらの組成物は、アルコールおよびオリゴサッカリドに分解され、両親媒性であることから無毒性である。
【0065】
また、本発明の表面活性物質にはサッカリドも含まれる。本明細書で用いる場合、「サッカリド」は、モノサッカリド、直鎖形態または環状形態にあるオリゴサッカリドもしくはポリサッカリド、またはサッカリド鎖を形成するそれらの組み合わせを含む。オリゴサッカリドは、2つまたはそれ以上のモノサッカリド残基を有するサッカリドのことである。サッカリドは、例えば、現在市販されている任意のサッカリド種から選択することもでき、または合成することもできる。用いうる可能性のある多くのサッカリドのいくつかの例には、グルコース、マルトース、マルトトリオース、マルトテトラオース、スクロースおよびトレハロースが含まれる。好ましいサッカリドには、マルトース、スクロースおよびグルコースが含まれる。
【0066】
本発明の表面活性物質は、同様にスクロースエステルからなることもできる。本明細書で用いる場合、「スクロースエステル」とは脂肪酸のスクロースエステルのことであり、これはスクロースおよび脂肪酸の複合体である。スクロースエステルは、スクロース中に反応に利用しうるヒドロキシル基が8つあり、酢酸基からより大きくより嵩の高い脂肪酸までに至る多くの脂肪酸基がスクロースと反応しうるため、多くの形態をとることができる。この融通性は、用いる脂肪酸モイエティーに基づき、多くの生成物および官能性を適応させることができることを意味する。スクロースエステルは、食品用および非食品用の用途を有し、特に表面活性物質および乳化剤としての用途を有し、医薬品、化粧品、表面活性物質および食品添加物における適用が増えている。それらは生分解性であり、皮膚に対して無毒性であるとともに刺激性が少ない。
【0067】
本発明の表面活性物質は、親水性サッカリドと連結した疎水性アルキル基を有する。疎水性アルキル基と親水性サッカリドとの間の結合には、他にも可能性はあるものの、グリコシド、チオグリコシド(Horton)、アミド(Carbohydrates as Organic Raw Materials, F. W. Lichtenthaler ed., VCH Publishers, New York, 1991)、ウレイド(オーストラリア特許第386,414号(1988);Chem. Abstr. 11O:137536p (1989);Gruber, H. and Greber, G., "Reactive Sucrose Derivatives" in Carbohydrates as Organic Raw Materials, pp. 95-116を参照)またはエステル結合(Sugar Esters: Preparation and Application, J. C. Colbert ed., (Noyes Data Corp., New Jersey), (1974))が含まれうる。さらに、好ましいグリコシドには、グリコシド結合によって炭素原子約9〜16個のアルキル鎖と連結されたマルトース、スクロースおよびグルコース、例えば、ノニル-、デシル-、ドデシル-およびテトラデシルスクロシド、グルコシドおよびマルトシドが含まれうる。この場合も、これらの組成物は両親媒性であり、それらはアルコールおよびオリゴサッカリドへと分解されることから無毒性である。
【0068】
上記の例は、本明細書において特許請求を行う方法に用いられるグリコシドの種類の例示であるが、このリストは網羅的ではない。特許請求の範囲の基準に当てはまる上記の化合物の誘導体も、グリコシドを選択する場合に考慮されるべきである。化合物はすべて、本明細書および実施例に教示された方法に従って有効性に関してスクリーニングすることができる。
【0069】
本発明の組成物は、錠剤、カプセル剤、坐薬、滴剤、噴霧剤、エアロゾル、および持続放出形式または遅延バースト形式からなる群より選択される形式で投与することができる。噴霧剤およびエアロゾルは、適切なディスペンサーの使用によって実現することができる。持続放出形式は、眼インサート(ocular insert)、侵食性(erodible)微粒子、膨張性粘膜付着剤粒子、pH感受性微粒子、ナノ粒子/ラテックス系、イオン交換樹脂ならびに他のポリマー性ゲルおよびインプラントでありうる(Ocusert, Alza Corp., California;Joshi, A., S. Ping and K. J. Himmelstein, 特許出願第WO 91/19481号)。これらの系は、吸収性表面との長期にわたる薬物接触を維持し、流出および非生産的な薬物喪失を防止する。長期にわたる薬物接触は皮膚および粘膜の表面に対して無毒性である。
【0070】
本発明の表面活性物質組成物は安定である。例えば、Baudysらは、米国特許第5,726,154号において、SDS(ドデシル硫酸ナトリウム、表面活性物質)および有機酸を含む水性液体組成物中のカルシトニンが、少なくとも6カ月にわたって安定であることを示している。同様に、本発明の表面活性物質組成物は、薬物と混和された場合に、改善された安定化特性を有する。これらの製剤中に有機酸は必要でない。例えば、本発明の組成物は、約4℃〜25℃に維持された場合に、タンパク質およびペプチド治療薬の安定性を約6カ月またはそれ以上にわたって維持する。
【0071】
表面活性物質組成物の安定性は、一部には、それらの高い最大無毒性レベル(NOAEL)に起因する。米国環境保護庁(Environmental Protection Agency)(EPA)は、最大無毒性レベル(NOAEL)を、曝露された集団とその適切な対照との間で有害作用の頻度または重症度に関して統計学的にも生物学的にも有意な増加がみられない曝露レベルと定義している。それ故に、「最大無毒性レベル」(またはNOAEL)という用語は、規定された条件下で標的生物の形態、機能的能力、成長、発達または寿命の検出可能で有害な変化を引き起こさない、実験または観察によって見いだされる、物質の最大の濃度または量のことである。
【0072】
世界保健機構(World Health Organization)(WHO)の国連食糧農業機関(Food and Agriculture Organization of the United Nations)(FAO)は、いくつかのアルキルグリコシドが非常に高いNOAELを有し、いかなる有害作用も伴わずにこれらのアルキルグリコシドの摂取増加が可能であることを示している。この報告は、ワールドワイドウェブ上のinchem.org/documents/jecfa/jecmono/v1Oje11.htmで見ることができる。例えば、食品中に用いられるスクロースエステルの1つであるドデカン酸スクロースのNOAELは、約20〜30グラム/キログラム/日であり、例えば、70キログラムの人(約154ポンド)は、1日当たり約1400〜2100グラム(または約3〜4.6ポンド)のドデカン酸スクロースを、いかなる観察可能な有害作用も伴わずに摂取することができる。典型的には、ヒトの1日許容摂取量はNOAELの約1%であり、これは無期限に1日当たり約14〜21グラムまたは1400万マイクログラム〜2100万マイクログラムに換算される。NOAELの定義および他の関連した定義は、ワールドワイドウェブ上のepa.gov/OCEPAtermsで見ることができる。このため、本発明において予期されるアルキルグリコシドのレベルで、ある程度の影響は生じる可能性はあるが、それらのレベルは有害であるとも有害作用の前兆であるとも考えられない。
【0073】
したがって、少なくとも1つのアルキルグリコシド、例えば、テトラデシルマルトシド(TDM;またはIntravail A)を、重量比で約0.125%の濃度のアルキルグリコシドを有する本発明の表面活性物質組成物により、治療レジメンに応じて1日2回または1日3回またはそれ以上治療される対象は、1日当たり合計で約200〜300マイクログラムのTDMを摂取する。そのため、TDMの有効量はNOAELの少なくとも1000分の1(すなわち、1/1000)であり、1日許容摂取量であるNOAELの1%よりもはるかに少ない;またはこの場合には1日許容摂取量の約1/50,000である。別の言い方をすれば、本発明のアルキルグリコシドは、本発明において用いられるアルキルグリコシドの量または濃度が有害作用を引き起こさず、いかなる有害作用も伴わずに安全に摂取しうるような高いNOAELを有する。
【0074】
本発明の表面活性物質組成物はまた、それらが生理的に無毒性および非刺激性であるため、安定でもある。「無毒性」という用語は、アルキルグリコシド分子が、ヒトへの投与および摂取のために適する十分に低い毒性を有することを意味する。好ましいアルキルグリコシドは、それらが適用される組織に対して非刺激性である。用いられる任意のアルキルグリコシドは、それが細胞に障害を引き起こさないように非常にわずかな毒性しか有しないかまたは全く毒性を有しないべきである。しかしながら、任意の所与のアルキルグリコシドの毒性は、用いられるアルキルグリコシドの濃度に応じて異なりうる。選択されたアルキルグリコシドが身体によって代謝または排出され、この代謝または排出が有害な毒性をもたらさない様式で行われることも有益である。「非刺激性」という用語は、その作用物質が、皮膚表面または粘膜との即時的、長期的または反復的な接触後に炎症を引き起こさないことを意味する。
【0075】
その上、表面活性物質組成物の、特にスクロースエステルの1つの態様は、抗菌作用薬として役立つ。ある作用物質は、その作用物質またはその等価物が細菌を破壊するかまたは細菌の増殖もしくは繁殖を抑制するならば、「抗菌」薬または物質である。スクロースエステルおよびそれらの脂肪酸の抗菌活性は報告されている。Tetsuaki et al. (1997) "Lysis of Bacillus subtilis cells by glycerol and sucrose esters of fatty acids," Applied and Environmental Microbiology, 53(3):505-508。Watanabeら(2000)は、ラウリン酸ガラクトースおよびラウリン酸フルクトース(galactose and fructose laureate)が特に有効な糖質モノエステルであることを記載している。Watanabe et al., (2000) "Antibacterial carbohydrate monoesters suppressing cell growth of Streptococcus mutan in the presence of sucrose," Curr Microbiol 41(3): 210-213。それ故に、本発明は、本明細書に記載されたスクロースエステルには限定されず、細菌の増殖および繁殖を抑制する、ガラクトースエステルおよびフルクトースエステルを含む他の糖質エステルも範囲に含む。Sutton and Porter (2002), "Development of the antimicrobial effectiveness test as USP Chapter <51>," 56(6): 300-311を参照、これはその全体が参照により本明細書に組み入れられる。例えば、塩化ベンザルコニウムのように一般的に用いられる抗菌薬は、鼻腔内製剤で一般的に用いられるよりもはるかに低いベンザルコニウム濃度で粘膜繊毛表面の破壊が観察されている電子顕微鏡検査によって示されているように、毒性が高度である。例えば、Sebahattin Cureoglu, Murat Akkus, Ustun Osma, Mehmet Yaldiz, Faruk Oktay, Belgin Can, Cengiz Guven, Muhanunet Tekin, and Faruk Menc (2002), "The effect of benzalkonium chloride an electron microscopy study," Eur Arch Otorhinolaryngol 259:362-364を参照。
【0076】
本発明の表面活性物質組成物は、典型的には、重量比で約0.01%〜20%のレベルで存在する。より好ましい組み込みレベルは、重量比で約0.01%〜5%、重量比で約0.01%〜2%、約0.01%〜1%、最も好ましくは重量比で約0.01%〜0.125%である。表面活性物質は好ましくは、組成物中に存在する他の成分と適合するように製剤化される。液体またはゲルまたはカプセルまたは注射用または噴霧薬の組成物において、表面活性物質は最も好ましくは、それがこれらの組成物中の任意のタンパク質または酵素の安定性を促進するか、または少なくともそれを劣化させないように製剤化される。さらに、本発明は、依然として所望の効果を維持しながら吸収強化因子の濃度をできるだけ低く保つことにより、濃度を最適化する。
【0077】
本発明の組成物は、対象に投与された場合、生物活性化合物または薬物の粘膜送達の強化をもたらし、対象への等価濃度の化合物の筋肉内注射後の組織(例えば、CNS)または体液または血漿における化合物のCmaxと比較して、対象の組織または体液または血漿中における化合物のピーク濃度(またはCmax)が約15%、20%または50%またはそれ以上である。
【0078】
薬物または化合物が設定期間、例えば、24時間で血流にどの程度達するかの指標を、24時間またはより長い期間の中のさまざまな時間で薬物血中濃度をプロットし、続いて0〜24時間の間の曲線下面積(AUC)を測定することによって算出することもできる。同様に、薬物有効性の指標を、約0.1〜1.0時間の間の、対象の組織(例えば、CNS)または体液または血漿における生物活性化合物の最大濃度到達時間(Tmax)から決定することもできる。本発明の治療用組成物は、薬物作用の開始の速さを約1.5倍〜2倍に高める(すなわち、Tmaxを減少させる)。
【0079】
また、本発明の治療用組成物または製剤を、必要とする対象に全身的または局所的に投与または送達することもできる。適した経路には、例えば、経口、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)、経粘膜投与、膣、直腸のほか、筋肉内、皮下、静脈内、腹腔内またはCSF送達を含む避腸的送達が含まれる。その上、送達の様式、例えば、液体、ゲル、錠剤、噴霧剤などは、対象への送達の方法にも依存すると考えられる。
【0080】
さらに、本発明の治療用組成物は、薬学的に許容される担体からなることもできる。「薬学的に許容される担体」とは、水性または非水性の作用物質、例えばアルコール性または油性のもの、またはそれらの混合物であり、表面活性物質、皮膚軟化剤、潤滑剤、安定剤、色素、香料、保存料、pHの調整のための酸もしくは塩基、溶媒、乳化剤、ゲル化剤、保湿剤(moisturizer)、安定剤、湿潤剤、持続放出剤、保湿剤(humectant)、または特定の薬学的組成物の形態に一般的に含まれる他の成分が含まれうる。薬学的に許容される担体は当技術分野において周知であり、これには例えば、水もしくは生理的緩衝食塩水などの水性溶液、またはグリコール、グリセロールなどの他の溶媒または媒体、およびオリーブ油または注射可能な有機エステルなどの油が含まれる。薬学的に許容される担体には、例えば、特定の阻害薬の吸収を安定化または増加させるように作用する生理的に許容される化合物、例えば、グルコース、スクロースもしくはデキストランなどの糖質、アスコルビン酸もしくはグルタチオンなどの酸化防止剤、キレート剤、低分子量タンパク質、または他の安定剤もしくは添加剤が含まれうる。また、薬学的に許容される担体を、蒸留水、ベンジルアルコール、ラクトース、デンプン、タルク、ステアリン酸マグネシウム、ポリビニルピロリドン、アルギン酸、コロイドシリカ、二酸化チタンおよび香味剤などの物質から選択することもできる。
【0081】
さらに、薬物の加水分解に対するアルキルサッカリドまたはサッカリドアルキルエステルの感受性を低下させるために、薬物内部のさまざまな酸素原子をイオウで置換することもできる(Defaye, J. and Gelas, J. in Studies in Natural Product Chemistry (Atta-ur-Rahman, ed.) Vol. 8, pp. 315-357, Elsevier, Amsterdam, 1991)。例えば、糖環のヘテロ原子は酸素もしくはイオウのいずれであってもよく、またはオリゴサッカリド中のモノサッカリド間の結合が酸素もしくはイオウであってもよい(Horton, D. and Wander, J. D., "Thio Sugars and Derivatives," The Carbohydrates: Chemistry and Biochemistry, 2d. Ed. Vol. IB, (W. Reyman and D. Horton eds.), pp. 799-842, (Academic Press, New York), (1972))。オリゴサッカリドはα(アルファ)またはβ(ベータ)アノマー立体配置のいずれであってもよい(Pacsu, E., et al. in Methods in Carbohydrate Chemistry (R. L. Whistler, et al., eds.) Vol. 2, pp. 376-385, Academic Press, New York 1963)を参照。
【0082】
本発明の組成物は、治療薬または薬物、ならびに本発明による1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル、ならびに適切な薬学的担体または添加剤、例えばマンニトール、コーンスターチ、ポリビニルピロリドンなどを混合し、その混合物を顆粒化し、最後にそれをコーンスターチ、ステアリン酸マグネシウムなどの薬学的担体の存在下で圧縮することにより、錠剤形態に調製することができる。必要に応じて、そのようにして調製された製剤が、糖コーティングもしくは腸溶性コーティングを含んでもよく、または例えば適切なpH媒質中で活性成分が徐々に放出されるような様式で覆われていてもよい。
【0083】
「腸溶性コーティング」という用語は、治療用組成物またはコアを包み込む、取り囲む、またはその周りに層もしくは膜を形成するポリマーのことである。また、腸溶性コーティングが、コーティングに対して適合性または不適合性の薬物を含んでもよい。1つの錠剤組成物は、比較的高いpHレベル(例えば、4.0を上回るpH、4.5を上回るpH、5.0を上回るpH、またはそれ以上)では溶解するかもしくは薬物を放出するが低いpHレベル(例えば、pH 4またはそれ未満)ではそうでない適合性薬物;またはその反対である適合性薬物を有する腸溶性コーティングポリマーを含みうる。
【0084】
1つの好ましい態様において、本発明の用量依存的放出形態は、以下のものを含む錠剤である:
(a)以下のものを含むコア:
(i)治療薬または薬物;
(ii)少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む表面活性物質;ならびに
(b)コアを取り囲む少なくとも1つの膜コーティング、ここでこのコーティングは非透過性、透過性、半透過性または多孔性コーティングであり、規定されたpHの水性環境と接触すると、より透過性または多孔性になる。
【0085】
「膜」という用語は、「コーティング」またはその等価物と同義である。これらの用語は、医薬品、例えば錠剤の、水性溶液もしくは体液に対して、および/またはその中に封入された治療薬もしくは薬物に対して非透過性、透過性、半透過性または多孔性である領域を特定するために用いられる。膜が薬物に対して透過性、半透過性または多孔性であるならば、薬物は溶液中またはインビボで、膜の開口部または細孔を通って放出されうる。多孔性膜は、機械的に製造することもでき(例えば、膜層の中にレーザーを用いて微細な穴または細孔を開ける)、またはそれをコーティングポリマーの物理化学的性状によって付与することもできる。本発明の膜またはコーティングポリマーは当技術分野において周知であり、これにはセルロースエステル、セルロースジエステル、セルローストリエステル、セルロースエーテル、セルロースエステルエーテル、セルロースアシレート、セルロースジアシレート(cellulose diacylate)、セルローストリアシレート(cellulose triacylate)、酢酸セルロース、二酢酸セルロース、三酢酸セルロース、セルロースアセテートプロピオネートおよびセルロースアセテートブチレートが含まれる。
【0086】
その他の適したポリマーは、参照により本明細書に組み入れられる米国特許第3,845,770号、第3,916,899号、第4,008,719号、第4,036,228号および第4,11210号に記載されている。
【0087】
さらに、本発明による腸溶性コーティングは、可塑剤、および、溶液中またはインビボで懸濁液のpHを達成または調整するのに十分な量の水酸化ナトリウム(NaOH)を含みうる。可塑剤の例には、クエン酸トリエチル、トリアセチン、セバシン酸トリブチル(tributyl sebecate)またはポリエチレングリコールが含まれる。また、水酸化カリウム、炭酸カルシウム、カルボキシメチルセルロースナトリウム、酸化マグネシウムおよび水酸化マグネシウムを含む他のアルカリ化剤を、溶液中またはインビボで懸濁液のpHを達成または調整するために用いることもできる。
【0088】
したがって、1つの態様において、腸溶性コーティングは、ある特定のpHまたはpH範囲を有するある特定の媒質中で、1つまたは複数の薬物のある特定のパーセンテージを放出するように設計することができる。例えば、本発明の治療用組成物は、酸性環境(例えば、胃)において化学的に不安定な少なくとも1つの薬物を包み込むかまたは保護する少なくとも1つの腸溶性コーティングを含みうる。腸溶性コーティングは、その薬物を酸性環境(例えば、pH<3)から保護するとともに、より酸性度の低い場所、例えば、pHが3または4または5またはそれ以上である小腸および大腸の領域では、薬物を放出する。この性質を持つ医薬品は、胃腸管の1つの領域から別の領域に運ばれると考えられ、例えば、薬物が胃から小腸(十二指腸、空腸および回腸)に移動するまでに約2〜約4時間かかる。この通過または移行の間に、pHは約3(例えば、胃)、4または5から、約6または7またはそれ以上のpHに変化する。このため、腸溶性コーティングは、薬物を含むコアが実質的に元のままに保たれるのを可能にするとともに、時期尚早な薬物放出、または酸が浸透して薬物を不安定化することを防止する。
【0089】
適した腸溶性ポリマーの例には、酢酸フタル酸セルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸フタル酸ポリビニル、メタクリル酸コポリマー、セラック、酢酸トリメット酸セルロース、酢酸コハク酸ヒドロキシプロピルメチルセルロース、フタル酸ヒドロキシプロピルメチルセルロース、酢酸フタル酸セルロース、酢酸コハク酸セルロース、酢酸フマル酸セルロース、安息香酸フタル酸セルロース、プロピオン酸フタル酸セルロース、フタル酸メチルセルロース、カルボキシメチルエチルセルロース、フタル酸エチルヒドロキシエチルセルロース、セラック、スチレン-アクリル酸コポリマー、アクリル酸メチル-アクリル酸コポリマー、アクリル酸メチル-メタクリル酸コポリマー、アクリル酸ブチル-スチレン-アクリル酸コポリマー、メタクリル酸-メタクリル酸メチルコポリマー、メタクリル酸-アクリル酸エチルコポリマー、アクリル酸メチル-メタクリル酸-アクリル酸オクチルコポリマー、酢酸ビニル-無水マレイン酸コポリマー、スチレン-無水マレイン酸コポリマー、スチレン-マレイン酸モノエステルコポリマー、ビニルメチルエーテル-無水マレイン酸コポリマー、エチレン-無水マレイン酸コポリマー、ビニルブチルエーテル-無水マレイン酸コポリマー、アクリロニトリル-アクリル酸メチル-無水マレイン酸コポリマー、アクリル酸ブチル-スチレン-無水マレイン酸コポリマー、ポリビニルアルコールフタレート、ポリビニルアセタールフタレート、ポリビニルブチレートフタレートおよびポリビニルアセトアセタールフタレート、またはそれらの組み合わせが非限定的に含まれる。当業者は、本発明によるコーティングの全体または一部として、他の親水性、疎水性および腸溶性コーティングポリマーを単独または任意の組み合わせで容易に用いうることを認識しているであろう。
【0090】
錠剤の形態にある本発明の治療用組成物は、複数のコーティング、例えば、親水性コーティング(例えば、ヒドロキシプロピルメチルセルロース)、および/または疎水性コーティング(例えば、アルキルセルロース)、および/または腸溶性コーティングを有することができる。例えば、錠剤コアを、親水性、疎水性または腸溶性コーティングから選択される、同じ種類の複数のコーティングまたは複数の異なる種類のコーティングによって包み込むことができる。それ故に、錠剤は、標的組織、または1つもしくは複数の薬物の目的に応じて、少なくとも1つの層を有するが、同じまたは異なるコーティングからなる複数の層も有しうる設計することができると期待される。例えば、錠剤コア層が、第1のコーティング層(例えば、親水性、疎水性または腸溶性コーティング)によって囲まれた第1の組成物、および第2のコーティング層に囲まれた同じまたは異なる用量を有する第2の同じまたは異なる組成物または薬物、などを有してもよい。さまざまなコーティングによるこの多層化は、同じまたは異なる薬物を含む組成物の、第1、第2、第3またはそれ以上の段階的または用量依存的な放出を提供する。
【0091】
1つの好ましい態様においては、本発明の第1の組成物の第1の用量を錠剤コアの中に腸溶性コーティングとともに含め、その結果、腸溶性コーティングがその中に含まれる組成物を崩壊または胃内部への放出から保護し、それらを防止するようにする。もう1つの例では、治療用組成物の第1の負荷用量は第1の層内に含められ、製剤または錠剤の中に含まれる全組成物の総量の約10%〜約40%を占める。第2の負荷用量では、組成物の総用量の別のパーセンテージが放出される。本発明は、治療レジメンにおいて必要とされるだけの多数回の放出用量を想定している。したがって、ある局面において、単一のコーティングまたは複数のコーティング層は、コーティングされた単位剤形の重量比で約2%〜6%、好ましくは約2%〜約5%、さらにより好ましくは重量比で約2%〜約3%の範囲にわたる量にある。
【0092】
したがって、本発明の組成物調合物は、特定の領域のために適したpH溶解性ポリマーを選択することにより、硬カプセル剤または錠剤の内容物を、胃腸管(例えば、小腸および大腸)のより遠隔部にある所望の部位で選択的に放出させることを可能にする。また、組成物調合物の機械的圧出を、水を吸収すると半透過性硬カプセルの内部で膨張し、それ故に硬カプセル剤の開口部を通して組成物を排出させる吸水性ポリマーを含めることによって達成することもできる。
【0093】
用量依存的持続放出に特に適する薬物には、インスリン様増殖因子-I(IGF-I)、ソマトメジン-C(SM-C;糖尿病、神経機能、腎機能)、インスリン(糖尿病)、カルシトニン(骨粗鬆症)、レプチン(肥満;不妊症)、レプチン由来短ペプチド(OB-3)、hGH(エイズ性消耗、低身長症)、ヒト副甲状腺ホルモン(PTH)(骨粗鬆症)、メラトニン(睡眠)、GLP-1またはグルカゴン様ペプチド-1(糖尿病)、GiP(糖尿病)、下垂体アデニル酸シクラーゼ活性化ポリペプチド(PACAP)および膵島機能(糖尿病)、GM-1ガングリオシド(アルツハイマー病)、神経成長因子(NGF)(アルツハイマー病)、ナファレリン(子宮内膜症)、Synarel(登録商標)(酢酸ナファレリン点鼻液),(D-tryp6)-LHRH(妊孕性)、FGF(十二指腸潰瘍、黄斑変性症、熱傷、創傷、脊髄損傷、骨および軟骨損傷の修復)、VEGFアンタゴニスト(受容体を遮断するため)、VEGF(アゴニスト)新生児呼吸窮迫症候群;ALS)、ロイプロリド(前立腺癌および乳癌)、インターフェロン-α(慢性C型肝炎)、低分子量ヘパリン(血液凝固、深部静脈血栓症)、PYY(肥満)、LHRHアンタゴニスト(妊孕性)、LH(黄体形成ホルモン)、グレリンアンタゴニスト(肥満)、KGF(パーキンソン病)、GDNF(パーキンソン病)、G-CSF(癌における赤血球形成)、イミトレックス(Imitrex)(片頭痛)、インテグレリン(Integrelin)(抗凝固)、Natrecor(登録商標)(うっ血性心不全)、ヒトB型ナトリウム利尿ペプチド(hBNP)、SYNAREL(登録商標)(Searl;酢酸ナファレリン点鼻液)、サンドスタチン(Sandostatin)(成長ホルモン補充)、フォルテオ(Forteo)(骨粗鬆症)、DDAVP(登録商標)鼻噴霧薬(酢酸デスモプレシン)、Cetrotide(登録商標)(注射用酢酸セトロレリクス)、Antagon(商標)(酢酸ガニレリクス)、アンジオマックス(Angiomax)(ビバリルジン;トロンビン阻害薬)、Accolate(登録商標)(ザフィルルカスト;注射用)、エキセンディン-4(Exanatide;糖尿病)、SYMLIN(登録商標)(酢酸プラムリンチド;合成アミリン;糖尿病)、デスモプレシン、グルカゴン、ACTH(コルチコトロピン)、インスリンCペプチド、GHRHおよび類似体(GnRHa)、成長ホルモン放出ホルモン、オキシトシン、コルチコトロピン放出ホルモン(CRH)、心房性ナトリウム利尿ペプチド(ANP)、チロキシン放出ホルモン(TRHrh)、濾胞刺激ホルモン(FSH)、プロラクチン、眼用トブラマイシン(角膜感染)、バソプレシン、デスモプレシン、フューゼオン(Fuzeon)(Roche;HIV融合阻害薬 MW 4492)およびエプチフィバチド(Eptifibatide)が非限定的に含まれる。
【0094】
さらに、治療を必要とする任意の特定の対象に対する具体的な用量レベルおよび投薬の頻度はさまざまであってよく、それは用いる具体的な化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全般的健康状態、性別、食事、投与の様式および回数、排泄速度、薬物併用、特定の病状の重症度、ならびに治療法を受ける宿主を含む種々の要因に依存することが、当業者には理解されるであろう。
【0095】
アルキルグリコシド、特にアルキルマルトシド、より具体的にはドデシルマルトシド(DDM)およびテトラデシルマルトシド(TDM)は、溶液中のインスリンを安定化し、ペプチドの凝集を防止することが示されている。Hovgaard et al., "Insulin Stabilization and GI absorption," J. Control. Rel, 19 (1992) 458-463、これはHovgaard et al., "Stabilization of insulin by alkylmaltosides: A spectroscopic evaluation," Int. J. Pharmaceutics 132 (1996) 107-113中に引用(本明細書では以後「Hovgaard-1」)。さらに、Hovgaard-1は、57日後でさえもDDM-インスリン複合体は安定に保たれ、ほぼ完全な生物活性を有していた。この複合体の安定性はアルキル基の長さ(炭素原子の数)に起因していて、DDMとインスリンとの比が高いほどより優れるという仮説が立てられた(例えば、4:1および16:1;Hovgaard 1における図1参照)。しかし、Hovgaard-1によれば、DDM-インスリン複合体は安定であったものの、他のマルトシドについては同じ安定性は示されなかった。なおかつ、1件の関連した研究において、Hovgaardら(1996)は、DDM-インスリンを動物にインビボで経口投与した場合には、この複合体の生物学的利用能は低い(例えば、0.5%〜1%という生物学的利用能)ことを示している。Hovgaard et al., "Stabilization of insulin by alkylmaltoside. B. Oral absorption in vivo in rats," Int. J. Pharmaceutics 132 (1996) 115-121(Hovgaard-2)。それ故に、本発明の改良された1つの局面は、表面活性物質が標的組織、臓器、系などに対する薬物の生物学的利用能を高めること、さらには薬物安定性も高めることである。
【0096】
したがって、本発明の1つの局面は、少なくとも1つの薬物および1つの表面活性物質を有する治療用組成物であって、表面活性物質がさらに、その薬物の生物学的利用能を強化する少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステル製剤からなる、治療用組成物を提供することである。薬物製剤の生物学的利用能の決定については本明細書に記載している。本明細書で用いる場合、「生物学的利用能」とは、活性物質またはモイエティーが、元のままの薬物として体循環に到達する比率および程度のことである。あらゆる薬物の生物学的利用能は、いかに良く吸着するか、およびそのうちどれだけ多くが肝臓からの除去を免れるかに依存すると考えられる。
【0097】
絶対的生物学的利用能を決定するためには、被験薬物および投与様式の測定を、静脈内参照用量と対比して行う。静脈内用量の生物学的利用能は定義上100%である。例えば、動物または志願者に対して薬物の静脈内注射および対応する経口用量を投与する。尿試料または血漿試料をある期間にわたって採取し、その期間にわたる薬物のレベルを決定する。
【0098】
血漿中薬物濃度-時間曲線の曲線下面積(AUC)を、静脈内用量および経口用量の両方についてプロットし、両方の製剤の生物学的利用能の計算を単比例によって行う。例えば、同一の静脈内用量および経口用量を投与して、経口AUCが静脈内AUCの50%であるならば、その経口製剤の生物学的利用能は50%である。任意の薬物の生物学的利用能は、不完全な吸収、初回通過クリアランスまたはこれらの組み合わせを含む、多くの要因に依存する(以下でより詳細に考察する)。さらに、血漿中薬物濃度のピーク濃度(またはCmax)も測定し、その薬物の等価濃度の筋肉内(IM)注射後の血漿中薬物濃度のピーク濃度(Cmax)と対比する。その上、血漿中薬物の最大濃度到達時間(またはtmax)は約0.1〜1.0時間である。
【0099】
薬物の複数の製剤(例えば、アルキルグリコシドまたはサッカリドアルキルエステルの薬物製剤)の相対的生物学的利用能を決定するためには、一方または両方の薬物が初回通過クリアランス(以下でより詳細に考察)を受け、それ故に検出されない可能性があるため、その製剤の生物学的利用能を互いと対比して評価する。例えば、第1の経口製剤を第2の経口製剤と対比して評価する。第2の製剤は、第1のものの生物学的利用能を評価するための参照として用いる。この種の検討は、薬物が吸収されることについての2つの製剤の相対的成績の指標を提供する。
【0100】
薬物の生物学的利用能は一致せず、薬物毎に大きく異なる。例えば、女性の閉経後骨粗鬆症の治療のための処方医薬品であるMIACALCIN(登録商標)(Novartis社のサケカルシトニン)鼻噴霧薬の生物学的利用能は、約3%という平均生物学的利用能を有する(0.3%〜30.6%の範囲;図1参照)。MIACALCIN(登録商標)の製品情報シートは、ワールドワイドウェブ上のmiacalcin.com/info/howWorks/index.jspおよびdrugs.com/PDR/Miacalcin_Nasal_Spray.htmlに見ることができる。異なる方法および被験者を用いてさまざまな研究者によって得られたMIACALCIN(登録商標)に関するデータは、薬物の生物学的利用能の大きなばらつきを示しており、例えば、健常志願者では経鼻投与した用量のわずか3%ほどしか生物学的に利用されず、これは筋肉内注射によって投与した同じ用量(MIACALCIN(登録商標)製品挿入物)とは対照的であった。このことは、2桁のばらつきがあり、消費者にとって望ましくないことを表している。
【0101】
薬物の生物学的利用能の低さは、筋肉内ビタミンB12療法後の寛解期にある患者の治療および維持のために用いられる、NASCOBAL(登録商標)(Nastech)またはシアノコバラミンでも観察される。このゲル製剤を鼻腔内に投与し、B12の生物学的利用能を筋肉内B12注射と比較した。B12のピーク濃度(またはTmax)は鼻腔内投与から12時間後に到達し、筋肉内注射と対比した相対的な、B12鼻腔用ゲルの生物学的利用能は約8.9%であった(90%信頼区間、7.1%〜11.2%)。
【0102】
本発明のアルキルグリコシドまたはスクロースエステルには、現在知られている、または将来発見される、あらゆる化合物が含まれる。本発明のアルキルグリコシドおよび/またはサッカリドアルキルエステルとの混和に特によく適している薬物には、他の方法によって投与するのが難しいもの、例えば、胃腸(GI)管の中で分解される薬物、またはGI管から吸収されないもの、または注射などの伝統的な方法の代わりに眼、鼻、鼻涙管、吸入もしくはCSF送達経路を介して自己投与することが可能な薬物がある。いくつかの具体的な例には、ペプチド、ポリペプチド、タンパク質、核酸および他の高分子、例えば、インスリンおよびカルシトニン、エンケファリン、グルカゴンなどのペプチドホルモン、ならびにトルブタミドおよびグリブリドなどの血糖降下薬、ならびに経腸経路によってはほとんど吸収されない作用物質、例えば抗真菌薬の1つであるグリセオフルビンなどが含まれる。他の化合物には、例えば、ニコチン、インターフェロン(例えば、α、β、γ)、PYY、GLP-1、合成エキセンディン-4(Exenatide)、副甲状腺ホルモンおよびヒト成長ホルモン、または他の低分子量ペプチドおよびタンパク質が含まれる。
【0103】
または、薬物の生物学的利用能を、肝臓による薬物の初回通過クリアランスのレベルを測定することによって決定することもできる。鼻腔内に、または口腔(舌下細胞もしくは頬粘膜細胞)を介して投与された、本発明のアルキルグリコシドおよび/またはサッカリドアルキルエステル組成物は、肝門脈血系には入らず、その結果、肝臓による初回通過クリアランスを回避する。肝臓によるこれらの製剤の初回通過クリアランスの回避については本明細書で記載している。「初回通過肝クリアランス」という用語は、薬物が、門脈血から肝臓を通って体循環へと至るその初回通過の間に肝臓によって除去される程度のことである。これはまた、初回通過代謝または初回通過抽出とも呼ばれる。
【0104】
身体からの薬物排出の経路には、薬物が変化することのない、腎臓による排泄;および、薬物が代謝される、肝臓による排出という主に2つがある。これらの2つの経路間のバランスは、この2つの過程の相対的効率に依存する。本発明は本明細書において肝臓または肝クリアランスによる排出について述べる。初回通過肝クリアランスは、Birkett et al. (1990 and 1991)によって記載されており、これはその全体が参照により組み入れられる。Birkett et al., Aust Prescr, 13(1990):88-9;およびBirkett et al., Austra Prescr 14:14-16 (1991)。
【0105】
体循環からの薬物を運ぶ血液は門脈を介して肝臓に入り、続いて肝臓が、その薬物のある特定のパーセンテージまたは比(すなわち、0.5または50%)を抽出する。残った残留物(すなわち、0.2または20%)は肝静脈を介して再び体循環に入る。薬物のこのクリアランスの比率は肝抽出比と呼ばれる。これは肝臓を通っての血液の初回通過の間に非可逆的に除去(または抽出)される、血液中の薬物の割合である。薬物が全く抽出されないならば肝抽出比はゼロである。その反対に、薬物が肝臓を通る初回通過において高度に抽出されるならば、肝抽出比は100%または1.0という高さになることもある。一般に、肝臓による薬物のクリアランスは、その場合、その薬物の肝臓への送達(または肝血流)の速度およびその薬物の除去の効率(または抽出比)に依存する。
【0106】
したがって、肝クリアランスを決定するために用いられる最終的な式は、以下である:
(肝クリアランス−血流)=(非結合率*固有クリアランス)/血流+(非結合率*固有クリアランス)(1)
【0107】
薬物の「非結合率」は、薬物が血液中のタンパク質および細胞とどの程度強固に結合するかに依存する。一般に、血液から肝細胞内への拡散に供されうるのは、この非結合(または遊離)薬物のみである。肝血流およびタンパク質結合がない場合、「固有クリアランス」とは、肝臓がその薬物を除去(または代謝)する能力のことである。生化学の用語では、これは特定の薬物基質に対する肝酵素活性の指標である。ところが、固有クリアランスが高くても、薬物は肝臓内に送られたものよりも迅速には取り除かれない可能性がある。簡単に言えば、2つの状況がある:肝酵素活性が非常に高い場合または非常に低い場合(すなわち、高い抽出比または低い抽出比)。
【0108】
肝酵素活性が低い場合、この式は以下のものに単純化される:
肝クリアランス=非結合率*固有クリアランス(2)
【0109】
クリアランスはこの場合、血流には依存せず、その代わり、血液中でのタンパク質結合の度合いおよびその薬物に向かう薬物代謝酵素の活性に直接依存する。
【0110】
対照的に、肝酵素活性が高い場合、この式は以下になる:
肝クリアランス=肝血流(3)
【0111】
このケースでは、酵素の活性が非常に高いため、肝臓はそこに送られた薬物の大部分を除去し、抽出比が高くなる。したがって、実際の肝クリアランスを決定する唯一の要因は、肝臓への薬物の供給(または肝血流)の速度である。
【0112】
初回通過肝クリアランスは、薬物の抽出の小さな変化でさえも生物学的利用能の大きな変化を引き起こす可能性があるため、重要である。例えば、薬物Aの、それが体循環に到達する時点までの経口投与による生物学的利用能が20%であり、静脈内投与による同じ薬物Aのそれが100%であり、他の複雑化要因が存在しなければ、故に、同程度の血漿中濃度を達成するためには経口用量は静脈内用量の5倍でなければならないと考えられる。
【0113】
第2に、肝酵素活性が非常に高いいくつかの場合には、体循環へと直接に薬物通過が行われ、初回通過肝クリアランスがすべて回避されるように薬物製剤を設計すべきである。例えば、鼻腔内、舌下、頬側、直腸、膣などに投与された薬物は、体循環に直接入り、肝臓によって部分的または完全に抽出されることになる肝門脈血循環には入らない。または、薬物を上記の手段によって投与することができない場合には、胃の中(すなわち、高度の酸性環境)での薬物の放出を防止する少なくとも1つの腸溶性コーティング層を有する錠剤を提供する。したがって、本発明の1つの目的は、薬物をこれらの代替的経路を用いて投与することである。
【0114】
さらに、初回通過肝クリアランスは、多くの患者が複数の薬物レジメンを受けており、このことが肝酵素活性を上昇または低下させる薬物相互作用を引き起こし;その結果、関心対象の薬物の代謝を増加または減少させる(肝抽出比を増加または減少させる)可能性があることからも、重要な要因である。
【0115】
それ故に、本発明の治療用組成物は、体循環系に直接投与し、初回通過肝クリアランスを回避させることができる。初回通過クリアランスを回避することで、より多く薬物が系に供されることが保証される。言い方を変えれば、初回通過肝クリアランスを回避することにより、薬物の生物学的利用能は高くなる。
【0116】
本発明はまた、対象の循環器系への低分子化合物の吸収を増加させる方法であって、経口、眼、鼻、鼻涙管、吸入またはCSF送達経路を介して、化合物、および親水性サッカリドとの結合によって連結された疎水性アルキルを有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を投与する段階を含む方法にも関する。
【0117】
組成物製剤は、経口投与(経口調合物)、外部投与(例えば、軟膏)、注射(注射用調合物)および粘膜投与(例えば、頬側および坐薬)などといった投与経路に従って適切に選択される。例えば、添加剤(例えば、デンプン、ラクトース、結晶セルロース、乳酸カルシウム、メタケイ酸アルミン酸マグネシウムおよび無水ケイ酸塩)、崩壊剤(例えば、カルボキシメチルセルロースおよびカルボキシメチルセルロースカルシウム)、潤滑剤(例えば、ステアリン酸マグネシウムおよびタルク)、コーティング剤(例えば、ヒドロキシエチルセルロース)および香味剤を、経口製剤および粘膜用製剤に対して用いることができる;一方、水性注射剤を形成させることのできる溶解補助剤および補助的溶解補助剤(例えば、注射用蒸留水、生理的食塩水およびプロピレングリコール)、懸濁剤(例えば、ポリソルベート80などの表面活性物質)、pH調節剤(例えば、有機酸およびその金属塩)ならびに安定剤が注射剤に対して用いられる;ならびに、水性または油性の溶解補助剤および補助的溶解補助剤(例えば、アルコールおよび脂肪酸エステル)、粘着防止剤(例えば、カルボキシビニルポリマーおよびポリサッカリド)ならびに乳化剤(例えば、表面活性物質)が、外部用薬剤に対して用いられる。薬物およびアルキルグリコシドは、投与の前に、上記の添加剤、崩壊剤、コーティング用ポリマー、溶解補助剤、懸濁剤などと混和、混合もしくは配合することもでき、またはそれらを任意の順序で逐次的に投与することもできる。それらを投与前に混合することが好ましい。
【0118】
「粘膜送達強化剤」という用語には、化合物(例えば、生物活性化合物)の放出または溶解性(例えば、製剤送達媒体からの)、拡散速度、透過の能力およびタイミング、取り込み、滞留時間、安定性、有効半減期、ピーク濃度または持続濃度レベル、クリアランスならびに他の所望の粘膜送達特性(例えば、血流または中枢神経系といった、送達部位、または選択された活性標的部位で測定されるもの)を強化する作用物質が含まれる。粘膜送達の強化は、例えば、化合物の拡散、輸送、存続性もしくは安定性を高めること、膜流動性を高めること、細胞内または傍細胞透過を調節するカルシウムおよび他のイオンの利用能または作用をモジュレートすること、粘膜成分(例えば、脂質)を可溶化すること、粘膜組織における非タンパク質およびタンパク質のスルフヒドリルレベルを変化させること、粘膜表面を越える水の流れを増加させること、上皮結合生理機能をモジュレートすること、粘膜上皮を覆う粘液の粘度を低下させること、粘液線毛クリアランス速度を低下させること、ならびに他の機序によるものを含む、種々の機構の任意のものによって生じうる。
【0119】
例示的な粘膜送達強化剤には、以下の作用物質およびそれらの任意の組み合わせが含まれる:
(a)凝集阻害剤;
(b)電荷修飾剤;
(c)pH制御剤;
(d)分解酵素阻害剤;
(e)粘液溶解剤または粘液除去剤;
(f)繊毛静止剤;
(g)以下から選択される膜透過強化剤:
(i)表面活性物質;(ii)胆汁酸塩;(ii)リン脂質添加物、混合ミセル、リポソームまたは担体;(iii)アルコール;(iv)エナミン;(v)NOドナー化合物;(vi)長鎖両親媒性分子;(vii)小型の疎水性透過強化物質;(viii)ナトリウムまたはサリチル酸誘導体;(ix)アセト酢酸のグリセロールエステル;(x)シクロデキストリンまたはβ-シクロデキストリン誘導体;(xi)中鎖脂肪酸;(xii)キレート剤;(xiii)アミノ酸またはその塩;(xiv)N-アセチルアミノ酸またはその塩;(xv)選択された膜成分に対する分解作用のある酵素;(ix)脂肪酸合成の阻害薬;(x)コレステロール合成の阻害薬;および(xi)(i)〜(x)に列挙された膜透過強化剤の任意の組み合わせ;
(h)上皮結合生理機能の調節剤;
(i)血管拡張薬;
(j)選択的輸送強化剤;ならびに
(k)化合物と有効に組み合わされ、会合し、含有され、封入され、または結合し、その結果、鼻粘膜送達の強化のための化合物の安定化をもたらす、安定化性の送達媒体、担体、粘膜付着剤、支持体または複合体形成種であって、化合物と鼻腔内送達強化剤との製剤化が対象の血漿における化合物の生物学的利用能の増大をもたらすもの。
【0120】
そのほかの粘膜送達強化剤には、例えば、クエン酸、クエン酸ナトリウム、プロピレングリコール、グリセリン、アスコルビン酸(例えば、L-アスコルビン酸)、二亜硫酸ナトリウム、エチレンジアミン四酢酸(EDTA)二ナトリウム、塩化ベンザルコニウム、水酸化ナトリウム、およびそれらの混合物が含まれる。例えば、EDTAまたはその塩(例えば、ナトリウム塩またはカリウム塩)を、アルキルサッカリド保存料を含む組成物の重量比で約0.01%〜2%の範囲にわたる量で用いる。
【0121】
本発明の治療薬または薬物は、医学的または診断的に有用で、サイズが小さいかもしくは中程度である、例えば、最大で約15kD、30kD、50kD、75kDなどであるペプチドもしくはタンパク質、または約1〜300個もしくはそれ以上のアミノ酸を有するタンパク質でありうる。本発明の方法はまた、小分子、例えば、3kD未満または1.5kD未満の分子量を有する有機化合物の使用も予期している。
【0122】
本発明による薬物吸収の改善の機序は一般に該当性があり、そのようなすべてのペプチドまたはタンパク質に当てはまるべきであるが、それらの吸収が改善される度合いは、ペプチドまたはタンパク質の分子量(MW)および物理化学的性状、ならびに用いる特定の強化物質によって異なりうる。ペプチドまたはタンパク質の例には、バソプレシン、バソプレシンポリペプチド類似体、デスモプレシン、グルカゴン、コルチコトロピン(ACTH)、ゴナドトロピン、カルシトニン、インスリンのC-ペプチド、副甲状腺ホルモン(PTH)、成長ホルモン(HG)、ヒト成長ホルモン(hGH)、成長ホルモン放出ホルモン(GHRH)、オキシトシン、コルチコトロピン放出ホルモン(CRH)、ソマトスタチンまたはソマトスタチンポリペプチド類似体、ゴナドトロピンアゴニストまたはゴナドトロフィンアゴニストポリペプチド類似体、ヒト心房性ナトリウム利尿ペプチド(ANP)、ヒトチロキシン放出ホルモン(TRH)、濾胞刺激ホルモン(FSH)およびプロラクチンが含まれる。
【0123】
本発明の1つの好ましい組成物は、Exenatide(またはエキセンディン-4)およびアルキルグリコシドであるペプチド薬物である。Exenatideは合成型のエキセンディン-4であり、Amylin(商標)Pharmaceuticalsによる臨床試験に用いられている。エキセンディン-4は、インクレチン模倣薬またはホルモンとして知られる新たなクラスの治療用医薬品の最初のものである低分子量ペプチドである。インクレチンホルモンとは、インスリン分泌の強力な刺激物質として、例えば、胃抑制ポリペプチド(GIP)、グルカゴン様ペプチド-1(GLP-1)、またはExenatideもしくはエキセンディン-4、またはそれらの等価物などとして作用する、さまざまな胃腸(GI)ホルモンおよび因子の任意のもののことである。
【0124】
エキセンディン-4は、アメリカドクトカゲ(Gila Monster Lizard)の唾液分泌物から単離された、天然に存在する39アミノ酸のペプチドである。Eng et al., "Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas," J. Biol. Chem. 267(15):7402-7405(1992)。Exenatideは、グルカゴン様ペプチドまたはGLP-1と類似したグルコース低下作用を示す。Exenatideは、2型糖尿病を有する多くの人々の、未だに満たされていない重要な医学的需要に対処する能力に関して試験が実施中である。臨床試験により、Exenatide治療が血糖を標的レベルに向けて低下させること、および体重減少を伴うことが示唆されている。Exenatide治療によって観察された血糖コントロールに対する効果は、天然に存在するインクレチンホルモンGLP-1のものに類似したいくつかの作用に起因する可能性が高い(実施例7参照)。これらの作用には、血糖レベルの上昇に応答してインスリンを産生する身体の能力を刺激すること、食後のグルカゴン放出を阻害すること、および栄養分が血流中に吸収される速度を遅らせることが含まれる。動物試験において、Exenatide投与は、2型糖尿病が進行するにつれて機能しなくなる膵臓内のインスリン産生細胞であるβ細胞の保持および新たなβ細胞の形成をもたらした。
【0125】
Exenatide、インクレチン模倣薬またはそれらの等価物は、不安定型糖尿病、化学的糖尿病もしくは耐糖能障害、妊娠性糖尿病、尿崩症、中枢性尿崩症、腎性尿崩症、下垂体性尿崩症、潜在性糖尿病、脂肪萎縮性糖尿病、若年発症成人型糖尿病(MODY)、真性糖尿病(DM)、成人発症型真性糖尿病(2型DM)、インスリン依存性真性糖尿病(IDDMまたは1型DM)、インスリン非依存性真性糖尿病(NIDDM)、若年性もしくは若年発症型真性糖尿病、ケトーシスを起こしやすい真性糖尿病、ケトーシス抵抗性真性糖尿病、栄養不良関連真性糖尿病(MRDM)、熱帯性もしくは熱帯性膵性真性糖尿病、真性糖尿病、発症前糖尿病、またはさまざまな薬物によって誘発される糖尿病、例えばサイアザイド糖尿病、ステロイド糖尿病など、またはアロキサン糖尿病および穿刺性糖尿病を非限定的に含むさまざまな糖尿病動物モデルを非限定的に含む、さまざまな型の糖尿病を治療するために用いることができる。
【0126】
もう1つの局面において、本発明の治療用組成物は、肥満を治療するために用いられる。肥満は、成人および青少年の両方においてよくみられる問題である。例えば、PYY3-36(またはAC162352)は、食欲を低下させる上で決定的な役割を果たすホルモンである。消化管ホルモン断片ペプチドPYY3-36(PYY)は、正常体重の対象に注入されると食欲および食物摂取を低下させる。脂肪細胞ホルモンであるレプチンと同様に、PYYは視床下部における食欲回路をモジュレートすることによって食物摂取を低下させる。しかし、肥満患者では、レプチンの作用に対する抵抗性があり、その結果、レプチンの治療有効性が制限される。さらに他の研究も、PYYが食物摂取を低下させることを示している。PYYの注射は、彼らが食事するのを通常よりも平均30%減らし、その結果として体重減少が起こることが明らかになっている。それ故に、PYY 3-36は、肥満に対する治療薬として可能性がある。Amylin(商標)Pharmaceuticalsは、2003年にPYY 3-36の新薬臨床試験開始申請を提出している。
【0127】
本発明の方法によって吸収を増加させることのできる化合物には、他の方法によって投与することが困難な、特定の薬物または治療用の化合物、分子もしくは作用物質の中の、現在知られている、または将来発見される、任意の化合物が含まれる。例えば、胃腸(GI)管の中で分解される薬物、またはGI管からはあまり吸収されないもの、または対象が、眼、鼻、鼻涙管、吸入もしくは肺、口腔(舌下細胞もしくは頬粘膜細胞)またはCSF送達経路を介して、注射などの伝統的な自己投与方法よりも容易に自分で投与しうると考えられる薬物が含まれる。いくつかの具体的な例には、ペプチド、ポリペプチド、タンパク質および他の高分子、例えば、インスリンおよびカルシトニン、エンケファリン、グルカゴンなどのペプチドホルモン、ならびにトルブタミドおよびグリブリドなどの血糖降下薬、ならびに経腸経路によってはほとんど吸収されない作用物質、例えば抗真菌薬の1つであるグリセオフルビンなどが含まれる。他の化合物には、例えば、ニコチン、インターフェロン(例えば、α、β、γ)、PYY、GLP-1、合成エキセンディン-4(Exenatide)、副甲状腺ホルモン(PTH)、およびヒト成長ホルモン、または他の低分子量ペプチドおよびタンパク質が含まれる。
【0128】
本明細書において考察した通り、薬物が消化管の頬側、舌下、口咽頭および食道前胃部分を通過する際に、さまざまな量の薬物が吸収される可能性がある。薬物の大部分は胃内に移行し、錠剤、カプセル剤または液剤などの腸溶性剤形が吸収される通常の胃吸収様式で吸収される。薬物が腸から吸収されると、薬物は肝臓内に直接運ばれ、そこで、その固有の化学構造に応じて、肝細胞における通常の解毒プロセスを遂行する酵素によって代謝されるかまたは排出される。以前に考察したように、この排出は、肝臓における「初回通過」代謝または「初回通過」効果と呼ばれる。ほとんどの場合は元の薬物と比較して実質的または完全に不活性である、その結果生じた代謝産物は、多くの場合は血流中を循環している状態で認められ、その後に尿中および/または便中に排出される。
【0129】
本発明の諸局面は、急速分散性剤形に含められた場合に、ある種のアルキルサッカリドの添加が、初回通過効果を受ける薬物の割合をモジュレートし、それ故に、一定量の薬物がより大きな臨床的有益性を発揮することを可能にする、またはより少量の薬物が、他の点では同様なより高い用量と比較して同程度の臨床的有益性を達成することを可能にするという発見に基づく。
【0130】
本発明の別の諸局面は、急速分散性剤形に含められた特定のアルキルサッカリドの量を増加させるかまたは減少させることが、薬物の吸収部位を変更またはモジュレートさせて、頬側組織を通して吸収される薬物の割合を、消化管の他の部位と比較してそれぞれ増加させるかまたは減少させるという発見に基づく。薬物作用の開始を早めながらも、標準的な経口錠剤でみられる通常の比較的長いTmaxは保持することが望ましい場合には、薬物の一部分は頬側で直ちに吸収されて迅速開始をもたらすが、その残りはより緩徐な胃吸収過程で吸収されるように頬側吸収を弱めるために、アルキルサッカリドの含有量を減らすことができる。このようにして、最大または最大に近い頬側吸収を生じることが実験によって見いだされているアルキルサッカリドの濃度よりも低い、例えば20%低いアルキルサッカリド濃度を選択することによって、「全身薬物レベル」-時間グラフにおいて、臨床的に望ましいと判断される、全体的にみてより幅広い吸収ピークを達成することができることが見いだされた。
【0131】
以下の実施例においてさらに考察するように、急速分散性錠剤に対する、特定のアルキル鎖長を有するある種のアルキルサッカリドの添加は、前胃薬物吸収の薬物動態を有益な様式で変化させる。具体的には、約0.2%〜0.3%、0.3%〜0.4%、0.4%〜0.5%、0.5%〜1.0%、1.0%〜2.0%、2.0%〜3.0%、3.0%〜4.0%、4.0%〜5.0%、5.0%〜6.0%、6.0%〜7.0%、7.0%〜8.0%、9.0%〜10.0%、および10%を上回るアルキルグリコシドの組み込みは、前胃薬物吸収の薬物動態を有益な様式で変化させる。例示的な態様において、アルキルサッカリドはドデシルマルトシド、テトラデシルマルトシドおよび/またはドデカン酸スクロースであり、これらは急速分散性錠剤形式中に組み込まれると、体循環に入る薬物を増加させて、肝臓における「初回通過」効果によって排出される薬物を減少させる。さらに、最大薬物レベル到達時間は、典型的には1〜6時間からおよそ15〜45分へと著しく短縮される。精神病エピソードを来している攻撃的患者の治療に用いる場合、薬物のこのより迅速な吸収はより迅速な作用開始をもたらし、それは非常に有益であると考えられる。
【0132】
さらに、本発明の他の諸局面は、ある特定の種類の急速溶解性または急速分散性の錠剤を頬と歯肉との間、または口腔内に頬側組織に近接させて置くと、薬物のさらに多くの割合が体循環に直接吸収され、より少ない量がその後に肝臓において初回通過排出を受けるという発見に基づく。最後に、この効果に関して特に好都合な口腔内の位置は、鼻のすぐ下の、上唇の中央部の内側の、唇と歯肉との間であることが発見された。例示的な局面において、これらの種類の急速溶解性投薬製剤は、凍結乾燥または真空乾燥によって調製される。1つの例示的な局面において、投薬製剤は、実質的に多孔性である投薬製剤がもたらされる様式で調製される。
【0133】
「急速分散性剤形」という用語は、口腔内ですべてまたは一部が溶解しうる、あらゆる種類の剤形を範囲に含むものとする。しかし、例示的な局面において、急速分散性剤形は、有効成分の固体の急速分散性ネットワークであり、有効成分および添加剤に対して不活性である水溶性または水分散性の担体マトリックスである。さまざまな態様において、このネットワークは、固体状態にある組成物から溶媒を凍結乾燥または昇華させることによって得ることができ、この組成物は有効成分、アルキルサッカリド、および溶媒中にある担体の溶液を含む。種々の溶媒がこの用途に適することが当技術分野において公知であるが、本発明とともに用いるのに特に適した1つの溶媒は水である。また、水-アルコール混合物を、この混合溶媒中で薬物溶解性が強化される場合には用いることもできる。水難溶性の薬物の場合は、小さな薬物粒子の分散体を、凍結乾燥または昇華の過程において実質的に不溶性の薬の均一な分布を維持させる水性ゲル中に懸濁させることができる。
【0134】
1つの態様において、水性ゲルは、モノ-およびジ-ステアリン酸スクロースならびに/またはテトラデシル-マルトシドなどの選択されたアルキルサッカリドを用いて形成される、参照により本明細書に組み入れられる米国特許出願第60/957,960号に記載された自己集合性ヒドロゲルであってよい。さまざまな局面において、本発明の急速溶解性組成物は、口腔内に置かれてから20秒以内に、好ましくは10秒未満で崩壊する。
【0135】
本発明の急速溶解性製剤に用いるのに適したマトリックス形成性物質は、本出願の全体を通じて記載される。そのような作用物質には、ゼラチン、コラーゲン、デキストリンならびにダイズ、コムギおよびアメリカオオバコの種子タンパク質などの動物性または植物性タンパク質に由来する材料;アラビアゴム、グアーガム、寒天およびキサンタンなどのゴム類;ポリサッカリド;アルギナート;カラゲナン;デキストラン;カルボキシメチルセルロース;ペクチン;ポリビニルピロリドンなどの合成ポリマー;ならびにゼラチン-アラビアゴム複合体などのポリペプチド/タンパク質またはポリサッカリド複合体が含まれる。例示的な局面においては、ゼラチン、特に魚肉ゼラチンまたはブタゼラチンが用いられる。
【0136】
本明細書に記載された急速溶解性投薬製剤には事実上あらゆる薬物を組み込むことができると考えているが、特によく適した薬物には、メラトニン、ラロキシフェン、オランザピンおよびジフェンヒドラミンが含まれる。
【0137】
さらに、本発明の治療用組成物は、非ペプチド性の薬物または治療薬も想定している。例えば、米国特許第5,552,534号には、種々のペプチドの化学的および/または生物的活性を模倣または阻害する非ペプチド性化合物が開示されている。そのような化合物は、テトラヒドロピラニル環などのある特定の種に対して、化合物をペプチドに対して少なくとも部分的に交差反応性にする化学官能基を付属させることにより、生成させることができる。認識されているであろうが、ペプチドを模倣または阻害する化合物は、それに対してさまざまな度合いの交差反応性を有する。ペプチド模倣物を調製するための他の手法は、米国特許第5,550,251号および第5,288,707号に開示されている。上記の米国特許は、それらの全体が参照により組み入れられる。
【0138】
本発明の方法はまた、アルキルグリコシドおよびタンパク質またはペプチドとともに、タンパク質またはペプチドが体内のその活性部位に活性状態で到達するのを助けるために(すなわち、タンパク質が依然として適正に機能しうる程度に分解が最小限になるように)、プロテアーゼ阻害薬またはペプチダーゼ阻害薬、アプロチニン、ベスタチン、α1プロテイナーゼ阻害薬、ダイズトリプシン阻害薬、組換え分泌性白血球プロテアーゼ阻害薬、カプトプリルおよび他のアンジオテンシン変換酵素(ACE)阻害薬、ならびにチオルファンの投与も含むことができる。プロテアーゼ阻害薬またはペプチダーゼ阻害薬をアルキルグリコシドおよび薬物と混合し、続いて投与することもでき、またはそれをグリコシドもしくは薬物の投与の前もしくは後のいずれかに別個に投与することもできる。
【0139】
本発明はまた、対象における血糖レベルを低下させる方法であって、インスリン、および親水性サッカリドとの結合によって連結された疎水性アルキル基を有する適した無毒性で非イオン性のアルキルグリコシドの吸収増加量を含む組成物の血糖低下量を投与し、それにより、インスリンの吸収を増加させ、かつ血糖レベルを低下させる段階を含む方法も提供する。そのような組成物の「血糖低下量」とは、本明細書において教示されるように、血糖レベルを低下させる効果を生じさせることのできる量のことである。血糖を正常血糖範囲または正常血糖に近い範囲に減少させる量が好ましい。血糖レベルの持続的低下を引き起こす量も好ましい。さらにより好ましいのは、血糖レベルを低下させることによって真性糖尿病(DM)を含む糖尿病を治療するのに十分な量である。したがって、本方法は真性糖尿病を治療するために用いることができる。好ましいアルキルグリコシドは、上記のものおよび実施例において例示しているものと同じである。
【0140】
グルカゴン、ならびに少なくとも1つのアルキルグリコシドおよび/またはサッカリドアルキルエステルを含む血糖上昇量を投与することにより、対象における血糖レベルを上昇させる方法も同じく提供される。組成物がインスリンを含む場合には、血流中でインスリンの公知の効果を引き起こすため、すなわち、対象における血糖レベルを低下させるためにそれを用いることができる。そのような投与は、真性糖尿病または関連疾患を治療するために用いることができる。そのような組成物におけるグルカゴンの「血糖上昇量」とは、血糖レベルを上昇させる効果を生じさせることのできる量のことである。ある好ましい量は、血糖を正常血糖範囲または正常血糖に近い範囲に増加させるものである。もう1つの好ましい量は、血糖レベルの持続的上昇を引き起こすものである。さらにより好ましいのは、血糖レベルを上昇させることによって低血糖症を治療するのに十分な量である。したがって、この方法は低血糖症を治療するために用いることができる。好ましいアルキルグリコシドは、上記のものおよび実施例において例示しているものと同じである。
【0141】
同様に、この組成物がグルカゴンを含む場合には、血流中でグルカゴンの公知の効果を引き起こすため、すなわち、対象における血糖レベルを上昇させるためにそれを用いることができる。したがって、そのような投与は、低血糖クリーゼを含む低血糖症を治療するために用いることができる。
【0142】
本発明はまた、脳脊髄液(CSF)に治療薬を投与する段階を含む、神経障害を改善するための方法も提供する。「神経障害」という用語は、適切な治療薬に反応する、脳、脊柱、および髄膜などの関連組織における任意の障害を表す。神経障害を改善する本発明の治療薬の驚くべき能力は、治療薬の提示を脳室腔内に存続させることに起因する。治療薬を神経障害の領域に存続させることを可能にする本発明の方法の能力は、そのような障害を治療するための特に有効な手段を提供する。
【0143】
しかし、治療を必要とする任意の特定の対象に対する具体的な用量レベルおよび投薬頻度はさまざまであってよく、用いる具体的な化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全般的健康状態、性別、食事、投与の様式および回数、排泄速度、薬物併用、特定の病状の重症度、ならびに治療法を受ける宿主を含む種々の要因に依存することは理解されるであろう。しかし、一般に、投薬は、その具体的な化合物の公知の投与方法にとって典型的であるものに近似すると考えられる。例えば、インスリンの鼻腔内投与に関して、近似的な投与量は約0.5単位/kgのブタ・レギュラーインスリンであると考えられる(Moses et al.)。血糖レベルに影響を及ぼす化合物の投与量は、最適には、適正なグルコースレベル、例えば約5〜6.7mMの正常範囲を達成するために必要なものであると考えられる。さらに、適切な量が、本明細書における教示を考慮して、定型的なものに留まる検査のみを用いて、当業者によって決定されてもよい(実施例を参照)。
【0144】
さらに、本発明の組成物を、滴剤、噴霧剤、エアロゾルおよび持続放出形式からなる群より選択される形式で投与することもできる。噴霧剤およびエアロゾルは、適切なディスペンサーの使用によって実現することができる。持続放出形式は、眼インサート、侵食性微粒子、膨張性粘膜付着剤粒子、pH感受性微粒子、ナノ粒子/ラテックス系、イオン交換樹脂ならびに他のポリマー性ゲルおよびインプラントでありうる(Ocusert, Alza Corp., California;Joshi, A., S. Ping and K. J. Himmelstein, 特許出願第WO 91/19481号)。これらの系は、吸収性表面との長期にわたる薬物接触を維持し、流出および非生産的な薬物喪失を防止する。
【0145】
さまざまな局面において、5-HT受容体アゴニストを含む本組成物の特性をIMITREX(登録商標)溶液と比較する。本明細書で用いる場合、溶液は以下の通りである。IMITREX(登録商標)(コハク酸スマトリプタン)注射剤は、選択的5-ヒドロキシトリプタミン1受容体サブタイプアゴニストである。コハク酸スマトリプタンは、化学的にはコハク酸3-[2-(ジメチルアミノ)エチル]-N-メチル-インドール-5-メタンスルホンアミド(1:1)と称される。IMITREX(登録商標)注射剤は、透明で、無色ないし淡黄色で、無菌性で、発熱物質を含まない皮下注射用の溶液である。IMITREX(登録商標)注射剤の8mg/mL溶液の各0.5mLは、コハク酸塩としての4mgのスマトリプタン(基剤)および3.8mgのUSP塩化ナトリウムを、USP注射用蒸留水の中に含む。IMITREX(登録商標)注射剤の12mg/mL溶液の各0.5mLは、コハク酸塩としての6mgのスマトリプタン(基剤)および3.5mgのUSP塩化ナトリウムを、USP注射用蒸留水の中に含む。どちらの溶液のpH範囲もおよそ4.2〜5.3である。どちらの注射剤のオスモル濃度も291mOsmolである。IMITREX(登録商標)(コハク酸スマトリプタン)鼻噴霧薬は、5mgまたは20mgのスマトリプタンを、NFリン酸二水素カリウム、USP無水リン酸二ナトリウム、NF硫酸、NF水酸化ナトリウムおよびUSP精製水を含む100μL単位用量の水性緩衝溶液中に含む。溶液のpHはおよそ5.5である。溶液のオスモル濃度は、5mgおよび20mgのIMITREX(登録商標)鼻噴霧薬でそれぞれ372または742mOsmolである。
【0146】
本明細書で用いる場合、5-HT受容体には、5-HT1から5-HT7までの受容体ファミリーの任意の受容体が含まれる。そのような受容体には、5-HT1A、5-HT1B、5-HT1D、5-HT1E、5-HT1F、5-HT2A、5-HT2B、5-HT2C、5-HT3、5-HT4、5-HT5A、5-HT6および5-HT7が含まれる。
【0147】
本発明を以下の実施例においてより詳細に説明するが、当業者にはさまざまな変更物および変形物が明らかであると考えられるため、これらは例示に過ぎないことを意図している。以下の実施例は、本発明を限定するのではなく、それを例示することを意図している。
【実施例】
【0148】
実施例1
アルキルグリコシドおよび/またはスクロースエステル製剤は粘膜の刺激も破壊も引き起こさない
鼻粘膜は高度に血管が発達しており、それ故に高度の薬物透過のために最適である。その上、鼻粘膜を通しての薬物の吸収は、中枢神経系(CNS)にとって利用可能である。薬物の局所適用は望ましいものの、この投与方法の課題は粘膜刺激性である。
【0149】
市販の一般用医薬品(OTC)鼻腔用食塩水中のアルキルグリコシド(0.125% TDM)からなる製剤を、インビボでヒト鼻上皮に対して1カ月を上回る期間にわたって投与した。この0.125% TDM製剤を、対照、すなわち同じ市販(OTC)の鼻腔用食塩水と同じ期間にわたって比較した。その結果は、33日間にわたる毎日のTDM投与(すなわち、試験の継続期間)の間および後に、鼻粘膜に対して観察可能な刺激はみられなかったことを示している(非提示データ)。したがって、本発明の組成物は無毒性および非刺激性であり、慢性および進行中の疾患を有する患者にとって有益である反復的および長期的な鼻腔内投与を実現させる。
【0150】
同様の試験を、スクロースエステルの1つであるドデカン酸スクロースを用いて行った。ドデカン酸スクロースをインビボでヒト鼻上皮に対して投与したところ、47日間(すなわち、試験の継続期間)の間および後に、観察可能な刺激は検出されなかった(非提示データ)。したがって、これらの結果は、本発明のアルキルグリコシドおよびスクロースエステルが無毒性であり、長期間にわたって毎日投与した場合に粘膜刺激を引き起こさないことを示している。
【0151】
実施例2
アルキルグリコシドおよび/またはスクロースエステル組成物は、薬物の生物学的利用能を高め、薬物の生物学的利用能のばらつきを減少させることにより、薬物を安定化する
アルキルグリコシドの安定性は、一部には、炭素原子の数、またはアルキル鎖および他の長いアルキル鎖の長さに依存し、テトラデシルマルトシド(TDM)が最大の効果を有する;しかし、DDMを含む他の高度に分枝したアルキル鎖も安定化効果を有する。高いアルキルグリコシド-薬物比の好ましさを記載しているHovgaard-1とは対照的に、本発明はこの比がはるかに低いことを示している。例えば、重量比で約0.01%〜約6%の範囲にあるアルキルグリコシドは、薬物の優れた安定化をもたらす;一方、Hovgaard-1は、はるかに高いアルキルグリコシド-薬物比(10:1および16:1)でのみ安定化が達成されたことを示している。さらにより興味深いことに、約0.01%〜約6%の範囲にある本発明のアルキルグリコシドでは、生物学的利用能が高まっていた(図1参照)。これは、高いアルキルグリコシド比(10:1および16:1)で相対的に低い生物学的利用能(0.5〜1%)を示したHovgaard-2とは極めて対照的である。
【0152】
図1は、アルキルグリコシド(TDM)の存在下および非存在下における薬物MIACALCIN(登録商標)(Novartis社のサケカルシトニン)の生物学的利用能を比較しているグラフである。MIACALCIN(登録商標)は鼻噴霧薬であり、鼻上皮または鼻粘膜に対して直接投与される。図1は、アルキルグリコシドを伴うMIACALCIN(登録商標)をラットに投与した場合と比較して、アルキルグリコシドを伴わないMIACALCIN(登録商標)がヒトにおいて極めて低い生物学的利用能レベルを有する(MIACALCIN(登録商標)製品仕様書挿入物)ことを示している。より具体的には、0.125%および0.250%のアルキルグリコシド(TDM)を伴うMIACALCIN(登録商標)の鼻腔内送達は、それぞれ約43%〜約90%の生物学的利用能をもたらした。アルキルグリコシドを伴わないMIACALCIN(登録商標)の鼻腔内投与の生物学的利用能は、ヒトでは約3%に過ぎず、ラットでは検出不能であり、このことはラットがヒトにおける鼻腔内薬物吸収を評価するための厳格なモデルであることを示唆している。このように、本発明のアルキルグリコシドは薬物の吸収を強化し、その生物学的利用能を高める。
【0153】
さらに、薬物の生物学的利用能を高めることに加えて、本発明のアルキルグリコシド組成物は、薬物の生物学的利用能のばらつきを効果的に低下させる。図1は、アルキルグリコシド(0.125%または0.25%)を伴うMIACALCIN(登録商標)の鼻腔内への投与は+/-8%という生物学的利用能のばらつきを有し、一方、アルキルグリコシドを伴わない場合の生物学的利用能のばらつきは0.3%〜30%すなわち2桁の変化であることを示している。生物学的利用能の増大および生物学的利用能のばらつきの低下は、患者間変動も同じく減少することを確かなものにする。図1に示されている結果は鼻腔内への投与であるが、同様の結果は、経口、頬側、膣、直腸送達などについても、さらには異なるアルキルグリコシド濃度でも期待される。
【0154】
このように、当技術分野とは対照的に、本発明のアルキルグリコシド組成物は、約0.01%〜約6%の範囲で、生物学的利用能の増大および生物学的利用能のばらつきの低下をもたらす。このことは他には報告されていない。
【0155】
実施例3
アルキルサッカリド+インスリンの眼投与はインビボで血糖低下効果を生じさせる
正常ラットに、それらの血糖レベルを上昇させるためにキシラジン/ケタミンの混合物による麻酔を施した。麻酔に反応して起こるD-グルコースレベルの上昇は、薬物投与、例えばインスリンを含む点眼薬の全身血糖低下作用を測定するための最適な系を与える。この動物モデルは、糖尿病性の動物およびヒトで認められる高血糖状態によく似ている。実験動物群では、麻酔ラットにインスリンを含む点眼薬を投与する。実験群による血糖レベルを、インスリンを伴わない点眼薬を投与した麻酔ラットと比較する。血糖レベルの変化および全身反応の違いは、投与経路、例えば眼経路を介して吸収されたインスリンの降下を反映する。
【0156】
成体雄性Sprague-Dawleyラット(250〜350g)に飼料を自由に摂取させ、10:00a.m.〜3:00p.m.の間に実験を実施した。腹腔内(IP)に投与したキシラジン(7.5mg/kg)およびケタミン(50mg/kg)混合物によってラットに麻酔を施し、50〜90分間かけて安定させた後に点眼薬を投与した。キシラジン/ケタミンによる正常ラットの麻酔は血糖値の上昇を生じさせ、それはインスリンを含む点眼薬の全身血糖低下作用を判定するための最適な状態をもたらす。血中D-グルコース値は、実験全体を通じて尾静脈から5〜10分間隔で血液小滴を採取し、その血液を、グルコメーター試験紙(Chemstrip bG)に、装置(Accu-Chek II, Boehringer Mannheim Diagnostics;Indianapolis, Ind.)に用意された指示に従って適用することによって測定した。麻酔された非糖尿病性ラットにおける血中D-グルコース値は200〜400mg/dlの範囲にわたった。
【0157】
50〜90分間の安定化期間の後に、時間0の時点で、0.2%ブタ・レギュラーインスリンを含むかまたは含まず、試験しようとする0.125%〜0.5%の吸収強化性アルキルグリコシド(例えば、TDM)を含むリン酸緩衝食塩水(PBS)で構成される20μlの点眼薬をラットに投与した。点眼薬は、時間0の時点で、眼を開けたままにしてプラスチック製使い捨てピペットチップを用いて滴下し、プロトコールの全体を通じてラットを加温パッド(37℃)上で水平位に保った。ラットが覚醒の徴候を示した場合には追加の麻酔を施した。ラットの各眼に、各個体につき合計2Uの0.2%(50U/ml)ブタ・レギュラーインスリン(Squibb-Novo, Inc.)を含む(実験)かまたは含まない(対照)リン酸緩衝食塩水、pH 7.4中にある0.125〜0.5%吸収強化物質20μlを投与した。オクチル-β-D-マルトシド、デシル-β-D-マルトシド、ドデシル-μ-D-マルトシド、トリデシル-β-D-マルトシドおよびテトラデシル-β-D-マルトシドは、Anatrace, Inc.(Maumee, Ohio)から入手した。ヘキシルグリコピラノシド、ヘプチルグルコピラノシド、ノニルグルコピラノシド、デシルスクロースおよびドデシルスクロースはCalbiochem, Inc.(San Diego, Calif.)から入手した;サポニン、BL-9およびBrij 78はSigma Chemical Co.(St. Louis, Mo)から入手した。
【0158】
1)食塩水のみ;2)食塩水中の0.2%ブタ・レギュラーインスリンのみ;または3)吸収強化物質のみを含む点眼薬をラットに投与したところ、血中D-グルコースレベルは上昇したまま保たれた。しかし、0.2%ブタ・レギュラーインスリンおよびいくつかのアルキルマルトシド化合物またはアルキルスクロース化合物を含む点眼薬を投与した場合には、血中D-グルコース値の顕著な低下が生じ、これは最長2時間にわたって維持された。0.5%ドデシル-β-D-マルトシド(表I参照)または0.5%デシル-β-Dマルトシド(表III参照)とともに眼投与したインスリンは、血糖レベルの即時的かつ持続な降下をもたらし、これは実験の2時間の継続期間にわたって正常血糖(80〜120mg/dl)または正常血糖に近い(120〜160mg/dl)範囲に維持される。それ故に、少なくとも2種のアルキルマルトシドは、眼経路を介して送達されたインスリンの十分な吸収を達成させて、実験的高血糖動物における血糖レベルの即時的かつ持続的な降下を生じさせるのに有効である。したがって、本発明の表面活性物質組成物は、点眼薬の形態で眼経路を介して送達されたインスリンならびに他のペプチド/タンパク質、例えばグルカゴンおよび高分子薬物およびヘパリンなどの全身吸収を達成するために有用である。
【0159】
他のいくつかのアルキルマルトシドも、0.5%トリデシルマルトシド(表III参照)ならびに0.125%(表II)および0.5%のテトラデシルマルトシドを含むインスリンの眼投与に対する吸収強化物質として有効である。これらの試験は、より長いアルキル鎖(または炭素原子数)を有するアルキルマルトシド、例えば、ドデシル-、トリデシル-およびテトラデシル-β-D-マルトシドがより有効なことを示している。炭素原子数の増加も、より大きな疎水性/親水性構造バランスおよび吸収強化効果に寄与する。アルキル鎖がより短い(炭素原子がより少ない)もの、例えばデシルマルトシド、または全く有しないもの(or no)、例えばオクチルマルトシドは、より弱い吸収強化活性を生じさせる。最も有効なアルキルマルトシドはサポニンなどの他の吸収強化物質で認められるものと同等かまたはそれを上回る効果を生じさせる上、全身吸収後に無毒性生成物に代謝されるというさらなる利点があることが注目される。
【0160】
吸収強化物質としてのアルキルマルトシドが、インスリンと併用された場合に血糖低下効果を生じさせる効果は、0.125〜0.5%の範囲にわたる異なる濃度の効果を検討することによって認められるように用量依存的である。一方、0.5%および0.375%のドデシルマルトシドはインスリンの全身吸収および血糖レベルの低下を達成する上で等しく有効であり、0.25%はより小さくかつより一過性の効果を有し、0.125%は無効であるように思われる(表I)。同様に、トリデシルマルトシドもインスリンと併用された場合に血糖濃度を低下させる上で用量依存的効果を示すが、0.25%の吸収強化物質によって達成された効果でさえも実験の2時間の時間経過にわたって持続する。このように、アルキルマルトシドの用量依存的効果は、それらが眼経路を介したタンパク質吸収の強化を、その作用物質の濃度に比例した段階的な様式で達成することを示唆する。
【0161】
(表I)インスリン+さまざまな濃度のドデシルマルトシドを含む点眼薬の、ラットにおける血糖値(mg/dl)に対する効果
【0162】
アルキルサッカリドの吸収強化効果は、ドデシルスクロース(0.125%、0.25%、0.375%)もインスリンの眼吸収および血糖レベルの低下を生じさせる上で用量依存的効果を示すことから、アルキルマルトシドのみには限定されない。この効果は0.125%アルキルサッカリドでさえも観察される(0分時点の335mg/dl.+-.26mg/dlから、120分時点では150mg/dl+-.44mg/dlに)。0.5%デシルスクロースも血糖レベルを低下させる上では有効であったが、アルキルマルトシドに関して示されているように、分子のアルキル鎖の長さおよびそれ故に疎水性の減少はアルキルスクロース化合物の効力を低下させるように思われる。しかし、0.5%デシルスクロースにより、有意かつ持続的な血糖レベルの低下が達成されている(0分時点の313mg/dl.+-.15mg/dlから、120分時点では164mg/dl+-.51mg/dlに)。2つの異なる二糖モイエティーを有するアルキルサッカリドの吸収強化能力は、それがそれらの活性にとって決定的な化合物の物理化学的性状であること、ならびに他のアルキルサッカリド、例えばドデシルラクトースが、アルキルサッカリド強化剤の代謝性状および無毒性性状を保ちながら吸収強化物質として等しくまたはより有効であるという正しい性状バランスを有することを示唆する。これらのアルキルサッカリドは本発明によって予期されている。
【0163】
アルキルグルコシドを用いた試験も実施した;0.5%ヘキシルグルコシドおよび0.5%ヘプチルグルコシドは眼からのインスリン吸収を促進させるのに無効であったが、0.5%ノニルグルコシドはインスリン吸収および血糖レベル低下(297mg/dlから150mg/dlに)を有効に刺激した。この結果は、アルキル鎖の長さならびに糖質モイエティーがインスリン吸収を有効に強化する上で決定的な役割を果たすことをさらに裏づける。
【0164】
これらの試験に用いたアルキルマルトシドまたはアルキルスクロース作用物質のいずれにおいても、眼表面に対する損傷作用が観察されなかったこと(すなわち、非刺激性)に注目すべきである。さらに、インスリンとの併用下でこれらの作用物質によって生じた即時的かつ持続な血糖低下効果は、これらの吸収強化物質が、それらの非変性的で穏和な表面活性物質性状に一致して、そのホルモンの生物活性に有害な影響を及ぼさないことを示唆する。
【0165】
このように、少なくとも1つのアルキルグリコシドおよび薬物からなる本発明の治療用組成物は安定であり、アルキルグリコシドは薬物の吸収を強化する。
【0166】
実施例4
TDM+グルカゴンの眼投与および鼻腔内投与はインビボで血糖低下効果を生じさせる
以前の実施例により、インスリンなどの薬物を伴った吸収強化物質の点眼薬を介した投与が鼻涙管排液系を介した薬物の有意な吸収をもたらすことが示されたことから、ここでは、鼻腔内投与によるインスリンのアルキルマルトシド、アルキルスクロースおよび同様の作用物質との治療的に有効な投与に関して検討した。
【0167】
インスリンと併用したテトラデシルマルトシド(TDM)は、眼経路による点眼薬を介した場合と同じく、点鼻薬の形態で鼻腔内に投与した場合にも血中D-グルコースレベルの低下を生じさせた。0.2%ブタ・レギュラーインスリンを0.125%テトラデシルマルトシドとともに含む点眼薬を、以前に記載した通りにラットに投与する。この組成物の投与は、血糖レベルの即時的かつ顕著な低下を生じさせる。血糖レベルの低下は、同じ濃度のインスリンを0.5%テトラデシルマルトシドとともに含む点鼻薬の投与によってより一層大きくなる(表II)。このように、アルキルサッカリドと薬物との鼻腔内送達および投与は血糖レベルの低下をもたらす。
【0168】
(表II)0.125%テトラデシルマルトシドを含むインスリン点眼薬および0.5%テトラデシルマルトシドを含む点鼻薬の、ラットにおける血糖値に対する効果
【0169】
実施例5
アルキルサッカリド+インスリンの眼投与はインビボで血糖低下効果を生じさせる
以前の研究により、眼からのインスリン吸収がサポニン、BL-9およびBrij-78によって刺激されることが示されている。BL-9およびBrij-78は眼からのグルカゴンの吸収を刺激するのには無効であり、一方、サポニンは有効である。さまざまな表面活性物質+グルカゴン(30μg)(Eli Lilly, Indianapolis, Indiana)を含む点眼薬を投与したラットにおいて、血中D-グルコースレベルの上昇をモニターすることにより、眼からのグルカゴン吸収を測定した。これらの実験では、キシラジン/ケタミンではなくペントバルビタールナトリウムによってラットに麻酔を施した。この手順の変更は、正常血糖範囲にある基礎血糖レベルをもたらし、眼から吸収されたグルカゴンの血糖上昇作用を容易にモニターすることを可能にした。
【0170】
表面活性物質のみまたはグルカゴンのみを含む点眼薬を投与した対のラットを、表面活性物質+グルカゴンを含む点眼薬を投与したラットと比較した。0.5%サポニン+グルカゴンを含む点眼薬をラットに投与すると血中のD-グルコースのレベルは有意に上昇するが、0.5% BL-9または0.5% Brij-78+グルカゴンを含む点眼薬ではそのような効果は観察されない。興味深いことに、ドデシルスクロース、デシルマルトースまたはトリデシルマルトース+グルカゴンを含む点眼薬を、これらの表面活性物質作用物質+インスリンを含む点眼薬による処置を前もって行ったラットに投与すると、グルカゴンが吸収され、血中D-グルコース値が有意に上昇する(表III)。この結果は、ある種のアルキルサッカリドの眼投与が、グルカゴンおよびインスリンを含む薬物の吸収を強化することを裏づけている。その上、本発明の少なくとも1つのアルキルサッカリドを有する製剤を用いて、低血糖クリーゼを治療することも今や可能である。
【0171】
(表III)インスリンまたはグルカゴン、および0.5%デシルマルトシド、0.5%ドデシルスクロースまたは0.5%トリデシルマルトシドを含む点眼薬の、ラットにおける血糖値に対する効果
【0172】
実施例6
0.25% TDM+インスリンの鼻腔内投与はインビボで血糖レベルを低下させる
薬物または作用物質の鼻腔内投与は、例えばマウスおよびラットなどの動物モデルにおいて、鼻の開口は極めて狭いが、可能である。ここに記載した実験および結果では、麻酔誘発性高血糖モデルを用いた(上記の実施例に記載)。キシラジン-ケタミンを含む腹腔内(IP)注射によって高血糖動物を誘発し、血糖レベルをある期間にわたってモニターした。キシラジン-ケタミン注射の直後に、図2に示されているように血糖レベルの上昇がみられ(●)、血糖レベルは約450mg/dlとなった。血糖レベルの上昇は膵インスリン分泌の阻害が原因で起こる。血糖レベルは、キシラジン-ケタミン注射から30分後に約482mg/dlのピークとなった(図2)。続いて、キシラジン-ケタミン注射のおよそ33分後に、0.25%テトラデシルマルトシド(TDM;またはIntravail A)中の6μLのインスリン(Humalog)を、細長いマイクロピペットチップを用いて鼻腔内に投与し、血糖レベルを約15分間隔でモニターした。0.25% TDM/インスリン組成物の投与後に血糖レベルの急速な低下がみられ、約60分の時点、またはインスリン投与から約30分後には約80mg/dlという低さに達した(図2)。約75分の時点には、血糖レベルは正常血糖マウスにおけるベースラインレベル、または約80〜100mg/dlに段階的に復帰した。
【0173】
以上の結果を、0.25% TDMを含まないインスリンのみ(同じ投与量)によって処置した動物と比較した(図2、○)。インスリンのみによる処置は、約120分のタイムマーク、またはインスリン投与の約110分後までは低下を始めない血糖レベルを示した。さらに、インスリンのみによって処置した動物において観察された血糖レベルは、インスリン+0.25% TDMを投与した動物において観察されたような正常血糖レベルには復帰することはなかった(図2)。
【0174】
このように、これらの結果はやはり、ある種のアルキルグリコシドまたはアルキルサッカリド+インスリンなどの薬物からなる本発明の組成物が血糖レベルを有効に低下させること、およびこれらの効果が薬物投与のすぐ後に測定可能であることを示している。
【0175】
実施例7
0.25% TDM(Intravail A)+エキセンディン-4の鼻腔内投与はインビボで血糖レベルを低下させる
ここに記載した試験には、ob/obマウスモデルを利用した。Friedman, J. M., Nature 404, 632-634 (2000)。すべてのマウスに、耐糖能を判定する目的で2g/kgグルコースのボーラスを腹腔内(IP)注射した。時間0の時点で、実験動物に、約100マイクログラム/kgのエキセンディン-4/0.25% TDM(American Peptide社のエキセンディン-4)を10μlの点鼻薬(図3;▲)として、もしくはIP注射(図3;●)により、または食塩水のみのIP注射(薬物を含まずTDMも含まない;図3;○)による投与を行った。対照動物については事前に実施し、薬物は投与しなかった。この試験の結果は図3に示されている。
【0176】
図3は、マウスにグルコースボーラスを投与した時間0の時点での血糖レベルがさまざまであることから、これらの動物の耐糖能が異なることを示している。時間0時点での耐糖能レベルにかかわらず、グルコースボーラスの注射直後に、3匹のマウスすべてで血糖レベルが上昇した。食塩水のみのIP注射を受けた動物の血糖レベルは、薬物を投与された実験動物ほど迅速には低下しなかった。その上、食塩水のみのIP注射を受けた動物は正常血糖レベルに到達することはなかった(図3、○)。対照的に、実験動物は、エキセンディン-4/TDMの点鼻薬の投与またはエキセンディン-4/TDMのIP注射の後に、血糖レベルの迅速かつ即座の低下を示した。
【0177】
グルコースボーラスの約15〜30分前に投与されたエキセンディン-4(図3中の時間0の前;非提示データ)も、さらにより顕著な血糖低下効果を生じさせたが、これはこのホルモンが吸収されて活性化するまでにはある程度の時間がかかるためである。このように、ヒトの臨床試験が現在行われているエキセンディン-4(またはExenatide)は、本発明のアルキルグリコシドと併用した場合に、高血糖対象の血糖レベルを低下させることにより、高血糖状態を有効に治療する。
【0178】
実施例8
アルキルグリコシド(alkyglycoside)は細菌対数増殖を低下させることによって抗菌活性を有する
カンジダ・アルビカンス(Candida albicans)(ATCC No. 10231)、黒色アスペルギルス(Aspergillus niger)(ATCC No. 16404)、大腸菌(Escherichia coli)(ATCC No. 8739)、緑膿菌(Pseudomonas aeruginosa)(ATCC No. 9027)および黄色ブドウ球菌(Staphylococcus aureus)(ATCC No. 6538)の培養物を、American Type Culture Collection, 10801 University Boulevard, Manassas, VA 20110-2209から入手した。本発明に用いた生菌は、元のATCC培養物から取り出して5回を上回る継代を行っていないものとした。本明細書において記載する場合、1回の継代は樹立培養物からの微生物の新たな培地への移行と定義し、すべての移行を算入した。
【0179】
ATCCから受け取った培養物を、ATTCによって用意された指示に従って蘇生させた。ブロス中で増殖させた細胞を遠心によってペレット化し、1/20倍容積の新たな維持用ブロス中に再懸濁させ、同じ容積の20%(v/v、水中)滅菌グリセロールと混ぜ合わせた。寒天培地上で増殖させた細胞を表面から掻き取り、同じく10%グリセロールブロスを含む維持用ブロス中に入れた。懸濁液の少量アリコートを滅菌バイアルに小分けし、バイアルを液体窒素中または温度が約-50℃を上回らない冷凍庫内で貯蔵した。新たな播種用ストックバイアルが必要になった時点で、それを取り出して、一連の作業用ストック培養物の接種に用いた。続いて、これらの作業用ストック培養物を定期的に用いて(細菌および酵母の場合には毎日)、培養物の接種を開始した。
【0180】
本明細書に記載した培地はすべて、被験微生物として上記に指し示した微生物を用いて増殖促進に関して検査すべきである。
【0181】
本発明のアルキルサッカリドが増殖を阻害するか、すなわち抗菌活性を有するか否かを判定するために、適した体積の固形寒天培地の表面に、指定された微生物のそれぞれの新たに復活させたストック培養物からの接種を行った。接種物培養のための培養条件は、表IVに記載されたものと実質的に同じである。例えば、適した培地には、ダイズ-カゼイン消化物またはサブローデキストロース寒天培地が非限定的に含まれうる。細菌およびC.アルビカンスの培養物は、TS滅菌食塩水を用いて表面増殖物を洗浄し、それを適した容器内に集めて、1mL当たり約1×108コロニー形成単位(cfu)の微生物数を得るために十分なTS滅菌食塩水を添加することによって収集した。黒色アスペルギルスの細胞を収集するためには、0.05%のポリソルベート80を含むTS滅菌食塩水を用い、続いて1mL当たり約1×1O8cfuの数を得るために十分なTS滅菌食塩水を添加した。
【0182】
または、ストック培養生物を任意の適した液体培地(例えば、ダイズ-カゼイン消化ブロスまたはサブローデキストロースブロス)中で増殖させて、細胞を遠心によって収集して洗浄し、TS滅菌食塩水中に希釈して1mL当たり約1×108cfuの微生物を得ることもできる。接種物濃度の推定値は、チャレンジ微生物の濁度測定によって決定した。懸濁液は、それを2時間以内に用いない場合には冷蔵すべきである。1mL当たりの初期cfuの推定値を確かめるためには、各懸濁液における1mL当たりのcfu数を、表IVに列記された培地および微生物回収のインキュベーション時間の条件(例えば、約3〜約7日)を用いて決定した。この値は、試験に用いる接種物のサイズを較正するために役立つ。細菌および酵母の懸濁液は収集から24時間以内に用いた;一方、真菌調製物は冷蔵下で最長7日間貯蔵することができる。
【0183】
(表IV)接種物調製のための培養条件
【0184】
製剤をpH 7のリン酸緩衝食塩水(PBS)中にて調製し、どのアルキルグリコシド製剤が抗菌活性を有するかを判定した。栄養分の供給源として、1.5mg/mLのウシ血清アルブミン(BSA;表VおよびVIを参照)または1mg/mLのPYYを培地に添加した(表VIIを参照)。BSA(CAS Number:9048-46-8)はSigma-Aldrich, St. Louis, MO, USAから入手し、n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドおよびn-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドはAnatrace Inc., Maumee, OH, USAから入手し、PYYはBachem California Inc., Torrance, CA, USAから入手した。
【0185】
アルキルグリコシドの抗菌活性を、十分な容積のアルキルグリコシド溶液を移し入れた適したサイズの4つの滅菌蓋付き細菌用容器において調べた。調製して標準化した接種物を各容器に1つずつ接種し、混合した。懸濁液接種物の容積は、アルキルグリコシド溶液の容積の約0.5%〜約1.0%であった。アルキルグリコシド溶液に添加する被験微生物の濃度は、接種後の被験調製物の濃度がアルキルグリコシド溶液1mL当たり約1×105および1×106cfuとなるようにした。増殖阻害または増殖の低下のレベルを対数尺度に基づいて決定するために、各被験調製物中の生菌の初期濃度を、プレート計数法によって決定される、標準化された接種物のそれぞれにおける微生物の濃度に基づいて推定した。接種した容器を、続いて約22.5℃±2.5でインキュベートした。各培養物/容器における微生物の増殖または非増殖を、第14日および第28日に再び判定した。各計数物中に存在するcfu数を、当技術分野において標準的なプレート計数手順によって適用可能な間隔で決定した。続いて、細菌および/または真菌の桁数の変化を、適用可能な試験間隔(例えば、第14日および第28日;表V、VIおよびVIIを参照)での各微生物に関する1mL当たりのcfu濃度のlog10値から、出発時または開始時(例えば、第0日)に存在した1mL当たりのcfu濃度の第1の算出log10値を差し引くことによって求めた。
【0186】
(表V)0.125% n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物における微生物の対数減少値
【0187】
(表VI)0.2% n-テトラデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物における対数減少値
【0188】
(表VII)0.25% n-ドデシル-4-O-α-D-グルコピラノシル-β-D-グルコピラノシドを含む培養物の対数減少値
【0189】
他のアルキルグリコシドの抗菌活性の決定は、実質的に本明細書に記載された通りに行われると考えられる。
【0190】
実施例9
霊長動物に対するアルキルグリコシド(alkyglycoside)とアンチセンスオリゴヌクレオチドとの投与
修飾された骨格を有するおよそ7,000ダルトンのアンチセンスオリゴヌクレオチド(ASO)(米国特許第7,132,530号に記載されたようなホスホロチエオートオリゴヌクレオチド)をアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標))と混合したものを、空腸内にカニューレを挿入した6匹のカニクイザルに用量10mg/kgで投与した。これらの動物は投与前に絶食させた。被験作用物質をPBS緩衝液中に溶解させ、表VIIIに記載したように、カニューレを通して各動物の空腸内に1.5mL容積を注入するか、または皮下投与(s.c.)した。
【0191】
(表VIII)テトラデシル-β-D-マルトシドとともに投与したアンチセンス薬物の生物学的利用能
【0192】
このプロトコールは、各動物に対して、表VIII中の最初の3つの被験作用物質を3つの異なる日付に投与するという3者間クロスオーバーを伴った。投薬日の間には1週間のウォッシュアウト期間を置いた。これらの動物のうち2匹にはその後、吸収強化物質として5%カプリン酸ナトリウムを含む第4の被験作用物質を投与した。血中レベルの分析は、カチオン性ポリスチレンナノ粒子を用いた固相抽出を伴う定量分析を用いて実施した。
【0193】
血液試料の固相抽出をまず行った。既知量のオリゴヌクレオチドを各試料のアリコート(200〜400μl)に添加し、脱イオン水中の50mM Tris-HCl(pH 9)800μlで希釈したものを用いて、ナノ粒子-オリゴヌクレオチド結合物を形成させた。この混合物を短時間ボルテックス処理した後に、正の表面電荷を誘導するために水溶性カチオン性開始剤を用いて、表面活性物質を含まないエマルション重合によって調製した、200μlのポリスチレンナノ粒子懸濁液を添加した(固形分:およそ10mg/ml)。この混合物をその後に再びボルテックス処理した。5〜10分間のインキュベーション後に懸濁液を遠心し、上清を除去した。粒子を脱イオン水/エタノール(1:1)中の0.5M酢酸の1ml溶液中に再懸濁させ、遠心によって洗浄溶液から分離した。上清を除去した後に、粒子を1mlの脱イオン水中に再懸濁させ、別の遠心段階によって分離した。アンモニア水(25%)/アセトニトリル(60/40)中にある150μM SDS溶液200μlをナノ粒子-オリゴヌクレオチド結合物に添加し、遊離したオリゴヌクレオチドを遠心によって担体から分離した。試料への残留粒子の混入をなくすために、上清を別の1.5mlチューブに入れて再び遠心した。その後に、試料を回転蒸発または凍結乾燥によって乾燥させ、分析時まで-20℃で貯蔵した。
【0194】
抽出した試料のキャピラリーゲル電気泳動によって定量分析を行った。キャピラリーゲル電気泳動(CGE)は、キャピラリー電気泳動システムを用いて行った。ポリビニルアルコール(PVA)でコーティングしたキャピラリー、ポリマー溶液Bおよびオリゴヌクレオチド緩衝液を含むオリゴヌクレオチド分析キットを入手した。PVAでコーティングしたキャピラリーを用い、製造元のプロトコールを用いて分析を行った。
【0195】
CGE分析から得られたデータを用いて、ホスホロチエオートオリゴヌクレオチドの定量を行った。試料中のオリゴヌクレオチドの量(nON)は、以下の式を用いて算出した:
nON=nStd(εStd/εON)((A0N/TON)/(AStd/TStd))、
式中、nStdは試料に添加された標準的なオリゴヌクレオチドの量であり、εStdおよびεONはモル吸光係数であり、AStd/TStdおよびAON/TONはそれぞれ、標準物質および調べようとする化合物の補正ピーク面積(ピーク面積および移動時間の商)である。分析物および標準物質の補正ピーク面積の商は正規化面積と呼ばれる。
【0196】
AUCは、240分間の期間にわたる濃度-時間曲線(cure)から算出した。相対的生物学的利用能は、各AUCを静脈内投与した薬物のAUCによって除算した比として求めた。Intravail(商標)(テトラデシル-β-D-マルトシド)添加剤は最大18%の生物学的利用能をもたらした。表面活性物質添加剤を伴わない対照は検出可能な吸収を示さなかった。カプリン酸ナトリウム製剤は9%という平均生物学的利用能を示した。
【0197】
実施例10
オランザピンの急速分散性剤形の調製
オランザピンの急速分散性剤形を以下の通りに調製した。オランザピン、CAS# 132539-06-1は、SynFine(Ontario, Canada)から入手する。10mM酢酸ナトリウム緩衝液、pH 5.0およびpH 6.5を、以下の通りに調製する。容積測定用の目印の付いた適切なサイズの清浄容器に、495mLの滅菌注射用蒸留水を入れる。0.286mLの酢酸を添加する。1N NaOHを添加してpHを5.00(またはpH 6.5)にする。適正なpHが得られたところでさらに水を添加して総容積を500mLにし、pHを再び確かめる。
【0198】
過程の全体を通じて撹拌を行いながら、魚肉ゼラチンまたは豚皮ゼラチンを酢酸緩衝液にゆっくりと添加し、溶解するのに十分な時間を置くことにより、以下の表IXに含まれた組成物を有する液体製剤を作る。魚肉ゼラチンまたは豚皮ゼラチンが完全に溶解したところで、マンニトールを添加して溶解させる。続いて、甘味料を添加する。これが十分に分散したところで、本発明の化合物のための例の1つである有効成分であるオランザピンを添加し、最終的な溶液を作製する。保存料、酸化防止剤、表面活性物質、粘性強化物質、着色剤、香味剤、甘味料または味遮蔽剤などの二次的成分を組成物に組み込んでもよい。適した着色剤には、赤色、黒色および黄色の酸化鉄、ならびにEllis & Everardから入手しうるFD & C blue No. 2およびFD & C red No. 40などのFD & C色素が含まれうる。適した香味剤には、ミント、ラズベリー、カンゾウ、オレンジ、レモン、グレープフルーツ、キャラメル、バニラ、チェリーおよびグレープの着香料ならびにこれらの組み合わせが含まれうる。適した甘味料には、アスパルテーム、アセスルファムKおよびタウマチンが含まれる。適した味遮蔽剤には炭酸水素ナトリウムが含まれる。シクロデキストリンは、これらの添加剤の有効性を低下させる包接化合物をアルキルサッカリドと形成するため、避けるべきである。
【0199】
上記の薬物溶液の1mLずつのアリコートを、24ウェル使い捨てマイクロウェルプラスチックプレートのウェルに入れる。液体アリコートを含むマイクロウェルプレートを凍結プレートにて-70゜で凍結させ、LabConco Freezone Model 4.5卓上凍結乾燥器に取り付けたガラス製凍結乾燥フラスコに入れて、真空下で凍結乾燥させる。凍結乾燥の後に、迅速分散性錠剤を試験時までマイクロウェルプレート中にて乾燥環境で貯蔵する。モノ-およびジ-ステアリン酸スクロース混合物はCroda Inc.によって寄贈され、これはCRODESTA F-110と命名されている。ドデシルマルトシド、テトラデシルマルトシドおよびスクロースモノデカノエートはAnatrace Inc., Maumee, OHから入手する。
【0200】
(表IX)オランザピン(olanzopine)製剤
1 SynFine, Ontario, Canada
2 Croda Colloids Ltd(加水分解されていない、噴霧乾燥させた魚肉ゼラチン)
4 ドデカン酸スクロース(モノエステル)-Anatrace Inc.
5 Sigma Aldrich(ゼラチンA型、豚皮‐G6144)
Qs=下記量にするために十分な量
【0201】
薬物オランザピンは、Zyprexaとも呼ばれ、「丸飲み(whole-swallowed)」錠剤として投与された場合によく吸収され、経口投与からおよそ6時間後にピーク濃度に達することが知られている。これは初回通過代謝によって高度に排出され、用量のおよそ40%が体循環に達する前に代謝される。薬物動態試験により、口腔内に置いた場合に約3秒〜10秒で崩壊する「丸飲み」オランザピン錠剤、およびこの実施例に上述した様式で凍結乾燥によって調製された迅速分散性オランザピン錠剤は生物学的に同等であり、投与から約6時間後にピーク濃度を示すことが示されている。同様に、肝臓における初回通過効果により、用量のおよそ40%が体循環に達する前に排出される。
【0202】
本実施例において、10mgのオランザピンを含む急速分散性錠剤は、この実施例において上述した通りに凍結乾燥によって調製される。頬側組織と接触させることによって急速分散性オランザピン錠剤を投与することにより、急速分散性オランザピン錠剤に対する特定のアルキル鎖長を有するある種のアルキルサッカリドの添加は、代謝されていない活性薬物と比較した体循環中のオランザピン代謝産物の相対的割合の低下によって認められるように、オランザピンの初回通過効果代謝の実質的な低下をもたらすことが発見されている。血清中または血漿中のオランザピンおよびオランザピン代謝産物の相対的割合は、HPLC Chromatograph, Perkin Elmer 200を、恒温セルを装着したRefractive Index Detectorとともに用いて決定することができる。Lichro sorb RP-18(Merck, Darmstadt, Germany)250mmなどの適した固相吸着剤を、アセトニトリル:水勾配からなる移動相とともに用いることができる。Perkin Elmer 200 auto-samplerを用いた20μLの注射容積および0.8mL/分の流速がこの目的には好適である。具体的には、0.2%から最大10%までのドデシルマルトシドまたはテトラデシルマルトシドまたはドデカン酸スクロースを急速分散性錠剤形式中に組み入れることで、体循環に入る薬物が増加し、肝臓における「初回通過」効果によって排出される薬物が減少する。さらに、最大薬物レベル到達時間は、典型的には1〜6時間からおよそ15〜45分へと著しく短縮される。精神病エピソードを来している攻撃的患者の治療に用いる場合、薬物のこのより迅速な吸収はより迅速な作用開始をもたらし、それは非常に有益であると考えられる。
【0203】
実施例11
メラトニンの急速分散性剤形の調製
メラトニンまたは5-メトキシ-N-アセチルトリプタミンは、睡眠障害の患者における睡眠覚醒サイクルを調節するために用いられる神経ホルモンである。内因性メラトニンは、概日または概年リズムを示すすべての動物で松果体によって分泌される。メラトニンは睡眠覚醒リズムの維持に役割を果たすことが実証されており、その補充は、時差ぼけ、交代制勤務、うつ病およびさまざまな神経学的機能異常に伴って起こる睡眠障害を調節する助けになる可能性がある。
【0204】
市販されているメラトニン製剤には、経口および舌下錠剤、カプセル剤、茶剤、ロゼンジ剤および経口噴霧剤送達系が含まれる。経口メラトニン投与は、内因性ホルモンのものとは異なる薬物動態プロファイルをたどる。経口投与後に、メラトニンはかなりの初回通過肝代謝を受けて6-スルファトキシメラトニン(6-sulfaoxy melatonin)となることから、30〜50%と推定されるメラトニンの生物学的利用能がもたらされる。DeMuroら(2000)は、健常志願者において試験された経口メラトニン錠剤の絶対的生物学的利用能はやや低く、およそ15%であると報告している。メラトニンの平均排出半減期は概ね45分である。
【0205】
1mg、5mg、10mgおよび20mgを含む急速分散性メラトニン錠剤は、上記の実施例10に記載された方法に従い、実施例10に記載されたような1%〜2%のアルキルサッカリドを伴うかまたは伴わずに調製される。投薬が容易になるように、麻酔をかけるニュージーランドホワイトウサギを制限ボックスに入れ、耳介静脈への注射によって投与されるアセプロマジン/ケタミン(0.7mg/0.O3mg、0.1mL中)の単回投与を用いて麻酔を施す。これによって約10分間の麻酔が得られ、その間にウサギに被験物質を投与する。その後にウサギは意識を回復する。2時間の期間にわたる個々の時点で、1mLの血液試料を中心耳動脈から収集する。収集後に、リチウム/ヘパリンを抗凝固薬として用いて、各血液試料から血漿を直ちに調製する。試料はすべて、メラトニンに関してアッセイするまで-70℃で貯蔵する。メラトニンは、GenWay Biotech Inc., San Diego, CAによって製造されている市販のELISAキットを用いて測定する。急速崩壊性錠剤を口腔上部の頬側組織と接触させることによって投与した場合、メラトニンは、アルキルサッカリドの存在下における曲線下面積による測定では少なくとも75%の生物学的利用能で、アルキルサッカリドの非存在下では50%未満で吸収される。メラトニンは、GenWay Biotech Inc., San Diego, CAによって製造されている市販のELISA kit(No. 40-371-25005)を用いて測定する。加えて、アルキルサッカリドを含む錠剤の場合には、メラトニンの最大濃度に、アルキルサッカリドを含まない錠剤の場合に要する時間のおよそ半分で到達する。
【0206】
実施例12
ラロキシフェンの急速分散性剤形の調製
ラロキシフェンは、Evista(登録商標)とも呼ばれ、閉経後女性における骨粗鬆症の治療および予防、骨粗鬆症を有する閉経後女性における浸潤性乳癌のリスク低減、ならびに浸潤性乳癌のリスクが高い閉経後女性における浸潤性乳癌のリスク低減のために用いられている。推奨投与量は60mg錠剤の1日1回投与である。ラロキシフェンの経口用量のおよそ60%は経口投与後に迅速に吸収されるが、全身循環以前の(presystemic)グルクロニド抱合が高度であり、その結果、ラロキシフェンの絶対的生物学的利用能は2%に過ぎない。米国特許第5,576,014号または第6,696,085 B2号または第6,024,981号に記載された通りに調製された急速分散性60mgラロキシフェン錠剤は、およそ2%の絶対的生物学的利用能という極めて類似した薬物動態を有することが見いだされている。しかし、噴霧乾燥分散(Bend Research Inc., Bend Oregon、またはAzoPharma, Miramar, FL)によって、またはより一般的に用いられる標準的な薬学的粉砕もしくは製粉過程によって調製される、10mgまたはそれ未満の微粉化ラロキシフェン、および0.5%〜5%のドデシルマルトシドを含む急速分散性錠剤は、頬側投与された場合、60mg経口錠剤によって達成されるものに類似した全身薬物レベルを達成し、同時に循環中の不活性ラロキシフェングルクロニドがより少なくなる。
【0207】
臨床的有益性は主として非抱合型薬物に起因するが、副作用は、活性薬物、およびグルクロニド抱合を受けた実質的に不活性な薬物のいずれかまたは両方によって媒介される可能性がある。このため、この場合には活性薬物の30倍もの高さの濃度である不活性薬物抱合体に対する曝露を減らすことで、副作用の恐れを低下させる有意な臨床的有益性が得られる可能性がある。ラロキシフェンの水溶解度はおよそ0.25mg/Lである。その結果として、実施例10に記載したような急速分散性製剤を調製するための凍結乾燥のための準備においてラロキシフェンを水中に溶解させることは不可能である。
【0208】
この場合には、1%〜30% w/wのCRODESTA F-110を適した緩衝液に添加し、それをボルテックス処理して、45℃に1時間加熱することによって自己集合性ヒドロゲルを形成させることができる。続いて、微粒子または微粉の形態にあるラロキシフェンをこの温かい液体に添加して、懸濁下で60mg/mLの濃度を達成させ、固体が均一に懸濁化および分散されるまで、それを再びボルテックス処理によって混合する。室温に冷却すると、小分けすることができ、それでも均一な懸濁を維持している安定なチキソトロープヒドロゲルが形成される。pH 2〜pH 7のpH範囲にある酢酸緩衝液は、この目的に特によく適することが見いだされている。ラロキシフェンのゲル懸濁液の1mLアリコートを24ウェル使い捨てマイクロウェルプラスチックプレートのプレートに入れ、実施例1に記載した通りに凍結乾燥する。
【0209】
頬側組織に対する提示によるこの急速分散性製剤の投与は、絶対的生物学的利用能(absolute bioavailabilty)の少なくとも4%への増大(倍加)、および、それに対応した、循環中のラロキシフェングルクロニド抱合体濃度と非抱合型ラロキシフェンとの比の測定可能な低下をもたらす。
【0210】
実施例13
ジフェンヒドラミンの急速分散性剤形の調製
ジフェンヒドラミンは、顕著な中枢鎮静性を有する鎮静性抗ヒスタミン薬であり、不眠症の短期的管理、蕁麻疹および血管性浮腫、鼻炎および結膜炎、掻痒性皮膚障害、悪心および嘔吐を含むアレルギー性病状の症状緩和、乗り物酔い、めまい、ある種の精神科薬物の副作用に起因する不随意運動の予防および治療、ならびにその抗ムスカリン性状によるパーキンソニズムの管理において、睡眠薬として用いられている。ジフェンヒドラミンの特に望ましい特徴は、それが依存症を生み出すという証拠が見たところ存在しないことである。その優れた安全性プロフィールのため、これは一般用医薬品として販売されており、Ambien(登録商標)およびLunesta(登録商標)といったより新たな睡眠薬のいくつかが、夢遊症および睡眠中の食事-むちゃ食いなどの奇妙な挙動を、時として起こる重度のアレルギー反応および顔面腫脹とともに引き起こす恐れがあり、これらのより新たな処方医薬品のこれらの副作用についてFDAが警告表示を求める原因となったこととは異なる。
【0211】
塩酸ジフェンヒドラミンは、25〜50mgの常用量で1日3回または4回投与される。成人および小児における最大用量は1日約300mgである。20〜50mgの用量は、成人および12歳以上の小児において睡眠薬として用いることができる。この薬物は胃腸管からよく吸収される;しかし、これは全身薬物レベルに影響を及ぼすように思われる高度の初回通過代謝を受ける。ピーク血漿中濃度は経口投与から約1〜4時間後に達成される。ジフェンヒドラミンはCNSを含む全身に広く分布し、肝臓における高度の代謝のために、この薬物は主として尿中に代謝産物として排泄され、少量の未変化薬物が存在することが認められる。
【0212】
ジフェンヒドラミンは不眠症および他の障害の治療のために安全かつ有効であると考えられるが、ピーク血漿中濃度の達成までの1〜4時間の遅れに起因する比較的遅い作用開始は不都合であり、安全かつ有効なこの薬物の実用性を減じさせている。静脈内に投与されたジフェンヒドラミンは迅速な作用開始を及ぼす;しかし、静脈内投与は、外来患者の使用にとっても、重篤でない医学的適応症にとっても現実的ではない。ジフェンヒドラミンの迅速作用開始製剤の必要性は明らかである。不眠症の場合には、患者は、長時間の落ち着かない不眠の恐れを最小限に抑えるために、この薬物がその所望の薬理効果を発揮するのに十分な全身薬物レベルを達成するのを待ちながら、この薬物の現行の経口剤形を就眠よりもはるかに前に服用する必要がある。ジフェンヒドラミンの制吐的適用の場合にも、悪心および嘔吐をできるだけ早く緩和するために、迅速作用開始が非常に望ましい。これは乗り物酔いおよびめまいの治療の場合も、これらの症状は予期せずに起こることがあるという理由から同様であり、経口投与した薬物がその有益な効果を達成するのに十分な全身薬物レベルを達成するまでに1〜4時間待たねばならないことは不都合であり、かつ望ましくない。
【0213】
ジフェンヒドラミンの水溶解度はおよそ3.06mg/mLである。このため、実施例12に記載した方法を用いて、50mgの薬物および1%〜2%のアルキルサッカリドを含む急速分散性ジフェンヒドラミン錠剤を調製することができる。ジフェンヒドラミンはやや苦いため、味を改善するために、薬学的に許容される着香料および甘味料の味遮蔽量を添加してもよい。このようにして調製された急速分散性錠剤は、「丸飲み」錠剤シロップ、チュアブル錠、ロゼンジ剤または可食フィルムストリップと比較して作用開始がより迅速であり、より少ない初回通過代謝も示す。
【0214】
実施例14
マウスに対するアルキルグリコシドと抗肥満ペプチド・マウス[D-Leu4]OB3との投与
この実施例は、雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の抗肥満ペプチド、マウス[D-Leu-4]OB3の取り込みを示す。0.3%アルキルグリコシドテトラデシルβ-D-マルトシド(Intravail(商標)A3)と混合した合成レプチンアゴニスト[D-Leu-4]OB3を、6週齢雄性Swiss Websterマウスに対して、胃管により1mgの用量で投与した。
【0215】
マウス[D-Leu-4]OB3(1mg/200ulの濃度)を、PBS(pH 7.2)、またはPBS(pH 7.2)中に再構成した0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)のいずれかに溶解させて、各時点につき4匹ずつのマウスのそれぞれに、麻酔を行わずに胃管によって投与した。10、30、50、70、90または120分後の時点で、イソフルラン(5%)の吸入によってマウスを安楽死させ、尾部大静脈の穿刺によって全瀉血を行った。ペプチドを投与されていない4匹のマウスからも血液を収集した(前採血)。この期間内の4匹のマウスのそれぞれからの血液をプールし、血清試料を調製した。プールした試料のマウス[D-Leu-4]OB3含有量は競合的ELISAによって測定した。
【0216】
これらの実験を2回繰り返した。1回の実験から収集されたデータを表Xおよび図4に提示している。このデータは高度に再現性があると判定された。取り込み曲線をMicrosoft(商標) Excelを用いてプロットし、画像用プログラムSigmaPlot 8.0(商標)(SPSS Science, Chicage, IL)の機能を用いてAUCを算出した。得られた最小のAUC値を1.0に任意に設定した。他のすべてのAUC値を1.0と比較することにより、相対的生物学的利用能を求めた。
【0217】
(表X)胃管による投与後の雄性Swiss Websterマウスによる、0.3%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中の1mgのマウスp-Leu-4]OB3の取り込み
【0218】
表Xおよび図4から明らかなように、0.3%のアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)の添加は、PBSのみの中のペプチドと比較して、OB-3ペプチドの相対的吸収を4倍に増加させた。
【0219】
実施例15
イヌに対するアルキルグリコシドとスマトリプタンとの投与
この実施例は、イヌによる、0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)中のスマトリプタンの取り込みを示す。0.5%アルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)と混合したスマトリプタンを、イヌに対して、25mgの用量で、経口投与および直腸投与の両方によって投与した。
【0220】
図5から明らかなように、0.5%のアルキルグリコシドテトラデシル-β-D-マルトシド(Intravail(商標)A3)の添加は、現在利用可能な25mg経口錠剤と比較して、経口投与および直腸投与の両方で、スマトリプタンのCmaxを増加させた。現在利用可能な錠剤のCmaxは、図5中の横向きの点線によって表されているように、イヌでは104ng/mlと決定された。
【0221】
実施例16
アルキルグリコシドとトリプタンとの投与は生物学的利用能を高める
硫酸スマトリプタン、ナラトリプタン-HCl;または安息香酸リザトリプタンを、0%、0.02%、0.05%、0.1%、0.2%または1.0%のアルキルサッカリドを含む20mM酢酸ナトリウム緩衝液、pH 5.5中に溶解させる。薬物溶液の各セットを、各ラットの1つの鼻孔への2OuLの点滴注入により、ラット8匹ずつからなる6つの群に投与する。3時間の期間にわたり、0、5、10、15、30、60、120および180分の時点で、200μlの血液試料を眼窩出血によって採取する。最後の血液試料を収集した後に、各ラットをCO2によって安楽死させる。採血後に、リチウム/ヘパリンを抗凝固薬として用いて、各血液試料から血漿を直ちに調製する。血漿試料は分析するまで-70℃で貯蔵する。血漿中薬物レベルを、Boultonによって記載された方法を用いるHPLC、または類似のHPLC法によって決定する。濃度-時間データをプロットして、各アルキルサッカリド濃度でのCmaxを求め、アルキルサッカリドが存在する場合と存在しない場合のCmaxの比を算出し、表XIに示されているように記録する。各プロットからのTmax観測値を、濃度-時間プロットの検査によって求め、表XIIに示されているように記録する。この表に明示されている用量は、いずれの場合もトリプタン遊離塩基の量を反映している。
【0222】
(表XI)トリプタン類似物投与のCmax比
【0223】
(表XII)トリプタン類似物投与のTmax
【0224】
実施例17
スマトリプタンとアルキルグリコシドとの鼻投与は生物学的利用能およびCMAXを高め、全身吸収の開始を早める
硫酸スマトリプタンを、0.18%ドデシルマルトシド(「製剤A」)を含むかまたはドデシルマルトシド添加剤を含まない(「製剤B」)、水1L中に0.2gのリン酸二ナトリウムおよび10.0gのリン酸一カリウムを溶解させることによって調製し、pH 5.5に調整したリン酸緩衝液中に溶解させ、100マイクロリットルの噴霧薬当たり20mgという最終的なスマトリプタン濃度にする。最終的なpH調整は、硫酸または水酸化ナトリウム溶液を用いて行う。第3の製剤である、GlaxoSmithKlineによって製造されている100マイクロリットル当たり20mgのImitrex(登録商標)スマトリプタン鼻噴霧薬を「参照」とする。18人の患者に対して、各薬物溶液を、Ing. Erich Pfeiffer GmbH, Radolfzell, Germany、Valois Pharma, Le Neubourg, FranceまたはBecton Dickinson, New Jersey, USAによって製造されているもののような標準的な定量鼻噴霧薬装置を用いた100マイクロリットルの定量鼻噴霧薬として、投薬間に少なくとも3日間のウォッシュアウト期間を置いた3者間クロスオーバー試験の形で投与する。K2 EDTA(EDTA二カリウム)を抗凝固薬として用いる各血液試料からの血漿の調製のために、各患者から、血液試料を、時間指定した間隔で、例えば、図6および7に示されているように0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間の時点で収集する。スマトリプタンの血漿レベルを、高速液体クロマトグラフィー(Ge, Tessier et al. 2004)によって決定する。
【0225】
図6は、Imitrex(登録商標)鼻噴霧薬参照、およびアルキルサッカリド添加剤を含まない製剤Bに関する、図示したさまざまな時点での全患者の平均血漿レベルの比較を標準偏差とともに示している。参照および製剤Bは、Cmaxが1mL当たりおよそ15ng、およびTmaxが1〜2時間というほぼ等しい成績を示した。
【0226】
図7は、Imitrex(登録商標)鼻噴霧薬参照、および0.18%ドデシルマルトシド添加剤を含む製剤Aに関する、図示したさまざまな時点での全患者の平均血漿レベルの比較を標準偏差とともに示している。製剤AのCmaxは1mL当たりおよそ60ng、またはImitrex鼻噴霧薬参照で観察されたCmaxのおよそ4倍である。T-maxは、参照での1〜2時間から、アルキルサッカリドを含む製剤Aではおよそ8〜10分に短縮しており、参照製剤によって1〜2時間で達成される15ng/mLという治療的に意味があると推定されるCmax値に、製剤Aの場合にはおよそ2分で到達した。
【0227】
得られたデータから以下のパラメーターを算出する:
AUC0-t=投与時間から最後の測定可能な濃度の時間までの薬物血漿中濃度-時間曲線下面積。
AUC0-∞=投与時間から無限時までの薬物血漿中濃度-時間曲線下面積。
Cmax=最大薬物血漿中濃度。
Tmax=最大薬物血漿中濃度到達時間。
半減期=投与後に薬物血漿中濃度がその最大濃度の半分になるまでの時間
Kel=排出速度定数。
薬物動態成績の平均を以下に表としてまとめている:
【0228】
(表XIII)
【0229】
すべての対象に関する、0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間といった個々の時点での平均Cmaxデータを、図7にプロットしている。
【0230】
実施例18
リザトリプタン(安息香酸塩)と重量比で最大2%までのアルキルグリコシドとの舌下/頬側投与は、生物学的利用能を有意には高めず、全身吸収の開始も早めない
鼻腔内の粘膜を越えるスマトリプタンの生物学的利用能はドデシルマルトシドの存在下で著しく高まるため、他のトリプタンについても、アルキルサッカリドの存在下で、類似の構成を有する口腔の舌下または頬側の膜を越える同等の生物学的利用能が同様に得られる可能性が高いと考えられる。
【0231】
(表XIV)頬側/舌下送達用のリザトリプタンの製剤
【0232】
製造手順:すべての添加剤を形状的に混合し、適した丸形の浅い凹状工具を用いて圧縮して錠剤にする。または、リザトリプタンおよびアルキルグリコシドを水中または水性アルコール混合物中に溶解させ、その溶液を用いて添加剤を顆粒化し、続いて顆粒を乾燥させ、製粉し、圧縮して錠剤にする。Maxalt-MLT(安息香酸塩リザトリプタン)経口崩壊性錠剤10mgを「参照」とする。製剤Aはアルキルグリコシドを含まない。製剤Bは0.5%アルキルグリコシドを含む。製剤Cは2%アルキルグリコシドを含む。参照、ならびにそれぞれ0%、0.5%および2%のドデシルマルトシドを含む、上記の通りに調製した崩壊性錠剤の3種の製剤を、20人の患者に対して、投薬間に少なくとも3日間のウォッシュアウト期間を置いた4者間4期間クロスオーバー試験の形で投与する。錠剤を舌下または頬と歯肉との間(頬側)のいずれかとして口腔内に置き、唾液との接触によって崩壊させる。血液試料を、K2 EDTA(EDTA二カリウム)を抗凝固薬として用いる各血液試料からの血漿の調製のために、各患者から、血液試料を、時間指定した間隔で、例えば0.08、0.17、0.25、0.33、0.42、0.6、0.67、0.83、1、2、3、4、6、8、12、24時間の時点で収集する。リザトリプタンの血漿レベルを、適切なリザトリプタン標準物質を含める(Ge, Tessier et al. 2004)の方法の変法により、高速液体クロマトグラフィーによって決定する。
【0233】
驚いたことに、以下の表XVに見られるように、アルキルサッカリドの添加による舌下または頬側生物学的利用能の増大はほとんどみられないか、または検出不能である。同様に、CmaxおよびTmax値もわずかな変化しか示さない。
【0234】
(表XV)
【0235】
実施例19
ナラトリプタンと重量比で最大0.2%までのアルキルグリコシドとの舌下噴霧投与は、生物学的利用能を有意には高めず、全身吸収の開始も早めない
【0236】
(表XVI)頬側/舌下送達用のナラトリプタン舌下噴霧薬の製剤
【0237】
製造過程:ナラトリプタン塩酸塩を水中に溶解させて、必要な数量のポリエチレングリコールまたはエタノールを添加する。次に、アルキルグリコシド、着香料、アスパルテームをこの溶液に溶解させる。容積の最終的な調整は水で行う。ドデシルマルトシドアルキルサッカリドを伴うかまたは伴わない舌下製剤を、20人の患者に対して、定量噴霧ポンプを用いて2者間2期間クロスオーバー試験の形で投与する。驚いたことに、実施例18の場合と同様に、0.18〜0.2%アルキルサッカリドの存在下で、舌下粘膜を越えるナラトリプタン生物学的利用能の増大はほとんど観察されない。
【0238】
本出願の全体を通じて、さまざまな刊行物を参照している。これらの刊行物の開示内容は、本発明の属する最先端の技術水準をより十分に説明するために、それらの全体が参照により本出願に組み入れられる。
【0239】
【0240】
上記の実施例において、本過程をその特定の態様の具体的な詳細を参照しながら説明してきたが、その変更物および変形物が本発明の趣旨および範囲に含まれることは理解されるであろう。したがって、本発明は以下の特許請求の範囲のみによって限定される。
【特許請求の範囲】
【請求項1】
以下のものを含む組成物:
a)5-HT受容体アゴニストの治療的有効量;および
b)アルキルサッカリド。
【請求項2】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項1記載の組成物。
【請求項3】
アルキルサッカリド濃度が約0.05%〜2%である、請求項1記載の組成物。
【請求項4】
アルキルサッカリド濃度が約0.1%〜0.25%である、請求項3記載の組成物。
【請求項5】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項1記載の組成物。
【請求項6】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項5記載の組成物。
【請求項7】
対象の循環器系への、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側、またはCSF送達経路を介した投与のために製剤化されている、請求項1記載の組成物。
【請求項8】
鼻腔内送達のために製剤化されている、請求項7記載の組成物。
【請求項9】
アルキルグリコシドの臨界ミセル濃度(CMC)が約1mM未満である、請求項1記載の組成物。
【請求項10】
アルキルグリコシドのCMCが約0.5mM未満である、請求項9記載の組成物。
【請求項11】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項1記載の組成物。
【請求項12】
EDTAが重量比で約0.01%〜2%の濃度を有する、請求項11記載の組成物。
【請求項13】
約7.0またはそれ未満のpHを有する、請求項1記載の組成物。
【請求項14】
ヒトにおいて5-HTアゴニストに関して約30分間またはそれ未満のTmaxをもたらす、請求項1記載の組成物。
【請求項15】
アルキルサッカリドの非存在下でもたらされる対応するAUC 0-1hrと比較して、5-HTアゴニストに関して約1.3倍、1.5倍またはそれ以上の大きさであるAUC 0-1hrをもたらす、請求項1記載の組成物。
【請求項16】
アルキルサッカリドの非存在下でもたらされる対応するCmaxと比較して、5-HTアゴニストに関して約1.3倍またはそれ以上の大きさであるCmaxをもたらす、請求項1記載の組成物。
【請求項17】
ヒトへの投与後約10〜15分の時点で、投与約60分後の時点でのアゴニストの血漿中濃度または血中濃度と比較して少なくとも約1.3倍、1.5倍またはそれ以上の高さである5-HTアゴニストの血漿中濃度または血中濃度をもたらす、請求項1記載の組成物。
【請求項18】
ヒトへの投与後約20分未満の時点で、投与約60分後の時点でのアンタゴニストの血漿中濃度または血中濃度と比較して少なくとも約1.3倍、1.5倍またはそれ以上の高さである5-HTアゴニストの最大血漿中濃度または血中濃度をもたらす、請求項1記載の組成物。
【請求項19】
ヒトへの投与後約20分未満のうちに血漿中または血液中の5-HTアゴニストに関するCmaxをもたらし、アルキルサッカリド濃度が0.05〜0.2%である、請求項1記載の組成物。
【請求項20】
ヒトへの投与後約20分未満のうちに血漿中または血液中の5-HTアゴニストに関するCmaxをもたらし、かつCmaxの少なくとも約0.25倍である60分時点での持続的5-HTアゴニスト濃度をもたらす、請求項1記載の組成物。
【請求項21】
5-HT受容体アゴニストがスマトリプタンである、鼻腔内投与のために製剤化されている、請求項1記載の組成物。
【請求項22】
IMITREX(登録商標)注射剤と比較して少なくとも約17%の生物学的利用能を有し、かつAUC0-1hrが約10ng.hr/mLまたはそれ以上である、請求項21記載の組成物。
【請求項23】
IMITREX(登録商標)鼻噴霧薬と比較して少なくとも約120%の生物学的利用能を有し、かつAUC0-1hrが約10ng.hr/mLまたはそれ以上である、請求項21記載の組成物。
【請求項24】
15ng/mLを上回るCmaxを有する、請求項21記載の組成物。
【請求項25】
少なくとも約17ng/mLであるCmaxを有する、請求項21記載の組成物。
【請求項26】
約1×10(exp 6)mL(exp-1)(1×106mL-1)を上回る用量/Cmax比を有する、請求項21記載の組成物。
【請求項27】
少なくとも約1.15×10(exp 6)mL(exp-1)(1.15×106mL-1)である用量/Cmax比を有する、請求項21記載の組成物。
【請求項28】
約20分未満のTmaxを有する、請求項21記載の組成物。
【請求項29】
約15分未満のTmaxを有する、請求項21記載の組成物。
【請求項30】
スマトリプタン濃度が5mg〜100mgである、請求項21記載の組成物。
【請求項31】
約10ng*hr/mLを上回るAUC0-1hrをもたらす、請求項21記載の組成物。
【請求項32】
約5分またはそれ未満で、約5ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項33】
約20mgのスマトリプタンを含み、約20分またはそれ未満で、約16ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項34】
約2分またはそれ未満で、約5ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項35】
約15分またはそれ未満で、約10ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項36】
組成物を投与する段階を含む、5-HTアゴニストの減量されているが治療的には有効な量を対象に与える方法であって、組成物が:
i)5-HT受容体アゴニストの治療的有効量;および
ii)アルキルサッカリド、
を含み、AUCが、アルキルサッカリドの非存在下で投与された5-HTアゴニストの増量された治療的有効量によってもたらされるAUCと比較してほぼ等しい、方法。
【請求項37】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項36記載の方法。
【請求項38】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項36記載の方法。
【請求項39】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項38記載の方法。
【請求項40】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側またはCSF送達経路を介して投与される、請求項36記載の方法。
【請求項41】
組成物が鼻腔内に投与される、請求項40記載の方法。
【請求項42】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項36記載の方法。
【請求項43】
約7.0またはそれ未満のpHを有する、請求項36記載の組成物。
【請求項44】
組成物がスマトリプタンを約20mgの量で含み、さらに組成物が約10ng*hr/mLまたはそれ以上のAUC0-1hrおよび約40ng*hr/mLのAUC0-4hrをもたらす、請求項41記載の方法。
【請求項45】
組成物を投与する段階を含む、対象における片頭痛緩和の迅速開始をもたらす方法であって、組成物が:
i)5-HT受容体アゴニストの治療的有効量;および
ii)アルキルサッカリド、
を含み、組成物が対象において約30分またはそれ未満のTmaxを示し、それにより、片頭痛緩和の迅速開始をもたらす方法。
【請求項46】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項45記載の方法。
【請求項47】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項45記載の方法。
【請求項48】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項47記載の方法。
【請求項49】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側またはCSF送達経路を介して投与される、請求項45記載の方法。
【請求項50】
組成物が鼻腔内に投与される、請求項49記載の方法。
【請求項51】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項45記載の方法。
【請求項52】
約7.0またはそれ未満のpHを有する、請求項45記載の組成物。
【請求項53】
組成物を投与する段階を含む、対象における片頭痛再発の発生率の低下をもたらす方法であって、組成物が:
a)5-HT受容体アゴニストの治療的有効量;および
b)アルキルサッカリド、
を含み、組成物が約20分未満のTmaxをもたらし、それにより、対象における片頭痛再発の発生率の低下を提供する方法。
【請求項54】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項53記載の方法。
【請求項55】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項53記載の方法。
【請求項56】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項55記載の方法。
【請求項57】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側、またはCSF送達経路を介して投与される、請求項53記載の方法。
【請求項58】
組成物が鼻腔内に投与される、請求項57記載の方法。
【請求項59】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項53記載の方法。
【請求項60】
約7.0またはそれ未満のpHを有する、請求項53記載の組成物。
【請求項1】
以下のものを含む組成物:
a)5-HT受容体アゴニストの治療的有効量;および
b)アルキルサッカリド。
【請求項2】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項1記載の組成物。
【請求項3】
アルキルサッカリド濃度が約0.05%〜2%である、請求項1記載の組成物。
【請求項4】
アルキルサッカリド濃度が約0.1%〜0.25%である、請求項3記載の組成物。
【請求項5】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項1記載の組成物。
【請求項6】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項5記載の組成物。
【請求項7】
対象の循環器系への、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側、またはCSF送達経路を介した投与のために製剤化されている、請求項1記載の組成物。
【請求項8】
鼻腔内送達のために製剤化されている、請求項7記載の組成物。
【請求項9】
アルキルグリコシドの臨界ミセル濃度(CMC)が約1mM未満である、請求項1記載の組成物。
【請求項10】
アルキルグリコシドのCMCが約0.5mM未満である、請求項9記載の組成物。
【請求項11】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項1記載の組成物。
【請求項12】
EDTAが重量比で約0.01%〜2%の濃度を有する、請求項11記載の組成物。
【請求項13】
約7.0またはそれ未満のpHを有する、請求項1記載の組成物。
【請求項14】
ヒトにおいて5-HTアゴニストに関して約30分間またはそれ未満のTmaxをもたらす、請求項1記載の組成物。
【請求項15】
アルキルサッカリドの非存在下でもたらされる対応するAUC 0-1hrと比較して、5-HTアゴニストに関して約1.3倍、1.5倍またはそれ以上の大きさであるAUC 0-1hrをもたらす、請求項1記載の組成物。
【請求項16】
アルキルサッカリドの非存在下でもたらされる対応するCmaxと比較して、5-HTアゴニストに関して約1.3倍またはそれ以上の大きさであるCmaxをもたらす、請求項1記載の組成物。
【請求項17】
ヒトへの投与後約10〜15分の時点で、投与約60分後の時点でのアゴニストの血漿中濃度または血中濃度と比較して少なくとも約1.3倍、1.5倍またはそれ以上の高さである5-HTアゴニストの血漿中濃度または血中濃度をもたらす、請求項1記載の組成物。
【請求項18】
ヒトへの投与後約20分未満の時点で、投与約60分後の時点でのアンタゴニストの血漿中濃度または血中濃度と比較して少なくとも約1.3倍、1.5倍またはそれ以上の高さである5-HTアゴニストの最大血漿中濃度または血中濃度をもたらす、請求項1記載の組成物。
【請求項19】
ヒトへの投与後約20分未満のうちに血漿中または血液中の5-HTアゴニストに関するCmaxをもたらし、アルキルサッカリド濃度が0.05〜0.2%である、請求項1記載の組成物。
【請求項20】
ヒトへの投与後約20分未満のうちに血漿中または血液中の5-HTアゴニストに関するCmaxをもたらし、かつCmaxの少なくとも約0.25倍である60分時点での持続的5-HTアゴニスト濃度をもたらす、請求項1記載の組成物。
【請求項21】
5-HT受容体アゴニストがスマトリプタンである、鼻腔内投与のために製剤化されている、請求項1記載の組成物。
【請求項22】
IMITREX(登録商標)注射剤と比較して少なくとも約17%の生物学的利用能を有し、かつAUC0-1hrが約10ng.hr/mLまたはそれ以上である、請求項21記載の組成物。
【請求項23】
IMITREX(登録商標)鼻噴霧薬と比較して少なくとも約120%の生物学的利用能を有し、かつAUC0-1hrが約10ng.hr/mLまたはそれ以上である、請求項21記載の組成物。
【請求項24】
15ng/mLを上回るCmaxを有する、請求項21記載の組成物。
【請求項25】
少なくとも約17ng/mLであるCmaxを有する、請求項21記載の組成物。
【請求項26】
約1×10(exp 6)mL(exp-1)(1×106mL-1)を上回る用量/Cmax比を有する、請求項21記載の組成物。
【請求項27】
少なくとも約1.15×10(exp 6)mL(exp-1)(1.15×106mL-1)である用量/Cmax比を有する、請求項21記載の組成物。
【請求項28】
約20分未満のTmaxを有する、請求項21記載の組成物。
【請求項29】
約15分未満のTmaxを有する、請求項21記載の組成物。
【請求項30】
スマトリプタン濃度が5mg〜100mgである、請求項21記載の組成物。
【請求項31】
約10ng*hr/mLを上回るAUC0-1hrをもたらす、請求項21記載の組成物。
【請求項32】
約5分またはそれ未満で、約5ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項33】
約20mgのスマトリプタンを含み、約20分またはそれ未満で、約16ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項34】
約2分またはそれ未満で、約5ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項35】
約15分またはそれ未満で、約10ng/mLを上回るかまたはそれと等しいスマトリプタンの血漿レベルをもたらす、請求項21記載の組成物。
【請求項36】
組成物を投与する段階を含む、5-HTアゴニストの減量されているが治療的には有効な量を対象に与える方法であって、組成物が:
i)5-HT受容体アゴニストの治療的有効量;および
ii)アルキルサッカリド、
を含み、AUCが、アルキルサッカリドの非存在下で投与された5-HTアゴニストの増量された治療的有効量によってもたらされるAUCと比較してほぼ等しい、方法。
【請求項37】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項36記載の方法。
【請求項38】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項36記載の方法。
【請求項39】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項38記載の方法。
【請求項40】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側またはCSF送達経路を介して投与される、請求項36記載の方法。
【請求項41】
組成物が鼻腔内に投与される、請求項40記載の方法。
【請求項42】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項36記載の方法。
【請求項43】
約7.0またはそれ未満のpHを有する、請求項36記載の組成物。
【請求項44】
組成物がスマトリプタンを約20mgの量で含み、さらに組成物が約10ng*hr/mLまたはそれ以上のAUC0-1hrおよび約40ng*hr/mLのAUC0-4hrをもたらす、請求項41記載の方法。
【請求項45】
組成物を投与する段階を含む、対象における片頭痛緩和の迅速開始をもたらす方法であって、組成物が:
i)5-HT受容体アゴニストの治療的有効量;および
ii)アルキルサッカリド、
を含み、組成物が対象において約30分またはそれ未満のTmaxを示し、それにより、片頭痛緩和の迅速開始をもたらす方法。
【請求項46】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項45記載の方法。
【請求項47】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項45記載の方法。
【請求項48】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項47記載の方法。
【請求項49】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側またはCSF送達経路を介して投与される、請求項45記載の方法。
【請求項50】
組成物が鼻腔内に投与される、請求項49記載の方法。
【請求項51】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項45記載の方法。
【請求項52】
約7.0またはそれ未満のpHを有する、請求項45記載の組成物。
【請求項53】
組成物を投与する段階を含む、対象における片頭痛再発の発生率の低下をもたらす方法であって、組成物が:
a)5-HT受容体アゴニストの治療的有効量;および
b)アルキルサッカリド、
を含み、組成物が約20分未満のTmaxをもたらし、それにより、対象における片頭痛再発の発生率の低下を提供する方法。
【請求項54】
5-HTアゴニストが、スマトリプタン、ナラトリプタン、リザトリプタン、エレトリプタン、フロバトリプタン、アルモトリプタン、ゾルミトリプタン、それらの塩、またはそれらの組み合わせである、請求項53記載の方法。
【請求項55】
アルキルサッカリドが、10〜16個の炭素を含むアルキル鎖を有する、請求項53記載の方法。
【請求項56】
アルキルサッカリドが、ウンデシル-β-D-マルトシド、ドデシル-β-D-マルトシド、トリデシル-β-D-マルトシド、テトラデシル-β-D-マルトシド、スクロースモノドデカノエート、またはそれらの組み合わせである、請求項55記載の方法。
【請求項57】
組成物が対象の循環器系に、経口、眼、鼻腔内、鼻涙管、吸入、肺、舌下、頬側、またはCSF送達経路を介して投与される、請求項53記載の方法。
【請求項58】
組成物が鼻腔内に投与される、請求項57記載の方法。
【請求項59】
エチレンジアミン四酢酸(EDTA)またはその塩をさらに含む、請求項53記載の方法。
【請求項60】
約7.0またはそれ未満のpHを有する、請求項53記載の組成物。
【図1】

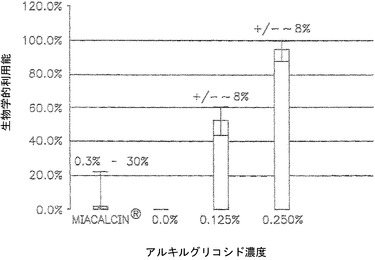
【図2】
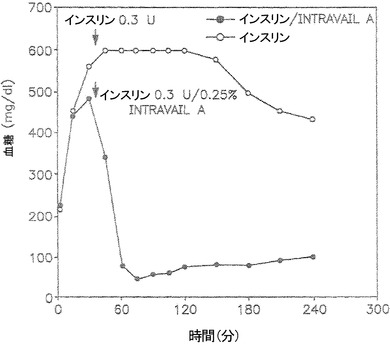
【図3】
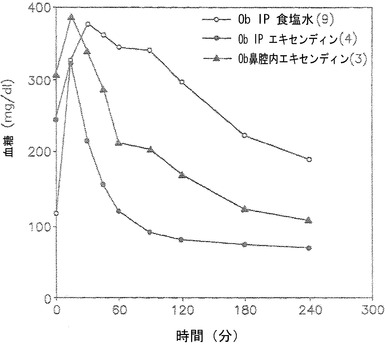
【図4】
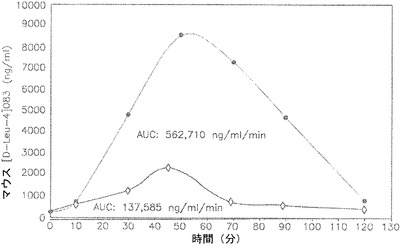
【図5】
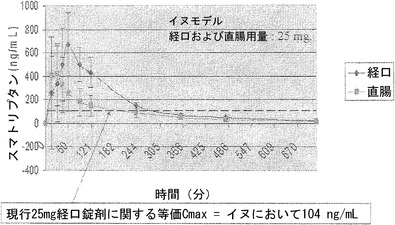
【図6】
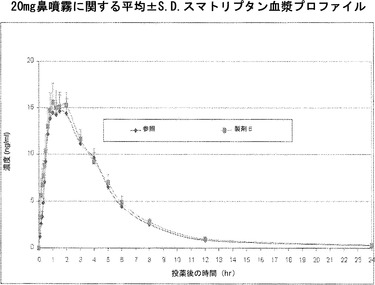
【図7】
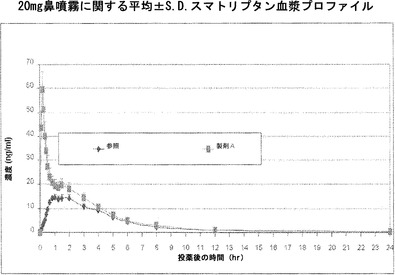
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2012−513412(P2012−513412A)
【公表日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願番号】特願2011−542586(P2011−542586)
【出願日】平成21年12月22日(2009.12.22)
【国際出願番号】PCT/US2009/069326
【国際公開番号】WO2010/075465
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(511151008)イージス セラピューティクス リミテッド ライアビリティ カンパニー (2)
【Fターム(参考)】
【公表日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願日】平成21年12月22日(2009.12.22)
【国際出願番号】PCT/US2009/069326
【国際公開番号】WO2010/075465
【国際公開日】平成22年7月1日(2010.7.1)
【出願人】(511151008)イージス セラピューティクス リミテッド ライアビリティ カンパニー (2)
【Fターム(参考)】
[ Back to top ]