
薬理学的活性ペプチド/タンパク質の血清中半減期を上昇させるためのトランスサイレチンペプチド/タンパク質融合物の使用
【課題】トランスサイレチン(TTR)を生物学的活性物質との融合パートナーとして使用することにより、選択した生物学的活性物質の血清中半減期を上昇させるための手段を提供する。
【解決手段】TTR(又はTTR変異体)−生物学的活性物質融合物及びPEG−TTR(PEG−TTR変異体)−生物学的活性物質融合物の実質的に均質な製剤。製剤を製造する方法は、(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得る(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTRを得(c)上記PEG−TTRを対象ペプチドに融合してPEG−TTR−ペプチド融合物を得る(d)上記PEG−TTR−ペプチド融合物を単離する。
【解決手段】TTR(又はTTR変異体)−生物学的活性物質融合物及びPEG−TTR(PEG−TTR変異体)−生物学的活性物質融合物の実質的に均質な製剤。製剤を製造する方法は、(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得る(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTRを得(c)上記PEG−TTRを対象ペプチドに融合してPEG−TTR−ペプチド融合物を得る(d)上記PEG−TTR−ペプチド融合物を単離する。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、本明細書中に参考として援用されている、2002年4月4日出願の米国出願第10/117,109号の一部継続出願である。
【背景技術】
【0002】
治療用途のためのタンパク質、ペプチド及び他の薬剤分子は、現在、主として組換えDNAテクノロジーの進歩の結果として適切な形態で十分な量が入手可能である。そのようなペプチド及びタンパク質が入手できることは、タンパク質の製剤及び化学的修飾における進歩を生じさせた。生物学的に活性なペプチド、タンパク質、オリゴヌクレオチド及び他の薬剤の血清中半減期を延長するために、そのような生物学的活性物質の化学的修飾が広汎に検討されてきた。そのような物質の血清中半減期を延長する能力は、薬剤の治療上の潜在的可能性が高用量及び高頻度の投与を必要とせずに実現されることを可能にする。
【0003】
インビボでタンパク質の半減期を延長するために使用される化学的修飾は、対象タンパク質へのポリエチレングリコール(PEG)などの水溶性ポリマーの化学的複合体化を包含する。ポリエチレングリコール分子をタンパク質に結合するために(ペグ化)様々なアプローチが使用されてきた。例えば、Royer(米国特許第4,002,531号)は、酵素へのポリエチレングリコール分子の結合のために還元的アルキル化を使用したと述べている。Davisら(米国特許第4,179,337号)は、例えば酵素とインスリンを含むPEG:タンパク質複合体を開示している。Shaw(米国特許第4,904,584号)は、反応性アミン基によるポリエチレングリコール分子の結合のための、タンパク質中のリシン残基数の変更を開示している。Hakimiら(米国特許第5,834,594号)は、例えばタンパク質である、IL−2、インターフェロンα及びIL−1raを含む、実質的に非免疫原性の水溶性PEG:タンパク質複合体を開示している。Hakimiらの方法は、タンパク質中の様々な遊離アミノ基をPEGに連結するためのユニークなリンカーの使用を含む。Kinstlerら(米国特許第5,824,784号及び同第5,985,265号)は、G−CSF及びコンセンサスインターフェロンを含む、選択的N末端化学的修飾タンパク質及びその類似体を可能にする方法を教示している。
【0004】
生物学的活性物質の血清中半減期を延長するために設計された他のアプローチは次のものを含む:あまりに大きすぎて腎ではろ過されない、大きく安定なタンパク質(例えば血清アルブミン)へのペプチドの複合体化;G.D.Maoら、Biomat.Art.Cells,Art.Org.17:229−244(1989);輸送ビヒクルとして及び血清中半減期を上昇させるための低密度及び高密度リポタンパク質の使用;P.Chris de Smidtら、Nuc.Acids.Res.,19(17):4695−4700(1991);Fc−タンパク質融合物を生産するための免疫グロブリンのFc領域の使用;PCT国際公開公報第98/28427号(Mannら及びその中で引用される参考文献);及び1以上の生物学的活性ペプチドのインビボでの半減期を上昇させるためのFcドメインの使用;PCT国際公開公報第00/24782号(Feigeら及びその中で引用される参考文献)。
【0005】
トランスサイレチン(TTR)(以前はプレアルブミンと呼ばれた)は、サイロキシン及びレチノール結合タンパク質の輸送体として重要な生理的役割を果たす56kDaの四量体血清タンパク質である;HamiltonとBenson,Cell.Mol.Life Sci.,58:1491−1521(2001)及びその中で引用される参考文献。米国特許第5,714,142号においてBlaneyらは、投与する薬剤に、該タンパク質に特異的に結合しうる機能を付与することによるTTRの利用を述べている。詳細には、Blaneyらは、トランスサイレチン選択的リガンドへのペプチド、タンパク質、ヌクレオチド、オリゴヌクレオチド、オリゴ糖又は他の薬剤の共有結合が、薬剤をTTRに可逆的に結合させ、それによってTTRに関するリガンドの親和性に基づき該薬剤の血清中半減期を上昇させることを明らかにしている。薬剤の内因性作用は有害な影響を受けず、生じる薬剤−TTRリガンド複合体はなお経口吸収されるのに十分なほど小さいと述べられている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第4,002,531号明細書
【特許文献2】米国特許第4,179,337号明細書
【特許文献3】米国特許第4,904,584号明細書
【特許文献4】米国特許第5,834,594号明細書
【特許文献5】米国特許第5,824,784号明細書
【特許文献6】米国特許第5,985,265号明細書
【特許文献7】国際公開第98/28427号
【特許文献8】国際公開第00/24782号
【特許文献9】米国特許第5,714,142号明細書
【非特許文献】
【0007】
【非特許文献1】G.D.Maoら、Biomat.Art.Cells,Art.Org.17:229−244(1989)
【非特許文献2】P.Chris de Smidtら、Nuc.Acids.Res.,19(17):4695−4700(1991)
【非特許文献3】HamiltonとBenson,Cell.Mol.Life Sci.,58:1491−1521(2001)
【発明の概要】
【発明が解決しようとする課題】
【0008】
驚くべきことに及び重要なこととして、TTR(又はTTR変異体)、特に、例えば水溶性ポリマーへの複合体化によって化学的に修飾されたTTR又はTTR変異体は、生物学的活性物質の血清中半減期を上昇させるために生物学的活性物質との融合パートナーとして使用できることが見出されている。従って、本発明は、選択した生物学的活性物質の血清中半減期を上昇させるための手段を提供する。
【課題を解決するための手段】
【0009】
本発明は、それ故、TTR(又はTTR変異体)−生物学的活性物質融合物及びPEG−TTR(PEG−TTR変異体)−生物学的活性物質融合物の実質的に均質な製剤に関する。生物学的活性物質単独と比較して、TTR−生物学的活性物質融合物及び/又はPEG−TTR−生物学的活性物質融合物は実質的に上昇した血清中半減期を有する。
【0010】
本発明はさらに、薬理的活性化合物を提供するための、医薬適合性の担体中のTTR−生物学的活性物質融合物及びPEG−TTR−生物学的活性物質融合物に関する。
【0011】
本発明はさらに、TTR変異体の調製に関する。詳細には、TTRタンパク質を、システイン残基がTTRタンパク質配列内に工作されるように修飾する。該TTR変異体は高収率で回収可能であり、その後システイン残基での水溶性ポリマーの複合によって化学的修飾して、化学的修飾されたTTR変異体を提供し、次に選択した生物学的活性物質にそれを融合することができる。
【0012】
本発明はさらに、薬理的活性化合物を調製する方法に関する。例えば、PEG−TTR−ペプチド融合物の実質的に均質な製剤を製造する方法の主要実施態様は、(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTRを得ること;(c)上記PEG−TTRを対象ペプチドに融合してPEG−TTR−ペプチド融合物を得ること;及び(d)上記PEG−TTR−ペプチド融合物を単離することを含む。
【0013】
本発明はまた、上記のような薬理的活性化合物を使用した個体の治療方法に関する。
【図面の簡単な説明】
【0014】
【図1】大腸菌(E.coli)で発現された組換えヒトトランスサイレチン(TTR)変異体(C10A/G83C)の、TTRのC末端に融合したブラジキニンペプチドによる精製を示すSDSゲルである。レーン1は、Novex Mark 12分子量標準物質を含み、レーン2−7はそれぞれ次のものを含む:細胞溶解産物、加熱後上清、Qセファロースクロマトグラフィー段階からのプール、フェニルセファロースクロマトグラフィー段階からのプール、ヒドロキシアパタイトクロマトグラフィー段階からのプール、及びソースQクロマトグラフィー段階からのプール。
【図2】TTR変異体、TTR(C10A/G83C)のアミノ末端又はカルボキシ末端へのペプチドの融合がそのオリゴマー構造を変化させないことをサイズ排除クロマトグラフィーによって明らかにする。実線はTTR(C10A/G83C)、破線はTTR(C10A/G83C)のアミノ末端に融合した副甲状腺ホルモン(PTH)、及び点線はTTR(C10A/G83C)のカルボキシ末端に融合したブラジキニンである。
【図3】TR変異体、TTR(C10A)のアミノ末端又はカルボキシ末端へのタンパク質の融合がそのオリゴマー構造を変化させないことをサイズ排除クロマトグラフィーによって明らかにする。実線はTTR(C10A)、破線はTTR(C10A)のカルボキシ末端に融合したIL−1−ra、及び点線はTTR(C10A)のアミノ末端に融合したIL−1−raである。
【図4】BIAcoreを使用して認められた、ヒトMPL受容体への様々なTPO−ミメティックペプチド(TMP)構築物の結合を示す:
【化1】

【図5】TMP(m)−TTR−PEG5Kの注入がマウスにおいて血小板形成を誘導することを示す。次の記号はその対応する構築物に相当する:
【化2】
【図6】天然TTR及びTTR(C10A)が同様のオリゴマー立体配置(見かけ上の四量体)を維持することをサイズ排除クロマトグラフィーによって明らかにしている。実線は天然TTRであり、破線はTTR(C10A)である。
【図7】TTRへのPEGの複合体化が予測可能に一様にその分子サイズを上昇させることをサイズ排除クロマトグラフィーによって明らかにする。実線はPEG複合体化なし、破線は5K PEG融合、及び点線は20K PEG融合を示す。次の構築物を使用した:A)TMP−TTR(C10A/A37C)、B)TMP−TTR(C10A/D38C)、C)TMP−TTR(C10A/A81C)、及びD)TMP−TTR(C10A/G83C)。
【図8】4つの異なる位置の1つに工作された非天然システインを有するTTR変異体を含む様々なTMP−TTR構築物のペグ化の程度を示すSDSゲルである。レーン1はNovex Mark 12分子量標準物質を含む;レーン2は非ペグ化TMP−TTR(C10A/A37C)である;レーン3−6は、それぞれTMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)及びTMP−TTR(C10A/G83C)の5Kペグ化型である;レーン7−10は、それぞれTMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)及びTMP−TTR(C10A/G83C)の20Kペグ化型である。
【図9】図9A−Cは、BIAcore分析によるヒトMPLへのFc−TMPとTMP−TTRの競合的結合を比較している。
【化3】
【図10】図10A−Cは、工作したシステインに複合体化したPEGを伴うTMP−TTRの注入がマウスにおいて血小板形成を誘導することを示す。
【化4】
【図11】工作されたシステインに複合体化したPEGを伴うPTH−TTRの注入がマウスにおいてイオン化カルシウム放出を誘導することを示す。次の記号はその対応する構築物に相当する:
【化5】
【図12】工作されたシステインに複合体化したPEGを伴うグルカゴン様ペプチド1(GLP1)−TTRの注入がマウスにおいて血中グルコースレベルを低下させることを示す。次の記号はその対応する構築物に相当する:
【化6】
【図13】融合CH2ドメインを有するTMP−TTR複合体の注入がマウスにおいて血清中の血小板レベルを上昇させることを示す。次の記号はその対応する構築物に相当する:
【化7】
【図14】ペグ化TTRとTMPのカルボキシ末端融合物の注入がマウスにおいて血中血小板数を上昇させることを示す。次の記号はその対応する構築物に相当する:
【化8】
【図15】図15A−Cは、K15A変化を含むペグ化TMP−TTR融合物の注入がマウスにおいて血中血小板数を上昇させることを示す。次の記号はその対応する構築物に相当する:
【化9】
【発明を実施するための形態】
【0015】
本発明を説明するために、次の用語は下記で述べるように定義される。
【0016】
「生物学的活性物質」という用語は、予防、治療又は診断的適用のために有用な化学物質又は化合物を指す。「薬理的活性化合物」という用語は、所望の局所又は全身作用を誘導する、哺乳動物、好ましくはヒト個体への投与に適した化合物を指す。
【0017】
「ペプチド」、「ポリペプチド」及び「タンパク質」という用語は、生物学的活性物質の種類を表わし、それらの用語は、天然に生じる、組換え生産された又は化学合成されたアミノ酸のポリマーを指すためにここでは交換可能に使用される。それらの用語は、2個という低い数のアミノ酸を含むペプチド分子、化学的修飾されたポリペプチド、コンセンサス分子、それらの類似体、誘導体又は組合せを包含することが意図される。
【0018】
本発明に関連して任意の数のペプチドが使用しうる。特に興味深いのは、エリスロポエチン(EPO)、トロンボポエチン(TPO)、グルカゴン様ペプチド1(GLP−1)、副甲状腺ホルモン(PTH)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、インターロイキン−1受容体アンタゴニスト(IL−1ra)、レプチン、細胞傷害性Tリンパ球抗原4(CTLA4)、TNF関連アポトーシス誘導リガンド(TRAIL)、腫瘍増殖因子α及びβ(それぞれTGF−α及びTGF−β)、及び成長ホルモンの作用を模倣するペプチドである。「ミメティックペプチド」及び「アゴニストペプチド」という用語は、対象とするタンパク質と相互作用するタンパク質(例えばGLP−1、PTH、EPO、TPO、G−CSF等)に匹敵する生物学的活性を有するペプチドを指す。これらの用語はさらに、対象とするタンパク質の天然リガンドの作用を増強することなどによって、対象とするタンパク質の作用を間接的に模倣するペプチドを包含する。それ故、「EPOミメティックペプチド」という用語は、EPOミメティックの性質(EPO−mimetic subject matter)を有すると同定できる又は推論できるいかなるペプチドも包含する;例えばWrightonら、Science,273:458−63(1996);及びNarandaら、Proc.Natl.Acad.Sci.USA 96:7569−74(1999)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0019】
「TPOミメティックペプチド」(TMP)という用語は、TPOミメティックの性質を有すると同定できる又は推論できるペプチドを包含する;例えば、その全体が参考として本明細書中に援用されている、Cwirlaら、Science,276:1696−9(1997);米国特許第5,869,451号及び同第5,932,946号;及びPCT国際公開公報第00/24782号(Liuら、及びその中で引用される参考文献)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0020】
「G−CSFミメティックペプチド」という用語は、G−CSFミメティックの性質を有すると同定できるいかなるペプチドも包含する;例えばPaukovitsら、Hoppe−Seylers Z.Physiol.Chem.365:303−11(1984)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0021】
「CTLA4−ミメティックペプチド」という用語は、Fukumotoら、Nature Biotech.16:267−70(1998)に述べられているように同定できる又は推論できるいかなるペプチドも包含する。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0022】
ペプチドアンタゴニストも興味の対象であり、特にTNF、レプチン、インターロイキンのいずれか及び補体活性化に関与するタンパク質(例えばC3b)の作用に拮抗するものが興味深い。「アンタゴニストペプチド」又は「阻害剤ペプチド」という用語は、対象とする関連タンパク質の生物学的活性をブロックする又は何らかの方法で生物学的活性に干渉する、あるいは対象関連タンパク質の既知のアンタゴニスト又は阻害剤に匹敵する生物学的活性を有するペプチドを指す。それ故、「TNFアンタゴニストペプチド」という用語は、TNF拮抗性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Takasakiら、Nature Biotech.,15:1266−70(1997)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0023】
「IL−1アンタゴニスト」及び「IL−1raミメティックペプチド」という用語は、IL−1によるIL−1受容体の活性化を阻害する又は下方調節するペプチドを包含する。IL−1受容体の活性化は、IL−1、IL−1受容体及びIL−1受容体補助タンパク質(accessory protein)の間での複合体の形成から生じる。IL−1アンタゴニスト及びIL−1raミメティックペプチドは、IL−1、IL−1受容体又はIL−1受容体補助タンパク質に結合して、複合体の任意の2つ又は3つの成分の間での複合体形成を妨げる。例示的なIL−1アンタゴニスト及びIL−1raミメティックペプチドは、米国特許第5,608,035号、同第5,786,331号、同第5,880,096号に述べられているように同定する又は推論することができる。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0024】
「VEGFアンタゴニストペプチド」という用語は、VEGF拮抗性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Fairbrother,Biochem.,37:17754−64(1998)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0025】
「MMP阻害剤ペプチド」という用語は、MMP阻害性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Koivunen,Nature Biotech.,17:768−74(1999)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0026】
腫瘍ホーミングペプチド、膜輸送ペプチド等を含む、標的化ペプチドも興味深い。
【0027】
例示的ペプチドは当技術分野において既知の様々な手法によってランダムに生成しうる。例えば、固相合成手法は当技術分野において周知であり、Merrifield,Chem.Polypeptides,p.335−61(KatsoyannisとPanayotis編集)(1973);Merrifield,J.Am.Chem.Soc.,85:2149(1963);Davisら、Biochem.Intl.,10:394−414(1985);StewartとYoung,Solid Phase Peptide Synthesis(1969);米国特許第3,941,763号;Finnら、The Proteins,第3版、2:105−253(1976);及びEricksonら、The Proteins,第3版、2:257−527(1976)に述べられているものを包含する。固相合成は、小さなペプチドを作製する最もコスト効果的な方法であるので、個々のペプチドを作製する好ましい手法である。
【0028】
ファージディスプレイは、本発明において使用するためのペプチドを作製する上でのもう1つの有用な方法である。ランダムなペプチドのライブラリーからのアフィニティー選択が、任意の遺伝子産物の任意の部位についてのペプチドリガンドを同定するために使用できることが記述されている;Dedmanら、J.Biol.Chem.,268:23025−30(1993)。ファージディスプレイは、細胞表面受容体又は線状エピトープを有する任意のタンパク質のような対象タンパク質に結合するペプチドを同定するのに特に適する;Wilsonら、Can.J.Microbiol.,44:313−29(1998);Kayら、Drug Disc.Today,3:370−8(1998)。そのようなタンパク質は、本明細書中に参考として援用されている、Herzら、J.Receptor & Signal Transduction Res.,17(5):671−776(1997)において広く概説されている。
【0029】
該ペプチドはまた、組換えDNA手法を用いて形質転換宿主細胞においても作製しうる。そのために、該ペプチドをコードする組換えDNA分子を調製する。そのようなDNA及び/又はRNA分子を調製する方法は当技術分野において周知である。例えば、適切な制限酵素を使用して該ペプチドをコードする配列をDNAから切り出すことができた。その後のクローニングのために有用な制限部位を含んだ関連する配列を、ポリメラーゼ連鎖反応(PCR)を用いて創製することができた。あるいは、ホスホルアミダイト法などの化学合成手法を用いてDNA/RNA分子を合成することができよう。また、これらの手法の組合せも使用しうる。
【0030】
使用のために考慮されるさらなる生物学的活性物質は、ヒト又は動物の組換え又は天然のタンパク質、ホルモン、サイトカイン、造血因子、成長因子、抗肥満因子、栄養因子、抗炎症因子及び酵素を含む。そのようなタンパク質は、インターフェロン(図面を含めて本明細書中に参考として援用されている、米国特許第5,372,808号、同第5,541,293号、同第4,897,471号及び同第4,695,623号参照)、インターロイキン(図面を含めて本明細書中に参考として援用されている、米国特許第5,075,222号参照)、エリスロポエチン(図面を含めて本明細書中に参考として援用されている、米国特許第4,703,008号、同第5,441,868号、同第5,618,698号、同第5,547,933号及び同第5,621,080号参照)、顆粒球コロニー刺激因子(図面を含めて本明細書中に参考として援用されている、米国特許第4,810,643号、同第4,999,291号、同第5,581,476号、同第5,582,823号及びPCT国際公開公報第94/17185号参照)、幹細胞因子(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第91/05795号、同第92/17505号及び同第95/17206号)、NESP(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第US94/02957号)、オステオプロテゲリン(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第97/23614号)、インターロイキン−1受容体アンタゴニスト(L−1ra)(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第91/08285号及び同第92/16221号)及びレプチン(OBタンパク質)(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第96/40912号、同第96/05309号、同第97/00128号、同第97/01010号及び同第97/06816号)を含むが、これらに限定されない。
【0031】
さらに、生物学的活性物質はまた、インスリン、ガストリン、プロラクチン、副腎皮質刺激ホルモン(ACTH)、甲状腺刺激ホルモン(TSH)、黄体形成ホルモン(LH)、卵胞刺激ホルモン(FSH)、ヒト絨毛性性腺刺激ホルモン(HCG)、モチリン、インターフェロン(α、β、γ)、インターロイキン類(IL−1からIL−12まで)、腫瘍壊死因子(TNF)、腫瘍壊死因子結合タンパク質(TNF−bp)、脳由来神経栄養因子(BDNF)、神経膠由来神経栄養因子(GNDF)、神経栄養因子3(NT3)、線維芽細胞増殖因子(FGF)、神経栄養増殖因子(NGF)、オステオプロテゲリン(OPG)などの骨成長因子、インスリン様増殖因子(GF)、マクロファージコロニー刺激因子(M−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、巨核球由来増殖因子(MGDF)、角質細胞増殖因子(KGF)、トロンボポエチン、血小板由来増殖因子(PDGF)、コロニー刺激増殖因子(CSF)、骨誘導因子(BMP)、スーパーオキシドジスムターゼ(SOD)、組織プラスミノーゲン活性化因子(TPA)、ウロキナーゼ、ストレプトキナーゼ及びカリクレインを含みうるが、これらに限定されない。
【0032】
本発明における使用のための考慮されるトランスサイレチン(TTR)は、Mitaら、Biochem.Biophys.Res.Commun.,124(2):558−564(1984)に報告されているTTRのDNA及びアミノ酸配列を有する。これらの配列は、受入番号K02091としてGenbankに寄託されている。ここで使用する127アミノ酸のTTR配列は、K02091配列のシグナル配列(アミノ酸1−20)を含まず、配列番号1として下記に示す:
【0033】
【化10】
【0034】
「TTR変異体」という用語は、天然TTRの修飾形態である分子又は配列を指す。例えば、天然TTRは除去しうる部位を含む。それらは、構造的特徴を与えるか又は本発明の融合分子には必要とされない生物学的活性を提供するからである。それ故、「TTR変異体」という用語は、1以上の天然TTR部位又は残基を欠く、あるいは異なるアミノ酸で置換された1以上の天然TTR部位又は残基を有する、あるいは該配列に付加された1以上の残基を有する分子又は配列を包含する。一例として、アミノ酸配列37位のアラニン残基がシステイン残基で置換されたTTR変異体をTTR変異体(A37C)と称する;及びアミノ酸配列37位のアラニン残基及びアミノ酸配列83位のグリシン残基の両方がシステイン残基で置換されたTTR変異体をTTR変異体(A37C/G83C)と称する。
【0035】
1つの実施態様では、生物学的活性物質に融合したTTR又はTTR変異体を、TTR−生物学的活性物質融合タンパク質をさらに安定化し、それによって、生じる融合物の血清中半減期を上昇させる、第三のタンパク質又はタンパク質フラグメントに融合しうる。そのような方法及び組成物において使用できる付加的タンパク質又はそのフラグメントの例は、免疫グロブリンのFcドメイン又はCH2ドメイン、あるいは当業者がタンパク質の安定性を上昇させる性質を有すると認識する他の何らかのタンパク質ドメイン(例えば下記の実施例29)を包含する。そのようなタンパク質群は、TTR−生物学的活性物質融合タンパク質のカルボキシ又はアミノ末端に融合することができ、あるいはTTRと生物学的活性物質の間に配置することができる。所望の活性を促進するために該融合タンパク質の各々のドメインの間に、必要に応じて、リンカー又はスペーサーを配置できることが意図される。
【0036】
もう1つの実施態様では、本発明のTTR又はTTR変異体を生物学的活性物質に化学的に架橋することができる。タンパク質の架橋は、例えば確立された公表手順に従い、N−スクシニミジル3−(2−ピリジルジチオ)プロピオネート(SPDP)を使用することによって実施できる。さらなる架橋剤は容易に入手可能であり、当業者によって同定されうる。上記手順の詳細については、例えばKarpovskyら、J.Exp.Med.160.1686−1701(1984);Perezら、Nature,316、354−356(1985)又はTitusら、Journal of Immunology,139,3153−3158(1987)参照。
【0037】
もう1つの実施態様では、安定性及び/又は血清中の半減期を上昇させるように、分子を該融合タンパク質に共有結合することができる。例えば、好ましいTTR又はTTR変異体は、ポリエチレングリコール(PEG)などの水溶性ポリマーを用いて化学的修飾されうる。PEG基は、好都合ないかなる分子量であってもよく、直鎖又は分枝鎖のいずれでもよい。PEGの平均分子量は、好ましくは約2kDaから約100kDa、より好ましくは約5kDa−約50kDaの範囲であり、最も好ましくは約20kDaである。
【0038】
PEG基は一般に、アシル化、還元的アルキル化、マイケル付加、チオールアルキル化あるいはペグ部分の反応基(例えばアルデヒド、アミノ、エステル、チオール、ハロアセチル、マレイミド又はヒドラジノ基)を通しての標的化合物上の反応基(例えばアルデヒド、アミノ、エステル、チオール、ハロアセチル、マレイミド又はヒドラジノ基)への他の化学選択的複合体化/連結法によって、本発明の化合物に結合される。
【0039】
使用される他の水溶性ポリマーは、エチレングリコール/プロピレングリコールのコポリマー、カルボキシメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリ−1,3−ジオキソラン、ポリ−1,3,6−トリオキサン、エチレン/無水マレイン酸コポリマー、ポリアミノ酸(ホモポリマー又はランダムコポリマーのいずれか)及びデキストランを含む。
【0040】
対象ペプチド、対象タンパク質、TTR又はTTR変異体をコードするDNA分子は、Sambrookら(Molecular Cloning:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY[1989])及び/又はAusubelら編集、Current Protocols in Molecular Biology,Green Publishers Inc.及びWiley and Sons,NY(1994)に述べられているような周知の組換えDNAテクノロジーを使用して作製できる。対象タンパク質又はそのフラグメントをコードする遺伝子又はcDNAは、例えばゲノム又はcDNAライブラリーを適切なプローブでスクリーニングすることによって入手しうる。適切なプローブは、例えば、該プローブが選択されたハイブリダイゼーション条件下で対象タンパク質をコードする遺伝子とハイブリダイズするように、対象タンパク質をコードする遺伝子とある程度の相同性を有すると予想されるオリゴヌクレオチド、cDNAフラグメント又はゲノムDNAフラグメントを含む。DNAライブラリーをスクリーニングする代替的手段は、対象タンパク質をコードする遺伝子のポリメラーゼ連鎖反応「PCR」増幅によってである。PCRは、典型的には、少なくともプライマーの十分な部分が該遺伝子とハイブリダイズするように、増幅すべき遺伝子と十分な相同性を有すると考えられる配列を有するオリゴヌクレオチド「プライマー」を使用して実施される。
【0041】
あるいは、対象ペプチド又は対象タンパク質をコードする遺伝子は、Engelsら、Angew.Chem.Intl.Ed.,28:716−734(1989)によって述べられているような当業者に周知の方法を用いた化学合成によって作製しうる。これらの方法は、中でも特に、核酸合成のためのホスホトリエステル、ホスホルアミダイト及びH−ホスホネート法を含む。そのような化学合成のための好ましい方法は、標準ホスホルアミダイト化学を使用したポリマー支持合成である。典型的には、対象タンパク質をコードするDNAは数百ヌクレオチドの長さである。約100ヌクレオチド以上の大きさの核酸は、これらの方法を用いていくつかのフラグメントとして合成できる。次にそれらのフラグメントを連結して、対象とする完全長タンパク質をコードする遺伝子を形成することができる。通常、ポリペプチドのアミノ末端をコードするDNAフラグメントは、メチオニン残基をコードするATGを有する。このメチオニンは成熟形態の対象タンパク質上に存在してもよく又は存在しなくてもよい。該メチオニンは、細胞の内部で又は分泌過程の間に除去することができる。好ましいTTRポリペプチドは、大腸菌における発現を最適化し、好都合な制限部位を導入するように変化した核酸配列を有するTTRを含みうる。大腸菌における発現のためのコドン最適化についての一般的考察は、Kane,Curr.Opin.Biotechnol.,6:494−500(1995)に述べられている。
【0042】
ひとたび対象タンパク質とTTRポリペプチドをコードする遺伝子が得られれば、5’及び/又は3’末端で制限エンドヌクレアーゼ部位を創製する標準的方法を用いてそれらを修飾しうる。制限部位の創製は、該遺伝子が増幅及び/又は発現ベクターに適切に挿入されることを可能にする。制限部位の付加は典型的にはPCRを用いて実施され、そこでは各々のPCR反応の1つのプライマーは、典型的には、中でも特に所望の制限部位のヌクレオチド配列を含む。
【0043】
対象ペプチド又は対象タンパク質をコードする遺伝子又はcDNAは、宿主細胞における発現のために適切な発現ベクターに挿入することができる。該ベクターは、特に使用する宿主細胞において機能性であるように選択される(すなわち、該ベクターは、対象タンパク質をコードする遺伝子の増幅及び/又は発現が起こりうるように宿主細胞機構と適合性である)。
【0044】
典型的には、宿主細胞のいずれかにおいて使用されるベクターは、プロモーター(「5’側隣接配列」とも称される)及び他の調節エレメントならびにエンハンサー、複製起点エレメント、転写終結エレメント、リボソーム結合部位エレメント、発現しようとするポリペプチドをコードする核酸を挿入するためのポリリンカー領域、及び選択マーカーエレメントを含む。これらのエレメントの各々は下記で論じる。場合により、該ベクターは「タグ」DNA配列、すなわち、融合DNA構築物の5’又は3’末端のいずれかに位置するオリゴヌクレオチド配列を含みうる。タグDNAは、ヘキサHis、c−myc、FLAG(Invitrogen,San Diego,CA)などの分子又は別の小さな免疫原性配列をコードする。適切なリーディングフレーム内に置くとき、このタグは融合タンパク質と共に発現され、宿主細胞からの融合タンパク質の精製のためにアフィニティータグとして使用できる。場合により、該タグはその後、例えば選択したペプチダーゼを使用することなどの様々な手段によって精製融合タンパク質から除去することができる。
【0045】
該プロモーターは、同種(すなわち宿主細胞と同じ種及び/又は株に由来)、異種(すなわち宿主細胞の種又は株以外の種に由来)、ハイブリッド(すなわち2つ以上のソースからのプロモーターの組合せ)、合成でありえ、あるいは天然の対象タンパク質プロモーターでありうる。さらに、該プロモーターは構成的又は誘導的プロモーターでありうる。それ故に、該プロモーターのソースは、該プロモーターが宿主細胞機構において機能性であり、宿主細胞機構によって活性化されうることを条件として、いかなる単細胞原核又は真核の生物、脊椎又は無脊椎生物、あるいはいかなる植物であってもよい。
【0046】
本発明のベクターにおいて有用なプロモーターは、当技術分野において周知のいくつかの方法のいずれかによって入手しうる。典型的には、ここで有用なプロモーターは、マッピングによって及び/又は制限エンドヌクレアーゼ消化によってあらかじめ同定され、それ故適切な制限エンドヌクレアーゼを使用して適切な組織ソースから単離されうる。一部の場合には、プロモーターの完全なヌクレオチド配列が既知であり得る。その場合は、核酸合成又はクローニングについて上述した方法を用いてプロモーターを合成しうる。
【0047】
プロモーター配列の全部又は一部だけが既知である場合は、PCRを用いて及び/又は適切なオリゴヌクレオチド及び/又は同じ又は別の種からの5’側隣接配列フラグメントでゲノムライブラリーをスクリーニングすることによって、完全なプロモーターを入手しうる。
【0048】
本発明を実施するための適切なプロモーターは、luxプロモーター、lacプロモーター、アラビノースプロモーター、trpプロモーター、tacプロモーター、tnaプロモーター、合成λプロモーター(バクテリオファージλから)、及びT5又はT7プロモーターなどの誘導的プロモーターである。好ましいプロモーターは、lux及びlacプロモーターを含む。
【0049】
複製起点エレメントは、典型的には、市販のものを購入するか又は使用者によって構築された原核生物発現ベクターの一部である。一部の場合には、ある一定コピー数へのベクターの増幅が対象タンパク質又はポリペプチドの最適発現のために重要でありうる。また別の場合には、不変のコピー数が好ましい。いずれの場合も、必要条件を満たす複製起点を有するベクターは当業者によって容易に選択されうる。選択ベクターが複製起点部位を含まない場合は、既知の配列に基づいて化学合成し、ベクターに連結しうる。
【0050】
転写終結エレメントは、典型的には融合タンパク質DNA構築物の末端の3’側に位置し、融合ポリペプチドをコードするRNAメッセージの転写を終結させる働きをする。通常は、原核細胞内の転写終結エレメントは、G−Cに富むフラグメントとそれに続くポリT配列である。該エレメントはライブラリーから容易にクローン化されるか又はベクターの一部として市販のものが購入されるが、上述したような核酸合成のための方法を用いて容易に合成することもできる。
【0051】
発現ベクターは、典型的には選択マーカーをコードする遺伝子を含む。この遺伝子は、選択培地において増殖する宿主細胞の生存と増殖のために必要なタンパク質をコードする。典型的な選択マーカー遺伝子は、(a)抗生物質又は他の毒素、例えば原核生物宿主細胞についてはアンピシリン、テトラサイクリン、クロラムフェニコール又はカナマイシンに対する耐性を付与する、(b)該細胞の栄養要求性欠損を補う、又は(c)複合培地からは得られない必須栄養素を供給するタンパク質をコードする。好ましい選択マーカーは、カナマシン耐性遺伝子、アンピシリン耐性遺伝子、クロラムフェニコール耐性遺伝子及びテトラサイクリン耐性遺伝子である。
【0052】
原核生物では一般にシャイン−ダルガーノ配列と呼ばれる、リボソーム結合エレメントは、mRNAの翻訳の開始のために必要である。このエレメントは、典型的にはプロモーターの3’側及び融合タンパク質DNA構築物のコード配列の5’側に位置する。シャイン−ダルガーノ配列は多様であるが、典型的にポリプリンである(すなわち高いA−G含量を有する)。多くのシャイン−ダルガーノ配列が同定されており、それらの各々が上述した方法を用いて容易に合成でき、原核生物ベクターにおいて使用される。
【0053】
上述したエレメントの1つ以上が、使用するベクター内に既に存在しない場合は、それらを個々に入手して、ベクターに連結しうる。各々のエレメントを入手するために使用する方法は当業者に周知であり、上述した方法(すなわちDNAの合成、ライブラリースクリーニング等)に匹敵する。
【0054】
各々のエレメントは、連結するエレメントの末端とベクターの末端が連結のために適合性であるように、ベクターを適切な制限エンドヌクレアーゼで切断することによって個別にベクターに連結しうる。一部の場合には、十分な連結を得るために、連結する末端を「平滑化する」必要がありうる。平滑化は、最初に4つのヌクレオチドすべての存在下にクレノウDNAポリメラーゼ又はT4 DNAポリメラーゼなどの酵素を使用して「粘着末端」を充填することによって実施できる。この手順は当技術分野において周知であり、例えばSambrookら、前出に述べられている。
【0055】
あるいは、ベクターに挿入する2以上のエレメントを最初に互いに連結して(それらを互いに隣接して位置付けようとする場合)、その後ベクターに連結しうる。
【0056】
ベクターを構築するためのもう1つの方法は、様々なエレメントのすべての連結を1つの反応混合物中で同時に実施することである。この場合、エレメントの不適切な連結又は挿入により多くのナンセンス又は非機能性ベクターを生じることがあるが、選択マーカーの発現によって機能性ベクターを同定しうる。制限エンドヌクレアーゼによる消化又はDNA配列決定によって連結産物の正しい配列を確認することができる。
【0057】
ベクターを構築し、融合タンパク質DNA構築物をベクターの適切な部位に挿入した後、完成したベクターを融合タンパク質発現のために適切な宿主細胞に挿入しうる。
【0058】
本発明に適する宿主細胞は細菌細胞である。例えば、大腸菌の様々な菌株(例えばHB101、JM109、DH5α、DH10及びMC1061)は、組換えポリペプチドの作製において使用するための周知の宿主細胞である。細菌株の選択は、典型的には菌株と使用する発現ベクターが適合性であるように行われる。枯草菌(B.subtilis)、シュードモナス種、他のバチルス種、ストレプトミセス種等の様々な菌株も、適切な発現ベクターと共に本発明の実施において使用しうる。
【0059】
選択宿主細胞へのベクターの挿入(「形質転換」又は「トランスフェクション」とも称される)は、リン酸カルシウム沈殿又は電気穿孔などの方法を用いて実施しうる。選択される方法は、一部には使用する宿主細胞の種類に依存する。これらの方法及び他の適切な方法は当業者に周知であり、例えばSambrookら、前出に述べられている。
【0060】
ベクターを含む宿主細胞(すなわち形質転換された又はトランスフェクトされた宿主細胞)は、当業者に周知の1以上の標準培地を使用して培養しうる。選択された培地は、典型的には宿主細胞の増殖と生存のために必要なすべての栄養素を含む。大腸菌細胞を培養するための適切な培地は、例えばルリア肉汁(「LB」)、YT肉汁、SOB、SOC及び/又はテリフィック肉汁(「TB」)である。
【0061】
TTR遺伝子、対象ペプチド又はタンパク質をコードする遺伝子及び場合により、それら2個の遺伝子の間に位置するリンカーペプチドをコードするDNA分子を含む融合タンパク質をコードするDNA構築物を作製するいくつかの方法が存在する。
【0062】
1つの手順では、TTR遺伝子と対象タンパク質をコードする遺伝子(「融合パートナー遺伝子」)をいずれかの方向で連結することができる(例えば構築物の5’又は3’末端でTTR遺伝子)。リンカーDNA分子を含めようとする場合は、最初にそれを融合パートナー遺伝子の1つに連結し、次にその構築物を他の融合パートナー遺伝子に連結することができる。連結は、典型的にはDNAリガーゼ酵素を製造者の指示に従って使用して実施される。
【0063】
別個の手順によって、選択されたベクターへの1つの融合パートナー遺伝子の最初の連結を行い、その後で他の融合パートナー遺伝子を、最初の融合パートナー遺伝子の3’側又は5’側のいずれかの位置でベクターに連結することができる。リンカーDNA分子を含めようとする場合は、その遺伝子をベクターに連結する前又は連結した後に、そのリンカーDNA分子をいずれかの融合パートナー遺伝子に連結しうる。
【0064】
本発明のTTR−TMPは、既存の巨核球/血小板欠損又は予想される巨核球/血小板欠損(例えば予定されている手術又は血小板提供のため)を含むものとして一般的に知られる状態を治療するために使用できる。そのような状態は、通常、インビボでの活性Mp1リガンドの欠損(一時的又は永続的)の結果である。血小板欠損についての一般的用語は血小板減少症(thrombocytopenia)であり、それ故本発明の方法及び組成物は一般に、それを必要とする患者において血小板減少症を治療するために使用しうる。血小板減少症(血小板欠損)は、化学療法及び様々な薬剤による他の療法、放射線治療、手術、偶発的血液損失及び他の同定疾患状態を含む、様々な理由のために存在しうる。
【0065】
血小板減少症を含み、本発明に従って治療しうる例示的な具体的な疾患状態は:再生不良性貧血、特発性血小板減少症、血小板減少症をもたらす転移性腫瘍、全身性エリテマトーデス、巨脾腫、ファンコーニ症候群、ビタミンB12欠損症、葉酸欠損症、メイ−ヘグリン異常、ヴィスコット−オールドリッチ症候群及び発作性夜間血色素尿症である。また、AIDSのためのある種の治療も血小板減少症を生じさせる(例えばAZT)。ある種の創傷治癒障害も、血小板数の上昇に起因すると考えられる。
【0066】
例えば将来の手術による、予想される血小板欠損に関しては、血小板が必要となる数日前から数時間前に本発明の化合物を投与することができる。急性状況、例えば偶発的な大量失血に関しては、本発明の化合物を血液又は精製血小板と共に投与することができる。
【0067】
本発明のTMP化合物はまた、そのような細胞がMp1受容体を発現することが認められる場合には、巨核球以外のある種の細胞型を刺激する上でも有用でありうる。Mp1リガンドによる刺激に対して応答性である、Mp1受容体を発現するそのような細胞に関連する状態も、本発明の範囲内である。
【0068】
本発明のTMP化合物は、血小板又は血小板前駆細胞の産生が所望される、又はc−Mp1受容体の刺激が所望されるいかなる状況においても使用しうる。それ故例えば、本発明の化合物は、血小板、巨核球等の必要性が存在する哺乳動物において何らかの状態を治療するために使用しうる。そのような状態は次の例示的ソース:国際公開公報第95/26746号、国際公開公報第95/21919号、国際公開公報第95/18858号、国際公開公報第95/21910号に詳細に記述されており、本明細書中で援用されている。
【0069】
本発明の化合物はまた、血小板及び/又は巨核球及び関連細胞の生存性又は保存寿命を維持する上でも有用でありうる。従って、そのような細胞を含む組成物中に1以上のそのような化合物の有効量を含めることは有用であり得る。
【0070】
本発明の治療方法、組成物及び化合物はまた、血小板欠損のほかに他の症状によって特徴付けられる疾患状態の治療においても、単独であるいは他のサイトカイン、可溶性Mp1受容体、造血因子、インターロイキン、増殖因子又は抗体と組み合わせて使用しうる。本発明の化合物は、IL−3又はGM−CSFなどの一般的な造血刺激物質と組み合わせて、一部の形態の血小板減少症を治療する上で有用であることが明らかになると予想される。他の巨核球刺激因子、すなわちmeg−CSF、幹細胞因子(SCF)、白血病阻害剤(LIF)、オンコスタチンM(OSM)又は巨核球刺激作用を有する他の分子も、Mp1リガンドと共に使用しうる。そのような同時投与のためのさらなる例示的サイトカイン又は造血因子は、IL−1α、IL−1β、IL−2、IL−3、IL−4、IL−5、IL−6、IL−11、コロニー刺激因子−1(CSF−1)、SCF、GM−CSF、顆粒球コロニー刺激因子(G−CSF)、EPO、インターフェロン−α(IFN−α)、コンセンサスインターフェロン、IFN−β又はIFN−γを含む。さらに、ひとたび巨核球が成熟形態に達すれば、巨核球を血小板へと細分する作用を有すると思われる、可溶性の哺乳動物Mp1受容体の有効量を同時に又は連続的に投与することは有用であると考えられる。それ故、本発明の化合物の投与(成熟巨核球の数を高めるため)とそれに続く可溶性Mp1受容体の投与(リガンドを不活性化して成熟巨核球が血小板を産生することを可能にするため)は、血小板産生を刺激する特に有効な手段であると予想される。適切な用量は、治療組成物中のそのような付加的成分を相殺するように調整する。治療される患者の経過は従来の方法によってモニターすることができる。
【0071】
2型糖尿病患者としても知られる、インスリン非依存性糖尿病(NIDDM)においては、グルカゴン様ペプチド−1(GLP−1)の投与は抗糖尿病特性を有する。しかし、GLP−1は、インビボでのその放出後にジペプチジルペプチダーゼIV(DPPIV)によって速やかに分解される。それ故、GLP−1ペプチド又はその変異体を本発明のTTRポリペプチドに融合してGLP−1を安定化し、インビボでその半減期を上昇させうることは本発明の利点である。従って、本発明のもう1つの実施態様では、ここで述べるTTR−GLP1融合タンパク質を、II型糖尿病としても知られるインスリン非依存性糖尿病(NIDDM)を含むことが一般的に知られる状態を治療するために使用することができる。
【0072】
当業者は、GLP−1ペプチドの配列は、そのインスリン向性作用を保持するように変化させうることを認識する。当技術分野で既知のそのような変異型の同定例は、例えば、GLP−1(7−34)、(7−35)、(7−36)又は(7−37)、Gln9−GLP−1(7−37)、D−Gln9−GLP−1(7−37)、Thr16−Lys18−GLP−1(7−37)及びLys18−GLP−1(7−37)を含む。GLP−1変異体のさらなる例は、本明細書中に参考として援用されている、米国特許第5,118,666号、同第5,545,618号、同第5,977,071号及び国際公開公報第02/46227号及びAdelhorstら、J.Biol.Chem.269:6275(1994)に述べられている。従って、いかなるGLP−1ペプチドも、該GLP−1融合タンパク質がそのコグネイト受容体を通してシグナルに結合し、シグナルを誘導することができる限り、本発明の融合タンパク質を生成するために使用できる。受容体結合及び活性化は標準的アッセイによって測定できる(米国特許第5,120,712号)。
【0073】
患者の血中グルコースを正常化するために有効な融合タンパク質の用量は、下記でより詳細に論じるように、被験者の体重、年齢、血中グルコースを調節する能力欠如の重症度、融合タンパク質の投与経路、バイオアベイラビリティー、薬物動態プロフィール及び製剤を含む多くの因子に依存する。
【0074】
本発明の治療方法、組成物及び化合物はまた、単独で、あるいはインスリン、DPPIV阻害剤等を含むがこれらに限定されない他の糖尿病治療と組み合わせて使用しうる。GLP−1融合タンパク質の用量は、治療組成物中のそのような付加的成分を相殺するように調整する。治療される患者の経過は、例えば血中グルコースレベルのモニターなどの従来の方法によってモニターすることができる。
【0075】
本発明はまた、本発明の化合物の医薬組成物を提供する。そのような医薬組成物は、注入投与用であるか、あるいは経口、経鼻、経皮あるいは、例えば静脈内、皮内、筋肉内、乳房内、腹腔内、髄腔内、眼内、眼球後、肺内(例えばエーロゾル投与薬剤)又は皮下注射(長期放出のためのデポー投与を含む)による投与;舌下、肛門、膣による投与、あるいは例えば脾臓被膜下、脳又は角膜内に包埋する外科的移植による投与を含む他の形態の投与用でありうる。治療は、単回用量又は一定期間にわたる多回用量で構成されうる。一般に、医薬適合性の希釈剤、防腐剤、安定剤、可溶化剤、乳化剤、アジュバント及び/又は担体と共に本発明の化合物の有効量を含有する医薬組成物は、本発明に包含される。そのような組成物は、様々な緩衝剤含量、pH及びイオン強度の希釈剤(例えばTris−HCl、酢酸塩、リン酸塩、クエン酸塩等);界面活性剤及び可溶化剤(例えばツイーン80、ポリソルベート80等)、抗酸化剤(例えばアルコルビン酸、メタ重亜硫酸ナトリウム)、防腐剤(例えばチメロサール、ベンジルアルコール)及び充填物質(例えばラクトース、マンニトール)などの添加物;ポリ乳酸、ポリグリコール酸等のような高分子化合物の微粒子製剤又はリポソームへの物質の組込みを含む。ヒアルロン酸も使用でき、これは循環中の存在期間の持続を促進する作用を及ぼしうる。該医薬組成物は、場合により、モノラウリン酸ポリオキシエチレンソルビタン、ステアリン酸マグネシウム、メチル及びプロピルヒドロキシベンゾエート、デンプン、スクロース、デキストロース、アカシアゴム、リン酸カルシウム、鉱物油、ココアバター及びカカオ脂を含むがこれらに限定されない、製薬ビヒクル、賦形剤又は媒質として働くさらなる他の医薬適合性の液体、半固体又は固体希釈剤を含有しうる。そのような組成物は、本発明のタンパク質及び誘導体の物理的状態、安定性、インビボでの放出速度及びインビボでのクリアランス速度に影響しうる。例えば、本明細書中に参考として援用されている、Remington’s Pharmaceutical Sciences,第18版(1990,Mack Publishing Co.,Easton,PA 18042)p.1435−1712参照。該組成物は、液体形態で製造されるか、又は凍結乾燥形態のような乾燥粉末でありうる。経皮的製剤と同様に、移植可能な持続放出性製剤も考慮される。
【0076】
制御放出製剤は望ましいと考えられる。拡散又は浸出機序による放出を可能にする不活性マトリックス、例えばゴムに薬剤を組み込むことができる。緩やかに変性するマトリックス、例えばアルギン酸塩、多糖類も製剤に組み込みうる。この治療薬の制御放出のもう1つの形態は、Oros治療システム(Alza Corp.)に基づく方法による、すなわち水が入るのを許容し、浸透作用によって単一の小さな開口部を通して薬剤を押し出すことができる半透性膜に薬剤を入れる。一部の腸溶剤皮も遅延放出作用を有する。
【0077】
またここでは、本発明のタンパク質(又はその誘導体)の肺への送達も考慮される。該タンパク質(又は誘導体)は、吸入される間に哺乳類の肺へと送達され、肺上皮内層を横切って血流に入る。(これについての他の報告は、Adjeiら、Pharmaceutical Research 7:565−569(1990);Adjeiら、International Journal of Pharmaceutics 63:135−144(1990)(酢酸ロイプロリド);Braquetら、Journal of Cardiovascular Pharmacology 13(補遺5):s.143−146(1989)(エンドセリン−1);Hubbardら、Annals of Internal Medicine 3:206−212(1989)(1−アンチトリプシン);Smithら、J.Clin.Invest.84:1145−1146(1989)(1−プロテイナーゼ);Osweinら、「Aerosolization of Proteins」,Proceedings of Symposium on Respiratory Drug Delivery II,Keystone,Colorado,1990年3月(組換えヒト成長ホルモン);Debsら、The Journal of Immunology 140:3482−3488(1988)(インターフェロン及び腫瘍壊死因子)及びPlatzら、米国特許第5,284,656号(顆粒球コロニー刺激因子)を含む)。
【0078】
それらのすべてが当業者に周知である、ネブライザ、定量噴霧式吸入器及びパウダー式吸入器を含むがこれらに限定されない、治療製品の肺送達用に設計された広範囲の機械的装置が、本発明の実施における使用のために考慮される。
【0079】
本発明の実施に適する市販装置の一部の同定例は、Mallinckrodt,Inc.,St.Louis,Missouriによって製造される、Ultraventネブライザ;Marquest Medical Products,Englewood,Coloradoによって製造される、Acorn IIネブライザ;Glaxo Inc.,Research Triangle Park,North Carolinaによって製造される、Ventolin定量噴霧式吸入器;及びFisons Corp.,Bedford,Massachusettsによって製造される、Spinhalerパウダー式吸入器である。
【0080】
そのような装置はすべて、本発明の化合物を配分するのに適した製剤の使用を必要とする。典型的には、各々の製剤は使用する装置の種類に特異的であり、治療において有用な希釈剤、アジュバント及び/又は担体に加えて、適切な推進剤の使用を含みうる。
【0081】
本発明の化合物は、最も好都合には、遠位肺への最も有効な送達のために10μm(又はミクロン)未満、最も好ましくは0.5から5μmの平均粒径を有する微粒子形態で製造すべきである。
【0082】
担体は、トレハロース、マンニトール、キシリトール、スクロース、ラクトース及びソルビトールなどの炭水化物を含む。製剤中での使用のための他の成分は、DPPC、DOPE、DSPC及びDOPCを含みうる。天然又は合成界面活性剤を使用してもよい。ポリエチレングリコールを使用してもよい(該タンパク質又は類似体を誘導体化する上でのその使用は別として)。シクロデキストランなどのデキストランを使用してもよい。胆汁酸塩及び他の関連エンハンサーが使用しうる。セルロース及びセルロース誘導体も使用しうる。緩衝製剤における使用のような、アミノ酸も使用しうる。
【0083】
上述した状態を治療するための方法に関わる投与レジメは、薬剤の作用を変化させる様々な因子、例えば患者の年齢、状態、体重、性別及び食事、感染の重症度、投与時間及び他の臨床的因子を考慮して、主治医によって決定される。一般に、用量は、1日1回投与又は等しい用量をより長い又は短い間隔で、例えば1日おき、週に2回、週に1回、あるいは1日2回又は3回投与される、1日当り体重1キログラムにつき本発明の化合物0.1μgから100mg、好ましくは0.1から1000μg/kg、より好ましくは0.1から150μg/kgの範囲内とすべきである。
【0084】
本発明の化合物は、初回ボーラス及びそれに続く、薬剤製品の治療循環レベルを維持するための持続注入によって投与しうる。もう1つの例として、本発明の化合物は単回用量として投与しうる。当業者は、良質の医療のための原則及び個々の患者の臨床状態によって決定される、有効な用量及び投与レジメを容易に最適化する。投与頻度は、薬剤の薬物動態パラメータ及び投与経路に依存する。最適医薬製剤は、投与経路と所望用量に依存して当業者によって決定される。例えば、その開示が本明細書中に参考として援用されている、Remington’s Pharmaceutical Sciences,第18版(1990,Mack Publishing Co.,Easton,PA 18042)p.1435−1712参照。投与経路に依存して、体重、体表面積又は器官の大きさに従って適切な用量を算定しうる。
【0085】
適切な投与量は、適切な用量−反応データと共に、血清中レベルを測定するための確立されたアッセイの使用を通して確認しうる。最終的な投与レジメは、薬剤の作用を変化させる様々な因子、例えば薬剤の特異的活性、損傷の重症度及び患者の応答性、患者の年齢、状態、体重、性別及び食事、感染の重症度、投与時間及び他の臨床的因子を考慮して、主治医によって決定される。試験が実施されると共に、様々な疾患及び状態についての適切な用量レベル及び治療期間に関するさらなる情報がもたらされる。
【0086】
下記の実施例は説明だけを目的とするものであり、いかなる意味においても本発明を限定すると解釈されるべきではない。
【実施例1】
【0087】
この実施例は、天然組換えヒトトランスサイレチン(TTR)及び次のTTR変異体:TTR(C10A)、TTR(C10A/A37C)、TTR(C10A/D38C)、TTR(C10A/A81C)、TTR(C10A/G83C)、及びTTR(C10A/K15A/G83C)についてのDNAの作製を述べる。
【0088】
発現プラスミドpAMG21は、1996年7月24日に寄託された、受入番号98113の下にATCCより入手可能である(pAMG21の説明については1997年7月3日公開のPCT国際公開公報第97/23614号参照)。TTR、TTR変異体又はTTR−ペプチド融合物をコードするDNA配列を、pAMG21内のLuxPRプロモーターの制御下に置いた。
【0089】
細菌宿主GM221は、初期ebg領域内の温度感受性λリプレッサーcI857s7及び後期ebg領域(68分)内のlacIQリプレッサーの両方を含むように改変された大腸菌K−12株である。これら2個のリプレッサー遺伝子の存在は、この宿主を多様な発現系と共に使用することを可能にするが、これらのリプレッサーは共にluxPRからの発現には無関係である。非形質転換宿主は抗生物質耐性を持たない。cI857s7遺伝子のリボソーム結合部位を、強化(enhanced)RBSを含むように修飾した。それを、介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド1170位と1411位の間のebgオペロンに挿入した。この構築物を、F’tet/393へのMMebg−cI857s7強化RBS No.4と呼ばれる組換えファージを使用して染色体に送達した。組換えと分離(resolution)後、上述した染色体挿入物だけが細胞内に残る。これを新たにF’tet/GM101と命名した。次に介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド2493位と2937位の間のebgオペロンにlacIQ構築物を送達することによってF’tet/GM101を修飾した。その構築物を、F’tet/GM101へのAGebg−lacIQ No.5と呼ばれる組換えファージを使用して染色体に送達した。組換えと分離後、上述した染色体挿入物だけが細胞内に残る。これを新たにF’tet/GM221と命名した。F’tetエピソームを、LB中25μg/mlの濃度のアクリジンオレンジを使用して該菌株からキュアリングした。キュアリングした菌株をテトラサイクリン感受性と同定し、GM221として保存した。
【0090】
オリゴヌクレオチド(各々1.0nm)をホスホルアミダイト法によって合成した。ヌクレオチドを、一部の場合は、大腸菌における最適発現のために変性させた。これらのコドン変化はアミノ酸配列の変化を生じさせなかった。この実施例において使用した各々のオリゴヌクレオチドを表1に列記する。
【0091】
Expand Long Polymeraseを製造者(Boehringer Mannheim)のプロトコールに従って使用してPCRを実施した。PCR産物をアガロースゲル電気泳動によって確認し、精製して、NdeI及びXhoI(New England Biolabs)で消化した。発現ベクターpAMG21を同様に消化して、その後で子ウシ腸ホスファターゼ(Boehringer Mannheim)で処理した。ベクターと挿入物をアガロースゲルから精製し、その後で混合して、T4 DNAリガーゼ(New England Biolabs)によって連結した。連結は4℃で2時間実施した。各々の連結混合物を、Biorad GenePulser(Biorad Laboratories)をギャップ長約2mmのキュベット中2.5V、25μFD及び200オームで使用して、電気穿孔によって上述した宿主菌株GM221に形質転換した。電気穿孔後、ルリア肉汁(LB)1ml中37℃で約1時間、静かに振とうしながら細胞を回収した。形質転換混合物の全体を、50μg/mlカナマイシンを含むLB寒天上にプレートした。隣接ベクター配列に対するオリゴヌクレオチドを使用したPCRにより、所望される分子量の存在に関してコロニーをスクリーニングした。そのPCR産物をアガロースゲル電気泳動によって評価した。陽性クローンを、組換えタンパク質産物を生産する能力に関してさらにスクリーニングし、ヌクレオチド配列決定によって最終的に確認した。
【0092】
TTRのDNA及びアミノ酸配列は既知である(Mita,S.ら、Biochem.Biophys.Res.Commun.,124(2):558−564[1984])。これらの配列は受入番号K02091としてGenbankに寄託されている。シグナルペプチドを除く天然TTRのcDNAを、ヒト肝から誘導したcDNAライブラリー(Clontech)からクローン化した。詳細には、TTRの5’末端の8個のコドンをコードするオリゴヌクレオチド(オリゴ2693−79)及び終止コドンを含むTTR3’末端の7個のコドンをコードするオリゴヌクレオチド(オリゴ2693−80)を合成し、ヒト肝cDNAライブラリーを鋳型として使用してExpand Longポリメラーゼで完全長成熟TTRを増幅するために使用した。生じたPCRフラグメントをNdeI及びXhoIで消化し、ゲル精製して、NdeI/XhoI制限発現ベクターpAMG21と連結した。4℃で2時間後、連結混合物を電気穿孔によってGM221細胞に導入した。単一コロニーを採取し、プラスミドDNAを作製して配列決定した。1個の生じたプラスミド(菌株No.5316)は、天然TTRの正しいDNA配列(プラスN末端のメチオニン)を有することを示し、それを発現のために使用した。このDNA配列を配列番号2に同定する。
【0093】
上記オリゴヌクレオチド2693−80とオリゴヌクレオチド2820−88を使用することによって突然変異体TTR(C10A)を作製した(10番目の位置のコドンCysがAlaに置換された天然TTRの最初の11個のコドンを含む)。PCR手順及び発現菌株を選択するための工程は上述したのと同様であった。生じた菌株(菌株No.5619)は、配列番号3に同定するDNA配列を有していた。
【0094】
プラスミド5619を、次の位置:A37、D38、A81及びG83のアミノ酸をシステインのアミノ酸で置換することによってさらに修飾した。下記で述べるように、所望突然変異を内包する相補的オリゴヌクレオチドの各々の対を、部位特異的突然変異誘発のために設計された標準的な2段階PCR手順において、上述したTTR5’及び3’プライマーと共に使用した。菌株5619由来のプラスミドを鋳型として使用する20サイクルのPCRにおいて、フォワードプライマーの各々をTTR3’プライマーと共に使用し、リバースプライマーの各々をTTR5’プライマーと共に使用した。生じたPCR増幅5’及び3’フラグメントを混合し、完全長突然変異体を生成するための第二段階PCRのための鋳型として使用した。その後のクローニング及び配列決定手順は既に述べたものと同様であった。次のオリゴヌクレオチドを使用した:TTR(A37C)フォワード(オリゴ2823−91);TTR(A37C)リバース(オリゴ2823−92);TTR(D38C)フォワード(オリゴ2823−93);TTR(D38C)リバース(オリゴ2823−94);TTR(A81C)フォワード(オリゴ2823−95);TTR(A81C)リバース(オリゴ2823−96);TTR(G83C)フォワード(オリゴ2823−97);TTR(G83C)リバース(オリゴ2823−98)。該プラスミドを含む生じた大腸菌株は次のように説明される:TTR(C10A/A37C)(菌株5641)は配列番号4に同定するDNA配列を有していた。TTR(C10A/D38C)(菌株5642)は配列番号5に同定するDNA配列を有していた。TTR(C10A/A81C)(菌株5643)は配列番号6に同定するDNA配列を有していた。TTR(C10A/G83C)(菌株5651)は配列番号7に同定するDNA配列を有していた。
【0095】
菌株5651の15位のLysを、菌株5641、5642、5643及び5651について述べたのと同様の手順により、オリゴヌクレオチド2953−67及び2953−68を使用してさらにAlaに突然変異誘発した。生じた菌株、TTR(C10A/K15A/G83C)(菌株5895)は配列番号8に同定するDNA配列を有していた。
【0096】
【表1】
【実施例2】
【0097】
この実施例は、様々なTMP−TTR融合物の作製を述べる。TTRとTMPを含むいくつかの融合タンパク質を作製した。この実施例において使用した各々のオリゴヌクレオチドを表2に列記する。
【0098】
TMPを含むフラグメントを、最初に、TMPの最初の7個のコドンに加えてNdeI部位を含む12ヌクレオチドの5’延長部をコードするオリゴヌクレオチド2743−96、及び天然TTRの最初の7個のコドンと対象TMPの最後の7個のコドンをコードするオリゴヌクレオチド2743−97を使用して、完全長TMP−Fc融合物をコードするプラスミドを内包する菌株から増幅した(PCT国際公開公報第00/24770号参照)。生じたPCRフラグメントを菌株5619由来のプラスミドと混合し、その混合物をオリゴヌクレオチドプライマー2743−96及び2693−80のための鋳型として使用して、完全長TMP−TTRを増幅した。クローニング及び発現については上述したのと同様の手順を用いた。生じた菌株、TTR−TMP(菌株5513)は配列番号9に同定するDNA配列を有していた。
【0099】
次に、TMPをそれぞれ菌株5641、5642、5643及び5651のN末端に導入した。プラスミド5513をXbaIで消化し、生じたTMP及びTTR(C10A)の最初の18個のコドンを含むXbaI/XbaI挿入物をゲル精製し、5641、5642、5643及び5651由来の、XbaIで切断し、ホスファターゼ処理してゲル精製したベクターと連結した。各々の融合物について正しい方向を選択するためにDNA塩基配列決定を実施した。該プラスミドを含む生じた大腸菌株は次のように説明される:TMP−TTR(C10A/A37C)(菌株5704)は配列番号10に同定するDNA配列を有していた。TMP−TTR(C10A/D38C)(菌株5705)は配列番号11に同定するDNA配列を有していた。TMP−TTR(C10A/A81C)(菌株5706)は配列番号12に同定するDNA配列を有していた。TMP−TTR(C10A/G83C)(菌株5707)は配列番号13に同定するDNA配列を有していた。
【0100】
【表2】
【実施例3】
【0101】
この実施例は、PTH(1−34)−TTR(C10A/K15A/G83C)融合物の作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表3に列記する。
【0102】
2個の新しいオリゴヌクレオチド、ヒトPTHの最初の7個のコドンをコードするオリゴヌクレオチド2694−01、及びTTRの最初の7個のコドンとPTHのアミノ酸28−34をコードするオリゴヌクレオチド2694−03を合成して、融合物を作製した。オリゴヌクレオチド2694−01及び2694−03を上述したように20サイクルのPCR手順において使用して、TTRリンカーを有するPTH(1−34)を増幅した。この反応のための鋳型は、PTH1−34−Fc融合物をコードするプラスミドを内包する菌株であった(PCT国際公開公報第01/81415号参照)。生じたPCR混合物を菌株5895と混合して、プライマー2694−01及び2693−80を用いて完全長PTH(1−34)−TTR(C10A/K15A/G83C)を増幅するための鋳型として使用した。配列確認後、この新しいプラスミドを含む生じた発現菌株をPTH−TTR(C10A/K15A/G83C)(菌株5920)と称した。この菌株は配列番号14に同定するDNA配列を有していた。
【0103】
【表3】
【実施例4】
【0104】
この実施例は、IL−1ra−TTR(C10A)融合物及びTTR(C10A)−GSGS−IL−1ra融合物の作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表4に列記する。
【0105】
IL−1ra−TTR(C10A)融合物を作製するために、2個のオリゴヌクレオチド、ヒトタンパク質IL−1raの最初の7個のコドンをコードするオリゴヌクレオチド2823−13、及びIL−1raの最後の7個のアミノ酸とTTRの最初の7個のアミノ酸をコードするオリゴヌクレオチド2823−14を合成した。IL−1raを発現する菌株由来のプラスミド(PCT国際公開公報第91/08285号参照)を、オリゴヌクレオチド2823−13及び2823−14を用いて増幅した。生じたPCR産物を、菌株5619から精製したプラスミドと混合し、オリゴヌクレオチドプライマー2823−13及び2693−80を用いて完全長IL−1−ra−TTR(C10A)を増幅するための鋳型として使用した。PCR産物を上述したようにクローン化し、配列決定して、発現させた。この新しいプラスミドを含む生じた菌株をIL−1ra−TTR(C10A)(菌株5644)と称した。この菌株は配列番号15に同定するDNA配列を有していた。
【0106】
TTR(C10A)−IL−1raを作製するために、次の2個のオリゴヌクレオチド、すなわち、その間にGSGSリンカーを導入した、TTRの最後の7個のアミノ酸とIL−1−raの最初の7個のアミノ酸をコードするオリゴヌクレオチド2787−32及びIL−1−raの最後の7個のコドンをコードするオリゴヌクレオチド2787−33を合成した。これら2個のオリゴヌクレオチドプライマーを用いてプラスミド2693を増幅し、生じたPCR産物をプラスミド5619と混合して、これらを共に、プライマー2787−33及び2693−79を用いて完全長TTR(C10A)−IL−1raを増幅するための鋳型として使用した。PCR産物を上述したようにクローン化し、配列決定して、発現させた。この新しいプラスミドを含む生じた菌株をTTR(C10A)−IL−1ra(菌株5645)と称した。この菌株は配列番号16に同定するDNA配列を有していた。
【0107】
【表4】
【実施例5】
【0108】
この実施例は、TTR(C10A/G83C)−ブラジキニンの作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表5に列記する。
【0109】
菌株5651から精製したプラスミドを、オリゴヌクレオチドプライマー2693−79及びPstI制限部位を含むTTR3’プライマーであるオリゴヌクレオチドプライマー2943−47によるPCRのために使用した。このPCR産物をゲル精製し、NdeI及びPstIで制限消化した。生じたDNAフラグメントを、NdeI及びXhoIで消化したAMG21、及びGSGSGリンカーをコードするアニーリングしたオリゴヌクレオチド2943−48、及びPstI5’とXhoI3’重複末端を有するブラジキニンアンタゴニストペプチドKRPPGFSPLをコードするオリゴヌクレオチド2943−49を含む連結混合物中で使用した。GM121をこの連結産物で形質転換し、DNAをカナマイシン耐性コロニーから精製した。次に該耐性コロニーにおいてDNA配列を確認した。確認した菌株を30℃で増殖させ、下記に述べる10リットル発酵物において発現を誘導した。その新しい菌株をTTR(C10A/G83C)−ブラジキニン(菌株5914)と称した。この菌株は配列番号17に同定するDNA配列を有していた。
【0110】
【表5】
【実施例6】
【0111】
この実施例は、大腸菌におけるTTR及びTTR融合構築物の組換え発現を述べる。新たに構築したTTR又はTTR融合物の各々を、最初に16℃から37℃の範囲の温度で可溶性発現に関して検査した。この目的のために、TTR又はTTR融合物の各々を発現するGM221の培養物(25ml)を、50μg/mlカナマイシンを補足したLB肉汁中37℃で、600nmでの光学密度(OD)が0.5から1.0に達するまで増殖させた。次に培養物を、それぞれ16℃、20℃、25℃、30℃、34℃及び37℃の温度設定で振とう機に入れた。合成自己誘導物質N−(3−オキソヘキサノイル)−DL−ホモセリンラクトンを最終濃度20ng/mlまで培地に添加した後、luxPRプロモーターからの遺伝子産物発現の誘導を実施した。6時間後、細菌培養物を封入体の存在に関して顕微鏡で検査した。TTR及びその融合物のために培養物を30℃未満の温度で増殖させることにより、しばしば可溶性又は部分的可溶性の発現を達成することができ、この温度を大量発現のために使用した。可溶性発現が達成できなかった場合は、発現のレベルが最も高かった温度を大量振とう機又は発酵槽のために使用した。
【0112】
大量発現は、通常は4リットルフラスコで実施した。LB1リットルを含む4〜8台の4リットル振とう機にTTR又はその融合株の一晩培養物を接種した。発現は基本的に上述したように実施した。細胞を遠心分離によって収集した。
【0113】
無菌手法を用いる発酵段階は、無菌ルリア肉汁500mLを含む振とうフラスコにおいて生産した菌株の種培養(seed culture)からの接種で開始する。この培養物が適切な細胞密度(600nmで0.8−2)に達したとき、その内容物を使用して、複合ベースの増殖培地10リットルを含む20リットル発酵槽に接種した。発酵槽を30℃、pH7に維持し、溶解酸素レベルを30%飽和に保持する。細胞密度が600nmで10−12OD単位の光学密度に達したとき、N−(3−オキソヘキサノイル)ホモセリンラクトンの添加によって培養物を誘導した。誘導後6時間目に遠心分離によって発酵槽から細胞を収集した。
【実施例7】
【0114】
この実施例は、TTR(C10A/G83C)−ブラジキニンの精製を述べる。−80℃で保存したクローン5914からの大腸菌ペースト約193gを、50mMトリスHCl、5mM EDTA、pH8.0 1447ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、該懸濁液を12,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物(図1、レーン2)を11,325×g、4℃で50分間遠心分離した。上清を可溶性分画として採取した。該可溶性分画をポリプロピレンボトル中65℃水浴で30分間加熱し、その時点で内容物の温度は63℃であった。該可溶性分画を11,325×g、4℃で50分間遠心分離した。上清を熱可溶物(Heat Soluble)として採取した(図1、レーン3)。該熱可溶性分画を、2つのプレフィルターを備えた0.45μm酢酸セルロースフィルターでろ過し、室温(約23℃)で、Q緩衝液A(20mMトリスHCl、2.5mM EDTA、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(Q−sepharose fast flow)(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に20ml/分で負荷した。カラムを20ml/分のQ緩衝液A約2300mlで洗った。Qカラムを、60%Q緩衝液B(20mMトリスHCl、1M NaCl、2.5mM EDTA、pH8.0)への15カラム容積の直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(1150ml)(図1、レーン4)に貯留し、DTT1.77gを加えた。そのQプールを室温(約23℃)で30分間静かに攪拌した。該Qプールに、3.8M硫酸アンモニウムpH7.0 410mlを緩やかに加え、1M HClの緩やかな添加によってpHを約7.5から7.0に低下させた。次に該Qプールの約2分の1を、10ml/分でP緩衝液A(50mM NaH2PO4、1M硫酸アンモニウム、pH7.0)中84mlフェニルセファロース高速カラム(内径2.6cm)(Amersham Pharmacia Biotech)に負荷した。カラムをP緩衝液A約170mlで洗い、次いで50%、80%及び100%P緩衝液B(50mM NaH2PO4、pH7.0)を用いて3段階溶出させた。次に、該Qプールの残りの半分を最初の半分と同じプロトコールを用いて処理した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Pプール(260ml)(図1、レーン5)に貯留し、そのPプールを、直径20.4mmの8kDaカットオフ透析チューブ(Spectrum Laboratories Inc.,Rancho Dominguez,CA)を使用して室温(約23℃)で2時間、HA緩衝液A(10mM NaH2PO4、pH7.0)4Lに透析した。透析緩衝液を新鮮なHA緩衝液A4Lに交換し、さらに約15時間透析を継続した。該Pプールを透析から取り出し、1M DTT 600μlを加え、その後室温(約23℃)で約1時間インキュベートした。Pプールを、HA緩衝液A中10ml/分で105ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを10ml/分のHA緩衝液A約210mlで洗い、次いで12.5%、25%、50%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の4段階で溶出させた。そのフロースルーをHAプール(340ml)(図1、レーン6)として貯留し、DTT 524mgを加えて、その後室温(約23℃)で1時間インキュベートした。
【0115】
該HAプールの約2分の1を、10ml/分で47mlソース15Qカラム(内径2.6cm)(Amersham Pharmacia Biotech)に負荷し、次にQ−緩衝液A約250mlで洗った。10%から50%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。該HAプールの残りの半分を最初の半分と同じプロトコールを用いて処理した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Q2プール(260ml)に貯留し、10kDa膜を有する攪拌式セルを用いて約75mlに濃縮した。次にQ2プール(図1、レーン7)を0.22μm酢酸セルロースフィルターでろ過し、算定比吸光係数18,450 M−1 cm−1を用いてタンパク質濃度を16.9mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.52と測定されたので、無視しうると判定した。
【実施例8】
【0116】
この実施例は、TTR(C10A/G83C)のC末端又はN末端のいずれかにペプチドを融合することがそのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。20mMトリスpH8.0及び約250mM NaCl中のTTR(C10A/G83C)、PTH−TTR(C10A/K15A/G83C)及びTTR(C10A/G83C)−ブラジキニンを室温(約23℃)で約1時間、9mM DTTで還元した。還元したTTR約50μgを、1ml/分でSEC緩衝液(50mM NaH2PO4、500mM NaCl、pH6.7)中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex,Torrance,CA)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図2からわかるように、TTR(C10A/G83C)は、49kDaの分子サイズに相当する約8.8分で溶出し、これは55kDaの四量体の算定分子量に匹敵する。PTH−TTR(C10A/K15A/G83C)は、67kDaの分子サイズに相当する約8.6分で溶出し、これは該四量体について算定される71kDaに近い。TTR(C10A/G83C)−ブラジキニンは、57kDaの分子サイズに相当する約8.7分で溶出し、これも該四量体について算定される60kDaに近い。
【実施例9】
【0117】
この実施例は、ジスルフィド結合を含むタンパク質をTTR(C10A)のC末端又はN末端のいずれかに融合することがそのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。TTR(C10A)、IL−1−ra−TTR(C10A)及びTTR(C10A)−IL−1−raの各々約50μgを、1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図3からわかるように、TTR(C10A)は、55kDaの四量体の算定分子量に匹敵する49kDaの分子サイズに相当する、約8.8分で溶出する。IL−1−ra−TTR(C10A)融合物は、188kDaの分子サイズに相当する約7.9分で溶出し、これは124kDaという該四量体についての予想分子量よりも著しく大きい。同様に、TTR(C10A)−IL−1−raは、該四量体について予想される124kDaと比較して、やはり188kDaの分子サイズに相当する約7.9分で溶出した。サイズ排除クロマトグラフィーは形状依存的であり、標準物質を球状タンパク質についてカリブレートしているので、これらの大きさのずれはおそらく分子の形状の相違によるものである。
【実施例10】
【0118】
この実施例は、ヒト免疫グロブリンFc(Fc−TMP)のカルボキシ末端に融合したTMP配列とTMP(m)−TTRの、可溶性ヒト骨髄増殖性白血病(MPL)受容体への結合を比較する。加えて、この実施例は、天然TTRシステインのペグ化がTMP融合物のMPL受容体への結合に及ぼす作用を示す。ペグ化TTR融合物の作製は実施例13において詳細に述べる。
【0119】
この実施例のために、ヒトMPL受容体を、EDC/NHS化学を製造者(BIAcore,Uppsula,Sweden)の指示に従って使用して、RL=1300RUでBIAcore CM5チップに共有結合した。すべての試料を、0.1mg/mlウシ血清アルブミン及び0.005%P20(ポリオキシエチレンソルビタン)を含むダルベッコのPBS(Gibco BRL,Gaithersburg,MD)中50μl/分で該チップに通した。平衡エンドポイントは注入後3分とした。図4からわかるように、Fc−TMPはTMP(m)−TTRと比較してより優れた結合特性を示す。さらに、この図は、天然TTRシステイン(Cys10)のペグ化がTMPのMPL受容体への結合を妨げることを明らかにする。TMP(m)−TTR−PEG5Kの結合は、その非ペグ化対応物と比較して有意に低い結合応答を示し、TMP(m)−TTR−PEG20Kはさらに一層強い阻害を示した。これは、システイン10上のPEGの存在が、おそらく融合TMPからMPL受容体の結合に関して立体障害を引き起こし、より大きなPEGはより大きな障害を生じさせることを示唆する。
【実施例11】
【0120】
この実施例は、マウスへのTMP(m)−TTRの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために、50匹のBDF1マウス(Charles River Laboratories,Wilmington,Massachusetts)を5群に分け、希釈剤(0.1%ウシ血清アルブミンを含むダルベッコのPBS)又は希釈剤と供試タンパク質50μg/kg動物のいずれかを皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、11日目、12日目、14日目及び17日目)採血した(140μl)。採血の前にマウスをイソフルランで麻酔した。
【0121】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図5からわかるように、Fc−TMPは、5日目に4.3×1012血小板 L−1の血小板数ピークに達する最大応答を示し、これは基線値である1.2×1012血小板 L−1の3.4倍以上である。TMP(m)−TTR−PEG5Kは、基線値の2倍を少しだけ下回る2.3×1012血小板 L−1でピークに達する中等度の応答を示した。非ペグ化形態のTMP(m)−TTRは、基線値を20%だけ上回る1.5×1012血小板 L−1という極めて低い応答を示す。非ペグ化形態のTMP(m)−TTRは、インビトロではそのペグ化対応物よりも良好な結合を示すが(図4)、インビボではTMP(m)−TTR−PEG5Kと比較して低い成績である。これは、TTR構築物の生物学的半減期を改善するためにはPEGが必要であること、及びそれは受容体に対する低い親和性を補って余りあることを示唆する。
【実施例12】
【0122】
この実施例は、TTR上のシステイン10のアラニンへの突然変異、TTR(C10A)がそのオリゴマー構造に有意に影響を及ぼさないことを明らかにする。TTR及びTTR(C10A)の各々約50μgを1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図6からわかるように、TTR(C10A)は、55kDaの四量体の算定分子量と同様の57kDaの分子サイズに相当する、約8.8分で溶出する。両方の形態のTTRが65℃で沈殿に対して抵抗性であるという所見(データは示していない)と合わせてこのデータは、システイン10のアラニンへの突然変異はTTRの構造又は安定性に有意の影響を及ぼさないことを示唆する。
【実施例13】
【0123】
この実施例は、システイン10からアラニンへの突然変異のバックグラウンドにおけるアラニン37のシステインへの突然変異、TMP−TTR(C10A/A37C)、アスパラギン酸38のシステインへの突然変異、TMP−TTR(C10A/D38C)、アラニン81のシステインへの突然変異、TMP−TTR(C10A/A81C)、又はグリシン83のシステインへの突然変異、TMP−TTR(C10A/G83C)がTTRのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。加えて、この実施例は、これらの突然変異体形態のTTRの5K又は20K PEGによるペグ化が、非ペグ化形態よりも有意に大きい分子サイズを有する2つの異なる種のTTRを生じることを明らかにする。TTRのペグ化は、最初にTTR(7.28g/ml)約8mlを、50mMトリスHCl、pH8.5の存在下に30℃で30分間、10mM DTTで還元することによって実施した。還元したTTRを、次に、20mMトリスHCl、pH8.5中2.5ml/分で26ml SEPHADEX(商標)G25ミディアムカラム(内径2.6cm)(Amersham Pharmacia Biotech)を使用して脱塩した。次に、還元TTRの吸光度を280nmで測定し、算定比吸光係数(TMP−TTR(C10A/A37C)については29,450 M−1)を使用することによって濃度を判定した(5.14mg/ml)。その後、還元試料の2分の1(4.6ml)を直ちに5mMメトキシ−PEG−マレイミド5K(Shearwater Corporation,Huntsville,AL)810μlと混合し、残りの半分を2.5mMメトキシ−PEG−マレイミド20K(Shearwater Corporation)1620μlと混合した。この反応を30℃で30分間進行させ、1M DTT46μlを加えてクエンチした。次に各々のペグ化試料を2.5ml/分で5ml HiTrap Q−セファロースカラムに負荷し、5ml/分で数カラム容積のQ緩衝液A(20mMトリスHCl、pH8.0)で洗った。カラムを、40%Q緩衝液B(20mMトリスHCl、1M NaCl、pH8.0)への直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。ピーク分画をプールし、280nmでプールの吸光度を測定することによって濃度を判定した。各々の試料約50μgを1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図7からわかるように、4つの非ペグ化TMP−TTR構築物の見かけ分子サイズは、予想される70kDa四量体よりも著しく低い、40−45kDaの間である。この遅延した溶出時間はおそらく、いくつかの他のTMP構築物に関して認められた(データは示していない)、サイズ排除樹脂とTMP−TTR構築物のわずかな相互作用によるものと考えられる。5K PEGとの複合後、見かけ分子サイズは、予想される90kDaよりもはるかに大きい、421−428kDaの間に上昇する(非ペグ化対応物よりも1.53−1.64分早い溶出)。サイズ排除クロマトグラフィーでのペグ化分子の誇張された分子量の所見はしばしば認められる現象である(データは示していない)。20K PEG構築物は、最大カリブレート標準物質(670kDa)よりも早く溶出し、それらの5Kペグ化対応物よりも1.28−1.40分早い溶出を示した。総合してこのデータは、4つの工作した突然変異体形態のTMP−TTRすべてが、ペグ化されてそれらの見かけ分子サイズを劇的に上昇させうることを明らかにしている。
【0124】
ペグ化TMP−TTR構築物約2μgをSDS−PAGEによって分析した(図8)。この図は、TMP−TTR単量体の大部分が1個のみのメトキシ−PEG−マレイミドによって修飾されたこと、及びその反応はほぼ完全であって非修飾単量体をほとんど残さなかったことをゲルシフトによって明らかにしている。
【実施例14】
【0125】
この実施例は、Fc−TMP、TMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)、及びTMP−TTR(C10A/G83C)がインビトロでのヒトMPL受容体への結合に関して類似の親和性を有することを明らかにする。この実施例のために、Fc−TMPを、製造者(BIAcore,Uppsula,Sweden)の指示に従って高密度でBIAcoreプロテインGチップに結合した。供試タンパク質を、0.1mg/mlウシ血清アルブミン及び0.005%P20(ポリオキシエチレンソルビタン)を含む結合緩衝液(ダルベッコのPBS(Gibco BRL,Gaithersburg,MD))中で5nM MPL受容体と共に室温(約23℃)で2時間以上プレインキュベートした。非ペグ化タンパク質については、非特異的結合を防ぐために0.1mg/mlヘパリンを添加した。すべての試料を、結合緩衝液中50μl/分で該チップに通した。平衡エンドポイントは注入後3分とした。図9からわかるように、すべてのTTR構築物はMPL受容体に対して同様の親和性を示し、0.881−2.333nmの範囲の親和性であったのに対し、Fc−TMP構築物は3.276−5.369nmの範囲の親和性を有していた。
【実施例15】
【0126】
この実施例は、ペグ化TMP−TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために、170匹のBDF1マウスを17群に分け、供試タンパク質50μg/kg動物(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目、12日目及び14日目)採血した(140μl)。採血の前にマウスをイソフルランで麻酔した。
【0127】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図10Aに認められるように、Fc−TMPは、5日目に4.2×1012血小板 L−1以上に達する血小板数で最大応答を示し、これは基線値である1.4×1012血小板 L−1の3倍である。4つの非ペグ化TMP−TTR構築物はすべて対照よりも良好な成績であったが、Fc−TMPほど良好ではなく、Fc−TMPは、5日目に基線値に比べて29%から107%の改善である1.8から2.9×1012血小板 L−1の血小板数を示した。図10Bに認められるように、4つのTMP−TTR構築物すべての工作されたシステインへの5K PEG基の付加は、3.7から4.4×1012血小板 L−1(基線値の2.8から3.4倍)の血小板数を伴って実質的に効果を改善する。
【0128】
同様に図10Cに認められるように、TMP−TTRへの20K PEGの複合体化は、4.2から4.6×1012血小板 L−1(基線値の3.2から3.5倍)の血小板数を伴って、付加的な、しかしそれほど劇的ではない効果の改善を生じさせる。TMP融合構築物のすべてがインビトロでMPLに関して類似の結合親和性を有していたので、この差はおそらく、構築物の有効生物学的半減期を上昇させるPEG複合体化の影響によるものであると考えられる。
【実施例16】
【0129】
この実施例は、ペグ化PTH−TTR構築物のマウスへの注入が血液中のイオン化カルシウム放出に及ぼす作用を示す。この実施例のために、60匹の雄性BDF1、4週齢マウスを12群に分け、供試タンパク質8.91mg/kg動物(PTH融合構築物、PTH−Fc又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を0、24、48及び72時間目の時点で採血した(75μl)。採血の前にマウスをイソフルランで麻酔した。
【0130】
採集した血液を、Ciba*Corning 634 Ca++/pH分析器を用いてイオン化カルシウムに関して分析した。図11に認められるように、PTH−Fc、PTH−TTR(C10A/K15A/A37C)(PEG5K)、PTH−TTR(C10A/K15A/A37C)(PEG20K)、PTH−TTR(C10A/K15A/G83C)(PEG5K)及びPTH−TTR(C10A/K15A/G83C)(PEG20K)は、注射後24時間目に2.2から2.7mmol L−1に上るイオン化カルシウムレベルの最大応答を示し、これは基線値である1.3mmol L−1の1.7倍である。注射後72時間目に、1.8から1.9mmol L−1の増大したイオン化カルシウムレベルを維持したPTH−TTR(C10A/K15A/A37C)(PEG5K)、PTH−TTR(C10A/K15A/G83C)(PEG5K)及びPTH−TTR(C10A/K15A/G83C)(PEG20K)処置群を除いて、すべての群のイオン化カルシウムレベルが基線値に戻った。非ペグ化PTH−TTR構築物は、血清中イオン化カルシウムレベルの上昇においてTTR(C10A)対照と同等であるか又はそれよりわずかに良好であった。
【実施例17】
【0131】
この実施例は、プラスミドを含むPTH−TTR(C10A/K15A/A81C)の構築を述べる。5920のXbaI/XbaIフラグメントを、プラスミド5643(実施例1に述べられている)をXbaIで消化して誘導した精製ベクターに連結した。生じたプラスミドを含む大腸菌株を5933 PTH−TTRC10A/K15A/A81Cと称する。
【0132】
【化11】
【実施例18】
【0133】
この実施例は、GLP−1−TTR(C10A/G83C)融合物及びGLP−1−TTR(C10A/K15A/G83C)融合物の作製を述べる。実施例1に述べられているプラスミドpAMG21を使用してこれらの構築物をクローン化した。この実施例において使用した各々のオリゴヌクレオチドを表6に列記する。
【0134】
細菌宿主GM121は、後期ebg領域(68分)内のlacIQリプレッサーを含むように改変された大腸菌K−12株である。このリプレッサー遺伝子の存在は、この宿主を様々な発現系と共に使用することを可能にするが、このリプレッサーはluxPRからの発現には無関係である。非形質転換宿主は抗生物質耐性を持たない。詳細には、lacIQ構築物を、介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド2493位と2937位の間のebgオペロンに送達することによってF’tet/393を修飾した。この構築物を、AGebg−lacIQ No.5と呼ばれる組換えファージを用いて染色体に送達した。
【0135】
組換えと分離後、上述した染色体挿入物だけが細胞内にとどまる。これを新たにF’tet/GM120と命名した。次にF’tet/GM120を、hsdR遺伝子においてそれを不活性化するように突然変異させた。これを新たにF’tet/GM121と命名した。F’tetエピソームを該菌株からキュアリングし、テトラサイクリン感受性であることを確認して、GM121(ATCC No.202174)として保存した。
【0136】
GLP−1配列及びリンカーを増幅するために、ミニプレッププラスミドDNA鋳型2−4μl、各々のオリゴヌクレオチド1μM、各々のオリゴヌクレオチド0.2mM、5%DMSO(Sigma)及びTaq DNAポリメラーゼ2Uを含む反応物80μl中、Roche PCR Core Kit(カタログ番号1 578 553)でPCRを実施した。反応サイクルは、94℃、5分間、次いで[94℃、20秒間、45℃、30秒間、72℃、1分間]の35サイクルであった。PCR産物を、製造者(QIAGEN)のプロトコールに従ってQIAquick(登録商標)PCR Purification Kitで精製した。次にPCR産物とベクターをNdeI及びKpnI(New England Biolabs)で消化した。
【0137】
消化したDNAをアガロースゲルから精製し、その後混合して、T4 DNAリガーゼ(New England Biolabs)によって室温で1.5−2時間連結した。各々の連結混合物を、ギャップ長2mmのキュベット中2.5KVでBiorad大腸菌Pulserを使用して、電気穿孔によって上述した宿主菌株GM121に形質転換した。テリフィック肉汁(TB)2ml中37℃、250rpmで約3時間、細胞を回収した。回収培養物70−100μlを、40μg/mlカナマイシンを含むLB寒天上にプレートした。DNAミニプレップを作製し、ヌクレオチド配列決定によって正しいクローンを確認した。
【0138】
GLP−1−TTR(C10A/G83C)融合物を作製するために、2個のオリゴヌクレオチド、すなわち、luxRプロモーター領域に結合するオリゴヌクレオチド1209−85、及び融合リンカーの最後の12個のアミノ酸とTTRの最初の8個のアミノ酸をコードするオリゴヌクレオチド3131−63を合成した。N末端Met−Lys開始点とそれに続くニッケルカラム精製のための7ヒスチジン配列を有するGLP−1配列、カスパーゼによるGLP−1の最初のヒスチジンの前での開裂のためのアスパラギン酸−グルタミン酸−バリン−アスパラギン酸配列、GLP−1(A2G)配列、及び27アミノ酸の融合リンカーを発現する菌株由来のpAMG21プラスミドを、オリゴヌクレオチド1209−85及び3131−63を用いて増幅した。そのPCR産物を上述したようにクローン化し、配列決定した。新しいプラスミドを含む生じた菌株をGLP−1−TTR(C10A/G83C)(菌株6298)と称した。この菌株は配列番号47に同定するDNA配列を有していた。
【0139】
GLP−1−TTR(C10A/K15A/G83C)融合物を作製するために、2個のオリゴヌクレオチド、すなわち、NdeI部位を含み、上述した精製及び開裂配列に加えてGLP−1(A2G)の最初の6個のアミノ酸をコードするオリゴヌクレオチド3183−83、及び融合リンカーの最後の6個のアミノ酸とTTRの最初の8個のアミノ酸をコードするオリゴヌクレオチド3183−84を合成した。
【0140】
N末端Met−Lys開始点とそれに続くニッケルカラム精製のための7ヒスチジン配列を有するGLP−1配列、カスパーゼによるGLP−1の最初のヒスチジンの前での開裂のためのアスパラギン酸−グルタミン酸−バリン−アスパラギン酸配列、GLP−1(A2G)配列、及び25アミノ酸の融合リンカーを発現する菌株由来のpAMG21プラスミドを、オリゴヌクレオチド3183−83及び3183−84を用いて増幅した。そのPCR産物を上述したようにクローン化し、配列決定した。新しいプラスミドを含む生じた菌株をGLP−1−TTR(C10A/K15A/G83C)(菌株6450)と称した。この菌株は配列番号48に同定するDNA配列を有していた。
【0141】
【表6】
【0142】
【化12】
【実施例19】
【0143】
この実施例は、GLP−1(A2G)−K−Fc融合物の作製を述べる。この構築物は、luxタンパク質及びプロモーターがlacI結合部位及びIPTG誘導プロモーターで置換されていること及びリボソーム結合部位配列がより短いこと(AatIIとClaI認識部位の間の配列が
【0144】
【化13】
で置換されており、SacII認識部位の後の配列の一部が欠失していること(
【0145】
【化14】
が残存する)においてpAMG21と異なる、プラスミドpAMG33*を使用してクローン化した。この実施例において使用した各々のオリゴヌクレオチドを表7に列記する。
【0146】
GLP−1(A2G)−Fc融合物を作製するために、2個のオリゴヌクレオチド、すなわち、NdeI部位を含み、上述した精製及び開裂配列に加えてGLP−1(A2G)の最初の8個のアミノ酸をコードするオリゴヌクレオチド2985−92、及びFcのアミノ酸18から23をコードするオリゴヌクレオチド2687−50を合成した。N末端Met開始点を有するGLP−1(A2G)配列、27アミノ酸リンカー及びFc配列を発現する菌株由来のpAMG33*プラスミドを、オリゴヌクレオチド2985−92及び2687−50を用いて増幅した。そのPCR産物を、使用した酵素がNdeI及びEcoRIであったことを除いて上述したようにクローン化し、配列決定した。上流ベクター配列及び挿入物を導入した5His−アスパラギン酸配列に対するオリゴヌクレオチドを用いたPCRによって挿入物の存在を確認する、コロニースクリーニング段階を含めた。新しいプラスミドを含む生じた菌株をGLP−1(A2G)−K−Fc(菌株5945)と称した。この菌株は配列番号51に同定するDNA配列を有していた。
【0147】
【表7】
【0148】
【化15】
【実施例20】
【0149】
この実施例は、TMP−CH2−TTRC10A及びTTRC10A−CH2−TMPを作製するための、免疫グロブリン分子のCH2ドメインのTTR(C10A)へのクローニングを述べる。
【0150】
TMP−Fcから誘導したCH2ドメインを、2段階PCR手順によってTTR(C10A)、すなわち菌株5619のC末端に連結した。そのCH2ドメイン(5’から3’まで:TTRの最後の7個のコドン、CH2及びBamHI−XhoIリンカーを含む)を最初に次のオリゴヌクレオチドによって増幅した:
2933−77:
GTC GTC ACC AAT CCC AAG GAA GGT TCT GGC TCC GGA TCA GGG GGA CCG TCA GTT TTC CTC(配列番号53)、及び
2973−78:
CCG CGG ATC CTC GAG ATT AGG ATC CAG AAC CCC CTT TGG CTT TGG AGA TGG T(配列番号54)。
【0151】
次にこのフラグメントを、オリゴヌクレオチド2973−78及び
2973−79:
GAG GAA TAA CAT ATG GGT CCA ACT GGT ACC GGT GAA TCC AAG(配列番号55)
によるその後のPCRにおいて5619に融合し、次いでNdeI/XhoIで消化して、同様に制限切断したpAMG21にクローニングした。生じたプラスミドを6017(TTRC10A−CH2)と称する:
【0152】
【化16】
【0153】
6017のXbaI/XbaIフラグメントを上述した5704の対応するフラグメントで置換し、TMP−TTRC10A−CH2(菌株6024)を構築した:
【0154】
【化17】
【0155】
TTRC10A−CH2−TMPの構築を次のように実施した:5’BamHIリンカーと3’XhoIリンカーを含むTMPフラグメントを、オリゴヌクレオチド2694−19及び
2974−70:
GAG GAA TAA GGA TCC ATC GAA GGT CCG ACT CTG CG(配列番号58)
によって増幅した。
【0156】
増幅したフラグメントをBamHI及びXhoIで消化し、その後同様に制限切断した6017と連結した。生じたクローンを菌株6104(TTRC10A−CH2−TMP)と称する。
【0157】
【化18】
【0158】
この融合物のもう1つの立体配置をTMP−CH2−TTR2として作製した。TMP−Fcから誘導したCH2ドメインを、最初に2段階PCRによってTTRC10AのN末端に連結した。そのCH2ドメイン(5’から3’まで:NdeI−BamHIリンカー、CH2及びTTRC10Aの最初の7個のコドンを含む)を、最初にオリゴヌクレオチド:
【0159】
【化19】
によって増幅した。
【0160】
このフラグメントをオリゴヌクレオチド2974−66及び2693−80によるその後のPCRにおいて5619に融合し(実施例1)、次いでNdeI/XhoIで制限消化して、同様に制限切断したpAMG21にクローニングした。生じたクローンを6016(CH2−TTRC10A)と称する:
【0161】
【化20】
【0162】
5’末端にNdeIリンカー及び3’末端にBamHIリンカーを含むTMPフラグメントを、オリゴヌクレオチド:
【0163】
【化21】
によって増幅した。
【0164】
次にこのフラグメントをNdeI/BamHIで消化し、菌株6016由来の同様に制限切断してゲル精製したベクターと連結した。生じたクローンを6110(TMP−CH2−TTRC10A)と称する:
【0165】
【化22】
【実施例21】
【0166】
この実施例は、TTRC10A/K15A−TMP、TTRC10A/K15A/A81C−TMP及びTTRC10A/K15A/G83C−TMPの構築を述べる。
【0167】
TMPをTTR及びその変異体のC末端でもクローン化した。そのN末端に5アミノ酸リンカー(gsgsg)プラスwt TTRの最後の7個のアミノ酸を含む完全長TMPを、標準PCR手順において次のセットのオリゴヌクレオチドによって増幅した。
【0168】
【化23】
【0169】
このPCRフラグメントを、さらに、実施例1で述べたようにオリゴヌクレオチド2694−19及び2693−79を用いる第二のPCRによってwt TTRの3’末端に連結した。生じたクローンを配列確認し、菌株5365(TTR−TMP)と称する:
【0170】
【化24】
【0171】
5365のXbaI/XbaIフラグメントを、上述したように菌株5895の対応するXbaI/XbaIフラグメントで置換して、菌株5921(TTRC10A/K15A−TMP)を作製した:
【0172】
【化25】
【0173】
プラスミド5921を、その後、次の位置:A37、A81及びG83のアミノ酸を、実施例1で突然変異オリゴヌクレオチド(2693−80)と共に使用したTTR3’オリゴヌクレオチドを2694−19に置き換えたことを除いて、実施例1で述べたようにシステインアミノ酸で置換することによって修飾し、TTRC10A/K15A/A37C−TMP(配列番号70)を含む菌株5982、TTRC10A/K15A/A81C−TMP(配列番号71)を含む菌株5983、及びTTRC10A/K15A/G83C−TMP(配列番号72)を含む菌株5984を生成した。
【0174】
【化26】
【実施例22】
【0175】
この実施例は、TMP−TTRC10A/K15A/A81C及びTMP−TTRC10A/K15A/A37Cの構築を述べる。次の方法により、菌株5704、5706及び5707においてTTRの15位のLysをAlaに突然変異させた。プラスミド5513をNdeI/KpnIで消化し、TMPフラグメントとTTRの最初の6個のアミノ酸を内包する挿入物を精製して、NdeI/KpnIで制限消化してゲル精製した菌株5895由来のベクターと連結した。生じたプラスミドを含む細菌株を5919(TMP−TTRC10A/K15A/G83C)と称する。次にプラスミド5919をXbaIで消化し、TMP及びC10AとK15A突然変異を含むTTRの最初の18個のコドンを含む、生じたXbaI/XbaIフラグメントをゲル精製し、XbaI消化してホスファターゼ処理し、ゲル精製した菌株5704及び5706由来のベクターと連結した。新しい菌株を5918(TMP−TTRC10A/K15A/A81C)及び6023(TMP−TTRC10A/K15A/A37C)と称する。
【0176】
【化27】
【実施例23】
【0177】
この実施例は、大腸菌におけるGLP−1融合タンパク質の発現を述べる。飽和一晩培養物25−100mlを使用して、250mlバフルフラスコ中で20μg/mlカナマイシンを含むTB50mlに接種し、37℃、250rpmで一晩インキュベートした。これらの一晩培養物10−35mlを使用して、2Lバフルフラスコ中で20μg/mlカナマイシンを含むTB1Lに接種し、600nmでの光学密度が約0.7に達するまで37℃、250rpmでインキュベートした。次にその培養物を、pAMG21の場合は30μg/ml N−(B−ケトカプロイル)−DL−ホモセリンラクトン(Sigma)を含むエタノール1mlの添加、あるいはpAMG33*の場合は0.1mMまでのIPTGの添加によって、組換えタンパク質を発現するように誘導した。さらに2−4時間インキュベーションを継続し、遠心分離によって細胞を収集した。
【実施例24】
【0178】
この実施例は、PTH−TTR(C10A/K15A/A81C)の精製を述べる。−80℃で保存したクローン5933からの大腸菌ペースト約197gを、50mMトリスHCl、5mM EDTA、pH8.0 1480ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)60錠を細胞懸濁液に溶解し、その懸濁液を14,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を11,325×g、4℃で50分間遠心分離した。上清を可溶性分画として採取した。その可溶性分画をポリプロピレンボトル中65℃水浴で30分間加熱し、その時点で内容物の温度は63℃であった。該可溶性分画を11,325×g、4℃で50分間遠心分離した。上清を熱可溶物(Heat Soluble)として採取した。該熱可溶性分画を、2つのプレフィルターを備えた0.45μm酢酸セルロースフィルターでろ過し、室温(約23℃)で、Q緩衝液A(20mMトリスHCl、2.5mM EDTA、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に25ml/分で負荷した。カラムを30ml/分のQ緩衝液A約2200mlで洗った。Qカラムを、60%Q緩衝液B(20mMトリスHCl、1M NaCl、2.5mM EDTA、pH8.0)への15カラム容積の直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(1300ml)に貯留した。そのQプールに、3.8M硫酸アンモニウムpH7.2 464mlを緩やかに加えた。その溶液を11,325×g、4℃で50分間遠心分離した。上清を硫酸アンモニウム可溶性分画として採取し、廃棄して、ペレットを、室温で約30分間静かに攪拌することによって10mM NaH2PO4、pH7.0 450mlに再懸濁した。その溶液を11,325×g、4℃で50分間遠心分離した。上清をリン酸緩衝液可溶性分画として採取し、0.45μm酢酸セルロースフィルターでろ過した。該リン酸緩衝液可溶性分画に1Mジチオトレイトール240μlを加え、HA緩衝液A中10ml/分で105ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを10ml/分のHA緩衝液A約210mlで洗い、次いで25%、50%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の3段階で溶出させた。50%溶出からの分画をHAプール(725ml)として貯留し、0.22μm酢酸セルロースフィルターでろ過した。ジチオトレイトール1.16gをHAプールに加え、トリス塩基を使用してそのpHを8.0に上げ、その後室温で約30分間インキュベートした。水750mlで希釈したHAプールを、10ml/分で50mlソース15Q(内径2.6cm)カラム(Amersham Pharmacia Biotech)に負荷し、その後Q緩衝液A約250mlで洗った。10%から60%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Q2プール(170ml)に貯留し、0.22μm酢酸セルロースフィルターでろ過した。算定比吸光係数23,950 M−1 cm−1を用いてタンパク質濃度を3.7mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.61と測定されたので、無視しうると判定した。
【実施例25】
【0179】
この実施例は、TMP−TTR(C10A/D38C)の精製を述べる。−80℃で保存したクローン5705からの大腸菌ペースト約170gを、50mMトリスHCl、5mM EDTA、pH8.0 1275ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、その懸濁液を14,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を11,325×g、4℃で30分間遠心分離した。上清を可溶性分画として採取し、廃棄した。組織粉砕器を使用してペレットを水1200mlに再懸濁し、さらに20錠のSigmaプロテアーゼ阻害剤錠剤を加えた。その懸濁液を11,325×g、4℃で30分間遠心分離した。上清をWhatman GF/Aフィルターでろ過し、ジチオトレイトール2.1gを加えて、その後7℃で30分間インキュベートした。還元した試料を、7℃で、Q緩衝液A(20mMトリスHCl、0.02%アジ化ナトリウム、pH8.0)中で平衡させた30ml/分の240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷した。カラムを30ml/分のQ緩衝液A約1920mlで洗った。Qカラムを、20%、35%及び100%Q緩衝液B(20mMトリスHCl、1M NaCl、0.02%アジ化ナトリウム、pH8.0)の3段階で溶出させた。500mM EDTA pH8.0 13mlをQカラムからのフロースルーに加え、11,325×g、4℃で30分間遠心分離した。上清を廃棄し、ペレットを4M尿素700ml、20mMトリスHCl、pH8.0に再懸濁した。尿素で可溶化したペレットをWhatman GF/Aフィルターでろ過し、7℃でQ緩衝液A(20mMトリスHCl、0.02%アジ化ナトリウム、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に30ml/分で負荷した。カラムを30ml/分のQ緩衝液A約1920mlで洗った。Qカラムを、15ml/分の20%、35%及び100%Q緩衝液B(20mMトリスHCl、1M NaCl、0.02%アジ化ナトリウム、pH8.0)の3段階で溶出させた。35%溶出ピークを含む分画を貯留し、0.22μm酢酸セルロースフィルターでろ過し、ジチオトレイトール(最終濃度10mM)0.5gを加えて、その後7℃で30分間インキュベートした。次にその35%Qプールを、7℃で20mMトリスHCl、350mM NaCl、pH8.0中、5ml/分で45ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを、5ml/分の20mMトリスHCl、350mM NaCl、pH8.0 約70ml、次いで2.5%、25%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の3段階で洗った。2.5%溶出からの分画をHAプール(80ml)として貯留し、0.22μm酢酸セルロースフィルターでろ過した。算定比吸光係数29,450 M−1 cm−1を用いてタンパク質濃度を6.8mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.54と測定されたので、無視しうると判定した。
【実施例26】
【0180】
この実施例は、TTR(C10A)−CH2−TMPのリフォールディングと精製を述べる。−80℃で保存したクローン6104からの大腸菌ペースト約23gを、50mMトリスHCl、5mM EDTA、pH8.0 200ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、その懸濁液を12,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として採取し、廃棄した。組織粉砕器を使用してペレットを50mMトリスHCl、5mM EDTA、pH8.0 50mlに再懸濁した。その懸濁液を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として採取し、廃棄した。組織粉砕器を使用してペレットを50mMトリスHCl、5mM EDTA、pH8.0 50mlに再懸濁した。その懸濁液を14,000×g、室温で10分間遠心分離した。上清を洗浄液として採取し、廃棄した。超音波処理装置を約1分間使用してペレットを8MグアニジンHCl、50mMトリスHCl、pH8.0 50mlに溶解した。溶解したタンパク質を、1M DTT 500μlを加えることによって室温で30分間還元した。還元したタンパク質を20℃、27,216×gで30分間遠心分離した。その後、上清を2ml/分で50mMトリス塩基、160mMアルギニン塩基、1M尿素、1mMシスタミン、4mMシステイン、pH9.5 4Lに加え、4℃で約16時間インキュベートした。次に再生したタンパク質をGellman SUPORCAP(登録商標)50でろ過し、その後Pall Firtron3平方フィートYM10膜接線型フローシステム(tangential flow system)を用いて約500mlに濃縮して、20mMトリスHCl、pH8.0 2Lにろ過透析した。次に濃縮タンパク質を18ml/分で45mlソース15Q(内径2.6cm)カラム(Amersham Pharmacia Biotech)に負荷し、その後Q緩衝液A(20mMトリスHCl、pH8.0)約150mlで洗った。0%から60%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(29ml)に貯留した。次にそのQプールをMillipore CENTRIPREP(登録商標)10を用いて約6.3mlに濃縮し、1ml/分でPall ACRODISC(商標)MUSTANG(登録商標)E膜フィルターに通した。算定比吸光係数46,410 M−1 cm−1を用いてタンパク質濃度を10.5mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.51と測定されたので、無視しうると判定した。
【実施例27】
【0181】
この実施例は、GLP−1−TTR(C10A/K15A/G83C)の精製を述べる。−80℃で保存したクローン6450からの大腸菌ペースト約30gを、50mM NaH2PO4、pH7.0 250ml中で解凍した。細胞懸濁液を12,000PSIでマイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を15,344×g、4℃で50分間遠心分離した。上清を可溶性分画として廃棄し、組織粉砕器を使用してペレットをデオキシコレート200mlに再懸濁した。その懸濁液を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として廃棄し、組織粉砕器を使用してペレットを水200mlに再懸濁した。上清を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として廃棄し、組織粉砕器を使用してペレットを水100mlに再懸濁した。上清を27,216×g、室温で30分間遠心分離した。上清を洗浄液として廃棄し、ペレットの約2/3を約15分間攪拌することによって8MグアニジンHCl、50mMトリスHCl、pH8.0 75mlに溶解した。上清を27,216×g、室温で30分間遠心分離し、その上清を水18mlで希釈した。次に試料を、5ml/分で50mlキレート化セファロースファーストフローカラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷し、NiCl2で充填した。10ml/分のNi緩衝液A(6MグアニジンHCl、37.5mlトリスHCl、pH8.0)約150mlで洗浄した後、10%及び100%Ni緩衝液B(6MグアニジンHCl、37.5mMトリスHCl、400mMイミダゾール、pH8.0)の2段階で溶出させた。融合構築物を含むピークをNiプール(40ml)として混合し、比吸光係数25,440 M−1を用いて8MグアニジンHCl中280nmでの吸光度を観察することにより、タンパク質濃度を6.4mg/mlと判定した。500mM EDTA、pH8.0 800μlを加え、ペグ化反応のためにタンパク質80mgを採取した。1M DTT 230μlを加え、30℃で30分間インキュベートした。20mMトリスHCl、6M尿素、pH8.5中8ml/分で、130ml SEPHADEX(登録商標)G25ミディアムカラム(内径2.6cm)に負荷した。280nmでの吸光度によって測定したタンパク質ピークをプールし(22ml)、比吸光係数25,440 M−1を用いて20mMトリスHCl、6M尿素、pH8.5中280nmでの吸光度を観察することにより、タンパク質濃度を3.2mg/mlと判定した。緩衝液を交換した該物質の45%を、30℃で140分間、5mMメトキシ−PEG−マレイミド5K(Shearwater Corporation,Huntsville,AL)950μlと反応させた。各々の反応物に1M 2−メルカプトエタノール100μlを加えてクエンチした。Pierce 10kDa Slidealyzerを使用して室温で2時間、反応物を25mM NaH2PO4、3M尿素、pH7.25 1Lに透析した。透析緩衝液を25mM NaH2PO4、10%スクロース、2mM EDTA、pH7.25に交換し、室温で約16時間インキュベートした。5%CHAPS140μl及び2−メルカプトエタノール7.28μl及び3mg/mlカスパーゼ3 0.475mlを加え、その後室温で2時間インキュベートした。反応混合物を、20mMトリスHCl、pH8.0中1ml/分で、5ml HiTrap Q−sepharose HPカラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷し、その後同じ緩衝液約15mlで洗った。次に60%20mMトリスHCl、1M NaCl、pH8.0への直線勾配、次いで100%溶出緩衝液への段階を使用して5ml/分でカラムを展開した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプールに貯留した(9.5ml)。Millipore CENTRIPREP(登録商標)30kDaを用いてQプールを3.2mlに濃縮し、約1ml/分でPall MUSTANG(登録商標)E膜に通してろ過した。Qプールを水で6.5mlに希釈し、アセトニトリル375μlを加えた。95%RP緩衝液A(0.1%トリフルオロ酢酸)及び5%RP緩衝液B(95%アセトニトリル、0.1%トリフルオロ酢酸)中、5ml/分でVydac Protein/Peptide 10×250mm C4カラム(Vydac,Hisperia,CA)に注入した。100%RP緩衝液Bへの直線勾配に通してカラムを展開した。Millipore CENTRIPREP(登録商標)30kDaを用いてタンパク質ピークを約3mlに濃縮し、20mMトリスHCl、pH8.0を使用して15mlに希釈した。緩衝液交換をさらに3回反復した後、約1ml/分でPall MUSTANG(登録商標)E膜に通した。算定比吸光係数25,440 M−1 cm−1を用いてタンパク質濃度を7.7mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.54と測定されたので、無視しうると判定した。
【実施例28】
【0182】
この実施例は、マウスへのペグ化GLP1−TTR構築物の注入が血中グルコースレベルに及ぼす作用を示す。この実施例のために40匹の雄性、db/db、9週齢マウスを4群に分け、各動物当り7.4−16.6mgの供試タンパク質(すべての群について538pmol単量体)(5Kペグ化GLP1−TTR融合構築物10mg、20Kペグ化GLP1−TTR融合構築物10mg、GLP1−Fc16.6mg及びTTR(C10A)対照7.4mg)を腹腔内注射した(0時間目)。各々の群を注射後0時間目(基線測定)、1、4、6、12、24及び48時間目に採血した。実験の最初の6時間はマウスに食餌を与えず、6時間目の採血後に食餌を再開した。
【0183】
各時点で採取した各々の血液を、One Touch Profileグルコース測定器を用いてグルコース含量に関して分析し、その結果を図12に示す。
【実施例29】
【0184】
この実施例は、融合抗体CH2ドメインを有するTMP−TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために50匹の雌性BDF1マウスを5群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目及び10日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0185】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図13からわかるように、Fc−TMPは、5日目に4.2×1012血小板 L−1以上に達する血小板数で最大応答を示し、これは基線値の1.4×1012血小板 L−1の3倍である。CH2融合TMP−TTR構築物はすべて対照よりも良好な成績であったが、5日目に、基線値に比べて64%から86%の改善にあたる、2.3×1012から2.6×1012血小板 L−1の血小板数を示したFc−TMPほどではなかった。
【実施例30】
【0186】
この実施例は、ペグ化TTRのカルボキシ末端に融合したTMPとのペグ化TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために80匹のBDF1マウスを8群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目及び12日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0187】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図14からわかるように、Fc−TMP及び3つのアミノ末端(TMP−TTR)融合物は、5日目に、基線値の1.3×1012血小板 L−1の3倍以上である、4.3×1012から4.6×1012血小板 L−1に達する血小板数で最大応答を示した。3つのカルボキシ末端(TTR−TMP)構築物はすべて対照よりも良好な成績であった。
【実施例31】
【0188】
この実施例は、K15A変化を含むペグ化TTR−TMP構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために120匹のBDF1マウスを12群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)(この試験は2つのバッチ(一方においてはPEG20K及び他方ではPEG5K及び非ペグ化試料)に分け、対照を反復して別々の時点で実施した)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目及び12日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0189】
採取した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図15Aからわかるように、2つの非ペグ化構築物は基線(1.3×1012血小板 L−1)よりも良好な成績であり、5日目に1.8×1012から2.0×1012血小板 L−1に上る血小板応答を示した。図15Bからわかるように、Fc−TMPと3つの5Kペグ化融合物は、基線(1.3×1012血小板 L−1)の少なくとも2.7倍である、3.5×1012から4.4×1012血小板 L−1に達する血小板数で、5日目に同等の応答を示した。図15Cからわかるように、Fc−TMPと3つの20Kペグ化融合物は、基線値の1.3×1012血小板 L−1の3倍以上である、4.3×1012から4.6×1012血小板 L−1に達する血小板数で、5日目に同等の応答を示した。
【0190】
加えて、20Kペグ化TTR構築物は、3.1×1012血小板 L−1のFc−TMPに比べて、7日目に3.7×1012から4.9×1012血小板 L−1の範囲の血小板数を示し、改善された持続性応答を有すると思われる。この持続性応答は、3つの20Kペグ化TTR構築物に関しては10日目にも維持され、血小板数は、2.0×1012血小板 L−1のFc−TMPに比べて2.3×1012から3.1×1012血小板 L−1の範囲であった。
【技術分野】
【0001】
本出願は、本明細書中に参考として援用されている、2002年4月4日出願の米国出願第10/117,109号の一部継続出願である。
【背景技術】
【0002】
治療用途のためのタンパク質、ペプチド及び他の薬剤分子は、現在、主として組換えDNAテクノロジーの進歩の結果として適切な形態で十分な量が入手可能である。そのようなペプチド及びタンパク質が入手できることは、タンパク質の製剤及び化学的修飾における進歩を生じさせた。生物学的に活性なペプチド、タンパク質、オリゴヌクレオチド及び他の薬剤の血清中半減期を延長するために、そのような生物学的活性物質の化学的修飾が広汎に検討されてきた。そのような物質の血清中半減期を延長する能力は、薬剤の治療上の潜在的可能性が高用量及び高頻度の投与を必要とせずに実現されることを可能にする。
【0003】
インビボでタンパク質の半減期を延長するために使用される化学的修飾は、対象タンパク質へのポリエチレングリコール(PEG)などの水溶性ポリマーの化学的複合体化を包含する。ポリエチレングリコール分子をタンパク質に結合するために(ペグ化)様々なアプローチが使用されてきた。例えば、Royer(米国特許第4,002,531号)は、酵素へのポリエチレングリコール分子の結合のために還元的アルキル化を使用したと述べている。Davisら(米国特許第4,179,337号)は、例えば酵素とインスリンを含むPEG:タンパク質複合体を開示している。Shaw(米国特許第4,904,584号)は、反応性アミン基によるポリエチレングリコール分子の結合のための、タンパク質中のリシン残基数の変更を開示している。Hakimiら(米国特許第5,834,594号)は、例えばタンパク質である、IL−2、インターフェロンα及びIL−1raを含む、実質的に非免疫原性の水溶性PEG:タンパク質複合体を開示している。Hakimiらの方法は、タンパク質中の様々な遊離アミノ基をPEGに連結するためのユニークなリンカーの使用を含む。Kinstlerら(米国特許第5,824,784号及び同第5,985,265号)は、G−CSF及びコンセンサスインターフェロンを含む、選択的N末端化学的修飾タンパク質及びその類似体を可能にする方法を教示している。
【0004】
生物学的活性物質の血清中半減期を延長するために設計された他のアプローチは次のものを含む:あまりに大きすぎて腎ではろ過されない、大きく安定なタンパク質(例えば血清アルブミン)へのペプチドの複合体化;G.D.Maoら、Biomat.Art.Cells,Art.Org.17:229−244(1989);輸送ビヒクルとして及び血清中半減期を上昇させるための低密度及び高密度リポタンパク質の使用;P.Chris de Smidtら、Nuc.Acids.Res.,19(17):4695−4700(1991);Fc−タンパク質融合物を生産するための免疫グロブリンのFc領域の使用;PCT国際公開公報第98/28427号(Mannら及びその中で引用される参考文献);及び1以上の生物学的活性ペプチドのインビボでの半減期を上昇させるためのFcドメインの使用;PCT国際公開公報第00/24782号(Feigeら及びその中で引用される参考文献)。
【0005】
トランスサイレチン(TTR)(以前はプレアルブミンと呼ばれた)は、サイロキシン及びレチノール結合タンパク質の輸送体として重要な生理的役割を果たす56kDaの四量体血清タンパク質である;HamiltonとBenson,Cell.Mol.Life Sci.,58:1491−1521(2001)及びその中で引用される参考文献。米国特許第5,714,142号においてBlaneyらは、投与する薬剤に、該タンパク質に特異的に結合しうる機能を付与することによるTTRの利用を述べている。詳細には、Blaneyらは、トランスサイレチン選択的リガンドへのペプチド、タンパク質、ヌクレオチド、オリゴヌクレオチド、オリゴ糖又は他の薬剤の共有結合が、薬剤をTTRに可逆的に結合させ、それによってTTRに関するリガンドの親和性に基づき該薬剤の血清中半減期を上昇させることを明らかにしている。薬剤の内因性作用は有害な影響を受けず、生じる薬剤−TTRリガンド複合体はなお経口吸収されるのに十分なほど小さいと述べられている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第4,002,531号明細書
【特許文献2】米国特許第4,179,337号明細書
【特許文献3】米国特許第4,904,584号明細書
【特許文献4】米国特許第5,834,594号明細書
【特許文献5】米国特許第5,824,784号明細書
【特許文献6】米国特許第5,985,265号明細書
【特許文献7】国際公開第98/28427号
【特許文献8】国際公開第00/24782号
【特許文献9】米国特許第5,714,142号明細書
【非特許文献】
【0007】
【非特許文献1】G.D.Maoら、Biomat.Art.Cells,Art.Org.17:229−244(1989)
【非特許文献2】P.Chris de Smidtら、Nuc.Acids.Res.,19(17):4695−4700(1991)
【非特許文献3】HamiltonとBenson,Cell.Mol.Life Sci.,58:1491−1521(2001)
【発明の概要】
【発明が解決しようとする課題】
【0008】
驚くべきことに及び重要なこととして、TTR(又はTTR変異体)、特に、例えば水溶性ポリマーへの複合体化によって化学的に修飾されたTTR又はTTR変異体は、生物学的活性物質の血清中半減期を上昇させるために生物学的活性物質との融合パートナーとして使用できることが見出されている。従って、本発明は、選択した生物学的活性物質の血清中半減期を上昇させるための手段を提供する。
【課題を解決するための手段】
【0009】
本発明は、それ故、TTR(又はTTR変異体)−生物学的活性物質融合物及びPEG−TTR(PEG−TTR変異体)−生物学的活性物質融合物の実質的に均質な製剤に関する。生物学的活性物質単独と比較して、TTR−生物学的活性物質融合物及び/又はPEG−TTR−生物学的活性物質融合物は実質的に上昇した血清中半減期を有する。
【0010】
本発明はさらに、薬理的活性化合物を提供するための、医薬適合性の担体中のTTR−生物学的活性物質融合物及びPEG−TTR−生物学的活性物質融合物に関する。
【0011】
本発明はさらに、TTR変異体の調製に関する。詳細には、TTRタンパク質を、システイン残基がTTRタンパク質配列内に工作されるように修飾する。該TTR変異体は高収率で回収可能であり、その後システイン残基での水溶性ポリマーの複合によって化学的修飾して、化学的修飾されたTTR変異体を提供し、次に選択した生物学的活性物質にそれを融合することができる。
【0012】
本発明はさらに、薬理的活性化合物を調製する方法に関する。例えば、PEG−TTR−ペプチド融合物の実質的に均質な製剤を製造する方法の主要実施態様は、(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTRを得ること;(c)上記PEG−TTRを対象ペプチドに融合してPEG−TTR−ペプチド融合物を得ること;及び(d)上記PEG−TTR−ペプチド融合物を単離することを含む。
【0013】
本発明はまた、上記のような薬理的活性化合物を使用した個体の治療方法に関する。
【図面の簡単な説明】
【0014】
【図1】大腸菌(E.coli)で発現された組換えヒトトランスサイレチン(TTR)変異体(C10A/G83C)の、TTRのC末端に融合したブラジキニンペプチドによる精製を示すSDSゲルである。レーン1は、Novex Mark 12分子量標準物質を含み、レーン2−7はそれぞれ次のものを含む:細胞溶解産物、加熱後上清、Qセファロースクロマトグラフィー段階からのプール、フェニルセファロースクロマトグラフィー段階からのプール、ヒドロキシアパタイトクロマトグラフィー段階からのプール、及びソースQクロマトグラフィー段階からのプール。
【図2】TTR変異体、TTR(C10A/G83C)のアミノ末端又はカルボキシ末端へのペプチドの融合がそのオリゴマー構造を変化させないことをサイズ排除クロマトグラフィーによって明らかにする。実線はTTR(C10A/G83C)、破線はTTR(C10A/G83C)のアミノ末端に融合した副甲状腺ホルモン(PTH)、及び点線はTTR(C10A/G83C)のカルボキシ末端に融合したブラジキニンである。
【図3】TR変異体、TTR(C10A)のアミノ末端又はカルボキシ末端へのタンパク質の融合がそのオリゴマー構造を変化させないことをサイズ排除クロマトグラフィーによって明らかにする。実線はTTR(C10A)、破線はTTR(C10A)のカルボキシ末端に融合したIL−1−ra、及び点線はTTR(C10A)のアミノ末端に融合したIL−1−raである。
【図4】BIAcoreを使用して認められた、ヒトMPL受容体への様々なTPO−ミメティックペプチド(TMP)構築物の結合を示す:
【化1】
【図5】TMP(m)−TTR−PEG5Kの注入がマウスにおいて血小板形成を誘導することを示す。次の記号はその対応する構築物に相当する:
【化2】
【図6】天然TTR及びTTR(C10A)が同様のオリゴマー立体配置(見かけ上の四量体)を維持することをサイズ排除クロマトグラフィーによって明らかにしている。実線は天然TTRであり、破線はTTR(C10A)である。
【図7】TTRへのPEGの複合体化が予測可能に一様にその分子サイズを上昇させることをサイズ排除クロマトグラフィーによって明らかにする。実線はPEG複合体化なし、破線は5K PEG融合、及び点線は20K PEG融合を示す。次の構築物を使用した:A)TMP−TTR(C10A/A37C)、B)TMP−TTR(C10A/D38C)、C)TMP−TTR(C10A/A81C)、及びD)TMP−TTR(C10A/G83C)。
【図8】4つの異なる位置の1つに工作された非天然システインを有するTTR変異体を含む様々なTMP−TTR構築物のペグ化の程度を示すSDSゲルである。レーン1はNovex Mark 12分子量標準物質を含む;レーン2は非ペグ化TMP−TTR(C10A/A37C)である;レーン3−6は、それぞれTMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)及びTMP−TTR(C10A/G83C)の5Kペグ化型である;レーン7−10は、それぞれTMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)及びTMP−TTR(C10A/G83C)の20Kペグ化型である。
【図9】図9A−Cは、BIAcore分析によるヒトMPLへのFc−TMPとTMP−TTRの競合的結合を比較している。
【化3】
【図10】図10A−Cは、工作したシステインに複合体化したPEGを伴うTMP−TTRの注入がマウスにおいて血小板形成を誘導することを示す。
【化4】
【図11】工作されたシステインに複合体化したPEGを伴うPTH−TTRの注入がマウスにおいてイオン化カルシウム放出を誘導することを示す。次の記号はその対応する構築物に相当する:
【化5】
【図12】工作されたシステインに複合体化したPEGを伴うグルカゴン様ペプチド1(GLP1)−TTRの注入がマウスにおいて血中グルコースレベルを低下させることを示す。次の記号はその対応する構築物に相当する:
【化6】
【図13】融合CH2ドメインを有するTMP−TTR複合体の注入がマウスにおいて血清中の血小板レベルを上昇させることを示す。次の記号はその対応する構築物に相当する:
【化7】
【図14】ペグ化TTRとTMPのカルボキシ末端融合物の注入がマウスにおいて血中血小板数を上昇させることを示す。次の記号はその対応する構築物に相当する:
【化8】
【図15】図15A−Cは、K15A変化を含むペグ化TMP−TTR融合物の注入がマウスにおいて血中血小板数を上昇させることを示す。次の記号はその対応する構築物に相当する:
【化9】
【発明を実施するための形態】
【0015】
本発明を説明するために、次の用語は下記で述べるように定義される。
【0016】
「生物学的活性物質」という用語は、予防、治療又は診断的適用のために有用な化学物質又は化合物を指す。「薬理的活性化合物」という用語は、所望の局所又は全身作用を誘導する、哺乳動物、好ましくはヒト個体への投与に適した化合物を指す。
【0017】
「ペプチド」、「ポリペプチド」及び「タンパク質」という用語は、生物学的活性物質の種類を表わし、それらの用語は、天然に生じる、組換え生産された又は化学合成されたアミノ酸のポリマーを指すためにここでは交換可能に使用される。それらの用語は、2個という低い数のアミノ酸を含むペプチド分子、化学的修飾されたポリペプチド、コンセンサス分子、それらの類似体、誘導体又は組合せを包含することが意図される。
【0018】
本発明に関連して任意の数のペプチドが使用しうる。特に興味深いのは、エリスロポエチン(EPO)、トロンボポエチン(TPO)、グルカゴン様ペプチド1(GLP−1)、副甲状腺ホルモン(PTH)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、インターロイキン−1受容体アンタゴニスト(IL−1ra)、レプチン、細胞傷害性Tリンパ球抗原4(CTLA4)、TNF関連アポトーシス誘導リガンド(TRAIL)、腫瘍増殖因子α及びβ(それぞれTGF−α及びTGF−β)、及び成長ホルモンの作用を模倣するペプチドである。「ミメティックペプチド」及び「アゴニストペプチド」という用語は、対象とするタンパク質と相互作用するタンパク質(例えばGLP−1、PTH、EPO、TPO、G−CSF等)に匹敵する生物学的活性を有するペプチドを指す。これらの用語はさらに、対象とするタンパク質の天然リガンドの作用を増強することなどによって、対象とするタンパク質の作用を間接的に模倣するペプチドを包含する。それ故、「EPOミメティックペプチド」という用語は、EPOミメティックの性質(EPO−mimetic subject matter)を有すると同定できる又は推論できるいかなるペプチドも包含する;例えばWrightonら、Science,273:458−63(1996);及びNarandaら、Proc.Natl.Acad.Sci.USA 96:7569−74(1999)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0019】
「TPOミメティックペプチド」(TMP)という用語は、TPOミメティックの性質を有すると同定できる又は推論できるペプチドを包含する;例えば、その全体が参考として本明細書中に援用されている、Cwirlaら、Science,276:1696−9(1997);米国特許第5,869,451号及び同第5,932,946号;及びPCT国際公開公報第00/24782号(Liuら、及びその中で引用される参考文献)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0020】
「G−CSFミメティックペプチド」という用語は、G−CSFミメティックの性質を有すると同定できるいかなるペプチドも包含する;例えばPaukovitsら、Hoppe−Seylers Z.Physiol.Chem.365:303−11(1984)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0021】
「CTLA4−ミメティックペプチド」という用語は、Fukumotoら、Nature Biotech.16:267−70(1998)に述べられているように同定できる又は推論できるいかなるペプチドも包含する。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0022】
ペプチドアンタゴニストも興味の対象であり、特にTNF、レプチン、インターロイキンのいずれか及び補体活性化に関与するタンパク質(例えばC3b)の作用に拮抗するものが興味深い。「アンタゴニストペプチド」又は「阻害剤ペプチド」という用語は、対象とする関連タンパク質の生物学的活性をブロックする又は何らかの方法で生物学的活性に干渉する、あるいは対象関連タンパク質の既知のアンタゴニスト又は阻害剤に匹敵する生物学的活性を有するペプチドを指す。それ故、「TNFアンタゴニストペプチド」という用語は、TNF拮抗性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Takasakiら、Nature Biotech.,15:1266−70(1997)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0023】
「IL−1アンタゴニスト」及び「IL−1raミメティックペプチド」という用語は、IL−1によるIL−1受容体の活性化を阻害する又は下方調節するペプチドを包含する。IL−1受容体の活性化は、IL−1、IL−1受容体及びIL−1受容体補助タンパク質(accessory protein)の間での複合体の形成から生じる。IL−1アンタゴニスト及びIL−1raミメティックペプチドは、IL−1、IL−1受容体又はIL−1受容体補助タンパク質に結合して、複合体の任意の2つ又は3つの成分の間での複合体形成を妨げる。例示的なIL−1アンタゴニスト及びIL−1raミメティックペプチドは、米国特許第5,608,035号、同第5,786,331号、同第5,880,096号に述べられているように同定する又は推論することができる。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0024】
「VEGFアンタゴニストペプチド」という用語は、VEGF拮抗性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Fairbrother,Biochem.,37:17754−64(1998)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0025】
「MMP阻害剤ペプチド」という用語は、MMP阻害性の性質を有すると同定できる又は推論できるペプチドを包含する;例えば、Koivunen,Nature Biotech.,17:768−74(1999)参照。当業者は、これらの参考文献の各々が、様々なペプチドライブラリーを用いて開示されている手順に従うことにより、その中で実際に開示されている以上の種々のペプチドを選択することを可能にすることを認識する。
【0026】
腫瘍ホーミングペプチド、膜輸送ペプチド等を含む、標的化ペプチドも興味深い。
【0027】
例示的ペプチドは当技術分野において既知の様々な手法によってランダムに生成しうる。例えば、固相合成手法は当技術分野において周知であり、Merrifield,Chem.Polypeptides,p.335−61(KatsoyannisとPanayotis編集)(1973);Merrifield,J.Am.Chem.Soc.,85:2149(1963);Davisら、Biochem.Intl.,10:394−414(1985);StewartとYoung,Solid Phase Peptide Synthesis(1969);米国特許第3,941,763号;Finnら、The Proteins,第3版、2:105−253(1976);及びEricksonら、The Proteins,第3版、2:257−527(1976)に述べられているものを包含する。固相合成は、小さなペプチドを作製する最もコスト効果的な方法であるので、個々のペプチドを作製する好ましい手法である。
【0028】
ファージディスプレイは、本発明において使用するためのペプチドを作製する上でのもう1つの有用な方法である。ランダムなペプチドのライブラリーからのアフィニティー選択が、任意の遺伝子産物の任意の部位についてのペプチドリガンドを同定するために使用できることが記述されている;Dedmanら、J.Biol.Chem.,268:23025−30(1993)。ファージディスプレイは、細胞表面受容体又は線状エピトープを有する任意のタンパク質のような対象タンパク質に結合するペプチドを同定するのに特に適する;Wilsonら、Can.J.Microbiol.,44:313−29(1998);Kayら、Drug Disc.Today,3:370−8(1998)。そのようなタンパク質は、本明細書中に参考として援用されている、Herzら、J.Receptor & Signal Transduction Res.,17(5):671−776(1997)において広く概説されている。
【0029】
該ペプチドはまた、組換えDNA手法を用いて形質転換宿主細胞においても作製しうる。そのために、該ペプチドをコードする組換えDNA分子を調製する。そのようなDNA及び/又はRNA分子を調製する方法は当技術分野において周知である。例えば、適切な制限酵素を使用して該ペプチドをコードする配列をDNAから切り出すことができた。その後のクローニングのために有用な制限部位を含んだ関連する配列を、ポリメラーゼ連鎖反応(PCR)を用いて創製することができた。あるいは、ホスホルアミダイト法などの化学合成手法を用いてDNA/RNA分子を合成することができよう。また、これらの手法の組合せも使用しうる。
【0030】
使用のために考慮されるさらなる生物学的活性物質は、ヒト又は動物の組換え又は天然のタンパク質、ホルモン、サイトカイン、造血因子、成長因子、抗肥満因子、栄養因子、抗炎症因子及び酵素を含む。そのようなタンパク質は、インターフェロン(図面を含めて本明細書中に参考として援用されている、米国特許第5,372,808号、同第5,541,293号、同第4,897,471号及び同第4,695,623号参照)、インターロイキン(図面を含めて本明細書中に参考として援用されている、米国特許第5,075,222号参照)、エリスロポエチン(図面を含めて本明細書中に参考として援用されている、米国特許第4,703,008号、同第5,441,868号、同第5,618,698号、同第5,547,933号及び同第5,621,080号参照)、顆粒球コロニー刺激因子(図面を含めて本明細書中に参考として援用されている、米国特許第4,810,643号、同第4,999,291号、同第5,581,476号、同第5,582,823号及びPCT国際公開公報第94/17185号参照)、幹細胞因子(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第91/05795号、同第92/17505号及び同第95/17206号)、NESP(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第US94/02957号)、オステオプロテゲリン(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第97/23614号)、インターロイキン−1受容体アンタゴニスト(L−1ra)(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第91/08285号及び同第92/16221号)及びレプチン(OBタンパク質)(図面を含めて本明細書中に参考として援用されている、PCT国際公開公報第96/40912号、同第96/05309号、同第97/00128号、同第97/01010号及び同第97/06816号)を含むが、これらに限定されない。
【0031】
さらに、生物学的活性物質はまた、インスリン、ガストリン、プロラクチン、副腎皮質刺激ホルモン(ACTH)、甲状腺刺激ホルモン(TSH)、黄体形成ホルモン(LH)、卵胞刺激ホルモン(FSH)、ヒト絨毛性性腺刺激ホルモン(HCG)、モチリン、インターフェロン(α、β、γ)、インターロイキン類(IL−1からIL−12まで)、腫瘍壊死因子(TNF)、腫瘍壊死因子結合タンパク質(TNF−bp)、脳由来神経栄養因子(BDNF)、神経膠由来神経栄養因子(GNDF)、神経栄養因子3(NT3)、線維芽細胞増殖因子(FGF)、神経栄養増殖因子(NGF)、オステオプロテゲリン(OPG)などの骨成長因子、インスリン様増殖因子(GF)、マクロファージコロニー刺激因子(M−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、巨核球由来増殖因子(MGDF)、角質細胞増殖因子(KGF)、トロンボポエチン、血小板由来増殖因子(PDGF)、コロニー刺激増殖因子(CSF)、骨誘導因子(BMP)、スーパーオキシドジスムターゼ(SOD)、組織プラスミノーゲン活性化因子(TPA)、ウロキナーゼ、ストレプトキナーゼ及びカリクレインを含みうるが、これらに限定されない。
【0032】
本発明における使用のための考慮されるトランスサイレチン(TTR)は、Mitaら、Biochem.Biophys.Res.Commun.,124(2):558−564(1984)に報告されているTTRのDNA及びアミノ酸配列を有する。これらの配列は、受入番号K02091としてGenbankに寄託されている。ここで使用する127アミノ酸のTTR配列は、K02091配列のシグナル配列(アミノ酸1−20)を含まず、配列番号1として下記に示す:
【0033】
【化10】
【0034】
「TTR変異体」という用語は、天然TTRの修飾形態である分子又は配列を指す。例えば、天然TTRは除去しうる部位を含む。それらは、構造的特徴を与えるか又は本発明の融合分子には必要とされない生物学的活性を提供するからである。それ故、「TTR変異体」という用語は、1以上の天然TTR部位又は残基を欠く、あるいは異なるアミノ酸で置換された1以上の天然TTR部位又は残基を有する、あるいは該配列に付加された1以上の残基を有する分子又は配列を包含する。一例として、アミノ酸配列37位のアラニン残基がシステイン残基で置換されたTTR変異体をTTR変異体(A37C)と称する;及びアミノ酸配列37位のアラニン残基及びアミノ酸配列83位のグリシン残基の両方がシステイン残基で置換されたTTR変異体をTTR変異体(A37C/G83C)と称する。
【0035】
1つの実施態様では、生物学的活性物質に融合したTTR又はTTR変異体を、TTR−生物学的活性物質融合タンパク質をさらに安定化し、それによって、生じる融合物の血清中半減期を上昇させる、第三のタンパク質又はタンパク質フラグメントに融合しうる。そのような方法及び組成物において使用できる付加的タンパク質又はそのフラグメントの例は、免疫グロブリンのFcドメイン又はCH2ドメイン、あるいは当業者がタンパク質の安定性を上昇させる性質を有すると認識する他の何らかのタンパク質ドメイン(例えば下記の実施例29)を包含する。そのようなタンパク質群は、TTR−生物学的活性物質融合タンパク質のカルボキシ又はアミノ末端に融合することができ、あるいはTTRと生物学的活性物質の間に配置することができる。所望の活性を促進するために該融合タンパク質の各々のドメインの間に、必要に応じて、リンカー又はスペーサーを配置できることが意図される。
【0036】
もう1つの実施態様では、本発明のTTR又はTTR変異体を生物学的活性物質に化学的に架橋することができる。タンパク質の架橋は、例えば確立された公表手順に従い、N−スクシニミジル3−(2−ピリジルジチオ)プロピオネート(SPDP)を使用することによって実施できる。さらなる架橋剤は容易に入手可能であり、当業者によって同定されうる。上記手順の詳細については、例えばKarpovskyら、J.Exp.Med.160.1686−1701(1984);Perezら、Nature,316、354−356(1985)又はTitusら、Journal of Immunology,139,3153−3158(1987)参照。
【0037】
もう1つの実施態様では、安定性及び/又は血清中の半減期を上昇させるように、分子を該融合タンパク質に共有結合することができる。例えば、好ましいTTR又はTTR変異体は、ポリエチレングリコール(PEG)などの水溶性ポリマーを用いて化学的修飾されうる。PEG基は、好都合ないかなる分子量であってもよく、直鎖又は分枝鎖のいずれでもよい。PEGの平均分子量は、好ましくは約2kDaから約100kDa、より好ましくは約5kDa−約50kDaの範囲であり、最も好ましくは約20kDaである。
【0038】
PEG基は一般に、アシル化、還元的アルキル化、マイケル付加、チオールアルキル化あるいはペグ部分の反応基(例えばアルデヒド、アミノ、エステル、チオール、ハロアセチル、マレイミド又はヒドラジノ基)を通しての標的化合物上の反応基(例えばアルデヒド、アミノ、エステル、チオール、ハロアセチル、マレイミド又はヒドラジノ基)への他の化学選択的複合体化/連結法によって、本発明の化合物に結合される。
【0039】
使用される他の水溶性ポリマーは、エチレングリコール/プロピレングリコールのコポリマー、カルボキシメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリ−1,3−ジオキソラン、ポリ−1,3,6−トリオキサン、エチレン/無水マレイン酸コポリマー、ポリアミノ酸(ホモポリマー又はランダムコポリマーのいずれか)及びデキストランを含む。
【0040】
対象ペプチド、対象タンパク質、TTR又はTTR変異体をコードするDNA分子は、Sambrookら(Molecular Cloning:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY[1989])及び/又はAusubelら編集、Current Protocols in Molecular Biology,Green Publishers Inc.及びWiley and Sons,NY(1994)に述べられているような周知の組換えDNAテクノロジーを使用して作製できる。対象タンパク質又はそのフラグメントをコードする遺伝子又はcDNAは、例えばゲノム又はcDNAライブラリーを適切なプローブでスクリーニングすることによって入手しうる。適切なプローブは、例えば、該プローブが選択されたハイブリダイゼーション条件下で対象タンパク質をコードする遺伝子とハイブリダイズするように、対象タンパク質をコードする遺伝子とある程度の相同性を有すると予想されるオリゴヌクレオチド、cDNAフラグメント又はゲノムDNAフラグメントを含む。DNAライブラリーをスクリーニングする代替的手段は、対象タンパク質をコードする遺伝子のポリメラーゼ連鎖反応「PCR」増幅によってである。PCRは、典型的には、少なくともプライマーの十分な部分が該遺伝子とハイブリダイズするように、増幅すべき遺伝子と十分な相同性を有すると考えられる配列を有するオリゴヌクレオチド「プライマー」を使用して実施される。
【0041】
あるいは、対象ペプチド又は対象タンパク質をコードする遺伝子は、Engelsら、Angew.Chem.Intl.Ed.,28:716−734(1989)によって述べられているような当業者に周知の方法を用いた化学合成によって作製しうる。これらの方法は、中でも特に、核酸合成のためのホスホトリエステル、ホスホルアミダイト及びH−ホスホネート法を含む。そのような化学合成のための好ましい方法は、標準ホスホルアミダイト化学を使用したポリマー支持合成である。典型的には、対象タンパク質をコードするDNAは数百ヌクレオチドの長さである。約100ヌクレオチド以上の大きさの核酸は、これらの方法を用いていくつかのフラグメントとして合成できる。次にそれらのフラグメントを連結して、対象とする完全長タンパク質をコードする遺伝子を形成することができる。通常、ポリペプチドのアミノ末端をコードするDNAフラグメントは、メチオニン残基をコードするATGを有する。このメチオニンは成熟形態の対象タンパク質上に存在してもよく又は存在しなくてもよい。該メチオニンは、細胞の内部で又は分泌過程の間に除去することができる。好ましいTTRポリペプチドは、大腸菌における発現を最適化し、好都合な制限部位を導入するように変化した核酸配列を有するTTRを含みうる。大腸菌における発現のためのコドン最適化についての一般的考察は、Kane,Curr.Opin.Biotechnol.,6:494−500(1995)に述べられている。
【0042】
ひとたび対象タンパク質とTTRポリペプチドをコードする遺伝子が得られれば、5’及び/又は3’末端で制限エンドヌクレアーゼ部位を創製する標準的方法を用いてそれらを修飾しうる。制限部位の創製は、該遺伝子が増幅及び/又は発現ベクターに適切に挿入されることを可能にする。制限部位の付加は典型的にはPCRを用いて実施され、そこでは各々のPCR反応の1つのプライマーは、典型的には、中でも特に所望の制限部位のヌクレオチド配列を含む。
【0043】
対象ペプチド又は対象タンパク質をコードする遺伝子又はcDNAは、宿主細胞における発現のために適切な発現ベクターに挿入することができる。該ベクターは、特に使用する宿主細胞において機能性であるように選択される(すなわち、該ベクターは、対象タンパク質をコードする遺伝子の増幅及び/又は発現が起こりうるように宿主細胞機構と適合性である)。
【0044】
典型的には、宿主細胞のいずれかにおいて使用されるベクターは、プロモーター(「5’側隣接配列」とも称される)及び他の調節エレメントならびにエンハンサー、複製起点エレメント、転写終結エレメント、リボソーム結合部位エレメント、発現しようとするポリペプチドをコードする核酸を挿入するためのポリリンカー領域、及び選択マーカーエレメントを含む。これらのエレメントの各々は下記で論じる。場合により、該ベクターは「タグ」DNA配列、すなわち、融合DNA構築物の5’又は3’末端のいずれかに位置するオリゴヌクレオチド配列を含みうる。タグDNAは、ヘキサHis、c−myc、FLAG(Invitrogen,San Diego,CA)などの分子又は別の小さな免疫原性配列をコードする。適切なリーディングフレーム内に置くとき、このタグは融合タンパク質と共に発現され、宿主細胞からの融合タンパク質の精製のためにアフィニティータグとして使用できる。場合により、該タグはその後、例えば選択したペプチダーゼを使用することなどの様々な手段によって精製融合タンパク質から除去することができる。
【0045】
該プロモーターは、同種(すなわち宿主細胞と同じ種及び/又は株に由来)、異種(すなわち宿主細胞の種又は株以外の種に由来)、ハイブリッド(すなわち2つ以上のソースからのプロモーターの組合せ)、合成でありえ、あるいは天然の対象タンパク質プロモーターでありうる。さらに、該プロモーターは構成的又は誘導的プロモーターでありうる。それ故に、該プロモーターのソースは、該プロモーターが宿主細胞機構において機能性であり、宿主細胞機構によって活性化されうることを条件として、いかなる単細胞原核又は真核の生物、脊椎又は無脊椎生物、あるいはいかなる植物であってもよい。
【0046】
本発明のベクターにおいて有用なプロモーターは、当技術分野において周知のいくつかの方法のいずれかによって入手しうる。典型的には、ここで有用なプロモーターは、マッピングによって及び/又は制限エンドヌクレアーゼ消化によってあらかじめ同定され、それ故適切な制限エンドヌクレアーゼを使用して適切な組織ソースから単離されうる。一部の場合には、プロモーターの完全なヌクレオチド配列が既知であり得る。その場合は、核酸合成又はクローニングについて上述した方法を用いてプロモーターを合成しうる。
【0047】
プロモーター配列の全部又は一部だけが既知である場合は、PCRを用いて及び/又は適切なオリゴヌクレオチド及び/又は同じ又は別の種からの5’側隣接配列フラグメントでゲノムライブラリーをスクリーニングすることによって、完全なプロモーターを入手しうる。
【0048】
本発明を実施するための適切なプロモーターは、luxプロモーター、lacプロモーター、アラビノースプロモーター、trpプロモーター、tacプロモーター、tnaプロモーター、合成λプロモーター(バクテリオファージλから)、及びT5又はT7プロモーターなどの誘導的プロモーターである。好ましいプロモーターは、lux及びlacプロモーターを含む。
【0049】
複製起点エレメントは、典型的には、市販のものを購入するか又は使用者によって構築された原核生物発現ベクターの一部である。一部の場合には、ある一定コピー数へのベクターの増幅が対象タンパク質又はポリペプチドの最適発現のために重要でありうる。また別の場合には、不変のコピー数が好ましい。いずれの場合も、必要条件を満たす複製起点を有するベクターは当業者によって容易に選択されうる。選択ベクターが複製起点部位を含まない場合は、既知の配列に基づいて化学合成し、ベクターに連結しうる。
【0050】
転写終結エレメントは、典型的には融合タンパク質DNA構築物の末端の3’側に位置し、融合ポリペプチドをコードするRNAメッセージの転写を終結させる働きをする。通常は、原核細胞内の転写終結エレメントは、G−Cに富むフラグメントとそれに続くポリT配列である。該エレメントはライブラリーから容易にクローン化されるか又はベクターの一部として市販のものが購入されるが、上述したような核酸合成のための方法を用いて容易に合成することもできる。
【0051】
発現ベクターは、典型的には選択マーカーをコードする遺伝子を含む。この遺伝子は、選択培地において増殖する宿主細胞の生存と増殖のために必要なタンパク質をコードする。典型的な選択マーカー遺伝子は、(a)抗生物質又は他の毒素、例えば原核生物宿主細胞についてはアンピシリン、テトラサイクリン、クロラムフェニコール又はカナマイシンに対する耐性を付与する、(b)該細胞の栄養要求性欠損を補う、又は(c)複合培地からは得られない必須栄養素を供給するタンパク質をコードする。好ましい選択マーカーは、カナマシン耐性遺伝子、アンピシリン耐性遺伝子、クロラムフェニコール耐性遺伝子及びテトラサイクリン耐性遺伝子である。
【0052】
原核生物では一般にシャイン−ダルガーノ配列と呼ばれる、リボソーム結合エレメントは、mRNAの翻訳の開始のために必要である。このエレメントは、典型的にはプロモーターの3’側及び融合タンパク質DNA構築物のコード配列の5’側に位置する。シャイン−ダルガーノ配列は多様であるが、典型的にポリプリンである(すなわち高いA−G含量を有する)。多くのシャイン−ダルガーノ配列が同定されており、それらの各々が上述した方法を用いて容易に合成でき、原核生物ベクターにおいて使用される。
【0053】
上述したエレメントの1つ以上が、使用するベクター内に既に存在しない場合は、それらを個々に入手して、ベクターに連結しうる。各々のエレメントを入手するために使用する方法は当業者に周知であり、上述した方法(すなわちDNAの合成、ライブラリースクリーニング等)に匹敵する。
【0054】
各々のエレメントは、連結するエレメントの末端とベクターの末端が連結のために適合性であるように、ベクターを適切な制限エンドヌクレアーゼで切断することによって個別にベクターに連結しうる。一部の場合には、十分な連結を得るために、連結する末端を「平滑化する」必要がありうる。平滑化は、最初に4つのヌクレオチドすべての存在下にクレノウDNAポリメラーゼ又はT4 DNAポリメラーゼなどの酵素を使用して「粘着末端」を充填することによって実施できる。この手順は当技術分野において周知であり、例えばSambrookら、前出に述べられている。
【0055】
あるいは、ベクターに挿入する2以上のエレメントを最初に互いに連結して(それらを互いに隣接して位置付けようとする場合)、その後ベクターに連結しうる。
【0056】
ベクターを構築するためのもう1つの方法は、様々なエレメントのすべての連結を1つの反応混合物中で同時に実施することである。この場合、エレメントの不適切な連結又は挿入により多くのナンセンス又は非機能性ベクターを生じることがあるが、選択マーカーの発現によって機能性ベクターを同定しうる。制限エンドヌクレアーゼによる消化又はDNA配列決定によって連結産物の正しい配列を確認することができる。
【0057】
ベクターを構築し、融合タンパク質DNA構築物をベクターの適切な部位に挿入した後、完成したベクターを融合タンパク質発現のために適切な宿主細胞に挿入しうる。
【0058】
本発明に適する宿主細胞は細菌細胞である。例えば、大腸菌の様々な菌株(例えばHB101、JM109、DH5α、DH10及びMC1061)は、組換えポリペプチドの作製において使用するための周知の宿主細胞である。細菌株の選択は、典型的には菌株と使用する発現ベクターが適合性であるように行われる。枯草菌(B.subtilis)、シュードモナス種、他のバチルス種、ストレプトミセス種等の様々な菌株も、適切な発現ベクターと共に本発明の実施において使用しうる。
【0059】
選択宿主細胞へのベクターの挿入(「形質転換」又は「トランスフェクション」とも称される)は、リン酸カルシウム沈殿又は電気穿孔などの方法を用いて実施しうる。選択される方法は、一部には使用する宿主細胞の種類に依存する。これらの方法及び他の適切な方法は当業者に周知であり、例えばSambrookら、前出に述べられている。
【0060】
ベクターを含む宿主細胞(すなわち形質転換された又はトランスフェクトされた宿主細胞)は、当業者に周知の1以上の標準培地を使用して培養しうる。選択された培地は、典型的には宿主細胞の増殖と生存のために必要なすべての栄養素を含む。大腸菌細胞を培養するための適切な培地は、例えばルリア肉汁(「LB」)、YT肉汁、SOB、SOC及び/又はテリフィック肉汁(「TB」)である。
【0061】
TTR遺伝子、対象ペプチド又はタンパク質をコードする遺伝子及び場合により、それら2個の遺伝子の間に位置するリンカーペプチドをコードするDNA分子を含む融合タンパク質をコードするDNA構築物を作製するいくつかの方法が存在する。
【0062】
1つの手順では、TTR遺伝子と対象タンパク質をコードする遺伝子(「融合パートナー遺伝子」)をいずれかの方向で連結することができる(例えば構築物の5’又は3’末端でTTR遺伝子)。リンカーDNA分子を含めようとする場合は、最初にそれを融合パートナー遺伝子の1つに連結し、次にその構築物を他の融合パートナー遺伝子に連結することができる。連結は、典型的にはDNAリガーゼ酵素を製造者の指示に従って使用して実施される。
【0063】
別個の手順によって、選択されたベクターへの1つの融合パートナー遺伝子の最初の連結を行い、その後で他の融合パートナー遺伝子を、最初の融合パートナー遺伝子の3’側又は5’側のいずれかの位置でベクターに連結することができる。リンカーDNA分子を含めようとする場合は、その遺伝子をベクターに連結する前又は連結した後に、そのリンカーDNA分子をいずれかの融合パートナー遺伝子に連結しうる。
【0064】
本発明のTTR−TMPは、既存の巨核球/血小板欠損又は予想される巨核球/血小板欠損(例えば予定されている手術又は血小板提供のため)を含むものとして一般的に知られる状態を治療するために使用できる。そのような状態は、通常、インビボでの活性Mp1リガンドの欠損(一時的又は永続的)の結果である。血小板欠損についての一般的用語は血小板減少症(thrombocytopenia)であり、それ故本発明の方法及び組成物は一般に、それを必要とする患者において血小板減少症を治療するために使用しうる。血小板減少症(血小板欠損)は、化学療法及び様々な薬剤による他の療法、放射線治療、手術、偶発的血液損失及び他の同定疾患状態を含む、様々な理由のために存在しうる。
【0065】
血小板減少症を含み、本発明に従って治療しうる例示的な具体的な疾患状態は:再生不良性貧血、特発性血小板減少症、血小板減少症をもたらす転移性腫瘍、全身性エリテマトーデス、巨脾腫、ファンコーニ症候群、ビタミンB12欠損症、葉酸欠損症、メイ−ヘグリン異常、ヴィスコット−オールドリッチ症候群及び発作性夜間血色素尿症である。また、AIDSのためのある種の治療も血小板減少症を生じさせる(例えばAZT)。ある種の創傷治癒障害も、血小板数の上昇に起因すると考えられる。
【0066】
例えば将来の手術による、予想される血小板欠損に関しては、血小板が必要となる数日前から数時間前に本発明の化合物を投与することができる。急性状況、例えば偶発的な大量失血に関しては、本発明の化合物を血液又は精製血小板と共に投与することができる。
【0067】
本発明のTMP化合物はまた、そのような細胞がMp1受容体を発現することが認められる場合には、巨核球以外のある種の細胞型を刺激する上でも有用でありうる。Mp1リガンドによる刺激に対して応答性である、Mp1受容体を発現するそのような細胞に関連する状態も、本発明の範囲内である。
【0068】
本発明のTMP化合物は、血小板又は血小板前駆細胞の産生が所望される、又はc−Mp1受容体の刺激が所望されるいかなる状況においても使用しうる。それ故例えば、本発明の化合物は、血小板、巨核球等の必要性が存在する哺乳動物において何らかの状態を治療するために使用しうる。そのような状態は次の例示的ソース:国際公開公報第95/26746号、国際公開公報第95/21919号、国際公開公報第95/18858号、国際公開公報第95/21910号に詳細に記述されており、本明細書中で援用されている。
【0069】
本発明の化合物はまた、血小板及び/又は巨核球及び関連細胞の生存性又は保存寿命を維持する上でも有用でありうる。従って、そのような細胞を含む組成物中に1以上のそのような化合物の有効量を含めることは有用であり得る。
【0070】
本発明の治療方法、組成物及び化合物はまた、血小板欠損のほかに他の症状によって特徴付けられる疾患状態の治療においても、単独であるいは他のサイトカイン、可溶性Mp1受容体、造血因子、インターロイキン、増殖因子又は抗体と組み合わせて使用しうる。本発明の化合物は、IL−3又はGM−CSFなどの一般的な造血刺激物質と組み合わせて、一部の形態の血小板減少症を治療する上で有用であることが明らかになると予想される。他の巨核球刺激因子、すなわちmeg−CSF、幹細胞因子(SCF)、白血病阻害剤(LIF)、オンコスタチンM(OSM)又は巨核球刺激作用を有する他の分子も、Mp1リガンドと共に使用しうる。そのような同時投与のためのさらなる例示的サイトカイン又は造血因子は、IL−1α、IL−1β、IL−2、IL−3、IL−4、IL−5、IL−6、IL−11、コロニー刺激因子−1(CSF−1)、SCF、GM−CSF、顆粒球コロニー刺激因子(G−CSF)、EPO、インターフェロン−α(IFN−α)、コンセンサスインターフェロン、IFN−β又はIFN−γを含む。さらに、ひとたび巨核球が成熟形態に達すれば、巨核球を血小板へと細分する作用を有すると思われる、可溶性の哺乳動物Mp1受容体の有効量を同時に又は連続的に投与することは有用であると考えられる。それ故、本発明の化合物の投与(成熟巨核球の数を高めるため)とそれに続く可溶性Mp1受容体の投与(リガンドを不活性化して成熟巨核球が血小板を産生することを可能にするため)は、血小板産生を刺激する特に有効な手段であると予想される。適切な用量は、治療組成物中のそのような付加的成分を相殺するように調整する。治療される患者の経過は従来の方法によってモニターすることができる。
【0071】
2型糖尿病患者としても知られる、インスリン非依存性糖尿病(NIDDM)においては、グルカゴン様ペプチド−1(GLP−1)の投与は抗糖尿病特性を有する。しかし、GLP−1は、インビボでのその放出後にジペプチジルペプチダーゼIV(DPPIV)によって速やかに分解される。それ故、GLP−1ペプチド又はその変異体を本発明のTTRポリペプチドに融合してGLP−1を安定化し、インビボでその半減期を上昇させうることは本発明の利点である。従って、本発明のもう1つの実施態様では、ここで述べるTTR−GLP1融合タンパク質を、II型糖尿病としても知られるインスリン非依存性糖尿病(NIDDM)を含むことが一般的に知られる状態を治療するために使用することができる。
【0072】
当業者は、GLP−1ペプチドの配列は、そのインスリン向性作用を保持するように変化させうることを認識する。当技術分野で既知のそのような変異型の同定例は、例えば、GLP−1(7−34)、(7−35)、(7−36)又は(7−37)、Gln9−GLP−1(7−37)、D−Gln9−GLP−1(7−37)、Thr16−Lys18−GLP−1(7−37)及びLys18−GLP−1(7−37)を含む。GLP−1変異体のさらなる例は、本明細書中に参考として援用されている、米国特許第5,118,666号、同第5,545,618号、同第5,977,071号及び国際公開公報第02/46227号及びAdelhorstら、J.Biol.Chem.269:6275(1994)に述べられている。従って、いかなるGLP−1ペプチドも、該GLP−1融合タンパク質がそのコグネイト受容体を通してシグナルに結合し、シグナルを誘導することができる限り、本発明の融合タンパク質を生成するために使用できる。受容体結合及び活性化は標準的アッセイによって測定できる(米国特許第5,120,712号)。
【0073】
患者の血中グルコースを正常化するために有効な融合タンパク質の用量は、下記でより詳細に論じるように、被験者の体重、年齢、血中グルコースを調節する能力欠如の重症度、融合タンパク質の投与経路、バイオアベイラビリティー、薬物動態プロフィール及び製剤を含む多くの因子に依存する。
【0074】
本発明の治療方法、組成物及び化合物はまた、単独で、あるいはインスリン、DPPIV阻害剤等を含むがこれらに限定されない他の糖尿病治療と組み合わせて使用しうる。GLP−1融合タンパク質の用量は、治療組成物中のそのような付加的成分を相殺するように調整する。治療される患者の経過は、例えば血中グルコースレベルのモニターなどの従来の方法によってモニターすることができる。
【0075】
本発明はまた、本発明の化合物の医薬組成物を提供する。そのような医薬組成物は、注入投与用であるか、あるいは経口、経鼻、経皮あるいは、例えば静脈内、皮内、筋肉内、乳房内、腹腔内、髄腔内、眼内、眼球後、肺内(例えばエーロゾル投与薬剤)又は皮下注射(長期放出のためのデポー投与を含む)による投与;舌下、肛門、膣による投与、あるいは例えば脾臓被膜下、脳又は角膜内に包埋する外科的移植による投与を含む他の形態の投与用でありうる。治療は、単回用量又は一定期間にわたる多回用量で構成されうる。一般に、医薬適合性の希釈剤、防腐剤、安定剤、可溶化剤、乳化剤、アジュバント及び/又は担体と共に本発明の化合物の有効量を含有する医薬組成物は、本発明に包含される。そのような組成物は、様々な緩衝剤含量、pH及びイオン強度の希釈剤(例えばTris−HCl、酢酸塩、リン酸塩、クエン酸塩等);界面活性剤及び可溶化剤(例えばツイーン80、ポリソルベート80等)、抗酸化剤(例えばアルコルビン酸、メタ重亜硫酸ナトリウム)、防腐剤(例えばチメロサール、ベンジルアルコール)及び充填物質(例えばラクトース、マンニトール)などの添加物;ポリ乳酸、ポリグリコール酸等のような高分子化合物の微粒子製剤又はリポソームへの物質の組込みを含む。ヒアルロン酸も使用でき、これは循環中の存在期間の持続を促進する作用を及ぼしうる。該医薬組成物は、場合により、モノラウリン酸ポリオキシエチレンソルビタン、ステアリン酸マグネシウム、メチル及びプロピルヒドロキシベンゾエート、デンプン、スクロース、デキストロース、アカシアゴム、リン酸カルシウム、鉱物油、ココアバター及びカカオ脂を含むがこれらに限定されない、製薬ビヒクル、賦形剤又は媒質として働くさらなる他の医薬適合性の液体、半固体又は固体希釈剤を含有しうる。そのような組成物は、本発明のタンパク質及び誘導体の物理的状態、安定性、インビボでの放出速度及びインビボでのクリアランス速度に影響しうる。例えば、本明細書中に参考として援用されている、Remington’s Pharmaceutical Sciences,第18版(1990,Mack Publishing Co.,Easton,PA 18042)p.1435−1712参照。該組成物は、液体形態で製造されるか、又は凍結乾燥形態のような乾燥粉末でありうる。経皮的製剤と同様に、移植可能な持続放出性製剤も考慮される。
【0076】
制御放出製剤は望ましいと考えられる。拡散又は浸出機序による放出を可能にする不活性マトリックス、例えばゴムに薬剤を組み込むことができる。緩やかに変性するマトリックス、例えばアルギン酸塩、多糖類も製剤に組み込みうる。この治療薬の制御放出のもう1つの形態は、Oros治療システム(Alza Corp.)に基づく方法による、すなわち水が入るのを許容し、浸透作用によって単一の小さな開口部を通して薬剤を押し出すことができる半透性膜に薬剤を入れる。一部の腸溶剤皮も遅延放出作用を有する。
【0077】
またここでは、本発明のタンパク質(又はその誘導体)の肺への送達も考慮される。該タンパク質(又は誘導体)は、吸入される間に哺乳類の肺へと送達され、肺上皮内層を横切って血流に入る。(これについての他の報告は、Adjeiら、Pharmaceutical Research 7:565−569(1990);Adjeiら、International Journal of Pharmaceutics 63:135−144(1990)(酢酸ロイプロリド);Braquetら、Journal of Cardiovascular Pharmacology 13(補遺5):s.143−146(1989)(エンドセリン−1);Hubbardら、Annals of Internal Medicine 3:206−212(1989)(1−アンチトリプシン);Smithら、J.Clin.Invest.84:1145−1146(1989)(1−プロテイナーゼ);Osweinら、「Aerosolization of Proteins」,Proceedings of Symposium on Respiratory Drug Delivery II,Keystone,Colorado,1990年3月(組換えヒト成長ホルモン);Debsら、The Journal of Immunology 140:3482−3488(1988)(インターフェロン及び腫瘍壊死因子)及びPlatzら、米国特許第5,284,656号(顆粒球コロニー刺激因子)を含む)。
【0078】
それらのすべてが当業者に周知である、ネブライザ、定量噴霧式吸入器及びパウダー式吸入器を含むがこれらに限定されない、治療製品の肺送達用に設計された広範囲の機械的装置が、本発明の実施における使用のために考慮される。
【0079】
本発明の実施に適する市販装置の一部の同定例は、Mallinckrodt,Inc.,St.Louis,Missouriによって製造される、Ultraventネブライザ;Marquest Medical Products,Englewood,Coloradoによって製造される、Acorn IIネブライザ;Glaxo Inc.,Research Triangle Park,North Carolinaによって製造される、Ventolin定量噴霧式吸入器;及びFisons Corp.,Bedford,Massachusettsによって製造される、Spinhalerパウダー式吸入器である。
【0080】
そのような装置はすべて、本発明の化合物を配分するのに適した製剤の使用を必要とする。典型的には、各々の製剤は使用する装置の種類に特異的であり、治療において有用な希釈剤、アジュバント及び/又は担体に加えて、適切な推進剤の使用を含みうる。
【0081】
本発明の化合物は、最も好都合には、遠位肺への最も有効な送達のために10μm(又はミクロン)未満、最も好ましくは0.5から5μmの平均粒径を有する微粒子形態で製造すべきである。
【0082】
担体は、トレハロース、マンニトール、キシリトール、スクロース、ラクトース及びソルビトールなどの炭水化物を含む。製剤中での使用のための他の成分は、DPPC、DOPE、DSPC及びDOPCを含みうる。天然又は合成界面活性剤を使用してもよい。ポリエチレングリコールを使用してもよい(該タンパク質又は類似体を誘導体化する上でのその使用は別として)。シクロデキストランなどのデキストランを使用してもよい。胆汁酸塩及び他の関連エンハンサーが使用しうる。セルロース及びセルロース誘導体も使用しうる。緩衝製剤における使用のような、アミノ酸も使用しうる。
【0083】
上述した状態を治療するための方法に関わる投与レジメは、薬剤の作用を変化させる様々な因子、例えば患者の年齢、状態、体重、性別及び食事、感染の重症度、投与時間及び他の臨床的因子を考慮して、主治医によって決定される。一般に、用量は、1日1回投与又は等しい用量をより長い又は短い間隔で、例えば1日おき、週に2回、週に1回、あるいは1日2回又は3回投与される、1日当り体重1キログラムにつき本発明の化合物0.1μgから100mg、好ましくは0.1から1000μg/kg、より好ましくは0.1から150μg/kgの範囲内とすべきである。
【0084】
本発明の化合物は、初回ボーラス及びそれに続く、薬剤製品の治療循環レベルを維持するための持続注入によって投与しうる。もう1つの例として、本発明の化合物は単回用量として投与しうる。当業者は、良質の医療のための原則及び個々の患者の臨床状態によって決定される、有効な用量及び投与レジメを容易に最適化する。投与頻度は、薬剤の薬物動態パラメータ及び投与経路に依存する。最適医薬製剤は、投与経路と所望用量に依存して当業者によって決定される。例えば、その開示が本明細書中に参考として援用されている、Remington’s Pharmaceutical Sciences,第18版(1990,Mack Publishing Co.,Easton,PA 18042)p.1435−1712参照。投与経路に依存して、体重、体表面積又は器官の大きさに従って適切な用量を算定しうる。
【0085】
適切な投与量は、適切な用量−反応データと共に、血清中レベルを測定するための確立されたアッセイの使用を通して確認しうる。最終的な投与レジメは、薬剤の作用を変化させる様々な因子、例えば薬剤の特異的活性、損傷の重症度及び患者の応答性、患者の年齢、状態、体重、性別及び食事、感染の重症度、投与時間及び他の臨床的因子を考慮して、主治医によって決定される。試験が実施されると共に、様々な疾患及び状態についての適切な用量レベル及び治療期間に関するさらなる情報がもたらされる。
【0086】
下記の実施例は説明だけを目的とするものであり、いかなる意味においても本発明を限定すると解釈されるべきではない。
【実施例1】
【0087】
この実施例は、天然組換えヒトトランスサイレチン(TTR)及び次のTTR変異体:TTR(C10A)、TTR(C10A/A37C)、TTR(C10A/D38C)、TTR(C10A/A81C)、TTR(C10A/G83C)、及びTTR(C10A/K15A/G83C)についてのDNAの作製を述べる。
【0088】
発現プラスミドpAMG21は、1996年7月24日に寄託された、受入番号98113の下にATCCより入手可能である(pAMG21の説明については1997年7月3日公開のPCT国際公開公報第97/23614号参照)。TTR、TTR変異体又はTTR−ペプチド融合物をコードするDNA配列を、pAMG21内のLuxPRプロモーターの制御下に置いた。
【0089】
細菌宿主GM221は、初期ebg領域内の温度感受性λリプレッサーcI857s7及び後期ebg領域(68分)内のlacIQリプレッサーの両方を含むように改変された大腸菌K−12株である。これら2個のリプレッサー遺伝子の存在は、この宿主を多様な発現系と共に使用することを可能にするが、これらのリプレッサーは共にluxPRからの発現には無関係である。非形質転換宿主は抗生物質耐性を持たない。cI857s7遺伝子のリボソーム結合部位を、強化(enhanced)RBSを含むように修飾した。それを、介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド1170位と1411位の間のebgオペロンに挿入した。この構築物を、F’tet/393へのMMebg−cI857s7強化RBS No.4と呼ばれる組換えファージを使用して染色体に送達した。組換えと分離(resolution)後、上述した染色体挿入物だけが細胞内に残る。これを新たにF’tet/GM101と命名した。次に介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド2493位と2937位の間のebgオペロンにlacIQ構築物を送達することによってF’tet/GM101を修飾した。その構築物を、F’tet/GM101へのAGebg−lacIQ No.5と呼ばれる組換えファージを使用して染色体に送達した。組換えと分離後、上述した染色体挿入物だけが細胞内に残る。これを新たにF’tet/GM221と命名した。F’tetエピソームを、LB中25μg/mlの濃度のアクリジンオレンジを使用して該菌株からキュアリングした。キュアリングした菌株をテトラサイクリン感受性と同定し、GM221として保存した。
【0090】
オリゴヌクレオチド(各々1.0nm)をホスホルアミダイト法によって合成した。ヌクレオチドを、一部の場合は、大腸菌における最適発現のために変性させた。これらのコドン変化はアミノ酸配列の変化を生じさせなかった。この実施例において使用した各々のオリゴヌクレオチドを表1に列記する。
【0091】
Expand Long Polymeraseを製造者(Boehringer Mannheim)のプロトコールに従って使用してPCRを実施した。PCR産物をアガロースゲル電気泳動によって確認し、精製して、NdeI及びXhoI(New England Biolabs)で消化した。発現ベクターpAMG21を同様に消化して、その後で子ウシ腸ホスファターゼ(Boehringer Mannheim)で処理した。ベクターと挿入物をアガロースゲルから精製し、その後で混合して、T4 DNAリガーゼ(New England Biolabs)によって連結した。連結は4℃で2時間実施した。各々の連結混合物を、Biorad GenePulser(Biorad Laboratories)をギャップ長約2mmのキュベット中2.5V、25μFD及び200オームで使用して、電気穿孔によって上述した宿主菌株GM221に形質転換した。電気穿孔後、ルリア肉汁(LB)1ml中37℃で約1時間、静かに振とうしながら細胞を回収した。形質転換混合物の全体を、50μg/mlカナマイシンを含むLB寒天上にプレートした。隣接ベクター配列に対するオリゴヌクレオチドを使用したPCRにより、所望される分子量の存在に関してコロニーをスクリーニングした。そのPCR産物をアガロースゲル電気泳動によって評価した。陽性クローンを、組換えタンパク質産物を生産する能力に関してさらにスクリーニングし、ヌクレオチド配列決定によって最終的に確認した。
【0092】
TTRのDNA及びアミノ酸配列は既知である(Mita,S.ら、Biochem.Biophys.Res.Commun.,124(2):558−564[1984])。これらの配列は受入番号K02091としてGenbankに寄託されている。シグナルペプチドを除く天然TTRのcDNAを、ヒト肝から誘導したcDNAライブラリー(Clontech)からクローン化した。詳細には、TTRの5’末端の8個のコドンをコードするオリゴヌクレオチド(オリゴ2693−79)及び終止コドンを含むTTR3’末端の7個のコドンをコードするオリゴヌクレオチド(オリゴ2693−80)を合成し、ヒト肝cDNAライブラリーを鋳型として使用してExpand Longポリメラーゼで完全長成熟TTRを増幅するために使用した。生じたPCRフラグメントをNdeI及びXhoIで消化し、ゲル精製して、NdeI/XhoI制限発現ベクターpAMG21と連結した。4℃で2時間後、連結混合物を電気穿孔によってGM221細胞に導入した。単一コロニーを採取し、プラスミドDNAを作製して配列決定した。1個の生じたプラスミド(菌株No.5316)は、天然TTRの正しいDNA配列(プラスN末端のメチオニン)を有することを示し、それを発現のために使用した。このDNA配列を配列番号2に同定する。
【0093】
上記オリゴヌクレオチド2693−80とオリゴヌクレオチド2820−88を使用することによって突然変異体TTR(C10A)を作製した(10番目の位置のコドンCysがAlaに置換された天然TTRの最初の11個のコドンを含む)。PCR手順及び発現菌株を選択するための工程は上述したのと同様であった。生じた菌株(菌株No.5619)は、配列番号3に同定するDNA配列を有していた。
【0094】
プラスミド5619を、次の位置:A37、D38、A81及びG83のアミノ酸をシステインのアミノ酸で置換することによってさらに修飾した。下記で述べるように、所望突然変異を内包する相補的オリゴヌクレオチドの各々の対を、部位特異的突然変異誘発のために設計された標準的な2段階PCR手順において、上述したTTR5’及び3’プライマーと共に使用した。菌株5619由来のプラスミドを鋳型として使用する20サイクルのPCRにおいて、フォワードプライマーの各々をTTR3’プライマーと共に使用し、リバースプライマーの各々をTTR5’プライマーと共に使用した。生じたPCR増幅5’及び3’フラグメントを混合し、完全長突然変異体を生成するための第二段階PCRのための鋳型として使用した。その後のクローニング及び配列決定手順は既に述べたものと同様であった。次のオリゴヌクレオチドを使用した:TTR(A37C)フォワード(オリゴ2823−91);TTR(A37C)リバース(オリゴ2823−92);TTR(D38C)フォワード(オリゴ2823−93);TTR(D38C)リバース(オリゴ2823−94);TTR(A81C)フォワード(オリゴ2823−95);TTR(A81C)リバース(オリゴ2823−96);TTR(G83C)フォワード(オリゴ2823−97);TTR(G83C)リバース(オリゴ2823−98)。該プラスミドを含む生じた大腸菌株は次のように説明される:TTR(C10A/A37C)(菌株5641)は配列番号4に同定するDNA配列を有していた。TTR(C10A/D38C)(菌株5642)は配列番号5に同定するDNA配列を有していた。TTR(C10A/A81C)(菌株5643)は配列番号6に同定するDNA配列を有していた。TTR(C10A/G83C)(菌株5651)は配列番号7に同定するDNA配列を有していた。
【0095】
菌株5651の15位のLysを、菌株5641、5642、5643及び5651について述べたのと同様の手順により、オリゴヌクレオチド2953−67及び2953−68を使用してさらにAlaに突然変異誘発した。生じた菌株、TTR(C10A/K15A/G83C)(菌株5895)は配列番号8に同定するDNA配列を有していた。
【0096】
【表1】
【実施例2】
【0097】
この実施例は、様々なTMP−TTR融合物の作製を述べる。TTRとTMPを含むいくつかの融合タンパク質を作製した。この実施例において使用した各々のオリゴヌクレオチドを表2に列記する。
【0098】
TMPを含むフラグメントを、最初に、TMPの最初の7個のコドンに加えてNdeI部位を含む12ヌクレオチドの5’延長部をコードするオリゴヌクレオチド2743−96、及び天然TTRの最初の7個のコドンと対象TMPの最後の7個のコドンをコードするオリゴヌクレオチド2743−97を使用して、完全長TMP−Fc融合物をコードするプラスミドを内包する菌株から増幅した(PCT国際公開公報第00/24770号参照)。生じたPCRフラグメントを菌株5619由来のプラスミドと混合し、その混合物をオリゴヌクレオチドプライマー2743−96及び2693−80のための鋳型として使用して、完全長TMP−TTRを増幅した。クローニング及び発現については上述したのと同様の手順を用いた。生じた菌株、TTR−TMP(菌株5513)は配列番号9に同定するDNA配列を有していた。
【0099】
次に、TMPをそれぞれ菌株5641、5642、5643及び5651のN末端に導入した。プラスミド5513をXbaIで消化し、生じたTMP及びTTR(C10A)の最初の18個のコドンを含むXbaI/XbaI挿入物をゲル精製し、5641、5642、5643及び5651由来の、XbaIで切断し、ホスファターゼ処理してゲル精製したベクターと連結した。各々の融合物について正しい方向を選択するためにDNA塩基配列決定を実施した。該プラスミドを含む生じた大腸菌株は次のように説明される:TMP−TTR(C10A/A37C)(菌株5704)は配列番号10に同定するDNA配列を有していた。TMP−TTR(C10A/D38C)(菌株5705)は配列番号11に同定するDNA配列を有していた。TMP−TTR(C10A/A81C)(菌株5706)は配列番号12に同定するDNA配列を有していた。TMP−TTR(C10A/G83C)(菌株5707)は配列番号13に同定するDNA配列を有していた。
【0100】
【表2】
【実施例3】
【0101】
この実施例は、PTH(1−34)−TTR(C10A/K15A/G83C)融合物の作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表3に列記する。
【0102】
2個の新しいオリゴヌクレオチド、ヒトPTHの最初の7個のコドンをコードするオリゴヌクレオチド2694−01、及びTTRの最初の7個のコドンとPTHのアミノ酸28−34をコードするオリゴヌクレオチド2694−03を合成して、融合物を作製した。オリゴヌクレオチド2694−01及び2694−03を上述したように20サイクルのPCR手順において使用して、TTRリンカーを有するPTH(1−34)を増幅した。この反応のための鋳型は、PTH1−34−Fc融合物をコードするプラスミドを内包する菌株であった(PCT国際公開公報第01/81415号参照)。生じたPCR混合物を菌株5895と混合して、プライマー2694−01及び2693−80を用いて完全長PTH(1−34)−TTR(C10A/K15A/G83C)を増幅するための鋳型として使用した。配列確認後、この新しいプラスミドを含む生じた発現菌株をPTH−TTR(C10A/K15A/G83C)(菌株5920)と称した。この菌株は配列番号14に同定するDNA配列を有していた。
【0103】
【表3】
【実施例4】
【0104】
この実施例は、IL−1ra−TTR(C10A)融合物及びTTR(C10A)−GSGS−IL−1ra融合物の作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表4に列記する。
【0105】
IL−1ra−TTR(C10A)融合物を作製するために、2個のオリゴヌクレオチド、ヒトタンパク質IL−1raの最初の7個のコドンをコードするオリゴヌクレオチド2823−13、及びIL−1raの最後の7個のアミノ酸とTTRの最初の7個のアミノ酸をコードするオリゴヌクレオチド2823−14を合成した。IL−1raを発現する菌株由来のプラスミド(PCT国際公開公報第91/08285号参照)を、オリゴヌクレオチド2823−13及び2823−14を用いて増幅した。生じたPCR産物を、菌株5619から精製したプラスミドと混合し、オリゴヌクレオチドプライマー2823−13及び2693−80を用いて完全長IL−1−ra−TTR(C10A)を増幅するための鋳型として使用した。PCR産物を上述したようにクローン化し、配列決定して、発現させた。この新しいプラスミドを含む生じた菌株をIL−1ra−TTR(C10A)(菌株5644)と称した。この菌株は配列番号15に同定するDNA配列を有していた。
【0106】
TTR(C10A)−IL−1raを作製するために、次の2個のオリゴヌクレオチド、すなわち、その間にGSGSリンカーを導入した、TTRの最後の7個のアミノ酸とIL−1−raの最初の7個のアミノ酸をコードするオリゴヌクレオチド2787−32及びIL−1−raの最後の7個のコドンをコードするオリゴヌクレオチド2787−33を合成した。これら2個のオリゴヌクレオチドプライマーを用いてプラスミド2693を増幅し、生じたPCR産物をプラスミド5619と混合して、これらを共に、プライマー2787−33及び2693−79を用いて完全長TTR(C10A)−IL−1raを増幅するための鋳型として使用した。PCR産物を上述したようにクローン化し、配列決定して、発現させた。この新しいプラスミドを含む生じた菌株をTTR(C10A)−IL−1ra(菌株5645)と称した。この菌株は配列番号16に同定するDNA配列を有していた。
【0107】
【表4】
【実施例5】
【0108】
この実施例は、TTR(C10A/G83C)−ブラジキニンの作製を述べる。この実施例において使用した各々のオリゴヌクレオチドを表5に列記する。
【0109】
菌株5651から精製したプラスミドを、オリゴヌクレオチドプライマー2693−79及びPstI制限部位を含むTTR3’プライマーであるオリゴヌクレオチドプライマー2943−47によるPCRのために使用した。このPCR産物をゲル精製し、NdeI及びPstIで制限消化した。生じたDNAフラグメントを、NdeI及びXhoIで消化したAMG21、及びGSGSGリンカーをコードするアニーリングしたオリゴヌクレオチド2943−48、及びPstI5’とXhoI3’重複末端を有するブラジキニンアンタゴニストペプチドKRPPGFSPLをコードするオリゴヌクレオチド2943−49を含む連結混合物中で使用した。GM121をこの連結産物で形質転換し、DNAをカナマイシン耐性コロニーから精製した。次に該耐性コロニーにおいてDNA配列を確認した。確認した菌株を30℃で増殖させ、下記に述べる10リットル発酵物において発現を誘導した。その新しい菌株をTTR(C10A/G83C)−ブラジキニン(菌株5914)と称した。この菌株は配列番号17に同定するDNA配列を有していた。
【0110】
【表5】
【実施例6】
【0111】
この実施例は、大腸菌におけるTTR及びTTR融合構築物の組換え発現を述べる。新たに構築したTTR又はTTR融合物の各々を、最初に16℃から37℃の範囲の温度で可溶性発現に関して検査した。この目的のために、TTR又はTTR融合物の各々を発現するGM221の培養物(25ml)を、50μg/mlカナマイシンを補足したLB肉汁中37℃で、600nmでの光学密度(OD)が0.5から1.0に達するまで増殖させた。次に培養物を、それぞれ16℃、20℃、25℃、30℃、34℃及び37℃の温度設定で振とう機に入れた。合成自己誘導物質N−(3−オキソヘキサノイル)−DL−ホモセリンラクトンを最終濃度20ng/mlまで培地に添加した後、luxPRプロモーターからの遺伝子産物発現の誘導を実施した。6時間後、細菌培養物を封入体の存在に関して顕微鏡で検査した。TTR及びその融合物のために培養物を30℃未満の温度で増殖させることにより、しばしば可溶性又は部分的可溶性の発現を達成することができ、この温度を大量発現のために使用した。可溶性発現が達成できなかった場合は、発現のレベルが最も高かった温度を大量振とう機又は発酵槽のために使用した。
【0112】
大量発現は、通常は4リットルフラスコで実施した。LB1リットルを含む4〜8台の4リットル振とう機にTTR又はその融合株の一晩培養物を接種した。発現は基本的に上述したように実施した。細胞を遠心分離によって収集した。
【0113】
無菌手法を用いる発酵段階は、無菌ルリア肉汁500mLを含む振とうフラスコにおいて生産した菌株の種培養(seed culture)からの接種で開始する。この培養物が適切な細胞密度(600nmで0.8−2)に達したとき、その内容物を使用して、複合ベースの増殖培地10リットルを含む20リットル発酵槽に接種した。発酵槽を30℃、pH7に維持し、溶解酸素レベルを30%飽和に保持する。細胞密度が600nmで10−12OD単位の光学密度に達したとき、N−(3−オキソヘキサノイル)ホモセリンラクトンの添加によって培養物を誘導した。誘導後6時間目に遠心分離によって発酵槽から細胞を収集した。
【実施例7】
【0114】
この実施例は、TTR(C10A/G83C)−ブラジキニンの精製を述べる。−80℃で保存したクローン5914からの大腸菌ペースト約193gを、50mMトリスHCl、5mM EDTA、pH8.0 1447ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、該懸濁液を12,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物(図1、レーン2)を11,325×g、4℃で50分間遠心分離した。上清を可溶性分画として採取した。該可溶性分画をポリプロピレンボトル中65℃水浴で30分間加熱し、その時点で内容物の温度は63℃であった。該可溶性分画を11,325×g、4℃で50分間遠心分離した。上清を熱可溶物(Heat Soluble)として採取した(図1、レーン3)。該熱可溶性分画を、2つのプレフィルターを備えた0.45μm酢酸セルロースフィルターでろ過し、室温(約23℃)で、Q緩衝液A(20mMトリスHCl、2.5mM EDTA、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(Q−sepharose fast flow)(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に20ml/分で負荷した。カラムを20ml/分のQ緩衝液A約2300mlで洗った。Qカラムを、60%Q緩衝液B(20mMトリスHCl、1M NaCl、2.5mM EDTA、pH8.0)への15カラム容積の直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(1150ml)(図1、レーン4)に貯留し、DTT1.77gを加えた。そのQプールを室温(約23℃)で30分間静かに攪拌した。該Qプールに、3.8M硫酸アンモニウムpH7.0 410mlを緩やかに加え、1M HClの緩やかな添加によってpHを約7.5から7.0に低下させた。次に該Qプールの約2分の1を、10ml/分でP緩衝液A(50mM NaH2PO4、1M硫酸アンモニウム、pH7.0)中84mlフェニルセファロース高速カラム(内径2.6cm)(Amersham Pharmacia Biotech)に負荷した。カラムをP緩衝液A約170mlで洗い、次いで50%、80%及び100%P緩衝液B(50mM NaH2PO4、pH7.0)を用いて3段階溶出させた。次に、該Qプールの残りの半分を最初の半分と同じプロトコールを用いて処理した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Pプール(260ml)(図1、レーン5)に貯留し、そのPプールを、直径20.4mmの8kDaカットオフ透析チューブ(Spectrum Laboratories Inc.,Rancho Dominguez,CA)を使用して室温(約23℃)で2時間、HA緩衝液A(10mM NaH2PO4、pH7.0)4Lに透析した。透析緩衝液を新鮮なHA緩衝液A4Lに交換し、さらに約15時間透析を継続した。該Pプールを透析から取り出し、1M DTT 600μlを加え、その後室温(約23℃)で約1時間インキュベートした。Pプールを、HA緩衝液A中10ml/分で105ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを10ml/分のHA緩衝液A約210mlで洗い、次いで12.5%、25%、50%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の4段階で溶出させた。そのフロースルーをHAプール(340ml)(図1、レーン6)として貯留し、DTT 524mgを加えて、その後室温(約23℃)で1時間インキュベートした。
【0115】
該HAプールの約2分の1を、10ml/分で47mlソース15Qカラム(内径2.6cm)(Amersham Pharmacia Biotech)に負荷し、次にQ−緩衝液A約250mlで洗った。10%から50%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。該HAプールの残りの半分を最初の半分と同じプロトコールを用いて処理した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Q2プール(260ml)に貯留し、10kDa膜を有する攪拌式セルを用いて約75mlに濃縮した。次にQ2プール(図1、レーン7)を0.22μm酢酸セルロースフィルターでろ過し、算定比吸光係数18,450 M−1 cm−1を用いてタンパク質濃度を16.9mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.52と測定されたので、無視しうると判定した。
【実施例8】
【0116】
この実施例は、TTR(C10A/G83C)のC末端又はN末端のいずれかにペプチドを融合することがそのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。20mMトリスpH8.0及び約250mM NaCl中のTTR(C10A/G83C)、PTH−TTR(C10A/K15A/G83C)及びTTR(C10A/G83C)−ブラジキニンを室温(約23℃)で約1時間、9mM DTTで還元した。還元したTTR約50μgを、1ml/分でSEC緩衝液(50mM NaH2PO4、500mM NaCl、pH6.7)中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex,Torrance,CA)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図2からわかるように、TTR(C10A/G83C)は、49kDaの分子サイズに相当する約8.8分で溶出し、これは55kDaの四量体の算定分子量に匹敵する。PTH−TTR(C10A/K15A/G83C)は、67kDaの分子サイズに相当する約8.6分で溶出し、これは該四量体について算定される71kDaに近い。TTR(C10A/G83C)−ブラジキニンは、57kDaの分子サイズに相当する約8.7分で溶出し、これも該四量体について算定される60kDaに近い。
【実施例9】
【0117】
この実施例は、ジスルフィド結合を含むタンパク質をTTR(C10A)のC末端又はN末端のいずれかに融合することがそのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。TTR(C10A)、IL−1−ra−TTR(C10A)及びTTR(C10A)−IL−1−raの各々約50μgを、1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図3からわかるように、TTR(C10A)は、55kDaの四量体の算定分子量に匹敵する49kDaの分子サイズに相当する、約8.8分で溶出する。IL−1−ra−TTR(C10A)融合物は、188kDaの分子サイズに相当する約7.9分で溶出し、これは124kDaという該四量体についての予想分子量よりも著しく大きい。同様に、TTR(C10A)−IL−1−raは、該四量体について予想される124kDaと比較して、やはり188kDaの分子サイズに相当する約7.9分で溶出した。サイズ排除クロマトグラフィーは形状依存的であり、標準物質を球状タンパク質についてカリブレートしているので、これらの大きさのずれはおそらく分子の形状の相違によるものである。
【実施例10】
【0118】
この実施例は、ヒト免疫グロブリンFc(Fc−TMP)のカルボキシ末端に融合したTMP配列とTMP(m)−TTRの、可溶性ヒト骨髄増殖性白血病(MPL)受容体への結合を比較する。加えて、この実施例は、天然TTRシステインのペグ化がTMP融合物のMPL受容体への結合に及ぼす作用を示す。ペグ化TTR融合物の作製は実施例13において詳細に述べる。
【0119】
この実施例のために、ヒトMPL受容体を、EDC/NHS化学を製造者(BIAcore,Uppsula,Sweden)の指示に従って使用して、RL=1300RUでBIAcore CM5チップに共有結合した。すべての試料を、0.1mg/mlウシ血清アルブミン及び0.005%P20(ポリオキシエチレンソルビタン)を含むダルベッコのPBS(Gibco BRL,Gaithersburg,MD)中50μl/分で該チップに通した。平衡エンドポイントは注入後3分とした。図4からわかるように、Fc−TMPはTMP(m)−TTRと比較してより優れた結合特性を示す。さらに、この図は、天然TTRシステイン(Cys10)のペグ化がTMPのMPL受容体への結合を妨げることを明らかにする。TMP(m)−TTR−PEG5Kの結合は、その非ペグ化対応物と比較して有意に低い結合応答を示し、TMP(m)−TTR−PEG20Kはさらに一層強い阻害を示した。これは、システイン10上のPEGの存在が、おそらく融合TMPからMPL受容体の結合に関して立体障害を引き起こし、より大きなPEGはより大きな障害を生じさせることを示唆する。
【実施例11】
【0120】
この実施例は、マウスへのTMP(m)−TTRの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために、50匹のBDF1マウス(Charles River Laboratories,Wilmington,Massachusetts)を5群に分け、希釈剤(0.1%ウシ血清アルブミンを含むダルベッコのPBS)又は希釈剤と供試タンパク質50μg/kg動物のいずれかを皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、11日目、12日目、14日目及び17日目)採血した(140μl)。採血の前にマウスをイソフルランで麻酔した。
【0121】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図5からわかるように、Fc−TMPは、5日目に4.3×1012血小板 L−1の血小板数ピークに達する最大応答を示し、これは基線値である1.2×1012血小板 L−1の3.4倍以上である。TMP(m)−TTR−PEG5Kは、基線値の2倍を少しだけ下回る2.3×1012血小板 L−1でピークに達する中等度の応答を示した。非ペグ化形態のTMP(m)−TTRは、基線値を20%だけ上回る1.5×1012血小板 L−1という極めて低い応答を示す。非ペグ化形態のTMP(m)−TTRは、インビトロではそのペグ化対応物よりも良好な結合を示すが(図4)、インビボではTMP(m)−TTR−PEG5Kと比較して低い成績である。これは、TTR構築物の生物学的半減期を改善するためにはPEGが必要であること、及びそれは受容体に対する低い親和性を補って余りあることを示唆する。
【実施例12】
【0122】
この実施例は、TTR上のシステイン10のアラニンへの突然変異、TTR(C10A)がそのオリゴマー構造に有意に影響を及ぼさないことを明らかにする。TTR及びTTR(C10A)の各々約50μgを1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図6からわかるように、TTR(C10A)は、55kDaの四量体の算定分子量と同様の57kDaの分子サイズに相当する、約8.8分で溶出する。両方の形態のTTRが65℃で沈殿に対して抵抗性であるという所見(データは示していない)と合わせてこのデータは、システイン10のアラニンへの突然変異はTTRの構造又は安定性に有意の影響を及ぼさないことを示唆する。
【実施例13】
【0123】
この実施例は、システイン10からアラニンへの突然変異のバックグラウンドにおけるアラニン37のシステインへの突然変異、TMP−TTR(C10A/A37C)、アスパラギン酸38のシステインへの突然変異、TMP−TTR(C10A/D38C)、アラニン81のシステインへの突然変異、TMP−TTR(C10A/A81C)、又はグリシン83のシステインへの突然変異、TMP−TTR(C10A/G83C)がTTRのオリゴマー構造に有意の影響を及ぼさないことを明らかにする。加えて、この実施例は、これらの突然変異体形態のTTRの5K又は20K PEGによるペグ化が、非ペグ化形態よりも有意に大きい分子サイズを有する2つの異なる種のTTRを生じることを明らかにする。TTRのペグ化は、最初にTTR(7.28g/ml)約8mlを、50mMトリスHCl、pH8.5の存在下に30℃で30分間、10mM DTTで還元することによって実施した。還元したTTRを、次に、20mMトリスHCl、pH8.5中2.5ml/分で26ml SEPHADEX(商標)G25ミディアムカラム(内径2.6cm)(Amersham Pharmacia Biotech)を使用して脱塩した。次に、還元TTRの吸光度を280nmで測定し、算定比吸光係数(TMP−TTR(C10A/A37C)については29,450 M−1)を使用することによって濃度を判定した(5.14mg/ml)。その後、還元試料の2分の1(4.6ml)を直ちに5mMメトキシ−PEG−マレイミド5K(Shearwater Corporation,Huntsville,AL)810μlと混合し、残りの半分を2.5mMメトキシ−PEG−マレイミド20K(Shearwater Corporation)1620μlと混合した。この反応を30℃で30分間進行させ、1M DTT46μlを加えてクエンチした。次に各々のペグ化試料を2.5ml/分で5ml HiTrap Q−セファロースカラムに負荷し、5ml/分で数カラム容積のQ緩衝液A(20mMトリスHCl、pH8.0)で洗った。カラムを、40%Q緩衝液B(20mMトリスHCl、1M NaCl、pH8.0)への直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。ピーク分画をプールし、280nmでプールの吸光度を測定することによって濃度を判定した。各々の試料約50μgを1ml/分でSEC緩衝液中Biosep−Sec−S 3000カラム(内径7.8mm×300mm)(Phenomenex)に注入した。Bio−Rad分子量標準物質(151−1901)を使用してカラムをカリブレートし、注入した試料のおよその分子サイズを算定した。図7からわかるように、4つの非ペグ化TMP−TTR構築物の見かけ分子サイズは、予想される70kDa四量体よりも著しく低い、40−45kDaの間である。この遅延した溶出時間はおそらく、いくつかの他のTMP構築物に関して認められた(データは示していない)、サイズ排除樹脂とTMP−TTR構築物のわずかな相互作用によるものと考えられる。5K PEGとの複合後、見かけ分子サイズは、予想される90kDaよりもはるかに大きい、421−428kDaの間に上昇する(非ペグ化対応物よりも1.53−1.64分早い溶出)。サイズ排除クロマトグラフィーでのペグ化分子の誇張された分子量の所見はしばしば認められる現象である(データは示していない)。20K PEG構築物は、最大カリブレート標準物質(670kDa)よりも早く溶出し、それらの5Kペグ化対応物よりも1.28−1.40分早い溶出を示した。総合してこのデータは、4つの工作した突然変異体形態のTMP−TTRすべてが、ペグ化されてそれらの見かけ分子サイズを劇的に上昇させうることを明らかにしている。
【0124】
ペグ化TMP−TTR構築物約2μgをSDS−PAGEによって分析した(図8)。この図は、TMP−TTR単量体の大部分が1個のみのメトキシ−PEG−マレイミドによって修飾されたこと、及びその反応はほぼ完全であって非修飾単量体をほとんど残さなかったことをゲルシフトによって明らかにしている。
【実施例14】
【0125】
この実施例は、Fc−TMP、TMP−TTR(C10A/A37C)、TMP−TTR(C10A/D38C)、TMP−TTR(C10A/A81C)、及びTMP−TTR(C10A/G83C)がインビトロでのヒトMPL受容体への結合に関して類似の親和性を有することを明らかにする。この実施例のために、Fc−TMPを、製造者(BIAcore,Uppsula,Sweden)の指示に従って高密度でBIAcoreプロテインGチップに結合した。供試タンパク質を、0.1mg/mlウシ血清アルブミン及び0.005%P20(ポリオキシエチレンソルビタン)を含む結合緩衝液(ダルベッコのPBS(Gibco BRL,Gaithersburg,MD))中で5nM MPL受容体と共に室温(約23℃)で2時間以上プレインキュベートした。非ペグ化タンパク質については、非特異的結合を防ぐために0.1mg/mlヘパリンを添加した。すべての試料を、結合緩衝液中50μl/分で該チップに通した。平衡エンドポイントは注入後3分とした。図9からわかるように、すべてのTTR構築物はMPL受容体に対して同様の親和性を示し、0.881−2.333nmの範囲の親和性であったのに対し、Fc−TMP構築物は3.276−5.369nmの範囲の親和性を有していた。
【実施例15】
【0126】
この実施例は、ペグ化TMP−TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために、170匹のBDF1マウスを17群に分け、供試タンパク質50μg/kg動物(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目、12日目及び14日目)採血した(140μl)。採血の前にマウスをイソフルランで麻酔した。
【0127】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図10Aに認められるように、Fc−TMPは、5日目に4.2×1012血小板 L−1以上に達する血小板数で最大応答を示し、これは基線値である1.4×1012血小板 L−1の3倍である。4つの非ペグ化TMP−TTR構築物はすべて対照よりも良好な成績であったが、Fc−TMPほど良好ではなく、Fc−TMPは、5日目に基線値に比べて29%から107%の改善である1.8から2.9×1012血小板 L−1の血小板数を示した。図10Bに認められるように、4つのTMP−TTR構築物すべての工作されたシステインへの5K PEG基の付加は、3.7から4.4×1012血小板 L−1(基線値の2.8から3.4倍)の血小板数を伴って実質的に効果を改善する。
【0128】
同様に図10Cに認められるように、TMP−TTRへの20K PEGの複合体化は、4.2から4.6×1012血小板 L−1(基線値の3.2から3.5倍)の血小板数を伴って、付加的な、しかしそれほど劇的ではない効果の改善を生じさせる。TMP融合構築物のすべてがインビトロでMPLに関して類似の結合親和性を有していたので、この差はおそらく、構築物の有効生物学的半減期を上昇させるPEG複合体化の影響によるものであると考えられる。
【実施例16】
【0129】
この実施例は、ペグ化PTH−TTR構築物のマウスへの注入が血液中のイオン化カルシウム放出に及ぼす作用を示す。この実施例のために、60匹の雄性BDF1、4週齢マウスを12群に分け、供試タンパク質8.91mg/kg動物(PTH融合構築物、PTH−Fc又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を0、24、48及び72時間目の時点で採血した(75μl)。採血の前にマウスをイソフルランで麻酔した。
【0130】
採集した血液を、Ciba*Corning 634 Ca++/pH分析器を用いてイオン化カルシウムに関して分析した。図11に認められるように、PTH−Fc、PTH−TTR(C10A/K15A/A37C)(PEG5K)、PTH−TTR(C10A/K15A/A37C)(PEG20K)、PTH−TTR(C10A/K15A/G83C)(PEG5K)及びPTH−TTR(C10A/K15A/G83C)(PEG20K)は、注射後24時間目に2.2から2.7mmol L−1に上るイオン化カルシウムレベルの最大応答を示し、これは基線値である1.3mmol L−1の1.7倍である。注射後72時間目に、1.8から1.9mmol L−1の増大したイオン化カルシウムレベルを維持したPTH−TTR(C10A/K15A/A37C)(PEG5K)、PTH−TTR(C10A/K15A/G83C)(PEG5K)及びPTH−TTR(C10A/K15A/G83C)(PEG20K)処置群を除いて、すべての群のイオン化カルシウムレベルが基線値に戻った。非ペグ化PTH−TTR構築物は、血清中イオン化カルシウムレベルの上昇においてTTR(C10A)対照と同等であるか又はそれよりわずかに良好であった。
【実施例17】
【0131】
この実施例は、プラスミドを含むPTH−TTR(C10A/K15A/A81C)の構築を述べる。5920のXbaI/XbaIフラグメントを、プラスミド5643(実施例1に述べられている)をXbaIで消化して誘導した精製ベクターに連結した。生じたプラスミドを含む大腸菌株を5933 PTH−TTRC10A/K15A/A81Cと称する。
【0132】
【化11】
【実施例18】
【0133】
この実施例は、GLP−1−TTR(C10A/G83C)融合物及びGLP−1−TTR(C10A/K15A/G83C)融合物の作製を述べる。実施例1に述べられているプラスミドpAMG21を使用してこれらの構築物をクローン化した。この実施例において使用した各々のオリゴヌクレオチドを表6に列記する。
【0134】
細菌宿主GM121は、後期ebg領域(68分)内のlacIQリプレッサーを含むように改変された大腸菌K−12株である。このリプレッサー遺伝子の存在は、この宿主を様々な発現系と共に使用することを可能にするが、このリプレッサーはluxPRからの発現には無関係である。非形質転換宿主は抗生物質耐性を持たない。詳細には、lacIQ構築物を、介在ebg配列を欠失させてGenbank受入番号M64441Gb_Baにおいて番号付けられているヌクレオチド2493位と2937位の間のebgオペロンに送達することによってF’tet/393を修飾した。この構築物を、AGebg−lacIQ No.5と呼ばれる組換えファージを用いて染色体に送達した。
【0135】
組換えと分離後、上述した染色体挿入物だけが細胞内にとどまる。これを新たにF’tet/GM120と命名した。次にF’tet/GM120を、hsdR遺伝子においてそれを不活性化するように突然変異させた。これを新たにF’tet/GM121と命名した。F’tetエピソームを該菌株からキュアリングし、テトラサイクリン感受性であることを確認して、GM121(ATCC No.202174)として保存した。
【0136】
GLP−1配列及びリンカーを増幅するために、ミニプレッププラスミドDNA鋳型2−4μl、各々のオリゴヌクレオチド1μM、各々のオリゴヌクレオチド0.2mM、5%DMSO(Sigma)及びTaq DNAポリメラーゼ2Uを含む反応物80μl中、Roche PCR Core Kit(カタログ番号1 578 553)でPCRを実施した。反応サイクルは、94℃、5分間、次いで[94℃、20秒間、45℃、30秒間、72℃、1分間]の35サイクルであった。PCR産物を、製造者(QIAGEN)のプロトコールに従ってQIAquick(登録商標)PCR Purification Kitで精製した。次にPCR産物とベクターをNdeI及びKpnI(New England Biolabs)で消化した。
【0137】
消化したDNAをアガロースゲルから精製し、その後混合して、T4 DNAリガーゼ(New England Biolabs)によって室温で1.5−2時間連結した。各々の連結混合物を、ギャップ長2mmのキュベット中2.5KVでBiorad大腸菌Pulserを使用して、電気穿孔によって上述した宿主菌株GM121に形質転換した。テリフィック肉汁(TB)2ml中37℃、250rpmで約3時間、細胞を回収した。回収培養物70−100μlを、40μg/mlカナマイシンを含むLB寒天上にプレートした。DNAミニプレップを作製し、ヌクレオチド配列決定によって正しいクローンを確認した。
【0138】
GLP−1−TTR(C10A/G83C)融合物を作製するために、2個のオリゴヌクレオチド、すなわち、luxRプロモーター領域に結合するオリゴヌクレオチド1209−85、及び融合リンカーの最後の12個のアミノ酸とTTRの最初の8個のアミノ酸をコードするオリゴヌクレオチド3131−63を合成した。N末端Met−Lys開始点とそれに続くニッケルカラム精製のための7ヒスチジン配列を有するGLP−1配列、カスパーゼによるGLP−1の最初のヒスチジンの前での開裂のためのアスパラギン酸−グルタミン酸−バリン−アスパラギン酸配列、GLP−1(A2G)配列、及び27アミノ酸の融合リンカーを発現する菌株由来のpAMG21プラスミドを、オリゴヌクレオチド1209−85及び3131−63を用いて増幅した。そのPCR産物を上述したようにクローン化し、配列決定した。新しいプラスミドを含む生じた菌株をGLP−1−TTR(C10A/G83C)(菌株6298)と称した。この菌株は配列番号47に同定するDNA配列を有していた。
【0139】
GLP−1−TTR(C10A/K15A/G83C)融合物を作製するために、2個のオリゴヌクレオチド、すなわち、NdeI部位を含み、上述した精製及び開裂配列に加えてGLP−1(A2G)の最初の6個のアミノ酸をコードするオリゴヌクレオチド3183−83、及び融合リンカーの最後の6個のアミノ酸とTTRの最初の8個のアミノ酸をコードするオリゴヌクレオチド3183−84を合成した。
【0140】
N末端Met−Lys開始点とそれに続くニッケルカラム精製のための7ヒスチジン配列を有するGLP−1配列、カスパーゼによるGLP−1の最初のヒスチジンの前での開裂のためのアスパラギン酸−グルタミン酸−バリン−アスパラギン酸配列、GLP−1(A2G)配列、及び25アミノ酸の融合リンカーを発現する菌株由来のpAMG21プラスミドを、オリゴヌクレオチド3183−83及び3183−84を用いて増幅した。そのPCR産物を上述したようにクローン化し、配列決定した。新しいプラスミドを含む生じた菌株をGLP−1−TTR(C10A/K15A/G83C)(菌株6450)と称した。この菌株は配列番号48に同定するDNA配列を有していた。
【0141】
【表6】
【0142】
【化12】
【実施例19】
【0143】
この実施例は、GLP−1(A2G)−K−Fc融合物の作製を述べる。この構築物は、luxタンパク質及びプロモーターがlacI結合部位及びIPTG誘導プロモーターで置換されていること及びリボソーム結合部位配列がより短いこと(AatIIとClaI認識部位の間の配列が
【0144】
【化13】
で置換されており、SacII認識部位の後の配列の一部が欠失していること(
【0145】
【化14】
が残存する)においてpAMG21と異なる、プラスミドpAMG33*を使用してクローン化した。この実施例において使用した各々のオリゴヌクレオチドを表7に列記する。
【0146】
GLP−1(A2G)−Fc融合物を作製するために、2個のオリゴヌクレオチド、すなわち、NdeI部位を含み、上述した精製及び開裂配列に加えてGLP−1(A2G)の最初の8個のアミノ酸をコードするオリゴヌクレオチド2985−92、及びFcのアミノ酸18から23をコードするオリゴヌクレオチド2687−50を合成した。N末端Met開始点を有するGLP−1(A2G)配列、27アミノ酸リンカー及びFc配列を発現する菌株由来のpAMG33*プラスミドを、オリゴヌクレオチド2985−92及び2687−50を用いて増幅した。そのPCR産物を、使用した酵素がNdeI及びEcoRIであったことを除いて上述したようにクローン化し、配列決定した。上流ベクター配列及び挿入物を導入した5His−アスパラギン酸配列に対するオリゴヌクレオチドを用いたPCRによって挿入物の存在を確認する、コロニースクリーニング段階を含めた。新しいプラスミドを含む生じた菌株をGLP−1(A2G)−K−Fc(菌株5945)と称した。この菌株は配列番号51に同定するDNA配列を有していた。
【0147】
【表7】
【0148】
【化15】
【実施例20】
【0149】
この実施例は、TMP−CH2−TTRC10A及びTTRC10A−CH2−TMPを作製するための、免疫グロブリン分子のCH2ドメインのTTR(C10A)へのクローニングを述べる。
【0150】
TMP−Fcから誘導したCH2ドメインを、2段階PCR手順によってTTR(C10A)、すなわち菌株5619のC末端に連結した。そのCH2ドメイン(5’から3’まで:TTRの最後の7個のコドン、CH2及びBamHI−XhoIリンカーを含む)を最初に次のオリゴヌクレオチドによって増幅した:
2933−77:
GTC GTC ACC AAT CCC AAG GAA GGT TCT GGC TCC GGA TCA GGG GGA CCG TCA GTT TTC CTC(配列番号53)、及び
2973−78:
CCG CGG ATC CTC GAG ATT AGG ATC CAG AAC CCC CTT TGG CTT TGG AGA TGG T(配列番号54)。
【0151】
次にこのフラグメントを、オリゴヌクレオチド2973−78及び
2973−79:
GAG GAA TAA CAT ATG GGT CCA ACT GGT ACC GGT GAA TCC AAG(配列番号55)
によるその後のPCRにおいて5619に融合し、次いでNdeI/XhoIで消化して、同様に制限切断したpAMG21にクローニングした。生じたプラスミドを6017(TTRC10A−CH2)と称する:
【0152】
【化16】
【0153】
6017のXbaI/XbaIフラグメントを上述した5704の対応するフラグメントで置換し、TMP−TTRC10A−CH2(菌株6024)を構築した:
【0154】
【化17】
【0155】
TTRC10A−CH2−TMPの構築を次のように実施した:5’BamHIリンカーと3’XhoIリンカーを含むTMPフラグメントを、オリゴヌクレオチド2694−19及び
2974−70:
GAG GAA TAA GGA TCC ATC GAA GGT CCG ACT CTG CG(配列番号58)
によって増幅した。
【0156】
増幅したフラグメントをBamHI及びXhoIで消化し、その後同様に制限切断した6017と連結した。生じたクローンを菌株6104(TTRC10A−CH2−TMP)と称する。
【0157】
【化18】
【0158】
この融合物のもう1つの立体配置をTMP−CH2−TTR2として作製した。TMP−Fcから誘導したCH2ドメインを、最初に2段階PCRによってTTRC10AのN末端に連結した。そのCH2ドメイン(5’から3’まで:NdeI−BamHIリンカー、CH2及びTTRC10Aの最初の7個のコドンを含む)を、最初にオリゴヌクレオチド:
【0159】
【化19】
によって増幅した。
【0160】
このフラグメントをオリゴヌクレオチド2974−66及び2693−80によるその後のPCRにおいて5619に融合し(実施例1)、次いでNdeI/XhoIで制限消化して、同様に制限切断したpAMG21にクローニングした。生じたクローンを6016(CH2−TTRC10A)と称する:
【0161】
【化20】
【0162】
5’末端にNdeIリンカー及び3’末端にBamHIリンカーを含むTMPフラグメントを、オリゴヌクレオチド:
【0163】
【化21】
によって増幅した。
【0164】
次にこのフラグメントをNdeI/BamHIで消化し、菌株6016由来の同様に制限切断してゲル精製したベクターと連結した。生じたクローンを6110(TMP−CH2−TTRC10A)と称する:
【0165】
【化22】
【実施例21】
【0166】
この実施例は、TTRC10A/K15A−TMP、TTRC10A/K15A/A81C−TMP及びTTRC10A/K15A/G83C−TMPの構築を述べる。
【0167】
TMPをTTR及びその変異体のC末端でもクローン化した。そのN末端に5アミノ酸リンカー(gsgsg)プラスwt TTRの最後の7個のアミノ酸を含む完全長TMPを、標準PCR手順において次のセットのオリゴヌクレオチドによって増幅した。
【0168】
【化23】
【0169】
このPCRフラグメントを、さらに、実施例1で述べたようにオリゴヌクレオチド2694−19及び2693−79を用いる第二のPCRによってwt TTRの3’末端に連結した。生じたクローンを配列確認し、菌株5365(TTR−TMP)と称する:
【0170】
【化24】
【0171】
5365のXbaI/XbaIフラグメントを、上述したように菌株5895の対応するXbaI/XbaIフラグメントで置換して、菌株5921(TTRC10A/K15A−TMP)を作製した:
【0172】
【化25】
【0173】
プラスミド5921を、その後、次の位置:A37、A81及びG83のアミノ酸を、実施例1で突然変異オリゴヌクレオチド(2693−80)と共に使用したTTR3’オリゴヌクレオチドを2694−19に置き換えたことを除いて、実施例1で述べたようにシステインアミノ酸で置換することによって修飾し、TTRC10A/K15A/A37C−TMP(配列番号70)を含む菌株5982、TTRC10A/K15A/A81C−TMP(配列番号71)を含む菌株5983、及びTTRC10A/K15A/G83C−TMP(配列番号72)を含む菌株5984を生成した。
【0174】
【化26】
【実施例22】
【0175】
この実施例は、TMP−TTRC10A/K15A/A81C及びTMP−TTRC10A/K15A/A37Cの構築を述べる。次の方法により、菌株5704、5706及び5707においてTTRの15位のLysをAlaに突然変異させた。プラスミド5513をNdeI/KpnIで消化し、TMPフラグメントとTTRの最初の6個のアミノ酸を内包する挿入物を精製して、NdeI/KpnIで制限消化してゲル精製した菌株5895由来のベクターと連結した。生じたプラスミドを含む細菌株を5919(TMP−TTRC10A/K15A/G83C)と称する。次にプラスミド5919をXbaIで消化し、TMP及びC10AとK15A突然変異を含むTTRの最初の18個のコドンを含む、生じたXbaI/XbaIフラグメントをゲル精製し、XbaI消化してホスファターゼ処理し、ゲル精製した菌株5704及び5706由来のベクターと連結した。新しい菌株を5918(TMP−TTRC10A/K15A/A81C)及び6023(TMP−TTRC10A/K15A/A37C)と称する。
【0176】
【化27】
【実施例23】
【0177】
この実施例は、大腸菌におけるGLP−1融合タンパク質の発現を述べる。飽和一晩培養物25−100mlを使用して、250mlバフルフラスコ中で20μg/mlカナマイシンを含むTB50mlに接種し、37℃、250rpmで一晩インキュベートした。これらの一晩培養物10−35mlを使用して、2Lバフルフラスコ中で20μg/mlカナマイシンを含むTB1Lに接種し、600nmでの光学密度が約0.7に達するまで37℃、250rpmでインキュベートした。次にその培養物を、pAMG21の場合は30μg/ml N−(B−ケトカプロイル)−DL−ホモセリンラクトン(Sigma)を含むエタノール1mlの添加、あるいはpAMG33*の場合は0.1mMまでのIPTGの添加によって、組換えタンパク質を発現するように誘導した。さらに2−4時間インキュベーションを継続し、遠心分離によって細胞を収集した。
【実施例24】
【0178】
この実施例は、PTH−TTR(C10A/K15A/A81C)の精製を述べる。−80℃で保存したクローン5933からの大腸菌ペースト約197gを、50mMトリスHCl、5mM EDTA、pH8.0 1480ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)60錠を細胞懸濁液に溶解し、その懸濁液を14,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を11,325×g、4℃で50分間遠心分離した。上清を可溶性分画として採取した。その可溶性分画をポリプロピレンボトル中65℃水浴で30分間加熱し、その時点で内容物の温度は63℃であった。該可溶性分画を11,325×g、4℃で50分間遠心分離した。上清を熱可溶物(Heat Soluble)として採取した。該熱可溶性分画を、2つのプレフィルターを備えた0.45μm酢酸セルロースフィルターでろ過し、室温(約23℃)で、Q緩衝液A(20mMトリスHCl、2.5mM EDTA、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に25ml/分で負荷した。カラムを30ml/分のQ緩衝液A約2200mlで洗った。Qカラムを、60%Q緩衝液B(20mMトリスHCl、1M NaCl、2.5mM EDTA、pH8.0)への15カラム容積の直線勾配、次いで100%Q緩衝液Bへの2カラム容積段階で溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(1300ml)に貯留した。そのQプールに、3.8M硫酸アンモニウムpH7.2 464mlを緩やかに加えた。その溶液を11,325×g、4℃で50分間遠心分離した。上清を硫酸アンモニウム可溶性分画として採取し、廃棄して、ペレットを、室温で約30分間静かに攪拌することによって10mM NaH2PO4、pH7.0 450mlに再懸濁した。その溶液を11,325×g、4℃で50分間遠心分離した。上清をリン酸緩衝液可溶性分画として採取し、0.45μm酢酸セルロースフィルターでろ過した。該リン酸緩衝液可溶性分画に1Mジチオトレイトール240μlを加え、HA緩衝液A中10ml/分で105ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを10ml/分のHA緩衝液A約210mlで洗い、次いで25%、50%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の3段階で溶出させた。50%溶出からの分画をHAプール(725ml)として貯留し、0.22μm酢酸セルロースフィルターでろ過した。ジチオトレイトール1.16gをHAプールに加え、トリス塩基を使用してそのpHを8.0に上げ、その後室温で約30分間インキュベートした。水750mlで希釈したHAプールを、10ml/分で50mlソース15Q(内径2.6cm)カラム(Amersham Pharmacia Biotech)に負荷し、その後Q緩衝液A約250mlで洗った。10%から60%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Q2プール(170ml)に貯留し、0.22μm酢酸セルロースフィルターでろ過した。算定比吸光係数23,950 M−1 cm−1を用いてタンパク質濃度を3.7mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.61と測定されたので、無視しうると判定した。
【実施例25】
【0179】
この実施例は、TMP−TTR(C10A/D38C)の精製を述べる。−80℃で保存したクローン5705からの大腸菌ペースト約170gを、50mMトリスHCl、5mM EDTA、pH8.0 1275ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、その懸濁液を14,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を11,325×g、4℃で30分間遠心分離した。上清を可溶性分画として採取し、廃棄した。組織粉砕器を使用してペレットを水1200mlに再懸濁し、さらに20錠のSigmaプロテアーゼ阻害剤錠剤を加えた。その懸濁液を11,325×g、4℃で30分間遠心分離した。上清をWhatman GF/Aフィルターでろ過し、ジチオトレイトール2.1gを加えて、その後7℃で30分間インキュベートした。還元した試料を、7℃で、Q緩衝液A(20mMトリスHCl、0.02%アジ化ナトリウム、pH8.0)中で平衡させた30ml/分の240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷した。カラムを30ml/分のQ緩衝液A約1920mlで洗った。Qカラムを、20%、35%及び100%Q緩衝液B(20mMトリスHCl、1M NaCl、0.02%アジ化ナトリウム、pH8.0)の3段階で溶出させた。500mM EDTA pH8.0 13mlをQカラムからのフロースルーに加え、11,325×g、4℃で30分間遠心分離した。上清を廃棄し、ペレットを4M尿素700ml、20mMトリスHCl、pH8.0に再懸濁した。尿素で可溶化したペレットをWhatman GF/Aフィルターでろ過し、7℃でQ緩衝液A(20mMトリスHCl、0.02%アジ化ナトリウム、pH8.0)中で平衡させた240ml Qセファロース・ファーストフロー(内径5cm)カラム(Amersham Pharmacia Biotech,Piscataway,NJ)に30ml/分で負荷した。カラムを30ml/分のQ緩衝液A約1920mlで洗った。Qカラムを、15ml/分の20%、35%及び100%Q緩衝液B(20mMトリスHCl、1M NaCl、0.02%アジ化ナトリウム、pH8.0)の3段階で溶出させた。35%溶出ピークを含む分画を貯留し、0.22μm酢酸セルロースフィルターでろ過し、ジチオトレイトール(最終濃度10mM)0.5gを加えて、その後7℃で30分間インキュベートした。次にその35%Qプールを、7℃で20mMトリスHCl、350mM NaCl、pH8.0中、5ml/分で45ml(2.6cm)1型セラミックヒドロキシアパタイトカラム(Bio−Rad Inc.,Hercules,CA)に負荷した。カラムを、5ml/分の20mMトリスHCl、350mM NaCl、pH8.0 約70ml、次いで2.5%、25%及び100%HA緩衝液B(400mM NaH2PO4、pH7.0)の3段階で洗った。2.5%溶出からの分画をHAプール(80ml)として貯留し、0.22μm酢酸セルロースフィルターでろ過した。算定比吸光係数29,450 M−1 cm−1を用いてタンパク質濃度を6.8mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.54と測定されたので、無視しうると判定した。
【実施例26】
【0180】
この実施例は、TTR(C10A)−CH2−TMPのリフォールディングと精製を述べる。−80℃で保存したクローン6104からの大腸菌ペースト約23gを、50mMトリスHCl、5mM EDTA、pH8.0 200ml中で解凍した。Sigmaプロテアーゼ阻害剤カクテル1−873−580(Saint Louis,MO)50錠を細胞懸濁液に溶解し、その懸濁液を12,000PSIで110−Y型マイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として採取し、廃棄した。組織粉砕器を使用してペレットを50mMトリスHCl、5mM EDTA、pH8.0 50mlに再懸濁した。その懸濁液を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として採取し、廃棄した。組織粉砕器を使用してペレットを50mMトリスHCl、5mM EDTA、pH8.0 50mlに再懸濁した。その懸濁液を14,000×g、室温で10分間遠心分離した。上清を洗浄液として採取し、廃棄した。超音波処理装置を約1分間使用してペレットを8MグアニジンHCl、50mMトリスHCl、pH8.0 50mlに溶解した。溶解したタンパク質を、1M DTT 500μlを加えることによって室温で30分間還元した。還元したタンパク質を20℃、27,216×gで30分間遠心分離した。その後、上清を2ml/分で50mMトリス塩基、160mMアルギニン塩基、1M尿素、1mMシスタミン、4mMシステイン、pH9.5 4Lに加え、4℃で約16時間インキュベートした。次に再生したタンパク質をGellman SUPORCAP(登録商標)50でろ過し、その後Pall Firtron3平方フィートYM10膜接線型フローシステム(tangential flow system)を用いて約500mlに濃縮して、20mMトリスHCl、pH8.0 2Lにろ過透析した。次に濃縮タンパク質を18ml/分で45mlソース15Q(内径2.6cm)カラム(Amersham Pharmacia Biotech)に負荷し、その後Q緩衝液A(20mMトリスHCl、pH8.0)約150mlで洗った。0%から60%Q緩衝液Bへの20カラム容積の直線勾配、次いで2カラム容積の100%Q緩衝液Bの段階でカラムを溶出させた。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプール(29ml)に貯留した。次にそのQプールをMillipore CENTRIPREP(登録商標)10を用いて約6.3mlに濃縮し、1ml/分でPall ACRODISC(商標)MUSTANG(登録商標)E膜フィルターに通した。算定比吸光係数46,410 M−1 cm−1を用いてタンパク質濃度を10.5mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.51と測定されたので、無視しうると判定した。
【実施例27】
【0181】
この実施例は、GLP−1−TTR(C10A/K15A/G83C)の精製を述べる。−80℃で保存したクローン6450からの大腸菌ペースト約30gを、50mM NaH2PO4、pH7.0 250ml中で解凍した。細胞懸濁液を12,000PSIでマイクロフルイダイザー(Microfluidics,Newton,MA)に2回通した。その溶解産物を15,344×g、4℃で50分間遠心分離した。上清を可溶性分画として廃棄し、組織粉砕器を使用してペレットをデオキシコレート200mlに再懸濁した。その懸濁液を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として廃棄し、組織粉砕器を使用してペレットを水200mlに再懸濁した。上清を15,344×g、4℃で50分間遠心分離した。上清を洗浄液として廃棄し、組織粉砕器を使用してペレットを水100mlに再懸濁した。上清を27,216×g、室温で30分間遠心分離した。上清を洗浄液として廃棄し、ペレットの約2/3を約15分間攪拌することによって8MグアニジンHCl、50mMトリスHCl、pH8.0 75mlに溶解した。上清を27,216×g、室温で30分間遠心分離し、その上清を水18mlで希釈した。次に試料を、5ml/分で50mlキレート化セファロースファーストフローカラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷し、NiCl2で充填した。10ml/分のNi緩衝液A(6MグアニジンHCl、37.5mlトリスHCl、pH8.0)約150mlで洗浄した後、10%及び100%Ni緩衝液B(6MグアニジンHCl、37.5mMトリスHCl、400mMイミダゾール、pH8.0)の2段階で溶出させた。融合構築物を含むピークをNiプール(40ml)として混合し、比吸光係数25,440 M−1を用いて8MグアニジンHCl中280nmでの吸光度を観察することにより、タンパク質濃度を6.4mg/mlと判定した。500mM EDTA、pH8.0 800μlを加え、ペグ化反応のためにタンパク質80mgを採取した。1M DTT 230μlを加え、30℃で30分間インキュベートした。20mMトリスHCl、6M尿素、pH8.5中8ml/分で、130ml SEPHADEX(登録商標)G25ミディアムカラム(内径2.6cm)に負荷した。280nmでの吸光度によって測定したタンパク質ピークをプールし(22ml)、比吸光係数25,440 M−1を用いて20mMトリスHCl、6M尿素、pH8.5中280nmでの吸光度を観察することにより、タンパク質濃度を3.2mg/mlと判定した。緩衝液を交換した該物質の45%を、30℃で140分間、5mMメトキシ−PEG−マレイミド5K(Shearwater Corporation,Huntsville,AL)950μlと反応させた。各々の反応物に1M 2−メルカプトエタノール100μlを加えてクエンチした。Pierce 10kDa Slidealyzerを使用して室温で2時間、反応物を25mM NaH2PO4、3M尿素、pH7.25 1Lに透析した。透析緩衝液を25mM NaH2PO4、10%スクロース、2mM EDTA、pH7.25に交換し、室温で約16時間インキュベートした。5%CHAPS140μl及び2−メルカプトエタノール7.28μl及び3mg/mlカスパーゼ3 0.475mlを加え、その後室温で2時間インキュベートした。反応混合物を、20mMトリスHCl、pH8.0中1ml/分で、5ml HiTrap Q−sepharose HPカラム(Amersham Pharmacia Biotech,Piscataway,NJ)に負荷し、その後同じ緩衝液約15mlで洗った。次に60%20mMトリスHCl、1M NaCl、pH8.0への直線勾配、次いで100%溶出緩衝液への段階を使用して5ml/分でカラムを展開した。SDS−PAGEによって測定したTTR融合物を含む分画を単一Qプールに貯留した(9.5ml)。Millipore CENTRIPREP(登録商標)30kDaを用いてQプールを3.2mlに濃縮し、約1ml/分でPall MUSTANG(登録商標)E膜に通してろ過した。Qプールを水で6.5mlに希釈し、アセトニトリル375μlを加えた。95%RP緩衝液A(0.1%トリフルオロ酢酸)及び5%RP緩衝液B(95%アセトニトリル、0.1%トリフルオロ酢酸)中、5ml/分でVydac Protein/Peptide 10×250mm C4カラム(Vydac,Hisperia,CA)に注入した。100%RP緩衝液Bへの直線勾配に通してカラムを展開した。Millipore CENTRIPREP(登録商標)30kDaを用いてタンパク質ピークを約3mlに濃縮し、20mMトリスHCl、pH8.0を使用して15mlに希釈した。緩衝液交換をさらに3回反復した後、約1ml/分でPall MUSTANG(登録商標)E膜に通した。算定比吸光係数25,440 M−1 cm−1を用いてタンパク質濃度を7.7mg/mlと判定した。発熱物質レベルは、Limulus Ameboycyte Lysateアッセイ(Associates of Cape Cod,Falmouth,MA)を用いて<1EU/mgタンパク質と判定された。核酸含量は、260nm対280nmの吸光度比が0.54と測定されたので、無視しうると判定した。
【実施例28】
【0182】
この実施例は、マウスへのペグ化GLP1−TTR構築物の注入が血中グルコースレベルに及ぼす作用を示す。この実施例のために40匹の雄性、db/db、9週齢マウスを4群に分け、各動物当り7.4−16.6mgの供試タンパク質(すべての群について538pmol単量体)(5Kペグ化GLP1−TTR融合構築物10mg、20Kペグ化GLP1−TTR融合構築物10mg、GLP1−Fc16.6mg及びTTR(C10A)対照7.4mg)を腹腔内注射した(0時間目)。各々の群を注射後0時間目(基線測定)、1、4、6、12、24及び48時間目に採血した。実験の最初の6時間はマウスに食餌を与えず、6時間目の採血後に食餌を再開した。
【0183】
各時点で採取した各々の血液を、One Touch Profileグルコース測定器を用いてグルコース含量に関して分析し、その結果を図12に示す。
【実施例29】
【0184】
この実施例は、融合抗体CH2ドメインを有するTMP−TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために50匹の雌性BDF1マウスを5群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目及び10日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0185】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図13からわかるように、Fc−TMPは、5日目に4.2×1012血小板 L−1以上に達する血小板数で最大応答を示し、これは基線値の1.4×1012血小板 L−1の3倍である。CH2融合TMP−TTR構築物はすべて対照よりも良好な成績であったが、5日目に、基線値に比べて64%から86%の改善にあたる、2.3×1012から2.6×1012血小板 L−1の血小板数を示したFc−TMPほどではなかった。
【実施例30】
【0186】
この実施例は、ペグ化TTRのカルボキシ末端に融合したTMPとのペグ化TTR構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために80匹のBDF1マウスを8群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目及び12日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0187】
採集した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図14からわかるように、Fc−TMP及び3つのアミノ末端(TMP−TTR)融合物は、5日目に、基線値の1.3×1012血小板 L−1の3倍以上である、4.3×1012から4.6×1012血小板 L−1に達する血小板数で最大応答を示した。3つのカルボキシ末端(TTR−TMP)構築物はすべて対照よりも良好な成績であった。
【実施例31】
【0188】
この実施例は、K15A変化を含むペグ化TTR−TMP構築物のマウスへの注入が血液中の血小板数に及ぼす作用を示す。この実施例のために120匹のBDF1マウスを12群に分け、各動物kg当り供試タンパク質50mg(TMP融合構築物、Fc−TMP又はTTR(C10A)対照)を皮下注射した(0日目)(この試験は2つのバッチ(一方においてはPEG20K及び他方ではPEG5K及び非ペグ化試料)に分け、対照を反復して別々の時点で実施した)。各々の群を半分ずつに分けて、交互の時点で(0日目、3日目、5日目、7日目、10日目及び12日目)採血した(140ml)。採血の前にマウスをイソフルランで麻酔した。
【0189】
採取した血液を、ADVIA 120自動血液分析器をネズミ用ソフトウエア(Bayer Diagnostics,New York,NY)と共に使用して完全血球算定及び白血球分画に関して分析した。図15Aからわかるように、2つの非ペグ化構築物は基線(1.3×1012血小板 L−1)よりも良好な成績であり、5日目に1.8×1012から2.0×1012血小板 L−1に上る血小板応答を示した。図15Bからわかるように、Fc−TMPと3つの5Kペグ化融合物は、基線(1.3×1012血小板 L−1)の少なくとも2.7倍である、3.5×1012から4.4×1012血小板 L−1に達する血小板数で、5日目に同等の応答を示した。図15Cからわかるように、Fc−TMPと3つの20Kペグ化融合物は、基線値の1.3×1012血小板 L−1の3倍以上である、4.3×1012から4.6×1012血小板 L−1に達する血小板数で、5日目に同等の応答を示した。
【0190】
加えて、20Kペグ化TTR構築物は、3.1×1012血小板 L−1のFc−TMPに比べて、7日目に3.7×1012から4.9×1012血小板 L−1の範囲の血小板数を示し、改善された持続性応答を有すると思われる。この持続性応答は、3つの20Kペグ化TTR構築物に関しては10日目にも維持され、血小板数は、2.0×1012血小板 L−1のFc−TMPに比べて2.3×1012から3.1×1012血小板 L−1の範囲であった。
【特許請求の範囲】
【請求項1】
生物学的活性物質をトランスサイレチン(TTR)又はTTR変異体に融合することを含む、該生物学的活性物質の血清中半減期を上昇させる方法。
【請求項2】
上記TTR又はTTR変異体が、デキストラン、ポリ(n−ビニルピロリドン)、ポリエチレングリコール、プロプロピレングリコールホモポリマー、ポリプロピレンオキシド/エチレンオキシドコポリマー、ポリオキシエチル化ポリオール及びポリビニルアルコールから成る群より選択される化学物質で化学的修飾されている、請求項1に記載の方法。
【請求項3】
上記TTR又はTTR変異体がポリエチレングルコールで化学的修飾されている、請求項2に記載の方法。
【請求項4】
上記ポリエチレングルコールが約1kD〜100kDの分子量を有する、請求項3に記載の方法。
【請求項5】
上記ポリエチレングルコールが約5kD〜30kDの分子量を有する、請求項4に記載の方法。
【請求項6】
上記TTRが配列番号2の核酸によってコードされる、請求項1に記載の方法。
【請求項7】
上記TTR変異体が配列番号8の核酸によってコードされる、請求項1に記載の方法。
【請求項8】
上記生物学的活性物質がタンパク質である、請求項1に記載の方法。
【請求項9】
上記生物学的活性物質がペプチドである、請求項1に記載の方法。
【請求項10】
上記ペプチドがTPOミメティックペプチド(TMP)である、請求項9に記載の方法。
【請求項11】
上記生物学的活性物質がグルカゴン様ペプチド−1(GLP−1)である、請求項9に記載の方法。
【請求項12】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、TTR−生物学的活性物質融合物の実質的に均質な製剤。
【請求項13】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、PEG−TTR−生物学的活性物質融合物の実質的に均質な製剤。
【請求項14】
上記生物学的活性物質がタンパク質である、請求項13に記載の製剤。
【請求項15】
上記生物学的活性物質がペプチドである、請求項13に記載の製剤。
【請求項16】
上記ペプチドがTMPである、請求項15に記載の製剤。
【請求項17】
上記ペプチドがGLP−1である、請求項15に記載の製剤。
【請求項18】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、TTR変異体−生物学的活性物質融合物の実質的に均質な製剤。
【請求項19】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、PEG−TTR変異体−生物学的活性物質融合物の実質的に均質な製剤。
【請求項20】
上記生物学的活性物質がタンパク質である、請求項19に記載の製剤。
【請求項21】
上記生物学的活性物質がペプチドである、請求項19に記載の製剤。
【請求項22】
上記ペプチドがTMPである、請求項21に記載の製剤。
【請求項23】
上記ペプチドがGLP−1である、請求項21に記載の製剤。
【請求項24】
上記融合物がリンカーペプチドを含む、請求項10〜23のいずれかに記載の製剤。
【請求項25】
(a)TTRを生物学的活性物質に融合してTTR−生物学的活性物質融合物を得ること;及び(b)上記TTR−生物学的活性物質融合物を単離することを含む、TTR−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項26】
(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記TTR変異体を生物学的活性物質に融合してTTR変異体−生物学的活性物質融合物を得ること;及び(c)上記TTR変異体−生物学的活性物質融合物を単離することを含む、TTR変異体−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項27】
(a)ポリエチレングリコールをTTRに複合体化してPEG−TTRを得ること;(b)上記PEG−TTRを生物学的活性物質に融合してPEG−TTR−生物学的活性物質融合物を得ること;及び(c)上記PEG−TTR−生物学的活性物質融合物を単離することを含む、PEG−TTR−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項28】
(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTR変異体を得ること;(c)上記PEG−TTR変異体を生物学的活性物質に融合してPEG−TTR−生物学的活性物質融合物を得ること;及び(d)上記PEG−TTR−生物学的活性物質融合物を単離することを含む、PEG−TTR変異体−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項29】
請求項16に記載の製剤の治療上有効な用量を投与することを含む、血小板減少症を治療する方法。
【請求項30】
請求項22に記載の製剤の治療上有効な用量を投与することを含む、血小板減少症を治療する方法。
【請求項31】
請求項17に記載の製剤の治療上有効な用量を投与することを含む、インスリン非依存性糖尿病を治療する方法。
【請求項32】
請求項23に記載の製剤の治療上有効な用量を投与することを含む、インスリン非依存性糖尿病を治療する方法。
【請求項33】
異種配列に融合したTTRタンパク質を含む融合タンパク質。
【請求項34】
上記異種配列がTMPである、請求項33に記載の融合タンパク質。
【請求項35】
上記異種配列がGLP−1である、請求項33に記載の融合タンパク質。
【請求項36】
上記TTRタンパク質と上記異種配列の間にリンカー配列をさらに含む、請求項33、34又は35のいずれか1つに記載の融合タンパク質。
【請求項37】
請求項33、34又は35のいずれか1つに記載の融合タンパク質をコードする核酸。
【請求項1】
生物学的活性物質をトランスサイレチン(TTR)又はTTR変異体に融合することを含む、該生物学的活性物質の血清中半減期を上昇させる方法。
【請求項2】
上記TTR又はTTR変異体が、デキストラン、ポリ(n−ビニルピロリドン)、ポリエチレングリコール、プロプロピレングリコールホモポリマー、ポリプロピレンオキシド/エチレンオキシドコポリマー、ポリオキシエチル化ポリオール及びポリビニルアルコールから成る群より選択される化学物質で化学的修飾されている、請求項1に記載の方法。
【請求項3】
上記TTR又はTTR変異体がポリエチレングルコールで化学的修飾されている、請求項2に記載の方法。
【請求項4】
上記ポリエチレングルコールが約1kD〜100kDの分子量を有する、請求項3に記載の方法。
【請求項5】
上記ポリエチレングルコールが約5kD〜30kDの分子量を有する、請求項4に記載の方法。
【請求項6】
上記TTRが配列番号2の核酸によってコードされる、請求項1に記載の方法。
【請求項7】
上記TTR変異体が配列番号8の核酸によってコードされる、請求項1に記載の方法。
【請求項8】
上記生物学的活性物質がタンパク質である、請求項1に記載の方法。
【請求項9】
上記生物学的活性物質がペプチドである、請求項1に記載の方法。
【請求項10】
上記ペプチドがTPOミメティックペプチド(TMP)である、請求項9に記載の方法。
【請求項11】
上記生物学的活性物質がグルカゴン様ペプチド−1(GLP−1)である、請求項9に記載の方法。
【請求項12】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、TTR−生物学的活性物質融合物の実質的に均質な製剤。
【請求項13】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、PEG−TTR−生物学的活性物質融合物の実質的に均質な製剤。
【請求項14】
上記生物学的活性物質がタンパク質である、請求項13に記載の製剤。
【請求項15】
上記生物学的活性物質がペプチドである、請求項13に記載の製剤。
【請求項16】
上記ペプチドがTMPである、請求項15に記載の製剤。
【請求項17】
上記ペプチドがGLP−1である、請求項15に記載の製剤。
【請求項18】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、TTR変異体−生物学的活性物質融合物の実質的に均質な製剤。
【請求項19】
場合により、医薬適合性の希釈剤、担体又はアジュバント中の、PEG−TTR変異体−生物学的活性物質融合物の実質的に均質な製剤。
【請求項20】
上記生物学的活性物質がタンパク質である、請求項19に記載の製剤。
【請求項21】
上記生物学的活性物質がペプチドである、請求項19に記載の製剤。
【請求項22】
上記ペプチドがTMPである、請求項21に記載の製剤。
【請求項23】
上記ペプチドがGLP−1である、請求項21に記載の製剤。
【請求項24】
上記融合物がリンカーペプチドを含む、請求項10〜23のいずれかに記載の製剤。
【請求項25】
(a)TTRを生物学的活性物質に融合してTTR−生物学的活性物質融合物を得ること;及び(b)上記TTR−生物学的活性物質融合物を単離することを含む、TTR−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項26】
(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記TTR変異体を生物学的活性物質に融合してTTR変異体−生物学的活性物質融合物を得ること;及び(c)上記TTR変異体−生物学的活性物質融合物を単離することを含む、TTR変異体−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項27】
(a)ポリエチレングリコールをTTRに複合体化してPEG−TTRを得ること;(b)上記PEG−TTRを生物学的活性物質に融合してPEG−TTR−生物学的活性物質融合物を得ること;及び(c)上記PEG−TTR−生物学的活性物質融合物を単離することを含む、PEG−TTR−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項28】
(a)TTRのアミノ酸配列内の特定アミノ酸位置にシステイン残基を工作して上記TTRの変異体を得ること;(b)上記システイン残基での上記TTR変異体にポリエチレングリコールを複合体化してPEG−TTR変異体を得ること;(c)上記PEG−TTR変異体を生物学的活性物質に融合してPEG−TTR−生物学的活性物質融合物を得ること;及び(d)上記PEG−TTR−生物学的活性物質融合物を単離することを含む、PEG−TTR変異体−生物学的活性物質融合物の実質的に均質な製剤を調製する方法。
【請求項29】
請求項16に記載の製剤の治療上有効な用量を投与することを含む、血小板減少症を治療する方法。
【請求項30】
請求項22に記載の製剤の治療上有効な用量を投与することを含む、血小板減少症を治療する方法。
【請求項31】
請求項17に記載の製剤の治療上有効な用量を投与することを含む、インスリン非依存性糖尿病を治療する方法。
【請求項32】
請求項23に記載の製剤の治療上有効な用量を投与することを含む、インスリン非依存性糖尿病を治療する方法。
【請求項33】
異種配列に融合したTTRタンパク質を含む融合タンパク質。
【請求項34】
上記異種配列がTMPである、請求項33に記載の融合タンパク質。
【請求項35】
上記異種配列がGLP−1である、請求項33に記載の融合タンパク質。
【請求項36】
上記TTRタンパク質と上記異種配列の間にリンカー配列をさらに含む、請求項33、34又は35のいずれか1つに記載の融合タンパク質。
【請求項37】
請求項33、34又は35のいずれか1つに記載の融合タンパク質をコードする核酸。
【図1】

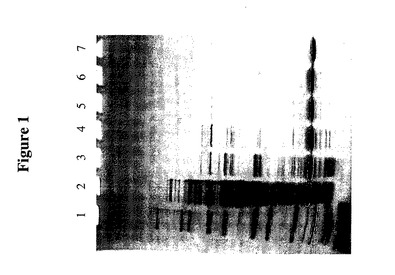
【図2】
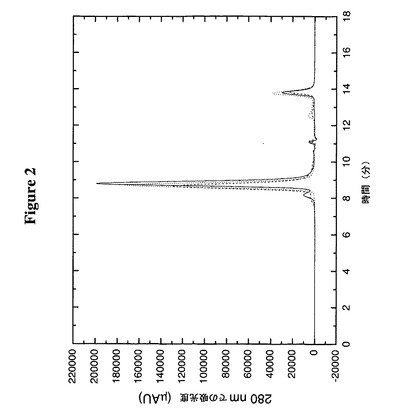
【図3】
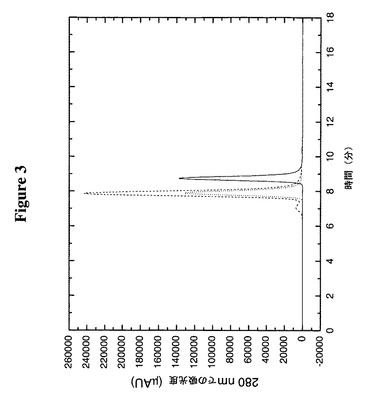
【図4】
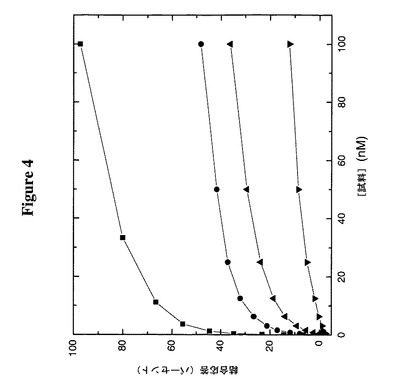
【図5】
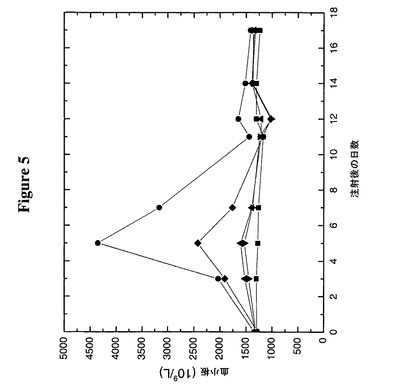
【図6】
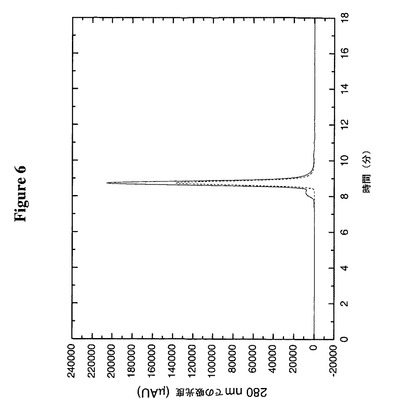
【図7】
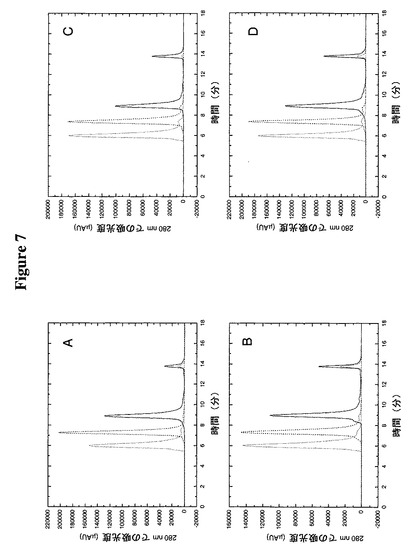
【図8】
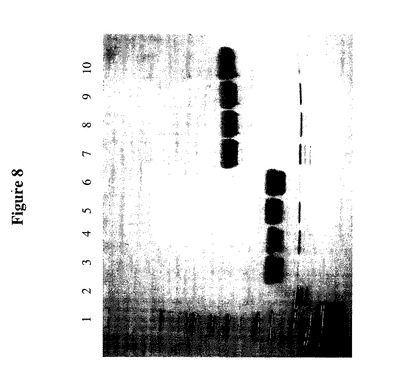
【図9】
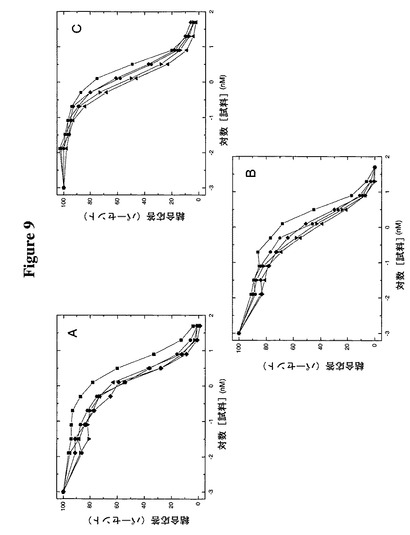
【図10】
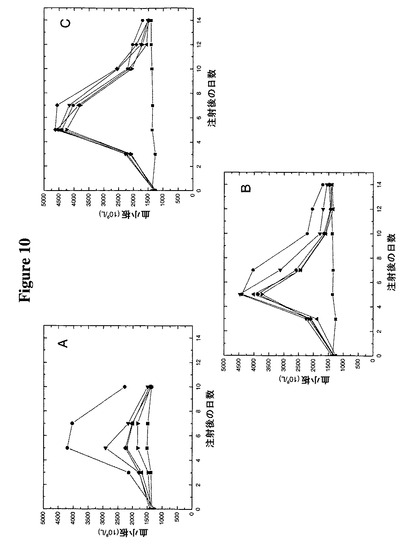
【図11】
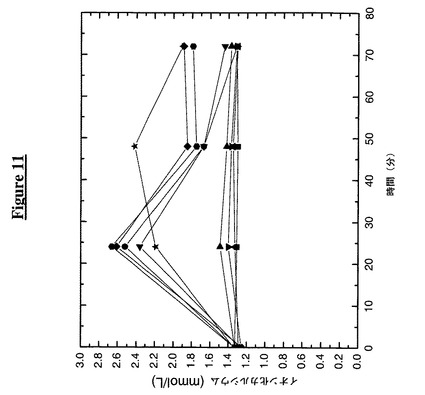
【図12】
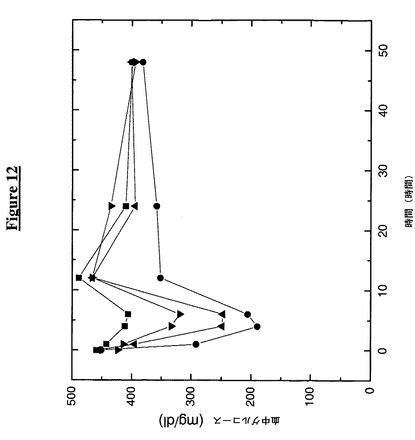
【図13】
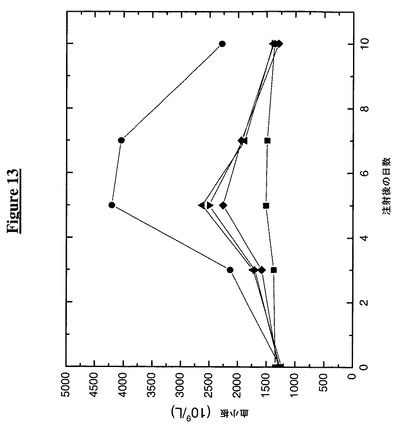
【図14】
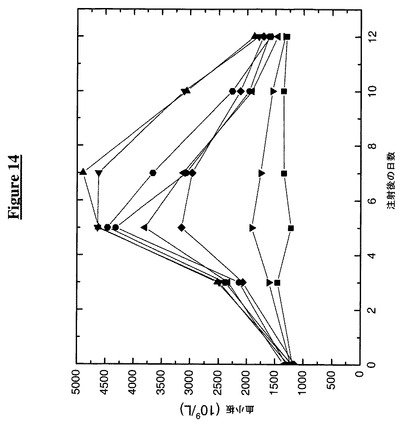
【図15】
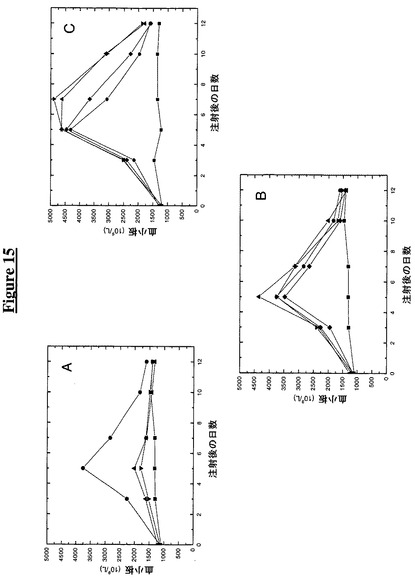
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2011−136981(P2011−136981A)
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−264759(P2010−264759)
【出願日】平成22年11月29日(2010.11.29)
【分割の表示】特願2003−583462(P2003−583462)の分割
【原出願日】平成15年4月3日(2003.4.3)
【出願人】(500049716)アムジエン・インコーポレーテツド (242)
【Fターム(参考)】
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願番号】特願2010−264759(P2010−264759)
【出願日】平成22年11月29日(2010.11.29)
【分割の表示】特願2003−583462(P2003−583462)の分割
【原出願日】平成15年4月3日(2003.4.3)
【出願人】(500049716)アムジエン・インコーポレーテツド (242)
【Fターム(参考)】
[ Back to top ]