
蛋白質の発現または抑制のためのシステム、方法、および装置
本発明は、蛋白質発現の誘導および/または抑制のためのシステム、方法、および装置に関する。より具体的には、本発明は色素体中で蛋白質の発現を誘導および/または抑制するシステム、方法、および装置に関する。代表的な実施形態には、本明細書に記載された方法、システム、および装置を用いて水素ガスの産生を促進するための、水素産生に関与する蛋白質の発現制御が含まれる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、蛋白質の発現の誘導および/または抑制のためのシステム、方法、および装置に関する。より具体的には、本発明は色素体中での蛋白質の発現の誘導および/または抑制のためのシステム、方法、および装置に関する。
【背景技術】
【0002】
蛋白質(ペプチド、オリゴペプチド、およびポリペプチドなど)は、触媒作用、情報伝達、防御、運動、輸送など、細胞の多くの活動に関与している。蛋白質の生物活性の基礎となるのは、アミノ酸配列および/またはその立体構造である。したがって、蛋白質の生物活性部位は原則的にそのままの形を維持し、生物学的に機能する立体構造をとらなければならない。蛋白質発現に関する遺伝子工学技術の進歩は、蛋白質の生物活性を維持した形のまま様々なシステムで内在性および外来性の蛋白質を制御下に発現するための方法の開発に繋がっている。このような遺伝子工学により作られた制御発現システムは、蛋白質発現を制御することができる結果、蛋白質の不活性化や分解が抑えられ、正しく折り畳まれた安定な蛋白質を発現することにより高い蛋白質収量をもたらす。
【発明の概要】
【発明が解決しようとする課題】
【0003】
微生物、菌類や酵母を含む真核細胞、そして哺乳類細胞、昆虫細胞など、様々な宿主細胞の細胞小器官で多数の異なる様式の発現システムが開発されているが、色素体での蛋白質発現を制御するために核由来の安定化因子を利用する蛋白質制御発現のための発現システムはこれまでに開発されていない。色素体とは光合成に関与する細胞小器官であり、一般に葉緑体、白色体、アミロプラスト、または有色体に分類される。色素体はこれらの形態間で分化または再分化が可能である。
【課題を解決するための手段】
【0004】
一実施形態では、細胞の色素体内で蛋白質の産生を誘導するための発現システムを作成する方法が提供される。本方法は、誘導性プロモーターをコードする第一の核酸を細胞核に導入するステップと、第一の核酸を安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、インデューサーの導入またはリプレッサーの除去が安定化因子の発現を誘導し、発現した安定化因子が色素体内で安定化因子で安定化され第三の核酸から転写されるmRNAの非翻訳領域と結合し、第三の核酸が色素体に内在性または外来性で第三の核酸が蛋白質をコードし、mRNAの発現が蛋白質の産生をもたらすことを特徴とするステップとを含む。
【0005】
別の例示的な実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制するための発現システムを作成する方法が提供される。本方法は、抑制性プロモーターをコードする第一の核酸を細胞核に導入するステップと、第一の核酸を安定化因子をコードする第二の核酸に作動的に連結して組換え核酸を形成するステップであって、リプレッサーの導入またはインデューサーの除去が安定化因子の発現を抑制し、安定化因子の発現抑制が安定化因子で安定化され第三の核酸から転写されるmRNAの発現抑制をもたらし、第三の核酸が色素体に内在性または外来性であり、第三の核酸が蛋白質をコードし、蛋白質の発現が抑制されることを特徴とするステップとを含む。
【0006】
さらに別の例示的な形態では、細胞の色素体内で色素体蛋白質を発現させる方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現させるステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現させるステップと、色素体内で蛋白質を産生させるステップとを含む。
【0007】
別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制する方法が提供される。本方法は、細胞をリプレッサーと接触させるかインデューサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップとを含む。
【0008】
さらに別の実施形態では、組換え宿主細胞の色素体内で色素体蛋白質を発現させるシステムが提供される。本システムは、外から加えられる核蛋白質の発現を誘導するインデューサーと、核に組換え核酸を含む組換え宿主細胞であって、組換え核酸が第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、第一の核酸が誘導性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、色素体蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体とを含む。
【0009】
別の例示的な実施形態では、組換え宿主細胞の色素体内で色素体蛋白質の発現を抑制するシステムが提供される。本システムは、外から加えられる核蛋白質の発現を抑制するリプレッサーと、核に組換え核酸を含む組換え宿主細胞であって、組換え核酸が第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、第一の核酸が抑制性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、色素体に内在性または外来性の第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体とを含む。
【0010】
別の実施形態では、細胞の色素体内で色素体蛋白質を発現させることにより水素ガスの産生を促進する方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現させるステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現させるステップと、色素体内で蛋白質を産生させるステップと、水素ガスを産生するステップとを含む。
【0011】
さらに別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制することにより水素ガスの産生を阻害する方法が提供される。本方法は、細胞をリプレッサーと接触させるかまたはインデューサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップと、水素ガスの産生を阻害するステップとを含む。
【0012】
さらに別の実施形態では、細胞の色素体内で色素体蛋白質の発現を誘導および抑制することにより水素ガスの産生を促進する方法が提供される。本方法は、順次的にi)細胞をインデューサーと接触させるかまたはリプレッサーの除去をもたらす条件で細胞を処理してから、ii)細胞をリプレッサーと接触させるかまたはインデューサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、順次的に安定化因子を発現させてから抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、順次的にmRNAを発現させてから抑制するステップと、色素体内で蛋白質を産生させるステップと、水素ガスを産生するステップとを含む。
【0013】
上記のいずれかの実施形態において、組換え核酸を核に導入する前に、第一の核酸は第二の核酸と作動的に連結して組換え核酸を形成することができ、細胞は第二または第三の核酸の非作動型コピーを有するかまたはそれらのコピーやホモログを欠失することができ、細胞は植物細胞または藻類細胞であってもよく、色素体は葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択することができ、誘導性プロモーターはCyc6プロモーターと少なくとも90%の配列類似性を有することができ、第三の核酸はpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることができる。
【0014】
上記のいずれかの実施形態において、インデューサーまたはリプレッサーは化学物質または環境条件であってもよく、化学物質は銅であってもよく、環境条件は所定レベルまでの酸素濃度の低下であってもよく、インデューサーはその添加と除去を一サイクルとして複数サイクルの添加と除去を行うことができ、蛋白質は光合成または水素ガス産生に関与する蛋白質であってもよく、蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、栄養補助食品から選択することができ、医薬品は抗体、ワクチン抗原、抗菌剤、またはその他の宿主細胞防御産物から選択され、安定化因子はNac2およびMbb1から選択することができる。別の例示的な実施形態では、第二の核酸は例えばTbc2やTca1などの翻訳活性化因子をコードすることができる。
【0015】
本発明の別の実施形態では、色素体内で内在性または外来性遺伝子の発現または抑制を制御するシステムおよび方法が提供される。一実施形態では、本発明は核にコードされる葉緑体転写因子でありその発現がCyc6遺伝子の誘導性プロモーターによって制御されるNac2を採用する発現システムに関する。別の実施形態では、低い酸素レベルなど、インデューサー(すなわち、薬剤または環境条件の変化)によるNac2発現の誘導が葉緑体内でのpsbD遺伝子の発現をもたらす。さらに別の実施形態では、薬剤または銅の除去などの環境条件がCyc6プロモーターの誘導をもたらし、psbD遺伝子の発現ももたらす。別の例示的な実施形態では、高い酸素レベルなど、リプレッサー(すなわち、薬剤または環境条件の変化)によるNac2遺伝子の抑制がpsbD遺伝子の発現阻害または低下をもたらす。関連した実施形態では、誘導性Cyc6プロモーターを抑制する薬剤または環境条件の変化もpsbD遺伝子の発現低下をもたらす。
【0016】
別の例示的な実施形態では、本発明は葉緑体内での外来遺伝子の誘導性発現または抑制に関するので、葉緑体内のpsbD遺伝子を外来遺伝子で置換することで、Nac2発現制御により葉緑体内での外来遺伝子の誘導性発現または抑制が促進される。
【0017】
別の実施形態では、本発明はNac2の発現またはNac2の発現抑制によるpsbD遺伝子の制御を介して葉緑体内で水素ガスを産生する方法に関する。本発明のこの例示的な形態では、Nac2遺伝子の誘導および抑制を促進する環境条件(例えば、酸素レベルを低下させて発現を誘導する、酸素レベルを上昇させて発現を抑制するなど)が、psbD遺伝子発現の周期的な誘導と抑制をもたらし、光合成速度の低下およびその結果としての酸素濃度の低下をもたらす。この実施形態では、酸素濃度の低下が水素産生を促進する。よって、一実施形態では、本発明はNac2およびpsbD遺伝子発現の周期的な誘導と抑制を制御することにより水素ガスを産生する方法に関する。
【0018】
さらに別の実施形態では、本発明は葉緑体内での他の遺伝子の組換え発現を介して水素発生システムを促進する方法に関する。例としてヒドロゲナーゼや、他の組換え蛋白質またはホスホリブロースキナーゼなどの内在性蛋白質の抑制がある。
【0019】
さらに別の実施形態では水素産生のための装置が提供される。本装置は、実質的に酸素のない環境下に細胞培養液を保持するように構成された第一の容器と、第一の容器と液体連通があり所定の速度で第一の容器に培地を供給するように構成された第一のポンプと、第一の容器に連結され細胞培養液によって産生される水素量を測定するように構成された測定器を備える。
【0020】
この実施形態では、第一のポンプは細胞培養の増殖速度と実質的に同じ速度で一定量の培地を第一の容器に供給するように構成することができ、第一のポンプはペリスタ型ポンプを含むことができ、細胞培養液はcy6Nac2.49培養液を含むことができ、測定器は質量分析器を含むことができ、本装置はさらに第一の容器に連結され細胞培養液を撹拌するように操作できる撹拌器を備えることができ、撹拌器は磁気撹拌子を含むことができる。本装置はさらに一定量の培地を保持するように構成された第二の容器を備えることができ、第一のポンプは第二の容器と液体的に連結して第二の容器から所定速度で培地を汲み上げるように構成されることを特徴とする。本装置はさらに第一の容器と液体連通して第一の容器からあふれた培地が入るように構成された第三の容器を備えることができ、本装置はさらに第三の容器および第二の容器と液体連通したフィルターと第二のポンプを備えることができ、第二のポンプは第三の容器から一定量の培地を汲み上げフィルターを通して第二の容器へと供給するように構成される。
【0021】
本発明は以下の図面を参照することでより理解しやすい。
【0022】
<関連出願の相互参照>
本出願は35 U.S.C § 119(e)により、本明細書に参照として含まれる米国仮出願No. 60/837,001(2006年8月11日出願)に対する優先権を主張する。
【図面の簡単な説明】
【0023】
【図1】図1は好気的および嫌気的条件下におけるC. reinhardtii葉緑体内の水素および電子の流れを示す(Kruse et al., 2005より)。
【図2】図2はインデューサーの存在下に核内の誘導性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の誘導性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図3】図3はインデューサーの非存在下に核の誘導性プロモーターが抑制されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の誘導性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図4】図4はリプレッサーの存在下に核の抑制性プロモーターが抑制されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の抑制性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図5】図5はリプレッサーの非存在下に核の抑制性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の抑制性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図6】図6はpsbD遺伝子の位置が示されたChlamydomonas葉緑体ゲノムの概要図を示す。矢印はpSK108の挿入部位を示す。
【図7】図7は水素産生装置の作業工程図を示す。
【図8】図8は水素産生装置を示す。
【図9】図9は核Nac2突然変異体の形質転換並びにCyc6プロモーターおよびNac2遺伝子の導入に使用される核発現ベクターpSL17の概要図を示す。マップはプロモーター、HSP70Aプロモーターのエンハンサーエレメント、そしてCyc6プロモーターおよびNac2を挿入する制限酵素部位の配置を示す。
【図10】図10はcy6Nac2(paroR)の概要図を示す。
【図11】図11a〜cはNac2 midi遺伝子のゲノム配列を示す。開始コドンは下線を引いた部分の最初のATGである。推定される輸送ペプチドにも下線を引いてある。
【図12】図12はCyc6ゲノム配列を示す。Nac2 midi遺伝子との融合コンストラクトを作成するのに使用したゲノム配列に下線を引いてある。NdeI制限酵素部位を作り出したcyc6Nac2コンストラクトでの3塩基対の相違も示している(二重下線)。
【図13】図13はcy6Nac2.49遺伝子導入株の増殖特性を示す。
【図14】図14はcy6Nac2.49遺伝子導入株のウェスタンブロット解析を示す。
【図15】図15はcy6Nac2.49遺伝子導入株の水素産生を示す。
【図16】図16はpSK108ベクターのマップを示す。pSK108ベクターは、psbD遺伝子周辺領域へ導く葉緑体DNA隣接領域を有する。
【図17】図17はpcg12のマップを示す。
【図18】図18は葉緑体発現プラスミドpcg12_IBDV_Flagのマップを示す。
【図19】図19は葉緑体発現プラスミドpcg12_VP28_Flagのマップを示す。
【図20】図20はpsbDの葉緑体DNA配列を示す。下線を引いた配列はcyc6Nac2システムを用いて葉緑体内での遺伝子発現を駆動するのに使用された。
【図21】図21はIND_aadA_X 遺伝子導入株の単離と各種培地上での増殖を示す。
【図22】図22は野性型およびIND_aadA_117単離株から抽出した全RNAのノーザンブロット解析を示す。
【図23】図23は野生型およびIND_aadA_117から抽出した総蛋白質および可溶性蛋白質(α-Nac2)の解析を示す。
【図24】図24は3つの外来性蛋白質(DILP、IBDV、VP28)の発現を誘導する遺伝子導入株のスクリーニングを示す。
【図25】図25はNac2誘導性葉緑体遺伝子発現システムを用いた外来性蛋白質(DILP)の誘導性産生を示す。
【図26】図26はcy6Nac2.49でのpsbD RNA、D2およびNac2蛋白質の蓄積を示す。
【図27】図27はpsbD 5' UTRをpetA 5' UTRで置換することによるcy6Nac2.49での構成的PSII蓄積の回復を示す。
【図28】図28は葉緑体psbD遺伝子の誘導性発現を示す。
【図29】図29はcy6Nac2.49での銅を介したPSII合成阻害の経時変化を示す。
【図30】図30はcy6Nac2.49でのPSII蓄積の経時変化を示す。
【図31】図31はIND_aadA_117形質転換体にてpsbD-aadA遺伝子の発現が銅枯渇で誘導され銅で抑制されることを示す。
【図32】図32はcy6Nac2-49株での水素産生を示す。
【発明を実施するための形態】
【0024】
本明細書で用いられる用語「外来性遺伝子」または「外来性核酸」(すなわち、導入遺伝子)とは、組換えDNA技術を用いて細胞の核酸に挿入される核酸を意味し、外来性遺伝子または外来性核酸は細胞の当該位置に通常は存在しない。外来性遺伝子または外来性核酸はコーディングおよび非コーディング核酸配列を含むことができる。外来性遺伝子または外来性核酸は、組換えDNA技術を用いて改変され細胞に再導入された内在性核酸を含むか、または細胞内のある位置から別の位置に移動された内在性核酸を含むことができる。
【0025】
本明細書で用いられる用語「内在性核酸」または「内在性遺伝子」とは、細胞内で天然の配列(自然突然変異を含む)と位置を有する核酸を意味する。
【0026】
本明細書で用いられる用語「誘導性」とは、mRNA転写物が産生されるよう制御されることが可能なプロモーターを意味する。mRNA転写物レベルの決定方法はノーザンブロットおよびリアルタイムPCRを含む。
【0027】
本明細書で用いられる用語「発現」とは、DNAからRNAへの転写またはRNAから蛋白質への翻訳を意味することができる。
【0028】
本明細書で用いられる用語「安定化因子」とは、転写、転写後、翻訳、翻訳後、蛋白質ターゲティング、蛋白質リクルートメントなどの活性に限らないがこれらを含む活性を示し、葉緑体蛋白質の発現または活性を増強する核蛋白質を意味する。
【0029】
本発明は、有用産物を産生する目的で細胞(例えば藻類または植物の細胞)の色素体内で遺伝子発現を制御するためのシステム、方法、および装置に関する。色素体内での遺伝子発現の誘導または抑制は、葉緑体移行蛋白質をコードする遺伝子と作動的に連結した誘導性または抑制性プロモーターで細胞の核ゲノムを形質転換することにより達成される。葉緑体移行蛋白質は、色素体で発現するmRNAの非翻訳領域と直接または間接的に(例えば、アクセサリー蛋白質を介して)結合する。葉緑体移行蛋白質は色素体で発現するmRNAの安定性および/または翻訳に必要であり、よって色素体遺伝子の発現に必要である。
【0030】
一実施形態では、核プロモーターは誘導性プロモーターであり、化学物質(例えば銅、炭化水素、蛋白質など)の添加または除去あるいは環境条件の変化(例えば低酸素濃度、または光、温度、栄養状態の変化など)がプロモーターを活性化し、葉緑体mRNAの発現をもたらし、続いてmRNAがコードする蛋白質の発現をもたらす。別の実施形態では、核プロモーターは抑制性プロモーターであり、化学物質(例えば銅、炭化水素、蛋白質など)の添加または除去あるいは環境条件の変化(例えば高酸素濃度、または光、温度、栄養状態の変化など)がプロモーターを抑制し、安定化因子が発現されなくなって葉緑体mRNAの発現阻害をもたらし、続いてmRNAがコードする蛋白質の発現阻害をもたらす。
【0031】
一実施形態では、本発明は細胞の色素体内で蛋白質を発現する方法に関する。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が第二の核酸に作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現するステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性の第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現するステップと、色素体内で蛋白質を産生するステップとを含む。
【0032】
別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制する方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性の第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップとを含む。
【0033】
上記の実施形態では、蛋白質が細胞の色素体内で発現される。例示的に、細胞は植物細胞もしくは藻類細胞、または色素体を含むいかなる細胞種でもよい。色素体は色素体ゲノムを含む細胞小器官であり、複数コピー含まれることが多い。色素体は、例えば植物や藻類に見られ、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体が含まれる。
【0034】
本明細書に記載される方法およびシステムの実施形態では、第三の核酸が目的の蛋白質をコードする。例示的な一実施形態では、発現される蛋白質をコードする第三の核酸は、色素体に内在性または外来性(すなわち導入遺伝子)のいずれでもよい。この実施形態では、発現される蛋白質(例えばペプチド、オリゴペプチド、ポリペプチドなど)は誘導性または抑制性プロモーターの制御下に発現することができ、光化学系IまたはIIの構成成分(例えばpsbAおよびpsbD、光化学系IIのD1およびD2サブユニットなど)、CO2固定に関与する蛋白質(例えばホスホリブロースキナーゼなど)、ヒドロゲナーゼ(例えばHydA1、HydA2など)、そしてこれらの蛋白質のうちいずれかの活性を制御する蛋白質(例えばこれら蛋白質のいずれかの集合(HydEF、HydGなど))などの光合成に関与する蛋白質、または色素体に内在性の他の蛋白質を含む蛋白質を誘導する。誘導性または抑制性プロモーターの制御下に発現することのできる例示的な内在性蛋白質としては、図1に描写または示唆される光合成過程または炭素同化過程の制御に関与するすべての蛋白質、あるいは色素体に内在性の他の蛋白質が含まれる。別の実施形態では、芳香族アミノ酸などのアミノ酸合成経路に関与する色素体に内在性の蛋白質の発現制御によって、芳香族アミノ酸などのアミノ酸またはアミノ酸前駆体が産生されてもよい。
【0035】
別の例示的実施形態では、本明細書で記載した方法、システム、および装置を使用して、誘導性または抑制性プロモーターあるいは両方の制御下に、水素ガス産生に関与する蛋白質が発現されてもよい。この実施形態では、水素ガス産生に関与する蛋白質は、本項または前項で記載した蛋白質のいずれでも、または図1に描写または示唆される光合成過程または炭素同化過程の制御に関与する蛋白質のいずれでもよい。この実施形態では、インデューサーまたはリプレッサーは、その添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことができる。
【0036】
別の実施形態では、第三の核酸は色素体に外来性である(すなわち外来性核酸または外来性遺伝子である)。この実施形態では、蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、または栄養補助食品であってもよい。この実施形態では、発現蛋白質は、例えば抗体、ワクチン抗原(例えばワクチンに使用するための)、抗菌剤、またはその他の宿主細胞防御産物、成長ホルモン、インターロイキンやインターフェロンなどのサイトカイン、インシュリン、コロニー刺激因子、凝固因子、エリスロポエチン、上皮成長因子などの成長因子、ソマトトロピン、線維芽細胞増殖因子、血小板由来増殖因子、および同類のもの、アミラーゼ、プロテアーゼ、リパーゼ、ペクチナーゼ、セルラーゼ、ヘミセルラーゼ、ペントサナーゼ、キシラナーゼ、およびフィターゼ、殺虫性蛋白質、フェニルアンモニアリアーゼ、またはその他のあらゆる蛋白質性の医薬品、工業用酵素、または栄養補助食品であってもよい。
【0037】
さらに別の例示的な実施形態では、発現蛋白質をコードする追加の核酸(例えば第四の核酸)を葉緑体内で発現してもよく、それは色素体に内在性または外来性(すなわち外来性核酸または外来性遺伝子)であってもよい。これらの実施形態では、追加の核酸の発現は、核内で追加の核酸がコードするそれ自身の安定化因子によって制御されるか(すなわち第二の核酸と同様に)、またはこれらの追加の核酸の発現は第二の核酸がコードする安定化因子によって制御されてもよい。一つの例示的な実施形態では、一つの安定化因子が色素体mRNA内の安定化因子結合エレメントと結合し、第三の核酸および第三の核酸と作動的に連結した追加の核酸(例えば第四の核酸など)の発現を促進する。これらの実施形態では、発現される蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、または栄養補助食品であってもよい。この実施形態では、発現蛋白質は、例えば抗体、ワクチン抗原(例えばワクチンに使用)、抗菌剤、またはその他の宿主細胞防御産物、成長ホルモン、インターロイキンやインターフェロンなどのサイトカイン、インシュリン、コロニー刺激因子、凝固因子、エリスロポエチン、上皮成長因子などの成長因子、ソマトトロピン、線維芽細胞増殖因子、血小板由来増殖因子、および同類のもの、アミラーゼ、プロテアーゼ、リパーゼ、ペクチナーゼ、セルラーゼ、ヘミセルラーゼ、ペントサナーゼ、キシラナーゼ、およびフィターゼ、殺虫性蛋白質、フェニルアンモニアリアーゼ、またはその他のあらゆる蛋白質性の医薬品、工業用酵素、または栄養補助食品であってもよい。
【0038】
発現蛋白質がワクチンとして使用するワクチン抗原である実施形態では、発現蛋白質、またはその一部は、藻類細胞や植物細胞などの細胞の細胞小器官上または細胞小器官内に位置することができる。もしワクチン抗原が少なくとも部分的には病原体に由来する場合、例えば藻類はその後溶解し、病原体ワクチン抗原を使って宿主動物の病原体への免疫応答を誘導することができる。
【0039】
一実施形態では、ワクチン抗原を有する藻類を食物材料として投与する。ワクチンを投与できる代表的な動物は、哺乳類、鳥類、水産養殖種を含むがこれらに限らない。特に、ワクチンはすべての脊椎動物魚類などの水生脊椎動物に投与でき、これはサケ科(マス、サケ、北極イワナなど)、コイ、ナマズ、ブリ、タイ、スズキなどに限らないがこれらを含む骨性または軟骨性魚類であってもよい。このようなワクチンは、ハマグリやアサリ、ロブスター、エビ、カニ、カキに限らないがこれらを含む貝類や甲殻類に投与することもできる。代表的な送達方法は、乾燥粉末として、通常飼料の一成分として、そしてワクチンを含む懸濁液に動物を浸水させることでの経口投与を含む。
【0040】
水生脊椎動物の場合、本明細書に記載した方法およびシステムを用いて、その抗原決定基を細胞表面にワクチン抗原として発現させる病原体の例は、Rennibacterium salmoninarum(サケ、マス、イワナ、ホワイトフィッシュなどサケ科の魚における細菌性腎疾患の病原体)、Aeromonas salmonicida、Aeromonas hydrophila、Vibrio種(V. anguillarumとV. ordaliiを含む)、Pasteurella種(P. piscicidaを含む)、Yersinia種、Streptococcus種、Edwardsiella tardaおよびEdwardsiella ictaluria、ウイルス性出血性敗血症、伝染性膵臓壊死症、コイのウイルス血症の原因ウイルス、伝染性造血器壊死症ウイルス、アメリカナマズウイルス、グラスカープ出血性ウイルス、神経壊死症ウイルスやシマアジ神経壊死症ウイルスなどのノダウイルス、伝染性サケ貧血ウイルス、そして寄生虫のCeratomyxa shasta、Ichthyophthirius multifillius、Cryptobia salmositica、Lepeophtheirus salmonis、Tetrahymena種、Trichodina種およびEpistylus種などを含むが、但しこれらに限らない。
【0041】
蛋白質を藻類で発現させる実施形態では、藻類は例えば緑藻類であってもよい。例えば、使用できる藻類にはCharoidesなどのChlorophyta(Charoides、Lamprothamnium、Nitellopsis、Nitellaなど)、Zynematales(Zygnema、Closterium、Netriumなど)、Codials(Codium fragile、Helimida opunta、Caulerpaなど)、Bryopsis plumosa(Bryopsis、Pseudobryopsis、Bryopsidella、Derbesis、Pedobesiaなど)、Acetabularia ryukyuensis(Acetabularia ryukyuensis、Halicoryne wrightii、Neomeris annulata、Cymopolia van bossei、Bornettella ovalis、Acetabularia calyculusなど)、Siphonocladales(ValoniaceaeおよびBoodleaceae)、Cladophora(Anadyomene writii、Cladophora、Cladophora sauteri、およびChaetomorpha)、Ulva(UlvaおよびFnteromorpha)、Ulotrichales(Acrosiphoniaceae、Collinsiellaceae、Monostromaceae、およびChlorocystidaceae)、Prasiola、Chlorella、Chlorococcales(PediastrumおよびHydrodictyon)、およびVolvocales(Chlamydomonus、Pandorina、Pleodorina、およびVolvox)が含まれる。
【0042】
この適用で記述されるいずれかの実施形態で通常使用される代表的な藻類には、Chlamydomonas種、特にChlamydomonas reinhardtii、Chlorella種、およびVolvox種が含まれる。単細胞真核緑藻のChlamydomonas reinhardtiiは特に有利である。Chlamydomonas株は例えばデューク大学クラミドモナス遺伝ストックセンター(ノースカロライナ州ダーラム)から入手できる。Chlamydomonas reinhardtiiの栄養要求性変異株(一つ以上の成長に必要な栄養成分を有する点で野生株と異なる変異株)はクラミドモナス遺伝ストックセンターから入手可能であり、そうした変異株はDNAで形質転換(すなわち細胞に外来性DNAを導入)することにより遺伝的に相補でき、これにより目的とする導入遺伝子を含む藻類の選択が容易となる。他の実施形態では無能化した藻類を使用できる。無能化した藻類とは、非常に特殊な制御環境以外では増殖しないように遺伝子操作したものである(すなわち、このような藻類株は野生では生育したり遺伝子を移動させたりしない)。本開示中では、このような藻類を「無能化した」という。このような無能化した株を使用することにより、本発明で用いられる遺伝子導入藻類株が環境中に広まることが阻止されるか制限される。
【0043】
本明細書に記載される方法およびシステムでの使用に適した代表的な植物は、培養植物細胞(プロトプラストおよびカルス細胞)や全植物体(単細胞および多細胞植物)を含む。様々な実施形態において、植物細胞(すなわち培養植物細胞または全植物体の細胞)は燕麦、小麦、大麦、米、紅花、トウモロコシ、アルファルファなどの豆類、大豆、トマト、甜菜、ジャガイモなどの植物から得られる。その他の有用な植物には、例えば、リンゴ、ナシ、サクランボ、ブドウ、柑橘果実、パイナップル、バナナなどの果実をつける植物、そしてカラマツなどの樹木がある。その他の適した植物には、油ヤシ、茶、カカオ、コーヒーなどの潅木、タバコ、綿花、フラックス、ヒマワリ、牧草、飼料用穀類、飼料用植物、そして落花生やレンズ豆植物などがある。その他の有用な植物には、シロイヌナズナ、サボンソウ(Saponaria)、ウキクサ(Lemnaceae)、シダ類、コケ類、ゼニゴケ類などがある。この実施形態では、植物の遺伝子操作に通常用いられるベクターを使って、本発明に従い核酸分子を植物細胞に移行させる。
【0044】
本明細書に記載される方法およびシステムでは、第一および第二の核酸を細胞(例えば藻類または植物の細胞)内に導入する。この実施形態では、第一の核酸は誘導性、抑制性、または誘導性かつ抑制性のプロモーターをコードし、第二の核酸は色素体mRNAの発現を制御する安定化因子(例えばNac2またはMbb1)をコードする。別の例示的な実施形態では、第二の核酸は例えばTbc2やTca1などの翻訳活性化因子をコードしてもよい。
【0045】
誘導性または抑制性のプロモーターは安定化因子の発現を制御する。様々な実施形態において、使用する核プロモーターに応じてあらゆる適切な形のインデューサーまたはリプレッサー(化学物質または改変した環境条件)が使用できる。本明細書に記載される方法およびシステムでの使用に適した代表的なプロモーターはCyc6プロモーターである(図12参照)。Cyc6プロモーターと配列類似性を有するプロモーター、例えばCyc6プロモーターと60%、70%、80%、85%、90%、95%、98%の配列類似性を有するものなど、その他の適切なプロモーターのいずれでも使用できる。また、Cyc6プロモーターの相補配列に高ストリンジェンシー条件でハイブリダイズする配列も使用できる。その他の使用に適した誘導性または抑制性プロモーターには、環境条件(無酸素、熱、干ばつ、または光など)、化学物質、栄養素、ホルモン、病原体、損傷、摂食、発生段階、組織型などの因子に応答するプロモーターなどがある。このようなプロモーターは当業者に知られている。プロモーターが複数の因子に応答することもあり、単一因子が複数のプロモーターを活性化または抑制することもある。
【0046】
Cyc6以外では、クラミドモナスのその他の誘導性プロモーターには、CO2誘導性細胞膜蛋白質遺伝子のプロモーター(Genbankアクセション番号U31976)、鉄利用能で緊密に制御されるFEA1遺伝子のプロモーター(Sasaki et al., 1998; Rubinelli et al., 2002)、そしてアンモニアやグルタミン酸の存在下に抑制されアンモニアを含まない培地中で誘導されるNit1遺伝子のプロモーター(Fernandez 1989)などがある。高等植物での誘導性プロモーターの例には、リブロース1,5-ビスリン酸カルボキシラーゼ/オキシゲナーゼ小サブユニットの光誘導性プロモーター、カルコンシンターゼ遺伝子のUV誘導性プロモーター、カルコンシンターゼ遺伝子のクマル酸誘導性プロモーター、アルコールでヒドロゲナーゼ遺伝子の低酸素誘導性プロモーター、そしてタバコ、トマト、キュウリおよびアラビドプシスの病原誘導性プロモーター(PR-1-14)などがある。
【0047】
安定化因子は誘導性および/または抑制性プロモーターの制御下に発現し、色素体内に導入され、色素体内で安定化因子はmRNAの非翻訳領域と直接または間接的に(例えばアクセサリー蛋白質を介して)結合しmRNAを安定化する。様々な実施形態において、mRNAの非翻訳領域はmRNAの5'または3'末端に位置しうる。mRNAは発現蛋白質をコードする第三の核酸から転写される。別の例示的な実施形態では、mRNAは、第三の核酸に作動的に連結しているかまたは第三の核酸に連結しておらず、それ自身の安定化因子で制御される追加の核酸から転写されてもよい。よって、発現蛋白質は安定化因子の制御下に色素体内で産生され、安定化因子の発現は誘導性および/または抑制性核プロモーターにより制御される。
【0048】
一つの例示的な形態では安定化因子は、mRNAの5'末端か3'末端あるいは両方にある非翻訳領域に直接または間接的に結合するだけでなく、mRNAのコード領域にも直接または間接的に結合してもよい。別の例示的な形態では、安定化因子は例えば他の蛋白質がmRNAの非翻訳領域および/またはコード領域に直接または間接的に結合している複合体中に含まれる他のアクセサリー蛋白質との結合を介してmRNAと直接または間接的に結合してもよい。
【0049】
一実施形態では、第一および第二の核酸は、例えば組込みまたは組換え(例えば相同組換えや他の種類の組換えなど)を行う核DNAに組み入れられる。別の実施形態では、第一および第二の核酸は細胞内に導入される発現ベクターを用いて発現される。この実施形態では、ベクター内の第一および第二の核酸の挿入断片は核DNAと組換えを行わず、むしろ安定化因子をコードする第二の核酸はベクター内に存在する、第一の核酸にコードされる誘導性および/または抑制性プロモーターなどの制御配列を使って自律的に発現される。本明細書の実施例1〜4で記載されるベクターを含め、当業者に知られるあらゆる適切なベクターが使用できる。
【0050】
これらの実施形態のそれぞれにおいて、第一の核酸は第二の核酸に作動的に連結して組換え核酸を形成する。本明細書に記載される代表的な核酸は、Nac2コード配列(すなわち第二の核酸)と作動的に連結したCyc6プロモーター(すなわち第一の核酸)である。第一および第二の核酸は、当業者によく知られるクローニング方法を用いて互いに作動的に連結でき、この方法には制限酵素で核酸を切断し、切断したベクターの末端に第一および第二の核酸をリガーゼを使って互いに連結することを含む。このようなクローニング方法は、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0051】
第二の核酸が第一の核酸にコードされるプロモーターの制御下に自律的に発現する実施形態では、発現コンストラクト(すなわち、ベクター‐挿入断片コンストラクト)は、第二の核酸に存在するコード配列の転写を終止させるための転写ターミネーターを通常含み、その他の5'および3'制御配列を含んでもよい。転写ターミネーターは第二の核酸内に通常存在するが、ベクターに組み入れることもできる。
【0052】
第一および第二の核酸が核DNAに安定に組み入れられる実施形態では、転写ターミネーターは第二の核酸に通常存在するが、核DNA配列の一部でもよい。第一または第二の核酸、ベクター、および/または核DNAには、例えば転写エンハンサーエレメントやmRNA安定化に関与する配列などのさらなる5'および3'制御配列が存在してもよい。この実施形態では、ベクターは、組換え核酸が核DNAに組込まれるのを促進する核ターゲティング配列も含んでもよい。一実施形態では、核は第二の核酸の非作動型コピーを有してもよく、第二の核酸のコピーやホモログを欠失してもよい。
【0053】
本明細書に記載される様々な実施形態において、安定化因子の自律的発現または組換え核酸の核DNAへの組み入れのためのベクターは、ベクターコンストラクトの複製のための細菌の複製起点を有し、組換え核酸(すなわち、作動的に連結した第一および第二の核酸)の挿入の有無にかかわらず目的とするベクターをクローニング用に大量調製することができる。ベクターは通常、DNA断片挿入のための制限エンドヌクレアーゼ切断部位(例えば多重クローニング部位)、そして形質転換体選択のための選択遺伝子マーカーも有する。選択マーカーは、抗生物質を含む培地上での形質転換細胞の増殖を可能にするaadA遺伝子やnptIIなどのマーカーである(Goldschmidt-Clermont, Nucl. Acids Res., vol. 19, pages 4083-4089 (1991))。内在性遺伝子(arg7、nit1)と外来性遺伝子(ble、aphVIII、aadA、nptII)の両方の選択マーカーが核形質転換のレポーター遺伝子として開発されている。
【0054】
組換え核酸(すなわち、第一および第二の核酸を含む挿入断片)を有するベクターは、当業者によく知られる標準的な形質転換技術により細胞(例えば藻類または植物の細胞など)に導入される。代表的な形質転換方法には、エレクトロポレーション、ガラスビーズを用いたDNA送達、ポリエチレングリコールを用いた形質転換、遺伝子銃、および同類のものがある。
【0055】
一実施形態では、藻類の形質転換のために、接合中に放出され細胞壁を分解する酵素であるオートリシンを使用して、形質転換の前に細胞壁を分解する。別の実施形態では、細胞壁の合成能を欠失する突然変異株(例えばcw15、cw10など)が作出されており、効率的な形質転換にこれらを使用できる。
【0056】
一つの例示的な形態において、形質転換体はPCRおよびサザンブロット法により検出できる。抗生物質添加、銅添加、抗体検出(例えばELISAおよびウェスタンブロット法)、および配列決定などの、当業者に知られる他の方法も使用できる。これらの方法は使用するコンストラクトによって選択する。
【0057】
もし導入遺伝子が色素体中で発現される場合、第三の核酸をベクターに組み入れ、第一および第二の核酸について上述したのと同じようにベクターコンストラクトを作成して一般的に複製できる。例示的に、遺伝子銃で色素体の形質転換ができ、これは導入遺伝子を含むベクターを金やタングステンの微粒子上に付着させてヘリウムなどの不活性ガスで加速させる。この方法は細胞の損傷が少ない。関連する例示的な実施形態では、形質転換する細胞を選択用固形寒天培地上にのせ、表面にDNAをコートしたタングステンビーズをヘリウムガスまたは火薬を使って加速させて色素体中に送達する。この技術を使用して、組換えDNAが色素体中に効率的に送達される。
【0058】
一実施形態では、細菌のアミノグリコシドアデニルトランスフェラーゼ遺伝子(aadA)をコードする異種DNAの発現により第三の核酸の色素体DNAへの組込みを検出できる。この実施形態では、抗生物質スペクチノマイシンまたはストレプトマイシンを使って形質転換体を選択する方法が可能となる。このレポーターコンストラクトを使用して、あらゆる非必須の遺伝子またはシス作動性エレメントを色素体中で特異的に挿入、破壊、修飾、変異または削除することが可能である。
【0059】
別の例示的な実施形態では、導入遺伝子の葉緑体DNAへの組込みを促進する相同的葉緑体ターゲティング配列を導入遺伝子の両末端に隣接させて、導入遺伝子をもつ葉緑体形質転換体を得ることができる。一実施形態では、ベクター上の隣接葉緑体配列と葉緑体ゲノム内の対応する相同配列との間での二つの相同組換えイベントにより導入遺伝子の組込みが起こる。一実施形態では、葉緑体は第三の核酸の非作動型コピーを有してもよく、第三の核酸のコピーやホモログを欠失してもよい。
【0060】
核酸を外来性にする代表的な修飾法には、コドンを最適化するように核酸を修飾する、他の遺伝子の5'または3'非翻訳領域を核酸に連結する、プロモーターまたは終止配列を核酸に付加する、相同組換えに有効な配列を核酸に付加する、そして例えば遺伝子発現、ターゲティング、安定化に必要な他のエレメントを核酸に付加することが含まれるがこれらに限らない。さらなる修飾法には、核酸を他の内在性または導入遺伝子の核酸と融合させて融合蛋白質を形成することも含まれる。
【0061】
本発明の範囲に含まれる色素体遺伝子制御システムの代表的な概要図を図2〜5に示す。図1はインデューサーの存在下に核内の誘導性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。この実施形態では、インデューサー(例えば低酸素レベルなどの環境条件など)が安定化因子の発現を制御するプロモーターを誘導し、安定化因子が発現する。翻訳後、安定化因子は色素体に移行し、発現される安定化因子に応じた特異的mRNAに結合し、mRNAとそれがコードする蛋白質が発現する。
【0062】
図3はインデューサーの非存在下に核内の誘導性プロモーターが抑制されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、インデューサーが存在せず、誘導性プロモーターは活性化されず安定化因子は発現しない。安定化因子がないので、色素体内のmRNAは分解されるかまたは翻訳されず、蛋白質は発現しない。
【0063】
図4はリプレッサー(例えば培地中の銅の所定レベルなど)の存在下に核内の抑制性プロモーターが抑制されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、リプレッサーが存在し、抑制性プロモーターは活性化されず安定化因子は発現しない。安定化因子がないので、色素体内のmRNAは分解されるかまたは翻訳されず、蛋白質は発現しない。
【0064】
図5はリプレッサー(例えば培地中の銅の低レベルまたは銅の非存在)の非存在下に核内の抑制性プロモーターが誘導されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、リプレッサーが存在せず、安定化因子の発現を制御する核プロモーターが誘導され、安定化因子が発現する。翻訳後、安定化因子は色素体に移行し、発現される安定化因子に応じた特異的mRNAに結合し、mRNAとそれがコードする蛋白質が発現する。
【0065】
本明細書に記載される方法およびシステムを使って得られた発現蛋白質を単離精製することが望ましい実施形態では、蛋白質を発現した後に従来技術を用いて精製する。例えば、純度約40%、純度約50%、純度約60%、純度約70-80%、純度約90%、純度約95%、または純度約98%の蛋白質が得られる。細胞からの精製では、例えば細胞溶解物を硫安沈殿にかけてからDEAE-セファロースカラムクロマトグラフィーを行う。ゲルろ過、イオン交換クロマトグラフィー、DEAE-セファロースカラムクロマトグラフィー、アフィニティークロマトグラフィー(以下の実施例2に記載されるFLAGタグシステムを使用するなど)、溶媒-溶媒抽出、限外ろ過、HPLCなど、当業者に知られる他の従来技術も使用できる。または、蛋白質が非常に高濃度に存在して細胞溶解物中の蛋白質が実質的に純粋(例えば純度70-80%)であれば、精製段階が必要でないこともある。発現蛋白質は、例えば限外ろ過や接線流ろ過などの技術により濃縮できる。
【0066】
一実施形態では、例えば超音波処理、加熱、または化学処理などにより細胞を溶解し、ホモジネートを遠心分離して細胞片を除いてもよい。その後上清を硫安沈殿にかけ、さらに必要に応じてゲルろ過、イオン交換クロマトグラフィー、DEAE-セファロースカラムクロマトグラフィー、アフィニティークロマトグラフィー、溶媒-溶媒抽出、限外ろ過、HPLCなどの分画技術を使って発現蛋白質を精製できる。培地または細胞から発現蛋白質を精製する方法は上述した精製方法に限らず、実質的に純粋な蛋白質を得るのに必要であれば、当業者に知られるいずれかの精製技術を用いて発現蛋白質が精製できることを理解すべきである。
【0067】
細胞(例えば藻類または植物の細胞)は、蛋白質発現を促進するために様々な技術を用いて培養できる。藻類や植物細胞を含む細胞の培養培地は、当技術分野で知られており、炭素源(例えばブドウ糖または酢酸)が通常添加される。細胞は、例えば後述のように一例として水素ガスの産生に有用な培養システムおよび装置を使用して、望ましい濃度を維持しながら培養してもよい。
【0068】
上記に詳細を記したとおり、本明細書に記載される発現方法およびシステムは水素産生に使用できる。よって、別の実施形態では水素産生のための装置が提供される。本装置は、実質的に酸素のない環境下に細胞培養液を保持するように構成された第一の容器と、第一の容器と液体連通があり所定の速度で第一の容器に培地を供給するように構成された第一のポンプと、第一の容器に連結され細胞培養によって産生される水素量を測定するように構成された測定器を備える。
【0069】
図7は、一部の実施形態で使用することのできる水素産生のための装置10を図解する。装置10は新鮮培地保存容器12、ポンプ14、反応システム16、そしてオーバーフロー容器18を含む。新鮮培地保存容器12は、実質的に密閉された環境下にTAP(酢酸培地)またはHSM(最小培地)などの培地を一定量保持することのできる任意の種類の容器を用いればよい。一部の実施形態では、アルゴンガスなど一定量のガス24を導管26を介して新鮮培地保存容器12に送り込んで容器12内の酸素を追い出し、それにより実質的に酸素のない環境を作り出す。導管26には、新鮮培地保存容器への流体(ガスなど)の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。
【0070】
ポンプ14は導管28を介して新鮮培地保存容器12と、導管30を介して反応システム16と液体的に連結する。導管28(そして含まれる場合は導管26)は、新鮮培地保存容器12の実質的に密閉された環境が維持されるように容器12と連結する。導管28には、ポンプ14によるポンプ作用を介して流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。ポンプ14には、新鮮培地保存容器12から反応システム16に一定量の培地を所定の速度で送り込むことができ、新鮮培地に悪影響を及ぼさない任意の種類のポンプを用いればよい。例えば、一実施形態では、ポンプ14はポンプ過程で培地が損傷する可能性が低いペリスタ型ポンプを用いる。
【0071】
反応システム16は反応容器20および撹拌器22を含む。反応容器20は、新鮮培地保存容器12と実質的に同様であり、実質的に密閉された環境下に培地および藻類培養液、または他の種類の宿主細胞を一定量保持することのできる任意の種類の容器を用いればよい。新鮮培地は、導管28、30およびポンプ14を介して新鮮培地保存容器12から反応容器16に送り込まれる。新鮮培地は実質的に酸素のない環境下に保存されるため、反応容器16に不注意に酸素が入り込む可能性が低い。導管30は、反応容器12の実質的に密閉された環境が維持されるように反応容器20と連結する。
【0072】
撹拌器22には、反応容器20と作動的に連結し、反応容器20に保持された培養液の撹拌状態を維持することのできる任意の種類の機器を用いればよい。例えば、撹拌器22は磁気撹拌子アセンブリまたは同種のものなど自動撹拌器を用いてもよい。
【0073】
導管32は反応容器20からあふれた培養液をオーバーフロー容器18に排出する。オーバーフロー容器18は、容器12、20と実質的に同様であってもよく、一定量の培養液を保持することのできる任意の種類の容器を用いればよい。導管32は、導管28、30と実質的に同様であり、反応容器20からオーバーフロー容器18への流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。導管30と同様に、導管28は、反応容器20の実質的に密閉された環境が維持されるように反応容器20と連結する。導管30は、一部の培養液をオーバーフロー容器18に排出することにより反応容器20に入った培養液のレベルが所定レベル(またはそれ以内)に維持されるような配置で、反応容器20と連結されてもよい。
【0074】
一部の実施形態では、容器20内で培養液により産生された水素または他のガスの量が測定できるように、測定器34は通信リンク36を介して反応容器20と連結されてもよい。測定器34には、目的とするガスの量を測定することができる任意の種類の機器を用いればよい。一実施形態では、測定器34は質量分析器を用いるが、他の実施形態では他の種類の測定器を用いてもよい。通信リンク36は、例えば任意の本数のワイヤ、ケーブル、光ファイバーケーブル、チューブ、導管、または同種のものなど、測定器34へのデータ通信を促進することのできる任意の種類の通信リンクであればよい。一実施形態では、通信リンク36は反応容器20内に置かれた電極部分などである。電極部分は例えば銀電極であってもよい。
【0075】
一部の実施形態では、装置10にはフィルターシステム40および二次ポンプ38が含まれてもよい。フィルターシステム40は導管42を介してオーバーフロー容器18と、導管44を介してポンプ38と連結する。ポンプ38は導管46を介して新鮮培地保存容器12と連結する。導管42、44、46は導管28、30、32と実質的に同様であり、流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。ポンプ38はポンプ14と同様で、オーバーフロー容器18から新鮮培地保存容器12に一定量の「使用済み」培養液を所定の速度で送り込むことができ、新鮮培地に悪影響を及ぼさない任意の種類のポンプを用いればよい。例えば、一実施形態では、ポンプ38はペリスタ型ポンプである。フィルターシステム40は、オーバーフロー容器18に保持された培養液をろ過することのできる任意の数および種類のフィルターおよび関連する連結部品を用いればよい。
【0076】
ポンプ14は、稼働時に新鮮培地保存容器12から反応システム16の反応容器20に新鮮培地を所定の速度で送り込むように構成される。一実施形態では、ポンプ14は反応容器20に保持されている藻類の細胞増殖速度と実質的に同じ速度で新鮮培地を反応容器20に供給するように構成される。上記に詳細を示したとおり、実質的に酸素のない環境下で藻類または他の種類の宿主細胞は反応容器20に保持されるので、藻類または他の種類の宿主細胞の中では、水素産生が誘導されるように望ましい遺伝子発現が誘導される。上述したとおり、新鮮培地も実質的に酸素のない環境下で新鮮培地保存容器12に保存されるため、新鮮培地保存容器12から新鮮培地が送り込まれる間も反応容器20の実質的に酸素のない環境は維持される。または、他の実施形態では、反応容器20に保持される藻類または他の種類の宿主細胞は自己誘導される。いずれにせよ、装置10は単相装置であることを理解すべきである。すなわち、藻類または他の種類の宿主細胞は反応容器20に保持され、同じ容器内で増殖し誘導される。
【0077】
新鮮培地が反応容器20に送り込まれる際、反応容器内に保持された培養液が連続撹拌されるように撹拌器22は構成される。また、新鮮培地が送り込まれる際、既存の培養液の一部が反応容器からオーバーフロー容器18に移る。こうして、反応容器20に入っている藻類または他の種類の宿主細胞の量は実質的に一定値に維持される。ポンプ38およびフィルターシステム40を含む実施形態では、ポンプ38はオーバーフロー容器から一定量の「使用済み」培養液を送り出し、それをフィルターシステム40でろ過してその中に含まれる藻類や他の種類の宿主細胞を除去した後で新鮮培地保存容器へと戻す。
【0078】
次に図8を参照すると、一実施形態では、水素産生のための装置50は新鮮培地保存容器52、ペリスタ型ポンプ54、反応システム56、およびオーバーフロー容器62を含む。新鮮培地保存容器52は、新鮮培地保存容器12と実質的に同様であり、例として0.25L容器が用いられる。例としてTAP(酢酸培地)またはHSM(最小培地)など、一定量の新鮮培地64を容器52に保持する。容器52の内腔68が外環境から実質的に密閉されるように、キャップ66を容器52に連結する。導管70はキャップ66と連結し、容器52の内腔68内に位置する末端部72を含む。一定量のアルゴンガス74を導管70を介して内腔68へ送り込み、これにより内腔内の酸素を実質的に追い出す。
【0079】
ペリスタ型ポンプ54は導管76を介して新鮮培地保存容器52と、導管78を介して反応システム56と連結する。ペリスタ型ポンプ54は例としてモデルIP4ペリスタ型ポンプが用いられ、これはIsmatec(スイス、グラットブルグ)から購入できる。ポンプ54は、反応システム56に保持されている藻類培養液または他の種類の宿主細胞の増殖速度と実質的に同じ速度で一定量の新鮮培地を新鮮培地保存容器52から反応システム56に送り込むように構成される。
【0080】
反応システム56は、例として0.25L容器などの反応容器88、および反応容器88に保持された一定量の培地および藻類培養液94または他の種類の宿主細胞を連続撹拌するように構成された、磁気撹拌子システム90を含む。磁気撹拌子システム90は例としてモデルKM02磁気撹拌子が用いられ、これはMilian(スイス、ジュネーブ)から購入できる。反応容器88の内腔92が外環境から実質的に密閉されるように、キャップ92を容器88に連結する。導管78はキャップ92と連結し、反応容器88の内腔94内に位置する末端部96を含む。
【0081】
質量分析器100も通信リンク102を介して反応容器88と連結する。質量分析器100は例としてモデルMM8-80質量分析器が用いられ、これはVG Instruments(英国、チエスター)から購入できる。通信リンク102は容器88の内腔94内に位置する電極104を含む。質量分析器100は、反応容器88に保持されている培地および藻類培養液または他の種類の宿主細胞によって産生される水素量(および一部の実施形態では酸素量)を測定するように設定される。
【0082】
さらに、導管106はキャップ92と連結し、容器88の内腔94内に位置する末端部108を含む。導管106の先端部110はオーバーフロー容器62の内腔内に位置する。導管106は、反応溶液88に保持されている藻類または他の種類の宿主細胞の増殖速度と実質的に同じ速度で、培地および藻類培養液または他の種類の宿主細胞培養液が反応容器88から送り出されるように配置される。
【0083】
上記は本明細書に記載されるすべての方法およびシステムに適用される。以下の例は説明目的のみであり、本文書に添付する特許請求の範囲に記載される本発明の範囲を限定するものではない。本文書で引用した参考資料は本明細書に具体的な参照として含まれる。
【0084】
〔実施例1 内在性遺伝子のための誘導性色素体発現システム〕
Cyc6プロモーターの制御下にNac2遺伝子を含むベクターは、当業者に知られる分子クローニング技術を用いて作成した。クローニング方法は、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0085】
Nac2遺伝子をCyc6プロモーターエレメントの制御下に置くため、Cyc6プロモーターエレメントおよびNac2ゲノムDNAに特異的な4つのオリゴヌクレオチドを用いたオーバーラップ伸長PCRにより、psbDコード配列に融合させたCyc6プロモーターを含むキメラDNA断片を作出した。得られたPCR断片は、833塩基対のNac2ゲノム配列断片にインフレーム融合した428塩基対のCyc6プロモーター配列からなる。PCR断片はサブクローニングして配列を決定した。PCR断片は次に、XbaIおよびAatIIユニーク制限部位を用いてpNac2(midi)プラスミドにクローニングした。pNac(midi)プラスミドは、Nac2の5.1kbキメラmidi遺伝子を含み、これはBoudreauら(2000)により記載されており、本明細書に参照として含まれる。この遺伝子は、3'側cDNA配列に融合した5'側Nac2ゲノム配列からなり、C末端がトリプルHAエピトープでタグされた全長Nac2蛋白質をコードするオープンリーディングフレーム(ORF)がもたらされる。pNac2(midi)をXbaIおよびAatIIで制限分解した。次にこのプラスミドにPCR断片を方向性に連結した。得られたプラスミドpcy6Nac2(midi)は、Nac2 midi遺伝子にインフレーム融合した428塩基対のCyc6プロモーター配列を含む。
【0086】
最後に、pSL17の多重クローニング部位内のユニーク制限部位(EcoRIおよびXbaI、図9参照)を使って、5.8kb cyc6Nac2導入遺伝子をpSL17プラスミドにクローニングした。pSL17は抗生物質パラモマイシンへの耐性を付与するaphVIIカセットおよびクローニングのための多重クローニング部位を含む。得られた10.8kbプラスミドpcy2(paroR)(図10参照)は、nac2-26突然変異細胞の形質転換に用いた。ベクターの構築に用いたNac2およびCyc6の配列をそれぞれ図11と12に示す。
【0087】
pcy6Nac2(paroR)ベクターを、エレクトロポレーションによりnac2ヌル変異体であるnac2-26に導入した。藻類および植物の形質転換法は当業者に知られており、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0088】
Cyc6誘導性Nac2遺伝子を含むChlamydomonas遺伝子導入株を単離するため、nac2-26細胞をまずオートリシン処理して次にpcyc6Nac2(paroR)を用いたエレクトロポレーションにより形質転換した。得られた形質転換株を抗生物質パロモマイシン(20μg/ml)を含むTAP培地にのせた。パロモマイシン耐性コロニーは、150μMニッケル(II)(Cyc6転写インデューサー)を添加した最少培地ならびに銅を含まない最少培地(HSM -Cu+2、図13参照)の上で光独立栄養的に増殖するものを選択した。光独立栄養株は次に、インデューサーを含まない最小培地(HSM)上での増殖の有無を調べた。
【0089】
この方法を使って、遺伝子導入株cy6Nac2.49を単離した(図13参照)。この株はHSM -Cu+2上で光独立栄養的に増殖できるが、銅を添加したHSM上では光独立栄養的に増殖できない。得られたcy6Nac2.49株は、以下の二通りの方法で増殖する:1)銅を含まない培地中で光独立栄養的に増殖、または2)酸素と銅が存在する培地中では細胞内にPSII複合体がないため嫌気的に増殖(図1を参照)。よって、遺伝子導入株cy6Nac2.49では、光合成による酸素産生は低酸素に応答する核プロモーターを介して制御される。
【0090】
さらに、非誘導条件下に増殖するcy6Nac2.49の密閉培養は、光合成による酸素発生がなくミトコンドリアの呼吸による酸素消費は一定に維持されるので、急速に嫌気的となる。cy6Nac2.49の密閉培養では、低酸素が光合成酸素産生を誘導し、それがPSII合成と光合成酸素発生を再び抑制するという、阻害のフィードバックループが存在する。
【0091】
実際に、完全培地中でのcy6Nac2.49の光照射密閉培養ではまさしくこの現象が観察された。培養液中の酸素濃度は、最初は大気中酸素濃度と同じで、その後急速に消費され、嫌気的増殖と水素発生の誘導に至った。一定の嫌気的増殖期間の後、培養液の酸素レベルが回復して容器内酸素濃度が大気中の2倍となり、水素発生が阻害された(データ示さず)。通常の密閉容器では、酸素消費と水素産生が一度ずつしか観測されず、これはおそらく還元炭素がミトコンドリアの呼吸により消費されてしまうと酸素消費は止まるが、光合成による酸素産生は一定に維持されるためと見られる。
【0092】
野生型、nac2-26(cy6Nac2.49の親株)およびcy6Nac2.49細胞について、完全培地(TAP)、抗生物質パロモマイシン添加完全培地(TAP + Paro)、最少培地(HSM)、および銅非含有培地(HSM-Cu+2)上での増殖能を調べた。非誘導条件下(TAP)および誘導条件下(TAP-Cu+2およびTAP-O2)に増殖した野生型およびcy6Nac2.49細胞の全細胞抽出物についてのウェスタンブロット解析を図14に示す。ウェスタンブロットは、嫌気的増殖の指標としてα-HydA、α-D2およびα-RbcLを認識する抗体でプローブした。
【0093】
図15に水素発生を測定した実験結果を示す。Cy6Nac2.49細胞は、培養液中の溶存ガスを質量分析器で測定できる(酸素-点線、水素-直線)光照射容器に密閉し、培地に銅を添加することで誘導条件から非誘導条件に移行させた(TAP-Cu+2からTAPへ)。液相中の水素濃度を測定し、mBar単位で表した。
【0094】
通常の密閉容器は、cy6Nac2.49遺伝子導入株を使って水素産生を持続するのに適さないため、増殖するcy6Nac2.49培養液に新鮮な無酸素培地を一定速度で供給し、対数増殖期を維持できる密閉無酸素システムを設計した(図7、8参照)。培養液を対数増殖期に維持することで酸素消費/水素産生/酸素産生のサイクルを確立できるとする仮説を立てた。cy6Nac2.49細胞をこのシステムで増殖させたところ、培養液が低酸素になったすぐ後に水素発生が観測された。ついでPSII複合体の合成が誘導されるとともに容器内の酸素がわずかに増加した。通常の密閉容器とは異なり、光合成による酸素産生は水素産生を阻害せず、これはおそらく光合成による酸素産生がミトコンドリアの呼吸による酸素消費を決して上回らないためと思われる。このシステムでは、水素産生は光エネルギーと直接連関しており、したがって水素産生のための直接バイオ光分解を表している。無酸素システムを使用して、気相の約0.5%に達する一定速度の水素発生が得られた。
【0095】
光合成の副産物として酸素が発生する。結果として、光エネルギーを使った藻類での水素発生維持(「バイオ光分解」とも呼ばれる過程)における中心課題は、ヒドロゲナーゼ酵素の酸素感受性を克服することであった。本発明で開発した直接バイオ光分解法とは対照的に、間接バイオ光分解法(または光合成と水素産生の二段階法)は光合成による酸素産生と水素産生を時間的または/および空間的に分けることでヒドロゲナーゼ酵素の酸素感受性を克服する(Benemann 1996; Melis 2000)。第一段階は通常の酸素発生型光合成、つまり酸素放出、CO2固定、バイオマス蓄積が見られる。第二段階では、硫黄などの重要な栄養素が枯渇して、酸素発生型光合成が生理学的に阻害される。硫黄欠乏状態が約22時間続くと酸素発生型光合成が急激に減速するので、ミトコンドリアの呼吸による総酸素消費のせいで、密閉培養液は嫌気的になる(Melis 2000)。嫌気的増殖が確立すると、ヒドロゲナーゼ経路が誘導され、主として残存する水の光酸化から、また蛋白質やデンプンなどの内在基質の異化からも得られる電子を使って、水素が発生する(Ghirardi 2000)。
【0096】
無酸素システム、すなわち直接バイオ光分解法と、間接光分解法の違いを見ると、無酸素システムを使用することの大きな利点がいくつか明らかとなる。第一に、無酸素システムの特長として、単一の光照射密閉容器内で酸素産生と水素発生が行われるというだけの理由で、水素産生能は50%高い。光合成と水素産生の二段階法または間接バイオ光分解法は、酸素産生と水素産生を時間的もしくは空間的に分けるので、結果として2つの容器のうち一方が水素産生には使用されず産生能の50%が失われる。第二に、光合成と水素産生の二段階法は硫黄の生理学的枯渇が酸素発生型光合成を阻害することに依存する。硫黄が極度に枯渇すると様々な細胞過程に広く影響する。水素放出を触媒するヒドロゲナーゼ酵素にはFe-Sクラスターが含まれ、その会合が機能性に必要である((Posewitz, et al. (2004))。また、硫黄の枯渇は、水素発生に重要なPSIなどの葉緑体中の他の複合体に悪影響を及ぼす。明らかに、硫黄の生理学的枯渇は水素発生装置の重要な部分に重大な影響を及ぼす。これに対し無酸素システムは、水素産生を誘導するのに必要な微量栄養素の枯渇に依存しない。事実、水素産生は最適な生理学的条件下に(完全培地中での対数増殖期に)起こる。
【0097】
最後に、例えば藻類を使った大規模な水素産生において主な障害となるのは大量培養液に光エネルギーを供給することである。藻類の高濃度大量培養では、集光性複合体中のクロロフィル量が多いために、容器中心付近の細胞に光エネルギーが到達しにくい。本発明の直接バイオ光分解法では、光吸収および水素産生が最大となるような最適細胞濃度の確立されるシステムで水素産生が起こる。
【0098】
〔実施例2 外来性遺伝子のための誘導性色素体発現システム〕
葉緑体形質転換ベクター。プラスミドpSK108は、psbD遺伝子と、psbD遺伝子周辺領域への挿入を導く5'隣接配列を含む3kb葉緑体DNA断片を含む。このコンストラクトはまた、psbD遺伝子の上流に挿入されたaadAカセットも含む。導入遺伝子はpSK108のNcoIおよびSphI部位を使ってインフレームに挿入できる。連結後の新規ベクターは、その発現を担うatpAの5'末端およびターミネーターとして機能するrbcLの3'配列をもつ導入遺伝子(加えて3塩基対も)を含む。atpAプロモーターは葉緑体のATP産生プロトンポンプをコードする遺伝子の発現を担うので、D1修復機構に影響されない。
【0099】
FLAGタグをつけた3種類の外来性遺伝子、VP28、DILPおよびIBDVをそれぞれ葉緑体形質転換・発現ベクターpCG12にサブクローニングした(図17)。このベクターは葉緑体のatpB遺伝子座の下流に挿入される。導入遺伝子はatpAプロモーターの制御下に発現し、psbDの5'UTRの安定化因子結合部位をもつ(図18、19)。
【0100】
これらのベクターを、Chlamydomonasの16S rRNAを有しスペクチノマイシン耐性を付与するp228ベクターとともに、cyc6/Nac2誘導性株(制御プロモーターとしてCyc6プロモーター、安定化因子としてNac2をもつ)へ同時形質転換した。
【0101】
〔実施例3 C. reinhardtii葉緑体でのHydA1、HydEF、およびHydG遺伝子の過剰発現〕
ヒドロゲナーゼがC. reinhardtiiで過剰発現されるかどうか、そしてこれが水素産生を促進するかどうかを検討する。大腸菌でHydEF、HydGおよびHydA1を過剰発現させると活性型Feヒドロゲナーゼが産生されることが、最近の実験で示された(Posewitz, et al, (2004))。C. reinhardtiiの核内で蛋白質を過剰発現させることは、導入遺伝子がサイレンシングを受けることが多いためほとんど成功していない。したがって、遺伝子銃による形質転換を用いてこれらの遺伝子を葉緑体中で発現させる。この方法にはいくつかの利点がある。第一に、葉緑体では遺伝子のサイレンシングが起こらない。第二に、C. reinhardtiiには各葉緑体遺伝子が80コピーずつ存在する。第三に、外来性遺伝子を葉緑体のコドンバイアスに合うように再合成すれば、葉緑体プロモーター、5'および3'UTRの制御下に、葉緑体中で外来性蛋白質を高レベルに発現させることが可能であることが近年の実験で判明している(Mayfield, et al. (2001))。葉緑体遺伝子配列には強いATバイアスがある。
【0102】
HydA1、HydEFおよびHydGのそれぞれのコード配列をまず、C. reinhardtiiの葉緑体コドン使用頻度に従って再合成する。これは、外来性遺伝子を葉緑体中で発現させるのに成功した既報の方法を用いて行う。例として、近年我々はC. reinhardtiiの葉緑体中で2つのウイルス蛋白質を過剰発現させることに成功した(下記参照)。導入遺伝子は、コピー数を2倍にするため葉緑体の逆方向反復配列内に挿入する。宿主株としてcyc6Nac2株を使用する。この株は核nac2変異と、Cyc6プロモーター制御下にNac2遺伝子を有し、Nac2遺伝子は銅枯渇または嫌気的条件下で誘導される。
【0103】
この時点では、HydA1p、HydEFp、HydGpのうちどの蛋白質が水素産生を律速しているかは不明である。これを調べるため、3つの遺伝子のそれぞれにpsbDプロモーターと5'UTRを融合させたものを、遺伝子銃形質転換を使って葉緑体逆方向反復配列に個別に挿入する。可能性があるのは、リボソームオペロンの16Sと23S rRNA遺伝子間のスペーサー配列内に遺伝子を挿入することで、この方法により高等植物の葉緑体で高レベル発現が成功している。導入遺伝子は、Nac2を介したCyc6プロモーターの制御を受けるpsbD 5' URTの制御下にあるため、導入遺伝子は嫌気的条件でのみ発現する。RNAブロットハイブリダイゼーションまたはリアルタイムRT-PCRにより発現を調べる。検討する3つの遺伝子のそれぞれについて、Zhang and Melis (2002)の記載のとおり、上下逆にして水を満たしたビュレットにチューブで接続した密閉瓶内で形質転換細胞を培養し、移動する水の体積から水素産生量を直接測定する。cyc6Nac2の制御と相関して水素産生が増加すれば、過剰発現させた蛋白質が律速であることが示される。これらの実験により複数の蛋白質が水素生産の律速となっていることが分かれば、それらの蛋白質を同じ方法で過剰発現させる。各蛋白質を過剰発現させて水素産生量を高めるように、野生型細胞ではこれら3つの蛋白質の発現が調整されている可能性がある。この場合は3つの蛋白質を過剰発現させる必要がある。高等植物の葉緑体内で複数の導入遺伝子を発現させることは成功している(Quesada-Vargas, et al. (2005))。psbD 5'UTRによる発現が不十分であれば、psbA、atpA、リボソームプロモーターなど、他の強力な葉緑体プロモーター-5'UTRを検討する。
【0104】
〔実施例4 水素産生に影響を及ぼす遺伝子の発現〕
上記に加え、他のいくつかの遺伝子が水素産生を増加させるかどうか、色素体内での蛋白質発現のための誘導性および/または抑制性システムを使って検討する。酸素非感受性ヒドロゲナーゼとアンテナサイズの縮小を2つの例として挙げる(Melias, et al. (2004); Ghirardi, et al. (2005))。簡単に言うと、前者はChlamydomonasや他の生物からの野生型および突然変異型ヒドロゲナーゼ遺伝子をクローニングして酸素感受性が低くなるか解析する。アンテナサイズ縮小という後者の目的は、藻類細胞が捕捉する光子量を減らすことにより、光が光反応器の内部まで貫通し、通常であれば光の当たらない細胞に光を供給することである。藻類細胞は効果的に光を捕捉するがその利用効率は低く、吸収した光子の80%ほどが無駄になる。DNA挿入突然変異法によりアンテナサイズを制御する遺伝子が同定されている。変異体tla1のPSIIおよびPSIアンテナサイズはそれぞれ野生株の50%および65%であった。最後に、呼吸速度が上昇した結果ヒドロゲナーゼを阻害する酸素量が低下していると見られる変異株が複数同定されている(Krause, et al. (2005))。これらの遺伝子をcy6Nac2.49遺伝子導入株に挿入し、水素産生が増加するか検討する。
【0105】
水素産生を律速する重要な因子の一つはカルビンベンソン回路との競合である。ヒドロゲナーゼへの電子伝達を促進する可能性の一つは、カルビンベンソン回路に関与する酵素量を低くしてこの回路の活性を下げることである。例えば、ホスホリブロースキナーゼ(PRK)活性を持たないChlamydomonasの2つの変異株、F60とac214が入手可能なので、PRKを利用することができる。PRK遺伝子を誘導性Cyc6プロモーターに融合し、このコンストラクトをF60に挿入する。この方法の利点は、PSIIとカルビンベンソン回路の両方が銅(または酸素)の非存在下に機能するので還元力を蓄積できる。銅(または酸素)の存在下に両遺伝子の発現は止まる。よってヒドロゲナーゼが誘導される条件下で電子の流れはカルビンベンソン回路からヒドロゲナーゼへと移り、水素産生が促進される。
【0106】
別の例示的な実施形態では、温度感受性PRK変異体をスクリーニングする。この方法では、制限温度でカルビンベンソン回路が止まり、ヒドロゲナーゼへと電子が流れる。PRK変異体を、PCRで作成したPRK変異遺伝子ライブラリーで形質転換する。形質転換体を許容温度(24℃)で最小培地上で選別する。コロニーを別の最小培地にレプリカし、制限温度(32℃)で培養し、この温度で増殖しない変異体を同定する。PRK遺伝子に変異があるかどうかを、PCR増幅と配列決定により確認する。これらの変異体ではカルビン経路を阻害すると蛍光収量が上昇すると見られることから、蛍光によるスクリーニングも可能である。
【0107】
別の実施形態では、ヒドロゲナーゼからの電子の分岐経路として別に循環的電子伝達があり、状態遷移が状態1でブロックされている欠損変異株を用いる。状態遷移は、光条件の変化に伴い光合成収量を最適化するために、LHCII可動部がPSIIからPSIへと可逆的に移動することによりPSII(LHCII)とPSIのアンテナ間での光励起エネルギーを再バランス化することが関与する。状態1では、LHCII可動部がPSIIと結合しており、状態2ではPSIと結合している。さらにChlamydomonasでは、状態1で主に直線的電子伝達が起こるのに対し、状態2では循環的電子伝達が起こる(Finazzi et al., 2002)。よって、状態1でブロックされた変異体では循環的電子伝達が見られない。状態1でブロックされているstt7変異体(Depege et al., 2003)を、Cyc6-Nac2およびCyc6-PRKコンストラクトを含むnac2-26と交配し、PSII活性が抑制された状態で水素産生が増加するかどうか調べる。もしくは、温度感受性PRK変異体が得られれば、これをnac2-26 stt7 Cyc6-Nac2株と交配させる。これらの株について上記の条件下で水素産生を調べる。
【0108】
stt7以外の他の状態遷移欠損変異体を解析する。光合成酸素発生に対するミトコンドリア呼吸の比率が変化するような光合成電子伝達およびミトコンドリア電子伝達の制御変化によっても、状態遷移の変化が間接的に起こりうるので、これらの変異体は特に興味深い。この種の変異体は、野生型細胞に比べて水素産生が上昇していることが報告されている(Kruse et al., 2005)。
【0109】
〔実施例5 外来性遺伝子のための誘導性色素体発現システム〕
誘導性葉緑体遺伝子発現には選択マーカー遺伝子aadAを使用した。この遺伝子は、Nac2依存性に発現させることができ、抗生物質スペクチノマイシンを用いて容易にスクリーニングができ、高い特異活性をもつことが知られている。低レベルのaadA発現でもある程度の抗生物質耐性が得られ、従ってInd41_18でのcy6Nac2制御の「堅固さ」の指標となる(下記)。psbDのプロモーターおよび5'UTRによりaadA遺伝子の発現を制御する葉緑体挿入ベクター挿入が構築されている。
【0110】
nac2-26変異株はすでに記載されている(Kuchka, et al. EMBO J. 7, 319-324; Nickelsen, et al. EMBO J. 13, 3182-3191)。cyc6Nac2.49株は、Nac2 midi遺伝子と融合したCyc6プロモーターを含む導入遺伝子がnac2-2変異体の核ゲノムに挿入されている(Δnac2::cy6proNac2)。Ind41はcy6Nac2.49のpsbDプロモーターおよび5'UTRをpetAのプロモーターおよび5'UTRを含む675bp断片に置換したものである(Δnac2::cy6pro Nac2::5'petA-psbD)。Ind41_18はInd41株と関連しているが、Ind41_18ではaadAカセットが葉緑体DNAから完全に削除されているためスペクチノマイシンに感受性である(Δnac2::cy6proNac2::5'petA-psbD[SpcS])。Ind_aadA_117はInd41_18から得られたもので、atpB遺伝子の下流に挿入されたpsbDプロモーターおよび5'UTRの制御下にaadAカセットを含む(Δnac2::cy6proNac2::5'petA-psbD::5'psbD-aadA)。
【0111】
<Ind_aadA遺伝子導入株のスクリーニング>
Ind41_18細胞の葉緑体ゲノムへの5'psbD-aadAカセットの挿入は、遺伝子銃形質転換を用いて銅枯渇状態のInd41_18細胞にpcg12葉緑体挿入ベクターを導入することにより得られた(図21)。銅を含まないスペクチノマイシン100μg/mL含有TAP培地上で形質転換株を選択した。形質転換で得られたコロニーを拾って、スペクチノマイシン100μg/mL含有TAP-Cu+2培地に3回移植し直し、5'psbD-aadAカセットを含む細胞を確実に分離した。
【0112】
実験に使用したpcg12ベクターの概要図を図21に示す。このベクター内のaadAカセットはpsbD 5'UTRを使って発現する。野生型Ind41_18およびInd_aadA_X遺伝子導入株の生育も示す。これらの細胞を段階希釈し、固形TAP培地、銅非含有TAP培地(TAP-Cu+2)、スペクチノマイシン100-1000μg/mL含有TAP培地(TAP-Cu+2+Spc)、およびスペクチノマイシン100-1000μg/mL含有TAP-Cu+2培地上に滴下し、光強度100 μE m-2s-1の照射下に7-10日間培養した。
【0113】
誘導性aadA形質転換株の候補をTAP + Spc100およびTAP-Cu+2 +Spc100寒天培地上にレプリカし、スクリーニングを行った。試験したコロニーはすべて両培地で同様に増殖し、Cyc6Nac2導入遺伝子のプロモーター制御が失われていることを示した(図21A)。しかし、高濃度のスペクチノマイシンで増殖を調べたところ、これらの株の85%は培地中に銅が含まれるとスペクチノマイシンに感受性を示し、培地中に銅が含まれないと耐性を示した(図21B)。こうした株のいくつかをInd_aadAと命名し、さらに解析を行った。これらの株の遺伝子型はnac2::cy6ProNac2::5'petA-psbD::5'psbD-aadAである。
【0114】
<Ind_aadA形質導入株の増殖解析>
一次スクリーニングからのいくつかの誘導性株の増殖解析では、段階希釈した後、野生型、Ind41_18、Ind_aadA細胞をTAP、TAP-Cu+2、HSM、HSM-Cu+2、および一定範囲内で各種濃度のスペクチノマイシン(0、100、250、500、750、1000、2000 μg/mL)を含む寒天培地に滴下して行った(図21B)。この方法で試験した11株中の1株、Ind_aadA_36は、500 μg/mLより高濃度のスペクチノマイシンを含むTAP培地で増殖を示した。一方、11株中6株は銅を含まないTAP培地で低濃度のスペクチノマイシンでは増殖したが、250 μg/mLを超えるスペクチノマイシンでは増殖しなかった(図21B)。Ind_aadA_117と命名した株は、TAP培地では250 μg/mLのスペクチノマイシンに感受性を示したが、銅を含まないTAP培地では試験したスペクチノマイシン濃度範囲のすべてで増殖した。
【0115】
<Ind_aadA_117から抽出した全RNAのノーザン解析>
Ind_aadA_117でのaadA発現誘導を解析するために、非誘導性(TAP)または誘導性条件(TAP-Cu+2、TAP-Cu+2 + Spc、TAP-O2、HSM-Cu+2)で培養したInd_aadA_117細胞から全RNAを抽出した。これらの試料をノーザン解析した結果、psbD転写物の蓄積は親株Ind41_18と同等、もしくは野生型のpsbD RNAの約25%であることが判明した。予想どおり、Ind_aadA_117のpsbD RNAは野生型psbD転写物よりも大きく、親株Ind41_18からの遺伝形質として、真のpsbD遺伝子はもはや存在しないことを示した(図22)。重要な点として、Cyc6転写が誘導されているすべての培養細胞でaadA転写物の蓄積が認められた。TAP培養したInd_aadA_117細胞でaadA RNAが少量検出されたことから、Ind_aadAのリーキーな表現型が確認された(図22)。驚くべきことに、銅枯渇培養したInd_aadA_117細胞よりも嫌気培養したInd_aadA_117細胞の方がaadA転写物は多く、psbD/D2発現に関して「親株の親株」であるcy6Nac2.49には見られない特徴であった(図22)。図22に示した分析では、TAP、TAP-Cu+2、TAP-Cu+2 +Spc、TAP-O2 +Spc、HSM-Cu+2およびHSM-Cu+2+Spc液体培地中で培養した野生型、およびInd_aadA_117細胞から全RNAを抽出した。図22の左側に使用プローブを示す。
【0116】
<Ind_aadA_117蛋白質抽出物の解析>
Ind_aadA_117細胞から抽出した総蛋白質を、葉緑体にコードされる重要な蛋白質のいくつかに特異的な抗血清を使って免疫ブロット解析した。検討したすべてのInd_aadA_117培養細胞でD2蛋白質レベルは野生型の約25%であり、これはD2蛋白質を50%蓄積した親株Ind41_18よりも少なかった(図23)。一方、検討したほとんどのInd_aadA_117蛋白質抽出物でD1およびCP47蛋白質の蓄積はほぼ完全に回復しており、D2蛋白質の蓄積は75%低下していると推定された。よって、驚くべきことに、D1とCP47の蓄積はD2蛋白質レベルに対して調整されなかった。各種条件で培養したInd_aadA_117から調製した可溶性抽出物を使って、Ind_aadA_117でのNac2蛋白質の蓄積を調べた。嫌気培養および銅枯渇培養したInd_aadA_117からの可溶性抽出物ではCyc6Nac2の誘導が認められたが、TAP培養したInd_aadA_117で少量のNac2が検出された。Cyc6の発現が抑制される条件でのTAP培養細胞で微量のNac2が検出されたことから、このシステムはInd_aadAではわずかに「リーキー」であることが示された(図23)。したがって、TAP培養されたInd_aadA形質導入株が低濃度のスペクチノマイシンで増殖したという当初の観察結果は、Cyc6Nac2のややリーキーな性質による。図23では、TAP、TAP-Cu+2、TAP-Cu+2 +Spc、TAP-O2 +Spc、HSM-Cu+2およびHSM-Cu+2+Spc液体培地中で培養した野生型、およびInd_aadA_117細胞から総蛋白質を抽出し、PVDFメンブレンに固定した。図23の左側に使用プローブを示す。
【0117】
<Ind_aadA_117のaadA分析>
aadAの量は活性測定により間接的に推定した(表1)。活性測定はaadA酵素がATP分子のアデニル基をスペクチノマイシンに転移し、スペクチノマイシン分子に正の電荷を与えることに基づく。正の電荷をもつスペクチノマイシン分子は、負の電荷をもつリン酸セルロース膜に結合する。よって、もしaadAを発現するChlamydomonasの粗抽出物をスペクチノマイシンおよびα32P標識化dATPとともにインキュベートした場合、その反応液をリン酸セルロース膜上に滴下してから非特異的産物を洗い落とした後に膜に残る放射能量から、aadA酵素活性の相対的測定が可能である。TAP、TAP-Cu+2、またはTAP-Cu+2 +Spc液体培地で培養した細胞の粗抽出物について、この遺伝子導入株中のaadA活性を測定し、親株および野生型バックグランドで従来のaadAカセットを発現する他の株と比較した。いくつかの個別の分析結果を合わせて表1に示す。Ind41_18ではaadA活性が検出されず、この株ではカセットが完全に除去されていることを示した。一方、従来のaadAカセットを発現する野生型およびTAP-Cu+2やTAP-O2で培養したInd_aadA_117のどちらも、既報のaadA発現遺伝子導入株と同等のaadA活性を有した。TAP培地で培養したInd_aadA_117の粗抽出物中に、少量だが有意なaadA活性が存在した。この結果から、誘導されていないInd_aadA_117にも、誘導されたInd_aadA_117の活性よりはかなり少ないが、少量のaadA活性が存在することが確認された。
【0118】
cy6Nac2葉緑体誘導性遺伝子発現システムを使ったaadA遺伝子発現を誘導できる株のスクリーニングでは、常に「リーキー」な表現型を示す株が得られた。親株ではCyc6Nac2導入遺伝子が緊密に制御されていることが明らかなので、Ind_aadAではCyc6Nac2導入遺伝子が脱抑制されることがスクリーニングを生き残るのに必要であると推測される。この観測結果の解釈の一つとして、遺伝子銃によるInd41_18のpcg12形質転換に使用した培地を含め、スクリーニングの各段階で使用した銅枯渇培地には初めから微量の銅が混入していた可能性がある。もしこれが正しければ、銅の混在濃度が1細胞あたり2x106イオンを下回るまでCyc6転写が一時的に抑制されると予想され、結果としてCyc6Nac2が脱抑制された形質転換株のみが、pcg12プラスミドによる最初の形質転換を生き残ると考えられる。言い換えると、Cyc6Nac2の転写抑制が十分に長くてスペクチノマイシン含有培地で細胞が生存するのに負の影響を及ぼしたとすれば、最初の形質転換を生き残ったすべてのコロニーは、Cyc6Nac2が脱抑制されていなければ分裂しなかっただろう単一の細胞から派生したと考えられる。微量の銅が混入しただけでもCyc6の転写が抑制されたことからも明らかなように、培地への混入は避けられないと考えられる。事実、cy6Nac2.49での銅を介した抑制のキネティクスを検討するために計画した実験では、銅枯渇cy6Nac2.49細胞を銅を含まない培地に再懸濁すると、細胞分裂が1、2回行われる間に、真のCyc6遺伝子座の転写と共にCyc5Nac2の転写が一時的に抑制された。
【0119】
それでもなお、Ind_aad_117遺伝子導入株は、Chlamydomonasにおいて核にコードされるNac2蛋白質に基づいた誘導性葉緑体遺伝子発現システムの設計が可能であることを示す。psbD 5' UTRで目的とする遺伝子の発現を制御できるならば、Ind41_18遺伝子導入株を使って、葉緑体ゲノムに導入されたpsbD 5'UTR制御下にあるキメラ遺伝子の発現を誘導することができる。
【0120】
表1の分析では、誘導性および抑制性条件下にInd41_aadA-117のアミノグリコシドアデニルトランスフェラーゼ活性を測定した。野生型-aadA、Ind41_18およびInd41_aadA-117株からの抽出物について、aadA活性および総蛋白質含量を測定した。活性は蛋白質1μgあたりに取り込まれた放射能cpmを示す。個別の測定の数をカッコ内に示す。
【0121】
〔表1〕

【0122】
<VP28_FLAG、IBVD_FLAG、およびDILP_FLAGの誘導性発現>
本明細書に記載した誘導性葉緑体遺伝子発現システムが外来性蛋白質の産生に応用できるか調べるため、3つの異なる外来性蛋白質VP28、DILP、およびIBDVを用いて5'psbDの制御下に導入遺伝子の異種発現を行った。IBDV(またはVP2)は家禽での伝染性ファブリキウス嚢病ウイルス(IBDV)対策用ワクチンの作成に使用されている(Mundt 1995)。ブラックタイガー(Penaeus monodon)のエビホワイトスポット病ウイルスの主要構造エンベロープ蛋白質の23kD断片VP28は、サブユニットワクチンとして飼料添加するとエビを保護することが示された(Witteveldt 2004)。
【0123】
C. reinhardtiiの葉緑体ゲノム機構のコドンバイアスに最適化してFLAGタグをつけた3種類の外来性遺伝子VP28、DILPおよびIBDVは、Stefan Surzycki博士(インディアナ大学ブルーミントン校)から提供を受け、psbD 5'UTRの制御下にrbcL 3'UTRをターミネーターとして外来蛋白質が発現するように個別に葉緑体形質転換・発現ベクターpcg12にサブクローニングした(図24A)。これらの新規コンストラクトは選択マーカーを持たないため、形質転換体を作出するには選択マーカーをもつ別の葉緑体挿入ベクターと同時形質転換する必要があった(図24A)。
【0124】
ショウジョウバエのインシュリン様ペプチドの発現を誘導する実験で使用したpcg12_DILPベクターの概要図を図24Aに示す。黒塗り部分はDILPコード配列を示す。矢印はpsbD遺伝子の5'リーダーを示す。星印は3HA-11エピトープの挿入を示す。図24Bでは、Ind_VP28(左上)、Ind_IBVD(右上)、Ind_DILP(下)からの総蛋白質抽出物を使った免疫ブロットを、FLAGエピトープを認識する抗体でプローブしたものを示す。蛋白質の予測分子量は、VP28が23kD、IBDVが49kD、DILPが12kDであり、星印で示してある。
【0125】
これらのベクターと、ycf1遺伝子および隣接する葉緑体配列およびスペクチノマイシン耐性を付与するaadAカセットを有するpY1_INTベクターを、Ind41_18株に同時形質転換した。スペクチノマイシン添加TAP寒天培地上で選択した後、外来性遺伝子を増幅するオリゴヌクレオチドを使ったPCRにより、形質転換体に外来性遺伝子が存在するかどうか調べた。導入遺伝子の挿入についてPCRで試験した10コロニーのうち、7つがVP28およびDILPを保有しており、5つがIBDV遺伝子を保有していた。これらの細胞株はIndVP28_x、IndDILP_xおよびIndIBVD_xと命名され、nac2.Cyc6pro Nac2::5'petA-psbD::5'psbD-VP28/DILP/IBVDの遺伝子型を有していた。
【0126】
葉緑体ゲノム内に外来性遺伝子の挿入が確認されたコロニーは、銅枯渇による遺伝子誘導後にFlag(登録商標)抗体を用いた免疫ブロット解析で蛋白質産生を調べた。この方法で調べた22の形質転換株のうち、8株中8株でVP28蛋白質が蓄積していると見られ、IndVP28からの抽出物中に野生型や非誘導コントロールには見られない23kD蛋白質が誘導されていた(図24B、左上)。分析したIndDILP株6株のうち4株で、発現誘導した場合の総蛋白質抽出物で>25kD蛋白質が蓄積した(図24B、下)。この方法で分析したIndIBVD株7株のうち3株で50kD蛋白質が蓄積した(図24B、右上)。
【0127】
図25は、一次スクリーニング実験でDILP_FLAGの発現誘導が見られたIndDILP株からの総蛋白質抽出物の免疫ブロットを示す。IndDILP株を、50 μg/mlスペクチノマイシン含有TAP(ui)またはTAP-Cu+2(i)液体培地で培養し、15%SDS-PAGEゲルで大きさによる分画を行った。得られた免疫ブロットは次にFLAGエピトープを認識する抗体とともにインキュベートした。実験ではFLAGタグAlb3.1蛋白質を発現する遺伝子導入株からの総蛋白質を、FLAGエピトープ検出の陽性コントロールとして用いた。星印は、pcg12_DILPベクターに挿入されたDILP_FLAGペプチドの予想サイズを示す。
【0128】
DILP蛋白質を発現する3つのIndDILP株から抽出した蛋白質はさらに、Cyc6転写を抑制した状態と誘導した状態の細胞から抽出した蛋白質を比較した。調べた4つのIndDILP株のうち2つはDILP_FLAGを12kD蛋白質として正しく誘導していると見られた。したがって、本発明で確立した葉緑体誘導性発現システムはDILPの発現を誘導した。Chlamydomonas reinhardtiiの葉緑体中で構成的にDILP蛋白質を発現する今までの試みでは高レベル発現が得らていなかったことから、この結果は特に興味深い(Stefan Surzycki、私信)。よって、この誘導性葉緑体遺伝子発現システムは、外来性蛋白質、特に構成的発現が得られにくい蛋白質の誘導性発現のための商業的に重要なツールとなりうる。
【0129】
〔実施例6 水素産生に影響を及ぼす遺伝子の発現〕
<細胞株および培地>
これまでにnac2-26変異株が報告されている(Kuchka, et al. EMBO J. 7, 319-324; Nickelsen, et al. EMBO J. 13, 3182-3191)。cyc6Nac2.49株は、Nac2 midi遺伝子と融合したCyc6プロモーターを含む導入遺伝子がnac2-26変異体の核ゲノムに挿入されている(Δnac2::cy6proNac2)。Ind41はcy6Nac2.49のpsbDプロモーターおよび5'UTRをpetAのプロモーターおよび5'UTRを含む675塩基対断片で置換したものである(Δnac2::cy6pro Nac2::5'petA-psbD)。Ind41_18はInd41株と関連しているが、Ind41_18ではaadAカセットが葉緑体DNAから完全に削除されているためスペクチノマイシンに感受性である(Δnac2::cy6proNac2::5'petA-psbD[SpcS])。Ind_aadA_117はInd41_18から得られたもので、atpB遺伝子の下流に挿入されたpsbDプロモーターおよび5'UTRの制御下にaadAカセットを含む(Δnac2::cy6proNac2::5'petA-psbD::5'psbD-aadA)。細胞株はすべて、1.5%バクトアガー含有TAP(トリス-酢酸-リン酸)培地上で弱光下、25℃で維持した。銅含有または銅非含有(-Cu+2)固形寒天および液体TAPおよびHSM培地を使用した実験では、QuinnおよびMerchant(Methods in Enzymol. 297, 263-279)の方法に従い培地を調製した。TAPおよびTAP-Cu+2培地は、必要に応じて100μg/mLスペクチノマイシン(シグマ-アルドリッチ)または20μg/mLパラモマイシン(シグマ-アルドリッチ)を添加した。酸素を欠乏させた細胞を使う実験では、液体培養に窒素ガスを通気しながら150rpm/minで撹拌し連続光照射(20 μE/m-1s-2)を行った。細胞濃度は血球計数板を使って計測した。
【0130】
<プラスミドの構築>
標準的な方法を使ってすべてのプラスミドコンストラクトの操作および分析を行った。コンストラクトの配列決定はBigDyeターミネーター配列決定キット(Applied Biosystems、ラホーヤ、CA)およびABI Prism 377自動配列解析装置(ABI)を使用して行った。クローニングに使用した細菌宿主は大腸菌DH10B(Amersham Biosciences)である。すべてのオリゴヌクレオチドの作成はMicrosynth GmbH(バルガッハ、CH)に発注した。
【0131】
<Chlamydomonas細胞の形質転換>
Chlamydomonas reinhardtii nac2-26株の核形質転換は、基本的にShimogawaraら(Genetics 148, 1821-8)のエレクトロポレーション法により行った。
【0132】
<クロロフィルおよび酸素発生速度の測定>
酸素発生および呼吸速度は、X型光源に接続したクラーク型酸素電極を使用して25℃で測定した(Hansatech Instruments Ltd.、ノーフォーク、英国)。
【0133】
<水素測定および算出>
N2、O2、H2およびCO2の液相および気相測定は以下のとおり行った。溶存ガスの連続モニタリングは、密閉できる温度自動調節式のクラーク型容器を使用し、連続撹拌下、光ファイバー照明器(モデルKL 1500、Schott、マインツ、ドイツ)による連続照射にて、ガスをポリプロピレン膜を通し質量分析器(モデルMM880、VG Instruments、チェシア、英国)のイオン源に供給して測定した。Cy6Nac2導入遺伝子の転写を抑制する実験では、容器内の培地に12μMの銅を添加した(TAP-Cu+2からTAP)。すべての気相計測点の前に、空気および純粋水素ガス試料を直接イオン源に注入して質量分析器の較正を行った。
【0134】
<aadA活性の測定>
aadA活性の測定は野生型、Ind41_18、およびInd_aadA_117株について行い、基本的にはGoldschmidt-Clermont(Nucleic Acids Res 19, 4083-9)の方法に従ったが、元の実験で用いられた放射性標識化rATPの代わりに32P-標識化dATPを使用した。
【0135】
<プラスミド構築‐pRS1_rcy_aadAの構築>
プラスミドpRS1_rcy_aadAの構築にはプラスミドpKS-108#14を使用した。pKS-108#14プラスミドは、C. reinhardtii葉緑体DNA EcoRI R3断片を含み、aadAカセットはpsbD ATG開始コドンから-263塩基対の位置に挿入されている。psbD 5'UTRをpetA 5'UTRで置換するために、オリゴヌクレオチドRS1、RS2、RS3およびRS4を用いたオーバーラップ伸長PCRにより、psbDコード配列に融合したpetA 5'UTRを含む943塩基対キメラDNA断片を作成した。得られたPCR産物をPvuII/ClaI制限エンドヌクレアーゼで制限分解し、同じ酵素で制限分解したプラスミドpKS-108#14に連結してpRS1プラスミドを作成した。葉緑体誘導性発現システムの設計は、2つの連続した葉緑体形質転換を伴い、それには2つの異なる選択マーカーが必要となる。もしくはaadAの再利用が可能であり、、483bp直接反復配列を両末端に隣接させた改変型aadAカセットを用いて、選択圧を取り除いた後に相同組換えによりカセットが効率よく除去されるようにする。再利用可能なaadAカセットをpRS1にクローニングするため、まずpRS1をClaIとSphIで切断し、得られた6.3kbプラスミドの5'および3'末端をT4 DNAポリメラーゼで平滑化した。この結果、pRS1プラスミドからatpAプロモーターおよびaadAコード配列に融合した5'UTRが切り出されるが、rbcLの3'配列は除かれなかった。再利用可能なカセットをpRS1ΔaadAに挿入するため、まず制限エンドヌクレアーゼを使ってpKS-483-aadA-483プラスミドから再利用可能なaadAカセットを切り出し、両端をT4 DNAポリメラーゼとPNKキナーゼで平滑化した。2.8kbの再利用可能なaadAカセットをpRS1ΔaadAに平滑末端連結し、pRS1_rcy_aadAを作出した(図27)。
【0136】
<pcy6Nac2(paroR)の構築>
Nac2コード配列にインフレーム融合した428bpのCyc6プロモーター配列を含むプラスミドの構築には、Nac2の5.1kbキメラmidi遺伝子を用いた。手短に言うと、プラスミドpKS(-)nac2(midi)は、3HA、6Hisおよび9Mycエピトープでタグした3' cDNA配列を含む1.96kbのSfrI/XhoI断片を、Nac2コード配列とインフレームになるよう終止コドンのすぐ上流に導入したNac2コード配列内のSfrI制限部位で終わる3.0kbの5' Nac2ゲノム配列を含む。Nac2遺伝子をCyc6プロモーターの制御下に置くため、Cyc6プロモーターエレメントおよびNac2ゲノムDNAに特異的な4つのオリゴヌクレオチドを用いたオーバーラップ伸長PCRにより、Nac2コード配列に融合させたCyc6プロモーターを含むキメラDNA断片を作出した。得られたPCR断片は、833bpゲノムNac2断片に融合した428bpのCyc6プロモーター断片からなる。このPCR断片は次に、pKS(-)Nac2(midi)のXbaIおよびAatIIユニーク制限部位を用いてpNac2(midi)プラスミドにクローニングした。最後に、pSL17のユニーク制限部位EcoRIおよびXbaIを使って、5.8kb Cyc6Nac2導入遺伝子をpSL17プラスミドにクローニングした。このプラスミドはパロマイシン耐性を付与するaphVIIカセットを含む。得られた10.8kbプラスミド、pcy2(paroR)は、nac2-26突然変異細胞の形質転換に用いた。
【0137】
<Chlamydomonas細胞の形質転換>
nac2-26細胞はTAP培地で培養し、対数増殖期中期(2-4 x 106 cells/mL)で回収し、配偶子オートリシンで処理し、40 mMショ糖添加TAP培地に懸濁した。各エレクトロポレーションでは、処理細胞108個を2.5 μgの直線化したpcyc6Nac2(paroR)またはpSL17プラスミド(エレクトロポレーション効率の対照)、サケ精子DNA 50 μgとインキュベートし、0.2mLエレクトロポレーションキュベット(バイオラッド、米国)に入れてバイオラッド(SIC)を使用し0.75kV、25μF、抵抗ゼロの設定で形質転換した(バイオラッド、米国)。処理した細胞は1mLの新鮮TAP、40mMショ糖、0.4%PEG-8000、20%デンプン培地中で10分間回復させ、抗生物質パロモマイシン(20μg/mL)含有TAP培地にのせた。パロモマイシン耐性コロニーは、銅非含有最少培地(HSM-Cu+2)上で強光下、25℃で光独立栄養的な増殖能についてスクリーニングした。光独立栄養株は次に、最小培地(HSM)上での増殖の有無を調べた。
【0138】
ヘリウム加圧式微粒子銃を用いて、Chlamydomonas葉緑体の遺伝子銃による形質転換を行った。TAP培養したcy6Nac2.49細胞108個を100μg/mLスペクチノマイシン含有TAP寒天培地(TAP + Spc100)にのせ、1μgのpRS1_rcy_aadAプラスミドDNAをコートしたタングステン粒子を射入した。弱光下(5 μEm-2s-1)で2週間後、単一コロニーを拾ってTAP + Spec 100培地で4回クローン化し、弱光下(5 μEm-2s-1)25℃で培養し、細胞株が選択マーカーについてホモプラスミックであることを確実にした。光独立栄養的な増殖を調べるため、細胞を固形HSM培地にのせ、中光下(45 μEm-2s-1)25℃で培養した。Ind41_18の形質転換の場合は、TAP-Cu+2で培養した細胞108個を100μg/mLスペクチノマイシン含有TAP-Cu+2寒天培地(TAP-Cu+2 + Spc100)にのせ、1μgのpcg12プラスミドDNAをコートしたタングステン粒子を射入した。弱光下(5 μEm-2s-1)で2週間後、単一コロニーを拾ってTAP-Cu+2 + Spec 100培地で3回クローン化し、弱光下(5 μEm-2s-1)25℃で培養した。
【0139】
<増殖解析>
野生型、nac2-26、cy6Nac2.49株の増殖解析では、細胞をTAP-Cu+2培地にて濃度2-4 x 106 細胞/mLまで培養し、ついで濃度1 x 106 細胞/mLに希釈し、10倍段階希釈を繰り返して最終的に1プレートあたり100個の細胞が含まれるようにした。10μLの希釈液を適切な固形寒天培地にのせ、強光下に25℃で10日間培養した。Ind41およびInd41-18株の場合は、適切な培地に細胞103個ずつのせて連続光(100 μEm-2s-1)下に25℃で10日間培養した。誘導性aadA遺伝子導入株の増殖解析では以下のような実験を行った。手短に言うと、Ind41-18およびInd_aadA遺伝子導入株をTAP-Cu+2またはTAP液体培地で培養し、つぎに新鮮なTAP-Cu+2またはTAP液体培地に3回移植し直した。TAPおよびTAP-Cu+2での培養液を段階希釈し、0-1000 μg/mLの範囲で段階的に濃度を変えたスペクチノマイシン含有TAPまたはTAP-Cu+2固形寒天培地にのせ、連続光(100 μEm-1s-2)下に25℃で10日間培養した。
【0140】
<蛍光遷移>
蛍光遷移解析を行った。TAP寒天培地で暗所にて培養した細胞を、5分間暗順応させてから植物効率分析計(Plant Efficiency Analyzer、PEA、Hansatech Instruments、英国)で測定した。
【0141】
<RNA解析>
野生型、nac2-26、cy6Nac2.49、Ind41_18およびInd_aadA_117株からの全RNA単離は、RNA植物ミニRNA抽出キット(キアゲン Ghmb、ドイツ)を用いて使用説明書に従い行った。経時変化を調べるためのRNA試料の場合、細胞108個を3000gで遠心分離して、RNAeasy RNAプロテクション溶液(アンビオン、米国)を用いて使用説明書に従い処理した。
【0142】
RNAブロット解析を行った。RNA(2μg)を1.2%アガロース‐4%ホルムアルデヒドゲルにて1X MOPS緩衝液中で電気泳動し、つぎに20X SSC緩衝溶液中でHybond N+ナイロン膜(アマシャム、米国)に転写し、ストラタリンカーUVクロスリンカーを用いてRNAを膜に固定した。膜のプリハイブリダイゼーションおよびハイブリダイゼーションは変法チャーチハイブリダイゼーション溶液(0.5Mリン酸緩衝液pH7.2、7w/v%SDS、10mM EDTA)中65℃で行った。プラスミドpks-108#14をAccIおよびStyIで制限分解してpsbDの380bpDNA断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。Cyc6 cDNAの685bpDNA断片をランダムプライミング法で[α32P]dCTP標識した。pcg12プラスミドを制限分解してaadAコード配列の804bp NcoI/SphI断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。pcg12をEcoRVおよびHpaIIで制限分解してatpBの693bp断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。ハイブリダイゼーション後に、膜を高ストリンジェント洗浄緩衝液[0.1% SDS、0.1% SSC]で10分間洗浄した。
【0143】
<蛋白質解析>
Chlamydomonasの野生型、nac2-26、cy6Nac2.49、Ind41_18およびInd_aadA_117株からの総蛋白質抽出物は、1.5mLエッペンドルフチューブに細胞3 x 106個を採取し、ペレットをシグマプロテアーゼ阻害剤カクテル(シグマ-アルドリッチ、米国)2倍溶液に懸濁し、同量の細胞溶解緩衝液(100 mM Tris-HCl pH 6.8、4% SDS)を添加して37℃で30分間置き溶解させた。細胞片を沈降させるため、試料を10,0000gで5分間遠心分離し、上清を総蛋白質抽出物として使用した。蛋白質濃度の測定は、5μLの上清をブラッドフォード法(バイオラッド蛋白質アッセイ、バイオラッド、米国)で分析した。
【0144】
免疫ブロット解析では、20μgの総蛋白質を12%SDSポリアクリルアミドゲルで泳動分離し、Protran 0.45μmニトロセルロース膜(Schleicher and Schuell)に転写した。Nac2抗体を使用する際は、80μgの蛋白質を8%ポリアクリルアミドゲルで泳動分離した。トリス緩衝生理食塩水(5%脱脂粉乳、0.1%Tween-20含有)(TBS-T)中で膜をブロッキングした。一次抗体反応では、以下のようにTBS-Tに抗体を希釈した。D2抗体、1:10,000希釈;D2抗体1:10,000希釈;Nac2抗体、1:10,000希釈;PsaA抗体1:10,000希釈;AtpB抗体、1:10,000希釈;RUBISCO-ホロ酵素抗体、1:50,000希釈。インキュベートは室温で1時間行った。つづいて、膜を1%脱脂粉乳含有TBS-Tで5分間洗浄し、これを5回繰り返した。二次抗体反応では、10,000倍希釈したペルオキシダーゼ結合抗ウサギIgG(1%脱脂粉乳含有TBS)と膜を室温で1時間インキュベートした。膜をTBSで5分間洗浄することを5回繰り返し、増強化学発光法でシグナルを検出した。
【0145】
<銅を介した抑制および経時変化実験>
cy6Nac2.49遺伝子導入株での銅を介したCy6Nac2抑制の経時変化を調べるため、cy6Nac2.49細胞をTAP-Cu+2培地で濃度4 x 106 細胞/mLまで培養し、新鮮TAP-Cu+2培地で濃度5 x 105 細胞/mLに希釈し、ついで2つの独立した培養液に分けて、一方はそのまま、他方には銅を最終濃度6 μMとなるよう添加した。それぞれ8時間ごと40時間後まで経時的に分析した。FV/FM測定には2つの独立した試料を使用し、各培養液での平均を各時点で算出した。
【0146】
銅を介したcy6Nac2.49の誘導を調べる実験は、銅を介した抑制実験と同様に行ったが、ただしcy6Nac2.49をTAP培地で培養し、細胞を銅非含有培地で2回洗浄してからTAP-Cu+2培地で濃度5 x 105 細胞/mLに希釈した。経時変化実験を開始する際は、細胞を2つの独立した培養液に分けて、一方に銅を最終濃度6 μMとなるよう添加した。
【0147】
<水素測定>
硫黄枯渇下にcy6Nac2.49株での水素産生を野生型と比較した。図32で使用したcy6Nac2.49培養液では、1サイクルの間に20 μmol/Lの水素が産生され、これは最大速度1 mmol H2 mol-1 Chl s-1に相当した。水素産生速度は実験ごとに異なり、3.1 mmol H2 mol-1 Chl s-1に達する場合もあった。光合成の許される条件下(Cu非含有培地)では、Cy6Nac2.49細胞での総酸素発生速度は23 mmol O2 mol-1 Chl s-1であった。これは野生型細胞の1.5〜2倍であった。よって、水素産生の最大速度は総酸素産生の4から13%の範囲だった。100時間硫黄枯渇状態においた培養液の場合、平均値は4 mmol H2 mol-1 Chl s-1と推定される。cy6Nac2.49細胞で1サイクルの間の水素産生20 μmol H2/Lと、野生型で100時間硫黄枯渇状態の水素産生4 mmol H2/Lとを比べた場合、同等の水素産生を達成するためにはcy6Nac2.49システムには、1サイクルでの効率向上または複数サイクルの連続など、さらに改良が必要である。原理的にはChlamydomonasに水素産生を向上させるような遺伝的改変を加えることが可能である。例えば、循環的電子伝達を行えないよう状態1でブロックされた状態遷移変異体を用いる、またはカルビン-ベンソン酵素の1つをCyc6プロモーターの制御下に置くなどである。これにより、PSII活性と同時に炭素同化も現象し、水素産生段階での電子の競合が低下する。
【0148】
〔表2 オリゴヌクレオチド一覧〕
【0149】
Chlamydomonasでは天然の誘導性葉緑体遺伝子発現システムがない。そうしたシステムを、核にコードされる葉緑体Nac2蛋白質の特性を利用して開発した。この蛋白質はPSIIのD2反応中心ポリペプチドをコードするpsbD mRNAのプロセシングおよび安定蓄積に必要である。Nac2のターゲット部位は74塩基psbD 5'UTR配列内に含まれる。この5'UTRを別のコード配列に融合することでその遺伝子の発現はNac2依存性になる。Nac2コード配列を、チトクロームC6遺伝子のCyc6プロモーターに融合した。C6遺伝子の発現は銅枯渇、嫌気的増殖、そしてニッケル添加によって誘導されるが、銅が充分に存在する条件下では抑制される。Nac2はpsbD 5'UTRに特異的なので、どんな葉緑体遺伝子でもコード配列をpsbD 5'UTRに融合させれば、原則的にはこのシステムで誘導性発現ができる。
【0150】
上述したとおり、パロモマイシン耐性を付与するaphVIII遺伝子を含むプラスミドに、Cyc6-Nac2コンストラクトを挿入した(図28A)。このプラスミドを使ってChlamydomonasnac2-26変異体の形質転換し、パロモマイシン耐性で選択した。調べた55の形質転換株のうち2つが、銅によるNac2発現の適切な制御を示した。これらの形質転換株の1つであるcy6Nac2.49、野生型、そしてnac2-26変異体の増殖特性は上述している(図28B)。予想したとおり、いずれも銅の有無にかかわらずTAP培地上で増殖し、形質転換株は選択マーカーaphVIIIを含むためパロモマイシンの存在下でも増殖する。野生型細胞だけが銅を含む最小培地で増殖する。しかし、cy6Nac2.49株は銅を含まない最小培地で増殖するようになった。培地にニッケルを添加しても増殖するが、これはCyc6プロモーターがニッケルで誘導されるからである。異なる増殖条件におけるpsbD発現レベルをRNAブロットハイブリダイゼーションで調べた(図28C)。予想どおり、nac2-26変異株ではpsbD RNAが検出されない。対するcy6Nac2.49株では、Cyc6の発現につづきpsbDの発現が見られ、銅非存在下または嫌気的条件下に誘導される(図28C)。psbD産物であるD2のレベルをD2抗血清を使った免疫ブロットで調べた(図28D)。すべての条件下にTAP寒天培地で増殖したnac2-26細胞でD2蛋白質は検出されない。しかしcy6Nac2.49を銅非存在下または嫌気的条件下に増殖した場合、D2が野生型の20%まで蓄積する。最小培地ではD2の誘導はわずかに低い。予想どおり、CP47の他のPSII蛋白質はD2と同様のパターンを示し、これはD2蛋白質がないとこれらの蛋白質が不安定なためである。これに対しnac2-26ではRubisco蛋白質(RbcL)のレベルは影響を受けない(図28D)。
【0151】
Nac2合成が止まってから細胞内のPSIIが消失するまでの時間を調べるため、cy6Nac2.49細胞をまず銅非含有TAP培地で培養した。この条件ではPSIIは合成され蓄積する。培養液を半分に分け、一方は銅非存在化のまま、他方は銅を添加して培養した。細胞濃度およびPSII量子収量の推定となるFV/FM比(可変蛍光/最大蛍光)の経時変化を調べた(図29A)。銅存在下では32時間以内にFV/FM比が最小値まで低下した。この期間内に、いずれの培養条件下でも細胞は3〜4倍に分裂し定常期に達した。各種時点でRNAおよび蛋白質解析のための細胞抽出物を調製した。Cyc6およびpsbD RNAのレベルは銅添加8時間後に著しく低下し、その後は検出できなかった。他の葉緑体RNA(atpB、rRNA)はいずれの条件でも安定だった。免疫ブロットの結果、銅添加後にmRNAの減少から少し遅れてD2の量が低下し(図29C)、他のPSIIコア蛋白質D1も減少した。期待どおりNac2の減少も認められた。これに対して葉緑体蛋白質PSI(PsaA)およびRubiscoは安定だった(図29C)。
【0152】
逆の実験では、銅存在下に培養した細胞を銅非含有TAP培地に移し、細胞濃度およびFV/FM比の経時変化を調べた。FV/FM比は25時間遅れて増加し始めたが、これはおそらく細胞内の銅が枯渇するまでに時間がかかるためである(図30A)。異なる時点で採取した細胞抽出物からのRNAおよび蛋白質を、RNAブロット解析および蛋白質免疫ブロットにより調べた(図30B、C)。Cyc6 RNAは16時間後に検出されたものの、psbD RNA蓄積およびPSII活性の検出には遅れが見られ、これはおそらくpsbD mRNA蓄積およびD2合成にはある程度のNac2レベルが必要であるためと思われる。
【0153】
<PSIIに関連しない葉緑体遺伝子の誘導性発現>
Nac2システムを使ってPSIIを可逆的に消失させることが可能だが、他の葉緑体遺伝子すべてに応用することができるか調べた。Nac2蛋白質はpsbD 5'UTRに特異的に働きキメラpsbD 5'UTRレポーター遺伝子を制御することができることから、これは原理的には可能である。よってpsbDプロモーターと5'UTRを対象となる遺伝子に融合するだけでよいはずである。しかし、psbD RNAの欠失をもたらすnac2-26変異のため、こうした条件下ではPSIIは蓄積しない。この問題を回避するため、petAプロモーターと5' UTRをpsbDコード配列に融合し、このコンストラクトを修飾型p108-14葉緑体形質転換ベクターに挿入した。このベクターでは、petAプロモーターと5'UTRの制御下にあるpsbD遺伝子の上流に再利用可能なaadAカセットが挿入されている。このDNAは、選択マーカーとしてaadAカセットを用いた遺伝子銃形質転換により葉緑体ゲノムに挿入した。このように内在性psbD遺伝子がpetA-psbDコンストラクトで置換されたため、その転写はもはやNac2に依存しなくなった。形質転換株をスペクチノマイシン含有寒天培地に3回移し換え、DNAブロットおよびPCR解析によりホモプラスミックかどうかを調べ、形質転換株のうちの1つInd41を選択した。使用したaadAカセットの両側は2つの反復配列に隣接していた。このカセットが切り出されるように、ホモプラスミックな形質転換株Ind41をスペクチノマイシンを含まない培地上で繰りかえし移し換えた。このようにして得たInd41_18は、aadAカセットを含まないためスペクチノマイシンに感受性である。Ind41、Ind41_18およびcy6Nac2.49について、異なる培地上での増殖特性を調べた。予想どおり、Ind41_18がスペクチノマイシンに感受性であるのに対し、Ind41はスペクチノマイシン存在下に増殖した。さらに、Ind41とInd41_18の両方が銅の有無にかかわらずHSM最少培地で増殖した。RNAブロット解析の結果、Ind41_18ではCyc6 RNAレベルにかかわらずすべての条件でキメラpetA-psbD RNAが蓄積した(図27C)。psbD 5'UTRよりもpetA 5'UTRの方が大きいので、psbD RNAはより大きい。免疫ブロット解析の結果、試験したすべての条件、特にNac2が発現しない条件でD2およびD1蛋白質が同等レベルに蓄積した(図27D)。
【0154】
次に、cg12ベクターを使った形質転換により、psbDプロモーターおよび5'UTRに融合したaadAカセットを、この株に導入した(図31A)。形質転換株Ind_aadA-117およびInd41_18の増殖を、銅含有および銅非含有の濃度漸増したスペクチノマイシン含有TAP寒天培地で調べた(図31B)。すべての株が銅非存在下でも(図31B)銅存在下でも増殖した。予想どおり、Ind_aadA-117はスペクチノマイシン250 μg/ml以上を含む培地では銅が存在する場合のみ増殖した。スペクチノマイシン100 μg/mlを含む培地でも銅の存在下にわずかな増殖が見られた。RNAブロット解析の結果、Cyc6プロモーターが誘導される条件でのみaadA RNAが蓄積した(図31C)。D2、D1、CP47、およびRbcLの蛋白質レベルはあまり変化しなかったが、Nac2は誘導条件でのみ検出された(図31D)。適正なaadA抗体がないため、この蛋白質の定量はアミノグリコシドアデニルトランスフェラーゼ活性の測定により行った。Nac2が発現する条件では活性が著しく上昇していた(表3)。
【0155】
〔表3 誘導条件下および抑制条件下におけるInd41_aadA-117中のアミノグリコシドアデニルトランスフェラーゼ活性〕
上述のとおり、野生型-aadA、Ind41_18およびInd41_aadA-117株からの抽出物について、aadA活性および総蛋白質含量を測定した。活性は蛋白質1μgあたりに取り込まれた放射能cpmを示す。独立した測定の数をカッコ内に示す。
【0156】
〔誘導性葉緑体遺伝子発現システムを使って水素産生を引き起こすことができる〕
Chlamydomonasは明所で嫌気的条件下にヒドロゲナーゼを誘導し水素を産生することができる。そこで、呼吸により水素産生誘導に適した嫌気的条件になるよう、誘導性Nac2システムを使ってPSII活性と酸素発生を止められるかどうか調べた。cy6Nac2.49細胞を銅を含まないTAP培地で濃度2 x 106 細胞/mLまで培養した。銅を添加し、質量分析器に接続し白色光を照射したチャンバーに細胞を移した。このシステムでは、チャンバーの底はポリプロピレン膜で塞がれ、溶存ガスが膜を通して質量分析器のイオン源に直接拡散する。このようにして酸素と水素の量を一定間隔で測定できる。チャンバーは密閉され銅がPSII合成を抑制しているので、酸素発生は減少した。200分以内に酸素は呼吸により消費された。嫌気的状態に達し、活性型ヒドロゲナーゼの合成と水素産生が見られた(図32A)。水素産生の最大速度は1から3.1 mmol H2 mol-1 Chl sec-1の範囲であり、硫黄枯渇細胞での速度よりもやや遅く、持続時間はずっと短かった(3〜4日間に対して約1.5時間)。
【0157】
Cyc6プロモーターの興味深い特徴として、このプロモーターは銅存在下であっても嫌気的条件で誘導される。したがって、cy6Nac2.49細胞では一度嫌気的条件になればヒドロゲナーゼが誘導され、Nac2合成が再開し、PSIIが合成され、酸素レベルが上昇してヒドロゲナーゼが不活性化されて水素産生が止まる。これが観測された。嫌気的水素産生段階では酸素発生およびヒドロゲナーゼ不活性化と同時にPSII合成が行われ、水素レベルは一定を保った(図32A)。対照として同じ細胞培養液に銅を添加せずに調べた。この条件では、銅処理細胞で見られたように、水素が産生されず(図32B)、一定の酸素産生と二酸化炭素の漸減を伴った。Cyc6プロモーターは銅非含有培地で好気条件下にオフになると予想されるので、新たな水素産生サイクルが期待される。この可能性を検証するため、密閉容器内の銅非含有TAP培地でcy6Nac2.49細胞を50時間培養し、質量分析器で水素および酸素量を測定した。その結果は、2度の連続した水素産生段階と酸素産生段階があったことを示す。
【0158】
本発明は特定の望ましい実施形態に関して詳細に記述してきたが、以下の特許請求の範囲に記載する本発明の範囲及び主旨内で、変更及び修正が可能である。
【技術分野】
【0001】
本発明は、蛋白質の発現の誘導および/または抑制のためのシステム、方法、および装置に関する。より具体的には、本発明は色素体中での蛋白質の発現の誘導および/または抑制のためのシステム、方法、および装置に関する。
【背景技術】
【0002】
蛋白質(ペプチド、オリゴペプチド、およびポリペプチドなど)は、触媒作用、情報伝達、防御、運動、輸送など、細胞の多くの活動に関与している。蛋白質の生物活性の基礎となるのは、アミノ酸配列および/またはその立体構造である。したがって、蛋白質の生物活性部位は原則的にそのままの形を維持し、生物学的に機能する立体構造をとらなければならない。蛋白質発現に関する遺伝子工学技術の進歩は、蛋白質の生物活性を維持した形のまま様々なシステムで内在性および外来性の蛋白質を制御下に発現するための方法の開発に繋がっている。このような遺伝子工学により作られた制御発現システムは、蛋白質発現を制御することができる結果、蛋白質の不活性化や分解が抑えられ、正しく折り畳まれた安定な蛋白質を発現することにより高い蛋白質収量をもたらす。
【発明の概要】
【発明が解決しようとする課題】
【0003】
微生物、菌類や酵母を含む真核細胞、そして哺乳類細胞、昆虫細胞など、様々な宿主細胞の細胞小器官で多数の異なる様式の発現システムが開発されているが、色素体での蛋白質発現を制御するために核由来の安定化因子を利用する蛋白質制御発現のための発現システムはこれまでに開発されていない。色素体とは光合成に関与する細胞小器官であり、一般に葉緑体、白色体、アミロプラスト、または有色体に分類される。色素体はこれらの形態間で分化または再分化が可能である。
【課題を解決するための手段】
【0004】
一実施形態では、細胞の色素体内で蛋白質の産生を誘導するための発現システムを作成する方法が提供される。本方法は、誘導性プロモーターをコードする第一の核酸を細胞核に導入するステップと、第一の核酸を安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、インデューサーの導入またはリプレッサーの除去が安定化因子の発現を誘導し、発現した安定化因子が色素体内で安定化因子で安定化され第三の核酸から転写されるmRNAの非翻訳領域と結合し、第三の核酸が色素体に内在性または外来性で第三の核酸が蛋白質をコードし、mRNAの発現が蛋白質の産生をもたらすことを特徴とするステップとを含む。
【0005】
別の例示的な実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制するための発現システムを作成する方法が提供される。本方法は、抑制性プロモーターをコードする第一の核酸を細胞核に導入するステップと、第一の核酸を安定化因子をコードする第二の核酸に作動的に連結して組換え核酸を形成するステップであって、リプレッサーの導入またはインデューサーの除去が安定化因子の発現を抑制し、安定化因子の発現抑制が安定化因子で安定化され第三の核酸から転写されるmRNAの発現抑制をもたらし、第三の核酸が色素体に内在性または外来性であり、第三の核酸が蛋白質をコードし、蛋白質の発現が抑制されることを特徴とするステップとを含む。
【0006】
さらに別の例示的な形態では、細胞の色素体内で色素体蛋白質を発現させる方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現させるステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現させるステップと、色素体内で蛋白質を産生させるステップとを含む。
【0007】
別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制する方法が提供される。本方法は、細胞をリプレッサーと接触させるかインデューサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップとを含む。
【0008】
さらに別の実施形態では、組換え宿主細胞の色素体内で色素体蛋白質を発現させるシステムが提供される。本システムは、外から加えられる核蛋白質の発現を誘導するインデューサーと、核に組換え核酸を含む組換え宿主細胞であって、組換え核酸が第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、第一の核酸が誘導性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、色素体蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体とを含む。
【0009】
別の例示的な実施形態では、組換え宿主細胞の色素体内で色素体蛋白質の発現を抑制するシステムが提供される。本システムは、外から加えられる核蛋白質の発現を抑制するリプレッサーと、核に組換え核酸を含む組換え宿主細胞であって、組換え核酸が第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、第一の核酸が抑制性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、色素体に内在性または外来性の第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体とを含む。
【0010】
別の実施形態では、細胞の色素体内で色素体蛋白質を発現させることにより水素ガスの産生を促進する方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現させるステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現させるステップと、色素体内で蛋白質を産生させるステップと、水素ガスを産生するステップとを含む。
【0011】
さらに別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制することにより水素ガスの産生を阻害する方法が提供される。本方法は、細胞をリプレッサーと接触させるかまたはインデューサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップと、水素ガスの産生を阻害するステップとを含む。
【0012】
さらに別の実施形態では、細胞の色素体内で色素体蛋白質の発現を誘導および抑制することにより水素ガスの産生を促進する方法が提供される。本方法は、順次的にi)細胞をインデューサーと接触させるかまたはリプレッサーの除去をもたらす条件で細胞を処理してから、ii)細胞をリプレッサーと接触させるかまたはインデューサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、順次的に安定化因子を発現させてから抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、順次的にmRNAを発現させてから抑制するステップと、色素体内で蛋白質を産生させるステップと、水素ガスを産生するステップとを含む。
【0013】
上記のいずれかの実施形態において、組換え核酸を核に導入する前に、第一の核酸は第二の核酸と作動的に連結して組換え核酸を形成することができ、細胞は第二または第三の核酸の非作動型コピーを有するかまたはそれらのコピーやホモログを欠失することができ、細胞は植物細胞または藻類細胞であってもよく、色素体は葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択することができ、誘導性プロモーターはCyc6プロモーターと少なくとも90%の配列類似性を有することができ、第三の核酸はpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることができる。
【0014】
上記のいずれかの実施形態において、インデューサーまたはリプレッサーは化学物質または環境条件であってもよく、化学物質は銅であってもよく、環境条件は所定レベルまでの酸素濃度の低下であってもよく、インデューサーはその添加と除去を一サイクルとして複数サイクルの添加と除去を行うことができ、蛋白質は光合成または水素ガス産生に関与する蛋白質であってもよく、蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、栄養補助食品から選択することができ、医薬品は抗体、ワクチン抗原、抗菌剤、またはその他の宿主細胞防御産物から選択され、安定化因子はNac2およびMbb1から選択することができる。別の例示的な実施形態では、第二の核酸は例えばTbc2やTca1などの翻訳活性化因子をコードすることができる。
【0015】
本発明の別の実施形態では、色素体内で内在性または外来性遺伝子の発現または抑制を制御するシステムおよび方法が提供される。一実施形態では、本発明は核にコードされる葉緑体転写因子でありその発現がCyc6遺伝子の誘導性プロモーターによって制御されるNac2を採用する発現システムに関する。別の実施形態では、低い酸素レベルなど、インデューサー(すなわち、薬剤または環境条件の変化)によるNac2発現の誘導が葉緑体内でのpsbD遺伝子の発現をもたらす。さらに別の実施形態では、薬剤または銅の除去などの環境条件がCyc6プロモーターの誘導をもたらし、psbD遺伝子の発現ももたらす。別の例示的な実施形態では、高い酸素レベルなど、リプレッサー(すなわち、薬剤または環境条件の変化)によるNac2遺伝子の抑制がpsbD遺伝子の発現阻害または低下をもたらす。関連した実施形態では、誘導性Cyc6プロモーターを抑制する薬剤または環境条件の変化もpsbD遺伝子の発現低下をもたらす。
【0016】
別の例示的な実施形態では、本発明は葉緑体内での外来遺伝子の誘導性発現または抑制に関するので、葉緑体内のpsbD遺伝子を外来遺伝子で置換することで、Nac2発現制御により葉緑体内での外来遺伝子の誘導性発現または抑制が促進される。
【0017】
別の実施形態では、本発明はNac2の発現またはNac2の発現抑制によるpsbD遺伝子の制御を介して葉緑体内で水素ガスを産生する方法に関する。本発明のこの例示的な形態では、Nac2遺伝子の誘導および抑制を促進する環境条件(例えば、酸素レベルを低下させて発現を誘導する、酸素レベルを上昇させて発現を抑制するなど)が、psbD遺伝子発現の周期的な誘導と抑制をもたらし、光合成速度の低下およびその結果としての酸素濃度の低下をもたらす。この実施形態では、酸素濃度の低下が水素産生を促進する。よって、一実施形態では、本発明はNac2およびpsbD遺伝子発現の周期的な誘導と抑制を制御することにより水素ガスを産生する方法に関する。
【0018】
さらに別の実施形態では、本発明は葉緑体内での他の遺伝子の組換え発現を介して水素発生システムを促進する方法に関する。例としてヒドロゲナーゼや、他の組換え蛋白質またはホスホリブロースキナーゼなどの内在性蛋白質の抑制がある。
【0019】
さらに別の実施形態では水素産生のための装置が提供される。本装置は、実質的に酸素のない環境下に細胞培養液を保持するように構成された第一の容器と、第一の容器と液体連通があり所定の速度で第一の容器に培地を供給するように構成された第一のポンプと、第一の容器に連結され細胞培養液によって産生される水素量を測定するように構成された測定器を備える。
【0020】
この実施形態では、第一のポンプは細胞培養の増殖速度と実質的に同じ速度で一定量の培地を第一の容器に供給するように構成することができ、第一のポンプはペリスタ型ポンプを含むことができ、細胞培養液はcy6Nac2.49培養液を含むことができ、測定器は質量分析器を含むことができ、本装置はさらに第一の容器に連結され細胞培養液を撹拌するように操作できる撹拌器を備えることができ、撹拌器は磁気撹拌子を含むことができる。本装置はさらに一定量の培地を保持するように構成された第二の容器を備えることができ、第一のポンプは第二の容器と液体的に連結して第二の容器から所定速度で培地を汲み上げるように構成されることを特徴とする。本装置はさらに第一の容器と液体連通して第一の容器からあふれた培地が入るように構成された第三の容器を備えることができ、本装置はさらに第三の容器および第二の容器と液体連通したフィルターと第二のポンプを備えることができ、第二のポンプは第三の容器から一定量の培地を汲み上げフィルターを通して第二の容器へと供給するように構成される。
【0021】
本発明は以下の図面を参照することでより理解しやすい。
【0022】
<関連出願の相互参照>
本出願は35 U.S.C § 119(e)により、本明細書に参照として含まれる米国仮出願No. 60/837,001(2006年8月11日出願)に対する優先権を主張する。
【図面の簡単な説明】
【0023】
【図1】図1は好気的および嫌気的条件下におけるC. reinhardtii葉緑体内の水素および電子の流れを示す(Kruse et al., 2005より)。
【図2】図2はインデューサーの存在下に核内の誘導性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の誘導性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図3】図3はインデューサーの非存在下に核の誘導性プロモーターが抑制されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の誘導性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図4】図4はリプレッサーの存在下に核の抑制性プロモーターが抑制されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の抑制性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図5】図5はリプレッサーの非存在下に核の抑制性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。左斜線で示した部分は核の抑制性プロモーター、右斜線で示した部分は安定化因子の遺伝子を表す。黒塗りの部分は安定化因子結合エレメントを表し、本実施形態では色素体mRNAの5'非翻訳領域内に位置する。白抜きの部分は色素体の内在性遺伝子から産生されるmRNAを表す。網斜線の部分は色素体中の外来性遺伝子から産生されるmRNAを表す。
【図6】図6はpsbD遺伝子の位置が示されたChlamydomonas葉緑体ゲノムの概要図を示す。矢印はpSK108の挿入部位を示す。
【図7】図7は水素産生装置の作業工程図を示す。
【図8】図8は水素産生装置を示す。
【図9】図9は核Nac2突然変異体の形質転換並びにCyc6プロモーターおよびNac2遺伝子の導入に使用される核発現ベクターpSL17の概要図を示す。マップはプロモーター、HSP70Aプロモーターのエンハンサーエレメント、そしてCyc6プロモーターおよびNac2を挿入する制限酵素部位の配置を示す。
【図10】図10はcy6Nac2(paroR)の概要図を示す。
【図11】図11a〜cはNac2 midi遺伝子のゲノム配列を示す。開始コドンは下線を引いた部分の最初のATGである。推定される輸送ペプチドにも下線を引いてある。
【図12】図12はCyc6ゲノム配列を示す。Nac2 midi遺伝子との融合コンストラクトを作成するのに使用したゲノム配列に下線を引いてある。NdeI制限酵素部位を作り出したcyc6Nac2コンストラクトでの3塩基対の相違も示している(二重下線)。
【図13】図13はcy6Nac2.49遺伝子導入株の増殖特性を示す。
【図14】図14はcy6Nac2.49遺伝子導入株のウェスタンブロット解析を示す。
【図15】図15はcy6Nac2.49遺伝子導入株の水素産生を示す。
【図16】図16はpSK108ベクターのマップを示す。pSK108ベクターは、psbD遺伝子周辺領域へ導く葉緑体DNA隣接領域を有する。
【図17】図17はpcg12のマップを示す。
【図18】図18は葉緑体発現プラスミドpcg12_IBDV_Flagのマップを示す。
【図19】図19は葉緑体発現プラスミドpcg12_VP28_Flagのマップを示す。
【図20】図20はpsbDの葉緑体DNA配列を示す。下線を引いた配列はcyc6Nac2システムを用いて葉緑体内での遺伝子発現を駆動するのに使用された。
【図21】図21はIND_aadA_X 遺伝子導入株の単離と各種培地上での増殖を示す。
【図22】図22は野性型およびIND_aadA_117単離株から抽出した全RNAのノーザンブロット解析を示す。
【図23】図23は野生型およびIND_aadA_117から抽出した総蛋白質および可溶性蛋白質(α-Nac2)の解析を示す。
【図24】図24は3つの外来性蛋白質(DILP、IBDV、VP28)の発現を誘導する遺伝子導入株のスクリーニングを示す。
【図25】図25はNac2誘導性葉緑体遺伝子発現システムを用いた外来性蛋白質(DILP)の誘導性産生を示す。
【図26】図26はcy6Nac2.49でのpsbD RNA、D2およびNac2蛋白質の蓄積を示す。
【図27】図27はpsbD 5' UTRをpetA 5' UTRで置換することによるcy6Nac2.49での構成的PSII蓄積の回復を示す。
【図28】図28は葉緑体psbD遺伝子の誘導性発現を示す。
【図29】図29はcy6Nac2.49での銅を介したPSII合成阻害の経時変化を示す。
【図30】図30はcy6Nac2.49でのPSII蓄積の経時変化を示す。
【図31】図31はIND_aadA_117形質転換体にてpsbD-aadA遺伝子の発現が銅枯渇で誘導され銅で抑制されることを示す。
【図32】図32はcy6Nac2-49株での水素産生を示す。
【発明を実施するための形態】
【0024】
本明細書で用いられる用語「外来性遺伝子」または「外来性核酸」(すなわち、導入遺伝子)とは、組換えDNA技術を用いて細胞の核酸に挿入される核酸を意味し、外来性遺伝子または外来性核酸は細胞の当該位置に通常は存在しない。外来性遺伝子または外来性核酸はコーディングおよび非コーディング核酸配列を含むことができる。外来性遺伝子または外来性核酸は、組換えDNA技術を用いて改変され細胞に再導入された内在性核酸を含むか、または細胞内のある位置から別の位置に移動された内在性核酸を含むことができる。
【0025】
本明細書で用いられる用語「内在性核酸」または「内在性遺伝子」とは、細胞内で天然の配列(自然突然変異を含む)と位置を有する核酸を意味する。
【0026】
本明細書で用いられる用語「誘導性」とは、mRNA転写物が産生されるよう制御されることが可能なプロモーターを意味する。mRNA転写物レベルの決定方法はノーザンブロットおよびリアルタイムPCRを含む。
【0027】
本明細書で用いられる用語「発現」とは、DNAからRNAへの転写またはRNAから蛋白質への翻訳を意味することができる。
【0028】
本明細書で用いられる用語「安定化因子」とは、転写、転写後、翻訳、翻訳後、蛋白質ターゲティング、蛋白質リクルートメントなどの活性に限らないがこれらを含む活性を示し、葉緑体蛋白質の発現または活性を増強する核蛋白質を意味する。
【0029】
本発明は、有用産物を産生する目的で細胞(例えば藻類または植物の細胞)の色素体内で遺伝子発現を制御するためのシステム、方法、および装置に関する。色素体内での遺伝子発現の誘導または抑制は、葉緑体移行蛋白質をコードする遺伝子と作動的に連結した誘導性または抑制性プロモーターで細胞の核ゲノムを形質転換することにより達成される。葉緑体移行蛋白質は、色素体で発現するmRNAの非翻訳領域と直接または間接的に(例えば、アクセサリー蛋白質を介して)結合する。葉緑体移行蛋白質は色素体で発現するmRNAの安定性および/または翻訳に必要であり、よって色素体遺伝子の発現に必要である。
【0030】
一実施形態では、核プロモーターは誘導性プロモーターであり、化学物質(例えば銅、炭化水素、蛋白質など)の添加または除去あるいは環境条件の変化(例えば低酸素濃度、または光、温度、栄養状態の変化など)がプロモーターを活性化し、葉緑体mRNAの発現をもたらし、続いてmRNAがコードする蛋白質の発現をもたらす。別の実施形態では、核プロモーターは抑制性プロモーターであり、化学物質(例えば銅、炭化水素、蛋白質など)の添加または除去あるいは環境条件の変化(例えば高酸素濃度、または光、温度、栄養状態の変化など)がプロモーターを抑制し、安定化因子が発現されなくなって葉緑体mRNAの発現阻害をもたらし、続いてmRNAがコードする蛋白質の発現阻害をもたらす。
【0031】
一実施形態では、本発明は細胞の色素体内で蛋白質を発現する方法に関する。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、インデューサーまたはリプレッサーが核内の第一の核酸と結合し、第一の核酸が誘導性プロモーターをコードし、第一の核酸が第二の核酸に作動的に連結して組換え核酸を形成することを特徴とするステップと、安定化因子を発現するステップと、安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性の第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAを発現するステップと、色素体内で蛋白質を産生するステップとを含む。
【0032】
別の実施形態では、細胞の色素体内で色素体蛋白質の発現を抑制する方法が提供される。本方法は、細胞をインデューサーと接触させるかリプレッサーの除去をもたらす条件で細胞を処理するステップであって、リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、第一の核酸が抑制性プロモーターをコードし、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、第二の核酸が安定化因子をコードすることを特徴とするステップと、安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性の第三の核酸から転写され、第三の核酸が蛋白質をコードすることを特徴とするステップと、mRNAの発現を抑制するステップと、蛋白質の発現を抑制するステップとを含む。
【0033】
上記の実施形態では、蛋白質が細胞の色素体内で発現される。例示的に、細胞は植物細胞もしくは藻類細胞、または色素体を含むいかなる細胞種でもよい。色素体は色素体ゲノムを含む細胞小器官であり、複数コピー含まれることが多い。色素体は、例えば植物や藻類に見られ、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体が含まれる。
【0034】
本明細書に記載される方法およびシステムの実施形態では、第三の核酸が目的の蛋白質をコードする。例示的な一実施形態では、発現される蛋白質をコードする第三の核酸は、色素体に内在性または外来性(すなわち導入遺伝子)のいずれでもよい。この実施形態では、発現される蛋白質(例えばペプチド、オリゴペプチド、ポリペプチドなど)は誘導性または抑制性プロモーターの制御下に発現することができ、光化学系IまたはIIの構成成分(例えばpsbAおよびpsbD、光化学系IIのD1およびD2サブユニットなど)、CO2固定に関与する蛋白質(例えばホスホリブロースキナーゼなど)、ヒドロゲナーゼ(例えばHydA1、HydA2など)、そしてこれらの蛋白質のうちいずれかの活性を制御する蛋白質(例えばこれら蛋白質のいずれかの集合(HydEF、HydGなど))などの光合成に関与する蛋白質、または色素体に内在性の他の蛋白質を含む蛋白質を誘導する。誘導性または抑制性プロモーターの制御下に発現することのできる例示的な内在性蛋白質としては、図1に描写または示唆される光合成過程または炭素同化過程の制御に関与するすべての蛋白質、あるいは色素体に内在性の他の蛋白質が含まれる。別の実施形態では、芳香族アミノ酸などのアミノ酸合成経路に関与する色素体に内在性の蛋白質の発現制御によって、芳香族アミノ酸などのアミノ酸またはアミノ酸前駆体が産生されてもよい。
【0035】
別の例示的実施形態では、本明細書で記載した方法、システム、および装置を使用して、誘導性または抑制性プロモーターあるいは両方の制御下に、水素ガス産生に関与する蛋白質が発現されてもよい。この実施形態では、水素ガス産生に関与する蛋白質は、本項または前項で記載した蛋白質のいずれでも、または図1に描写または示唆される光合成過程または炭素同化過程の制御に関与する蛋白質のいずれでもよい。この実施形態では、インデューサーまたはリプレッサーは、その添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことができる。
【0036】
別の実施形態では、第三の核酸は色素体に外来性である(すなわち外来性核酸または外来性遺伝子である)。この実施形態では、蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、または栄養補助食品であってもよい。この実施形態では、発現蛋白質は、例えば抗体、ワクチン抗原(例えばワクチンに使用するための)、抗菌剤、またはその他の宿主細胞防御産物、成長ホルモン、インターロイキンやインターフェロンなどのサイトカイン、インシュリン、コロニー刺激因子、凝固因子、エリスロポエチン、上皮成長因子などの成長因子、ソマトトロピン、線維芽細胞増殖因子、血小板由来増殖因子、および同類のもの、アミラーゼ、プロテアーゼ、リパーゼ、ペクチナーゼ、セルラーゼ、ヘミセルラーゼ、ペントサナーゼ、キシラナーゼ、およびフィターゼ、殺虫性蛋白質、フェニルアンモニアリアーゼ、またはその他のあらゆる蛋白質性の医薬品、工業用酵素、または栄養補助食品であってもよい。
【0037】
さらに別の例示的な実施形態では、発現蛋白質をコードする追加の核酸(例えば第四の核酸)を葉緑体内で発現してもよく、それは色素体に内在性または外来性(すなわち外来性核酸または外来性遺伝子)であってもよい。これらの実施形態では、追加の核酸の発現は、核内で追加の核酸がコードするそれ自身の安定化因子によって制御されるか(すなわち第二の核酸と同様に)、またはこれらの追加の核酸の発現は第二の核酸がコードする安定化因子によって制御されてもよい。一つの例示的な実施形態では、一つの安定化因子が色素体mRNA内の安定化因子結合エレメントと結合し、第三の核酸および第三の核酸と作動的に連結した追加の核酸(例えば第四の核酸など)の発現を促進する。これらの実施形態では、発現される蛋白質は医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、または栄養補助食品であってもよい。この実施形態では、発現蛋白質は、例えば抗体、ワクチン抗原(例えばワクチンに使用)、抗菌剤、またはその他の宿主細胞防御産物、成長ホルモン、インターロイキンやインターフェロンなどのサイトカイン、インシュリン、コロニー刺激因子、凝固因子、エリスロポエチン、上皮成長因子などの成長因子、ソマトトロピン、線維芽細胞増殖因子、血小板由来増殖因子、および同類のもの、アミラーゼ、プロテアーゼ、リパーゼ、ペクチナーゼ、セルラーゼ、ヘミセルラーゼ、ペントサナーゼ、キシラナーゼ、およびフィターゼ、殺虫性蛋白質、フェニルアンモニアリアーゼ、またはその他のあらゆる蛋白質性の医薬品、工業用酵素、または栄養補助食品であってもよい。
【0038】
発現蛋白質がワクチンとして使用するワクチン抗原である実施形態では、発現蛋白質、またはその一部は、藻類細胞や植物細胞などの細胞の細胞小器官上または細胞小器官内に位置することができる。もしワクチン抗原が少なくとも部分的には病原体に由来する場合、例えば藻類はその後溶解し、病原体ワクチン抗原を使って宿主動物の病原体への免疫応答を誘導することができる。
【0039】
一実施形態では、ワクチン抗原を有する藻類を食物材料として投与する。ワクチンを投与できる代表的な動物は、哺乳類、鳥類、水産養殖種を含むがこれらに限らない。特に、ワクチンはすべての脊椎動物魚類などの水生脊椎動物に投与でき、これはサケ科(マス、サケ、北極イワナなど)、コイ、ナマズ、ブリ、タイ、スズキなどに限らないがこれらを含む骨性または軟骨性魚類であってもよい。このようなワクチンは、ハマグリやアサリ、ロブスター、エビ、カニ、カキに限らないがこれらを含む貝類や甲殻類に投与することもできる。代表的な送達方法は、乾燥粉末として、通常飼料の一成分として、そしてワクチンを含む懸濁液に動物を浸水させることでの経口投与を含む。
【0040】
水生脊椎動物の場合、本明細書に記載した方法およびシステムを用いて、その抗原決定基を細胞表面にワクチン抗原として発現させる病原体の例は、Rennibacterium salmoninarum(サケ、マス、イワナ、ホワイトフィッシュなどサケ科の魚における細菌性腎疾患の病原体)、Aeromonas salmonicida、Aeromonas hydrophila、Vibrio種(V. anguillarumとV. ordaliiを含む)、Pasteurella種(P. piscicidaを含む)、Yersinia種、Streptococcus種、Edwardsiella tardaおよびEdwardsiella ictaluria、ウイルス性出血性敗血症、伝染性膵臓壊死症、コイのウイルス血症の原因ウイルス、伝染性造血器壊死症ウイルス、アメリカナマズウイルス、グラスカープ出血性ウイルス、神経壊死症ウイルスやシマアジ神経壊死症ウイルスなどのノダウイルス、伝染性サケ貧血ウイルス、そして寄生虫のCeratomyxa shasta、Ichthyophthirius multifillius、Cryptobia salmositica、Lepeophtheirus salmonis、Tetrahymena種、Trichodina種およびEpistylus種などを含むが、但しこれらに限らない。
【0041】
蛋白質を藻類で発現させる実施形態では、藻類は例えば緑藻類であってもよい。例えば、使用できる藻類にはCharoidesなどのChlorophyta(Charoides、Lamprothamnium、Nitellopsis、Nitellaなど)、Zynematales(Zygnema、Closterium、Netriumなど)、Codials(Codium fragile、Helimida opunta、Caulerpaなど)、Bryopsis plumosa(Bryopsis、Pseudobryopsis、Bryopsidella、Derbesis、Pedobesiaなど)、Acetabularia ryukyuensis(Acetabularia ryukyuensis、Halicoryne wrightii、Neomeris annulata、Cymopolia van bossei、Bornettella ovalis、Acetabularia calyculusなど)、Siphonocladales(ValoniaceaeおよびBoodleaceae)、Cladophora(Anadyomene writii、Cladophora、Cladophora sauteri、およびChaetomorpha)、Ulva(UlvaおよびFnteromorpha)、Ulotrichales(Acrosiphoniaceae、Collinsiellaceae、Monostromaceae、およびChlorocystidaceae)、Prasiola、Chlorella、Chlorococcales(PediastrumおよびHydrodictyon)、およびVolvocales(Chlamydomonus、Pandorina、Pleodorina、およびVolvox)が含まれる。
【0042】
この適用で記述されるいずれかの実施形態で通常使用される代表的な藻類には、Chlamydomonas種、特にChlamydomonas reinhardtii、Chlorella種、およびVolvox種が含まれる。単細胞真核緑藻のChlamydomonas reinhardtiiは特に有利である。Chlamydomonas株は例えばデューク大学クラミドモナス遺伝ストックセンター(ノースカロライナ州ダーラム)から入手できる。Chlamydomonas reinhardtiiの栄養要求性変異株(一つ以上の成長に必要な栄養成分を有する点で野生株と異なる変異株)はクラミドモナス遺伝ストックセンターから入手可能であり、そうした変異株はDNAで形質転換(すなわち細胞に外来性DNAを導入)することにより遺伝的に相補でき、これにより目的とする導入遺伝子を含む藻類の選択が容易となる。他の実施形態では無能化した藻類を使用できる。無能化した藻類とは、非常に特殊な制御環境以外では増殖しないように遺伝子操作したものである(すなわち、このような藻類株は野生では生育したり遺伝子を移動させたりしない)。本開示中では、このような藻類を「無能化した」という。このような無能化した株を使用することにより、本発明で用いられる遺伝子導入藻類株が環境中に広まることが阻止されるか制限される。
【0043】
本明細書に記載される方法およびシステムでの使用に適した代表的な植物は、培養植物細胞(プロトプラストおよびカルス細胞)や全植物体(単細胞および多細胞植物)を含む。様々な実施形態において、植物細胞(すなわち培養植物細胞または全植物体の細胞)は燕麦、小麦、大麦、米、紅花、トウモロコシ、アルファルファなどの豆類、大豆、トマト、甜菜、ジャガイモなどの植物から得られる。その他の有用な植物には、例えば、リンゴ、ナシ、サクランボ、ブドウ、柑橘果実、パイナップル、バナナなどの果実をつける植物、そしてカラマツなどの樹木がある。その他の適した植物には、油ヤシ、茶、カカオ、コーヒーなどの潅木、タバコ、綿花、フラックス、ヒマワリ、牧草、飼料用穀類、飼料用植物、そして落花生やレンズ豆植物などがある。その他の有用な植物には、シロイヌナズナ、サボンソウ(Saponaria)、ウキクサ(Lemnaceae)、シダ類、コケ類、ゼニゴケ類などがある。この実施形態では、植物の遺伝子操作に通常用いられるベクターを使って、本発明に従い核酸分子を植物細胞に移行させる。
【0044】
本明細書に記載される方法およびシステムでは、第一および第二の核酸を細胞(例えば藻類または植物の細胞)内に導入する。この実施形態では、第一の核酸は誘導性、抑制性、または誘導性かつ抑制性のプロモーターをコードし、第二の核酸は色素体mRNAの発現を制御する安定化因子(例えばNac2またはMbb1)をコードする。別の例示的な実施形態では、第二の核酸は例えばTbc2やTca1などの翻訳活性化因子をコードしてもよい。
【0045】
誘導性または抑制性のプロモーターは安定化因子の発現を制御する。様々な実施形態において、使用する核プロモーターに応じてあらゆる適切な形のインデューサーまたはリプレッサー(化学物質または改変した環境条件)が使用できる。本明細書に記載される方法およびシステムでの使用に適した代表的なプロモーターはCyc6プロモーターである(図12参照)。Cyc6プロモーターと配列類似性を有するプロモーター、例えばCyc6プロモーターと60%、70%、80%、85%、90%、95%、98%の配列類似性を有するものなど、その他の適切なプロモーターのいずれでも使用できる。また、Cyc6プロモーターの相補配列に高ストリンジェンシー条件でハイブリダイズする配列も使用できる。その他の使用に適した誘導性または抑制性プロモーターには、環境条件(無酸素、熱、干ばつ、または光など)、化学物質、栄養素、ホルモン、病原体、損傷、摂食、発生段階、組織型などの因子に応答するプロモーターなどがある。このようなプロモーターは当業者に知られている。プロモーターが複数の因子に応答することもあり、単一因子が複数のプロモーターを活性化または抑制することもある。
【0046】
Cyc6以外では、クラミドモナスのその他の誘導性プロモーターには、CO2誘導性細胞膜蛋白質遺伝子のプロモーター(Genbankアクセション番号U31976)、鉄利用能で緊密に制御されるFEA1遺伝子のプロモーター(Sasaki et al., 1998; Rubinelli et al., 2002)、そしてアンモニアやグルタミン酸の存在下に抑制されアンモニアを含まない培地中で誘導されるNit1遺伝子のプロモーター(Fernandez 1989)などがある。高等植物での誘導性プロモーターの例には、リブロース1,5-ビスリン酸カルボキシラーゼ/オキシゲナーゼ小サブユニットの光誘導性プロモーター、カルコンシンターゼ遺伝子のUV誘導性プロモーター、カルコンシンターゼ遺伝子のクマル酸誘導性プロモーター、アルコールでヒドロゲナーゼ遺伝子の低酸素誘導性プロモーター、そしてタバコ、トマト、キュウリおよびアラビドプシスの病原誘導性プロモーター(PR-1-14)などがある。
【0047】
安定化因子は誘導性および/または抑制性プロモーターの制御下に発現し、色素体内に導入され、色素体内で安定化因子はmRNAの非翻訳領域と直接または間接的に(例えばアクセサリー蛋白質を介して)結合しmRNAを安定化する。様々な実施形態において、mRNAの非翻訳領域はmRNAの5'または3'末端に位置しうる。mRNAは発現蛋白質をコードする第三の核酸から転写される。別の例示的な実施形態では、mRNAは、第三の核酸に作動的に連結しているかまたは第三の核酸に連結しておらず、それ自身の安定化因子で制御される追加の核酸から転写されてもよい。よって、発現蛋白質は安定化因子の制御下に色素体内で産生され、安定化因子の発現は誘導性および/または抑制性核プロモーターにより制御される。
【0048】
一つの例示的な形態では安定化因子は、mRNAの5'末端か3'末端あるいは両方にある非翻訳領域に直接または間接的に結合するだけでなく、mRNAのコード領域にも直接または間接的に結合してもよい。別の例示的な形態では、安定化因子は例えば他の蛋白質がmRNAの非翻訳領域および/またはコード領域に直接または間接的に結合している複合体中に含まれる他のアクセサリー蛋白質との結合を介してmRNAと直接または間接的に結合してもよい。
【0049】
一実施形態では、第一および第二の核酸は、例えば組込みまたは組換え(例えば相同組換えや他の種類の組換えなど)を行う核DNAに組み入れられる。別の実施形態では、第一および第二の核酸は細胞内に導入される発現ベクターを用いて発現される。この実施形態では、ベクター内の第一および第二の核酸の挿入断片は核DNAと組換えを行わず、むしろ安定化因子をコードする第二の核酸はベクター内に存在する、第一の核酸にコードされる誘導性および/または抑制性プロモーターなどの制御配列を使って自律的に発現される。本明細書の実施例1〜4で記載されるベクターを含め、当業者に知られるあらゆる適切なベクターが使用できる。
【0050】
これらの実施形態のそれぞれにおいて、第一の核酸は第二の核酸に作動的に連結して組換え核酸を形成する。本明細書に記載される代表的な核酸は、Nac2コード配列(すなわち第二の核酸)と作動的に連結したCyc6プロモーター(すなわち第一の核酸)である。第一および第二の核酸は、当業者によく知られるクローニング方法を用いて互いに作動的に連結でき、この方法には制限酵素で核酸を切断し、切断したベクターの末端に第一および第二の核酸をリガーゼを使って互いに連結することを含む。このようなクローニング方法は、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0051】
第二の核酸が第一の核酸にコードされるプロモーターの制御下に自律的に発現する実施形態では、発現コンストラクト(すなわち、ベクター‐挿入断片コンストラクト)は、第二の核酸に存在するコード配列の転写を終止させるための転写ターミネーターを通常含み、その他の5'および3'制御配列を含んでもよい。転写ターミネーターは第二の核酸内に通常存在するが、ベクターに組み入れることもできる。
【0052】
第一および第二の核酸が核DNAに安定に組み入れられる実施形態では、転写ターミネーターは第二の核酸に通常存在するが、核DNA配列の一部でもよい。第一または第二の核酸、ベクター、および/または核DNAには、例えば転写エンハンサーエレメントやmRNA安定化に関与する配列などのさらなる5'および3'制御配列が存在してもよい。この実施形態では、ベクターは、組換え核酸が核DNAに組込まれるのを促進する核ターゲティング配列も含んでもよい。一実施形態では、核は第二の核酸の非作動型コピーを有してもよく、第二の核酸のコピーやホモログを欠失してもよい。
【0053】
本明細書に記載される様々な実施形態において、安定化因子の自律的発現または組換え核酸の核DNAへの組み入れのためのベクターは、ベクターコンストラクトの複製のための細菌の複製起点を有し、組換え核酸(すなわち、作動的に連結した第一および第二の核酸)の挿入の有無にかかわらず目的とするベクターをクローニング用に大量調製することができる。ベクターは通常、DNA断片挿入のための制限エンドヌクレアーゼ切断部位(例えば多重クローニング部位)、そして形質転換体選択のための選択遺伝子マーカーも有する。選択マーカーは、抗生物質を含む培地上での形質転換細胞の増殖を可能にするaadA遺伝子やnptIIなどのマーカーである(Goldschmidt-Clermont, Nucl. Acids Res., vol. 19, pages 4083-4089 (1991))。内在性遺伝子(arg7、nit1)と外来性遺伝子(ble、aphVIII、aadA、nptII)の両方の選択マーカーが核形質転換のレポーター遺伝子として開発されている。
【0054】
組換え核酸(すなわち、第一および第二の核酸を含む挿入断片)を有するベクターは、当業者によく知られる標準的な形質転換技術により細胞(例えば藻類または植物の細胞など)に導入される。代表的な形質転換方法には、エレクトロポレーション、ガラスビーズを用いたDNA送達、ポリエチレングリコールを用いた形質転換、遺伝子銃、および同類のものがある。
【0055】
一実施形態では、藻類の形質転換のために、接合中に放出され細胞壁を分解する酵素であるオートリシンを使用して、形質転換の前に細胞壁を分解する。別の実施形態では、細胞壁の合成能を欠失する突然変異株(例えばcw15、cw10など)が作出されており、効率的な形質転換にこれらを使用できる。
【0056】
一つの例示的な形態において、形質転換体はPCRおよびサザンブロット法により検出できる。抗生物質添加、銅添加、抗体検出(例えばELISAおよびウェスタンブロット法)、および配列決定などの、当業者に知られる他の方法も使用できる。これらの方法は使用するコンストラクトによって選択する。
【0057】
もし導入遺伝子が色素体中で発現される場合、第三の核酸をベクターに組み入れ、第一および第二の核酸について上述したのと同じようにベクターコンストラクトを作成して一般的に複製できる。例示的に、遺伝子銃で色素体の形質転換ができ、これは導入遺伝子を含むベクターを金やタングステンの微粒子上に付着させてヘリウムなどの不活性ガスで加速させる。この方法は細胞の損傷が少ない。関連する例示的な実施形態では、形質転換する細胞を選択用固形寒天培地上にのせ、表面にDNAをコートしたタングステンビーズをヘリウムガスまたは火薬を使って加速させて色素体中に送達する。この技術を使用して、組換えDNAが色素体中に効率的に送達される。
【0058】
一実施形態では、細菌のアミノグリコシドアデニルトランスフェラーゼ遺伝子(aadA)をコードする異種DNAの発現により第三の核酸の色素体DNAへの組込みを検出できる。この実施形態では、抗生物質スペクチノマイシンまたはストレプトマイシンを使って形質転換体を選択する方法が可能となる。このレポーターコンストラクトを使用して、あらゆる非必須の遺伝子またはシス作動性エレメントを色素体中で特異的に挿入、破壊、修飾、変異または削除することが可能である。
【0059】
別の例示的な実施形態では、導入遺伝子の葉緑体DNAへの組込みを促進する相同的葉緑体ターゲティング配列を導入遺伝子の両末端に隣接させて、導入遺伝子をもつ葉緑体形質転換体を得ることができる。一実施形態では、ベクター上の隣接葉緑体配列と葉緑体ゲノム内の対応する相同配列との間での二つの相同組換えイベントにより導入遺伝子の組込みが起こる。一実施形態では、葉緑体は第三の核酸の非作動型コピーを有してもよく、第三の核酸のコピーやホモログを欠失してもよい。
【0060】
核酸を外来性にする代表的な修飾法には、コドンを最適化するように核酸を修飾する、他の遺伝子の5'または3'非翻訳領域を核酸に連結する、プロモーターまたは終止配列を核酸に付加する、相同組換えに有効な配列を核酸に付加する、そして例えば遺伝子発現、ターゲティング、安定化に必要な他のエレメントを核酸に付加することが含まれるがこれらに限らない。さらなる修飾法には、核酸を他の内在性または導入遺伝子の核酸と融合させて融合蛋白質を形成することも含まれる。
【0061】
本発明の範囲に含まれる色素体遺伝子制御システムの代表的な概要図を図2〜5に示す。図1はインデューサーの存在下に核内の誘導性プロモーターが誘導されている色素体遺伝子制御システムの概要図を示す。この実施形態では、インデューサー(例えば低酸素レベルなどの環境条件など)が安定化因子の発現を制御するプロモーターを誘導し、安定化因子が発現する。翻訳後、安定化因子は色素体に移行し、発現される安定化因子に応じた特異的mRNAに結合し、mRNAとそれがコードする蛋白質が発現する。
【0062】
図3はインデューサーの非存在下に核内の誘導性プロモーターが抑制されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、インデューサーが存在せず、誘導性プロモーターは活性化されず安定化因子は発現しない。安定化因子がないので、色素体内のmRNAは分解されるかまたは翻訳されず、蛋白質は発現しない。
【0063】
図4はリプレッサー(例えば培地中の銅の所定レベルなど)の存在下に核内の抑制性プロモーターが抑制されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、リプレッサーが存在し、抑制性プロモーターは活性化されず安定化因子は発現しない。安定化因子がないので、色素体内のmRNAは分解されるかまたは翻訳されず、蛋白質は発現しない。
【0064】
図5はリプレッサー(例えば培地中の銅の低レベルまたは銅の非存在)の非存在下に核内の抑制性プロモーターが誘導されている代表的な色素体遺伝子制御システムの概要図を示す。この実施形態では、リプレッサーが存在せず、安定化因子の発現を制御する核プロモーターが誘導され、安定化因子が発現する。翻訳後、安定化因子は色素体に移行し、発現される安定化因子に応じた特異的mRNAに結合し、mRNAとそれがコードする蛋白質が発現する。
【0065】
本明細書に記載される方法およびシステムを使って得られた発現蛋白質を単離精製することが望ましい実施形態では、蛋白質を発現した後に従来技術を用いて精製する。例えば、純度約40%、純度約50%、純度約60%、純度約70-80%、純度約90%、純度約95%、または純度約98%の蛋白質が得られる。細胞からの精製では、例えば細胞溶解物を硫安沈殿にかけてからDEAE-セファロースカラムクロマトグラフィーを行う。ゲルろ過、イオン交換クロマトグラフィー、DEAE-セファロースカラムクロマトグラフィー、アフィニティークロマトグラフィー(以下の実施例2に記載されるFLAGタグシステムを使用するなど)、溶媒-溶媒抽出、限外ろ過、HPLCなど、当業者に知られる他の従来技術も使用できる。または、蛋白質が非常に高濃度に存在して細胞溶解物中の蛋白質が実質的に純粋(例えば純度70-80%)であれば、精製段階が必要でないこともある。発現蛋白質は、例えば限外ろ過や接線流ろ過などの技術により濃縮できる。
【0066】
一実施形態では、例えば超音波処理、加熱、または化学処理などにより細胞を溶解し、ホモジネートを遠心分離して細胞片を除いてもよい。その後上清を硫安沈殿にかけ、さらに必要に応じてゲルろ過、イオン交換クロマトグラフィー、DEAE-セファロースカラムクロマトグラフィー、アフィニティークロマトグラフィー、溶媒-溶媒抽出、限外ろ過、HPLCなどの分画技術を使って発現蛋白質を精製できる。培地または細胞から発現蛋白質を精製する方法は上述した精製方法に限らず、実質的に純粋な蛋白質を得るのに必要であれば、当業者に知られるいずれかの精製技術を用いて発現蛋白質が精製できることを理解すべきである。
【0067】
細胞(例えば藻類または植物の細胞)は、蛋白質発現を促進するために様々な技術を用いて培養できる。藻類や植物細胞を含む細胞の培養培地は、当技術分野で知られており、炭素源(例えばブドウ糖または酢酸)が通常添加される。細胞は、例えば後述のように一例として水素ガスの産生に有用な培養システムおよび装置を使用して、望ましい濃度を維持しながら培養してもよい。
【0068】
上記に詳細を記したとおり、本明細書に記載される発現方法およびシステムは水素産生に使用できる。よって、別の実施形態では水素産生のための装置が提供される。本装置は、実質的に酸素のない環境下に細胞培養液を保持するように構成された第一の容器と、第一の容器と液体連通があり所定の速度で第一の容器に培地を供給するように構成された第一のポンプと、第一の容器に連結され細胞培養によって産生される水素量を測定するように構成された測定器を備える。
【0069】
図7は、一部の実施形態で使用することのできる水素産生のための装置10を図解する。装置10は新鮮培地保存容器12、ポンプ14、反応システム16、そしてオーバーフロー容器18を含む。新鮮培地保存容器12は、実質的に密閉された環境下にTAP(酢酸培地)またはHSM(最小培地)などの培地を一定量保持することのできる任意の種類の容器を用いればよい。一部の実施形態では、アルゴンガスなど一定量のガス24を導管26を介して新鮮培地保存容器12に送り込んで容器12内の酸素を追い出し、それにより実質的に酸素のない環境を作り出す。導管26には、新鮮培地保存容器への流体(ガスなど)の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。
【0070】
ポンプ14は導管28を介して新鮮培地保存容器12と、導管30を介して反応システム16と液体的に連結する。導管28(そして含まれる場合は導管26)は、新鮮培地保存容器12の実質的に密閉された環境が維持されるように容器12と連結する。導管28には、ポンプ14によるポンプ作用を介して流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。ポンプ14には、新鮮培地保存容器12から反応システム16に一定量の培地を所定の速度で送り込むことができ、新鮮培地に悪影響を及ぼさない任意の種類のポンプを用いればよい。例えば、一実施形態では、ポンプ14はポンプ過程で培地が損傷する可能性が低いペリスタ型ポンプを用いる。
【0071】
反応システム16は反応容器20および撹拌器22を含む。反応容器20は、新鮮培地保存容器12と実質的に同様であり、実質的に密閉された環境下に培地および藻類培養液、または他の種類の宿主細胞を一定量保持することのできる任意の種類の容器を用いればよい。新鮮培地は、導管28、30およびポンプ14を介して新鮮培地保存容器12から反応容器16に送り込まれる。新鮮培地は実質的に酸素のない環境下に保存されるため、反応容器16に不注意に酸素が入り込む可能性が低い。導管30は、反応容器12の実質的に密閉された環境が維持されるように反応容器20と連結する。
【0072】
撹拌器22には、反応容器20と作動的に連結し、反応容器20に保持された培養液の撹拌状態を維持することのできる任意の種類の機器を用いればよい。例えば、撹拌器22は磁気撹拌子アセンブリまたは同種のものなど自動撹拌器を用いてもよい。
【0073】
導管32は反応容器20からあふれた培養液をオーバーフロー容器18に排出する。オーバーフロー容器18は、容器12、20と実質的に同様であってもよく、一定量の培養液を保持することのできる任意の種類の容器を用いればよい。導管32は、導管28、30と実質的に同様であり、反応容器20からオーバーフロー容器18への流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。導管30と同様に、導管28は、反応容器20の実質的に密閉された環境が維持されるように反応容器20と連結する。導管30は、一部の培養液をオーバーフロー容器18に排出することにより反応容器20に入った培養液のレベルが所定レベル(またはそれ以内)に維持されるような配置で、反応容器20と連結されてもよい。
【0074】
一部の実施形態では、容器20内で培養液により産生された水素または他のガスの量が測定できるように、測定器34は通信リンク36を介して反応容器20と連結されてもよい。測定器34には、目的とするガスの量を測定することができる任意の種類の機器を用いればよい。一実施形態では、測定器34は質量分析器を用いるが、他の実施形態では他の種類の測定器を用いてもよい。通信リンク36は、例えば任意の本数のワイヤ、ケーブル、光ファイバーケーブル、チューブ、導管、または同種のものなど、測定器34へのデータ通信を促進することのできる任意の種類の通信リンクであればよい。一実施形態では、通信リンク36は反応容器20内に置かれた電極部分などである。電極部分は例えば銀電極であってもよい。
【0075】
一部の実施形態では、装置10にはフィルターシステム40および二次ポンプ38が含まれてもよい。フィルターシステム40は導管42を介してオーバーフロー容器18と、導管44を介してポンプ38と連結する。ポンプ38は導管46を介して新鮮培地保存容器12と連結する。導管42、44、46は導管28、30、32と実質的に同様であり、流体の通過を促進することのできる任意の種類のチューブ、ライン、または他の導管を用いればよい。ポンプ38はポンプ14と同様で、オーバーフロー容器18から新鮮培地保存容器12に一定量の「使用済み」培養液を所定の速度で送り込むことができ、新鮮培地に悪影響を及ぼさない任意の種類のポンプを用いればよい。例えば、一実施形態では、ポンプ38はペリスタ型ポンプである。フィルターシステム40は、オーバーフロー容器18に保持された培養液をろ過することのできる任意の数および種類のフィルターおよび関連する連結部品を用いればよい。
【0076】
ポンプ14は、稼働時に新鮮培地保存容器12から反応システム16の反応容器20に新鮮培地を所定の速度で送り込むように構成される。一実施形態では、ポンプ14は反応容器20に保持されている藻類の細胞増殖速度と実質的に同じ速度で新鮮培地を反応容器20に供給するように構成される。上記に詳細を示したとおり、実質的に酸素のない環境下で藻類または他の種類の宿主細胞は反応容器20に保持されるので、藻類または他の種類の宿主細胞の中では、水素産生が誘導されるように望ましい遺伝子発現が誘導される。上述したとおり、新鮮培地も実質的に酸素のない環境下で新鮮培地保存容器12に保存されるため、新鮮培地保存容器12から新鮮培地が送り込まれる間も反応容器20の実質的に酸素のない環境は維持される。または、他の実施形態では、反応容器20に保持される藻類または他の種類の宿主細胞は自己誘導される。いずれにせよ、装置10は単相装置であることを理解すべきである。すなわち、藻類または他の種類の宿主細胞は反応容器20に保持され、同じ容器内で増殖し誘導される。
【0077】
新鮮培地が反応容器20に送り込まれる際、反応容器内に保持された培養液が連続撹拌されるように撹拌器22は構成される。また、新鮮培地が送り込まれる際、既存の培養液の一部が反応容器からオーバーフロー容器18に移る。こうして、反応容器20に入っている藻類または他の種類の宿主細胞の量は実質的に一定値に維持される。ポンプ38およびフィルターシステム40を含む実施形態では、ポンプ38はオーバーフロー容器から一定量の「使用済み」培養液を送り出し、それをフィルターシステム40でろ過してその中に含まれる藻類や他の種類の宿主細胞を除去した後で新鮮培地保存容器へと戻す。
【0078】
次に図8を参照すると、一実施形態では、水素産生のための装置50は新鮮培地保存容器52、ペリスタ型ポンプ54、反応システム56、およびオーバーフロー容器62を含む。新鮮培地保存容器52は、新鮮培地保存容器12と実質的に同様であり、例として0.25L容器が用いられる。例としてTAP(酢酸培地)またはHSM(最小培地)など、一定量の新鮮培地64を容器52に保持する。容器52の内腔68が外環境から実質的に密閉されるように、キャップ66を容器52に連結する。導管70はキャップ66と連結し、容器52の内腔68内に位置する末端部72を含む。一定量のアルゴンガス74を導管70を介して内腔68へ送り込み、これにより内腔内の酸素を実質的に追い出す。
【0079】
ペリスタ型ポンプ54は導管76を介して新鮮培地保存容器52と、導管78を介して反応システム56と連結する。ペリスタ型ポンプ54は例としてモデルIP4ペリスタ型ポンプが用いられ、これはIsmatec(スイス、グラットブルグ)から購入できる。ポンプ54は、反応システム56に保持されている藻類培養液または他の種類の宿主細胞の増殖速度と実質的に同じ速度で一定量の新鮮培地を新鮮培地保存容器52から反応システム56に送り込むように構成される。
【0080】
反応システム56は、例として0.25L容器などの反応容器88、および反応容器88に保持された一定量の培地および藻類培養液94または他の種類の宿主細胞を連続撹拌するように構成された、磁気撹拌子システム90を含む。磁気撹拌子システム90は例としてモデルKM02磁気撹拌子が用いられ、これはMilian(スイス、ジュネーブ)から購入できる。反応容器88の内腔92が外環境から実質的に密閉されるように、キャップ92を容器88に連結する。導管78はキャップ92と連結し、反応容器88の内腔94内に位置する末端部96を含む。
【0081】
質量分析器100も通信リンク102を介して反応容器88と連結する。質量分析器100は例としてモデルMM8-80質量分析器が用いられ、これはVG Instruments(英国、チエスター)から購入できる。通信リンク102は容器88の内腔94内に位置する電極104を含む。質量分析器100は、反応容器88に保持されている培地および藻類培養液または他の種類の宿主細胞によって産生される水素量(および一部の実施形態では酸素量)を測定するように設定される。
【0082】
さらに、導管106はキャップ92と連結し、容器88の内腔94内に位置する末端部108を含む。導管106の先端部110はオーバーフロー容器62の内腔内に位置する。導管106は、反応溶液88に保持されている藻類または他の種類の宿主細胞の増殖速度と実質的に同じ速度で、培地および藻類培養液または他の種類の宿主細胞培養液が反応容器88から送り出されるように配置される。
【0083】
上記は本明細書に記載されるすべての方法およびシステムに適用される。以下の例は説明目的のみであり、本文書に添付する特許請求の範囲に記載される本発明の範囲を限定するものではない。本文書で引用した参考資料は本明細書に具体的な参照として含まれる。
【0084】
〔実施例1 内在性遺伝子のための誘導性色素体発現システム〕
Cyc6プロモーターの制御下にNac2遺伝子を含むベクターは、当業者に知られる分子クローニング技術を用いて作成した。クローニング方法は、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0085】
Nac2遺伝子をCyc6プロモーターエレメントの制御下に置くため、Cyc6プロモーターエレメントおよびNac2ゲノムDNAに特異的な4つのオリゴヌクレオチドを用いたオーバーラップ伸長PCRにより、psbDコード配列に融合させたCyc6プロモーターを含むキメラDNA断片を作出した。得られたPCR断片は、833塩基対のNac2ゲノム配列断片にインフレーム融合した428塩基対のCyc6プロモーター配列からなる。PCR断片はサブクローニングして配列を決定した。PCR断片は次に、XbaIおよびAatIIユニーク制限部位を用いてpNac2(midi)プラスミドにクローニングした。pNac(midi)プラスミドは、Nac2の5.1kbキメラmidi遺伝子を含み、これはBoudreauら(2000)により記載されており、本明細書に参照として含まれる。この遺伝子は、3'側cDNA配列に融合した5'側Nac2ゲノム配列からなり、C末端がトリプルHAエピトープでタグされた全長Nac2蛋白質をコードするオープンリーディングフレーム(ORF)がもたらされる。pNac2(midi)をXbaIおよびAatIIで制限分解した。次にこのプラスミドにPCR断片を方向性に連結した。得られたプラスミドpcy6Nac2(midi)は、Nac2 midi遺伝子にインフレーム融合した428塩基対のCyc6プロモーター配列を含む。
【0086】
最後に、pSL17の多重クローニング部位内のユニーク制限部位(EcoRIおよびXbaI、図9参照)を使って、5.8kb cyc6Nac2導入遺伝子をpSL17プラスミドにクローニングした。pSL17は抗生物質パラモマイシンへの耐性を付与するaphVIIカセットおよびクローニングのための多重クローニング部位を含む。得られた10.8kbプラスミドpcy2(paroR)(図10参照)は、nac2-26突然変異細胞の形質転換に用いた。ベクターの構築に用いたNac2およびCyc6の配列をそれぞれ図11と12に示す。
【0087】
pcy6Nac2(paroR)ベクターを、エレクトロポレーションによりnac2ヌル変異体であるnac2-26に導入した。藻類および植物の形質転換法は当業者に知られており、例えば本明細書に参照として含まれるSambrookらの「分子クローニング:実験室マニュアル」(第3版、Cold Spring Harbor Laboratory Press、2001)またはS. Surzyckiの「分子生物学の基本技術」(Springer-Verlag、2000)などに記載されている。
【0088】
Cyc6誘導性Nac2遺伝子を含むChlamydomonas遺伝子導入株を単離するため、nac2-26細胞をまずオートリシン処理して次にpcyc6Nac2(paroR)を用いたエレクトロポレーションにより形質転換した。得られた形質転換株を抗生物質パロモマイシン(20μg/ml)を含むTAP培地にのせた。パロモマイシン耐性コロニーは、150μMニッケル(II)(Cyc6転写インデューサー)を添加した最少培地ならびに銅を含まない最少培地(HSM -Cu+2、図13参照)の上で光独立栄養的に増殖するものを選択した。光独立栄養株は次に、インデューサーを含まない最小培地(HSM)上での増殖の有無を調べた。
【0089】
この方法を使って、遺伝子導入株cy6Nac2.49を単離した(図13参照)。この株はHSM -Cu+2上で光独立栄養的に増殖できるが、銅を添加したHSM上では光独立栄養的に増殖できない。得られたcy6Nac2.49株は、以下の二通りの方法で増殖する:1)銅を含まない培地中で光独立栄養的に増殖、または2)酸素と銅が存在する培地中では細胞内にPSII複合体がないため嫌気的に増殖(図1を参照)。よって、遺伝子導入株cy6Nac2.49では、光合成による酸素産生は低酸素に応答する核プロモーターを介して制御される。
【0090】
さらに、非誘導条件下に増殖するcy6Nac2.49の密閉培養は、光合成による酸素発生がなくミトコンドリアの呼吸による酸素消費は一定に維持されるので、急速に嫌気的となる。cy6Nac2.49の密閉培養では、低酸素が光合成酸素産生を誘導し、それがPSII合成と光合成酸素発生を再び抑制するという、阻害のフィードバックループが存在する。
【0091】
実際に、完全培地中でのcy6Nac2.49の光照射密閉培養ではまさしくこの現象が観察された。培養液中の酸素濃度は、最初は大気中酸素濃度と同じで、その後急速に消費され、嫌気的増殖と水素発生の誘導に至った。一定の嫌気的増殖期間の後、培養液の酸素レベルが回復して容器内酸素濃度が大気中の2倍となり、水素発生が阻害された(データ示さず)。通常の密閉容器では、酸素消費と水素産生が一度ずつしか観測されず、これはおそらく還元炭素がミトコンドリアの呼吸により消費されてしまうと酸素消費は止まるが、光合成による酸素産生は一定に維持されるためと見られる。
【0092】
野生型、nac2-26(cy6Nac2.49の親株)およびcy6Nac2.49細胞について、完全培地(TAP)、抗生物質パロモマイシン添加完全培地(TAP + Paro)、最少培地(HSM)、および銅非含有培地(HSM-Cu+2)上での増殖能を調べた。非誘導条件下(TAP)および誘導条件下(TAP-Cu+2およびTAP-O2)に増殖した野生型およびcy6Nac2.49細胞の全細胞抽出物についてのウェスタンブロット解析を図14に示す。ウェスタンブロットは、嫌気的増殖の指標としてα-HydA、α-D2およびα-RbcLを認識する抗体でプローブした。
【0093】
図15に水素発生を測定した実験結果を示す。Cy6Nac2.49細胞は、培養液中の溶存ガスを質量分析器で測定できる(酸素-点線、水素-直線)光照射容器に密閉し、培地に銅を添加することで誘導条件から非誘導条件に移行させた(TAP-Cu+2からTAPへ)。液相中の水素濃度を測定し、mBar単位で表した。
【0094】
通常の密閉容器は、cy6Nac2.49遺伝子導入株を使って水素産生を持続するのに適さないため、増殖するcy6Nac2.49培養液に新鮮な無酸素培地を一定速度で供給し、対数増殖期を維持できる密閉無酸素システムを設計した(図7、8参照)。培養液を対数増殖期に維持することで酸素消費/水素産生/酸素産生のサイクルを確立できるとする仮説を立てた。cy6Nac2.49細胞をこのシステムで増殖させたところ、培養液が低酸素になったすぐ後に水素発生が観測された。ついでPSII複合体の合成が誘導されるとともに容器内の酸素がわずかに増加した。通常の密閉容器とは異なり、光合成による酸素産生は水素産生を阻害せず、これはおそらく光合成による酸素産生がミトコンドリアの呼吸による酸素消費を決して上回らないためと思われる。このシステムでは、水素産生は光エネルギーと直接連関しており、したがって水素産生のための直接バイオ光分解を表している。無酸素システムを使用して、気相の約0.5%に達する一定速度の水素発生が得られた。
【0095】
光合成の副産物として酸素が発生する。結果として、光エネルギーを使った藻類での水素発生維持(「バイオ光分解」とも呼ばれる過程)における中心課題は、ヒドロゲナーゼ酵素の酸素感受性を克服することであった。本発明で開発した直接バイオ光分解法とは対照的に、間接バイオ光分解法(または光合成と水素産生の二段階法)は光合成による酸素産生と水素産生を時間的または/および空間的に分けることでヒドロゲナーゼ酵素の酸素感受性を克服する(Benemann 1996; Melis 2000)。第一段階は通常の酸素発生型光合成、つまり酸素放出、CO2固定、バイオマス蓄積が見られる。第二段階では、硫黄などの重要な栄養素が枯渇して、酸素発生型光合成が生理学的に阻害される。硫黄欠乏状態が約22時間続くと酸素発生型光合成が急激に減速するので、ミトコンドリアの呼吸による総酸素消費のせいで、密閉培養液は嫌気的になる(Melis 2000)。嫌気的増殖が確立すると、ヒドロゲナーゼ経路が誘導され、主として残存する水の光酸化から、また蛋白質やデンプンなどの内在基質の異化からも得られる電子を使って、水素が発生する(Ghirardi 2000)。
【0096】
無酸素システム、すなわち直接バイオ光分解法と、間接光分解法の違いを見ると、無酸素システムを使用することの大きな利点がいくつか明らかとなる。第一に、無酸素システムの特長として、単一の光照射密閉容器内で酸素産生と水素発生が行われるというだけの理由で、水素産生能は50%高い。光合成と水素産生の二段階法または間接バイオ光分解法は、酸素産生と水素産生を時間的もしくは空間的に分けるので、結果として2つの容器のうち一方が水素産生には使用されず産生能の50%が失われる。第二に、光合成と水素産生の二段階法は硫黄の生理学的枯渇が酸素発生型光合成を阻害することに依存する。硫黄が極度に枯渇すると様々な細胞過程に広く影響する。水素放出を触媒するヒドロゲナーゼ酵素にはFe-Sクラスターが含まれ、その会合が機能性に必要である((Posewitz, et al. (2004))。また、硫黄の枯渇は、水素発生に重要なPSIなどの葉緑体中の他の複合体に悪影響を及ぼす。明らかに、硫黄の生理学的枯渇は水素発生装置の重要な部分に重大な影響を及ぼす。これに対し無酸素システムは、水素産生を誘導するのに必要な微量栄養素の枯渇に依存しない。事実、水素産生は最適な生理学的条件下に(完全培地中での対数増殖期に)起こる。
【0097】
最後に、例えば藻類を使った大規模な水素産生において主な障害となるのは大量培養液に光エネルギーを供給することである。藻類の高濃度大量培養では、集光性複合体中のクロロフィル量が多いために、容器中心付近の細胞に光エネルギーが到達しにくい。本発明の直接バイオ光分解法では、光吸収および水素産生が最大となるような最適細胞濃度の確立されるシステムで水素産生が起こる。
【0098】
〔実施例2 外来性遺伝子のための誘導性色素体発現システム〕
葉緑体形質転換ベクター。プラスミドpSK108は、psbD遺伝子と、psbD遺伝子周辺領域への挿入を導く5'隣接配列を含む3kb葉緑体DNA断片を含む。このコンストラクトはまた、psbD遺伝子の上流に挿入されたaadAカセットも含む。導入遺伝子はpSK108のNcoIおよびSphI部位を使ってインフレームに挿入できる。連結後の新規ベクターは、その発現を担うatpAの5'末端およびターミネーターとして機能するrbcLの3'配列をもつ導入遺伝子(加えて3塩基対も)を含む。atpAプロモーターは葉緑体のATP産生プロトンポンプをコードする遺伝子の発現を担うので、D1修復機構に影響されない。
【0099】
FLAGタグをつけた3種類の外来性遺伝子、VP28、DILPおよびIBDVをそれぞれ葉緑体形質転換・発現ベクターpCG12にサブクローニングした(図17)。このベクターは葉緑体のatpB遺伝子座の下流に挿入される。導入遺伝子はatpAプロモーターの制御下に発現し、psbDの5'UTRの安定化因子結合部位をもつ(図18、19)。
【0100】
これらのベクターを、Chlamydomonasの16S rRNAを有しスペクチノマイシン耐性を付与するp228ベクターとともに、cyc6/Nac2誘導性株(制御プロモーターとしてCyc6プロモーター、安定化因子としてNac2をもつ)へ同時形質転換した。
【0101】
〔実施例3 C. reinhardtii葉緑体でのHydA1、HydEF、およびHydG遺伝子の過剰発現〕
ヒドロゲナーゼがC. reinhardtiiで過剰発現されるかどうか、そしてこれが水素産生を促進するかどうかを検討する。大腸菌でHydEF、HydGおよびHydA1を過剰発現させると活性型Feヒドロゲナーゼが産生されることが、最近の実験で示された(Posewitz, et al, (2004))。C. reinhardtiiの核内で蛋白質を過剰発現させることは、導入遺伝子がサイレンシングを受けることが多いためほとんど成功していない。したがって、遺伝子銃による形質転換を用いてこれらの遺伝子を葉緑体中で発現させる。この方法にはいくつかの利点がある。第一に、葉緑体では遺伝子のサイレンシングが起こらない。第二に、C. reinhardtiiには各葉緑体遺伝子が80コピーずつ存在する。第三に、外来性遺伝子を葉緑体のコドンバイアスに合うように再合成すれば、葉緑体プロモーター、5'および3'UTRの制御下に、葉緑体中で外来性蛋白質を高レベルに発現させることが可能であることが近年の実験で判明している(Mayfield, et al. (2001))。葉緑体遺伝子配列には強いATバイアスがある。
【0102】
HydA1、HydEFおよびHydGのそれぞれのコード配列をまず、C. reinhardtiiの葉緑体コドン使用頻度に従って再合成する。これは、外来性遺伝子を葉緑体中で発現させるのに成功した既報の方法を用いて行う。例として、近年我々はC. reinhardtiiの葉緑体中で2つのウイルス蛋白質を過剰発現させることに成功した(下記参照)。導入遺伝子は、コピー数を2倍にするため葉緑体の逆方向反復配列内に挿入する。宿主株としてcyc6Nac2株を使用する。この株は核nac2変異と、Cyc6プロモーター制御下にNac2遺伝子を有し、Nac2遺伝子は銅枯渇または嫌気的条件下で誘導される。
【0103】
この時点では、HydA1p、HydEFp、HydGpのうちどの蛋白質が水素産生を律速しているかは不明である。これを調べるため、3つの遺伝子のそれぞれにpsbDプロモーターと5'UTRを融合させたものを、遺伝子銃形質転換を使って葉緑体逆方向反復配列に個別に挿入する。可能性があるのは、リボソームオペロンの16Sと23S rRNA遺伝子間のスペーサー配列内に遺伝子を挿入することで、この方法により高等植物の葉緑体で高レベル発現が成功している。導入遺伝子は、Nac2を介したCyc6プロモーターの制御を受けるpsbD 5' URTの制御下にあるため、導入遺伝子は嫌気的条件でのみ発現する。RNAブロットハイブリダイゼーションまたはリアルタイムRT-PCRにより発現を調べる。検討する3つの遺伝子のそれぞれについて、Zhang and Melis (2002)の記載のとおり、上下逆にして水を満たしたビュレットにチューブで接続した密閉瓶内で形質転換細胞を培養し、移動する水の体積から水素産生量を直接測定する。cyc6Nac2の制御と相関して水素産生が増加すれば、過剰発現させた蛋白質が律速であることが示される。これらの実験により複数の蛋白質が水素生産の律速となっていることが分かれば、それらの蛋白質を同じ方法で過剰発現させる。各蛋白質を過剰発現させて水素産生量を高めるように、野生型細胞ではこれら3つの蛋白質の発現が調整されている可能性がある。この場合は3つの蛋白質を過剰発現させる必要がある。高等植物の葉緑体内で複数の導入遺伝子を発現させることは成功している(Quesada-Vargas, et al. (2005))。psbD 5'UTRによる発現が不十分であれば、psbA、atpA、リボソームプロモーターなど、他の強力な葉緑体プロモーター-5'UTRを検討する。
【0104】
〔実施例4 水素産生に影響を及ぼす遺伝子の発現〕
上記に加え、他のいくつかの遺伝子が水素産生を増加させるかどうか、色素体内での蛋白質発現のための誘導性および/または抑制性システムを使って検討する。酸素非感受性ヒドロゲナーゼとアンテナサイズの縮小を2つの例として挙げる(Melias, et al. (2004); Ghirardi, et al. (2005))。簡単に言うと、前者はChlamydomonasや他の生物からの野生型および突然変異型ヒドロゲナーゼ遺伝子をクローニングして酸素感受性が低くなるか解析する。アンテナサイズ縮小という後者の目的は、藻類細胞が捕捉する光子量を減らすことにより、光が光反応器の内部まで貫通し、通常であれば光の当たらない細胞に光を供給することである。藻類細胞は効果的に光を捕捉するがその利用効率は低く、吸収した光子の80%ほどが無駄になる。DNA挿入突然変異法によりアンテナサイズを制御する遺伝子が同定されている。変異体tla1のPSIIおよびPSIアンテナサイズはそれぞれ野生株の50%および65%であった。最後に、呼吸速度が上昇した結果ヒドロゲナーゼを阻害する酸素量が低下していると見られる変異株が複数同定されている(Krause, et al. (2005))。これらの遺伝子をcy6Nac2.49遺伝子導入株に挿入し、水素産生が増加するか検討する。
【0105】
水素産生を律速する重要な因子の一つはカルビンベンソン回路との競合である。ヒドロゲナーゼへの電子伝達を促進する可能性の一つは、カルビンベンソン回路に関与する酵素量を低くしてこの回路の活性を下げることである。例えば、ホスホリブロースキナーゼ(PRK)活性を持たないChlamydomonasの2つの変異株、F60とac214が入手可能なので、PRKを利用することができる。PRK遺伝子を誘導性Cyc6プロモーターに融合し、このコンストラクトをF60に挿入する。この方法の利点は、PSIIとカルビンベンソン回路の両方が銅(または酸素)の非存在下に機能するので還元力を蓄積できる。銅(または酸素)の存在下に両遺伝子の発現は止まる。よってヒドロゲナーゼが誘導される条件下で電子の流れはカルビンベンソン回路からヒドロゲナーゼへと移り、水素産生が促進される。
【0106】
別の例示的な実施形態では、温度感受性PRK変異体をスクリーニングする。この方法では、制限温度でカルビンベンソン回路が止まり、ヒドロゲナーゼへと電子が流れる。PRK変異体を、PCRで作成したPRK変異遺伝子ライブラリーで形質転換する。形質転換体を許容温度(24℃)で最小培地上で選別する。コロニーを別の最小培地にレプリカし、制限温度(32℃)で培養し、この温度で増殖しない変異体を同定する。PRK遺伝子に変異があるかどうかを、PCR増幅と配列決定により確認する。これらの変異体ではカルビン経路を阻害すると蛍光収量が上昇すると見られることから、蛍光によるスクリーニングも可能である。
【0107】
別の実施形態では、ヒドロゲナーゼからの電子の分岐経路として別に循環的電子伝達があり、状態遷移が状態1でブロックされている欠損変異株を用いる。状態遷移は、光条件の変化に伴い光合成収量を最適化するために、LHCII可動部がPSIIからPSIへと可逆的に移動することによりPSII(LHCII)とPSIのアンテナ間での光励起エネルギーを再バランス化することが関与する。状態1では、LHCII可動部がPSIIと結合しており、状態2ではPSIと結合している。さらにChlamydomonasでは、状態1で主に直線的電子伝達が起こるのに対し、状態2では循環的電子伝達が起こる(Finazzi et al., 2002)。よって、状態1でブロックされた変異体では循環的電子伝達が見られない。状態1でブロックされているstt7変異体(Depege et al., 2003)を、Cyc6-Nac2およびCyc6-PRKコンストラクトを含むnac2-26と交配し、PSII活性が抑制された状態で水素産生が増加するかどうか調べる。もしくは、温度感受性PRK変異体が得られれば、これをnac2-26 stt7 Cyc6-Nac2株と交配させる。これらの株について上記の条件下で水素産生を調べる。
【0108】
stt7以外の他の状態遷移欠損変異体を解析する。光合成酸素発生に対するミトコンドリア呼吸の比率が変化するような光合成電子伝達およびミトコンドリア電子伝達の制御変化によっても、状態遷移の変化が間接的に起こりうるので、これらの変異体は特に興味深い。この種の変異体は、野生型細胞に比べて水素産生が上昇していることが報告されている(Kruse et al., 2005)。
【0109】
〔実施例5 外来性遺伝子のための誘導性色素体発現システム〕
誘導性葉緑体遺伝子発現には選択マーカー遺伝子aadAを使用した。この遺伝子は、Nac2依存性に発現させることができ、抗生物質スペクチノマイシンを用いて容易にスクリーニングができ、高い特異活性をもつことが知られている。低レベルのaadA発現でもある程度の抗生物質耐性が得られ、従ってInd41_18でのcy6Nac2制御の「堅固さ」の指標となる(下記)。psbDのプロモーターおよび5'UTRによりaadA遺伝子の発現を制御する葉緑体挿入ベクター挿入が構築されている。
【0110】
nac2-26変異株はすでに記載されている(Kuchka, et al. EMBO J. 7, 319-324; Nickelsen, et al. EMBO J. 13, 3182-3191)。cyc6Nac2.49株は、Nac2 midi遺伝子と融合したCyc6プロモーターを含む導入遺伝子がnac2-2変異体の核ゲノムに挿入されている(Δnac2::cy6proNac2)。Ind41はcy6Nac2.49のpsbDプロモーターおよび5'UTRをpetAのプロモーターおよび5'UTRを含む675bp断片に置換したものである(Δnac2::cy6pro Nac2::5'petA-psbD)。Ind41_18はInd41株と関連しているが、Ind41_18ではaadAカセットが葉緑体DNAから完全に削除されているためスペクチノマイシンに感受性である(Δnac2::cy6proNac2::5'petA-psbD[SpcS])。Ind_aadA_117はInd41_18から得られたもので、atpB遺伝子の下流に挿入されたpsbDプロモーターおよび5'UTRの制御下にaadAカセットを含む(Δnac2::cy6proNac2::5'petA-psbD::5'psbD-aadA)。
【0111】
<Ind_aadA遺伝子導入株のスクリーニング>
Ind41_18細胞の葉緑体ゲノムへの5'psbD-aadAカセットの挿入は、遺伝子銃形質転換を用いて銅枯渇状態のInd41_18細胞にpcg12葉緑体挿入ベクターを導入することにより得られた(図21)。銅を含まないスペクチノマイシン100μg/mL含有TAP培地上で形質転換株を選択した。形質転換で得られたコロニーを拾って、スペクチノマイシン100μg/mL含有TAP-Cu+2培地に3回移植し直し、5'psbD-aadAカセットを含む細胞を確実に分離した。
【0112】
実験に使用したpcg12ベクターの概要図を図21に示す。このベクター内のaadAカセットはpsbD 5'UTRを使って発現する。野生型Ind41_18およびInd_aadA_X遺伝子導入株の生育も示す。これらの細胞を段階希釈し、固形TAP培地、銅非含有TAP培地(TAP-Cu+2)、スペクチノマイシン100-1000μg/mL含有TAP培地(TAP-Cu+2+Spc)、およびスペクチノマイシン100-1000μg/mL含有TAP-Cu+2培地上に滴下し、光強度100 μE m-2s-1の照射下に7-10日間培養した。
【0113】
誘導性aadA形質転換株の候補をTAP + Spc100およびTAP-Cu+2 +Spc100寒天培地上にレプリカし、スクリーニングを行った。試験したコロニーはすべて両培地で同様に増殖し、Cyc6Nac2導入遺伝子のプロモーター制御が失われていることを示した(図21A)。しかし、高濃度のスペクチノマイシンで増殖を調べたところ、これらの株の85%は培地中に銅が含まれるとスペクチノマイシンに感受性を示し、培地中に銅が含まれないと耐性を示した(図21B)。こうした株のいくつかをInd_aadAと命名し、さらに解析を行った。これらの株の遺伝子型はnac2::cy6ProNac2::5'petA-psbD::5'psbD-aadAである。
【0114】
<Ind_aadA形質導入株の増殖解析>
一次スクリーニングからのいくつかの誘導性株の増殖解析では、段階希釈した後、野生型、Ind41_18、Ind_aadA細胞をTAP、TAP-Cu+2、HSM、HSM-Cu+2、および一定範囲内で各種濃度のスペクチノマイシン(0、100、250、500、750、1000、2000 μg/mL)を含む寒天培地に滴下して行った(図21B)。この方法で試験した11株中の1株、Ind_aadA_36は、500 μg/mLより高濃度のスペクチノマイシンを含むTAP培地で増殖を示した。一方、11株中6株は銅を含まないTAP培地で低濃度のスペクチノマイシンでは増殖したが、250 μg/mLを超えるスペクチノマイシンでは増殖しなかった(図21B)。Ind_aadA_117と命名した株は、TAP培地では250 μg/mLのスペクチノマイシンに感受性を示したが、銅を含まないTAP培地では試験したスペクチノマイシン濃度範囲のすべてで増殖した。
【0115】
<Ind_aadA_117から抽出した全RNAのノーザン解析>
Ind_aadA_117でのaadA発現誘導を解析するために、非誘導性(TAP)または誘導性条件(TAP-Cu+2、TAP-Cu+2 + Spc、TAP-O2、HSM-Cu+2)で培養したInd_aadA_117細胞から全RNAを抽出した。これらの試料をノーザン解析した結果、psbD転写物の蓄積は親株Ind41_18と同等、もしくは野生型のpsbD RNAの約25%であることが判明した。予想どおり、Ind_aadA_117のpsbD RNAは野生型psbD転写物よりも大きく、親株Ind41_18からの遺伝形質として、真のpsbD遺伝子はもはや存在しないことを示した(図22)。重要な点として、Cyc6転写が誘導されているすべての培養細胞でaadA転写物の蓄積が認められた。TAP培養したInd_aadA_117細胞でaadA RNAが少量検出されたことから、Ind_aadAのリーキーな表現型が確認された(図22)。驚くべきことに、銅枯渇培養したInd_aadA_117細胞よりも嫌気培養したInd_aadA_117細胞の方がaadA転写物は多く、psbD/D2発現に関して「親株の親株」であるcy6Nac2.49には見られない特徴であった(図22)。図22に示した分析では、TAP、TAP-Cu+2、TAP-Cu+2 +Spc、TAP-O2 +Spc、HSM-Cu+2およびHSM-Cu+2+Spc液体培地中で培養した野生型、およびInd_aadA_117細胞から全RNAを抽出した。図22の左側に使用プローブを示す。
【0116】
<Ind_aadA_117蛋白質抽出物の解析>
Ind_aadA_117細胞から抽出した総蛋白質を、葉緑体にコードされる重要な蛋白質のいくつかに特異的な抗血清を使って免疫ブロット解析した。検討したすべてのInd_aadA_117培養細胞でD2蛋白質レベルは野生型の約25%であり、これはD2蛋白質を50%蓄積した親株Ind41_18よりも少なかった(図23)。一方、検討したほとんどのInd_aadA_117蛋白質抽出物でD1およびCP47蛋白質の蓄積はほぼ完全に回復しており、D2蛋白質の蓄積は75%低下していると推定された。よって、驚くべきことに、D1とCP47の蓄積はD2蛋白質レベルに対して調整されなかった。各種条件で培養したInd_aadA_117から調製した可溶性抽出物を使って、Ind_aadA_117でのNac2蛋白質の蓄積を調べた。嫌気培養および銅枯渇培養したInd_aadA_117からの可溶性抽出物ではCyc6Nac2の誘導が認められたが、TAP培養したInd_aadA_117で少量のNac2が検出された。Cyc6の発現が抑制される条件でのTAP培養細胞で微量のNac2が検出されたことから、このシステムはInd_aadAではわずかに「リーキー」であることが示された(図23)。したがって、TAP培養されたInd_aadA形質導入株が低濃度のスペクチノマイシンで増殖したという当初の観察結果は、Cyc6Nac2のややリーキーな性質による。図23では、TAP、TAP-Cu+2、TAP-Cu+2 +Spc、TAP-O2 +Spc、HSM-Cu+2およびHSM-Cu+2+Spc液体培地中で培養した野生型、およびInd_aadA_117細胞から総蛋白質を抽出し、PVDFメンブレンに固定した。図23の左側に使用プローブを示す。
【0117】
<Ind_aadA_117のaadA分析>
aadAの量は活性測定により間接的に推定した(表1)。活性測定はaadA酵素がATP分子のアデニル基をスペクチノマイシンに転移し、スペクチノマイシン分子に正の電荷を与えることに基づく。正の電荷をもつスペクチノマイシン分子は、負の電荷をもつリン酸セルロース膜に結合する。よって、もしaadAを発現するChlamydomonasの粗抽出物をスペクチノマイシンおよびα32P標識化dATPとともにインキュベートした場合、その反応液をリン酸セルロース膜上に滴下してから非特異的産物を洗い落とした後に膜に残る放射能量から、aadA酵素活性の相対的測定が可能である。TAP、TAP-Cu+2、またはTAP-Cu+2 +Spc液体培地で培養した細胞の粗抽出物について、この遺伝子導入株中のaadA活性を測定し、親株および野生型バックグランドで従来のaadAカセットを発現する他の株と比較した。いくつかの個別の分析結果を合わせて表1に示す。Ind41_18ではaadA活性が検出されず、この株ではカセットが完全に除去されていることを示した。一方、従来のaadAカセットを発現する野生型およびTAP-Cu+2やTAP-O2で培養したInd_aadA_117のどちらも、既報のaadA発現遺伝子導入株と同等のaadA活性を有した。TAP培地で培養したInd_aadA_117の粗抽出物中に、少量だが有意なaadA活性が存在した。この結果から、誘導されていないInd_aadA_117にも、誘導されたInd_aadA_117の活性よりはかなり少ないが、少量のaadA活性が存在することが確認された。
【0118】
cy6Nac2葉緑体誘導性遺伝子発現システムを使ったaadA遺伝子発現を誘導できる株のスクリーニングでは、常に「リーキー」な表現型を示す株が得られた。親株ではCyc6Nac2導入遺伝子が緊密に制御されていることが明らかなので、Ind_aadAではCyc6Nac2導入遺伝子が脱抑制されることがスクリーニングを生き残るのに必要であると推測される。この観測結果の解釈の一つとして、遺伝子銃によるInd41_18のpcg12形質転換に使用した培地を含め、スクリーニングの各段階で使用した銅枯渇培地には初めから微量の銅が混入していた可能性がある。もしこれが正しければ、銅の混在濃度が1細胞あたり2x106イオンを下回るまでCyc6転写が一時的に抑制されると予想され、結果としてCyc6Nac2が脱抑制された形質転換株のみが、pcg12プラスミドによる最初の形質転換を生き残ると考えられる。言い換えると、Cyc6Nac2の転写抑制が十分に長くてスペクチノマイシン含有培地で細胞が生存するのに負の影響を及ぼしたとすれば、最初の形質転換を生き残ったすべてのコロニーは、Cyc6Nac2が脱抑制されていなければ分裂しなかっただろう単一の細胞から派生したと考えられる。微量の銅が混入しただけでもCyc6の転写が抑制されたことからも明らかなように、培地への混入は避けられないと考えられる。事実、cy6Nac2.49での銅を介した抑制のキネティクスを検討するために計画した実験では、銅枯渇cy6Nac2.49細胞を銅を含まない培地に再懸濁すると、細胞分裂が1、2回行われる間に、真のCyc6遺伝子座の転写と共にCyc5Nac2の転写が一時的に抑制された。
【0119】
それでもなお、Ind_aad_117遺伝子導入株は、Chlamydomonasにおいて核にコードされるNac2蛋白質に基づいた誘導性葉緑体遺伝子発現システムの設計が可能であることを示す。psbD 5' UTRで目的とする遺伝子の発現を制御できるならば、Ind41_18遺伝子導入株を使って、葉緑体ゲノムに導入されたpsbD 5'UTR制御下にあるキメラ遺伝子の発現を誘導することができる。
【0120】
表1の分析では、誘導性および抑制性条件下にInd41_aadA-117のアミノグリコシドアデニルトランスフェラーゼ活性を測定した。野生型-aadA、Ind41_18およびInd41_aadA-117株からの抽出物について、aadA活性および総蛋白質含量を測定した。活性は蛋白質1μgあたりに取り込まれた放射能cpmを示す。個別の測定の数をカッコ内に示す。
【0121】
〔表1〕
【0122】
<VP28_FLAG、IBVD_FLAG、およびDILP_FLAGの誘導性発現>
本明細書に記載した誘導性葉緑体遺伝子発現システムが外来性蛋白質の産生に応用できるか調べるため、3つの異なる外来性蛋白質VP28、DILP、およびIBDVを用いて5'psbDの制御下に導入遺伝子の異種発現を行った。IBDV(またはVP2)は家禽での伝染性ファブリキウス嚢病ウイルス(IBDV)対策用ワクチンの作成に使用されている(Mundt 1995)。ブラックタイガー(Penaeus monodon)のエビホワイトスポット病ウイルスの主要構造エンベロープ蛋白質の23kD断片VP28は、サブユニットワクチンとして飼料添加するとエビを保護することが示された(Witteveldt 2004)。
【0123】
C. reinhardtiiの葉緑体ゲノム機構のコドンバイアスに最適化してFLAGタグをつけた3種類の外来性遺伝子VP28、DILPおよびIBDVは、Stefan Surzycki博士(インディアナ大学ブルーミントン校)から提供を受け、psbD 5'UTRの制御下にrbcL 3'UTRをターミネーターとして外来蛋白質が発現するように個別に葉緑体形質転換・発現ベクターpcg12にサブクローニングした(図24A)。これらの新規コンストラクトは選択マーカーを持たないため、形質転換体を作出するには選択マーカーをもつ別の葉緑体挿入ベクターと同時形質転換する必要があった(図24A)。
【0124】
ショウジョウバエのインシュリン様ペプチドの発現を誘導する実験で使用したpcg12_DILPベクターの概要図を図24Aに示す。黒塗り部分はDILPコード配列を示す。矢印はpsbD遺伝子の5'リーダーを示す。星印は3HA-11エピトープの挿入を示す。図24Bでは、Ind_VP28(左上)、Ind_IBVD(右上)、Ind_DILP(下)からの総蛋白質抽出物を使った免疫ブロットを、FLAGエピトープを認識する抗体でプローブしたものを示す。蛋白質の予測分子量は、VP28が23kD、IBDVが49kD、DILPが12kDであり、星印で示してある。
【0125】
これらのベクターと、ycf1遺伝子および隣接する葉緑体配列およびスペクチノマイシン耐性を付与するaadAカセットを有するpY1_INTベクターを、Ind41_18株に同時形質転換した。スペクチノマイシン添加TAP寒天培地上で選択した後、外来性遺伝子を増幅するオリゴヌクレオチドを使ったPCRにより、形質転換体に外来性遺伝子が存在するかどうか調べた。導入遺伝子の挿入についてPCRで試験した10コロニーのうち、7つがVP28およびDILPを保有しており、5つがIBDV遺伝子を保有していた。これらの細胞株はIndVP28_x、IndDILP_xおよびIndIBVD_xと命名され、nac2.Cyc6pro Nac2::5'petA-psbD::5'psbD-VP28/DILP/IBVDの遺伝子型を有していた。
【0126】
葉緑体ゲノム内に外来性遺伝子の挿入が確認されたコロニーは、銅枯渇による遺伝子誘導後にFlag(登録商標)抗体を用いた免疫ブロット解析で蛋白質産生を調べた。この方法で調べた22の形質転換株のうち、8株中8株でVP28蛋白質が蓄積していると見られ、IndVP28からの抽出物中に野生型や非誘導コントロールには見られない23kD蛋白質が誘導されていた(図24B、左上)。分析したIndDILP株6株のうち4株で、発現誘導した場合の総蛋白質抽出物で>25kD蛋白質が蓄積した(図24B、下)。この方法で分析したIndIBVD株7株のうち3株で50kD蛋白質が蓄積した(図24B、右上)。
【0127】
図25は、一次スクリーニング実験でDILP_FLAGの発現誘導が見られたIndDILP株からの総蛋白質抽出物の免疫ブロットを示す。IndDILP株を、50 μg/mlスペクチノマイシン含有TAP(ui)またはTAP-Cu+2(i)液体培地で培養し、15%SDS-PAGEゲルで大きさによる分画を行った。得られた免疫ブロットは次にFLAGエピトープを認識する抗体とともにインキュベートした。実験ではFLAGタグAlb3.1蛋白質を発現する遺伝子導入株からの総蛋白質を、FLAGエピトープ検出の陽性コントロールとして用いた。星印は、pcg12_DILPベクターに挿入されたDILP_FLAGペプチドの予想サイズを示す。
【0128】
DILP蛋白質を発現する3つのIndDILP株から抽出した蛋白質はさらに、Cyc6転写を抑制した状態と誘導した状態の細胞から抽出した蛋白質を比較した。調べた4つのIndDILP株のうち2つはDILP_FLAGを12kD蛋白質として正しく誘導していると見られた。したがって、本発明で確立した葉緑体誘導性発現システムはDILPの発現を誘導した。Chlamydomonas reinhardtiiの葉緑体中で構成的にDILP蛋白質を発現する今までの試みでは高レベル発現が得らていなかったことから、この結果は特に興味深い(Stefan Surzycki、私信)。よって、この誘導性葉緑体遺伝子発現システムは、外来性蛋白質、特に構成的発現が得られにくい蛋白質の誘導性発現のための商業的に重要なツールとなりうる。
【0129】
〔実施例6 水素産生に影響を及ぼす遺伝子の発現〕
<細胞株および培地>
これまでにnac2-26変異株が報告されている(Kuchka, et al. EMBO J. 7, 319-324; Nickelsen, et al. EMBO J. 13, 3182-3191)。cyc6Nac2.49株は、Nac2 midi遺伝子と融合したCyc6プロモーターを含む導入遺伝子がnac2-26変異体の核ゲノムに挿入されている(Δnac2::cy6proNac2)。Ind41はcy6Nac2.49のpsbDプロモーターおよび5'UTRをpetAのプロモーターおよび5'UTRを含む675塩基対断片で置換したものである(Δnac2::cy6pro Nac2::5'petA-psbD)。Ind41_18はInd41株と関連しているが、Ind41_18ではaadAカセットが葉緑体DNAから完全に削除されているためスペクチノマイシンに感受性である(Δnac2::cy6proNac2::5'petA-psbD[SpcS])。Ind_aadA_117はInd41_18から得られたもので、atpB遺伝子の下流に挿入されたpsbDプロモーターおよび5'UTRの制御下にaadAカセットを含む(Δnac2::cy6proNac2::5'petA-psbD::5'psbD-aadA)。細胞株はすべて、1.5%バクトアガー含有TAP(トリス-酢酸-リン酸)培地上で弱光下、25℃で維持した。銅含有または銅非含有(-Cu+2)固形寒天および液体TAPおよびHSM培地を使用した実験では、QuinnおよびMerchant(Methods in Enzymol. 297, 263-279)の方法に従い培地を調製した。TAPおよびTAP-Cu+2培地は、必要に応じて100μg/mLスペクチノマイシン(シグマ-アルドリッチ)または20μg/mLパラモマイシン(シグマ-アルドリッチ)を添加した。酸素を欠乏させた細胞を使う実験では、液体培養に窒素ガスを通気しながら150rpm/minで撹拌し連続光照射(20 μE/m-1s-2)を行った。細胞濃度は血球計数板を使って計測した。
【0130】
<プラスミドの構築>
標準的な方法を使ってすべてのプラスミドコンストラクトの操作および分析を行った。コンストラクトの配列決定はBigDyeターミネーター配列決定キット(Applied Biosystems、ラホーヤ、CA)およびABI Prism 377自動配列解析装置(ABI)を使用して行った。クローニングに使用した細菌宿主は大腸菌DH10B(Amersham Biosciences)である。すべてのオリゴヌクレオチドの作成はMicrosynth GmbH(バルガッハ、CH)に発注した。
【0131】
<Chlamydomonas細胞の形質転換>
Chlamydomonas reinhardtii nac2-26株の核形質転換は、基本的にShimogawaraら(Genetics 148, 1821-8)のエレクトロポレーション法により行った。
【0132】
<クロロフィルおよび酸素発生速度の測定>
酸素発生および呼吸速度は、X型光源に接続したクラーク型酸素電極を使用して25℃で測定した(Hansatech Instruments Ltd.、ノーフォーク、英国)。
【0133】
<水素測定および算出>
N2、O2、H2およびCO2の液相および気相測定は以下のとおり行った。溶存ガスの連続モニタリングは、密閉できる温度自動調節式のクラーク型容器を使用し、連続撹拌下、光ファイバー照明器(モデルKL 1500、Schott、マインツ、ドイツ)による連続照射にて、ガスをポリプロピレン膜を通し質量分析器(モデルMM880、VG Instruments、チェシア、英国)のイオン源に供給して測定した。Cy6Nac2導入遺伝子の転写を抑制する実験では、容器内の培地に12μMの銅を添加した(TAP-Cu+2からTAP)。すべての気相計測点の前に、空気および純粋水素ガス試料を直接イオン源に注入して質量分析器の較正を行った。
【0134】
<aadA活性の測定>
aadA活性の測定は野生型、Ind41_18、およびInd_aadA_117株について行い、基本的にはGoldschmidt-Clermont(Nucleic Acids Res 19, 4083-9)の方法に従ったが、元の実験で用いられた放射性標識化rATPの代わりに32P-標識化dATPを使用した。
【0135】
<プラスミド構築‐pRS1_rcy_aadAの構築>
プラスミドpRS1_rcy_aadAの構築にはプラスミドpKS-108#14を使用した。pKS-108#14プラスミドは、C. reinhardtii葉緑体DNA EcoRI R3断片を含み、aadAカセットはpsbD ATG開始コドンから-263塩基対の位置に挿入されている。psbD 5'UTRをpetA 5'UTRで置換するために、オリゴヌクレオチドRS1、RS2、RS3およびRS4を用いたオーバーラップ伸長PCRにより、psbDコード配列に融合したpetA 5'UTRを含む943塩基対キメラDNA断片を作成した。得られたPCR産物をPvuII/ClaI制限エンドヌクレアーゼで制限分解し、同じ酵素で制限分解したプラスミドpKS-108#14に連結してpRS1プラスミドを作成した。葉緑体誘導性発現システムの設計は、2つの連続した葉緑体形質転換を伴い、それには2つの異なる選択マーカーが必要となる。もしくはaadAの再利用が可能であり、、483bp直接反復配列を両末端に隣接させた改変型aadAカセットを用いて、選択圧を取り除いた後に相同組換えによりカセットが効率よく除去されるようにする。再利用可能なaadAカセットをpRS1にクローニングするため、まずpRS1をClaIとSphIで切断し、得られた6.3kbプラスミドの5'および3'末端をT4 DNAポリメラーゼで平滑化した。この結果、pRS1プラスミドからatpAプロモーターおよびaadAコード配列に融合した5'UTRが切り出されるが、rbcLの3'配列は除かれなかった。再利用可能なカセットをpRS1ΔaadAに挿入するため、まず制限エンドヌクレアーゼを使ってpKS-483-aadA-483プラスミドから再利用可能なaadAカセットを切り出し、両端をT4 DNAポリメラーゼとPNKキナーゼで平滑化した。2.8kbの再利用可能なaadAカセットをpRS1ΔaadAに平滑末端連結し、pRS1_rcy_aadAを作出した(図27)。
【0136】
<pcy6Nac2(paroR)の構築>
Nac2コード配列にインフレーム融合した428bpのCyc6プロモーター配列を含むプラスミドの構築には、Nac2の5.1kbキメラmidi遺伝子を用いた。手短に言うと、プラスミドpKS(-)nac2(midi)は、3HA、6Hisおよび9Mycエピトープでタグした3' cDNA配列を含む1.96kbのSfrI/XhoI断片を、Nac2コード配列とインフレームになるよう終止コドンのすぐ上流に導入したNac2コード配列内のSfrI制限部位で終わる3.0kbの5' Nac2ゲノム配列を含む。Nac2遺伝子をCyc6プロモーターの制御下に置くため、Cyc6プロモーターエレメントおよびNac2ゲノムDNAに特異的な4つのオリゴヌクレオチドを用いたオーバーラップ伸長PCRにより、Nac2コード配列に融合させたCyc6プロモーターを含むキメラDNA断片を作出した。得られたPCR断片は、833bpゲノムNac2断片に融合した428bpのCyc6プロモーター断片からなる。このPCR断片は次に、pKS(-)Nac2(midi)のXbaIおよびAatIIユニーク制限部位を用いてpNac2(midi)プラスミドにクローニングした。最後に、pSL17のユニーク制限部位EcoRIおよびXbaIを使って、5.8kb Cyc6Nac2導入遺伝子をpSL17プラスミドにクローニングした。このプラスミドはパロマイシン耐性を付与するaphVIIカセットを含む。得られた10.8kbプラスミド、pcy2(paroR)は、nac2-26突然変異細胞の形質転換に用いた。
【0137】
<Chlamydomonas細胞の形質転換>
nac2-26細胞はTAP培地で培養し、対数増殖期中期(2-4 x 106 cells/mL)で回収し、配偶子オートリシンで処理し、40 mMショ糖添加TAP培地に懸濁した。各エレクトロポレーションでは、処理細胞108個を2.5 μgの直線化したpcyc6Nac2(paroR)またはpSL17プラスミド(エレクトロポレーション効率の対照)、サケ精子DNA 50 μgとインキュベートし、0.2mLエレクトロポレーションキュベット(バイオラッド、米国)に入れてバイオラッド(SIC)を使用し0.75kV、25μF、抵抗ゼロの設定で形質転換した(バイオラッド、米国)。処理した細胞は1mLの新鮮TAP、40mMショ糖、0.4%PEG-8000、20%デンプン培地中で10分間回復させ、抗生物質パロモマイシン(20μg/mL)含有TAP培地にのせた。パロモマイシン耐性コロニーは、銅非含有最少培地(HSM-Cu+2)上で強光下、25℃で光独立栄養的な増殖能についてスクリーニングした。光独立栄養株は次に、最小培地(HSM)上での増殖の有無を調べた。
【0138】
ヘリウム加圧式微粒子銃を用いて、Chlamydomonas葉緑体の遺伝子銃による形質転換を行った。TAP培養したcy6Nac2.49細胞108個を100μg/mLスペクチノマイシン含有TAP寒天培地(TAP + Spc100)にのせ、1μgのpRS1_rcy_aadAプラスミドDNAをコートしたタングステン粒子を射入した。弱光下(5 μEm-2s-1)で2週間後、単一コロニーを拾ってTAP + Spec 100培地で4回クローン化し、弱光下(5 μEm-2s-1)25℃で培養し、細胞株が選択マーカーについてホモプラスミックであることを確実にした。光独立栄養的な増殖を調べるため、細胞を固形HSM培地にのせ、中光下(45 μEm-2s-1)25℃で培養した。Ind41_18の形質転換の場合は、TAP-Cu+2で培養した細胞108個を100μg/mLスペクチノマイシン含有TAP-Cu+2寒天培地(TAP-Cu+2 + Spc100)にのせ、1μgのpcg12プラスミドDNAをコートしたタングステン粒子を射入した。弱光下(5 μEm-2s-1)で2週間後、単一コロニーを拾ってTAP-Cu+2 + Spec 100培地で3回クローン化し、弱光下(5 μEm-2s-1)25℃で培養した。
【0139】
<増殖解析>
野生型、nac2-26、cy6Nac2.49株の増殖解析では、細胞をTAP-Cu+2培地にて濃度2-4 x 106 細胞/mLまで培養し、ついで濃度1 x 106 細胞/mLに希釈し、10倍段階希釈を繰り返して最終的に1プレートあたり100個の細胞が含まれるようにした。10μLの希釈液を適切な固形寒天培地にのせ、強光下に25℃で10日間培養した。Ind41およびInd41-18株の場合は、適切な培地に細胞103個ずつのせて連続光(100 μEm-2s-1)下に25℃で10日間培養した。誘導性aadA遺伝子導入株の増殖解析では以下のような実験を行った。手短に言うと、Ind41-18およびInd_aadA遺伝子導入株をTAP-Cu+2またはTAP液体培地で培養し、つぎに新鮮なTAP-Cu+2またはTAP液体培地に3回移植し直した。TAPおよびTAP-Cu+2での培養液を段階希釈し、0-1000 μg/mLの範囲で段階的に濃度を変えたスペクチノマイシン含有TAPまたはTAP-Cu+2固形寒天培地にのせ、連続光(100 μEm-1s-2)下に25℃で10日間培養した。
【0140】
<蛍光遷移>
蛍光遷移解析を行った。TAP寒天培地で暗所にて培養した細胞を、5分間暗順応させてから植物効率分析計(Plant Efficiency Analyzer、PEA、Hansatech Instruments、英国)で測定した。
【0141】
<RNA解析>
野生型、nac2-26、cy6Nac2.49、Ind41_18およびInd_aadA_117株からの全RNA単離は、RNA植物ミニRNA抽出キット(キアゲン Ghmb、ドイツ)を用いて使用説明書に従い行った。経時変化を調べるためのRNA試料の場合、細胞108個を3000gで遠心分離して、RNAeasy RNAプロテクション溶液(アンビオン、米国)を用いて使用説明書に従い処理した。
【0142】
RNAブロット解析を行った。RNA(2μg)を1.2%アガロース‐4%ホルムアルデヒドゲルにて1X MOPS緩衝液中で電気泳動し、つぎに20X SSC緩衝溶液中でHybond N+ナイロン膜(アマシャム、米国)に転写し、ストラタリンカーUVクロスリンカーを用いてRNAを膜に固定した。膜のプリハイブリダイゼーションおよびハイブリダイゼーションは変法チャーチハイブリダイゼーション溶液(0.5Mリン酸緩衝液pH7.2、7w/v%SDS、10mM EDTA)中65℃で行った。プラスミドpks-108#14をAccIおよびStyIで制限分解してpsbDの380bpDNA断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。Cyc6 cDNAの685bpDNA断片をランダムプライミング法で[α32P]dCTP標識した。pcg12プラスミドを制限分解してaadAコード配列の804bp NcoI/SphI断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。pcg12をEcoRVおよびHpaIIで制限分解してatpBの693bp断片を単離し、これをランダムプライミング法で[α32P]dATP標識してプローブに使用した。ハイブリダイゼーション後に、膜を高ストリンジェント洗浄緩衝液[0.1% SDS、0.1% SSC]で10分間洗浄した。
【0143】
<蛋白質解析>
Chlamydomonasの野生型、nac2-26、cy6Nac2.49、Ind41_18およびInd_aadA_117株からの総蛋白質抽出物は、1.5mLエッペンドルフチューブに細胞3 x 106個を採取し、ペレットをシグマプロテアーゼ阻害剤カクテル(シグマ-アルドリッチ、米国)2倍溶液に懸濁し、同量の細胞溶解緩衝液(100 mM Tris-HCl pH 6.8、4% SDS)を添加して37℃で30分間置き溶解させた。細胞片を沈降させるため、試料を10,0000gで5分間遠心分離し、上清を総蛋白質抽出物として使用した。蛋白質濃度の測定は、5μLの上清をブラッドフォード法(バイオラッド蛋白質アッセイ、バイオラッド、米国)で分析した。
【0144】
免疫ブロット解析では、20μgの総蛋白質を12%SDSポリアクリルアミドゲルで泳動分離し、Protran 0.45μmニトロセルロース膜(Schleicher and Schuell)に転写した。Nac2抗体を使用する際は、80μgの蛋白質を8%ポリアクリルアミドゲルで泳動分離した。トリス緩衝生理食塩水(5%脱脂粉乳、0.1%Tween-20含有)(TBS-T)中で膜をブロッキングした。一次抗体反応では、以下のようにTBS-Tに抗体を希釈した。D2抗体、1:10,000希釈;D2抗体1:10,000希釈;Nac2抗体、1:10,000希釈;PsaA抗体1:10,000希釈;AtpB抗体、1:10,000希釈;RUBISCO-ホロ酵素抗体、1:50,000希釈。インキュベートは室温で1時間行った。つづいて、膜を1%脱脂粉乳含有TBS-Tで5分間洗浄し、これを5回繰り返した。二次抗体反応では、10,000倍希釈したペルオキシダーゼ結合抗ウサギIgG(1%脱脂粉乳含有TBS)と膜を室温で1時間インキュベートした。膜をTBSで5分間洗浄することを5回繰り返し、増強化学発光法でシグナルを検出した。
【0145】
<銅を介した抑制および経時変化実験>
cy6Nac2.49遺伝子導入株での銅を介したCy6Nac2抑制の経時変化を調べるため、cy6Nac2.49細胞をTAP-Cu+2培地で濃度4 x 106 細胞/mLまで培養し、新鮮TAP-Cu+2培地で濃度5 x 105 細胞/mLに希釈し、ついで2つの独立した培養液に分けて、一方はそのまま、他方には銅を最終濃度6 μMとなるよう添加した。それぞれ8時間ごと40時間後まで経時的に分析した。FV/FM測定には2つの独立した試料を使用し、各培養液での平均を各時点で算出した。
【0146】
銅を介したcy6Nac2.49の誘導を調べる実験は、銅を介した抑制実験と同様に行ったが、ただしcy6Nac2.49をTAP培地で培養し、細胞を銅非含有培地で2回洗浄してからTAP-Cu+2培地で濃度5 x 105 細胞/mLに希釈した。経時変化実験を開始する際は、細胞を2つの独立した培養液に分けて、一方に銅を最終濃度6 μMとなるよう添加した。
【0147】
<水素測定>
硫黄枯渇下にcy6Nac2.49株での水素産生を野生型と比較した。図32で使用したcy6Nac2.49培養液では、1サイクルの間に20 μmol/Lの水素が産生され、これは最大速度1 mmol H2 mol-1 Chl s-1に相当した。水素産生速度は実験ごとに異なり、3.1 mmol H2 mol-1 Chl s-1に達する場合もあった。光合成の許される条件下(Cu非含有培地)では、Cy6Nac2.49細胞での総酸素発生速度は23 mmol O2 mol-1 Chl s-1であった。これは野生型細胞の1.5〜2倍であった。よって、水素産生の最大速度は総酸素産生の4から13%の範囲だった。100時間硫黄枯渇状態においた培養液の場合、平均値は4 mmol H2 mol-1 Chl s-1と推定される。cy6Nac2.49細胞で1サイクルの間の水素産生20 μmol H2/Lと、野生型で100時間硫黄枯渇状態の水素産生4 mmol H2/Lとを比べた場合、同等の水素産生を達成するためにはcy6Nac2.49システムには、1サイクルでの効率向上または複数サイクルの連続など、さらに改良が必要である。原理的にはChlamydomonasに水素産生を向上させるような遺伝的改変を加えることが可能である。例えば、循環的電子伝達を行えないよう状態1でブロックされた状態遷移変異体を用いる、またはカルビン-ベンソン酵素の1つをCyc6プロモーターの制御下に置くなどである。これにより、PSII活性と同時に炭素同化も現象し、水素産生段階での電子の競合が低下する。
【0148】
〔表2 オリゴヌクレオチド一覧〕
【0149】
Chlamydomonasでは天然の誘導性葉緑体遺伝子発現システムがない。そうしたシステムを、核にコードされる葉緑体Nac2蛋白質の特性を利用して開発した。この蛋白質はPSIIのD2反応中心ポリペプチドをコードするpsbD mRNAのプロセシングおよび安定蓄積に必要である。Nac2のターゲット部位は74塩基psbD 5'UTR配列内に含まれる。この5'UTRを別のコード配列に融合することでその遺伝子の発現はNac2依存性になる。Nac2コード配列を、チトクロームC6遺伝子のCyc6プロモーターに融合した。C6遺伝子の発現は銅枯渇、嫌気的増殖、そしてニッケル添加によって誘導されるが、銅が充分に存在する条件下では抑制される。Nac2はpsbD 5'UTRに特異的なので、どんな葉緑体遺伝子でもコード配列をpsbD 5'UTRに融合させれば、原則的にはこのシステムで誘導性発現ができる。
【0150】
上述したとおり、パロモマイシン耐性を付与するaphVIII遺伝子を含むプラスミドに、Cyc6-Nac2コンストラクトを挿入した(図28A)。このプラスミドを使ってChlamydomonasnac2-26変異体の形質転換し、パロモマイシン耐性で選択した。調べた55の形質転換株のうち2つが、銅によるNac2発現の適切な制御を示した。これらの形質転換株の1つであるcy6Nac2.49、野生型、そしてnac2-26変異体の増殖特性は上述している(図28B)。予想したとおり、いずれも銅の有無にかかわらずTAP培地上で増殖し、形質転換株は選択マーカーaphVIIIを含むためパロモマイシンの存在下でも増殖する。野生型細胞だけが銅を含む最小培地で増殖する。しかし、cy6Nac2.49株は銅を含まない最小培地で増殖するようになった。培地にニッケルを添加しても増殖するが、これはCyc6プロモーターがニッケルで誘導されるからである。異なる増殖条件におけるpsbD発現レベルをRNAブロットハイブリダイゼーションで調べた(図28C)。予想どおり、nac2-26変異株ではpsbD RNAが検出されない。対するcy6Nac2.49株では、Cyc6の発現につづきpsbDの発現が見られ、銅非存在下または嫌気的条件下に誘導される(図28C)。psbD産物であるD2のレベルをD2抗血清を使った免疫ブロットで調べた(図28D)。すべての条件下にTAP寒天培地で増殖したnac2-26細胞でD2蛋白質は検出されない。しかしcy6Nac2.49を銅非存在下または嫌気的条件下に増殖した場合、D2が野生型の20%まで蓄積する。最小培地ではD2の誘導はわずかに低い。予想どおり、CP47の他のPSII蛋白質はD2と同様のパターンを示し、これはD2蛋白質がないとこれらの蛋白質が不安定なためである。これに対しnac2-26ではRubisco蛋白質(RbcL)のレベルは影響を受けない(図28D)。
【0151】
Nac2合成が止まってから細胞内のPSIIが消失するまでの時間を調べるため、cy6Nac2.49細胞をまず銅非含有TAP培地で培養した。この条件ではPSIIは合成され蓄積する。培養液を半分に分け、一方は銅非存在化のまま、他方は銅を添加して培養した。細胞濃度およびPSII量子収量の推定となるFV/FM比(可変蛍光/最大蛍光)の経時変化を調べた(図29A)。銅存在下では32時間以内にFV/FM比が最小値まで低下した。この期間内に、いずれの培養条件下でも細胞は3〜4倍に分裂し定常期に達した。各種時点でRNAおよび蛋白質解析のための細胞抽出物を調製した。Cyc6およびpsbD RNAのレベルは銅添加8時間後に著しく低下し、その後は検出できなかった。他の葉緑体RNA(atpB、rRNA)はいずれの条件でも安定だった。免疫ブロットの結果、銅添加後にmRNAの減少から少し遅れてD2の量が低下し(図29C)、他のPSIIコア蛋白質D1も減少した。期待どおりNac2の減少も認められた。これに対して葉緑体蛋白質PSI(PsaA)およびRubiscoは安定だった(図29C)。
【0152】
逆の実験では、銅存在下に培養した細胞を銅非含有TAP培地に移し、細胞濃度およびFV/FM比の経時変化を調べた。FV/FM比は25時間遅れて増加し始めたが、これはおそらく細胞内の銅が枯渇するまでに時間がかかるためである(図30A)。異なる時点で採取した細胞抽出物からのRNAおよび蛋白質を、RNAブロット解析および蛋白質免疫ブロットにより調べた(図30B、C)。Cyc6 RNAは16時間後に検出されたものの、psbD RNA蓄積およびPSII活性の検出には遅れが見られ、これはおそらくpsbD mRNA蓄積およびD2合成にはある程度のNac2レベルが必要であるためと思われる。
【0153】
<PSIIに関連しない葉緑体遺伝子の誘導性発現>
Nac2システムを使ってPSIIを可逆的に消失させることが可能だが、他の葉緑体遺伝子すべてに応用することができるか調べた。Nac2蛋白質はpsbD 5'UTRに特異的に働きキメラpsbD 5'UTRレポーター遺伝子を制御することができることから、これは原理的には可能である。よってpsbDプロモーターと5'UTRを対象となる遺伝子に融合するだけでよいはずである。しかし、psbD RNAの欠失をもたらすnac2-26変異のため、こうした条件下ではPSIIは蓄積しない。この問題を回避するため、petAプロモーターと5' UTRをpsbDコード配列に融合し、このコンストラクトを修飾型p108-14葉緑体形質転換ベクターに挿入した。このベクターでは、petAプロモーターと5'UTRの制御下にあるpsbD遺伝子の上流に再利用可能なaadAカセットが挿入されている。このDNAは、選択マーカーとしてaadAカセットを用いた遺伝子銃形質転換により葉緑体ゲノムに挿入した。このように内在性psbD遺伝子がpetA-psbDコンストラクトで置換されたため、その転写はもはやNac2に依存しなくなった。形質転換株をスペクチノマイシン含有寒天培地に3回移し換え、DNAブロットおよびPCR解析によりホモプラスミックかどうかを調べ、形質転換株のうちの1つInd41を選択した。使用したaadAカセットの両側は2つの反復配列に隣接していた。このカセットが切り出されるように、ホモプラスミックな形質転換株Ind41をスペクチノマイシンを含まない培地上で繰りかえし移し換えた。このようにして得たInd41_18は、aadAカセットを含まないためスペクチノマイシンに感受性である。Ind41、Ind41_18およびcy6Nac2.49について、異なる培地上での増殖特性を調べた。予想どおり、Ind41_18がスペクチノマイシンに感受性であるのに対し、Ind41はスペクチノマイシン存在下に増殖した。さらに、Ind41とInd41_18の両方が銅の有無にかかわらずHSM最少培地で増殖した。RNAブロット解析の結果、Ind41_18ではCyc6 RNAレベルにかかわらずすべての条件でキメラpetA-psbD RNAが蓄積した(図27C)。psbD 5'UTRよりもpetA 5'UTRの方が大きいので、psbD RNAはより大きい。免疫ブロット解析の結果、試験したすべての条件、特にNac2が発現しない条件でD2およびD1蛋白質が同等レベルに蓄積した(図27D)。
【0154】
次に、cg12ベクターを使った形質転換により、psbDプロモーターおよび5'UTRに融合したaadAカセットを、この株に導入した(図31A)。形質転換株Ind_aadA-117およびInd41_18の増殖を、銅含有および銅非含有の濃度漸増したスペクチノマイシン含有TAP寒天培地で調べた(図31B)。すべての株が銅非存在下でも(図31B)銅存在下でも増殖した。予想どおり、Ind_aadA-117はスペクチノマイシン250 μg/ml以上を含む培地では銅が存在する場合のみ増殖した。スペクチノマイシン100 μg/mlを含む培地でも銅の存在下にわずかな増殖が見られた。RNAブロット解析の結果、Cyc6プロモーターが誘導される条件でのみaadA RNAが蓄積した(図31C)。D2、D1、CP47、およびRbcLの蛋白質レベルはあまり変化しなかったが、Nac2は誘導条件でのみ検出された(図31D)。適正なaadA抗体がないため、この蛋白質の定量はアミノグリコシドアデニルトランスフェラーゼ活性の測定により行った。Nac2が発現する条件では活性が著しく上昇していた(表3)。
【0155】
〔表3 誘導条件下および抑制条件下におけるInd41_aadA-117中のアミノグリコシドアデニルトランスフェラーゼ活性〕
上述のとおり、野生型-aadA、Ind41_18およびInd41_aadA-117株からの抽出物について、aadA活性および総蛋白質含量を測定した。活性は蛋白質1μgあたりに取り込まれた放射能cpmを示す。独立した測定の数をカッコ内に示す。
【0156】
〔誘導性葉緑体遺伝子発現システムを使って水素産生を引き起こすことができる〕
Chlamydomonasは明所で嫌気的条件下にヒドロゲナーゼを誘導し水素を産生することができる。そこで、呼吸により水素産生誘導に適した嫌気的条件になるよう、誘導性Nac2システムを使ってPSII活性と酸素発生を止められるかどうか調べた。cy6Nac2.49細胞を銅を含まないTAP培地で濃度2 x 106 細胞/mLまで培養した。銅を添加し、質量分析器に接続し白色光を照射したチャンバーに細胞を移した。このシステムでは、チャンバーの底はポリプロピレン膜で塞がれ、溶存ガスが膜を通して質量分析器のイオン源に直接拡散する。このようにして酸素と水素の量を一定間隔で測定できる。チャンバーは密閉され銅がPSII合成を抑制しているので、酸素発生は減少した。200分以内に酸素は呼吸により消費された。嫌気的状態に達し、活性型ヒドロゲナーゼの合成と水素産生が見られた(図32A)。水素産生の最大速度は1から3.1 mmol H2 mol-1 Chl sec-1の範囲であり、硫黄枯渇細胞での速度よりもやや遅く、持続時間はずっと短かった(3〜4日間に対して約1.5時間)。
【0157】
Cyc6プロモーターの興味深い特徴として、このプロモーターは銅存在下であっても嫌気的条件で誘導される。したがって、cy6Nac2.49細胞では一度嫌気的条件になればヒドロゲナーゼが誘導され、Nac2合成が再開し、PSIIが合成され、酸素レベルが上昇してヒドロゲナーゼが不活性化されて水素産生が止まる。これが観測された。嫌気的水素産生段階では酸素発生およびヒドロゲナーゼ不活性化と同時にPSII合成が行われ、水素レベルは一定を保った(図32A)。対照として同じ細胞培養液に銅を添加せずに調べた。この条件では、銅処理細胞で見られたように、水素が産生されず(図32B)、一定の酸素産生と二酸化炭素の漸減を伴った。Cyc6プロモーターは銅非含有培地で好気条件下にオフになると予想されるので、新たな水素産生サイクルが期待される。この可能性を検証するため、密閉容器内の銅非含有TAP培地でcy6Nac2.49細胞を50時間培養し、質量分析器で水素および酸素量を測定した。その結果は、2度の連続した水素産生段階と酸素産生段階があったことを示す。
【0158】
本発明は特定の望ましい実施形態に関して詳細に記述してきたが、以下の特許請求の範囲に記載する本発明の範囲及び主旨内で、変更及び修正が可能である。
【特許請求の範囲】
【請求項1】
細胞の色素体内で蛋白質の産生を誘導するための発現システムを作成する方法であって、
誘導性プロモーターをコードする第一の核酸を細胞核に導入するステップと、
第一の核酸を安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、
インデューサーの導入またはリプレッサーの除去が安定化因子の発現を誘導し、
発現した安定化因子が色素体内で、安定化因子で安定化され第三の核酸から転写されるmRNAの非翻訳領域と結合し、
第三の核酸が色素体に内在性または外来性であって、第三の核酸が前記蛋白質をコードし、
前記mRNAの発現が前記蛋白質の産生をもたらすことを特徴とするステップと
を含む方法。
【請求項2】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項1に記載の方法。
【請求項3】
細胞が、第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項2に記載の方法。
【請求項4】
細胞が、第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項1に記載の方法。
【請求項5】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項1に記載の方法。
【請求項6】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項5に記載の方法。
【請求項7】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項1に記載の方法。
【請求項8】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項1に記載の方法。
【請求項9】
インデューサーが化学物質または環境条件であることを特徴とする、請求項1に記載の方法。
【請求項10】
化学物質が銅であり、銅の枯渇がmRNA発現と蛋白質の産生を誘導することを特徴とする、請求項9に記載の方法。
【請求項11】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項9に記載の方法。
【請求項12】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項1に記載の方法。
【請求項13】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項1に記載の方法。
【請求項14】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項1に記載の方法。
【請求項15】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項14に記載の方法。
【請求項16】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項1に記載の方法。
【請求項17】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項1に記載の方法。
【請求項18】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項14に記載の方法。
【請求項19】
細胞の色素体内で色素体蛋白質の発現を抑制するための発現システムを作成する方法であって、
抑制性プロモーターをコードする第一の核酸を細胞核に導入するステップと、
第一の核酸を、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、
リプレッサーの導入またはインデューサーの除去が安定化因子の発現を抑制し、
安定化因子の発現抑制が、安定化因子で安定化され第三の核酸から転写されるmRNAの発現抑制をもたらし、
第三の核酸が色素体に内在性または外来性であり、第三の核酸が前記蛋白質をコードし、
前記蛋白質の発現が抑制されることを特徴とするステップと
を含む方法。
【請求項20】
細胞の色素体内で色素体蛋白質を発現させる方法であって、
細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
安定化因子を発現させるステップと、
安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAを発現させるステップと、
色素体内で前記蛋白質を産生させるステップと
を含む方法。
【請求項21】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項20に記載の方法。
【請求項22】
細胞が第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項20に記載の方法。
【請求項23】
細胞が第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項20に記載の方法。
【請求項24】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項20に記載の方法。
【請求項25】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項24に記載の方法。
【請求項26】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項20に記載の方法。
【請求項27】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項20に記載の方法。
【請求項28】
インデューサーが化学物質または環境条件であることを特徴とする、請求項20に記載の方法。
【請求項29】
化学物質が銅であり、銅の枯渇がmRNA発現と蛋白質の産生を誘導することを特徴とする、請求項28に記載の方法。
【請求項30】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項28に記載の方法。
【請求項31】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項20に記載の方法。
【請求項32】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項31に記載の方法。
【請求項33】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項20に記載の方法。
【請求項34】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項33に記載の方法。
【請求項35】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項20に記載の方法。
【請求項36】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項20に記載の方法。
【請求項37】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項33に記載の方法。
【請求項38】
細胞の色素体内で色素体蛋白質の発現を抑制する方法であって、
細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、
第一の核酸が抑制性プロモーターをコードし、
第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、
第二の核酸が安定化因子をコードすることを特徴とするステップと、
安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAの発現を抑制するステップと、
前記蛋白質の発現を抑制するステップと
を含む方法。
【請求項39】
組換え宿主細胞の色素体内で色素体蛋白質を発現させるシステムであって、
外から加えるか除去される、核蛋白質の発現を誘導するインデューサーと、
核に組換え核酸を含む組換え宿主細胞であって、
組換え核酸が、第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、
第一の核酸が誘導性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、
色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、色素体蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体と
を含むシステム。
【請求項40】
細胞が第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項39に記載のシステム。
【請求項41】
細胞が第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項39に記載のシステム。
【請求項42】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項39に記載のシステム。
【請求項43】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項42に記載のシステム。
【請求項44】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項39に記載のシステム。
【請求項45】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項39に記載のシステム。
【請求項46】
インデューサーが化学物質または環境条件であることを特徴とする、請求項39に記載のシステム。
【請求項47】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項39に記載のシステム。
【請求項48】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項39に記載のシステム。
【請求項49】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項48に記載のシステム。
【請求項50】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項39に記載のシステム。
【請求項51】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内に存在することを特徴とする、請求項39に記載の方法。
【請求項52】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項48に記載のシステム。
【請求項53】
組換え宿主細胞の色素体内で色素体蛋白質の発現を抑制するシステムであって、
外から加えられる、核蛋白質の発現を抑制するリプレッサーと、
核に組換え核酸を含む組換え宿主細胞であって、
組換え核酸が、第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、
第一の核酸が抑制性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする細胞と、
色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、前記蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体と
を含むシステム。
【請求項54】
細胞の色素体内で色素体蛋白質を発現させることにより水素ガスの産生を促進する方法であって、
細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
安定化因子を発現させるステップと、
安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAを発現させるステップと、
色素体内で前記蛋白質を産生させるステップと、
水素ガスを産生するステップと
を含む方法。
【請求項55】
細胞の色素体内で色素体蛋白質の発現を抑制することにより水素ガスの産生を阻害する方法であって、
細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、
第一の核酸が抑制性プロモーターをコードし、
第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、
第二の核酸が安定化因子をコードすることを特徴とするステップと、
安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAの発現を抑制するステップと、
前記蛋白質の発現を抑制するステップと、
水素ガスの産生を阻害するステップと
を含む方法。
【請求項56】
細胞の色素体内で色素体蛋白質の発現を促進および抑制することにより水素ガスの産生を促進する方法であって、
順次的にi)細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理してから、ii)細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
順次的に安定化因子を発現させてから抑制するステップであって、
安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
順次的にmRNAを発現させてから抑制するステップと、
色素体内で前記蛋白質を産生させるステップと、
水素ガスを産生するステップと
を含む方法。
【請求項57】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項56に記載の方法。
【請求項58】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項56に記載の方法。
【請求項59】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項58に記載の方法。
【請求項60】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項56に記載の方法。
【請求項61】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項56に記載の方法。
【請求項62】
インデューサーが化学物質または環境条件であることを特徴とする、請求項56に記載の方法。
【請求項63】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項62に記載の方法。
【請求項64】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項56に記載の方法。
【請求項65】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項56に記載の方法。
【請求項66】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項56に記載の方法。
【請求項1】
細胞の色素体内で蛋白質の産生を誘導するための発現システムを作成する方法であって、
誘導性プロモーターをコードする第一の核酸を細胞核に導入するステップと、
第一の核酸を安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、
インデューサーの導入またはリプレッサーの除去が安定化因子の発現を誘導し、
発現した安定化因子が色素体内で、安定化因子で安定化され第三の核酸から転写されるmRNAの非翻訳領域と結合し、
第三の核酸が色素体に内在性または外来性であって、第三の核酸が前記蛋白質をコードし、
前記mRNAの発現が前記蛋白質の産生をもたらすことを特徴とするステップと
を含む方法。
【請求項2】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項1に記載の方法。
【請求項3】
細胞が、第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項2に記載の方法。
【請求項4】
細胞が、第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項1に記載の方法。
【請求項5】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項1に記載の方法。
【請求項6】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項5に記載の方法。
【請求項7】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項1に記載の方法。
【請求項8】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項1に記載の方法。
【請求項9】
インデューサーが化学物質または環境条件であることを特徴とする、請求項1に記載の方法。
【請求項10】
化学物質が銅であり、銅の枯渇がmRNA発現と蛋白質の産生を誘導することを特徴とする、請求項9に記載の方法。
【請求項11】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項9に記載の方法。
【請求項12】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項1に記載の方法。
【請求項13】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項1に記載の方法。
【請求項14】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項1に記載の方法。
【請求項15】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項14に記載の方法。
【請求項16】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項1に記載の方法。
【請求項17】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項1に記載の方法。
【請求項18】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項14に記載の方法。
【請求項19】
細胞の色素体内で色素体蛋白質の発現を抑制するための発現システムを作成する方法であって、
抑制性プロモーターをコードする第一の核酸を細胞核に導入するステップと、
第一の核酸を、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成するステップであって、
リプレッサーの導入またはインデューサーの除去が安定化因子の発現を抑制し、
安定化因子の発現抑制が、安定化因子で安定化され第三の核酸から転写されるmRNAの発現抑制をもたらし、
第三の核酸が色素体に内在性または外来性であり、第三の核酸が前記蛋白質をコードし、
前記蛋白質の発現が抑制されることを特徴とするステップと
を含む方法。
【請求項20】
細胞の色素体内で色素体蛋白質を発現させる方法であって、
細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
安定化因子を発現させるステップと、
安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAを発現させるステップと、
色素体内で前記蛋白質を産生させるステップと
を含む方法。
【請求項21】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項20に記載の方法。
【請求項22】
細胞が第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項20に記載の方法。
【請求項23】
細胞が第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項20に記載の方法。
【請求項24】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項20に記載の方法。
【請求項25】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項24に記載の方法。
【請求項26】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項20に記載の方法。
【請求項27】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項20に記載の方法。
【請求項28】
インデューサーが化学物質または環境条件であることを特徴とする、請求項20に記載の方法。
【請求項29】
化学物質が銅であり、銅の枯渇がmRNA発現と蛋白質の産生を誘導することを特徴とする、請求項28に記載の方法。
【請求項30】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項28に記載の方法。
【請求項31】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項20に記載の方法。
【請求項32】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項31に記載の方法。
【請求項33】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項20に記載の方法。
【請求項34】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項33に記載の方法。
【請求項35】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項20に記載の方法。
【請求項36】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項20に記載の方法。
【請求項37】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項33に記載の方法。
【請求項38】
細胞の色素体内で色素体蛋白質の発現を抑制する方法であって、
細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、
第一の核酸が抑制性プロモーターをコードし、
第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、
第二の核酸が安定化因子をコードすることを特徴とするステップと、
安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAの発現を抑制するステップと、
前記蛋白質の発現を抑制するステップと
を含む方法。
【請求項39】
組換え宿主細胞の色素体内で色素体蛋白質を発現させるシステムであって、
外から加えるか除去される、核蛋白質の発現を誘導するインデューサーと、
核に組換え核酸を含む組換え宿主細胞であって、
組換え核酸が、第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、
第一の核酸が誘導性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする組換え宿主細胞と、
色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、色素体蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体と
を含むシステム。
【請求項40】
細胞が第二の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項39に記載のシステム。
【請求項41】
細胞が第三の核酸の非作動型コピーを有するかまたはコピー若しくはホモログを欠失することを特徴とする、請求項39に記載のシステム。
【請求項42】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項39に記載のシステム。
【請求項43】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項42に記載のシステム。
【請求項44】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項39に記載のシステム。
【請求項45】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項39に記載のシステム。
【請求項46】
インデューサーが化学物質または環境条件であることを特徴とする、請求項39に記載のシステム。
【請求項47】
蛋白質が水素ガスの産生に関与することを特徴とする、請求項39に記載のシステム。
【請求項48】
蛋白質が、医薬品、工業用酵素、葉緑体の成熟または分解に関与する酵素、および栄養補助食品から選択されることを特徴とする、請求項39に記載のシステム。
【請求項49】
医薬品が、抗体、ワクチン、および抗菌剤から選択されることを特徴とする、請求項48に記載のシステム。
【請求項50】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項39に記載のシステム。
【請求項51】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内に存在することを特徴とする、請求項39に記載の方法。
【請求項52】
蛋白質が、宿主細胞のための防御産物、アミノ酸合成に関与する酵素、およびプロテアーゼから選択されることを特徴とする、請求項48に記載のシステム。
【請求項53】
組換え宿主細胞の色素体内で色素体蛋白質の発現を抑制するシステムであって、
外から加えられる、核蛋白質の発現を抑制するリプレッサーと、
核に組換え核酸を含む組換え宿主細胞であって、
組換え核酸が、第二の核酸と作動的に連結して組換え核酸を形成する第一の核酸を含み、
第一の核酸が抑制性プロモーターをコードし、第二の核酸が安定化因子をコードすることを特徴とする細胞と、
色素体に内在性または外来性である第三の核酸を含む色素体であって、第三の核酸が発現される色素体蛋白質をコードし、前記蛋白質をコードするmRNAが安定化因子で制御されることを特徴とする色素体と
を含むシステム。
【請求項54】
細胞の色素体内で色素体蛋白質を発現させることにより水素ガスの産生を促進する方法であって、
細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
安定化因子を発現させるステップと、
安定化因子を色素体に導入するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAを発現させるステップと、
色素体内で前記蛋白質を産生させるステップと、
水素ガスを産生するステップと
を含む方法。
【請求項55】
細胞の色素体内で色素体蛋白質の発現を抑制することにより水素ガスの産生を阻害する方法であって、
細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
リプレッサーまたはインデューサーが細胞核内の第一の核酸と結合し、
第一の核酸が抑制性プロモーターをコードし、
第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成し、
第二の核酸が安定化因子をコードすることを特徴とするステップと、
安定化因子の発現を抑制するステップであって、安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
mRNAの発現を抑制するステップと、
前記蛋白質の発現を抑制するステップと、
水素ガスの産生を阻害するステップと
を含む方法。
【請求項56】
細胞の色素体内で色素体蛋白質の発現を促進および抑制することにより水素ガスの産生を促進する方法であって、
順次的にi)細胞をインデューサーと接触させるか、またはリプレッサーの除去をもたらす条件で細胞を処理してから、ii)細胞をリプレッサーと接触させるか、またはインデューサーの除去をもたらす条件で細胞を処理するステップであって、
インデューサーまたはリプレッサーが核内の第一の核酸と結合し、
第一の核酸が誘導性プロモーターをコードし、
第一の核酸が、安定化因子をコードする第二の核酸と作動的に連結して組換え核酸を形成することを特徴とするステップと、
順次的に安定化因子を発現させてから抑制するステップであって、
安定化因子が色素体内でmRNAの非翻訳領域と結合してmRNAを安定化し、mRNAが色素体に内在性または外来性である第三の核酸から転写され、第三の核酸が前記蛋白質をコードすることを特徴とするステップと、
順次的にmRNAを発現させてから抑制するステップと、
色素体内で前記蛋白質を産生させるステップと、
水素ガスを産生するステップと
を含む方法。
【請求項57】
組換え核酸を核に導入する前に、第一の核酸が第二の核酸と作動的に連結して組換え核酸を形成することを特徴とする、請求項56に記載の方法。
【請求項58】
細胞が植物細胞または藻類細胞であることを特徴とする、請求項56に記載の方法。
【請求項59】
色素体が、葉緑体、白色体、アミロプラスト、エチオプラスト、エライオプラスト、および有色体から選択されることを特徴とする、請求項58に記載の方法。
【請求項60】
誘導性プロモーターがCyc6プロモーターと少なくとも90%の配列類似性を有することを特徴とする、請求項56に記載の方法。
【請求項61】
第三の核酸がpsbD遺伝子と少なくとも90%の配列類似性を有する遺伝子をコードすることを特徴とする、請求項56に記載の方法。
【請求項62】
インデューサーが化学物質または環境条件であることを特徴とする、請求項56に記載の方法。
【請求項63】
環境条件が所定レベルまでの酸素濃度の低下であることを特徴とする、請求項62に記載の方法。
【請求項64】
インデューサーはその添加と除去を一サイクルとして、複数サイクルの添加と除去を行うことを特徴とする、請求項56に記載の方法。
【請求項65】
安定化因子がNac2およびMbb1から選択されることを特徴とする、請求項56に記載の方法。
【請求項66】
第三の核酸に作動的に連結しているかまたは第三の核酸に連結していない、追加の核酸が色素体内で発現されることを特徴とする、請求項56に記載の方法。
【図1】

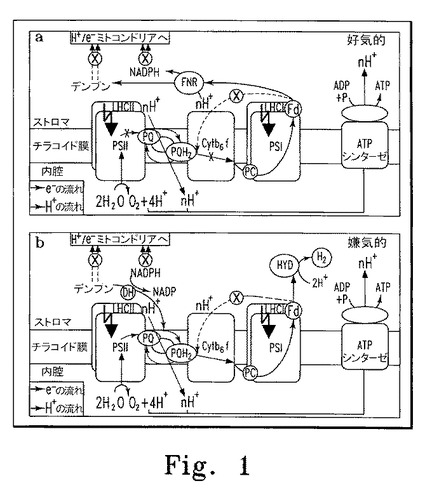
【図2】
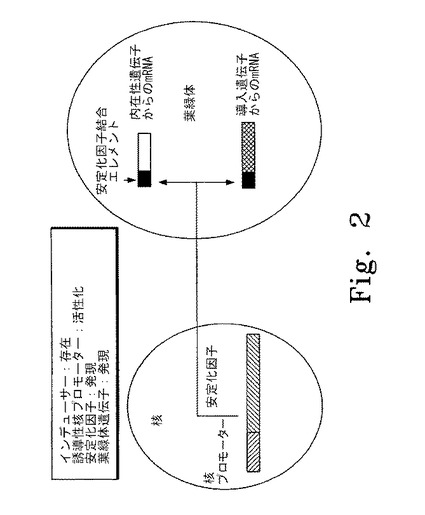
【図3】
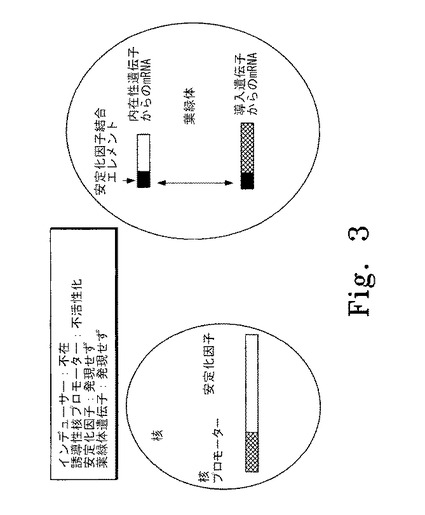
【図4】
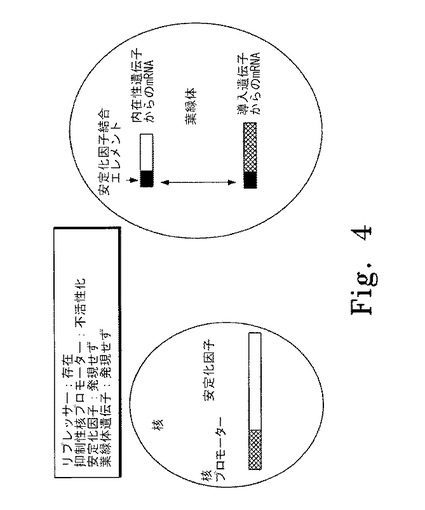
【図5】
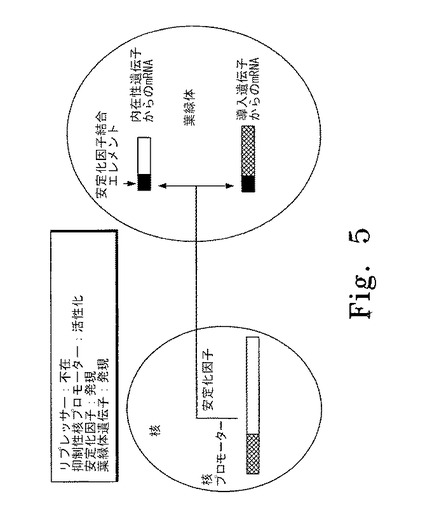
【図6】
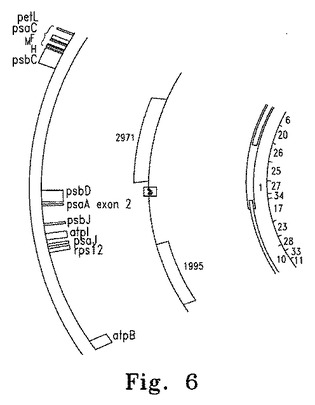
【図7】
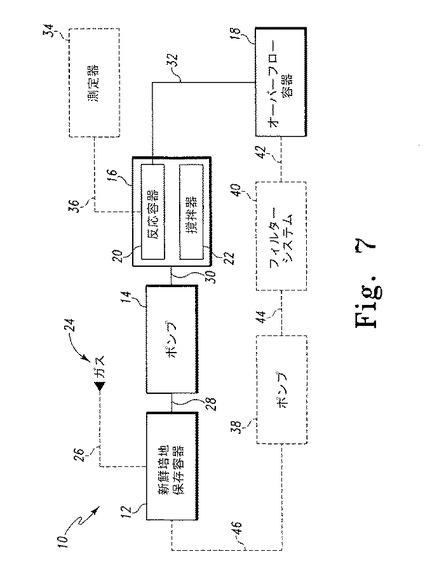
【図8】
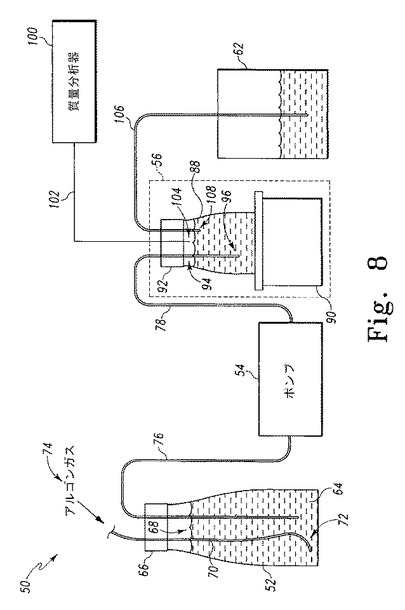
【図9】
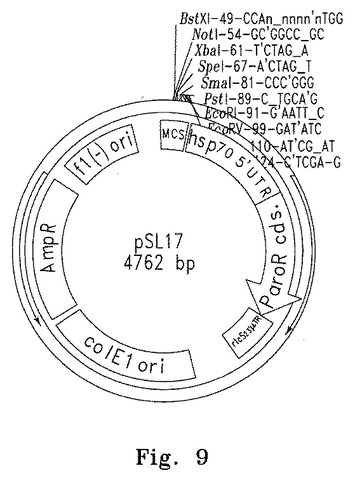
【図10】
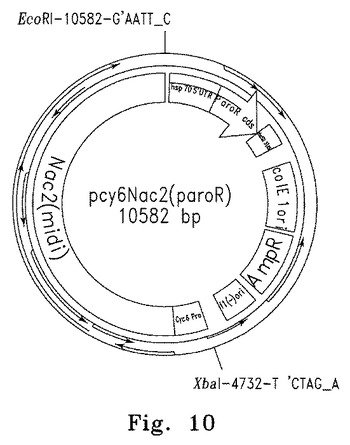
【図11A】

【図11B】

【図11C】
【図12】

【図13】
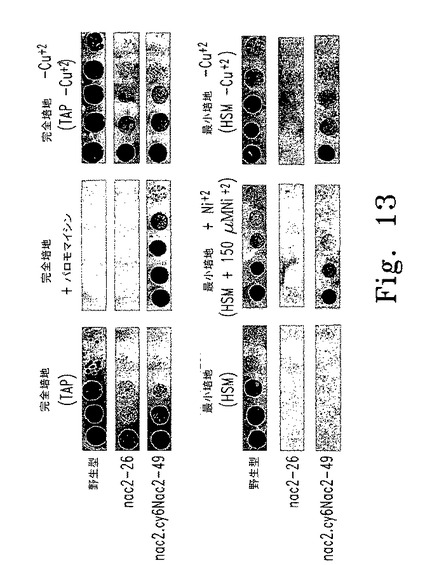
【図14】
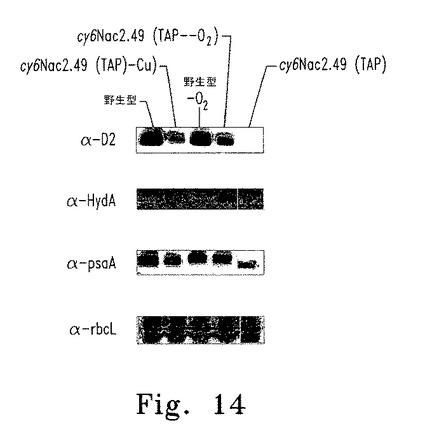
【図15】
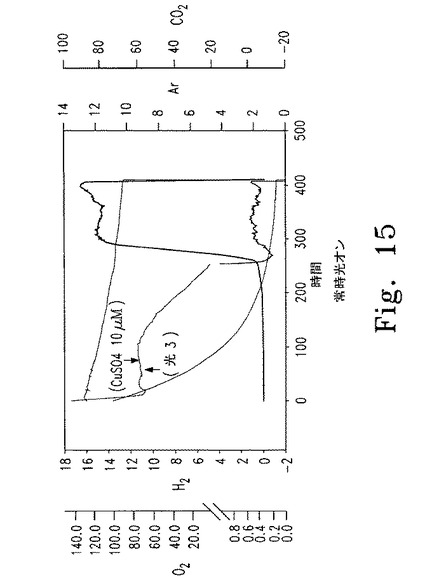
【図16】
【図17】
【図18】
【図19】
【図20】

【図21】
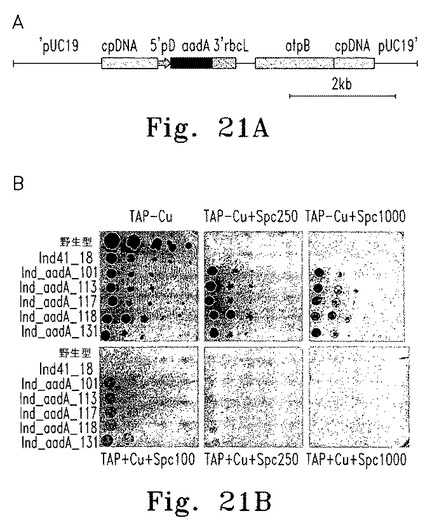
【図22】
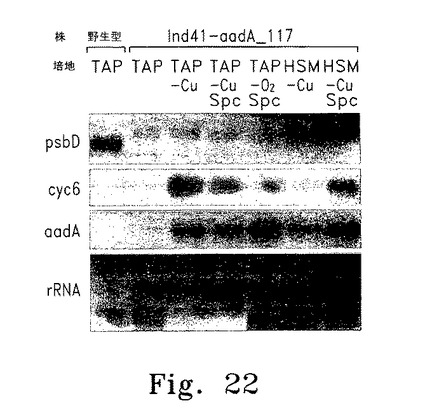
【図23】
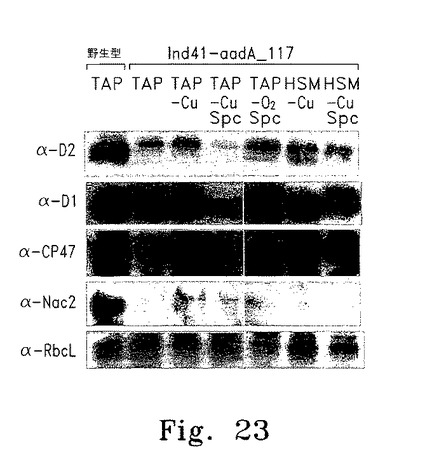
【図24】
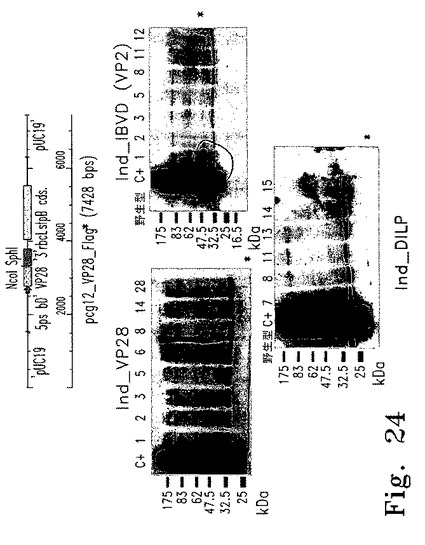
【図25】
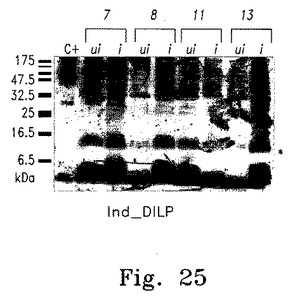
【図26】
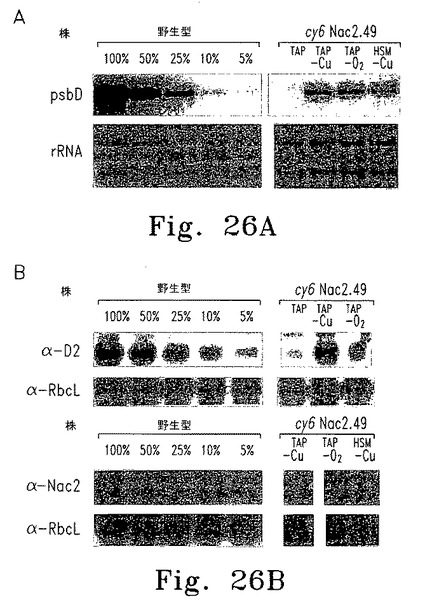
【図27】
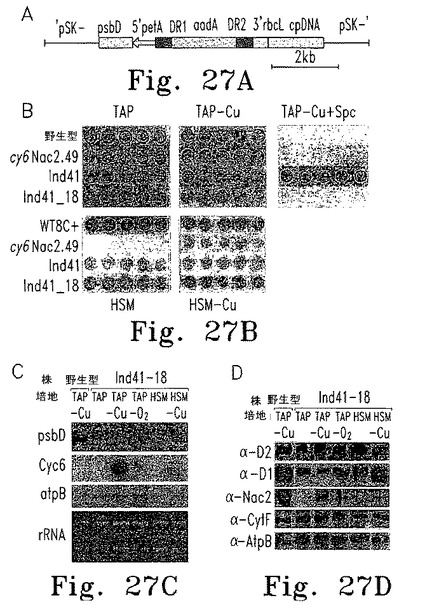
【図28A】
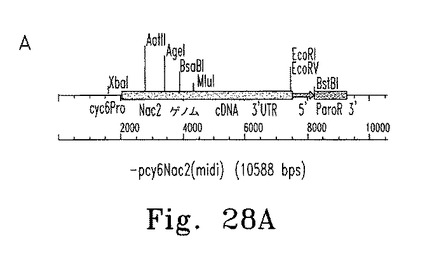
【図28B】
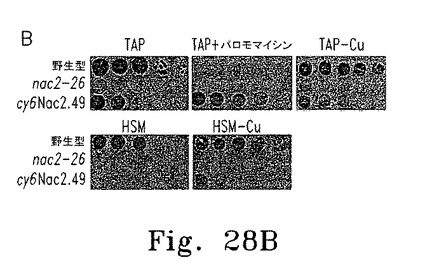
【図28C】
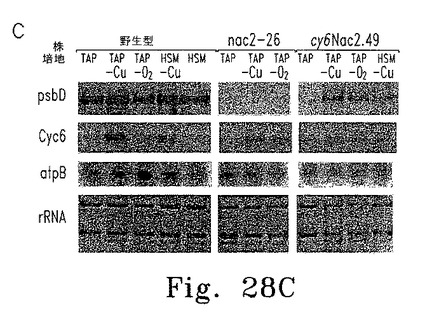
【図28D】
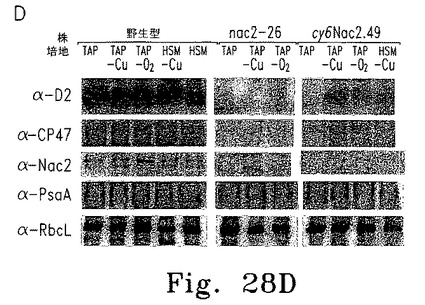
【図29A】
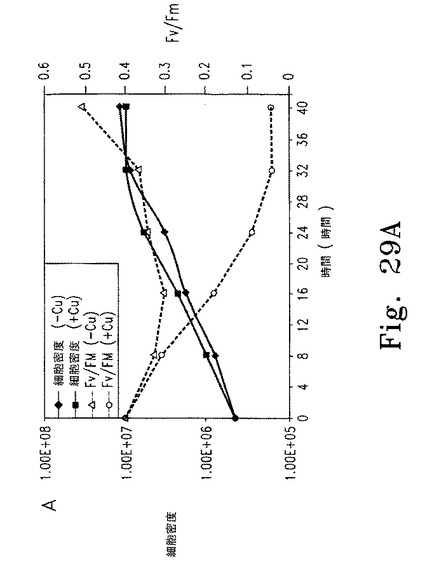
【図29B】
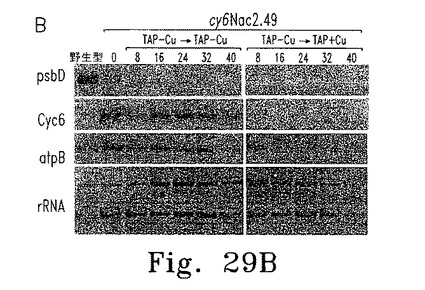
【図29C】
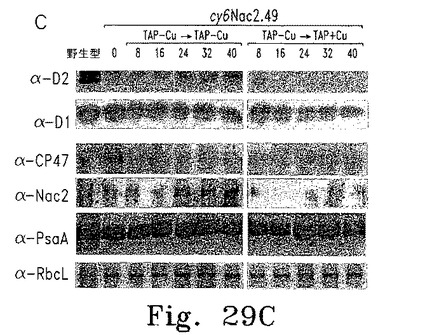
【図30A】
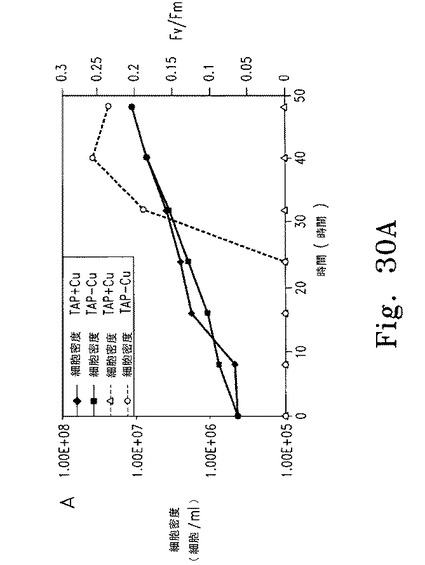
【図30B】
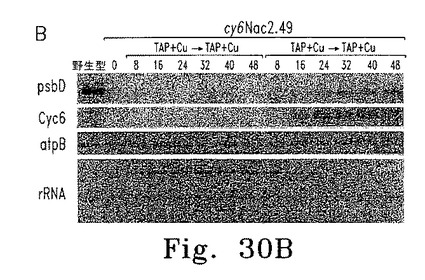
【図30C】
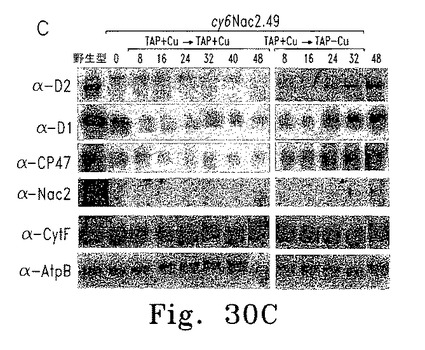
【図31】
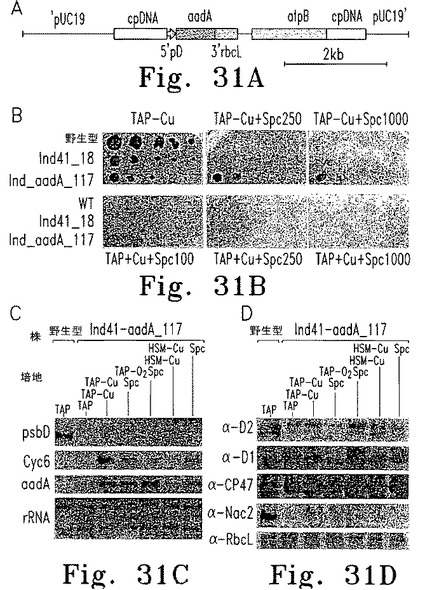
【図32A】
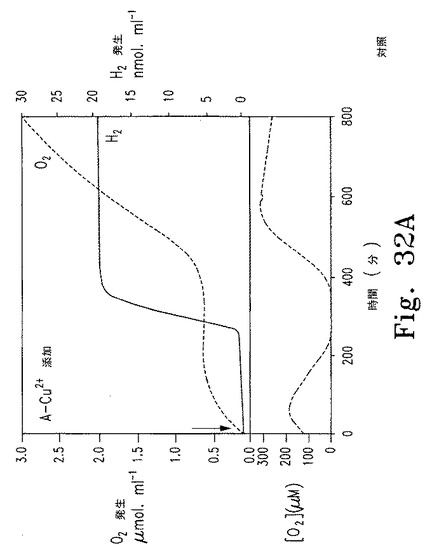
【図32B】
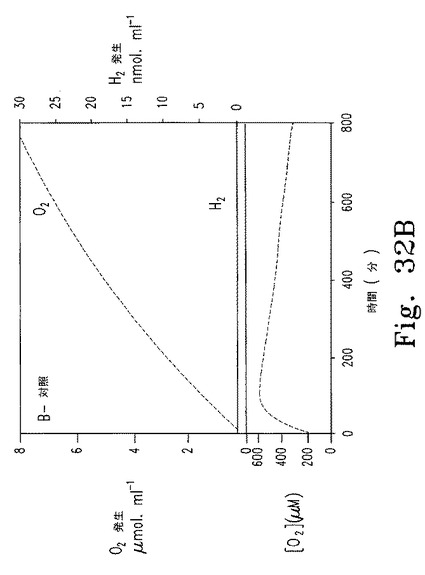
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11A】
【図11B】
【図11C】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28A】
【図28B】
【図28C】
【図28D】
【図29A】
【図29B】
【図29C】
【図30A】
【図30B】
【図30C】
【図31】
【図32A】
【図32B】
【公表番号】特表2010−500048(P2010−500048A)
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願番号】特願2009−524627(P2009−524627)
【出願日】平成19年8月11日(2007.8.11)
【国際出願番号】PCT/US2007/017774
【国際公開番号】WO2008/021223
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(509041898)ユニバーシティ オブ ジュネーブ (1)
【氏名又は名称原語表記】UNIVERSITY OF GENEVA
【住所又は居所原語表記】24,rue du General−Dufour,CH−1211 Geneve 4 (CH).
【出願人】(509041418)
【氏名又は名称原語表記】WAGNER,Richard,E.
【住所又は居所原語表記】3416 Ashwood Drive,Bloomington,IN 47401 (US).
【Fターム(参考)】
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願日】平成19年8月11日(2007.8.11)
【国際出願番号】PCT/US2007/017774
【国際公開番号】WO2008/021223
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(509041898)ユニバーシティ オブ ジュネーブ (1)
【氏名又は名称原語表記】UNIVERSITY OF GENEVA
【住所又は居所原語表記】24,rue du General−Dufour,CH−1211 Geneve 4 (CH).
【出願人】(509041418)
【氏名又は名称原語表記】WAGNER,Richard,E.
【住所又は居所原語表記】3416 Ashwood Drive,Bloomington,IN 47401 (US).
【Fターム(参考)】
[ Back to top ]