
蛍光タンパク質
【課題】新規の性質を有した有用な蛍光タンパク質およびその利用方法を提供する。
【解決手段】刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する蛍光タンパク質であり、該蛍光タンパク質は、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する。
【解決手段】刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する蛍光タンパク質であり、該蛍光タンパク質は、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はpH依存的に蛍光活性が変化する蛍光タンパク質およびその利用方法に関する。
【背景技術】
【0002】
蛍光タンパク質は、細胞のイメージング等の生物学的研究において重要な役割を果たしている。特に、オワンクラゲ(Aequorea victoria)に由来する緑色蛍光タンパク質(Green Fluorescent Protein:GFP)が広く利用されている。
【0003】
また、異なる性質を有した蛍光タンパク質を取得する目的で、様々な試みが行われている。例えば、オワンクラゲ以外の生物から様々な蛍光タンパク質が取得されている。あるいは、既に取得された蛍光タンパク質にアミノ酸置換等の改変を加えることで、異なる性質を付与しようとしている。
【0004】
非特許文献1には、オワンクラゲGFPの立体構造情報に基づいて、部位特異的に203番目のスレオニンをチロシンに置換する変異を導入することで、野生型に比べて吸収波長および蛍光波長が長波長側にシフトした黄色蛍光を発する変異体(YFP)を作製したことが記載されている。非特許文献1には、長波長側へのシフトは、203番目の残基と発色団とのπ電子間の相互作用により、発色団のπ電子がより広く非局在化することに起因すると記載されている。この変異体は513nm付近に吸収ピークを有し、525nm付近に蛍光ピークを有する。このように取得されたYFPは、細胞内現象のモニタリングに幅広く利用され、また蛍光共鳴エネルギー移動(FRET)における蛍光を発する受容体としても利用されている。
【0005】
非特許文献2には、オワンクラゲGFPに変異を導入し(65番目のセリンがスレオニン、48番目のシステインがセリン、148番目のヒスチジンがシステインおよび203番目のスレオニンがシステインにそれぞれ置換される)、pHによって蛍光波長が変化する変異体を作製したことが記載されている。この変異体は、400nm付近および500nm付近の吸収スペクトルがpH7付近で変化し、酸性側において400nm付近が、アルカリ性側において500nm付近の吸収がそれぞれ大きくなる。さらに、このような吸収スペクトルの変化に依存して、酸性側において460nm付近の蛍光強度が、アルカリ性側において510nm付近の蛍光強度がそれぞれ増加する。非特許文献2は、この性質を利用した細胞内のセンサーとしての有用性を示唆している。また、各pHにおけるGFPのX線結晶構造解析の結果から、このようなpH依存的な蛍光波長の変化は、pHが高くなると147番目のセリンの側鎖の向きが変化することに起因すると記載している。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Ormo,M. et al.(1996), Science, 273:1392-1395
【非特許文献2】Green Fluorescent Protein Variants as Rationmetric Dual Emission pH Sensors. 1. Structural Characterization and Preliminary Application. Hanson GT., McAnaney TB., Park ES., Rendell MEP., Yarbrough DK., Chu S., Xi L., Boxer SG., Montrose MH., and Remington SJ. Biochemistry 2002, 41, 15477-15488
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、新規の性質を有した有用な蛍光タンパク質およびその利用方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明の実施態様によれば、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質が提供される。
【図面の簡単な説明】
【0009】
【図1】野生型ウミサボテンGFPおよびA−H198Y変異体について、pHによる蛍光の変化を示す図。
【図2】A−H198Y変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図3】野生型ウミサボテンGFPおよびH198T変異体について、pHによる蛍光の変化を示す図。
【図4】H198T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図5】野生型ウミサボテンGFPおよびH198T−S147T変異体について、pHによる蛍光の変化を示す図。
【図6】H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図7】野生型ウミサボテンGFPおよびA−H198T−S147T変異体について、pHによる蛍光の変化を示す図。
【図8】A−H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図9】各蛍光タンパク質の常温培養時および37℃培養時における蛍光を示す図。
【発明を実施するための形態】
【0010】
<蛍光タンパク質>
本発明は、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質に関する。
【0011】
ここにおいて、強アルカリ性とはpHが10から11付近以上であることを意味し、弱アルカリ性とはpHが9から10付近以下であることを意味する。
【0012】
本発明に係る蛍光タンパク質は、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来するものであってよい。特に、カベルヌラリア・オベサ(Cavernularia obesa)(和名:ウミサボテン)に由来するものであってよい。本願において「由来する」という表現は、その生物から取得された野生型の蛍光タンパク質だけでなく、当該野生型蛍光タンパク質を改変して得られたものも含むことを意味する。
【0013】
ウミサボテンはGFPを発現するが、これを改変することで本発明に係る蛍光タンパク質を得ることができる。ウミサボテンGFPは、221アミノ酸(配列番号9)からなる緑色蛍光タンパク質である。このGFPはpHによって蛍光波長が変化する特徴を有している。すなわち、pH4では458nm付近の青色蛍光を有し、pH5以上では507nm付近の緑色蛍光を有する。このウミサボテンGFPに対して、蛍光波長が長波長側にシフトする改変、蛍光強度が増大する改変等を行うことで本発明に係る蛍光タンパク質を得ることができる。このような改変には、野生型ウミサボテンGFPの198番目のヒスチジンをチロシンまたはスレオニンに置換する変異が含まれる。さらに、198番目のヒスチジンをチロシンまたはスレオニンに置換し且つ147番目のセリンをスレオニンに置換する変異が含まれる。
【0014】
本発明に係る蛍光タンパク質に対して、本発明に係る蛍光タンパク質の性質(強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する)をもたらす変異の他に、当該タンパク質の研究等への利用性を高めるような変異を導入することができる。例えば、タンパク質を単量体化する変異または特定の温度で安定化する変異を導入することができる。単量体化する変異とは、野生型蛍光タンパク質が本来の状態で多量体を形成する場合に、そのような多量体の形成を回避して単量体で存在できるようにする変異である。ウミサボテンGFPは、野生型では二量体を形成するが、154番目のシステインをセリンに置換(C154S)することで単量体化させることができる。特定の温度で安定化させる変異とは、その温度下でもタンパク質が望ましい状態を維持するような変異を意味し、特に蛍光タンパク質の場合、例えば、その温度においても蛍光活性を維持する変異を意味する。この変異により、蛍光タンパク質を導入する細胞の培養温度においても、十分な強度の蛍光を発するようになる。ウミサボテンGFPの場合、S129G、D156G、K204IおよびN209Yの4箇所を置換することで、またはN126YおよびY166Fの2箇所を置換することで、37℃において蛍光強度を高い状態で維持する変異体を得ることができる。
【0015】
以下に、本発明に係る蛍光タンパク質の具体的な例を説明する。
1つ目は、野生型のウミサボテンGFPに、198番目のヒスチジンをチロシンに置換する変異、単量体化する変異および温度安定化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198Y、C154S、S129G、D156G、K204IおよびN209Yのアミノ酸置換を導入している(配列番号1)。本願では、この変異体を「A−H198Y変異体」と称する。この変異体は、pH4〜9で516nm付近、pH10〜11で512nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしている。
【0016】
2つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異および単量体化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198TおよびC154Sのアミノ酸置換を導入している(配列番号3)。本願では、この変異体を「H198T変異体」と称する。この変異体は、pH4〜9で515nm付近、pH10〜11で507nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしている。さらに、当該変異体は37℃で培養した場合にも蛍光を発する。よって哺乳細胞などの培養に適した温度条件でも用いることが可能である。
【0017】
3つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異、147番目のセリンをスレオニンに置換する変異および単量体化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198T、S147TおよびC154Sのアミノ酸置換を導入している(配列番号5)。本願では、この変異体を「H198T−S147T変異体」と称する。この変異体は、pH4〜10で515nm付近、pH11で508nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしており、蛍光強度も高まっている。また、H198T変異体がpH10付近で蛍光波長が変化するのに対し、当該変異体はpH11付近で蛍光波長の変化が生じる。
【0018】
4つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異、147番目のセリンをスレオニンに置換する変異、単量体化する変異および温度安定化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198T、S147T、C154S、S129G、D156G、K204IおよびN209Yのアミノ酸置換を導入している(配列番号7)。本願では、この変異体を「A−H198T−S147T変異体」と称する。この変異体は、pH4〜10で514nm付近、pH11で508nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしており、蛍光強度も高まっている。また、H198T変異体がpH10付近で蛍光波長が変化するのに対し、当該変異体はpH11付近で蛍光波長の変化が生じる。さらに、当該変異体は37℃で培養した場合にも蛍光を発する。よって哺乳細胞などの培養に適した温度条件でも用いることが可能である。
【0019】
本発明に係る蛍光タンパク質により、一般的なGFPよりも長波長側の蛍光色を用いて細胞内現象をイメージングすることが可能となる。特に、野生型ウミサボテンGFPは、酸性側で青色、中性〜アルカリ性側で緑色の蛍光活性を有するため、これらとは異なる蛍光色を利用することができる。また、本発明に係る蛍光タンパク質は、より短波長側の蛍光を発するタンパク質(CFP等)を供与体としたFRET系等の受容体としても用いることができる。さらに、本発明に係る蛍光タンパク質は、pHによって蛍光波長が変化する性質を有するため、細胞内等のpHの測定が可能となる。特に、野生型ウミサボテンGFPとは蛍光色が変化するpHの値が異なるため、本発明に係る蛍光タンパク質と野生型ウミサボテンGFPとを組み合わせて使用することで、より精度の高いpH測定が可能となる。
【0020】
<核酸>
本発明は、本発明に係る蛍光タンパク質をコードする塩基配列を含む核酸に関する。このような塩基配列は、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する塩基配列であってよい。ここにおける「由来する」とは、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物が本来有する野生型の塩基配列に変異が生じた塩基配列を含むことを意味する。また、ここにおける変異とは、塩基配列中の特定の塩基の置換、欠失および/または付加等を指す。塩基配列の変異には、コードされるアミノ酸配列に変化を生じさせない変異をも含む。また、核酸とは、特に、DNAまたはRNAを指す。
【0021】
本発明に係る核酸の好ましい例は、配列番号2に示される塩基配列を含む核酸(A−H198Y変異体をコード)、配列番号4に示される塩基配列を含む核酸(H198T変異体をコード)、配列番号6に示される塩基配列を含む核酸(H198T−S147T変異体をコード)、または配列番号8に示される塩基配列を含む核酸(A−H198T−S147T変異体をコード)である。
【0022】
また、本発明は、これらの核酸を含むベクターを含む。当該ベクターには、蛍光タンパク質をコードする核酸以外に、発現を調節するための配列またはマーカー遺伝子の配列を含む核酸等を含んでよい。
【0023】
<イメージング方法>
本発明は、本発明に係る蛍光タンパク質または本発明に係る核酸を用いた生物試料のイメージング方法に関する。
【0024】
イメージング方法とは、観察手段を用いて対象を測定または観察することを意味する。その具体的な例は、本発明に係る蛍光タンパク質と特定のタンパク質とを融合させたタンパク質を細胞に発現させ、当該細胞中の動態を蛍光顕微鏡により観察する方法である。
【0025】
生物試料は、バクテリアの細胞、酵母細胞、真菌細胞、昆虫細胞または哺乳細胞といった細胞であってよい。さらに、生物試料は、組織または個体であってよい。細胞は、固定したものであってよいし、生きた状態のものであってよい。固定は、ホルマリン、メタノール等による一般的な方法で行うことができる。また、生物試料としての個体は、ヒトを除く個体であってよい。生物試料とする細胞、組織および個体に特に限定はなく、従来の取得方法によって得られるものを使用することができる。
【0026】
生物試料に蛍光タンパク質を導入するために、細胞等へタンパク質を導入するための当該分野で一般的ないずれの方法を使用してもよい。例えば、生物試料が細胞である場合、マイクロインジェクション法によって蛍光タンパク質を直接細胞内へ注入してもよい。あるいは、蛍光タンパク質の遺伝子を含む発現ベクターを細胞に導入し、適切に発現させることで導入してもよい。発現ベクター等による遺伝子の導入は、リン酸カルシウム法、リポフェクションまたはエレクトロポレーション等を使用することができる。
【0027】
観察手段として、特定の波長の蛍光を検出することが可能な、従来の方法または装置を使用することができる。例えば蛍光顕微鏡を使用することができる。測定または観察とは、顕微鏡を介して目視により確認すること、顕微鏡に設置したカメラを利用して撮像すること等を含む。このような測定および観察は、単一の細胞に対して行うことが可能である。
【0028】
<pH測定法>
本発明は、本発明に係る蛍光タンパク質または本発明に係る核酸を用いた生物試料内のpHを測定する方法に関する。
【0029】
例えば、本発明に係る蛍光タンパク質を対象に導入し、前記蛍光タンパク質が発する蛍光に基づいて前記対象内部のpHを測定する方法に関する。すなわち、本発明に係る方法では、対象の内部に蛍光タンパク質を導入した後、励起光を照射して蛍光タンパク質が発する蛍光を検出し、当該蛍光の波長に基づいてpHを特定する。
【0030】
生物試料としては、イメージング方法と同様な試料を使用することができる。
【0031】
蛍光の検出は、目視で行うことも可能であるが、特定の波長の蛍光を測定することが可能な、従来の方法または装置を使用して行うことができる。例えば蛍光顕微鏡を使用することができる。励起光を照射して蛍光の色を目視で確認してpHを判断してもよいが、検出装置を用いることで、より詳細にpHを特定することが可能となる。例えば、蛍光タンパク質が発する2種の蛍光に対応したフィルターセットの蛍光キューブを倒立型蛍光顕微鏡に設置し、それぞれの波長の蛍光画像をCCDカメラによって撮像する。撮像した画像内のpH測定対象領域の蛍光強度を数値化しpHを求めることができる。このとき、細胞を種々の条件で刺激し、その後一定時間ごとに撮像することで、系時的に蛍光画像を取得することもできる。また、特定の波長だけの測定ではなく、蛍光スペクトルを測定することもできる。この場合、具体的には、顕微鏡のカメラポートに光ファイバーを接続し分光光度計にて測定対象領域の蛍光スペクトルを測定することで行うことができる。
【実施例】
【0032】
[実施例1:野生型ウミサボテン蛍光タンパク遺伝子のクローニング]
ウミサボテン(Cavernularia obesa)から野生型蛍光タンパク質を抽出および精製し、その部分的なアミノ酸配列を読み取った後、当該配列をもとに遺伝子配列を決定することで蛍光タンパク質遺伝子のクローニングを行った。
【0033】
蛍光タンパク質の抽出精製およびアミノ酸配列分析
島根近海で採取したウミサボテン15個体(約200g)を500mlのSDS−グリシンバッファー中ですり潰し、蛍光活性を持つタンパクを含む可溶性タンパクを抽出した。遠心機を用いて残渣を取り除き、蛍光活性を持つタンパク質を含む溶液を抽出した。この抽出溶液に、最終濃度が80%になるように硫酸アンモニウムを加えて硫安沈殿を行った。具体的には、抽出溶液500mlに262gの硫酸アンモニウムを加えた。得られた沈殿を、50mlのトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)に再溶解させた。これに2倍量のエタノールを加え、タンパク質を再度沈殿させた。さらに、この沈殿にトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を加えて再溶解させた。この溶液50mlを、十分量のトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)および透析膜27/32(三光純薬株式会社)を用いて透析した。透析後の溶液を、DEAE SepharoseCL−6Bカラム(GEヘルスケアバイオサイエンス)を装着したクロマトグラフィーシステムAKTAexplorer(GEヘルスケアバイオサイエンス)を用いてイオン交換分離精製した。このイオン交換は、低塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)で十分に平衡化した後、サンプルを添加し、高塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、1M NaCl)でNaCl濃度を0.4Mまで上昇させることで行い、その結果、蛍光活性のあるフラクションを分取した。蛍光活性を持つタンパクを含むフラクションの選択は、UV/BLUE CONVERTER PLATE(UVP)を用いて行った。得られたフラクションを、十分量のトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)および透析膜27/32(三光純薬株式会社)を用いて透析した。透析後の溶液を、限外ろ過アミコンウルトラ:分画分子量30kDa(日本ミリポア株式会社)を用いて濃縮した。次に、Sephacryl S−200 High Resolution(GEヘルスケアバイオサイエンス)およびトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を用いてゲルろ過を行った。ゲルろ過後、得られた蛍光活性のあるフラクションをmonoQ 5/50(GEヘルスケアバイオサイエンス)によってイオン交換分離した。このイオン交換は、低塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)で十分に平衡化した後、サンプルを添加し、高塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、1M NaCl)でNaCl濃度を0.4Mまで上昇させることで行い、その結果、蛍光活性のあるフラクションを得た。この活性のあるフラクションを限外ろ過アミコンウルトラ:分画分子量30kDa(日本ミリポア株式会社)を用いて濃縮し、その後、Superdex 75 10/300 GL(GEヘルスケアバイオサイエンス)およびトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を用いてゲルろ過分離を行い、蛍光活性のあるフラクションを得た。このサンプルをSDS−ポリアクリルアミドゲル電気泳動によって分離してアミノ酸配列を分析した。分析の結果、22残基のアミノ酸配列(IPD YFV QSF PEG FTF ERT LSF E:配列番号17)を決定することができた。
【0034】
蛍光タンパク質遺伝子のクローニング
蛍光タンパク質遺伝子全長のクローニングのために、3’−RACE PCRを下記のとおり実施した。Rapid Amplification of cDNA End法(以下、RACEと略す)により、蛍光タンパク質遺伝子をクローニングするための混合プライマーを作成した。アミノ酸配列解析によって得られたアミノ酸配列IPD YFV QSF PEG FTF ERT LSF E(配列番号17)のうち、コドンの塩基組み合わせが少ないIPDYFVおよびEGFTFERのアミノ酸領域に注目した。これらのアミノ酸領域をコードする塩基配列を予測し、3’末端RACE polymerase chain reaction(以下PCRと略す)に用いる蛍光タンパク質特異的混合プライマー(合計12種類)を以下のように作成した。IPDYFVアミノ酸領域に結合するプライマーとして、COGFP−TTT(5’−ATH CCN GAT TAT TTT GT−3’)(配列番号18)、COGFP−TTC(5’−ATH CCN GAT TAT TTC GT−3’)(配列番号19)、COGFP−TCT(5’−ATH CCN GAT TAC TTT GT−3’)(配列番号20)、COGFP−TCC(5’−ATH CCN GAT TAC TTC GT−3’)(配列番号21)、COGFP−CTT(5’−ATH CCN GAC TAT TTT GT−3’)(配列番号22)、COGFP−CTC(5’−ATH CCN GAC TAT TTC GT−3’)(配列番号23)、COGFP−CCT(5’−ATH CCN GAC TAC TTT GT−3’)(配列番号24)およびCOGFP−CCC(5’−ATH CCN GAC TAC TTC GT−3’)(配列番号25)を作成し、ならびに、EGFTFERアミノ酸領域に結合するプライマーとして、COGFP−ATTAA(5’−GAA GGN TTT ACN TTT GAA AG−3’)(配列番号26)、COGFP−ACCAA(5’−GAA GGN TTC ACN TTC GAA AG−3’)(配列番号27)、COGFP−ATTGA(5’−GAG GGN TTT ACN TTT GAG AG−3’)(配列番号28)およびCOGFP−ACCGA(5’−GAG GGN TTC ACN TTC GAG AG−3’)(配列番号29)を作成した。プライマー中のHおよびNは、混合塩基を示す。
【0035】
完全長cDNA合成試薬GeneRacer(インビトロジェン)を用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、蛍光タンパク質のアミノ酸配列から予測して作成した12種類の特異的混合プライマーおよび3’末端特異的プライマーであるGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’)(配列番号30)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’)(配列番号31)を用いて3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれており、これを使用した。3’−RACE PCRによって効果的に蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型とし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRは、ポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0036】
一度目のPCRは、IPDYFVアミノ酸領域で作成した8種類プライマー(COGFP−TTT、COGFP−TTC、COGFP−TCT、COGFP−TCC、COGFP−CTT、COGFP−CTC、COGFP−CCT、COGFP−CCC)のいずれかとGeneRacer3’ Primerとの計8つのプライマー対で蛍光タンパク質遺伝子の増幅を行った。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍、dNTP Mixture(各2.5mM)を最終濃度各0.2mM、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μl、8種類のプライマーのうちの1つを最終濃度0.4μMおよびGeneRacer3’Primerを最終濃度0.4μMとして20μlのPCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μl加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%トリス酢酸緩衝液(以下、TAEと略す)アガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。8つの反応溶液でわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0037】
nested PCRは、EGFTFERアミノ酸領域で作成した4種類プライマー(COGFP−ATTAA、COGFP−ACCAA、COGFP−ATTGA、COGFP−ACCGA)の何れかとGeneRacer3’Nested Primerとの計4つのプライマー対で蛍光タンパク質遺伝子の増幅を行った。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍、dNTP Mixture(各2.5mM)を最終濃度各0.2mM、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μl、4種類のプライマーのうちの1つを最終濃度0.4μMおよびGeneRacer3’ Nested Primerを最終濃度0.4μMとして10μlのnested PCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0038】
その結果、1度目のPCR反応として、COGFP−TTCとGeneRacer3’ Primerとのプライマー対を用いて実施し、nested PCR反応として、得られたPCR反応溶液を鋳型としてCOGFP−ACCAAとGeneRacer3’ Nested Primerとのプライマー対を用いて実施した場合に、顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定し、ウミサボテン蛍光タンパク質遺伝子のウミサボテン3’末端側の塩基配列(配列番号32)とした。
【0039】
蛍光タンパク質の5’末端クローニングのための5’−RACEを下記の通り行った。一連の3’−RACE解析によって得られたウミサボテン蛍光タンパク質遺伝子の3’末端側の塩基配列をもとに、5’末端クローニングのための5’−RACEに用いるプライマーを作成した。作成した5’末端クローニング用のプライマーは、COGFP−A−R1(5’−GCT ATA GCC GTC TCA TGT TGC TCG T−3’)(配列番号33)、COGFP−A−R2(5’−AGC CGT CTC ATG TTG CTC GTA GTA G−3’)(配列番号34)およびCOGFP−A−R3(5’−ATG TTG CTC GTA GTA GTT GCC TTC CTC GAC−3’)(配列番号35)の3種類である。COGFP−A−R1、COGFP−A−R2、COGFP−A−R3の順で5’末端に近い位置に結合し、順次nested PCR反応のプライマーとして用いる。
【0040】
完全長cDNA合成試薬GeneRacerを用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、3種類の5’末端クローニング用のプライマーならびに5’末端特異的プライマーであるGeneRacer5’ Primer(5’−CGA CTG GAG CAC GAG GAC ACT GA−3’)(配列番号36)およびGeneRacer5’ Nested Primer(5’−GGA CAC TGA CAT GGA CTG AAG GAG TA−3’)(配列番号37)を用いて、5’−RACE PCRを行った。GeneRacer5’ PrimerおよびGeneRacer5’ Nested Primerは完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれており、これを使用した。5’−RACE PCRによって効果的に蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taqを用いて、マニュアルに従って実施した。
【0041】
一度目の5’−RACE PCRとして、3’−RACEで増幅した遺伝子の塩基配列をもとに作成したCOGFP−A−R1とGeneRacer5’Primerとのプライマー対を用いて、蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R1を最終濃度0.4μMとし、GeneRacer5’ Primerを最終濃度0.4μMとして、10μlのPCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。この5’−RACEでわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0042】
nested PCRとして、COGFP−A−R2とGeneRacer5’ Nested Primerとのプライマー対を用いて蛍光タンパク質GFP遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R2を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、10μlのPCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0043】
その結果、1度目のPCR反応として、COGFP−A−R1とGeneRacer5’ Primerとのプライマー対を用いて実施し、nested PCR反応として、得られたPCR反応溶液を鋳型としてCOGFP−A−R2とGeneRacer5’ Nested Primerとのプライマー対を用いて実施した場合に、顕著な遺伝子増幅を確認できた。しかし、非特異的な複数の遺伝子の増幅が認められたため、このPCR反応溶液を鋳型として、異なるプライマー対を用いて再度nested PCR反応を実施した。
【0044】
二度目のnested PCRとして、COGFP−A−R3とGeneRacer5’ Nested Primerとのプライマー対を用いて、GFP遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R3を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、20μlのnested PCR反応溶液を作製し、1度目のnested PCR反応溶液を鋳型として0.4μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。その結果、一連のnested PCR反応で顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定し、ウミサボテン蛍光タンパク質遺伝子の5’末端側の塩基配列(配列番号38)とした。
【0045】
次に、蛍光タンパク質遺伝子の完全長の増幅を下記のとおりに行った。上述した3’−RACEおよび5’−RACE解析によって得られたウミサボテン由来の蛍光タンパク質遺伝子の5’末端側の塩基配列をもとに、完全長蛍光タンパク質遺伝子クローニングのための3’−RACEに用いるプライマーを作成した。作成した完全長GFP遺伝子クローニング用のプライマーは、COGFP−A−Full−F3(5’−ATT TAG GTG GCT GCG TAC AG−3’)(配列番号39)、COGFP−A−Full−F4(5’−ATT TAG GTG GCT GCG TAC AGT TAA CAC−3’)(配列番号40)の2種類である。COGFP−A−Full−F3、COGFP−A−Full−F4の順で3’末端に近い位置に結合する。COGFP−A−Full−F4をnested PCR反応のプライマーとして用いる。
【0046】
完全長cDNA合成試薬GeneRacerを用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、2種類の完全長蛍光タンパク質遺伝子クローニング用のプライマーならびに3’末端特異的プライマーであるGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’)(配列番号30)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’)(配列番号31)を用いて、3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているので、これを使用した。3’−RACE PCRによって効果的に完全長蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taqを用いてマニュアルに従って実施した。
【0047】
一度目のPCRとして、COGFP−A−Full−F3とGeneRacer3’ Primerとのプライマー対を用いて蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−Full−F3を最終濃度0.4μMとし、GeneRacer3’ Primerを最終濃度0.4μMとして、10μlのnested PCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、50℃30秒および72℃1分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。一度目の5’−RACEでわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0048】
nested PCRとしてCOGFP−A−Full−F4とGeneRacer5’ Nested Primerとのプライマー対を用いて、蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−Full−F4を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、10μlのnested PCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、50℃30秒および72℃1分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。その結果、顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定して、完全長ウミサボテン蛍光タンパク質遺伝子の塩基配列(配列番号41)とした。
【0049】
得られた完全長ウミサボテン蛍光タンパク質の塩基配列から、配列情報解析ソフトウェアDNASIS Proを用いてウミサボテンGFP遺伝子のオープンリーディングフレームの塩基配列を予想した(配列番号10)。さらに、このオープンリーディングフレームを翻訳してウミサボテン蛍光タンパク質のアミノ酸配列を得た(配列番号9)。このアミノ酸配列は、ウミサボテンから精製し、アミノ酸分析によって得られた22残基の配列(配列番号17)を完全一致の状態で含むことから、ウミサボテンに由来する野生型蛍光タンパク質遺伝子であると決定した。この遺伝子をpRSETベクター(インビトロジェン)に組み込み、大腸菌にトランスフォーメーションし、UV/BLUE CONVERTER PLATE(UVP)を用いて観察したところ顕著な蛍光活性が示された。
【0050】
[実施例2:C154S変異(単量体化)の導入]
実施例1にて得られた野生型ウミサボテンGFPの遺伝子に対して、C154S変異を導入した。この変異を導入することで、野生型ウミサボテンGFPを単量体化することができる。
【0051】
以下の方法によって、野生型蛍光タンパク質のアミノ酸配列の154番目のシステインをセリンに置換した。変異の導入には、QuickChange II Site−Directed Mutagenesis Kit (Stratagene社)を用いた。pRSET−A(インビトロジェン社)にウミサボテン蛍光タンパク質遺伝子をクローニングしたプラスミドを鋳型とし、Pirmer1(CAATGTATGTATCGGACGACACTTTGG)(配列番号42)およびPrimer2(CCAAAGTGTCGTCCGATACATACATTG)(配列番号43)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、37度にてDpnIで処理し、大腸菌JM109(DE3)株にトランスフォームした。
【0052】
トランスフォームされた菌を培養してベクターを取り出し、蛍光タンパク質遺伝子の塩基配列をシーケンサーによって読み取った。その結果、配列番号14の配列が得られ、狙い通りに変異が導入できたことが確認できた。この遺伝子から、154番目がセリンに置換された蛍光タンパク質(配列番号11)が発現する。
【0053】
また、トランスフォーム後の大腸菌を、28度にて培養した後、溶菌してライセートを作製した。これにSDS−PAGEサンプルバッファーを加え、熱を加えずにサンプルを調製して、SDS−PAGEを行った。その結果、野生型蛍光タンパク質のレーンで出現する2量体のバンドが、変異体蛍光タンパク質のレーンで消失していた。このことから、蛍光タンパク質が単量体化されたことが確認された。
【0054】
[実施例3:温度安定化変異の導入]
温度安定化変異の導入について以下に述べる。具体的には、S129G、D156G、K204IおよびN209Yの4箇所が変異した変異体を得ることができる。これらの変異の導入よって、37℃において発色団形成を安定化させることができる。
【0055】
実施例2で得られた単量体化した変異体蛍光タンパク質に対してランダムに変異を導入し、37℃において発色団形成が安定化する変異体をスクリーニングした。
【0056】
変異の導入は、GeneMorph II EZClone Domain Mutagenesis Kit(Stratagene社)を用いた。pRSET−A(インビトロジェン社)に単量体化変異体(154番目のシステインがセリンに置換)の遺伝子がクローニングされたプラスミドを鋳型とし、Pirmer1(ATGAGTATTCCAGAGAATTCGGGCTTAACAG)(配列番号44)およびPrimer2(TCATGGTTTAGCTATGGCCGTCTCATG)(配列番号45)を加えて、キットに添付されているマニュアルに準じてPCR反応を行った。PCR反応後、1%アガロースゲルを用いて電気泳動を行い、目的とするPCR産物をWizard SV Gel and PCR Clean−Up(Promega社)を用いて精製した。精製後、マニュアルに準じてPCR反応を行い、37℃にてDpnIで処理した後に、エタノール沈殿を行った。沈殿後、少量の蒸留水にDNAを溶かして、MicroPulser(BioRad社)を用いてエレクトロポレーション法にてJM109(DE3)にトランスフォームした。トランスフォーム後の大腸菌を、25cm四方のLB(50ng/mlのアンピシリンを添加済み)プレートに播種して、37℃で培養してコロニーを形成させた。コロニー形成後、UV/BLUE CONVERTER PLATE(UVP)を使用して、野生型蛍光タンパク質を発現するコロニーと比べて、強い蛍光を発するコロニー(それぞれ変異体1および変異体2と名付ける)を2つ選択し、再度培養した。その後、それらが保持するプラスミドの塩基配列を決定した。それぞれの配列は配列番号15および配列番号16に示される。
【0057】
さらに、これらの塩基配列をアミノ酸配列に変換し(変異体1は配列番号12、変異体2は配列番号13)、野生型のアミノ酸配列と比較した。変異体1は、126番目のアスパラギンがチロシンに置換され、154番目のシステインがセリンに置換され、166番目のチロシンがフェニルアラニンに置換されていることがわかった。変異体2は、129番目のセリンがグリシンに置換され、154番目のシステインがセリンに置換され、156番目のアスパラギン酸がグリシンに置換され、204番目のリジンがイソロイシンに置換され、209番目のアスパラギンがチロシンに置換されていることがわかった。
【0058】
また、変異体1および2の37℃における発色団形成の安定性を、野生型蛍光タンパク質および単量体化蛍光タンパク質と比較した。pRSET−A(インビトロジェン社)に、野生型遺伝子、実施例2で作製した単量体化変異体の遺伝子、変異体1の遺伝子および変異体2の遺伝子をそれぞれクローニングし、それぞれJM109(DE3)株にトランスフォームした。これらの株をプレートに塗布し、37℃で培養して蛍光タンパク質を発現させた。その状態をUV/BLUE CONVERTER PLATE(UVP)で観察して、画像を撮影した。その結果、野生型および単量体化変異体と比較して、変異体1および2は非常に強い蛍光を発していた。特に、変異体1に比べて、変異体2のほうが強い蛍光を発していた。
【0059】
次に、ゲルろ過クロマトグラフィーにて、蛍光タンパク質の会合状態を調べた。野生型蛍光タンパク質を発現する大腸菌および変異体2を発現する大腸菌から、それぞれ蛍光タンパク質を精製し解析した。野生型では66kDal付近にピークが得られ、一方、変異体2では37kDal付近でピークが得られた。これらの結果から、野生型蛍光タンパク質が2量体を形成しているのに対し、変異体2の蛍光タンパク質が単量体を形成していることが示唆された。
【0060】
[実施例4:H198Y変異の導入]
実施例3で得られた遺伝子に対し、以下に述べるH198Yのアミノ酸置換の変異を導入し、A−H198Y変異体の遺伝子を得た。この変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0061】
pRSET−B(インビトロジェン社)に、変異体2(129番目のセリンがグリシンに置換、154番目のシステインがセリンに置換、156番目のアスパラギン酸がグリシンに置換、204番目のリジンがイソロイシンに置換、209番目のアスパラギンがチロシンに置換)の遺伝子を挿入したプラスミドを鋳型として、H198Yプライマー1(CATTATGTTCATTATCGCCTCGAAAAG、配列番号46)およびH198Yプライマー2(CTTTTCGAGGCGATAATGAACATAATG、配列番号47)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0062】
[実施例5:H198T変異の導入]
実施例2で得られた遺伝子に対し、以下に述べるH198Tのアミノ酸置換の変異を導入し、H198T変異体の遺伝子を得た。この変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0063】
pRSET−B(インビトロジェン社)に、単量体化変異体(154番目のシステインがセリンに置換)の遺伝子を挿入したプラスミドを鋳型として、H198Tプライマー1(CATTATGTTCATACCCGCCTCGAAAAG、配列番号48)およびH198Tプライマー2(CTTTTCGAGGCGGGTATGAACATAATG、配列番号49)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0064】
[実施例6:S147T変異の導入]
実施例5で得られた遺伝子に対し、以下に述べるS147Tのアミノ酸置換の変異を導入し、H198T−S147T変異体の遺伝子を得た。また、実施例3で得られた遺伝子に対し実施例5に記載の通りにH198T変異を導入した後、さらに以下に述べるS147Tのアミノ酸置換の変異を導入することで、A−H198T−S147T変異体の遺伝子を得た。これらの変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0065】
pRSET−B(インビトロジェン社)に、H198T変異体(198番目のヒスチジンがスレオニンに置換および154番目のシステインがセリンに置換)の遺伝子を挿入したプラスミド、または単量体化および温度安定化され、さらにH198T変異が導入された変異体(198番目のヒスチジンがスレオニン、154番目のシステインがセリンに置換、129番目のセリンがグリシンに置換、156番目のアスパラギン酸がグリシンに置換、204番目のリジンがイソロイシンに置換および209番目のアスパラギンがチロシンに置換)の遺伝子を挿入したプラスミドを鋳型として、S147Tプライマー1(CCAAACTAGAACCGACCAGTGAGTCAATG、配列番号50)およびS147Tプライマー2(CATTGACTCACTGGTCGGTTCTAGTTTGG、配列番号51)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0066】
[実施例7:pHによる蛍光の変化並びに蛍光および吸収スペクトルの測定]
A−H198Y変異体、H198T変異体、H198T−S147T変異体およびA−H198T−S147T変異体の4種の変異型蛍光タンパク質について、pHによる蛍光の変化並びに蛍光スペクトルおよび吸収スペクトルをそれぞれ測定した。
【0067】
実施例4から6により取得した、4種の変異体の内いずれかの遺伝子を有するプラスミドを、大腸菌に導入し、37℃で培養して各種変異型蛍光タンパク質を発現させ、溶菌後、Ni−NTA(Quiagen社)カラムクロマトグラフィーにより蛍光タンパク質をそれぞれ精製した。
【0068】
精製された変異体タンパク質を異なるpHの溶液に添加し、UVハンディライト(365nm)を用いて蛍光色を確認した。また、分光光度計(HITACHI U−3010)および蛍光光度計(HITACHI F−2500)を用いて、吸収スペクトルおよび蛍光スペクトルを測定した。
【0069】
以下に、それぞれの変異体の測定結果を説明する。
(A−H198Y変異体)
図1にpHによる蛍光の変化の結果をモノクロ画像として示し、図2に蛍光スペクトルおよび吸収スペクトルを示す。
【0070】
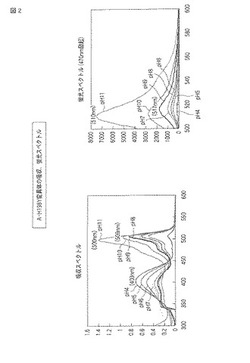
図1中、暗視野画像はUVハンディライトを照射したときに発せられる蛍光の画像であり、明視野画像は通常の照明下で確認される画像である。それぞれの上段のチューブには野生型ウミサボテンGFPが、下段のチューブにはA−H198Y変異体が、各pHの緩衝液とともに添加されている。なお、後述する図3、5および7についても同様に、上段に野生型が、下段に変異型がそれぞれ配置される。
【0071】
図1の元となるカラー画像によれば、野生型ウミサボテンGFPでは、pH4の環境下で青色の蛍光を発し、pH6以上の環境下で緑色の蛍光を発し、pH5において中間的な蛍光を発した。また、明視野において、pH4および5の環境下で緩衝液は透明であり、pH6の環境下で黄緑色を示し、pH7以上の環境下で緑色を示した。
【0072】
これに対し、A−H198Y変異体では、pH5から10の環境下で黄色の蛍光を発し、pH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から10の環境下で緩衝液は赤みがかった黄色を示し、pH11の環境下で黄色を示した。
【0073】
図2には、A−H198Y変異体の吸収スペクトルおよび蛍光スペクトル(470nm励起)が示される。吸収スペクトルでは、全体として400nm付近および500nm付近の2つのピークを示した。特に、pH4から5で403nm付近のピークが高く、pH6から10で509nm付近のピークが高く、pH11では500nm付近のピークが高かった。蛍光スペクトルでは、pH4から9で517nm付近にピークを示し、pH10および11で510nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表1にまとめる。
【0074】
【表1】

【0075】
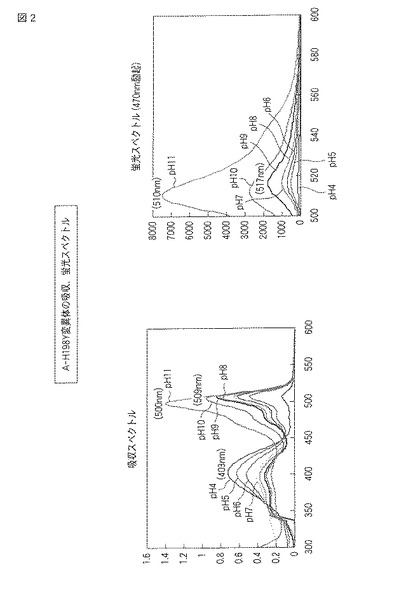
(H198T変異体)
図3にpHによる蛍光の変化の結果をモノクロ画像として示し、図4に蛍光スペクトルおよび吸収スペクトルを示す。
【0076】
図3の元となるカラー画像によれば、H198T変異体は、pH4から9の環境下で黄色の蛍光を発し、pH10および11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から9の環境下で緩衝液は赤みがかった黄色を示し、pH10および11の環境下で黄緑色を示した。
【0077】
図4には、H198T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルは、pH4から10で507nm付近のピークを、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から9で515nm付近にピークを示し、pH10および11で507nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表2にまとめる。
【0078】
【表2】
【0079】
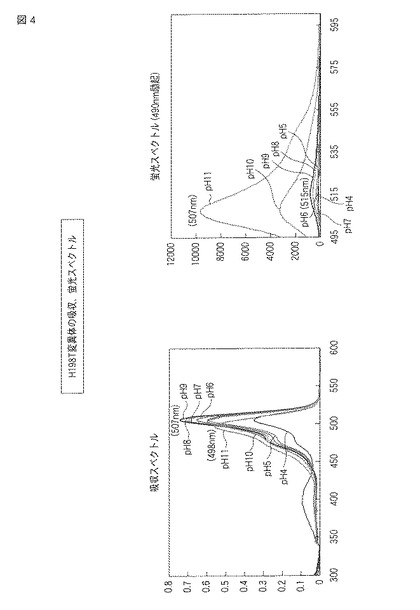
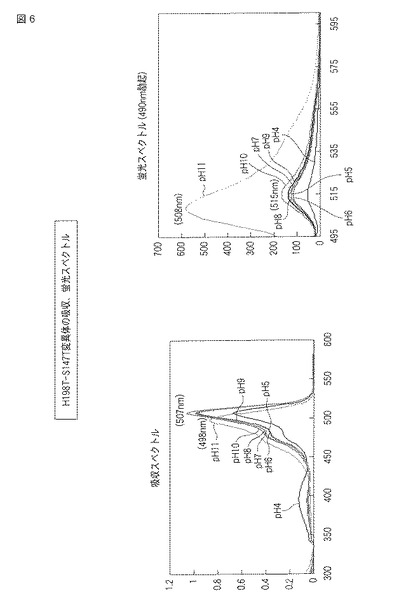
(H198T−S147T変異体)
図5にpHによる蛍光の変化の結果をモノクロ画像として示し、図6に蛍光スペクトルおよび吸収スペクトルを示す。
【0080】
図5の元となるカラー画像によれば、H198T−S147T変異体では、pH4から9の環境下で黄色の蛍光を発し、pH10およびpH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から9の環境下で緩衝液はオレンジよりの黄色を示し、pH10の環境下で黄色、pH11の環境下で黄緑色を示した。
【0081】
図6には、H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルでは、pH4から10で507nm付近のピークを示し、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から10で515nm付近にピークを示し、pH11で508nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。また、pH7における蛍光強度を同濃度の野生型ウミサボテンGFPと比較した場合、H198T−S147T変異体の方が約1.6倍高かった。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表3にまとめる。
【0082】
【表3】
【0083】
(A−H198T−S147T変異体)
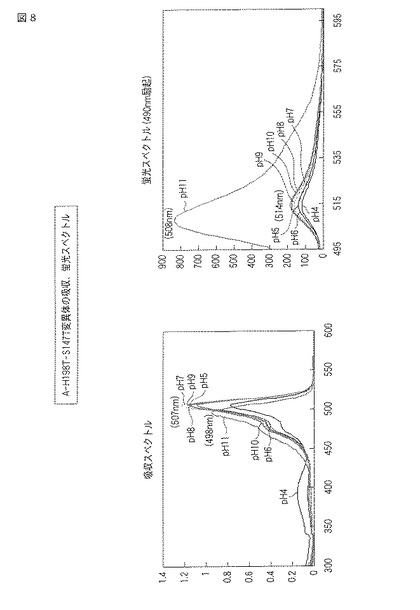
図7にpHによる蛍光の変化の結果をモノクロ画像として示し、図8に蛍光スペクトルおよび吸収スペクトルを示す。
【0084】
図7の元となるカラー画像によれば、A−H198T−S147T変異体では、pH4から10の環境下で黄色の蛍光を発し、pH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から10の環境下で緩衝液はオレンジよりの黄色を示し、pH11の環境下で黄緑色を示した。
【0085】
図8には、A−H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルでは、pH4から10で507nm付近のピークを示し、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から10で514nm付近にピークを示し、pH11で508nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。また、pH7における蛍光強度を同濃度の野生型ウミサボテンGFPと比較した場合、A−H198T−S147T変異体の方が約1.8倍高かった。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表4にまとめる。
【0086】
【表4】
【0087】
[実施例8:蛍光強度の比較]
4種の変異型蛍光タンパク質の蛍光強度を確認した。
【0088】
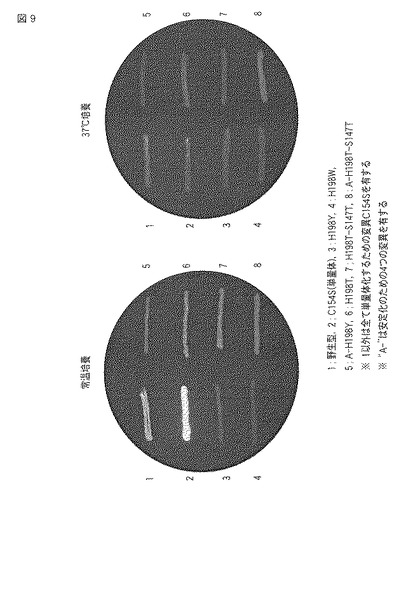
1:野生型ウミサボテンGFP、2:C154S変異体(単量体化のみを行った変異体)、3:H198Y変異体、4:H198W変異体、5:A−H198Y変異体、6:H198T変異体、7:H198T−S147T変異体および8:A−H198T−S147T変異体について、それぞれの遺伝子を形質転換させた8つの大腸菌を2つのプレートに塗布し、一方を常温で培養し、他方を37℃にて培養して、各蛍光タンパク質を発現させた。各プレートをUV/BLUE CONVERTER PLATE(UVP)で観察して、画像を撮影した。図9は、それらをモノクロで表した画像である。
【0089】
3:H198Y変異体および4:H198W変異体は、先行技術に倣って作製した。非特許文献1によれば、野生型オワンクラゲGFPの203番目のスレオニンをチロシンに置換することでYFPを作製している。この203番目のアミノ酸に立体構造上対応するウミサボテンGFPの198番目のアミノ酸について、先行技術と同様にチロシンまたはトリプトファンに置換した。
【0090】
図9の常温培養の結果によると、1:野生型および2:C154S変異体は緑色の蛍光を示した。特に、2:C154S変異体の方が強い蛍光を示した。3:H198Y変異体および4:H198W変異体は蛍光を示さなかった。本発明に係る5から8の変異体は黄色から黄緑色の蛍光を示した。特に、6から8の変異体は蛍光が強かった。
【0091】
37℃培養の結果によると、1:野生型および2:C154S変異体は常温培養の場合より弱いものの同色の蛍光を示した。また、3:H198Y変異体、4:H198W変異体、5:A−H198Y変異体および7:H198T−S147T変異体は蛍光を示さなかった。これらに対し、6:H198T変異体および8:A−H198T−S147T変異体は常温培養の場合よりも弱いものの同色の蛍光を示した。
【技術分野】
【0001】
本発明はpH依存的に蛍光活性が変化する蛍光タンパク質およびその利用方法に関する。
【背景技術】
【0002】
蛍光タンパク質は、細胞のイメージング等の生物学的研究において重要な役割を果たしている。特に、オワンクラゲ(Aequorea victoria)に由来する緑色蛍光タンパク質(Green Fluorescent Protein:GFP)が広く利用されている。
【0003】
また、異なる性質を有した蛍光タンパク質を取得する目的で、様々な試みが行われている。例えば、オワンクラゲ以外の生物から様々な蛍光タンパク質が取得されている。あるいは、既に取得された蛍光タンパク質にアミノ酸置換等の改変を加えることで、異なる性質を付与しようとしている。
【0004】
非特許文献1には、オワンクラゲGFPの立体構造情報に基づいて、部位特異的に203番目のスレオニンをチロシンに置換する変異を導入することで、野生型に比べて吸収波長および蛍光波長が長波長側にシフトした黄色蛍光を発する変異体(YFP)を作製したことが記載されている。非特許文献1には、長波長側へのシフトは、203番目の残基と発色団とのπ電子間の相互作用により、発色団のπ電子がより広く非局在化することに起因すると記載されている。この変異体は513nm付近に吸収ピークを有し、525nm付近に蛍光ピークを有する。このように取得されたYFPは、細胞内現象のモニタリングに幅広く利用され、また蛍光共鳴エネルギー移動(FRET)における蛍光を発する受容体としても利用されている。
【0005】
非特許文献2には、オワンクラゲGFPに変異を導入し(65番目のセリンがスレオニン、48番目のシステインがセリン、148番目のヒスチジンがシステインおよび203番目のスレオニンがシステインにそれぞれ置換される)、pHによって蛍光波長が変化する変異体を作製したことが記載されている。この変異体は、400nm付近および500nm付近の吸収スペクトルがpH7付近で変化し、酸性側において400nm付近が、アルカリ性側において500nm付近の吸収がそれぞれ大きくなる。さらに、このような吸収スペクトルの変化に依存して、酸性側において460nm付近の蛍光強度が、アルカリ性側において510nm付近の蛍光強度がそれぞれ増加する。非特許文献2は、この性質を利用した細胞内のセンサーとしての有用性を示唆している。また、各pHにおけるGFPのX線結晶構造解析の結果から、このようなpH依存的な蛍光波長の変化は、pHが高くなると147番目のセリンの側鎖の向きが変化することに起因すると記載している。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Ormo,M. et al.(1996), Science, 273:1392-1395
【非特許文献2】Green Fluorescent Protein Variants as Rationmetric Dual Emission pH Sensors. 1. Structural Characterization and Preliminary Application. Hanson GT., McAnaney TB., Park ES., Rendell MEP., Yarbrough DK., Chu S., Xi L., Boxer SG., Montrose MH., and Remington SJ. Biochemistry 2002, 41, 15477-15488
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、新規の性質を有した有用な蛍光タンパク質およびその利用方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明の実施態様によれば、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質が提供される。
【図面の簡単な説明】
【0009】
【図1】野生型ウミサボテンGFPおよびA−H198Y変異体について、pHによる蛍光の変化を示す図。
【図2】A−H198Y変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図3】野生型ウミサボテンGFPおよびH198T変異体について、pHによる蛍光の変化を示す図。
【図4】H198T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図5】野生型ウミサボテンGFPおよびH198T−S147T変異体について、pHによる蛍光の変化を示す図。
【図6】H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図7】野生型ウミサボテンGFPおよびA−H198T−S147T変異体について、pHによる蛍光の変化を示す図。
【図8】A−H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトルを示す図。
【図9】各蛍光タンパク質の常温培養時および37℃培養時における蛍光を示す図。
【発明を実施するための形態】
【0010】
<蛍光タンパク質>
本発明は、強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質に関する。
【0011】
ここにおいて、強アルカリ性とはpHが10から11付近以上であることを意味し、弱アルカリ性とはpHが9から10付近以下であることを意味する。
【0012】
本発明に係る蛍光タンパク質は、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来するものであってよい。特に、カベルヌラリア・オベサ(Cavernularia obesa)(和名:ウミサボテン)に由来するものであってよい。本願において「由来する」という表現は、その生物から取得された野生型の蛍光タンパク質だけでなく、当該野生型蛍光タンパク質を改変して得られたものも含むことを意味する。
【0013】
ウミサボテンはGFPを発現するが、これを改変することで本発明に係る蛍光タンパク質を得ることができる。ウミサボテンGFPは、221アミノ酸(配列番号9)からなる緑色蛍光タンパク質である。このGFPはpHによって蛍光波長が変化する特徴を有している。すなわち、pH4では458nm付近の青色蛍光を有し、pH5以上では507nm付近の緑色蛍光を有する。このウミサボテンGFPに対して、蛍光波長が長波長側にシフトする改変、蛍光強度が増大する改変等を行うことで本発明に係る蛍光タンパク質を得ることができる。このような改変には、野生型ウミサボテンGFPの198番目のヒスチジンをチロシンまたはスレオニンに置換する変異が含まれる。さらに、198番目のヒスチジンをチロシンまたはスレオニンに置換し且つ147番目のセリンをスレオニンに置換する変異が含まれる。
【0014】
本発明に係る蛍光タンパク質に対して、本発明に係る蛍光タンパク質の性質(強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する)をもたらす変異の他に、当該タンパク質の研究等への利用性を高めるような変異を導入することができる。例えば、タンパク質を単量体化する変異または特定の温度で安定化する変異を導入することができる。単量体化する変異とは、野生型蛍光タンパク質が本来の状態で多量体を形成する場合に、そのような多量体の形成を回避して単量体で存在できるようにする変異である。ウミサボテンGFPは、野生型では二量体を形成するが、154番目のシステインをセリンに置換(C154S)することで単量体化させることができる。特定の温度で安定化させる変異とは、その温度下でもタンパク質が望ましい状態を維持するような変異を意味し、特に蛍光タンパク質の場合、例えば、その温度においても蛍光活性を維持する変異を意味する。この変異により、蛍光タンパク質を導入する細胞の培養温度においても、十分な強度の蛍光を発するようになる。ウミサボテンGFPの場合、S129G、D156G、K204IおよびN209Yの4箇所を置換することで、またはN126YおよびY166Fの2箇所を置換することで、37℃において蛍光強度を高い状態で維持する変異体を得ることができる。
【0015】
以下に、本発明に係る蛍光タンパク質の具体的な例を説明する。
1つ目は、野生型のウミサボテンGFPに、198番目のヒスチジンをチロシンに置換する変異、単量体化する変異および温度安定化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198Y、C154S、S129G、D156G、K204IおよびN209Yのアミノ酸置換を導入している(配列番号1)。本願では、この変異体を「A−H198Y変異体」と称する。この変異体は、pH4〜9で516nm付近、pH10〜11で512nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしている。
【0016】
2つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異および単量体化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198TおよびC154Sのアミノ酸置換を導入している(配列番号3)。本願では、この変異体を「H198T変異体」と称する。この変異体は、pH4〜9で515nm付近、pH10〜11で507nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしている。さらに、当該変異体は37℃で培養した場合にも蛍光を発する。よって哺乳細胞などの培養に適した温度条件でも用いることが可能である。
【0017】
3つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異、147番目のセリンをスレオニンに置換する変異および単量体化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198T、S147TおよびC154Sのアミノ酸置換を導入している(配列番号5)。本願では、この変異体を「H198T−S147T変異体」と称する。この変異体は、pH4〜10で515nm付近、pH11で508nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしており、蛍光強度も高まっている。また、H198T変異体がpH10付近で蛍光波長が変化するのに対し、当該変異体はpH11付近で蛍光波長の変化が生じる。
【0018】
4つめは、野生型のウミサボテンGFPに、198番目のヒスチジンをスレオニンに置換する変異、147番目のセリンをスレオニンに置換する変異、単量体化する変異および温度安定化する変異を導入した変異体である。具体的には、野生型ウミサボテンGFPに対して、H198T、S147T、C154S、S129G、D156G、K204IおよびN209Yのアミノ酸置換を導入している(配列番号7)。本願では、この変異体を「A−H198T−S147T変異体」と称する。この変異体は、pH4〜10で514nm付近、pH11で508nm付近の蛍光を示し、野生型ウミサボテンGFPと比べて蛍光波長が長波長側にシフトしており、蛍光強度も高まっている。また、H198T変異体がpH10付近で蛍光波長が変化するのに対し、当該変異体はpH11付近で蛍光波長の変化が生じる。さらに、当該変異体は37℃で培養した場合にも蛍光を発する。よって哺乳細胞などの培養に適した温度条件でも用いることが可能である。
【0019】
本発明に係る蛍光タンパク質により、一般的なGFPよりも長波長側の蛍光色を用いて細胞内現象をイメージングすることが可能となる。特に、野生型ウミサボテンGFPは、酸性側で青色、中性〜アルカリ性側で緑色の蛍光活性を有するため、これらとは異なる蛍光色を利用することができる。また、本発明に係る蛍光タンパク質は、より短波長側の蛍光を発するタンパク質(CFP等)を供与体としたFRET系等の受容体としても用いることができる。さらに、本発明に係る蛍光タンパク質は、pHによって蛍光波長が変化する性質を有するため、細胞内等のpHの測定が可能となる。特に、野生型ウミサボテンGFPとは蛍光色が変化するpHの値が異なるため、本発明に係る蛍光タンパク質と野生型ウミサボテンGFPとを組み合わせて使用することで、より精度の高いpH測定が可能となる。
【0020】
<核酸>
本発明は、本発明に係る蛍光タンパク質をコードする塩基配列を含む核酸に関する。このような塩基配列は、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する塩基配列であってよい。ここにおける「由来する」とは、刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物が本来有する野生型の塩基配列に変異が生じた塩基配列を含むことを意味する。また、ここにおける変異とは、塩基配列中の特定の塩基の置換、欠失および/または付加等を指す。塩基配列の変異には、コードされるアミノ酸配列に変化を生じさせない変異をも含む。また、核酸とは、特に、DNAまたはRNAを指す。
【0021】
本発明に係る核酸の好ましい例は、配列番号2に示される塩基配列を含む核酸(A−H198Y変異体をコード)、配列番号4に示される塩基配列を含む核酸(H198T変異体をコード)、配列番号6に示される塩基配列を含む核酸(H198T−S147T変異体をコード)、または配列番号8に示される塩基配列を含む核酸(A−H198T−S147T変異体をコード)である。
【0022】
また、本発明は、これらの核酸を含むベクターを含む。当該ベクターには、蛍光タンパク質をコードする核酸以外に、発現を調節するための配列またはマーカー遺伝子の配列を含む核酸等を含んでよい。
【0023】
<イメージング方法>
本発明は、本発明に係る蛍光タンパク質または本発明に係る核酸を用いた生物試料のイメージング方法に関する。
【0024】
イメージング方法とは、観察手段を用いて対象を測定または観察することを意味する。その具体的な例は、本発明に係る蛍光タンパク質と特定のタンパク質とを融合させたタンパク質を細胞に発現させ、当該細胞中の動態を蛍光顕微鏡により観察する方法である。
【0025】
生物試料は、バクテリアの細胞、酵母細胞、真菌細胞、昆虫細胞または哺乳細胞といった細胞であってよい。さらに、生物試料は、組織または個体であってよい。細胞は、固定したものであってよいし、生きた状態のものであってよい。固定は、ホルマリン、メタノール等による一般的な方法で行うことができる。また、生物試料としての個体は、ヒトを除く個体であってよい。生物試料とする細胞、組織および個体に特に限定はなく、従来の取得方法によって得られるものを使用することができる。
【0026】
生物試料に蛍光タンパク質を導入するために、細胞等へタンパク質を導入するための当該分野で一般的ないずれの方法を使用してもよい。例えば、生物試料が細胞である場合、マイクロインジェクション法によって蛍光タンパク質を直接細胞内へ注入してもよい。あるいは、蛍光タンパク質の遺伝子を含む発現ベクターを細胞に導入し、適切に発現させることで導入してもよい。発現ベクター等による遺伝子の導入は、リン酸カルシウム法、リポフェクションまたはエレクトロポレーション等を使用することができる。
【0027】
観察手段として、特定の波長の蛍光を検出することが可能な、従来の方法または装置を使用することができる。例えば蛍光顕微鏡を使用することができる。測定または観察とは、顕微鏡を介して目視により確認すること、顕微鏡に設置したカメラを利用して撮像すること等を含む。このような測定および観察は、単一の細胞に対して行うことが可能である。
【0028】
<pH測定法>
本発明は、本発明に係る蛍光タンパク質または本発明に係る核酸を用いた生物試料内のpHを測定する方法に関する。
【0029】
例えば、本発明に係る蛍光タンパク質を対象に導入し、前記蛍光タンパク質が発する蛍光に基づいて前記対象内部のpHを測定する方法に関する。すなわち、本発明に係る方法では、対象の内部に蛍光タンパク質を導入した後、励起光を照射して蛍光タンパク質が発する蛍光を検出し、当該蛍光の波長に基づいてpHを特定する。
【0030】
生物試料としては、イメージング方法と同様な試料を使用することができる。
【0031】
蛍光の検出は、目視で行うことも可能であるが、特定の波長の蛍光を測定することが可能な、従来の方法または装置を使用して行うことができる。例えば蛍光顕微鏡を使用することができる。励起光を照射して蛍光の色を目視で確認してpHを判断してもよいが、検出装置を用いることで、より詳細にpHを特定することが可能となる。例えば、蛍光タンパク質が発する2種の蛍光に対応したフィルターセットの蛍光キューブを倒立型蛍光顕微鏡に設置し、それぞれの波長の蛍光画像をCCDカメラによって撮像する。撮像した画像内のpH測定対象領域の蛍光強度を数値化しpHを求めることができる。このとき、細胞を種々の条件で刺激し、その後一定時間ごとに撮像することで、系時的に蛍光画像を取得することもできる。また、特定の波長だけの測定ではなく、蛍光スペクトルを測定することもできる。この場合、具体的には、顕微鏡のカメラポートに光ファイバーを接続し分光光度計にて測定対象領域の蛍光スペクトルを測定することで行うことができる。
【実施例】
【0032】
[実施例1:野生型ウミサボテン蛍光タンパク遺伝子のクローニング]
ウミサボテン(Cavernularia obesa)から野生型蛍光タンパク質を抽出および精製し、その部分的なアミノ酸配列を読み取った後、当該配列をもとに遺伝子配列を決定することで蛍光タンパク質遺伝子のクローニングを行った。
【0033】
蛍光タンパク質の抽出精製およびアミノ酸配列分析
島根近海で採取したウミサボテン15個体(約200g)を500mlのSDS−グリシンバッファー中ですり潰し、蛍光活性を持つタンパクを含む可溶性タンパクを抽出した。遠心機を用いて残渣を取り除き、蛍光活性を持つタンパク質を含む溶液を抽出した。この抽出溶液に、最終濃度が80%になるように硫酸アンモニウムを加えて硫安沈殿を行った。具体的には、抽出溶液500mlに262gの硫酸アンモニウムを加えた。得られた沈殿を、50mlのトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)に再溶解させた。これに2倍量のエタノールを加え、タンパク質を再度沈殿させた。さらに、この沈殿にトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を加えて再溶解させた。この溶液50mlを、十分量のトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)および透析膜27/32(三光純薬株式会社)を用いて透析した。透析後の溶液を、DEAE SepharoseCL−6Bカラム(GEヘルスケアバイオサイエンス)を装着したクロマトグラフィーシステムAKTAexplorer(GEヘルスケアバイオサイエンス)を用いてイオン交換分離精製した。このイオン交換は、低塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)で十分に平衡化した後、サンプルを添加し、高塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、1M NaCl)でNaCl濃度を0.4Mまで上昇させることで行い、その結果、蛍光活性のあるフラクションを分取した。蛍光活性を持つタンパクを含むフラクションの選択は、UV/BLUE CONVERTER PLATE(UVP)を用いて行った。得られたフラクションを、十分量のトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)および透析膜27/32(三光純薬株式会社)を用いて透析した。透析後の溶液を、限外ろ過アミコンウルトラ:分画分子量30kDa(日本ミリポア株式会社)を用いて濃縮した。次に、Sephacryl S−200 High Resolution(GEヘルスケアバイオサイエンス)およびトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を用いてゲルろ過を行った。ゲルろ過後、得られた蛍光活性のあるフラクションをmonoQ 5/50(GEヘルスケアバイオサイエンス)によってイオン交換分離した。このイオン交換は、低塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)で十分に平衡化した後、サンプルを添加し、高塩濃度トリスバッファー(20mM Tris−HCl(pH7.0)、1M NaCl)でNaCl濃度を0.4Mまで上昇させることで行い、その結果、蛍光活性のあるフラクションを得た。この活性のあるフラクションを限外ろ過アミコンウルトラ:分画分子量30kDa(日本ミリポア株式会社)を用いて濃縮し、その後、Superdex 75 10/300 GL(GEヘルスケアバイオサイエンス)およびトリスバッファー(20mM Tris−HCl(pH7.0)、20mM NaCl)を用いてゲルろ過分離を行い、蛍光活性のあるフラクションを得た。このサンプルをSDS−ポリアクリルアミドゲル電気泳動によって分離してアミノ酸配列を分析した。分析の結果、22残基のアミノ酸配列(IPD YFV QSF PEG FTF ERT LSF E:配列番号17)を決定することができた。
【0034】
蛍光タンパク質遺伝子のクローニング
蛍光タンパク質遺伝子全長のクローニングのために、3’−RACE PCRを下記のとおり実施した。Rapid Amplification of cDNA End法(以下、RACEと略す)により、蛍光タンパク質遺伝子をクローニングするための混合プライマーを作成した。アミノ酸配列解析によって得られたアミノ酸配列IPD YFV QSF PEG FTF ERT LSF E(配列番号17)のうち、コドンの塩基組み合わせが少ないIPDYFVおよびEGFTFERのアミノ酸領域に注目した。これらのアミノ酸領域をコードする塩基配列を予測し、3’末端RACE polymerase chain reaction(以下PCRと略す)に用いる蛍光タンパク質特異的混合プライマー(合計12種類)を以下のように作成した。IPDYFVアミノ酸領域に結合するプライマーとして、COGFP−TTT(5’−ATH CCN GAT TAT TTT GT−3’)(配列番号18)、COGFP−TTC(5’−ATH CCN GAT TAT TTC GT−3’)(配列番号19)、COGFP−TCT(5’−ATH CCN GAT TAC TTT GT−3’)(配列番号20)、COGFP−TCC(5’−ATH CCN GAT TAC TTC GT−3’)(配列番号21)、COGFP−CTT(5’−ATH CCN GAC TAT TTT GT−3’)(配列番号22)、COGFP−CTC(5’−ATH CCN GAC TAT TTC GT−3’)(配列番号23)、COGFP−CCT(5’−ATH CCN GAC TAC TTT GT−3’)(配列番号24)およびCOGFP−CCC(5’−ATH CCN GAC TAC TTC GT−3’)(配列番号25)を作成し、ならびに、EGFTFERアミノ酸領域に結合するプライマーとして、COGFP−ATTAA(5’−GAA GGN TTT ACN TTT GAA AG−3’)(配列番号26)、COGFP−ACCAA(5’−GAA GGN TTC ACN TTC GAA AG−3’)(配列番号27)、COGFP−ATTGA(5’−GAG GGN TTT ACN TTT GAG AG−3’)(配列番号28)およびCOGFP−ACCGA(5’−GAG GGN TTC ACN TTC GAG AG−3’)(配列番号29)を作成した。プライマー中のHおよびNは、混合塩基を示す。
【0035】
完全長cDNA合成試薬GeneRacer(インビトロジェン)を用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、蛍光タンパク質のアミノ酸配列から予測して作成した12種類の特異的混合プライマーおよび3’末端特異的プライマーであるGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’)(配列番号30)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’)(配列番号31)を用いて3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれており、これを使用した。3’−RACE PCRによって効果的に蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型とし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRは、ポリメラーゼEx−Taq(タカラバイオ株式会社)を用いて、マニュアルに従って実施した。
【0036】
一度目のPCRは、IPDYFVアミノ酸領域で作成した8種類プライマー(COGFP−TTT、COGFP−TTC、COGFP−TCT、COGFP−TCC、COGFP−CTT、COGFP−CTC、COGFP−CCT、COGFP−CCC)のいずれかとGeneRacer3’ Primerとの計8つのプライマー対で蛍光タンパク質遺伝子の増幅を行った。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍、dNTP Mixture(各2.5mM)を最終濃度各0.2mM、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μl、8種類のプライマーのうちの1つを最終濃度0.4μMおよびGeneRacer3’Primerを最終濃度0.4μMとして20μlのPCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μl加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%トリス酢酸緩衝液(以下、TAEと略す)アガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。8つの反応溶液でわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0037】
nested PCRは、EGFTFERアミノ酸領域で作成した4種類プライマー(COGFP−ATTAA、COGFP−ACCAA、COGFP−ATTGA、COGFP−ACCGA)の何れかとGeneRacer3’Nested Primerとの計4つのプライマー対で蛍光タンパク質遺伝子の増幅を行った。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍、dNTP Mixture(各2.5mM)を最終濃度各0.2mM、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μl、4種類のプライマーのうちの1つを最終濃度0.4μMおよびGeneRacer3’ Nested Primerを最終濃度0.4μMとして10μlのnested PCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0038】
その結果、1度目のPCR反応として、COGFP−TTCとGeneRacer3’ Primerとのプライマー対を用いて実施し、nested PCR反応として、得られたPCR反応溶液を鋳型としてCOGFP−ACCAAとGeneRacer3’ Nested Primerとのプライマー対を用いて実施した場合に、顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定し、ウミサボテン蛍光タンパク質遺伝子のウミサボテン3’末端側の塩基配列(配列番号32)とした。
【0039】
蛍光タンパク質の5’末端クローニングのための5’−RACEを下記の通り行った。一連の3’−RACE解析によって得られたウミサボテン蛍光タンパク質遺伝子の3’末端側の塩基配列をもとに、5’末端クローニングのための5’−RACEに用いるプライマーを作成した。作成した5’末端クローニング用のプライマーは、COGFP−A−R1(5’−GCT ATA GCC GTC TCA TGT TGC TCG T−3’)(配列番号33)、COGFP−A−R2(5’−AGC CGT CTC ATG TTG CTC GTA GTA G−3’)(配列番号34)およびCOGFP−A−R3(5’−ATG TTG CTC GTA GTA GTT GCC TTC CTC GAC−3’)(配列番号35)の3種類である。COGFP−A−R1、COGFP−A−R2、COGFP−A−R3の順で5’末端に近い位置に結合し、順次nested PCR反応のプライマーとして用いる。
【0040】
完全長cDNA合成試薬GeneRacerを用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、3種類の5’末端クローニング用のプライマーならびに5’末端特異的プライマーであるGeneRacer5’ Primer(5’−CGA CTG GAG CAC GAG GAC ACT GA−3’)(配列番号36)およびGeneRacer5’ Nested Primer(5’−GGA CAC TGA CAT GGA CTG AAG GAG TA−3’)(配列番号37)を用いて、5’−RACE PCRを行った。GeneRacer5’ PrimerおよびGeneRacer5’ Nested Primerは完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれており、これを使用した。5’−RACE PCRによって効果的に蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taqを用いて、マニュアルに従って実施した。
【0041】
一度目の5’−RACE PCRとして、3’−RACEで増幅した遺伝子の塩基配列をもとに作成したCOGFP−A−R1とGeneRacer5’Primerとのプライマー対を用いて、蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R1を最終濃度0.4μMとし、GeneRacer5’ Primerを最終濃度0.4μMとして、10μlのPCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。この5’−RACEでわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0042】
nested PCRとして、COGFP−A−R2とGeneRacer5’ Nested Primerとのプライマー対を用いて蛍光タンパク質GFP遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R2を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、10μlのPCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。
【0043】
その結果、1度目のPCR反応として、COGFP−A−R1とGeneRacer5’ Primerとのプライマー対を用いて実施し、nested PCR反応として、得られたPCR反応溶液を鋳型としてCOGFP−A−R2とGeneRacer5’ Nested Primerとのプライマー対を用いて実施した場合に、顕著な遺伝子増幅を確認できた。しかし、非特異的な複数の遺伝子の増幅が認められたため、このPCR反応溶液を鋳型として、異なるプライマー対を用いて再度nested PCR反応を実施した。
【0044】
二度目のnested PCRとして、COGFP−A−R3とGeneRacer5’ Nested Primerとのプライマー対を用いて、GFP遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−R3を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、20μlのnested PCR反応溶液を作製し、1度目のnested PCR反応溶液を鋳型として0.4μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、45℃30秒および72℃2分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを、1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。その結果、一連のnested PCR反応で顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定し、ウミサボテン蛍光タンパク質遺伝子の5’末端側の塩基配列(配列番号38)とした。
【0045】
次に、蛍光タンパク質遺伝子の完全長の増幅を下記のとおりに行った。上述した3’−RACEおよび5’−RACE解析によって得られたウミサボテン由来の蛍光タンパク質遺伝子の5’末端側の塩基配列をもとに、完全長蛍光タンパク質遺伝子クローニングのための3’−RACEに用いるプライマーを作成した。作成した完全長GFP遺伝子クローニング用のプライマーは、COGFP−A−Full−F3(5’−ATT TAG GTG GCT GCG TAC AG−3’)(配列番号39)、COGFP−A−Full−F4(5’−ATT TAG GTG GCT GCG TAC AGT TAA CAC−3’)(配列番号40)の2種類である。COGFP−A−Full−F3、COGFP−A−Full−F4の順で3’末端に近い位置に結合する。COGFP−A−Full−F4をnested PCR反応のプライマーとして用いる。
【0046】
完全長cDNA合成試薬GeneRacerを用いて作成したウミサボテン完全長cDNAライブラリーを鋳型とし、2種類の完全長蛍光タンパク質遺伝子クローニング用のプライマーならびに3’末端特異的プライマーであるGeneRacer3’ Primer(5’−GCT GTC AAC GAT ACG CTA CGT AAC G−3’)(配列番号30)およびGeneRacer3’ Nested Primer(5’−CGC TAC GTA ACG GCA TGA CAG TG−3’)(配列番号31)を用いて、3’−RACE PCRを行った。GeneRacer3’ PrimerおよびGeneRacer3’ Nested Primerは、完全長cDNA合成試薬GeneRacerキット(インビトロジェン社)に含まれているので、これを使用した。3’−RACE PCRによって効果的に完全長蛍光タンパク質遺伝子を増幅させるため、一度PCRによって増幅した遺伝子を鋳型にし、内側のプライマー対でさらに特異的に遺伝子増幅させるnested PCRを行った。PCRにはポリメラーゼEx−Taqを用いてマニュアルに従って実施した。
【0047】
一度目のPCRとして、COGFP−A−Full−F3とGeneRacer3’ Primerとのプライマー対を用いて蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−Full−F3を最終濃度0.4μMとし、GeneRacer3’ Primerを最終濃度0.4μMとして、10μlのnested PCR反応溶液を作製し、ウミサボテン完全長cDNAライブラリー溶液を0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、50℃30秒および72℃1分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。一度目の5’−RACEでわずかに遺伝子増幅が認められたため、このPCR反応溶液を鋳型としてnested PCR反応を実施した。
【0048】
nested PCRとしてCOGFP−A−Full−F4とGeneRacer5’ Nested Primerとのプライマー対を用いて、蛍光タンパク質遺伝子を増幅した。10×Ex Taq Buffer(20mM Mg2+添加)を最終濃度等倍とし、dNTP Mixture(各2.5mM)を最終濃度各0.2mMとし、TaKaRa Ex Taq(5U/μl)を最終濃度0.05U/μlとし、COGFP−A−Full−F4を最終濃度0.4μMとし、GeneRacer5’ Nested Primerを最終濃度0.4μMとして、10μlのnested PCR反応溶液を作製し、1度目のPCR反応溶液を鋳型として0.2μlを加えた。PCR反応条件として、最初に94℃1分間の熱変性を行い、次に、94℃30秒、50℃30秒および72℃1分のサイクルを30回行い、最後に、72℃5分間の伸長反応を行った。PCR反応後、PCR反応溶液2μlを1%TAEアガロースゲルを用いて電気泳動し、エチジウムブロマイドで染色後、紫外線照射下で増幅遺伝子のバンドを観察した。その結果、顕著な遺伝子増幅を確認できた。この増幅した遺伝子の塩基配列を決定して、完全長ウミサボテン蛍光タンパク質遺伝子の塩基配列(配列番号41)とした。
【0049】
得られた完全長ウミサボテン蛍光タンパク質の塩基配列から、配列情報解析ソフトウェアDNASIS Proを用いてウミサボテンGFP遺伝子のオープンリーディングフレームの塩基配列を予想した(配列番号10)。さらに、このオープンリーディングフレームを翻訳してウミサボテン蛍光タンパク質のアミノ酸配列を得た(配列番号9)。このアミノ酸配列は、ウミサボテンから精製し、アミノ酸分析によって得られた22残基の配列(配列番号17)を完全一致の状態で含むことから、ウミサボテンに由来する野生型蛍光タンパク質遺伝子であると決定した。この遺伝子をpRSETベクター(インビトロジェン)に組み込み、大腸菌にトランスフォーメーションし、UV/BLUE CONVERTER PLATE(UVP)を用いて観察したところ顕著な蛍光活性が示された。
【0050】
[実施例2:C154S変異(単量体化)の導入]
実施例1にて得られた野生型ウミサボテンGFPの遺伝子に対して、C154S変異を導入した。この変異を導入することで、野生型ウミサボテンGFPを単量体化することができる。
【0051】
以下の方法によって、野生型蛍光タンパク質のアミノ酸配列の154番目のシステインをセリンに置換した。変異の導入には、QuickChange II Site−Directed Mutagenesis Kit (Stratagene社)を用いた。pRSET−A(インビトロジェン社)にウミサボテン蛍光タンパク質遺伝子をクローニングしたプラスミドを鋳型とし、Pirmer1(CAATGTATGTATCGGACGACACTTTGG)(配列番号42)およびPrimer2(CCAAAGTGTCGTCCGATACATACATTG)(配列番号43)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、37度にてDpnIで処理し、大腸菌JM109(DE3)株にトランスフォームした。
【0052】
トランスフォームされた菌を培養してベクターを取り出し、蛍光タンパク質遺伝子の塩基配列をシーケンサーによって読み取った。その結果、配列番号14の配列が得られ、狙い通りに変異が導入できたことが確認できた。この遺伝子から、154番目がセリンに置換された蛍光タンパク質(配列番号11)が発現する。
【0053】
また、トランスフォーム後の大腸菌を、28度にて培養した後、溶菌してライセートを作製した。これにSDS−PAGEサンプルバッファーを加え、熱を加えずにサンプルを調製して、SDS−PAGEを行った。その結果、野生型蛍光タンパク質のレーンで出現する2量体のバンドが、変異体蛍光タンパク質のレーンで消失していた。このことから、蛍光タンパク質が単量体化されたことが確認された。
【0054】
[実施例3:温度安定化変異の導入]
温度安定化変異の導入について以下に述べる。具体的には、S129G、D156G、K204IおよびN209Yの4箇所が変異した変異体を得ることができる。これらの変異の導入よって、37℃において発色団形成を安定化させることができる。
【0055】
実施例2で得られた単量体化した変異体蛍光タンパク質に対してランダムに変異を導入し、37℃において発色団形成が安定化する変異体をスクリーニングした。
【0056】
変異の導入は、GeneMorph II EZClone Domain Mutagenesis Kit(Stratagene社)を用いた。pRSET−A(インビトロジェン社)に単量体化変異体(154番目のシステインがセリンに置換)の遺伝子がクローニングされたプラスミドを鋳型とし、Pirmer1(ATGAGTATTCCAGAGAATTCGGGCTTAACAG)(配列番号44)およびPrimer2(TCATGGTTTAGCTATGGCCGTCTCATG)(配列番号45)を加えて、キットに添付されているマニュアルに準じてPCR反応を行った。PCR反応後、1%アガロースゲルを用いて電気泳動を行い、目的とするPCR産物をWizard SV Gel and PCR Clean−Up(Promega社)を用いて精製した。精製後、マニュアルに準じてPCR反応を行い、37℃にてDpnIで処理した後に、エタノール沈殿を行った。沈殿後、少量の蒸留水にDNAを溶かして、MicroPulser(BioRad社)を用いてエレクトロポレーション法にてJM109(DE3)にトランスフォームした。トランスフォーム後の大腸菌を、25cm四方のLB(50ng/mlのアンピシリンを添加済み)プレートに播種して、37℃で培養してコロニーを形成させた。コロニー形成後、UV/BLUE CONVERTER PLATE(UVP)を使用して、野生型蛍光タンパク質を発現するコロニーと比べて、強い蛍光を発するコロニー(それぞれ変異体1および変異体2と名付ける)を2つ選択し、再度培養した。その後、それらが保持するプラスミドの塩基配列を決定した。それぞれの配列は配列番号15および配列番号16に示される。
【0057】
さらに、これらの塩基配列をアミノ酸配列に変換し(変異体1は配列番号12、変異体2は配列番号13)、野生型のアミノ酸配列と比較した。変異体1は、126番目のアスパラギンがチロシンに置換され、154番目のシステインがセリンに置換され、166番目のチロシンがフェニルアラニンに置換されていることがわかった。変異体2は、129番目のセリンがグリシンに置換され、154番目のシステインがセリンに置換され、156番目のアスパラギン酸がグリシンに置換され、204番目のリジンがイソロイシンに置換され、209番目のアスパラギンがチロシンに置換されていることがわかった。
【0058】
また、変異体1および2の37℃における発色団形成の安定性を、野生型蛍光タンパク質および単量体化蛍光タンパク質と比較した。pRSET−A(インビトロジェン社)に、野生型遺伝子、実施例2で作製した単量体化変異体の遺伝子、変異体1の遺伝子および変異体2の遺伝子をそれぞれクローニングし、それぞれJM109(DE3)株にトランスフォームした。これらの株をプレートに塗布し、37℃で培養して蛍光タンパク質を発現させた。その状態をUV/BLUE CONVERTER PLATE(UVP)で観察して、画像を撮影した。その結果、野生型および単量体化変異体と比較して、変異体1および2は非常に強い蛍光を発していた。特に、変異体1に比べて、変異体2のほうが強い蛍光を発していた。
【0059】
次に、ゲルろ過クロマトグラフィーにて、蛍光タンパク質の会合状態を調べた。野生型蛍光タンパク質を発現する大腸菌および変異体2を発現する大腸菌から、それぞれ蛍光タンパク質を精製し解析した。野生型では66kDal付近にピークが得られ、一方、変異体2では37kDal付近でピークが得られた。これらの結果から、野生型蛍光タンパク質が2量体を形成しているのに対し、変異体2の蛍光タンパク質が単量体を形成していることが示唆された。
【0060】
[実施例4:H198Y変異の導入]
実施例3で得られた遺伝子に対し、以下に述べるH198Yのアミノ酸置換の変異を導入し、A−H198Y変異体の遺伝子を得た。この変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0061】
pRSET−B(インビトロジェン社)に、変異体2(129番目のセリンがグリシンに置換、154番目のシステインがセリンに置換、156番目のアスパラギン酸がグリシンに置換、204番目のリジンがイソロイシンに置換、209番目のアスパラギンがチロシンに置換)の遺伝子を挿入したプラスミドを鋳型として、H198Yプライマー1(CATTATGTTCATTATCGCCTCGAAAAG、配列番号46)およびH198Yプライマー2(CTTTTCGAGGCGATAATGAACATAATG、配列番号47)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0062】
[実施例5:H198T変異の導入]
実施例2で得られた遺伝子に対し、以下に述べるH198Tのアミノ酸置換の変異を導入し、H198T変異体の遺伝子を得た。この変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0063】
pRSET−B(インビトロジェン社)に、単量体化変異体(154番目のシステインがセリンに置換)の遺伝子を挿入したプラスミドを鋳型として、H198Tプライマー1(CATTATGTTCATACCCGCCTCGAAAAG、配列番号48)およびH198Tプライマー2(CTTTTCGAGGCGGGTATGAACATAATG、配列番号49)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0064】
[実施例6:S147T変異の導入]
実施例5で得られた遺伝子に対し、以下に述べるS147Tのアミノ酸置換の変異を導入し、H198T−S147T変異体の遺伝子を得た。また、実施例3で得られた遺伝子に対し実施例5に記載の通りにH198T変異を導入した後、さらに以下に述べるS147Tのアミノ酸置換の変異を導入することで、A−H198T−S147T変異体の遺伝子を得た。これらの変異は、Quick Change II Site−Directed Mutagenesis Kit(Stratagene社)を用いて導入した。
【0065】
pRSET−B(インビトロジェン社)に、H198T変異体(198番目のヒスチジンがスレオニンに置換および154番目のシステインがセリンに置換)の遺伝子を挿入したプラスミド、または単量体化および温度安定化され、さらにH198T変異が導入された変異体(198番目のヒスチジンがスレオニン、154番目のシステインがセリンに置換、129番目のセリンがグリシンに置換、156番目のアスパラギン酸がグリシンに置換、204番目のリジンがイソロイシンに置換および209番目のアスパラギンがチロシンに置換)の遺伝子を挿入したプラスミドを鋳型として、S147Tプライマー1(CCAAACTAGAACCGACCAGTGAGTCAATG、配列番号50)およびS147Tプライマー2(CATTGACTCACTGGTCGGTTCTAGTTTGG、配列番号51)を使用して、キットに添付されているマニュアルに準じてPCR反応を行った。反応後、反応産物にDpnIを加えて37度で処理した後に、大腸菌JM109(DE3)株に形質転換した。形質転換された大腸菌を増殖させてプラスミドDNAを精製し、目的の変異が導入された遺伝子が作製されたことを確認した。
【0066】
[実施例7:pHによる蛍光の変化並びに蛍光および吸収スペクトルの測定]
A−H198Y変異体、H198T変異体、H198T−S147T変異体およびA−H198T−S147T変異体の4種の変異型蛍光タンパク質について、pHによる蛍光の変化並びに蛍光スペクトルおよび吸収スペクトルをそれぞれ測定した。
【0067】
実施例4から6により取得した、4種の変異体の内いずれかの遺伝子を有するプラスミドを、大腸菌に導入し、37℃で培養して各種変異型蛍光タンパク質を発現させ、溶菌後、Ni−NTA(Quiagen社)カラムクロマトグラフィーにより蛍光タンパク質をそれぞれ精製した。
【0068】
精製された変異体タンパク質を異なるpHの溶液に添加し、UVハンディライト(365nm)を用いて蛍光色を確認した。また、分光光度計(HITACHI U−3010)および蛍光光度計(HITACHI F−2500)を用いて、吸収スペクトルおよび蛍光スペクトルを測定した。
【0069】
以下に、それぞれの変異体の測定結果を説明する。
(A−H198Y変異体)
図1にpHによる蛍光の変化の結果をモノクロ画像として示し、図2に蛍光スペクトルおよび吸収スペクトルを示す。
【0070】
図1中、暗視野画像はUVハンディライトを照射したときに発せられる蛍光の画像であり、明視野画像は通常の照明下で確認される画像である。それぞれの上段のチューブには野生型ウミサボテンGFPが、下段のチューブにはA−H198Y変異体が、各pHの緩衝液とともに添加されている。なお、後述する図3、5および7についても同様に、上段に野生型が、下段に変異型がそれぞれ配置される。
【0071】
図1の元となるカラー画像によれば、野生型ウミサボテンGFPでは、pH4の環境下で青色の蛍光を発し、pH6以上の環境下で緑色の蛍光を発し、pH5において中間的な蛍光を発した。また、明視野において、pH4および5の環境下で緩衝液は透明であり、pH6の環境下で黄緑色を示し、pH7以上の環境下で緑色を示した。
【0072】
これに対し、A−H198Y変異体では、pH5から10の環境下で黄色の蛍光を発し、pH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から10の環境下で緩衝液は赤みがかった黄色を示し、pH11の環境下で黄色を示した。
【0073】
図2には、A−H198Y変異体の吸収スペクトルおよび蛍光スペクトル(470nm励起)が示される。吸収スペクトルでは、全体として400nm付近および500nm付近の2つのピークを示した。特に、pH4から5で403nm付近のピークが高く、pH6から10で509nm付近のピークが高く、pH11では500nm付近のピークが高かった。蛍光スペクトルでは、pH4から9で517nm付近にピークを示し、pH10および11で510nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表1にまとめる。
【0074】
【表1】
【0075】
(H198T変異体)
図3にpHによる蛍光の変化の結果をモノクロ画像として示し、図4に蛍光スペクトルおよび吸収スペクトルを示す。
【0076】
図3の元となるカラー画像によれば、H198T変異体は、pH4から9の環境下で黄色の蛍光を発し、pH10および11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から9の環境下で緩衝液は赤みがかった黄色を示し、pH10および11の環境下で黄緑色を示した。
【0077】
図4には、H198T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルは、pH4から10で507nm付近のピークを、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から9で515nm付近にピークを示し、pH10および11で507nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表2にまとめる。
【0078】
【表2】
【0079】
(H198T−S147T変異体)
図5にpHによる蛍光の変化の結果をモノクロ画像として示し、図6に蛍光スペクトルおよび吸収スペクトルを示す。
【0080】
図5の元となるカラー画像によれば、H198T−S147T変異体では、pH4から9の環境下で黄色の蛍光を発し、pH10およびpH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から9の環境下で緩衝液はオレンジよりの黄色を示し、pH10の環境下で黄色、pH11の環境下で黄緑色を示した。
【0081】
図6には、H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルでは、pH4から10で507nm付近のピークを示し、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から10で515nm付近にピークを示し、pH11で508nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。また、pH7における蛍光強度を同濃度の野生型ウミサボテンGFPと比較した場合、H198T−S147T変異体の方が約1.6倍高かった。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表3にまとめる。
【0082】
【表3】
【0083】
(A−H198T−S147T変異体)
図7にpHによる蛍光の変化の結果をモノクロ画像として示し、図8に蛍光スペクトルおよび吸収スペクトルを示す。
【0084】
図7の元となるカラー画像によれば、A−H198T−S147T変異体では、pH4から10の環境下で黄色の蛍光を発し、pH11の環境下で黄緑色の蛍光を発した。また、明視野において、pH4から10の環境下で緩衝液はオレンジよりの黄色を示し、pH11の環境下で黄緑色を示した。
【0085】
図8には、A−H198T−S147T変異体の吸収スペクトルおよび蛍光スペクトル(490nm励起)が示される。吸収スペクトルでは、pH4から10で507nm付近のピークを示し、pH11で498nm付近のピークを示した。蛍光スペクトルでは、pH4から10で514nm付近にピークを示し、pH11で508nm付近にピークを示した。蛍光強度は、pHの値が大きいほど高い傾向を示した。また、pH7における蛍光強度を同濃度の野生型ウミサボテンGFPと比較した場合、A−H198T−S147T変異体の方が約1.8倍高かった。それぞれのスペクトルについて、pHごとに最大強度を示した波長を以下の表4にまとめる。
【0086】
【表4】
【0087】
[実施例8:蛍光強度の比較]
4種の変異型蛍光タンパク質の蛍光強度を確認した。
【0088】
1:野生型ウミサボテンGFP、2:C154S変異体(単量体化のみを行った変異体)、3:H198Y変異体、4:H198W変異体、5:A−H198Y変異体、6:H198T変異体、7:H198T−S147T変異体および8:A−H198T−S147T変異体について、それぞれの遺伝子を形質転換させた8つの大腸菌を2つのプレートに塗布し、一方を常温で培養し、他方を37℃にて培養して、各蛍光タンパク質を発現させた。各プレートをUV/BLUE CONVERTER PLATE(UVP)で観察して、画像を撮影した。図9は、それらをモノクロで表した画像である。
【0089】
3:H198Y変異体および4:H198W変異体は、先行技術に倣って作製した。非特許文献1によれば、野生型オワンクラゲGFPの203番目のスレオニンをチロシンに置換することでYFPを作製している。この203番目のアミノ酸に立体構造上対応するウミサボテンGFPの198番目のアミノ酸について、先行技術と同様にチロシンまたはトリプトファンに置換した。
【0090】
図9の常温培養の結果によると、1:野生型および2:C154S変異体は緑色の蛍光を示した。特に、2:C154S変異体の方が強い蛍光を示した。3:H198Y変異体および4:H198W変異体は蛍光を示さなかった。本発明に係る5から8の変異体は黄色から黄緑色の蛍光を示した。特に、6から8の変異体は蛍光が強かった。
【0091】
37℃培養の結果によると、1:野生型および2:C154S変異体は常温培養の場合より弱いものの同色の蛍光を示した。また、3:H198Y変異体、4:H198W変異体、5:A−H198Y変異体および7:H198T−S147T変異体は蛍光を示さなかった。これらに対し、6:H198T変異体および8:A−H198T−S147T変異体は常温培養の場合よりも弱いものの同色の蛍光を示した。
【特許請求の範囲】
【請求項1】
強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質。
【請求項2】
刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する請求項1に記載の蛍光タンパク質。
【請求項3】
pH10〜11の環境下で512nm付近の蛍光活性を有し、pH4〜9の環境下で516nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項4】
pH10〜11の環境下で507nm付近の蛍光活性を有し、pH4〜9の環境下で515nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項5】
pH11の環境下で508nm付近の蛍光活性を有し、pH4〜10の環境下で515nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項6】
pH11の環境下で508nm付近の蛍光活性を有し、pH4〜10の環境下で514nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項7】
配列番号1、3、5または7に示されたアミノ酸配列を含む請求項1に記載の蛍光タンパク質。
【請求項8】
請求項1から7の何れか1項に記載の蛍光タンパク質をコードする塩基配列を含む核酸。
【請求項9】
前記塩基配列が配列番号2、4、6または8に示された塩基配列である請求項8に記載の核酸。
【請求項10】
請求項1から7の何れか1項に記載の蛍光タンパク質または請求項8または9に記載の核酸を用いた生物試料のイメージング方法。
【請求項11】
請求項1から7の何れか1項に記載の蛍光タンパク質または請求項8または9に記載の核酸を用いた生物試料内のpHを測定する方法。
【請求項1】
強アルカリ性環境下で緑色蛍光を発し、弱アルカリ性〜酸性環境下で前記緑色蛍光よりも長波長の蛍光を発する蛍光タンパク質。
【請求項2】
刺胞動物門花虫綱ウミエラ目ウミサボテン科に属す生物に由来する請求項1に記載の蛍光タンパク質。
【請求項3】
pH10〜11の環境下で512nm付近の蛍光活性を有し、pH4〜9の環境下で516nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項4】
pH10〜11の環境下で507nm付近の蛍光活性を有し、pH4〜9の環境下で515nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項5】
pH11の環境下で508nm付近の蛍光活性を有し、pH4〜10の環境下で515nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項6】
pH11の環境下で508nm付近の蛍光活性を有し、pH4〜10の環境下で514nm付近の蛍光活性を有する、請求項1または2に記載の蛍光タンパク質。
【請求項7】
配列番号1、3、5または7に示されたアミノ酸配列を含む請求項1に記載の蛍光タンパク質。
【請求項8】
請求項1から7の何れか1項に記載の蛍光タンパク質をコードする塩基配列を含む核酸。
【請求項9】
前記塩基配列が配列番号2、4、6または8に示された塩基配列である請求項8に記載の核酸。
【請求項10】
請求項1から7の何れか1項に記載の蛍光タンパク質または請求項8または9に記載の核酸を用いた生物試料のイメージング方法。
【請求項11】
請求項1から7の何れか1項に記載の蛍光タンパク質または請求項8または9に記載の核酸を用いた生物試料内のpHを測定する方法。
【図1】


【図2】
【図3】

【図4】
【図5】

【図6】
【図7】

【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−105589(P2012−105589A)
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願番号】特願2010−256999(P2010−256999)
【出願日】平成22年11月17日(2010.11.17)
【出願人】(000000376)オリンパス株式会社 (11,466)
【Fターム(参考)】
【公開日】平成24年6月7日(2012.6.7)
【国際特許分類】
【出願日】平成22年11月17日(2010.11.17)
【出願人】(000000376)オリンパス株式会社 (11,466)
【Fターム(参考)】
[ Back to top ]