
蛍光共鳴エネルギー移動酵素基質
式(I)の化合物が開示されている。
【化1】

式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、Mは、D1の蛍光特性を調節するように選択される酵素切断可能基である。式(I)の化合物は、蛍光共鳴エネルギー移動を利用するアッセイにおいて生化学的切断現象を検出するためのレポーター分子として使用することができる。
【化1】
式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、Mは、D1の蛍光特性を調節するように選択される酵素切断可能基である。式(I)の化合物は、蛍光共鳴エネルギー移動を利用するアッセイにおいて生化学的切断現象を検出するためのレポーター分子として使用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、蛍光ベースのアッセイに関し、また酵素活性を測定するための試薬及び方法、特に酵素切断アッセイに関する。
【背景技術】
【0002】
酵素活性、特に加水分解などの酵素切断活性を測定するためのアッセイは、生物科学及び薬学において広く利用される。コンビナトリアルケミストリー及びハイスループットスクリーニングの到来とともに、酵素活性の潜在的なモジュレータをスクリーニングするための簡単で、感度が高く、かつ費用効率の高いアッセイの必要性が高まっている。製薬業界が特に関心があるのは、タンパク分解酵素による切断及びリン酸の切断を検出するための方法である。
【0003】
蛍光ベースのアッセイは、扱いやすさ、感度、コスト及び自動化の容易さの点で、酵素の切断活性を測定するための放射化学法、ELISA、抗体法及びより伝統的な技法に勝る有意な利点を提供する。蛍光原基質を均一系酵素アッセイに定期的に使用して、酵素活性を、あるいは創薬における潜在的な薬物の酵素活性への作用を決定する。これらの酵素の多くの基質が市販され、又は文献で報告されている。例えば、米国特許第4812409号(Babb,B.,et al)は、フェナレノン又はベンズフェナレノンから誘導されるブロックされた色素部分を含んでなる加水分解性蛍光基質を開示している。これらの色素部分は、酵素加水分解中に基質から切断されると、約530nmより上の蛍光を有する蛍光色素を形成する。加水分解酵素に対応する基質も、フルオレセイン及びローダミンの誘導体を基に入手可能である。例えば、フルオレセイン二リン酸は、アルカリホスファターゼ(PP2A)用の、広く使用されている無色の非蛍光基質であり、この酵素によって加水分解されると、フルオレセインを形成する(Vieytes M.,et al,Anal.Biochem.,(1997),248,258−64)。しかしながら、フルオレセインに基づく基質には、通常2つの酵素切断可能部位が組み込まれており、二相の酵素反応速度が、切断を通して、まずモノ置換蛍光類似体に対して、次いで遊離蛍光体に対して起こる。このモノ置換蛍光基質は、遊離蛍光体と同じ波長を吸収し同じ波長で発光するので、この酵素反応速度の解釈はより難しくなる。4−メチルウンベリフェロンのフッ素化誘導体に基づく蛍光原基質が、β−ガラクトシダーゼ活性及び酸性ホスファターゼ活性のアッセイに関して記載されている(Gee,K.R.,et al,(1999),273,41−8)。
【0004】
蛍光ベースの酵素アッセイで使用される色素の多くは、350nm〜550nmの範囲で発光する。より高波長で発光する色素のいくつか、例えばレゾルフィンやDDAOは、チオール類(チオール官能基を有するペプチド類/タンパク類を含む)に対して反応性があり、それらの色素は蛍光特性を失う。したがって、これらの色素は、生体内の用途では有用ではなさそうである。スペクトルの赤又は赤外領域で発光する有用な蛍光特性の色素を有する蛍光ベースの酵素基質は、ニトロ置換されたシアニン色素誘導体である(欧州特許第1086179号:Hamilton,A.et al)。これらの色素の蛍光は、分子内の置換ニトロ基の存在により消光される。このニトロ基がニトロ還元酵素により還元されると、色素は蛍光性となる。これらの蛍光原基質が有用であることが判明し、現在は生細胞アッセイで使用されているが、それらは限られた用途しかない。したがって、関心のある酵素に対して以外は非反応性で、高いSBR(signal to background ratio)を提供する、より高波長(赤又は赤外)で発光する新たな蛍光原酵素基質が明らかに必要である。
【0005】
本発明では、一連の異なる酵素の基質であり、赤及び近赤外を含めた異なる波長で発光する新たな蛍光原酵素基質について説明する。2つの酵素活性の比を決定するための二重酵素基質も提供する。これは、個々の単一酵素基質の吸収特性、分布特性、及び排出特性に差があるために、2つの別々の酵素基質を利用して相対酵素活性を測定することができない生体内システムにより適切である。最後に、酵素活性を決定する方法、ならびに生体外又は生体内の代謝状態を決定する方法も提供する。
【特許文献1】米国特許第4812409号明細書
【特許文献2】欧州特許第1086179号明細書
【非特許文献1】Vieytes M.,et al,Anal.Biochem.,(1997),248,258−64
【非特許文献2】Gee,K.R.,et al,(1999),273,41−8
【非特許文献3】Forster,T.,“Intermolecular Energy Transfer and Fluorescence”,Ann.Physik.,Vol.2,p.55,(1948)
【非特許文献4】Freshney,R.I.,Culture of Animal Cells:A Manual of Basic Technique,2nd Edition,Alan R.Liss Inc.1987
【非特許文献5】“Non−Radioactive Labelling,a Practical Introduction”,Garman,A.J.Academic Press,1997
【非特許文献6】“Bioconjugation−Protein Coupling Techniques for the Biomedical Sciences”,Aslam,M.and Dent,A.,Macmillan Reference Ltd,(1998)
【非特許文献7】Hermanson,G.T.,Bioconjugate Techniques,Academic Press(1996)
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって、本発明は、蛍光標識酵素基質、特に蛍光共鳴エネルギー移動(FRET)標識基質、ならびにFRET標識及び酵素切断可能基を含んでなる基質を切断する際の酵素の活性を測定する方法を提供することを一目的とする。
【0007】
酵素切断活性に影響を及ぼすことがある試験剤をスクリーニングする方法を提供することも本発明の一目的である。
【課題を解決するための手段】
【0008】
したがって、本発明の第1の態様では、式(I)の化合物が提供される。
【0009】
【化1】
式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、
D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、
Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、
Mは、D1の蛍光特性を調節するように選択される酵素切断可能基である。
【0010】
適切には、D1とD2は、適切な条件下で蛍光共鳴エネルギー移動(FRET)が互いに起こるように、連結基Lによってリンクされる。FRETは、2種の色素分子の電子励起状態がフォトンの放出なしに相互作用する、距離に関係するプロセスである。Forster,T.,“Intermolecular Energy Transfer and Fluorescence”,Ann.Physik.,Vol.2,p.55,(1948)を参照のこと。この相互作用による1つの結果は、ドナー分子の励起がアクセプター分子の蛍光発光を促進することである。ドナーの蛍光量子効率は、それに対応して減少する。エネルギー移動効率はドナー分子とアクセプター分子の間の距離(r)の6乗に反比例して減少するので、FRETが起こるためには、適切にはドナー色素分子とアクセプター色素分子が近接近していなければならない(通常10〜100Å)。適切には、本発明において、D1がドナー分子である場合にD2がアクセプターとなるように、また逆も同様となるように、FRETの関係においてD1又はD2をドナー又はアクセプターとすることができる。ドナーとは、その色素部分が光からのエネルギーを吸収する能力があり、一部分以上がアクセプターの吸収スペクトル内にある波長周波数の光を放出することを意味する。アクセプターとは、その色素部分がドナー色素部分によって放出される波長のエネルギーを吸収する能力があることを意味する。適切には、ドナー色素分子の発光スペクトルの一部分以上とアクセプター色素分子の吸収スペクトルとの間に重なりがある。適切には、アクセプター色素の最大発光波長が、ドナー色素の最大発光波長よりも長い。一形式では、ドナー及びアクセプター分子の一方が、第2の蛍光性アクセプター又はドナー蛍光体に近接近している非蛍光性(又は消光性)の蛍光体であることがある。非蛍光色素が励起されると、蛍光エネルギーではなく熱としてエネルギーが散逸され、共鳴エネルギー移動も蛍光発光も起こり得ない。
【0011】
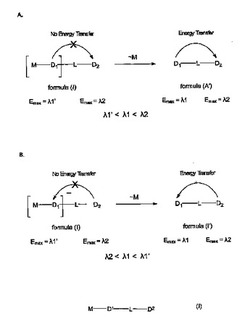
MがD1と共有結合している場合に、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大発光波長Emax=λ1’で蛍光発光する第1の蛍光状態となるように、基Mは適切にはD1の蛍光特性を調節する能力がある酵素切断可能基から選択される。適切には、MをD1から酵素的に切断すると式(I’)の化合物が形成されるように、Mは酵素の基質を含み、(図1参照)、D1が最大発光波長Emax=λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。適切には、D2は最大発光波長がEmax=λ2である蛍光色素である。適切には、式(I)の化合物において、λ1’≠λ1≠λ2である。
【0012】
適切には、連結基Lは、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−、フェニレニル及び下記の基から選択される1種以上の基を適宜含むことができる炭素原子から選択される2〜200個の連結原子を含有する基を含んでなる。
【0013】
【化2】
式中、R’は水素及びC1〜C4アルキルから選択され、mは1〜3の整数である。
【0014】
好ましい一実施形態では、連結基Lは2〜50個の連結原子を含有し、下記構造を有する。
【0015】
−{(CHR’)p−Q−(CHR’)r}s−
式中、Qは、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−及びフェニレニル基から選択され、R’は水素又はC1〜C4アルキルであり、各pは独立して0〜10であり、各rは独立して0〜10であり、sは1、2、又は3である。
【0016】
好ましくは、Qは−C(O)−、−CHR’−、−O−及び−CO−NH−から選択され、その場合のR’、p、r及びsは上に定義されている。
【0017】
別の実施形態では、リンカー基Lはペプチド又はオリゴヌクレオチド断片を含むこともできる。このリンカーがペプチドである、又はペプチド断片を含有する場合、リンカーは2〜20個の自然又は非自然アミノ酸、あるいはそれらの組合せを含有することができる。Lは、より好ましくは2〜12個のアミノ酸を、最も好ましくは2〜8個のアミノ酸を含有する。このリンカー基Lがオリゴヌクレオチドである、又はオリゴヌクレオチド断片を含有する場合、リンカー基Lは2〜20個のヌクレオシド塩基又は修飾塩基を含有することができる。好ましくは、塩基の個数は4〜15個の範囲であり、より好ましくは4〜10個の範囲である。
【0018】
このリンカーは、蛍光色素から延びる成分の一部を含むことができる。言い換えると、このリンカーを色素発色団に結合するが、その一部ではない。本発明の化合物では、リンカーはいずれもドナーとアクセプターの共役を可能にする二重結合のネットワークを含んでいない。最適なエネルギー移動が起こるように、ドナー及びアクセプター蛍光色素の遷移モーメントは、互いに垂直でない方向に配向させる、例えば互いに概ね平行又は直列に位置する。比較的短いリンカー及び最適な配向により、スペクトルの重なりが小さくなっても、効率的な共鳴エネルギー移動が起こり得る。
【発明を実施するための最良の形態】
【0019】
本発明の第1の態様による一実施形態では、D1がドナー色素であり、D2がアクセプター色素である。この実施形態(図1Aに示す)では、D1の最大発光波長(λ1)がD2の最大発光波長(λ2)よりも小さく、D1の発光スペクトルの一部分以上とD2の吸収スペクトルとの間に重なりがある。MがD1と共有結合している場合には、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大蛍光発光波長λ1’で蛍光発光する。ここで、λ1’はλ1よりも小さい。この状態においては、D1の発光スペクトルとD2の吸収スペクトルとの間には重なりがなく、又は実質的には重なりがなく、したがってD1とD2との間のエネルギー移動はない。MをD1から切断すると、D1が最大発光波長λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。その結果、D1の発光スペクトルとD2の吸収スペクトルとの間の重なりが再生され、D1とD2との間のエネルギー移動が起こり得る。したがって、ドナー色素D1を適当な励起波長で励起すると、D2からの蛍光発光の増加を検出及び/又は定量化することができる。
【0020】
第2の実施形態では、D2がドナー色素であり、D1がアクセプター色素である。この実施形態(図1Bに示す)では、D2の最大発光波長(λ2)がD1の最大発光波長(λ1)よりも小さく、D2の発光スペクトルの一部分以上とD1の吸収スペクトルとの間に重なりがある。MがD1と共有結合している場合には、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大蛍光発光波長=λ1’で蛍光発光する。ここで、λ1’はλ1よりも大きい。この状態においては、D2の発光スペクトルとD1の吸収スペクトルとの間には重なりがなく、又は実質的には重なりがなく、したがってD2とD1との間のエネルギー移動はない。MをD1から切断すると、D1が最大発光波長λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。その結果、D2の発光スペクトルとD1の吸収スペクトルとの間の重なりが再生され、エネルギー移動が起こり得る。ドナー色素D2を適当な励起波長で励起すると、D1からの蛍光発光の増加を検出及び/又は定量化することができる。
【0021】
D2がドナー色素であり、D1がアクセプターであるこの実施形態においては、エネルギー移動がドナー色素とアクセプター色素の2つの蛍光状態との間で代わりに起こることがある。この構成では、第1の蛍光状態におけるアクセプター色素がある波長λ1’で発光し、Mが切断されると、第2の波長λ1で発光する第2の蛍光状態を生じる。
【0022】
さらなる実施形態では、D1の第1及び第2の蛍光状態が、それぞれの最大発光波長λ1及びλ1’によってではなく、これらの2つの状態から放出される蛍光の強度によって異なることがある。
【0023】
式(I)の化合物は、蛍光共鳴エネルギー移動を利用するアッセイにおける生化学的切断現象を検出するためのレポーター分子として使用することができる。適切には、基Mは、好ましくはペプチダーゼ、プロテアーゼ、ホスファターゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される切断酵素の基質を含んでなる。MをD1から切断するのに適した条件下でこの基質を酵素で処理することにより、D1の蛍光特性を調節し、それによりD1を上述したように第1の蛍光状態から第2の蛍光状態へ切り替える。
【0024】
好ましい一実施形態では、Mが、D1と共有結合している1つ以上のリン酸基を有し、かつ下記構造を有するリン酸エステル結合を含む。
【0025】
【化3】
式中、nは1〜4の整数である。この実施形態では、色素D1のアルコール誘導体及び無機リン酸を生成するために、細菌アルカリホスファターゼや酸性ホスファターゼなどのホスファターゼによって基質を切断することができる。結果として、D1は第1の蛍光状態から第2の蛍光状態へと切り替えられ、それによりドナー部分とアクセプター部分との間で励起エネルギーの移動が可能となる。ドナー色素がその励起波長で励起されると、アクセプター色素の発光波長における蛍光強度が増加する。リン酸エステルは、予め合成した基質でもよいし、あるいは、化学的加水分解によって、又は以下の構造を有する終端リン酸標識ヌクレオシドポリリン酸からの酵素触媒ヌクレオシド一リン酸もしくはヌクレオシドポリリン酸の移動によってその場で生成することもできる。
【0026】
【化4】
式中、R1及びR2は、独立してH及びOHから選択され、Raはアデニン、グアニン、シトシン、チミン、ウラシル、ヒポキサチン及びキサチンから選択されるヌクレオシド塩基であり、kは1〜6の整数である。
【0027】
別の好ましい実施形態では、Mが、D1に共有結合しているペプチド結合(−CO−NH−)を1つ以上含む。この実施形態では、Mが通常下記構造を有する。
【0028】
【化5】
式中、Rbはペプチド又はタンパクの残基である。ペプチダーゼ又はプロテアーゼによって加水分解すると、Mが蛍光エネルギー移動標識から切断される。ドナー部分をその励起波長で励起すると、ドナー色素とアクセプター色素との間でエネルギーが移動し、それによりアクセプターからの蛍光発光の増加を検出することが可能となる。
【0029】
さらなる実施形態では、Mはグリコシド結合を含み、α−グリコシダーゼ(例えばα−アミラーゼ)やβ−グリコシダーゼ(例えばβ−グルコシダーゼ)などのグリコシダーゼの基質であり、下記構造を有する。
【0030】
【化6】
さらなる実施形態では、Mが、デアルキラーゼの基質であり下記構造を有するエーテル結合を含んでなる。
【0031】
【化7】
式中、RcはC1〜C20の直鎖又は分枝鎖アルキルである。
【0032】
さらなる一実施形態では、基Mがさらに細胞膜透過性の基を含んでなり、この基は次式のエーテル、エステル、アミド、ホスホジエステル基などの基から選択することができる。
【0033】
【化8】
式中、Rdは、非置換又は1つ以上のハロゲン原子で置換されたC1〜C10の直鎖又は分枝鎖アルキル、非置換又は1つ以上のハロゲン原子で置換されたフェニル、あるいはペプチド鎖である。適切には、ハロゲン原子は、フッ素、塩素、臭素及びヨウ素から選択することができる。このような膜透過性の基の有用な例には、酢酸エステル基、ピバロイルエステル基、アセトキシメチルエステル基、ペンタフルオロベンジルエーテル基、ペンタフルオロベンジルアミド基、ペンタフルオロベンジルエステル基、フェニルリン酸基、パーフルオロ−(C1〜C6)アルキルエーテル基、パーフルオロ−(C1〜C6)アルキルエステル基及びパーフルオロ−(C1〜C6)アルキルアミド基が含まれる。このような基は、デアルキラーゼ、プロテアーゼなどのエステラーゼ、及び細胞内に見られるようなホスホジエステラーゼの基質であり、それらによって加水分解される。レポーター分子の細胞透過性の可変性は当業者によって理解されるであろうし、また当業者が試験することが可能である。
【0034】
ドナー色素は、適切にはクマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から、好ましくはキサンチン色素及びシアニン色素から選択することができ、このドナー色素はエネルギーをアクセプター色素に移動させる能力がある。
【0035】
アクセプター色素は、適切にはクマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から選択することができ、このアクセプター色素は、ドナー色素と共にエネルギーを移動させる能力がある。
【0036】
好ましくは、ドナー色素がキサンチン色素又はシアニン色素であり、アクセプター色素がローダミン又はシアニン色素である。
【0037】
好ましい一実施形態では、前記ドナー及びアクセプター色素部分の1つ以上がシアニン色素である。別の好ましい実施形態では、ドナー色素がキサンチン色素であり、アクセプター色素がローダミン色素である。
【0038】
適切なキサンチン色素には、5−カルボキシフルオレセイン、6−カルボキシフルオレセイン、6−カルボキシ−4’,5’−ジクロロ−2’,7’−ジメトキシフルオレセインなどのフルオレセイン及びその誘導体が含まれるが、それだけには限らない。
【0039】
適切なシアニン色素には、CyA(3−(ε−カルボキシペンチル)−3’−エチル−5,5’−ジメチルオキサカルボシアニン)、Cy2(3−(ε−カルボキシペンチル)−3’−エチル−オキサ−カルボシアニン)、Cy3(3−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−カルボシアニン)、Cy3.5(3−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−4,5,4’,5’−(1,3−ジスルフォナト)ジベンゾ−カルボシアニン)、Cy5(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−ジカルボシアニン、Cy5.5(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−4,5,4’,5’−(1,3−ジスルフォナト)−ジベンゾ−ジカルボシアニン)、及びCy7(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−トリカルボシアニンが含まれるが、それだけには限らない。
【0040】
適切なローダミンアクセプター色素には、5−カルボキシローダミン(ローダミン110−5)、6−カルボキシローダミン(ローダミン110−6)、5−カルボキシローダミン−6G(R6G−5又はREG−5)、6−カルボキシローダミン−6G(R6G−6又はREG−6)、N,N,N’,N’−テトラメチル−5−カルボキシローダミン、N,N,N’,N’−テトラメチル−6−カルボキシローダミン(TAMRA又はTMR)、5−カルボキシ−X−ローダミン、及び6−カルボキシ−X−ローダミン(ROX)が含まれるがそれだけには限らない。他のクラスの色素には、BODIPY(登録商標)、ポルフィリン色素、rhodol色素及びペリレン色素が含まれる。
【0041】
この第1の態様による蛍光標識酵素基質は、化合物に親水特性を与えるためにそれら基質に結合した水溶化成分を含むこともできる。それら水溶化成分は、好ましくはドナー又はアクセプター色素部分の芳香族環系に結合する。あるいは、連結基Lがこの水可溶化基を含有することもできる。適切な可溶化成分は、アミド、スルホン酸塩、硫酸塩、リン酸塩、四級アンモニウム、ヒドロキシル、グアニジン及びホスホン酸塩からなる群から選択することができる。ドナー及び/又はアクセプター蛍光色素の芳香族環に直接結合したスルホン酸塩又はスルホン酸基が特に好ましい。
【0042】
本発明によるエネルギー移動レポーター分子の例を表1に示す。
【0043】
【表1】
【0044】
【表2】
【0045】
【表3】
例(II)、(III)及び(IV)では、レポーターが消光フルオレセインドナー色素を含有し、またCy5、Cy3及びローダミンアクセプター色素をそれぞれ含有する非蛍光エネルギー移動錯体であり、フルオレセイン部分は、それに結合した酵素的に切断可能なリン酸基を有する。このリン酸基は、ホスファターゼの作用によって切断することができ、それによりドナー色素の蛍光が、フルオレセインをその励起波長で励起すると回復する。この切断現象の検出及びホスファターゼの活性の測定は、アクセプター色素の発光波長での蛍光発光を検出することによって実現することができる。
【0046】
例(V)及び(VI)では、レポーターが、消光フルオレセインドナー色素を含有し、またCy5及びローダミンアクセプター色素をそれぞれ含有する非蛍光エネルギー移動錯体であり、フルオレセイン部分は、それに結合したヌクレオシドポリリン酸塩部分を有する。ヌクレオシジルホスホトランスフェラーゼの作用によりその場でホスファターゼ基質が生成され、ホスファターゼの存在下でリン酸が除去される。フルオレセインドナー色素の蛍光が回復する。続いて起こるアクセプター色素へのエネルギー移動により、赤色にシフトした発光が生じる。このヌクレオシジルホスホトランスフェラーゼ酵素が核酸ポリメラーゼである場合、既知及び未知の核酸の検出、特性決定、定量化に本発明の組成物を使用することができる。
【0047】
一実施形態では、Lが切断可能リンカーであり、酵素基質基Mとは異なる酵素切断可能基Pを含む。Pは、エーテル基、エステル基、アミド基、リン酸ジエステル基などの基から選択することができる。このような基は、それぞれデアルキラーゼ、プロテアーゼなどのエステラーゼ、及びホスホジエステラーゼの基質であり、それらによって水性緩衝媒体中で切断される。この実施形態では、D2がLと結合しているときは第1の発光波長λ2で、D2がLから切断されると第2の発光波長λ2’でD2が発光することが好ましい。ここでλ2≠λ2’である。この実施形態のレポーター化合物は、2つの別々の酵素基質を確実に等しく供給することが困難である生体内及び細胞系内で2つの酵素の相対活性を測定するのに有用である。また、Mと結合しているときは第1の発光波長(λ1)で、Mが切断されると異なる波長(λ1’)でD1が発光することも可能である。ここでλ1≠λ1’である。例(VII)は、アリルスルファターゼ及びカテプシンGの活性に対するこのような二重酵素レポーターを示しており、この二重酵素レポーターは、炎症を検出する際には潜在的に有用である。酵素活性がない場合、例(VII)の化合物を360nmで励起すると610nmで弱い発光がある。645nmの励起では発光は認められない。スルファターゼ活性のみの場合、645nmで励起すると660nmで強い発光が認められ、360nmで励起すると450nm及び660nmで弱い発光がある。カテプシンG活性のみの場合、360nmで励起すると450nmで強い発光が認められるが、360nm又は645nmで励起しても660nmでは発光しない。両方の酵素が存在する場合には、360nmで励起すると450nmで強い発光が認められ、645nmで励起すると660nmで別の強い発光がある。360nmの励起では、660nmの発光は認められない。したがって、660nm及び450nmでの発光を測定し、既知の基準と比較することにより、生体系内における特定の時点又は位置での両酵素の相対酵素活性を決定することが可能である。このような二重酵素活性用基質は、異なる病状を区別する、ならびに病状の臨床病期又は病原力を決定するのに有用である。
【0048】
MMP−2もMMP−9も共に腫瘍の検出及び治療を目標としている。例えば、メラノーマでは、これらのタンパクの発現は大きく変化する。非常に攻撃的な腫瘍ではMMP−9が高レベルで発現するが、早期腫瘍では、過剰発現は認められない。良性腫瘍はMMP−9活性を示さない。同様に、高い活性を示す湿潤がんではMMP−2の活性も大きく変化する。これは、患者の大部分に当てはまるが、すべての患者に当てはまるわけではない。したがって、MMP−2又はMMP−9の活性のみを測定すると、攻撃的な腫瘍を良性であると誤診してしまうことがある。骨粗しょう症では、カテプシンKが、破骨細胞に過剰発現するので、治療標的に認定されているが、この活性は、骨粗しょう症とは関連のない通常の骨代謝回転中にも認められる。骨粗しょう症であっても、高レベルのMMP−9が認められる。しかしながら、MMP−9の活性を測定することは、それほど骨粗しょう症の特異的な指標ではない。したがって、両方の活性及びそれらの比を測定することにより、単一の酵素活性を測定するよりも優れた診断ツールが提供される。
【0049】
本発明の第2の態様では、基質分子に作用する酵素の活性を決定する方法が提供され、この基質は式(I)の化合物を含んでなる。式中のD1、D2及びLは上に定義されており、Mは切断酵素の基質を含んでなる。この方法は、i)蛍光標識基質の蛍光強度を測定する段階と、ii)MをD1から切断するための条件下で、活性を決定すべき酵素を基質と複合させる段階と、iii)段階ii)の複合に続いて、蛍光標識の蛍光強度の変化を測定する段階とを含んでなり、蛍光標識の蛍光強度の変化を使用して酵素の活性を決定する。
【0050】
適切には、式(I)の化合物は、活性を決定すべき酵素の基質であり、Mは、上に定義されているように好ましくはリン酸エステル結合、1つ以上のペプチド結合、エーテル結合及びグリコシド結合から選択される酵素切断可能基を含んでなる。この酵素は好ましくは、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される加水分解酵素である。
【0051】
第2の態様による好ましい一実施形態では、酵素が、PTPase、PPTase−2A、PPTase−2B、又はPPTase−2Cを含めた、E.C.Class3.1から選択されるホスファターゼである。例には、チロシンホスファターゼ、PP1及びPP−2B(セリン/トレオニンホスファターゼ)がある。
【0052】
第2の態様による第2の好ましい実施形態では、酵素がE.C.Class3.4から選択されるペプチダーゼである。例には、アンギオテンシン変換酵素(ACE)、カスパーゼ、カテプシンD、キモトリプシン、ペプシン、サブチリシン、プロテイナーゼK、エラスターゼ、ネプリライシン、サーモライシン、asp−n、マトリックスメタロプロテイナーゼ1〜20、パパイン、プラスミン、トリプシン、エンテロキナーゼ及びウロキナーゼが含まれるが、それだけには限らない。
【0053】
別の実施形態では、酵素が好ましくはE.C.Class3.2からなる群から選択されるグリコシダーゼ、例えば、α−アミラーゼ、β−アミラーゼ、グルカン1,4−α−グルコシダーゼ、セルラーゼ、エンド−1,3−β−グルカナーゼ、オリゴ−1,6−グルコシダーゼ、リゾチームである。
【0054】
さらなる実施形態では、酵素がデアルキラーゼ、例えばシトクロムP−450の異なるイソ酵素である。
【0055】
ペルオキシダーゼ、β−ラクタマーゼ、ニトロ還元酵素など他の酵素を、適当なM又はリンカー構造とともに使用することもできる。これらの酵素の蛍光原基質、例えば、(欧州特許第1086179号,Hamilyton,A.et al)に記載されているような、Amplex red、フルオレセイン及び6−クロロ−7−ヒドロキシクマリン置換セロファロスポリン、ならびにニトロシアニンがよく知られている。同様の構造を、上述の組成物に組み込むこともできる。
【0056】
調査中の酵素反応への試験剤の阻害作用、増強作用、アゴニスト又はアンタゴニスト作用をスクリーニングする用途を含めたハイスループットスクリーニング用途では、本発明に従ってアッセイを行うことができる。したがって、第2の態様による好ましい実施形態では、基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法が提供される。この方法は、(a)試験剤がある状態とない状態とで第2の態様による方法を実施する段階と、(b)この剤がある状態とない状態とで酵素の活性を決定する段階とを含んでなり、前記剤がある状態とない状態との間の酵素の活性の差は、酵素の活性への試験剤の作用を示す。あるいは、試験剤がある状態でこの方法を行い、酵素の活性の値を試験剤のない状態における酵素活性の対照値と比較することによりスクリーニングを実施することもできる。この対照値は、データベースに又は他の電子形式で都合よく電子的に格納することができる。
【0057】
本発明の第3の態様では、式(I)の化合物を含んでなる基質分子に作用する2つの酵素の相対活性を決定する方法が提供される。式中、上に定義されているようにD1は第1の色素部分、D2は第2の色素部分であり、Mは第1の切断酵素の基質を含んでなり、Lは第1の切断酵素とは異なる第2の切断酵素の基質である酵素切断可能基Pを含む。この方法は、i)蛍光標識基質の蛍光発光強度を測定する段階と、ii)MをD1から、またD2をD1から切断するための条件下で、第1及び第2の酵素を基質と複合させる段階と、iii)段階ii)の複合に続いて、第1及び第2の色素部分の蛍光発光強度を測定する段階と、iv)第1及び第2の色素部分の蛍光強度の変化を利用してこれらの酵素の相対活性を決定する段階とを含んでなる。
【0058】
適切には、第1及び第2の酵素を、いずれかの順序で経時的に、あるいは同時に基質分子と複合させることができる。好ましくは、第1及び第2の酵素は、上述したように、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される。
【0059】
本発明の第3の態様による方法を使用して、基質分子に作用する2つの酵素の相対活性への試験剤の作用を決定することもできる。その場合、試験剤がない状態とある状態とで複合段階ii)を実施する。前記剤がある状態とない状態との間の第1及び第2の酵素の相対活性の差は、第1及び第2の酵素の相対活性への試験剤の作用を示す。
【0060】
この試験剤は、例えば、合成分子や天然物(例えばペプチド、オリゴヌクレオチド)など任意の有機又は無機化合物、あるいはエネルギー形態(例えば光又は熱、あるいは他の形態の電磁放射線)でよい。この好ましい実施形態による方法を用いて、酵素活性の阻害剤を検出することもできる。例えば、公知の阻害剤ペプスタチンAがカテプシンDの酵素活性を阻害することは、本発明の方法を用いることで容易に示すことができる。したがって、12ナノモルのペプスタチンAがある状態では、カテプシンDの活性は、阻害剤がない状態の約5分の1である。
【0061】
適切には、剤がない状態とある状態との間の酵素活性の差は、規格化され、電子的に格納され、基準値と比較される。したがって、例えば、活性の差を阻害率(%)(又は刺激率(%))として電子データベースに格納し、この値を問題となっている酵素の基準阻害剤での対応する値と比較することができる。このようにして、ある所定のしきい値(例えば、基準化合物と同等以上に有効であること)を満たす試験剤のみを、さらなる試験の対象として選択することができる。
【0062】
一例としては、タンパク分解酵素の活性を検出するためのアッセイを以下のように構成することができる。酵素と式(I)による蛍光エネルギー移動レポーター化合物を複合させることにより、反応混合物を調製する。式中、基Mはタンパク分解酵素切断部位を含んでなる。このアッセイは、24個、96個、384個又はより高密度のウェル、例えば1536個のウェルを有するマルチウェルプレート、例えばマイクロタイタプレートのウェル内で適切に行われる。この反応は、酵素基質が最初に存在する水性アッセイ緩衝液中で、適切には5mMのMgCl2を含有する10mMのMOPs、50mMのTris又は50mMのHEPES中で行うことができる。既知の又は推定上の阻害剤などの試験剤を、この反応混合物に適宜含めることができる。酵素を添加すると、通常反応を最後まで進行させることができ、蛍光光度計を用いて記録される蛍光性アクセプター色素による定常状態の蛍光発光を観察することにより進行がモニターされる。あるいは、反応を所定の時間進行させ、その後、多くの場合非特異的である停止試薬、通常酵素活性の阻害剤で停止させることができる「停止」条件下で、このアッセイを行うこともできる。停止試薬の例にはEDTAがあり、この試薬を使用して通常酵素活性に必要とされる金属イオンを分離する。
【0063】
本発明による方法を利用して、細胞環境における、本発明による化合物を含んでなる基質に作用する酵素の活性を測定することもできる。したがって、第2及び第3の態様による特定の実施形態では、その方法が、流動媒体中の1つ以上の細胞に基質を加える段階を段階i)の前に含んでなる。
【0064】
通常、培養細胞は、細胞増殖に適した条件下で、0.5時間から24時間までに及び得るある期間、適切な細胞培養液中0.1〜100μMの濃度のFRET結合酵素基質で培養される。細胞は、標準的な細胞培養技術に従って培養される。例えば、加湿した空気95%/CO25%の雰囲気を含む培養器内の、37℃の無菌環境における適切な容器内で細胞が培養される。多種多様な細胞型の培養に利用可能なプロトコールが確立されている(例えば、Freshney,R.I.,Culture of Animal Cells:A Manual of Basic Technique,2nd Edition,Alan R.Liss Inc.1987を参照のこと。)。細胞培養又は組織培養において培養される細胞に基質を導入する必要がある場合、基質を単に培養液に添加する。酵素活性への作用を決定すべき試験剤がある状態で、細胞を基質に接触させることもできる。この実施形態では、検出段階により、調査中の酵素の活性への試験剤の作用が測定される。
【0065】
光電子倍増管が検出器として組み込まれた器具を用いて蛍光強度の測定を行うことができる。荷電結合素子(CCD)撮像装置(走査型撮像装置や面走査撮像素子など)によって蛍光強度の変化を測定して、マイクロタイタプレートのウェルのすべてを撮像することができる。LEADseeker(登録商標)システムは、CCDカメラを特徴とし、単一パスで高密度マイクロタイタプレートの蛍光画像化を可能にする。画像化は定量的かつ高速であり、画像化用途に適した器具使用により、現在マルチウェルプレート全体を同時に撮像することができる。酵素活性に対する試験剤の活性を検出するためにアッセイの形式を定めようとする場合、基質の蛍光の連続測定下でアッセイを行うことができる。この形式では、蛍光標識基質の強度は連続的に変化する。この標識基質は、酵素反応の生成物から分離する必要がなく、したがって反応の時間経過を得ることができ、反応速度研究をリアルタイムで行うことが可能となる。
【0066】
本発明の第4の態様では、1種以上の酵素の活性を測定するための試験キットが提供される。この試験キットは1種以上の異なる酵素基質を含んでなり、前記基質の各々は式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる。好ましくは、この試験キットは、各酵素が異なる前記基質に特異的な1種以上の異なる酵素をさらに含んでなる。
【0067】
当業者にはよく知られている直接化学結合法を用いて蛍光エネルギー移動部分D1−L−D2を基Mと共有結合することによって、式(I)の化合物を調製することができる。ここで、D1、D2、M及びLは上に定義されている。あるいは、実施例に示すようなデノボ合成法によって本発明の化合物を調製することもできる。最初に基MをD1に結合して中間化合物M−D1を形成し、続いてリンカー基Lを介してD2を結合することができる。D2を共有結合する前に、リンカー基LをM−D1に都合よく共有結合してもよく、あるいはLを前もってD2に結合してもよい。本発明で用いるペプチド、タンパク及びオリゴヌクレオチド基質を終端位置で、あるいは1つ以上の内部位置で標識化することができる。蛍光色素標識試薬を用いたタンパク標識の検討及び例には、“Non−Radioactive Labelling,a Practical Introduction”,Garman,A.J.Academic Press,1997、“Bioconjugation−Protein Coupling Techniques for the Biomedical Sciences”,Aslam,M.and Dent,A.,Macmillan Reference Ltd,(1998)を参照のこと。合成ペプチドにおける部位特異的な標識を得るためのプロトコールが利用可能である。例えば、Hermanson,G.T.,Bioconjugate Techniques,Academic Press(1996)を参照のこと。
【0068】
本発明の原理及び機能を明確化するために、以下の実施例についてこれから言及するが、添付の特許請求の範囲及び本発明の範囲を限定すると解釈すべきではない。本発明は、本明細書に提供した教示から明らかとなる任意の及びすべての変形形態を包含すべきものである。
【実施例】
【0069】
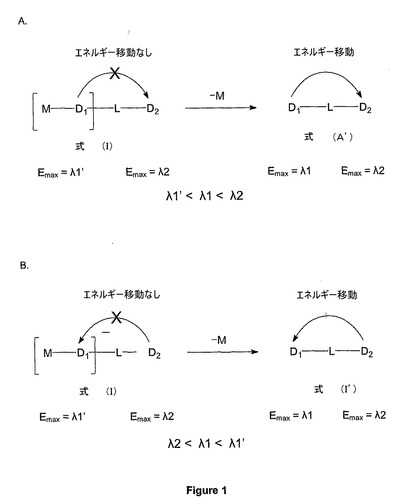
1. FRET結合レポーター分子の調製(化合物(V)及び化合物(VI)(図2参照)
1.1. 4’(5’)−フルオレセインジエチルエーテル−エステル:化合物(2)
NaOH(1.44g)のメタノール(145ml)溶液に、フルオレセイン(6.0g)を加えて暗色溶液を生成し、この溶液を真空中で濃縮乾燥した。これを無水DMF(2×50ml)と同時蒸発させ、無水DMF(150ml)に再溶解した。ヨウ化エチル(11.6ml、8eq)を加え、反応物を室温で一晩中攪拌した。この反応混合物を濃縮した後、重炭酸ナトリウム飽和溶液(150ml)に溶解し、ジエチルエーテルで洗浄した。この溶液を冷却すると黄色い沈殿物が形成され、この沈殿物をろ過し、最後にエーテル(2×25ml)及びエーテル/ペンタン(1:1、50ml)で洗浄した。
【0070】
1.2. 4’(5’)−クロロアセチルアミノメチル−3’(6’)−エチルフルオレセインジエチルエーテル−エステル:化合物(3)
エチルフルオレセインエステル(化合物(2)、2.67g)の50ml硫酸攪拌溶液に、クロロアセチルアセトアミド(850mg)をゆっくりと5分間かけて加えた。この反応混合物を、室温で一晩攪拌した。その反応混合物を砕氷上に注ぎ、形成された沈殿物をろ過し、冷水で洗浄した。TLC及び質量分析により、モノ及びビスクロロアセトアミド化合物の形成が示された。これらの生成物を、塩化メチレン中2〜15%のメタノールを用いたシリカゲルカラムクロマトグラフィーによって精製した。まずビスアルキル化生成物を溶出し、その後2種のモノアルキル化生成物を溶出した。主生成物であるより低い分子量のモノアルキル化生成物をさらなる反応に使用した。
【0071】
1.3. 4’(5’)−アミノメチル−3’(6’)−エチルフルオレセイン:化合物(5)
主生成物のモノアルキル化生成物(化合物(3)、1g)を、2.5時間の加熱によって6NのHCl(15ml)で加水分解した。この生成物をCH2Cl2で抽出し、水で洗浄し、無水硫酸ナトリウムで乾燥した後、シリカゲルカラムクロマトグラフィーによって精製して500mgの純粋化合物を得た。MS ES+:390.3
1.4. 4’(5’)−トリフルオロアセチルアミドメチル−3’(6’)−エチルフルオレセイン:化合物(6)
化合物(5)(390mg)を無水ピリジン(5ml)中に入れ、TFA−NHS(650mg)を加えた。この混合物を室温で一晩攪拌した。真空中で濃縮し、塩化メチレンで抽出した後、この生成物をシリカゲルクロマトグラフィーによって精製して350mgの純粋生成物を得た。MS ES+:486.3
1.5. 4’(5’)−トリフルオロアセチルアミドメチル−3’(6’)−エチルフルオレセイン−6’(3’)−リン酸:化合物(7)
化合物(6)(200mg)のアセトニトリル(5ml)溶液に、POCl3(189mg、3eq)を加え、この反応混合物を室温で1時間攪拌した。ピリジン(290μl、9eq)を加え、この反応物をさらに2時間攪拌した。TEAB(0.1M、10ml)を加えることによってこの反応物を失活させた。1時間後、この混合物を真空中で濃縮し、CH2Cl2/メタノールのグラジェントを用いたシリカゲルクロマトグラフィーによって精製して130mgの一リン酸を得た。MS ES−:564.5
1.6. 4’(5’)−アミノメチル−3’(6’)−エチルフルオレセイン−6’(3’)−四リン酸塩−チミジン:化合物(8)
1.6.1. 化合物(7)イミダゾライド誘導体の合成
化合物(7)(60mg)の無水DMF(2ml)溶液に、トリブチルアミン(30μl、3eq)を加え、この混合物を真空中で濃縮乾燥した。その混合物を無水DMF(2ml)に再溶解し、攪拌しながらカルボニルジイミダゾール(85mg、5eq)を加えた。この混合物を室温で2時間攪拌し、HPLC−MSによって反応終了を確認した。この混合物をメタノール(100μl)で失活させ、室温で1時間攪拌し、真空中で濃縮乾燥し、無水DMF(2ml)に再溶解した。MS ES−:614.6
1.6.2. 化合物(7)イミダゾライド誘導体のチミジン−5’−三リン酸への付加
チミジン−5’−三リン酸(トリエチルアンモニウム塩、100μmol)を、無水DMF(2×2ml)中のトリブチルアミン(400μmol)と同時蒸発させ、最終的に無水DMF(2ml)に再溶解した。
【0072】
上記チミジン−5’−三リン酸(TTP)のトリブチルアンモニウム塩溶液に、イミダゾライド誘導体(段階1.6.1による)を加え、この混合物を室温で一晩攪拌した。HPLC−MSにより、>90%の完全な反応であることが示された。これをさらに5時間攪拌した。
【0073】
1.6.3. 段階1.6.2による反応混合物の濃縮及びNH4OH(30%溶液、10ml)の添加
この混合物を室温で2.5時間攪拌し、冷蔵庫に一晩放置した。その混合物を真空中で濃縮乾燥し、アニオン交換クロマトグラフィーによって精製した。この混合物をヘビ毒ホスホジエステラーゼ(SVD)及びアルカリホスファターゼで一晩処理し(未反応TTPを除去するため)、さらに0.1MのTEAB中25%のアセトニトリルに対して0.1MのTEAB(緩衝液A)を用いた逆相クロマトグラフィーによって精製した。HPLC−MSによって2つの主なピークを収集し、分析した。第1の主なピークは931.8で質量を示し、第2のピークでは所要の932.8で質量を示した。
【0074】
トリフルオロアセチル基の加水分解中のアンモニア処理ではスピロラクトンとも反応し、結果として不要なアミド化合物が形成されたことが明らかであった。ピーク2を真空中で濃縮乾燥し、メタノール(2×5ml)と同時蒸発させ、H2O(1ml)に再溶解した。その濃度はUVによって測定した。全部で5.5μmolの純粋化合物を得た。
【0075】
1.7. 化合物(8)へのアクセプター色素の付加:化合物(V)及び(VI)の合成
化合物(8)(2.2μmol、400μlの5.5mM溶液)2つの別々の反応容器に、400μlの0.5M炭酸ナトリウム/重炭酸ナトリウム緩衝液(pH8.5)を加え、一方の容器には5−ROX−NHSエステル(2mlのDMF中5.5mg)を、もう一方の容器にはCy5−NHSエステル(2mlのDMF中5mg)を加えた。両方の反応混合物を室温で一晩攪拌し、HPLC−MSによって反応の終了をモニターした。この反応混合物を真空中で濃縮し、0.1MのTEAB中の25%アセトニトリル(緩衝液A)及び1MのTEAB中の25%アセトニトリル(緩衝液B)を用いたイオン交換カラムで最初に精製した。生成物を含有するピークを真空中で濃縮し、さらに逆相HPLCカラムによって再精製した。正確な画分を収集し、真空中で濃縮し、紫外分光法によって定量化した。化合物(V):MS ES−:1449;化合物(VI):MS ES−:1572
2. 化合物(II)、(III)及び(IV)の合成
出発原料が(化合物(7)をメタノール中でK2CO3により加水分解することによって得られた)4’(5’)−アミノメチル−エトキシフルオレセインリン酸であることを除いては、化合物(V)及び(VI)のための上述のやり方で合成を行った。生成物をHPLCによって精製し、紫外分光法によって定量化した。
【0076】
化合物(II):MS ES−:1107.3;
化合物(III):MS ES−:1342.4;
化合物(IV):MS ES−:881
3. アルキルホスファターゼの化合物(II)、(III)及び(IV)に対する作用
【0077】
【化9】
化合物II、III及びIVを、25mMのHEPES及び5mMのMgCl2緩衝液(pH8.5)中に入れ、室温で2時間アルカリホスファターゼにより処理した。反応を分光法で追跡した結果、反応が30分以内にほとんど終了したことが示された。
【0078】
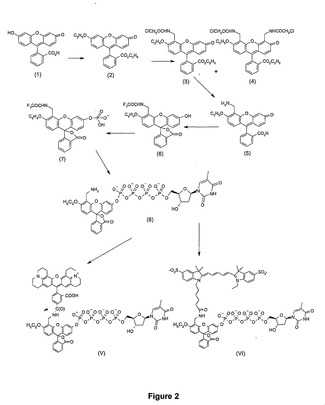
4. 化合物(II)、(III)、(IV)、(VIII)、(IX)及び(X)のUV、蛍光及びエネルギー移動測定
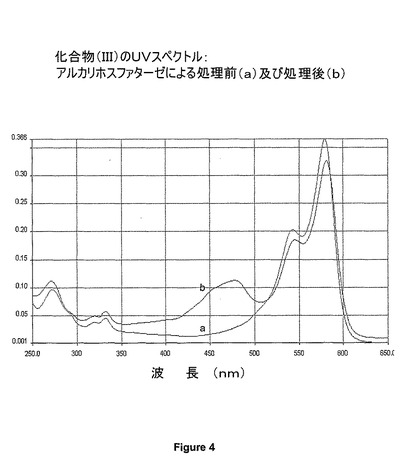
リン酸化色素(化合物(II)、(III)及び(IV))及び非リン酸化色素(化合物(VIII)、(IX)及び(X))のUV及び蛍光スペクトルを図2〜8に示す。アルカリホスファターゼで処理すると約472nmにピークが出現することを、UVスペクトルがはっきりと示している。
【0079】
本発明の特定の望ましい諸実施形態を本明細書で説明してきたが、企図した本発明の範囲から逸脱することなくそれらの実施形態に変更を加えることができることを理解されたい。本発明の真の範囲は、添付の特許請求の範囲に示される。
【図面の簡単な説明】
【0080】
【図1】本発明の好ましい諸実施形態を示す概略図である。
【図2】化合物(V)及び(VI)を調製するための合成反応スキームを示す図である。
【図3】アルカリホスファターゼによる処理前及び処理後の化合物(II)のUVスペクトルを示す図である。
【図4】アルカリホスファターゼによる処理前及び処理後の化合物(III)のUVスペクトルを示す図である。
【図5】アルカリホスファターゼによる処理前及び処理後の化合物(IV)のUVスペクトルを示す図である。
【図6】アルカリホスファターゼによる処理前及び処理後の化合物(II)の、455nm及び475nm励起による発光スペクトルを示す図である。
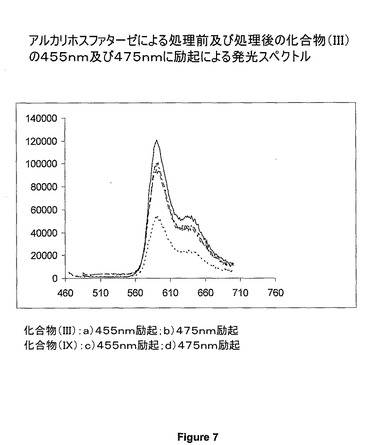
【図7】アルカリホスファターゼによる処理前及び処理後の化合物(III)の、455nm及び475nm励起による発光スペクトルを示す図である。
【図8】アルカリホスファターゼによる処理前及び処理後の化合物(IV)の、455nm及び475nm励起による発光スペクトルを示す図である。
【技術分野】
【0001】
本発明は、蛍光ベースのアッセイに関し、また酵素活性を測定するための試薬及び方法、特に酵素切断アッセイに関する。
【背景技術】
【0002】
酵素活性、特に加水分解などの酵素切断活性を測定するためのアッセイは、生物科学及び薬学において広く利用される。コンビナトリアルケミストリー及びハイスループットスクリーニングの到来とともに、酵素活性の潜在的なモジュレータをスクリーニングするための簡単で、感度が高く、かつ費用効率の高いアッセイの必要性が高まっている。製薬業界が特に関心があるのは、タンパク分解酵素による切断及びリン酸の切断を検出するための方法である。
【0003】
蛍光ベースのアッセイは、扱いやすさ、感度、コスト及び自動化の容易さの点で、酵素の切断活性を測定するための放射化学法、ELISA、抗体法及びより伝統的な技法に勝る有意な利点を提供する。蛍光原基質を均一系酵素アッセイに定期的に使用して、酵素活性を、あるいは創薬における潜在的な薬物の酵素活性への作用を決定する。これらの酵素の多くの基質が市販され、又は文献で報告されている。例えば、米国特許第4812409号(Babb,B.,et al)は、フェナレノン又はベンズフェナレノンから誘導されるブロックされた色素部分を含んでなる加水分解性蛍光基質を開示している。これらの色素部分は、酵素加水分解中に基質から切断されると、約530nmより上の蛍光を有する蛍光色素を形成する。加水分解酵素に対応する基質も、フルオレセイン及びローダミンの誘導体を基に入手可能である。例えば、フルオレセイン二リン酸は、アルカリホスファターゼ(PP2A)用の、広く使用されている無色の非蛍光基質であり、この酵素によって加水分解されると、フルオレセインを形成する(Vieytes M.,et al,Anal.Biochem.,(1997),248,258−64)。しかしながら、フルオレセインに基づく基質には、通常2つの酵素切断可能部位が組み込まれており、二相の酵素反応速度が、切断を通して、まずモノ置換蛍光類似体に対して、次いで遊離蛍光体に対して起こる。このモノ置換蛍光基質は、遊離蛍光体と同じ波長を吸収し同じ波長で発光するので、この酵素反応速度の解釈はより難しくなる。4−メチルウンベリフェロンのフッ素化誘導体に基づく蛍光原基質が、β−ガラクトシダーゼ活性及び酸性ホスファターゼ活性のアッセイに関して記載されている(Gee,K.R.,et al,(1999),273,41−8)。
【0004】
蛍光ベースの酵素アッセイで使用される色素の多くは、350nm〜550nmの範囲で発光する。より高波長で発光する色素のいくつか、例えばレゾルフィンやDDAOは、チオール類(チオール官能基を有するペプチド類/タンパク類を含む)に対して反応性があり、それらの色素は蛍光特性を失う。したがって、これらの色素は、生体内の用途では有用ではなさそうである。スペクトルの赤又は赤外領域で発光する有用な蛍光特性の色素を有する蛍光ベースの酵素基質は、ニトロ置換されたシアニン色素誘導体である(欧州特許第1086179号:Hamilton,A.et al)。これらの色素の蛍光は、分子内の置換ニトロ基の存在により消光される。このニトロ基がニトロ還元酵素により還元されると、色素は蛍光性となる。これらの蛍光原基質が有用であることが判明し、現在は生細胞アッセイで使用されているが、それらは限られた用途しかない。したがって、関心のある酵素に対して以外は非反応性で、高いSBR(signal to background ratio)を提供する、より高波長(赤又は赤外)で発光する新たな蛍光原酵素基質が明らかに必要である。
【0005】
本発明では、一連の異なる酵素の基質であり、赤及び近赤外を含めた異なる波長で発光する新たな蛍光原酵素基質について説明する。2つの酵素活性の比を決定するための二重酵素基質も提供する。これは、個々の単一酵素基質の吸収特性、分布特性、及び排出特性に差があるために、2つの別々の酵素基質を利用して相対酵素活性を測定することができない生体内システムにより適切である。最後に、酵素活性を決定する方法、ならびに生体外又は生体内の代謝状態を決定する方法も提供する。
【特許文献1】米国特許第4812409号明細書
【特許文献2】欧州特許第1086179号明細書
【非特許文献1】Vieytes M.,et al,Anal.Biochem.,(1997),248,258−64
【非特許文献2】Gee,K.R.,et al,(1999),273,41−8
【非特許文献3】Forster,T.,“Intermolecular Energy Transfer and Fluorescence”,Ann.Physik.,Vol.2,p.55,(1948)
【非特許文献4】Freshney,R.I.,Culture of Animal Cells:A Manual of Basic Technique,2nd Edition,Alan R.Liss Inc.1987
【非特許文献5】“Non−Radioactive Labelling,a Practical Introduction”,Garman,A.J.Academic Press,1997
【非特許文献6】“Bioconjugation−Protein Coupling Techniques for the Biomedical Sciences”,Aslam,M.and Dent,A.,Macmillan Reference Ltd,(1998)
【非特許文献7】Hermanson,G.T.,Bioconjugate Techniques,Academic Press(1996)
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって、本発明は、蛍光標識酵素基質、特に蛍光共鳴エネルギー移動(FRET)標識基質、ならびにFRET標識及び酵素切断可能基を含んでなる基質を切断する際の酵素の活性を測定する方法を提供することを一目的とする。
【0007】
酵素切断活性に影響を及ぼすことがある試験剤をスクリーニングする方法を提供することも本発明の一目的である。
【課題を解決するための手段】
【0008】
したがって、本発明の第1の態様では、式(I)の化合物が提供される。
【0009】
【化1】
式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、
D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、
Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、
Mは、D1の蛍光特性を調節するように選択される酵素切断可能基である。
【0010】
適切には、D1とD2は、適切な条件下で蛍光共鳴エネルギー移動(FRET)が互いに起こるように、連結基Lによってリンクされる。FRETは、2種の色素分子の電子励起状態がフォトンの放出なしに相互作用する、距離に関係するプロセスである。Forster,T.,“Intermolecular Energy Transfer and Fluorescence”,Ann.Physik.,Vol.2,p.55,(1948)を参照のこと。この相互作用による1つの結果は、ドナー分子の励起がアクセプター分子の蛍光発光を促進することである。ドナーの蛍光量子効率は、それに対応して減少する。エネルギー移動効率はドナー分子とアクセプター分子の間の距離(r)の6乗に反比例して減少するので、FRETが起こるためには、適切にはドナー色素分子とアクセプター色素分子が近接近していなければならない(通常10〜100Å)。適切には、本発明において、D1がドナー分子である場合にD2がアクセプターとなるように、また逆も同様となるように、FRETの関係においてD1又はD2をドナー又はアクセプターとすることができる。ドナーとは、その色素部分が光からのエネルギーを吸収する能力があり、一部分以上がアクセプターの吸収スペクトル内にある波長周波数の光を放出することを意味する。アクセプターとは、その色素部分がドナー色素部分によって放出される波長のエネルギーを吸収する能力があることを意味する。適切には、ドナー色素分子の発光スペクトルの一部分以上とアクセプター色素分子の吸収スペクトルとの間に重なりがある。適切には、アクセプター色素の最大発光波長が、ドナー色素の最大発光波長よりも長い。一形式では、ドナー及びアクセプター分子の一方が、第2の蛍光性アクセプター又はドナー蛍光体に近接近している非蛍光性(又は消光性)の蛍光体であることがある。非蛍光色素が励起されると、蛍光エネルギーではなく熱としてエネルギーが散逸され、共鳴エネルギー移動も蛍光発光も起こり得ない。
【0011】
MがD1と共有結合している場合に、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大発光波長Emax=λ1’で蛍光発光する第1の蛍光状態となるように、基Mは適切にはD1の蛍光特性を調節する能力がある酵素切断可能基から選択される。適切には、MをD1から酵素的に切断すると式(I’)の化合物が形成されるように、Mは酵素の基質を含み、(図1参照)、D1が最大発光波長Emax=λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。適切には、D2は最大発光波長がEmax=λ2である蛍光色素である。適切には、式(I)の化合物において、λ1’≠λ1≠λ2である。
【0012】
適切には、連結基Lは、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−、フェニレニル及び下記の基から選択される1種以上の基を適宜含むことができる炭素原子から選択される2〜200個の連結原子を含有する基を含んでなる。
【0013】
【化2】
式中、R’は水素及びC1〜C4アルキルから選択され、mは1〜3の整数である。
【0014】
好ましい一実施形態では、連結基Lは2〜50個の連結原子を含有し、下記構造を有する。
【0015】
−{(CHR’)p−Q−(CHR’)r}s−
式中、Qは、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−及びフェニレニル基から選択され、R’は水素又はC1〜C4アルキルであり、各pは独立して0〜10であり、各rは独立して0〜10であり、sは1、2、又は3である。
【0016】
好ましくは、Qは−C(O)−、−CHR’−、−O−及び−CO−NH−から選択され、その場合のR’、p、r及びsは上に定義されている。
【0017】
別の実施形態では、リンカー基Lはペプチド又はオリゴヌクレオチド断片を含むこともできる。このリンカーがペプチドである、又はペプチド断片を含有する場合、リンカーは2〜20個の自然又は非自然アミノ酸、あるいはそれらの組合せを含有することができる。Lは、より好ましくは2〜12個のアミノ酸を、最も好ましくは2〜8個のアミノ酸を含有する。このリンカー基Lがオリゴヌクレオチドである、又はオリゴヌクレオチド断片を含有する場合、リンカー基Lは2〜20個のヌクレオシド塩基又は修飾塩基を含有することができる。好ましくは、塩基の個数は4〜15個の範囲であり、より好ましくは4〜10個の範囲である。
【0018】
このリンカーは、蛍光色素から延びる成分の一部を含むことができる。言い換えると、このリンカーを色素発色団に結合するが、その一部ではない。本発明の化合物では、リンカーはいずれもドナーとアクセプターの共役を可能にする二重結合のネットワークを含んでいない。最適なエネルギー移動が起こるように、ドナー及びアクセプター蛍光色素の遷移モーメントは、互いに垂直でない方向に配向させる、例えば互いに概ね平行又は直列に位置する。比較的短いリンカー及び最適な配向により、スペクトルの重なりが小さくなっても、効率的な共鳴エネルギー移動が起こり得る。
【発明を実施するための最良の形態】
【0019】
本発明の第1の態様による一実施形態では、D1がドナー色素であり、D2がアクセプター色素である。この実施形態(図1Aに示す)では、D1の最大発光波長(λ1)がD2の最大発光波長(λ2)よりも小さく、D1の発光スペクトルの一部分以上とD2の吸収スペクトルとの間に重なりがある。MがD1と共有結合している場合には、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大蛍光発光波長λ1’で蛍光発光する。ここで、λ1’はλ1よりも小さい。この状態においては、D1の発光スペクトルとD2の吸収スペクトルとの間には重なりがなく、又は実質的には重なりがなく、したがってD1とD2との間のエネルギー移動はない。MをD1から切断すると、D1が最大発光波長λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。その結果、D1の発光スペクトルとD2の吸収スペクトルとの間の重なりが再生され、D1とD2との間のエネルギー移動が起こり得る。したがって、ドナー色素D1を適当な励起波長で励起すると、D2からの蛍光発光の増加を検出及び/又は定量化することができる。
【0020】
第2の実施形態では、D2がドナー色素であり、D1がアクセプター色素である。この実施形態(図1Bに示す)では、D2の最大発光波長(λ2)がD1の最大発光波長(λ1)よりも小さく、D2の発光スペクトルの一部分以上とD1の吸収スペクトルとの間に重なりがある。MがD1と共有結合している場合には、D1が蛍光発光しない又は実質的に蛍光発光しない、あるいは最大蛍光発光波長=λ1’で蛍光発光する。ここで、λ1’はλ1よりも大きい。この状態においては、D2の発光スペクトルとD1の吸収スペクトルとの間には重なりがなく、又は実質的には重なりがなく、したがってD2とD1との間のエネルギー移動はない。MをD1から切断すると、D1が最大発光波長λ1で蛍光発光するようにD1の蛍光特性を第2の蛍光状態へと変化させる。その結果、D2の発光スペクトルとD1の吸収スペクトルとの間の重なりが再生され、エネルギー移動が起こり得る。ドナー色素D2を適当な励起波長で励起すると、D1からの蛍光発光の増加を検出及び/又は定量化することができる。
【0021】
D2がドナー色素であり、D1がアクセプターであるこの実施形態においては、エネルギー移動がドナー色素とアクセプター色素の2つの蛍光状態との間で代わりに起こることがある。この構成では、第1の蛍光状態におけるアクセプター色素がある波長λ1’で発光し、Mが切断されると、第2の波長λ1で発光する第2の蛍光状態を生じる。
【0022】
さらなる実施形態では、D1の第1及び第2の蛍光状態が、それぞれの最大発光波長λ1及びλ1’によってではなく、これらの2つの状態から放出される蛍光の強度によって異なることがある。
【0023】
式(I)の化合物は、蛍光共鳴エネルギー移動を利用するアッセイにおける生化学的切断現象を検出するためのレポーター分子として使用することができる。適切には、基Mは、好ましくはペプチダーゼ、プロテアーゼ、ホスファターゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される切断酵素の基質を含んでなる。MをD1から切断するのに適した条件下でこの基質を酵素で処理することにより、D1の蛍光特性を調節し、それによりD1を上述したように第1の蛍光状態から第2の蛍光状態へ切り替える。
【0024】
好ましい一実施形態では、Mが、D1と共有結合している1つ以上のリン酸基を有し、かつ下記構造を有するリン酸エステル結合を含む。
【0025】
【化3】
式中、nは1〜4の整数である。この実施形態では、色素D1のアルコール誘導体及び無機リン酸を生成するために、細菌アルカリホスファターゼや酸性ホスファターゼなどのホスファターゼによって基質を切断することができる。結果として、D1は第1の蛍光状態から第2の蛍光状態へと切り替えられ、それによりドナー部分とアクセプター部分との間で励起エネルギーの移動が可能となる。ドナー色素がその励起波長で励起されると、アクセプター色素の発光波長における蛍光強度が増加する。リン酸エステルは、予め合成した基質でもよいし、あるいは、化学的加水分解によって、又は以下の構造を有する終端リン酸標識ヌクレオシドポリリン酸からの酵素触媒ヌクレオシド一リン酸もしくはヌクレオシドポリリン酸の移動によってその場で生成することもできる。
【0026】
【化4】
式中、R1及びR2は、独立してH及びOHから選択され、Raはアデニン、グアニン、シトシン、チミン、ウラシル、ヒポキサチン及びキサチンから選択されるヌクレオシド塩基であり、kは1〜6の整数である。
【0027】
別の好ましい実施形態では、Mが、D1に共有結合しているペプチド結合(−CO−NH−)を1つ以上含む。この実施形態では、Mが通常下記構造を有する。
【0028】
【化5】
式中、Rbはペプチド又はタンパクの残基である。ペプチダーゼ又はプロテアーゼによって加水分解すると、Mが蛍光エネルギー移動標識から切断される。ドナー部分をその励起波長で励起すると、ドナー色素とアクセプター色素との間でエネルギーが移動し、それによりアクセプターからの蛍光発光の増加を検出することが可能となる。
【0029】
さらなる実施形態では、Mはグリコシド結合を含み、α−グリコシダーゼ(例えばα−アミラーゼ)やβ−グリコシダーゼ(例えばβ−グルコシダーゼ)などのグリコシダーゼの基質であり、下記構造を有する。
【0030】
【化6】
さらなる実施形態では、Mが、デアルキラーゼの基質であり下記構造を有するエーテル結合を含んでなる。
【0031】
【化7】
式中、RcはC1〜C20の直鎖又は分枝鎖アルキルである。
【0032】
さらなる一実施形態では、基Mがさらに細胞膜透過性の基を含んでなり、この基は次式のエーテル、エステル、アミド、ホスホジエステル基などの基から選択することができる。
【0033】
【化8】
式中、Rdは、非置換又は1つ以上のハロゲン原子で置換されたC1〜C10の直鎖又は分枝鎖アルキル、非置換又は1つ以上のハロゲン原子で置換されたフェニル、あるいはペプチド鎖である。適切には、ハロゲン原子は、フッ素、塩素、臭素及びヨウ素から選択することができる。このような膜透過性の基の有用な例には、酢酸エステル基、ピバロイルエステル基、アセトキシメチルエステル基、ペンタフルオロベンジルエーテル基、ペンタフルオロベンジルアミド基、ペンタフルオロベンジルエステル基、フェニルリン酸基、パーフルオロ−(C1〜C6)アルキルエーテル基、パーフルオロ−(C1〜C6)アルキルエステル基及びパーフルオロ−(C1〜C6)アルキルアミド基が含まれる。このような基は、デアルキラーゼ、プロテアーゼなどのエステラーゼ、及び細胞内に見られるようなホスホジエステラーゼの基質であり、それらによって加水分解される。レポーター分子の細胞透過性の可変性は当業者によって理解されるであろうし、また当業者が試験することが可能である。
【0034】
ドナー色素は、適切にはクマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から、好ましくはキサンチン色素及びシアニン色素から選択することができ、このドナー色素はエネルギーをアクセプター色素に移動させる能力がある。
【0035】
アクセプター色素は、適切にはクマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から選択することができ、このアクセプター色素は、ドナー色素と共にエネルギーを移動させる能力がある。
【0036】
好ましくは、ドナー色素がキサンチン色素又はシアニン色素であり、アクセプター色素がローダミン又はシアニン色素である。
【0037】
好ましい一実施形態では、前記ドナー及びアクセプター色素部分の1つ以上がシアニン色素である。別の好ましい実施形態では、ドナー色素がキサンチン色素であり、アクセプター色素がローダミン色素である。
【0038】
適切なキサンチン色素には、5−カルボキシフルオレセイン、6−カルボキシフルオレセイン、6−カルボキシ−4’,5’−ジクロロ−2’,7’−ジメトキシフルオレセインなどのフルオレセイン及びその誘導体が含まれるが、それだけには限らない。
【0039】
適切なシアニン色素には、CyA(3−(ε−カルボキシペンチル)−3’−エチル−5,5’−ジメチルオキサカルボシアニン)、Cy2(3−(ε−カルボキシペンチル)−3’−エチル−オキサ−カルボシアニン)、Cy3(3−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−カルボシアニン)、Cy3.5(3−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−4,5,4’,5’−(1,3−ジスルフォナト)ジベンゾ−カルボシアニン)、Cy5(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−ジカルボシアニン、Cy5.5(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−4,5,4’,5’−(1,3−ジスルフォナト)−ジベンゾ−ジカルボシアニン)、及びCy7(1−(ε−カルボキシペンチル)−1’−エチル−3,3,3’,3’−テトラメチル−5,5’−ジスルフォナト−トリカルボシアニンが含まれるが、それだけには限らない。
【0040】
適切なローダミンアクセプター色素には、5−カルボキシローダミン(ローダミン110−5)、6−カルボキシローダミン(ローダミン110−6)、5−カルボキシローダミン−6G(R6G−5又はREG−5)、6−カルボキシローダミン−6G(R6G−6又はREG−6)、N,N,N’,N’−テトラメチル−5−カルボキシローダミン、N,N,N’,N’−テトラメチル−6−カルボキシローダミン(TAMRA又はTMR)、5−カルボキシ−X−ローダミン、及び6−カルボキシ−X−ローダミン(ROX)が含まれるがそれだけには限らない。他のクラスの色素には、BODIPY(登録商標)、ポルフィリン色素、rhodol色素及びペリレン色素が含まれる。
【0041】
この第1の態様による蛍光標識酵素基質は、化合物に親水特性を与えるためにそれら基質に結合した水溶化成分を含むこともできる。それら水溶化成分は、好ましくはドナー又はアクセプター色素部分の芳香族環系に結合する。あるいは、連結基Lがこの水可溶化基を含有することもできる。適切な可溶化成分は、アミド、スルホン酸塩、硫酸塩、リン酸塩、四級アンモニウム、ヒドロキシル、グアニジン及びホスホン酸塩からなる群から選択することができる。ドナー及び/又はアクセプター蛍光色素の芳香族環に直接結合したスルホン酸塩又はスルホン酸基が特に好ましい。
【0042】
本発明によるエネルギー移動レポーター分子の例を表1に示す。
【0043】
【表1】
【0044】
【表2】
【0045】
【表3】
例(II)、(III)及び(IV)では、レポーターが消光フルオレセインドナー色素を含有し、またCy5、Cy3及びローダミンアクセプター色素をそれぞれ含有する非蛍光エネルギー移動錯体であり、フルオレセイン部分は、それに結合した酵素的に切断可能なリン酸基を有する。このリン酸基は、ホスファターゼの作用によって切断することができ、それによりドナー色素の蛍光が、フルオレセインをその励起波長で励起すると回復する。この切断現象の検出及びホスファターゼの活性の測定は、アクセプター色素の発光波長での蛍光発光を検出することによって実現することができる。
【0046】
例(V)及び(VI)では、レポーターが、消光フルオレセインドナー色素を含有し、またCy5及びローダミンアクセプター色素をそれぞれ含有する非蛍光エネルギー移動錯体であり、フルオレセイン部分は、それに結合したヌクレオシドポリリン酸塩部分を有する。ヌクレオシジルホスホトランスフェラーゼの作用によりその場でホスファターゼ基質が生成され、ホスファターゼの存在下でリン酸が除去される。フルオレセインドナー色素の蛍光が回復する。続いて起こるアクセプター色素へのエネルギー移動により、赤色にシフトした発光が生じる。このヌクレオシジルホスホトランスフェラーゼ酵素が核酸ポリメラーゼである場合、既知及び未知の核酸の検出、特性決定、定量化に本発明の組成物を使用することができる。
【0047】
一実施形態では、Lが切断可能リンカーであり、酵素基質基Mとは異なる酵素切断可能基Pを含む。Pは、エーテル基、エステル基、アミド基、リン酸ジエステル基などの基から選択することができる。このような基は、それぞれデアルキラーゼ、プロテアーゼなどのエステラーゼ、及びホスホジエステラーゼの基質であり、それらによって水性緩衝媒体中で切断される。この実施形態では、D2がLと結合しているときは第1の発光波長λ2で、D2がLから切断されると第2の発光波長λ2’でD2が発光することが好ましい。ここでλ2≠λ2’である。この実施形態のレポーター化合物は、2つの別々の酵素基質を確実に等しく供給することが困難である生体内及び細胞系内で2つの酵素の相対活性を測定するのに有用である。また、Mと結合しているときは第1の発光波長(λ1)で、Mが切断されると異なる波長(λ1’)でD1が発光することも可能である。ここでλ1≠λ1’である。例(VII)は、アリルスルファターゼ及びカテプシンGの活性に対するこのような二重酵素レポーターを示しており、この二重酵素レポーターは、炎症を検出する際には潜在的に有用である。酵素活性がない場合、例(VII)の化合物を360nmで励起すると610nmで弱い発光がある。645nmの励起では発光は認められない。スルファターゼ活性のみの場合、645nmで励起すると660nmで強い発光が認められ、360nmで励起すると450nm及び660nmで弱い発光がある。カテプシンG活性のみの場合、360nmで励起すると450nmで強い発光が認められるが、360nm又は645nmで励起しても660nmでは発光しない。両方の酵素が存在する場合には、360nmで励起すると450nmで強い発光が認められ、645nmで励起すると660nmで別の強い発光がある。360nmの励起では、660nmの発光は認められない。したがって、660nm及び450nmでの発光を測定し、既知の基準と比較することにより、生体系内における特定の時点又は位置での両酵素の相対酵素活性を決定することが可能である。このような二重酵素活性用基質は、異なる病状を区別する、ならびに病状の臨床病期又は病原力を決定するのに有用である。
【0048】
MMP−2もMMP−9も共に腫瘍の検出及び治療を目標としている。例えば、メラノーマでは、これらのタンパクの発現は大きく変化する。非常に攻撃的な腫瘍ではMMP−9が高レベルで発現するが、早期腫瘍では、過剰発現は認められない。良性腫瘍はMMP−9活性を示さない。同様に、高い活性を示す湿潤がんではMMP−2の活性も大きく変化する。これは、患者の大部分に当てはまるが、すべての患者に当てはまるわけではない。したがって、MMP−2又はMMP−9の活性のみを測定すると、攻撃的な腫瘍を良性であると誤診してしまうことがある。骨粗しょう症では、カテプシンKが、破骨細胞に過剰発現するので、治療標的に認定されているが、この活性は、骨粗しょう症とは関連のない通常の骨代謝回転中にも認められる。骨粗しょう症であっても、高レベルのMMP−9が認められる。しかしながら、MMP−9の活性を測定することは、それほど骨粗しょう症の特異的な指標ではない。したがって、両方の活性及びそれらの比を測定することにより、単一の酵素活性を測定するよりも優れた診断ツールが提供される。
【0049】
本発明の第2の態様では、基質分子に作用する酵素の活性を決定する方法が提供され、この基質は式(I)の化合物を含んでなる。式中のD1、D2及びLは上に定義されており、Mは切断酵素の基質を含んでなる。この方法は、i)蛍光標識基質の蛍光強度を測定する段階と、ii)MをD1から切断するための条件下で、活性を決定すべき酵素を基質と複合させる段階と、iii)段階ii)の複合に続いて、蛍光標識の蛍光強度の変化を測定する段階とを含んでなり、蛍光標識の蛍光強度の変化を使用して酵素の活性を決定する。
【0050】
適切には、式(I)の化合物は、活性を決定すべき酵素の基質であり、Mは、上に定義されているように好ましくはリン酸エステル結合、1つ以上のペプチド結合、エーテル結合及びグリコシド結合から選択される酵素切断可能基を含んでなる。この酵素は好ましくは、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される加水分解酵素である。
【0051】
第2の態様による好ましい一実施形態では、酵素が、PTPase、PPTase−2A、PPTase−2B、又はPPTase−2Cを含めた、E.C.Class3.1から選択されるホスファターゼである。例には、チロシンホスファターゼ、PP1及びPP−2B(セリン/トレオニンホスファターゼ)がある。
【0052】
第2の態様による第2の好ましい実施形態では、酵素がE.C.Class3.4から選択されるペプチダーゼである。例には、アンギオテンシン変換酵素(ACE)、カスパーゼ、カテプシンD、キモトリプシン、ペプシン、サブチリシン、プロテイナーゼK、エラスターゼ、ネプリライシン、サーモライシン、asp−n、マトリックスメタロプロテイナーゼ1〜20、パパイン、プラスミン、トリプシン、エンテロキナーゼ及びウロキナーゼが含まれるが、それだけには限らない。
【0053】
別の実施形態では、酵素が好ましくはE.C.Class3.2からなる群から選択されるグリコシダーゼ、例えば、α−アミラーゼ、β−アミラーゼ、グルカン1,4−α−グルコシダーゼ、セルラーゼ、エンド−1,3−β−グルカナーゼ、オリゴ−1,6−グルコシダーゼ、リゾチームである。
【0054】
さらなる実施形態では、酵素がデアルキラーゼ、例えばシトクロムP−450の異なるイソ酵素である。
【0055】
ペルオキシダーゼ、β−ラクタマーゼ、ニトロ還元酵素など他の酵素を、適当なM又はリンカー構造とともに使用することもできる。これらの酵素の蛍光原基質、例えば、(欧州特許第1086179号,Hamilyton,A.et al)に記載されているような、Amplex red、フルオレセイン及び6−クロロ−7−ヒドロキシクマリン置換セロファロスポリン、ならびにニトロシアニンがよく知られている。同様の構造を、上述の組成物に組み込むこともできる。
【0056】
調査中の酵素反応への試験剤の阻害作用、増強作用、アゴニスト又はアンタゴニスト作用をスクリーニングする用途を含めたハイスループットスクリーニング用途では、本発明に従ってアッセイを行うことができる。したがって、第2の態様による好ましい実施形態では、基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法が提供される。この方法は、(a)試験剤がある状態とない状態とで第2の態様による方法を実施する段階と、(b)この剤がある状態とない状態とで酵素の活性を決定する段階とを含んでなり、前記剤がある状態とない状態との間の酵素の活性の差は、酵素の活性への試験剤の作用を示す。あるいは、試験剤がある状態でこの方法を行い、酵素の活性の値を試験剤のない状態における酵素活性の対照値と比較することによりスクリーニングを実施することもできる。この対照値は、データベースに又は他の電子形式で都合よく電子的に格納することができる。
【0057】
本発明の第3の態様では、式(I)の化合物を含んでなる基質分子に作用する2つの酵素の相対活性を決定する方法が提供される。式中、上に定義されているようにD1は第1の色素部分、D2は第2の色素部分であり、Mは第1の切断酵素の基質を含んでなり、Lは第1の切断酵素とは異なる第2の切断酵素の基質である酵素切断可能基Pを含む。この方法は、i)蛍光標識基質の蛍光発光強度を測定する段階と、ii)MをD1から、またD2をD1から切断するための条件下で、第1及び第2の酵素を基質と複合させる段階と、iii)段階ii)の複合に続いて、第1及び第2の色素部分の蛍光発光強度を測定する段階と、iv)第1及び第2の色素部分の蛍光強度の変化を利用してこれらの酵素の相対活性を決定する段階とを含んでなる。
【0058】
適切には、第1及び第2の酵素を、いずれかの順序で経時的に、あるいは同時に基質分子と複合させることができる。好ましくは、第1及び第2の酵素は、上述したように、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される。
【0059】
本発明の第3の態様による方法を使用して、基質分子に作用する2つの酵素の相対活性への試験剤の作用を決定することもできる。その場合、試験剤がない状態とある状態とで複合段階ii)を実施する。前記剤がある状態とない状態との間の第1及び第2の酵素の相対活性の差は、第1及び第2の酵素の相対活性への試験剤の作用を示す。
【0060】
この試験剤は、例えば、合成分子や天然物(例えばペプチド、オリゴヌクレオチド)など任意の有機又は無機化合物、あるいはエネルギー形態(例えば光又は熱、あるいは他の形態の電磁放射線)でよい。この好ましい実施形態による方法を用いて、酵素活性の阻害剤を検出することもできる。例えば、公知の阻害剤ペプスタチンAがカテプシンDの酵素活性を阻害することは、本発明の方法を用いることで容易に示すことができる。したがって、12ナノモルのペプスタチンAがある状態では、カテプシンDの活性は、阻害剤がない状態の約5分の1である。
【0061】
適切には、剤がない状態とある状態との間の酵素活性の差は、規格化され、電子的に格納され、基準値と比較される。したがって、例えば、活性の差を阻害率(%)(又は刺激率(%))として電子データベースに格納し、この値を問題となっている酵素の基準阻害剤での対応する値と比較することができる。このようにして、ある所定のしきい値(例えば、基準化合物と同等以上に有効であること)を満たす試験剤のみを、さらなる試験の対象として選択することができる。
【0062】
一例としては、タンパク分解酵素の活性を検出するためのアッセイを以下のように構成することができる。酵素と式(I)による蛍光エネルギー移動レポーター化合物を複合させることにより、反応混合物を調製する。式中、基Mはタンパク分解酵素切断部位を含んでなる。このアッセイは、24個、96個、384個又はより高密度のウェル、例えば1536個のウェルを有するマルチウェルプレート、例えばマイクロタイタプレートのウェル内で適切に行われる。この反応は、酵素基質が最初に存在する水性アッセイ緩衝液中で、適切には5mMのMgCl2を含有する10mMのMOPs、50mMのTris又は50mMのHEPES中で行うことができる。既知の又は推定上の阻害剤などの試験剤を、この反応混合物に適宜含めることができる。酵素を添加すると、通常反応を最後まで進行させることができ、蛍光光度計を用いて記録される蛍光性アクセプター色素による定常状態の蛍光発光を観察することにより進行がモニターされる。あるいは、反応を所定の時間進行させ、その後、多くの場合非特異的である停止試薬、通常酵素活性の阻害剤で停止させることができる「停止」条件下で、このアッセイを行うこともできる。停止試薬の例にはEDTAがあり、この試薬を使用して通常酵素活性に必要とされる金属イオンを分離する。
【0063】
本発明による方法を利用して、細胞環境における、本発明による化合物を含んでなる基質に作用する酵素の活性を測定することもできる。したがって、第2及び第3の態様による特定の実施形態では、その方法が、流動媒体中の1つ以上の細胞に基質を加える段階を段階i)の前に含んでなる。
【0064】
通常、培養細胞は、細胞増殖に適した条件下で、0.5時間から24時間までに及び得るある期間、適切な細胞培養液中0.1〜100μMの濃度のFRET結合酵素基質で培養される。細胞は、標準的な細胞培養技術に従って培養される。例えば、加湿した空気95%/CO25%の雰囲気を含む培養器内の、37℃の無菌環境における適切な容器内で細胞が培養される。多種多様な細胞型の培養に利用可能なプロトコールが確立されている(例えば、Freshney,R.I.,Culture of Animal Cells:A Manual of Basic Technique,2nd Edition,Alan R.Liss Inc.1987を参照のこと。)。細胞培養又は組織培養において培養される細胞に基質を導入する必要がある場合、基質を単に培養液に添加する。酵素活性への作用を決定すべき試験剤がある状態で、細胞を基質に接触させることもできる。この実施形態では、検出段階により、調査中の酵素の活性への試験剤の作用が測定される。
【0065】
光電子倍増管が検出器として組み込まれた器具を用いて蛍光強度の測定を行うことができる。荷電結合素子(CCD)撮像装置(走査型撮像装置や面走査撮像素子など)によって蛍光強度の変化を測定して、マイクロタイタプレートのウェルのすべてを撮像することができる。LEADseeker(登録商標)システムは、CCDカメラを特徴とし、単一パスで高密度マイクロタイタプレートの蛍光画像化を可能にする。画像化は定量的かつ高速であり、画像化用途に適した器具使用により、現在マルチウェルプレート全体を同時に撮像することができる。酵素活性に対する試験剤の活性を検出するためにアッセイの形式を定めようとする場合、基質の蛍光の連続測定下でアッセイを行うことができる。この形式では、蛍光標識基質の強度は連続的に変化する。この標識基質は、酵素反応の生成物から分離する必要がなく、したがって反応の時間経過を得ることができ、反応速度研究をリアルタイムで行うことが可能となる。
【0066】
本発明の第4の態様では、1種以上の酵素の活性を測定するための試験キットが提供される。この試験キットは1種以上の異なる酵素基質を含んでなり、前記基質の各々は式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる。好ましくは、この試験キットは、各酵素が異なる前記基質に特異的な1種以上の異なる酵素をさらに含んでなる。
【0067】
当業者にはよく知られている直接化学結合法を用いて蛍光エネルギー移動部分D1−L−D2を基Mと共有結合することによって、式(I)の化合物を調製することができる。ここで、D1、D2、M及びLは上に定義されている。あるいは、実施例に示すようなデノボ合成法によって本発明の化合物を調製することもできる。最初に基MをD1に結合して中間化合物M−D1を形成し、続いてリンカー基Lを介してD2を結合することができる。D2を共有結合する前に、リンカー基LをM−D1に都合よく共有結合してもよく、あるいはLを前もってD2に結合してもよい。本発明で用いるペプチド、タンパク及びオリゴヌクレオチド基質を終端位置で、あるいは1つ以上の内部位置で標識化することができる。蛍光色素標識試薬を用いたタンパク標識の検討及び例には、“Non−Radioactive Labelling,a Practical Introduction”,Garman,A.J.Academic Press,1997、“Bioconjugation−Protein Coupling Techniques for the Biomedical Sciences”,Aslam,M.and Dent,A.,Macmillan Reference Ltd,(1998)を参照のこと。合成ペプチドにおける部位特異的な標識を得るためのプロトコールが利用可能である。例えば、Hermanson,G.T.,Bioconjugate Techniques,Academic Press(1996)を参照のこと。
【0068】
本発明の原理及び機能を明確化するために、以下の実施例についてこれから言及するが、添付の特許請求の範囲及び本発明の範囲を限定すると解釈すべきではない。本発明は、本明細書に提供した教示から明らかとなる任意の及びすべての変形形態を包含すべきものである。
【実施例】
【0069】
1. FRET結合レポーター分子の調製(化合物(V)及び化合物(VI)(図2参照)
1.1. 4’(5’)−フルオレセインジエチルエーテル−エステル:化合物(2)
NaOH(1.44g)のメタノール(145ml)溶液に、フルオレセイン(6.0g)を加えて暗色溶液を生成し、この溶液を真空中で濃縮乾燥した。これを無水DMF(2×50ml)と同時蒸発させ、無水DMF(150ml)に再溶解した。ヨウ化エチル(11.6ml、8eq)を加え、反応物を室温で一晩中攪拌した。この反応混合物を濃縮した後、重炭酸ナトリウム飽和溶液(150ml)に溶解し、ジエチルエーテルで洗浄した。この溶液を冷却すると黄色い沈殿物が形成され、この沈殿物をろ過し、最後にエーテル(2×25ml)及びエーテル/ペンタン(1:1、50ml)で洗浄した。
【0070】
1.2. 4’(5’)−クロロアセチルアミノメチル−3’(6’)−エチルフルオレセインジエチルエーテル−エステル:化合物(3)
エチルフルオレセインエステル(化合物(2)、2.67g)の50ml硫酸攪拌溶液に、クロロアセチルアセトアミド(850mg)をゆっくりと5分間かけて加えた。この反応混合物を、室温で一晩攪拌した。その反応混合物を砕氷上に注ぎ、形成された沈殿物をろ過し、冷水で洗浄した。TLC及び質量分析により、モノ及びビスクロロアセトアミド化合物の形成が示された。これらの生成物を、塩化メチレン中2〜15%のメタノールを用いたシリカゲルカラムクロマトグラフィーによって精製した。まずビスアルキル化生成物を溶出し、その後2種のモノアルキル化生成物を溶出した。主生成物であるより低い分子量のモノアルキル化生成物をさらなる反応に使用した。
【0071】
1.3. 4’(5’)−アミノメチル−3’(6’)−エチルフルオレセイン:化合物(5)
主生成物のモノアルキル化生成物(化合物(3)、1g)を、2.5時間の加熱によって6NのHCl(15ml)で加水分解した。この生成物をCH2Cl2で抽出し、水で洗浄し、無水硫酸ナトリウムで乾燥した後、シリカゲルカラムクロマトグラフィーによって精製して500mgの純粋化合物を得た。MS ES+:390.3
1.4. 4’(5’)−トリフルオロアセチルアミドメチル−3’(6’)−エチルフルオレセイン:化合物(6)
化合物(5)(390mg)を無水ピリジン(5ml)中に入れ、TFA−NHS(650mg)を加えた。この混合物を室温で一晩攪拌した。真空中で濃縮し、塩化メチレンで抽出した後、この生成物をシリカゲルクロマトグラフィーによって精製して350mgの純粋生成物を得た。MS ES+:486.3
1.5. 4’(5’)−トリフルオロアセチルアミドメチル−3’(6’)−エチルフルオレセイン−6’(3’)−リン酸:化合物(7)
化合物(6)(200mg)のアセトニトリル(5ml)溶液に、POCl3(189mg、3eq)を加え、この反応混合物を室温で1時間攪拌した。ピリジン(290μl、9eq)を加え、この反応物をさらに2時間攪拌した。TEAB(0.1M、10ml)を加えることによってこの反応物を失活させた。1時間後、この混合物を真空中で濃縮し、CH2Cl2/メタノールのグラジェントを用いたシリカゲルクロマトグラフィーによって精製して130mgの一リン酸を得た。MS ES−:564.5
1.6. 4’(5’)−アミノメチル−3’(6’)−エチルフルオレセイン−6’(3’)−四リン酸塩−チミジン:化合物(8)
1.6.1. 化合物(7)イミダゾライド誘導体の合成
化合物(7)(60mg)の無水DMF(2ml)溶液に、トリブチルアミン(30μl、3eq)を加え、この混合物を真空中で濃縮乾燥した。その混合物を無水DMF(2ml)に再溶解し、攪拌しながらカルボニルジイミダゾール(85mg、5eq)を加えた。この混合物を室温で2時間攪拌し、HPLC−MSによって反応終了を確認した。この混合物をメタノール(100μl)で失活させ、室温で1時間攪拌し、真空中で濃縮乾燥し、無水DMF(2ml)に再溶解した。MS ES−:614.6
1.6.2. 化合物(7)イミダゾライド誘導体のチミジン−5’−三リン酸への付加
チミジン−5’−三リン酸(トリエチルアンモニウム塩、100μmol)を、無水DMF(2×2ml)中のトリブチルアミン(400μmol)と同時蒸発させ、最終的に無水DMF(2ml)に再溶解した。
【0072】
上記チミジン−5’−三リン酸(TTP)のトリブチルアンモニウム塩溶液に、イミダゾライド誘導体(段階1.6.1による)を加え、この混合物を室温で一晩攪拌した。HPLC−MSにより、>90%の完全な反応であることが示された。これをさらに5時間攪拌した。
【0073】
1.6.3. 段階1.6.2による反応混合物の濃縮及びNH4OH(30%溶液、10ml)の添加
この混合物を室温で2.5時間攪拌し、冷蔵庫に一晩放置した。その混合物を真空中で濃縮乾燥し、アニオン交換クロマトグラフィーによって精製した。この混合物をヘビ毒ホスホジエステラーゼ(SVD)及びアルカリホスファターゼで一晩処理し(未反応TTPを除去するため)、さらに0.1MのTEAB中25%のアセトニトリルに対して0.1MのTEAB(緩衝液A)を用いた逆相クロマトグラフィーによって精製した。HPLC−MSによって2つの主なピークを収集し、分析した。第1の主なピークは931.8で質量を示し、第2のピークでは所要の932.8で質量を示した。
【0074】
トリフルオロアセチル基の加水分解中のアンモニア処理ではスピロラクトンとも反応し、結果として不要なアミド化合物が形成されたことが明らかであった。ピーク2を真空中で濃縮乾燥し、メタノール(2×5ml)と同時蒸発させ、H2O(1ml)に再溶解した。その濃度はUVによって測定した。全部で5.5μmolの純粋化合物を得た。
【0075】
1.7. 化合物(8)へのアクセプター色素の付加:化合物(V)及び(VI)の合成
化合物(8)(2.2μmol、400μlの5.5mM溶液)2つの別々の反応容器に、400μlの0.5M炭酸ナトリウム/重炭酸ナトリウム緩衝液(pH8.5)を加え、一方の容器には5−ROX−NHSエステル(2mlのDMF中5.5mg)を、もう一方の容器にはCy5−NHSエステル(2mlのDMF中5mg)を加えた。両方の反応混合物を室温で一晩攪拌し、HPLC−MSによって反応の終了をモニターした。この反応混合物を真空中で濃縮し、0.1MのTEAB中の25%アセトニトリル(緩衝液A)及び1MのTEAB中の25%アセトニトリル(緩衝液B)を用いたイオン交換カラムで最初に精製した。生成物を含有するピークを真空中で濃縮し、さらに逆相HPLCカラムによって再精製した。正確な画分を収集し、真空中で濃縮し、紫外分光法によって定量化した。化合物(V):MS ES−:1449;化合物(VI):MS ES−:1572
2. 化合物(II)、(III)及び(IV)の合成
出発原料が(化合物(7)をメタノール中でK2CO3により加水分解することによって得られた)4’(5’)−アミノメチル−エトキシフルオレセインリン酸であることを除いては、化合物(V)及び(VI)のための上述のやり方で合成を行った。生成物をHPLCによって精製し、紫外分光法によって定量化した。
【0076】
化合物(II):MS ES−:1107.3;
化合物(III):MS ES−:1342.4;
化合物(IV):MS ES−:881
3. アルキルホスファターゼの化合物(II)、(III)及び(IV)に対する作用
【0077】
【化9】
化合物II、III及びIVを、25mMのHEPES及び5mMのMgCl2緩衝液(pH8.5)中に入れ、室温で2時間アルカリホスファターゼにより処理した。反応を分光法で追跡した結果、反応が30分以内にほとんど終了したことが示された。
【0078】
4. 化合物(II)、(III)、(IV)、(VIII)、(IX)及び(X)のUV、蛍光及びエネルギー移動測定
リン酸化色素(化合物(II)、(III)及び(IV))及び非リン酸化色素(化合物(VIII)、(IX)及び(X))のUV及び蛍光スペクトルを図2〜8に示す。アルカリホスファターゼで処理すると約472nmにピークが出現することを、UVスペクトルがはっきりと示している。
【0079】
本発明の特定の望ましい諸実施形態を本明細書で説明してきたが、企図した本発明の範囲から逸脱することなくそれらの実施形態に変更を加えることができることを理解されたい。本発明の真の範囲は、添付の特許請求の範囲に示される。
【図面の簡単な説明】
【0080】
【図1】本発明の好ましい諸実施形態を示す概略図である。
【図2】化合物(V)及び(VI)を調製するための合成反応スキームを示す図である。
【図3】アルカリホスファターゼによる処理前及び処理後の化合物(II)のUVスペクトルを示す図である。
【図4】アルカリホスファターゼによる処理前及び処理後の化合物(III)のUVスペクトルを示す図である。
【図5】アルカリホスファターゼによる処理前及び処理後の化合物(IV)のUVスペクトルを示す図である。
【図6】アルカリホスファターゼによる処理前及び処理後の化合物(II)の、455nm及び475nm励起による発光スペクトルを示す図である。
【図7】アルカリホスファターゼによる処理前及び処理後の化合物(III)の、455nm及び475nm励起による発光スペクトルを示す図である。
【図8】アルカリホスファターゼによる処理前及び処理後の化合物(IV)の、455nm及び475nm励起による発光スペクトルを示す図である。
【特許請求の範囲】
【請求項1】
次式の化合物。
【化1】
(式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、
D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、
Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、
Mは、D1の前記蛍光特性を調節するように選択される酵素切断可能基である。)
【請求項2】
Mが酵素の基質を含んでなる、請求項1記載の化合物。
【請求項3】
前記酵素が、ペプチダーゼ、プロテアーゼ、ホスファターゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される、請求項2記載の化合物。
【請求項4】
Mが下記の基である、請求項1乃至請求項3記載の化合物。
【化2】
(式中、nは1〜4の整数である。)
【請求項5】
Mが、D1に共有結合しているペプチド結合(−CO−NH−)を1つ以上含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項6】
Mが、グリコシダーゼの基質であるグリコシド結合を含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項7】
Mが、デアルキラーゼの基質であるエーテル結合を含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項8】
Mが、細胞膜透過性の基をさらに含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項9】
前記細胞膜透過性の基が下記の基から選択される、請求項8記載の化合物。
【化3】
(式中、Rdは非置換又は1つ以上のハロゲン原子で置換されたC1〜C10の直鎖又は分枝鎖アルキル、非置換又は1つ以上のハロゲン原子で置換されたフェニル、あるいはペプチド鎖である。)
【請求項10】
連結基Lが、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−、フェニレニル及び下記の基から選択される1種以上の基を適宜含むことができる炭素原子から選択される、請求項1乃至請求項9のいずれか1項記載の化合物。
【化4】
(式中、R’は水素及びC1〜C4アルキルから選択され、mは1〜3の整数である。)
【請求項11】
連結基Lがペプチド又はオリゴヌクレオチド断片を含んでなる、請求項1乃至請求項9のいずれか1項記載の化合物。
【請求項12】
Lが切断可能リンカーであり、前記酵素基質基Mとは異なる酵素切断可能基Pを含む、請求項1乃至請求項9のいずれか1項記載の化合物。
【請求項13】
前記ドナー色素が、クマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から、好ましくはキサンチン色素及びシアニン色素から選択され、前記ドナー色素がエネルギーを前記アクセプター色素に移動させる能力がある、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項14】
前記アクセプター色素が、クマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から選択され、前記アクセプター色素が前記ドナー色素と共にエネルギー移動能力がある、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項15】
前記ドナー色素がキサンチン色素又はシアニン色素であり、前記アクセプター色素がローダミン又はシアニン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項16】
前記ドナー及びアクセプター色素部分の1つ以上がシアニン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項17】
前記ドナー色素がキサンチン色素であり、前記アクセプター色素がローダミン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項18】
スルホン酸塩、スルホン酸、リン酸塩、ホスホン酸塩、四級アンモニウム、及びヒドロキシルからなる群から選択される、結合した水溶化成分をさらに含む、請求項1乃至請求項17のいずれか1項記載の化合物。
【請求項19】
基質分子に作用する酵素の活性を決定する方法において、前記基質が式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる方法であって、
i)前記蛍光標識基質の蛍光強度を測定する段階と、
ii)MをD1から切断するための条件下で、活性を決定すべき酵素を前記基質と組み合わせる段階と、
iii)前記段階ii)の組合せに続いて、前記蛍光標識の蛍光強度の変化を測定する段階とを含んでなり、
前記蛍光標識の蛍光強度の前記変化を使用して前記酵素の活性を決定する方法。
【請求項20】
Mが、リン酸エステル連結部、1つ以上のペプチド連結部、エーテル連結部及びグリコシド連結部から選択される酵素切断可能基を含んでなる、請求項19記載の方法。
【請求項21】
前記酵素が、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される加水分解酵素である、請求項19記載の方法。
【請求項22】
基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法であって、
(a)前記試験剤がある状態とない状態とで請求項19乃至請求項21のいずれか1項記載の前記方法を実施する段階と、
(b)前記試験剤がある状態とない状態とで前記酵素の活性を決定する段階とを含んでなり、
前記試験剤がある状態とない状態との間の前記酵素の活性の差が、前記酵素の活性への前記試験剤の作用を示す方法。
【請求項23】
基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法であって、
(a)前記試験剤がある状態で請求項19乃至請求項21のいずれか1項記載の前記方法を実施する段階と、
(b)前記酵素の活性の値を前記試験剤のない状態における酵素活性の対照値と比較する段階とを含んでなる方法。
【請求項24】
前記対照値を、データベースに又は他の電子形式で電子的に格納する、請求項23記載の方法。
【請求項25】
式(I)の化合物を含んでなる基質分子に作用する2つの酵素の相対活性を決定する方法において、式中、上に定義されているようにD1は第1の色素部分、D2は第2の色素部分であり、Mは第1の切断酵素の基質を含んでなり、Lは前記第1の切断酵素とは異なる第2の切断酵素の基質である酵素切断可能基Pを含む方法であって、
i)前記蛍光標識基質の蛍光発光強度を測定する段階と、
ii)MをD1から、またD2をD1から切断するための条件下で、前記第1及び第2の酵素を前記基質と組み合わせる段階と、
iii)前記段階ii)の組合せに続いて、前記第1及び第2の色素部分の蛍光発光強度を測定する段階と、
iv)前記第1及び第2の色素部分の蛍光強度の変化を利用して前記酵素の相対活性を決定する段階とを含んでなる方法。
【請求項26】
前記第1及び第2の酵素が、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される、請求項25記載の方法。
【請求項27】
前記第1及び第2の酵素の相対活性への作用を決定すべき試験剤がない状態とある状態とで前記組合せ段階ii)が実施され、前記試験剤がある状態とない状態との間の前記第1及び第2の酵素の相対活性の差が、前記第1及び第2の酵素の相対活性への前記試験剤の作用を示す、請求項25又は請求項26記載の方法。
【請求項28】
1種以上の異なる酵素基質を含んでなる、酵素の活性を測定するための試験キットであって、前記基質の各々が式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる試験キット。
【請求項29】
各酵素が異なる前記基質に特異的な1種以上の異なる前記酵素をさらに含んでなる、請求項28記載の試験キット。
【請求項1】
次式の化合物。
【化1】
(式中、D1は第1の色素部分であり、その蛍光特性は、エネルギー移動構成におけるドナー又はアクセプターとして適するよう調節することができ、
D2は、前記第1の色素とのエネルギー移動構成においてアクセプター又はドナーとして適切な第2の色素部分であり、
Lは、2〜200個の連結原子を含んでなり、また酵素切断部位を適宜含む連結基であり、
Mは、D1の前記蛍光特性を調節するように選択される酵素切断可能基である。)
【請求項2】
Mが酵素の基質を含んでなる、請求項1記載の化合物。
【請求項3】
前記酵素が、ペプチダーゼ、プロテアーゼ、ホスファターゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される、請求項2記載の化合物。
【請求項4】
Mが下記の基である、請求項1乃至請求項3記載の化合物。
【化2】
(式中、nは1〜4の整数である。)
【請求項5】
Mが、D1に共有結合しているペプチド結合(−CO−NH−)を1つ以上含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項6】
Mが、グリコシダーゼの基質であるグリコシド結合を含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項7】
Mが、デアルキラーゼの基質であるエーテル結合を含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項8】
Mが、細胞膜透過性の基をさらに含んでなる、請求項1乃至請求項3のいずれか1項記載の化合物。
【請求項9】
前記細胞膜透過性の基が下記の基から選択される、請求項8記載の化合物。
【化3】
(式中、Rdは非置換又は1つ以上のハロゲン原子で置換されたC1〜C10の直鎖又は分枝鎖アルキル、非置換又は1つ以上のハロゲン原子で置換されたフェニル、あるいはペプチド鎖である。)
【請求項10】
連結基Lが、−C(O)−、−NR’−、−O−、−S−、−CH=CH−、−CO−NH−、フェニレニル及び下記の基から選択される1種以上の基を適宜含むことができる炭素原子から選択される、請求項1乃至請求項9のいずれか1項記載の化合物。
【化4】
(式中、R’は水素及びC1〜C4アルキルから選択され、mは1〜3の整数である。)
【請求項11】
連結基Lがペプチド又はオリゴヌクレオチド断片を含んでなる、請求項1乃至請求項9のいずれか1項記載の化合物。
【請求項12】
Lが切断可能リンカーであり、前記酵素基質基Mとは異なる酵素切断可能基Pを含む、請求項1乃至請求項9のいずれか1項記載の化合物。
【請求項13】
前記ドナー色素が、クマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から、好ましくはキサンチン色素及びシアニン色素から選択され、前記ドナー色素がエネルギーを前記アクセプター色素に移動させる能力がある、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項14】
前記アクセプター色素が、クマリン色素、ベンゾクマリン色素、アクリドン色素、キサンチン色素、フェノキサジン色素、ローダミン色素、メロシアニン色素、及びシアニン色素から選択され、前記アクセプター色素が前記ドナー色素と共にエネルギー移動能力がある、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項15】
前記ドナー色素がキサンチン色素又はシアニン色素であり、前記アクセプター色素がローダミン又はシアニン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項16】
前記ドナー及びアクセプター色素部分の1つ以上がシアニン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項17】
前記ドナー色素がキサンチン色素であり、前記アクセプター色素がローダミン色素である、請求項1乃至請求項12のいずれか1項記載の化合物。
【請求項18】
スルホン酸塩、スルホン酸、リン酸塩、ホスホン酸塩、四級アンモニウム、及びヒドロキシルからなる群から選択される、結合した水溶化成分をさらに含む、請求項1乃至請求項17のいずれか1項記載の化合物。
【請求項19】
基質分子に作用する酵素の活性を決定する方法において、前記基質が式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる方法であって、
i)前記蛍光標識基質の蛍光強度を測定する段階と、
ii)MをD1から切断するための条件下で、活性を決定すべき酵素を前記基質と組み合わせる段階と、
iii)前記段階ii)の組合せに続いて、前記蛍光標識の蛍光強度の変化を測定する段階とを含んでなり、
前記蛍光標識の蛍光強度の前記変化を使用して前記酵素の活性を決定する方法。
【請求項20】
Mが、リン酸エステル連結部、1つ以上のペプチド連結部、エーテル連結部及びグリコシド連結部から選択される酵素切断可能基を含んでなる、請求項19記載の方法。
【請求項21】
前記酵素が、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される加水分解酵素である、請求項19記載の方法。
【請求項22】
基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法であって、
(a)前記試験剤がある状態とない状態とで請求項19乃至請求項21のいずれか1項記載の前記方法を実施する段階と、
(b)前記試験剤がある状態とない状態とで前記酵素の活性を決定する段階とを含んでなり、
前記試験剤がある状態とない状態との間の前記酵素の活性の差が、前記酵素の活性への前記試験剤の作用を示す方法。
【請求項23】
基質を切断する際の酵素の活性への作用を決定すべき試験剤をスクリーニングする方法であって、
(a)前記試験剤がある状態で請求項19乃至請求項21のいずれか1項記載の前記方法を実施する段階と、
(b)前記酵素の活性の値を前記試験剤のない状態における酵素活性の対照値と比較する段階とを含んでなる方法。
【請求項24】
前記対照値を、データベースに又は他の電子形式で電子的に格納する、請求項23記載の方法。
【請求項25】
式(I)の化合物を含んでなる基質分子に作用する2つの酵素の相対活性を決定する方法において、式中、上に定義されているようにD1は第1の色素部分、D2は第2の色素部分であり、Mは第1の切断酵素の基質を含んでなり、Lは前記第1の切断酵素とは異なる第2の切断酵素の基質である酵素切断可能基Pを含む方法であって、
i)前記蛍光標識基質の蛍光発光強度を測定する段階と、
ii)MをD1から、またD2をD1から切断するための条件下で、前記第1及び第2の酵素を前記基質と組み合わせる段階と、
iii)前記段階ii)の組合せに続いて、前記第1及び第2の色素部分の蛍光発光強度を測定する段階と、
iv)前記第1及び第2の色素部分の蛍光強度の変化を利用して前記酵素の相対活性を決定する段階とを含んでなる方法。
【請求項26】
前記第1及び第2の酵素が、ホスファターゼ、ペプチダーゼ、プロテアーゼ、デアルキラーゼ及びグリコシダーゼからなる群から選択される、請求項25記載の方法。
【請求項27】
前記第1及び第2の酵素の相対活性への作用を決定すべき試験剤がない状態とある状態とで前記組合せ段階ii)が実施され、前記試験剤がある状態とない状態との間の前記第1及び第2の酵素の相対活性の差が、前記第1及び第2の酵素の相対活性への前記試験剤の作用を示す、請求項25又は請求項26記載の方法。
【請求項28】
1種以上の異なる酵素基質を含んでなる、酵素の活性を測定するための試験キットであって、前記基質の各々が式(I)の化合物を含んでなり、式中のD1、D2、Lは上に定義されており、Mは切断酵素の基質を含んでなる試験キット。
【請求項29】
各酵素が異なる前記基質に特異的な1種以上の異なる前記酵素をさらに含んでなる、請求項28記載の試験キット。
【図1】

【図2】
【図3】
【図4】
【図5】

【図6】

【図7】
【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2007−533828(P2007−533828A)
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願番号】特願2007−509542(P2007−509542)
【出願日】平成17年4月18日(2005.4.18)
【国際出願番号】PCT/US2005/013141
【国際公開番号】WO2005/108994
【国際公開日】平成17年11月17日(2005.11.17)
【出願人】(396019387)ジーイー・ヘルスケア・アクスイェ・セルスカプ (82)
【Fターム(参考)】
【公表日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願日】平成17年4月18日(2005.4.18)
【国際出願番号】PCT/US2005/013141
【国際公開番号】WO2005/108994
【国際公開日】平成17年11月17日(2005.11.17)
【出願人】(396019387)ジーイー・ヘルスケア・アクスイェ・セルスカプ (82)
【Fターム(参考)】
[ Back to top ]