
蛍光性アミラーゼ基質及び該基質を用いたアミラーゼ活性測定方法
【課題】本発明は、新規アミラーゼ活性測定用基質及び該基質を含有したアミラーゼ活性測定試薬、並びに該基質又は試薬を使用したアミラーゼ活性測定方法の提供を目的とする。
【解決手段】
一般式(I)

(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質及び該基質を含有する試薬、並びに該基質又は試薬を用いたアミラーゼ活性の測定方法。
【解決手段】
一般式(I)
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質及び該基質を含有する試薬、並びに該基質又は試薬を用いたアミラーゼ活性の測定方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規アミラーゼ活性測定用基質及び該基質を含有したアミラーゼ活性測定試薬、並びに該基質又は試薬を使用したアミラーゼ活性測定方法に関する。
【背景技術】
【0002】
α−アミラーゼは、デンプンをはじめとするマルトオリゴ糖のα1,4結合を加水分解する酵素である。法医学分野においては唾液であることの証明、大便であることの証明、膣分泌液であることの証明等に用いられている。また、医療分野においては血液検査の指標の一つとして、膵臓疾患等のマーカーとして用いられてきた。法医学分野においては、一般的に、劣悪な環境に置かれた試料からのアミラーゼ活性の測定となるため、酵素の多くが失活していることが想定され、従って、できるだけ高感度の検査法が必要とされている。
【0003】
また、法医学、医療両分野において、アミラーゼのアイソザイム分析は重要である。アミラーゼは、主として唾液腺細胞、膵腺房細胞から分泌されており、免疫活性、酵素の安定性などの違いにより、S型(唾液由来)とP型(唾液以外、例えば膵液由来)の2つのアイソザイムに分類されている。医療分野においては、アミラーゼ活性値の異常が認められ場合、活性値の異常を示すアイソザイム活性を測定することにより、疾患の原因臓器の特定が可能となる。例えば、体液又は尿中のP型アミラーゼ活性が高ければ膵炎などの膵臓疾患が疑われ、S型アミラーゼ活性が高ければ耳下腺炎又は顎下腺炎など唾液腺疾患の可能性が示唆される。
【0004】
現在、アミラーゼ活性を測定する方法として、ブルースターチ法、ヨードデンプン法、酵素法(特許文献1)などが使用されている。しかし、これらの方法は、低感度性、操作の煩雑さ、基質構造の不明確さ、などの問題点が指摘されている。また、FRETを利用したマルトオリゴ糖誘導体を基質とするアミラーゼ活性測定方法(非特許文献1)も報告されており、測定感度の改善が期待されているが、蛍光のバックグラウンドの影響により低いアミラーゼ活性の測定における困難性が未だ存在しており、さらなる改善が望まれている。一方、アミラーゼアイソザイムを識別する方法としては、等電点電気泳動、ELISA、基質抗体阻害法(特許文献2)等が報告されているが、いずれの方法も操作が煩雑であり、結果を得るまでに長時間を要していた。
【0005】
【非特許文献1】Nishimura,Carbohydr.Let.,4:77-84 2001
【特許文献1】特許第3901990号
【特許文献2】特許第3862058号
【発明の開示】
【発明が解決しようとする課題】
【0006】
以上のように、アミラーゼ活性の高感度測定方法並びにアミラーゼアイソザイムの識別に関する簡便で正確な測定方法を開発すべく多くの研究が行われている。
本発明は、簡便で高感度にアミラーゼ活性を検出することが可能であり、また、アミラーゼアイソザイムの識別をも可能とする基質及び試薬の提供、並びに、該基質又は試薬を用いたアミラーゼ活性の測定方法の提供を目的とする。
【課題を解決するための手段】
【0007】
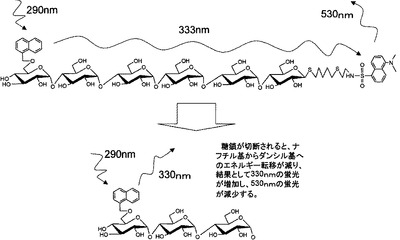
本発明者らは、マルトオリゴ糖の非還元末端をナフチル基、還元末端をダンシル基で化学修飾した基質により、FRETを利用した測定方法の感度を向上させ、さらに、該基質をアミラーゼアイソザイムの識別に効果的に利用できることを見出し、本発明を完成させた。
FRETを利用したアミラーゼ活性の測定法に関する従来技術(非特許文献1)においては、ナフチル基からの蛍光(333nm)のバックグラウンドが元々高いことから、微量のアミラーゼ活性が測定誤差の範囲に入ってしまい検出が困難であった。この点について、本発明の基質を用いると、333nmの蛍光のバックグラウンドを従来技術(非特許文献1)の約10分の1に抑えることが可能となり、微量なアミラーゼ活性の測定が可能となった。さらに、本発明の基質を用いると、測定感度が向上されることから、アミラーゼアイソザイムを識別することも可能となる。
【0008】
すなわち、本発明は、一般式(I)
【化3】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質及び該基質を含有する試薬、並びに該基質又は試薬を用いたアミラーゼ活性の測定方法を提供する。
さらに、本発明の基質又は該基質を含有する試薬は、アミラーゼアイソザイムの識別にも使用することができる。
【発明の効果】
【0009】
本発明の基質又は該基質を含有する試薬を用いると、従来法に比較して、アミラーゼの検出感度を顕著に向上させることができる(例えば、ブルースターチ法と比較して、約10倍程度)。
【0010】
また、本発明のアミラーゼ活性測定方法を用いると、アミラーゼアイソザイム(例えば、P型、S型)の識別が可能となる。
【発明を実施するための最良の形態】
【0011】
本発明により、一般式(I)で示されるアミラーゼ活性測定用の基質及び該基質を含有アミラーゼ活性測定用試薬が提供される。
一般式(I)において、nは、マルトオリゴ糖の、糖の数に依存する整数であり、例えば、0〜8、1〜7、2〜6、2〜5であり、好ましくは、2、4、5である。本発明の基質は、当業者において周知の方法により合成することができる。また、一般式(I)で表される本発明に係るアミラーゼ測定用の基質には、特に断らない限り、その構造異性体(例えば、幾何異性体及び光学異性体など)も含まれる。例えば、限定はしないが、ナフチル基への結合位置は、1位又は2位が好ましく、特に、2位が好ましい。本発明に係るアミラーゼ活性測定用基質は、例えば、nが4、2−ナフチル基を蛍光ドナーとする場合には、以下に示すスキーム1のように合成することができる。
【0012】
【化4】
スキーム1
【0013】
すなわち、α-シクロデキストリンをO−アセチル化後、アセトリシス反応を用いて直鎖状のマルトオリゴ6糖を合成する。還元末端のブロモ化、チオアセチル化を経由しチオペンテニルグリコシドとする。脱O−アセチル化、非還元末端のナフチリデン化、O−アセチル化、ナフチリデンの還元開裂反応、脱O−アセチル化を行い、非還元末端の6位の水酸基のみがナフチルエーテル化された蛍光性マルトオリゴ糖を合成する。次いで、還元末端の2重結合にアミノエタンチオールのラジカル付加反応とダンシル化を行うことにより、還元末端及び非還元末端の両方に蛍光官能基を持った蛍光性マルトオリゴ6糖を合成することができる。
【0014】
本発明の基質又は試薬を用いたアミラーゼ活性の測定は、アミラーゼ、特にα−アミラーゼを含む試料に対して適用することができる。該試料は、溶液状態であっても、乾燥状態であってもよく、乾燥状態にある場合には、該乾燥試料に測定用緩衝液(本発明の基質を含んでも含んでなくてもよい)などを噴霧する方法等により、容易に測定可能な条件を整えることができる。従って、本発明に係るアミラーゼ活性の測定方法は、唾液、大便、膣分泌液などを含む体内由来の試料の他、家具や什器表面上に付着した試料中のアミラーゼの活性測定にも適用することができる。
【0015】
本発明のアミラーゼ活性の測定方法は、一般式(I)で示されるマルトオリゴ糖誘導体の基質を、アミラーゼを含む試料と接触させ、一般式(I)の基質とアミラーゼとを反応させることによって生じる、基質由来の蛍光共鳴エネルギー移動(FRET)反応の変化を検出し、該変化をアミラーゼの量に換算することで、アミラーゼ活性を測定するものである(図1)。アミラーゼの活性測定条件は、当業者によって従来行われている測定条件であれば、いかなる条件であっても測定可能であり、例えば、反応温度を約25℃〜約40℃、好ましくは37℃、pHを約6〜約8、好ましくはpH7程度とし、反応時間は、約1分〜約10時間程度の間に、適当な時間間隔(例えば、約1、2、3、4、5、6、7、8、9、10、15、20、25、30、50、100、150、200分間隔等)で数回蛍光強度変化を検出する条件などにより測定することができる。
【0016】
本発明のアミラーゼ活性測定用試薬は、本発明の基質以外にも、当業者においてアミラーゼ活性測定に必要と思われる物質、又は、アミラーゼ活性測定に効果的に作用する物質など、その他の添加物質を含んでもよい。このような添加物質としては、限定はしないが、例えば、界面活性剤、安定化剤、防腐剤、キレート剤などを使用することができる。また、本発明のアミラーゼ活性測定試薬は、適当な緩衝液中に溶解させた状態で提供することもできる。こうような緩衝液としては、当業者が容易に選択される範囲に含まれる緩衝液が使用可能であり、例えば、PIPES,HEPES,MESなどのグッド緩衝液のように、pHが弱酸性から弱アルカリ性の測定条件下において干渉効果を示すものであれば、如何なるものを使用してもよい。
【0017】
本発明の試薬は、キットの形態で、適当な容器又はパック中に使用説明書と共に含めることができる。本発明に係る試薬がキットとして供給される場合、基質を含む該試薬の構成成分を、活性構成成分の機能を低下することを回避するなどの必要に応じて、各構成成分を別々に包装してもよい。キット中に含まれる試薬は、構成成分が活性を長期間有効に持続し、構成成分を吸着せず、また、変質させない材質の容器中に供給される。また、キットに使用説明書が添付される場合、その使用説明は、紙又は他の材質上に印刷されて供給されてもよく、及び/又はフロッピー(登録商標)ディスク、CD−ROM、DVD−ROM、Zipディスク、ビデオテープ、オーディオテープなどの電気的又は電磁的に読み取り可能な媒体として供給されてもよい。あるいは、キットの製造者によって指定され又は電子メール等で通知されるウェブサイトに掲載されていてもよい。
以下の実施例は、本発明のアミラーゼ活性測定用基質の合成例とアミラーゼ活性測定方法及び測定結果を示すものである。本発明の実施例は、あくまでも例示にすぎず、本発明の範囲を限定するものではない。
【実施例】
【0018】
〔合成例1〕マルトオリゴ6糖誘導体の基質
α−シクロデキストリン(CD)完全アセチル化体
無水酢酸 (14mL)に酢酸ナトリウム(1.00g,12.2mmol)を加え、120℃に加熱した。α−シクロデキストリン(2.00g,2.06mmol)を徐々に加え、反応液を120℃で3時間攪拌した。反応液を室温に戻し、氷水を加え、一晩攪拌した。反応液に大量の水を加え、析出物をろ取した。メタノールによる再結晶化で精製し、結晶状のα−シクロデキストリン完全アセチル化体(2.43g,68.24%)を得た。
【0019】
G6完全アセチル化体
α−シクロデキストリン完全アセチル化体(1.0g,0.58mmol)を無水酢酸 (9.8mL)に溶解し、濃硫酸(0.2mL)を加え、50℃で17時間攪拌した。反応液を0℃に冷却し、ピリジン(1.0mL)を加え中和した。反応液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:3(v/v))で精製し、直鎖状のマルトオリゴ6糖完全アセチル化体(670mg,63.2%)を得た。
【0020】
G6チオアセチル化体
マルトオリゴ6糖完全アセチル化体(30.0g,16.3mmol)を氷酢酸(60.0mL)に溶解し、無水酢酸(6.0mL)を加えた。反応容器を水浴(室温)に設置し、30% 臭化水素/酢酸溶液(30.0mL)を加え、密栓をして、室温で4時間攪拌した。反応液を濃縮し、臭化糖の粗精製物を得た。得られた、臭化糖の粗精製物をDMF(30mL)に溶解し、反応液を、0℃に冷却後、チオ酢酸カリウム(5.60g,48.9mmol)を加え、反応液を室温に戻し、一晩攪拌した。反応液を酢酸エチルで希釈し、冷水、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ6糖チオアセチル化体(1.34g,62.2%)を得た。後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ6糖チオアセテート(18.1g,59.9%)を得た。
【表1】
【0021】
G6チオペンテニルアセチル化体
マルトオリゴ6糖チオアセテート(550mg,0.30mmol)をDMF(2.8mL)に溶解し、5−ブロモ−1−ペンテン(40μL,0.45mmol)を加えた。反応液を、−30℃に冷却し、ジエチルアミン(0.62mL,6.0mmol)を滴下し、滴下終了後、反応液を室温に戻し、2時間攪拌した。反応液を酢酸エチルで希釈し、冷水、飽和食塩水の順に抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ6糖チオペンテニルグリコシドアセチル化体(458mg,81.5%)を得た。
【表2】
【0022】
G6チオペンテニルOH体
マルトオリゴ6糖チオペンテニルグリコシド完全アセチル化体(11.0g,5.87mmol)をメタノール(220mL)に溶解し、ナトリウムメトキシド(317mg,5.87mmol)を加え、室温で4時間攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(water)による精製を行い、脱保護体を定量的に得た。
【表3】
【0023】
G6 4,6−O−ナフチリデン体
2−ナフトアルデヒド−ジイソブチルアセタール(1.38g,4.64mmol)をDMF(25.0mL)に溶解し、マルトオリゴ6糖チオペンテニルグリコシド(2.50g,2.32mmol)、10−(+)−カンファスルホン酸(265mg,1.16mmol)を加え、水流アスピレーターによる減圧下、50℃で2時間攪拌した。反応液を、0℃に冷却し、トリエチルアミン(1.6mL)を加え中和した。反応液を濃縮後、ピリジン(15.0mL)、無水酢酸(15.0mL)を加え、室温で3日間攪拌した。反応液を濃縮後、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:1(v/v))で精製し、4,6−O−ナフチリデンマルトオリゴ6糖アセチル化体(984mg,22.0%)を得た。また、原料の完全アセチル化体(2.31g,53.1%)を回収した。
【表4】
【0024】
G6ナフチルAc体
4,6−O−ナフチリデンマルトオリゴ6糖(1.70g,0.88mmol)をTHF(25.0mL)に溶解し、MS4Å(3.40g)、ボラン−トリメチルアミン錯体(897mg,12.3mmol)を加え、室温で1時間攪拌した。反応容器を氷浴(0℃)に設置し、塩化アルミニウム(1.60g,12.3mmol)を加え、室温で4時間攪拌した。反応液を綿濾過し、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、6−O−ナフチルマルトオリゴ6糖アセチル化体(1.30g,76.5%)を得た。
【表5】
【0025】
G6ナフチルOH体
6−O−ナフチルマルトオリゴ6糖アセチル化体(1.18g,0.61mmol)をメタノール(24.0mL)に溶解し、ナトリウムメトキシド(33mg,0.61mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解し、セファデックスLH−20(MeOH)によるゲルろ過精製を行い、脱保護体を定量的に得た。
【表6】
【0026】
G6アミン体
6−O−ナフチルマルトオリゴ6糖チオペンテニルグリコシド体(300mg,0.247mmol)をメタノール(1.0mL)に溶解し、水(1.0mL)、2−メルカプトエチルアミン塩酸塩(283mg,2.47mmol)を加え、完全溶解させた。室温で紫外線を2時間照射後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、6−O−ナフチルマルトオリゴ6糖アミン体塩酸塩を定量的に得た。
【表7】
【0027】
G6ダンシル体
6−O−ナフチルマルトオリゴ6糖アミン体(100mg,0.077mmol)をメタノール(5.0mL)に溶解し、塩化ダンシル(83mg,0.308mmol)を加えた。反応容器を水浴(室温)に設置し、トリエチルアミン(21μL,0.154mmol)を滴下した。3時間攪拌後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、次いでGPC(MeOH)による精製を行い、ダンシルマルトオリゴ6糖を定量的に得た。
【表8】
【0028】
〔合成例2〕マルトオリゴ7糖誘導体の基質
β−シクロデキストリン(CD)完全アセチル化体
無水酢酸(14mL)に酢酸ナトリウム(1.00g,12.2mmol)を加え、60℃に加熱した。β−シクロデキストリン(2.00g,1.76mmol)を徐々に加え、反応液を140℃で2時間攪拌した。反応液を室温に戻し、氷水を加え、一晩攪拌した。反応液に大量の水を加え、析出物をろ取した。メタノールによる再結晶化で精製し、結晶状のβ−シクロデキストリン完全アセチル化体(2.11g,59.4%)を得た。
【0029】
G7完全アセチル化体
β−シクロデキストリン完全アセチル化体(10.2g,5.06mmol)を無水酢酸(102mL)に溶解し、濃硫酸(2.0mL)を加え、50℃で12時間攪拌した。反応液を0℃に冷却し、ピリジン(10mL)を加え中和した。反応液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:7(v/v))で精製し、直鎖状のマルトオリゴ7糖完全アセチル化体(5.98g,55.9%)を得た。
【0030】
G7チオアセチル化体
マルトオリゴ7糖完全アセチル化体(22.7g,10.7mmol)を氷酢酸(45.0mL)に溶解し、無水酢酸(4.5mL)を加えた。反応容器を水浴(室温)に設置し、33%臭化水素/酢酸溶液(23.0mL)を加え、密栓をして、室温で4時間攪拌した。反応液をクロロホルムで希釈し、冷純水、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮し、臭化糖の粗精製物を得た。得られた、臭化糖の粗精製物をDMF(220mL)に溶解し、反応液を、0℃に冷却後、チオ酢酸カリウム(3.66g,32.1mmol)を加え、反応液を室温に戻し、一晩攪拌した。反応液を酢酸エチルで希釈し、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ7糖チオアセチル化体(1.34g,62.2%)を得た。後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ7糖チオアセテート(15.8g,69.1%)を得た。
【表9】
【0031】
G7チオペンテニルアセチル化体
マルトオリゴ7糖チオアセテート(2.13g,1.00mmol)をDMF(21.0mL)に溶解し、5−ブロモ−1−ペンテン(173μL,2.00mmol)を加えた。反応液を、−15℃に冷却し、ジエチルアミン(2.1mL,20mmol)を滴下し、滴下終了後、反応液を室温に戻し、5時間攪拌した。反応液を酢酸エチルで希釈し、飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ7糖チオペンテニルグリコシドアセチル化体(1.34g,62.2%)を得た。
【表10】
【0032】
G7チオペンテニルOH体
マルトオリゴ7糖チオペンテニルグリコシド完全アセチル化体(8.00g,3.70mmol)をメタノール(160mL)に溶解し、ナトリウムメトキシド(199mg,3.70mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(MeOH)による精製を行い、脱保護体を定量的に得た。
【表11】
【0033】
G7 4,6−O−ナフチリデン体
2−ナフトアルデヒド−ジイソブチルアセタール(850mg,2.8mmol)をDMF(18.0mL)に溶解し、マルトオリゴ7糖チオペンテニルグリコシド(1.77mg,1.4mmol)、10−(+)−カンファスルホン酸(162mg,0.7mmol)を加え、水流アスピレーターによる減圧下、50℃で2時間攪拌した。反応液を室温に戻し、反応容器を水浴(室温)に設置し、トリエチルアミン(1.7mL)を加え中和した。反応液を濃縮後、ピリジン(10.0mL)、無水酢酸(10.0mL)を加え、室温で一晩攪拌した。反応液を濃縮後、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に酢酸エチルで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:1(v/v))で精製し、4,6−O−ナフチリデンマルトオリゴ7糖アセチル化体(518mg,16.3%)を得た。また、原料の完全アセチル化体(1.84g,59.3%)を回収した。
【表12】
【0034】
G7ナフチルAc体
4,6−O−ナフチリデンマルトオリゴ7糖(518mg,0.233mmol)をTHF(7.5mL)に溶解し、MS4Å(1.03g)、ボラン−トリメチルアミン錯体(239mg,3.27mmol)を加え、室温で30分間攪拌した。反応容器を水浴(室温)に設置し、塩化アルミニウム(435mL,3.27mmol)を加え、室温で3時間攪拌した。反応液を綿濾過し、冷1 M 塩酸、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(4:5(v/v))で精製し、6−O−ナフチルマルトオリゴ7糖アセチル化体(230mg,44.6%)を得た。
【表13】
【0035】
G7ナフチルOH体
6−O−ナフチルマルトオリゴ7糖アセチル化体(110mg,0.050mmol)をメタノール(2.0mL)に溶解し、ナトリウムメトキシド(2.7mg,0.050mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(MeOH)による精製を行い、脱保護体(70mg)を定量的に得た。
【表14】
【0036】
G7アミン体
6−O−ナフチルマルトオリゴ7糖チオペンテニルグリコシド体(136mg,0.0946mmol)をメタノール(0.45mL)に溶解し、水(0.45mL)、2−メルカプトエチルアミン塩酸塩(107mg,0.946mmol)を加え、完全溶解させた。室温で紫外線を3時間照射後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、6−O−ナフチルマルトオリゴ7糖アミン体塩酸塩(114mg,80.9%)を得た。
【表15】
【0037】
G7ダンシル体
6−O−ナフチルマルトオリゴ7糖アミン体(50mg,0.033mmol)をメタノール(2.5mL)に溶解し、塩化ダンシル(36mg,0.134mmol)を加えた。反応容器を水浴(室温)に設置し、トリエチルアミン(9.3mL,0.067mmol)を滴下した。3時間攪拌後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、次いでGPC(MeOH)による精製を行い、ダンシルマルトオリゴ7糖(47mg,83.2%)を得た。
【表16】
【0038】
〔実施例1〕蛍光性マルトオリゴ6糖を用いたアミラーゼ活性の測定
上記のように合成した蛍光性マルトオリゴ6糖(合成例1)を用いて、アミラーゼ活性の測定を行った。
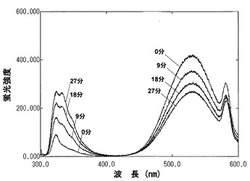
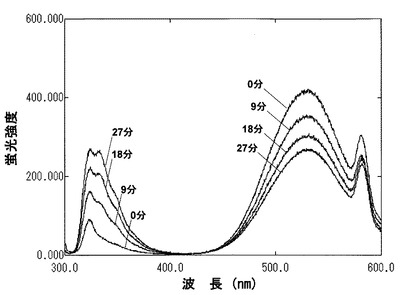
合成した蛍光性マルトオリゴ6糖を、8.3μMになるようにHEPESバッファー(10mM HEPES pH7.2,10mM CaCl2,0.01% NaN3,)、3mL中にあらかじめ溶解しておき、そこに検査サンプルであるAmylase(a-amylase from human saliva (Lee bio solutions inc., St. Louis, Missouri, USA)、a-amylase from human pancreas (Elastin products co. inc., Owensville, Missouri, USA))0.03U/vial(caraway method(Caraway, W T. Am. J. Clin. Path., 32 97-99 (1959))を加え、37℃、30分加温後、蛍光強度を測定した。測定は、9分毎に行った。その結果、検査サンプルにアミラーゼが存在していると、333nmの蛍光強度が増大し、530nmの蛍光強度が減弱したのに対し(図2)、検査サンプルにアミラーゼが存在しないと蛍光強度の変化は認められなかった。また、蛍光強度の増減は酵素量が多い程大きかった。
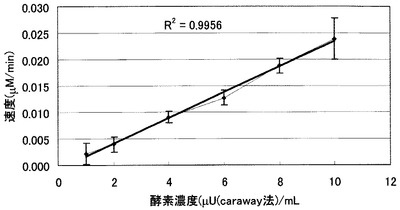
本方法による測定において、基質の加水分解速度とアミラーゼ濃度(酵素活性)との間には一次相関が認められたことから(図3)、蛍光強度の変化から、酵素活性の定量も可能である。
【0039】
〔実施例2〕アミラーゼアイソザイム(P型、S型)の識別
次に、本発明の方法により、アミラーゼアイソザイムの識別が可能であることを以下に示す。
膵由来のアミラーゼP型及び唾液由来のアミラーゼS型の酵素活性は、ブルースターチ法(Phadebas(登録商標)、 (Magle Life Sciences, Lund, Sweden)(Myersら,J Forensic Sci, 53:862-867, 2008)で同一の活性になるように希釈したものを使用した。ブルースターチ法では、同等の酵素活性であったが、本基質(6糖)では約2倍の分解速度(酵素活性)の差が認められた(図4)。従って、本発明の方法では、唾液由来のアミラーゼ(S型)活性を強く検出することができ、同量の酵素活性の強さの差により、アイソザイムの識別を行うことが可能となる。具体的には、caraway法で標準品のS型、P型アミラーゼの活性が、各々、1μUになるように濃度調製行った後、既知の混合比率で調製したS型P型標準酵素について、本発明の基質を用いて活性測定を行う。本発明の基質はS型とP型で反応速度が異なるため、S型P型標準酵素の検量線を作成することができる(図5)。次に検査試料のアミラーゼ酵素活性を測定した後、作成した検量線を用いてS型及びP型アイソザイムの混合比率を算出することができる。
【産業上の利用可能性】
【0040】
本発明のアミラーゼ活性測定基質及び該基質を含有する試薬は、法医学分野及び医療分野において、証拠物の特定あるいは疾患原因の特定を行うにあたり、優れた効果を発揮することが期待される。
【図面の簡単な説明】
【0041】
【図1】図1は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)によるFRETについて説明した図である。
【図2】図2は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)溶液にアミラーゼを添加した際の経時的な蛍光強度の変化を示した図である.各線はそれぞれアミラーゼ添加後、0分、9分、18分、27分を示す。時間の経過と共に、330nmの蛍光強度が増大し、530nmの蛍光強度が減少している。
【図3】図3は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)溶液に各濃度のアミラーゼを添加した際の酵素反応速度(基質分解速度)を示した図である。caraway法にて検定した酵素をそれぞれの濃度になるように反応液に加え、酵素添加後3分後に蛍光測定を行い、その蛍光強度変化より速度(初速度)を算出した。X軸は酵素濃度、Y軸は該当濃度に対する速度をプロットしたものである(測定は各4回行い、点は平均、エラーバーは標準偏差を示す。)。酵素濃度の増加と共に酵素反応速度が増加している。
【図4】本発明の方法により検出されるP型及びS型の酵素活性の差を示す。 ブルースターチ法で同一の活性を示すP型(膵臓由来)及びS型(唾液腺由来)アイソザイムについて、本発明の基質を用いて酵素活性(加水分解速度)を測定したところ、S型アミラーゼの酵素活性が、P型の約2倍の強さで検出された。
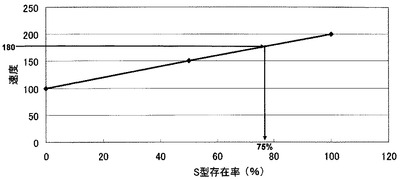
【図5】本発明の基質を用いて作成したP型S型混合標準酵素の検量線を示す。S型及びP型アイソザイムを、各々、100%及び0%、50%及び50%、0%及び100%の混合比率になるように調製したP型S型混合標準酵素サンプルの活性を測定し、S型アイソザイムの存在比率(X軸)に対し、酵素活性(蛍光強度)をプロットした。検査試料の酵素活性が、例えば速度が180の場合、図5の検量線から、S型75%、P型25%の混合比率であると評価することができる。
【技術分野】
【0001】
本発明は、新規アミラーゼ活性測定用基質及び該基質を含有したアミラーゼ活性測定試薬、並びに該基質又は試薬を使用したアミラーゼ活性測定方法に関する。
【背景技術】
【0002】
α−アミラーゼは、デンプンをはじめとするマルトオリゴ糖のα1,4結合を加水分解する酵素である。法医学分野においては唾液であることの証明、大便であることの証明、膣分泌液であることの証明等に用いられている。また、医療分野においては血液検査の指標の一つとして、膵臓疾患等のマーカーとして用いられてきた。法医学分野においては、一般的に、劣悪な環境に置かれた試料からのアミラーゼ活性の測定となるため、酵素の多くが失活していることが想定され、従って、できるだけ高感度の検査法が必要とされている。
【0003】
また、法医学、医療両分野において、アミラーゼのアイソザイム分析は重要である。アミラーゼは、主として唾液腺細胞、膵腺房細胞から分泌されており、免疫活性、酵素の安定性などの違いにより、S型(唾液由来)とP型(唾液以外、例えば膵液由来)の2つのアイソザイムに分類されている。医療分野においては、アミラーゼ活性値の異常が認められ場合、活性値の異常を示すアイソザイム活性を測定することにより、疾患の原因臓器の特定が可能となる。例えば、体液又は尿中のP型アミラーゼ活性が高ければ膵炎などの膵臓疾患が疑われ、S型アミラーゼ活性が高ければ耳下腺炎又は顎下腺炎など唾液腺疾患の可能性が示唆される。
【0004】
現在、アミラーゼ活性を測定する方法として、ブルースターチ法、ヨードデンプン法、酵素法(特許文献1)などが使用されている。しかし、これらの方法は、低感度性、操作の煩雑さ、基質構造の不明確さ、などの問題点が指摘されている。また、FRETを利用したマルトオリゴ糖誘導体を基質とするアミラーゼ活性測定方法(非特許文献1)も報告されており、測定感度の改善が期待されているが、蛍光のバックグラウンドの影響により低いアミラーゼ活性の測定における困難性が未だ存在しており、さらなる改善が望まれている。一方、アミラーゼアイソザイムを識別する方法としては、等電点電気泳動、ELISA、基質抗体阻害法(特許文献2)等が報告されているが、いずれの方法も操作が煩雑であり、結果を得るまでに長時間を要していた。
【0005】
【非特許文献1】Nishimura,Carbohydr.Let.,4:77-84 2001
【特許文献1】特許第3901990号
【特許文献2】特許第3862058号
【発明の開示】
【発明が解決しようとする課題】
【0006】
以上のように、アミラーゼ活性の高感度測定方法並びにアミラーゼアイソザイムの識別に関する簡便で正確な測定方法を開発すべく多くの研究が行われている。
本発明は、簡便で高感度にアミラーゼ活性を検出することが可能であり、また、アミラーゼアイソザイムの識別をも可能とする基質及び試薬の提供、並びに、該基質又は試薬を用いたアミラーゼ活性の測定方法の提供を目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、マルトオリゴ糖の非還元末端をナフチル基、還元末端をダンシル基で化学修飾した基質により、FRETを利用した測定方法の感度を向上させ、さらに、該基質をアミラーゼアイソザイムの識別に効果的に利用できることを見出し、本発明を完成させた。
FRETを利用したアミラーゼ活性の測定法に関する従来技術(非特許文献1)においては、ナフチル基からの蛍光(333nm)のバックグラウンドが元々高いことから、微量のアミラーゼ活性が測定誤差の範囲に入ってしまい検出が困難であった。この点について、本発明の基質を用いると、333nmの蛍光のバックグラウンドを従来技術(非特許文献1)の約10分の1に抑えることが可能となり、微量なアミラーゼ活性の測定が可能となった。さらに、本発明の基質を用いると、測定感度が向上されることから、アミラーゼアイソザイムを識別することも可能となる。
【0008】
すなわち、本発明は、一般式(I)
【化3】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質及び該基質を含有する試薬、並びに該基質又は試薬を用いたアミラーゼ活性の測定方法を提供する。
さらに、本発明の基質又は該基質を含有する試薬は、アミラーゼアイソザイムの識別にも使用することができる。
【発明の効果】
【0009】
本発明の基質又は該基質を含有する試薬を用いると、従来法に比較して、アミラーゼの検出感度を顕著に向上させることができる(例えば、ブルースターチ法と比較して、約10倍程度)。
【0010】
また、本発明のアミラーゼ活性測定方法を用いると、アミラーゼアイソザイム(例えば、P型、S型)の識別が可能となる。
【発明を実施するための最良の形態】
【0011】
本発明により、一般式(I)で示されるアミラーゼ活性測定用の基質及び該基質を含有アミラーゼ活性測定用試薬が提供される。
一般式(I)において、nは、マルトオリゴ糖の、糖の数に依存する整数であり、例えば、0〜8、1〜7、2〜6、2〜5であり、好ましくは、2、4、5である。本発明の基質は、当業者において周知の方法により合成することができる。また、一般式(I)で表される本発明に係るアミラーゼ測定用の基質には、特に断らない限り、その構造異性体(例えば、幾何異性体及び光学異性体など)も含まれる。例えば、限定はしないが、ナフチル基への結合位置は、1位又は2位が好ましく、特に、2位が好ましい。本発明に係るアミラーゼ活性測定用基質は、例えば、nが4、2−ナフチル基を蛍光ドナーとする場合には、以下に示すスキーム1のように合成することができる。
【0012】
【化4】
スキーム1
【0013】
すなわち、α-シクロデキストリンをO−アセチル化後、アセトリシス反応を用いて直鎖状のマルトオリゴ6糖を合成する。還元末端のブロモ化、チオアセチル化を経由しチオペンテニルグリコシドとする。脱O−アセチル化、非還元末端のナフチリデン化、O−アセチル化、ナフチリデンの還元開裂反応、脱O−アセチル化を行い、非還元末端の6位の水酸基のみがナフチルエーテル化された蛍光性マルトオリゴ糖を合成する。次いで、還元末端の2重結合にアミノエタンチオールのラジカル付加反応とダンシル化を行うことにより、還元末端及び非還元末端の両方に蛍光官能基を持った蛍光性マルトオリゴ6糖を合成することができる。
【0014】
本発明の基質又は試薬を用いたアミラーゼ活性の測定は、アミラーゼ、特にα−アミラーゼを含む試料に対して適用することができる。該試料は、溶液状態であっても、乾燥状態であってもよく、乾燥状態にある場合には、該乾燥試料に測定用緩衝液(本発明の基質を含んでも含んでなくてもよい)などを噴霧する方法等により、容易に測定可能な条件を整えることができる。従って、本発明に係るアミラーゼ活性の測定方法は、唾液、大便、膣分泌液などを含む体内由来の試料の他、家具や什器表面上に付着した試料中のアミラーゼの活性測定にも適用することができる。
【0015】
本発明のアミラーゼ活性の測定方法は、一般式(I)で示されるマルトオリゴ糖誘導体の基質を、アミラーゼを含む試料と接触させ、一般式(I)の基質とアミラーゼとを反応させることによって生じる、基質由来の蛍光共鳴エネルギー移動(FRET)反応の変化を検出し、該変化をアミラーゼの量に換算することで、アミラーゼ活性を測定するものである(図1)。アミラーゼの活性測定条件は、当業者によって従来行われている測定条件であれば、いかなる条件であっても測定可能であり、例えば、反応温度を約25℃〜約40℃、好ましくは37℃、pHを約6〜約8、好ましくはpH7程度とし、反応時間は、約1分〜約10時間程度の間に、適当な時間間隔(例えば、約1、2、3、4、5、6、7、8、9、10、15、20、25、30、50、100、150、200分間隔等)で数回蛍光強度変化を検出する条件などにより測定することができる。
【0016】
本発明のアミラーゼ活性測定用試薬は、本発明の基質以外にも、当業者においてアミラーゼ活性測定に必要と思われる物質、又は、アミラーゼ活性測定に効果的に作用する物質など、その他の添加物質を含んでもよい。このような添加物質としては、限定はしないが、例えば、界面活性剤、安定化剤、防腐剤、キレート剤などを使用することができる。また、本発明のアミラーゼ活性測定試薬は、適当な緩衝液中に溶解させた状態で提供することもできる。こうような緩衝液としては、当業者が容易に選択される範囲に含まれる緩衝液が使用可能であり、例えば、PIPES,HEPES,MESなどのグッド緩衝液のように、pHが弱酸性から弱アルカリ性の測定条件下において干渉効果を示すものであれば、如何なるものを使用してもよい。
【0017】
本発明の試薬は、キットの形態で、適当な容器又はパック中に使用説明書と共に含めることができる。本発明に係る試薬がキットとして供給される場合、基質を含む該試薬の構成成分を、活性構成成分の機能を低下することを回避するなどの必要に応じて、各構成成分を別々に包装してもよい。キット中に含まれる試薬は、構成成分が活性を長期間有効に持続し、構成成分を吸着せず、また、変質させない材質の容器中に供給される。また、キットに使用説明書が添付される場合、その使用説明は、紙又は他の材質上に印刷されて供給されてもよく、及び/又はフロッピー(登録商標)ディスク、CD−ROM、DVD−ROM、Zipディスク、ビデオテープ、オーディオテープなどの電気的又は電磁的に読み取り可能な媒体として供給されてもよい。あるいは、キットの製造者によって指定され又は電子メール等で通知されるウェブサイトに掲載されていてもよい。
以下の実施例は、本発明のアミラーゼ活性測定用基質の合成例とアミラーゼ活性測定方法及び測定結果を示すものである。本発明の実施例は、あくまでも例示にすぎず、本発明の範囲を限定するものではない。
【実施例】
【0018】
〔合成例1〕マルトオリゴ6糖誘導体の基質
α−シクロデキストリン(CD)完全アセチル化体
無水酢酸 (14mL)に酢酸ナトリウム(1.00g,12.2mmol)を加え、120℃に加熱した。α−シクロデキストリン(2.00g,2.06mmol)を徐々に加え、反応液を120℃で3時間攪拌した。反応液を室温に戻し、氷水を加え、一晩攪拌した。反応液に大量の水を加え、析出物をろ取した。メタノールによる再結晶化で精製し、結晶状のα−シクロデキストリン完全アセチル化体(2.43g,68.24%)を得た。
【0019】
G6完全アセチル化体
α−シクロデキストリン完全アセチル化体(1.0g,0.58mmol)を無水酢酸 (9.8mL)に溶解し、濃硫酸(0.2mL)を加え、50℃で17時間攪拌した。反応液を0℃に冷却し、ピリジン(1.0mL)を加え中和した。反応液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:3(v/v))で精製し、直鎖状のマルトオリゴ6糖完全アセチル化体(670mg,63.2%)を得た。
【0020】
G6チオアセチル化体
マルトオリゴ6糖完全アセチル化体(30.0g,16.3mmol)を氷酢酸(60.0mL)に溶解し、無水酢酸(6.0mL)を加えた。反応容器を水浴(室温)に設置し、30% 臭化水素/酢酸溶液(30.0mL)を加え、密栓をして、室温で4時間攪拌した。反応液を濃縮し、臭化糖の粗精製物を得た。得られた、臭化糖の粗精製物をDMF(30mL)に溶解し、反応液を、0℃に冷却後、チオ酢酸カリウム(5.60g,48.9mmol)を加え、反応液を室温に戻し、一晩攪拌した。反応液を酢酸エチルで希釈し、冷水、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ6糖チオアセチル化体(1.34g,62.2%)を得た。後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ6糖チオアセテート(18.1g,59.9%)を得た。
【表1】
【0021】
G6チオペンテニルアセチル化体
マルトオリゴ6糖チオアセテート(550mg,0.30mmol)をDMF(2.8mL)に溶解し、5−ブロモ−1−ペンテン(40μL,0.45mmol)を加えた。反応液を、−30℃に冷却し、ジエチルアミン(0.62mL,6.0mmol)を滴下し、滴下終了後、反応液を室温に戻し、2時間攪拌した。反応液を酢酸エチルで希釈し、冷水、飽和食塩水の順に抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ6糖チオペンテニルグリコシドアセチル化体(458mg,81.5%)を得た。
【表2】
【0022】
G6チオペンテニルOH体
マルトオリゴ6糖チオペンテニルグリコシド完全アセチル化体(11.0g,5.87mmol)をメタノール(220mL)に溶解し、ナトリウムメトキシド(317mg,5.87mmol)を加え、室温で4時間攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(water)による精製を行い、脱保護体を定量的に得た。
【表3】
【0023】
G6 4,6−O−ナフチリデン体
2−ナフトアルデヒド−ジイソブチルアセタール(1.38g,4.64mmol)をDMF(25.0mL)に溶解し、マルトオリゴ6糖チオペンテニルグリコシド(2.50g,2.32mmol)、10−(+)−カンファスルホン酸(265mg,1.16mmol)を加え、水流アスピレーターによる減圧下、50℃で2時間攪拌した。反応液を、0℃に冷却し、トリエチルアミン(1.6mL)を加え中和した。反応液を濃縮後、ピリジン(15.0mL)、無水酢酸(15.0mL)を加え、室温で3日間攪拌した。反応液を濃縮後、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:1(v/v))で精製し、4,6−O−ナフチリデンマルトオリゴ6糖アセチル化体(984mg,22.0%)を得た。また、原料の完全アセチル化体(2.31g,53.1%)を回収した。
【表4】
【0024】
G6ナフチルAc体
4,6−O−ナフチリデンマルトオリゴ6糖(1.70g,0.88mmol)をTHF(25.0mL)に溶解し、MS4Å(3.40g)、ボラン−トリメチルアミン錯体(897mg,12.3mmol)を加え、室温で1時間攪拌した。反応容器を氷浴(0℃)に設置し、塩化アルミニウム(1.60g,12.3mmol)を加え、室温で4時間攪拌した。反応液を綿濾過し、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、6−O−ナフチルマルトオリゴ6糖アセチル化体(1.30g,76.5%)を得た。
【表5】
【0025】
G6ナフチルOH体
6−O−ナフチルマルトオリゴ6糖アセチル化体(1.18g,0.61mmol)をメタノール(24.0mL)に溶解し、ナトリウムメトキシド(33mg,0.61mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解し、セファデックスLH−20(MeOH)によるゲルろ過精製を行い、脱保護体を定量的に得た。
【表6】
【0026】
G6アミン体
6−O−ナフチルマルトオリゴ6糖チオペンテニルグリコシド体(300mg,0.247mmol)をメタノール(1.0mL)に溶解し、水(1.0mL)、2−メルカプトエチルアミン塩酸塩(283mg,2.47mmol)を加え、完全溶解させた。室温で紫外線を2時間照射後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、6−O−ナフチルマルトオリゴ6糖アミン体塩酸塩を定量的に得た。
【表7】
【0027】
G6ダンシル体
6−O−ナフチルマルトオリゴ6糖アミン体(100mg,0.077mmol)をメタノール(5.0mL)に溶解し、塩化ダンシル(83mg,0.308mmol)を加えた。反応容器を水浴(室温)に設置し、トリエチルアミン(21μL,0.154mmol)を滴下した。3時間攪拌後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、次いでGPC(MeOH)による精製を行い、ダンシルマルトオリゴ6糖を定量的に得た。
【表8】
【0028】
〔合成例2〕マルトオリゴ7糖誘導体の基質
β−シクロデキストリン(CD)完全アセチル化体
無水酢酸(14mL)に酢酸ナトリウム(1.00g,12.2mmol)を加え、60℃に加熱した。β−シクロデキストリン(2.00g,1.76mmol)を徐々に加え、反応液を140℃で2時間攪拌した。反応液を室温に戻し、氷水を加え、一晩攪拌した。反応液に大量の水を加え、析出物をろ取した。メタノールによる再結晶化で精製し、結晶状のβ−シクロデキストリン完全アセチル化体(2.11g,59.4%)を得た。
【0029】
G7完全アセチル化体
β−シクロデキストリン完全アセチル化体(10.2g,5.06mmol)を無水酢酸(102mL)に溶解し、濃硫酸(2.0mL)を加え、50℃で12時間攪拌した。反応液を0℃に冷却し、ピリジン(10mL)を加え中和した。反応液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:7(v/v))で精製し、直鎖状のマルトオリゴ7糖完全アセチル化体(5.98g,55.9%)を得た。
【0030】
G7チオアセチル化体
マルトオリゴ7糖完全アセチル化体(22.7g,10.7mmol)を氷酢酸(45.0mL)に溶解し、無水酢酸(4.5mL)を加えた。反応容器を水浴(室温)に設置し、33%臭化水素/酢酸溶液(23.0mL)を加え、密栓をして、室温で4時間攪拌した。反応液をクロロホルムで希釈し、冷純水、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮し、臭化糖の粗精製物を得た。得られた、臭化糖の粗精製物をDMF(220mL)に溶解し、反応液を、0℃に冷却後、チオ酢酸カリウム(3.66g,32.1mmol)を加え、反応液を室温に戻し、一晩攪拌した。反応液を酢酸エチルで希釈し、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:2(v/v))で精製し、マルトオリゴ7糖チオアセチル化体(1.34g,62.2%)を得た。後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ7糖チオアセテート(15.8g,69.1%)を得た。
【表9】
【0031】
G7チオペンテニルアセチル化体
マルトオリゴ7糖チオアセテート(2.13g,1.00mmol)をDMF(21.0mL)に溶解し、5−ブロモ−1−ペンテン(173μL,2.00mmol)を加えた。反応液を、−15℃に冷却し、ジエチルアミン(2.1mL,20mmol)を滴下し、滴下終了後、反応液を室温に戻し、5時間攪拌した。反応液を酢酸エチルで希釈し、飽和食塩水で抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(2:3(v/v))で精製し、マルトオリゴ7糖チオペンテニルグリコシドアセチル化体(1.34g,62.2%)を得た。
【表10】
【0032】
G7チオペンテニルOH体
マルトオリゴ7糖チオペンテニルグリコシド完全アセチル化体(8.00g,3.70mmol)をメタノール(160mL)に溶解し、ナトリウムメトキシド(199mg,3.70mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(MeOH)による精製を行い、脱保護体を定量的に得た。
【表11】
【0033】
G7 4,6−O−ナフチリデン体
2−ナフトアルデヒド−ジイソブチルアセタール(850mg,2.8mmol)をDMF(18.0mL)に溶解し、マルトオリゴ7糖チオペンテニルグリコシド(1.77mg,1.4mmol)、10−(+)−カンファスルホン酸(162mg,0.7mmol)を加え、水流アスピレーターによる減圧下、50℃で2時間攪拌した。反応液を室温に戻し、反応容器を水浴(室温)に設置し、トリエチルアミン(1.7mL)を加え中和した。反応液を濃縮後、ピリジン(10.0mL)、無水酢酸(10.0mL)を加え、室温で一晩攪拌した。反応液を濃縮後、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順に酢酸エチルで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(1:1(v/v))で精製し、4,6−O−ナフチリデンマルトオリゴ7糖アセチル化体(518mg,16.3%)を得た。また、原料の完全アセチル化体(1.84g,59.3%)を回収した。
【表12】
【0034】
G7ナフチルAc体
4,6−O−ナフチリデンマルトオリゴ7糖(518mg,0.233mmol)をTHF(7.5mL)に溶解し、MS4Å(1.03g)、ボラン−トリメチルアミン錯体(239mg,3.27mmol)を加え、室温で30分間攪拌した。反応容器を水浴(室温)に設置し、塩化アルミニウム(435mL,3.27mmol)を加え、室温で3時間攪拌した。反応液を綿濾過し、冷1 M 塩酸、冷飽和炭酸水素ナトリウム水溶液、飽和食塩水の順にクロロホルムで抽出を行い、有機層を無水硫酸ナトリウムで乾燥した。この有機層をセライトろ過し、ろ液を濃縮後、シリカゲルカラムクロマトグラフィー[トルエン−酢酸エチル(4:5(v/v))で精製し、6−O−ナフチルマルトオリゴ7糖アセチル化体(230mg,44.6%)を得た。
【表13】
【0035】
G7ナフチルOH体
6−O−ナフチルマルトオリゴ7糖アセチル化体(110mg,0.050mmol)をメタノール(2.0mL)に溶解し、ナトリウムメトキシド(2.7mg,0.050mmol)を加え、室温で一晩攪拌した。反応液を強酸性陽イオン交換樹脂IR−120B(H+型)で中和後、イオン交換樹脂をガラスフィルターでろ取し、ろ液を濃縮した。残渣をメタノールで溶解しGPC(MeOH)による精製を行い、脱保護体(70mg)を定量的に得た。
【表14】
【0036】
G7アミン体
6−O−ナフチルマルトオリゴ7糖チオペンテニルグリコシド体(136mg,0.0946mmol)をメタノール(0.45mL)に溶解し、水(0.45mL)、2−メルカプトエチルアミン塩酸塩(107mg,0.946mmol)を加え、完全溶解させた。室温で紫外線を3時間照射後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、6−O−ナフチルマルトオリゴ7糖アミン体塩酸塩(114mg,80.9%)を得た。
【表15】
【0037】
G7ダンシル体
6−O−ナフチルマルトオリゴ7糖アミン体(50mg,0.033mmol)をメタノール(2.5mL)に溶解し、塩化ダンシル(36mg,0.134mmol)を加えた。反応容器を水浴(室温)に設置し、トリエチルアミン(9.3mL,0.067mmol)を滴下した。3時間攪拌後、反応液をそのままセファデックスLH−20(MeOH)によりゲルろ過精製を行い、次いでGPC(MeOH)による精製を行い、ダンシルマルトオリゴ7糖(47mg,83.2%)を得た。
【表16】
【0038】
〔実施例1〕蛍光性マルトオリゴ6糖を用いたアミラーゼ活性の測定
上記のように合成した蛍光性マルトオリゴ6糖(合成例1)を用いて、アミラーゼ活性の測定を行った。
合成した蛍光性マルトオリゴ6糖を、8.3μMになるようにHEPESバッファー(10mM HEPES pH7.2,10mM CaCl2,0.01% NaN3,)、3mL中にあらかじめ溶解しておき、そこに検査サンプルであるAmylase(a-amylase from human saliva (Lee bio solutions inc., St. Louis, Missouri, USA)、a-amylase from human pancreas (Elastin products co. inc., Owensville, Missouri, USA))0.03U/vial(caraway method(Caraway, W T. Am. J. Clin. Path., 32 97-99 (1959))を加え、37℃、30分加温後、蛍光強度を測定した。測定は、9分毎に行った。その結果、検査サンプルにアミラーゼが存在していると、333nmの蛍光強度が増大し、530nmの蛍光強度が減弱したのに対し(図2)、検査サンプルにアミラーゼが存在しないと蛍光強度の変化は認められなかった。また、蛍光強度の増減は酵素量が多い程大きかった。
本方法による測定において、基質の加水分解速度とアミラーゼ濃度(酵素活性)との間には一次相関が認められたことから(図3)、蛍光強度の変化から、酵素活性の定量も可能である。
【0039】
〔実施例2〕アミラーゼアイソザイム(P型、S型)の識別
次に、本発明の方法により、アミラーゼアイソザイムの識別が可能であることを以下に示す。
膵由来のアミラーゼP型及び唾液由来のアミラーゼS型の酵素活性は、ブルースターチ法(Phadebas(登録商標)、 (Magle Life Sciences, Lund, Sweden)(Myersら,J Forensic Sci, 53:862-867, 2008)で同一の活性になるように希釈したものを使用した。ブルースターチ法では、同等の酵素活性であったが、本基質(6糖)では約2倍の分解速度(酵素活性)の差が認められた(図4)。従って、本発明の方法では、唾液由来のアミラーゼ(S型)活性を強く検出することができ、同量の酵素活性の強さの差により、アイソザイムの識別を行うことが可能となる。具体的には、caraway法で標準品のS型、P型アミラーゼの活性が、各々、1μUになるように濃度調製行った後、既知の混合比率で調製したS型P型標準酵素について、本発明の基質を用いて活性測定を行う。本発明の基質はS型とP型で反応速度が異なるため、S型P型標準酵素の検量線を作成することができる(図5)。次に検査試料のアミラーゼ酵素活性を測定した後、作成した検量線を用いてS型及びP型アイソザイムの混合比率を算出することができる。
【産業上の利用可能性】
【0040】
本発明のアミラーゼ活性測定基質及び該基質を含有する試薬は、法医学分野及び医療分野において、証拠物の特定あるいは疾患原因の特定を行うにあたり、優れた効果を発揮することが期待される。
【図面の簡単な説明】
【0041】
【図1】図1は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)によるFRETについて説明した図である。
【図2】図2は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)溶液にアミラーゼを添加した際の経時的な蛍光強度の変化を示した図である.各線はそれぞれアミラーゼ添加後、0分、9分、18分、27分を示す。時間の経過と共に、330nmの蛍光強度が増大し、530nmの蛍光強度が減少している。
【図3】図3は、本発明のアミラーゼ測定用基質(マルトオリゴ糖6糖の誘導体の場合)溶液に各濃度のアミラーゼを添加した際の酵素反応速度(基質分解速度)を示した図である。caraway法にて検定した酵素をそれぞれの濃度になるように反応液に加え、酵素添加後3分後に蛍光測定を行い、その蛍光強度変化より速度(初速度)を算出した。X軸は酵素濃度、Y軸は該当濃度に対する速度をプロットしたものである(測定は各4回行い、点は平均、エラーバーは標準偏差を示す。)。酵素濃度の増加と共に酵素反応速度が増加している。
【図4】本発明の方法により検出されるP型及びS型の酵素活性の差を示す。 ブルースターチ法で同一の活性を示すP型(膵臓由来)及びS型(唾液腺由来)アイソザイムについて、本発明の基質を用いて酵素活性(加水分解速度)を測定したところ、S型アミラーゼの酵素活性が、P型の約2倍の強さで検出された。
【図5】本発明の基質を用いて作成したP型S型混合標準酵素の検量線を示す。S型及びP型アイソザイムを、各々、100%及び0%、50%及び50%、0%及び100%の混合比率になるように調製したP型S型混合標準酵素サンプルの活性を測定し、S型アイソザイムの存在比率(X軸)に対し、酵素活性(蛍光強度)をプロットした。検査試料の酵素活性が、例えば速度が180の場合、図5の検量線から、S型75%、P型25%の混合比率であると評価することができる。
【特許請求の範囲】
【請求項1】
一般式(I)
【化1】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質。
【請求項2】
前記試料中に複数のアミラーゼアイソザイムが存在し、該アミラーゼアイソザイムを識別するための請求項1に記載のアミラーゼ活性測定基質。
【請求項3】
nが2の4糖から構成される請求項1又は2に記載のアミラーゼ活性測定基質。
【請求項4】
請求項1乃至3のいずれかの基質を含有するアミラーゼ活性測定試薬。
【請求項5】
一般式(I)
【化2】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を、アミラーゼを含有する試料と接触させて反応させ、該アミラーゼ活性を測定する方法。
【請求項6】
前記試料中に複数存在するアミラーゼアイソザイムを識別するための請求項5に記載のアミラーゼ活性を測定する方法。
【請求項1】
一般式(I)
【化1】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を含有し、試料中に存在するアミラーゼ活性を測定するためのアミラーゼ活性測定基質。
【請求項2】
前記試料中に複数のアミラーゼアイソザイムが存在し、該アミラーゼアイソザイムを識別するための請求項1に記載のアミラーゼ活性測定基質。
【請求項3】
nが2の4糖から構成される請求項1又は2に記載のアミラーゼ活性測定基質。
【請求項4】
請求項1乃至3のいずれかの基質を含有するアミラーゼ活性測定試薬。
【請求項5】
一般式(I)
【化2】
(式中、nは、0〜8の整数)で表されるマルトオリゴ糖誘導体を、アミラーゼを含有する試料と接触させて反応させ、該アミラーゼ活性を測定する方法。
【請求項6】
前記試料中に複数存在するアミラーゼアイソザイムを識別するための請求項5に記載のアミラーゼ活性を測定する方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2010−68777(P2010−68777A)
【公開日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願番号】特願2008−242083(P2008−242083)
【出願日】平成20年9月22日(2008.9.22)
【出願人】(504190548)国立大学法人埼玉大学 (292)
【出願人】(391041062)福島県 (42)
【Fターム(参考)】
【公開日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願日】平成20年9月22日(2008.9.22)
【出願人】(504190548)国立大学法人埼玉大学 (292)
【出願人】(391041062)福島県 (42)
【Fターム(参考)】
[ Back to top ]