
蛍光標識としてのスルホン化ジアリールローダミン色素
【課題】スルホン化ジアリールローダミン化合物として、ヌクレオシド、ヌクレオチド、ポリヌクレオチド、およびポリペプチドの蛍光標識として有用であるものを提供すること。
【解決手段】本発明の化合物は、蛍光核酸分析(例えば、自動化DNA配列決定)およびフラグメント分析、ハイブリダイゼーションアレイにおけるプローブハイブリダイゼーションの検出、核酸増幅産物の検出などの分野における特定の用途を見出している。本発明の別の局面は、エネルギー移動色素化合物を包含し、この化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、このドナー色素およびアクセプター色素を連結するためのリンカー、を含有する。
【解決手段】本発明の化合物は、蛍光核酸分析(例えば、自動化DNA配列決定)およびフラグメント分析、ハイブリダイゼーションアレイにおけるプローブハイブリダイゼーションの検出、核酸増幅産物の検出などの分野における特定の用途を見出している。本発明の別の局面は、エネルギー移動色素化合物を包含し、この化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、このドナー色素およびアクセプター色素を連結するためのリンカー、を含有する。
【発明の詳細な説明】
【技術分野】
【0001】
(I.発明の分野)
本発明は、一般に、蛍光ローダミン色素化合物に関する。より詳細には、本発明は、蛍光標識試薬として有用な、スルホン化ジアリールローダミン色素に関する。
【背景技術】
【0002】
(II.背景)
蛍光標識を利用する生物学的分析の非放射活性検出は、現代の分子生物学において重要な技術である。放射活性標識の必要性を排除することによって、安全性が高められ、そして試薬の処理に関連した環境影響およびコストは、大いに減少する。このような非放射活性蛍光検出を利用する方法の例としては、4色自動化DNA配列決定、オリゴヌクレオチドハイブリダイゼーション方法、ポリメラーゼ連鎖反応生成物の検出、免疫アッセイなどが挙げられる。
【0003】
多くの適用において、空間的に重複した複数の分析物の非依存的な検出(例えば、単管の多重化DNAプローブアッセイおよび4色自動化DNA配列決定方法)を達成するために、スペクトルにより識別可能な複数の蛍光標識を使用することは、有益である。多重化DNAプローブアッセイの場合、スペクトルにより識別可能な蛍光標識を使用することによって、反応管の数は減少され得、これによって、実験プロトコルが単純化され、適用に特異的な試薬キットの作製が容易になる。4色自動化DNA配列決定の場合、多色蛍光標識によって、単一レーンにおいて複数塩基の分析が可能となり、これによって、単色方法を超えてスループットが増大し、レーン間の電気泳動の移動度変化に関連した不確実度が減少する。
【発明の概要】
【発明が解決しようとする課題】
【0004】
スペクトルにより識別可能な複数の蛍光標識のセットを構築することは、問題である。多色蛍光検出は、色素標識の選択について(特に、単一の励起光源、電気泳動分離、および/または酵素を用いた処理を必要とする適用(例えば、自動化DNA配列決定)について)、少なくとも6つの厳格な制限を課している。第一に、発光スペクトルがスペクトルにより分解される、構造的に類似の色素のセットを見出すことは、困難である。なぜなら、有機蛍光色素に関する代表的な発光バンドの半値幅が、約40〜80ナノメートル(nm)であるからである。第二に、たとえ非重複発光スペクトルを有する色素が同定されたとしても、それぞれの蛍光量子効率が低すぎる場合、このセットは依然として適切であり得ない。第三に、数種の蛍光色素が同時に使用される場合、同時励起は困難になる。なぜなら、色素の吸収バンドは、通常は、広範に分離されているからである。第四に、電荷、分子サイズ、および色素のコンホメーションは、分析物の電気泳動移動度に悪影響を与えてはいけいない。第五に、蛍光色素は、分析物を作製または操作するために使用される化学物質(例えば、DNA合成溶媒およびDNA合成試薬、緩衝液、ポリメラーゼ酵素、リガーゼ酵素など)と適合性でなければならない。第六に、色素は、レーザ励起に抵抗するのに十分な光安定性を有さなければならない。
【0005】
4色自動化DNA配列決定の適用において適切な、現在利用可能な多重化色素セットは、これらのセットを構成する全ての色素から蛍光発光を十分に励起するために青色または青緑色のレーザ光(例えば、アルゴン−イオンレーザ)を必要とする。商業的な自動化DNA配列決定システムにおいて青色または青緑色のレーザを使用することは、このようなレーザの高コストおよび制限された寿命に起因して、不利益である。
【課題を解決するための手段】
【0006】
(III.要旨)
したがって、本発明は以下を提供する。
(1)以下の構造:
【化1】

を有する、スルホン化ジアリールローダミン化合物であって、ここで、 R2、R2’、R12およびR12’は、水素、C1〜C12アルキル、C1〜C12アルキルジイル、フェニル、置換フェニル、ベンジル、置換ベンジル、ビフェニル、置換ビフェニル、ナフチル、置換ナフチル、複素環、置換複素環、水溶性基または連結部分であり;そして、
R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21は、水素、フッ素、塩素、C1〜C8アルキル、カルボキシレート、スルフェート、スルホネート、アルキルスルホネート、アミノメチル(−CH2NH2)、アミノアルキル、4−ジアルキルアミノピリジニウム、ヒドロキシメチル(−CH2OH)、メトキシ(−OCH3)、ヒドロキシアルキル(−ROH)、チオメチル(−CH2SH)、チオアルキル(−RSH)、アルキルスルホン(−SO2R)、アリールチオ(−SAr)、アリールスルホン(−SO2Ar)、スルホンアミド(−SO2NR2)、アルキルスルホキシド(−SOR)、アリールスルホキシド(−SOAr)、アミノ(−NH2)、アンモニウム(−NH3+)、アミド(−CONR2)、ニトリル(−CN)、C1〜C8アルコキシ(−OR)、フェノキシ、フェノール類、トリル、フェニル、アリール、ベンジル、複素環、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリエチレンオキシ、水溶性基、または連結部分であり;
但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21のうちの少なくとも1つがスルホネートである、スルホン化ジアリールローダミン化合物。
(2)R2、R2’、R12およびR12’の少なくとも1つがC1〜C6アルキルスルホネートまたはC4〜C10アリールスルホネートである、項目1に記載の化合物。
(3)前記アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルがスルホネートで置換されている、請求項1に記載の化合物。
(4)前記アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルがカルボキシルで置換されている、請求項1に記載の化合物。
(5)前記連結部分が、アジド、一置換の第一級アミン、二置換の第二級アミン、チオール、ヒドロキシル、ハライド、エポキシド、N−ヒドロキシスクシンイミジルエステル、カルボキシル、イソチオシアネート、塩化スルホニル、スルホネートエステル、シリルハライド、クロロトリアジニル、スクシンイミジルエステル、ペンタフルオロフェニルエステル、マレイミド、ハロアセチル、エポキシド、アルキルハライド、アリルハライド、アルデヒド、ケトン、アシルアジド、無水物、ヨードアセトアミドまたは活性化エステルである、項目1に記載の化合物。
(6)前記水溶性基が、カルボキシレート、スルホネート、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリヒドロキシル、または水溶性ポリマーである、項目1に記載の化合物。
(7)前記複素環が、ピロール、インドール、フラン、ベンゾフラン、チオフェン、ベンゾチオフェン、2−ピリジル、3−ピリジル、4−ピリジル、2−キノリル、3−キノリル、4−キノリル、2−イミダゾール、4−イミダゾール、3−ピラゾール、4−ピラゾール、ピリダジン、ピリミジン、ピラジン、シンノリン、フタラジン、キナゾリン、キノキサリン、3−(1,2,4−N)−トリアゾリル、5−(1,2,4−N)−トリアゾリル、5−テトラゾリル、4−(1−O,3−N)−オキサゾール、5−(1−O,3−N)−オキサゾール、4−(1−S,3−N)−トリアゾール、5−(1−S,3−N)−チアゾール、2−ベンゾオキサゾール、2−ベンゾチアゾール、4−(1,2,3−N)−ベンゾトリアゾール、またはベンゾイミダゾールである、項目1に記載の化合物。
(8)R1、R3、R6、R8、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21が水素である、請求項1に記載の化合物。
(9)以下:
C−12に結合した窒素とC−12炭素およびC−13炭素が一緒になる場合に4〜7員を有する第一の環構造を形成する、第一の架橋基;ならびに/または、
C−2に結合した窒素とC−1炭素およびC−2炭素が一緒になる場合に4〜7員を有する第二の環構造を形成する、第二の架橋基、を含有する、項目1に記載の化合物。
(10)前記第一および第二の環構造の一方または両方が5員を有する、項目9に記載の化合物。
(11)前記5員の環構造が1つのgem二置換炭素を含む、請求項10に記載の化合物。
(12)前記gem置換基が(C1〜C8)アルキルである、請求項11に記載の化合物。
(13)前記gem置換基がメチルである、項目11に記載の化合物。
(14)前記5員環が連結部分または水溶性基で置換されている、項目10に記載の化合物。
(15)以下:
C−12に結合した窒素とC−11炭素およびC−12炭素が一緒になる場合に5〜7員を有する第三の環構造を形成する、第三の架橋基;ならびに/または、
C−2に結合した窒素とC−2炭素およびC−3炭素が一緒になる場合に5〜7員を有する第四の環構造を形成する、第四の架橋基、を含有する、項目1に記載の化合物。
(16)前記第三および第四の環構造の一方または両方が6員を有する、項目15に記載の化合物。
(17)前記6員環構造が1つのgem二置換炭素を含む、項目16に記載の化合物。
(18)前記gem置換基が(C1〜C8)アルキルである、請求項17に記載の化合物。
(19)前記gem置換基がメチルである、項目18に記載の化合物。
(20)以下の構造:
【化2】
を有する、項目1に記載の化合物であって、ここで、
L2およびL12は、アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルからなる群より選択されるリンカーである、化合物。
(21)C−3炭素およびC−4炭素、C−4炭素およびC−5炭素、C−9炭素およびC−10炭素、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含有する、項目1に記載の化合物。
(22)C−3炭素およびC−4炭素ならびにC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含有する、項目1に記載の化合物。
(23)エネルギー移動色素であって、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;および、 該ドナー色素および該アクセプター色素を連結するための、リンカー、を含み、ここで、
該ドナー色素およびアクセプター色素の少なくとも一方が、以下の構造:
【化3】
のうちの1つを有するスルホン化ジアリールローダミン化合物(それらの窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートである、エネルギー移動色素。
(24)前記アクセプター色素がスルホン化ジアリールローダミン化合物であり、前記ドナー色素がシアニン、フタロシアニン、スクアライン、ボディピー、ベンゾフェノキサジン、フルオレセイン、ジベンゾローダミン、またはローダミン色素である、項目23に記載のエネルギー移動色素。
(25)前記アクセプター色素がスルホン化ジアリールローダミン化合物であり、前記ドナー色素が、該アクセプター色素およびポリヌクレオチドに連結した、フルオレセインまたはローダミンである、項目24に記載のエネルギー移動色素。
(26)前記ドナー色素が前記ポリヌクレオチドの5’末端に連結されている、項目25に記載のエネルギー移動色素。
(27)前記ドナー色素が前記ポリヌクレオチドの3’末端に連結されている、項目25に記載のエネルギー移動色素。
(28)前記ドナー色素が前記ポリヌクレオチドの核酸塩基に連結されており、ここで、該核酸塩基がプリンの場合、前記リンカーは、8位で結合しており;該核酸塩基が7−デアザプリンの場合、該リンカーは、7位または8位で結合しており;該核酸塩基がピリミジンの場合、該リンカーは、5位で結合している、項目25に記載のエネルギー移動色素。
(29)項目23に記載のエネルギー移動色素であって、ここで、
前記リンカーは、以下の構造:
【化4】
を有し、ここで、
Zは、NH、SおよびOからなる群より選択され;
R21は、前記ドナー色素に結合したC1〜C12アルキルであり;
R22は、C1〜C12アルキルジイル、少なくとも1つの不飽和結合を有する5員環および6員環、ならびにカルボニル炭素に結合した縮合環構造からなる群より選択される置換基であり;
R23は、該リンカーを前記アクセプター色素に結合する官能基を含む、エネルギー移動色素。
(30)前記リンカーが以下の構造:
【化5】
を有し、nが2〜10の範囲である、項目29に記載の化合物。
(31)R23が以下の構造:
【化6】
を有し、ここで、R24がC1〜C12アルキルである、項目29に記載のエネルギー移動色素。
(32)前記リンカーが以下の構造:
【化7】
を有する、項目23に記載のエネルギー移動色素。
(33)前記リンカーが以下の構造:
【化8】
を有する、項目23に記載のエネルギー移動色素。
(34)前記リンカーが以下の構造:
【化9】
を有する、項目23に記載のエネルギー移動色素。
(35)前記リンカーが以下の構造:
【化10】
を有し、ここで、Dがドナー色素であり、Aがアクセプター色素であり、そしてnが1または2である、項目23に記載のエネルギー移動色素。
(36)前記リンカーが前記スルホン化ジアリールローダミン化合物のR2、R2’、R12またはR12’で結合している、項目23に記載のエネルギー移動色素。
(37)前記ドナーまたは前記アクセプターに対するリンカーがR2またはR2’に結合しており、ポリヌクレオチドに対するリンカーがR12またはR12’に結合している、項目36に記載のエネルギー移動色素。
(38)以下の式:
【化11】
を有する、標識されたヌクレオシドまたはヌクレオチドであって、ここで、
DYEは、以下の構造:
【化12】
の1つを有する、スルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
DYEは、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、
該ドナー色素および該アクセプター色素を連結するための、リンカー、を含む、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物であり;
Bは、核酸塩基であり;
Lは、リンカーであり;
R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはホスフェートアナログであり;そして、
R26およびR27は、単独である場合、各々独立して、H、HO、F、およびポリメラーゼ媒介性の標的指向性重合をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、標識されたヌクレオシドまたはヌクレオチド。
(39)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(40)Lが、以下:
【化13】
であり、nが0、1、または2である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(41)酵素学的に組み込み可能である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(42)ターミネーターである、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(43)以下の構造:
【化14】
を有する、項目42に記載の標識されたヌクレオシドまたはヌクレオチドであって、ここで、
R26およびR27は、単独である場合、各々独立して、H、F、およびポリメラーゼ媒介性の標的指向性重合をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、標識されたヌクレオシドまたはヌクレオチド。
(44)酵素学的に伸長可能である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(45)標識に共有結合したポリヌクレオチドを含有する、標識されたポリヌクレオチドであって、ここで、該標識は、以下の構造:
【化15】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物である、標識されたポリヌクレオチド。
(46)以下の式:
【化16】
を含有する、項目45に記載の標識されたポリヌクレオチドであって、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、核酸塩基であり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28およびR29は、単独である場合、各々独立して、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログであり;ここで、 該ポリヌクレオチドは、2〜1000ヌクレオチドを含む、標識されたポリヌクレオチド。
(47)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目46に記載の標識されたポリヌクレオチド。
(48)以下の式:
【化17】
を含有する、項目46に記載の標識されたポリヌクレオチドであって、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、核酸塩基であり;
Xは、O、NH、またはSであり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28は、ヌクレオチド間ホスホジエステルまたはヌクレオチド間アナログであり;ここで、
該ポリヌクレオチドは、2〜1000ヌクレオチドを含む、標識されたポリヌクレオチド。
(49)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目48に記載の標識されたポリヌクレオチド。
(50)Lが、C1〜C12アルキルジイルまたは−(CH2CH2O)n−であり、ここで、nが1〜100である、項目48に記載の標識されたポリヌクレオチド。
(51)以下の構造:
【化18】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)に共有結合したポリペプチドを含有する、標識されたポリペプチドであって、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、 第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素に共有結合したポリペプチドであり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物である、標識されたポリペプチド。
(52)以下の式:
【化19】
を有する、ホスホラミダイト化合物であって、ここで、
DYEは、以下の構造:
【化20】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
DYEは、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、
該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物であり、
Lは、リンカーであり;
別個になったR30およびR31は、C1〜C12アルキル、C1〜C12シクロアルキル、およびアリールからなる群より選択されるか;または、窒素原子と一緒になったR30およびR31は、飽和窒素複素環を形成し;そして、
R32は、亜リン酸エステル保護基である、ホスホラミダイト化合物。
(53)R32が、メチル、2−シアノエチル、および2−(4−ニトロフェニル)エチルからなる群より選択される、項目52に記載のホスホラミダイト化合物。
(54)R30およびR31が、各々イソプロピルである、項目52に記載のホスホラミダイト化合物。
(55)一緒になったR30およびR31がモルホリノである、請求項52に記載のホスホラミダイト化合物。
(56)LがC1〜C12アルキルジイルである、項目52に記載のホスホラミダイト化合物。
(57)Lが、前記DYEのR2、R2’、R12またはR12’で結合している、項目52に記載のホスホラミダイト化合物。
(58)以下の構造:
【化21】
を有する、項目57に記載のホスホラミダイト化合物。
(59)Lが、
【化22】
であり、nが1〜10の範囲である、項目57に記載のホスホラミダイト化合物。
(60)標識された基質を形成する方法であって、該方法は、請求項1に記載の化合物または項目23に記載のエネルギー移動色素の連結部分と、ポリヌクレオチド、ヌクレオチド、ヌクレオシド、ポリペプチド、炭水化物、リガンド、粒子、および表面からなる群より選択される基質とを反応させ、これによって標識された基質を形成する工程を包含する、方法。
(61)前記連結部分がN−ヒドロキシスクシンイミドである、請求項60に記載の方法。
(62)前記連結部分がホスホラミダイトである、項目60に記載の方法。
(63)前記粒子が、ナノ粒子、ミクロスフェア、ビーズ、またはリポソームである、項目60に記載の方法。
(64)前記表面がガラスである、項目60に記載の方法。
(65)標識されたポリヌクレオチドを合成する方法であって、該方法は、項目52に記載のホスホラミダイト化合物をポリヌクレオチドにカップリングする工程を包含し、ここで、該ポリヌクレオチドは、固体支持体に結合し、これによって、標識されたポリヌクレオチドを形成する、方法。
(66)標識されたプライマー伸長産物を作製する方法であって、該方法は、酵素学的に組み込み可能なヌクレオチドを用いてプライマー標識ハイブリッドを伸長させる工程を包含し、ここで、該プライマーまたは該ヌクレオチドは、項目1に記載の化合物または項目23に記載のエネルギー移動化合物で標識され、これによって、該プライマーが伸長される、方法。
(67)前記ヌクレオチドが酵素学的に伸長可能である、項目66に記載の方法。
(68)前記プライマーが標識されたポリヌクレオチドである、請求項66に記載の方法であって、該標識されたポリヌクレオチドは、以下の式:
【化23】
を含み、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される核酸塩基であり;
Xは、O、NH、またはSであり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28は、ヌクレオチド間ホスホジエステルまたはヌクレオチド間アナログであり;ここで、
該ポリヌクレオチドは、2〜100ヌクレオチドを含有する、方法。
(69)前記酵素学的に組み込み可能なヌクレオチドが、以下の式:
【化24】
を有する標識されたヌクレオシドまたはヌクレオチドである、項目66に記載の方法であって;ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される核酸塩基であり;
Lは、リンカーであり;
R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはホスフェートアナログであり;そして、
R26およびR27は、単独である場合、各々独立して、H、HO、およびFである、方法。
(70)ターミネーターヌクレオチドをさらに含む、項目66に記載の方法。
(71)R26およびR27が、単独である場合、各々独立して、
H、F、ポリメラーゼ媒介性の標的指向性プライマー伸長をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、項目69に記載の方法。
(72)ポリヌクレオチド配列決定の方法であって、該方法は、以下の工程:
第一、第二、第三、および第四のクラスのポリヌクレオチドの混合物を形成する工程であり、該クラスは、以下のようであり:
該第一のクラスの各ポリヌクレオチドは、3’末端ジデオキシアデノシンを含み、かつ第一の色素で標識されており;
該第二のクラスの各ポリヌクレオチドは、3’末端ジデオキシシチジンを含み、かつ第二の色素で標識されており;
該第三のクラスの各ポリヌクレオチドは、3’末端ジデオキシグアノシンを含み、かつ第三の色素で標識されており;そして、
該第四のクラスの各ポリヌクレオチドは、3’末端ジデオキシチミジンを含
み、かつ第四の色素で標識されており;ここで、
該第一、第二、第三、または第四の色素の1つ以上は、項目1に記載のスルホン化ジアリールローダミン化合物または項目23に記載のエネルギー移動色素であり、他の色素は、スペクトルにより互いに分解可能である、工程;ならびに、
該ポリヌクレオチドを電気泳動によって分離する工程、を包含する、方法。
(73)蛍光検出によって前記分離したポリヌクレオチドを検出する工程をさらに包含する、項目72に記載の方法。
(74)前記色素の蛍光スペクトルによって前記ポリヌクレオチドの3’末端ヌクレオチドを同定する工程をさらに包含する、項目72に記載の方法。
(75)オリゴヌクレオチドのライゲーション方法であって、該方法は、2つのプローブを標的配列にアニーリングする工程、および、一方のプローブの5’末端と他方のプローブの3’末端との間にホスホジエステル結合を形成する工程を包含し;ここで、
プローブの1つまたは両方が、項目1に記載の化合物または項目23に記載のエネルギー移動色素で標識されている、
方法。
(76)フラグメントの分析方法であって、該方法は、サイズ依存性分離プロセスによって、標識されたポリヌクレオチドフラグメントを分離する工程、および、該分離プロセスの後に、該分離した標識されたポリヌクレオチドフラグメントを検出する工程を包含し;ここで、
該フラグメントが、項目1に記載の化合物または項目23に記載のエネルギー移動色素で標識されている、方法。
(77)前記フラグメントが移動度変更標識で標識されている、請求項76に記載の方法。
(78)前記フラグメントがライゲーションによって形成される、項目76に記載の方法。
(79)前記サイズ依存性分離プロセスが電気泳動であり、前記標識されたポリヌクレオチドフラグメントが蛍光によって検出される、項目76に記載の方法。
(80)増幅方法であって、該方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該プライマーの少なくとも1つが、項目45に記載の標識されたポリヌクレオチドである、方法。
(81)増幅方法であって、該方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該ヌクレオチドの少なくとも1つが、項目38に記載の標識されたヌクレオチドである、方法。
(82)増幅方法であって、該方法は、2つ以上のプライマーおよび蛍光色素消光剤プローブを標的核酸にアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該プローブが、項目45に記載の標識されたポリヌクレオチドである、方法。
(83)ハイブリダイゼーション方法であって、該方法は、標的ポリヌクレオチドを固定化プローブにアニーリングする工程、ならびに、標的−プローブ複合体からの蛍光を検出する工程を包含し;ここで;
該標的ポリヌクレオチドが項目45に記載の標識されたポリヌクレオチドであり、該固定化プローブが平坦表面に共有結合している、方法。
(84)前記平坦表面がアドレス可能なアレイである、項目83に記載の方法。
(85)項目5に記載の連結部分を含むスルホン化ジアリールローダミン化合物およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(86)項目23に記載のエネルギー移動色素およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(87)項目52に記載のホスホラミダイト化合物およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(88)1つ以上の酵素学的に組み込み可能なヌクレオチドおよびプライマーを備える、標識したプライマー伸長産物を作製するためのキットであって、該プライマーが項目45に記載の標識されたポリヌクレオチドである、キット。
(89)1つ以上の酵素学的に組み込み可能なヌクレオチドおよびプライマーを備える、標識したプライマー伸長産物を作製するためのキットであって、該ヌクレオチドの少なくとも1つが項目38に記載の標識されたヌクレオチドである、キット。
(90)前記標識されたヌクレオチドがターミネーターである、請求項89に記載のキット。
(91)4つの異なるターミネーターを備える、項目90に記載のキットであって、これらの1つは標的Aで終結し、これらの1つは標的Gで終結し、これらの1つは標的Cで終結し、そしてこれらの1つは標的TまたはUで終結する、キット。
(92)スルホン化アミノナフタレン化合物およびアミノアントラセン化合物を合成する方法であって、該方法は、以下の工程:
アミノ基を保護基試薬で保護する工程;
該保護したアミノ化合物をクロルスルホン酸と反応させる工程;および、
該アミノ保護基を除去する工程、を包含する、方法。
また、第一の局面において、本発明は、以下の構造:
【0007】
【化25】
を有するスルホン化ジアリールローダミン色素化合物(その窒素置換形態およびアリール置換形態を含む)を包含する。R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つは、スルホネートである。
【0008】
窒素置換基、R2、R2’、R12およびR12’は、C1〜C6アルキルスルホネートまたはC4〜C10アリールスルホネートであり得る。特定の実施形態において、アルキルスルホネートは、−(CH2)n−SO3Hであり、nは、1〜6の整数であり、そしてアリールスルホネートは、以下:
【0009】
【化26】
であり、ここで、nは、0または1である。
【0010】
他の実施形態において、窒素置換基、R2、R2’、R12およびR12’は、C1〜C6アルキルカルボキシレートまたはC4〜C10アリールカルボキシレート:
【0011】
【化27】
(ここでnは、0または1である)であり得る。
【0012】
本発明の別の局面は、エネルギー移動色素化合物を包含し、この化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、このドナー色素およびアクセプター色素を連結するためのリンカー、を含有し;ここで、このドナー色素およびアクセプター色素の少なくとも一方は、スルホン化ジアリールローダミン化合物である。
【0013】
本発明の別の局面は、標識されたヌクレオシド、ヌクレオチド、ポリヌクレオチドまたはポリペプチドであり、ここで、この標識は、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素である。
【0014】
本発明の別の局面は、標識試薬および基質を試薬で標識する方法であり、これらとしては、基質と共有結合を形成する、ホスホラミダイトまたはスルホン化ジアリールローダミン化合物の活性エステル連結部分が挙げられる。
【0015】
本発明の別の局面は、標識した基質を形成するための方法であり、この方法は、基質を、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素の連結部分と反応させる工程を包含する。
【0016】
本発明の別の局面は、プライマー標的ハイブリッドを酵素学的に組み込み可能なヌクレオチドで伸長させることによって、標識プライマー伸長産物を作製する方法である。このプライマーまたはヌクレオチドは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識され得る。
【0017】
本発明の別の局面は、4つのクラスのポリヌクレオチドの混合物を形成することによる、ポリヌクレオチド配列決定方法であり、ここで、各クラスは、3’末端ヌクレオチドにおいて、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されており、かつこの標識は、スペクトルにより分解可能である。
【0018】
本発明の別の局面は、オリゴヌクレオチドライゲーション方法であり、この方法は、2つのプローブを標的配列にアニーリングして、一方のプローブの5’末端と他方のプローブの3’末端との間にホスホジエステル結合を形成することにより、ここで、プローブの1つまたは両方は、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されている。
【0019】
本発明の別の局面は、増幅方法であり、この方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングして、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によってこのプライマーを伸長させることにより、ここで、このプライマーの少なくとも1つまたはこのヌクレオチドの1つは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されている。
【0020】
本発明の別の局面は、試薬のキットであり、このキットは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素を備える。
【0021】
本発明のこれらおよび他の特徴および利点は、以下の説明、図面、および添付の特許請求の範囲を参照して、より理解されるようになる。
【図面の簡単な説明】
【0022】
【図1】図1は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図2】図2は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図3】図3は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図4】図4は、本発明のジベンゾローダミン色素化合物の合成のための一般化された合成経路を示す。
【図5】図5は、本発明のジベンゾローダミン色素化合物の合成のための例示的な合成経路を示す。
【図6】図6は、本発明のジベンゾローダミン色素化合物の合成のための例示的な合成経路を示す。
【図7】図7は、本発明の数種の例示的なジベンゾローダミン色素化合物の構造を示す。
【図8】図8は、化合物18の合成を示す。
【図9】図9は、化合物57〜61の合成を示す。
【図10】図10は、化合物67の合成を示す。
【図11】図11は、化合物72の合成を示す。
【図12】図12は、化合物76の合成を示す。
【図13】図13は、化合物81の合成を示す。
【図14】図14は、化合物82〜84の合成を示す。
【図15】図15は、化合物85の合成を示す。
【図16】図16は、化合物89の合成を示す。
【図17A】図17aは、化合物90の合成を示す。
【図17B】図17bは、化合物93の合成を示す。
【図18】図18は、化合物94〜99を示す。
【図19】図19は、化合物100〜105を示す。
【図20】図20は、化合物106〜109を示す。
【図21】図21は、スルホン化ジベンゾローダミン化合物(上段)、スルホン化ベンゾ−ナフトローダミン化合物(中段)、およびスルホン化ジナフトローダミン化合物(下段)を形成するための環化反応を示す。
【図22A】図22aは、スルホン化ジベンゾローダミン化合物110〜113を示す。
【図22B】図22bは、スルホン化ベンゾ−ナフトローダミン化合物114〜116を示す。
【図22C】図22cは、スルホン化ジナフトローダミン化合物117〜120を示す。
【発明を実施するための形態】
【0023】
(V.例示的な実施形態の詳細な説明)
ここで、本発明の特定の実施形態に対して、参照が詳細になされており、これらの例は、添付の図面において例示されている。本発明は、例示した実施形態と共に記載されているが、本発明がこれらの実施形態に限定されることは意図されないことが理解される。これとは対照的に、本発明は、全ての代替物、変更、および等価物を含むことが意図されており、これらは、添付の特許請求の範囲によって規定されるような本発明の範囲内に含まれ得る。
【0024】
(V.1.定義)
他に記載がなければ、以下の用語および句は、本明細書中で使用される場合、以下の意味を有することが意図される。
【0025】
一連の色素を参照して、「スペクトル分解能」とは、色素の蛍光発光バンドが十分に明確であり(すなわち、十分に非重複である)、それぞれの色素が結合した試薬(例えば、ポリヌクレオチド)が、標準的な光検出システムを用いて(例えば、米国特許第4,230,558号、同第4,811,218号、またはWheelessら、21〜76頁、Flow Cytometry:Instrumentation and Data Analysis(Acaemic Press,New York,1985)に記載されるシステムによって例示されるような、バンド通過フィルターおよび光電子増倍管のシステム、荷電連結デバイスおよび分光器などを使用して)、それぞれの色素によって発生される蛍光シグナルに基づいて識別され得ることを意味する。
【0026】
「複素環」とは、1つ以上の環原子が炭素ではない(すなわち、ヘテロ原子である)、環式化合物を意味する。例示的な複素環としては、ピロール、インドール、フラン、ベンゾフラン、チオフェン、ベンゾチオフェン、2−ピリジル、3−ピリジル、4−ピリジル、2−キノリル、3−キノリル、4−キノリル、2−イミダゾール、4−イミダゾール、3−ピラゾール、4−ピラゾール、ピリダジン、ピリミジン、ピラジン、シンノリン、フタラジン(pthalazine)、キナゾリン、キノキサリン、3−(1,2,4−N)−トリアゾリル、5−(1,2,4−N)−トリアゾリル、5−テトラゾリル、4−(1−O,3−N)−オキサゾール、5−(1−O,3−N)−オキサゾール、4−(1−S,3−N)−チアゾール、5−(1−S,3−N)−チアゾール、2−ベンズオキサゾール、2−ベンゾチアゾール、4−(1,2,3−N)−ベンゾトリアゾール、およびベンズイミダゾールが挙げられるが、これらに限定されない。
【0027】
「リンカー」とは、共有結合、または標識をポリヌクレオチドに共有結合するか、もしくは1つの標識を別の標識に共有結合する原子鎖を含む、化学的部分を意味する。
【0028】
「連結部分」とは、別の分子と反応して共有結合、または連鎖を形成し得る、化学的反応性基、置換基または部分(例えば、求核原子または求電子)を意味する。
【0029】
用語「標識」とは、本明細書中で使用される場合、基質(例えば、オリゴヌクレオチド、ヌクレオチドまたはヌクレオチド5’−トリホスフェート)に結合し得、かつ以下の(i)〜(v)のように機能する任意の部分を意味する:(i)検出可能なシグナルを提供する;(ii)第二の標識と相互作用して第一または第二の標識によって提供される検出可能なシグナル(例えば、FRET)を変更する;(iii)ハイブリダイゼーション(すなわち、二重鎖形成)を安定化する;(iv)電荷、疎水性、形状、または他の物理的パラメータによって移動度(例えば、電気泳動移動度または細胞透過性)に影響を与える;あるいは、(v)捕獲部分(例えば、親和性、抗体/抗原、またはイオン錯体)を提供する。
【0030】
「基質」とは、本発明の色素化合物が結合する実体である。基質としては、以下が挙げられるがこれらに限定されない:(i)ポリヌクレオチド、(ii)ヌクレオシドおよびヌクレオチド、(iii)ポリペプチド、(iv)炭水化物、(v)リガンド、ならびに(vi)前述の(i)〜(v)の任意のアナログ。
【0031】
「置換(された)」とは、本明細書中で使用される場合、1つ以上の水素原子が1つ以上の非水素原子、官能基または部分で置換されている分子をいう。例えば、非置換窒素は、−NH2であり、一方、置換窒素は、−NHCH3である。例示的な置換基としては、以下が挙げられるがこれらに限定されない:ハロ(例えば、フッ素および塩素)、(C1〜C8)アルキル、スルフェート、スルホネート、スルホン、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、複素環、水溶性基、および連結部分。
【0032】
「多環式芳香族」とは、複数の環構造を有する芳香族炭化水素を意味し、これには、ビアリールおよび縮合ベンゼノイド炭化水素が挙げられる。ビアリールは、2つ以上の環が単結合によって一緒に連結しているベンゼノイド化合物である。このクラスの親系は、ビフェニルである。縮合ベンゼノイド化合物は、環の各対が2つの炭素を共有するような様式においてオルト位にて一緒に縮合した2つ以上のベンゼン環によって特徴付けられる。この群の最も単純なメンバーは、2つの環を有するナフタレン、および3つの環を有するアントラセンおよびフェナントレンである。
【0033】
「アルキル」とは、親アルカン、アルケン、またはアルキンの単一の炭素原子から1つの水素原子を除去することによって誘導される、飽和または不飽和の、分枝鎖、直鎖、または環式の炭化水素基を意味する。代表的なアルキル基は、1〜12個の飽和および/または不飽和の炭素からなり、これらとしては、メチル、エチル、プロピル、ブチルなどが挙げられるがこれらに限定されない。
【0034】
「アルコキシ」とは、−OR(ここで、Rは、(C1〜C6)アルキルである)を意味する。
【0035】
「アルキルジイル」とは、飽和または不飽和の、分枝鎖、直鎖または環式の、1〜20個の炭素原子の炭化水素基であって、親アルカン、アルケンまたはアルキンの同じかまたは2つの異なる炭素原子から2つの水素原子を除去することによって誘導される、2つの一価のラジカル中心(radical center)を有する炭化水素基を意味する。代表的なアルキルジイル基としては、1,2−エチルジイル、1,3−プロピルジイル、1,4−ブチルジイルなどが挙げられるがこれらに限定されない。
【0036】
「アリール」とは、親芳香族環系の単一炭素原子から1つの水素原子を除去することによって誘導される、6〜20個の炭素原子の、一価の芳香族炭化水素基を意味する。代表的なアリール基としては、ベンゼン、置換ベンゼン、ナフタレン、アントラセン、ビフェニルなどから誘導される基が挙げられるがこれらに限定されない。
【0037】
「アリールジイル」とは、共役共鳴電子系、および親アリール化合物の2つの異なる炭素原子から2つの水素原子を除去することによって誘導される少なくとも2つの一価のラジカル中心を有する、6〜20個の炭素原子の、不飽和環式基または多環式炭化水素基を意味する。
【0038】
「置換アルキル」、「置換アルキルジイル」、「置換アリール」および「置換アリールジイル」とは、それぞれ、1つ以上の水素原子が各々独立して別の置換基で置換されている、アルキル、アルキルジイル、アリールおよびアリールジイルを意味する。代表的な置換基としては、以下が挙げられるがこれらに限定されない:−X、−R、−O−、−OR、−SR、−S−、−NR2、−NR3、=NR、−CX3、−CN、−OCN、−SCN、−NCO、−NCS、−NO、−NO2、=N2、−N3、NC(O)R、−C(O)R、−C(O)NRR、−S(O)2O−、−S(O)2OH、−S(O)2R,−OS(O)2OR、−S(O)2NR、−S(O)R、−OP(O)O2RR、−P(O)O2RR、−P(O)(O−)2、−P(O)(OH)2、−C(O)R、−C(O)X、−C(S)R、−C(O)OR、−C(O)O−、−C(S)OR、−C(O)SR、−C(S)SR、−C(O)NRR、−C(S)NRR、−C(NR)NRR(ここで、各Xは、独立してハロゲンであり、各Rは、独立して−H、アルキル、アリール、複素環、または連結基である)。
【0039】
「ヌクレオチド間アナログ」とは、以下のようなオリゴヌクレオチドのリン酸エステルアナログを意味する:(i)アルキルホスホネート(例えば、C1〜C4アルキルホスホネート(特に、メチルホスホネート));(ii)ホスホラミダイト;(iii)アルキルホスホトリエステル(例えば、C1〜C4アルキルホスホトリエステル);(iv)ホスホロチオエート;および(v)ホスホロジチオエート。ヌクレオチド間アナログとしてはまた、非ホスフェートアナログが挙げられ、ここで、糖/ホスフェートのサブユニットは、非ホスフェート含有骨格構造によって置換されている。非ホスフェートオリゴヌクレオチドアナログの1つの型は、アミド結合(例えば、PNA(Nielsen(1991)「Sequence−selective recognition of DNA by strand displacement with a thymidinesubstituted polyamide」Science 254:1497〜1500)と通常称される、2−アミノエチルグリシンユニット)を有する。
【0040】
「核酸塩基」とは、相補的な核酸塩基または核酸塩基アナログ(例えば、プリン、7−デアザプリン、またはピリミジン)との対形成においてワトソン−クリック型水素結合を形成し得る窒素含有複素環式部分を意味する。代表的な核酸塩基は、天然に存在する核酸塩基であるアデニン、グアニン、シトシン、ウラシル、チミン、および天然に存在する核酸塩基のアナログ(例えば、7−デアザアデニン、7−デアザグアニン、7−デアザ−8−アザグアニン、7−デアザ−8−アザアデニン(Kutyavin,米国特許第5,912,340号)、イノシン、ネブラリン(nebularine)、ニトロピロール、ニトロインドール、2−アミノプリン、2,6−ジアミノプリン、ヒポキサンチン、偽ウリジン(pseudouridine)、偽シトシン、偽イソシトシン、5−プロピニルシトシン、イソシトシン、イソグアニン、7−デアザグアニン、2−チオピリミジン、6−チオグアニン、4−チオチミン、4−チオウラシル、O6−メチルグアニン、N6−メチルアデニン、O4−メチルチミン、5,6−ジヒドロチミン、5,6−ジヒドロウラシル、4−メチルインドール、およびエテノアデニンである(Fasman(1989)Practical Handbook of Biochemistry and Molecular Biology,385〜394頁,CRC Press,Boca Raton,Fl)。
【0041】
「ヌクレオシド」は、リボース糖のC−1’炭素に連結した核酸塩基を含む化合物を意味する。このリボースは、置換されていても置換されていなくてもよい。置換されたリボース糖としては、1つ以上の炭素原子(例えば、3’炭素原子)が1つ以上の同じかまたは異なる、−R、−OR、−NRRまたはハロゲン基(ここで、各R基は、独立して、水素、C1〜C6アルキルまたはC5〜Cl4アリールである)で置換されたリボースが挙げられるが、これらに限定されない。リボースとしては、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、3’−ハロリボース、3’−フルオロリボース、3’−クロロリボース、3’−アルキルリボース(例えば、2’−O−メチル、4’−α−アノマーヌクレオチド、1’−α−アノマーヌクレオチド、ならびに2’−4’−連結および他のロックド(locked)二環式糖修飾物)(Imanishi WO 98/22489;Imanishi WO 98/39352;Wengel WO 99/14226)が挙げられる。核酸塩基がプリン(例えば、AまたはG)である場合、リボース糖は、核酸塩基のN9位に結合される。核酸塩基がピリミジン(例えば、C、TまたはU)である場合、ペントース糖は、核酸塩基のN1位に結合される(KornbergおよびBaker,(1992)DNA Replication,第2版,Freeman,San Francisco,CA)。
【0042】
「ヌクレオチド」とは、モノマー単位としてまたは核酸内の、ヌクレオシドのリン酸エステルを意味する。ヌクレオチドは、ときどき、特にリボース糖の構造的特徴を示すために、「NTP」、または「dNTP」および「ddNTP」として記載される。「ヌクレオチド5’三リン酸」は、5’位に三リン酸エステル基を有するヌクレオチドをいう。三リン酸エステル基は、種々の酸素の硫黄での置換がを含み得る(例えば、α−チオヌクレオチド5’三リン酸)。
【0043】
「酵素学的に組み込み可能な」とは、ヌクレオチドが、ポリメラーゼ酵素の作用を介して、新生ポリヌクレオチド鎖の末端(例えば、3’末端)に、酵素学的に組み込まれることが可能であるヌクレオチドの特性を意味する。
【0044】
「ターミネーター」とは、生じるポリヌクレオチド鎖へのヌクレオチドのそれ以降の組み込みを妨害し、それによってポリメラーゼ媒介伸長を停止させる、酵素学的に組み込み可能なヌクレオチドを意味する。代表的なターミネーターは、3’−ヒドロキシル置換基を欠き、そしてターミネーターとしては、2’,3’−ジデオキシリボース、2’,3’−ジデヒドロリボース、および2’,3’−ジデオキシ、3’−ハロリボース(例えば、3’−フルオロ)が挙げられる。あるいは、リボフラノースアナログ(例えば、アラビロース)が、使用され得る。例示的なヌクレオチドターミネーターとしては、以下が挙げられる:2’,3’−ジデオキシ−β−D−リボフラノシル、β−D−アラビノフラノシル、3’−デオキシ−β−D−アラビノフラノシル、3’−アミノ−2’,3’−ジデオキシ−β−D−リボフラノシル、および2’,3’−ジデオキシ−3’−フルオロ−β−D−リボフラノシル(Chidgeavadze(1984)Nucleic Acids Res.,12:1671〜1686;およびChidgeavadze(1985)FEB.Lett.,183:275〜278)。ヌクレオチドターミネーターとしてはまた、リバーシブルヌクレオチドターミネーターが挙げられる(Metzker(1994)Nucleic Acids Res.,22(20):4259)。
【0045】
「酵素学的に伸長可能な」は、ポリヌクレオチドの末端において酵素学的に組み込み可能であり、かつ得られる伸長ポリヌクレオチドが、ヌクレオチドまたはヌクレオチドアナログの引き続く組み込みを行い得る、ヌクレオチドの特性である。
【0046】
用語「標的配列」および「標的ポリヌクレオチド」とは、相補的なポリヌクレオチドとのハイブリダイゼーションの対象であるポリヌクレオチド配列(例えば、プライマーまたはプローブ)を意味する。この配列は、DNA、RNA、それらのアナログ(それらの組み合わせを含む)から構成され得る。
【0047】
「水溶性基」とは、水溶液中での本発明の化合物の溶解度を増加させる置換基を意味する。例示的な水溶性基としては、第四級アミン、スルフェート、スルホネート、カルボキシレート、ホスホネート、ホスフェート、ポリエーテル、ポリヒドロキシル、およびボロネートが挙げられるがこれらに限定されない。
【0048】
本明細書中で使用される場合、用語「オリゴヌクレオチド」および「ポリヌクレオチド」は、交換可能に使用され、ヌクレオチドモノマーの一本鎖ポリマーおよび二本鎖ポリマーを意味し、これらとしては、ヌクレオチド間ホスホジエステル結合の連鎖によって連結されている、2’−デオキシリボヌクレオチド(DNA)およびリボヌクレオチド(RNA)、またはヌクレオチド間アナログ、および付随する対イオン(例えば、H+、NH4+、トリアルキルアンモニウム、Mg2+、Na+など)が挙げられる。ポリヌクレオチドは、デオキシリボヌクレオチド、リボヌクレオチドから完全に構成されても、それらのキメラ混合物から構成されてもよい。ポリヌクレオチドは、ヌクレオチド間アナログ、核酸塩基アナログおよび糖アナログから構成され得る。ポリヌクレオチドのサイズは、これらがしばしばオリゴヌクレオチドと呼ばれる場合、代表的には、数個のモノマー単位(例えば、5〜40)〜数千個のモノマーヌクレオチド単位の範囲である。他に記載されない限り、ポリヌクレオチド配列が示される場合はいつでも、このヌクレオチドが左から右に5’→3’の順であり、そして「A」がデオキシアデノシンを示し、「C」がデオキシシチジンを示し、「G」がデオキシグアノシンを示し、そして「T」がチミジンを示すことが、理解される。
【0049】
「ローダミン色素」とは、以下の多環式の一般構造:
【0050】
【化28】
を含む色素(それらの任意および全ての置換バージョンを含む)をいう。
【0051】
(V.2 1−アミノ−3−メトキシナフタレンおよび1−アミノ−3−メトキシアントラセンの中間体)
1−アミノ−3−メトキシナフタレン(式I)および1−アミノ−3−メトキシアントラセン(式II)化合物のクラスは、スルホン化ジアリールローダミン色素の合成における中間体として有用である。式Iおよび式IIの化合物はさらに、それらのアリール置換形態および窒素置換形態を含む(本明細書中に提供される全ての分子構造は、示される正確な電子構造だけを含むのではなく、それらの全ての共鳴構造、プロトン化状態および付随する対イオンも含むことに注意のこと)。
【0052】
【化29】
式IおよびIIの化合物の1つの実施形態において、C−12に結合した窒素ならびにC−12炭素およびC−13炭素は、4〜7員を有する第一の環構造を形成し得、そして/またはC−12に結合した窒素ならびにC−11炭素およびC−12炭素は、5〜7員を有する第二の環構造を形成する。この第一および第二の環構造は、5員を有し得、ここで、この5員環構造は、1つのgem二置換炭素を含み得る。このgem置換基は、アルキル(例えば、メチル)であり得る。別の実施形態において、この5員環は、連結部分で置換されている。この実施形態において、C−12に結合した窒素ならびにC−12炭素およびC−13炭素は、4〜7員を有する第一の環構造を形成し、そして/またはC−12に結合した窒素ならびにC−11炭素およびC−12炭素は、5〜7員を有する第二の環構造を形成する。
【0053】
窒素置換基としては、アルキル、フェニル、芳香族、複素環、多環式芳香族、水溶性基、連結部部分、およびそれらの置換形態が挙げられ得る。窒素置換基は、アルキル、フェニル、またはそれらの置換形態であり得、ここで、置換基は、連結部分、スルホネートまたは水溶性基であり得る。例示的な水溶性基は、カルボキシレート、スルホネート、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリヒドロキシル、および水溶性ポリマーである。1つの実施形態において、窒素置換基は、−L−Rであり、ここで、Lは、任意のリンカーであり、Rは、連結部分または水溶性基であり得る。特定の実施形態において、Lは、以下:
【0054】
【化30】
であり、ここで、nは、1〜8の範囲である。
【0055】
式Iおよび式IIの化合物は、1つ以上のアリール位(例えば、C−8〜C−11、およびC−13)に1つ以上の置換基を含み得る。置換基としては、ホルミル、ヒドロキシル、フッ素、塩素、アルキル、スルフェート、スルホネート、スルホン、スルホンアミド、スルホキシド、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、水溶性基、複素環、および連結部分(それらの置換形態を含む)が挙げられ得る。1つの実施形態において、この化合物は、C−9炭素およびC−10炭素にわたって、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態(ここで、置換基はスルホネート基である)を含む)を含有する。
【0056】
式IおよびIIの非ホルミル化バーションは、以下に詳述されるスルホン化ジアリールローダミン色素を形成するための、環化反応における中間体である。
【0057】
本発明の代表的な1−アミノ−3−ヒドロキシナフタレン化合物および1−アミノ−3−ヒドロキシアントラセン化合物は、図1〜3(すなわち、化合物4、化合物9、化合物15、化合物17、化合物22、化合物27および化合物29);図10〜13(すなわち、化合物62、化合物63、化合物68、化合物73および化合物80);ならびに図16〜20(すなわち、化合物86〜109)に示される。
【0058】
当業者はまた、特に、周囲のpHに依存して、本発明の化合物が多数の異なるプロトン化状態で存在し得ることを認識している。本明細書中に提供される構造式は、数種の可能なプロトン化状態のうちの1種の化合物のみを示しているが、これらの構造は単に例示であり、本発明は任意の特定のプロトン化状態に制限されない、すなわち、任意および全てのプロトン化形態の化合物が本発明の範囲内に包含されることが意図されることが理解される。
【0059】
本発明の化合物は、複数の正電荷または負電荷を有し得る。本発明の色素の正味の電荷は、正または負のいずれかであり得る。色素に付随する対イオンは、代表的には、化合物が得られる合成方法および/または単離方法によって決定される。代表的な対イオンとしては、アンモニウム、ナトリウム、カリウム、リチウム、ハライド、アアセテート、トリフルオロアセテートなど、およびこれらの混合物が挙げられるがこれらに限定されない。任意の付随する対イオンの同定は、本発明の重要な特徴ではなく、そして本発明は、対イオンの任意の型に付随する色素を包含することが理解される。さらに、化合物が種々の異なる形態で存在する場合、本発明は、対イオンに付随する色素の形態(例えば、乾燥塩)だけでなく、対イオンに付随しない形態(例えば、水溶液または有機溶液)も包含することが意図される。
【0060】
上記発明の種々の局面は、多重化蛍光検出のために有用な公知の蛍光色素化合物に対する、予測できない、驚くべき、以下の重要な利点の1つ以上を達成する:(1)目的の色素化合物は、約630nmの波長を使用する低コストの赤色レーザによって、効率的に励起され得る;(2)目的の色素化合物の発光スペクトルは、窒素置換基および/またはアリール置換基の型および位置のモニター変化によって調節され得、類似の吸収特性を有するが、なお分解可能な蛍光発光スペクトルを有する色素のセットを作製するのを可能にする;(3)目的の色素化合物は、有利な蛍光特性を損なうことなく、ヌクレオシド、ヌクレオチドまたはポリヌクレオチドに容易に結合され得る;(4)目的の色素化合物は、狭い発光バンド幅を有する(すなわち、この発光バンド幅は、約70nmよりも低い、最大発光強度の半分の全幅を有する);(5)目的の色素化合物は、緩衝化水溶液に非常に可溶性であるが、高量子収率を維持する;(6)目的の色素化合物は、相対的に光安定性である;そして、(7)目的の色素化合物は、相対的に高い吸光係数(すなわち、約50,000よりも大きい)を有する。
【0061】
いくつかの合成方法が、上記の1−アミノ−3−ヒドロキシナフタレン化合物の合成のために利用可能であり、合成される特定の化合物の環構造の性質および窒素置換基の性質に依存する、種々の方法が好ましい。
【0062】
1−置換アミノ−3−ヒドロキシナフタレン化合物(例えば、1−シエチルアミノ−3−ヒドロキシナフタレン(4))の合成に適切な、第一の合成方法が、図1に示される。この第一の方法において、3−メトキシ−1−ヒドロキシナフタレン(1)を乾燥トリエチルアミンおよびトリフルオロメタンスルホン酸無水物と反応させて、粗製の3−メトキシナフタレン−1−トリフレート(2)が形成される。次いで、トリフレート(2)を、パラジウムで触媒したトリフレート/アミンのカップリングを用いて、アミン(例えば、第二級アミン(例えば、ジエチルアミン))と反応させて、置換アミン化合物(3)が形成される。次いで、化合物(3)は、三臭化ホウ素脱保護手順を使用して脱保護されて、1−アミノ−3−ヒドロキシナフタレン生成物(例えば、1−ジエチルアミノ−3−ヒドロキシナフタレン(4))が生成される。この合成の例は、以下の実施例1に提供される。
【0063】
ベンゾインドリン化合物(例えば、N−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(9))の合成に適した第二の合成方法もまた、図1に示される。この方法において、3−メトキシナフタレン−1−トリフレート(2)は、パラジウムで触媒したトリフレートカップリング反応を使用して第一級アミン(例えば、アニリン)から誘導体化され、第二級アミン(例えば、1−アニリン−3−メトキシナフタレン(5))が得られる。この第二級アミン(5)は、酸塩化物(例えば、ハロ塩化アセチル)を用いてアセチル化され、二置換アミド(例えば、1−アミド−3−メトキシナフタレン(6))が得られる。この第三級アミン(6)は、ルイス酸を触媒したフリーデル−クラフツ環化手順を使用して(例えば、AlCl3を使用して)環化され、化合物(7)が得られる。次いで、化合物(7)は、例えば、(水素化アルミニウムリチウム)LAHを使用して還元されて化合物(8)が得られる。三臭化ホウ素脱保護手順によってメトキシ基を脱保護した後、ベンゾインドリン(例えば、N−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(9))が得られる。この合成の例は、以下の実施例2に提供される。
【0064】
N−置換−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン化合物(例えば、N−メチル−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン(15))の合成に適した第三の合成方法が、図2に示される。この方法において、化合物10は、ピペリジン触媒を含有するピリジンを使用してマロン酸と縮合することによって、メトキシナフトアルデヒドから合成される。次いで、化合物10は、LAH還元の前に水素で還元され、そして、トリフルオロメタンスルホン酸無水物と反応させてトリフレート(11)が得られる。このトリフレート(11)は、NaN3と反応され、化合物(12)が得られる。化合物(12)を、ルイス酸(例えば、AlCl3)と錯化して還流し、環化ベンゾキノリン誘導体(13)が得られる。次に、窒素置換基を加える(例えば、窒素を、従来のアルキル化手順を使用してアルキル化する(例えば、ベンゾキノリン誘導体(13)を、n−ブチルリチウムおよびアルキル化剤(例えば、MeI)と反応させて化合物(14)が得られるか、またはプロパンスルホン(propane sultone)と反応させて化合物(16)が得られる))。次いで、メトキシ基が、三臭化ホウ素手順によって除去されて、N−アルキルベンゾキノリン誘導体(例えば、化合物15または化合物17)が得られる。この合成の例は、以下の実施例3に提供される。
【0065】
N−置換−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン化合物(例えば、N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン(22))の合成に適した第四の合成方法が、図3に示される。この方法において、Rosowsky(1965)Jour.Org.Chem.30:1832、および本明細書中の参考文献の手順に従って、1−アミノ−3−メトキシナフタレン(18)を、ヨウ素によって触媒されたアセトンと反応させ、次いで、飽和Na2S2O3でクエンチして、ベンゾキノリン化合物(19)が得られる。次いで、化合物(19)を、一般的なアルキル化手順に従ってアルキル化剤(例えば、MeI)でアルキル化し、化合物20が得られる。このアルキル化した化合物(20)を、Pd/Cによって触媒されたH2で還元して、N−メチルメトキシキノリン中間体(21)が得られ、続いて、一般的な三臭化ホウ素手順によってメトキシ基を脱保護して、N−置換−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン化合物(例えば、N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン(22)が得られる。この合成の例は、以下の実施例4に提供される。
【0066】
N−置換−3,3−ジメチル−4−ヒドロキシベンゾインドリン化合物(例えば、N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(27))の合成に適した第五の合成方法もまた、図3に提供される。この方法において、1−アミノ−3−メトキシナフタレン(18)は、酸塩化物(例えば、2−ブロモ−2−メチルプロピオニルクロライド)でアセチル化されて、化合物(23)が得られる。化合物(23)は、AlCl3と反応させることによって環化されて、化合物(24)が得られる。次いで、化合物(24)は、LAHで還元されて、3,3−ジメチル−4−メトキシベンゾインドリン(25)が得られる。次いで、化合物(25)は、アルキル化剤(例えば、ヨウ化メチル)でアルキル化されて、N−メチル−3,3−ジメチル−4−メトキシベンゾインドリン(例えば、化合物(26))が得られる。三臭化ホウ素によるメトキシ基脱保護の後に、化合物(27)が得られる。この合成の例は、実施例5に提供される。
【0067】
アミノアントラセン化合物の合成に適した第六の合成方法が、図17aに示される。この方法において、アミノアントラセン化合物(メトキシナフトインドリン)90は、1,3−ジメトキシアントラセン121を脱メチル化し、ヒドロキシル基のうちの1つをモノメチル化して調製して123を得、トルフレート124を形成し、そして、ヒドラジンによって置換して125を得る。イソブチルアルデヒドとのヒドラゾンの形成、フッシャーインドリン環化(Fischer indoline cyclization)およびシアノホウ化水素ナトリウムでの還元によって90が得られる(実施例15)。
【0068】
アミノナフタレン化合物およびアミノアントラセン化合物は、スルホン化剤(例えば、クロロスルホン酸)でスルホン化され得る。例えば、アミノナフタレン化合物86は、初めに、N−スルホンアミオ化合物87(図16、実施例14)のように窒素保護され、次いで、低温度にて、クロロスルホン酸を含有する酢酸およびジクロロメタンと反応させ、脱保護してスルホン化化合物89が得られる。同様に、アミノアントラセン化合物90は、トルフルオロアセチル化化合物91のように保護され、次いで、スルホン化され、そして脱保護されて、93(図17b、実施例15)が得られる。中間体89および93は、スルホン化ジアリールローダミン化合物を合成する際に有用である。
【0069】
(V.3 スルホン化ジアリールローダミン色素化合物)
新規なクラスのスルホン化ジアリールローダミン色素化合物は、以下の式IIIabcに示されるような一般構造(それらのアリール置換形態および窒素置換形態を含む)を有する分子標識として有用である。
【0070】
【化31】
式IIIabcにおいて、少なくとも1つのアリール置換基は、スルホネートである。少なくともアリール−スルホネート基の存在によって、ジアリールローダミン化合物に、予想外の有利な特性(例えば、調節された溶解度、電気泳動移動度、およびスペクトル増強)が与えられ得る。
【0071】
式IIIabcの化合物の1つの実施形態において、この化合物は、第一結合基および/または第二結合基を含有する。第一結合基は、C−12に結合した窒素ならびにC−12炭素およびC−13炭素が一緒になる場合に、4〜7員を有する第一の環構造を形成し得、第二結合基は、C−2に結合した窒素ならびにC−1炭素およびC−2炭素が一緒になる場合に、4〜7員を有する第二の環構造を形成する。この第一および第二の環構造の一方または両方が、5員を有し得る。別の実施形態において、この5員環構造は、1つのgem二置換炭素を含み、ここで、このgem置換基は、アルキル(例えば、メチル)であり得る。代替の実施形態において、この5員環は、連結基で置換されている。別の実施形態において、この5員環は、以下に記載される1つ以上の窒素置換基を含む。架橋環構造を有する例示的なスルホン化ジアリールローダミン化合物としては、以下が挙げられる:
【0072】
【化32】
L2およびL12は、アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、および置換ナフチルを含むリンカーである。
【0073】
本発明のなお別の実施形態において、式IIIabcの化合物は、1つ以上の窒素置換基、R2、R2’、R12およびR12’を含む。このような置換基は、以下からなる群より選択される:アルキル、フェニル、芳香族、複素環、多環式芳香族、水溶性基、連結基、およびそれらの置換形態。また、窒素置換基は、アルキル、フェニル、多環式芳香族、またはそれらの置換形態であり、ここで、例示的な置換基としては、連結基および水溶性基が挙げられる。
【0074】
本発明の、この第二の局面の別の実施形態において、式IIIabcの化合物は、第三結合基および/または第四結合基を含有する。第三結合基は、C−12に結合した窒素ならびにC−11炭素およびC−12炭素が一緒になる場合に、5〜7員を有する第三の環構造を形成し得、第四結合基は、C−2に結合した窒素ならびにC−2炭素およびC−3炭素が一緒になる場合に、5〜7員を有する第四の環構造を形成する。この第三および第四の環構造の一方または両方が、6員を有し得る。この6員環構造は、1つのgem二置換炭素を含み得、ここで、このgem置換基は、アルキル(例えば、メチル)であり得る。
【0075】
本発明の別の実施形態において、式IIIabcの化合物は、1つ以上の炭素(C−1、C−3〜C−6、C−8〜C−11、およびC14〜C−21)にてアリール置換基を含む。例示的なアリール置換基としては、以下が挙げられる:水素、フッ素、塩素、アルキル、スルフェート、スルホネート、スルホン、スルホンアミド、スルホキシド、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、水溶性基、複素環、および連結部分(それらの置換形態を含む)。少なくとも1つのアリール−置換基は、スルホネートである。別の実施形態において、式IIIabcの化合物は、C−3炭素およびC−4炭素、C−4炭素およびC−5炭素、C−9炭素およびC−10炭素、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含む。この縮合芳香族環は、C−3炭素およびC−4炭素ならびにC−10炭素およびC−11炭素、またはC−9炭素およびC−10炭素ならびにC−4炭素およびC−5炭素(それらの置換形態を含む)にわたって結合し得る。
【0076】
本発明のこの第二の局面に従う数種の例示的な色素化合物は、以下に示される:図7および図10〜13(すなわち、化合物41〜47および化合物67、化合物72、化合物76および化合物81);ならびに、図21、図22a、図22b、図22c(すなわち、予測外の有利なスペクトル特性を有する化合物110〜120)。吸光率、量子収量および輝度は、非スルホン化アナログジアリールローダミン色素化合物と比較して、スルホン化ジアリールローダミン色素化合物111、114、118、119、120の全てにおいて増加する。
【0077】
(アリールスルホン化/非アリールスルホン化)
【0078】
【化33】
全ての測定を、8Mの尿素および1×Tris/EDTA緩衝液中で行った。
【0079】
上表は、非アリール−スルホネートアナログに対する、5つの例示的な色素のうちの2つのアリール−スルホネート基によって生じる吸光率、量子収量および輝度の差を測定している。輝度増加は、吸光率および量子収量を掛けることによって算出される。
【0080】
一般的に、本発明のスルホン化ジアリールローダミン色素は、以下(図4)のように合成される:無水物誘導体(例えば、無水フタル酸)を、1−アミノ−3−メトキシ中間体31および32、ならびにルイス酸(例えば、ZnCl2)と混合する(ここで、化合物30のR置換基は、同じであっても異なっていてもよいが、好ましくは、同じである)。例示的なR置換基としては、アセチレン、アルキル、フェニル、複素環、およびそれらの置換形態が挙げられるがこれらに限定されない。この混合物を、融解が観察されるまで、手短に加熱する。溶媒(例えば、1,2−ジクロロベンゼン)を、この反応混合物に加え、そして、不均一な混合物を、約130℃〜約180℃まで加熱する。粗製反応混合物を冷却し、そして、順相のフラッシュクロマトグラフィーによって精製して、色素化合物33を得る。無水物が置換フタル酸無水物の一部(例えば、化合物34)である場合、2つの異性体が形成される(図5)。これらの異性体35および36は、PTLCによって分離される。異性学的に(isomerically)純粋な色素は、順相TLCおよび逆相TLCでの単一スポットによって、ならびにそれらのUV/可視吸収スペクトルおよびそれらの長い波長蛍光励起スペクトルおよび発光スペクトルによって同定される。
【0081】
非対称性色素化合物の合成のための代替の手順が、図6に示される。このプロセスにおいて、無水物誘導体(例えば、フタル酸無水物34)を、乾燥ニトロベンゼンと混合して、加熱する。この混合物を、室温まで冷却し、無水AlCl3を、攪拌しながら加える。続いて、1−アミノ−3−メトキシ中間体31を、攪拌しながら加え、この反応物を加熱する。この反応物を冷却し、EtOAc中に懸濁する。有機層を、飽和NH4Cl、ブラインで洗浄し、Na2SO4で乾燥し、濾過し、そして溶媒を減圧下で除去する。得られたケトン中間体37/38を精製し、そして、フラッシュクロマトグラフィーまたは再結晶化によって、異なる異性体37および38に分離する(C−14およびC−17における置換基が同じであり、かつC−15およびC−16における置換基が同じである場合を除く)。異性学的に純粋なケトン中間体37または38のメトキシ基を、一般的な三臭化ホウ素脱保護手順に従って除去する。これによって、化合物38は、アミノヒドロキシナフタレンケトン中間体39を与える。次いで、アミノヒドロキシナフタレンケトン中間体39を、1−アミノ−3−メトキシ中間体32と反応させる。この反応物を冷却して、PTLCによってさらに精製され得る、異性学的に純粋でかつ非対称的に置換された生成物40が得られる。
【0082】
本発明のジベンゾローダミン色素を合成し、かつC−7位で置換されていない色素(例えば、ピロニン色素)の合成に適している別の方法において、色素は、O−保護してN−置換された3−ヒドロキシベンゾインドリン化合物から生成されるヒドロキシベンゾインドリン中間体から合成され、続いて、酸素保護基(例えば、メチル基)を脱保護し、脱メチル化試薬(例えば、塩化アルミニウム)によって脱保護し、そして順相クロマトグラフィーによる精製によって単離する。この合成に従って、色素分子の半分に対応するヒドロキシベンゾインドリン中間体を、最初にホルミル化剤(例えば、メチルホルムアニリド/POCl3)と反応し、その後、ホルミル化したヒドロキシベンゾインドリン中間体を、色素分子の半分に対応する異なる(または、同じ)ヒドロキシベンゾインドリン中間体と直接反応させる。この反応を、酸性脱水条件下(例えば、POCl3)で行い、そして例えば、120〜160℃に加熱して、粗製カルボン酸エステル誘導体化ベンゾピロニン色素が得られる。いくつかの場合において、メトキシベンゾインドリン中間体を、メチル基を脱保護する前にホルミル化し、次いで、第2当量のヒドロキシインドリンと反応させてピロニン色素を得る前に、このホルミル化したメトキシインドリン誘導体のメチル基を脱保護して、ホルミル化したヒドロキシベンゾインドリン中間体を得る。純粋な色素を、水性ワークアップおよび順相クロマトグラフィーの後に単離する。次いで、中間体色素のカルボン酸エステルを、酸(例えば、HBr)で加水分解して、水性ワークアップおよび順相クロマトグラフィーの後に遊離酸色素誘導体を得る。その後、N−フェニル置換色素を、スルホン化剤(例えば、ClSO3H)でスルホン化して、水性ワークアップおよび順相クロマトグラフィーの後に最終色素が得られ得る。
【0083】
(V.4 スルホン化ジアリールローダミン色素を組み込むエネルギー移動色素)
本発明は、式IIIabcのスルホン化ジアリールローダミン色素化合物を組み込むエネルギー移動色素化合物を含む。一般的に、本発明のエネルギー移動色素化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射する、ドナー色素、このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素を含む。ドナー色素は、リンカーによってアクセプターに結合しており、このリンカーは、ドナー色素とアクセプター色素との間の効率的なエネルギー移動を促進するのに有効である(Lee「Energy−transfer dyes with enhanced fluorescence」、米国特許第5,800,996号; Lee「Energy−transfer dyes with enhanced fluorescence」、米国特許第5,945,526号;Mathies「Fluorescent labels and their use in separations」;米国特許第5,654,419号;Lee(1997)Nucleic Acids Res.25:2816−22)。あるいは、ドナー色素およびアクセプター色素は、基質上の異なる結合部位において標識され得る。例えば、オリゴヌクレオチドは、5’末端においてドナー色素で標識され得、3’末端においてアクセプター色素で標識され得る。ポリペプチドは、カルボキシル末端においてドナー色素で標識され得、内部システインまたはリジン側鎖においてアクセプター色素で標識され得る(Komoriya「Compositions for the detection of proteases in biological samples and methods of use thereof」、米国特許第5,605,809号)。本発明のエネルギー移動色素において、基質を標識する、少なくとも1つのドナー色素またはアクセプター色素は、スルホン化ジアリールローダミン色素化合物である。エネルギー移動色素を含有する他の色素は、任意の蛍光部分であり得、この蛍光部分は、スルホン化ジアリールローダミン色素化合物(フルオレセイン、ロドル(rhodol)、およびローダミンを含む)を用いてエネルギー移動プロセスを行う。他の色素としては、蛍光色素(例えば、シアニン、フタロシアニン、スクアライン(squaraine)、ボディピー(bodipy)、ベンゾフェノキサジン、フルオレセイン、ジアリールローダミン、またはローダミン)のクラスが挙げられる。
【0084】
エネルギー移動色素は、混合物中の複数の標識された基質を同時に検出(例えば、DNA配列決定)する際に使用するのに利点を有する。各色素が通常の波長において強力な吸収を有するように、単一のドナー色素が、エネルギー移動色素のセットにおいて使用され得る。次いで、エネルギー移動セットのアクセプター色素を変更することによって、アクセプター色素は、それらそれぞれの発光最大値によってスペクトルにより分解され得る。エネルギー移動色素はまた、非エネルギー移動色素よりも大きい有効なストークスシフトを提供する。このストロークシフトは、励起最大値(ドナー色素が最大に光を吸収する波長)と発光最大値(アクセプターが最大に光を発する波長)との間で異なる。
【0085】
一般的に、ドナー色素とアクセプター色素との間のリンカーは、以下の構造:
【0086】
【化34】
を有し、ここで、Zは、NH、SおよびOからなる群より選択され;R21は、ドナー色素に結合されたC1〜C12アルキルであり;R22は、C1〜C12アルキルジイル、少なくとも1つの不飽和結合を有する、5員環および6員環、ならびにカルボニル炭素に結合した縮合環構造からなる群より選択される置換基であり;そして、R23は、アクセプター色素にリンカーを結合している官能基を含む。R22は、5員環または6員環(例えば、シクロペンテン、シクロヘキセン、シクロペンタジエン、シクロヘキサジエン、フラン、チオフラン、ピロール、イソピロール、イソアゾール、ピラゾール、イソイミダゾール、ピラン、ピロン、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、オキサジン、インデン、ベンゾフラン、チオナフテン、インドールおよびナフタレン)であり得る。詳細には、このリンカーは、以下の構造:
【0087】
【化35】
を有し得、ここで、nは、2〜10の範囲である。
【0088】
一般的にまた、R23は、以下の構造:
【0089】
【化36】
を有し得、ここで、R24は、C1〜C12である。
【0090】
1つの実施形態において、ドナー色素とアクセプター色素との間のリンカーは、このリンカーにある程度の構造的な剛性を与える官能基(例えば、アルケン、ジエン、アルキン、少なくとも1つの不飽和結合を有する5員環および6員環、または縮合環構造)を含む(米国特許第5,821,356号;同第5,770,716号;同第5,948,648号;同第6,096,875号)。エネルギー移動色素のドナー色素およびアクセプター色素は、以下の例示的な構造:
【0091】
【化37】
を有するリンカーによって結合されており、ここで、Dはドナー色素であり、Aはアクセプター色素であり、nは1または2である。フェニル環は、スルホネート基、ホスホネート基、および他の荷電した基のような基で置換され得る。
【0092】
エネルギー移動色素のドナー色素とアクセプター色素との間のリンカーの結合部位は、ドナー色素およびアクセプター色素のうちの一方または両方が本発明の化合物である、任意の位置であり得る。例示的な結合部位としては、R2、R2’、R12およびR12’が挙げられる。
【0093】
エネルギー移動色素化合物は、リンカーによって基質に共有結合している。リンカーは、結合、C1〜C12アルキルジイルまたはC6〜C20アリールジイルであり得る。このリンカーは、アミド、カルバメート、尿素、チオ尿素、ホスフェート、ホスホネート、スルホネート、ホスホロチオエートなどを含む官能基を有し得る。例示的なリンカーとしては、1,2−エチルジイルおよび1,6−ヘキシジイルが挙げられる。エネルギー移動色素と基質との間のリンカーの結合部位は、ドナー色素およびアクセプター色素のうちの一方または両方が本発明のスルホン化ジアリールローダミン色素である、エネルギー移動色素上の任意の位置であり得る。基質がヌクレオシドまたはヌクレオチドである場合、エネルギー移動色素に対する1つの結合部位は、核酸塩基上にある。この核酸塩基がプリンである場合、リンカーは、8位で結合し得る。核酸塩基が7−デアザプリンである場合、リンカーは、7位または8位で結合し得る。核酸塩基がピリミジンである場合、リンカーは、5位で結合し得る。基質がオリゴヌクレオチドである場合、結合部位は、3’末端および5’末端を含む。他のオリゴヌクレオチド結合部位としては、ヌクレオチド間ホスフェートまたはホスフェートアナログ結合、あるいは、糖上の位置(2’または4’)が挙げられる。基質がポリペプチド(ペプチドまたはタンパク質)である場合、結合部位は、アミノ末端およびカルボキシル末端、ならびにリジン残基のアミノ基を含む。
【0094】
(V.5 標識方法)
本発明は、標識試薬を含み、ここで、スルホン化ジアリールローダミン化合物は、基質と反応するように反応性形態である(すなわち、連結部分を有する)。本発明はまた、本発明の化合物(式IIIabc)で標識された(すなわち、結合体化された)基質を含む。基質は、実質的に任意の分子であり得るか、または本発明の色素が結合体化され得る物質(例としては、ポリヌクレオチド、ヌクレオチド、ヌクレオシド、ポリペプチド、炭水化物、リガンド、実質的に鏡像異性的に純粋な化合物、粒子、表面、脂質、固体支持体、有機ポリマーおよび無機ポリマー、ならびにそれらの組み合わせおよび集合(例えば、染色体、核、生きた細胞(例えば、細菌または他の微生物、哺乳動物細胞、組織など))などが挙げられるがこれらに限定されない)であり得る。粒子としては、ナノ粒子、微粒子、ビーズ、またはリポソームが挙げられ得る。表面は、ガラスまたは他の非多孔性平面材料であり得る。本発明の化合物は、種々の手法(疎水性引力、イオン引力、および共有結合を含む)によって、必要に応じて、リンカーを介して基質に結合体化される。
【0095】
標識は、代表的には、当該分野で周知の結合体化方法(Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.pp.40−55,643−71)を使用して、連結部分、基質、および適切な溶媒を有するスルホン化ジアリールローダミンを混合することによって生じ、その後、標識された基質、結合体を、任意の非結合開始材料または望ましくない副産物から分離する。結合体は、後に使用するために、乾燥状態でまたは溶液中で保存され得る。
【0096】
スルホン化ジアリールローダミンは、置換基位置の1つで連結部分を含み得る。この連結部分は、代表的には、求電子性官能基であり、これは、基質上の求核性官能基と反応することによって共有結合を形成し得る。求核性官能基としては、例えば、アルコール、アルコキシド、アミン、ヒドロキシルアミン、およびチオールが挙げられ得る。あるいは、連結部分としては、基質上の求電子性基と反応する求核性官能基が挙げられ得る。連結基の例としては、アジド、一置換の第一級アミン、二置換の第二級アミン、チオール、ヒドロキシル、ハライド、エポキシド、N−ヒドロキシスクシンイミジルエステル、カルボキシル、イソチオシアネート、塩化スルホニル、スルホン酸エステル、シリルハライド、クロロトリアジニル、スクシンイミジルエステル、ペンタフルオロフェニルエステル、マレイミド、ハロアセチル、エポキシド、アルキルハライド、アリルハライド、アルデヒド、ケトン、アシルアジド、無水物、ヨードアセトアミドおよび活性化エステルが挙げられる。
【0097】
1つの連結部分は、スルホン化ジアリールローダミン化合物のカルボキシル基のN−ヒドロキシスクシンイミジルエステル(NHS)である(図14)。この化合物のNHSエステル形態は、標識試薬である。色素のNHSエステルは、予備形成され得、単離され得、精製され得、そして/または特徴付けされ得るか、あるいは、インサイチュで形成されて、基質の求核性基(例えば、オリゴヌクレオチド、ヌクレオシド、ヌクレオチド、ポリペプチドなど)と反応され得る(Brinkley,M.(1992)Bioconjugate Chem.3:2〜13)。代表的には、色素のカルボキシル形態は、以下の(1)〜(3)のいくつかの組み合わせと反応させることによって活性化され、色素のNHSエステル(例えば、図14の化合物82、化合物83、化合物84および図15の化合物85)が得られる:(1)カルボジイミド試薬(例えば、DCC(ジシクロヘキシルカルボジイミド)、DIPCDI(ジイソプロピルカルボジイミド))、またはウロニウム(uronium)試薬(例えば、TSTU(O−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート)、HBTU(O−ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート)、HATU(O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート));(2)活性化剤(例えば、1−ヒドロキシベンゾトリアゾール(HOBt));ならびに、(3)N−ヒドロキシスクシンイミド。
【0098】
いくつかの場合において、スルホン化ジアリールローダミン化合物および基質は、1つの工程において、化合物をインサイチュでの活性化し、そして基質と反応させて、スルホン化ジアリールローダミン−基質結合体を形成することによって結合され得る。他の活性化剤およびカップリング剤としては、以下が挙げられる:TBTU(2−(1H−ベンゾトリアゾール−1−イル)−1−1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)、TFFH(N,N’,N’’,N’’’−テトラメチルウロニウム2−フルオロヘキサフルオロホスフェート)、PyBOP(ベンゾトリアゾール−1−イル−オキシ−トリス−ピロリジニノ−ホスホニウムヘキサフルオロホスフェート)、EEDQ(2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロ−キノリン)、DCC、DIPCDI、MSNT(1−(メシチレン−2−スルホニル)−3−ニトロ−1H−1,2,4−トリアゾール)、およびアリールスルホニルハライド(例えば、塩化トリイソプロピルベンゼンスルホン酸)。
【0099】
別の反応性連結基は、スルホン化ジアリールローダミン化合物のホスホラミダイト形態である。ホスホラミダイト色素試薬は、本発明の色素で標識されるオリゴヌクレオチドの自動化合成のために、特に有用である。最も好都合なことに、ホスホラミダイト色素試薬は、固相合成の通常の過程の間に固体支持体に結合されたオリゴヌクレオチドに結合し得る。オリゴヌクレオチドは通常、ホスホラミダイト方法(Caruthers,M.およびBeaucage,S.「Phosphoramidite compounds and processes」、米国特許第4,415,732号;Caruthers,M.およびMatteucci,M.「Process for preparing polynucleotides」、米国特許第4,458,066号;Beaucage,S.およびIyer,R.(1992)「Advances in the synthesis of oligonucleotides by the phosphoramidite approach」、Tetrahedron 48:2223〜2311)によって、固体支持体上で合成される。
【0100】
ホスホラミダイトスルホン化ジアリールローダミン試薬は、ヌクレオシドまたは非ヌクレオシドであり得る。ホスホラミダイト試薬の非ヌクレオシド形態は、一般式IV:
【0101】
【化38】
を有し、ここで、DYEは、スルホン化ジアリールローダミンIIIabc(エネルギー移動色素を含む)の保護形態または非保護形態である。Lは、リンカーである。別個になっているR30およびR31は、C1〜C12アルキル、C4〜C10アリール、および10個までの炭素原子を含むシクロアルキルであるか、またはホスホラミダイト窒素原子と一緒になったR30およびR31は、飽和窒素複素環を形成し得る。R32は、オリゴヌクレオチドの望ましくない伸長を防止する、亜リン酸エステル保護基である。一般的に、R32は、オリゴヌクレオチド合成条件に対して安定であり、オリゴヌクレオチドまたは色素の完全性に悪影響を及ぼさない試薬を用いて、合成オリゴヌクレオチド生成物から除去され得る。R32は、以下であり得る:(i)メチル、(ii)2−シアノエチル;−CH2CH2CN、または(iii)2−(4−ニトロフェニル)エチル;−CH2CH2(p−NO2Ph)。ホスホラミダイト試薬の実施形態は、以下を含む:(i)R30およびR31は、各々イソプロピルである;(ii)一緒になったR30およびR31は、モルホリノである;(iii)Lは、C1〜C12アルキルである;(iv)R32は、2−シアノエチルである;そして(v)DYEは、リンカーによってR18またはR19に結合している。あるいは、リンカーLは、以下であり得る:
【0102】
【化39】
ここで、nは、1〜10の範囲である。ホスホラミダイト試薬IVの例は、以下の構造:
【0103】
【化40】
を有する。
【0104】
ホスホラミダイト色素試薬IVは、本発明のスルホン化ジアリールローダミン化合物での基質の標識に影響を与える。基質がオリゴヌクレオチドである場合、色素は、3’から5’方向への代表的な合成の結果として、オリゴヌクレオチドの5’末端で結合しているか、または5’から3’方向への合成方法が行われる場合、オリゴヌクレオチドの3’末端で結合している(Wagner(1997)Nucleosides & Nucleotides 16:1657−60)。試薬IVは、例えば、3’末端を介して固体支持体に結合しているポリヌクレオチドに連結され得る。ヌクレオシドおよび非ヌクレオシドの他のホスホラミダイト色素試薬により、オリゴヌクレオチドの他の部位(例えば、3’末端、核酸塩基、ヌクレオチド間結合、糖)での標識が可能になる。核酸塩基、ヌクレオチド間結合、および糖部位での標識により、蛍光色素での内部標識および複数標識が可能になる。
【0105】
本発明のスルホン化ジアリールローダミン化合物は、求核官能基を三価の亜リン酸化試薬で亜リン酸化(phosphitylation)する、任意の公知の方法によって、非ヌクレオシドのホスホラミダイト標識試薬に転換され得る。例えば、化合物がカルボキシル基(例えば、110〜120)を含む場合、カルボキシルは、例えば、NHSに対して活性化され得、そして、6−アミノ−1−ヘキサノールでアミド化され得る。得られたヒドロキルは、ビス(ジイソプロピルアミノ)シアノエチルホスファイトまたはクロロジイソプロピルアミノシアノエチルホスファイトで亜リン酸化されて、ホスホラミダイト色素標識試薬IVが得られ得る(Theisen(1992)「Fluorescent dye phosphoramidite labelling of oligonucleotides」、Nucleic Acid Symposium Series No.27,Oxford University Press,Oxford,99〜100頁)。あるいは、化合物のカルボキシル基は、ヒドロキシルに還元され、亜リン酸化され得る。
【0106】
ホスホラミダイト試薬IVは、穏やかな酸活性化の下でヒドロキシル基(例えば、固体支持体に結合したオリゴヌクレオチドの5’末端のOH)と反応して、ヌクレオチド間亜リン酸基(これは、次いで、ヌクレオチド間ホスフェート基に酸化される)を形成する。いくつかの例において、スルホン化ジアリールローダミン化合物は、官能基を含有し得、この官能基は、ホスホラミダイト試薬を合成する間または分子(例えば、オリゴヌクレオチド)を標識するために続いて使用する間のいずれかで、保護を必要とする。使用される保護基は、官能基の性質に依存し、そして当業者に明らかである(Greene,T.およびWuts,P. Protective Groups in Organic Synthesis,第2版、John Wiley & Sons,New York,1991)。一般的に、使用される保護基は、5’−ヒドロキシル保護基(例えば、ジメトキシトリチル)を除去するためにオリゴヌクレオチドの合成において通常使用される酸性条件下(例えば、トリクロロ酢酸、ジクロロ酢酸)で安定でなければならなず、かつ合成オリゴヌクレオチドを固体支持体から脱保護および/または切断するために使用される塩基性条件下(例えば、水酸化アンモニウム、水性メチルアミン)で不安定でなければならない。
【0107】
ポリペプチド、抗体、ならびにアミノ酸およびアミノ酸アナログから構成される他の生体高分子は、本発明のスルホン化ジアリールローダミン化合物との結合体化によって共有結合的に標識され得る。代表的に、この化合物は、求電子形態(例えば、NHS反応性連結基)であり、この化合物は、ペプチドの求核基(例えば、アミノ末端、またはアミノ酸(例えば、リジン)のアミノ側鎖)と反応する。あるいは、この色素は、求核形態(例えば、アミノ反応性連結基またはチオール反応性連結基)であり得、この色素は、ペプチドの求電子基(例えば、カルボキシル末端のNHSまたはアミノ酸のカルボキシル側鎖)と反応し得る。標識されたポリペプチドは、好ましくは、細胞表面および細胞内成分との相互作用の際にそれらの特異的な結合特性および認識特性を維持する。色素として作用するスルホン化ジアリールローダミン化合物により、結合事象または認識事象を局在化、可視化および定量化するための検出エレメントが提供される。ポリペプチドはまた、蛍光共鳴エネルギー移動(FRET)を一緒に行う、2つの部分(蛍光レポーターおよび消光剤)で標識され得る。この蛍光レポーターは、インタクトなポリペプチドの消光剤部分によって部分的にかまたは有意にクエンチされ得る。ペプチダーゼまたはプロテアーゼによってポリペプチドを切断する際に、蛍光の検出可能な増加が測定され得る(Knight,C.(1995)「Fluorimetric Assays of Proteolytic Enzymes」、Methods in Enzymology,Academic Press,248:18〜34)。
【0108】
(V.6 標識されたヌクレオチド)
1つのクラスの標識された基質は、本発明のスルホン化ジアリールローダミン化合物で標識される、ヌクレオシドおよびヌクレオチドの結合体を含む。このような標識されたヌクレオシドおよびヌクレオチドは、酵素学的合成によって形成されるポリヌクレオチド(例えば、PCR増幅、サンガー型ポリヌクレオチド配列決定、およびニック翻訳反応の状況において使用される、標識されたヌクレオチドの5’トリホスフェート)を標識するために、特に有用である。
【0109】
ヌクレオシドおよびヌクレオチドは、糖部分または核酸塩基部分上の部位で標識され得る。核酸塩基標識部位としては、プリン核酸塩基の8−C、7−デアザプリン核酸塩基のC−7またはC−8、およびピリミジン核酸塩基の5位が挙げられる。ヌクレオチシドまたはヌクレオチドと色素との間で、リンカーは、任意の位置において、スルホン化ジアリールローダミン化合物に結合し得る。
【0110】
標識されたヌクレオシドまたはヌクレオチドは、酵素学的に組み込み可能かつ酵素学的に伸長可能であり得る。本発明の化合物で標識されるヌクレオシドまたはヌクレオチドは、以下の式V:
【0111】
【化41】
を有し得、ここで、DYEは、化合物IIIabcの保護形態または非保護形態(エネルギー移動色素を含む)である。Bは、任意の核酸塩基(例えば、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および8−デアザグアノシン)であり得る。R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはリン酸エステルアナログである。R26およびR27は、単独である場合、各々独立して、H、HO、Fおよびホスホラミダイトである。R26またはR27がホスホラミダイトである場合、R25は、自動化合成条件下で後のモノマーカップリングを可能にする、酸で切断可能なヒドロキシル保護基(例えば、ジメトキシトリチル)である(米国特許第4,415,732号および同第4,458,066号;Beaucage,S.and Iyer,R.(1992)「Advances in the synthesis of oligonucleotides by the phosphoramidite approach」,Tetrahedron 48:2223〜2311)。
【0112】
標識されたヌクレオシまたはヌクレオチドがターミネーターである場合、R26およびR27は、ポリメラーゼ媒介テンプレート指向性重合を遮断するように選択される。ターミネーターヌクレオチドにおいて、R26およびR27は、単独である場合、各々独立して、H、F、およびポリメラーゼ媒介テンプレート指向性重合を遮断する部分であるか、または一緒になる場合、2’−3’−ジデヒドロリボースを形成する。式Vにおいて、R26およびR27の両方がヒドロキシルである場合、得られる化合物は、標識されたリボヌクレオシドまたはリボヌクレオチド(NTP)である。R27が水素であり、かつR26がヒドロキシルである場合、得られる化合物は、標識された2’−デオキシリボヌクレオシドおよび2’−デオキシリボヌクレオチド(dNTP)である。R26およびR27が各々水素である場合、得られる化合物は、2’,3’−ジデオキシリボヌクレオシドおよび2’,3’−ジデオキシリボヌクレオチド(ddNTP)である。標識されたddNTPは、蛍光検出を使用するサンガー型DNA配列決定方法におけるターミネーターとしての特定の用途が見出される。標識された2’−デオキシリボヌクレオシド−5’−トリホスフェート(dNTP)は、DNAポリメラーゼ伸長産物を標識するための試薬としての特定の用途(例えば、ポリメラーゼ連鎖反応またはニック翻訳において)が見出される。標識されたリボヌクレオシド−5’−トリホスフェート(NTP)は、RNAポリメラーゼ伸長産物を標識するための試薬としての特定の用途が見出される。
【0113】
【化42】
アルキニルアミノ連結化合物V(ここで、Lは、アルキンジイル(alkyndiyl)基を含む)は、スルホン化ジアリールローダミン化合物をヌクレオシド、ヌクレオチドおよびそれらのアナログに結合体化するのに有用である。これらの合成は、EP 87305844.0およびHobbs,(1989)J.Org.Chem.54:3420に教示されている。対応するヌクレオシドのモノホスフェート、ジホスフェートおよびトリホスフェートは、標準技術(例えば、米国特許第5,821,356号;同第5,770,716号;同第5,948,648号;同第6,096,875号に記載される方法)によって得られる。化合物Vを改変したプロパルギルエトキシアミド(propargylethoxyamido)リンカーLを用いて合成するための方法もまた、これらの特許において見出され得る。構造式Vに従う化合物の合成に使用するのに適切な、さらなる合成手順が、例えば、以下に記載される:Gibson(1987)Nucl.Acids Res.15:6455〜6467;Gebeyehu(1987)Nucl.Acids Res.15:4513〜4535;Haralambidis(1987)Nucl.Acids Res.15:4856〜4876;Nelson(1986)Nucleosides and Nucleotides.5(3):233〜241;Bergstrom(1989)J.Am.Chem.Soc.111:374〜375;米国特許第4,855,225号;同第5,231,191号および同第5,449,767号(これらは、本明細書中で参考として援用される)。これらの方法のいずれかは、全範囲の標識されたヌクレオシド、ヌクレオチド、および本明細書中に記載されるアナログを合成するために必要な方法として、日常的に適用または変更され得る。
【0114】
アルキニルリンカーLの1つの実施形態は、以下:
【0115】
【化43】
であり得、ここで、nは、0、1または2である。
【0116】
エネルギー移動色素対は、核酸塩基アミノ基を介して、以下:(i)エネルギー移動色素対の活性化エステルに連結するか、または(ii)1つの色素(例えば、R11保護アミノメチル、R18カルボキシルフルオレセイン)にカップリングし、次いで、非保護R11アミノメチルを対の第二色素にカップリングして、段階的に連結することによって、ヌクレオチドの5’トリホスフェートに結合体化され得る。
【0117】
異なる輻合性スキームを用いた、エネルギー移動ヌクレオチドおよびポリヌクレオチドに対する代替的な合成経路が、用いられ得る。基質、色素およびリンカーサブユニット、またはシントンが、任意の順序でカップリングするために構築され得る。例えば、ドナー色素およびアクセプター色素のエネルギー移動対は、リンカーによって共有結合され得、次いで、ヌクレオチドまたはポリヌクレオチドにカップリングされ得る。本発明の色素でのヌクレオチドの標識を生じる、多数の異なる合成経路が、用いられ得る。反応性官能基(例えば、カルボン酸基、アミノ基、ヒドロキシル基)は、広範な有機合成分野の方法論を使用する際に、保護を必要とし得る。
【0118】
(V.7 標識されたオリゴヌクレオチド)
オリゴヌクレオチドは、市販のホスホラミダイトヌクレオシド、支持体(例えば、シリカ)、制御された有孔ガラス(米国特許第4,458,066号)およびポリスチレン(米国特許第5,047,524号および同第5,262,530号)および自動化合成機(Models 392,394,3948 DNA/RNA Synthesizers,Applied Biosystems)を使用するホスホラミダイト方法(米国特許第4,415,732号;同第4,973,679号;同第4,458,066号;Beaucage,S.およびIyer,R.(1992)Tetrahedron 48:2223〜2311)によって、通常、固体支持体上に合成される。
【0119】
別のクラスの標識された基質としては、オリゴヌクレオチドと本発明の化合物との結合体が挙げられる。このような結合体は、DNA配列決定プライマー、PCRプライマー、オリゴヌクレオチドハイブリダイゼーションプローブ、オリゴヌクレオチドライゲーションプローブ、二重標識5’エキソヌクレアーゼ(TaqManTM)プローブ、電気泳動のためのサイズ標準(すなわち、「レーン標準」または「レーンマーカ」)などとしての有用性が見出され得る(Fung、米国特許第4,757,141号;Andrus「Chemical methods for 5’non−isotopic labelling of PCR probes and primers」(1995)、PCR 2:A Practical Approach,Oxford University Press,Oxford,39〜54項;Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.40〜55項,643−71;Mullah(1998)「Efficient synthesis of double dye−labelled oligodeoxyribonucleotide probes and their application in a real time PCR assay」Nucl.Acids Res.26:1026〜1031)。標識されたオリゴヌクレオチドは、以下の式VI:
【0120】
【化44】
を有し得、ここで、このオリゴヌクレオチドは、2〜1000ヌクレオチドを含む。DYEは、化合物Iまたは化合物IIの保護形態または非保護形態(エネルギー移動色素を含む)である。Bは、任意の核酸塩基(例えば、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および8−デアザグアニシン)である。Lは、リンカーである。R27は、H、OH、ハライド、アジド、アミン、C1〜C6アミノアルキル、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2である。R22は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログである。R29は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログである。この実施形態において、核酸塩基で標識されたオリゴヌクレオチドVIは、核酸塩基によって結合された本発明の複数の色素を有し得る。この核酸塩基で標識されたオリゴヌクレオチドVIは、以下によって形成され得る:(i)DNAポリメラーゼまたはリガーゼによる、酵素学的に組み込み可能なヌクレオチド試薬Vの酵素学的組み込み(ここで、R25は、トリホスフェートである)、および(ii)自動化合成によるヌクレオシドホスホラミダイト試薬のカップリング。これによって、核酸塩基で標識されたオリゴヌクレオチドVIは、1つより多くの組み込み可能なヌクレオチドVの組み込みによって複数標識され得、1つずつ5’標識されたオリゴヌクレオチドを導く、以下の式VII:
【0121】
【化45】
に従う色素標識試薬(例えば、IV)で標識され、ここで、Xは、O、NHまたはSであり;R27は、H、OH、ハライド、アジド、アミン、C1〜C6アミノアルキル、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3またはOCH2CH=CH2であり;R28は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログであり;そして、Lは、C1〜C12アルキル、アリール、または100までのエチレンオキシユニットのポリエチレンオキシである。
【0122】
式VIまたはVII中のリンカーLは、任意の部位にて、本発明のスルホン化ジアリールローダミン化合物IIIabc(DYE)に結合され得る。
【0123】
合成オリゴヌクレオチドを標識するための第一の方法において、求核官能基(例えば、第一級脂肪族アミン)は、オリゴヌクレオチド上の標識結合部位(例えば、5’末端)に導入される。自動化された固体支持体合成が終了した後、オリゴヌクレオチドが、この支持体から切断され、そして全ての保護基が除去される。求核オリゴヌクレオチドは、求電子部分(例えば、イソチオシアネートまたは活性化エステル(例えば、N−ヒドロキシスクシンイミド(NHS)))を含有する過剰な標識試薬と、均一溶液条件下で反応される(Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.pp.40−55,643−71;Andrus,A.「Chemical methods for 5’non−isotopic labelling of PCR probes and primers」(1995)、PCR 2:A Practical Approach,Oxford University Press,Oxford,39〜54頁)。標識されたオリゴヌクレオチドVIIは、色素の反応性連結基形態(例えば、NHS)を、5’−アミノアルキルオリゴヌクレオチドと反応させることによって形成され得る(米国特許第4,757,141号)。
【0124】
第二の方法において、標識は、自動化合成の間または自動化合成前に、例えば、支持試薬として(Mullah,「Solid support reagents for the direct synthesis of 3’−labelled polynucleotides」、米国特許第5,736,626号;Nelson,「Multifunctional controlled pore glass reagent for solid phase oligonucleotide synthesis」、米国特許第5,141,813号)、またはホスホラミダイト試薬IVとして、オリゴヌクレオチドに直接組み込まれる。特定の蛍光色素および他の標識は、5’標識のためのホスホラミダイト試薬として機能化される(Theisen(1992)Nucleic Acid Symposium Series No.27,Oxford University Press,Oxford,99〜100頁)。
【0125】
一般的に、標識されたオリゴヌクレオチドが酵素学的合成によって作製される場合、以下の手順が使用され得る。標的DNAは、変性され、そしてオリゴヌクレオチドプライマーは、テンプレートDNAにアニーリングされる。初回刺激された標的(例えば、dGTP、dATP、dCTPおよびdTTPまたはdUTPを含む混合物)の連続的なテンプレート指向性酵素学的伸長を支持し得る酵素学的に組み込み可能なヌクレオチドまたはヌクレオチドアナログの混合物が、この初回刺激された標的に添加される。ヌクレオチドの少なくとも1つの画分は、標識されたターミネーターV(例えば、ddNTPまたは3’F−dNTP)であり、これは、スルホン化ジアリールローダミン色素IIIabcで標識される。次に、ポリメラーゼ酵素は、このポリメラーゼ酵素が活性である条件下でこの混合物に添加される。標識されたオリゴヌクレオチドは、ポリメラーゼ媒介性鎖合成の間に、標識されたヌクレオチドまたはターミネーターの組み込みによって形成される。代替的な酵素学的合成方法において、2つのプライマー(標的の(+)鎖に相補的なプライマーおよび標的の(−)鎖に相補的な別のプライマー)
が、代わりに使用され、ポリメラーゼは、熱安定性ポリメラーゼであり、反応温度は、変性温度と伸長温度との間を循環し、それによって、標的配列に対する標識された相補体がPCRによって指数関数的に合成される(Innis(1990)PCR Protocols,編、Academic Press)。
【0126】
本発明のスルホン化ジアリールローダミン化合物で標識されたポリヌクレオチドは、求電子遊走速度に影響を与える部分(すなわち、移動度変更標識)でさらに標識され得る。移動度変更標識は、ポリエチレンオキシユニット、−(CH2CH2O)n−を含み、ここで、nは、1〜100であり得(米国特許第5,624,800号)、好ましくは、nは、2〜20である。ポリエチレンオキシユニットは、ホスフェート基で引き離され得る。別個でありかつ公知のサイズであるポリエチレンオキシのさらなる標識を伴う、特異的に標識されたスルホン化ジアリールローダミン標識ポリヌクレオチドによって、ポリヌクレオチド中のヌクレオチドの数に実質的に非依存性の電気泳動による分離が可能になる。すなわち、同じ長さのポリヌクレオチドは、スペクトルにより分解可能な色素標識および移動度変更標識の存在によって識別され得る。色素標識と移動度変更標識との両方を有するポリヌクレオチドは、単一標識されたポリヌクレオチドまたはヌクレオチド構成成分のライゲーションまたはポリメラーゼ伸長によって酵素学的に形成され得る。
【0127】
(V.8 スルホン化ジアリールローダミン色素を使用する方法)
スペクトルにより重複した複数の分析物を同時検出するのに必要な方法は、標識としてのスルホン化ジアリールローダミン色素から利益を獲得し得る。本発明のスルホン化ジアリールローダミン化合物は、以下のような蛍光検出を使用する任意の方法に十分適している:ポリメラーゼ連鎖反応(PCR)増幅、DNA配列決定、遺伝子発現のアンチセンス転写性制御および翻訳制御、遺伝分析、およびDNAプローブベースの診断試験(Kricka,L.(1992)Nonisotopic DNA Probe Techniques,Academic Press,San Diego,3〜28頁)。蛍光色素により標識されたオリゴヌクレオチドの蛍光検出は、以下のような核酸配列検出アッセイに基づく:5’エキソヌクレアーゼアッセイ(Livak,米国特許第5,723,591号)、FRETハイブリダイゼーション(Tyagi,S.およびKramer,F.(1996)「Molecular Beacons:Probes that fluoresce upon hybridization」、Nature Biotechnology,14:303−08)、遺伝連鎖マッピング(Dib(1996)「A comprehensive genetic map of the human genome based on 5,264 microsatellites」Nature 380:152−54)、およびオリゴヌクレオチドライゲーションアッセイ(Grossman(1994)「High−density multiplex detection of nucleic acid sequences: oligonucleotide ligation assay and sequence−coded separation」Nucl.Acids Res.22:4527−34)。
【0128】
本発明は、特に、差次的に標識されたポリヌクレオチドのクラスを検出するのに非常に適しており、これらのポリヌクレオチドは、生化学的分離手順(例えば、電気泳動(RickwoodおよびHames、編、Gel Electrophoresis of Nucleic Acids:A Practical Approach,IRL Press Limited,London,1981))に供される。電気泳動マトリクスは、シービング(sieving)ポリマー(例えば、架橋もしくは非架橋のポリアクリルアミド、または約2〜20重量%の間の濃度(重量/体積)を有する他のアミド含有ポリマー)であり得る(Madabhushi,米国特許第5,552,028号;同第5,567,292号;同第5,916,426号)。この電気泳動マトリクスは、スラブゲルまたはキャピラリーフォーマットで構成され得る(Rosenblum,(1997)Nucleic Acids Res.25:3925−29;Mathies,米国特許第5,274,240号)。
【0129】
電気泳動分離の後、色素−ポリヌクレオチド結合体は、例えば、高輝度照射の水銀放電灯、レーザなどによって、色素標識ポリヌクレオチドからの蛍光発光を測定することによって検出され得る。この照射手段は、約600nmより大きい波長の照射ビームを有するレーザであり得る。また、この色素−ポリヌクレオチドは、He−Neガスレーザまたは固体状態のダイオードレーザによって生じるレーザ光によって照射され得る。次いで、蛍光が、光感受性検出器(例えば、光電子増倍管、荷電結合デバイス(charged coupled device)など)によって検出される。例示的な電気泳動検出システムは、他の箇所(例えば、米国特許第5,543,026号;同第5,274,240号;同第4,879,012号;同第5,091,652号および同第4,811,218号)に記載される。
【0130】
(V.8A プライマー伸長)
「フラグメント分析」方法または「遺伝子分析」方法として本明細書中で参照される方法の1つのカテゴリーにおいて、蛍光色素(スルホン化ジアリールローダミン化合物を含む)で標識されたポリヌクレオチドフラグメントは、例えば、ライゲーションまたはポリメラーゼ指向性プライマー伸長によって、標識されたプライマーまたはヌクレオチドを使用するテンプレート指向性酵素学的合成によって生じる。プライマーオリゴヌクレオチドは、標的配列との相補的な塩基対形成によってハイブリダイズし、プライマー−標的ハイブリッドを形成する。プライマー伸長産物は、ハイブリッド中のプライマーの3’末端におけるヌクレオチドの酵素学的組み込みによって生じる。プライマーまたはヌクレオチドが本発明のスルホン化ジアリールローダミン色素で標識される場合、標識されたプライマー伸長産物が生じる。これらのポリヌクレオチドフラグメントは、サイズ依存性分離プロセス(例えば、電気泳動またはクロマトグラフィー)に供され得、分離されたフラグメントは、例えば、レーザ誘導化蛍光(Hunkapiller,米国特許第4,811,218号)によって分離後に検出される。複数のクラスのポリヌクレオチドが、同時に分離され得、そして種々のクラスが、スペクトルにより分解可能な標識(本発明の色素を含む)によって識別される。電気泳動において、これらのクラスは、電気泳動遊走速度に基づいて分離する。
【0131】
(V.8B DNA配列決定)
スルホン化ジアリールローダミン化合物は、複数色の蛍光検出が可能である、自動化4色DNA配列決定システムにおける使用に十分適している。このシステムは、約630nmより大きい波長を有する励起光源(例えば、ヘリウム−ネオンガスレーザまたは固体状態のダイオードレーザ)を使用し得る。
【0132】
DNA配列決定(すなわち、ジデオキシDNA配列決定、またはサンガー型配列決定)の連鎖停止方法、およびフラグメント分析が、使用され得る(Sanger(1977)「DNA sequencing with chain−terminating inhibitors」Proc.Natl.Acad.Sci.USA 74:5463〜5467)。例示的な連鎖停止ヌクレオチドアナログは、2’,3’−ジデオキシヌクレオチド5’−トリホスフェート(ddNTP)(これは、3’〜5’DNA鎖伸長に必要な3’−OH基を欠失している)を含む。プライマーまたはddNTPは、本発明のスルホン化ジアリールローダミン色素で標識され得、そして高分解能電気泳動によってフラグメントを分離した後に、蛍光によって検出され得る。色素は、プライマーの5’末端上の官能基(例えば、アミノ(米国特許第4,757,141号))、オリゴヌクレオチドプライマーの核酸塩基上の官能基;またはジデオキシヌクレオチドの核酸塩基上の官能基(米国特許第5,770,716号;同第5,821,356号;同第5,151,507号)に連結され得る。
【0133】
ターミネーターの各々は、異なる蛍光色素を有し、かつこの実験のターミネーターは、集合的に、特異的に分離可能な蛍光標識のセット(1種以上の本発明のスルホン化ジアリールローダミン色素を含む)を有する。例示的なフラグメント分析方法において、色素で標識されたフラグメントは、電気泳動条件下で相対サイズ(すなわち、配列の長さ)によって同定される。フラグメントサイズと配列との間の対応は、4つの可能な終結ヌクレオチド(「ターミネーター」)およびスペクトルにより分解可能な色素のセットのメンバーの組み込みによって確立される(米国特許第5,366,860号)。スペクトルにより分解可能な色素のセットとしては、少なくとも1つのスルホン化ジアリールローダミン化合物が挙げられる。十分に分解された発光を伴う4つのスルホン化ジアリールローダミン色素の1つのセットは、110、114、115および119からなる(図22a、22b、22c)。別のセットは、110、114、116および119からなる。さらに別のセットは、110、114、119および120からなる。色素のセットは、色素110〜120のいずれかの組み合わせ、または一般的に、任意のスルホン化ジアリールローダミン色素を含み得る。1つのセットは、1種以上のスルホン化ジアリールローダミン色素および他の構造クラス由来の1種以上の色素(例えば、フルオレセイン、非スルホン化ジアリールローダミン、シアニン、スクアラニンなど)を含み得る。
【0134】
標識されたヌクレオチドVは、4つの核酸塩基(B=A、G、C、T(またはU))およびそれらのアナログを含む4つのターミネーターのセットとして、配列決定する際に使用され得る。B、リンカーLおよびDYEの組み合わせは、ポリヌクレオチドフラグメントの電気泳動移動度、スペクトル分解能、およびプライマー伸長の間のポリメラーゼの組み込み速度に影響を及ぼす。
【0135】
(V.8C ライゲーション)
リガーゼ酵素による核酸プローブの共有結合は、分子生物学者に利用可能な最も有用なツールの1つである。2つのプローブがテンプレート核酸にアニーリングされる場合(ここで、これら2つのプローブは隣接しており、かつ介在性ギャップがない)、リガーゼ酵素によって、一方のプローブの5’末端ともう一方のプローブの3’末端との間に、ホスホジエステル結合が形成され得る(Whiteley、米国特許第4,883,750号;Landegren,(1988)「A ligase mediated gene detection technique」、Science 241:1077−80;Nickerson,「Automated DNA diagnostics using an ELISA−based oligonucleotide assay」(1990)Proc.Natl.Acad.Sci USA 87:8923−27)。オリゴヌクレオチドライゲーションアッセイは、標的DNAサンプル中の特定の配列の存在を検出する。一方または両方のプローブが色素で標識される場合、ライゲーション産物が蛍光によって検出され得る。一方または両方のプローブは、スルホン化ジアリールローダミン色素で標識され得る。ライゲーション産物は、電気泳動、クロマトグラフィー、または他のサイズに基づく分離方法もしくは電荷に基づく分離方法によって検出され得る。
【0136】
(V.8D 増幅)
本発明のスルホン化ジアリールローダミン化合物は、ポリメラーゼ連鎖反応(PCR)ならびに他の核酸増幅方法および選択方法のための、5’標識されたオリゴヌクレオチドプライマー上の標識としての用途が見出されている。PCR増幅は、種々の可変数のタンデム反復(VNTR)、短タンデム反復(STR)、および二重鎖DNA(特定の配列の隣接した複数のコピーを含む)の反復領域を増幅するマイクロサテライト法(microsatellite method)によって遺伝子型を決定する(genotyping)のための、標識されたオリゴヌクレオチドの使用を含み、反復単位の数は、可変である。このようなPCR遺伝子型決定方法において、PCRプライマーは、本発明のスルホン化ジアリールローダミンで標識され得る。
【0137】
1つの実施形態において、スルホン化ジアリールローダミン化合物は、定量方法およびPCRの間の増幅産物の実時間測定または終点測定を提供する試薬において使用され得る(米国特許第5,210,015号;同第5,538,848号)。蛍光色素−消光剤プローブを使用するエキソヌクレアーゼアッセイ(Taqman(登録商標))(米国特許第5,723,591号;Mullah(1998)「Efficient synthesis of double dye−labelled oligodeoxyribonucleotide probes and their application in a real time PCR assay、Nucl.Acids Res.26:1026〜1031)は、閉管システムにおいてポリメラーゼ連鎖反応(PCR)産物の直接検出を与え、PCRを行うのに必要とされるサンプル以上のサンプル処理を伴わない。Taqman(登録商標)アッセイにおいて、プライマー伸長を行いポリヌクレオチドを増幅するポリメラーゼはまた、5’〜3’エキソヌクレアーゼ活性によって標的配列にアニーリングされたプローブを置換および切断する。Taqman(登録商標)型アッセイにおいて、プローブは、自己消光し、蛍光色素および消光剤部分で標識され、これらのいずれかは、本発明の色素であり得る。スペクトルの重複によって、プローブがインタクトである場合に、効率的なエネルギー移動(FRET)が可能となる(Clegg(1992)「Fluorescence resonance energy transfer and nucleic acids」、Meth.Enzymol.211:353〜388)。標的配列にハイブリダイズされる場合、プローブはPCRの間に切断されて、標的プローブハイブリッドの存在量に比例した蛍光シグナルが放射される米国特許第5,538,848号;同第5,723,591号)。
【0138】
増幅の進行は、連続的にモニタリングされ得る(すなわち、実時間検出)。スペクトルにより分解可能な、本発明のスルホン化ジアリールローダミン色素は、標的をPCR増幅した後の遺伝子型決定実験において有用である。詳細には、各々異なる色素を用いて5’末端で標識されたプライマーオリゴヌクレオチドのセットは、複数の遺伝子座を増幅し、そして一塩基多型(SNP)および対立遺伝子を識別し得る。色素で標識された増幅産物の、サイズ基準を用いた電気泳動分離は、プライマー配列のセットに依存した特定の遺伝子型を示す、プロファイル、すなわち特徴的データセットを確立する。
【0139】
(V.8E ハイブリダイゼーションアッセイ)
標的核酸にハイブリダイズする特定の蛍光色素−消光剤プローブは、ハイブリダイゼーションアッセイにおいて有用である。プローブが標的にハイブリダイズされない場合、このプローブは、蛍光色素と蛍光消光を生じる消光剤部分との間を空間的に近位にするコンホメーションを維持し得る。標的へのハイブリダイゼーションの際に、この部分が物理的に分離され、消光が中断または減少され、そして蛍光が増加する。蛍光が検出可能であるかまたは定量化される場合、サンプル中の標的配列の存在が推定される。本発明のスルホン化ジアリールローダミン色素はまた、蛍光色素または消光剤部分として使用され得る。「ヘアピン」領域(いわゆる、「分子ビーコン(Molecular beacons)」(Tyagi and Kramer))を形成する自己相補的配列を有する蛍光色素−消光剤プローブは、それらの相補的な標的配列へのハイブリダイゼーション(例えば、生存細胞中のmRNAのインサイチュでの定量化)の際に蛍光変化を起こす。異なる蛍光色素で標識されたハイブリダイゼーションプローブ(本発明のスルホン化ジアリールローダミン色素を含む)によって、多重化した均一なハイブリダイゼーションアッセイが密閉された反応管中で行われるのが可能になる。
【0140】
ハイブリダイゼーションアッセイはまた、予め決定された位置において固定化されたプローブの固相アレイ上で実施され得、これらの位置は、これらの特異的な試薬の送達またはインタロゲーション/検出によってアドレス可能であり得る。例えば、スルホン化ジアリールローダミン色素で標識された標的ポリヌクレオチドを含有するサンプルは、固定されたプローブのアレイに送達され得る。ポリヌクレオチドが、塩基対によって固定されたプローブにハイブリダイズする場合、その位置での標的−プローブ複合体からの蛍光が検出され得る。蛍光検出パターンおよび/または強度から、ポリヌクレオチドの配列が推定され得、そして、サンプル中の特定のポリヌクレオチド配列の存在が決定され得る。
【0141】
(V.9 キット)
本発明はキットを含み、このキットは、本発明のスルホン化ジアリールローダミン化合物および/またはそれらの標識された結合体を備える。1つの実施形態において、このキットは、連結部分を有するスルホン化ジアリールローダミン化合物を別の分子(例えば、基質)に結合体化するために有用である。このようなキットは、一般に、色素を別の分子または基質に結合体化するのに適切な、本発明のスルホン化ジアリールローダミン(任意の連結部分および試薬、酵素、緩衝液、溶媒などを含む)を備える。スルホン化ジアリールローダミンは、エネルギー移動色素のアクセプターまたはドナーであり得る。
【0142】
1つの実施形態において、このキットは、酵素学的に合成されたオリゴヌクレオチドおよびポリヌクレオチドを、本発明のスルホン化ジアリールローダミンで標識するために有用である。このようなキットは、一般に、本発明に従う、酵素学的に組み込み可能な、標識されたクレオチドまたはヌクレオチドアナログ、連続したプライマー伸長およびポリメラーゼ酵素を支持し得る、酵素学的に組み込み可能なヌクレオチドまたはヌクレオチドアナログの混合物を備える。酵素学的に組み込み可能な、標識されたヌクレオチドまたはヌクレオチドアナログは、構造Vに従う化合物(例えば、標識されたターミネーター)である。ポリメラーゼは、熱安定性であり、これらは、例えば、AMPLITAQ(登録商標)DNAポリメラーゼFS(Applied Biosystems,Foster City,CA)である。
【0143】
あるいは、このキットは、1つ以上のオリゴヌクレオチドプライマーを備え得る。このプライマーは、スルホン化ジアリールローダミンおよびスルホン化ジアリールローダミンを含むエネルギー移動色素で標識され得る。
【0144】
(VI. 実施例)
本発明は、以下の実施例を考慮することによってさらに明瞭になり、これらの実施例は、本発明の単なる例示であり、いかなる様式においても本発明の範囲を制限しないことが意図される。
【0145】
(実施例1)
(1−ジエチルアミノ−3−ヒドロキシナフタレン4の合成(図1))
K.H.BellおよびL.F.McCaffery(1993),Aust.J.Chem.46:731の方法によって、1,3−ジヒドロキシナフタレンから合成した3−メトキシ−1−ヒドロキシナフタレン1(1gm)を、無水CH2Cl2(30ml)中に懸濁した。乾燥トリエチルアミン(1.2当量)を加え、この反応物を−5℃まで冷却した。CH2Cl2(15ml)中に懸濁したトリフルオロメタンスルホン酸無水物(1.1当量)を、激しく攪拌しながら2時間にわたって滴下した。この反応物を室温にし、5% HClおよびCH2Cl2を使用する水性ワークアップ(aqueous work up)に供した。得られた粗製3−メトキシナフタレン−1−トリフレート2を、移動相としてEtOAc/ヘキサン(1:10)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0146】
精製した3−メトキシナフタレン−1−トリフレート2を、J.P.WolfeおよびS.L.Buchwald(1996)Jour.Org.Chem.61:1133のパラジウムで触媒したトリフレート/アミンカップリング手順を使用して、1−ジエチルアミノ−3−メトキシナフタレン3に転換した。この3−メトキシ−ナフタレン−1−トリフレート2(1g)を、100mlの乾燥トルエン(0.015当量の(S)−(−)−2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフトル(BINAP)、0.005当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、および3当量の無水ジエチルアミンを含む)中に懸濁した。この反応物を、アルゴンでパージし、そして、3.3当量の固体ナトリウムt−ブトキシドを、攪拌しながら加えた。次いで、この反応物を加熱し、そして、油浴中で80℃にて16時間攪拌した。この反応物を室温にし、5%HClおよびCH2Cl2を使用する水性ワークアップに供して、粗製1−ジエチルアミノ−3−メトキシナフタレン3を得、これを、移動相としてEtOAc:ヘキサン(1:49)を使用する順相フラッシュクロマトグラフィーによって精製した(1HNMR:CDCl3 d 8.20(ブロードなd,1H,J=9Hz),7.72(ブロードなd,1H,J=7.8Hz),7.43(dt,1H,J=7.2,1.2Hz),7.34(dt,1H,J=7.7,1.2Hz),6.88(d,1H,J=2.4Hz),),6.82(d,1H,J=2.4Hz),3.93(s,3H),3.21(q,4H,J=7.2Hz),1.08(t,6H,J=7.2Hz))。
【0147】
次に、1−ジエチルアミノ−3−メトキシ−ナフタレン3のメチル基を、以下のような三臭化ホウ素脱保護によって除去した。1−アミノ−3−メトキシナフタレン(100mg)を、無水CH2C12(5ml)に懸濁し、そして、この混合物を、ドライアイス/アセトン浴中で−70℃まで冷却した。三臭化ホウ素(10当量)を滴下し、そしてこの反応物を、30分間攪拌し、次いで、冷蔵庫内(0℃)に一晩置いた。慎重にMeOH(10ml)を加えることによって、この反応物を−70℃にてクエンチした。固体NaHCO3(30当量)を加え、そして、この反応物を室温まで温め、次いで、手短に加熱還流した。この混合物を冷却し、そして濾過し、この濾液をAcOHで酸性化し、そして減圧下で溶媒を除去して、粗製1−ジエチルアミノ−3−ヒドロキシナフタレン4を得、これを、移動相としてEtOAc:ヘキサン(1:4)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0148】
(実施例2)
(N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン9の合成(図1))
上記実施例1のパラジウムで触媒したトリフレート/アミンカップリング反応に従って、アニリンを用いて、3−メトキシナフタレン−1−トリフレート2を誘導体化して、1−アニリノ−3−メトキシナフタレン5を得た。
【0149】
この1−アニリノ−3−メトキシナフタレン5を、以下のようなアミノ基のアセチル化手順によってアセチル化した。この1−アミノ−3−メトキシナフタレン5(500mg)および1.2当量の無水Et3Nを、10mlの無水CH2Cl2中に懸濁し、氷/NaCl浴を使用して−5℃まで冷却した。1.1当量の2−ブロモ−2−塩化メチルプロピニルを滴下し、そしてこの反応物を、−5℃にて1時間攪拌し、そして室温にてさらに1時間攪拌した。この反応物を室温にし、そして、5% HClおよび酢酸エチルを使用する水性ワークアップに供し、粗製中間体1−(ブロモアルキル)アミド−3−メトキシナフタレン6を得、これを、移動相としてEtOAc:ヘキサン(1:9)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0150】
この1−(ブロモアルキル)アミド−3−メトキシナフタレン6を、以下のようなAlCl3を触媒したフリーデル−クラフツ環化手順を使用して環化した。1〜3当量のAlCl3を含有するニトロベンゼンを、この1−(ブロモアルキル)アミド−3−ヒドロキシナフタレン6に加えた。この反応物を130℃まで加熱し、そして1時間反応させた。NH4C1およびEtOAcを使用する水性ワークアップによって、粗製N−フェニルベンゾインドリノン(N−phenyl−benzoindolinone)中間体7を得、これを、移動相としてEtOAc:ヘキサン(1:4)を使用する順相フラッシュクロマトグラフィーによって精製した。次いで、N−フェニルベンゾインドリン中間体7のアミドカルボニル基をLAHで還元して、化合物8を得た(1HNMR:CDCl3 d 7.71(d,1H,J=7.8Hz),7.32(m,2H),7.24(m,2H),7.07(bt,1H,J=6.6Hz),6.96(m,3H),6.84(s,1H),3.97(s,3H),3.92(s,2H),1.44(s,6H)。
【0151】
実施例1に記載される三臭化ホウ素脱保護手順を使用して、化合物8のメトキシ基を脱保護し、N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン9を得た。
【0152】
(実施例3)
(N−メチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン15の合成(図2))
ピリジン中でピペラジンの触媒作用を使用したメトキシナフトアルデヒドおよびマロン酸の縮合によって、化合物10を合成した。化合物10を、10% Pd/炭素上で水素で還元し、続いてLAHで還元し、そして上記化合物2の合成の概要のように、トリフルオロメタンスルホン酸無水物と反応させて、トリフレート11を得た。次いで、トリフレート11を、DMF中のNaN3(3当量)と、100℃にて6時間反応させた。次いで、この反応物を室温にし、そして、純水およびEtOAcを使用する水性ワークアップに供し、化合物12を得た。化合物12を、無水CH2Cl2中に懸濁し、3〜5当量の固体AlCl3と複合体化し、そして2時間還流してコーティング13を得た。
【0153】
化合物13を、以下のような一般的なアミノ基のアルキル化手順に従ってMeIでアルキル化した。この3−メトキシベンゾキノリン誘導体(100mg)13を、5mlの乾燥THF中に懸濁し、そして−5℃まで冷却した(氷/NaCl)。1.1当量のn−ブチルリチウム(1M)を滴下し、そして、この反応物を、1時間攪拌した。3当量のMeIアルキル化剤をゆっくりと加え、この反応物を、室温にて2時間攪拌した。NH4ClおよびEtOAcを使用する水性ワークアップによって、粗製アルキル化3−メトキシベンゾキノリン中間体14を得た。次いで、中間体14を、移動相としてEtOAc:ヘキサン(1:19)を使用する順相フラッシュクロマトグラフィーによって精製した(1HNMR:CDCl3 d 8.1(ブロードなd,1H,J=8.1Hz),7.68(dd,1H,J=8.1,1.8Hz),7.34(m,2H),6.8(s,1H),3.92(s,3H),3.21(m,2H),2.94(s,3H),2.77(t,2H,J=6.6Hz),1.92(m,2H))。上記実施例1の一般的な三臭化ホウ素手順によって化合物のメトキシ基を脱保護した後、N−メチルヒドロキシベンゾキノリン誘導体15を得た。
【0154】
(実施例4)
(3−(5−ヒドロキシベンゾキノリン−1−イル)プロパンスルホン酸17の合成(図2))
N−メチルヒドロキシベンゾキノリン誘導体15を合成するために、実施例3における上記概要の手順に従って、化合物13を合成した。次いで、今回は、MeIではなく1,3−プロパンスルホン(1,3−propane sultone)をアルキル化剤として使用して、上記実施例3の一般的なアミノ基のアルキル化手順に従って、化合物13を合成し、5−メトキシベンゾキノリン−N−プロパンスルホン酸中間体16を得た(1HNMR:CD3OD d 7.94(d,1H,J=8.7Hz),7.65(d,1H,J=8.4Hz),7.32(t,1H),7.27(t,1H),6.85(s,1H),4.89(s,3H),3.20(m,2H),3.08(bt,2H,J=6Hz),2.91(m,2H),2.72(t,2H,J=6.6Hz),2.33(m,2H),1.89(m,2H)。上記実施例1の一般的な三臭化ホウ素手順によって化合物16のメトキシ基を脱保護した後、3−(5−ヒドロキシベンゾキノリン−1−イル)プロパンスルホン酸17を得た。
【0155】
(実施例5)
(N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン22の合成(図3)) G.T.MorganおよびE.D.Evans(1919),J.Chem.Soc.115:1126の手順に従ってか、または実施例10のように、1−アミノ−3−メトキシナフタレン18を合成した。A.Rosowsky and E.J.Modest(1965)Jour.Org.Chem.30:1832の手順に従って、18(1gm)を、乾燥アセトン(50ml)中に懸濁し、そして、0.01当量のヨウ素をこの溶液に加えた。この反応物を加熱し、そして16時間攪拌し、冷却し、次いで、飽和Na2S2O3でクエンチした。次いで、この反応混合物を、飽和Na2S2O3およびEtOAcを使用する水性ワークアップに供して、粗製メトキシベンゾキノリン19を得た。このメトキシベンゼンキノリン19を、EtOAc/ヘキサン 1:9の移動相を使用するフラッシュクロマトグラフィーによって精製した。次いで、化合物19を、上記実施例3の一般的なアミノ基のアルキル化手順に従って、MeIでアルキル化し、化合物20を得た。化合物20を、70psiにて(Parr hydrogenator)中のH2および10% Pd/C 触媒で還元して、N−メチル−2,2,4−トリメチル−5−メトキシベンゼンキノリン中間体21を得た(1HNMR:CDCl3 d 8.20(bd,1H,J=7.5Hz),7.65(bd,1H,J=7.5Hz),7.33(m,2H),6.89(s,1H),3.94(s,3H),3.14(b sextet,1H,J=6.6Hz),2.80(3,3H),1.89(d,2H,J=8.7),1.42(d,3H,J=6.9Hz),1.34(s,3H),1.05(s,3H)。上記実施例1の一般的な三臭化ホウ素手順によって化合物21のメトキシ基を脱保護した後、N−メチル−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン22を得た。
【0156】
(実施例6)
(N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン27の合成(図3))
上記実施例2の一般的なアミノ基のアセチル化手順に従って、1−アミノ−3−メトキシナフタレン18を、2−ブロモ−2−塩化メチルプロピニルでアセチル化して、化合物23を得た。化合物23を、上記実施例2のフリーデル−クラフツ環化手順によって環化して、化合物24を得た。次に、化合物24を、3当量のLAHを含有するTHFで還元して4−メトキシベンゾインドリン25を得た。アルキル化試薬としてヨードメチル使用する、上記実施例3の一般的なアミノ基のアルキル化手順を使用して、化合物25をアルキル化し、N−メチル−3,3−ジメチル−4−メトキシベンゾインドリン中間体26を得た(1HNMR CDCl3 d 8.07(bd,1H,J=8.4Hz),7.69(bd,1H,J=8.1Hz),7.33(bt,1H,J=7.8Hz),7.22(bt,1H,J=8.1Hz),6.70(s,1H),3.92(s,3H),3.32(s,2H),3.32(s,3H),1.44(s,6H)。実施例1の一般的な三臭化ホウ素手順によって化合物26のメトキシ基を脱保護した後、N−メチル−3,3−ジメチル−4ヒドロキシベンゾインドリン27を得た。
【0157】
(実施例7)
(N−エチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン29の合成(図3))
4−メトキシベンゾインドリン25を、上記実施例6のように合成した。
【0158】
化合物25を、アルキル化剤としてヨードメチルを使用する実施例3に記載の一般的なアルキル化手順によってアルキル化して、N−エチル−3,3−ジメチル−4−メトキシベンゾインドリン中間体28を得た(1HNMR:CDCl3 d 7.90(d,1H,J=8.7Hz),7.68(d,1H,J=8.1Hz),7.32(bt,1H,J=7.5Hz),7.22(bt,1H,J=6.9Hz),6.69(s,1H),3.83(s,3H),3.52(q,2H J=7.5Hz),3.38(s,2H),1.46(s,6H),1.27(t,3H,J=7.5Hz)。実施例1の一般的な三臭化ホウ素手順によって化合物28のメトキシ基を脱保護した後、N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン29を得た。
【0159】
(実施例8)
(選択したジベンゾローダミン色素化合物の合成)
(一般手順A(図5))
固体無水フタル酸誘導体34を、1.4当量のアミノヒドロキシ中間体31および2.8当量のZnCl2と混合した。オーブン乾燥反応用容器を、ゴム隔膜でキャップし、アルゴンでパージした。この固体混合物を、融解が観察されるまで(例えば、約15分後)130℃にて手短に加熱した。1,2−ジクロロベンゼン(約10当量)を、この反応混合物にシリンジによって加え、この異種混合物を、4時間の間130〜170℃まで加熱した。粗製反応混合物を冷却し、最小量のMeOH:CH2Cl2(1:19)中に懸濁し、順相フラッシュクロマトグラフィーカラムに直接充填し、そしてこの粗製色素を、MeOH:CH2Cl2(1:19)移動相で溶出した。必要に応じて、この色素を精製し、そして、MeOH:CH2Cl2(1:9)を用いて展開するPTLCによって、別個の異性体35および36に分離した。この異性学的に純粋な色素(これは、1:9 MeOH:CH2Cl2で溶出するシリカのTLC上の単一スポットとして遊走される)を、そのUV/可視吸収スペクトル、ならびにその長波長蛍光励起スペクトルおよび発光スペクトルによって同定した。
【0160】
(一般手順B(図6))
図6に概略を示した一般的な手順において、固体無水フタル酸誘導体34(100mg)を、ゴム隔膜でキャップをした丸底フラスコ内に置き、アルゴンでパージした。無水ニトロベンゼン(2ml)を加え、そして加熱して無水物を溶解した。この混合物を、室温まで冷却し、3〜6当量の無水AlCl3を攪拌しながら加えて固体を溶解した。その後、1当量の1−アミノ−3−メトキシナフタレン中間体31を、攪拌しながら加え、この反応物を、1時間130℃まで加熱した。次いで、この反応物を冷却し、そしてEtOAc中に懸濁した。有機層を、飽和NH4Clおよびブラインで洗浄し、Na2SO4で乾燥し、濾過し、そして、溶媒を、減圧下で除去した。必要に応じて、この得られたケトン中間体37/38を精製し、移動相として(MeOH:CH2C12、1:19)を使用する順相フラッシュクロマトグラフィーを使用してかまたは再結晶によって、別個の異性体37および38に分離した。異性学的に純粋な中間体37または38のメトキシ基を、実施例1に記載される一般的な三臭化ホウ素脱保護手順に従って除去して、アミノヒドロキシナフタレンケトン39を得た。次いで、このアミノヒドロキシナフタレンケトン39(100mg)を、1当量の1−アミノ−3−ナフタレン中間体32を含有する無水1,2−ジクロロベンゼン(2ml)と2時間130℃にて反応させた。この反応物を冷却し、異性学的に純粋でかつ非対称的に置換された生成物40を得、これを、上記一般手順Aによって精製した。
【0161】
(ジベンゾローダミン色素41の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31として1−ジエチルアミノ−3−ヒドロキシナフタレン4を使用して、一般手順Aに従った。
【0162】
(ジベンゾローダミン色素42の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−5−ヒドロキシベンゾキノリンを使用して、一般手順Aに従った。
【0163】
(ジベンゾローダミン色素43の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31として5−ヒドロキシベンゾキノリン17を使用して、一般手順Aに従った。
【0164】
(ジベンゾローダミン色素44の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン22を使用して、一般手順Aに従った。
【0165】
(ジベンゾローダミン色素45の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン27を使用して、一般手順Aに従った。
【0166】
(ジベンゾローダミン色素46の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はFである)、そしてアミノヒドロキシ中間体31としてN−エチル−3,3−ジメチル−4ヒドロキシベンゾインドリン29を使用して、一般手順Aに従った。
【0167】
(ジベンゾローダミン色素47の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン9を使用して、一般手順Aに従った。
【0168】
(実施例9)
(選択されたジベンゾローダミン色素化合物のスペクトル特性)
以下の表は、本発明の数種の代表的なジベンゾローダミン色素化合物の重要なスペクトル特性を示す。全てのスペクトルは、色素のλmax,absにおいて0.05の吸収を有する色素のついて、1×TBE緩衝液および8M尿素中で、室温にて記録されている。色素濃度は、約10−6Mであった。
【0169】
【表1】
(実施例10)
(3,3−ジメチルヒドロキシベンゾインドリン25の合成(図8および9))
化合物2を、以下のようにパラジウムで触媒したトリフレート/イミンカップリングおよびBuchwaldの加水分解手順(Buchwald,S.L.ら、Tetrahedron Letters,1997,38/36,6367〜6370)を使用して、1−アミノ−3−メトキシナフタレン18に転換した。(図8)。
【0170】
3−メトキシナフタレン−1−トリフレート2(25g)を、0.013当量のトリス(ジベンジリデンアセトン)−ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフトル(±BINAP)、1.3当量の炭酸カリウム、1.3当量の炭酸セシウム、および1.5当量のベンゾフェノンイミンと混合した。あるいは、カリウムまたはナトリウムt−ブトキシド(2.6当量)を、この反応のための置換塩基として使用した。この反応混合物を、75mlの無水トルエンおよび75mlの無水テトラヒドロフラン中に懸濁し、そして油浴中で24時間120℃にて攪拌した。75mlのヘキサン/酢酸エチル(9:1)の混合物を、この反応物混合物に加え、そしてこの混合物を、ヘキサン/酢酸エチル(3:2)を用いてシルカゲルによって溶出した。この溶出液を収集し、次いで、減圧下で濃縮した。濃縮したオイルに、ヘキサン/酢酸エチル(9:1)を加えて、ベンゾフェノンイミノ−3メトキシナフタレン49を黄色結晶として結晶化した(20.0g、72.6%)。母液を濃縮し、ヘキサン/酢酸エチル(9:1)を加えて49の第二群(second crop)を黄色結晶として結晶化した(3.0g、10.9%)(全収率:83.5%)。
【0171】
化合物49を、酸性条件下にて加水分解して以下のように18を得た。化合物49(27gm)を、150mlの1,4−ジオキサンおよび100mlの5% 硫酸(H2SO4)中に懸濁した。次いで、この反応物を加熱し、そして油浴中で1時間50℃にて攪拌した。この反応混合物を室温まで冷却し、そして150mlのヘキサン/酢酸エチル(3:2)で洗浄した。次いで、この溶液を、氷冷水性NaOHでpH10〜11まで塩基性化して、酢酸エチルで3回抽出した。合わせた有機抽出物を、硫酸ナトリウム(Na2SO4)で乾燥し、そして減圧下でエバポレートして、淡褐色オイルとして18を得、これを、放置したまま結晶化させた(13.9g、100%)。
【0172】
化合物25(3,3−ジメチルヒドロキシベンゾインドリン)を、上記実施例6の手順に従って、18から合成した。
【0173】
(実施例11)
(N置換メトキシベンゾインドリン中間体57〜61の合成(図9))
化合物25を通常の中間体として用いて、アルキル置換またはパラジウムで触媒した第2級アミン基へのカップリングのいずれかによって、N−置換誘導体57〜61を合成した(図9)。
【0174】
(N−(6−ヘキサン酸)−3,3−ジメチルヒドロキシベンゾインドリン57の合成)
化合物25(2.3g)を3当量の市販のメチル−6−ヨードヘキサン酸52および3当量のジイソプロピルエチルアミンで処理することによってアルキル化した。この混合物を23mlの無水トルエン中に懸濁し、18時間攪拌しながら油浴中で130℃で加熱した。溶媒を減圧下でエバポレートして、粗製N−(6−ヘキサン酸)−3,3−ジメチルヒドロキシベンゾインドリン57を得た。粗製57をジクロロメタン中に溶解し、ヘキサン/酢酸エチル(99:1)で溶出する順相フラッシュクロマトグラフィーによって精製した(収率2.55g,74%)。
【0175】
(N−(ビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン58の合成)
化合物25(2.3g)を以下のように、確立された手順(Buchwald,ら,J.Org.,1997,62,6066−6068)に従ってパラジウムで触媒したカップリングによってN−アリール化した;化合物25(2g)を1.4当量の4−ブロモビフェニル7、ナトリウムt−ブトキシド(1.3当量)、0.013当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(±BINAP)と混合した。この混合物を100mlの無水トルエン中に懸濁し、十分攪拌し、そして油浴中で130℃にて一晩(16時間)加熱した。この反応混合物を水性塩化アンモニウム(NH4Cl)でクエンチし、酢酸エチルで抽出した(3×)。有機抽出物を無水Na2SO4で乾燥した。ヘキサン(等量)をこの粗製反応混合物に加え、それを、ヘキサン/酢酸エチル(1:1)を用いてシリカゲルによって溶出した。溶媒を減圧下でエバポレートし、残渣をヘキサン/酢酸エチル(98:2)で溶出する順相フラッシュクロマトグラフィーによって精製して、N(ビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン58(収率85%)を白色粉末として得た。
【0176】
(N−(カルボキシビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン59の合成)
化合物54をハロホルム反応によって市販のブロモビフェニルメチルケトンから合成して、ブロモビフェニル酸を得、続いて、確立された手順を用いてエステル化した。化合物25を、以下のように、確立された手順(Buchwald,ら Tetrahedron Letters,1997,38/36,6359−6362)に従って54でN−アリール化した:化合物25(0.4g)を1.4当量の化合物54、0.013当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(±BINAP)、および3当量の炭酸セシウムと混合した。この反応混合物を100mlの無水トルエンおよび100mlの無水テトラヒドロフラン中に懸濁し、次いで、油浴中で130℃にて加熱し、そして18時間攪拌した。この反応物を冷却し、粗製混合物にヘキサン(30ml)を加えた。この粗製生成物をヘキサン/酢酸エチル(3:2)を用いて、シリカゲルによって溶出した。この溶媒を減圧下で除去し、N−(カルボキシビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン59をヘキサン/酢酸エチル:(95:5)を使用する順相フラッシュクロマトグラフィーによって精製した(明黄色固体収率0.25g、32%)。
【0177】
(N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン60の合成)
化合物25を、化合物25からの化合物58の合成について上述したようにヨードベンゼンまたはブロモベンゼンでN−アリール化した。純粋なN−フェニル−3,3−ジメチルヒドロキシベンゾインドリン8を、ヘキサン/酢酸エチル:(98:2)を使用する順相フラッシュクロマトグラフィーの後に単離した。
【0178】
(N−カルボキシフェニル−3,3−ジメチルヒドロキシベンゾインドリン61の合成)
化合物25を、化合物59の合成について上述したようにヨード安息香酸メチルまたは臭化安息香酸メチルでN−アリール化した。純粋なN−カルボキシフェニル−3,3−メチルヒドロキシベンゾインドリン61を、ヘキサン/酢酸エチル:(95:5)を使用する順相フラッシュクロマトグラフィーの後に単離した。
【0179】
(実施例12)
(ジベンゾローダミン色素67、72、76および81の合成(図10〜13)
(色素67の合成(図10))
ヒドロキシインドリン62を、以下の手順に従って塩化アルミニウム(AlCl3)を使用する化合物58の脱メチル化によって得た:化合物58(2g)および塩化アルミニウム(6g)を、アルゴンの強い流れの下で完全に混合し、そしてこの反応混合物を、アルゴンで10分間パージした。この固体混合物を、アルゴン下で、90℃〜120℃まで油浴中にて20分間にわたって加熱した。この反応混合物を冷却し、そしてこの固体をジクロロメタン中に懸濁し、氷および希H2SO4の溶液に移した。水層を、5回抽出し、合わせた有機抽出物を、飽和塩化ナトリウムで洗浄し、そして硫酸ナトリウムで乾燥した。濾過した後、この溶媒を、減圧下でエバポレートし、そして純粋な62(0.13g)を、ジクロロメタン/メタノール(99:1)を使用する順相フラッシュクロマトグラフィーの後に単離した。ヒドロキシインドリン63を、化合物62からの化合物58の転換と同じ手順を使用して、化合物59から生成した。
【0180】
次に、1当量の1:1のN−メチルホルムアニリドおよび無水オキシ塩化物の複合体を含有する無水ジクロオロメタンを、小西洋ナシ形(pear)フラスコ(5mlフラスコ中100mg)中の1当量の62に加えた。この反応混合物を、アルゴンの流れの下で、ジクロロメタンがエバポレートして暗赤色オイルが残るまで、油浴中で80℃にて加熱した。ニトロベンゼン(300μl)を加え、続いて2当量のPOCl3を加えた。この上記溶液に、ニトロベンゼン(500μl)に懸濁した1当量の63を加えた。この反応混合物を、25分間にわたって80℃〜155℃まで加熱し、次いで、15分間155℃に保った。この反応物を冷却し、ジクロロメタンと共に5%HClに移した。水層をブラインで飽和させ、そしてジクロロメタンで3回抽出した。合わせた有機抽出物を分離し、そして減圧下でエバポレートして、粗製の色素誘導体64を得た。粗製の64を、酢酸(3ml)および5%HCl(3ml)中に懸濁し、60℃にて1時間加熱した。次いで、この反応混合物を、氷冷した塩化ナトリウム飽和水溶液中に注ぎ、ジクロロメタンで3回抽出した。合わせた有機層をエバポレートした後、次いで、純粋なモノカルボキシル化非対称性色素誘導体65を、ジクロロメタン/メタノール(9:1)で溶出する順相フラッシュクロマトグラフィーによって、他の2種の対称性色素生成物(ビス−ビフェニル色素およびビス−カルボキシフェニル色素)から精製した。
【0181】
最後に、この精製した色素誘導体65をスルホン化して、以下のような色素67を得た。色素誘導体65(30mg)および3当量の無水硫酸ナトリウムを、アルゴン下で0℃にて、無水クロロメタン(50ml)中に懸濁した。上記溶液に、5当量の塩化スルホン酸(ClSO3H)を加え、そしてこの反応混合物を、室温にて18時間攪拌した。この反応混合物を、飽和水性塩化ナトリウム/5% HClでクエンチし、そして水層を、ジクロロメタンで3回抽出した。有機層を減圧下でエバポレートして、粗製色素66を得た。粗製色素66を、ジオキsンおよび5%HCl(2:1)の溶液中に懸濁し、そして室温にて20時間攪拌した。次いで、この溶液を減圧下で濃縮し、飽和水性塩化ナトリウム中に懸濁し、そしてジクロロメタンで3回抽出した。この溶媒をエバポレートして、粗製色素67を得た。純粋な色素67を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0182】
(色素72の合成(図11))
化合物62を化合物58に転換するための、上記塩化アルミニウム脱保護手順をともに使用して、化合物61を脱メチル化して化合物68を得、そして化合物8を脱メチル化して化合物9を得た。化合物68を、メチルホルムアニリド/POCl3複合体と反応させ、続いて、化合物9と反応させて、化合物62および63からの色素誘導体64の合成について記載したように、色素誘導体70を生成した。粗製色素誘導体70を、酸によって加水分解して、上記64からの色素誘導体65の生成手順に従って、色素誘導体71を得た。純粋な非対称性色素誘導体71を、2種の対称性ビスフェニルおよびビス−カルボキシフェニル色素生成物からのPTLCによって精製した。次いで、この精製した色素71をスルホン化および加水分解して、色素誘導体65からの色素67の生成について上記したように、粗製色素72を得た。純粋な色素72を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0183】
(色素76の合成(図12))
化合物57を脱メチル化して、三臭化ホウ素脱保護によって化合物73を得、そして、化合物8を脱メチル化して、化合物62からの化合物58の転換について上記した、塩化アルミニウム脱保護手順を使用して、9を得た。化合物57を、標準条件下でメチルホルムアニリド/POCl3複合体と反応させ、続いて、標準条件下で化合物9と反応させて、上記実施例7のように色素誘導体74を生成した。粗製色素誘導体74を、標準的な酸加水分解によって加水分解して、色素誘導体75を得た。この純粋な非対称性色素誘導体75を、2種の対称性ビスフェニルおよびビス−ヘキサン色素生成物からのPTLCによって精製した。次いで、この純粋な色素75を、65から67を形成するために記載された標準的な手順によってスルホン化して、粗製色素76を得た。純粋な色素76を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0184】
(色素81の合成(図13))
化合物57(0.313g)を、室温にて3時間にわたって、7当量の1M KOHを含有するメタノールで処理することによって鹸化した。この溶液を、乾燥するまで濃縮し、そしてオイルを酢酸エチルおよび5%塩酸で十分に抽出した。有機層を硫酸ナトリウムで乾燥し、そして無色オイルになるまで濃縮した。純粋な77を、5%メタノール/ジクロロメタン/0.1%酢酸を使用する順相クロマトグラフィーによって単離した(収率0.243g;84%)。
【0185】
次に、化合物77(0.360gm)を、無水ジクロロメタン(2ml)、ならびにジクロロメチルメチルエーテル(2.6当量)および四塩化スズ(4当量)の溶液中に溶解した。この溶液を、1.5時間還流した。この混合物を、酢酸エチルおよび3M塩酸溶液で抽出した。有機層を、水およびブラインで洗浄し、Na2SO4で乾燥し、オイルになるまで濃縮し、そして精製することなく使用した。薄層クロマトグラフィーによって、この時点で、ほぼ等モルの化合物77および78の混合物が存在した。このオイルを、無水ジクロロメタン(8ml)中に懸濁し、そして−78℃まで冷却した。この混合物の脱メチル化を、確立された三臭化ホウ素手順によって行って、化合物79と化合物80との混合物を得た。得られた粗製オイルを、無水ジクロロメタン(5ml)中に溶解し、そしてオキシ塩化リン(2当量)を加え;次いで、ニトロベンゼン(5ml)を加えた。この混合物を、45分間にわたって80〜150℃まで加熱し、そして、即座に加熱を停止した(remove)。ワークアップおよび単離を、化合物67の合成についての実施例12のように行って、粗製色素81を得た。純粋な色素81を、化合物67の合成について実施例12で記載したように、PTLCによって単離した(収率32mg)。
【0186】
(実施例13)
(化合物67、72、76および81の色素カルボキシN−ヒドロキスクシンイミド誘導体の合成、ならびにこの誘導体のヌクレオチドおよびポリヌクレオチドへのカップリング(図14および15))
カルボン酸色素67、72、76および81のN−ヒドロキシスクシンイミド(NHS)誘導体化を、2つの方法の1つによって行った。NHS色素82、83および84の合成については、方法Aを、O−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート試薬を用いて行い、一方、色素85については、方法Bを、ジシクロヘキシルカルボジイミド(DCC)およびN−ヒドロキシスクシンイミドを使用して行った。
【0187】
(方法A:)
色素を、6当量のジイソプロピルエチルアミンと共に、無水DMF(5mg色素:300μl DMF)中に懸濁した。この色素溶液に、15当量のO−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレートを加え、そしてこの反応物を室温にて15分間攪拌した。この反応物を、ジクロロメタンとともに5% HCl(20ml)中に移した。水層を、飽和NaClを加えることによって塩にし、そしてジクロロメタンで3回抽出した。合わせた有機層を、乾燥するまで濃縮し、そして粗製色素NHSを、MeOH/CH2Cl2(1:9)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0188】
(方法B:)
色素および20当量のN−ヒドロキシスクシンイミドを、無水ジクロロメタン(5mg:500μl CH2Cl2)中に懸濁した。9当量のジシクロヘキシルカルボジイミドを加え、そしてこの反応物を、室温にて1時間攪拌した。この反応物をクエンチし、そして純粋な色素NHSを、方法Aについてと同様に単離した。
【0189】
NHS誘導体のヌクレオチドおよびポリヌクレオチドへの結合を、以下のように行った。アミノ基置換オリゴマーまたはターミネーターを、ホルムアミド(5×10−4M)中に懸濁し、そして15当量のジイソプロピルエチルアミンを加えた。DMSO(5mg/60μl)中に懸濁した過剰な(5〜10当量)の無水NHSを、室温にて攪拌しながら加えた。この反応物を、室温にて4時間攪拌した。この反応混合物に3M NaOAcを加えて、0.5M NaOAcを得た。EtOHの反応容量を4回加え、そしてこの混合物を冷却した。沈殿した色素標識オリゴマーを、遠心分離によって単離し、そして30% AcCN/0.1×TEAA緩衝液で溶出する逆相HPLCによって精製した。
【0190】
(実施例14)
(スルホメトキシベンゾインドリン89の合成(図16))
メトキシベンゾインドリン86(1g、0.0044mol)を、50mlの無水CH2Cl2中に懸濁し、そしてこの溶液を、水/アセトンのドライアイス浴中で−20℃まで冷却し、そしてアルゴンで十分にパージした(図16)。この攪拌溶液に、1.1当量(0.82ml)の無水トリフルオロメタンスルホン酸を滴下した。この無水物の添加が終わった後、この反応物を、−20℃に維持し、そして、2当量の無水トリエチルアミン(1.2ml)を滴下しながら攪拌した。1時間後、この反応物を、5% HClを加えることによって、−20℃にてクエンチし、そして水相を、CH2Cl2で3回抽出した。合わせた有機層を、飽和NaHCO3、ブラインで洗浄し、そしてNaSO4で乾燥し、濾過し、そして溶媒を、減圧下で除去した。この粗製生成物を、ヘキサン:CH2Cl2(3:2)で溶出する順相クロマトグラフィーによって精製して、2.3gのスルホンアミド87を得た(68%収率)。
【0191】
250mlの丸底フラスコ中で、スルホンアミド87(1g、0.00279mol)および顆粒状の無水Na2SO4(0.1g)を、100mlの無水CH2Cl2中に懸濁した。この反応混合物をアルゴンでパージし、そして0℃まで冷却した。迅速に攪拌した溶液に、1当量のクロロスルホン酸(185μl)を滴下した。この反応物を室温まで温め、そしてアルゴン下で16時間攪拌した。この反応物を、50mlのTHF、75mlの1M NaOHを加えることによってクエンチし;そして2時間攪拌した。1当量の硫酸水素テトラブチルアンモニウム(1g)を、この混合物に加え、そして水相を、CH2Cl2で5回抽出した。有機相を減圧下で濃縮して、油状の88の固体混合物および他の位置異性体(regioisomers)を得た。MeOH:CH2Cl2(8:92)で溶出する順相クロマトグラフィーによる精製によって、1gの純粋な88(53%収率)のモノテトラブチルアンモニウム塩を得た。
【0192】
スルホンアミド88のテトラブチルアンモニウム塩(1g)を、50mlの無水THF中に懸濁した。水素化リチウムアルミニウム(20当量、1g)を慎重に加え、そして、この反応物を、アルゴン下で2時間還流した。この反応物を、氷、次いで、50mlの1M NaOHを慎重に加えることによってクエンチした。テトラブチル硫酸水素アンモニウム(1g)をこの混合物に加え、そしてこの混合物を、減圧下で濃縮して大部分のTHFを除去した。この混合物を、分液漏斗に移し、CH2Cl2で5回抽出した。合わせた有機層を、減圧下でオイルになるまで濃縮し、そしてMeOH:CH2Cl2(8:92)で溶出する順相クロマトグラフィーによって精製して、0.733gの純粋な89(89%収率)のテトラブチルアンモニウム塩を得た。
【0193】
(実施例15)
(スルホメトキシナフトインドリン93の合成(図17aおよび17b))
メトキシナフトインドリン90を、24.9gの1,3−ジメトキシアントラセン121(Fitzgerald(1992)Jour.Org.Chem.57:7122−26)を含有する48% HBr(175ml)および氷状酢酸(450ml)を、90℃にて1時間加熱することによって調製した(図17a)。この反応混合物を、攪拌しながら、氷および水にゆっくりと注いだ。1,3−ジヒドロキシアントラセン122の沈殿物が、直ちに形成され、これを濾過し、300mlの冷水で洗浄し、そして褐色固体として収集した。化合物122を、0.14M HC1を含有するメタノールに5℃にて加え、そしてアルゴン下で3時間5℃にて維持した。この反応混合物を、冷水(1000ml)で希釈し、そして3:2 ヘキサン/酢酸エチルの混合物で3回抽出した。合わせた有機抽出物を、飽和NaClで洗浄し、そしてNa2SO4で乾燥した。濾過した後、減圧下で溶媒を除去し、そして、ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって、123を明黄色固体として得た(6.93g、66%)。
【0194】
トリフルオロメタンスルホン酸無水物(6.8ml)を、123(7.0g、0.031mol)およびジクロロメタン(600ml)の攪拌溶液に、アルゴン下で−60℃にてゆっくりと加えた。20分後、トリエチルアミン(5.65ml)を、この反応物に滴下した。この反応混合物を、−30℃にて30分間攪拌し、次いで、5%硫酸中に注いだ。次いで、この混合物を、400mlのヘキサン:ジクロロメタン 2:3の混合物で抽出し、飽和NaClで洗浄し、Na2SO4で乾燥し、濾過し、そして減圧下で濃縮した。ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって、淡黄色オイルとして124を得、これを、冷却して凝固させた。ヒドラジン(6ml)を、124(6g、0.017mol)、Cs2CO3(30g)、Pd2(dba)3(0.75g)、BINAP(1.5g)、トルエン(100ml)、およびTHF(100ml)の攪拌溶液に、アルゴン下で室温にて加え、次いで、90〜100℃にて1時間加熱した。冷却した溶液を濾過し、そして溶媒を減圧下で除去して、褐色固体して125を得た。125を含有する無水エタノールおよびイソブチルアルデヒド(14ml)の溶液を、90〜100℃にて加熱した。この溶液を、乾燥するまでエバポレートし、そしてヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって精製して、淡黄色固体として126を得た。
【0195】
126を含有する氷酢酸(150ml)の溶液を、アルゴン下で攪拌しながら、88℃にて1時間加熱した。この溶液を、氷浴中で冷却し、そしてNaCNBH3(3g)を、15分間にわたって一部ずつ加え、室温にてさらに15分間攪拌した。酢酸を減圧下で除去し、残渣をジクロロメタン中に懸濁し、そして1N NaOH、飽和NH4Cl、および飽和NaClで洗浄した。溶媒を、減圧下で除去し、そして残渣を、ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって精製して、淡黄色固体として90を得た(2.5g、53%)。
【0196】
メトキシナフトインドリン90(1g、3.6mmol)を、40mlの無水CH2Cl2中に懸濁し、溶媒を、氷浴中で0℃まで冷却し、そしてアルゴンでパージした(図17)。この攪拌溶液に、1.5当量のトリフルオロ酢酸無水物(0.76ml)を滴下した。この無水物の添加が終了した後、この反応物を0℃に維持し、2当量の無水トリエチルアミン(1ml)を滴下しながら攪拌した。1時間後、この反応物を、5% HClを加えることによってクエンチし、そして水相を、飽和CH2Cl2で3回抽出した。合わせた有機層を、NaHCO3、ブラインで洗浄し、そして有機層を、NaSO4で乾燥し、濾過し、そして溶媒を減圧下で除去した。粗製生成物を、1:4 EtOAc:ヘキサンを使用する順相クロマトグラフィーによって精製して、黄色固体として1gの91を得た(収率74%)。
【0197】
トリフルオロアセトアミド91(1g、2.7mmol)を、60mlの氷酢酸および6mlの無水CH2Cl2中に懸濁した。溶媒を、アルゴンで十分にパージし、そして0℃まで冷却した。この溶液を、十分に攪拌し、そしてクロロスルホン酸(4ml、20当量)を滴下した。この反応物を、室温まで温め、そして3日間攪拌した。この反応物を、最少量の氷(約1g)を加えることによってクエンチした。酢酸の除去を補助するためにトルエンを加えながら、この溶媒を減圧下(en vacuo)で除去した。この粗製反応混合物を、最少量の1:4 MeOH:CH2Cl2中に懸濁し、そして最初にCH2Cl2で溶出し、次いで、1:20 MeOH:CH2Cl2まで増加させるシリカゲルプラグを通して部分的に精製して、92と他のスルホン化位置異性体との混合物を得た。
【0198】
92を含有する混合物を、100mlのMeOH(1mlの水および1.5gのK2CO3を含有する)中に懸濁することによって脱保護した。この反応物を、室温にて30分間攪拌した。この粗製反応混合物を、MeOHで溶出するシリカゲルのプラグに通し、そして溶媒を、減圧下で除去した。所望の異性体であるスルホメトキシナフトインドリン93(0.350g、収率37%)を、1:9〜1:6 MeOH:CH2Cl2の勾配溶出を使用する順相クロマトグラフィーによって、副生成物異性体から分離した。(図17b)。
【0199】
(実施例16)
(ホルミルスルホメトキシベンゾインドリン106の合成(図20))
メチル−6−ブロモヘキサノエートでの、89の加熱しながらアルキル化よって、N−メチルカルボキシヘキシルスルホメトキシベンゾインドリン94(図18)を調製した。100mlの丸底フラスコ中で、テトラブチルアンモニウム塩の混合物として、1g(918、1.1mmol)の94を、25mlの無水CH2Cl2中に懸濁した。この溶液を、アルゴンで十分にパージし、そして氷浴中で0℃まで冷却した。この攪拌溶液に、5当量の新たに作製したPOCl3/DMFの1:1 Vilsmeyer複合体を含有するCH2Cl2(5当量の複合体を含有する約1mlのCH2Cl2、以下を参照のこと)を滴下した。この反応物を、氷浴から取り除き、そして室温にて1時間攪拌した。この反応物を、飽和NaHCO3(10ml)およびTHF(20ml)を加えることによってクエンチし、そして室温にて30分間攪拌した。この混合物を、CH2Cl2ともに、分液漏斗に移し、有機層を除去し、水層を、CH2Cl2で3回抽出し、そして合わせた有機層を濃縮して、粗製の106を得た(図20)。この粗製オイルから、純粋なアルデヒド106を、EtOAc:ヘキサン(1:2)を使用する順相クロマトグラフィーによって単離して、黄色固体を得た(0.222g、42%収率)。
【0200】
(実施例17)
(ホルミルスルホメトキシナフトインドリン108の合成(図20))
メチル4−(ブロモメチル)ベンゾエートでの、93(図18)の加熱しながらのアルキル化によって、スルホメトキシナフトインドリン101(図19)を調製した。100mlの丸底フラスコ中で、テトラブチルアンモニウム塩として、1g(1mmol)の101を、25mlの無水ジクロロメタン中に懸濁した。この溶液を、アルゴンで十分にパージし、そして氷浴中で0℃まで冷却した。この攪拌溶液に、5当量の新たに作製したPOCl3/DMFの1:1 Vilsmeyer複合体を含有するCH2Cl2(5当量を含有する約1mlのCH2Cl2、以下を参照のこと)を滴下した。室温にて1時間攪拌した後、この反応物を、飽和NaHCO3(10ml)およびTHF(20ml)を加えることによってクエンチし、そして室温にて30分間攪拌した。この混合物を、CH2Cl2ともに、分液漏斗に移し、有機層を除去し、水層を、CH2Cl2で3回抽出した。合わせた有機層濃縮して、粗製の108を得た(図20)。この粗製生成物を、EtOAc:ヘキサン(1:2)を使用する順相クロマトグラフィーに供して、0.497gの純粋なアルデヒド生成物108を黄色固体として得た(90%収率)。
【0201】
(Vilsmeyer試薬)
10mlのフラスコを、8.5mlの無水CH2Cl2および660μlの無水DMFでチャージした。この溶液を0℃まで冷却し、そしてアルゴンで十分にパージした。この攪拌溶液に、790μ1のPOCl3を滴下した。この反応物を、使用前に0℃にて15分間攪拌した。次いで、必要量の複合体を、必要に応じてシリンジで取り出した。
【0202】
(実施例18)
(スルホン化ジベンゾローダミン色素111の合成(図22a))
1,3−プロパンスルホン(1,3−propane sultone)での、89(図16)の熱しながらのアルキル化によって、N−プロピルスルホネートベンゾインドリン96(図18)を調製した。20mlの西洋ナシ形フラスコ中に、50mgのホルミルベンゾインドリン106(図20)、1.2当量の96、5ml 無水CH2Cl2および3ml 無水ニトロベンゼンを加えた。この混合物を攪拌し、大部分のCH2Cl2がエバポレートするまで、アルゴン蒸気下で、110℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、2時間攪拌しながら加熱した。
【0203】
この反応物を、手短に冷却し、次いで、5mlのMeOHを加え、そしてこの反応物を、5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素111を、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。エステル化色素111を含有する画分を、可視吸収スペクトル(MeOH中において638nmで吸収最大値)、蛍光発光スペクトル(MeOH中において651nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、871.5に相当する、算出値:871.3)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml 水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そして水層を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相HPLCを行って、20%の全体の収率において純粋な色素111を得た(図22a)。
【0204】
(実施例19)
(スルホン化ベンゾナフトローダミン色素115の合成(図22b))
1,3−プロパンスルホンでの、93(図17)の加熱しながらのアルキル化によって、N−プロピルスルホネートナフトインドリン102(図19)を調製した。ホルミルベンゾインドリン誘導体107(図20)を、95(図18)を実施例17の手順によってホルミル化することによって調製した。
【0205】
20mlの洋ナシ形フラスコ中に、50mgの107、1.2当量の102、5mlのCH2Cl2および3mlのニトロベンゼンを加えた。この混合物を攪拌し、そしてアルゴン蒸気下で大部分のCH2Cl2がエバポレートするまで、120℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、攪拌しながら2時間加熱した。この反応物を、手短に冷却し、次いで、10mlのMeOHを加え、この反応物を5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素115のメチルエステルを、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。115のメチルエステルを含有する画分を、可視吸収スペクトル(MeOH中において656nmで吸収最大値)、蛍光発光スペクトル(MeOH中において672nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、885.3に相当する、算出値:885.2)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そして水層を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相クロマトグラフィーを行って、20%の全体の収率において純粋な色素115を得た(図22b)。
【0206】
(実施例20)
(スルホン化ジナフトローダミン色素120の合成(図22c))
1,3−プロパンスルホンでの、93(図17)の加熱しながらのアルキル化によって、N−プロピルスルホネートナフトインドリン102(図19)を調製した。ホルミルベンゾインドリン誘導体108(図20)を、101(図191)を実施例17の手順によってホルミル化することによって調製した。
【0207】
20mlの洋ナシ形フラスコ中に、50mgの108、1.2当量の102、5mlのCH2Cl2および3mlのニトロベンゼンを加えた。この混合物を攪拌し、そしてアルゴン蒸気下で大部分のCH2Cl2がエバポレートするまで、110℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、攪拌しながら2時間加熱した。この反応物を、手短に冷却し、次いで、5mlのMeOHを加え、この反応物を5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素120を、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。エステル化色素120を含有する画分を、可視吸収スペクトル(MeOH中において686nmで吸収最大値)、蛍光発光スペクトル(695nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、991.5に相当する、算出値:991.3)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml 水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そしてこの反応物を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相クロマトグラフィーを行って、20%の全体の収率において純粋な色素120を得た(図22c)。
【0208】
(実施例21)
(スルホン化ジアリールローダミン−ターミネーターを用いたpGEMの配列決定)
米国特許第5,770,716号;同第5,948,648号;および同第6,096,875号の条件に従って、本発明のスルホン化ジアリールローダミン色素で標識されたターミネーターを、サンガー型の4色自動化DNA配列決定実験において他の標準試薬とともに使用する。
【0209】
色素−ターミネーター配列決定反応を、ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Manual(Applied Biosystems p/n 402116)によって提供されるプロトコルに従って、AmpliTaq DNA Polymerase、FSを使用して行う。pGEM−3Zf(+)テンプレートの配列決定を、未標識の−21 M13配列決定プライマー(順方向)を用いて行った。試薬(緩衝液、未標識プライマー、AmpliTaq DNA Polymerase(FS)を含む)は、ABI PRISMTM Dye Terminator Core Kit(Applied Biosystems p/n 402117)由来であり得る。dNTPミックスは、各々2mMのdATP、dCTP、dITP、およびdUTPまたはdTTPからなり得る。以下を含有する反応組成物のプレミックスを調製する:5×緩衝液4.0μL;dNTPミックス 1.0μL;テンプレート:pGEM(登録商標)−3Zf(+)、0.2μg/μL、2.0μL;プライマー:−21 M13(順方向)、0.8pmol/μL、4.0μL;AmpliTaq DNA Polymerase(FS)、0.5μL;および、H2O 3.5μL(ここで、全ての量は、反応に基づいて与えられる)。
【0210】
例示的な反応物を、Perkin Elmer 480 DNA Thermal Cycler(Applied Biosystems p/n N801−100またはApplied Biosystems Gene Amp PCR System 9700用の0.2mlチューブ)に適用される0.5mlのチューブ内に構築した。1〜250pmolの色素ターミネーターを、各反応物に加えた。30μlの鉱油を、各反応物の上面に加え、エバポレーションを防止する(Applied Biosystems 480 Thermal Cyclerを使用する場合)。反応物容量は、20μLであり、これは、15μLの上記反応プレミックス、変量の色素標識ターミネーター、および全反応物容量を20μLにするのに十分な容量の水を含有する。反応を、以下のように熱サイクルする:96℃で30秒間、50℃で15秒間、および60℃で4分間を25サイクル;続いて、4℃での保持サイクル。
【0211】
全ての反応物を、製造業者の指示書に従って(Princeton Separations p/n CS−901)、Centri−Sepスピンカラムによるスピンカラム精製によって精製する。カラム中のゲル材料を、0.8ml 脱イオン水で、少なくとも30分間室温にて水和させる。カラムを水和させ、ゲル材料中に気泡が閉じ込められていない状態にした後、カラムの上端および下端のキャップを取り外し、このカラムを、重力によって排水させる。次いで、カラムを、キットに備えられている洗浄チューブに挿入し、可変速度のミクロ遠心分離機で1300gにて2分間遠心分離し、洗浄チューブから取り除き、そしてサンプル収集チューブに挿入する。この反応混合物を、オイルから慎重に取り除き、そしてゲル材料に充填し、そしてチューブを再遠心分離する。次いで、溶出したサンプルを、減圧遠心分離機内で乾燥する。
【0212】
配列決定ゲルに充填する前に、乾燥したサンプルを、25μlのTemplate Suppression Reagent(Applied Biosystems p/n 401674)中に再懸濁し、ボルテックスし、95℃まで2分間加熱し、氷上で冷却し、再度ボルテックスし、そして遠心分離する(13,000×g)。10μLの懸濁したサンプルを、ABI PRISMTM 310 Genetic Analyzer(Applied Biosystems p/n 310−00−100/120)に適用されるサンプルバイアル(Applied Biosystems p/n 401957)にアリコートする。310 Genetic Analyzer上での電気泳動を、DNA配列決定分析(Applied Biosystems p/n 402837または4313087(ポリマー)およびp/n 402840(キャピラリー))に特異的に適用される、シービング(sieving)ポリマーおよびキャピラリーを用いて行う。このシービングポリマーは、核酸変性剤を含む。サンプルを、30秒間、2.5kVにてキャピラリーに動電学的に注入し、10〜12.2kVにて2時間まで泳動させ、この時、キャピラリーの外壁は、50℃で維持され、配列決定データとしてエレクトロフェログラムを作成する。
【0213】
スルホン化ジアリールローダミンターミネーターは、プライマー伸長の3’末端、4色配列決定反応間のポリヌクレオチドフラグメントに特異的に組み込まれている。1〜約1000塩基対の溶出フラグメントが、検出およびプロットされる。エレクトロフェログラムは、長さごとに溶出される標識されたフラグメントのスルホン化ジアリールローダミン色素によって発せられる蛍光強度を、ABI PRISMTM 310 Genetic Analyzerによる電気泳動間の時間の関数として、プロットする。
【0214】
全ての文献、特許、および本明細書中で参照される特許出願は、各個々の文献、特許、または特許出願が参考として援用されることを特異的かつ個々に示すかのように、同じ程度まで、参考として本明細書中で援用される。
【0215】
わずかな実施形態のみが上に詳細に記載されているが、化学分野の当業者は、多くの改変がこれらの実施形態においてその教示から逸脱することなく可能であることを、明確に理解している。このような全ての改変は、添付の特許請求の範囲の範囲内に含まれることが意図される。
【技術分野】
【0001】
(I.発明の分野)
本発明は、一般に、蛍光ローダミン色素化合物に関する。より詳細には、本発明は、蛍光標識試薬として有用な、スルホン化ジアリールローダミン色素に関する。
【背景技術】
【0002】
(II.背景)
蛍光標識を利用する生物学的分析の非放射活性検出は、現代の分子生物学において重要な技術である。放射活性標識の必要性を排除することによって、安全性が高められ、そして試薬の処理に関連した環境影響およびコストは、大いに減少する。このような非放射活性蛍光検出を利用する方法の例としては、4色自動化DNA配列決定、オリゴヌクレオチドハイブリダイゼーション方法、ポリメラーゼ連鎖反応生成物の検出、免疫アッセイなどが挙げられる。
【0003】
多くの適用において、空間的に重複した複数の分析物の非依存的な検出(例えば、単管の多重化DNAプローブアッセイおよび4色自動化DNA配列決定方法)を達成するために、スペクトルにより識別可能な複数の蛍光標識を使用することは、有益である。多重化DNAプローブアッセイの場合、スペクトルにより識別可能な蛍光標識を使用することによって、反応管の数は減少され得、これによって、実験プロトコルが単純化され、適用に特異的な試薬キットの作製が容易になる。4色自動化DNA配列決定の場合、多色蛍光標識によって、単一レーンにおいて複数塩基の分析が可能となり、これによって、単色方法を超えてスループットが増大し、レーン間の電気泳動の移動度変化に関連した不確実度が減少する。
【発明の概要】
【発明が解決しようとする課題】
【0004】
スペクトルにより識別可能な複数の蛍光標識のセットを構築することは、問題である。多色蛍光検出は、色素標識の選択について(特に、単一の励起光源、電気泳動分離、および/または酵素を用いた処理を必要とする適用(例えば、自動化DNA配列決定)について)、少なくとも6つの厳格な制限を課している。第一に、発光スペクトルがスペクトルにより分解される、構造的に類似の色素のセットを見出すことは、困難である。なぜなら、有機蛍光色素に関する代表的な発光バンドの半値幅が、約40〜80ナノメートル(nm)であるからである。第二に、たとえ非重複発光スペクトルを有する色素が同定されたとしても、それぞれの蛍光量子効率が低すぎる場合、このセットは依然として適切であり得ない。第三に、数種の蛍光色素が同時に使用される場合、同時励起は困難になる。なぜなら、色素の吸収バンドは、通常は、広範に分離されているからである。第四に、電荷、分子サイズ、および色素のコンホメーションは、分析物の電気泳動移動度に悪影響を与えてはいけいない。第五に、蛍光色素は、分析物を作製または操作するために使用される化学物質(例えば、DNA合成溶媒およびDNA合成試薬、緩衝液、ポリメラーゼ酵素、リガーゼ酵素など)と適合性でなければならない。第六に、色素は、レーザ励起に抵抗するのに十分な光安定性を有さなければならない。
【0005】
4色自動化DNA配列決定の適用において適切な、現在利用可能な多重化色素セットは、これらのセットを構成する全ての色素から蛍光発光を十分に励起するために青色または青緑色のレーザ光(例えば、アルゴン−イオンレーザ)を必要とする。商業的な自動化DNA配列決定システムにおいて青色または青緑色のレーザを使用することは、このようなレーザの高コストおよび制限された寿命に起因して、不利益である。
【課題を解決するための手段】
【0006】
(III.要旨)
したがって、本発明は以下を提供する。
(1)以下の構造:
【化1】
を有する、スルホン化ジアリールローダミン化合物であって、ここで、 R2、R2’、R12およびR12’は、水素、C1〜C12アルキル、C1〜C12アルキルジイル、フェニル、置換フェニル、ベンジル、置換ベンジル、ビフェニル、置換ビフェニル、ナフチル、置換ナフチル、複素環、置換複素環、水溶性基または連結部分であり;そして、
R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21は、水素、フッ素、塩素、C1〜C8アルキル、カルボキシレート、スルフェート、スルホネート、アルキルスルホネート、アミノメチル(−CH2NH2)、アミノアルキル、4−ジアルキルアミノピリジニウム、ヒドロキシメチル(−CH2OH)、メトキシ(−OCH3)、ヒドロキシアルキル(−ROH)、チオメチル(−CH2SH)、チオアルキル(−RSH)、アルキルスルホン(−SO2R)、アリールチオ(−SAr)、アリールスルホン(−SO2Ar)、スルホンアミド(−SO2NR2)、アルキルスルホキシド(−SOR)、アリールスルホキシド(−SOAr)、アミノ(−NH2)、アンモニウム(−NH3+)、アミド(−CONR2)、ニトリル(−CN)、C1〜C8アルコキシ(−OR)、フェノキシ、フェノール類、トリル、フェニル、アリール、ベンジル、複素環、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリエチレンオキシ、水溶性基、または連結部分であり;
但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21のうちの少なくとも1つがスルホネートである、スルホン化ジアリールローダミン化合物。
(2)R2、R2’、R12およびR12’の少なくとも1つがC1〜C6アルキルスルホネートまたはC4〜C10アリールスルホネートである、項目1に記載の化合物。
(3)前記アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルがスルホネートで置換されている、請求項1に記載の化合物。
(4)前記アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルがカルボキシルで置換されている、請求項1に記載の化合物。
(5)前記連結部分が、アジド、一置換の第一級アミン、二置換の第二級アミン、チオール、ヒドロキシル、ハライド、エポキシド、N−ヒドロキシスクシンイミジルエステル、カルボキシル、イソチオシアネート、塩化スルホニル、スルホネートエステル、シリルハライド、クロロトリアジニル、スクシンイミジルエステル、ペンタフルオロフェニルエステル、マレイミド、ハロアセチル、エポキシド、アルキルハライド、アリルハライド、アルデヒド、ケトン、アシルアジド、無水物、ヨードアセトアミドまたは活性化エステルである、項目1に記載の化合物。
(6)前記水溶性基が、カルボキシレート、スルホネート、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリヒドロキシル、または水溶性ポリマーである、項目1に記載の化合物。
(7)前記複素環が、ピロール、インドール、フラン、ベンゾフラン、チオフェン、ベンゾチオフェン、2−ピリジル、3−ピリジル、4−ピリジル、2−キノリル、3−キノリル、4−キノリル、2−イミダゾール、4−イミダゾール、3−ピラゾール、4−ピラゾール、ピリダジン、ピリミジン、ピラジン、シンノリン、フタラジン、キナゾリン、キノキサリン、3−(1,2,4−N)−トリアゾリル、5−(1,2,4−N)−トリアゾリル、5−テトラゾリル、4−(1−O,3−N)−オキサゾール、5−(1−O,3−N)−オキサゾール、4−(1−S,3−N)−トリアゾール、5−(1−S,3−N)−チアゾール、2−ベンゾオキサゾール、2−ベンゾチアゾール、4−(1,2,3−N)−ベンゾトリアゾール、またはベンゾイミダゾールである、項目1に記載の化合物。
(8)R1、R3、R6、R8、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21が水素である、請求項1に記載の化合物。
(9)以下:
C−12に結合した窒素とC−12炭素およびC−13炭素が一緒になる場合に4〜7員を有する第一の環構造を形成する、第一の架橋基;ならびに/または、
C−2に結合した窒素とC−1炭素およびC−2炭素が一緒になる場合に4〜7員を有する第二の環構造を形成する、第二の架橋基、を含有する、項目1に記載の化合物。
(10)前記第一および第二の環構造の一方または両方が5員を有する、項目9に記載の化合物。
(11)前記5員の環構造が1つのgem二置換炭素を含む、請求項10に記載の化合物。
(12)前記gem置換基が(C1〜C8)アルキルである、請求項11に記載の化合物。
(13)前記gem置換基がメチルである、項目11に記載の化合物。
(14)前記5員環が連結部分または水溶性基で置換されている、項目10に記載の化合物。
(15)以下:
C−12に結合した窒素とC−11炭素およびC−12炭素が一緒になる場合に5〜7員を有する第三の環構造を形成する、第三の架橋基;ならびに/または、
C−2に結合した窒素とC−2炭素およびC−3炭素が一緒になる場合に5〜7員を有する第四の環構造を形成する、第四の架橋基、を含有する、項目1に記載の化合物。
(16)前記第三および第四の環構造の一方または両方が6員を有する、項目15に記載の化合物。
(17)前記6員環構造が1つのgem二置換炭素を含む、項目16に記載の化合物。
(18)前記gem置換基が(C1〜C8)アルキルである、請求項17に記載の化合物。
(19)前記gem置換基がメチルである、項目18に記載の化合物。
(20)以下の構造:
【化2】
を有する、項目1に記載の化合物であって、ここで、
L2およびL12は、アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、置換複素環および置換ナフチルからなる群より選択されるリンカーである、化合物。
(21)C−3炭素およびC−4炭素、C−4炭素およびC−5炭素、C−9炭素およびC−10炭素、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含有する、項目1に記載の化合物。
(22)C−3炭素およびC−4炭素ならびにC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含有する、項目1に記載の化合物。
(23)エネルギー移動色素であって、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;および、 該ドナー色素および該アクセプター色素を連結するための、リンカー、を含み、ここで、
該ドナー色素およびアクセプター色素の少なくとも一方が、以下の構造:
【化3】
のうちの1つを有するスルホン化ジアリールローダミン化合物(それらの窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートである、エネルギー移動色素。
(24)前記アクセプター色素がスルホン化ジアリールローダミン化合物であり、前記ドナー色素がシアニン、フタロシアニン、スクアライン、ボディピー、ベンゾフェノキサジン、フルオレセイン、ジベンゾローダミン、またはローダミン色素である、項目23に記載のエネルギー移動色素。
(25)前記アクセプター色素がスルホン化ジアリールローダミン化合物であり、前記ドナー色素が、該アクセプター色素およびポリヌクレオチドに連結した、フルオレセインまたはローダミンである、項目24に記載のエネルギー移動色素。
(26)前記ドナー色素が前記ポリヌクレオチドの5’末端に連結されている、項目25に記載のエネルギー移動色素。
(27)前記ドナー色素が前記ポリヌクレオチドの3’末端に連結されている、項目25に記載のエネルギー移動色素。
(28)前記ドナー色素が前記ポリヌクレオチドの核酸塩基に連結されており、ここで、該核酸塩基がプリンの場合、前記リンカーは、8位で結合しており;該核酸塩基が7−デアザプリンの場合、該リンカーは、7位または8位で結合しており;該核酸塩基がピリミジンの場合、該リンカーは、5位で結合している、項目25に記載のエネルギー移動色素。
(29)項目23に記載のエネルギー移動色素であって、ここで、
前記リンカーは、以下の構造:
【化4】
を有し、ここで、
Zは、NH、SおよびOからなる群より選択され;
R21は、前記ドナー色素に結合したC1〜C12アルキルであり;
R22は、C1〜C12アルキルジイル、少なくとも1つの不飽和結合を有する5員環および6員環、ならびにカルボニル炭素に結合した縮合環構造からなる群より選択される置換基であり;
R23は、該リンカーを前記アクセプター色素に結合する官能基を含む、エネルギー移動色素。
(30)前記リンカーが以下の構造:
【化5】
を有し、nが2〜10の範囲である、項目29に記載の化合物。
(31)R23が以下の構造:
【化6】
を有し、ここで、R24がC1〜C12アルキルである、項目29に記載のエネルギー移動色素。
(32)前記リンカーが以下の構造:
【化7】
を有する、項目23に記載のエネルギー移動色素。
(33)前記リンカーが以下の構造:
【化8】
を有する、項目23に記載のエネルギー移動色素。
(34)前記リンカーが以下の構造:
【化9】
を有する、項目23に記載のエネルギー移動色素。
(35)前記リンカーが以下の構造:
【化10】
を有し、ここで、Dがドナー色素であり、Aがアクセプター色素であり、そしてnが1または2である、項目23に記載のエネルギー移動色素。
(36)前記リンカーが前記スルホン化ジアリールローダミン化合物のR2、R2’、R12またはR12’で結合している、項目23に記載のエネルギー移動色素。
(37)前記ドナーまたは前記アクセプターに対するリンカーがR2またはR2’に結合しており、ポリヌクレオチドに対するリンカーがR12またはR12’に結合している、項目36に記載のエネルギー移動色素。
(38)以下の式:
【化11】
を有する、標識されたヌクレオシドまたはヌクレオチドであって、ここで、
DYEは、以下の構造:
【化12】
の1つを有する、スルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
DYEは、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、
該ドナー色素および該アクセプター色素を連結するための、リンカー、を含む、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物であり;
Bは、核酸塩基であり;
Lは、リンカーであり;
R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはホスフェートアナログであり;そして、
R26およびR27は、単独である場合、各々独立して、H、HO、F、およびポリメラーゼ媒介性の標的指向性重合をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、標識されたヌクレオシドまたはヌクレオチド。
(39)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(40)Lが、以下:
【化13】
であり、nが0、1、または2である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(41)酵素学的に組み込み可能である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(42)ターミネーターである、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(43)以下の構造:
【化14】
を有する、項目42に記載の標識されたヌクレオシドまたはヌクレオチドであって、ここで、
R26およびR27は、単独である場合、各々独立して、H、F、およびポリメラーゼ媒介性の標的指向性重合をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、標識されたヌクレオシドまたはヌクレオチド。
(44)酵素学的に伸長可能である、項目38に記載の標識されたヌクレオシドまたはヌクレオチド。
(45)標識に共有結合したポリヌクレオチドを含有する、標識されたポリヌクレオチドであって、ここで、該標識は、以下の構造:
【化15】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物である、標識されたポリヌクレオチド。
(46)以下の式:
【化16】
を含有する、項目45に記載の標識されたポリヌクレオチドであって、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、核酸塩基であり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28およびR29は、単独である場合、各々独立して、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログであり;ここで、 該ポリヌクレオチドは、2〜1000ヌクレオチドを含む、標識されたポリヌクレオチド。
(47)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目46に記載の標識されたポリヌクレオチド。
(48)以下の式:
【化17】
を含有する、項目46に記載の標識されたポリヌクレオチドであって、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、核酸塩基であり;
Xは、O、NH、またはSであり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28は、ヌクレオチド間ホスホジエステルまたはヌクレオチド間アナログであり;ここで、
該ポリヌクレオチドは、2〜1000ヌクレオチドを含む、標識されたポリヌクレオチド。
(49)Bがウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される、項目48に記載の標識されたポリヌクレオチド。
(50)Lが、C1〜C12アルキルジイルまたは−(CH2CH2O)n−であり、ここで、nが1〜100である、項目48に記載の標識されたポリヌクレオチド。
(51)以下の構造:
【化18】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)に共有結合したポリペプチドを含有する、標識されたポリペプチドであって、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、 第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素に共有結合したポリペプチドであり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物である、標識されたポリペプチド。
(52)以下の式:
【化19】
を有する、ホスホラミダイト化合物であって、ここで、
DYEは、以下の構造:
【化20】
の1つを有するスルホン化ジアリールローダミン化合物(その窒素置換形態およびアリール置換形態を含む)であり、但し、R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つがスルホネートであるか、または、
DYEは、以下:
第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;
該ドナー色素によって放射された該励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、
該ドナー色素および該アクセプター色素を連結するための、リンカー、を含有する、エネルギー移動色素であり;ここで、
該ドナー色素および該アクセプター色素の少なくとも1つは、該スルホン化ジアリールローダミン化合物であり、
Lは、リンカーであり;
別個になったR30およびR31は、C1〜C12アルキル、C1〜C12シクロアルキル、およびアリールからなる群より選択されるか;または、窒素原子と一緒になったR30およびR31は、飽和窒素複素環を形成し;そして、
R32は、亜リン酸エステル保護基である、ホスホラミダイト化合物。
(53)R32が、メチル、2−シアノエチル、および2−(4−ニトロフェニル)エチルからなる群より選択される、項目52に記載のホスホラミダイト化合物。
(54)R30およびR31が、各々イソプロピルである、項目52に記載のホスホラミダイト化合物。
(55)一緒になったR30およびR31がモルホリノである、請求項52に記載のホスホラミダイト化合物。
(56)LがC1〜C12アルキルジイルである、項目52に記載のホスホラミダイト化合物。
(57)Lが、前記DYEのR2、R2’、R12またはR12’で結合している、項目52に記載のホスホラミダイト化合物。
(58)以下の構造:
【化21】
を有する、項目57に記載のホスホラミダイト化合物。
(59)Lが、
【化22】
であり、nが1〜10の範囲である、項目57に記載のホスホラミダイト化合物。
(60)標識された基質を形成する方法であって、該方法は、請求項1に記載の化合物または項目23に記載のエネルギー移動色素の連結部分と、ポリヌクレオチド、ヌクレオチド、ヌクレオシド、ポリペプチド、炭水化物、リガンド、粒子、および表面からなる群より選択される基質とを反応させ、これによって標識された基質を形成する工程を包含する、方法。
(61)前記連結部分がN−ヒドロキシスクシンイミドである、請求項60に記載の方法。
(62)前記連結部分がホスホラミダイトである、項目60に記載の方法。
(63)前記粒子が、ナノ粒子、ミクロスフェア、ビーズ、またはリポソームである、項目60に記載の方法。
(64)前記表面がガラスである、項目60に記載の方法。
(65)標識されたポリヌクレオチドを合成する方法であって、該方法は、項目52に記載のホスホラミダイト化合物をポリヌクレオチドにカップリングする工程を包含し、ここで、該ポリヌクレオチドは、固体支持体に結合し、これによって、標識されたポリヌクレオチドを形成する、方法。
(66)標識されたプライマー伸長産物を作製する方法であって、該方法は、酵素学的に組み込み可能なヌクレオチドを用いてプライマー標識ハイブリッドを伸長させる工程を包含し、ここで、該プライマーまたは該ヌクレオチドは、項目1に記載の化合物または項目23に記載のエネルギー移動化合物で標識され、これによって、該プライマーが伸長される、方法。
(67)前記ヌクレオチドが酵素学的に伸長可能である、項目66に記載の方法。
(68)前記プライマーが標識されたポリヌクレオチドである、請求項66に記載の方法であって、該標識されたポリヌクレオチドは、以下の式:
【化23】
を含み、ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される核酸塩基であり;
Xは、O、NH、またはSであり;
Lは、リンカーであり;
R27は、H、OH、ハライド、アジド、アミン、アルキルアミン、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2であり;そして、
R28は、ヌクレオチド間ホスホジエステルまたはヌクレオチド間アナログであり;ここで、
該ポリヌクレオチドは、2〜100ヌクレオチドを含有する、方法。
(69)前記酵素学的に組み込み可能なヌクレオチドが、以下の式:
【化24】
を有する標識されたヌクレオシドまたはヌクレオチドである、項目66に記載の方法であって;ここで、
DYEは、スルホン化ジアリールローダミン化合物またはエネルギー移動色素であり;
Bは、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および7−デアザグアノシンからなる群より選択される核酸塩基であり;
Lは、リンカーであり;
R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはホスフェートアナログであり;そして、
R26およびR27は、単独である場合、各々独立して、H、HO、およびFである、方法。
(70)ターミネーターヌクレオチドをさらに含む、項目66に記載の方法。
(71)R26およびR27が、単独である場合、各々独立して、
H、F、ポリメラーゼ媒介性の標的指向性プライマー伸長をブロックする部分であるか、または一緒になる場合に2’−3’−ジデヒドロリボースを形成する、項目69に記載の方法。
(72)ポリヌクレオチド配列決定の方法であって、該方法は、以下の工程:
第一、第二、第三、および第四のクラスのポリヌクレオチドの混合物を形成する工程であり、該クラスは、以下のようであり:
該第一のクラスの各ポリヌクレオチドは、3’末端ジデオキシアデノシンを含み、かつ第一の色素で標識されており;
該第二のクラスの各ポリヌクレオチドは、3’末端ジデオキシシチジンを含み、かつ第二の色素で標識されており;
該第三のクラスの各ポリヌクレオチドは、3’末端ジデオキシグアノシンを含み、かつ第三の色素で標識されており;そして、
該第四のクラスの各ポリヌクレオチドは、3’末端ジデオキシチミジンを含
み、かつ第四の色素で標識されており;ここで、
該第一、第二、第三、または第四の色素の1つ以上は、項目1に記載のスルホン化ジアリールローダミン化合物または項目23に記載のエネルギー移動色素であり、他の色素は、スペクトルにより互いに分解可能である、工程;ならびに、
該ポリヌクレオチドを電気泳動によって分離する工程、を包含する、方法。
(73)蛍光検出によって前記分離したポリヌクレオチドを検出する工程をさらに包含する、項目72に記載の方法。
(74)前記色素の蛍光スペクトルによって前記ポリヌクレオチドの3’末端ヌクレオチドを同定する工程をさらに包含する、項目72に記載の方法。
(75)オリゴヌクレオチドのライゲーション方法であって、該方法は、2つのプローブを標的配列にアニーリングする工程、および、一方のプローブの5’末端と他方のプローブの3’末端との間にホスホジエステル結合を形成する工程を包含し;ここで、
プローブの1つまたは両方が、項目1に記載の化合物または項目23に記載のエネルギー移動色素で標識されている、
方法。
(76)フラグメントの分析方法であって、該方法は、サイズ依存性分離プロセスによって、標識されたポリヌクレオチドフラグメントを分離する工程、および、該分離プロセスの後に、該分離した標識されたポリヌクレオチドフラグメントを検出する工程を包含し;ここで、
該フラグメントが、項目1に記載の化合物または項目23に記載のエネルギー移動色素で標識されている、方法。
(77)前記フラグメントが移動度変更標識で標識されている、請求項76に記載の方法。
(78)前記フラグメントがライゲーションによって形成される、項目76に記載の方法。
(79)前記サイズ依存性分離プロセスが電気泳動であり、前記標識されたポリヌクレオチドフラグメントが蛍光によって検出される、項目76に記載の方法。
(80)増幅方法であって、該方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該プライマーの少なくとも1つが、項目45に記載の標識されたポリヌクレオチドである、方法。
(81)増幅方法であって、該方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該ヌクレオチドの少なくとも1つが、項目38に記載の標識されたヌクレオチドである、方法。
(82)増幅方法であって、該方法は、2つ以上のプライマーおよび蛍光色素消光剤プローブを標的核酸にアニーリングする工程、ならびに、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によって該プライマーを伸長させる工程を包含し;ここで、
該プローブが、項目45に記載の標識されたポリヌクレオチドである、方法。
(83)ハイブリダイゼーション方法であって、該方法は、標的ポリヌクレオチドを固定化プローブにアニーリングする工程、ならびに、標的−プローブ複合体からの蛍光を検出する工程を包含し;ここで;
該標的ポリヌクレオチドが項目45に記載の標識されたポリヌクレオチドであり、該固定化プローブが平坦表面に共有結合している、方法。
(84)前記平坦表面がアドレス可能なアレイである、項目83に記載の方法。
(85)項目5に記載の連結部分を含むスルホン化ジアリールローダミン化合物およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(86)項目23に記載のエネルギー移動色素およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(87)項目52に記載のホスホラミダイト化合物およびポリヌクレオチドを備える、ポリヌクレオチドを標識するためのキット。
(88)1つ以上の酵素学的に組み込み可能なヌクレオチドおよびプライマーを備える、標識したプライマー伸長産物を作製するためのキットであって、該プライマーが項目45に記載の標識されたポリヌクレオチドである、キット。
(89)1つ以上の酵素学的に組み込み可能なヌクレオチドおよびプライマーを備える、標識したプライマー伸長産物を作製するためのキットであって、該ヌクレオチドの少なくとも1つが項目38に記載の標識されたヌクレオチドである、キット。
(90)前記標識されたヌクレオチドがターミネーターである、請求項89に記載のキット。
(91)4つの異なるターミネーターを備える、項目90に記載のキットであって、これらの1つは標的Aで終結し、これらの1つは標的Gで終結し、これらの1つは標的Cで終結し、そしてこれらの1つは標的TまたはUで終結する、キット。
(92)スルホン化アミノナフタレン化合物およびアミノアントラセン化合物を合成する方法であって、該方法は、以下の工程:
アミノ基を保護基試薬で保護する工程;
該保護したアミノ化合物をクロルスルホン酸と反応させる工程;および、
該アミノ保護基を除去する工程、を包含する、方法。
また、第一の局面において、本発明は、以下の構造:
【0007】
【化25】
を有するスルホン化ジアリールローダミン色素化合物(その窒素置換形態およびアリール置換形態を含む)を包含する。R1、R3、R4、R5、R6、R8、R9、R10、R11、R13、R14、R15、R16、R17、R18、R19、R20、およびR21の少なくとも1つは、スルホネートである。
【0008】
窒素置換基、R2、R2’、R12およびR12’は、C1〜C6アルキルスルホネートまたはC4〜C10アリールスルホネートであり得る。特定の実施形態において、アルキルスルホネートは、−(CH2)n−SO3Hであり、nは、1〜6の整数であり、そしてアリールスルホネートは、以下:
【0009】
【化26】
であり、ここで、nは、0または1である。
【0010】
他の実施形態において、窒素置換基、R2、R2’、R12およびR12’は、C1〜C6アルキルカルボキシレートまたはC4〜C10アリールカルボキシレート:
【0011】
【化27】
(ここでnは、0または1である)であり得る。
【0012】
本発明の別の局面は、エネルギー移動色素化合物を包含し、この化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射し得る、ドナー色素;このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素;ならびに、このドナー色素およびアクセプター色素を連結するためのリンカー、を含有し;ここで、このドナー色素およびアクセプター色素の少なくとも一方は、スルホン化ジアリールローダミン化合物である。
【0013】
本発明の別の局面は、標識されたヌクレオシド、ヌクレオチド、ポリヌクレオチドまたはポリペプチドであり、ここで、この標識は、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素である。
【0014】
本発明の別の局面は、標識試薬および基質を試薬で標識する方法であり、これらとしては、基質と共有結合を形成する、ホスホラミダイトまたはスルホン化ジアリールローダミン化合物の活性エステル連結部分が挙げられる。
【0015】
本発明の別の局面は、標識した基質を形成するための方法であり、この方法は、基質を、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素の連結部分と反応させる工程を包含する。
【0016】
本発明の別の局面は、プライマー標的ハイブリッドを酵素学的に組み込み可能なヌクレオチドで伸長させることによって、標識プライマー伸長産物を作製する方法である。このプライマーまたはヌクレオチドは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識され得る。
【0017】
本発明の別の局面は、4つのクラスのポリヌクレオチドの混合物を形成することによる、ポリヌクレオチド配列決定方法であり、ここで、各クラスは、3’末端ヌクレオチドにおいて、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されており、かつこの標識は、スペクトルにより分解可能である。
【0018】
本発明の別の局面は、オリゴヌクレオチドライゲーション方法であり、この方法は、2つのプローブを標的配列にアニーリングして、一方のプローブの5’末端と他方のプローブの3’末端との間にホスホジエステル結合を形成することにより、ここで、プローブの1つまたは両方は、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されている。
【0019】
本発明の別の局面は、増幅方法であり、この方法は、2つ以上のプライマーを標的ポリヌクレオチドにアニーリングして、ポリメラーゼおよび酵素学的に伸長可能なヌクレオチドの混合物によってこのプライマーを伸長させることにより、ここで、このプライマーの少なくとも1つまたはこのヌクレオチドの1つは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素で標識されている。
【0020】
本発明の別の局面は、試薬のキットであり、このキットは、スルホン化ジアリールローダミン化合物またはスルホン化ジアリールローダミン化合物を含有するエネルギー移動色素を備える。
【0021】
本発明のこれらおよび他の特徴および利点は、以下の説明、図面、および添付の特許請求の範囲を参照して、より理解されるようになる。
【図面の簡単な説明】
【0022】
【図1】図1は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図2】図2は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図3】図3は、本発明の1−アミノ−3−ヒドロキシナフタレン中間体の合成のための例示的な合成経路を示す。
【図4】図4は、本発明のジベンゾローダミン色素化合物の合成のための一般化された合成経路を示す。
【図5】図5は、本発明のジベンゾローダミン色素化合物の合成のための例示的な合成経路を示す。
【図6】図6は、本発明のジベンゾローダミン色素化合物の合成のための例示的な合成経路を示す。
【図7】図7は、本発明の数種の例示的なジベンゾローダミン色素化合物の構造を示す。
【図8】図8は、化合物18の合成を示す。
【図9】図9は、化合物57〜61の合成を示す。
【図10】図10は、化合物67の合成を示す。
【図11】図11は、化合物72の合成を示す。
【図12】図12は、化合物76の合成を示す。
【図13】図13は、化合物81の合成を示す。
【図14】図14は、化合物82〜84の合成を示す。
【図15】図15は、化合物85の合成を示す。
【図16】図16は、化合物89の合成を示す。
【図17A】図17aは、化合物90の合成を示す。
【図17B】図17bは、化合物93の合成を示す。
【図18】図18は、化合物94〜99を示す。
【図19】図19は、化合物100〜105を示す。
【図20】図20は、化合物106〜109を示す。
【図21】図21は、スルホン化ジベンゾローダミン化合物(上段)、スルホン化ベンゾ−ナフトローダミン化合物(中段)、およびスルホン化ジナフトローダミン化合物(下段)を形成するための環化反応を示す。
【図22A】図22aは、スルホン化ジベンゾローダミン化合物110〜113を示す。
【図22B】図22bは、スルホン化ベンゾ−ナフトローダミン化合物114〜116を示す。
【図22C】図22cは、スルホン化ジナフトローダミン化合物117〜120を示す。
【発明を実施するための形態】
【0023】
(V.例示的な実施形態の詳細な説明)
ここで、本発明の特定の実施形態に対して、参照が詳細になされており、これらの例は、添付の図面において例示されている。本発明は、例示した実施形態と共に記載されているが、本発明がこれらの実施形態に限定されることは意図されないことが理解される。これとは対照的に、本発明は、全ての代替物、変更、および等価物を含むことが意図されており、これらは、添付の特許請求の範囲によって規定されるような本発明の範囲内に含まれ得る。
【0024】
(V.1.定義)
他に記載がなければ、以下の用語および句は、本明細書中で使用される場合、以下の意味を有することが意図される。
【0025】
一連の色素を参照して、「スペクトル分解能」とは、色素の蛍光発光バンドが十分に明確であり(すなわち、十分に非重複である)、それぞれの色素が結合した試薬(例えば、ポリヌクレオチド)が、標準的な光検出システムを用いて(例えば、米国特許第4,230,558号、同第4,811,218号、またはWheelessら、21〜76頁、Flow Cytometry:Instrumentation and Data Analysis(Acaemic Press,New York,1985)に記載されるシステムによって例示されるような、バンド通過フィルターおよび光電子増倍管のシステム、荷電連結デバイスおよび分光器などを使用して)、それぞれの色素によって発生される蛍光シグナルに基づいて識別され得ることを意味する。
【0026】
「複素環」とは、1つ以上の環原子が炭素ではない(すなわち、ヘテロ原子である)、環式化合物を意味する。例示的な複素環としては、ピロール、インドール、フラン、ベンゾフラン、チオフェン、ベンゾチオフェン、2−ピリジル、3−ピリジル、4−ピリジル、2−キノリル、3−キノリル、4−キノリル、2−イミダゾール、4−イミダゾール、3−ピラゾール、4−ピラゾール、ピリダジン、ピリミジン、ピラジン、シンノリン、フタラジン(pthalazine)、キナゾリン、キノキサリン、3−(1,2,4−N)−トリアゾリル、5−(1,2,4−N)−トリアゾリル、5−テトラゾリル、4−(1−O,3−N)−オキサゾール、5−(1−O,3−N)−オキサゾール、4−(1−S,3−N)−チアゾール、5−(1−S,3−N)−チアゾール、2−ベンズオキサゾール、2−ベンゾチアゾール、4−(1,2,3−N)−ベンゾトリアゾール、およびベンズイミダゾールが挙げられるが、これらに限定されない。
【0027】
「リンカー」とは、共有結合、または標識をポリヌクレオチドに共有結合するか、もしくは1つの標識を別の標識に共有結合する原子鎖を含む、化学的部分を意味する。
【0028】
「連結部分」とは、別の分子と反応して共有結合、または連鎖を形成し得る、化学的反応性基、置換基または部分(例えば、求核原子または求電子)を意味する。
【0029】
用語「標識」とは、本明細書中で使用される場合、基質(例えば、オリゴヌクレオチド、ヌクレオチドまたはヌクレオチド5’−トリホスフェート)に結合し得、かつ以下の(i)〜(v)のように機能する任意の部分を意味する:(i)検出可能なシグナルを提供する;(ii)第二の標識と相互作用して第一または第二の標識によって提供される検出可能なシグナル(例えば、FRET)を変更する;(iii)ハイブリダイゼーション(すなわち、二重鎖形成)を安定化する;(iv)電荷、疎水性、形状、または他の物理的パラメータによって移動度(例えば、電気泳動移動度または細胞透過性)に影響を与える;あるいは、(v)捕獲部分(例えば、親和性、抗体/抗原、またはイオン錯体)を提供する。
【0030】
「基質」とは、本発明の色素化合物が結合する実体である。基質としては、以下が挙げられるがこれらに限定されない:(i)ポリヌクレオチド、(ii)ヌクレオシドおよびヌクレオチド、(iii)ポリペプチド、(iv)炭水化物、(v)リガンド、ならびに(vi)前述の(i)〜(v)の任意のアナログ。
【0031】
「置換(された)」とは、本明細書中で使用される場合、1つ以上の水素原子が1つ以上の非水素原子、官能基または部分で置換されている分子をいう。例えば、非置換窒素は、−NH2であり、一方、置換窒素は、−NHCH3である。例示的な置換基としては、以下が挙げられるがこれらに限定されない:ハロ(例えば、フッ素および塩素)、(C1〜C8)アルキル、スルフェート、スルホネート、スルホン、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、複素環、水溶性基、および連結部分。
【0032】
「多環式芳香族」とは、複数の環構造を有する芳香族炭化水素を意味し、これには、ビアリールおよび縮合ベンゼノイド炭化水素が挙げられる。ビアリールは、2つ以上の環が単結合によって一緒に連結しているベンゼノイド化合物である。このクラスの親系は、ビフェニルである。縮合ベンゼノイド化合物は、環の各対が2つの炭素を共有するような様式においてオルト位にて一緒に縮合した2つ以上のベンゼン環によって特徴付けられる。この群の最も単純なメンバーは、2つの環を有するナフタレン、および3つの環を有するアントラセンおよびフェナントレンである。
【0033】
「アルキル」とは、親アルカン、アルケン、またはアルキンの単一の炭素原子から1つの水素原子を除去することによって誘導される、飽和または不飽和の、分枝鎖、直鎖、または環式の炭化水素基を意味する。代表的なアルキル基は、1〜12個の飽和および/または不飽和の炭素からなり、これらとしては、メチル、エチル、プロピル、ブチルなどが挙げられるがこれらに限定されない。
【0034】
「アルコキシ」とは、−OR(ここで、Rは、(C1〜C6)アルキルである)を意味する。
【0035】
「アルキルジイル」とは、飽和または不飽和の、分枝鎖、直鎖または環式の、1〜20個の炭素原子の炭化水素基であって、親アルカン、アルケンまたはアルキンの同じかまたは2つの異なる炭素原子から2つの水素原子を除去することによって誘導される、2つの一価のラジカル中心(radical center)を有する炭化水素基を意味する。代表的なアルキルジイル基としては、1,2−エチルジイル、1,3−プロピルジイル、1,4−ブチルジイルなどが挙げられるがこれらに限定されない。
【0036】
「アリール」とは、親芳香族環系の単一炭素原子から1つの水素原子を除去することによって誘導される、6〜20個の炭素原子の、一価の芳香族炭化水素基を意味する。代表的なアリール基としては、ベンゼン、置換ベンゼン、ナフタレン、アントラセン、ビフェニルなどから誘導される基が挙げられるがこれらに限定されない。
【0037】
「アリールジイル」とは、共役共鳴電子系、および親アリール化合物の2つの異なる炭素原子から2つの水素原子を除去することによって誘導される少なくとも2つの一価のラジカル中心を有する、6〜20個の炭素原子の、不飽和環式基または多環式炭化水素基を意味する。
【0038】
「置換アルキル」、「置換アルキルジイル」、「置換アリール」および「置換アリールジイル」とは、それぞれ、1つ以上の水素原子が各々独立して別の置換基で置換されている、アルキル、アルキルジイル、アリールおよびアリールジイルを意味する。代表的な置換基としては、以下が挙げられるがこれらに限定されない:−X、−R、−O−、−OR、−SR、−S−、−NR2、−NR3、=NR、−CX3、−CN、−OCN、−SCN、−NCO、−NCS、−NO、−NO2、=N2、−N3、NC(O)R、−C(O)R、−C(O)NRR、−S(O)2O−、−S(O)2OH、−S(O)2R,−OS(O)2OR、−S(O)2NR、−S(O)R、−OP(O)O2RR、−P(O)O2RR、−P(O)(O−)2、−P(O)(OH)2、−C(O)R、−C(O)X、−C(S)R、−C(O)OR、−C(O)O−、−C(S)OR、−C(O)SR、−C(S)SR、−C(O)NRR、−C(S)NRR、−C(NR)NRR(ここで、各Xは、独立してハロゲンであり、各Rは、独立して−H、アルキル、アリール、複素環、または連結基である)。
【0039】
「ヌクレオチド間アナログ」とは、以下のようなオリゴヌクレオチドのリン酸エステルアナログを意味する:(i)アルキルホスホネート(例えば、C1〜C4アルキルホスホネート(特に、メチルホスホネート));(ii)ホスホラミダイト;(iii)アルキルホスホトリエステル(例えば、C1〜C4アルキルホスホトリエステル);(iv)ホスホロチオエート;および(v)ホスホロジチオエート。ヌクレオチド間アナログとしてはまた、非ホスフェートアナログが挙げられ、ここで、糖/ホスフェートのサブユニットは、非ホスフェート含有骨格構造によって置換されている。非ホスフェートオリゴヌクレオチドアナログの1つの型は、アミド結合(例えば、PNA(Nielsen(1991)「Sequence−selective recognition of DNA by strand displacement with a thymidinesubstituted polyamide」Science 254:1497〜1500)と通常称される、2−アミノエチルグリシンユニット)を有する。
【0040】
「核酸塩基」とは、相補的な核酸塩基または核酸塩基アナログ(例えば、プリン、7−デアザプリン、またはピリミジン)との対形成においてワトソン−クリック型水素結合を形成し得る窒素含有複素環式部分を意味する。代表的な核酸塩基は、天然に存在する核酸塩基であるアデニン、グアニン、シトシン、ウラシル、チミン、および天然に存在する核酸塩基のアナログ(例えば、7−デアザアデニン、7−デアザグアニン、7−デアザ−8−アザグアニン、7−デアザ−8−アザアデニン(Kutyavin,米国特許第5,912,340号)、イノシン、ネブラリン(nebularine)、ニトロピロール、ニトロインドール、2−アミノプリン、2,6−ジアミノプリン、ヒポキサンチン、偽ウリジン(pseudouridine)、偽シトシン、偽イソシトシン、5−プロピニルシトシン、イソシトシン、イソグアニン、7−デアザグアニン、2−チオピリミジン、6−チオグアニン、4−チオチミン、4−チオウラシル、O6−メチルグアニン、N6−メチルアデニン、O4−メチルチミン、5,6−ジヒドロチミン、5,6−ジヒドロウラシル、4−メチルインドール、およびエテノアデニンである(Fasman(1989)Practical Handbook of Biochemistry and Molecular Biology,385〜394頁,CRC Press,Boca Raton,Fl)。
【0041】
「ヌクレオシド」は、リボース糖のC−1’炭素に連結した核酸塩基を含む化合物を意味する。このリボースは、置換されていても置換されていなくてもよい。置換されたリボース糖としては、1つ以上の炭素原子(例えば、3’炭素原子)が1つ以上の同じかまたは異なる、−R、−OR、−NRRまたはハロゲン基(ここで、各R基は、独立して、水素、C1〜C6アルキルまたはC5〜Cl4アリールである)で置換されたリボースが挙げられるが、これらに限定されない。リボースとしては、リボース、2’−デオキシリボース、2’,3’−ジデオキシリボース、3’−ハロリボース、3’−フルオロリボース、3’−クロロリボース、3’−アルキルリボース(例えば、2’−O−メチル、4’−α−アノマーヌクレオチド、1’−α−アノマーヌクレオチド、ならびに2’−4’−連結および他のロックド(locked)二環式糖修飾物)(Imanishi WO 98/22489;Imanishi WO 98/39352;Wengel WO 99/14226)が挙げられる。核酸塩基がプリン(例えば、AまたはG)である場合、リボース糖は、核酸塩基のN9位に結合される。核酸塩基がピリミジン(例えば、C、TまたはU)である場合、ペントース糖は、核酸塩基のN1位に結合される(KornbergおよびBaker,(1992)DNA Replication,第2版,Freeman,San Francisco,CA)。
【0042】
「ヌクレオチド」とは、モノマー単位としてまたは核酸内の、ヌクレオシドのリン酸エステルを意味する。ヌクレオチドは、ときどき、特にリボース糖の構造的特徴を示すために、「NTP」、または「dNTP」および「ddNTP」として記載される。「ヌクレオチド5’三リン酸」は、5’位に三リン酸エステル基を有するヌクレオチドをいう。三リン酸エステル基は、種々の酸素の硫黄での置換がを含み得る(例えば、α−チオヌクレオチド5’三リン酸)。
【0043】
「酵素学的に組み込み可能な」とは、ヌクレオチドが、ポリメラーゼ酵素の作用を介して、新生ポリヌクレオチド鎖の末端(例えば、3’末端)に、酵素学的に組み込まれることが可能であるヌクレオチドの特性を意味する。
【0044】
「ターミネーター」とは、生じるポリヌクレオチド鎖へのヌクレオチドのそれ以降の組み込みを妨害し、それによってポリメラーゼ媒介伸長を停止させる、酵素学的に組み込み可能なヌクレオチドを意味する。代表的なターミネーターは、3’−ヒドロキシル置換基を欠き、そしてターミネーターとしては、2’,3’−ジデオキシリボース、2’,3’−ジデヒドロリボース、および2’,3’−ジデオキシ、3’−ハロリボース(例えば、3’−フルオロ)が挙げられる。あるいは、リボフラノースアナログ(例えば、アラビロース)が、使用され得る。例示的なヌクレオチドターミネーターとしては、以下が挙げられる:2’,3’−ジデオキシ−β−D−リボフラノシル、β−D−アラビノフラノシル、3’−デオキシ−β−D−アラビノフラノシル、3’−アミノ−2’,3’−ジデオキシ−β−D−リボフラノシル、および2’,3’−ジデオキシ−3’−フルオロ−β−D−リボフラノシル(Chidgeavadze(1984)Nucleic Acids Res.,12:1671〜1686;およびChidgeavadze(1985)FEB.Lett.,183:275〜278)。ヌクレオチドターミネーターとしてはまた、リバーシブルヌクレオチドターミネーターが挙げられる(Metzker(1994)Nucleic Acids Res.,22(20):4259)。
【0045】
「酵素学的に伸長可能な」は、ポリヌクレオチドの末端において酵素学的に組み込み可能であり、かつ得られる伸長ポリヌクレオチドが、ヌクレオチドまたはヌクレオチドアナログの引き続く組み込みを行い得る、ヌクレオチドの特性である。
【0046】
用語「標的配列」および「標的ポリヌクレオチド」とは、相補的なポリヌクレオチドとのハイブリダイゼーションの対象であるポリヌクレオチド配列(例えば、プライマーまたはプローブ)を意味する。この配列は、DNA、RNA、それらのアナログ(それらの組み合わせを含む)から構成され得る。
【0047】
「水溶性基」とは、水溶液中での本発明の化合物の溶解度を増加させる置換基を意味する。例示的な水溶性基としては、第四級アミン、スルフェート、スルホネート、カルボキシレート、ホスホネート、ホスフェート、ポリエーテル、ポリヒドロキシル、およびボロネートが挙げられるがこれらに限定されない。
【0048】
本明細書中で使用される場合、用語「オリゴヌクレオチド」および「ポリヌクレオチド」は、交換可能に使用され、ヌクレオチドモノマーの一本鎖ポリマーおよび二本鎖ポリマーを意味し、これらとしては、ヌクレオチド間ホスホジエステル結合の連鎖によって連結されている、2’−デオキシリボヌクレオチド(DNA)およびリボヌクレオチド(RNA)、またはヌクレオチド間アナログ、および付随する対イオン(例えば、H+、NH4+、トリアルキルアンモニウム、Mg2+、Na+など)が挙げられる。ポリヌクレオチドは、デオキシリボヌクレオチド、リボヌクレオチドから完全に構成されても、それらのキメラ混合物から構成されてもよい。ポリヌクレオチドは、ヌクレオチド間アナログ、核酸塩基アナログおよび糖アナログから構成され得る。ポリヌクレオチドのサイズは、これらがしばしばオリゴヌクレオチドと呼ばれる場合、代表的には、数個のモノマー単位(例えば、5〜40)〜数千個のモノマーヌクレオチド単位の範囲である。他に記載されない限り、ポリヌクレオチド配列が示される場合はいつでも、このヌクレオチドが左から右に5’→3’の順であり、そして「A」がデオキシアデノシンを示し、「C」がデオキシシチジンを示し、「G」がデオキシグアノシンを示し、そして「T」がチミジンを示すことが、理解される。
【0049】
「ローダミン色素」とは、以下の多環式の一般構造:
【0050】
【化28】
を含む色素(それらの任意および全ての置換バージョンを含む)をいう。
【0051】
(V.2 1−アミノ−3−メトキシナフタレンおよび1−アミノ−3−メトキシアントラセンの中間体)
1−アミノ−3−メトキシナフタレン(式I)および1−アミノ−3−メトキシアントラセン(式II)化合物のクラスは、スルホン化ジアリールローダミン色素の合成における中間体として有用である。式Iおよび式IIの化合物はさらに、それらのアリール置換形態および窒素置換形態を含む(本明細書中に提供される全ての分子構造は、示される正確な電子構造だけを含むのではなく、それらの全ての共鳴構造、プロトン化状態および付随する対イオンも含むことに注意のこと)。
【0052】
【化29】
式IおよびIIの化合物の1つの実施形態において、C−12に結合した窒素ならびにC−12炭素およびC−13炭素は、4〜7員を有する第一の環構造を形成し得、そして/またはC−12に結合した窒素ならびにC−11炭素およびC−12炭素は、5〜7員を有する第二の環構造を形成する。この第一および第二の環構造は、5員を有し得、ここで、この5員環構造は、1つのgem二置換炭素を含み得る。このgem置換基は、アルキル(例えば、メチル)であり得る。別の実施形態において、この5員環は、連結部分で置換されている。この実施形態において、C−12に結合した窒素ならびにC−12炭素およびC−13炭素は、4〜7員を有する第一の環構造を形成し、そして/またはC−12に結合した窒素ならびにC−11炭素およびC−12炭素は、5〜7員を有する第二の環構造を形成する。
【0053】
窒素置換基としては、アルキル、フェニル、芳香族、複素環、多環式芳香族、水溶性基、連結部部分、およびそれらの置換形態が挙げられ得る。窒素置換基は、アルキル、フェニル、またはそれらの置換形態であり得、ここで、置換基は、連結部分、スルホネートまたは水溶性基であり得る。例示的な水溶性基は、カルボキシレート、スルホネート、ホスホネート、ホスフェート、第四級アミン、スルフェート、ポリヒドロキシル、および水溶性ポリマーである。1つの実施形態において、窒素置換基は、−L−Rであり、ここで、Lは、任意のリンカーであり、Rは、連結部分または水溶性基であり得る。特定の実施形態において、Lは、以下:
【0054】
【化30】
であり、ここで、nは、1〜8の範囲である。
【0055】
式Iおよび式IIの化合物は、1つ以上のアリール位(例えば、C−8〜C−11、およびC−13)に1つ以上の置換基を含み得る。置換基としては、ホルミル、ヒドロキシル、フッ素、塩素、アルキル、スルフェート、スルホネート、スルホン、スルホンアミド、スルホキシド、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、水溶性基、複素環、および連結部分(それらの置換形態を含む)が挙げられ得る。1つの実施形態において、この化合物は、C−9炭素およびC−10炭素にわたって、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態(ここで、置換基はスルホネート基である)を含む)を含有する。
【0056】
式IおよびIIの非ホルミル化バーションは、以下に詳述されるスルホン化ジアリールローダミン色素を形成するための、環化反応における中間体である。
【0057】
本発明の代表的な1−アミノ−3−ヒドロキシナフタレン化合物および1−アミノ−3−ヒドロキシアントラセン化合物は、図1〜3(すなわち、化合物4、化合物9、化合物15、化合物17、化合物22、化合物27および化合物29);図10〜13(すなわち、化合物62、化合物63、化合物68、化合物73および化合物80);ならびに図16〜20(すなわち、化合物86〜109)に示される。
【0058】
当業者はまた、特に、周囲のpHに依存して、本発明の化合物が多数の異なるプロトン化状態で存在し得ることを認識している。本明細書中に提供される構造式は、数種の可能なプロトン化状態のうちの1種の化合物のみを示しているが、これらの構造は単に例示であり、本発明は任意の特定のプロトン化状態に制限されない、すなわち、任意および全てのプロトン化形態の化合物が本発明の範囲内に包含されることが意図されることが理解される。
【0059】
本発明の化合物は、複数の正電荷または負電荷を有し得る。本発明の色素の正味の電荷は、正または負のいずれかであり得る。色素に付随する対イオンは、代表的には、化合物が得られる合成方法および/または単離方法によって決定される。代表的な対イオンとしては、アンモニウム、ナトリウム、カリウム、リチウム、ハライド、アアセテート、トリフルオロアセテートなど、およびこれらの混合物が挙げられるがこれらに限定されない。任意の付随する対イオンの同定は、本発明の重要な特徴ではなく、そして本発明は、対イオンの任意の型に付随する色素を包含することが理解される。さらに、化合物が種々の異なる形態で存在する場合、本発明は、対イオンに付随する色素の形態(例えば、乾燥塩)だけでなく、対イオンに付随しない形態(例えば、水溶液または有機溶液)も包含することが意図される。
【0060】
上記発明の種々の局面は、多重化蛍光検出のために有用な公知の蛍光色素化合物に対する、予測できない、驚くべき、以下の重要な利点の1つ以上を達成する:(1)目的の色素化合物は、約630nmの波長を使用する低コストの赤色レーザによって、効率的に励起され得る;(2)目的の色素化合物の発光スペクトルは、窒素置換基および/またはアリール置換基の型および位置のモニター変化によって調節され得、類似の吸収特性を有するが、なお分解可能な蛍光発光スペクトルを有する色素のセットを作製するのを可能にする;(3)目的の色素化合物は、有利な蛍光特性を損なうことなく、ヌクレオシド、ヌクレオチドまたはポリヌクレオチドに容易に結合され得る;(4)目的の色素化合物は、狭い発光バンド幅を有する(すなわち、この発光バンド幅は、約70nmよりも低い、最大発光強度の半分の全幅を有する);(5)目的の色素化合物は、緩衝化水溶液に非常に可溶性であるが、高量子収率を維持する;(6)目的の色素化合物は、相対的に光安定性である;そして、(7)目的の色素化合物は、相対的に高い吸光係数(すなわち、約50,000よりも大きい)を有する。
【0061】
いくつかの合成方法が、上記の1−アミノ−3−ヒドロキシナフタレン化合物の合成のために利用可能であり、合成される特定の化合物の環構造の性質および窒素置換基の性質に依存する、種々の方法が好ましい。
【0062】
1−置換アミノ−3−ヒドロキシナフタレン化合物(例えば、1−シエチルアミノ−3−ヒドロキシナフタレン(4))の合成に適切な、第一の合成方法が、図1に示される。この第一の方法において、3−メトキシ−1−ヒドロキシナフタレン(1)を乾燥トリエチルアミンおよびトリフルオロメタンスルホン酸無水物と反応させて、粗製の3−メトキシナフタレン−1−トリフレート(2)が形成される。次いで、トリフレート(2)を、パラジウムで触媒したトリフレート/アミンのカップリングを用いて、アミン(例えば、第二級アミン(例えば、ジエチルアミン))と反応させて、置換アミン化合物(3)が形成される。次いで、化合物(3)は、三臭化ホウ素脱保護手順を使用して脱保護されて、1−アミノ−3−ヒドロキシナフタレン生成物(例えば、1−ジエチルアミノ−3−ヒドロキシナフタレン(4))が生成される。この合成の例は、以下の実施例1に提供される。
【0063】
ベンゾインドリン化合物(例えば、N−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(9))の合成に適した第二の合成方法もまた、図1に示される。この方法において、3−メトキシナフタレン−1−トリフレート(2)は、パラジウムで触媒したトリフレートカップリング反応を使用して第一級アミン(例えば、アニリン)から誘導体化され、第二級アミン(例えば、1−アニリン−3−メトキシナフタレン(5))が得られる。この第二級アミン(5)は、酸塩化物(例えば、ハロ塩化アセチル)を用いてアセチル化され、二置換アミド(例えば、1−アミド−3−メトキシナフタレン(6))が得られる。この第三級アミン(6)は、ルイス酸を触媒したフリーデル−クラフツ環化手順を使用して(例えば、AlCl3を使用して)環化され、化合物(7)が得られる。次いで、化合物(7)は、例えば、(水素化アルミニウムリチウム)LAHを使用して還元されて化合物(8)が得られる。三臭化ホウ素脱保護手順によってメトキシ基を脱保護した後、ベンゾインドリン(例えば、N−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(9))が得られる。この合成の例は、以下の実施例2に提供される。
【0064】
N−置換−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン化合物(例えば、N−メチル−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン(15))の合成に適した第三の合成方法が、図2に示される。この方法において、化合物10は、ピペリジン触媒を含有するピリジンを使用してマロン酸と縮合することによって、メトキシナフトアルデヒドから合成される。次いで、化合物10は、LAH還元の前に水素で還元され、そして、トリフルオロメタンスルホン酸無水物と反応させてトリフレート(11)が得られる。このトリフレート(11)は、NaN3と反応され、化合物(12)が得られる。化合物(12)を、ルイス酸(例えば、AlCl3)と錯化して還流し、環化ベンゾキノリン誘導体(13)が得られる。次に、窒素置換基を加える(例えば、窒素を、従来のアルキル化手順を使用してアルキル化する(例えば、ベンゾキノリン誘導体(13)を、n−ブチルリチウムおよびアルキル化剤(例えば、MeI)と反応させて化合物(14)が得られるか、またはプロパンスルホン(propane sultone)と反応させて化合物(16)が得られる))。次いで、メトキシ基が、三臭化ホウ素手順によって除去されて、N−アルキルベンゾキノリン誘導体(例えば、化合物15または化合物17)が得られる。この合成の例は、以下の実施例3に提供される。
【0065】
N−置換−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン化合物(例えば、N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン(22))の合成に適した第四の合成方法が、図3に示される。この方法において、Rosowsky(1965)Jour.Org.Chem.30:1832、および本明細書中の参考文献の手順に従って、1−アミノ−3−メトキシナフタレン(18)を、ヨウ素によって触媒されたアセトンと反応させ、次いで、飽和Na2S2O3でクエンチして、ベンゾキノリン化合物(19)が得られる。次いで、化合物(19)を、一般的なアルキル化手順に従ってアルキル化剤(例えば、MeI)でアルキル化し、化合物20が得られる。このアルキル化した化合物(20)を、Pd/Cによって触媒されたH2で還元して、N−メチルメトキシキノリン中間体(21)が得られ、続いて、一般的な三臭化ホウ素手順によってメトキシ基を脱保護して、N−置換−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン化合物(例えば、N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン(22)が得られる。この合成の例は、以下の実施例4に提供される。
【0066】
N−置換−3,3−ジメチル−4−ヒドロキシベンゾインドリン化合物(例えば、N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン(27))の合成に適した第五の合成方法もまた、図3に提供される。この方法において、1−アミノ−3−メトキシナフタレン(18)は、酸塩化物(例えば、2−ブロモ−2−メチルプロピオニルクロライド)でアセチル化されて、化合物(23)が得られる。化合物(23)は、AlCl3と反応させることによって環化されて、化合物(24)が得られる。次いで、化合物(24)は、LAHで還元されて、3,3−ジメチル−4−メトキシベンゾインドリン(25)が得られる。次いで、化合物(25)は、アルキル化剤(例えば、ヨウ化メチル)でアルキル化されて、N−メチル−3,3−ジメチル−4−メトキシベンゾインドリン(例えば、化合物(26))が得られる。三臭化ホウ素によるメトキシ基脱保護の後に、化合物(27)が得られる。この合成の例は、実施例5に提供される。
【0067】
アミノアントラセン化合物の合成に適した第六の合成方法が、図17aに示される。この方法において、アミノアントラセン化合物(メトキシナフトインドリン)90は、1,3−ジメトキシアントラセン121を脱メチル化し、ヒドロキシル基のうちの1つをモノメチル化して調製して123を得、トルフレート124を形成し、そして、ヒドラジンによって置換して125を得る。イソブチルアルデヒドとのヒドラゾンの形成、フッシャーインドリン環化(Fischer indoline cyclization)およびシアノホウ化水素ナトリウムでの還元によって90が得られる(実施例15)。
【0068】
アミノナフタレン化合物およびアミノアントラセン化合物は、スルホン化剤(例えば、クロロスルホン酸)でスルホン化され得る。例えば、アミノナフタレン化合物86は、初めに、N−スルホンアミオ化合物87(図16、実施例14)のように窒素保護され、次いで、低温度にて、クロロスルホン酸を含有する酢酸およびジクロロメタンと反応させ、脱保護してスルホン化化合物89が得られる。同様に、アミノアントラセン化合物90は、トルフルオロアセチル化化合物91のように保護され、次いで、スルホン化され、そして脱保護されて、93(図17b、実施例15)が得られる。中間体89および93は、スルホン化ジアリールローダミン化合物を合成する際に有用である。
【0069】
(V.3 スルホン化ジアリールローダミン色素化合物)
新規なクラスのスルホン化ジアリールローダミン色素化合物は、以下の式IIIabcに示されるような一般構造(それらのアリール置換形態および窒素置換形態を含む)を有する分子標識として有用である。
【0070】
【化31】
式IIIabcにおいて、少なくとも1つのアリール置換基は、スルホネートである。少なくともアリール−スルホネート基の存在によって、ジアリールローダミン化合物に、予想外の有利な特性(例えば、調節された溶解度、電気泳動移動度、およびスペクトル増強)が与えられ得る。
【0071】
式IIIabcの化合物の1つの実施形態において、この化合物は、第一結合基および/または第二結合基を含有する。第一結合基は、C−12に結合した窒素ならびにC−12炭素およびC−13炭素が一緒になる場合に、4〜7員を有する第一の環構造を形成し得、第二結合基は、C−2に結合した窒素ならびにC−1炭素およびC−2炭素が一緒になる場合に、4〜7員を有する第二の環構造を形成する。この第一および第二の環構造の一方または両方が、5員を有し得る。別の実施形態において、この5員環構造は、1つのgem二置換炭素を含み、ここで、このgem置換基は、アルキル(例えば、メチル)であり得る。代替の実施形態において、この5員環は、連結基で置換されている。別の実施形態において、この5員環は、以下に記載される1つ以上の窒素置換基を含む。架橋環構造を有する例示的なスルホン化ジアリールローダミン化合物としては、以下が挙げられる:
【0072】
【化32】
L2およびL12は、アルキルジイル、置換フェニル、置換ベンジル、置換ビフェニル、および置換ナフチルを含むリンカーである。
【0073】
本発明のなお別の実施形態において、式IIIabcの化合物は、1つ以上の窒素置換基、R2、R2’、R12およびR12’を含む。このような置換基は、以下からなる群より選択される:アルキル、フェニル、芳香族、複素環、多環式芳香族、水溶性基、連結基、およびそれらの置換形態。また、窒素置換基は、アルキル、フェニル、多環式芳香族、またはそれらの置換形態であり、ここで、例示的な置換基としては、連結基および水溶性基が挙げられる。
【0074】
本発明の、この第二の局面の別の実施形態において、式IIIabcの化合物は、第三結合基および/または第四結合基を含有する。第三結合基は、C−12に結合した窒素ならびにC−11炭素およびC−12炭素が一緒になる場合に、5〜7員を有する第三の環構造を形成し得、第四結合基は、C−2に結合した窒素ならびにC−2炭素およびC−3炭素が一緒になる場合に、5〜7員を有する第四の環構造を形成する。この第三および第四の環構造の一方または両方が、6員を有し得る。この6員環構造は、1つのgem二置換炭素を含み得、ここで、このgem置換基は、アルキル(例えば、メチル)であり得る。
【0075】
本発明の別の実施形態において、式IIIabcの化合物は、1つ以上の炭素(C−1、C−3〜C−6、C−8〜C−11、およびC14〜C−21)にてアリール置換基を含む。例示的なアリール置換基としては、以下が挙げられる:水素、フッ素、塩素、アルキル、スルフェート、スルホネート、スルホン、スルホンアミド、スルホキシド、アミノ、アンモニウム、アミド、ニトリル、低級アルコキシ、フェノキシ、芳香族、フェニル、多環式芳香族、水溶性基、複素環、および連結部分(それらの置換形態を含む)。少なくとも1つのアリール−置換基は、スルホネートである。別の実施形態において、式IIIabcの化合物は、C−3炭素およびC−4炭素、C−4炭素およびC−5炭素、C−9炭素およびC−10炭素、またはC−10炭素およびC−11炭素にわたって結合した縮合芳香族環(それらの置換形態を含む)を含む。この縮合芳香族環は、C−3炭素およびC−4炭素ならびにC−10炭素およびC−11炭素、またはC−9炭素およびC−10炭素ならびにC−4炭素およびC−5炭素(それらの置換形態を含む)にわたって結合し得る。
【0076】
本発明のこの第二の局面に従う数種の例示的な色素化合物は、以下に示される:図7および図10〜13(すなわち、化合物41〜47および化合物67、化合物72、化合物76および化合物81);ならびに、図21、図22a、図22b、図22c(すなわち、予測外の有利なスペクトル特性を有する化合物110〜120)。吸光率、量子収量および輝度は、非スルホン化アナログジアリールローダミン色素化合物と比較して、スルホン化ジアリールローダミン色素化合物111、114、118、119、120の全てにおいて増加する。
【0077】
(アリールスルホン化/非アリールスルホン化)
【0078】
【化33】
全ての測定を、8Mの尿素および1×Tris/EDTA緩衝液中で行った。
【0079】
上表は、非アリール−スルホネートアナログに対する、5つの例示的な色素のうちの2つのアリール−スルホネート基によって生じる吸光率、量子収量および輝度の差を測定している。輝度増加は、吸光率および量子収量を掛けることによって算出される。
【0080】
一般的に、本発明のスルホン化ジアリールローダミン色素は、以下(図4)のように合成される:無水物誘導体(例えば、無水フタル酸)を、1−アミノ−3−メトキシ中間体31および32、ならびにルイス酸(例えば、ZnCl2)と混合する(ここで、化合物30のR置換基は、同じであっても異なっていてもよいが、好ましくは、同じである)。例示的なR置換基としては、アセチレン、アルキル、フェニル、複素環、およびそれらの置換形態が挙げられるがこれらに限定されない。この混合物を、融解が観察されるまで、手短に加熱する。溶媒(例えば、1,2−ジクロロベンゼン)を、この反応混合物に加え、そして、不均一な混合物を、約130℃〜約180℃まで加熱する。粗製反応混合物を冷却し、そして、順相のフラッシュクロマトグラフィーによって精製して、色素化合物33を得る。無水物が置換フタル酸無水物の一部(例えば、化合物34)である場合、2つの異性体が形成される(図5)。これらの異性体35および36は、PTLCによって分離される。異性学的に(isomerically)純粋な色素は、順相TLCおよび逆相TLCでの単一スポットによって、ならびにそれらのUV/可視吸収スペクトルおよびそれらの長い波長蛍光励起スペクトルおよび発光スペクトルによって同定される。
【0081】
非対称性色素化合物の合成のための代替の手順が、図6に示される。このプロセスにおいて、無水物誘導体(例えば、フタル酸無水物34)を、乾燥ニトロベンゼンと混合して、加熱する。この混合物を、室温まで冷却し、無水AlCl3を、攪拌しながら加える。続いて、1−アミノ−3−メトキシ中間体31を、攪拌しながら加え、この反応物を加熱する。この反応物を冷却し、EtOAc中に懸濁する。有機層を、飽和NH4Cl、ブラインで洗浄し、Na2SO4で乾燥し、濾過し、そして溶媒を減圧下で除去する。得られたケトン中間体37/38を精製し、そして、フラッシュクロマトグラフィーまたは再結晶化によって、異なる異性体37および38に分離する(C−14およびC−17における置換基が同じであり、かつC−15およびC−16における置換基が同じである場合を除く)。異性学的に純粋なケトン中間体37または38のメトキシ基を、一般的な三臭化ホウ素脱保護手順に従って除去する。これによって、化合物38は、アミノヒドロキシナフタレンケトン中間体39を与える。次いで、アミノヒドロキシナフタレンケトン中間体39を、1−アミノ−3−メトキシ中間体32と反応させる。この反応物を冷却して、PTLCによってさらに精製され得る、異性学的に純粋でかつ非対称的に置換された生成物40が得られる。
【0082】
本発明のジベンゾローダミン色素を合成し、かつC−7位で置換されていない色素(例えば、ピロニン色素)の合成に適している別の方法において、色素は、O−保護してN−置換された3−ヒドロキシベンゾインドリン化合物から生成されるヒドロキシベンゾインドリン中間体から合成され、続いて、酸素保護基(例えば、メチル基)を脱保護し、脱メチル化試薬(例えば、塩化アルミニウム)によって脱保護し、そして順相クロマトグラフィーによる精製によって単離する。この合成に従って、色素分子の半分に対応するヒドロキシベンゾインドリン中間体を、最初にホルミル化剤(例えば、メチルホルムアニリド/POCl3)と反応し、その後、ホルミル化したヒドロキシベンゾインドリン中間体を、色素分子の半分に対応する異なる(または、同じ)ヒドロキシベンゾインドリン中間体と直接反応させる。この反応を、酸性脱水条件下(例えば、POCl3)で行い、そして例えば、120〜160℃に加熱して、粗製カルボン酸エステル誘導体化ベンゾピロニン色素が得られる。いくつかの場合において、メトキシベンゾインドリン中間体を、メチル基を脱保護する前にホルミル化し、次いで、第2当量のヒドロキシインドリンと反応させてピロニン色素を得る前に、このホルミル化したメトキシインドリン誘導体のメチル基を脱保護して、ホルミル化したヒドロキシベンゾインドリン中間体を得る。純粋な色素を、水性ワークアップおよび順相クロマトグラフィーの後に単離する。次いで、中間体色素のカルボン酸エステルを、酸(例えば、HBr)で加水分解して、水性ワークアップおよび順相クロマトグラフィーの後に遊離酸色素誘導体を得る。その後、N−フェニル置換色素を、スルホン化剤(例えば、ClSO3H)でスルホン化して、水性ワークアップおよび順相クロマトグラフィーの後に最終色素が得られ得る。
【0083】
(V.4 スルホン化ジアリールローダミン色素を組み込むエネルギー移動色素)
本発明は、式IIIabcのスルホン化ジアリールローダミン色素化合物を組み込むエネルギー移動色素化合物を含む。一般的に、本発明のエネルギー移動色素化合物は、第一の波長において光を吸収し、かつそれに応答して励起エネルギーを放射する、ドナー色素、このドナー色素によって放射された励起エネルギーを吸収し、かつそれに応答して第二の波長において蛍光を発し得る、アクセプター色素を含む。ドナー色素は、リンカーによってアクセプターに結合しており、このリンカーは、ドナー色素とアクセプター色素との間の効率的なエネルギー移動を促進するのに有効である(Lee「Energy−transfer dyes with enhanced fluorescence」、米国特許第5,800,996号; Lee「Energy−transfer dyes with enhanced fluorescence」、米国特許第5,945,526号;Mathies「Fluorescent labels and their use in separations」;米国特許第5,654,419号;Lee(1997)Nucleic Acids Res.25:2816−22)。あるいは、ドナー色素およびアクセプター色素は、基質上の異なる結合部位において標識され得る。例えば、オリゴヌクレオチドは、5’末端においてドナー色素で標識され得、3’末端においてアクセプター色素で標識され得る。ポリペプチドは、カルボキシル末端においてドナー色素で標識され得、内部システインまたはリジン側鎖においてアクセプター色素で標識され得る(Komoriya「Compositions for the detection of proteases in biological samples and methods of use thereof」、米国特許第5,605,809号)。本発明のエネルギー移動色素において、基質を標識する、少なくとも1つのドナー色素またはアクセプター色素は、スルホン化ジアリールローダミン色素化合物である。エネルギー移動色素を含有する他の色素は、任意の蛍光部分であり得、この蛍光部分は、スルホン化ジアリールローダミン色素化合物(フルオレセイン、ロドル(rhodol)、およびローダミンを含む)を用いてエネルギー移動プロセスを行う。他の色素としては、蛍光色素(例えば、シアニン、フタロシアニン、スクアライン(squaraine)、ボディピー(bodipy)、ベンゾフェノキサジン、フルオレセイン、ジアリールローダミン、またはローダミン)のクラスが挙げられる。
【0084】
エネルギー移動色素は、混合物中の複数の標識された基質を同時に検出(例えば、DNA配列決定)する際に使用するのに利点を有する。各色素が通常の波長において強力な吸収を有するように、単一のドナー色素が、エネルギー移動色素のセットにおいて使用され得る。次いで、エネルギー移動セットのアクセプター色素を変更することによって、アクセプター色素は、それらそれぞれの発光最大値によってスペクトルにより分解され得る。エネルギー移動色素はまた、非エネルギー移動色素よりも大きい有効なストークスシフトを提供する。このストロークシフトは、励起最大値(ドナー色素が最大に光を吸収する波長)と発光最大値(アクセプターが最大に光を発する波長)との間で異なる。
【0085】
一般的に、ドナー色素とアクセプター色素との間のリンカーは、以下の構造:
【0086】
【化34】
を有し、ここで、Zは、NH、SおよびOからなる群より選択され;R21は、ドナー色素に結合されたC1〜C12アルキルであり;R22は、C1〜C12アルキルジイル、少なくとも1つの不飽和結合を有する、5員環および6員環、ならびにカルボニル炭素に結合した縮合環構造からなる群より選択される置換基であり;そして、R23は、アクセプター色素にリンカーを結合している官能基を含む。R22は、5員環または6員環(例えば、シクロペンテン、シクロヘキセン、シクロペンタジエン、シクロヘキサジエン、フラン、チオフラン、ピロール、イソピロール、イソアゾール、ピラゾール、イソイミダゾール、ピラン、ピロン、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、オキサジン、インデン、ベンゾフラン、チオナフテン、インドールおよびナフタレン)であり得る。詳細には、このリンカーは、以下の構造:
【0087】
【化35】
を有し得、ここで、nは、2〜10の範囲である。
【0088】
一般的にまた、R23は、以下の構造:
【0089】
【化36】
を有し得、ここで、R24は、C1〜C12である。
【0090】
1つの実施形態において、ドナー色素とアクセプター色素との間のリンカーは、このリンカーにある程度の構造的な剛性を与える官能基(例えば、アルケン、ジエン、アルキン、少なくとも1つの不飽和結合を有する5員環および6員環、または縮合環構造)を含む(米国特許第5,821,356号;同第5,770,716号;同第5,948,648号;同第6,096,875号)。エネルギー移動色素のドナー色素およびアクセプター色素は、以下の例示的な構造:
【0091】
【化37】
を有するリンカーによって結合されており、ここで、Dはドナー色素であり、Aはアクセプター色素であり、nは1または2である。フェニル環は、スルホネート基、ホスホネート基、および他の荷電した基のような基で置換され得る。
【0092】
エネルギー移動色素のドナー色素とアクセプター色素との間のリンカーの結合部位は、ドナー色素およびアクセプター色素のうちの一方または両方が本発明の化合物である、任意の位置であり得る。例示的な結合部位としては、R2、R2’、R12およびR12’が挙げられる。
【0093】
エネルギー移動色素化合物は、リンカーによって基質に共有結合している。リンカーは、結合、C1〜C12アルキルジイルまたはC6〜C20アリールジイルであり得る。このリンカーは、アミド、カルバメート、尿素、チオ尿素、ホスフェート、ホスホネート、スルホネート、ホスホロチオエートなどを含む官能基を有し得る。例示的なリンカーとしては、1,2−エチルジイルおよび1,6−ヘキシジイルが挙げられる。エネルギー移動色素と基質との間のリンカーの結合部位は、ドナー色素およびアクセプター色素のうちの一方または両方が本発明のスルホン化ジアリールローダミン色素である、エネルギー移動色素上の任意の位置であり得る。基質がヌクレオシドまたはヌクレオチドである場合、エネルギー移動色素に対する1つの結合部位は、核酸塩基上にある。この核酸塩基がプリンである場合、リンカーは、8位で結合し得る。核酸塩基が7−デアザプリンである場合、リンカーは、7位または8位で結合し得る。核酸塩基がピリミジンである場合、リンカーは、5位で結合し得る。基質がオリゴヌクレオチドである場合、結合部位は、3’末端および5’末端を含む。他のオリゴヌクレオチド結合部位としては、ヌクレオチド間ホスフェートまたはホスフェートアナログ結合、あるいは、糖上の位置(2’または4’)が挙げられる。基質がポリペプチド(ペプチドまたはタンパク質)である場合、結合部位は、アミノ末端およびカルボキシル末端、ならびにリジン残基のアミノ基を含む。
【0094】
(V.5 標識方法)
本発明は、標識試薬を含み、ここで、スルホン化ジアリールローダミン化合物は、基質と反応するように反応性形態である(すなわち、連結部分を有する)。本発明はまた、本発明の化合物(式IIIabc)で標識された(すなわち、結合体化された)基質を含む。基質は、実質的に任意の分子であり得るか、または本発明の色素が結合体化され得る物質(例としては、ポリヌクレオチド、ヌクレオチド、ヌクレオシド、ポリペプチド、炭水化物、リガンド、実質的に鏡像異性的に純粋な化合物、粒子、表面、脂質、固体支持体、有機ポリマーおよび無機ポリマー、ならびにそれらの組み合わせおよび集合(例えば、染色体、核、生きた細胞(例えば、細菌または他の微生物、哺乳動物細胞、組織など))などが挙げられるがこれらに限定されない)であり得る。粒子としては、ナノ粒子、微粒子、ビーズ、またはリポソームが挙げられ得る。表面は、ガラスまたは他の非多孔性平面材料であり得る。本発明の化合物は、種々の手法(疎水性引力、イオン引力、および共有結合を含む)によって、必要に応じて、リンカーを介して基質に結合体化される。
【0095】
標識は、代表的には、当該分野で周知の結合体化方法(Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.pp.40−55,643−71)を使用して、連結部分、基質、および適切な溶媒を有するスルホン化ジアリールローダミンを混合することによって生じ、その後、標識された基質、結合体を、任意の非結合開始材料または望ましくない副産物から分離する。結合体は、後に使用するために、乾燥状態でまたは溶液中で保存され得る。
【0096】
スルホン化ジアリールローダミンは、置換基位置の1つで連結部分を含み得る。この連結部分は、代表的には、求電子性官能基であり、これは、基質上の求核性官能基と反応することによって共有結合を形成し得る。求核性官能基としては、例えば、アルコール、アルコキシド、アミン、ヒドロキシルアミン、およびチオールが挙げられ得る。あるいは、連結部分としては、基質上の求電子性基と反応する求核性官能基が挙げられ得る。連結基の例としては、アジド、一置換の第一級アミン、二置換の第二級アミン、チオール、ヒドロキシル、ハライド、エポキシド、N−ヒドロキシスクシンイミジルエステル、カルボキシル、イソチオシアネート、塩化スルホニル、スルホン酸エステル、シリルハライド、クロロトリアジニル、スクシンイミジルエステル、ペンタフルオロフェニルエステル、マレイミド、ハロアセチル、エポキシド、アルキルハライド、アリルハライド、アルデヒド、ケトン、アシルアジド、無水物、ヨードアセトアミドおよび活性化エステルが挙げられる。
【0097】
1つの連結部分は、スルホン化ジアリールローダミン化合物のカルボキシル基のN−ヒドロキシスクシンイミジルエステル(NHS)である(図14)。この化合物のNHSエステル形態は、標識試薬である。色素のNHSエステルは、予備形成され得、単離され得、精製され得、そして/または特徴付けされ得るか、あるいは、インサイチュで形成されて、基質の求核性基(例えば、オリゴヌクレオチド、ヌクレオシド、ヌクレオチド、ポリペプチドなど)と反応され得る(Brinkley,M.(1992)Bioconjugate Chem.3:2〜13)。代表的には、色素のカルボキシル形態は、以下の(1)〜(3)のいくつかの組み合わせと反応させることによって活性化され、色素のNHSエステル(例えば、図14の化合物82、化合物83、化合物84および図15の化合物85)が得られる:(1)カルボジイミド試薬(例えば、DCC(ジシクロヘキシルカルボジイミド)、DIPCDI(ジイソプロピルカルボジイミド))、またはウロニウム(uronium)試薬(例えば、TSTU(O−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート)、HBTU(O−ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート)、HATU(O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート));(2)活性化剤(例えば、1−ヒドロキシベンゾトリアゾール(HOBt));ならびに、(3)N−ヒドロキシスクシンイミド。
【0098】
いくつかの場合において、スルホン化ジアリールローダミン化合物および基質は、1つの工程において、化合物をインサイチュでの活性化し、そして基質と反応させて、スルホン化ジアリールローダミン−基質結合体を形成することによって結合され得る。他の活性化剤およびカップリング剤としては、以下が挙げられる:TBTU(2−(1H−ベンゾトリアゾール−1−イル)−1−1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)、TFFH(N,N’,N’’,N’’’−テトラメチルウロニウム2−フルオロヘキサフルオロホスフェート)、PyBOP(ベンゾトリアゾール−1−イル−オキシ−トリス−ピロリジニノ−ホスホニウムヘキサフルオロホスフェート)、EEDQ(2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロ−キノリン)、DCC、DIPCDI、MSNT(1−(メシチレン−2−スルホニル)−3−ニトロ−1H−1,2,4−トリアゾール)、およびアリールスルホニルハライド(例えば、塩化トリイソプロピルベンゼンスルホン酸)。
【0099】
別の反応性連結基は、スルホン化ジアリールローダミン化合物のホスホラミダイト形態である。ホスホラミダイト色素試薬は、本発明の色素で標識されるオリゴヌクレオチドの自動化合成のために、特に有用である。最も好都合なことに、ホスホラミダイト色素試薬は、固相合成の通常の過程の間に固体支持体に結合されたオリゴヌクレオチドに結合し得る。オリゴヌクレオチドは通常、ホスホラミダイト方法(Caruthers,M.およびBeaucage,S.「Phosphoramidite compounds and processes」、米国特許第4,415,732号;Caruthers,M.およびMatteucci,M.「Process for preparing polynucleotides」、米国特許第4,458,066号;Beaucage,S.およびIyer,R.(1992)「Advances in the synthesis of oligonucleotides by the phosphoramidite approach」、Tetrahedron 48:2223〜2311)によって、固体支持体上で合成される。
【0100】
ホスホラミダイトスルホン化ジアリールローダミン試薬は、ヌクレオシドまたは非ヌクレオシドであり得る。ホスホラミダイト試薬の非ヌクレオシド形態は、一般式IV:
【0101】
【化38】
を有し、ここで、DYEは、スルホン化ジアリールローダミンIIIabc(エネルギー移動色素を含む)の保護形態または非保護形態である。Lは、リンカーである。別個になっているR30およびR31は、C1〜C12アルキル、C4〜C10アリール、および10個までの炭素原子を含むシクロアルキルであるか、またはホスホラミダイト窒素原子と一緒になったR30およびR31は、飽和窒素複素環を形成し得る。R32は、オリゴヌクレオチドの望ましくない伸長を防止する、亜リン酸エステル保護基である。一般的に、R32は、オリゴヌクレオチド合成条件に対して安定であり、オリゴヌクレオチドまたは色素の完全性に悪影響を及ぼさない試薬を用いて、合成オリゴヌクレオチド生成物から除去され得る。R32は、以下であり得る:(i)メチル、(ii)2−シアノエチル;−CH2CH2CN、または(iii)2−(4−ニトロフェニル)エチル;−CH2CH2(p−NO2Ph)。ホスホラミダイト試薬の実施形態は、以下を含む:(i)R30およびR31は、各々イソプロピルである;(ii)一緒になったR30およびR31は、モルホリノである;(iii)Lは、C1〜C12アルキルである;(iv)R32は、2−シアノエチルである;そして(v)DYEは、リンカーによってR18またはR19に結合している。あるいは、リンカーLは、以下であり得る:
【0102】
【化39】
ここで、nは、1〜10の範囲である。ホスホラミダイト試薬IVの例は、以下の構造:
【0103】
【化40】
を有する。
【0104】
ホスホラミダイト色素試薬IVは、本発明のスルホン化ジアリールローダミン化合物での基質の標識に影響を与える。基質がオリゴヌクレオチドである場合、色素は、3’から5’方向への代表的な合成の結果として、オリゴヌクレオチドの5’末端で結合しているか、または5’から3’方向への合成方法が行われる場合、オリゴヌクレオチドの3’末端で結合している(Wagner(1997)Nucleosides & Nucleotides 16:1657−60)。試薬IVは、例えば、3’末端を介して固体支持体に結合しているポリヌクレオチドに連結され得る。ヌクレオシドおよび非ヌクレオシドの他のホスホラミダイト色素試薬により、オリゴヌクレオチドの他の部位(例えば、3’末端、核酸塩基、ヌクレオチド間結合、糖)での標識が可能になる。核酸塩基、ヌクレオチド間結合、および糖部位での標識により、蛍光色素での内部標識および複数標識が可能になる。
【0105】
本発明のスルホン化ジアリールローダミン化合物は、求核官能基を三価の亜リン酸化試薬で亜リン酸化(phosphitylation)する、任意の公知の方法によって、非ヌクレオシドのホスホラミダイト標識試薬に転換され得る。例えば、化合物がカルボキシル基(例えば、110〜120)を含む場合、カルボキシルは、例えば、NHSに対して活性化され得、そして、6−アミノ−1−ヘキサノールでアミド化され得る。得られたヒドロキルは、ビス(ジイソプロピルアミノ)シアノエチルホスファイトまたはクロロジイソプロピルアミノシアノエチルホスファイトで亜リン酸化されて、ホスホラミダイト色素標識試薬IVが得られ得る(Theisen(1992)「Fluorescent dye phosphoramidite labelling of oligonucleotides」、Nucleic Acid Symposium Series No.27,Oxford University Press,Oxford,99〜100頁)。あるいは、化合物のカルボキシル基は、ヒドロキシルに還元され、亜リン酸化され得る。
【0106】
ホスホラミダイト試薬IVは、穏やかな酸活性化の下でヒドロキシル基(例えば、固体支持体に結合したオリゴヌクレオチドの5’末端のOH)と反応して、ヌクレオチド間亜リン酸基(これは、次いで、ヌクレオチド間ホスフェート基に酸化される)を形成する。いくつかの例において、スルホン化ジアリールローダミン化合物は、官能基を含有し得、この官能基は、ホスホラミダイト試薬を合成する間または分子(例えば、オリゴヌクレオチド)を標識するために続いて使用する間のいずれかで、保護を必要とする。使用される保護基は、官能基の性質に依存し、そして当業者に明らかである(Greene,T.およびWuts,P. Protective Groups in Organic Synthesis,第2版、John Wiley & Sons,New York,1991)。一般的に、使用される保護基は、5’−ヒドロキシル保護基(例えば、ジメトキシトリチル)を除去するためにオリゴヌクレオチドの合成において通常使用される酸性条件下(例えば、トリクロロ酢酸、ジクロロ酢酸)で安定でなければならなず、かつ合成オリゴヌクレオチドを固体支持体から脱保護および/または切断するために使用される塩基性条件下(例えば、水酸化アンモニウム、水性メチルアミン)で不安定でなければならない。
【0107】
ポリペプチド、抗体、ならびにアミノ酸およびアミノ酸アナログから構成される他の生体高分子は、本発明のスルホン化ジアリールローダミン化合物との結合体化によって共有結合的に標識され得る。代表的に、この化合物は、求電子形態(例えば、NHS反応性連結基)であり、この化合物は、ペプチドの求核基(例えば、アミノ末端、またはアミノ酸(例えば、リジン)のアミノ側鎖)と反応する。あるいは、この色素は、求核形態(例えば、アミノ反応性連結基またはチオール反応性連結基)であり得、この色素は、ペプチドの求電子基(例えば、カルボキシル末端のNHSまたはアミノ酸のカルボキシル側鎖)と反応し得る。標識されたポリペプチドは、好ましくは、細胞表面および細胞内成分との相互作用の際にそれらの特異的な結合特性および認識特性を維持する。色素として作用するスルホン化ジアリールローダミン化合物により、結合事象または認識事象を局在化、可視化および定量化するための検出エレメントが提供される。ポリペプチドはまた、蛍光共鳴エネルギー移動(FRET)を一緒に行う、2つの部分(蛍光レポーターおよび消光剤)で標識され得る。この蛍光レポーターは、インタクトなポリペプチドの消光剤部分によって部分的にかまたは有意にクエンチされ得る。ペプチダーゼまたはプロテアーゼによってポリペプチドを切断する際に、蛍光の検出可能な増加が測定され得る(Knight,C.(1995)「Fluorimetric Assays of Proteolytic Enzymes」、Methods in Enzymology,Academic Press,248:18〜34)。
【0108】
(V.6 標識されたヌクレオチド)
1つのクラスの標識された基質は、本発明のスルホン化ジアリールローダミン化合物で標識される、ヌクレオシドおよびヌクレオチドの結合体を含む。このような標識されたヌクレオシドおよびヌクレオチドは、酵素学的合成によって形成されるポリヌクレオチド(例えば、PCR増幅、サンガー型ポリヌクレオチド配列決定、およびニック翻訳反応の状況において使用される、標識されたヌクレオチドの5’トリホスフェート)を標識するために、特に有用である。
【0109】
ヌクレオシドおよびヌクレオチドは、糖部分または核酸塩基部分上の部位で標識され得る。核酸塩基標識部位としては、プリン核酸塩基の8−C、7−デアザプリン核酸塩基のC−7またはC−8、およびピリミジン核酸塩基の5位が挙げられる。ヌクレオチシドまたはヌクレオチドと色素との間で、リンカーは、任意の位置において、スルホン化ジアリールローダミン化合物に結合し得る。
【0110】
標識されたヌクレオシドまたはヌクレオチドは、酵素学的に組み込み可能かつ酵素学的に伸長可能であり得る。本発明の化合物で標識されるヌクレオシドまたはヌクレオチドは、以下の式V:
【0111】
【化41】
を有し得、ここで、DYEは、化合物IIIabcの保護形態または非保護形態(エネルギー移動色素を含む)である。Bは、任意の核酸塩基(例えば、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および8−デアザグアノシン)であり得る。R25は、H、モノホスフェート、ジホスフェート、トリホスフェート、チオホスフェート、またはリン酸エステルアナログである。R26およびR27は、単独である場合、各々独立して、H、HO、Fおよびホスホラミダイトである。R26またはR27がホスホラミダイトである場合、R25は、自動化合成条件下で後のモノマーカップリングを可能にする、酸で切断可能なヒドロキシル保護基(例えば、ジメトキシトリチル)である(米国特許第4,415,732号および同第4,458,066号;Beaucage,S.and Iyer,R.(1992)「Advances in the synthesis of oligonucleotides by the phosphoramidite approach」,Tetrahedron 48:2223〜2311)。
【0112】
標識されたヌクレオシまたはヌクレオチドがターミネーターである場合、R26およびR27は、ポリメラーゼ媒介テンプレート指向性重合を遮断するように選択される。ターミネーターヌクレオチドにおいて、R26およびR27は、単独である場合、各々独立して、H、F、およびポリメラーゼ媒介テンプレート指向性重合を遮断する部分であるか、または一緒になる場合、2’−3’−ジデヒドロリボースを形成する。式Vにおいて、R26およびR27の両方がヒドロキシルである場合、得られる化合物は、標識されたリボヌクレオシドまたはリボヌクレオチド(NTP)である。R27が水素であり、かつR26がヒドロキシルである場合、得られる化合物は、標識された2’−デオキシリボヌクレオシドおよび2’−デオキシリボヌクレオチド(dNTP)である。R26およびR27が各々水素である場合、得られる化合物は、2’,3’−ジデオキシリボヌクレオシドおよび2’,3’−ジデオキシリボヌクレオチド(ddNTP)である。標識されたddNTPは、蛍光検出を使用するサンガー型DNA配列決定方法におけるターミネーターとしての特定の用途が見出される。標識された2’−デオキシリボヌクレオシド−5’−トリホスフェート(dNTP)は、DNAポリメラーゼ伸長産物を標識するための試薬としての特定の用途(例えば、ポリメラーゼ連鎖反応またはニック翻訳において)が見出される。標識されたリボヌクレオシド−5’−トリホスフェート(NTP)は、RNAポリメラーゼ伸長産物を標識するための試薬としての特定の用途が見出される。
【0113】
【化42】
アルキニルアミノ連結化合物V(ここで、Lは、アルキンジイル(alkyndiyl)基を含む)は、スルホン化ジアリールローダミン化合物をヌクレオシド、ヌクレオチドおよびそれらのアナログに結合体化するのに有用である。これらの合成は、EP 87305844.0およびHobbs,(1989)J.Org.Chem.54:3420に教示されている。対応するヌクレオシドのモノホスフェート、ジホスフェートおよびトリホスフェートは、標準技術(例えば、米国特許第5,821,356号;同第5,770,716号;同第5,948,648号;同第6,096,875号に記載される方法)によって得られる。化合物Vを改変したプロパルギルエトキシアミド(propargylethoxyamido)リンカーLを用いて合成するための方法もまた、これらの特許において見出され得る。構造式Vに従う化合物の合成に使用するのに適切な、さらなる合成手順が、例えば、以下に記載される:Gibson(1987)Nucl.Acids Res.15:6455〜6467;Gebeyehu(1987)Nucl.Acids Res.15:4513〜4535;Haralambidis(1987)Nucl.Acids Res.15:4856〜4876;Nelson(1986)Nucleosides and Nucleotides.5(3):233〜241;Bergstrom(1989)J.Am.Chem.Soc.111:374〜375;米国特許第4,855,225号;同第5,231,191号および同第5,449,767号(これらは、本明細書中で参考として援用される)。これらの方法のいずれかは、全範囲の標識されたヌクレオシド、ヌクレオチド、および本明細書中に記載されるアナログを合成するために必要な方法として、日常的に適用または変更され得る。
【0114】
アルキニルリンカーLの1つの実施形態は、以下:
【0115】
【化43】
であり得、ここで、nは、0、1または2である。
【0116】
エネルギー移動色素対は、核酸塩基アミノ基を介して、以下:(i)エネルギー移動色素対の活性化エステルに連結するか、または(ii)1つの色素(例えば、R11保護アミノメチル、R18カルボキシルフルオレセイン)にカップリングし、次いで、非保護R11アミノメチルを対の第二色素にカップリングして、段階的に連結することによって、ヌクレオチドの5’トリホスフェートに結合体化され得る。
【0117】
異なる輻合性スキームを用いた、エネルギー移動ヌクレオチドおよびポリヌクレオチドに対する代替的な合成経路が、用いられ得る。基質、色素およびリンカーサブユニット、またはシントンが、任意の順序でカップリングするために構築され得る。例えば、ドナー色素およびアクセプター色素のエネルギー移動対は、リンカーによって共有結合され得、次いで、ヌクレオチドまたはポリヌクレオチドにカップリングされ得る。本発明の色素でのヌクレオチドの標識を生じる、多数の異なる合成経路が、用いられ得る。反応性官能基(例えば、カルボン酸基、アミノ基、ヒドロキシル基)は、広範な有機合成分野の方法論を使用する際に、保護を必要とし得る。
【0118】
(V.7 標識されたオリゴヌクレオチド)
オリゴヌクレオチドは、市販のホスホラミダイトヌクレオシド、支持体(例えば、シリカ)、制御された有孔ガラス(米国特許第4,458,066号)およびポリスチレン(米国特許第5,047,524号および同第5,262,530号)および自動化合成機(Models 392,394,3948 DNA/RNA Synthesizers,Applied Biosystems)を使用するホスホラミダイト方法(米国特許第4,415,732号;同第4,973,679号;同第4,458,066号;Beaucage,S.およびIyer,R.(1992)Tetrahedron 48:2223〜2311)によって、通常、固体支持体上に合成される。
【0119】
別のクラスの標識された基質としては、オリゴヌクレオチドと本発明の化合物との結合体が挙げられる。このような結合体は、DNA配列決定プライマー、PCRプライマー、オリゴヌクレオチドハイブリダイゼーションプローブ、オリゴヌクレオチドライゲーションプローブ、二重標識5’エキソヌクレアーゼ(TaqManTM)プローブ、電気泳動のためのサイズ標準(すなわち、「レーン標準」または「レーンマーカ」)などとしての有用性が見出され得る(Fung、米国特許第4,757,141号;Andrus「Chemical methods for 5’non−isotopic labelling of PCR probes and primers」(1995)、PCR 2:A Practical Approach,Oxford University Press,Oxford,39〜54項;Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.40〜55項,643−71;Mullah(1998)「Efficient synthesis of double dye−labelled oligodeoxyribonucleotide probes and their application in a real time PCR assay」Nucl.Acids Res.26:1026〜1031)。標識されたオリゴヌクレオチドは、以下の式VI:
【0120】
【化44】
を有し得、ここで、このオリゴヌクレオチドは、2〜1000ヌクレオチドを含む。DYEは、化合物Iまたは化合物IIの保護形態または非保護形態(エネルギー移動色素を含む)である。Bは、任意の核酸塩基(例えば、ウラシル、チミン、シトシン、アデニン、7−デアザアデニン、グアニン、および8−デアザグアニシン)である。Lは、リンカーである。R27は、H、OH、ハライド、アジド、アミン、C1〜C6アミノアルキル、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3、またはOCH2CH=CH2である。R22は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログである。R29は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログである。この実施形態において、核酸塩基で標識されたオリゴヌクレオチドVIは、核酸塩基によって結合された本発明の複数の色素を有し得る。この核酸塩基で標識されたオリゴヌクレオチドVIは、以下によって形成され得る:(i)DNAポリメラーゼまたはリガーゼによる、酵素学的に組み込み可能なヌクレオチド試薬Vの酵素学的組み込み(ここで、R25は、トリホスフェートである)、および(ii)自動化合成によるヌクレオシドホスホラミダイト試薬のカップリング。これによって、核酸塩基で標識されたオリゴヌクレオチドVIは、1つより多くの組み込み可能なヌクレオチドVの組み込みによって複数標識され得、1つずつ5’標識されたオリゴヌクレオチドを導く、以下の式VII:
【0121】
【化45】
に従う色素標識試薬(例えば、IV)で標識され、ここで、Xは、O、NHまたはSであり;R27は、H、OH、ハライド、アジド、アミン、C1〜C6アミノアルキル、C1〜C6アルキル、アリル、C1〜C6アルコキシ、OCH3またはOCH2CH=CH2であり;R28は、H、ホスフェート、ヌクレオチド間ホスホジエステル、またはヌクレオチド間アナログであり;そして、Lは、C1〜C12アルキル、アリール、または100までのエチレンオキシユニットのポリエチレンオキシである。
【0122】
式VIまたはVII中のリンカーLは、任意の部位にて、本発明のスルホン化ジアリールローダミン化合物IIIabc(DYE)に結合され得る。
【0123】
合成オリゴヌクレオチドを標識するための第一の方法において、求核官能基(例えば、第一級脂肪族アミン)は、オリゴヌクレオチド上の標識結合部位(例えば、5’末端)に導入される。自動化された固体支持体合成が終了した後、オリゴヌクレオチドが、この支持体から切断され、そして全ての保護基が除去される。求核オリゴヌクレオチドは、求電子部分(例えば、イソチオシアネートまたは活性化エステル(例えば、N−ヒドロキシスクシンイミド(NHS)))を含有する過剰な標識試薬と、均一溶液条件下で反応される(Hermanson,Bioconjugate Techniques,(1996)Academic Press,San Diego,CA.pp.40−55,643−71;Andrus,A.「Chemical methods for 5’non−isotopic labelling of PCR probes and primers」(1995)、PCR 2:A Practical Approach,Oxford University Press,Oxford,39〜54頁)。標識されたオリゴヌクレオチドVIIは、色素の反応性連結基形態(例えば、NHS)を、5’−アミノアルキルオリゴヌクレオチドと反応させることによって形成され得る(米国特許第4,757,141号)。
【0124】
第二の方法において、標識は、自動化合成の間または自動化合成前に、例えば、支持試薬として(Mullah,「Solid support reagents for the direct synthesis of 3’−labelled polynucleotides」、米国特許第5,736,626号;Nelson,「Multifunctional controlled pore glass reagent for solid phase oligonucleotide synthesis」、米国特許第5,141,813号)、またはホスホラミダイト試薬IVとして、オリゴヌクレオチドに直接組み込まれる。特定の蛍光色素および他の標識は、5’標識のためのホスホラミダイト試薬として機能化される(Theisen(1992)Nucleic Acid Symposium Series No.27,Oxford University Press,Oxford,99〜100頁)。
【0125】
一般的に、標識されたオリゴヌクレオチドが酵素学的合成によって作製される場合、以下の手順が使用され得る。標的DNAは、変性され、そしてオリゴヌクレオチドプライマーは、テンプレートDNAにアニーリングされる。初回刺激された標的(例えば、dGTP、dATP、dCTPおよびdTTPまたはdUTPを含む混合物)の連続的なテンプレート指向性酵素学的伸長を支持し得る酵素学的に組み込み可能なヌクレオチドまたはヌクレオチドアナログの混合物が、この初回刺激された標的に添加される。ヌクレオチドの少なくとも1つの画分は、標識されたターミネーターV(例えば、ddNTPまたは3’F−dNTP)であり、これは、スルホン化ジアリールローダミン色素IIIabcで標識される。次に、ポリメラーゼ酵素は、このポリメラーゼ酵素が活性である条件下でこの混合物に添加される。標識されたオリゴヌクレオチドは、ポリメラーゼ媒介性鎖合成の間に、標識されたヌクレオチドまたはターミネーターの組み込みによって形成される。代替的な酵素学的合成方法において、2つのプライマー(標的の(+)鎖に相補的なプライマーおよび標的の(−)鎖に相補的な別のプライマー)
が、代わりに使用され、ポリメラーゼは、熱安定性ポリメラーゼであり、反応温度は、変性温度と伸長温度との間を循環し、それによって、標的配列に対する標識された相補体がPCRによって指数関数的に合成される(Innis(1990)PCR Protocols,編、Academic Press)。
【0126】
本発明のスルホン化ジアリールローダミン化合物で標識されたポリヌクレオチドは、求電子遊走速度に影響を与える部分(すなわち、移動度変更標識)でさらに標識され得る。移動度変更標識は、ポリエチレンオキシユニット、−(CH2CH2O)n−を含み、ここで、nは、1〜100であり得(米国特許第5,624,800号)、好ましくは、nは、2〜20である。ポリエチレンオキシユニットは、ホスフェート基で引き離され得る。別個でありかつ公知のサイズであるポリエチレンオキシのさらなる標識を伴う、特異的に標識されたスルホン化ジアリールローダミン標識ポリヌクレオチドによって、ポリヌクレオチド中のヌクレオチドの数に実質的に非依存性の電気泳動による分離が可能になる。すなわち、同じ長さのポリヌクレオチドは、スペクトルにより分解可能な色素標識および移動度変更標識の存在によって識別され得る。色素標識と移動度変更標識との両方を有するポリヌクレオチドは、単一標識されたポリヌクレオチドまたはヌクレオチド構成成分のライゲーションまたはポリメラーゼ伸長によって酵素学的に形成され得る。
【0127】
(V.8 スルホン化ジアリールローダミン色素を使用する方法)
スペクトルにより重複した複数の分析物を同時検出するのに必要な方法は、標識としてのスルホン化ジアリールローダミン色素から利益を獲得し得る。本発明のスルホン化ジアリールローダミン化合物は、以下のような蛍光検出を使用する任意の方法に十分適している:ポリメラーゼ連鎖反応(PCR)増幅、DNA配列決定、遺伝子発現のアンチセンス転写性制御および翻訳制御、遺伝分析、およびDNAプローブベースの診断試験(Kricka,L.(1992)Nonisotopic DNA Probe Techniques,Academic Press,San Diego,3〜28頁)。蛍光色素により標識されたオリゴヌクレオチドの蛍光検出は、以下のような核酸配列検出アッセイに基づく:5’エキソヌクレアーゼアッセイ(Livak,米国特許第5,723,591号)、FRETハイブリダイゼーション(Tyagi,S.およびKramer,F.(1996)「Molecular Beacons:Probes that fluoresce upon hybridization」、Nature Biotechnology,14:303−08)、遺伝連鎖マッピング(Dib(1996)「A comprehensive genetic map of the human genome based on 5,264 microsatellites」Nature 380:152−54)、およびオリゴヌクレオチドライゲーションアッセイ(Grossman(1994)「High−density multiplex detection of nucleic acid sequences: oligonucleotide ligation assay and sequence−coded separation」Nucl.Acids Res.22:4527−34)。
【0128】
本発明は、特に、差次的に標識されたポリヌクレオチドのクラスを検出するのに非常に適しており、これらのポリヌクレオチドは、生化学的分離手順(例えば、電気泳動(RickwoodおよびHames、編、Gel Electrophoresis of Nucleic Acids:A Practical Approach,IRL Press Limited,London,1981))に供される。電気泳動マトリクスは、シービング(sieving)ポリマー(例えば、架橋もしくは非架橋のポリアクリルアミド、または約2〜20重量%の間の濃度(重量/体積)を有する他のアミド含有ポリマー)であり得る(Madabhushi,米国特許第5,552,028号;同第5,567,292号;同第5,916,426号)。この電気泳動マトリクスは、スラブゲルまたはキャピラリーフォーマットで構成され得る(Rosenblum,(1997)Nucleic Acids Res.25:3925−29;Mathies,米国特許第5,274,240号)。
【0129】
電気泳動分離の後、色素−ポリヌクレオチド結合体は、例えば、高輝度照射の水銀放電灯、レーザなどによって、色素標識ポリヌクレオチドからの蛍光発光を測定することによって検出され得る。この照射手段は、約600nmより大きい波長の照射ビームを有するレーザであり得る。また、この色素−ポリヌクレオチドは、He−Neガスレーザまたは固体状態のダイオードレーザによって生じるレーザ光によって照射され得る。次いで、蛍光が、光感受性検出器(例えば、光電子増倍管、荷電結合デバイス(charged coupled device)など)によって検出される。例示的な電気泳動検出システムは、他の箇所(例えば、米国特許第5,543,026号;同第5,274,240号;同第4,879,012号;同第5,091,652号および同第4,811,218号)に記載される。
【0130】
(V.8A プライマー伸長)
「フラグメント分析」方法または「遺伝子分析」方法として本明細書中で参照される方法の1つのカテゴリーにおいて、蛍光色素(スルホン化ジアリールローダミン化合物を含む)で標識されたポリヌクレオチドフラグメントは、例えば、ライゲーションまたはポリメラーゼ指向性プライマー伸長によって、標識されたプライマーまたはヌクレオチドを使用するテンプレート指向性酵素学的合成によって生じる。プライマーオリゴヌクレオチドは、標的配列との相補的な塩基対形成によってハイブリダイズし、プライマー−標的ハイブリッドを形成する。プライマー伸長産物は、ハイブリッド中のプライマーの3’末端におけるヌクレオチドの酵素学的組み込みによって生じる。プライマーまたはヌクレオチドが本発明のスルホン化ジアリールローダミン色素で標識される場合、標識されたプライマー伸長産物が生じる。これらのポリヌクレオチドフラグメントは、サイズ依存性分離プロセス(例えば、電気泳動またはクロマトグラフィー)に供され得、分離されたフラグメントは、例えば、レーザ誘導化蛍光(Hunkapiller,米国特許第4,811,218号)によって分離後に検出される。複数のクラスのポリヌクレオチドが、同時に分離され得、そして種々のクラスが、スペクトルにより分解可能な標識(本発明の色素を含む)によって識別される。電気泳動において、これらのクラスは、電気泳動遊走速度に基づいて分離する。
【0131】
(V.8B DNA配列決定)
スルホン化ジアリールローダミン化合物は、複数色の蛍光検出が可能である、自動化4色DNA配列決定システムにおける使用に十分適している。このシステムは、約630nmより大きい波長を有する励起光源(例えば、ヘリウム−ネオンガスレーザまたは固体状態のダイオードレーザ)を使用し得る。
【0132】
DNA配列決定(すなわち、ジデオキシDNA配列決定、またはサンガー型配列決定)の連鎖停止方法、およびフラグメント分析が、使用され得る(Sanger(1977)「DNA sequencing with chain−terminating inhibitors」Proc.Natl.Acad.Sci.USA 74:5463〜5467)。例示的な連鎖停止ヌクレオチドアナログは、2’,3’−ジデオキシヌクレオチド5’−トリホスフェート(ddNTP)(これは、3’〜5’DNA鎖伸長に必要な3’−OH基を欠失している)を含む。プライマーまたはddNTPは、本発明のスルホン化ジアリールローダミン色素で標識され得、そして高分解能電気泳動によってフラグメントを分離した後に、蛍光によって検出され得る。色素は、プライマーの5’末端上の官能基(例えば、アミノ(米国特許第4,757,141号))、オリゴヌクレオチドプライマーの核酸塩基上の官能基;またはジデオキシヌクレオチドの核酸塩基上の官能基(米国特許第5,770,716号;同第5,821,356号;同第5,151,507号)に連結され得る。
【0133】
ターミネーターの各々は、異なる蛍光色素を有し、かつこの実験のターミネーターは、集合的に、特異的に分離可能な蛍光標識のセット(1種以上の本発明のスルホン化ジアリールローダミン色素を含む)を有する。例示的なフラグメント分析方法において、色素で標識されたフラグメントは、電気泳動条件下で相対サイズ(すなわち、配列の長さ)によって同定される。フラグメントサイズと配列との間の対応は、4つの可能な終結ヌクレオチド(「ターミネーター」)およびスペクトルにより分解可能な色素のセットのメンバーの組み込みによって確立される(米国特許第5,366,860号)。スペクトルにより分解可能な色素のセットとしては、少なくとも1つのスルホン化ジアリールローダミン化合物が挙げられる。十分に分解された発光を伴う4つのスルホン化ジアリールローダミン色素の1つのセットは、110、114、115および119からなる(図22a、22b、22c)。別のセットは、110、114、116および119からなる。さらに別のセットは、110、114、119および120からなる。色素のセットは、色素110〜120のいずれかの組み合わせ、または一般的に、任意のスルホン化ジアリールローダミン色素を含み得る。1つのセットは、1種以上のスルホン化ジアリールローダミン色素および他の構造クラス由来の1種以上の色素(例えば、フルオレセイン、非スルホン化ジアリールローダミン、シアニン、スクアラニンなど)を含み得る。
【0134】
標識されたヌクレオチドVは、4つの核酸塩基(B=A、G、C、T(またはU))およびそれらのアナログを含む4つのターミネーターのセットとして、配列決定する際に使用され得る。B、リンカーLおよびDYEの組み合わせは、ポリヌクレオチドフラグメントの電気泳動移動度、スペクトル分解能、およびプライマー伸長の間のポリメラーゼの組み込み速度に影響を及ぼす。
【0135】
(V.8C ライゲーション)
リガーゼ酵素による核酸プローブの共有結合は、分子生物学者に利用可能な最も有用なツールの1つである。2つのプローブがテンプレート核酸にアニーリングされる場合(ここで、これら2つのプローブは隣接しており、かつ介在性ギャップがない)、リガーゼ酵素によって、一方のプローブの5’末端ともう一方のプローブの3’末端との間に、ホスホジエステル結合が形成され得る(Whiteley、米国特許第4,883,750号;Landegren,(1988)「A ligase mediated gene detection technique」、Science 241:1077−80;Nickerson,「Automated DNA diagnostics using an ELISA−based oligonucleotide assay」(1990)Proc.Natl.Acad.Sci USA 87:8923−27)。オリゴヌクレオチドライゲーションアッセイは、標的DNAサンプル中の特定の配列の存在を検出する。一方または両方のプローブが色素で標識される場合、ライゲーション産物が蛍光によって検出され得る。一方または両方のプローブは、スルホン化ジアリールローダミン色素で標識され得る。ライゲーション産物は、電気泳動、クロマトグラフィー、または他のサイズに基づく分離方法もしくは電荷に基づく分離方法によって検出され得る。
【0136】
(V.8D 増幅)
本発明のスルホン化ジアリールローダミン化合物は、ポリメラーゼ連鎖反応(PCR)ならびに他の核酸増幅方法および選択方法のための、5’標識されたオリゴヌクレオチドプライマー上の標識としての用途が見出されている。PCR増幅は、種々の可変数のタンデム反復(VNTR)、短タンデム反復(STR)、および二重鎖DNA(特定の配列の隣接した複数のコピーを含む)の反復領域を増幅するマイクロサテライト法(microsatellite method)によって遺伝子型を決定する(genotyping)のための、標識されたオリゴヌクレオチドの使用を含み、反復単位の数は、可変である。このようなPCR遺伝子型決定方法において、PCRプライマーは、本発明のスルホン化ジアリールローダミンで標識され得る。
【0137】
1つの実施形態において、スルホン化ジアリールローダミン化合物は、定量方法およびPCRの間の増幅産物の実時間測定または終点測定を提供する試薬において使用され得る(米国特許第5,210,015号;同第5,538,848号)。蛍光色素−消光剤プローブを使用するエキソヌクレアーゼアッセイ(Taqman(登録商標))(米国特許第5,723,591号;Mullah(1998)「Efficient synthesis of double dye−labelled oligodeoxyribonucleotide probes and their application in a real time PCR assay、Nucl.Acids Res.26:1026〜1031)は、閉管システムにおいてポリメラーゼ連鎖反応(PCR)産物の直接検出を与え、PCRを行うのに必要とされるサンプル以上のサンプル処理を伴わない。Taqman(登録商標)アッセイにおいて、プライマー伸長を行いポリヌクレオチドを増幅するポリメラーゼはまた、5’〜3’エキソヌクレアーゼ活性によって標的配列にアニーリングされたプローブを置換および切断する。Taqman(登録商標)型アッセイにおいて、プローブは、自己消光し、蛍光色素および消光剤部分で標識され、これらのいずれかは、本発明の色素であり得る。スペクトルの重複によって、プローブがインタクトである場合に、効率的なエネルギー移動(FRET)が可能となる(Clegg(1992)「Fluorescence resonance energy transfer and nucleic acids」、Meth.Enzymol.211:353〜388)。標的配列にハイブリダイズされる場合、プローブはPCRの間に切断されて、標的プローブハイブリッドの存在量に比例した蛍光シグナルが放射される米国特許第5,538,848号;同第5,723,591号)。
【0138】
増幅の進行は、連続的にモニタリングされ得る(すなわち、実時間検出)。スペクトルにより分解可能な、本発明のスルホン化ジアリールローダミン色素は、標的をPCR増幅した後の遺伝子型決定実験において有用である。詳細には、各々異なる色素を用いて5’末端で標識されたプライマーオリゴヌクレオチドのセットは、複数の遺伝子座を増幅し、そして一塩基多型(SNP)および対立遺伝子を識別し得る。色素で標識された増幅産物の、サイズ基準を用いた電気泳動分離は、プライマー配列のセットに依存した特定の遺伝子型を示す、プロファイル、すなわち特徴的データセットを確立する。
【0139】
(V.8E ハイブリダイゼーションアッセイ)
標的核酸にハイブリダイズする特定の蛍光色素−消光剤プローブは、ハイブリダイゼーションアッセイにおいて有用である。プローブが標的にハイブリダイズされない場合、このプローブは、蛍光色素と蛍光消光を生じる消光剤部分との間を空間的に近位にするコンホメーションを維持し得る。標的へのハイブリダイゼーションの際に、この部分が物理的に分離され、消光が中断または減少され、そして蛍光が増加する。蛍光が検出可能であるかまたは定量化される場合、サンプル中の標的配列の存在が推定される。本発明のスルホン化ジアリールローダミン色素はまた、蛍光色素または消光剤部分として使用され得る。「ヘアピン」領域(いわゆる、「分子ビーコン(Molecular beacons)」(Tyagi and Kramer))を形成する自己相補的配列を有する蛍光色素−消光剤プローブは、それらの相補的な標的配列へのハイブリダイゼーション(例えば、生存細胞中のmRNAのインサイチュでの定量化)の際に蛍光変化を起こす。異なる蛍光色素で標識されたハイブリダイゼーションプローブ(本発明のスルホン化ジアリールローダミン色素を含む)によって、多重化した均一なハイブリダイゼーションアッセイが密閉された反応管中で行われるのが可能になる。
【0140】
ハイブリダイゼーションアッセイはまた、予め決定された位置において固定化されたプローブの固相アレイ上で実施され得、これらの位置は、これらの特異的な試薬の送達またはインタロゲーション/検出によってアドレス可能であり得る。例えば、スルホン化ジアリールローダミン色素で標識された標的ポリヌクレオチドを含有するサンプルは、固定されたプローブのアレイに送達され得る。ポリヌクレオチドが、塩基対によって固定されたプローブにハイブリダイズする場合、その位置での標的−プローブ複合体からの蛍光が検出され得る。蛍光検出パターンおよび/または強度から、ポリヌクレオチドの配列が推定され得、そして、サンプル中の特定のポリヌクレオチド配列の存在が決定され得る。
【0141】
(V.9 キット)
本発明はキットを含み、このキットは、本発明のスルホン化ジアリールローダミン化合物および/またはそれらの標識された結合体を備える。1つの実施形態において、このキットは、連結部分を有するスルホン化ジアリールローダミン化合物を別の分子(例えば、基質)に結合体化するために有用である。このようなキットは、一般に、色素を別の分子または基質に結合体化するのに適切な、本発明のスルホン化ジアリールローダミン(任意の連結部分および試薬、酵素、緩衝液、溶媒などを含む)を備える。スルホン化ジアリールローダミンは、エネルギー移動色素のアクセプターまたはドナーであり得る。
【0142】
1つの実施形態において、このキットは、酵素学的に合成されたオリゴヌクレオチドおよびポリヌクレオチドを、本発明のスルホン化ジアリールローダミンで標識するために有用である。このようなキットは、一般に、本発明に従う、酵素学的に組み込み可能な、標識されたクレオチドまたはヌクレオチドアナログ、連続したプライマー伸長およびポリメラーゼ酵素を支持し得る、酵素学的に組み込み可能なヌクレオチドまたはヌクレオチドアナログの混合物を備える。酵素学的に組み込み可能な、標識されたヌクレオチドまたはヌクレオチドアナログは、構造Vに従う化合物(例えば、標識されたターミネーター)である。ポリメラーゼは、熱安定性であり、これらは、例えば、AMPLITAQ(登録商標)DNAポリメラーゼFS(Applied Biosystems,Foster City,CA)である。
【0143】
あるいは、このキットは、1つ以上のオリゴヌクレオチドプライマーを備え得る。このプライマーは、スルホン化ジアリールローダミンおよびスルホン化ジアリールローダミンを含むエネルギー移動色素で標識され得る。
【0144】
(VI. 実施例)
本発明は、以下の実施例を考慮することによってさらに明瞭になり、これらの実施例は、本発明の単なる例示であり、いかなる様式においても本発明の範囲を制限しないことが意図される。
【0145】
(実施例1)
(1−ジエチルアミノ−3−ヒドロキシナフタレン4の合成(図1))
K.H.BellおよびL.F.McCaffery(1993),Aust.J.Chem.46:731の方法によって、1,3−ジヒドロキシナフタレンから合成した3−メトキシ−1−ヒドロキシナフタレン1(1gm)を、無水CH2Cl2(30ml)中に懸濁した。乾燥トリエチルアミン(1.2当量)を加え、この反応物を−5℃まで冷却した。CH2Cl2(15ml)中に懸濁したトリフルオロメタンスルホン酸無水物(1.1当量)を、激しく攪拌しながら2時間にわたって滴下した。この反応物を室温にし、5% HClおよびCH2Cl2を使用する水性ワークアップ(aqueous work up)に供した。得られた粗製3−メトキシナフタレン−1−トリフレート2を、移動相としてEtOAc/ヘキサン(1:10)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0146】
精製した3−メトキシナフタレン−1−トリフレート2を、J.P.WolfeおよびS.L.Buchwald(1996)Jour.Org.Chem.61:1133のパラジウムで触媒したトリフレート/アミンカップリング手順を使用して、1−ジエチルアミノ−3−メトキシナフタレン3に転換した。この3−メトキシ−ナフタレン−1−トリフレート2(1g)を、100mlの乾燥トルエン(0.015当量の(S)−(−)−2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフトル(BINAP)、0.005当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、および3当量の無水ジエチルアミンを含む)中に懸濁した。この反応物を、アルゴンでパージし、そして、3.3当量の固体ナトリウムt−ブトキシドを、攪拌しながら加えた。次いで、この反応物を加熱し、そして、油浴中で80℃にて16時間攪拌した。この反応物を室温にし、5%HClおよびCH2Cl2を使用する水性ワークアップに供して、粗製1−ジエチルアミノ−3−メトキシナフタレン3を得、これを、移動相としてEtOAc:ヘキサン(1:49)を使用する順相フラッシュクロマトグラフィーによって精製した(1HNMR:CDCl3 d 8.20(ブロードなd,1H,J=9Hz),7.72(ブロードなd,1H,J=7.8Hz),7.43(dt,1H,J=7.2,1.2Hz),7.34(dt,1H,J=7.7,1.2Hz),6.88(d,1H,J=2.4Hz),),6.82(d,1H,J=2.4Hz),3.93(s,3H),3.21(q,4H,J=7.2Hz),1.08(t,6H,J=7.2Hz))。
【0147】
次に、1−ジエチルアミノ−3−メトキシ−ナフタレン3のメチル基を、以下のような三臭化ホウ素脱保護によって除去した。1−アミノ−3−メトキシナフタレン(100mg)を、無水CH2C12(5ml)に懸濁し、そして、この混合物を、ドライアイス/アセトン浴中で−70℃まで冷却した。三臭化ホウ素(10当量)を滴下し、そしてこの反応物を、30分間攪拌し、次いで、冷蔵庫内(0℃)に一晩置いた。慎重にMeOH(10ml)を加えることによって、この反応物を−70℃にてクエンチした。固体NaHCO3(30当量)を加え、そして、この反応物を室温まで温め、次いで、手短に加熱還流した。この混合物を冷却し、そして濾過し、この濾液をAcOHで酸性化し、そして減圧下で溶媒を除去して、粗製1−ジエチルアミノ−3−ヒドロキシナフタレン4を得、これを、移動相としてEtOAc:ヘキサン(1:4)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0148】
(実施例2)
(N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン9の合成(図1))
上記実施例1のパラジウムで触媒したトリフレート/アミンカップリング反応に従って、アニリンを用いて、3−メトキシナフタレン−1−トリフレート2を誘導体化して、1−アニリノ−3−メトキシナフタレン5を得た。
【0149】
この1−アニリノ−3−メトキシナフタレン5を、以下のようなアミノ基のアセチル化手順によってアセチル化した。この1−アミノ−3−メトキシナフタレン5(500mg)および1.2当量の無水Et3Nを、10mlの無水CH2Cl2中に懸濁し、氷/NaCl浴を使用して−5℃まで冷却した。1.1当量の2−ブロモ−2−塩化メチルプロピニルを滴下し、そしてこの反応物を、−5℃にて1時間攪拌し、そして室温にてさらに1時間攪拌した。この反応物を室温にし、そして、5% HClおよび酢酸エチルを使用する水性ワークアップに供し、粗製中間体1−(ブロモアルキル)アミド−3−メトキシナフタレン6を得、これを、移動相としてEtOAc:ヘキサン(1:9)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0150】
この1−(ブロモアルキル)アミド−3−メトキシナフタレン6を、以下のようなAlCl3を触媒したフリーデル−クラフツ環化手順を使用して環化した。1〜3当量のAlCl3を含有するニトロベンゼンを、この1−(ブロモアルキル)アミド−3−ヒドロキシナフタレン6に加えた。この反応物を130℃まで加熱し、そして1時間反応させた。NH4C1およびEtOAcを使用する水性ワークアップによって、粗製N−フェニルベンゾインドリノン(N−phenyl−benzoindolinone)中間体7を得、これを、移動相としてEtOAc:ヘキサン(1:4)を使用する順相フラッシュクロマトグラフィーによって精製した。次いで、N−フェニルベンゾインドリン中間体7のアミドカルボニル基をLAHで還元して、化合物8を得た(1HNMR:CDCl3 d 7.71(d,1H,J=7.8Hz),7.32(m,2H),7.24(m,2H),7.07(bt,1H,J=6.6Hz),6.96(m,3H),6.84(s,1H),3.97(s,3H),3.92(s,2H),1.44(s,6H)。
【0151】
実施例1に記載される三臭化ホウ素脱保護手順を使用して、化合物8のメトキシ基を脱保護し、N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン9を得た。
【0152】
(実施例3)
(N−メチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン15の合成(図2))
ピリジン中でピペラジンの触媒作用を使用したメトキシナフトアルデヒドおよびマロン酸の縮合によって、化合物10を合成した。化合物10を、10% Pd/炭素上で水素で還元し、続いてLAHで還元し、そして上記化合物2の合成の概要のように、トリフルオロメタンスルホン酸無水物と反応させて、トリフレート11を得た。次いで、トリフレート11を、DMF中のNaN3(3当量)と、100℃にて6時間反応させた。次いで、この反応物を室温にし、そして、純水およびEtOAcを使用する水性ワークアップに供し、化合物12を得た。化合物12を、無水CH2Cl2中に懸濁し、3〜5当量の固体AlCl3と複合体化し、そして2時間還流してコーティング13を得た。
【0153】
化合物13を、以下のような一般的なアミノ基のアルキル化手順に従ってMeIでアルキル化した。この3−メトキシベンゾキノリン誘導体(100mg)13を、5mlの乾燥THF中に懸濁し、そして−5℃まで冷却した(氷/NaCl)。1.1当量のn−ブチルリチウム(1M)を滴下し、そして、この反応物を、1時間攪拌した。3当量のMeIアルキル化剤をゆっくりと加え、この反応物を、室温にて2時間攪拌した。NH4ClおよびEtOAcを使用する水性ワークアップによって、粗製アルキル化3−メトキシベンゾキノリン中間体14を得た。次いで、中間体14を、移動相としてEtOAc:ヘキサン(1:19)を使用する順相フラッシュクロマトグラフィーによって精製した(1HNMR:CDCl3 d 8.1(ブロードなd,1H,J=8.1Hz),7.68(dd,1H,J=8.1,1.8Hz),7.34(m,2H),6.8(s,1H),3.92(s,3H),3.21(m,2H),2.94(s,3H),2.77(t,2H,J=6.6Hz),1.92(m,2H))。上記実施例1の一般的な三臭化ホウ素手順によって化合物のメトキシ基を脱保護した後、N−メチルヒドロキシベンゾキノリン誘導体15を得た。
【0154】
(実施例4)
(3−(5−ヒドロキシベンゾキノリン−1−イル)プロパンスルホン酸17の合成(図2))
N−メチルヒドロキシベンゾキノリン誘導体15を合成するために、実施例3における上記概要の手順に従って、化合物13を合成した。次いで、今回は、MeIではなく1,3−プロパンスルホン(1,3−propane sultone)をアルキル化剤として使用して、上記実施例3の一般的なアミノ基のアルキル化手順に従って、化合物13を合成し、5−メトキシベンゾキノリン−N−プロパンスルホン酸中間体16を得た(1HNMR:CD3OD d 7.94(d,1H,J=8.7Hz),7.65(d,1H,J=8.4Hz),7.32(t,1H),7.27(t,1H),6.85(s,1H),4.89(s,3H),3.20(m,2H),3.08(bt,2H,J=6Hz),2.91(m,2H),2.72(t,2H,J=6.6Hz),2.33(m,2H),1.89(m,2H)。上記実施例1の一般的な三臭化ホウ素手順によって化合物16のメトキシ基を脱保護した後、3−(5−ヒドロキシベンゾキノリン−1−イル)プロパンスルホン酸17を得た。
【0155】
(実施例5)
(N−メチル−2,2,4−トリメチル−5−ヒドロキシ−(テトラヒドロ)ベンゾキノリン22の合成(図3)) G.T.MorganおよびE.D.Evans(1919),J.Chem.Soc.115:1126の手順に従ってか、または実施例10のように、1−アミノ−3−メトキシナフタレン18を合成した。A.Rosowsky and E.J.Modest(1965)Jour.Org.Chem.30:1832の手順に従って、18(1gm)を、乾燥アセトン(50ml)中に懸濁し、そして、0.01当量のヨウ素をこの溶液に加えた。この反応物を加熱し、そして16時間攪拌し、冷却し、次いで、飽和Na2S2O3でクエンチした。次いで、この反応混合物を、飽和Na2S2O3およびEtOAcを使用する水性ワークアップに供して、粗製メトキシベンゾキノリン19を得た。このメトキシベンゼンキノリン19を、EtOAc/ヘキサン 1:9の移動相を使用するフラッシュクロマトグラフィーによって精製した。次いで、化合物19を、上記実施例3の一般的なアミノ基のアルキル化手順に従って、MeIでアルキル化し、化合物20を得た。化合物20を、70psiにて(Parr hydrogenator)中のH2および10% Pd/C 触媒で還元して、N−メチル−2,2,4−トリメチル−5−メトキシベンゼンキノリン中間体21を得た(1HNMR:CDCl3 d 8.20(bd,1H,J=7.5Hz),7.65(bd,1H,J=7.5Hz),7.33(m,2H),6.89(s,1H),3.94(s,3H),3.14(b sextet,1H,J=6.6Hz),2.80(3,3H),1.89(d,2H,J=8.7),1.42(d,3H,J=6.9Hz),1.34(s,3H),1.05(s,3H)。上記実施例1の一般的な三臭化ホウ素手順によって化合物21のメトキシ基を脱保護した後、N−メチル−5−ヒドロキシ(テトラヒドロ)ベンゾキノリン22を得た。
【0156】
(実施例6)
(N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン27の合成(図3))
上記実施例2の一般的なアミノ基のアセチル化手順に従って、1−アミノ−3−メトキシナフタレン18を、2−ブロモ−2−塩化メチルプロピニルでアセチル化して、化合物23を得た。化合物23を、上記実施例2のフリーデル−クラフツ環化手順によって環化して、化合物24を得た。次に、化合物24を、3当量のLAHを含有するTHFで還元して4−メトキシベンゾインドリン25を得た。アルキル化試薬としてヨードメチル使用する、上記実施例3の一般的なアミノ基のアルキル化手順を使用して、化合物25をアルキル化し、N−メチル−3,3−ジメチル−4−メトキシベンゾインドリン中間体26を得た(1HNMR CDCl3 d 8.07(bd,1H,J=8.4Hz),7.69(bd,1H,J=8.1Hz),7.33(bt,1H,J=7.8Hz),7.22(bt,1H,J=8.1Hz),6.70(s,1H),3.92(s,3H),3.32(s,2H),3.32(s,3H),1.44(s,6H)。実施例1の一般的な三臭化ホウ素手順によって化合物26のメトキシ基を脱保護した後、N−メチル−3,3−ジメチル−4ヒドロキシベンゾインドリン27を得た。
【0157】
(実施例7)
(N−エチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン29の合成(図3))
4−メトキシベンゾインドリン25を、上記実施例6のように合成した。
【0158】
化合物25を、アルキル化剤としてヨードメチルを使用する実施例3に記載の一般的なアルキル化手順によってアルキル化して、N−エチル−3,3−ジメチル−4−メトキシベンゾインドリン中間体28を得た(1HNMR:CDCl3 d 7.90(d,1H,J=8.7Hz),7.68(d,1H,J=8.1Hz),7.32(bt,1H,J=7.5Hz),7.22(bt,1H,J=6.9Hz),6.69(s,1H),3.83(s,3H),3.52(q,2H J=7.5Hz),3.38(s,2H),1.46(s,6H),1.27(t,3H,J=7.5Hz)。実施例1の一般的な三臭化ホウ素手順によって化合物28のメトキシ基を脱保護した後、N−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン29を得た。
【0159】
(実施例8)
(選択したジベンゾローダミン色素化合物の合成)
(一般手順A(図5))
固体無水フタル酸誘導体34を、1.4当量のアミノヒドロキシ中間体31および2.8当量のZnCl2と混合した。オーブン乾燥反応用容器を、ゴム隔膜でキャップし、アルゴンでパージした。この固体混合物を、融解が観察されるまで(例えば、約15分後)130℃にて手短に加熱した。1,2−ジクロロベンゼン(約10当量)を、この反応混合物にシリンジによって加え、この異種混合物を、4時間の間130〜170℃まで加熱した。粗製反応混合物を冷却し、最小量のMeOH:CH2Cl2(1:19)中に懸濁し、順相フラッシュクロマトグラフィーカラムに直接充填し、そしてこの粗製色素を、MeOH:CH2Cl2(1:19)移動相で溶出した。必要に応じて、この色素を精製し、そして、MeOH:CH2Cl2(1:9)を用いて展開するPTLCによって、別個の異性体35および36に分離した。この異性学的に純粋な色素(これは、1:9 MeOH:CH2Cl2で溶出するシリカのTLC上の単一スポットとして遊走される)を、そのUV/可視吸収スペクトル、ならびにその長波長蛍光励起スペクトルおよび発光スペクトルによって同定した。
【0160】
(一般手順B(図6))
図6に概略を示した一般的な手順において、固体無水フタル酸誘導体34(100mg)を、ゴム隔膜でキャップをした丸底フラスコ内に置き、アルゴンでパージした。無水ニトロベンゼン(2ml)を加え、そして加熱して無水物を溶解した。この混合物を、室温まで冷却し、3〜6当量の無水AlCl3を攪拌しながら加えて固体を溶解した。その後、1当量の1−アミノ−3−メトキシナフタレン中間体31を、攪拌しながら加え、この反応物を、1時間130℃まで加熱した。次いで、この反応物を冷却し、そしてEtOAc中に懸濁した。有機層を、飽和NH4Clおよびブラインで洗浄し、Na2SO4で乾燥し、濾過し、そして、溶媒を、減圧下で除去した。必要に応じて、この得られたケトン中間体37/38を精製し、移動相として(MeOH:CH2C12、1:19)を使用する順相フラッシュクロマトグラフィーを使用してかまたは再結晶によって、別個の異性体37および38に分離した。異性学的に純粋な中間体37または38のメトキシ基を、実施例1に記載される一般的な三臭化ホウ素脱保護手順に従って除去して、アミノヒドロキシナフタレンケトン39を得た。次いで、このアミノヒドロキシナフタレンケトン39(100mg)を、1当量の1−アミノ−3−ナフタレン中間体32を含有する無水1,2−ジクロロベンゼン(2ml)と2時間130℃にて反応させた。この反応物を冷却し、異性学的に純粋でかつ非対称的に置換された生成物40を得、これを、上記一般手順Aによって精製した。
【0161】
(ジベンゾローダミン色素41の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31として1−ジエチルアミノ−3−ヒドロキシナフタレン4を使用して、一般手順Aに従った。
【0162】
(ジベンゾローダミン色素42の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−5−ヒドロキシベンゾキノリンを使用して、一般手順Aに従った。
【0163】
(ジベンゾローダミン色素43の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31として5−ヒドロキシベンゾキノリン17を使用して、一般手順Aに従った。
【0164】
(ジベンゾローダミン色素44の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−2,2,4−トリメチル−5−ヒドロキシベンゾキノリン22を使用して、一般手順Aに従った。
【0165】
(ジベンゾローダミン色素45の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−メチル−3,3−ジメチル−4−ヒドロキシベンゾインドリン27を使用して、一般手順Aに従った。
【0166】
(ジベンゾローダミン色素46の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はFである)、そしてアミノヒドロキシ中間体31としてN−エチル−3,3−ジメチル−4ヒドロキシベンゾインドリン29を使用して、一般手順Aに従った。
【0167】
(ジベンゾローダミン色素47の合成(図7))
無水フタル酸誘導体としてジクロロトリメチル酸無水物(すなわち、化合物34(ここで、C−14およびC−17での置換基はClであり、C−15での置換基はCO2Hである))、そしてアミノヒドロキシ中間体31としてN−フェニル−3,3−ジメチル−4−ヒドロキシベンゾインドリン9を使用して、一般手順Aに従った。
【0168】
(実施例9)
(選択されたジベンゾローダミン色素化合物のスペクトル特性)
以下の表は、本発明の数種の代表的なジベンゾローダミン色素化合物の重要なスペクトル特性を示す。全てのスペクトルは、色素のλmax,absにおいて0.05の吸収を有する色素のついて、1×TBE緩衝液および8M尿素中で、室温にて記録されている。色素濃度は、約10−6Mであった。
【0169】
【表1】
(実施例10)
(3,3−ジメチルヒドロキシベンゾインドリン25の合成(図8および9))
化合物2を、以下のようにパラジウムで触媒したトリフレート/イミンカップリングおよびBuchwaldの加水分解手順(Buchwald,S.L.ら、Tetrahedron Letters,1997,38/36,6367〜6370)を使用して、1−アミノ−3−メトキシナフタレン18に転換した。(図8)。
【0170】
3−メトキシナフタレン−1−トリフレート2(25g)を、0.013当量のトリス(ジベンジリデンアセトン)−ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフトル(±BINAP)、1.3当量の炭酸カリウム、1.3当量の炭酸セシウム、および1.5当量のベンゾフェノンイミンと混合した。あるいは、カリウムまたはナトリウムt−ブトキシド(2.6当量)を、この反応のための置換塩基として使用した。この反応混合物を、75mlの無水トルエンおよび75mlの無水テトラヒドロフラン中に懸濁し、そして油浴中で24時間120℃にて攪拌した。75mlのヘキサン/酢酸エチル(9:1)の混合物を、この反応物混合物に加え、そしてこの混合物を、ヘキサン/酢酸エチル(3:2)を用いてシルカゲルによって溶出した。この溶出液を収集し、次いで、減圧下で濃縮した。濃縮したオイルに、ヘキサン/酢酸エチル(9:1)を加えて、ベンゾフェノンイミノ−3メトキシナフタレン49を黄色結晶として結晶化した(20.0g、72.6%)。母液を濃縮し、ヘキサン/酢酸エチル(9:1)を加えて49の第二群(second crop)を黄色結晶として結晶化した(3.0g、10.9%)(全収率:83.5%)。
【0171】
化合物49を、酸性条件下にて加水分解して以下のように18を得た。化合物49(27gm)を、150mlの1,4−ジオキサンおよび100mlの5% 硫酸(H2SO4)中に懸濁した。次いで、この反応物を加熱し、そして油浴中で1時間50℃にて攪拌した。この反応混合物を室温まで冷却し、そして150mlのヘキサン/酢酸エチル(3:2)で洗浄した。次いで、この溶液を、氷冷水性NaOHでpH10〜11まで塩基性化して、酢酸エチルで3回抽出した。合わせた有機抽出物を、硫酸ナトリウム(Na2SO4)で乾燥し、そして減圧下でエバポレートして、淡褐色オイルとして18を得、これを、放置したまま結晶化させた(13.9g、100%)。
【0172】
化合物25(3,3−ジメチルヒドロキシベンゾインドリン)を、上記実施例6の手順に従って、18から合成した。
【0173】
(実施例11)
(N置換メトキシベンゾインドリン中間体57〜61の合成(図9))
化合物25を通常の中間体として用いて、アルキル置換またはパラジウムで触媒した第2級アミン基へのカップリングのいずれかによって、N−置換誘導体57〜61を合成した(図9)。
【0174】
(N−(6−ヘキサン酸)−3,3−ジメチルヒドロキシベンゾインドリン57の合成)
化合物25(2.3g)を3当量の市販のメチル−6−ヨードヘキサン酸52および3当量のジイソプロピルエチルアミンで処理することによってアルキル化した。この混合物を23mlの無水トルエン中に懸濁し、18時間攪拌しながら油浴中で130℃で加熱した。溶媒を減圧下でエバポレートして、粗製N−(6−ヘキサン酸)−3,3−ジメチルヒドロキシベンゾインドリン57を得た。粗製57をジクロロメタン中に溶解し、ヘキサン/酢酸エチル(99:1)で溶出する順相フラッシュクロマトグラフィーによって精製した(収率2.55g,74%)。
【0175】
(N−(ビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン58の合成)
化合物25(2.3g)を以下のように、確立された手順(Buchwald,ら,J.Org.,1997,62,6066−6068)に従ってパラジウムで触媒したカップリングによってN−アリール化した;化合物25(2g)を1.4当量の4−ブロモビフェニル7、ナトリウムt−ブトキシド(1.3当量)、0.013当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(±BINAP)と混合した。この混合物を100mlの無水トルエン中に懸濁し、十分攪拌し、そして油浴中で130℃にて一晩(16時間)加熱した。この反応混合物を水性塩化アンモニウム(NH4Cl)でクエンチし、酢酸エチルで抽出した(3×)。有機抽出物を無水Na2SO4で乾燥した。ヘキサン(等量)をこの粗製反応混合物に加え、それを、ヘキサン/酢酸エチル(1:1)を用いてシリカゲルによって溶出した。溶媒を減圧下でエバポレートし、残渣をヘキサン/酢酸エチル(98:2)で溶出する順相フラッシュクロマトグラフィーによって精製して、N(ビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン58(収率85%)を白色粉末として得た。
【0176】
(N−(カルボキシビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン59の合成)
化合物54をハロホルム反応によって市販のブロモビフェニルメチルケトンから合成して、ブロモビフェニル酸を得、続いて、確立された手順を用いてエステル化した。化合物25を、以下のように、確立された手順(Buchwald,ら Tetrahedron Letters,1997,38/36,6359−6362)に従って54でN−アリール化した:化合物25(0.4g)を1.4当量の化合物54、0.013当量のトリス(ジベンジリデンアセトン)ジパラジウム(Pd2(dba)3)、0.04当量のラセミ酸2,2’−ビス(ジフェニルホスフィノ)−1,1’−ビナフチル(±BINAP)、および3当量の炭酸セシウムと混合した。この反応混合物を100mlの無水トルエンおよび100mlの無水テトラヒドロフラン中に懸濁し、次いで、油浴中で130℃にて加熱し、そして18時間攪拌した。この反応物を冷却し、粗製混合物にヘキサン(30ml)を加えた。この粗製生成物をヘキサン/酢酸エチル(3:2)を用いて、シリカゲルによって溶出した。この溶媒を減圧下で除去し、N−(カルボキシビフェニル)−3,3−ジメチルヒドロキシベンゾインドリン59をヘキサン/酢酸エチル:(95:5)を使用する順相フラッシュクロマトグラフィーによって精製した(明黄色固体収率0.25g、32%)。
【0177】
(N−フェニル−3,3−ジメチルヒドロキシベンゾインドリン60の合成)
化合物25を、化合物25からの化合物58の合成について上述したようにヨードベンゼンまたはブロモベンゼンでN−アリール化した。純粋なN−フェニル−3,3−ジメチルヒドロキシベンゾインドリン8を、ヘキサン/酢酸エチル:(98:2)を使用する順相フラッシュクロマトグラフィーの後に単離した。
【0178】
(N−カルボキシフェニル−3,3−ジメチルヒドロキシベンゾインドリン61の合成)
化合物25を、化合物59の合成について上述したようにヨード安息香酸メチルまたは臭化安息香酸メチルでN−アリール化した。純粋なN−カルボキシフェニル−3,3−メチルヒドロキシベンゾインドリン61を、ヘキサン/酢酸エチル:(95:5)を使用する順相フラッシュクロマトグラフィーの後に単離した。
【0179】
(実施例12)
(ジベンゾローダミン色素67、72、76および81の合成(図10〜13)
(色素67の合成(図10))
ヒドロキシインドリン62を、以下の手順に従って塩化アルミニウム(AlCl3)を使用する化合物58の脱メチル化によって得た:化合物58(2g)および塩化アルミニウム(6g)を、アルゴンの強い流れの下で完全に混合し、そしてこの反応混合物を、アルゴンで10分間パージした。この固体混合物を、アルゴン下で、90℃〜120℃まで油浴中にて20分間にわたって加熱した。この反応混合物を冷却し、そしてこの固体をジクロロメタン中に懸濁し、氷および希H2SO4の溶液に移した。水層を、5回抽出し、合わせた有機抽出物を、飽和塩化ナトリウムで洗浄し、そして硫酸ナトリウムで乾燥した。濾過した後、この溶媒を、減圧下でエバポレートし、そして純粋な62(0.13g)を、ジクロロメタン/メタノール(99:1)を使用する順相フラッシュクロマトグラフィーの後に単離した。ヒドロキシインドリン63を、化合物62からの化合物58の転換と同じ手順を使用して、化合物59から生成した。
【0180】
次に、1当量の1:1のN−メチルホルムアニリドおよび無水オキシ塩化物の複合体を含有する無水ジクロオロメタンを、小西洋ナシ形(pear)フラスコ(5mlフラスコ中100mg)中の1当量の62に加えた。この反応混合物を、アルゴンの流れの下で、ジクロロメタンがエバポレートして暗赤色オイルが残るまで、油浴中で80℃にて加熱した。ニトロベンゼン(300μl)を加え、続いて2当量のPOCl3を加えた。この上記溶液に、ニトロベンゼン(500μl)に懸濁した1当量の63を加えた。この反応混合物を、25分間にわたって80℃〜155℃まで加熱し、次いで、15分間155℃に保った。この反応物を冷却し、ジクロロメタンと共に5%HClに移した。水層をブラインで飽和させ、そしてジクロロメタンで3回抽出した。合わせた有機抽出物を分離し、そして減圧下でエバポレートして、粗製の色素誘導体64を得た。粗製の64を、酢酸(3ml)および5%HCl(3ml)中に懸濁し、60℃にて1時間加熱した。次いで、この反応混合物を、氷冷した塩化ナトリウム飽和水溶液中に注ぎ、ジクロロメタンで3回抽出した。合わせた有機層をエバポレートした後、次いで、純粋なモノカルボキシル化非対称性色素誘導体65を、ジクロロメタン/メタノール(9:1)で溶出する順相フラッシュクロマトグラフィーによって、他の2種の対称性色素生成物(ビス−ビフェニル色素およびビス−カルボキシフェニル色素)から精製した。
【0181】
最後に、この精製した色素誘導体65をスルホン化して、以下のような色素67を得た。色素誘導体65(30mg)および3当量の無水硫酸ナトリウムを、アルゴン下で0℃にて、無水クロロメタン(50ml)中に懸濁した。上記溶液に、5当量の塩化スルホン酸(ClSO3H)を加え、そしてこの反応混合物を、室温にて18時間攪拌した。この反応混合物を、飽和水性塩化ナトリウム/5% HClでクエンチし、そして水層を、ジクロロメタンで3回抽出した。有機層を減圧下でエバポレートして、粗製色素66を得た。粗製色素66を、ジオキsンおよび5%HCl(2:1)の溶液中に懸濁し、そして室温にて20時間攪拌した。次いで、この溶液を減圧下で濃縮し、飽和水性塩化ナトリウム中に懸濁し、そしてジクロロメタンで3回抽出した。この溶媒をエバポレートして、粗製色素67を得た。純粋な色素67を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0182】
(色素72の合成(図11))
化合物62を化合物58に転換するための、上記塩化アルミニウム脱保護手順をともに使用して、化合物61を脱メチル化して化合物68を得、そして化合物8を脱メチル化して化合物9を得た。化合物68を、メチルホルムアニリド/POCl3複合体と反応させ、続いて、化合物9と反応させて、化合物62および63からの色素誘導体64の合成について記載したように、色素誘導体70を生成した。粗製色素誘導体70を、酸によって加水分解して、上記64からの色素誘導体65の生成手順に従って、色素誘導体71を得た。純粋な非対称性色素誘導体71を、2種の対称性ビスフェニルおよびビス−カルボキシフェニル色素生成物からのPTLCによって精製した。次いで、この精製した色素71をスルホン化および加水分解して、色素誘導体65からの色素67の生成について上記したように、粗製色素72を得た。純粋な色素72を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0183】
(色素76の合成(図12))
化合物57を脱メチル化して、三臭化ホウ素脱保護によって化合物73を得、そして、化合物8を脱メチル化して、化合物62からの化合物58の転換について上記した、塩化アルミニウム脱保護手順を使用して、9を得た。化合物57を、標準条件下でメチルホルムアニリド/POCl3複合体と反応させ、続いて、標準条件下で化合物9と反応させて、上記実施例7のように色素誘導体74を生成した。粗製色素誘導体74を、標準的な酸加水分解によって加水分解して、色素誘導体75を得た。この純粋な非対称性色素誘導体75を、2種の対称性ビスフェニルおよびビス−ヘキサン色素生成物からのPTLCによって精製した。次いで、この純粋な色素75を、65から67を形成するために記載された標準的な手順によってスルホン化して、粗製色素76を得た。純粋な色素76を、メタノール/ジクロロメタン(1:4)を用いて展開する順相の分取薄層クロマトグラフィー(PTLC)、メタノール/ジクロロメタン(1:9)を用いるフラッシュカラムクロマトグラフィーによって単離した。
【0184】
(色素81の合成(図13))
化合物57(0.313g)を、室温にて3時間にわたって、7当量の1M KOHを含有するメタノールで処理することによって鹸化した。この溶液を、乾燥するまで濃縮し、そしてオイルを酢酸エチルおよび5%塩酸で十分に抽出した。有機層を硫酸ナトリウムで乾燥し、そして無色オイルになるまで濃縮した。純粋な77を、5%メタノール/ジクロロメタン/0.1%酢酸を使用する順相クロマトグラフィーによって単離した(収率0.243g;84%)。
【0185】
次に、化合物77(0.360gm)を、無水ジクロロメタン(2ml)、ならびにジクロロメチルメチルエーテル(2.6当量)および四塩化スズ(4当量)の溶液中に溶解した。この溶液を、1.5時間還流した。この混合物を、酢酸エチルおよび3M塩酸溶液で抽出した。有機層を、水およびブラインで洗浄し、Na2SO4で乾燥し、オイルになるまで濃縮し、そして精製することなく使用した。薄層クロマトグラフィーによって、この時点で、ほぼ等モルの化合物77および78の混合物が存在した。このオイルを、無水ジクロロメタン(8ml)中に懸濁し、そして−78℃まで冷却した。この混合物の脱メチル化を、確立された三臭化ホウ素手順によって行って、化合物79と化合物80との混合物を得た。得られた粗製オイルを、無水ジクロロメタン(5ml)中に溶解し、そしてオキシ塩化リン(2当量)を加え;次いで、ニトロベンゼン(5ml)を加えた。この混合物を、45分間にわたって80〜150℃まで加熱し、そして、即座に加熱を停止した(remove)。ワークアップおよび単離を、化合物67の合成についての実施例12のように行って、粗製色素81を得た。純粋な色素81を、化合物67の合成について実施例12で記載したように、PTLCによって単離した(収率32mg)。
【0186】
(実施例13)
(化合物67、72、76および81の色素カルボキシN−ヒドロキスクシンイミド誘導体の合成、ならびにこの誘導体のヌクレオチドおよびポリヌクレオチドへのカップリング(図14および15))
カルボン酸色素67、72、76および81のN−ヒドロキシスクシンイミド(NHS)誘導体化を、2つの方法の1つによって行った。NHS色素82、83および84の合成については、方法Aを、O−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート試薬を用いて行い、一方、色素85については、方法Bを、ジシクロヘキシルカルボジイミド(DCC)およびN−ヒドロキシスクシンイミドを使用して行った。
【0187】
(方法A:)
色素を、6当量のジイソプロピルエチルアミンと共に、無水DMF(5mg色素:300μl DMF)中に懸濁した。この色素溶液に、15当量のO−(N−スクシンイミジル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレートを加え、そしてこの反応物を室温にて15分間攪拌した。この反応物を、ジクロロメタンとともに5% HCl(20ml)中に移した。水層を、飽和NaClを加えることによって塩にし、そしてジクロロメタンで3回抽出した。合わせた有機層を、乾燥するまで濃縮し、そして粗製色素NHSを、MeOH/CH2Cl2(1:9)を使用する順相フラッシュクロマトグラフィーによって精製した。
【0188】
(方法B:)
色素および20当量のN−ヒドロキシスクシンイミドを、無水ジクロロメタン(5mg:500μl CH2Cl2)中に懸濁した。9当量のジシクロヘキシルカルボジイミドを加え、そしてこの反応物を、室温にて1時間攪拌した。この反応物をクエンチし、そして純粋な色素NHSを、方法Aについてと同様に単離した。
【0189】
NHS誘導体のヌクレオチドおよびポリヌクレオチドへの結合を、以下のように行った。アミノ基置換オリゴマーまたはターミネーターを、ホルムアミド(5×10−4M)中に懸濁し、そして15当量のジイソプロピルエチルアミンを加えた。DMSO(5mg/60μl)中に懸濁した過剰な(5〜10当量)の無水NHSを、室温にて攪拌しながら加えた。この反応物を、室温にて4時間攪拌した。この反応混合物に3M NaOAcを加えて、0.5M NaOAcを得た。EtOHの反応容量を4回加え、そしてこの混合物を冷却した。沈殿した色素標識オリゴマーを、遠心分離によって単離し、そして30% AcCN/0.1×TEAA緩衝液で溶出する逆相HPLCによって精製した。
【0190】
(実施例14)
(スルホメトキシベンゾインドリン89の合成(図16))
メトキシベンゾインドリン86(1g、0.0044mol)を、50mlの無水CH2Cl2中に懸濁し、そしてこの溶液を、水/アセトンのドライアイス浴中で−20℃まで冷却し、そしてアルゴンで十分にパージした(図16)。この攪拌溶液に、1.1当量(0.82ml)の無水トリフルオロメタンスルホン酸を滴下した。この無水物の添加が終わった後、この反応物を、−20℃に維持し、そして、2当量の無水トリエチルアミン(1.2ml)を滴下しながら攪拌した。1時間後、この反応物を、5% HClを加えることによって、−20℃にてクエンチし、そして水相を、CH2Cl2で3回抽出した。合わせた有機層を、飽和NaHCO3、ブラインで洗浄し、そしてNaSO4で乾燥し、濾過し、そして溶媒を、減圧下で除去した。この粗製生成物を、ヘキサン:CH2Cl2(3:2)で溶出する順相クロマトグラフィーによって精製して、2.3gのスルホンアミド87を得た(68%収率)。
【0191】
250mlの丸底フラスコ中で、スルホンアミド87(1g、0.00279mol)および顆粒状の無水Na2SO4(0.1g)を、100mlの無水CH2Cl2中に懸濁した。この反応混合物をアルゴンでパージし、そして0℃まで冷却した。迅速に攪拌した溶液に、1当量のクロロスルホン酸(185μl)を滴下した。この反応物を室温まで温め、そしてアルゴン下で16時間攪拌した。この反応物を、50mlのTHF、75mlの1M NaOHを加えることによってクエンチし;そして2時間攪拌した。1当量の硫酸水素テトラブチルアンモニウム(1g)を、この混合物に加え、そして水相を、CH2Cl2で5回抽出した。有機相を減圧下で濃縮して、油状の88の固体混合物および他の位置異性体(regioisomers)を得た。MeOH:CH2Cl2(8:92)で溶出する順相クロマトグラフィーによる精製によって、1gの純粋な88(53%収率)のモノテトラブチルアンモニウム塩を得た。
【0192】
スルホンアミド88のテトラブチルアンモニウム塩(1g)を、50mlの無水THF中に懸濁した。水素化リチウムアルミニウム(20当量、1g)を慎重に加え、そして、この反応物を、アルゴン下で2時間還流した。この反応物を、氷、次いで、50mlの1M NaOHを慎重に加えることによってクエンチした。テトラブチル硫酸水素アンモニウム(1g)をこの混合物に加え、そしてこの混合物を、減圧下で濃縮して大部分のTHFを除去した。この混合物を、分液漏斗に移し、CH2Cl2で5回抽出した。合わせた有機層を、減圧下でオイルになるまで濃縮し、そしてMeOH:CH2Cl2(8:92)で溶出する順相クロマトグラフィーによって精製して、0.733gの純粋な89(89%収率)のテトラブチルアンモニウム塩を得た。
【0193】
(実施例15)
(スルホメトキシナフトインドリン93の合成(図17aおよび17b))
メトキシナフトインドリン90を、24.9gの1,3−ジメトキシアントラセン121(Fitzgerald(1992)Jour.Org.Chem.57:7122−26)を含有する48% HBr(175ml)および氷状酢酸(450ml)を、90℃にて1時間加熱することによって調製した(図17a)。この反応混合物を、攪拌しながら、氷および水にゆっくりと注いだ。1,3−ジヒドロキシアントラセン122の沈殿物が、直ちに形成され、これを濾過し、300mlの冷水で洗浄し、そして褐色固体として収集した。化合物122を、0.14M HC1を含有するメタノールに5℃にて加え、そしてアルゴン下で3時間5℃にて維持した。この反応混合物を、冷水(1000ml)で希釈し、そして3:2 ヘキサン/酢酸エチルの混合物で3回抽出した。合わせた有機抽出物を、飽和NaClで洗浄し、そしてNa2SO4で乾燥した。濾過した後、減圧下で溶媒を除去し、そして、ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって、123を明黄色固体として得た(6.93g、66%)。
【0194】
トリフルオロメタンスルホン酸無水物(6.8ml)を、123(7.0g、0.031mol)およびジクロロメタン(600ml)の攪拌溶液に、アルゴン下で−60℃にてゆっくりと加えた。20分後、トリエチルアミン(5.65ml)を、この反応物に滴下した。この反応混合物を、−30℃にて30分間攪拌し、次いで、5%硫酸中に注いだ。次いで、この混合物を、400mlのヘキサン:ジクロロメタン 2:3の混合物で抽出し、飽和NaClで洗浄し、Na2SO4で乾燥し、濾過し、そして減圧下で濃縮した。ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって、淡黄色オイルとして124を得、これを、冷却して凝固させた。ヒドラジン(6ml)を、124(6g、0.017mol)、Cs2CO3(30g)、Pd2(dba)3(0.75g)、BINAP(1.5g)、トルエン(100ml)、およびTHF(100ml)の攪拌溶液に、アルゴン下で室温にて加え、次いで、90〜100℃にて1時間加熱した。冷却した溶液を濾過し、そして溶媒を減圧下で除去して、褐色固体して125を得た。125を含有する無水エタノールおよびイソブチルアルデヒド(14ml)の溶液を、90〜100℃にて加熱した。この溶液を、乾燥するまでエバポレートし、そしてヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって精製して、淡黄色固体として126を得た。
【0195】
126を含有する氷酢酸(150ml)の溶液を、アルゴン下で攪拌しながら、88℃にて1時間加熱した。この溶液を、氷浴中で冷却し、そしてNaCNBH3(3g)を、15分間にわたって一部ずつ加え、室温にてさらに15分間攪拌した。酢酸を減圧下で除去し、残渣をジクロロメタン中に懸濁し、そして1N NaOH、飽和NH4Cl、および飽和NaClで洗浄した。溶媒を、減圧下で除去し、そして残渣を、ヘキサンおよび酢酸エチルで溶出する順相シリカゲルクロマトグラフィーによって精製して、淡黄色固体として90を得た(2.5g、53%)。
【0196】
メトキシナフトインドリン90(1g、3.6mmol)を、40mlの無水CH2Cl2中に懸濁し、溶媒を、氷浴中で0℃まで冷却し、そしてアルゴンでパージした(図17)。この攪拌溶液に、1.5当量のトリフルオロ酢酸無水物(0.76ml)を滴下した。この無水物の添加が終了した後、この反応物を0℃に維持し、2当量の無水トリエチルアミン(1ml)を滴下しながら攪拌した。1時間後、この反応物を、5% HClを加えることによってクエンチし、そして水相を、飽和CH2Cl2で3回抽出した。合わせた有機層を、NaHCO3、ブラインで洗浄し、そして有機層を、NaSO4で乾燥し、濾過し、そして溶媒を減圧下で除去した。粗製生成物を、1:4 EtOAc:ヘキサンを使用する順相クロマトグラフィーによって精製して、黄色固体として1gの91を得た(収率74%)。
【0197】
トリフルオロアセトアミド91(1g、2.7mmol)を、60mlの氷酢酸および6mlの無水CH2Cl2中に懸濁した。溶媒を、アルゴンで十分にパージし、そして0℃まで冷却した。この溶液を、十分に攪拌し、そしてクロロスルホン酸(4ml、20当量)を滴下した。この反応物を、室温まで温め、そして3日間攪拌した。この反応物を、最少量の氷(約1g)を加えることによってクエンチした。酢酸の除去を補助するためにトルエンを加えながら、この溶媒を減圧下(en vacuo)で除去した。この粗製反応混合物を、最少量の1:4 MeOH:CH2Cl2中に懸濁し、そして最初にCH2Cl2で溶出し、次いで、1:20 MeOH:CH2Cl2まで増加させるシリカゲルプラグを通して部分的に精製して、92と他のスルホン化位置異性体との混合物を得た。
【0198】
92を含有する混合物を、100mlのMeOH(1mlの水および1.5gのK2CO3を含有する)中に懸濁することによって脱保護した。この反応物を、室温にて30分間攪拌した。この粗製反応混合物を、MeOHで溶出するシリカゲルのプラグに通し、そして溶媒を、減圧下で除去した。所望の異性体であるスルホメトキシナフトインドリン93(0.350g、収率37%)を、1:9〜1:6 MeOH:CH2Cl2の勾配溶出を使用する順相クロマトグラフィーによって、副生成物異性体から分離した。(図17b)。
【0199】
(実施例16)
(ホルミルスルホメトキシベンゾインドリン106の合成(図20))
メチル−6−ブロモヘキサノエートでの、89の加熱しながらアルキル化よって、N−メチルカルボキシヘキシルスルホメトキシベンゾインドリン94(図18)を調製した。100mlの丸底フラスコ中で、テトラブチルアンモニウム塩の混合物として、1g(918、1.1mmol)の94を、25mlの無水CH2Cl2中に懸濁した。この溶液を、アルゴンで十分にパージし、そして氷浴中で0℃まで冷却した。この攪拌溶液に、5当量の新たに作製したPOCl3/DMFの1:1 Vilsmeyer複合体を含有するCH2Cl2(5当量の複合体を含有する約1mlのCH2Cl2、以下を参照のこと)を滴下した。この反応物を、氷浴から取り除き、そして室温にて1時間攪拌した。この反応物を、飽和NaHCO3(10ml)およびTHF(20ml)を加えることによってクエンチし、そして室温にて30分間攪拌した。この混合物を、CH2Cl2ともに、分液漏斗に移し、有機層を除去し、水層を、CH2Cl2で3回抽出し、そして合わせた有機層を濃縮して、粗製の106を得た(図20)。この粗製オイルから、純粋なアルデヒド106を、EtOAc:ヘキサン(1:2)を使用する順相クロマトグラフィーによって単離して、黄色固体を得た(0.222g、42%収率)。
【0200】
(実施例17)
(ホルミルスルホメトキシナフトインドリン108の合成(図20))
メチル4−(ブロモメチル)ベンゾエートでの、93(図18)の加熱しながらのアルキル化によって、スルホメトキシナフトインドリン101(図19)を調製した。100mlの丸底フラスコ中で、テトラブチルアンモニウム塩として、1g(1mmol)の101を、25mlの無水ジクロロメタン中に懸濁した。この溶液を、アルゴンで十分にパージし、そして氷浴中で0℃まで冷却した。この攪拌溶液に、5当量の新たに作製したPOCl3/DMFの1:1 Vilsmeyer複合体を含有するCH2Cl2(5当量を含有する約1mlのCH2Cl2、以下を参照のこと)を滴下した。室温にて1時間攪拌した後、この反応物を、飽和NaHCO3(10ml)およびTHF(20ml)を加えることによってクエンチし、そして室温にて30分間攪拌した。この混合物を、CH2Cl2ともに、分液漏斗に移し、有機層を除去し、水層を、CH2Cl2で3回抽出した。合わせた有機層濃縮して、粗製の108を得た(図20)。この粗製生成物を、EtOAc:ヘキサン(1:2)を使用する順相クロマトグラフィーに供して、0.497gの純粋なアルデヒド生成物108を黄色固体として得た(90%収率)。
【0201】
(Vilsmeyer試薬)
10mlのフラスコを、8.5mlの無水CH2Cl2および660μlの無水DMFでチャージした。この溶液を0℃まで冷却し、そしてアルゴンで十分にパージした。この攪拌溶液に、790μ1のPOCl3を滴下した。この反応物を、使用前に0℃にて15分間攪拌した。次いで、必要量の複合体を、必要に応じてシリンジで取り出した。
【0202】
(実施例18)
(スルホン化ジベンゾローダミン色素111の合成(図22a))
1,3−プロパンスルホン(1,3−propane sultone)での、89(図16)の熱しながらのアルキル化によって、N−プロピルスルホネートベンゾインドリン96(図18)を調製した。20mlの西洋ナシ形フラスコ中に、50mgのホルミルベンゾインドリン106(図20)、1.2当量の96、5ml 無水CH2Cl2および3ml 無水ニトロベンゼンを加えた。この混合物を攪拌し、大部分のCH2Cl2がエバポレートするまで、アルゴン蒸気下で、110℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、2時間攪拌しながら加熱した。
【0203】
この反応物を、手短に冷却し、次いで、5mlのMeOHを加え、そしてこの反応物を、5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素111を、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。エステル化色素111を含有する画分を、可視吸収スペクトル(MeOH中において638nmで吸収最大値)、蛍光発光スペクトル(MeOH中において651nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、871.5に相当する、算出値:871.3)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml 水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そして水層を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相HPLCを行って、20%の全体の収率において純粋な色素111を得た(図22a)。
【0204】
(実施例19)
(スルホン化ベンゾナフトローダミン色素115の合成(図22b))
1,3−プロパンスルホンでの、93(図17)の加熱しながらのアルキル化によって、N−プロピルスルホネートナフトインドリン102(図19)を調製した。ホルミルベンゾインドリン誘導体107(図20)を、95(図18)を実施例17の手順によってホルミル化することによって調製した。
【0205】
20mlの洋ナシ形フラスコ中に、50mgの107、1.2当量の102、5mlのCH2Cl2および3mlのニトロベンゼンを加えた。この混合物を攪拌し、そしてアルゴン蒸気下で大部分のCH2Cl2がエバポレートするまで、120℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、攪拌しながら2時間加熱した。この反応物を、手短に冷却し、次いで、10mlのMeOHを加え、この反応物を5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素115のメチルエステルを、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。115のメチルエステルを含有する画分を、可視吸収スペクトル(MeOH中において656nmで吸収最大値)、蛍光発光スペクトル(MeOH中において672nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、885.3に相当する、算出値:885.2)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そして水層を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相クロマトグラフィーを行って、20%の全体の収率において純粋な色素115を得た(図22b)。
【0206】
(実施例20)
(スルホン化ジナフトローダミン色素120の合成(図22c))
1,3−プロパンスルホンでの、93(図17)の加熱しながらのアルキル化によって、N−プロピルスルホネートナフトインドリン102(図19)を調製した。ホルミルベンゾインドリン誘導体108(図20)を、101(図191)を実施例17の手順によってホルミル化することによって調製した。
【0207】
20mlの洋ナシ形フラスコ中に、50mgの108、1.2当量の102、5mlのCH2Cl2および3mlのニトロベンゼンを加えた。この混合物を攪拌し、そしてアルゴン蒸気下で大部分のCH2Cl2がエバポレートするまで、110℃まで加熱した。過剰なオキシ塩化リン(300μl、60当量)を、一部ずつ加え、そしてこの反応物を、攪拌しながら2時間加熱した。この反応物を、手短に冷却し、次いで、5mlのMeOHを加え、この反応物を5分間加熱還流した。MeOHおよびいくらか過剰なニトロベンゼンを、減圧下で除去し、そしてこの粗製混合物を、順相シルカゲルカラムに充填した。色素120を、100% CH2Cl2〜1:9 MeOH:CH2Cl2の勾配溶出によって溶出した。エステル化色素120を含有する画分を、可視吸収スペクトル(MeOH中において686nmで吸収最大値)、蛍光発光スペクトル(695nmで発光最大値)、および質量スペクトル(分子イオン+3メチル基は、991.5に相当する、算出値:991.3)によって示されるように収集し、オイルになるまで濃縮した。スルホン酸メチルエステル基およびカルボン酸メチルエステル基を、100ml MeOH、20ml 水、0.85g LiOH水和物中に懸濁することによって加水分解し、そして80℃にて1時間加熱した。メタノールのバルクを、減圧下で取り除いた。3当量のテトラブチルアンモニウム塩を加え、そしてこの反応物を、CH2Cl2で5回抽出した。合わせた有機層を、オイルになるまで濃縮し、そして1:3 CH3CN:H2Oを使用する逆相クロマトグラフィーを行って、20%の全体の収率において純粋な色素120を得た(図22c)。
【0208】
(実施例21)
(スルホン化ジアリールローダミン−ターミネーターを用いたpGEMの配列決定)
米国特許第5,770,716号;同第5,948,648号;および同第6,096,875号の条件に従って、本発明のスルホン化ジアリールローダミン色素で標識されたターミネーターを、サンガー型の4色自動化DNA配列決定実験において他の標準試薬とともに使用する。
【0209】
色素−ターミネーター配列決定反応を、ABI PRISMTM Dye Terminator Cycle Sequencing Core Kit Manual(Applied Biosystems p/n 402116)によって提供されるプロトコルに従って、AmpliTaq DNA Polymerase、FSを使用して行う。pGEM−3Zf(+)テンプレートの配列決定を、未標識の−21 M13配列決定プライマー(順方向)を用いて行った。試薬(緩衝液、未標識プライマー、AmpliTaq DNA Polymerase(FS)を含む)は、ABI PRISMTM Dye Terminator Core Kit(Applied Biosystems p/n 402117)由来であり得る。dNTPミックスは、各々2mMのdATP、dCTP、dITP、およびdUTPまたはdTTPからなり得る。以下を含有する反応組成物のプレミックスを調製する:5×緩衝液4.0μL;dNTPミックス 1.0μL;テンプレート:pGEM(登録商標)−3Zf(+)、0.2μg/μL、2.0μL;プライマー:−21 M13(順方向)、0.8pmol/μL、4.0μL;AmpliTaq DNA Polymerase(FS)、0.5μL;および、H2O 3.5μL(ここで、全ての量は、反応に基づいて与えられる)。
【0210】
例示的な反応物を、Perkin Elmer 480 DNA Thermal Cycler(Applied Biosystems p/n N801−100またはApplied Biosystems Gene Amp PCR System 9700用の0.2mlチューブ)に適用される0.5mlのチューブ内に構築した。1〜250pmolの色素ターミネーターを、各反応物に加えた。30μlの鉱油を、各反応物の上面に加え、エバポレーションを防止する(Applied Biosystems 480 Thermal Cyclerを使用する場合)。反応物容量は、20μLであり、これは、15μLの上記反応プレミックス、変量の色素標識ターミネーター、および全反応物容量を20μLにするのに十分な容量の水を含有する。反応を、以下のように熱サイクルする:96℃で30秒間、50℃で15秒間、および60℃で4分間を25サイクル;続いて、4℃での保持サイクル。
【0211】
全ての反応物を、製造業者の指示書に従って(Princeton Separations p/n CS−901)、Centri−Sepスピンカラムによるスピンカラム精製によって精製する。カラム中のゲル材料を、0.8ml 脱イオン水で、少なくとも30分間室温にて水和させる。カラムを水和させ、ゲル材料中に気泡が閉じ込められていない状態にした後、カラムの上端および下端のキャップを取り外し、このカラムを、重力によって排水させる。次いで、カラムを、キットに備えられている洗浄チューブに挿入し、可変速度のミクロ遠心分離機で1300gにて2分間遠心分離し、洗浄チューブから取り除き、そしてサンプル収集チューブに挿入する。この反応混合物を、オイルから慎重に取り除き、そしてゲル材料に充填し、そしてチューブを再遠心分離する。次いで、溶出したサンプルを、減圧遠心分離機内で乾燥する。
【0212】
配列決定ゲルに充填する前に、乾燥したサンプルを、25μlのTemplate Suppression Reagent(Applied Biosystems p/n 401674)中に再懸濁し、ボルテックスし、95℃まで2分間加熱し、氷上で冷却し、再度ボルテックスし、そして遠心分離する(13,000×g)。10μLの懸濁したサンプルを、ABI PRISMTM 310 Genetic Analyzer(Applied Biosystems p/n 310−00−100/120)に適用されるサンプルバイアル(Applied Biosystems p/n 401957)にアリコートする。310 Genetic Analyzer上での電気泳動を、DNA配列決定分析(Applied Biosystems p/n 402837または4313087(ポリマー)およびp/n 402840(キャピラリー))に特異的に適用される、シービング(sieving)ポリマーおよびキャピラリーを用いて行う。このシービングポリマーは、核酸変性剤を含む。サンプルを、30秒間、2.5kVにてキャピラリーに動電学的に注入し、10〜12.2kVにて2時間まで泳動させ、この時、キャピラリーの外壁は、50℃で維持され、配列決定データとしてエレクトロフェログラムを作成する。
【0213】
スルホン化ジアリールローダミンターミネーターは、プライマー伸長の3’末端、4色配列決定反応間のポリヌクレオチドフラグメントに特異的に組み込まれている。1〜約1000塩基対の溶出フラグメントが、検出およびプロットされる。エレクトロフェログラムは、長さごとに溶出される標識されたフラグメントのスルホン化ジアリールローダミン色素によって発せられる蛍光強度を、ABI PRISMTM 310 Genetic Analyzerによる電気泳動間の時間の関数として、プロットする。
【0214】
全ての文献、特許、および本明細書中で参照される特許出願は、各個々の文献、特許、または特許出願が参考として援用されることを特異的かつ個々に示すかのように、同じ程度まで、参考として本明細書中で援用される。
【0215】
わずかな実施形態のみが上に詳細に記載されているが、化学分野の当業者は、多くの改変がこれらの実施形態においてその教示から逸脱することなく可能であることを、明確に理解している。このような全ての改変は、添付の特許請求の範囲の範囲内に含まれることが意図される。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

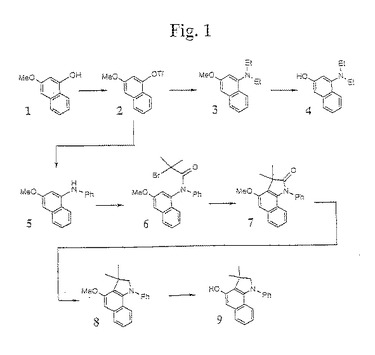
【図2】
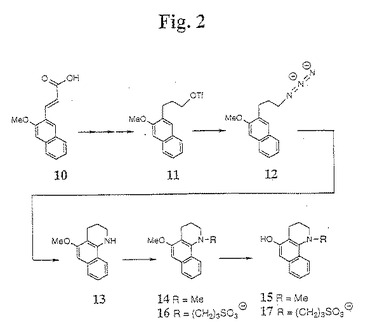
【図3】
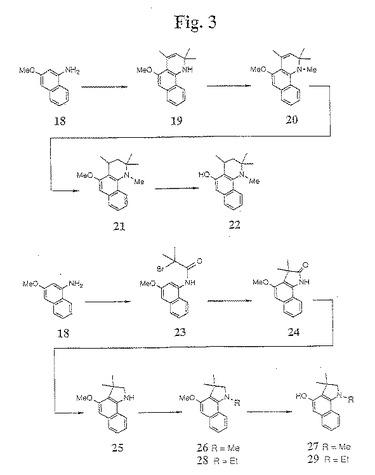
【図4】
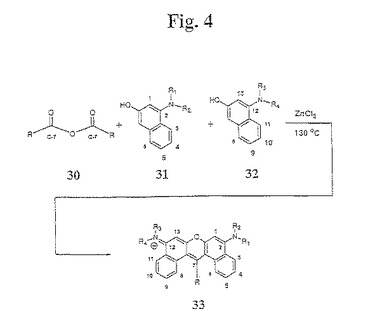
【図5】
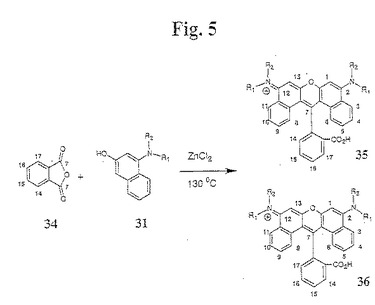
【図6】
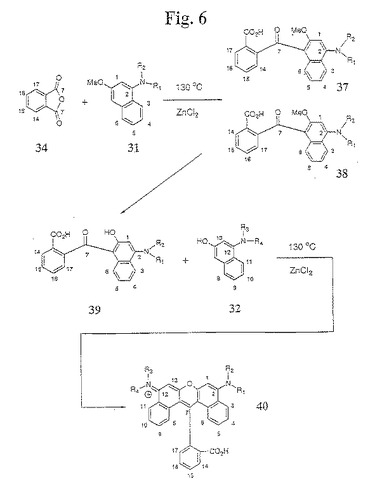
【図7】
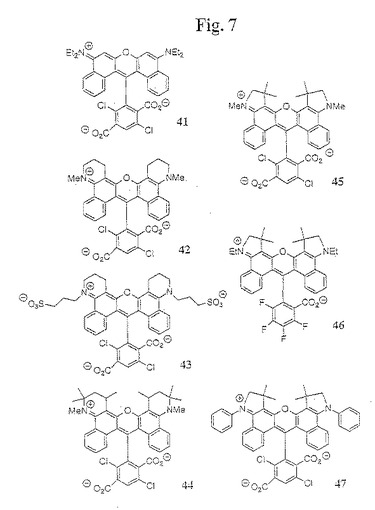
【図8】
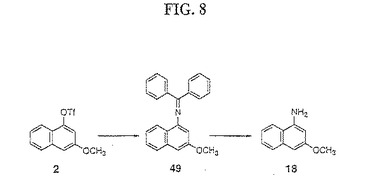
【図9】
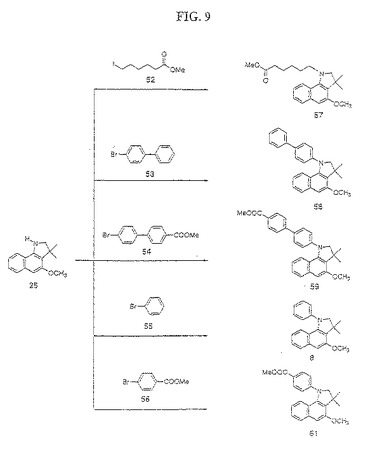
【図10】
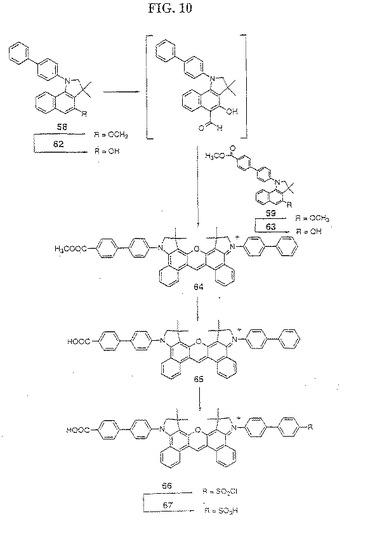
【図11】
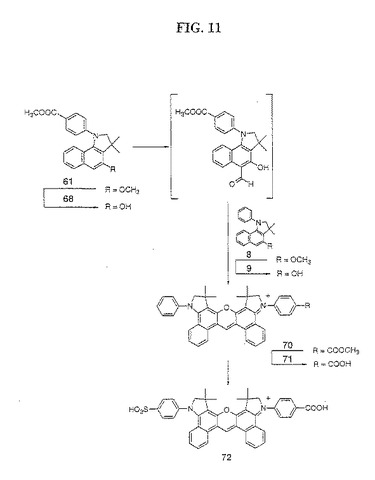
【図12】
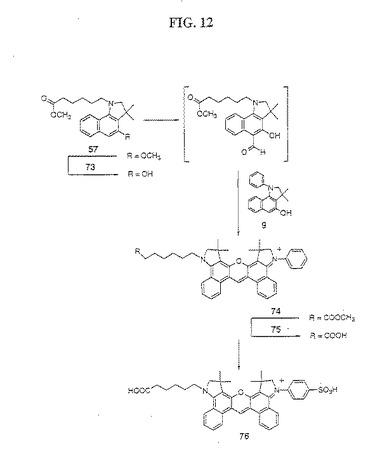
【図13】
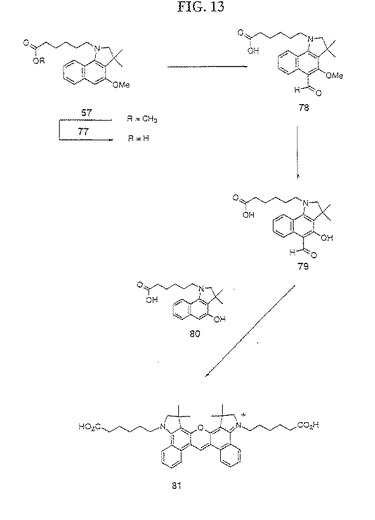
【図14】
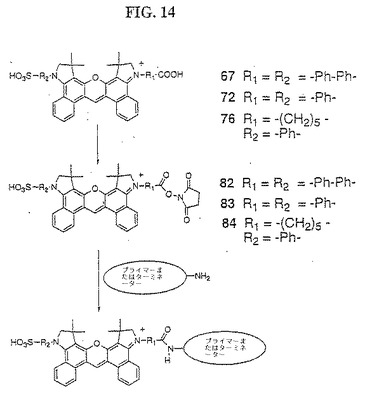
【図15】
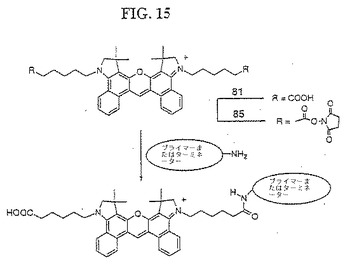
【図16】
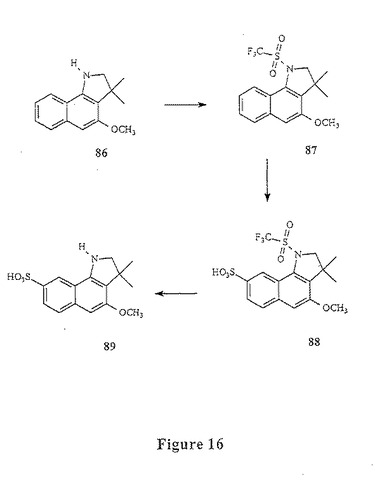
【図17A】
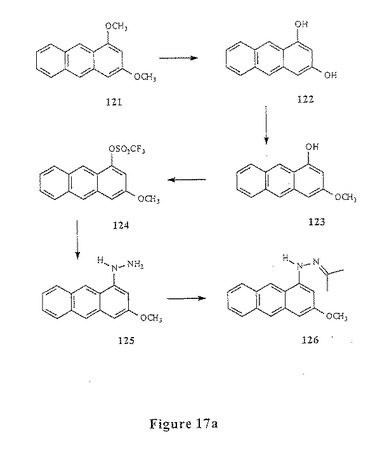
【図17B】
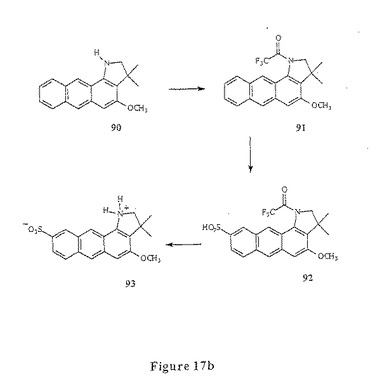
【図18】
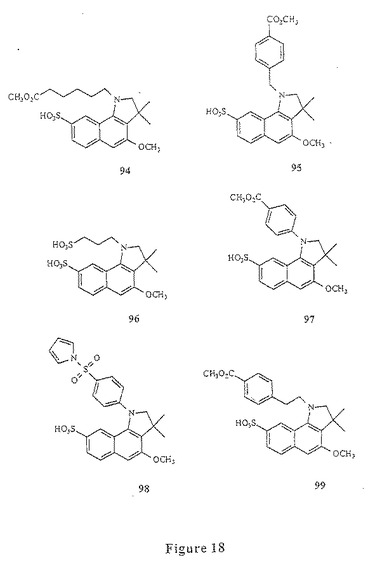
【図19】
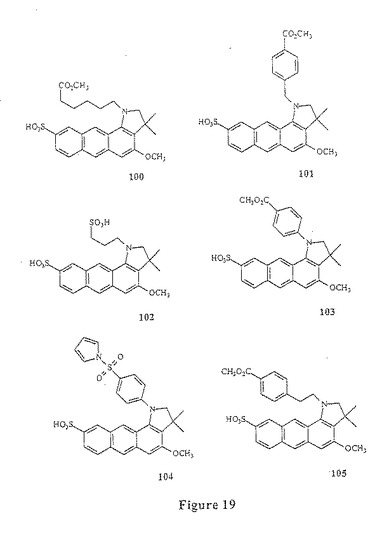
【図20】
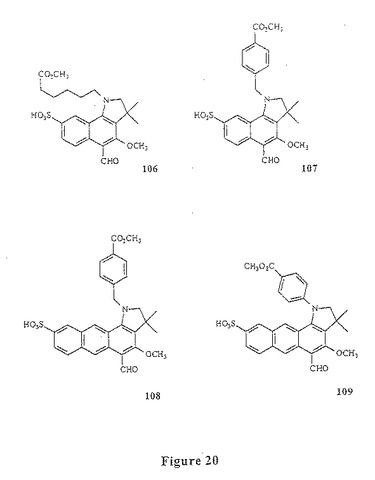
【図21】
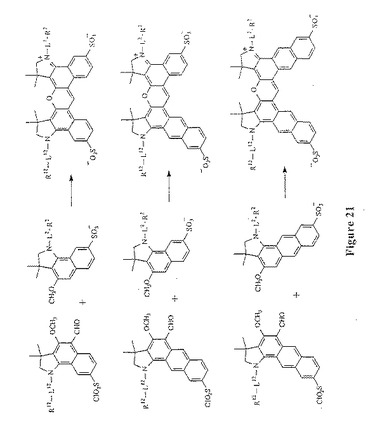
【図22A】
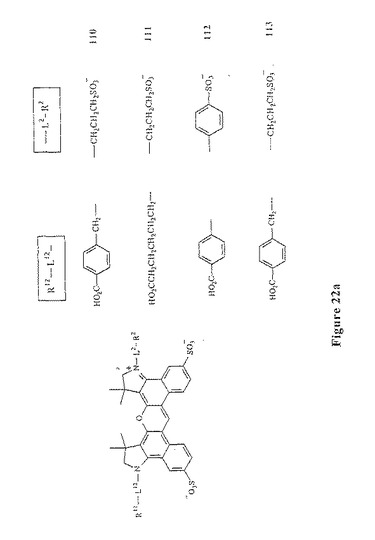
【図22B】
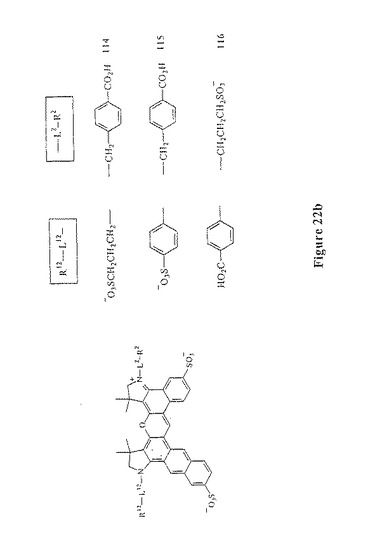
【図22C】
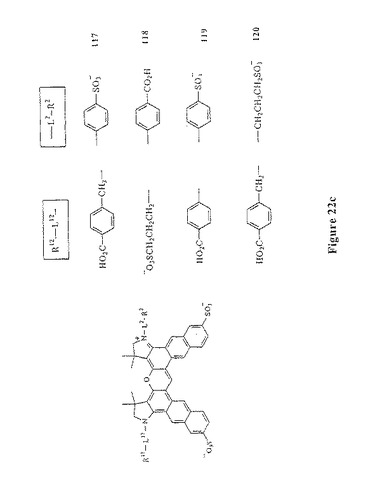
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17A】
【図17B】
【図18】
【図19】
【図20】
【図21】
【図22A】
【図22B】
【図22C】
【公開番号】特開2010−285625(P2010−285625A)
【公開日】平成22年12月24日(2010.12.24)
【国際特許分類】
【出願番号】特願2010−161112(P2010−161112)
【出願日】平成22年7月15日(2010.7.15)
【分割の表示】特願2008−335693(P2008−335693)の分割
【原出願日】平成13年11月27日(2001.11.27)
【出願人】(509130413)アプライド バイオシステムズ, エルエルシー (48)
【Fターム(参考)】
【公開日】平成22年12月24日(2010.12.24)
【国際特許分類】
【出願日】平成22年7月15日(2010.7.15)
【分割の表示】特願2008−335693(P2008−335693)の分割
【原出願日】平成13年11月27日(2001.11.27)
【出願人】(509130413)アプライド バイオシステムズ, エルエルシー (48)
【Fターム(参考)】
[ Back to top ]