
蛍光蛋白質
【課題】励起のピーク値(吸収極大波長)と蛍光のピーク値(蛍光極大波長)の差(ストークスシフト)を大きくすることにより、最大の励起で最大の蛍光を得ることができることを特徴とする赤色又は橙色の蛍光蛋白質を提供すること。
【解決手段】クサビライシ(Fungia sp.)由来の蛍光蛋白質に変異を導入することにより単量体化した新規な蛍光蛋白質及びその利用。コモンサンゴ(Montipora. sp)由来の新規な色素蛋白質及び蛍光蛋白質、並びにその利用。特定の配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【解決手段】クサビライシ(Fungia sp.)由来の蛍光蛋白質に変異を導入することにより単量体化した新規な蛍光蛋白質及びその利用。コモンサンゴ(Montipora. sp)由来の新規な色素蛋白質及び蛍光蛋白質、並びにその利用。特定の配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、単量体で存在する新規な蛍光蛋白質に関する。より詳細には、本発明は、クサビライシ(Fungia sp.)由来の蛍光蛋白質に変異を導入することにより単量体化した新規な蛍光蛋白質及びその利用に関する。さらに本発明は、新規な色素蛋白質並びに蛍光蛋白質に関する。より詳細には、本発明は、コモンサンゴ(Montipora. sp)由来の新規な色素蛋白質及び蛍光蛋白質、並びにその利用に関する。
【背景技術】
【0002】
クラゲのエクオレア・ビクトリア(Aequorea victoria)に由来する緑色蛍光蛋白質(GFP)は、生物系において多くの用途を有する。最近、ランダム突然変異誘発法および半合理的(semi-rational)突然変異誘発法に基づいて、色を変化させたり、折りたたみ特性を改善したり、輝度を高めたり、あるいはpH感受性を改変したといった様々なGFP変異体が作製されている。遺伝子組み換え技術により他の蛋白質をGFP等の蛍光蛋白質に融合させて、それらの発現および輸送のモニタリングを行うことが行われている。
【0003】
最もよく使用されるGFP変異体の一つとして黄色蛍光蛋白質(YFP)が挙げられる。YFPは、クラゲ(Aequorea)GFP変異体の中でも最長波長の蛍光を示す。大部分のYFPのεおよびΦは、それぞれ60,000〜100,000M-1cm-1および0.6〜0.8であり(Tsien, R. Y. (1998). Ann. Rev. Biochem. 67, 509-544)、これらの値は、一般的な蛍光団(フルオレセインおよびローダミンなど)の値に匹敵する。従ってYFPの絶対的輝度の改善は、ほぼ限界に達しつつある。
【0004】
また、GFP変異体の他の例として、シアン色蛍光蛋白質(CFP)があり、ECFP(enhanced cyan fluorescent protein)が知られている。また、イソギンチャク(Discoma sp.)からは赤色蛍光蛋白質(RFP)も単離されており、DasRedが知られている。このように蛍光蛋白質は、緑色、黄色、シアン色、赤色の4種が次々と開発されスペクトルの範囲は大幅に広がっている。
【0005】
先に本発明者らは、クサビライシ(Fungia sp.)のcDNAライブラリーから、既知の蛍光蛋白のアミノ酸配列に基づいて設計した好適なプライマーを用いて蛍光蛋白質遺伝子を増幅してクローニングすることに成功し、得られたクサビライシ(Fungia sp.)由来の蛍光蛋白質の蛍光特性を調べた結果、当該蛍光蛋白質が所望の蛍光特性を有することを見出している(国際公開WO03/54191号公報)。
【0006】
また、オワンクラゲ由来のGFPホモログの中には、ストークスシフト(励起のピーク値と蛍光のピーク値の差)の大きいタイプのもの(GFPuv、sapphire)があるが、380nmのUV光で励起して緑色蛍光を取得するため、生物内での観察には毒性をもつUV光の使用は適さない。赤色蛍光蛋白質についてはストークスシフトの大きなものは存在せず、蛍光観察においては、励起もしくは蛍光のどちらかを犠牲にしなければならないのが現状である。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Tsien, R. Y. (1998). Ann. Rev. Biochem. 67, 509-544
【特許文献】
【0008】
【特許文献1】国際公開WO03/54191号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
国際公開WO03/54191号公報に記載されたイシサンゴ目のクサビライシより単離された蛍光蛋白質Kusabira-Orange(KO)は分子量測定の結果、70kDa(アミノ酸配列から計算される分子量は26kDa)を示し、通常は二量体を形成していると考えられる。近年、蛍光蛋白質をもちいて細胞や分子のラベルする需要が急速に高まっている。細胞をラベルする際には蛍光蛋白質が多量体を形成しようと、蛍光蛋白質自身は細胞質中に漂っているだけなので問題は起こらないが、分子をラベルする際には問題が生じてくる。例えば、ラベルしたい分子が多量体を形成する場合、ターゲット分子と蛍光蛋白質分子が互いに多量体を形成し合い、巨大なポリマーを形成してしまう可能性がある。また、どちらかの多量体形成が阻害された時には、その多量体形成できない分子が本来の性質を失うことになる。蛍光蛋白質を複数用いた分子内FRET(蛍光エネルギー共鳴移動)のプローブにおいても同様に、多量体形成蛍光蛋白質同士を一本のペプチド鎖として発現させた場合に、互いが多量体形成をしあうためにFRETの観測は困難となる。本発明は、上記した問題を解消することを解決すべき課題とするものであり、具体的には、多量体を形成することなく単量体で存在する新規な蛍光蛋白質を提供することを解決すべき課題とした。
【0010】
また、蛍光蛋白質は低分子の蛍光物質に比して励起と蛍光のスペクトルがブロードである。そして、多くの蛍光蛋白質では励起スペクトルと蛍光スペクトルの重なりがあるため、励起のピーク値で励起して蛍光のピーク値で観測することが非常に困難である。本発明は、上記した問題点を解消した蛍光蛋白質を提供することを解決すべき課題とした。即ち、本発明は、励起のピーク値(吸収極大波長)と蛍光のピーク値(蛍光極大波長)の差(ストークスシフト)を大きくすることにより、最大の励起で最大の蛍光を得ることができることを特徴とする赤色又は橙色の蛍光蛋白質を提供することを解決すべき課題とした。
【課題を解決するための手段】
【0011】
本発明者らは上記課題を解決するために鋭意検討し、国際公開WO03/54191号公報に記載された蛋白質KOのアミノ酸配列から多量体形成界面を予測し、多量体形成界面のアミノ酸を置換し、なおかつ蛍光特性を保持するようKOの単量体化を行うことに成功した。さらに本発明者らは、得られた単量体蛍光蛋白質の蛍光特性を調べた結果、所望の蛍光特性を有することを見出した。本発明はこれらの知見に基づいて完成したものである。
【0012】
さらに本発明者らは上記課題を解決するために鋭意検討し、材料としてコモンサンゴ(Montipora. sp)を用いて新規色素蛋白質をコードする遺伝子の単離を試み、色素蛋白質COCPを取得した。次いで、COCP蛋白質の94番目のヒスチジンをアスパラギンに、142番目のアスパラギンをセリンに、157番目のアスパラギンをアスパラギン酸に、201番目のリジンをアルギニンに、206番目のフェニルアラニンをセリンに置き換えることにより蛍光性を獲得した蛍光蛋白質COCP-FLを作成した。COCP-FLは560nmに励起のピークを持ち、この励起によって蛍光スペクトルは600nmにピークした。さらに、本発明者者らは、上記COCP-FLの61番目のセリンをフェニルアラニンに、92番目のイソロイシンをトレオニンに、123番目のバリンをトレオニンに、158番目のフェニルアラニンをチロシンに、191番目のバリンをイソロイシンに、213番目のセリンをアラニンに置き換えることによりCOCP-FLとは異なる蛍光特性をもつ蛋白質keima616を作成した。keima616は、440nmに励起のピークをもち、この励起によって蛍光スペクトルは616nmにピークを持ち、ストークスシフトは176nmと非常に大きな値であった。さらに、本発明者らは、Keima616の61番目のフェニルアラニンをメチオニンに、62番目のグルタミンをシステインに置き換えることにより蛍光蛋白質Keima570を作成した。このKeima570はKeima616と同様440nmに励起のピークを持ち、この励起により570nmの蛍光のピークを示し、ストークスシフトは130nmと大きな値であった。本発明はこれらの知見に基づいて完成したものである。
【0013】
即ち、本発明によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質;
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0014】
本発明の別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0015】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0016】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0017】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0018】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質:
【0019】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号2に記載の塩基配列を有するDNA
(b)配列番号2に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ配列番号2に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質であって、単量体で存在する蛋白質をコードする塩基配列を有するDNA。
【0020】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号4、6、8又は10に記載の塩基配列を有するDNA
(b)配列番号4、6、8又は10に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつそれぞれ配列番号4、6、8又は10に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質。
【0021】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号12、14、16、18、20、22、24、26、28又は30に記載の塩基配列を有するDNA。
(b)配列番号12、14、16、18、20、22、24、26、28又は30に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつそれぞれ12、14、16、18、20、22、24、26、28又は30に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質。
【0022】
さらにまた本発明によれば、以下の(a)又は(b)に示す色素蛋白質が提供される。
(a)配列番号37に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質。
【0023】
本発明の別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号39に記載のアミノ酸配列を有する蛋白質;
(b)配列番号39に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質。
【0024】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【0025】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す色素蛋白質をコードするDNAが提供される。
(a)配列番号37に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質。
【0026】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号39に記載のアミノ酸配列を有する蛋白質;
(b)配列番号39に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質。
【0027】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【0028】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号38に記載の塩基配列を有するDNA;
(b)配列番号38に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ吸光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0029】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号40に記載の塩基配列を有するDNA;
(b)配列番号40に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ蛍光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0030】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ、蛍光特性を有し、100nm以上のストークスシフトを有する蛋白質をコードする塩基配列を有するDNA。
【0031】
本発明のさらに別の態様によれば、上記した本発明のDNAを有する組み換えベクターが提供される。
本発明のさらに別の態様によれば、上記した本発明のDNA又は組み換えベクターを有する形質転換体が提供される。
本発明のさらに別の態様によれば、上記した本発明の蛍光蛋白質と他の蛋白質とから成る融合蛍光蛋白質が提供される。好ましくは、他の蛋白質は細胞内に局在する蛋白質であり、さらに好ましくは、他の蛋白質は細胞内小器官に特異的な蛋白質である。好ましくは、他の蛋白質が蛍光蛋白質である。この場合、好ましくは、融合蛋白質は分子内FRETを生じることができる。
【0032】
本発明のさらに別の態様によれば、上記した本発明の融合蛋白質を細胞内で発現させることを特徴とする、細胞内における蛋白質の局在または動態を分析する方法が提供される。
本発明のさらに別の態様によれば、上記した本発明の蛍光蛋白質、DNA、組み換えベクター、形質転換体、又は融合蛋白質を含む、蛍光試薬キットが提供される。
【発明の効果】
【0033】
本発明により、単量体で存在することができる新規な蛍光蛋白質(mKO)が提供されることになった。二量体の蛍光蛋白質KOによるHeLa細胞でのミトコンドリアラベルにおいて、ミトコンドリアが粒々にラベルされ、本来のミトコンドリア像は得られない。しかし、単量体の蛍光蛋白質mKOでミトコンドリアをラベルした場合には正常な細長いひも状のミトコンドリア像が得られ、ダイナミックなミトコンドリアの動きも観察される。このような単量体化による有効性がミトコンドリア分子のラベルにより確認された。
【0034】
また本発明の蛍光蛋白質(keima616,keima570)は、赤色、橙色の蛍光を放ち、励起のピークが440nm(青色)である。ストークスシフト(励起のピーク値と蛍光のピーク値の差)は従来の赤色蛍光蛋白質(DsRed、HcRed)では20nm〜30nmであるのに対し、本発明の赤色蛍光蛋白質が176nm、橙色蛍光蛋白質が130nmと非常に大きい。故に本発明の蛍光蛋白質は最大の励起で最大の蛍光を得ることができることを特徴とする。また、励起のピークが440nmであるため、青緑蛍光蛋白質(CFP)や緑色蛍光蛋白質(GFP)との同時励起染色において両者の蛍光を非常に有効に取得することが可能である。さらに従来の赤色蛍光蛋白質の励起ピークが560nmから590nmであるのに対し、本発明の蛍光蛋白質は励起のピークが440nmであるので、励起光を変えて従来の赤色蛍光蛋白質と同時に染色することも可能とした。
【図面の簡単な説明】
【0035】
【図1】図1は、mKOの吸収スペクトルを示す。
【図2】図2は、mKOの励起スペクトル(点線)及び蛍光スペクトル(実線)を示す。
【図3】図3は、超遠心による分子量測定の結果を示す。測定結果より分子量は28kDaであることが分かった。
【図4】図4は、HeLa細胞でKO(二量体)を用いてミトコンドリアをラベルした結果を示す。粒状になり正常なミトコンドリアの形態とは異なる。
【図5】図5は、HeLa細胞でmKO(単量体)を用いてミトコンドリアをラベルした結果を示す。ひも状の正常なミトコンドリアの形態として観察される。
【図6】図6は、UV励起緑色蛍光変異体mKVU-1の吸収スペクトルを示す。
【図7】図7は、UV励起緑色蛍光変異体mKVU-1の励起スペクトル及び蛍光スペクトルを示す。
【図8】図8は、青色蛍光変異体mKUV-2の吸収スペクトルを示す。
【図9】図9は、青色蛍光変異体mKUV-2の励起スペクトル及び蛍光スペクトルを示す。
【図10】図10は、緑色蛍光変異体mKO-FM32の吸収スペクトルを示す。
【図11】図11は、緑色蛍光変異体mKO-FM32の励起スペクトル及び蛍光スペクトルを示す。
【図12】図12は、赤色蛍光変異体mKO-F90の吸収スペクトルを示す。
【図13】図13は、赤色蛍光変異体mKO-F90の励起スペクトル及び蛍光スペクトルを示す。
【図14】図14は、mKO時間経過変異体の580nmの励起スペクトルを示す。
【図15】図15は、mKO時間経過変異体の580nmの励起スペクトルを示す。
【図16】図16は、mKO時間経過変異体の蛍光スペクトルを示す。
【図17】図17は、mKO時間経過変異体の蛍光スペクトルを示す。
【図18】図18は、mKO時間経過変異体の蛍光スペクトルを示す。
【図19】図19は、mKO時間経過変異体の蛍光スペクトルを示す。
【図20】図20は、mKO時間経過変異体の蛍光スペクトルを示す。
【図21】図21は、mKO時間経過変異体の蛍光スペクトルを示す。
【図22】図22は、mKO時間経過変異体の蛍光スペクトルを示す。
【図23】図23は、mKOの合成後25時間まで580nmの励起スペクトルを示す。
【図24】図24は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図25】図25は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図26】図26は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図27】図27は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図28】図28は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図29】図29は、mKO時間経過変異体について、緑蛍光の励起ピーク部分の500nmとオレンジ蛍光の励起ピークである548nmの値の比のプロットを示す。
【図30】図30は、mKO-FM14のN末端にTauを連結した融合蛋白質遺伝子をHeLa−S3細胞に遺伝子導入して、イメージングを行った結果を示す。
【図31】図31は、mKO蛋白質に強い緑色光を照射し、照射前後の吸収スペクトルを測定した結果を示す。
【図32】図32は、mKO-FM14蛋白質に強い緑色光を照射し、照射前後の吸収スペクトルを測定した結果を示す。
【図33】図33は、BDNF-mKO-FM14発現遺伝子ベクターの導入後、約12時間から2日間に細胞に発現した融合蛋白質を蛍光顕微鏡で検出し、強い緑色光を照射して、特定の領域のみオレンジ色蛍光を褪色させた結果を示す。
【図34】図34は、BDNF-mKO-FM14発現遺伝子ベクターの導入後、約12時間から2日間に細胞に発現した融合蛋白質を蛍光顕微鏡で検出し、色の変化から分子(BDNF-mKO-FM14)を追跡した結果を示す。
【図35】図35は、分子内FRETを行うための蛍光蛋白質の模式図を示す。
【図36】図36は、単量体蛍光蛋白質mKOと2量体蛍光蛋白質MiCyの蛍光スペクトルと吸収スペクトルを示す。
【図37】図37は、MiCy−linker−mKOを用いて、Caspase3との反応前と反応後の反応液の蛍光スペクトルを440nmで励起して測定した結果を示す。
【図38】図38は、MiCy−linker−mKOを用いてin vivoでCaspase3の活性を測定した結果を示す。
【図39】図39は、COCPの吸収スペクトルを示す。
【図40】図40は、COCPのpH感受性の測定結果を示す。
【図41】図41は、Keima616の励起スペクトルと蛍光スペクトルを示す。
【図42】図42は、Keima570の励起スペクトルと蛍光スペクトルを示す。
【図43】図43は、Keima616のpH感受性の測定結果を示す。
【図44】図44は、Keima570のpH感受性の測定結果を示す。
【図45】図45は、cmkeima620の超遠心分子量測定の結果を示す。
【図46】図46は、cmkeima620の吸収スペクトルを示す。
【図47】図47は、mkeima620の吸収スペクトルを示す。
【図48】図48は、keima616とECFPの励起スペクトルと蛍光スペクトルを示す。
【図49】図49は、Caspase-3の活性測定に用いた蛋白質モチーフを示す。
【図50】図50は、In vitro プロテアーゼ活性と相互相関を示す。リンカー部分にDEVDの配列を挿入したタンデム蛍光蛋白質のサンプルは3種ECFP-Keima616、Keima616-ECFP、EGFP-mRFP1(x2)。 (上段)caspase-3を加える前の自己相関、相互相関関数。 (中段)caspase-3添加後の相互相関関数。 (下段)同添加後の蛍光強度。
【図51】図51は、各融合蛋白モチーフにおけるRelative amplitudeを示す。
【図52】図52は、Caspase-3によるペプチド鎖切断の検出(SDS-PAGE)を示す。
【図53】図53は、タンパク質間相互作用の検出に用いた融合蛋白質モチーフを示す。
【図54】図54は、CaCl2(+)時のECFP−CaMとM13-Keima616の蛍光相互相関関数を示す。
【図55】図55は、CaCl2(−)時のECFP−CaMとM13-Keima616の蛍光相互相関関数を示す。
【発明を実施するための形態】
【0036】
以下、本発明の実施の形態について詳細に説明する。
(1)本発明の蛋白質
(i)本発明の第1の型の蛍光蛋白質
本発明の第1の型の蛍光蛋白質は、以下の(a)又は(b)の何れかに示す蛋白質である。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質;
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
本発明の蛍光蛋白質は、下記の特性を有することを特徴とする。
(1)励起極大波長が548nmであり、蛍光極大波長は559nmである;
(2)548nmにおけるモル吸光係数が、51600である;
(3)量子収率が0.6である;及び
(4)蛍光特性のpH感受性がpKa=5.0である
【0037】
クサビライシ(Fungia sp.)はサンゴの1種で、主に西部大西洋に生息し、群体の外形は多角形で触手が長く、全体が鮮やかなオレンジ色を呈することを特徴とする。
なお、本書中以下の実施例では、クサビライシ(Fungia sp.)を出発材料として上記特性を有する本発明の蛍光蛋白質を取得したが、クサビライシ(Fungia sp.)以外の蛍光を発するサンゴから本発明の蛍光蛋白質を取得することができる場合もあり、そのような蛍光蛋白質も本発明の範囲内である。
【0038】
本明細書で言う「1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
本明細書で言う「同等の蛍光特性」とは、同等の蛍光強度、同等の励起波長、同等の蛍光波長、同等のpH感受性などを有することを意味する。
【0039】
本発明の蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号1から30に記載したアミノ酸配列並びに塩基配列の情報を利用することにより適当なプライマーを設計し、それらを用いて上記した国際公開WO03/54191号公報に記載の蛍光蛋白質のcDNAクローンを鋳型にしてPCRを行うことにより、本発明の蛍光蛋白質をコードするDNAを取得することができる。本発明の蛍光蛋白質をコードするDNAの一部の断片を上記したPCRにより得た場合には、作製したDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の蛍光蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の蛍光蛋白質を産生することができる。発現系での発現については本明細書中後記する。
【0040】
さらに本発明によれば、上記した本発明の蛋白質(mKO)の変異体蛋白質も提供される。具体的には、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0041】
さらに別の具体例としては、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0042】
(ii)本発明の第2の型の蛋白質
本発明の第2の型の蛋白質は、配列番号37、39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質;並びに配列番号37、39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性又は蛍光特性を有する蛋白質である。配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質のストークスシフト(吸収極大波長と蛍光極大波長の差)はそれぞれ176nm、130nm、180nm、及び180nmである。配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質については、そのストークスシフトは100nm以上、より好ましくは120nm以上になるものとする。
【0043】
本発明の蛋白質は、下記の特性を有することを特徴とする。
(1)COCP(アミノ酸配列を配列番号37に示し、塩基配列を配列番号38に示す)
励起極大波長(吸収極大波長):576nm
576nmにおけるモル吸光係数:64000
pH感受性:なし
(2)COCP-FL(アミノ酸配列を配列番号39に示し、塩基配列を配列番号40に示す)
励起極大波長(吸収極大波長):560nm
蛍光極大波長:600nm
(3)keima616(アミノ酸配列を配列番号41に示し、塩基配列を配列番号42に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長:616nm
pH感受性:pH7.5〜10で蛍光強度は安定
【0044】
(4)keima570(アミノ酸配列を配列番号43に示し、塩基配列を配列番号44に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長:570nm
pH感受性:pH7.5〜10で蛍光強度は安定
(5)cmkeima620(アミノ酸配列を配列番号45に示し、塩基配列を配列番号46に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長: 620nm
(6)mkeima620(アミノ酸配列を配列番号47に示し、塩基配列を配列番号48に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長: 620nm
【0045】
本明細書中の実施例においては、本発明の蛋白質をコードするDNAは、コモンサンゴ(Montipora. sp)を出発材料としてクローニングされた。コモンサンゴ(Montipora. sp)は、刺胞動物門花虫綱六放サンゴ亜綱イシサンゴ目ミドリイシ科に属するサンゴの1種であり、塊状や被覆状の群体を形成することが多い。なお、コモンサンゴ(Montipora. sp)以外の蛍光を発するサンゴから本発明の蛋白質を取得することができる場合もあり、そのような蛋白質も本発明の範囲内である。
【0046】
本明細書で言う「1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
【0047】
本明細書において、「吸光特性を有する蛋白質」とは一定の波長の光を吸収できる性質を有する蛋白質を意味する。「配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質」の吸光特性は、配列番号37に記載のアミノ酸配列を有する蛋白質の吸光特性と実質的に同一でもよいし、異なっていてもよい。吸光特性は、例えば、吸光強度、励起波長(吸収波長)、pH感受性などにより評価することができる。本発明の蛋白質のうち吸光特性を有し、蛍光を発しない色素蛋白質は、(1)FRETのアクセプター分子(エネルギー受容体)として用いたり、(2)照射した光のエネルギーを光以外のエネルギーに変換させるシステムの開発に利用したり、あるいは(3)蛋白質のアミノ酸配列に変異を導入して蛍光を発するように改変することなどに用いることができる。
【0048】
本明細書において、「蛍光特性を有する蛋白質」とは、一定の波長の光で励起することにより蛍光を発することができる性質を有する蛋白質を意味する。「配列番号39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質」の蛍光特性はそれぞれ、配列番号39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質の蛍光特性と実質的に同一でもよいし、異なっていてもよい。蛍光特性は、例えば、蛍光強度、励起波長、蛍光波長、pH感受性などにより評価することができる。
【0049】
本発明の色素蛋白質又は蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号37、39、41、43、45又は47に記載したアミノ酸配列並びに配列番号38、40、42、44、46又は48に記載した塩基配列の情報を利用することにより適当なプライマーを設計し、それらを用いてコモンサンゴ(Montipora sp.)由来のcDNAライブラリーを鋳型にしてPCRを行うことにより、本発明の蛋白質をコードするDNAを取得することができる。本発明の蛋白質をコードするDNAの一部の断片を上記したPCRにより得た場合には、作製したDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の蛋白質を産生することができる。発現系での発現については本明細書中後記する。
【0050】
(2)本発明のDNA
本発明によれば、本発明の第1の型の蛍光蛋白質をコードするDNAが提供される。
本発明の第1の型の蛍光蛋白質をコードするDNAの具体例としては、以下の(a)又は(b)に示す蛋白質をコードするDNAが挙げられる。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0051】
本発明の蛍光蛋白質をコードするDNAの更なる具体例としては、以下の(a)又は(b)に示すDNAもまた挙げられる。
(a)配列番号2に記載の塩基配列を有するDNA
(b)配列番号2に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ配列番号2に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質であって、単量体で存在する蛋白質をコードする塩基配列を有するDNA。
また、上記した(1)に記載した本発明の蛋白質(mKO)の変異体蛋白質をコードするDNAも本発明の範囲内である。
【0052】
さらに本発明によれば、本発明の第2の型の蛋白質をコードするDNAが提供される。
本発明の蛋白質をコードするDNAの具体例としては、以下の(a)又は(b)に示す蛋白質をコードするDNAが挙げられる。
(a)配列番号37、39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37、39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性又は蛍光特性を有する蛋白質。
【0053】
本発明の色素蛋白質又は蛍光蛋白質をコードするDNAの更なる具体例としては、以下の(a)又は(b)に示すDNAもまた挙げられる。
(a)配列番号38、40、42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号38、40、42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ吸光特性又は蛍光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0054】
本明細書で言う「1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列」における「1から数個」の範囲は特には限定されないが、例えば、1から50個、好ましくは1から30個、より好ましくは1から20個、さらに好ましくは1から10個、特に好ましくは1から5個程度を意味する。
本発明のDNAは、例えばホスホアミダイト法などにより合成することができるし、特異的プライマーを用いたポリメラーゼ連鎖反応(PCR)によって製造することもできる。本発明のDNA又はその断片の作製方法については、本明細書中上述した通りである。
【0055】
また、所定の核酸配列に所望の変異を導入する方法は当業者に公知である。例えば、部位特異的変異誘発法、縮重オリゴヌクレオチドを用いるPCR、核酸を含む細胞の変異誘発剤又は放射線への露出等の公知の技術を適宜使用することによって、変異を有するDNAを構築することができる。このような公知の技術は、例えば、Molecular Cloning: A laboratory Mannual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.,1989、並びにCurrent Protocols in Molecular Biology, Supplement 1〜38, John Wiley & Sons (1987-1997)に記載されている。
【0056】
(3)本発明の組み換えベクター
本発明のDNAは適当なベクター中に挿入して使用することができる。本発明で用いるベクターの種類は特に限定されず、例えば、自立的に複製するベクター(例えばプラスミド等)でもよいし、あるいは、宿主細胞に導入された際に宿主細胞のゲノムに組み込まれ、組み込まれた染色体と共に複製されるものであってもよい。
【0057】
好ましくは、本発明で用いるベクターは発現ベクターである。発現ベクターにおいて本発明のDNAは、転写に必要な要素(例えば、プロモータ等)が機能的に連結されている。プロモータは宿主細胞において転写活性を示すDNA配列であり、宿主の種類に応じて適宜することができる。
細菌細胞で作動可能なプロモータとしては、バチルス・ステアロテルモフィルス・マルトジェニック・アミラーゼ遺伝子(Bacillusstearothermophilus maltogenic amylase gene)、バチルス・リケニホルミスαアミラーゼ遺伝子(Bacillus licheniformis alpha-amylase gene)、バチルス・アミロリケファチエンス・BANアミラーゼ遺伝子(Bacillus amyloliquefaciens BAN amylase gene)、バチルス・サブチリス・アルカリプロテアーゼ遺伝子(Bacillus Subtilis alkaline protease gene)もしくはバチルス・プミルス・キシロシダーゼ遺伝子(Bacillus pumilus xylosldase gene)のプロモータ、またはファージ・ラムダのPR若しくはPLプロモータ、大腸菌の lac、trp若しくはtacプロモータなどが挙げられる。
【0058】
哺乳動物細胞で作動可能なプロモータの例としては、SV40プロモータ、MT−1(メタロチオネイン遺伝子)プロモータ、またはアデノウイルス2主後期プロモータなどがある。昆虫細胞で作動可能なプロモータの例としては、ポリヘドリンプロモータ、P10プロモータ、オートグラファ・カリホルニカ・ポリヘドロシス塩基性蛋白プロモータ、バキュウロウイルス即時型初期遺伝子1プロモータ、またはバキュウロウイルス39K遅延型初期遺伝子プロモータ等がある。酵母宿主細胞で作動可能なプロモータの例としては、酵母解糖系遺伝子由来のプロモータ、アルコールデヒドロゲナーゼ遺伝子プロモータ、TPI1プロモータ、ADH2-4cプロモータなどが挙げられる。
糸状菌細胞で作動可能なプロモータの例としては、ADH3プロモータまたはtpiAプロモータなどがある。
【0059】
また、本発明のDNAは必要に応じて、例えばヒト成長ホルモンターミネータまたは真菌宿主についてはTPI1ターミネータ若しくはADH3ターミネータのような適切なターミネータに機能的に結合されてもよい。本発明の組み換えベクターは更に、ポリアデニレーションシグナル(例えばSV40またはアデノウイルス5E1b領域由来のもの)、転写エンハンサ配列(例えばSV40エンハンサ)および翻訳エンハンサ配列(例えばアデノウイルス VA RNA をコードするもの)のような要素を有していてもよい。
本発明の組み換えベクターは更に、該ベクターが宿主細胞内で複製することを可能にするDNA配列を具備してもよく、その一例としてはSV40複製起点(宿主細胞が哺乳類細胞のとき)が挙げられる。
【0060】
本発明の組み換えベクターはさらに選択マーカーを含有してもよい。選択マーカーとしては、例えば、ジヒドロ葉酸レダクターゼ(DHFR)またはシゾサッカロマイセス・ポンベTPI遺伝子等のようなその補体が宿主細胞に欠けている遺伝子、または例えばアンピシリン、カナマイシン、テトラサイクリン、クロラムフェニコール、ネオマイシン若しくはヒグロマイシンのような薬剤耐性遺伝子を挙げることができる。
本発明のDNA、プロモータ、および所望によりターミネータおよび/または分泌シグナル配列をそれぞれ連結し、これらを適切なベクターに挿入する方法は当業者に周知である。
【0061】
(4)本発明の形質転換体
本発明のDNA又は組み換えベクターを適当な宿主に導入することによって形質転換体を作製することができる。
本発明のDNAまたは組み換えベクターを導入される宿主細胞は、本発明のDNA構築物を発現できれば任意の細胞でよく、細菌、酵母、真菌および高等真核細胞等が挙げられる。
【0062】
細菌細胞の例としては、バチルスまたはストレプトマイセス等のグラム陽性菌又は大腸菌等のグラム陰性菌が挙げられる。これら細菌の形質転換は、プロトプラスト法、または公知の方法でコンピテント細胞を用いることにより行えばよい。
哺乳類細胞の例としては、HEK293細胞、HeLa細胞、COS細胞、BHK細胞、CHL細胞またはCHO細胞等が挙げられる。哺乳類細胞を形質転換し、該細胞に導入されたDNA配列を発現させる方法も公知であり、例えば、エレクトロポーレーション法、リン酸カルシウム法、リポフェクション法等を用いることができる。
【0063】
酵母細胞の例としては、サッカロマイセスまたはシゾサッカロマイセスに属する細胞が挙げられ、例えば、サッカロマイセス・セレビシエ(Saccharomyces cerevis1ae)またはサッカロマイセス・クルイベリ(Saccharomyces kluyveri)等が挙げられる。酵母宿主への組み換えベクターの導入方法としては、例えば、エレクトロポレーション法、スフェロブラスト法、酢酸リチウム法等を挙げることができる。
他の真菌細胞の例は、糸状菌、例えばアスペルギルス、ニューロスポラ、フザリウム、またはトリコデルマに属する細胞である。宿主細胞として糸状菌を用いる場合、DNA構築物を宿主染色体に組み込んで組換え宿主細胞を得ることにより形質転換を行うことができる。DNA構築物の宿主染色体への組み込みは、公知の方法に従い、例えば相同組換えまたは異種組換えにより行うことができる。
【0064】
昆虫細胞を宿主として用いる場合には、組換え遺伝子導入ベクターおよびバキュロウイルスを昆虫細胞に共導入して昆虫細胞培養上清中に組換えウイルスを得た後、さらに組換えウイルスを昆虫細胞に感染させ、蛋白質を発現させることができる(例えば、Baculovirus Expression Vectors, A Laboratory Manua1;及びカレント・プロトコールズ・イン・モレキュラー・バイオロジー、Bio/Technology, 6, 47(1988)等に記載)。
【0065】
バキュロウイルスとしては、例えば、ヨトウガ科昆虫に感染するウイルスであるアウトグラファ・カリフォルニカ・ヌクレアー・ポリヘドロシス・ウイルス(Autographa californica nuclear polyhedrosis virus)等を用いることができる。
昆虫細胞としては、Spodoptera frugiperdaの卵巣細胞であるSf9、Sf21〔バキュロウイルス・エクスプレッション・ベクターズ、ア・ラボラトリー・マニュアル、ダブリュー・エイチ・フリーマン・アンド・カンパニー(W. H. Freeman and Company)、ニューヨーク(New York)、(1992)〕、Trichoplusia niの卵巣細胞であるHiFive(インビトロジェン社製)等を用いることができる。
組換えウイルスを調製するための、昆虫細胞への組換え遺伝子導入ベクターと上記バキュロウイルスの共導入方法としては、例えば、リン酸カルシウム法又はリポフェクション法等を挙げることができる。
【0066】
上記の形質転換体は、導入されたDNA構築物の発現を可能にする条件下で適切な栄養培地中で培養する。形質転換体の培養物から、本発明の蛍光融合蛋白質を単離精製するには、通常の蛋白質の単離、精製法を用いればよい。
例えば、本発明の蛋白質が、細胞内に溶解状態で発現した場合には、培養終了後、細胞を遠心分離により回収し水系緩衝液に懸濁後、超音波破砕機等により細胞を破砕し、無細胞抽出液を得る。該無細胞抽出液を遠心分離することにより得られた上清から、通常の蛋白質の単離精製法、即ち、溶媒抽出法、硫安等による塩析法、脱塩法、有機溶媒による沈殿法、ジエチルアミノエチル(DEAE)セファロース等のレジンを用いた陰イオン交換クロマトグラフィー法、S-Sepharose FF(ファルマシア社製)等のレジンを用いた陽イオン交換クロマトグラフィー法、ブチルセファロース、フェニルセファロース等のレジンを用いた疎水性クロマトグラフィー法、分子篩を用いたゲルろ過法、アフィニティークロマトグラフィ一法、クロマトフォーカシング法、等電点電気泳動等の電気泳動法等の手法を単独あるいは組み合わせて用い、精製標品を得ることができる。
【0067】
(5)本発明の蛍光蛋白質及びそれを含む融合蛍光蛋白質の利用
本発明は蛍光蛋白質を他の蛋白質と融合させることにより、融合蛍光蛋白質を構築することができる。
本発明の融合蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
【0068】
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号1から30に記載したアミノ酸配列及び塩基配列の情報を利用することにより適当なプライマーを設計し、本発明の蛍光蛋白質の遺伝子を含むDNA断片を鋳型にしてPCRを行うことにより、本発明の蛍光蛋白質をコードするDNAを構築するのに必要なDNA断片を作製することができる。また同様に、融合すべき蛋白質をコードするDNA断片も入手する。
次いで、これらのDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の融合蛍光蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の融合蛍光蛋白質を産生することができる。
【0069】
本発明の蛍光蛋白質は、特に、標識としての利用価値が高い。即ち、本発明の蛍光蛋白質を被検アミノ酸配列との融合蛋白質として精製し、マイクロインジェクション法などの手法により細胞内に導入し、該融合蛋白質の分布を経時的に観察すれば、被検アミノ酸配列の細胞内におけるターゲッティング活性を検出することが可能である。
【0070】
本発明の蛍光蛋白質を融合させる他の蛋白質(被検アミノ酸配列)の種類は特に限定されるものではないが、例えば、細胞内に局在する蛋白質、細胞内小器官に特異的な蛋白質、ターゲティングシグナル(例えば、核移行シグナル、ミトコンドリアプレ配列)等が好適である。なお、本発明の蛍光蛋白質は、マイクロインジェクション法などにより細胞内に導入する以外に、細胞内で発現させて用いることも可能である。この場合には、本発明の蛍光蛋白質をコードするDNAが発現可能に挿入されたベクターが宿主細胞に導入される。
【0071】
また、本発明の蛍光蛋白質は、レポーター蛋白質としてプロモータ活性の測定に用いることも可能である。即ち、被検プロモータの下流に、本発明の蛍光蛋白質をコードするDNAが配置されたベクターを構築し、これを宿主細胞に導入し、該細胞から発せられる本発明の蛍光蛋白質の蛍光を検出することにより、被検プロモータの活性を測定することが可能である。被検プロモータとしては、宿主細胞内で機能するものであれば、特に制限はない。
【0072】
上記アミノ酸配列のターゲティング活性の検出やプロモータ活性の測定において用いられるベクターとしては、特に制限はないが、例えば、動物細胞用ベクターでは、「pNEO」(P. Southern, and P. Berg (1982) J. MOl. Appl. Genet. 1:327)、「pCAGGS」(H.Niwa,K.Yamamura,and J.Miyazaki. Gene 108,193-200(1991))、「pRc/CMV」(インビトロゲン社製)、「pCDM8」(インビトロゲン社製)などが、酵母用ベクターでは、「pRS303」,「pRS304」,「pRS305」,「pRS306」,「pRS313」,「pRS314」,「pRS315」,[pRS316](R.S.Sikorski and P.Hieter (1989) Genetics 122: 19-27)、「pRS423」,「pRS424」,「pRS425」,「pRS426」(T.W.Christianson, R.S.Sikorski, M.Dante, J.H.Shero, and P. Hieter (1992) Gene 110: 119-122)などが好適に用いられる。
【0073】
また、使用可能な細胞の種類も特に限定されず、各種の動物細胞、例えば、L細胞、BalbC-3T3細胞、NIH3T3細胞、CHO(Chinese hamster ovary)細胞、HeLa細胞、NRK(normal rat kidney)細胞、「Saccharomyces cerevisiae」などの酵母細胞や大腸菌(E. coli)細胞などを使用することができる。ベクターの宿主細胞への導入は、例えば、リン酸カルシウム法やエレクトロポレーション法などの常法により行うことができる。
【0074】
上記のようにして得た、本発明の蛍光蛋白質と他の蛋白質(蛋白質Xとする)とを融合させた融合蛍光蛋白質を細胞内で発現させ、発する蛍光をモニターすることにより、細胞内における蛋白質Xの局在や動態を分析することが可能になる。即ち、本発明の融合蛍光蛋白質をコードするDNAで形質転換またはトランスフェクトした細胞を蛍光顕微鏡で観察することにより細胞内における蛋白質Xの局在や動態を可視化して分析することができる。
【0075】
例えば、蛋白質Xとして細胞内オルガネラに特異的な蛋白質を利用することにより、核、ミトコンドリア、小胞体、ゴルジ体、分泌小胞、ペルオキソームなどの分布や動きを観察できる。
また、例えば、神経細胞の軸索、樹状突起などは発生途中の個体の中で著しく複雑な走向の変化を示すので、こういった部位を蛍光ラベルすることにより動的解析が可能になる。
【0076】
本発明の蛍光蛋白質の蛍光は、生細胞のまま検出することが可能である。この検出は、例えば、蛍光顕微鏡(カールツァイス社 アキシオフォト フィルターセット09)や画像解析装置(ATTO デジタルイメージアナライザー)などを用いて行うことが可能である。
顕微鏡の種類は目的に応じて適宜選択できる。経時変化を追跡するなど頻回の観察を必要とする場合には、通常の落射型蛍光顕微鏡が好ましい。細胞内の詳細な局在を追及したい場合など、解像度を重視する場合は、共焦点レーザー顕微鏡の方が好ましい。顕微鏡システムとしては、細胞の生理状態を保ち、コンタミネーションを防止する観点から、倒立型顕微鏡が好ましい。正立顕微鏡を使用する場合、高倍率レンズを用いる際には水浸レンズを用いることができる。
【0077】
フィルターセットは蛍光蛋白質の蛍光波長に応じて適切なものを選択できる。本発明の蛍光蛋白質は、励起極大波長が548nmであり、蛍光極大波長が559nmであることから、励起光530〜550nm、蛍光550〜600nm程度のフィルターを使用することが好ましい。
また、蛍光顕微鏡を用いた生細胞での経時観察を行う場合には、短時間で撮影を行うべきなので、高感度冷却CCDカメラを使用する。冷却CCDカメラは、CCDを冷却することにより熱雑音を下げ、微弱な蛍光像を短時間露光で鮮明に撮影することができる。
【0078】
また、分子間の相互作用を分析する手法の一つとして、FRET(蛍光共鳴エネルギー転移)が知られている。FRETでは、例えば、第一の蛍光蛋白質としてのシアン蛍光蛋白質(CFP)で標識した第一の分子と、第二の蛍光蛋白質としての黄色蛍光蛋白質(YFP)で標識した第二の分子とを共存させることにより、黄色蛍光蛋白質(YFP)をアクセプター分子として作用させ、シアン蛍光蛋白質(CFP)をドナー分子として作用させ、両者の間でFRET(蛍光共鳴エネルギー転移)を生じさせることにより、第一の分子と第二の分子との間の相互作用を可視化することができる。即ち、FRETでは2種類の分子にそれぞれ異なる色素を導入し、エネルギーレベルの高い方の色素(ドナー分子)を選択的に励起し、その色素の蛍光を測定し、もう一方の色素(アクセプター分子)からの長波長蛍光も測定して、それらの蛍光変化量によって分子間の相互作用を可視化する。両方の色素が、2種類の分子の相互作用によって近接したときのみドナー分子の蛍光の減少とアクセプター分子の蛍光の増加が1波長励起2波長測光法により観測される。しかし、アクセプター分子に色素蛋白質を用いた場合は、両方の色素が、2種類の分子の相互作用によって近接したときのみドナー分子の蛍光の減少を生じ1波長励起1波長測光法により観測することができる。即ち、測定機器の簡易化が可能となる。
【0079】
本発明の蛍光蛋白質及び色素蛋白質は、特に、FRET(蛍光共鳴エネルギー転移)におけるドナー分子及びアクセプター分子としての利用価値が高い。即ち、本発明の色素蛋白質と被験物質との融合体(第一の融合体)を作製する。次いで、該被験物質と相互作用する別の被験物質と別の蛍光蛋白質との融合体(第2の融合体)を作製する。そして、第一の融合体と第2の融合体とを相互作用させ、発する蛍光を分析することにより、上記2種類の被験物質間の相互作用を分析することができる。なお、本発明の色素蛋白質を用いたFRET(蛍光共鳴エネルギー転移)は、試験管内で行ってもよいし、細胞内で行ってもよい。
【0080】
さらにまた、本発明の蛍光蛋白質又は色素蛋白質の何れか1種以上をドナー蛋白質又はアクセプター蛋白質として使用することによって、分析物質の標的配列の両端にドナー蛍光蛋白質とアクセプター蛍光蛋白質が結合している構造を有する蛍光指示薬を作成することもできる。分析物質の該標的配列への結合又は作用の有無により、指示薬の立体構造が変化し、これにより蛍光共鳴エネルギー転移(FRET)の有無が生じさせることができる。
【0081】
(6)本発明のキット
本発明によれば、本明細書に記載した蛍光蛋白質、融合蛍光蛋白質、DNA、組み換えベクター又は形質転換体から選択される少なくとも1種以上を含むことを特徴とする、細胞内成分の局在の分析及び/又は生理活性物質の分析のためのキットが提供される。本発明のキットは、それ自体既知の通常用いられる材料及び手法で調製することができる。
【0082】
蛍光蛋白質又はDNAなどの試薬は、適当な溶媒に溶解することにより保存に適した形態に調製することができる。溶媒としては、水、エタノール、各種緩衝液などを用いることができる。
以下の実施例により本発明を具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例】
【0083】
実施例1:点変異導入による多量体形成阻害変異体の作製
KO−1のアミノ酸配列から多量体形成界面を予測し、多量体形成界面のアミノ酸を置換し、なおかつ蛍光特性を保持するようKO−1の単量体化を行った。点変異導入はKO−1を挿入した大腸菌発現ベクター(pRSET B)(国際公開WO03/54191号公報に記載のKO-1をコードするDNAを有する発現ベクター)で点変異導入プライマーを用いて行った。具体的には鋳型プラスミドの片側鎖に複数の変異導入プライマーを同時にアニールさせ、ポリメラーゼで伸長させる。各プライマーにより伸長された各DNA断片を同反応液中でDNAリガーゼを用いてつなぎ、変異導入された部分以外が鋳型と相補的なものを得るという手法を行った。DNAリガーゼで各DNA断片をつなぐ際にDNAの末端にリン酸基を必要とするため、用いたプライマーは5'側のリン酸化を行った。
【0084】
(1)プライマーの5'リン酸化
100μM プライマー 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
上記混合物を37℃で30分間インキュベートした。ここでプライマーとしては、以下の配列番号3から17に記載の塩基配列を有するプライマーを使用した。
【0085】
K11R, F13Y
CCAGAGATGAAGATGAGGTACTACATGGACGGC(配列番号59)
V25I
CATGAGTTCACAATTGAAGGTGAAGGC(配列番号60)
K32R
GAAGGCACAGGCAGACCTTACGAGGGA(配列番号61)
S55A
CCAATGCCTTTCGCGTTTGACTTAGTG(配列番号62)
T62V
TTAGTGTCACACGTGTTCTGTTACGGC(配列番号63)
Q96E
GAAAGGTCGTTGGAGTTCGAAGATGGT(配列番号64)
F102S, A104S
GAAGATGGTGGGTCCGCTTCAGTCAGTGCG(配列番号65)
C115T, E117Y
AGCCTTAGAGGAAACACCTTCTACCACAAATCCA(配列番号66)
V123T
CAAATCCAAATTTACTGGGGTTAACTTTCCTG(配列番号67)
V133I
GCCGATGGTCCTATCATGCAAAACCAAAGT(配列番号68)
S139V
GCCGATGGTCCTATCATGCAAAACCAAAGTGTTGATTGGGAGCCA(配列番号69)
T150A, C151S
GAGAAAATTACTGCCAGCGACGGAGTTCTGAAG(配列番号70)
F162Y, A166E
GATGTTACGATGTACCTAAAACTTGAAGGAGGCGGCAATCAC(配列番号71)
Q190G, F193Y, G195S
CTTAAAATGCCAGGAAGCCATTACATCAGCCATCGCCTCGTCAGG(配列番号72)
C217S
GATGCAGTAGCTCATTCCCTCGAGCACCACCACC(配列番号73)
【0086】
(2)点変異導入PCR
5'リン酸化プライマー 4μl
template(KO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0087】
プログラム:
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
上記の3)〜5)を25サイクル繰り返す
6)75℃ 7 min
7)4℃ hold
【0088】
(3)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0089】
(4)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109に形質転換して変異導入後のKO-1を発現させた。
【0090】
(5)単量体化Kusabira-Orange(mKO)のアミノ酸配列
変異導入後のKO変異体の塩基配列を解析し、アミノ酸配列を決定した。その結果、11番目のリジン(K)をアルギニン(R)に、13番目のフェニルアラニン(F)をチロシン(Y)に、25番目のバリン(V)をイソロイシン(I)に、32番目のリジン(K)をアルギニン(R)に、55番目のセリン(S)をアラニン(A)に、62番目のトレオニン(T)をバリン(V)に、96番目のグルタミン(Q)をグルタミン酸(E)に、102番目のフェニルアラニン(F)をセリン(S)に、104番目のアラニン(A)をセリン(S)に、115番目のシステイン(C)をトレオニン(T)に、117番目のグルタミン酸(E)をチロシン(Y)に、123番目のバリン(V)をトレオニン(T)に、133番目のバリン(V)をイソロイシン(I)に、139番目のセリン(S)をバリン(V)に、150番目のトレオニン(T)をアラニン(A)に、151番目のシステイン(C)をセリン(S)に、162番目のフェニルアラニン(F)をチロシン(Y)に、166番目のアラニン(A)をグルタミン酸(E)に、190番目のグルタミン(Q)をグリシン(G)に、193番目のフェニルアラニン(F)をチロシン(Y)に、195番目のグリシン(G)をセリン(S)に、217番目のシステイン(C)をセリン(S)に置換されていた。さらにKozak配列付加のため2番目のセリン(S)の前にバリン(V)を導入した。この変異体をmKOとした。mKOのアミノ酸配列を配列表の配列番号1に記載し、塩基配列を配列表の配列番号2に記載する。
大腸菌を用いてmKOにHis-Tagを付加した蛋白質を常法により発現させ、Ni-Agaroseを用いて精製した。
【0091】
実施例2:蛍光特性の解析
実施例1で精製したmKO蛋白質の蛍光及び吸収スペクトルを以下の通り測定し、量子収率およびモル吸光係数を算出した。
20μM蛍光蛋白、50mM HEPES pH7.5溶液を用いて吸収スペクトルを測定した。このスペクトルのピークの値よりモル吸光係数を計算した。mKOでは548nmに吸収のピークが認められ、500nmにおける吸収が0.0025となるように蛍光蛋白を上記の緩衝液で希釈して、500nmで励起した時の蛍光スペクトルと590nmにおける蛍光による励起スペクトルを測定した。DsRed(CLONTECH)を同様に500nmにおける吸収が0.0025となるようにして蛍光スペクトルを測定し、DsRedの量子収率を0.29としてmKOの量子収率を求めた。
結果を表1、図1及び図2に示す。表1には、国際公開WO03/54191号公報に記載のKO蛋白質(二量体蛋白質)のデータも併記する。
【0092】
【表1】

【0093】
実施例3:超遠心分析による分子量の測定
mKO蛋白質溶液を150mM KCl,50mM HEPES-KOH pH7.4とした。mKOの分子量決定のため超遠心分析をおこなった。超遠心機XL-1(ベックマン・コールター)を用いて25,000rpm、22時間遠心して、mKOの吸収極大(548nm)付近の540nmの吸収を測定した。その測定結果からmKOの分子量は28kDaと計算された(図3)。これはアミノ酸配列から予測される26kDaとほぼ一致し、mKOが単量体として存在することが確認された。
【0094】
実施例4:ミトコンドリアへのターゲティング
KOおよびmKOのN末端に、Yeast由来のcytochrome oxidaseサブユニット4のN末端12アミノ酸(MLSLRQSIRFFK)を付加し、HeLa細胞のミトコンドリアへのターゲティングを行い、ミトコンドリアのラベルを行った。KO(二量体)は正確にターゲティングされずに、ミトコンドリアが粒々に染色されているのが確認された(図4)。一方、mKO(単量体)は正確にミトコンドリアにターゲティングされ、細長い糸状のミトコンドリアが観察され、単量体化による有効性が確認された(図5)。
【0095】
実施例5:蛍光特性の異なるmKOの変異体の作製
(1)変異導入
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。点変異導入はmKOを挿入した大腸菌発現ベクター(pRSETB)に点変異導入プライマーをもちいてPCRをかけることにより行った。PCRに用いたプライマーは5'側のリン酸化を行った。
【0096】
(a)プライマーの5'リン酸化
100μM primer 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
37℃で30分間インキュベートした。
【0097】
(b)点変異導入PCR
5'リン酸化プライマー 4μl
template(mKO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0098】
プログラム
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
6)75℃ 7 min
7)4℃ hold
3)〜5)を25サイクル
【0099】
(c)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0100】
(d)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109(DE3)に形質転換して変異導入後のmKOを発現させ解析を行なった。
【0101】
(2)mKO変異体のアミノ酸置換部位および蛍光特性
蛍光測定には蛍光分光光度計F-2500(HITACHI)を使用した。吸収測定には分光光度計U-3310(HITACHI)を使用した。
【0102】
(i)UV励起緑色蛍光変異体mKVU-1(アミノ酸配列を配列番号3に示し、塩基配列を配列番号4に示す)
mKOの70番目のプロリン(P)をシステイン(C)に、160番目のバリン(V)をアスパラギン酸(D)に、162番目のメチオニン(M)をロイシン(L)に、176番目のフェニルアラニン(F)をメチオニン(M)にアミノ酸置換することにより、505nmに蛍光ピークを持ち、398nmに励起のピークを持つ緑色蛍光蛋白質となった(図6、7)。モル吸光係数は10000で、蛍光の量子収率は0.27となった。
【0103】
(ii) 青色蛍光変異体mKUV-2(アミノ酸配列を配列番号5に示し、塩基配列を配列番号6に示す)
mKOの65番目のシステイン(C)をグリシン(G)に、70番目のプロリン(P)をグリシン(G)に、160番目のバリン(V)をアスパラギン酸(D)に、176番目のフェニルアラニン(F)をメチオニン(M)にアミノ酸置換することにより、469nmに蛍光ピークを持ち、322nmに励起のピークを持つ青色蛍光蛋白質となった(図8、9)。モル吸光係数は12500で、蛍光の量子収率は0.2となった。
【0104】
(iii) 緑色蛍光変異体mKO-FM32(アミノ酸配列を配列番号7に示し、塩基配列を配列番号8に示す)
mKOの65番目のシステイン(C)をアラニン(A)に、70番目のプロリン(P)をグリシン(G)にアミノ酸置換することにより、506nmに蛍光ピークを持ち、493nmに励起のピークを持つ緑色蛍光蛋白質となった(図10、11)。モル吸光係数は27500で、蛍光の量子収率は0.44となった。
【0105】
(iv)赤色蛍光変異体mKO-F90(アミノ酸配列を配列番号9に示し、塩基配列を配列番号10に示す)
mKOの41番目のメチオニン(M)をロイシン(L)に、49番目のリジン(K)をグルタミン酸(E)に、69番目のアルギニン(R)をリジン(K)に、145番目のセリン(S)をトリプトファン(W)に、185番目のリジン(K)をグルタミン酸(E)に、188番目のリジン(K)をグルタミン酸(E)に、192番目のセリン(S)をアスパラギン酸(D)にアミノ酸置換することにより、582nmに蛍光ピークを持ち、564nmに励起のピークを持つ赤色蛍光蛋白質となった(図12、13)。モル吸光係数は25000で、蛍光の量子収率は0.05となった。
【0106】
実施例6:緑色とオレンジ色の2蛍光を発するmKOの変異体の作製(時間経過測定プローブおよび追跡プローブ)
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。mKOは翻訳されてからすぐは緑の蛍光を放ち、その後オレンジ色の蛍光を放つようになる。しかし、緑色蛍光からオレンジ色蛍光への移行はすばやく完了するために、通常はほとんど見られない。そこで、いろいろな時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質を作製した。この変異体を使用することによって蛋白質発現からの時間を緑色蛍光とオレンジ色蛍光の比で測定することができる。また、この変異体は緑色蛍光とオレンジ色蛍光が独立しているために、オレンジ色蛍光のみを消光させることができた。つまり、オレンジ色蛍光のみを消光させて、オレンジ色蛍光の増加を測定すれば、時間経過測定のリセットも可能となる。さらに、同じくオレンジ色のみ任意の部分を消光して、緑色蛍光とオレンジ色蛍光の比で測定すれば消光した部分のラベルした分子や細胞などの挙動を測定することもできる。結果としてわかったことは70番目のプロリン(P)をアミノ酸置換することにより、多様な、時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質を作製できることであった。
【0107】
(1)変異導入
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。点変異導入はmKOを挿入した大腸菌発現ベクター(pRSETB)に点変異導入プライマーをもちいてPCRをかけることにより行った。PCRに用いたプライマーは5'側のリン酸化を行った。
【0108】
(a)プライマーの5'リン酸化
100μM primer 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
37℃で30分間インキュベートした。
【0109】
(b)点変異導入PCR
5'リン酸化プライマー 4μl
template(mKO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0110】
プログラム
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
6)75℃ 7 min
7)4℃ hold
3)〜5)を25サイクル
【0111】
(c)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0112】
(d)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109(DE3)に形質転換して変異導入後のmKOを発現させ解析を行なった。
【0113】
(2)mKO時間経過変異体の解析
作製されたmKOの変異体は塩基配列の解析により、49番目のリジン(K)をグルタミン酸(E)に、70番目のプロリン(P)をグリシン(G)に、185番目のリジン(K)をグルタミン酸(E)に、188番目のリジン(K)をグルタミン酸(E)に、192番目のセリン(S)をアスパラギン酸(D)に、196番目のセリン(S)をグリシン(G)にアミノ酸置換されていた。このmKOの変異体は時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質であった。このmKOの変異体の70番目のプロリン(P)をいろいろなアミノ酸に置換することにより、時間経過に伴う緑色蛍光とオレンジ色蛍光の比が変化する速度が変わった。
【0114】
グリシン(G)に置換された変異体をmKO-FM9とした(アミノ酸配列を配列番号11に示し、塩基配列を配列番号12に示す)。
アラニン(A)に置換された変異体をmKO-FM5とした(アミノ酸配列を配列番号13に示し、塩基配列を配列番号14に示す)。
セリン(S)に置換された変異体をmKO-FM3とした(アミノ酸配列を配列番号15に示し、塩基配列を配列番号16に示す)。
システイン(C)に置換された変異体をmKO-FM20とした(アミノ酸配列を配列番号17に示し、塩基配列を配列番号18に示す)。
トレオニン(T)に置換された変異体をmKO-FM24とした(アミノ酸配列を配列番号19に示し、塩基配列を配列番号20に示す)。
バリン(V)に置換された変異体をmKO-FM14とした(アミノ酸配列を配列番号21に示し、塩基配列を配列番号22に示す)。
ロイシン(L)に置換された変異体をmKO-FM19とした(アミノ酸配列を配列番号23に示し、塩基配列を配列番号24に示す)。
チロシン(Y)に置換された変異体をmKO-FM23とした(アミノ酸配列を配列番号25に示し、塩基配列を配列番号26に示す)。
グルタミン(Q)に置換された変異体をmKO-FM21とした(アミノ酸配列を配列番号27に示し、塩基配列を配列番号28に示す)。
アスパラギン(N)に置換された変異体をmKO-FM25とした(アミノ酸配列を配列番号29に示し、塩基配列を配列番号30に示す)。
【0115】
それぞれのmKO時間経過変異体の測定は大腸菌JM109(DE3)で発現させたリコンビナント蛍光蛋白質でおこなうか、in vitroトランスレーションシステムPURE SYSTEM CLASSIC MINI(ポストゲノム研究所)を使用した。大腸菌での測定は各変異体を発現させた培養プレートを37℃に保温し、時間を追ってサンプリングして580nmの励起スペクトルを測定した(図14、15)。その結果、緑蛍光の励起ピークである約500nmのピークにくらべ、オレンジ蛍光の励起ピークである548nmのピークが時間により増加し、各変異体によってその増加率は違った。緑蛍光のピークは509nm、オレンジ蛍光のピークは560nmであった(図16、17、18、19、20、21、22;それぞれカッコ内の波長で励起)。蛍光測定には蛍光分光光度計F-2500(HITACHI)を使用した。大腸菌内では新たな蛋白質が断続的に生産されるために、緑からオレンジへの推移に必要とされる時間が見かけ上長くなってしまう。そこで、in vitroトランスレーションシステムを使用することによって蛋白質の生産時間を限定し、より正確な時間に伴う緑からオレンジへの推移を測定した。蛋白質合成時間は1時間とした。その直後にATPなどのタンパク質合成に必要なエネルギー源をゲルろ過で除き、37℃に保温して合成後25時間まで580nmの励起スペクトルを測定した(図23、24、25、26、27、28)。緑蛍光の励起ピーク部分の500nmとオレンジ蛍光の励起ピークである548nmの値の比をプロットすると、これらが比較的にアミノ酸の側鎖が大きくなるに従い(G→A→S→C→T→V→P)、オレンジ色蛍光成分への推移が速くなる傾向があることが分かった(図29)。
【0116】
mKO-FM14のN末端にTau(チューブリンなどに結合し微小管重合を促進し安定化させる蛋白質)を遺伝子的に繋いだ融合蛋白質遺伝子(アミノ酸配列を配列番号31に示し、塩基配列を配列番号32に示す)を動物細胞発現ベクターpCDNA3のBamH1-Xho1サイトにサブクローニングした。作製したベクターをPolyfect(キアゲン)を用いてHeLa−S3細胞に遺伝子導入した。遺伝子導入23時間後に培養液からHBSS(Hanks' Balanced Salt Solution)溶液に置換してイメージングをおこなった。その結果、HeLa−S3細胞がベクターを細胞内に取り込んだ時間差によって、緑色〜オレンジ色まで様々な色調の細胞が観察された。オレンジ色/緑色の比から細胞4と細胞5はベクターを早い時間に細胞内に取り込んで、つづいて細胞1、後に細胞2と細胞3がベクターを細胞内に取り込んでいることが確認できた(図30)。顕微鏡はIX-70(OLYMPUS)を用いた。緑色成分検出のために、励起フィルターは470DF35(OMEGA)、蛍光フィルターはHQ525/50M(CHROMA)、ダイクロイックミラーは505DRLP(OMEGA)を用いた。オレンジ色成分検出のために、励起フィルターはHQ500/40X(CHROMA)、蛍光フィルターはOG550(OMEGA)、ダイクロイックミラーはQ530LP(CHROMA)を用いた。
【0117】
(3)mKO時間経過変異体による分子の追跡
リコンビナントmKO-FM14蛋白質に強い緑色光を照射して、リコンビナントmKO-FM14蛋白質のオレンジ色蛍光成分のみを退色させることができるかを実験した。100Wのキセノンランプに直接フィルターを装着して、強い緑色光をリコンビナントmKO-FM14蛋白質に照射した。フィルターは546DF20(OMEGA)を使用した。コントロールとしてリコンビナントmKO蛋白質も同時に強い緑色光を照射して、照射前後の吸収スペクトルを測定し、548nmの吸収値が低下するかを調べた。吸収測定には分光光度計U-3310(HITACHI)を使用した。その結果、コントロールに用いたリコンビナントmKO蛋白質の548nmの吸収値は変化しなかった。それに対してリコンビナントmKO-FM14蛋白質の548nmの吸収値は優位に低下した。しかし、緑色蛍光成分を発するのに必要である500nmの吸収ピークに変化はなかった(図31、32)。これは、mKO-FM14蛋白質に強い緑色光を照射することによって、オレンジ蛍光成分のみを無くす、もしくは低下させることができることを示す。また、mKO-FM14蛋白質またはmKO-FM14蛋白質を付加したものが満たされた空間において、局所のみの強い緑色光照射によるオレンジ色蛍光の消光または低下により、オレンジ色蛍光シグナルと緑色蛍光シグナルの比を計算すれば、その部位をラベルすることができる。そこでmKO-FM14のN末端にBDNF(brain derived neurotrophic factor)を融合した融合蛋白質遺伝子(アミノ酸配列を配列番号33に示し、塩基配列を配列番号34に示す)をpEGFP-N1(clontech)からEGFP部分を抜き出したものにサブクローニングした。ラット海馬のニューロンに発現させてイメージングを行なった。
【0118】
ラット海馬のニューロンを用意した。妊娠ラット(17-19日目)の胎仔、あるいは生後1-3日目のラットの新生仔より顕微鏡下で海馬(約10匹分)を摘出した。次いで海馬を消化酵素パパインで十数分間、加温処理を行い、さらにピペットを用いて機械的に分散させ神経細胞に富む海馬細胞懸濁液を得た。必要に応じてこの懸濁液を培地で希釈し、ポリーリジンなどの細胞接着基質をコーティングした直径35 mmの培養皿の表面に播種した。播種密度2-4万細胞/cm2程度とし、これらの細胞を培養皿の表面に接着させ、牛胎仔血清およびN2-supplement(神経細胞用添加物)を含むイーグル培地を用いて高密度での初代培養を行なった。培養開始後6-7日目の細胞に対して35 mmの培養皿1枚当たり2-4マイクログラムのDNAをリン酸カルシウム法により37度で30分間、BDNF―mKO-FM14発現遺伝子ベクターの導入を行なった。この遺伝子導入の後、約12時間から2日間に細胞に発現した蛍光タンパクを蛍光顕微鏡で検出し色の変化を追跡する実験に用いた。緑色蛍光シグナル励起には490DF20(OMEGA)に10%減光フィルターを装着したものを、緑色蛍光シグナル検出フィルターは535DF35(OMEGA)を用いた。オレンジ色蛍光シグナル励起には546DF10(OMEGA)を、オレンジ色蛍光シグナル検出フィルターは595RDF60(OMEGA)を用いた。ダイクロイックミラーは505DRLPXR(OMEGA)を使用した。視野絞りを調節して、ラット海馬ニューロンのソーマ部分(細胞体)のみを、550DF30(OMEGA)を使用して強い緑色光でオレンジ色蛍光のみを退色させた。オレンジ蛍光シグナル/緑色蛍光シグナルの比(Ratio)から計算しソーマから神経突起へのBDNF―mKO-FM14の移動を観察した(図33、34)。図34の白矢印はBDNF―mKO-FM14がソーマ部分から神経突起の先端へ向かって移動している様子を示す。
【0119】
実施例7:単量体蛍光蛋白質mKOと2量体(多量体)蛍光蛋白質MiCyを用いたCaspase3活性測定プローブ
分子内FRETを行う際は少なくとも一種は単量体であるべきである。(A)単量体(白)と2量体(黒)の組み合わせ(図35A)。2量体(多量体)蛍光蛋白質MiCyと単量体蛍光蛋白質mKOの組み合わせはこのパターンとなる。例えば、2量体(白)と2量体(黒)の組み合わせはポリマーのように連なってしまうことが考えられる(図35B)。単量体蛍光蛋白質mKOと2量体蛍光蛋白質MiCyはMiCyの蛍光スペクトルとmKOの吸収スペクトルに重なりがあるため、両者を用いたFRET(蛍光共鳴エネルギー移動法)測定が可能である(図36)。そこで、MiCyとmKOをCaspase3の認識配列であるDEVD(Asp-Glu-Val-Asp)を含んだリンカーでつなぎ(アミノ酸配列を配列番号35に示し、塩基配列を配列番号36に示す)、Caspase3の活性化に伴うリンカー配列の切断をFRETにより測定した。
【0120】
(1)in vitroでのCaspase3活性測定
MiCy−linker−mKOの順に繋ぎ、大腸菌発現ベクターpRSETBのBamH1-EcoR1サイトにサブクローニングして大腸菌JM109(DE3)に発現させた。リンカーの配列はGGSGGDEVDGTGGS(Gly-Gly-Ser-Gly-Gly-Asp-Glu-Val-Asp-Gly-Thr-Gly-Gly-Ser)を用いた。これのコンストラクトをMiCy-DEVD-mKOとした。発現したリコンビナント融合蛋白質はNi-NTAアガロースで精製した。精製したリコンビナント融合蛋白質をセファデックスG-25カラムでゲルろ過を行い150mM KCl、50mM HEPES-KOH pH7.4溶液にバッファー置換した。活性測定にはリコンビナントActive−Caspase3(MBL:BV-1083-9)を用いた。20mM HEPES-KOH pH7.4、100mM NaCl、0.1% CHAPS、10% sucrose溶液中に各リコンビナント融合蛋白質を1mg/mlになるようにして、リコンビナントActive−Caspase3を1unit加えて30度で3時間反応させた。反応前と反応後の反応液の蛍光スペクトルを440nmで励起して測定した。測定には蛍光分光光度計F-2500(HITACHI)を使用した。その結果、Caspase3添加まえはFRETがおこってmKOの蛍光ピーク(559nm)が現れているが、添加後にはリンカーの切断によるFRETの解消によりmKOの蛍光ピーク(559nm)は消失し、MiCyの蛍光ピーク(495nm)のみとなった(図37)。
【0121】
(2)in vivoでのCaspase3活性測定
MiCy-DEVD-mKOを動物細胞での発現ベクターpCS2+のBamH1-EcoR1サイトにサブクローニングした。作製したベクターをPolyfect(キアゲン)を用いてHeLa−S3細胞に遺伝子導入した。遺伝子導入24時間後に培養液から500ng/ml 抗Fas抗体(CH-11:MBL)、10μg/ml サイクロヘキシミド、HBSS(Hanks' Balanced Salt Solution)溶液に置換してアポトーシスを誘導し、Caspase3活性測定のイメージングをおこなった。
【0122】
顕微鏡はIX-70(OLYMPUS)を用いた。励起フィルターは440AF21(OMEGA)、ダイクロイックミラーは455DRLP(OMEGA)を用いた。蛍光シグナルの検出は480ALP(OMEGA)のフィルターを通してカラー3CCDカメラASHURA(浜松ホトニクス)で行い、GreenチャンネルでMiCyの蛍光シグナルを、RedチャンネルでmKOの蛍光シグナルを検出した。その結果、HeLa細胞でのアポトーシスに伴い、Caspase3が活性化されて導入遺伝子の翻訳産物のリンカーが切断され、FRETが解消してRedチャンネルのシグナルが低下し、Greenチャンネルのシグナルが上昇する現象が観察された。Red/GreenのRatio(比)はCaspase3の活性化に伴い低下した。また、HeLa細胞のアポトーシスによる形態変化も観察された(図38)。
【0123】
実施例8:イシサンゴからの新規色素蛋白遺伝子の単離、新規蛍光蛋白の作製、及び特性解析
(1)total RNAの抽出
珊瑚より色素蛋白質の遺伝子の単離を行った。材料にはコモンサンゴ(Montipora. sp)を用いた。凍結したコモンサンゴを乳鉢で砕き、湿重量1グラムに"TRIzol"(GIBCO BRL)を7.5ml加えてホモジナイズし、1500×gで10分間遠心した。上清にクロロホルム1.5mlを加え、15秒間攪拌した後、3分間静置した。7500×gで15分間遠心した。上清にイソプロパノール3.75mlを加え、15秒間攪拌した後、10分間静置した。17000×gで10分間遠心した。上清を捨て70%エタノールを6ml加えて17000×gで10分間遠心した。上清を捨て、沈殿をDEPC水200μlで溶解した。DEPC水で溶解したtotal RNAを100倍に希釈して、O.D.260とO.D.280の値を測定してRNA濃度を測った。53μgのtotal RNAを得た。
【0124】
(2)First strand cDNAの合成
total RNA 4μgを使用し、First strand cDNAの合成キット"Ready To Go"(Amersham Pharmacia)によりcDNA(33μl)を合成した。
【0125】
(3)Degenerated PCR
合成したFirst strand cDNA(33μl)のうち3μlを鋳型としてPCRを行った。プライマーのデザインは既知の蛍光蛋白のアミノ酸配列を見比べて、似ている部分を抜き出し、塩基配列に変換し直し作製した。
使用プライマー
5'- GAAGGRTGYGTCAAYGGRCAY -3' (primer1)(配列番号74)
5'- ACVGGDCCATYDGVAAGAAARTT -3'(primer2)(配列番号75)
Iはイノシン、RはA又はG、YはC又はT、VはA,C又はG、DはA,G又はT SはC又はG、HはA,T又はCを示す。
【0126】
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 taq バッファー 5μl
2.5mM dNTPs 4μl
100μM primer1 1μl
100μM primer2 1μl
ミリQ 35μl
taq polymerase(5U/ul) 1μl
【0127】
PCR反応条件
94℃ 1分(PAD)
94℃ 30秒(変性)
52℃ 30秒(鋳型へのプライマーのアニーリング)
72℃ 1分 (プライマーの伸長)
上記3ステップを35サイクル行った。
72℃ 7分(最後の伸長)
4℃ 保持
【0128】
一回目のPCR反応で得られた増幅産物1μlをテンプレートとして、もう一度同じ条件でPCRを行った。アガロースゲル電気泳動で、350bpを切り出し、精製した。
【0129】
(4)サブクローニング及び塩基配列の決定
精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。得られた塩基配列を他の蛍光蛋白遺伝子の塩基配列と比較してそのDNA塩基配列が蛍光蛋白由来のものであるかを判断した。蛍光蛋白遺伝子の一部であると判断したものに関して、5'-RACE法および3'-RACE法による遺伝子全長のクローニングを行った。
【0130】
(5)5'-RACE法
Degenerated PCRで得られたDNA断片の5'側の塩基配列を決定するために5'-RACE System for Rapid Amplification of cDNA Ends,Version 2.0(GIBCO BRL)を用いて、5'-RACE法を行った。鋳型として(1)で調整したtotal RNAを5μg使用した。
【0131】
dC-tailed cDNAの一回目の増幅には
5'-GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG-3' (primer3)(配列番号76)
5'- CTCAGGGAATGACTGCTTTACAT -3' (primer4)(配列番号77)
のプライマーを用いた。
Iはイノシンを示す。
【0132】
二回目の増幅には
5'-GGCCACGCGTCGACTAGTAC-3' (primer5)(配列番号78)
5'- GTCTTCAGGGTACTTGGTGA -3' (primer6)(配列番号79)
のプライマーを用いた。PCR反応条件等はキットのプロトコールに準じた。
アガロースゲル電気泳動で、増幅された350bpのバンドを切り出し、精製した。精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。
【0133】
(6)3'-RACE法
Degenerated PCRで得られたDNA断片の3'側部分は、(4)の塩基配列決定で得られた情報を基に作製したプライマーとオリゴdTプライマーのPCRで得た。鋳型として(2)で調整したfirst strand cDNAを3μl使用した。
【0134】
作成したプライマーは、
5'- ATGTAAAGCAGTCATTCCCTGAG -3' (primer7)(配列番号80)
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 taq バッファー 5μl
2.5mM dNTPs 4μl
20μM primer7 1μl
10μM オリゴdTprimer 1μl
ミリQ 35μl
taq polymerase(5U/μl) 1μl
【0135】
PCR反応条件
94℃ 1min(PAD)
94℃ 30sec (変性)
52℃ 30sec (鋳型へのプライマーのアニーリング)
72℃ 1min (プライマーの伸長)
上記3ステップを30サイクル行った。
72℃ 7min (最後の伸長)
4℃ 保持
【0136】
アガロースゲル電気泳動で、増幅された約650bpのバンドを切り出し、精製した。精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。
【0137】
(7)大腸菌での蛋白発現
得られた全長の塩基配列より、蛋白のN末端に相当する部分でプライマーを作製し、C末端側はオリゴdTプライマーを使用して、(2)で調整したFirst strand cDNAを鋳型としてPCRを行った。全アミノ酸配列および全塩基配列を配列表の配列番号37及び38に示す。配列番号37に記載のアミノ酸配列を有する蛋白質をCOCPと称する。
【0138】
使用プライマー
5'- CCCGGATCCGACCATGGCTACCTTGGTTAAAGA -3' (primer8)(配列番号81)
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 pyrobest バッファー 5μl
2.5mM dNTPs 4μl
100uM primer8 1μl
100uM オリゴdTプライマー 1μl
ミリQ 35μl
pyrobest polymerase(5U/μl) 1μl
【0139】
PCR反応条件
94℃ 1min(PAD)
94℃ 30sec (変性)
52℃ 30sec (鋳型へのプライマーのアニーリング)
72℃ 1min (プライマー伸長)
上記3ステップを30サイクル行った。
72℃ 7min (最後の伸長)
4℃ 保持
【0140】
アガロースゲルの電気泳動で、増幅された約800bpのバンドを切り出し、精製してpRSET vector(Invitrogen)のBamHI、EcoRI部位にサブクローニングして、大腸菌株(JM109-DE3)で発現させた。発現蛋白はN末端にHis-tagが付くようにコンストラクトしたので発現蛋白はNi-Agarose gel(QIAGEN)で精製した。精製の方法は付属のプロトコールに準じた。次に精製した蛋白の性質を解析した。
【0141】
(8)光吸収特性の解析
20μM色素蛋白、50mM HEPES pH7.9溶液を用いて吸収スペクトルを測定した。このスペクトルのピークの値よりモル吸光係数を計算した。コモンサンゴ由来色素蛋白(COCP)では576nmに吸収のピークが認められた(表2、図39)。また、pH4〜10で安定していた。(図40)
【0142】
【表2】
【0143】
(9)色素蛋白質から蛍光蛋白質への改変
COCPは蛍光蛋白質ではない。しかしCOCPの1番目のメチオニンと2番目のセリンの間にバリンを挿入し、94番目のヒスチジンをアスパラギンに、142番目のアスパラギンをセリンに、157番目のアスパラギンをアスパラギン酸に、202番目のリジンをアルギニンに、206番目のフェニルアラニンをセリンに置き換えることにより蛍光性を獲得した。この改変蛍光蛋白質をCOCP-FLとした(アミノ酸配列を配列番号39に示し、塩基配列を配列番号40に示す)。COCP-FLは560nmに励起のピークを持つ。この励起によって蛍光スペクトルは600nmにピークを示す。
【0144】
(10)ストークスシフトの大きな赤色蛍光蛋白質の作製
COCP-FLの62番目のセリンをフェニルアラニンに、93番目のイソロイシンをトレオニンに、124番目のバリンをトレオニンに、159番目のフェニルアラニンをチロシンに、192番目のバリンをイソロイシンに、214番目のセリンをアラニンに置き換えることによりCOCP-FLとは異なる蛍光をもつ蛋白質を獲得した。この改変蛍光蛋白質をkeima616(アミノ酸配列を配列番号41に示し、塩基配列を配列番号42に示す)とした。440nmに励起のピークをもち、この励起によって蛍光スペクトルは616nmにピークを持つ(図41、表2)。ストークスシフトは176nmと非常に大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないためできなかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。
【0145】
(11)ストークスシフトの大きな橙色蛍光蛋白質の作製
Keima616の62番目のフェニルアラニンをメチオニンに、63番目のグルタミンをシステインに置き換えることにより蛍光蛋白質を獲得した。この改変蛍光蛋白質をKeima570(アミノ酸配列を配列番号43に示し、塩基配列を配列番号44に示す)とした。このKeima570はKeima616と同様440nmに励起のピークを持ち、この励起により570nmの蛍光のピークを示す(図42)。ストークスシフトは130nmと大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないためできなかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。
【0146】
(12)pH感受性の測定
50mMの下記の緩衝液中で蛋白質(Keima616及びKeima570)の吸収スペクトルを測定した(図43及び44)。
各pHの緩衝液は次の通り、
pH4、5、5.5 : 酢酸バッファー
pH6 : リン酸バッファー
pH6.6 : MOPSバッファー
pH7、7.5、8 : HEPESバッファー
pH9、10 : グリシンバッファー
pH7.5〜10でピークの値は安定していた。(図43及び図44)
【0147】
実施例9
(1) ストークスシフトの大きな単量体赤色蛍光蛋白質の作製
keim616の61番目のロイシンをグルタミンに、93番目のトレオニンをセリンに、124番目のトレオニンをグルタミン酸に、189番目のチロシンをアルギニンに、191番目のチロシンをグルタミン酸に置き換えることにより超遠心分子量測定により分子量30.1kDaの結果から、アミノ酸配列から予想されるおよそ29kDaとほぼ一致することから単量体となったkeima616蛋白を獲得した。本改変蛍光蛋白質をcmkeima620とした(図45)(アミノ酸配列を配列表の配列番号45に示し、塩基配列を配列番号46に示す)。440nmに励起のピークをもち、この励起によって蛍光スペクトルは620nmにピークを持つ(図46)。ストークスシフトは180nmと非常に大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないため出来なかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。また、全体の分子量をおさえ且つ蛍光蛋白質自身の間で多量体形成による相互作用がないため、ラベル分子の性質変化を最小限にとどめることができる。
【0148】
(2)ストークスシフトの大きな単量体赤色蛍光蛋白質の改良
cmkeim616の62番目のフェニルアラニンをロイシンに置き換えることによりcmkeima620のフォールディング効率が上昇したものを得た。本改変蛍光蛋白質をmkeima620とした(アミノ酸配列を配列表の配列番号47に示し、塩基配列を配列番号48に示す)。440nmに励起のピークをもち、この励起によって蛍光スペクトルは620nmにピークを持つ。ストークスシフトは180nmと非常に大きな値である。cmkeima620よりも相対的な蛍光強度が高いため(図46、47)、単量体でもkeima616と同様に十分使いやすくなっている。
【0149】
実施例10:ストークスシフトの大きな蛋白質を用いた一波長励起二波長測光型蛍光相互相関分光法の測定系の開発
分子間相互作用を測定するための手法として、蛍光分子を利用した蛍光相互相関分光法(FCCS) がある。これは2つの蛍光分子をプローブとして用いることにより分子間相互作用をモニタリングできる。
【0150】
現在用いられている2つの異なる蛍光分子を用いた2 波長励起FCCS 測定において相互相関の感度を下げる大きな要員として色収差による2 波長の測定領域の重なりのずれがあげられる。Keima616とECFPのような一つの波長で励起できしかも蛍光が分離できる蛍光蛋白質の組み合わせを用いた一波長励起FCCS ではこれを回避する事ができるため、FCCS 測定の感度の上昇が期待される(図48)。また、Fluorescence Resonance energy transfer(FRET)を回避できるため、FCCSでの測定が簡便化され、蛋白質間相互作用のFCCSによる検出に適している。従ってストークスシフトが大きい蛍光蛋白質であるkeima616を用いることによりFCCSによる蛋白質間相互作用の検出が簡便かつ強力なものになると思われる。
【0151】
(1) Caspase-3の活性検出
(a)蛍光相互相関測定における装置
蛍光相互相関測定にはTCS SP2 SOBS(Leica)とFCCSシステムを用いた。EGFP-(スペーサー)DEVD-mRFP1には、458 nm Argon ion Laserと594 nm HeNe Laserを用いて2 波長励起を行なった。またECFPとKeima616の組み合わせの蛋白質には458nm Argon Laserを用いた。受光用バンドパスフィルタはEGFP:500-550、mRFP1:607-683を、ECFP :470-500、keima616:535-585を用いた。
【0152】
(b)蛍光相互相関測定の解析
Caspase-3 により切断されるアミノ酸配列DEVDをEGFPとmRFPの間、keima616とECFPの間に導入し (図49)、リコンビナントEGFP-DEVDmRFP1(x2)(アミノ酸配列を配列表の配列番号49に示し、塩基配列を配列番号50に示す) 、ECFP-(スペーサー)DEVD-keima616(アミノ酸配列を配列表の配列番号51に示し、塩基配列を配列番号52に示す)、keima616-(スペーサー)DEVD- ECFP(アミノ酸配列を配列表の配列番号53に示し、塩基配列を配列番号54に示す)を作製した。発現蛋白質はN末端にHis-tagがつくようにコンストラクトしたので発現蛋白質はNi-Agarose gel(QIAGEN)で精製した。精製方法は付属のプロトコールに準じた。次にそれらの蛋白質を用いて相関作用を解析した。
【0153】
相互相関の定量的な評価は、relative amplitudeという相互相関関数の振幅(Gcross(0))を自己相関関数の振幅(Glower(0)) で割算した値を用いた。EGFP-DEVD-mRFP1(x2) では、Gcross(0) /Glower(0) は約0.4 であった(図51)。Caspase-3の添加によりGcross(0) の減少が観られた(図50)。
ECFPとkeima616の組み合わせではGcross(0) /Glower(0)は0.4であった(図51)。Caspase-3の添加によりGcross(0)の迅速な減少が見られた。Gcross(0)の減少はCaspase-3添加により蛍光相関が無くなっていることを示している。EGFP-DEVD-mRFPよりもECFPとKeima616を組み合わせた蛋白質がより短時間で相関が無くなっておりこれにより後者の組み合わせがより蛍光相互相関法により簡便かつ迅速に蛋白質の相互作用を示すことが明らかになった。
【0154】
(c) SDS-PAGEによる蛋白質間相互作用の解析
keima616-DEVD-ECFPをCaspase-3で反応させるとkeima616とECFPの大きさのバンドが確認出来た。これらの蛋白質はCaspase-3によってDEVDが切断されたことを意味する(図52)。Native-PAGEにおいても同様に反応後に2つのバンドが確認され、それぞれがkeima616とECFPであることが同定され、蛍光検出でもCaspase-3の活性があることが検出できた(図52)。
【0155】
(2)カルモジュリンとの相互作用
(a)蛋白質の合成・発現
カルモジュリンにはECFPを、M13にはKeima616を繋いだ(図53)。ECFP−カルモジュリンのアミノ酸配列を配列番号55に示し、塩基配列を配列番号56に示す。また、M13−Keima616のアミノ酸配列を配列番号57に示し、塩基配列を配列番号58に示す。それらの融合蛋白質は大腸菌株(JM109-DE3)で発現させた。発現蛋白はN末端にHis-tagがつくようにコンストラクトしたので発現蛋白はNi-Agarose gel(QIAGEN)で精製した。精製方法は付属のプロトコールに準じた。次にそれらの蛋白質を用いて相関作用を解析した。
【0156】
(b)蛍光相互相関測定における装置
蛍光相互相関測定にはConfoCor2(Carl Zeiss)とLSM510 version3.2を用いた。458 nm Argon ion Laserを用いた。受光用バンドパスフィルタはECFP:475-525、Keima616:LP610を用いた。
【0157】
(c)蛍光相互相関測定の解析
相互相関の定量的な評価は、relative amplitudeという相互相関関数の振幅(Gcross(0))を自己相関関数の振幅(Glower(0)) で割算した値を用いた。EGTAによりカルシウムイオンをキレートしたサンプルではGcross(0) /Glower(0) は約0.005であった(図54)。しかしカルシウムイオンの添加によりGcross(0) の値の上昇が確認できた(図55)。この結果はカルシウム依存的な蛋白質の相互作用を検出したことを示している。これにより蛍光相互相関法により蛋白間相互作用が迅速且つ簡便に測定できることが明らかになった。
【技術分野】
【0001】
本発明は、単量体で存在する新規な蛍光蛋白質に関する。より詳細には、本発明は、クサビライシ(Fungia sp.)由来の蛍光蛋白質に変異を導入することにより単量体化した新規な蛍光蛋白質及びその利用に関する。さらに本発明は、新規な色素蛋白質並びに蛍光蛋白質に関する。より詳細には、本発明は、コモンサンゴ(Montipora. sp)由来の新規な色素蛋白質及び蛍光蛋白質、並びにその利用に関する。
【背景技術】
【0002】
クラゲのエクオレア・ビクトリア(Aequorea victoria)に由来する緑色蛍光蛋白質(GFP)は、生物系において多くの用途を有する。最近、ランダム突然変異誘発法および半合理的(semi-rational)突然変異誘発法に基づいて、色を変化させたり、折りたたみ特性を改善したり、輝度を高めたり、あるいはpH感受性を改変したといった様々なGFP変異体が作製されている。遺伝子組み換え技術により他の蛋白質をGFP等の蛍光蛋白質に融合させて、それらの発現および輸送のモニタリングを行うことが行われている。
【0003】
最もよく使用されるGFP変異体の一つとして黄色蛍光蛋白質(YFP)が挙げられる。YFPは、クラゲ(Aequorea)GFP変異体の中でも最長波長の蛍光を示す。大部分のYFPのεおよびΦは、それぞれ60,000〜100,000M-1cm-1および0.6〜0.8であり(Tsien, R. Y. (1998). Ann. Rev. Biochem. 67, 509-544)、これらの値は、一般的な蛍光団(フルオレセインおよびローダミンなど)の値に匹敵する。従ってYFPの絶対的輝度の改善は、ほぼ限界に達しつつある。
【0004】
また、GFP変異体の他の例として、シアン色蛍光蛋白質(CFP)があり、ECFP(enhanced cyan fluorescent protein)が知られている。また、イソギンチャク(Discoma sp.)からは赤色蛍光蛋白質(RFP)も単離されており、DasRedが知られている。このように蛍光蛋白質は、緑色、黄色、シアン色、赤色の4種が次々と開発されスペクトルの範囲は大幅に広がっている。
【0005】
先に本発明者らは、クサビライシ(Fungia sp.)のcDNAライブラリーから、既知の蛍光蛋白のアミノ酸配列に基づいて設計した好適なプライマーを用いて蛍光蛋白質遺伝子を増幅してクローニングすることに成功し、得られたクサビライシ(Fungia sp.)由来の蛍光蛋白質の蛍光特性を調べた結果、当該蛍光蛋白質が所望の蛍光特性を有することを見出している(国際公開WO03/54191号公報)。
【0006】
また、オワンクラゲ由来のGFPホモログの中には、ストークスシフト(励起のピーク値と蛍光のピーク値の差)の大きいタイプのもの(GFPuv、sapphire)があるが、380nmのUV光で励起して緑色蛍光を取得するため、生物内での観察には毒性をもつUV光の使用は適さない。赤色蛍光蛋白質についてはストークスシフトの大きなものは存在せず、蛍光観察においては、励起もしくは蛍光のどちらかを犠牲にしなければならないのが現状である。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Tsien, R. Y. (1998). Ann. Rev. Biochem. 67, 509-544
【特許文献】
【0008】
【特許文献1】国際公開WO03/54191号公報
【発明の概要】
【発明が解決しようとする課題】
【0009】
国際公開WO03/54191号公報に記載されたイシサンゴ目のクサビライシより単離された蛍光蛋白質Kusabira-Orange(KO)は分子量測定の結果、70kDa(アミノ酸配列から計算される分子量は26kDa)を示し、通常は二量体を形成していると考えられる。近年、蛍光蛋白質をもちいて細胞や分子のラベルする需要が急速に高まっている。細胞をラベルする際には蛍光蛋白質が多量体を形成しようと、蛍光蛋白質自身は細胞質中に漂っているだけなので問題は起こらないが、分子をラベルする際には問題が生じてくる。例えば、ラベルしたい分子が多量体を形成する場合、ターゲット分子と蛍光蛋白質分子が互いに多量体を形成し合い、巨大なポリマーを形成してしまう可能性がある。また、どちらかの多量体形成が阻害された時には、その多量体形成できない分子が本来の性質を失うことになる。蛍光蛋白質を複数用いた分子内FRET(蛍光エネルギー共鳴移動)のプローブにおいても同様に、多量体形成蛍光蛋白質同士を一本のペプチド鎖として発現させた場合に、互いが多量体形成をしあうためにFRETの観測は困難となる。本発明は、上記した問題を解消することを解決すべき課題とするものであり、具体的には、多量体を形成することなく単量体で存在する新規な蛍光蛋白質を提供することを解決すべき課題とした。
【0010】
また、蛍光蛋白質は低分子の蛍光物質に比して励起と蛍光のスペクトルがブロードである。そして、多くの蛍光蛋白質では励起スペクトルと蛍光スペクトルの重なりがあるため、励起のピーク値で励起して蛍光のピーク値で観測することが非常に困難である。本発明は、上記した問題点を解消した蛍光蛋白質を提供することを解決すべき課題とした。即ち、本発明は、励起のピーク値(吸収極大波長)と蛍光のピーク値(蛍光極大波長)の差(ストークスシフト)を大きくすることにより、最大の励起で最大の蛍光を得ることができることを特徴とする赤色又は橙色の蛍光蛋白質を提供することを解決すべき課題とした。
【課題を解決するための手段】
【0011】
本発明者らは上記課題を解決するために鋭意検討し、国際公開WO03/54191号公報に記載された蛋白質KOのアミノ酸配列から多量体形成界面を予測し、多量体形成界面のアミノ酸を置換し、なおかつ蛍光特性を保持するようKOの単量体化を行うことに成功した。さらに本発明者らは、得られた単量体蛍光蛋白質の蛍光特性を調べた結果、所望の蛍光特性を有することを見出した。本発明はこれらの知見に基づいて完成したものである。
【0012】
さらに本発明者らは上記課題を解決するために鋭意検討し、材料としてコモンサンゴ(Montipora. sp)を用いて新規色素蛋白質をコードする遺伝子の単離を試み、色素蛋白質COCPを取得した。次いで、COCP蛋白質の94番目のヒスチジンをアスパラギンに、142番目のアスパラギンをセリンに、157番目のアスパラギンをアスパラギン酸に、201番目のリジンをアルギニンに、206番目のフェニルアラニンをセリンに置き換えることにより蛍光性を獲得した蛍光蛋白質COCP-FLを作成した。COCP-FLは560nmに励起のピークを持ち、この励起によって蛍光スペクトルは600nmにピークした。さらに、本発明者者らは、上記COCP-FLの61番目のセリンをフェニルアラニンに、92番目のイソロイシンをトレオニンに、123番目のバリンをトレオニンに、158番目のフェニルアラニンをチロシンに、191番目のバリンをイソロイシンに、213番目のセリンをアラニンに置き換えることによりCOCP-FLとは異なる蛍光特性をもつ蛋白質keima616を作成した。keima616は、440nmに励起のピークをもち、この励起によって蛍光スペクトルは616nmにピークを持ち、ストークスシフトは176nmと非常に大きな値であった。さらに、本発明者らは、Keima616の61番目のフェニルアラニンをメチオニンに、62番目のグルタミンをシステインに置き換えることにより蛍光蛋白質Keima570を作成した。このKeima570はKeima616と同様440nmに励起のピークを持ち、この励起により570nmの蛍光のピークを示し、ストークスシフトは130nmと大きな値であった。本発明はこれらの知見に基づいて完成したものである。
【0013】
即ち、本発明によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質;
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0014】
本発明の別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0015】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0016】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0017】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0018】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質:
【0019】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号2に記載の塩基配列を有するDNA
(b)配列番号2に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ配列番号2に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質であって、単量体で存在する蛋白質をコードする塩基配列を有するDNA。
【0020】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号4、6、8又は10に記載の塩基配列を有するDNA
(b)配列番号4、6、8又は10に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつそれぞれ配列番号4、6、8又は10に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質。
【0021】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号12、14、16、18、20、22、24、26、28又は30に記載の塩基配列を有するDNA。
(b)配列番号12、14、16、18、20、22、24、26、28又は30に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつそれぞれ12、14、16、18、20、22、24、26、28又は30に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質。
【0022】
さらにまた本発明によれば、以下の(a)又は(b)に示す色素蛋白質が提供される。
(a)配列番号37に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質。
【0023】
本発明の別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号39に記載のアミノ酸配列を有する蛋白質;
(b)配列番号39に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質。
【0024】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【0025】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す色素蛋白質をコードするDNAが提供される。
(a)配列番号37に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質。
【0026】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号39に記載のアミノ酸配列を有する蛋白質;
(b)配列番号39に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質。
【0027】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示す蛍光蛋白質をコードするDNAが提供される。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【0028】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号38に記載の塩基配列を有するDNA;
(b)配列番号38に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ吸光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0029】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号40に記載の塩基配列を有するDNA;
(b)配列番号40に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ蛍光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0030】
本発明のさらに別の態様によれば、以下の(a)又は(b)に示すDNAが提供される。
(a)配列番号42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ、蛍光特性を有し、100nm以上のストークスシフトを有する蛋白質をコードする塩基配列を有するDNA。
【0031】
本発明のさらに別の態様によれば、上記した本発明のDNAを有する組み換えベクターが提供される。
本発明のさらに別の態様によれば、上記した本発明のDNA又は組み換えベクターを有する形質転換体が提供される。
本発明のさらに別の態様によれば、上記した本発明の蛍光蛋白質と他の蛋白質とから成る融合蛍光蛋白質が提供される。好ましくは、他の蛋白質は細胞内に局在する蛋白質であり、さらに好ましくは、他の蛋白質は細胞内小器官に特異的な蛋白質である。好ましくは、他の蛋白質が蛍光蛋白質である。この場合、好ましくは、融合蛋白質は分子内FRETを生じることができる。
【0032】
本発明のさらに別の態様によれば、上記した本発明の融合蛋白質を細胞内で発現させることを特徴とする、細胞内における蛋白質の局在または動態を分析する方法が提供される。
本発明のさらに別の態様によれば、上記した本発明の蛍光蛋白質、DNA、組み換えベクター、形質転換体、又は融合蛋白質を含む、蛍光試薬キットが提供される。
【発明の効果】
【0033】
本発明により、単量体で存在することができる新規な蛍光蛋白質(mKO)が提供されることになった。二量体の蛍光蛋白質KOによるHeLa細胞でのミトコンドリアラベルにおいて、ミトコンドリアが粒々にラベルされ、本来のミトコンドリア像は得られない。しかし、単量体の蛍光蛋白質mKOでミトコンドリアをラベルした場合には正常な細長いひも状のミトコンドリア像が得られ、ダイナミックなミトコンドリアの動きも観察される。このような単量体化による有効性がミトコンドリア分子のラベルにより確認された。
【0034】
また本発明の蛍光蛋白質(keima616,keima570)は、赤色、橙色の蛍光を放ち、励起のピークが440nm(青色)である。ストークスシフト(励起のピーク値と蛍光のピーク値の差)は従来の赤色蛍光蛋白質(DsRed、HcRed)では20nm〜30nmであるのに対し、本発明の赤色蛍光蛋白質が176nm、橙色蛍光蛋白質が130nmと非常に大きい。故に本発明の蛍光蛋白質は最大の励起で最大の蛍光を得ることができることを特徴とする。また、励起のピークが440nmであるため、青緑蛍光蛋白質(CFP)や緑色蛍光蛋白質(GFP)との同時励起染色において両者の蛍光を非常に有効に取得することが可能である。さらに従来の赤色蛍光蛋白質の励起ピークが560nmから590nmであるのに対し、本発明の蛍光蛋白質は励起のピークが440nmであるので、励起光を変えて従来の赤色蛍光蛋白質と同時に染色することも可能とした。
【図面の簡単な説明】
【0035】
【図1】図1は、mKOの吸収スペクトルを示す。
【図2】図2は、mKOの励起スペクトル(点線)及び蛍光スペクトル(実線)を示す。
【図3】図3は、超遠心による分子量測定の結果を示す。測定結果より分子量は28kDaであることが分かった。
【図4】図4は、HeLa細胞でKO(二量体)を用いてミトコンドリアをラベルした結果を示す。粒状になり正常なミトコンドリアの形態とは異なる。
【図5】図5は、HeLa細胞でmKO(単量体)を用いてミトコンドリアをラベルした結果を示す。ひも状の正常なミトコンドリアの形態として観察される。
【図6】図6は、UV励起緑色蛍光変異体mKVU-1の吸収スペクトルを示す。
【図7】図7は、UV励起緑色蛍光変異体mKVU-1の励起スペクトル及び蛍光スペクトルを示す。
【図8】図8は、青色蛍光変異体mKUV-2の吸収スペクトルを示す。
【図9】図9は、青色蛍光変異体mKUV-2の励起スペクトル及び蛍光スペクトルを示す。
【図10】図10は、緑色蛍光変異体mKO-FM32の吸収スペクトルを示す。
【図11】図11は、緑色蛍光変異体mKO-FM32の励起スペクトル及び蛍光スペクトルを示す。
【図12】図12は、赤色蛍光変異体mKO-F90の吸収スペクトルを示す。
【図13】図13は、赤色蛍光変異体mKO-F90の励起スペクトル及び蛍光スペクトルを示す。
【図14】図14は、mKO時間経過変異体の580nmの励起スペクトルを示す。
【図15】図15は、mKO時間経過変異体の580nmの励起スペクトルを示す。
【図16】図16は、mKO時間経過変異体の蛍光スペクトルを示す。
【図17】図17は、mKO時間経過変異体の蛍光スペクトルを示す。
【図18】図18は、mKO時間経過変異体の蛍光スペクトルを示す。
【図19】図19は、mKO時間経過変異体の蛍光スペクトルを示す。
【図20】図20は、mKO時間経過変異体の蛍光スペクトルを示す。
【図21】図21は、mKO時間経過変異体の蛍光スペクトルを示す。
【図22】図22は、mKO時間経過変異体の蛍光スペクトルを示す。
【図23】図23は、mKOの合成後25時間まで580nmの励起スペクトルを示す。
【図24】図24は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図25】図25は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図26】図26は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図27】図27は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図28】図28は、mKO時間経過変異体の合成後25時間まで580nmの励起スペクトルを示す。
【図29】図29は、mKO時間経過変異体について、緑蛍光の励起ピーク部分の500nmとオレンジ蛍光の励起ピークである548nmの値の比のプロットを示す。
【図30】図30は、mKO-FM14のN末端にTauを連結した融合蛋白質遺伝子をHeLa−S3細胞に遺伝子導入して、イメージングを行った結果を示す。
【図31】図31は、mKO蛋白質に強い緑色光を照射し、照射前後の吸収スペクトルを測定した結果を示す。
【図32】図32は、mKO-FM14蛋白質に強い緑色光を照射し、照射前後の吸収スペクトルを測定した結果を示す。
【図33】図33は、BDNF-mKO-FM14発現遺伝子ベクターの導入後、約12時間から2日間に細胞に発現した融合蛋白質を蛍光顕微鏡で検出し、強い緑色光を照射して、特定の領域のみオレンジ色蛍光を褪色させた結果を示す。
【図34】図34は、BDNF-mKO-FM14発現遺伝子ベクターの導入後、約12時間から2日間に細胞に発現した融合蛋白質を蛍光顕微鏡で検出し、色の変化から分子(BDNF-mKO-FM14)を追跡した結果を示す。
【図35】図35は、分子内FRETを行うための蛍光蛋白質の模式図を示す。
【図36】図36は、単量体蛍光蛋白質mKOと2量体蛍光蛋白質MiCyの蛍光スペクトルと吸収スペクトルを示す。
【図37】図37は、MiCy−linker−mKOを用いて、Caspase3との反応前と反応後の反応液の蛍光スペクトルを440nmで励起して測定した結果を示す。
【図38】図38は、MiCy−linker−mKOを用いてin vivoでCaspase3の活性を測定した結果を示す。
【図39】図39は、COCPの吸収スペクトルを示す。
【図40】図40は、COCPのpH感受性の測定結果を示す。
【図41】図41は、Keima616の励起スペクトルと蛍光スペクトルを示す。
【図42】図42は、Keima570の励起スペクトルと蛍光スペクトルを示す。
【図43】図43は、Keima616のpH感受性の測定結果を示す。
【図44】図44は、Keima570のpH感受性の測定結果を示す。
【図45】図45は、cmkeima620の超遠心分子量測定の結果を示す。
【図46】図46は、cmkeima620の吸収スペクトルを示す。
【図47】図47は、mkeima620の吸収スペクトルを示す。
【図48】図48は、keima616とECFPの励起スペクトルと蛍光スペクトルを示す。
【図49】図49は、Caspase-3の活性測定に用いた蛋白質モチーフを示す。
【図50】図50は、In vitro プロテアーゼ活性と相互相関を示す。リンカー部分にDEVDの配列を挿入したタンデム蛍光蛋白質のサンプルは3種ECFP-Keima616、Keima616-ECFP、EGFP-mRFP1(x2)。 (上段)caspase-3を加える前の自己相関、相互相関関数。 (中段)caspase-3添加後の相互相関関数。 (下段)同添加後の蛍光強度。
【図51】図51は、各融合蛋白モチーフにおけるRelative amplitudeを示す。
【図52】図52は、Caspase-3によるペプチド鎖切断の検出(SDS-PAGE)を示す。
【図53】図53は、タンパク質間相互作用の検出に用いた融合蛋白質モチーフを示す。
【図54】図54は、CaCl2(+)時のECFP−CaMとM13-Keima616の蛍光相互相関関数を示す。
【図55】図55は、CaCl2(−)時のECFP−CaMとM13-Keima616の蛍光相互相関関数を示す。
【発明を実施するための形態】
【0036】
以下、本発明の実施の形態について詳細に説明する。
(1)本発明の蛋白質
(i)本発明の第1の型の蛍光蛋白質
本発明の第1の型の蛍光蛋白質は、以下の(a)又は(b)の何れかに示す蛋白質である。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質;
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
本発明の蛍光蛋白質は、下記の特性を有することを特徴とする。
(1)励起極大波長が548nmであり、蛍光極大波長は559nmである;
(2)548nmにおけるモル吸光係数が、51600である;
(3)量子収率が0.6である;及び
(4)蛍光特性のpH感受性がpKa=5.0である
【0037】
クサビライシ(Fungia sp.)はサンゴの1種で、主に西部大西洋に生息し、群体の外形は多角形で触手が長く、全体が鮮やかなオレンジ色を呈することを特徴とする。
なお、本書中以下の実施例では、クサビライシ(Fungia sp.)を出発材料として上記特性を有する本発明の蛍光蛋白質を取得したが、クサビライシ(Fungia sp.)以外の蛍光を発するサンゴから本発明の蛍光蛋白質を取得することができる場合もあり、そのような蛍光蛋白質も本発明の範囲内である。
【0038】
本明細書で言う「1から数個のアミノ酸の欠失、置換及び/又は付加を有するアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
本明細書で言う「同等の蛍光特性」とは、同等の蛍光強度、同等の励起波長、同等の蛍光波長、同等のpH感受性などを有することを意味する。
【0039】
本発明の蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号1から30に記載したアミノ酸配列並びに塩基配列の情報を利用することにより適当なプライマーを設計し、それらを用いて上記した国際公開WO03/54191号公報に記載の蛍光蛋白質のcDNAクローンを鋳型にしてPCRを行うことにより、本発明の蛍光蛋白質をコードするDNAを取得することができる。本発明の蛍光蛋白質をコードするDNAの一部の断片を上記したPCRにより得た場合には、作製したDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の蛍光蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の蛍光蛋白質を産生することができる。発現系での発現については本明細書中後記する。
【0040】
さらに本発明によれば、上記した本発明の蛋白質(mKO)の変異体蛋白質も提供される。具体的には、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質;
(b)配列番号3、5、7又は9に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号3、5、7又は9に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0041】
さらに別の具体例としては、以下の(a)又は(b)に示す蛍光蛋白質が提供される。
(a)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛍光蛋白質;
(b)配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、それぞれ配列番号11、13、15、17、19、21、23、25、27又は29に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有する蛋白質。
【0042】
(ii)本発明の第2の型の蛋白質
本発明の第2の型の蛋白質は、配列番号37、39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質;並びに配列番号37、39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性又は蛍光特性を有する蛋白質である。配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質のストークスシフト(吸収極大波長と蛍光極大波長の差)はそれぞれ176nm、130nm、180nm、及び180nmである。配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質については、そのストークスシフトは100nm以上、より好ましくは120nm以上になるものとする。
【0043】
本発明の蛋白質は、下記の特性を有することを特徴とする。
(1)COCP(アミノ酸配列を配列番号37に示し、塩基配列を配列番号38に示す)
励起極大波長(吸収極大波長):576nm
576nmにおけるモル吸光係数:64000
pH感受性:なし
(2)COCP-FL(アミノ酸配列を配列番号39に示し、塩基配列を配列番号40に示す)
励起極大波長(吸収極大波長):560nm
蛍光極大波長:600nm
(3)keima616(アミノ酸配列を配列番号41に示し、塩基配列を配列番号42に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長:616nm
pH感受性:pH7.5〜10で蛍光強度は安定
【0044】
(4)keima570(アミノ酸配列を配列番号43に示し、塩基配列を配列番号44に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長:570nm
pH感受性:pH7.5〜10で蛍光強度は安定
(5)cmkeima620(アミノ酸配列を配列番号45に示し、塩基配列を配列番号46に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長: 620nm
(6)mkeima620(アミノ酸配列を配列番号47に示し、塩基配列を配列番号48に示す)
励起極大波長(吸収極大波長):440nm
蛍光極大波長: 620nm
【0045】
本明細書中の実施例においては、本発明の蛋白質をコードするDNAは、コモンサンゴ(Montipora. sp)を出発材料としてクローニングされた。コモンサンゴ(Montipora. sp)は、刺胞動物門花虫綱六放サンゴ亜綱イシサンゴ目ミドリイシ科に属するサンゴの1種であり、塊状や被覆状の群体を形成することが多い。なお、コモンサンゴ(Montipora. sp)以外の蛍光を発するサンゴから本発明の蛋白質を取得することができる場合もあり、そのような蛋白質も本発明の範囲内である。
【0046】
本明細書で言う「1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列」における「1から数個」の範囲は特には限定されないが、例えば、1から20個、好ましくは1から10個、より好ましくは1から7個、さらに好ましくは1から5個、特に好ましくは1から3個程度を意味する。
【0047】
本明細書において、「吸光特性を有する蛋白質」とは一定の波長の光を吸収できる性質を有する蛋白質を意味する。「配列番号37に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性を有する蛋白質」の吸光特性は、配列番号37に記載のアミノ酸配列を有する蛋白質の吸光特性と実質的に同一でもよいし、異なっていてもよい。吸光特性は、例えば、吸光強度、励起波長(吸収波長)、pH感受性などにより評価することができる。本発明の蛋白質のうち吸光特性を有し、蛍光を発しない色素蛋白質は、(1)FRETのアクセプター分子(エネルギー受容体)として用いたり、(2)照射した光のエネルギーを光以外のエネルギーに変換させるシステムの開発に利用したり、あるいは(3)蛋白質のアミノ酸配列に変異を導入して蛍光を発するように改変することなどに用いることができる。
【0048】
本明細書において、「蛍光特性を有する蛋白質」とは、一定の波長の光で励起することにより蛍光を発することができる性質を有する蛋白質を意味する。「配列番号39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有する蛋白質」の蛍光特性はそれぞれ、配列番号39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質の蛍光特性と実質的に同一でもよいし、異なっていてもよい。蛍光特性は、例えば、蛍光強度、励起波長、蛍光波長、pH感受性などにより評価することができる。
【0049】
本発明の色素蛋白質又は蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号37、39、41、43、45又は47に記載したアミノ酸配列並びに配列番号38、40、42、44、46又は48に記載した塩基配列の情報を利用することにより適当なプライマーを設計し、それらを用いてコモンサンゴ(Montipora sp.)由来のcDNAライブラリーを鋳型にしてPCRを行うことにより、本発明の蛋白質をコードするDNAを取得することができる。本発明の蛋白質をコードするDNAの一部の断片を上記したPCRにより得た場合には、作製したDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の蛋白質を産生することができる。発現系での発現については本明細書中後記する。
【0050】
(2)本発明のDNA
本発明によれば、本発明の第1の型の蛍光蛋白質をコードするDNAが提供される。
本発明の第1の型の蛍光蛋白質をコードするDNAの具体例としては、以下の(a)又は(b)に示す蛋白質をコードするDNAが挙げられる。
(a)配列番号1に記載のアミノ酸配列を有する蛋白質
(b)配列番号1に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、配列番号1に記載のアミノ酸配列を有する蛋白質と同等の蛍光特性を有し、かつ単量体で存在する蛋白質。
【0051】
本発明の蛍光蛋白質をコードするDNAの更なる具体例としては、以下の(a)又は(b)に示すDNAもまた挙げられる。
(a)配列番号2に記載の塩基配列を有するDNA
(b)配列番号2に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ配列番号2に記載の塩基配列がコードする蛋白質と同等の蛍光特性を有する蛋白質であって、単量体で存在する蛋白質をコードする塩基配列を有するDNA。
また、上記した(1)に記載した本発明の蛋白質(mKO)の変異体蛋白質をコードするDNAも本発明の範囲内である。
【0052】
さらに本発明によれば、本発明の第2の型の蛋白質をコードするDNAが提供される。
本発明の蛋白質をコードするDNAの具体例としては、以下の(a)又は(b)に示す蛋白質をコードするDNAが挙げられる。
(a)配列番号37、39、41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号37、39、41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、吸光特性又は蛍光特性を有する蛋白質。
【0053】
本発明の色素蛋白質又は蛍光蛋白質をコードするDNAの更なる具体例としては、以下の(a)又は(b)に示すDNAもまた挙げられる。
(a)配列番号38、40、42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号38、40、42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ吸光特性又は蛍光特性を有する蛋白質をコードする塩基配列を有するDNA。
【0054】
本明細書で言う「1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列」における「1から数個」の範囲は特には限定されないが、例えば、1から50個、好ましくは1から30個、より好ましくは1から20個、さらに好ましくは1から10個、特に好ましくは1から5個程度を意味する。
本発明のDNAは、例えばホスホアミダイト法などにより合成することができるし、特異的プライマーを用いたポリメラーゼ連鎖反応(PCR)によって製造することもできる。本発明のDNA又はその断片の作製方法については、本明細書中上述した通りである。
【0055】
また、所定の核酸配列に所望の変異を導入する方法は当業者に公知である。例えば、部位特異的変異誘発法、縮重オリゴヌクレオチドを用いるPCR、核酸を含む細胞の変異誘発剤又は放射線への露出等の公知の技術を適宜使用することによって、変異を有するDNAを構築することができる。このような公知の技術は、例えば、Molecular Cloning: A laboratory Mannual, 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.,1989、並びにCurrent Protocols in Molecular Biology, Supplement 1〜38, John Wiley & Sons (1987-1997)に記載されている。
【0056】
(3)本発明の組み換えベクター
本発明のDNAは適当なベクター中に挿入して使用することができる。本発明で用いるベクターの種類は特に限定されず、例えば、自立的に複製するベクター(例えばプラスミド等)でもよいし、あるいは、宿主細胞に導入された際に宿主細胞のゲノムに組み込まれ、組み込まれた染色体と共に複製されるものであってもよい。
【0057】
好ましくは、本発明で用いるベクターは発現ベクターである。発現ベクターにおいて本発明のDNAは、転写に必要な要素(例えば、プロモータ等)が機能的に連結されている。プロモータは宿主細胞において転写活性を示すDNA配列であり、宿主の種類に応じて適宜することができる。
細菌細胞で作動可能なプロモータとしては、バチルス・ステアロテルモフィルス・マルトジェニック・アミラーゼ遺伝子(Bacillusstearothermophilus maltogenic amylase gene)、バチルス・リケニホルミスαアミラーゼ遺伝子(Bacillus licheniformis alpha-amylase gene)、バチルス・アミロリケファチエンス・BANアミラーゼ遺伝子(Bacillus amyloliquefaciens BAN amylase gene)、バチルス・サブチリス・アルカリプロテアーゼ遺伝子(Bacillus Subtilis alkaline protease gene)もしくはバチルス・プミルス・キシロシダーゼ遺伝子(Bacillus pumilus xylosldase gene)のプロモータ、またはファージ・ラムダのPR若しくはPLプロモータ、大腸菌の lac、trp若しくはtacプロモータなどが挙げられる。
【0058】
哺乳動物細胞で作動可能なプロモータの例としては、SV40プロモータ、MT−1(メタロチオネイン遺伝子)プロモータ、またはアデノウイルス2主後期プロモータなどがある。昆虫細胞で作動可能なプロモータの例としては、ポリヘドリンプロモータ、P10プロモータ、オートグラファ・カリホルニカ・ポリヘドロシス塩基性蛋白プロモータ、バキュウロウイルス即時型初期遺伝子1プロモータ、またはバキュウロウイルス39K遅延型初期遺伝子プロモータ等がある。酵母宿主細胞で作動可能なプロモータの例としては、酵母解糖系遺伝子由来のプロモータ、アルコールデヒドロゲナーゼ遺伝子プロモータ、TPI1プロモータ、ADH2-4cプロモータなどが挙げられる。
糸状菌細胞で作動可能なプロモータの例としては、ADH3プロモータまたはtpiAプロモータなどがある。
【0059】
また、本発明のDNAは必要に応じて、例えばヒト成長ホルモンターミネータまたは真菌宿主についてはTPI1ターミネータ若しくはADH3ターミネータのような適切なターミネータに機能的に結合されてもよい。本発明の組み換えベクターは更に、ポリアデニレーションシグナル(例えばSV40またはアデノウイルス5E1b領域由来のもの)、転写エンハンサ配列(例えばSV40エンハンサ)および翻訳エンハンサ配列(例えばアデノウイルス VA RNA をコードするもの)のような要素を有していてもよい。
本発明の組み換えベクターは更に、該ベクターが宿主細胞内で複製することを可能にするDNA配列を具備してもよく、その一例としてはSV40複製起点(宿主細胞が哺乳類細胞のとき)が挙げられる。
【0060】
本発明の組み換えベクターはさらに選択マーカーを含有してもよい。選択マーカーとしては、例えば、ジヒドロ葉酸レダクターゼ(DHFR)またはシゾサッカロマイセス・ポンベTPI遺伝子等のようなその補体が宿主細胞に欠けている遺伝子、または例えばアンピシリン、カナマイシン、テトラサイクリン、クロラムフェニコール、ネオマイシン若しくはヒグロマイシンのような薬剤耐性遺伝子を挙げることができる。
本発明のDNA、プロモータ、および所望によりターミネータおよび/または分泌シグナル配列をそれぞれ連結し、これらを適切なベクターに挿入する方法は当業者に周知である。
【0061】
(4)本発明の形質転換体
本発明のDNA又は組み換えベクターを適当な宿主に導入することによって形質転換体を作製することができる。
本発明のDNAまたは組み換えベクターを導入される宿主細胞は、本発明のDNA構築物を発現できれば任意の細胞でよく、細菌、酵母、真菌および高等真核細胞等が挙げられる。
【0062】
細菌細胞の例としては、バチルスまたはストレプトマイセス等のグラム陽性菌又は大腸菌等のグラム陰性菌が挙げられる。これら細菌の形質転換は、プロトプラスト法、または公知の方法でコンピテント細胞を用いることにより行えばよい。
哺乳類細胞の例としては、HEK293細胞、HeLa細胞、COS細胞、BHK細胞、CHL細胞またはCHO細胞等が挙げられる。哺乳類細胞を形質転換し、該細胞に導入されたDNA配列を発現させる方法も公知であり、例えば、エレクトロポーレーション法、リン酸カルシウム法、リポフェクション法等を用いることができる。
【0063】
酵母細胞の例としては、サッカロマイセスまたはシゾサッカロマイセスに属する細胞が挙げられ、例えば、サッカロマイセス・セレビシエ(Saccharomyces cerevis1ae)またはサッカロマイセス・クルイベリ(Saccharomyces kluyveri)等が挙げられる。酵母宿主への組み換えベクターの導入方法としては、例えば、エレクトロポレーション法、スフェロブラスト法、酢酸リチウム法等を挙げることができる。
他の真菌細胞の例は、糸状菌、例えばアスペルギルス、ニューロスポラ、フザリウム、またはトリコデルマに属する細胞である。宿主細胞として糸状菌を用いる場合、DNA構築物を宿主染色体に組み込んで組換え宿主細胞を得ることにより形質転換を行うことができる。DNA構築物の宿主染色体への組み込みは、公知の方法に従い、例えば相同組換えまたは異種組換えにより行うことができる。
【0064】
昆虫細胞を宿主として用いる場合には、組換え遺伝子導入ベクターおよびバキュロウイルスを昆虫細胞に共導入して昆虫細胞培養上清中に組換えウイルスを得た後、さらに組換えウイルスを昆虫細胞に感染させ、蛋白質を発現させることができる(例えば、Baculovirus Expression Vectors, A Laboratory Manua1;及びカレント・プロトコールズ・イン・モレキュラー・バイオロジー、Bio/Technology, 6, 47(1988)等に記載)。
【0065】
バキュロウイルスとしては、例えば、ヨトウガ科昆虫に感染するウイルスであるアウトグラファ・カリフォルニカ・ヌクレアー・ポリヘドロシス・ウイルス(Autographa californica nuclear polyhedrosis virus)等を用いることができる。
昆虫細胞としては、Spodoptera frugiperdaの卵巣細胞であるSf9、Sf21〔バキュロウイルス・エクスプレッション・ベクターズ、ア・ラボラトリー・マニュアル、ダブリュー・エイチ・フリーマン・アンド・カンパニー(W. H. Freeman and Company)、ニューヨーク(New York)、(1992)〕、Trichoplusia niの卵巣細胞であるHiFive(インビトロジェン社製)等を用いることができる。
組換えウイルスを調製するための、昆虫細胞への組換え遺伝子導入ベクターと上記バキュロウイルスの共導入方法としては、例えば、リン酸カルシウム法又はリポフェクション法等を挙げることができる。
【0066】
上記の形質転換体は、導入されたDNA構築物の発現を可能にする条件下で適切な栄養培地中で培養する。形質転換体の培養物から、本発明の蛍光融合蛋白質を単離精製するには、通常の蛋白質の単離、精製法を用いればよい。
例えば、本発明の蛋白質が、細胞内に溶解状態で発現した場合には、培養終了後、細胞を遠心分離により回収し水系緩衝液に懸濁後、超音波破砕機等により細胞を破砕し、無細胞抽出液を得る。該無細胞抽出液を遠心分離することにより得られた上清から、通常の蛋白質の単離精製法、即ち、溶媒抽出法、硫安等による塩析法、脱塩法、有機溶媒による沈殿法、ジエチルアミノエチル(DEAE)セファロース等のレジンを用いた陰イオン交換クロマトグラフィー法、S-Sepharose FF(ファルマシア社製)等のレジンを用いた陽イオン交換クロマトグラフィー法、ブチルセファロース、フェニルセファロース等のレジンを用いた疎水性クロマトグラフィー法、分子篩を用いたゲルろ過法、アフィニティークロマトグラフィ一法、クロマトフォーカシング法、等電点電気泳動等の電気泳動法等の手法を単独あるいは組み合わせて用い、精製標品を得ることができる。
【0067】
(5)本発明の蛍光蛋白質及びそれを含む融合蛍光蛋白質の利用
本発明は蛍光蛋白質を他の蛋白質と融合させることにより、融合蛍光蛋白質を構築することができる。
本発明の融合蛍光蛋白質の取得方法については特に制限はなく、化学合成により合成した蛋白質でもよいし、遺伝子組み換え技術による作製した組み換え蛋白質でもよい。
【0068】
組み換え蛋白質を作製する場合には、先ず当該蛋白質をコードするDNAを入手することが必要である。本明細書の配列表の配列番号1から30に記載したアミノ酸配列及び塩基配列の情報を利用することにより適当なプライマーを設計し、本発明の蛍光蛋白質の遺伝子を含むDNA断片を鋳型にしてPCRを行うことにより、本発明の蛍光蛋白質をコードするDNAを構築するのに必要なDNA断片を作製することができる。また同様に、融合すべき蛋白質をコードするDNA断片も入手する。
次いで、これらのDNA断片を順番に遺伝子組み換え技術により連結することにより、所望の融合蛍光蛋白質をコードするDNAを得ることができる。このDNAを適当な発現系に導入することにより、本発明の融合蛍光蛋白質を産生することができる。
【0069】
本発明の蛍光蛋白質は、特に、標識としての利用価値が高い。即ち、本発明の蛍光蛋白質を被検アミノ酸配列との融合蛋白質として精製し、マイクロインジェクション法などの手法により細胞内に導入し、該融合蛋白質の分布を経時的に観察すれば、被検アミノ酸配列の細胞内におけるターゲッティング活性を検出することが可能である。
【0070】
本発明の蛍光蛋白質を融合させる他の蛋白質(被検アミノ酸配列)の種類は特に限定されるものではないが、例えば、細胞内に局在する蛋白質、細胞内小器官に特異的な蛋白質、ターゲティングシグナル(例えば、核移行シグナル、ミトコンドリアプレ配列)等が好適である。なお、本発明の蛍光蛋白質は、マイクロインジェクション法などにより細胞内に導入する以外に、細胞内で発現させて用いることも可能である。この場合には、本発明の蛍光蛋白質をコードするDNAが発現可能に挿入されたベクターが宿主細胞に導入される。
【0071】
また、本発明の蛍光蛋白質は、レポーター蛋白質としてプロモータ活性の測定に用いることも可能である。即ち、被検プロモータの下流に、本発明の蛍光蛋白質をコードするDNAが配置されたベクターを構築し、これを宿主細胞に導入し、該細胞から発せられる本発明の蛍光蛋白質の蛍光を検出することにより、被検プロモータの活性を測定することが可能である。被検プロモータとしては、宿主細胞内で機能するものであれば、特に制限はない。
【0072】
上記アミノ酸配列のターゲティング活性の検出やプロモータ活性の測定において用いられるベクターとしては、特に制限はないが、例えば、動物細胞用ベクターでは、「pNEO」(P. Southern, and P. Berg (1982) J. MOl. Appl. Genet. 1:327)、「pCAGGS」(H.Niwa,K.Yamamura,and J.Miyazaki. Gene 108,193-200(1991))、「pRc/CMV」(インビトロゲン社製)、「pCDM8」(インビトロゲン社製)などが、酵母用ベクターでは、「pRS303」,「pRS304」,「pRS305」,「pRS306」,「pRS313」,「pRS314」,「pRS315」,[pRS316](R.S.Sikorski and P.Hieter (1989) Genetics 122: 19-27)、「pRS423」,「pRS424」,「pRS425」,「pRS426」(T.W.Christianson, R.S.Sikorski, M.Dante, J.H.Shero, and P. Hieter (1992) Gene 110: 119-122)などが好適に用いられる。
【0073】
また、使用可能な細胞の種類も特に限定されず、各種の動物細胞、例えば、L細胞、BalbC-3T3細胞、NIH3T3細胞、CHO(Chinese hamster ovary)細胞、HeLa細胞、NRK(normal rat kidney)細胞、「Saccharomyces cerevisiae」などの酵母細胞や大腸菌(E. coli)細胞などを使用することができる。ベクターの宿主細胞への導入は、例えば、リン酸カルシウム法やエレクトロポレーション法などの常法により行うことができる。
【0074】
上記のようにして得た、本発明の蛍光蛋白質と他の蛋白質(蛋白質Xとする)とを融合させた融合蛍光蛋白質を細胞内で発現させ、発する蛍光をモニターすることにより、細胞内における蛋白質Xの局在や動態を分析することが可能になる。即ち、本発明の融合蛍光蛋白質をコードするDNAで形質転換またはトランスフェクトした細胞を蛍光顕微鏡で観察することにより細胞内における蛋白質Xの局在や動態を可視化して分析することができる。
【0075】
例えば、蛋白質Xとして細胞内オルガネラに特異的な蛋白質を利用することにより、核、ミトコンドリア、小胞体、ゴルジ体、分泌小胞、ペルオキソームなどの分布や動きを観察できる。
また、例えば、神経細胞の軸索、樹状突起などは発生途中の個体の中で著しく複雑な走向の変化を示すので、こういった部位を蛍光ラベルすることにより動的解析が可能になる。
【0076】
本発明の蛍光蛋白質の蛍光は、生細胞のまま検出することが可能である。この検出は、例えば、蛍光顕微鏡(カールツァイス社 アキシオフォト フィルターセット09)や画像解析装置(ATTO デジタルイメージアナライザー)などを用いて行うことが可能である。
顕微鏡の種類は目的に応じて適宜選択できる。経時変化を追跡するなど頻回の観察を必要とする場合には、通常の落射型蛍光顕微鏡が好ましい。細胞内の詳細な局在を追及したい場合など、解像度を重視する場合は、共焦点レーザー顕微鏡の方が好ましい。顕微鏡システムとしては、細胞の生理状態を保ち、コンタミネーションを防止する観点から、倒立型顕微鏡が好ましい。正立顕微鏡を使用する場合、高倍率レンズを用いる際には水浸レンズを用いることができる。
【0077】
フィルターセットは蛍光蛋白質の蛍光波長に応じて適切なものを選択できる。本発明の蛍光蛋白質は、励起極大波長が548nmであり、蛍光極大波長が559nmであることから、励起光530〜550nm、蛍光550〜600nm程度のフィルターを使用することが好ましい。
また、蛍光顕微鏡を用いた生細胞での経時観察を行う場合には、短時間で撮影を行うべきなので、高感度冷却CCDカメラを使用する。冷却CCDカメラは、CCDを冷却することにより熱雑音を下げ、微弱な蛍光像を短時間露光で鮮明に撮影することができる。
【0078】
また、分子間の相互作用を分析する手法の一つとして、FRET(蛍光共鳴エネルギー転移)が知られている。FRETでは、例えば、第一の蛍光蛋白質としてのシアン蛍光蛋白質(CFP)で標識した第一の分子と、第二の蛍光蛋白質としての黄色蛍光蛋白質(YFP)で標識した第二の分子とを共存させることにより、黄色蛍光蛋白質(YFP)をアクセプター分子として作用させ、シアン蛍光蛋白質(CFP)をドナー分子として作用させ、両者の間でFRET(蛍光共鳴エネルギー転移)を生じさせることにより、第一の分子と第二の分子との間の相互作用を可視化することができる。即ち、FRETでは2種類の分子にそれぞれ異なる色素を導入し、エネルギーレベルの高い方の色素(ドナー分子)を選択的に励起し、その色素の蛍光を測定し、もう一方の色素(アクセプター分子)からの長波長蛍光も測定して、それらの蛍光変化量によって分子間の相互作用を可視化する。両方の色素が、2種類の分子の相互作用によって近接したときのみドナー分子の蛍光の減少とアクセプター分子の蛍光の増加が1波長励起2波長測光法により観測される。しかし、アクセプター分子に色素蛋白質を用いた場合は、両方の色素が、2種類の分子の相互作用によって近接したときのみドナー分子の蛍光の減少を生じ1波長励起1波長測光法により観測することができる。即ち、測定機器の簡易化が可能となる。
【0079】
本発明の蛍光蛋白質及び色素蛋白質は、特に、FRET(蛍光共鳴エネルギー転移)におけるドナー分子及びアクセプター分子としての利用価値が高い。即ち、本発明の色素蛋白質と被験物質との融合体(第一の融合体)を作製する。次いで、該被験物質と相互作用する別の被験物質と別の蛍光蛋白質との融合体(第2の融合体)を作製する。そして、第一の融合体と第2の融合体とを相互作用させ、発する蛍光を分析することにより、上記2種類の被験物質間の相互作用を分析することができる。なお、本発明の色素蛋白質を用いたFRET(蛍光共鳴エネルギー転移)は、試験管内で行ってもよいし、細胞内で行ってもよい。
【0080】
さらにまた、本発明の蛍光蛋白質又は色素蛋白質の何れか1種以上をドナー蛋白質又はアクセプター蛋白質として使用することによって、分析物質の標的配列の両端にドナー蛍光蛋白質とアクセプター蛍光蛋白質が結合している構造を有する蛍光指示薬を作成することもできる。分析物質の該標的配列への結合又は作用の有無により、指示薬の立体構造が変化し、これにより蛍光共鳴エネルギー転移(FRET)の有無が生じさせることができる。
【0081】
(6)本発明のキット
本発明によれば、本明細書に記載した蛍光蛋白質、融合蛍光蛋白質、DNA、組み換えベクター又は形質転換体から選択される少なくとも1種以上を含むことを特徴とする、細胞内成分の局在の分析及び/又は生理活性物質の分析のためのキットが提供される。本発明のキットは、それ自体既知の通常用いられる材料及び手法で調製することができる。
【0082】
蛍光蛋白質又はDNAなどの試薬は、適当な溶媒に溶解することにより保存に適した形態に調製することができる。溶媒としては、水、エタノール、各種緩衝液などを用いることができる。
以下の実施例により本発明を具体的に説明するが、本発明は実施例によって限定されるものではない。
【実施例】
【0083】
実施例1:点変異導入による多量体形成阻害変異体の作製
KO−1のアミノ酸配列から多量体形成界面を予測し、多量体形成界面のアミノ酸を置換し、なおかつ蛍光特性を保持するようKO−1の単量体化を行った。点変異導入はKO−1を挿入した大腸菌発現ベクター(pRSET B)(国際公開WO03/54191号公報に記載のKO-1をコードするDNAを有する発現ベクター)で点変異導入プライマーを用いて行った。具体的には鋳型プラスミドの片側鎖に複数の変異導入プライマーを同時にアニールさせ、ポリメラーゼで伸長させる。各プライマーにより伸長された各DNA断片を同反応液中でDNAリガーゼを用いてつなぎ、変異導入された部分以外が鋳型と相補的なものを得るという手法を行った。DNAリガーゼで各DNA断片をつなぐ際にDNAの末端にリン酸基を必要とするため、用いたプライマーは5'側のリン酸化を行った。
【0084】
(1)プライマーの5'リン酸化
100μM プライマー 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
上記混合物を37℃で30分間インキュベートした。ここでプライマーとしては、以下の配列番号3から17に記載の塩基配列を有するプライマーを使用した。
【0085】
K11R, F13Y
CCAGAGATGAAGATGAGGTACTACATGGACGGC(配列番号59)
V25I
CATGAGTTCACAATTGAAGGTGAAGGC(配列番号60)
K32R
GAAGGCACAGGCAGACCTTACGAGGGA(配列番号61)
S55A
CCAATGCCTTTCGCGTTTGACTTAGTG(配列番号62)
T62V
TTAGTGTCACACGTGTTCTGTTACGGC(配列番号63)
Q96E
GAAAGGTCGTTGGAGTTCGAAGATGGT(配列番号64)
F102S, A104S
GAAGATGGTGGGTCCGCTTCAGTCAGTGCG(配列番号65)
C115T, E117Y
AGCCTTAGAGGAAACACCTTCTACCACAAATCCA(配列番号66)
V123T
CAAATCCAAATTTACTGGGGTTAACTTTCCTG(配列番号67)
V133I
GCCGATGGTCCTATCATGCAAAACCAAAGT(配列番号68)
S139V
GCCGATGGTCCTATCATGCAAAACCAAAGTGTTGATTGGGAGCCA(配列番号69)
T150A, C151S
GAGAAAATTACTGCCAGCGACGGAGTTCTGAAG(配列番号70)
F162Y, A166E
GATGTTACGATGTACCTAAAACTTGAAGGAGGCGGCAATCAC(配列番号71)
Q190G, F193Y, G195S
CTTAAAATGCCAGGAAGCCATTACATCAGCCATCGCCTCGTCAGG(配列番号72)
C217S
GATGCAGTAGCTCATTCCCTCGAGCACCACCACC(配列番号73)
【0086】
(2)点変異導入PCR
5'リン酸化プライマー 4μl
template(KO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0087】
プログラム:
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
上記の3)〜5)を25サイクル繰り返す
6)75℃ 7 min
7)4℃ hold
【0088】
(3)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0089】
(4)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109に形質転換して変異導入後のKO-1を発現させた。
【0090】
(5)単量体化Kusabira-Orange(mKO)のアミノ酸配列
変異導入後のKO変異体の塩基配列を解析し、アミノ酸配列を決定した。その結果、11番目のリジン(K)をアルギニン(R)に、13番目のフェニルアラニン(F)をチロシン(Y)に、25番目のバリン(V)をイソロイシン(I)に、32番目のリジン(K)をアルギニン(R)に、55番目のセリン(S)をアラニン(A)に、62番目のトレオニン(T)をバリン(V)に、96番目のグルタミン(Q)をグルタミン酸(E)に、102番目のフェニルアラニン(F)をセリン(S)に、104番目のアラニン(A)をセリン(S)に、115番目のシステイン(C)をトレオニン(T)に、117番目のグルタミン酸(E)をチロシン(Y)に、123番目のバリン(V)をトレオニン(T)に、133番目のバリン(V)をイソロイシン(I)に、139番目のセリン(S)をバリン(V)に、150番目のトレオニン(T)をアラニン(A)に、151番目のシステイン(C)をセリン(S)に、162番目のフェニルアラニン(F)をチロシン(Y)に、166番目のアラニン(A)をグルタミン酸(E)に、190番目のグルタミン(Q)をグリシン(G)に、193番目のフェニルアラニン(F)をチロシン(Y)に、195番目のグリシン(G)をセリン(S)に、217番目のシステイン(C)をセリン(S)に置換されていた。さらにKozak配列付加のため2番目のセリン(S)の前にバリン(V)を導入した。この変異体をmKOとした。mKOのアミノ酸配列を配列表の配列番号1に記載し、塩基配列を配列表の配列番号2に記載する。
大腸菌を用いてmKOにHis-Tagを付加した蛋白質を常法により発現させ、Ni-Agaroseを用いて精製した。
【0091】
実施例2:蛍光特性の解析
実施例1で精製したmKO蛋白質の蛍光及び吸収スペクトルを以下の通り測定し、量子収率およびモル吸光係数を算出した。
20μM蛍光蛋白、50mM HEPES pH7.5溶液を用いて吸収スペクトルを測定した。このスペクトルのピークの値よりモル吸光係数を計算した。mKOでは548nmに吸収のピークが認められ、500nmにおける吸収が0.0025となるように蛍光蛋白を上記の緩衝液で希釈して、500nmで励起した時の蛍光スペクトルと590nmにおける蛍光による励起スペクトルを測定した。DsRed(CLONTECH)を同様に500nmにおける吸収が0.0025となるようにして蛍光スペクトルを測定し、DsRedの量子収率を0.29としてmKOの量子収率を求めた。
結果を表1、図1及び図2に示す。表1には、国際公開WO03/54191号公報に記載のKO蛋白質(二量体蛋白質)のデータも併記する。
【0092】
【表1】
【0093】
実施例3:超遠心分析による分子量の測定
mKO蛋白質溶液を150mM KCl,50mM HEPES-KOH pH7.4とした。mKOの分子量決定のため超遠心分析をおこなった。超遠心機XL-1(ベックマン・コールター)を用いて25,000rpm、22時間遠心して、mKOの吸収極大(548nm)付近の540nmの吸収を測定した。その測定結果からmKOの分子量は28kDaと計算された(図3)。これはアミノ酸配列から予測される26kDaとほぼ一致し、mKOが単量体として存在することが確認された。
【0094】
実施例4:ミトコンドリアへのターゲティング
KOおよびmKOのN末端に、Yeast由来のcytochrome oxidaseサブユニット4のN末端12アミノ酸(MLSLRQSIRFFK)を付加し、HeLa細胞のミトコンドリアへのターゲティングを行い、ミトコンドリアのラベルを行った。KO(二量体)は正確にターゲティングされずに、ミトコンドリアが粒々に染色されているのが確認された(図4)。一方、mKO(単量体)は正確にミトコンドリアにターゲティングされ、細長い糸状のミトコンドリアが観察され、単量体化による有効性が確認された(図5)。
【0095】
実施例5:蛍光特性の異なるmKOの変異体の作製
(1)変異導入
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。点変異導入はmKOを挿入した大腸菌発現ベクター(pRSETB)に点変異導入プライマーをもちいてPCRをかけることにより行った。PCRに用いたプライマーは5'側のリン酸化を行った。
【0096】
(a)プライマーの5'リン酸化
100μM primer 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
37℃で30分間インキュベートした。
【0097】
(b)点変異導入PCR
5'リン酸化プライマー 4μl
template(mKO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0098】
プログラム
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
6)75℃ 7 min
7)4℃ hold
3)〜5)を25サイクル
【0099】
(c)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0100】
(d)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109(DE3)に形質転換して変異導入後のmKOを発現させ解析を行なった。
【0101】
(2)mKO変異体のアミノ酸置換部位および蛍光特性
蛍光測定には蛍光分光光度計F-2500(HITACHI)を使用した。吸収測定には分光光度計U-3310(HITACHI)を使用した。
【0102】
(i)UV励起緑色蛍光変異体mKVU-1(アミノ酸配列を配列番号3に示し、塩基配列を配列番号4に示す)
mKOの70番目のプロリン(P)をシステイン(C)に、160番目のバリン(V)をアスパラギン酸(D)に、162番目のメチオニン(M)をロイシン(L)に、176番目のフェニルアラニン(F)をメチオニン(M)にアミノ酸置換することにより、505nmに蛍光ピークを持ち、398nmに励起のピークを持つ緑色蛍光蛋白質となった(図6、7)。モル吸光係数は10000で、蛍光の量子収率は0.27となった。
【0103】
(ii) 青色蛍光変異体mKUV-2(アミノ酸配列を配列番号5に示し、塩基配列を配列番号6に示す)
mKOの65番目のシステイン(C)をグリシン(G)に、70番目のプロリン(P)をグリシン(G)に、160番目のバリン(V)をアスパラギン酸(D)に、176番目のフェニルアラニン(F)をメチオニン(M)にアミノ酸置換することにより、469nmに蛍光ピークを持ち、322nmに励起のピークを持つ青色蛍光蛋白質となった(図8、9)。モル吸光係数は12500で、蛍光の量子収率は0.2となった。
【0104】
(iii) 緑色蛍光変異体mKO-FM32(アミノ酸配列を配列番号7に示し、塩基配列を配列番号8に示す)
mKOの65番目のシステイン(C)をアラニン(A)に、70番目のプロリン(P)をグリシン(G)にアミノ酸置換することにより、506nmに蛍光ピークを持ち、493nmに励起のピークを持つ緑色蛍光蛋白質となった(図10、11)。モル吸光係数は27500で、蛍光の量子収率は0.44となった。
【0105】
(iv)赤色蛍光変異体mKO-F90(アミノ酸配列を配列番号9に示し、塩基配列を配列番号10に示す)
mKOの41番目のメチオニン(M)をロイシン(L)に、49番目のリジン(K)をグルタミン酸(E)に、69番目のアルギニン(R)をリジン(K)に、145番目のセリン(S)をトリプトファン(W)に、185番目のリジン(K)をグルタミン酸(E)に、188番目のリジン(K)をグルタミン酸(E)に、192番目のセリン(S)をアスパラギン酸(D)にアミノ酸置換することにより、582nmに蛍光ピークを持ち、564nmに励起のピークを持つ赤色蛍光蛋白質となった(図12、13)。モル吸光係数は25000で、蛍光の量子収率は0.05となった。
【0106】
実施例6:緑色とオレンジ色の2蛍光を発するmKOの変異体の作製(時間経過測定プローブおよび追跡プローブ)
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。mKOは翻訳されてからすぐは緑の蛍光を放ち、その後オレンジ色の蛍光を放つようになる。しかし、緑色蛍光からオレンジ色蛍光への移行はすばやく完了するために、通常はほとんど見られない。そこで、いろいろな時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質を作製した。この変異体を使用することによって蛋白質発現からの時間を緑色蛍光とオレンジ色蛍光の比で測定することができる。また、この変異体は緑色蛍光とオレンジ色蛍光が独立しているために、オレンジ色蛍光のみを消光させることができた。つまり、オレンジ色蛍光のみを消光させて、オレンジ色蛍光の増加を測定すれば、時間経過測定のリセットも可能となる。さらに、同じくオレンジ色のみ任意の部分を消光して、緑色蛍光とオレンジ色蛍光の比で測定すれば消光した部分のラベルした分子や細胞などの挙動を測定することもできる。結果としてわかったことは70番目のプロリン(P)をアミノ酸置換することにより、多様な、時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質を作製できることであった。
【0107】
(1)変異導入
mKOのアミノ酸を置換し、mKOとは異なった蛍光特性を持つ蛍光蛋白質の作製を行った。点変異導入はmKOを挿入した大腸菌発現ベクター(pRSETB)に点変異導入プライマーをもちいてPCRをかけることにより行った。PCRに用いたプライマーは5'側のリン酸化を行った。
【0108】
(a)プライマーの5'リン酸化
100μM primer 2μl
10× T4 polynucleotide kinase buffer 5μl
100μM ATP 0.5μl
滅菌水 41.5μl
T4 polynucleotide kinase (10 U/μl) 1μl
37℃で30分間インキュベートした。
【0109】
(b)点変異導入PCR
5'リン酸化プライマー 4μl
template(mKO−pRSET B) 100ng
10× polymerase buffer 2.5μl
10× DNA ligase buffer 2.5μl
2.5mM dNTPs 1μl
polymerase(pfu)2.5U/μl 1μl
Taq DNA ligase 40U/μl 0.5μl
滅菌水で計50μlとする。
【0110】
プログラム
サーマルサイクラーはGeneAmp PCR system 9700を使用した。
1)65℃ 5 min
2)95℃ 2 min
3)95℃ 20 sec
4)52℃ 20 sec
5)65℃ 8 min
6)75℃ 7 min
7)4℃ hold
3)〜5)を25サイクル
【0111】
(c)Dpn1処理
PCR後のサンプルにDpn1を1μl加えて37℃に1時間インキュベートしてテンプレートプラスミドを切断した。
【0112】
(d)大腸菌への形質転換
Dpn1処理後のサンプルを大腸菌JM109(DE3)に形質転換して変異導入後のmKOを発現させ解析を行なった。
【0113】
(2)mKO時間経過変異体の解析
作製されたmKOの変異体は塩基配列の解析により、49番目のリジン(K)をグルタミン酸(E)に、70番目のプロリン(P)をグリシン(G)に、185番目のリジン(K)をグルタミン酸(E)に、188番目のリジン(K)をグルタミン酸(E)に、192番目のセリン(S)をアスパラギン酸(D)に、196番目のセリン(S)をグリシン(G)にアミノ酸置換されていた。このmKOの変異体は時間経過に伴って緑色蛍光とオレンジ色蛍光の比の異なる蛍光蛋白質であった。このmKOの変異体の70番目のプロリン(P)をいろいろなアミノ酸に置換することにより、時間経過に伴う緑色蛍光とオレンジ色蛍光の比が変化する速度が変わった。
【0114】
グリシン(G)に置換された変異体をmKO-FM9とした(アミノ酸配列を配列番号11に示し、塩基配列を配列番号12に示す)。
アラニン(A)に置換された変異体をmKO-FM5とした(アミノ酸配列を配列番号13に示し、塩基配列を配列番号14に示す)。
セリン(S)に置換された変異体をmKO-FM3とした(アミノ酸配列を配列番号15に示し、塩基配列を配列番号16に示す)。
システイン(C)に置換された変異体をmKO-FM20とした(アミノ酸配列を配列番号17に示し、塩基配列を配列番号18に示す)。
トレオニン(T)に置換された変異体をmKO-FM24とした(アミノ酸配列を配列番号19に示し、塩基配列を配列番号20に示す)。
バリン(V)に置換された変異体をmKO-FM14とした(アミノ酸配列を配列番号21に示し、塩基配列を配列番号22に示す)。
ロイシン(L)に置換された変異体をmKO-FM19とした(アミノ酸配列を配列番号23に示し、塩基配列を配列番号24に示す)。
チロシン(Y)に置換された変異体をmKO-FM23とした(アミノ酸配列を配列番号25に示し、塩基配列を配列番号26に示す)。
グルタミン(Q)に置換された変異体をmKO-FM21とした(アミノ酸配列を配列番号27に示し、塩基配列を配列番号28に示す)。
アスパラギン(N)に置換された変異体をmKO-FM25とした(アミノ酸配列を配列番号29に示し、塩基配列を配列番号30に示す)。
【0115】
それぞれのmKO時間経過変異体の測定は大腸菌JM109(DE3)で発現させたリコンビナント蛍光蛋白質でおこなうか、in vitroトランスレーションシステムPURE SYSTEM CLASSIC MINI(ポストゲノム研究所)を使用した。大腸菌での測定は各変異体を発現させた培養プレートを37℃に保温し、時間を追ってサンプリングして580nmの励起スペクトルを測定した(図14、15)。その結果、緑蛍光の励起ピークである約500nmのピークにくらべ、オレンジ蛍光の励起ピークである548nmのピークが時間により増加し、各変異体によってその増加率は違った。緑蛍光のピークは509nm、オレンジ蛍光のピークは560nmであった(図16、17、18、19、20、21、22;それぞれカッコ内の波長で励起)。蛍光測定には蛍光分光光度計F-2500(HITACHI)を使用した。大腸菌内では新たな蛋白質が断続的に生産されるために、緑からオレンジへの推移に必要とされる時間が見かけ上長くなってしまう。そこで、in vitroトランスレーションシステムを使用することによって蛋白質の生産時間を限定し、より正確な時間に伴う緑からオレンジへの推移を測定した。蛋白質合成時間は1時間とした。その直後にATPなどのタンパク質合成に必要なエネルギー源をゲルろ過で除き、37℃に保温して合成後25時間まで580nmの励起スペクトルを測定した(図23、24、25、26、27、28)。緑蛍光の励起ピーク部分の500nmとオレンジ蛍光の励起ピークである548nmの値の比をプロットすると、これらが比較的にアミノ酸の側鎖が大きくなるに従い(G→A→S→C→T→V→P)、オレンジ色蛍光成分への推移が速くなる傾向があることが分かった(図29)。
【0116】
mKO-FM14のN末端にTau(チューブリンなどに結合し微小管重合を促進し安定化させる蛋白質)を遺伝子的に繋いだ融合蛋白質遺伝子(アミノ酸配列を配列番号31に示し、塩基配列を配列番号32に示す)を動物細胞発現ベクターpCDNA3のBamH1-Xho1サイトにサブクローニングした。作製したベクターをPolyfect(キアゲン)を用いてHeLa−S3細胞に遺伝子導入した。遺伝子導入23時間後に培養液からHBSS(Hanks' Balanced Salt Solution)溶液に置換してイメージングをおこなった。その結果、HeLa−S3細胞がベクターを細胞内に取り込んだ時間差によって、緑色〜オレンジ色まで様々な色調の細胞が観察された。オレンジ色/緑色の比から細胞4と細胞5はベクターを早い時間に細胞内に取り込んで、つづいて細胞1、後に細胞2と細胞3がベクターを細胞内に取り込んでいることが確認できた(図30)。顕微鏡はIX-70(OLYMPUS)を用いた。緑色成分検出のために、励起フィルターは470DF35(OMEGA)、蛍光フィルターはHQ525/50M(CHROMA)、ダイクロイックミラーは505DRLP(OMEGA)を用いた。オレンジ色成分検出のために、励起フィルターはHQ500/40X(CHROMA)、蛍光フィルターはOG550(OMEGA)、ダイクロイックミラーはQ530LP(CHROMA)を用いた。
【0117】
(3)mKO時間経過変異体による分子の追跡
リコンビナントmKO-FM14蛋白質に強い緑色光を照射して、リコンビナントmKO-FM14蛋白質のオレンジ色蛍光成分のみを退色させることができるかを実験した。100Wのキセノンランプに直接フィルターを装着して、強い緑色光をリコンビナントmKO-FM14蛋白質に照射した。フィルターは546DF20(OMEGA)を使用した。コントロールとしてリコンビナントmKO蛋白質も同時に強い緑色光を照射して、照射前後の吸収スペクトルを測定し、548nmの吸収値が低下するかを調べた。吸収測定には分光光度計U-3310(HITACHI)を使用した。その結果、コントロールに用いたリコンビナントmKO蛋白質の548nmの吸収値は変化しなかった。それに対してリコンビナントmKO-FM14蛋白質の548nmの吸収値は優位に低下した。しかし、緑色蛍光成分を発するのに必要である500nmの吸収ピークに変化はなかった(図31、32)。これは、mKO-FM14蛋白質に強い緑色光を照射することによって、オレンジ蛍光成分のみを無くす、もしくは低下させることができることを示す。また、mKO-FM14蛋白質またはmKO-FM14蛋白質を付加したものが満たされた空間において、局所のみの強い緑色光照射によるオレンジ色蛍光の消光または低下により、オレンジ色蛍光シグナルと緑色蛍光シグナルの比を計算すれば、その部位をラベルすることができる。そこでmKO-FM14のN末端にBDNF(brain derived neurotrophic factor)を融合した融合蛋白質遺伝子(アミノ酸配列を配列番号33に示し、塩基配列を配列番号34に示す)をpEGFP-N1(clontech)からEGFP部分を抜き出したものにサブクローニングした。ラット海馬のニューロンに発現させてイメージングを行なった。
【0118】
ラット海馬のニューロンを用意した。妊娠ラット(17-19日目)の胎仔、あるいは生後1-3日目のラットの新生仔より顕微鏡下で海馬(約10匹分)を摘出した。次いで海馬を消化酵素パパインで十数分間、加温処理を行い、さらにピペットを用いて機械的に分散させ神経細胞に富む海馬細胞懸濁液を得た。必要に応じてこの懸濁液を培地で希釈し、ポリーリジンなどの細胞接着基質をコーティングした直径35 mmの培養皿の表面に播種した。播種密度2-4万細胞/cm2程度とし、これらの細胞を培養皿の表面に接着させ、牛胎仔血清およびN2-supplement(神経細胞用添加物)を含むイーグル培地を用いて高密度での初代培養を行なった。培養開始後6-7日目の細胞に対して35 mmの培養皿1枚当たり2-4マイクログラムのDNAをリン酸カルシウム法により37度で30分間、BDNF―mKO-FM14発現遺伝子ベクターの導入を行なった。この遺伝子導入の後、約12時間から2日間に細胞に発現した蛍光タンパクを蛍光顕微鏡で検出し色の変化を追跡する実験に用いた。緑色蛍光シグナル励起には490DF20(OMEGA)に10%減光フィルターを装着したものを、緑色蛍光シグナル検出フィルターは535DF35(OMEGA)を用いた。オレンジ色蛍光シグナル励起には546DF10(OMEGA)を、オレンジ色蛍光シグナル検出フィルターは595RDF60(OMEGA)を用いた。ダイクロイックミラーは505DRLPXR(OMEGA)を使用した。視野絞りを調節して、ラット海馬ニューロンのソーマ部分(細胞体)のみを、550DF30(OMEGA)を使用して強い緑色光でオレンジ色蛍光のみを退色させた。オレンジ蛍光シグナル/緑色蛍光シグナルの比(Ratio)から計算しソーマから神経突起へのBDNF―mKO-FM14の移動を観察した(図33、34)。図34の白矢印はBDNF―mKO-FM14がソーマ部分から神経突起の先端へ向かって移動している様子を示す。
【0119】
実施例7:単量体蛍光蛋白質mKOと2量体(多量体)蛍光蛋白質MiCyを用いたCaspase3活性測定プローブ
分子内FRETを行う際は少なくとも一種は単量体であるべきである。(A)単量体(白)と2量体(黒)の組み合わせ(図35A)。2量体(多量体)蛍光蛋白質MiCyと単量体蛍光蛋白質mKOの組み合わせはこのパターンとなる。例えば、2量体(白)と2量体(黒)の組み合わせはポリマーのように連なってしまうことが考えられる(図35B)。単量体蛍光蛋白質mKOと2量体蛍光蛋白質MiCyはMiCyの蛍光スペクトルとmKOの吸収スペクトルに重なりがあるため、両者を用いたFRET(蛍光共鳴エネルギー移動法)測定が可能である(図36)。そこで、MiCyとmKOをCaspase3の認識配列であるDEVD(Asp-Glu-Val-Asp)を含んだリンカーでつなぎ(アミノ酸配列を配列番号35に示し、塩基配列を配列番号36に示す)、Caspase3の活性化に伴うリンカー配列の切断をFRETにより測定した。
【0120】
(1)in vitroでのCaspase3活性測定
MiCy−linker−mKOの順に繋ぎ、大腸菌発現ベクターpRSETBのBamH1-EcoR1サイトにサブクローニングして大腸菌JM109(DE3)に発現させた。リンカーの配列はGGSGGDEVDGTGGS(Gly-Gly-Ser-Gly-Gly-Asp-Glu-Val-Asp-Gly-Thr-Gly-Gly-Ser)を用いた。これのコンストラクトをMiCy-DEVD-mKOとした。発現したリコンビナント融合蛋白質はNi-NTAアガロースで精製した。精製したリコンビナント融合蛋白質をセファデックスG-25カラムでゲルろ過を行い150mM KCl、50mM HEPES-KOH pH7.4溶液にバッファー置換した。活性測定にはリコンビナントActive−Caspase3(MBL:BV-1083-9)を用いた。20mM HEPES-KOH pH7.4、100mM NaCl、0.1% CHAPS、10% sucrose溶液中に各リコンビナント融合蛋白質を1mg/mlになるようにして、リコンビナントActive−Caspase3を1unit加えて30度で3時間反応させた。反応前と反応後の反応液の蛍光スペクトルを440nmで励起して測定した。測定には蛍光分光光度計F-2500(HITACHI)を使用した。その結果、Caspase3添加まえはFRETがおこってmKOの蛍光ピーク(559nm)が現れているが、添加後にはリンカーの切断によるFRETの解消によりmKOの蛍光ピーク(559nm)は消失し、MiCyの蛍光ピーク(495nm)のみとなった(図37)。
【0121】
(2)in vivoでのCaspase3活性測定
MiCy-DEVD-mKOを動物細胞での発現ベクターpCS2+のBamH1-EcoR1サイトにサブクローニングした。作製したベクターをPolyfect(キアゲン)を用いてHeLa−S3細胞に遺伝子導入した。遺伝子導入24時間後に培養液から500ng/ml 抗Fas抗体(CH-11:MBL)、10μg/ml サイクロヘキシミド、HBSS(Hanks' Balanced Salt Solution)溶液に置換してアポトーシスを誘導し、Caspase3活性測定のイメージングをおこなった。
【0122】
顕微鏡はIX-70(OLYMPUS)を用いた。励起フィルターは440AF21(OMEGA)、ダイクロイックミラーは455DRLP(OMEGA)を用いた。蛍光シグナルの検出は480ALP(OMEGA)のフィルターを通してカラー3CCDカメラASHURA(浜松ホトニクス)で行い、GreenチャンネルでMiCyの蛍光シグナルを、RedチャンネルでmKOの蛍光シグナルを検出した。その結果、HeLa細胞でのアポトーシスに伴い、Caspase3が活性化されて導入遺伝子の翻訳産物のリンカーが切断され、FRETが解消してRedチャンネルのシグナルが低下し、Greenチャンネルのシグナルが上昇する現象が観察された。Red/GreenのRatio(比)はCaspase3の活性化に伴い低下した。また、HeLa細胞のアポトーシスによる形態変化も観察された(図38)。
【0123】
実施例8:イシサンゴからの新規色素蛋白遺伝子の単離、新規蛍光蛋白の作製、及び特性解析
(1)total RNAの抽出
珊瑚より色素蛋白質の遺伝子の単離を行った。材料にはコモンサンゴ(Montipora. sp)を用いた。凍結したコモンサンゴを乳鉢で砕き、湿重量1グラムに"TRIzol"(GIBCO BRL)を7.5ml加えてホモジナイズし、1500×gで10分間遠心した。上清にクロロホルム1.5mlを加え、15秒間攪拌した後、3分間静置した。7500×gで15分間遠心した。上清にイソプロパノール3.75mlを加え、15秒間攪拌した後、10分間静置した。17000×gで10分間遠心した。上清を捨て70%エタノールを6ml加えて17000×gで10分間遠心した。上清を捨て、沈殿をDEPC水200μlで溶解した。DEPC水で溶解したtotal RNAを100倍に希釈して、O.D.260とO.D.280の値を測定してRNA濃度を測った。53μgのtotal RNAを得た。
【0124】
(2)First strand cDNAの合成
total RNA 4μgを使用し、First strand cDNAの合成キット"Ready To Go"(Amersham Pharmacia)によりcDNA(33μl)を合成した。
【0125】
(3)Degenerated PCR
合成したFirst strand cDNA(33μl)のうち3μlを鋳型としてPCRを行った。プライマーのデザインは既知の蛍光蛋白のアミノ酸配列を見比べて、似ている部分を抜き出し、塩基配列に変換し直し作製した。
使用プライマー
5'- GAAGGRTGYGTCAAYGGRCAY -3' (primer1)(配列番号74)
5'- ACVGGDCCATYDGVAAGAAARTT -3'(primer2)(配列番号75)
Iはイノシン、RはA又はG、YはC又はT、VはA,C又はG、DはA,G又はT SはC又はG、HはA,T又はCを示す。
【0126】
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 taq バッファー 5μl
2.5mM dNTPs 4μl
100μM primer1 1μl
100μM primer2 1μl
ミリQ 35μl
taq polymerase(5U/ul) 1μl
【0127】
PCR反応条件
94℃ 1分(PAD)
94℃ 30秒(変性)
52℃ 30秒(鋳型へのプライマーのアニーリング)
72℃ 1分 (プライマーの伸長)
上記3ステップを35サイクル行った。
72℃ 7分(最後の伸長)
4℃ 保持
【0128】
一回目のPCR反応で得られた増幅産物1μlをテンプレートとして、もう一度同じ条件でPCRを行った。アガロースゲル電気泳動で、350bpを切り出し、精製した。
【0129】
(4)サブクローニング及び塩基配列の決定
精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。得られた塩基配列を他の蛍光蛋白遺伝子の塩基配列と比較してそのDNA塩基配列が蛍光蛋白由来のものであるかを判断した。蛍光蛋白遺伝子の一部であると判断したものに関して、5'-RACE法および3'-RACE法による遺伝子全長のクローニングを行った。
【0130】
(5)5'-RACE法
Degenerated PCRで得られたDNA断片の5'側の塩基配列を決定するために5'-RACE System for Rapid Amplification of cDNA Ends,Version 2.0(GIBCO BRL)を用いて、5'-RACE法を行った。鋳型として(1)で調整したtotal RNAを5μg使用した。
【0131】
dC-tailed cDNAの一回目の増幅には
5'-GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG-3' (primer3)(配列番号76)
5'- CTCAGGGAATGACTGCTTTACAT -3' (primer4)(配列番号77)
のプライマーを用いた。
Iはイノシンを示す。
【0132】
二回目の増幅には
5'-GGCCACGCGTCGACTAGTAC-3' (primer5)(配列番号78)
5'- GTCTTCAGGGTACTTGGTGA -3' (primer6)(配列番号79)
のプライマーを用いた。PCR反応条件等はキットのプロトコールに準じた。
アガロースゲル電気泳動で、増幅された350bpのバンドを切り出し、精製した。精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。
【0133】
(6)3'-RACE法
Degenerated PCRで得られたDNA断片の3'側部分は、(4)の塩基配列決定で得られた情報を基に作製したプライマーとオリゴdTプライマーのPCRで得た。鋳型として(2)で調整したfirst strand cDNAを3μl使用した。
【0134】
作成したプライマーは、
5'- ATGTAAAGCAGTCATTCCCTGAG -3' (primer7)(配列番号80)
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 taq バッファー 5μl
2.5mM dNTPs 4μl
20μM primer7 1μl
10μM オリゴdTprimer 1μl
ミリQ 35μl
taq polymerase(5U/μl) 1μl
【0135】
PCR反応条件
94℃ 1min(PAD)
94℃ 30sec (変性)
52℃ 30sec (鋳型へのプライマーのアニーリング)
72℃ 1min (プライマーの伸長)
上記3ステップを30サイクル行った。
72℃ 7min (最後の伸長)
4℃ 保持
【0136】
アガロースゲル電気泳動で、増幅された約650bpのバンドを切り出し、精製した。精製したDNA断片をpT7-blue vector(Novagen)にライゲーションした。大腸菌株(TG1)にトランスフォーメーションしてブルーホワイトセレクションを行い、白いコロニーの大腸菌よりplasmid DNAを精製して、挿入されたDNA断片の塩基配列をDNAシークエンサーにより決定した。
【0137】
(7)大腸菌での蛋白発現
得られた全長の塩基配列より、蛋白のN末端に相当する部分でプライマーを作製し、C末端側はオリゴdTプライマーを使用して、(2)で調整したFirst strand cDNAを鋳型としてPCRを行った。全アミノ酸配列および全塩基配列を配列表の配列番号37及び38に示す。配列番号37に記載のアミノ酸配列を有する蛋白質をCOCPと称する。
【0138】
使用プライマー
5'- CCCGGATCCGACCATGGCTACCTTGGTTAAAGA -3' (primer8)(配列番号81)
PCR反応液組成
テンプレート(first strand cDNA) 3μl
X10 pyrobest バッファー 5μl
2.5mM dNTPs 4μl
100uM primer8 1μl
100uM オリゴdTプライマー 1μl
ミリQ 35μl
pyrobest polymerase(5U/μl) 1μl
【0139】
PCR反応条件
94℃ 1min(PAD)
94℃ 30sec (変性)
52℃ 30sec (鋳型へのプライマーのアニーリング)
72℃ 1min (プライマー伸長)
上記3ステップを30サイクル行った。
72℃ 7min (最後の伸長)
4℃ 保持
【0140】
アガロースゲルの電気泳動で、増幅された約800bpのバンドを切り出し、精製してpRSET vector(Invitrogen)のBamHI、EcoRI部位にサブクローニングして、大腸菌株(JM109-DE3)で発現させた。発現蛋白はN末端にHis-tagが付くようにコンストラクトしたので発現蛋白はNi-Agarose gel(QIAGEN)で精製した。精製の方法は付属のプロトコールに準じた。次に精製した蛋白の性質を解析した。
【0141】
(8)光吸収特性の解析
20μM色素蛋白、50mM HEPES pH7.9溶液を用いて吸収スペクトルを測定した。このスペクトルのピークの値よりモル吸光係数を計算した。コモンサンゴ由来色素蛋白(COCP)では576nmに吸収のピークが認められた(表2、図39)。また、pH4〜10で安定していた。(図40)
【0142】
【表2】
【0143】
(9)色素蛋白質から蛍光蛋白質への改変
COCPは蛍光蛋白質ではない。しかしCOCPの1番目のメチオニンと2番目のセリンの間にバリンを挿入し、94番目のヒスチジンをアスパラギンに、142番目のアスパラギンをセリンに、157番目のアスパラギンをアスパラギン酸に、202番目のリジンをアルギニンに、206番目のフェニルアラニンをセリンに置き換えることにより蛍光性を獲得した。この改変蛍光蛋白質をCOCP-FLとした(アミノ酸配列を配列番号39に示し、塩基配列を配列番号40に示す)。COCP-FLは560nmに励起のピークを持つ。この励起によって蛍光スペクトルは600nmにピークを示す。
【0144】
(10)ストークスシフトの大きな赤色蛍光蛋白質の作製
COCP-FLの62番目のセリンをフェニルアラニンに、93番目のイソロイシンをトレオニンに、124番目のバリンをトレオニンに、159番目のフェニルアラニンをチロシンに、192番目のバリンをイソロイシンに、214番目のセリンをアラニンに置き換えることによりCOCP-FLとは異なる蛍光をもつ蛋白質を獲得した。この改変蛍光蛋白質をkeima616(アミノ酸配列を配列番号41に示し、塩基配列を配列番号42に示す)とした。440nmに励起のピークをもち、この励起によって蛍光スペクトルは616nmにピークを持つ(図41、表2)。ストークスシフトは176nmと非常に大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないためできなかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。
【0145】
(11)ストークスシフトの大きな橙色蛍光蛋白質の作製
Keima616の62番目のフェニルアラニンをメチオニンに、63番目のグルタミンをシステインに置き換えることにより蛍光蛋白質を獲得した。この改変蛍光蛋白質をKeima570(アミノ酸配列を配列番号43に示し、塩基配列を配列番号44に示す)とした。このKeima570はKeima616と同様440nmに励起のピークを持ち、この励起により570nmの蛍光のピークを示す(図42)。ストークスシフトは130nmと大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないためできなかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。
【0146】
(12)pH感受性の測定
50mMの下記の緩衝液中で蛋白質(Keima616及びKeima570)の吸収スペクトルを測定した(図43及び44)。
各pHの緩衝液は次の通り、
pH4、5、5.5 : 酢酸バッファー
pH6 : リン酸バッファー
pH6.6 : MOPSバッファー
pH7、7.5、8 : HEPESバッファー
pH9、10 : グリシンバッファー
pH7.5〜10でピークの値は安定していた。(図43及び図44)
【0147】
実施例9
(1) ストークスシフトの大きな単量体赤色蛍光蛋白質の作製
keim616の61番目のロイシンをグルタミンに、93番目のトレオニンをセリンに、124番目のトレオニンをグルタミン酸に、189番目のチロシンをアルギニンに、191番目のチロシンをグルタミン酸に置き換えることにより超遠心分子量測定により分子量30.1kDaの結果から、アミノ酸配列から予想されるおよそ29kDaとほぼ一致することから単量体となったkeima616蛋白を獲得した。本改変蛍光蛋白質をcmkeima620とした(図45)(アミノ酸配列を配列表の配列番号45に示し、塩基配列を配列番号46に示す)。440nmに励起のピークをもち、この励起によって蛍光スペクトルは620nmにピークを持つ(図46)。ストークスシフトは180nmと非常に大きな値である。従来の蛍光蛋白質に比べ励起波長域と蛍光波長域を大きくとることができ、蛍光測定時に効率よく測定できる。また、同時多色蛍光測定も可能である。同一励起波長をもつ蛍光色素を用いることによりレーザーなど単一波長での励起による二つの波長での測光ができる。いままでの蛍光蛋白では同じ励起スペクトルをもつ蛋白がないため出来なかったことで、これらの蛋白を用いることで励起のちがいによる測定のぶれという問題を解決できる。また、全体の分子量をおさえ且つ蛍光蛋白質自身の間で多量体形成による相互作用がないため、ラベル分子の性質変化を最小限にとどめることができる。
【0148】
(2)ストークスシフトの大きな単量体赤色蛍光蛋白質の改良
cmkeim616の62番目のフェニルアラニンをロイシンに置き換えることによりcmkeima620のフォールディング効率が上昇したものを得た。本改変蛍光蛋白質をmkeima620とした(アミノ酸配列を配列表の配列番号47に示し、塩基配列を配列番号48に示す)。440nmに励起のピークをもち、この励起によって蛍光スペクトルは620nmにピークを持つ。ストークスシフトは180nmと非常に大きな値である。cmkeima620よりも相対的な蛍光強度が高いため(図46、47)、単量体でもkeima616と同様に十分使いやすくなっている。
【0149】
実施例10:ストークスシフトの大きな蛋白質を用いた一波長励起二波長測光型蛍光相互相関分光法の測定系の開発
分子間相互作用を測定するための手法として、蛍光分子を利用した蛍光相互相関分光法(FCCS) がある。これは2つの蛍光分子をプローブとして用いることにより分子間相互作用をモニタリングできる。
【0150】
現在用いられている2つの異なる蛍光分子を用いた2 波長励起FCCS 測定において相互相関の感度を下げる大きな要員として色収差による2 波長の測定領域の重なりのずれがあげられる。Keima616とECFPのような一つの波長で励起できしかも蛍光が分離できる蛍光蛋白質の組み合わせを用いた一波長励起FCCS ではこれを回避する事ができるため、FCCS 測定の感度の上昇が期待される(図48)。また、Fluorescence Resonance energy transfer(FRET)を回避できるため、FCCSでの測定が簡便化され、蛋白質間相互作用のFCCSによる検出に適している。従ってストークスシフトが大きい蛍光蛋白質であるkeima616を用いることによりFCCSによる蛋白質間相互作用の検出が簡便かつ強力なものになると思われる。
【0151】
(1) Caspase-3の活性検出
(a)蛍光相互相関測定における装置
蛍光相互相関測定にはTCS SP2 SOBS(Leica)とFCCSシステムを用いた。EGFP-(スペーサー)DEVD-mRFP1には、458 nm Argon ion Laserと594 nm HeNe Laserを用いて2 波長励起を行なった。またECFPとKeima616の組み合わせの蛋白質には458nm Argon Laserを用いた。受光用バンドパスフィルタはEGFP:500-550、mRFP1:607-683を、ECFP :470-500、keima616:535-585を用いた。
【0152】
(b)蛍光相互相関測定の解析
Caspase-3 により切断されるアミノ酸配列DEVDをEGFPとmRFPの間、keima616とECFPの間に導入し (図49)、リコンビナントEGFP-DEVDmRFP1(x2)(アミノ酸配列を配列表の配列番号49に示し、塩基配列を配列番号50に示す) 、ECFP-(スペーサー)DEVD-keima616(アミノ酸配列を配列表の配列番号51に示し、塩基配列を配列番号52に示す)、keima616-(スペーサー)DEVD- ECFP(アミノ酸配列を配列表の配列番号53に示し、塩基配列を配列番号54に示す)を作製した。発現蛋白質はN末端にHis-tagがつくようにコンストラクトしたので発現蛋白質はNi-Agarose gel(QIAGEN)で精製した。精製方法は付属のプロトコールに準じた。次にそれらの蛋白質を用いて相関作用を解析した。
【0153】
相互相関の定量的な評価は、relative amplitudeという相互相関関数の振幅(Gcross(0))を自己相関関数の振幅(Glower(0)) で割算した値を用いた。EGFP-DEVD-mRFP1(x2) では、Gcross(0) /Glower(0) は約0.4 であった(図51)。Caspase-3の添加によりGcross(0) の減少が観られた(図50)。
ECFPとkeima616の組み合わせではGcross(0) /Glower(0)は0.4であった(図51)。Caspase-3の添加によりGcross(0)の迅速な減少が見られた。Gcross(0)の減少はCaspase-3添加により蛍光相関が無くなっていることを示している。EGFP-DEVD-mRFPよりもECFPとKeima616を組み合わせた蛋白質がより短時間で相関が無くなっておりこれにより後者の組み合わせがより蛍光相互相関法により簡便かつ迅速に蛋白質の相互作用を示すことが明らかになった。
【0154】
(c) SDS-PAGEによる蛋白質間相互作用の解析
keima616-DEVD-ECFPをCaspase-3で反応させるとkeima616とECFPの大きさのバンドが確認出来た。これらの蛋白質はCaspase-3によってDEVDが切断されたことを意味する(図52)。Native-PAGEにおいても同様に反応後に2つのバンドが確認され、それぞれがkeima616とECFPであることが同定され、蛍光検出でもCaspase-3の活性があることが検出できた(図52)。
【0155】
(2)カルモジュリンとの相互作用
(a)蛋白質の合成・発現
カルモジュリンにはECFPを、M13にはKeima616を繋いだ(図53)。ECFP−カルモジュリンのアミノ酸配列を配列番号55に示し、塩基配列を配列番号56に示す。また、M13−Keima616のアミノ酸配列を配列番号57に示し、塩基配列を配列番号58に示す。それらの融合蛋白質は大腸菌株(JM109-DE3)で発現させた。発現蛋白はN末端にHis-tagがつくようにコンストラクトしたので発現蛋白はNi-Agarose gel(QIAGEN)で精製した。精製方法は付属のプロトコールに準じた。次にそれらの蛋白質を用いて相関作用を解析した。
【0156】
(b)蛍光相互相関測定における装置
蛍光相互相関測定にはConfoCor2(Carl Zeiss)とLSM510 version3.2を用いた。458 nm Argon ion Laserを用いた。受光用バンドパスフィルタはECFP:475-525、Keima616:LP610を用いた。
【0157】
(c)蛍光相互相関測定の解析
相互相関の定量的な評価は、relative amplitudeという相互相関関数の振幅(Gcross(0))を自己相関関数の振幅(Glower(0)) で割算した値を用いた。EGTAによりカルシウムイオンをキレートしたサンプルではGcross(0) /Glower(0) は約0.005であった(図54)。しかしカルシウムイオンの添加によりGcross(0) の値の上昇が確認できた(図55)。この結果はカルシウム依存的な蛋白質の相互作用を検出したことを示している。これにより蛍光相互相関法により蛋白間相互作用が迅速且つ簡便に測定できることが明らかになった。
【特許請求の範囲】
【請求項1】
以下の(a)又は(b)に示す蛍光蛋白質。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【請求項2】
以下の(a)又は(b)に示す蛍光蛋白質をコードするDNA。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【請求項3】
以下の(a)又は(b)に示すDNA。
(a)配列番号42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ、蛍光特性を有し、100nm以上のストークスシフトを有する蛋白質をコードする塩基配列を有するDNA。
【請求項4】
請求項2又は3記載のDNAを有する組み換えベクター。
【請求項5】
請求項2又は3記載のDNA又は請求項4に記載の組み換えベクターを有する形質転換体。
【請求項6】
請求項1に記載の蛋白質と他の蛋白質とから成る融合蛋白質。
【請求項7】
他の蛋白質が細胞内に局在する蛋白質である、請求項6に記載の融合蛋白質。
【請求項8】
他の蛋白質が細胞内小器官に特異的な蛋白質である、請求項6又は7に記載の融合蛋白質。
【請求項9】
他の蛋白質が蛍光蛋白質である、請求項6に記載の融合蛋白質。
【請求項10】
分子内FRETを生じる、請求項9に記載の融合蛋白質。
【請求項11】
請求項6から8の何れか1項に記載の融合蛋白質を細胞内で発現させることを特徴とする、細胞内における蛋白質の局在または動態を分析する方法。
【請求項12】
請求項1に記載の蛍光蛋白質、請求項2又は3に記載のDNA、請求項4に記載の組み換えベクター、請求項5に記載の形質転換体、又は請求項6から10の何れか1項に記載の融合蛋白質を含む、試薬キット。
【請求項1】
以下の(a)又は(b)に示す蛍光蛋白質。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【請求項2】
以下の(a)又は(b)に示す蛍光蛋白質をコードするDNA。
(a)配列番号41、43、45又は47に記載のアミノ酸配列を有する蛋白質;
(b)配列番号41、43、45又は47に記載のアミノ酸配列において1から数個のアミノ酸が欠失、置換、及び/又は付加されたアミノ酸配列を有し、蛍光特性を有し、かつ100nm以上のストークスシフトを有する蛋白質。
【請求項3】
以下の(a)又は(b)に示すDNA。
(a)配列番号42、44、46又は48に記載の塩基配列を有するDNA;
(b)配列番号42、44、46又は48に記載の塩基配列において、1から数個の塩基の欠失、置換及び/又は付加を有する塩基配列を有し、かつ、蛍光特性を有し、100nm以上のストークスシフトを有する蛋白質をコードする塩基配列を有するDNA。
【請求項4】
請求項2又は3記載のDNAを有する組み換えベクター。
【請求項5】
請求項2又は3記載のDNA又は請求項4に記載の組み換えベクターを有する形質転換体。
【請求項6】
請求項1に記載の蛋白質と他の蛋白質とから成る融合蛋白質。
【請求項7】
他の蛋白質が細胞内に局在する蛋白質である、請求項6に記載の融合蛋白質。
【請求項8】
他の蛋白質が細胞内小器官に特異的な蛋白質である、請求項6又は7に記載の融合蛋白質。
【請求項9】
他の蛋白質が蛍光蛋白質である、請求項6に記載の融合蛋白質。
【請求項10】
分子内FRETを生じる、請求項9に記載の融合蛋白質。
【請求項11】
請求項6から8の何れか1項に記載の融合蛋白質を細胞内で発現させることを特徴とする、細胞内における蛋白質の局在または動態を分析する方法。
【請求項12】
請求項1に記載の蛍光蛋白質、請求項2又は3に記載のDNA、請求項4に記載の組み換えベクター、請求項5に記載の形質転換体、又は請求項6から10の何れか1項に記載の融合蛋白質を含む、試薬キット。
【図1】

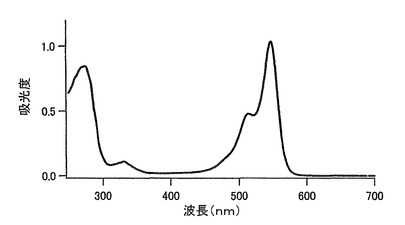
【図2】
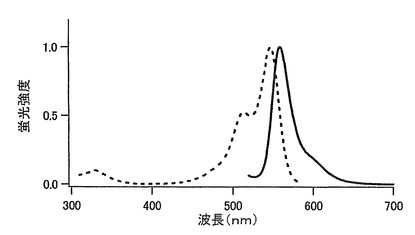
【図3】
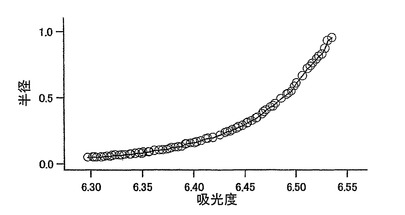
【図6】
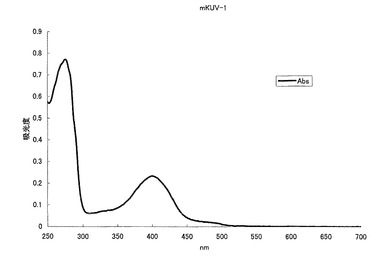
【図7】
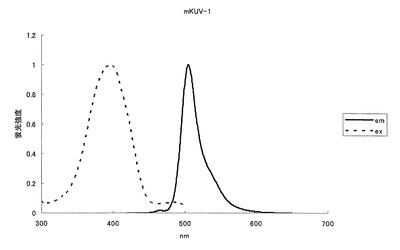
【図8】
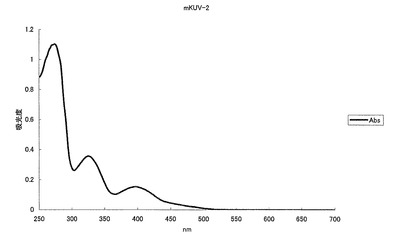
【図9】
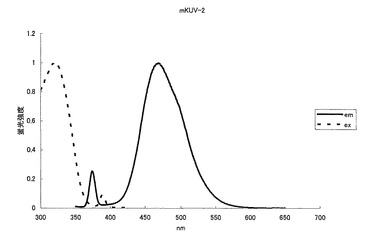
【図10】
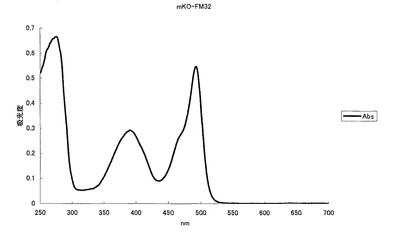
【図11】
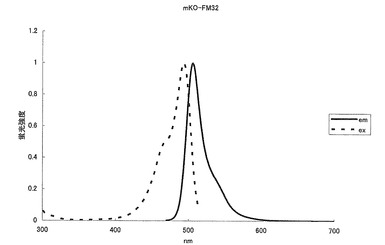
【図12】
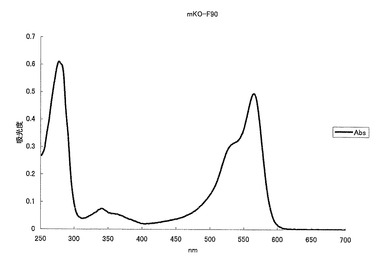
【図13】
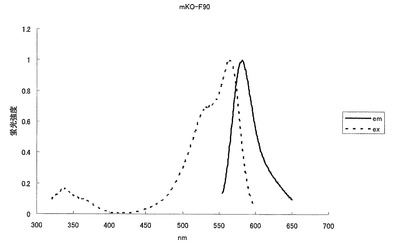
【図14】
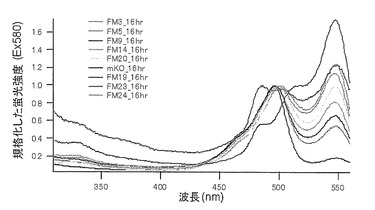
【図15】
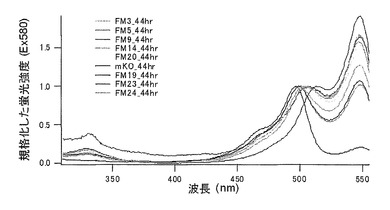
【図16】
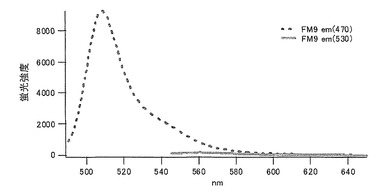
【図17】
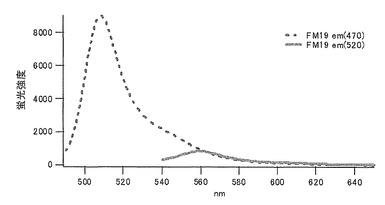
【図18】
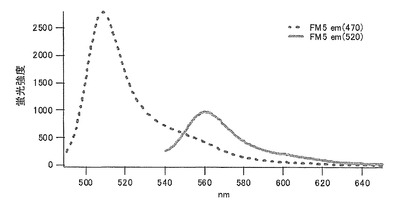
【図19】
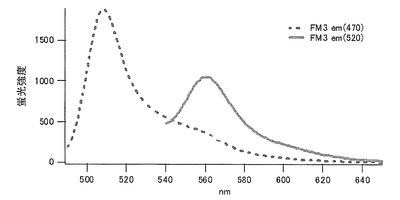
【図20】
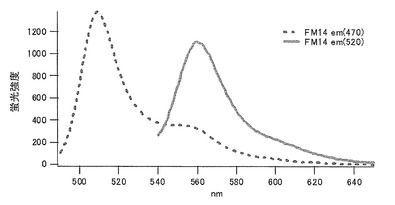
【図21】
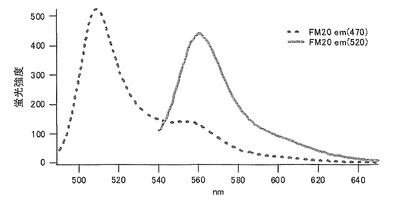
【図22】
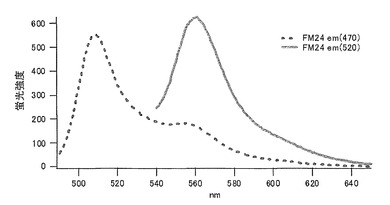
【図23】
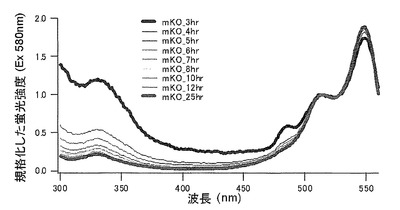
【図24】
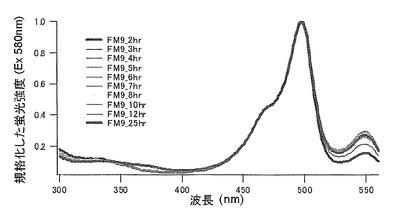
【図25】
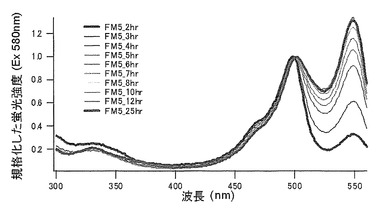
【図26】
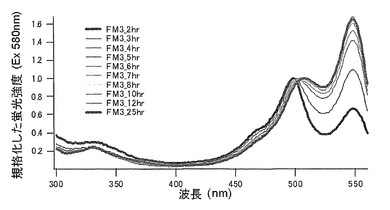
【図27】
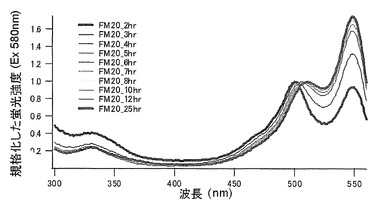
【図28】
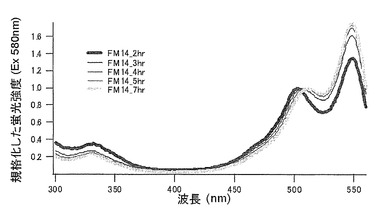
【図29】
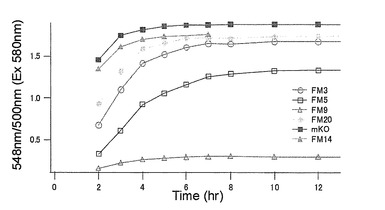
【図31】
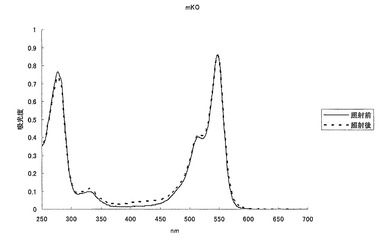
【図32】
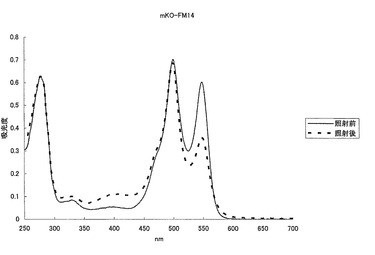
【図35】
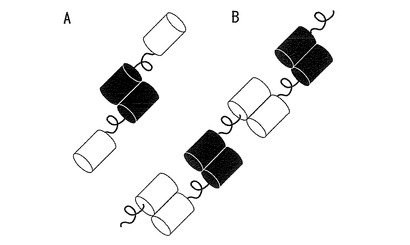
【図36】
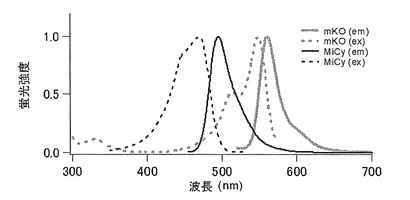
【図37】
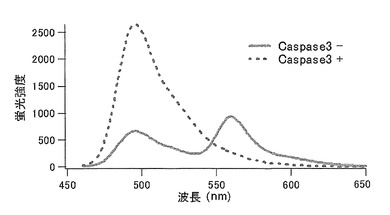
【図39】
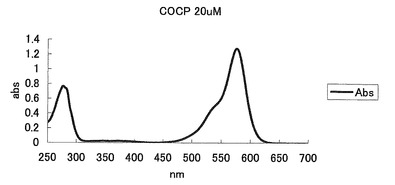
【図40】
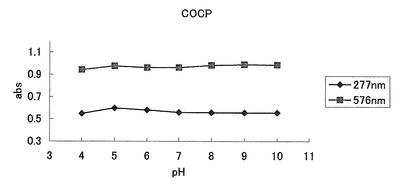
【図41】
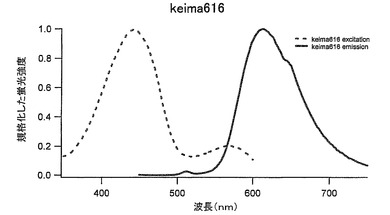
【図42】
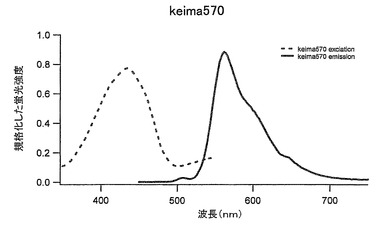
【図43】
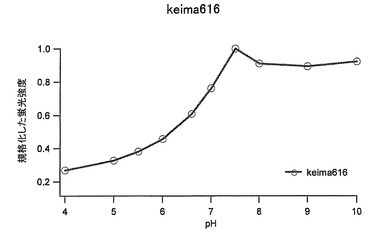
【図44】
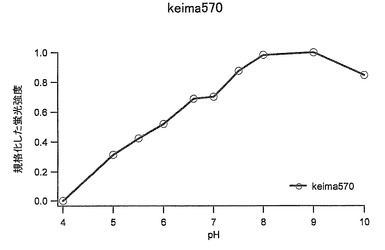
【図45】
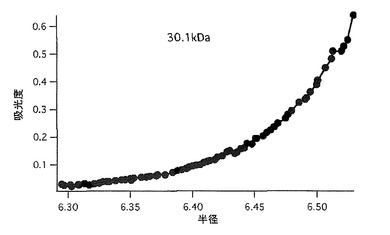
【図46】
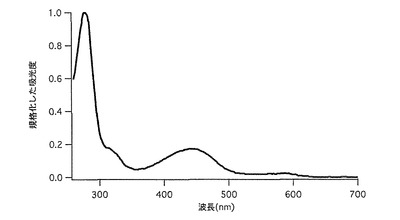
【図47】
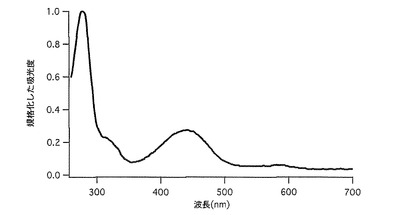
【図48】
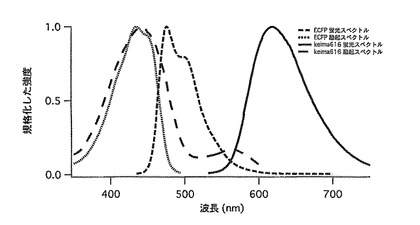
【図49】
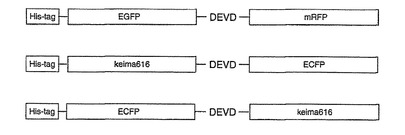
【図50】
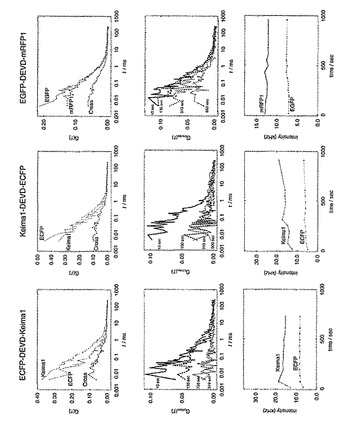
【図51】
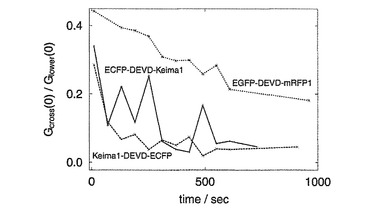
【図53】
【図54】
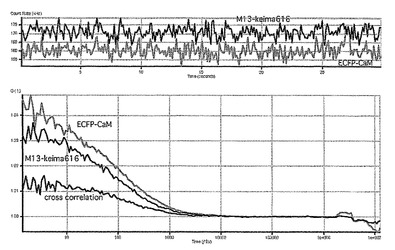
【図55】
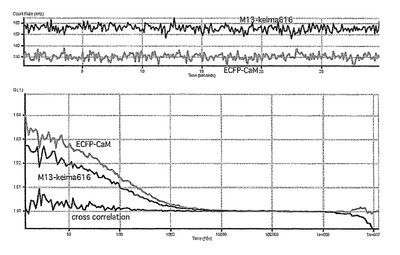
【図4】

【図5】
【図30】
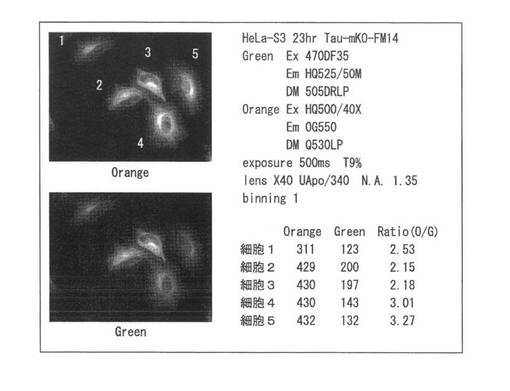
【図33】
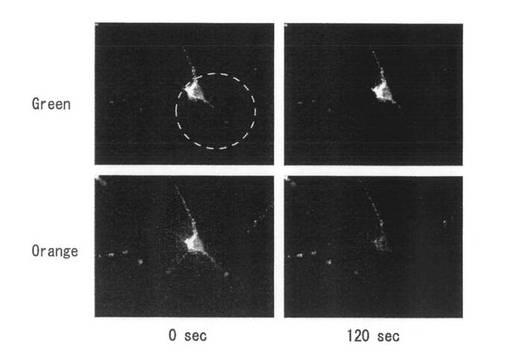
【図34】
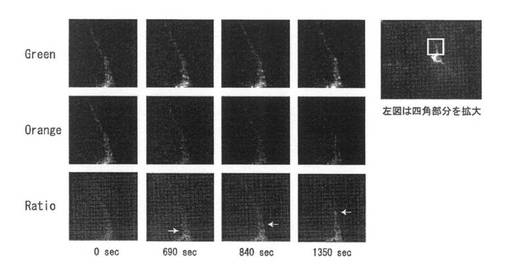
【図38】
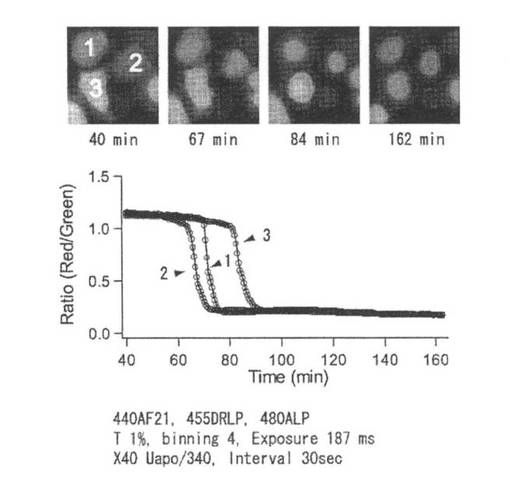
【図52】

【図2】
【図3】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図31】
【図32】
【図35】
【図36】
【図37】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図53】
【図54】
【図55】
【図4】
【図5】
【図30】
【図33】
【図34】
【図38】
【図52】
【公開番号】特開2011−36262(P2011−36262A)
【公開日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願番号】特願2010−214870(P2010−214870)
【出願日】平成22年9月27日(2010.9.27)
【分割の表示】特願2005−516041(P2005−516041)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
【公開日】平成23年2月24日(2011.2.24)
【国際特許分類】
【出願日】平成22年9月27日(2010.9.27)
【分割の表示】特願2005−516041(P2005−516041)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
[ Back to top ]