
融合タンパク質発現ベクター
【課題】 難溶性タンパク質を効率的に生産できる手段を提供すること。
【解決手段】 好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【解決手段】 好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、融合タンパク質発現ベクター、詳しくは、目的とする難溶性タンパク質を可溶化状態で発現させるための融合タンパク質発現ベクター、及び該融合タンパク質発現ベクターを用いるタンパク質の製造方法に関する。
【背景技術】
【0002】
現在、ヒトをはじめとする数多くの生物のゲノム塩基配列が解読され、多くの遺伝子情報がデータベースにて公開されている。また、これらの遺伝子情報を元に発現させたタンパク質の網羅的な機能解析も急速に進みつつある。タンパク質の発現は、それをコードする遺伝子をベクターに組み込み、細菌、酵母、昆虫細胞等の宿主に導入し、培養することにより行われる。しかしながら、遺伝子産物であるタンパク質によっては、その高次構造形成がうまくいかず、お互いに変性会合し、不溶性になってしまうパターン、あるいは、折りたたみ(フォールディング)異常によって、立体構造の異なった異常型タンパク質しか得られないパターンがある。このようなタンパク質は宿主内で封入体と呼ばれる凝集体として発現したり、宿主細胞のプロテアーゼにより分解されたりすることが知られている。
【0003】
この問題を克服するためのひとつの手法として、融合タンパク質発現法がある。この方法は、目的の難溶性タンパク質を特定の容易に発現するタンパク質に結合させ、融合タンパク質として可溶化状態で発現させ、その後、目的の難溶性タンパク質のみをプロテアーゼ処理によって切り離し取得する方法である。ところが、この方法は、比較的軽度の難溶性タンパク質については一定の成功をおさめてはいるものの、これまで用いられている融合タンパク質はいずれも可溶性に限界があり、高度な難溶性タンパク質については、依然として正しい生理活性を持つタンパク質として得られていないのが現状である。
【0004】
一方、ヒトをはじめ通常生物が生育できないような高濃度塩の環境下で生育可能な好塩性細菌由来のタンパク質は、極めて高い可溶性を有し、一度変性しても直ちに可逆的に本来の構造が回復し、活性状態に戻るという性質を持っていることが知られている。本発明者らが発見した中度好塩菌由来タンパク質であるβ−ラクタマーゼ (非特許文献1)や、高度好塩菌由来タンパク質であるヌクレオシド二リン酸キナーゼ (非特許文献2、3)もまた上記のような特性を有する。例えば、β−ラクタマーゼは、5分間煮沸しても常温に戻すと瞬時に80%活性を回復し、また、ヌクレオシド二リン酸キナーゼは、95℃で熱処理しても100%活性を回復する。これらの好塩性酵素タンパク質の構造を調べると、活性中心等の触媒機能に重要な部分は通常の酵素タンパク質の構造をよく保存しており、それ以外の部分、特にタンパク質表面に高頻度で酸性アミノ酸残基が局在しており、この酸性アミノ酸に起因する大量のマイナス荷電がその高い可溶性の維持を可能にしていることが示唆される。
【0005】
【非特許文献1】Tokunaga, H., et al., Highly efficient renaturation of β-lactamase isolated from mederately palophilic bacteria, FEBS Letters 558, 7-12, 2004
【非特許文献2】Ishibashi, M., et al., et al., Facilitated folding and subunit assembly in E.coli and in vitro of nucleoside diphosphate kinase from halophilic archaeon conferred by amino-terminal extension containing Hexa-His-tag, FEBS Letters 570, 87-92, 2004
【非特許文献3】Ishibashi, M., et al., NaCl-activated nucleoside diphosphate kinase from extremetly halophilic archaeon, H.salinarum, maitains native confirmation without salt, FEBS Letters 493, 134-138, 2001
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明の課題は、難溶性タンパク質を効率的に生産できる手段を提供することにある。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決するため鋭意研究を重ねた結果、好塩性細菌由来のタンパク質を目的とする難溶性タンパク質の融合パートナーとして用いることにより、宿主細胞内で可溶性の融合タンパク質の形態で発現することに成功し、本発明を完成させるに至った。
【0008】
すなわち、本発明は以下の発明を包含する。
(1) 好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【0009】
(2) 好塩性細菌由来のタンパク質が、クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼである、(1)に記載の融合タンパク質発現ベクター。
(3) 好塩性細菌由来のタンパク質が、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼである、(1)に記載の融合タンパク質発現ベクター。
【0010】
(4) (1)〜(3)のいずれかに記載の融合タンパク質発現ベクターに、目的タンパク質をコードする遺伝子を組み込んだことを特徴とする、融合タンパク質発現ベクター。
(5) 目的タンパク質が、難溶性タンパク質である、(4)に記載の融合タンパク質発現ベクター。
【0011】
(6) (4)に記載の融合タンパク質発現ベクターにより形質転換された形質転換体。
(7) (6)に記載の形質転換体を培地に培養し、得られる培養物から好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質を採取し、プロテアーゼで消化することを含む、タンパク質の製造方法。
【発明の効果】
【0012】
本発明によれば、これまで正しい構造と機能をもって発現・生合成させることが不可能であった難溶性タンパク質を効率よく製造できる融合タンパク質発現ベクターが提供される。従って、本発明の融合タンパク質発現ベクターを利用すれば、医薬品として有用な種々の生理活性を有する機能性タンパク質を、活性を維持したまま大量に製造することができる。
【発明を実施するための最良の形態】
【0013】
1.融合タンパク質発現ベクター
本発明の融合タンパク質発現ベクターは、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させるものであり、好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流に目的タンパク質をコードする遺伝子を挿入するための制限酵素部位(マルチクローニングサイトという)、および発現した融合タンパク質から目的タンパク質を切り出すために必要なプロテアーゼ切断部位とを含む。
【0014】
上記の好塩性細菌由来のタンパク質としては、0.2M以上のNaCl濃度下において生育・増殖可能な細菌由来、例えば、中度好塩性細菌(至適NaCl濃度:0.5-2.5M)または高度好塩性細菌(至適NaCl濃度:2.5-5.2M)由来のタンパク質が含まれる。例えば、中度好塩性細菌であるクロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼ(β-lactamase:BLA)、高度好塩性細菌であるハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼ(nucleoside diphosphate kinase:NDK)が好適に用いることができる。クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼのアミノ酸配列を配列番号2に、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼのアミノ酸配列は配列番号4に示す。
【0015】
好塩性細菌由来のタンパク質をコードする遺伝子として、具体的には、配列番号1又は3に示す塩基配列からなるDNAが用いられる。また、上記遺伝子には、所望の酵素活性を有する限り変異遺伝子も含まれ、例えば、配列番号1又は3に示す塩基配列からなるDNAに相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAが含まれる。
【0016】
上記の「配列番号1又は3に示す塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズできるDNA」としては、配列番号1又は3に示す塩基配列と約70%以上、好ましくは約80%以上、より好ましくは約90%以上、最も好ましくは約95%以上の相同性を有する塩基配列からなるDNA等が挙げられる。ここで、
ストリンジェントな条件とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいい、例えば、ナトリウム濃度が、10mM〜300mM、好ましくは20〜100mMであり、温度が25℃〜70℃、好ましくは42℃〜55℃での条件をいう。
【0017】
遺伝子に変異を導入するには、Kunkel法又は Gapped duplex法等の公知手法又はこれに準ずる方法により行うことができ、例えば部位特異的突然変異誘発法を利用した変異導入用キット(例えばMutant-K(TAKARA社製)やMutant-G(TAKARA社製))などを用いて、あるいは、TAKARA社のLA PCR in vitro Mutagenesis シリーズキットを用いて変異が導入される。
【0018】
一方、目的タンパク質としては、通常の発現系ではインクルージョンボディ(inclusion body, 封入体)と呼ばれる不溶性で不活性な凝集体(変性タンパク質)としてしか回収することのできない難溶性タンパク質が好ましい。このような難溶性タンパク質としては、例えば、ヒトαデフェンシン1(hαD1)、ヒトインターロイキン1α(hIL1α)、ヒトセリンラセマーゼ(hSR)、膜埋没型タンパク質(porin)、プロテインキナーゼ等が挙げられる。
【0019】
プロテアーゼ切断部位は、上記好塩性細菌由来のタンパク質をコードする遺伝子と、上記目的タンパク質をコードする遺伝子を挿入することができる制限酵素部位(マルチクローニングサイト)との間に設ける。
【0020】
上記プロテアーゼ切断部位は、本融合タンパク質発現ベクターにより発現される好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質において、両タンパク質をつなぐペプチドリンカーとなる。このペプチドリンカー内にあるプロテアーゼ切断部位にプロテアーゼを作用させることによって、容易に融合タンパク質から目的タンパク質を切り出すことができる。
【0021】
上記プロテアーゼとしては特に限定されず、例えば、トロンビン、ファクターXa、エンテロキナーゼ等が挙げられる。上記ペプチドリンカーとなるべき塩基配列の長さは特に限定されないが、12〜18塩基程度であることが好ましく、翻訳されてグリシンやセリン等の中性アミノ酸となる塩基配列を多く含むことが好ましい。
【0022】
また、本発明において上記の好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質をコードする遺伝子を組み込むために用いるベクターとしては、宿主内で該融合タンパク質遺伝子を発現することが可能であり、宿主内で複製可能なものであれば特に限定されず、例えば、プラスミドDNA、ファージDNA等のいずれであってもよい。プラスミド DNAとしては、大腸菌由来のプラスミド(例えばpBR322, pUC118等)、枯草菌由来のプラスミド(例えばpUB110, pTP5等)、酵母由来のプラスミド(例えばYEp13, YEp24, YCp50等)などが挙げられ、ファージDNAとしてはλファージが挙げられる。さらに、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0023】
上記ベクターには複製開始点、選択マーカー、プロモーターを含み、必要に応じてエンハンサー、ターミネーター、リボソーム結合部位、ポリアデニル化シグナル等を含んでいてもよい。
【0024】
複製開始点としては、大腸菌用ベクターには、例えばColE1、R因子、F因子由来のものが、酵母用ベクターには、例えば2μm DNA、ARS1由来のものが、動物細胞用ベクターには、SV40、アデノウイルス由来のものが用いられる。
【0025】
プロモーターとしては、大腸菌用ベクターには、trpプロモーター、lacプロモーター、PLプロモーター、PRプロモーター等が、酵母用ベクターには、gal1プロモーター、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーター、AOX1プロモーター等が、動物細胞用ベクターには、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMVプロモーター等が用いられる。
【0026】
選択マーカーとしては、大腸菌用ベクターには、カナマイシン耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子等が、酵母用ベクターには、Leu2、Trp1、Ura3遺伝子等が、動物細胞用ベクターには、ネオマイシン耐性遺伝子、チミジンキナーゼ遺伝子、ジヒドロ葉酸還元酵素遺伝子等が用いられる。
【0027】
ベクターは商業的に入手可能なものを使用することができるが、そのようなベクターには、宿主細胞が大腸菌である場合は、例えばpETベクター(Novagen社製) 、pTrxFUSベクター(Invitrogen社製) 、pCYBベクター(NEW ENGLAMD Bio Labs社製) 等が、宿主細胞が酵母である場合は、例えばpESP-1発現ベクター(STRATAGENE社製) 、pAUR123ベクター(宝酒造社製)、pPICベクター(Invitrogen社製) 等が、また宿主細胞が動物細胞である場合は、例えばpMAM-neo発現ベクター (CLONTECH社製) 、pCDNA3.1ベクター(Invitrogen社製) 、pBK-CMVベクター (STRATAGENE社製) 等が、宿主細胞が昆虫細胞である場合は、例えばpBacPAKベクター (CLONTECH社製) 、pAcUW31ベクター(CLONTECH社製) 、pAcP(+)IE1ベクター(Novagen社製) 等がそれぞれ挙げられる。
【0028】
また、本発明の融合タンパク質発現ベクターには、発現される融合タンパク質の検出および精製を容易にするためにタグを組み込んでもよい。タグの例としては、例えば、グルタチオン-S-トランスフェラーゼ、マルトース結合プロテイン、ヒスチジン(His6)等が挙げられる。これらのタグに親和性のある担体(ヒスチジンであればニッケルをキレートした担体)を用いることにより、アフィニティー精製が可能となる。
【0029】
2.形質転換体
本発明の形質転換体は、上記の融合タンパク質発現ベクターを宿主中に導入することにより得ることができる。ここで、宿主としては、融合タンパク質遺伝子を発現できるものであれば特に限定されるものではない。例えば、大腸菌(Escherichia coli)等のエシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)等のバチルス属、シュードモナス・プチダ(Pseudomonas putida)等のシュードモナス属に属する細菌;サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の酵母;サル細胞COS-7、Vero、チャイニーズハムスター卵巣細胞(CHO細胞)、マウスL細胞、ヒトGH3、ヒトFL細胞等の動物細胞;あるいはSf9、Sf21等の昆虫細胞が挙げられる。
【0030】
宿主細胞への発現ベクターの導入は、宿主の種類に応じて公知の方法で行うことができ、たとえば、リン酸カルシウム法、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法、リポフェクション法等が挙げられる。また、上記の各宿主細胞への遺伝子導入は、組換えベクターによらない方法、例えばパーティクルガン法なども用いることができる。
【0031】
3.目的タンパク質の製造
目的タンパク質は、前記形質転換体を培養して好塩性細菌由来のタンパク質との融合タンパク質として発現させ、その培養物から融合タンパク質を採取した後、プロテアーゼで消化することにより製造することができる。ここで、「培養物」とは、培養上清のほか、培養細胞若しくは培養菌体又は細胞若しくは菌体の破砕物のいずれをも意味するものである。
【0032】
本発明の形質転換体を培地に培養する方法は、その宿主細胞の培養に用いられる通常の方法に従って行うことができる。
【0033】
大腸菌や酵母菌等の微生物を宿主として得られた形質転換体を培養する培地としては、微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれを用いてもよい。炭素源としては、該生物が資化し得るものであればよく、グルコース、フラクトース、スクロース、デンプン等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコール類が用いられる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸若しくは有機酸のアンモニウム塩又はその他の含窒素化合物のほか、ペプトン、肉エキス、コーンスチープリカー等が用いられる。無機塩類としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
【0034】
培養は、通常、振盪培養又は通気攪拌培養などの好気的条件下、30〜37℃で6〜62時間程度行う。培養期間中、pHは7.0〜7.5に保持する。pHの調整は、無機又は有機酸、アルカリ溶液等を用いて行う。培養中は必要に応じてアンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
【0035】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養する場合は、必要に応じてインデューサーを培地に添加してもよい。例えば、Lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル-β-D-チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドール酢酸(IAA)等を培地に添加してもよい。
【0036】
動物細胞を宿主として得られた形質転換体を培養する培地としては、一般に使用されているRPMI1640培地、DMEM培地又はこれらの培地に牛胎児血清等を添加した培地等が用いられる。培養は、通常、5%CO2存在下、37℃で1〜30日行う。培養中は必要に応じてカナマイシン、ペニシリン等の抗生物質を培地に添加してもよい。
【0037】
培養後、融合タンパク質が菌体内又は細胞内に生産される場合には、菌体又は細胞を破砕することにより該融合タンパク質を抽出する。また、融合タンパク質が菌体外又は細胞外に生産される場合には、培養液をそのまま使用するか、遠心分離等により菌体又は細胞を除去する。その後、タンパク質の単離精製に用いられる一般的な生化学的方法、例えば硫酸アンモニウム沈殿、ゲルろ過、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー等を単独で又は適宜組み合わせて用いることにより、前記培養物中から融合タンパク質を単離精製することができる。
【0038】
本発明において好ましい精製方法は、例えば、融合タンパク質を前記のタグ(ヒスチジン等)によって担体に結合させて回収し、その後、担体に結合した融合タンパク質を、プロテアーゼで消化することによって、目的タンパク質のみを担体から遊離させる方法である。あるいは、イミダゾールで溶出することにより融合タンパク質のまま担体から遊離させることも可能である。
【実施例】
【0039】
以下、実施例により本発明をさらに具体的に説明する。但し、本発明はこれら実施例に限定されるものではない。
【0040】
(実施例1) 融合タンパク質発現ベクター(pBF-Th及びpBF)の構築
目的タンパク質をChromohalobacter sp.由来β−ラクタマーゼ(β-lactamase:BLA)との融合タンパク質として発現させるためのベクターを以下のようにして構築した。
【0041】
まず、クローン化したbla遺伝子(配列番号1)をフォワードプライマー:5'-GGGCATATGCAAGACGACGCGTCGGAC-3'(配列番号5)とリバースプライマー:5'-GGGGATCCTTACGGCACGTCGATCGC-3'(配列番号6)を用いて増幅した。増幅断片をpET15bプラスミド(Novagen)のNdeI/end-filled BamHI部位にライゲートし、N-末端にHis-tagを付加したpET-blaを構築した(Tokunaga et al., FEBS Letters 558 (2004) 7-12)。
【0042】
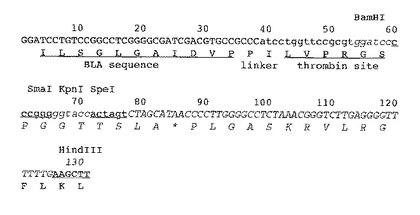
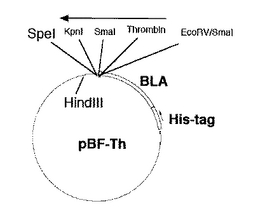
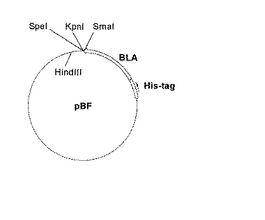
このpET-blaにコードされたBLAタンパク質のC-末端付近にあるBamHI部位とベクターのHindIII部位の間に、目的遺伝子を挿入するためのマルチクローニングサイトとトロンビン切断部位を含む113塩基からなるDNA断片(図1、配列番号7)を挿入し、pBF-Thベクター(図2)を構築した。またすでにトロンビン切断部位を持った目的遺伝子発現用に、トロンビン切断部位を持たず、マルチクローニングサイトのみを持ったpBFベクター(図3)も構築した。
【0043】
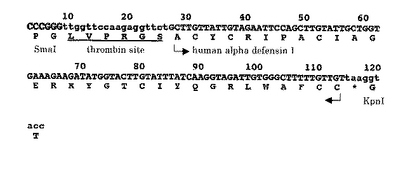
(実施例2) BLA-ヒトαデフェンシン1(BLA-hαD1)融合タンパク質の発現
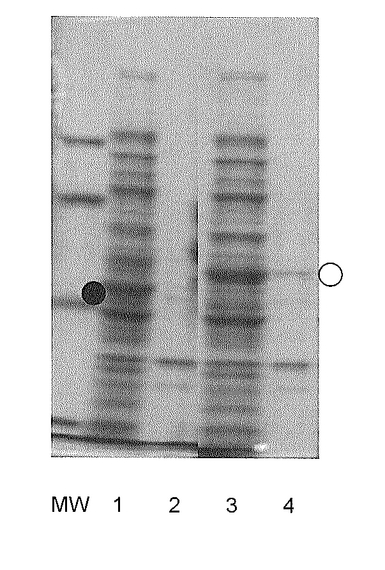
ヒトαデフェンシン1(hαD1)遺伝子をベースに、酵母において出現頻度の高いコドンを用いて合成した配列番号8に示す塩基配列を有する遺伝子の5'側にSmaI-トロンビン切断部位、3'側にKpnI部位を付加したDNA断片(図4)を、フォワードプライマー:5'- aaaCCCGGGttggttccaagaggtt-3’(配列番号9)とリバースプライマー:5'- GGGggtaccttaACAACAAAAAGCC-3’(配列番号10)とを用いて増幅し、実施例1で構築したpBFベクターのSmaI/KpnI部位に挿入して、pBLA-hαD1を得た。この pBLA-hαD1又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、菌体を集め、50mM Tris-HCl buffer(pH8.0)に懸濁し、超音波処理にて菌体を破砕し、細胞粗抽出液を得た。該抽出液を10000×gにて約15分間遠心し、可溶性(上清)画分と不溶性(沈殿)画分に分け、各画分をSDS−PAGEに供した。図5に可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hαD1導入大腸菌の可溶性画分、レーン4:pBLA-hαD1導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hαD1)。
【0044】
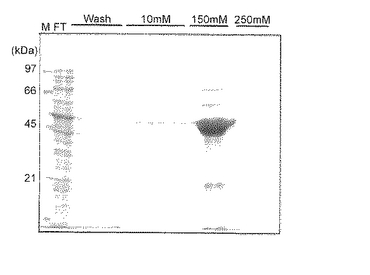
BLA-hαD1融合タンパク質の発現は可溶性画分に認められたので、この可溶性画分をNi-カラムにて精製した。Ni-カラム溶出画分のSDS-PAGE分析結果を図6に示す。
【0045】
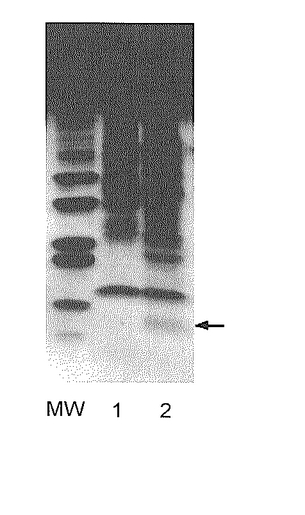
次に、精製したBLA-hαD1融合タンパク質、およびそのトロンビンで消化物に対してTricin-SDS-PAGEを行った。図7に、その結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hαD1融合タンパク質、レーン2:トロンビン消化BLA-hαD1融合タンパク質。矢印:分離したhαD1)。
【0046】
図7において検出された矢印のバンドが目的のBLA-hαD1であることを確かめるために、Problott膜に転写し、N-末端アミノ酸配列を調べたところ、GSAXYXRIPAX(X決定できなかった残基)となった。Cysは通常の分析では解析できないのでXはCys残基と予想され、解析した配列はDNA配列からの予想配列であるGSACYCRIPACと一致し、目的のhαD1が生成していることが確かめられた。
【0047】
また、TOF MS分析によると、この質量は3589Daとなり、予想質量3592Daと誤差の範囲で一致し、hαD1が正しく発現されていることが証明された。
【0048】
(実施例3) BLA-ヒトインターロイキン1α(BLA-hIL1α)融合タンパク質の発現
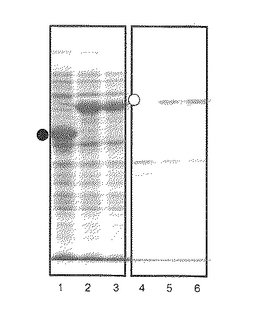
配列番号11に示す塩基配列を有するヒトインターロイキン1α(hIL1α)遺伝子の5’末端にSmaI、3’末端にSpeIの制限酵素部位を付加したDNA断片をフォワードプライマー:5'- ggCCCgggTCATCACCTTTTAgCTT-3’(配列番号12)とリバースプライマー:5'-: gggACTAgTCTACgCCTggTTTTCCA-3’(配列番号13)とを用いてPCRで増幅して、実施例1で構築した発現ベクターpBF-ThのSmaI/SpeI部位に挿入し、pBLA-hIL1αを得た。このpBLA-hIL1α又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、実施例2と同様にして菌体から可溶性画分と不溶性画分を得、各画分をSDS−PAGEに供した。図8にその結果を示す(レーン1:pET-bla導入大腸菌の可溶性画分、レーン2、3:pBLA-hIL1α導入大腸菌の可溶性画分、レーン4:pET-bla導入大腸菌の不溶性画分、レーン5、6:pBLA-hIL1α導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hIL1α)。
【0049】
BLA-hIL1α融合タンパク質の発現は可溶性画分に認められた。一方、hIL1αの大腸菌における直接発現では、hIL1αはインクルージョンボディ(IBs)を形成して不溶性画分に発現した。
【0050】
(実施例4) BLA-ヒトセリンラセマーゼ(BLA-hSR)融合タンパク質の発現
配列番号14に示す塩基配列を有するヒトセリンラセマーゼ (hSR)遺伝子の5'側にSmaI-リンカー-トロンビン切断部位-リンカー、3’側にSpeI部位を付加したDNA断片(図9)を、フォワードプライマー:5'- AAAACCCGGGAGCAGCGGCCTGGT-3’(配列番号15)とリバースプライマー:5'- CCCACTAGTTTAAACAGAAACAGACTGA-3’(配列番号16)とを用いてPCRで増幅して、実施例1で構築した発現ベクターpBFのSmaI/SpeI部位に挿入し、pBLA-hSRを得た。このpBLA-hSR又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、実施例2と同様にして菌体から可溶性画分と不溶性画分を得、各画分をSDS−PAGEに供した。図10にその結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hSR導入大腸菌の可溶性画分、レーン4:pBLA-hSR導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hSR)。
【0051】
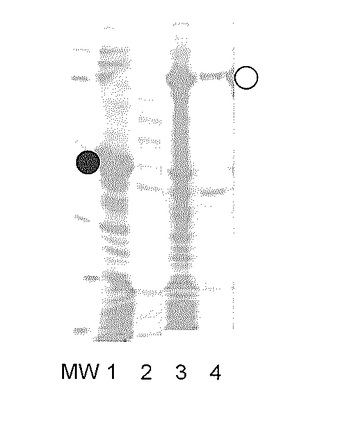
BLA-hSR融合タンパク質の発現が可溶性画分に認められたので、この可溶性画分をNi-カラムにて精製した。精製したBLA-hSR融合タンパク質、およびそのトロンビンで消化物に対してTricin-SDS-PAGEを行った。図11に、その結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hSR融合タンパク質、レーン2:トロンビン消化BLA-hSR融合タンパク質。●印:BLA、○印:分離したhSR)。
【図面の簡単な説明】
【0052】
【図1】目的遺伝子を挿入するマルチクローニングサイトとトロンビン切断部位を含むDNA断片の配列を示す。
【図2】融合タンパク発現ベクター:pBF-Thの構造(制限酵素地図)を示す。
【図3】融合タンパク発現ベクター:pBFの構造(制限酵素地図)を示す。
【図4】「SmaI-トロンビン切断部位-ヒトαデフェンシン1(hαD1)-KpnI」をコードするDNA断片の構造を示す。
【図5】コントロールベクター、融合タンパク質発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hαD1導入大腸菌の可溶性画分、レーン4:pBLA-hαD1導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hαD1)。
【図6】Ni-カラム溶出画分のSDS-PAGE分析結果を示す。
【図7】精製したBLA-hαD1融合タンパク質、およびそのトロンビン消化物のTricin-SDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hαD1融合タンパク質、レーン2:トロンビン消化BLA-hαD1融合タンパク質。矢印:分離したhαD1)。
【図8】コントロールベクター、融合タンパク発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(レーン1:pET-bla導入大腸菌の可溶性画分、レーン2、3:pBLA-hIL1α導入大腸菌の可溶性画分、レーン4:pET-bla導入大腸菌の不溶性画分、レーン5、6:pBLA-hIL1α導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hIL1α)。
【図9】「SmaI-リンカー-トロンビン切断部位リンカー-ヒトセリンラセマーゼ(hSR)-SpeI」をコードするDNA断片」の構造を示す。
【図10】コントロールベクター、融合タンパク発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hSR導入大腸菌の可溶性画分、レーン4:pBLA-hSR導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hSR)。
【図11】BLA-hSR融合タンパク質、およびそのトロンビン消化物のTricin-SDS-PAGE分析結果を示す (MW:分子量マーカー、レーン1:トロンビン未消化BLA-hSR融合タンパク質、レーン2:トロンビン消化BLA-hSR融合タンパク質。●印:BLA、○印:分離したhSR)。
【技術分野】
【0001】
本発明は、融合タンパク質発現ベクター、詳しくは、目的とする難溶性タンパク質を可溶化状態で発現させるための融合タンパク質発現ベクター、及び該融合タンパク質発現ベクターを用いるタンパク質の製造方法に関する。
【背景技術】
【0002】
現在、ヒトをはじめとする数多くの生物のゲノム塩基配列が解読され、多くの遺伝子情報がデータベースにて公開されている。また、これらの遺伝子情報を元に発現させたタンパク質の網羅的な機能解析も急速に進みつつある。タンパク質の発現は、それをコードする遺伝子をベクターに組み込み、細菌、酵母、昆虫細胞等の宿主に導入し、培養することにより行われる。しかしながら、遺伝子産物であるタンパク質によっては、その高次構造形成がうまくいかず、お互いに変性会合し、不溶性になってしまうパターン、あるいは、折りたたみ(フォールディング)異常によって、立体構造の異なった異常型タンパク質しか得られないパターンがある。このようなタンパク質は宿主内で封入体と呼ばれる凝集体として発現したり、宿主細胞のプロテアーゼにより分解されたりすることが知られている。
【0003】
この問題を克服するためのひとつの手法として、融合タンパク質発現法がある。この方法は、目的の難溶性タンパク質を特定の容易に発現するタンパク質に結合させ、融合タンパク質として可溶化状態で発現させ、その後、目的の難溶性タンパク質のみをプロテアーゼ処理によって切り離し取得する方法である。ところが、この方法は、比較的軽度の難溶性タンパク質については一定の成功をおさめてはいるものの、これまで用いられている融合タンパク質はいずれも可溶性に限界があり、高度な難溶性タンパク質については、依然として正しい生理活性を持つタンパク質として得られていないのが現状である。
【0004】
一方、ヒトをはじめ通常生物が生育できないような高濃度塩の環境下で生育可能な好塩性細菌由来のタンパク質は、極めて高い可溶性を有し、一度変性しても直ちに可逆的に本来の構造が回復し、活性状態に戻るという性質を持っていることが知られている。本発明者らが発見した中度好塩菌由来タンパク質であるβ−ラクタマーゼ (非特許文献1)や、高度好塩菌由来タンパク質であるヌクレオシド二リン酸キナーゼ (非特許文献2、3)もまた上記のような特性を有する。例えば、β−ラクタマーゼは、5分間煮沸しても常温に戻すと瞬時に80%活性を回復し、また、ヌクレオシド二リン酸キナーゼは、95℃で熱処理しても100%活性を回復する。これらの好塩性酵素タンパク質の構造を調べると、活性中心等の触媒機能に重要な部分は通常の酵素タンパク質の構造をよく保存しており、それ以外の部分、特にタンパク質表面に高頻度で酸性アミノ酸残基が局在しており、この酸性アミノ酸に起因する大量のマイナス荷電がその高い可溶性の維持を可能にしていることが示唆される。
【0005】
【非特許文献1】Tokunaga, H., et al., Highly efficient renaturation of β-lactamase isolated from mederately palophilic bacteria, FEBS Letters 558, 7-12, 2004
【非特許文献2】Ishibashi, M., et al., et al., Facilitated folding and subunit assembly in E.coli and in vitro of nucleoside diphosphate kinase from halophilic archaeon conferred by amino-terminal extension containing Hexa-His-tag, FEBS Letters 570, 87-92, 2004
【非特許文献3】Ishibashi, M., et al., NaCl-activated nucleoside diphosphate kinase from extremetly halophilic archaeon, H.salinarum, maitains native confirmation without salt, FEBS Letters 493, 134-138, 2001
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明の課題は、難溶性タンパク質を効率的に生産できる手段を提供することにある。
【課題を解決するための手段】
【0007】
本発明者らは、上記課題を解決するため鋭意研究を重ねた結果、好塩性細菌由来のタンパク質を目的とする難溶性タンパク質の融合パートナーとして用いることにより、宿主細胞内で可溶性の融合タンパク質の形態で発現することに成功し、本発明を完成させるに至った。
【0008】
すなわち、本発明は以下の発明を包含する。
(1) 好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【0009】
(2) 好塩性細菌由来のタンパク質が、クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼである、(1)に記載の融合タンパク質発現ベクター。
(3) 好塩性細菌由来のタンパク質が、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼである、(1)に記載の融合タンパク質発現ベクター。
【0010】
(4) (1)〜(3)のいずれかに記載の融合タンパク質発現ベクターに、目的タンパク質をコードする遺伝子を組み込んだことを特徴とする、融合タンパク質発現ベクター。
(5) 目的タンパク質が、難溶性タンパク質である、(4)に記載の融合タンパク質発現ベクター。
【0011】
(6) (4)に記載の融合タンパク質発現ベクターにより形質転換された形質転換体。
(7) (6)に記載の形質転換体を培地に培養し、得られる培養物から好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質を採取し、プロテアーゼで消化することを含む、タンパク質の製造方法。
【発明の効果】
【0012】
本発明によれば、これまで正しい構造と機能をもって発現・生合成させることが不可能であった難溶性タンパク質を効率よく製造できる融合タンパク質発現ベクターが提供される。従って、本発明の融合タンパク質発現ベクターを利用すれば、医薬品として有用な種々の生理活性を有する機能性タンパク質を、活性を維持したまま大量に製造することができる。
【発明を実施するための最良の形態】
【0013】
1.融合タンパク質発現ベクター
本発明の融合タンパク質発現ベクターは、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させるものであり、好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流に目的タンパク質をコードする遺伝子を挿入するための制限酵素部位(マルチクローニングサイトという)、および発現した融合タンパク質から目的タンパク質を切り出すために必要なプロテアーゼ切断部位とを含む。
【0014】
上記の好塩性細菌由来のタンパク質としては、0.2M以上のNaCl濃度下において生育・増殖可能な細菌由来、例えば、中度好塩性細菌(至適NaCl濃度:0.5-2.5M)または高度好塩性細菌(至適NaCl濃度:2.5-5.2M)由来のタンパク質が含まれる。例えば、中度好塩性細菌であるクロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼ(β-lactamase:BLA)、高度好塩性細菌であるハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼ(nucleoside diphosphate kinase:NDK)が好適に用いることができる。クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼのアミノ酸配列を配列番号2に、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼのアミノ酸配列は配列番号4に示す。
【0015】
好塩性細菌由来のタンパク質をコードする遺伝子として、具体的には、配列番号1又は3に示す塩基配列からなるDNAが用いられる。また、上記遺伝子には、所望の酵素活性を有する限り変異遺伝子も含まれ、例えば、配列番号1又は3に示す塩基配列からなるDNAに相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAが含まれる。
【0016】
上記の「配列番号1又は3に示す塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズできるDNA」としては、配列番号1又は3に示す塩基配列と約70%以上、好ましくは約80%以上、より好ましくは約90%以上、最も好ましくは約95%以上の相同性を有する塩基配列からなるDNA等が挙げられる。ここで、
ストリンジェントな条件とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいい、例えば、ナトリウム濃度が、10mM〜300mM、好ましくは20〜100mMであり、温度が25℃〜70℃、好ましくは42℃〜55℃での条件をいう。
【0017】
遺伝子に変異を導入するには、Kunkel法又は Gapped duplex法等の公知手法又はこれに準ずる方法により行うことができ、例えば部位特異的突然変異誘発法を利用した変異導入用キット(例えばMutant-K(TAKARA社製)やMutant-G(TAKARA社製))などを用いて、あるいは、TAKARA社のLA PCR in vitro Mutagenesis シリーズキットを用いて変異が導入される。
【0018】
一方、目的タンパク質としては、通常の発現系ではインクルージョンボディ(inclusion body, 封入体)と呼ばれる不溶性で不活性な凝集体(変性タンパク質)としてしか回収することのできない難溶性タンパク質が好ましい。このような難溶性タンパク質としては、例えば、ヒトαデフェンシン1(hαD1)、ヒトインターロイキン1α(hIL1α)、ヒトセリンラセマーゼ(hSR)、膜埋没型タンパク質(porin)、プロテインキナーゼ等が挙げられる。
【0019】
プロテアーゼ切断部位は、上記好塩性細菌由来のタンパク質をコードする遺伝子と、上記目的タンパク質をコードする遺伝子を挿入することができる制限酵素部位(マルチクローニングサイト)との間に設ける。
【0020】
上記プロテアーゼ切断部位は、本融合タンパク質発現ベクターにより発現される好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質において、両タンパク質をつなぐペプチドリンカーとなる。このペプチドリンカー内にあるプロテアーゼ切断部位にプロテアーゼを作用させることによって、容易に融合タンパク質から目的タンパク質を切り出すことができる。
【0021】
上記プロテアーゼとしては特に限定されず、例えば、トロンビン、ファクターXa、エンテロキナーゼ等が挙げられる。上記ペプチドリンカーとなるべき塩基配列の長さは特に限定されないが、12〜18塩基程度であることが好ましく、翻訳されてグリシンやセリン等の中性アミノ酸となる塩基配列を多く含むことが好ましい。
【0022】
また、本発明において上記の好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質をコードする遺伝子を組み込むために用いるベクターとしては、宿主内で該融合タンパク質遺伝子を発現することが可能であり、宿主内で複製可能なものであれば特に限定されず、例えば、プラスミドDNA、ファージDNA等のいずれであってもよい。プラスミド DNAとしては、大腸菌由来のプラスミド(例えばpBR322, pUC118等)、枯草菌由来のプラスミド(例えばpUB110, pTP5等)、酵母由来のプラスミド(例えばYEp13, YEp24, YCp50等)などが挙げられ、ファージDNAとしてはλファージが挙げられる。さらに、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0023】
上記ベクターには複製開始点、選択マーカー、プロモーターを含み、必要に応じてエンハンサー、ターミネーター、リボソーム結合部位、ポリアデニル化シグナル等を含んでいてもよい。
【0024】
複製開始点としては、大腸菌用ベクターには、例えばColE1、R因子、F因子由来のものが、酵母用ベクターには、例えば2μm DNA、ARS1由来のものが、動物細胞用ベクターには、SV40、アデノウイルス由来のものが用いられる。
【0025】
プロモーターとしては、大腸菌用ベクターには、trpプロモーター、lacプロモーター、PLプロモーター、PRプロモーター等が、酵母用ベクターには、gal1プロモーター、PHO5プロモーター、PGKプロモーター、GAPプロモーター、ADHプロモーター、AOX1プロモーター等が、動物細胞用ベクターには、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMVプロモーター等が用いられる。
【0026】
選択マーカーとしては、大腸菌用ベクターには、カナマイシン耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子等が、酵母用ベクターには、Leu2、Trp1、Ura3遺伝子等が、動物細胞用ベクターには、ネオマイシン耐性遺伝子、チミジンキナーゼ遺伝子、ジヒドロ葉酸還元酵素遺伝子等が用いられる。
【0027】
ベクターは商業的に入手可能なものを使用することができるが、そのようなベクターには、宿主細胞が大腸菌である場合は、例えばpETベクター(Novagen社製) 、pTrxFUSベクター(Invitrogen社製) 、pCYBベクター(NEW ENGLAMD Bio Labs社製) 等が、宿主細胞が酵母である場合は、例えばpESP-1発現ベクター(STRATAGENE社製) 、pAUR123ベクター(宝酒造社製)、pPICベクター(Invitrogen社製) 等が、また宿主細胞が動物細胞である場合は、例えばpMAM-neo発現ベクター (CLONTECH社製) 、pCDNA3.1ベクター(Invitrogen社製) 、pBK-CMVベクター (STRATAGENE社製) 等が、宿主細胞が昆虫細胞である場合は、例えばpBacPAKベクター (CLONTECH社製) 、pAcUW31ベクター(CLONTECH社製) 、pAcP(+)IE1ベクター(Novagen社製) 等がそれぞれ挙げられる。
【0028】
また、本発明の融合タンパク質発現ベクターには、発現される融合タンパク質の検出および精製を容易にするためにタグを組み込んでもよい。タグの例としては、例えば、グルタチオン-S-トランスフェラーゼ、マルトース結合プロテイン、ヒスチジン(His6)等が挙げられる。これらのタグに親和性のある担体(ヒスチジンであればニッケルをキレートした担体)を用いることにより、アフィニティー精製が可能となる。
【0029】
2.形質転換体
本発明の形質転換体は、上記の融合タンパク質発現ベクターを宿主中に導入することにより得ることができる。ここで、宿主としては、融合タンパク質遺伝子を発現できるものであれば特に限定されるものではない。例えば、大腸菌(Escherichia coli)等のエシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)等のバチルス属、シュードモナス・プチダ(Pseudomonas putida)等のシュードモナス属に属する細菌;サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の酵母;サル細胞COS-7、Vero、チャイニーズハムスター卵巣細胞(CHO細胞)、マウスL細胞、ヒトGH3、ヒトFL細胞等の動物細胞;あるいはSf9、Sf21等の昆虫細胞が挙げられる。
【0030】
宿主細胞への発現ベクターの導入は、宿主の種類に応じて公知の方法で行うことができ、たとえば、リン酸カルシウム法、エレクトロポレーション法、スフェロプラスト法、酢酸リチウム法、リポフェクション法等が挙げられる。また、上記の各宿主細胞への遺伝子導入は、組換えベクターによらない方法、例えばパーティクルガン法なども用いることができる。
【0031】
3.目的タンパク質の製造
目的タンパク質は、前記形質転換体を培養して好塩性細菌由来のタンパク質との融合タンパク質として発現させ、その培養物から融合タンパク質を採取した後、プロテアーゼで消化することにより製造することができる。ここで、「培養物」とは、培養上清のほか、培養細胞若しくは培養菌体又は細胞若しくは菌体の破砕物のいずれをも意味するものである。
【0032】
本発明の形質転換体を培地に培養する方法は、その宿主細胞の培養に用いられる通常の方法に従って行うことができる。
【0033】
大腸菌や酵母菌等の微生物を宿主として得られた形質転換体を培養する培地としては、微生物が資化し得る炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれを用いてもよい。炭素源としては、該生物が資化し得るものであればよく、グルコース、フラクトース、スクロース、デンプン等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコール類が用いられる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸若しくは有機酸のアンモニウム塩又はその他の含窒素化合物のほか、ペプトン、肉エキス、コーンスチープリカー等が用いられる。無機塩類としては、リン酸第一カリウム、リン酸第二カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウム等が用いられる。
【0034】
培養は、通常、振盪培養又は通気攪拌培養などの好気的条件下、30〜37℃で6〜62時間程度行う。培養期間中、pHは7.0〜7.5に保持する。pHの調整は、無機又は有機酸、アルカリ溶液等を用いて行う。培養中は必要に応じてアンピシリンやテトラサイクリン等の抗生物質を培地に添加してもよい。
【0035】
プロモーターとして誘導性のプロモーターを用いた発現ベクターで形質転換した微生物を培養する場合は、必要に応じてインデューサーを培地に添加してもよい。例えば、Lacプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはイソプロピル-β-D-チオガラクトピラノシド(IPTG)等を、trpプロモーターを用いた発現ベクターで形質転換した微生物を培養するときにはインドール酢酸(IAA)等を培地に添加してもよい。
【0036】
動物細胞を宿主として得られた形質転換体を培養する培地としては、一般に使用されているRPMI1640培地、DMEM培地又はこれらの培地に牛胎児血清等を添加した培地等が用いられる。培養は、通常、5%CO2存在下、37℃で1〜30日行う。培養中は必要に応じてカナマイシン、ペニシリン等の抗生物質を培地に添加してもよい。
【0037】
培養後、融合タンパク質が菌体内又は細胞内に生産される場合には、菌体又は細胞を破砕することにより該融合タンパク質を抽出する。また、融合タンパク質が菌体外又は細胞外に生産される場合には、培養液をそのまま使用するか、遠心分離等により菌体又は細胞を除去する。その後、タンパク質の単離精製に用いられる一般的な生化学的方法、例えば硫酸アンモニウム沈殿、ゲルろ過、イオン交換クロマトグラフィー、アフィニティークロマトグラフィー等を単独で又は適宜組み合わせて用いることにより、前記培養物中から融合タンパク質を単離精製することができる。
【0038】
本発明において好ましい精製方法は、例えば、融合タンパク質を前記のタグ(ヒスチジン等)によって担体に結合させて回収し、その後、担体に結合した融合タンパク質を、プロテアーゼで消化することによって、目的タンパク質のみを担体から遊離させる方法である。あるいは、イミダゾールで溶出することにより融合タンパク質のまま担体から遊離させることも可能である。
【実施例】
【0039】
以下、実施例により本発明をさらに具体的に説明する。但し、本発明はこれら実施例に限定されるものではない。
【0040】
(実施例1) 融合タンパク質発現ベクター(pBF-Th及びpBF)の構築
目的タンパク質をChromohalobacter sp.由来β−ラクタマーゼ(β-lactamase:BLA)との融合タンパク質として発現させるためのベクターを以下のようにして構築した。
【0041】
まず、クローン化したbla遺伝子(配列番号1)をフォワードプライマー:5'-GGGCATATGCAAGACGACGCGTCGGAC-3'(配列番号5)とリバースプライマー:5'-GGGGATCCTTACGGCACGTCGATCGC-3'(配列番号6)を用いて増幅した。増幅断片をpET15bプラスミド(Novagen)のNdeI/end-filled BamHI部位にライゲートし、N-末端にHis-tagを付加したpET-blaを構築した(Tokunaga et al., FEBS Letters 558 (2004) 7-12)。
【0042】
このpET-blaにコードされたBLAタンパク質のC-末端付近にあるBamHI部位とベクターのHindIII部位の間に、目的遺伝子を挿入するためのマルチクローニングサイトとトロンビン切断部位を含む113塩基からなるDNA断片(図1、配列番号7)を挿入し、pBF-Thベクター(図2)を構築した。またすでにトロンビン切断部位を持った目的遺伝子発現用に、トロンビン切断部位を持たず、マルチクローニングサイトのみを持ったpBFベクター(図3)も構築した。
【0043】
(実施例2) BLA-ヒトαデフェンシン1(BLA-hαD1)融合タンパク質の発現
ヒトαデフェンシン1(hαD1)遺伝子をベースに、酵母において出現頻度の高いコドンを用いて合成した配列番号8に示す塩基配列を有する遺伝子の5'側にSmaI-トロンビン切断部位、3'側にKpnI部位を付加したDNA断片(図4)を、フォワードプライマー:5'- aaaCCCGGGttggttccaagaggtt-3’(配列番号9)とリバースプライマー:5'- GGGggtaccttaACAACAAAAAGCC-3’(配列番号10)とを用いて増幅し、実施例1で構築したpBFベクターのSmaI/KpnI部位に挿入して、pBLA-hαD1を得た。この pBLA-hαD1又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、菌体を集め、50mM Tris-HCl buffer(pH8.0)に懸濁し、超音波処理にて菌体を破砕し、細胞粗抽出液を得た。該抽出液を10000×gにて約15分間遠心し、可溶性(上清)画分と不溶性(沈殿)画分に分け、各画分をSDS−PAGEに供した。図5に可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hαD1導入大腸菌の可溶性画分、レーン4:pBLA-hαD1導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hαD1)。
【0044】
BLA-hαD1融合タンパク質の発現は可溶性画分に認められたので、この可溶性画分をNi-カラムにて精製した。Ni-カラム溶出画分のSDS-PAGE分析結果を図6に示す。
【0045】
次に、精製したBLA-hαD1融合タンパク質、およびそのトロンビンで消化物に対してTricin-SDS-PAGEを行った。図7に、その結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hαD1融合タンパク質、レーン2:トロンビン消化BLA-hαD1融合タンパク質。矢印:分離したhαD1)。
【0046】
図7において検出された矢印のバンドが目的のBLA-hαD1であることを確かめるために、Problott膜に転写し、N-末端アミノ酸配列を調べたところ、GSAXYXRIPAX(X決定できなかった残基)となった。Cysは通常の分析では解析できないのでXはCys残基と予想され、解析した配列はDNA配列からの予想配列であるGSACYCRIPACと一致し、目的のhαD1が生成していることが確かめられた。
【0047】
また、TOF MS分析によると、この質量は3589Daとなり、予想質量3592Daと誤差の範囲で一致し、hαD1が正しく発現されていることが証明された。
【0048】
(実施例3) BLA-ヒトインターロイキン1α(BLA-hIL1α)融合タンパク質の発現
配列番号11に示す塩基配列を有するヒトインターロイキン1α(hIL1α)遺伝子の5’末端にSmaI、3’末端にSpeIの制限酵素部位を付加したDNA断片をフォワードプライマー:5'- ggCCCgggTCATCACCTTTTAgCTT-3’(配列番号12)とリバースプライマー:5'-: gggACTAgTCTACgCCTggTTTTCCA-3’(配列番号13)とを用いてPCRで増幅して、実施例1で構築した発現ベクターpBF-ThのSmaI/SpeI部位に挿入し、pBLA-hIL1αを得た。このpBLA-hIL1α又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、実施例2と同様にして菌体から可溶性画分と不溶性画分を得、各画分をSDS−PAGEに供した。図8にその結果を示す(レーン1:pET-bla導入大腸菌の可溶性画分、レーン2、3:pBLA-hIL1α導入大腸菌の可溶性画分、レーン4:pET-bla導入大腸菌の不溶性画分、レーン5、6:pBLA-hIL1α導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hIL1α)。
【0049】
BLA-hIL1α融合タンパク質の発現は可溶性画分に認められた。一方、hIL1αの大腸菌における直接発現では、hIL1αはインクルージョンボディ(IBs)を形成して不溶性画分に発現した。
【0050】
(実施例4) BLA-ヒトセリンラセマーゼ(BLA-hSR)融合タンパク質の発現
配列番号14に示す塩基配列を有するヒトセリンラセマーゼ (hSR)遺伝子の5'側にSmaI-リンカー-トロンビン切断部位-リンカー、3’側にSpeI部位を付加したDNA断片(図9)を、フォワードプライマー:5'- AAAACCCGGGAGCAGCGGCCTGGT-3’(配列番号15)とリバースプライマー:5'- CCCACTAGTTTAAACAGAAACAGACTGA-3’(配列番号16)とを用いてPCRで増幅して、実施例1で構築した発現ベクターpBFのSmaI/SpeI部位に挿入し、pBLA-hSRを得た。このpBLA-hSR又はpET-bla(コントロールベクター)をそれぞれ大腸菌BL21(DE3)株に導入し、培養した。培養後、実施例2と同様にして菌体から可溶性画分と不溶性画分を得、各画分をSDS−PAGEに供した。図10にその結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hSR導入大腸菌の可溶性画分、レーン4:pBLA-hSR導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hSR)。
【0051】
BLA-hSR融合タンパク質の発現が可溶性画分に認められたので、この可溶性画分をNi-カラムにて精製した。精製したBLA-hSR融合タンパク質、およびそのトロンビンで消化物に対してTricin-SDS-PAGEを行った。図11に、その結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hSR融合タンパク質、レーン2:トロンビン消化BLA-hSR融合タンパク質。●印:BLA、○印:分離したhSR)。
【図面の簡単な説明】
【0052】
【図1】目的遺伝子を挿入するマルチクローニングサイトとトロンビン切断部位を含むDNA断片の配列を示す。
【図2】融合タンパク発現ベクター:pBF-Thの構造(制限酵素地図)を示す。
【図3】融合タンパク発現ベクター:pBFの構造(制限酵素地図)を示す。
【図4】「SmaI-トロンビン切断部位-ヒトαデフェンシン1(hαD1)-KpnI」をコードするDNA断片の構造を示す。
【図5】コントロールベクター、融合タンパク質発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hαD1導入大腸菌の可溶性画分、レーン4:pBLA-hαD1導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hαD1)。
【図6】Ni-カラム溶出画分のSDS-PAGE分析結果を示す。
【図7】精製したBLA-hαD1融合タンパク質、およびそのトロンビン消化物のTricin-SDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:トロンビン未消化BLA-hαD1融合タンパク質、レーン2:トロンビン消化BLA-hαD1融合タンパク質。矢印:分離したhαD1)。
【図8】コントロールベクター、融合タンパク発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(レーン1:pET-bla導入大腸菌の可溶性画分、レーン2、3:pBLA-hIL1α導入大腸菌の可溶性画分、レーン4:pET-bla導入大腸菌の不溶性画分、レーン5、6:pBLA-hIL1α導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hIL1α)。
【図9】「SmaI-リンカー-トロンビン切断部位リンカー-ヒトセリンラセマーゼ(hSR)-SpeI」をコードするDNA断片」の構造を示す。
【図10】コントロールベクター、融合タンパク発現ベクターをそれぞれ導入した大腸菌の可溶性画分と不溶性画分のSDS-PAGE分析結果を示す(MW:分子量マーカー、レーン1:pET-bla導入大腸菌の可溶性画分、レーン2:pET-bla導入大腸菌の不溶性画分、レーン3:pBLA-hSR導入大腸菌の可溶性画分、レーン4:pBLA-hSR導入大腸菌の不溶性画分。●印:BLA、○印:BLA-hSR)。
【図11】BLA-hSR融合タンパク質、およびそのトロンビン消化物のTricin-SDS-PAGE分析結果を示す (MW:分子量マーカー、レーン1:トロンビン未消化BLA-hSR融合タンパク質、レーン2:トロンビン消化BLA-hSR融合タンパク質。●印:BLA、○印:分離したhSR)。
【特許請求の範囲】
【請求項1】
好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【請求項2】
好塩性細菌由来のタンパク質が、クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼである、請求項1に記載の融合タンパク質発現ベクター。
【請求項3】
好塩性細菌由来のタンパク質が、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼである、請求項1に記載の融合タンパク質発現ベクター。
【請求項4】
請求項1〜3のいずれかに記載の融合タンパク質発現ベクターに、目的タンパク質をコードする遺伝子を組み込んだことを特徴とする、融合タンパク質発現ベクター。
【請求項5】
目的タンパク質が、難溶性タンパク質である、請求項4に記載の融合タンパク質発現ベクター。
【請求項6】
請求項4に記載の融合タンパク質発現ベクターにより形質転換された形質転換体。
【請求項7】
請求項6に記載の形質転換体を培地に培養し、得られる培養物から好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質を採取し、プロテアーゼで消化することを含む、タンパク質の製造方法。
【請求項1】
好塩性細菌由来のタンパク質をコードする遺伝子と、該遺伝子の下流にプロテアーゼ切断部位と目的タンパク質をコードする遺伝子を挿入するための制限酵素部位とを含み、目的タンパク質を好塩性細菌由来のタンパク質との融合タンパク質として発現させることを特徴とする、融合タンパク質発現ベクター。
【請求項2】
好塩性細菌由来のタンパク質が、クロモハロバクター・エスピー(Chromohalobacter sp.)由来のβ−ラクタマーゼである、請求項1に記載の融合タンパク質発現ベクター。
【請求項3】
好塩性細菌由来のタンパク質が、ハロバクテリウム・サリナーラム(Halobacterium salinarum)由来のヌクレオシド二リン酸キナーゼである、請求項1に記載の融合タンパク質発現ベクター。
【請求項4】
請求項1〜3のいずれかに記載の融合タンパク質発現ベクターに、目的タンパク質をコードする遺伝子を組み込んだことを特徴とする、融合タンパク質発現ベクター。
【請求項5】
目的タンパク質が、難溶性タンパク質である、請求項4に記載の融合タンパク質発現ベクター。
【請求項6】
請求項4に記載の融合タンパク質発現ベクターにより形質転換された形質転換体。
【請求項7】
請求項6に記載の形質転換体を培地に培養し、得られる培養物から好塩性細菌由来のタンパク質と目的タンパク質との融合タンパク質を採取し、プロテアーゼで消化することを含む、タンパク質の製造方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】

【図10】
【図11】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2006−230251(P2006−230251A)
【公開日】平成18年9月7日(2006.9.7)
【国際特許分類】
【出願番号】特願2005−47809(P2005−47809)
【出願日】平成17年2月23日(2005.2.23)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
【公開日】平成18年9月7日(2006.9.7)
【国際特許分類】
【出願日】平成17年2月23日(2005.2.23)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
[ Back to top ]