
血栓形成または血液凝固疾患に対する薬剤の製造のためのLIMK−1、その類似体およびリガンドの使用
本発明は、血栓形成または血液凝固疾患の予防または治療用の薬剤の製造用のGPIIbへの結合および/またはGPIIb/IIIa下流シグナル伝達の活性化もしくは阻害用のLIMK−1、LIMK−1類似体またはLIMK−1リガンド、LIMK−1類似体またはLIMK−1リガンドをスクリーニングするスクリーニング方法、ならびに血栓形成または血液凝固疾患の治療用の薬剤を製造する方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、血栓形成または血液凝固疾患の予防または治療用の薬剤の製造用のGPIIbへの結合および/またはGPIIb/IIIa下流シグナル伝達の活性化もしくは阻害用のLIMK−1、LIMK−1類似体またはLIMK−1リガンド、LIMK−1類似体またはLIMK−1リガンドをスクリーニングするスクリーニング方法の方法、ならびに血栓形成または血液凝固疾患の治療用の薬剤を製造する方法に関する。
【背景技術】
【0002】
血小板は、最小の血液細胞であり、巨核球細胞質の断片にすぎないが、正常な止血に重要な役割を有し、血栓性疾患への重要な寄与因子である。血小板付着、活性化および凝集は、止血の開始のための欠くことができない反応であり、様々な冠動脈、脳および末梢血管疾患の病理に重要な役割を果たす(Bhattら、2003年、George、2000年、Moroiら、1998年、Raoら、2000年、Ruggeri、2000年)。
【0003】
血小板膜は、細胞外マトリックス成分への血小板付着に関与する高濃度のインテグリンおよび他の糖タンパク質を含む(Lopezら、1988年、Phillipsら、1988年、Shattil、1999年)。コラーゲン線維およびヴォン・ヴィレブランド因子(vWF)は、曝露された内皮下層への血小板の付着のための重要な部位を与え、血管損傷の部位にそれらを捕捉し、損傷した範囲にわたる細胞の単層の形成を可能にする(Ruggeri、2002年、Watson、1999年)。コラーゲン線維およびvWFは、Gタンパク質結合受容体(GPCRs)を介して作用するシグナルとともに、インテグリン糖タンパク質GPIIb/IIIa(□IIb□3としても知られている)の「裏返し(inside−out)」調節、高密度およびα顆粒からの分泌、トロンボキサンの生成および前凝固活性の発現につながる血小板の活性化も刺激する(Parise、1999年、Ruggeri、2002年、Shattil、1999年)。また、血小板は、活性化により、通常の円盤状から付着を促進する長い樹状延長部分を有する緻密な球体に変化する。これらのすべての事象が止血過程を支えている。膜タンパク質GPIIb(CD41)およびGPIIIa(CD61)は、フィブリノーゲン、ヴォン・ヴィレブランド因子(vWF)およびフィブロネクチンに結合する血小板の表面上の最も豊富なヘテロ二量体複合体を形成する。GPIIb/IIIaは、可溶性フィブリノーゲンに対する低い親和力を有する不活性構造の休止血小板の表面上に発現する。コラーゲン、ADPまたはトロンビンのような多数の重要なアゴニストによる血小板の活性化の後に、GPIIb/IIIaは、可溶性フィブリノーゲンに対するその結合親和力を増加させる構造の変化を受け、血小板凝集をもたらす(Parise、1999年、Ruggeri、2002年、Shattil、1999年)。GPIIb/IIIaによるシグナル伝達は、細胞質側末端のいくつかの領域に関係する。エンドネキシン、カルシウムおよびインテグリン結合タンパク質(CIB)、ShcおよびGrb2などの数種のシグナル伝達タンパク質がGPIIb/IIIaインテグリンの細胞質ドメインに結合する。さらに、αサブユニットの細胞質側末端における配列KVGFFKRがシグナル伝達に関与しており、この領域に対応する脂質修飾ペプチドが血小板を完全に活性化する(Stephensら、1998年)。しかし、血小板活性化からフィブリノーゲン受容体曝露までの正確な一連のシグナル伝達は、知られていない。
【0004】
アスピリンが血小板に対して、また心筋梗塞の一次予防に有効であることが実証されて以来、血栓性障害に対するアスピリンの予防的使用は著しく増加した。したがって、数十年間にわたって、抗血小板療法は、トロンボキサン経路およびアスピリンによるその阻害に重点が置かれている。
【0005】
その関連性を考えると、付着は抗血栓薬の開発の魅力的な標的であると予想される。しかし、血小板凝集を妨げるように設計された最初の一連の化合物であるαIIbβ3アンタゴニストは、経口長期治療薬として成功しなかった。これは、天然リガンドの場合と同様に、αIIbβ3アンタゴニスト(多くがフィブリノーゲンの受容体認識配列に基づく)が「細胞内への(outside−in)」シグナル伝達を実際に誘発することに起因すると想定される(Coxら、2000年)。したがって、血小板を活性化せずに、リガンドと受容体との相互作用を直接妨害することは不可能かもしれない。
【0006】
近年、いくつかの抗血小板療法が導入され、血栓塞栓性障害におけるアスピリンから、チクロピジンおよびクロピドロゲルにおよぶ抗血小板療法の有用性が実証されている(Konstantopoulosら、2001年、Mousaら、2002年、Weksler、2000年)。しかし、これらの各種療法はすべて血栓塞栓性障害の治療に付加的な利点を加えるものであるが、種々の薬剤を用いた併用療法の有効性が十分でないことが判明する多くの症例は依然として存在する。さらに、多くの場合にこれらの抗血小板療法による治療は、望ましくない副作用を伴う。したがって、血栓塞栓性障害の治療のための新規なアプローチが緊急に求められている。
【非特許文献1】Mizunoら、Oncogene、第9巻、1605〜1612頁、1994年
【非特許文献2】R.P.Haugland、Molucular Probes、Handbook of Fluorescent Probes and Research Chemicals、第5版、Molecular Probes,Inc.、Eugene、Oreg.、1992年
【非特許文献3】Mujumdarら、Bioconjuga Chem.、第4巻、105頁、1992年
【非特許文献4】Ponczら、J.Biol.Chem.、第262巻、8476〜8482頁、1987年
【非特許文献5】Schobel U.ら、(1999年)Bioconjugate Chem.、第10巻、1107〜1114頁
【特許文献1】米国特許第5744305号
【発明の開示】
【発明が解決しようとする課題】
【0007】
したがって、本発明の目的は、血栓形成または血液凝固疾患の予防および治療改善のための新規のアプローチならびに例えば、抗血栓療法に対する新規の標的を特定する方法を提供することであった。
【0008】
驚くべきことに、LIMK−1タンパク質がGPIIb/IIIaに結合することが今回発見された。LIMK−1は、72.6kDaのタンパク質(ヒト:647アミノ酸)であり、LIMモチーフ含有タンパク質キナーゼファミリーのメンバーである。その遺伝子は、7q11.23における第7染色体上に位置する。該タンパク質は、2つのN末端システインリッチLIM/二重ジンクフィンガーモチーフ、いくつかの推定上のカゼインキナーゼおよびMAPキナーゼ認識部位を含むプロリンセリンリッチ領域、PDZドメイン(PSD95/disc large/ZO−1)およびC末端セリン/トレオニンキナーゼドメインを含む。ジンクフィンガー様LIMドメインは、タンパク質−タンパク質相互作用を仲介すると推定されており、核および細胞骨格タンパク質内に存在すると記載された。LIMK−1のC末端キナーゼ断片は、LIMドメインに結合し、LIM断片は、LIMK−1のキナーゼコア断片のキナーゼ触媒活性を用量依存的に阻害することが示された。したがって、N末端LIMドメインは、C末端キナーゼドメインとの直接的相互作用によって、LIMK−1のキナーゼ活性を負に調節する(Nagataら、1999年)。LIMドメインキナーゼ1(LIMK−1)は、コフィリンのリン酸化によってアクチン細胞骨格の再組織化を調節する。活性なフィリンは、アクチンの解重合をもたらす。結果としてLIMK−1によるコフィリンのリン酸化は、コフィリンのアクチン解重合活性の失活につながる(Bierneら、2001年、Gohlaら、2002年、Yangら、1998年)。しかし、これまでに血小板におけるLIMK−1(またはLIMK−2)の潜在的役割に関する報告はなかった。LIMK−1は血小板インテグリンGPIIbに結合するので、LIMK−1は、血小板の活性化のために誘発され、また必要な血小板の形状の変化を誘発する。したがって、LIMK−1の阻害は、催血栓刺激による血小板の活性化の阻害につながる。したがって、LIMK−1の阻害は、血小板の活性化と凝集、およびしたがって血栓形成を妨げる。
【課題を解決するための手段】
【0009】
したがって、本発明の第一の態様は、特に、インテグリンGPIIb/IIIaのシグナル伝達下流を調節、好ましくは阻害するための、GPIIbへの結合のためのLIMK−1またはLIMK−1類似体のインビトロおよびインビボでの使用を対象とする。
【0010】
本発明によるLIMK−1は、天然のあらゆるLIMK−1タンパク質である。これは、LIMK−1タンパク質および種々の動物種、好ましくは脊椎動物、より好ましくは哺乳類のその変異型ならびにスプライス変異型を含む。好ましいLIMK−1タンパク質は、ヒトLIMK−1タンパク質である。ヒトLIMK−1タンパク質のアミノ酸配列は、Mizunoら、Oncogene、第9巻、1605〜1612頁、1994年に開示されている。
【0011】
本発明による「LIMK−1類似体」という用語は、GPIIbに結合するが、LIMK−1ではないLIMK−1由来のタンパク質を指す。より詳細には、LIMK−1類似体は、天然のLIMK−1配列(すなわち、野生型ポリペプチド)と1つまたはそれ以上のアミノ酸が異なっているアミノ酸配列を指す。そのような類似体は、1つまたはそれ以上のアミノ酸の置換、挿入または欠失の点で野生型ポリペプチドと異なっている。好ましいものは半保存的置換であり、より好ましいものは保存的アミノ酸置換であり、それにより、残基が同様な特性を有する他のものによって置換されている。一般的な置換は、脂肪族アミノ酸の間、脂肪族ヒドロキシル側鎖を有するアミノ酸の間、酸性残基を有するアミノ酸の間、アミド誘導体の間、塩基性残基を有するアミノ酸の間または芳香族残基を有するアミノ酸の間の置換である。一般的な半保存的および保存的置換は、以下のとおりである。
【0012】
【表1】

【0013】
A、F、H、I、L、M、P、V、WまたはYからCへの変化は、新たなシステインが遊離のチオールとして留まる場合、半保存的である。さらに、当業者は、立体的に必要な位置におけるグリシンは置換すべきでなく、Pはαらせんまたはβシート構造を有するタンパク質の部分に導入すべきでないことを認識するであろう。
【0014】
変異型ポリペプチドは、野生型と比べて、一次構造(アミノ酸配列)が異なっているが、二次もしくは三次構造または機能はさほど異なっていてもまたはいなくてもよい。いずれにせよ、類似体は、少なくとも75%、好ましくは少なくとも80%、より好ましくは少なくとも90%、さらにより好ましくは少なくとも95%、最も好ましくは少なくとも99%の野生型LIMK−1との同一性(相同)を示す。
【0015】
類似体は、GPIIbに結合するために十分なLIMK−1の部分であってもよい。この部分は、少なくとも30アミノ酸、好ましくは少なくとも100アミノ酸、より好ましくは少なくとも300アミノ酸、さらにより好ましくは少なくとも450アミノ酸、最も好ましくは少なくとも600アミノ酸を含む。類似体のこの部分は、上で詳述したように1つまたはそれ以上のアミノ酸の置換、挿入または欠失において野生型ポリペプチドの部分と異なっていてよい。1つの実施形態において、LIMK−1またはLIMK−1類似体は、他の分子、例えば、タンパク質および/またはマーカー(例えば、上で詳述したような)に融合されていてよい。
【0016】
「GPIIbへの結合」または「GPIIbに結合する」という用語は、GPIIbタンパク質への優勢な化合物(LIMK−1またはLIMK−1類似体)の特異的結合を指す。本発明によるGPIIbタンパク質への特異的結合は、限定なしに、10-4mol/lを超えない、好ましくは10-5mol/lを超えない、より好ましくは10-6mol/lを超えない解離定数KDでの結合を含む。解離定数KDは、例えば、実施例(例えば、図1を参照)に示すように免疫沈降を用いて、種々の濃度の供試化合物、例えば、LIMK−1またはLIMK−1類似体ならびに一定の濃度のGPIIbを用いることにより測定することができる。GPIIbに結合している供試化合物の濃度は、例えば、LIMK−1またはLIMK−1類似体に対する特異抗体を用いることにより測定される。KDは、次の式により求められる。
B[L]=[L]/([L]+KD)
ここで、[L]は化合物の濃度を表す。KDは供試化合物の解離定数であり、B[L]は供試化合物の特定の濃度における結合(%)である。
【0017】
検出のために、抗体は標識することができ、これは適宜、フルオロフォア、発色団、放射性標識、金属コロイド、酵素、または化学発光もしくは生物発光分子であってよい。適切なフルオロフォアおよび発色団は、R.P.Haugland、Molucular Probes、Handbook of Fluorescent Probes and Research Chemicals、第5版、Molecular Probes,Inc.、Eugene、Oreg.、1992年に開示されている。好ましいフルオロフォアの例としては、フルオレセイン、ローダミンおよびスルホインドシアニン色素Cy5などがある(Mujumdarら、Bioconjuga Chem.、第4巻、105頁、1992年)。好ましい放射性標識としては、3H、14C、32P、33P、35S、99mTcまたは125Iなどがある。好ましい酵素としては、西洋ワサビペルオキシダーゼ、アリカリホスファターゼ、グルコースオキシダーゼおよびウレアーゼなどがある。
【0018】
本発明によるGPIIbは、任意の天然に存在するPIIbタンパク質である。これは、種々の動物種、好ましくは脊椎動物、より好ましくは哺乳類のGPIIbタンパク質およびその変異型ならびにスプライス変異型を含む。好ましいGPIIbタンパク質は、ヒトGPIIbタンパク質であり、そのアミノ酸配列は、Ponczら、J.Biol.Chem.、第262巻、8476〜8482頁、1987年に開示されている。
【0019】
本発明の1つの好ましい実施形態において、LIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の活性化物質または阻害物質、好ましくはその阻害物質である。
【0020】
GPIIb/IIIa下流シグナル伝達の活性化物質は、検出可能なシグナルをもたらす各シグナル伝達経路を活性化する。阻害物質は、GPIIb/IIIa下流シグナル伝達を少なくとも部分的に遮断する。活性化および不活性化は、以下で定義する。活性化物質および不活性化物質は、下記のように特定される。
【0021】
本発明の他の好ましい実施形態において、LIMK−1またはLIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の活性化または阻害、より好ましくはその阻害のために用いる。
【0022】
上で詳述したように、GPIIb/IIIa下流シグナル伝達の活性化は、とりわけ通常の円盤状から長い樹状延長部分を有する小型の球体への血小板の外観における構造変化を特徴とする血小板の活性化につながる。本発明によるGPIIb/IIIa下流シグナル伝達の活性化は、LIMK−1またはアゴニスト性LIMK−1類似体で刺激したとき、血小板の少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%の活性化に対応する。したがって、GPIIb/IIIa下流シグナル伝達の阻害は、血小板の活性化の阻害につながる。本発明によるGPIIb/IIIa下流シグナル伝達の阻害は、場合によりGPIIb/IIIa活性化物質の存在下で、阻害性LIMK−1類似体で阻害したとき、血小板の少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%の阻害に対応する。
【0023】
本発明の他の対象は、GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のためのLIMK−1リガンドの使用である。
【0024】
LIMK−1リガンドは、LIMK−1に特異的に結合するあらゆる化合物分子である。本発明によるLIMK−1への特異的結合は、限定なしに、10-4mol/lを超えない、好ましくは10-5mol/lを超えない解離定数KDでの結合を含む。解離定数KDは、GPIIbを用いて詳述したように、またはLIMK−1タンパク質および適切な標識LIMK−1リガンドを用いることにより測定することができる。適切なマーカーは、上で詳述した。さらに、LIMK−1アゴニスト性またはアンタゴニスト性LIMK−1リガンドの結合は、それぞれLIMK−1のキナーゼ機能の活性化または不活性化により検出することができる。キナーゼ機能は、当業者に知られている方法により容易に検出することができる。それらのうちの一部は以下で詳述する。アンタゴニスト性LIMK−1リガンドの検出のためには、例えば、PAK(p21活性化キナーゼ)またはROCK(Rhoキナーゼ)でLIMK−1を活性化することが必要である場合がある。あるいは、LIMK−1リガンドの結合は、シグナル伝達経路のより下流のシグナル、例えば、血小板の活性化もしくは不活性化またはそれらの構造変化の誘導を測定することによって検出することができる。本発明によるLIMK−1への特異的結合は、限定なしに、アゴニスト性およびアンタゴニスト性LIMK−1リガンドについてそれぞれ、10-4mol/lを超えない、好ましくは10-5mol/lを超えないEC50値およびIC50値での結合を含む。EC50およびIC50値の測定および計算は、当業者に知られている。
【0025】
LIMK−1は、非重合有機化合物、脂質、炭水化物、ペプチド、好ましくは約10〜約300アミノ酸、特に10〜50アミノ酸を有するペプチドであってよい。特に好ましいものは、約200g/mol〜約1500g/mol、特に400g/mol〜1000g/molの好ましい分子量を有する実験室で合成されるかまたは天然に発見される小化合物分子、特に非重合有機化合物である。
【0026】
あるいは、本発明のLIMK−1リガンドは、粗製または精製形態の天然産物抽出物であってよい。抽出物は、ヘビ毒、葉または微生物発酵ブロスのような動物、植物、真菌または細菌源からの水および/またはアルコールおよび/または有機溶媒抽出および/またはカラムクロマトグラフィーおよび/または沈降などの標準的な方法により製造することができる。
【0027】
好ましい実施形態において、LIMK−1リガンドは、LIMK−1アゴニストまたはLIMK−1アンタゴニスト、より好ましくはLIMK−1アンタゴニストである。
【0028】
アゴニストは、LIMK−1に結合し、LIMK−1の変化、例えば、構造変化を誘発し、各シグナル伝達を活性化、例えば、検出可能なシグナルをもたらす、LIMK−1のキナーゼ機能を活性化する。アンタゴニストまたはブロッカーもLIMK−1に結合するが、一般的にシグナル伝達を誘導しない。アゴニストの存在下では、アンタゴニストは、アゴニストにより誘導されるシグナル伝達を用量依存的に阻害する。
【0029】
本発明の他の対象は、血栓形成または血液凝固疾患の予防または治療用の薬剤の製造のための本発明によるLIMK−1、LIMK−1類似体またはLIMK−1リガンドの使用である。血栓形成または血液凝固疾患は、血栓形成または血液凝固の変化を伴う疾患である。健常者と比較して、血栓形成または血液凝固(の素因)が増加または減少し得る。
【0030】
薬剤の生産のために、LIMK−1、LIMK−1類似体またはLIMK−1リガンドまたはその製薬上許容できる塩は、一般的に所望の特性を得るために混合された製薬上許容できる担体または補助物質のような成分の混合物からなる薬剤剤形中に混入しなければならない。
【0031】
製剤は、少なくとも1つの適切な製薬上許容できる担体または補助物質を含む。そのような物質の例は、脱塩水、等張食塩水、リンゲル液、緩衝液、有機または無機酸および塩基ならびにそれらの塩、塩化ナトリウム、炭酸水素ナトリウム、クエン酸ナトリウムまたはリン酸二カルシウム、プロピレングリコールなどのグリコール、オレイン酸エチルおよびラウリン酸エチルなどのエステル、グルコース、スクロースおよびラクトースなどの糖、トウモロコシデンプンおよびジャガイモデンプンなどのデンプン、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、ジメチルホルムアミドなどの可溶化剤および乳化剤、落花生油、綿実油、トウモロコシ油、大豆油、ひまし油などの油、オレイン酸エチル、ミリスチン酸イソプロピルなどの合成脂肪酸エステル、ゼラチン、デキストラン、セルロースおよびその誘導体などのポリマーアジュバント、アルブミン、有機溶媒、クエン酸塩および尿素などの錯化剤、プロテアーゼまたはヌクレアーゼ阻害剤、好ましくはアプロチニン、ε−アミノカプロン酸またはペプスタチンAなどの安定化剤、ベンジルアルコールなどの保存剤、亜硫酸ナトリウムなどの酸化防止剤、ワックスおよびEDTAなどの安定化剤である。着色剤、放出剤、コーティング剤、甘味料、着香および芳香剤、保存剤および抗酸化剤も組成物中に存在していてよい。生理緩衝液は、好ましくは、約6.0〜8.0のpH、特に約6.8〜7.8のpH、特に約7.4のpHおよび/または約200〜400ミリオスモル/リットル、好ましくは約290〜310ミリオスモル/リットルのオスモル濃度を有する。薬剤のpHは、一般的に適切な有機または無機緩衝液、例えば、好ましくはリン酸緩衝液、トリス緩衝液(トリス(ヒドロキシメチル)アミノメタン)、HEPES緩衝液([4−(2−ヒドロキシエチル)ピペラジノ]エタンスルホン酸)またはMOPS緩衝液(3−モルホリノ−1−プロパンスルホン酸)などを用いて調整する。各緩衝液の選択は、一般的に所望の緩衝液のモル濃度に依存する。例えば、注射および注入液剤には、リン酸緩衝液が適切である。薬剤ならびに適切な製剤上許容できる担体および補助物質を調合する方法は、当業者によく知られている。製剤上許容できる担体および補助物質は、とりわけ、広く用いられている剤形および特定された化合物に応じて選択する。
【0032】
経口、鼻内、直腸、非経口、膣、局所または膣投与用の薬剤組成物を製造することができる。非経口投与は、皮下、皮内、筋肉内、静脈内または腹腔内投与などである。
【0033】
薬剤は、カプセル剤、錠剤、丸薬、散剤および顆粒剤などの経口投与用の固形剤形、製薬上許容できる乳剤、マイクロエマルジョン剤、液剤、懸濁剤、シロップ剤およびエリキシル剤などの経口投与用の液体剤形、注射用製剤、例えば、滅菌注射用水性または油性懸濁剤、直腸または膣投与用組成物、好ましくは坐剤、ならびに軟膏剤、ペースト剤、クリーム剤、ローション剤、ゲル剤、散剤、液剤、噴霧剤、吸入剤またはパッチ剤などの局所または経皮投与用の剤形を含む様々な剤形として調合することができる。
【0034】
個々の患者における特定の治療的に有効な用量レベルは、特定された化合物の活性、剤形、患者の年齢、体重および性別、治療期間ならびに医療技術分野でよく知られている同様な因子を含む様々な因子に依存する。
【0035】
1回または分割量でヒトまたは他の哺乳類に投与する本発明の化合物の総1日量は、例えば、約0.01〜約50mg/kg体重またはより好ましくは約0.1〜約25mg/kg体重の量であってよい。1回量組成物は、そのような量または1日量を構成するその分量を含んでいてよい。一般的に、本発明による治療法は、1日当たり本発明の化合物の約10mg〜約1000mgを1回または複数回そのような治療を必要とする患者に投与することを含む。
【0036】
好ましい実施形態において、LIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の阻害物質である。他の好ましい実施形態において、LIMK−1リガンドは、LIMK−1アンタゴニストである。
【0037】
血栓形成または血液凝固疾患を有する患者のほとんどが血栓形成または血液凝固(の素因)の亢進を伴う疾患に罹患している。したがって、それらの患者は、血栓形成または血液凝固を抑制する薬剤を特に必要とする。したがって、阻害性LIMK−1類似体およびアンタゴニスト性LIMK−1リガンドのような血栓形成または血液凝固を抑制する薬物が、薬剤の製造のための使用に好ましい。
【0038】
他の好ましい実施形態において、血栓形成または血液凝固疾患は、血栓形成または血液凝固の亢進を伴う疾患である。
【0039】
血栓形成または血液凝固の亢進を伴う疾患は、当業者に知られており、例えば、アテローム血栓性疾患を含む。
【0040】
より好ましい実施形態において、疾患は、アテローム血栓性疾患であり、より好ましくは、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬(comedication to vascular interventional strategy)からなる群から選択される疾患である。
【0041】
さらに、本発明の他の実施形態は、
(a)GPIIbタンパク質および場合によりその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(b)試験化合物を供給する工程、および
(c)GPIIbタンパク質への試験化合物の結合を検出する工程
を含み、試験化合物がLIMK−1由来であるLIMK−1類似体をスクリーニングする方法である。
【0042】
一般的に、GPIIbおよび場合によりその下流シグナル伝達の少なくとも1つの要素を、例えば、検定システムに供給し、LIMK−1由来の試験化合物と直接または間接的に接触させる。「LIMK−1由来の試験化合物」という表現は、上記で定義したようにLIMK−1類似体を指す。次いで、LIMK−1タンパク質への試験化合物の結合を検出する。この場合、結合は、GPIIbと試験化合物との相互作用を測定するか、または下流シグナル伝達に対するその影響を測定することによって検出することができる。
【0043】
「GPIIb/GPIIIaまたはGPIIbの下流シグナル伝達の要素」という用語は、GPIIb/GPIIIaまたはGPIIbの下流のシグナル伝達の一部である各分子またはイオンを指す。これは、シグナル伝達カスケードにおけるいずれかの段階のいずれかの要素であってよい。好ましくは、要素それ自体が測定可能なシグナルであるか、またはそれが測定可能なシグナルを発生させる。要素は、例えば、二次メッセンジャーまたは酵素であってよい。シグナルは、例えば、物質の濃度の変化または構造変化であってよい。
【0044】
結合は、直接的に、すなわち、例えばシグナル伝達経路における下流シグナルであり得る構築複合体を検出、または間接的に、すなわち、複合体の構築の影響を検出することにより、検出することができる。その後、適切なリガンドを分析し、かつ/または分離することができる。GPIIbへのLIMK−1類似体の結合を直接測定する方法は、上に詳述した。
【0045】
本発明の他の実施形態において、本発明によるLIMK−1類似体をスクリーニングする方法は、工程(c)の代わりに次の工程を含む。
(c’)GPIIb/IIIaおよび/またはその下流シグナル伝達の活性化または阻害を検出する。
【0046】
LIMK−1の活性化または阻害を検出するための適切な機能検定法は、例えば、GPIIb/GPIIIaの下流シグナル伝達を含むものであってよい。活性化または阻害は、例えば、下流シグナル、例えば、血小板の活性化またはそれらの構造変化を解析することによって上記で詳述したように検出することができる。シグナルを阻害性LIMK−1類似体により阻害させ、GPIIbおよび/またはLIMK−1刺激物質の存在下でGPIIb/GPIIIaシグナル伝達の阻害を検出することが必要であろう。
【0047】
本発明の他の実施形態は、
(a)LIMK−1タンパク質を供給する工程、
(b)試験化合物を供給する工程、および
(c)LIMK−1タンパク質への試験化合物の結合を検出する工程
を含むLIMK−1リガンドをスクリーニングする方法である。
【0048】
一般的に、LIMK−1を例えば検定システムに供給し、試験化合物、特に生化学または化学試験化合物、例えば、化合物ライブラリーの形で直接または間接的に接触させる。次いで、LIMK−1タンパク質への試験化合物の結合を検出する。結合は、直接的に、すなわち、例えばシグナル伝達経路における下流シグナルであり得る構築複合体を検出、または間接的に、すなわち、複合体の構築の影響を検出することにより、検出することができる。LIMK−1へのLIMK−1リガンドの結合を直接測定する方法は、上記で詳述した。その後、適切なリガンドを分析し、かつ/または分離することができる。
【0049】
本発明の他の実施形態において、本発明によるLIMK−1リガンドをスクリーニングする方法は、工程(c)の代わりに次の工程を含む。
(c’)LIMK−1またはGPII/IIIa下流シグナル伝達の活性化または阻害、好ましくはその活性化を検出する工程。
【0050】
LIMK−1の活性化または不活性化を検出するための適切な機能検定は、例えば、LIMK−1のキナーゼ機能に関するものである。キナーゼ活性を検出する方法は、当業者に知られている(以下を参照)。一般的に、これらの方法は、リン酸基、例えば、32Pまたは33P標識リン酸基のような標識リン酸基の受容体への移動に関するものである。したがって、LIMK−1の活性化はリン酸の移動の増加を伴い、不活性化はリン酸の移動の減少を伴う。シグナルをアンタゴニスト性LIMK−1リガンドにより阻害させ、LIMK−1刺激物質の存在下でLIMK−1の不活性化を検出することが必要であろう。活性化または不活性化は、下流シグナル、例えば、血小板の活性化またはそれらの構造変化を測定することによっても検出することができる。
【0051】
本発明の他の対象は、
(a)LIMK−1タンパク質を供給する工程、
(b)GPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(c)試験化合物を供給する工程、および
(d)GPIIb/GPIIIa下流シグナル伝達に対する試験化合物の影響を検出する工程
を含むGPIIb/IIIaおよび/またはその下流シグナル伝達と相互作用する試験化合物をスクリーニングする方法である。
【0052】
一般的に、LIMK−1およびGPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を例えば検定システムに供給し、試験化合物、特に生化学または化学試験化合物、例えば、化合物ライブラリーの形で直接または間接的に接触させる。次いで、GPIIb/GPIIIaシグナル伝達への試験化合物の影響を上記のように検出する。その後、適切なリガンドを分析し、かつ/または分離することができる。
【0053】
1つの好ましい実施形態において、全細胞、例えば、血小板を本発明のスクリーニング方法に用い、それにより、GPIIb/GPIIIaおよびその下流シグナル伝達のさらなる要素を提供する。この場合、検出されるシグナルは、血小板の形状の変化であり得る。
【0054】
試験化合物の存在下でのLIMK−1またはその類似体のGPIIbへの結合が試験化合物の非存在下での結合と異なる場合、試験化合物はGPIIbおよび/またはその下流シグナル伝達と相互作用している。シグナル伝達は、増加または減少、好ましくは減少し得る。下流シグナル伝達の検出される差は、少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%であり、試験化合物の存在下および非存在下でのシグナルの比として計算される。
【0055】
本発明のスクリーニング方法の他の実施形態において、試験化合物を化合物ライブラリーの形で供給する。化合物ライブラリーは、複数の化合物を含み、化学的に合成された分子および天然産物を含む複数の源のいずれかから集められ、あるいはコンビナトリアルケミストリー技術により得られたものであった。それらは、高処理スクリーニングに特に適している。それらは、特定の構造の化合物または植物のような特定の生物の化合物から構成されていてよい。本発明の状況において、化合物ライブラリーは、好ましくは、タンパク質およびポリペプチドまたは小分子を含むライブラリーである。
【0056】
本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法の他の実施形態において、前記GPIIbタンパク質または前記LIMK−1タンパク質への試験化合物の結合あるいはGPIIbおよび/またはその下流シグナル伝達に対するその影響を不均一または均一検定法により検出する。本明細書で用いているように、不均一検定法は、1つまたはそれ以上の洗浄工程を含む検定法であるが、均一検定法では、そのような洗浄工程は必要ではない。試薬および化合物を混合し、測定するのみである。
【0057】
好ましい実施形態において、不均一検定法は、ELISA(酵素結合免疫吸着検定法)、DELFIA(解離増強ランタニド蛍光免疫検定法)、SPA(シンチレーション近接検定法)またはフラッシュプレート検定法である。
【0058】
ELISA(酵素結合免疫吸着検定法)は、種々の会社によって供給されている。該検定法は、LIMK−1のようなキナーゼによりリン酸化することができるランダムペプチドを用いる。キナーゼを含む試料を通常、例えばATPおよび必須の陽イオンを含む反応緩衝液で希釈し、次いで、プレートウエルに加える。単に混合物を除去して、反応を停止させる。その後、プレートを洗浄する。例えば、ビオチン化基質をキナーゼに加えて、反応を開始させる。反応後、特異的抗体を加える。通常、試料をあらかじめブロックしたプロテインGプレートに移し、洗浄した後、例えば、ストレプトアビジン−HRPを加える。その後、非結合ストレプトアビジン−HRP(西洋ワサビペルオキシダーゼ)を除去し、ペルオキシダーゼ基質を加えて、ペルオキシダーゼ呈色反応を開始させ、適切なデンシトメーターで光学濃度を測定する。
【0059】
DELFIA(解離増強ランタニド蛍光免疫検定法)を用いる検定法は、固相検定法である。通常、抗体をユウロピウムまたは他のランタニドで標識し、非結合ユウロピウム標識抗体を洗い流した後、ユウロピウム蛍光を検出する。
【0060】
SPA(シンチレーション近接検定法)およびフラッシュプレート検定法は、通常、放射性標識基質を捕捉するためにビオチン/アビジン相互作用を利用する。一般的に、反応混合物は、キナーゼ、ビオチン化ペプチド基質およびγ−[P33]ATPを含む。反応後、ビオチン化ペプチドをストレプトアビジンにより捕捉する。SPA検出においては、ストレプトアビジンはシンチラントを含むビーズに結合し、一方、フラッシュプレート検出においては、ストレプトアビジンはシンチラントを含むマイクロプレートのウエルの内側に結合する。免疫化されたならば、放射性標識基質は、発光を刺激するのに十分にシンチラントに近い。
【0061】
他の好ましい実施形態において、均一検定法は、TR−FRET(時間分解蛍光共鳴エネルギー移動)検定法、FP(蛍光偏光)検定法、ALPHA(増幅ルミネセンス近接均一検定法)、EFC(酵素断片相補性)検定法または遺伝子検定法である。
【0062】
TR−FRET(時間分解蛍光共鳴エネルギー移動)に基づく検定法は、通常、Cy3/Cy5またはCy5/Cy7のような重複スペクトルを伴うユウロピウムとAPC、修飾アロフィコシアニンまたは他の色素との間の蛍光共鳴エネルギー移動を利用する検定法である(Schobel U.ら、(1999年)Bioconjugate Chem.、第10巻、1107〜1114頁)。例えば、337nmの光によるユウロピウムの励起後に、分子は620nmの蛍光を発する。しかし、この蛍光がAPCに十分に近い場合、ユウロピウムはその励起エネルギーをAPCに移動させ、APCは665nmの蛍光を発する。キナーゼ基質は、通常ビオチン標識基質である。キナーゼ反応の後に、ユウロピウム標識(P)特異抗体をストレプトアビジン−APCとともに加える。リン酸化ペプチドがユウロピウム標識抗体とストレプトアビジン−APCとを密接に接触させる。ユウロピウムフルオロフォアへのAPCの密接な近接により、APC蛍光のためにユウロピウム蛍光の消光がもたらされる(FRET)。
【0063】
蛍光偏光(FP)を用いる検定法は、溶液中の蛍光性基質ペプチドを励起するために偏光を用いる検定法である。これらの蛍光性ペプチドは、溶液中で遊離であり、回転して、放射光を偏光解消させる。しかし、基質ペプチドが(P)−Tyrのようなより大きい分子に結合しているときには、その回転率は著しく減少し、放射光は高度に偏光したままである。キナーゼの検定のために、一般的に以下の2つの選択肢がある。
(a)蛍光性ホスホペプチドトレーサーを(P)特異抗体に結合させる。リン酸化生成物が抗体における蛍光性ホスホペプチドと競合して、高から低への偏光の変化をもたらす。
(b)リン酸化基質ペプチドがリン特異的抗体に結合して、低から高への偏光の変化をもたらす。
【0064】
ALPHA(増幅ルミネセンス近接均一)に基づく検定法は、リン酸化ペプチドにより近接させたドナービーズとアクセプタービーズとの間の一重項酸素の移動に依拠する検定法である。680nmでの励起により、ドナービーズにおける光増感剤が周囲の酸素を一重項状態酸素に変換し、一重項状態酸素が200nmまでの距離に拡散する。アクセプタービーズにおけるケミルミネセンス基がビーズ内の蛍光性受容体にエネルギーを移動させ、次いで、蛍光性受容体が約600nmの光を放射する。
【0065】
EFC(酵素断片相補性)に基づく検定法または同等の検定法は、特に、化合物の高処理スクリーニングに用いることができる。EFC検定法は、酵素アクセプター(EA)と酵素ドナー(ED)の2つの断片からなる工学処理β−ガラクトシダーゼ酵素に基づいている。断片を分離すると、β−ガラクトシダーゼ活性はないが、断片が共存すると、結合(相補)して活性な酵素を形成する。EFC検定法は、分析物が抗体または受容体のような特異的結合性タンパク質によって認識され得る、ED−分析物複合体を用いるものである。特異的結合性タンパク質が存在しない場合、ED−分析物複合体は、EAを相補結合して活性なβ−ガラクトシダーゼを形成し、正のルミネセンスシグナルを発生することができる。ED−分析物複合体が特異的結合性タンパク質によって結合されている場合、EAとの相補結合が妨げられて、シグナルは存在しない。遊離分析物が供給される(試料中に)場合、遊離分析物は、特異的結合性タンパク質への結合についてED−分析物複合体と競合する。遊離分析物は、EAとの相補結合のためにED−分析物複合体を遊離させ、試料中に存在する遊離分析物の量に依存するシグナルを発生させる。
【0066】
また、他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、アレイにGPIIbまたはLIMK−1を結合させることによりアレイについて実施する。固相化学および光不安定性保護基を用いてそのようなアレイを調製する方法は、例えば、米国特許第5744305号明細書に開示されている。これらのアレイは、試験化合物または化合物ライブラリーと接触させ、相互作用、例えば、結合や構造変化について試験することもできる。
【0067】
なお、他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、全細胞を用いて実施する。血小板は血液ドナーから容易に入手することができるため、血小板をそのような試験に用いることが好ましい。あるいは、細胞株、場合によりトランスフェクト細胞株を用いることができる。そのような細胞株は、巨核球細胞株(例えば、DAMIまたはMEG−1)、HEK293細胞(初代ヒト胚腎)、3T3細胞(マウス胚線維芽細胞)、CHO細胞(チャイニーズハムスター卵巣)、COS−7細胞(アフリカミドリザル細胞株)、HeLa細胞(ヒト類上皮子宮頸癌)、JURKAT細胞(ヒトT細胞白血病)、BHK21細胞(ハムスター正常腎、線維芽細胞)およびMCF−7細胞(ヒト乳癌)を含むが、これらに限定されない。全細胞を用いることは、細胞内物質、酵素等を試験系に加える必要がないため、膜と比べて有利である。さらに、マルチウエルプレートに加えた全細胞は、高処理スクリーニング試験および自動化試験系に特に適している。
【0068】
他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、ロボット工学システムにおいて実施する。本発明の方法を、例えば、マイクロフルイディクス、すなわち、チャンネル構造を用いた、ロボットによる平板培養およびロボットによる液体輸送を含むロボット工学システムで実施することが有利である。
【0069】
他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、高処理スクリーニングシステムの形で実施する。そのようなシステムでは、スクリーニング方法は、自動化かつ小型化されていることが有利である。特に、システムは、ロボットにより制御される小型化ウエルおよびマイクロフルイディクスを用いている。
【0070】
本発明の他の対象は、
(a)本発明によるスクリーニングする方法を実施する工程、
(b)検出された試験化合物の適切な量を供給する工程、および
(c)検出された試験化合物を上記で詳述したような1つまたはそれ以上の製薬上許容できる担体または補助物質を用いて製剤化する工程
を含む血栓形成または血液凝固疾患の予防または治療用の薬剤を製造する方法である。
【0071】
血栓形成または血液凝固疾患は、血栓形成または血液凝固の変化を伴う疾患である。健常者と比較して、血栓形成または血液凝固(の素因)が増加または減少し得る。血栓形成または血液凝固の亢進を伴う疾患は、当業者に知られており、例えば、アテローム血栓性疾患を含む。
【0072】
好ましい実施形態において、疾患はアテローム血栓性疾患である。
【0073】
本発明のより好ましい実施形態において、アテローム血栓性疾患は、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される。
【0074】
以下の図および表は、本発明の範囲を限定することなく、本発明をより詳細に説明するものである。
【発明を実施するための最良の形態】
【0075】
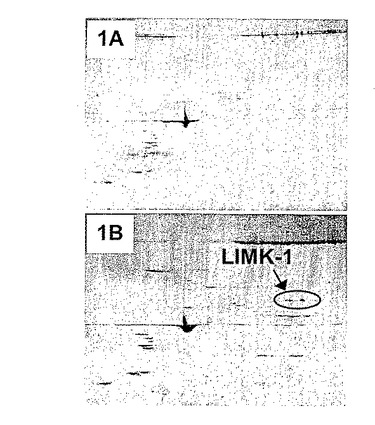
図1にGPIIb免疫沈降物の2D電気泳動によるLIMK1の検出を示す。対照IgG(図1A)またはGPIIbに対する特異抗体(図1B)によるヒト休止血小板抽出物(3.9mg)の免疫沈降物を、pH3〜10勾配で最初の段階においてIEF(等電点電気泳動)により分離した。その後、タンパク質を10%SDS−PAGE上で二次元で分離した。ゲルをコロイダルクマシー染色し、分析した。特異GPIIb抗体のみを含むゲル上の可視タンパク質スポットを切除し、トリプシンで消化し(ゲル中消化)、抽出し、MALDI−TOF MSにより分析した。
【0076】
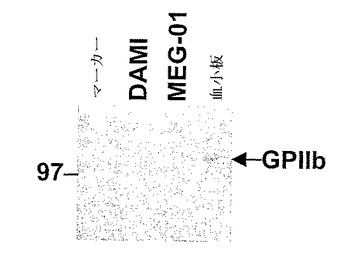
図2にLIMK−1に対する抗体による免疫沈降後のインテグリンGPIIbの検出を示す。LIMK−1に対する特異抗体によるヒト血小板抽出物(2mg)ならびにヒト巨核球細胞株DAMIおよびMEG−01の免疫沈降物を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。インテグリンGPIIbを特異抗体を用いて検出した。
【0077】
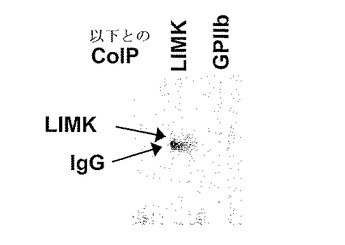
図3にLIMK−1およびGPIIbに対する抗体による免疫沈降後のLIMK−1の検出を示す。LIMK−1およびGPIIbに対する特異抗体によるヒト血小板抽出物の免疫沈降物を10%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0078】
図4に血小板ならびにDAMIおよびMeg−01細胞の抽出物中のLIMK−1の検出を示す。ヒト血小板の抽出物からの等量のタンパク質(50μg)を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0079】
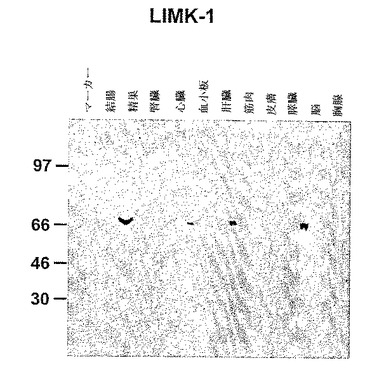
図5に種々のヒト組織の抽出物中のLIMK−1の検出を示す。ヒトの結腸、精巣、腎臓、心臓、血小板、肝臓、筋肉、皮膚、膵臓、脳および胸腺組織の抽出物からの等量のタンパク質を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0080】
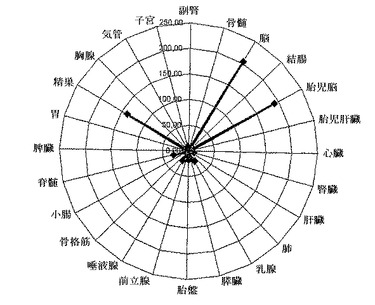
図6に種々のヒト組織における定量的Taqman分析によるLIMK−1転写物の検出を示す。ヒト骨髄、結腸、精巣、腎臓、心臓、血小板、肝臓、胎児肝臓、肺、乳腺、胎盤、前立腺、唾液腺、腸、脊髄、脾臓、胃、気管、子宮、筋肉、膵臓、脳、胎児脳および胸腺組織のmRNAをTaqman分析により分析した。
【実施例】
【0081】
1.材料および方法
1.1 血小板の準備
血小板は、血液銀行からの(4人のドナー)血小板濃縮製剤から準備した。すべての操作段階を室温で実施した。血小板濃縮製剤をタイロード緩衝液(pH7.4;137mmol/l NaCl、2.7mmol/l KCl、12mmol/l NaHCO3、0.36mmol/l NaH2PO4、1mmol/l MgCl2、10mmol/l HEPES、5.6mmol/lデキストロース)で希釈し、得られた懸濁液を制動せずに120×gで15分間遠心分離した。次いで、プロスタグランジンE1(PGE1 0.5μg/ml)を上清に加え、混合物を室温で5分間インキュベートし、制動せずに650×gで15分間遠心分離した。上清を捨て、ペレットをタイロード緩衝液(0.1%BSAを含む)に再懸濁し、PGE1(0.5μg/ml)を加えた。混合物を室温で5分間インキュベートし、その後、制動せずに650×gで5分間遠心分離した。上清を捨て、ペレットをタイロード緩衝液(BSAを含まない)に再懸濁し、PGE1(0.25μg/ml)を加え、混合物をインキュベートし(室温で5分間)、遠心分離した(制動せずに650×gで15分間)。再び上清を捨て、ペレットをタイロード緩衝液(BSAを含まない)に再懸濁し、細胞を液体窒素で凍結し、次いで、使用時まで−80℃で保存した。
【0082】
1.2 細胞培養
ヒトDAMIおよびMeg−01巨核細胞株の培養物を10%ウシ胎児血清(FCS)およびペニシリン/ストレプトマイシンを含むダルベッコの変法イーグル培地(DMEM)(37℃、5%CO2)中で増殖させた。細胞を洗浄し、収集し、細胞のペレットを、抽出物の調製の前に−80℃で保存した。
【0083】
1.3 免疫沈降のための血小板の処理
凍結血小板ペレットを1〜2mlの溶解緩衝液(下を参照)中で解凍し、次いで、ピペッティングにより上下にかき混ぜた後、音波処理(10パルス、30%)により再懸濁した。血小板を遠心分離して(5分、3000×g)、上清中の可溶性たんぱく質の濃縮のために前分画を行った。
【0084】
図1で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH8):10mmol/lトリス、140mmol/lNaCl、1%トリトン、0.05%SDS、1tab.Complete、200μmol/lペファブロック(4−(2−アミノエチル)−ベンゼンスルホニルフルオリド塩酸塩))
洗浄緩衝液(pH8):10mmol/lトリス、140mmol/lNaCl、1%トリトン
図2で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH7.5):50mmol/lトリス、50mmol/lNaCl、0.5%トリトン、0.05%SDS、1mmol/lNa3VO4、1tab.Complete、1μg/mlペプスタチン、5mmol/lNaF
洗浄緩衝液(pH7.5):50mmol/lトリス、50mmol/lNaCl、0.1%トリトン
図3で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH7.5):50mmol/lトリス、150mmol/lNaCl、1%トリトン、1mmol/lNa3VO4、1tab.Complete、1μg/mlペプスタチン、5mmol/lNaF
洗浄緩衝液(pH7.5):50mmol/lトリス、150mmol/lNaCl、0.1%トリトン
【0085】
1.4 免疫沈降
プロテインA−セファロース(Amersham Biosciences、Uppsala、Sweden)ならびにプロテインG−アガロース(Roche Diagnostics GmbH、Mannheim、Germany)を、抗体の種類によって免疫沈降に用いた。すべての操作段階を4℃で行った。
【0086】
ビーズを使用前に洗浄緩衝液(上を参照)で3回洗浄した。血小板溶解物をビーズ材料とともに1時間インキュベートしてあらかじめ清澄にし(図1および2、図3ではなし)、その後、遠心分離した(3分、500×g)。血小板溶解物を抗体とともに1時間インキュベートし、免疫沈降のためにビーズを一夜加えた。その後、ビーズを洗浄緩衝液(上記参照)で3回洗浄し、IEF再水和緩衝液(2Dゲル分析用)または溶解緩衝液(1Dゲル分析用)とともにインキュベートして溶出を行った。
【0087】
1.5 抗体の仕様
GPIIb(Santa Cruz Biotecnology,Inc.、Santa Cruz、Califronia、USA、カタログ番号sc−7310)、免疫沈降用(プロテインA)
GPIIb(Biotrend Chemikalien GmbH、Cologne、Germany、カタログ番号6065)、ウエスタンブロット(1:1000)用 LIMK(Santa Cruz Biotechnology,Inc.、Santa Cruz、Califronia、USA、カタログ番号sc−5576)、免疫沈降用(プロテインG)およびウエスタンブロット(1:250)用
【0088】
1.6 2Dゲル電気泳動
1次元ゲル電気泳動は、Biorad Protean IEF Cell(IPGストリップ:11cm、pH3〜10)(Biorad Laboratories、Hercules、CA、USA)および次のIEF再水和緩衝液(8mol/l尿素、0.5%CHAPS、10mmol/lDTT、0.2%Biolyte、0.001%ブロモフェノールブルー)を用いて行った。能動的再水和のために、185μlの容積の試料を用いてIPGストリップを50Vで一夜放置した。IEF(等電点電気泳動)は、直線ランプ、最終電圧8000Vおよび合計少なくとも40000Vhrsで行った。2次元ゲル電気泳動の前に還元/アルキル化を各平衡緩衝液(pH8.8、6mol/l尿素、2%SDS、0.375mol/lトリス、20%グリセロール、130mmol/lDTTまたは135mmol/lヨードアセタミド)中で10分間行った。
2次元ゲル電気泳動は、Criterion4〜20%ゲルを用い、ランニング緩衝液として25mmol/lトリス、192mmol/lグリシン、0.1%SDSを用いて150Vで行った。
【0089】
1.7 1Dゲル電気泳動
1Dゲル電気泳動は、X−Cell Sure Lock System(Novex−system、Invitrogen GmbH、Karlsruhe、Germany)ならびにランニング緩衝液としてのMOPS−SDSおよび150Vの電圧を用いて行った。
【0090】
1.8 エレクトロトランスファー/ウエスタンブロット(Semidry)
エレクトロトランスファーのために、Multiphor II System(不連続緩衝液システム)(Amersham、Biosciencies、Uppsala、Sweden)を用いた。陽極緩衝液1は0.3oil/lトリス、20%メタノール、陽極緩衝液2は25mmol/lトリス、20%メタノール、陰極緩衝液は40mmol/lアミノカプロン酸、0.01%SDS、20%メタノールであった。
タンパク質を0.8mA/cm2を用いてニトロセルロースシート(Schleicher&Schuell BioScience GmbH、Dassel/Relliehausen、Germany、Protran BA85 0.45μm)上にブロットした。
【0091】
1.9 染色技術
ゲルはコロイダルクマシー(Neuhoffら、1988年)または銀(Blumら、1987年の後に修正)で染色した。修正された銀染色は、ゲルを40%エタノール/10%酢酸で1時間固定し、30%エタノールで2回、水で1回各20分間洗浄し、0.02%Na2S2O3で1分間増感し、水(3×20秒)で洗浄し、冷0.1%AgNO3中で4℃で20分間インキュベートし、水(1×1分よりも3×20秒)で洗浄し、3%NaCO3/0.05%ホルマリンで現像し、水中でもう1回20秒間洗浄した。最後に5%酢酸で染色を完了した。
【0092】
1.10 画像化
抗体は、Hyperfilm ECL(Amersham Biosciences、Uppsala、Sweden、カタログ番号RPN2132)への5分間の曝露後にケミルミネセンス(ECL plus、Amersham Biosciences、Uppsala、Sweden、カタログ番号RPN2132)により検出し、Adefo−Developer(00009)およびAdefo−Fixator(00062)(ADEFO−CHEMIE GmbH、Nuernberg、Germany)により現像した。
【0093】
1.11 ゲル中消化
ゲル中消化は、Pandeyら、2000年に従って行った。簡単に述べると、ゲルスポットを手作業により切り取り、分解のためにトリプシン(ブタ、Promega GmbH、Mannheim、Germany、カタログ番号V511A)を用いた。
【0094】
1.12 MALDI−TOF質量分析
MALDI−TOF質量分析は、Voyager DE−STR MALDI−TOFワークステーション(Applied Biosystems、Foster City、CA、USA)を用いて行った。試料を乾燥小滴法により調製し、□−シアノヒドロキシケイ皮酸(アセトニトリル/トリフルオロ酢酸中3mg/ml)をマトリックスとして用いて2×96ウエルテフロン被覆試料プレート上にスポットした。700〜4000Daの検出範囲で行った。
【0095】
1.13 タンパク質の定量
試料のタンパク質含量は、製造業者の指示(562nmでの検出)に従ってBCAキット(Pierce Chemical Company、Rockford、Illinois、USA)を用いて測定した。
【0096】
1.14 組織分布パターン
レーンにつき50μgのタンパク質をゲル上にのせた。種々のヒト組織をBioCat GmbH(Heidelberg、Germany)から購入した。血小板および巨核球からのLIMKの検出は、上清中であらかじめ濃縮して促進した(図4および図5についてそれぞれ15000×g、15分間または3000×g、5分間遠心分離)。
【0097】
1.15 Taqman分析
増幅のために、以下のPCRプロトコールを用いた。増幅のための次の試薬のすべてがApplied Biosystems(Foster City、USA)により供給された。20ngのゲノムDNA、1単位のTaqGoldポリメラーゼ、1×Taqポリメラーゼ緩衝液、500μMのdNTP、2.5mmol/lのMgCl2、200nmol/lの各増幅プライマー対、H2O ad5μl
PCRの増幅プログラム
95℃×10分 ×1サイクル
95℃×30秒
70℃×30秒 ×2サイクル
95℃×30秒
65℃×30秒 ×2サイクル
95℃×30秒
60℃×30秒 ×2サイクル
95℃×30秒
56℃×30秒
72℃×30秒 ×40サイクル
72℃×10分
4℃×30秒 ×1サイクル
【0098】
1.16 装置
実験は、CM5チップおよびNi−NTAチップを装着したBiacore 3000最高性能研究システム(Biacore International SA、79111 Freiburg、Germany)を用いて行った。
【0099】
結果
2.1 LIMK−1とインテグリンGPIIbとの相互作用
本発明者らは、GPIIbに対する市販の特異抗体との共同免疫沈降を用いて、GPIIbとのタンパク質複合体を濃縮した。これらの複合体は、2次元ゲル電気泳動により分離した(図1)。この実験で得られた結果は、LIMK−1はヒト血小板抽出物中のインテグリンGPIIbと共同沈降することを示している。
【0100】
これらの結果を検証するために、本発明者らは、ウエスタン分析によるGPIIbのその後の検出のため、LIMK−1に対する市販の特異抗体による血小板抽出物ならびにヒト巨核球細胞株DAMIおよびMEG−01の免疫沈降を用いた(図2)。これらの結果は、インテグリンGPIIbがヒト血小板中のLIMK−1と会合するという知見を確認するものであった。ヒト巨核球細胞株DAMIおよびMEG−01においてはGPIIbとLIMK−1との相互作用は示されなかった。これらの巨核球細胞株は、分化後に血小板を生産する前駆細胞株である(Fugmanら、1990年、George、2000年、Greenbergら、1988年、Oguraら、1988年、Schickら、1998年、Takeuchiら、1998年、Vittetら、1992年)。しかし、本発明者らの実験のために本発明者らはこれらの細胞株を未分化形として用いた。これらの未分化巨核球細胞株におけるGPIIbインテグリンの発現レベルを分析したとき、本発明者らはGPIIbの発現はこれらの細胞株においては血小板における発現と比較してはるかに低いことを見いだした。これは、本発明者らがこれらの細胞株におけるインテグリンとLIMK−1との相互作用を検出しなかった理由の説明となっている。
【0101】
本発明者らの結果をさらに検証するために、本発明者らは、後続のウエスタン分析によるLIMK−1の検出のためにLIMK−1およびGPIIbに対する市販の特異抗体による血小板抽出物の免疫沈降を用いた(図3)。再び、これらの結果は、インテグリンGPIIbがヒト血小板中のLIMK−1と会合しているという本発明者らの以前の知見を確認するものであった。
【0102】
2.2 LIMK−1の組織分布
本発明者らは、LIMK−1に対する特異抗体を用いて血小板ならびに巨核球細胞株DAMIおよびMEG−01におけるLIMK−1の発現を分析した(図4)。このように、本発明者らはLIMK−1に対する特異抗体を用いて、血小板ならびに巨核球細胞株DAMIおよびMEG−01におけるLIMK−1の発現を確認した。
【0103】
さらに、種々のヒト組織におけるLIMK−1の発現を分析するために、本発明者らは後続のウエスタン分析のためにヒト組織抽出物をゲル電気泳動により分離した(図5)。これらの結果は、LIMK−1が精巣、心臓、血小板、筋肉および脳組織において発現することを示している。発現は血小板に対して絶対的に特異的でないが、LIMK−1はすべての組織に普遍的には発現しない。この知見は、脳、胎児脳および精巣におけるLIMK−1の発現を示しているTaqman分析(図6)により確認されている。血小板は、このTaqmanパネルには示さない。
【図面の簡単な説明】
【0104】
【図1】GPIIb免疫沈降物の2D電気泳動によるLIMK1の検出を示す。
【図2】LIMK−1に対する抗体による免疫沈降後のインテグリンGPIIbの検出を示す。
【図3】LIMK−1およびGPIIbに対する抗体による免疫沈降後のLIMK−1の検出を示す。
【図4】血小板ならびにDAMIおよびMeg−01細胞の抽出物中のLIMK−1の検出を示す。
【図5】種々のヒト組織の抽出物中のLIMK−1の検出を示す。
【図6】種々のヒト組織における定量的Taqman分析によるLIMK−1転写物の検出を示す。
【技術分野】
【0001】
本発明は、血栓形成または血液凝固疾患の予防または治療用の薬剤の製造用のGPIIbへの結合および/またはGPIIb/IIIa下流シグナル伝達の活性化もしくは阻害用のLIMK−1、LIMK−1類似体またはLIMK−1リガンド、LIMK−1類似体またはLIMK−1リガンドをスクリーニングするスクリーニング方法の方法、ならびに血栓形成または血液凝固疾患の治療用の薬剤を製造する方法に関する。
【背景技術】
【0002】
血小板は、最小の血液細胞であり、巨核球細胞質の断片にすぎないが、正常な止血に重要な役割を有し、血栓性疾患への重要な寄与因子である。血小板付着、活性化および凝集は、止血の開始のための欠くことができない反応であり、様々な冠動脈、脳および末梢血管疾患の病理に重要な役割を果たす(Bhattら、2003年、George、2000年、Moroiら、1998年、Raoら、2000年、Ruggeri、2000年)。
【0003】
血小板膜は、細胞外マトリックス成分への血小板付着に関与する高濃度のインテグリンおよび他の糖タンパク質を含む(Lopezら、1988年、Phillipsら、1988年、Shattil、1999年)。コラーゲン線維およびヴォン・ヴィレブランド因子(vWF)は、曝露された内皮下層への血小板の付着のための重要な部位を与え、血管損傷の部位にそれらを捕捉し、損傷した範囲にわたる細胞の単層の形成を可能にする(Ruggeri、2002年、Watson、1999年)。コラーゲン線維およびvWFは、Gタンパク質結合受容体(GPCRs)を介して作用するシグナルとともに、インテグリン糖タンパク質GPIIb/IIIa(□IIb□3としても知られている)の「裏返し(inside−out)」調節、高密度およびα顆粒からの分泌、トロンボキサンの生成および前凝固活性の発現につながる血小板の活性化も刺激する(Parise、1999年、Ruggeri、2002年、Shattil、1999年)。また、血小板は、活性化により、通常の円盤状から付着を促進する長い樹状延長部分を有する緻密な球体に変化する。これらのすべての事象が止血過程を支えている。膜タンパク質GPIIb(CD41)およびGPIIIa(CD61)は、フィブリノーゲン、ヴォン・ヴィレブランド因子(vWF)およびフィブロネクチンに結合する血小板の表面上の最も豊富なヘテロ二量体複合体を形成する。GPIIb/IIIaは、可溶性フィブリノーゲンに対する低い親和力を有する不活性構造の休止血小板の表面上に発現する。コラーゲン、ADPまたはトロンビンのような多数の重要なアゴニストによる血小板の活性化の後に、GPIIb/IIIaは、可溶性フィブリノーゲンに対するその結合親和力を増加させる構造の変化を受け、血小板凝集をもたらす(Parise、1999年、Ruggeri、2002年、Shattil、1999年)。GPIIb/IIIaによるシグナル伝達は、細胞質側末端のいくつかの領域に関係する。エンドネキシン、カルシウムおよびインテグリン結合タンパク質(CIB)、ShcおよびGrb2などの数種のシグナル伝達タンパク質がGPIIb/IIIaインテグリンの細胞質ドメインに結合する。さらに、αサブユニットの細胞質側末端における配列KVGFFKRがシグナル伝達に関与しており、この領域に対応する脂質修飾ペプチドが血小板を完全に活性化する(Stephensら、1998年)。しかし、血小板活性化からフィブリノーゲン受容体曝露までの正確な一連のシグナル伝達は、知られていない。
【0004】
アスピリンが血小板に対して、また心筋梗塞の一次予防に有効であることが実証されて以来、血栓性障害に対するアスピリンの予防的使用は著しく増加した。したがって、数十年間にわたって、抗血小板療法は、トロンボキサン経路およびアスピリンによるその阻害に重点が置かれている。
【0005】
その関連性を考えると、付着は抗血栓薬の開発の魅力的な標的であると予想される。しかし、血小板凝集を妨げるように設計された最初の一連の化合物であるαIIbβ3アンタゴニストは、経口長期治療薬として成功しなかった。これは、天然リガンドの場合と同様に、αIIbβ3アンタゴニスト(多くがフィブリノーゲンの受容体認識配列に基づく)が「細胞内への(outside−in)」シグナル伝達を実際に誘発することに起因すると想定される(Coxら、2000年)。したがって、血小板を活性化せずに、リガンドと受容体との相互作用を直接妨害することは不可能かもしれない。
【0006】
近年、いくつかの抗血小板療法が導入され、血栓塞栓性障害におけるアスピリンから、チクロピジンおよびクロピドロゲルにおよぶ抗血小板療法の有用性が実証されている(Konstantopoulosら、2001年、Mousaら、2002年、Weksler、2000年)。しかし、これらの各種療法はすべて血栓塞栓性障害の治療に付加的な利点を加えるものであるが、種々の薬剤を用いた併用療法の有効性が十分でないことが判明する多くの症例は依然として存在する。さらに、多くの場合にこれらの抗血小板療法による治療は、望ましくない副作用を伴う。したがって、血栓塞栓性障害の治療のための新規なアプローチが緊急に求められている。
【非特許文献1】Mizunoら、Oncogene、第9巻、1605〜1612頁、1994年
【非特許文献2】R.P.Haugland、Molucular Probes、Handbook of Fluorescent Probes and Research Chemicals、第5版、Molecular Probes,Inc.、Eugene、Oreg.、1992年
【非特許文献3】Mujumdarら、Bioconjuga Chem.、第4巻、105頁、1992年
【非特許文献4】Ponczら、J.Biol.Chem.、第262巻、8476〜8482頁、1987年
【非特許文献5】Schobel U.ら、(1999年)Bioconjugate Chem.、第10巻、1107〜1114頁
【特許文献1】米国特許第5744305号
【発明の開示】
【発明が解決しようとする課題】
【0007】
したがって、本発明の目的は、血栓形成または血液凝固疾患の予防および治療改善のための新規のアプローチならびに例えば、抗血栓療法に対する新規の標的を特定する方法を提供することであった。
【0008】
驚くべきことに、LIMK−1タンパク質がGPIIb/IIIaに結合することが今回発見された。LIMK−1は、72.6kDaのタンパク質(ヒト:647アミノ酸)であり、LIMモチーフ含有タンパク質キナーゼファミリーのメンバーである。その遺伝子は、7q11.23における第7染色体上に位置する。該タンパク質は、2つのN末端システインリッチLIM/二重ジンクフィンガーモチーフ、いくつかの推定上のカゼインキナーゼおよびMAPキナーゼ認識部位を含むプロリンセリンリッチ領域、PDZドメイン(PSD95/disc large/ZO−1)およびC末端セリン/トレオニンキナーゼドメインを含む。ジンクフィンガー様LIMドメインは、タンパク質−タンパク質相互作用を仲介すると推定されており、核および細胞骨格タンパク質内に存在すると記載された。LIMK−1のC末端キナーゼ断片は、LIMドメインに結合し、LIM断片は、LIMK−1のキナーゼコア断片のキナーゼ触媒活性を用量依存的に阻害することが示された。したがって、N末端LIMドメインは、C末端キナーゼドメインとの直接的相互作用によって、LIMK−1のキナーゼ活性を負に調節する(Nagataら、1999年)。LIMドメインキナーゼ1(LIMK−1)は、コフィリンのリン酸化によってアクチン細胞骨格の再組織化を調節する。活性なフィリンは、アクチンの解重合をもたらす。結果としてLIMK−1によるコフィリンのリン酸化は、コフィリンのアクチン解重合活性の失活につながる(Bierneら、2001年、Gohlaら、2002年、Yangら、1998年)。しかし、これまでに血小板におけるLIMK−1(またはLIMK−2)の潜在的役割に関する報告はなかった。LIMK−1は血小板インテグリンGPIIbに結合するので、LIMK−1は、血小板の活性化のために誘発され、また必要な血小板の形状の変化を誘発する。したがって、LIMK−1の阻害は、催血栓刺激による血小板の活性化の阻害につながる。したがって、LIMK−1の阻害は、血小板の活性化と凝集、およびしたがって血栓形成を妨げる。
【課題を解決するための手段】
【0009】
したがって、本発明の第一の態様は、特に、インテグリンGPIIb/IIIaのシグナル伝達下流を調節、好ましくは阻害するための、GPIIbへの結合のためのLIMK−1またはLIMK−1類似体のインビトロおよびインビボでの使用を対象とする。
【0010】
本発明によるLIMK−1は、天然のあらゆるLIMK−1タンパク質である。これは、LIMK−1タンパク質および種々の動物種、好ましくは脊椎動物、より好ましくは哺乳類のその変異型ならびにスプライス変異型を含む。好ましいLIMK−1タンパク質は、ヒトLIMK−1タンパク質である。ヒトLIMK−1タンパク質のアミノ酸配列は、Mizunoら、Oncogene、第9巻、1605〜1612頁、1994年に開示されている。
【0011】
本発明による「LIMK−1類似体」という用語は、GPIIbに結合するが、LIMK−1ではないLIMK−1由来のタンパク質を指す。より詳細には、LIMK−1類似体は、天然のLIMK−1配列(すなわち、野生型ポリペプチド)と1つまたはそれ以上のアミノ酸が異なっているアミノ酸配列を指す。そのような類似体は、1つまたはそれ以上のアミノ酸の置換、挿入または欠失の点で野生型ポリペプチドと異なっている。好ましいものは半保存的置換であり、より好ましいものは保存的アミノ酸置換であり、それにより、残基が同様な特性を有する他のものによって置換されている。一般的な置換は、脂肪族アミノ酸の間、脂肪族ヒドロキシル側鎖を有するアミノ酸の間、酸性残基を有するアミノ酸の間、アミド誘導体の間、塩基性残基を有するアミノ酸の間または芳香族残基を有するアミノ酸の間の置換である。一般的な半保存的および保存的置換は、以下のとおりである。
【0012】
【表1】
【0013】
A、F、H、I、L、M、P、V、WまたはYからCへの変化は、新たなシステインが遊離のチオールとして留まる場合、半保存的である。さらに、当業者は、立体的に必要な位置におけるグリシンは置換すべきでなく、Pはαらせんまたはβシート構造を有するタンパク質の部分に導入すべきでないことを認識するであろう。
【0014】
変異型ポリペプチドは、野生型と比べて、一次構造(アミノ酸配列)が異なっているが、二次もしくは三次構造または機能はさほど異なっていてもまたはいなくてもよい。いずれにせよ、類似体は、少なくとも75%、好ましくは少なくとも80%、より好ましくは少なくとも90%、さらにより好ましくは少なくとも95%、最も好ましくは少なくとも99%の野生型LIMK−1との同一性(相同)を示す。
【0015】
類似体は、GPIIbに結合するために十分なLIMK−1の部分であってもよい。この部分は、少なくとも30アミノ酸、好ましくは少なくとも100アミノ酸、より好ましくは少なくとも300アミノ酸、さらにより好ましくは少なくとも450アミノ酸、最も好ましくは少なくとも600アミノ酸を含む。類似体のこの部分は、上で詳述したように1つまたはそれ以上のアミノ酸の置換、挿入または欠失において野生型ポリペプチドの部分と異なっていてよい。1つの実施形態において、LIMK−1またはLIMK−1類似体は、他の分子、例えば、タンパク質および/またはマーカー(例えば、上で詳述したような)に融合されていてよい。
【0016】
「GPIIbへの結合」または「GPIIbに結合する」という用語は、GPIIbタンパク質への優勢な化合物(LIMK−1またはLIMK−1類似体)の特異的結合を指す。本発明によるGPIIbタンパク質への特異的結合は、限定なしに、10-4mol/lを超えない、好ましくは10-5mol/lを超えない、より好ましくは10-6mol/lを超えない解離定数KDでの結合を含む。解離定数KDは、例えば、実施例(例えば、図1を参照)に示すように免疫沈降を用いて、種々の濃度の供試化合物、例えば、LIMK−1またはLIMK−1類似体ならびに一定の濃度のGPIIbを用いることにより測定することができる。GPIIbに結合している供試化合物の濃度は、例えば、LIMK−1またはLIMK−1類似体に対する特異抗体を用いることにより測定される。KDは、次の式により求められる。
B[L]=[L]/([L]+KD)
ここで、[L]は化合物の濃度を表す。KDは供試化合物の解離定数であり、B[L]は供試化合物の特定の濃度における結合(%)である。
【0017】
検出のために、抗体は標識することができ、これは適宜、フルオロフォア、発色団、放射性標識、金属コロイド、酵素、または化学発光もしくは生物発光分子であってよい。適切なフルオロフォアおよび発色団は、R.P.Haugland、Molucular Probes、Handbook of Fluorescent Probes and Research Chemicals、第5版、Molecular Probes,Inc.、Eugene、Oreg.、1992年に開示されている。好ましいフルオロフォアの例としては、フルオレセイン、ローダミンおよびスルホインドシアニン色素Cy5などがある(Mujumdarら、Bioconjuga Chem.、第4巻、105頁、1992年)。好ましい放射性標識としては、3H、14C、32P、33P、35S、99mTcまたは125Iなどがある。好ましい酵素としては、西洋ワサビペルオキシダーゼ、アリカリホスファターゼ、グルコースオキシダーゼおよびウレアーゼなどがある。
【0018】
本発明によるGPIIbは、任意の天然に存在するPIIbタンパク質である。これは、種々の動物種、好ましくは脊椎動物、より好ましくは哺乳類のGPIIbタンパク質およびその変異型ならびにスプライス変異型を含む。好ましいGPIIbタンパク質は、ヒトGPIIbタンパク質であり、そのアミノ酸配列は、Ponczら、J.Biol.Chem.、第262巻、8476〜8482頁、1987年に開示されている。
【0019】
本発明の1つの好ましい実施形態において、LIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の活性化物質または阻害物質、好ましくはその阻害物質である。
【0020】
GPIIb/IIIa下流シグナル伝達の活性化物質は、検出可能なシグナルをもたらす各シグナル伝達経路を活性化する。阻害物質は、GPIIb/IIIa下流シグナル伝達を少なくとも部分的に遮断する。活性化および不活性化は、以下で定義する。活性化物質および不活性化物質は、下記のように特定される。
【0021】
本発明の他の好ましい実施形態において、LIMK−1またはLIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の活性化または阻害、より好ましくはその阻害のために用いる。
【0022】
上で詳述したように、GPIIb/IIIa下流シグナル伝達の活性化は、とりわけ通常の円盤状から長い樹状延長部分を有する小型の球体への血小板の外観における構造変化を特徴とする血小板の活性化につながる。本発明によるGPIIb/IIIa下流シグナル伝達の活性化は、LIMK−1またはアゴニスト性LIMK−1類似体で刺激したとき、血小板の少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%の活性化に対応する。したがって、GPIIb/IIIa下流シグナル伝達の阻害は、血小板の活性化の阻害につながる。本発明によるGPIIb/IIIa下流シグナル伝達の阻害は、場合によりGPIIb/IIIa活性化物質の存在下で、阻害性LIMK−1類似体で阻害したとき、血小板の少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%の阻害に対応する。
【0023】
本発明の他の対象は、GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のためのLIMK−1リガンドの使用である。
【0024】
LIMK−1リガンドは、LIMK−1に特異的に結合するあらゆる化合物分子である。本発明によるLIMK−1への特異的結合は、限定なしに、10-4mol/lを超えない、好ましくは10-5mol/lを超えない解離定数KDでの結合を含む。解離定数KDは、GPIIbを用いて詳述したように、またはLIMK−1タンパク質および適切な標識LIMK−1リガンドを用いることにより測定することができる。適切なマーカーは、上で詳述した。さらに、LIMK−1アゴニスト性またはアンタゴニスト性LIMK−1リガンドの結合は、それぞれLIMK−1のキナーゼ機能の活性化または不活性化により検出することができる。キナーゼ機能は、当業者に知られている方法により容易に検出することができる。それらのうちの一部は以下で詳述する。アンタゴニスト性LIMK−1リガンドの検出のためには、例えば、PAK(p21活性化キナーゼ)またはROCK(Rhoキナーゼ)でLIMK−1を活性化することが必要である場合がある。あるいは、LIMK−1リガンドの結合は、シグナル伝達経路のより下流のシグナル、例えば、血小板の活性化もしくは不活性化またはそれらの構造変化の誘導を測定することによって検出することができる。本発明によるLIMK−1への特異的結合は、限定なしに、アゴニスト性およびアンタゴニスト性LIMK−1リガンドについてそれぞれ、10-4mol/lを超えない、好ましくは10-5mol/lを超えないEC50値およびIC50値での結合を含む。EC50およびIC50値の測定および計算は、当業者に知られている。
【0025】
LIMK−1は、非重合有機化合物、脂質、炭水化物、ペプチド、好ましくは約10〜約300アミノ酸、特に10〜50アミノ酸を有するペプチドであってよい。特に好ましいものは、約200g/mol〜約1500g/mol、特に400g/mol〜1000g/molの好ましい分子量を有する実験室で合成されるかまたは天然に発見される小化合物分子、特に非重合有機化合物である。
【0026】
あるいは、本発明のLIMK−1リガンドは、粗製または精製形態の天然産物抽出物であってよい。抽出物は、ヘビ毒、葉または微生物発酵ブロスのような動物、植物、真菌または細菌源からの水および/またはアルコールおよび/または有機溶媒抽出および/またはカラムクロマトグラフィーおよび/または沈降などの標準的な方法により製造することができる。
【0027】
好ましい実施形態において、LIMK−1リガンドは、LIMK−1アゴニストまたはLIMK−1アンタゴニスト、より好ましくはLIMK−1アンタゴニストである。
【0028】
アゴニストは、LIMK−1に結合し、LIMK−1の変化、例えば、構造変化を誘発し、各シグナル伝達を活性化、例えば、検出可能なシグナルをもたらす、LIMK−1のキナーゼ機能を活性化する。アンタゴニストまたはブロッカーもLIMK−1に結合するが、一般的にシグナル伝達を誘導しない。アゴニストの存在下では、アンタゴニストは、アゴニストにより誘導されるシグナル伝達を用量依存的に阻害する。
【0029】
本発明の他の対象は、血栓形成または血液凝固疾患の予防または治療用の薬剤の製造のための本発明によるLIMK−1、LIMK−1類似体またはLIMK−1リガンドの使用である。血栓形成または血液凝固疾患は、血栓形成または血液凝固の変化を伴う疾患である。健常者と比較して、血栓形成または血液凝固(の素因)が増加または減少し得る。
【0030】
薬剤の生産のために、LIMK−1、LIMK−1類似体またはLIMK−1リガンドまたはその製薬上許容できる塩は、一般的に所望の特性を得るために混合された製薬上許容できる担体または補助物質のような成分の混合物からなる薬剤剤形中に混入しなければならない。
【0031】
製剤は、少なくとも1つの適切な製薬上許容できる担体または補助物質を含む。そのような物質の例は、脱塩水、等張食塩水、リンゲル液、緩衝液、有機または無機酸および塩基ならびにそれらの塩、塩化ナトリウム、炭酸水素ナトリウム、クエン酸ナトリウムまたはリン酸二カルシウム、プロピレングリコールなどのグリコール、オレイン酸エチルおよびラウリン酸エチルなどのエステル、グルコース、スクロースおよびラクトースなどの糖、トウモロコシデンプンおよびジャガイモデンプンなどのデンプン、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、ジメチルホルムアミドなどの可溶化剤および乳化剤、落花生油、綿実油、トウモロコシ油、大豆油、ひまし油などの油、オレイン酸エチル、ミリスチン酸イソプロピルなどの合成脂肪酸エステル、ゼラチン、デキストラン、セルロースおよびその誘導体などのポリマーアジュバント、アルブミン、有機溶媒、クエン酸塩および尿素などの錯化剤、プロテアーゼまたはヌクレアーゼ阻害剤、好ましくはアプロチニン、ε−アミノカプロン酸またはペプスタチンAなどの安定化剤、ベンジルアルコールなどの保存剤、亜硫酸ナトリウムなどの酸化防止剤、ワックスおよびEDTAなどの安定化剤である。着色剤、放出剤、コーティング剤、甘味料、着香および芳香剤、保存剤および抗酸化剤も組成物中に存在していてよい。生理緩衝液は、好ましくは、約6.0〜8.0のpH、特に約6.8〜7.8のpH、特に約7.4のpHおよび/または約200〜400ミリオスモル/リットル、好ましくは約290〜310ミリオスモル/リットルのオスモル濃度を有する。薬剤のpHは、一般的に適切な有機または無機緩衝液、例えば、好ましくはリン酸緩衝液、トリス緩衝液(トリス(ヒドロキシメチル)アミノメタン)、HEPES緩衝液([4−(2−ヒドロキシエチル)ピペラジノ]エタンスルホン酸)またはMOPS緩衝液(3−モルホリノ−1−プロパンスルホン酸)などを用いて調整する。各緩衝液の選択は、一般的に所望の緩衝液のモル濃度に依存する。例えば、注射および注入液剤には、リン酸緩衝液が適切である。薬剤ならびに適切な製剤上許容できる担体および補助物質を調合する方法は、当業者によく知られている。製剤上許容できる担体および補助物質は、とりわけ、広く用いられている剤形および特定された化合物に応じて選択する。
【0032】
経口、鼻内、直腸、非経口、膣、局所または膣投与用の薬剤組成物を製造することができる。非経口投与は、皮下、皮内、筋肉内、静脈内または腹腔内投与などである。
【0033】
薬剤は、カプセル剤、錠剤、丸薬、散剤および顆粒剤などの経口投与用の固形剤形、製薬上許容できる乳剤、マイクロエマルジョン剤、液剤、懸濁剤、シロップ剤およびエリキシル剤などの経口投与用の液体剤形、注射用製剤、例えば、滅菌注射用水性または油性懸濁剤、直腸または膣投与用組成物、好ましくは坐剤、ならびに軟膏剤、ペースト剤、クリーム剤、ローション剤、ゲル剤、散剤、液剤、噴霧剤、吸入剤またはパッチ剤などの局所または経皮投与用の剤形を含む様々な剤形として調合することができる。
【0034】
個々の患者における特定の治療的に有効な用量レベルは、特定された化合物の活性、剤形、患者の年齢、体重および性別、治療期間ならびに医療技術分野でよく知られている同様な因子を含む様々な因子に依存する。
【0035】
1回または分割量でヒトまたは他の哺乳類に投与する本発明の化合物の総1日量は、例えば、約0.01〜約50mg/kg体重またはより好ましくは約0.1〜約25mg/kg体重の量であってよい。1回量組成物は、そのような量または1日量を構成するその分量を含んでいてよい。一般的に、本発明による治療法は、1日当たり本発明の化合物の約10mg〜約1000mgを1回または複数回そのような治療を必要とする患者に投与することを含む。
【0036】
好ましい実施形態において、LIMK−1類似体は、GPIIb/IIIa下流シグナル伝達の阻害物質である。他の好ましい実施形態において、LIMK−1リガンドは、LIMK−1アンタゴニストである。
【0037】
血栓形成または血液凝固疾患を有する患者のほとんどが血栓形成または血液凝固(の素因)の亢進を伴う疾患に罹患している。したがって、それらの患者は、血栓形成または血液凝固を抑制する薬剤を特に必要とする。したがって、阻害性LIMK−1類似体およびアンタゴニスト性LIMK−1リガンドのような血栓形成または血液凝固を抑制する薬物が、薬剤の製造のための使用に好ましい。
【0038】
他の好ましい実施形態において、血栓形成または血液凝固疾患は、血栓形成または血液凝固の亢進を伴う疾患である。
【0039】
血栓形成または血液凝固の亢進を伴う疾患は、当業者に知られており、例えば、アテローム血栓性疾患を含む。
【0040】
より好ましい実施形態において、疾患は、アテローム血栓性疾患であり、より好ましくは、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬(comedication to vascular interventional strategy)からなる群から選択される疾患である。
【0041】
さらに、本発明の他の実施形態は、
(a)GPIIbタンパク質および場合によりその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(b)試験化合物を供給する工程、および
(c)GPIIbタンパク質への試験化合物の結合を検出する工程
を含み、試験化合物がLIMK−1由来であるLIMK−1類似体をスクリーニングする方法である。
【0042】
一般的に、GPIIbおよび場合によりその下流シグナル伝達の少なくとも1つの要素を、例えば、検定システムに供給し、LIMK−1由来の試験化合物と直接または間接的に接触させる。「LIMK−1由来の試験化合物」という表現は、上記で定義したようにLIMK−1類似体を指す。次いで、LIMK−1タンパク質への試験化合物の結合を検出する。この場合、結合は、GPIIbと試験化合物との相互作用を測定するか、または下流シグナル伝達に対するその影響を測定することによって検出することができる。
【0043】
「GPIIb/GPIIIaまたはGPIIbの下流シグナル伝達の要素」という用語は、GPIIb/GPIIIaまたはGPIIbの下流のシグナル伝達の一部である各分子またはイオンを指す。これは、シグナル伝達カスケードにおけるいずれかの段階のいずれかの要素であってよい。好ましくは、要素それ自体が測定可能なシグナルであるか、またはそれが測定可能なシグナルを発生させる。要素は、例えば、二次メッセンジャーまたは酵素であってよい。シグナルは、例えば、物質の濃度の変化または構造変化であってよい。
【0044】
結合は、直接的に、すなわち、例えばシグナル伝達経路における下流シグナルであり得る構築複合体を検出、または間接的に、すなわち、複合体の構築の影響を検出することにより、検出することができる。その後、適切なリガンドを分析し、かつ/または分離することができる。GPIIbへのLIMK−1類似体の結合を直接測定する方法は、上に詳述した。
【0045】
本発明の他の実施形態において、本発明によるLIMK−1類似体をスクリーニングする方法は、工程(c)の代わりに次の工程を含む。
(c’)GPIIb/IIIaおよび/またはその下流シグナル伝達の活性化または阻害を検出する。
【0046】
LIMK−1の活性化または阻害を検出するための適切な機能検定法は、例えば、GPIIb/GPIIIaの下流シグナル伝達を含むものであってよい。活性化または阻害は、例えば、下流シグナル、例えば、血小板の活性化またはそれらの構造変化を解析することによって上記で詳述したように検出することができる。シグナルを阻害性LIMK−1類似体により阻害させ、GPIIbおよび/またはLIMK−1刺激物質の存在下でGPIIb/GPIIIaシグナル伝達の阻害を検出することが必要であろう。
【0047】
本発明の他の実施形態は、
(a)LIMK−1タンパク質を供給する工程、
(b)試験化合物を供給する工程、および
(c)LIMK−1タンパク質への試験化合物の結合を検出する工程
を含むLIMK−1リガンドをスクリーニングする方法である。
【0048】
一般的に、LIMK−1を例えば検定システムに供給し、試験化合物、特に生化学または化学試験化合物、例えば、化合物ライブラリーの形で直接または間接的に接触させる。次いで、LIMK−1タンパク質への試験化合物の結合を検出する。結合は、直接的に、すなわち、例えばシグナル伝達経路における下流シグナルであり得る構築複合体を検出、または間接的に、すなわち、複合体の構築の影響を検出することにより、検出することができる。LIMK−1へのLIMK−1リガンドの結合を直接測定する方法は、上記で詳述した。その後、適切なリガンドを分析し、かつ/または分離することができる。
【0049】
本発明の他の実施形態において、本発明によるLIMK−1リガンドをスクリーニングする方法は、工程(c)の代わりに次の工程を含む。
(c’)LIMK−1またはGPII/IIIa下流シグナル伝達の活性化または阻害、好ましくはその活性化を検出する工程。
【0050】
LIMK−1の活性化または不活性化を検出するための適切な機能検定は、例えば、LIMK−1のキナーゼ機能に関するものである。キナーゼ活性を検出する方法は、当業者に知られている(以下を参照)。一般的に、これらの方法は、リン酸基、例えば、32Pまたは33P標識リン酸基のような標識リン酸基の受容体への移動に関するものである。したがって、LIMK−1の活性化はリン酸の移動の増加を伴い、不活性化はリン酸の移動の減少を伴う。シグナルをアンタゴニスト性LIMK−1リガンドにより阻害させ、LIMK−1刺激物質の存在下でLIMK−1の不活性化を検出することが必要であろう。活性化または不活性化は、下流シグナル、例えば、血小板の活性化またはそれらの構造変化を測定することによっても検出することができる。
【0051】
本発明の他の対象は、
(a)LIMK−1タンパク質を供給する工程、
(b)GPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(c)試験化合物を供給する工程、および
(d)GPIIb/GPIIIa下流シグナル伝達に対する試験化合物の影響を検出する工程
を含むGPIIb/IIIaおよび/またはその下流シグナル伝達と相互作用する試験化合物をスクリーニングする方法である。
【0052】
一般的に、LIMK−1およびGPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を例えば検定システムに供給し、試験化合物、特に生化学または化学試験化合物、例えば、化合物ライブラリーの形で直接または間接的に接触させる。次いで、GPIIb/GPIIIaシグナル伝達への試験化合物の影響を上記のように検出する。その後、適切なリガンドを分析し、かつ/または分離することができる。
【0053】
1つの好ましい実施形態において、全細胞、例えば、血小板を本発明のスクリーニング方法に用い、それにより、GPIIb/GPIIIaおよびその下流シグナル伝達のさらなる要素を提供する。この場合、検出されるシグナルは、血小板の形状の変化であり得る。
【0054】
試験化合物の存在下でのLIMK−1またはその類似体のGPIIbへの結合が試験化合物の非存在下での結合と異なる場合、試験化合物はGPIIbおよび/またはその下流シグナル伝達と相互作用している。シグナル伝達は、増加または減少、好ましくは減少し得る。下流シグナル伝達の検出される差は、少なくとも10%、好ましくは少なくとも20%、より好ましくは少なくとも50%、最も好ましくは少なくとも80%であり、試験化合物の存在下および非存在下でのシグナルの比として計算される。
【0055】
本発明のスクリーニング方法の他の実施形態において、試験化合物を化合物ライブラリーの形で供給する。化合物ライブラリーは、複数の化合物を含み、化学的に合成された分子および天然産物を含む複数の源のいずれかから集められ、あるいはコンビナトリアルケミストリー技術により得られたものであった。それらは、高処理スクリーニングに特に適している。それらは、特定の構造の化合物または植物のような特定の生物の化合物から構成されていてよい。本発明の状況において、化合物ライブラリーは、好ましくは、タンパク質およびポリペプチドまたは小分子を含むライブラリーである。
【0056】
本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法の他の実施形態において、前記GPIIbタンパク質または前記LIMK−1タンパク質への試験化合物の結合あるいはGPIIbおよび/またはその下流シグナル伝達に対するその影響を不均一または均一検定法により検出する。本明細書で用いているように、不均一検定法は、1つまたはそれ以上の洗浄工程を含む検定法であるが、均一検定法では、そのような洗浄工程は必要ではない。試薬および化合物を混合し、測定するのみである。
【0057】
好ましい実施形態において、不均一検定法は、ELISA(酵素結合免疫吸着検定法)、DELFIA(解離増強ランタニド蛍光免疫検定法)、SPA(シンチレーション近接検定法)またはフラッシュプレート検定法である。
【0058】
ELISA(酵素結合免疫吸着検定法)は、種々の会社によって供給されている。該検定法は、LIMK−1のようなキナーゼによりリン酸化することができるランダムペプチドを用いる。キナーゼを含む試料を通常、例えばATPおよび必須の陽イオンを含む反応緩衝液で希釈し、次いで、プレートウエルに加える。単に混合物を除去して、反応を停止させる。その後、プレートを洗浄する。例えば、ビオチン化基質をキナーゼに加えて、反応を開始させる。反応後、特異的抗体を加える。通常、試料をあらかじめブロックしたプロテインGプレートに移し、洗浄した後、例えば、ストレプトアビジン−HRPを加える。その後、非結合ストレプトアビジン−HRP(西洋ワサビペルオキシダーゼ)を除去し、ペルオキシダーゼ基質を加えて、ペルオキシダーゼ呈色反応を開始させ、適切なデンシトメーターで光学濃度を測定する。
【0059】
DELFIA(解離増強ランタニド蛍光免疫検定法)を用いる検定法は、固相検定法である。通常、抗体をユウロピウムまたは他のランタニドで標識し、非結合ユウロピウム標識抗体を洗い流した後、ユウロピウム蛍光を検出する。
【0060】
SPA(シンチレーション近接検定法)およびフラッシュプレート検定法は、通常、放射性標識基質を捕捉するためにビオチン/アビジン相互作用を利用する。一般的に、反応混合物は、キナーゼ、ビオチン化ペプチド基質およびγ−[P33]ATPを含む。反応後、ビオチン化ペプチドをストレプトアビジンにより捕捉する。SPA検出においては、ストレプトアビジンはシンチラントを含むビーズに結合し、一方、フラッシュプレート検出においては、ストレプトアビジンはシンチラントを含むマイクロプレートのウエルの内側に結合する。免疫化されたならば、放射性標識基質は、発光を刺激するのに十分にシンチラントに近い。
【0061】
他の好ましい実施形態において、均一検定法は、TR−FRET(時間分解蛍光共鳴エネルギー移動)検定法、FP(蛍光偏光)検定法、ALPHA(増幅ルミネセンス近接均一検定法)、EFC(酵素断片相補性)検定法または遺伝子検定法である。
【0062】
TR−FRET(時間分解蛍光共鳴エネルギー移動)に基づく検定法は、通常、Cy3/Cy5またはCy5/Cy7のような重複スペクトルを伴うユウロピウムとAPC、修飾アロフィコシアニンまたは他の色素との間の蛍光共鳴エネルギー移動を利用する検定法である(Schobel U.ら、(1999年)Bioconjugate Chem.、第10巻、1107〜1114頁)。例えば、337nmの光によるユウロピウムの励起後に、分子は620nmの蛍光を発する。しかし、この蛍光がAPCに十分に近い場合、ユウロピウムはその励起エネルギーをAPCに移動させ、APCは665nmの蛍光を発する。キナーゼ基質は、通常ビオチン標識基質である。キナーゼ反応の後に、ユウロピウム標識(P)特異抗体をストレプトアビジン−APCとともに加える。リン酸化ペプチドがユウロピウム標識抗体とストレプトアビジン−APCとを密接に接触させる。ユウロピウムフルオロフォアへのAPCの密接な近接により、APC蛍光のためにユウロピウム蛍光の消光がもたらされる(FRET)。
【0063】
蛍光偏光(FP)を用いる検定法は、溶液中の蛍光性基質ペプチドを励起するために偏光を用いる検定法である。これらの蛍光性ペプチドは、溶液中で遊離であり、回転して、放射光を偏光解消させる。しかし、基質ペプチドが(P)−Tyrのようなより大きい分子に結合しているときには、その回転率は著しく減少し、放射光は高度に偏光したままである。キナーゼの検定のために、一般的に以下の2つの選択肢がある。
(a)蛍光性ホスホペプチドトレーサーを(P)特異抗体に結合させる。リン酸化生成物が抗体における蛍光性ホスホペプチドと競合して、高から低への偏光の変化をもたらす。
(b)リン酸化基質ペプチドがリン特異的抗体に結合して、低から高への偏光の変化をもたらす。
【0064】
ALPHA(増幅ルミネセンス近接均一)に基づく検定法は、リン酸化ペプチドにより近接させたドナービーズとアクセプタービーズとの間の一重項酸素の移動に依拠する検定法である。680nmでの励起により、ドナービーズにおける光増感剤が周囲の酸素を一重項状態酸素に変換し、一重項状態酸素が200nmまでの距離に拡散する。アクセプタービーズにおけるケミルミネセンス基がビーズ内の蛍光性受容体にエネルギーを移動させ、次いで、蛍光性受容体が約600nmの光を放射する。
【0065】
EFC(酵素断片相補性)に基づく検定法または同等の検定法は、特に、化合物の高処理スクリーニングに用いることができる。EFC検定法は、酵素アクセプター(EA)と酵素ドナー(ED)の2つの断片からなる工学処理β−ガラクトシダーゼ酵素に基づいている。断片を分離すると、β−ガラクトシダーゼ活性はないが、断片が共存すると、結合(相補)して活性な酵素を形成する。EFC検定法は、分析物が抗体または受容体のような特異的結合性タンパク質によって認識され得る、ED−分析物複合体を用いるものである。特異的結合性タンパク質が存在しない場合、ED−分析物複合体は、EAを相補結合して活性なβ−ガラクトシダーゼを形成し、正のルミネセンスシグナルを発生することができる。ED−分析物複合体が特異的結合性タンパク質によって結合されている場合、EAとの相補結合が妨げられて、シグナルは存在しない。遊離分析物が供給される(試料中に)場合、遊離分析物は、特異的結合性タンパク質への結合についてED−分析物複合体と競合する。遊離分析物は、EAとの相補結合のためにED−分析物複合体を遊離させ、試料中に存在する遊離分析物の量に依存するシグナルを発生させる。
【0066】
また、他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、アレイにGPIIbまたはLIMK−1を結合させることによりアレイについて実施する。固相化学および光不安定性保護基を用いてそのようなアレイを調製する方法は、例えば、米国特許第5744305号明細書に開示されている。これらのアレイは、試験化合物または化合物ライブラリーと接触させ、相互作用、例えば、結合や構造変化について試験することもできる。
【0067】
なお、他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、全細胞を用いて実施する。血小板は血液ドナーから容易に入手することができるため、血小板をそのような試験に用いることが好ましい。あるいは、細胞株、場合によりトランスフェクト細胞株を用いることができる。そのような細胞株は、巨核球細胞株(例えば、DAMIまたはMEG−1)、HEK293細胞(初代ヒト胚腎)、3T3細胞(マウス胚線維芽細胞)、CHO細胞(チャイニーズハムスター卵巣)、COS−7細胞(アフリカミドリザル細胞株)、HeLa細胞(ヒト類上皮子宮頸癌)、JURKAT細胞(ヒトT細胞白血病)、BHK21細胞(ハムスター正常腎、線維芽細胞)およびMCF−7細胞(ヒト乳癌)を含むが、これらに限定されない。全細胞を用いることは、細胞内物質、酵素等を試験系に加える必要がないため、膜と比べて有利である。さらに、マルチウエルプレートに加えた全細胞は、高処理スクリーニング試験および自動化試験系に特に適している。
【0068】
他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、ロボット工学システムにおいて実施する。本発明の方法を、例えば、マイクロフルイディクス、すなわち、チャンネル構造を用いた、ロボットによる平板培養およびロボットによる液体輸送を含むロボット工学システムで実施することが有利である。
【0069】
他の実施形態において、本発明によるLIMK−1類似体またはLIMK−1リガンドをスクリーニングするためのスクリーニング方法は、高処理スクリーニングシステムの形で実施する。そのようなシステムでは、スクリーニング方法は、自動化かつ小型化されていることが有利である。特に、システムは、ロボットにより制御される小型化ウエルおよびマイクロフルイディクスを用いている。
【0070】
本発明の他の対象は、
(a)本発明によるスクリーニングする方法を実施する工程、
(b)検出された試験化合物の適切な量を供給する工程、および
(c)検出された試験化合物を上記で詳述したような1つまたはそれ以上の製薬上許容できる担体または補助物質を用いて製剤化する工程
を含む血栓形成または血液凝固疾患の予防または治療用の薬剤を製造する方法である。
【0071】
血栓形成または血液凝固疾患は、血栓形成または血液凝固の変化を伴う疾患である。健常者と比較して、血栓形成または血液凝固(の素因)が増加または減少し得る。血栓形成または血液凝固の亢進を伴う疾患は、当業者に知られており、例えば、アテローム血栓性疾患を含む。
【0072】
好ましい実施形態において、疾患はアテローム血栓性疾患である。
【0073】
本発明のより好ましい実施形態において、アテローム血栓性疾患は、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される。
【0074】
以下の図および表は、本発明の範囲を限定することなく、本発明をより詳細に説明するものである。
【発明を実施するための最良の形態】
【0075】
図1にGPIIb免疫沈降物の2D電気泳動によるLIMK1の検出を示す。対照IgG(図1A)またはGPIIbに対する特異抗体(図1B)によるヒト休止血小板抽出物(3.9mg)の免疫沈降物を、pH3〜10勾配で最初の段階においてIEF(等電点電気泳動)により分離した。その後、タンパク質を10%SDS−PAGE上で二次元で分離した。ゲルをコロイダルクマシー染色し、分析した。特異GPIIb抗体のみを含むゲル上の可視タンパク質スポットを切除し、トリプシンで消化し(ゲル中消化)、抽出し、MALDI−TOF MSにより分析した。
【0076】
図2にLIMK−1に対する抗体による免疫沈降後のインテグリンGPIIbの検出を示す。LIMK−1に対する特異抗体によるヒト血小板抽出物(2mg)ならびにヒト巨核球細胞株DAMIおよびMEG−01の免疫沈降物を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。インテグリンGPIIbを特異抗体を用いて検出した。
【0077】
図3にLIMK−1およびGPIIbに対する抗体による免疫沈降後のLIMK−1の検出を示す。LIMK−1およびGPIIbに対する特異抗体によるヒト血小板抽出物の免疫沈降物を10%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0078】
図4に血小板ならびにDAMIおよびMeg−01細胞の抽出物中のLIMK−1の検出を示す。ヒト血小板の抽出物からの等量のタンパク質(50μg)を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0079】
図5に種々のヒト組織の抽出物中のLIMK−1の検出を示す。ヒトの結腸、精巣、腎臓、心臓、血小板、肝臓、筋肉、皮膚、膵臓、脳および胸腺組織の抽出物からの等量のタンパク質を4〜12%SDS−PAGEにより分離した。タンパク質をエレクトロトランスファーによりニトロセルロース膜に移した。LIMK−1を特異抗体を用いて検出した。
【0080】
図6に種々のヒト組織における定量的Taqman分析によるLIMK−1転写物の検出を示す。ヒト骨髄、結腸、精巣、腎臓、心臓、血小板、肝臓、胎児肝臓、肺、乳腺、胎盤、前立腺、唾液腺、腸、脊髄、脾臓、胃、気管、子宮、筋肉、膵臓、脳、胎児脳および胸腺組織のmRNAをTaqman分析により分析した。
【実施例】
【0081】
1.材料および方法
1.1 血小板の準備
血小板は、血液銀行からの(4人のドナー)血小板濃縮製剤から準備した。すべての操作段階を室温で実施した。血小板濃縮製剤をタイロード緩衝液(pH7.4;137mmol/l NaCl、2.7mmol/l KCl、12mmol/l NaHCO3、0.36mmol/l NaH2PO4、1mmol/l MgCl2、10mmol/l HEPES、5.6mmol/lデキストロース)で希釈し、得られた懸濁液を制動せずに120×gで15分間遠心分離した。次いで、プロスタグランジンE1(PGE1 0.5μg/ml)を上清に加え、混合物を室温で5分間インキュベートし、制動せずに650×gで15分間遠心分離した。上清を捨て、ペレットをタイロード緩衝液(0.1%BSAを含む)に再懸濁し、PGE1(0.5μg/ml)を加えた。混合物を室温で5分間インキュベートし、その後、制動せずに650×gで5分間遠心分離した。上清を捨て、ペレットをタイロード緩衝液(BSAを含まない)に再懸濁し、PGE1(0.25μg/ml)を加え、混合物をインキュベートし(室温で5分間)、遠心分離した(制動せずに650×gで15分間)。再び上清を捨て、ペレットをタイロード緩衝液(BSAを含まない)に再懸濁し、細胞を液体窒素で凍結し、次いで、使用時まで−80℃で保存した。
【0082】
1.2 細胞培養
ヒトDAMIおよびMeg−01巨核細胞株の培養物を10%ウシ胎児血清(FCS)およびペニシリン/ストレプトマイシンを含むダルベッコの変法イーグル培地(DMEM)(37℃、5%CO2)中で増殖させた。細胞を洗浄し、収集し、細胞のペレットを、抽出物の調製の前に−80℃で保存した。
【0083】
1.3 免疫沈降のための血小板の処理
凍結血小板ペレットを1〜2mlの溶解緩衝液(下を参照)中で解凍し、次いで、ピペッティングにより上下にかき混ぜた後、音波処理(10パルス、30%)により再懸濁した。血小板を遠心分離して(5分、3000×g)、上清中の可溶性たんぱく質の濃縮のために前分画を行った。
【0084】
図1で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH8):10mmol/lトリス、140mmol/lNaCl、1%トリトン、0.05%SDS、1tab.Complete、200μmol/lペファブロック(4−(2−アミノエチル)−ベンゼンスルホニルフルオリド塩酸塩))
洗浄緩衝液(pH8):10mmol/lトリス、140mmol/lNaCl、1%トリトン
図2で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH7.5):50mmol/lトリス、50mmol/lNaCl、0.5%トリトン、0.05%SDS、1mmol/lNa3VO4、1tab.Complete、1μg/mlペプスタチン、5mmol/lNaF
洗浄緩衝液(pH7.5):50mmol/lトリス、50mmol/lNaCl、0.1%トリトン
図3で用いた溶解および洗浄緩衝液:
溶解緩衝液(pH7.5):50mmol/lトリス、150mmol/lNaCl、1%トリトン、1mmol/lNa3VO4、1tab.Complete、1μg/mlペプスタチン、5mmol/lNaF
洗浄緩衝液(pH7.5):50mmol/lトリス、150mmol/lNaCl、0.1%トリトン
【0085】
1.4 免疫沈降
プロテインA−セファロース(Amersham Biosciences、Uppsala、Sweden)ならびにプロテインG−アガロース(Roche Diagnostics GmbH、Mannheim、Germany)を、抗体の種類によって免疫沈降に用いた。すべての操作段階を4℃で行った。
【0086】
ビーズを使用前に洗浄緩衝液(上を参照)で3回洗浄した。血小板溶解物をビーズ材料とともに1時間インキュベートしてあらかじめ清澄にし(図1および2、図3ではなし)、その後、遠心分離した(3分、500×g)。血小板溶解物を抗体とともに1時間インキュベートし、免疫沈降のためにビーズを一夜加えた。その後、ビーズを洗浄緩衝液(上記参照)で3回洗浄し、IEF再水和緩衝液(2Dゲル分析用)または溶解緩衝液(1Dゲル分析用)とともにインキュベートして溶出を行った。
【0087】
1.5 抗体の仕様
GPIIb(Santa Cruz Biotecnology,Inc.、Santa Cruz、Califronia、USA、カタログ番号sc−7310)、免疫沈降用(プロテインA)
GPIIb(Biotrend Chemikalien GmbH、Cologne、Germany、カタログ番号6065)、ウエスタンブロット(1:1000)用 LIMK(Santa Cruz Biotechnology,Inc.、Santa Cruz、Califronia、USA、カタログ番号sc−5576)、免疫沈降用(プロテインG)およびウエスタンブロット(1:250)用
【0088】
1.6 2Dゲル電気泳動
1次元ゲル電気泳動は、Biorad Protean IEF Cell(IPGストリップ:11cm、pH3〜10)(Biorad Laboratories、Hercules、CA、USA)および次のIEF再水和緩衝液(8mol/l尿素、0.5%CHAPS、10mmol/lDTT、0.2%Biolyte、0.001%ブロモフェノールブルー)を用いて行った。能動的再水和のために、185μlの容積の試料を用いてIPGストリップを50Vで一夜放置した。IEF(等電点電気泳動)は、直線ランプ、最終電圧8000Vおよび合計少なくとも40000Vhrsで行った。2次元ゲル電気泳動の前に還元/アルキル化を各平衡緩衝液(pH8.8、6mol/l尿素、2%SDS、0.375mol/lトリス、20%グリセロール、130mmol/lDTTまたは135mmol/lヨードアセタミド)中で10分間行った。
2次元ゲル電気泳動は、Criterion4〜20%ゲルを用い、ランニング緩衝液として25mmol/lトリス、192mmol/lグリシン、0.1%SDSを用いて150Vで行った。
【0089】
1.7 1Dゲル電気泳動
1Dゲル電気泳動は、X−Cell Sure Lock System(Novex−system、Invitrogen GmbH、Karlsruhe、Germany)ならびにランニング緩衝液としてのMOPS−SDSおよび150Vの電圧を用いて行った。
【0090】
1.8 エレクトロトランスファー/ウエスタンブロット(Semidry)
エレクトロトランスファーのために、Multiphor II System(不連続緩衝液システム)(Amersham、Biosciencies、Uppsala、Sweden)を用いた。陽極緩衝液1は0.3oil/lトリス、20%メタノール、陽極緩衝液2は25mmol/lトリス、20%メタノール、陰極緩衝液は40mmol/lアミノカプロン酸、0.01%SDS、20%メタノールであった。
タンパク質を0.8mA/cm2を用いてニトロセルロースシート(Schleicher&Schuell BioScience GmbH、Dassel/Relliehausen、Germany、Protran BA85 0.45μm)上にブロットした。
【0091】
1.9 染色技術
ゲルはコロイダルクマシー(Neuhoffら、1988年)または銀(Blumら、1987年の後に修正)で染色した。修正された銀染色は、ゲルを40%エタノール/10%酢酸で1時間固定し、30%エタノールで2回、水で1回各20分間洗浄し、0.02%Na2S2O3で1分間増感し、水(3×20秒)で洗浄し、冷0.1%AgNO3中で4℃で20分間インキュベートし、水(1×1分よりも3×20秒)で洗浄し、3%NaCO3/0.05%ホルマリンで現像し、水中でもう1回20秒間洗浄した。最後に5%酢酸で染色を完了した。
【0092】
1.10 画像化
抗体は、Hyperfilm ECL(Amersham Biosciences、Uppsala、Sweden、カタログ番号RPN2132)への5分間の曝露後にケミルミネセンス(ECL plus、Amersham Biosciences、Uppsala、Sweden、カタログ番号RPN2132)により検出し、Adefo−Developer(00009)およびAdefo−Fixator(00062)(ADEFO−CHEMIE GmbH、Nuernberg、Germany)により現像した。
【0093】
1.11 ゲル中消化
ゲル中消化は、Pandeyら、2000年に従って行った。簡単に述べると、ゲルスポットを手作業により切り取り、分解のためにトリプシン(ブタ、Promega GmbH、Mannheim、Germany、カタログ番号V511A)を用いた。
【0094】
1.12 MALDI−TOF質量分析
MALDI−TOF質量分析は、Voyager DE−STR MALDI−TOFワークステーション(Applied Biosystems、Foster City、CA、USA)を用いて行った。試料を乾燥小滴法により調製し、□−シアノヒドロキシケイ皮酸(アセトニトリル/トリフルオロ酢酸中3mg/ml)をマトリックスとして用いて2×96ウエルテフロン被覆試料プレート上にスポットした。700〜4000Daの検出範囲で行った。
【0095】
1.13 タンパク質の定量
試料のタンパク質含量は、製造業者の指示(562nmでの検出)に従ってBCAキット(Pierce Chemical Company、Rockford、Illinois、USA)を用いて測定した。
【0096】
1.14 組織分布パターン
レーンにつき50μgのタンパク質をゲル上にのせた。種々のヒト組織をBioCat GmbH(Heidelberg、Germany)から購入した。血小板および巨核球からのLIMKの検出は、上清中であらかじめ濃縮して促進した(図4および図5についてそれぞれ15000×g、15分間または3000×g、5分間遠心分離)。
【0097】
1.15 Taqman分析
増幅のために、以下のPCRプロトコールを用いた。増幅のための次の試薬のすべてがApplied Biosystems(Foster City、USA)により供給された。20ngのゲノムDNA、1単位のTaqGoldポリメラーゼ、1×Taqポリメラーゼ緩衝液、500μMのdNTP、2.5mmol/lのMgCl2、200nmol/lの各増幅プライマー対、H2O ad5μl
PCRの増幅プログラム
95℃×10分 ×1サイクル
95℃×30秒
70℃×30秒 ×2サイクル
95℃×30秒
65℃×30秒 ×2サイクル
95℃×30秒
60℃×30秒 ×2サイクル
95℃×30秒
56℃×30秒
72℃×30秒 ×40サイクル
72℃×10分
4℃×30秒 ×1サイクル
【0098】
1.16 装置
実験は、CM5チップおよびNi−NTAチップを装着したBiacore 3000最高性能研究システム(Biacore International SA、79111 Freiburg、Germany)を用いて行った。
【0099】
結果
2.1 LIMK−1とインテグリンGPIIbとの相互作用
本発明者らは、GPIIbに対する市販の特異抗体との共同免疫沈降を用いて、GPIIbとのタンパク質複合体を濃縮した。これらの複合体は、2次元ゲル電気泳動により分離した(図1)。この実験で得られた結果は、LIMK−1はヒト血小板抽出物中のインテグリンGPIIbと共同沈降することを示している。
【0100】
これらの結果を検証するために、本発明者らは、ウエスタン分析によるGPIIbのその後の検出のため、LIMK−1に対する市販の特異抗体による血小板抽出物ならびにヒト巨核球細胞株DAMIおよびMEG−01の免疫沈降を用いた(図2)。これらの結果は、インテグリンGPIIbがヒト血小板中のLIMK−1と会合するという知見を確認するものであった。ヒト巨核球細胞株DAMIおよびMEG−01においてはGPIIbとLIMK−1との相互作用は示されなかった。これらの巨核球細胞株は、分化後に血小板を生産する前駆細胞株である(Fugmanら、1990年、George、2000年、Greenbergら、1988年、Oguraら、1988年、Schickら、1998年、Takeuchiら、1998年、Vittetら、1992年)。しかし、本発明者らの実験のために本発明者らはこれらの細胞株を未分化形として用いた。これらの未分化巨核球細胞株におけるGPIIbインテグリンの発現レベルを分析したとき、本発明者らはGPIIbの発現はこれらの細胞株においては血小板における発現と比較してはるかに低いことを見いだした。これは、本発明者らがこれらの細胞株におけるインテグリンとLIMK−1との相互作用を検出しなかった理由の説明となっている。
【0101】
本発明者らの結果をさらに検証するために、本発明者らは、後続のウエスタン分析によるLIMK−1の検出のためにLIMK−1およびGPIIbに対する市販の特異抗体による血小板抽出物の免疫沈降を用いた(図3)。再び、これらの結果は、インテグリンGPIIbがヒト血小板中のLIMK−1と会合しているという本発明者らの以前の知見を確認するものであった。
【0102】
2.2 LIMK−1の組織分布
本発明者らは、LIMK−1に対する特異抗体を用いて血小板ならびに巨核球細胞株DAMIおよびMEG−01におけるLIMK−1の発現を分析した(図4)。このように、本発明者らはLIMK−1に対する特異抗体を用いて、血小板ならびに巨核球細胞株DAMIおよびMEG−01におけるLIMK−1の発現を確認した。
【0103】
さらに、種々のヒト組織におけるLIMK−1の発現を分析するために、本発明者らは後続のウエスタン分析のためにヒト組織抽出物をゲル電気泳動により分離した(図5)。これらの結果は、LIMK−1が精巣、心臓、血小板、筋肉および脳組織において発現することを示している。発現は血小板に対して絶対的に特異的でないが、LIMK−1はすべての組織に普遍的には発現しない。この知見は、脳、胎児脳および精巣におけるLIMK−1の発現を示しているTaqman分析(図6)により確認されている。血小板は、このTaqmanパネルには示さない。
【図面の簡単な説明】
【0104】
【図1】GPIIb免疫沈降物の2D電気泳動によるLIMK1の検出を示す。
【図2】LIMK−1に対する抗体による免疫沈降後のインテグリンGPIIbの検出を示す。
【図3】LIMK−1およびGPIIbに対する抗体による免疫沈降後のLIMK−1の検出を示す。
【図4】血小板ならびにDAMIおよびMeg−01細胞の抽出物中のLIMK−1の検出を示す。
【図5】種々のヒト組織の抽出物中のLIMK−1の検出を示す。
【図6】種々のヒト組織における定量的Taqman分析によるLIMK−1転写物の検出を示す。
【特許請求の範囲】
【請求項1】
GPIIbへの結合のためのLIMK−1またはLIMK−1類似体の使用。
【請求項2】
LIMK−1類似体がGPIIb/IIIa下流シグナル伝達の活性化物質または阻害物質、好ましくはその阻害物質である請求項1に記載の使用。
【請求項3】
GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のための請求項1または2に記載の使用。
【請求項4】
GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のためのLIMK−1リガンドの使用。
【請求項5】
LIMK−1リガンドがLIMK−1アゴニストまたはLIMK−1アンタゴニスト、好ましくはLIMK−1アンタゴニストである請求項4に記載の使用。
【請求項6】
血栓形成または血液凝固疾患の予防または治療用薬剤の製造のためのLIMK−1、LIMK−1類似体またはLIMK−1リガンドの使用。
【請求項7】
LIMK−1類似体がGPIIb/IIIa下流シグナル伝達の阻害物質であるか、あるいはLIMK−1リガンドがLIMK−1アンタゴニストである請求項6に記載の使用。
【請求項8】
疾患が血栓形成または血液凝固の増加に関連する疾患である請求項6または7に記載の使用。
【請求項9】
疾患がアテローム血栓性疾患である請求項6〜8のいずれか1項に記載の使用。
【請求項10】
疾患が、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される請求項6〜9のいずれか1項に記載の使用。
【請求項11】
(a)GPIIbタンパク質および場合によりその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(b)試験化合物を供給する工程、および
(c)GPIIbタンパク質への試験化合物の結合を検出する工程
を含み、試験化合物がLIMK−1由来である、LIMK−1類似体をスクリーニングする方法。
【請求項12】
工程(c)の代わりに
(c’)GPIIb/IIIaおよび/またはその下流シグナル伝達の活性化または阻害を検出する工程
を含む、請求項11に記載の方法。
【請求項13】
(a)LIMK−1タンパク質を供給する工程、
(b)試験化合物を供給する工程、および
(c)LIMK−1タンパク質への試験化合物の結合を検出する工程
を含む、LIMK−1リガンドをスクリーニングする方法。
【請求項14】
工程(c)の代わりに、
(c’)LIMK−1またはGPII/IIIa下流シグナル伝達の活性化または阻害、好ましくは、その活性化を検出する工程
をさらに含む請求項13に記載の方法。
【請求項15】
(a)LIMK−1タンパク質を供給する工程、
(b)GPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(c)試験化合物を供給する工程、および
(d)試験化合物とLIMK−1タンパク質との相互作用を検出する工程
を含む、GPIIb/IIIaおよび/またはその下流シグナル伝達カスケードと相互作用する試験化合物をスクリーニングする方法。
【請求項16】
試験化合物を化合物ライブラリーの形で供給する請求項13〜15のいずれか1項に記載の方法。
【請求項17】
GPIIbタンパク質もしくはLIMK−1タンパク質への試験化合物の結合またはGPIIbおよび/もしくはその下流シグナル伝達に対するその影響を不均一または均一検定法で検出する請求項11〜16のいずれか1項に記載の方法。
【請求項18】
不均一検定法がELISA(酵素結合免疫吸着検定法)、DELFIA(解離増強ランタニド蛍光免疫検定法)またはSPA(シンチレーション近接検定法)である請求項17に記載の方法。
【請求項19】
均一検定法がTR−FRET(時間分解蛍光共鳴エネルギー移動)検定法、FP(蛍光偏光)検定法、ALPHA(増幅ルミネセンス近接均一検定法)、EFC(酵素断片相補性)検定法である請求項17に記載の方法。
【請求項20】
アレイで実施する請求項11〜19のいずれか1項に記載の方法。
【請求項21】
全細胞を用いて実施する請求項11〜17のいずれか1項に記載の方法。
【請求項22】
ロボット工学システムで実施する請求項11〜21のいずれか1項に記載の方法。
【請求項23】
マイクロフルイディクスを用いて実施する請求項11〜22のいずれか1項に記載の方法。
【請求項24】
LIMK−1類似体またはLIMK−1リガンドの高処理スクリーニングの方法である請求項11〜23のいずれか1項に記載の方法。
【請求項25】
(a)請求項11〜24のいずれか1項に記載の方法を実施する工程、
(b)検出された化合物の適切な量を供給する工程、および
(c)検出された試験化合物を1つまたはそれ以上の製薬上許容できる担体または補助物質を用いて製剤化する工程
を含む血栓形成または血液凝固疾患の予防または治療用の薬剤を製造する方法。
【請求項26】
疾患がアテローム血栓性疾患である請求項25に記載の方法。
【請求項27】
アテローム血栓性疾患が心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される請求項26に記載の方法。
【請求項1】
GPIIbへの結合のためのLIMK−1またはLIMK−1類似体の使用。
【請求項2】
LIMK−1類似体がGPIIb/IIIa下流シグナル伝達の活性化物質または阻害物質、好ましくはその阻害物質である請求項1に記載の使用。
【請求項3】
GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のための請求項1または2に記載の使用。
【請求項4】
GPIIb/IIIa下流シグナル伝達の活性化または阻害、好ましくはその阻害のためのLIMK−1リガンドの使用。
【請求項5】
LIMK−1リガンドがLIMK−1アゴニストまたはLIMK−1アンタゴニスト、好ましくはLIMK−1アンタゴニストである請求項4に記載の使用。
【請求項6】
血栓形成または血液凝固疾患の予防または治療用薬剤の製造のためのLIMK−1、LIMK−1類似体またはLIMK−1リガンドの使用。
【請求項7】
LIMK−1類似体がGPIIb/IIIa下流シグナル伝達の阻害物質であるか、あるいはLIMK−1リガンドがLIMK−1アンタゴニストである請求項6に記載の使用。
【請求項8】
疾患が血栓形成または血液凝固の増加に関連する疾患である請求項6または7に記載の使用。
【請求項9】
疾患がアテローム血栓性疾患である請求項6〜8のいずれか1項に記載の使用。
【請求項10】
疾患が、心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される請求項6〜9のいずれか1項に記載の使用。
【請求項11】
(a)GPIIbタンパク質および場合によりその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(b)試験化合物を供給する工程、および
(c)GPIIbタンパク質への試験化合物の結合を検出する工程
を含み、試験化合物がLIMK−1由来である、LIMK−1類似体をスクリーニングする方法。
【請求項12】
工程(c)の代わりに
(c’)GPIIb/IIIaおよび/またはその下流シグナル伝達の活性化または阻害を検出する工程
を含む、請求項11に記載の方法。
【請求項13】
(a)LIMK−1タンパク質を供給する工程、
(b)試験化合物を供給する工程、および
(c)LIMK−1タンパク質への試験化合物の結合を検出する工程
を含む、LIMK−1リガンドをスクリーニングする方法。
【請求項14】
工程(c)の代わりに、
(c’)LIMK−1またはGPII/IIIa下流シグナル伝達の活性化または阻害、好ましくは、その活性化を検出する工程
をさらに含む請求項13に記載の方法。
【請求項15】
(a)LIMK−1タンパク質を供給する工程、
(b)GPIIbおよび/またはその下流シグナル伝達の少なくとも1つの要素を供給する工程、
(c)試験化合物を供給する工程、および
(d)試験化合物とLIMK−1タンパク質との相互作用を検出する工程
を含む、GPIIb/IIIaおよび/またはその下流シグナル伝達カスケードと相互作用する試験化合物をスクリーニングする方法。
【請求項16】
試験化合物を化合物ライブラリーの形で供給する請求項13〜15のいずれか1項に記載の方法。
【請求項17】
GPIIbタンパク質もしくはLIMK−1タンパク質への試験化合物の結合またはGPIIbおよび/もしくはその下流シグナル伝達に対するその影響を不均一または均一検定法で検出する請求項11〜16のいずれか1項に記載の方法。
【請求項18】
不均一検定法がELISA(酵素結合免疫吸着検定法)、DELFIA(解離増強ランタニド蛍光免疫検定法)またはSPA(シンチレーション近接検定法)である請求項17に記載の方法。
【請求項19】
均一検定法がTR−FRET(時間分解蛍光共鳴エネルギー移動)検定法、FP(蛍光偏光)検定法、ALPHA(増幅ルミネセンス近接均一検定法)、EFC(酵素断片相補性)検定法である請求項17に記載の方法。
【請求項20】
アレイで実施する請求項11〜19のいずれか1項に記載の方法。
【請求項21】
全細胞を用いて実施する請求項11〜17のいずれか1項に記載の方法。
【請求項22】
ロボット工学システムで実施する請求項11〜21のいずれか1項に記載の方法。
【請求項23】
マイクロフルイディクスを用いて実施する請求項11〜22のいずれか1項に記載の方法。
【請求項24】
LIMK−1類似体またはLIMK−1リガンドの高処理スクリーニングの方法である請求項11〜23のいずれか1項に記載の方法。
【請求項25】
(a)請求項11〜24のいずれか1項に記載の方法を実施する工程、
(b)検出された化合物の適切な量を供給する工程、および
(c)検出された試験化合物を1つまたはそれ以上の製薬上許容できる担体または補助物質を用いて製剤化する工程
を含む血栓形成または血液凝固疾患の予防または治療用の薬剤を製造する方法。
【請求項26】
疾患がアテローム血栓性疾患である請求項25に記載の方法。
【請求項27】
アテローム血栓性疾患が心筋梗塞、不安定狭心症、急性冠動脈症候群、冠動脈疾患、冠動脈血栓溶解後の再閉塞、血栓形成術中の閉塞および冠動脈再狭窄、卒中、一過性虚血性発作、肺塞栓症、左室機能不全、心血管および脳血管疾患を有する患者における臨床的血管性合併症の二次的予防、アテローム性動脈硬化症、血管インターベンショナル戦略に対する同時投薬からなる群から選択される請求項26に記載の方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2007−520456(P2007−520456A)
【公表日】平成19年7月26日(2007.7.26)
【国際特許分類】
【出願番号】特願2006−541840(P2006−541840)
【出願日】平成16年11月23日(2004.11.23)
【国際出願番号】PCT/EP2004/013274
【国際公開番号】WO2005/053734
【国際公開日】平成17年6月16日(2005.6.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
【公表日】平成19年7月26日(2007.7.26)
【国際特許分類】
【出願日】平成16年11月23日(2004.11.23)
【国際出願番号】PCT/EP2004/013274
【国際公開番号】WO2005/053734
【国際公開日】平成17年6月16日(2005.6.16)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
[ Back to top ]