
血液型抗原融合ポリペプチドおよびその使用方法
【課題】本発明は、抗体媒介性移植拒絶を処置または予防するための、組成物および方法を提供することを課題とする。
【解決手段】本発明は、血液型エピトープが、高密度で、かつムチン型タンパク質骨格の異なるコア糖鎖によって特異的に発現され得るという発見に一部基づく。ポリペプチドは、本明細書中でABO融合ポリペプチドと称される。1つの局面において、本発明は、第1ポリペプチドを含む融合ポリペプチドを提供し、この第1ポリペプチドは、ムチンポリペプチドの少なくとも1領域を含み、第2ポリペプチドに作動可能に連結されるα1,2フコシルトランスフェラーゼによってグリコシル化される。第1ポリペプチドは、α1,3 N−アセチルガラクトサミニルトランスフェラーゼおよび/またはα1,3 ガラクトシルトランスフェラーゼによって連続的にグリコシル化される。ムチンポリペプチドは、例えば、PSGL−1である。
【解決手段】本発明は、血液型エピトープが、高密度で、かつムチン型タンパク質骨格の異なるコア糖鎖によって特異的に発現され得るという発見に一部基づく。ポリペプチドは、本明細書中でABO融合ポリペプチドと称される。1つの局面において、本発明は、第1ポリペプチドを含む融合ポリペプチドを提供し、この第1ポリペプチドは、ムチンポリペプチドの少なくとも1領域を含み、第2ポリペプチドに作動可能に連結されるα1,2フコシルトランスフェラーゼによってグリコシル化される。第1ポリペプチドは、α1,3 N−アセチルガラクトサミニルトランスフェラーゼおよび/またはα1,3 ガラクトシルトランスフェラーゼによって連続的にグリコシル化される。ムチンポリペプチドは、例えば、PSGL−1である。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、概して抗体媒介性移植拒絶を処置または予防するための組成物および方法に
関し、より具体的には、血液型(blood group)決定基を含む融合ポリペプチ
ドを含む組成物に関する。
【背景技術】
【0002】
(発明の背景)
主要なヒトの血液型系(組織−血液型ABO系)は、3つの糖質決定基(血液型Aエピ
トープ、BエピトープおよびHエピトープ)によって規定される。ABH決定基を有する
グリカンは、糖タンパク質、糖脂質、または遊離のオリゴ糖において見出される。ABH
抗原は、N連結グリカンまたはO連結グリカンにおいて見出され得る。O連結グリカンに
関してこれまで記載された最も共通なコア構造は、コア1(Galβ3GalNAc)、
コア2(Galβ3(GlcNAcβ6)GalNAc)、コア3(GlcNAcβ3G
alNAc)およびコア4(GlcNAcβ3(GlcNAcβ6)GalNAc)であ
る。これらは、1型(Galβ3GlcNAc)構造、2型(Galβ4GlcNAc)
構造、および3型(Galβ3GalNAcα)構造を保有することが示されている。1
型構造は主に、コア3構造およびコア4構造の伸長物として見出されるが、2型鎖(ポリ
ラクトサミン)は、コア2構造のGlcNAcβ1,6分枝の伸長物として発見された。
【0003】
ABO障壁を越える移植(Tx)は、通常、組織Txを避ける。なぜなら、実施する抗
体に起因する抗体媒介性拒絶(AMR)の危険性がしばしば高いからである。これはまた
、骨髄移植において適用され得るが、血液型ABHの不一致はその結果に影響しないと長
い間考えられてきた。しかし、いくつかの場合、ABO障害を越えて移植することが依然
として所望される。これは、ドナーの総数が増加しなくても、特定のレシピエントに対し
て利用可能なドナーのプールを広げるからである。
【0004】
体外免疫吸収(EIA)または血漿交換(PP)による抗A型抗体または抗B型抗体の
除去は、ABO不適合性組織Tx後の移植片の生存を改善することが証明された。ABO
不適合性Txおよび異種Txの両方におけるAMRの予防のために使用される別の方法は
、遊離オリゴ糖の注入であるが、遊離糖に対する抗体の低い親和性および循環における低
分子量のオリゴ糖の短い半減期(Yeら、1994;Simonら、1998)は、より
広い使用を抑制する。
【発明の概要】
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、血液型エピトープが、高密度で、かつムチン型タンパク質骨格の異なるコア
糖鎖によって特異的に発現され得るという発見に一部基づく。ポリペプチドは、本明細書
中でABO融合ポリペプチドと称される。したがって、本発明は以下を提供する。
(1)
第2ポリペプチドに作動可能に連結される第1ポリペプチドを含む融合ポリペプチドであ
って、該第1ポリペプチドは:
(a)ムチンポリペプチドであり、そして
(b)α1,2フコシルトランスフェラーゼによってグリコシル化され、
該第2ポリペプチドは、免疫グロブリンポリペプチドの少なくとも1領域を含む、融合
ポリペプチド。
(2)
前記第1ポリペプチドが、α1,3 N−アセチルガラクトサミニルトランスフェラーゼ
またはα1,3 ガラクトシルトランスフェラーゼによってさらにグリコシル化される、
請求項1に記載の融合ポリペプチド。
(3)
前記α1,2フコシルトランスフェラーゼが、血液型Hまたは分泌型α1,2フコシルト
ランスフェラーゼである、請求項1に記載の融合ポリペプチド。
(4)
前記第1ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも1領域
を含む、請求項1に記載の融合ポリペプチド。
(5)
前記第1ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含む
、請求項4に記載の融合ポリペプチド。
(6)
前記第2ポリペプチドが、重鎖免疫グロブリンポリペプチドの領域を含む、請求項1に記
載の融合ポリペプチド。
(7)
前記第2ポリペプチドが、免疫グロブリン重鎖のFc領域を含む、請求項6に記載の融合
ポリペプチド。
(8)
ダイマーである、請求項1に記載の融合ポリペプチド。
(9)
請求項1に記載のペプチドをコードする、単離された核酸。
(10)
請求項9に記載の核酸を含む、ベクター。
(11)
請求項11に記載のベクターを含む、細胞。
(12)
請求項1に記載の融合ポリペプチドを含む、吸収体。
(13)
抗体媒介性拒絶を処置または予防する必要のある被験体において、抗体媒介性拒絶を処置
または予防するための方法であって、該方法は、以下:
(a)融合ポリペプチド複合体を形成するために、該被験体に由来する生物学的サンプ
ルと、請求項1に記載の融合ポリペプチドとを接触させる工程;および
(b)該生物学的サンプルから該複合体を除去することによって、該被験体における抗
体媒介性拒絶を処置または予防する工程
を包含する、方法。
(14)
生物学的サンプルから抗体を除去する方法であって、該方法は、以下:
(a)融合ポリペプチド複合体を形成するために、生物学的サンプルと、請求項1に記
載の融合ポリペプチドとを接触させる工程;
(b)該生物学的サンプルから該複合体を除去することによって、該生物学的サンプル
から該抗体を除去する工程
を包含する、方法。
【0006】
1つの局面において、本発明は、第1ポリペプチドを含む融合ポリペプチドを提供し、
この第1ポリペプチドは、ムチンポリペプチドの少なくとも1領域を含み、第2ポリペプ
チドに作動可能に連結されるα1,2フコシルトランスフェラーゼによってグリコシル化
される。第1ポリペプチドは、α1,3 N−アセチルガラクトサミニルトランスフェラ
ーゼおよび/またはα1,3 ガラクトシルトランスフェラーゼによって連続的にグリコ
シル化される。ムチンポリペプチドは、例えば、PSGL−1である。好ましくは、この
ムチンポリペプチドは、PSGL−1の細胞外部分である。α1,2,フコシルトランス
フェラーゼは、例えば、血液型Hまたは分泌型α1,2,フクシルトランスフェラーゼ(
例えば、FUT1またはFUT2)である。
【0007】
好ましい実施形態において、第2ポリペプチドは、免疫グロブリンポリペプチドの少な
くとも1領域を含む。例えば、第2ポリペプチドは、重鎖免疫グロブリンポリペプチドの
1領域を含む。あるいは、第2ポリペプチドは、免疫グロブリン重鎖のFC領域を含む。
【0008】
ABO融合ポリペプチドは多量体(mutimer)である。好ましくは、ABO融合
ポリペプチドはダイマーである。
【0009】
本発明はまた、ABO融合ポリペプチドをコードする核酸、ならびに本明細書中に記載
される核酸をコードするABO融合ポリペプチドを含むベクター、および本明細書中に記
載されるベクターまたは核酸を含む細胞を含む。あるいは、ベクターは、α1,2,フコ
シルトランスフェラーゼおよび/またはα1,3 Nアセチルガラクトサミニルトランス
フェラーゼまたはα1,3 ガラクトシルトランスフェラーゼをコードする核酸をさらに
含む。
【0010】
別の局面において、本発明は、被験体における抗体媒介性拒絶を処置する方法を提供す
る。この方法は、融合ポリペプチド−抗体複合体を形成させるために、生物学的サンプル
(例えば、患者に由来する全血または血漿)と、本発明のABO融合ポリペプチドとを接
触させる工程を包含する。この複合体は、生物学的サンプルから除去され、そしてこの生
物学的サンプルは被験体に再融合される。
【0011】
抗体融合ペプチド複合体を形成し、そして生物学的サンプルからこの複合体を除去する
ために、サンプルと本発明のABO融合ペプチドとを接触させる工程によりこのサンプル
から抗体を除去する方法もまた、本発明に含まれる。
【0012】
ABO融合ポリペプチドを含む薬学的組成物もまた、本発明に含まれる。
【0013】
別の記載がない限り、本明細書中で使用される全ての技術および専門用語は、本発明に
属する当業者によって一般的に理解されるのと同じ意味を有する。本明細書中に記載され
るのと類似または等価な方法および材料は、本発明の実施または試験において使用され得
るが、適切な方法および材料は、以下に記載される。本明細書中に記載される全ての特許
刊行物、特許出願、特許および他の参考文献は、その全体が参考として援用される。矛盾
する場合、明細書(定義を含む)が制御する。さらに、材料、方法および実施例は、例示
のみであり、制限を意図するものではない。本発明の他の特徴および利点は、以下の詳細な説明、および特許請求の範囲から示され
る。
【図面の簡単な説明】
【0014】
【図1】図1A〜Dは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされた293T細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型H鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、CDM8(レーン1および5)、PSGL−1/mIgG2b(レーン2および6)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン7)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Eにおいて、CDM8およびPSGL−1/mIgG2bでトランスフェクトされた細胞由来の重複したサンプルは、省略した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図2】図2A〜Dは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされた293T細胞において産生された、PNGaseF処理された、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。免疫精製したPSGL−1/mIgG2bのPNGaseF処理あり(+)またはなし(−)の後、このPSGL−1/mIgG2bを、8%SDS−PAGEで分離し、そしてニトロセルロース膜へブロッティングした。抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(C)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(D)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、PSGL−1/mIgG2bおよびH遺伝子(レーン1および2)、PSGL−1/mIgG2b、ならびにH遺伝子およびA遺伝子(レーン3および4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン5および6)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン7および8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。
【図3】図3A〜Eは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされたCOS−7m6細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型H鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、CDM8(レーン1および5)、PSGL−1/mIgG2b(レーン2および6)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン7)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Eにおいて、CDM8およびPSGL−1/mIgG2bでトランスフェクトされた細胞由来の重複したサンプルは、省略した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図4】図4A〜Eは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされたCHO−K1細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型A鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。CDM8(レーン1)、PSGL−1/mIgG2b(レーン2)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン5)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン6)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図5】FUT1またはFUT2をα1,3GalNAcTと同時発現するCOS細胞、CHO細胞および293T(2931)細胞において産生された組換えPSGL−1/mIgG2bに対する相対的血液型A密度を示す、棒グラフである。密度は、血液型AとmIgGとの反応性についての容積の比として計算し、そしてこの値を、FUT1およびα1,3GalNAcTを同時発現するCOS細胞において産生されるA置換PSGL−1/mIgG2bについて得られた値に対して正規化した。
【図6】異なる血液型A置換吸収剤に対する、吸収後に血清中に残った抗血液型Aの反応性を示す折れ線グラフである。A−PAA−ビオチンコーティングされたELISAによって測定されるような、非吸収血液型O血清の割合での、残存する反応性を、使用した吸収剤の量に対してプロットした。傾向線を、対数回帰を使用して計算した。円は、FUT2およびα1,3GalNAcTを有するCHO細胞において産生されたPSGL−1/mIgG2bを示し、そして四角は、A−PAA−MPGを介して連結されたAトリサッカリドを示す。B−PAA−MPGを介して連結されたBトリサッカリドを使用して、全ての吸収において同じ量のPAA−MPGを得た。
【発明を実施するための形態】
【0015】
(発明の詳細な説明)
本発明は、血液型エピトープが、高密度で特異的に、そしてムチン型タンパク質骨格上
の異なるコアサッカリド鎖で発現され得るという発見に一部基づく。より高い密度の血液
型エピトープは、遊離のサッカリドまたは固相に結合したAB決定因子と比較して、抗血
液型抗体の結合の増加または除去(すなわち、吸収)を生じる。
【0016】
本発明は、抗血液型抗体の吸収剤として有用な複数の血液型エピトープを含む、ムチン
−免疫グロブリン融合タンパク質(本明細書中で「ABO融合タンパク質」という)を提
供する。ABO融合ペプチドはまた、グリコシル化についての研究のためのモデルタンパ
ク質として有用である。例えば、このABO融合タンパク質は、ABO不適合性の臓器移
植または骨髄移植の前に、血液または血漿由来のレシピエント抗血液型ABO抗体を排除
する際に、有用である。このABO融合タンパク質は、レシピエントの血液または血漿由
来の抗血液型ABO抗体の50%、60%、70%、80%、90%、95%、98%、
または100%を吸収する。
【0017】
ABO融合ペプチドは、野生型AB決定因子の遊離サッカリドと比較して、抗血液型抗
体を除去または結合する際に、炭水化物モル濃度基準で、より有効である。このABO融
合ペプチドは、等量の野生型AB決定因子の遊離サッカリドと比較して、2倍、4倍、1
0倍、20倍、50倍、80倍、100倍またはそれ以上多い数の抗血液型抗体と結合す
る。
【0018】
本発明のABO融合タンパク質は、血液型決定因子に特異的なエピトープを保有する。
例えば、ABO融合タンパク質は、Aエピトープ、BエピトープまたはHエピトープのい
ずれかを保有する。あるいは、このABO融合物は、血液型抗原についての2つのエピト
ープを保有する。例えば、このABO融合タンパク質は、AエピトープおよびBエピトー
プの両方を保有する。いくつかの局面において、このABO融合タンパク質は、3つ全て
のエピトープ(すなわち、A、BおよびH)を保有する。
【0019】
(融合ポリペプチド)
種々の局面において、本発明は、第二のポリペプチドと作動可能に連結した、糖タンパ
ク質(例えば、ムチンポリペプチド)の少なくとも一部を含む第一のポリペプチドを含む
融合タンパク質を提供する。本明細書中で使用される場合、「融合タンパク質」または「
キメラタンパク質」は、非ムチンポリペプチドと作動可能に連結したムチンポリペプチド
の少なくとも一部を含む。「ムチンポリペプチド」は、ムチンドメインを有するポリペプ
チドをいう。このムチンポリペプチドは、1個、2個、3個、5個、10個、20個また
はそれ以上のムチンドメインを有する。このムチンポリペプチドは、O−グリカンで置換
されたアミノ酸配列によって特徴付けられる任意の糖タンパク質である。例えば、ムチン
ポリペプチドは、2アミノ酸毎または3アミノ酸毎に、セリンまたはスレオニンであるア
ミノ酸を有する。このムチンポリペプチドは、分泌タンパク質である。あるいは、ムチン
ポリペプチドは、細胞表面タンパク質である。
【0020】
ムチンドメインは、アミノ酸のスレオニン、セリンおよびプロリンに富んでおり、ここ
で、オリゴサッカリドは、ヒドロキシアミノ酸(O−グリカン)に、N−アセチルガラク
トサミンを介して連結される。ムチンドメインは、O−連結グリコシル化部位を含むか、
またはO−連結グリコシル化部位からなる。ムチンドメインは、1個、2個、3個、5個
、10個、20個、50個、100個またはそれ以上のO−連結グリコシル化部位を有す
る。あるいは、ムチンドメインは、N−連結グリコシル化部位を含むか、またはO−連結
グリコシル化部位からなる。ムチンポリペプチドは、その質量の50%、60%、80%
、90%、95%または100%がグリカンに帰する。ムチンポリペプチドは、MUC遺
伝子(すなわち、MUC1、MUC2、MUC3など)によりコードされる任意のポリペ
プチドである。あるいは、ムチンポリペプチドは、P−セレクチン糖タンパク質リガンド
1(PSGL−1)、CD34、CD43、CD45、CD96、GlyCAM−1、M
AdCAMまたは赤血球糖タンパク質である。好ましくは、このムチンは、PSGL−1
である。一方、「非ムチンポリペプチド」は、その質量の少なくとも40%未満がグリカ
ンに帰するポリペプチドをいう。
【0021】
本発明のABO融合タンパク質において、このムチンポリペプチドは、ムチンタンパク
質の全てまたは一部に対応し得る。1実施形態において、ABO融合タンパク質は、ムチ
ンタンパク質の少なくとも一部を含む。「少なくとも一部」は、このムチンポリペプチド
が、少なくとも1つのムチンドメイン(例えば、O−連結グリコシル化部位)を含むこと
を意味する。1実施形態において、このムチンタンパク質は、このポリペプチドの細胞外
部分を含む。例えば、このムチンポリペプチドは、PSGL−1の細胞外部分を含む。
【0022】
第一のポリペプチドは、1以上の血液型トランスフェラーゼによってグリコシル化され
る。第一のポリペプチドは、2個、3個、5個またはそれ以上の血液型トランスフェラー
ゼによってグリコシル化される。グリコシル化は、連続的または継続的である。あるいは
、グリコシル化は、同時発生的またはランダム(すなわち、特定の順序はない)である。
例えば、第一のポリペプチドは、α1,2フコシルトランスフェラーゼ(例えば、H遺伝
子またはSe遺伝子によってコードされるα1,2フコシルトランスフェラーゼ)によっ
てグリコシル化される。例示的なα1,2フコシルトランスフェラーゼは、FUT1(G
enBank登録番号Q10984;O10983;O10981;AT455028お
よびNM00148)およびFUT2(GenBank登録番号P19526;BAA1
1638;D82933およびA56098)である。あるいは、第一のポリペプチドは
、1,3N−アセチルガラクトサミニルトランスフェラーゼまたはα1,3アセチルガラ
クトサミニルトランスフェラーゼによってグリコシル化される。いくつかの局面において
、第一のポリペプチドは、α1,2フコシルトランスフェラーゼおよび1,3N−アセチ
ルガラクトサミニルトランスフェラーゼまたはα1,3アセチルガラクトサミニルトラン
スフェラーゼの両方によってグリコシル化される。
【0023】
融合タンパク質において、用語「作動可能に連結される」は、第一および第二のポリペ
プチドが、第一のポリペプチドのO−連結グリコシル化を可能にする様式で、(最も代表
的にはペプチド結合のような共有結合を介して)化学的に連結されることを示すことが意
図される。融合ポリペプチドをコードする核酸をいうために使用される場合、用語、作動
可能に連結される、は、ムチンポリペプチドおよび非ムチンポリペプチドをコードする核
酸が、互いにインフレームで融合していることを意味する。この非ムチンポリペプチドは
、ムチンポリペプチドのN末端またはC末端に融合され得る。
【0024】
さらなる実施形態において、このABO融合タンパク質は、1以上のさらなる部分に連
結され得る。例えば、このABO融合タンパク質は、ABO融合タンパク質配列がGST
(すなわち、グルタチオンS−トランスフェラーゼ)配列のN末端に連結された、GST
融合タンパク質にさらに連結され得る。このような融合タンパク質は、ABO融合タンパ
ク質の精製を容易にし得る。あるいは、ABO融合タンパク質は、固体支持体にさらに連
結され得る。種々の固体支持体が当業者に公知である。このような組成物は、抗血液型抗
体の除去を容易にし得る。例えば、ABO融合タンパク質は、例えば、金属化合物、シリ
カ、ラテックス、ポリマー物質から作製される粒子;マイクロタイタープレート;ニトロ
セルロースもしくはナイロンまたはこれらの組み合わせに連結され得る。固体支持体に連
結されたABO融合タンパク質は、吸収剤として使用されて、生物学的サンプル(例えば
、血液または血漿)から、抗血液型抗体を除去する。
【0025】
別の実施形態では、融合タンパク質は、異種シグナル配列(すなわち、ムチン核酸によ
ってコードされるポリペプチド中に存在しないポリペプチド配列)をそのN末端に含む。
例えば、ネイティブなムチンシグナル配列は除去され得、そして別のタンパク質由来のシ
グナル配列で置き換えられ得る。特定の宿主細胞(例えば、哺乳動物宿主細胞)では、ポ
リペプチドの発現および/または分泌は、異種シグナル配列の使用を通して増大され得る
。
【0026】
本発明のキメラタンパク質または融合タンパク質は、標準的な組換えDNA技術によっ
て産生され得る。例えば、種々のポリペプチド配列をコードするDNAフラグメントは、
従来技術に従って、例えば、連結のための平滑末端化末端もしくは付着末端化末端、適切
な末端を提供する制限酵素消化、必要に応じて粘着末端のフィルイン、望ましくない連結
を回避するアルカリホスファターゼ処理、および酵素的連結を用いることによって、イン
フレームで一緒に連結される。別の実施形態では、融合遺伝子は、自動化DNA合成機を
含む、従来技術によって合成され得る。あるいは、遺伝子フラグメントのPCR増幅は、
その後アニーリングおよび再増幅されてキメラ遺伝子配列を作製し得る、2つの連続した
遺伝子フラグメントの間での相補的突出部を生じる、アンカープライマーを用いて実施さ
れ得る(例えば、Ausubelら(編)CURRENT PROTOCOLS IN
MOLECULAR BIOLOGY,John Wiley & Sons,1992
を参照のこと)。さらに、融合部分(例えば、免疫グロブリン重鎖のFc領域)をコード
する多くの発現ベクターが市販されている。糖タンパク質Ibαコード核酸は、その融合
部分が、免疫グロブリンタンパク質に対してインフレームで連結されるように、このよう
な発現ベクター中にクローニングされ得る。
【0027】
ABO融合ポリペプチドは、オリゴマー(例えば、ダイマー、トリマーまたはペンタマ
ー)として存在し得る。好ましくは、ABO融合ポリペプチドは、ダイマーである。
【0028】
第1ポリペプチド(および/または第1ポリペプチドをコードする核酸)は、当該分野
で公知であるムチンコード配列を用いて構築され得る。ムチンポリペプチドおよびムチン
ポリペプチドをコードする核酸についての適切な供給源としては、GenBank登録番
号NP663625およびNM145650、CAD10625およびAJ417815
、XP140694およびXM140694、XP006867およびXM006867
、ならびにNP00331777およびNM009151がそれぞれ挙げられ、そしてそ
れらの全体が本明細書中に参考として援用される。
【0029】
いくつかの実施形態では、ムチンポリペプチド部分は、天然に存在するムチン配列(野
生型)中に(非変異配列と比較して)増大した糖質含量をもたらす変異を有する改変体ム
チンポリペプチドとして提供される。例えば、改変体ムチンポリペプチドは、野生型ムチ
ンと比較してさらなるO結合型グリコシル化部位を含んでいた。あるいは、改変体ムチン
ポリペプチドは、野生型ムチンポリペプチドと比較して増加した数のセリン残基、トレオ
ニン残基またはプロリン残基をもたらす、アミノ酸配列変異を含む。この増加した糖質含
量は、当業者に公知の方法によって、ムチンのタンパク質対糖質の比率を決定することに
よって評価され得る。
【0030】
いくつかの実施形態では、ムチンポリペプチド部分は、天然に存在するムチン配列(野
生型)中に、タンパク質分解に対して(非変異配列と比較して)より耐性のムチン配列を
もたらす変異を有する改変体ムチンポリペプチドとして提供される。
【0031】
いくつかの実施形態では、第1ポリペプチドは、全長PSGL−1を含む。あるいは、
第1ポリペプチドは、全長未満のPSGL−1ポリペプチド(例えば、PSGL−1の細
胞外部分)を含む。例えば、400アミノ酸長未満(例えば、300アミノ酸長以下、2
50アミノ酸長以下、150アミノ酸長以下、100アミノ酸長以下、50アミノ酸長以
下または25アミノ酸長以下)の第1ポリペプチド。例示的なPSGL−1ポリペプチド
配列およびPSGL−1核酸配列としては、GenBank登録番号XP006867;
XM006867;XP140694およびXM140694が挙げられる。
【0032】
第2ポリペプチドは好ましくは可溶性である。いくつかの実施形態では、第2ポリペプ
チドは、第2ムチンポリペプチドとのABO融合ポリペプチドの会合を容易にする配列を
含む。好ましい実施形態では、第2ポリペプチドは、免疫グロブリンポリペプチドの少な
くとも一領域を含む。「少なくとも一領域」は、免疫グロブリン分子の任意の部分(例え
ば、軽鎖、重鎖、FC領域、Fab領域、Fv領域またはそれらの任意のフラグメント)
を含むことを意味する。免疫グロブリン融合ポリペプチドは、当該分野で公知であり、そ
して例えば、米国特許第5,516,964号;同第5,225,538号;同第5,4
28,130号;同第5,514,582号;同第5,714,147号;および同第5
,455,165号に記載される。
【0033】
いくつかの実施形態では、第2ポリペプチドは、全長免疫グロブリンポリペプチドを含
む。あるいは、第2ポリペプチドは、全長未満の免疫グロブリンポリペプチド(例えば、
重鎖、軽鎖、Fab、Fab2、Fv、またはFc)を含む。好ましくは、第2ポリペプ
チドは、免疫グロブリンポリペプチドの重鎖を含む。より好ましくは、第2ポリペプチド
は、免疫グロブリンポリペプチドのFc領域を含む。
【0034】
本発明の別の局面では、第2ポリペプチドは、野生型免疫グロブリン重鎖のFc領域の
エフェクター機能よりも劣ったエフェクター機能を有する。Fcエフェクター機能として
は、例えば、Fcレセプター結合、補体結合およびT細胞枯渇活性が挙げられる(例えば
、米国特許第6,136,310号を参照のこと)。T細胞枯渇活性、Fcエフェクター
機能、および抗体安定性をアッセイする方法は、当該分野で公知である。1つの実施形態
では、第2ポリペプチドは、Fcレセプターに対する親和性が低いかまたはない。代替的
な実施形態では、第2ポリペプチドは、補体タンパク質C1qに対する親和性が低いかま
たはない。
【0035】
本発明の別の局面は、ムチンポリペプチド、またはその誘導体、フラグメント、アナロ
グもしくはホモログをコードする核酸を含む、ベクター(好ましくは、発現ベクター)に
関する。種々の局面では、このベクターは、免疫グロブリンポリペプチド、またはその誘
導体、フラグメント、アナログもしくはホモログをコードする核酸に作動可能に連結した
、ムチンポリペプチドをコードする核酸を含む。さらに、このベクターは、血液型トラン
スフェラーゼ(例えば、α1,2フコシルトランスフェラーゼ、α1,3Nアセチルガラ
クトサミニルトランスフェラーゼ(α1,3N acetylgalactosamin
inytransferase)、α1,3ガラクトシルトランスフェラーゼ、またはこ
れらの任意の組み合わせ)をコードする核酸を含む。血液型トランスフェラーゼは、AB
O融合タンパク質のムチン部分のペプチド(peptode)骨格に対する血液型決定因
子の付加を促進する。本明細書中で用いられる場合、用語「ベクター」とは、連結されて
いる別の核酸分子を輸送し得る核酸分子をいう。ベクターの1つの型は、「プラスミド」
であり、プラスミドとは、さらなるDNAセグメントが連結され得る環状二本鎖DNAル
ープをいう。ベクターの別の型は、ウイルスベクターであり、ここで、さらなるDNAセ
グメントがウイルスゲノムに連結され得る。特定のベクターは、これらが導入される宿主
細胞中で自律的複製をし得る(例えば、細菌の複製起点を有する細菌ベクターおよびエピ
ソームの哺乳動物ベクターを有する)。他のベクター(例えば、非エピソームの哺乳動物
ベクター)は、宿主細胞へと導入される際に、宿主細胞のゲノムへと組み込まれ、それに
よって、宿主ゲノムと共に複製される。さらに、特定のベクターは、それらが作動可能に
連結される遺伝子の発現を指向し得る。このようなベクターは、本明細書中で、「発現ベ
クター」といわれる。一般に、組換えDNA技術において有用な発現ベクターは、頻繁に
、プラスミドの形態である。本明細書中で、「プラスミド」および「ベクター」は、交換
可能に用いられ得る。なぜならば、プラスミドは、最も一般的に用いられるベクターの形
態だからである。しかし、本発明は、等価な機能を果たす、ウイルスベクター(例えば、
複製欠損のレトロウイルス、アデノウイルスおよびアデノ随伴ウイルス)のような、発現
ベクターのこのような他の形態を含むことを意図する。
【0036】
本発明の組換え発現ベクターは、宿主における核酸の発現に適切な形態で、本発明の核
酸分子を含み、このことは、組換え発現ベクターが、発現に用いられる宿主細胞に基づい
て選択された(すなわち、発現される核酸配列に作動可能に連結された)、1つ以上の調
節配列を含むことを意味する。組換え発現ベクターにおいて、「作動可能に連結した(さ
れる)」とは、目的のヌクレオチド配列が、このヌクレオチド配列の発現を可能にする様
式で(例えば、インビトロ転写/翻訳システムにおいて、またはベクターが宿主細胞に導
入される場合、宿主細胞において)、調節配列に連結されるという意味が意図される。
【0037】
用語「調節配列」は、プロモーター、エンハンサーおよび他の発現制御エレメント(例
えば、ポリアデニル化シグナル)を含むことが意図される。このような調節配列は、例え
ば、Goeddl,GENE EXPRESSION TECHNOLOGY:METH
ODS IN ENZYMOLOGY 185,Academic Press,San
Diego,Calif.(1990)に記載される。調節配列としては、宿主細胞の
多くの型においてヌクレオチド配列の構成的発現を指向する配列、および特定の宿主細胞
においてのみヌクレオチド配列の発現を指向する配列(例えば、組織特異的調節配列)が
挙げられる。発現ベクターの設計は、例えば、形質転換される宿主細胞の選択、所望され
るタンパク質の発現のレベルなどの因子に依存し得ることが当業者により理解される。本
発明の発現ベクターは、宿主細胞へと導入されて、それにより、本明細書中に記載される
ような核酸によりコードされる、融合タンパク質もしくは融合ペプチド(例えば、ABO
融合ポリペプチド、ABO融合ポリペプチドの変異形態など)を含む、タンパク質もしく
はペプチドを産生し得る。
【0038】
本発明の組換え発現ベクターは、原核生物細胞または真核生物細胞におけるABO融合
ポリペプチドの発現のために設計され得る。例えば、ABO融合ポリペプチドは、Esc
herichia coliのような細菌細胞、昆虫細胞(バキュロウイルス発現ベクタ
ーを用いる)、酵母細胞または哺乳動物細胞において発現され得る。適切な宿主細胞は、
Goeddle,GENE EXPRESSION TECHNOLOGY:METHO
DS IN ENZYMOLOGY 185,Academic Press,San
Diego,Calif.(1990)にさらに考察される。あるいは、組換え発現ベク
ターは、例えば、T7プロモーター調節配列およびT7ポリメラーゼを用いて、インビト
ロで転写および翻訳され得る。
【0039】
原核生物におけるタンパク質の発現は、最も頻繁に、融合タンパク質または非融合タン
パク質のいずれかの発現を指向する構成的プロモーターまたは誘導性プロモーターを含む
ベクターを用いて、Escherichia coliで実施される。融合ベクターは、
それらの中でコードされるタンパク質に、通常は、組換えタンパク質のアミノ末端に、多
数のアミノ酸を付加する。このような融合ベクターは、代表的には、以下の3つの目的を
果たす:(i)組換えタンパク質の発現を増大させること;(ii)組換えタンパク質の
可溶性を増大させること;および(iii)アフィニティー精製におけるリガンドとして
作用することにより、組換えタンパク質の精製を補助すること。頻繁に、融合発現ベクタ
ーにおいて、タンパク質分解切断部位が、融合部分と組み換えタンパク質との接合部に導
入されて、融合タンパク質の精製、引き続く、この融合部分からの組換えタンパク質の分
離を可能にする。このような酵素、およびこれらの同族の認識配列としては、第Xa因子
、トロンビンおよびエンテロキナーゼが挙げられる。代表的な融合発現ベクターとしては
、pGEX(Pharmacia Biotech Inc;SmithおよびJohn
son,1988.Gene 67:31−40)、pMAL(New England
Biolabs,Beverly,Mass.)、およびpRIT5(Pharmac
ia,Piscataway,N.J.)が挙げられ、これらは、標的組換えタンパク質
に対して、それぞれ、グルタチオンS−トランスフェラーゼ(GST)、マルトースE結
合タンパク質、またはプロテインAを融合する。
【0040】
適切な誘導性非融合E.coli発現ベクターの例としては、pTrc(Amrann
ら(1988)Gene 69:301−305)およびpET 11d(Studie
rら、GENE EXPRESSION TECHNOLOGY:METHODS IN
ENZYMOLOGY 185,Academic Press,San Diego
,Calif.(1990)60−89)が挙げられる。
【0041】
E.coli中の組換えタンパク質発現を最小にする1つのストラテジーは、組み換え
タンパク質をタンパク質分解により切断する能力が損なわれた宿主細菌において、タンパ
ク質を発現することである。例えば、Gottesman,GENE EXPRESSI
ON TECHNOLOGY:METHODS IN ENZYMOLOGY 185,
Academic Press,San Diego,Calif.(1990)119
−128を参照のこと。別のストラテジーは、各アミノ酸についての個々のコドンが、E
.coliにおいて優先的に利用されるものであるように、発現ベクターへと挿入される
核酸の核酸配列を変化させることである(例えば、Wadaら、1992.Nucl.A
cids Res.20:2111−2118を参照のこと)。本発明の核酸配列のこの
ような変更は、標準的なDNA合成技術により実行され得る。
【0042】
別の実施形態において、ABO融合ポリペプチド発現ベクターは、酵母発現ベクターで
ある。酵母Saccharomyces cerivisaeにおける発現のためのベク
ターの例としては、pYepSec1(Baldariら、1987.EMBO J.6
:229−234)、pMFa(KurjanおよびHerskowitz,1982.
Cell 30:933−943)、pJRY88(Schultzら、1987.Ge
ne 54:113−123)、pYES2(Invitrogen Corporat
ion,San Diego,Calif.)およびpicZ(InVitrogen
Corp,San Diego,Calif.)が挙げられる。
【0043】
あるいは、ABO融合ポリペプチドは、バキュロウイルス発現ベクターを使用して、昆
虫細胞において発現され得る。培養昆虫細胞(例えば、SF9細胞)におけるタンパク質
の発現のために利用可能なバキュロウイルスベクターとしては、pAcシリーズ(Smi
thら、1983.Mol.Cell.Biol.3:2156−2165)およびpV
Lシリーズ(LucklowおよびSummers,1989.Virology 17
0:31−39)が挙げられる。
【0044】
さらに別の実施形態において、本発明の核酸は、哺乳動物発現ベクターを使用して、哺
乳動物細胞において発現される。哺乳動物発現ベクターの例としては、pCDM8(Se
ed,1987.Nature 329:840)およびpMT2PC(Kaufman
ら、1987.EMBO J.6:187−195)が挙げられる。哺乳動物細胞におい
て使用される場合、この発現ベクターの制御機能は、しばしば、ウイルス調節エレメント
により提供される。例えば、一般に使用されるプロモーターは、ポリオーマ、アデノウイ
ルス2、サイトメガロウイルスおよびシミアンウイルス40由来である。原核生物細胞お
よび真核生物細胞の両方に適切な他の発現系については、例えば、Sambrookら、
MOLECULAR CLONING:A LABORATORY MANUAL.第2
版、Cold Spring Harbor Laboratory,Cold Spr
ing Harbor Laboratory Press,Cold Spring
Harbor,N.Y.,1989の第16章および17章を参照のこと。
【0045】
本発明の別の局面は、本発明の組換え発現ベクターが導入された宿主細胞に関する。用
語「宿主細胞」および「組換え宿主細胞」とは、本明細書中で交換可能に使用される。こ
のような用語は、特定の被験体細胞を言及するだけでなく、このような細胞の子孫または
潜在的な子孫も言及することが理解される。特定の改変は、変異の影響または環境的な影
響のいずれかに起因して、次世代で生じ得るので、このような子孫は、実際のところ、親
細胞と同一ではないかもしれないが、本明細書中で使用されるような用語の範囲内になお
含まれる。
【0046】
宿主細胞は、任意の原核動物細胞または真核生物細胞であり得る。例えば、糖タンパク
質Ibα融合ポリペプチドは、細菌細胞(例えば、E.coli)、昆虫細胞、酵母細胞
または哺乳動物細胞(例えば、ヒト細胞、チャイニーズハムスター卵巣細胞(CHO)も
しくはCOS細胞)において発現され得る。他の適切な宿主細胞が、当業者に公知である
。
【0047】
ベクターDNAは、従来の形質転換技術またはトランスフェクション技術によって、原
核生物細胞または真核生物細胞に導入され得る。本明細書中で使用される場合、用語「形
質転換」および「トランスフェクション」は、異種核酸(例えば、DNA)を宿主細胞に
導入するための当該分野で認められた種々の技術をいうことが意図され、これらの技術と
しては、リン酸カルシウム共沈殿もしくは塩化カルシウム共沈殿、DEAE−デキストラ
ン媒介性トランスフェクション、リポフェクションまたはエレクトロポレーションが挙げ
られる。宿主細胞を形質転換またはトランスフェクションするための適切な方法は、Sa
mbrookら(MOLECULAR CLONING:A LABORATORY M
ANUAL.第2版、Cold Spring Harbor Laboratory,
Cold Spring Harbor Laboratory Press,Cold
Spring Harbor,N.Y.,1989)、および他の研究用マニュアルに
見出され得る。
【0048】
哺乳動物細胞の安定なトランスフェクションについて、使用される発現ベクターおよび
トランスフェクション技術に依存して、わずかな細胞しか異種DNAをそのゲノムに組み
込み得ないことが知られている。これらの要素を同定および選択するために、選択マーカ
ー(例えば、抗生物質に対する耐性)をコードする遺伝子は、一般的に、目的の遺伝子と
共に宿主細胞に導入される。種々の選択マーカーとしては、薬物に対する耐性を与えるマ
ーカー(例えば、G418、ハイグロマイシンおよびメトトレキサート)が挙げられる。
選択マーカーをコードする核酸は、糖タンパク質Ibα融合ポリペプチドをコードするベ
クターと同じベクターを用いて宿主細胞に導入され得るか、または別個のベクターを用い
て導入され得る。導入された核酸で安定にトランスフェクトされた細胞は、薬物選択によ
り同定され得る(例えば、選択マーカー遺伝子を組み込まれた細胞は生き残り、一方、他
の細胞は、死ぬ)。
【0049】
培養物中の本発明の宿主細胞(例えば、原核生物宿主細胞または真核生物宿主細胞)は
、ABO融合ポリペプチドを産生(すなわち、発現)するために使用され得る。従って、
本発明はさらに、本発明の宿主細胞を使用して、ABO融合ポリペプチドを産生するため
の方法を提供する。一実施形態において、この方法は、本発明の宿主細胞(この宿主細胞
に、ABO融合ポリペプチドをコードする組換え発現ベクターが導入されている)を、適
切な培地中で、ABO融合ポリペプチドが産生するように、培養する工程を包含する。別
の実施形態において、この方法はさらに、この培地または宿主細胞からABOポリペプチ
ドを単離する工程を包含する。
【0050】
ABO融合ポリペプチドは、従来の条件(例えば、抽出、沈殿、クロマトグラフィー、
アフィニティクロマトグラフィー、電気泳動など)に従って、単離および精製され得る。
例えば、免疫グロブリン融合タンパク質は、この融合タンパク質のFc部分に選択的に結
合する固定化されたプロテインAまたはプロテインGを含むカラムに溶液を通すことによ
り、精製され得る。例えば、Reis,K.J.ら、J.Immunol.132:30
98−3102(1984)PCT出願公開番号WO87/00329を参照のこと。こ
の融合ポリペプチドは、カオトロピック塩での処理によって、または酢酸水溶液(1M)
での溶出によって、溶出され得る。
【0051】
あるいは、本発明に従うABO融合ポリペプチドは、当該分野で公知の方法を使用して
、化学的に合成され得る。ポリペプチドの化学合成は、例えば、以下に記載される。種々
のタンパク質合成方法が、当該分野で公知であり、これにはペプチド合成機を使用する合
成が挙げられる。例えば、Peptide Chemistry,A Practica
l Textbook,Bodasnsky編、Springer−Verlag,19
88;Merrifield,Science 232:241−247(1986);
Baranyら、Intl.J.Peptide Protein Res.30:70
5−739(1987);Kent,Ann.Rev.Biochem.57:957−
989(1988)、およびKaiserら、Science 243:187−198
(1989)を参照のこと。このポリペプチドは、化学前駆体も他の化学物質も実質的に
含まなくなるように、標準的なペプチド精製技術を使用して精製される。用語「化学前駆
体も他の化学物質も実質的に含まない」は、ペプチドが、このペプチドの合成に関与する
化学前駆体または他の化学物質から分離されているペプチドの調製物を含む。一実施形態
において、用語「化学前駆体も他の化学物質も実質的に含まない」は、約30%(乾燥重
量)未満の化学前駆体または非ペプチド化学物質、より好ましくは約20%未満の化学前
駆体または非ペプチド化学物質、さらにより好ましくは約10%未満の化学前駆体または
非ペプチド化学物質、最も好ましくは約5%未満の化学前駆体または非ペプチド化学物質
を有するペプチドの調製物を含む。
【0052】
ポリペプチドの化学合成は、改変されたかまたは非天然のアミノ酸(D−アミノ酸が挙
げられる)および他の有機低分子の組み込みを容易にする。対応するD−アミノ酸アイソ
フォームでの、ペプチド中の1つ以上のL−アミノ酸の置換は、酵素的加水分解に対する
ペプチドの耐性を増加するため、および生物学的に活性なペプチドの1つ以上の特性(す
なわち、レセプター結合、機能的効力または作用の持続時間)を増強するために、使用さ
れ得る。例えば、Dohertyら、1993.J.Med.Chem.36:2585
−2594;Kirbyら、1993.J.Med.Chem.36:3802−380
8;Moritaら、1994.FEBS Lett.353:84−88;Wangら
、1993.Int.J.Pept.Protein Res.42:392−399;
FauchereおよびThiunieau,1992.Adv.Drug Res.2
3:127−159を参照のこと。
【0053】
ペプチド配列への供給架橋の導入は、ポリペプチド骨格を立体配置的かつ組織分布的に
制約し得る。このストラテジーは、増加した効力、選択性および安定性を有する融合ポリ
ペプチドのペプチドアナログを開発するために使用され得る。環状ペプチドの構造エント
ロピーは、その直線状対応物の構造エントロピーより低いので、特定のコンフォメーショ
ンの採用は、非環式アナログのエントロピーの減少よりも小さい環状アナログのエントロ
ピーの減少を伴って生じ、それにより、結合の自由エネルギーをより有利にする。大環状
化は、しばしば、ペプチドのN末端とC末端との間、側鎖とN末端もしくはC末端との間
[例えば、pH8.5においてK3Fe(CN)6を用いた場合](Samsonら、E
ndocrinology,137:5182−5185(1996))、または2つの
アミノ酸側鎖の間のアミド結合の形成によって達成される。例えば、DeGrado,A
dv Protein Chem,39:51−124(1988)を参照のこと。ジス
ルフィド結合もまた、直線状配列に組み込まれ、その可撓性を低下させる。例えば、Ro
seら、Adv Protein Chem,37:1−109(1985);Mosb
ergら、Biochem Biophys Res Commun,106:505−
512(1982)を参照のこと。さらに、ペニシラミン(Pen,3−メルカプト−(
D)バリン)でのシステイン残基の置換は、あるオピオイド−レセプター相互作用の選択
性を増加するために使用されている。LipkowskiおよびCarr,Peptid
es:Synthesis,Structures,and Applications
,Gutte編、Academic Press 287−320頁(1995)。
【0054】
(抗体媒介性移植片拒絶の処置または予防の方法)
抗体媒介性移植片拒絶(AMR)(例えば、臓器移植拒絶)を処置または予防する方法
もまた、本発明に含まれる。このような移植片としては、腎臓、肝臓、皮膚、膵臓、角膜
または心臓が挙げられるが、これらに限定されない。AMRとは、レシピエントによる任
意の抗体媒介性移植片拒絶を含むことを意味する。この方法は、被験体由来の生物学的サ
ンプルを本発明のABO融合ペプチドと接触させる工程を包含する。この生物学的サンプ
ルは、例えば、血液(すなわち、全血または血漿)である。このサンプルは、抗体(例え
ば、抗血液型抗体)を含むことが知られているか、またはこのような抗体を含むと疑われ
ている。いくつかの局面において、この生物学的サンプルは、ABO融合ポリペプチドと
このサンプルとを接触させる前に、被験体から取り出される。この生物学的サンプルは、
ABO融合ペプチド−抗血液型抗体複合体の形成を可能にする条件下で、ABO融合ペプ
チドと接触される。このABO融合ペプチド複合体は、存在する場合、抗血液型抗体を排
除するためにこの生物学的サンプルから分離され、そしてこの生物学的サンプルは、被験
体に再注入される。AMRはまた、本発明のABO融合ポリペプチドを被験体に投与する
ことによって、処置または予防される。
【0055】
被験体は、例えば、任意の哺乳動物(例えば、ヒト、霊長類、マウス、ラット、イヌ、
ネコ、ウシ、ウマ、ブタ)であり得る。処置は、被験体がABO不適合性移植片を受ける
前に、施される。あるいは、処置は、被験体がABO不適合性移植片を受けた後に、施さ
れる。
【0056】
生物学的サンプルは、当業者に公知の方法によって、ABO融合タンパク質と接触され
る。例えば、血漿しゃ血または体外免疫吸収。
【0057】
本質的に、抗体媒介性反応と病原学的に関連する任意の障害は、予防または処置を受け
やすいとみなされる。AMRは、臓器移植の生存率が、本発明の方法により処置されない
臓器移植の生存率よりも大きい場合、処置または予防される。移植の生存率とは、移植片
がレシピエントにより拒絶される前の時間を意味する。例えば、AMRは、移植片が、移
植後少なくとも1、2、4または8週間生存する場合、処置または予防される。好ましく
は、この移植片は、3、6、13ヶ月生存する。より好ましくは、この移植片は、2、3
、5年またはそれ以上生存する。
【0058】
(サンプルから抗血液型抗体を除去する方法)
サンプルから抗血液型抗体を除去または枯渇する方法もまた、本発明に含まれる。この
サンプルは、生物学的流体(例えば、血液または血漿)である。あるいは、このサンプル
は、生物学的組織(例えば、心臓組織、肝臓組織、皮膚または腎臓組織)である。この方
法は、サンプルと本発明のABO融合ペプチドを接触させる工程を包含する。このサンプ
ルは、ABO融合ペプチド−抗血液型抗体複合体の形成を可能にする条件下で、ABO融
合ペプチドと接触される。このABO融合ペプチド−抗体複合体は、存在する場合、抗血
液型抗体を除去または枯渇するために、この生物学的サンプルから分離される。
【0059】
(ABO融合ポリペプチドまたはこれらをコードする核酸を含む薬学的組成物)
本発明のABO融合タンパク質、またはこれらの融合タンパク質をコードする核酸分子
(本明細書中において、「治療剤」または「活性化合物」とも呼ばれる)、ならびにこれ
らの誘導体、フラグメント、アナログおよびホモログは、投与のために適切な薬学的組成
物に混合され得る。このような組成物は、代表的に、核酸分子、タンパク質、または抗体
および薬学的に受容可能なキャリアを含む。本明細書中において使用される場合、「薬学
的に受容可能なキャリア」は、薬学的投与に適合性の、任意の全ての溶媒、分散媒体、コ
ーティング、抗菌剤および抗真菌剤、等張性剤および吸収遅延剤などを含むことが意図さ
れる。適切なキャリアは、当該分野における標準的な参考教科書である、Remingt
on’s Pharmaceutical Sciencesの最新版(本明細書中にお
いて参考として援用される)に記載される。このようなキャリアまたは希釈剤の好ましい
例としては、限定しないが、水、生理食塩水、フィンガー溶液(finger’s so
lution)、デキストロース溶液、および5%ヒト血清アルブミンが挙げられる。リ
ポソームおよび非水性ビヒクル(例えば、不揮発性油)もまた使用され得る。薬学的に活
性な物質のためのこのような媒体および薬剤の使用は、当該分野において周知である。任
意の従来の媒体または薬剤が活性化合物と不適合性である場合を除いて、組成物における
それらの使用が企図される。補助的な活性化合物もまた、組成物に組み込まれ得る。
【0060】
本明細書中に開示される活性剤はまた、リポソームとして処方され得る。リポソームは
、当該分野で公知の方法(例えば、Epsteinら、Proc.Natl.Acad.
Sci.USA,82:3688(1985);Hwangら、Proc.Natl.A
cad.Sci.USA,77:4030(1980);および米国特許第4,485,
045号および同第4,544,545号に記載される)によって調製される。向上した
循環時間を有するリポソームは、米国特許第5,013,556号に開示される。
【0061】
特に有用なリポソームは、ホスファチジルコリン、コレステロール、およびPEG誘導
体化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物を用いる、逆相
エバポレーション法によって作製され得る。リポソームは、所望の直径を有するリポソー
ムを生じるように、規定された孔サイズのフィルターを通して押し出される。
【0062】
本発明の薬学的組成物は、その意図された投与の経路と適合性であるように処方される
。投与の経路の例としては、非経口的投与、例えば、静脈内投与、皮内投与、皮下投与、
経口(例えば、吸入)投与、経皮(すなわち、局所的)投与、経粘膜投与、および直腸投
与が挙げられる。非経口適用、皮内適用、または皮下適用のために使用される溶液または
懸濁液は、以下の成分を含み得る:注射用水、生理食塩水溶液、不揮発性油、ポリエチレ
ングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒のような滅菌希釈
剤;ベンジルアルコールまたはメチルパラベンのような抗菌剤;アスコルビン酸または重
亜硫酸ナトリウムのような抗酸化剤;エチレンジアミン四酢酸(EDTA)のようなキレ
ート剤;アセテート、シトレートまたはホスフェートのような緩衝剤、および塩化ナトリ
ウムまたはデキストロースのような張度の調節のための薬剤。pHは、酸または塩基(例
えば、塩酸または水酸化ナトリウム)を用いて調節され得る。非経口調製物は、ガラスま
たはプラスチックから作製されたアンプル、使い捨て可能シリンジまたは複数用量のバイ
アル中に含まれ得る。
【0063】
注射可能用途に適切な薬学的組成物としては、滅菌水溶液(水溶性である場合)または
分散物および滅菌注射可能溶液または分散物の即席調製物のための滅菌粉末が挙げられる
。静脈内投与のために、適切なキャリアとしては、生理学的生理食塩水、静菌水、Cre
mophor ELTM(BASF、Parsippany,N.J.)またはリン酸緩
衝化生理食塩水(PBS)が挙げられる。全ての場合において、組成物は、滅菌でなけれ
ばならず、そして容易に注射可能である程度まで流体であるべきである。組成物は、製造
および保存の条件下において安定でなければならず、そして微生物(例えば、細菌および
真菌)の混入作用に対して保存されなければならない。キャリアは、例えば、水、エタノ
ール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチ
レングリコールなど)、およびこれらの適切な混合物を含む溶媒または分散媒体であり得
る。適切な流動性は、例えば、レシチンのようなコーティングの使用によって、分散の場
合、必要とされる粒子サイズの維持によって、そして界面活性剤の使用によって、維持さ
れ得る。微生物の作用の防止は、種々の抗菌剤および抗真菌剤(例えば、パラベン、クロ
ロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成され得る
。多くの場合において、等張性剤(例えば、糖、ポリアルコール(例えば、マンニトール
)、ソルビトール、塩化ナトリウム)を組成物中に含むことが好ましい。注射可能組成物
の長期の吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよび
ゼラチン)を組成物中に含むことによってもたらされ得る。
【0064】
滅菌注射可能溶液は、活性化合物(例えば、ABO融合タンパク質)を必要とされる量
で、適切な溶媒中に、上記成分の1つまたは組合せと混合し、必要に応じて、続いて、滅
菌濾過することによって、調製され得る。一般的に、分散物は、ベーシックな分散媒体お
よび必要とされる上記成分からの他の成分を含む、滅菌ビヒクルに、活性化合物を混合す
ることによって調製される。滅菌注射可能溶液の調製のための滅菌粉末の場合、調製方法
は、真空乾燥および凍結乾燥であり、これは、先に滅菌濾過された溶液からの、活性成分
および任意のさらなる所望の成分の粉末を生じる。
【0065】
経口組成物は、一般的に、不活性な希釈剤または食用キャリアを含む。これらは、ゼラ
チンカプセルに含まれ得るかまたは錠剤に圧縮され得る。経口治療投与のために、活性化
合物は、賦形剤と混合され得、そして錠剤、トローチ、またはカプセルの形態で使用され
得る。経口組成物はまた、マウスウオッシュとしての使用のために流体キャリアを使用し
て調製され得、ここで、流体キャリア中の化合物は、経口適用され、スウィッシュされ、
そして吐き出されるかまたは嚥下される。薬学的に適合性の結合剤、および/またはアジ
ュバント物質は、組成物の一部として含まれ得る。錠剤、丸剤、カプセル剤、トローチ剤
などは、類似の性質の以下の成分、または化合物のいずれかを含み得る:結合剤(例えば
、微結晶セルロース、トラガカントガムまたはゼラチン);賦形剤(例えば、デンプンま
たはラクトース)、崩壊剤(例えば、アルギン酸、Primogel、またはコーンスタ
ーチ);滑沢剤(例えば、ステアリン酸マグネシウムまたはSterote);グリダン
ト(glidant)(例えば、コロイド状二酸化ケイ素);甘味剤(例えば、スクロー
スまたはサッカリン);または香料(例えば、ペパーミント、サリチル酸メチル、または
オレンジ香料)。
【0066】
吸入による投与のために、化合物は、適切な噴霧剤(例えば、二酸化炭素のような気体
)を含む加圧容器または分散器、あるいは噴霧器からエアロゾル噴霧の形態で送達される
。
【0067】
全身投与はまた、経粘膜的手段または経皮的手段によってであり得る。経粘膜的投与ま
たは経皮的投与のために、浸透される障壁に対して適切な浸透剤は、処方物で使用される
。このような浸透剤は、一般的に、当該分野で公知であり、そして例えば、経粘膜的投与
のため、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜的投与は
、経鼻スプレーまたは坐剤の使用によって達成され得る。経皮投与のために、活性化合物
は、当該分野において一般的に公知な、軟膏(ointment)、軟膏(salve)
、ゲル、またはクリームに処方される。
【0068】
化合物はまた、坐剤の形態で(例えば、ココアバターおよび他のグリセリドのような従
来の坐剤ベースとともに)または直腸送達のための保持浣腸で、調製され得る。
【0069】
1つの実施形態において、活性化合物は、身体からの迅速な排除に対して化合物を保護
するキャリア(例えば、徐放処方物(移植物およびマイクロカプセル化送達システムを含
む))を用いて調製される。生分解性、生体適合性ポリマー(例えば、エチレンビニルア
セテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポ
リ乳酸)が使用され得る。このような処方物の調製のための方法は、当業者に明らかであ
る。これらの材料はまた、Alza CorporationおよびNova Phar
maceuticals,Inc.から商業的に入手され得る。リポソーム懸濁物(ウイ
ルス抗原に対するモノクローナル抗体を用いて感染した細胞に標的化されたリポソームを
含む)もまた、薬学的に受容可能なキャリアとして使用され得る。これらは、例えば、米
国特許第4,522,811号に記載されるように、当業者に公知の方法に従って調製さ
れ得る。
【0070】
いくつかの実施形態において、経口組成物または非経口組成物は、投与の容易さおよび
均一な投与のために投薬単位形態で処方される。本明細書中で使用される投薬単位形態は
、処置される被験体に対して単位投薬として適切な物理的に異なる単位をいい;それぞれ
の単位は、必要とされる薬学的キャリアとともに、所望の治療的効果を生成するように計
算された所定量の活性化合物を含む。本発明の投薬単位形態のための詳細は、活性化合物
の独特の特徴および達成されるべき特定の治療効果、ならびに個体の処置のためのこのよ
うな活性化合物を配合する当該分野に固有の制限によって示され、そして直接的に依存す
る。
【0071】
本発明の核酸分子は、ベクターに挿入され得、そして遺伝子治療ベクターとして使用さ
れ得る。遺伝子治療ベクターは、被験体に、例えば、静脈内注射、局所投与(例えば、米
国特許第5,328,470号を参照のこと)によって、または定位的な注射(例えば、
Chenら、1994.Proc.Natl.Acad.Sci.USA 91:305
4−3057を参照のこと)によって被験体に送達され得る。遺伝子治療ベクターの薬学
的調製物は、受容可能な希釈剤中に遺伝子治療ベクターを含み得るか、または遺伝子治療
ビヒクルが埋め込まれる徐放マトリクスを含み得る。あるいは、完全な遺伝子送達ベクタ
ーが、組換え細胞からインタクトに作製され得る場合(例えば、レトロウイルスベクター
)、薬学的調製物は、遺伝子送達システムを作製する1つ以上の細胞を含み得る。
【0072】
持続放出調製物は、所望であれば、調製され得る。持続放出調製物の適切な例としては
、抗体を含む固体疎水性ポリマーの半透過性マトリクスが挙げられ、このマトリクスは、
形付けられた物品(例えば、フィルムまたはマイクロカプセル)の形態である。持続放出
マトリクスの例としては、ポリエステル、ヒドロゲル(例えば、ポリ(2−ヒドロキシエ
チル−メタクリレート)、またはポリ(ビニルアルコール))、ポリラクチド(米国特許
第3,773,919号)、L−グルタミン酸およびγエチル−L−グルタメートのコポ
リマー、非分解性エチレン−ビニルアセテート、分解性乳酸−グリコール酸コポリマー(
例えば、LUPRON DEPOTTM(乳酸−グリコール酸コポリマーおよび酢酸ロイ
プロリドから構成される注射可能なミクロスフェア)、およびポリ−D−(−)−3−ヒ
ドロキシ酪酸が挙げられる。エチレンビニルアセテートおよび乳酸−グリコール酸のよう
なポリマーが100日にわたる間、分子の放出を可能にする一方で、特定のヒドロゲルが
より短い時間の間、タンパク質を放出する。
【0073】
薬学的組成物は、投与のための説明書とともに、容器、パックまたは分散器中に含まれ
得る。
【0074】
本発明はさらに、以下の非制限的な実施例で例示される。
【実施例】
【0075】
(実施例1:一般的方法)
本明細書中に記載されるデータは、以下の試薬および方法を使用して作製された。
【0076】
(細胞培養)
COS−7 m6細胞(Seed,1987)、CHO−K1(ATCC CCL−6
1)、およびSV40ラージT抗原を発現する293ヒト胚性腎臓細胞株(293T;B
.Seedによって親切にも提供された)を、Dulbeccoの改変Eagle培地(
GibcoBrl,Life Technologies,Paisley,Scotl
and)(10%ウシ胎仔血清(GibcoBrl,Life Technologie
s)、25μg/mlゲンタマイシンスルフェート(Sigma、St.Louis、M
O)および2mMグルタミン(GibcoBrl,Life Technologies
)を補充した)において培養した。細胞を、2〜4日毎に継代させた。HH14ハイブリ
ドーマ(ATCC HB−9299;米国特許第4,857,639号)を、RPMI
1640(GibcoBrl,Life Technologies)(10%ウシ胎仔
血清、100U/mlペニシリン、100μg/μlストレプトマイシンおよび2mMグ
ルタミンを補充した)において培養した。
【0077】
(HH14抗体の精製)
上清を、培養したHH14細胞から収集した。2リットルの上清を、ヤギ抗マウスIg
M(Sigma)カラムで、Bio−Rad LPクロマトグラフ(Bio−Rad,H
ercules,CA)を用いてアフィニティー精製した。結合したタンパク質を、0.
1MグリシンCl(pH2.5)によって溶出させ、そして溶出物を、1M Tris−
Cl(pH7.5)を用いて直ちに中和した。溶出物を、1%リン酸緩衝化生理食塩水(
PBS)に対して透析し、そして凍結乾燥した。凍結乾燥したタンパク質を、BCAアッ
セイによって測定した場合に3μg/μlの最終濃度になるように、蒸留H2O中に溶解
させた。
【0078】
(発現ベクターの構築)
ヒト血液型A遺伝子を、MKN−45細胞株から単離された総RNAから作製されたc
DNAから、5’−cgc ggg aag ctt gcc gag acc aga
cgc gga−3’(配列番号1)を正方向プライマーとして、そして5’−cgc
ggg cgg ccg ctc acg ggt tcc gga ccg c−3
’(配列番号2)を逆方向プライマーとして用いたポリメラーゼ連鎖反応(PCR)によ
って増幅した。増幅されたcDNA(A遺伝子)を、Hind IIIおよびNot I
を用いて、CDM8(Seed,1987)のポリリンカー中にサブクローン化した。血
液型H遺伝子を、ヒト扁桃支質ライブラリーをテンプレートとして用いてPCR増幅して
2片とした。内部Sse I部位を、ヌクレオチド775(GenBank登録番号M3
5531)をCへと変化させて、Pst I部位からSse I部位を作製する内部重複
プライマーを用いたPCRによって作製した。FUT1のカルボキシ末端をコードするc
DNAを、5’−ggg gac tac ctg cag gtt atg cct
cag cgc−3’(配列番号3)を正方向プライマーとして、そして5’−cgc
ggg gcg gcc gct tca agg ctt agc caa tgt−
3’(配列番号4)を逆方向プライマーとして用いて増幅し、Sse IおよびNot
Iによって切断し、そしてPst IおよびNot Iで消化したCDM8発現ベクター
中にサブクローン化した。FUT1のアミノ末端をコードするcDNAを、5’−cgc
ggg aag ctt acc atg tgg ctc cgg agc cat
−3’(配列番号5)を正方向プライマーとし、そして5’−cca gcg ctg
agg cat aac ctg cag gta gtc−3’(配列番号6)を逆方
向プライマーとして用いて増幅し、Hind IIIおよびSse Iによって切断し、
そしてHind IIIおよびSse I切断後の、カルボキシ末端を保有するCDM8
中にサブクローン化した。
【0079】
Se遺伝子を、血液型A2Le(a−b+)Se個体によって提供された末梢血単核細
胞から単離された総RNAから逆転写されたcDNAから、5’−cgc ggg aa
g ctt acc atg ctg gtc gtt cag atg−3’(配列番
号7)を正方向プライマーとして、そして5’−cgc ggg cgg ccg ct
t agt gct tga gta agg g−3’(配列番号8)を逆方向プライ
マーとして用いて同様にPCR増幅した。このSe遺伝子cDNAを、Hind III
およびNot Iを用いてCDM8中にサブクローン化した。グリコシルトランスフェラ
ーゼcDNAを配列決定し、そしてこれらがコードする酵素活性を、血液型HおよびA特
異的モノクローナル抗体を用いた、一過的にトランスフェクトされた細胞のフローサイト
メトリー分析によってチェックした。PSGL−1/mIgG2bキメラを、以前(Li
uら,1997)に記載されたとおりに構築した。
【0080】
(トランスフェクションおよび分泌されたPSGL−1/mIgG2bキメラの産生)
トランスフェクションカクテルを、5mlポリスチレンチューブ中で39μlの20%
グルコース、39μgのプラスミドDNA、127μlのdH2O、および15.2μl
の0.1Mポリエチレンイミン(25kDa;Aldrich,Milwaukee,W
I)を混合することによって調製した。全てのトランスフェクション混合物において、1
3μgのPSGL‐1/mIgG2bプラスミドを用いた。種々のグリコシルトランスフ
ェラーゼ(glycosyitransferase)に対して、13μgのこのプラス
ミドを添加し、そして必要な場合、合計39μgのプラスミドDNAに達するようにCD
M8プラスミドを添加した。この混合物を室温で10分間静置し、その後、10mlの培
養培地中に入れて、約70%コンフルエンシーな細胞に添加した。7日後、細胞上清を収
集し、細片をスピンダウン(1400×g、15分間)し、そしてNaN3を、0.02
%(w/v)の最終濃度になるように添加した。
【0081】
(SDS−PAGEおよびウェスタンブロット分析のための、分泌されたPSGL−1
/mIgG2bの精製)
PSGL−1/mIgG2b融合タンパク質を、4℃で一晩回転させることによって、
50μlヤギ抗mIgGアガロースビーズ(100:1のスラリー;Sigma)によっ
て、収集した上清から精製した。融合タンパク質を有するビーズをPBS中で3回洗浄し
、そしてその後の分析のために用いた。代表的には、サンプルを50μlの2×還元サン
プル緩衝液中に溶解し、そして10:1のサンプルを各ウェルにローディングした。
【0082】
(アフィニティー精製したPSGL−1/mIgG2bのPNGaseF処理)
PNGaseFキット(Roche Diagnostics,Indianapol
is,IN)を、N−グリカンの脱グリコシル化のために用いた。製造業者によって提供
されたプロトコルのわずかな改変版を用いた。1.5mlのEppendorfチューブ
中で、20μlの反応緩衝液を、アガロースビーズ上の精製したPSGL−1/mIgG
2bと混合し、そして3分間煮沸した。この混合物をスピンダウンし、そして10μlの
上清を新たなEppendorfチューブに移した。10μlのPNGaseFまたはネ
ガティブコントロールとして10μlの反応緩衝液を添加した。チューブを、1.5時間
にわたって37℃でインキュベートした。インキュベーション後、20μlの2×還元サ
ンプル緩衝液および10μlのH2Oを添加し、そしてサンプルを3分間煮沸した。
【0083】
(上清中のPSGL−1/mIgG2b濃度の決定のためのELISA)
96ウェルELISAプレート(Costar 3590,Corning,NY)を
、50μlの50mM炭酸緩衝液(pH9.6)中の0.5μg/ウェルのアフィニティ
ー精製したヤギ抗mIgG特異性抗体(Sigma)で、室温で2時間にわたってコーテ
ィングした。0.05% Tween(PBS−T)を含むPBS中の3%ウシ血清アル
ブミン(BSA)300μlを用いて4℃で一晩ブロッキングし、続いて洗浄した後、培
養培地中に連続希釈した、50μlのサンプル上清を添加した。洗浄後、これらのプレー
トを、ブロッキング緩衝液中に10,000:1希釈したヤギ抗mIgM−HRP(Si
gma)50μlとともに2時間にわたってインキュベートした。発色溶液のために、1
錠の3,3’,5,5’−テトラメチルベンジジン(Sigma)を、3μlの30%(
w/v)H2O2を含む11mlの0.05Mクエン酸/リン酸緩衝液中に溶解した。1
00μlの発色溶液を添加した。反応を、25μlの2M H2SO4を用いて停止させ
た。これらのプレートを、自動化マイクロプレートリーダー(Bio−Tek Inst
ruments,Winooski,VT)において450nmおよび540nmで読み
取った。標準として、培養培地中の精製mIgG Feフラグメント(Sigma)の希
釈シリーズを三連で用いた。
【0084】
(SDS−PAGEおよびウェスタンブロッティング)
SDS−PAGEを、5%スタッキングゲルおよび8%分離ゲルを用いるLaemml
i(1970)の方法によって行い、そして分離されたタンパク質を、以前(Liuら,
1997)に記載されるとおりに、HybondTM−Cエキストラメンブレン上に電気
泳動的にブロッティングした。3% BSAを含む、0.05% Tween−20を含
むTris緩衝化生理食塩水(TBS−T)中での一晩のブロッキング後、これらのメン
ブレンを、TBS−Tで3回洗浄した。次いで、これらを、マウス抗ヒト血液型A全種類
(mIgM,Dako,Carpinteria,CA)または抗ヒトH型1(mIgG
3,Signet;Dedham,MA)、抗ヒトH型2(mIgM,Dako)もしく
は抗ヒトH型3(mIgM,ハイブリドーマHH14,ATCC HB9299)ととも
に、1時間にわたって室温でインキュベートした。H型3抗体以外の全ての抗体を、TB
S−T中の3% BSA中に1:200希釈した。H型3抗体を、TBS−T中の3%
BSAにおいて1μg/mlの濃度になるように希釈した。これらのメンブレンを、TB
S−Tで3回洗浄し、その後、TBS−T中の3% BSAに2000:1希釈した、西
洋ワサビペルオキシダーゼ(HRP)結合体化二次抗体である、ヤギ抗mIgM(Cap
pel,Durham,NC)またはヤギ抗mIgG3(Serotec,Oxford
,England)とともに1時間にわたって室温でインキュベートした。結合した二次
抗体を、製造業者の指示に従って、ECLキット(Amersham Pharmaci
a Biotech,Uppsala,Sweden)を用いた化学発光によって可視化
した。PSGL−1/mIgG2b自体の検出のために、HRP標識ヤギ抗mIgG(S
igma)を、二次抗体とのインキュベーションを行わないこと以外は上記のとおりに、
TBS−T中の3% BSAにおける1:10,000の希釈で用いた。
【0085】
(PSGL−1/mIgG2bによる相対血液型Aエピトープ密度の決定)
ウェスタンブロットを、上記の通りに行った。メンブレンを、−35℃で作動するCC
Dカメラを備えるFluor−S Max Muitlmager(BioRad)にお
いて可視化した。Quantity Oneソフトウェア(BioRad)の分析ウィン
ドウにおいて量ツールを用いて、血液型A反応性についての量(量境界xピクセル領域内
のピクセルの強度の合計)を、COS細胞、CHO細胞および293T細胞中で作製した
PSGL−1/mIgG2bについてのmIgG反応性量で除算した。異なる宿主細胞中
で作製したPSGL−1/mIgGb2b間でAエピトープ/マウスIgG比を比較する
ために、比を、H遺伝子およびA遺伝子でトランスフェクトされたCOS細胞中で作製さ
れたA置換PSGL−1/mIgG2bから得られた比に対して正規化した。
【0086】
(血清の吸収)
ヤギ抗mIgGアガロースビーズ(Sigma)の600マイクロリットルのスラリー
を、1.5mlのEppendorf微量遠心管に移した。これらのビーズを、400×
gでのすばやいスピンによってスピンダウンした。次いで、上清を除去し、そしてこれら
のビーズを1ml PBSで1回洗浄し、再度スピンダウンし、そしてPSGL−1/m
IgG2bをコードするcDNA(Se遺伝子およびA遺伝子)でトランスフェクトされ
たCHO細胞由来の180mlの上清に移した。アガロースビーズを含む上清を、4℃で
一晩、回転させながらインキュベートした。収集のために、これらのビーズを400×g
で15分間、室温でスピンダウンし、そして1.5mlのEppendorf微量遠心管
に移した。PBSでの洗浄を3回行った。これらのビーズは、Aムチンビーズと呼ばれる
。600マイクロリットルの抗mIgGアガロースビーズをまた、Aムチン−ビーズの希
釈のために用いて吸収剤としてのAムチン−ビーズの希釈系列を得たこと以外は、同じ様
式で調製した。これらのビーズは、ヤギ抗mIgGビーズと呼ばれる。これらのビーズを
、4mlのEllermanチューブ中に、表Iに従ってアリコートに分けた。A−PA
A−MPG(Syntesome,Munich,Germany)およびB−PAA−
MPG(Syntesome)を計量し、そしてEllermanチューブ中に表Iに従
ってアリコートに分け、その後、PBSで1回洗浄した。
【0087】
血液型O型の5人の患者由来のプールした血清を、Huddinge Univers
ity Hospitalの血液銀行(倫理的許可、Dnr.392/99、承認日08
/15/2000)から入手した。細胞破片を、Jouan A−14微量遠心機での1
4,000rpmでの5分間の遠心分離によって血清から除去した。透明にされた血清を
別のチューブに移し、そして水浴中で56℃で1時間インキュベートして、補体を不活化
した。血清を、使用するまでアリコートにして−20℃で保存した。
【0088】
【表1】

500マイクロリットルの血清を、各チューブに添加し、そしてこれらのビーズと、ロ
ーリングテーブル上で4℃で4時間混同した。吸収後、これらのビーズをスピンダウンし
、そして吸収された血清を新たなEllermanチューブに移した。吸収された血清を
、さらなる分析まで、−20℃で保存した。
【0089】
Aムチン−ビーズ上でのPSGL−1/mIgG2bの量を決定するために、ヤギ抗m
IgG Fc ELISA(上記の手順を参照のこと)を、アガロースビーズとのインキ
ュベーション前の上清およびインキュベーション後の上清について行った。
【0090】
(抗A抗体の定量のためのELISA)
96ウェルELISAプレート(Costar 3590,Corning)を、1ウ
ェルあたり50μlの50mM炭酸緩衝液(pH9.6)中0.05μlのA−PAA−
ビオチン(Syntesome)で2時間、室温でコーティングした。PBS−T中の3
% BSA(300μl)を用いた4℃での一晩のブロッキングおよびその後の洗浄後、
PBS中に連続希釈した50μlの血清を添加し、そしてこのプレートを室温で2時間イ
ンキュベートした。洗浄後、ブロッキング緩衝液中に10,000:1希釈したマウス抗
ヒトIgA−HRP、マウス抗ヒトIgG−HRPおよびマウス抗ヒトIgM−HRP(
Jackson,PA)(50μl)とともにインキュベーションを行った。このプレー
トの発色および読取りを上記の通りに行った。
【0091】
(血清中における総タンパク質濃度の決定)
血清中における総タンパク質濃度を、抗A抗体の吸収の前後に決定した。BCAタンパ
ク質アッセイ試薬のマイクロタイタープレートプロトコール(Pierce,Rockf
ord,IL)を製造業者の指針書に従って使用し、そしてこのサンプルを二連または三
連で実施した。
【0092】
(実施例2:血液型HおよびAを、種々の宿主細胞中で作製した組換えPSGL−1/
mIgG20において決定する)
293T細胞。免疫親和性により精製したPSGL−1/mIgG2b(H遺伝子、S
e遺伝子、またはA遺伝子をコードするプラスミドを含むか、または含まないPSGL−
1/mIgG2b cDNAで一過性でトランスフェクトした293T細胞により産生し
た)を、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS−PAGE
)ならびに検出のために抗体に特異的な抗mIgG、抗血液型A、H1型、H2型、およ
びH3型を使用するウエスタンブロットによって分析した(図1A〜E)。この融合タン
パク質を、見掛け上の分子量約100および140〜160kDa(A)を用いる二連の
還元状態下で移動させた。このPSGL−1/mIgG2bは、高度にグリコシル化され
たムチン型タンパク質の挙動に関して、上記観察に従って銀でわずかに染色した(示さず
)(CarrawayおよびHull,1991;ShimizuおよびShaw,19
93)。上に示されるように(Liuら,1997)、この融合タンパク質は、ホモダイ
マーとして産生した(データは示さず)。65kDaおよび35kDa付近の二本のバン
ドは、抗mIgG抗体で決定された。これらのバンドは、エンプティーベクターのみでト
ランスフェクトされた293T細胞の上清には見られず、従って、融合タンパク質由来の
タンパク質フラグメントであるようであった(CarrawayおよびHull,199
1)。細胞フラクション中においてみられた、この47kDaおよび20kDaのバンド
は、サンプル緩衝液中で沸騰させることでアガロースビーズから脱離させた、ヤギ抗マウ
ス抗体の重鎖Igおよび軽鎖Ig由来である。
【0093】
αl,3GalNAcTを有するFUT1およびFUT2の両方は、PSGL−1/m
IgG2b(B)上の血液型A鎖の生合成を支持する。FUT2とαl,3GalNAc
Tとの組み合わせは、FUT1とαl,3GalNAcTとの組み合わせよりも、PSG
L−1/mIgG2bにおいてよりAエピトープを産生するようであった(Bにおけるこ
れらのレーンを参照)。一方、FUT1のみは、大量のH2型構造(D)の発現を支持し
たが、FUT2は、ムチン/Ig(D)において、わずかなH2型エピトープを生じた。
PSGL−1/mIgG2bにおいて、検出可能なH1型構造は、存在しか生じなかった
。FUT2により産生された、このHエピトープは、ほとんど排他的に型3(すなわち、
Fucα2Galβ3GalNAcα−R)に基づく。なぜならば、H1型構造およびわ
ずかなH2型構造が、この酵素(E)によってPSGL−1/mIgG2bにおいて作製
されたからである。興味深いことに、A遺伝子でのH遺伝子またはSe遺伝子の両方の同
時トランスフェクションは、抗H3型抗体(E)と反応性のPSGL−1/mIgG2b
において大量のエピトープを生じる。血液型A、H2型およびH3型エピトープは、主に
O連結された。なぜならば、PSGL−1/mIgG2bのペプチドNグリコシダーゼF
(PNGaseF)処置は、これらのエピトープに対して特異的な抗体を使用する、抗体
染色を減少させたからである(図2B〜D)。効率的なN−グルカン脱グリコシル化は、
PSGL−1/mIgG2bの完全な移動シフトによって示された(図2A〜D)。
【0094】
COS細胞。COS細胞において作製されたPSGL−1/mIgG2bのウエスタン
ブロット分析は、図3A〜Eにおいて示される。抗A特異的抗体および抗H特異的抗体を
使用して染色するPSGL−1/mIgG2bのパターンは、293T細胞において作製
されたPSGL−1/mIgG2bに対して見出されたパターンに、弱いがいくらか類似
していた。α1,3GalNacTを一緒に含むFUT1およびFUT2の両方は、Aエ
ピトープ発現を支持し、より多量のエピトープが、FUT2およびα1,3GalNAc
T(B)によって産生された。FUT1のみが、H2型構造(D)を作製し得、そしてF
UT1もFUT2も、COS細胞(C)において、H1型構造の発現を支持しなかった。
293T細胞において作製されたPSGL−1/mIgG2bに対して、非常に低レベル
のH3型エピトープが、FUT1およびα1,3GalNAcT、またはFUT2酵素の
み(E)を発現するCOS細胞において産生されたPSGL−1/mIgG2bにおいて
見られた。
【0095】
しかし、FUT2およびα1,3GalNAcTのジョイント発現(joint ex
pression)は、H3型抗体(E)との増加した反応性を生じた。さらに、弱いバ
ンドは、α1,2FTのみが発現された場合、抗A抗体で見出され、上記で報告したよう
に、COS細胞において、α1,3GalNAcTの弱い内因性の活性を示した(Cla
rkeおよびWatkins,1999)。PSGL−1/mIgG2bのPNGase
F処理は、抗血液型A、H2型またはH3型抗体の染色における任意の検出可能な減少を
示さず(データは示さず)、このことは、これらのエピトープが、PSGL−1/mIg
G2bOグリカン上で主に保持されることを示す。
【0096】
(チャイニーズハムスター卵巣(CHO)細胞)。図4A〜Eは、CHO細胞において
作製されたPSGL−1/mIgG2bの染色、FUT1またはFUT2 cDNAとα
1,3GalNAcT(B)との同時発現後のPSGL−1/mIgG2b保持血液型A
エピトープの染色を示す。H1型(C)構造もH2型(D)構造も、FUT1またはFU
T2のいずれかとの同時トランスフェクション後に、CHO細胞において作製されたPS
GL−1/mIgG2bキメラ上で検出され得ず、このことは、H3型構造が、α1,3
GalNAcTに対して利用可能な唯一の前駆体であることを示した。抗H3型抗体を用
いた染色は、FUT1およびFUT2(B)を用いて見出され、Aエピトープが、CHO
細胞におけるコア1構造に唯一基づくという理論を支持する。さらに、このことに対する
支持は、PNGaseF処置後に得られ、このことは、抗体染色強度に影響を及ぼさず、
Oグリカン制限Aエピトープ発現を示す(データは示さず)。PSGL−1/mIgG2
bキメラ上におけるAエピトープおよびH3型エピトープの数は、FUT1と比較した場
合、FUT2で明らかにより多く、このことは、このSe遺伝子産物が、コア1(Gal
β3GalNAcα−Ser/Thr)構造のα1,2−フコシル化の点において、H遺
伝子産物よりも優れていることを示唆する。
【0097】
(実施例3.異なる宿主細胞において産生されたPSGL−1/mIgG2bにおける
、血液型Aエピトープの相対密度)
A遺伝子をα1,2FTのいずれかと発現する種々の宿主細胞において作製したPSG
L−1/mIgG2bキメラにおけるAエピトープの相対数を半定量するために、抗A抗
体および抗mIgG抗体を用いたウエスタンブロット、続いてFluor−S(登録商標
)Max MultImagerでの化学ルミネセンス検出を使用した。各PSGL−1
/mIgG2bに対する、血液型AおよびmIgGの反応性の比を、図5に示す(3つの
代表的な実験のうちの1つを示した)。理解されるように、Aエピトープの密度は、α1
,2FTをコードするSe遺伝子とともにA遺伝子を発現するCHO細胞において作製さ
れたPSGL−1/mIgG2bにおいて最も高かった。このPSGL−1/mIgG2
bは、FUT2およびA遺伝子を用いてトランスフェクトされた293T細胞において製
造されたPSGL−1/mIgG2bよりも約3倍多量のAエピトープを保持する;PS
GL−1/mIgG2bは、2番目に高いAエピトープ密度を有する(図5)。各細胞レ
ーンにおいて、A遺伝子をともに有するFUT1よりも、A遺伝子をともに有するFUT
2は、より高いAエピトープ密度を与えた。
【0098】
(実施例4:血液型Aエピトープを保持するPSGL−1/mIgG2bにおける抗A
抗体の吸収)
血液型Aエピトープを有するか、または有さない組換えPSGL−1/mIgG2bに
おける抗A抗体吸収の効率を、マクロ孔質ガラスビーズに対するポリ[N−(2−ヒドロ
キシエチル)アクリルアミド]介して連結されたAトリサッカライド(A−PAA−MP
G)上での吸収の効率と比較した。抗A抗体の吸収前および吸収後のレベルを、酵素結合
イムノソルベント検定法(ELISA)において評価し、このプレートを、ビオチンでも
また置換したポリ[N−(2−ヒドロキシエチル)アクリルアミド]に連結させたAトリ
サッカライド(A−PAA−ビオチン)でコーティングした。この結果を、図6に示す。
20pmolの組換えAエピトープ置換PSGL−1/mIgG2bが、A−PAA−ビ
オチンELISAにおいて検出される場合に、抗A抗体の60%を吸収するために必要と
され、一方、A−PAA−MPGのような164,000pmolのA決定基は、同量の
抗A抗体を吸収するために必要とされた。このPSGL−1/mIgG2bダイマーは、
106ヶ所の強力なO連結グリコシル化部位および8ヶ所の強力なN連結グリコシル化部
位を有し(Wilkinsら,1996;Aeedら,1998,2001)、この後者
の8ヶ所は、分枝構造を保持し得る。各PSGL−1/mIgG2bが、約100のAエ
ピトープを保持すると推定する場合、これらのほとんどは、推定を超えるようであり、F
UT2およびα1,3GalNacTを有するCHO細胞において作製されたムチンは、
A−PAAF−MAPよりも、カルボキシレートモルを基準として約80倍以上の効率を
有する。
【0099】
(実施例5:抗体媒介性移植拒絶の処置または予防)
ABO障壁を横切る組織移植を、ドナー組織血液型に対して予め形成されるか、または
誘導された抗体により媒介されたAMRによって特徴付する(Porter,1963;
Sanchez−Uradazpalら,1993;Fargesら,1995;Tan
abeら,1998;Alkhunaiziら,1999)。抗ABO抗体を、EIA設
定における吸収剤としてA決定基を保持するPSGL−1/mIgG2b融合タンパク質
を使用して、除去する。結果は、(100のA決定基/molのPSGL−1/mIgG
2bに基づく)2nmolのA決定基に対応する、FUT2およびα1,3GalNAc
Tを含むCHO−K1中で作製された約20pmolの組換えPSGL−1/mIgG2
b(分子量300kDaとして計算された)(Wilkinsら,1996;Aeedら
,1998,2001)は、A−PAA−反応性抗体の60%を吸収する。約164,0
00pmolのAトリサッカリドに対応するA−PAA−MPG(2μmol/μgとし
て計算された)が、同量の抗A抗体を吸収するために必要とされた。両方の化合物にして
非特異的なタンパク質吸収量もまた評価し、そしてこの非特異的な吸収が、PAA−MP
Gベース化合物よりほぼ4倍高かった。ムチンベース吸収の比較的高度な吸収効率は、(
1)多価カルボキシレート置換、(2)カルボキシレートエピトープの密接した空間、お
よび(3)主要抗原決定基を保持するコアサッカライド鎖の構造多能性に依存し得る。さ
らに、例えば、最適化されたムチンタンデム反復によって構成された合成ムチン型タンパ
ク質を作製することによる、タンパク質骨格の変化は、吸収体を改善し得る(Silve
rmanら,2001)。また、GalNAc:ポリペプチドトランスフェラーゼの異な
る組み合わせを発現するように操作された細胞は、Oグリカン置換密度を最適化すること
によって吸収効率を改善し得る。
【0100】
(略語)
AMR(抗体媒介拒絶(antibody−mediated rejection)
);BSA(ウシ血清アルブミン);CHO(チャイニーズハムスターの卵巣);COS
;ETA(体外免疫吸収);ELISA(酵素結合イムノソルベント検定法);FT(フ
ルコシルトランスフェラーゼ);GalNAcT(N−アセチルガラクトサミニルトラン
スフェラーゼ);HRP(西洋ワサビペルオキシダーゼ);mIgG(マウスIgG);
mIgM(マウスIgM);MPG(マクロ孔質ガラスビーズ);PAA(ポリ[N−(
2−Hドロキシエチル)アクリルアミド]);PBS(リン酸緩衝生理食塩水);PBS
−T(0.05% Tweenを含むPBS);PCR(ポリメラーゼ連鎖反応);PN
GaseF(ペプチド:N−グリコシダーゼF);PP(血漿瀉血);PSGL−1(P
セレクチン糖タンパク質リガンド−1);SDS−PAGE(ドデシル硫酸ナトリウム−
ポリアクリルアミドゲル電気泳動);TBS−T(0.05%のTween−20を含む
Tris緩衝化生理食塩水);Tx(移植)。
【0101】
(参考文献)
【0102】
【表2−1】
【0103】
【表2−2】
【0104】
【表2−3】
【0105】
【表2−4】
【0106】
【表2−5】
【0107】
【表2−6】
【0108】
【表2−7】
【0109】
【表2−8】
(他の実施形態)
本発明は、その詳細な説明と組み合わせて議論されるが、上記の記載は、本発明を例示
するものであって、その範囲を限定するものではないことを意図し、この範囲は、添付の
特許請求の範囲によって規定される。他の局面、利点および改変は、以下の特許請求の範
囲内である。
【技術分野】
【0001】
(発明の分野)
本発明は、概して抗体媒介性移植拒絶を処置または予防するための組成物および方法に
関し、より具体的には、血液型(blood group)決定基を含む融合ポリペプチ
ドを含む組成物に関する。
【背景技術】
【0002】
(発明の背景)
主要なヒトの血液型系(組織−血液型ABO系)は、3つの糖質決定基(血液型Aエピ
トープ、BエピトープおよびHエピトープ)によって規定される。ABH決定基を有する
グリカンは、糖タンパク質、糖脂質、または遊離のオリゴ糖において見出される。ABH
抗原は、N連結グリカンまたはO連結グリカンにおいて見出され得る。O連結グリカンに
関してこれまで記載された最も共通なコア構造は、コア1(Galβ3GalNAc)、
コア2(Galβ3(GlcNAcβ6)GalNAc)、コア3(GlcNAcβ3G
alNAc)およびコア4(GlcNAcβ3(GlcNAcβ6)GalNAc)であ
る。これらは、1型(Galβ3GlcNAc)構造、2型(Galβ4GlcNAc)
構造、および3型(Galβ3GalNAcα)構造を保有することが示されている。1
型構造は主に、コア3構造およびコア4構造の伸長物として見出されるが、2型鎖(ポリ
ラクトサミン)は、コア2構造のGlcNAcβ1,6分枝の伸長物として発見された。
【0003】
ABO障壁を越える移植(Tx)は、通常、組織Txを避ける。なぜなら、実施する抗
体に起因する抗体媒介性拒絶(AMR)の危険性がしばしば高いからである。これはまた
、骨髄移植において適用され得るが、血液型ABHの不一致はその結果に影響しないと長
い間考えられてきた。しかし、いくつかの場合、ABO障害を越えて移植することが依然
として所望される。これは、ドナーの総数が増加しなくても、特定のレシピエントに対し
て利用可能なドナーのプールを広げるからである。
【0004】
体外免疫吸収(EIA)または血漿交換(PP)による抗A型抗体または抗B型抗体の
除去は、ABO不適合性組織Tx後の移植片の生存を改善することが証明された。ABO
不適合性Txおよび異種Txの両方におけるAMRの予防のために使用される別の方法は
、遊離オリゴ糖の注入であるが、遊離糖に対する抗体の低い親和性および循環における低
分子量のオリゴ糖の短い半減期(Yeら、1994;Simonら、1998)は、より
広い使用を抑制する。
【発明の概要】
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明は、血液型エピトープが、高密度で、かつムチン型タンパク質骨格の異なるコア
糖鎖によって特異的に発現され得るという発見に一部基づく。ポリペプチドは、本明細書
中でABO融合ポリペプチドと称される。したがって、本発明は以下を提供する。
(1)
第2ポリペプチドに作動可能に連結される第1ポリペプチドを含む融合ポリペプチドであ
って、該第1ポリペプチドは:
(a)ムチンポリペプチドであり、そして
(b)α1,2フコシルトランスフェラーゼによってグリコシル化され、
該第2ポリペプチドは、免疫グロブリンポリペプチドの少なくとも1領域を含む、融合
ポリペプチド。
(2)
前記第1ポリペプチドが、α1,3 N−アセチルガラクトサミニルトランスフェラーゼ
またはα1,3 ガラクトシルトランスフェラーゼによってさらにグリコシル化される、
請求項1に記載の融合ポリペプチド。
(3)
前記α1,2フコシルトランスフェラーゼが、血液型Hまたは分泌型α1,2フコシルト
ランスフェラーゼである、請求項1に記載の融合ポリペプチド。
(4)
前記第1ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも1領域
を含む、請求項1に記載の融合ポリペプチド。
(5)
前記第1ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含む
、請求項4に記載の融合ポリペプチド。
(6)
前記第2ポリペプチドが、重鎖免疫グロブリンポリペプチドの領域を含む、請求項1に記
載の融合ポリペプチド。
(7)
前記第2ポリペプチドが、免疫グロブリン重鎖のFc領域を含む、請求項6に記載の融合
ポリペプチド。
(8)
ダイマーである、請求項1に記載の融合ポリペプチド。
(9)
請求項1に記載のペプチドをコードする、単離された核酸。
(10)
請求項9に記載の核酸を含む、ベクター。
(11)
請求項11に記載のベクターを含む、細胞。
(12)
請求項1に記載の融合ポリペプチドを含む、吸収体。
(13)
抗体媒介性拒絶を処置または予防する必要のある被験体において、抗体媒介性拒絶を処置
または予防するための方法であって、該方法は、以下:
(a)融合ポリペプチド複合体を形成するために、該被験体に由来する生物学的サンプ
ルと、請求項1に記載の融合ポリペプチドとを接触させる工程;および
(b)該生物学的サンプルから該複合体を除去することによって、該被験体における抗
体媒介性拒絶を処置または予防する工程
を包含する、方法。
(14)
生物学的サンプルから抗体を除去する方法であって、該方法は、以下:
(a)融合ポリペプチド複合体を形成するために、生物学的サンプルと、請求項1に記
載の融合ポリペプチドとを接触させる工程;
(b)該生物学的サンプルから該複合体を除去することによって、該生物学的サンプル
から該抗体を除去する工程
を包含する、方法。
【0006】
1つの局面において、本発明は、第1ポリペプチドを含む融合ポリペプチドを提供し、
この第1ポリペプチドは、ムチンポリペプチドの少なくとも1領域を含み、第2ポリペプ
チドに作動可能に連結されるα1,2フコシルトランスフェラーゼによってグリコシル化
される。第1ポリペプチドは、α1,3 N−アセチルガラクトサミニルトランスフェラ
ーゼおよび/またはα1,3 ガラクトシルトランスフェラーゼによって連続的にグリコ
シル化される。ムチンポリペプチドは、例えば、PSGL−1である。好ましくは、この
ムチンポリペプチドは、PSGL−1の細胞外部分である。α1,2,フコシルトランス
フェラーゼは、例えば、血液型Hまたは分泌型α1,2,フクシルトランスフェラーゼ(
例えば、FUT1またはFUT2)である。
【0007】
好ましい実施形態において、第2ポリペプチドは、免疫グロブリンポリペプチドの少な
くとも1領域を含む。例えば、第2ポリペプチドは、重鎖免疫グロブリンポリペプチドの
1領域を含む。あるいは、第2ポリペプチドは、免疫グロブリン重鎖のFC領域を含む。
【0008】
ABO融合ポリペプチドは多量体(mutimer)である。好ましくは、ABO融合
ポリペプチドはダイマーである。
【0009】
本発明はまた、ABO融合ポリペプチドをコードする核酸、ならびに本明細書中に記載
される核酸をコードするABO融合ポリペプチドを含むベクター、および本明細書中に記
載されるベクターまたは核酸を含む細胞を含む。あるいは、ベクターは、α1,2,フコ
シルトランスフェラーゼおよび/またはα1,3 Nアセチルガラクトサミニルトランス
フェラーゼまたはα1,3 ガラクトシルトランスフェラーゼをコードする核酸をさらに
含む。
【0010】
別の局面において、本発明は、被験体における抗体媒介性拒絶を処置する方法を提供す
る。この方法は、融合ポリペプチド−抗体複合体を形成させるために、生物学的サンプル
(例えば、患者に由来する全血または血漿)と、本発明のABO融合ポリペプチドとを接
触させる工程を包含する。この複合体は、生物学的サンプルから除去され、そしてこの生
物学的サンプルは被験体に再融合される。
【0011】
抗体融合ペプチド複合体を形成し、そして生物学的サンプルからこの複合体を除去する
ために、サンプルと本発明のABO融合ペプチドとを接触させる工程によりこのサンプル
から抗体を除去する方法もまた、本発明に含まれる。
【0012】
ABO融合ポリペプチドを含む薬学的組成物もまた、本発明に含まれる。
【0013】
別の記載がない限り、本明細書中で使用される全ての技術および専門用語は、本発明に
属する当業者によって一般的に理解されるのと同じ意味を有する。本明細書中に記載され
るのと類似または等価な方法および材料は、本発明の実施または試験において使用され得
るが、適切な方法および材料は、以下に記載される。本明細書中に記載される全ての特許
刊行物、特許出願、特許および他の参考文献は、その全体が参考として援用される。矛盾
する場合、明細書(定義を含む)が制御する。さらに、材料、方法および実施例は、例示
のみであり、制限を意図するものではない。本発明の他の特徴および利点は、以下の詳細な説明、および特許請求の範囲から示され
る。
【図面の簡単な説明】
【0014】
【図1】図1A〜Dは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされた293T細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型H鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、CDM8(レーン1および5)、PSGL−1/mIgG2b(レーン2および6)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン7)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Eにおいて、CDM8およびPSGL−1/mIgG2bでトランスフェクトされた細胞由来の重複したサンプルは、省略した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図2】図2A〜Dは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされた293T細胞において産生された、PNGaseF処理された、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。免疫精製したPSGL−1/mIgG2bのPNGaseF処理あり(+)またはなし(−)の後、このPSGL−1/mIgG2bを、8%SDS−PAGEで分離し、そしてニトロセルロース膜へブロッティングした。抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(C)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(D)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、PSGL−1/mIgG2bおよびH遺伝子(レーン1および2)、PSGL−1/mIgG2b、ならびにH遺伝子およびA遺伝子(レーン3および4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン5および6)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン7および8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。
【図3】図3A〜Eは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされたCOS−7m6細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型H鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。パネルA〜Dにおいて、CDM8(レーン1および5)、PSGL−1/mIgG2b(レーン2および6)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン7)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン8)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Eにおいて、CDM8およびPSGL−1/mIgG2bでトランスフェクトされた細胞由来の重複したサンプルは、省略した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図4】図4A〜Eは、H遺伝子もしくはSe遺伝子単独でか、またはA遺伝子によりコードされたα1,3GalNAcTと組み合わせてトランスフェクトされたCHO−K1細胞において産生されたキメラである、免疫精製したPSGL−1/mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。8%SDS−PAGEでの分離およびニトロセルロース膜へのブロッティングの後、抗マウスIgG抗体により(A)、抗血液型A抗体の後でヤギ抗マウスIgM抗体により(B)、抗1型A鎖特異的抗体の後でHRP標識ヤギ抗マウスIgG3抗体により(C)、抗2型H鎖特異的抗体の後でヤギ抗マウスIgM−HRP抗体により(D)、そして抗3型H鎖特異的抗体の後にHRP標識したヤギ抗マウスIgM抗体により(E)、PSGL−1/mIgG2bキメラを、プローブした。CDM8(レーン1)、PSGL−1/mIgG2b(レーン2)、PSGL−1/mIgG2bおよびH遺伝子(レーン3)、PSGL−1/mIgG2b、H遺伝子およびA遺伝子(レーン4)、PSGL−1/mIgG2bおよびSe遺伝子(レーン5)、またはPSGL−1/mIgG2b、ならびにSe遺伝子およびA遺伝子(レーン6)をコードするプラスミドでトランスフェクトした細胞由来のサンプルを分析した。Cにおいて、250ngの1型H鎖−BSAを、ポジティブコントロールとして使用した。
【図5】FUT1またはFUT2をα1,3GalNAcTと同時発現するCOS細胞、CHO細胞および293T(2931)細胞において産生された組換えPSGL−1/mIgG2bに対する相対的血液型A密度を示す、棒グラフである。密度は、血液型AとmIgGとの反応性についての容積の比として計算し、そしてこの値を、FUT1およびα1,3GalNAcTを同時発現するCOS細胞において産生されるA置換PSGL−1/mIgG2bについて得られた値に対して正規化した。
【図6】異なる血液型A置換吸収剤に対する、吸収後に血清中に残った抗血液型Aの反応性を示す折れ線グラフである。A−PAA−ビオチンコーティングされたELISAによって測定されるような、非吸収血液型O血清の割合での、残存する反応性を、使用した吸収剤の量に対してプロットした。傾向線を、対数回帰を使用して計算した。円は、FUT2およびα1,3GalNAcTを有するCHO細胞において産生されたPSGL−1/mIgG2bを示し、そして四角は、A−PAA−MPGを介して連結されたAトリサッカリドを示す。B−PAA−MPGを介して連結されたBトリサッカリドを使用して、全ての吸収において同じ量のPAA−MPGを得た。
【発明を実施するための形態】
【0015】
(発明の詳細な説明)
本発明は、血液型エピトープが、高密度で特異的に、そしてムチン型タンパク質骨格上
の異なるコアサッカリド鎖で発現され得るという発見に一部基づく。より高い密度の血液
型エピトープは、遊離のサッカリドまたは固相に結合したAB決定因子と比較して、抗血
液型抗体の結合の増加または除去(すなわち、吸収)を生じる。
【0016】
本発明は、抗血液型抗体の吸収剤として有用な複数の血液型エピトープを含む、ムチン
−免疫グロブリン融合タンパク質(本明細書中で「ABO融合タンパク質」という)を提
供する。ABO融合ペプチドはまた、グリコシル化についての研究のためのモデルタンパ
ク質として有用である。例えば、このABO融合タンパク質は、ABO不適合性の臓器移
植または骨髄移植の前に、血液または血漿由来のレシピエント抗血液型ABO抗体を排除
する際に、有用である。このABO融合タンパク質は、レシピエントの血液または血漿由
来の抗血液型ABO抗体の50%、60%、70%、80%、90%、95%、98%、
または100%を吸収する。
【0017】
ABO融合ペプチドは、野生型AB決定因子の遊離サッカリドと比較して、抗血液型抗
体を除去または結合する際に、炭水化物モル濃度基準で、より有効である。このABO融
合ペプチドは、等量の野生型AB決定因子の遊離サッカリドと比較して、2倍、4倍、1
0倍、20倍、50倍、80倍、100倍またはそれ以上多い数の抗血液型抗体と結合す
る。
【0018】
本発明のABO融合タンパク質は、血液型決定因子に特異的なエピトープを保有する。
例えば、ABO融合タンパク質は、Aエピトープ、BエピトープまたはHエピトープのい
ずれかを保有する。あるいは、このABO融合物は、血液型抗原についての2つのエピト
ープを保有する。例えば、このABO融合タンパク質は、AエピトープおよびBエピトー
プの両方を保有する。いくつかの局面において、このABO融合タンパク質は、3つ全て
のエピトープ(すなわち、A、BおよびH)を保有する。
【0019】
(融合ポリペプチド)
種々の局面において、本発明は、第二のポリペプチドと作動可能に連結した、糖タンパ
ク質(例えば、ムチンポリペプチド)の少なくとも一部を含む第一のポリペプチドを含む
融合タンパク質を提供する。本明細書中で使用される場合、「融合タンパク質」または「
キメラタンパク質」は、非ムチンポリペプチドと作動可能に連結したムチンポリペプチド
の少なくとも一部を含む。「ムチンポリペプチド」は、ムチンドメインを有するポリペプ
チドをいう。このムチンポリペプチドは、1個、2個、3個、5個、10個、20個また
はそれ以上のムチンドメインを有する。このムチンポリペプチドは、O−グリカンで置換
されたアミノ酸配列によって特徴付けられる任意の糖タンパク質である。例えば、ムチン
ポリペプチドは、2アミノ酸毎または3アミノ酸毎に、セリンまたはスレオニンであるア
ミノ酸を有する。このムチンポリペプチドは、分泌タンパク質である。あるいは、ムチン
ポリペプチドは、細胞表面タンパク質である。
【0020】
ムチンドメインは、アミノ酸のスレオニン、セリンおよびプロリンに富んでおり、ここ
で、オリゴサッカリドは、ヒドロキシアミノ酸(O−グリカン)に、N−アセチルガラク
トサミンを介して連結される。ムチンドメインは、O−連結グリコシル化部位を含むか、
またはO−連結グリコシル化部位からなる。ムチンドメインは、1個、2個、3個、5個
、10個、20個、50個、100個またはそれ以上のO−連結グリコシル化部位を有す
る。あるいは、ムチンドメインは、N−連結グリコシル化部位を含むか、またはO−連結
グリコシル化部位からなる。ムチンポリペプチドは、その質量の50%、60%、80%
、90%、95%または100%がグリカンに帰する。ムチンポリペプチドは、MUC遺
伝子(すなわち、MUC1、MUC2、MUC3など)によりコードされる任意のポリペ
プチドである。あるいは、ムチンポリペプチドは、P−セレクチン糖タンパク質リガンド
1(PSGL−1)、CD34、CD43、CD45、CD96、GlyCAM−1、M
AdCAMまたは赤血球糖タンパク質である。好ましくは、このムチンは、PSGL−1
である。一方、「非ムチンポリペプチド」は、その質量の少なくとも40%未満がグリカ
ンに帰するポリペプチドをいう。
【0021】
本発明のABO融合タンパク質において、このムチンポリペプチドは、ムチンタンパク
質の全てまたは一部に対応し得る。1実施形態において、ABO融合タンパク質は、ムチ
ンタンパク質の少なくとも一部を含む。「少なくとも一部」は、このムチンポリペプチド
が、少なくとも1つのムチンドメイン(例えば、O−連結グリコシル化部位)を含むこと
を意味する。1実施形態において、このムチンタンパク質は、このポリペプチドの細胞外
部分を含む。例えば、このムチンポリペプチドは、PSGL−1の細胞外部分を含む。
【0022】
第一のポリペプチドは、1以上の血液型トランスフェラーゼによってグリコシル化され
る。第一のポリペプチドは、2個、3個、5個またはそれ以上の血液型トランスフェラー
ゼによってグリコシル化される。グリコシル化は、連続的または継続的である。あるいは
、グリコシル化は、同時発生的またはランダム(すなわち、特定の順序はない)である。
例えば、第一のポリペプチドは、α1,2フコシルトランスフェラーゼ(例えば、H遺伝
子またはSe遺伝子によってコードされるα1,2フコシルトランスフェラーゼ)によっ
てグリコシル化される。例示的なα1,2フコシルトランスフェラーゼは、FUT1(G
enBank登録番号Q10984;O10983;O10981;AT455028お
よびNM00148)およびFUT2(GenBank登録番号P19526;BAA1
1638;D82933およびA56098)である。あるいは、第一のポリペプチドは
、1,3N−アセチルガラクトサミニルトランスフェラーゼまたはα1,3アセチルガラ
クトサミニルトランスフェラーゼによってグリコシル化される。いくつかの局面において
、第一のポリペプチドは、α1,2フコシルトランスフェラーゼおよび1,3N−アセチ
ルガラクトサミニルトランスフェラーゼまたはα1,3アセチルガラクトサミニルトラン
スフェラーゼの両方によってグリコシル化される。
【0023】
融合タンパク質において、用語「作動可能に連結される」は、第一および第二のポリペ
プチドが、第一のポリペプチドのO−連結グリコシル化を可能にする様式で、(最も代表
的にはペプチド結合のような共有結合を介して)化学的に連結されることを示すことが意
図される。融合ポリペプチドをコードする核酸をいうために使用される場合、用語、作動
可能に連結される、は、ムチンポリペプチドおよび非ムチンポリペプチドをコードする核
酸が、互いにインフレームで融合していることを意味する。この非ムチンポリペプチドは
、ムチンポリペプチドのN末端またはC末端に融合され得る。
【0024】
さらなる実施形態において、このABO融合タンパク質は、1以上のさらなる部分に連
結され得る。例えば、このABO融合タンパク質は、ABO融合タンパク質配列がGST
(すなわち、グルタチオンS−トランスフェラーゼ)配列のN末端に連結された、GST
融合タンパク質にさらに連結され得る。このような融合タンパク質は、ABO融合タンパ
ク質の精製を容易にし得る。あるいは、ABO融合タンパク質は、固体支持体にさらに連
結され得る。種々の固体支持体が当業者に公知である。このような組成物は、抗血液型抗
体の除去を容易にし得る。例えば、ABO融合タンパク質は、例えば、金属化合物、シリ
カ、ラテックス、ポリマー物質から作製される粒子;マイクロタイタープレート;ニトロ
セルロースもしくはナイロンまたはこれらの組み合わせに連結され得る。固体支持体に連
結されたABO融合タンパク質は、吸収剤として使用されて、生物学的サンプル(例えば
、血液または血漿)から、抗血液型抗体を除去する。
【0025】
別の実施形態では、融合タンパク質は、異種シグナル配列(すなわち、ムチン核酸によ
ってコードされるポリペプチド中に存在しないポリペプチド配列)をそのN末端に含む。
例えば、ネイティブなムチンシグナル配列は除去され得、そして別のタンパク質由来のシ
グナル配列で置き換えられ得る。特定の宿主細胞(例えば、哺乳動物宿主細胞)では、ポ
リペプチドの発現および/または分泌は、異種シグナル配列の使用を通して増大され得る
。
【0026】
本発明のキメラタンパク質または融合タンパク質は、標準的な組換えDNA技術によっ
て産生され得る。例えば、種々のポリペプチド配列をコードするDNAフラグメントは、
従来技術に従って、例えば、連結のための平滑末端化末端もしくは付着末端化末端、適切
な末端を提供する制限酵素消化、必要に応じて粘着末端のフィルイン、望ましくない連結
を回避するアルカリホスファターゼ処理、および酵素的連結を用いることによって、イン
フレームで一緒に連結される。別の実施形態では、融合遺伝子は、自動化DNA合成機を
含む、従来技術によって合成され得る。あるいは、遺伝子フラグメントのPCR増幅は、
その後アニーリングおよび再増幅されてキメラ遺伝子配列を作製し得る、2つの連続した
遺伝子フラグメントの間での相補的突出部を生じる、アンカープライマーを用いて実施さ
れ得る(例えば、Ausubelら(編)CURRENT PROTOCOLS IN
MOLECULAR BIOLOGY,John Wiley & Sons,1992
を参照のこと)。さらに、融合部分(例えば、免疫グロブリン重鎖のFc領域)をコード
する多くの発現ベクターが市販されている。糖タンパク質Ibαコード核酸は、その融合
部分が、免疫グロブリンタンパク質に対してインフレームで連結されるように、このよう
な発現ベクター中にクローニングされ得る。
【0027】
ABO融合ポリペプチドは、オリゴマー(例えば、ダイマー、トリマーまたはペンタマ
ー)として存在し得る。好ましくは、ABO融合ポリペプチドは、ダイマーである。
【0028】
第1ポリペプチド(および/または第1ポリペプチドをコードする核酸)は、当該分野
で公知であるムチンコード配列を用いて構築され得る。ムチンポリペプチドおよびムチン
ポリペプチドをコードする核酸についての適切な供給源としては、GenBank登録番
号NP663625およびNM145650、CAD10625およびAJ417815
、XP140694およびXM140694、XP006867およびXM006867
、ならびにNP00331777およびNM009151がそれぞれ挙げられ、そしてそ
れらの全体が本明細書中に参考として援用される。
【0029】
いくつかの実施形態では、ムチンポリペプチド部分は、天然に存在するムチン配列(野
生型)中に(非変異配列と比較して)増大した糖質含量をもたらす変異を有する改変体ム
チンポリペプチドとして提供される。例えば、改変体ムチンポリペプチドは、野生型ムチ
ンと比較してさらなるO結合型グリコシル化部位を含んでいた。あるいは、改変体ムチン
ポリペプチドは、野生型ムチンポリペプチドと比較して増加した数のセリン残基、トレオ
ニン残基またはプロリン残基をもたらす、アミノ酸配列変異を含む。この増加した糖質含
量は、当業者に公知の方法によって、ムチンのタンパク質対糖質の比率を決定することに
よって評価され得る。
【0030】
いくつかの実施形態では、ムチンポリペプチド部分は、天然に存在するムチン配列(野
生型)中に、タンパク質分解に対して(非変異配列と比較して)より耐性のムチン配列を
もたらす変異を有する改変体ムチンポリペプチドとして提供される。
【0031】
いくつかの実施形態では、第1ポリペプチドは、全長PSGL−1を含む。あるいは、
第1ポリペプチドは、全長未満のPSGL−1ポリペプチド(例えば、PSGL−1の細
胞外部分)を含む。例えば、400アミノ酸長未満(例えば、300アミノ酸長以下、2
50アミノ酸長以下、150アミノ酸長以下、100アミノ酸長以下、50アミノ酸長以
下または25アミノ酸長以下)の第1ポリペプチド。例示的なPSGL−1ポリペプチド
配列およびPSGL−1核酸配列としては、GenBank登録番号XP006867;
XM006867;XP140694およびXM140694が挙げられる。
【0032】
第2ポリペプチドは好ましくは可溶性である。いくつかの実施形態では、第2ポリペプ
チドは、第2ムチンポリペプチドとのABO融合ポリペプチドの会合を容易にする配列を
含む。好ましい実施形態では、第2ポリペプチドは、免疫グロブリンポリペプチドの少な
くとも一領域を含む。「少なくとも一領域」は、免疫グロブリン分子の任意の部分(例え
ば、軽鎖、重鎖、FC領域、Fab領域、Fv領域またはそれらの任意のフラグメント)
を含むことを意味する。免疫グロブリン融合ポリペプチドは、当該分野で公知であり、そ
して例えば、米国特許第5,516,964号;同第5,225,538号;同第5,4
28,130号;同第5,514,582号;同第5,714,147号;および同第5
,455,165号に記載される。
【0033】
いくつかの実施形態では、第2ポリペプチドは、全長免疫グロブリンポリペプチドを含
む。あるいは、第2ポリペプチドは、全長未満の免疫グロブリンポリペプチド(例えば、
重鎖、軽鎖、Fab、Fab2、Fv、またはFc)を含む。好ましくは、第2ポリペプ
チドは、免疫グロブリンポリペプチドの重鎖を含む。より好ましくは、第2ポリペプチド
は、免疫グロブリンポリペプチドのFc領域を含む。
【0034】
本発明の別の局面では、第2ポリペプチドは、野生型免疫グロブリン重鎖のFc領域の
エフェクター機能よりも劣ったエフェクター機能を有する。Fcエフェクター機能として
は、例えば、Fcレセプター結合、補体結合およびT細胞枯渇活性が挙げられる(例えば
、米国特許第6,136,310号を参照のこと)。T細胞枯渇活性、Fcエフェクター
機能、および抗体安定性をアッセイする方法は、当該分野で公知である。1つの実施形態
では、第2ポリペプチドは、Fcレセプターに対する親和性が低いかまたはない。代替的
な実施形態では、第2ポリペプチドは、補体タンパク質C1qに対する親和性が低いかま
たはない。
【0035】
本発明の別の局面は、ムチンポリペプチド、またはその誘導体、フラグメント、アナロ
グもしくはホモログをコードする核酸を含む、ベクター(好ましくは、発現ベクター)に
関する。種々の局面では、このベクターは、免疫グロブリンポリペプチド、またはその誘
導体、フラグメント、アナログもしくはホモログをコードする核酸に作動可能に連結した
、ムチンポリペプチドをコードする核酸を含む。さらに、このベクターは、血液型トラン
スフェラーゼ(例えば、α1,2フコシルトランスフェラーゼ、α1,3Nアセチルガラ
クトサミニルトランスフェラーゼ(α1,3N acetylgalactosamin
inytransferase)、α1,3ガラクトシルトランスフェラーゼ、またはこ
れらの任意の組み合わせ)をコードする核酸を含む。血液型トランスフェラーゼは、AB
O融合タンパク質のムチン部分のペプチド(peptode)骨格に対する血液型決定因
子の付加を促進する。本明細書中で用いられる場合、用語「ベクター」とは、連結されて
いる別の核酸分子を輸送し得る核酸分子をいう。ベクターの1つの型は、「プラスミド」
であり、プラスミドとは、さらなるDNAセグメントが連結され得る環状二本鎖DNAル
ープをいう。ベクターの別の型は、ウイルスベクターであり、ここで、さらなるDNAセ
グメントがウイルスゲノムに連結され得る。特定のベクターは、これらが導入される宿主
細胞中で自律的複製をし得る(例えば、細菌の複製起点を有する細菌ベクターおよびエピ
ソームの哺乳動物ベクターを有する)。他のベクター(例えば、非エピソームの哺乳動物
ベクター)は、宿主細胞へと導入される際に、宿主細胞のゲノムへと組み込まれ、それに
よって、宿主ゲノムと共に複製される。さらに、特定のベクターは、それらが作動可能に
連結される遺伝子の発現を指向し得る。このようなベクターは、本明細書中で、「発現ベ
クター」といわれる。一般に、組換えDNA技術において有用な発現ベクターは、頻繁に
、プラスミドの形態である。本明細書中で、「プラスミド」および「ベクター」は、交換
可能に用いられ得る。なぜならば、プラスミドは、最も一般的に用いられるベクターの形
態だからである。しかし、本発明は、等価な機能を果たす、ウイルスベクター(例えば、
複製欠損のレトロウイルス、アデノウイルスおよびアデノ随伴ウイルス)のような、発現
ベクターのこのような他の形態を含むことを意図する。
【0036】
本発明の組換え発現ベクターは、宿主における核酸の発現に適切な形態で、本発明の核
酸分子を含み、このことは、組換え発現ベクターが、発現に用いられる宿主細胞に基づい
て選択された(すなわち、発現される核酸配列に作動可能に連結された)、1つ以上の調
節配列を含むことを意味する。組換え発現ベクターにおいて、「作動可能に連結した(さ
れる)」とは、目的のヌクレオチド配列が、このヌクレオチド配列の発現を可能にする様
式で(例えば、インビトロ転写/翻訳システムにおいて、またはベクターが宿主細胞に導
入される場合、宿主細胞において)、調節配列に連結されるという意味が意図される。
【0037】
用語「調節配列」は、プロモーター、エンハンサーおよび他の発現制御エレメント(例
えば、ポリアデニル化シグナル)を含むことが意図される。このような調節配列は、例え
ば、Goeddl,GENE EXPRESSION TECHNOLOGY:METH
ODS IN ENZYMOLOGY 185,Academic Press,San
Diego,Calif.(1990)に記載される。調節配列としては、宿主細胞の
多くの型においてヌクレオチド配列の構成的発現を指向する配列、および特定の宿主細胞
においてのみヌクレオチド配列の発現を指向する配列(例えば、組織特異的調節配列)が
挙げられる。発現ベクターの設計は、例えば、形質転換される宿主細胞の選択、所望され
るタンパク質の発現のレベルなどの因子に依存し得ることが当業者により理解される。本
発明の発現ベクターは、宿主細胞へと導入されて、それにより、本明細書中に記載される
ような核酸によりコードされる、融合タンパク質もしくは融合ペプチド(例えば、ABO
融合ポリペプチド、ABO融合ポリペプチドの変異形態など)を含む、タンパク質もしく
はペプチドを産生し得る。
【0038】
本発明の組換え発現ベクターは、原核生物細胞または真核生物細胞におけるABO融合
ポリペプチドの発現のために設計され得る。例えば、ABO融合ポリペプチドは、Esc
herichia coliのような細菌細胞、昆虫細胞(バキュロウイルス発現ベクタ
ーを用いる)、酵母細胞または哺乳動物細胞において発現され得る。適切な宿主細胞は、
Goeddle,GENE EXPRESSION TECHNOLOGY:METHO
DS IN ENZYMOLOGY 185,Academic Press,San
Diego,Calif.(1990)にさらに考察される。あるいは、組換え発現ベク
ターは、例えば、T7プロモーター調節配列およびT7ポリメラーゼを用いて、インビト
ロで転写および翻訳され得る。
【0039】
原核生物におけるタンパク質の発現は、最も頻繁に、融合タンパク質または非融合タン
パク質のいずれかの発現を指向する構成的プロモーターまたは誘導性プロモーターを含む
ベクターを用いて、Escherichia coliで実施される。融合ベクターは、
それらの中でコードされるタンパク質に、通常は、組換えタンパク質のアミノ末端に、多
数のアミノ酸を付加する。このような融合ベクターは、代表的には、以下の3つの目的を
果たす:(i)組換えタンパク質の発現を増大させること;(ii)組換えタンパク質の
可溶性を増大させること;および(iii)アフィニティー精製におけるリガンドとして
作用することにより、組換えタンパク質の精製を補助すること。頻繁に、融合発現ベクタ
ーにおいて、タンパク質分解切断部位が、融合部分と組み換えタンパク質との接合部に導
入されて、融合タンパク質の精製、引き続く、この融合部分からの組換えタンパク質の分
離を可能にする。このような酵素、およびこれらの同族の認識配列としては、第Xa因子
、トロンビンおよびエンテロキナーゼが挙げられる。代表的な融合発現ベクターとしては
、pGEX(Pharmacia Biotech Inc;SmithおよびJohn
son,1988.Gene 67:31−40)、pMAL(New England
Biolabs,Beverly,Mass.)、およびpRIT5(Pharmac
ia,Piscataway,N.J.)が挙げられ、これらは、標的組換えタンパク質
に対して、それぞれ、グルタチオンS−トランスフェラーゼ(GST)、マルトースE結
合タンパク質、またはプロテインAを融合する。
【0040】
適切な誘導性非融合E.coli発現ベクターの例としては、pTrc(Amrann
ら(1988)Gene 69:301−305)およびpET 11d(Studie
rら、GENE EXPRESSION TECHNOLOGY:METHODS IN
ENZYMOLOGY 185,Academic Press,San Diego
,Calif.(1990)60−89)が挙げられる。
【0041】
E.coli中の組換えタンパク質発現を最小にする1つのストラテジーは、組み換え
タンパク質をタンパク質分解により切断する能力が損なわれた宿主細菌において、タンパ
ク質を発現することである。例えば、Gottesman,GENE EXPRESSI
ON TECHNOLOGY:METHODS IN ENZYMOLOGY 185,
Academic Press,San Diego,Calif.(1990)119
−128を参照のこと。別のストラテジーは、各アミノ酸についての個々のコドンが、E
.coliにおいて優先的に利用されるものであるように、発現ベクターへと挿入される
核酸の核酸配列を変化させることである(例えば、Wadaら、1992.Nucl.A
cids Res.20:2111−2118を参照のこと)。本発明の核酸配列のこの
ような変更は、標準的なDNA合成技術により実行され得る。
【0042】
別の実施形態において、ABO融合ポリペプチド発現ベクターは、酵母発現ベクターで
ある。酵母Saccharomyces cerivisaeにおける発現のためのベク
ターの例としては、pYepSec1(Baldariら、1987.EMBO J.6
:229−234)、pMFa(KurjanおよびHerskowitz,1982.
Cell 30:933−943)、pJRY88(Schultzら、1987.Ge
ne 54:113−123)、pYES2(Invitrogen Corporat
ion,San Diego,Calif.)およびpicZ(InVitrogen
Corp,San Diego,Calif.)が挙げられる。
【0043】
あるいは、ABO融合ポリペプチドは、バキュロウイルス発現ベクターを使用して、昆
虫細胞において発現され得る。培養昆虫細胞(例えば、SF9細胞)におけるタンパク質
の発現のために利用可能なバキュロウイルスベクターとしては、pAcシリーズ(Smi
thら、1983.Mol.Cell.Biol.3:2156−2165)およびpV
Lシリーズ(LucklowおよびSummers,1989.Virology 17
0:31−39)が挙げられる。
【0044】
さらに別の実施形態において、本発明の核酸は、哺乳動物発現ベクターを使用して、哺
乳動物細胞において発現される。哺乳動物発現ベクターの例としては、pCDM8(Se
ed,1987.Nature 329:840)およびpMT2PC(Kaufman
ら、1987.EMBO J.6:187−195)が挙げられる。哺乳動物細胞におい
て使用される場合、この発現ベクターの制御機能は、しばしば、ウイルス調節エレメント
により提供される。例えば、一般に使用されるプロモーターは、ポリオーマ、アデノウイ
ルス2、サイトメガロウイルスおよびシミアンウイルス40由来である。原核生物細胞お
よび真核生物細胞の両方に適切な他の発現系については、例えば、Sambrookら、
MOLECULAR CLONING:A LABORATORY MANUAL.第2
版、Cold Spring Harbor Laboratory,Cold Spr
ing Harbor Laboratory Press,Cold Spring
Harbor,N.Y.,1989の第16章および17章を参照のこと。
【0045】
本発明の別の局面は、本発明の組換え発現ベクターが導入された宿主細胞に関する。用
語「宿主細胞」および「組換え宿主細胞」とは、本明細書中で交換可能に使用される。こ
のような用語は、特定の被験体細胞を言及するだけでなく、このような細胞の子孫または
潜在的な子孫も言及することが理解される。特定の改変は、変異の影響または環境的な影
響のいずれかに起因して、次世代で生じ得るので、このような子孫は、実際のところ、親
細胞と同一ではないかもしれないが、本明細書中で使用されるような用語の範囲内になお
含まれる。
【0046】
宿主細胞は、任意の原核動物細胞または真核生物細胞であり得る。例えば、糖タンパク
質Ibα融合ポリペプチドは、細菌細胞(例えば、E.coli)、昆虫細胞、酵母細胞
または哺乳動物細胞(例えば、ヒト細胞、チャイニーズハムスター卵巣細胞(CHO)も
しくはCOS細胞)において発現され得る。他の適切な宿主細胞が、当業者に公知である
。
【0047】
ベクターDNAは、従来の形質転換技術またはトランスフェクション技術によって、原
核生物細胞または真核生物細胞に導入され得る。本明細書中で使用される場合、用語「形
質転換」および「トランスフェクション」は、異種核酸(例えば、DNA)を宿主細胞に
導入するための当該分野で認められた種々の技術をいうことが意図され、これらの技術と
しては、リン酸カルシウム共沈殿もしくは塩化カルシウム共沈殿、DEAE−デキストラ
ン媒介性トランスフェクション、リポフェクションまたはエレクトロポレーションが挙げ
られる。宿主細胞を形質転換またはトランスフェクションするための適切な方法は、Sa
mbrookら(MOLECULAR CLONING:A LABORATORY M
ANUAL.第2版、Cold Spring Harbor Laboratory,
Cold Spring Harbor Laboratory Press,Cold
Spring Harbor,N.Y.,1989)、および他の研究用マニュアルに
見出され得る。
【0048】
哺乳動物細胞の安定なトランスフェクションについて、使用される発現ベクターおよび
トランスフェクション技術に依存して、わずかな細胞しか異種DNAをそのゲノムに組み
込み得ないことが知られている。これらの要素を同定および選択するために、選択マーカ
ー(例えば、抗生物質に対する耐性)をコードする遺伝子は、一般的に、目的の遺伝子と
共に宿主細胞に導入される。種々の選択マーカーとしては、薬物に対する耐性を与えるマ
ーカー(例えば、G418、ハイグロマイシンおよびメトトレキサート)が挙げられる。
選択マーカーをコードする核酸は、糖タンパク質Ibα融合ポリペプチドをコードするベ
クターと同じベクターを用いて宿主細胞に導入され得るか、または別個のベクターを用い
て導入され得る。導入された核酸で安定にトランスフェクトされた細胞は、薬物選択によ
り同定され得る(例えば、選択マーカー遺伝子を組み込まれた細胞は生き残り、一方、他
の細胞は、死ぬ)。
【0049】
培養物中の本発明の宿主細胞(例えば、原核生物宿主細胞または真核生物宿主細胞)は
、ABO融合ポリペプチドを産生(すなわち、発現)するために使用され得る。従って、
本発明はさらに、本発明の宿主細胞を使用して、ABO融合ポリペプチドを産生するため
の方法を提供する。一実施形態において、この方法は、本発明の宿主細胞(この宿主細胞
に、ABO融合ポリペプチドをコードする組換え発現ベクターが導入されている)を、適
切な培地中で、ABO融合ポリペプチドが産生するように、培養する工程を包含する。別
の実施形態において、この方法はさらに、この培地または宿主細胞からABOポリペプチ
ドを単離する工程を包含する。
【0050】
ABO融合ポリペプチドは、従来の条件(例えば、抽出、沈殿、クロマトグラフィー、
アフィニティクロマトグラフィー、電気泳動など)に従って、単離および精製され得る。
例えば、免疫グロブリン融合タンパク質は、この融合タンパク質のFc部分に選択的に結
合する固定化されたプロテインAまたはプロテインGを含むカラムに溶液を通すことによ
り、精製され得る。例えば、Reis,K.J.ら、J.Immunol.132:30
98−3102(1984)PCT出願公開番号WO87/00329を参照のこと。こ
の融合ポリペプチドは、カオトロピック塩での処理によって、または酢酸水溶液(1M)
での溶出によって、溶出され得る。
【0051】
あるいは、本発明に従うABO融合ポリペプチドは、当該分野で公知の方法を使用して
、化学的に合成され得る。ポリペプチドの化学合成は、例えば、以下に記載される。種々
のタンパク質合成方法が、当該分野で公知であり、これにはペプチド合成機を使用する合
成が挙げられる。例えば、Peptide Chemistry,A Practica
l Textbook,Bodasnsky編、Springer−Verlag,19
88;Merrifield,Science 232:241−247(1986);
Baranyら、Intl.J.Peptide Protein Res.30:70
5−739(1987);Kent,Ann.Rev.Biochem.57:957−
989(1988)、およびKaiserら、Science 243:187−198
(1989)を参照のこと。このポリペプチドは、化学前駆体も他の化学物質も実質的に
含まなくなるように、標準的なペプチド精製技術を使用して精製される。用語「化学前駆
体も他の化学物質も実質的に含まない」は、ペプチドが、このペプチドの合成に関与する
化学前駆体または他の化学物質から分離されているペプチドの調製物を含む。一実施形態
において、用語「化学前駆体も他の化学物質も実質的に含まない」は、約30%(乾燥重
量)未満の化学前駆体または非ペプチド化学物質、より好ましくは約20%未満の化学前
駆体または非ペプチド化学物質、さらにより好ましくは約10%未満の化学前駆体または
非ペプチド化学物質、最も好ましくは約5%未満の化学前駆体または非ペプチド化学物質
を有するペプチドの調製物を含む。
【0052】
ポリペプチドの化学合成は、改変されたかまたは非天然のアミノ酸(D−アミノ酸が挙
げられる)および他の有機低分子の組み込みを容易にする。対応するD−アミノ酸アイソ
フォームでの、ペプチド中の1つ以上のL−アミノ酸の置換は、酵素的加水分解に対する
ペプチドの耐性を増加するため、および生物学的に活性なペプチドの1つ以上の特性(す
なわち、レセプター結合、機能的効力または作用の持続時間)を増強するために、使用さ
れ得る。例えば、Dohertyら、1993.J.Med.Chem.36:2585
−2594;Kirbyら、1993.J.Med.Chem.36:3802−380
8;Moritaら、1994.FEBS Lett.353:84−88;Wangら
、1993.Int.J.Pept.Protein Res.42:392−399;
FauchereおよびThiunieau,1992.Adv.Drug Res.2
3:127−159を参照のこと。
【0053】
ペプチド配列への供給架橋の導入は、ポリペプチド骨格を立体配置的かつ組織分布的に
制約し得る。このストラテジーは、増加した効力、選択性および安定性を有する融合ポリ
ペプチドのペプチドアナログを開発するために使用され得る。環状ペプチドの構造エント
ロピーは、その直線状対応物の構造エントロピーより低いので、特定のコンフォメーショ
ンの採用は、非環式アナログのエントロピーの減少よりも小さい環状アナログのエントロ
ピーの減少を伴って生じ、それにより、結合の自由エネルギーをより有利にする。大環状
化は、しばしば、ペプチドのN末端とC末端との間、側鎖とN末端もしくはC末端との間
[例えば、pH8.5においてK3Fe(CN)6を用いた場合](Samsonら、E
ndocrinology,137:5182−5185(1996))、または2つの
アミノ酸側鎖の間のアミド結合の形成によって達成される。例えば、DeGrado,A
dv Protein Chem,39:51−124(1988)を参照のこと。ジス
ルフィド結合もまた、直線状配列に組み込まれ、その可撓性を低下させる。例えば、Ro
seら、Adv Protein Chem,37:1−109(1985);Mosb
ergら、Biochem Biophys Res Commun,106:505−
512(1982)を参照のこと。さらに、ペニシラミン(Pen,3−メルカプト−(
D)バリン)でのシステイン残基の置換は、あるオピオイド−レセプター相互作用の選択
性を増加するために使用されている。LipkowskiおよびCarr,Peptid
es:Synthesis,Structures,and Applications
,Gutte編、Academic Press 287−320頁(1995)。
【0054】
(抗体媒介性移植片拒絶の処置または予防の方法)
抗体媒介性移植片拒絶(AMR)(例えば、臓器移植拒絶)を処置または予防する方法
もまた、本発明に含まれる。このような移植片としては、腎臓、肝臓、皮膚、膵臓、角膜
または心臓が挙げられるが、これらに限定されない。AMRとは、レシピエントによる任
意の抗体媒介性移植片拒絶を含むことを意味する。この方法は、被験体由来の生物学的サ
ンプルを本発明のABO融合ペプチドと接触させる工程を包含する。この生物学的サンプ
ルは、例えば、血液(すなわち、全血または血漿)である。このサンプルは、抗体(例え
ば、抗血液型抗体)を含むことが知られているか、またはこのような抗体を含むと疑われ
ている。いくつかの局面において、この生物学的サンプルは、ABO融合ポリペプチドと
このサンプルとを接触させる前に、被験体から取り出される。この生物学的サンプルは、
ABO融合ペプチド−抗血液型抗体複合体の形成を可能にする条件下で、ABO融合ペプ
チドと接触される。このABO融合ペプチド複合体は、存在する場合、抗血液型抗体を排
除するためにこの生物学的サンプルから分離され、そしてこの生物学的サンプルは、被験
体に再注入される。AMRはまた、本発明のABO融合ポリペプチドを被験体に投与する
ことによって、処置または予防される。
【0055】
被験体は、例えば、任意の哺乳動物(例えば、ヒト、霊長類、マウス、ラット、イヌ、
ネコ、ウシ、ウマ、ブタ)であり得る。処置は、被験体がABO不適合性移植片を受ける
前に、施される。あるいは、処置は、被験体がABO不適合性移植片を受けた後に、施さ
れる。
【0056】
生物学的サンプルは、当業者に公知の方法によって、ABO融合タンパク質と接触され
る。例えば、血漿しゃ血または体外免疫吸収。
【0057】
本質的に、抗体媒介性反応と病原学的に関連する任意の障害は、予防または処置を受け
やすいとみなされる。AMRは、臓器移植の生存率が、本発明の方法により処置されない
臓器移植の生存率よりも大きい場合、処置または予防される。移植の生存率とは、移植片
がレシピエントにより拒絶される前の時間を意味する。例えば、AMRは、移植片が、移
植後少なくとも1、2、4または8週間生存する場合、処置または予防される。好ましく
は、この移植片は、3、6、13ヶ月生存する。より好ましくは、この移植片は、2、3
、5年またはそれ以上生存する。
【0058】
(サンプルから抗血液型抗体を除去する方法)
サンプルから抗血液型抗体を除去または枯渇する方法もまた、本発明に含まれる。この
サンプルは、生物学的流体(例えば、血液または血漿)である。あるいは、このサンプル
は、生物学的組織(例えば、心臓組織、肝臓組織、皮膚または腎臓組織)である。この方
法は、サンプルと本発明のABO融合ペプチドを接触させる工程を包含する。このサンプ
ルは、ABO融合ペプチド−抗血液型抗体複合体の形成を可能にする条件下で、ABO融
合ペプチドと接触される。このABO融合ペプチド−抗体複合体は、存在する場合、抗血
液型抗体を除去または枯渇するために、この生物学的サンプルから分離される。
【0059】
(ABO融合ポリペプチドまたはこれらをコードする核酸を含む薬学的組成物)
本発明のABO融合タンパク質、またはこれらの融合タンパク質をコードする核酸分子
(本明細書中において、「治療剤」または「活性化合物」とも呼ばれる)、ならびにこれ
らの誘導体、フラグメント、アナログおよびホモログは、投与のために適切な薬学的組成
物に混合され得る。このような組成物は、代表的に、核酸分子、タンパク質、または抗体
および薬学的に受容可能なキャリアを含む。本明細書中において使用される場合、「薬学
的に受容可能なキャリア」は、薬学的投与に適合性の、任意の全ての溶媒、分散媒体、コ
ーティング、抗菌剤および抗真菌剤、等張性剤および吸収遅延剤などを含むことが意図さ
れる。適切なキャリアは、当該分野における標準的な参考教科書である、Remingt
on’s Pharmaceutical Sciencesの最新版(本明細書中にお
いて参考として援用される)に記載される。このようなキャリアまたは希釈剤の好ましい
例としては、限定しないが、水、生理食塩水、フィンガー溶液(finger’s so
lution)、デキストロース溶液、および5%ヒト血清アルブミンが挙げられる。リ
ポソームおよび非水性ビヒクル(例えば、不揮発性油)もまた使用され得る。薬学的に活
性な物質のためのこのような媒体および薬剤の使用は、当該分野において周知である。任
意の従来の媒体または薬剤が活性化合物と不適合性である場合を除いて、組成物における
それらの使用が企図される。補助的な活性化合物もまた、組成物に組み込まれ得る。
【0060】
本明細書中に開示される活性剤はまた、リポソームとして処方され得る。リポソームは
、当該分野で公知の方法(例えば、Epsteinら、Proc.Natl.Acad.
Sci.USA,82:3688(1985);Hwangら、Proc.Natl.A
cad.Sci.USA,77:4030(1980);および米国特許第4,485,
045号および同第4,544,545号に記載される)によって調製される。向上した
循環時間を有するリポソームは、米国特許第5,013,556号に開示される。
【0061】
特に有用なリポソームは、ホスファチジルコリン、コレステロール、およびPEG誘導
体化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物を用いる、逆相
エバポレーション法によって作製され得る。リポソームは、所望の直径を有するリポソー
ムを生じるように、規定された孔サイズのフィルターを通して押し出される。
【0062】
本発明の薬学的組成物は、その意図された投与の経路と適合性であるように処方される
。投与の経路の例としては、非経口的投与、例えば、静脈内投与、皮内投与、皮下投与、
経口(例えば、吸入)投与、経皮(すなわち、局所的)投与、経粘膜投与、および直腸投
与が挙げられる。非経口適用、皮内適用、または皮下適用のために使用される溶液または
懸濁液は、以下の成分を含み得る:注射用水、生理食塩水溶液、不揮発性油、ポリエチレ
ングリコール、グリセリン、プロピレングリコールまたは他の合成溶媒のような滅菌希釈
剤;ベンジルアルコールまたはメチルパラベンのような抗菌剤;アスコルビン酸または重
亜硫酸ナトリウムのような抗酸化剤;エチレンジアミン四酢酸(EDTA)のようなキレ
ート剤;アセテート、シトレートまたはホスフェートのような緩衝剤、および塩化ナトリ
ウムまたはデキストロースのような張度の調節のための薬剤。pHは、酸または塩基(例
えば、塩酸または水酸化ナトリウム)を用いて調節され得る。非経口調製物は、ガラスま
たはプラスチックから作製されたアンプル、使い捨て可能シリンジまたは複数用量のバイ
アル中に含まれ得る。
【0063】
注射可能用途に適切な薬学的組成物としては、滅菌水溶液(水溶性である場合)または
分散物および滅菌注射可能溶液または分散物の即席調製物のための滅菌粉末が挙げられる
。静脈内投与のために、適切なキャリアとしては、生理学的生理食塩水、静菌水、Cre
mophor ELTM(BASF、Parsippany,N.J.)またはリン酸緩
衝化生理食塩水(PBS)が挙げられる。全ての場合において、組成物は、滅菌でなけれ
ばならず、そして容易に注射可能である程度まで流体であるべきである。組成物は、製造
および保存の条件下において安定でなければならず、そして微生物(例えば、細菌および
真菌)の混入作用に対して保存されなければならない。キャリアは、例えば、水、エタノ
ール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチ
レングリコールなど)、およびこれらの適切な混合物を含む溶媒または分散媒体であり得
る。適切な流動性は、例えば、レシチンのようなコーティングの使用によって、分散の場
合、必要とされる粒子サイズの維持によって、そして界面活性剤の使用によって、維持さ
れ得る。微生物の作用の防止は、種々の抗菌剤および抗真菌剤(例えば、パラベン、クロ
ロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成され得る
。多くの場合において、等張性剤(例えば、糖、ポリアルコール(例えば、マンニトール
)、ソルビトール、塩化ナトリウム)を組成物中に含むことが好ましい。注射可能組成物
の長期の吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよび
ゼラチン)を組成物中に含むことによってもたらされ得る。
【0064】
滅菌注射可能溶液は、活性化合物(例えば、ABO融合タンパク質)を必要とされる量
で、適切な溶媒中に、上記成分の1つまたは組合せと混合し、必要に応じて、続いて、滅
菌濾過することによって、調製され得る。一般的に、分散物は、ベーシックな分散媒体お
よび必要とされる上記成分からの他の成分を含む、滅菌ビヒクルに、活性化合物を混合す
ることによって調製される。滅菌注射可能溶液の調製のための滅菌粉末の場合、調製方法
は、真空乾燥および凍結乾燥であり、これは、先に滅菌濾過された溶液からの、活性成分
および任意のさらなる所望の成分の粉末を生じる。
【0065】
経口組成物は、一般的に、不活性な希釈剤または食用キャリアを含む。これらは、ゼラ
チンカプセルに含まれ得るかまたは錠剤に圧縮され得る。経口治療投与のために、活性化
合物は、賦形剤と混合され得、そして錠剤、トローチ、またはカプセルの形態で使用され
得る。経口組成物はまた、マウスウオッシュとしての使用のために流体キャリアを使用し
て調製され得、ここで、流体キャリア中の化合物は、経口適用され、スウィッシュされ、
そして吐き出されるかまたは嚥下される。薬学的に適合性の結合剤、および/またはアジ
ュバント物質は、組成物の一部として含まれ得る。錠剤、丸剤、カプセル剤、トローチ剤
などは、類似の性質の以下の成分、または化合物のいずれかを含み得る:結合剤(例えば
、微結晶セルロース、トラガカントガムまたはゼラチン);賦形剤(例えば、デンプンま
たはラクトース)、崩壊剤(例えば、アルギン酸、Primogel、またはコーンスタ
ーチ);滑沢剤(例えば、ステアリン酸マグネシウムまたはSterote);グリダン
ト(glidant)(例えば、コロイド状二酸化ケイ素);甘味剤(例えば、スクロー
スまたはサッカリン);または香料(例えば、ペパーミント、サリチル酸メチル、または
オレンジ香料)。
【0066】
吸入による投与のために、化合物は、適切な噴霧剤(例えば、二酸化炭素のような気体
)を含む加圧容器または分散器、あるいは噴霧器からエアロゾル噴霧の形態で送達される
。
【0067】
全身投与はまた、経粘膜的手段または経皮的手段によってであり得る。経粘膜的投与ま
たは経皮的投与のために、浸透される障壁に対して適切な浸透剤は、処方物で使用される
。このような浸透剤は、一般的に、当該分野で公知であり、そして例えば、経粘膜的投与
のため、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜的投与は
、経鼻スプレーまたは坐剤の使用によって達成され得る。経皮投与のために、活性化合物
は、当該分野において一般的に公知な、軟膏(ointment)、軟膏(salve)
、ゲル、またはクリームに処方される。
【0068】
化合物はまた、坐剤の形態で(例えば、ココアバターおよび他のグリセリドのような従
来の坐剤ベースとともに)または直腸送達のための保持浣腸で、調製され得る。
【0069】
1つの実施形態において、活性化合物は、身体からの迅速な排除に対して化合物を保護
するキャリア(例えば、徐放処方物(移植物およびマイクロカプセル化送達システムを含
む))を用いて調製される。生分解性、生体適合性ポリマー(例えば、エチレンビニルア
セテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポ
リ乳酸)が使用され得る。このような処方物の調製のための方法は、当業者に明らかであ
る。これらの材料はまた、Alza CorporationおよびNova Phar
maceuticals,Inc.から商業的に入手され得る。リポソーム懸濁物(ウイ
ルス抗原に対するモノクローナル抗体を用いて感染した細胞に標的化されたリポソームを
含む)もまた、薬学的に受容可能なキャリアとして使用され得る。これらは、例えば、米
国特許第4,522,811号に記載されるように、当業者に公知の方法に従って調製さ
れ得る。
【0070】
いくつかの実施形態において、経口組成物または非経口組成物は、投与の容易さおよび
均一な投与のために投薬単位形態で処方される。本明細書中で使用される投薬単位形態は
、処置される被験体に対して単位投薬として適切な物理的に異なる単位をいい;それぞれ
の単位は、必要とされる薬学的キャリアとともに、所望の治療的効果を生成するように計
算された所定量の活性化合物を含む。本発明の投薬単位形態のための詳細は、活性化合物
の独特の特徴および達成されるべき特定の治療効果、ならびに個体の処置のためのこのよ
うな活性化合物を配合する当該分野に固有の制限によって示され、そして直接的に依存す
る。
【0071】
本発明の核酸分子は、ベクターに挿入され得、そして遺伝子治療ベクターとして使用さ
れ得る。遺伝子治療ベクターは、被験体に、例えば、静脈内注射、局所投与(例えば、米
国特許第5,328,470号を参照のこと)によって、または定位的な注射(例えば、
Chenら、1994.Proc.Natl.Acad.Sci.USA 91:305
4−3057を参照のこと)によって被験体に送達され得る。遺伝子治療ベクターの薬学
的調製物は、受容可能な希釈剤中に遺伝子治療ベクターを含み得るか、または遺伝子治療
ビヒクルが埋め込まれる徐放マトリクスを含み得る。あるいは、完全な遺伝子送達ベクタ
ーが、組換え細胞からインタクトに作製され得る場合(例えば、レトロウイルスベクター
)、薬学的調製物は、遺伝子送達システムを作製する1つ以上の細胞を含み得る。
【0072】
持続放出調製物は、所望であれば、調製され得る。持続放出調製物の適切な例としては
、抗体を含む固体疎水性ポリマーの半透過性マトリクスが挙げられ、このマトリクスは、
形付けられた物品(例えば、フィルムまたはマイクロカプセル)の形態である。持続放出
マトリクスの例としては、ポリエステル、ヒドロゲル(例えば、ポリ(2−ヒドロキシエ
チル−メタクリレート)、またはポリ(ビニルアルコール))、ポリラクチド(米国特許
第3,773,919号)、L−グルタミン酸およびγエチル−L−グルタメートのコポ
リマー、非分解性エチレン−ビニルアセテート、分解性乳酸−グリコール酸コポリマー(
例えば、LUPRON DEPOTTM(乳酸−グリコール酸コポリマーおよび酢酸ロイ
プロリドから構成される注射可能なミクロスフェア)、およびポリ−D−(−)−3−ヒ
ドロキシ酪酸が挙げられる。エチレンビニルアセテートおよび乳酸−グリコール酸のよう
なポリマーが100日にわたる間、分子の放出を可能にする一方で、特定のヒドロゲルが
より短い時間の間、タンパク質を放出する。
【0073】
薬学的組成物は、投与のための説明書とともに、容器、パックまたは分散器中に含まれ
得る。
【0074】
本発明はさらに、以下の非制限的な実施例で例示される。
【実施例】
【0075】
(実施例1:一般的方法)
本明細書中に記載されるデータは、以下の試薬および方法を使用して作製された。
【0076】
(細胞培養)
COS−7 m6細胞(Seed,1987)、CHO−K1(ATCC CCL−6
1)、およびSV40ラージT抗原を発現する293ヒト胚性腎臓細胞株(293T;B
.Seedによって親切にも提供された)を、Dulbeccoの改変Eagle培地(
GibcoBrl,Life Technologies,Paisley,Scotl
and)(10%ウシ胎仔血清(GibcoBrl,Life Technologie
s)、25μg/mlゲンタマイシンスルフェート(Sigma、St.Louis、M
O)および2mMグルタミン(GibcoBrl,Life Technologies
)を補充した)において培養した。細胞を、2〜4日毎に継代させた。HH14ハイブリ
ドーマ(ATCC HB−9299;米国特許第4,857,639号)を、RPMI
1640(GibcoBrl,Life Technologies)(10%ウシ胎仔
血清、100U/mlペニシリン、100μg/μlストレプトマイシンおよび2mMグ
ルタミンを補充した)において培養した。
【0077】
(HH14抗体の精製)
上清を、培養したHH14細胞から収集した。2リットルの上清を、ヤギ抗マウスIg
M(Sigma)カラムで、Bio−Rad LPクロマトグラフ(Bio−Rad,H
ercules,CA)を用いてアフィニティー精製した。結合したタンパク質を、0.
1MグリシンCl(pH2.5)によって溶出させ、そして溶出物を、1M Tris−
Cl(pH7.5)を用いて直ちに中和した。溶出物を、1%リン酸緩衝化生理食塩水(
PBS)に対して透析し、そして凍結乾燥した。凍結乾燥したタンパク質を、BCAアッ
セイによって測定した場合に3μg/μlの最終濃度になるように、蒸留H2O中に溶解
させた。
【0078】
(発現ベクターの構築)
ヒト血液型A遺伝子を、MKN−45細胞株から単離された総RNAから作製されたc
DNAから、5’−cgc ggg aag ctt gcc gag acc aga
cgc gga−3’(配列番号1)を正方向プライマーとして、そして5’−cgc
ggg cgg ccg ctc acg ggt tcc gga ccg c−3
’(配列番号2)を逆方向プライマーとして用いたポリメラーゼ連鎖反応(PCR)によ
って増幅した。増幅されたcDNA(A遺伝子)を、Hind IIIおよびNot I
を用いて、CDM8(Seed,1987)のポリリンカー中にサブクローン化した。血
液型H遺伝子を、ヒト扁桃支質ライブラリーをテンプレートとして用いてPCR増幅して
2片とした。内部Sse I部位を、ヌクレオチド775(GenBank登録番号M3
5531)をCへと変化させて、Pst I部位からSse I部位を作製する内部重複
プライマーを用いたPCRによって作製した。FUT1のカルボキシ末端をコードするc
DNAを、5’−ggg gac tac ctg cag gtt atg cct
cag cgc−3’(配列番号3)を正方向プライマーとして、そして5’−cgc
ggg gcg gcc gct tca agg ctt agc caa tgt−
3’(配列番号4)を逆方向プライマーとして用いて増幅し、Sse IおよびNot
Iによって切断し、そしてPst IおよびNot Iで消化したCDM8発現ベクター
中にサブクローン化した。FUT1のアミノ末端をコードするcDNAを、5’−cgc
ggg aag ctt acc atg tgg ctc cgg agc cat
−3’(配列番号5)を正方向プライマーとし、そして5’−cca gcg ctg
agg cat aac ctg cag gta gtc−3’(配列番号6)を逆方
向プライマーとして用いて増幅し、Hind IIIおよびSse Iによって切断し、
そしてHind IIIおよびSse I切断後の、カルボキシ末端を保有するCDM8
中にサブクローン化した。
【0079】
Se遺伝子を、血液型A2Le(a−b+)Se個体によって提供された末梢血単核細
胞から単離された総RNAから逆転写されたcDNAから、5’−cgc ggg aa
g ctt acc atg ctg gtc gtt cag atg−3’(配列番
号7)を正方向プライマーとして、そして5’−cgc ggg cgg ccg ct
t agt gct tga gta agg g−3’(配列番号8)を逆方向プライ
マーとして用いて同様にPCR増幅した。このSe遺伝子cDNAを、Hind III
およびNot Iを用いてCDM8中にサブクローン化した。グリコシルトランスフェラ
ーゼcDNAを配列決定し、そしてこれらがコードする酵素活性を、血液型HおよびA特
異的モノクローナル抗体を用いた、一過的にトランスフェクトされた細胞のフローサイト
メトリー分析によってチェックした。PSGL−1/mIgG2bキメラを、以前(Li
uら,1997)に記載されたとおりに構築した。
【0080】
(トランスフェクションおよび分泌されたPSGL−1/mIgG2bキメラの産生)
トランスフェクションカクテルを、5mlポリスチレンチューブ中で39μlの20%
グルコース、39μgのプラスミドDNA、127μlのdH2O、および15.2μl
の0.1Mポリエチレンイミン(25kDa;Aldrich,Milwaukee,W
I)を混合することによって調製した。全てのトランスフェクション混合物において、1
3μgのPSGL‐1/mIgG2bプラスミドを用いた。種々のグリコシルトランスフ
ェラーゼ(glycosyitransferase)に対して、13μgのこのプラス
ミドを添加し、そして必要な場合、合計39μgのプラスミドDNAに達するようにCD
M8プラスミドを添加した。この混合物を室温で10分間静置し、その後、10mlの培
養培地中に入れて、約70%コンフルエンシーな細胞に添加した。7日後、細胞上清を収
集し、細片をスピンダウン(1400×g、15分間)し、そしてNaN3を、0.02
%(w/v)の最終濃度になるように添加した。
【0081】
(SDS−PAGEおよびウェスタンブロット分析のための、分泌されたPSGL−1
/mIgG2bの精製)
PSGL−1/mIgG2b融合タンパク質を、4℃で一晩回転させることによって、
50μlヤギ抗mIgGアガロースビーズ(100:1のスラリー;Sigma)によっ
て、収集した上清から精製した。融合タンパク質を有するビーズをPBS中で3回洗浄し
、そしてその後の分析のために用いた。代表的には、サンプルを50μlの2×還元サン
プル緩衝液中に溶解し、そして10:1のサンプルを各ウェルにローディングした。
【0082】
(アフィニティー精製したPSGL−1/mIgG2bのPNGaseF処理)
PNGaseFキット(Roche Diagnostics,Indianapol
is,IN)を、N−グリカンの脱グリコシル化のために用いた。製造業者によって提供
されたプロトコルのわずかな改変版を用いた。1.5mlのEppendorfチューブ
中で、20μlの反応緩衝液を、アガロースビーズ上の精製したPSGL−1/mIgG
2bと混合し、そして3分間煮沸した。この混合物をスピンダウンし、そして10μlの
上清を新たなEppendorfチューブに移した。10μlのPNGaseFまたはネ
ガティブコントロールとして10μlの反応緩衝液を添加した。チューブを、1.5時間
にわたって37℃でインキュベートした。インキュベーション後、20μlの2×還元サ
ンプル緩衝液および10μlのH2Oを添加し、そしてサンプルを3分間煮沸した。
【0083】
(上清中のPSGL−1/mIgG2b濃度の決定のためのELISA)
96ウェルELISAプレート(Costar 3590,Corning,NY)を
、50μlの50mM炭酸緩衝液(pH9.6)中の0.5μg/ウェルのアフィニティ
ー精製したヤギ抗mIgG特異性抗体(Sigma)で、室温で2時間にわたってコーテ
ィングした。0.05% Tween(PBS−T)を含むPBS中の3%ウシ血清アル
ブミン(BSA)300μlを用いて4℃で一晩ブロッキングし、続いて洗浄した後、培
養培地中に連続希釈した、50μlのサンプル上清を添加した。洗浄後、これらのプレー
トを、ブロッキング緩衝液中に10,000:1希釈したヤギ抗mIgM−HRP(Si
gma)50μlとともに2時間にわたってインキュベートした。発色溶液のために、1
錠の3,3’,5,5’−テトラメチルベンジジン(Sigma)を、3μlの30%(
w/v)H2O2を含む11mlの0.05Mクエン酸/リン酸緩衝液中に溶解した。1
00μlの発色溶液を添加した。反応を、25μlの2M H2SO4を用いて停止させ
た。これらのプレートを、自動化マイクロプレートリーダー(Bio−Tek Inst
ruments,Winooski,VT)において450nmおよび540nmで読み
取った。標準として、培養培地中の精製mIgG Feフラグメント(Sigma)の希
釈シリーズを三連で用いた。
【0084】
(SDS−PAGEおよびウェスタンブロッティング)
SDS−PAGEを、5%スタッキングゲルおよび8%分離ゲルを用いるLaemml
i(1970)の方法によって行い、そして分離されたタンパク質を、以前(Liuら,
1997)に記載されるとおりに、HybondTM−Cエキストラメンブレン上に電気
泳動的にブロッティングした。3% BSAを含む、0.05% Tween−20を含
むTris緩衝化生理食塩水(TBS−T)中での一晩のブロッキング後、これらのメン
ブレンを、TBS−Tで3回洗浄した。次いで、これらを、マウス抗ヒト血液型A全種類
(mIgM,Dako,Carpinteria,CA)または抗ヒトH型1(mIgG
3,Signet;Dedham,MA)、抗ヒトH型2(mIgM,Dako)もしく
は抗ヒトH型3(mIgM,ハイブリドーマHH14,ATCC HB9299)ととも
に、1時間にわたって室温でインキュベートした。H型3抗体以外の全ての抗体を、TB
S−T中の3% BSA中に1:200希釈した。H型3抗体を、TBS−T中の3%
BSAにおいて1μg/mlの濃度になるように希釈した。これらのメンブレンを、TB
S−Tで3回洗浄し、その後、TBS−T中の3% BSAに2000:1希釈した、西
洋ワサビペルオキシダーゼ(HRP)結合体化二次抗体である、ヤギ抗mIgM(Cap
pel,Durham,NC)またはヤギ抗mIgG3(Serotec,Oxford
,England)とともに1時間にわたって室温でインキュベートした。結合した二次
抗体を、製造業者の指示に従って、ECLキット(Amersham Pharmaci
a Biotech,Uppsala,Sweden)を用いた化学発光によって可視化
した。PSGL−1/mIgG2b自体の検出のために、HRP標識ヤギ抗mIgG(S
igma)を、二次抗体とのインキュベーションを行わないこと以外は上記のとおりに、
TBS−T中の3% BSAにおける1:10,000の希釈で用いた。
【0085】
(PSGL−1/mIgG2bによる相対血液型Aエピトープ密度の決定)
ウェスタンブロットを、上記の通りに行った。メンブレンを、−35℃で作動するCC
Dカメラを備えるFluor−S Max Muitlmager(BioRad)にお
いて可視化した。Quantity Oneソフトウェア(BioRad)の分析ウィン
ドウにおいて量ツールを用いて、血液型A反応性についての量(量境界xピクセル領域内
のピクセルの強度の合計)を、COS細胞、CHO細胞および293T細胞中で作製した
PSGL−1/mIgG2bについてのmIgG反応性量で除算した。異なる宿主細胞中
で作製したPSGL−1/mIgGb2b間でAエピトープ/マウスIgG比を比較する
ために、比を、H遺伝子およびA遺伝子でトランスフェクトされたCOS細胞中で作製さ
れたA置換PSGL−1/mIgG2bから得られた比に対して正規化した。
【0086】
(血清の吸収)
ヤギ抗mIgGアガロースビーズ(Sigma)の600マイクロリットルのスラリー
を、1.5mlのEppendorf微量遠心管に移した。これらのビーズを、400×
gでのすばやいスピンによってスピンダウンした。次いで、上清を除去し、そしてこれら
のビーズを1ml PBSで1回洗浄し、再度スピンダウンし、そしてPSGL−1/m
IgG2bをコードするcDNA(Se遺伝子およびA遺伝子)でトランスフェクトされ
たCHO細胞由来の180mlの上清に移した。アガロースビーズを含む上清を、4℃で
一晩、回転させながらインキュベートした。収集のために、これらのビーズを400×g
で15分間、室温でスピンダウンし、そして1.5mlのEppendorf微量遠心管
に移した。PBSでの洗浄を3回行った。これらのビーズは、Aムチンビーズと呼ばれる
。600マイクロリットルの抗mIgGアガロースビーズをまた、Aムチン−ビーズの希
釈のために用いて吸収剤としてのAムチン−ビーズの希釈系列を得たこと以外は、同じ様
式で調製した。これらのビーズは、ヤギ抗mIgGビーズと呼ばれる。これらのビーズを
、4mlのEllermanチューブ中に、表Iに従ってアリコートに分けた。A−PA
A−MPG(Syntesome,Munich,Germany)およびB−PAA−
MPG(Syntesome)を計量し、そしてEllermanチューブ中に表Iに従
ってアリコートに分け、その後、PBSで1回洗浄した。
【0087】
血液型O型の5人の患者由来のプールした血清を、Huddinge Univers
ity Hospitalの血液銀行(倫理的許可、Dnr.392/99、承認日08
/15/2000)から入手した。細胞破片を、Jouan A−14微量遠心機での1
4,000rpmでの5分間の遠心分離によって血清から除去した。透明にされた血清を
別のチューブに移し、そして水浴中で56℃で1時間インキュベートして、補体を不活化
した。血清を、使用するまでアリコートにして−20℃で保存した。
【0088】
【表1】
500マイクロリットルの血清を、各チューブに添加し、そしてこれらのビーズと、ロ
ーリングテーブル上で4℃で4時間混同した。吸収後、これらのビーズをスピンダウンし
、そして吸収された血清を新たなEllermanチューブに移した。吸収された血清を
、さらなる分析まで、−20℃で保存した。
【0089】
Aムチン−ビーズ上でのPSGL−1/mIgG2bの量を決定するために、ヤギ抗m
IgG Fc ELISA(上記の手順を参照のこと)を、アガロースビーズとのインキ
ュベーション前の上清およびインキュベーション後の上清について行った。
【0090】
(抗A抗体の定量のためのELISA)
96ウェルELISAプレート(Costar 3590,Corning)を、1ウ
ェルあたり50μlの50mM炭酸緩衝液(pH9.6)中0.05μlのA−PAA−
ビオチン(Syntesome)で2時間、室温でコーティングした。PBS−T中の3
% BSA(300μl)を用いた4℃での一晩のブロッキングおよびその後の洗浄後、
PBS中に連続希釈した50μlの血清を添加し、そしてこのプレートを室温で2時間イ
ンキュベートした。洗浄後、ブロッキング緩衝液中に10,000:1希釈したマウス抗
ヒトIgA−HRP、マウス抗ヒトIgG−HRPおよびマウス抗ヒトIgM−HRP(
Jackson,PA)(50μl)とともにインキュベーションを行った。このプレー
トの発色および読取りを上記の通りに行った。
【0091】
(血清中における総タンパク質濃度の決定)
血清中における総タンパク質濃度を、抗A抗体の吸収の前後に決定した。BCAタンパ
ク質アッセイ試薬のマイクロタイタープレートプロトコール(Pierce,Rockf
ord,IL)を製造業者の指針書に従って使用し、そしてこのサンプルを二連または三
連で実施した。
【0092】
(実施例2:血液型HおよびAを、種々の宿主細胞中で作製した組換えPSGL−1/
mIgG20において決定する)
293T細胞。免疫親和性により精製したPSGL−1/mIgG2b(H遺伝子、S
e遺伝子、またはA遺伝子をコードするプラスミドを含むか、または含まないPSGL−
1/mIgG2b cDNAで一過性でトランスフェクトした293T細胞により産生し
た)を、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS−PAGE
)ならびに検出のために抗体に特異的な抗mIgG、抗血液型A、H1型、H2型、およ
びH3型を使用するウエスタンブロットによって分析した(図1A〜E)。この融合タン
パク質を、見掛け上の分子量約100および140〜160kDa(A)を用いる二連の
還元状態下で移動させた。このPSGL−1/mIgG2bは、高度にグリコシル化され
たムチン型タンパク質の挙動に関して、上記観察に従って銀でわずかに染色した(示さず
)(CarrawayおよびHull,1991;ShimizuおよびShaw,19
93)。上に示されるように(Liuら,1997)、この融合タンパク質は、ホモダイ
マーとして産生した(データは示さず)。65kDaおよび35kDa付近の二本のバン
ドは、抗mIgG抗体で決定された。これらのバンドは、エンプティーベクターのみでト
ランスフェクトされた293T細胞の上清には見られず、従って、融合タンパク質由来の
タンパク質フラグメントであるようであった(CarrawayおよびHull,199
1)。細胞フラクション中においてみられた、この47kDaおよび20kDaのバンド
は、サンプル緩衝液中で沸騰させることでアガロースビーズから脱離させた、ヤギ抗マウ
ス抗体の重鎖Igおよび軽鎖Ig由来である。
【0093】
αl,3GalNAcTを有するFUT1およびFUT2の両方は、PSGL−1/m
IgG2b(B)上の血液型A鎖の生合成を支持する。FUT2とαl,3GalNAc
Tとの組み合わせは、FUT1とαl,3GalNAcTとの組み合わせよりも、PSG
L−1/mIgG2bにおいてよりAエピトープを産生するようであった(Bにおけるこ
れらのレーンを参照)。一方、FUT1のみは、大量のH2型構造(D)の発現を支持し
たが、FUT2は、ムチン/Ig(D)において、わずかなH2型エピトープを生じた。
PSGL−1/mIgG2bにおいて、検出可能なH1型構造は、存在しか生じなかった
。FUT2により産生された、このHエピトープは、ほとんど排他的に型3(すなわち、
Fucα2Galβ3GalNAcα−R)に基づく。なぜならば、H1型構造およびわ
ずかなH2型構造が、この酵素(E)によってPSGL−1/mIgG2bにおいて作製
されたからである。興味深いことに、A遺伝子でのH遺伝子またはSe遺伝子の両方の同
時トランスフェクションは、抗H3型抗体(E)と反応性のPSGL−1/mIgG2b
において大量のエピトープを生じる。血液型A、H2型およびH3型エピトープは、主に
O連結された。なぜならば、PSGL−1/mIgG2bのペプチドNグリコシダーゼF
(PNGaseF)処置は、これらのエピトープに対して特異的な抗体を使用する、抗体
染色を減少させたからである(図2B〜D)。効率的なN−グルカン脱グリコシル化は、
PSGL−1/mIgG2bの完全な移動シフトによって示された(図2A〜D)。
【0094】
COS細胞。COS細胞において作製されたPSGL−1/mIgG2bのウエスタン
ブロット分析は、図3A〜Eにおいて示される。抗A特異的抗体および抗H特異的抗体を
使用して染色するPSGL−1/mIgG2bのパターンは、293T細胞において作製
されたPSGL−1/mIgG2bに対して見出されたパターンに、弱いがいくらか類似
していた。α1,3GalNacTを一緒に含むFUT1およびFUT2の両方は、Aエ
ピトープ発現を支持し、より多量のエピトープが、FUT2およびα1,3GalNAc
T(B)によって産生された。FUT1のみが、H2型構造(D)を作製し得、そしてF
UT1もFUT2も、COS細胞(C)において、H1型構造の発現を支持しなかった。
293T細胞において作製されたPSGL−1/mIgG2bに対して、非常に低レベル
のH3型エピトープが、FUT1およびα1,3GalNAcT、またはFUT2酵素の
み(E)を発現するCOS細胞において産生されたPSGL−1/mIgG2bにおいて
見られた。
【0095】
しかし、FUT2およびα1,3GalNAcTのジョイント発現(joint ex
pression)は、H3型抗体(E)との増加した反応性を生じた。さらに、弱いバ
ンドは、α1,2FTのみが発現された場合、抗A抗体で見出され、上記で報告したよう
に、COS細胞において、α1,3GalNAcTの弱い内因性の活性を示した(Cla
rkeおよびWatkins,1999)。PSGL−1/mIgG2bのPNGase
F処理は、抗血液型A、H2型またはH3型抗体の染色における任意の検出可能な減少を
示さず(データは示さず)、このことは、これらのエピトープが、PSGL−1/mIg
G2bOグリカン上で主に保持されることを示す。
【0096】
(チャイニーズハムスター卵巣(CHO)細胞)。図4A〜Eは、CHO細胞において
作製されたPSGL−1/mIgG2bの染色、FUT1またはFUT2 cDNAとα
1,3GalNAcT(B)との同時発現後のPSGL−1/mIgG2b保持血液型A
エピトープの染色を示す。H1型(C)構造もH2型(D)構造も、FUT1またはFU
T2のいずれかとの同時トランスフェクション後に、CHO細胞において作製されたPS
GL−1/mIgG2bキメラ上で検出され得ず、このことは、H3型構造が、α1,3
GalNAcTに対して利用可能な唯一の前駆体であることを示した。抗H3型抗体を用
いた染色は、FUT1およびFUT2(B)を用いて見出され、Aエピトープが、CHO
細胞におけるコア1構造に唯一基づくという理論を支持する。さらに、このことに対する
支持は、PNGaseF処置後に得られ、このことは、抗体染色強度に影響を及ぼさず、
Oグリカン制限Aエピトープ発現を示す(データは示さず)。PSGL−1/mIgG2
bキメラ上におけるAエピトープおよびH3型エピトープの数は、FUT1と比較した場
合、FUT2で明らかにより多く、このことは、このSe遺伝子産物が、コア1(Gal
β3GalNAcα−Ser/Thr)構造のα1,2−フコシル化の点において、H遺
伝子産物よりも優れていることを示唆する。
【0097】
(実施例3.異なる宿主細胞において産生されたPSGL−1/mIgG2bにおける
、血液型Aエピトープの相対密度)
A遺伝子をα1,2FTのいずれかと発現する種々の宿主細胞において作製したPSG
L−1/mIgG2bキメラにおけるAエピトープの相対数を半定量するために、抗A抗
体および抗mIgG抗体を用いたウエスタンブロット、続いてFluor−S(登録商標
)Max MultImagerでの化学ルミネセンス検出を使用した。各PSGL−1
/mIgG2bに対する、血液型AおよびmIgGの反応性の比を、図5に示す(3つの
代表的な実験のうちの1つを示した)。理解されるように、Aエピトープの密度は、α1
,2FTをコードするSe遺伝子とともにA遺伝子を発現するCHO細胞において作製さ
れたPSGL−1/mIgG2bにおいて最も高かった。このPSGL−1/mIgG2
bは、FUT2およびA遺伝子を用いてトランスフェクトされた293T細胞において製
造されたPSGL−1/mIgG2bよりも約3倍多量のAエピトープを保持する;PS
GL−1/mIgG2bは、2番目に高いAエピトープ密度を有する(図5)。各細胞レ
ーンにおいて、A遺伝子をともに有するFUT1よりも、A遺伝子をともに有するFUT
2は、より高いAエピトープ密度を与えた。
【0098】
(実施例4:血液型Aエピトープを保持するPSGL−1/mIgG2bにおける抗A
抗体の吸収)
血液型Aエピトープを有するか、または有さない組換えPSGL−1/mIgG2bに
おける抗A抗体吸収の効率を、マクロ孔質ガラスビーズに対するポリ[N−(2−ヒドロ
キシエチル)アクリルアミド]介して連結されたAトリサッカライド(A−PAA−MP
G)上での吸収の効率と比較した。抗A抗体の吸収前および吸収後のレベルを、酵素結合
イムノソルベント検定法(ELISA)において評価し、このプレートを、ビオチンでも
また置換したポリ[N−(2−ヒドロキシエチル)アクリルアミド]に連結させたAトリ
サッカライド(A−PAA−ビオチン)でコーティングした。この結果を、図6に示す。
20pmolの組換えAエピトープ置換PSGL−1/mIgG2bが、A−PAA−ビ
オチンELISAにおいて検出される場合に、抗A抗体の60%を吸収するために必要と
され、一方、A−PAA−MPGのような164,000pmolのA決定基は、同量の
抗A抗体を吸収するために必要とされた。このPSGL−1/mIgG2bダイマーは、
106ヶ所の強力なO連結グリコシル化部位および8ヶ所の強力なN連結グリコシル化部
位を有し(Wilkinsら,1996;Aeedら,1998,2001)、この後者
の8ヶ所は、分枝構造を保持し得る。各PSGL−1/mIgG2bが、約100のAエ
ピトープを保持すると推定する場合、これらのほとんどは、推定を超えるようであり、F
UT2およびα1,3GalNacTを有するCHO細胞において作製されたムチンは、
A−PAAF−MAPよりも、カルボキシレートモルを基準として約80倍以上の効率を
有する。
【0099】
(実施例5:抗体媒介性移植拒絶の処置または予防)
ABO障壁を横切る組織移植を、ドナー組織血液型に対して予め形成されるか、または
誘導された抗体により媒介されたAMRによって特徴付する(Porter,1963;
Sanchez−Uradazpalら,1993;Fargesら,1995;Tan
abeら,1998;Alkhunaiziら,1999)。抗ABO抗体を、EIA設
定における吸収剤としてA決定基を保持するPSGL−1/mIgG2b融合タンパク質
を使用して、除去する。結果は、(100のA決定基/molのPSGL−1/mIgG
2bに基づく)2nmolのA決定基に対応する、FUT2およびα1,3GalNAc
Tを含むCHO−K1中で作製された約20pmolの組換えPSGL−1/mIgG2
b(分子量300kDaとして計算された)(Wilkinsら,1996;Aeedら
,1998,2001)は、A−PAA−反応性抗体の60%を吸収する。約164,0
00pmolのAトリサッカリドに対応するA−PAA−MPG(2μmol/μgとし
て計算された)が、同量の抗A抗体を吸収するために必要とされた。両方の化合物にして
非特異的なタンパク質吸収量もまた評価し、そしてこの非特異的な吸収が、PAA−MP
Gベース化合物よりほぼ4倍高かった。ムチンベース吸収の比較的高度な吸収効率は、(
1)多価カルボキシレート置換、(2)カルボキシレートエピトープの密接した空間、お
よび(3)主要抗原決定基を保持するコアサッカライド鎖の構造多能性に依存し得る。さ
らに、例えば、最適化されたムチンタンデム反復によって構成された合成ムチン型タンパ
ク質を作製することによる、タンパク質骨格の変化は、吸収体を改善し得る(Silve
rmanら,2001)。また、GalNAc:ポリペプチドトランスフェラーゼの異な
る組み合わせを発現するように操作された細胞は、Oグリカン置換密度を最適化すること
によって吸収効率を改善し得る。
【0100】
(略語)
AMR(抗体媒介拒絶(antibody−mediated rejection)
);BSA(ウシ血清アルブミン);CHO(チャイニーズハムスターの卵巣);COS
;ETA(体外免疫吸収);ELISA(酵素結合イムノソルベント検定法);FT(フ
ルコシルトランスフェラーゼ);GalNAcT(N−アセチルガラクトサミニルトラン
スフェラーゼ);HRP(西洋ワサビペルオキシダーゼ);mIgG(マウスIgG);
mIgM(マウスIgM);MPG(マクロ孔質ガラスビーズ);PAA(ポリ[N−(
2−Hドロキシエチル)アクリルアミド]);PBS(リン酸緩衝生理食塩水);PBS
−T(0.05% Tweenを含むPBS);PCR(ポリメラーゼ連鎖反応);PN
GaseF(ペプチド:N−グリコシダーゼF);PP(血漿瀉血);PSGL−1(P
セレクチン糖タンパク質リガンド−1);SDS−PAGE(ドデシル硫酸ナトリウム−
ポリアクリルアミドゲル電気泳動);TBS−T(0.05%のTween−20を含む
Tris緩衝化生理食塩水);Tx(移植)。
【0101】
(参考文献)
【0102】
【表2−1】
【0103】
【表2−2】
【0104】
【表2−3】
【0105】
【表2−4】
【0106】
【表2−5】
【0107】
【表2−6】
【0108】
【表2−7】
【0109】
【表2−8】
(他の実施形態)
本発明は、その詳細な説明と組み合わせて議論されるが、上記の記載は、本発明を例示
するものであって、その範囲を限定するものではないことを意図し、この範囲は、添付の
特許請求の範囲によって規定される。他の局面、利点および改変は、以下の特許請求の範
囲内である。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

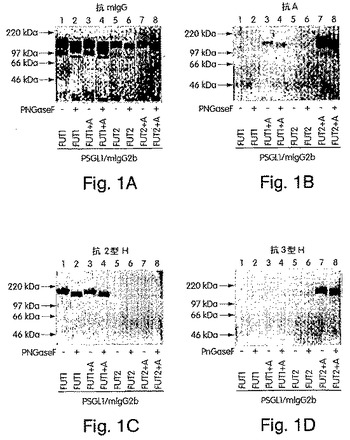
【図2】
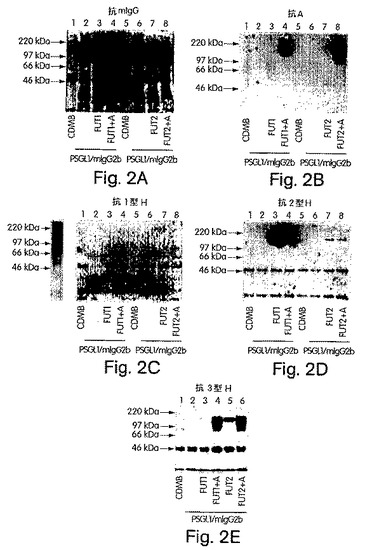
【図3】
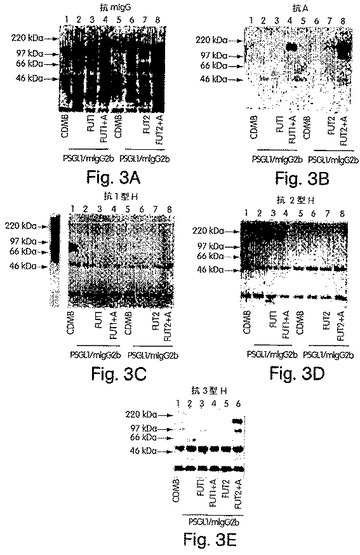
【図4】
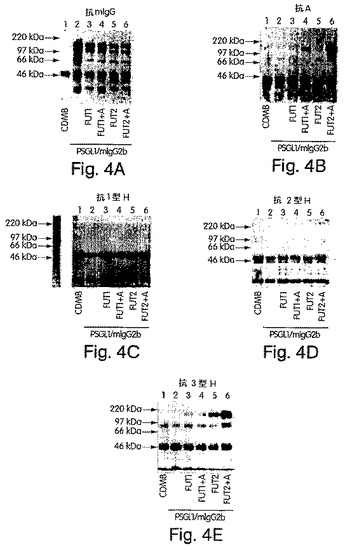
【図5】
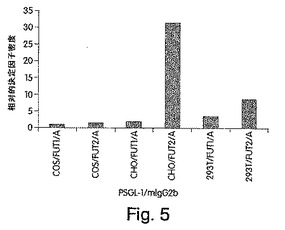
【図6】
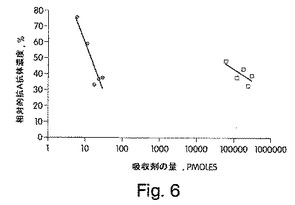
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2009−131279(P2009−131279A)
【公開日】平成21年6月18日(2009.6.18)
【国際特許分類】
【出願番号】特願2009−62104(P2009−62104)
【出願日】平成21年3月13日(2009.3.13)
【分割の表示】特願2003−515560(P2003−515560)の分割
【原出願日】平成14年7月19日(2002.7.19)
【出願人】(502070521)アブソーバー アクチボラゲット (2)
【Fターム(参考)】
【公開日】平成21年6月18日(2009.6.18)
【国際特許分類】
【出願日】平成21年3月13日(2009.3.13)
【分割の表示】特願2003−515560(P2003−515560)の分割
【原出願日】平成14年7月19日(2002.7.19)
【出願人】(502070521)アブソーバー アクチボラゲット (2)
【Fターム(参考)】
[ Back to top ]